/































































































































































































































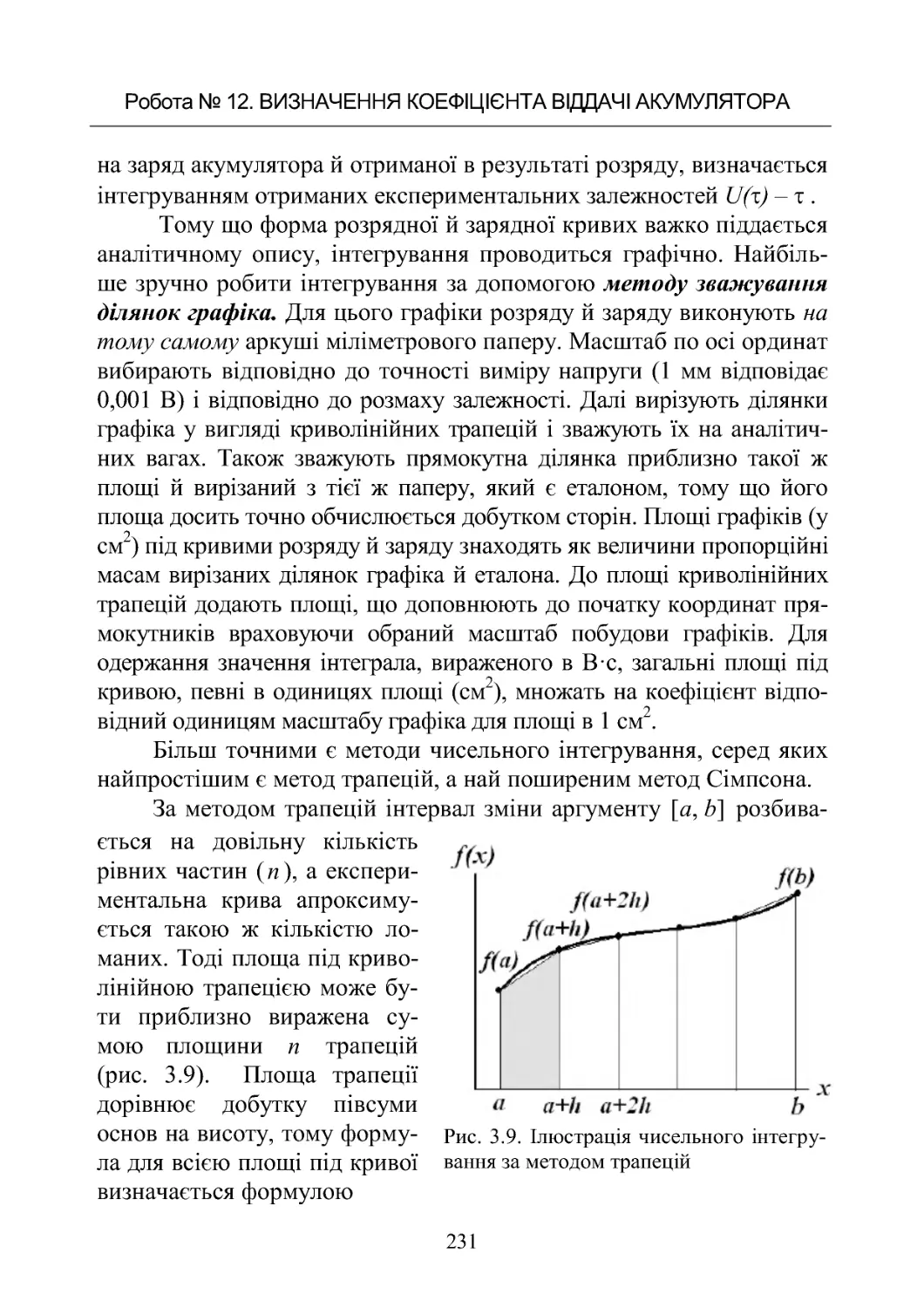




















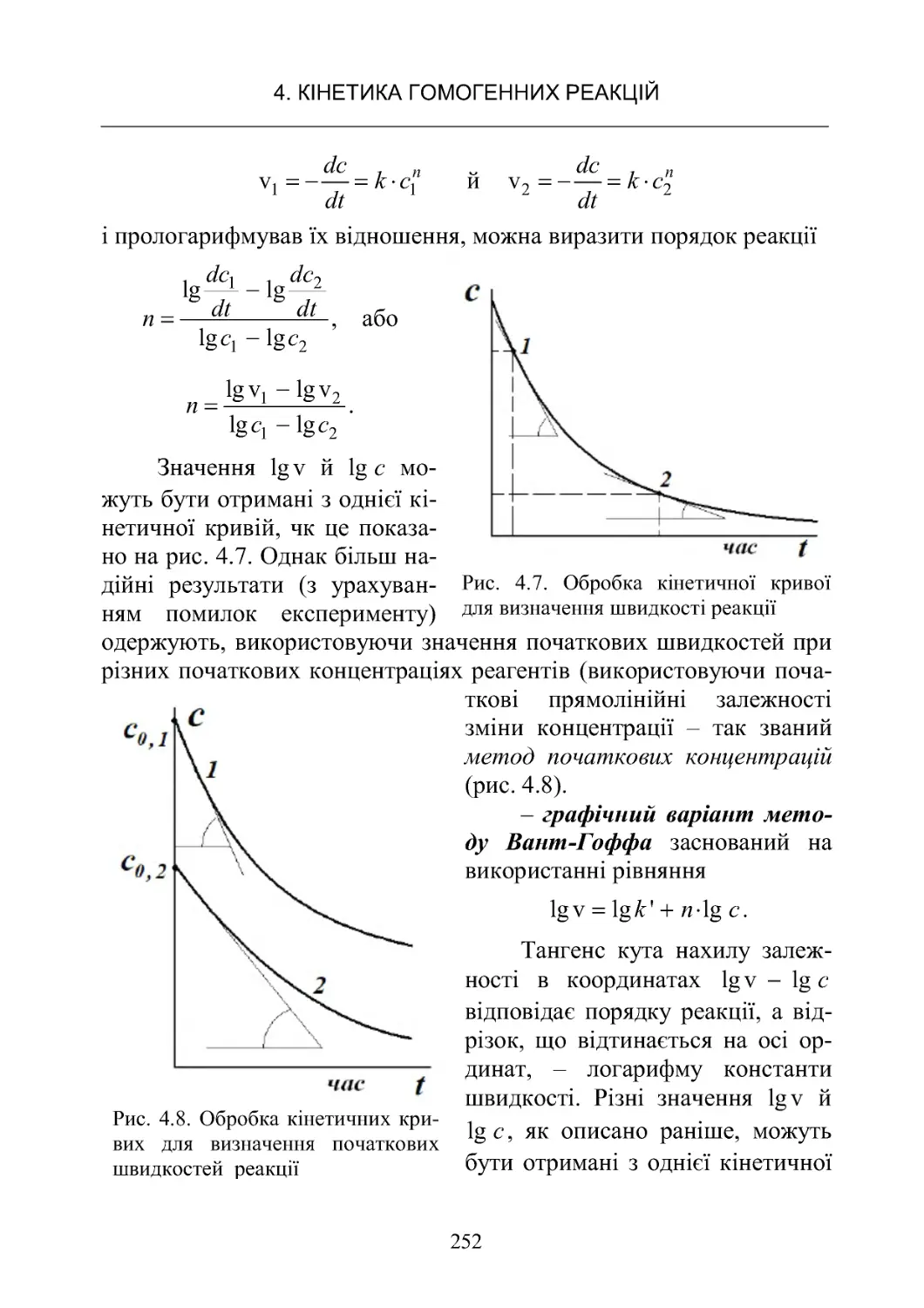






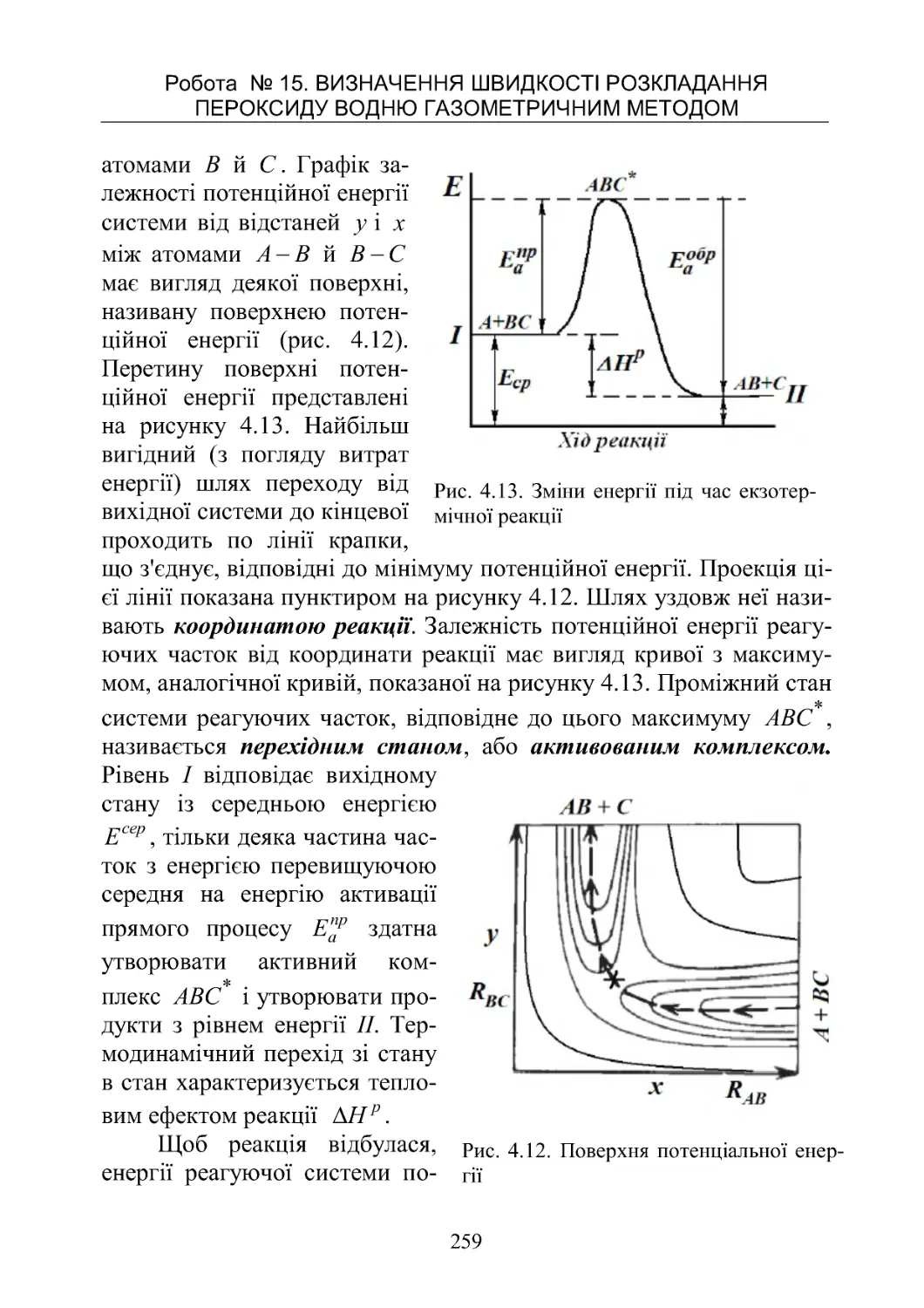




























































































Текст
Харківський національний університет
імені В. Н . Каразіна
В. І. Рубцов
ЛАБОРАТОРНИЙ ПРАКТИКУМ
З ФІЗИЧНОЇ ХІМІЇ
Частина 2
Розчини електролітів
Електропровідність
Гальванічні елементи
Хімічні джерела струму
Хімічна кінетика
Каталіз
Навчальний посібник
МІНІСТЕРСТВО ОСВІТИ Й НАУКИ УКРАЇНИ
Харківський національний університет імені В. Н. Каразіна
В. І. Рубцов
Частина 2
Розчини електролітів
Електропровідність розчинів
Гальванічні елементи
Хімічні джерела струму
Хімічна кінетика
Каталіз
Харків – 2020
УДК 544.6(075.8)
ББК 24.57я73
Рецензенти:
Затверджене до друку рішенням Вченої ради Харківського національного
університету імені В.Н . Каразіна (протокол No від 2020 р.).
Р82 Рубцов В. І. Лабораторний практикум з фізичної хімії : навчальний
посібник. В 2–х кн . Ч. 2. – Харків : Вид. ХНУ імені В. Н. Каразіна, 2020.
–
372 с., іл., бібл. –
55 найм.
Навчальний посібник призначений для виконання лабораторних робіт
загального курсу фізичної хімії на хімічних факультетах університетів. Друга
частина посібника включає розділи: розчини електролітів, електропровідність
розчинів, гальванічні елементи, хімічні джерела струму, хімічну кінетику й
каталіз У кожному розділі викладені базові поняття й теоретичні основи,
порядок проведення досліджень, описані прилади для фізико -хімічних
досліджень, порядок проведення розрахунків і оформлення результатів.
Наведені сучасні методи обробки експериментальних даних фізико-хімічних
досліджень із використанням персональних комп'ютерів.
Посібник складений відповідно до програми курсу фізичної хімії для
хімічних факультетів університетів і буде корисним для студентів хімічних
і хіміко-технологічних спеціальностей вищих навчальних закладів, аспірантів і
викладачів.
УДК 544.6(075.8)
ББК 24.57я73
ISBN 978–966 –285–276–9 © В. І. Рубцов. 2020
© Харківський національний університет імені
В. Н. Каразіна, 2020
ЗМІСТ
3
ЗМІСТ
ЗМІСТ................................................. 3
ПЕРЕДМОВА.......................................... 7
ПРАВИЛА РОБОТИ В ХІМІЧНІЙ ЛАБОРАТОРІЇ Й ТЕХНІКА
БЕЗПЕКИ.............................................. 8
ВИМОГИ ДО ВИКОНАННЯ Й ОФОРМЛЕННЮ ЛАБОРАТОР-
НИХРОБІТ............................................. 10
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ
РОЗЧИНІВЕЛЕКТРОЛІТІВ............................. 12
1.1 . Електролітична дисоціація, іонні рівноваги, термодина-
мічнівластивості...................................... 12
1.2. Електрична проівність розіинів електроліиів . . . . . . . . . . . 27
1.2.1. Вимір електричної провідності розчинів електролітів 39
1.2.2. Готування розчинів для електрохімічних досліджень 45
Робота No 1. Визначення розчинності малорозчинних спо-
луккондуктометричнимметодом .....................
51
Робота No 2. Визначення граничної еквівалентної електро-
провідностісильнихелектролітів...................... 55
Робота No3.Кондуктометричнетитрування............ 59
Робота No 4. Визначення константи дисоціації слабкої кис-
лоти методом електропровідності її розчинів . . . . . . . . . . . . 64
Робота No 5. Визначення чисел переносу іонів у розчині
сульфатноїкислоти ................................. 73
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЕЛЕКТРОРУШІЙНІ СИЛИ 84
2.1.Класифікаціягальванічнихелементів................ 84
2.2.Типиелектродів................................... 93
2.3 . Вимір електрорушійної сили гальванічних елементів. . . . 105
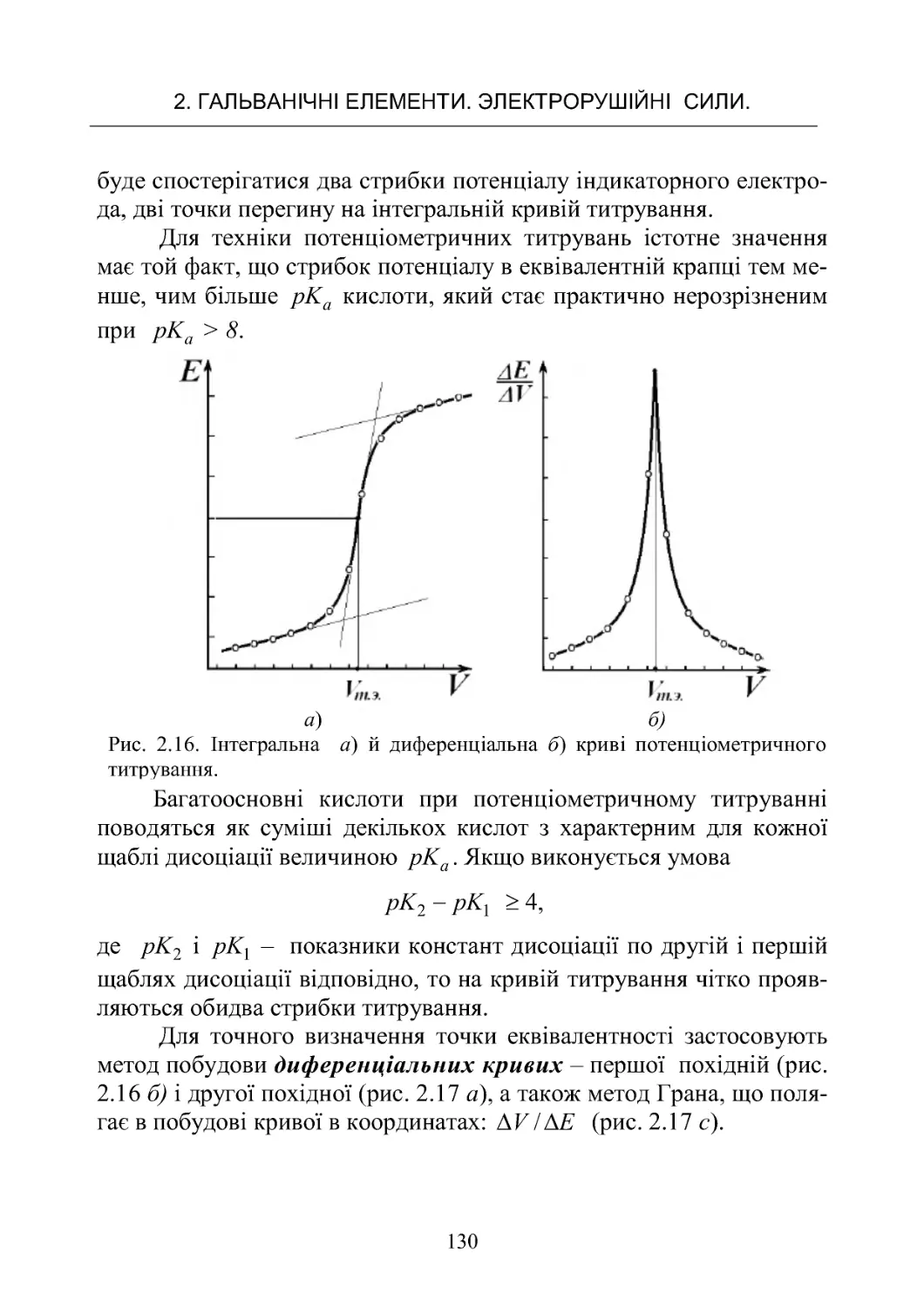
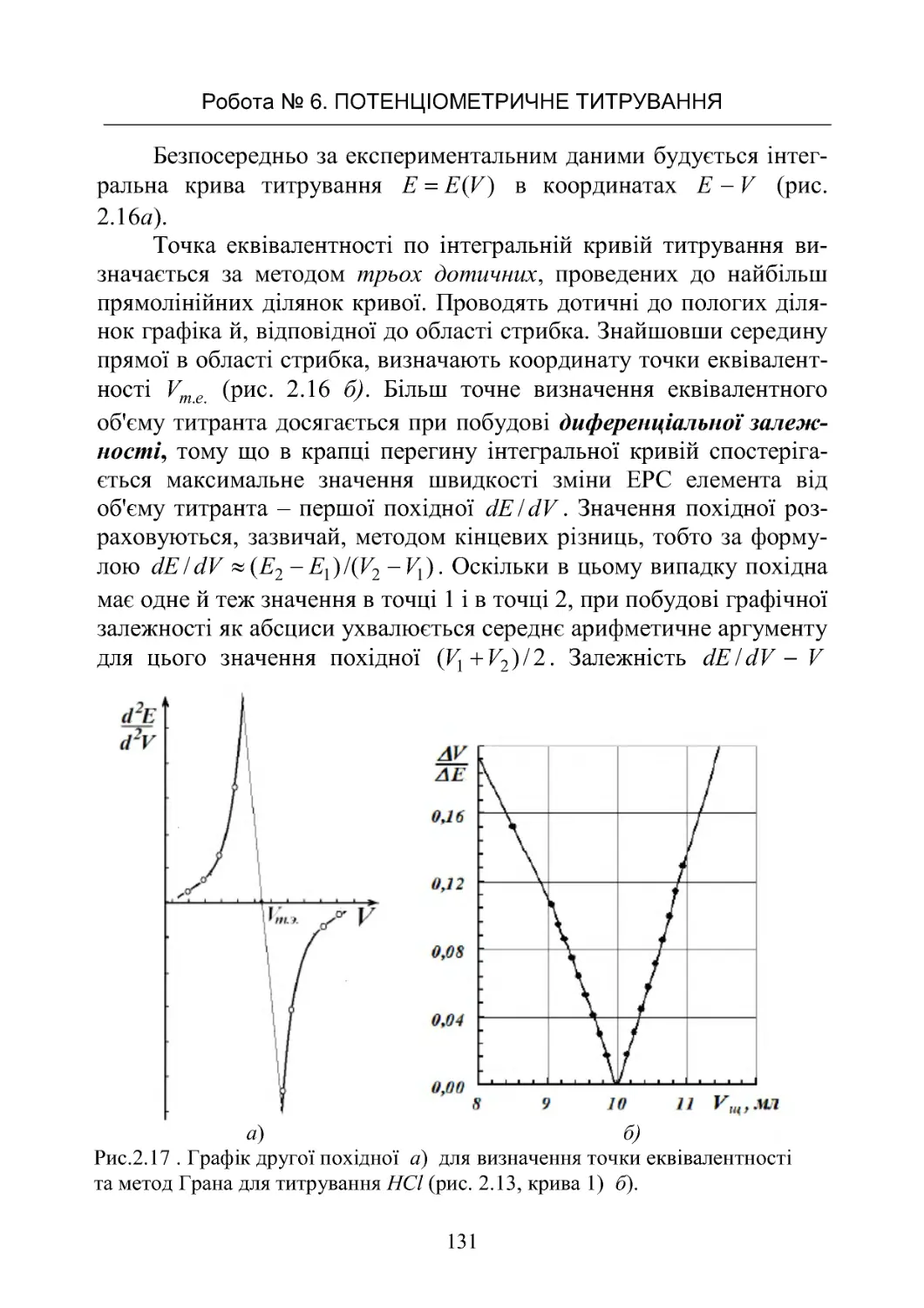
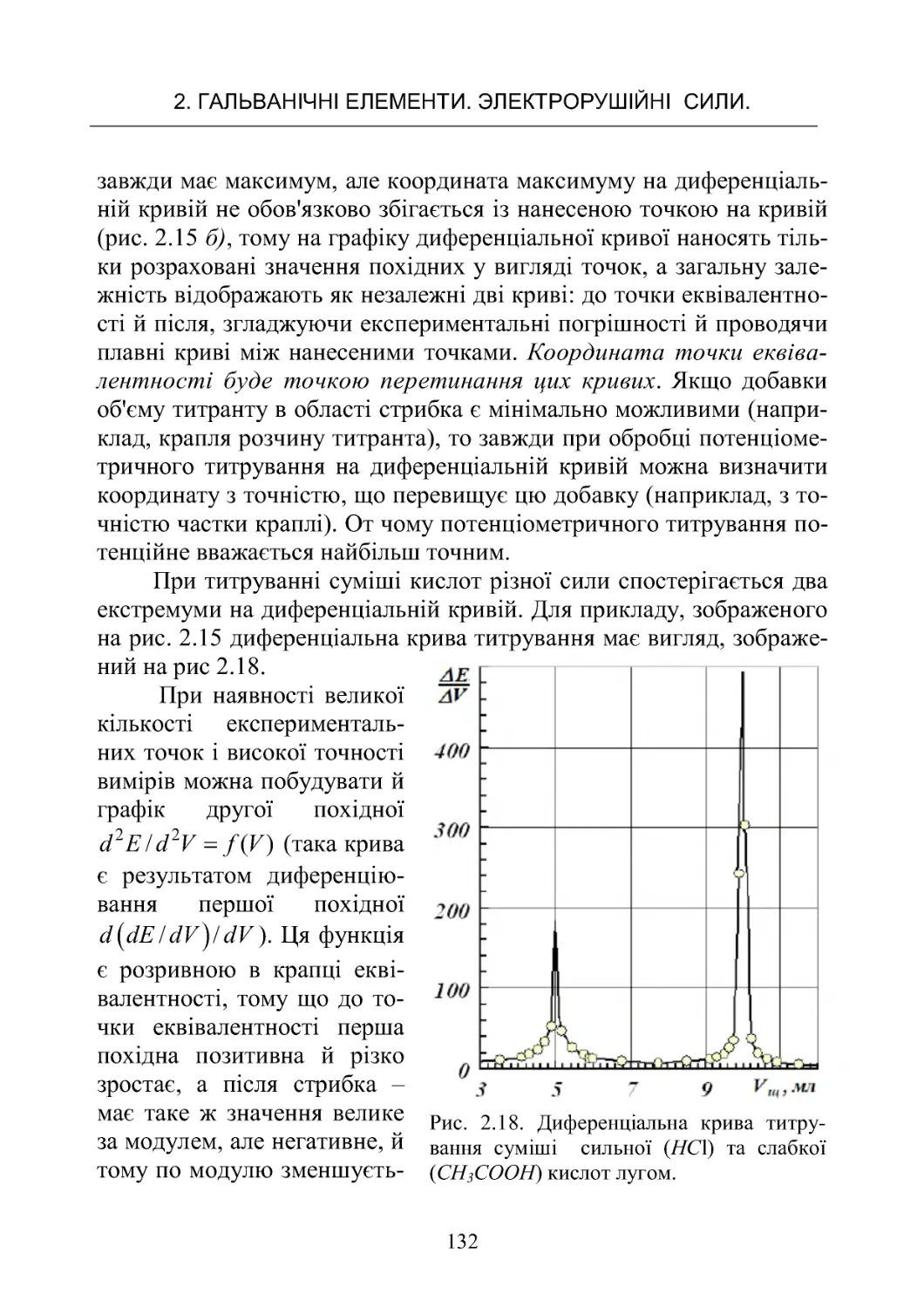
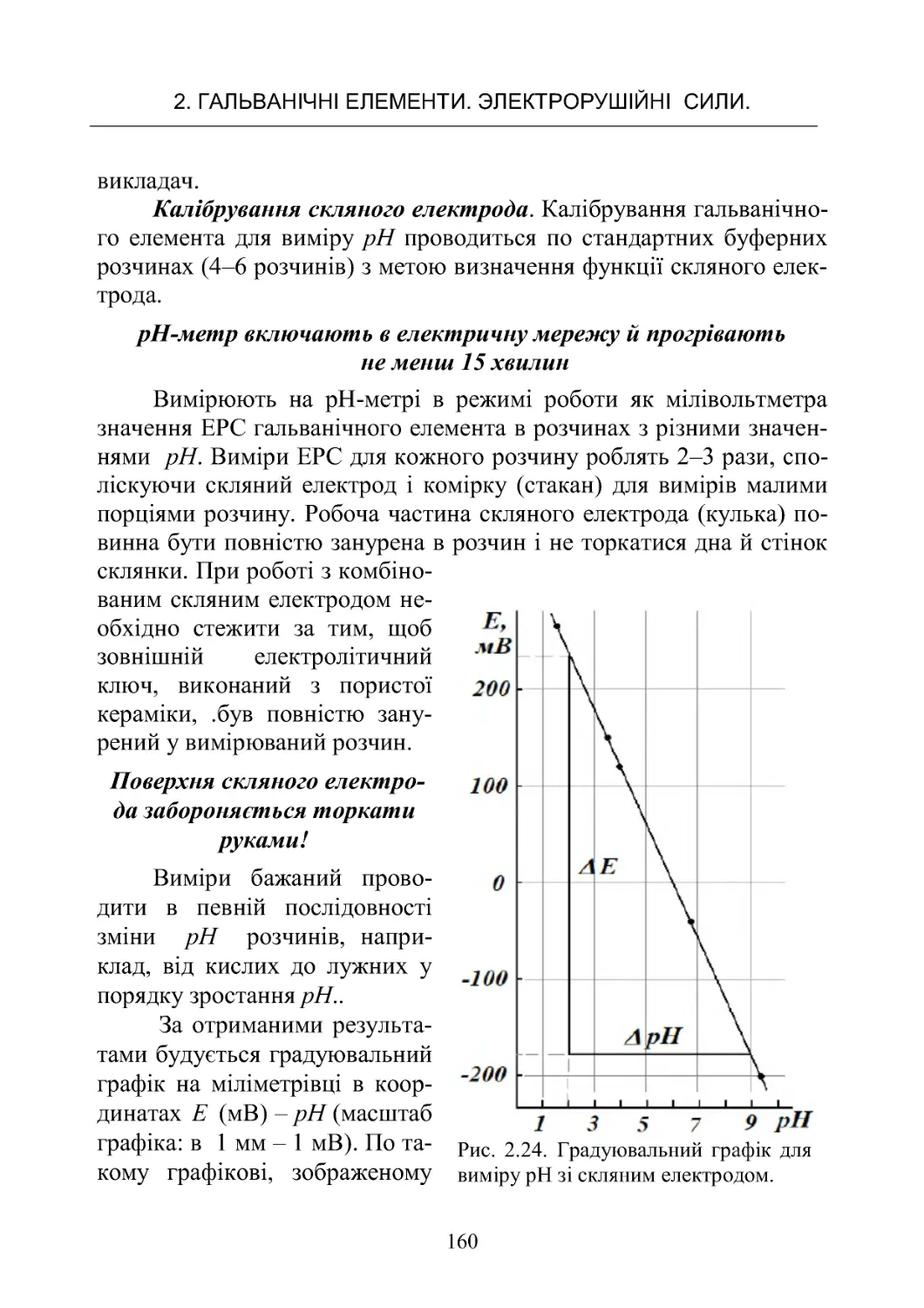
РоботаNo6.Потенціометричнетитрування ............. 125
Робота No 7. Визначення іонного добутку води в ланцюгах
безпереносу........................................ 142
Робота No 8. Потенціометричне визначення рН і досліджен-
нябуфернихвластивостейрозчинів.................... 151
Робота No 9. Визначення коефіцієнтів активності сильних
електролітівпотенціометричнимметодом............... 168
ЗМІСТ
4
Робота No 10. Визначення крнстанти дисоціації слабкої кис-
лоти в буферних розчинах у ланцюгах без переносу . . . . . 182
Робота No 11. Визначення добутку розчинності малороз-
чинної солі срібла
z
AgA.............................. 190
3.ХІМІЧНІДЖЕРЕЛАСТРУМУ......................... 198
3.1.Типийбудовахімічнихджерелструму................198
3.2.ЕлектричніхарактеристикиХДС..................... 201
3.3.Первинніджереластруму.......................... 207
3.4.Вторинніджереластруму(акумулятори).............. 214
Робота No 12. Визначення коефіцієнта віддачі акумулятора 223
4.КІНЕТИКАГОМОГЕННИХРЕАКЦІЙ................. 236
4.1.Формальнакінетика................................ 236
4.2.Методивизначенняпорядкуреакції.................. 249
4.3 . Залежність швидкості хімічної реакції від температури. . . 253
Робота No 13. Визначення константи швидкості й енергії
активаціїреакціїйодуванняацетону................... 262
Робота No 14. Визначення константи швидкості й енергії
активаціїреакціїомиленняестеру..................... 268
Робота No 15. Визначення швидкості розкладання перок-
сидуводнюгазометричнимметодом................... 272
Робота No 16. Вивчення швидкості реакції гідратації оцто-
вого ангідриду методом електропровідності . . . . . . . . . . . . . 272
Робота No 17. Кінетика окислювання йодид іонів перокси-
домводню......................................... 282
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ. . . . 289
5.1 Загальні поняття й закономірності гетерогенних процесів 289
Робота No 18. Вивчення кінетики розчинення малорозчин-
нихречовин........................................ 299
Робота No 19. Дослідження кінетики розчинення мало-
розчинної солі методом електропровідності . . . . . . . . . . . . 302
5.2.Каталіз.......................................... 306
Робота No 20. Вивчення реакції окислення йодоводнової
кислоти пероксилом водню, що каталізується молібдатом
амонію............................................ 313
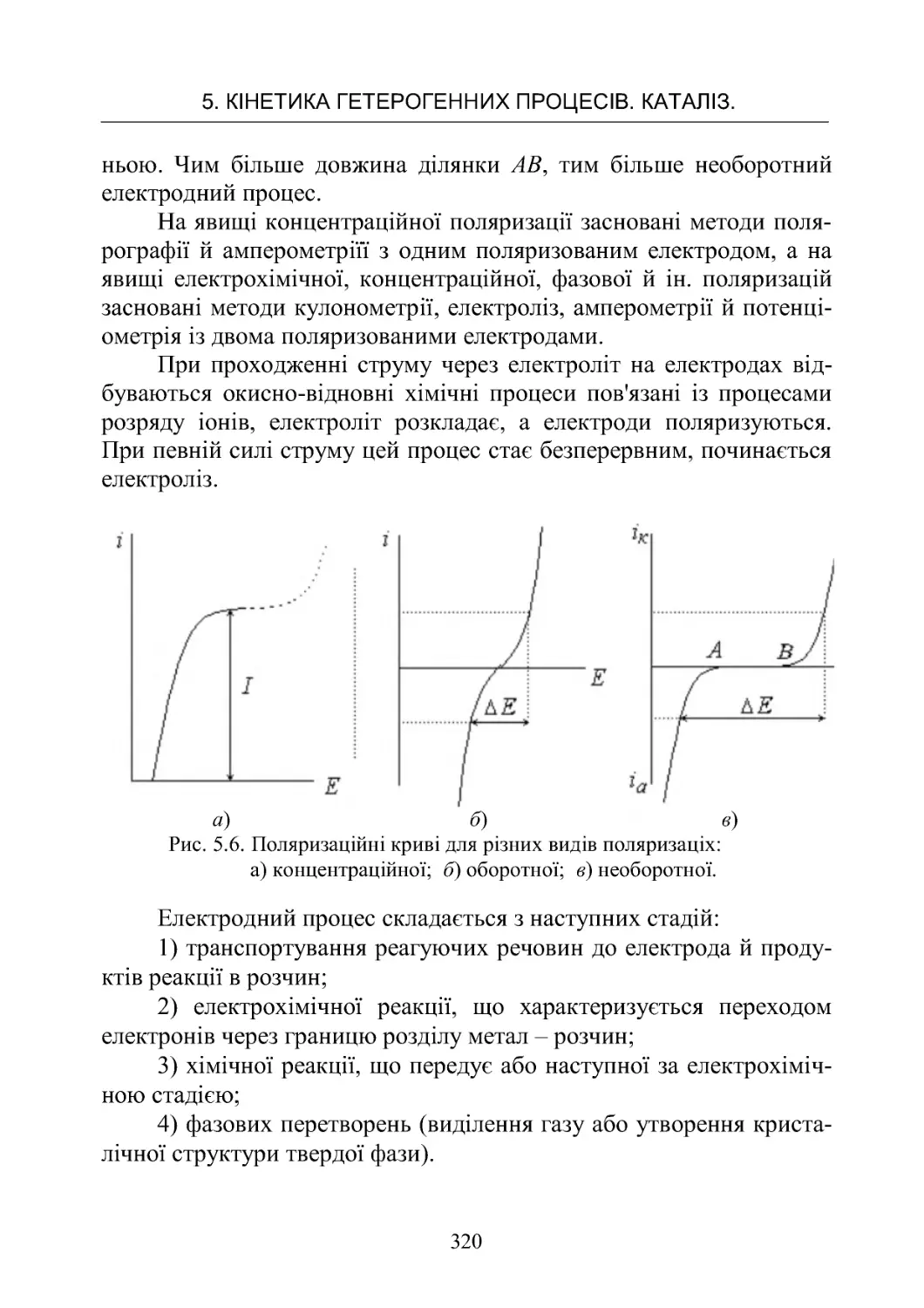
5.3.Кінетикаелектроднихпроцесів ..................... 317
ЗМІСТ
5
Робота No 21. Визначення напруги розкладання при елект-
ролітичномуосадженнінікелюзрозчину ............... 325
БІБЛІОГРАФІЧНИЙ СПИСОК РЕКОМЕНДОВАНОЇ
ЛІТЕРАТУРИ.......................................... 330
ДОДАТОК............................................. 330
Таблиця1.Основніфундаментальніпостійні............... 338
Таблиця2.Деякіперекладнімножники................... 338
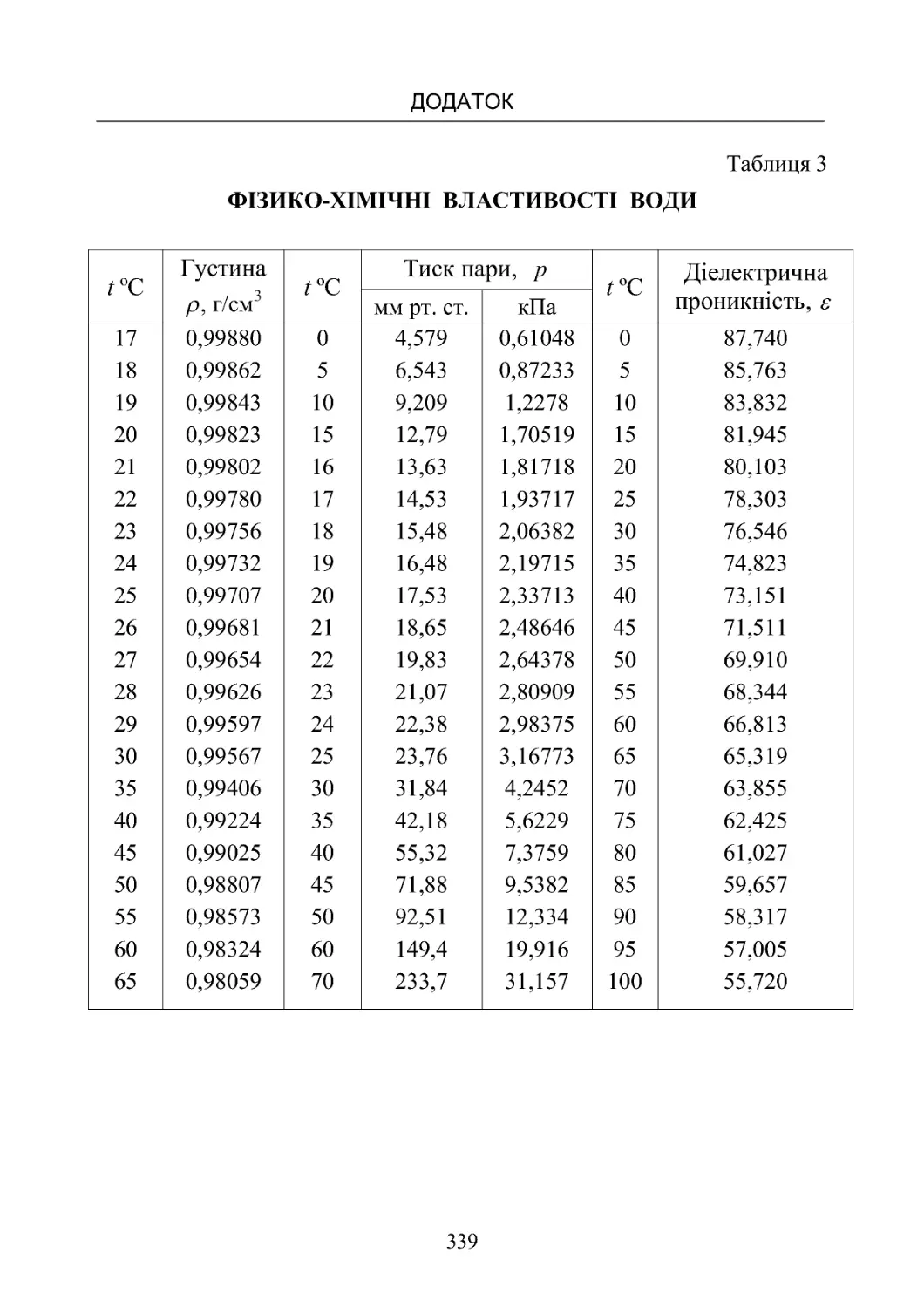
Таблиця3.Фізико-хімічнівластивостіводи............... 339
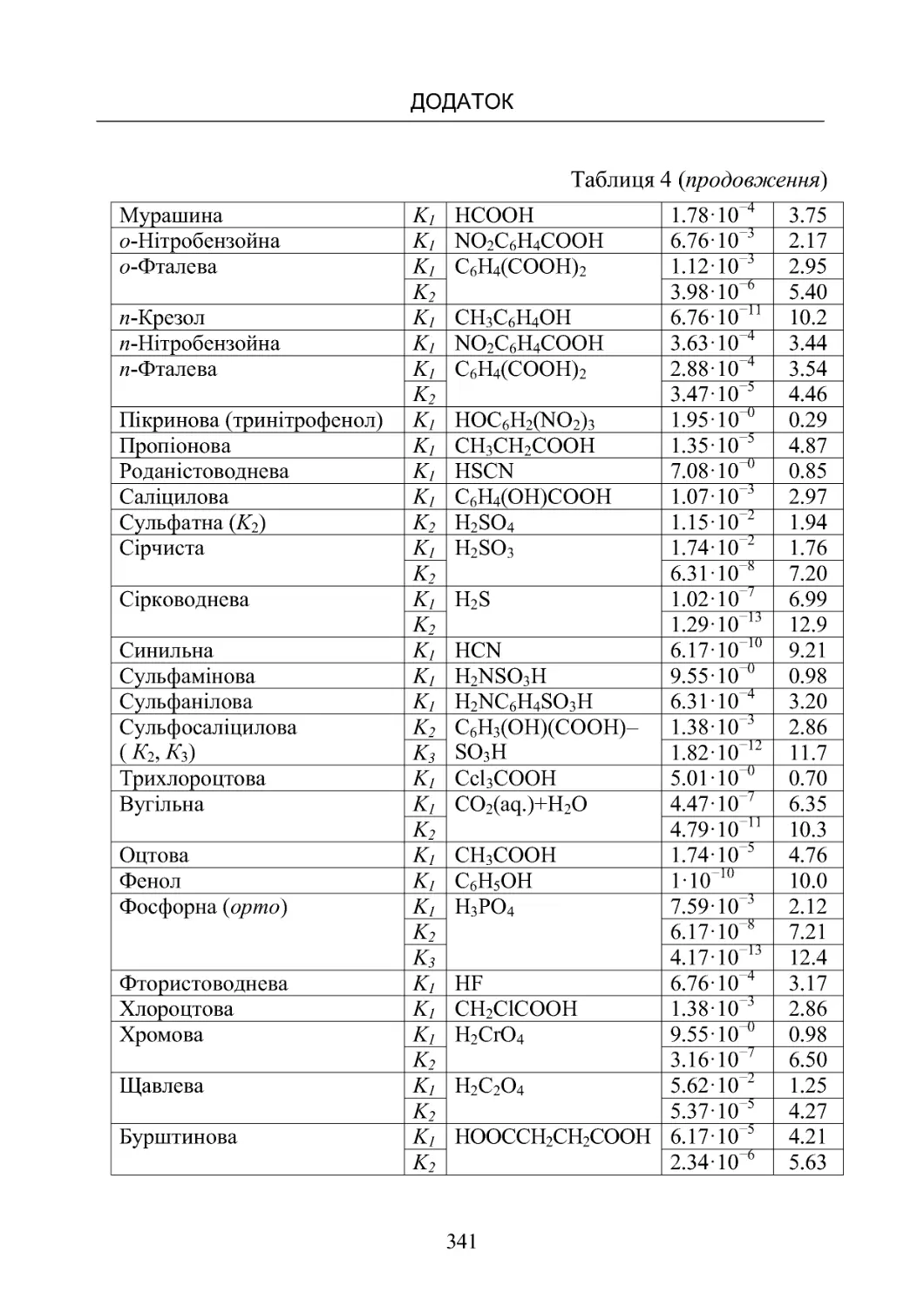
Таблиця 4. Константи дисоціації слабких кислот у водних роз-
чинахзатемператури298К............................ 340
Таблиця 5. Іонний добуток, в'язкість та питома електропровід-
ністьводиприрізнихтемпературах..................... 342
Таблиця 6. Добуток розчинності деяких сполук за температу-
ри298К ............................................. 343
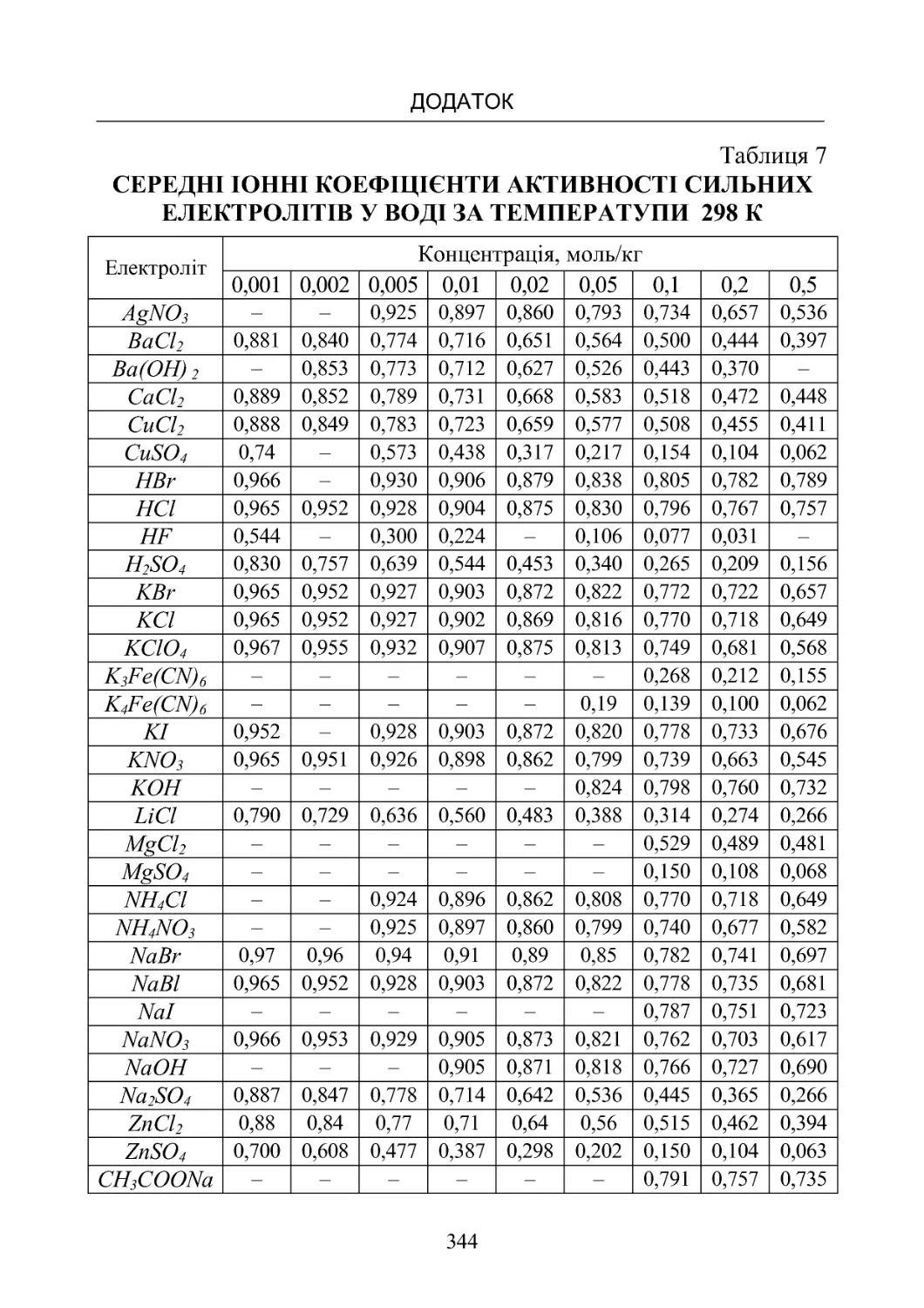
Таблиця 7. Середні іонні коефіцієнти активності сильних
електролітівуводізатемператури298К.................. 344
Таблиця 8. Питома електропровідність (, Ом
–1
см
–1
) водяних
розчинів хлористого калію при різних температурах . . . . . . . 345
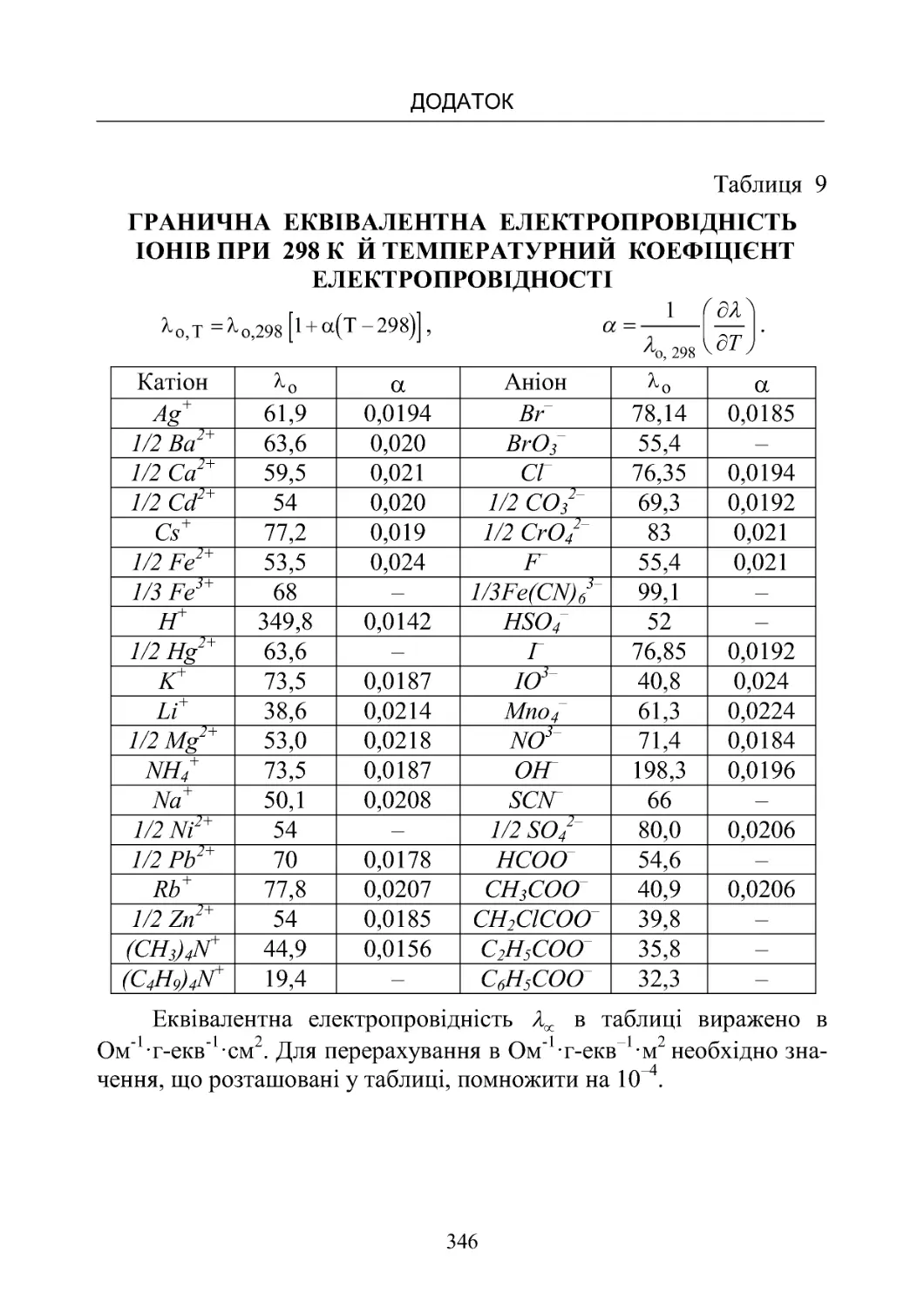
Таблиця 9. Гранична еквівалентна електропровідність іонів
при 298 К и температурний коефіцієнт електропровідності . 346
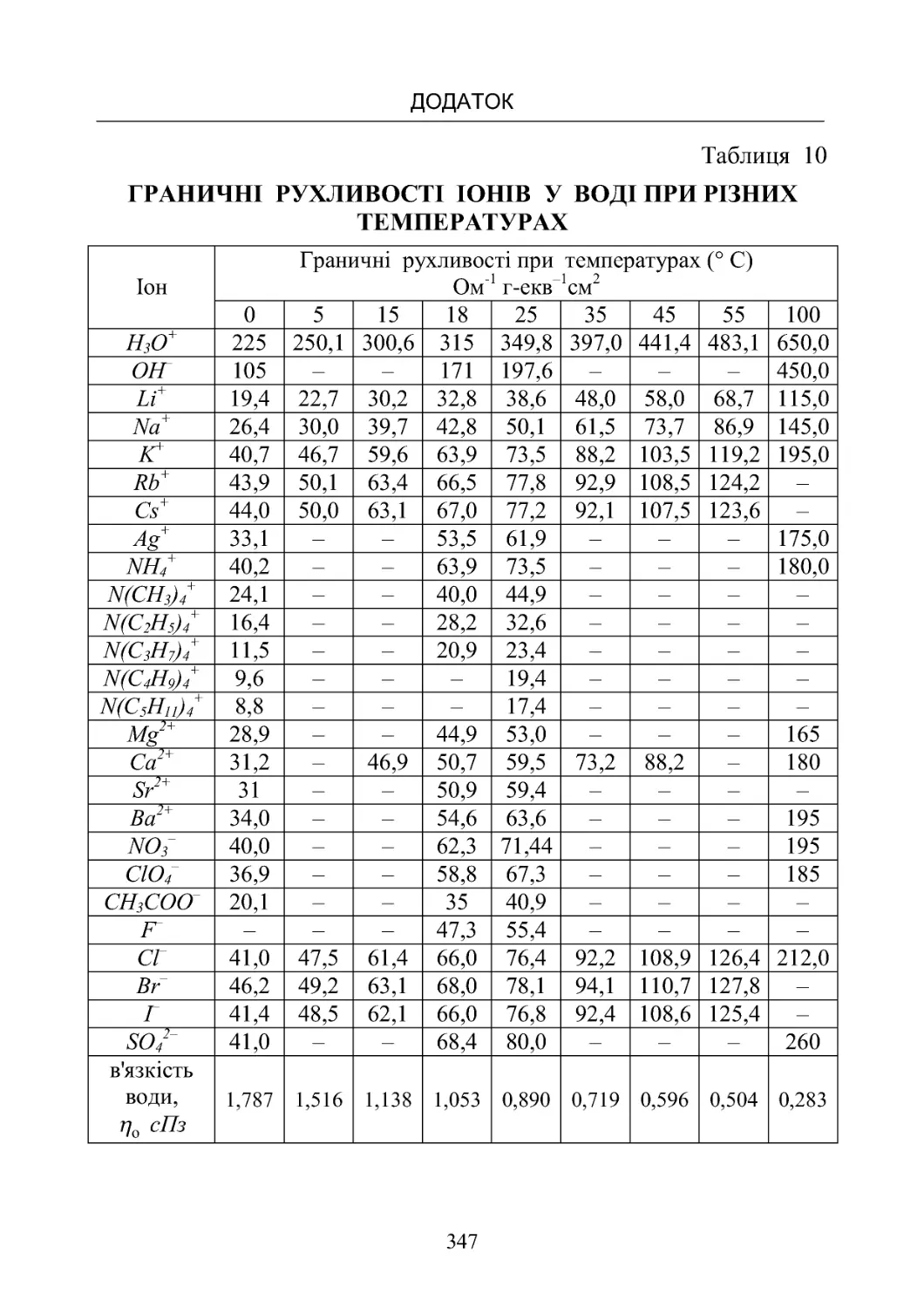
Таблиця 10. Граничні рухливості іонів у воді при різних тем-
пературах............................................ 347
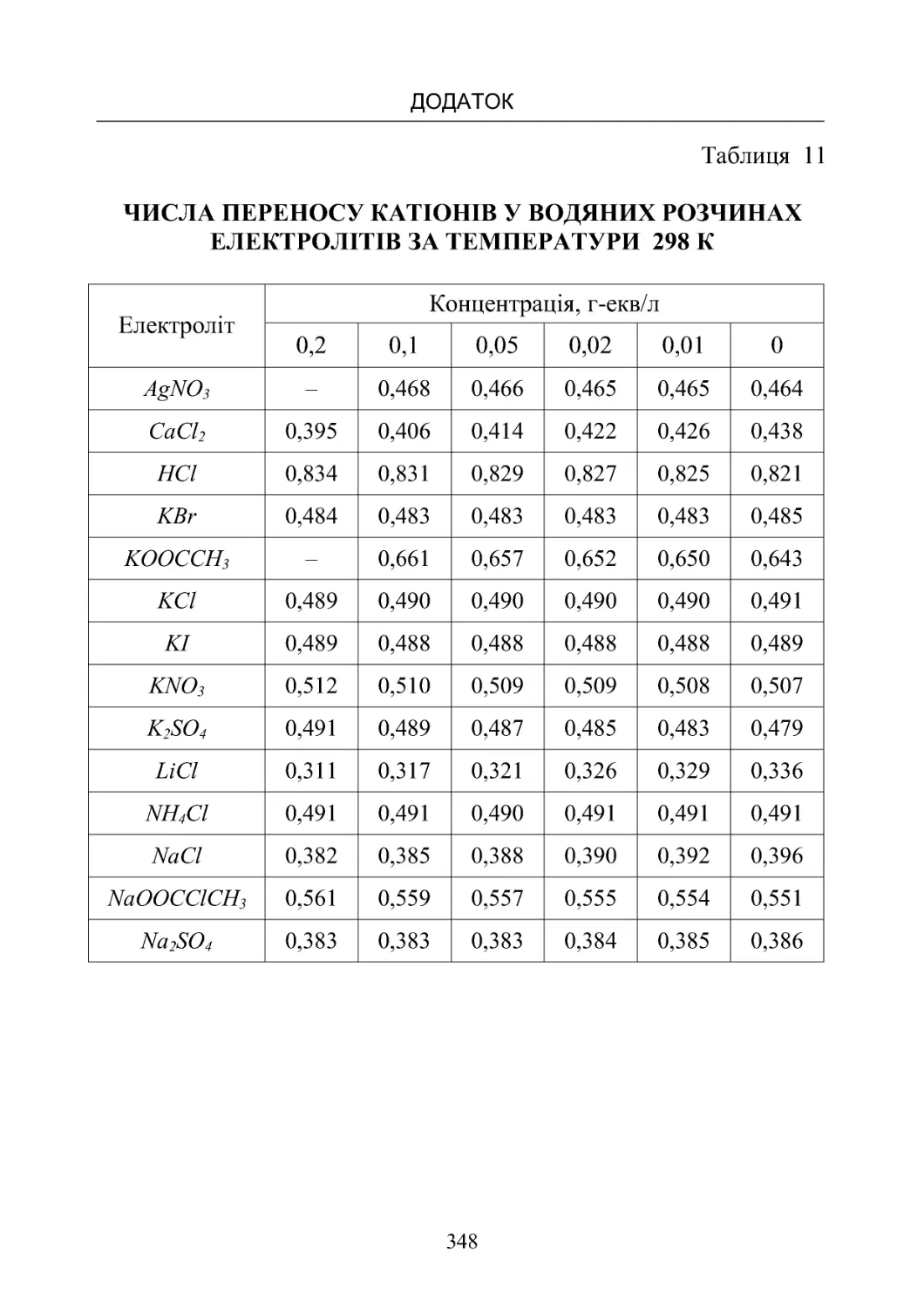
Таблиця 11. Числа переносу катіонів у водяних розчинах елек-
тролітівпри 298К.................................... 348
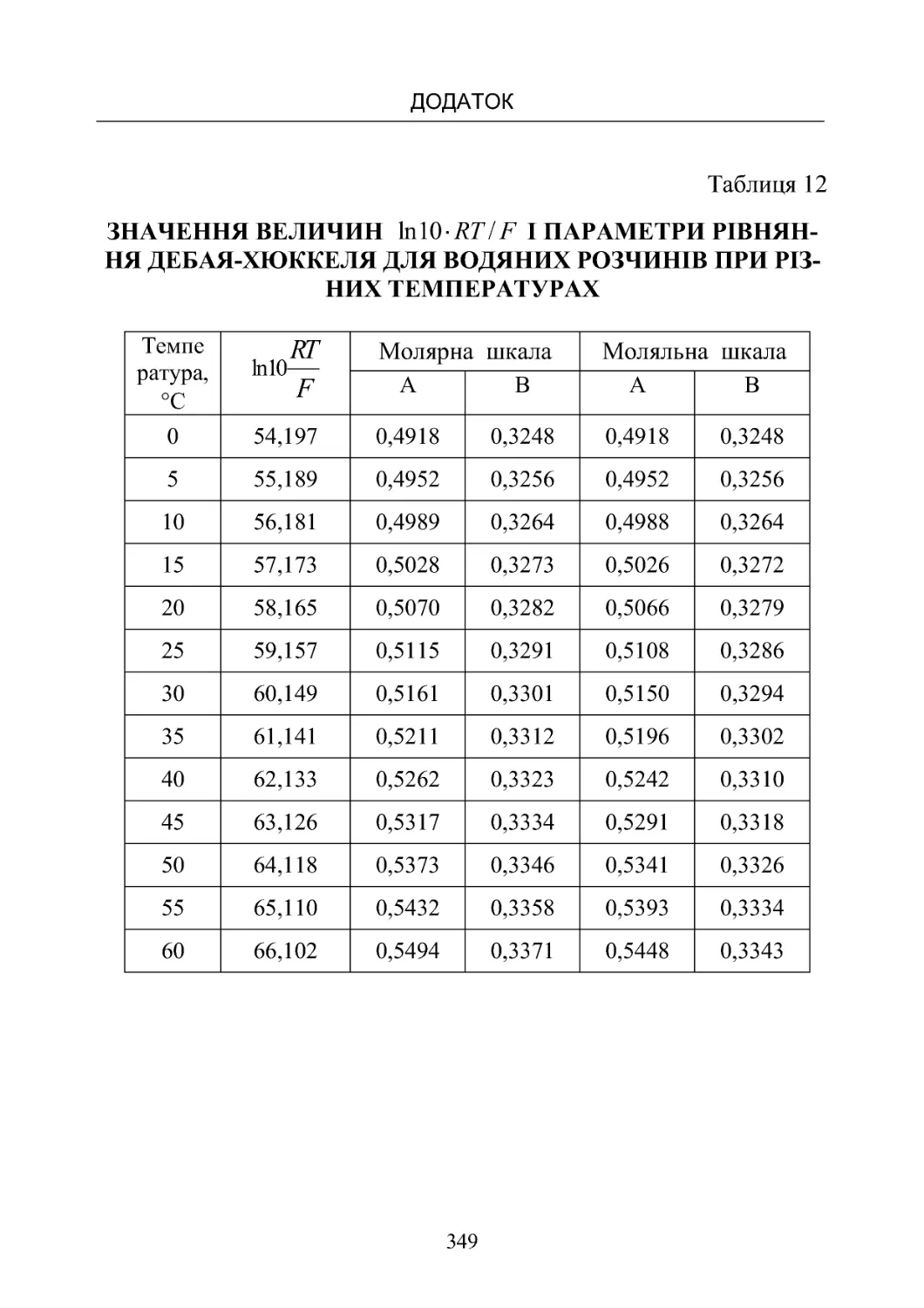
Таблиця 12. Значення величин ln10 /
RTF
і параметри рів-
няння Дебая – Хюккеля для водяних розчинів при різних те-
мпературах .......................................... 349
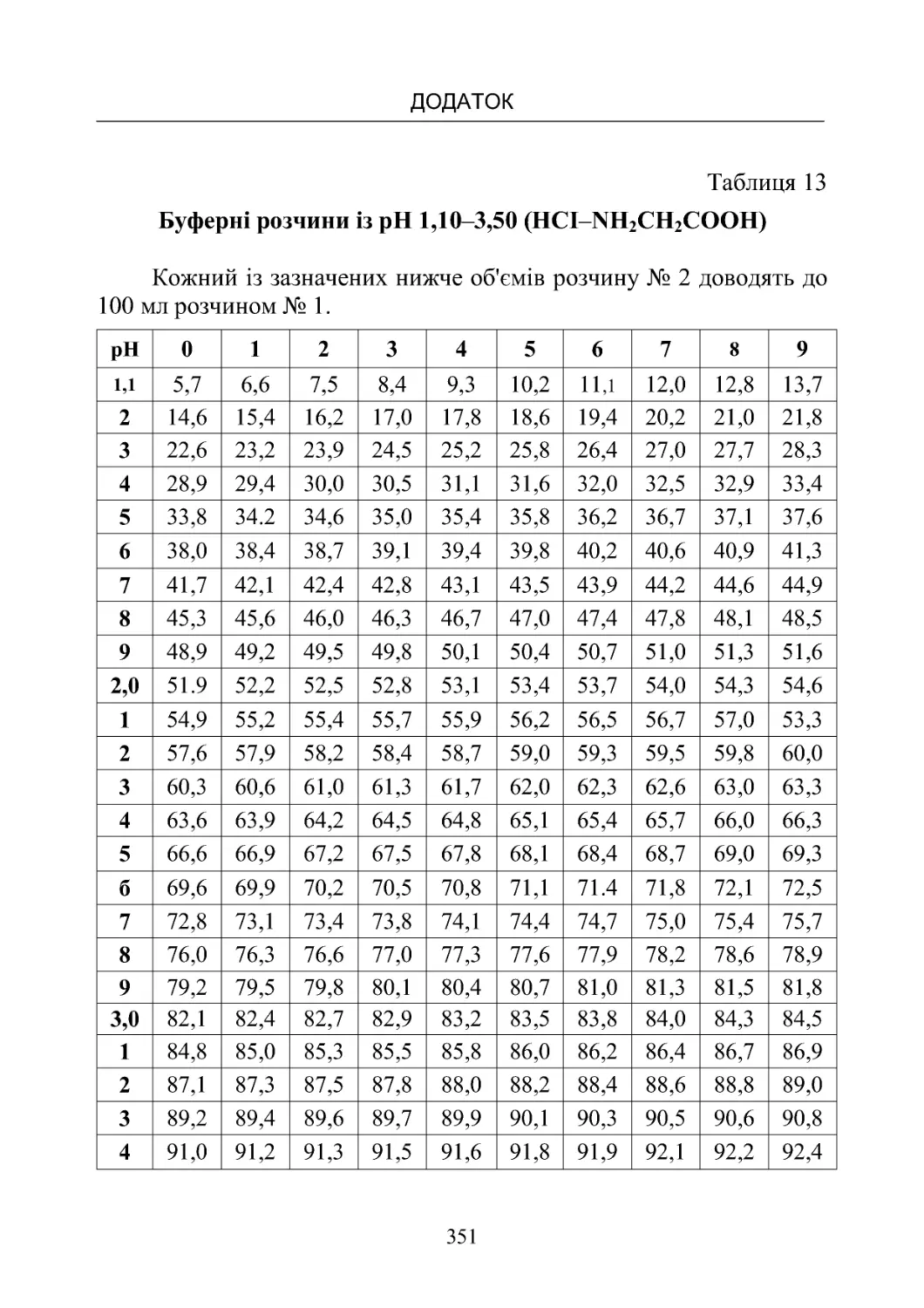
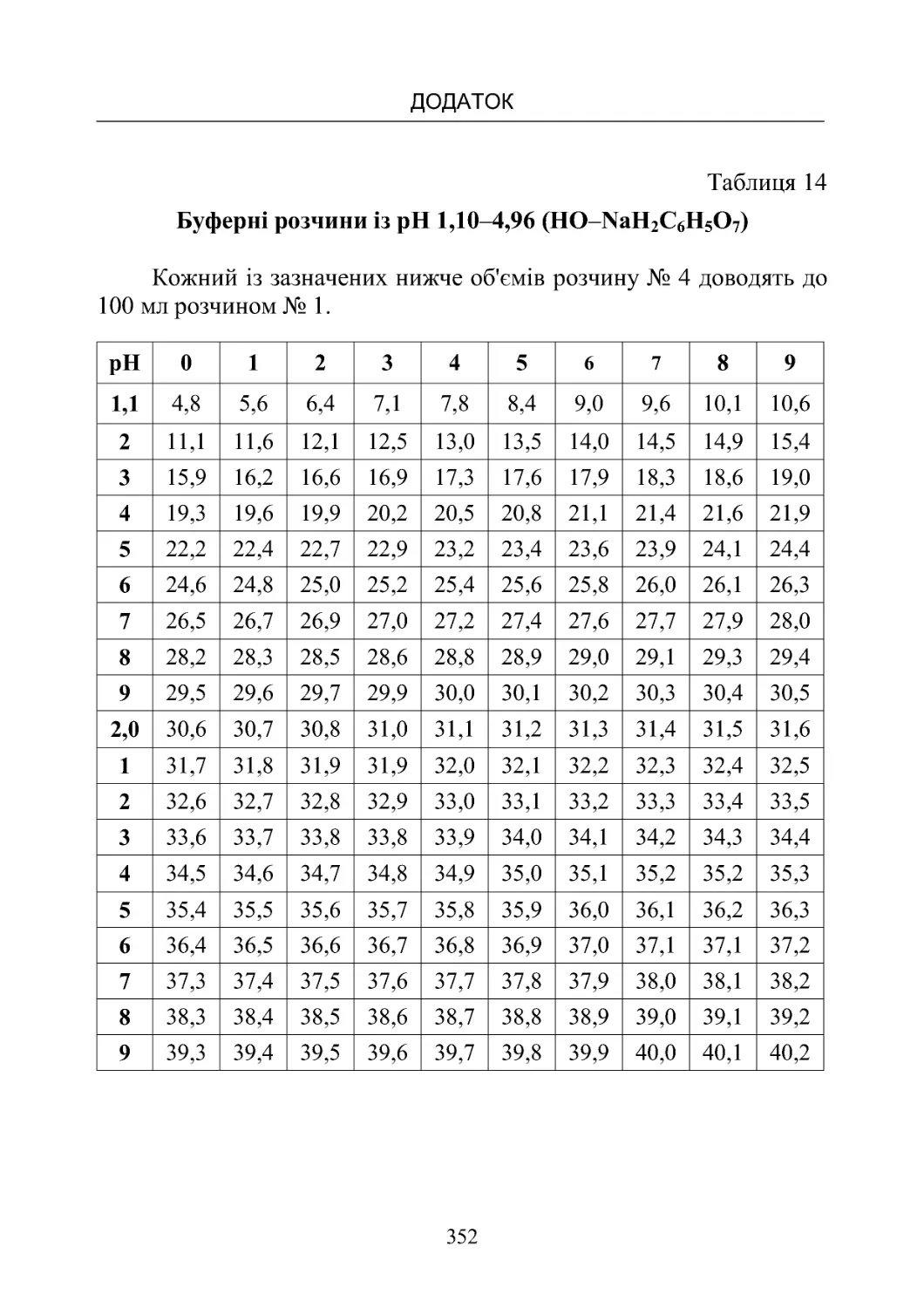
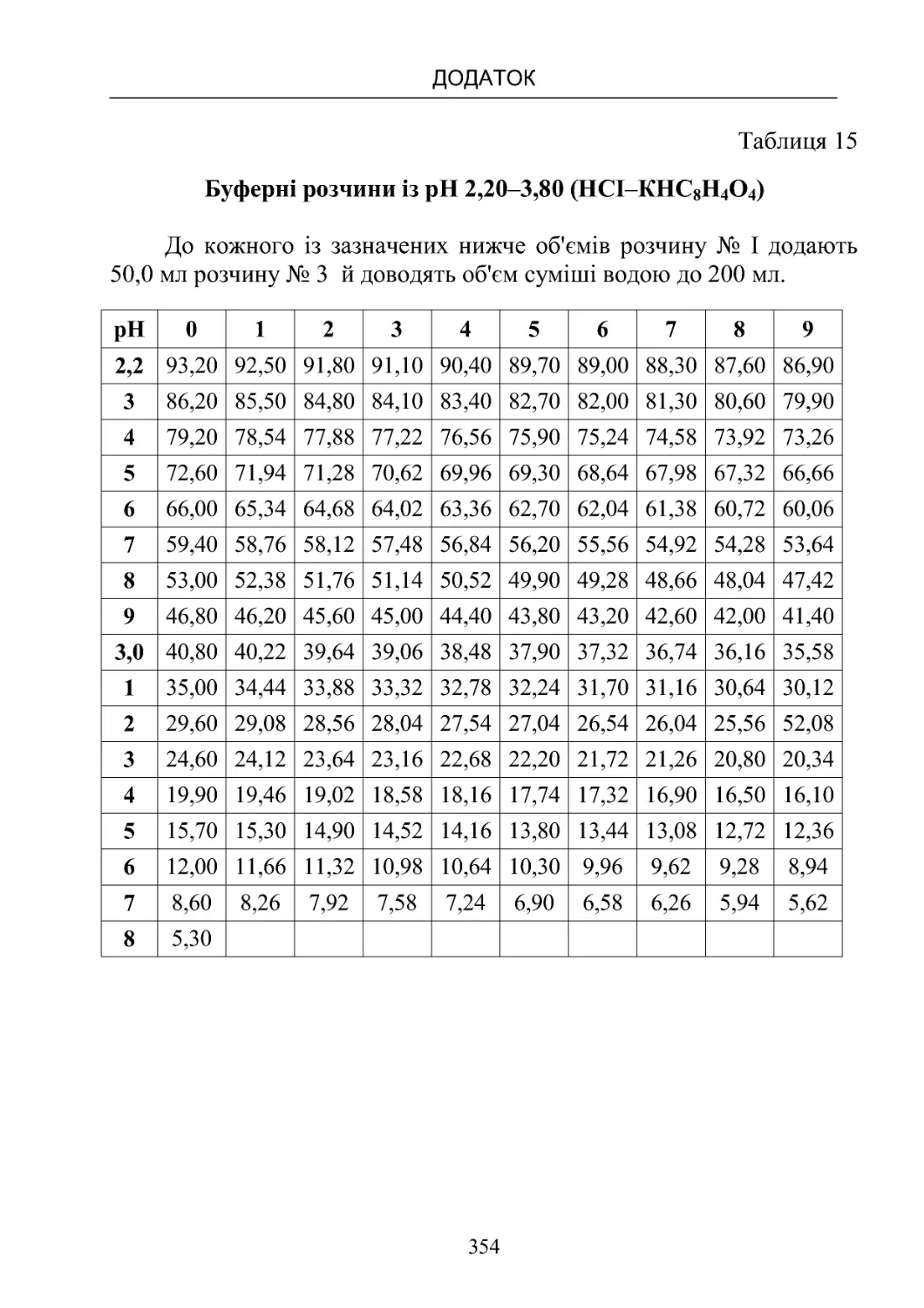
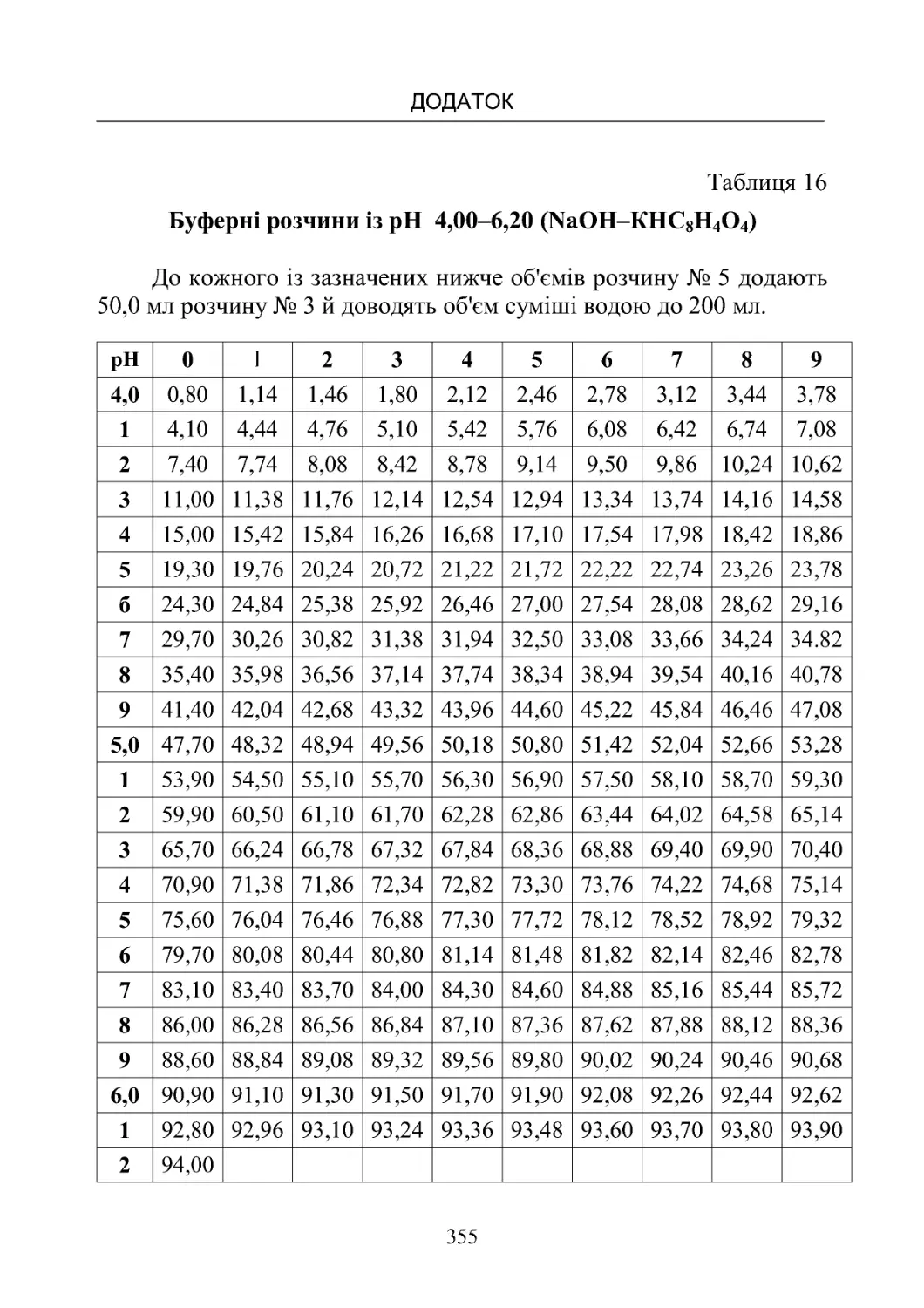
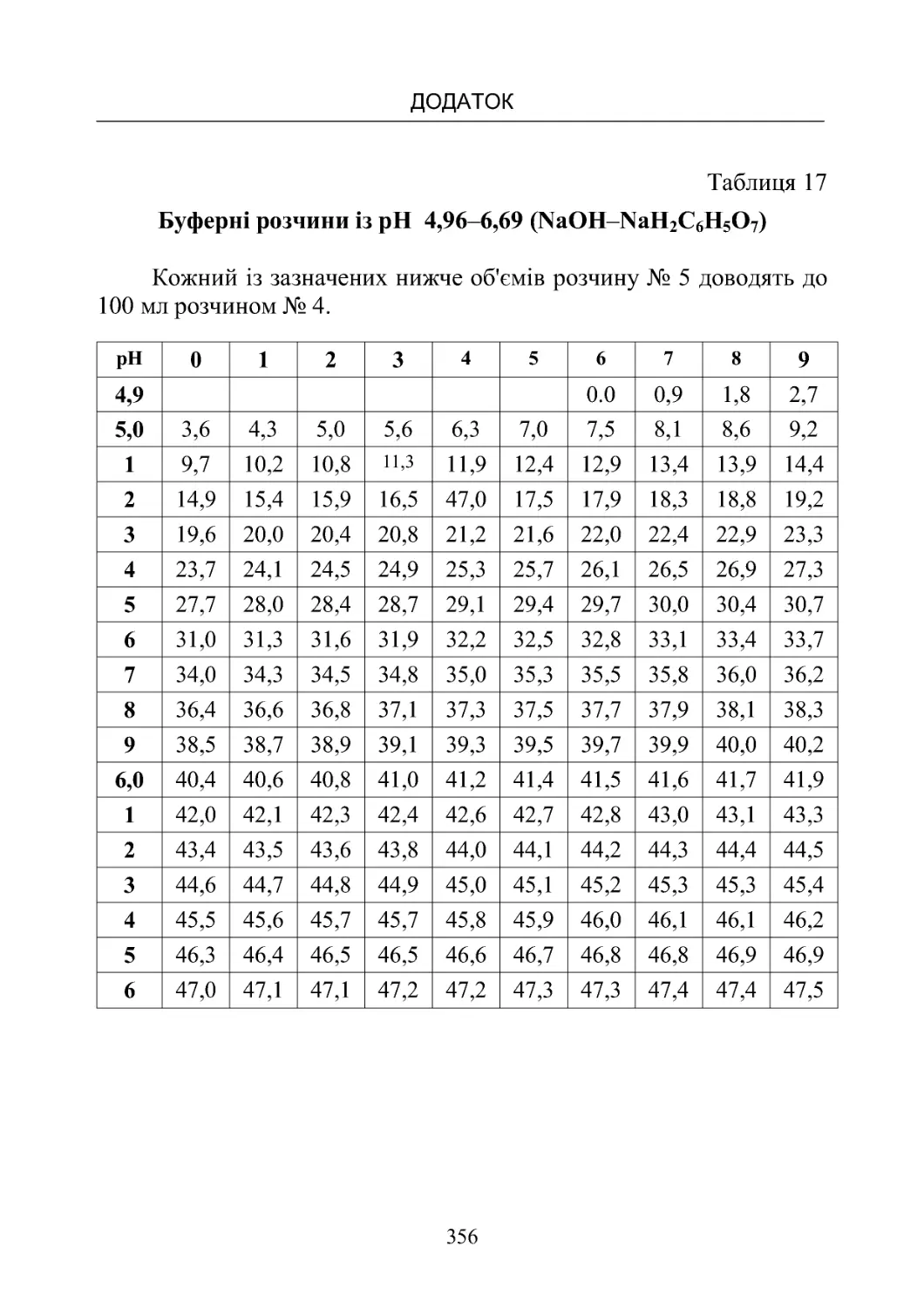
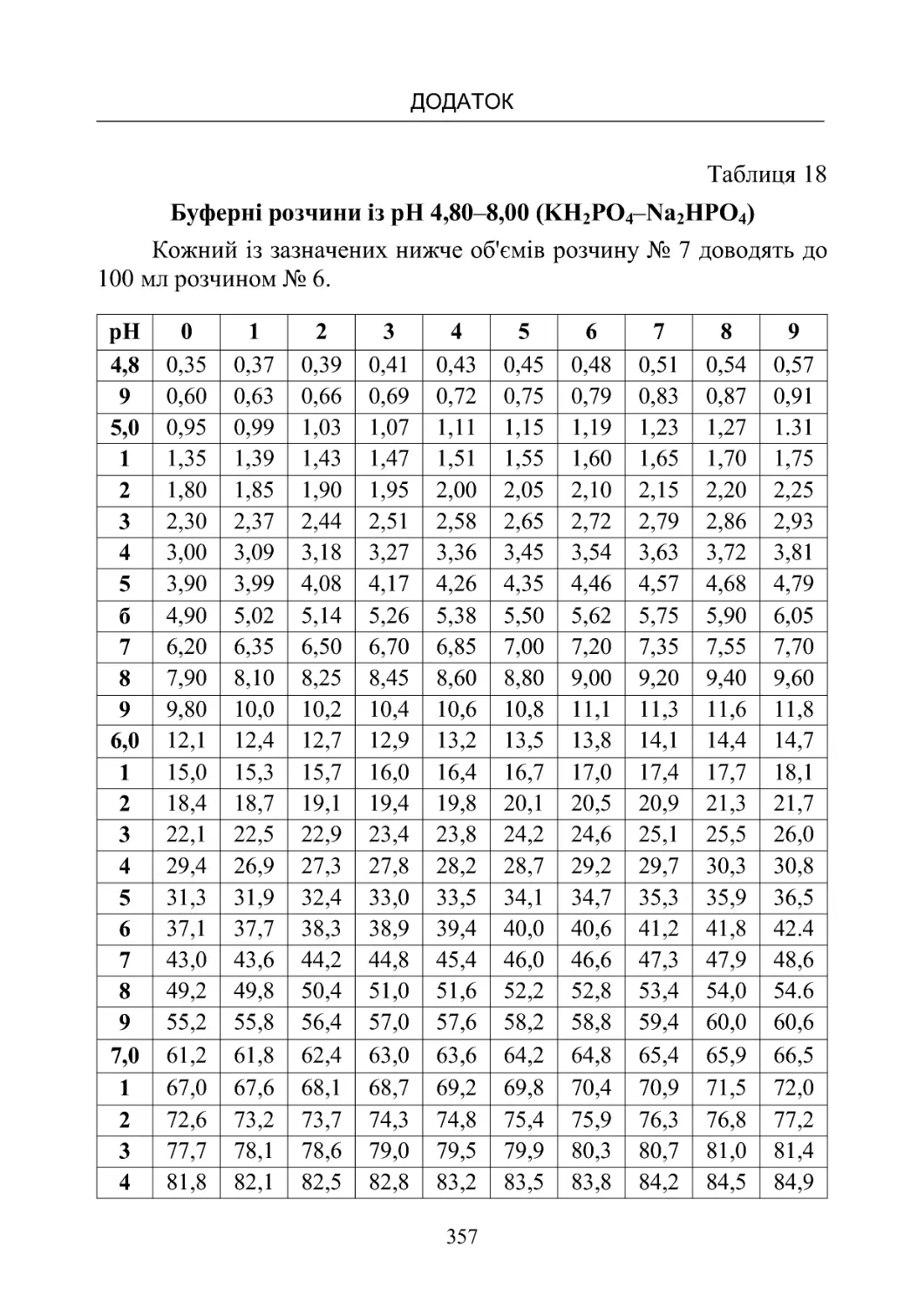
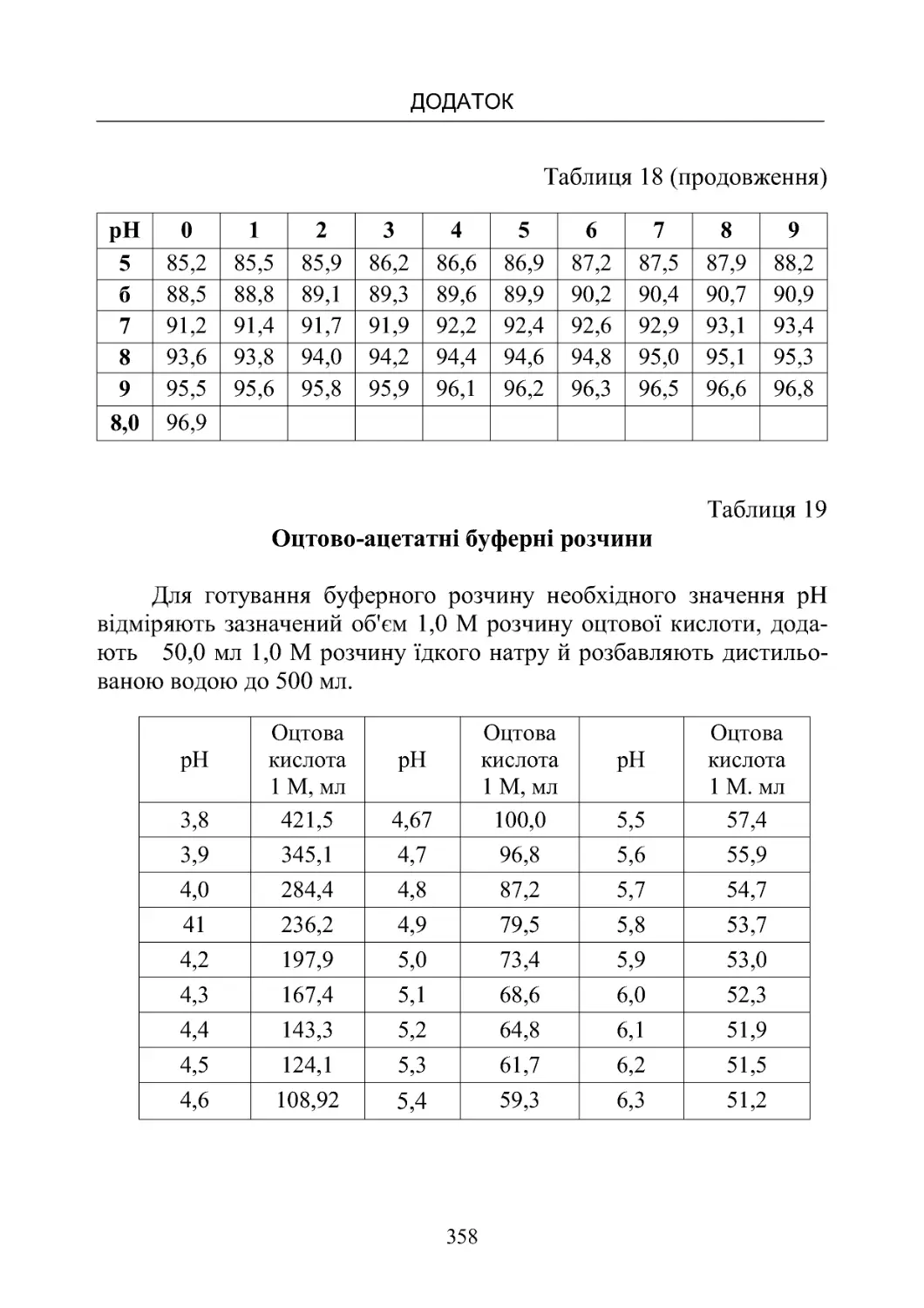
Таблиця 13-19. Склад буферних розчинів (рН від 1,10 до
8,00)температура20oС.................................350
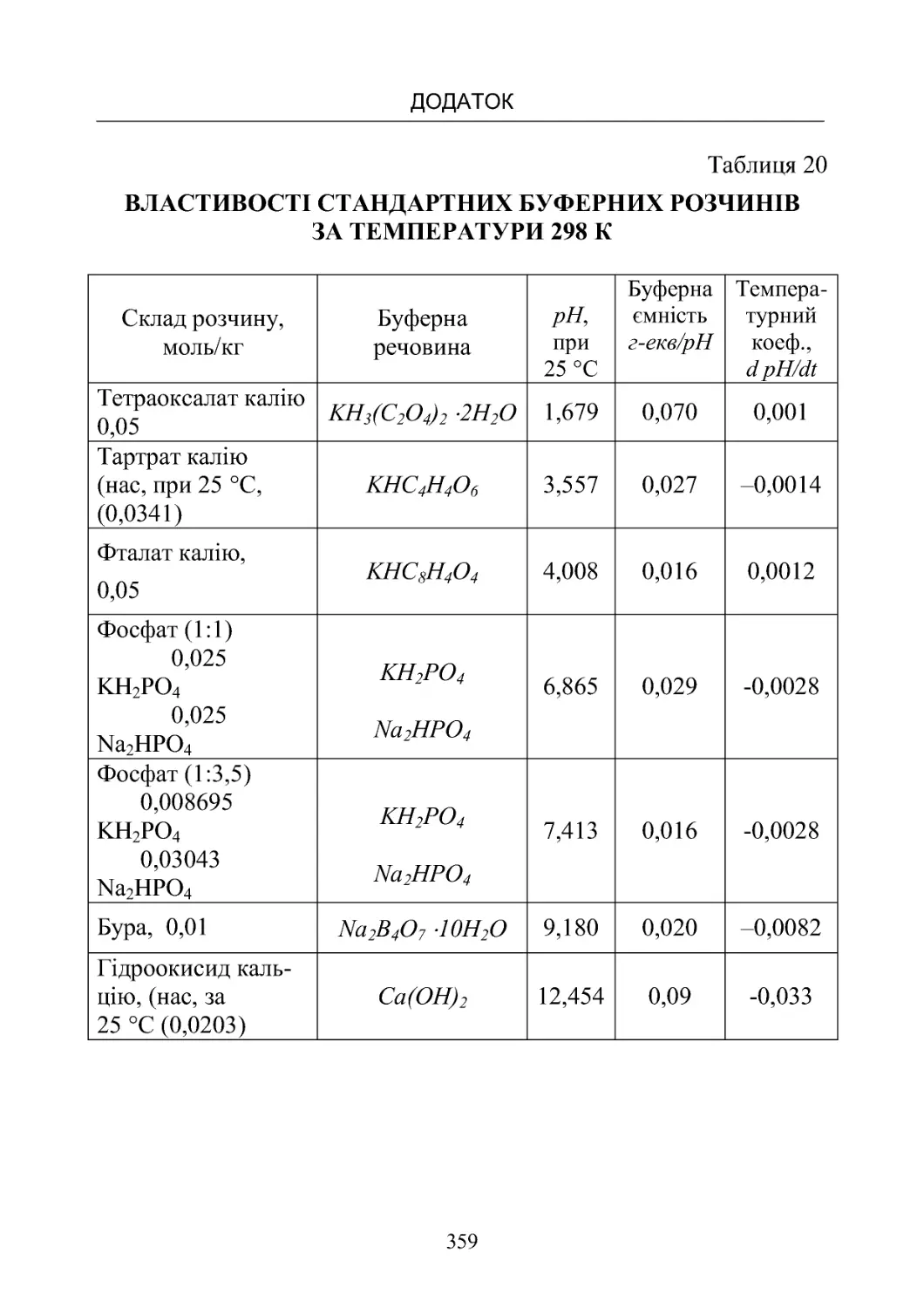
Таблиця 20. Властивості стандартних буферних розчинів за те-
мператури 298К......................................... 359
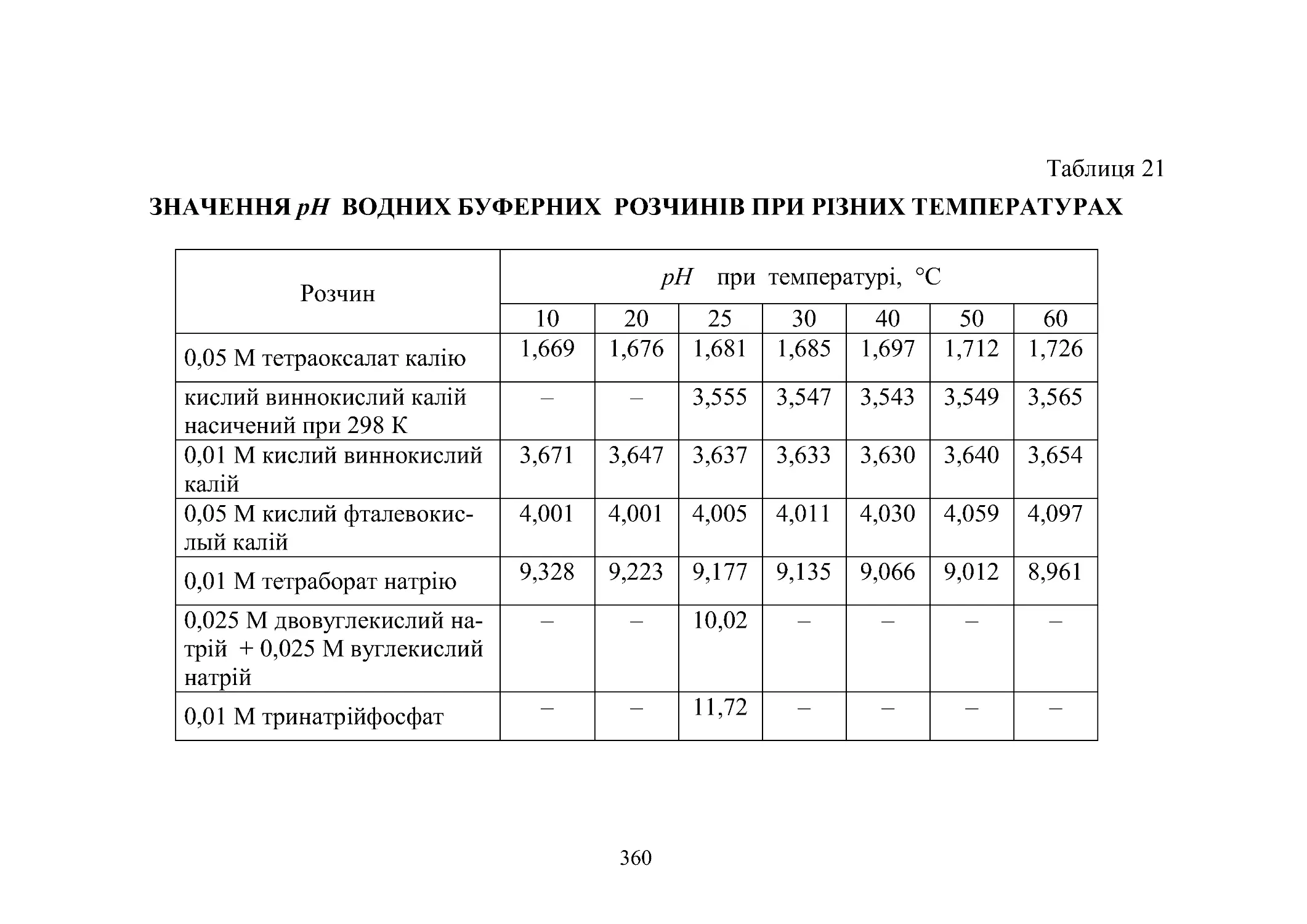
Таблиця 21. Значення pН водяних буферних розчинів при рі-
знихтемпературах.................................... 360
Таблиця 22. Універсальна буферна суміш Бриттона. . . . . . . . . 361
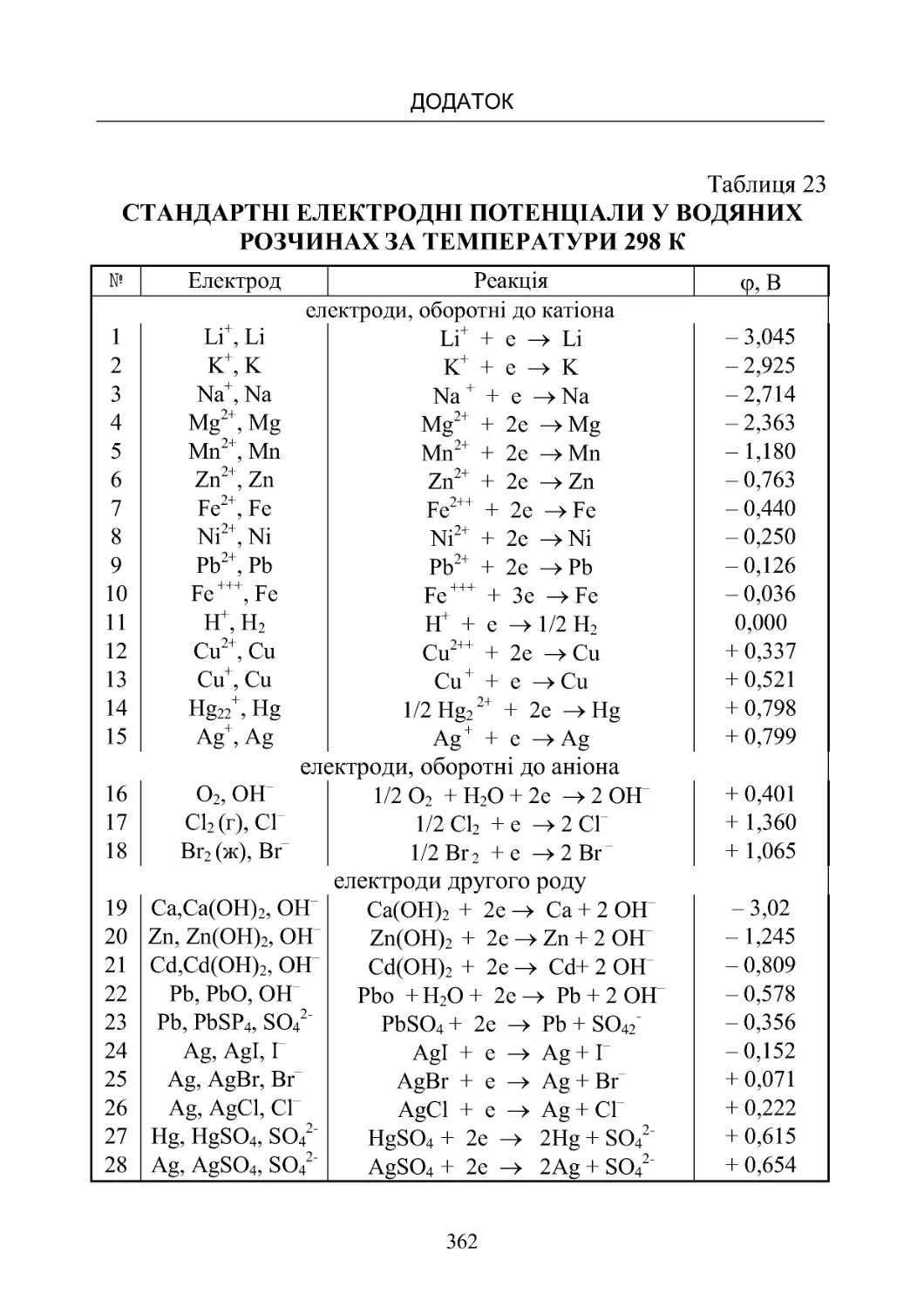
Таблиця 23. Стандартні електродні потенціали у водяних роз-
чинахпри298К...................................... 362
ЗМІСТ
6
Таблиця 24. Електрорушійна сила нормального (насиченого)
елементаВестонаприрізнихтемпературах................ 364
Таблиця 25. Стандартні потенціали електродів другого роду у
водянихрозчинахпри температурі298К.................. 364
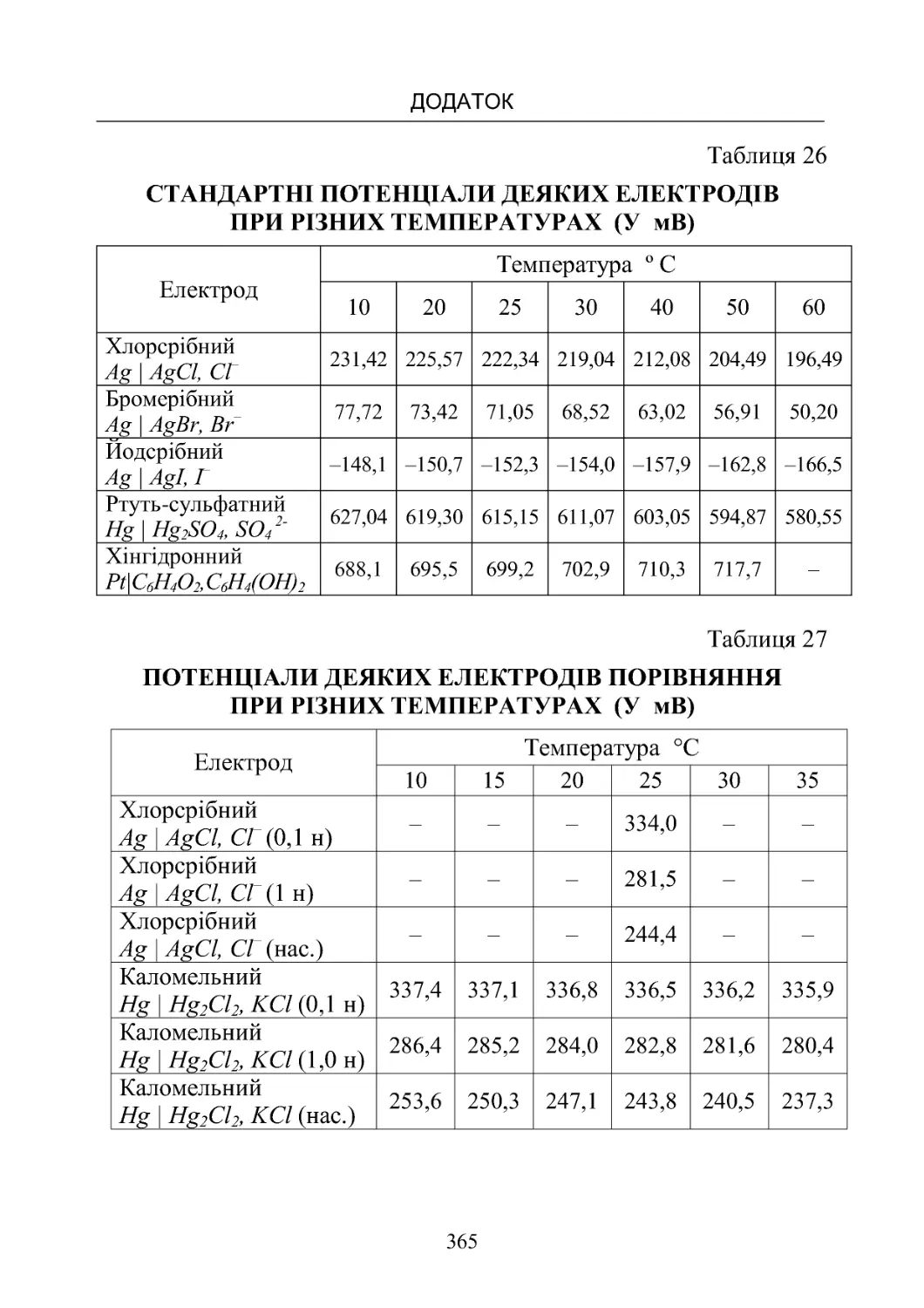
Таблиця 26. Стандартні потенціали деяких електродів при рі-
знихтемпературах (умВ)............................
