/





































































































































































































































































































































































































































Текст
Б В К 24,2я73
У Д К 577.1 (075)
Г 93
Рекомен до ван о
Міністерством охорони здоров ‘я України як підручник для студентів
вищих медичних та фармацевтичних закладів освіти Ш -ІУ рівнів
акредитації. Протокол №1 від 10.02.2004 р.
Р е ц е н зе н т и :
Зімеиковський Б. С., з а с л у ж е н и й
п р а ц ів н и к В и щ о ї ш к о л и У к р а їн и , л а у р е а т
Д е р ж а в н о ї п р е м ії У к р а їн и , а к а д е м ік А Н В Ш У к р а їн и , д о к т о р ф а р м а ц е в т и ч н и х
наук, п р о ф есо р ;
Кучеренко М. Є., а к а д е м ік Н А Н У к р а їн и , д о к т о р б іо л о г іч н и х н а у к , п р о ф е с о р ;
Кресюн В. Й., ч л е н - к о р е с п о н д е н т А М Н У к р а їн и , д о к т о р м е д и ч н и х н а у к ,
проф есор;
КалібабчукВ. О., д о к т о р
Г 93
х ім іч н и х н а у к , п р о ф е с о р .
Г у б с ь к и й Ю . І.
Б іо о р г а н іч н а х ім ія . / В и д а н н я 2 -е , д о о п р а ц ь о в а н е і д о п о в н е н е . - К и їв В ін н и ц я , 2 0 0 7 . - 4 3 2 с . Іл .
9 7 8 -9 6 6 -3 8 2 -0 4 5 -3
У підручнику викладен о будову, реакційн у здатність, хім ічні перетворен ня т а
біологічне значення органічних сполук, які входять до складу ж ивих організм ів; низько
молекулярні біом олекули, б іополім ери {білки, нуклеїнові кислота, полісахариди), бю регулятори (ф ерм енти, гормони, вітам іни, регуляторні м олекули ім унної систем и тощ о),
природні і синтетичні ф ізіологічн о активні сполуки, в том у числі лікарські засоби та
речовини з токсичною дією .
Б Б К 24.2я73
ISBN 978-966-382-045-3
© Губський Ю.І., 2004
© ПП Нова Книга, 2004
З м іс т
П ередм ова...................................................................................................... .7
Вступ. Біоорганічна хімія як наука......................................................... 11
Ч а с т и н а ! Будова та реакційна здатність
біоорганічних с п о л у к ................................... ......................................... 16
Розділ 1. Загальні положення біоорганічної х і м і ї ..................................... 16
1.1. Способи зображення органічних м олекул............................... 16
1.2. Класифікація та номенклатура органічних сп о л у к................ 18
1.2.1. Класифікація органічних сполук..................................... 18
1.2.2. Номенклатура органічних с п о л у к
......................... 22
1.3. Будова біоорганічних сполук. Ізомерія..........................-......... 28
1.3.1. Електронна структура атома вуглецю в органічних
сполуках ............................................................................. 28
1.3.2. Природа хімічних зв’язків в органічних сполуках.... 31
1.3.3. Взаємний вплив атомів в органічних сполуках......... .39
1.3.4. Ізомерія в органічних сполуках.................................... .42
1.4. Реакції в біоорганічній хім ії..........................................................62
1.4.1. Характеристика типів реакцій..................... -.................62
1.4.2. Реагенти та субстрати. Нуклеофіли, електрофіли ....68
1.4.3. Кислоти та основи в біоорганічній хім ії................. .69
Розділ 2. Характеристика окремих класів біоорганічних сполук
2.1
Вуглеводні та їх похідні......................... *...............................
2.1.1. А л кан и ........... ............................................ .................
.72
.72
.73
З
2.1.2. Алкени. А лкадіени.............................................................. 77
2.1.3. А лкін и .....................
84
2.2. Ароматичні вуглеводні (арени)....................................................86
2.2.1. Загальна характеристика ароматичних вуглеводнів .... 86
2.2.2. Хімічні властивості ар с н ів ................................................ 90
2.2.3. Багатоядерні арени: будова, представники...................95
2.3. Гідроксисполуки (спирти, феноли). Т іо л и .................................98
2.3.1. Спирти (алкоголі): будова, властивості, представники ... 98
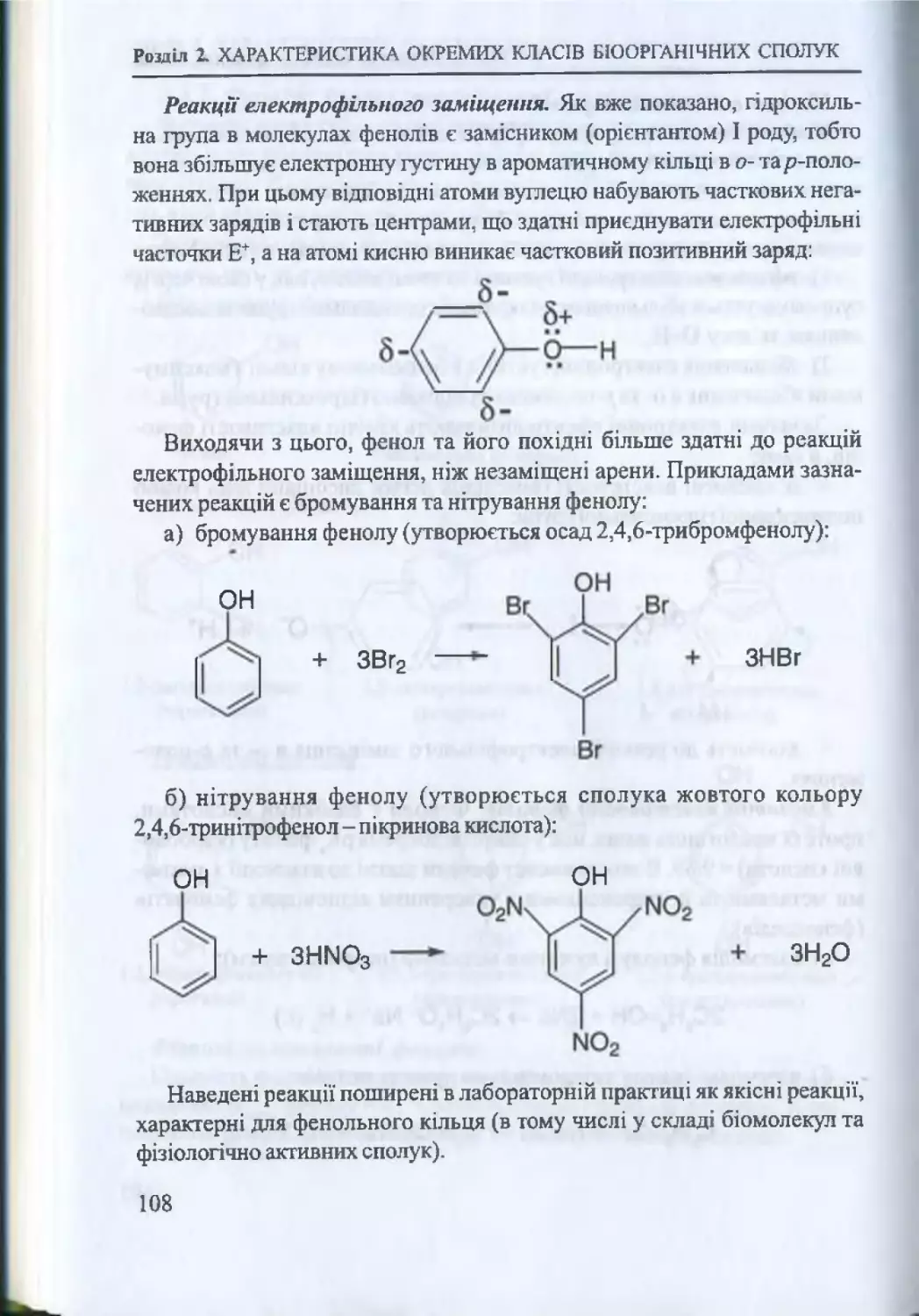
2.3.2. Феноли: будова, властивості, представники................ 106
2.3.3. Тіоли (меркаптани)............................................................ 109
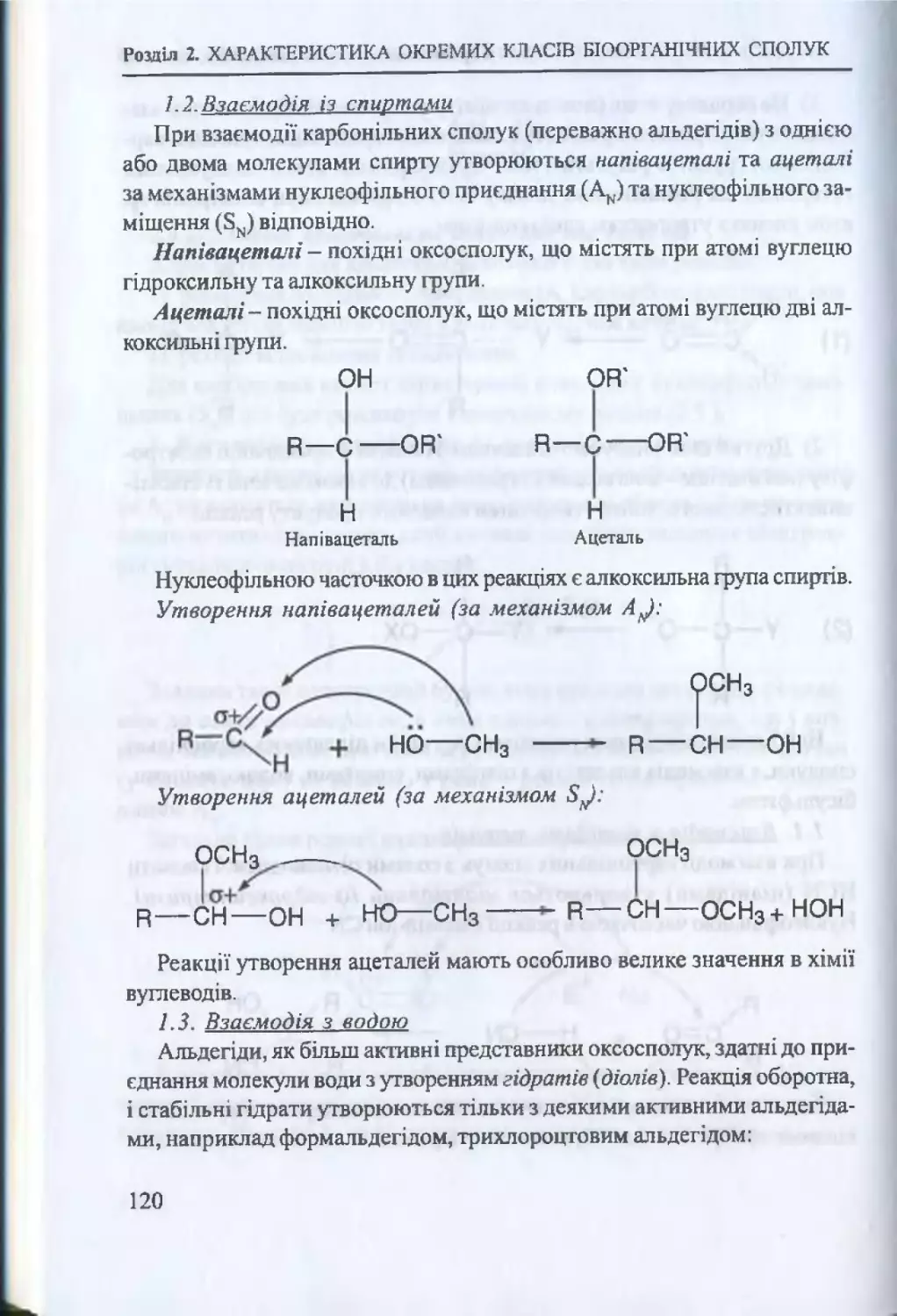
2.4. Карбонільні сполуки (альдегіди, кетони)............................... 114
2.4.1. Загальна характеристика карбонільних сполук
114
2.4.2. Хімічні властивості альдегідів та кетонів................... 118
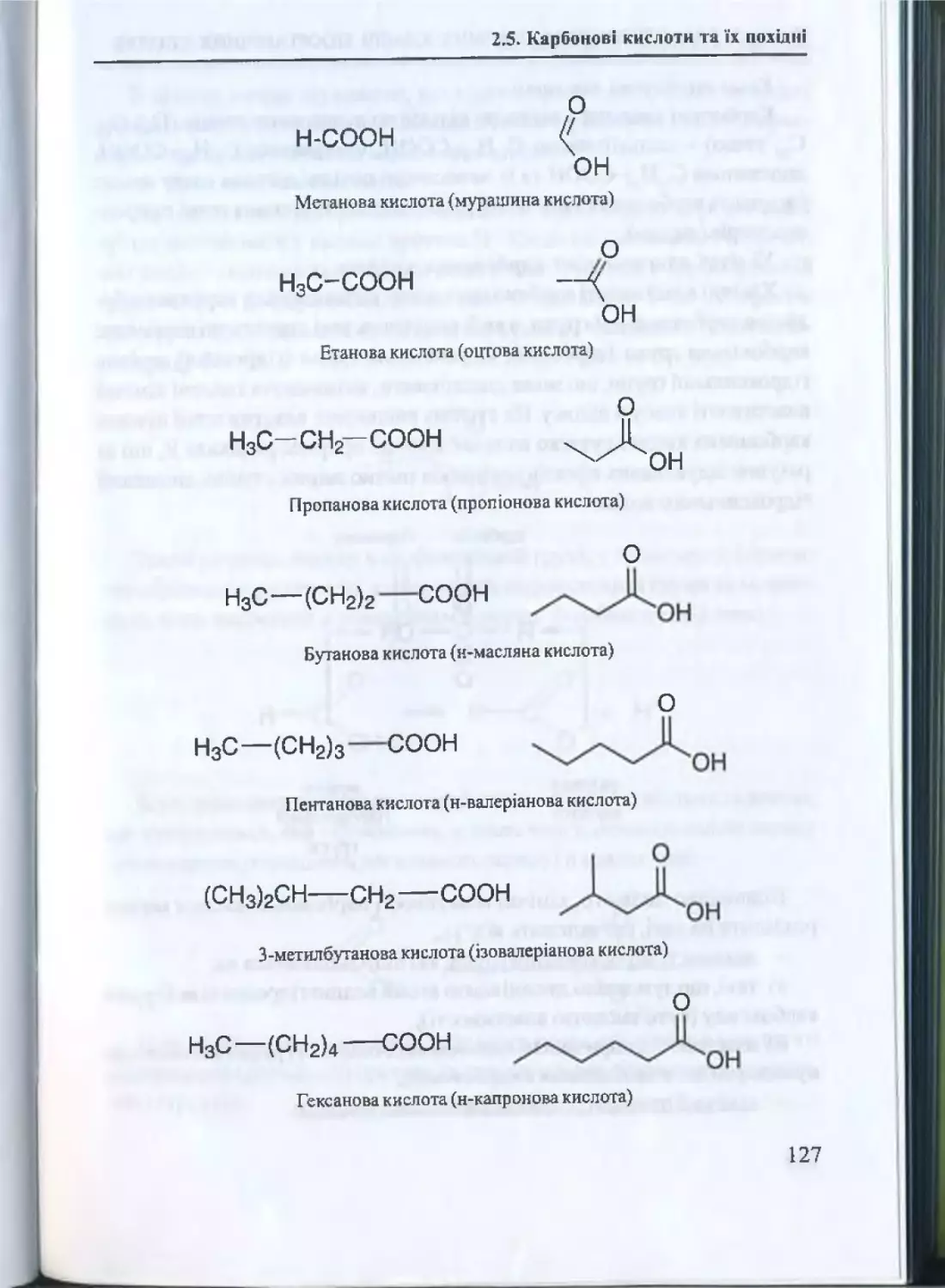
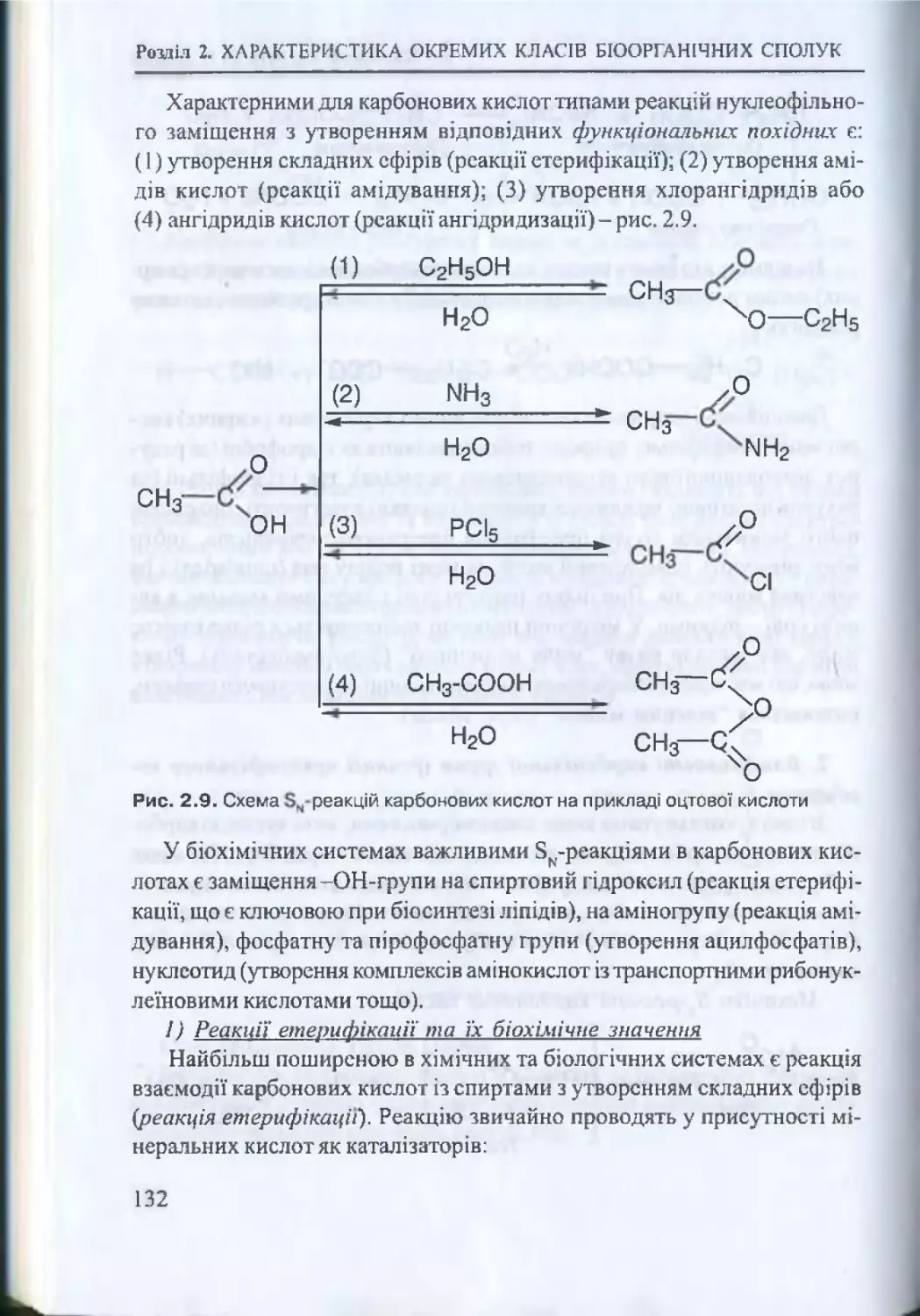
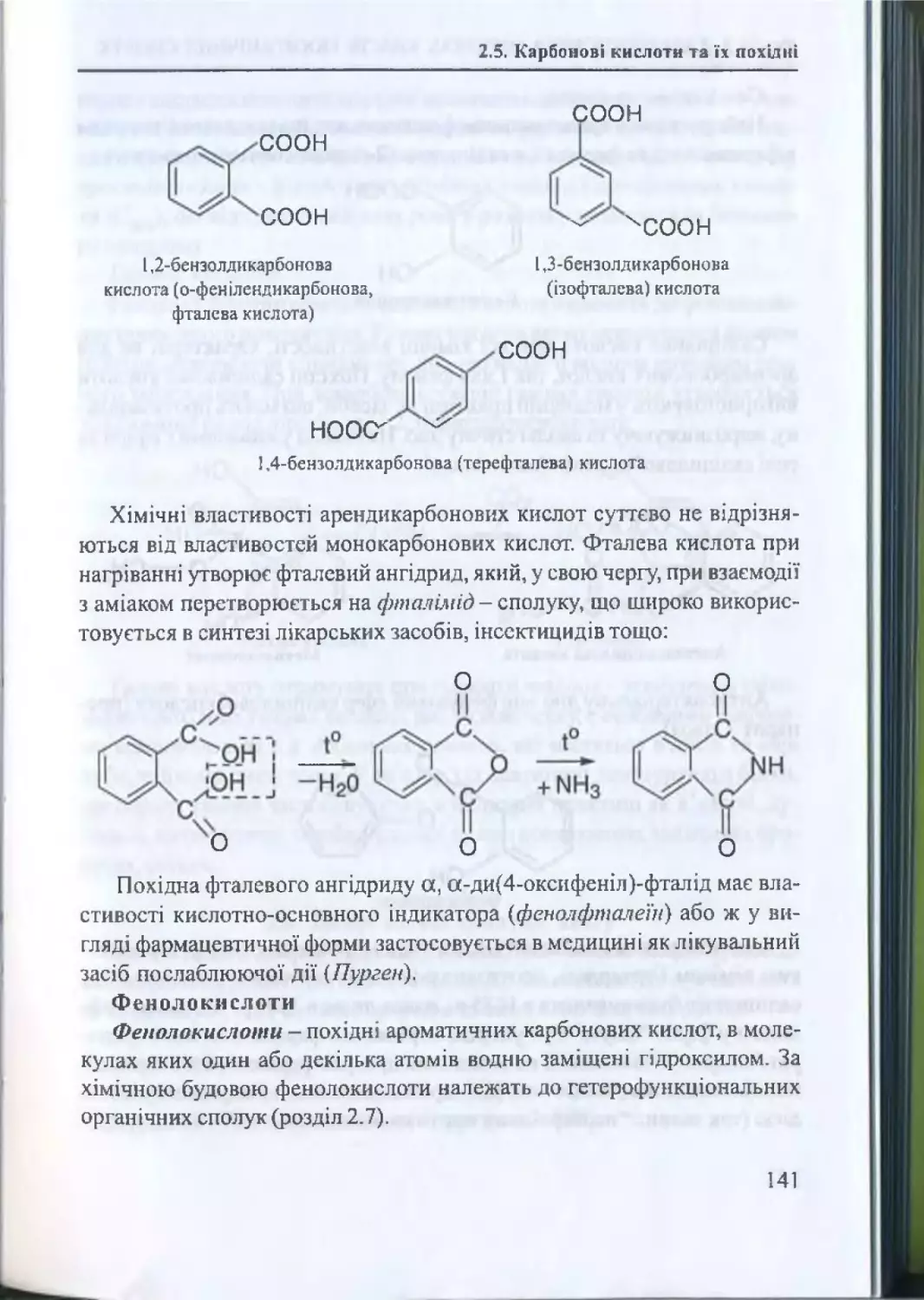
2.5. Карбонові кислоти та їх похідні................................................. 126
2.5.1. Загальна характеристика карбонових кислот
126
2.5.2. Монокарбонові аліфатичні кислоти.............................. 126
2.5.3. Дикарбонові та трикарбонові аліфатичні кислоти .... 137
2.5.4. Ароматичні карбонові кислоти. Ф енолокислоти
139
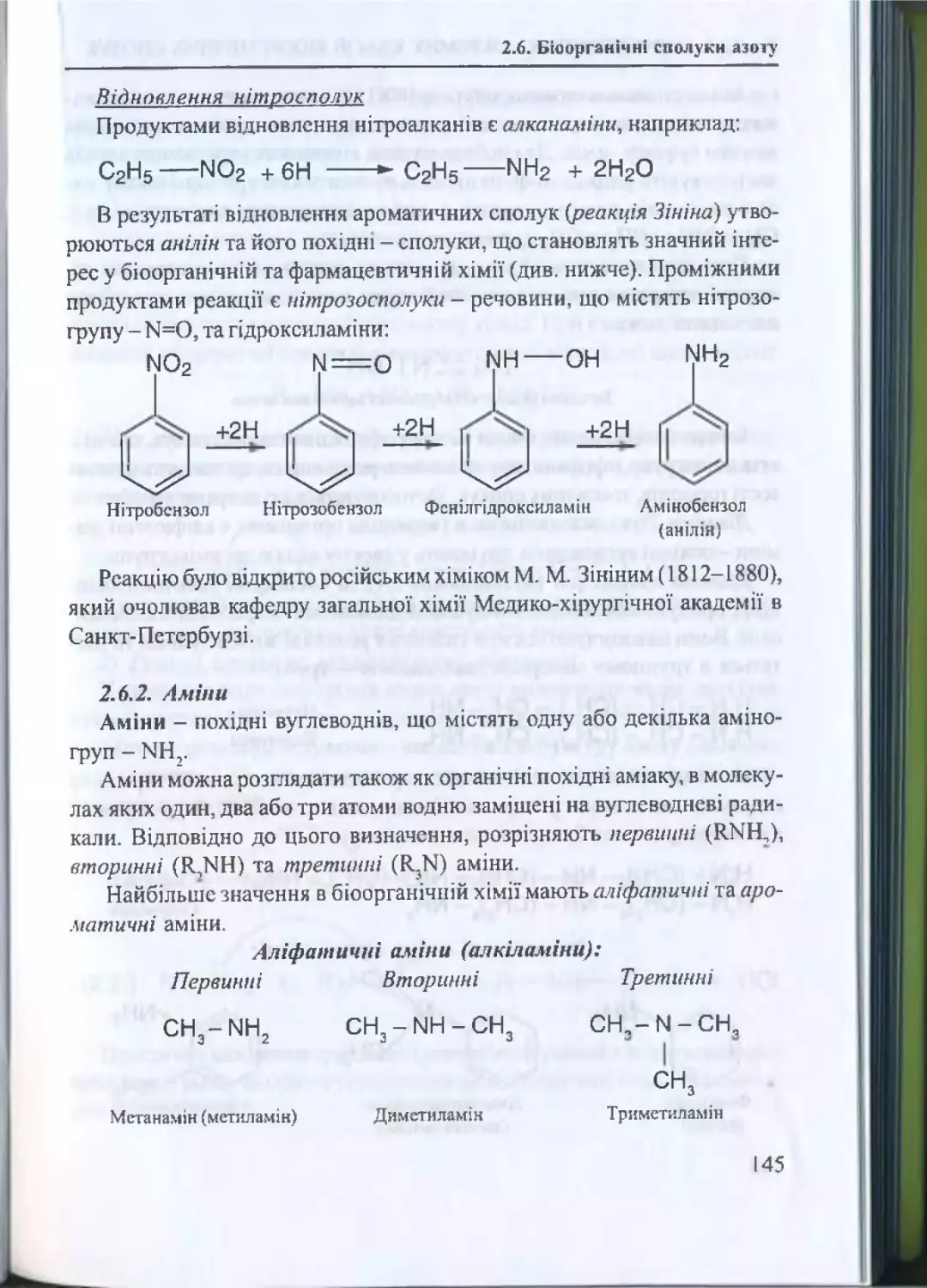
2.6. Біоорганічні сполуки азоту..........................................................143
2.6.1. Нітросполуки................................................................
143
2.6.2. А м ін и .................................................................................... 145
2.6.3. Аміди к и с л о т ..........................................
151
2.7. Гетерофункціональні сполуки.................................................... 153
2.7.1. Амінокислоти та їх властивості.....................................154
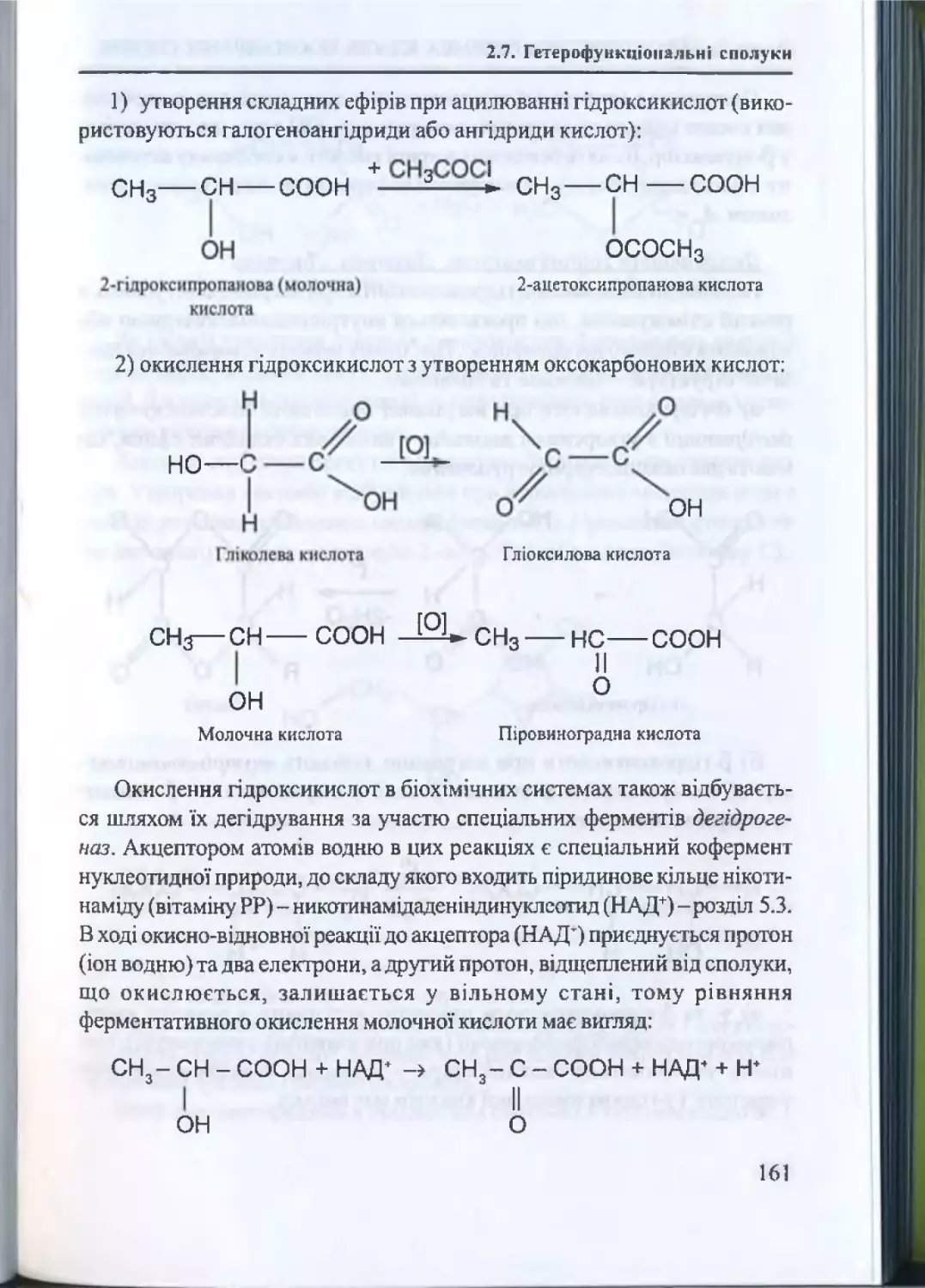
2.7.2. Пдроксикислоти та їх властивості............................... 159
2.7.3. Оксокислоти та їх властивості....................................... 163
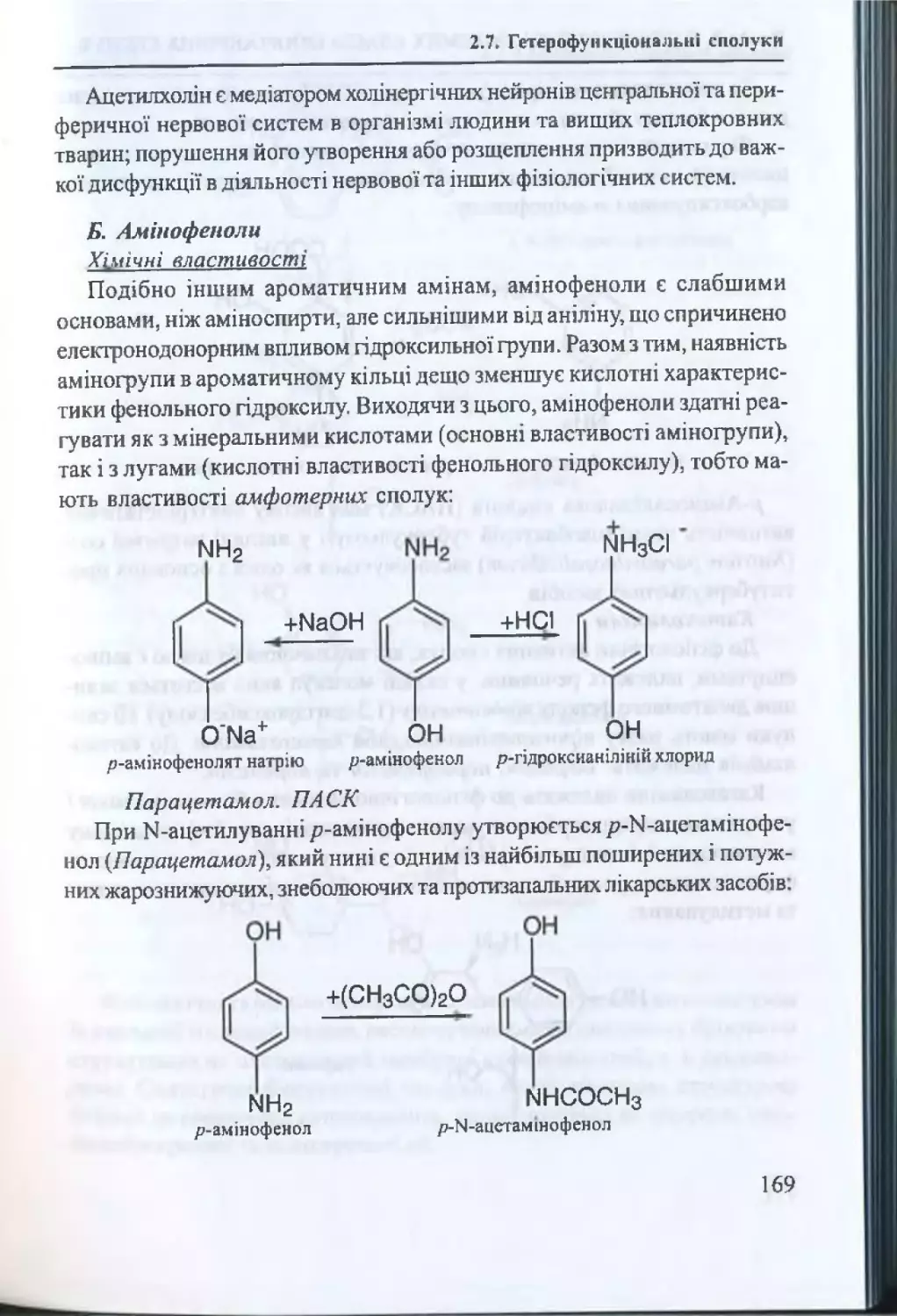
2.7.4. Аміноспирти. А мінофеноли.............................................167
Ч а с т и н а II. Б іоорганічні сполуки я к метаболіти
та л ік ар ські за с о б и ..................................................................................172
Розділ 3. Гетероциклічні сполуки та їх похідні.....................................172
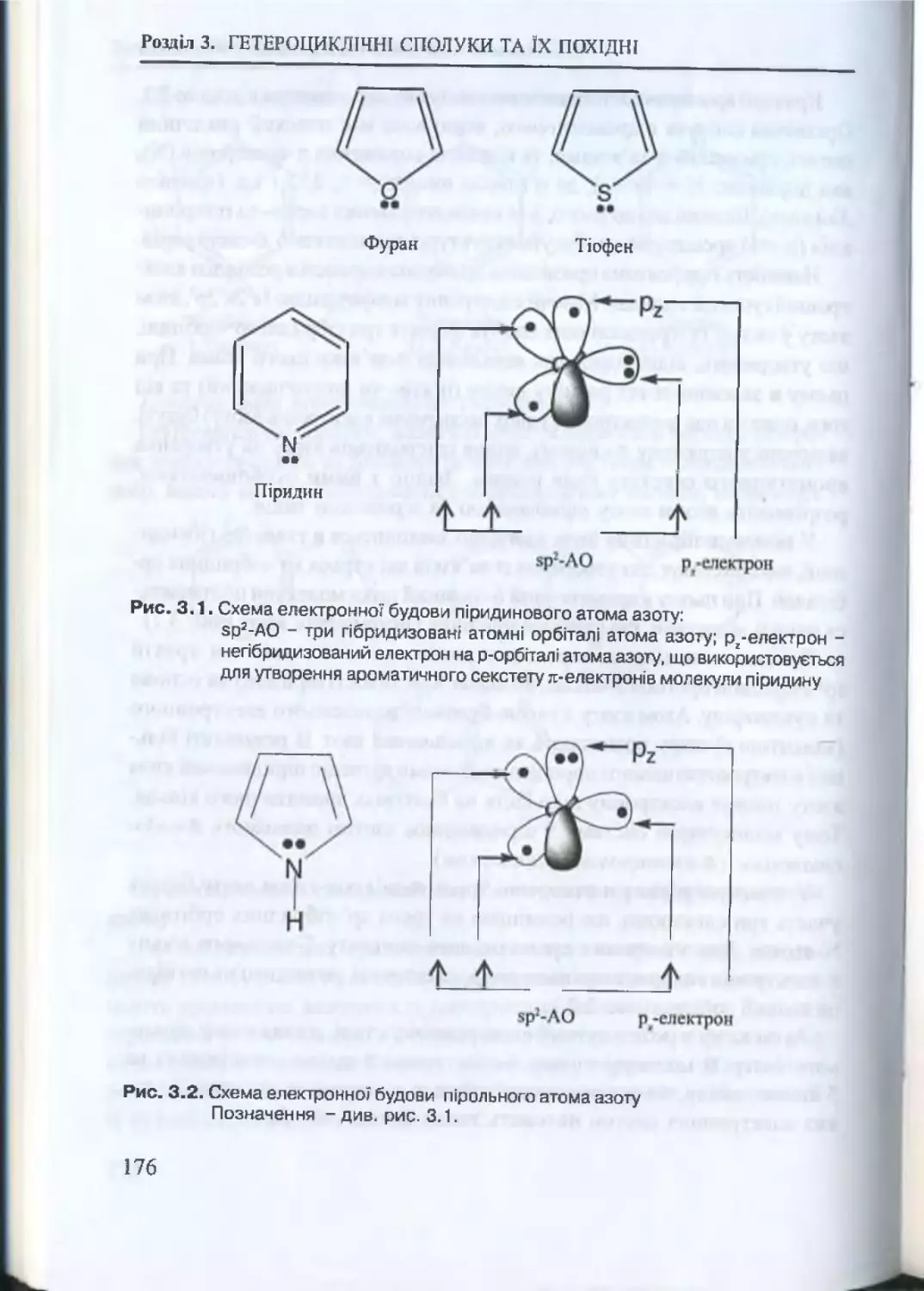
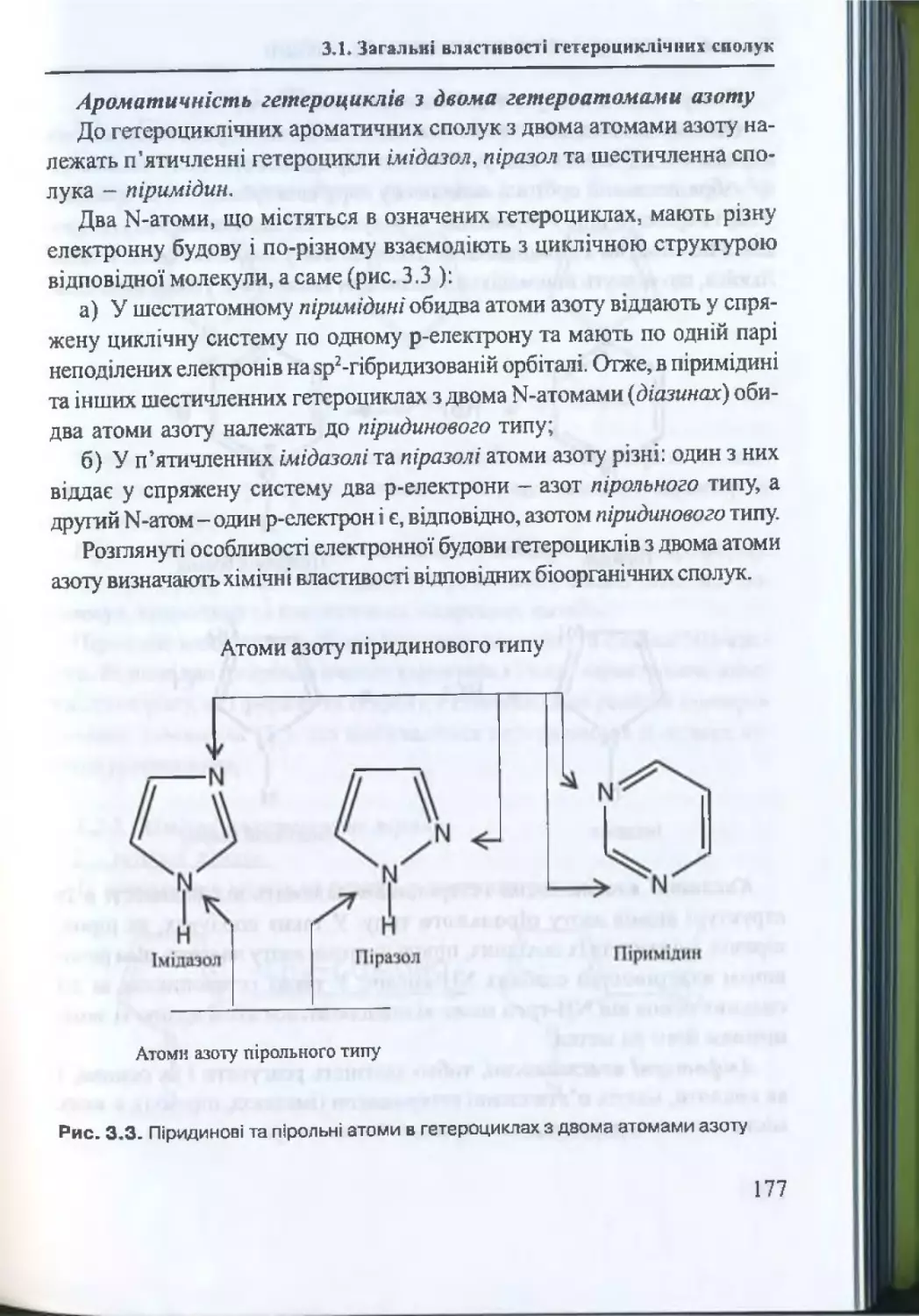
3.1. Загальні властивості гетероциклічних с п о л у к ..................... 172
4
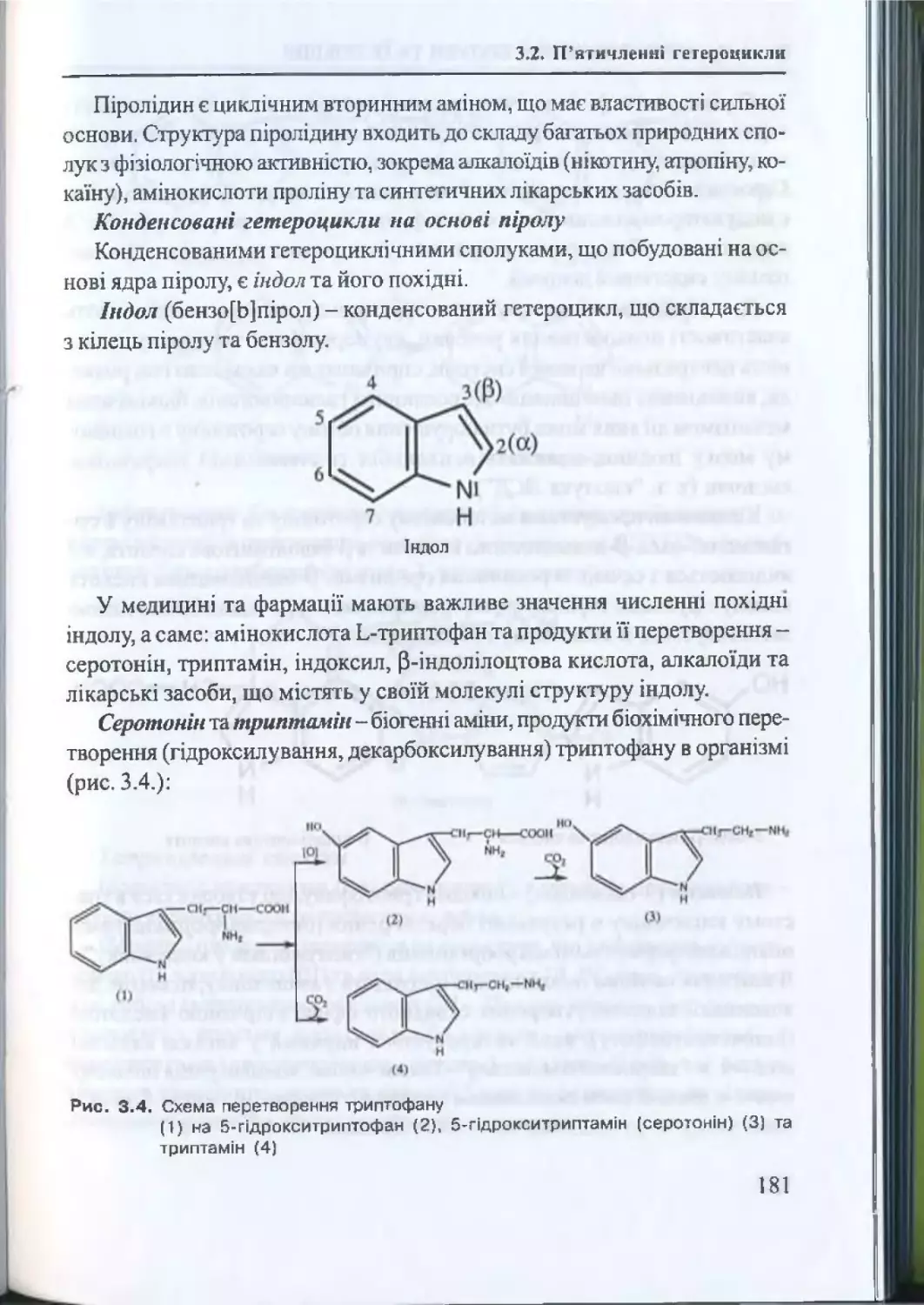
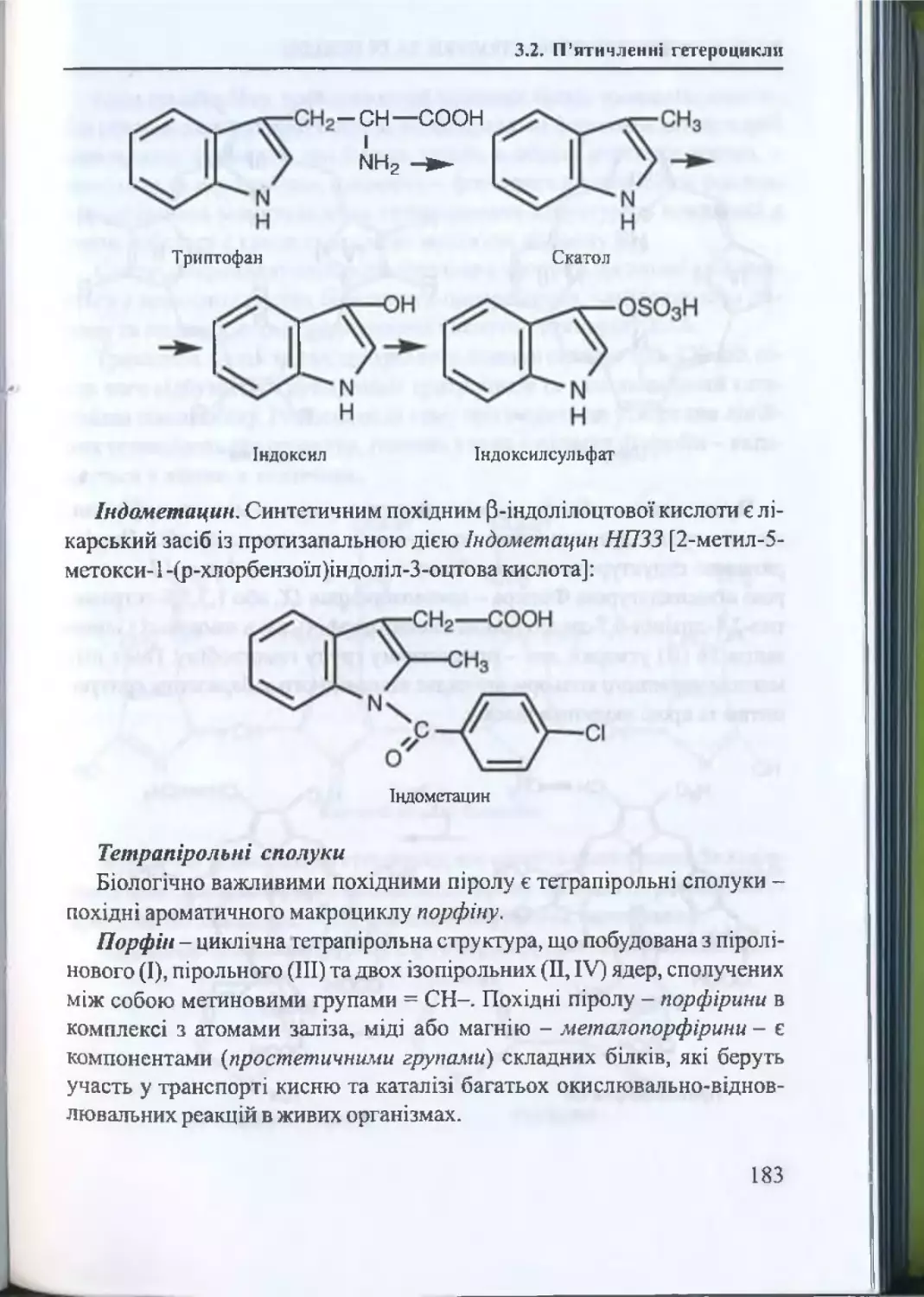
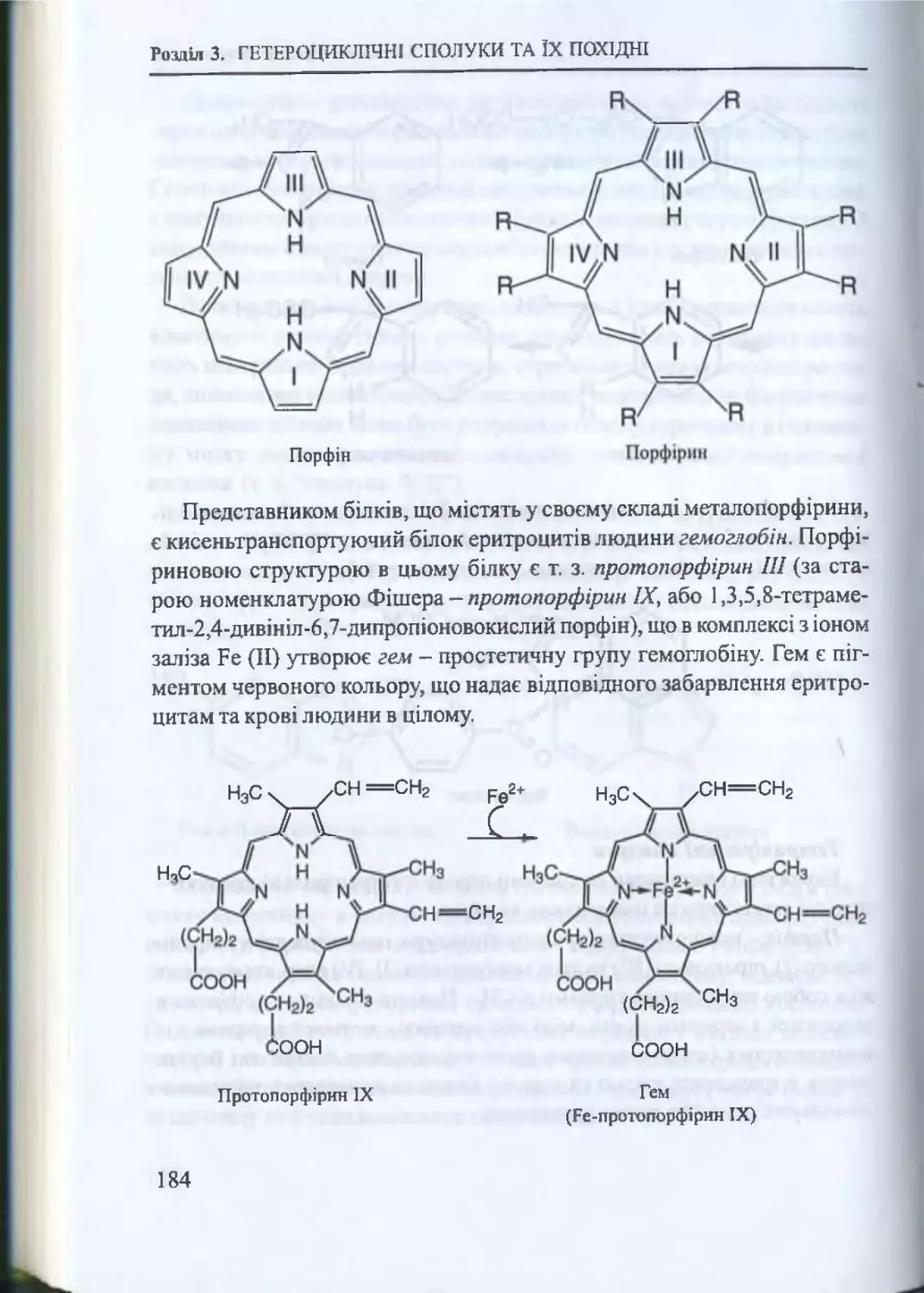
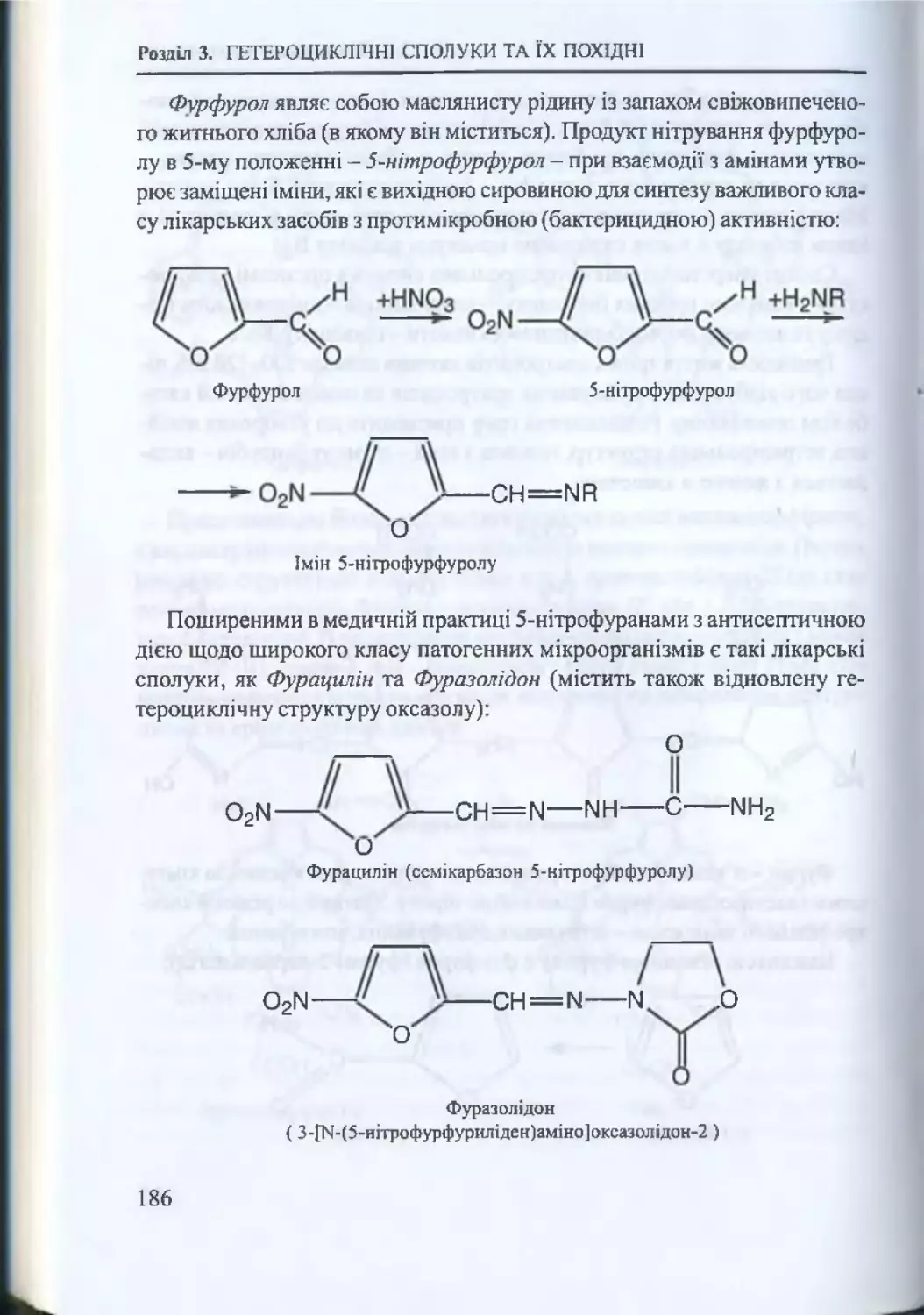
3.2. П ’ятичленнігстероцикли............................................................179
3.2.1. П ’ятичленні гетероцикли з одним гетероатомом .... 179
3.2.2. Хімічні властивості п іролу............................................... 179
3.2.3. П ’ятичленні гетероцикли з двома гетероатомами ..187
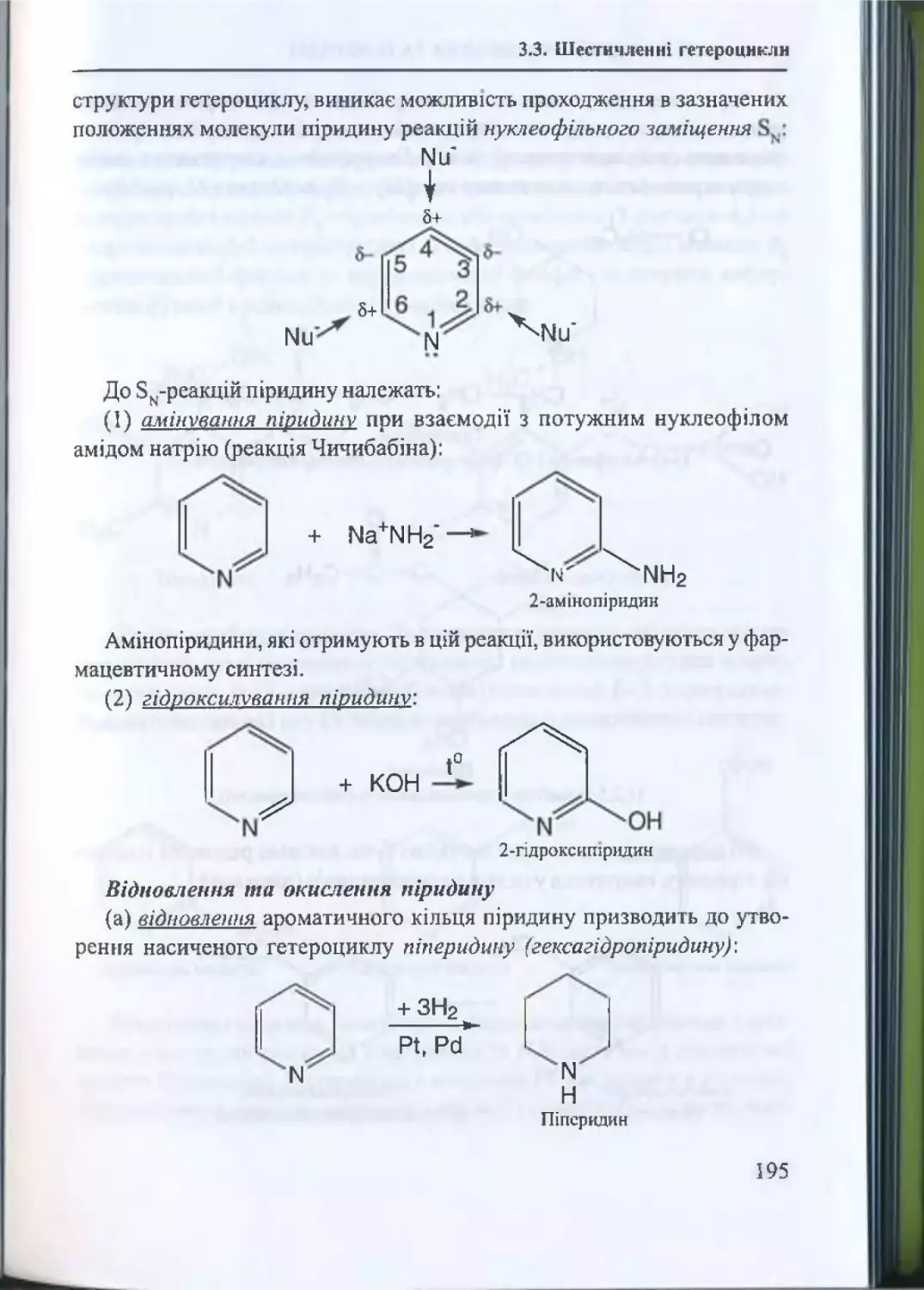
3.3. Шестичленні гетероцикли...........................................................193
3.3.1. Шестичленні гетероцикли з одним гетероатомом... 193
3.3.2. Шестичленні гетероцикли з двома гетероатомами
206
3.4. Семичленні гетероцикли.....................................