365
Таблиця 27. Потенціали деяких електродів порівняння при
різнихтемпературах (умВ)............................ 365
Таблиця 28. Виправлення для приведення потенціалу водне-
вогоелектродадотискводню101,325кПа................ 366
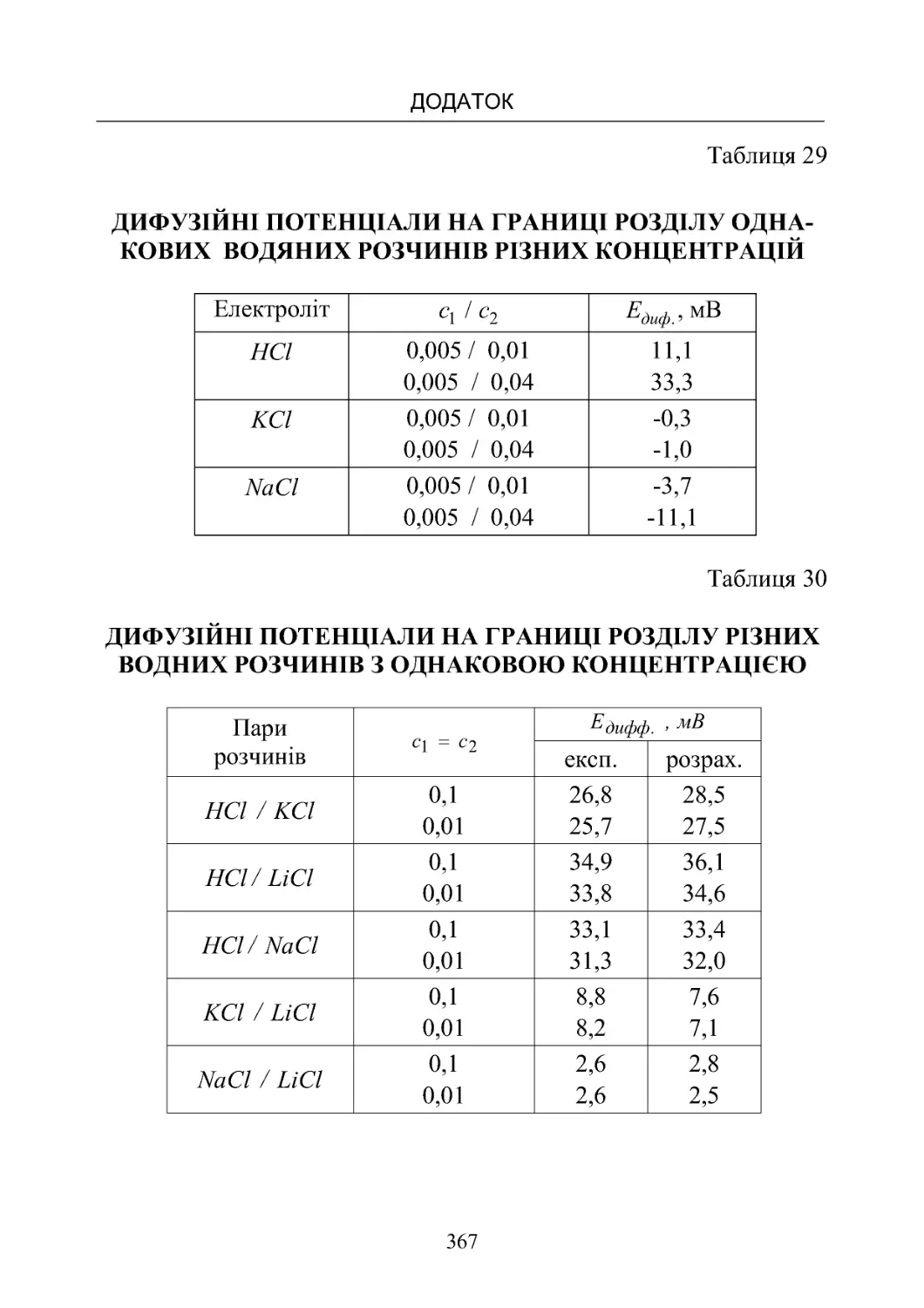
Таблиця 29. Дифузійні потенціали на границі розділу одна-
ковихводянихрозчиніврізнихконцентрацій.............. 367
Таблиця 30. Дифузійні потенціали на границі розділу різних
водянихрозчинівзоднаковоюконцентрацією............. 367
Таблиця 31. Константи швидкості лужного омилення естерів 368
Платинуванняелектродів............................... 369
Очищенняртуті....................................... 370
Готуванняхлорсрібнихелектродів....................... 370
7
ПЕРЕДМОВА
Фізична хімія, яка виникла на стику двох фундаментальних розді-
лів природознавства – фізики й хімії, займається проблемами протікан-
ня хімічних реакцій, пов'язаної з ними перетвореннями енергії, будовою
речовин, а також властивостями речовини в різних станах. Хімічна тер-
модинаміка, кінетика, квантова хімія, каталітична хімія, електрохімія й
ін. будучи цілком самостійними пов'язані з іншими областями хімічно-
го знання входить систему хімічних наук, які утворюють сучасну хімію.
Єдиним універсальним зв'язуванням цих наук є методи фізичної хімії.
При вивченні фізичної хімії важливу роль займає лабораторний
практикум, метою якого є закріплення на практиці отриманих при ви-
вченні курсу фізичної хімії теоретичних знань, оволодіння експеримен-
тальними звичками роботи в хімічній лабораторії з дослідницькими
приладами. При виконанні робіт лабораторного практикуму у фізико-
хімічній лабораторії кожний студент має можливість ознайомитися з
теоретичними положеннями фізичної хімії, які отримані під час теоре-
тичного вивчення курсу, навчитися ставити експеримент, працювати із
приладами, проводити виміру й протоколювати їх у лабораторному жу-
рналі, обробляти результати експерименту й оформляти у вигляді ви-
сновків, осмислюючи спостереження й отримані кількісні характерис-
тики. При оформленні роботи й осмислюванні результатів досліджень
необхідно навчитися користуватися підручниками, довідковою літера-
турою, інтернетом.
Даний навчальний посібник складений на основі лабораторного
практикуму, який проводиться протягом багатьох років на кафедрі фі-
зичної хімії Харківського національного університету. У посібник
включені вже відомі роботи, для яких дані поліпшені й більш точні ме-
тоди виміру шуканих величин. Друга частина посібника включає розді-
ли фізичної хімії: розчини електролітів, електропровідність розчинів,
гальванічні елементи, хімічна кінетика та каталіз. В роботах дані реко-
мендації щодо використання при обробці експериментальних даних пе-
рсонального комп'ютера. Кожній темі посібника передує теоретичне
введення, необхідне для освоєння змісту роботи й грамотного її вико-
нання. В кожному розділі пропонується список рекомендованої літера-
тури, наданий список контрольних питань для перевірки студентом
глибини своїх знань та готовності до здачі результатів роботи.
Навчальний посібник призначений для студентів хімічних факуль-
тетів університетів, які вивчають курс фізичної хімії за програмою для
вищих навчальних закладів.
Автор вважає своїм обов'язком висловити подяку викладачам і
співробітникам кафедри фізичної хімії за висловлені зауваження й про-
позиції.
8
ПРАВИЛА РОБОТИ В ХІМІЧНІЙ ЛАБОРАТОРІЇ
І ТЕХНІКА БЕЗПЕКИ
ЗАГАЛЬНІ ПРАВИЛА ПОВЕДІНКИ В ЛАБОРАТОРІЇ
Перед початком роботи студенти повинні пройти інструктаж з
техніки безпеки й розписатися в спеціальному журналі. Усі роботи
виконуються в більшості випадків індивідуально або групою не бі-
льше двох студентів за завданням викладача.
Тому при роботі в хімічній лабораторії слід дотримуватися на-
ступних правил:
–
забороняється знімати й розвішувати верхній одяг, ухвалю-
вати їжу, пити, включати й виключати рубильники, торкати прила-
ди й проводити досвіди, які не ставляться до лабораторної роботи.
–
робоче місце тримати в чистоті, не захаращувати сторонній
предметами. Оберігайте методичні вказівки, інструкції й книги від
влучення на них реактивів.
–
у лабораторії слід перебувати в халаті, дотримуватися тиші,
не користуватися мобільними телефонами;
–
акуратно використовувати прилади загального користування
й реактиви, не нести їх на своє робоче місце;
–
категорично заборонено ухвалювати їжу в лабораторії, після
завершення експериментальних досліджень мити руки;
–
речовини й розчини для проведення експерименту брати в
кількості, які рекомендовані в описі. Узяті в надлишку реактиви або
розчини не можна зливати назад у склянки, – зливати необхідно в
спеціальні ємності для зливу.
ЗАПОБІЖНІ ЗАХОДИ ПРИ РОБОТІ
У ХІМІЧНІЙ ЛАБОРАТОРІЇ Й НАДАННЯ ПЕРШОЇ
ДОПОМОГИ
1. Працюючі в лабораторії повинні знати, де перебуває най-
ближчий евакуаційний вихід, на випадок позаштатних або надзви-
чайних ситуацій.
2. Необхідно знати, де перебуває рубильник вимикання елек-
троживлення. При загорянні в лабораторії приладів, матеріалів, у
випадку короткого замикання необхідно лабораторію знеструмити.
Для гасіння загоряння електропроводки використовувати тільки по-
рошкові вогнегасники.
9
3. Кожний працюючий у лабораторії повинен знати, де в ла-
бораторії перебувають засоби пожежогасіння: вода, пісок, спеціаль-
на ковдра, вогнегасник, а також уміти користуватися цими засобами.
4. При запаленні горючої рідини, наприклад, на одязі працю-
ючого, необхідно закрити джерело загоряння спеціальною негорю-
чою ковдрою.
5. Кожний зобов'язано знати, де в лабораторії перебуває апте-
чка для надання першої допомоги.
6. При опіках гарячим металом або склом обпалене місце ба-
гаторазове змочите розчином перманганату калію, а потім змажте
маззю від опіків.
7. При розведенні концентрованих кислот (особливо сарною)
і лугів необхідно невеликими порціями вливати реактив у воду з
безперервним перемішуванням розчину.
8. У випадку влучення будь-якої кислоти або луги на одяг
уражене місце промийте більшою кількістю проточної води, а потім
нейтралізуйте або розчином гідрокарбонату натрію (кислоту), або
слабким розчином борної кислоти (луг).
9. Для випробування газу або рідини на наявність тримаєте по-
судину (пробірку) в одній руці так, щоб отвір перебував нижче рівня
носа, а іншою рукою направляйте до себе слабкий струм повітря.
10. Дотримуйте особливої обережності при роботі із прилада-
ми, які містять ртуть, особливо з термометром Бекмана, що містить
ртуть у відносно великій кількості. Не відволікайтеся, тримаєте
прилад над столом.
11. Дотримуйте обережності при роботі з посудинами Дьюара
щоб уникнути їх ушкодження. Контролюйте свої дії при зануренні з
такі посудини термометра, піпеток і інших приладів. При роботі з
посудинами високого або низького тиску користуйтеся захисними
окулярами.
12. Не допускайте випадкового влучення на руки розчинів хі-
мічних реактивів, особливо їдких кислот і лугів, органічних розчин-
ників, особливо бензолу. Органічні розчинники неприпустимо ви-
ливати в раковини – для них також призначені спеціальні ємності.
13. Будьте обережні при роботі із солями срібла, навіть розве-
деними – їх влучення на шкіру через якийсь час викликає появи чо-
рних плям диспергованого металевого срібла, яке важко вилучити.
Користуйтеся захисними гумовими рукавичками. Зливи солей сріб-
ла проводяться в спеціальні ємності.
10
14. Не відволікайтеся при роботі з вимірювальними прилада-
ми, не підбудовуйте й не калібруйте прилади самі без участі викла-
дача або інженера. Акуратно працюйте з аналітичними або елект-
ронними вагами. Перевіряйте оцінку нульової ваги перед зважуван-
ням. При користуванні важками беріть гирі тільки пінцетом.
15. Електричні прилади, які використовуються при виконанні
роботи, виключає тільки інженерно-технічний персонал лаборато-
рії.
У всіх випадках при виникненні позаштатної ситуації в лабо-
раторії необхідно відразу ж привести до популярність викладача й
навчально-допоміжний персонал.
ВИМОГИ ДО ВИКОНАННЯ Й ОФОРМЛЕННЮ
ЛАБОРАТОРНИХ РОБІТ
1. Виконанню роботи передує бесіда з викладачем, до якої сту-
дент готується заздалегідь. Перед виконанням роботи необхідно чі-
тко знати ціль дослідження, основні теоретичні положення, поняття,
розрахункові формули, представляти послідовність виконання за-
вдання, принцип роботи й обладнання приладів і експериментальної
установки. На висновку опису кожної лабораторної роботи в дано-
му посібнику приводиться список контрольних питань, на які сту-
дент повинен уміти відповісти. Робота виконується тільки з дозволу
викладача.
2. Усі записи під час роботи проводяться тільки в лаборатор-
ному журналі, який призначений тільки для даного предмета. Лабо-
раторні роботи оформляються в тому ж лабораторному журналі.
Перед початком ведення протоколу досліджень записується назва
лабораторної роботи, проставляється дата, умови проведення експе-
рименту (температура, тиск і інші відомості), формується мета ро-
боти, записується індивідуальне завдання, дане викладачем під час
попередньої бесіди. Бажане заздалегідь скласти план роботи й про-
думати форму ведення протоколу дослідження.
3. Експериментальні результати, отримані під час виконання
роботи в лабораторії, обов'язково аналізуються й підписуються ви-
кладачем наприкінці заняття. Після закінчення лабораторної роботи
робоче місце приводиться в порядок, миється використаний посуд.
4. Усі записи, які ставляться до виконуваної роботи, прово-
дяться в лабораторному журналі тільки ручкою. Записи олівцем не
допускаються. Категорично забороняється користуватися чернетка-
11
ми, виправляти дані експерименту (при необхідності невірні резуль-
тати перекреслюються лінією й поруч записується правильне зна-
чення).
5. Усі необхідні обчислення проводяться в робочому журналі;
при чисельних розрахунках записується формула, підставляються
відповідні значення для обчислення; округлення проводиться тіль-
ки кінцевого результату обчислень. Статистична обробка результа-
тів для оцінки можливої погрішності, побудова графіків є частиною
лабораторної роботи. Графіки слід будувати на міліметрівці й вкле-
ювати в лабораторний журнал. При виконанні лабораторних робіт
практикуму можна використовувати програмне забезпечення на ос-
нові електронних таблиць Microsoft Excel для:
–
побудови лінійних графічних залежностей з використанням
методу найменших квадратів;
–
розрахунків статистичних параметрів для набору експери-
ментальних даних (середнє арифметичне, середнє й стандартне від-
хилення, дисперсія, довірчий інтервал для різного рівня довірчої
ймовірності й ін.);
–
проведення Q-тесту для виключення грубих промахів, якщо
є достатня вибірка – три й більш результатів паралельних вимірів.
Оформлення лабораторної роботи завершується висновками.
6. Після виконання роботи й проведення відповідних розра-
хунків у позааудиторний час, викладачем проводиться бесіда з ви-
конавцем роботи для оцінки як теоретичних знань виконавця, так і
якості отриманих результатів.
7. У випадку використання для проведення розрахунків і офо-
рмлення лабораторної роботи персонального комп'ютера при здачі
результатів приводяться роздруківки таблиць і графіків відредаго-
ваних за всіма правилами оформлення документів. Проводиться
форматування стовпців таблиці із приведенням результатів з необ-
хідною кількістю значущих цифр, форматуються заголовки таб-
лиць. На графіках редагується розмах по обом осях, приводяться за-
головки таблиць і найменування осей графіка. При кількісному опи-
сі залежності у вигляді апроксимуючого рівняння приводяться па-
раметри лінії регресії, а також розраховуються статистичні характе-
ристики апроксимації: коефіцієнт множинної кореляції й довірчі ін-
тервали параметрів рівняння регресії.
12
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ.
ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ.
1.1. ЕЛЕКТРОЛІТИЧНА ДИСОЦІАЦІЯ, ІОННІ РІВНОВАГИ,
ТЕРМОДИНАМІЧНІ ВЛАСТИВОСТІ
Рідкі системи, які складаються з іонів, або містять іони, є іонні
системи до яких відносять розплави та розчини. Головною ознакою
іонних систем є спроможність проводити електричний струм, який
викликає пересування речовини у вигляді іонів і викликає хімічні
перетворення в місцях входу й виходу струму. Такі іонні системи
мають назву, електролітів й ставляться до провідників другого ро-
ду, на відміну від провідників першого роду, носіями електрики в
яких є електрони, прикладам яких є метали. До провідників другого
роду ставляться розчини солей, кислот і основ у воді й в інших іоні-
зуючих розчинниках, а також розплави солей і деякі тверді солі.
При наявності іонів у системі виникають значні відхилення від іде-
альності за рахунок далеко діючих сил Кулонівської взаємодії між
зарядженими частинками.
Мимовільний процес розпаду електроліту в розчинах на іони
називається електролітичною дисоціацією. Уперше теорія дисоціа-
ції була сформульована в 1887 році шведським хіміком Сванте Ар-
реніусом. Ця теорія, що одержала назву класичної теорії дисоціації
електролітів, складається з наступних основних положень:
–
молекули кислот, основ і солей при розчиненні мимовільно
дисоціюють на іони. При цьому іони поводяться як молекули ідеа-
льного газу, тобто не взаємодіють між собою.
–
не весь електроліт, а тільки його частина розпадається на іо-
ни. Інша його частина перебуває в розчині в недисоційованому
1.1. ЕЛЕКТРОЛІТИЧНА ДИСОЦІАЦІЯ, ІОННІ РІВНОВАГИ,
ТЕРМОДИНАМІЧНІ ВЛАСТИВОСТІ
13
стані. Частка електроліту, яка розпадається на іони, називається
ступенем дисоціації електроліту.
–
до рівноважного процесу електролітичної дисоціації застосу-
ємий закон діючих мас.
Умовно поділяють електроліти на сильні електроліти, які іс-
нують в розчині тільки у вигляді іонів, тому що вони повністю дис-
соційовані; слабкі електроліти – електроліти, які в не дуже розве-
дених розчинах присутні частково у вигляді іонів, частково у вигля-
ді недіссоціірованних молекул. Крім закону діючих мас в розчинах
електролітів застосовують основні закони:
–
рівняння матеріального балансу – число атомів даного типу в
ізольованій системі незмінно;
–
рівняння електронейтральності – сумарний заряд гомогенної
рідкої системи, що містить дисоційовані електроліти, дорівнює нулю.
Електроліти за своєю природою бувають іонофорні й іоногенні.
Іонофорні електроліти до розчинення складаються з іонів (іон-
ні кристали). Перехід іонів в розчин відбувається в результаті взає-
модії їх з полярними молекулами розчинника внаслідок утворення
зв'язків між іонами і молекулами розчинника. Під дією електрично-
го поля іонів полярні молекули розчинника притягуються та орієн-
туються близько іонів, які в свою чергу, притягаючи іони до себе,
послаблюють зв'язок між ними. Проводиться робота роз'єднання іо-
нів при розчиненні за рахунок енергії сольватації.
У розчинниках з низкою діелектричною проникністю іони мо-
жуть взаємодіяти між собою та утворювати асоціати – іонні пари й
рівновага буде здійснюватися між іонами й іонними парами.
Крім того, у розчині можливі рівноваги між частками електро-
літу й молекулами (або іонами) розчинника.
Іоногенні електроліти в чистому вигляді складаються з недіс-
соціірованних молекул. Наприклад, молекула хлористого водню,
оцтової кислоти й ін. У полярному розчиннику, наприклад, у воді ці
речовини гідратуються, причому вони реагують з водою як з осно-
вою (за протолітичною теорією Бренстеда – Лоурі), пов'язані з кис-
лотою – іоном гідроксонію 3
HO
.
За сучасними уявленнями, іони в розчинах можуть утворюва-
тися в результаті протікання наступних типів реакцій:
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
14
–
дисоціація молекул розчинених речовин. Ці реакції характе-
ри в основному для водних розчинів солей і лугів, тобто для речо-
вин з іонним типом зв'язку:
NaCl Na Cl
,
NaOH Na OH
;
–
взаємодія молекул розчиненої речовини з молекулами роз-
чинника. Ці процеси характерні для розчинів слабких кислот і основ
(для речовин з ковалентним полярної зв'язком):
2
3
HCOOH HO HCOO HO
?
3
2
4
NHHONHOH
;
–
взаємодія іонів з молекулами розчинника:
2
HCOO
HO HCOOH OН
,
4
2
3
3
NH
HONHHO
.
У двох останніх випадках процес утворення іонів зводиться до
передачі протонів від однієї частинки до іншої. Такі реакції назива-
ються протолітичними.
Взаємодія іонів з молекулами розчинника визначає практично
всі властивості розчинів, однак кількісно ця взаємодія однозначна
виразити важко. Для цього використовують поняття чисел гідратації
–
кількість молекул зв'язаних іоном, однак виходячи з розмитості
границь близької й далекої сольватації величини, знайдені різними
способами, відрізняються часом у десятки раз. Єдиною характерис-
тикою, що однозначно кількісно характеризує процес сольватації, є
термодинамічні характеристики сольватації. Розуміючи під сольва-
тацією сукупність усіх енергетичних змін, пов'язаних з переходом
іонів зі стандартного стану у вакуумі в стандартний розчин, сольва-
тацію можна характеризувати зміною енергії Гіббса в цьому пере-
ході. Енергія сольватації являє собою виграш в енергії, який вихо-
дить при перенесенні моля досліджуваних іонів з вакууму в даний
розчинник. При цьому передбачається, що такий виграш енергії не
містить у собі електростатична взаємодія іонів, яка неминуче поз-
начилася б уже при введенні в розчинник другого й кожного насту-
пного іонів.
1.1. ЕЛЕКТРОЛІТИЧНА ДИСОЦІАЦІЯ, ІОННІ РІВНОВАГИ,
ТЕРМОДИНАМІЧНІ ВЛАСТИВОСТІ
15
Розрахунки енергій і тепло-
ти гідратації, виконані Борному й
Габером на підставі термохіміч-
них даних за допомогою циклів,
показують, що вони залежать від
радіуса іонів і їх заряду.
Можливість теоретичного
розрахунків енергії сольватації
вперше почата М. Борном у
(1920). Згідно із цією моделлю,
іон розглядається як заряджена
сфера радіуса ir , а розчинник, як суцільне однорідне середовище
(континуум) з діелектричної проникністю .
Процес переносу зарядженого кульки з вакууму в середовище
розбивається на три етапи:
1) розряд кульки у вакуумі;
2) перенос незарядженої кульки з вакууму в розчинник;
3) зарядження кульки в середовищі (рис. 1 .1).
При цьому передбачається, що робота на другому етапі 2А =
0, а для розрахунків роботи на етапах 1 і 3 ( 1А і 3А ) використову-
ються основні закони електростатики. Так, згідно із законом Куло-
на, сила, що діє на кожний із двох зарядів 1
q і 2q ( з урахуванням
знака), що перебувають у середовищі з діелектричної постійної на
відстані r , рівна
12
2
0
4
qq
F
r
.
Тому напруженість поля Х , тобто
сила, яка діє на заряд +1, що перебуває в середовищі на відстані r
від заряду q , становить
2
0
4
q
Х
r
.
Тому що напруженість поля пов'язана з електричним потенціалом
загальною формулою
d
Х
dr
, то для потенціалу на поверхні
сфери радіуса ir одержуємо:
Рис 1.1 . Схема для розрахунку
енергії сольватації іона за модел-
лю Борна
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
16
2
0
0
0
4
4
i
i
r
r
q
q
Xdr
dr
r
r
,
де нижня межа інтегрування відповідає вибору нульового потенціа-
лу на нескінченно великій відстані від розглянутої сфери. Тоді для
роботи зарядження сфери слушне рівняння
A=
2
0
0
0
0
4
8
q
q
i
i
q
q
dq
dq
r
r
,
а для іона із зарядом
i
qze
, для робіт зарядження у вакуумі 1А й у
середовищі розчинника 3
А маємо
22
1
0
8
i
i
ze
А
r
й
22
2
0
8
i
i
ze
А
r
.
Враховуючи, що вільна енергія сольватації s
A
i
i
GN
A
для
енергії сольватації моля іонів маємо шукане рівняння відоме як рів-
няння Борна для сольватації
22
22
22
1
1
8
8
8
i
i
i
A
A
A
s
oi
oi
oi
zeN
zeN
zeN
G
r
r
r
.
део
– діелектрична проникність вакууму 8,854·10–12
Ф/м, NА – чис-
ло Авогадро 6,022·1023
моль
–1
; e – елементарний електричний заряд
1,602·10–19
Кл; ir і iz – радіус і заряд іона відповідно; – відносна
діелектрична проникність розчинника.
У цім рівнянні розчинник характеризується основним параме-
тром – відносною діелектричною проникністю , а взаємодія іон –
молекула розчинника розглядається з позицій електростатичної вза-
ємодії,
Якщо скористатися рівнянням Гіббса – Гельмгольца, згідно з
яким
()
s
s
s
dG
H
GT
dT
, можна одержати вираження для ента-
льпії сольватації. Ентальпія й енергія сольватації зв'язані між собою
1.1. ЕЛЕКТРОЛІТИЧНА ДИСОЦІАЦІЯ, ІОННІ РІВНОВАГИ,
ТЕРМОДИНАМІЧНІ ВЛАСТИВОСТІ
17
рівнянням Борна – Б'єррума, згідно з яким у перерахуванні на один
моль
2
1
1
.
8
i
A
s
oi
zeN
T
H
r
T
Аналітичні методи розрахунків дають хімічну енергію сольва-
тації іона
,
siх
G
, а експериментально одержують тільки реальну
,
sip
G
, між якими існує зв'язок
,
sip
G
=
,
siр
i
i
G zF
.
Середнє число молекул розчинника, міцно пов'язане з іоном,
називається числом сольватації s
n.
Безумовно, висновки теорії Борна є наближеними, тому що не
враховують хімічну природу й структуру розчинника й електронну
конфігурацію іонів, але якісно правильно описує зміни енергії соль-
ватації при зміні температури фізико-хімічних властивостей роз-
чинника й заряду іонів. Енергії гідратації, розраховані по моделі
Борна, досить значні й достатні для руйнування кристалічної решіт-
ки при утворі розчинів електролітів. Деталізація мікроскопічної бу-
дови розчинів, розвиток молекулярно-статистичних теорій і розра-
хункових методів дозволяють у цей час використовувати значно
більш складні моделі й у багатьох випадках усунути відмінності
між розрахованими й експериментальними значеннями.
Для розчинів електролітів слушне застосування закону діючих
мас і кількісно рівновага між компонентами розчину характеризу-
ється константою рівноваги. Так, для електроліту, що дисоціює не
повністю K A
за рівнянням
KA
z
K
+
z
A
с утворенням катіонів
z
K
c зарядом z і аніонів
z
Aіззаря-
дом z
, можна записати
[][].
[
]
z
z
c
K
A
K
KA
У вираженні в квадратних дужках позначені рівноважні кон-
центрації часток у розчині й константа c
K є концентраційною конс-
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
18
тантою рівноваги. Концентраційна константа рівноваги має постій-
не значень тільки в дуже розведених розчинах, коли властивості ро-
зчинів наближаються по поведінці до ідеального. Тому що розчини
електролітів містять заряджені частки – іони, між якими навіть у ро-
зведених розчинах здійснюється сильна електростатична взаємодія,
концентраційна константа залежить від концентрації розчину.
Для приведення у відповідність теорії із практикою й збере-
ження багатьох зручних співвідношень, отриманих на підставі тео-
рії Арреніуса, Г. Льюіс і М. Рендалл запропонували використовува-
ти замість концентрацій активності. Тоді всі термодинамічні спів-
відношення, записані у формі рівнянь для ідеальних розчинів, але
утримуючі не концентрації, а активності, строго узгодяться з експе-
риментальними даними. Це дозволило формально врахувати все рі-
зноманіття взаємодій у розчинах без обліку їх фізичної природи.
Для опису поведінки реальних розчинів користуються термо-
динамічною константою дисоціації, яка має ідентичний вигляд, але
виражається через рівноважні активності частинок у розчині:
.
z
z
K
A
a
KA
a
a
K
a
Відхилення властивостей розчинів електролітів від властивос-
тей ідеального розчину обумовлене наявністю сил електростатичної
взаємодії, яка проявляється на набагато більших відстанях, чому
сили ван-дер-ваальсівської взаємодії в молекулярних розчинах. Цим
обумовлена наявність відхилень навіть у дуже розведених розчинах,
з концентрацією менш 0,001 моль/л.
Для розведених розчинів електролітів задовільна згода з екс-
периментом дає теорія розчинів електролітів, створена на основі ро-
згляду електростатичної взаємодії між іонами в розчині Дебаєм і
Хюккелем (1923). У теорії передбачається, що:
–
електроліт дисоційований повністю;
–
характер розподілу іонів у розчині обумовлений електроста-
тичною взаємодією іонів різних знаків і їх тепловим рухом, що при-
водить до утвору так званої іонної атмосфери навколо будь-якого
іона, обраного за центральний;
1.1. ЕЛЕКТРОЛІТИЧНА ДИСОЦІАЦІЯ, ІОННІ РІВНОВАГИ,
ТЕРМОДИНАМІЧНІ ВЛАСТИВОСТІ
19
–
розчинник представляється якоїсь безперервним середовищем з
єдиною характеристикою – його діелектричною проникністю ;
–
енергія міжчастинкової взаємодії розглядається як енергія
електростатичної взаємодії іона з його іонною атмосферою.
Енергія взаємодії j-го іона з іонною атмосферою визначається
вираженням
22
,
8
j
j
o
ze
U
де
1
2
2
2
jj
o
e
nz
kT
.
Величина 1 / ототожнюється з радіусом іонної атмосфери й у
молярній шкалі концентрацій обчислюється по формулі
3
o
2
10
1
.
2
A
kT
eNI
Енергія взаємодії пов'язана з коефіцієнтом активності іона j
співвідношенням
ln.
j
j
UkT
Теорія Дебая –Хюккеля дає рівняння для розрахунків коефіці-
єнтів активності окремого виду заряджених часток, у першому на-
ближенні
2
lg
,
j
j
AzI
і в другому наближенні теорії, що враховує формально середній ді-
аметр іонів електроліту, виражається рівнянням
2
o
lg
,
1
j
j
zAI
BaI
у якому A і B – теоретичні коефіцієнти, що залежать від температу-
ри й діелектричної проникності середовища, для водних розчинів за
температури 298 К вони мають значення 0,5115 та 0,329·1010
, відпо-
відно;
o
a – напівемпіричний параметр, що має зміст відстані найбі-
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
20
льшого зближення іонів, близький до середнього розміру іонів: 3–
5·10–10
м; I – іонна сила (іонність) розчину, функція іонної концент-
рації всіх заряджених форм у розчині, що й розраховується як напі-
всума добутку концентрації кожного іона на квадрат його заряду за
рівнянням
2
1
.
2ii
I
zm
Якщо прийняти відстань найбільшого зближення іонів рівним
3,04 А° (1 А° – Ангстрем, дорівнює 10–10
м), то середні іонні коефі-
цієнти активності іоніd можна розрахувати за рівнянням Гюнтель-
берга
lg
,
1
zzAI
I
Згідно з теорією вплив концентрації на коефіцієнти активності
іонів відбувається тільки через іонну силу розчину. Рівняння Дебая
–
Хюккеля підтверджують експериментально встановлену раніше
Льюісом і Рендаллом закономірність, яка зветься правила іонної си-
ли: у розчині з даною іонною силою всі електроліти характеризують-
ся значенням коефіцієнта активності, що не залежить від природи й
концентрації речовини, а залежні тільки від заряду його іонів.
Рівняння Дебая – Хюккеля для коефіцієнтів активності засто-
совне для опису властивостей розчинів при низьких значеннях іон-
них сил: до 0,02–0,05 моль/л; у концентрованих розчинах спостері-
гаються істотні відхилення, пов'язані із проявом близькодіючих сил,
які теорія не враховує. У рамках другого наближення неможливо
пояснити збільшення коефіцієнтів активності при високих концент-
раціях. У зв'язку із цим Дебай і Хюккель доповнили рівняння чисто
емпіричним доданком і одержали так зване третє наближення те-
орії, яке для середніх іонних коефіцієнтів активності має вигляд
2
o
lg
,
1
j
j
b
zAI
I
BaI
де b – емпірична константа. Як було показано надалі Р. Робінсоном
і Р. Стоксом (1948), фізичний зміст параметра b можна обґрунтува-
ти, якщо в якості діелектричного середовища, у яке поміщені іони,
1.1. ЕЛЕКТРОЛІТИЧНА ДИСОЦІАЦІЯ, ІОННІ РІВНОВАГИ,
ТЕРМОДИНАМІЧНІ ВЛАСТИВОСТІ
21
розглядати не весь розчинник, а лише ту його частину, яка не вхо-
дить у сольватні оболонки іонів.
Константи дисоціації кислот варіюють у дуже широких межах.
Тому часто використовують не самі константи, а взяті зі знаком мі-
нус їх десяткові логарифми (негативне значення показника ступені
константи). Ці величини прийнято позначати символом pK :
lg
pK
K
..
Уявлення про рівновагу в розчині можуть бути застосовані й
безпосередньо до самого розчинника, якщо його молекули здатні
розпадатися на іони. Так у таких однокомпонентних системах мож-
ливі протолітичні рівноваги, що полягають у передачі протонів від
одних молекул до інших. Це відбувається в рідкому аміаку, нітрат-
ній кислоті
3
3
4
2,
NH NH
NH NH
3
3
23
3
2
2
3,
HNO HNO
HNO NO
HONONO
а також у воді
2
2
3
HO HO
HO OH
с утворенням іона гідроксонію 3
H O й гідроксилу OH
.
Загальним для всіх цих реакцій є те, що одна молекула є доно-
ром протонів (кислотою), а інша акцептором (підставою). Таким
чином, у кожній протолітичній рівновазі бере участь сполучена па-
ра кислота – основа. Такі системи (розчинники) називаються само-
протонуючими, а рівноваги, у яких протон переходить від однієї
молекули до такої ж іншої, – автопротолітичними.
У двокомпонентних системах компоненти, що містять, з різ-
ними протонодонорними й протоноакцепторними властивостями,
також можливий перенос протона; у такій рівновазі також беруть
участь сполучені пари, наприклад,
реакція дисоціації
3
2
3
3,
CHCOOH HO
CHCOO HO
3
2
3
3
HNO HO
HO NO
;
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
22
реакція гідролізу
2
4
2
3
3
4
3
HO
NHHONHHO
NHOH HO
,
3
2
3
CHCOO HO
CH COOH OH
;
реакція нейтралізації
3
3
3
4,
CH COOH NH
CH COO NH
2
.
HCl NaOH
HOHCl
Вода по своїй хімічній природі є дуже слабким електролітом і
нівелює (вирівнює) донорну здатність мінеральних кислот, тобто у
воді всі вони є донорами протонів. Відмінність донорної здатності
проявляється в сумішах кислот і деякі з них можна розташувати в
ряд, що характеризує властивості, що диференціюють, кислот:
4
24
653
3.
HClO HSO CHSOH HCl HBr HI HNO
Термодинамічна константа реакції дисоціації води
2
2
3
HO HO
HO OH
,
яку часто записують спрощено у вигляді
2
HO
HOH
записується через рівноважні активності вираженням
2
.
H
OH
a
HO
a
a
K
a
Оскільки активність чистої води
2
1,
HO
a
то, добуток активно-
сті іонів гідроксонію й гідроксилу є величиною постійної й зветься
іонним добутком води w
K . Значення іонного добутку води залежить
тільки від температури й дорівнює за температури 298 К 1,008·10–14
.
Звідси рівноважна концентрація (активність) іонів дуже мала й до-
рівнює
7
10
H
w
a
K
г-іон/л.
1.1. ЕЛЕКТРОЛІТИЧНА ДИСОЦІАЦІЯ, ІОННІ РІВНОВАГИ,
ТЕРМОДИНАМІЧНІ ВЛАСТИВОСТІ
23
Збільшення температури приводить до росту числа як іонів гі-
дроксонію, так і гідроксилу, тому з ростом температури pH змен-
шується. Так, при 100 С: w
K = 10–12
,аpH=6.
При розчиненні у воді кислот або підстав концентрація іонів
водню й гідроксильних іонів змінюється й взаємно визначається від-
повідно до вираження іонного добутку води.
Багатоосновні кислоти дисоціюють послідовно і відщіплення
кожного з іонів водню характеризується відповідною константою
дисоціації. Наприклад, при дисоціації фосфорної кислоти маємо при
291 К
3
34
2
3
24
,1
,
7,610 ;
a
HPO HO
HOHPOK
2
8
24
2
3
4
,2
,
5,910 ;
a
HPO HO
HO HPO
K
2
3
13
4
2
3
4
,3
,
3,510 .
a
HPO HO
HO PO
K
Відщіплення першого іона водню походить від кислотного за-
лишку, що несе один заряд, другого, – несучого вже два негативні
заряди і т. д. Тому для відриву кожного наступного іона водню від
кислотного залишку потрібно все більшої енергії й константа дисо-
ціації відповідно зменшується.
За допомогою константи дисоціації кислоти й іонного добутку
води можна охарактеризувати всі інші кислотно-основні перетво-
рення сполученої пари кислота – основа у водяних розчинах. Так,
для основи B при розчиненні у воді характерне утворення іонів
OH у реакції
2
BHO
BH OH
.
Константа рівноваги
BH
OH
b
B
a
a
K
a
називається константою основності основи B . Неважко показати,
що вона виражається через константу дисоціації a
K сполученої ки-
слоти BH
й іонний добуток води
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
24
3
3
.
HO
OH
BH
OH
w
b
B
B
a
HO
BH
a
a
a
a
K
K
a
aa
a
K
Деякі іони металів з'єднуються з аніонами або нейтральними
молекулами й утворюють так звані комплексні іони. Наприклад, ро-
зчини
2
CdI містить іони й молекули CdI
,
2
CdI ,
3
CdI і
2
4
CdI
, які
перебувають у рівновазі з іонами
2
CdіI
. У якості інших прик-
ладів комплексних іонів можна привести
2
()
AgCN
,
2
34
()
CuNH
,
2
()
FeOH
.
Константи дисоціації комплексних іонів називаються конста-
нтами нестійкості комплексу, а константи рівноваги зворотного
процесу – константою утвору комплексного іона. Рівноваги утвору
комплексних іонів і дисоціації багатоосновних кислот ідентичні.
У розчинах можливі й гетерогенні рівноваги між малорозчин-
ною сіллю (основою), що утворюють іони в розчині. Наприклад, над
осадом
()
тв
AgCl у рівновазі з його насиченим розчином перебуває
деяка кількість розчиненого хлориду срібла, які практично повністю
дисоційовані на іони й, отже, має місце рівновага
()
тв
AgCl
Ag Cl
.
При розчиненні твердих речовин затрачається енергія, що йде
на руйнування кристалічної решітки. Енергія решітки визначається
за рівнянням Борна
2
кр.гр.
4
1
1,
o
o
AM
e
r
zz
U
NK
n
де
4
2
18
1
;
о
M
r
n
Ke
– коефіцієнт стискальності кристалу, KM –
константа Моделунга.
У розчинах може мати місце й окиснювально-відновна рівно-
вага, наприклад
2
2
2
2
Br Cl
Br Cl
,
до якого також застосуємо закон діючих мас
1.1. ЕЛЕКТРОЛІТИЧНА ДИСОЦІАЦІЯ, ІОННІ РІВНОВАГИ,
ТЕРМОДИНАМІЧНІ ВЛАСТИВОСТІ
25
2
2
2
2
.
Br
Cl
Cl
Br
a
a
K
a
a
КОНТРОЛЬНІ ПИТАННЯ ПО ТЕМІ
1. Чи можна передбачити, у якого з іонів Li
,
Rb
,Cs
енер-
гія сольватації більша?
2. Яку роль відіграють процеси гідратації (сольватації) при ро-
згляді рівноважних властивостей розчинів електролітів?
3. Як експериментальним шляхом визначити енергію гідрата-
ції? Чи можна визначити енергію сольватації кожного виду іонів?
4. Які властивості розчинника є визначальними в його здатнос-
ті іонізувати розчинена речовина?
5. За рахунок чого виходить головним чином більша енергія,
необхідна для руйнування порівняно міцних кристалічних решіток
іонних кристалів при розчиненні електролітів?
6. Чи змінюється константа дисоціації електроліту з ростом ді-
електричної проникності розчинника, якщо розчинники близькі по
своїх хімічних властивостях?
7. У чому особливості застосування закону діючих мас до роз-
чинів електролітів?
8. Чим пояснюється значне зменшення констант послідовної
дисоціації багатоосновних кислот?
9. Що таке коефіцієнт активності, у яких випадках можна за-
мість активності використовувати концентрацію?
10. У чому сутність явища гідролізу (сольволізу)?
11.Що може бути кількісним заходом основності (кислотності)
розчинника?
12. Які допущення використовуються при висновку основного
рівняння Дебая – Хюккеля?
13. У чому полягає правило іонної сили?
14. Що таке активність електроліту, іона і як вона може бути
визначена?
15. На чому заснована ідея розрахунків активності згідно з те-
орією Дебая – Хюккеля?
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
26
16. Охарактеризуйте рівняння трьох наближень теорії Дебая –
Хюккеля.
17. Що називають константою нестійкості комплексу?
18. Чому з ростом температури збільшується іонний добуток
води?
19. Що можна сказати про вплив будови органічних кислот і
підстав на їхню силу? Для відповіді скористайтеся даними таблиці 5
додатка.
20. Як кількісно характеризують сольватацію іонів?
ЛІТЕРАТУРА
1. Курс физической химии, т. 2 под ред. чл. – корр. АН СССР
проф. Я . И. Герасимова. Издание 2, испр.,
–М. : «Химия», 1973.
–
624 с.
2. Практические работы по физической химии: Учебное посо-
бие для вузов / Под ред. К. П. Мищенко, А. А. Равделя и А. М. По-
номаревой. – Л. : Химия, 1982. – С . 153–173.
3. Батлер Дж. Ионные равновесия (математическое описание) /
Дж. Батлер. – Л. : Химия. 1973. – 448 с.
4. Ротинян А. Л., Тихонов К.И., Шошина И.А. Теоретическая
электрохимия / Под. ред. А. Л. Ротиняна. – М. : Студент, 2013. – 496 с.
5. Дамаскин Б. Б. и др. Электрохимия / Б. Б. Дамаскин, О. А.
Петрий, Г. А. Цирлина. – М. : Химия, КолосС, 2006. – 672 с.
6. Измайлов Н. А. Электрохимия растворов / Н. А. Измайлов. –
М. : Химия, 1986. – 576 с.
7. Ионная сольватация / Г. А. Крестов [и др.].
–
М. : Наука,
1987. – 320 с.
8. Ковальчук Є. П., Решетняк О. В . Фізична хімія : Підручник.
–
Львів : Видавничий центр ЛНУ Імені Івана Франка, 2007. – 800 с.
9. Крестов Г.А. Термодинамика ионных процессов в раство-
рах. – 2–е изд. перераб. и доп. – Л. : Химия, 1984. – 272 с.
10. Рабинович В. А. Термодинамическая активность ионов в ра-
створах электролитов / В. А. Рабинович. – Л. : Химия, 1985. – 176 с.
11. Скорчеллетти В. В . Теоретическая электрохимия / В. В .
Скорчеллетти.
–
Л. : Химия, 1974. – 608 с.
1.2. ЕЛЕКТРИЧНА ПРОВІДНІСТЬ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ
27
12. Теоретические и экспериментальные методы химии раство-
ров (Проблемы химии растворов) / Рос. акад. наук. Ин–т химии раст-
воров; отв. ред. А. Ю. Цивадзе. – М. : Проспект, 2011. – 684 с.
13. Бугаевский А. А. Расчет равновесий в растворе / А. А. Бу-
гаевский. – Х. : Вища школа. 1980. – 156 с.
14. Кукоз Ф. И. Равновесие и энергетика электрохимических си-
стем : учебное пособие / Ф. И. Кукоз. – Новочеркасск : Новочеркасс-
кий политехн. ин–т, 1993. – 134 с.
15. Миомандр Ф. Электрохимия / Ф. Миомандр, С. Садки, П.
Одебер, Р. Меале–Рено. пер. с фр. – М. : Техносфера, 2008. – 359 с.
1.2. ЕЛЕКТРИЧНА ПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ
Електроліти – це речовини, що дисоціюють в розчинах (розп-
лавах) на іони, які обумовлюють під впливом прикладеного зовніш-
нього електричного поля електричну провідність. Електроліти став-
ляться до провідників другого роду, на відміну від металів – прові-
дників першого роду, електрична провідність у яких обумовлена
спрямованим рухом електронів. Електроліти можна розділити на
сильні й слабкі. Перші – з іонним межчастинковим зв'язком (крис-
тали солей) – іонофор и – дисоціюють у розчині практично повніс-
тю; другі – переважно з ковалентними зв'язками (органічні й деякі
мінеральні кислоти й основи) – іоногени – дисоціюють частково.
Кількісно процес дисоціації характеризують ступенем дисоціації
( – частка продисоційованих молекул).
При накладенням на гніздо з розчином електричного поля
термодинамічна рівновага, що встановилася, у якому перебувала
система, порушується, відбувається спрямоване переміщення заря-
джених часток (електричний струм). Здатність провідника проводи-
ти електричний струм характеризується його електричною провід-
ністю L, яку можна визначити як кількість електрики, стерпного че-
рез перетин провідника в одиницю часу при одиничній напрузі.
Опір провідника R будь-якого типу незалежно від способу пе-
реносу зарядів, пропорційно його довжині l і назад пропорційно
його перетину S
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
28
,
l
R
S
де – питомий опір, рівний опору R провідника за умови одинич-
них довжині й перетину.
Електрична провідність L обернено пропорційна опору провід-
ника R, виміряється в Сименсах (1 См = 1 Ом
–1
):
11
,
S
S
L
R
l
l
де – питома електрична провідність, являє собою електроп-
ровідність одиничного об'єму розчину (1 см
3
), поміщеного між па-
ралельними електродами одиничної площі (1 см
2
), що перебувають
на відстані, рівному 1 см.
На відміну від провідників першого роду, у яких носіями елект-
рики служать електрони, а питома електропровідність при даній те-
мпературі постійна й залежить тільки від природи металу, у провід-
никах другого роду питома електрична провідність залежить ще й
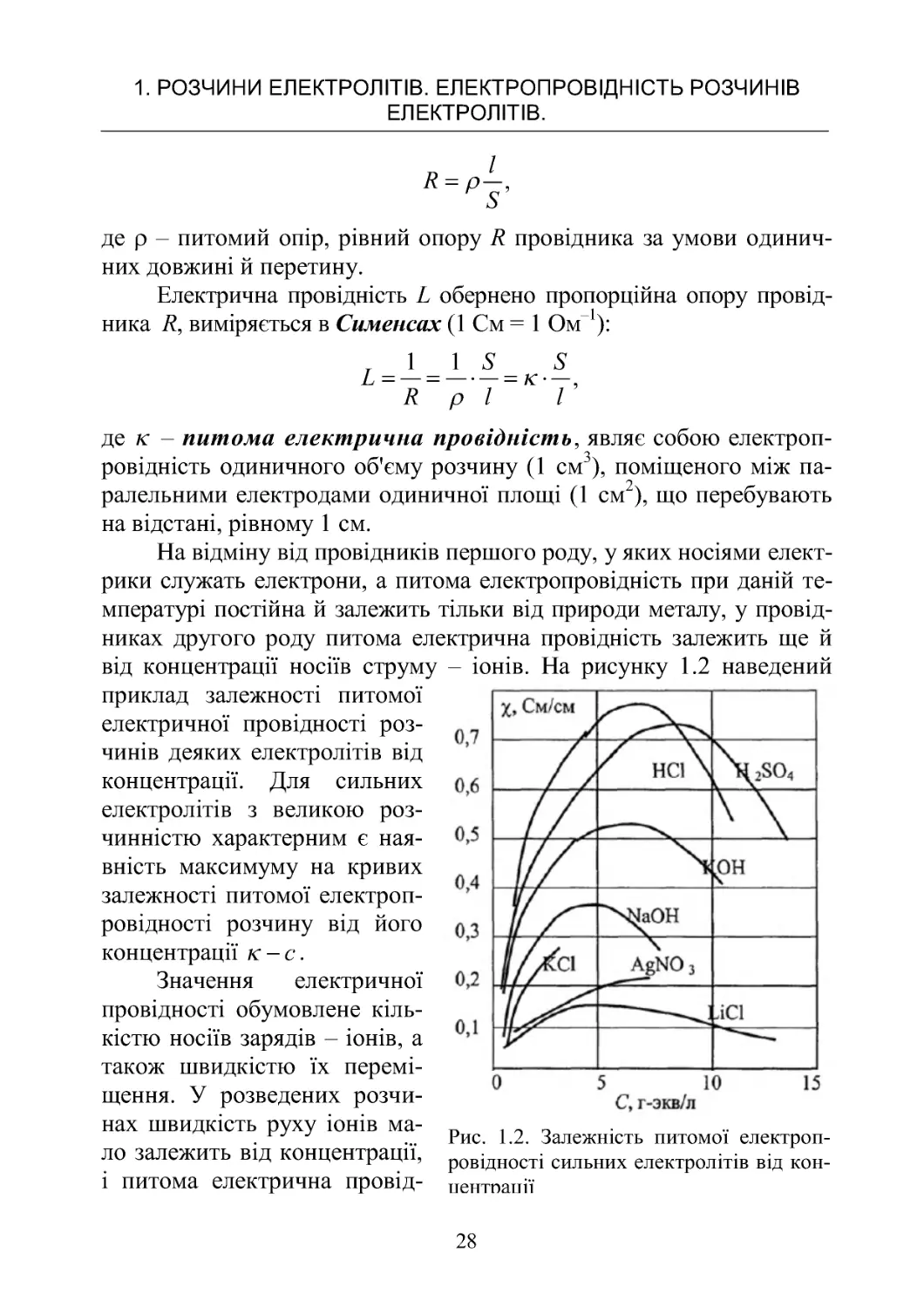
від концентрації носіїв струму – іонів. На рисунку 1.2 наведений
приклад залежності питомої
електричної провідності роз-
чинів деяких електролітів від
концентрації. Для сильних
електролітів з великою роз-
чинністю характерним є ная-
вність максимуму на кривих
залежності питомої електроп-
ровідності розчину від його
концентрації c
.
Значення
електричної
провідності обумовлене кіль-
кістю носіїв зарядів – іонів, а
також швидкістю їх перемі-
щення. У розведених розчи-
нах швидкість руху іонів ма-
ло залежить від концентрації,
і питома електрична провід-
Рис. 1 .2 . Залежність питомої електроп-
ровідності сильних електролітів від кон-
центрації
1.2. ЕЛЕКТРИЧНА ПРОВІДНІСТЬ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ
29
ність зростає майже прямо пропорційно кількості часток в одиниці
об'єму розчину – концентрації. У міру збільшення концентрації під-
силюється міжіонна взаємодія, зростає щільність іонної атмосфери,
збільшується ймовірність утвору асоціатів, що приводить до змен-
шення швидкості руху іонів. Перевага цього ефекту над збільшенням
числа іонів обумовлює при високих концентраціях зменшення пито-
мої електричної провідності й поява екстремуму на кривій.
У розчинах слабких електролітів іонна концентрація невелика
й швидкість руху іонів мало залежить від концентрації, але зі змі-
ною концентрації змінюється ступінь дисоціації електроліту, а, от-
же, і концентрація іонів, якої й визначається величина питомої еле-
ктричної провідності.
Для розчинів електролітів виконується закон Ома, згідно з
яким сила струму, що проходить через провідник, пропорційна при-
кладеній напрузі й обернено пропорційна опору провідника, тобто
виражається рівнянням
.
U
S
I
ULU
R
l
Тоді
l
U
i
S
l
виражає густину струму (А/см
2
);U
l
–
градієнт по-
тенціалу або напруженість поля (В/см).
Іони в розчині перебувають у стані хаотичного теплового руху:
при накладенні зовнішнього електричного поля рух стає спрямова-
ним з якоїсь постійною швидкістю v пропорційної градієнту потен-
ціалу
.
U
vu
l
Рух іонів в електричному полі є рівномірним, тому що її збі-
льшення під дією зовнішнього електричного поля викликає й збі-
льшення опору руху в грузлому середовищу. Коефіцієнт пропор-
ційності u зветься електричної абсолютної рухливістю іона й
дорівнює швидкості руху при одиничному градієнті потенціалу.
Густина струму, стерпного катіонами й аніонами i й i
, про-
порційна їхнім зарядам iz , концентрації ic (моль/см
3
) і швидкості
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
30
спрямованого руху
U
u
l
:
,
U
izcu
F
l
,
U
izcu
F
l
де F – число Фарадея (кількість електрики, стерпна 1 моль елект-
ронів,
A
FNe
; NA – число Авогадро, e – заряд електрона.
Сумарна густина струму іонів обох знаків дорівнює
(
),
ii
U
l
iiizc
Fuu
оскільки
.
ii
zc
zc
zc
Дорівнюючи отримане вираження для щільності струму з вира-
женням закону Ома, одержимо для питомої електропровідності роз-
чину
(
),
ii
zcFuu
Для сильних електролітів концентрацію іонів будь-якого знака
можна виразити через концентрацію електроліту в розчині ic
(моль/л);
/1000
i
i
cvc
, де iv – число іонів даного знака в молекулі
електроліту. Тоді маємо
(
) /1000,
zcFu u
де
ii
zvz
–
число г-екв в одному молі.
З рівняння видне, що питома електропровідність залежить від
концентрації електроліту й рухливості іонів.
Концентрація іонів у розчинах слабких електролітів залежить
від ступеня дисоціації Виразивши концентрацію іонів як
/1000
i
i
c
c
, одержимо вираження питомої електропровідності
для слабких електролітів
(
)
1000
ii
cFuu
z
Поряд з питомою електричною провідністю використовують
також величини молярної електричної провідності, частіше екві-
валентної електропровідності.
1.2. ЕЛЕКТРИЧНА ПРОВІДНІСТЬ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ
31
Остання – це відношення питомої електропровідності до кон-
центрації,
c
.
У системі CІ молярну електропровідність виражають у
См·м
2
·моль
–1
, а еквівалентну См·м
2
·г–екв. При цьому концентрацію
електроліту c виражають у моль/м
3
або екв/м
3
. Якщо виражати в
См·см
3
(СГС), а концентрацію, як звичайно, моль/л або екв/л, то
1000
(
)
,
FuuUU
c
де
,
UuF
UuF
–
рухливості іонів.
Еквівалентна електропровідність може бути виражена через
питому електропровідність і розведення :
,
де – розведення тобто – об'єм, у якому втримується 1 г-екв речо-
вини.
Хоча еквівалентна електрична провідність характеризує такий
об'єм розчину, у якому перебуває
завжди 1 г-екв електроліту, вона
залежить від концентрації – змен-
шується при збільшенні концент-
рації електроліту.
У розчинах сильних електро-
літів це пов'язане з тим, що при
збільшенні концентрації підсилю-
ється міжіонна взаємодія й швид-
кість руху іонів при цьому змен-
шується. У випадку слабких елект-
ролітів при зростанні концентрації
зменшується ступінь дисоціації, і
число іонів в об'ємі розчину змен-
шується. Залежність еквівалентної
електропровідності сильних і сла-
бких електролітів від концентрації
й кореня квадратного з концентра-
Рис. 1 .3 . Залежність еквівалент-
ної електропровідності електро-
літів (см
2
Ом
–1
моль
–1
) від концент-
рації
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
32
ції представлено на рисунках 1.3 і
1.4, відповідно.
Кольраушем експеримента-
льно встановлено, що в області
розведених розчинів молярна
електрична провідність лінійно
зменшується зі збільшенням ко-
реня квадратного з концентрації,
що виражається емпіричним рів-
нянням Кольрауша:
,
Ac
де
–
гранична молярна елек-
трична провідність, тобто елек-
трична провідність при нескін-
ченному розведенні (
при
0,
c томущо
1
);A–емпі-
рична константа, що залежить від
природи розчину.
Для слабких електролітів залежність еквівалентної електроп-
ровідності практично визначається зміною ступеня дисоціації
електроліту
(
)
,
UU
звідки
.
При нескінченному розведенні, коли = 1,
,
UU
що виражає закон Кольрауша – закон незалежного руху іонів: у
нескінченно розведених розчинах еквівалентна електропровідність
дорівнює сумі рухливостей іонів.
Якщо для даного електроліту відсутні табличні дані для іонів,
що становлять електроліт, значення для слабкого електроліту
Рис. 1 .4 . Залежність еквівалентної
електропровідності електролітів від
кореня з концентрації
1.2. ЕЛЕКТРИЧНА ПРОВІДНІСТЬ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ
33
можна знайти комбінацією значень сильних електролітів, обу-
мовлених досить просто – екстраполяцією по рівнянню Кольрауша.
Так, для визначення слабкої оцтової кислоти визначають експе-
риментально граничні електропровідності солей – сильних елек-
тролітів
3
CH COONa , HCl і NaCl і далі обчислюють
3
3
,
,
,
CH COOH
H
CH COO
=
3
,
,
,
.
CH COONa
HCl
NaCl
Гранична електрична провідність або граничні рухливості іо-
нів є важливими константами, що характеризують здатність даного
електроліту проводити електричний струм у розчині.
Рухливість j-го іона
,j
пов'язана з його радіусом jr рівнян-
ням Стокса
,
6
j
o
j
zeF
r
деo
– в'язкість розчинника, з якого випливає, що для того самого
іона або електроліту при нескінченному розведенні
,
6
j
jo
j
zeF
const
r
.
Це рівняння є кількісним вираженням правила Вальдена –
Писаржевського.
Значення граничної еквівалентної електричної провідності іо-
нів у розчині залежать від їхнього радіуса й заряду.
Для іонів з однаковим по величині зарядом рухливість тим бі-
льше, чим менше їх розмір (радіус). При цьому необхідно врахову-
вати, що у водяних розчинах іони гідратовані й, отже, мова йде про
розмір (радіусі) гідратованого іона. Через те, що маленькі іони гід-
ратуються сильніше, чим великі, співвідношення радіусів гідрато-
ваних іонів притилежно співвідношенню радіусів самих іонів:
,
K
Na
Li
r
r
r
але
,
.
,
.
,
.
,
K гiдр
Na гiдр
Li гiдр
r
r
r
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
34
внаслідок чого
,
,
,
K
Na
Li
.
Близькість розмірів однозарядних органічних аніонів
3
2
,
.
,
.
,
.
CH COOH гiдр CH CNOO гiдр
HOOCCOO гiдр
r
r
r
обумовлює близькість значень їх граничної еквівалентної електрич-
ної провідності:
3
2
,
,
,
CH COOH
CH CNOO
HOOCCOO
Збільшення заряду іона повинне приводити до збільшення йо-
го рухливості, однак у цьому випадку слід ураховувати той факт,
що зі збільшенням заряду іона збільшується розмір його гідратної
оболонки. Останнє негативно позначається на рухливості іона й
приводить у ряді випадків до того, що значення граничної еквівале-
нтної електропровідності багатозарядних іонів близькі до величин
однозарядних іонів, а іноді навіть нижче останніх:
3
2
,
,
,
Fe
Fe
Na
,
але
3
,
,
Al
Ag
и.
2
,
,
Cu
Cs
Електрична провідність розчинів закономірно збільшується з
ростом температури, внаслідок зменшення в'язкості середовища,
яке змінюється відповідно до рівняння
,
T
o
E
RT
e
у якому E – енергія активації в'язкої течії.
Зі зменшенням в'язкості падає опір руху іонів і швидкість їх
переміщення до електродів збільшується. Залежність еквівалентної
електропровідності розчину від температури може бути виражена
рівнянням
,
,
T
E
RT
Ae
де A – константа, що не залежить від температури; E – енергія ак-
тивації процесу, що визначає швидкість руху іона.
1.2. ЕЛЕКТРИЧНА ПРОВІДНІСТЬ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ
35
Логарифмуючи й диференціюючи рівняння по температурі,
одержимо
ln
1
E
d
d
dT
dT RT
або в інтегральній формі
1
ln
,
E
const
RT
що відбиває лінійну залежність ln від температури T .
Залежність питомої електричної провідності від температури
проходить через максимум, який визначається концентрацією, ти-
пом електроліту й природою розчинника.
Зменшення рухливості іонів і еквівалентної електропровіднос-
ті сильних електролітів з ростом концентрації пояснюється наявніс-
тю іонної атмосфери. Електростатична взаємодія іонів частково
впорядковує їхнє розташування, так, що поблизу даного (централь-
ного) іона більш імовірне знаходження протилежне заряджених іо-
нів, що утворюють симетричну іонну атмосферу із зарядом, рівним
по величині, але протилежним за знаком заряду центрального іона.
В електричному полі іони і їх іонні атмосфери рухаються в проти-
лежні сторони, що створює зустрічний потік, що захоплюються іон-
ною атмосферою молекул розчинника, тому середовище, у якому
рухається іон, переміщається йому назустріч. Внаслідок цього ви-
никає ефект електрофоретичного гальмування. У міру збільшення
концентрації збільшується й щільність іонної атмосфери, а, отже, і
прояв електрофоретичного ефекту.
Іонна атмосфера не рухається, як єдине ціле, а створюється й
руйнується в міру руху центрального іона. Цей процес відбувається
не миттєво, а вимагає деякого часу релаксації. Внаслідок цього по-
рушується симетричність іонної атмосфери: за іоном, що рухається,
виявляється надлишок іонів протилежного знака, електростатично
гальмуючих центральний іон. Таке гальмування називається релак-
саційним (релаксаційний ефект).
Ці уявлення дозволили П. Дебаю, Е. Хюккелю й Л. Онзагеру
одержати залежність еквівалентної електричної провідності від
концентрації електроліту
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
36
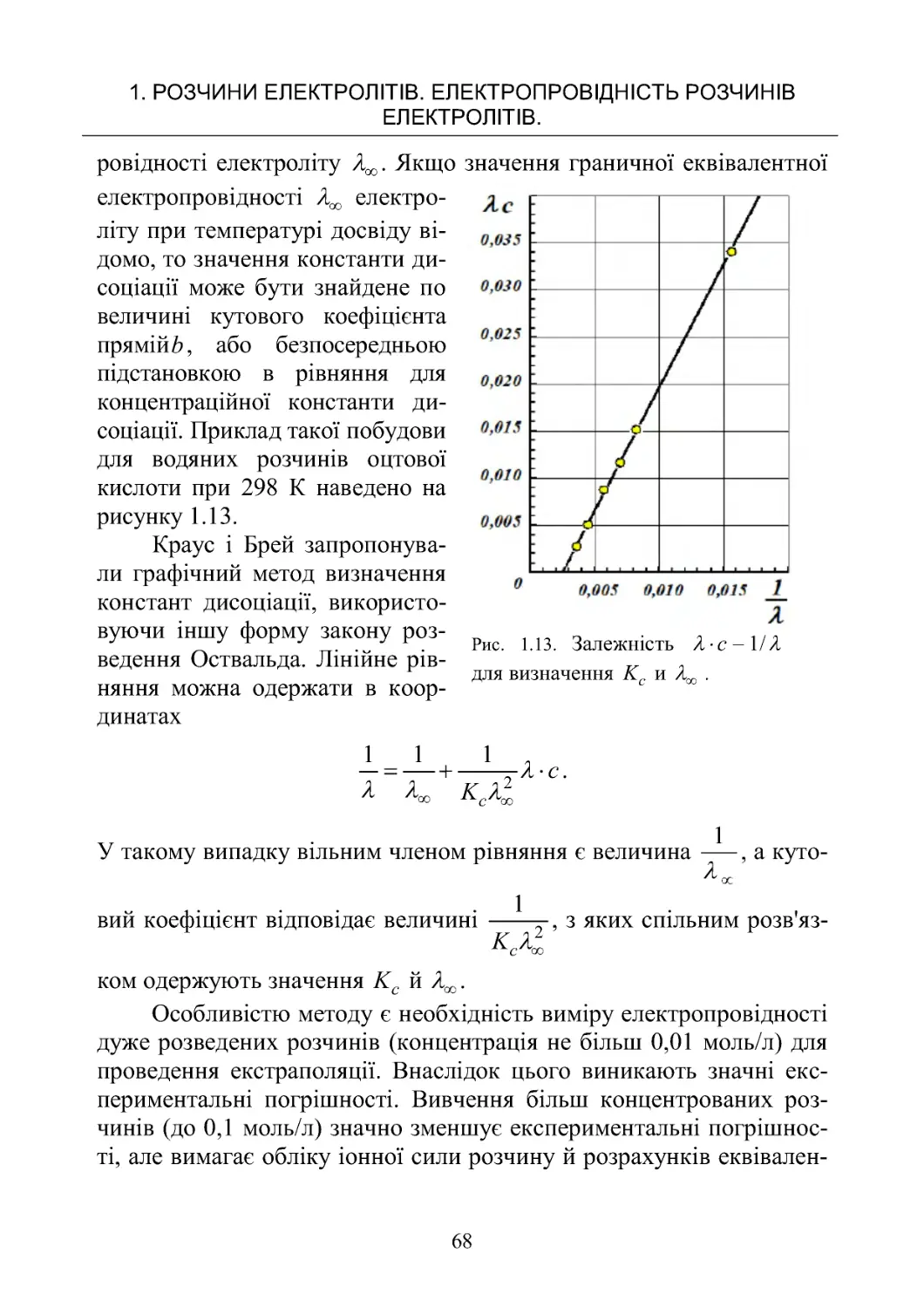
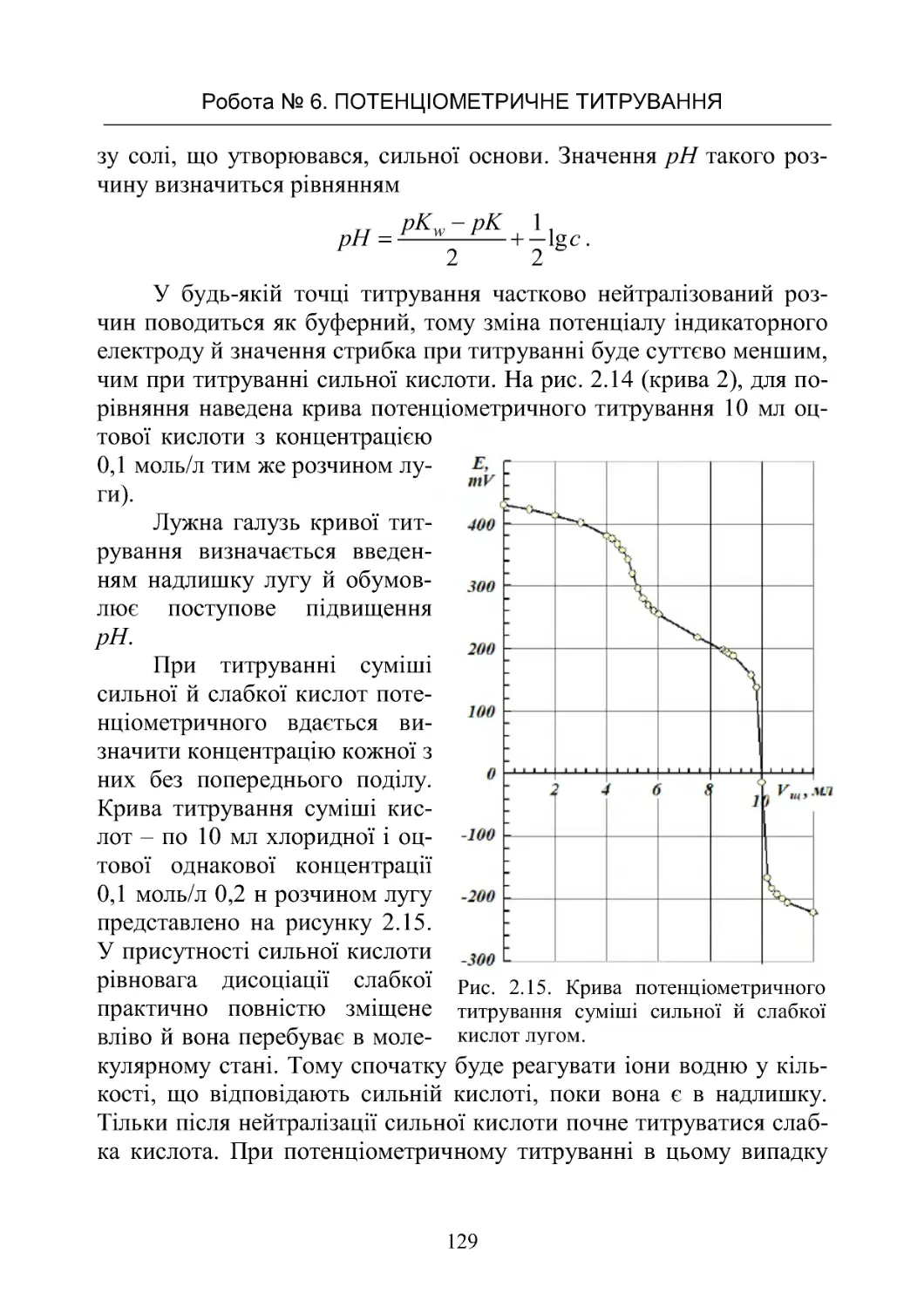
1
2
()
BB
cfc
,
де 1B й 2B – постійні, що залежать від температури, в'язкості й діе-
лектричної проникності розчинника й заряду іонів. Параметр 1B
ураховує електрофоретичний ефект, а 2
B – релаксаційний, f(c) –
деяка функція від концентрації c .
Для слабких електролітів, для яких дуже мала, другим і тре-
тім доданком можна зневажити в порівнянні з Тоді рівняння пе-
ретвориться у відоме співвідношення Арреніуса
.
Для сильних електролітів, для яких ( = 1, рівняння приймає
вигляд рівняння Фуосса - Онзагера
1
2
()
BB
cfc
.
У сильно розведених розчинах, у яких іони можна ототожнити
з матеріальними точками, останнім доданком можна зневажити й
рівняння перетвориться в рівняння Дебая – Хюккеля – Онзагера
1
2
BB
c
.
Коефіцієнти 1
Bиа 2
B , що характеризують електорофоретич-
ний та релаксаційний ефекти можуть бути розраховані згідно до
теоретичної моделі за равнянням
2
1/2
4
1
1/2
2
1/2
1
Смм
К
8, 248 10
г-екв
()
(г-екв/л)
Нс
B
Т
м
;
1/2
5
3/2
2
3/2
1
г-екв
8, 204 10
К
л
()
B
Т
.
У водних возчинах за температури 298 К за діелектричною
проникністю =78,3 та в'язкістю 8,937·10–4
Н·с/м
2
, рівняння Дебая
–
Хюккеля – Онзагера набуває вигляду
4
60,4 10 0,23
c
,
причому електрофоретичний ефект дає близько 60% від загального
ефекту гальмування.
1.2. ЕЛЕКТРИЧНА ПРОВІДНІСТЬ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ
37
Переконливим підтвердженням правильності уявлень Дебая й
Хюккеля про іонну атмосферу є ефект Вина (1927 р.). Тому що
швидкість утворення іонної атмосфери кінцева, то при великих на-
пруженостях поля (порядку 200 000 В/см) швидкість руху іонів буде
настільки великою, що іонна атмосфері не буде встигати утворюва-
тися, іони будуть рухатися без неї й еквівалентна електропровід-
ність повинна бути максимальної. Дійсно, за цих умов вона досягає
значення
.
Інше підтвердження (Дебай, Фалькенгаген, 1928 р.)
–
явище
дисперсії електропровідності (частотний ефект), що полягає в
помітному зростанні електропровідності при збільшенні частоти
струму вище деякої межі. При великих частотах змінного струму
взаємні зсуви іонної атмосфери й іона настільки малі, що іонна ат-
мосфера стає симетричною й гальмуючий ефект релаксації, обумов-
лений асиметрією іонної атмосфери, зникає. При цьому електрофо-
ретичний ефект залишається, тому що іонна атмосфера не зникає.
Кондуктометричний метод знайшов широке застосування у
фундаментальних дослідженнях розчинів електролітів, а також при
розв'язку багатьох прикладних завдань. Метод дає можливість про-
водити дослідження при різних температурах, тисках і концентра-
ціях електроліту ( від сильно розведених до розплавів) і практично в
будь-яких розчинниках. Метод простий і в той же час точний навіть
при низьких концентраціях, що дозволяє застосовувати досить
строгі теорії й модельні уявлення. Залежність електропровідності
від концентрації електроліту завжди описується рівнянням на осно-
ві фізичної моделі. Рівняння Онзагера заснована на примітивній мо-
делі, яка розглядає поведінку твердої зарядженої неполяризуємої
сфери радіуса j
a в континуумі c діелектричною проникністю й
в'язкістю , які такі самі, як в об'ємі розчинника. У рівняннях, за-
снованих на примітивній моделі, спочатку припускають, що елект-
роліт повністю дисоційований, а асоціація враховується згодом за
допомогою співвідношення діючих мас. Пізніше Фуосс, Лі й Уітон
запропонували рівняння електропровідності, засновані на більш ре-
алістичній фізичній картині, чому примітивна модель. Так, у моделі
Лі й Уітона навколо іона виділяються три області: у першій
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
38
(j
j
a
rR
),де j
a – кристалографічний радіус іона) молекули роз-
чинника орієнтуються в електричному полі іона і є ефект діелектри-
чного насичення; у другий ( ' j
j
RrR
), де jR – косфера Герні іо-
на, структура розчинника ще модифікована під впливом поля іона й
значення діелектричної постійної не збігається з об'ємної; а в третій
(j
rR
) властивості розчинника такі ж, як в об'ємі.
Враховуючи ефекти діелектричного насичення й включаючи
концепцію про іонну асоціацію, можливий опис електропровідності
сумішей електролітів, багатоосновних кислот і їх солей, хоча реалі-
зація можливостей вимагає розв'язку досить складних і громіздких
рівнянь, доступних для розв'язку тільки із застосуванням сучасних
комп’ютерних засобів обчислень.
Сучасні підходи в описі концентраційної залежності електроп-
ровідності дозволяють одержати інформацію про стан часток у роз-
чині, їх ефективному розмірі, рухливості й асоціації. Комбінація
кондуктометричного методу дослідження з виміром чисел переносу
дають можливість без яких-небудь допущень одержати транспортні
характеристики індивідуальних іонів. Експериментальні дані про
електропровідність сумішей багатоосновних кислот і їх солей при
використанні повного рівняння Лі й Уітона дозволяють одержати
індивідуальні транспортні характеристики іонів методом математи-
чної оптимізації параметрів математичної моделі рівноваги.
Наприкінці ХХ й початку XXI століття одержані значні успіхи
в теорії електропровідності. Є. М. Кузнєцовою зроблений опис кон-
центраційної залежності електропровідності розчинів електролітів
від розведених до насичених розчинів реалізований без традиційних
уявлень Дебая та Хюккеля на основі квазікристалічної моделі силь-
ного електроліту. М . М . Балдановим й Б. Б. Тангановим запропоно-
вана плазмоподібна теорія електролітів, яка описує розчин іоногену
як систему зарядів, які вагаються з плазмовою частотою, що зале-
жить як від властивостей самого електроліту, так і від макроскопіч-
них параметрів середовища. Це дозволило теоретично пояснити
зміну електропровідності розчинів сильних електролітів практично
у всьому діапазоні зміни концентрації іонів.
1.2.1. ВИМІР ЕЛЕКТРИЧНОЇ ПРОВІДНОСТІ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ
39
1.2.1. ВИМІР ЕЛЕКТРИЧНОЇ ПРОВІДНОСТІ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ
Вимір електричної провідності розчинів проводять у посуди-
нах (комірках) з міцно закріпленими нерухливими електродами, що
перебувають у розчині в момент виміру. Така конструкція при фік-
сованому об'ємі рідини, що заповнює гніздо, забезпечує сталість ві-
дстані між електродами й площі перетину провідника.
Вимірюваною експериментально величиною є опір розчину R,
яке пов'язане з питомою електропровідністю розчину співвідно-
шенням
1
,
c
k
l
l
R
S
S
де – питомий опір провідника, c
k – коефіцієнт пропорційності,
що залежить від геометричних особливостей гнізда: відстані між
електродами l і площею перетину S і називаний постійного гнізда
(cell). Чисельно c
k показує, у скільки раз опір стовпа розчину, що
перебуває між електродами, більше або менше питомого опору.
Значення c
k визначається як
c
lR
k
S
[см
–1
].
Постійну кондуктометричнох комірки (рис. 1 .5) неможливо
точно визначити прямим виміром довжини посудини й площини
його поперечного перерізу внаслідок :
–
розсіювання силових ліній струму, які не обмежуються стов-
пом розчину електроліту, що пере-
буває між електродами;
–
неможливості витримування
строгої паралельності електродів і
певної їхньої форми;
–
складної форми вимірюваль-
ної посудини, що обмежує розподіл
силових ліній.
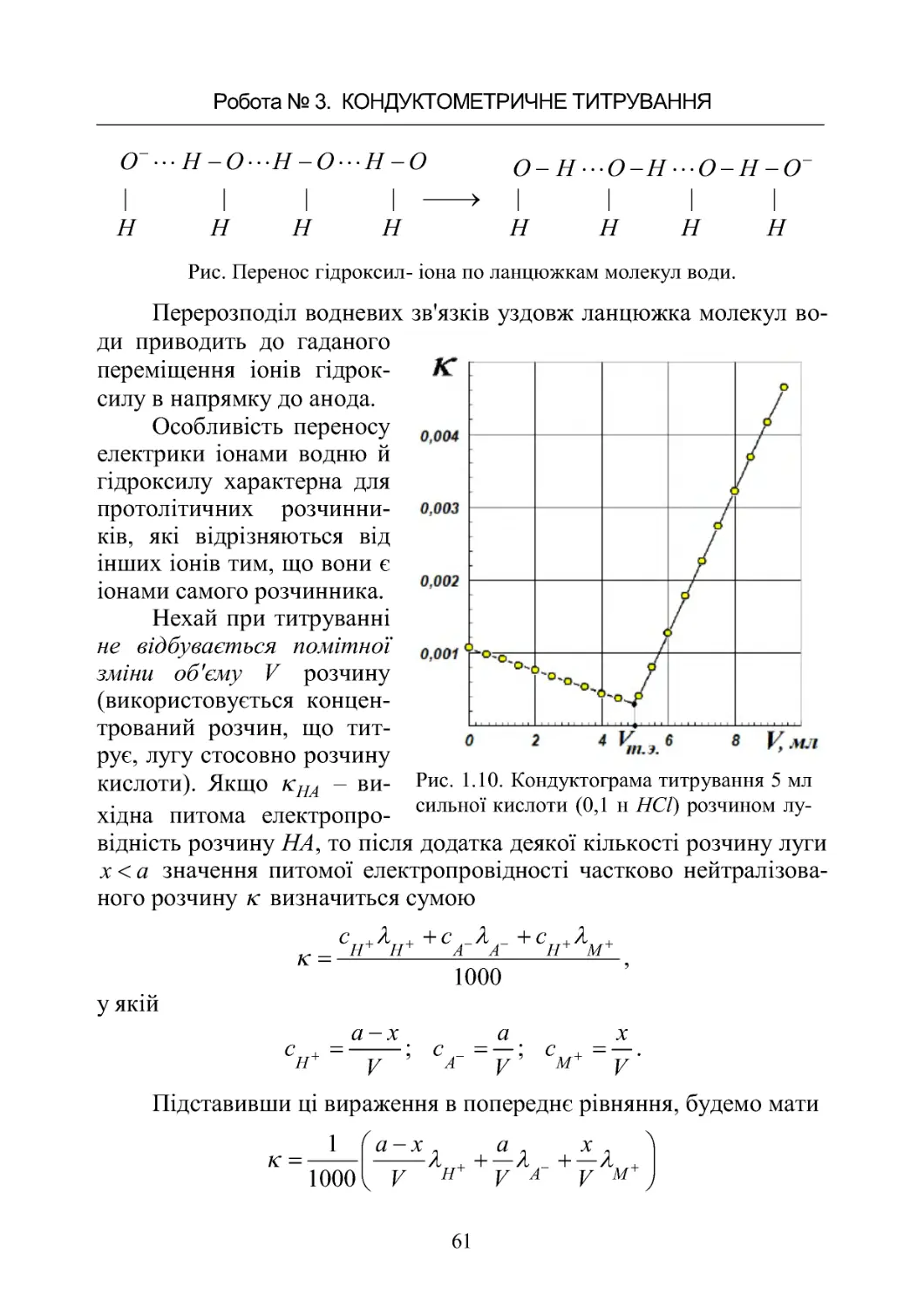
Чисельне значення постійної
посудини визначають по даним ви-
міру опору стандартних розчинів –
Рис. 1.5 . Напрямок поширення
струму в двоелектродній кондук-
тометричній комірці.
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
40
розчинів з точно відомими значеннями питомої електропровідності
при даній температурі. Такими найчастіше є водяні розчини хлори-
ду калію з концентрацією солі 0,01 або 0,02 моль/л, величина елект-
ропровідності яких при різних температурах відома з великою точ-
ністю. На рисунку 1.6 представлені деякі конструкції кондуктомет-
ричних комірок. Комірки а) і б) для виміру розведених і концент-
рованих розчинів, комірки в) і г) для кондуктометричного титру-
вання.
Процес виміру електропровідності будь-якого розчину зво-
диться до порівняння експериментально вимірюваних опорів даного
розчину р ну
R й опору стандартного розчину хлористого калію
KCl
R з відомою питомою електропровідністю KCl
водніййтієїж
вимірювальній комірці
KCl
рну
KCl
рну
R
R
.
Питома електропровідність ( KCl
,Ом
–1
·см
–1
) стандартних во-
дяних розчинів хлориду калію при деяких температурах наведена в
таблиці додатка.
Вимір опору розчинів, як правило, проводиться за допомогою
змінного струму, тому що зміна напрямку струму є кращим засобом
для усунення поляризаційного опору. Чим вище частота струму, тем
а)
б)
в)
г)
Рис. 1.6 . Деякі конструкції кондуктометричних комірок
1.2.1. ВИМІР ЕЛЕКТРИЧНОЇ ПРОВІДНОСТІ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ
41
менше будуть позначатися поляризаційні явища. Найбільш частий
вимір опору розчинів проводять при частоті змінного струму 1000
Гц. Робота більшості приладів для виміру опору заснована на мос-
товій схемі. Принцип виміру опору зрозумілий при розгляді схеми
мосту Уітстона (рис. 1.7).
Нехай струм від джерела надходить у точку A, де він розгалу-
жується до точок B і D і потім через точку C знову вертається до
джерела. Опору відрізків AB, BC, AD і DC відповідно рівні R1, R2,
R3 і R4 . Якщо в точках B і D включити "нуль інструмент", то він
покаже відсутність струму тільки в тому випадку, коли спадання
напруги на ділянках AB, AD і відповідно BC і DC будуть одна-
кові. Це означає, що
1133
IRIR
і2244
IRIR
,
Де 1I , 2I , 3I і 4I – сили струмів, що протікають через відповідні
опори. Але тому що нульовий прилад показує відсутність різниці
потенціалів між точками B і D, то 1I = 2I і 3I = 4I . Тоді, побрав-
ши відносини спадання напруги на плечах мосту, будемо мати
33
11
22
44
IR
IR
IRIR
або
3
1
2
4
R
R
RR
.
Це рівняння виражає баланс
мосту. Практично на місце опору R1
ставлять посудину з розчином, опір
якого необхідно визначити, у якості
R2 – магазин опорів. Опори R3 і R4
вибираються таким чином, щоб вони
були одного порядку з вимірюваним
опором. Знаючи величини R2, R3 і
R4 у момент балансу мосту неважко
визначити й опір посудини з розчи-
ном електроліту. Якщо в якості дже-
Рис. 1.7 . Схема моста Уітстона
для виміру опору.
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
42
рела змінного струму використовувати звуковий генератор, то в
якості "нуль інструмента" може бути використаний телефон, у зага-
льному випадку використовують вібраційний гальванометр або ос-
цилограф.
На практиці для виміру опору застосовують спеціальні прила-
ди – мости змінного струму, наприклад, типу Р–38 або кондукто-
метри (Е7–11, Е7–17, Р–5010, Р–5079 й ін.). У цей час широке по-
ширення одержали універсальні прилади для виміру ряду електрич-
них параметрів RLC–Метри (RLC–100 – RLC–300, BR 2817, BR
2820 й ін.).
Принцип виміру всіх вимірників повного опору засновані на
аналізі проходження тестового сигналу із заданою частотою через
ланцюг, що володіє комплексним опором і наступним порівнянням
з опорною напругою. Напруга робочої частоти із внутрішнього ге-
нератора подається на вимірюваний об'єкт і на об'єкті виміряється
напруга. Струм, що протікає через об'єкт, за допомогою внутріш-
нього перетворювача струм-напруга перетвориться в напругу. Ви-
мір відносини цих двох напруг і дає повний опір ланцюга.
Обладнання й зовнішній вигляд сучасних портативних вимір-
ників LCR (мають вбудоване джерело живлення) і лабораторних
приладів з живленням від мережі ідентичний. Найбільш популярні й
доступні в цей час моделі серії BR–2822 компанією MCP, DE–5000
компанії DER ЕЕ.
ВИМІР ОПОРУ НА LCR–МЕТРІ DE –5000.
Для виміру комплексних параметрів ланцюгів на різних час-
тотах або комплексного опору призначені прилади, які називаються
вимірники імпедансу. Вимірник іммітансу – прилад, що вимірює
комплексну провідність. Найчастіше ці прилади називають вимір-
ники RLC.
Вимірник іммітансу DE–5000 призначений для визначення па-
раметрів повного опору або повної провідності електричного лан-
цюга. Прилад дозволяє також вимірювати: ємність, індуктивність,
еквівалентний послідовний опір (ESR), еквівалентний паралельний
опір, d–factor, добротність, фазове зрушення. Прилад постачений
1.2.1. ВИМІР ЕЛЕКТРИЧНОЇ ПРОВІДНОСТІ РОЗЧИНІВ ЕЛЕКТРОЛІТІВ
43
подвійним дисплеєм (мак-
симальне відображене чис-
ло 19999 / 9999). Точність
виміру опору змінному
струму на частоті 1000 Гц
становить на діапазонах:
20Ом–1%;200Ом20
кому – 0,3%; 200 кому –
0,5%;2МОм200МОм–
2%. Вимірник DE–5000
працює в режимі автома-
тичного вибору діапазону
повного опору змінного
струму й опору постійного
струму. Параметри L/C/R
можна вимірювати прямо
за допомогою інтелектуа-
льного режиму «AUTO–
LCR», не використовуючи
функціональну кнопку.
Прилад дозволяє ви-
бирати наступні значення
частоти виміру: 100 Гц /
120Гц/1кГц/10кГц/
100 кГц. Вимір опору роз-
чинів електролітів проводять на частоті змінної напруги 1кГц =
1000 Гц.
Обмірювані дані можна передавати в комп'ютер через інтерфейс
USB–IR.
Зовнішній вигляд і призначення елементів керування наведені
на рис. 1.8.
Для досягнення оптимальної точності вимірів опору, особливо
на найбільших і маленьких діапазонах, перед виміром потрібно
зменшити вплив паразитних складових іспитової арматур. Для цьо-
го виконується калібрування приладу (проводиться інженером пе-
ред початком роботи із приладом).
Рис. 1 .8 . Зовнішний вигляд й кнопки
управління LCR–метру DE–5000.
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
44
1 Рідкокристалічний дисплей
2 POWER
Кнопка включення/вимикання харчування при-
ладу
3 LCR AUTO
Режим автоматичного виміру параметрів LCR,
кнопка вибору виміру індуктивності, ємності,
опору й опору постійного струму
4 FREQ
Кнопка вибору частоти виміру
5☼
Підсвічування дисплея
6 SORTING Кнопка керування режимом сортування
7PC
Кнопка керування передачею даних
8 CAL
Кнопка запуску калібрування в режимі холостого
ходу/короткого замикання
9 D/Q/ESR/ Θ Кнопка вибору вторинних параметрів D/Q/ESR/Θ
10 SETUP
Кнопка виклику меню настроювання (у режим
сортування)
11 SER / PAL Кнопка вибору послідовної й паралельної схеми
12 ENTER
Кнопка керування меню настроювання (у режимі
сортування)
13 REL%
Кнопка режиму відносного виміру
14 HOLD
Кнопка режиму втримання даних
У режимі автоматичного відключення харчування (АОП) на
дисплеї буде відображатися індикація «АРО». Через п'ять хвилин
простою прилад буде автоматично вимикатися. Троєкратний звуко-
вий сигнал зумера попередить про те, що харчування буде виклю-
чено, потім на дисплеї з'явиться індикація «OFF» і прилад виклю-
читися.
Кнопка «LCR AUTO» вибирає функцію виміру первинного
параметра. Натискаючи цю кнопку можна по черзі вибрати режим
Auto–LCR, Auto–L Auto–C, Auto–R або Auto–DCR.
Включите харчування вимірника DE–5000. За замовчуванням
увімкнутися режим LCR Auto, який автоматично запустить вимір
параметрів L / C / R.
1.2.2. ГОТУВАННЯ РОЗЧИНІВ ДЛЯ ЕЛЕКТРОХІМІЧНИХ ДОСЛІДЖЕНЬ
45
Функція <CAL> включає калібрування внутрішніх параметрів
вимірника й залишкових явищ на зовнішніх розніманнях для вико-
нання нормального й точного виміру.
Кнопка <FREQ> вибирає частоту виміру. Натискаючи підряд
цю кнопку, можна вибрати одне з
п'яти значень: 1 кГц, 10 кГц, 100
кГц, 100 Гц або 120 Гц. За замовчу-
ванням установлена частота виміру
1 кГц.
Вимірювальне гніздо приєднує
через блок вимірних проводів і за-
тисками типу «крокодил». Результа-
ти виміру представляються на рід-
кокристалічному дисплеї (рис. 1 .9).
Таким чином, для проведення
вимірів опору досить приєднати до затисків провідники від елект-
родів гнізда, далі після встановлення постійного значення показання
зафіксувати в журналі. Додаткове настроювання й калібрування при-
ладу, заміна джерела автономного харчування або приєднання адап-
тера змінної напруги проводиться тільки інженером!
1.2.2. ГОТУВАННЯ РОЗЧИНІВ ДЛЯ ЕЛЕКТРОХІМІЧНИХ
ДОСЛІДЖЕНЬ
Електрохімічні виміри, проведені на сучасних приладах, до-
зволяють визначати електричні параметри (опір, електрорушійну
силу, силу струму) з погрішністю 0,01–0,1%. Це забезпечує 4–и зна-
чущих цифри результату виміру. Для одержання шуканих фізико-
хімічних величин з порівнянною точністю, у рівняння розрахунків
яких завжди входить концентрація розчину, виникає необхідність у
готуванні розчинів з погрішністю, що не перевищує погрішність
виміру електричних параметрів, тому що сумарна погрішність роз-
рахункових величин визначається погрішністю всіх вхідних у роз-
рахункову формулу експериментальних даних. Найбільшим обра-
зом вплив точності концентрації розчинів проявляється в кондукто-
метрії, тому що основною характеристикою є еквівалентна електро-
провідність розчинів, яка не виміряється безпосередньо на досвіді, а
Рис. 1.9 . Рідкокристалічний
дисплей показань виміру.
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
46
розраховується на підставі обмірюваних опорів розчинів (досліджу-
ваного й стандартного) і відповідних їм концентрацій. Відносна по-
грішність розрахованої еквівалентної електропровідності
c
єсу-
мою відносних погрішностей опорів і концентрацій виходячи зі
співвідношень:
o
1000
1000
c
KCl
c
KCl
c
R
с
Rc
,
o
c
KCl
c
KCl
R
R
c
.
Погрішність характеристик стандартного розчину хлориду ка-
лію
o
KCl
йKCl
R
дає систематичну погрішність (погрішність пос-
тійного гнізда), а вимірів
c
R
і c робочих розчинів може бути різ-
ної для різних розчинів. Ця погрішність може носити випадковий
характер, але також може систематично мінятися при зміні концен-
трації, вносячи закономірні функціональні викривлення в кінцевий
результат.
Аналіз рівняння для
c
показує, що щоб уникнути втрати точ-
ності необхідно вимірювати з однаковою відносною погрішністю як
концентрацію, так і опір досліджуваних розчинів – однаково точно
значить як малі, так і більші концентрації й опору. Бажаним є дося-
гнення рівноточності величин у ході вивчення концентраційної за-
лежності електропровідності розчинів.
У кондуктометрії в силу необхідності використання теоретич-
них рівнянь, використовується мольно-об'ємне вираження концент-
рації розчинів (моль/л), отже, готування розчинів пов'язане з необ-
хідністю точного виміру об'єму. Погрішність у визначенні кількості
розчиненого речовини зводиться до мінімуму використанням зва-
жування на аналітичних вагах. Тому основну погрішність вносить
об'єм використовуваного мірного посуду – мірних колб, піпеток і
бюреток.
У навчальній лабораторії рідко користуються каліброваним
мірним посудом, тому що калібрування вимагає підтримки постій-
ної температури, внаслідок зміни об'єму зі зміною температури ото-
чення. Тому об'єм розчину рідко відомий точніше 0,1%. Крім того
готування серії розчинів убутної концентрації для вивчень концент-
1.2.2. ГОТУВАННЯ РОЗЧИНІВ ДЛЯ ЕЛЕКТРОХІМІЧНИХ ДОСЛІДЖЕНЬ
47
раційної залежності розведенням вихідного розчину приводить до
явно вираженої нерівноточності даних, при відборі різних об'ємів
розчину при однаковій абсолютній погрішності. Так, при погрішно-
сті стандартної бюретки 0,05 мол відбір для розведення в мірній ко-
лбі 2,5 мол і 25 мол дає відмінність у відносній погрішності в 10 раз.
Як правило, розведення проводиться в 100 і 1000 раз, що, безсумні-
вно, дасть ще більші погрішності.
Найменш вимогливим до зовнішніх умов роботи, що й дають
найменшу погрішність і нерівноточність є метод готування розчинів
послідовним розведенням удвічі.
Основний розчин для роботи готується з максимальною
точністю: навішення речовини береться на аналітичних вагах у
кількості, щоб забезпечити задану погрішність, наприклад, при
відносній помилці 0,1% і точності зважування ±0,0005 г наві-
шення повинна бути не менш 0,5 г. Робочі розчини готуються
в сухих чистих колбах об'ємом в 2 рази перевищуючим зада-
ний об'єм розчинів. Піпеткою об'ємом, рівним передбачувано-
му об'єму робочих розчинів, у колби наливається чистий роз-
чинник, з точним набором рідини до мітки. Після цього піпет-
ка промивається основним розчином від розчинника, що зали-
шився на стінках піпетки. Відбирається основний розчин і пе-
реноситься в першу колбу. Розчин ретельно (1–2 хв) перемі-
шується, після чого набирається в піпетку для промивання її
стінок розведеним точно вдвічі розчином. Для готування серії
розведених розчинів цю процедуру повторюють багаторазово,
обов'язково перемішуючи, що розбавляється розчин і проми-
ваючи піпетку. У підсумку при бага торазовому розведенні на-
копичується відносна погрішність, яка зростає систематично й
визначається тільки погрішністю відліку об'єму піпетки, який
дуже незначний, на стільки, що можна вважати концентрації
розчинів рівноточними. Єдиним недоліком методу є незначна
швидкість убування концентрації – для розведення в 1000 раз
необхідно послідовно приготувати 10 розчинів (розведення
210). Але цього розведення часто цілком достатньо для прове-
дення досліджень, а кількість розчинів не є надлишковою.
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
48
Таким чином, метод послідовного розведення вдвічі має
ряд переваг:
–
не вимагає калібрування мірного посуду;
–
не залежить від температури готування розчинів;
–
погрішність готування розчинів мінімальна й концент-
рації приготовлених розчинів практично рівноточні.
КОНТРОЛЬНІ ПИТАННЯ ДО ТЕМИ
1. Чим обумовлена електрична провідність розчинів елек т-
ролітів?
2. За рахунок чого головним чином виходить більша енергія,
необхідна для руйнування порівняно міцних іонних кристалічних
решіток при розчиненні електроліту?
3. Що називається питомою електричною провідністю? Пояс-
ните зміну питомої електричної провідності сильних і слабких елек-
тролітів від концентрації й розведення.
4. чи Залежить питома електрична провідність електролітів від
температури й чому?