215
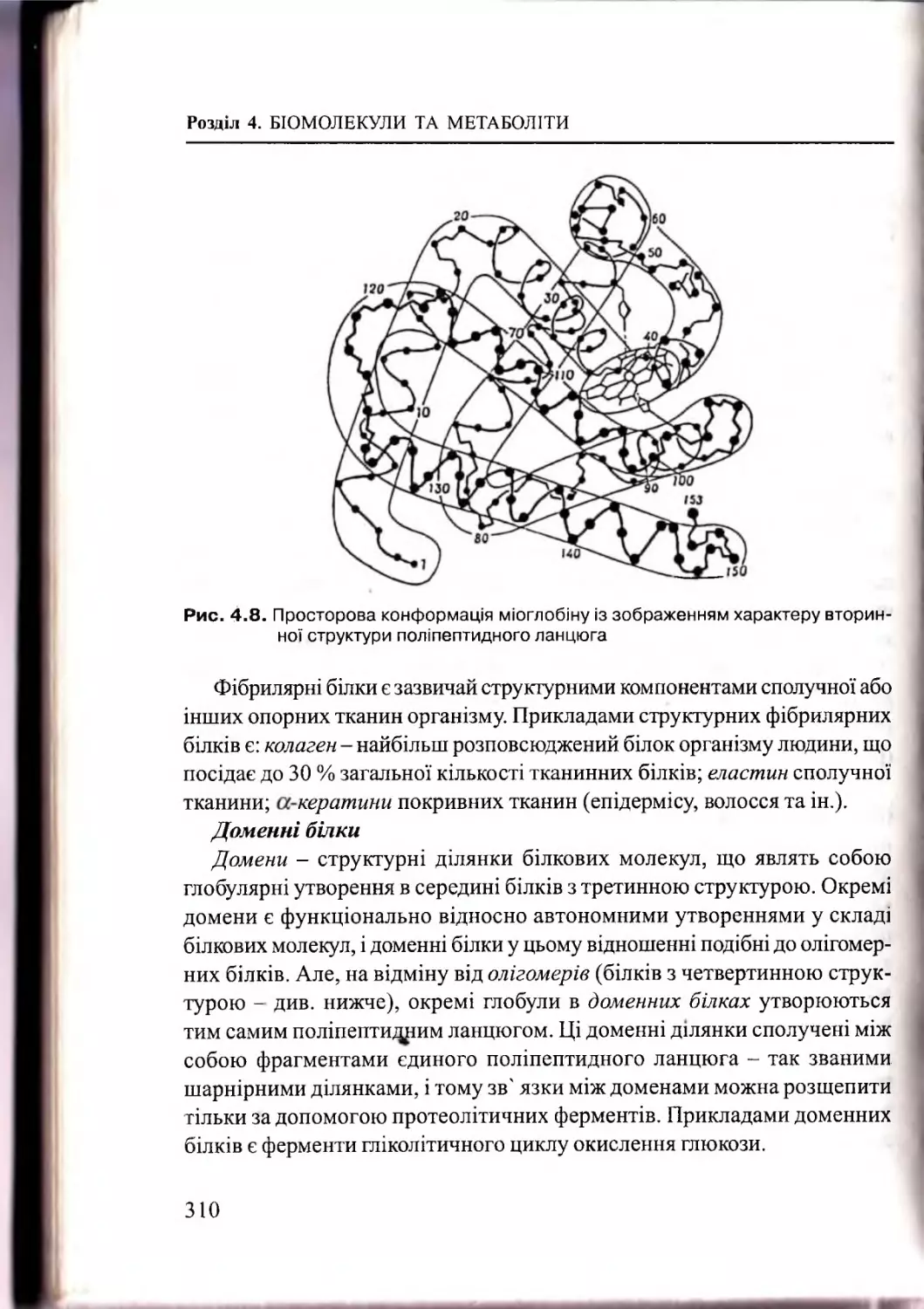
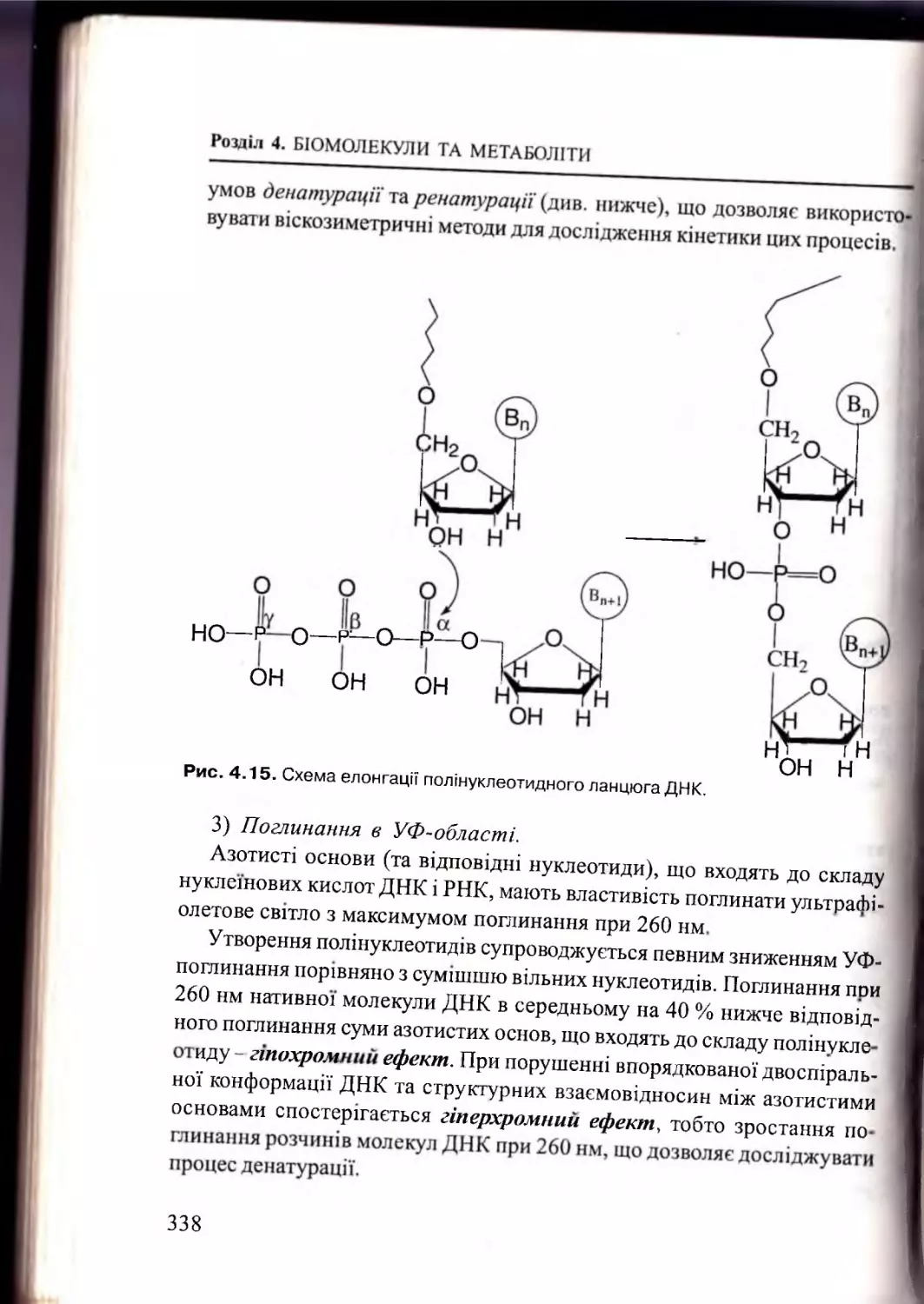
Розділ 4. Біомолекули та м етаболіти.................................
217
4.1.
Вуглеводи та їх похідні.........................
217
4.1.1. Загальна характеристика вуглеводів........................... 217
4.1.2. Моносахариди та їх похідні............................................. 218
4.1.3. Олігосахариди. Гомополісахариди.............................. 244
4.1.4. Гетерополісахариди......................
249
4.2.
Ліпіди. Стероїди. Т ерпени.........................................................251
4.2.1. Ліпіди: будова, властивості. Жирні ки сл оти ............... 251
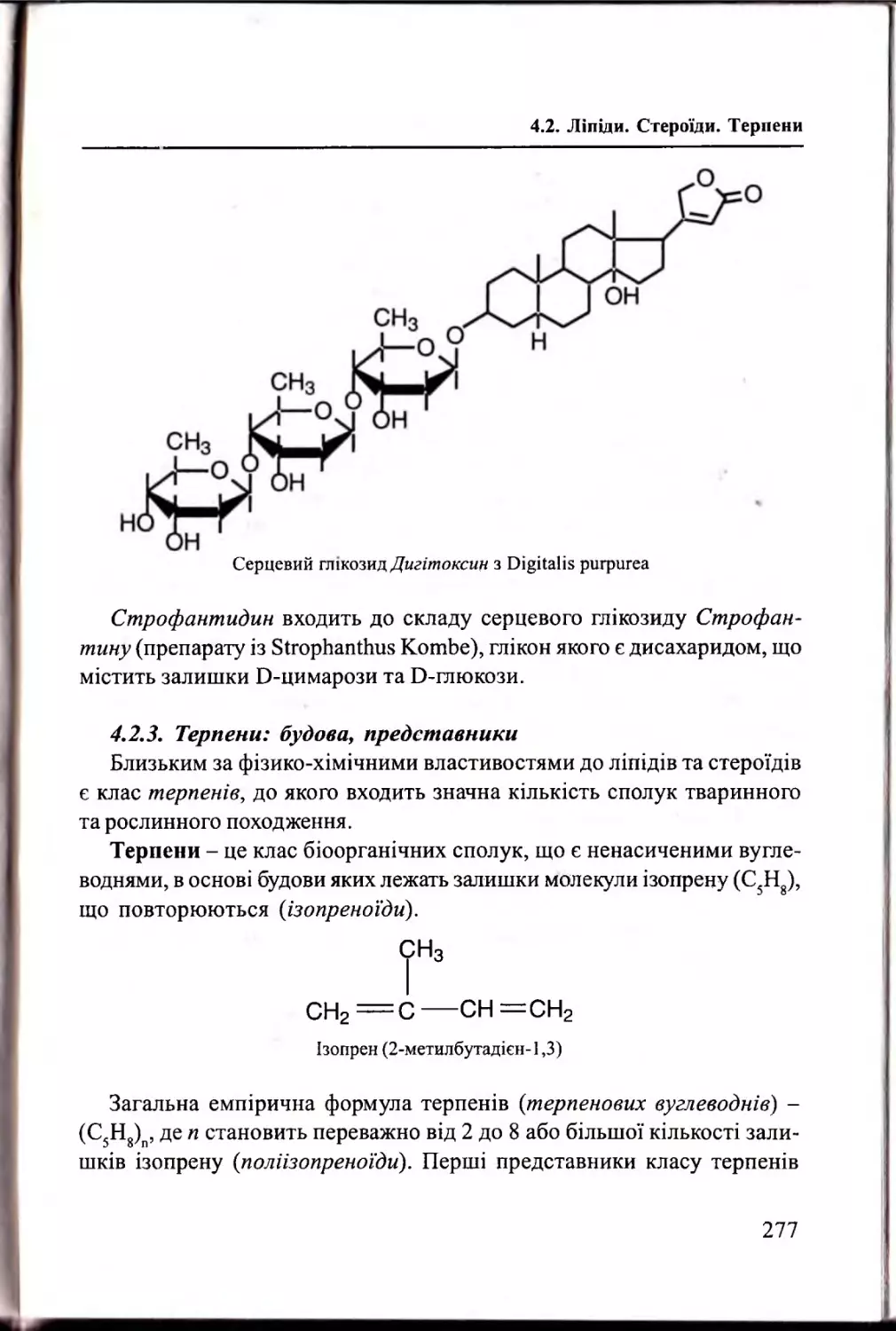
4.2.2. Стероїди та їх п охідн і....................................................... 268
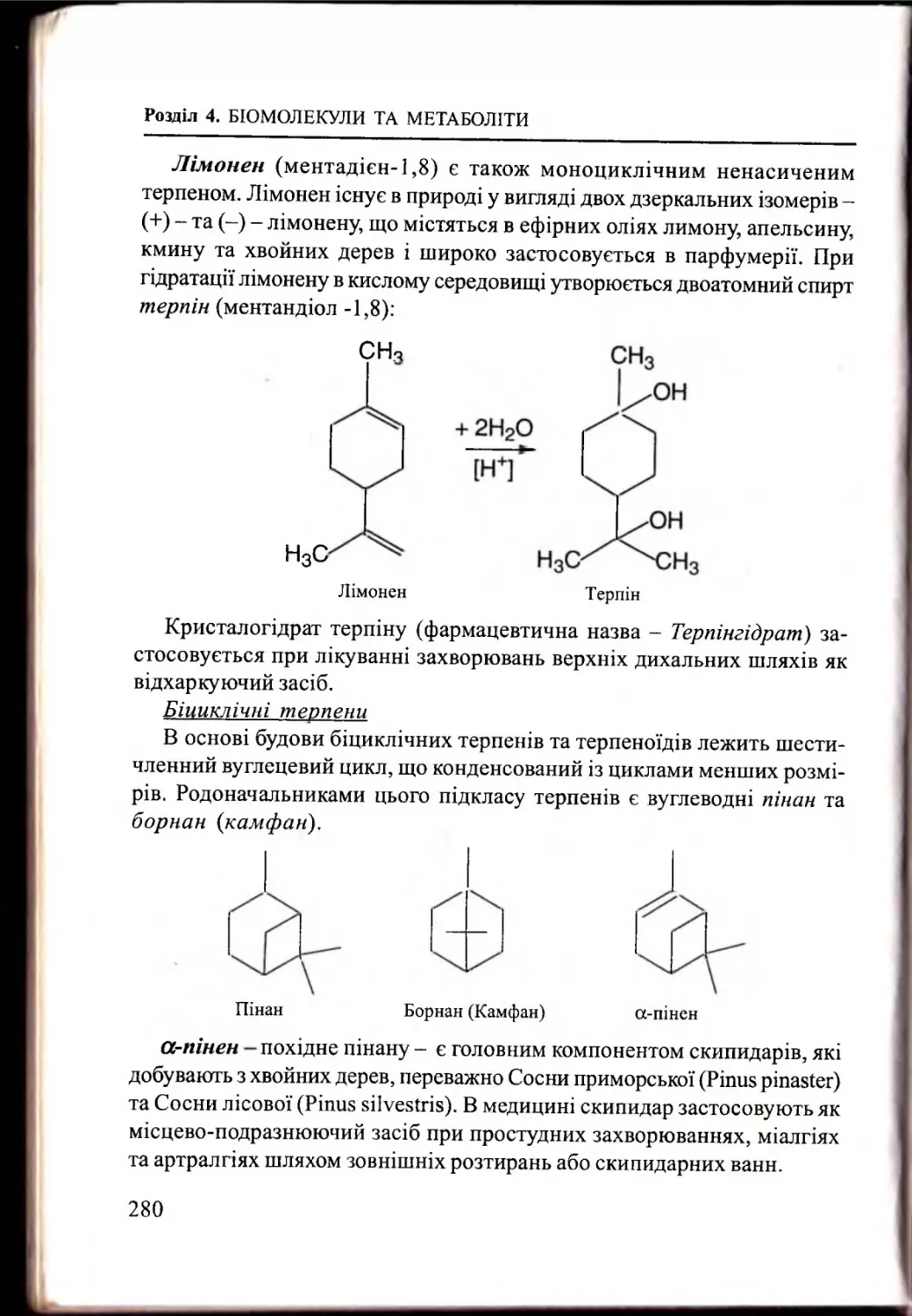
4.2.3. Терпени: будова, представники...................................... 277
4.3.
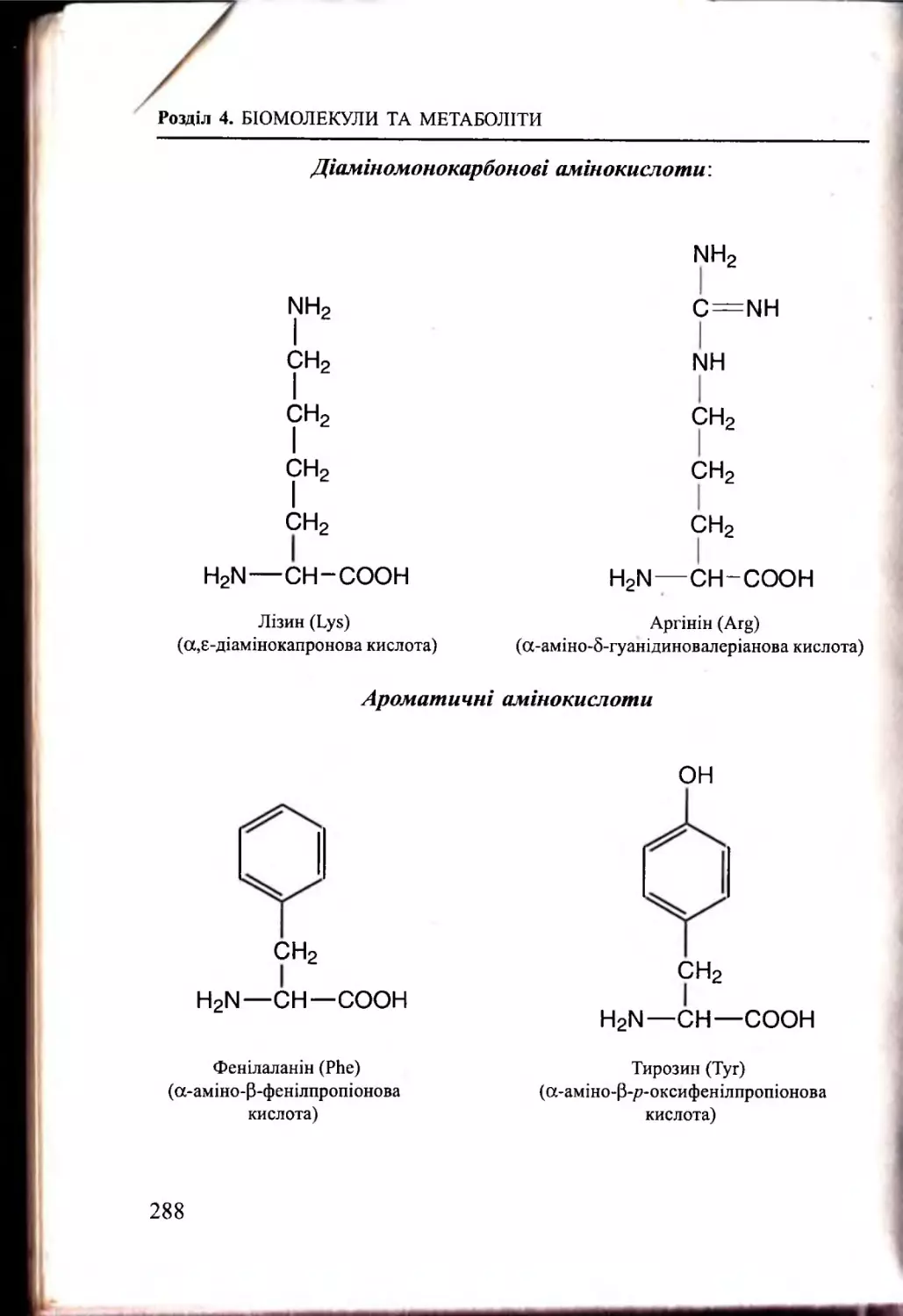
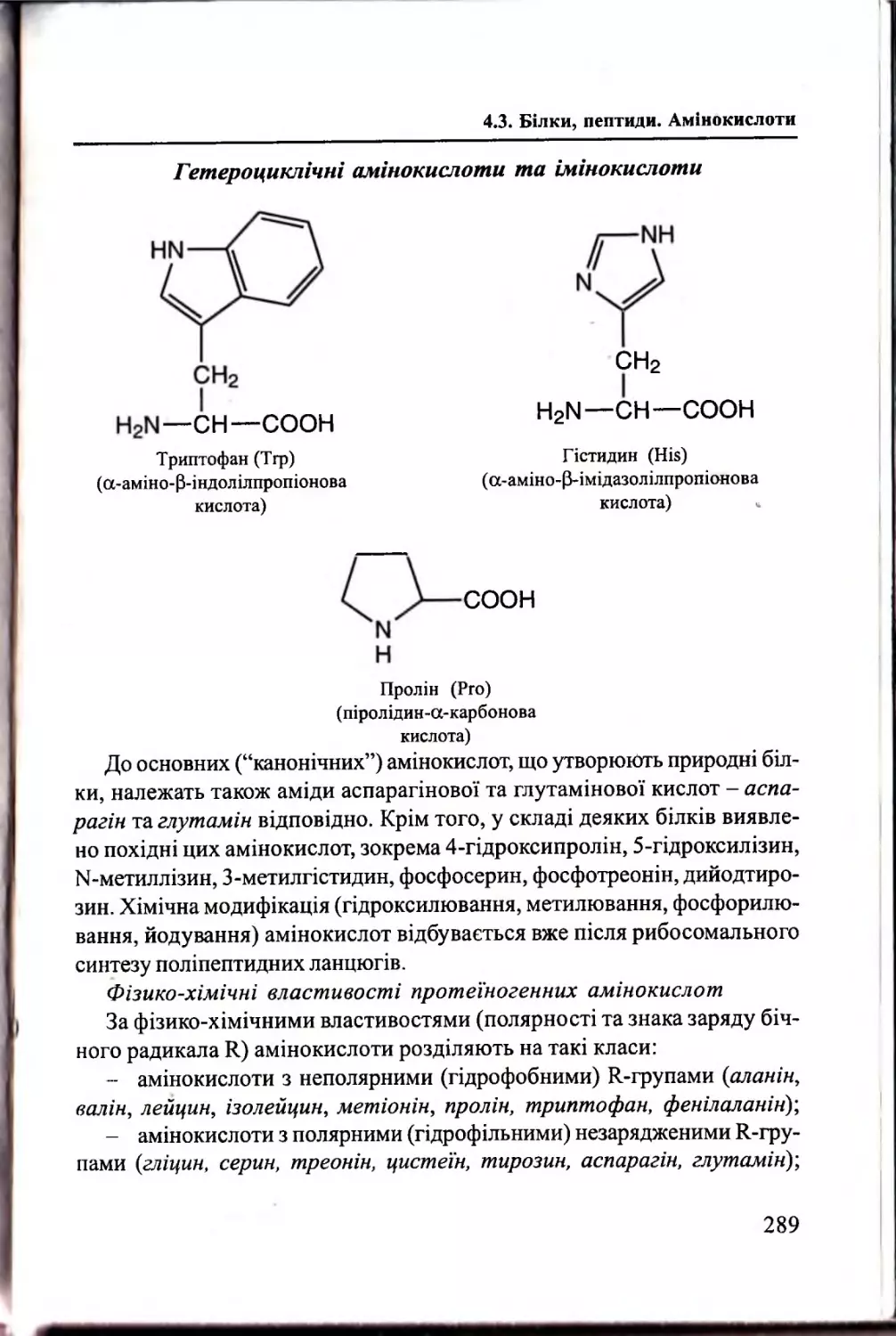
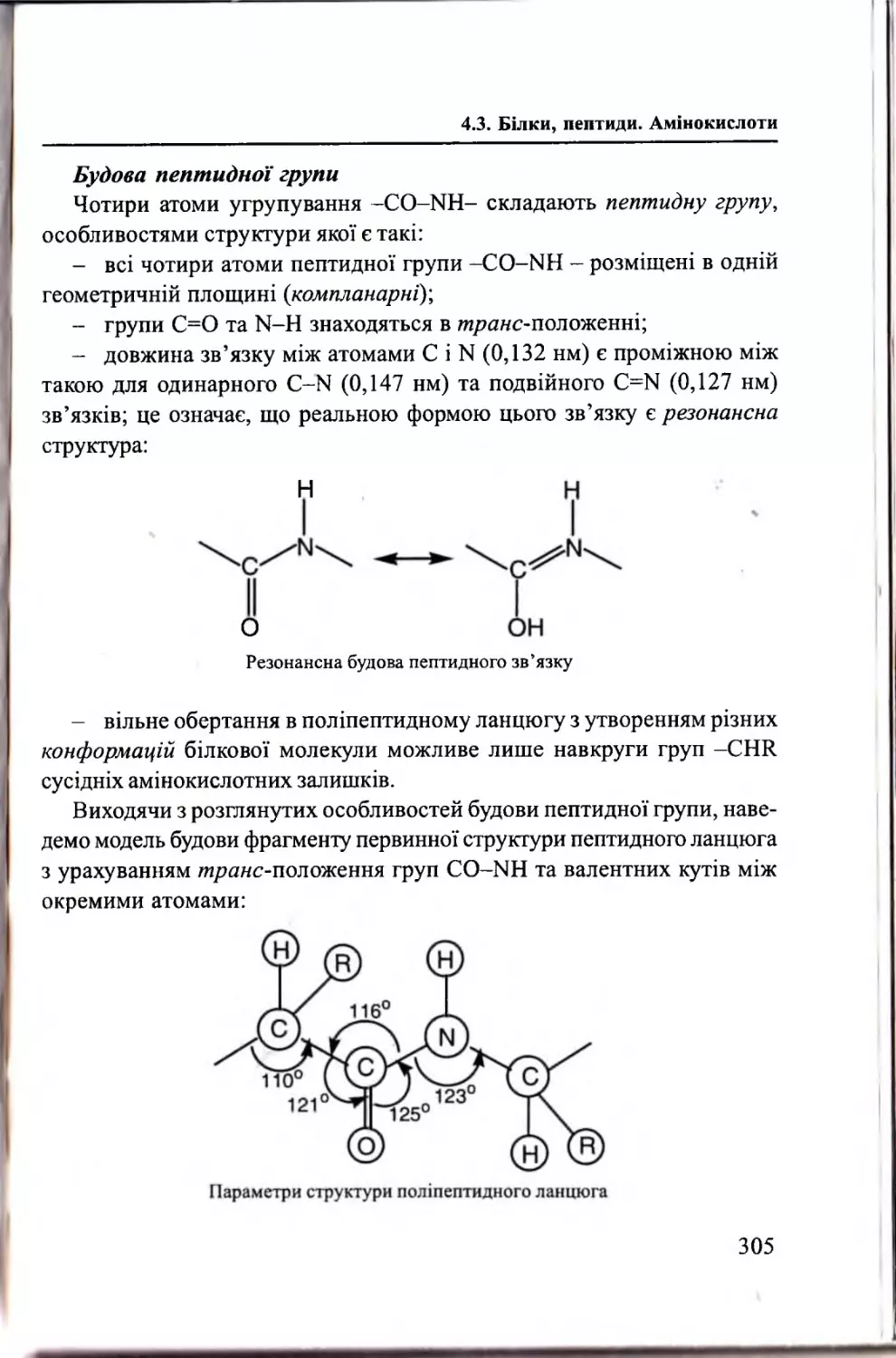
Білки, пептиди. Амінокислоти................................................. 283
4.3.1. Білки: визначення, загальна характеристика............. 283
4.3.2. Амінокислотний склад білків та пептидів...................284
4.3.3. Структурна організація білків та пеп ти д ів..................302
4.3.4. Методи виділення та аналізу білків і пептидів........... 314
4.4.
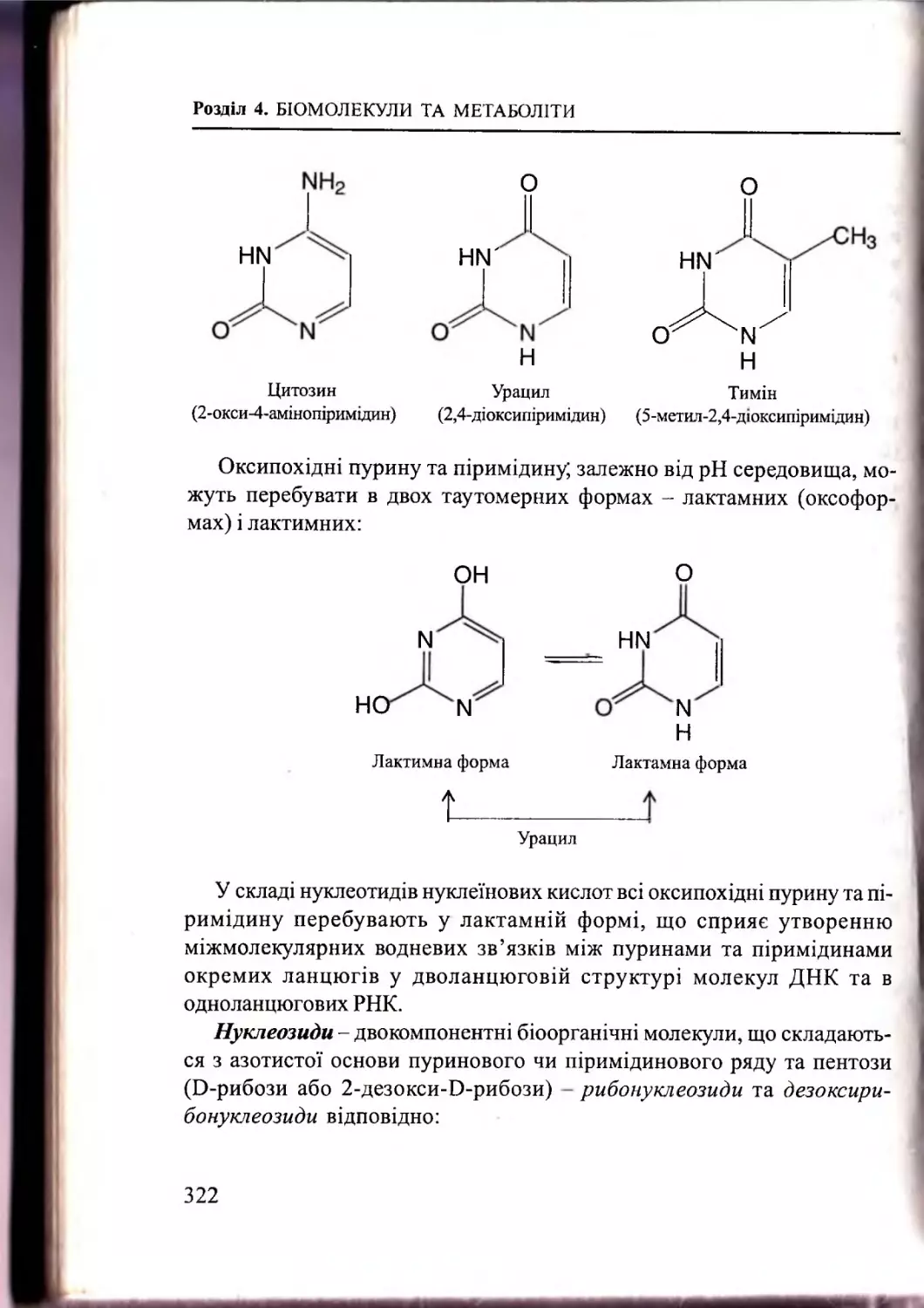
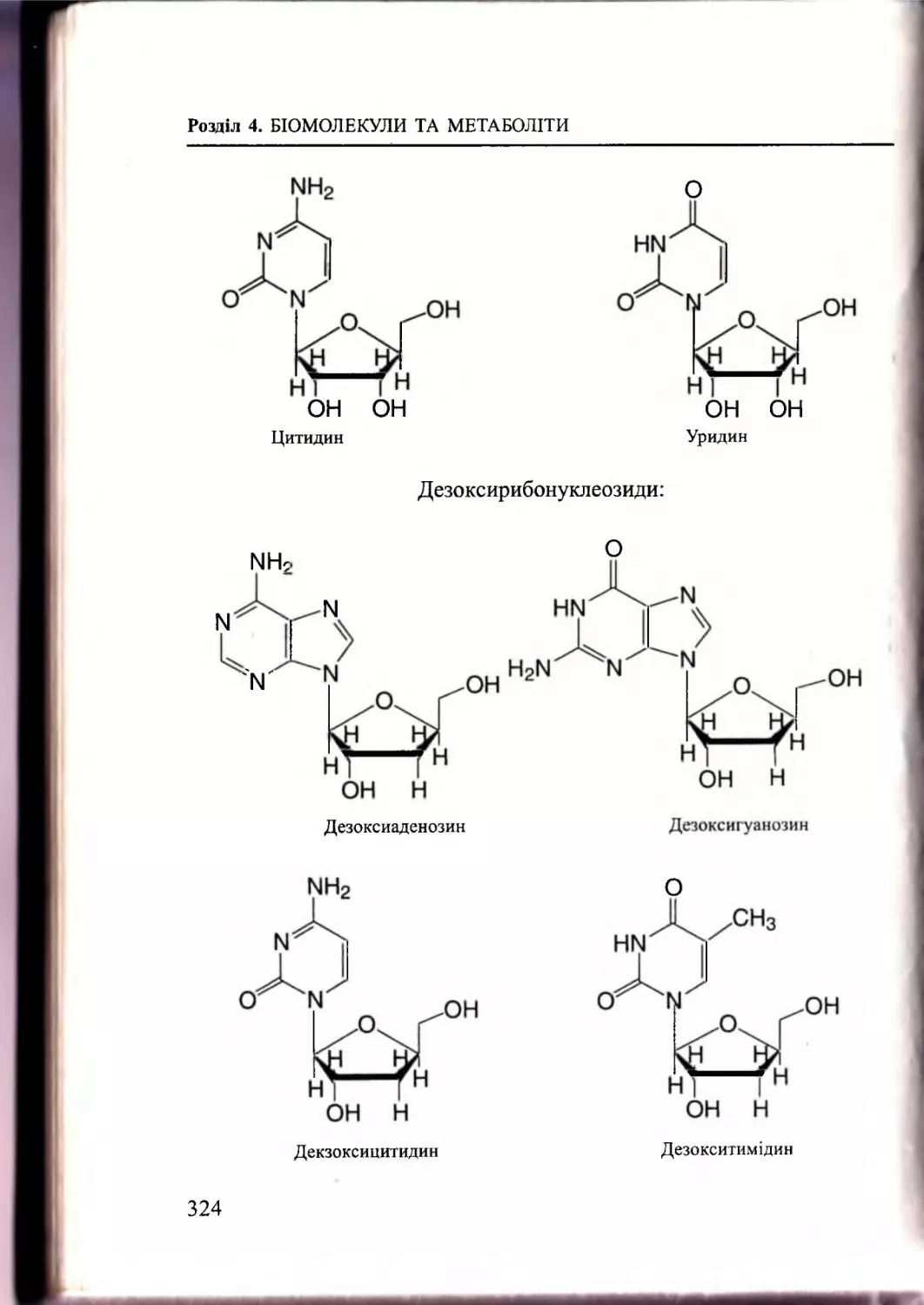
Нуклеотиди. Нуклеїнові кислоти............................................. 320
4.4.1. Будова, властивості та функції нуклеотидів............... 321
4.4.2. Первинна структура нуклеїнових кислот;
полярність полінуклеотидів............................................. 330
4.4.3. Будова, властивості та функції Д Н К .............................332
4.4.4. Будова, властивості та функції Р Н К .............................339
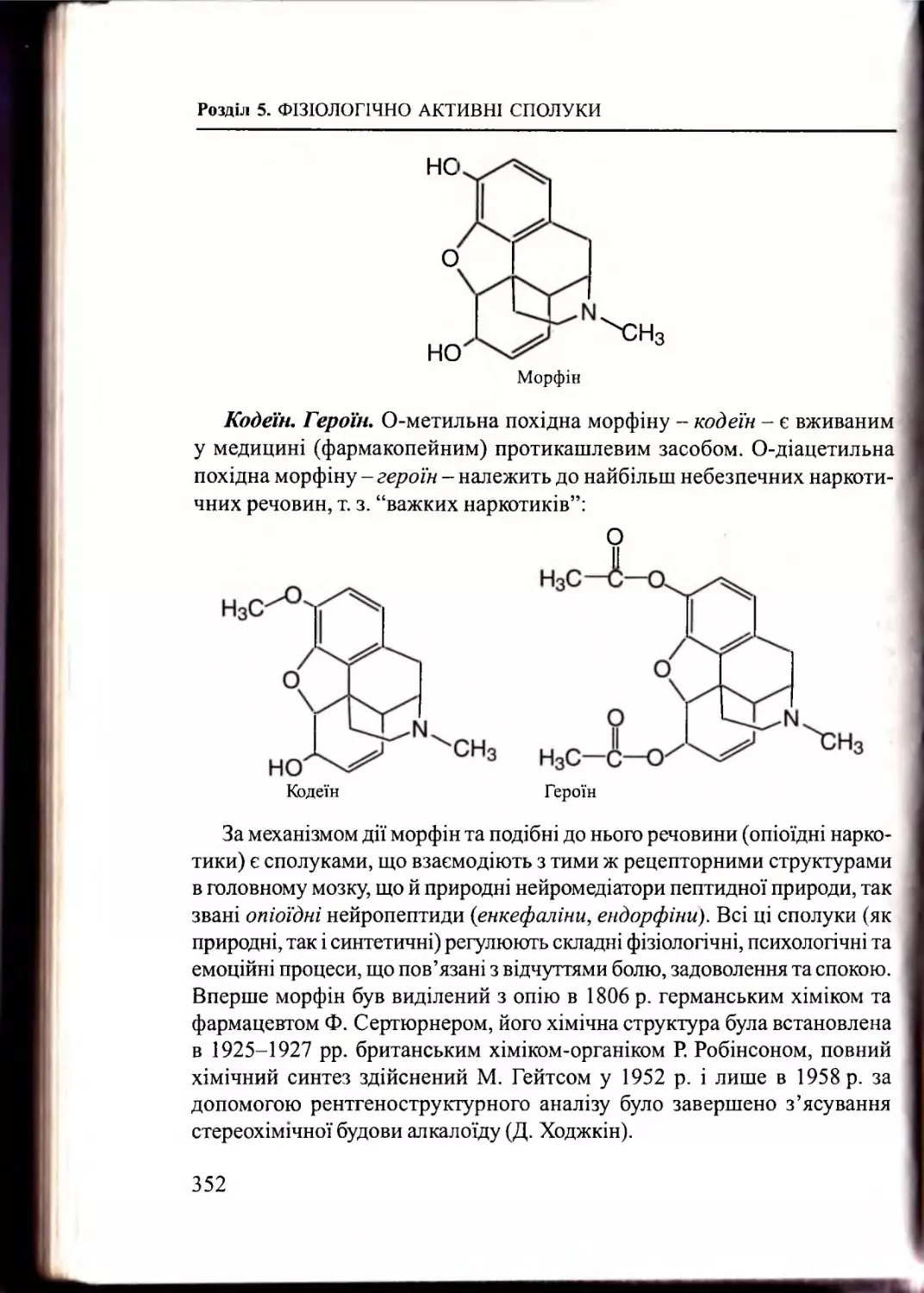
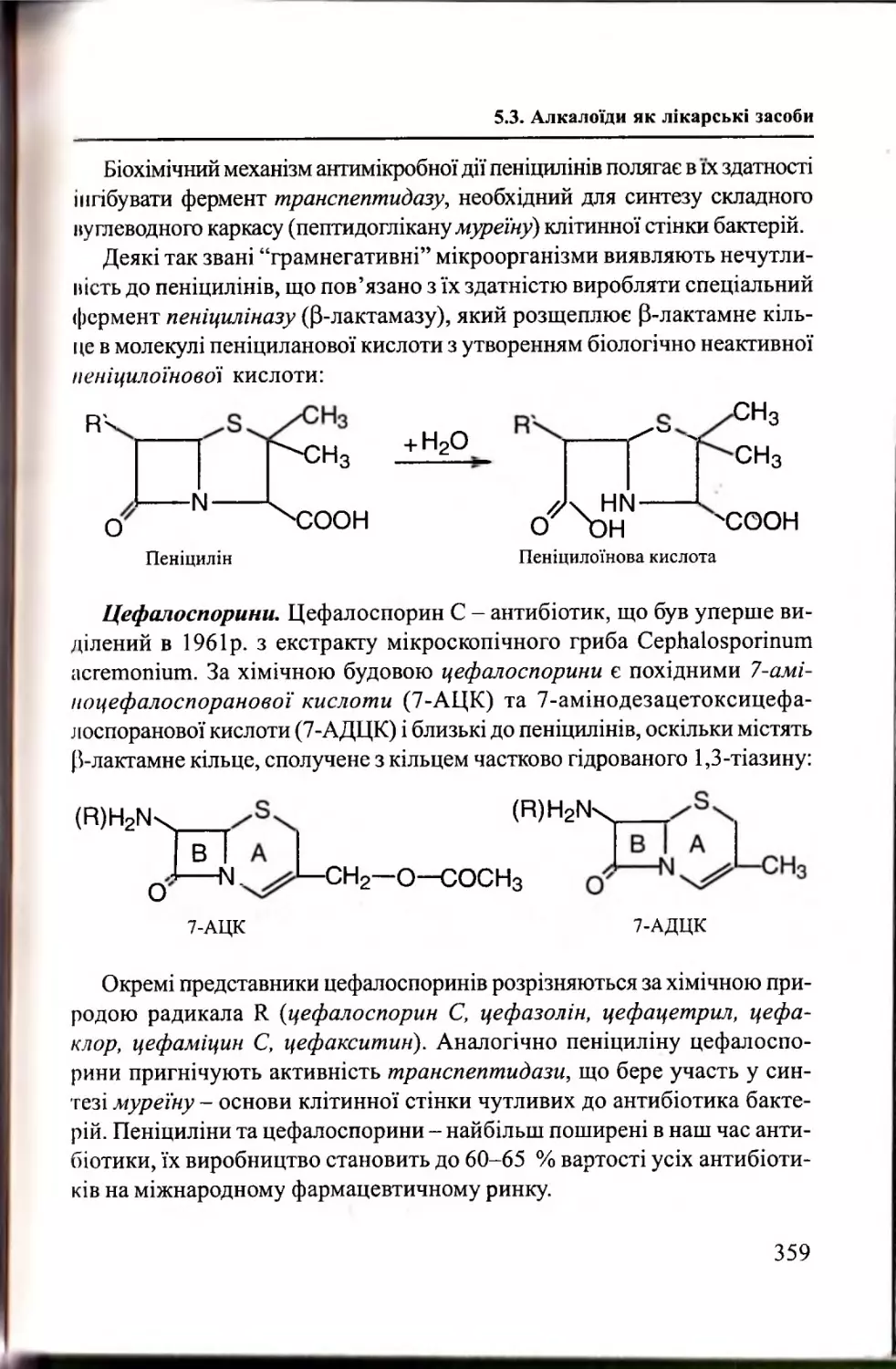
Розділ 5. Фізіологічно активні сп о л у ки
..........................................344
5. і . Загальна характеристика фізіологічно активних сполук.... 344
5.2.
Алкалоїди як лікарські з а с о б и ................................................. 346
5.3.
Антибіотики як лікарські засоби
5.4.
Вітаміни. Коферменти............................................................... 361
5.5.
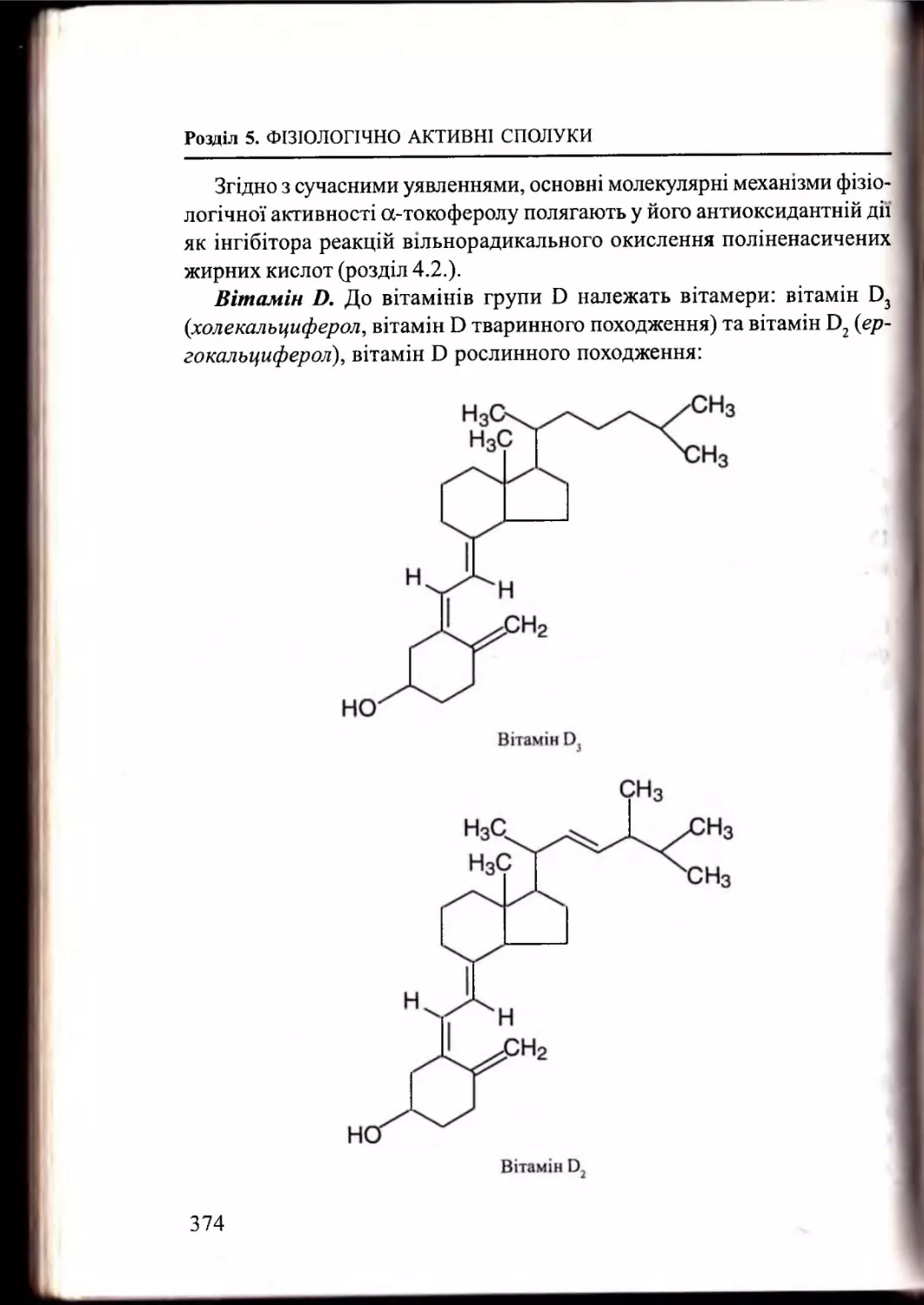
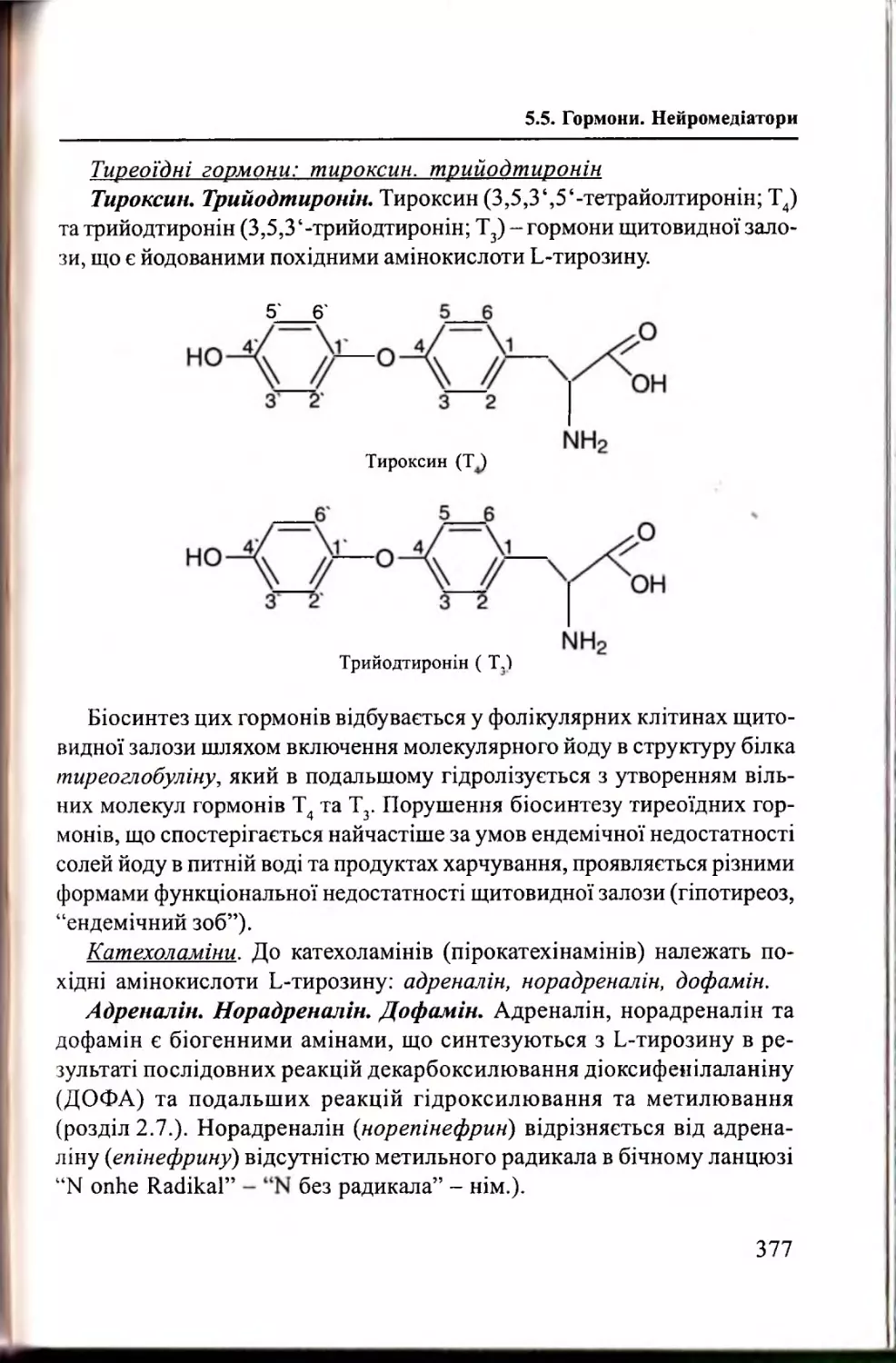
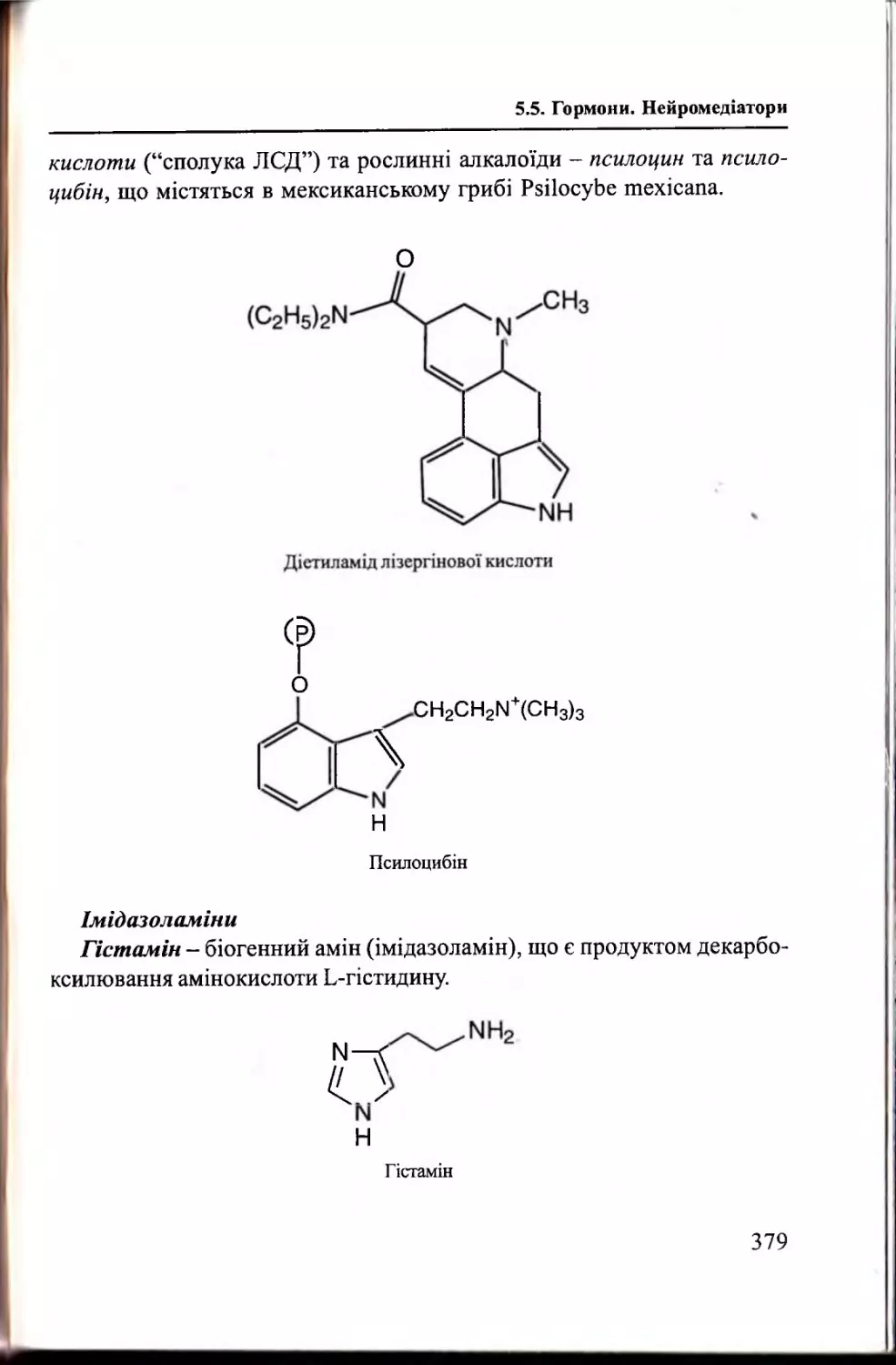
Гормони. Н ейромедіатори........................................................375
5.6.