5. Що називається еквівалентною електричною провідністю?
Яка розмірність цієї величини? Яка залежність еквівалентної елект-
ричної провідності від концентрації для слабких і сильних електро-
літів?
6. Чому по величині еквівалентної електричної провідності во-
дяних розчинів хлориди металів першої групи розташовуються в
порядку, зворотному порядку розташування цих же солей у розпла-
вах?
7. Як зв'язані між собою величини молярної й еквівалентної
електричної провідності для несиметричних електролітів?
8. Якими способами можна виміряти абсолютну швидкість ру-
ху іонів у розчині? Від яких факторів вона залежить?
9. Яка зв'язок між рухливістю іонів і їх абсолютною швидкіс-
тю? Укажіть їхньої розмірності.
10. По яких ознаках відносять електроліти до сильних і слаб-
ких? Якому закону підкоряються слабкі електроліти й у чому його
зміст?
1.2.2. ГОТУВАННЯ РОЗЧИНІВ ДЛЯ ЕЛЕКТРОХІМІЧНИХ ДОСЛІДЖЕНЬ
49
11. Чому методом виміру електропровідності електролітів мож-
на визначити для слабких електролітів їх дійсний ступінь дисоціа-
ції, а для сильних тільки гадану? Чим обумовлюється збільшення
дійсного ступеня дисоціації з розведенням у слабких електролітів і
гаданої в сильних?
12. Чому для виміру опору розчинів використовують змінний
струм?
13. Чим обумовлена наявність максимуму на кривої залежності
питомої електричної провідності від розведення для деяких елект-
ролітів?
14. чи Можна визначити теплоту дисоціації води й оцтової кис-
лоти методом електропровідності?
15. Як впливає в'язкість розчину на його електричну провід-
ність?
16. В чому полягає електрофоретичний і релаксаційний ефекти.
17. Що ви знаєте про ефект Дебая – Фалькенгагена? При якій час-
тоті спостерігається цей ефект? чи Залежить він від концентрації ро-
зчину?
18. У чому полягає ефект Вина? Як можна його пояснити? чи
Спостерігається ефект Вина в слабких електролітах? При яких зна-
ченнях напруженості поля виявляється цей ефект?
19. Як готуються розчини методом послідовного розведення
удвічі. У чому гідності методу ?
20. Дайте теоретичне обґрунтування правила Вальдена – Писар-
жевського.
21. Які ви знаєте сучасні теорії електропровідності розчинів еле-
ктролітів?
ЛІТЕРАТУРА
1. Физическая химия. (Теоретическое и практическое руковод-
ство) // Под ред. Никольского Б. П. – Л.: Химия, 1987, – 880 с.
2. Дамаскин Б. Б. и др. Электрохимия / Б. Б. Дамаскин, О. А.
Петрий, Г. А. Цирлина. – М. : Химия, КолосС, 2006. – 672 с.
3. Герасимов Я. И. и др. Курс физической химии. Т . 2.
–
М.:
Химия, 1966. – 623 с.
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
50
4. Практикум по физической химии. Кинетика и катализ. Эле-
ктрохимия : учеб. пособие для студ. учреждений высш. проф. обра-
зования / А. В. Абраменков и др. ; под ред. В. В.Лунина, Е. П. Агее-
ва. – М. : Издательский центр «Академия», 2012. – 304 с.
5. Теоретические и экспериментальные методы химии раство-
ров (Проблемы химии растворов) /Рос. акад. наук. Ин–т химии рас-
творов; отв. ред. А. Ю. Цивадзе. – М. : Проспект, 2011. – 684 с.
6. Практикум по физической химии: Учебное пособие для ву-
зов. Изд. 3 -е. / Под ред. С . В . Горбачева. – М .: Высшая школа, 1974.
–
495 с.
7. Робинсон Р., Стокс Р. Растворы электролитов.
–М. : ИЛ.
1963. – 646 с.
8. Практикум по физической химии. Кинетика и катализ. Эле-
ктрохимия : учеб. пособие для студ. учреждений высш. проф. обра-
зования / А. В.Абраменков и др.; под ред. В. В. Лунина, Е. П. Агее-
ва. – М. : Издательский центр «Академия», 2012. – 304 с.
9. Методы измерения в электрохимии. т. 1 / пер. с англ. – М . :
1977. – 586 с.
10. Байрамов В. М. Основы электрохимии / В. М. Байрамов. – М.
: ACADEMIA, 2005. – 238 с.
11. Антропов Л. И. Теоретическая электрохимия / Л. И. Антро-
пов. – М. : Высшая школа, 1984. – 519 с.
12. Измайлов Н. А. Электрохимия растворов. Изд. 3 –е, испр., –
М.: «Химия», 1976. – 488 с.
13. Лопатин Б. А. Кондуктометрия : (Измерение электропрово-
дности электролитов) / Акад. наук СССР. Сиб. отд-ние.
–
Новоси-
бирск : 1964. – 280 с.
14. Грилихес М. С ., Филановский Б. К. Контактная кондуктоме-
трия : Теория и практика метода. –Л. : Химия, 1980. – 176 с.
Робота No 1. ВИЗНАЧЕННЯ РОЗЧИННОСТІ МАЛОРОЗЧИННИХ СПОЛУК
КОНДУКТОМЕТРИЧНИМ МЕТОДОМ
51
Робота No 1.
ВИЗНАЧЕННЯ РОЗЧИННОСТІ МАЛОРОЗЧИННИХ СПОЛУК
КОНДУКТОМЕТРИЧНИМ МЕТОДОМ
Ціль роботи: Визначити розчинність і оцінити добуток роз-
чинності малорозчинної сполуки при різних температурах і оцінити
термодинамічні характеристики розчинення.
Прилади й реактиви: прилад для виміру електропровідності
із гніздом, магнітна мішалка, дистильована вода, малорозчинна
сіль (галогеніди срібла й т. п.) .
У насиченому розчині речовини існує рівновага між твердою
фазою і її розчином. У випадку малорозчинної сполуки ти-
пуM A
, що повністю дисоціює в розчині, маємо
z
z
MA
M
A
.
Для цього процесу константа рівноваги виражається рівнянням
.
M
M
A
A
MA
тв
a
a
a
a
K
a
a
,
де
z
M
aй
z
A
a – активності катіона й аніона,
.
тв
a – активність тве-
рдої фази, яка при постійній температурі є постійної, а під час від-
сутності утвору кристалогідратів дорівнює одиниці. Таким чином,
при даній температурі добуток активностей іонів є величина пос-
тійна та має назву добутка розчинності sp
K (solubility product).
При дуже малій розчинності й під час відсутності інших електро-
літів постійною можна назвати й величину добутку концентрацій
іонів, тобто
z
z
z
z
sp
M
A
M
A
K
a
a
c
c
,
враховуючи, що при цих умовах коефіцієнти активності іонів мо-
жуть бути прийняті рівними одиниці.
Залежність розчинності від температури виражається рівнянням
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
52
.
2
ln нас
dc
H
dT
RT
,
деH
– диференціальна теплота розчинення,
.
нас
c – концентрація
насиченого розчину в моль/л. Інтегральна форма цього рівняння до-
зволяє розрахувати диференціальну теплоту розчинення, якщо ві-
дома розчинність даного речовини, принаймні, при двох температу-
рах. Для електроліту 1:1 валентного типу рівняння набуває вигляду
1
2
,
,
1
2
11
ln
spT
spT
K
H
K
RTT
.
При наявності даних про розчинність при декількох темпера-
турах диференціальна теплота розчинення визначається по кутово-
му коефіцієнту прямої залежності
(1/ )
sp
lgK
fT
. Тангенс кута на-
хилу прямій у цих координатах рівний
2, 303
H
tg
R
.
За отриманими значенням добутку розчинності можна визна-
чити енергії Гіббса процесу розчинення
ln
o
sp
GRTK
і зміну ентропії S
, використовуючи рівняння Гіббса – Гельмголь-
ца.
Концентрацію насиченого розчину малорозчинної сполуки s
c
(розчинність) можна знайти по величині питомої електричної про-
відності. Оскільки концентрація дуже мала, еквівалентна електроп-
ровідність розчину практично збігається з величиною еквівалентної
електропровідності при нескінченному розведенні
. Тоді у випа-
дку 1:1 валентного електроліту концентрація s
c рівна ( виражено
вОм
–1
·см
2
г
-
·екв
–1
)
1000
c
,
у якому
()()
M
A
.
Робота No 1. ВИЗНАЧЕННЯ РОЗЧИННОСТІ МАЛОРОЗЧИННИХ СПОЛУК
КОНДУКТОМЕТРИЧНИМ МЕТОДОМ
53
Значення іонних рухомостей знаходять у таблиці 10 додатка, а
для визначення цих значень при інших температурах можна скорис-
татися значеннями температурних коефіцієнтів рухомостей M
і
A
(додаток, таблиця 9), скориставшись рівнянням
[() (18)][()(18)]
M
A
M
t
A
t
.
Через малу електропровідність розчину необхідно в розрахун-
ках ураховувати електропровідність самого розчинника (води) при
температурі проведення вимірів. Тому остаточно розрахунки роз-
чинності проводиться по формулі
2
1000 (
)
HO
s
c
і для 1:1 валентного електроліту
2
sp
Kc
.
ВИКОНАННЯ РОБОТИ
Для визначення розчинності малорозчинної сполуки особлива
вимоги пред'являються до води. Її питома електропровідність не по-
винна перевищувати 2·10–6
Ом
–1
·см
–1
.
Воду необхідно зберігати в
посудині з кварцу або скла пірекс. Перед проведенням вимірів воду
кип'ятять для видалення вуглекислого газу і швидко охолоджують в
посудині, закритій пробкою з трубкою, що містить натронне вапно
(суміш гідроксиду кальцію з невеликою кількістю їдкого натру).
Досліджувану сіль ретельно очищають від домішок, для цього
її розтирають у ступці з невеликою кількістю чистої води, а потім
кілька раз промивають декантацією. Сіль поміщають у посудину
для виміру провідності, який термостатують з точністю ±0,05 oС і
витримують не менш 20-25 хвилин. Виміру проводять 2- 3 рази до
одержання відтворених результатів. При вивченні температурної
залежності виміру проводять аналогічно, змінивши температуру до
заданої. Результати вимірів записують у таблицю.
Сполука..............Температура............
No
п/п
Опір комір-
ки, R ,Ом
Постійна
посудини,
c
k
Питома
ел-сть,
розчину,
Питома
ел-сть
води,
Розчин-
ність,
s
c
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
54
2
HO
Потім вимірюють постійну посудини, як описано раніше, а та-
кож вимірюють електропровідність води, застосовуваної для готу-
вання насичених розчинів при всіх температурах. По питомій елек-
тропровідності розчину й електропровідності при нескінченному
розведенні розраховують концентрацію насиченого розчину дослі-
джуваного речовини при кожній температурі. Знайдену концентра-
цію в моль/л переводять у г-іон/л і розраховують добуток розчинно-
сті. По вивченій температурній залежності визначають розрахунка-
ми або графічно диференціальну теплоту розчинення, за значення-
ми добутку розчинності розраховують G
розчинення й визнача-
ють зміна ентропії розчинення S
. Залежність розчинності сполуки
від температури представляють у вигляді графіка. Усі отримані ха-
рактеристики порівнюють із довідковими даними.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Чому при визначенні розчинності кондуктометричним мето-
дом до води пред'являються особливі вимоги?
2. Чому необхідно обов'язкове термостатування вимірювальної
комірки й визначення постійної комірки при кожній температурі?
3. Яким чином ураховується власна провідність розчинника
при розрахунках еквівалентної електропровідності розведених роз-
чинів?
4. Як за температурній залежності розчинності малорозчинної
сполуки визначають диференціальні теплоти розчинення?
5. Покажіть, як зв'язані розчинність несиметричної малороз-
чинної сполуки з його добутком розчинності.
6. Які допущення роблять при розрахунках розчинності за да-
ними про електропровідність насичених розчинів малорозчинної
сполуки?
7. Як змінюється розчинність малорозчинної речовини при до-
даванні індиферентного електроліту.
8. Чи є пенс говорити при добуток розчинності добре розчин-
них солей.
9. Чи можна за методом електропровідності визначити розчин-
ність малорозчинної сполуки в присутності інших електролітів.
Робота No 2. ВИЗНАЧЕННЯ ГРАНИЧНОЇ ЕКВІВАЛЕНТНОЇ
ЕЛЕКТРОПРОВІДНОСТІ СИЛЬНИХ ЕЛЕКТРОЛІТІВ
55
Робота No 2.
ВИЗНАЧЕННЯ ГРАНИЧНОЇ ЕКВІВАЛЕНТНОЇ
ЕЛЕКТРОПРОВІДНОСТІ СИЛЬНИХ ЕЛЕКТРОЛІТІВ
Ціль роботи: Визначити еквівалентну електропровідність
сильних електролітів (хлориду натрію, хлористого водню й ацета-
ту натрію) і зрівняти з літературними даними. Оцінити значення
коефіцієнта електропровідності f й граничної рухливості іонів
електролітів. Перевірити закон Кольрауша із залученням власного
експерименту ( при необхідності – довідкових даних).
Прилади й реактиви: прилад для виміру електропровідності
із гніздом, дистильована вода, розчини сильних електролітів точної
концентрації ( HCl , NaCl ,
3
CH COONa ), колби для послідовного ро-
зведення на 100 мл, піпетка 50 мл, колба 800 мл для дистильованої
води.
Виміряти граничну еквівалентну електропровідність при не-
скінченному розведенні розчину електроліту безпосередньо екс-
периментально неможливо. Для визначення найбільш придатний
метод екстраполяції еквівалентної електропровідності розчинів на
нескінченне розведення. Згідно з рівнянням Кольрауша, залежність
від c має лінійний характер, що використовується для екстра-
поляції. Для цього знайдені експериментально значення еквівалент-
ної електропровідності наносять на графік у вигляді функції від ко-
реня квадратного з концентрації. Якщо отриману прямолінійну за-
лежність екстраполювати на нульову іонну силу (концентрацію), то
відрізок, що відтинається на осі ординат, безпосередньо дає значен-
ня граничної електропровідності
.
За розрахованими значенням
і знайденим розраховують значення коефіцієнтів електропро-
відності f
f
,
який може ухвалювати значення менші одиниці не тільки у зв'язку
із проявом асоціації іонів, як у випадку слабких електролітів при
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
56
високих концентраціях, а й за рахунок впливу сил міжіонної взає-
модії.
Величина коефіцієнта електропровідності залежить від конце-
нтрації й валентного типу електроліту. Так, в 0,1 н розчині електро-
літу 1:1 валентного типу f = 0,8; для 1:2 валентного електроліту
f = 0,75; для 1:3 валентного електроліту тієї ж концентрації f =
0,4. У міру розведення ці відмінності поступово зникають.
Значення граничних еквівалентних електропровідностей силь-
них електролітів можна використовувати для визначення граничних
еквівалентних електропровідностей слабких електролітів, скориста-
вшись законом Кольрауша. Наприклад, для оцтової кислоти, типо-
вого слабкого електроліту, можна розрахувати граничну еквівален-
тниу електропровідність за рівнянням
3
3
,
,
,
,
CH COOH
CH COONa
HCl
NaCl
3
,
,
,
,
,
,
Na
CH COO
H
Cl
Na
Cl
3
,
,
H
CH COO
.
У правій частині перебувають граничні електропровідності си-
льних електролітів, кожна з яких може бути визначена методом екс-
траполяції з використанням рівняння Кольрауша.
ВИКОНАННЯ РОБОТИ
Усі виміри електропровідностей розчинів проводяться в тер-
мостаті за постійною температурою.
Для готування розчинів в окрему ємність відбирають необхід-
на кількість води, щоб у наслідку виміряти її питому електропрові-
дність. Для визначення граничної еквівалентної електропровідності
сильних електролітів (солей: хлориду натрію, ацетату натрію й хло-
ристого водню) готовлять 5–6 розчинів різної концентрації в інтер-
валі від 0,05 до 0,0005 моль/л методом послідовного розведення
(див. стор. 45 –48). Розчини готуються в кількості 25–50 мл. Не ре-
комендується готовити розчини з найбільш концентрованих розчи-
нів електролітів, тому що при цьому можливі більші помилки, по-
в'язані з невиконанням теоретичних передумов. Починаючи з кон-
Робота No 2. ВИЗНАЧЕННЯ ГРАНИЧНОЇ ЕКВІВАЛЕНТНОЇ
ЕЛЕКТРОПРОВІДНОСТІ СИЛЬНИХ ЕЛЕКТРОЛІТІВ
57
центрації 0,01 моль/л і нижче, випливає при обчисленні питомої
електропровідності розчину в с
експериментальному значенні
.
вим
врахувати власну провідність розчинника – води
2
HO
,зякої
були приготовлені всі розчини, тому що вона стає порівнянною з
електропровідністю розчинів
2
2
.
1
1
с
вим
HO
c
c
HO
k
RR
.
Дослідження починають із виміру електропровідності розчин-
ника, а далі вимірюють електропровідність розчинів у порядку зро-
стання концентрації електроліту. Опір кожного розчину виміряється
не менш 2-х раз, при відмінності результатів вимірів більш, ніж на
0,5% проводиться третє заповнення комірки. Визначення постійної
комірки по стандартному розчину хлориду калію роблять на завер-
шення всього експерименту. Вимір опору комірки з кожним розчи-
ном роблять не менш трьох раз до відтворюваності результатів з то-
чністю не гірше 0,1%.
Методика виміру й розрахунків постійної кондуктометричної
комірки й електропровідності розчинів наведена на стор. 39 цього
посібника.
Вимірявши питому електропровідність, розраховують еквіва-
лентну електропровідність розчинів. Результати розрахунків нано-
сяться на графік з координатами
c
, екстраполяцією визнача-
ється гранична електропровідність. Обчислення граничної еквівале-
нтної електропровідності електроліту можна здійснити й аналітич-
но, визначивши по методу найменших квадратів параметри лінійно-
го рівняння
abc
, у якому вільний член рівняння рівний
a
.
Для проведення аналітичних розрахунків зручно скориста-
тися програмним забезпеченням Microsoft Excel застосовуючи по-
будову лінії тренда з виведенням параметрів лінійного рівняння. У
цьому випадку необхідно оцінити погрішності визначення парамет-
рів рівняння прямій, зокрема, вільного члена рівняння для подаль-
ших висновків роботи.
Результати вимірів і розрахунків рекомендується вести в про-
токолі дослідження з форми
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
58
Електроліт................
Температура,oС...........
No
Концентрація
електроліту,
c , моль/л
c
Опір
комірки,
R, Ом
Середнє
значення,
R,Ом
,
Ом
–1
см
–1
f
1
Після завершення розрахунків і обробки результатів розрахо-
вують граничну еквівалентну електропровідність слабкого електро-
літу – оцтової кислоти відповідно до закону Кольрауша. Оцінюють
експериментальну погрішність визначення
3
(
)
CH COOH
й ступінь
збігу з довідковими даними за температури досліду.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Обґрунтуйте лінійну залежність еквівалентної електропро-
відності від кореня квадратного з концентрації для сильних елект-
ролітів.
2. У чому полягає принцип виміру опору розчинів?
3. Який фізичний зміст постійної кондуктометричної комірки.
4. Чому коефіцієнт електропровідності сильних електролітів
може відрізнятися від одиниці?
5. У якому порядку й чому слід вимірювати опір серії розчинів
різної концентрації?
6. Чому в обов'язковому порядку треба термостату вати комір-
ку при вимірі опору розчину (електропровідності)?
7. Чи можна теоретично розрахувати значення постійної кон-
дуктометричної комірки?
8. Як по даним граничної електропровідності розчинів сильних
електролітів розраховують еквівалентну електропровідність слаб-
ких електролітів при нескінченному розведенні? Відповідь обґрун-
туйте.
9. Якими теоретичними положеннями можна обґрунтувати ем-
піричне рівняння Кольрауша
10. Які методи застосовують для визначення граничної еквіва-
лентної електричної провідності слабких електролітів.
11. Чому вимірювання опору розчинів починають з вивчення
розчинника, а закінчують розчином хлориду калію?
Робота No 3. КОНДУКТОМЕТРИЧНЕ ТИТРУВАННЯ
59
Робота No 3.
КОНДУКТОМЕТРИЧНЕ ТИТРУВАННЯ
Ціль роботи: Переконатися в можливості визначення конце-
нтрації кислот у суміші сильної й слабкої кислот методом кондук-
тометричного титрування. Вивчити види кривих кислотно-
основного титрування.
Прилади й реактиви: прилад для виміру електропровідності
із гніздом для кондуктометричного титрування, дистильована во-
да, бюретки з розчинами хлоридної, оцтової кислот і лугу (концен-
трації ≈0,1 моль/л), магнітна мішалка.
Метод кондуктометричного титрування застосовується у випа-
дках, коли між аналізованим, що й титрують розчинами можуть
протікати обмінні іонні або окисно-відновні реакції, у результаті
яких змінюється електропровідність розчинів, як наслідок зміни
сполуки розчину при титруванні. Як правило, це пов'язане з утвору
малодисоціюючих сполук, наприклад, води у випадку нейтралізації,
або малорозчинних сполук (реакції осадження). Електропровідність
розчину змінюється внаслідок еквівалентної заміни одних іонів ін-
шими відповідно до реакції, що йде при титруванні. При цьому до
крапки еквівалентності спостерігається різний характер зміни елек-
тропровідності залежно від співвідношення значень рухомостей іо-
нів, які є індивідуальними характеристиками іонів.
Розглянемо процес кондуктометричного титрування a еквіва-
лентів сильної кислоти HA сильною підставою MOH.
Рівняння реакції нейтралізації має вигляд:
2
2
HO
HAMOH
HOMA
.
Кислотно-основне титрування характеризується зміною елект-
ропровідності, викликаним заміною іонів водню (гідроксилу), що
володіють аномально високими рухомостями еквівалентною кількі-
стю іонів металу, що мають значно менші рухливості у водяному
розчині.
Іони водню у водяних розчинах гідратовані, існують у вигляді
тригідрата іона гідроксонію 3
H O й мають наступну структуру
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
60
|
...
...
|
|
...
|
H
HOHOHOH
H
HOH
H
Відповідно до такої структури рухливість іонів водню повин-
на бути низкою, однак насправді вона в 5–7 раз перевищує рухливо-
сті інших іонів, за винятком іонів гідроксилу. Висока електрична
рухливість іонів водню обумовлена тим, що за рахунок перерозпо-
ділу водневих зв'язків фактично відбувається перенос протона уз-
довж ланцюжка з молекул води, з'єднаних водневим зв'язком. Ниж-
че показаний початковий розподіл зв'язків у групі орієнтованих мо-
лекул води (а) і остаточне (б) при накладенні електричного поля
|
|
|
|
HOHOHOHOH
H
H
H
H
|
|
|
|
HOHOHOHOH
H
H
H
H
Рис. Перенос іону водню по ланцюжкам молекул води
Для переносу іншого іона водню вправо через усю цю послі-
довність молекул води необхідно, щоб відбувалося молекулярне
обертання, за рахунок чого створювалася б орієнтація, сприятлива
для переносу заряду. Необхідно також ураховувати, що водневі зв'я-
зки між молекулами води безупинно руйнуються й утворюються
знову, що сприяє створенню ланцюжків для переносу заряду в пот-
рібному напрямку. Такий механізм провідності називають ест а-
фетним, або ланцюговим.
Аналогічно можна пояснити більшу рухливість і іонів гідрок-
силу, який можна представити схемою
Робота No 3. КОНДУКТОМЕТРИЧНЕ ТИТРУВАННЯ
61
|
|
|
|
OHOHOHO
H
H
H
H
|
|
|
|
OHOHOHO
H
H
H
H
Рис. Перенос гідроксил - іона по ланцюжкам молекул води.
Перерозподіл водневих зв'язків уздовж ланцюжка молекул во-
ди приводить до гаданого
переміщення іонів гідрок-
силу в напрямку до анода.
Особливість переносу
електрики іонами водню й
гідроксилу характерна для
протолітичних розчинни-
ків, які відрізняються від
інших іонів тим, що вони є
іонами самого розчинника.
Нехай при титруванні
не відбувається помітної
зміни об'єму V розчину
(використовується концен-
трований розчин, що тит-
рує, лугу стосовно розчину
кислоти). Якщо HA
–
ви-
хідна питома електропро-
відність розчину HA, то після додатка деякої кількості розчину луги
xa
значення питомої електропровідності частково нейтралізова-
ного розчину визначиться сумою
1000
HH
AA
HM
c
c
c
,
у якій
H
ax
c
V
;A
a
c
V
;M
x
c
V
.
Підставивши ці вираження в попереднє рівняння, будемо мати
1
1000
H
A
M
ax
a
x
V
V
V
Рис. 1.10. Кондуктограма титрування 5 мл
сильної кислоти (0,1 н HCl) розчином лу-
гу (0.1 н NaOH).
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
62
Тому що
1000
HA
H
Ma
V
після перетворення одержимо
.
1000
HA
H
Ma
V
Залежність питомої електропровідності від об'єму долитого
лугу показане на рисунку. 1 .10. Згідно з рівнянням при додаванні
лугу питома електропровідність розчину безупинно зменшується,
випливаючи лінійної залежності від кількості лугу, що пішов на тит-
рування. Причиною цієї зміни є заміна більш рухливих іонів водню
на значно(в 5–6 раз) менш рухливі іони металу M
+
.
При додатку надлишку луги ( x a
) електропровідність зрос-
тає за рахунок збільшення кількості провідних часток (M+ і OH–) та-
кож лінійно від концентрації лугу. При графічному зображенні цих
прямих, останні перетинаються в точці b , що відповідає крапці екві-
валентності, x a
. Тангенси
кутів нахилу визначаються
рухомостями іонів H
+
+A
–
+M
+
і OH– і залежать від
концентрації реагентів.
Крива зміни електроп-
ровідності при кондуктоме-
тричному титруванні слаб-
кої кислоти сильною підста-
вою представлена на рис.
1.11.
На початку електроп-
ровідність розчину росте
внаслідок заміщення слабо-
дисоційованої кислоти її
сіллю. Після еквівалентної
крапки b електропровід-
ність розчину зростає ще бі-
льше, тому що в розчині з'являється надлишок іонів гідроксилу OH–
, що володіють великою рухливістю. На рис. 1 .12 приводиться кри-
ва титрування суміші сильної й слабкої кислот сильною підставою.
У першу чергу в реакцію нейтралізації вступає сильна кислота, тому
Рис. 1 .11 . Кондуктограмма титрования 5
мл слабой кислоты (0,1 н HAc) щелочью
(0.1 н NaOH).
Робота No 3. КОНДУКТОМЕТРИЧНЕ ТИТРУВАННЯ
63
що слабка перебу-
ває в молекуляр-
ному стані за раху-
нок практично по-
вного зрушення рі-
вноваги дисоціації
слабкої
кислоти
вліво – убік моле-
зації. Тільки після
нейтралізації силь-
ної кислоти почи-
нає
титруватися
слабка кислота. Ві-
дповідно крапки b
і c є крапками ек-
вівалентності при
титруванні.
Чутливість
кондуктометричного титрування в дуже сильному ступені залежить
від різниці в рухомостях іонів, що зв'язуються, і з'являються іонів,
причому вона буде тем вище, чим більше ця різниця. При кондукто-
метричному титруванні слід ураховувати не тільки наскільки повно
протікає основна реакція, але також рухливість іонів, що не ухвалю-
ють у ній прямої участі.
ВИКОНАННЯ РОБОТИ
Для кондуктометричного титрування користуються заглибни-
ми електродами (рис. 1 .6в, 1.6г), що опускаються в будь-яку посу-
дину для титрування, постачений магнітною мішалкою. Досліджу-
ваний розчин (кислоту або суміш кислот і їх кількості для титру-
вання в мл указує викладач) наливають у посудину й розбавляють
дистильованою водою не менш, чим в 20 разів (або до позначеної
мітки на посудині). Слід ураховувати, що в ході титрування вихід-
ний об'єм повинен залишатися практично постійним. Для одержан-
ня прямих ліній необхідно, щоб об'єм титранту, що додається, в ек-
вівалентній точці не перевищував 2% від аналізованого об'єму. Це
досягається розведенням вихідного розчину в 15–20 разів або кон-
центрація титранту повинна на порядок бути вище концентрації
Рис. 1 .12 . Кондуктограмма титрування суміші
сильної (0,1 н HCl) й слабкої (0,1 н HAc) кислот лу-
гом (0.1 н NaOH).
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
64
аналізованого розчину. Для одержання прямих ліній на кондуктог-
рамі необхідно уникнути розведення розчину в ході титрування,
тому що позначається вплив не тільки заміни іонів при титрування,
але й зміна загальної іонної концентрації.
Розчин лугу доливають із бюретки такими однаковими порці-
ями, щоб до крапки еквівалентності одержати не менш 8-10 вимірів
опору розчину, достатніх для побудови лінійних ділянок графіка
титрування. Опір розчину визначають за допомогою мосту змінного
струму, кондуктометра або LCR-метра.
При кондуктометричному титруванні немає необхідності у
визначенні постійної посудини c
k й перерахування обмірюваних
значень опору розчину в питому електропровідність, тому що згідно
з визначенням
/ck R
і замість значень можна для побудови
кривих брати величини електропровідності розчину:
1/
LR
і бу-
дувати графік залежності цієї величини від об'єму лугу, що пішов на
титрування.
По отриманих графіках титрування сильної (слабкої) кислоти й
суміші сильної й слабкої кислот визначають еквівалентні крапки,
розраховують концентрації кислот, узятих для дослідження. Порів-
нюють результати титрування окремо взятих кислот і дані, розрахо-
вані по титруванню їх суміші. При точних визначеннях координати
крапок еквівалентності можуть бути знайдені спільним розв'язком
рівнянь прямих, коефіцієнти яких знаходять із використанням ме-
тоду найменших квадратів.
Нехай рівняння для аналітичного опису залежності
()
LLV
на прямолінійних ділянках кондуктограми представлені рівняннями
()
L' L'V
a' b'V
,
()
L'' L'' V
a'' b'' V
.
Тоді крапка еквівалентності, як крапка перетинання цих прямих,
належить кожному з рівнянь і її координата ( . .
.
.
,
те
те
L V ) визначаєть-
ся з рівності L' L''
, звідки об'єм у крапці еквівалентності визнача-
ється параметрами рівнянь
.
.
тэ
a'' a'
V
b' b''
.
Робота No 3. КОНДУКТОМЕТРИЧНЕ ТИТРУВАННЯ
65
Якщо при обробці прямих були оцінені погрішності визначен-
ня параметрів, то можна оцінити й погрішність визначення об'єму в
крапці еквівалентності, а, отже, і погрішність визначення шуканої
концентрації. Слід помітити, що в таких розрахунках не врахована
систематична помилка, викликана зміною об'єму розчину в ході ти-
трування графіка, що впливає на лінійність.
Результати вимірів у ході кондуктометричного титрування
зводять у таблицю
Об'ємкислоти(кислот)длятитрування............
Концентраціялугу.............................
No
Об'єм лугу,
мл
Опір
розчину, R , Ом
Електропровідність
розчину, 1/
LR
,Ом
–1
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Чому при титруванні кислоти лугом змінюється електропро-
відність розчину?
2. Для чого при кондуктометричному титруванні перед прове-
денням титрування проводиться значне розведення розчину?
3. Поясните характер зміни електропровідності залежно від кі-
лькості долитої до кислоти лугу при титруванні сильної, слабкої ки-
слот і їх суміші.
4. Як визначити оптимальний об'єм порцій лугу, що долива-
ється, при кондуктометричному титруванні кислоти, що необхідно
при цьому знать?
5. Чому рухливість іонів гідроксонію й гідроксилу різко відрі-
зняється від рухливості інших іонів?
6. Як аналітично визначити координати крапки еквівалентності
по кривим кондуктометричного титрування?
7. Перелічить переваги кондуктометричного титрування в по-
рівнянні з іншими видами титрування, наприклад, індикаторним і
потенціометричним.
8. Чим визначається вид залежності електропровідності від
об'єму титранта при титруванні за осадженням?
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
66
9. У яких розчинниках можна чекати аномально високу рухли-
вість іонів водню?
10. Чому при проведенні кондуктометричного титрування доли-
вається значний надлишок титранта після досягнення точки еквіва-
лентності?
11. Чи є необхідним калібрування кондуктометричної комірки
при проведенні кондуктометричного титрування й чому?
12. Чому при кондуктометричному титруванні бажане викорис-
товувати концентровані розчини титранта?
13. Як змінюється вид кондуктограм при зміні порядку зливання
розчинів?
14. Як правильно вибрати титрант для одержання максимальної
точності у визначенні концентрації?
15. Побудуйте математичну модель зміни електропровідності
розчину й зобразите кондуктограми при титруванні сильної, слабкої
й суміші сильної й слабкої кислот користуючись електронними таб-
лицями Excel.
16. Поясните, що краще вибрати в якості титранта при осаджу-
вальному кондуктометричному титруванні сульфату калію: хлорид
або гідроксид барію?
Робота No 4.
ВИЗНАЧЕННЯ КОНСТАНТИ ДИСОЦІАЦІЇ
СЛАБКОЇ КИСЛОТИ МЕТОДОМ ВИМІРУ
ЕЛЕКТРОПРОВІДНОСТІ ЇЇ РОЗЧИНІВ
Ціль роботи: Визначити константу дисоціації слабкої кисло-
ти й порівняти її з літературними даними.
Прилади й реактиви: прилад для виміру електропровідності
із коміркою, дистильована вода, розчин слабкої кислоти відомої
концентрації для розведення, мірна колба (200 мл), 6 колб на 100 мл
для послідовного розведення, колба 500 мл для дистильованої води,
термометр.
Дисоціацію слабкої одноосновної кислоти на підставі закону
дії мас і відповідно до рівняння реакції дисоціації можна предста-
вити схемою
Робота No 4. ВИЗНАЧЕННЯ КОНСТАНТИ ДИСОЦІАЦІЇ СЛАБКОЇ
КИСЛОТИ МЕТОДОМ ВИМІРУ ЕЛЕКТРОПРОВІДНОСТІ
67
[](1)
,
HA
H
A
c
c
c
c
де – ступінь дисоціації слабкої кислоти.
Вираження для константи дисоціації має вигляд
2
2
2
[][]
[]
(1)1
a
c
c
HA
cc
c
KK
K
HA
c
,
у якому коефіцієнти активності іонів прийняті рівними одиниці, то-
му що з відсутність інших електролітів іонна сила розведених роз-
чинів слабкої кислоти вкрай мала. Підстановка вираження для сту-
пеня дисоціації, як відношення еквівалентних електропровідностей
у розчині даної концентрації й при нескінченному розведенні
/
, у комбінації із законом розведення Оствальда, дає рів-
няння
2
2
/
.
1/
()
c
c
c
K
Вирішуючи це рівняння щодо добутку
с
, після математич-
ного перетворень одержимо рівняння в модифікації Крауса – Брея
21
c
c
c
K
K
,
з якого випливає, що добуток
с
є лінійною функцією величини,
зворотної еквівалентної електропровідності 1/
1
cab
,
де параметри рівняння прямій мають наступний сенс:
c
а
K
і
2
c
bK
.
Значення величин а і b можуть бути знайдені або за
графіком прямої в координатах
1
c
, або математичною оброб-
кою даних по методу найменших квадратів. Спільним розв'язком
рівнянь для параметрів а і b визначають константу дисоціації c
K
слабкого електроліту й значення граничної еквівалентної електроп-
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
68
ровідності електроліту
. Якщо значення граничної еквівалентної
електропровідності електро-
літу при температурі досвіду ві-
домо, то значення константи ди-
соціації може бути знайдене по
величині кутового коефіцієнта
прямійb , або безпосередньою
підстановкою в рівняння для
концентраційної константи ди-
соціації. Приклад такої побудови
для водяних розчинів оцтової
кислоти при 298 К наведено на
рисунку 1.13.
Краус і Брей запропонува-
ли графічний метод визначення
констант дисоціації, використо-
вуючи іншу форму закону роз-
ведення Оствальда. Лінійне рів-
няння можна одержати в коор-
динатах
2
11
1
c
c
K
.
У такому випадку вільним членом рівняння є величина
1
, а куто-
вий коефіцієнт відповідає величині
2
1
c
K
, з яких спільним розв'яз-
ком одержують значення c
K й
.
Особливістю методу є необхідність виміру електропровідності
дуже розведених розчинів (концентрація не більш 0,01 моль/л) для
проведення екстраполяції. Внаслідок цього виникають значні екс-
периментальні погрішності. Вивчення більш концентрованих роз-
чинів (до 0,1 моль/л) значно зменшує експериментальні погрішнос-
ті, але вимагає обліку іонної сили розчину й розрахунків еквівален-
Рис. 1.13. Залежність
1/
c
для визначення c
K и
.
Робота No 4. ВИЗНАЧЕННЯ КОНСТАНТИ ДИСОЦІАЦІЇ СЛАБКОЇ
КИСЛОТИ МЕТОДОМ ВИМІРУ ЕЛЕКТРОПРОВІДНОСТІ
69
тної електропровідності за рівнянням Онзагера. Тоді ступінь дисо-
ціації виражається рівнянням
(1)
()
1
2
k
k
BB
c
.
Тому що в правій частині рівняння також утримується ступінь
дисоціації , класичним способом розв'язку є метод простої ітерації
й у якості першого наближення (номер ітерації, k =0) можна прийн-
яти, уважаючи, що єдиною причиною відмінності від є непов-
на дисоціація електроліту
.
Як правило, досить провести 1–2 ітерації для збіжності у всіх
експериментальних крапках.
Значення коефіцієнтів рівняння Дебая – Хюжкеля – Онзагера
1
B й 2B розраховуються відповідно до положень електростатичної
теорії сильних електролітів і виставі про іонну атмосферу й наявно-
сті електрофоретичного й релаксаційного ефектів гальмування іонів
2
1
6
A
FС
B
N
,
2
2
o
8
A
FС
B
NRT
,
де
o
2
СF
RT
.
У цих рівняннях – числовий коефіцієнт, який для симетрич-
ного електроліту рівний 0,1953; F – число Фарадея, 96487 Кл; і
–
в'язкість розчинника та його відносна діелектрична проникність,
відповідно, С – параметр. Інші позначення – загальноприйняті.
Для води за температури 298 К С =3,29·109 м
–1
(л/моль)
1/2
;
1
B = 6,07·10–3
(См·м
2
·моль
–1
·(л/моль)
1/2
); 2B =2,3·10–1
(л/моль)
1/2
.
Константа дисоціації слабкої кислоти в цьому випадку розра-
ховується по повному рівнянню
2
2
2
1
a
c
c
KK
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
70
або
2
o
2
lg
lg
1
1
D
a
D
Ac
c
K
Bac
,
деD
AйD
B – коефіцієнти рівняння Дебая – Хюккеля (див. таблицю
12 додатка).
Фуосс і Краус запропонували для визначення констант дисоці-
ації рівняння
2
2
()1
()
a
c
FZ
KFZ
,
де
()1
Sc
FZ
;
3/2
ZS
c
;
1
2
SB
B
.
Розрахунки ведуться методом послідовних наближень. Спочатку,
побудувавши графік
1
–
c
, знаходять наближене значення
, далі
розраховують Z і по інтерполяційних таблицях Фуосса знаходять
функцію ( )
F Z . Потім розраховують ступінь дисоціації
()
FZ
й коефіцієнт активності по рівнянню Дебая – Хюккеля при іонній
концентрації c
. За отриманим даними будують графік залежності
()
FZ
–
2
()
c
FZ
і знаходять більш точне значення
.
Розрахунки
повторюють до одержання прямолінійної залежності й за тангенсом
кута нахилу прямій обчислюють константу дисоціації.
ВИКОНАННЯ РОБОТИ
У мірній колбі на 50 мл готовлять вихідний розчин слабкої
кислоти з концентрацією не більш 0,01 моль/л (концентрацію й кис-
лоту вказує викладач). Методом розведення вдвічі з використанням
піпетки на 25 мл у чистих і сухих колбах готовлять серію розчинів
убутної концентрації (4–6 розчинів). Воду для готування розчинів
використовують ту саму, попередньо відібрану в колбу (близько 300
мл). Перед вимірами опору знайомляться із правилами роботи на
кондуктометрі (LCR-метрі). Виміру починають із вивчення елект-
Робота No 4. ВИЗНАЧЕННЯ КОНСТАНТИ ДИСОЦІАЦІЇ СЛАБКОЇ
КИСЛОТИ МЕТОДОМ ВИМІРУ ЕЛЕКТРОПРОВІДНОСТІ
71
ропровідності чистого розчинника – води, яка використовувалася
для готування розчинів кислоти. Тому що електропровідність роз-
ведених розчинів порівнянна з електропровідністю самого розчин-
ника р ка
, при розрахунках питомої електропровідності розчину
рну
електропровідність розчинника обов'язково враховується від-
повідно до рівняння
,
.
рну
рнувим
рка
.
Виміру електропровідності розчинів проводять, починаючи із
самих розведених, тому що чистота гнізда здійснюється її проми-
ванням досліджуваним розчином. Комірку заповнюють до мітки або
однаковою кількістю розчину за допомогою піпетки, але електроди
обов'язково повинні бути занурені не менш, чим на 0,5–1 см.
Вимірювальна комірка під час виміру опору перебуває в тер-
мостаті, де витримується при заданій температурі не менш 5–10 хв.
Виміру опору розчинів роблять не менш 3-х разів, проводячи нові
заповнення кондуктометричної комірки тим же самим розчином до
одержання відтворених результатів. Після дослідження всіх приго-
товлених розчинів слабкого електроліту аналогічно роблять виміри
опору стандартного розчину хлориду калію з точно відомою конце-
нтрацією: 0,01; 0,02 або 0,1 моль/л, (див. табл. 8 додатку) для визна-
чення постійної посудини й подальшого розрахунків питомої й ек-
вівалентної електропровідності розчинів.
Результати вимірів і розрахунків зводять у таблицю
Кислота ....................
Температура С...............
No
Концент-
рація,
c , моль/л
Опір
R, Ом
.
cp
R
Питома
ел-сть,
Екві-
вал.
ел-сть,
Ступінь
дисоціа-
ції,
c
K
За отриманими результатами будують графіки залежності пи-
томої й еквівалентної електропровідності від концентрації електро-
літу й розведення, а по графічній залежності
1/
c
визначають
значення константи дисоціації. Отримане значення порівнюють із
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
72
середнім арифметичним значенням константи дисоціації, отриманої
розрахунками по знайдених у довідковій літературі значеннях гра-
ничної рухливості іонів.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. З яких міркувань вибирається концентраційний інтервал ро-
зчинів кислоти для дослідження?
2. Чому визначення постійної посудини робиться на заверша-
льному етапі роботи?
3. Чому для проведення вимірів опору розчину використову-
ють платиновані платинові електроди?
4. Як здійснити розрахунок значень константи дисоціації слаб-
кого електроліту при обмеженому числі вимірів – при відсутності
можливості побудови достовірної залежності
c
від1/?
5. Поясните характер концентраційної залежності питомої й
еквівалентної електропровідності для слабких електролітів.
6. Як оцінити значення іонної сили розчинів по даним виміру
електропровідності?
7. Чому при кондуктометричному дослідженні розчинів обо-
в'язковим є термостатування розчинів?
8. Можна чи за отриманими значенням константи дисоціації
одержати термодинамічну константу?
9. Чому при калібруванні й проведенні вимірів опору розчинів
необхідно комірку заповнювати однаковою кількістю розчину?
10. Який вигляд має бути залежність еквівалентої електропрові-
дності розчинів оцтової кислоти від квадратного корня х іонної сили
розчину? Чому?
11. Вода є дуже слабким електролітом, але в роблті наполегливо
рекомендують враховувати її власну електропровідність під час роз-
рахунків. Чому?
12. В чому полягають уточнення розрахунків константи дисоці-
ації слабкого електроліту за методом Фуосса та Крауса в порівнянні з
методом Крауса – Брея?
13. Як перерахувати погрішність визначення параметрів рів-
няння лінійного рівняння до погрішночті константи дисоціаціх та
граничної електропровідності електроліту?
Робота No 5. ВИЗНАЧЕННЯ ЧИСЕЛ ПЕРЕНОСУ ІОНІВ У РОЗЧИНІ
СІРЧАНОЇ КИСЛОТИ
73
Робота No 5.
ВИЗНАЧЕННЯ ЧИСЕЛ ПЕРЕНОСУ ІОНІВ У РОЗЧИНІ
СУЛЬФАТНОЇ КИСЛОТИ
Ціль роботи: визначити числа переносу іонів 3
HOі
2
4
SO у
розчині сульфатної кислоти та порівняти отримані результати з
довідковими даними.
Прилади й реактиви: мідний кулонометр, чутливий ампер-
метр (міліамперметр), аналітичні ваги, дистильована вода, мірна
колба (200 мл), 6–8 колб ємністю 100 мл для послідовного розведен-
ня, колба 500 мл для дистильованої води, термометр, сульфатно
кислота, спирт або ацетон.
Перенос електрики в розчинах електролітів здійснюється іона-
ми обох знаків: катіонами й аніонами. Залежно від змісту даних іонів
у розчині, їх валентності й швидкості руху в електричному полі вони
переносять різну кількість електрики.
На підставі закону Фарадея кількість, що виділився або розпа-
вся на кожному електроді речовини пропорційно кількості минуло-
го електрики й хімічному еквіваленту цієї речовини.
Для виділення або розчинення одного грам-еквівалента речо-
вини потрібно 96487 кулона (число Фарадея, F ). Позначивши через
I – силу струму в амперах, g – кількість речовини, що виділилася,
у г, a – еквівалентну масу його в г-екв, – тривалість електролізу в
секундах, n – число грам-еквівалентів, одержуємо рівняння, що
представляє собою кількісне вираження для закону Фарадея
96487
gI
n
a
.
При електролізі іони переносять електричні заряди. Через ко-
жний електрод проходить однакова кількість електрики, але кожний
вид іонів переносить неоднакову частку електрики.
Загальна кількість електрики Q , що пройшла через розчин,
складається з кількості електрики, перенесеного всіма іонами, тобто
1
2 ...
m
m
i
i
QQQ
Q
Q
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
74
Числом переносу називають частку кількості електрики, пере-
несеного іонами даного виду (i),
i
i
i
Q
t
Q
.
Для електроліту, що дисоціює за схемою
AB
AB
,
числа переносу іонів виражаються рівностями
Q
t
QQ
й
Q
t
QQ
.
Швидкість руху іонів в електричному полі залежить від на-
пруженості поля. Щоб одержати порівнянні швидкості іонів, що за-
лежать від їхньої природи, розглядаються швидкості іонів при па-
дінні потенціалу 1 В на 1 см, тобто величину см/(с·В/см) = см
2
/(В·с),
яку називають абсолютною швидкістю й позначають через v і
v відповідно для катіона й аніона. Помноживши абсолютні швид-
кості на число Фарадея, одержують іонні електропровідності, або
рухливості й
.
Таким чином, частку кількості електрики, стерпну кожним іо-
ном, можна виразити через відношення абсолютної швидкості іона
до суми абсолютних швидкостей усіх іонів або відповідно через ві-
дношення іонних електропровідностей (рухомостей):
u
t
u
u
й
u
t
u
u
;
t
йt
.
Яким би не була загальна кількість електрики, що пройшов че-
рез розчин, сума чисел переносу t й t електроліту завжди дорів-
нює одиниці.
Числа переносу залежать від рухливості всіх іонів у розчині,
природи розчинника, концентрації й температури. Концентрація
Робота No 5. ВИЗНАЧЕННЯ ЧИСЕЛ ПЕРЕНОСУ ІОНІВ У РОЗЧИНІ
СІРЧАНОЇ КИСЛОТИ
75
впливає й при низьких концентраціях числа переносу практично по-
стійні. Знаючи число переносу й еквівалентну електропровідність
при нескінченному розведенні, можна розрахувати рухливість іонів
,
,
і
,
,
,
t
Схематично перенос електрики в розчині можна представити
схемою, на якій простір між електродами умовно розділене на три
частини: катодну, анодну й середню
Катод
Анод
При пропущенні електричного струму відбуваються наступні
зміни. Нехай швидкість рух катіонів перевищує швидкість руху ані-
онів у три рази. Коли аніони перемістяться до анода тільки на одне
місце, катіони перемістяться до катода на три місця, у результаті
чого розподіл їх у просторах зміниться, а саме.
Катод
Анод
Зі схеми видне, що в катодній і анодній частині залишаться по
чотири іони, заряди яких не компенсовані. Ці іони й розряджаються
на електродах відповідно до закону Фарадея.
Катод
Анод
Причому відмінність у швидкостях руху катіонів і аніонів не
приводить до появи в катодному або анодному просторах надлишку
іонів якого або одного знака. Таким чином, перенос електрики при
прийнятому співвідношенні швидкостей іонів приводить до збіль-
шення концентрації розчину в катода й зменшенню концентрації
розчину в анода.
Якщо через розчин пропущено один фарадей електрики
(1F), то на електродах відбудеться розряд одного грам-еквівалента
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
76
аніонів і катіонів. Коли кількість електрики рівно одному фарадею,
величини t й t визначають відповідно кількість грам-еквівалентів
катіонів і аніонів, стерпних струмом.
Зміни в анодному просторі визначаються: розрядом одного
грам-еквівалента аніонів, вступу з катодного простору t г -екв аніо-
нів, переносом у катодний простір t г-екв катіонів. У підсумку за-
гальний збиток концентрації аніонів рівна (1 )
t
t
г-екв і така ж
збиток концентрації катіонів. Таким чином, у цілому загальна концен-
трація електроліту в анодному просторі знижується на t г-екв.
У катодному просторі 1 грам-еквівалент катіонів розряджається
на катоді, t г-екв катіонів надходить із анодного простору, t
г-екв
аніонів іде в анодний простір. Збиток концентрації катіонів у катод-
ному просторі рівна (1 )
t
t
г-екв. Зменшення концентрації ані-
онів близько катода також рівна t г -екв. У цілому загальна концен-
трація електроліту в катодному просторі зменшується на t г -екв.
Позначивши концентраційні зміни близько електродів
a
c
і
к
c
для анодного й катодного просторів, можна записати наступне
співвідношення
a
k
c
t
c
t
.
Отримане співвідношення дає можливість роздільного визна-
чення чисел переносу аніонів і катіонів. Додавши до обом частинам
рівності по одиниці, одержимо
1
1
a
k
c
t
c
t
або
a
к
к
c
c
tt
c
t
.
Тому що
1
t
t
, у підсумку маємо
к
a
к
c
t
c
c
й
a
a
к
c
t
c
c
.
Робота No 5. ВИЗНАЧЕННЯ ЧИСЕЛ ПЕРЕНОСУ ІОНІВ У РОЗЧИНІ
СІРЧАНОЇ КИСЛОТИ
77
Таким чином, число переносу аніона дорівнює відношенню
убули концентрації електроліту близько катода до загального зни-
ження концентрації електроліту в обох електродів.
Необхідно пам'ятати, що отримане співвідношення вірне тіль-
ки в тому випадку, коли в процесі електролізу не відбувається роз-
чинення електродів. А якщо ні, то ця формула не вірна.
Визначення чисел переносу іонів сірчаної кислоти засноване
на визначенні кількості електрики, що пройшов через розчин, і змі-
ни концентрації електроліту в просторах біля електродів.
Іони водню у водяних розчинах гідратовані й перенос електри-
ки у водяному розчині сірчаної кислоти здійснюється іонами 3
HO
й2
4
SO
, тому що концентрація гідросульфат- іонів
4
HSO і, тим бі-
льше, гідроксильних іонів OH
мізерно мала.
При електролізі сірчаної кислоти з інертними електродами ві-
дбуваються наступні електрохімічні процеси:
На катоді:
3
2
2
2
22
HOe
HOH
.
На аноді:
2
3
2
1
3
22
2
HOe
HO
O
.
По суті, електролізу зазнає не сірчана кислота, а вода. Тому в
катодному просторі під час досліду зміст сірчаної кислоти зменшу-
ється на величину
к
ntQ
, де Q – кількість пропущеної елект-
рики. Число переносу аніона сірчаної кислоти дорівнює
2
4
к
SO
n
t
Q
.
Навпаки, в анодному просторі зміст сірчаної кислоти при елек-
тролізі збільшиться на таку ж величину:
a
ntQ
.
Тому число
переносу аніонів сірчаної кислоти можна визначити також по збі-
льшенню змісту кислоти в анодному просторі:
2
4
a
SO
n
t
Q
.
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
78
Таким чином, визначивши експериментально кількість елект-
рики, що пройшов через розчин, і зміна концентрації в анодному
a
с
або катодному
a
с
просторі, можна знайти числа переносу іо-
нів досліджуваних електролітів за умови, що не відбуваються побі-
чні електрохімічні процеси.
Зміни кількісного змісту електроліту в просторах навколо еле-
ктродів визначаються аналітичним шляхом. Кількість електрикиQ ,
що пройшов через розчин, визначається за допомогою кулономет-
ра – приладу, заснованого на електрохімічному принципі дії. Розра-
хунки кількості електрики проводиться на підставі законів електро-
лізу Фарадея. Одним з таких приладів є мідний кулонометр. Він
складається із трьох мідних електродів, занурених у розчин сульфа-
ту міді. Два електроди 1 є анодами й електрод 2, розташований між
ними, – катодом. На катоді відбувається процес відновлення іонів
міді
2
2
Cu
e
Cu
,
а на аноді – процес окиснення
2
2
CueCu
.
Визначення кількості минулого через кулонометр електрики
визначається по прирості ваги катода за рахунок міді, що виділився
на ньому
31, 77
g
Q
,
деg
– маса міді, що виділився, а 31,77 – електрохімічний еквіва-
лент міді.
Точність показань кулонометра становить 0,1–0,5 % і досяга-
ється використанням кислого електроліту сполуки: мідний купорос
4
2
5
CuSO HO
–
150 г/л; концентрована сірчана кислота 2 4
HSO–50
г/л; етиловий спирт 2 5
C H OH – 50 г/л. Добавки етанолу сприяє
утвору щільного осаду міді й перешкоджає утвору оксиду міді (I) у
присутності кисню повітря за рахунок реакції на електроді
2
CueCu
.
Робота No 5. ВИЗНАЧЕННЯ ЧИСЕЛ ПЕРЕНОСУ ІОНІВ У РОЗЧИНІ
СІРЧАНОЇ КИСЛОТИ
79
ВИКОНАННЯ РОБОТИ
Установка для визначення чисел переносу іонів працює на по-
стійному струмі. Схема установки представлено на рисунку 1.14 .
Живлення від джерела постійного струму 1 подається на елек-
тролітичне гніздо – електролізер 3 через реостат 5. Послідовно із
гніздом у ланцюг включений мідний кулонометр 2. Ланцюг замика-
ється ключем 6. Сила струму фіксується амперметром 4. Електролі-
зер 3 складається із двох посудин: A – анодного й K –катодного, які
з'єднані електролітичним містком M.
Перед початком досвіду катод мідного кулонометра поперед-
ньо покривається шаром електролітичної міді. Для цього збирають
установку без електролітичної комірки, катод кулонометра зачища-
ють дрібним наждаковим папером, промивають дистильованою во-
дою, спиртом і висушують на повітрі протягом 10–15 хв. Потім за-
кріплюють електроди, заливають у склянку електроліт і занурюють
у нього електроди. Замикають ланцюг, і реостатом установлюють
густину струму на катоді 2–20 мА/см
2
. Електроліз ведуть протягом
години, потім катод виймають, промивають дистильованою водою,
спиртом, висушують і зважують на аналітичних вагах.
Зважений катод установлюється на колишнє місце, електроди
Рис. 1.14 . Прилад для виміру чисел переносу іонів
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
80
поринають в електроліт, і кулонометр готовий до роботи. Після
цього підготовляють до роботи електролітичне гніздо. Стаканчики
комірки зважують (сухими), заповнюють обоє склянки 0,01 н роз-
чином сірчаної кислоти в кількості, достатньому для повного зану-
рення електродів, і поміщають у них попередньо очищені свинцеві
електроди. Склянки з'єднують електролітичним містком і заповню-
ють його розчином.
Для виконання вимірів збирають установку, зображену на ри-
сунку 1.13, тобто включають у ланцюг кулонометр і електролітичну
комірку. При цьому стежать, щоб зважений електрод кулонометра й
електрод електролітичної комірки були включені катодами. Коли
вся установка зібрана, включають струм і реостатом установлюють
силу струму 30–50 мА. Миліамперметр призначений тільки для
встановлення необхідної сили струму, для визначення кількості ми-
нулого електрики його не використовують через можливі коливання
сили струму під час електролізу. Електроліз ведуть протягом годи-
ни. Ці умови вибирають так, щоб концентрація в просторах навколо
електродів змінилася достатньо для точного аналізу й щоб не встиг-
нула відбутися дифузія з більш концентрованого розчину в більш
розведений. За час електролізу визначають початкову концентрацію
розчину сірчаної кислоти. Для цього піпеткою відбирають пробу
розчину кислоти (об'єм 10 мл) і титрують 0,05 н розчином лугу з
метилоранжем (або метиловим червоним) у якості індикатору. Із
трьох титрувань беруть середнє значення об'єму луги.
По закінченню досліду виключають струм, відкривають кран 7
в електролітичному містку й дають стекти розчину в обидва стакан-
чики. Виймаючи місток і катод з електролітичної комірки, теж да-
ють можливість стекти розчину. Катодний і анодний склянки знову
зважують із кислотою й визначають вага катодної й анодної рідини.
Для розведених розчинів густину розчинів можна прийняти рівній
одиниці, точніше її визначають по довідникові для температури до-
сліду. По масі розчину визначають його об'єм. Для визначення кон-
центрації кислоти після досліду знову проводять титрування. Знаю-
чи концентрацію лугу л
с і її об'єм, що пішов на титрування кислоти
до досліду o
к
V й після досліду лV , а також загальна кількість кисло-
Робота No 5. ВИЗНАЧЕННЯ ЧИСЕЛ ПЕРЕНОСУ ІОНІВ У РОЗЧИНІ
СІРЧАНОЇ КИСЛОТИ
81
ти в катодному просторі кV , можна розрахувати зміна кількості кис-
лоти в катодному просторі за формулою
o
3
(
)10
к
к
к
к
к
gVV
n
c
V
г-екв,
де V – об'єм проби, що пішла на титрування.
Для визначення кількості електрики, що пройшов через роз-
чин при електролізі сірчаної кислоти, катод мідного кулонометра
промивають, висушують і зважують на аналітичних вагах. По різ-
ниці двох зважувань катода до й послу досвіду визначають масу об-
ложеної міді g
й розраховують кількість електрики Q .
Результати експерименту рекомендується записувати у вигляді
таблиці
Концентраціялугу,моль/л.................
У кулонометрі
Вага катоду, г
Приріст ваги катоду
після досліду
до досліду
g
В електролітичній комірці
Катодна склянка
Анодна склянка
Титрування
вага з
розчи-
ном
вага
пус-
тої
об'єм,
мл
вага з
роз-
чином
вага
пус-
тої
об’єм,
мл
No
о
л
V
л
V
За отриманим даними здійснюють розрахунок чисел переносу
сульфат- іонів і іонів водню. Розрахунки чисел переносу роблять по
католіту й аноліту, а потім визначають середнє значення, тому що
2
4
a
к
SO
n
n
t
n
n
,
2
3
4
1
HO
SO
t
t
.
Розраховані за даними експеримену числа переносу порівню-
ють з літературними даними, та аналізують причини погрішності їх
визначення.
1. РОЗЧИНИ ЕЛЕКТРОЛІТІВ. ЕЛЕКТРОПРОВІДНІСТЬ РОЗЧИНІВ
ЕЛЕКТРОЛІТІВ.
82
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Чим визначаються числа переносу іонів електроліту?
2. Чому в мідному кулонометрі використовується два аноди й
один катод?
3. Яке призначення електролітичного ключа в установці для
визначення чисел переносу?
4. Чому змінюється концентрація сірчаної кислоти в катодно-
му й анодному просторі при електролізі?
5. Навіщо проводиться зважування катодної й анодної скля-
нок?
6. Чому в якості електроліту в кулонометрі використовується
кислий розчин?
7. Які ви знаєте методи визначення чисел переносу? Дайте їх-
ню коротку характеристику.
8. Як на підставі чисел переносу визначити еквівалентні елект-
ропровідності окремих іонів.
9. Які власні характеристики іонів визнвчають їх числа пере-
носу?
10. Чому для матеріалу електродів використовують мідь? Які
метали та з якої причини ще можна застосувати в якості електродів
куорноетра?
11. Чому числа переносу іонів мало залежать від зміни темпера-
тури розчину?
12. Сформулюйте перший та другий закони Фарадея, за якими
визначають кількість електрики, що пройшла через прилад.
ЛІТЕРАТУРА
1. Антропов Л. И. Теоретическая электрохимия / Л. И. Антро-
пов. – М. : Высшая школа, 1984. – 519 с.
2. Практикум по прикладной электрохимии: Учеб. пособие для
вузов / Н. Г . Бахчисарайцьян, Ю. В . Борисоглебский, Г. К. Буркат и
др,; Под ред. В. Н. Варыпаева, В. Н. Кудрявцева. —
3-е изд., пере-
раб. – Л. : Химия, 1990. — 304 с.
3. Практикум по физико-химическим методам анализа : учеб-
ное пособие / И. Я. Гурецкий [и др.]. – М. : ООО «Путь»: ООО ИД
«Альянс», 2006. – 248 с.
Робота No 5. ВИЗНАЧЕННЯ ЧИСЕЛ ПЕРЕНОСУ ІОНІВ У РОЗЧИНІ
СІРЧАНОЇ КИСЛОТИ
83
4. Практикум по физической химии / Под ред. В . В. Буданова
и Н. К. Воробьева. – М. : Химия, 1986. – 351 с.
5. Практикум по физической химии / Под ред. И. В . Кудряшо-
ва. –М. : Высшая школа, 1986. – 495 с.
6. Практикум по физической химии. Кинетика и катализ.
Электрохимия : учеб. пособие для студ. учреждений высш. проф.
образования / [А. В .Абраменков и др.]; под ред. В. В. Лунина,
Е.П.Агеева. – М. : Издательский центр «Академия», 2012. – 304 с.
7. Байрамов В. М. Основы электрохимии / В. М. Байрамов. – М.
: ACADEMIA, 2005. – 238 с.
8. Дамаскин Б. Б. и др. Электрохимия / Б. Б. Дамаскин, О. А.
Петрий, Г. А. Цирлина. – М. : Химия, КолосС, 2006. – 672 с.
9. Методы измерения в электрохимии, т. 1, 2 / Под ред. Э. Еге-
раиА.Залкинда. – М.:Мир,1977. – 566с. – 476с.
10. Практикум по электрохимии / Под ред. Б. Б. Дамаскина.
–
М. : Высшая школа, 1991. – 288 с.
11. Практические работы по физической химии : Учебное посо-
бие для вузов / Под ред. К . П. Мищенко, А. А. Равделя, A. M. Поно-
маревой. – СПб. : Изд-во «Профессия», 2002. – 384 с.
12. Робинсон Р., Стокс Р. Растворы электролитов / Р. Робинсон,
Р. Стокс. – М. : Издатинлит, 1963. – 646 с.
13. Скорчеллетти В. В . Теоретическая электрохимия / В. В .
Скорчеллетти.
–
Л. : Химия, 1974. – 608 с.
14. Физическая химия. Теоретическое и практическое руково-
дство. Изд. 2 –е, переработанное и дополненное. / Под ред. акаде-
мика Б. П. Никольского. – Л. : Химия, 1987. – 880 с.
84
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ.
ЕЛЕКТРОРУШІЙНІ СИЛИ.
2.1. КЛАСИФІКАЦІЯ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
Гальванічний елемент являє собою систему, що полягає із
провідників першого й другого роду, що перебувають у контакті
один з одним. Це прилад, що дає можливість одержувати електрич-
ний струм за рахунок проведення в ньому тієї або іншої хімічної ре-
акції. На границі зіткнення цих провідників виникає стрибок потен-
ціалу. Алгебраїчна сума стрибків рывноважних потенціалів у галь-
ванічному елементі називається його електрорушійною силою, або
ЕРС. Послідовну сукупність усіх стрибків потенціалів на різних по-
верхнях роздылу, що відповідають даному гальванічному елементу,
називають гальванічним ланцюгом.
Механізм виникнення стрибків потенціалів на границі розділу
фаз різний. Залежно від природи контактуючих фаз стрибки потен-
ціалів можна розділити на три типи:
1) стрибки контактних потенціалів, що виникають на гра-
ниці роздягнула двох металів за рахунок переходу вільних електро-
нів з одного металу в іншій (контактні потенціали, як правило, малі
й ними можна зневажити);
2) стрибки електродних потенціалів, що виникають на гра-
ниці роздягнула металу й електроліту, електродні потенціали й ви-
значають в основному ЕРС гальванічного елемента;
3) стрибки дифузійних потенціалів, що виникають на границі
зіткнення двох різних електролітів або двох розчинів, що містять
той самий електроліт різної концентрації. Причиною його виник-
2.1. КЛАСИФІКАЦІЯ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
85
нення є різна швидкість дифузії катіонів і аніонів через границю зі-
ткнення фаз унаслідок відмінності їх рухомість. При цьому розчини
в поверхні роздягнула здобувають заряди, відповідно до надлишку
тих іонів, кількість яких збільшилася за рахунок дифузії. Це почи-
нає перешкоджати подальшій дифузії іонів цього знака й у підсумку
встановлюється рівновага, що характеризується утворенням подвій-
ного електричного шару з певним стрибком потенціалу між фазами.
Механізм виникнення стрибка потенціалу на границі роздяг-
нула метал – розчин можна пояснити в такий спосіб. При зануренні
металу в полярний розчинник (воду) послабляється зв'язок між ато-
мами поверхневого шару металу під дією полярних молекул роз-
чинника. Іони сольватуються (гідратуються), їхній зв'язок з іншими
іонами кристалічної решітки металу послабляється й деяка кількість
атомів металу у вигляді іонів переходить у прилягаючий до поверх-
ні металу шар розчину, при цьому метал заряджається негативно.
Одночасно йде зворотний процес повернення іонів з розчину на по-
верхню металу. Обоє ці процесу йдуть безупинно й із часом устано-
влюється рухлива рівновага, у результаті якого на границі зіткнення
металу й розчину утворюється подвійний електричний шар (ПЕШ) і
виникає стрибок потенціалу.
Метали залежно від будови атомів і міцності зв'язки між ними
мають різну здатність до переходу в розчин у вигляді іонів у даному
конкретному розчиннику. Кожному металу відповідає свій рівнова-
жний стрибок потенціалу й рівноважна концентрація іонів у розчи-
ні. При зануренні металу в розчин солі цього металу, тобто в сере-
довище зі свідомо більшою концентрацією іонів даного металу,
процес переходу іонів цього металу в розчин буде порушений, рів-
новага виявиться зрушеним убік переходу іонів на поверхню мета-
лу, стрибок потенціалу на границі фаз буде знижений. У металів з
невеликою здатністю посилати свої іони в розчин рівновага буде
порушена в такому ступені, що стрибок потенціалу може виявитися
рівним нулю й навіть змінити свій знак. Поверхня металу зарядить-
ся позитивно за рахунок осідання на неї іонів металу.
При схематичному позначенні гальванічного елемента кожна
границя розділу фаз, на якій виникає стрибок потенціалу, зображу-
ється суцільною вертикальною рискою, штриховою лінією прийня-
то позначати границю розділу розчинів.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
86
Гальванічний елемент розділяють на два напівелементи (елек-
тродні комірки), які можуть бути з'єднані з утвором границі розділу
між розчинами й без неї. Розрізняють два основні типи гальваніч-
них елементів:
1) без рідинного з’єднання (без переносу), наприклад,
,
Me MeA AgA Ag.
2) з рідинним з’єднанням (з переносом), наприклад,
2
22
():
,
PtH H
KClHgCl Hg
Тому що розчини в елементах з переносом розділено один від
іншого, концентрацію одного з них можна підтримувати постійної,
у такому випадку потенціал електрода не буде мінятися. Такий еле-
ктрод називається електродом порівняння. Зміни ЕРС елемента
будуть визначатися процесами, що проходять в іншому напівелеме-
нті, потенціал якого буде змінюватися відповідно до зміни активно-
сті відповідних потенціалвизначальних іонів. Такий електрод нази-
вається індикаторним електродом.
Електрохімічні ланцюги звичайно класифікують по двох озна-
ках: 1) за джерелом електричної енергії (фізичні, концентраційні,
хімічні); 2) по наявності або відсутності в ланцюзі границі двох
різних розчинів (відповідно, ланцюга з переносом і без переносу).
У фізичних ланцюгах джерелом електричної енергії служить
розходження у фізичному стані двох однакових по своєму хімічно-
му складу електродів. Ці електроди занурені в той самий розчин і
при роботі ланцюга електрод, що перебуває в менш стійкому стані,
переходить у більше стійкий стан. Фізичні ланцюги – ланцюги без
переносу – підрозділяються на алотропічні й гравітаційні.
Алотропічні ланцюги – це ланцюги, у яких менш стійкий стан
одного електрода обумовлений тим, що він виготовлений з метаста-
більної модифікації даного матеріалу.
Гравітаційні ланцюги, які вперше були реалізовані Р. А. Колі
(1875 р.) використовують єнергія гравітаційного поля. Наприклад,
гравітаційний ланцюг із двох ртутних електродів у розчині нітрату
ртуті(1): лівий електрод з більше високим рівнем ртуті має більший
запас потенційної енергії в порівнянні із правим електродом. У ре-
2.1. КЛАСИФІКАЦІЯ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
87
зультаті цих двох електродних процесів відбувається перенос мета-
левої ртуті з лівої частини в праву, котрий спрямований на вирів-
нювання рівнів ртуті.
Через дуже малі величини ЕРС (мкВ) фізичні ланцюги не ма-
ють практичного значення, а становлять інтерес як приклад, що
ілюструє закони перетворення енергії.
У хімічних ланцюгах джерелом електричної енергії є вільна
енергія хімічної реакції, що протікає в електрохімічній системі і
ЕРС визначається максимально корисною роботою реакції, напри-
клад,
1) хімічний елемент без переносу
2
()
,
Pt H HCl AgCl Ag.
2) хімічний елемент із переносом
2
22
()
,
PtH H KClHgCl Hg
.
Концентраційні елементи складаються з однакових електродів,
занурених у розчини з різною активністю потенціалвизначальних
іонів, наприклад,
3) концентраційний елемент із переносом
3
1
32
1
,(
)
(
)
Ag AgNO a
AgNOaaAg
або з різною активністю металу в електроді, наприклад,
4) концентраційний елемент без переносу (амальгамний)
1
2
(,
)
(
,
)
Na
Na
NaHgaaNaClaaHgNa
.
Очевидно, що ніякої хімічної реакції в елементах не відбува-
ється. Так, нерівність потенціалів у ланцюзі 4) обумовлене відмінні-
стю в активностях іонів срібла, що й обумовлює виникнення струму
при замиканні ланцюга. Результатом роботи елемента є процес ви-
рівнювання концентрацій іонів срібла в обох напівкомірок: збіль-
шення в лівого електрода за рахунок реакції Ag Ag+
+e
–
і зме-
ншення концентрації в правого за рахунок реакції Ag+
+e
–
Ag.
Корисна максимальна робота цього процесу визначається роботою
ізотермічного вирівнювання активностей. Такий процес іде з погли-
нанням теплоти із зовнішнього середовища, отже, джерелом елект-
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
88
ричної енергії концентраційного елемента є енергія навколишнього
середовища.
ЕРС будь-якого гальванічного елемента виникає за рахунок
хімічних реакцій, що протікають у ньому, або за рахунок зміни ак-
тивності іонів поблизу електродів. При роботі гальванічного елеме-
нта хімічна енергія реакції переходить в електричну енергію – еле-
мент робить електричну роботу. В основі роботи будь-якому галь-
ванічному ланцюга лежить окисно-відновна реакція, проведена так,
що на одному з електродів (негативному) відбувається окиснення, а
на іншому (позитивному) – відновлення.
Як приклад оборотного гальванічного елемента можна приве-
сти мідно-цинковий елемент Даніеля – Якобіи
4
4
Zn
j
Cu
Zn ZnSO CuSO Cu
.
У місцях зіткнення різних провідників виникають перегони
потенціалу: Zn
іCu
– перегони на границі метал – розчин, j
–
дифузійний стрибок потенціалу на границі розчин – розчин. Алгеб-
раїчна сума стрибків потенціалів і становить ЕРС даного елемента.
Дифузійний стрибок потенціалу можна елімінувати (зменшити
практично до нуля) включенням між двома розчинами електроліти-
чного сольового мосту з концентрованим розчином електроліту, ру-
хливості катіона й аніона якого практично однакові. Таким чином,
ЕРС мідно-цинкового елемента практично рівна
E= Cu
–Zn
.
Цинк легко віддає свої іони в розчин, на його поверхні утво-
рюється надлишок електроновий, він заряджається негативно. Мідь,
навпаки, заряджається позитивно, тому що вона із труднощами по-
силає свої іони в розчин і легше притягає іони з розчину. Тому при
роботі елемента Якобі цинковий електрод є джерелом електронів,
на його поверхні протікає реакція розчинення цинку
2
2
Zn
Zn
e
.
У це ж час мідний електрод споживає електрони, тому що на
його поверхні розряджаються іони міді й осаджується металева мідь
2.1. КЛАСИФІКАЦІЯ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
89
2
2
Cu
e
Cu
.
Таким чином, у цьому елементі електрична енергія виходить у
результаті хімічної реакції
4
4
Zn CuSO
Cu ZnSO
.
Ця реакція у звичайних умовах може протікати мимовільно,
що видне, наприклад, по витисненню металевої міді на поверхні
цинку, зануреного в розчин сульфату міді. У цьому випадку елект-
рони переходять безпосередньо від атомів цинку до іонів міді. При
такій взаємодії процес необоротний і робота, яка могла б відбувати-
ся за рахунок цієї реакції, не використовується. Та ж реакція, але
проведена в гальванічному елементі, де розділені процеси окиснен-
ня й відновлення на різних електродах, дозволяє одержати електри-
чну роботу.
Гальванічний елемент працює оборотно, якщо:
1) його ЕРС лише на нескінченно малу величину перевищує
прикладену до нього ззовні й протилежно спрямовану ЕРС (оборот-
ність умов роботи);
2) якщо реакція в елементі може бути повністю звернена в
протилежному напрямку при додатку до нього ззовні протилежно
спрямованої ЕРС, яка на нескінченно малу величину перевищує
ЕРС даного елемента (оборотність хімічного процесу). Ланцюг є
оборотному, якщо виконується друга умова.
Прикладом необоротного елемента може служити елемент
Вольта
24
Zn
Cu
Zn HSO Cu
.
Електрохімічні реакції в цьому елементі йдуть і за умови розі-
мкнутого зовнішнього ланцюга, тобто під час відсутності струму
цинковий електрод реагує з кислотою мимовільно. Ланцюги такого
типу в термодинамічному змісті необоротні, тому що в кожний мо-
мент часу система не є рівноважної, G < 0. Коли елемент дає
струм, (
.
.
зовн
внутр
E
E
), відбуваються реакції
2
2
Zn
Zn
e
і
2
2
2
H
e
H
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
90
При зворотному процесі, якщо (
.
.
зовн
внутр
E
E
)
2
2
Cu
e
Cu
і
2
2
2
H
e
H
,
тобто, процеси не є оборотними.
При оборотному протіканні хімічної реакції в умовах сталості
температури й тиску чинена елементом робота буде максимальною
корисною роботою реакції G. Віднесена до перетворення 1 г-іона
електрична робота рівна ел
А nFE
, де n – число електронів, що
брав участь в елементарному акті реакції, F – число Фарадея (96487
Кл/моль), E – різниця потенціалів (ЕРС) елемента. Тому що макси-
мальна корисна робота дорівнює збитку ізобарно-ізотермічного по-
тенціалу електродної реакції rG
, можна записати
r
A
GnFE
.
Якщо в елементі протікає реакція
A+B=C+D,
то величину максимальної роботи реакції можна розрахувати за рі-
внянням
ln
lnCD
r
a
AB
aa
GRTKRT
aa
,
де R – універсальна газова постійна, T – абсолютна температура,
a
K
–
константа рівноваги,
,
,
,
ABCD
a a a a – активності всіх речовин, що
брав участь у реакції. Активність кожного компонента пов'язана з
його концентрацією m і коефіцієнтом активності i
ii
am
. Ці спів-
відношення дозволяють вивести рівняння для ЕРС гальванічного
елемента у зв'язку з концентраціями речовин. Розділивши обидві
частини рівняння ізотерми реакції на nF , одержимо
ln
lnCD
a
AB
aa
RT
RT
E
K
nF
nF
aa
позначивши через
o
E ЕРС елемента, що ставиться до стандартних
умов,
1
A
B
C
D
a
a
a
a
, одержимо
2.1. КЛАСИФІКАЦІЯ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
91
ln
o
CD
AB
aa
RT
EE
nF
aa
,
де
ln
o
a
RT
E
K
nF
–
стандартна ЕРС елемента, рівна ЕРС цього еле-
мента при такому співвідношенні концентрацій, коли активності
речовин дорівнюють одиниці. Вирішуючи спільно це рівняння з рі-
внянням Гіббса-Гельмгольца
p
G
GHT
T
,
де H – тепловий ефект реакції при постійному тиску, одержимо
для ЕРС елемента рівняння
p
H
E
E
nF
T
,
де(/)p
ET
температурний коефіцієнт ЕРС ланцюга.
Це рівняння має велике значення, тому що знаючи E і
(/)p
ET
, можна визначити тепловий ефект реакції в елементі.
Згідно з рівнянням Гіббса-Гельмгольца
GHTS
,
випливає, що зміна ентропії елемента пов'язане з його ЕРС рівнян-
ням
p
E
SnF
T
Якщо ЕРС елемента на залежить від температури ( /E Т
)=0,
то зміна ентальпії реакції H рівно зробленій електричній роботі –
елемент працює без теплообміну. При ( /E Т
)>0,колиЕРСеле-
мента зростає з підвищенням температури, зроблена робота більше
енергії, що звільняється в результаті реакції. Надлишок енергії чер-
пається з навколишнього середовища або із внутрішньої енергії
елемента, реакція в елементі ендотермічна (елемент прохолоджу-
ється). Якщо ж ( /E Т
< 0, ЕРС елемента знижується з підвищен-
ням температури, TS < 0, зроблена робота менше теплового ефекту
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
92
відповідного хімічного процесу, частина енергії буде виділятися у
вигляді теплоти в навколишнє середовище або перетворюватися у
внутрішню енергію елемента (елемент нагрівається).
ЕРС будь-якого гальванічного елемента є величиною позитив-
ної, тому що відповідає певному процесу, що мимовільно протікає й
дає позитивну роботу,
0
G
,G
zFE
). Зворотний процес
можливий, якщо інший гальванічний елемент, приєднаний до дано-
го, змінює напрямок реакції.
ЕРС електрохімічному ланцюга вважається позитивної, якщо
катіони при роботі ланцюга переходять у розчині від електрода, за-
писаного в схемі ланцюга ліворуч, у напрямку до електрода, запи-
саного праворуч, і в тому ж напрямку рухаються в зовнішньому ла-
нцюзі електрони. При цьому правий електрод заряджений позитив-
но відносно лівого. За цим правилом можна знайти суму ЕРС галь-
ванічних елементів, приєднаних послідовно.
Оскільки досвідченим шляхом можна визначити тільки ЕРС
електрохімічному ланцюга, значення потенціалів окремих електро-
дів визначаються як відносні величини, обмірювані стосовно деяко-
го стандартного електрода, потенціал якого ухвалюють умовно рів-
ним нулю. Стандартним електродом, або електродом порівняння є
оборотний водневий електрод, у якому газоподібний водень пере-
буває під тиском 1 атм. (1,01325·105 Па) і насичує платиновий елек-
трод, занурений у розчин з активністю іонів водню (
H
a,апракти-
чно з a , рівній одиниці). Електродним потенціалом електрода
називають ЕРС елемента, складеного із цього електрода (праворуч)
і стандартного водневого електрода (ліворуч), наприклад,
2
2
()( ,1атм)
,
,
()
PtH
HaqZn
Zn Pt
.
ЕРС такого елемента негативна, тому що ліворуч перебуває
позитивно заряджений електрод. Якщо активність іонів цинку буде
дорівнює одиниці, то ЕРС елемента буде рівна стандартному елект-
родному потенціалу цинку, при 298 К 2 /
Zn Zn
= –0,763 В. Уважа-
ється, що дифузійний потенціал на границі двох розчинів еліміно-
ваний. Нормальні електродні потенціали при 298 К для деяких
2.2. ТИПИ ЕЛЕКТРОДІВ
93
електродів наведено в таблиці 23 додатка. Нормальний потенціал
залежить від природи металу й від заряду його іонів у розчині. Не-
гативний нормальний потенціал належить тем електродам, які сто-
совно нормального водневого електрода заряджаються негативно,
тобто на цьому електроді при роботі відповідного елемента іони ме-
талу переходять із електрода в розчин. Отже, перехід іонів з розчи-
ну в метал можливо тільки шляхом електролізу – при додатку ззовні
ЕРС, що перевищує нормальний потенціал електрода. Позитивний
нормальний потенціал означає, що стосовно нормального водневого
електрода даний електрод заряджається позитивно, тобто при робо-
ті елемента іони переходять із розчину на електрод. Знаючи норма-
льні електродні потенціали
o
неважко, користуючись рівнянням
Нернста, розрахувати й електродні потенціали даного металу й у
розчинах інших концентрацій, якщо для них відома активність іонів
цього металу.
Метали, записані в порядку зростання їх нормальних потенці-
алів, являють собою ряд активності металів (ряд напруг), який хара-
ктеризує їхні відбудовні властивості, що зменшуються в порядку
зростання їх нормальних електродних потенціалів.
2.2. ТИПИ ЕЛЕКТРОДІВ
Електроди першого роду. До них відносять електроди, оборо-
тні щодо катіона, і металоїдні, оборотні щодо аніона. Це метали, що
перебувають у рівновазі з іонами даного металу в розчині. Узагаль-
нена схема електродів такого типу записується
z
Me Me
, напри-
клад,
цинковий електрод
2
Zn Zn
,
срібний електрод
Ag Ag
.
На електродах протікає оборотна електродна реакція, напри-
клад, для цинкового електрода
2
2
Zn
e
Zn
. Потенціал ци-
нкового електрода записується рівнянням Нернста
2
2
2
o
/
/
ln
2
Zn Zn
Zn Zn
Zn
RT
a
F
.
Для електрода, оборотного стосовно аніона, електрод запису-
ється схемою
z
AA
, наприклад,
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
94
селеновий електрод
2
Se Se
,
а відповідна електродна реакція представляється рівнянням
2
2
See
Se
.
До електродів першого роду ставляться також амальгамні
електроди, потенціал яких визначається рівновагою між іонами ме-
талу в розчині з атомами металу, розчиненого в ртуті:
(),
z
Me MeHg
наприклад, натрієвий
(),
Na NaHg Pt
Потенціал амальгамного електрода залежить від активності іонів
натрію в розчині
,
Na
a й від активності натрію в амальгамі
()
Na Hg
a
o
/
/
()
ln Na
Na Na
Na Na
Na Hg
a
RT
F
a
.
Газові електроди являють собою трифазні електроди – пла-
тиновану платинову пластину, що перебуває в струмі газу й помі-
щену в розчин, що містить іони, що обмінюються з газом, абсорбо-
ваним металом, електронами, наприклад,
водневий електрод 3
2
()
HOHPt,
кисневий електрод
2
()
PtO OH
,
хлорний електрод
2
()
Pt Cl Cl
.
У вираження для потенціалу електрода входить безрозмірний
тиск газу p, що представляє собою відношення парціального тиску
газу в системі до тиску, що відповідає однієї атмосфері (1 атм =
=1,01325·105 Па).
У газовому водневому електроді протікає реакція
2
1
2
He
H
,
а його стандартний потенціал
2
о
/
HH
умовно ухвалюється рівним
нулю при будь-якій температурі. Він є основою побудови шкали
електродних потенціалів. Потенціал водневого електрода запису-
ється згідно з рівнянням Нернста вираженням
2.2. ТИПИ ЕЛЕКТРОДІВ
95
2
2
2
2
o
1/2
1/2
/ 0.5
/ 0.5
ln
ln
H
H
H
H
H
H
H
H
a
a
RT
RT
F
F
p
p
.
Потенціал електрода зі збільшенням активності іонів водню в
розчині збільшується й зменшується зі збільшенням активності га-
зоподібного водню. Приведення потенціалу водневого електрода до
тиску водню 1 атм. здійснюється по формулі
2
2
o
/0.5 (1
.)
/ 0.5
760
ln
2
H
H атм
H
H
s
RT
FPp
,
де P – атмосферний тиск у момент виміру, у мм. рт. ст.
s
p – тиск пари над розчином при температурі T.
Електроди другого роду – електроди, що представляють со-
бою метал, покритий малорозчинною сполукою цього металу (сіль,
оксид), що й перебуває в розчині, що містять загальний аніон із ма-
лорозчинною сполукою цього металу. До таких електродів можна
віднести напівелементи
хлорсрібний електрод
,
,
Cl AgCl Ag Pt
,
окисно-ртутний електрод
,
,
OH HgOHgPt
,
каломельний електрод
22,,
Cl HgClHgPt
.
На електроді другого роду
,
z
A MeAMe
відбуваються наступні електрохімічні процеси:
z
MeA
ze
MeA
,
z
Me
ze
Me
.
У першому випадку його потенціал (
o
/,
z
A MeAMe
) буде оборот-
ний стосовно аніона
z
A
o
/,
/,
ln
z
z
AMeAM
A MeAMe
A
RT
a
zF
,
активність якої практично визначається концентрацією добре роз-
чинної солі, тому що концентрація MeA в розчині мізерно мала.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
96
У другому випадку потенціал електрода (
/
z
Me Me
) визнача-
ється його оборотністю стосовно катіонів у розчині
z
Me
/
/
ln
z
z
z
o
Me Me
Me Me
Me
RT
a
zF
.
Тому що розчин стосовно MeA є насиченим, те
z
z
sp
Me
A
a
a
K
де sp
K – добуток розчинності малорозчинної сполуки. Підставивши
z
Me
a із цього рівняння в рівняння для потенціалу електрода, одер-
жимо
/
/,
/
ln
ln
z
z
z
z
o
sp
Me Me
A MeAMe
Me Me
A
RT
RT
K
a
zF
zF
Звідси випливає рівняння зв'язку між стандартними потенціа-
лами металевого електрода й електрода другого роду із цим мета-
лом
o
o
,
/,
/
ln
z
z
sp MA
A MeAMe
Me Me
RT
K
zF
.
Наприклад, для хроматсрібного електроду, зв'язок між станда-
ртними потенціалами срібного й хромат срібного електродами й до-
бутком розчинності
2
4
(
)
sp
K Ag CrO хромату срібла виражається рів-
нянням
2
2
24
24
24
4
4
o
o
,
/
,
/
,
ln
2
sp Ag CrO
CrO Ag CrO Ag
CrO Ag CrO Ag
RT
K
F
.
Потенціал електрода другого роду зі збільшенням активності
аніона стає менш позитивним.
Хлорсрібний напівелемент широко використовується на
практиці як електрод оборотний до іонів хлору. Стандартний поте-
нціал хлорсрібного електрода при різних температурах (T К) (таб-
лиця 20 додатка) дається рівнянням
o
,
/
Ag AgCl Cl
=
-4
-6
2
0,22237 - 6,4553·10 ( -298) - 3,284·10 ( -298) +
T
T
-9
3
+ 9,948·10 ( -298)
T
.
2.2. ТИПИ ЕЛЕКТРОДІВ
97
Значення потенціалу хлорсрібного елект-
рода в розчинах хлориду калію різної концент-
рації при різних температурах наведено в таб-
лиці 21 додатка.
Насичений хлорсрібний електрод порів-
няння часто служить електродом порівняння в
комплекті зі скляним і платиновим електрода-
ми. Хлорсрібний електрод складається зі сріб-
ного дроту 8, зануреної в пасту із хлориду сріб-
ла 7 у насиченому розчині хлориду калію 4,
який повідомляється з насиченим розчином по
трубці з азбестовими нитками 2. При готуванні
хлорид срібла попередньо розплавляють і сріб-
ний дріт занурюють у розплавлену масу. Після
охолодження на електроді виходить рівномір-
ний шар хлориду срібла, який добре проводить
електричний струм. Розчин хлориду калію за-
ливають через отвір 5 у скляному корпусі 3, що
закривається пробкою 6. Контакт із досліджу-
ваним розчином відбувається через азбестову
нитку, упаяну в капіляр 1.
Діапазон робочих температур зразкового
електрода – від 15 до 35 °С. Температурний коефіцієнт потенціалу
зразкового електрода — від –0,10 до –0,20 мВ/°С у діапазоні темпе-
ратур 15–35 С. Нестабільність потенціалу зразкового електрода – не
більш ±0,5 мВ. Потенціал зразкового електрода щодо нормального
водневого електрода при 20°С повинен перебувати в межах від 199,5
до 204,5 мВ.
Подібно електродам другого роду можна виготовити електро-
ди третього роду, що полягають із металу, зануреного в насичений
розчин своєї малорозчинної солі, і малорозчинної солі з тим же ані-
оном, що й малорозчинна сіль. Розчин повинен містити також добре
розчинну сіль, що має загальний катіон із малорозчинною сіллю.
Прикладом такого електрода є свинцевий електрод, занурений у ро-
зчин солей хлориду кальцію, оксалату кальцію (малорозчинна сіль)
і оксалату свинцю (малорозчинна сіль, розчинність якої багато ме-
Рис. 2 .1 . Устрій на-
сиченого хлорсріб-
ного електроду
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
98
нше, чим розчинність CaС2O4). Потенціал такого електрода можна
виразити рівнянням Нернста
2
2
2
/
/
ln
2
o
Pb Pb
Pb Pb
Pb
RT
a
F
,
у якому активність іонів Pb2+
повинна бути замінена активностями
інших іонів за допомогою виражень, що випливають із добутку роз-
чинності солей
24
2
2
24
,
spPbCO
Pb
CO
K
a
a
;
24
2
24
2
,
spCaCO
CO
Ca
K
a
a
.
Після підстановок і об'єднання постійних величин, одержуємо
рівняння для потенціалу такої електродної системи
2
o
'
ln
2
Ca
RT
a
F
,
де
24
2
24
,
o
o
/
,
'
ln
2
spPbCO
Pb Pb
spCaCO
K
RT
FK
.
Потенціал такого електрода ( Pb ) оборотне реагує на зміну ак-
тивності чужорідних катіонів (
2
Са
).
Окисно-відновні (редокс) електроди – це електроди з інертно-
го металу, що є переносником електронів, зануреного в розчин, що
містить одночасно як окиснену, так і відновлену форми, наприклад,
іони
2
3
/
Fe Fe
,
3
4
/
Ce Ce
,
2
4
/
Sn Sn
і т. п . Матеріал електрода,
на відміну від електродів раніше описаних типів, в окисно-
відновній реакції участі не ухвалює.
Потенціал редокс електродів, наприклад, ферро-феррі елект-
рода
2
3
,
PtFeFe
,
у якому відбувається реакція на електроді
3
2
Fe
e
Fe
,
виражається рівнянням
2
3
2
3
2
3
o
/
/
ln Fe
Fe Fe
Fe Fe
Fe
a
RT
Fa
,
2.2. ТИПИ ЕЛЕКТРОДІВ
99
де
3
2
o
/
Fe Fe
– стандартний електродний потенціал.
Якщо окисно-відновні реакції протікають за участю інших іо-
нів, часто за участю іонів водню або гідроксильних іонів, то окисно-
відновний потенціал системи залежить і від активності цих іонів,
наприклад, для системи
2
4
2
8
5
4
MnO
H
e
Mn
HO
потенціал буде рівний
4
4
2
4
2
2
2
o
/
/
ln
5
MnO
H
MnO MnO
MnO MnO
Mn
a
a
RT
F
a
Хінгідронний електрод – ставиться до окисно-відновного ти-
пу електродів і є важливим для практичного використання. Хінгід-
ронний електрод являє собою платинову пластинку (дріт), опущену
в розчин, насичений хінгідроном (еквімолекулярна суміш хінону й
гідрохінону 6 4 2 6 4
2
()
CHO CHOH
). На поверхні металевої платини
при рН розчину менш ніж 8 протікає реакція
642
64
2
2
2
()
CHO
H
e
CH OH
.
Електродний потенціал визначається вираженням
642
64
2
2
o
()
ln
2
CHO
H
хг
хг
CH OH
a
a
RT
F
a
.
У насиченому розчині малорозчинного хінгідрону співвідно-
шення активностей хінону й гідрохінону дорівнює одиниці й елект-
родний потенціал залежить тільки від активності іонів водню, тобто
так само, як і потенціал водневого електрода
ln
o
хг
хг
H
RT
a
F
.
Стандартний потенціал хінгідронного електрода виражається
залежно від температури рівнянням
o
-4
0,6992 + 7,4·10 (298 - ).
хг
T
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
100
Значення стандартного потенціалу хінгідронного електрода
при різних температурах представлено в таблиці 20 додатка.
Іоноселективні електроди. Принцип роботи цих електродів
заснований на іонному обміні. Історично першим іоноселективним
електродом був електрод, оборотний до іонів водню, – скляний еле-
ктрод. іони, що втримуються в склі, натрію обмінюються в розчині з
іонами водню, які з аніонним залишком утворюють слабкодисоці-
юючі кремнієві кислоти. Цей обмін іде до встановлення рівноваги.
На границі стекло-розчин виникає потенціал, ве-
личина якого залежить від активності іонів вод-
ню. Не існує електрода такого типу, абсолютно
специфічного стосовно якого-небудь одному іону:
його роботі часто заважає присутність сторонніх
іонів. Крім того, суттєво впливають розчинення
активного матеріалу, отруєння поверхні й т.п.
Схематично обладнання скляного електрода
з водневою функцією записується
,
стекло
, раствор
Ag AgCl HCl
H
і презентовано на малюнку. Скляний електрод з
водневою функцією являє собою тонкостінна ку-
лька 1 з електродного скла, упаяний у скляну
трубку 2, заповнену розчином електроліту ( HCl )
3, у який вставлений електрод 4 (найчастіше хло-
рсрібний) для відводу струму, який проводиться
за допомогою коаксіального екранованого кабе-
лю 5.
Стандартний потенціал скляного електрода
визначається типом внутрішнього електрода й розчину й сортом
скла. Якби внутрішня й зовнішня поверхні скляного електрода були
зовсім однакові, то потенціал електрода визначався б тільки відмін-
ністю рН розчинів по обидва боки мембрани, тобто ЕРС ланцюги, у
якому по обидві сторони мембрани перебувають однакові розчини
була б дорівнює нулю
,
()
()
,
Ag AgCl HCl m стекло HCl m AgCl Ag.
Рис. 2 .2 . Устрій
скляного елект-
роду
2.2. ТИПИ ЕЛЕКТРОДІВ
101
Однак у силу відмінності властивостей поверхонь скла це зна-
чення відмінне від нуля й зветься потенціалу асиметрії. Для гар-
них електродів його значення невелике й досягає лише 2–3 мВ.
Перед уживанням нові скляні електроди потребують вимочу-
вання в розчині кислоти (0,1 н HCl ). При цьому з поверхні поверх-
невого шару скла вилуговуються іони натрію, у невеликому ступені
іони кальцію й на поверхні скла утворюється гелеподібна плівка з
набряклого SiО2. Така гелеподібна плівка діє як твердий буферний
розчин, що містить постійну кількість іонів водню, а підготовлений
у такий спосіб електрод оборотний до іонів водню. Скляні електро-
ди зберігають у воді або в розчині KСl. Висихання порушує стабі-
льну роботу електрода й вимагає попереднього тривалого замочу-
вання.
Основний недолік скляного електрода – його великий внутрі-
шній опір, до 10–1000 МОм, що вимагає використання підсилюва-
чів з більшим вхідним опором для виміру його потенціалу. Залеж-
ність потенціалу електрода від рН розчину трохи відрізняється від
теоретичної внаслідок наявності струмів витоку при його великому
опорі. Це вимагає калібрування кожного окремого електрода для
його практичного використання.
Переваги скляного електрода в порівнянні з водневим і хінгід-
ронним електродами наступні:
–
у систему не вводять сторонні речовини (хінгідрон);
–
потенціал електрода не залежить від присутності окиснюва-
чів або відновників;
–
електрод можна використовувати в широкій області рН;
–
електрод не отруюється;
–
час установлення рівноважного значення потенціалу дуже
невелике.
Сучасні електроди для визначення рН, як правило, є комбіно-
ваними, тобто сполучені в одному корпусі із зовнішнім електродом
порівняння, і дозволяють визначати рН у діапазоні від −2 до 14.
Комбіновані скляні електроди. Комбінований скляний
електрод являє собою вимірювальне обладнання, що поєднує в од-
ному корпусі скляний електрод і електрод порівняння. Такі елект-
роди широко поширені в лабораторній практиці в усьому світі,
оскільки виявилися дуже зручними в обігу. Схема обладнання елек-
трода представлена на рис. 2 .3 .
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
102
Основою електрода (рис.
2.3а) є скляний корпус 2, у ниж-
ній частині якого розташована
сферична робоча мембрана (ку-
лька) 5, діаметром 10 мм із тов-
щиною стінок 0,06–0.1 мм, виго-
товлений зі скла особливого
складу (Me2O·Аl2O3·SiO2, де Me –
Li, Na),. У верхній частині скля-
ного корпуса встановлений плас-
тмасовий ковпачок 7 зі сполуч-
ним кабелем 6. Усередині скля-
ного корпуса 2 установлений
двохключовий електрод порів-
няння. Цей електрод складається
із пластмасової трубки 3, запов-
неної 3 М розчином КCl. У трубці
перебуває хлорсрібний напівеле-
мент, а в нижній торець трубки
встановлений пористий кераміч-
ний електролітичний ключ 4
(внутрішній). Порожнина корпу-
са 2 заповнена електролітом 3 М КCl, а в його нижній частині пере-
буває другий (зовнішній) електролітичний ключ 9, також виконаний
з пористої кераміки.
Таким чином, вбудований електрод порівняння складається із
двох ємностей, – внутрішньої й зовнішньої, заповнених електролі-
том і розділених пористої керамічною перегородкою. Контакт із
аналізованим розчином здійснюється через другу пористу перего-
родку 9. Внутрішня частина електрода порівняння заповнюється
при його виготовленні, зовнішня частина (сольовий місток) допус-
кає дозаповнення або використання інших електролітів замість роз-
чину хлористого калію (KNO3, NH4Cl, NH4NO3). Для цього в корпусі
комбінованого електрода є отвір 8, закрите захисним кільцем (1) або
спеціальною пробкою. Звичайно електрод порівняння в комбінова-
ному електроді заповнюється не насиченим розчином хлористого
Рис. 2 .3 . Комбіновані скляні елек-
троди а) двохключові й б) одноклю-
чові.
2.2. ТИПИ ЕЛЕКТРОДІВ
103
калію а його 3 М розчином, оскільки в ньому не утворюється осад
твердого хлористого калію.
Існують різні варіанти обладнання комбінованих електродів.
Серед них слід назвати одноключовий комбінований електрод (типу
ЭСК–10603), у якого внутрішній (вбудований) хлорсрібний електрод
порівняння поміщений безпосередньо в об'єм трубки 2. У цьому ви-
падку є тільки одна пориста перегородка 9. Об'єм у трубці 2 повинен
бути постійно заповнений розчином хлористого калію (рис. 2.3б).
Діапазон визначення рН комбінованими електродами при тем-
пературі розчину 20 °C – від 0 до 12. (у лужних розчинах з концент-
рацією іонів Na
+
, що не перевищує 0,1 моль/л). Відхилення водневої
характеристики від лінійності в діапазоні визначення рН і темпера-
турі розчину 20 °C не більше ±0,2 рН. Діапазон температур аналізо-
ваного середовища від 0 °C до 100 °C.
Глибина занурення електрода в розчин при вимірі рН повинна
бути не менш 16 мм. Рівень електроліту в електролітичному містку
внутрішнього електрода порівняння при вимірах повинен бути вище
рівня аналізованого розчину. Не допускається застосування елект-
рода в розчинах, що містять фтори- іони й речовини, що утворюють
опади й плівки на поверхні електрода, а також експлуатація й збері-
гання електрода з незаповненим електролітичним містком внутріш-
нього електрода порівняння.
Іоноселективні електроди (ІСЕ). Усі ІСЕ в основі своєї конс-
трукції мають іоночутливу мембрану , проникну для конкретного
типу іонів. Для створення подібних мембранних електродів викори-
стовуються різні електродноактивні речовини, такі як моно- і полік-
ристали, рідкі й тверді іоніти, природні й синтетичні циклічні й
ациклічні органічні сполуки, що селективно зв'язують ті або інші
іони. Мембрана – основний компонент будь-якого ІСЕ має іонооб-
мінні властивості, причому проникність її до іонів різного типу різ-
на. Вона розділяє внутрішній розчин з постійною концентрацією
обумовленого іона й досліджуваний розчин. Одночасно мембрана
служить як електролітичний контакт між ними. Незалежно від типу
мембрани поведінка ІСЕ підкоряється загальним закономірностям:
на поверхні мембрани встановлюється динамічна рівновага, при
якому виникаючий потенціал відповідає величині, необхідної для
запобігання подальшого руху іонів. Існують іоноселективні елект-
роди наступних типів:
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
104
–
твердотельні електроди, у яких мембраною служить мо-
нокристал; або таблетка, спресована з нерозчинної солі, що володіє
іонною провідністю (наприклад, AgCl ), і запаяна в трубку. У цих
мембранах один із двох утворюючих сіль іонів здатний переміщати-
ся по дефектах кристалічної решітки під дією електричного поля.