Пестициди та інші біоцидні ксенобіотики..............................383
...................................... 357
Д о д а т о к .................................................................
389
Рекомендована л ітература..................................................................410
Предметний п ок аж ч и к ....................
412
ПЕРЕДМ ОВА
Будь-яке вчення, якщо воно с e u e т е м о ю,
тобто певиою сукупністю положень, що
пізнаються, та є упорядкованим згідно з
принципами, зветься н а у к о ю .
Імануїл Кант,
“М етафізичні засади природознавст ва” (1786)
Біоорганічна хімія є наукою та навчальною дисципліною, що разом
з біохімією, молекулярною біологією, мікробіологією та фізіологією скла
дає фундамент сучасної теоретичної медицини та медичної біології.
Завданнями біоорганічної хімії є вивчення хімічної структури і властиво
стей органічних сполук вуглецю, що входять до складу живих організмів
і становлять фундамент молекулярної організації і функціонування жи
вих клітин. На відміну від органічної хімії, біоорганічна хімія розглядає
закономірності будови і реакцій окремих класів вуглецевих сполук у зв’яз
ку з їх біохімічними функціями та впливом на фізіологічні процеси, що
відбуваються в біологічних системах.
Призначення біоорганічної хімії як науки полягає у з’ясуванні хіміч
них та фізико-хімічних основ функціонування молекул, що складають
живу клітину. Виходячи з цього, біоорганічна хімія нерозривно зв’язана і
є пропедевтичною дисципліною в опануванні засадами біохімії та м о
лекулярної б іо ло гії- наук, які вивчають хімічний склад живих організ
мів, властивості і механізми перетворень молекул, що входять до їх скла
ду, та біофізики - науки про фізичні закономірності, що властиві живим
системам. Таким чином, успіхи біоорганічної хімії значною мірою ви
значають подальший розвиток усього комплексу біомедичних наук,
оскільки без знання будови і хімічних перетворень органічних речовин,
які беруть участь в обміні речовин (метаболізмі), не можна зрозуміти
його суті, розробити методи регуляції таких біологічних явищ, як ріст і
розмноження клітини, генерація мембранного потенціалу, передача
нервового імпульсу, м ’язове скорочення та ін.
Першочергове значення біоорганічна хімія має також для оволодін
ня загальною та молекулярною фармакологією, предметами фармацев
тичного комплексу - ф арм ацевт ичною хім ією , фармакогнозією,
Передмова
технологією ліків. Більшість лікарських засобів (фармацевтичних пре
паратів) - це різноманітні органічні сполуки, тому біоорганічна хімія, поряд
з біохімією та фізіологією, є теоретичною основою фармакології, зокре
ма методологічною основою клінічної фармакокінетики. Хімічні реакції,
характерні для певних класів органічних сполук (вуглеводнів та їх похід
них, гетеро циклів, алкалоїдів тощо), широко застосовуються у фармацев
тичній хімії для ідентифікації лікарських речовин та аналізу якості фар
мацевтичних препаратів.
Органічні речовини широко застосовуються в промисловості, сільсь
кому господарстві, побуті, зокрема як промислові розчинники, барвники,
фосфорорганічні та хлорорганічні пестициди тощо. Біоорганічна хімія не
тільки з ’ясовує механізми шкідливого впливу токсичних органічних
сполук, а й розробляє методи їх виявлення та кількісного визначення в
біологічних рідинах та тканинах організму людини і тварин (токсиколо
гічна хім ія, медична хімія).
Виходячи із зазначеного, біоорганічна хімія є сучасною фундаменталь
ною наукою, яка повинна обов’язково входити як окрема дисципліна в
робочі плани медичних та фармацевтичних вищих навчальних закладів
(університетів, академій, інститутів, окремих факультетів) та профільних
факультетів класичних університетів. Відповідно до цієї освітньої кон
цепції кафедрою біоорганічної, біологічної та фармацевтичної хімії Націо
нального медичного університету імені О. О. Богомольця було вперше
складено навчальну Програму з біоорганічної хімії, яку затверджено
Центральним методичним кабінетом з вищої медичної освіти МОЗ Украї
ни (директор ЦМК професор І. С. Вітенко) як програму для студентів
вищих медичних закладів освіти III—IV рівнів акредитації (Київ, 2000).
Програму було складено виходячи з робочого плану з біоорганічної хімії,
що має такий розподіл робочих годин: всього навчальних годин - 60, у
тому числі лекційних - 16, лабораторно-практичних та контрольних за
нять - 24, самостійної та позааудиторної роботи студентів - 20; кінце
вим контролем якості знань повинен бути іспит або диференційований
залік. Загалом позитивно сприймаючи запропоноване МОН Україии спря
мування розподілу навчальних планів на збільшення годин самостійної
роботи студентів, вважаємо, що такі зміни можуть реалізуватися лише
за умов докорінних та якісних позитивних змін в організації навчального
процесу шляхом суцільної його інформатизації, впровадження сучасних
комп’ютерних технологій, доступних для кожного викладача і студента.
8
_____________ Передмова
В основу даного підручника, що пропонується як навчальний посіб
ник для студентів вищих медичних закладів освіти III—IV рівнів акреди
тації, покладена вищезазначена діюча Програма з біоорганічної хімії
(2000), при його підготовці використано багаторічний (понад т р идцять
років) досвід викладання дисципліни на кафедрі біоорганічної та біологічної
хімії Національного медичного університету імені О. О. Богомольця та
враховано матеріали виданого раніше за участю проф. Ю. В. Хмелевського, доц. Л. Г. Сударикової та к.м.н. О. К. Усатенка навчального
посібника “Біоорганічна хімія. Навчальний посібник' (Ю. !. Губськийта
співавт. - К.: “Вища школа”, 199 7).
При відборі навчального матеріалу і формуванні структури підруч
ника автором насамперед враховувалося, що теоретичною основою біо
органічної хімії є класична органічна хімія, і опанування закономірностя
ми останньої є обов’язковою передумовою вивчення біоорганічної хімії.
Разом з тим, оскільки основи органічної хімії входять до сучасних про
грам середніх загальноосвітніх шкіл, коледжів та ліцеїв, у програмі вищої
медичної освіти викладанню біоорганічної хімії не передує вивчення суто
органічної хімії. З огляду на ці обставини підручник, що пропонується,
побудований з двох частин. У першій частині розглядаються загальні
закономірності будови та реакційної здатності органічних молекул, які є
необхідними для подальшого розгляду хімічних властивостей та хіміч
них перетворень окремих класів біоорганічних речовин. Друга частина
підручника присвячена біоорганічній хімії найбільш розповсюджених
похідних вуглеводнів, карбоциклічних та гетероциклічних речовин, що є
метаболітами (вуглеводів, ліпідів, амінокислот та пептидів, нуклеотидів
та нуклеїнових кислот тощо) та структурною основою багатьох природ
них лікарських засобів (гормонів, вітамінів, алкалоїдів, антибіотиків) і
біоцндних ксенобіотиків.
До складу підручника включено як додаток Програму з біоорганічної
хімії (Київ, 2000 р.), яка містить орієнтовні тематичні плани лекцій та
лабораторно-практичних занять, а також перелік контрольних питань для
іспиту або заліку з біоорганічної хімії на медичних та стоматологічних
факультетах.
Підручник призначений для студентів медичних факультетів вищих
навчальних закладів, у яких, залежно від конкретних особливостей
навчальних планів, біоорганічна хімія викладається на першому або дру
9
Передмова
гому семестрах, передуючи вивченню біологічної хімії (біохімії-). В якості
додаткового навчального посібника видання рекомендується також для
студентів вищих фармацевтичних закладів освіти (факультетів, універ
ситетів). Підготовкою цього видання завершується формування навчаль
но-методичного комплексу, першим етапом якого була публікація та
впровадження у навчальний процес підручника: Ю. І. Губський “Біоло
гічна хімія” (Укрмедкнига, 2000 р.) та розробка відповідних робочих
програм. На думку автора, підручник, що пропонується читачеві, може
бути також використаний студентами біологічних факультетів універси
тетів, аспірантами, науковцями, що працюють в галузі біоорганічної хімії,
біохімії, медичної та фармацевтичної хімії.
В обговоренні концептуальних засад викладання біоорганічної хімії у
вищих медичних навчальних закладах України у зв’язку з реалізацією
положень Болонської декларації та введенням кредитно-модульної сис
теми навчання (Наказ МОН України від 23.01.2004 р. № 49) і відповід
ної структури підручника взяли участь співробітники кафедри біоорган
ічної, біологічної та фармацевтичної хімії НМУ - доктор медичних наук
професор Ю. В. Хмелевський, доценти Л. Г. Сударикова, О. В. Задорина, Л. В. Гайова, А. С. Ягупова, А. Б. Гладчук, ст. викладач О. К. Усатенко, асистенти О. В. Слесаренко, О. В. Стеченко, керівники та викла
дачі інших профільних кафедр медичних та фармацевтичного факуль
тету НМУ - член-кореспондент НАН та АМ Н України професор
І. С. Чекман, професор В.О.Калібабчук, професор Д. С. Волох, профе
сор П. І. Середа, члени Проблемної комісії МОЗ та АМН України “Біоло
гічна та медична хімія” - член-кореспондент НАН та АМН Н. М. Гула,
професор Я. І. Гонський, професор О. О. Мардашко, професор І.Ф.Мещишен, яким автор висловлює глибоку подяку. Підручник такого спря
мування створений у нашій країні вперше, і автор із вдячністю сприйме
всі зауваження та побажання.
10
ВСТУП
Біоорганічна хімія як наука
Найзначнішим прогресом >’ біології
нашого сторіччя було обернення до
вивчення процесів на молекулярному
рівні. Наступним кроком буде, очевидно,
перехід на субмолекулярний, електрон
ний рівень.
Альберт Септ-Дьйордьї,
“Біоел ект роніка ”
Біоорганічна хімія - наука, що вивчає будову, реакційну здатність,
хімічні перетворення та біологічне значення органічних сполук, які вхо
дять до складу живих організмів. Об’єктами вивчення біоорганічної хімії
є низькомолекулярні біомолекули, біополімери (білки, нуклеїнові кисло
ти, полісахариди), біорегулятори (ферменти, гормони, вітаміни, регуля
торні молекули імунної системи тощо), природні і синтетичні фізіологіч
но активні сполуки, в тому числі лікарські засоби та речовини з токсич
ною дією.
Біомолекули - біоорганічні сполуки, що входять до складу живих
організмів і спеціалізовані для утворення клітинних структур і участі в
біохімічних реакціях, що становлять основу обміну речовин (метаболіз
м у) та фізіологічних функцій живих клітин і багатоклітинних організмів в
цілому. Головними класами біомолекул є: білки та амінокислоти, нуклеї
нові кислоти та нуклеотиди, вуглеводи та їх похідні, жирні кислоти, ліпіди
та їх похідні, вітаміни, гормони та інші біорегулятори. Крім означених
біоорганічних молекул, до складу всіх живих організмів входить певна
кількість низькомолекулярних моно-, ди- і трикарбонових кислот, окси
кислот, спиртів, амінів тощо, які є проміжними продуктами обміну речо
вин (метаболітами, інтермедіатами).
Біоелементи. До складу біоорганічних сполук, крім атомів вуглецю
(С), які становлять основу будь-яких органічних молекул, входять також
кисень (О), водень (Н), азот (Ы), фосфор (Р) та сірка (5). Ці біоелемен
ти (“органогени”) сконцентровані в живих організмах у кількості, що в
20-200 разів перевищує їх вміст в об’єктах неживої природи, становля
чи понад 99% елементного складу біомолекул.
11
Вступ
Наукові напрямки та методи біоорганічної хімії
Біоорганічна хімія ґрунтується на наукових та методичних принципах
органічної х і м і ї - хімії сполук вуглецю. Разом з тим, біоорганічна хімія
є особливим розділом органічної хімії, що вирішує фундаментальні і при
кладні проблеми біохімії, молекулярної біології та фармакології. Конкрет
ними завданнями цієї дисципліни є визначення структури біомолекул
(амінокислот, білків та пептидів, нуклеотидів та нуклеїнових кислот, ву
глеводів, ліпідів, карбонових кислот, спиртів тощо) та продуктів їх мета
болізму, механізмів ферментативного каталізу, молекулярних основ вза
ємодії з клітинними рецепторами біорегуляторів різної хімічної природи.
Вивчаючи перебіг та механізми органічних реакцій, які є моделями та
ких, що мають місце в біохімічних системах не тільки in vitro, а й in
vivo, біоорганічна хімія створює фундамент теоретичної, молекулярної
біохімії.
Другою складовою, важливим розділом біоорганічної хімії є дослід
ження структури та молекулярних основ взаємодії з б to структурами
синтетичних фізіологічно активних сполук (ФАС), у тому числі лікар
ських засобів та інших речовин, що надходять до організму людини з
оточуючого середовища —промислових отрут, пестицидів, засобів по
бутової хімії, харчових домішок та ін. Ці речовини є чужорідними для
організму людини і вищих тварин - “ксенобіотиками” , тобто сполуками,
з якими біосистеми на попередніх етапах еволюції не зустрічалися, і
тому багато з них виявляють біоцидні та екологічно небезпечні ефекти
(Ю. І. Губський, 1993). У завдання біоорганічної хімії входять вивчення
будови, властивостей та шляхів синтезу численних ФАС, виявлення ко
реляцій між їх будовою і біологічною дією, вивчення хімічних перетво
рень біоорганічних сполук в організмі.
Саме завдяки застосуванню методів та теоретичних підходів біоор
ганічної хімії стало можливим з ’ясування залежностей між молекуляр
ною, електронною будовою фізіологічно активних речовин (ліків, токси
нів) та їх фізіологічними, зокрема фармакологічними, ефектами, вивчен
ня закономірностей перетворення, або біотрансформації, цих речовин
в організмі людини і тварин.
Цей напрямок біоорганічної хімії має фундаментальне значення для
розвитку загальної та молекулярної фармакології, токсикології, фарма
цевтичної хімії.
12
Вступ
Розв’язання означених наукових проблем стало можливим лише за
вдяки розробці та застосуванню в біоорганічній хімії високочутливих
фізичних та фізико-хімічних методів розділення, очищення органічних
сполук (хроматографія, електрофорез) та дослідження їх будови (інфра
червона та ультрафіолетова спектроскопія, спектроскопія електронного
парамагнітного та ядерного магнітного резонансу, мас-спектрометрія,
рентгеноструктурний аналіз).
Етапи розвитку біоорганічної хімії
Біоорганічна хімія належить до наук, які стрімко розвиваються. За
вдяки досягнутим нею успіхам встановлено структуру багатьох фізіо
логічно активних пептидів, ферментних та гормональних білків, нуклеї
нових кислот, вітамінів, алкалоїдів. Опановано методи хімічного та біотехнологічного синтезу багатьох складних сполук, що використовують
ся в медичній практиці як лікарські препарати та матеріали для виготов
лення ендопротезів та штучних органів людини. Аналітичні методи біо
органічної хімії широко застосовуються в медичній біохімії при клініколабораторному дослідженні крові, ссчі, спинномозкової рідини, слини,
травних соків з метою встановлення діагнозу захворювання. Вивчення
структури та хімічних властивостей головних класів біополімерів - біл
ків та нуклеїнових кислот ДНК і РНК, що відповідальні за фундамен
тальні прояви життєдіяльності (обмін речовин, спадковість), робить су
часну біоорганічну хімію також теоретичною основою молекулярної біо
логії, генної інженерії та біотехнології.
У своєму історичному розвиткові біоорганічна хімія пройшла довгий
шлях як складова органічної хімії від перших теорій чотиривалентності
вуглецю, будови сполук цього елементу та ізомерії - Фрідріх А. Кекуле
(F. Kekule; 1829-1896), А рчибалд Купер (A. Couper; 1831-1892),
О. М. Бутлеров (1828-1886) - до сучасних квантово-механічних розра
хунків і прогнозування структури та функцій біополімерів і низькомоле
кулярних фізіологічно активних сполук шляхом їх комп’ютерного моде
лювання. Наріжними каменями у становленні цієї науки стали такі кла
сичні наукові досягнення, як здійснений Фрідріхом Вьолером (F. Wohler)
перший штучний (поза організмом) синтез органічної речовини сечовини, та Марселеном Бертло (М. Berthelot) - жироподібних сполук,
відкриття Луї Пастером (L. Pasteur) асиметричної будови органічних
молекул, піонерські дослідження з хімії білків, вуглеводів та пептидів
13
Вступ
Альбрєхта Косселя {A. Kossel), Франца Гофмейстера (F. Hofineister) та
Еміля Ф ішера(Е. Fischer), перші роботи Фрідріха Мішера (F, Miescher)
щодо виділення та хімічної характеристики нуклеїнових кислот.
Важливим етапом у сучасному розумінні генезису біоорганічних
сполук стали дослідження С. Міллера (S. Miller) з абіогенного синтезу
головних класів біомолекул, що входять до складу живих організмів. У
своїх класичних експериментах С. Міллер (1953) вперше довів можли
вість утворення амінокислоти аланіну - однієї з «-амінокислот, що є скла
довою в біосинтезі природних білків - за умов дії електричних розрядів
на газову суміш метану, аміаку, водню та водяної пари. Цей процес можна
схематично подати такою послідовністю рівнянь:
С Н 4 + NH3—> H CN + ЗНг
Ц іанід водню
HCN + С 2Н4 -> C H 3- C H a- C N
П роп іон ітрил
С Н - С Н - C N + NHg-> C H 3 - C H N H - C N
Амінопропіонітрил
ch
3- c h n h 2- c n + н го -> c h 3- c h n h 2- c o o h
а -ал а н ін
Пізніше була доведена можливість утворення в умовах, які моделю
ють первісну атмосферу Землі, також і азотистих основ нуклеотидів,
тобто попередників нуклеїнових кислот, що має фундаментальне зна
чення для сучасного розуміння проблеми виникнення життя.
Як окрема галузь знань, яка поєднує концептуальні засади та мето
дологію органічної хімії, з одного боку, та молекулярної біохімії і моле
кулярної фармакології, —з іншого, біоорганічна хімія сформувалася в 60-х—
70-х роках XX сторіччя на підставі розробок у галузі хімії природних
сполук та біополімерів —M. М. Шемякін (1908-1970), Ю. А. Овчинников (1934-1988). Сучасне обличчя біоорганічної хімії значною мірою
формують наукові розробки, що стосуються хімічних аспектів проблем
біохімії та молекулярної біології - насамперед структури і функцій білків
та інформаційних нуклеїнових кислот ДНК і РНК,
14
Вступ
Відповідно до цього фундаментальний внесок у формування сучас
ної біоорганічної хімії було зроблено завдяки розробці вже в середині
XX століття відповідної методичної бази, а потому - й класичні™ дослід
ж енням У. С тейна (W. Stein), С. М ура (S. M oore) та Ф. С енгера
(F. Sanger) з аналізу амінокислотного складу та визначення первинної
структури пептидів і білків, Л. П олінга (L. Pauling) та У. А стбері
(W. Astbury) із з ’ясування будови а-спіралі та ß-структури та їх значен
ня в реалізації біологічних функцій білкових молекул, Е. Чаргаффа
(E. Chargaff) з розшифрування особливостей нуклеотидного складу ну
клеїнових кислот, Дж. Уотсона (J. Watson), Фр. К ріка(Е Crick), М. Уілкінса(М . Wilkins), Р. Франклін (R. Franklin)- з і встановлення закономір
ностей просторової структури молекули ДНК, Г. Корани (H. Khorana) з хімічного синтезу гена тощо. Весь цей комплекс досліджень дав змогу
перейти наприкінці X X - на початку XXI століття до створення принципово
нових напрямків і методів у молекулярній діагностиці хвороб людини,
розшифровці нуклеотидних послідовностей геному вищих організмів та
розробки цих питань як конкретної біотехнологічної проблеми. У наш
час найбільший внесок в розвиток біоорганічної хімії, молекулярної біо
хімії та молекулярної біології в Україні створюють наукові школи акаде
міка НАНУ В. П. Кухаря (Інститут біоорганічної хімії та нафтохімії),
академіка НАНУ та АМНУ С. В. Комісаренка (Інститут біохімії імені
О. В. Палладіна), академіка НАНУ Г. X. Мацуки (Інститут молекуляр
ної біології та генетики).
15
Частина І
БУДОВА ТА РЕАКЦІЙНА ЗДАТНІСТЬ
БІООРГАНІЧНИХ СПОЛУК
Розділ І ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
1.1. Способи зображення органічних молекул
Структурну та хімічну характеристику будь-якої молекули органіч
ної (та біоорганічної зокрема) сполуки надають її молекулярна (емпі
рична) та структурна формули, які являють собою умовне, формалі
зоване зображення атомного складу та будови молекули.
М олекулярна формула - формула органічної сполуки, що вказує на
кількість атомів кожного елемента в молекулі. Наприклад: С,Н 6,С }НВмолекулярні формули вуглеводнів етану та бутану відповідно, С пН20М4Обмолекулярна формула рибофлавіну (вітаміну В 2), С 15Н [|І4І'ТО4 - моле
кулярна формула гормону щитовидної залози тироксину. Разом з тим,
С6Н|г0 6- молекулярна формула не тільки глюкози, але й також декіль
кох інших моносахаридів (галактози, манози, фруктози та ін.), що є ізо
мерами глюкози, тобто мають однаковий атомний склад, але відрізня
ються одне від одного порядком сполучення елементів в молекулі. Подіб
ним чином, молекулярна формула С2Н60 відповідає двом різним хіміч
ним речовинам - етиловому спирту (етанолу) та диметиловому ефіру,
що різняться своїми структурними формулами і мають відмінні не
тільки фізико-хімічні, а й біологічні властивості.
С т рукт урн а ф орм ула. Д окладніш у інф орм ацію про будову
органічної молекули надає її структурна формула. Структурна форму
ла - це графічне зображення будови молекули органічної сполуки на
н
н
1
1
н-с -с -н
1 1
І 1
н н
Етан
н
н
н
1 1 1
н -с-с -с-н
11 11 11
н
н
н
П роп ан
Рис. 1 .1 . Структурні формули етану (молекулярна формула С2Н6) та пропану
(молекулярна формула С3На)
16
1.1. С п особі! зобр аж ен н я ор ганічн их м олекул
площині або в просторі. В структурній формулі хімічними символами
позначають природу атомів (С, О, ЬІ, Б, Р та Ін.), їх число та послідов
ність зв'язування в молекулі. Хімічні зв’язки між окремими атомами в
молекулі позначають рисочками, кількість яких відповідає валентності
атома (вуглець у складі всіх органічних сполук є чотиривалентним). З
точки зору теорії атомної будови, рисочка між символами атомів відпо
відає електронній парі, що утворює хімічний зв ’язок.
Скорочена структурна формула
При скороченому написанні структурної формули частина зв’язків у
молекулі не позначається (найчастіше - між атомами вуглецю, азоту чи
кисню, з одного боку, та атомами водню - з іншого); у деяких випадках
не позначаються і зв’язки між окремими атомами вуглецю, наприклад:
Н
Н
Н
Н
Н
і
і
І
і
І
с н 3- с н 2- с н 2- с н 2- с н 3
с н 3с н 2с н 2с н 2с н 3
н -с -с -с -с -с -н
I
н
I
н
!
н
!
н
!
н
С тр у кту р н а ф орм ул а
п ен тан у
С корочені структурні
ф орм ули пен тан у
Спрощена структурна формула
Для графічного зображення структурних формул органічних молекул
з великою кількістю (іноді - кількома десятками) вуглецевих атомів
застосовують спрощений спосіб написання структурних формул, при
якому атоми вуглецю та водню взагалі не вказуються, але натомість
позначаються валентні кути між окремими атомами:
Б утан
Е танол
Бензол
ОН
199 Ш
П урин
О
ГУ)
N
Н
”
І
УКРАЇНСЬКОЇ МЕДИ ЧНОЇ
І
бібліотека
С Т О М А Т О Л О Г ІЧ Н О Ї
А К А Д Е М ІЇ
ІДЕНТИФІКАЦІЙНИЙ КОД 0 2 0 1 0 8 2 4
м .П ол тав а
___
17
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
О
П альм іти нова ки сл ота
Як уже зазначено, для багатьох органічних молекул, які мають одна
кові молекулярні формули, їх структурні формули не співпадають, що
свідчить про відмінності в площинній або просторовій будові таких мо
лекул —ізомерів, що будуть розглянуті нижче. При зображенні просто
рового розташування окремих атомів та груп у молекулах просторо
вих ізомерів органічних сполук використовують спеціальні способи та
символи (розділ 1.3.4).
1.2. Класифікація та номенклатура органічних сполук
Завдяки практично необмеженій здатності атомів вуглецю утворю
вати лінійні, розгалужені та циклічні структури, в тому числі такі, що
включають гетероатоми, кількість органічних сполук різної будови, які
існують у живій природі або синтезовані штучно, налічує нині декілька
мільйонів назв. У зв’язку з цим, надзвичайно важливим для вивчення
органічної та біоорганічної хімії стало створення науково обґрунтованих
класифікації та номенклатури, враховують будову вуглецевих ланцюгів
та н аявн ість певних ф ун к ц іо н а льн и х груп, які значною мірою
визначають хімічні властивості окремої сполуки.
1.2.1. Класифікація органічних сполук
Всі органічні сполуки поділяються натри великі угруповання-ряди,
а саме:
1.
А ц и к ліч н і сполуки, тобто сполуки з відкритим (незамкненим)
ланцюгом вуглецевих атомів. їх називають також аліфатичними спо
луками, або сполуками жирного ряду. Ациклічні сполуки можуть мати
лінійні або розгалужені вуглецеві ланцюги:
с н 3- с н 2- с н 2- с н 2- с н 2- с н 3
н-гексан
18
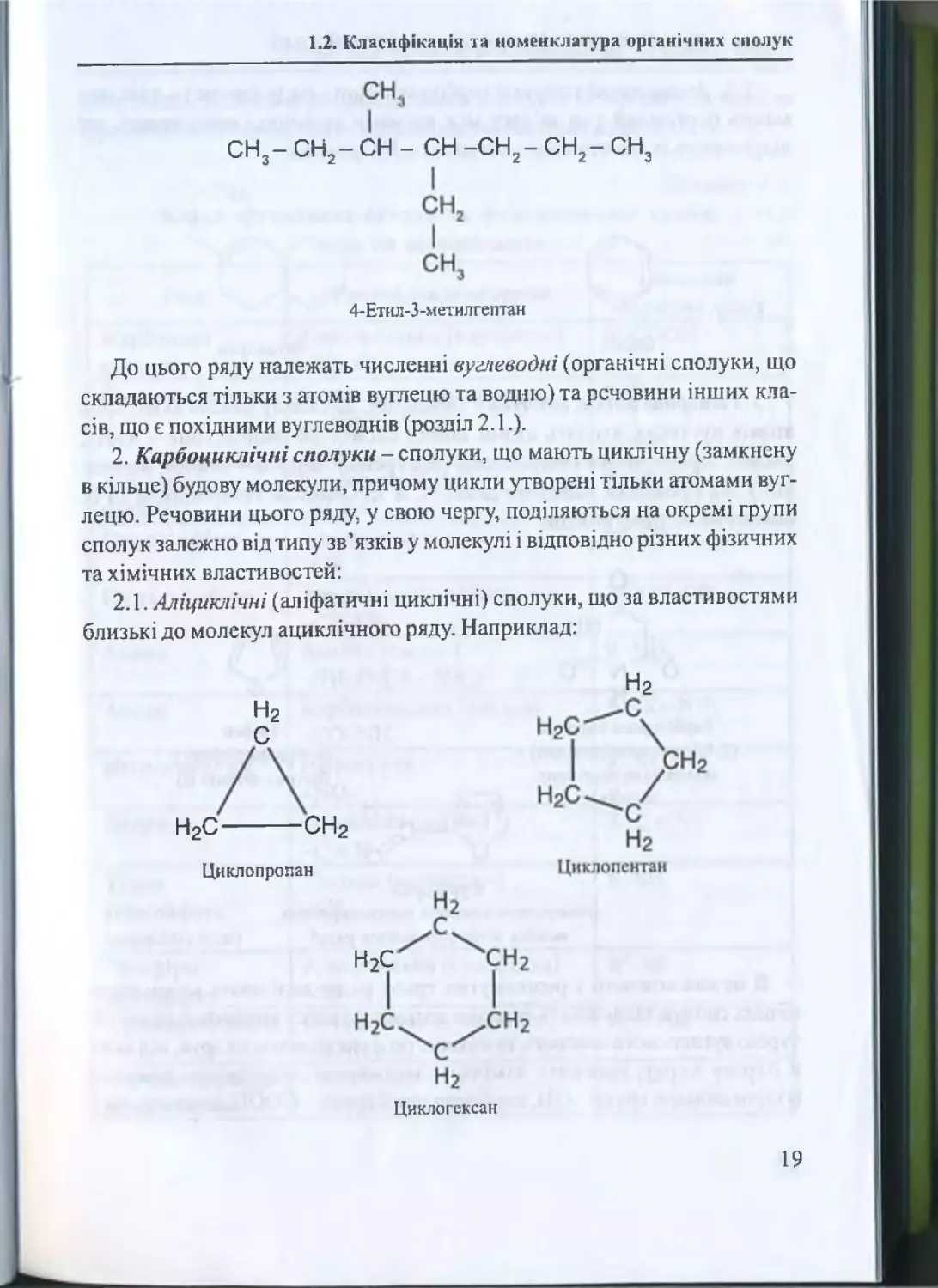
1.2. К ласи ф ік ац ія та ном ен к латура ор ган іч н и х сп ол ук
с н 3- с н 2- сн - сн - с н 2- с н 2- с н
3
4-Етил-З -метилгептан
До цього ряду належать численні вуглеводні (органічні сполуки, що
складаються тільки з атомів вуглецю та водню) та речовини інших кла
сів, що є похідними вуглеводнів (розділ 2.1.).
2.
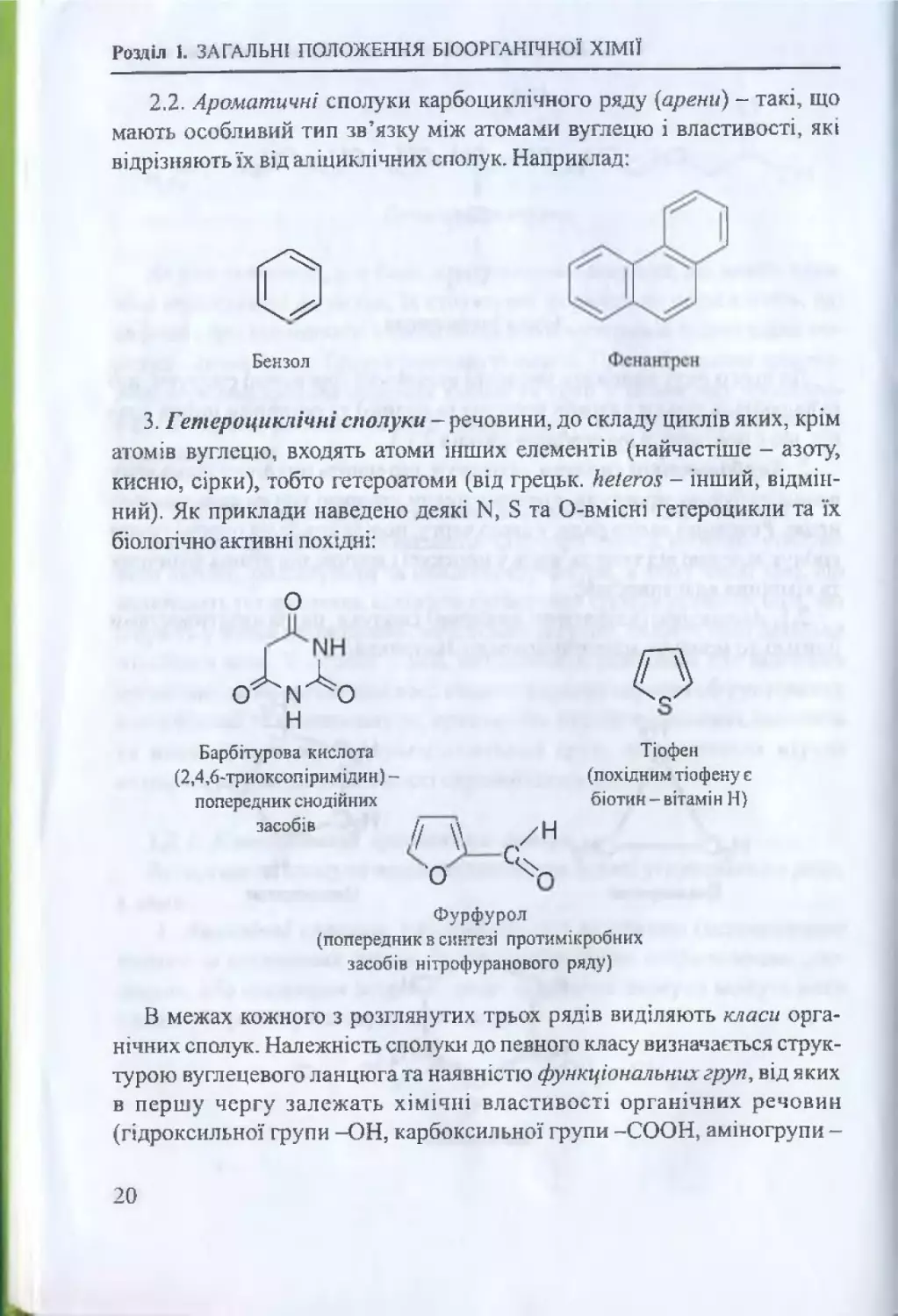
Карбоцикпічні сполуки - сполуки, що мають циклічну (замкнену
в кільце) будову молекули, причому цикли утворені тільки атомами вуг
лецю. Речовини цього ряду, у свою чергу, поділяються на окремі групи
сполук залежно від типу зв’язків у молекулі і відповідно різних фізичних
та хімічних властивостей:
2.1.
Аліциклічні (аліфатичні циклічні) сполуки, що за властивостями
близькі до молекул ациклічного ряду. Наприклад:
Н2
н2
с
Н2С
СН2
Ц иклопропан
н2с
Ц иклогексан
19
Розділ І. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
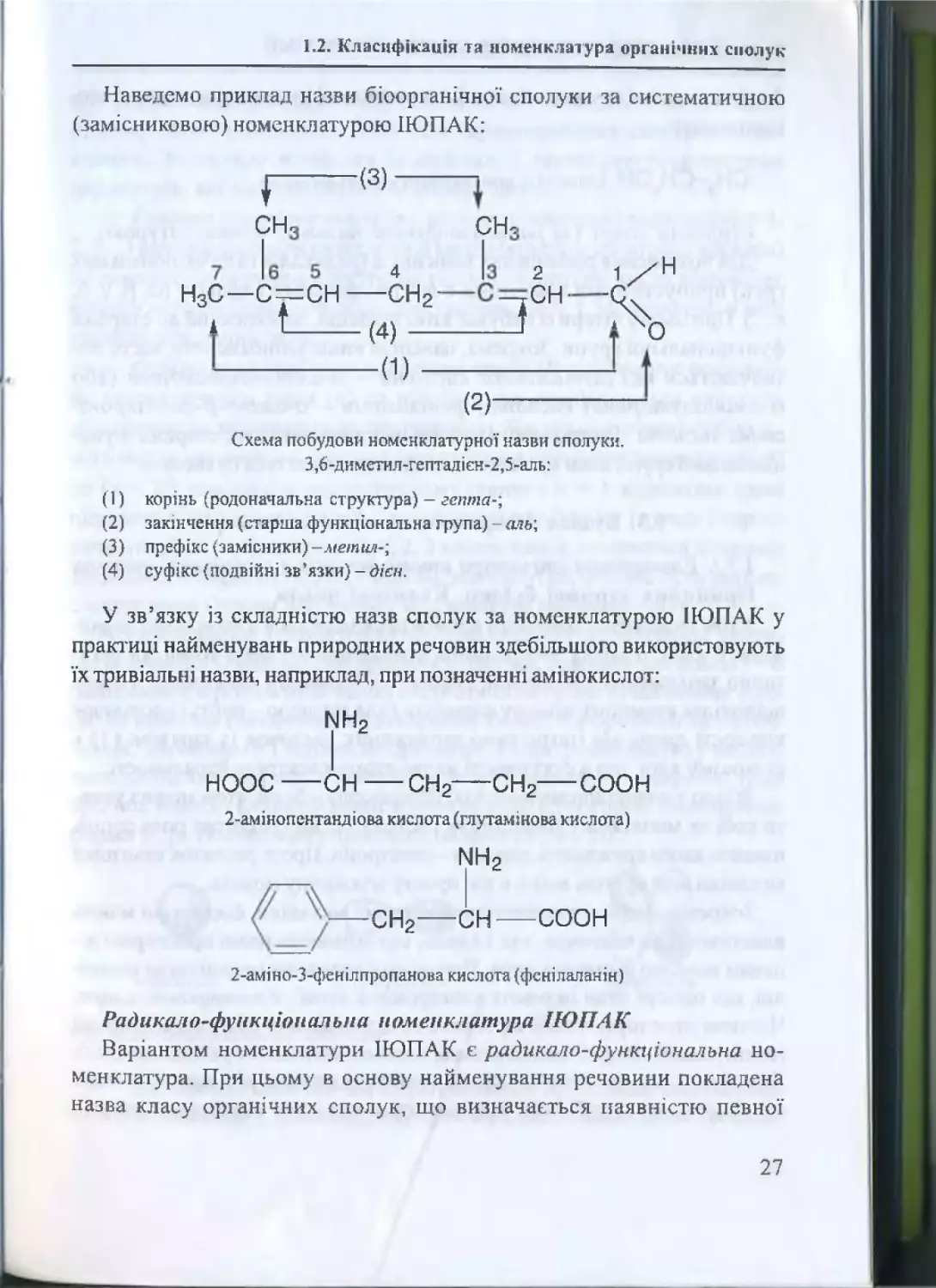
2.2.
Ароматичні сполуки карбоциклічного ряду (арени) - такі, що
мають особливий тип зв’язку між атомами вуглецю і властивості, які
відрізняють їх від аліциклічних сполук. Наприклад:
О
Бензол
3.
Гетероциклічні сполуки - речовини, до складу циклів яких, крім
атомів вуглецю, входять атоми інших елементів (найчастіше - азоту,
кисню, сірки), тобто гетероатоми (від грецьк. кеіегок - інший, відмін
ний). Як приклади наведено деякі N. Б та О-вмісні гетероцикли та їх
біологічно активні похідні:
О
О
О
А '!Ч'
.д О
Н
Тіофен
(похідним ті о фену є
біоти н -в іт ам ін Н)
Барбітурова кислота
(2,4,6-триоксопірим ідин) попередник снодійних
засобів
// V
•о
°С
Ф урф урол
(попередни к в синтезі протим ікробних
засоб ів нітроф уран ового ряду)
В межах кожного з розглянутих трьох рядів виділяють класи орга
нічних сполук. Належність сполуки до певного класу визначається струк
турою вуглецевого ланцюга та наявністю функціональних груп, від яких
в першу чергу залеж ать хімічні властивості органічних речовин
(гідроксильної групи -О Н , карбоксильної групи -СООН, аміногрупи20
1.2. К ласи ф ік ац ія та ном ен клатура ор ганічн их сполук
ІЧН, тощо). Найбільш поширені класи сполук в біоорганічній хімії та
функціональні групи, що їм відповідають, наведені в таблиці 1.1.
Таблиця 1.1.
Класи органічних сполук та функціональні групи,
що їм відповідають
К лас
Карбонові
кислоти
Спирти, феноли
Альдегіди,
кетони
Прості ефіри
Складні ефіри
Аміни
Аміди
Нітросполуки
Нітрили
Тіоли
(тіоснирти,
меркаптани)
Тіоефіри
(сульфіди)
Сульфокислоти
Г алогсшшохідні
вуглеводнів
Ф ункціональна група
Карбоксильна (карбокси-)
-СООН
Гідроксильна (гідрокси-,
0КСИ-)
-ОН
Карбонільна (оксо-)
>(С)=0
Алкоксильна
-011
Алкоксикарбонільна
-С О -О К
Амінна (аміно-)
-МН2( Ш К 2, N1*,)
Карбоксамідна (амідна)
-С О -Ш 2
Нітрогрупа
-N 0 ,
Нітрнльна (ціано-)
Загальна
формула класу
я -со о н
и -о н
Я (Г )С = 0
Г -О И
Г -С О -О ІІ
к -> щ 2
І1 -С 0 -Ш 2
я -и о 2
Тіольна (меркапто-)
-ЗН
Я-ЗН
Алкілтіольна (сульфідна)
-Б Я
Сульфонова (сульфо-)
- 3 0 3Н
Г алогенова
-СІ, -Вг, -Б , - І
Г -Б Я
Я-БОзН
а-Н аї
21
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
У складі молекули органічної речовини може міститися декілька
функціональних груп. Якщо ці групи однакові (дві чи три гідроксильні
або галогенові групи тощо), така сполука є п оліфункц іопально ю. Ор
ганічні сполуки, що містять у молекулі декілька різних функціональних
груп, називаються гетеро фуикц іопал ьним и. До гетерофупкціональних сполук належать, зокрема, амінокислоти, окси- та оксокислоти, ву
глеводи, аміноспиртита більшість біологічно активних гетероциклів при
родного походження. В цьому випадку окремі функціональні групи мо
жуть бути відповідальними за різні прояви фізіологічної (біохімічної або
фармакологічної) дії молекули.
1.2.2. Номенклатура органічних сполук
Номенклатура - це сукупність правил, згідно з якими будують од
нозначну назву індивідуальної органічної сполуки. Розрізняють триві
альну та систематичну номенклатури, кожна з яких має в наш час
певні особливості та межі застосування.
1. Тривіальна номенклатура. Під тривіальною номенклатурою
розуміють фактично сукупність, перелік назв органічних сполук, що скла
лися історично та не відбивають, як правило, його хімічної будови. Триві
альні назви лежать в основі коренів похідних перших чотирьох членів
гомологічного ряду вуглеводнів (метан, етан, пропан, бутан) і найчасті
ше використовуються в найменуваннях природних сполук - амінокис
лот, вуглеводів (цукрів), жирних кислот (олеїнова кислота, пальмітинова
кислота), інтермедіатів обміну речовин (яблучна кислота, янтарна кис
лота тощо).
Найбільш уживаними тривіальні назви є в фармакології та фармації,
де вони позначають лікарські засоби (особливо рослинного походженнятак звані алкалоїди), що мають складні найменування за систематич
ною номенклатурою. Наприклад: атропін (тропіновий ефір d, 1-тропо вої
кислоти), папаверин (6,7-диметокси-1-(3 ',4'-диметоксибснзил)-ізохінолін), новокаїн (гідрохлорид ЬШ -діетиламіноетилового ефіру параамінобензойної кислоти).
2. Систематична номенклатура (міжнародна номенклатура
ІЮ П А К). Сукупність понять, термінів та правил, що застосовуються в
систематичній номенклатурі, визначена Міжнародною спілкою теоретич
ної та прикладної хімії (International Union of Pure and Applied Chemie IUPAC, англ., або “ІЮПАК” ).
22
1.2. К ласи ф ік ація та ном ен клатура ор ганічн их сполук
Назви органічних сполук за систематичною номенклатурою одно
значно визначають їх молекулярну будову, тобто площинну та просторо
ву структуру.
Н ом енклат урним и понят т ями ІЮ ПАК є : органічний радикал
(вуглеводневий радикал), функціональна (характеристична) група,
замісник, локант, родоначальна структура, головний ланцюг. Роз
глянемо ці терміни докладніше.
О рганічний р а д и ка л (вуглеводневий р а д и к а л) - залишок вуглевод
ню, з якого умовно видалено один або декілька атомів водню. Залежно
від кількості вільних валентностей, що утворюються при атомі вуглецю,
розрізняють одно-, дво- і тривалентні радикали.
О дновалент ні радикали називаю ть також алкільним и групами
(алкільними радикалами) і позначають літерою Я (II-). їх назви утво
рюються шляхом заміни закінчення -ан у відповідному насиченому ву
глеводні (алкані) на -ил (-іл), або додавання -ил (-іл) до закінчення -єн
ненасиченош вуглеводню (алкену). Іноді використовуються тривіальні
назви радикалів (вініл, аліл, феніл тощо). Наприклад:
СН 3- (метил)-,
СН з - СН2 - (етил);
СН3 - СН2- СН г -(пропіл);
(СН3)2С Н - СН2 — (ізобутил);
СН г = СН - (етеніл; вініл)
СН2 = СН - СН2- (пропен-2-іл; аліл)
С6Н5 - (феніл);
С 6Н5— СН2 - (бензил).
Двовалентні радикали, що мають вільні валентності при різних ато
мах вуглецю (нарізних кінцях вуглецю) мають закінчення -илен (-імен);
такі, що мають дві валентності при одному атомі вуглецю -иліден
(-іліден), наприклад:
/
- СН 2- (метилен);
- СН3- СН^ (еМиліден);
- СН 2- СН2- (етилен);
/
С 6Н5- СН^
(бензиліден)
Ф у н к ц іо н а л ь н а (х а р а к т е р и с т и ч н а ) група - замісник (замісни
ки), що (на відміну від органічних радикалів) містить невуглецевІ атоми.
Існує також поняття старшинства функціональних груп. Старшою
(відносно іншої, молоШиоїгруищ) є функціональна група, що розташова
23
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
на в переліку таблиці 1.2. вище. Функціональну групу, що сполучена з
родоначальною структурою (див. нижче) називають характеристич
ною групою. У найменуванні органічної сполуки функціональні (харак
теристичні) групи стають префіксами або закінченнями. Деякі функціо
нальні групи (хлор-, бром-, нітро- тощо) позначають тільки у префіксі.
Таблиця 1.2.
Функціональні (характеристичні) групи, що вказуються в
префіксах або закінченнях органічних сполук (перелічені в
порядку зменшення старшинства)
Функціональна група
-С О О Н
-в О д Н
- (С)= N
-н о о
/(С )= о
-о н
-в н
-х н ,
Назва у префіксі
КарбоксиСульфо-
ОксоОксоГідрокси- (окси-)
МеркаптоАміно-
Назва в закінченні
-ова кислота
-сульфонова кислота
-нітрил
-аль
-он
-ол
-тіол
-амін
З ам існик- поняття, що охоплює як функціональні групи, так і вугле
водневі радикали.
Родоначальна структура - хімічна структура, що є основою на
йменування даної органічної речовини. Родоначальною структурою для
ациклічних сполук є їх головний ланцюг, для карбо- та гетероциклічних
сполук - відповідний цикл. В якості головного ланцюга в ациклічних
вуглеводнях та їх похідних обирають зазвичай найдовший вуглецевий
ланцюг; в разі наявності у вуглеводні декількох кратних зв’язків або
характеристичних (функціональних) груп головним стає ланцюг, що
містить найбільшу їх кількість.
Замісникова номенклатура ІІОПАК
Раціональним базисом систематичної номенклатури ІЮПАК є залисниковий принцип, згідно з яким будь-яка органічна сполука розгляда
ється як похідне вуглеводню (речовини, що складається тільки з атомів
вуглецю і водню), в молекулі якого один або декілька водневих атомів
заміщені на інші атоми або групи атомів (замісники).
24
1.2. К ласи ф ік ація га ном ен клатура ор ганічн их сполук
Формування назви органічної сполуки ациклічного ряду за зам ісма
ковою номенклатурою ІЮ ПАК здійснюють згідно з такою послідовні
стю логічних операцій:
1) обирають родоначальну структуру (головний ланцю г) за кри
теріями, розглянутими вище;
2) формулюють назву родоначальної структури за такими правилами:
- за корінь слова беруть назву насиченого вуглеводню з такою кіль
кістю вуглецевих атомів, яку має обраний головний ланцюг;
- найменування старшої функціональної групи (якщо вона є) стає
суфіксом або (закінченням) назви родоначальної структури, наприклад*:
старша функціональна група
корінь
\
і
▼
н 3с - с н 2- о н
Етан-ол
стариш функціональна група
корінь
1
н 3с - с н 2- с о о н
П роп ан-ова кислота
- наявність кратних зв ’язків позначають суфіксами -єн (-єн) або
-ин -(ін )-я п я подвійних або потрійних зв’язків відповідно;
3)
проводять нумерацію атомів вуглецю головного ланцюга, вико
ристовуючи такі критерії:
а) найменший номер набуває старша функціональна група; в разі
її відсутності враховують критерії за пп. б та в);
б) найменший номер отримує той атом вуглецю головного лан
цюга, до якого найближче розташований замісник або подвій
ний зв’язок;
в) при наявності декількох замісників їх нумерацію проводять з
урахуванням алфавітного порядку їх назв;
* у прикладах найм енуван ня зак ін чен ь подано через дефіс
25
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
4) назви зам ісників - вуглеводневих радикалів стають префікса
ми в назві сполуки; назви замісників - функціональних груп {крім стар
шої функціональної групи!) стають префіксами або суфіксами (чи за
кінченнями)'. номери (локанти) відповідних замісників позначаються
перед їх назвами (префіксами), наприклад:
т2
сн3
4
3
Н зС -С Н
12
— сн
1
— соон
БН
2-амін о-З -мсркапто-З -метилбутано ва кислота
Використання функціональної групи в якості префікса або суфікса
можна проілюструвати таким прикладом:
ОН-група як префікс (гідрокси-):
ОН
4
3
Н3С — С Н
2
1
СН2------ СООН
3-гідроксиб утанова кислота
ОН-група як суфікс (-ол):
З
н 3с —
2
с н 2—
1
с н — ОН
П р о п ан о л -1
5) полож ення кратного з в ’язку позначають таким чином: цифра,
що ставиться після відповідного суфікса (-єн (-єн) або -ин (-ін)), вказує
на номер першого з двох вуглецевих атомів, які утворюють кратний зв ’язок, наприклад:
26
5
4
3
2
1
СН 3- СН 2- СН 2- С Н = СН2
Пентен-і
5
4
3
2
1
СН, - СН . - СН = СН - СН_
Пентен-2
1.2. К ласи ф ік ац ія та ном ен к латура ор ганічн их сполук
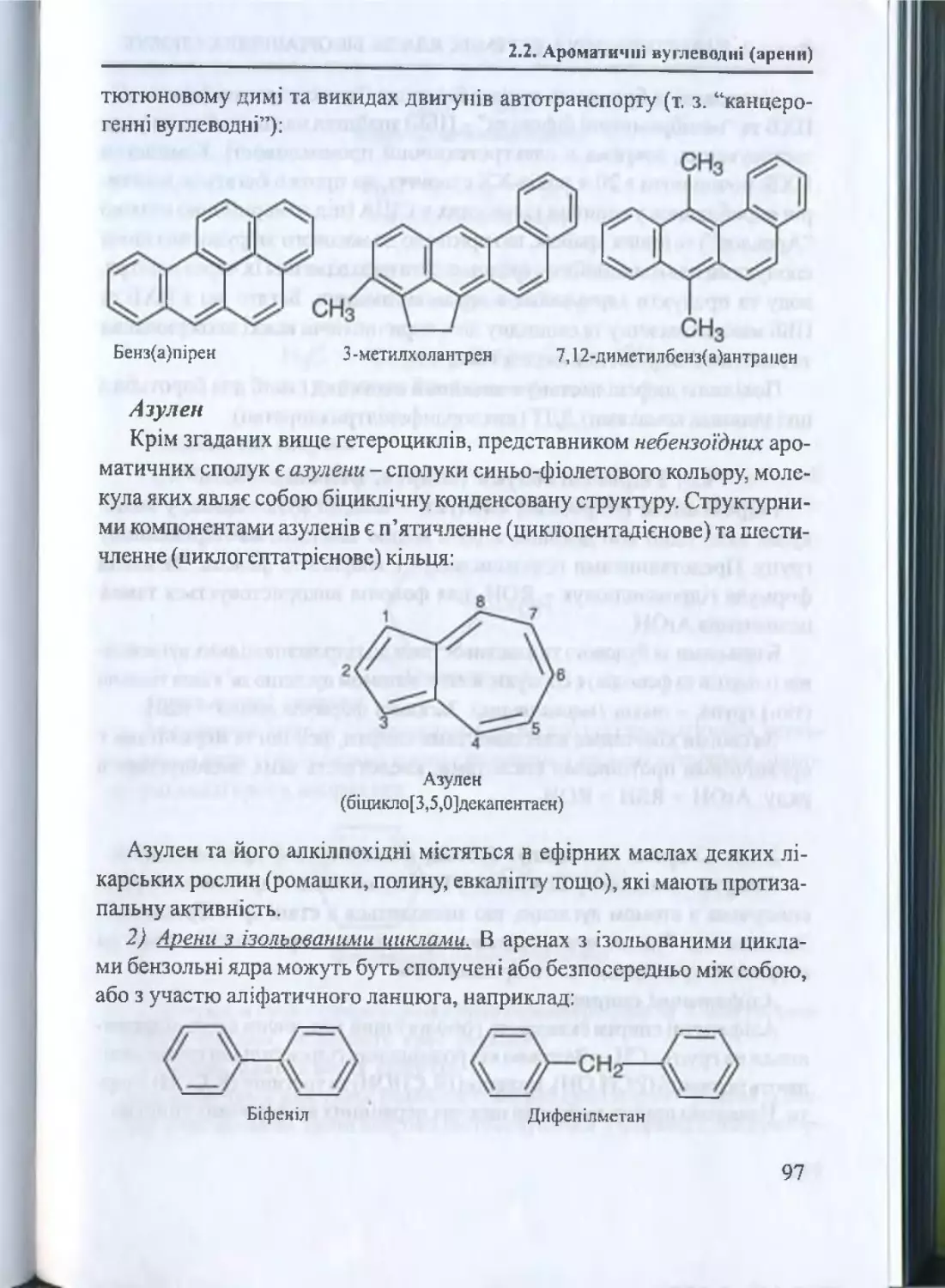
Наведемо приклад назви біоорганічної сполуки за систематичною
(замісниковою) номенклатурою ІЮПАК:
Г
-(3)
сн.
сн.
4
Н 3С — с = = с н
і
{_____
-с н 2(4) -
2
з
сн-
у *
%о
- ( 1)
( 2 )------------------------Схема побудови ном енклатурної назви сполуки.
3,6 -диметил-гептад існ-2,5 -аль:
гепта-;
(1)
корінь (родоначальна структура) -
(2 )
закінчення {старш а ф ун кц іон ал ьн а група) преф ікс {зам існи ки ) -метил-;
суф ікс (подвійні з в ’язки) -дієн.
(3)
(4)
аль;
У зв’язку із складністю назв сполук за номенклатурою ІЮПАК у
практиці найменувань природних речовин здебільшого використовують
їх тривіальні назви, наприклад, при позначенні амінокислот:
!МН2
НООС — С Н ------ СН2— СН2-----СООН
2-амінопентандіова кислота (глутам і нова кислота)
т 2
с н 2— с н — с о о н
2-ам іно-З-ф енілпропанова кислота {фенілаланін)
Рад икало-функціонал ьп а номенклатура ІЮ ПАК
Варіантом номенклатури ІЮПАК є радикало-функціональна но
менклатура. При цьому в основу найменування речовини покладена
назва класу органічних сполук, що визначається наявністю певної
27
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
функціональної групи (таблиця 1 .1 ) - спирт, ефір, альдегід, кетон тощо.
Наприклад:
СН3-С Н 2ОН
Е танол (за
замісниковою ном енклатурою )
Етиловий спирт (за радикало-ф упкціональною номенклатурою)
Для позначення розміщення замісників (радикалів та функціональних
груп) припустимими локантами є літери грецького алфавіту (ос, [3, у, 5,
є ...). При цьому літери а набуває атом вуглецю, найближчий до старшої
функціональної групи. Зокрема, наведені вище амінокислоти часто по
значаю ться як: гл ут а м ін о ва ки сло т а — а -а м ін о п ен т а н д іо ва (або
СС-аміноглутарова) кислот а; ф енілаланін - а - ам шо-(5-фе н іл про пі онова кислота. Дистальний (найбільш віддалений) від старшої функ
ціональної групи атом вуглецю звичайно позначається буквою со.