Наприклад, електрод з мембраною із фторида лантану дозволяє
проводити пряме визначення іона фтору розчинах при його концен-
траціях 10–1
до 10–6
моль/л.. Такий електрод дає високу відтворюва-
ність результатів, причому тільки іон ВІН
–
суттєво заважає визна-
ченню.
–
рідинні іонообмінні електроди, у яких іон, що цікавить
нас, входить до складу великої органічної молекули, погано розчин-
ної у воді. Органічні молекули, розчинені в органічному розчиннику,
звичайно відділяються від досліджуваних водяних розчинів електро-
літів пористою перегородкою. На відміну від твердих мембран, де
активні центри закріплені в просторі силами хімічному зв'язку, у рід-
ких мембранах органофільні активні компоненти рухливі в мембран-
ній фазі. Наприклад, в електроді, селективному до іонів Са
2+
у кон-
центраціях 1–10–4
моль/л, використовують кальцієву сіль додецил-
фосфорної кислоти [(С10Н21)2РО2]2Са, розчинену в ди-n-ацетилфеніл-
фосфонаті. Визначенню кальцію заважають іони водню, магнію й
барію.
–
скляні електроди займають проміжне положення між тве-
рдими й рідкими мембранами.. Іонообмінні процеси протікають у
тонкому гелеподібному шарі скла, утвореному на зовнішній і внут-
рішньої поверхнях скла після витримування у відповідному водя-
ному розчині. Катіоноселективні скляні електроди виготовлені ана-
логічно воденьселективному скляному електроду. Електроди, чут-
ливі до іонів Na
,K
,
4
NH
, Ag
, одержують, змінюючи склад
електродного скла. У присутності інших катіонів селективність та-
ких електродів, однак, невисока.
До рідких близькі пластифіковані мембрани, отримані в ре-
зультаті введення рідкого активного компонента в інертну, напри-
клад, полівінилхлоридну матрицю, еластичність і механічну міц-
ність якої надає присутність пластифікаторів – ефірів фталевої, фо-
сфорної, себацинової і інших кислот.
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
105
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ
ЕЛЕМЕНТІВ
Виміряти електрорушійну силу (ЕРС, E) гальванічного елеме-
нта за допомогою стрілочного вольтметра не можна, тому що такий
прилад показує не ЕРС елемента, а спадання напруги, що залежить
від опору вольтметра. Відповідно до закону Ома струм у ланцюзі I в
момент виміру визначається загальним опором ланцюга, рівним су-
мі опорів приладу, що вимірює, –
.
пр
R вольтметра й кR елемента:
.
/(
).
к
пр
IERR
Звідси
.
пр
к
ЕIR
IR
й стрілка вольтметра покаже тільки
падання напруги на приладі
.
пр
E , яке складе тільки частину дійсної
ЕРС,E:
.
.
.
.
1
1
1/
к
пр
к
к
пр
к
к
пр
R
EEEEIRE
E
RR
RR
.
Таким чином, чим вище опір вимірюваного приладу – тем ближче
його показання до дійсної різниці потенціалів вимірюваного джере-
ла струму. Але при цьому сила струму в загальному ланцюзі стає
мізерно малою. При прагненні сили струму у вимірюваному ланцю-
зі до нуля вимірюване значення наближається до ЕРС.
Внаслідок вищевикладеного, для виміру електрорушійної си-
ли гальванічних елементів використовують спеціальні методи вимі-
рів. У тих випадках, коли важливим є саме абсолютне значення
ЕРС, наприклад, при вимірі стандартних потенціалів електродів,
термодинамічних характеристик електродних реакцій, і т. п., вико-
ристовують компенсаційний метод виміру, що дозволяє виміряти
ЕРС елемента практично під час відсутності проходження струму че-
рез нього (практично сила струму стає незначною – порядку 10–10
А).
У тих випадках, коли важливим є зміна ЕРС, наприклад, спостере-
ження стрибка потенціалу індикаторного електрода в ході потенці-
ометричному титрування, порівняння потенціалу електрода у двох
різних розчинах і т.п . – в икористовують електронні прилади з висо-
ким вхідним опором: рН-мілівольтметри або цифрові вольтметри.
Високий вхідний опір сучасних приладів, а, отже, великий коефіці-
єнт підсилення по струму, дає можливість виміряти напругу, яка
практично збігається зі значенням ЕРС.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
106
КОМПЕНСАЦІЙНИЙ МЕТОД ВИМІРУ ЕРС
ЕРС електрохімічного гнізда в потенціометрії необхідно вимі-
ряти в умовах виконання рівняння Нернста, тобто потенціали елек-
тродів повинні бути рівноважними. Для цього необхідно забезпе-
чити відсутність протікання струму в ланцюзі з вимірювальним гні-
здом щоб уникнути поляризації електродів і викривлення результа-
тів. Вимір ЕРС практично під час відсутності струму здійснюється в
приладах – потенціометрах різних типів.
Класичним методом виміру ЕРС під час відсутності струму є
компенсаційний .метод. Компенсаційний метод виміру ЕРС із ви-
користанням компенсаційної схеми Поггендорфа використовується
у високоомних потенціометрах постійного струму типу Р–307, Р–
37/1, Р–363/1 і т.п . У цьому методі вимірювана ЕРС вирівняється з
ЕРС, що регулюється від зовнішнього джерела: при їхнім вирів-
нюванні струм у ланцюзі з досліджуваним гальванічним елементом,
спостережуваний за допомогою чутливого гальванометра G, дорів-
нює нулю.
Для створення однакової різниці потенціалів на обох гальва-
нічних елементах використовують дільник напруги для зовнішнього
джерела постійного струму (батарея гальванічних елементів, акуму-
лятор або стабілізоване джерело харчування постійного струму) Б
(мал. 2 .4). При постійній силі струму спадання напруги на послідо-
вно з'єднаних опорах, згідно закону Ома, прямо пропорційно вели-
чинам опорів. Принципова схема для компенсаційного методу ви-
міру ЕРС наведена рис. 2.4 .
Струм від акумулятора Б підводить до реохорда АВ (рис. 2 .4а),
який протягом досвіду залишається постійним і рівний відповідно
до закону Ома
Б
AB
U
I
R
.
Реохордом у найпростішому виді служить дріт постійного пе-
ретину, натягнута уздовж шкали. При пересуванні движка реохорда
праворуч ліворуч напруга на ділянці АС падає рівномірно – від на-
пруги, рівного ЕРС акумулятора ( aE > x
E ) до нуля. З’єднання аку-
мулятора з кінцями реохорда A утворює більший ланцюг. Назустріч
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
107
електрорушійній силі акумулятора до кінця реохорда А и ковзному
контакту із приєднується випробуваний гальванічний елемент X. Ця
сполука називається малою або бічним ланцюгом. У малий ланцюг
для фіксування компенсації струмів включається чутливий гальва-
нометр і ключ К, яким на короткий час можна замкнути ланцюг.
Пересуваючи движок реохорда й замикаючи час від часу ключ К,
домагаються того, що стрілка гальванометра встановлюється в ну-
льовому положенні, показуючи відсутність струму з бічного ланцю-
га. У цьому випадку ЕРС досліджуваного елемента Ex скомпенсова-
на різницею потенціалів, що встановився на реохорді на ділянці АС:
Б
x
AC
AC
AC
AB
U
EU
IR
R
R
.
Для визначення x
E , крім значення відносини Опорів AC
Rі
AB
R , які легко знайти по відношенню відповідних довжин відрізків
реохорда, необхідно знати величину напруги акумулятора Б
U.Зна-
чення напруги зовнішнього джерела струму визначають за допомо-
гою тієї ж схеми, включаючи в малий ланцюг замість випробуваного
гальванічного елемента х
Е еталон – нормальний елемент Вестона з
точно відомої ЕРС N
Е . Спадання напруги на реохорді пропорційно
його довжині. Якщо позначити положення движка, відповідне до
компенсації нормального елемента, через N
С , і опір ділянки ланцю-
ги
N
AC через
N
AC
R то:
а)
б)
Рис. 2 .4 . Принципова схема для виміру ЕРС компенсаційним методом:
а) з реохордом; б) з трьома магазинами опорів.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
108
N
N
N
Б
N
AC
AC
AC
AB
U
EU
IR
R
R
,
звідки ЕРС шуканого гальванічного елемента розраховується через
ЕРС еталона й довжини ділянок реохорда AC
lйN
AC
l
пропорційні
їхнім опорам AC
RіN
AC
R:
N
N
AC
AC
x
N
N
AC
AC
l
R
EE
E
l
R
.
Реохорд має невисокий опір, внаслідок чого струм, що йде че-
рез більший ланцюг і напруга батареї не залишаються постійними
через розряд батареї. Тому реохорд може бути замінений на магази-
ни опорів 1
R і 2R (рис. 2.4б) з опорами сотень кому. У цьому випа-
дку розряд батареї виключається й схема працює стабільно. Голов-
ною умовою є збереження постійної суми опорів 1
R і 2Rдляза-
безпечення сталості робочого струму. Змінюють тільки їх співвід-
ношення (імітація пересування движка реохорда).
Для того, щоб зібраний прилад став прямопоказуючим, потрі-
бно так настроїти робочий струм великого ланцюга, щоб спадання
напруги на ділянці
1
АС
R
R
чисельно була дорівнює падінню на-
пруги на ньому. Для цього на магазині опорів виставляють значення
чисельно рівне ЕРС нормального елемента, приєднують бічний ла-
нцюг з нормальним елементом і міняючи опір пR , так міняють робо-
чий струм, щоб дотягтися моменту компенсації, тобто спадання на-
пруги на магазині рівне ЕРС еталону. Після цього можна виміряти
ЕРС і шуканого гальванічного елемента, підбираючи опору магази-
нів 1
R і 2R таким чином, щоб при заданій їхній сумі опорів добити-
ся моменту компенсації. При цьому, значення опору 1R чисельно
буде рівним ЕРС шуканого елемента. Чим менше струм у бічному
ланцюзі, тем, що ближче компенсує напруги на 1
R до ЕРС елемента.
Тому бажана чутливість гальванометра – нуль інструмента повинна
бути 10–7
– 10–9
А/діл.
Викладений компенсаційний метод виміру ЕРС, розглянутий
на схемі з магазинами опорів реалізований у вигляді приладів для
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
109
виміру ЕРС, які звуться високоомних потенціометрів постійного
струму. Найпоширеніші потенціометри типу Р–305, Р–307, Р–37/1 і
ін. Високий клас точності цих приладів дозволяє вимірювати ЕРС із
точністю 0,000001 В. Конструктивний опір настроювання робочого
струму винесене у вигляді групи опору для точного настроювання, а
одночасна зміна опорів 1
R і 2R сполучена.
Відзначимо деякі особливості обладнання й роботи потенціо-
метра Р–307. Спрощена схема потенціометра зображена на мал. 2 .5 .
Струм через потенціометр створюється батареєю акумуляторів
"Б" і регулюється ручками "ГРУБО" і "ТОНКО". (На схемі зазна-
чені тільки дві ручки із чотирьох.) Для установки струму в схему
компенсації включається елемент із відомою величиною ЕРС – но-
рмальний елемент Вестона («НЕ»). У потенціометрі Р–307 урахову-
ється зміну ЕРС нормального елемента зі зміною температури. Для
цієї мети резистор роблять змінним (резистор RНЭ, ручка «НЕ» на
потенціометрі).
За допомогою перемикача роду робіт «В» у схему компенсації
можна включати нормальний елемент НЕ або вимірювана напруга
(положення «X»). Є й проміжне положення, у якому потенціометр
Рис. 2 .5. Спрощена схема потенціометра постійного струму Р-307.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
110
виключений. У потенціометр Р–307 можна включати дві вимірювані
напруги «X1» і «X2». При вимірі невідомої напруги перемикач У
стає в положення X, а компенсація здійснюється двома робочими
декадами: 0,1 V, 0,01 V, як показано спрощено на малюнку (У
потенціометрі Р–307 робочих декад шість і застосована більш скла-
дна схема комутації).
Перевірка компенсації проводиться натисканням кнопки "0".
Для зменшення чутливості гальванометра, при сильної розкомпен-
сованості призначена кнопка "430 кОм".
Можливі три стани:
1. Найменша чутливість – усі кнопки у верхньому положенні.
2. Проміжна чутливість – натиснута тільки кнопка "430 кОм".
3. Найбільша чутливість – натиснута кнопка "0" (положення кно-
пки "430 кОм" у цьому випадку не має значення).
Кнопка "Заспокоєння" замикає рамку гальванометра накорот-
ко й швидко заспокоює коливання покажчика гальванометра.
Зовнішня панель потенціометра зображена на мал. 2 .6 .
Раціонально в якості чутливого (до 10–9
А/діл.) гальванометра
використовувати мікроамперметри типу М–95, М–193 зі світловим
покажчиком, які йдуть у комплекті із шунтом, що забезпечують за-
побігання від більших струмів у відсутності повної компенсації.
ПОРЯДОК ПРОВЕДЕННЯ ВИМІРІВ
Зовнішня панель потенціометра Р–307 показана на рис. 2 .6 .
Нормальний елемент Вестона (клеми НЕ), гальванометр (клеми Г) і
акумуляторна батарея (клеми Б) приєднана до відповідних до зати-
сків потенціометра, дотримуючи полярності. До затисків «X1» або
«X2» приєднується джерело вимірюваного напруги, дотримуючи
полярності ( а якщо ні, то компенсувати ланцюг не вдасться !).
1. На потенціометрі ручкою «НЕ» виставляється значення, ві-
дповідне до значення ЕРС нормального елемента при даній темпе-
ратурі в кімнаті. Перемикач роду робіт поставити в положення НЕ.
Проводиться настроювання робочого струму. Для цього спочатку
ручками "Грубо", а потім "Тонко" виставляється положення нульо-
вого струму на гальванометрі. Струм набудовувати спочатку при
віджатих кнопках "430 кОм" і "0". Потім послідовно включаючи
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
111
кнопки "430 кОм" і
"0", тобто послідовно
збільшуючи
чутли-
вість гальванометра.
2. Перемикач
роду робіт «В» ста-
виться в положенні
«X1» або «X2» ( зале-
жно від того, до яких
клем підключена ви-
мірювана величина), і
ручками декад пере-
микачів,.
починаючи
з 0,1V до 0,000001
V зробити компенсацію вимірюваної величини при послідовному
збільшенні чутливості гальванометра. На практиці, у зв'язку з відт-
ворюваністю вимірюваних ЕРС елементів ±0,,0001 В, досить вико-
ристовувати для компенсації перші чотири декади.
Значення вимірюваної величини відлічується по цифрах у ві-
концях верхньої панелі приладу. Настроювання робочого струму
періодично перевіряють і, якщо буде потреба, підбудовують, особ-
ливо при нестабільній температурі оточення в процесі вимірів.
Для того, щоб переконається в настанні рівноваги в електрод-
ній системі, періодично через 5 хвилин роблять записи ЕРС елемен-
та. При незмінності значень рівноважної різниці потенціалів протя-
гом установленого часу (часто 10 хв) із заданою точністю (часто 0,1
мВ) уважають рівновага досягнутою.
Обов'язковим елементом компенсаційної установки є еталон
ЕРС, у якості якого використовують нормальний насичений еле-
мент Вестона.
Нормальний елемент являє собою гальванічний елемент
4
4
(),()
,
,
()
PtHgCd CdSO HgSOHgPt
,
ЕРС якого відрізняється малим температурним коефіцієнтом, біль-
шим значенням, сталістю, рівна при 293 К EN = 1,0183 В, що дозво-
ляє його використовувати як еталон при потенціометричних вимі-
рах. Нормальний насичений елемент Вестона прийнятий Міжнаро-
Рис. 2.6 . Зовнішня панель потенціометра Р-307.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
112
дною конференцією по електричних одиницях і еталонам у Лондоні
( 1938 р.) у якості міжнародного еталона ЕРС.
Сумарне рівняння струмоутворюючої реакції в елементі
Вестона
2
2
24
4
2
HgSO Cd
Hg SO
Cd
.
Конструкція нормального елемента представлено на рисунку 2.7 .
Негативним електродом нормального елемента є 12,5 % амальгама
кадмію, яка перебуває в конта-
кті з насиченим розчином су-
льфату кадмію CdSO4, що пе-
ребуває в рівновазі із криста-
логідратом
4
2
3
CdSO HO
,що
заповнює більшу частину по-
судини й забезпечує насичення
розчину при зміні температури.
Позитивним електродом
служить ртуть – ртутьсульфат-
ний електрод, що представляє
собою ртуть у контакті із су-
льфатом ртуті (I) у розчині су-
льфату кадмію. Залежно від
температури ЕРС насиченого елемента Вестона передається рівнян-
ням
()
N
ET
, 293
N
E – 4,06·10–5
(T– 293) – 9,5·10–7
(T–293)2
+
+ 1,0·10–7
(T– 293)3
.
Відповідно до міжнародних правил, ЕРС елементів Вестона,
виготовлених у різних місцях, повинні відрізнятися не більше ніж
на 10–5
В. Значення ЕРС нормального насиченого елемента Вестона
при різних температурах наведено в таблиці 18 додатка.
Постійне певне значення ЕРС зберігається тільки у випадку
короткочасного відбору незначних токів. не перевищуючих 10–6
А.
Щоб уникнути зсуву рівноваги, неприпустиме замикання норма-
льного елемента на будь-який опір, навіть на вольтметр.
Рис. 2 .7 . Устрій нормального насиче-
ного елементу Вестона.
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
113
ВИМІР ЕРС ЗА ДОПОМОГОЮ ЕЛЕКТРОННИХ ПРИЛАДІВ.
Компенсаційну установку часто заміняють високоомними на-
півпровідниковими вольтметрами
8
9
10 10
R
Ом), тому що через
нього йде струм, що не перевищує струму, що реєструється нуль–
інструментом. Прилади виробництва ВАТ «Гомельський завод ви-
мірювальних приладів» зі стрілочним покажчиком, типу застарілих
рН–121, рН–340, ИЭ–74 і ін. менш зручні, чому сучасні прилади із
цифровим дисплеєм серії рН–150М. Придатні для виміру ЕРС і ла-
бораторні цифрові електронні вольтметри з маркуванням від В2–xx
до В7–xx, Щ–xx, Ф–xx., наприклад, В2–34, В–7 –73/1 Щ–301/1, (Ро-
сія) і т.п ., а також багатофункціональні прилади: Вольтметр АКИП–
2405 ((АКИП, Росія), GDM–78341 Вольтметр універсальний цифро-
вий True RMS (GOOD WILL INSTRUMENT CO., LTD), U3402A
Мультиметр універсальний KEYSIGHT (Keysight Technologies Inc.
(ex Agilent Technologies), Fluke 8508A/01 (США), і ін.
Нижче приводяться основні характеристики, коротке облад-
нання й принцип роботи деяких приладів, а також методика прове-
дення вимірів, які стосуються виконання лабораторних робіт. Більш
повний опис роботи в інших режимах і методи настроювання при-
ладів можна знайти в прикладені до них паспортам. Дозволяється
проводити виміру, які стосуються тільки виконуваної роботи й опи-
сані в даному посібнику !
рН–МІЛІВОЛЬТМЕТР рН–121
Широкорозповсюджений прямопоказуючий прилад, градуйо-
ваний в одиницях pН і мілівольтах, для визначення величин рН,
pNa,pAg, pK,
4
pNH і окисно-відновних потенціалів; може також
використовуватися в якості високоомного нуль-індикатору й мілі-
вольтметра. Його можна використовувати для ручного й автомати-
чного потенціометричного титрування. За допомогою рН–121 мож-
на робити виміри в комірці для мікровимірів і безпосередньо в ла-
бораторних установках. Зовнішній вигляд приладу представлений
на рис. 2.8.
Межі виміру величини рН: від –1 до 14 з діапазонами –1 14; і
піддіапазонами: –1 4; 4 9; 9 14. Межі виміру ЕРС, відповідно із
множником 100: –100 1400; 100 400; 400 900; 900 1400.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
114
Основна погрішність вимі-
рів 0,05 одиниці рН. Тем-
пературна компенсація ру-
чна й автоматична від 0 до
100 °С. Живлення приладу
від мережі змінного струму
напругою 220 В. Підсилю-
вач постійної напруги хар-
чується від напівпровідни-
кового стабілізатора на-
пруги компенсаційного ти-
пу на напівпровідникових
елементах. Для харчування
вимірювальної схеми в
блоці живлення є два стабілізовані джерела живлення, що мають
малий коефіцієнт пульсації.
Електродом порівняння в приладі рН–121 служить проточний
хлорсрібний електрод ЕВЛ–1МЗ, один для всіх видів потенціометри-
чних вимірів. Його потенціал при 20 °С рівний 202 ± 2 мВ; область
робочої температури 0–100 °С.
У якості індикаторного електрода для виміру величини рН
служать скляні електроди ЭСЛ–43–07 і ЭСЛ–63 –07. Діапазон пря-
молінійної водневої характеристики електрода ЭСЛ–43–07 за тем-
ператури 25 °С від 0 до 12 рН, а при найбільшій робочій температу-
рі (40 °С) від 0 до 10 pН. Тому цим електродом слід користуватися
для вимірів рН у межах 0–10 рН при температурі аналізованого се-
редовища від 0 до +40°С.
Електрод ЭСЛ–63–07 використовується для вимірів рН у ме-
жах 0–14 одиниць рН, при температурі аналізованого середовища
від +25 до +100 °С.
На лицьовій панелі рН-метра розташовуються органі керуван-
ня приладу, що показує, 1 і органі настроювання й регулювання: ре-
зистори – «КАЛІБРУВАННЯ» – 2 «рНі» – 3, «КРУТІСТЬ» – 4 і «НІ»
–
5 і ручка ручної установки температури розчину – 6 . призначені
для оперативного настроювання приладу на дану електродну систе-
Рис. 2.8 . рН-метр-мілівольтметр рН-121 .
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
115
му. При включенні приладу загоряється індикатор 7. Отвір 8 приз-
начене для механічної корекції нуля приладу. На шкалі приладу, що
показує, є показання в цифрах, відповідні до діапазонів виміру pН
(ЕРС). Блок виміру приладу забезпечує його настроювання для ро-
біт на різних діапазонах виміру й корекцію показань при зміні тем-
ператури досліджуваного розчину від 0 до + 100 °С.
Настроювання рН-метра здійснює інженер, положення
ручок керування 2–6 змінювати не дозволяється!
Підготовка приладу до роботи й виміру:
1. Залежно від виду вимірів підбирають необхідні електроди й
приналежності й збирають гальванічний елемент. Вимірювальний
електрод підключають до гнізда «ИЗМ», а допоміжний – до гнізда
«ВСП», обоє гнізда розташовані на задній стінці приладу. Є два гні-
зда для іоноселективний електродом, їх комутація здійснюється
клавішею «ИЗМ1/ИЗМ2» на бічній панелі (рис. 2 .9).
Є три режиму роботи:
–
вимір рН, для чого на бічній панелі натискається кнопка
«рН»;
–
вимір ЕРС, для чого на бічній панелі натискається кнопка
«+mv» або «––mv»;
–
режим роботи в якості «0»–
інструменту для чого натискається кнопка
«НІ».
Для виміру рН або ЕРС натискають на
бічній панелі спочатку кнопку широких меж
виміру –1–14. Після встановлення показань і
з'ясування орієнтовного значення натискають
одну із кнопок "вузького" діапазону й запи-
сують після встановлення постійних показань
приладу: рН – за показниками на відповідної
до діапазону шкалі, – ЕРС за аналогічними
показниками із множником « ». Точність ві-
дліку з використанням дзеркальної шкали:
ЕРС: ±1 мВ, рН: ±0,01 од. Н
Характерною рисою цієї моделі рН-
метра є його здатність роботи в режимі «0»–
Рис. 2 .9 . Бокова па-
нель управліния рН-
мілівольтметра рН-
121.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
116
інструмента. Це дозволяє його використовувати разом з високоом-
ним потенціометром у компенсаційній схемі при вимірах зі скляним
електродом. Для цього спочатку при натиснутій клавіші «0,t» біч-
ної панелі ручкою «НІ» на головній панелі виставляють нульовий
значення на приладі, користуючись дзеркальною шкалою. Після
цього переводять роботу приладу в режим «0»–інструмента натис-
канням кнопки «НІ» бічної панелі. Вимір ЕРС проводиться подачею
з потенціометра відповідної напруги, що компенсує, приводить
стрілку приладу в нульове положення. Потенціометр у такій схемі
підключається до клем "ПОТЕНЦІОМЕТР" на задній панелі.
ІОНОМІР УНІВЕРСАЛЬНИЙ ЭВ–74.
По призначенню й обладнанню іономір ЭВ–74 конструктивно
близький до рН+121. На приладі можна вимірювати рН, рХ іонів з
використанням вхідних з комплект іоноселективних електродів.
Межі виміру величини pХ – від 1 до 19 з діапазонами –1 19; і під-
діапазонами: –1 4; 4 9; 9 14, 14 19. Межі виміру ЕРС, від-
повідно із множником 100: –100 1400; 100 400; 400 900; 900
1400, 1400 1900.
Для виконання вимірів рН-метр установлюють на робочому
місці й перевіряють механічний нуль приладу, що показує, "0". При
необхідності корекції нуля викруткою встановлюють стрілку на по-
чаткову оцінку (мал. 2.10).
Приладу дають прогрітися протягом 25–30 хв і ручкою з напи-
сом «Температура розчину» установлюють значення температури
контрольованого розчину. Для вимірів рН або рХ необхідно відко-
ригувати шкалу рН (рХ) приладу по стандартних буферних розчи-
нах (для рН включаючи 0,1 н. розчин HСl). Для цього в склянку або
термостатуєму комірку наливають стандартний буферний розчин,
опускають у нього вимірювальний скляний електрод і хлорсрібний
електрод порівняння й перемикачем установлюють відповідний діа-
пазон, що показує прилад, для вимірів. Потім ручкою потенціометра
«Калібрування» установлюють стрілку приладу, що показує, на
розподіл шкали, що відповідає значенню pН стандартного буферно-
го розчину при даній температурі. По закінченню калібрування ви-
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
117
ключають прилад (контрольна лампочка гасне) і відключають його
від мережі. Електроди обережно виймають, промивають дистильо-
ваною водою й залишають зануреними в 0,1 н. HCl – вимірюваль-
ний електрод і в насичений розчин KCl – електрод порівняння.
Під час виміру pН електроди поміщають в посудину з випро-
буваним розчином,
приєднують їх до
відповідних клем,
установлюють ав-
токомпенсатор те-
мператури, вклю-
чають прилад у ме-
режу й через 25–30
хв відзначають по-
казання стрілки по
шкалі 1–14. Далі
встановлюють пе-
ремикач на відпо-
відний
діапазон
вимірів і роблять
відлік показань приладу.
Для роботи приладу в режимі міліамперметра на бічній пане-
лі натискають кнопку «mv» а відліки по шкалі приладу множать на
100. Дана модель не має режиму роботи «0»–інструмент.
Широке поширення одержують портативні обладнання, які
при необхідності можуть працювати автономно на вбудованому ни-
зьковольтному джерелі харчування.
Прилади такого типу раціонально використовувати для прове-
дення потенціометричного титрування, виміру рН розчинів. Прила-
ди такого типу вимагають калібрування, перетворювача напруги,
яку виконують по державних стандартних зразках розчину сполуки
іонів або стандарт-титрам з кожним вимірювальним електродом.
Найбільш популярні моделі, що й добре зарекомендували себе,
рН-метрів: рН–150М и рН–410.
Рис. 2.9 . Іономір універсальний ЭВ-74 .
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
118
РН-МЕТР – МІЛІВОЛЬТМЕТР РН–150М
Прилад призначений для виміру показника активності іонів
водню (рН), окисно-відновного потенціалу (Eh) і температури (t)
водяних розчинів. Прилад представляє собою мілівольтметр, особ-
ливістю якого є дуже маленький вхідний струм, так що він може
вимірювати напругу із самих різ-
них електродів, у тому числі
скляних, що мають дисить вели-
кий (до 1 МОм) опір. рН-метр пе-
рераховує напругу в одиниці рН.
Зовнішній вигляд приладу пред-
ставлений на рис. 2 .11.
рН-метр не може виміряти
потенціал одного електроду. Він
вимірює напругу, тобто різниця
потенціалів двох електродів. То-
му необхідні як індикаторний
електрод, так і електрод порів-
няння, потенціал якого подається
на другий вхід рН-метра.
За допомогою портативного
рН-метра – мілівольтметра pН–
150М можна виміряти:
–
величину pН у діапазоні
1,00 14,00 м дискретністю 0,01
од. Погрішність перетворювача
±0,02, погрішність приладу ±0,05.
–
ЕРС (Eh) з діапазоні від –1999 1999 мВ із дискретністю
1 мВ. Погрішність приладу ±3 мВ.
–
температуру T у діапазоні –10..100 °С с дискретність 1 °С.
При вимірі рН (або Eh) розчинів використовується первинний
вимірювальний перетворювач – електродна система, що полягає з
вимірювального електрода й електрода порівняння. Ці електроди
можуть являти собою як роздільні обладнання, так і бути об'єднані в
одному корпусі (комбінований електрод).
Рис. 2.11 . Зовнішній вигляд рН-
метра типа рН-150М.
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
119
ЕРС електродної системи залежить від температури аналізова-
ного розчину. Для виміру температури й обліку її впливу на елект-
родну систему (термокомпенсації) використовується первинний пе-
ретворювач – датчик температури, що представляє собою чутливий
терморезистор.
Для електродних систем, застосовуваних для визначення рН
розчинів, існує крапка (значення рН) у якій їх ЕРС не залежить від
температури. Ця крапка зветься ізопотенціально ю, а відповідні їй
значення «
i
pX » і « iE » називаються координатами ізопотенціальної
точки. На основі обмірюваної величини ЕРС вторинний перетворю-
вач здійснює розрахунки значення рН за наступній формулі:
( )/ (54,1 0,198 )
i
i
S
pHpXEEK
t
.
де E – обмірювана ЕРС електродної системи, мВ; i
pX – координата
ізопотенціальної точки електродної системи; iE – координата ізопо-
тенціальної точки електродної систе-
ми, мВ; S
K – частка, яку становить
реальна крутість електродної харак-
теристики від теоретичного значення,
рівного (54,1 + 0,198· t ); t – темпера-
тура розчину, обмірювана за допомо-
гою термодатчика або введена вруч-
ну, °С.
Значення рН виводиться на дис-
плей 5 перетворювача. Крім цього на
дисплей можуть виводитися резуль-
тати виміру ЕРС (мВ) електродної пари й температури середовища
(°С) (рис. 2.12).
Виміру проводять із комбінованим скляним електродом, типу
ЭСК–10603, верхня частина електрода закінчується втулкою, з якої
виходить кабель із розніманням для підключення до перетворювача.
Термодатчик ТДЛ–1000–06 являє собою пустотілий стрижень,
виготовлений з нержавіючої сталі, усередині якого встановлений
термоелемент. З верхньої частини датчика виходить кабель із розні-
манням для підключення до перетворювача. При роботі датчик
Рис. 2 .12 . Цифровий дисплей
рН-метра.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
120
установлюється на штатив разом з електродною системою й пори-
нає в аналізований розчин на глибину не менш 30 мм.
На лицьовій панелі розташовані рідкокристалічний дисплей і
панель керування. Рознімання для підключення зовнішніх електри-
чних сполук розташовані з торця перетворювача у верхній його час-
тині.
1. Гніздо «6V...14V» – для підключення блоку мережного харчу-
вання;
2. Клеми «СРАВН.» – для підключення електрода порівняння;
3. Клеми «ИЗМ.»
–
для підключення комбінованого або вимі-
рювального електрода;
4. Клеми «ТД» – для підключення датчика температури;
5. Рідкокристалічний дисплей;
6. Панель керування;
На панелі керування розташовано сім кнопок, що служать для
керування приладом (рис. 2 .13), зміст яких наведено нижче.
1. Включення / вимикання приладу
2. Тимчасова зупинка процесу виміру з утриманням на
дисплеї поточного результату
3. Вибір режиму роботи приладу.
4. Вибір одиниць виміру в режимі виміру. Вибір розряду
змінюваного числа або знака при редагуванні (зміні) чис-
лових значень.
5. Підтвердження введення даних, обраного режиму, зна-
ка або числового значення.
6. Збільшення числа або зміна знака при редагуванні чис-
лових значень.
Витяг умісту гнізд блокнота на дисплей.
7. Зменшення числа або зміна знака при редагуванні чис-
лових значень.
Переклад приладу в стан готовності до збереження ре-
зультату виміру в обраному гнізді блокнота.
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
121
ПІДГОТОВКА ПРИЛАДУ ДО РОБОТИ Й ПРОВЕДЕННЯ
ВИМІРІВ.
Комбінований електрод (наприклад, ЭСК–10603) слід закріпи-
ти в штативі й підключити до гнізда «ИЗМ.». Допускається викори-
стовувати роздільну електродну пару, що полягає з вимірювального
електрода (наприклад, ЭС–10601) і електрода порівняння (напри-
клад, ЭСр–10103). При цьому обоє електрода закріплюються в шта-
тиві, рознімання кабелю вимірювального електрода підключається
до гнізда «ИЗМ.», а електрода порівняння до гнізда «СРАВН.» пе-
ретворювача. Термодатчик закріпити в штативі й підключити до
гнізда «ТД». Термодатчик можна не
підключати до приладу, але темпера-
туру в цьому випадку необхідно ввес-
ти вручну.
Для включення приладу необхід-
но нажати кнопку 1 включення прила-
ду й утримувати її протягом 1–2 се-
кунд. При включенні на дисплеї коро-
ткочасно висвітлюється номер версії
програмного забезпечення приладу,
наприклад «v 1.12», після чого прилад
переходить у режим вимірів у тих
одиницях, які були встановлені при
попередньому його вимиканні. У поле
режимів роботи на дисплеї висвітлю-
ється знак «ВИМІР». На основному
цифровому полі дисплея відобража-
ються результати поточного виміру.
Прилад має наступні режими ро-
боти:
–
«ВИМІР» – основний режим
роботи;
–
«НАСТРОЮВАННЯ» – гра-
дуювання приладу (сукупність опера-
цій по доведенню погрішності вимірів
рН-метра до нормованих значень);
–
«НАСТРОЮВАННЯ pXi » –
Рис. 2.13. Зовнішній вигляд
портативного цифрового рН-
мілівольтметра рН-150М.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
122
градуювання приладу з уточненням координат ізопотенціальної то-
чки;
–
«КОНТРОЛЬ» – контроль і редагування параметрів елект-
родної системи.
Працюючи із приладом використовувати тільки режим «ВИ-
МІР», включений за замовчуванням. У поле режимів роботи на ди-
сплеї висвітлюється знак «ВИМІР» (рис 2.13). Іншими режимами
роботи приладу використовують тільки інженерний персонал !
Вимір ЕРС.
Режим вимірів «mv», установлюється кнопкою ВИБІР, при
цьому в правій частині дисплея висвітлюється символ «mv ». Режим
використовується:
–
для виміру ЕРС електродної пари;
–
для побудови градуювального графіка й визначення по ньо-
му концентрації (показника активності) різних іонів при викорис-
танні іоноселективних електродів у якості.
Вимір рН.
Режим виміру «рН». установлюється кнопкою ВИБІР. При
цьому в правій частині дисплея висвітлюється символ «рН».
Промити електроди й інші застосовувані обладнання (напри-
клад, термодатчик або термометр) дистильованою водою, осушити
їхнім фільтрувальним папером і занурити в аналізований розчин.
При використанні термодатчика глибина його занурення в аналізо-
ваний розчин повинна бути не менш 30 мм. Після встановлення
стабільних показань уважати результат виміру з дисплея (рис. 2.12).
Якщо до приладу не підключений термодатчик (визначається
автоматично) необхідно ввести температуру вручну. На дисплеї при
цьому індикується знак «ТР» і значення температури, установлене
раніше вручну. Для редагування значення температури слід нажати
одну із кнопок 6 або 7. На дисплей виводиться тризначне число те-
мператури. Молодший знак числа мигає, показуючи, що він може
бути змінений. Вибір розряду числа здійснюється послідовним на-
тисканням кнопки ВИБІР. Змінювати можна ту цифру, яка мигає в
цей момент на дисплеї. Збільшення або зменшення проводиться ві-
дповідними кнопками 6 або 7. Для завершення редагування слід
2.3. ВИМІР ЕЛЕКТРОРУШІЙНОЇ СИЛИ ГАЛЬВАНІЧНИХ ЕЛЕМЕНТІВ
123
нажати кнопку ВВЕДЕННЯ. Ручне введення температури прово-
диться з дискретністю 1 °С.
При необхідності використовувати інші режими роботи прила-
ду, необхідно ознайомитися з описом.
ТЕОРЕТИЧНІ ПИТАННЯ ПО ТЕМІ
1. У чому причина виникнення різниці потенціалів на границі
двох фаз, наприклад: а) метал – метал; б) метал – розчин; в) метал
–
вакуум; г) розчин – розчин.
2. Які процеси, що відбуваються на границі роздягнула метал –
розчин, приводять до появи подвійного електричного шару?
3. Що являють собою електроди першого й другого роду?
4. Виведіть рівняння залежності електродного потенціалу: а)
цинкового електрода від активності іонів цинку; б) каломельного
електрода від активності іонів хлору в розчині; в) редокс електрода
від співвідношення іонів олова різної валентності.
5. Які процеси в гальванічному ланцюзі можуть бути причи-
ною виникнення ЕРС? Які типи ланцюгів ви знаєте?
6. Приведіть приклади різних концентраційних елементів.
7. На підставі яких даних можна розрахувати при заданій тем-
пературі константу рівноваги електрохімічної реакції?
8. Приведіть приклади гальванічних ланцюгів з переносом і
без переносу.
9. Приведіть приклади оборотних і необоротних гальванічних
ланцюгів.
10. Що таке ряд напруг? Значення яких електродних потенціа-
лів зазначені в ньому?
11. Як впливає тиск водню в газовій фазі на потенціал водне-
вого електрода?
12. Яке обладнання й особливості нормального елемента Вес-
тона?
13. До якого типу електродів ставиться хінгідронний електрод?
Чому й у яких розчинах ним користуються для виміру рН ?
14. Який механізм виникнення потенціалу в скляному елект-
роді? Які його переваги й недоліки при визначенні активності іонів
водню?
15. У чому полягає принцип компенсаційного методу виміру
ЕРС гальванічних елементів?
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
124
16. Температурний коефіцієнт оборотного гальванічного еле-
мента менше нуля. Визначите, виділяється або поглинається тепло-
та при роботі цього елемента.
17. Від чого залежить величина стандартної ЕРС гальванічного
елемента?
18. У якому випадку знак ЕРС гальванічного елемента вважа-
ється позитивним?
19. Яка природа дифузійного потенціалу і яким способом мо-
жна зменшити його величину?
20. Поясните оборотність електродів другого роду стосовно
катіонів і аніонам.
21. Що називають потенціалом асиметрії скляного електрода і
які причини його виникнення?
ЛІТЕРАТУРА
1. Герасимов Я. И. и др. Курс физической химии. Т. 2. – М. :
Химия, 1973. – 625 с.
2. Лебідь В. І. Фізична хімія : підруч. для студ. вищ. навч. за-
кл. / В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
3. Практикум по физической химии. Кинетика и катализ.
Электрохимия : учеб. пособие для студ. учреждений высш. проф.
образования / под ред. В . В . Лунина, Е. П. Агеева. — М . : Издатель-
ский центр «Академия», 2012. – 304 с.
4. Практикум по физической химии. Учебное пособие для ву-
зов. Изд. 3–е. / Под ред. С. В. Горбачева, – М. : Высшая школа,
1974. – 495 с.
5. Практические работы по физической химии : Учебное по-
собие для вузов / Под ред. К. П. Мищенко, А. А. Равделя, A. M. По-
номаревой. – СПб. : Изд-во «Профессия», 2002. – 384 с.
6. Практикум по прикладной электрохимии: Учеб. пособие
для вузов / Н. Г . Бахчисарайцьян, Ю. В . Борисоглебский, Г. К . Бур-
кат и др,; Под ред. В. Н. Варыпаева, В. Н. Кудрявцева.
–
3-е изд.,
перераб. – Л. : Химия, 1990. — 304 с.
7. Багоцкий В. С. Основы электрохимии. –М. : Химия, 1988. –
С. 33 –97.
8. Ротинян А. Л., Тихонов К.И., Шошина И.А. Теоретическая
электрохимия / Под. ред. А. Л. Ротиняна. – М. : Студент, 2013. – 496 с.
Робота No 6. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
125
9. Скорчеллетти В. В . Теоретическая электрохимия / В. В .
Скорчеллетти.
–
Л. : Химия, 1974. – 608 с.
10. Антропов Л. И. Теоретическая электрохимия / Л. И. Антро-
пов. – М. : Высшая школа, 1984. – 519 с.
11. Афанасьев Б. Н., Акулова Ю. П. Физическая химия : Учеб-
ное пособие.
–
СПб. : Издательство «Лань», 2012.
–
464 с: ил.
–
(Учебники для вузов. Специальная литература).
12. Дамаскин Б. Б. и др. Электрохимия / Б. Б. Дамаскин, О. А.
Петрий, Г. А. Цирлина. – М. : Химия, КолосС, 2006. – 672 с.
13. Измайлов Н. А. Электрохимия растворов / Н. А. Измайлов.
–
М. : Химия, 1986. – 576 с.
14. Ковальчук Є. П., Решетняк О. В. Фізична хімія : Підручник.
–
Львів : Видавничий центр ЛНУ Імені Івана Франка, 2007. – 800 с.
15. Методы измерения в электрохимии, т. 1, 2 / Под ред. Э.
ЕгераиА.Залкинда. – М.:Мир,1977. – 566с. – 476с.
16. Практикум по электрохимии / Под ред. Б. Б. Дамаскина.
–
М. : Высшая школа, 1991. – 288 с.
Робота No 6.
ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
Ціль роботи: методом потенціометричне титрування пока-
зати можливість визначення концентрацій сильної й слабкої кис-
лот у них суміші шляхом порівняння результатів спільного й розді-
льного титрування.
Прилади й реактиви: рН-мілівольтметр, хінгідронниый і на-
сичений хлорсрібний електроди, бюретка з розчином лугу, набір пі-
петок, магнітна мішалка, комірка для титрування.
Сутність методу потенціометричного титрування полягає в
тому, що кінцева точка титрування визначається не фіксацією то-
чки еквівалентності за деякою ознакою, наприклад зміні кольору
індикатора, як це робиться при звичайному титруванні з викорис-
танням індикаторів, а по різкій зміні потенціалу індикаторного еле-
ктрода в деякій області добавок титранта. Точне значення об'єму
реагенту, що пішов на титрування, визначається не інструменталь-
ним шляхом, а інтерполяцією вивченої закономірності зміни по-
тенціалу на область його максимальної зміни, що в значній мірі
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
126
збільшує точність титрування.
Потенціометричне титрування проводиться в ланцюгах з рі-
динною сполукою, тому що стрибок ЕРС вимірюваного ланцюга
обумовлений різкою зміною потенціалу індикаторного електрода, у
якості якого використовують електроди, оборотні стосовно іонів
речовини, що титрують, або до іонів реагенту, що додається. Залеж-
но від того, до яких іонів оборотний індикаторний електрод, потен-
ціал його буде відповідно або зменшуватися, або збільшуватися.
Потенціал другого електрода – електрода порівняння – залишається
під час вимірів постійним, тому що сполука розчину не змінюється.
Виникнення стрибка ЕРС пояснюється нерівномірною зміною
концентрації обумовленого речовини при додаванні однакових пор-
цій титранту й заміною однієї потенціалвизначальної реакції, що
протікає до точки еквівалентності, на іншу після її досягнення.
Потенціометричне титрування застосовують для визначення
концентрації речовини, використовуючи реакції ней тр а лі заці ї,
осадження, комплексоутворення й окисно-відновні.
У випадку кислотно-основного титрування в якості індикатор-
ного електрода може служити будь-який електрод, оборотний до іо-
нів водню: водневий, скляний з водневою функцією, хінгідронний,
металоксидний і ін. Прикладом гальванічного елемента для кислот-
но-основного потенціометричного титрування може служити еле-
мент із хінгідронним електродом у якості індикаторного й насиче-
ного хлорсрібного електрода – у якості електрода порівняння
64
264
2
(),
,
(.)
,
(),()()
PtAg AgClKClнас H CHOH CHOH Pt
.
ЕРС такого гальванічного елемента визначається різницею їх
потенціалів
/
хг
Ag AgCl
E
,
де хг
– потенціал хінгідронного електрода, оборотного стосовно
іонів водню:
o
o
lg
хг
хг
хг
H
a
pH
,
а
/
Ag AgCl
–
потенціал хлорсрібного електрода, незмінний протягом
часу титрування.
Робота No 6. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
127
Таким чином, ЕРС елемента змінюється тільки внаслідок змі-
ни концентрації (активності) іонів водню симбатно зміні рН роз-
чину
*
/
o
хг
Ag AgCl
E
pH
E
pH
,де
*
/
o
хг
Ag AgCl
E
.
Рівняння для кривій потенціометричного титрування сильної
кислоти HA сильною основою MOH може бути отримане в такий
спосіб. Нехай до a еквівалентам кислоти HA додане x еквівален-
тів основи MOH. Якщо V об'єм розчину (змінюється при дода-
ваннф в розчин кислоти розчину лугу), у якому проводиться нейт-
ралізація, то концентрація водневих іонів у розчині залежно від кі-
лькості доданого лугу виразиться рівнянням
[]ax
H
V
і водневий показник рН розчину в ході титрування буде змінюва-
тися, згідно із залежністю
lg
ax
pH
V
.
Тому
2, 303
dpH
dx
ax
.
На початку титрування при x 0 нахил кривій pН – x дуже
малий і сама крива повільно піднімається в міру збільшення x. Од-
нак, при наближенні до точки еквівалентності, коли x a значен-
ня dpН/dx , тобто крива титрування поблизу еквівалентної то-
чки піднімається нагору різким стрибком. При x > a останнє рів-
няння втрачає зміст. Тому другу половину кривої титрування, що
ставиться до лужної області, можна побудувати, розглядаючи нейт-
ралізацію a еквівалентів сильної основи при додаванні y еквіва-
лентів кислоти HA. Концентрація гідроксильних іонів у розчині змі-
нюється за рівнянням
[]ay
OH
V
,
з якого визначається значення pН розчину
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
128
lg
w
w
ay
pHpKpOHpK
V
й.
2,303
dpH
dx
ay
Із цього рівняння випливає, що pН розчину сильної основи в
ході його титрування кислотою HA зменшується спочатку повіль-
но, а поблизу еквівалентної точки – різким стрибком.
Повна крива титруван-
ня, (інтегральна крива), що
відповідає знайденим зако-
номірностям, наведена на
рисунку 2.14 (крива 1) – ти-
трування 10 мл хлоридної
кислоти 0,1 н розчином лугу
в елементі з хінгідронним
електродом.
Рівняння кривій потен-
ціометричного титрування
слабкої кислоти (
4
a
pK)
суттєво відрізняється. Тому
що ступінь дисоціації кис-
лоти мала ( << 1, то вира-
ження закону розведення
Оствальда
2
1
c
K
може бути приблизно замі-
нене рівністю
2
K
c
з яко-
го випливає вираження для
концентрації водневих іонів і для pН розчину для початкової точки
кривої титрування
[]
H
c
Kc
й
1
1
lg
2
2
pH
pK
c
.
В еквівалентній крапці титрування, тобто при повній нейтра-
лізації кислоти, розчин буде мати лужну реакцію внаслідок гідролі-
Рис. 2.14 . Крива потенціометричного тит-
рування сильної (I) й слабкої (2) кислоти
лугом.
Робота No 6. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
129
зу солі, що утворювався, сильної основи. Значення pН такого роз-
чину визначиться рівнянням
1
lg
2
2
w
pK pK
pH
c
.
У будь-якій точці титрування частково нейтралізований роз-
чин поводиться як буферний, тому зміна потенціалу індикаторного
електроду й значення стрибка при титруванні буде суттєво меншим,
чим при титруванні сильної кислоти. На рис. 2.14 (крива 2), для по-
рівняння наведена крива потенціометричного титрування 10 мл оц-
тової кислоти з концентрацією
0,1 моль/л тим же розчином лу-
ги).
Лужна галузь кривої тит-
рування визначається введен-
ням надлишку лугу й обумов-
лює поступове підвищення
рН.
При титруванні суміші
сильної й слабкої кислот поте-
нціометричного вдається ви-
значити концентрацію кожної з
них без попереднього поділу.
Крива титрування суміші кис-
лот – по 10 мл хлоридної і оц-
тової однакової концентрації
0,1 моль/л 0,2 н розчином лугу
представлено на рисунку 2.15 .
У присутності сильної кислоти
рівновага дисоціації слабкої
практично повністю зміщене
вліво й вона перебуває в моле-
кулярному стані. Тому спочатку буде реагувати іони водню у кіль-
кості, що відповідають сильній кислоті, поки вона є в надлишку.
Тільки після нейтралізації сильної кислоти почне титруватися слаб-
ка кислота. При потенціометричному титруванні в цьому випадку
Рис. 2.15 . Крива потенціометричного
титрування суміші сильної й слабкої
кислот лугом.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
130
буде спостерігатися два стрибки потенціалу індикаторного електро-
да, дві точки перегину на інтегральній кривій титрування.
Для техніки потенціометричних титрувань істотне значення
має той факт, що стрибок потенціалу в еквівалентній крапці тем ме-
нше, чим більше
a
pK кислоти, який стає практично нерозрізненим
при
a
pK >8.
Багатоосновні кислоти при потенціометричному титруванні
поводяться як суміші декількох кислот з характерним для кожної
щаблі дисоціації величиною
a
pK . Якщо виконується умова
2
1
pK pK
4,
де
2
pKі 1
pK – показники констант дисоціації по другій і першій
щаблях дисоціації відповідно, то на кривій титрування чітко прояв-
ляються обидва стрибки титрування.
Для точного визначення точки еквівалентності застосовують
метод побудови диференціальних кривих – першої похідній (рис.
2.16 б) і другої похідної (рис. 2 .17 а), а також метод Грана, що поля-
гає в побудові кривої в координатах:
/
VE
(рис. 2.17 c).
а)
б)
Рис. 2 .16. Інтегральна а) й диференціальна б) криві потенціометричного
титрування.
Робота No 6. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
131
Безпосередньо за експериментальним даними будується інтег-
ральна крива титрування
()
EЕV
в координатах E V
(рис.
2.16а).
Точка еквівалентності по інтегральній кривій титрування ви-
значається за методом трьох дотичних, проведених до найбільш
прямолінійних ділянок кривої. Проводять дотичні до пологих діля-
нок графіка й, відповідної до області стрибка. Знайшовши середину
прямої в області стрибка, визначають координату точки еквівалент-
ності
.
.
те
V (рис. 2 .16 б). Більш точне визначення еквівалентного
об'єму титранта досягається при побудові диференціальної залеж-
ності, тому що в крапці перегину інтегральної кривій спостеріга-
ється максимальне значення швидкості зміни ЕРС елемента від
об'єму титранта – першої похідної /
dE dV . Значення похідної роз-
раховуються, зазвичай, методом кінцевих різниць, тобто за форму-
лою
2
1
21
/
(
)/( )
dEdVEEVV
. Оскільки в цьому випадку похідна
має одне й теж значення в точці 1 і в точці 2, при побудові графічної
залежності як абсциси ухвалюється середнє арифметичне аргументу
для цього значення похідної 1 2
( )/2
VV
.
Залежність
/
dEdV V
а)
б)
Рис.2.17 . Графік другої похідної а) для визначення точки еквівалентності
та метод Грана для титрування HCl (рис. 2 .13, крива 1) б).
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
132
завжди має максимум, але координата максимуму на диференціаль-
ній кривій не обов'язково збігається із нанесеною точкою на кривій
(рис. 2 .15 б), тому на графіку диференціальної кривої наносять тіль-
ки розраховані значення похідних у вигляді точок, а загальну зале-
жність відображають як незалежні дві криві: до точки еквівалентно-
сті й після, згладжуючи експериментальні погрішності й проводячи
плавні криві між нанесеними точками. Координата точки еквіва-
лентності буде точкою перетинання цих кривих. Якщо добавки
об'єму титранту в області стрибка є мінімально можливими (напри-
клад, крапля розчину титранта), то завжди при обробці потенціоме-
тричного титрування на диференціальній кривій можна визначити
координату з точністю, що перевищує цю добавку (наприклад, з то-
чністю частки краплі). От чому потенціометричного титрування по-
тенційне вважається найбільш точним.
При титруванні суміші кислот різної сили спостерігається два
екстремуми на диференціальній кривій. Для прикладу, зображеного
на рис. 2 .15 диференціальна крива титрування має вигляд, зображе-
ний на рис 2.18.
При наявності великої
кількості експерименталь-
них точок і високої точності
вимірів можна побудувати й
графік другої
похідної
2
2
/
()
dEdV fV
(така крива
є результатом диференцію-
вання
першої
похідної
//
d dE dV dV).Цяфункція
є розривною в крапці екві-
валентності, тому що до то-
чки еквівалентності перша
похідна позитивна й різко
зростає, а після стрибка –
має таке ж значення велике
за модулем, але негативне, й
тому по модулю зменшуєть-
Рис. 2 .18. Диференціальна крива титру-
вання суміші сильної (HCl) та слабкої
(CH3COOH) кислот лугом.
Робота No 6. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
133
ся. Крапка еквівалентності визначається як крапка п еретинання
асимптот цих двох кривих (рис. 17 а).
Загальним недоліком усіх кривих титрування в потенціометрії
є їх S-подібна форма. Як наслідок – кінцева точка титрування роз-
ташовується в області максимального нахилу кривої, де точність
виміру є найменшим. Найменш точні результати точки еквівалент-
ності одержують по інтегральній кривій титрування. При побудові
диференціальної кривої титрування результати трохи краще, але
виникають погрішності, що викликані диференціюванням. Однак і в
цьому випадку найбільш важливі для знаходження координати точ-
ки еквівалентності експериментальні значення, як і раніше, перебу-
вають поблизу точки перегину, де виникають значні погрішності
виміру. Із цієї причини Гран запропонував використовувати в якості
кривої титрування залежності, що отримані лінеаризацією кривій
титрування.
/
VE
(рис. 2.17б). У цих координатах на графіку ви-
ходять дві лінійні залежності, точка перетинання яких лежить на осі
абсцис і відповідає кінцевій точці титрування. Точками, що розта-
шовані поблизу від кінцевої точки титрування, можна тепер знева-
жити й у такий спосіб добитися підвищення правильності й точнос-
ті визначення. Переваги й зручності методу Грана особливо помітні
при аналізі розведених розчинів, оскільки
.
.
тe
V визначається з біль-
шою точністю.
Метод Грана заснований на лінеаризації кривій потенціомет-
ричного (рН-метричного) титрування. Метод застосовується при
кислотно-основному титруванні, коли титрантом є сильна кислота
або сильна основа. Припустимо, сильна кислота (наприклад, хлори-
дна) з концентрацією к
c й початковим об'ємом к
V титрується силь-
ним лугом (гідроксидом натрію) з концентрацією л
c . За умови еле-
ктронейтральності випливає, що:
[][][][]
H
OH
Cl
Na
.
Концентрація гідроксид- іонів пов'язана з концентрацією іо-
нів водню через іонний добуток води:, тобто [
]/[]
w
OHKH
.
Концентрації іонів [ ]
Clі[]
Na
з врахуванням зміни об'єму в
процесі титрування описується рівняннями:
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
134
[]кк
o
щ
cV
Cl
VV
і[]лл
к
л
cV
Cl
VV
,
дел
V – об'єм доданого титранта – розчину лугу.
До крапки еквівалентності концентрація іонів водню при до-
даванні будь-якого об'єму титранта буде описуватися рівнянням:
кк
лл
w
Н
к
л
Н
cVcVК
с
VV
с
.
Це рівняння є правильним й після точки еквівалентності. Пі-
сля точки еквівалентності, коли в надлишку перебуває луг, спра-
ведливим буде рівняння
лл
кк
w
OН
к
л
OН
cVcVК
с
VV
с
.
Якщо концентрацію гідроксид- іонів виразити через іонний
добуток води й концентрацію іонів водню, рівняння набуває ви-
гляду:
w
лл
кк
Н
к
л
H
КcVcV
с
с
VV
.
Очевидно, що обидва рівняння тотожні.
Значення
Н
сй
w
К у рівнянні можна представити як 10 pH
й10 w
pK
або через ЕРС гальванічного елемента
*
10
10
EE
E
.
Тоді рівняння може бути перетворене в такий спосіб:
()(10 10
)(
)
w
E
E
pK
л
к
л
кк
лл
GV
VVсVсV
.
Вираження в лівій частині рівняння зветься функцією Грана
(()л
G V ). Якщо в кожній точці титрування розрахувати функцію
Грана й побудувати залежність її від об'єму титранта, графік буде
являти собою лінійну залежність, що перетинає вісь абсцис за
умови к к
лл
сV сV
, тобто в точці еквівалентності.
Робота No 6. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
135
Таким чином, точку еквівалентності можна визначити вико-
ристовуючи початкову ділянку кривої титрування сильної кис-
лоти або лугу.
Більш проста інтерпретація методу Грана полягає в побудові
залежності в координатах
/
VE
(рис. 2.16 б). У цих координатах
на графіку також виходять дві лінійні залежності, точка перетинан-
ня яких лежить на осі абсцис і відповідає кінцевій точці титрування.
Точками, що розташовані поблизу від кінцевої точки титрування, де
спостерігаються найбільші погрішності, можна тепер зневажити й у
такий спосіб добитися підвищення правильності й точності визна-
чення.
У порівнянні з індикаторним потенціометричне титрування
має низку переваг:
–
вимір можна проводити в пофарбованих розчинах і в розчи-
нах з осадом;
–
можливо безпосередньо й кількісно визначити спільно ком-
поненти розчину;
–
висока чутливість, що перевершує звичайний метод об'ємно-
го титрування;
–
можливість повної або часткової автоматизації процесу тит-
рування.
–
крім аналітичних завдань потенціометричне титрування до-
зволяє вирішувати й фізико-хімічні: визначення добутку розчиннос-
ті, констант стійкості, констант протолітичної дисоціації.
Аналітичні реакції, які використовуються при потенціометри-
чному титруванні, повинні задовольняти наступним умовам:
–
речовина, яку титрують, й титрант повинні реагувати між со-
бою в стехіометричному співвідношенні;
–
повинен застосовуватися цілком доступний індикаторний
електрод, потенціал якого повинен мати певне значення й оборотне
визначатися концентрацією одного з іонів у реакції;
–
реакція повинна протікати кількісно (константа рівноваги
повинна мати як можна більш високе значення);
–
рівновага між реагуючими компонентами й індикаторним
електродом повинне встановлюватися швидко.
До недоліків потенціометричного титрування можна віднести
не завжди швидке встановлення рівноважного значення потенціалу
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
136
після кожного додавання титранта й необхідність у багатьох випад-
ках робити при титруванні велику кількість відліків.
У цей час у наукових дослідженнях криві титрування поперед-
ньо згладжують за допомогою поліномів невисокого ступеня
(сплайнів), що забезпечують згладженість у точках розбивки, а вна-
слідок цього вищу якість розрахованих значень першої й другої по-
хідних.
ВИКОНАННЯ РОБОТИ
Для виконання потенціометричного титрування складають га-
льванічний елемент з напівелемента з хінгідронним (I) або скляним
(II) електродом як індикаторним й електродом порівняння, у якості
якого зручно використовувати насичений хлорсрібний електрод
64
264
2
(),,(
.)
,
(),()()
PtAgClKClнас H CHOH CHOH Pt
(I)
(),,(
.)
(),()
PtAgClKClнас H СЭH Pt
(II)
Установка для потенціометричного титрування з хінгідронним
електродом схематично наведено на рисунку 2.19, де: 1 – магнітна
мішалка з магнітом 2; 3 – склянка з розчином що титрують; 4 –
бюретка з лугом для
титрування; 5 – хінгі-
дронний електрод; 6 –
хлорсрібний
елект-
род, заповнений на-
сиченим
розчином
KCl з рідинним кон-
тактом, що утворю-
ється з титруємим ро-
зчином через азбесто-
ву нитку, що впаянау
в капіляр; 7 – рН-
метр-мілівольтметр.
При викорис-
танні хінгідронного
електрода як індика-
Рис. 2 .19 . Прилад для проведення потенціомет-
ричного титрування
Робота No 6. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
137
торного в розчин насипають невелику кількість хінгідрону (еквімо-
лекулярна суміш хінону й гідрохінону) і занурюють платинову пла-
стинку 5 зі струмовідводом, що упаяний в скляну трубку.
Розчин для аналізу (кислоту й її об'єм указує викладач) розба-
вляють дистильованою водою до такого об'єму, щоб електроди були
повністю занурені в розчин і не заважали обертанню магнітної мі-
шалки. Спочатку роблять грубе титрування для встановлення об'єму
лугу, необхідного до досягнення стрибка (стрибків) титрування. Для
цього розчин лугу з бюретки доливають такими однаковими порці-
ями (0,5–1 мл), щоб до стрибка титрування одержати 5–8 точок.
Електрорушійну силу гальванічного елемента зручно вимірювати на
рН-мілівольтметрі (типу pН–121 або іономірі типу ЭВ–74 або з ви-
користанням цифрового вольтметра з високим вхідним опором).
При наявності стрілочного приладу стрибок титрування спостеріга-
ється особливо наочно по значних відхиленнях стрілки приладу по
досягненню точки еквівалентності.
Після проведення грубого титрування роблять точне титру-
вання. Для його проведення титрування повторюють так само, із ті-
єю лише відмінністю, що в області стрибка потенціалу індикаторно-
го електрода розчин луги доливають дрібними порціями по 0,1 мл,
відслідковуючи зміну ЕРС в області стрибка титрування більш де-
тально. В обох випадках важливою умовою є додавання розчину лу-
ги й після досягнення крапки еквівалентності для забезпечення мо-
жливості побудови диференціальних кривих титрування, а саме –
їхніх правих галузей. При вимірюванні дочікуються рівноважного
значення потенціалу.
Результати дослідження записують у вигляді таблиці
Концентраціялугу с,моль/л.................
Об'ємкислотидлятитруванняV,мл.............
Грубе титрування
Точне титрування
Об'єм лугу,
V, мл
ЕРС,
E, мВ
Об'єм лугу,
V, мл
ЕРС,
E, мВ
Е/V
Аналогічно проводять титрування суміші сильної й слабкої
кислот, обравши для титрування об'єми кислот, дані викладачем.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
138
По даним потенціометричного титрування будують інтегра-
льні й диференціальні криві титрування, по яких розраховують зна-
чення концентрації кислот, порівнюють результати розрахунків,
отримані за даними титрування суміші й роздільному титруванні
однієї з кислот. Робляться висновки про можливість і точності спі-
льного визначення концентрації сильної й слабкої кислот методом
потенціометричного титрування.
ДИФЕРЕНЦІАЛЬНЕ ТИТРУВАННЯ
Диференціальні криві титрування можна побудувати безпосе-
редньо, а не розрахувати їх із графіка E – об'єм титранта V. Прилад
для проведення диференціального титрування представлений на ри-
сунку. Таким чином, можна провести як кислотно-основне, так і ти-
трування за осадженням або окисно-відновне титрування.
Для проведення титрування використовуються два ідентичні
індикаторні електроди 1 і 1', оборотних до іонів, що змінюють
свою концентрацію в ході реакції титрування. Наприклад, при кис-
лотно-основному титруванні можна використовувати два хінгід-
ронних електрода (рис. 2 .20). Один з
електродів 1' поміщають у трубку 2,
яка якийсь час утримує невелику части-
ну розчину, що титрують, а основна кі-
лькість розчину перебуває в посудині 3.
Спочатку ЕРС між електродами дорів-
нює нулю, бо розчини однакові. При
додаванні агента, що титрує, потенціал
електрода 1 змінюється внаслідок ви-
далення деякої кількості потенціалвиз-
начальних іонів з розчину при титру-
ванні, у той час як потенціал електрода
1' залишається колишнім, тому що роз-
чин у цього електрода ізольований від
розчину в основній посудині і його
склад не змінюється. Реєструється ЕРС,
причому кожному об'єму V доданого
титранта відповідає своя величина E.
Рис. 2 .20. Установка для ек-
спериментального проведення
дифференціального титруван-
ня.
Робота No 6. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
139
Виштовхуючи й затягуючи розчин із трубки 2 в основний розчин за
допомогою груші 4 (або шприца), уміст змішується й різниця поте-
нціалів між електродами знову стає рівною нулю. Додаючи порція-
ми титрант, процедуру виміру повторюють і будують графік залеж-
ності E /V від доданого об'єму титранта. Максимум кривої від-
повідає кінцевій точці титрування.
ПИТАННЯ ДО ЛАБОРАТОРНОЇ РОБОТИ
1. Чому потенціометричне титрування проводять у гальваніч-
них елементах з рідинною сполукою?
2. Чим обумовлений стрибок ЕРС при титруванні?
3. Які електроди можна використовувати як індикаторні й еле-
ктроди порівняння при кислотно-основному титруванні?
4. Як визначається крапка еквівалентності на інтегральній і
диференціальної кривих титрування?
5. Яка кислота й чому титрується лугом першої в суміші силь-
ної й слабкої кислот?
6. Яке умова визначення концентрації сильної й слабкої кисло-
ти при потенціометричному титруванні їх суміші?
7. У якій послідовності будуть титруватися галогенід іони в
суміші їх солей нітратом срібла й чому ця послідовність обумовлена?
8. Від чого залежить висота стрибка потенціалу на кривій по-
тенціометричному титрування?
9. Приведіть приклад окисно-відновного титрування.
10. Поясните принцип експериментальної побудови диференці-
альних кривих титрування.
11. Чому при проведенні потенціометричного титрування обо-
в'язково доливається надлишок титранта й після досягнення крапки
еквівалентності?
12. чи Можна використовувати ланцюги без переносу для поте-
нціометричного титрування?
13. Що необхідно знати, щоб розрахувати рН у кожній крапці
титрування по кривій потенціометричного титрування?
14. Як можна, використовуючи криву потенціометричного тит-
рування слабкої кислоти лугом, розрахувати константу дисоціації
кислоти?
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
140
15. Яким образом можна впливати на константу дисоціації сла-
бкого електроліту?
16. У чому проявляється дія, що диференціює, розчинника на
силу кислот?
ТЕОРЕТИЧНІ ПИТАННЯ ПО ТЕМІ
Електрохімічні процеси. Теорії виникнення електродного по-
тенціалу й електрорушійної сили.. Застосування концентраційних
елементів з переносом і без переносу. Методи виміру ЕРС, компен-
саційний метод. Елемент Вестона. Дифузійний потенціал, його при-
рода й методи елімінування. Рівняння Нернста для потенціалу елек-
трода й ЕРС гальванічного елемента. Стандартний потенціал і стан-
дартна ЕРС гальванічного елемента. Типи електродів: першого,
другого й третього роду, окисно-відновні, іонообмінні (скляний)
електроди. Теорія скляного електрода, потенціал асиметрії. Іоносе-
лективні електроди. Переваги й недоліки скляного електрода. Ме-
тоди перевірки електродної функції іоноселективних електродів.
Хінгідронний електрод, лужна й сольова помилки хінгідронного
електрода.
Буферна дія й буферна розчини, буферна ємність і її визначен-
ня. Кислотність розчинів. Термодинамічний зміст рН. Стандартні
буферні розчини, їх сполука. Потенціометричне визначення рН ста-
ндартних буферних розчинів. Використання електрохімічних лан-
цюгів для вивчення розчинів електролітів: визначення констант ди-
соціації слабких електролітів, іонного добутку розчинника, добутку
розчинності; визначення рН, коефіцієнтів активності електролітів,
потенціометричне титрування, визначення рН стандартних буфер-
них розчинів, термодинамічних характеристик реакцій і речовин.
Потенціометричне титрування й способи визначення крапки еквіва-
лентності.
ЛІТЕРАТУРА
1. Лебідь, В. І. Фізична хімія : підруч. для студ. вищ. навч. за-
кл. / В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
2. Герасимов Я. И. и др. Курс физической химии. Т. 2. – М. :
Химия, 1973. – 625 с.
Робота No 6. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
141
3. Дамаскин Б. Б. и др. Электрохимия / Б. Б. Дамаскин, О. А.
Петрий, Г. А. Цирлина. – М. : Химия, КолосС, 2006. – 672 с.
4. Никольский Б. П. Ионоселективные электроды / Б. П. Ни-
кольский, Е.А. Матерова. – Л. : Химия, 1980. – 240 с..
5. Практикум по физической химии / Под ред. В. В. Буданова
и Н. К. Воробьева. – М. : Химия, 1986. – 351 с
6. Практикум по электрохимии / Под ред. Б. Б. Дамаскина.
–
М. : Высшая школа, 1991. – 288 с.
7. Рабинович В. А. Термодинамическая активность ионов в рас-
творах электролитов / В. А. Рабинович. – Л. : Химия, 1985. – 176 с.
8. Ротинян А. Л., Тихонов К.И., Шошина И.А. Теоретическая
электрохимия / Под. ред. А. Л. Ротиняна. – М. : Студент, 2013. – 496 с
9. Измайлов Н. А. Электрохимия растворов / Н. А. Измайлов.
–
М. : Химия, 1986. – 576 с.
10. Александров В. В . Кислотность неводных растворов.
–
Харьков: Вища школа. Изд-во при Харьковском ун-те, 1981. – 152 с.
11. Александров В. В. Кислотность неводных растворов. В сб.
Научное наследие Н.А.Измайлова и актуальные проблемы физиче-
ской химии под. ред. В. И. Лебедя, Н. О. Мчедлова-Петросяна и Ю. В.
Холина. – Х. : ХНУ имени В.Н. Каразина, 2007. – 675 с. С. 227–407.
12. Батлер Дж. Ионные равновесия (математическое описание) /
Дж. Батлер. – Л. : Химия. 1973. – 448 с.
13. Бейтс Р. Определение рН. Теория и практика / Р. Бейтс. – Л.
: Химия, 1971. – 400 с.
14. Бугаевский А. А. Расчет равновесий в растворе / А. А. Буга-
евский. – Х. : Вища школа. 1980. – 156 с.
15. Практические работы по физической химии: Учебное посо-
бие для вузов / Под ред. К .П. Мищенко, А.А. Равделя и А.М . Поно-
маревой. – Л.: Химия, 1982. – с. 128–178.
16. Хартли Ф., Бергес К., Олкок Р. Равновесия в растворах / Ф.
Хартли, К. Бергес, Р. Олкок. – М. : Мир, 1983. – 360 с.
17. Шульц М. М . Стеклянный электрод. Теория и применение.
– Соросовский образовательный журнал. – 1998. No 1. с . 33 –39.
18. Кузнецов В. В . Определение рН // Соросовский образова-
тельный журнал. 2001. Т. 7(4). С . 44 –51.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
142
Робота No 7.
ВИЗНАЧЕННЯ ІОННОГО ДОБУТКУ ВОДИ
У ЛАНЦЮГАХ БЕЗ ПЕРЕНОСУ
Ціль роботи: визначити іонний добуток води при заданій те-
мпературі з даних виміру ЕРС у ланцюзі без переносу. Зрівняти
отримані результати з довідковими даними.
Прилади й реактиви. Установка для виміру ЕРС із потенці-
ометром і гальванометром (типу М-95). Термостат (точність
термостатування ±0,05°; електролізер з випрямлячем і системою
очищення водню, комірка U-подібна з водневим платинованим еле-
ктродом, хлорсрібний електрод термоелектролітичного типу; ро-
зчин лугу точної концентрації, хлорид натрію (дрібнокристалічний,
х.ч .); мірні колби 500 мл, 100 мл), піпетки (100, 50, 20, 15, 10 мл);
барометр – анероїд; "собачка" – ємність для зважування наважки
солі; терези аналітичні типу ВЛА–200.
Виміру іонного добутку розчинника проводять у гальванічно-
му елементі без рідинної сполуки типу
2
1
2
( , ,101325 )
(), ()
,
()
PtH г
Па KOHm NaClm AgClAgPt,
В розчинах лугу точної концентрації з додаванням хлориду натрію
для забезпечення роботи хлорсрібного електрода.
Значення стандартного потенціалу хлорсрібного електрода, яке
необхідне для розрахунків іонного добутку, необхідно знати зазда-
легідь. Його визначають:
–
екстраполяцією на нескінченне розведення ЕРС елемента на-
веденого до стандартної концентрації 1 моль/л за експерименталь-
ним даними концентраційної залежності ЕРС у розчинах хлористо-
го водню в області розведених розчинів (до 0,1 моль/л), використо-
вуючи рівняння Дебая – Хюккеля;
–
калібруванням електродної системи виміром ЕРС у тому ж
ланцюзі зі стандартним розчином хлористого водню (краще точної
концентрації 0,0100 або 0,1000 моль/л) і використанням для розра-
хунків стандартного потенціалу довідковими даними за коефіцієн-
тами активності НCl ;
Робота No 7. ВИЗНАЧЕННЯ ІОННОГО ДОБУТКУ ВОДИ У ЛАНЦЮГАХ
БЕЗ ПЕРЕНОСУ
143
–
використовуючи довідкові дані (за умови попередньої пере-
вірці й відбору електродів, які використовуються для роботи).
Електрорушійна сила досліджуваного гальванічного елемента
визначається відповідно до рівняння Нернста
o
lnH
Cl
RT
EE
a
a
F
або
o
/
lg
AgCl Ag
H
Cl
E
a
a
,
Активність іонів водню
H
a може бути виражена з рівняння для
іонного добутку води
2
HO
HOH
,
w
K,
w
H
OH
Kaa
,
w
H
OH
K
a
a
.
Підстановка
H
a в рівняння для ЕРС ланцюга дає вираження
o
o
/
/
lg
lg
Cl
Cl
AgCl Ag
w
AgCl Ag
w
OH
OH
a
a
E
K
pK
a
a
.
Тому що луг KOH і сіль NaCl є сильними електролітами, рі-
вняння для ЕРС гальванічного елемента може бути виражене через
відомі на досліді величини концентрацій компонентів розчину
(12
и
m
m ) і коефіцієнти активності хлорид і гідроксид іонів
o
2
/
1
lg
lg Cl
AgCl Ag
w
OH
m
E
pK
m
.
З даного рівняння виразимо функцію
w
pK , яка відрізняється
від шуканого термодинамічного значення
w
pK наявністю другого
доданка
o
/
2
1
lg
lg
AgCl Ag
Cl
w
w
OH
E
m
pK
pK
m
.
Відношення коефіцієнтів активності однозарядних іонів, що
знаходяться в правій частині рівняння, близько до одиниці й мало
залежить від іонної сили розчину. Це дозволяє використовувати ро-
зчини зі значною іонною силою, зберігаючи лінійність залежності
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
144
екстраполяційної функції pK від іонної сили. Так, описуючи від-
ношення коефіцієнтів активності у відповідності із другим набли-
женням теорії Дебая – Хюккеля, для логарифма відносини коефіці-
єнтів активності аніонів маємо вираз
lg
1
1
Cl
Cl
OH
OH
AI
AI
bI
bI
BaI
BaI
(
)
Cl
OH
bb
IbI
,
де іонна сила розчину дорівнює сумарної концентрації лугу й солі в
розчині
2
1
NaCl
NaOH
Im
m
m
m
.
Таким чином, екстраполюючи функцію іонної сили
w
pK на
нескінченне розведення, можна оцінити термодинамічне значення
іонного добутку води.
При роботі з водневим електродом обов'язково приводять по-
тенціал водневого електрода до парціального тиску водню 1 атм.
(101325 Па). Таблиця виправлень на тиск водню залежно від зовні-
шнього тиску й температури у водяних розчинах наведена в таблиці
2 додатка.
ВИКОНАННЯ РОБОТИ
Готування розчинів. Істотний вплив на вірогідність результа-
тів виявляє спосіб готування, стандартизації й зберігання розчинів
лугу. Необхідно передбачити захист основного розчину лугу від ат-
мосферного вуглекислого газу. Розчини лугу використовують сві-
жоприготовленими, усі дослідження проводяться протягом одного
заняття. Приготовлені розчини лугу не зберігати!
Основний розчин лугу (краще KOH ) готується з вихідного
стандартизованого й захищеного розчину лугу розведенням свіжо
перегнаної, прокип'яченої й швидко охолодженою дистильованою
водою. Концентрація вихідного розчину готується в межах 0,02–
0,05 моль/л (концентрацію вказує викладач). Для готування розве-
денням використовується мірна колба на 500 мл і піпетки. Частину
приготовленого розчину використовують для готування робочих
розчинів лугу з добавками солі – хлориду натрію. У мірну колбу на
Робота No 7. ВИЗНАЧЕННЯ ІОННОГО ДОБУТКУ ВОДИ У ЛАНЦЮГАХ
БЕЗ ПЕРЕНОСУ
145
200 мл методом відсипання (використо-
вувати "собачку") містять наважку солі –
хлориду натрію для одержання розчину
заданої концентрації за вказівкою викла-
дача (приблизно 0,1–0,2 моль/л). Мірна
колба доводиться до мітки приготовле-
ним розчином лугу й ретельно перемішу-
ється.
Серія робочих розчинів лугу з убу-
тною концентрацією хлориду натрію го-
тується змішанням основного розчину
лугу й розчину з добавками хлориду на-
трію. Співвідношення об'ємів розчинів
указує викладач. Для змішання розчинів
використовують мірну колбу на 100 мл і
набір піпеток: від 10 мл до 50 мл.
Кількості розчинів по 100 мл ціл-
ком достатньо для проведення вимірів у
вимірювальній комірці й при необхідно-
сті виконання декількох заповнень.
Для проведення вимірів ЕРС необхідно дотримуватися насту-
пного ходу роботи.
Перед роботою інженером включається випрямляч генератора
водню (электролізера) для повного заповнення внутрішнього прос-
тору електролізера воднем. При повному заповненні внутрішнього
простору електролізера воднем автоматично розривається контакт
електрода (катода) з розчином лугу й електроліз припиняється.
Проводиться настроювання потенціометра. Виміряється тем-
пературу нормального елемента Вестона, визначається по паспорт-
них таблицях ЕРС нормального елемента й виставляється це значен-
ня на потенціометрі. Далі проводиться настроювання робочого стру-
му потенціометра, контролюючи процес на гальванометрі. Чутли-
вість гальванометра послідовно збільшується за допомогою шунта
досягаючи максимального значення..
Першим для дослідження береться самий розведений щодо хло-
риду натрію розчин. Комірка разом з водневим 2 і хлорсеребряным 3
Рис. 2.21 . Комірка для ви-
міру ЕРС в гальванічному
елементі з водневим елект-
родом
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
146
електродами (рис. 2.21) промивається споліскуванням на менш 3-х ра-
зів і заповнюється цим же розчином. Частиною досліджуваного роз-
чину заповнюється рідинний затвор 1 і комірка міститься в термостат,
а електроди приєднують до потенціометра.
Простір над розчином у комірці інтенсивно продувається стру-
мом водню (50–100 мл газу), звільняючи його від повітря. Далі на-
строюється потік водню через розчин комірки повільніше – зі шви-
дкістю 1–2 пухирця в секунду.
Змінюючи, напругу на потенціометрі компенсують ЕРС галь-
ванічного елемента, встановлюючи нульове значення струму в лан-
цюзі контролюючи процес компенсації на гальванометрі. Спочатку
загрублюють чутливість гальванометра за допомогою шунта, а при
наближенні до рівноважної різниці потенціалів чутливість збіль-
шують до максимальної. Кожні 5 хвилин проводять операцію ком-
пенсації й записують показання потенціометра доти, поки протягом
10 хвилин (три показання підряд) значення не будуть відтворювати-
ся з точність ±0,1 мВ. Гальванометр включати тільки для пере-
вірки моменту компенсації ЕРС.
Як правило, заповнення комірки тим же розчином роблять не
менш двох разів, й при збіганні рівноважних значень ЕРС двох за-
повнень із точністю ±0,3 мВ переходять до дослідження інших роз-
чинів, що мають більшу концентрацію хлориду натрію. Останнім
виміряється вихідний розчин з найбільшою концентрацією хлориду
натрію, що залишився в мірній колбі.
При проведенні експерименту рекомендується вести записи,
дотримуючись наступної форми.
Вихіднаконцентраціялугу.......... Об'єм..........
солі........
Об'єм..........
Наважка хлоридунатрію: вагасолізтарою...........
вагапіслявідсипання.......
Концентраціяхлоридунатрію..........
Концентрації добавок хлориду натрію в розчинах лугу
1............
3............
2............
4. ...........
Робота No 7. ВИЗНАЧЕННЯ ІОННОГО ДОБУТКУ ВОДИ У ЛАНЦЮГАХ
БЕЗ ПЕРЕНОСУ
147
Виміри ЕРС:
Час
ЕРС, мВ
Час
ЕРС, мВ
с(NaСl)=.........
Перше заповнення
Друге заповнення
Для розрахунків беруть результати останнього заповнення
комірки як більш близького до дійсного значення. Значення ЕРС
елемента при роботі з водневим електродом необхідно привести до
парціального тиску водню рівному 1 атм (101325 Па). Для цього
вимірюють зовнішній тиск P на барометрі й рівноважні значення
ЕРС виправляють на величину виправлення на тиск пари розчинни-
каs
p за температури виміру потенціалу. Рівняння Нернста для вод-
невого електрода має вигляд
2
2
1/2
/
lnH
HH
H
a
RT
Fp
,
де
2
H
p=P–s
p,
у якому: P – зовнішній (атмосферний тиск),
s
p – тиск насиченої
пари розчинника за даною температурою. Приведення до парціаль-
ного тиску водню вимагає додатка до обмірюваних значень ЕРС ви-
правлення
760
ln
2
s
RT
E
FPp
(якщо тиск обмірюваний у мм. рт. ст.).
Таблиця виправлень E
на тиск водню в водних розчина за рі-
зних температур наведено в таблиці 28 додатку.
Після проведення вимірів і виконання попередніх обчислень
концентрації реагентів у розчині проводять розрахунки, пов'язані з
визначенням іонного добутку розчинника. Усі розрахунки проводять
у журналі, заносячи проміжні й кінцеві результати в таблицю за зра-
зком наведеному вище.
Графік залежності
w
pK – іонна сила розчину будують на мілі-
метрівці в масштабі, який відповідає точності вимірів ЕРС: 0,1 мВ =
1 мм. При відсутності викидів, проводять аналітичну екстраполя-
цію, використовуючи лінійний метод найменших квадратів. Розра-
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
148
хунки можна провести також з використанням електронних таб-
лиць, наприклад, у пакеті Microsoft Office Excel. У цьому випадку
результати представляються у вигляді відредагованих відповідним
чином електронних таблиць й екстраполяційного графіка. Приклад
обробки результатів визначення іонного добутку води по вимірах у
ланцюзі без переносу (I) при 298 К при постійній концентрації лугу
KOH 0,001524 моль/л наведено на рисунку 2.22 . Кутовий коефіці-
єнт залежності, обумовлений тільки відмінностями в коефіцієнтах
активності хлорид- і гідроксильних іонів, може виявитися статисти-
чно незначущим, а коефіцієнт кореляції менше 0,7. У такому випад-
ку регресія визнається незначущої, найбільш імовірним значенням
іонного добутку може вважатися середнє арифметичне значення.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1.Що називають іонним добутком розчинника? Яка зв'язок з
константою дисоціації розчинника.
2. Приведіть класифікацію розчинників. Приведіть приклади
протолітичних розчинників.
3. Основні теорії кислот і підстав.
4. Як і чому міняється з підвищенням температури іонний до-
буток води.
Рис.2.22 . Приклад екстраполяції функції
w
pK для визначення тер-
модинамічної константи іонного добутку води за температури 298 К.
Робота No 7. ВИЗНАЧЕННЯ ІОННОГО ДОБУТКУ ВОДИ У ЛАНЦЮГАХ
БЕЗ ПЕРЕНОСУ
149
5. Якими методами можна визначити іонний добуток води.
Дайте характеристику кожного з них.
6. Як розраховують іонний добуток розчинника за даними виміру
ЕРС ланцюгів без переносу. Які електроди можна використовувати
для роботи.
7. У чому сутність нівелюючої дії, що й диференціює, розчин-
ників.
8. Навіщо проводиться екстраполяція на нульову іонну силу?
9. Чому залежність екстраполяційної функції від іонної сили лі-
нійна?
10. Як розрахувати термодинамічні функції дисоціації води за
даними іонного добутку.
11. Проаналізуйте можливі джерела погрішностей визначення
іонного добутку розчинника методом ЕРС.
12. Яким образом можна оцінити стандартну ЕРС використову-
ваного гальванічного елемента, необхідну для розрахунків іонного
добутку.
13. У чому суть виправлень на тиск водню при роботі з водне-
вим електродом? Поясните розрахункову формулу.
14. Як змінюється характер дисоціації води при додаванні со-
лей? Чому.
15. У чому суть протонної теорії кислот і підстав Бренстеда-
Лоурі?
16. Як розрахувати іонний добуток води при заданій температурі
з термодинамічних даних?
17. Гідроліз солей. Види гідролізу. Константа гідролізу, ступінь
гідролізу.
18. Кислотність розчинів. Показник кислотності рН. Шкала рН і
зв'язок з іонним добутком розчинника.
19. Іонний добуток змішаних розчинників.
20. Опишіть визначення іонного добутку розчинника в ланцюгах
з переносом. Зрівняєте цей метод з методом ланцюгів без переносу.
21. Запропонуєте гальванічний елемент і компонента розчинів
для визначення іонного добутку етанолу.
22. Які електроди можна використовувати для визначення іон-
ного добутку в ланцюгах без переносу. Відповідь аргументуйте.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
150
23. Чому іонний добуток змінюється зі зміною температури.
24. Як і чому змінюється іонний добуток вода при добавці нево-
дного компонента
. ЛІТЕРАТУРА
1. Научное наследие Н. А. Измайлова и актуальные проблемы
физической химии (под ред. В. И. Лебедя, Н. О. Мчедлова-
Петросяна и Ю. В . Холина). – Х. : ХНУ им. В . Н. Каразина, 2007. –
675 с.
2. Александров В. В . Кислотность неводных растворов.
–
Харьков: Вища школа. Изд-во при Харьковском университете, 1981.
–
152 с.
3. Дамаскин Б. Б., Петрий О. А., Цирлина Г. А. Электрохимия.
–
М. : Химия, КолосС, 2006. – 672с.
4. Байрамов В. М . Основы электрохимии. – М . : ACADEM1A,
2005. – 238 с
5. Измайлов Н. А. Электрохимия растворов.
–
Х. : Изд-во
Харьк. ун-та, 1959. – 958 с.
6. Харнед Г., Оуэн. Б. Физическая химия растворов электроли-
тов. –М. : ИЛ, 1952. – 628 с.
7. Робинсон Р., Стокс Р. Растворы электролитов.
–
М.: ИЛ.
1963. – 646 с.
8. Бейтс Р. Определение рН. Теория и практика. – Л . : Химия,
1971. – 400 с.
9. Рабинович В. А. Термодинамическая активность ионов в
растворах электролитов. – Л. : Химия, 1985. – 176 с.
10. Шаталов А. Я . Введение в электрохимическую термодина-
мику. –М. : Высшая школа. 1984. – 215 с.
11. Скорчеллетти В. В . Теоретическая электрохимия.
–Л.:Хи-
мия, 1974. – 568 с.
Робота No 8. ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ РОЗЧИНІВ
151
Робота No 8.
ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ
РОЗЧИНІВ
Ціль роботи: провести калібрування скляного електрода, ви-
значити його електродну функцію й рН заданого розчину. Провести
дослідження буферної дії заданого буферного розчину. Визначити
значення рН розчину з використанням хінгідронного електроду.
Прилади й реактиви: рН–мілівольтметр, комірка для вимірів
зі скляним електродом, схема із трьома магазинами опорів або по-
тенціометр, платинові електроди для хінгідронного електрода, хі-
нгідрон, набір стандартних буферних розчинів, реактиви для готу-
вання робочих буферних розчинів, дистильована вода, мірні колби
на 100 мл, аналітичні ваги з важком.
Вода являє собою слабкий електроліт, що дисоціює з утвором
іонів водню, що перебувають у вигляді гідроксоній іонів 3
HO
,ігі-
дроксильних іонів OH
:
2
2
3
HO HO
HO OH
.
Застосування закону діючих мас до процесу дисоціації води з
обліком її винятково малої дисоціації й внаслідок цієї рівності акти-
вності води одиниці дає сталість добутку іонів, який залежить тіль-
ки від температури. За температури 298 К іонний добуток води
w
K = 1,008·10–14
, звідки за цієї температури активність іонів водню
дорівнює
7
10
w
H
a
K
г-іон/л.
При розчиненні у воді кислоти (луги) концентрація іонів вод-
ню в розчині збільшується (зменшується).
Величина
H
a характеризує активну кислотність розчину. Ки-
слотність прийнято виражати за допомогою водневого показника –
негативного логарифма активності водневих іонів:
lgH
pH
a
.У
нейтральному розчині при 298 К pН = 7, у кислому pН < 7, у луж-
ному pН > 7. Аналогічним образом можна ввести показник інших
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
152
іонів, наприклад, гідроксильний показник pОН. З вираження іон-
ного добутку води випливає за температури 298 К
w
pK pH pOH
= 14.
Експериментально визначити активність іонів одного виду не-
можливо, тому поняття активності іонів водню умовно й шкала зна-
чень рН завжди приводиться у відповідність із величинами, які
можуть бути обмірювані експериментально, наприклад, значення
констант дисоціації, ЕРС гальванічних елементів.
На практиці часто використовуються розчини, що володіють
буферною дією, тобто здатністю зберігати незмінним значення рН
при розведенні, додаванні невеликих кількостей кислоти або луги.
Такі розчини називаються буферними. Величина буферної дії кіль-
кісно характеризується буферною ємністю, що дорівнює кількості
еквівалентів сильної кислоти або основи, яку необхідно додати до
1 л буферного розчину, щоб змінити його рН на одиницю. Такі ро-
зчини можуть бути утворені зі слабкої кислоти та її солі, кислих
солей багатоосновних кислот, слабкої основи й її його солі із силь-
ною кислотою.
Розглянемо, наприклад, суміш оцтової кислоти з ацетатом на-
трію. Оцтова кислота дисоціює згідно з рівнянням
3
3
CH COOH
CHCOO H
Константа дисоціації її рівна
3
3
CH COO
H
a
CH COOH
a
a
K
a
.
Звідси
3
3
CH COOH
a
H
CH COO
a
a
K
a
.
Активність недисоційованої слабкої кислоти практично дорів-
нює її початкової концентрації o
к
c (моль/л), тому що при введенні
солі цієї кислоти рівновага дисоціації слабкої кислоти зміщується
вліво. Активність аніона практично визначається початковою кон-
Робота No 8. ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ РОЗЧИНІВ
153
центрацією солі
o
c
c . Тому активність іонів водню визначиться рів-
нянням константою дисоціації слабкої кислоти й співвідношенням
концентрацій слабкої кислоти
o
к
c іїїсолі
o
c
c
о
о
к
a
H
с
c
a
K
c
.
Тому що відношення початкових концентрацій кислоти й солі
не змінюється при розведенні буферного розчину, активність іонів
водню залишається при цьому практично постійній. Уведення в бу-
ферний розчин сильних кислот або лугів набагато менше підвищує
їхню кислотність або лужність, чим такий самий додаток їх до води,
тому що при цьому або аніони солі зв'язують, що додаються іони
водню з утворенням малодисоціюючої слабкої кислоти, або слабка
кислота частково взаємодіє з добавками лугу з утворенням солі.
Експериментально визначаються так звані «інструментальні»
величини рН, які засновані на порівнянні кислотності даного роз-
чину з еталоном рН – стандартним буферним розчином. Для цього
можна використовувати фотометричні або потенціометричний ме-
тоди.
Потенціометричний метод визначення активності іонів во-
дню заснований на використанні концентраційного гальванічного
елемента з рідинною сполукою з електродами, оборотними стосов-
но іонів водню (водневий електрод, хінгідронний або скляний).
Процес виміру рН досліджуваного розчину полягає в порівнянні
потенціалу оборотного до іонів водню електрода в цьому розчині з
потенціалом такого ж електрода, опущеного в еталонний, стандарт-
ний розчин, який є стандартом цієї властивості розчину.
Необхідно відзначити, що «абсолютні» значення рН не можна
визначити внаслідок неможливості визначення коефіцієнтів актив-
ності окремих іонів і наявності дифузійних стрибків потенціалу. У
цей час розроблені методи визначення рН з досить високою точні-
стю, зокрема по вимірах у ланцюгах без рідинної сполуки, які вико-
ристовуються для визначення рН стандартних буферних розчинів.
Прикладами еталонних стандартних водних буферних розчи-
нів є розчини первинних стандартів pН, склад й властивості яких за
температури 298 К наведені в таблиці
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
154
Склад розчину, (моль/кг)
рН
Тетраоксалат калію, KH3(C2H4)2(0,05)
1,68
Бітартрат калію, насичений
3,56
Біфталат калію (0,05)
4,01
KH2PO4, (0,025) + Na2HPO4 (0,025)
6,86
Бура Na2B4O7, (0,01)
9,18
При використанні водневого електрода елемент записується у
вигляді
2
2
стандарт-
розчин
розчин
,
, досліджува-
()
,
,
()
ний
ний
o
H
H
H
H
PtH
pa
pa
HPt
ЕРС такого елемента дорівнює
.
.
lg
lg
lgH
o
H
a
o
диф
диф
H
H
a
E
a
a
За умови елімінування залишкового дифузійного потенціалу
.
диф
, вирішуючи рівняння відносно
H
pa,маємо
o
H
H
E
pH pa
pa
.
Дійсне значення рН розчину відрізняється від «інструменталь-
ного» на величину
.
/
диф
,де
.
диф
–
залишковий дифузійний
потенціал, що практично завжди існує при вимірах у ланцюгах з пе-
реносом.
Значення ЕРС наведеного вище концентраційного елемента
може бути отримане шляхом комбінування потенціалів двох напі-
велементів з будь-яким оборотним до іонів водню електродом, об-
мірюваних у досліджуваному розчині (рН) і в стандартному ( o
pH)
проти того самого електрода порівняння, наприклад, насиченого
хлорсрібного напівелемента
2
()
) .),
PtH H KClнас AgClAg
.
На практиці замість водневого частіше використовують хінгі-
дронний або скляний індикаторні електроди.
Робота No 8. ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ РОЗЧИНІВ
155
Хінгідронний електрод ставиться до окисно-відновним елек-
тродам. Хінгідрон – слаборозчинна еквімолекулярна сполука хінону
C6H4O2 і гідрохінону C6H4(OH)2, які для спрощення тут позначимо
через Q і H2Q відповідно, утворюють із іонами водню оборотну
окисно-відновну систему
2
2
2
QH
e
HQ
.
Інертний електрод (платина, золото й т. п.), поміщений у роз-
чин, що містить усі компоненти реакції, приймає потенціал
2
/
QHQ
,
обумовлений їх активностями в розчині
2
2
2
2
o
/
/
lg
2
QH
QHQ QHQ
HQ
aa
a
2
2
2
o
/
lg
lg
2
QQ
QHQ
H
HQ HQ
m
a
m
.
У кислих розчинах концентрації хінону й гідрохінону практи-
чно рівні одна одній й хінгідронний електрод поводиться як водне-
вий електрод, тому що другий доданок у рівнянні дорівнює нулю.
2
2
2
/
/
/
lg
o
o
QHQ QHQ
QHQ
H
a
pH
.
При більших іонних силах виникає сольова помилка хінгід-
ронного електроду, пов'язана з відмінностями в коефіцієнтах актив-
ності хінону й гідрохінону ( Q і 2
HQ
). У лужних розчинах, внаслі-
док дисоціації гідрохінону як слабкої кислоти, виникає погрішність
за рахунок утвору лужної солі гідрохінону, внаслідок чого відно-
шення концентрації хінону й гідрохінону відрізняється від одиниці.
Хінгідронний електрод не можна застосовувати для вимірюван-
нярНврозчинахз pН>7–8
Скляний електрод являє собою тонку скляну мембрану у ви-
гляді кулі, упаяну в скляну трубку. Принцип роботи заснований на
обміні катіонів, що втримуються в структурі скла з іонами в розчи-
ні, у те час як аніони, міцно зв'язані у твердій фазі, не беруть участь
в обміні з аніонами розчину. При тривалому витримуванні скляної
мембрани, виготовленої зі спеціальним образом підібраного сполу-
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
156
ки скла, у воді іони водню обмінюються з катіонами в склі згідно з
рівноважними співвідношеннями їх концентрацій. Таким чином,
константа обміну має вираження
Na
H
ст
ст
H
Na
a
a
K
a
a
,
Де
H
a,
Na
aі
.
ст
H
a,
.
ст
Na
a – активності іонів водню й натрію в роз-
чині й склі відповідно. Звідси можна одержати, що
.
.
.
(
)
H
Na
H
ст
ст
ст
H
Na
H
H
Na
a
Ka
a
ka
Ka
a
a
a
,
де k' – деяка постійна, тому що сума активностей катіонів у склі по-
стійна.
Різниця потенціалів на межі поверхні стекло – розчин виникає
внаслідок різного розподілу іонів між розчином і склом. Стрибок
потенціалу буде рівний
.
lg
o
H
СЕ
СЕ
ст
H
a
a
або
o
lg(
)
СЕ
СЕ
H
Na
ka
Ka
.
Звідси потенціал скляного електрода рівний
o
lg(
)
СЕ
СЕ
H
Na
a
Ka
.
Потенціал СЕ
недоступний для виміру, а різниця потенціалів
між двома розчинами, що перебувають із двох сторін скляної мем-
брани, може бути обмірювана. Для цього у середину скляного елек-
трода заливають розчин HСl і поміщають хлорсрібний електрод, а
сам електрод міститься в розчин з активністю іонів водню x
a , з'єд-
наний через сольовий міст із електродом порівняння, наприклад,
насиченим хлорсрібного напівелементом. Запис такого гальванічно-
го елементу набуває вигляду
Робота No 8. ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ РОЗЧИНІВ
157
скляна мембрана
,
(нас.) ,
x
Ag AgCl HCl
HaKCl
AgCl Ag
,
де 1 – потенціал хлорсрібного електрода в розчині HCl внутріш-
нього заповнення скляного електрода;
2 – потенціал у поверхні розділу стекло – розчин HCl ;
3 – потенціал у поверхні розділу стекло – розчин з активністю
іонів водню x
a;
4 – залишковий дифузійний потенціал рідинного з’єднання;
.
.
ел пор
–
потенціал електроду порівняння.
Потенціали 1, 2 і
.
.
ел пор
–
постійні, значенням потенціалу
4 можна зневажити, а потенціал 3 залежить від активності іонів у
досліджуваному розчині x
a . Таким чином, об'єднавши постійні,
одержимо
.
.
lg(
o
ел пор
СЕ
H
Na
E
a
Ka
.
Константа обміну K – має дуже маленьке значення, порядку
10–10
– 10–14
.
У кислі й слабколужних середовищах, коли
H
Na
a
Ka
рівняння спрощується
.
.
lg
o
ел пор
СЕ
H
E
a
або
.
.
o
ел пор
СЕ
E
pH
.
ЕРС елемента є лінійною функцією рН розчину.
У сильнолужному середовищу величина
NaH
H
Kaa
a
й
електрод стає оборотним до іонів натрію, тобто змінює свою функ-
цію від водневої до натрієвої. Наслідком цього є зміна знака кутово-
го коефіцієнта залежності E – рН на протилежний.
У порівнянні з водневим і хінгідронним електродами скляний
електрод при вимірі рН має низку переваг:
–
електрод застосовується в широкому діапазоні рН;
–
електрод не отруюється й працює в окисно-відновному сере-
довищі;
–
рівноважне значення потенціалу встановлюється за дуже ко-
роткий час;
–
електрод працює в мутних, грузлих і пофарбованих середо-
вищах.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
158
При використанні для вимірів pН хінгідронного електрода ви-
користовують гальванічний елемент
64
2642
,
,
(.)
,
(),
PtAg АgClKClнас HCHOH CHO Pt
.
ЕРС такого елемента, обмірювана з застосуванням стандартно-
го буферного розчину, дорівнює
2
.
/
.
.
.
.
o
ст
QHQ
ст
ел пор
диф
E
pH
,
а обмірювана в елементі з досліджуваним розчином
2
/
.
.
.
o
x
QHQ
x
ел пор
диф
E
pH
.
Тоді рН досліджуваного розчину визначається спільним роз-
в'язком цих рівнянь, тому що стандартний потенціал індикаторного
електрода й електрода порівняння ідентичні
.
.
.
дифф
ст
x
x
ст
EE
pH pH
.
Дифузійний потенціал не може бути розрахований або визна-
чений експериментально з достатньою точністю, тому що він зале-
жить від характеристик окремих іонів. Застосування формул для ро-
зрахунків значень pН можливо, якщо дифузійний потенціал зроби-
ти дуже малим або постійним у двох послідовних вимірах: зі стан-
дартним і досліджуваним розчинами.. Це досягається використан-
ням сольових мостів з концентрованим розчином хлориду калію,
іони якого у водяних розчинах мають досить близьких рухомостях.
За елімінуванням в такий спосіб дифузійного потенціалу залишкове
його значення
.
диф
може бути виключено з розрахунку.
При використанні як індикаторного скляного електрода вико-
ристовують гальванічний елемент
,
,
(.)
(),
PtAg AgClKClнас H
СЕ Pt
й ЕРС такого елемента записується рівнянням Нернста
o
o*
.
.
.
СЕ
ел пор
диф
E
pH
E
pH
.
Робота No 8. ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ РОЗЧИНІВ
159
Однак застосувати описаний вище приймання розрахунків
значень рН досліджуваного розчину не можна, тому що скляний
електрод не має теоретичну оборотність до іонів водню – значення
коефіцієнта c
в рівнянні Нернста для потенціалу скляного елект-
рода часто не дорівнює величині 2,303·RT/F, а тільки близько до
нього. Внаслідок цього кожний конкретний електрод потребує калі-
брування – установленні значення коефіцієнта c
, яке являє собою
значення кутового коефіцієнта прямій у координатах E – рН згідно
з рівнянням Нернста для ЕРС, використовуваного для виміру галь-
ванічного елемента. Значення c
називають електродною функцією
скляного електрода. Відкалібрований по стандартних буферних
розчинах з відомими значеннями рН скляний електрод можна на-
далі використовувати для виміру рН будь-яких розчинів.
ВИКОНАННЯ РОБОТИ
Для проведення роботи збирають потенціометричну установку
з гальванічним елементом з рідинною сполукою
,
,
(.)
(),
PtAg АgClKClнас H СЕН Pt
.
Загальний вид установки для виміру рН представлено на рису-
нку 2.23, де: 1 – склянка з досліджуваним розчином; 2 – скляний
електрод; 3 – промисловий хлорсрібний електрод, заповнений на-
сиченим розчином хлористого калію рідинний контакт, що й має, з
досліджуваним роз-
чином через азбес-
тову нитку в капіля-
рі4;5–
термо-
метр; 6 – рН-метр-
мілівольтметр.
Для
дослі-
дження готовлять у
мірній колбі два бу-
ферні розчини в кі-
лькості 100 мл.
Склад розчинів та
значення рН указує
Рис. 2 .23 . Установка для виміру рН зі скляним
електродом
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
160
викладач.
Калібрування скляного електрода. Калібрування гальванічно-
го елемента для виміру рН проводиться по стандартних буферних
розчинах (4–6 розчинів) з метою визначення функції скляного елек-
трода.
рН-метр включають в електричну мережу й прогрівають
не менш 15 хвилин
Вимірюють на рН-метрі в режимі роботи як мілівольтметра
значення ЕРС гальванічного елемента в розчинах з різними значен-
нями рН. Виміри ЕРС для кожного розчину роблять 2–3 рази, спо-
ліскуючи скляний електрод і комірку (стакан) для вимірів малими
порціями розчину. Робоча частина скляного електрода (кулька) по-
винна бути повністю занурена в розчин і не торкатися дна й стінок
склянки. При роботі з комбіно-
ваним скляним електродом не-
обхідно стежити за тим, щоб
зовнішній електролітичний
ключ, виконаний з пористої
кераміки, .був повністю зану-
рений у вимірюваний розчин.
Поверхня скляного електро-
да забороняється торкати
руками!
Виміри бажаний прово-
дити в певній послідовності
зміни рН розчинів, напри-
клад, від кислих до лужних у
порядку зростання рН..
За отриманими результа-
тами будується градуювальний
графік на міліметрівці в коор-
динатах E (мВ) – рН (масштаб
графіка:в 1мм–1мВ).Пота-
кому графікові, зображеному
Рис. 2 .24 . Градуювальний графік для
виміру рН зі скляним електродом.
Робота No 8. ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ РОЗЧИНІВ
161
на рисунку 2.24, визначається кутовий коефіцієнт прямій, який чи-
сельно дорівнює похідній
.
/
cт
dEdpH
і відповідний до тангенса
кута нахилу прямій. Раціонально графік побудувати в програмі
Microsoft Excel і провести відповідну статистичну обробку лінійної
залежності.
Розраховане значення водневої функції скляного електрода
порівнюють із теоретичним значенням, певним для температури до-
сліду, яка виміряється термометром, що занурений у розчин.
Після калібрування в такий самий спосіб виміряється ЕРС
елемента із приготовленими буферними розчинами, дистильованою
водою й водопровідною водою. За графіком визначають значення
рН цих розчинів і роблять відповідні висновки. Якщо при обробці
даних на комп'ютері отримані статистичні дані про погрішності па-
раметрів рівняння, то можна оцінити погрішність виміру рН робо-
чих розчинів.
Визначення буферної дії. За описаною вище методиці вимі-
рюють рН розчинів 0.1 н НCl , і розчинів хлористого водню, ро-
зведених від вихідного в 10 і 100 раз. Для цього розведення прово-
дять із використанням мірної колби на 100 мл і піпетки на 10 мл.
Аналогічні виміри проводять із заданим викладачем буферним
розчином, вимірюючи його рН і значення рН розведеного розчи-
ну в 10 і 100 раз. За отриманими результатами зміни значень рН ро-
зчинів роблять висновки про буферну дію досліджених розчинів
при їхньому розведенні.
Далі досліджують вплив лугу на рН одного із приготовлених
буферних розчинів (побрати більш кислий з приготовлених). Для
цього вимірюють ЕРС елемента, у комірку якого зі скляним елект-
родом заливають 20 мл досліджуваного буферного розчину. Доли-
вають послідовно з бюретки по 1 мл 0,1 н розчину КОН, щораз ви-
мірюючи ЕРС елемента й вичікуючи не менш 5 хв для встановлення
рівноважного значення потенціалу скляного електрода. Після дода-
вання 10 мл розчину лугу, визначають значення рН у кожному ви-
падку використовуючи градуювальний графік. Для виявлення дії
лугу на рН розчину будують графік залежності концентрації лугу,
доданої в буферний розчин, залежно від рН розчину. Кутовий кое-
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
162
фіцієнт лінійної ділянки отриманої залежності дає можливість роз-
рахувати буферну ємність досліджуваного буферного розчину.
Рекомендована форма ведення протоколу дослідження
Температура (С................
Досліджуваний
розчин
ЕРС елемента,
Е, мВ
рН
E
ВИМІР рН ІЗ ХІНГІДРОННИМ ЕЛЕКТРОДОМ
Для виміру рН із хінгідронним електродом використовують
гальванічний елемент із переносом з насиченим хлорсрібним елект-
родом у якості електрода порівняння
64
2642
,
,
(),(),
PtAgAgClKClнас HCHOH CHO Pt
,
для чого використовують комірку, яка зображена на рисунку 2.25 .
У стаканчик 1 наливають досліджуваний розчин, додають не-
велика кількість хінгідрону й ретельно перемішують. Поміщають у
розчин гладкий платиновий електрод 2 і опускають насичений хло-
рсрібний електрод 3 проточного типу, попередньо знявши пробку 4
із заливного отвору в корпусі електрода. Через 5–7 хв роблять вимір
ЕРС елемента на компенсаційній установці із трьома магазинами
опорів або на високоомному потенціометрі.
ЕРС даного гальванічного елемента з переносом записується
рівнянням
2
2
/
.
.
/
.
.
.
o
х
QHQ
ел пор
QHQ
х
ел пор
диф
E
pH
,
де
2
/
QHQ
й
2
/
o
QHQ
–
значення потенціалу й стандартного потенці-
алу хінгідронного електрода,
.
.
эл ср
– потенціал насиченого хлорсрі-
бного електроду;
.
диф
–
залишковий дифузійний потенціал,
x
pH–
шукане значення рН розчину.
У даному гальванічному елементі вимірюють ЕРС, викорис-
товуючи кожний із приготовлених раніше буферних розчинів. Ви-
Робота No 8. ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ РОЗЧИНІВ
163
міри ЕРС для кожного розчину проводять двічі, заміняючи розчин у
напівкомірці з хінгідронним електродом.
Перший спосіб розрахунків значень рН заснований на безпо-
середньому використанні електрохімічних параметрів гальванічного
елемента. Вирішуючи рівняння відносно pН, і вважаючи еліміно-
ваним дифузійний потенціал, одержимо
2
/
.
.
o
QHQ
ел пор
х
Е
pH
.
Таким чином, для обчислення рН необхідно мати у своєму
розпорядженні значення
2
/
o
QHQ
й..
ел пор
за температури виміру, які
можна знайти в довідковій літературі.
Оскільки реально використовуваний електрод порівняння мо-
же мати значення потенціалу, що трохи відрізняється від довідково-
го, більш точні значення pН одержують і розраховують рН іншим
способом. Для цього проводять вимір ЕРС цього ланцюга з тими ж
електродами й сольовим мостом, але з розчином, рН якого точно
відомо – зі стандартним буферним розчином з відомим значенням
.
ст
pH . Спільний розв'язок рівнянь для ЕРС елемента, що містить
розчини із шуканим і стандартним значеннями,
дає рівняння для розрахунків щодо стандартно-
го буферного розчину
ст
x
x
ст
EE
pH pH
.
У якості стандартного розчину беруть бу-
ферний розчин з рН < 8 (за вказівкою виклада-
ча). Розрахунки значень рН роблять обома спо-
собами й порівнюють результати між собою.
Готування стандартних буферних розчинів
по наявних методиках дозволяє одержувати
більш відтворені характеристики розчину й з
меншою погрішністю, ніж виготовлення елект-
родів порівняння з теоретичними значеннями
потенціалів. Внаслідок цього другий спосіб ви-
міру рН є кращим. Для одержання точних ре-
Рис. 2.25 . Гальваніч-
ний елемент для ви-
міру рН з хнгідрон-
ним електродом
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
164
зультатів виміру й розрахунку проводять щодо двох стандартних
буферних розчинів, рН яких найбільш близькі до рН досліджува-
ного розчину. Для кожного виміру проводиться два заповнення роз-
чином і для розрахунків беруть середнє арифметичне значення ЕРС
елемента.
При проведенні вимірів на компенсаційній установці із трьома
магазинами опорів результати записуються у вигляді таблиці
ЕРС зі стандартним
буферним розчином,
мВ
ЕРС із досліджуваним
буферним розчином,
мВ
No рНст
Е1
Е2
Е3
Е1
Е2
Е3
рНх
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Що таке кислотність розчинів, як кількісно вона виража-
ється?
2. Чим визначається довжина шкали рН?
3. Які електроди використовуються для визначення рН елект-
рометричним методом?
4. Як улаштований хінгідронний електрод і чому пояснюється
наявність у нього водневої функції.
5. Поясните сольову й лужну помилки хінгідронного електрода.
6. Що таке буферні розчини? Чим пояснюється буферна дія?
7. Який принцип роботи й обладнання скляного електрода?
8. У чому переваги і які недоліки скляного електрода?
9. Як проводиться калібрування скляного електрода й у чому
вона полягає?
10. У чому полягає принцип електрометричного визначення рН
розчинів?
11. Яким образом визначають pН еталонних стандартних бу-
ферних розчинів? Сутність методу АНБС.
12. Як змінюється pН води при збільшенні температури?
13. Поясните використання кислих солей для готування буфер-
них розчинів.
Робота No 8. ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ РОЗЧИНІВ
165
14. Яка природа виникнення погрішності при вимірі рН сильно-
лужних розчинів за допомогою скляного електрода?
15. Чому хінгідронний електрод не використовують для визна-
чення рН лужних розчинів?
16. Укажіть гальванічні ланцюги, у яких можна провести каліб-
рування й визначити водневу функцію скляного електрода. чи Обо-
в'язково необхідно розташовувати стандартними буферними розчи-
нами?
17. У яких ланцюгах можна провести перевірку іоноселектив-
них електродів з натрієвою функцією?
18. Як перевірити водневу функцію скляних електродів у розчин-
никах, для яких відсутні розроблені стандартні буферні розчини?
ТЕОРЕТИЧНІ ПИТАННЯ ПО ТЕМІ
1. У чому причина виникнення різниці потенціалів на границі
двох фаз, наприклад: а) метал – метал; б) метал – розчин; в) метал–
вакуум; г) розчин – розчин.
2. Які процеси, що відбуваються на границі роздягнула метал –
розчин, приводять до появи подвійного електричного шару?
3. Що являють собою електроди першого й другого роду?
4. Виведіть рівняння залежності електродного потенціалу:
а) цинкового електрода від активності іонів цинку; б) каломе-
льного електрода від активності іонів хлору в розчині; в) редокс
електрода від співвідношення іонів олова різної валентності.
5. Які процеси в гальванічному ланцюзі можуть бути причи-
ною виникнення ЕРС? Які типи ланцюгів ви знаєте?
6. Приведіть приклади різних концентраційних елементів.
7. На підставі яких даних можна розрахувати константу рівно-
ваги електрохімічної реакції при заданій температурі?
8. Приведіть приклади гальванічних ланцюгів з переносом і
без переносу.
9. Приведіть приклади оборотних і необоротних гальванічних
ланцюгів.
10. Що таке ряд напруг? Значення яких електродних потенціа-
лів зазначені в ньому?
11. Як впливає тиск водню в газовій фазі на потенціал воднево-
го електрода?
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ.
166
12. Яке обладнання й особливості нормального елемента Вестона?
13. До якого типу електродів ставиться хінгідронний електрод?
Чому й у яких розчинах ним користуються для виміру рН?
14. Який механізм виникнення потенціалу в скляному електро-
ді? Які його переваги й недоліки при визначенні активності іонів
водню?
15. У чому полягає принцип компенсаційного методу виміру
ЕРС гальванічних елементів?
16. Температурний коефіцієнт оборотного гальванічного елеме-
нта менше нуля. Визначите, виділяється або поглинається теплота
при роботі цього елемента.
17. Від чого залежить величина стандартної ЕРС гальванічного
елемента?
18. У якому випадку знак ЕРС гальванічного елемента вважа-
ється позитивним?
19. Яка природа дифузійного потенціалу і яким способом мож-
на зменшити його величину?
20. Поясните оборотність електродів другого роду стосовно ка-
тіонів і аніонам.
21. Що називають потенціалом асиметрії скляного електрода і
які причини його виникнення? Яким чином можна виміряти потен-
ціал асиметрії?
ЛИТЕРАТУРА
1. Лебідь, В. І. Фізична хімія : підруч. для студ. вищ. навч. за-
кл. / В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
2. Практикум по физической химии. Кинетика и катализ. Эле-
ктрохимия : учеб. пособие для студ. учреждений высш. проф. обра-
зования / |А. В .Абраменков и др.] ; под ред. В . В .Лунина, Е.П.Агеева.
—
М. : Издательский центр «Академия», 2012. – 304 с.
3. Научное наследие Н. А. Измайлова и актуальные проблемы
физической химии (под ред. В. И. Лебедя, Н. О. Мчедлова-Петросяна
и Ю. В. Холина). – Х. : ХНУ им. В. Н. Каразина, 2007. – 675 с.
4. Практикум по электрохимии / Под ред. Б. Б. Дамаскина.
–
М. : Высшая школа, 1991. – 288 с.
Робота No 8. ПОТЕНЦІОМЕТРИЧНЕ ВИЗНАЧЕННЯ рН
І ДОСЛІДЖЕННЯ БУФЕРНИХ ВЛАСТИВОСТЕЙ РОЗЧИНІВ
167
5. Практические работы по физической химии : Учебное посо-
бие для вузов / Под ред. К .П. Мищенко, А.А. Равделя, A.M . Понома-
ревой. – СПб. : Изд–во «Профессия», 2002. – 384 с.
6. Александров В. В . Кислотность неводных растворов.
–
Харьков: Вища школа. Изд–во при Харьковском университете, 1981.
–
152 с.
7. Дамаскин Б. Б., Петрий О. А., Цирлина Г. А. Электрохимия.
–
М. : Химия, КолосС, 2006. – 672с.
8. Байрамов В. М . Основы электрохимии. – М . : ACADEM1A,
2005. – 238 с
9. Ротинян А. Л., Тихонов К.И., Шошина И.А. Теоретическая
электрохимия / Под. ред. А. Л. Ротиняна. – М. : Студент, 2013. – 496 с.
10. Измайлов Н. А. Электрохимия растворов.
–
Х. : Изд–во
Харьк. ун–та, 1959. – 958 с.
11. Физическая химия. Теоретическое и практическое руко-
водство. Изд. 2 –е, переработанное и дополненное. / Под ред. акаде-
мика Б. П. Никольского. – Л. : Химия, 1987. – 880 с.
12. Введенский А. В . Равновесные электродные потенциалы,
потенциометрия // Соросовский образовательный журнал. 2000. Т.
6(10). С . 50–56.
13. Харнед Г., Оуэн. Б. Физическая химия растворов электро-
литов. –М. : ИЛ, 1952. – 628 с.
14. Робинсон Р., Стокс Р. Растворы электролитов.
–
М.: ИЛ.
1963. – 646 с.
15. Бейтс Р. Определение рН. Теория и практика. – Л. : Химия,
1971. – 400 с.
16. Рабинович В. А. Термодинамическая активность ионов в
растворах электролитов. – Л. : Химия, 1985. – 176 с.
17. Шаталов А. Я . Введение в электрохимическую термодина-
мику. –М. : Высшая школа. 1984. –215 с.
18. Шульц М. М. Стеклянный электрод. Теория и применение.
–Соросовский образовательный журнал. – 1998. No 1. с . 33 –39.
19. Кузнецов В. В . Определение рН // Соросовский образова-
тельный журнал. 2001. Т. 7(4). С . 44 –51.
20. Шведене Н. В . Ионоселективные электроды // Соросовский
образовательный журнал. 1999. No 5. С . 60 –65.
21. Скорчеллетти В. В . Теоретическая электрохимия. – Л. : Хи-
мия, 1974. – 568 с.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
168
Робота No 9.
ВИЗНАЧЕННЯ КОЕФІЦІЄНТІВ АКТИВНОСТІ СИЛЬНИХ
ЕЛЕКТРОЛІТІВ ПОТЕНЦІОМЕТРИЧНИМ МЕТОДОМ
Ціль роботи: визначити середні іонні коефіцієнти активнос-
ті сильного електроліту методом виміру ЕРС у гальванічному еле-
менті без переносу й порівняти результати з довідковими значен-
нями.
Прилади й реактиви: термостат, рН-мілівольтметр (скля-
ний електрод), електролізер (водневий електрод, потенціометрич-
на установка, комірка для вимірів з електродами, 6 колб 100 мл для
зберігання розчинів, піпетка на 50 мл, мірна колба на 250 мл, вихід-
ний розчин хлористого водню (0,1 моль/л), хлористий натрій крис-
талічний, аналітичні ваги з вагами, пробірка для взяття наважки
солі.
Сильні електроліти у водяних розчинах уважаються дисоційо-
ваними повністю та концентрація недисоційованих молекул не під-
дається визначенню. Активність електроліту може бути визначена,
якщо певним чином вибрати стандартний стан. Нехай електроліт
v
v
MA
дисоціює на іони за схемою
MA
=
z
M
+
z
A
,
де й – число катіонів і аніонів, що утворюються при дисоціа-
ції молекул електроліту, а z
+
іz
–
–
їх заряди.
У силу електронейтральності розчину маємо рівність
z
z
.
Застосування закону діючих мас до процесу дисоціації дає рів-
няння
2
a
a
ka
,
у якому
2
,
и
aa
a
–
активності катіонів, аніонів і недисоційова-
них молекул електроліту відповідно.
Для іонів як стандартного стану вибирається стан нескінчен-
ний розведеного розчину, у якому відсутнє іон-іонна взаємодія,
Робота No 9. ВИЗНАЧЕННЯ КОЕФІЦІЄНТІВ АКТИВНОСТІ СИЛЬНИХ
ЕЛЕКТРОЛІТІВ ПОТЕНЦІОМЕТРИЧНИМ МЕТОДОМ
169
тобто виконується умова
1
i
i
a
lim
m
приI0,
де m – концентрація іона, а I – іонна сила розчину.
Стандартний стан для недисоційованих молекул сильного еле-
ктроліту вибирають таким чином, щоб константа дисоціації в рів-
нянні закону діючих мас (ЗДМ) була дорівнюю одиниці. При тако-
му виборі стандартного стану рівняння ЗДМ прийме вигляд
2
aa
a
.
Введемо поняття середньої іонної активності електроліту a ,
як середнього геометричного активності окремих іонів
1
1
a
a
a
a
a
,
де
–
загальне число іонів, що виходить при дисоціації
однієї молекули солі. З цього одержуємо рівняння, що зв'язує акти-
вність електроліту, іонні активності й середній іонний коефіцієнт
активності електроліту
1
2
v
v
v
v
a
a
a
a
.
Аналогічно вводяться поняття середньої іонної концентрації
електроліту m й середнього іонного коефіцієнта активності елект-
роліту :
1
2
v
v
v
v
m
m
m
m
1
2
v
v
v
v
.
Таким чином, для одне-одновалентного електроліту (напри-
клад,
,
,
HCl NaOH NaCl і т. п.), для якого
1,
,
2
маємо
2
2
2
(
)
a
a
aa
m
m
mm
,
a
m
m
.
Активність електроліту 2
a або середня іонна активність a
може бути визначена різними методами, що відбивають термодинамі-
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
170
чні властивості розчинів: за зниженням температури замерзання роз-
чину, за тиском пари, методом виміру ЕРС гальванічних елементів.
Найбільше часто для визначення коефіцієнтів активності си-
льних електролітів використовують метод електрорушійних сил з
використанням хімічних ланцюгів без рідинної сполуки (без пере-
носу), наприклад, для визначення коефіцієнтів активності хлористо-
го водню в розчинах використовують ланцюг
5
2
( , газ;1,0110 Па)
()
,
()
PtH
HClm AgClAgPt
.
У гальванічному елементі протікає оборотна реакція
2
1
(газ)
(тв.)
(тв.)
(р)
2
H
AgCl
Аg
HCl н
Зміна енергії Гіббса реакції rG
виражається рівнянням ізоте-
рми реакції
2
1/2
ln
ln
AgH
Cl
r
a
AgCl H
a
a
a
GRTKRT
a
a
.
З іншого боку, зміна енергії Гіббса реакції rG
дорівнює еле-
ктричній роботі rG
= – zFE, де E – ЕРС елемента. Тоді для ЕРС
елемента, електродна реакція,яка характеризується значенням z = 1,
маємо рівняння
о
RT
EE
F
2
1/2
ln
AgH
Cl
AgCl H
a
a
a
a
a
,
у якому
o
E – стандартна ЕРС елемента, рівна
o
o
rG
E
F
.
Враховуючи, що активності чистих фаз дорівнюють одиниці
(
1
AgCl
Ag
a
a
) і привівши активність газоподібного водню на па-
рціальний тиск, рівне 1 атм, остаточно маємо
o
lgH
Cl
EE
a
a
,
де
2,303
RT
F
.
Уводячи середні активності електроліту a , середні іонні кон-
церації m m
й середні іонні коефіцієнти активності , рівняння
Робота No 9. ВИЗНАЧЕННЯ КОЕФІЦІЄНТІВ АКТИВНОСТІ СИЛЬНИХ
ЕЛЕКТРОЛІТІВ ПОТЕНЦІОМЕТРИЧНИМ МЕТОДОМ
171
набуває вигляду для кожного розчину (індекс i )
o
22
o
,
,
lg
2lg
i
ii
ii
EE
m
E
m
.
Таким чином, середні іонні коефіцієнти активності електроліту
визначаються з обмірюваних експериментально значень ЕРС елеме-
нта з концентрацією електроліту m
,
o
lg
,
,
lg
lg,
10
2
i
i
i
i
i
EE
m
.
Для обчислення значень необхідно мати у своєму розпоря-
дженні дані про стандартну ЕРС елемента
o
E , яка зв'язана зі стан-
дартною енергією Гіббса реакції й виражає стан, коли активності
всіх учасників реакції дорівнюють одиниці. Для цього використову-
ємо прийом екстраполяції на стандартний стан, що носить назву
методу Харнеда. Визначення властивостей у стандартному стані
йде в два етапи.
По-перше, приведемо значення обмірюваних при різних кон-
центраціях ЕРС E елементу до концентрації електроліту m = 1, для
чого визначимо функцію E’, відповідну до цього стану для кожного
розчину концентрації im
о
о
,
2lg
2lg
i
i
i
i
E'E
m
E
.
Якби розчини були ідеальними, тобто коефіцієнти активності
іонів = 1 для кожного з розчинів, а значення
о
i
E' тодібнезале-
жали від концентрації, тобто збігалися б зі значенням
о
E.Дляреа-
льних розчинів
о
i
E ' збігається зі значенням
o
E тільки в стандартно-
му стані – у стані нескінченно розведеного розчину з іонною силою
рівної нулю, тобто при m 0, коли розчин здобуває властивості
ідеальної в силу відсутності іон-іонної взаємодії. Таким чином, по-
друге, значення
o
E може бути отримане екстраполяцією функції
о
i
E ' на стан нескінченно розведеного розчину.
При вивченні розведених розчинів характер залежності
o
()
E'fm
може бути встановлений за допомогою теорії Дебая –
Хюккеля.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
172
Так, згідно з теорією для дуже розведених розчинів, залеж-
ність lg від I має вигляд
||
lg
1o
zz AI
aBI
,
а в межі прагне до залежності лінійної від іонної сили розчину
lg
||,
zz AI
тобто залежність
o
()
E'fm
повинні прагнути до лінійної в коор-
динатах
o
E'
m
при m 0, тому що в розчинах сильного 1:1
валентного електроліту I = m.
Значення теоретичних коефіцієнтів
(,)
AfT
і
(,)
B fT
–
функції температури T та діелектричної проникності розчинника
можуть бути розраховані по рівняннях відповідно до електростати-
чної теорії Дебая –Хюккеля
3
1/2 3/2
2
36283, 2
, ( л/моль)
2, 303
1000
A
N
e
B
A
К
kT
kT
T
,
2
о-1
1/2 1/2
8
2529, 2
,(
) (л/моль)
A
еN
B
А
К
kT
T
,
Враховуючи вираження для коефіцієнтів активності, функція
o
E ' прийме вигляд лінійного рівняння, використовуваного для екс-
траполяції.
o
o
2lg
2
E'E
m
E
Am
.
Граничне рівняння Дебая – Хюккеля застосовне тільки до кон-
центрацій не вище, ніж 0,05 моль/кг. Для розчинів більших концен-
трацій (до 0,1 моль/кг) для опису концентраційної залежності кое-
фіцієнтів активності можна використовувати напівемпіричне рів-
няння виду
lg
||
zzAIbI
,
де b – емпіричний параметр.
Робота No 9. ВИЗНАЧЕННЯ КОЕФІЦІЄНТІВ АКТИВНОСТІ СИЛЬНИХ
ЕЛЕКТРОЛІТІВ ПОТЕНЦІОМЕТРИЧНИМ МЕТОДОМ
173
Використання цього рівняння дає можливість побудувати нову
екстраполяційну функцію
o
E ' ' , що враховує внесок електростатич-
них взаємодій згідно із граничним рівнянням Дебая –Хюккеля пе-
реданому першим доданком рівняння для коефіцієнта активності.
Функція
o
E ' ' лінійна від іонної сили (концентрації) електроліту на
значно більшому інтервалі іонних сил
o
o
o
2
2lg
2
2
i
i
i
i
i
i
i
E''E'
AmE
m
AmE
bm
Обидві залежності
o
E'й
o
E '' , що побудовані на одному гра-
фіку кожний залежно від своїх аргументів, відтинають на осі орди-
нат значення рівне стандартної ЕРС елемента
o
E , що представлено
на рисунку 2.26 для гальванічного елемента з водневим і хлорсріб-
ним електродом у розчинах хлористого водню при 298 К..
Певне методом екстраполяції значення стандартної ЕРС еле-
мента дозволяє розрахувати середні коефіцієнти активності елект-
роліту при будь-якій концентрації m за обмірюваними значенням
ЕРС E елемента в цьому розчині електроліту
Рис. 2.26. Приклад екстраполяції функції o
'
Eйo
''
E на стандартний стан
розчину під час визначення коефіцієнтів активності хлористого водню в га-
льванічному елементі.
5
2
( , газ;1,01 10 Па)
()
,
()
PtH
HClm AgClAgPt
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
174
,
lg
lg
2
o
i
i
i
EE
m
.
Коефіцієнт активності електролітів є складною функцією від
концентрації (іонної сили), що пояснюється впливом на коефіцієнт
активності діелектричної проникності, величини іонного радіуса,
заряду іона, специфіки сольватації іона молекулами розчинника.
При визначенні коефіцієнтів активності електролітів з вико-
ристанням іоноселективних електродів (оборотних до іонів H
,
K
,Na
і т. п.) зверніть увагу на знаки електродів у гальванічному
елементі й правильно складіть запис для ЕРС ланцюга. Якщо отри-
мане вираження відрізняється від наведеної вище для водень – хло-
рсрібного елемента, виведіть вираження для екстраполяційних фун-
кцій o
E'і
o
E''.
Для визначення знака електродів у ланцюзі скористайтеся
аналізом ходу зміна ЕРС елемента від концентрації
електроліту.
ВИКОНАННЯ РОБОТИ
Середні іонні коефіцієнти активності електроліту визначають,
вимірюючи ЕРС гальванічного елемента без рідинного з’єднання.
Гальванічний елемент складається із двох електродів, один з яких
оборотний до катіона, інший до аніона досліджуваного електроліту.
Так, при дослідженні розчинів хлористого водню можна використо-
вувати водневий електрод або скляний електрод з водневою функ-
цією в парі із хлорсрібним електродом
5
2
( , газ;1,0110 Па)
()
,
()
PtH
HClm AgClAgPt
.
Рівняння Нернста для такого ланцюга, що містить шукані кое-
фіцієнтів активність хлористого водню має вигляд
,
o
o
lg
2lg
HCl
AgCl/Ag
AgCl/Ag
HCl
H
Cl
EE
a
a
E
m
.
При використанні водневого електроду стандартна ЕРС еле-
менту дорівнює стандартному потенціалу хлорсрібного електроду.
Для виміру коефіцієнтів активності сірчаної кислоти – гальванічний
елемент без переносу, складений з водневого електрод або скляного
Робота No 9. ВИЗНАЧЕННЯ КОЕФІЦІЄНТІВ АКТИВНОСТІ СИЛЬНИХ
ЕЛЕКТРОЛІТІВ ПОТЕНЦІОМЕТРИЧНИМ МЕТОДОМ
175
електрода з водневою функцією в парі із сульфатртутним електро-
дом має вигляд
5
2
24
24
( , газ;1,01 10 Па)
()
,
()
PtH
HSOm HgSOHgPt
.
В цьому випадку середні іонні коефіцієнти активності сірчаної
кислоти пов'язані з ЕРС елемента співвідношенням
2
24
4
o
2
lg
Hg SO /Hg
H
SO
EE
a
a
24
24
24
o
3/2
,
3
lg
4
2
Hg SO /Hg
HSO
HSO
E
m
.
Для визначення коефіцієнтів активності солей можна застосо-
вувати у якості електродів, оборотних до іонів металу, амальгамні
електроди. Наприклад для солей лужних металів
,
(())
()
,
()
Na
PtHgNaa
NaClm AgClAgPt.
ЕРС такого елемента має вигляд
o
o
lg
AgCl/Ag
Na /Ag
Na
Cl
E
a
a
o
o
,
()
2lg
2lg
AgCl/Ag
NaCl
NaCl
Na Hg
Na /Ag
m
a
,
уякому ( )
Na Hg
a
–
активність металевого натрію в амальгамі.
Значно зручніше при вивченні солей користуватися іоноселек-
тивними електродами з відповідними катіонними функціями, на-
приклад, використовувати скляний електрод (СЕ) з натрієвою фун-
кцією при визначенні коефіцієнтів активності бромистого натрію, у
парі із бромсрібним електродом
(),,
()()
PtAgAgBrNaBrm СЕNa
З електродами слід звертатися акуратно, не стосуватися
ними стінок посудини, не згинати й не торкати робочі частини
руками.
Робота складається із двох етапів: 1) визначення стандартної
ЕРС елемента методом екстраполяції шляхом вивченням концент-
раційної залежності ЕРС у розведених розчинах і 2) визначення ко-
ефіцієнта активності електроліту в розчині середньох концентрації
за даними виміру ЕРС елемента в цьому розчині.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
176
Для визначення значень стандартної ЕРС готовлять серію з 5–
7 розведених розчинів електроліту з концентрацією, що не переви-
щує 0,1 моль/л для забезпечення можливості використання для екс-
траполяції рівнянь Дебая –Хюккеля. Спочатку готовлять у мірній
колбі основний розчин, при дослідженні солей розчин готовлять із
наважки солі, кислот – шляхом розведення більш концентрованого
розчину. Концентрацію основного розчину вказує викладач. Наваж-
ку обов’язково беруть на аналітичних вагах. Інші робочі розчини
готовлять методом послідовного розведення основного розчину
вдвічі, тому що при цьому відносна погрішність концентрації при
готуванні розчинів стає практично однакової й не потрібна калібру-
вання мірного посуду (див. стор. 39). Для цього в чисті висушені
колби на 200 мл піпеткою наливають по 100 мл дистильованої води,
а потім після споліскування вихідним розчином електроліту тією ж
піпеткою вносять 100 мл самого концентрованого розчину в першу
колбу. Розчин ретельно перемішують на магнітній мішалці протя-
гом 2–3 хв, після чого споліскують піпетку цим розчином, набира-
ючи його в піпетку й виливаючи назад у колбу, і далі, відібравши
100 мл, переносять розчин у наступну колбу. Операцію повторюють
необхідну кількість разів, розводячи розчин щораз точно у два рази.
Наприкінці в останній колбі з розчином найменшої концентрації
виявляється найбільша кількість розчину – 200 мл.
Перед заповненням комірки електроди й саму комірку не
менш двох разів споліскуються досліджуваним розчином електролі-
ту. У зв'язку з тим, що електроди й комірка для виміру ЕРС очи-
щаються від попередніх розчинів тільки споліскуванням, виміри
починають із самого розведеного розчину, тому що при цьому при
недостатньому промиванні виникають найменші погрішності.
РОБОТА З ВОДНЕВИМ ЕЛЕКТРОДОМ
Для роботи використовують комірку, використану для дослі-
дження іонного добутку води в роботі No 7. (рис. 2 –21).
Вимір ЕРС проводиться на установці компенсаційного типу на
високоомному потенціометрі з використанням у якості нуль-
інструмента дзеркального гальванометра (чутливість порядку 10–9
–
10–10
А/діл.). Для роботи використовують чистий водень, одержува-
ний електролізом 20% розчину лугу з нікелевими електродами. Га-
Робота No 9. ВИЗНАЧЕННЯ КОЕФІЦІЄНТІВ АКТИВНОСТІ СИЛЬНИХ
ЕЛЕКТРОЛІТІВ ПОТЕНЦІОМЕТРИЧНИМ МЕТОДОМ
177
зоподібний водень очищається від аерозо-
лю розчину лугу пропущенням через скля-
ну вату й сатуратор з дистильованою во-
дою. Використовується комірка для робо-
ти з водневим електродом, аналогічна та-
кої, що використана в роботі No 7 при ви-
значенні іонного добутку води (стор. 135,
рис. 2.21). Частина розчину в комірці обо-
в'язково переноситься у рідинний затвор
для запобігання влучення повітря при
промиванні воднем. Кількість розчину в
комірці повинне бути достатньою для пок-
риття не менш половини поверхні плати-
нованої платини. Швидкість подачі водню
1–2 пухирця в секунду. В інше коліно U-
подібної комірки поміщають хлорсрібний
електрод термоелектролітичного типу. Рі-
вноважне значення ЕРС елемента встанов-
люється протягом 20–35 хв після початку
пропущення водню. Для прискорення про-
цесу промивання комірки воднем у почат-
ковий момент часу швидкість пропущення водню можна збільшити.
Для встановлення стану рівноваги на електродах протягом
усього досліду ведеться компенсація ЕРС і через кожні 5 хв запи-
суються показання потенціометра. Якщо протягом 10–15 хв зна-
чення ЕРС залишається постійним з точністю 0,1 мВ, то момент рі-
вноваги вважається досягнутим. Для розчину тієї ж концентрації
робиться повторне заповнення комірки й процес виміру повторю-
ється. Якщо результати виміру двох заповнень комірки різняться не
більше ніж на 0,2–0,3 мВ, переходять до вивчення більш концент-
рованого розчину, а якщо ні, то роблять третє заповнення. Потенці-
ал водневого електрода в водяних розчинах – негативний.
РОБОТА З ІОНОСЕЛЕКТИВНИМИ (СКЛЯНИМИ)
ЕЛЕКТРОДАМИ
При роботі зі скляними електродами вимір ЕРС роблять на
установці з високоомним потенціометром з використанням у якості
Рис. 2 .27 . Комірка для
виміру ЕРС в ланцюгах
без переносу із скляним
електродом.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
178
нуль-інструмента рН-мілівольтметра (іономіра) з великим вхідним
опором у зв'язку з великим опором електрода сягаючим десятків
МОм. Для вимірів використовують комірку, зображену на рис. 2.27 .
При роботі з іоноселективними електродами необхідно вста-
новити значення електродної функції, для чого проводиться їхнє
попереднє калібрування. Для цього роблять виміри мінімум у двох
розчинах з відомими активностями солі, до катіона якої оборотний
електрод. Далі порядок виміру ЕРС такий же, як при роботі з водне-
вим електродом:: виміри починають із найбільш розведеного роз-
чину, сполоснувши електроди й комірку розчином. Виміри кожного
розчину повторюють до відтворюваності з точністю 0,2–0,3 мВ, до-
магаючись у кожному вимірі рівноважного значення ЕРС, яке по-
винне бути постійним з точністю 0,1 мВ протягом 15–20 хвилин.
При вимірі слід звернути увагу на знаки електродів у комірці, щоб
згодом правильно записати рівняння Нернста для ЕРС гальванічно-
го елемента й правильно розрахувати значення функцій o
E'і
o
E''.
Знаки електродів можна визначити аналізуючи напрям зміни ЕРС
елемента із зміною концентрації електроліту.
Результати досліджень рекомендується зводити в таблицю
Електроліт............... Температура.............
Концентрація
розчину,
m
Час,
астроно-
мічний
ЕРС
заповнень
комірки, E
.
cp
E
m
o
E'
o
E''
123
У завершення готовлять 50–100 мл розчину електроліту висо-
кої концентрації x
m (вказується викладачем) і вимірюють ЕРС еле-
мента за методикою, викладеної вище.
Для середніх арифметичних значень ЕРС елемента двох
останніх заповнень комірки
.
cp
E , що отримані в області розведених
розчинів, розраховують значення функцій o
E'і
o
E '' й будують
графіки (масштаб 1 мм = 0,1 мВ) у координатах
o
E'
m
і
o
E'' m
, по яких екстраполяцією на
0
Im
визначають значення
стандартної ЕРС елемента
o
E . Обидві залежності наносять на той
Робота No 9. ВИЗНАЧЕННЯ КОЕФІЦІЄНТІВ АКТИВНОСТІ СИЛЬНИХ
ЕЛЕКТРОЛІТІВ ПОТЕНЦІОМЕТРИЧНИМ МЕТОДОМ
179
самий графік, що має загальну вісь для значень
o
E'і
o
E'',ізагаль-
ний початок координат на осі абсцис для зображення m й m , але
в різному масштабі. Залежність
o
E'' m
носить виражений ліній-
ний характер і може бути оброблена для визначення
o
E за методом
найменших квадратів, або побудовою линії тренду при застосуванні
програми Excel для обробки даних. При роботі з водневим електро-
дом значення
o
E елемента збігається зі стандартним потенціалом
хлорсрібного електрода. Залежнімть
o
E'
m
не носить виражено-
го лінійного характеру, цей графік стає лінійним тільки в області
високого розведення, тому використати емпірічні рівняння для ап-
роксимації цієї залежності недоцільно. Визначивши із графіка гра-
ничний кут нахилу в залежності
o
E'
m
(тангенс кута , рис.
2.26), можна з даних експерименту перевірити справедливість теорії
Дебая –Хюккеля – розраховане значення коефіцієнта
/2
Atg
.
При правильній будові обох залежностей вони повинні при значенні
0
Im
взаємно перехрещуватися визначаючи значення стандарт-
ної ЕРС елементу.
За наявними значенням ЕРС елемента з розчином високої кон-
центрації розраховують середні іонні коефіцієнти активності елект-
роліту, використовуючи знайдене екстраполяцією значення станда-
ртної ЕРС ланцюга
,
lg
lg
2
o
x
x
x
EE
m
,
яке порівнюють із літературними даними. Для порівняння з літера-
турними даними бажано побудувати якісний графік залежності ко-
ефіцієнтів активності даного електроліту від концентрації електро-
літу з використанням програмного забезпечення комп’ютера, виби-
раючи для інтерполяційної залежності відповідні координати:
lg
m
.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Що таке коефіцієнт активності? При яких умовах замість ак-
тивності можна використовувати концентрації?
2. Сформулюйте основні положення теорії сильних електролітів.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
180
3. Напишіть математичне вираження граничного рівняння Де-
бая – Хюккеля. Дайте визначення поняття іонної сили розчину.
4. Сформулюйте правило іонної сили.
5. Обґрунтуйте неможливість експериментального визначення
коефіцієнтів активності окремого виду іонів.
6. Яка зв'язок між активністю електроліту й середнім іонним
коефіцієнтом активності?
7. Що виражає значення стандартної ЕРС гальванічного еле-
мента?
8. У чому сутність методу екстраполяції при визначенні зна-
чень стандартної ЕРС гальванічного елемента?
9. Чому середні іонні коефіцієнти активності електролітів ви-
значають за допомогою ланцюгів без переносу?
10. Якими електродами слід скористатися для експерименталь-
ного визначення середніх іонних коефіцієнтів активності бромисто-
го натрію?
11. Чому коефіцієнти активності сильних електролітів можуть
бути більше одиниці?
12. Якими методами, крім електрометричного, можна визначити
коефіцієнти активності сильного електроліту?
13. Які допущення використовуються при висновку рівняння
Дебая –Хюккеля? Які допущення використовуються при висновку
рівняння для потенціалу іонної атмосфери в першому наближенні?
14. Яким образом радіус іонної атмосфери залежить від концен-
трації іонів, їх природи, природи розчинника й температури?
15. Запишіть гальванічний елемент, вивченням ЕРС якого мож-
на виміряти середні іонні коефіцієнти активності сірчаної кислоти.
16. Чому при роботі з іоноселективними електродами необхід-
не їхнє попереднє калібрування?
17. Запропонуєте варіанти перевірки електродної функції скля-
ного електроду з натрієвою функцією.
ТЕОРЕТИЧНІ ПИТАННЯ ПО ТЕМІ
Електрохімічні процеси. Теорії виникнення електродного по-
тенціалу й електрорушійної сили. Потенціали Гальвані й Вольта.
Сольватаційна теорія електродного потенціалу. Термодинаміка еле-
ктрохімічних елементів. Розрахунки ЕРС хімічних елементів за
Робота No 9. ВИЗНАЧЕННЯ КОЕФІЦІЄНТІВ АКТИВНОСТІ СИЛЬНИХ
ЕЛЕКТРОЛІТІВ ПОТЕНЦІОМЕТРИЧНИМ МЕТОДОМ
181
термодинамічним даними. Класифікація гальванічних елементів:
оборотні й необоротні, хімічні й концентраційні, з переносом і без
переносу. Застосування концентраційних елементів з переносом і
без переносу. Методи виміру ЕРС, компенсаційний метод, схеми з
реохордом, магазинами опорів та потенціометра. Дифузійний поте-
нціал, його природа й методи елімінування. Рівняння Нернста для
потенціалу електрода й ЕРС гальванічного елемента. Стандартний
потенціал і стандартна ЕРС елемента. Типи електродів. Викорис-
тання електрохімічний ланцюгів для вивчення розчинів електролі-
тів: визначення констант дисоціації слабких електролітів, іонного
добутку розчинника, добутку розчинності; визначення рН, коефіці-
єнтів активності електролітів, потенціометричне титрування, визна-
чення рН стандартних буферних розчинів, термодинамічних харак-
теристик реакцій і речовин.
Теорія електролітичної дисоціації. Активність і коефіцієнти
активності електролітів. Стандартизація активності. Іонна сила роз-
чинів електролітів, правило іонної сили Льюіса. Електростатистич-
на теорія електролітів Дебая – Хюккеля, основні положення й виве-
дення основного рівняння. Наближення теорії та полуемпірічні рів-
няння. Методи визначення активності та коефіцієнтів активності
електролітів. Осмотичний коефіцієнт. Сучасні теорії розчинів елек-
тролітів.
ЛІТЕРАТУРА
1. Герасимов Я. И. и др. Курс физической химии. Т.2.
–М. :
Химия, 1973. – 625 с.
2. Лебідь, В. І. Фізична хімія : підруч. для студ. вищ. навч. за-
кл. / В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
3. Измайлов Н. А. Электрохимия растворов. Изд. 3 –е, испр. –
М. : «Химия», 1976. – 488 с.
4. Дамаскин Б. Б. и др. Электрохимия / Б. Б. Дамаскин, О. А.
Петрий, Г. А. Цирлина. – М. : Химия, КолосС, 2006. – 672 с.
5. Рабинович В. А. Термодинамическая активность ионов в ра-
створах электролитов / В. А. Рабинович. – Л. : Химия, 1985. – 176 с.
6. Практикум по физической химии. Учебное пособие для ву-
зов. Изд. 3–е. / Под ред. С. В. Горбачева, – М. : Высшая школа,
1974. – 495 с.
7. Практикум по электрохимии / Под ред. Б. Б. Дамаскина.
–
М. : Высшая школа, 1991. – 288 с.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
182
8. Практические работы по физической химии : Учебное по-
собие для вузов / Под ред. К. П. Мищенко, А. А. Равделя, A. M. По-
номаревой. – СПб. : Изд–во «Профессия», 2002. – 384 с.
9. Харнед Г. Физическая химия растворов электролитов / Г.
Харнед, Б. Оуэн.
–
М.:ИЛ,1952. – 628с.
10. Робинсон Р., Стокс Р. Растворы электролитов / Р. Робин-
сон, Р. Стокс. – М. : Издатинлит, 1963. – 646 с.
11. Ротинян А. Л., Тихонов К.И., Шошина И.А. Теоретическая
электрохимия / Под. ред. А. Л. Ротиняна. – М. : Студент, 2013. – 496 с.
12. Физическая химия. Теоретическое и практическое руко-
водство. Изд. 2 –е, переработанное и дополненное. / Под ред. ака-
демика Б. П. Никольского. – Л. : Химия, 1987. – 880 с.
Робота No 10.
ВИЗНАЧЕННЯ КОНСТАНТИ ДИСОЦІАЦІЇ СЛАБКОЇ
КИСЛОТИ В БУФЕРНИХ РОЗЧИНАХ У ЛАНЦЮГАХ
З ПЕРЕНОСОМ
Ціль роботи: визначення константи дисоціації слабкої кис-
лоти по вимірах ЕРС ланцюга з переносом.
Прилади й реактиви: установка з потенціометром постій-
ного струму або рН–мілівольтметр, насичений хлорсрібний елект-
род, платиновий електрод (для хінгідронного електроду), хінгідрон,
розчин досліджуваної слабкої кислоти (≈0,1 моль/л), розчин стан-
дартної слабкої кислоти (≈0,1 моль/л), комірка (стаканчик) для ви-
мірів, бюретка з розчином лугу (0,1 моль/л), мірні колби на 50 мл,
розчин фонового електроліту (хлорид натрію 0,5 моль/л).
В основі застосування ланцюгів з переносом для визначення
константи дисоціації слабкої кислоти лежить використання концент-
раційних гальванічних елементів типу
(0)
(0)
()
()
2
2
( ,г.)|
,
,
()|| , ,()|(
, г.)
х
х
PtH
HAMAMXmHAMAMXmPtH
, (I)
у якому потенціал правого електрода, обумовлений рівноважною
активністю іонів водню в розчині з досліджуваною слабкою кисло-
тою
()x
HA порівнюється з потенціалом лівого електрода, обумовле-
Робота No 10. ВИЗНАЧЕННЯ КОНСТАНТИ ДИСОЦІАЦІЇ СЛАБКОЇ
КИСЛОТИ В БУФЕРНИХ РОЗЧИНАХ У ЛАНЦЮГАХ З ПЕРЕНОСОМ
183
ного рівноважною активністю іонів водню в розчині з деякою кис-
лотою
(о)
HA , з відомим значенням константи дисоціації й обраної
за стандарт.
(0)
MAі()x
MA – солі цих кислот – сильні електроліти. Ки-
слота
(о)
HA , що обрана за стандарт, підбирається як можна більш
близької за силою до досліджуваної кислоти
()x
HA . ЕРС концентра-
ційного елемента (І) виражається рівнянням ( при (0)
H
a<
()х
H
a)
()
()
(o)
lg
x
H
I
j
H
a
E
a
,
деj
– залишковий дифузійний потенціал.
Для елімінування залишкового дифузійного потенціалу j
ви-
користовують сольовий міст, а також сольову добавку з розчину деякого
сильного індиферентного (фонового) електроліту MX з такою концент-
рацією ( )
m у розчині в кожному напівелементі, щоб забезпечити рівні
значення іонних сил обох розчинів. Іонна сила розчинів визначається як
концентрацією електроліту МХ , так і присутніми в розчинах добавками
солей (0)
MAі()x
MA з однойменними стосовно кислот аніонами. Надалі
будемо вважати, що залишковий дифузійний потенціал елімінований,
тобто
j
=0.
З виражень закону діючих мас для дисоціації кислот маємо
(o)
(o) (o)
(o)
(o)
(o)
,
;
H
A
HA
HA
a
a
HA
H
A
K
a
()
()
()
()
(),
.
x
x
х
x
x
H
A
HA
HA
a
a
HA
H
A
K
a
З цих рівнянь закону дії мас можна виразити активності іонів
водню
(o)
(o)
(o) ;
HA HA
H
A
Ka
a
a
()
()
()
x
x
HA HA
х
H
A
Ka
a
a
,
й підставити в рівняння для ЕРС елемента
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
184
() () (0)
()
(o) (o) ()
lg
x
x
HAHAA
I
х
HAHAA
Kaa
E
Kaa
.
Активності незаряджених форм слабких кислот практично дорі-
внюють одиниці. При створеній в експерименті однаковій іонної силі
розведених розчинів коефіцієнти активності аніонів також рівні між
собою й можуть бути скорочені в рівнянні для ЕРС.
У присутності солей з однойменними аніонами рівноваги дисо-
ціації обох слабких кислот сильно зміщені убік утвору молекулярних
форм. Внаслідок цього рівноважні концентрації молекул кислот
практично рівні аналітичної (вихідної) концентрації кислот, а рівно-
важні концентрації аніонів – аналітичної концентрації добавок солей
з однойменними аніонами, тобто
()х
HA
a=
()x
HA
m;
(o)
HA
a=
(o)
HA
m
і ()х
A
a
=
()х
МA
m;
(o)
A
a
=
(o)
МA
m . Враховуючи вищевикладене, рівняння для ЕРС ланцюга
(I) може бути переписане у вигляді, що включає відомі й задані в ек-
сперименті концентрації компонентів розчину
() () (o)
()
(o) (o) ()
lg
x
x
HA
HA
A
I
х
HAHAA
Kmm
E
Kmm
,
або
()
()
(o)
(o)
(o)
()
()
lg
lg
x
x
HA
HA
A
I
х
HA
HA
A
m
m
K
E
K
m
m
,
звідки
()
()
(o)
()
(o)
(o) ()
lg
lg
x
x
HA
I
HA
A
х
HA
HAA
m
m
E
K
K
m
m
,
або
()
()
(o)
()
(o)
(o) ()
lg
x
x
HA
I
A
HA
HA
х
HAA
m
m
E
pK
pK
m
m
.
Робота No 10. ВИЗНАЧЕННЯ КОНСТАНТИ ДИСОЦІАЦІЇ СЛАБКОЇ
КИСЛОТИ В БУФЕРНИХ РОЗЧИНАХ У ЛАНЦЮГАХ З ПЕРЕНОСОМ
185
Якщо вихідні концентрації досліджуваної й стандартної кислот
рівні між собою, а також рівні між собою концентрації їх солей, тобто
()x
HA
m
=
(o)
HA
m
і
()x
МA
m=
(o)
МA
m , то останній доданок перетворюється в
нуль і рівняння для розрахунків показника константи дисоціації дос-
ліджуваної кислоти спрощується, тобто
()
()
(o)
I
x
HA
HA
E
pK
pK
.
Внаслідок відсутності перевірки на повноту елімінування за-
лишкового дифузійного потенціалу викладений метод не може вва-
жатися прецизійним.
Серед методів визначення констант дисоціації слабких кислот і
основ найбільш точним уважається потенціометричний метод з ви-
користанням буферних розчинів з елементах без рідинного з’єднання
–
в елементах без переносу.
Для проведення вимірів можуть бути використані будь-які елект-
роди оборотні до іонів водню. При використанні безпосередньо конце-
нтраційного елемента типу (I) скляні електроди не можуть бути вико-
ристані безпосередньо, тому що мають різні значення стандартних по-
тенціалів, більше того, вимір ЕРС такого елемента технічно не реалізо-
ваний на сучасних приладах. Крім того, скляні електроди не мають те-
оретичну оборотність і потребують експериментального визначення
електродної функції.
Найбільш простою реалізацією методу визначення константи ди-
соціації слабкої кислоти є використання окисно-відновних хінгідрон-
них електродів, теоретично оборотних до іонів водню в кислих розчи-
нах. Можна виміряти безпосередньо ЕРС концентраційного гальваніч-
ного елементу з переносом типу
64264
2
64264
2
()
()
(o)
(o)
1
2
3
11
2
3
,
()
,
()
,
,
,
,
()()()()()()
x
x
CHOCHOH
CHOCHOH
PtHAMAMX
HAMAMX
Pt
m
m
m
m
m
m
,
(II)
де
()x
HA – досліджувана слабка кислота,
(o)
HA – стандартна кислота,
для якої величина константи дисоціації відома,
(o)
MA,()
x
MA – солі цих
кислот. Концентрації кислот і солей повинні бути невеликі й відповіда-
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
186
ти наступним рекомендованим значенням: концентрація кислот: 1
m=
1
m 0,005–0,01 моль/л; солей 2
m=2
m 0,005–0,01 моль/л. Іонна сила
розчинів вирівнюється добавками індиферентного електроліту MX з
концентрацією більшою інших компонентів розчину: 3
m=3
m ≈ 0,05
моль/л для забезпечення рівності значень іонних сил розчинів і вирів-
нювання коефіцієнтів активності аніонів, близької до I 0,05–0,1.
На практиці зручніше визначати ЕРС даного концентраційного
гальванічного елемента шляхом виміру й наступної комбінації ЕРС
гальванічних елементів, складених з індикаторного хінгідронного й
насиченого хлорсрібного електроду як електроду порівняння. У
цьому випадку можливе застосування й скляного електроду тому,
що він буде однаковим при вимірі як стандартного, так й досліджу-
ваного розчинів. У випадку використання хінгідронного електрода
гальванічні елементи записуються у вигляді
64264
2
()
1
()
2
3
,
(),
(),
( .),
(),()
x
x
CHO CHOH
Pt
HAm
KCl нас AgCl Ag
MAmMXm
(III)
і зі стандартним розчином кислоти
64264
2
(o)
1
(o)
2
3
,
(),
(),
( .),
('),(
')
CHO CHOH
Pt
HAm
KCl нас AgCl Ag
MAmMXm
(IV)
Очевидно, що якщо при вимірах і ланцюгах (IІI) і (IV) викорис-
товується той самий насичений хлорсрібний електрод і той самий
електролітичний міст, величина потенціалу електрода порівняння й
дифузійного потенціалу в них буде та сама, а значення ЕРС ланцюга
(II) буде дорівнювати різниці ЕРС ланцюгів (III) і (IV), тобто
II
III
IV
EEE
.
Вираження для розрахунків константи дисоціації в такому випа-
дку прийме вигляд
()
(o)
()()
12
12
lg
x
III
IV
HA
HA
E
E
mm
pK
pK
mm
.
Робота No 10. ВИЗНАЧЕННЯ КОНСТАНТИ ДИСОЦІАЦІЇ СЛАБКОЇ
КИСЛОТИ В БУФЕРНИХ РОЗЧИНАХ У ЛАНЦЮГАХ З ПЕРЕНОСОМ
187
ВИКОНАННЯ РОБОТИ
Визначають точну концентрацію досліджуваної й стандартної
кислот (вихідні розчини беруть із концентрацією близько 0,1
моль/л) титруванням розчином лугу. Титрування проводять до оде-
ржання 3–х співпадаючих результатів.
У мірній колбі на 100 мл готовлять розчин хлориду натрію (або
хлориду калію) з концентрацією 0,1–0,5 моль/л з наважок солі.
Буферні розчини – по три для кожної кислоти, готовлять у мі-
рній колбі на 50 мл розведенням вихідних титрованих розчинів кис-
лот з наступною їхньою частковою нейтралізацією для одержання
солей заданої концентрації. Кислоти беруться з надлишком із зага-
льною концентрацією до введення розчину лугу, що дорівнює сумі
концентрації кислоти та її солі. Склад буферних розчинів і загальну
іонну силу розчинів указує викладач. По концентрації солі в кож-
ному розчині попередньо оцінюється іонна сила й відповідно до
отриманих результатів розраховують об'єм приготовленого розчину
фонового електроліту ( KCl або NaCl ) для добавки до кожного бу-
ферного розчину для створення однакової іонної сили, заданої ви-
кладачем. Після добавки компонентів уміст мірної колби доводять
до мітки розчинником. Бажане, щоб концентрація фонового елект-
роліту перевищувала концентрацію солі кислоти в 5–10 разів.
Гальванічний елемент являє собою хімічний стакан на 50 мл, у яко-
му втримується буферний розчин насичений хінгідроном і опущений
платиновий електрод (рис. 2.25). У розчин занурена капілярна частина з
азбестовою ниткою проточного лабораторного насиченого хлорсрібного
електрода. Виміри ЕРС роблять через кожні 5 хвилин і фіксують
значення, яке залишається незмінним протягом 5–10 хвилин з точ-
ністю ±0,5–1 мВ. Виміри повторюють із кожним буферним розчи-
ном досліджуваної й стандартної кислоти не менш двох раз, дома-
гаючись збіжності результатів виміру з точністю ±0,5–1 мВ.
Розрахунки проводять, поєднуючи виміри кожного буферного
розчину з досліджуваною кислотою з кожним з 3-х стандартних ро-
зчинів. Таким чином, для оцінки константи дисоціації досліджува-
ної кислоти можна одержати 9 значень рК.
Найбільш імовірне значення рК обчислюють як середнє ари-
фметичне зі знайдених значень і оцінюють довірчий інтервал. Якщо
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
188
значення рК значно відрізняються один від одного проводять стати-
стичний аналіз для виявлення промахів. Значення константи дисо-
ціації слабкої кислоти порівнюють із довідковим.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. У чому полягає сутність використання ланцюгів з переносом
для визначення константи дисоціації слабкої кислоти?
2. Для чого в розчини кислот додають солі з однойменними
аніонами?
3. Чому бажане вибирати кислоти близькими за силою?
4. Для чого використовують добавки солі фонового електроліту?
5. Як використовувати в даному методі визначення константи
дисоціації слабкої кислоти скляний (водневий) електроди? Дайте
докладний аналіз.
6. Чим обумовлений вибір концентрацій кислот, солей і фоно-
вого електроліту.
7. У чому полягають принципові недоліки описаного методу
визначення константи дисоціації слабкої кислоти?
8. Які ускладнення з'являться при реалізації даного методу визна-
чення константи дисоціації слабкої кислоти в неводних середовищах?
9. Як елімінують залишковий дифузійний потенціал?
10. Чому в якості стандартної кислоти беруть слабку кислоту, а
не стандартний розведений розчин сильної кислоти, наприклад,
хлористого водню?
11. Приведіть приклади концентраційних елементів без рідин-
ної сполуки.
12. Доведіть, що компіляція хімічних ланцюгів з переносом (III)
і (IV) дає той же результат, що й застосування концентраційного
елемента (II).
13. Зрівняєте метод визначення константи дисоціації слабкої ки-
слоти в ланцюгах з переносом з методом визначення в ланцюгах без
переносу.
14. У чому утруднення використання даного методу для дослі-
дження кислот помірної сили?
15. Проведіть порівняльний аналіз методів визначення констант дисо-
ціації слабких кислот в елементах з переносом та без переносу з викорис-
танням буіерних розчинів.
Робота No 10. ВИЗНАЧЕННЯ КОНСТАНТИ ДИСОЦІАЦІЇ СЛАБКОЇ
КИСЛОТИ В БУФЕРНИХ РОЗЧИНАХ У ЛАНЦЮГАХ З ПЕРЕНОСОМ
189
ЛІТЕРАТУРА
1.Лебідь, В. І. Фізична хімія : підруч. для студ. вищ. навч. закл.
/ В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
2. Герасимов Я. И. и др. Курс физической химии. Т.2.
–М. :
Химия, 1973. – 625 с.
3. Измайлов Н. А. Электрохимия растворов / Н. А. Измайлов. –
М. : Химия, 1986.
–
576 с. Антропов Л. И. Теоретическая электро-
химия / Л. И. Антропов. – М. : Высшая школа, 1984. – 519 с.
4. Робинсон Р., Стокс Р. Растворы электролитов / Р. Робинсон,
Р. Стокс. – М. : Издатин–лит, 1963. – 646 с.
5.Альберт А. Константы ионизации кислот и оснований / А.
Альберт, Е. Сержент. – М. – Л. : Химия, 1964. – 262 с.
6. Практикум по физической химии. Кинетика и каталіз. Элек-
трохимия : учеб. пособие для студ. учреждений высш. проф. обра-
зования / [А. В . Абраменков и др.]; под ред. В . В. Лунина, Е. П. Аге-
ева. – М. : Издательский центр «Академия», 2012. – 304 с.
7. Практикум по физической химии. Учебное пособие для ву-
зов. Изд. 3–е. / Под ред. С. В. Горбачева, – М. : Высшая школа,
1974. – 495 с.
8. Практикум по электрохимии / Под ред. Б. Б. Дамаскина.
–
М. : Высшая школа, 1991. – 288 с.
9. Практические работы по физической химии : Учебное по-
собие для вузов / Под ред. К .П. Мищенко, А.А. Равделя, A.M . По-
номаревой. – СПб. : Изд–во «Профессия», 2002. – 384 с.
10. Байрамов В.М . Основы электрохимии.
–
М.:
ACADEM1A, 2005. – 238 с
11. Дамаскин Б. Б., Петрий О. А., Цирлина Г. А. Электрохи-
мия. – М.: Химия, КолосС, 2006. –
672 с.
12. Робинсон Р., Стокс Р. Растворы электролитов / Р. Робинсон,
Р. Стокс. – М. : Издатин–лит, 1963. – 646 с.
13. Харнед Г. Физическая химия растворов электролитов / Г.
Харнед, Б. Оуэн.
–
М.:ИЛ,1952. – 628с.
14. Александров В. В. Кислотность неводных растворов.
–
Харьков: Вища школа. Изд–во при Харьк. университете, 1981. – 152 с.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
190
Робота No 11.
ВИЗНАЧЕННЯ ДОБУТКУ РОЗЧИННОСТІ
МАЛОРОЗЧИННОЇ СОЛІ СРІБЛА z
Ag A.
Ціль роботи: методом виміру ЕРС у концентраційному еле-
менті з рідинним з’єднанням визначити розчинність і добуток роз-
чинності малорозчинної солі срібла. Переконатися в сталості зна-
чень добутку розчинності солі незалежно від концентрації елект-
ролітів у розчині.
Прилади й реактиви: установка з потенціометром постій-
ного струму або компенсаційна схема із трьома магазинами опорів,
комірки зі срібними електродами, електролітичний ключ із розчи-
ном нітрату амонію, 3 робочих колби на 100 мл для розчинів гало-
геніду калію, мірна колба на 50 мл, градуйована піпетка 25 мл, роз-
чин нітрату срібла (0,05 або 0,01 моль/)л.
Виміром ЕРС концентраційних елементів можна визначити ак-
тивність (концентрацію) іонів металу в розчині біля одного з елект-
родів, знаючи активність іонів металу в розчині біля іншого елект-
роду, а отже, визначити розчинність і добуток розчинності малороз-
чинної солі цього металу. Термодинамічна константа добутку акти-
вностей солі залежить тільки від хімічної природи солі, температури
й властивостей розчинника. У випадку відсутності сторонніх елект-
ролітів у розчині й малої розчинності самої солі добуток розчинності
чисельно дорівнює добутку активностей.
Концентраційними гальванічними елементами називають
елементи, у яких електрична енергія проводиться за рахунок осмо-
тичного процесу. Концентраційні елементи можуть бути без рідин-
ного з’єднання (ланцюги без переносу) і з рідинним з’єднанням (ла-
нцюги з переносом). ЕРС концентраційних елементів залежить
тільки від відношення активностей компонентів системи. Так, для
гальванічного елемента з рідинним з’єднанням
31
32
()
()
j
AgAgNOa AgNOa Ag
ЕРС залежить тільки від різниці активностей іонів срібла 1
aй2
aв
розчинах нітрату срібла.
Робота No 11. ВИЗНАЧЕННЯ ДОБУТКУ РОЗЧИННОСТІ
МАЛОРОЗЧИННОЇ СОЛІ СРІБЛА z
AgA
191
Рівняння електрохімічних реакцій і відповідні перегони потен-
ціалів на границі метал – розчин за умови, що 1
2
aa
наступні:
Ag
Age
,
/
1,( )
ln
o
Ag Ag
Ag
RT
a
F
Age
Ag
,
/
2,( )
ln
o
Ag Ag
Ag
RT
a
F
.
ЕРС ланцюгу включає крім стрибків потенціалів і на межі
розподілу метал – розчин дифузійний потенціал на межі розподілу
двох розчинів різних концентрацій j
1
2
ln
j
j
a
RT
E
Fa
.
Розрахувати величину дифузійного потенціалу можна тільки в
деяких простих випадках; так для наведеного ланцюга
1
2
(21)ln
j
a
RT
t
Fa
,
де t – число переносу аніона.
На практиці прагнуть знизити дифузійний потенціал до нуля –
елімінувати його. Для цього використовують з’єднання розчинів за
допомогою так званих сольових мостів – концентрованих розчинів
електроліту, рухливості катіона й аніона в якому близькі між собою.
Тоді залишковий дифузійний потенціал – різниця між дифузійними
потенціалами на границях контакту розчину мосту з досліджувани-
ми розчинами стає близькою до нуля. У якості електролітів для за-
повнення електролітичних мостів найбільше часто використову-
ються: KCl , KNO3, NH4Cl, NH4NO3.
Добуток розчинності
sp
K являє собою константу рівноваги
між речовиною та його насиченим розчином. Якщо речовина являє
собою сильний електроліт, то зв'язок добутку розчинності й роз-
чинності під час відсутності інших електролітів передається рівнян-
ням ЗДМ
()()
sp
s
s
K
c
c
,
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
192
у якому s
c – розчинність, а й – стехіометричні коефіцієнти
для катіона й аніона в реакції дисоціації.
Для визначення добутку розчинності sp
K солі
z
Ag A збирають
концентраційний елемент
1
32
1
2
,
()
()
z
z
j
AgAgAKAm AgNOm Ag
,
деz
K A – добре розчинна сіль калію – сильний електроліт ( KCl ,
KBr,KI ,KCNS, 2
4
KCrO, 3
6
()
KFeCN ,іт.п.).
Малорозчинна сіль
z
Ag A дисоціює у розчині за рівнянням
z
z
AgA
zAg A
і добуток розчинності солі запишеться у вигляді
,
.
z
z
z
spAgA
Ag
A
K
a
a
ЕРС елемента E за умови елімінування дифузійного потенціа-
лу j дорівнюєй
2
1
21
2
1
,
1/
2.303
lg
lg
z
spAgA
z
a
a
RT
E
a
F
a
K
.
Після перетворень можна виразити величину добутку розчин-
ності
,
2
1
lg
lg
lg
z
spAgA
zE
K
z
a
a
.
Активність іонів срібла в розчині нітрату срібла (
3
AgNO) 2
a
визначається за даними про коефіцієнти активності 2
2
22
a
m
,
а активність іонів
z
A
згідно рівняння
1
1
1
11
()zz
a
z
a
.
Коефіцієнти активності електролітів 1
і 2 знаходять графіч-
ною інтерполяцією довідкових даних.
Робота No 11. ВИЗНАЧЕННЯ ДОБУТКУ РОЗЧИННОСТІ
МАЛОРОЗЧИННОЇ СОЛІ СРІБЛА z
AgA
193
ВИКОНАННЯ РОБОТИ
Розчинність солі срібла визначають дослідженням трьох роз-
чинів солі калію з осадом (суспензією) солі срібла (сіль та концент-
рації її розчинів вказує викладач).
Для проведення ви-
мірів користуються елеме-
нтом, що складається із
двох комірок зі срібними
електродами, які з'єднані за
допомогою електролітич-
ного мосту. Схематичне
зображення елемента наве-
дено на рисунку 2.28. Ко-
мірка 1 заповнюється ро-
зчином нітрату срібла ві-
домої, заданої викладачем
концентрації; комірка 2 –
заповнюється розчином за-
даної концентрації солі ка-
лію z
K A , відповідного до
малорозчинної солі срібла із суспензією цієї срібної солі. Для цього
до розчину солі калію в стаканчик 4 додають 1–2 краплі 0,05 М роз-
чину нітрату срібла до утворення суспензії відповідної до срібної
солі
z
AgA.
Розчини солей калію для дослідження готовлять розведенням
вихідного розчину більшої концентрації з використанням мірної ко-
лби об'ємом 50 мл. Поверхню срібних електродів перед викорис-
танням ретельно зачищають дрібним наждаковим папером, щільно
вставляють у вимірювальні комірки й за допомогою патрубків 5, ві-
дкриваючи крани 6 (або віджимаючи зажими), затягують розчини
нітрату срібла зі стаканчика 3 або із сіллю калію зі стаканчика 4 у
комірку до повного занурення срібних електродів. Необхідно сте-
жити за тим, щоб не утворювалися пухирці повітря в сполучних
трубках щоб уникнути розриву ланцюгів. Стаканчики з'єднують за
допомогою електролітичного мосту 7, заповненого концентрованим
Рис. 2.28. Концентраційний гальванічний
елемент з срібними електродами для виміру
розчинності малорозчинних солей срібла
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
194
розчином KNO3 або NH4NO3. (У якості електролітичного мосту мо-
жна використовувати стаканчик з концентрованим розчином KNO3
або NH4NO3, у який можна вилучити сполучні трубки обох комі-
рок).
При роботі використовувати комірки, що призначені тільки для
роботи з відповідними їм солями срібла!
Виміри ЕРС зібраного гальванічного елементу проводять на
схемі із трьома магазинами опорів або на високоомному потенціо-
метрі постійного току. Проводять не менш двох вимірів кожного
розчину, наготовлюючи заново розчин, що містить суспензію мало-
розчинної солі срібла в розчині солі калію. Після кожного виміру
перевіряють установку робочого струму і якщо буде потреба набу-
довують знову. Комірку із стандартним розчином нітрату срібла
можна застосовувати одну й ту саму.
Для розрахунків беруть середнє арифметичне значення цих
вимірів.
Рекомендована форма ведення протоколу дослідження
Температурадосліду ....... Сількалію( z
KA)...........
No
Концентрація
солі z
KA,
моль/л
ЕРС
елементу,
мВ
1
2
lg sp
K
sp
K
Значення добутку розчинності розраховують за рівнянням
2
1
lg
lg
lg
zE
Kz
a
a
,
визначаючи активності іонів відповідно до їхнього заряду за коефі-
цієнтами активності відповідних іонів, знайдених у довідковій літе-
ратурі. У випадку необхідності для визначення коефіцієнтів актив-
ності використаної в роботі солі калію за зазначеними концентраці-
ями проводять чисельну або граіфчну інтерполяцію. Для якісної ін-
терполяції доцільно її проводити в координатах lg
m
.
Для електроліту 1:1 валентного типу розрахункова формула
для обчислення добутку розчинності має вигляд
Робота No 11. ВИЗНАЧЕННЯ ДОБУТКУ РОЗЧИННОСТІ
МАЛОРОЗЧИННОЇ СОЛІ СРІБЛА z
AgA
195
3
3
,
,
lg
lg
sp AgA
AgNO
AgNO
KA
KA
E
K
m
m
.
звідки розчинність солі у випадку відсутності будь-яких інших еле-
ктролітів визначається як
,
()
s
sp AgA
c AgA
K
.
Значення коефіцієнтів активності електролітів для розчинів за-
даних концентрацій визначаються графічною інтерполяцією табли-
чних значень. Отримані за даними виміру ЕРС в елементах з різною
концентрацією солі калію значення константи добутку розчинності
порівнюють між собою й зі значеннями в довідковій літературі при
температурі досліду.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Як буде мінятися розчинність малорозчинної солі з додат-
ком до насиченого розчину електролітів а) що містять і б) не міс-
тять однойменний з малорозчинною сіллю аніон або катіон?
2. Як підбирають електроліт для заповнення сольового мосту
з метою елімінування дифузійного потенціалу?
3. Що є джерелом електричної роботи концентраційного галь-
ванічного елемента?
4. Приведіть приклади концентраційних гальванічних елемен-
тів з переносом і без переносу. Напишіть для них рівняння Нернста.
5. Виведіть рівняння зв'язку розчинності малорозчинного не-
симетричного електроліту (M2A) з його добутком розчинності.
6. Якими індивідуальними властивостями речовини й розчин-
ника визначається розчинність?
7. Як використовувати концентраційні елементи для визна-
чення змісту металу в сплавах? Приведіть приклади.
8. Який порядок роботи на компенсаційній установці із трьома
магазинами опорів?
9. Чи застосовне поняття добутку розчинності до добре роз-
чинних речовин?
10. Дайте термодинамічний висновок константи добутку роз-
чинності.
11. Як змінюється розчинність малорозчинної солі при дода-
ванні солі з однойменним аніоном (катіоном). Покажіть математич-
но зв'язок розчинності з добутком розчинності в цьому випадку.
2. ГАЛЬВАНІЧНІ ЕЛЕМЕНТИ. ЭЛЕКТРОРУШІЙНІ СИЛИ
196
12. Поясните факт збільшення розчинності кристалічних спо-
лук з ростом температури.
13. Як і чому впливає іонна сила розчину на розчинність мало-
розчинного сполуки.
14. Як впливає природа розчинника на розчинність іонних кри-
сталів?
15. Як використовують метод розчинності для визначення кое-
фіцієнтів активності іонів.
16. Як впливає на розчинність дисперсність осаду й час від по-
чатку його осадження.
17. Поясните зв'язок стандартних потенціалів електродів дру-
гого роду зі станндартними потенціалами металевих електродів че-
рез добуток розчинності відповідних малорозчинних солей.
ТЕОРЕТИЧНІ ПИТАННЯ ПО ТЕМІ
Електрохімічні процеси. Теорії виникнення електродного по-
тенціалу й електрорушійної сили. Розрахунки ЕРС хімічних елемен-
тів за термодинамічним даними. Класифікація гальванічних елемен-
тів: оборотні й необоротні, хімічні й концентраційні, з переносом і
без переносу. Застосування концентраційних елементів з переносом
і без переносу. Методи виміру ЕРС, компенсаційний метод. Еле-
мент Вестона. Дифузійний потенціал, його природа й методи елімі-
нування. Рівняння Нернста для потенціалу електрода й ЕРС гальва-
нічного елемента. Стандартний потенціал і стандартна ЕРС елемен-
та. Типи електродів: першого, другого й третього роду. Викорис-
тання електрохімічний ланцюгів для вивчення добутку розчинності.
Розчинність, розчини насичені, ненасичені, пересичені. Закони
розчинності твердих речовин у рідинах. Залежність розчинності від
температури й тиску. Вплив на розчинність добавок електролітів із
загальним іоном, індиферентних електролітів, зміни сполуки роз-
чинника.
Експериментальні фізико-хімічні методи визначення розчин-
ності й добутку розчинності малорозчинних сполук. Потенціомет-
ричне титрування за методом осадження малорозчинної сполуки.
Робота No 11. ВИЗНАЧЕННЯ ДОБУТКУ РОЗЧИННОСТІ
МАЛОРОЗЧИННОЇ СОЛІ СРІБЛА z
AgA
197
ЛІТЕРАТУРА
1. Герасимов Я. И. и др. Курс физической химии. Т.2.
–М. :
Химия, 1973. – 625 с.
2. Лебідь, В. І. Фізична хімія : підруч. для студ. вищ. навч.
закл. / В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
3. Дамаскин Б. Б. и др. Электрохимия / Б. Б. Дамаскин, О. А.
Петрий, Г. А. Цирлина. – М. : Химия, КолосС, 2006. – 672 с..
4. Методы измерения в электрохимии, т. 1, 2 / Под ред. Э.
ЕгераиА.Залкинда. – М.:Мир,1977. – 566с. – 476с.
5. Практические работы по физической химии: Учебное посо-
бие для вузов / Под ред. К .П. Мищенко, А.А. Равделя и А.М . Поно-
маревой. – Л.: Химия, 1982. – с. 128–178.
6. Практикум по физической химии. Кинетика и катализ.
Электрохимия : учеб. пособие для студ. учреждений высш. проф.
образования / [А. В . Абраменков и др.] ; под ред. В . В .Лунина, Е. П.
Агеева. — М. : Издательский центр «Академия», 2012. – 304 с.
7. Практикум по физической химии. Учебное пособие для ву-
зов. Изд. 3–е. / Под ред. С. В. Горбачева, – М. : Высшая школа,
1974. – 495 с.
8. Практикум по электрохимии / Под ред. Б. Б. Дамаскина.
–
М. : Высшая школа, 1991. – 288 с.
9. Физическая химия. Теоретическое и практическое руковод-
ство. Изд. 2 –е, переработанное и дополненное. / Под ред. академи-
ка Б. П. Никольского. – Л. : Химия, 1987. – 880 с.
10. Практические работы по физической химии : Учебное посо-
бие для вузов / Под ред. К .П. Мищенко, А.А. Равделя, A.M . Поно-
маревой. – СПб. : Изд–во «Профессия», 2002. – 384 с.
11. Ротинян А. Л., Тихонов К.И., Шошина И.А. Теоретическая
электрохимия / Под. ред. А. Л. Ротиняна. – М. : Студент, 2013. – 496 с.
12. Кумок В. Н. Произведения растворимости / В. Н. Кумок, О.
М. Кулешова, Л. А. Карабин. – Новосибирск : Наука, 1983. – 267 с.
198
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
3.1. ТИПИ Й БУДОВА ХІМІЧНИХ ДЖЕРЕЛ СТРУМУ
Хімічним джерелом струму (ХДС) називають обладнання для
безпосереднього перетворення хімічної енергії активних речовин в
електричну енергію. Виникнення електричного струму відбувається
за рахунок протікання на електродах електрохімічних реакцій –
утворення електричної енергії відбувається за рахунок спаду енергії
Гіббса струмоутворюючого електрохімічного процесу. Основою
ХДС є два електроди: анод, що містить окислювач, і катод утриму-
ючий відновник, які опущені в розчин електроліту. Між електрода-
ми встановлюється різниця потенціалів – електрорушійна сила
(ЕРС), яка відповідає вільній енергії окисно-відновної реакції. Дія
хімічних джерел струму засноване на протіканні в замкненому зов-
нішньому ланцюзі просторово-розділених процесів. На катоді від-
новник окислюється, при цьому вільні електрони, що утворюються,
переходять по зовнішньому ланцюгу до анода, утворюючи розряд-
ний струм. На аноді електрони беруть участь у реакції відновлення
окислювача.
Гальванічним елементом (елементом) називають джерело
струму, що полягає з одного електрохімічного гнізда. Два або більш
елементів, з'єднаних послідовно або паралельно, називають гальва-
нічною батареєю (батареєю). Основні позначення елементів і бата-
рей в електричних схемах представлені на рисунку
Гальванічний елемент являє собою обладнання, що складаєть-
ся із двох електродів з електронною провідністю, які різняться
3.1. ТИПИ Й БУДОВА ХІМІЧНИХ ДЖЕРЕЛ СТРУМУ
199
потенціалами, рідкого або твердого електроліту з іонною провідніс-
тю, металевого провідника, що утворює зовнішній ланцюг. В основі
кожного ХДС лежить електрохімічна система виду
( ) розчин електроліту ( )
A
К
,
наприклад,
4
4
Zn ZnSO CuSO Cu .
Форма запису відповідає позитивному значенню напруги в си-
стемі: ліворуч зазначений негативний електрод, праворуч – позити-
вний. У найменуванні ж джерела струму першим прийнято називати
позитивний електрод, наприклад, мідно-цинковий.
У ХДС використовують системи як з оборотними, так і з не-
оборотними електрохімічними реакціями. Якщо хоча б на одному з
електродів окисно-відновний процес протікає незворотно, то таке
джерело струму називають первинним хімічним джерелом стру-
му. Такий елемент здатний забезпечити лише один розряд, тому що
необоротність електрохімічного процесу не дозволяє привести еле-
ктроди в первісний стан. Первинні ХДС поділяють на дві групи:
елементи з рідким електролітом й сухі елементи, що місти елект-
роліт, який не виливається.
Всі первинні джерела струму можна розділити на активні й
активуєсі. Активні готові до розряду в будь-який момент часу, тому
що характеризуються прямим контактом електродів і електроліту.
Найбільше поширення одержали марганцево-цинкові, повітряно-
цинкові й ртутно-цинкові елементи.
Активуєиі (резервні) можуть тривалий час перебувати в неро-
бочому стані через ізоляцію електроліту від електродів, тому що
електроліт перебуває або у твердому стані, або взагалі відсутній.
Процес активації таких елементів проводиться заливанням водою
або електролітом. Найбільш відомі ХДС такого типу – мідно-
магнієві батареї, що приводяться в робочий стан природною водою.
В активуємих ХДС ампульного типу агресивний електроліт укладе-
ний у герметичну ампулу, яка розбивається для приведення ХДС у
робочий стан. Найкращими характеристиками такого типу елемен-
тів є срібно-цинкові.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
200
Вторинні хімічні джерела струму (або акумулятори) явля-
ють собою систему з оборотне працюючими електродами й можуть
бути використані багаторазово. Для цього виряджений елемент (ба-
тарею) піддають заряду – поляризації від зовнішнього джерела пос-
тійного струму, при якому на позитивному електроді здійснюється
анодна реакція окиснення, а на негативному – катодна реакція від-
новлення. У результаті такого процесу активні речовини електродів
(електроліту) вертаються у вихідний стан.
Паливні елементи (електрохімічні генератори) відрізняються
від первинних і вторинних ХДС тим, що функціонують безперерв-
но: речовина для електрохімічних реакцій подається з зовні, а про-
дукти реакції видаляються з приладу.
Конструктивне обладнання ХДС обов'язково передбачає
1) поділ електродів, що запобігає коротке замикання;
2) розвиток активної електродної поверхні;
3) придушення небажаних процесів, що приводять до побічних
реакцій, витоку струму;
4) механічну міцність при мінімальній матеріалоємності.
Сучасні ХДС надзвичайно різноманітні, але конструктивно
обов'язково складаються з електродів, електроліту й сепаратора,
корпуса із кришкою.
Типовий ХДС складається з активної маси й струмоведучого
каркаса. Активна маса – це суміш хімічних речовин, що забезпе-
чують протікання струмоутворюючих реакцій. Найпоширенішими
активними речовинами негативного електрода є такі метали, як
свинець, цинк, залізо, кадмій, магній, літій, що окислюються в ре-
зультаті розряду до оксидів, гідроксидів та їх солей. У якості актив-
ної речовини позитивного електрода найбільше часто використову-
ються оксиди або гідроксиди металів, які при розряді відновлюють-
ся до металу або оксиду (гідроксиду) більш низького ступеня окис-
нення. В активні маси вводять різні добавки: для збільшення елект-
ричної провідності: добавки, що активують, володіють депасувую-
чим ефектом, що стабілізують – перешкоджають процесу старіння
активної маси, що зв'язують – додають електроду механічної міц-
ності.
3.2. ЕЛЕКТРИЧНІ ХАРАКТЕРИСТИКИ ХДС
201
Струмоведучий каркас призначений для фіксації активних
мас, що представляють часто порошкоподібний або пастоподібний
матеріал, у габаритах електрода й забезпечення електричного зв'яз-
ку зерен активної речовини з контактними провыдниками джерела
струму.
Струмоутворюючі реакції протікають на електродах при без-
посередный участі компонентів розчину електроліту. Головне приз-
начення електроліту – забезпечити електродні реакції що беруть
участь у них, іонами й молекулами. Підведення іонів здійснюється,
зазвичай, за рахунок міграції й дифузії. Від природи електроліту за-
лежать розчинність і структура твердофазних продуктів розряду.
Друге призначення електроліту – утворювати внутрішнє електричне
коло між електродами, тому електроліт повинен мати високу іонну
провідність.
Для запобігання контакту різнойменних електродів щоб уник-
нути короткого замикання в ХДС використовують сепаратори, що
виготовляються з діелектричних матеріалів.
Основною класифікаційною ознакою для вторинних джерел
струму (акумуляторів) служить тип електроліту. Розрізняють аку-
мулятори з кислотним, лужним,. твердим і розплавленим електролі-
том. У промисловості випускають єдиний кислотний акумулятор –
свинцевий. Найпоширеніші лужні акумулятори: нікель-залізні й ні-
кель-кадмієві, далі, по масштабах виробництва – срібно-цинкові.
3.2. ЕЛЕКТРИЧНІ ХАРАКТЕРИСТИКИ ХДС
Напруга – одна з найважливіших характеристик ХДС. Слід ро-
зрізняти: напруга електрохімічної системи, напруга розімкнутого
ланцюга, розрядна напруга, номінальна напруга. Для акумуляторів
становить інтерес також зарядна напруга.
Напруга електрохімічної системи або ЕРС характеризує не
стільки реальне джерело струму, скільки саме електрохімічну сис-
тему, що лежить у його основі. Величина
o
E являє собою різницю
стандартних потенціалів електродних реакцій, що протікають при
розряді.
Напруга розімкнутого ланцюга
.
рл
U – являє собою різниця
безструмових електродних потенціалів ХДС, обмірювану при розі-
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
202
мкнутому зовнішньому ланцюзі.
.
рл
U може бути дорівнює електро-
рушійній силі E (ЕРС), якщо стаціонарні потенціали позитивного й
негативного електродів дорівнюють рівноважним потенціалам. У
більшості випадків через існування побічних реакцій на електродах
не існує рівноваги й стаціонарна потенціал є компромісним.
Розрядна напруга (напруга джерела струму) найбільше важ-
ливо для споживачів ХДС. Розрізняють початкова напруга
.
н
U,кін-
цева напруга
.
к
U й середня напруга
.
сер
U . У кожному випадку ма-
ється на увазі напруга U при розряді певним режимом, зміст цих
величин ясний при розгляді так званих розрядних характеристик
ХДС, які представляються розрядними кривими в координатах на-
пруга – час. Корисна для розрахунків величина середньої напруги
визначається за рівнянням
.
0
1
()
сер
U
Ud
або приблизно
.
1N
сер
i
io
U
U
N
,
де N – число вимірів через однакові проміжки часу.
Номінальна напруга
.
ном
U – умовна величина напруги. близь-
ка до
.
н
U Воно звичайно показане на торговельній етикетці і є дос-
тупною для споживача характеристикою.
Повним внутрішнім опором (r) ХДС називають опір, надава-
не їм при проходженні всередині нього постійного струму
п
o
o
п
E
rr
rr
I
,
де.
п
E – ЕРС поляризації; I –
струм. Перше з доданків – o
r
називається омічним опо-
ром і являє собою суму опо-
рів електродів і електроліту.
Другий доданок,
.
п
r обумов-
лений зміною електродних
потенціалів і при про-
ходженні струму, цей опір
Рис. 4 .1 . Співвідношення між різними
характеристиками напруги ХІТ.
3.2. ЕЛЕКТРИЧНІ ХАРАКТЕРИСТИКИ ХДС
203
поляризації, який не підкоряється закону Ома.
Наявністю внутрішнього опору обумовлене те, що розрядна
напруга
.
р
U ХДС ( тобто напруга при замкненому зовнішньому ла-
нцюзі) завжди менше його ЕРС
,
p
pp
п
op
p
UErIEE
r
I
.
При постійній силі розрядного струму й постійній температу-
рі електроліту розрядна напруга зменшується в часі внаслідок збі-
льшення п
Eй,
op
r.
Для оборотних систем (акумуляторів) характерна наявність
зарядної напруги .з
U , яка виражається рівнянням
.
.
.
.
,.
.
з
з
з
п
oз
з
UErIEE
r
I
.
При постійній силі зарядного струму й постійній температурі
електроліту зарядна напруга збільшується в часі внаслідок збіль-
шення
.
п
E . Наприкінці заряду, коли в основному йде процес елект-
ролізу води, значення
.
з
U стабілізується.
Розрядною ємністю C називають кількість електрики, яка
джерело струму віддає при розряді від початкового до кінцевої на-
пруги.. Одиницею виміру ємності ХДС є ампер-година.
Спосіб розрахунків ємності залежить від струмового режиму.
Якщо розряд ведеться при постійному струмі, то ємність рівна
C=I·,
а в загальному випадку визначається як
0
CId
.
Існує поняття номінальної ємності
.
ном
C , віднесеної до регла-
ментованих умов. Ємність ХДС прямо залежить від маси активних
речовин, однак останнє використовується не повністю, що визна-
чається коефіцієнтом використання активної речовини c
K<1.На
величину
.
исп
K істотний вплив виявляють конструкція й пористість
електродів, швидкість розряду.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
204
Швидкість розряду часто оцінюють часом віддачі номінальної
ємності. Так 10–ти годинний режим розряду означає, що струм чи-
сельно рівний 1/10 номінальної ємності ХДС, або I10 = 0,1·
.
ном
CА.
Число, зворотне тривалості розряду, називають нормованим стру-
мом розряду.
Розрізняють наступні режими розряду: тривалий I < 0,1·
.
ном
C;
середній 0,1·
.
ном
C <I<1·
.
ном
C ікороткийI>1·
.
ном
C . Іноді виді-
ляють понадкороткий, або форсований розряд I > 3·
.
ном
C.
Ємність
.
з
C , повідомлена вторинному джерелу струму при
заряді, завжди менше розрядної ємності C. Відмінність пов'язане із
протіканням при заряді побічних електродних реакцій, наприклад,
електролізу води, на які витрачається частина електричної енергії.
Ефективність зарядно-розрядного циклу оцінюється коефіцієнтом
віддачі по ємності C
K (%):
.
100 /
C
з
K
CC
.
Ємність ХДС важлива з погляду експлуатації, але недостатня
характеристика ХДС, тому що основне завдання ХДС полягає в ге-
неруванні електричної енергії.
Енергією ХДС називають енергію, яка віддається в зовнішній
ланцюг при розряді до заданої кінцевої напруги. Номінальну енер-
гію ХДС розраховують по формулі
.
.
.
ном
ном
ср
W
СU
.
Розрахунки енергії в умовах, відмінних від номінальних, ви-
значається режимом розряду. У загальному випадку
0
W IUd
.
При розряді на постійний зовнішній опір
0
1
ц
W
IUd
R
.
Одиницею енергії є кДж, але часто використовують і позасис-
темну одиницю Вт·год (1 Вт·год = 3,6 кДж).
3.2. ЕЛЕКТРИЧНІ ХАРАКТЕРИСТИКИ ХДС
205
Для порівняння енергоємності різних ХДС служить величина
питомої енергії. Масова питома енергія – енергія при розряді, від-
несена до одиниці маси ХДС (Вт·год/кг).
Поряд з коефіцієнтом віддачі C
K застосовують і коефіцієнт
віддачі по енергії, W
K (%):
.
.
,
.
100
ср
W
C
з
зср
U
W
K
K
W
U
,
деWй .з
W – енергія, відповідно віддана при розряді й повідомлена
при заряді. Очевидно, W
C
KK
, тому що
.
,
.
ср
зср
U
U
<1.
Потужність ХДС – кількість енергії, що віддається в одини-
цю часу
W
N
IU
.
При розряді сталим струмом, I = const, потужність ХДС бе-
зупинно падає в міру зниження розрядної напруги
.
р
U . Для кожного
джерела струму існує максимально можлива припустима розрядна
потужність
.
доп
N , вище якої розряд втрачає всякий практичний
зміст через надмірне зниження розрядної напруги, небезпечного пе-
регріву.
У процесі експлуатації електричні характеристики ХДС погі-
ршуються: падає середня напруга, зменшується ємність, знижується
коефіцієнт віддачі. Працездатність оцінюється терміном служби,
що виражається інтервалом часу, протягом якого ХДС зберігає ха-
рактеристики, передбачені технічними умовами.
Для акумуляторів контрольним показником вичерпання тер-
міну служби є ємність при розряді струмом номінального режиму.
Якщо, наприклад, стандартом установлене, що ємність у межах те-
рміну служби повинна бути не менше 0,8·
.
ном
С,теприС<
0,8·
.
ном
С джерело струму вважається непридатним до експлуатації.
Працездатність вторинних джерел струму оцінюється величи-
ною технічного ресурсу – числом зарядно-розрядних циклів.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
206
Схоронність заряду ХДС – це властивість джерела струму збе-
рігати ємність під час зберігання при розімкнутому зовнішньому
ланцюзі. Для первинних джерел струму схоронність заряду безпо-
середньо впливає на термін служби, для акумуляторів такого взає-
мозв'язку немає. Втрату ємності акумулятора при зберіганні легко
компенсувати підзарядом. Заходом втрати ємності за час зберігання
є величина саморозряду джерела струму S (%):
100,
o
o
CC
S
CC
деo
C – ємність свіжо зарядженого акумулятора, C – ємність після
закінчення часу . Залежність C – , як правило, нелінійна, саморо-
зряд протікає найбільше інтенсивно в початковий період зберігання.
Головною причиною саморозряду є взаємодія активних мас із ком-
понентами електроліту, а іноді й електрода.
Для малогабаритної техніки широке поширення одержують
літієві ХДС. Під цим терміном ховаються кілька груп первинних і
вторинних джерел, що мають різну хімічну начинку, різні рівні ви-
хідної напруги (3,0 В и 3,6 В) і одмінних друг від друга ще по ряду
ознак – електричної ємності, діапазону робочих температур, стро-
кам зберігання і т. д. До них ставляться:
–
літій / тіонілхлоридні (
2
/
Li SOCl ),
–
літій/ діоксид сірки (
2
/
LiSO),
–
літій / діоксид марганцю (
2
/
LiMnO)
–
літій / діоксид міді (
2
/
LiCuO)
–
літій / фторвуглецеві ( /( )x
LiCF.
Найбільш вивчений і технологічно відпрацьований тип літіє-
вих первинних хімічних джерел струму – елементи на основі систе-
ми літій/діоксид марганцю (
2
/
Li MnO ), тому вони із усієї групи самі
доступні за ціною. Елементи
2
/
Li SOCl системи – кращі по більшос-
ті параметрів. Вони характеризується найвищою вихідною напру-
гою (3,6 В), найбільшою електричною ємністю, самим широким ді-
апазоном температур, дуже малими струмами саморозряду й серед-
нім типовим струмом розряду. Найчастіше елементи саме цієї групи
3.3. ПЕРВИННІ ДЖЕРЕЛА СТРУМУ
207
застосовуються для харчування промислових обладнань. Оскільки
при значних струмах розряду на внутрішньому опорі може виділя-
тися додаткове тепло в межах перевищуючих припустимий рівень,
то в конструкцію елемента вводять запобіжник-обмежник струму
(терморезистор), що не допускає струмових перевантажень.
3.3. ПЕРВИННІ ДЖЕРЕЛА СТРУМУ
МАРГАНЦЕВО-ЦИНКОВІ ЕЛЕМЕНТИ (МЦ)
Марганцево-цинкові елементи (МЦ) ділять на сольові й лужні.
У сольові використана електрохімічна система Лекланше
4
2
ZnNHClMnO.
У лужних МЦ елементах використовується система
2
ZnKOHMnO.
Сольові елементи більш прості, дешеві й надійні; лужні мають
кращі питомі характеристики, але більш трудомісткі у виготовленні.
Основний компонент електроліту – хлорид амонію, кислотність
електроліту близька до pН = 5. Реакція на негативному електроді
зводиться до анодного розчинення цинку
2
2
Zn
Zn
e
,
0, 763
o
E
B
При рН = 5 в амонійно-хлоридному електроліті цинк утворює
аміакатні комплекси типу
322
[()]
Zn NH Cl . При цьому переважно
протікає друга реакція утвору діамінцинку
4
322
2
[()]
2
Zn
NH Cl
ZnNH Cl
H
.
У міру виснаження електроліту стає істотною реакція гідролізу
хлориду цинку з утвором слаборозчинного оксихлориду цинку
2
2
2
2
5
8
4()8
ZnCl
HO
ZnCl Zn OH
HCl
.
На позитивному електроді, активна маса якого містить
2
MnO ,
що володіє електронною провідністю, порівнянної з іонною провід-
ністю електроліту, додають графіт для збільшення провідності й за-
безпечення більш повного розряду й стабільності напруги в часі.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
208
У початковому стані в слабкокислому хлоридному розчині
проходить реакція катодного відновлення діоксиду марганцю з
утвором добре розчинного хлориду
2
2
2
2
2
4
MnO
HOe
Mn
OH
,
1, 23
o
E
B
.
Під час підлужування прикатодного шару механізм віднов-
лення змінюється, при pН > 7 розряд протікає у твердій фазі зерен
2
MnO по рівнянню
2
2
2
MnO
HOe
MnOOH OH
,
0,35
o
E
B
.
тому що в нейтральному й лужному середовищі діоксид марганцю
не розчинений.
Саморозряд сольового МЦ елемента визначається саморозря-
дом (корозією) цинкового аноду. Устрій циліндричного сольового
МЦ елемента показано на рисунку 3.2 . Еле-
менти випускаються відповідно до типороз-
мірів, встановлених міжнародної електротех-
нічної комісії (МЕК). Основні частини – не-
гативний і позитивний електроди, сепаратор і
вузол герметизації. Негативний електрод 6
виготовлений із чистого цинку або цинку, ле-
гованого свинцем (0,5%) і кадмієм (0,05%),
і має форму склянки, що виконує функцію
корпусу. Позитивний електрод складається з
активної маси 7 і струмовідводу у вигляді ву-
гільного стрижня 8. До складу активної маси
входять: природний діоксид марганцю – пі-
ролюзит, штучний діоксид марганцю, елект-
ропровідні домішки (сажа, графіт) й електро-
літ. Сепаратором 9 служить лугостійкий па-
пір, на який нанесений шар загущеного кро-
хмальним клейстером електроліту ( 4
NHCl–
7%,
2
ZnCl – 5%,
2
CaCl – 3%,
2
HgCl –5–10
г/л). Картонна прокладка 10 перешкоджає короткому замиканню,
металевий ковпачок 1 є зовнішнім контактом позитивного електро-
да, за допомогою шайб 10 і 11 фіксується простір для водню, що
виділяється при саморозряді. Бітумна композиція 3, прикрита деко-
ративною шайбою 2, призначена для герметизації елемента.
Рис. 3 .2 . Схема кон-
струкції марганцево-
цинкового елемента.
3.3. ПЕРВИННІ ДЖЕРЕЛА СТРУМУ
209
СРІБНО-ЦИНКОВІ ЕЛЕМЕНТИ (СЦ)
В основі роботи елемента лежить електрохімічна система
ZnKOHAgOAg.
СЦ елементи вигідно відрізняються від МЦ більш стабільною
розрядною характеристикою в комбінації із тривалим строком збе-
рігання й високою питомою
енергією. Їх використовують у
мікрокалькуляторах, годиннику
й т.п . У порівнянні із РЦ перева-
гами, що володіють тими ж, СЦ
мають більш пологу вольт-
амперну характеристику й менш
чутливі до підвищення струмо-
вого навантаження, наприклад,
при включенні підсвічування в електрогодинниках.
Срібно-цинкові елементи відрізняються стабільною розрядною
характеристикою, мають тривалий строк зберігання й високу пито-
му енергію. Саморозряд не перевищує 10% у рік, а схоронність ста-
новить 2 року. Діапазон робочих температур 0–40 С, напруга 1.50–
1.55 В, питома енергія при розряді струмом 100–годинного режиму
рівна 80–110 Вт·год/кг в залежності від розмірів елемента. Активна
маса позитивного електроду готується з оксиду срібла (Ag2O) й при
розряді протікає реакція за рівнянням
2
2
2
2
2
AgOHОе
Ag ОH
.
На негативному електроді протікає реакція окиснення цинку
2
2
2
Zn OH
e
ZnO HO
/
Найбільше часто використовувана дискова конструкція елеме-
нтів, зображена на рис. 3.3. Анод із цинкового порошку 2 запресова-
ного в сталеву позолочену кришку 4. Активна маса катода 1, що по-
лягає з оксиду срібла й електропровідної добавки графіту, перебуває
в сталевий нікельованому корпусі 6. Сепаратор 3 складається з декі-
лькох шарів гідратцелюлозної плівки. Електроліт гідроксид калію
герметично закритий ущільнювачем 5.
Рис. 3 .3 . Схема конструкции диско-
вого серебряно-цинкового элемента.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
210
РТУТНО-ЦИНКОВІ ЕЛЕМЕНТИ (РЦ)
Малогабаритні герметичні ртутно-цинкові (РЦ) елементи в по-
рівнянні з марганцево-цинковими (МЦ) мають більш високі харак-
теристики: терміном служби, питомою енергією, механічною міцні-
стю, стабільністю розрядної напруги. Використовуються як авто-
номні джерела харчування в малогабаритній апаратурі: контрольно-
вимірювальні прилади, годинник, фотоапарати й т.п.