1.3. Будова біоорганічних сполук. Ізомерія
1.3.1. Електронна структура атома вуглецю в органічних сполуках
Принципи атомної будови. Квантові числа
Атом будь-якого хімічного елемента складається з позитивно заряд
женого ядра, в якому зосереджена практично вся маса атома, та нега
тивно заряджених елект ронів (заряд -1). Кількість електронів в атомі
відповідає атомному номеру елемента (для вуглецю - шість) і дорівнює
кількості прот онів (позитивно заряджених часточок із зарядом +1) в
атомному ядрі, що в сукупності надає атому електронейтральності.
Згідно з планетарною моделлю Резерфорда —Бора, атом можна уяви
ти собі як мініатюрну планетарну систему, де ядро відіграє роль сонця,
навколо якого кружляють планети—електрони. Проте розвиток квантової
механіки вніс сутгеві зміни в цю просту механічну модель.
Зокрема, згідно з уявленнями квантової механіки, електрони мають
властивості як часточок, так і хвиль, що займають певні просторові ді
лянки навколо атомного ядра. При цьому квантово-механічною модел
лю, що описує стан окремих електронів в атомі, є елект ронна хмара.
Частина простору, в якій вірогідність знаходження певного електрона
(електронної хмари) максимальна, зветься атомною орбіт аллю (АО).
Електрони, що розміщуються на певних відстанях від атомного ядра
(посідають певні орбіталі), розрізняються не тільки значеннями енергії,
28
1.3. Будова біоорган іч н и х сп олук . Ізомерія
а й формами орбіталей, в яких розташована електронна хмара, та їх
просторовою орієнтацією. Тому для характеристики електронів, що
входять до складу атома, та їх орбіталей застосовується система
параметрів, які дістали назву квантових чисел:
- Гоповиє квантове число (п), що має цілочислові значення (п = 1,
2 ,3 ...) визначає розміри електронної хмари (відстань електрона від ядра)
та енергет ичний рівень електрона; головне квантове число відповідає
номеру періода Періодичної системи елементів Д. І. іМендслєєва, в якій
знаходиться даний атом.
- Орбітальне (побічне) квантове число (/) відображує розділен
ня енергетичного рівня (п) на підрівні, що розрізняються енергією
електронів, які знаходяться на цих підрівнях. Чисельні значення орбіталь
ного числа для певного енергетичного рівня п складають величини від 0
до (п - І); наприклад, енергетичному рівню з п = 1 відповідає один
підрівень {/ = 0), рівню з п = 2 - два підрівні з / = 0 та / = І тощо. Енерге
тичні підрівні (орбіталі) з /= 0, 1,2,3 мають також позначення літерами
латинського алфавіту: я, р, сі, ї відповідно. Орбіталі можуть бути зайняті
електронами (одним або двома) або вакантні [вакантні АО).
Атомні орбіталі з певним орбітальним числом (та електронні хмари,
що їх займають) мають різні просторові форми. Зокрема, значенню І = 0
(найнижча енергія на будь-якому енергетичному рівні п) відповідає сфе
рична орбіталь (та сферична електронна хмара) - з-орбіт алі та $-електропи; значенню /= 1 відповідає орбіталь (та електронна хмара) у вигляді
двох еліпсоїдів форми “гантелей” - р-орб іт алі та р-елект рони, інші
орбіталі мають складніші геометричні форми. Просторові геометричні
форми ї-, р- та сі-атомних орбіталей подано на рис. 1.2.
О
5-орбіталь
о з
р-орбіталь
(і-орбіталь
Рис. 1 .2 . Форми атомних орбіталей
- Магнітне квантове число (ш) визначає орієнтацію еліпсоїдних
електронпих хмар у тривимірному просторі. Відповідно до цього, еліп
29
Розділ t. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
соїдні р-слектронні хмари, що відіграють значну роль у валентних зв’яз
ках атома вуглецю, підрозділяються на енергетично еквівалентні ps, ру
та рг-електрони.
- Спінове квантове число (s) характеризує обертання електрона на
вколо власної осі за або проти годинникової стрілки, що позначається спінови
ми числами +1/2 та - 1/2 або протилежно спрямованими стрілками (Î, -і).
Розподіл електронів в атомі. Електронні формули
Електронна конфігурація будь-якого атома відповідає певним законо
мірностям, які є наслідком правил розподілу електронів між енергетич
ними рівнями та заповнення ними атомних орбіталей. Основними з цих
правил є:
1) напрямок заповнення - АО заповнюються електронами в поряд
ку збільшення енергетичних рівнів, тобто спочатку заповнюються орбіталі з найменшою енергією (“нижчі’ орбіталі, починаючи від ядра); в
межах певного енергетичного рівня спочатку заповнюються електро
нами енергетично нижчі підрівні (спочатку s- а потім р-, d-орбіталі і т.д.);
2) кількість електронів (N) на одному енергетичному рівні дорівнює
подвоєному квадрату номера рівня п, тобто N = 2 п2; відповідно до цього
правила на першому (найближчому до ядра) енергетичному рівні (п = і)
може знаходитися не більше двох електронів (один —в атомі водню та
два - у всіх інших елементів), на другому (п = 2) - від одного до восьми
електронів (чотири —у вуглецю, п’ять —у азоту, шість —у кисню),
3) в межах кожного підрівня (0 існують додаткові підрівні (орбіталі),
що мають назву енергет ичних , або квантових, комірок', кількість кван
тових комірок для підрівня / = 0 (в якому розміщені s-електрони) дорів
нює одиниці, для підрівня / = 1 (в якому розміщені р-електрони) дорівнює
трьом. У кожній квантовій комірці (на орбіталі) може знаходитися один
електрон (неспарений елект рон ) або максимально два електрони з
протилежними спінами ( спарені елект рони ):
Виходячи з наведеного, розподіл електронів в атомі (його елект рон
н у конф ігурацію ) можна подати у вигляді електронних формул або
квантових комірок, наприклад:
- елект ронна конфігурація атома водню:
ЗО
1.3. Будові) біоорган іч н и х сполук . Ізомерія
15і
або
"=10
Тобто 1-й енергетичний рівень атома водню має один підрівень, який
складається з однієї квантової комірки з одним електроном.
- електронна конфігурація атома вуглецю:
1з32з22р2 (1522э32рх2ру) або
р-орбіталі
з-орбіталі
п=2
Ті
П=І
Ті
т
т
Тобто в атомі вуглецю: 1-й енергетичний рівень (п = 1) має один
підрівень (/ = 0) з двома й-електронами (з протилежними спінами); 2-й
енергетичний рівень має два підрівні (/ = 0 та /= 1, на яких розміщені два
б- та два р-електрони відповідно); два електрони р-підрівня (р-орбіталі)
мають різну просторову орієнтацію (рхта р ) і розташовані в різних кван
тових комірках.
1.3.2.
Природа хімічних з в ’язків в органічних сполуках
Здатність атомів приєднувати певну кількість інших атомів або атом
них груп з утворенням хімічних зв’язків називається валент ніст ю ато
ма. В молекулах органічних сполук атом вуглецю завжди чотиривалентний
і може утворювати з сусідніми атомами одинарні та кратні (подвійні,
потрійні) хімічні зв’язки.
Основними типами хімічних зв’язків, що сполучають окремі атоми
(вуглецю та інших елементів) в молекулах органічних сполук, є кова
лентні, донорно-акцепторпі (зокрема, семіполярні), іонні (сольові)
та водневі зв’язки.
1.
Ковалентні зв’язки. Валентні стани атома вуглецю
Ковалентні зв’язки - це хімічні зв’язки, які утворюються шляхом
усуспільнення електронів (утворення електронних пар) та перекривання
З!
Р о зд іл 1, ЗА ГА Л Ь Н І ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
__________
відповідних АО атомів, що формують молекулярні орбіталі (МО). В
утворенні ковалентних зв’язків беруть участь нест реш електрони зов
нішнього е н е р г етичного рівня, зокрема: у атома водню - його зовнішній
(і єдиний) з-електрон; у атомів другого періоду, що найчастіше входя
до складу органічних сполук (вуглецю, кисню, азоту) - р-електрони 2-го
ЄНЄНапрИиклад, утворення молекули водню (Н2) здійснюється шляхом
усуспільнення двох неспарених електронів 15 від кожного атома Н (по
значаються крапкою поряд із символом атома) з утворенням е л е р о н
ної пари, що розміщується на спільній молекулярній орбіталі (МО).
Електронна пара позначається двома крапками, що еквівалентно позна
ченню хімічного зв’язку рискою:
Н* + Н»-> Н *• Н (Н - Н)
Валентні ста и н атома вуглецю
Як зазначено вище, здатність атомів утворювати ковалентні зв язки, тоото їх валентність, визначається числом неспарених електронів на зовнішньому
енергетичному рівні. Атом вуглецю має на зовнішній орбіталі (р-орбітал.)
два неспарених електрони, однак його валентність у складі органічних спо
лук дорівнює чотирьом. Електронна конфігурація а т о м а вуглецю має ви
гляд 1522э22р 2р - ц е основний, незбуджений стан атома. За умов зоудження яке має місце при взаємодії вуглецю з атомами інших елементів, від увається перехід одного електрона з 25- на 2р-орб,таль з утворенням нового
розподілу електронів на атомних орбіталях збудженого атома:
и 22$22 р 2 р у-+ 1 # 2 5 2 р 2 р 2 р г
Нова електронна конфігурація атома вуглецю має чотири неспаре
них електрони, які, проте, різняться за енер
властивостями (один 28- та три 2р-електрони - 2рх, 2рута 2 р > тоді
у
складі більшості вуглеводнів С -С та С -Н зв’язки є однаковими (напри
клад, у молекулі метану СН4). Це теоретичне протиріччя було з ясоване завдяки запропонованому Лайнусом Полінгом (Ь. Раиііщ ) уявленню
Г'Р°Під^гібридизацією атомних орбіталей розуміють змішування (комбі
націю) і вирівнювання за формою та енергією одного 28-електрона та
32
1.3. Будова біоорган іч н и х сполук . Ізомерія
кількох (від одного до трьох) неспарених 2р-електронів, що відбуваєть
ся при утворенні атомом вуглецю хімічних ковалентних зв’язків. Залеж
но від кількості 2р-електронів, які беруть участь у гібридизації орбіталей, розрізняють три типи гібридизації (зр3-, эр2- та эр-) - рис. 1.3 - і
відповідно три валентні стани атома вуглецю.
О
І-----------------------------
1
ї
эр2
Бр
Р ис. 1 .3 . Типи гібридизації валентних орбіталей (п = 2) атома вуглецю
Перший валентний стан атома вуглецю (;ір3-гібридизсшія) - утво
рюється шляхом комбінації чотирьох атомних орбіталей збудженого
атома вуглецю, на яких знаходяться неспарені електрони другого
(п = 2) енергетичного рівня - однієї 2э- та трьох 2р-орбіталєй. Результа
том цього процесу є формування чотирьох рівноцінних гібридних
єр3-орбіталей, що структурно являють собою асиметричну об’ємну
вісімку. П росторове розташ ування зр^-орбіталей в атомі вуглецю
відповідає осям правильного тетраедра, що спрямовані від його центра
до вершин під кутами 109,5°:
33
Р озділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
О »
Гібридна Бр’-орбіталь
П росторове розм іщ ення вр3-орбіталей в атомі вуглецю
Другий валентний стан атома вуглеию Ґ.чр2-гібридизаиія) - ви
никає в результаті комбінації однієї 2з- та двох 2р-атомних орбіталей ( р ,
ру). При цьому формуються три асиметричні Бр2-гібриди і орбіталі, що
розміщені в одній площині під кутами 120° одна до одної. Негібридизована 2р-орбіталь (рг) розміщується в площині, що є перпендикулярною
до площини зр2-орбіталей.
Третій валентний стан атома вуглеию Ґяр-гібридизашя) —є ре
зультатом комбінації однієї 2в- та однієї 2р- орбіталей (рх). При цьому
формуються дві гібридні Бр-орбіталі, розташовані лінійно, тобто під ку
том 180° одна до одної. Негібридизовані 2р-орбіталі (рута рг) розміщу
ються в площинах, що є перпендикулярними до площини вр-орбіталі.
Просторове розміщення атомних орбіталей вуглецю в станах яр2 та
зр-гібридизації подано на рис. 1.4.
Рис. 1.4. Другий та третій валентний стани атома вуглецю: зр г- та - зр-гібридні
орбіталі з негібридизовзними р-електронами, розміщеними в перпен
дикулярних площинах
34
1.3. Будова біоорган іч н и х сполук . Ізомерія
Ковалентні (Т- та р -з в ’язки
Тип усуспільнення двох неспарених електронів при утворенні ковалент
них зв’язків залежить від валентного стану атомів вуглецю в органічних
сполуках, що взаємодіють, та характеру перекривання відповідних атом
них орбіталей. Різновидами ковалентних зв’язків, що утворюються при
цьому, є ст-зв’язки та л-зв’язки.
а-зв'язки. Цей тип ковалентних зв ’язків утворюється в результаті
перекривання орбіталей атомів, які взаємодіють (С, Н, О та ін.), впро
довж осі, що зв ’язує ці два атоми. Інакше кажучи, перекривання
атомних орбіталей здійсню ється таким чином, що максим альна
електронна густина зосередж ується впродовж ум овної лінії, яка
проходить через центри двох атомів - т. з. “осьове” перекривання.
ст-зв’язки утворюю ться при осьовій взаємодії двох к-орбіталей,
Б-орбіталі з гібридизованою орбіталлю (зр3, єр2, зр) або взаємодії гібрид
них орбіталей різних типів між собою - рис. 1.5.
(1 )
( 2)
( 3)
Рис. 1 .5 . Утворення сг-зв’язків при взаємодії різних видів атомних орбіталей:
(1) Б-Б-взаємодія (утворення зв ’язків Н -Н ); (2) Б-БрЗ-взаємодія (утво
рення зв ’язків С -Н ); (3) Бр^-єр3-взаємодія (утворення зв ’язків С -С)
п-зв 'язки - ковалентні зв’язки, які утворюються при бічному пере
криванні атомних орбіталей. Тобто, в цьому випадку ось перекривання
(і зосередження максимуму електронної густини) є перпендикулярною
власним осям двох орбіталей, що усуспільнюються, я-зв’язки виника
ють при бічній взаємодії негібридизованих атомних орбіталей р-електронів - рута р7, які присутні в атомах вуглецю, що знаходяться в другому (зр2) та третьому (зр) валентному станах - рис. 1.6. Із точки зору
просторової форми я-зв’язок являє собою дві хмари максимальної елек
тронної щільності, що локалізуються по обидва боки від осі о-зв’язку
(див. розділи 2.1.2, 2.1.3.).
Розглянуті типи гібридизації та ковалентних зв ’язків беруть участь
в утворенні зв’язків атома вуглецю різної кратності. Одинарні зв’язки
вуглецю утворюються за рахунок а -зв ’язку (молекули вуглеводнів ад35
Р озділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
канів та їх похідних); подвійні зв ’язки складаються з одного а- та одного
п- (алкени та їх похідні); потрійні зв’язки - з одного ст- та двох я-зв’язків
(алкіни та їх похідні).
Р и с .1 .6. я -зв ’я з ш при атомах вуглецю, що знаходяться в станах:
(1) зрг-тібридизації (один р -зв’язок, спрямований впродовж осі г) та
(2) зр-гібридизації (два р-зв'язки, спрямовані впродовж осей г та у)
Неполярні та полярні ковалентні з в ’язки
В разі формування ковалентного зв’язку між однаковими атомами
або атомами з однаковою електронегативністю, зв’язок, що утворився,
є неполярним - усуспільнена електронна пара розміщується симетрично
стосовно ядер обох атомів. Неполярними є ковалентні зв’язки в молекулах
вуглеводнів між атомами вуглецю, що, у свою чергу, сполучені з
близькими за хімічною будовою радикалами.
Полярні ковалентні зв’язки утворюються між атомами з різною електронегативністю, в яких спільна електронна пара зсунута в бік більш
електронегативного елемента. В результаті такого розподілу електрон
ної щільності на відповідних атомах виникають часткові заряди (5+та 5').
Напрямок поляризації ковалентних зв’язків позначають стрілкою:
н 3с — с н 3
Н еполярни й
к овалентний з в ’язок
+
—
н 3с —►он
П олярний
ковалентний з в ’язок
Детальніше розгляд електронних ефектів в органічних молекулах буде
проведений у розділі 1.3.3.
36
1.3. Будова біоорган іч н и х сполук. Ізомерія
2. Донорно-акцепторні зв ’язки
Ковалентні зв’язки, що розглянуті, формуються шляхом перекриван
ня двох однослектронних АО та усуспільнення електронів, які належали
різним атомам. Разом з тим хімічний зв ’язок може утворюватися за
рахунок двох електронів (електронної пари, що розміщена на одній зай
нятій АО) тільки одного атома. При цьому виникає нова, спільна для
двох атомів молекулярна орбіталь (МО) і такий зв’язок називається дондрно-акцепт орним .
Найчастіше донорно-акцепторний зв’язок утворюється за рахунок
неподіленої пари електронів атома азоту 0^), що входить як гетероатом
до складу багатьох біоорганічних сполук. Електронна конфігурація ато
ма азоту має вигляд: 1э22з22р3, тобто на його зовнішній електронній обо
лонці (енергетичний рівень п = 2) міститься п ’ять електронів, Із цих п ’яти
валентних електронів в утворенні ковалентних зв’язків звичайо беруть
участь три неспарених електрони р-орбіталі (рх, ру, рг), що пояснює
тривалентність азоту, а два електрони к-орбіталі існують у вигляді вільної
неподіленої пари:
В
Р •N•
^
Н----- |\Г.
=
Я
При взаємодії сполук азоту з атомом (найчастіше у вигляді іона),
який має нестачу електронів (вакантну орбіталь), неподілена елект
ронна пара атома азоту усуспільнюється, тобто утворює спільну МО.
яка і являє собою донорно-акцепторний зв’язок.
В
и— гл
В
+ н+с г — 1
► я — г/: Н СГ =
СІ
Донорно-акцепторні зв ’язки є ковалентними зв ’язками з особливим
механізмом утворення спільної електронної пари.
37
Розділ І. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
Семіполярний (координаційний) зв ’язок
Різновидом донорно-акцепторних зв ’язків є семіполярний зв'язок.
Цей тип зв ’язку утворюється при взаємодії атомів азоту та кисню і на
явний в органічних нітратах К-Ж>,. Електронна будова кисню (1э22822р4)
характеризується наявністю на зовнішній електронній оболонці (п=2)
секстету електронів, два з яких (нсспарені р-електрони) визначають
двовалентність цього елемента. Разом з тим, атом кисню є електроне
гативним елементом і схильний до приєднання двох додаткових елект
ронів (завершуючи заповнення зовнішньої 8-електронної оболонки до
стану, характерного для інертного газу неону), що і відбувається за ра
хунок неподіленої пари електронів азоту:
И
1ГГ-*
+ О*.
—
і1
Я-----і Л х '
==
Я3Ы— О
Я
Зв’язок між атомами азоту й кисню має властивості ковалентного,
оскільки він сформований усуспільненою парою електронів. Разом з тим,
при утворенні координаційного зв’язку атом азоту (донор електронів)
набуває позитивного заряду, а атом кисню (акцептор електронів) - нега
тивного, внаслідок чого між цими двома атомами виникає додаткова
іонна взаємодія, тобто новий зв’язок є частково полярним (“сем іполяр
ним”), що може схематично позначатися у вигляді таких структур, що
передають будову нітрогрупи:
Я
+
N =0
о-
=
^
Я— N
V
=
я— г
&
\
3.
Іонні (слектровалентні, сольові) зв’язки - зв’язки, що утво
рюються між протилежно зарядженими іонами (катіонами та аніонами)
і найбільш характерні для солей неорганічних сполук. В біоорганічній
хімії іонні зв ’язки утворюються в розчинах карбонових кислот (між
карбоксилат-іоном та протоном), між аміно- та карбоксильними група
ми в молекулах амінокислот тощо. Наявність сольових зв’язків між функ
ціональними групами бічних радикалів амінокислотних залишків є од38
1.3. Куди на б іо о р г а н іч н ії* с п о л у к . Ізо м е р ія
ним із факторів стабілізації вищих рівнів структурної організації білків та
пептидів. Іонні зв’язки беруть також участь у створенні вторинної струк
тури нуклеїнових кислот та взаємодії з клітинними біополімерами різних
фізіологічно активних сполук.
4. Водневі зв’язки
Водневі зв ’язки виникають між двома електронегативними атома
ми за рахунок рухомого атома водню, що ковалентно сполучений з од
ним із електронегативних атомів (азоту, кисню, сірки). Найчастіше вод
неві зв’язки утворюються між воднем, що входить до складу груп =КН,
-О Н , -8Н , та сусіднім атомом кисню. В утворенні цих зв’язків бере участь
неподілена пара електронів атома, з яким взаємодіє водень, тому фактич
но водневий зв’язок є різновидом донорно-акцепторного зв’язку. Прикла
ди угворення водневих зв’язків (позначаються трьома крапками):
^ ІМ — н - - - : о = С ^
— О — Н ■■• :
Водневі зв ’язки належать до типу слабких з в ’я зків, що надзвичайно
розповсюджені в біологічних системах, беручи участь у міжмолекуляр
ній взаємодії біомолекул та ФАС різних класів, формуванні просторової
будови біомакромолекул, організації надмолекулярних клітинних струк
тур за участю білків та нуклеїнових кислот.
1.3.3. Взаємний вплив атомів в органічних сполуках
Реакційна здатність органічних сполук залежить від особливостей
розподілу електричного заряду (електронної густини) впродовж атом
ного ланцюга (статична поляризація) та змін цього розподілу в умо
вах впливу електричного поля інших молекул - реагентів (динамічна
поляризація). У свою чергу, нерівномірність електронних хмар, що при
таманна молекулярній структурі, пов’язана з різницею в електронегативиості атомів, які складають молекулу.
Електронегативність - здатність окремих атомів в молекулі ор
ганічної речовини притягувати до себе електрони. Кількісне вираження
електронегативності було запропоноване Л. Полінгом. Згідно із шка
лою електронегативності за Л. ГІолінгом, елементи, які входять до
складу органічних сполук, розміщуються за відносною величиною елек
тронегативності в такий ряд:
39
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
Р > 0 > С зр> С І , М > С 5р2, В г >
і
> С 5р3, 3 > Н
Таким чином, наявність в органічних молекулах атомів і замісників
з різною електро негативністю спричиняє явище статичної поляризації
та появу на певних групах атомів часткового електричного заряду, що
і зумовлює утворення розглянутих вище (розділ 1.3.2 ) полярних кова
лент них зв 'язків.
Електронні ефекти, що лежать в основі статичної поляризації, за
механізмом зміщення електронної густини впродовж ланцюга атомів,
які утворюють органічну молекулу, поділяються на індуктивний та
мезомерний ефекти.
Індуктивний еф ект (І-ефект) - перерозподіл електронної густини
впродовж системи а-зв ’язків.
Індуктивний ефект зумовлений здатністю окремого атома чи групи
атомів (замісника) при тягувати або відштовхувати електрони (електронні
хмари) від сусіднього атома (та атомів) упродовж насиченого ланцюга
атомів вуглецю, що сполучені сг-зв’язками. Н апрям ок зміщ ення
електронів вздовж ст-зв’язків позначається прямою стрілкою. Внаслі
док зміщення електронів між окремими атомами останні набувають
часткового негативного (5“ ) або позитивного (5 і) заряду.
Залежно від електронегативності атома (замісника) та напрямку
зсуву електронної хмари розрізняють негативний (-І)т а позитивний (+1)
індуктивні ефекти.
Н егат ивний індукт ивний ефект ( - 1-ефект) - зсув електронів
ст-зв’язку в бік (у напрямку) певної групи атомів:
Максимальний -І-ефект мають замісники, що несуть на собі позитив
ний заряд. Електроноакцепторними замісниками, тобто такими, що
дають -І-ефект, є галогени, амінна, гідроксильна, карбонільна, карбок
сильна групи.
Позит ивний індукт ивний ефект (+ І-еф ект ) — зсув електронів
ст-зв’язку в напрямку, протилежному розташуванню певної групи атомів:
40
1.3. Будова біоорган іч н и х сп ол ук . Ізомерія
Я-«—
Максимальний +І-ефект мають замісники, що несуть на собі негатив
ний заряд. Електронодоноршши замісниками, тобто такими, що да
ють + 1 -ефект, є алкільні радикали (Аік-) - метальний, етильний тощо.
Індуктивний ефект атома водню у зв’язку С-Н вважають таким, що
дорівнює нулю:
5+
я — ►сн2— '
6-
5-
Я
СН2
Н
5+
Я - — СН2 — У
С тандарт
-І
+ї
1=0
Поляризація електричного заряду, спричинена індуктивним ефектом,
не обмежується сусідніми атомами, а розповсюджується впродовж вуг
лецевого ланцюга на декілька атомів, що також призводить до виник
нення на них додаткових часткових зарядів:
(6+ Г
(5+)'
5+
8-
СН 3 — ^ С Н 2 — ►СНг— ►Я
(5+)
<
(8+)
< 8+
М езомерний еф ект (М-ефект) - різновид електронного ефекту,
що передається по системі я-зв’язків. Між замісником, що проявляє
мезомерний ефект, та відповідним атомом чи групою атомів, відносно
яких цей ефект проявляється, повинна бути розташована сукупність про
стих та кратних (звичайно подвійних) зв ’язків. Така послідовність кова
лентних зв’язків, що складається з чергування одинарних (сг-) та по
двійних (сг-, я-зв’язків), дістала назву спряж еної системи.
Найчастіше спряжена система я-зв’язків (спряження) зустрічається
у спряжених алкадієнах та ароматичних вуглеводнях (аренах). У разі
сполучення такої системи із замісником, що проявляє ефект статичної
поляризації, в молекулі виникає мезомерний ефект. Напрямок зсуву еле
ктронної густини в результаті М-ефекту позначають зігнутою стрілкою.
Негативний мезомерний ефект (-М-ефект) виникає при взаємодії
спряженої системи з електроноакцепторпими групами, що відтягу
41
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
ють на себе рухомі р-елекірони спряженої системи. В результаті мезомерного ефекту на кінцевих атомах молекули виникають часткові елек
тричні заряди. -М -ефект здійснюють такі замісники, як карбоксильна,
карбонільна, нітрогрупа, наприклад:
-М -е ф е к т в молекулі акролеїну
Позитивний мезомерний ефект (+М-ефект) здійснюється електронодонортши замісниками, які відштовхують від себе р-електрони
системи спряжених зв’язків. +М-ефект властивий замісникам, які ма
ють неподілену електронну пару (наприклад, аміногрупа) або цілий не
гативний заряд:
54-М -ефект в молекулі аніліну
(+М > -І)
Результуючий розподіл електронної густини в молекулі залежить від
співвідношення атомних груп, що здійснюють індуктивні та мезомерні
ефекти різного знаку та величини. Виходячи з розглянутого, характер
замісників (відмінних від водню), які сполучені з певним атомом (вугле
цю, азоту, кисню, сірки) в органічній речовині, визначає розподіл на цих
атомах електричних зарядів, тобто поляризацію та відповідно хімічну
поведінку молекули.
1.3.4. Ізомерія в органічних сполуках
Ізомерія - явище, що полягає у наявності органічних сполук, моле
кули яких мають однаковий якісний атомний склад (та молекулярну
формулу), але розрізняються за своїми фізичними та хімічними власти
востями. Пояснення цього явища полягає в тому, що ізомери певної ре
човини відрізняються один від одного молекулярною структурою, тоб
42
1.3. Будова біоорган іч н и х сполук . Ізомерія
то порядком зв'язування окремих атомів у складі молекули або просто
рові™ розташуванням атомів та атомних груп. Перші уявлення про ізо
мерію були надані в розділі 1,1. Розглянемо це питання докладніше.
Розрізняють такі типи ізомерії:
1. Ізомерія будови.
2. Просторова ізомерія (стереоізомерія).
1.3.4.1. Ізомерія будови
Ізомерія будови (структурна ізомерія) - такий тип ізомерії, при
якому окремі молекули - структурні ізомери {ізомери будови) відріз
няються один від одного послідовністю зв’язування атомів у молекулі.
У свою чергу, ізомери будови підрозділяються на такі різновиди:
1) Ізомери ланцю га - при цьому виді ізомерії окремі ізомери від
різняються різним порядком (послідовністю) сполучення атомів у лі
нійному ланцюгу або різними видами утворюваних циклів.
Наявність ізомерів ланцюга виникає вже у чотиривуглецевого вуг
леводню бутану - одній молекулярній формулі С4Н |0 відповідає дві різні
структурні формули нормального бутану (н-бутану) та ізобутану.
Ці два ізомери відображують існування двох реально існуючих речо
вин, що відрізняються як молекулярною будовою, так і хімічними вла
стивостями.