Основу становить електрохімічна система
Zn KOH HgO.
В елементі використаний пористий цинковий електрод і роз-
чин KOH у малому об'ємі. В елементі протікає реакція
2
2
()2
Zn OH
Zn OH
e
с наступним розкладанням гідроксиду цинку на оксид цинку й воду
2
2
2
HgOHOe
HgO OH
.
Для зниження швидкості саморозряду цинкового анода вико-
ристовують особливо чистий цинк, з метою підвищення водневої
перенапруги цинк амальгамують. Оксидно-ртутна маса береться в
надлишку й тому ємність елемента визначається цинковим елект-
родом. У якості електроліту використовують 7–10 М розчин KOH, у
який попередньо вводять оксид цинку для утвору цинкату калію.
Звичайна конструкція елемента – дискового типу, аналогічна конс-
трукції срібно-цинкових елементів. З екологічних міркувань елеме-
нти типу РЦ не є широко розповсюдженими.
ЛІТІЄВИЙ ЕЛЕМЕНТ
Літій відомий як сильний відновник, що володіє серед металів
найбільш високою питомою ємністю (3.83 А·год/г). Проте, створити
літієве джерело струму довгий час не вдавалося, тому що у водяних
розчинах літій активно реагує с протонами, піддаючись швидкому
саморозряду. Тільки після створення апротонних (безводних) елек-
тролітів вдалося виготовити літієві елементи з питомою енергією в
3–5 разів вище, ніж у срібно-цинкових елементів. Оскільки літій в
апротонних електролітах стійкий, термін зберігання літієвих елеме-
3.3. ПЕРВИННІ ДЖЕРЕЛА СТРУМУ
211
нтів надзвичайно високий (10 років). В даний час широке поширен-
ня знайшли елементи на основі літію і діоксиду марганцю. Такі
джерела струму мають ємність 0.17 –1.5 А·год.
Апротонний електроліт являє собою неорганічну сіль літію,
розчинену в апротрнному диполярному розчиннику. У якості солей
літію використовують
4
4
4
4
,
,
,
,
LiClO LiBr LiAlCl LiBF LiAsF . Апро-
тонний розчинник повинен мати такі властивості: малою рухливіс-
тю водню в молекулі розчинника, здатністю розчиняти солі літію,
наявністю дипольного моменту, необхідного для дисоціації солей.
Такі властивості властиві деяким простим і складним циклічним
ефірам (поліпропіленкарбонат, поліеэтиленкарбонат). Розрядна реа-
кція на літієвому аноді зводиться до іонізації літію
Lie
Li
.
У реальному елементі поверхня літієвого електрода завжди
покрита плівкою із продуктів його взаємодії із сіллю й (або) роз-
чинником. Ця плівка підтримує стаціонарний стан літію й охороняє
його від саморозряду, а її катіонна провідність зберігає здатність лі-
тію до розчинення.
Катодна реакція описується рівнянням
2
x
MnO
xLi
xe
MnOOLi
.
Реакція протікає по твердофазному механізму, як і у водяних
розчинах, тільки роль протона тут відіграє катіон літію, що має ма-
лий іонний радіус і відносно високий коефіцієнт дифузії. Швидкість
катодного відновлення діоксиду марганцю обмежується дифузією
літію у твердій фазі, швидкість якої невисока й тому літієві елемен-
ти розраховані на розряд малими струмами.
Літієві елементи системи Li / MnО2 випускаються у вигляді ди-
сків або циліндрів. Тривалість розряду, залежно від призначення,
становить від 20 год до 5 років. Потенціал 2.9 В, діапазон робочих
температур від –20 до +50 °С. Питома енергія лежить у діапазоні
120–225 Вт·год/кг или 400–600 Вт·год/см
3
и залежить від розмірів
елемента і режиму розряду. Літієві елементи характеризуються ни-
зьким саморазрядом – 2–3% в рік.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
212
РЕЗЕРВНІ ЕЛЕМЕНТИ
Хімічні джерела електроживлення, які проводяться й зберіга-
ються в не активованому стані й перед початком розряду активізу-
ються тем або іншим способом називаються резервними джерелами
струму. Основні особливості цих резервних батарей:
–
можливість тривалого зберігання батарей у не активованому
стані;
–
короткий рядків розряду, який проводиться одноразово й
безперервно;
–
використання енергоємних або високоактивних компонентів;
–
високі значення питомої потужності.
Водоактивуємі батареї використовують для харчування метео-
рологічної апаратури, що запускається у верхні шари атмосфери за
допомогою радіозондів або куль-пілотів. Їхнім також застосовують
для харчування освітлювальних обладнань, якими забезпечуються
рятувальні жилети, костюми, плоти й інші засоби для порятунку
людей, потерпілих аварію в море.
Залежно від способу активації всі резервні хімічні джерела то-
ки діляться на чотири типи:
–
джерела струму, що активуються водою (або наливні);
–
хімічні джерела струму, що активуються розчином електро-
літу (або ампульні батареї);
–
газоактивні джерела струму;
–
теплові батареї..
У резервних джерелах харчування, що активують водою, у
якості анодних матеріалів звичайно використовують магнієві спла-
ви, рідше цинк. Катодними реагентами є малорозчинні хлориди срі-
бла, міді й свинцю. У якості електроліту використовується хлорид
натрію, який при активації розчиняється у воді, тим самим забезпе-
чуючи електропровідність. Активація також може проводитися за-
ливанням резервної батарей морської води або зануренням батарей
у морську воду.
Найбільше значення мають три електрохімічні системи з маг-
нієвими електродами:
()/
/
()
Mg NaCl CuCl
,
2
()/
/
()
Mg NaCl PbCl
і
3.3. ПЕРВИННІ ДЖЕРЕЛА СТРУМУ
213
()/
/
()
Mg NaCl AgCl
Прикладом резервного елемента є мідно-магнієвий резервний
елемент
2
2
()
()
Mg MgCl CuCl Cu
Для
2
Mg CuCl
системи поява струму в елементі обумовлене
окисно-відновними реакціями
–
на аноді
(0)
2
()
2
Mg
e
Mg
;
–
на катоді
(0)
()Cu e
Cu
або сумарна реакція має вигляд
2
2
2
Mg CuCl
MgCl
Cu
.
Для резервного Mg AgCl
елемента
2
()
()
Mg MgCl AgCl Ag
поява струму в елементі обумовлене окислювально-відновними ре-
акціями
(0)
2
()
2
Mg
e
Mg
;
(0)
()Ag
e
Ag
с сумарним процесом
2
2
2
Mg AgCl
MgCl
Ag
До переваг наливних батарей резервного харчування став-
ляться простота обладнання й активації, здатність працювати при
низьких температурах після активації, високі (для Mg AgCl
)ісе-
редні (для інших систем) питомі енергія й потужність.
До недоліків батарей резервного харчування можна віднести
високий саморозряд після активації, витоку струму в резервних ба-
тареях і високу вартість джерел струму системи Mg–Agcl. Внаслідок
корозії магнію схоронність магнієвих резервних батарей у залитому
стані не перевищує 48 год, у той час як у герметичному контейнері
без електроліту їх схоронність практично необмежена.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
214
3.4. ВТОРИННІ ДЖЕРЕЛА СТРУМУ (АКУМУЛЯТОРИ)
СВИНЦЕВІ (КИСЛОТНІ) АКУМУЛЯТОРИ
Кислотні акумулятори знайшли найбільше застосування особ-
ливе в тих випадках, коли від акумулятора потрібний великий роз-
рядний струм. Кислотні свинцеві акумулятори є найпоширенішими
серед вторинних джерел струму. Вони мають порівняно високу пи-
тому потужність у комбінації з низькою вартістю й знаходять різ-
номанітне застосування. Своєю популярністю зобов'язані викорис-
танням у якості стартерних батарей у різних засобах пересування.
Електричні й експлуатаційні характеристики кислотного свин-
цевого акумулятора залежать від матеріалу електродів, сполуки еле-
ктролітів і становлять: висока питома потужність ( до 8 Вт/кг); здат-
ність розряджатися більшими струмами ( до 1000 А); теоретична
енергоємність – 133 Вт·год/кг; питома енергія 30–60 Вт·год/кг; Тео-
ретична питома енергощільність 1250 Вт·год/дм
3
; ЕРС зарядженого
акумулятора 2,11–2,17 В; питома потужність 150 Вт/кг; робоча на-
пруга 2 В (3 або 6 секції в результаті дають стандартні 6 В або 12 В);
напруга повністю розрядженого акумулятора 1,75–1,8 В (з розрахун-
ку на 1 секцію); ККД – 90%; тривало зберігають запас енергії; мають
відносно невелику вартість; робоча температура від –40 до +40 °С.
Критерієм працездатності свинцевого акумулятора є здатність заря-
дженої батареї віддавати взимку більше 50% своєї ємності, а в літній
> 75%.
Елемент свинцевого кислотного акумулятора (рис. 3 .4а) скла-
дається з позитивних 1 і негативних 2 електродів зібраних у блоки
(б) розташованих у баку 3, залитим мастикою 4 і розділових ізоля-
торів 5 (сепараторів), які опущені в електроліт: 25–30% водяний
розчин сірчаної кислоти (d = 1,275 г/см
3
), що заливається через за-
ливний отвір 6, що закривається кришкою 7. Блоки пластин з'єднано
з полюсними штирями 8, 9. Зібрані в батареї штирі з'єднуються пе-
ремичками 10, елементи в баку стоять на ребрах 11. На практиці, ча-
стіше в районах з холодним кліматом використовують і більш високі
концентрації сульфатної кислоти, до густини d = 1,29–1,31 г/см
3
.У
батареях для побутових джерел безперебійного живлення рідкий
електроліт загущують до пастоподібного стану водним лужним
3.4. ВТОРИННІ ДЖЕРЕЛА СТРУМУ (АКУМУЛЯТОРИ)
215
розчином силікату натрію Na2Si2O4. Електроди – свинцеві решітки. У
позитивних електродах активною речовиною є оксид свинцю РbO2, у
негативних – губчатий свинець. Електроди виготовлене не із чистого
свинцю, а зі сплаву з додаванням сурми в кількості 1–2% для підви-
щення потужності й інших домішках.
Потенціалутворюючий процес на негативному електроді свин-
цевого акумулятора в сірчаній кислоті має вигляд
4
4
2
Pb HSO PbSO H
e
.
Потенциалутворюючий процес на границі діоксид свинцю –
сульфат свинцю в розчині сірчаної кислоти має вигляд
2
4
4
2
3
2
2
PbO HSO
H
e
PbSO
HO
.
Потенціал свинцевого й діоксид свинцевого електрода збіль-
шується з ростом концентрації електроліту. Оптимальна концентра-
ція сірчаної кислоти 25–30%. Сумарний процес у свинцевому аку-
муляторі при його роботі описується рівнянням
2
2
24
4
2
2
разряд
заряд
PbO PbO HSO
PbSO
HO
і ЕРС акумулятора описується рівнянням
а)
б)
Рис. 3 .4 . Конструкція кислотного свинцевого акумулятора.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
216
24
2
2,303
lg
HSO
o
HO
a
RT
EE
F
a
.
Теорія подвійної сульфатації, згідно з якою кінцевим продук-
том розряду на обох електродах є сульфат свинцю, була запропоно-
вана Д. Гладстоном і А. Трайбом (1882).
Саморозряд свинцевого акумулятора в основному визначаєть-
ся швидкістю саморозчинення свинцю по реакції
24
4
2
2
Pb HSO PbSO
H
.
Під час відсутності сторонніх домішок реакція протікає пові-
льно через високу перенапругу виділення водню на свинці. Практи-
чно у всіх металів, що є легуючими добавками (домішок) значення
водневої перенапруги нижче, чим у чистого свинцю. Тому поява
цих металів на поверхні негативного електрода збільшує самороз-
ряд. Підсилюється він і при підвищенні температури й концентрації
електроліту. На практиці дія багатьох домішок перекривається дією
сурми, зміст якої у свинці доходить до 0,6%. Використання деяких
органічних речовин (α-нафтол) у якості інгібіторів саморозряду за-
сноване на їхній адсорбції на електроді, що приводить до росту во-
дневої перенапруги, що еквівалентно зниженню швидкості саморо-
зчинення свинцю. Росту саморозряду сприяє й розчинений кисень
2
24
4
2
1
2
Pb
O HSO PbSO HO
.
Саморозряд позитивного електрода обумовлений, як правило,
мимовільним відновленням діоксиду свинцю по реакції
2
24
4
2
2
1
2
2
PbO H SO PbSO
HO
O
.
Інша причина втрати ємності – прямий контакт позитивної ак-
тивної маси з матеріалом решітки, у результаті чого протікає процес
утворення сульфату свинцю.
Необоротна сульфатація пластин пов'язана з рекристалізацією
сульфату свинцю, у результаті чого утворюється суцільний крупно-
кристалічний шар сульфатного осаду. Заряд таких пластин відбува-
ється значно сутужніше, тому що швидкість розчинення таких кри-
сталів, на відміну від мікрокристаликів, мала. При цьому ємність
електродів різко падає.
3.4. ВТОРИННІ ДЖЕРЕЛА СТРУМУ (АКУМУЛЯТОРИ)
217
ЛУЖНІ НІКЕЛЬ-ЗАЛІЗНІ Й НІКЕЛЬ-КАДМІЄВІ
АКУМУЛЯТОРИ
Лужні акумулятори в багатьох випадках є більш зручними,
чому свинцеві. Вони більш стійки до тряски й поштовхам, розря-
джати їх можна струмами більшої сили, вони стійкі навіть до корот-
кочасного короткого замикання. Їх можна залишати довгочасно у
вирядженому стані. Однак, вони мають менший коефіцієнт корис-
ної дії й меншу ЕРС
Лужні нікель-залізні (НЖ) акумулятори посідають друге міс-
це серед вторинних джерел струму по масштабах виробництва. Во-
ни мають великий ресурс – 2000–3000 циклів, характеризуються ви-
сокою механічною міцністю, простотою обслуговування, дешеви-
ною сировини. Це забезпечило їхнє широке використання як тягові
акумулятори на електрокарах, навантажувачах, рудничних електро-
возах, залізничному транспорті.
Електрохімічна система залізо-нікелевого НЖ акумулятора яв-
ляє собою елемент
Fe KOH NiOOH Ni .
У сталевому нікельованому
корпусі 1 (рис. 3 .5) перебуває акти-
вна маса позитивних пластин 2, що
полягає з гідроксиду нікелю(III) з
домішкою певного кількості графіту
для збільшення електропровідності.
Активна маса негативних пластин 3
складається з губчатого заліза. Для
запобігання замикання пластин між
ними вставлені ебонітові палички 4.
Електролітом, який заливається че-
рез отвір 5 із запобіжною пробкою є
20 % розчин КІН з домішкою гідро-
ксиду літію (20–30 г/л), який збіль-
шує термін служби акумулятора й
розширює діапазон робочих темпе-
ратур ( від –20 до +40 °С).
Рис.3.5 . Схема конструкції залі-
зо-нікелевого акумулятора.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
218
Сумарне рівняння електродних процесів має вигляд
2
2
2
2()
()
розряд
заряд
Fe NiOOH
NiOH FeOH
.
Електричні й експлуатаційні характеристики залізо-нікелевого
акумулятора: теоретична енергоємність 350 Вт·год/кг; ЕРС – 1,4 В;
питома енергія 50 Вт·год/кг; питома потужність 100 Вт/кг; термін
служби до 40 лет; число циклів перезаряду 1000.
У сучасних залізо-нікелевих акумуляторах КПД рівняється 55–
65 %. З погляду вартості й питомої енергоємності, залізо-нікелеві
акумулятори близькі до літій-іонним акумуляторам, а з погляду са-
морозряду, ефективності й величині напруги – до нікель-
металгідридним. Це досить витривалі акумулятори, стійкі до грубо-
го обігу (перезаряд, глибокий розряд, коротке замикання, термічні
удари, вібрація й струсу), і з довгим терміном служби.
Електрохімічна система нікель-кадмієвого акумулятора має
вигляд
Cd KOH NiOOH Ni .
При розряді на негативному електроді протікає реакція
2
2
()2
Cd
OH
Cd OH
e
,
на позитивному
3
2
2()2
2()
2
Ni OH
e
Ni OH
OH
.
Сумарно процес зарядки-розрядки можна представити рівнян-
ням
2
2
2
2
2
2()
()
розряд
заряд
Cd NiOOH HO
NiOH CdOH
.
Лужні нікель-кадмієві (НК) акумулятори в порівнянні із НЖ
мають кращу працездатність в умовах підвищеного струмового на-
вантаження й малим саморозрядом.
Конструкція нікель-кадмієвих акумуляторів аналогічна конс-
трукції залізо-нікелевих. Електричні й експлуатаційні характерис-
тики нікель-кадмієвого акумулятора: можливість швидкого й прос-
того заряду, навіть після тривалого зберігання акумулятора; велика
кількість циклів заряд / розряд : при правильній експлуатації –
3.4. ВТОРИННІ ДЖЕРЕЛА СТРУМУ (АКУМУЛЯТОРИ)
219
більш 1000 циклів; гарна здатність до навантажень і можливість
експлуатації при низьких температурах; тривалий строк зберігання
при будь-якому ступені заряду; зберігання стандартної ємності при
низьких температурах; низька вартість. Номінальна напруга герме-
тичних нікель-кадмієвих акумуляторів – 1,2 В; питома енергія 50
Вт·год/кг; питома потужність 200 Вт / кг; термін служби 12 років;
число циклів перезарядження 1000. При зарядці лужних акумулято-
рів ЕРС на електродах швидко зростає до 1.65 В і потім повільно
піднімається до 1,8 В. ЕРС зарядженого акумулятора через 1–2 го-
дини після зарядки падає до 1,65 В. При розрядці вона швидко зни-
жується до 1,25 В, а потім повільно опускається до 1,0 В. Розрядку
лужних акумуляторів рекомендується вести до ЕРС не нижче 1,1 В.
Для нікель-кадмієвих акумуляторів необхідний повний періо-
дичний розряд. Якщо його не робити, на пластинах елементів фор-
муються великі кристали, що значно знижує їхня ємність (так зва-
ний «ефект пам'яті»).
Недоліки Ni–Cd акумулятора: відносно низька в порівнянні з
іншими типами акумуляторних батарей енергетична щільність; вла-
стивий цим акумуляторам ефект пам'яті й необхідність проведення
періодичних робіт з його усунення; токсичність застосовуваних ма-
теріалів, які негативно впливають на навколишнє природне середо-
вище. До недоліків можна віднести й відносно високий саморозряд
(після зберігання необхідний цикл заряду); неможливість розряджа-
тися більшими струмами, неможливість тривалий строк зберігати
запас енергії.
Лужні акумулятори дорожче свинцевих, при цьому питомі по-
казники в них нижче.
НІКЕЛЬ-МЕТАЛОГІДРИДНІ АКУМУЛЯТОРИ
Загальна схема Ni MH
акумулятора має вигляд
()
()
MH KOH NiOOH Ni
Нікель-металогідридні акумулятори представляють із себе со-
бою вдосконалену метал-водневу систему, у якій водень на негати-
вному електроді в процесі заряду абсорбується активним каталітич-
ним матеріалом і перетворюється в гідрид. Використання водню у
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
220
вигляді гідридів металів дозволило знизити вагу й об'єм акумулято-
рних батарей, а також значно зменшити й небезпека вибуху батареї
при перегріванні.
Нікель-металогідридні батареї в останні десятиліття суттєво
потіснили нікель-кадмієві акумулятори в багатьох галузях техніки.
Особливо широко вони використовуються в автономних джерелах
харчування портативної апаратури, збільшення їх питомих характе-
ристик в 1,5–2 раз у порівнянні з нікель-кадмієві привело до поліп-
шення споживчих властивостей апаратури. Однак Ni Cd
іMH
джерела струму мають багато загального, оскільки саме позитивний
оксидно-нікелевий електрод визначає як розрядну ємність акумуля-
тора, так і його властивості. Переваги Ni MH
батарей: більша єм-
ність (на 40 % більше, чим у звичайної Ni Cd
батареї); відсутність
«ефекту пам'яті»; просте транспортування; екологічно безпечні,
можлива повторна переробка.
Найбільше
поширення знайшли
циліндричні
нікель-
металогідридні акумулятори (рис.
3.6). Корпус 1 закритий герметизу-
ючою кришкою 2, яка має запобіж-
ний клапан 4 з ковпачком 3, який
спрацьовує при тиску 2–4 МПа за
умови неправильної експлуатації
акумулятора. Позитивний 9 і нега-
тивний 7 електроди розділені сепа-
ратором 8, який скручений у вигляді
рулону й установлений у корпус че-
рез ізолятор 10. Позитивні електро-
ди з'єднані колектором 5.
ВNiMH
акумуляторах у
якості позитивного електрода вико-
ристовується
оксидно-нікелевий
електрод, як і в нікель-кадмієвому
акумуляторі. На негативному елект-
роді метал з абсорбованим воднем перетворюється в металогідрид
2
заряд
рoзряд
MHO
MH OH
Рис. 3 .6 . Конструкція нікель-
металогідридних акумуляторів.
3.4. ВТОРИННІ ДЖЕРЕЛА СТРУМУ (АКУМУЛЯТОРИ)
221
На позитивному оксидно-нікелевому електроді Ni MH
аку-
мулятора протікає реакція
2
2
()
заряд
рoзряд
NiOH OH
NiOOH H O
Загальна реакція в Ni MH
акумуляторі записується в насту-
пному виді
2
()
заряд
рoзряд
Ni OH
M
NiOOH MH
Електроліт не бере участь у загальній струмоутворюючої реа-
кції. Після втрати 70–80 % ємності й при надмірному заряді на ок-
сидно-нікелевому електроді починається виділятися водень
2
2
2
2
1/2
перезаряд
OH
e
OHO
,
який відновлюється на негативному електроді
2
2
1/2
2
2
перезаряд
OHOe
OH
.
Дві останні реакції забезпечують замкнений кисневий цикл.
При відновленні кисню забезпечується ще й додаткове підвищення
ємності металогідридного електрода за рахунок утворення групи
OH
.
Головним матеріалом, який визначає характеристики NiMH
акумулятора, є абсорбуючий для водню сплав, який може поглина-
тися об'єм водню, який в 1000 раз перевищує свій власний об'єм. В
останні роки найбільшого поширення придбали поліметалеві гідри-
дні композиції
5
LaNi ,
2
Mg Ni,
2
ZnMn , які використовуються в якос-
ті негативного електрода.
Електричні й експлуатаційні
характеристики
нікель-
металогідридного акумулятора: теоретична енергоємність 300
Вт·год/кг; ЕРС – 1,25 В; питома енергія 80 Вт·год/кг; питома поту-
жність 240 Вт/кг; термін служби 12 років; число циклів перезаря-
дження – тисячі.
У той же час никель-металогідридні акумулятори мають ряд
характерних недоліків: високий саморозряд батарей; обмежену єм-
ність – при перевищуючих припустимих навантаженнях зменшуєть-
ся час життя батарей; необхідність спеціального зарядного облад-
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
222
нання зі стадійним алгоритмом заряду, оскільки при заряді виділя-
ється велика кількість тепла й нікель-металогідридні батареї погано
переносять перезаряд; погану працездатність при високих темпера-
турах (понад 25 ÷ 30 °С).
ЛУЖНІ СРІБНО-ЦИНКОВІ (СЦА) АКУМУЛЯТОРИ
Срібно-цинкові акумулятори вкрай привабливі для спожива-
чів, тому що посідають перше місце по питомій енергії серед дже-
рел струму з водним електролітом. Їхня теоретична (максимальна)
питома енергія становить 465 Вт·год/кг, а фактична: 80–100
Вт·год/кг. Однак ресурс таких акумуляторів дуже низький. При
тривалих режимах розряду 30–60 циклів, при коротких – 8 циклів.
Їх використовують в радіо і телеапаратурі, контрольно-
вимірювальних та медичних приладах, в аерокосмічній і військовій
техніці.
У СЦ акумуляторах використовується електрохімічна система
2
2
2
(или
)
Zn KOH KZnO AgO
AgO Ag
.
ЕРС цього акумулятора (1,85 В. Як і в подібних первинних
джерелах струму, у них застосовують пористий цинковий електрод,
що володіє високою електрохімічною оборотністю. Продуктами ро-
зряду є металеве срібло й оксид (гідроксид) цинку. Струмотворюю-
чий процес описується сумарною реакцією
рoзряд
заряд
AgO Zn
Ag ZnO
.
Розряд позитивного електрода протікає по складному механізму.
На першій стадії потенціалвизначаючою є реакція
2
2
2
2
2
AgOHOe
AgO OH
.
Після того як поверхня зерен активної речовини покриється
тонким шаром оксиду срібла(I), потенціалвизначаючою стає реакція
2
2
2
2
2
AgOHOe
Ag OH
,
хоча ємність електрода визначається як і раніше фазою оксиду сріб-
ла(II).
3.4. ВТОРИННІ ДЖЕРЕЛА СТРУМУ (АКУМУЛЯТОРИ)
223
Розряд негативного електрода складний і може бути описаний
багатоступінчасто за схемою
2
4
2
24
[()]
()
Zne
OH
Zn OH
Zn OH
ZnO
.
Таким чином, анодне розчинення цинку в лужних електролітах
супроводжується утвором цинкатних розчинів підвищеною проти
рівноважною концентрацією цинкатів. Утвір пересичених цинкат-
них розчинів приводить до розкладання цинкатного комплексу ок-
сид, що випадає в осад, цинку характеризується пухкою структурою.
Заряд срібного електрода протікає також двоступенево: на по-
чатковому етапі срібло окислиться до оксиду срібла (I)
2
2
2
2
2
Ag OH
AgOHOe
і після утвору оксидної плівки Ag2O відбувається процес утвору ок-
сиду срібла (II)
2
2
2
Ag OH
AgO HO
e
.
Цей процес при заряді є переважним.
Основна причина втрати ємності – мимовільне розчинення ци-
нку в лузі. Уведення в цинковий електрод металевої ртуті амальга-
муванням ( до 2%) різко знижує швидкість саморозряду. Строк схо-
ронності СЦА в сухому у вигляді становить 3–5 років. Однак після
зберігання ресурс акумулятора знижується на 20–30% через дегра-
дацію сепаратора. Саморозряд СЦА рівний 3–6 % на місяць.
Позитивною якістю СЦА слід уважати його працездатність в
області низьких температур і здатність до саморозігріву. При тем-
пературах –20, –30 і –40 °С СЦА зберігає відповідно 70, 40 і 20%
номінальної ємності.
НІКЕЛЬ-ЦИНКОВИЙ АКУМУЛЯТОР
Нікель-цинкові акумулятори (НЦА) по питомих характеристи-
ках посідають друге місце серед акумуляторів з водними електролі-
тами, уступаючи тільки срібно-цинковим акумуляторам. Практично
досягнута питома енергія НЦА становить 60–70 Вт·год/кг.
Струмоутворююча реакція в НЦА
2
2
2
2
2
2()
()
разряд
заряд
Zn
NiOOH H O
Ni OH
Zn OH
.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
224
На негативному електроді при розряді протікають реакції
2
2
4
4
2
()2
Zn OH
HO
Zn OH
е
,
2
4
2
()
()2
Zn OH
Zn OH
OH
При заряді ці реакції протікають у зворотному напрямку.
На позитивному електроді при розряді й заряді протікають ре-
акції
2
()
рoзряд
заряд
NiOOH H
Ni OH
,
Реакція на негативному електроді визначає його оборотність.
Враховуючи. що потенціал цинку на 0.5 В негативного потенціалу
водневого електрода, заряд цинкового електрода супроводжується
виділенням водню. З метою зниження швидкості виділення водню
до складу цинкового електрода вводять добавки інгібіторів Hg , Pb ,
Cd . Застосування таких інгібіторів дозволяє підвищити ефектив-
ність використання зарядного струму до 90–95%. З іншого боку, не-
обхідно виключити домішки з низькою водневою перенапругою
(Fe, Ni).
При заряді на нікелевому електроді виділяється кисень, при-
чому швидкість виділення кисню зростає в міру збільшення ступені
зарядженості позитивного електрода.
Так само як у СЦА, для забезпечення стабільної роботи НЦА й
компенсації втрати негативної активної маси в процесі циклювання
потрібен надлишок останньої. Співвідношення ємності позитивних і
негативних електродів і НЦА становить 1:3.
Як і всі акумулятори із цинковим електродом, НЦА мають не-
великий термін служби.
Розрядна напруга НЦА рівно 1.6 –1.65 В, а напруга розімкнуто-
го ланцюга – 1 .8 В. Номінальні струми 0.5 –1.0 Сном,. КПД по ємно-
сті становить 85–90%, а по енергії – 85–88%. Діапазон робочих тем-
ператур: від –40 до 50 °С. При –40°С ємність НЦА становить 30%
від номінальної ємності й обмежується пасивацією цинкового елек-
троду, питома енергія, що практично досягається, НЦА рівна 50–60
Вт·год/кг. Ресурс НЦА не великий, складає лише 50–60 циклів.
3.4. ВТОРИННІ ДЖЕРЕЛА СТРУМУ (АКУМУЛЯТОРИ)
225
ЛІТІЙ-ІОННИЙ АКУМУЛЯТОР
Використовувати літій як активний матеріал в акумуляторних
системах перспективно для використання його високого енергетич-
ного потенціалу. Теоретична питома ємність літію становить 3830
мА·год/г. Однак при заряді й катодному осадженні літію утворю-
ється дуже активна поверхня, на якій наростає пасивна плівка, що
поступово виводить літій з подальших електродних процесів. За-
кладка надлишку літію значно знижує питомі характеристики аку-
мулятора. Крім того, при осадженні літію утворюється дендритний
осад, що сприяє утвору коротких замикань і, як наслідок цього, ви-
никненню аварійних ситуацій (загоряння, зривши). З метою зни-
ження активності літію пропонувалося використовувати сплави, на-
приклад літій-алюміній. Однак такий розв'язок, поряд зі значним
зниженням питомих характеристик, створює нові проблеми, основ-
ний з яких було охрупчення й опадання електрода при розрядах.
Інший спосіб зниження активності літію пов'язаний із застосу-
ванням електродних матеріалів, здатних оборотне інтеркалірувати
іони літію. Такими матеріалами є вуглецеві композиції й оксиди ря-
ду металів (кобальт, марганець, нікель).
Основна струмоутворююча реакція в літій-іонному акумуля-
торі може бути представлена у вигляді
2
(1)
2
()
LiMeO Li C
LiMeOLiC
/
При заряді протікають процеси
2
(1)
2
заряд
x
LiCoO
xLi
xe
Li CoO
,
6
6
заряд
C xLi
xe
LiC
.
а при розряді :
(1)
2
2
рoзряд
Li CoO
xLi
xe
LiCoO
,
6
6
рoзряд
LiC xLi
xe
C
.
При розряді мають місце стадії розчинення іонів літію на ано-
ді, міграції їх в електроліті й впровадження їх у кристалічну струк-
туру катода. Одночасно відбувається процес міграції компенсацій-
них електронів через зовнішній ланцюг і їх впровадження в ту ж
структуру. При заряді процес іде в протилежному напрямку. Таким
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
226
чином, при заряді й розряді відбувається переміщення х-іонів літію
з одного електрода на іншій. За зовнішньою простотою процесів
криються величезні конструкторські й технологічні проблеми, ос-
новними з яких є вибір матеріалів, здатних оборотне інтеркалірува-
ти (впроваджувати) літій протягом великої кількості зарядно-
розрядних циклів, а також пошук електроліту з низьким омічним
опором, високою рухливістю іонів літію й електрохімічною стабі-
льністю.
До позитивних характеристик літій-іонних акумуляторів слід
віднести: високу питому енергією (100–150 Вт·год/кг и 150–200
Вт·год/л); великий ресурс (500–1000 циклів); високу розрядну на-
пругу 3,6 В; низький саморозряд (1–2% в місяць), широкий діапазон
робочих температур (–20 ÷ 60 ° С).
ЛІТІЄВИЙ АКУМУЛЯТОР З ПОЛІМЕРНИМИ
ЕЛЕКТРОЛІТАМИ
Переваги літієвих акумуляторів з полімерними електролітами
(ЛІАПЕ) полягають у тому, що такі акумулятори мають високу пи-
тому енергію, компактні, не містять рідких електролітів і можуть
бути виготовлені у формі тонких і гнучких аркушів.
Основним недоліком є непрацездатність при низьких темпера-
турах. Анод відділений від катода полімерною перегородкою, ком-
позитним матеріалом, таким як поліакрілонітрит, який містить літіє-
ву сіль. У результаті стає можливим спрощення конструкції елемен-
та, оскільки будь-який витік гелеподібного електроліту неможливо.
Питома енергія літій-іонних акумуляторів з полімерним елект-
ролітом становить 150–200 Вт·год/кг або 300 Вт·год/дм
3
.
Робоча
напруга – 2,7 В. Ресурс при глибинного цикліювання 80% складає
150 циклів. Саморозряд – 1 –2% на місяць.
Основою полімерного електроліту головним чином є поліети-
леноксид (ПЕО) і добавкою солей літію (
4,
4
LiClO LiBF і ін.). При
температурі нижче 30° система ПЕО-сіль літію є структурованою і
її провідність украй низька. При підвищенні температури відбува-
ється аморфізація ПЭО (часткове плавлення), і провідність системи
росте. Таким чином, система ПЕО-сіль літію в робочому стані скла-
дається із твердої структурованої частини, яка забезпечує механічну
міцність і аморфної частини, здатної розчиняти й дисоціювати солі
літію.
Робота No 12. ВИЗНАЧЕННЯ КОЕФІЦІЄНТА ВІДДАЧІ АКУМУЛЯТОРА
227
Робота No 12.
ВИЗНАЧЕННЯ КОЕФІЦІЄНТА ВІДДАЧІ АКУМУЛЯТОРА
Ціль роботи: Визначити розрядну й зарядну ємність і коефі-
цієнт віддачі (корисної дії) акумулятора по струму й по енергії.
Прилади й реактиви: акумулятор, джерело постійного
струму (випрямляч), реостат, вольтметр (точність виміру не гір-
ше 0,02 В), амперметр (точність виміру не гірше 0,02 А).
Для дослідження можуть бути взяті як кислотний, так і луж-
ний акумулятор. Ємність акумулятора й коефіцієнт віддачі визнача-
ється по експериментальних кривих заряду й розряду. Енергія,
отримана при заряді або віддана при розряді акумулятора виража-
ється через параметри ланцюга: напруга на клемах акумулятора U ,
силу струму, що проходить через акумулятор I і час процесу , у
секундах
0
() ()
WIUd
.
Напруга й сила струму змінюються в ході процесу, тому, щоб
розрахувати енергію заряду або розряду, одна з характеристик по-
винна підтримуватися постійної, наприклад, сила струму.
При постійній силі струму площа під кривою розряду MNCB
відповідає кількості електричної енергії, отриманої від акумулятора,
а)
б)
Рис. 3.7. Типові криві ( )
U заряду (а) и розряду (б) акумулятора.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
228
а площа під кривою заряду MNDA – енергії, поглиненої акумулято-
ром. Перша площа рівна
0
()
р
р
р
WI
Ud
.
Друга площа визначається
0
()
з
з
з
W
IUd
,
дер
I й зI – сила струму при розряді й заряді, відповідно; ( )
U–
функція зміни напруги від часу, обумовлені експериментально.
На малюнку 3.7 представлена залежність напруги акумулятора
як функція часу при постійній силі струму. Визначення коефіцієнта
віддачі акумулятора, як правило, проводять заряджаючи акумулятор
від початкової напруги до максимального зарядного, а розряджаючи
від кінцевої напруги до мінімального розрядного.
Якщо 1
=2
, то відношення
/рз
W
WWK
–
представляє кое-
фіцієнт віддачі акумулятора по енергії.
Коефіцієнт віддачі по струму може також бути визначений з
відношення кількості металу, виділеного на мідному або срібному
кулонометрі при заряді й розряді або при відомій фіксованій силі
струму в цих процесах.
ВИКОНАННЯ РОБОТИ
Роботу починають із огляду акумулятора, перевіряють ареоме-
тром густину електроліту. Для лужного акумулятора вона повинна
бути л
= 1,10 г/см
3
, для зарядженого кислотного акумулятора в
межах к
= 1,18–1,28 г/см
3
. Перевіряють рівень електроліту над вер-
хнім краєм пластини, який повинен бути не менш 15 мм. Рівень ви-
значають за допомогою занурення скляної трубки в електроліт до
зіткнення із пластинами. Закривши пальцем отвір трубки й вийняв-
ши її з акумулятора, оцінюють рівень електроліту. Для проведення
експерименту збирають схему, зображену на рисунку 3.8, у якій по-
слідовно приєднані зовнішнє джерело постійного струму 1, акуму-
Робота No 12. ВИЗНАЧЕННЯ КОЕФІЦІЄНТА ВІДДАЧІ АКУМУЛЯТОРА
229
лятор 2, амперметр 3 і реостат
4. Паралельно акумулятору пі-
дключається вольтметр 5. Пе-
ремикач 6 ставлять у положен-
ня розряд або заряд, строго до-
тримуючи полярності приєд-
нання.
Роботу починають із роз-
рядки акумулятора, якщо він
заряджений, й із зарядки – як-
що розряджений. Перевірку
стану акумулятора обов’язково
проводять в умовах його нава-
нтаження – при здійсненні роз-
ряду при навантаженні розрядним струмом. Повністю зарядженим
уважається кислотний акумулятор, якщо напруга на ньому більш 2,1
В, лужний, якщо напруга на ньому близько 1,45 В. Вирядженими
вважаються акумулятори з напругою менш 1,7–1,8 В и 1,0–1,1 В від-
повідно.
Припустима сила зарядного струму не повинна перевищувати
0,25 від ємності акумулятора в (А·годинах); сила розрядного струму
повинна бути вдвічі менше зарядного. Якщо в процесі розряду на-
пруга менше номінального й помітне падає, акумулятор уважається
вирядженим і роботу починають із зарядки. Якщо акумулятор заря-
джений і здатний підтримувати розрядний струм без істотного па-
дання напруги процес розряду продовжують і починають через ко-
жні 5 хвилин фіксувати вольтметром зміна напруги на клемах аку-
мулятора при підтримці постійної сили розрядного струму. Розряд
роблять до досягнення розрядної напруги
.
к
U.
Аналогічно проводять процес заряду. Для цього послідовно
акумулятору підключають випрямляч, строго дотримуючи поляр-
ності підключення. Напруга випрямляча перевищує ЕРС акумуля-
тора, а напрямок струму протилежно напрямку при його розряді у
випадку мимовільного протікання струмоутворюючої хімічної реа-
кції. У результаті протікання через акумулятор електричного стру-
му відбувається утвір із продуктів реакції вихідних активних мас.
Підключення амперметра вимагає зміни його полярності, тому що
Рис. 3 .8 . Принципова схема для ви-
вчення коефіцієнту віддачі акумуля-
тора.
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
230
при заряді міняється напрямок електричного струму. Характерною
ознакою закінчення зарядки є досягнення на клемах вольтметра на-
пруги 2,65–2,75 В (кислотний) або 1,7–1,8 В (лужний) і рясне газо-
виділення на обох електродах (акумулятори "киплять"). При зарядці
лужних акумуляторів на електродах незабаром після початку про-
цесу спостерігається газовиділення внаслідок розкладання води. У
випадку лужного акумулятора постійне збільшення напруги, виді-
лення газів раніше закінчення заряду й відсутність зміни щільності
електроліту в процесі заряду утрудняють визначення кінця зарядки.
Основною ознакою кінця заряду слід уважати кількість електрики,
обумовлена силою зарядного струму й часом його пропущення.
Визначення енергії, відданої при розряді й отриманої при за-
ряді акумулятора, роблять на підставі залежності напруги акумуля-
тора від часу при постійній силі струму, значення якого підтриму-
ється незмінним за допомогою реостата протягом усього досвіду.
Ознакою закінчення заряду є різке підвищення напруги в ланцюзі.
Рекомендована форма запису результатів
Сила зарядного струму, з
І А: ........
Сила розрядного струму, р
І,А:.......
Час
заряду
Напруга при
заряді,
з
U,В
Час
розряду
Напруга при
розряді, р
U,В
Коефіцієнт віддачі (корисної дії) по струму визначається від-
ношенням кількості електрики, отриманого при розряді до кількості
електрики, що витрачена на заряд. Якщо час процесів заряду й роз-
ряду взяте однаковим, а сила струму підтримувалася постійної, то
коефіцієнт корисної дії по струму IK дорівнює відношенню сили
струму розряду до сили струму заряду,
/
I
рз
KII
.
Коефіцієнт віддачі (корисної дії) акумулятора по енергії ви-
значається відношенням енергії, що виділився при розряді, до енер-
гії, витраченої на заряд акумулятора. Якщо сила струму під час цих
процесів підтримувалася постійної, то кількість енергії, витраченої
Робота No 12. ВИЗНАЧЕННЯ КОЕФІЦІЄНТА ВІДДАЧІ АКУМУЛЯТОРА
231
на заряд акумулятора й отриманої в результаті розряду, визначається
інтегруванням отриманих експериментальних залежностей U() – .
Тому що форма розрядної й зарядної кривих важко піддається
аналітичному опису, інтегрування проводиться графічно. Найбіль-
ше зручно робити інтегрування за допомогою методу зважування
ділянок графіка. Для цього графіки розряду й заряду виконують на
тому самому аркуші міліметрового паперу. Масштаб по осі ординат
вибирають відповідно до точності виміру напруги (1 мм відповідає
0,001 В) і відповідно до розмаху залежності. Далі вирізують ділянки
графіка у вигляді криволінійних трапецій і зважують їх на аналітич-
них вагах. Також зважують прямокутна ділянка приблизно такої ж
площі й вирізаний з тієї ж паперу, який є еталоном, тому що його
площа досить точно обчислюється добутком сторін. Площі графіків (у
см
2
) під кривими розряду й заряду знаходять як величини пропорційні
масам вирізаних ділянок графіка й еталона. До площі криволінійних
трапецій додають площі, що доповнюють до початку координат пря-
мокутників враховуючи обраний масштаб побудови графіків. Для
одержання значення інтеграла, вираженого в В·с, загальні площі під
кривою, певні в одиницях площі (см
2
), множать на коефіцієнт відпо-
відний одиницям масштабу графіка для площі в 1 см
2
.
Більш точними є методи чисельного інтегрування, серед яких
найпростішим є метод трапецій, а най поширеним метод Сімпсона.
За методом трапецій інтервал зміни аргументу [ , ]
a b розбива-
ється на довільну кількість
рівних частин ( n ), а експери-
ментальна крива апроксиму-
ється такою ж кількістю ло-
маних. Тоді площа під криво-
лінійною трапецією може бу-
ти приблизно виражена су-
мою площини n трапецій
(рис. 3 .9). Площа трапеції
дорівнює добутку півсуми
основ на висоту, тому форму-
ла для всією площі під кривої
визначається формулою
Рис. 3 .9 . Ілюстрація чисельного інтегру-
вання за методом трапецій
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
232
1
()
()()
2
b
n
i
i
i
a
h
fxdx
fxfxh
()()
()(2)...
2
2
h
h
fafah
fahfah
((1))()
2
h
fanhfb
1
() ()
(
)
2
n
i
h
fafbhfaih
,
де
ba
h
n
.
У методі Сімпсона певний інтеграл представляється у ви-
гляді площі під кривою, що відображає функціональну залежність.
Вона розбивається на n елементарних площ фігур, які є криволіній-
ними трапеціями, підставами яких є вісь абсцис, і які обмежені ду-
гою інтегрованої функції. Ці площі можна обчислити за формулою
1
0
21
2
2
()
4()2()()
3
b
n
n
i
i
n
i
i
a
h
fxdx
y
fx
fxfx
.
Наведені аналітичні методи інтегрування легко застосовують-
ся при обробці даних проведеного експерименту якщо виміри на-
пруги були проведені через однаковий проміжок часу.
–
з однако-
вим кроком інтегрування.
По рівняннях
0
()
р
р
р
WIUd
і
0
()
з
з
з
WIUd
,
дер
I,з
I – струми розряду й заряду, розраховують значення р
Wй
.
з
W – розрядної й зарядної ємності в Дж. Коефіцієнт віддачі акуму-
лятора по енергії розраховують зі співвідношення
p
W
з
W
K
W
.
Слід помітити, що дослідження дійсних характеристик акуму-
лятора вимагає тимчасових витрат, що значно перевищують ауди-
торний час лабораторного практикуму. Тому дане дослідження, що
проходить в обмеженому за часом процесах розряду й заряду тільки
імітує методику вивчення.
Робота No 12. ВИЗНАЧЕННЯ КОЕФІЦІЄНТА ВІДДАЧІ АКУМУЛЯТОРА
233
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Які вимоги пред'являють до гальванічних елементів, вико-
ристовуваних у якості хімічних джерел струму?
2. Дайте визначення хімічного джерела струму.
3. Чим різняться первинні й вторинні джерела струму. Приве-
діть приклади.
4. Які вимоги пред'являють до вторинних джерел струму?
5. Дайте визначення основних електричних характеристик хі-
мічних джерел струму.
6. Чим різняться поняття ЕРС і напруги хімічного джерела
струму.
7. Як змінюється напруга і ємність при послідовній і парале-
льній сполуці гальванічних елементів. Дайте пояснення.
8. Від яких параметрів джерела струму залежить його ємність
9. У чому принципова відмінність сольових і лужних марган-
цево-цинкових елементів.
10.Опишіть хімічні процеси, що йдуть при розряді ртутно-
цинкового та срібно-цинкового елементів.
11. Чому по щільності розчину сірчаної кислоти можна судити
про стан свинцевого акумулятора?
12.Що розуміють під сульфатацією кислотного акумулятора і
які її причини?
13. Дайте порівняльний аналіз характеристик свинцевого й за-
лізо-нікелевого акумуляторів. Чим обумовлена область їх застосу-
вання.
14. Які фактори впливають на саморозряд свинцевого акуму-
лятора?
15. Обґрунтуйте оптимальну густину електроліту свинцевого
акумулятора. Чому в стартерних батареях оптимальна густина кис-
лоти 1,28 г/см
3
, а в стаціонарних 1,20 г/см
3
?
16. Які умови забезпечують створення герметизированого
свинцевого акумулятора?
17. Що означає поняття «пам'яті» для нікель-кадмієвих акуму-
ляторів.
18. Якими перевагами й недоліками мають лужні срібно-
цинкові акумулятори?
3. ХІМІЧНІ ДЖЕРЕЛА СТРУМУ
234
19. Як по кривих заряду й розряду визначають кількість отри-
маної й відданої акумулятором енергії?
20. Поясните полярність приєднання зовнішнього джерела пос-
тійної напруги при заряді акумулятора.
21. Які ознаки закінчення заряду кислотного й лужного акуму-
ляторів?
22. Який зміст коефіцієнта віддачі акумулятора?
23. Укажіть відомі вам способи інтегрування експерименталь-
них залежностей.
24. Поясните, як при проведенні інтегрування за допомогою
методу зважування ділянок графіка визначають значення енергії,
що виділився при розряді, і енергії, отриманої при заряді акумуля-
тора, в одиницях енергії?
25. Зрівняєте між собою характеристики залізного й кадмієво-
го електродів лужних акумуляторів. Які переваги й недоліки має
кожний з них?
26. Які електроди визначають термін служби НЖ, НК, СЦ, НЦ
акумуляторів? Дайте обґрунтування.
27. Які вимоги пред'являються до розчинників і електролітам у
літієвих джерелах струму.
28. Перелічите основні переваги літій-іонних і літій полімер-
них джерел струму.
29. Опишіть принцип роботи паливних елементів. Чим визна-
чається ємність паливних елементів.
30. Які акумулятори можуть бути виконані в герметичній
конструкції.
ЛІТЕРАТУРА
1. Багоцкий B. C . Химические источники тока / B. C . Багоц-
кий, A. M. Скундин. – М.: Энергоиздат, 1981. – 360 с.
2. Варыпаев В. Н., Дасоян М. А., Никольский В. А. Химиче-
ские источники тока. – М. : Высш. школа, 1990. – 240 с.
3. Кромптон Т. Первичные источники тока – М. : Мир, 1986. –
328 с.
4. Кромптон Т. Вторичные источники тока. М . : Мир, 1985.
–
302 с.
Робота No 12. ВИЗНАЧЕННЯ КОЕФІЦІЄНТА ВІДДАЧІ АКУМУЛЯТОРА
235
5. Коровин, Н. В . Электрохимическая энергетика / H. В . Коро-
вин. – М. : Энергоатомиздат, 1991. – 264 с.
6. Химические источники тока : Справочник / Под ред. Н. В.
Коровина и A. M. Скундина. – М. : Изд–во МЭИ, 2003. – 739 с.
7. Скорчелетти В. В . Теоретическая электрохимия.
–
Л.:Хи-
мия. 1969.
–
С. 318–325.
8. Таганова А. А. Герметичные химические источники тока / А.
А. Таганова, Ю. И. Бубнов, С. Б. Орлов. – СПб. : Химиздат, 2005. –
264 с.
9. Электрохимия / Б. Б. Дамаскин, О. Л . Петрий.
–
М.: Высш.
школа, 1987. – 295 с.
10. Электрохимия топливных элементов: учебное пособие / В.
П. Кривобоков, Н. С. Сочугов, А. А. Соловьев.
–
Томск: Изд–во
Томского политехнического университета, 2008. – 155 с.
11. Эксплуатация аккумуляторных батарей / А. М . Вайлов, Ф.
И. Эйгель. – изд. "Талер", 2009. – 170 с.
12. Технічна електрохімія : піручник / Б. І. Байрачний.
–
X.:
НТУ «XПІI», 2003.
–
Ч. 2.: Xімічні джерела струму. – 174 с.
13. Батарейки и аккумуляторы / В. С . Лаврус. – Серия "Инфор-
мационное Издание", 1995. – Выпуск 1. – 195 с.
14. Химические источники тока : Справочник / под ред. Н. В.
Коровина, А. М . Скундина. – М. : МЭИ, 2003. – 648 c.
15. Электрохимия / Миомандр Ф., Садки С, Одебер П., Меал-
ле–Рено Р. – М. : Техносфера, 2008. – 360 с.
16. Аккумуляторы / Д. А. Хрусталев. – Изумруд, 2003. – 222 с.
17. Аккумуляторные батареи / П. И. Устинов.
–
М. : Госэнер-
гоиздат, 1982. – 264 с.
18. Кедринский И. А. Литиевые источники тока. / И. А. Кед-
ринский, Л. Е. Дмитренко, И. И. Грудянов. – М. : Энергоатомиздат,
1992. – 240 с.
19. Кедринский И. Е . Li–ионные аккумуляторы. / И. Е. Кедрин-
ский, В. Г . Яковлев. – Красноярск : ИПК ''Платина'', 2002. – 266 с.
20. Гинделис Я. Е. Химические источники тока (Курс лекций).
–
Саратов : Изд–во Саратовского ун–та, 1984. – 174 с.
236
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
4.1. ФОРМАЛЬНА КІНЕТИКА
Хімічна кінетика вивчає швидкість і механізм протікання хімі-
чних процесів, а також залежність їх від різних факторів. Якщо реа-
генти перебувають в одній фазі, то реакція називається гомогенної,
якщо в різних фазах – гетерогенної. Гомогенні реакції протікають в
об'ємі; гетерогенні на границі розділу фаз.
Найважливішою кількісної кінетичної характеристикою будь-
якої хімічної реакції є її швидкість. Знаючи закономірності (матема-
тичну модель) хімічної реакції і її кінетичні параметри оптимальні
умови проведення реакції в промисловому реакторі, тобто організу-
вати керування промисловим процесом. Швидкість хімічної реакції
визначається зміною кількості даного компонента в одиницю часу в
одиниці об'єму
1
v
dn
Vdt
,
де n – число молів даного компонента в об'ємі V даної фази в мо-
мент часу t .
За умови сталості об'єму вираження спрощується й швидкість
реакції визначається зміною концентрації реагуючого речовини c в
одиницю часу
v
dc
dt
.
Швидкість реакції завжди позитивна, однак при протіканні
реакцій у часі концентрації вихідних речовин зменшуються, а про-
дуктів реакції – зростають і похідна
/
dc dt може бути як позитивної,
так і негативної, залежно від того, по зміні концентрації якого
4.1. ФОРМАЛЬНА КІНЕТИКА
237
компонента вивчається процес: по зміні концентрації вихідних ре-
човин знак «–» або продуктів реакції – знак «+». Якщо в системі
протікає реакція за участю кількох речовин, наприклад
...
'
'
...
'
'
A
B
C
D
E
A
B
C
D
E
A
B
C
D
E
c
c
c
c
c
де ic – поточні концентрації речовин), то для однозначного визна-
чення швидкості досить стежити за зміною концентрації будь-якої
одної речовини. Зміна концентрацій інших учасників реакції завжди
можна знайти за співвідношенням стехиометрических коефіцієнтів
і
. Наприклад, швидкість за речовиною A : v
A
dc
dt
пов'язана зі
швидкостями за іншим речовинам співвідношенням
'
v
...
...
'
C
A
AB
A
A
D
B
C
D
dc
dc
dc
dc
dt
dt
dt
dt
Швидкість реакції можна визначити дослідним шляхом по кі-
нетичних кривих – залежності концентрації що брав участь у реак-
ції речовин у часі. Усі методи одержання кінетичних кривих можна
розділити на два класи:
1. Хімічні методи
З реакційної посудини періодично відбирають деяка кількість
розчину – пробу. Реакцію в пробі швидко зупиняють, наприклад,
прохолоджують розчин або відразу ж додають якісь реагенти для
подальшого аналізу, або розбавляють більшою кількістю розчинни-
ка. Проводять хімічний аналіз і визначають або кількість вихідної
речовини, або продукту.
Таким методом одержують кінетичні криві в лабораторних ро-
ботах No 13 при вивченні кінетики йодування ацетону, No 14 при
вивченні кінетики омилення складних ефірів або No при вивченні
кінетики розчинення речовини. Перевагою такого методу одержан-
ня кінетичних кривих є те, що відразу одержуємо залежність конце-
нтрації від часу.
2. Фізико-хімічні методи
Тут визначають не безпосередня кількість речовини, а вимі-
рюють мінливе в результаті реакції якась фізична властивість,
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
238
наприклад, тиск, об'єм, оптичну щільність, електропровідність і т.д.
Таким методом одержують кінетичні криві в лабораторних роботах
No 15 при вивченні кінетики розкладання пероксиду водню або No
16 при вивченні кінетики гідратації оцтового ангідриду.
Фізико-хімічні методи зручні тим, що нема необхідності у від-
борі проб, однак потрібно знати закон, що зв'язує дану використову-
вану фізичну властивість із концентрацією компонента. У деяких ви-
падках попередньо для цих цілей проводять калібрування – установ-
люють залежність між вимірюваною властивістю й концентрацією.
Для хімічної реакції, записаної в загальному виді
...
...
A
B
C
D
vAvB
vC vD
швидкість реакції в цілому дорівнює швидкості реакції по даних ре-
човинах, діленій на відповідні стехіометричні коефіцієнти, тобто
1
1
1
1
v
C
A
B
D
A
B
C
D
dc
dc
dc
dc
vdt
vdtvdtvdt
.
Якщо збиток концентрації реагенту віднесена до нескінченно
малого часу dt , то говорять про дійсну (миттєвої) швидкості реа-
кції, яка є швидкістю реакції в цей момент часу. Крім дійсної швид-
кості реакції, також використовують
поняття середньої швидкості, обу-
мовленої як відношення зміни конце-
нтрації реагенту (або продукту) за час
t
12
12
v
cc
c
tt
t
.
На рис. 4.1 . дійсна й середня
швидкості представлені як тангенси
кута нахилу дотичних у точці N до
кінетичної кривої й прямій AN між
крапками 1c й 2
c при часі від початку
реакції 1t й 2t . Найбільша швидкість
у початковий момент часу визнача-
ється тангенсом кута дотичній OM у
Рис. 4 .1 . Змінення концентрації
реагента за часом реакції: o
c,1c
й 2c , відповідно початкова, по-
точна концентрації реагенту в
моменти 1t та 2t .
4.1. ФОРМАЛЬНА КІНЕТИКА
239
точці 0
t
. Зі зменшенням проміжку часу між двома вимірами кон-
центрації: v→ v при t → 0. Початковою швидкістю реакції 0
v
називають величину
0
v
A
dc
dt
приt →0.
Згідно із законом діючих мас швидкість v реакції пропор-
ційна добутку концентрацій реагуючих речовин у ступенях, рівних
їх стехіометричним коефіцієнтам у рівнянні швидкості реакції.
Залежність швидкості хімічної реакції від концентрації її учас-
ників виражається основним законом хімічної кінетики: "... швид-
кість хімічної реакції в будь-який момент часу при постійній
температурі прямо пропорційна добутку концентрацій реагую-
чих речовин, причому кожна з концентрацій береться в ступені
рівній коефіцієнту перед формулою цієї речовини в рівнянні хімі-
чної реакції" (закон Гульдберга - Вааге, 1867 р.).
Назва закону діючих мас (а не закону дії мас), відображає суть,
щр налана до цього закону його авторами. Під терміном «діючі ма-
си» розуміли саме концентрації вступають у взаємодію частинок.
В основі кінетики лежать постулати.
1. Хімічна реакція протікає в результаті зіткнення, щонаймен-
ше, двох статистично незалежні часток.
2. Реакція можлива у випадку, якщо система співвдарюючих
частинок має граничну енергію й підходящої просторовою орієнта-
цією. Це граничне значення енергії (мінімальний надлишок енергії в
порівнянні із середньою енергією RT), називається енергією актива-
ції реакції aE .
3. Імовірність протікання реакції дорівнює одиниці й не зале-
жить від енергії часток, якщо вона більше a
E й дорівнює нулю, як-
що енергія часток менше a
E.
Для елементарної реакції
A
B
C
D
vAvB
vC vD
,
що протікає ліворуч праворуч (пряма реакція), швидкість реакції
виражається рівнянням
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
240
1
v
A
B
B
A
kcc
,
де k – коефіцієнт пропорційності – константа швидкості прямої ре-
акції; A
cіB
c – концентрації речовин A і B . Константа швидко-
сті залежить тільки від температури. Ця залежність називається
законом Арреніуса й, у відповідності із другим постулатом, має ви-
гляд:
0
a
E
kkexp
RT
.
Тут R – універсальна газова постійна й 0k – передекспонен-
ційний множник, пропорційний числу зіткнень молекул з підходя-
щої просторовою орієнтацією, а множник
/a
expERT
ураховує
частку молекул, що мають при зіткненні енергію
a
EE
(закон Бо-
льцмана).
Константа швидкості чисельно дорівнює швидкості реакції за
умови, що концентрації всіх реагуючих речовин дорівнюють оди-
ниці. Константа швидкості залежить від природи реагуючих речо-
вин, температури, каталізатора і його концентрації й від середови-
ща, у якому реакція протікає.
Механізмом хімічної реакції називають сукупність елемен-
тарних процесів, через які проходить ця реакція. Практично всі реа-
кції в природі є складними, тобто складаються із сукупності елеме-
нтарних стадій.
Кінетично хімічні реакції класифікують по молекулярності й
порядку реакції.
Молекулярність визначають по числу молекул (або інших ча-
сток), що брав участь в елементарному акті хімічного перетворення.
Розрізняють:
мономолекуляні реакції, у яких перетерплює перетворення
тільки один тип молекул, причому стехіометричний коефіцієнт у рі-
внянні дорівнює одиниці, наприклад,
A
B
;A
BC
або 2
2
I
I
;
бімолекулярні реакції, у яких беруть участь два різні типи
молекул або дві молекули одного типу, наприклад,
4.1. ФОРМАЛЬНА КІНЕТИКА
241
AB
C
;2A
C
або
2
2
2
H
I
HI
;
тримолекулярні реакції, у яких беруть участь молекули трьох
типів, або три молекули одного типу, наприклад,
3A
C
;ABC
DF
або
2
2
2
2
2
H
O
HO
.
Такі реакції зустрічаються вкрай рідко, тому що ймовірність
одночасного зіткнення трьох часток дуже мала. Реакції більш висо-
кої молекулярності малоймовірні.
Молекулярність реакції поняття теоретичне. Для того, щоб
знати молекулярність, необхідно представляти механізм протікання
реакції: через взаємодії яких молекул і через які стадії.
Більшість хімічних реакцій протікають у кілька стадій, тому
кінетично швидкість визначається емпіричним рівнянням виду
1
2
1
2
1
1
n
n
АА
v
kcc
,
де1
nй2
n – порядки реакції відповідно по речовинах 1
Aі2A.
Порядок хімічної реакції по даній речовині – це число, рівне
ступені in , у якій концентрація цієї речовини входить у кінетичне
рівняння реакції. Порядок реакції в цілому визначається сумою по-
казників
i
n
n
, у яких концентрації всіх речовин входять у кіне-
тичне рівняння. Реакції можуть бути нульового, першого, другого й
навіть дробового порядку.
На противагу молекулярності порядок реакції експеримента-
льна величина. Він пов'язаний з дослідженою залежністю швидкості
даної реакції від концентрації вихідних речовин. Для простих реак-
цій, що протікають у повній відповідностях з них стехіометричним
рівнянням, порядок і молекулярність чисельно збігаються.
Завданням кінетичних досліджень є визначення порядку й
константи швидкості реакції, енергії активації реакції, числа й хара-
ктеру проміжних продуктів, з'ясування впливу природи розчинника,
каталізатора й ін.
За ступенем складності реакції класифікуються на оборотні й
необоротні.
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
242
Оборотні реакції – такі, які, можуть протікати як в одному,
так і у зворотному напрямку. Однобічні, або необоротні, реакції –
такі, у яких кінцеві продукти присутні в незначних кількостях випа-
дають в осад, виділяються у вигляді газу, утворюють малорозчинні
сполуки), у таких реакціях швидкості прямого й зворотного проце-
сів не порівнянні ( .
.
пр
обр
k
k
). Для оборотної реакції швидкість ре-
акції визначається різницею швидкостей прямій і зворотної реакцій.
Ізольовані реакції – реакції, у ході яких утворюється продукт
тільки одного типу.
Паралельні реакції – реакції, у ході яких вихідні речовини ре-
агують у дві або більш напрямках з утвором різних продуктів реак-
ції, наприклад,
СD
AB
EF
.
Такі реакції часто мають місце в органічній хімії.
Послідовні (консекутивні) реакції – реакції із проміжними
стадіями, наприклад, типу
1
2k
k
A
B
C
.
Якщо одна зі складових процесу має явно меншу швидкість,
що інші, то загальна швидкість процесу визначається саме цієї (, що
лімітує) стадією. У цьому випадку в кінетичних рівняннях реакції,
що швидко протікають, можуть не враховуватися.
Сполучені реакції – це реакції типу
AB
M
,
(I)
AC
N
,
(II)
з яких одна, наприклад (II), протікає лише разом з інший, тобто йде
лише за умови, якщо взяті всі речовини A, B і C. Реакція (II) інду-
кується реакцією (I). Речовина B називається індуктором реакції
(II), речовина A, загальне для обох реакцій, – актором і речовина
C – акцептором.
Залежно від порядку реакції кінетичні рівняння для розрахунків
4.1. ФОРМАЛЬНА КІНЕТИКА
243
швидкості реакції різні. Порядок реакції часто є емпіричною вели-
чиною, він або рівний молекулярності реакції, якщо реакція елемен-
тарна, або, у більшості випадків, менше її. При великому надлишку
одного з реагуючих речовин порядок реакції може бути на одиницю
менше, чим варто було б зі стехіометричного рівняння. Наприклад,
реакції інверсії цукру або гідратації у великому надлишку води.
Таким чином, порядок реакції характеризує формальнокінети-
чну залежність швидкості реакції від концентрації реагуючих речо-
вин, а молекулярність – елементарний механізм окремих стадій
складного процесу.
Якщо в системі можливо кілька простих реакцій, то кожна з
них, незалежно від інших реакцій, буде протікати зі швидкістю, що
визначається своїм диференціальним рівнянням і своєю константою
швидкості. Це положення становить принцип незалежності швид-
костей хімічних реакцій, який дає можливість записувати для
складних хімічних реакцій систему диференціальних рівнянь, що
описують швидкості кожної з можливих простих реакцій. Швид-
кість кожної з них прямо пропорційна концентраціям реагуючих
компонентів. Цей принцип застосовується для опису оборотних, па-
ралельних, послідовних і інших реакцій.
Реакції нульового порядку. Для реакцій нульового порядку
( n = 0) швидкість реакції постійна й не залежить від концентрації
вихідних речовин. Наприклад, для реакції
k
A
B
відповідно до основного постулату хімічної кінетики
0
0
0
v
()
A
A
dc
kc
k
dt
,
деA
c – поточна концентрація речовини A .
Розділивши змінні й проінтегрував
0
A
dckdt
,
0
0
0
()
c
t
A
c
t
dc
kdt
одержимо результуюче рівняння, що описує залежність концентра-
ції речовини A від часу
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
244
00
cckt
або
0
0
cc
k
t
,
де0
c – початкова концентрація речовини A в початковий момент
часуt=0, A
c – концентрація A в
момент часу t (поточна концентра-
ція речрвини A).
Одиниці виміру константи
швидкості – [концентрація/час].
Якщо концентрація виражена, на-
приклад, у моль/л, а час – у секун-
дах,
то
0
k виражається
в
моль·(л·сек)
–1
.
Час, протягом якого концент-
рація вихідної речовини зменшуєть-
ся у два рази, називається періодом
(часом) напівперетворення. Воно
позначається 1/ 2
.
Для реакції нульового порядку
00
0
1/2
0
/2
2
cc
c
k
.
тобто час напівперетворення прямо пропорційно початкової конце-
нтрації речовини ( при збільшенні 0
c в2рази, 1/2
збільшується в 2
рази). Залежність поточної концентрації від часу показана на рис. 4.2.
Реакції нульового порядку мають місце в тих випадках, коли
збиток речовини в результаті протікання хімічної реакції заповнює
доставкою його з іншої фази. Наприклад, реакція омилення ефіру
водою може протікати за нульовим порядком за умови, що взаємна
розчинність ефіру й води малі,
3
25
2
3
25
CHCOOCH HO
CHCOOH CHOH
і при зменшенні концентрації ефіру у воді з ефірного шару щораз
надходить його нова порція.
Якщо одне з реагуючих речовин узяте у великому надлишку,
то реакція буде мати нульовий порядок по даній речовині.
Нульовий порядок спостерігається також, якщо швидкість процесу
Рис. 4.2 . Залежність поточної
концентрації від часу для реакції
нульового порядку
4.1. ФОРМАЛЬНА КІНЕТИКА
245
лімітується подачею енергії, необхідної для активації реагуючих
молекул (фотосинтез).
Однобічна хімічна необоротна реакція першого порядку
схематично може бути представлена рівнянням
k
A
В
.
Прикладом такої реакції
можуть бути реакція радіоактив-
ного розпаду, ізомеризації, розк-
ладання при високій температурі
й ін.
Швидкість необоротної реа-
кції першого порядку рівна
1
A
A
dc
kc
dt
,
деA
c – концентрація речовини A
до моменту часу t від початку
реакції.
Розділяючи змінні й інтегруючи, одержимо
0
0
1
0
ln
c
c
t
A
A
A
c
c
t
dc
dc
kdt
c
,
0
ln ln
c
ckt
,
або
0
1
ln
c
kt
с
,
де0
c – початкова концентрація речовини А в початковий момент
часу t = 0, с – поточна концентрація А в момент часу t .
Звідси
0
1kt
ссe
, тобто концентрація речовини А в ході ре-
акції зменшується експоненціально (рис. 4 .3). Відповідно, концент-
рація продуктів реакції наростає теж експоненціально.
Вираження для константи швидкості реакції має вигляд
1
1
ln
o
c
k
t
с
.
Рис. 4.3 . Залежність поточної кон-
центрації від часу для реакції пе-
ршого порядку.
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
246
Одиниця виміру константи швидкості реакції першого порядку
–
[час
–1
]
Час напівперетворення 1/ 2
для реакції першого порядку не за-
лежить від початкової концентрації речовини – цей факт характер-
ний тільки для реакцій першого порядку й може служити ознакою,
що визначають цей тип реакцій
0
1/2
1
0
1
1
ln2
ln
/2
c
kc
k
.
Залежність логарифма концен-
трації речовини A від часу має лі-
нійний характер і представлено на
рисунку 4.4, звідки легко можна ви-
значити константу швидкості через
кутовий коефіцієнт прямої.
До однобічних реакцій першо-
го порядку ставляться реакції радіо-
активного розпаду, реакції розкла-
дання складних речовин (карбона-
тів, кристалогідратів) при їхнім на-
гріванні, реакції ізомеризації й ін.
Однобічна необоротна реакція другого порядку схематично
представляється рівнянням
2
продукты реакции
k
AB
.
До таких реакцій ставляться, наприклад, реакції омилення
складного ефіру лугом.
При рівних початкових концентраціях вихідних речовин A і
B(A
B
o
c
c
c
), реакція може бути представлена у вигляді
2
2
продукти реакції
k
A
і швидкість реакції виражається рівнянням
2
2
v
A
dc
kc
dt
,
Рис. 4 .4 . Лінійна залежність ло-
гарифма концентрації від часу
4.1. ФОРМАЛЬНА КІНЕТИКА
247
Розділимо змінні
2
2
dc
kdt
c
і проінтегруємо
2
2
0
o
c
t
c
t
dc
kdt
c
.
Побравши визначений інтеграл (від 0
t до t), одержимо
2
0
11
kt
cc
, звідки 2
0
()
1111o
o
cc
k
tсс
tcс
.
Одиниця виміру константи швидкості реакції другого порядку –
[концентрація
–1
·час
–1
]. Тобто, якщо концентрація виражена в моль/л, а
час – у секундах, то 2
k виражається в [л·(моль·сек)
–1
].
Час напівперетворення для реакції другого порядку назад про-
порційно початкової концентрації речовин:
1/2
20
1
kс
.
Звідси випливає, що час напіврозпаду залежить від початкової
концентрації. У скільки разів зменшується концентрація, у стільки
раз збільшується півперіод реак-
ції.
Залежність зворотної конце-
нтрації речовини А від часу має
лінійний характер. Вона представ-
лена на рисунку 4.5 . Ця залежність
дає
можливість
визначити
2
k(2k tg
).
Якщо початкові концент-
рації реагуючих речовин А і В
різні, (
,
,
oA
oB
c
c
) рівняння шви-
дкості реакції другого порядку в
диференціальній формі ухвалює
Рис. 4 .5 . Графічне визначення
константи швидкості реакції дру-
гого порядку ( A
B
c
c
).
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
248
вид
2
v
AB
dc
kcc
dt
і після інтегрування до моменту часу t , розв'язок щодо константи
швидкості реакції 2k має вигляд
,
2
,
,
,
1
1
ln
(
)
oBA
oA
oB
oAB
c
c
k
tc
c
c
c
,
деA
cйВ
c – поточні концентрації речовин А і В в момент часу t .
Розмірність константи швидкості
11
[]
tc
, наприклад, (л·моль/с).
Різновидом реакцій другого порядку є автокаталітичні реа-
кції, у яких каталізатором являє собою один із продуктів реакції.
Прикладом такої реакції може служити реакція йодування ацетону.
У таких реакціях концентрація каталізатора збільшується згодом і
диференціальне рівняння швидкості перетвориться до вигляду
,
,
(
)(
)
oA
oB
dc
kcccc
dt
,
де c – концентрація речовини, яка прореагувала до моменту часу t .
Після інтегрування для константи швидкості маємо
,
,
,
,
,
,
(
)
1
ln
(
)
(
)
oB oA
oA
oB
oA oB
c
c
c
k
tc
c
c
c
c
.
Реакції можуть бути й дробового порядку, який указує на од-
ночасне протікання декількох етапів реакції, близьких по швидкос-
тях, або на протікання оборотної реакції.
Для реакції довільного n-го порядку ( крім першого)
...
продукти реакції
n
k
ABС
За умови рівності концентрацій усіх вихідних речовин ( 0, A
c
=
,
oB
c=
,
oC
c =. . . ) константа швидкості виражається рівнянням
1
1
0
11
1
1
(1)
n
n
n
k
tn
c
c
,
4.2. МЕТОДИ ВИЗНАЧЕННЯ ПОРЯДКУ РЕАКЦІЇ
249
а час напівреакції 1/ 2
дорівнює
1
1/2
1
0
1121
1
n
n
n
k
n
c
,
або
1/2
1
0
n
Const
c
.
що показує зв'язок між періодом напі-
вреакції й початковою концентрацією
компонентів реакції.
Вплив концентрації на швидкість
реакції характеризує величина порядку
реакції: чим більше порядок реакції,
тем сильніше цей вплив. Як видне з кі-
нетичних кривих на рис. 4.6 концент-
рація реагенту убуває швидше у випадку більш високого порядку
реакції. У результаті такої залежності при низьких концентраціях
переважно реалізуються процеси з малим значенням порядку реакції.
4.2. МЕТОДИ ВИЗНАЧЕННЯ ПОРЯДКУ РЕАКЦІЇ
Для визначення порядку реакції необхідно мати у своєму роз-
порядженні експериментальні дані про зміну концентрації речовин
у часі. Загальний порядок реакції перебуває підсумовуванням при-
ватних порядків реакції.
Приватний порядок визначають користуючись методом ізо-
лювання Оствальда або метол надлишку. Реакцію проводять пос-
лідовно у великому надлишку всіх реагентів, крім того, порядок по
якому визначають.
Так, у випадку реакції
...
k
ABС
D
швидкість реакції дорівнює
v
C
A
Bn
n
n
B
A
C
kccc
.
Загальний порядок
A
B
C
nn
n
n
.
Рис. 4 .6 . Кінетичні криві для
реакцій, що мають порядок
за речовинорю А: 1)
0;
А
n
2)
1;
А
n3)
2;
А
n
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
250
Якщо речовини B й С узяті у великому надлишку, то при про-
тіканні реакції їх концентрації практично не змінюються, тобто
'
C
Bn
n
B
C
kccconstk
йv
'
A
n
A
kc
.
Усі відомі методи визначення порядку реакції розділяють на
дві групи – інтегральні й диференціальні.
I. Інтегральні методи використовують кінетичні рівняння,
для швидкості хімічної реакції в інтегральній формі:
–
метод добору рівнянь – заснований на підстановці експери-
ментальних даних по концентраціях у різні проміжки часу в кінети-
чні рівняння різних порядків, і виборі того рівняння для якого конс-
танта швидкості буде незалежної від часу реакції;
–
графічний метод – заснований на визначенні такої функції
від концентрації, залежність якої від часу буде лінійною. Так, для
реакцій першого порядку лінійна залежність спостерігається в коор-
динатах lg c t
; для реакції другого порядку 0,
0,
A
B
c
c
–
у координа-
тах 1/c t
і т. п. (фактично збігається з методом добору рівняння й за-
стосуємо для цілих порядків реакції);
–
за часом напівперетворення 1/ 2
–
часу, протягом якого ви-
хідна концентрація речовин зменшується у два рази. Так, для реак-
ції першого порядку період напівперетворення не залежить від по-
чаткової концентрації, тобто
1/2
lg2
2,303
k
,
для реакції другого порядку цей час обернено пропорційний почат-
кової концентрації вихідної речовини, для реакції n-го порядку
співвідношення між часом напівреакції й початковою концентраці-
єю має вигляд
1/2
1
1
n
o
c
.
За методом Оствальда-Нойеса досліди проводять при двох
різних початкових концентраціях 0
c'і0
c'' за умови еквівалентного
4.2. МЕТОДИ ВИЗНАЧЕННЯ ПОРЯДКУ РЕАКЦІЇ
251
зміста всіх інших речовин. Логарифмування відносини рівнянь для
часу напівреакції дає вираження для обчислення порядку реакції
1/2
1/2
0
0
lg
lg
lg
1
lg lg
lg
'
1/2
''
'
''
1/2
''
'
''
o
'
o
n
c
c
c
c
,
де 1/2
'
й 1/2
''
–
час напівперетворення при початкових концентра-
ціях 0
'
cі0
''
c , відповідно.
Коли час напівперетворення великий, можна користуватися
більш загальним методом, визначення порядку реакціх за часом до-
сягнення деякої визначенної частки перетворення ( р ):
0
0
lg
lg lg
1
lg lg
lg
'
р
''
'
''
р
р
р
''
'
''
o
'
o
n
c
c
c
c
.
Якщо співставити час перетворення вихідної речовини в реак-
ціяї різного порядку при однаковій початкових концентраціях та
однакових константах швидкості отримаємо
Порядок реакції
Перетворення
на
0
1
2
1/3
0,33
0,40
0,5
1/2
0,50
0,69
1,0
2/3
0,66
1,10
2,0
Очевидно, що при малих ступенях перетворення неможливо
досягти великої точності у визначенні порядків реакції.
II. Диференціальні методи засновані на використанні рівнян-
ня для швидкості реакції в його диференціальній формі.
–
метод Вант-Гоффа – заснований на визначенні швидкостей
реакції у випадку її проведення при двох різних вихідних концент-
раціях 1c і 2
c.
Тоді, записавши для швидкостей реакції вираження
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
252
1
1
v
n
dc
kc
dt
й2
2
v
n
dc
kc
dt
і прологарифмував їх відношення, можна виразити порядок реакції
1
2
1
2
lg
lg
lg lg
dc
dc
dt
dt
n
c
c
,
або
1
2
1
2
lgv lgv
lg lg
n
c
c
.
Значення lgv й lgc мо-
жуть бути отримані з однієї кі-
нетичної кривій, чк це показа-
но на рис. 4.7. Однак більш на-
дійні результати (з урахуван-
ням помилок експерименту)
одержують, використовуючи значення початкових швидкостей при
різних початкових концентраціях реагентів (використовуючи поча-
ткові прямолінійні залежності
зміни концентрації – так званий
метод початкових концентрацій
(рис. 4 .8).
–
графічний варіант мето-
ду Вант-Гоффа заснований на
використанні рівняння
lgvlg' lg
knc
.
Тангенс кута нахилу залеж-
ності в координатах lg v lg c
відповідає порядку реакції, а від-
різок, що відтинається на осі ор-
динат,
–
логарифму константи
швидкості. Різні значення lg v й
lg c , як описано раніше, можуть
бути отримані з однієї кінетичної
Рис. 4 .7 . Обробка кінетичної кривої
для визначення швидкості реакції
Рис. 4.8 . Обробка кінетичних кри-
вих для визначення початкових
швидкостей реакції
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
253
кривої, або використовуючи
значення початкових швидкос-
тей при різних початкових
концентраціях реагентів. Роз-
рахунки швидкості реакції при
різних концентраціях реагенту
можна визначити графічним
диференціюванням, а також
аналітично. При проведенні
досліду, з чкого отримані декі-
лька значень залежності швид-
кості реакції від концентрації
будується графік, по якому й
визначається приватний поря-
док реакції з тангенсу кута на-
хилу залежності (рис. 4 .9).
4.3. ЗАЛЕЖНІСТЬ ШВИДКОСТІ ХІМІЧНОЇ РЕАКЦІЇ
ВІД ТЕМПЕРАТУРИ.
Швидкість хімічної реакції з підвищенням температури значно
зростає. Для більшості реакцій константа швидкості збільшується в
2–4 рази при зміні температури на кожні 10 (емпіричне правило
Вант-Гоффа), що може бути відображене у вигляді рівняння з від-
ношенням констант швидкостей
10
T
T
k
k
,
де – температурний коефіцієнт константи швидкості,
10
T
kі
10
T
k – константи швидкості за температури (T + 10) і T, відповідно.
Залежність константи швидкості одностадійної необоротної
реакції від температури описується рівнянням Вант-Гоффа – Ар-
реніуса
2
ln
a
E
dk
dT RT
,
деa
E – арреніусівська (експериментальна) енергія активації.
Р
ис. 4 .9 . Визначення порядку реакції
за речовиною по залежності швид-
кості реакції від концентрації
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
254
Якщо a
E постійна величина на деякому інтервалі температур,
інтегрування дає рівняння
0
a
E
RT
kke
,
де0
k – передекспоненційний множник. Величини 0
kй aE –найва-
жливіші характеристики хімічної реакції.
Арреніус пояснив це рівняння, виходячи з припущень про те,
що в реакцію можуть вступати не всі молекули, а лише їх активні
модифікації, що утворюються оборотно з нормальних. Закон Арре-
ніуса розглядає частку частинок в даній системі, запас енергії яких
достатній для здійснення елементарної реакції. Ця частка визнача-
ється передекспоненціальним множником в рівнянні.
З підвищенням температури середня енергія вихідної системи
зростає, збільшується число часток, що володіють необхідним над-
лишком енергії, що веде до підвищення швидкості реакції.
Поняття енергії активації, як і константи швидкості, подібно
самому закону Арреніуса, відноситься до елементарних реакцій.
При переході до багатостадійним реакцій, описуваних вже не зако-
ном діючих мас, а більш складними кінетичними рівняннями, навіть
якщо вони містять одну постійну, картина температурної залежнос-
ті швидкості процесу може ускладнюватися. Це пов'язано з тим, що
константа кінетичних рівнянь може являти собою складне вираз, що
містить кілька констант елементарних стадій.
У логарифмічній формі рівняння Арреніуса має вигляд
0
lg lg
2,303
a
E
k
k
RT
,
що нажаєможливості визначення енергії активації реакції за куто-
вим нахилом лінійної залежності lg k від 1/ T .
Під енергією активації розуміють той надлишок енергії в по-
рівнянні із середньою енергією молекул при даній температурі, яку
повинні мати молекули, щоб вони могли вступити в реакцію.
З рівняння Арреніуса випливає, що між логарифмом констан-
ти швидкості й величинами зворотною температурою існує лінійна
залежність, що дозволяє розраховувати енергію активації графічно
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
255
по залежності lg
1/
k
T
по кутовому коефіцієнту прямої. При на-
явності даних тільки при двох температурах 1
T і 2T енергія активації
може бути розрахована по рівнянню
12
21
21
2,303
lg(/)
a
RTT
kk
E
TT
.
Величина енергії активації реакції досягає десятків кілоджоу-
лів на моль. Вона практично не залежить від температури. Величина
анергії активації залежить від типу хімічної реакції, що протікає.
Для низькотемпературних некаталітичних реакцій величина енергії
активації лежить у межах 60–160 кДж/моль, для високотемператур-
них реакцій – у межах 180–300 кДж/моль. Для дуже великої кілько-
сті реакцій енергія активації перебуває в межах від 60 до 240
кДж/моль, тобто приблизно відповідає енергіям хімічних зв'язків.
Для реакцій за участю активних часток (вільні атоми, радикали)
енергія активації може бути приблизно рівної нулю.
Основними факторами, що впливають на швидкість хімічної
реакції, є температура й концентрація реагентів, а також наявність
розчинника й каталізатора.
Сильна (експонентна) залежність швидкості реакції від темпе-
ратури приводить до того, що при низьких температурах звичайно
реалізуються процеси з низькою енергією активації (наприклад, гід-
рування, алкілування, гідратація) і, навпаки, при високих темпера-
турах реалізуються процеси з підвищеною енергією активації (на-
приклад, дегідрування, крекінг, дегідратація). Відзначимо, що з під-
вищенням температури зростає ймовірність дифузійного обмеження
швидкості реакції: дифузія «не встигає» за реакцією, тому що темпе-
ратурний коефіцієнт дифузії ( D ≈
0,5
T ) суттєво менше, чим у реакції.
Умовою протікання реакції є зіткнення молекул, однак ще Ар-
реніус показав, що не кожне зіткнення приводить до хімічної взає-
модії. Це проявляється:
–
у невідповідності між числом зіткнень і реальними швидкос-
тями процесів, які значно менше;
–
у залежності швидкості реакції від природи компонентів і
тому їх відмінністю для однотипних процесів;
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
256
–
у набагато більшій залежності швидкості реакції від темпе-
ратури, чому це диктується впливом температури на число зіткнень;
–
у впливі каталізаторів на швидкість реакції.
Таким чином, до реакції приводять лише ефективні зіткнення,
що становлять малу частку від загального їхнього числа, що є акти-
вними, енергія часток яких більше або рівна
a
E . Ця енергія необ-
хідна для подолання частками енергетичного бар'єра реакції – енер-
гії відштовхування електронних хмар, що зустрічаються молекул.
Зіткнення ефективне, якщо сумарна величина енергії часток, що зу-
стрічаються, рівна або більше енергії активації.
ТЕОРІЯ АКТИВНИХ ЗІТКНЕНЬ
Теорія активних зіткнень заснована на молекулярній теорії га-
зів, згідно з якою в газовій фазі існує певний розподіл часток по
швидкостях (розподіл Максвелла) і енергіям (розподіл Больцмана).
Згідно із цими уявленнями, для взаємодії частки повинні зіштовх-
нутися. Розглянемо бімолекулярну реакцію:
AB
C
.
Загальне число зіткнень часток A і B в газовій фазі залежить
від температури, концентрації часток, розмірів і властивостей час-
ток, швидкості їх руху. Згідно з молекулярно-кінетичною теорією
газів, загальне число зіткнень AB
Z частинок A і B в одиниці об'єму
в одиницю часу дорівнює
o
AB
ABAB
Z
Znn
,
де
o
AB
Z – деякий параметр, що залежить від властивостей частинок,
що зустрічаються; n – число частинок в одиниці об'єму.
Значення експериментальних швидкостей реакцій значно ме-
нше, чим відповідає розрахункам із припущенням, що кожне зітк-
нення ефективне. Отже, не всі зіткнення часток A і B результатив-
ні, тобто приводять до продуктів реакції. Взаємодія часток відбува-
ється тільки в тому випадку, коли ці зіткнення виявляться активни-
ми, тобто якщо частки будуть мати певний достатній запас енергії
(.
акт
AB
Z <AB
Z ). Отже, необхідно враховувати розподіл часток по ене-
ргіях
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
257
.
E
акт
RT
AB
AB
Z
Ze
,
де
E
RT
e
–
больцманівський множник, що враховує розподіл часток
по енергіях (для молекул, що мають енергію Е ). Для реальних мо-
лекул необхідно враховувати й той факт, що при зіткненні частинки
повинні бути відповідним чином орієнтовані у просторі й їх надли-
шкова енергія повинна бути зосереджена по лініях зв'язків, що роз-
риваються. Для цього в рівняння для швидкості вводять додатковий
множник – стеричний фактор Р . При сприятливій орієнтації мо-
лекул по відношенню друг до друга фактор близький до одиниці,
при несприятливої Р <1. Тоді
.
o
E
E
акт
RT
RT
AB
AB
ABAB
Z
PZe
PZnne
,
а
.
v
акт
AB
A
Z
N
,
A
A
A
n
с
N
,
B
B
A
n
с
N
,
деA
N – число Авогадро.
Для швидкості реакції одержимо співвідношення
o
v
E
RT
A
ABAB
NPZссe
,
яке збігається за формою постулату хімічної кінетики
v
AB
kсс
,
отже, для бімолекулярної реакції можна записати
o
E
RT
A
AB
kNPZe
,
де
o
AB
Z – число подвійних зіткнень в 1 см
3
при
1
A
B
n
n
, моле-
кул/см
3
.
Отримане рівняння теорії активних зіткнень для константи
швидкості бімолекулярної реакції дуже схоже на емпіричне рівнян-
ня Арреніуса
E
RT
kAe
,
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
258
із чого випливає зміст передекспоненційного множника А в рівнян-
ні Арреніуса
o
A
AB
ANPZ
,
тобто він ураховує число зіткнень і орієнтацію молекул.
Ця теорія не позбавлена недоліків, які полягають у тому, що
вона:
–
не дозволяє розраховувати значення енергії активації з пара-
метрів молекул реагуючих речовин;
–
не пояснює вплив на швидкість реакції інертних добавок, ро-
зчинників і т. д.;
–
не може пояснити аномально повільний плин ряду реакцій,
тому що розглядає їхнє зіткнення й не враховує можливість моле-
кул брати участь в обертальному й коливальному рухах.
ТЕОРІЯ АКТИВОВАНОГО КОМПЛЕКСУ
Теорія перехідного стану (теорія активного комплексу), покла-
дена Ейрингом і Поляни (1935). У ній використані основні вистави
теорії активних зіткнень і необхідність подолання деякого енерге-
тичного бар'єра в ході хімічного перетворення.
Основним положенням теорії є припущення, що будь-яка хімі-
чна реакція протікає через утвір деякого активного комплексу, який
згодом розпадається з утвором кінцевих продуктів хімічної реакції.
Так, наприклад, реакція
ABС
ABC
представляється в такий спосіб:
*
()
()
ABС
ABC
AB
C
,
де
*
ABC – активований комплекс.
Інакше кажучи, у ході реакції реагуючі частки утворюють спо-
чатку деякий малостійкий комплекс атомів A , B і С , який розпада-
ється на частки кінцевих продуктів реакції. Активований комплекс
має максимальної потенційної енергії, внаслідок чого є досить не-
стійким, – час його життя становить у середньому 10–13
секунд.
Взаємодія атома A із двохатомною молекулою BС здійсню-
ється зменшенням відстані між атомами A і B й збільшенням між
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
259
атомами B й С . Графік за-
лежності потенційної енергії
системи від відстаней у і х
між атомами A В
йBС
має вигляд деякої поверхні,
називану поверхнею потен-
ційної енергії (рис. 4.12).
Перетину поверхні потен-
ційної енергії представлені
на рисунку 4.13. Найбільш
вигідний (з погляду витрат
енергії) шлях переходу від
вихідної системи до кінцевої
проходить по лінії крапки,
що з'єднує, відповідні до мінімуму потенційної енергії. Проекція ці-
єї лінії показана пунктиром на рисунку 4.12. Шлях уздовж неї нази-
вають координатою реакції. Залежність потенційної енергії реагу-
ючих часток від координати реакції має вигляд кривої з максиму-
мом, аналогічної кривій, показаної на рисунку 4.13. Проміжний стан
системи реагуючих часток, відповідне до цього максимуму
*
ABC ,
називається перехідним станом, або активованим комплексом.
Рівень I відповідає вихідному
стану із середньою енергією
сер
Е , тільки деяка частина час-
ток з енергією перевищуючою
середня на енергію активації
прямого процесу
пр
а
Е здатна
утворювати активний ком-
плекс
*
ABC і утворювати про-
дукти з рівнем енергії II. Тер-
модинамічний перехід зі стану
в стан характеризується тепло-
вим ефектом реакції
р
Н
.
Щоб реакція відбулася,
енергії реагуючої системи по-
Рис. 4 .12 . Поверхня потенціальної енер-
гії
Рис. 4 .13. Зміни енергії під час екзотер-
мічної реакції
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
260
винне бути досить для утвору перехідного стану й імовірність здій-
снення реакції визначається ймовірністю утвору перехідного стану.
Основний постулат теорії активованого комплексу полягає в
тому, що вихідні речовини завжди перебувають у рівновазі з акти-
вованим комплексом. Тому перехідний стан можна розглядати як
звичайну молекулу, що має крім звичайних трьох ступенів волі пос-
тупального руху четвертий ступінь волі, пов'язану з рухом уздовж
шляху (координати) реакції.
Теорія активного комплексу застосовна до реакцій, що проті-
кають у розчинах, тоді як теорія активних зіткнень добре описує
тільки реакції, що протікають у газовій фазі. Молекули реагентів у
рідині перебувають на більш близькій відстані, коли сили взаємодії
між ними не можна вважати малими або навіть відсутніми, що час-
то припустимо в газах. Часто розчинник сильно впливає на швид-
кість реакції. Тому швидкості реакцій у розчинах можуть сильно ві-
дрізнятися від розрахованих по теорії активних зіткнень як у ту, так
і в іншу сторону. Стеричний фактор при цьому може бути більше
одиниці як у реакціях між зарядженими частками, так і багато мен-
ше.
ТЕОРЕТИЧНІ ПИТАННЯ ПО ТЕМІ
Визначення середньої й дійсної швидкості хімічної реакції.
Основний постулат хімічної кінетики. Константа швидкості хіміч-
ної реакції і її фізичний зміст. Молекулярність і порядок реакції.
Прості й складні реакції. Принцип незалежності елементарних ста-
дій складної хімічної реакції. Період напіврозпаду і його зв'язок з
константою швидкості реакції.
Методи визначення порядку реакції. Необоротні реакції пер-
шого порядку. Необоротні реакції другого порядку при рівних і різ-
них кількостях компонентів. Реакції n-го порядку. Оборотні реакції
першого й другого порядку. Паралельні й послідовні реакції. Прин-
цип стаціонарності Боденштейна.
Залежність швидкості й константи швидкості реакції від темпе-
ратури. Рівняння Арреніуса і його висновок. Енергія активації. Лан-
цюгові реакції й особливості розгалужених ланцюгових процесів.
Теорія активних зіткнень, рівняння для константи швидкості. Теорія
перехідного стану (активного комплексу), основне рівняння теорії
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
261
активного комплексу. Мономолекулярні реакції, схема Ліндемана.
Реакції в розчинах, вплив розчинника на швидкість реакції. Ланцю-
гові реакції. Фотохімічні реакції, основне кінетичне рівняння.
ЛІТЕРАТУРА
1. Герасимов Я. И. и др. Курс физической химии. Т.2 .
–М. :
Химия, 1973. – 625 с.
2. Лебідь, В. І. Фізична хімія : підруч. для студ. вищ. навч. за-
кл. / В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
3. Пурмаль А.П. А, Б, В... химической кинетики / А.П . Пур-
маль. — М.: ИКЦ «Академкнига», 2004. — 277 с.
4. Физическая химия. Теоретическое и практическое руковод-
ство. Изд. 2 –е, переработанное и дополненное. / Под ред. академи-
ка Б. П. Никольского. – Л. : Химия, 1987. – 880 с.
5. Стромберг А. Г ., Семченко Л. П. Физическая химия, учеб-
ник для студентов вузов, обучающихся по химическим специально-
стям / А. Г. Стромберг, Д. П. Семченко. - 6 -е изд., стер. – М. : Выс-
шая школа, 2006. – 527 с.
6. Еремин Е.Н. Основы химической кинетики.
–
М. : Высшая
школа, 1976. – 541 с.
7. Эмануэль Н. М., Кнорре Д. Г . Курс химической кинетики:
Учебник для хим. фак-тов. – 4 -е изд., перерзб. и доп. – М. : Высш.
шк., 1984. – 463 с.
8. Физическая химия. В 2 кн. Кн. 2 . Электрохимия. Химиче-
ская кинетика и катализ: Учеб. для вузов / К. С . Краснов, Н. К . Во-
робьев, И. Н. Годнев и др. ; Под ред. К .С. Краснова –3 -е изд., испр.
–
М. : Высш. шк., 2001. – 319 с.
9. Вишняков А. В., Кизим Н. Ф. Физическая химия.
–
М.:
Химия, 2012. – 839 с.
10. Эмануэль Н. М ., Кнорре Д. Г. Курс химической кинетики:
Учебник для хим. фак, тов. –4 -е изд., перерзб. и доп. – М . : Высш.
шк., 1984 – 463 с,
11. Эткинс П. Физическая химия, Т.1
–
М. : Издательство
«Мир», 1980. – 583 с.
12. Эткинс П. Физическая химия, Т.2
–
М. : Издательство
«Мир», 1980. – 585 с.
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
262
Робота No 13.
ВИЗНАЧЕННЯ КОНСТАНТИ ШВИДКОСТІ Й ЕНЕРГІЇ
АКТИВАЦІЇ РЕАКЦІЇ ЙОДУВАННЯ АЦЕТОНУ
Ціль роботи: Підтвердити механізм реакції йодування аце-
тону, визначити константу швидкості реакції при заданих темпе-
ратурах, оцінити температурний коефіцієнт константи швидко-
сті й енергію активації реакції.
Прилади й реактиви: термостат, мірні колби на 250 мл, пі-
петка на 15 мл, крапельниця (або пікнометр Оствальда) з ацето-
ном, 6 колб 250 мл для титрування, свіжоприготовлений розчин
крохмалю, розчин хлористого водню 1 моль/л, 0,1 н розчин йоду в
4% розчині йодистого калію, розчин гідрокарбонату натрію.
Реакція йодування ацетону протікає за рівнянням
3
3
2
3
2
3
2
3
2
k
CHCOCH I HO HO
CHCOCHI I
HO
.
Процес йодування ацетону є автокаталітичним, тому що
прискорюється одним із продуктів реакції – іонами водню. У нейт-
ральному розчині реакція протікає вкрай повільно.
Йодування ацетону в кислому врдному середовищі протікає у
дві стадії:
1) оборотна реакція енолизація ацетону. У кислому середо-
вищі ацетон стає протофільним – здатним до приєднання іона гід-
роксонію
3
3
3
3
3
2,
|
|
CHCCH HО
CHCCHHО
O
OH
Водень метильної групи кетоенолу придбає рухомість й зєдну-
ється з молекулою води
3
3
2
3
2
3.
|
|
CHCCHHO
CHCCH HO
OH
OH
Таким чином, перша стадія реакції є реакцією перетворенння
кетону до енолу. На другій стадії енол приєднує молекулу йоду
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
263
2) на другій стадії енол приєднує молекулу йоду
3
3
2
2
3
2
3
2
.
|
||
CHCCHIHO
CHCCHIHOI
OH
O
Перша реакція протікає повільно, друга швидко й практично
до кінця. Тому загальна швидкість процесу визначається самою по-
вільною стадією – в даному випадку енолізації ацетону. Вона про-
порційна концентрації водневих іонів та не залежить від концентра-
ції йоду, тобто відповідно до кінетичного рівняння
o
o,
(
)()
H
dc
kc
cc
c
dt
,
де t – час від початку реакції до даного виміру; ,
.
oац
c
–
початкова
концентрація ацетону;
,
оH
c – початкова концентрація іонів водню;
c – концентрація ацетону, що перетворився за час t (збиток концен-
трації).
Розділяючи змінні, одержимо диференціальне рівняння
,
.
,
(
)(
)
oац
oH
dc
kdt
c
cc
c
.
Для інтегрування рівняння потрібно його спрощення. Рівнян-
ня, що представляє собою складний дріб, перетвориться до суми
простих дробів, для яких легко обчислюється інтеграл. Для розкла-
дання складного дробу на прості, використовується метод неви-
значених коефіцієнтів, позначених далі A і B і потребуючі ви-
значення:
,
.
o,
.
,
,
1
(
)
(
)(
)
(
)
oац
ац
oH
oH
A
B
c
c
c
cc
c
c
c
,
,
.
,
(
)(
)
o
oH
oац
oH
AcAcBc
Bc
c
cc
c
.
Дорівнюючи в чисельнику коефіцієнти, що коштують при од-
накових ступенях концентрації c , знайдемо, що
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
264
0
BА
абоАB
.
Дорівнюючи члени, незалежні від c, одержимо
,
.
,
.
,
,
(
)1
oац
oац
oH
oH
Ac
Bc
Ac
c
.
Звідси випливає, що
,
.
,
1
(
)
oац
oH
AB
c
c
.
Таким чином, інтегрування дає рівняння
,
.
,
.
,
.
,
,
,
1
(
)(
)
(
)(
)
oац
oац
oац
oH
oH
oH
dc
dc
dc
c
cc
c
c
c
c
c
c
c
=
,
.
,
,
.
,
1
ln(
) ln(
)
oац
oH
oац
oH
c
c
c
c
const
c
c
.
Звідси остаточно одержуємо
,
,
.
,
.
,
1
ln
oH
oац
oац
oH
c
c
kdt kt const
c
c
c
c
.
Постійну інтегрування const знаходимо з початкових умов:
при 0
t,
0
с і,отже,
const =
o,
.
o,
.
o,
o,
1
ln
ац
ац
H
H
c
c
c
c
.
Тому вираження для константи швидкості реакції прийме вид
o,
.
,
o,
.
o,
.
o,
o,
(
)
2,303
lg
(
)
(
)
ац
x
oH
ац
ац
x
H
H
c
c
c
k
tc
c
c
c
c
.
Хід реакції при проведенні експерименту контролюється по
аналізі проб, що періодично відбираються з реакційної суміші для
визначення концентрації ацетону x
c , еквівалентної кількості йоду,
що прореагував.
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
265
ВИКОНАННЯ РОБОТИ
Дослідження кінетики йодування ацетону проводять у двох
термостатах, установленими на температури, що різняться не менш,
ніж на 10°. Один з дослідів можна проводити при температурі, яка є
в лабораторії. Дослідження при різних температурах проводяться
ідентично, різниться тільки час відбору проб – при більшій темпе-
ратурі проби повинні відбиратися частіше, тому що швидкість реа-
кції більше. Починають із готування реакційної суміші для більш
високої температури досліду.
У мірну колбу ємністю 250 мл наливають 25 мл 0,1 н розчину
йоду в 4% розчині йодистого калію, додають 25 мл (1 М розчину
HCl (концентрацію розчину хлористого водню уточнює викладач) і
заповнюють дистильованою водою, не доливаючи до мітки 5–10 мл.
Колбу занурюють у термостат і витримують 15–20 хв для встанов-
лення заданої температури розчину. Поки колба термостатується
готовлять аналогічним образом реакційну суміш для більш низької
температури.
На аналітичних вагах беруть навішення ацетону (близько 1,5–
2,5 мл). Ацетон додають у реакційну суміш, після чого вміст колби
швидко доводять до мітки дистильованою водою, яка має ту ж тем-
пературу, енергійно перемішують і негайно відбирають піпеткою
першу пробу в кількості 25 мл, а колбу щоб уникнути зникнення
ацетону закривають пробкою. Навішення ацетону беруть із попере-
дньо зваженої крапельниці із притертою пробкою або пікнометра
Оствальда, визначаючи масу ацетону по збиткові маси посудини.
За початок реакції (t =0) ухвалюють час уливання ацетону в реа-
кційну суміш. Відібрану пробу вливають у колбу для титрування, що
містить 25 мл 0,1 н розчину гідрокарбонату натрію Nahco3, реакція з
яким припиняє каталітична дія кислоти й процес йодування ацетону.
Уся операція добавки ацетону, перемішування й узяття проби повинна
зайняти не більш 30–40 с.
Після повторного зважування пікнометра (крапельниці) анало-
гічно проводять уведення ацетону й аналіз першої проби в реакцій-
ній суміші при більш високій температурі.
Зміст йоду визначають титруванням 0,01 н розчином тіосуль-
фату натрію Na2S2O3 у присутності крохмалю як індикатору, при-
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
266
чому розчин крохмалю додають лише наприкінці титрування
(0,5 мл), коли розчин прийме солом'яно-жовтий колір.
За ходом реакції спостерігають шляхом відбору й аналізу
проб через певні проміжки часу, починаючи з 20 хв і поступово збі-
льшуючи до 30 хв до кінця дослідження, яке триває близько двох-
трьох годин. При температурі близько 35° відбір проб проводиться
через 15–20 хвилин.
Часом відбору проби вважають момент вливання її
у розчин бікарбонату натрію. Під час відбору проб колбу з тер-
мостата не виймають!
Результати експерименту рекомендується записувати у вигляді
таблиці
Температурадосліду(С)..............
КонцентраціяNa2S2O3...............
Наважкаацетону....................
Час
по
годин-
нику
Час від
початку
реакції,
t, хв
Кіл-сть
0,01 н
р-ну
Na2S2O3,
мл
,
.
oац
c
ацетону,
г-екв/л
,
oH
c
г-екв/л
x
c
г-екв/л k
Концентрацію ацетону x
c визначається по рівнянню
223
25
o
t
x
NaSO
nn
c
N
,
де tn – кількість 0,01 н Na2S2O3, для кожного відбору проби для ко-
жної температури. Для витрачене на титрування даної проби, мл;
o
n – кількість 0,01 н Na2S2O3, яке повинне було бути витрачений на
титрування в момент початку реакції, мл;
223
NaSO
N
–
нормальність
розчину.
За даними аналізу й розрахунків концентрацій для кожного ві-
дбору проби розраховують значення константи швидкості реакції.
Перевіряють сталість значень константи швидкості цього необхідно
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
267
з 5–6 значень, розрахованих для різних моментів реакції по відбору
проб, виявити значення, що випадають (проводячи Q-тест), розра-
хувати середнє арифметичне значення й знайти погрішність із якої
знайдене значення можна вважати постійною величиною. По зале-
жності константи швидкості реакції від температури, обчислюють
температурний коефіцієнт швидкості реакції :
10
T
T
k
k
поаівнюючи значення з правилом Вант-Гоффа.
Оцінюють енергію активації реакції aE :
2
1
12
21
2,303
(lg
lg)
T
T
a
RTT
k
k
E
TT
.
Оцінюють погрішність розрахованої енергії активації. При ві-
дсутності власних експериментальних даних про константу швид-
кості при іншій температурі значення константи при іншій темпера-
турі можна обрати з довідника.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Чому швидкість реакції йодування ацетону не залежить від
концентрації йоду?
2. Запропонуєте способи визначення приватних порядків реак-
ції йодування.
3. Чому реакцію йодування ацетону можна назвати автокаталі-
тичною?
4. Що називають порядком і молекулярністю реакції?
5. Від яких факторів залежить швидкість хімічної реакції?
6. Виведіть рівняння для обчислення константи швидкості реа-
кції першого порядку, реакції другого порядку за умови однакових
початкових концентрацій реагентів.
7. Що являє собою температурний коефіцієнт швидкості реак-
ції? Як обчислити температурний коефіцієнт швидкості реакції при
двох довільних температурах?
8. У чому сутність визначення змісту йоду тіосульфатом? На-
пишіть відповідні рівняння реакцій.
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
268
9. Який механізм йодування ацетону і яка стадія визначає
швидкість процесу?
10. Як довести, що однієї зі стадій процесу йодування можна
зневажити?
11. Дайте висновок кінетичного рівняння реакції другого по-
рядку за умови різних початкових концентрацій реагентів.
12. Чому відбір проб при вивченні кінетики йодування ацетону
проводиться в розчин бікарбонату натрію?
13. Поясните, як визначають концентрацію ацетону по даним
титрування йоду в суміші тіосульфатом натрію.
14. Чому крохмаль рекомендується додавати для індикації кра-
пки еквівалентності лише наприкінці титрування?
15. Як впливає концентрація ацетону в реакційній суміші на
швидкість хімічної реакції йодування?
Робота No 14.
ВИЗНАЧЕННЯ КОНСТАНТИ ШВИДКОСТІ Й ЕНЕРГІЇ
АКТИВАЦІЇ РЕАКЦІЇ ОМИЛЕННЯ ЕСТЕРУ
Ціль роботи: Визначити середнє значення константи швид-
кості реакції омилення естеру в присутності іонів водню при двох
температурах і оцінити енергію активації реакції омилення.
Прилади й реактиви: термостат, мірні колби на 250 мл, пі-
петка на 10 мл, крапельниця (пікнометр Оствальда) з етилацета-
том, 8 колб 250 мл для титрування, вода з льодом, бюретка з роз-
чином лугу для титрування.
Омилення естеру (метил- або етилацетату) у нейтральному во-
дяному розчині відбувається практично повністю при надлишку во-
ди, але реакція протікає вкрай повільно. У присутності іонів водню,
які каталізують процес омилення, реакція проходить у прийнятний
для дослідження проміжок часу
3
25
2
25
3
k
CH COOC H
HO
CHOH CHCOOH
.
Для типових ефірів механізм кислотного гідролізу включає ряд
стадій:
Робота No 14. ВИЗНАЧЕННЯ КОНСТАНТИ ШВИДКОСТІ Й ЕНЕРГІЇ
АКТИВАЦІЇ РЕАКЦІЇ ОМИЛЕННЯ ЕСТЕРУ
269
1
|
|
'
'
k
O
OH
RCOR
RCOR
(швидко)
2
|
|
'
'
k
OH
OH
RCOR
RCOH
ROH
(повільно)
3
|
||
k
OH
O
RCOH
RCOH
H
(швидко)
Рівновага на першій стадії встановлюється дуже швидко, друга
стадія лімітує загальний процес. Ця стадія й визначає загальну
швидкість процесу – пунктиром відзначений розрив зв'язку кисень –
ацил, стадія включає утвір проміжного продукту при сполуки води
до протонованому естеру:
|
'
|
OH
RCOR
HOH
.
Тому що в результаті самої реакції утворюється кислота, то
омилення естеру в кислому середовищі є автокаталітичним проце-
сом, що ускладнює контроль ходу реакції. Якщо початкова концен-
трація іонів водню велика, то відносна зміна концентрації іонів вод-
ню буде невеликим, тому що в присутності надлишку сильної кис-
лоти рівновага дисоціації продукту реакції – оцтової кислоти буде
повністю зміщене вліво. Таким чином, реакцію омилення проводять
у присутності значної кількості сильної кислоти (
,
HCl24
HSO,
3
HNO ). При великому надлишку води швидкість омилення буде за-
лежати тільки від концентрації естеру, тому що концентрація води
залишається практично постійної, тобто реакція буде першого по-
рядку (псевдомономолекулярна). Швидкість реакції й константа
швидкості визначаться рівняннями
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
270
.
(
)
ест
x
dc
kc
с
dt
,
,
.
,
.
1
ln
o ест
o ест
x
c
k
tc
c
,
де–
,
.
o ест
c
початкова концентрація естеру; x
c – концентрація естеру,
що прореагував (рівний концентрації кислоти, що утворюється); а
(,
.
o ест
c
–
x
c ) – концентрація естеру в цей момент часу t .
ВИКОНАННЯ РОБОТИ
У чисту суху колбу ємністю близько 250 мл наливають вимі-
ряний піпеткою або бюреткою 200 мл 1 М розчину соляної кис-
лоти. Колбу щільно закривають пробкою й термостатують при за-
даній температурі (указує викладач) близько 25 хв. Поки колба тер-
мостатується визначають концентрацію кислоти титруванням лугом
( NaOH ) у присутності фенолфталеїну в якості індикатору. Титру-
вання проводять не менш 3-х разів. У колбу вносять піпеткою бли-
зько 5 мл етилацетату, доводять до мітки дистильованою водою,
знову щільно закривають пробкою, ретельно перемішують. У мо-
мент перемішування включають секундомір, уважаючи цей момент
за час початок реакції.
Проби реагуючої суміші для титрування відбирають піпеткою
на 10 мл не виймаючи колбу з термостата. Пробу поміщають в кол-
бу, що містить 30–40 мл охолодженої на льоді дистильованої води
(0–5 oС) для гальмування реакції омилення в процесі титрування.
Пробу титрують лугом, результат титрування відносять до момен-
ту часу розведення проби в холодній воді. Колбу з сумішшю, що
залишилася щільно закривають пробкою щоб уникнути випару
ефіру під час досліду. Рекомендовані від початку реакції моменти
часу відбору проб для температури 25 °С ставлять: 6, 13, 25, 45, 75,
120, 180 хв. Швидкість реакції зменшується, тому інтервали часу
зростають. При проведенні омилення при більш високих темпера-
турах інтервали відбору проб зменшують, тому що швидкість реак-
ції зростає.
Тому що в міру плину реакції концентрація оцтової кислоти
збільшується, то при титруванні можна швидко доливати таку ж кі-
лькість лугу, який пішов на попереднє титрування, а потім уже обе-
режно дотитрувати розчином лугу.
Робота No 14. ВИЗНАЧЕННЯ КОНСТАНТИ ШВИДКОСТІ Й ЕНЕРГІЇ
АКТИВАЦІЇ РЕАКЦІЇ ОМИЛЕННЯ ЕСТЕРУ
271
Щоб визначити початкову концентрацію етилацетату, необ-
хідно відтитрувати оцтову кислоту в пробі, узятої наприкінці реак-
ції, коли весь ефір піддасться омиленню. Для повного завершення
реакції суміш поміщають на 1 год у гарячу воду (90–95 oС), прохо-
лоджують її до температури досліду й відтитровують останню про-
бу. Можна залишити суміш до наступного заняття й тоді провести
останнє титрування. Для більшої точності це титрування проводять
не менш 3-х разів та беруть середнє арифметичне значення.
При інших температурах дослід проводять аналогічно.
Рекомендована форма ведення протоколу дослідження
ТемпературадослідуC...............
Концентраціялугу................
Номер
проби
Час від
початку
реакції
Об'єм лугу,
що пішов на
титрування, мл
Константа
швидкості
реакції
Тому що при титруванні проб лугом однієї й тієї ж концент-
рації об'єм луги пропорційний концентрації кислоти, а зміна остан-
ньої пропорційне концентрації прореагованого естеру ,
.
o ест
c
(o
VV
);( ,
.
o ест
c
–
x
c )(
t
VV
), розрахунки константи швидкості
реакції при кожній температурі роблять по рівнянню, у якому за-
мість концентрації ефіру підставлені об'єми лугу, що пішов на тит-
рування проб
,
.
,
.
1
2, 303
ln
lg
o ест
o
o ест
x
t
c
VV
k
tc
c
t
VV
.
Визначивши середні значення констант швидкості при різних
температурах, обчислюють енергію активації aE
2
1
12
21
2,303
(lg
lg)
T
T
a
RTT
k
k
E
TT
.
Отримане значення енергії активації реакції омилення естеру
порівнюють з літературним.
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
272
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. У якому випадку можна використовувати рівняння реакції
першого порядку для бімолекулярних реакцій ?
2. Чому молекулярність реакції не завжди збігається з поряд-
ком реакції?
3. Що називається енергією активації реакції?
4. Чому для повного завершення реакції реакційну суміш по-
міщають у гарячу воду?
5. З якою метою проби омилення, що відбираються під час, ві-
дбирають у сильно охолоджену воду?
6. Чому омилення складного ефіру проводять у присутності
великого надлишку кислоти?
7. Опишіть механізм кислотного гідролізу складних ефірів.
8. Виведіть рівняння для константи швидкості реакції першого
порядку.
9. Як залежить період напіврозпаду від початкової концентра-
ції реагуючих речовин для реакції першого порядку? Яка розмір-
ність константи швидкості реакції першого порядку?
10. Приведіть приклади псевдомономолекулярних реакцій.
11. Яким образом визначається початкова концентрація ефіру і
його концентрація в різні проміжки часу для розрахунків значення
константи швидкості реакції?
Робота No 15.
ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ ПЕРОКСИДУ
ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
Ціль роботи: експериментально підтвердити порядок реакції
розкладання пероксиду водню в присутності каталізатора, визна-
чити константу швидкості реакції.
Прилади й реактиви: установка для вивчення швидкості га-
зовиділення, розчин пероксиду водню, розчин каталізатора.
Пероксид водню 2 2
H O мимовільно повільно розкладається з
виділенням
кисню
за
рівнянням
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
273
22
2
2
1
2
k
HO
HO
O
.
Каталізаторами процесу розкладання можуть бути іони
2
Fe
,
3
Fe
,
2
27
CrO
,
2
4
CrO
,
2
4
WO
,
2
4
MoO
і ін., а також змішані каталіза-
тори
4
CuSO +
2
4
MoO
,
4
CuSO +
4
NiSO і т. п. При відповідному до-
борі умов реакція може протікати по механізму, близькому до реак-
ції першого порядку. За ходом реакції зручно спостерігати по зміні
об'єму, що виділяється кисню через певні проміжки часу від почат-
ку реакції.
Гомогенний каталіз диспропорціювання перекиси водню в
присутності сполук заліза вивчений найбільше докладно й глибоко,
хоча й у цьому випадку на деякі питання, що стосуються механізму
реакції (1), не отримане однозначної відповіді. На сьогоднішній
день експериментально більш обґрунтованим уважається циклічний
іон-радикальний (не ланцюговий) механізм диспропорціювання пе-
рекиси водню в системі «аква- іони
2
Fe
–
22
H O » (реагент Фентона).
У даній роботі вивчають кінетику реакції розкладання 2 2
HOв
присутності аква- іонів
2
Fe
у гомогенних умовах при рН = 1–3.
Схематично процес розкладання а моль пероксиду водню можна
представити схемою
22
2
1
2
2
HO
K
M
K
O
a
x
x
,
де K – каталізатор. Рівновага встановлюється швидко, і швидкість
визначальною стадією є розпад M .
Виходячи із припущення, що реакція має перший порядок по 2 2
HO
одержуємо
(2)
'( 2)
dax
kax
dt
,
де( 2
a
x
) – кількість молів 2 2
H O урозчині в момент часуt, a –
вихідна кількість молів 2 2
H O у розчині, x – кількість молів кисню,
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
274
що утворювалося до цього моменту часу,
'
k – ефективна константа
швидкості реакції.
Після перетворень маємо
'(
)'()
2
dx
a
k
xkх
x
dt
,
де
/
dx dt – швидкість утвору кисню, х
–
число молів кисню, яке
можна одержати при повному розкладанні a молів 2 2
H O . Оскільки
при фіксованій температурі й постійному тиску величина x пропо-
рційна об'єму кисню V , що виділився, то
'(
)
dV
kVV
dt
або після інтегрування маємо
ln
'
t
V
kt
VV
.
Вираження для константи швидкості прийме вид
1
'
ln
V
k
tVV
,
звідки випливає, що залежність ln
V
VV
від часу досліду – лінійна,
з кутовим коефіцієнтом, рівним константі швидкості реакції.
ВИКОНАННЯ РОБОТИ
Дослідження кінетики розкладання пероксиду водню прово-
дять на установці, схематично представленої на рисунку 4.1 . Дослід
можна проводити при будь-якій температурі, що позначиться тільки
на загальному часі експерименту.
Перед початком експерименту слід перевірити герметичність
системи. Для цього за допомогою триходового крана 6 з'єднати ви-
мірювальну посудину 3 одночасно з реакційною посудиною 1 і з
атмосферою. Піднімають зрівняльна посудина 4, щоб заповнити
вимірювальну бюретку підфарбованою рідиною до нульової оцінки,
після чого з'єднують бюретку 3 тільки з реакційною посудиною 1 і
опускають посудина 4 приблизно на чверть висоти бюретки. Якщо
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
275
забезпечена герметичність, то рі-
вень затворної рідини в бюретці 3,
опустившись на невелику величину,
повинен зупинитися, не досягнувши
рівня рідини в зрівняльній посудині
й повинен залишатися незмінним
протягом 2–3 хвилин. Після цього
при підніманні зрівняльної посуди-
ни до рівня рідини в бюретці,
останній повинен знову повернути-
ся до нульової оцінки.
Заповнюють реакційна посу-
дина 1 розчином каталізатора (але
вказівці викладача) таким чином,
щоб між пробкою зі скляною труб-
кою й рівнем рідини висота повіт-
ряного простору не перевищувала 2
см. Температуру досліду й каталіза-
тор фіксують у журналі.
Вливають у реакційну посуди-
ну 0.2 –0.5 мл 20–10% водяного роз-
чину 22
H O . Необхідний об'єм роз-
чину 22
H O можна розрахувати по-
передньо з урахуванням об'єму газометричної трубки (50 мл), у яку
буде виділятися кисень при розкладанні пероксиду водню. Розчин
перемішують і реакційна посудину щільно закривають пробкою, що
з'єднує його з газометричною трубкою.
Установлюють однакові рівні рідин у бюретці й у зрівняльній
посудині й роблять перший вимір, зафіксувавши час і рівень у ріди-
ні в газометричній трубці з точністю до 0,1 мл. Це необхідно, щоб
тиск усередині газометричної трубки було рівно атмосферному в
процесі всіх вимірів. Кожний наступний вимір, проведене через 2–5
хвилин, проводять у такий же спосіб, причому рівні рідини в зрівня-
льній посудині й бюретці безупинно підтримуються постійними.
Рис. 4 .1 . Установка для вивчен-
ня кінетики розкладання перок-
сиду водню.
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
276
При проведенні досліду при високих температурах відлік показань
можна робити й частіше.
Необхідно зробити не менш 10–15 аналогічних вимірів, поки
реакція практично не припиниться. Після повного припинення реа-
кції реакційна посудина, з'єднаний з вимірювальною системою, міс-
титься у водяну лазню, нагріту до температури 80–100°С, і витри-
мується в ній до повного розкладання пероксиду водню (15–20 хв).
Реакція вважається закінченої, якщо рівень газу в трубці 3 не змі-
нюється. При кип'ятінні на водяній лазні зрівняльна посудина підт-
римується в найбільш високому положенні.
Реакційна посудина прохолоджується в термостаті до темпера-
тури проведення досліду, витримується 20–25 хвилин, після чого
виміряється рівень рідини в бюретці при рівності його з рівнем у
зрівняльній посудині.
Фіксацію змін об'єму в ході розкладання пероксиду водню по-
чинають на відразу. Спочатку рівень рідини в бюретці змінюється
повільно внаслідок розчинення кисню в рідині до насичення. Тільки
після цього об'єм кисню, що виділився, можна вважати пропорцій-
ним кількості, що розклався пероксиду водню. Тому при обробці
експериментальних даних уважно аналізують загальний хід проце-
су, ухвалюючи за початок реакції часто не перші обмірювані зна-
чення, починаючи зі значень, що характеризують процес, що вста-
новився.
За експериментальним даними будуються графіки залежності
1) різниці об'ємів кисню, що виділився, від (
)()
t
VVft
часу (на осі абсцис відкладати час у хвилинах);
2) логарифма швидкості реакції від логарифма різниці об'ємів
виділеного кисню lg
[lg(
)]t
vf
VV
; швидкість визначити по
тангенсу кута нахилу дотичній до кривої в координатах
(
)()
t
VVft
до осі часу;
3) логарифма різниці об'ємів кисню від часу
lg(
)()
t
VVft
.
Останні два графіки використовують для визначення порядку
реакції. Далі проводиться розрахунки константи швидкості реакції
Робота No 15. ВИЗНАЧЕННЯ ШВИДКОСТІ РОЗКЛАДАННЯ
ПЕРОКСИДУ ВОДНЮ ГАЗОМЕТРИЧНИМ МЕТОДОМ
277
за допомогою графіка lg(
)()
t
VVft
й аналітичним шляхом, ви-
користовуючи рівняння
1
2,303
ln
lg
a
a
k
tax
t
ax
,
де концентрація пероксиду водню в початковий момент часу й у
момент виміру t заміняється різницею об'ємів кисню
2,303
lg
t
V
k
t
VV
,
де V
–
об'єм кисню, що виділився після розкладання всього перок-
сиду водню (визначається як різниця рівнів у бюретці в момент,
прийнятий за початок реакції, і після кип'ятіння H2O2 до повного
розкладання).
Рекомендована форма ведення протоколу дослідження
Температурадосліду,°C ....... Каталізатор..........
Кількістьпероксидуводню.................
No
вимі-
ру
Час
виміру
Час
від поча-
тку
реакції
Рівень
рідини в
бюретці
Об'єм
виділ.
газу
Швид-
кість
реакції
lg(
)t
VV
k
За отриманими значенням константи швидкості, розрахованої
за різні проміжки часу, визначають середнє значення константи.
Використовуючи результати розрахунків і отримані графіки,
установлюють, чи протікає реакція по першому порядкові. При зна-
чних відхиленнях від першого порядку розрахувати константу шви-
дкості за допомогою графіка lg
()
cft
.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Чому пероксид водню схильний до розкладання?
2. Чому проведення вимірів об'єму, що виділяється кисню по-
чинають через деякий час від початку процесу?
3. Якими способами можна впевнитися про проходження реа-
кції по першому порядкові?
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
278
4. Чому порядок реакції розкладання пероксиду водню може
відрізнятися від першого?
5. Чому при роботі із приладом у момент виміру об'єму рівні
рідини в бюретці й зрівняльній посудині зрівнюють?
6. Яким чином можна визначити концентрацію пероксиду во-
дню в узятому зразку?
7. У чому полягає теорія гомогенно-каталітичних реакцій?
Приведіть приклади гомогенно-каталітичних реакцій.
8. Поясните причину зміни енергії активації процесу при вве-
денні в систему каталізатора, що перебуває в одній фазі з реагую-
чою речовиною.
9. Чому середовище реакційної суміші має бути кислим.
10. Як можна статистично затвердити сталість константи шви-
дкості реакції та відсутність тренда?
Робота No 16.
ВИВЧЕННЯ ШВИДКОСТІ РЕАКЦІЇ ГІДРАТАЦІЇ ОЦТОВОГО
АНГІДРИДУ МЕТОДОМ ЕЛЕКТРОПРОВІДНОСТІ
Ціль роботи: використовуючи метод електропровідності для
вивчення кінетики реакції, визначити аналітично й графічно серед-
ню константу швидкості, енергію активації й передекспоненційний
множник реакції гідратації оцтового ангідриду.
Прилади й реактиви: термостат, кондуктометр або LCR-
метр, колба 50 мл, мірна піпетка 10 мл, оцтовий ангідрид, дисти-
льована вода.
Метод вивчення електропровідності можна успішно викорис-
товувати для вивчення кінетики реакції в тих випадках, коли в ході
реакції змінюється електропровідність системи. Метод зручний тим,
що дозволяє безупинно стежити за ходом реакції без відбору проб.
Наприклад, у ході реакції гідратації оцтового ангідриду
3
2
2
3
(
)
2
k
CHCOO HO
CH COOH
електропровідність системи згодом значно зростає внаслідок утвору
оцтової кислоти. Тому що дана реакція є псевдомономолекулярної,
те вона підкоряється кінетичному рівнянню першого порядку
Робота No 16. ВИВЧЕННЯ ШВИДКОСТІ РЕАКЦІЇ ГІДРАТАЦІЇ
ОЦТОВОГО АНГІДРИДУ МЕТОДОМ ЕЛЕКТРОПРОВІДНОСТІ
279
2,303
lgo
o
x
c
k
t
cc
,
деo
cйx
c – концентрації оцтового ангідриду в початковий момент
часу й того, що прореагував за проміжок часу t відповідно.
Кількість оцтової кислоти, що утворювався, у ході реакції про-
порційно вихідної концентрації оцтового ангідриду. Тому що збі-
льшення електропровідності системи згодом пропорційно концент-
рації, що утворювався оцтової кислоти, то воно, отже, пропорційно
початкової концентрації оцтового ангідриду. Тому можна записати
(
)
o
o
c const
;
[(
)(
)]
(
)
o
x
o
t
o
t
cc
const
,
деo
,t
,
–
питома електропровідність системи в початковий
момент, у цей момент часу й наприкінці реакції відповідно.
Користуючись цими вираженнями можна привести кінетичне
рівняння до наступного виду
2,303
lg
o
t
k
t
.
Оскільки експериментально визначається не електропровідність
розчину, а його опір R, те, заміняючи питому електропровідність
на опір згідно з рівнянням
x
l
K
R
K
S
,
де l – відстань між електродами, S – площа електродів, K – постійне
гнізда, – питомий опір розчину, – питома електропровідність
розчину. Можна здійснювати розрахунок константи швидкості реа-
кції по рівнянню через електричні параметри
(
)
2,303
lg
(
)
to
o
t
RRR
k
t
RRR
.
Тому що перший вимір опору розчину проводиться після закін-
чення деякого часу від початку реакції, то величина опору oR ви-
значається екстраполяцією. Для цього будується графік у координа-
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
280
тах lg
(
)
t
t
R
t
RR
, на якому експериментальні крапки повинні ук-
ладатися на пряму. Екстраполяцією отриманої прямої на
0
t зна-
ходять lg
(
)
o
o
R
RR
і розраховують значення o
R , а з нахилу прямої
до осі часу t – середнє значення константи швидкості
.
cер
k . Обчис-
люючи значення константи швидкості реакції для кожного моменту
часу, визначають середнє значення й порівнюють його з
.
cер
k , отри-
маним графічно.
Якщо робити вимір зміни опору розчину при двох температурах,
то можна визначити енергію активації aE даної реакції за форму-
лою
2
1
21
12
lg
2,303
T
a
T
k
E
TT
k
RTT
,
а з рівняння Арреніуса
0
a
E
RT
kke
.
знайти значення передекспоненційного множника В.
ВИКОНАННЯ РОБОТИ
Термостат установлюють на зазначену викладачем температу-
ру й підготовляють до роботи прилад для виміру електропровіднос-
ті (або опору) розчину. Наливають у колбу ємністю 50 мл 6 мл оц-
тового ангідриду й доводять об'єм розчину до мітки дистильованою
водою, попередньо термостатованої за заданої температури. У мо-
мент початку додавання води включають секундомір і не виключа-
ють його до кінця всього експерименту. Відзначають за секундомі-
ром час початку й кінця розчинення оцтового ангідриду (суміш ене-
ргійно збовтується при додаванні води й фіксується момент зник-
нення каламуті, що з'явилася, – ознакою кінця розчинення). Серед-
ній час приймається за час початку реакції.
Посудина для виміру електропровідності двічі споліскується
досліджуваним розчином і заповнюється так, щоб електроди вияви-
Робота No 16. ВИВЧЕННЯ ШВИДКОСТІ РЕАКЦІЇ ГІДРАТАЦІЇ
ОЦТОВОГО АНГІДРИДУ МЕТОДОМ ЕЛЕКТРОПРОВІДНОСТІ
281
лися нижче рівня розчину на 0,5–1 см. Комірку поринають в термо-
стат і через 3–5 хв приступають до виміру електропровідності. При
температурі реакційної суміші близько 298 К перші 2–3 виміри роб-
лять із інтервалом 30 с, 4–5 вимірів з інтервалом 1 хв, 2–3 виміру з
інтервалом 5 хв. Наступні виміри роблять через 10 хв, через 1 год і
ще через 1 годину. Закінчення реакції визначається по сталості зна-
чень опору розчину. Чим вище температура досліду, тим частіше
необхідно проводити виміри електропровідності.
Результати експерименту заносять у таблицю
Температурадосліду...............
Концентраціярозчину............
No
Час
виміру
по го-
динни-
кові
Час від
початку
реакції
t
RR
t
t
R
RR
lg
t
t
R
RR
o
Rk
Для розрахунків енергії активації даної реакції виміру константи
швидкості проводять при іншій температурі, що відрізняється від
попередньої не менш, чим на 10–15o (указує викладач).
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Чому реакцію гідратації оцтового ангідриду відносять до
псевдомономолекулярнмх?
2. Чому за ходом реакції гідратації оцтового ангідриду можна
стежити по зміні електропровідності реакційної суміші?
3. Дайте висновок рівняння для константи швидкості реакції
гідратації оцтового ангідриду через значення питомої електропро-
відності розчину й через значення опору.
4. Який момент ухвалюють за час початок реакції гідратації
оцтового ангідриду?
5. Як визначається значення опору розчину в початковий мо-
мент часу?
6. За якою ознакою можна судити про закінчення процесу гід-
ратації оцтового ангідриду?
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
282
7. Як по залежності константи швидкості реакції від температу-
ри визначити передекспоненційний множник у рівнянні Арреніуса?
8. Приведіть приклади процесів, швидкість яких можна конт-
ролювати по зміні електропровідності реакційної суміші.
9. У чому полягає принцип виміру електропровідності розчинів?
10. Чому при проведенні реакції гідратації оцтового ангідриду
при більш високих температурах необхідно частіше проводити від-
ліки зміни опору реакційної суміші?
11. Поясніть причини вибору концентрації оцтового ангідриду
для вивчення реакції омилення кондуктометричним методом.
Робота No 17.
КІНЕТИКА ОКИСЛЮВАННЯ ЙОДИД-ІОНІВ
ПЕРОКСИДОМ ВОДНЮ
Ціль роботи: визначити константу швидкості реакції окис-
лювання йодид-іонів пероксидом водню за двома температурами,
визначити порядок реакції за йодид-іонами. За температурною за-
лежністю константи швидкості знайти енергію активації реакції.
Прилади й реактиви: колба на 250 мл, колба на 50 мл, мірний
циліндр на 100 мл, бюретка на 25 мл, мірний циліндр на 10 мл, секу-
ндомір, механічна пінетка, магнітна мішалка з нагрівачем, 0,1 н ро-
зчин KI (40–100 мл), 2 н розчин 2 4
HSO,0,033нрозчин 2 2
H O (10-20
мл), 0,5 % розчин крохмалю, 0,05 н розчин
223
Na S O (20 мл), насиче-
ний розчин молібдату амонію.
У водяному розчині в кислому середовищі між йодид іонами
на пероксидом водню протікає реакція згідно рівнянню:
22
2
2
2
2
2
IHO
H
I
HO
.
Механізм реакції включає кілька стадій, перша з яких є най-
більш повілбною та лімітуючоює:
22
2
HOI
HO IO
.
За нею слідкує друга стадія, що поєднує декілька швидких ре-
акцій, типу:
2
2
IOH
IHO
,
Робота No 17. КІНЕТИКА ОКИСЛЮВАННЯ ЙОДИД-ІОНІВ
ПЕРОКСИДОМ ВОДНЮ
283
які не є єдиними із запропонованих механізмів реакції.
Кінетичне дослідження проводиться шляхом хімічного аналізу
молекулярного йоду, що утворюється в ході реакції. Титрування
надлишком розчину тіосульфату натрію здійснюється безпосеред-
ньо в реакційної посудині методом внутрішнього титрування. У то-
чці еквівалентності розчин здобуває блакитного забарвлення через
утворення комплексної сполуки йоду із крохмалем. Блакитний ко-
лір виникає раптово й відразу, через строго певний період часу. То-
му ціла серія таких реакції відома під назвою «хімічних» або «йод-
них» годинників.
Йод, що утворюється, практично миттєво реагує з тіосульфа-
том:
2
2
2
23
46
2
2
I
SO
ISO
.
Концентрація вільного йоду дуже мала. Час, що проходить від
моменту додавання до розчину певної кількості тіосульфату до мо-
менту появи блакитного забарвлення, дорівнює часу витрати екві-
валентної кількості пероксиду водню в реакції з йодидами-іонами.
Як видно, йодиди-іони регенеруються під час титрування. Тому кон-
центрація I
в ході досліду не змінюється й дорівнює початковій.
У кислотному буферному розчині кінетичне рівняння дається
вираженням:
22
22
[]'[][]
dHO
kHOI
dt
,
де'
k – константа швидкості, – порядок реакції за йодид іонами.
Оскільки при наданому порядку експерименту [ ]
I
= const, то, по-
єднуючи '
kз[]
I
, одержимо рівняння
22
22
[][]
dHO
kHO
dt
,
де k – константа швидкості псевдопершого порядку, тобто
'[
]
kkI
.
Остаточно після інтегрування одержимо вираження для розра-
хунку константи швидкості реакції k :
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
284
0
22
22
[]
1
ln
[]
HO
k
t
HO
,
де
0
22
[]
H O – початкова концентрація пероксиду водню.
Позначимо V
–
об'єм розчину
223
NaSO,щопішовнатитру-
вання всього йоду, а V – об'єм, що витрачений до моменту часу t .
Тоді константу швидкості реакції k можна розрахувати через об'є-
ми за формулою:
1
ln
V
k
tVV
,
оскільки
0
22
[]
V
HO
,а
22
[]
VHO
.
ВИКОНАННЯ РОБОТИ
У колбу ємністю 50 мл налити 0,033 н розчин 2 2
H O (приблиз-
но2мл3% 22
H O в 120 мл води). У ретельно вимиту склянку ємні-
стю250млвлитирозчин,5мл2н 2 4
H SO і стільки води, щоб після
приливания розчину 2 2
H O об'єм реакційної суміші був рівним 100
мл. Об'єми розчинів KI і 2 2
H O задаються викладачем. Відзначають
температуру досліду.
Над колбою встановлююит бюретку з 0.05 н розчином тіосу-
льфату натрію 2 2 3
Na S O . Колбу ставлять на магнітну мішалку. До-
ливають 1 мл титранту й додають 5 крапель 0,5 % розчину крохма-
лю. Потім вливають в склянку розчин пероксиду водню
22
HO.
Включають магнітну мішалку на 600 об./хв. У момент уливання
22
H O включають секундомір. З появою стійкого синього забарв-
лення помічають час, швидко додають 1 мл
223
NaSO –другупор-
цію – і записують час появи забарвлення. Такі операції повторити
6–8 разів при включеному секундомірі.
Потрібно постійно стежити за реакційною посудиною, тому
що забарвлення розчину з'являється раптово. Не можна пропускати
час додавання чергової порції титранту. Для кращого відзначення
забарвлення розчину, рекомендується ставити за склянкою білий
папір.
Робота No 17. КІНЕТИКА ОКИСЛЮВАННЯ ЙОДИД-ІОНІВ
ПЕРОКСИДОМ ВОДНЮ
285
При кімнатній температурі реакція йде до кінця досить довго.
Тому для знаходження значення V
–
сумарного об'єму розчину
223
Na S O , що піде на титрування, реакцію прискорюють, додаючи
до реакційної суміші 5 крапель каталізатору – насиченого розчину
молібдату амонію. Йод, що виділився, відтитровують, звідки зна-
ходять V
.
Титрування ведуть тільки до знебарвлення розчину. Не
слід домагатися знебарвлення осаду йодкрахмального комплексу,
тому що це може привести до перетитрування.
Результати досліду й результати розрахунків заносять у таб-
лицю за формою:
Концентрація розчину KI . . . . . . . . моль/л;
Об'єм 2 2
HO ....................мл;
Температурадосліду.............°С
Загальний об'єм
223
NaSO,V∞......мл
No
п\п
V,мл
Час t від початку
моменту реакції до
появи забарвлення
VV
мл
ln(
)
VV
Константа
швидкості, k
Розрахунок константи швидкості k за даною темпертаурою
проводять за розрахунковою формулою, а також графічно – на підс-
таві залежності ln (
)
VV
від часу t (за кутом нахилу лінійної за-
лежності). Обчислюють середнє значення
.
cp
k , оцінюють погріш-
ність.
Для визначення порядку реакції проводять другий дослід із
удвічі меншою або вдвічі більшою концентрацією йодид іонів I
.
Загальний об'єм розчину (100 мл) і температура дослвду залиша-
ються як раніше. За даними аналогічного дослідження визначають
константу швидкості "k , потім розраховують – порядок реакції за
йодид-іоном:
ln"ln'
ln[ ]" ln[ ]'
k
k
I
I
,
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
286
де індекси « ' » й « " » позначають номери дослідів.
Знайдене значення округлююит до найближчого цілого числа
й приймають його у чкості параметра кінетичного рівняння окис-
лювання йодид-іонів пероксидом водню. Константу швидкості 'k
також обчислюють та враховуючи вказують її розмірність.
Третій дослід проводять при збільшенні температури на 20°.
Значення констант швидкості реакції k й 'k визначають аналогічно.
За результатами дослідів, що проведені за різними температу-
рами, розрахувують енергію активації реакції:
1
2
12
12
'
ln
'
Т
a
Т
k
RTT
E
TT
k
,
де 1'
kй2'
k – константи швидкості при температурах 1
Tй2T.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Чому реакцію окислювання йодид-іонів пероксидом водню
вважають реакцією псевдопершого порядку?
2. За рахунок чого вважають, що крнцентарція йодид іонів в
реакційній сцміші остається постійною?
3. Як визначають початкову концентрацію пероксиду водню?
4. Для чого наприкінці реакції в реакційну суміш уводять мо-
лібдат амонію?
5. Від яких факторів залежить час появи забарвлення розчину?
6. Про що свідчить відмінність від одиниці ступеня при конце-
нтрації йодид іонів в кінетичному рівнянні?
7. Як і чому змінюється час забарвлення при зміні температури?
8. За яким методом визначають порядок реакції в роботі?
9. Для чого негайно після забарвлення розчину вливають визна-
чену кількість розчину тіосульфату натрію?
10. Для чого в розчин додають сульфатну кислоту, що обумовлює
кисле середовище?
11. В якому напрямі впливає збільшення концентрації йодиду ка-
лію в розчині. Чи є однаковий вплив такого ж збільшення концентра-
ції пероксиду водню?
Робота No 17. КІНЕТИКА ОКИСЛЮВАННЯ ЙОДИД-ІОНІВ
ПЕРОКСИДОМ ВОДНЮ
287
ТЕОРЕТИЧНІ ПИТАННЯ ПО ТЕМІ
1. Дайте визначення середньої й істинної швидкості хімічної
реакції.
2. Як змінюється швидкість хімічної реакції й концентрації ре-
агуючих речовин у часі? Приведіть графічні залежності.
3. Що називають константою швидкості хімічної реакції і який
її фізичний зміст?
4. Що називається молекулярністю й порядком реакції?
5. У чому причини частої невідповідності порядку реакції її
молекулярності? Що відбиває молекулярність і порядок реакції?
6. Які реакції називають простими, а які складними? Назвіть
відомі вам типи складних реакцій,
7. У чому суть основного закону кінетики для найпростіших
реакцій.
8. Що називається періодом напіврозпаду і як він залежить від
початкової концентрації реагуючих речовин для реакцій 1-го, 2-го,
n-го порядку?
9. Які методу визначення порядку реакції вам відомі й на чому
вони засновані?
10. Виходячи з яких положень визначають швидкість складної
хімічної реакції?
11. У чому полягає принцип незалежності елементарних стадій
складної хімічної реакції?
12. Як розрахувати константу швидкості прямої реакції, якщо
відома константа швидкості зворотної? Чи Існує зв'язок між конста-
нтою швидкості прямій і зворотної реакцій і максимальною робо-
тою цього процесу?
13. Як можна вивести рівняння Арреніуса термодинамічним
методом?
14. Що таке енергія активації хімічної реакції й від чого вона
залежить?
ЛІТЕРАТУРА
1. Герасимов Я. И. и др. Курс физической химии. Т.2.
–
М.:
Химия, 1973. – 625 с.
2. Лебідь, В. І. Фізична хімія : підруч. для студ. вищ. навч.
закл. / В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
4. КІНЕТИКА ГОМОГЕННИХ РЕАКЦІЙ
288
3. Пурмаль А. П. А, Б, В... химической кинетики / А. П. Пур-
маль. — М.: ИКЦ «Академкнига», 2004. — 277 с.
4. Физическая химия. Теоретическое и практическое руковод-
ство. Изд. 2 –е, переработанное и дополненное. / Под ред. академи-
ка Б. П. Никольского. – Л. : Химия, 1987. – 880 с.
5. Практикум по физической химии / Под ред. В . В. Буданова
и Н. К. Воробьева. – М. : Химия, 1986. – 351 с.
6. Практикум по физической химии / Под ред. И. В . Кудряшо-
ва. –М. : Высшая школа, 1986. – 495 с.
7. Практикум по физической химии. Кинетика и катализ.
Электрохимия : учеб. пособие для студ. учреждений высш. проф.
образования / [А. В .Абраменков и др.] ; под ред. В . В . Лунина, Е. П.
Агеева. — М. : Издательский центр «Академия», 2012. – 304 с.
8. Практикум по физической химии. Учебное пособие для ву-
зов. Изд. 3 –е. / Под ред. С. В. Горбачева, – М. : Высшая школа,
1974. – 495 с.
9. Стромберг А. Г., Семченко Л. П. Физическая химия, учеб-
ник для студентов вузов, обучающихся по химическим специально-
стям / А. Г. Стромберг, Д. П. Семченко. - 6 -е изд., стер. – М. : Выс-
шая школа, 2006. – 527 с.
10. Практические работы по физической химии : Учебное по-
собие для вузов / Под ред. К. П. Мищенко, А. А. Равделя, A. M. По-
номаревой. – СПб. : Изд-во «Профессия», 2002. – 384 с.
11. Еремин Е. Н. Основы химической кинетики.
-М.: Высшая
школа, 1976. – 541 с.
12. Эмануэль Н. М., Кнорре Д. Г . Курс химической кинетики :
Учебник для хим. факультетов.
–
4-е изд., перераб. и доп.
–
М.:
Высш. шк ., 1984. – 463 с.
13. Физическая химия. В 2 кн. Кн. 2 . Электрохимия. Химиче-
ская кинетика и катализ: Учеб. для вузов / К. С . Краснов, Н. К . Во-
робьев, И. Н. Годнев и др.; Под ред. К . С. Краснова –3 -е изд., испр.
–
М. : Высш. шк., 2001. – 319 с.
14. Вишняков А. В. Физическая химия. / А. В . Вишняков, Н. Ф.
Кизим.
–
М. : Химия, 2012. – 839 с.
289
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ.
КАТАЛІЗ
5.1 ЗАГАЛЬНІ ПОНЯТТЯ Й ЗАКОНОМІРНОСТІ
ГЕТЕРОГЕННИХ ПРОЦЕСІВ
Фізико-хімічні процеси, у яких беруть участь речовини, що пе-
ребувають у різних фазах, називаються гетерогенними. До них ста-
вляться процеси випару й конденсації, кристалізації й розчинення,
хімічні реакції на границі двох фаз, гетерогенний каталіз, електрохі-
мічні процеси на границі електрод – розчин і ін.
Особливість гетерогенних процесів полягає в тому, що акт пе-
ретворення речовин відбувається на границі роздягнула фаз, напри-
клад, тверде тіло – розчин. Це вносить істотні відмінності в кінети-
чні закономірності в порівнянні з кінетикою гомогенних процесів.
Тому що в гетерогенній реакції беруть участь речовини, що перебу-
вають у різних фазах, те одночасно з хімічною реакцією повинні
протікати й процеси, пов'язані з переходом речовин з однієї фази в
іншу. Такі процеси протікають звичайно через стадію адсорбції на
поверхні роздягнула фаз із наступною десорбцією в об'єм іншої фа-
зи або розчиненням у ній. Часто буває значна й роль дифузійних
стадій, що забезпечують обмін частками між приповерхніми шара-
ми й іншим об'ємом фази. Таким чином, гетерогенна реакція зви-
чайно включає не тільки чисто хімічні реакції, але й ряд стадій ін-
шої природи (адсорбція, десорбція, розчинення, кристалізація, ди-
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
290
фузія). При кінетичному описі гетерогенної реакції доводиться вра-
ховувати вплив цих стадій.
Інша особливість гетерогенних реакцій – локалізація або на
поверхні розділу фаз або в об'ємі однієї з фаз. На поверхні роздяг-
нула фаз, як правило, протікають реакції за участю твердих речо-
вин, в об'ємі фази – реакції за участю рідини. Зустрічаються й більш
складні випадки, коли одні реакції протікають на поверхні роздяг-
нула фаз, а інші в об'ємі фази.
Тому що в гетерогенній реакції « беруть участь», як мінімум,
дві фази, те хоча б одна з них повинна бути конденсованої, тобто
являти собою тверде тіло або рідина. При звичайних способах про-
ведення реакцій за участю конденсованої фази потік реагентів з ру-
хливої фази (газ, рідину) пропускають через фіксовану кількість не-
рухливої фази (рідина, тверде тіло). Якщо нерухливою фазою є рі-
дина, то найбільш близький до цієї схеми барботажний реактор, у
якому пухирці газу барботують через рідину; якщо нерухлива фаза
–
тверде тіло, то реакцію звичайно проводять, пропускаючи потік
газу або рідини через шар зерен твердого реагенту. У всіх цих випа-
дках система не є повністю відкритою: сполука й властивості кон-
денсованої фази змінюються в часі, що обумовлює нестаціонарність
гетерогенного процесу. Тому гетерогенні реакції, як правило і їх кі-
нетичний опис фактично є описом еволюції системи – зміни її влас-
тивостей у часі.
Тому що взаємодія відбувається на межі розділу фаз, швид-
кість гетерогенних процесів залежить від площі поверхні розділу
фаз, її стану й природи.
У зв'язку із цим логічно перевизначити швидкість хімічної ре-
акції: замість об'ємної щільності швидкості реакції, як це прийняте
для гомогенних реакцій ( V
v ), уводять поверхневу щільність швид-
кості хімічного перетворення ( S
v):
1
S
d
v
Sd
,
де S – площа поверхні роздягнула фаз, на якій локалізована дослі-
джувана реакція.
Для частки, хоча й дуже розповсюдженого, случаючи границі
розділу фаз тверда речовина – рідина швидкість реакції зв'язують
5.1 ЗАГАЛЬНІ ПОНЯТТЯ Й ЗАКОНОМІРНОСТІ ГЕТЕРОГЕННИХ ПРОЦЕСІВ
291
зі швидкістю зменшення концентрації вихідного реагенту
.
р
dс
dt
.
У
цьому випадку поверхнева швидкість реакції при постійному об'ємі
жилкою фази буде рівна
.
.
v
р
S
р
dс
V
Sv dt
,
де V – об'єм рідкої фази;
.
р
v – стехіометричний коефіцієнт (який
ураховується зі знаком мінус, тому що речовина вихідна). Характе-
рним для рівняння є те, що швидкість гетерогенного процесу суттє-
во залежить від відношення /S V .
Особливістю гетерогенних процесів є їх багатостадійність, що
проявляється в наявності не менше чому трьох стадій: 1) доставки
речовини з розчину до поверхні твердої фази, 2) власно процесу на
поверхні роздягнула фаз і 3) відводу продуктів взаємодії від поверхні
в об'єм розчину. Ці стадії протікають одна за іншою – послідовно.
Швидкість гетерогенного процесу в цілому залежить від шви-
дкості окремих стадій і їх співвідношення. При послідовному проті-
канні процесів процес із найменшою швидкістю буде визначати за-
гальну швидкість процесу – таку
стадію процесу називають лімі-
туючою.
Доставка речовини до границі
фаз і відвід продуктів взаємодії в
об'єм розчину найбільше часто здій-
снюється за допомогою дифузії –
переміщення речовини в результаті
хаотичного теплового руху його ча-
стинок.
Якщо визначальною стадією є
сам процес на поверхні розділу фаз,
то прийнято говорити, що гетеро-
генний процес протікає в кінетич-
ній області (рис. 5.1, I). У тому
випадку, коли найбільш повільними
стадіями є стадії підведення й від-
Рис. 5 .1 . Області протікання ге-
терогенних хімічних процесів.
I – кінетична, II –дифузійна,
III – перехідна області.
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
292
воду відповідних речовин шляхом дифузії, гетерогенний процес
уважається, що протікають у дифузійній області (рис. 5.1.II). Те-
мпература набагато сильніше впливає на швидкість хімічних проце-
сів, чим на дифузію. Тому гетерогенні хімічні реакції при підви-
щенні температури можуть перейти з кінетичної області в дифузій-
ну. Існує й перехідна область, для якої швидкості реакції й дифузії
порівнянні між собою (рис. 5.1, III).
Процес дифузії є двостороннім. Напрямок і інтенсивність ди-
фузії визначається градієнтом концентрації /
dc dx або, точніше,
градієнтом хімічного потенціалу /
ddx
. Рушійною силою дифузії є
прагнення системи до термодинамічної рівноваги шляхом вирівню-
вання хімічного потенціалу при відповідному розподілі речовини.
Швидкістю дифузії
.
vдиф називається кількість речовини m ,
що проходить через даний поперечний переріз площі S в одиницю
часуt,( /)
dmdt.
Кількісні закономірності описує перший закон Фіка, який за-
тверджує, що маса речовини dm , стерпного дифузією в напрямку
деякої осі x через перпендикулярну цьому напрямку майданчик S
а час dt , пропорційна градієнту концентрацій /
dc dx уздовж цього
напрямку:
dc
dmDSdt
dx
,
де знак «–» показує, що дифузія відбувається в тому напрямку, у
якому убуває концентрація; D – коефіцієнт дифузії, що чисельно
дорівнює кількості речовини, яка дифундує в одиницю часу через
одиничну площу поверхні, перпендикулярної напрямку дифузії, при
градієнті концентрації, рівним одиниці.
Коефіцієнт дифузії D, або питома швидкість дифузії, залежить
від термодинамічних параметрів процесу, природи середовища ре-
човини, що й дифундує. Розмірність D = [довжина]
2
/[час]. За темпе-
ратур близьких до 273 К коефіцієнт дифузії в газах має порядок 10–1
см
2
/с, у рідинах 10–5
см
2
/с, у твердих тілах 10–20
см
2
/с. У деяких кон-
кретних випадках відмінності в значеннях коефіцієнта дифузії в ма-
сі того самого речовини можуть досягати 10–12 порядків і залежать
5.1 ЗАГАЛЬНІ ПОНЯТТЯ Й ЗАКОНОМІРНОСТІ ГЕТЕРОГЕННИХ ПРОЦЕСІВ
293
від маси й розмірів молекул речовин, що беруть участь у дифузії,
від температури й в'язкості середовища.
Коефіцієнт дифузії сферичних часток у безперервному середо-
вищі з в'язкістю визначається по рівнянню Стокса-Ейнштейна
6
A
RT
D
r
N
,
де r – радіус частинки, що рухається; – в'язкість середовища;
A
N – число Авогадро.
На підставі закону Фіка швидкість дифузії записується рівнян-
ням
.
диф
dm
dc
v
DS
dt
dx
.
Залежність концентрації від часу для фіксованого перетину
встановлюється за допомогою другого закону Фіка
2
2
dc
dc
D
dt
dx
.
Якщо зміна концентрації не залежить від часу, а змінюється
тільки з відстанню, то дифузія носить стаціонарний характер
/0
dcdt ,а
/
dc dx a const
. Після інтегрування можна одержати
o
ccax
,
деo
c відповідає координаті х = 0.
Звідси випливає, що при стаціонарній дифузії спостерігається
лінійна зміна концентрації уздовж напрямку дифузії, а градієнт
концентрації записується тоді за допомогою скінченних величин
o
cc
dc
a
dx
,
де – деяке кінцеве значення координати х.
Коефіцієнт дифузії збільшується з підвищенням температури
за законом, аналогічному рівнянню Арреніуса
exp
a
E
Dk
RT
,
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
294
деa
E – енергія активації процесу дифузії, що рідко перевищує
величину в 4–10 кДж/моль, тобто в багато разів менше енергій ак-
тивації більшості хімічних реакцій.
Розчинення твердих тіл у рідинах є складним процесом і кіне-
тично складається із двох послідовних стадій. Перш а – перехід мо-
лекул або іонів, які утворюють кристалічну решітку речовини, що
розчиняється, із твердої фази в рідкий розчин у результаті взаємодії
з молекулами розчинника – сольватації. Швидкість цього процесу
дуже велика й кількість частинок розчиненого речовини в розчині
поблизу поверхні кристала швидко збільшується, досягаючи грани-
чного стану, що характеризує стан насиченого розчину. Друга
стадія – зіткнення сольватованих частинок розчиненого речовини
з молекулами розчинника й дифузія їх в об'єм розчину. Процес ди-
фузії протікає значно повільніше, чим процеси міфазових переходів,
і швидкість процесу розчинення в більшості випадків визначається
швидкістю дифузії.
Дійсно, тонкий шар розчину, що безпосередньо прилягає до
поверхні твердої фази, практично насичений, додатково розчинити-
ся може тільки така кількість речовини, яка за такий самий час про-
дифундує із прикордонного шару вглиб розчину.
Схематично границя розділу кристал – розчин представлена на
рисунку 5.2 . Нехай площа поверхні кристала речовини, що розчиня-
а)
б)
Рис. 5 .2 . Залежність концентрації від відстані до твердої поверхні а) за ак-
тивним перемішуванням розчину; б) за відсутності перемішування – товщи-
на дифузійного шару максимальна й градієнт концентрації не є постійним.
5.1 ЗАГАЛЬНІ ПОНЯТТЯ Й ЗАКОНОМІРНОСТІ ГЕТЕРОГЕННИХ ПРОЦЕСІВ
295
ється, зануреного в ненасичений розчин, дорівнює S , об'єм розчи-
ну дорівнює V , а концентрація розчину c . Через деякий час приля-
гаючий до поверхні кристала шар розчину стане майже насиченим і
концентрація в ньому стане рівною максимальної
.
макс
c
.
Із цього
шару речовина дифундує вглиб розчину. Позначимо через (рис.
5.2 а), відстань від поверхні кристала, де концентрація
.
макс
c
,до
найближчої точки, у якій концентрація розчину ще не змінилася й
ще дорівнює концентрації вихідного розчину c . Ця відстань нази-
вається товщиною дифузійного шару . При перемішуванні рідини
з постійною швидкістю деякий шар, що прилягає до поверхні твер-
дого тіла, залишається у відносному спокої.
Градієнт концентрації буде рівний
.
нас
o
c
c
dc
dx
.
Швидкість розчинення звичайно кількісно характеризують
значенням похідної від концентрації речовини в розчині за часом
.
v розч
dc
dt
.
Якщо в початковий момент розчинення проводилося в чисто-
му розчиннику й концентрація розчиненого речовини була рівною
нулю, то концентрацію речовини в розчині в будь-який момент роз-
раховують як частка від розподілу кількості розчиненого речовини
на об'єм розчину
/
cnV
.
При цьому об'єм розчину залишається
практично постійним і тому швидкість розчинення можна виразити
за формулою
1
dc dn
dt Vdt
.
Оскільки швидкість розчинення визначається швидкістю ди-
фузії й дорівнює їй, те
/
dn dt можна визначити за законом Фіка
нас
c
c
dn
dc
DS
DS
dt
dx
,
тобто швидкість процесу дифузії прямо пропорційна різниці конце-
нтрацій речовини в об'ємі розчину й в поверхневому шарі.
Отримане рівняння зветься рівнянням Щукарева-Нернста
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
296
(
)
нас
dc
kcc
dt
,
де k – константа швидкості розчинення, що дорівнює
DS
k
V
.
Для обчислення константи швидкості розчинення необхідно
проінтегрувати це рівняння.
Розділимо змінні
нас
dc
kdt
c
c
.
Інтегруючи, знаходимо
ln(
)
нас
c
c ktconst
,
де const – константа інтегрування.
Якщо час відраховувати від моменту занурення речовини, що
розчиняється, в розчинник, то за умови t = 0, c = 0. Із цього випли-
ває, що значення сconst =
.
ln ’нас
c
і, таким чином,
1
ln
нас
нас
c
k
tc
c
.
Це рівняння отримане за умови, що константа k не змінюєть-
ся за часом. Однак з вираження для константи видне, що вона зале-
жить від коефіцієнта дифузії речовини, що розчиняється, від площі
поверхні, на якій відбувається розчинення, від об'єму розчину й то-
вщини дифузійного шару . Щоб це рівняння дотримувалося точно,
необхідно при експериментальному визначенні значення константи
швидкості строго дотримувати наступних умов:
–
температура під час досвіду повинна залишатися постійної;
–
об'єм розчину повинен залишатися постійним;
–
площа поверхні речовини, що розчиняється, не повинна по-
мітно мінятися за час досліду (наприклад, досліджувана речовина є
малорозчинною);
–
товщина дифузійного шару повинна залишатися постійною
(швидкість обертання зразка, або перемішування розчину повинна
бути однакова в часі).
5.2. КАТАЛІЗ
297
Товщина дифузійного шару залежить головним чином від
того, чи перебуває розчин у спокої або перемішується, а також від
режиму перемішування (ламінарний або турбулентний). Під час ві-
дсутності перемішування товщина дифузійного шару максимальна
й градієнт концентрації не буде величиною постійної (рис. 5.2 б).
Чим інтенсивніше перемішування, тем тонше шар , тим більше
градієнт концентрації в ньому й тим більше швидкість дифузії й
швидкість розчинення. При постійній швидкості перемішування
градієнт концентрації в кожний момент часу постійний (рис. 5 .2 б).
Уважається, що швидкість розчинення прямо пропорційна швидко-
сті перемішування в ступені n, причому приблизно n = 2/3.
Рівняння для константи швидкості розчинення ідентично за
формою рівнянню реакції першого порядку, однак у цьому випадку
воно не може служити показником порядку реакції, тому що було
виведено в припущенні, що сумарна швидкість реакції визначається
швидкістю дифузії.,
ТЕОРЕТИЧНІ ПИТАННЯ ПО ТЕМІ
1. Дайте визначення середньої й істинної швидкості гетеро-
генної хімічної реакції.
2. Як змінюється швидкість хімічної реакції й концентрації ре-
агуючих речовин у часі?
3. Що називають константою швидкості хімічної реакції і який
її фізичний зміст?
4. Які характеристики гетерогенної системи впливають на
швидкість реакції?
5. Дайте характеристику кінетичної стадії гетерогенної хіміч-
ної реакції.
6. За якими ознаками можна судити, яка стадія гетерогенного
процесу є лімітуюча : хімічна взаємодія або процес дифузії?
7. У чому суть основного закону кінетики для найпростіших
реакцій.
8. Сформулюйте перший та другий закон Фіка.
9. Виходячи з яких положень визначають швидкість складної
хімічної реакції?
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
298
10. У чому полягає принцип незалежності елементарних стадій
складної хімічної реакції?
11. Які гетерогенні реакції називають топохімічними?
12. Як залежить константа швидкості хімічної реакції від тем-
ператури?
13. Що таке енергія активації хімічної реакції й від чого вона
залежить?
14. Поясніть роль адсорбції в гетерогенних процесах.
15. Чим кільквсно характеризують процес адсорбціх?
16. На підставі кривій розподілу Максвелла-Больцмана дайте
якісне пояснення впливу температури й каталізатора на константу
швидкості хімічної реакції.
17. У яких випадках константи швидкості, розраховані по числу
активних зіткнень, будуть збігатися з константами, розрахованими
за допомогою теорії активного комплексу?
18. Які особливості гетерогенних реакцій? З яких стадій може
полягати гетерогенна реакція?
ЛІТЕРАТУРА
1. Герасимов Я. И. и др. Курс физической химии. Т.2.
–
М.:
Химия, 1973. – 625 с.
2. Лебідь, В. І. Фізична хімія : підруч. для студ. вищ. навч.
закл. / В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
3. Пурмаль А. П. А, Б, В... химической кинетики / А. П. Пур-
маль. — М.: ИКЦ «Академкнига», 2004. — 277 с.
4. Физическая химия. Теоретическое и практическое руковод-
ство. Изд. 2 –е, переработанное и дополненное. / Под ред. академи-
ка Б. П. Никольского. – Л. : Химия, 1987. – 880 с.
5. Практикум по физической химии / Под ред. В . В. Буданова
и Н. К. Воробьева. – М. : Химия, 1986. – 351 с.
6. Практикум по физической химии / Под ред. И. В . Кудряшо-
ва. –М. : Высшая школа, 1986. – 495 с.
7. Практикум по физической химии. Учебное пособие для ву-
зов. Изд. 3 –е. / Под ред. С . В. Горбачева, – М . : Высшая школа,
1974.
–
495
с.
Робота No 19. ВИВЧЕННЯ КІНЕТИКИ РОЗЧИНЕННЯ
МАЛОРОЗЧИННИХ РЕЧОВИН
299
8. Стромберг А. Г., Семченко Л. П. Физическая химия, учеб-
ник для студентов вузов, обучающихся по химическим специально-
стям / А. Г. Стромберг, Д. П. Семченко. - 6 -е изд., стер. – М. : Выс-
шая школа, 2006. – 527 с.
9. Практические работы по физической химии : Учебное по-
собие для вузов / Под ред. К. П. Мищенко, А. А. Равделя, A. M. По-
номаревой. – СПб. : Изд-во «Профессия», 2002. – 384 с.
10. Еремин Е. Н. Основы химической кинетики.
-М.: Высшая
школа, 1976. – 541 с.
11. Эмануэль Н. М ., Кнорре Д. Г . Курс химической кинетики:
Учебник для хим. фак-тов. – 4 -е изд., перераб. и доп.
–
М. : Высш.
шк., 1984. – 463 с.
12. Физическая химия. В 2 кн. Кн. 2 . Электрохимия. Химиче-
ская кинетика и катализ: Учеб. для вузов / К. С . Краснов, Н. К . Во-
робьев, И. Н. Годнев и др.; Под ред. К . С. Краснова –3 -е изд., испр.
–
М. : Высш. шк., 2001. – 319 с.
13. Вишняков А. В., Кизим Н. Ф. Физическая химия. – М. : Хи-
мия, 2012. – 839 с.
Робота No 18.
ВИВЧЕННЯ КІНЕТИКИ РОЗЧИНЕННЯ
МАЛОРОЗЧИННИХ РЕЧОВИН
Ціль роботи: досліджувати кінетику розчинення малорозчин-
ного речовини у воді й довести наявність стадії, що лімітує, проце-
су розчинення.
Прилади й реактиви: установка з мотором і редуктором, 4
склянки на 100 мл, піпетка 100 мл, термометр, бензойна кислота,
пробірка, скляна паличка, бюретка 25 мл з розчином 0,02 н Ba(OH)2.
ВИКОНАННЯ РОБОТИ
У якості об'єкта дослідження зручно використовувати криста-
лічну малорозчинну кислоту, тому що легко визначати її концент-
рацію титруванням розчином лугу. У даній роботі використовують
зразок у вигляді циліндра із кристалічної бензойної кислоти. Для
виготовлення зразка в тонкостінній пробірці діаметром близько 15–
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
300
20 мм необхідно розплавити кислоту,
поступово додаючи її до утворення ша-
ру товщиною 5–6 см. Після розплавлю-
вання в центральну частину пробірки
опускають майже до дна скляну палич-
ку (діаметр 5–6 мм) з розплющеним кі-
нцем. Після застигання бензойної кис-
лоти на пробірці роблять кілька надрізів
напилком, розбивають пробірку, витя-
гають циліндр зразка 3 (рис. 5 .3) і згла-
джують його напилком. Верхній кінець
скляної палички 2 вставляють у власни-
ка осі редуктора, що приводиться в обе-
ртання електромотором 1, що забезпе-
чує постійну швидкість обертання бли-
зько 100–120 об./хв.
У чотири склянки ємністю 150 мл
наливають однієї й тою же піпеткою по
100 мл дистильованої води й вимірюють
температуру рідини. Під обертову заготовку зразка, відставивши пі-
дставку 5, підставляють у склянку з водою 4 так, щоб зразок був по-
вністю занурений у рідину й проводять розчинення протягом зада-
ного часу. Час розчинення зразка в кожній склянці вказується ви-
кладачем. Аналогічно роблять дослідження розчинення зразка в ін-
ших трьох склянках, вважаючи стан зразка незмінним.
Визначення концентрації кислоти в кожній склянці роблять
шляхом титрування проб розчином луги так, щоб одержати най-
більш точний результат. Для цього з кожної склянки беруть не
менш 3-х проб для обчислення згодом середнього арифметичного
значення об'єму титранта. Об'єм аліквоти підбирають таким чином,
щоб витратити максимальний об'єм титранта, але не перевищуючий
об'єму бюретки – не більш 25 мл. Концентрацію лугу Ba(OH)2 бе-
руть малої концентрації, близько 0,02 н, для того, щоб при титру-
ванні проб розчину малорозчинної кислоти пішов найбільший об'єм
титранта. При цьому буде досягнута мінімальна відносна погріш-
Рис. 5 .3 . Установка для ви-
вчення кінетики розчинення
бензойної кислоти.
Робота No 19. ВИВЧЕННЯ КІНЕТИКИ РОЗЧИНЕННЯ
МАЛОРОЗЧИННИХ РЕЧОВИН
301
ність визначення концентрації кислоти. У якості індикатору вико-
ристовують фенолфталеин, тому що бензойна кислота є слабкою.
Час розчинення беруть не менш 15 хвилин, але не більш ніж
1,5 години. При малому часі розчинення кількість аліквоти для тит-
рування збільшують (максимальний об'єм проби в умовах досліду
становить 25 мл), так, щоб об'єм лугу, що йде на титрування був
максимальним, але не більш об'єму бюретки – 25 мл.
Якщо перерахувати середній об'єм лугу, що пішов на титру-
вання проб у кожному досліді ( iV ), на об'єм проби рівній 100 мл з
концентрацією лугу 0,100 н ( iV ), то для кінетичного аналізу можна
скористатися даними таблиці 5.1, у якій представлені значення об'є-
му 0,100 н Ba(OH)2, що йде на титрування насиченого розчину бен-
зойної кислоти, узятого за температури проведення досліду
.
нас
V.
Рівняння для розрахунків константи швидкості розчинення то-
ді прийме вид
2,303
lg нас
нас
t
V
k
t
VV
.
Таблиця 5.1 .
Значення
.
нас
V залежно від температури досліду, С.
Темпера-
тура,
С
Об'єм 0,1 н Ba(OH)2,
що йде на титрування
100 мл насиченого роз-
чину бензойної кисло-
ти
Темпе-
ратура,
С
Об'єм 0,1 н Ba(OH)2,
що йде на титрування
100 мл насиченого
розчину бензойної
кислоти
10
17,2
18
22,10
11
17,7
19
22,95
12
18,25
20
23,80
13
18,85
21
24,65
14
19,64
22
25,50
15
20,05
23
26,35
16
20,65
24
27,20
17
21,35
25
28,10
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
302
Рекомендована форма протоколу дослідження:
Температура досліду, С. . . . .
Концентрація Ba(OH)2 . . . . .
No
дос-
ліду
Час
розчи-
нення,
хв
Об'єм проби
розчину
для титрув.,
мл
Об'єм р-ну
Ba(OH)2,
що пішов на
титрування, мл
t
V,
мл
t
V,
мл
k
Значення константи швидкості розчинення, розраховані для
кожного досліду, порівнюють між собою, розраховують середнє
арифметичне й, порівнюючи відхилення значень константи від се-
реднього арифметичного з експериментальною погрішністю визна-
чення значення константи швидкості розчинення.
Визначають погрішність із якої встановлена сталість констан-
ти швидкості розчинення. Для цього оцінюють довірчий інтервал
для середнього значення з довірчою ймовірністю 0,95. Роблять ви-
сновки щодо незалежності константи швидкості розчинення від ча-
су й справедливості теоретичних положень
Робота No 19.
ДОСЛІДЖЕННЯ КІНЕТИКИ РОЗЧИНЕННЯ МАЛОРОЗ-
ЧИННОЇ СОЛІ МЕТОДОМ ЕЛЕКТРОПРОВІДНОСТІ
Ціль роботи: досліджувати кінетику розчинення малорозчин-
ного речовини у воді за допомогою виміру зміни опору розчину під
час розчинення. Довести наявність стадії, що лімітує, процесу роз-
чинення.
Прилади й реактиви: магнітна мішалка, комірка – склянка
100-150 мл, міст змінного струму для виміру опору або LCR-метр ,
зразок малорозчинної солі, секундомір, стандартний розчин хлори-
ду калію.
У роботі визначають константу швидкості розчинення мало-
розчинної солі (сульфат кальцію, карбонат кальцію, галогеніди срі-
бла, сульфат барію й т. п.) при заданій температурі й постійної
швидкості перемішування, а також оцінюють розчинність сполуки.
Робота No 19. ВИВЧЕННЯ КІНЕТИКИ РОЗЧИНЕННЯ
МАЛОРОЗЧИННИХ РЕЧОВИН
303
Швидкість розчинення визначають за даними виміру питомої елек-
тропровідності водяного розчину в різні моменти часу.
Схема приладу для проведення досвіду представлено на рису-
нку 5.4 . Виміри проводяться в комірці 1 для виміру електропровід-
ності ємністю близько 300 мл з фіксованими впаяними в скляні тру-
бки платиновими електродами 6. Гніздо перебуває на столику магні-
тної мішалки 3, якір якої 4 перебуває в комірці й служить для пере-
мішування розчину. Електричну провід-
ність вимірюють із використанням мосту
змінного струму типу Р-38, на кондуктоме-
трі або за допомогою цифрового LCR-
метра типу BR-2817 або BR-2820. При ви-
мірі фіксують температуру досліду.
У комірку наливають 100–150 мл ди-
стильованої води, включають магнітну
мішалку й поміщають зразок 5 малороз-
чинної солі, установлюючи оптимальну
швидкість перемішування, яка повинна
бути постійної під час усього досліду.
Відлік часу процесу розчинення роб-
лять відразу ж після приміщення зразка,
включаючи секундомір. Після закінчення 5
хв починають вимір опору розчину, робля-
чи виміру протягом першої години через 5
хв, протягом другої години через 10 хви-
лин. Виміру опору розчину роблять доти,
поки електрична провідність не досягне
постійної величини. Загальний час прове-
дення експерименту залежить від температури розчину, умов пере-
мішування, природи зразка, що розчиняється, і його дисперсності.
Константу швидкості розчинення обчислюють за рівнянням
першого порядку. Оскільки для малорозчинних сполук питома еле-
ктрична провідність (у першому наближенні пропорційна концент-
рації солі в розчині c , рівняння для константи швидкості розчинення
записується у вигляді
Рис. 5 .4 . Установка для
вивчення зміни електроп-
ровідності розчину під
час розчинення солі.
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
304
.
.
2,303
lg нас
нас
t
k
t
,
де
.
нас
–
питома електрична провідність насиченого розчину, t
–пи-
тома електрична провідність у момент часу t .
Оскільки вимірюване експериментально опір розчину
1
l
R
BB
S
,
де – питомий опір; B – постійна посудини; l – відстань між еле-
ктродами; S – площа поверхні електроду; то
.
.
.
.
/
2,303
2, 303
lg
lg
/
/
нас
нас
нам
t
нас
t
BR
k
t
t
BR
BR
.
Отже розрахунки можна проводити по рівнянню
.
.
1/
2,303
lg
1/
1/
нас
нас
t
R
k
t
R
R
.
Значення величини
.
’нас
R визначається як гранична величина
опору при необмеженому збільшенні часу експерименту. Цю вели-
чину можна одержати екстраполяцією значень електропровідності
розчину від зворотного часу
11
t
Rt
на нульове значення, тому що за
умови t значення
1
t
0.
Рекомендована форма ведення протоколу дослідження
Речовина,щорозчиняється............
Температура досліду, ( oС. . . . . )
Час
розчи-
нення, t
t
R
.
’нас
R
1
t
R
.
11
’нас
t
R
R
lg
.
1/
1/
1/
’нас
’нас
t
R
R
R
.
сер
k
Робота No 19. ВИВЧЕННЯ КІНЕТИКИ РОЗЧИНЕННЯ
МАЛОРОЗЧИННИХ РЕЧОВИН
305
Результати слід представити у вигляді графіка в координатах
1
t
R
й
.
11
lg
нас
t
R
R
.
Останній графік використовується для
визначення середнього значення константи швидкості розчинення
.
сер
k за тангенсом кута нахилу отриманої прямій до осі часу t. По-
будувати графіки зі проведенням статистичного аналізу рівняння
регресії у вигляді прямої лінії доцільно провести в программі Micro-
soft Excel.
Константу
.
сер
k порівнюють зі значенням
.
сер
k , що обчислена
як середнє арифметичне зі значень констант швидкостей, розрахо-
ваних для кожного моменту часу окремо.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. У чому полягають особливості кінетики гетерогенних про-
цесів?
2. Перелічите необхідні умови проведення експерименту, які
необхідно строго витримати, щоб для опису процесу розчинення за-
стосувати кінетичне рівняння, за формою співпадаюче з рівнянням
реакції першого порядку.
3. Від яких термодинамічних параметрів, властивостей речо-
вини й середовища залежить швидкість розчинення й константа
швидкості?
4. Яка розмірність швидкості розчинення?
5. Як перетвориться рівняння для константи швидкості роз-
чинення, виражене через концентрації кислоти до рівняння, вира-
женого через об'єми лугу, що йде на титрування проб?
6. Як оцінити погрішність визначення константи швидкості
розчинення й статистично показати її незалежність від часу процесу
розчинення?
7. З яких стадій може полягати гетерогенна реакція?
8. По яких ознаках можна судити, яка стадія є, що лімітує в
гетерогенному процесі – хімічна взаємодія або процес дифузії?
9. Від чого залежить швидкість дифузії?
10. Сформулюйте й запишіть перший закон Фіка.
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
306
11. Чому при проведенні експерименту необхідне перемішу-
вання розчину?
12. Можна чи для контролю швидкості розчинення використо-
вувати дані виміри опору розчину? Чому при використанні методу
електропровідності як об'єктів беруть малорозчинні сполуки?
13. Яким образом можна визначити електричний опір насиче-
ного розчину?
14. Який фізичний зміст коефіцієнта дифузії й чому він визна-
чається?
15. Як оцінити максимальну відносну погрішність визначення
константи швидкості розчинення?
5.2. КАТАЛІЗ
Каталіз – це прискорення або вповільнення швидкості хімі-
чної реакції речовинами (каталізаторами), які не входять у стехіо-
метричне рівняння реакції й залишаються після реакції хімічно не-
змінними. Каталітичне прискорення реакції досягається за рахунок
зміни її механізму. При цьому каталізатор входить до складу нових
проміжних продуктів, які утворюються, але потім переходять у кін-
цеві продукти реакції з більшою швидкістю, чому при відсутності
каталізатора. У процесі реакції здійснюється багаторазове повто-
рення циклів, що включають почергове зв'язування й регенерацію
каталізатора.
Якщо швидкість хімічної реакції в присутності каталізатора
зменшується (негативний каталіз), його називають інгібітором.
Іноді каталітичною дією мають продукти реакції, у такому випадку
реакція називається автокаталітичними.
Якщо всі взаємодіючі речовини й сам каталізатор перебувають
в одній фазі, каталіз називається гомогенним , якщо каталізатор пе-
ребуває в іншій фазі, чому взаємодіючі речовини, – гетерогенним.
Прикладом гомогенних каталітичних реакцій є етерифікація й
омилення естерів, що каталізуються кислотами, розкладання перок-
сиду водню під дією іонів у розчині, інверсія сахарози й мутарота-
ція глюкози в присутності кислот і основ, полімеризація олефінів у
рідкій фазі під дією сірчаної кислоти, полімеризація олефінів у рід-
кій або паровій фазі під дією трифториду бору й фториду водню,
5.2. КАТАЛІЗ
307
алкілування парафінів або бензолу олефінами в присутності три-
фториду бору або фториду водню й ін. Дуже широко поширені го-
могенні каталітичні реакції в природі. Синтез білків і обмін речовин
у біологічних об'єктах відбуваються в присутності біокаталізаторів,
що одержали назву ферментів або энзімів.
Каталізуємі й некаталізуємі процеси підкоряються тим самим
законам і протікають лише за термодинамічно можливим напрям-
кам, незалежно від присутності або відсутності каталізатора, відрі-
зняючись лише швидкістю досягнення рівноваги.
У гомогенному каталізі реагуючі молекули й каталізатор у
формі атомів, молекул або іонів перебувають в одній фазі й утво-
рюють гомогенну хімічну систему. У ході реакції утворюються
проміжні сполуки стехіометричного складу. У гет ерогенному ка-
талізі каталітична реакція відбувається на поверхні розділу фаз, на
якій утворюються проміжні сполуки за рахунок адсорбційних сил.
Каталітична реакція зосереджена на поверхні твердого тіла, тому
зазвичай домагаються збільшення поверхні каталізатора, наприклад,
шляхом здрібнювання. Важливою характеристикою каталізатора в
гетерогенному каталізі є його питома поверхня – поверхня, що
розрахована на одиницю маси й порист ість – відношення об'єму
пор у зразку стосовно його загального об'єму. Каталізатор бере
участь у самому хімічному процесі. Він бере участь у проміжних
стадіях реакції, виділяючись до кінця реакції знову в хімічно незмі-
неному виді. Тому тверді каталізатори в результаті реакції часто із
крупнокристалічної форми переходять у дрібнокристалічну. Крис-
талічний діоксид марганцю, що є каталізатором розкладання берто-
летової солі, після реакції перетворюється в дрібний порошок.
Пояснення дії гетерогенного каталізатора ще запропоноване
Д. І. Менделєєвим, який висловив ряд припущень за природу гете-
рогенного каталізу. Він указав, що властивості молекул на поверхні
розділу фаз в енергетичному відношенні відрізняються від власти-
востей молекул в об'ємі; процес утримання молекул на поверхні по-
в'язаний з виділенням тепла, яке може йти на активування молекул;
молекули на поверхні переходять у більш реакційноздатний стан,
тому для хімічних процесів велике значення має контакт молекул на
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
308
границі розділу фаз; реакції на границі розділу фаз ідуть із більши-
ми швидкостями при невисоких температурах.
Каталізатор утворює з одним з реагентів проміжну сполуку,
активуючи даний реагент і полегшуючи реакцію. Типовими промі-
жними сполуками є сорбційні сполуки. Звідси випливає й один з
основних принципів гетерогенного каталізу: каталізатор має фізич-
ну або хімічну спорідненість до одного або декількох реагентів. На-
приклад, гідрогенізаційні й дегідрогенізаційні каталізатори Pt, Pd,
Ni, Сu й ін. легко сорбують водень, утворюючи з ним проміжні спо-
луки (палладій навіть здатний розчиняти газоподібний водень).
Каталізатор у випадку оборотної реакції не зміщає положення
рівноваги: константа швидкості прямій і зворотної реакцій у прису-
тності каталізатора змінюється в однакове число раз.
Роль каталізатора в переважній більшості випадків зводиться
до зниження енергії активації й, отже, до прискорення реакції.
Участь каталізатора, як правило, приводить до ускладнення кінети-
чного закону протікання реакції й опису процесу новими кінети-
чними рівняннями. Гомогенно-каталітичні процеси особливо поши-
рені при проведенні процесів у рідкій фазі.
Фактична участь у гетерогенному каталізі обумовлюють особ-
ливі активні мікроструктури – активні центри , що складають тіль-
ки частину загальної поверхні каталізатора. Найчастіше ними бува-
ють: атоми, розташовані на ребрах і вершинах кристалів, іони ано-
мальної валентності, дефекти кристалічної структури й т. п. Часто
домішки деяких речовин, що не володіють самі по собі каталітич-
ною дією, приводять до значного підвищення каталітичної активно-
сті каталізатора. Такі речовини називаються пр о м о т о р а м и .
Істотну роль у гетерогенному каталізі відіграє транспорт речо-
вин до поверхні, швидкість масообміну. Якщо швидкість хімічної
реакції велика в порівнянні з масообміном, то швидкість реакції лі-
мітується дифузією й говорять про дифузійну область протікання
реакції. У зворотному випадку говорять про кінетичну область про-
тікання реакції.
Для каталітичних реакцій характерні наступні закономірності.
1. Каталізатор вступає в хімічну взаємодію з одним або обома
реагуючими речовинами, утворюючи проміжну сполуку й входячи
5.2. КАТАЛІЗ
309
до складу активованого комплексу. Після кожного елементарно-
го акту каталізатор регенерується. Внаслідок утворення активовано-
го комплексу каталізатор направляє хімічну реакцію по принципово
новому шляхові, який може відрізнятися від некаталітичного як чи-
слом, так і природою проміжних сполук, складом й будовою пере-
хідного комплексу. Специфічність дії каталізатора пояснюється не-
обхідністю певної відповідності молекулярних орбіталей реагуючих
молекул і каталізатора за енергією й симетрією. Каталізатор не ви-
трачається в ході реакції і його кількість залишається незмінною;
каталізатор не змінюється в ході реакції хімічно, але може мінятися
фізично (спікатися, розпушуватися, втрачати активність і т. п.).
2. Дія каталізатора проявляється при відносно малою, у порів-
нянні з реагуючими речовинами, кількістю, незначні кількості ката-
лізатора можуть на багато порядків побільшати швидкість хімічної
реакції. Це обумовлене різким зростанням концентрації активова-
них комплексів за рахунок зниження енергії активації. Швидкість
гомогенного каталізу пропорційна концентрації каталізатора. Однак
є реакції, у яких каталізатор майже не впливає на енергію активації,
але сильно змінює передекспоненційний множник.
3. Каталізатор змінює швидкість реакції, але не впливає на рі-
вновагу реакції. Він однаковою мірою прискорює як пряму, так і
зворотну реакції.
4. Більшість каталітичних процесів протікає через ряд послідо-
вних стадій. Загальна швидкість процесу визначається стадією, що
лімітує, тобто стадією, швидкість якої є найменшою.
5. Дія кожного каталізатора специфічно: він змінює швидкість
тільки однієї реакції або групи певних реакцій. Наприклад, у прису-
тності оксиду алюмінію етанол розкладається на воду й етилен, а в
присутності міді – на ацетальдегід і водень. Швидкість каталітичної
реакції може суттєво змінюватися при наявності в системі деяких
речовин, називаних активаторами. Характер їх дії різноманітний:
деякі змінюють склад каталізатора; так, наприклад, заміна лігандів у
складі комплексного каталізатора приводить до зміни його каталі-
тичної активності.
6. Активність каталізатора згодом може змінюватися. Це може
бути наслідком побічних процесів, у результаті яких каталітично
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
310
активний центр – атом, молекула, іон або каталітичний центр на по-
верхні – блокується, виходячи зі сфери реакції. Наприклад, при реа-
кції нейтралізації в кислотно-основному каталізі, утворення в сис-
темі нерозчинних сполук. Каталізатори чутливі до наявності сто-
ронніх речовин, які можуть збільшувати каталітичну активність
(пр о м от о р и), а можуть зменшувати її або взагалі припиняти його
дія (отрути).
Компенсація енергії, що необхідна для розриву хімічних зв'яз-
ків у молекулах реагентів, здійснюється стадійним або злит им
механізмом. У першому випадку каталітична реакція протікає у ви-
гляді послідовних стадій, у результаті чого утворюються проміжні
продукти, що представляють собою одне або кілька сполук вихід-
них речовин з каталізатором. Зміна енергії в ході такої реакції зо-
бражено на рисунку 5.5 (крива I) і характеризується наявністю декі-
лькох максимумів, відповідних до активних комплексів, і мінімумів,
що відповідають проміжним речовинам.
При злитому механізмі стадії каталітичного процесу сполуче-
ні. активний комплекс, Що утворюється, містить каталізатор і всі
реагуючі речовини. На реакційному шляху є лише один максимум
(рис. 55, крива II).
Закономірності кінетики найпростіших каталітичних реакцій
можуть бути пояснені на основі положень формальної кінетики й
законів термодинаміки. Теорія каталітичного процесу, що протікає
на поверхні каталізатора, повинна
включати особливості будови по-
верхні каталізатора й реагуючих
молекул. В основі сучасних вистав
про каталітичний акт лежить ви-
става про активний центр, що
сприяє хемосорбційному процесу.
Каталітично активними центрами
можуть бути дефекти кристалічної
решітки, вершини й піки поверхні,
що характеризуються ненасичені-
стю зв'язків в атомах (Тейлор). Уя-
влення про активні центри знайш-
ли свій розвиток у мультиплет-
Рис. 5 .5 . Стадійний й злитний меха-
нізми компенсації енергії під час
розриву хімічних зв’язків
5.2. КАТАЛІЗ
311
ній теорії каталізу Баландіна (1929). Суть теорії укладена в по-
ложенні, що адсорбція реагуючої молекули на каталізаторі обумов-
лена одночасною взаємодією молекули з поверхнею адсорбенту по
декільком силовим центрам, що приводить до деформації молекули,
до ослаблення зв'язків у молекулі й підвищенню її реакційної здат-
ності. Атоми або іони активного центру утворюють так званий му-
льт иплет , який залежно від числа вхідних у нього часток каталі-
затора може бути дублетом, триплетом, квадруплетом або сексте-
том. Окремі атоми мультиплету є центрами адсорбції, до яких мо-
жуть прикріплюватися індексні атоми реагуючих молекул. В основі
теорії лежать принципи геометричної й енергетичної відпові-
дност і . Вони полягає у вимозі відповідності розташування силових
центрів на поверхні каталізатора розташуванню атомів у молекулі й
оптимальному значенню енергії взаємодії молекул з каталізатором.
Наприклад, передбачається, що реакція дегідрування етилово-
го спирту протікає на дублеті й може бути пояснена наступною
схемою:
||
||
|||
|
HH
HH
HCCO
HCCO
HHH
HHH
,
на якій атоми дублету на поверхні каталізатора позначені чорними
крапками.
Кобозєвим (1939) запропонована теорія активних ансам б-
лів , що розглядає питання про кількість атомів металу в каталітич-
но активному центрі. Неоднорідна в загальному поверхню каталіза-
тора представляється областями, обмеженими потенційними бар'є-
рами – областями міграції. Розглянутий статистичний розподіл ато-
мів каталізатора по областях міграції в процесі осадження на інерт-
ному носії й установлена залежність активності каталізатора від чи-
сла атомів в ансамблі. Було встановлено, що для окисних процесів
активним є одноатомний ансамбль, для реакцій гідрування – двоха-
томний і т. п. Зміна структури поверхні й розмірів блоків, що відбу-
вається при введенні домішок або внаслідок термічної обробки й
рекристалізації, впливає на число атомів в ансамблях. Цим поясню-
ється промотування, дія отрути і вплив способу готування каталіза-
тора на його активність.
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
312
В електронній теорії каталізу особливість взаємодії реагу-
ючих молекул з поверхнею каталізатора розглядається на основі
зонної теорії розподілу електронів по енергетичних рівнях у твер-
дих тілах.
КОНТРОЛЬНІ ПИТАННЯ ПО ТЕМІ
1. Що називають каталізатором і чому він відрізняється від
ініціатора реакції?
2. Як впливає каталізатор (його кількість, структура, промото-
ри) на кінетику хімічних реакцій?
3. Як класифікують каталітичні реакції?
4. Яким кінетичним рівнянням описується гетерогенно- каталі-
тична реакція, якщо її швидкість визначається адсорбційно-
десорбційними процесами? Яка можлива межа зміни гаданого по-
рядку реакції?
5. Яка роль промотору й на чому заснована дія, що сприяє збі-
льшенню активності каталізатора?
6. Які теорії гетерогенного каталізу вам відомі?
7. У чому специфічність дії окремих каталізаторів?
8. У чому полягає мультиплетна теорія каталізу Баландіна й
можна чи на підставі цієї теорії передбачити структуру каталізато-
ра для даної реакції?
9. У чому полягає теорія Кобозєва (теорія активних ансамб-
лів)?
10. Приведіть приклади виборчої дії каталізаторів для реакції
того самого речовини, наприклад, етанолу.
11. Чи можна збільшити вихід продуктів реакції за допомогою
каталізатора. Чому.
12. Які стадії передбачає схема дії гетерогенного каталізатора.
13. Яка фізична природа активних центрів гетерогенного ката-
лізатора.
14. Поясніть зміст енергії активації хімічної реакції з боку тео-
рії активованого комплексу.
15. В якому випадку доцільно застосовувати високодисперсні
каталізатори. Яка стадія реакції при цьому виступає як лімітуюча?
16. Яким чином можна визначити область протікання реакції?
17. Запропонуйте методику експерименту, що дозволяє вивчити
вплив площини поверхні розділу фаз на швидкість реакції.
Робота No 20.ВИВЧЕННЯ РЕАКЦІЇ ОКИСНЕННЯ HI ПЕРОКСИДОМ
ВОДНЮ, ЩО КАТАЛІЗУЄТЬСЯ МОЛІБДАТОМ АМОНІЮ
313
Робота No 20.
ВИВЧЕННЯ РЕАКЦІЇ ОКИСНЕННЯ ЙОДОВОДНЕВОЇ
КИСЛОТИ ПЕРОКСИДОМ ВОДНЮ, ЩО КАТАЛІЗУЄТЬСЯ
МОЛІБДАТОМ АМОНІЮ
Ціль роботи: визначити константи швидкостей реакції оки-
снення йодистоводневої кислоти пероксидом водню при різних те-
мпературах і визначити енергію активації реакції.
Прилади й реактиви: термостат, колба 200 мл, розчин йоди-
ду калію 40 мас.%, розчин сульфатної кислоти 1 моль/л, пероксид
водню 0,025 моль/л, сульфатна кислота 0,02 моль/л, розчин перман-
ганату калію 0,02 моль/л, колби для титрування, бюретка з розчи-
ном тіосульфату натрію, свіжоприготовлений крохмаль.
Реагуючі речовини й каталізатор у даній реакції перебувають
в одній фазі й досліджуваний процес ставиться до гомогенного ка-
талізу. Каталізатори, володіючи властивістю впливати лише на
швидкість реакції, не впливають на значення константи швидкості й
константи хімічної рівноваги. Каталізатори, як правило, мають ви-
борчу дію, тобто прискорюють або сповільнюють тільки деякі реак-
ції; невеликі кількості каталізатора можуть значно побільшати шви-
дкість процесу, тому що знижують енергію активації, яка в рівнянні
для константи швидкості входить у показник ступеня
o
a
E
RT
kZe
,
деo
Z – загальне число подвійних зіткнень.
Для прискорення реакції окиснення йодоводневої кислоти пе-
роксидом водню
22
2
2
2
2
HI HO
HOI
досить увести 5–7 крапель розчину молібдату амонію з концентра-
цією близько 1 моль/л. У реакційній суміші протікають наступні
процеси:
24
24
2
2
2
NaI HSO
Na SO
HI
,
(a)
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
314
22
2
2
2
2
HI HO
HOI
,
(b)
2
223
246
2
2
2
I
NaSO
NaI
NaSO
.
(c)
Витрата пероксида водню визначають по еквівалентній кілько-
сті йоду, що виділився, який титрують розчином тіосульфату на-
трію. Реакції (a) і (c) протікають практично миттєво, тому швид-
кість сумарного процесу визначається швидкістю більш повільної
реакції (c). Оскільки йодид натрію в процесі титрування йоду тіосу-
льфатом натрію регенерується, концентрації NaI і HI практично
постійні й, отже, швидкість реакції (c) визначається тільки концен-
трацією 2 2
HO.
Процес окиснення йодистоводневої кислоти пероксидом вод-
ню йде по кінетичному рівнянню першого порядку. Експеримента-
льно реакцію здійснюють у такий спосіб. До реакційної суміші NaI
+24
H SO при заданій температурі додають певні об'єми розчинів
пероксида водню (
22
HO
V ) і тіосульфату натрію (
223
NaSO
V
) рівних
концентрацій, а потім розчин крохмалю, у якості індикатору. Суміш
ретельно перемішують і відзначають момент появи синього фарбу-
вання ( ot ). У цей момент увесь тіосульфат прореагував з йодом, що
виділився, і пероксиду водню,залишилося (
22
HO
V
–
223
NaSO
V
), тому
що до цього часу прореагував об'єм розчину пероксида, який екві-
валентний
223
NaSO
V
.
Додають ще розчин тіосульфату натрію
( 223
NaSO
V
) і знову відзначають момент появи синього фарбування
(час 1t ). Таку операцію повторюють 5–6 разів.
Кінетичне рівняння можна представити у формі
22
223
22
223
2,303
lg
HO
NaSO
HO
NaSO
V
V
k
t
V
V
x
,
де(
22
HO
V
–
223
NaSO
V
) – об'єм розчину 2 2
HO умоментчасу ot ,мл;x
–
об'єм розчину 2 2
HO,щопрореагував(для 1tx =
223
NaSO
V
,для 2tx=
=2
223
NaSO
V
і т. д.).
Робота No 20.ВИВЧЕННЯ РЕАКЦІЇ ОКИСНЕННЯ HI ПЕРОКСИДОМ
ВОДНЮ, ЩО КАТАЛІЗУЄТЬСЯ МОЛІБДАТОМ АМОНІЮ
315
ВИКОНАННЯ РОБОТИ
У колбу ємністю 200 мл наливають 100 мл розчину йодиду на-
трію з концентрацією 0,4 масової частки, %, і 5 мл розчину сульфа-
тної кислоти з концентрацією 1 моль/л. У другу колбу місткістю 50
мл поміщають 15 мл розчину пероксиду водню з концентрацією
0,025 моль/л. Обидві колби поміщають у водяний термостат, на-
строєний на задану температуру.
Для визначення дійсного змісту пероксиду водню (у моль/см
3
)
беруть три проби по 2 мл. Проби виливають у заздалегідь приготов-
лені конічні колби із сульфатною кислотою (20 мл 0,15 М розчину) і
титрують кожну пробу 0,02 М розчином
4
KMnO (3,1607 г в 1000 мл
розчину) до появи незникаючого протягом 1–2 хв рожевого фарбу-
вання. Після цього знаходять середнє із трьох значень витраченого
об'єму перманганату V (у мл).
З рівняння реакції при титруванні
2
4
22
2
2
2
6
5
2
8
5
MnO
H
HO
Mn
HOO
випливає, що 1 моль
4
KMnO відповідає 2,5 моль пероксиду водню.
Якщо на титрування 2 мл проби витрачається v мл титранта, то про-
ба містить
5
1 (/2)510
n
V
моль/мл пероксиду водню.
Бюретку ємністю 25 мл заповнюють титрованим розчином тіо-
сульфату натрію з молярною концентрацією 0,025 моль/л. Через 10
хв, коли розчини приймуть задану температуру, їх зливають разом,
додають із полумікробюретки 1 мл розчину Na2S2O3 і 5 крапель
крохмалю концентрації 0,5 мас. частки, %. Уміст колби ретельно
перемішують, включають секундомір і відзначають час появи си-
нього фарбування ( ot ).
Додають (ш в ид к о !) ще 1 мл розчину тіосульфату натрію, ро-
зчин швидко перемішують і знову відзначають момент появи си-
нього фарбування ( 1t ). Не виключаючи секундомір, операцію дода-
вання розчину
223
Na S O по 1 мл повторюють 6 раз. Додають у реа-
кційну суміш 5 крапель розчину молібдату амонію для швидкого
доведення реакції до кінця. Йод що виділився, відтитровують тіосу-
льфатом з тієї ж бюретки до знебарвлення розчину. Загальний об'єм
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
316
витраченого тіосульфату еквівалентний узятому об'єму розчину пе-
роксиду водню для доведення реакції до кінця.
Аналогічним чином проводять реакцію й при іншій температурі.
Результати дослідження зводять у таблицю за формою
Температурадосліду .............
Номер
виміру
Час
від поча-
тку
досліду
Об'єм
доданого розчину
Na2S2O3, мл
22
223
22
223
HO
NaSO
HO
NaSO
V
V
V
V
x
k
Для кожного моменту часу обчислюють значення константи
швидкості реакції. Розбіжність у значеннях констант не повинне пе-
ревищувати 10%. Розраховують середнє арифметичне константи
швидкості при кожній температурі й результати обробляють статис-
тично із застосуванням t-критерію.
За отриманими результатами константи швидкості реакції при
різних температурах розраховують енергію активації реакції.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Опишіть процес окиснення йодоводневої кислоти перокси-
дом водню.
2. Яка зі стадій реакції окиснення йодоводневої кислоти перо-
ксидом водню є лімітуючою?
3. По яких ознаках контролюють швидкість процесу окиснен-
ня йодоводневої кислоти пероксидом водню?
4. Як визначають дійсний зміст пероксиду водню в пробі? На-
пишіть рівняння реакцій.
5. Чому реакція окиснення йодоводневої кислоти пероксидом
водню описується кінетичним рівнянням першого порядку?
6. Яким чином реакцію окиснення йодоводневої кислоти перо-
ксидом водню швидко доводять до завершення?
7. Чому вважається, що швидкість реакції окиснення йодовод-
невої кислоти пероксидом водню залежить тільки від концентрації
персоксиду водню?
5.3. КІНЕТИКА ЕЛЕКТРОДНИХ ПРОЦЕСІВ
317
5.3. КІНЕТИКА ЕЛЕКТРОДНИХ ПРОЦЕСІВ
Електродний потенціал, обумовлений за результатами виміру
електрорушійної сили відповідного гальванічного елемента, є хара-
ктерною величиною для даного електрода (залежної від концентра-
ції потенціалвизначальних іонів і температури) тільки в тому випа-
дку, якщо складений гальванічний елемент працює оборотне. Це
можливо тільки за умови, коли сила струму, що протікає в елементі,
нескінченно мала й, отже, нескінченно мала (дорівнює нулю) і шви-
дкість електрохімічного процесу, що протікає в гальванічному еле-
менті. Обмірюваний у таких умовах електродний потенціал назива-
ється рівноважним потенціалом, а ЕРС елемента дорівнює різниці
рівноважних потенціалів – анода й катода.
На практиці, однак, електрохімічний процес протікає з поміт-
ною швидкістю (сила струму, що проходить через елемент, відмінна
від нуля) і стає необоротним. Це приводить до зміни електродного
потенціалу в порівнянні з його рівноважним значенням. Відхилення
від рівноваги, пов'язане із проходженням струму через елемент,
явище зрушення потенціалу електрода, називається еле кт роліт и-
чною поляризацією, а електроди, виведені зі стану рівноваги, –
поляризованими.
Будь-який електрохімічний процес являє собою ге тероге нну
реакцію, що протікає на границі метал – розчин, що полягає з ряду
послідовних стадій, наприклад:
1) доставка речовини;
2) хімічні перетворення в електродів;
3) розряд (іонізація);
4) відвід продуктів реакції або побудова нової фази.
В електрохімічному процесі стадія, яка чинить найбільший
опір процесу (сама повільна), називається стадією, що лімітує. Зна-
ючи закономірності окремих стадій і виявивши стадію, що лімітує,
можна управляти її швидкістю й швидкістю всього процесу.
В електрохімічній кінетиці заходом швидкості електродної
реакції v є густина струму i – сила струму, віднесена до одиниці
площі поверхні електрода,
vzFi
,
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
318
тому що кількість електрики пов'язане з кількістю перетвореної ре-
човини законом Фарадея.
Розрізняють катодну й анодну поляризацію.
Зсув потенціалу від рівноважного значення, необхідне для
протікання електрохімічного процесу з певною швидкістю, назива-
ється перенапругою й визначається рівністю
.
.
поляриз
рiвн
,
де
.
рiвн
й
.
поляриз
–
рівноважний потенціал і потенціал поляризова-
ного електроду, відповідно.
Величина перенапруги залежить від густини струму, природи
електрода, стану його поверхні, температури електроліту й концен-
трації потенціалвизначальних іонів у розчині.
Ідеально неполяризуємий електрод – це система, через яку
можуть проходити великі струми в будь-якому напрямку, не викли-
каючи відхилення потенціалу від рівноважного значення. Практич-
но це недосяжне. Однак електродні системи, які мають велику гус-
тину струму обміну, наприклад
/
Ag Ag
–
електрод, можуть витри-
мувати пропускання великих струмів при досить малій перенапрузі.
Навпаки, ідеально поляризуємий електрод – це такий елек-
трод, через який струм не проходить при будь-якому значенні поте-
нціалу, прикладеного до електроду. До цього випадку наближаєть-
ся ртутний електрод у водяних розчинах нітрату натрію й деяких
інших солей. У цій системі доданий заряд витрачається майже ви-
нятково на утворення подвійного електричного шару, поки потенці-
ал на досягне досить великої величини, після чого почнуть протіка-
ти вимірні струми.
При обробці експериментальних даних по вивченню перенап-
руги процесу виділення водню на ряді металів І. Тафелем виведене
рівняння, що зв'язує перенапругу з величиною густини струму
lg
abi
,
де постійні a й b звуться тафелівськими. Величина a залежить
від складу електроліту й природи електрода, константа b постійна
для даного електродного процесу й залежить від температури. При
густині струму, близької до нуля, величина lg i прагне до , от-
5.3. КІНЕТИКА ЕЛЕКТРОДНИХ ПРОЦЕСІВ
319
же, рівняння перестає бути слушним. У цьому випадку величина
підкоряється вираженню
ki
, де k – константа.
Залежність між густиною струму й потенціалом (поляризаці-
єю) виражається у вигляді кінетичних рівнянь або поляризаційних
кривих у координатах
i
, або відповідно до рівняння Тафеля в
координатах lg i
.
На наведених нижче рисунках показані поляризаційні криві
для різних видів поляризації.
Концентраційна поляризація (рис 4.6 а) характеризується гра-
ничною ділянкою струму, що не залежать від потенціалу, причому
цей граничний струм, називаний дифузійним, прямо пропорційний
концентрації іонів у розчині. Якщо в умовах концентраційної поля-
ризації стежити за потенціалом електрода, то при досягненні грани-
чного струму потенціал електрода різко зростає й теоретично пря-
мує до нескінченності, а практично на електроді починається новий
процес, наприклад, розряд іонів фону або води.
При електрохімічній поляризації дифузія не є лімітуючою,
тому при зміні потенціалу струм зростає за експонентним зак о-
ном (рис. 5 .6 b, 5.6 с). Вигляд цих кривих суттєво залежить від
природи електродного процесу – оборотного або необоротного
(рис. 5.6b і 5.6 с).
Оборотний процес характеризується великою швидкістю пе-
реносу електрона (більшим струмом обміну). Як видне з рис 5.6 b,
при протіканні струму потенціал його мало відрізняється від рівно-
важного, тобто оборотний процес характеризується малою перенап-
ругою при протіканні струму. Інакше кажучи, якщо процес оборот-
ний, то для протікання помітного струму через електрохімічну сис-
тему до електродів досить прикласти невелику різницю потенціалів.
Необоротний електродний процес характеризується малою
швидкістю переносу електрона (малим струмом обміну). Як видне з
рис. 5.6 с, при протіканні через систему такого ж струму, як на рис.
5.6 b, потенціал електрода сильно відхиляється від рівноважного
(виникає велика перенапруга). Інакше кажучи, якщо процес необо-
ротний, для протікання помітного струму до електрода необхідно
прикласти досить велику перенапругу. На ділянці AB струм взагалі
не протікає, тому що прикладена напруга виявляється недостат-
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
320
ньою. Чим більше довжина ділянки AB, тим більше необоротний
електродний процес.
На явищі концентраційної поляризації засновані методи поля-
рографії й амперометріїї з одним поляризованим електродом, а на
явищі електрохімічної, концентраційної, фазової й ін. поляризацій
засновані методи кулонометрії, електроліз, амперометрії й потенці-
ометрія із двома поляризованими електродами.
При проходженні струму через електроліт на електродах від-
буваються окисно-відновні хімічні процеси пов'язані із процесами
розряду іонів, електроліт розкладає, а електроди поляризуються.
При певній силі струму цей процес стає безперервним, починається
електроліз.
Електродний процес складається з наступних стадій:
1) транспортування реагуючих речовин до електрода й проду-
ктів реакції в розчин;
2) електрохімічної реакції, що характеризується переходом
електронів через границю розділу метал – розчин;
3) хімічної реакції, що передує або наступної за електрохіміч-
ною стадією;
4) фазових перетворень (виділення газу або утворення криста-
лічної структури твердої фази).
а)
б)
в)
Рис. 5.6 . Поляризаційні криві для різних видів поляризаціх:
а) концентраційної; б) оборотної; в) необоротної.
5.3. КІНЕТИКА ЕЛЕКТРОДНИХ ПРОЦЕСІВ
321
Стадії 1) і 2) властиві всім електрохімічним процесам; стадії
3) і 4) специфічні для певних реакцій.
Швидкість багатостадійного процесу визначається найбільш
повільної – стадією, що лімітує, яка і є причиною виникнення поля-
ризації. При відомій природі стадії, що лімітує, замість поляризації
використовують термін перенапруга. При вповільненні процесу в
стадії 1) виникає дифузійна перенапруга, у стадії 2) – електрохімі-
чна перенапруга, пов'язане з уповільненням процесу розряду або іо-
нізації, у стадії 3) – перенапруга хімічної реакції, у стадії 4) – фазо-
ва перенапруга. У загальному випадку електродна поляризація
складається з перенапруги всіх видів, у конкретних випадках домі-
нує один з видів перенапруги. стадіями, що часто лімітують, є від-
разу дві (або більш), тоді процес називається "змішаним".
При гарному перемішуванні й невеликої густині струму зру-
шення потенціалу, викликувані концентраційною поляризацією,
малі й кінцева швидкість самої електродної реакції досить часто
приводить до значної поляризації. Різні електроди при проходженні
струму поводяться по різному: виділення з розчину й перехід у роз-
чин іонів
2
2
,
,
ZnCdAg
у розчинах простих солей цих металів
відбувається з незначною поляризацією. Навпаки, виділення з роз-
чину й перехід у розчин іонів
2
2
,
,
FeNiH
супроводжується ду-
же великою поляризацією.
При катодній поляризації потенціал електрода зрушується в
негативну сторону, при анодної – у позитивну. Тому різниця між
потенціалом електрода під струмом і рівноважним значенням для
катодного процесу буде негативна, для анодного – позитивна. Для
того щоб оперувати позитивними величинами, вважають перенап-
ругу для анодного процесу
.
a
рiвн
, а для катодного процесу
.
к
рiвн
. Різниця потенціалів між потенціалами катода й анода
зменшується й тим більшою мірою, чим вище сила струму. ЕРС га-
льванічного елемента при його роботі внаслідок поляризації анода й
катода знижується в порівнянні з ЕРС розімкнутого ланцюга.
При роботі електролізера напрямку поляризаційних змін елек-
тродних потенціалів залишаються тими ж самими, що й у гальвані-
чному елементі. Однак позитивним електродом електролізера слу-
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
322
жить анод, а негативним електродом – катод. Під впливом анодної й
катодної поляризації величина
a
EEE
буде зростати разом зі
збільшенням сили струму, що протікає.
Електроліз із інертними електродами найчастіше супроводжу-
ється розрядом катіонів на катоді й аніонів на аноді. Мінімальна рі-
зниця потенціалів,
.
розкл
E , при якій починається безперервний роз-
ряд іонів, тобто електроліз, називається напругою розкладання, а
потенціали відповідних електродів – потенціалами розряду.
Якби при незначних опорах електродів і електроліту в ланцюзі
електроліз здійснювався при рівноважних потенціалах електродів,
то напруга розкладання визначалася б різницею рівноважних поте-
нціалів
.
(),
.
(),
.
мін
рівн
рівн
E
.
Однак у більшості випадків, внаслідок поляризації електродів,
напруга розкладання перевищує
.
мін
E й визначається різницею по-
тенціалів розряду
.
(),
.
(),
.
розкл
рівн
рівн
E
.
Для визначення потенціалу розряду й напруги розкладання
будуються так звані поляризаційні криві в координатах: сила стру-
му (i ) – потенціал електрода ( ) або сила струму ( i ) різниця потен-
ціалів ( E ). Типова крива зо-
бражена на рис. 5.7. Точка пе-
ретину ділянок кривих дозво-
ляє знайти шукані величини.
Вивченням впливу тем-
ператури на швидкість елект-
рохімічного процесу може бу-
ти встановлена природа елект-
родної поляризації. Тому що
швидкість обміну іонами між
металом і розчином є скінчен-
ною величиною, те це значить,
що в реакцію вступають не всі
іони речовини, що перебува- Рис. 5.7 . Поляризаційна крива.
5.3. КІНЕТИКА ЕЛЕКТРОДНИХ ПРОЦЕСІВ
323
ють у сфері протікання реакції, а тільки їх частина, що володіє де-
яким надлишком енергії, називаною енергією активації. Стосов-
но до опису електрохімічних процесів, рівняння Арреніуса для кон-
станти швидкості реакції k
E
RT
kAe
,
lg lg
2,303
E
kA
RT
.
перепишеться із заміною k на еквівалентну їй величину густини
струму i , що відповідає деякому значенню перенапруги; E – на
ефективну енергію активації електродної реакції при цьому ж зна-
ченні перенапруги
lg lg
2,303
E
i
A
RT
.
Із цього рівняння видна лінійна залежність між l g i й величи-
ною 1/ T , яка дійсно зберігається в широкому інтервалі температур.
Енергія активації електрохімічних процесів залежить від при-
роди електродної перенапруги. При дифузійній перенапрузі енергія
активації не залежить від перенапруги й становить 10–12 кДж/моль;
при електрохімічній перенапрузі вона становить більшу величину –
порядку 40–80 кДж/моль і залежить від величини перенапруги.
Поляризаційні явища відіграють істотну роль при здійсненні
електрохімічних процесів. Величина й вид перенапруги визначають
багато важливих характеристик електрохімічних процесів, напри-
клад, швидкість корозії металів, кристалічну структуру металевих
катодних опадів і т.п . Напруга на клемах хімічних джерел струму й
на електролізерах, а отже й енергетичні характеристики цих систем,
визначаються значеннями потенціалів поляризованих електродів.
КОНТРОЛЬНІ ПИТАННЯ ПО ТЕМІ
1. У чому особливість електродних реакцій у порівнянні зі
звичайними хімічними?
2. Покажіть, що швидкість електрохімічний реакції пропор-
ційна силі струму.
3. Назвіть основні стадії електрохімічної реакції.
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
324
4. Що називається перенапругою при електролізі і які причини
його виникнення?
5. Які види поляризації мають місце при електролізі, по яким
ознакам можна визначити вид поляризації?
6. Яку роль при вивченні електрохімічної кінетики відіграють
поверхня електрода, його хімічна природа, поляризуємість і здат-
ність до адсорбції?
7. У яких координатах будуються поляризаційні криві?
8. Що називається напругою розкладання?
9. На підставі яких експериментальних даних можна визначи-
ти енергію активації електрохімічної реакції?
10. По якій величині можна судити про швидкість реакції в
процесі електролізу?
11. Як визначається вихід за струмом?
12. По яких ознаках можна з'ясувати природу електродної по-
ляризації?
13. Дайте характеристику різних видів поляризації.
14. Дайте аналіз рівняння Тафеля.
ЛІТЕРАТУРА
1. Багоцкий В. С. Основы электрохимии. – М. : Химия. 1988. –
400 с.
2. Байрамов В. М . Основы электрохимии / В. М . Байрамов.
–
М. : ACADEMIA, 2005. – 238 с.
3. Антропов Л. И. Теоретическая электрохимия / Л. И. Антро-
пов. – М. : Высшая школа, 1984. – 519 с.
4. Практикум по электрохимии. /Б. Б. Дамаскин, О. А. Петрий,
Б. И . Подловченко и др. /Под ред. Б . Б. Дамаскина.
–
М. : Высш.
школа. 1991. – 288 с.
5. Даниэльс Ф., Олберти Р. Физическая химия. – Пер. с англ. –
М.:Мир.1978. – 646с.
6. Дамаскин Б. Б. и др. Электрохимия / Б. Б. Дамаскин, О. А.
Петрий, Г. А. Цирлина. – М. : Химия, КолосС, 2006. – 672 с.
7. Практикум по прикладной электрохимии : Учеб. пособие
для вузов / Н. Г. Бахчисарайцьян, Ю. В . Борисоглебский, Г. К . Бур-
Робота No 21. ВИЗНАЧЕННЯ НАПРУГИ РОЗКЛАДАННЯ ПРИ
ЕЛЕКТРОЛІТИЧНОМУ ОСАДЖЕННІ НІКЕЛЮ З РОЗЧИНУ
325
кат и др,; Под ред. В . Н. Варыпаева, В. Н. Кудрявцева. – Л. : Химия,
1990. – 304 с.
8. Практические работы по физической химии : Учебное посо-
бие для вузов / Под ред. К . П. Мищенко, А. А. Равделя и А. М. По-
номаревой. – Л. : Химия, 1982. - 400 с.
9. Практикум по физической химии. / Под ред. Н.К . Воробье-
ва. Изд. 4 -е. – М. : Химия, 1975.
10.Практикум по физической химии: Учебное пособие для ву-
зов. Изд. 3 -е. / Под ред. С .В . Горбачева. – М . : Высшая школа, 1974.
–
495 с.
11. Практикум по физической химии / Под ред. И.В. Кудря-
шова. – М. : Высшая школа, 1986. – 495 с.
12. 18. Практикум по физической химии / Под ред. В .В. Буда-
нова и Н. К. Воробьева. – М. : Химия, 1986. – 351 с.
13. Скорчелетти В. В. Теоретическая электрохимия. – Л. : Хи-
мия. 1974. – 586 с.
14. Физическая химия. Теоретическое и практическое руково-
дство. Изд. 2 -е, переработанное и дополненное. / Под ред. академи-
ка Б. П. Никольского. – Л. : Химия, 1987. –880 с.
15. Физическая химия: Учеб. пособие для хим. -тех. спец. ву-
зов / И. Н. Годнев, Л. С . Краснов, Н. Л. Воробьев и др.; Под ред. К .
С. Краснова. – М. : Высш. школа, 1982. – 6 87 с.
Робота No 21.
ВИЗНАЧЕННЯ НАПРУГИ РОЗКЛАДАННЯ ПРИ
ЕЛЕКТРОЛІТИЧНОМУ ОСАДЖЕННІ НІКЕЛЮ З РОЗЧИНУ
Ціль роботи: Визначити напруга розкладання розчину елект-
роліту й вивчити залежність катодного потенціалу від щільності
струму при електролітичному осадженні нікелю з розчину сульфа-
ту нікелю.
Прилади й реактиви: джерело постійної напруги, реостат,
амперметр, вольтметр, високоомний потенціометр постійного
струму Р-307, комірка в капіляром Лугіна, 0,5 н розчин сульфату
нікелю, нікелеві електроди.
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
326
Для виміру по-
тенціалу поляризо-
ваного електрода й
зняття поляризацій-
них кривих викори-
стовується установ-
ка, зображена на ри-
сунку 5.8 .
Від джерела
струму 1 через реос-
тат 2 і амперметр 3
струм подається в
електролізер 4, де
здійснюється елект-
роліз між досліджу-
ваним електродом 5 (катод) і допоміжним електродом 6 (анод). Для
виміру ЕРС гальванічної пари досліджуваний електрод 5 – електрод
порівняння 7 служить компенсаційна схема з високоомним потен-
ціометром типу Р-307. Струм через комірку, вимірюваний амперме-
тром 3, регулюють, змінюючи опір 2. При такому з’єднанні елект-
роліз проходить у гальваностатичному режимі, коли потенціал є
функцією струму.
Між досліджуваним елект-
родом і електродом порівняння 7
поміщають капіляр Лугіна – запов-
нену електролітом трубку, тонкий
кінчик якої підходить уводити, уве-
сти до ладу самій поверхні робочо-
го електрода як показано на рисун-
ку 5.9. Таким чином, створюється
самостійний ланцюг для виміру по-
тенціалу під час відсутності струму
в цьому ланцюзі.
У якості електролізера вико-
ристовують W-подібну посудину із
трьома електродами: 5 – досліджу-
Рис. 5 .8 . Установка для виміру потенціалу поля-
ризованого електроду й зняття поляризацій-
них кривих.
Рис. 5.9 . Капіляр Лугіна в робочій
комірці.
Робота No 21. ВИЗНАЧЕННЯ НАПРУГИ РОЗКЛАДАННЯ ПРИ
ЕЛЕКТРОЛІТИЧНОМУ ОСАДЖЕННІ НІКЕЛЮ З РОЗЧИНУ
327
ваний електрод і 6 – допоміжний електрод. Вимірюючи ЕРС ланцю-
гу при різній густині струму, будують поляризаційну криву й зна-
ходять потенціал електрода (потенціал розряду).
Визначення величини напруги розкладання проводиться на
установці, схематично представленої на рисунку 5.10. Електроліз
проводиться в посудині Арреніуса 5, у якому електродами служать
платинові пластинки поверхнею близько 2 см
2
. Змінюючи напругу,
визначають силу струму в ланцюзі й, зобразивши результати у ви-
гляді кривої в координатах i – E, визначають величину напруги ро-
зкладання.
ВИКОНАННЯ РОБОТИ
Вивчення катодної поляризації. В W-подібну посудину для
виміру електродних потенціалів, зображений на малюнку 5.9, нали-
вають 0,5 н розчин сульфату нікелю й поміщають три добре очище-
ні дрібному наждаковим папером і промитих дистильованою водою
нікелевих електрода. З нікелевих електродів і хлорсрібгого електро-
да як електрода порівняння становлять гальванічний ланцюг
4
(.),(
.)
Ni NiSO KCl нас AgCl тв Ag Ni
і підключають її в установку для зняття поляризаційних кривих так,
щоб середній нікелевий електрод 5 служив катодом, а крайні 6 –
анодом. ЕРС такому ланцюга можна
визначити формулою
/
AgCl Ag
Ni
E
.
Попередньо вимірюють ЕРС до-
сліджуваного елемента при відсутно-
сті струму в ланцюзі поляризації. По-
тім включають струм ключем 9 і ре-
остатом 2 регулюють подавану на
електролизер напругу так, щоб через
нього проходив струм силою 5 мА.
Через 2–3 хв, необхідні для стабіліза-
ції потенціалу, вимірюють ЕРС. Ви-
міру повторюють при силі струму 10,
Рис. 5 .10. Схема приладу для ви-
значення напруги розкладання
5. КІНЕТИКА ГЕТЕРОГЕННИХ ПРОЦЕСІВ. КАТАЛІЗ.
328
15, 20, 40 і 50 мА. За отриманим даними будують поляризаційну
криву й визначають потенціал виділення нікелю за формулою, ко-
ристуючись знайденим значенням ЕРС, відповідної до потенціалу
виділення нікелю
,
.
/
.
Ni рoзкл
AgCl Ag
розкл
E
.
Потім визначають перенапруга виділення нікелю, згідно з фо-
рмулою
,
.
,
.
Ni розкл
Ni розкл
,
де
,
.
/
0
Ni розкл
AgCl Ag
i
E
.
Досліджені й розрахункові дані заносять у таблицю
i,A
E,B
/
AgCl Ag
,
.
Ni розкл
Вимір напруги розкладання електроліту. Перед вимірами
необхідно попередньо оцінити величину напруги розкладання. Ви-
значення величини напруги розкладання проводиться на установці,
схематично представленої на рисунку 5.10. Електроліз проводиться
в посудині Арреніуса 5, у якому електродами служать платинові
пластинки поверхнею близько 2 см
2
.
Змінюючи напругу, визнача-
ють силу струму в ланцюзі й, зобразивши результати у вигляді кри-
вої в координатах i E
, визначають величину напруги розкладан-
ня. Посудину 5 заповнюють досліджуваним електролітом і пересу-
вають движок реостата 2 у крайнє ліве положення так, щоб струм,
що протікає через мікроамперметр 6, був мінімальним. Ключем 3
замикають ланцюг, пересувають движок реостата 2 праворуч, збі-
льшуючи напругу на електродах доти, поки не почнуть виділятися
пухирці газу. Напруга, відзначене при цьому на вольтметрі 4, мож-
на розглядати як наближене значення напруги розкладання
.
.
розкл перед
E
.
Потім підбирають шунт для мікроамперметра 6. Для
цього на посудину 5 задають напругу на 0,5 В більше, чим
Робота No 21. ВИЗНАЧЕННЯ НАПРУГИ РОЗКЛАДАННЯ ПРИ
ЕЛЕКТРОЛІТИЧНОМУ ОСАДЖЕННІ НІКЕЛЮ З РОЗЧИНУ
329
.
.
розкл перед
E
. Поступово підвищують опір шунта 7, переміщаючи його
движок праворуч доти, поки стрілка мікроамперметра 6 не відхи-
литься в крайнє праве положення. Це забезпечує використання всієї
шкали мікроамперметра при зміні подаваної напруги від нуля до ве-
личини (
.
.
0,5
розкл перед
E
)В.
Підібравши шунт до мікроамперметра, переміщають движок
реостата 2 у крайнє ліве положення й відключають джерело стру-
му. Після того як стрілка мікроамперметра повернеться в нульове
положення (для прискорення можна замкнути накоротко електроди
посудини), приступають до точних вимірів напруги розкладання.
Знову включають струм і подають на посудину 5 напруга за допо-
могою движка реостата 2, з інтервалами спочатку в 0,2 В, потім по-
близу точки розкладання 0,1 В и за нею 0,05 В. Щораз, установив-
ши напругу, вичікують 2–3 хв, після чого відзначають показання
вольтметра й амперметра. По закінченню досліду движок реостата
повертають у крайнє ліве положення.
Результати досліду записують у таблицю
E,B
i,A
.
.
розкл перед
E
.
.
розкл іст
E
За отриманим даними будується графік у координатах i E
,
по якому визначають величину напруги розкладання
.
.
розкл перед
E
.
КОНТРОЛЬНІ ПИТАННЯ ДО РОБОТИ
1. Що називається напругою розкладання?
2. Як визначається потенціал розряду?
3. Як по поляризаційній кривій визначається напругу розкла-
дання?
4. Від яких факторів залежить величина перенапруги?
5. Як попередньо оцінюють значення напруги розкладання?
6. Яким чином при визначенні перенапруги приєднують елект-
род порівняння?
7. Як регулюють силу струму та напругу під час досліду.
8. Для чого призначений капіляр Лугіна.
БІБЛІОГРАФІЧНИЙ СПИСОК РЕКОМЕНДОВАНОЇ ЛІТЕРАТУРИ
330
БІБЛІОГРАФІЧНИЙ СПИСОК
РЕКОМЕНДОВАНОЇ ЛІТЕРАТУРИ
Підручники
1. Антропов Л. И. Теоретическая электрохимия / Л. И. Антро-
пов. – М. : Высшая школа, 1984. – 519 с.
2. Афанасьев Б. Н. Физическая химия : Учебное пособие / Б.
Н. Афанасьев, Ю. П. Акулова. – СПб. : Издательство «Лань», 2012.
–
464 с.
3. Багоцкий В.С . Основы электрохимии. – М . : Химия. 1988. –
400 с.
4. Байрамов В. М . Основы электрохимии / В. М . Байрамов.
–
М. : ACADEMIA, 2005. – 238 с.
5. Вишняков А. В. Физическая химия / А. В. Вишняков, Н. Ф.
Кизим.
–
М. : Химия, 2012. – 839 с.
6. Герасимов Я. И. и др.. Курс физической химии. Т.2.
–
М.:
Химия; 1973. – 623 с.
7. Дамаскин Б. Б. и др. Электрохимия / Б. Б. Дамаскин, О. А.
Петрий, Г. А. Цирлина. – М. : Химия, КолосС, 2006. – 672 с.
8. Еремин Е.Н. Основы химической кинетики.
–
М.: Высшая
школа, 1976. — 541 с.
9. Зарубин Д.П. Физическая химия: учеб. пособие/Д.П. Зару-
бин.
—
М.: ИНФРА-М, 2017.
–474 с. + Доп. материалы [Электрон-
ный ресурс; Режим доступа http://www.znanium.comj. – (Высшее об-
разование: Бакалавриат). – ww w.dx.doi.oiB/I0.12737/20894.
10. Зимой А. Д . Физическая химия / А. Д . Зимой, Н.Ф . Лещен-
ко. – М.: Химия, 2000. – 320 с.
11. Киреев В.А. Курс физической химии. Изд. 3 -е. –
М.:
Химия, 1975. – 776 с.
12. Ковальчук Є. П. Фізична хімія : Підручник / Є. П. Коваль-
чук, О. В . Решетняк.
–
Львів : Видавничий центр ЛНУ Імені Івана
Франка, 2007. – 80 0 с
13. Лебідь, В. І. Фізична хімія : підруч. для студ. вищ. навч.
закл. / В. І. Лебідь. – Х. : Гімназія, 2008. – 478 с.
14. Миомандр Ф. Электрохимия / Ф. Миомандр, С. Садки, П.
Одебер, Р. Меале-Рено. пер. с фр. – М. : Техносфера, 2008. – 359 с.
БІБЛІОГРАФІЧНИЙ СПИСОК РЕКОМЕНДОВАНОЇ ЛІТЕРАТУРИ
331
15. Панченков Г. М . Химическая кинетика и катализ. Учебное
пособив для вузов / Г. М . Панченков, В. П. Лебедев. – 3 -е изд. испр.
идоп. – М.:Химия,1986. – 592с.
16. Романовский Б. В, Основы химической кинетики: учебник
/ Б. В . Романовский. – M. : Издательство «Экзамен», 2006. – 415 с.
17. Скорчелетти В. В. Теоретическая электрохимия. / В. В .
Скорчелетти. – Л. : Химия. 1974.
–
568 с.
18. Стромберг А. Г ., Семченко Д. П. Физическая химия: Учеб.
для хим. спец. вузов / Под ред. А. Г . Стромберга. – 3 -е изд., испр. и
доп– М. : Высш. шк., 1999. – 527 с:
19. Товбин М. В. Физическая химия. / М. В. Товбин. – Киев:
Вища школа, 1975. – 488 с.
20. Физическая химия. Теоретическое и практическое руко-
водство. Изд. 2 -е, переработанное и дополненное. / Под ред. ака-
демика Б. П. Никольского. – Л.: Химия, 1987. –880 с.
21. Физическая химия: Учеб. пособие для хим. -тех. спец. ву-
зов / Годнев И. Н., Краснов Л. С ., Воробьев Н. Л. и др.; Под ред. К .
С. Краснова. – М. : Высш. школа, 1982. – 687 с.
22. Фролов Ю. Г ., Велик В. В . Физическая химия. / Под ред.
проф. Ю . Г . Фролова. Учебное пособие для вузов.
–
М. : Химия,
1993. – 464 с.
23. Эмануэль Н. М ., Кнорре Д. Г . Курс химической кинетики:
Учебник для хим. фак, тов. –4 -е изд., перерзб. и доп. – М . : Высш.
шк., 1984 – 463 с,
24. Эткинс П. Физическая химия, Т.1 / П. Эткинс. – М . : Изда-
тельство «Мир», 1980. – 583 с.
25. Эткинс П. Физическая химия, Т.2 / П. Эткинс. – М . : Изда-
тельство «Мир», 1980. – 585 с.
Практикуми й збірники задач
1. Практикум по физической химии / Под ред. В . В. Буданова
и Н. К. Воробьева. – М.: Химия, 1986. – 351 с.
2. Кудряшов И. В. Сборник примеров и задач по физической
химии / И. В . Кудряшов, Г. С . Каретников. Изд. 6 -е, перераб. и до-
полн. – М. : Высшая школа, 1991. – 527 с.
БІБЛІОГРАФІЧНИЙ СПИСОК РЕКОМЕНДОВАНОЇ ЛІТЕРАТУРИ
332
3. Колпакова Н.А. Сборник задач по электрохимии : учебн.
пособие для ВУЗов / Н. А. Колпакова, Л. С . Анисимова, Н. А. Пику-
ла и др.. – М. : Высшая школа, 2003. – 143 с.
4. Практикум по прикладной электрохимии : Учеб. пособие
для вузов / Н. Г . Бахчисарайцьян, Ю. В . Борисоглебский, Г. К . Бур-
кат и др. Под ред. В. Н. Варыпаева, В. Н. Кудрявцева. – 3 -е изд., пе-
рераб.
–
Л. : Химия, 1990. – 304 с.
5. Практикум по физической химии / Под ред. И. В. Кудряшо-
ва. – М. : Высшая школа, 1986. – 495 с.
6. Практикум по физической химии / Под ред. Н. К. Воробье-
ва. Изд. 4 -е. – М. : Химия, 1975.
7. Практикум по физической химии: Учебное пособие для ву-
зов. Изд. 3 -е. / Под ред. С. В. Горбачева.
–
М. : Высшая школа,
1974. – 495 с.
8. Практикум по электрохимии. Б. Б. Дамаскин, О. А. Петрий,
Б. И . Подловченко и др. / Под ред. Б. Б. Дамаскина.
–
М. : Высш.
школа. 1991. – 288 с.
9. Практические работы по физической химии: Учебное посо-
бие для вузов / Под ред. К. П. Мищенко, А. А. Равделя и А. М. По-
номаревой. – Л. : Химия, 1982. – 400 с.
10. Рубцов В. І. Лабораторний практикум з фізичної хімії :
навчальний посібник. В 2–х кн. Кн. 1 . / В. I. Рубцов – Харків : Вид.
ХНУ імені В. Н. Каразіна, 2020. – 350 с.
11. Рубцов В. I. Потенціометричні методи дослідження розчи-
нів : навчальний nociбник / В. I. Рубцов – X. : ХНУ iмені В. Н.
Каразіна, 2016. – 252 с.
12. Рубцов В. I. Фізична хімія : задачі та вправи : навчальний
посібник / В. I. Рубцов.
-2 -ге вид., випр.
–
X.:ХНУiменіВ.Н.
Каразіна, 2016. – 416 с.
13. Сметанина Е. И. Лабораторный практикум по физической
химии : учебное пособие / Е. И. Сметанина, В. А. Колпаков. Том-
ский политехнический университет.
–
Томск : Изд-во Томского по-
литехнического университета, 2012. – 272 с.
14. Физическая химия. Теоретическое и практическое руково-
дство. Изд. 2 -е, переработанное и дополненное. / Под ред. акаде-
мика Б. П. Никольского. – Л. : Химия, 1987. – 880 с.
БІБЛІОГРАФІЧНИЙ СПИСОК РЕКОМЕНДОВАНОЇ ЛІТЕРАТУРИ
333
Додаткова література
1. Александров В. В . Кислотность неводных растворов.
–
Харьков: Вища школа. Изд-во при Харьковском ун-те, 1981. – 152 с.
2. Александров В. В . Кислотность неводных растворов. В сб.
Научное наследие Н.А.Измайлова и актуальные проблемы физиче-
ской химии под. ред. В. И. Лебедя, Н. О. Мчедлова-Петросяна и Ю.
В. Холина. – Х. : ХНУ имени В.Н. Каразина, 2007. – 675 с.
3. Батлер Дж. Ионные равновесия (математическое описание)
/ Дж. Батлер. – Л. : Химия. 1973. – 448 с.
4. Бейтс Р. Определение рН. Теория и практика / Р. Бейтс. – Л.
: Химия, 1971. – 400 с.
5. Бугаевский А. А. Расчет равновесий в растворе / А. А. Буга-
евский. – Х. : Вища школа. 1980. – 156 с.
6. Булатов М. И. Расчеты равновесий в аналитической химии /
М. И. Булатов. – Л. : Химия, 1984. – 184 с.
7. Варыпаев В. Н. Химические источники тока : учебн. посо-
бие / В.Н. Варыпаев, М. А. Дасоян, В. А. Никольский. –
М. : Выс-
шая школа, 1990. – 240 с.
8. Введенский А. В. Равновесные электродные потенциалы, по-
теи-циометрия // Соросовский образовательный журнал. 2000. Т.
6(10). С. 50–56.
9. Глесстон С. Теория абсолютных скоростей реакций. / С.
Глесстон, К. Лейдлер, Г. Эйринг Г.
–
М.: ИЛ,1948. – 583с.
10. Даниэльс Ф. Физическая химия / Ф. Даниэльс, Р. Олберти.
–
Пер.сангл. – М.:Мир.1978. – 646с.
11. Девис С., Джеймс А. Электрохимический словарь – М. :
Издательство «Мир», 1979. – 288 с.
12. Денисов Е. Т. Кинетика гомогенных химических реакций :
Учеб пособие для хим. Спец. вузов. – 2 -е изд. перераб и доп. – М . :
Высш. шк ., 1988. – 391 с.
13. Измайлов Н. А. Электрохимия растворов / Н. А. Измайлов.
–
М. : Химия, 1986. – 576 с.
14. Кедринский И.А. Литиевые источники тока. / И. А. Кед-
ринский, В. Е. Дмитренко, И. И. Грудянов. – М. : Энергоатомиздат,
1992, – 184 с.
БІБЛІОГРАФІЧНИЙ СПИСОК РЕКОМЕНДОВАНОЇ ЛІТЕРАТУРИ
334
15. Коровин Н. В. Топливные элементы и электрохимические
энергоустановки. – М. : изд. МЭИ, 2005. 280 с.
16. Крестов Г. А. Термодинамика ионных процессов в раство-
рах. -2 -е изд. перераб. и доп. – Л.: Химия, 1984. – 272 с.
17. Кромптон Т. Вторичные источники тока. – М. : Мир. 1985.
18. Кузнецов В. В. Определение рН // Соросовский образова-
тельный журнал. 2001. Т. 7(4). С . 44 -51.
19. Кузнецов В. В. Определение рН // Соросовский образова-
тельный журнал. 2001. Т. 7(4). С. 44–51.
20. Кумок В. Н. Произведения растворимости / В. Н. Кумок, О.
М. Кулешова, Л. А. Карабин. – Новосибирск : Наука, 1983. – 267 с.
21. Лукомский, Ю. Я . Физико-химические основы электрохи-
мии / Ю. Я . Лукомский, Ю. Д . Гамбург. — Долгопрудный : Интел-
лект, 2008. – 424 с
22. Методы измерения в электрохимии, т. 1, 2 / Под ред. Э.
ЕгераиА.Залкинда. – М.:Мир,1977. – 566с. – 476с.
23. Мудров В. И. Методы обработки результатов / В. И. Муд-
ров, В. Л. Кушко. – М. : Сов. радио, 1976. – 192 с.
24. Никольский Б. П. Ионоселективные электроды / Б. П. Ни-
кольский, Е.А. Матерова. – Л. : Химия, 1980. – 240 с.
25. Оксредметрия / Под ред. Б. П. Никольского и В. В . Паль-
чев-ского. – М. – Л. : Химия, 1975. – 254 с.
26. Пурмаль А.П. А, Б, В... химической кинетики / А.П. Пур-
маль. — М.: ИКЦ «Академкнига», 2004. — 277 с.
27. Рабинович В. А. Термодинамическая активность ионов в
растворах электролитов /В. А. Рабинович. – Л. : Химия, 1985. – 176 с.
28. Робинсон Р., Стокс Р. Растворы электролитов / Р. Робин-
сон, Р. Стокс. – М. : Издатинлит, 1963. – 646 с.
29. Ротинян А. Л., Тихонов К.И., Шошина И.А. Теоретическая
электрохимия / Под. ред. А. Л. Ротиняна. – М. : Студент, 2013. – 496 с
30. Румшиский Л. З. Математическая обработка результатов
эксперимента: Справочное пособие. – М. : Наука. 1971. – 192 с.
31. Таганова А. А. Герметичные химические источники тока
для портативной аппаратуры. / А. А. Таганова, И. А. Пак. и др. – М.
: МЭИ, 2003. – 275 с.
БІБЛІОГРАФІЧНИЙ СПИСОК РЕКОМЕНДОВАНОЇ ЛІТЕРАТУРИ
335
32. Таганова А.А., Семенов А.Е . Свинцовые аккумуляторы и
батареи. Справочник. – М. : МЭИ, 2004. – 348 с.
33. Теоретические и экспериментальные методы химии рас-
творов (Проблемы химии растворов) / Рос. акад. наук. Ин-т химии
растворов; отв. ред. А. Ю . Цивадзе. – М. : Проспект, 2011. – 684 с.
34. Фиалков Ю. Я . Физическая химия неводных растворов /
Ю. Я. Фиалков, А. Н. Житомирский, Ю. А. Тарасенко. – Л. : Химия,
1973. – 376 с.
35. Хартли Ф. Равновесия в растворах // Ф. Хартли, К. Бергес,
Р. Олкок. Пер. с англ., – М. : Мир, 1983. – 360 с.
36. Черепанов В. А. Химическая кинетика : [учеб. пособие] /
В. А. Черепанов, Т. В . Аксенова ; М-во образования и науки Рос.
Федерации, Урал, федер. ун-т. – Екатеринбург : Изд-во Урал, ун-та,
2016. – 132 с.
37. Шаронов В. Е. Компьютер для химика: Учеб.-метод, посо-
бие. – Новосибирск : НГУ, 2003. – 44 с.
38. Шауб Ю. Б. Кондуктометрия : монография / Ю. Б. Шауб;
Отв. ред. Г. Я. Волошин. – Владивосток, 1996. – 488 с.
39. Шведене Н. В. Ионоселективные электроды // Соросовский
образовательный журнал. 1999. No 5. С. 60-65.
40. Шелковников В. В. Расчеты ионных равновесий в химии :
учеб.-мет. пособие / В. В. Шелковников.
–
Томск : Изд-во Томского
ун-та, 2006. – 70 с.
41. Шульц М. М. Стеклянный электрод. Теория и применение. –
Соросовский образовательный журнал. – 1998. No 1. с. 33–39.
42. Экспериментальные методы химии растворов : денсиметрия,
вискозиметрия, кондуктометрия и другие методы / В. К. Абросимов.
В. В. Королев, В. Н. Афанасьев. – М. : Наука, 1997. – 351 с
Довідники й енциклопедії
1. Краткий справочник физико-химических величин. Издание
10-е, испр. и дополи. / Под ред. А. А. Равделя и А. М . Пономаревой
–
СПб. : «Иван Федоров», 2003. – 240 с.
2. Новый справочник химика и технолога. Электродные про-
цессы. Химическая кинетика и диффузия. Коллоидная химия под
общ. ред. Симановой С. А.
–
СПб. : АНО НПО «Профессионал»,
2004. – 838 с.
БІБЛІОГРАФІЧНИЙ СПИСОК РЕКОМЕНДОВАНОЇ ЛІТЕРАТУРИ
336
3. Нижниковский Е. А. Химические источники автономного
электропитания радиоэлектронной аппаратуры. – М. : МЭИ, 2004. –
228 с.
4. Рабинович В. А. Краткий химический справочник : Справ.
изд. / В. А. Рабинович, З. Я . Хавин 3. Я. под ред. А. А. Потехина и
А. И. Ефимова. – 3 -е изд. перераб. и доп. – Л. : Химия, 1991. – 432 с.
5. Справочник по химии // А. И. Гончаров, М. Ю . Корнилов.
–
К. : Вища школа, 1974. – 304 с.
6. Справочник по электрохимии / Под ред. А. М . Сухотина.
–
Л. : Химия, 1981. – 488 с.
7. Справочник химика. 2 -ое изд. перераб. и доп. Т.2 Основные
свойства неорганических и органических соединений. – Л. : Химия,
1971. – 1169 с.
8. Справочник химика. Том 3. Химическое равновесие и кине-
тика, свойства растворов, электродные процессы. 2 -е изд. перераб. и
доп. – Л. : Химия, 1965. – 1006 с.
9. Справочник химика. 2 -ое изд. перераб. и доп. Изд-е 2-е, Т.4
Аналитическая химия, спектральный анализ, показатели преломле-
ния. – Л. : Химия, 1967. – 920 с.
10. Химическая энциклопедия: В 5 т.: т. 1 –5 : / Ред. кол.: И. Л.
Кнунянц (гл. ред.) и др. – М. : Сов. энцикл., 1988–1998.
11. Химические источники тока : Справочник / Под редакцией
Н. В . Коровина и A. M . Скундина. – М. : Издательство МЭИ, 2003. –
740 с.
ДОДАТОК
ДОДАТОК
338
Таблиця 1
ОСНОВНІ ФУНДАМЕНТАЛЬНІ ПОСТІЙНІ
Постійна
Позначення
Значення із вказівкою
погрішності
Швидкість світла у вакуумі
Діелектрична
проникність вакууму
Елементарний заряд
(заряд електрона)
Універсальна газова
постійна
Постійна Авогадро
Постійна Больцмана
Постійна Фарадея
Постійна Планка
c
o
e
R
A
N
/A
kRN
A
FeN
h
/2
h
2,99792458(1) 8
10
м/с
8,85418782(5) 12
10
2
Кл /(Н·
2
м)
1,6021892(46) 19
10
Кл
8,31441(26) Дж/( К·моль)
6,022045(31) 23
10
моль
1
1,380662(44) 23
10
Дж/К
9,648456(27) 4
10
Кл/моль
6,626176(36) 34
10
Дж/с
1,0545887(57) 34
10
Дж/с
Позасистемні одиниці
Універсальна газова
постійна
R
82,0568(26) 3
см /(К·моль)
1,98716(6) кал/(К·моль)
Таблиця 2
ДЕЯКІ ПЕРЕКЛАДНІ МНОЖНИКИ
Фізична
величина
Найменування
одиниці
Значення
в одиницях СИ
Довжина
Енергія
Сила
Тиск
Ангстрем,
о
А
Електронвольт, еВ
Хвильове число,
1
см
Калорія (термохіміч.)
Ерг
Дина
Атмосфера
мм. рт. ст.
10
10
м(
1
10
нм)
1,6022·
19
10 Дж
1,986·
23
10 Дж
4,184 Дж
7
10 Дж
5
10 Н
101,325 кН/ 2
м (кПа)
133,322 Н/ 2
м (Па)
ДОДАТОК
339
Таблиця 3
ФІЗИКО-ХІМІЧНІ ВЛАСТИВОСТІ ВОДИ
Тиск пари, p
toC
Густина
3
, г/см
toC
мм рт. ст.
кПа
toC
Діелектрична
проникність,
17
18
19
20
21
22
23
24
25
26
27
28
29
30
35
40
45
50
55
60
65
0,99880
0,99862
0,99843
0,99823
0,99802
0,99780
0,99756
0,99732
0,99707
0,99681
0,99654
0,99626
0,99597
0,99567
0,99406
0,99224
0,99025
0,98807
0,98573
0,98324
0,98059
0
5
10
15
16
17
18
19
20
21
22
23
24
25
30
35
40
45
50
60
70
4,579
6,543
9,209
12,79
13,63
14,53
15,48
16,48
17,53
18,65
19,83
21,07
22,38
23,76
31,84
42,18
55,32
71,88
92,51
149,4
233,7
0,61048
0,87233
1,2278
1,70519
1,81718
1,93717
2,06382
2,19715
2,33713
2,48646
2,64378
2,80909
2,98375
3,16773
4,2452
5,6229
7,3759
9,5382
12,334
19,916
31,157
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
87,740
85,763
83,832
81,945
80,103
78,303
76,546
74,823
73,151
71,511
69,910
68,344
66,813
65,319
63,855
62,425
61,027
59,657
58,317
57,005
55,720
ДОДАТОК
340
Таблиця 4
КОНСТАНТИ ДИСОЦІАЦІЇ СЛАБКИХ КИСЛОТ У ВОДНИХ
РОЗЧИНАХ ЗА ТЕМПЕРАТУРИ 298 К
Назва
Формула
Ka
pКa
K1
5.5 ·10−5
4.26
Адипінова
K2
HOOC(CH2)4COOH
5.01·10−6
5.30
Азотистоводнева
K1 HN3
1.91·10−5
4.72
Амінооцтова (Гліцин) K1 NH2CH2COOH
1.66 ·10−10
9.78
K1
9.12 ·10−5
4.04
Аскорбінова
K2
CH2(OH)CH(OH)CHC–
(OH)=C(OH)COO
4.57 ·10−12
11.3
Бензойна
K1 C6H5COOH
6.17 ·10−5
4.21
Борна (орто-) К1
K1 H3BO3
5.75 ·10−10
9.24
K1
1.82·10−4
3.74
Борна (тетра-)
K2
H2B4O7
2·10−8
7.70
Бромновата
K1 HBrO
2.51 ·10−9
8.60
K1
9.12 ·10−4
3.04
Винна
K2
HOOCCH(OH)CH(OH)–
COOH
4.27 ·10−5
4.37
Вольфрамова
K1 H2WO4
6.31·10−5
4.20
Гідрохінон
K1 1,4 -C6H4(OH)2
1.1 ·10−10
9.96
Гліколева
K1 CH2(OH)COOH
1.32·10−4
3.88
Двухромова ( К2)
K2 H2Cr2O7
2.29·10−2
1.64
Дихлороцтова
K1 CHCl2COOH
5.01·10−2
1.30
K3
2.69 ·10−3
2.57
Залізосинєродиста
( К3, К4)
K4
H4Fe(CN)6
4.47 ·10−5
4.35
Йодновата
K1 HIO3
6.17 ·10−0
0.79
K1
1.26·10−10
9.90
K2
1.58·10−12
11.8
Кремінна (орто)
K3
H4SiO4
2·10−14
13.7
K1
7.41 ·10−4
3.13
K2
1.74 ·10−5
4.76
Лимонна
K3
HOOCCH2C(OH)–
(COOH)CH2COOH
3.98 ·10−7
6.40
м-Нітробензойна
K1 NO2C6H4COOH
3.55 ·10−4
3.45
K1
2·10−4
3.70
м-Фталева
K2
C6H4(COOH)2
2.51 ·10−5
4.60
K1
1.41 ·10−3
2.85
Малонова
K2
HOOCCH2COOH
2·10−6
5.70
Масляна (норм.)
K1 CH3CH2CH2COOH
1.51 ·10−5
4.82
Молочна
K1 CH3CH(OH)COOH
1.38 ·10−4
3.86
ДОДАТОК
341
Таблиця 4 (продовження)
Мурашина
K1 HCOOH
1.78·10−4
3.75
о-Нітробензойна
K1 NO2C6H4COOH
6.76·10−3
2.17
K1
1.12 ·10−3
2.95
о-Фталева
K2
C6H4(COOH)2
3.98 ·10−6
5.40
п-Крезол
K1 CH3C6H4OH
6.76·10−11
10.2
п-Нітробензойна
K1 NO2C6H4COOH
3.63 ·10−4
3.44
K1
2.88 ·10−4
3.54
п-Фталева
K2
C6H4(COOH)2
3.47 ·10−5
4.46
Пікринова (тринітрофенол) K1 HOC6H2(NO2)3
1.95·10−0
0.29
Пропіонова
K1 CH3CH2COOH
1.35·10−5
4.87
Роданістоводнева
K1 HSCN
7.08 ·10−0
0.85
Саліцилова
K1 C6H4(OH)COOH
1.07·10−3
2.97
Сульфатна (K2)
K2 H2SO4
1.15 ·10−2
1.94
K1
1.74 ·10−2
1.76
Сірчиста
K2
H2SO3
6.31·10−8
7.20
K1
1.02·10−7
6.99
Сірководнева
K2
H2S
1.29·10−13
12.9
Синильна
K1 HCN
6.17 ·10−10
9.21
Сульфамінова
K1 H2NSO3H
9.55 ·10−0
0.98
Сульфанілова
K1 H2NC6H4SO3H
6.31·10−4
3.20
K2
1.38 ·10−3
2.86
Сульфосаліцилова
( К2, К3)
K3
C6H3(OH)(COOH)–
SO3H
1.82·10−12
11.7
Трихлороцтова
K1 Ccl3COOH
5.01·10−0
0.70
K1
4.47 ·10−7
6.35
Вугільна
K2
CO2(aq.)+H2O
4.79·10−11
10.3
Оцтова
K1 CH3COOH
1.74 ·10−5
4.76
Фенол
K1 C6H5OH
1·10−10
10.0
K1
7.59·10−3
2.12
K2
6.17 ·10−8
7.21
Фосфорна (орто)
K3
H3PO4
4.17 ·10−13
12.4
Фтористоводнева
K1 HF
6.76·10−4
3.17
Хлороцтова
K1 CH2ClCOOH
1.38 ·10−3
2.86
K1
9.55 ·10−0
0.98
Хромова
K2
H2CrО4
3.16·10−7
6.50
K1
5.62·10−2
1.25
Щавлева
K2
H2C2O4
5.37·10−5
4.27
K1
6.17 ·10−5
4.21
Бурштинова
K2
HOOCCH2CH2COOH
2.34·10−6
5.63
ДОДАТОК
342
Таблиця 5
ІОННИЙ ДОБУТОК, В'ЯЗКІСТЬ ТА ПИТОМА ЕЛЕКТРОП-
РОВІДНІСТЬ ВОДИ ПРИ РІЗНИХ ТЕМПЕРАТУРАХ
tC
w
K 1014
w
pK
103
Па·с
108 Ом
-1
см
-1
0
0,1139 14,943 1,7921
–
5
0,1846 14,734 1,5188
–
10
0,2920 14,535 1,3077
2,85
15
0,4505 14,346 1,1404
–
18
0,5702 14,244 1,0559
4,41
20
0,6809 14,167 1,0050
4,85
21
0,742
14,130 0,9810
–
22
0,802
14,096 0,9579
–
23
0,868
14,061 0,9358
–
24
0,948
14,023 0,9142
–
25
1,008
13,996 0,8937
6,33
30
1,469
13,833 0,8007
8,15
35
2,089
13,680 0,7225
10,02
40
2,919
13,535 0,6560
–
45
4,018
13,396 0,5988
–
50
5,474
13,262 0,5494
18,9
55
7,297
13,137 0,5064
–
60
9,614
13,017 0,4688
–
100
59,0
12,229 0,2838
–
іонний добуток,
w
K і показник іонного добутку,
w
pK;
динамічна в'язкість ·103 Па·с;
питома електропровідність ·108 Ом
-1
см
-1
.
ДОДАТОК
343
Таблиця 6
ДОБУТОК РОЗЧИННОСТІ ДЕЯКИХ СПОЛУК
ЗА ТЕМПЕРАТУРИ 298 К
=++–
Тверда фаза
sp
K
( г-іон/ л)
Тверда фа-
за
sp
K
( г-іон/ л)
AgBr
5,0 10–13
CuI
1,1 10–12
AgCN
1,6 10 –14
Fe(OH) 2
1,4 10–15
AgCl
1,7310–10
Hg2Br2
4 10–23
AgI
8,1 10–17
Hg2Cl2
1 10–18
AgBrO3
610–5
Hg2J2
4 10–29
AgIO3
3 10–8
Hg2SO4
6,2 10–7
Ag2CrO4
4,410–12
Ni(OH) 2
1,3 10–16
Ag2SO4
1,23 10–5
PbBr2
4,5 10–6
Al(OH)3
4 10–33
PbCl2
1,5 10–5
Ag2S
2,510–25
PbI2
8 10–9
BaSO3
9,5 10–10
Pb(OH) 2
5 10–16
BaDO4
1,5 10–9
PbSO4
1,6 10–8
CaHPO4
2 10–8
PbS
1,1 10–29
Ca(OH) 2
6 10–6
RbClO4
2,5 10–3
CaSO4
2,4 10–5
SrC2O4
5,6 10–8
Cd(OH) 2
1,66 10–14
SrCrO4
3,6 10–5
Co(OH) 2
2,5 10–16
SrSO4
2,8 10–7
CuCl
3,2 10–7
TlBr
3,6 10–6
CaCrO4
2,310–2
TlCl
1,8 10–4
CaCO3
5 10–9
TlI
8,9 10–8
CuCO3
2 10–10
ZnCO3
6 10–11
CuS
3,5 10–38
Zn(OH) 2
4,3 10–17
CuC2O4
2,87 10–8
ZnS
1,3 10–23
ДОДАТОК
344
Таблиця 7
СЕРЕДНІ ІОННІ КОЕФІЦІЄНТИ АКТИВНОСТІ СИЛЬНИХ
ЕЛЕКТРОЛІТІВ У ВОДІ ЗА ТЕМПЕРАТУПИ 298 К
Концентрація, моль/кг
Електроліт
0,001 0,002 0,005 0,01 0,02 0,05 0,1 0,2 0,5
AgNO3
–
–
0,925 0,897 0,860 0,793 0,734 0,657 0,536
BaCl2
0,881 0,840 0,774 0,716 0,651 0,564 0,500 0,444 0,397
Ba(OH) 2
–
0,853 0,773 0,712 0,627 0,526 0,443 0,370 –
CaCl2
0,889 0,852 0,789 0,731 0,668 0,583 0,518 0,472 0,448
CuCl2
0,888 0,849 0,783 0,723 0,659 0,577 0,508 0,455 0,411
CuSO4
0,74
–
0,573 0,438 0,317 0,217 0,154 0,104 0,062
HBr
0,966
–
0,930 0,906 0,879 0,838 0,805 0,782 0,789
HCl
0,965 0,952 0,928 0,904 0,875 0,830 0,796 0,767 0,757
HF
0,544
–
0,300 0,224
–
0,106 0,077 0,031 –
H2SO4
0,830 0,757 0,639 0,544 0,453 0,340 0,265 0,209 0,156
KBr
0,965 0,952 0,927 0,903 0,872 0,822 0,772 0,722 0,657
KCl
0,965 0,952 0,927 0,902 0,869 0,816 0,770 0,718 0,649
KClO4
0,967 0,955 0,932 0,907 0,875 0,813 0,749 0,681 0,568
K3Fe(CN)6
–
–
–
–
–
–
0,268 0,212 0,155
K4Fe(CN)6
–
–
–
–
–
0,19 0,139 0,100 0,062
KI
0,952 – 0,928 0,903 0,872 0,820 0,778 0,733 0,676
KNO3
0,965 0,951 0,926 0,898 0,862 0,799 0,739 0,663 0,545
KOH
–
–
–
–
–
0,824 0,798 0,760 0,732
LiCl
0,790 0,729 0,636 0,560 0,483 0,388 0,314 0,274 0,266
MgCl2
–
–
–
–
–
–
0,529 0,489 0,481
MgSO4
–
–
–
–
–
–
0,150 0,108 0,068
NH4Cl
–
–
0,924 0,896 0,862 0,808 0,770 0,718 0,649
NH4NO3
–
–
0,925 0,897 0,860 0,799 0,740 0,677 0,582
NaBr
0,97 0,96 0,94 0,91 0,89 0,85 0,782 0,741 0,697
NaBl
0,965 0,952 0,928 0,903 0,872 0,822 0,778 0,735 0,681
NaI
–
–
–
–
–
–
0,787 0,751 0,723
NaNO3 0,966 0,953 0,929 0,905 0,873 0,821 0,762 0,703 0,617
NaOH
–
–
–
0,905 0,871 0,818 0,766 0,727 0,690
Na2SO4 0,887 0,847 0,778 0,714 0,642 0,536 0,445 0,365 0,266
ZnCl2
0,88 0,84 0,77 0,71 0,64 0,56 0,515 0,462 0,394
ZnSO4
0,700 0,608 0,477 0,387 0,298 0,202 0,150 0,104 0,063
CH3COONa
–
–
–
–
–
–
0,791 0,757 0,735
ДОДАТОК
345
Таблиця 8
ПИТОМА ЕЛЕКТРОПРОВІДНІСТЬ (, Ом
–1
см
–1
)
ВОДЯНИХ РОЗЧИНІВ ХЛОРИСТОГО КАЛІЮ
ПРИ РІЗНИХ ТЕМПЕРАТУРАХ
Концентрація c, г-екв/л
Темпе-
ратура,
С
0,1
0,02
0,01
15
0,01048
0,002243
0,001147
16
0,01072
0,002294
0,001173
17
0,01095
0,002345
0,001199
18
0,01119
0,002397
0,001225
19
0,01143
0,002449
0,001251
20
0,01167
0,002501
0,001278
21
0,01191
0,002553
0,001305
22
0,01215
0,002606
0,001332
23
0,01239
0,002659
0,001359
24
0,01264
0,002712
0,001386
25
0,01288
0,002765
0,001417
ДОДАТОК
346
Таблиця 9
ГРАНИЧНА ЕКВІВАЛЕНТНА ЕЛЕКТРОПРОВІДНІСТЬ
ІОНІВ ПРИ 298 К Й ТЕМПЕРАТУРНИЙ КОЕФІЦІЄНТ
ЕЛЕКТРОПРОВІДНОСТІ
oT
o
T
,
,
298 1
298 ,
о, 298
1
Т
.
Катіон
o
Аніон
o
Ag+
61,9
0,0194
Br
–
78,14 0,0185
1/2 Ba
2+
63,6
0,020
BrO3
–
55,4
–
1/2 Ca
2+
59,5
0,021
Cl–
76,35 0,0194
1/2 Cd2+
54
0,020
1/2 CO3
2–
69,3 0,0192
Cs
+
77,2
0,019
1/2 CrO4
2–
83
0,021
1/2 Fe
2+
53,5
0,024
F–
55,4
0,021
1/3 Fe3+
68
–
1/3Fe(CN)6
3–
99,1
–
H+
349,8 0,0142
HSO4
–
52
–
1/2 Hg2+
63,6
–
I–
76,85 0,0192
K+
73,5
0,0187
IO3–
40,8
0,024
Li+
38,6
0,0214
Mno4
–
61,3 0,0224
1/2 Mg2+
53,0
0,0218
NO3–
71,4 0,0184
NH4
+
73,5
0,0187
OH–
198,3 0,0196
Na
+
50,1
0,0208
SCN–
66
–
1/2 Ni2+
54
–
1/2 SO4
2–
80,0 0,0206
1/2 Pb2+
70
0,0178
HCOO–
54,6
–
Rb+
77,8
0,0207
CH3COO
–
40,9 0,0206
1/2 Zn
2+
54
0,0185 CH2ClCOO– 39,8
–
(CH3)4N
+
44,9
0,0156 C2H5COO
–
35,8
–
(C4H9)4N
+
19,4
–
C6H5COO
–
32,3
–
Еквівалентна електропровідність в таблиці виражено в
Ом
-1
·г-екв
-1
·см
2
. Для перерахування в Ом
-1
·г-екв
–1
·м
2
необхідно зна-
чення, що розташовані у таблиці, помножити на 10–4
.
ДОДАТОК
347
Таблиця 10
ГРАНИЧНІ РУХЛИВОСТІ ІОНІВ У ВОДІ ПРИ РІЗНИХ
ТЕМПЕРАТУРАХ
Граничні рухливості при температурах (° C)
Ом
-1
г-екв
–1
см
2
Іон
0
5
151825354555100
H3O
+
225 250,1 300,6 315 349,8 397,0 441,4 483,1 650,0
OH–
105 –
–
171 197,6
–
–
–
450,0
Li+
19,4 22,7 30,2 32,8 38,6 48,0 58,0 68,7 115,0
Na
+
26,4 30,0 39,7 42,8 50,1 61,5 73,7 86,9 145,0
K+
40,7 46,7 59,6 63,9 73,5 88,2 103,5 119,2 195,0
Rb+
43,9 50,1 63,4 66,5 77,8 92,9 108,5 124,2
–
Cs
+
44,0 50,0 63,1 67,0 77,2 92,1 107,5 123,6
–
Ag+
33,1
–
–
53,5 61,9
–
–
–
175,0
NH4
+
40,2
–
–
63,9 73,5
–
–
–
180,0
N(CH3)4
+
24,1
–
–
40,0 44,9
–
–
–
–
N(C2H5)4
+
16,4
–
–
28,2 32,6
–
–
–
–
N(C3H7)4
+
11,5
–
–
20,9 23,4
–
–
–
–
N(C4H9)4
+
9,6
–
–
–
19,4
–
–
–
–
N(C5H11)4
+
8,8
–
–
–
17,4
–
–
–
–
Mg2+
28,9
–
–
44,9 53,0
–
–
–
165
Ca
2+
31,2
–
46,9 50,7 59,5 73,2 88,2
–
180
Sr
2+
31
–
–
50,9 59,4
–
–
–
–
Ba
2+
34,0
–
–
54,6 63,6
–
–
–
195
NO3
–
40,0
–
–
62,3 71,44
–
–
–
195
ClО4
–
36,9
–
–
58,8 67,3
–
–
–
185
CH3COO
–
20,1
–
–
35 40,9
–
–
–
–
F–
–
–
–
47,3 55,4
–
–
–
–
Cl–
41,0 47,5 61,4 66,0 76,4 92,2 108,9 126,4 212,0
Br
–
46,2 49,2 63,1 68,0 78,1 94,1 110,7 127,8
–
I–
41,4 48,5 62,1 66,0 76,8 92,4 108,6 125,4
–
SO4
2–
41,0
–
–
68,4 80,0
–
–
–
260
в'язкість
води,
о
сПз
1,787 1,516 1,138 1,053 0,890 0,719 0,596 0,504 0,283
ДОДАТОК
348
Таблиця 11
ЧИСЛА ПЕРЕНОСУ КАТІОНІВ У ВОДЯНИХ РОЗЧИНАХ
ЕЛЕКТРОЛІТІВ ЗА ТЕМПЕРАТУРИ 298 К
Концентрація, г-екв/л
Електроліт
0,2
0,1
0,05 0,02 0,01
0
AgNO3
–
0,468 0,466 0,465 0,465 0,464
CaCl2
0,395 0,406 0,414 0,422 0,426 0,438
HCl
0,834 0,831 0,829 0,827 0,825 0,821
KBr
0,484 0,483 0,483 0,483 0,483 0,485
KOOCCH3
–
0,661 0,657 0,652 0,650 0,643
KCl
0,489 0,490 0,490 0,490 0,490 0,491
KI
0,489 0,488 0,488 0,488 0,488 0,489
KNO3
0,512 0,510 0,509 0,509 0,508 0,507
K2SO4
0,491 0,489 0,487 0,485 0,483 0,479
LiCl
0,311 0,317 0,321 0,326 0,329 0,336
NH4Cl
0,491 0,491 0,490 0,491 0,491 0,491
NaCl
0,382 0,385 0,388 0,390 0,392 0,396
NaOOCClCH3 0,561 0,559 0,557 0,555 0,554 0,551
Na2SO4
0,383 0,383 0,383 0,384 0,385 0,386
ДОДАТОК
349
Таблиця 12
ЗНАЧЕННЯ ВЕЛИЧИН ln10 /
RTF
І ПАРАМЕТРИ РІВНЯН-
НЯ ДЕБАЯ-ХЮККЕЛЯ ДЛЯ ВОДЯНИХ РОЗЧИНІВ ПРИ РІЗ-
НИХ ТЕМПЕРАТУРАХ
Молярна шкала
Моляльна шкала
Темпе
ратура,
С
ln10
RT
F
А
В
А
В
0
54,197
0,4918
0,3248
0,4918
0,3248
5
55,189
0,4952
0,3256
0,4952
0,3256
10
56,181
0,4989
0,3264
0,4988
0,3264
15
57,173
0,5028
0,3273
0,5026
0,3272
20
58,165
0,5070
0,3282
0,5066
0,3279
25
59,157
0,5115
0,3291
0,5108
0,3286
30
60,149
0,5161
0,3301
0,5150
0,3294
35
61,141
0,5211
0,3312
0,5196
0,3302
40
62,133
0,5262
0,3323
0,5242
0,3310
45
63,126
0,5317
0,3334
0,5291
0,3318
50
64,118
0,5373
0,3346
0,5341
0,3326
55
65,110
0,5432
0,3358
0,5393
0,3334
60
66,102
0,5494
0,3371
0,5448
0,3343
ДОДАТОК
350
Таблиці 13–19
СКЛАД БУФЕРНИХ РОЗЧИНІВ
(рН від 1,10 до 8,0; температура 20 °С)
Буферні розчини готують із вихідних розчинів, що змішуються в
різних співвідношеннях. Об'єми вихідних розчинів (мл) наведено в
таблицях 13-191. Значення рН задане з точністю до сотої частки: цілі
й десяті частки – у лівому стовпці, соті – у верхньому рядку.
У таблицях 25–31 наведені відомості про буферні розчини на ос-
нові оцтової кислоти; суміші оцтової, фосфатної й борної кислот; роз-
чинах з індивідуальних речовин.
Вихідні розчини
Розчин No 1 – 0,1 М соляна кислота.
Розчин No 2 – 0,1 М амінооцтова кислота; (глікоколь, гліцин)
NH2CH2COOH (7,507 г глікоколя + 5,85 г NaСl в 1 л).
Розчин No 3 – 0,2 М гідрофталат калію; КНС8Н4O4 (40,846 г в 1 л).
Розчин No 4 – 0,1 М цитрат натрію; 21,014 г лимонної кислоти
С6H8O7Н2О+200мл 1.0MрозчинуNaOHв1л
Розчин No 5 – 0,1 М гідроксид натрію.
Розчин No б – 1/15 М дигідрофосфат калію; КН2РO4 (9,073 г в 1л).
Розчин No 7 – 1/15 М гідрофосфат натрію; Na2HPО4 2Н2O (11,866 г в 1
л).
Розчин No 8 – 0,05 М тетраборат натрію; Н3ВO3 12,367 1 + 100 мл 1
М розчину NaОН в 1 л.
Хлорид натрію х.ч . (хімічно чистий) двічі перекристалізовують
і висушують при 120 °С;
борну кислоту х.ч. двічі перекристалізовують з киплячої води
й висушують при температурі не вище 80 °С;
дигідрофосфат калію х.ч. двічі перекристалізовують і висушу-
ють при 110–120 °С;
гідрофосфат натрію х.ч. двічі перекристалізовують (при остан-
ній кристалізації температура розчину не повинна бути вище 90 °С),
потім зволожують водою й висушують у термостаті при 36 °С протя-
гом 2 доби;
лимонну кислоту х.ч . двічі перекристалізовують (при останній
кристалізації температура не повинна перевищувати 60 °С);
гідрофталат калію двічі перекристалізовують і висушують при
110–120 °С.
ДОДАТОК
351
Таблиця 13
Буферні розчини із рН 1,10–3,50 (HCI–NH2CH2COOH)
Кожний із зазначених нижче об'ємів розчину No 2 доводять до
100 мл розчином No 1.
рН0
1
2
3
4
5
6
7
8
9
1,1 5,7 6,6 7,5 8,4 9,3 10,2 11,1 12,0 12,8 13,7
2 14,6 15,4 16,2 17,0 17,8 18,6 19,4 20,2 21,0 21,8
3 22,6 23,2 23,9 24,5 25,2 25,8 26,4 27,0 27,7 28,3
4 28,9 29,4 30,0 30,5 31,1 31,6 32,0 32,5 32,9 33,4
5 33,8 34.2 34,6 35,0 35,4 35,8 36,2 36,7 37,1 37,6
6 38,0 38,4 38,7 39,1 39,4 39,8 40,2 40,6 40,9 41,3
7 41,7 42,1 42,4 42,8 43,1 43,5 43,9 44,2 44,6 44,9
8 45,3 45,6 46,0 46,3 46,7 47,0 47,4 47,8 48,1 48,5
9 48,9 49,2 49,5 49,8 50,1 50,4 50,7 51,0 51,3 51,6
2,0 51.9 52,2 52,5 52,8 53,1 53,4 53,7 54,0 54,3 54,6
1 54,9 55,2 55,4 55,7 55,9 56,2 56,5 56,7 57,0 53,3
2 57,6 57,9 58,2 58,4 58,7 59,0 59,3 59,5 59,8 60,0
3 60,3 60,6 61,0 61,3 61,7 62,0 62,3 62,6 63,0 63,3
4 63,6 63,9 64,2 64,5 64,8 65,1 65,4 65,7 66,0 66,3
5 66,6 66,9 67,2 67,5 67,8 68,1 68,4 68,7 69,0 69,3
б 69,6 69,9 70,2 70,5 70,8 71,1 71.4 71,8 72,1 72,5
7 72,8 73,1 73,4 73,8 74,1 74,4 74,7 75,0 75,4 75,7
8 76,0 76,3 76,6 77,0 77,3 77,6 77,9 78,2 78,6 78,9
9 79,2 79,5 79,8 80,1 80,4 80,7 81,0 81,3 81,5 81,8
3,0 82,1 82,4 82,7 82,9 83,2 83,5 83,8 84,0 84,3 84,5
1 84,8 85,0 85,3 85,5 85,8 86,0 86,2 86,4 86,7 86,9
2 87,1 87,3 87,5 87,8 88,0 88,2 88,4 88,6 88,8 89,0
3 89,2 89,4 89,6 89,7 89,9 90,1 90,3 90,5 90,6 90,8
4 91,0 91,2 91,3 91,5 91,6 91,8 91,9 92,1 92,2 92,4
ДОДАТОК
352
Таблиця 14
Буферні розчини із рН 1,10–4,96 (HO–NaН2С6H5О7)
Кожний із зазначених нижче об'ємів розчину No 4 доводять до
100 мл розчином No 1.
рН0
1
2
3
4
5
6
7
8
9
1,1 4,8 5,6 6,4 7,1 7,8 8,4 9,0 9,6 10,1 10,6
2 11,1 11,6 12,1 12,5 13,0 13,5 14,0 14,5 14,9 15,4
3 15,9 16,2 16,6 16,9 17,3 17,6 17,9 18,3 18,6 19,0
4 19,3 19,6 19,9 20,2 20,5 20,8 21,1 21,4 21,6 21,9
5 22,2 22,4 22,7 22,9 23,2 23,4 23,6 23,9 24,1 24,4
6 24,6 24,8 25,0 25,2 25,4 25,6 25,8 26,0 26,1 26,3
7 26,5 26,7 26,9 27,0 27,2 27,4 27,6 27,7 27,9 28,0
8 28,2 28,3 28,5 28,6 28,8 28,9 29,0 29,1 29,3 29,4
9 29,5 29,6 29,7 29,9 30,0 30,1 30,2 30,3 30,4 30,5
2,0 30,6 30,7 30,8 31,0 31,1 31,2 31,3 31,4 31,5 31,6
1 31,7 31,8 31,9 31,9 32,0 32,1 32,2 32,3 32,4 32,5
2 32,6 32,7 32,8 32,9 33,0 33,1 33,2 33,3 33,4 33,5
3 33,6 33,7 33,8 33,8 33,9 34,0 34,1 34,2 34,3 34,4
4 34,5 34,6 34,7 34,8 34,9 35,0 35,1 35,2 35,2 35,3
5 35,4 35,5 35,6 35,7 35,8 35,9 36,0 36,1 36,2 36,3
6 36,4 36,5 36,6 36,7 36,8 36,9 37,0 37,1 37,1 37,2
7 37,3 37,4 37,5 37,6 37,7 37,8 37,9 38,0 38,1 38,2
8 38,3 38,4 38,5 38,6 38,7 38,8 38,9 39,0 39,1 39,2
9 39,3 39,4 39,5 39,6 39,7 39,8 39,9 40,0 40,1 40,2
ДОДАТОК
353
Таблиця 14 (продовження)
Буферні розчини із рН 1,10–4,96 (HO–NaН2С6H5О7)
рН0
1
2
3
4
5
6
7
8
9
3,0 40,3 40,4 40,5 40,7 40,8 40,9 41,0 41,1 41,3 41,4
] 41,5 41,6 41,7 41,8 41,9 42,0 42,1 42,3 42,4 42,6
2 42,7 42,8 42,9 43,1 43,2 43,3 43,4 43,6 43,7 43,9
3 44,0 44,1 44,3 44,4 44,6 44,7 44,8 45,0 45,1 45,3
4 45,4 45,5 45,7 45,8 46.0 46,1 46,2 46,4 46,5 46,7
5 46,8 47,0 47,1 47,3 47,4 47,6 47,8 47,9 48,1 48,2
6 48,4 48,6 48,8 48,9 49,1 49,3 49,5 49,6 49,8 49,9
7 50,1 50,3 50,4 50,6 50,8 51,0 51,2 51,4 51,5 51,7
8 51,9 52,1 52,3 52,5 52,7 52,9 53,1 53,3 53,4 53,6
9 53,8 54,0 54,2 54,5 54,7 54,9 55,1 55,3 55,6 55,8
4,0 56,0 56,3 56,5 56,8 57,0 57,3 57,5 57,8 58,0 58,3
1 58,5 58,7 59,0 59,2 59,5 59,7 60,0 60,3 60,5 60,8
2 61,1 61,4 61,7 62,0 62,3 62,6 62,9 63,3 63,6 64,0
3 64,3 64,7 65,1 65,4 65,7 66,0 66,4 66,8 67,1 67,5
4 67,9 68,3 68,7 69,0 69,4 69,8 70,2 70,6 71,1 71,5
5 71,9 72,4 72,9 73,4 73,9 74,4 74,9 75,4 75,9 76,4
6 76,9 77,4 78,0 78,5 79,1 79,6 80,1 80,6 81,2 81,7
7 82,2 82,8 83,3 83,9 84,4 85,0 85,6 86,2 86,6 87,4
8 88,0 88,7 89,4 90,0 90,7 91,4 92,2 93,1 93,9 94,8
9 95,6 96,3 97,1 97,8 98,5 99,3 100,0
ДОДАТОК
354
Таблиця 15
Буферні розчини із рН 2,20–3,80 (HCI–КНС8Н4О4)
До кожного із зазначених нижче об'ємів розчину No I додають
50,0 мл розчину No 3 й доводять об'єм суміші водою до 200 мл.
pН0
1
2
3
4
5
6
7
8
9
2,2 93,20 92,50 91,80 91,10 90,40 89,70 89,00 88,30 87,60 86,90
3 86,20 85,50 84,80 84,10 83,40 82,70 82,00 81,30 80,60 79,90
4 79,20 78,54 77,88 77,22 76,56 75,90 75,24 74,58 73,92 73,26
5 72,60 71,94 71,28 70,62 69,96 69,30 68,64 67,98 67,32 66,66
6 66,00 65,34 64,68 64,02 63,36 62,70 62,04 61,38 60,72 60,06
7 59,40 58,76 58,12 57,48 56,84 56,20 55,56 54,92 54,28 53,64
8 53,00 52,38 51,76 51,14 50,52 49,90 49,28 48,66 48,04 47,42
9 46,80 46,20 45,60 45,00 44,40 43,80 43,20 42,60 42,00 41,40
3,0 40,80 40,22 39,64 39,06 38,48 37,90 37,32 36,74 36,16 35,58
1 35,00 34,44 33,88 33,32 32,78 32,24 31,70 31,16 30,64 30,12
2 29,60 29,08 28,56 28,04 27,54 27,04 26,54 26,04 25,56 52,08
3 24,60 24,12 23,64 23,16 22,68 22,20 21,72 21,26 20,80 20,34
4 19,90 19,46 19,02 18,58 18,16 17,74 17,32 16,90 16,50 16,10
5 15,70 15,30 14,90 14,52 14,16 13,80 13,44 13,08 12,72 12,36
6 12,00 11,66 11,32 10,98 10,64 10,30 9,96 9,62 9,28 8,94
7 8,60 8,26 7,92 7,58 7,24 6,90 6,58 6,26 5,94 5,62
8 5,30
ДОДАТОК
355
Таблиця 16
Буферні розчини із рН 4,00–6,20 (NaОН–КНС8Н4О4)
До кожного із зазначених нижче об'ємів розчину No 5 додають
50,0 мл розчину No 3 й доводять об'єм суміші водою до 200 мл.
рН0
]
2
3
4
5
6
7
8
9
4,0 0,80 1,14 1,46 1,80 2,12 2,46 2,78 3,12 3,44 3,78
1 4,10 4,44 4,76 5,10 5,42 5,76 6,08 6,42 6,74 7,08
2 7,40 7,74 8,08 8,42 8,78 9,14 9,50 9,86 10,24 10,62
3 11,00 11,38 11,76 12,14 12,54 12,94 13,34 13,74 14,16 14,58
4 15,00 15,42 15,84 16,26 16,68 17,10 17,54 17,98 18,42 18,86
5 19,30 19,76 20,24 20,72 21,22 21,72 22,22 22,74 23,26 23,78
б 24,30 24,84 25,38 25,92 26,46 27,00 27,54 28,08 28,62 29,16
7 29,70 30,26 30,82 31,38 31,94 32,50 33,08 33,66 34,24 34.82
8 35,40 35,98 36,56 37,14 37,74 38,34 38,94 39,54 40,16 40,78
9 41,40 42,04 42,68 43,32 43,96 44,60 45,22 45,84 46,46 47,08
5,0 47,70 48,32 48,94 49,56 50,18 50,80 51,42 52,04 52,66 53,28
1 53,90 54,50 55,10 55,70 56,30 56,90 57,50 58,10 58,70 59,30
2 59,90 60,50 61,10 61,70 62,28 62,86 63,44 64,02 64,58 65,14
3 65,70 66,24 66,78 67,32 67,84 68,36 68,88 69,40 69,90 70,40
4 70,90 71,38 71,86 72,34 72,82 73,30 73,76 74,22 74,68 75,14
5 75,60 76,04 76,46 76,88 77,30 77,72 78,12 78,52 78,92 79,32
6 79,70 80,08 80,44 80,80 81,14 81,48 81,82 82,14 82,46 82,78
7 83,10 83,40 83,70 84,00 84,30 84,60 84,88 85,16 85,44 85,72
8 86,00 86,28 86,56 86,84 87,10 87,36 87,62 87,88 88,12 88,36
9 88,60 88,84 89,08 89,32 89,56 89,80 90,02 90,24 90,46 90,68
6,0 90,90 91,10 91,30 91,50 91,70 91,90 92,08 92,26 92,44 92,62
1 92,80 92,96 93,10 93,24 93,36 93,48 93,60 93,70 93,80 93,90
2 94,00
ДОДАТОК
356
Таблиця 17
Буферні розчини із рН 4,96–6,69 (NaОН–NaН2C6H5О7)
Кожний із зазначених нижче об'ємів розчину No 5 доводять до
100 мл розчином No 4.
pН
0
1
2
3
4
5
6
7
8
9
4,9
0.0 0,9 1,8 2,7
5,0 3,6 4,3 5,0 5,6 6,3 7,0 7,5 8,1 8,6 9,2
1 9,7 10,2 10,8 11,3 11,9 12,4 12,9 13,4 13,9 14,4
2 14,9 15,4 15,9 16,5 47,0 17,5 17,9 18,3 18,8 19,2
3 19,6 20,0 20,4 20,8 21,2 21,6 22,0 22,4 22,9 23,3
4 23,7 24,1 24,5 24,9 25,3 25,7 26,1 26,5 26,9 27,3
5 27,7 28,0 28,4 28,7 29,1 29,4 29,7 30,0 30,4 30,7
6 31,0 31,3 31,6 31,9 32,2 32,5 32,8 33,1 33,4 33,7
7 34,0 34,3 34,5 34,8 35,0 35,3 35,5 35,8 36,0 36,2
8 36,4 36,6 36,8 37,1 37,3 37,5 37,7 37,9 38,1 38,3
9 38,5 38,7 38,9 39,1 39,3 39,5 39,7 39,9 40,0 40,2
6,0 40,4 40,6 40,8 41,0 41,2 41,4 41,5 41,6 41,7 41,9
1 42,0 42,1 42,3 42,4 42,6 42,7 42,8 43,0 43,1 43,3
2 43,4 43,5 43,6 43,8 44,0 44,1 44,2 44,3 44,4 44,5
3 44,6 44,7 44,8 44,9 45,0 45,1 45,2 45,3 45,3 45,4
4 45,5 45,6 45,7 45,7 45,8 45,9 46,0 46,1 46,1 46,2
5 46,3 46,4 46,5 46,5 46,6 46,7 46,8 46,8 46,9 46,9
6 47,0 47,1 47,1 47,2 47,2 47,3 47,3 47,4 47,4 47,5
ДОДАТОК
357
Таблиця 18
Буферні розчини із рН 4,80–8,00 (KH2PО4–Na2HPО4)
Кожний із зазначених нижче об'ємів розчину No 7 доводять до
100 мл розчином No 6.
рН0
1
2
3
4
5
6
7
8
9
4,8 0,35 0,37 0,39 0,41 0,43 0,45 0,48 0,51 0,54 0,57
9 0,60 0,63 0,66 0,69 0,72 0,75 0,79 0,83 0,87 0,91
5,0 0,95 0,99 1,03 1,07 1,11 1,15 1,19 1,23 1,27 1.31
1 1,35 1,39 1,43 1,47 1,51 1,55 1,60 1,65 1,70 1,75
2 1,80 1,85 1,90 1,95 2,00 2,05 2,10 2,15 2,20 2,25
3 2,30 2,37 2,44 2,51 2,58 2,65 2,72 2,79 2,86 2,93
4 3,00 3,09 3,18 3,27 3,36 3,45 3,54 3,63 3,72 3,81
5 3,90 3,99 4,08 4,17 4,26 4,35 4,46 4,57 4,68 4,79
б 4,90 5,02 5,14 5,26 5,38 5,50 5,62 5,75 5,90 6,05
7 6,20 6,35 6,50 6,70 6,85 7,00 7,20 7,35 7,55 7,70
8 7,90 8,10 8,25 8,45 8,60 8,80 9,00 9,20 9,40 9,60
9 9,80 10,0 10,2 10,4 10,6 10,8 11,1 11,3 11,6 11,8
6,0 12,1 12,4 12,7 12,9 13,2 13,5 13,8 14,1 14,4 14,7
1 15,0 15,3 15,7 16,0 16,4 16,7 17,0 17,4 17,7 18,1
2 18,4 18,7 19,1 19,4 19,8 20,1 20,5 20,9 21,3 21,7
3 22,1 22,5 22,9 23,4 23,8 24,2 24,6 25,1 25,5 26,0
4 29,4 26,9 27,3 27,8 28,2 28,7 29,2 29,7 30,3 30,8
5 31,3 31,9 32,4 33,0 33,5 34,1 34,7 35,3 35,9 36,5
6 37,1 37,7 38,3 38,9 39,4 40,0 40,6 41,2 41,8 42.4
7 43,0 43,6 44,2 44,8 45,4 46,0 46,6 47,3 47,9 48,6
8 49,2 49,8 50,4 51,0 51,6 52,2 52,8 53,4 54,0 54.6
9 55,2 55,8 56,4 57,0 57,6 58,2 58,8 59,4 60,0 60,6
7,0 61,2 61,8 62,4 63,0 63,6 64,2 64,8 65,4 65,9 66,5
1 67,0 67,6 68,1 68,7 69,2 69,8 70,4 70,9 71,5 72,0
2 72,6 73,2 73,7 74,3 74,8 75,4 75,9 76,3 76,8 77,2
3 77,7 78,1 78,6 79,0 79,5 79,9 80,3 80,7 81,0 81,4
4 81,8 82,1 82,5 82,8 83,2 83,5 83,8 84,2 84,5 84,9
ДОДАТОК
358
Таблиця 18 (продовження)
рН0
1
2
3
4
5
6
7
8
9
5 85,2 85,5 85,9 86,2 86,6 86,9 87,2 87,5 87,9 88,2
б 88,5 88,8 89,1 89,3 89,6 89,9 90,2 90,4 90,7 90,9
7 91,2 91,4 91,7 91,9 92,2 92,4 92,6 92,9 93,1 93,4
8 93,6 93,8 94,0 94,2 94,4 94,6 94,8 95,0 95,1 95,3
9 95,5 95,6 95,8 95,9 96,1 96,2 96,3 96,5 96,6 96,8
8,0 96,9
Таблиця 19
Оцтово-ацетатні буферні розчини
Для готування буферного розчину необхідного значення рН
відміряють зазначений об'єм 1,0 M розчину оцтової кислоти, дода-
ють 50,0 мл 1,0 М розчину їдкого натру й розбавляють дистильо-
ваною водою до 500 мл.
рН
Оцтова
кислота
1М,мл
рН
Оцтова
кислота
1М,мл
рН
Оцтова
кислота
1М.мл
3,8
421,5
4,67
100,0
5,5
57,4
3,9
345,1
4,7
96,8
5,6
55,9
4,0
284,4
4,8
87,2
5,7
54,7
41
236,2
4,9
79,5
5,8
53,7
4,2
197,9
5,0
73,4
5,9
53,0
4,3
167,4
5,1
68,6
6,0
52,3
4,4
143,3
5,2
64,8
6,1
51,9
4,5
124,1
5,3
61,7
6,2
51,5
4,6
108,92
5,4
59,3
6,3
51,2
ДОДАТОК
359
Таблиця 20
ВЛАСТИВОСТІ СТАНДАРТНИХ БУФЕРНИХ РОЗЧИНІВ
ЗА ТЕМПЕРАТУРИ 298 К
Склад розчину,
моль/кг
Буферна
речовина
pН,
при
25 °С
Буферна
ємність
г-екв/pН
Темпера-
турний
коеф.,
d pН/dt
Тетраоксалат калію
0,05
KH3(C2O4)2 2H2O 1,679 0,070
0,001
Тартрат калію
(нас, при 25 °С,
(0,0341)
KHC4H4O6
3,557 0,027 –0,0014
Фталат калію,
0,05
KHC8H4O4
4,008 0,016 0,0012
Фосфат (1:1)
0,025
KH2PO4
0,025
Na2HPO4
KH2PO4
Na2HPO4
6,865 0,029 -0,0028
Фосфат (1:3,5)
0,008695
KH2PO4
0,03043
Na2HPO4
KH2PO4
Na2HPO4
7,413 0,016 -0,0028
Бура, 0,01
Na2B4O7 10H2O 9,180 0,020 –0,0082
Гідроокисид каль-
цію, (нас, за
25 °С (0,0203)
Ca(OH)2
12,454 0,09
-0,033
360
Таблиця 21
ЗНАЧЕННЯ pН ВОДНИХ БУФЕРНИХ РОЗЧИНІВ ПРИ РІЗНИХ ТЕМПЕРАТУРАХ
pH при температурі, C
Розчин
10
20
25
30
40
50
60
0,05 М тетраоксалат калію
1,669 1,676 1,681 1,685 1,697 1,712 1,726
кислий виннокислий калій
насичений при 298 К
–
–
3,555 3,547 3,543 3,549 3,565
0,01 М кислий виннокислий
калій
3,671 3,647 3,637 3,633 3,630 3,640 3,654
0,05 М кислий фталевокис-
лый калій
4,001 4,001 4,005 4,011 4,030 4,059 4,097
0,01 М тетраборат натрію
9,328 9,223 9,177 9,135 9,066 9,012 8,961
0,025 М двовуглекислий на-
трій + 0,025 М вуглекислий
натрій
–
–
10,02 –
–
–
–
0,01 М тринатрійфосфат
–
–
11,72
–
–
–
–
ДОДАТОК
361
Таблиця 22
УНІВЕРСАЛЬНА БУФЕРНА СУМІШ БРИТТОНА
Суміш містить рівні кількості 0,4 М розчинів фосфорної, оцтової й
борної кислот. Для одержання буферного розчину із заданим зна-
ченням pH до 100 мол суміші додають x мол 0,2 н розчину
NaOH .
pH
x
pH
x
pH
x
1,81
0
5,02
35,0
9,15
70,0
1,89
2,5
5,33
37,5
9,37
72,5
1,98
5,0
5,72
40,0
9,62
75,0
2,09
7,5
6,09
42,5
9,91
77,5
2,21
10,0
6,37
45,0
10,38
80,0
2,36
12,5
6,59
47,5
10,88
82,5
2,56
15,0
6,80
50,0
11,20
85,0
2,87
17,5
7,00
52,5
11,40
87,5
3,29
20,0
7,24
55,0
11,58
90,0
3,78
22,5
7,54
57,5
11,70
92,5
4,10
25,0
7,96
60,0
11,82
95,0
4,35
27,5
8,36
62,5
11,92
97,5
4,56
30,0
8,69
65,0
11,98 100,0
4,78
32,5
8,95
67,5
ДОДАТОК
362
Таблиця 23
СТАНДАРТНІ ЕЛЕКТРОДНІ ПОТЕНЦІАЛИ У ВОДЯНИХ
РОЗЧИНАХ ЗА ТЕМПЕРАТУРИ 298 К
No
Електрод
Реакція
,В
електроди, оборотні до катіона
1
Li+
,Li
Li+
+eLi
–
3,045
2
K+
,K
K+
+eK
–
2,925
3
Na
+
,Na
Na
+
+eNa
–
2,714
4
Mg2+
,Mg
Mg2+
+2eMg
–
2,363
5
Mn
2+
,Mn
Mn
2+
+2eMn
–
1,180
6
Zn
2+
,Zn
Zn
2+
+2eZn
–
0,763
7
Fe
2+
,Fe
Fe
2++
+2eFe
–
0,440
8
Ni2+
,Ni
Ni2+
+2eNi
–
0,250
9
Pb2+
,Pb
Pb2+
+2ePb
–
0,126
10
Fe
+++
,Fe
Fe
+++
+3eFe
–
0,036
11
H+
,H2
H+
+ e 1/2H2
0,000
12
Cu
2+
,Cu
Cu
2++
+2eCu
+ 0,337
13
Cu
+
,Cu
Cu
+
+eCu
+ 0,521
14
Hg22
+
,Hg
1/2 Hg2
2+
+2eHg
+ 0,798
15
Ag+
,Ag
Ag+
+eAg
+ 0,799
електроди, оборотні до аніона
16
O2, OH–
1/2O2 +H2O+2e 2OH–
+ 0,401
17
Cl2 (г), Cl
–
1/2Cl2 +e 2Cl–
+ 1,360
18
Br2 (ж), Br
–
1/2Br2 +e 2Br
–
+ 1,065
електроди другого роду
19 Ca,Ca(OH)2, OH–
Ca(OH)2+2e Ca+2OH–
–
3,02
20 Zn, Zn(OH)2, OH–
Zn(OH)2+2eZn+2OH–
–
1,245
21 Cd,Cd(OH)2, OH–
Cd(OH)2+ 2e Cd+2OH–
–
0,809
22 Pb, PbO, OH–
Pbo +H2O+2e Pb+2OH–
–
0,578
23 Pb, PbSP4, SO4
2-
PbSO4+ 2e Pb+SO42
-
–
0,356
24
Ag, AgI, I
–
AgI+eAg+I
–
–
0,152
25 Ag, AgBr, Br
–
AgBr+eAg+Br
–
+ 0,071
26 Ag, AgCl, Cl
–
AgCl+eAg+Cl
–
+ 0,222
27 Hg, HgSO4, SO4
2-
HgSO4+ 2e 2Hg+SO4
2-
+ 0,615
28 Ag, AgSO4, SO4
2-
AgSO4+ 2e 2Ag+SO4
2-
+ 0,654
ДОДАТОК
363
Продовження таблиці 17
No
Електрод
Реакція
,В
окисно-відновні електроди
29 Cr
2+
,Cr
3+
|Pt
Cr
3+
+eCr
2+
–
0,41
30 Sn
2+
,Sn
4+
|Pt
Sn
4+
+2eSn
2+
+ 0,15
31
Fe(CN)64
–
,Fe(CN)63
–
|Pt
Fe(CN)6
3–
+ e Fe(CN)6
4–
+ 0,36
32 Mno4
–
,Mno42
–
|pt
Mno4
–
+eMno42
–
|
+ 0,564
33
C6H4O2, H
+
C6H4(OH)2 | Pt
C6H4O2 + 2 H+
+ 2e C6H4(OH)2 + 0,6999
34 Fe
2+
,Fe
3+
|Pt
Fe
3+
+eFe
2+
+ 0,771
35
2Hg2+
|Pt
2Hg2+
+ 2e Hg2
2+
+ 0,920
36
Mn
2+
,Mn
4+
,
(+4OH–)|pt
MnO2 + 4H+
+2eMn
2+
+ 2H2O
+ 1,23
37
Pb2+
, Pb4+
,
(+4OH–)|pt
PbO2 + 4H+
+2e Pb2+
+ 2H2O
+ 1,455
38 AsO4
3–
, AsO2
–
,
H+| Pt
H3AsO4 + 2H+
+ 2e HAsO2+
2H2O
+ 0,56
39 Mno4
–
,Mn
2+
,
H+|Pt
MnO4
–
+8H+
+5eMn
2+
+ 4H2O
+ 1,51
40 Mn
2+
,Mn
4+
|Pt
Mn
4+
+2eMn
2+
+ 1,51
41 Pbo2,PbSO4 | Pt PbO2 + SO4
2-
+ 4H+
+2e PbSO4+
2H2O
+ 1,685
42 Pb4+
, Pb2+
|Pt
Pb4+
+ 2e Pb2+
+ 1,7
43 Co
2+
,Co
3+
|Pt
Co
3+
+eCo
2+
+ 1,82
ДОДАТОК
364
Таблиця 24
ЕЛЕКТРОРУШІЙНА СИЛА НОРМАЛЬНОГО НАСИЧЕНОГО
ЕЛЕМЕНТА ВЕСТОНА ЗА РІЗНИХ ТЕМПЕРАТУР
Темпера-
тура, C
ЕРС,
В
Темпера-
тура, С
ЕРС,
В
11
1,01874
21
1,01826
12
1,01868
22
1,01822
13
1,01863
23
1,01817
14
1,01858
24
1,01812
45
1,01853
25
1,01807
16
1,01848
26
1,01802
17
1,01843
27
1,01797
18
1,01839
28
1,01792
19
1,01834
29
1,01786
20
1,01830
30
1,01781
Таблиця 25
СТАНДАРТНІ ПОТЕНЦІАЛИ ЕЛЕКТРОДІВ ДРУГОГО РОДУ
У ВОДЯНИХ РОЗЧИНАХ ЗА ТЕМПЕРАТУРИ 298 К
Електрод
Eo (мВ)
Електрод
Eo (мВ)
Ag | AgCl, Cl– 222,34
Hg | HgO, OH–
97,6
Ag | AgBr, Br
–
71,3
Hg | Hg2SO4, SO4
2-
615,1
Ag|AgI,I
–
–
152,3
Pt | MnO2, MnO4
–
, OH–
587
Ag | AgN3, N3
–
291,9
Pt | MnO2, Mn
2+
, H3O
+
1236
Ag | AgSCN, SCN– 94,7
Pt | PbO, OH–
–
578,5
Hg |Hg2Cl2, Cl–
267
P t| PbO2, Pb2+
, H3O
+
1467
Hg | Hg2Br2, Br
–
139,2 Pt | PbO2,PbSO4,SO4
2-
, H3O
+
1684,9
Hg| Hg2I2, I
–
40,5
Pt | PbSO4,SO4
2-
–
355
ДОДАТОК
365
Таблиця 26
СТАНДАРТНІ ПОТЕНЦІАЛИ ДЕЯКИХ ЕЛЕКТРОДІВ
ПРИ РІЗНИХ ТЕМПЕРАТУРАХ (У мВ)
Температура o С
Електрод
10
20
25
30
40
50
60
Хлорсрібний
Ag | AgCl, Cl–
231,42 225,57 222,34 219,04 212,08 204,49 196,49
Бромерібний
Ag | AgBr, Br
–
77,72 73,42 71,05 68,52 63,02 56,91 50,20
Йодсрібний
Ag|AgI,I
–
–148,1 –150,7 –152,3 –154,0 –157,9 –162,8 –166,5
Ртуть-cульфатний
Hg | Hg2SO4, SO4
2-
627,04 619,30 615,15 611,07 603,05 594,87 580,55
Хінгідронний
Pt|C6H4O2,C6H4(OH)2
688,1 695,5 699,2 702,9 710,3 717,7
–
Таблиця 27
ПОТЕНЦІАЛИ ДЕЯКИХ ЕЛЕКТРОДІВ ПОРІВНЯННЯ
ПРИ РІЗНИХ ТЕМПЕРАТУРАХ (У мВ)
Температура °C
Електрод
10
15
20
25
30
35
Хлорсрібний
Ag | AgСl, Cl– (0,1 н)
–
–
–
334,0
–
–
Хлорсрібний
Ag|AgСl,Cl–(1н)
–
–
–
281,5
–
–
Хлорсрібний
Ag | AgСl, Cl– (нас.)
–
–
–
244,4
–
–
Каломельний
Hg | Hg2Cl2, KСl (0,1 н)
337,4 337,1 336,8 336,5 336,2 335,9
Каломельний
Hg | Hg2Cl2, KСl (1,0 н)
286,4 285,2 284,0 282,8 281,6 280,4
Каломельний
Hg | Hg2Cl2, KCl (нас.)
253,6 250,3 247,1 243,8 240,5 237,3
ДОДАТОК
366
ДОДАТОК
367
Таблиця 29
ДИФУЗІЙНІ ПОТЕНЦІАЛИ НА ГРАНИЦІ РОЗДІЛУ ОДНА-
КОВИХ ВОДЯНИХ РОЗЧИНІВ РІЗНИХ КОНЦЕНТРАЦІЙ
Електроліт
12
/
cc
.
диф
E ,мВ
HCl
0,005 / 0,01
0,005 / 0,04
11,1
33,3
KCl
0,005 / 0,01
0,005 / 0,04
-0,3
-1,0
NaCl
0,005 / 0,01
0,005 / 0,04
-3,7
-11,1
Таблиця 30
ДИФУЗІЙНІ ПОТЕНЦІАЛИ НА ГРАНИЦІ РОЗДІЛУ РІЗНИХ
ВОДНИХ РОЗЧИНІВ З ОДНАКОВОЮ КОНЦЕНТРАЦІЄЮ
E
мВ
дифф .
,
Пари
розчинів
c
c
1
2
експ.
розрах.
HCl / KCl
0,1
0,01
26,8
25,7
28,5
27,5
HCl / LiCl
0,1
0,01
34,9
33,8
36,1
34,6
HCl / NaCl
0,1
0,01
33,1
31,3
33,4
32,0
KCl / LiCl
0,1
0,01
8,8
8,2
7,6
7,1
NaCl / LiCl
0,1
0,01
2,6
2,6
2,8
2,5
ДОДАТОК
368
Таблиця 31
КОНСТАНТИ ШВИДКОСТІ ЛУЖНОГО
ОМИЛЕННЯ ЕСТЕРІВ
t, °С
k,
см
3
/(моль·с)
k,
л/(моль·хв)
Етилацетат CH3COOC2H5
0
19,5
1,17
20
84,7
5,08
25
109,3
6,56
Етилпропіонат CH3CH2COOC2H5
0
19
1,14
25
95,7
5,94
Пропілацетат CH3COOC3H7
10
35,8
2,15
20
70,5
4,23
Бутилацетат CH3COOC4H9
10
35,3
1,94
20
65,6
3,93
втор-Бутилацетат CH3COOC4H9
10
29,3
1,76
20
59
3,54
трет-Бутилацетат CH3COOC4H9
10
0,615
0,0369
20
1,35
0,0810
ДОДАТОК
369
Платинування електродів
Платинування платинових електродів проводиться в 1-3% роз-
чині хлорплатинової кислоти, що містить невелику кількість ацета-
ту свинцю (0,02–0,03 г на 100 г розчину для утвору високодисперс-
ного осаду). Електроліз може проводитися з будь-яким джерелом
постійного струму на 2 В. Допоміжним електродом служить плати-
нова пластина. Тривалість електроосадження платини в розчині
хлорплатинової кислоти не більш 1–2 хв за сили струму 200–400
мА. Перед платинуванням електрод очищається опусканням у га-
рячий розчин 50% царської горілки (1 ч. нітратної кислоти на 3 ч.
хлориднох), і електролізом в 10% розчині сірчаної кислоти попере-
мінно в якості анода й катода. Поверхня платини вважається чис-
тою, якщо газ, що виділяється, відривається без затримки дрібними
пухирцями. Про якість платинування судять по зовнішньому вигля-
ду електрода – поверхня його повинна бути однорідної, без смуг і
бархатистої-чорної. Електроди слід зберігати в дистильованій воді.
Міднення
Для міднення застосовують електроліт, що містить 15 г сірча-
нокислої міді, 5 мл етилового спирту на 100 мл води. Покриття
очищеної поверхні міді проводиться при щільності струму 2–20
мА/см
2
. Тривалість електролізу – до 1 години.
Сріблення
Сріблення проводять у ціаністій ванні сполуки: AgCl – 30 г/л,
NaCN – 56 г/л,
23
Na CO – 45 г/л проти платинового або срібного
анода із джерелом харчування постійного струму (акумулятор з на-
пругою близько 2 В). У ланцюг послідовно включають магазин
опорів на 1000 ом. Електроліз ведуть 15 хв при повністю введеному
опорі магазину й потім по 10 хв при опору 500, 200 і 100 Ом. Закін-
чують сріблення, замикаючи накоротко акумулятор на комірку на
протязі 2–3 хв. Електрод з осадом срібла ретельно промивається ди-
стильованою водою. Ціаністі сполуки – найсильніші отрути, пра-
цювати необхідно під тягою й украй обережно!
ДОДАТОК
370
Очищення ртуті
Найбільш зроблений спосіб очищення продажної ртуті для
електрохімічних цілей – подвійна перегонка під вакуумом. Однак
для багатьох випадків достатньо профільтрувати ртуть через сухий
складений фільтр зі звичайного фільтрувального паперу, у якому
тонкою голкою в нижній частині пророблені тонкі отвори. Ця опе-
рація дозволяє звільниться від механічних домішок. Для очищення
від домішок більш активних металів ртуть через фільєри пропуска-
ють через стовп розчину (довжина близько 1 м) з 1% розчином ніт-
рату ртуті(I) в 5% нітратній кислоті. Ця операція повторюється кіль-
ка разів.
Після відділення ртуті від розчину за допомогою ділильної
лійки ртуть просушують за допомогою шматочків фільтрувального
паперу, профільтровують через фільтрувальний папір зазначеним
вище способом у чисту суху посудину із притертою пробкою, у
якому й зберігається.
У випадку сильного забруднення ртуті її слід піддати відми-
ванню свіжоприготовленою хромовою сумішшю в ділильній лійці.
Усі операції по очищенню ртуті проводяться під добре пра-
цюючою тягою на спеціальних лотках з бортами. Пари ртуті –
отрутні!
Готування хлорсрібних електродів
Срібний електрод добре очищають дрібним наждаковим папе-
ром, протруюють у концентрованій азотній кислоті, промивають
дистильованою водою й потім поміщають в 0,1–0,2 М розчин соля-
ної кислоти. У цей же розчин уводять допоміжний електрод (плати-
новий), приєднують обоє електрода до акумулятора (на 2 або 4 В).
Протягом 2–3 хв пропускають електричний струм, кілька разів змі-
нюючи полюса так, щоб востаннє срібний електрод обов'язково був
підключений до негативного полюса, тому що інакше на електроді
адсорбується вільний хлор. При електролізі на електроді утворю-
ється шар AgCl . Приготовлений у такий спосіб електрод поміща-
ють у розчин хлориду калію необхідної концентрації.
Навчальне видання
Рубцов Володимир Іванович
ЛАБОАРТОРНИЙ ПРАКТИКУМ
З ФІЗИЧНІОЇ ХІМІЇ
Частина 2
Розчини електролітів
Електропровідність розчинів
Гальванічні елементи
Хімічні джерела струму
Кінетика гомогенних реакцій
Кінетика гетерогенних процесів
Каталіз
Навчальний посібник
для студентів хімічного факультету університетів
Коректор
Комп'ютерне верстання В.І. Рубцов
Макет обкладинки В.І. Рубцов
Формат 60х84/16. Ум. друк. аркуш. 21,2 Тираж 100 пр. Зак. No
Видавець виготовлювач
Харківський національний університет іиені В.Н. Каразіна
61022, Харків, майдан Сврбоди, 4.
Свідоцтво суб'єкта видавничої справи ДК No 3367 від 13.01.09
Видавництво ХНУ імені В.Н. Каразіна
тел. 705-24 -32