Н Н
1
Н
1
1
Н
С труктурн а ф орм ула
1
Н -С -С -С -С -Н
І
н
н
н
І
н
н
І
(м олекулярна формула С ,Н |0)
І
н
н
І і
І
Н -С -С -С -Н
І
І І
н
н-бутану
С труктурна форм ула
ізобутану
(м олекулярна форм ула С4Н 10)
с н 3н
2) Ізомери положення - цей вид ізомерії зумовлений різним поло
женням у складі молекули однакових функціональних груп або подвій
них зв’язків, наприклад:
43
Р оїділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
а) ізомери положення функціональних груп (молекулярна формула
С3Н7Вг):
СН3 - СН2 - СН2 - Вг
СН3 - СН - СН2
1-бром пропан
Вг
2-бром пропан
У цьому типі ізомерів замісники можуть бути сполучені з первин
ним, вторинним або третинним атомами вуглецю. Первинним нази
вають атом вуглецю, що зв ’язаний тільки з одним сусіднім С-атомом,
вторинним та третинним - з двома та трьома С-атомами відповідно.
б) ізомери положення подвійного зв ’язку (молекулярна формула С ^ ) :
сн2 = с н
— с н 2— с н 3
н 3с - с н = = с н — СН3
Бутен-1
Бутен-2
3) Ізомери функціональних груп - тип ізомерії, при якому молекули
однакового атомного складу мають різні функціональні групи, тобто на
лежать до різних класів. Наприклад, молекулярній формулі С2Н60 від
повідають дві речовини з різних функціональних класів: етиловий спирт
тадиметиловий ефір:
с 2н 6о
{ Н3С-С Н 2-О Н
етиловий спирт
{ Н3С - 0 - С Н 3
диметиловий ефір
4) Таутомери - при таутомерії іспує динамічна рівновага між окре
мими структурними ізомерами. Прикладом таутомерів можуть бути
два ізомери функціональних груп - кето- та енольна форма ацетальдегіду, які у водному розчині перебувають у стані динамічної рівноваги:
н 3с — о ^ Н
0
А ц етальдегід
(к етоф орм а)
44
-
Н 2С = С — о н
і
А ц етальдегід
(ен ол ьн а ф орм а)
1.3. Будова б іо о р г а н іч н і« сполук . Ізомерія
1.3.4.2. Просторова ізомерія
Просторова ізомерія (стереоізомерія) —такий тип ізомерії, при
якому окремі ізомери мають однаковий склад та порядок зв'язування
атомів у молекулі, але відрізняються один від одного розташуванням
окремих атомів та груп атомів у просторі.
Перед тим як докладно розглянути види та типи стереоізомерів,
доцільно ознайомитися із способами зображення просторової будови
органічних молекул.
Стереоформули
Стерсоформули застосовуються для графічного зображення просто
рової будови органічних молекул на площині креслення (паперу, дошки
тощо). Існують такі різновиди стереоформул та їх проекцій:
- стереохімічні формули;
- проекційні формули Фішера;
- перспективні формули;
- проекційні формули Ньюмена.
У стереохімічних формулах зв ’язки, розташовані в площині паперу,
подають звичайною рискою; зв’язки, розташовані перед площиною па
перу (спрямовані до спостерігача), - жирним клином, а такі, що містяться
за площею креслення (спрямовані від спостерігача), - штриховим кли
ном (рис. 1.7.).
/н
* **111,
н
Рис. 1 ,7 . Стереохімічна формула етану
Принципи побудови та приклади застосування інших типів стерео
формул, що відображують просторове розташування атомів та атомних
груп у молекулах органічних сполук, будуть розглянуті нижче.
М олекулярні моделі
Молекулярні моделі дають більш наочне уявлення стосовно просто
рової будови органічної молекули, що розглядається. Найбільш уживаними
є скелетні моделі (skeletal models - англ., або моделі Драйдинга) 45
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
рис. 1.8., кульост риж неві моделі (ball-and-stick-m odels - англ.) рис. 1.9. та напівсферичні моделі {space-filling models - англ.; також масштабні, або моделі Стюарта - Бріглеба) - рис. 1.10.
Н
Н
Рис. 1.8. Скелетна (паличкова) модель етану.
У скелетних, або паличкових, моделях, зображують міжатомні зв'язки
(скелет молекули) з довжиною та валентними кутами, які пропорційні
таким, що існують в реальних об’єктах
Р и с. 1 .9 . Кульострижнева модель етану
При побудові кульострижневих моделей враховують просторове розта
шування атомів, їх розміри та міжатомні відстані
46
1.3. Будова біоорган іч ни х сполук . Ізомерія
Рис. 1.10 . Напівсферична модель етану.
В моделях цього типу враховуються ван-дер-ваальсівські радіуси атомів,
міжатомні відстані та їх просторове розташування
Конфігураційні та конформаційні ізомери
Залежно від особливостей просторової організації окремих молекул,
виділяють конфігураційні ізомери та конформаційні ізомери.
А. Конфігураційні ізомери - такі просторові ізомери (стереоізоме
ри), що відрізняються відносним розташуванням у просторі окремих
атомів без врахування можливостей обертання вуглецевих атомів на
вколо ст-зв’язків. Конфігураційні ізомери не здатні взаємоперетворюватися без розриву хімічних зв’язків, тобто вони можуть існувати окремо,
як індивідуальні молекули.
Б. Конформаційні (поворотні) ізомери —такі стереоізомери, що
утворюються шляхом обертання атомів вуглецю навколо простих (оди
нарних) ст-зв’язків. На відміну від конфігураційних, конформаційні ізоме
ри не піддаються розділенню, їх наявність та кількісне співвідношення у
розчині або іншій фізико-хімічній чи біологічній системі (наприклад, у
біомембрані) може бути встановлена лише фізичними, зокрема спект
роскопічними методами.
А. Конфігураційні ізомери
Залежно від особливостей просторової орієнтації окремих атомних
груп у молекулі, конфігураційні ізомери підрозділяють на такі типи:
1. Оптичні ізомери.
2. Геометричні ізомери.
Залежно від просторового розташування окремих груп атомів, оп
тичні ізомери, в свою чергу, поділяються на енантіомери (або дзер
кальні ізомери) та діастереомери.
47
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
1. Оптичні ізомери, Енантіомери, діастереомери
О п ти ч н а ізом ерія - явище, пов’язане з оптичною активністю
розчинів певних сполук, тобто властивістю молекул окремих речовин
обертати площину поляризації площиннополяризованого світла. Залеж
но від будови органічної молекули, оптично активні сполуки здатні до
відхилення площини поляризованого світла праворуч (тобто за годин
никовою стрілкою при погляді спостерігача назустріч світловому
променю) або ліворуч (тобто проти годинникової стрілки). Праве
обертання позначається знаком (+), ліве - знаком (-).
Оптична активність (+) або (-) є результатом м олекулярної аси
метрії органічних сполук, тобто виникає в таких молекулах, просторова
будова яких асиметрична (вони не мають площини, осі або центру си
метрії). Відсутність симетрії (та відповідно наявність оптичної актив
ності) є властивостю органічних молекул, в структурі яких присутній
бодай один атом вуглецю, сполучений з чотирма різними замісниками,
тобто такий, що знаходиться у стані ер3-гібридизації. Такий атом вугле
цю носить назву асиметричного, або хірального, і в структурній фор
мулі позначається зірочкою - С*.
Хіральність - це властивість певного геометричного об’єкту бути
несумісним із своїм дзеркальним відображенням. Класичним прикла
дом хіральних об’єктів є права і ліва руки людини (с/іеіг (%єіро<;) рука; грецьк. - звідси найменування явища), які не можна сумістити
між собою, але які є дзеркальним відображенням одна одної.
Енантіамери
Дві асиметричні молекули, що мають однакову молекулярну форму
лу, але за своєю просторовою будовою відносяться одна до одної як
об’єкт та його віддзеркалення (тобто є хіральними об’єктами), дістали
назву енантіомерів (рис. 1.11 ). Такі ізомери обертають площину поля
ризованого світла на той же самий кут, але в протилежних напрямках (+) та (-) відповідно, тобто є оптичними ізомерами, або оптичними
ант иподами.
Будову оптичних ізомерів біоорганічних молекул на прикладі стереохіміч
них формул енантіомерів молочної кислоти (лактаїу) подано на рис. 1.12:
48
1,3. Будова біоорган іч н и х сполук . Ізомерія
В'
'В
Е
.
Е
Рис. 1 .1 1 . Загальна будова оптичних ізомерів (енантіомерів), що виникають внас
лідок наявності в молекулі одного асиметричного атома вуглецю
Замісники А, В на рис, 1.11. розташовані перед площиною паперу
(спрямовані до спостерігача), Э, Е - за нею (від спостерігача). Енантіомери, що зображені нижче, відносяться один до одного як несиметрич
ний предмет до свого дзеркального відображення.
СООН
СООН
'ОН
Н
СН3
0-{-)-м ол очн а
кислота
н
НО'
СНз
і-(+ )-м о л о ч н а
кислота
Рис. 1 .1 2 . Стереохімічні формули енантіомерів молочної кислоти
Д іаст ереомери
За рахунок можливості існування в органічній молекулі декількох аси
м етричних (хіральних) атомів вуглецю (наприклад, в молекулах
моносахаридів) виникає багато стереоізомерів, що не є дзеркальними
антиподами. Такі стереоізомери називають діастереомерами. Іншими
словами, діаст ереомери - це такі стереоізомери, що не є енантіомерами (дзеркальними антиподами) один відносно другого. Загальна кіль
кість стереоізомерів у асиметричної органічної сполуки (енантіомерів
та діастереомерів сумарно) визначають за формулою: N = 2" , де п кількість хіральних атомів вуглецю в молекулі.
49
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
Номенклатура оптичних ізомерів
Р/Ь-сист ема
Враховуючи складність стереохімічних формул, для зображення оп
тичних ізомерів на площині (паперу, дошки) звичайно використовують їх
проекції на площину - так звані проекційні формули Фішера. Побудову
проекційних формул Фішера на підставі стереохімічних формул здійс
нюють за такими правилами:
1) головний вуглецевий ланцюг молекули орієнтують і записують
вертикально;
2) зверху розташовують найбільш окислений атом вуглецю;
3) замісники, що в стереохімічній формулі були над площиною (спря
мовані до спостерігача), розміщують горизонтально, а ті, що були
під площиною (від спостерігача) - вертикально;
4) асиметричний атом вуглецю розміщується на перетині вертикаль
ної і горизонтальної ліній і звичайно не позначається.
Відповідно до цих правил наведемо проекційні формули Фішера енан
ті омерів молочної кислоти:
СООН
СООН
н-
— ОН
си.
□ -( )-м олочна
кислота
но
—Н
СН3
Ь-(+)-молочна
кислота
Для позначення конфігурації оптичних Ізомерів застосовується порів
няння їх проекційних формул Фішера з відповідними формулами спеці
ально обраної стандартної речовини. В якості такого стандарту викори
стовують гліцериновий альдегід, один з енанті омерів якого був позна
чений літерою О, а його дзеркальний ізомер - літерою Ь. У проекції
Фішера оптичного ізомера, що набув назви О-гліцеринового альдегіду,
група-О Н розташована праворуч, відповідно в Ь-гліцериновому альде
гіді - ліворуч. О-гліцериновий альдегід є правообертальним ізомером
(+), Ь-гліцериновий альдегід - лівообертальним (-). Таким чином, пере50
1.3. Бу.ю ва біоорган іч ни х сполук . Ізомерія
хід від стереохімічних до проекційних формул Фііиера для енантіомерів
гліцеринового альдегіду має вигляд:
СНО
СНО
н
ОН
н о — с * — 'Н
СН2ОН
СН2ОН
СНО
СНО
н— — он
СН2ОН
Б -(+ ) ■
-гл і цс рино вий
альдегід
но — — н
СН2ОН
Ь -(-}-гл іце рино вий
альдегід
Органічні речовини, що за рахунок послідовності хімічних реакцій, які
не займають будови хіральиого центру, можуть буш перетворені на один
з оптичних ізомерів гліцеринового альдегіду, відносять до О- або Ь-ряду
сполук відповідно (молочна кислота та інші гідроксикислоти, амінокис
лоти, моносахариди тощо). Слід при цьому зазначити, що належність
стереоізомеру до О- чи Ь-ряду не визначає знак їх оптичної активності,
тобто вони можуть бути право- (+) або ліво- (-) обертальними, що ви
значається експериментально.
Я/$-сіістема
ІШ -система позначення конфігурації оптичних ізомерів є більш уні
версальною номенклатурою, яка дозволяє однозначно ідентифікувати
абсолютну конфігурацію стереоізомерів у разі, якщо їх неможливо порів
няти з конфігурацією гліцеринового альдегіду.
51
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
К/З-система структурної характеристики асиметричних органічпих
сполук, що була запропонована Каном, Інгольдом та Прелогом (Я. СаЬп,
С. Ingold, V, Р г е ^ , 1956), ґрунтується на врахуванні старшинства ра
дикалів (замісників), сполучених з хіральним атомом вуглецю. Для опи
су конфігурації певної молекули зазначені чотири радикали розміщують
у послідовності, що відповідає їх “старшинству”. При цьому замісник з
найменшим старшинством (звичайно - Н або -С Н 3) спрямовують від
спостерігача (за площу паперу), три інші замісники - до спостерігача,
розміщуючи їх по колу в послідовності зменшення старшинства:
(R)-
(S)-
У разі, якщо зменшення старшинства замісників (R,, R3, R3) відбува
ється за годинникового стрілкою (а), - така конфігурація позначається
як R (лат. rectus - правий), проти годиіпгакової стрілки (б) - S (лаг. sinister лівий). Старшинство замісників визначається залежно від атомного но
мера елемента, який безпосередньо зв’язаний з хіральним центром (якщо
необхідно - також з урахуванням суми атомних номерів елементів, що
створюють наступні шари); це дає такий ряд зростання старшинства
замісників: Н < СН3 < СН,СН3 < СН=СН, < С Д < СН,ОН < СНО <
СООН <NH3 < ОН < ОСН3~< F < СІ < Вг < І.
Наведемо приклади органічних молекул з R- та S-конфігурацією:
(Й,)-2-аміно-2-фенілетаналь
(S }-2-гі дрокеи-2- м етилбутаво в а
кислота
Властивості оптичних ізомерів. Рацемати
Оптичні ізомери (енантіомери) певної хімічної сполуки (амінокислоти,
гідроксикислоти, моносахариду тощо) не відрізняються один від одного за
52
1.3. Будова біоорган іч н и х сп ол ук . І зд мерія
своїми основними хімічними та фізичними властивостями. Основна відмін
ність, за якою розрізняють оптичні ізомери, - це різний напрямок обертання
площини поляризованого світла, що проходить через їх розчини.
Еквімолярна суміш двох оптичних антиподів мас назву рацемічної
форми, або рацемату. Внаслідок компенсації двома енантіомерами
протилежного за знаком обертання площиннополяризованого світла, су
марна оптична активність рацематів дорівнює нулю, тобто вони оптич
но неактивні.
Оптично неактивні рацемати завжди утворюються при звичайних
реакціях органічних та біоорганічних сполук, в яких беруть участь хіральні
молекули. Це означає, що при таких реакціях утворюється еквімолярна
кількість енантіомерів, які мають однакові хімічні властивості.
Для розділення (розщеплення) рацематів на окремі енантіомери або
виділення певного енантіомеру з рацемату застосовуються хімічні, біо
хімічні - стереоселективні реакції та хроматографічні методи.
Стереоселективні реакц ії
Спеціальні методи хімічних перетворень, що призводять до утворен
ня одного з оптично активних енантіомерів, дістали назву асиметрич
ного синтезу, або стереоселективних реакцій. Стереоселективні ре
акції відбуваються насамперед у біохімічних системах у живих організ
мах або в модельних системах in vitro за дії стереоспецифічних фер
ментів. Ферменти є стереоспецифічними каталізаторами; вони здатні
до зв’язування і перетворення лише певних оптичних антиподів, напри
клад, L- або D-амінокислот, L- чи D-моносахаридів. Такі властивості
ферментів пов’язані з хіральністю самих молекул ферментних білків, які
побудовані з залишків L-амінокислот, тобто хіральних сполук, що і пояснує їх стереоселективність.
Хроматографічні методи, що застосовуються для розділення раце
матів, грунтуються на спорідненості окремих енантіомерів до оптично
активних сорбентів (афінна хроматографія).
Біологічні властивості оптичних ізомерів
Незважаючи на однакові хімічні властивості та більшість фізичних
характеристик, біоорганічні сполуки, що є енантіомерами, проявляють,
як правило, різну фізіологічну активність, тобто по-різному впливають
на перебіг біохімічних реакцій та фізіологічних процесів у живих оргаиіз53
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
мах. Цс відноситься, наприклад, до різних антиподів а-амінокислот амінокислоти L-ряду є “природними”, або протеїногенними амінокисло
тами, тільки вони {а не амінокислоти D -ряду) можуть використовувати
ся для біосинтезу природних білків. У метаболічних перетвореннях в
організмі беруть участь D- (а не L-) форми гексоз (глюкози, фруктози,
галактози) тощо. Первинні причини, з яких абіогенна еволюція на Землі
пішла шляхом використання для утворення природних білків саме
L-амінокислот, лишаються нез’ясованими. Можна лише уявити собі
імовірність існування дзеркального “антисвіту” з білковими організмами,
що побудовані з D-амінокислот.
Стереоспецифічність по відношенню до певних оптичних антиподів
мають також рецептори ФАС, що визначає фармакологічну активність
останніх та враховується при створенні нових лікарських засобів. На
приклад, гормональну та фізіологічну активність в організмі людини і
тварин має D -(-)-адреналін, його оптичний антипод Ь-(+)-адреналін не
здатен до взаємодії з функціональними групами плазматичних мембран
чутливих клітин і, відповідно, не діє як гормон та лікарський засіб. Залеж
ність фармакологічних властивостей біоорганічних сполук від їх стерео
хімічної конфігурації є загальною закономірністю для багатьох класів
лікарських засобів. Ці проблеми є об’єктом дослідження молекулярної
фармакології.
Я к вже зазначено, різна біологічна активність оптичних антиподів
амінокислот та їх похідних, моносахаридів, енантіомерів фізіологічно
активних речовин природного та синтетичного походження, тобто хіральних сполук, залежить від наявності в самих молекулах ферментів та
рецепторів ФАС хіральних центрів.
2. Геометричні ізомери
Геометричними ізомерами називають ізомери, що виникають внаслі
док різного просторового розташування атомів та груп атомів (замісни
ків) відносно площини подвійного зв ’язку в ациклічних сполуках
(я-діастереомери) або відносно площини аліциклічного кільця.
Пис-. транс-ізомери
Найбільш уживаною номенклатурою для позначення геометричних
ізомерів є цис- трянс-еистема. Префікс цис- надають тому геометрич
ному ізомеру, в якого однакові замісники знаходяться по один бік по
двійного зв’язку (або площини циклу), а префікс транс - тому, в якому
54
1.3. Будова біоорган іч н и х сполук . Ізомерія
вони знаходяться навхрест—по різні боки від подвійного зв’язку. Прин
цип віднесення сполук до цис- або трлнс-ізомсрів легко зрозуміти з
прикладу позначення ізомерів пропандіової кислоти:
М алеїнова кислота
(цис-ізом ер)
Ф ум арова кислота
(транс-ізомер)
Подвійний вуглець-вуглецевий зв ’язок вносить у молекулу значний
ступінь жорсткості. Відносно вільне обертання груп атомів, можливе
навколо простих ст-зв’язків, при наявності я-зв’язків не відбувається.
Саме тому цис- і туишс-ізомери є реально існуючими сполуками, що
різняться фізичними та хімічними властивостями. Так, малеїнова кис
лота є кристалічною речовиною з т. п л .1 31 °С, легкорозчинна у воді та
спиртах, тоді як фумарова кислота має т. пл. 287 °С, важко розчиняється
у воді. Фумарова кислота належить до біомолекул, вона є важливим
інтермедіатом реакцій внутрішньоклітинного окислення, тоді як малеї
нова кислота токсична для вищих організмів.
Цис- транс-номенкпатура ізомерів може застосовуватися також
при позначенні просторового розміщення двох замісників відносно пло
щини аліциклічного кільця. У цьому випадку ізомер, в якому обидва
однакові замісники розташовані по один бік кільця, носить назву цисізомеру, а інший (з протилежним розташуванням замісників) - трансізомеру. Такі ізомери також відрізняються один від одного за своїми
фізичними та хімічними властивостями. Геометричні ізомери 1,2-диметилциклопропану:
Н
Цис- і ,2-дим стилци клоп ропан
Н
Транс-1,2-дим етнлци клоп ропан
55
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
Е-. Z-ізомерц
В тому випадку, коли з двома атомами вуглецю, сполученими по
двійним зв ’язком, зв ’язані чотири різних замісники, цис-, транс-номен
клатура не дає можливості побудувати раціональні назви для різних гео
метричних ізомерів, В цих умовах пропонується більш універсальна си
стема позначень, яка ґрунтується на визначенні старшинства замісни
ків за Каном, Інгольдом, Прелогом. В разі розташування обох старших
замісників (з кожної пари) по один бік від подвійного зв ’язку, відповідна
конфігурація дістала назви Z (нім. zusammen —разом); при протилеж
ному розміщенні замісників - Е (нім. entgegen - навпроти).
Приклади Z- та Е-ізомерів:
Br
СІ
V
U ,
Н3с
/
°
г/
--------------
\
СНз
Z -2 -б р о м -З -хл о рбутен - 2
Вг
V __ г/
н г /
нзС
СН3
4 сі
Е-2 - бром -3 -х л орбутен -2
(СІ > СН3, Вг > СН3)
Б. Конформаиійпі ізомери
Конформаційні ізомери - це такі просторові форми органічної моле
кули, що виникають внаслідок обертання окремих фрагментів молекули
навколо простих одинарних (а-) зв’язків.
Таким чином, конформація - це наступний рівень різноманітності у
тривимірній структурі органічних молекул. Якщо конфігурація молекул це фіксоване розташування атомів або груп атомів один відносно одно
го в просторі, то конформація враховує можливість їх взаємного пере
творення за рахунок обертання навколо вугпець-вугаецевих та інших про
стих зв ’язків. Ці ізомери, як правило, нестійкі і існують протягом незнач
ного часу. Разом з тим, поряд з нескінченною кількістю окремих нестій
ких просторових структур існують такі крайні за енергетичними харак
теристиками конформації, що є термодинамічно стабільними і можуть
існувати у вигляді окремих хімічних сполук —конформерів.
Для характеристики конформації органічної молекули і наявності різ
них конформаційних ізомерів найчастіше застосовують перспективні
формули та проекційні формули Ньюмена.
56
1.3. Будова біоорган іч н и х сполук . Ізомерія
Перспективні формули для зображення конформерів
При зображенні конформерів за допомогою перспективних формул зв’я
зок С -С подають як такий, що віддаляється у перспективу; вуглець, роз
ташований зліва, є найближчим до спостерігача, справа - віддаленим.
При зображенні перспективними формулами стереоізомерів етану, що
утворюються за рахунок обертання груп -С Н 3 навколо одинарного
о-зв’язку, виникають такі дві крайні конформації (конформери);
И
у н
н /Н
(1) Заслонена конф орм ація
(2) Загальм ован а конформація
етану
етану
Молекулярна структура (1) дістала назву “заслоненої”, (2) - “загальмованої” конформації етану.
Проекційні формули Ньюмена для зображення конформерів
При побудові формул Ньюмена органічну молекулу розглядають
вздовж вуглець-вуглецевого зв’язку з “торця” таким чином, щоб обидва
атоми вуглецю знаходились один позаду другого. При цьому ближчий
до спостерігача атом вуглецю розташовується на перетині трьох пря
мих, що сходяться (і іноді позначається крапкою), а віддалений атом
вуглецю позначається колом, з-поза якого “виглядають” зв’язки С -Н
віддаленої метальної групи. “Заслонена” (1) та “загальмована” (2) кон
формації етану в проекціях Ньюмена:
( 1)
(2 )
57
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
Заслонені та загальмовані конформери
Як видно з наведених стереохімічних формул, в заслоненій конфор
мації етану атоми водню сусідніх груп -С Н , просторово найбільш збли
жені (порівняно з іншими варіантами просторового розташування ме
тальних груп), внаслідок чого між ними існує максимальне відштовху
вання. Виходячи з цього, заслонені конформації вуглеводнів та їх похідні
с термодинамічно найменш стійкими.
На відміну від заслонених, у загальмованих конформаціях етану вугле
воднів замісники максимально віддалені один від одного, сили відштовху
вання і потенційна енергія взаємодії радикалів значно зменшені, в цих конформерах внутрішній поворот навколо простих зв’язків ніби гальмується.
Тому з усіх поворотних ізомерів етану загальмована конформація є енер
гетично та структурно найбільш стійкою. Енергетичний бар’єр переходу
етану із загальмованої конформації в заслонену складає близько 12,5 кДж.
Такі ж закономірності існують і для інших вуглеводнів, в яких можливе
обертання навколо одинарних зв’язків та виникнення поворотних ізомерів.
Кути повороту при утворенні конформерів
Завдяки обертанню метальних груп навколо вуглець-вуглецевого
єг-зв’язку молекула етану набуває множини конформацій, що відрізня
ються величиною кута, який утворюється між зв ’язками С -С Н 3 сусід
ніх атомів. Для позначення окремих конформацій використовують зна
чення кута ф, що вказує на взаємну орієнтацію СН3-груп:
В молекулі н-бутану при повному обертанні на 360° навкруги централь
ного С -С -зв’язку виникає шість окремих конформацій, що відрізняють
ся на 60° (три - заслонені, одна - загальмована і дві - скошені, або гоїиконформації). Серед цих конформацій термодинамічно найбільш
стабільними є загальмована {анти-) та гош-конформації;
58
1.3. Будова біоор ган іч н и х сполук . Ізомерія
Загальм ована
(анти-)
С кош ена
конф орм ація бутану
(гош-)
конф орм ація бутан у
Розглянуті конформації характерні для невеликих молекулярних струк
тур. Ациклічні вуглеводні, що мають у своєму ланцюгу п’ять і більше
атомів вуглецю, можуть утворювати зигзагоподібну (1), клешнеподібну (2) та нерегулярну (3) конформації:
( і)
(2)
(3)
Для сполук з відкритим ланцюгом термодинамічно найбільш вигід
ною є клешнеподібна конформація, оскільки вона стабілізується додат
ковим формуванням слабких зв’язків між окремими ділянками ланцюга
із замкненням внутрішньомолекулярних циклів.
Конформації циклічних вуглеводнів
Залежно від розмірів вуглецевого циклу циклічні вуглеводні поділя
ють на цнклоалкани з малими ( 3 ^ атоми вуглецю), звичайними (5-7
атомів вуглецю), середніми циклами (8-10 атомів вуглецю) та макроциклами (понад 10 атомів вуглецю).
Завдяки структурним обмеженням для вільного обертання атомних
груп навколо о-зв’язків у циклічних вуглеводнях, специфічні конформації
останніх залежать від розмірів циклу. Серед сил міжмолекулярних взає
модій, які формують певні види напружень у циклоалкаиах, виділяють:
- торсійну напругу, яка зумовлена взаємодією (електростатичним
відштовхуванням) атомних груп, що знаходяться в заслонених або близь
ких до заслонених гош-конформаціях;
- кутову (байєрівську) напругу, яка виникає внаслідок відхилення
валентних кутів між атомами вуглецю при утворенні циклів від їх нормаль
59
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
них значень (А. Байєр, 1885). Цей тип напруги валентних зв’язків є
специфічним для циклоалканів, напрямок валентних кутів у яких відріз
няється від їх нормального розташування в Бр3-гібридизованому атомі
вуглецю{109,5°).
Конформаиії ииклопентану та ииклогексану
Вуглецеві ядра циклопентану та циклогексану входять до складу вуг
леводів, нуклеїнових кислот, багатьох фізіологічно активних сполук вітамінів, гормонів, алкалоїдів, синтетичних лікарських засобів. Тому
вивчення будови конформерів циклоалканів, що входять до їх структури,
має неабияке значення для розуміння тривимірної будови та механізмів
біологічних і фармакологічних ефектів цих біомолекул і ФАС.
Циклопентан
В молекулі циклопентану валентні кути між атомами вуглецю дорів
нюють 108°, що близько до їх нормальних значень. Разом з тим, завдя
ки взаємодії між атомами водню, сполученими із сусідніми атомами
вуглецю, в циклопентані виникає торсійна напруга. Зменшення цієї на
пруги досягається шляхом утворення специфічної для цього циклоалісану і енергетично стабільнішої конформації конверта (рис. 1.13 ):
( 1)
(2)
Р ис. 1 .1 3 . Конформації циклопентану: (1) - плоска форма молекули; (2) - форма
конверта
Циклогексан
У плоскій структурі циклогексану валентні кути дорівнюють 120°,
тому в цьому циклоалкані присутні сили як кутової, так і торсійної на
пруги, що призводить до переходу молекули в термодинамічно більш
вигідні конформації. Такими для циклогексану є конформації крісла, ван
ни ("човна") та тв/сте-конформація (рис. 1.14 ).
0)
(2)
(3)
(4)
Рис. 1 .1 4 . Конформації циклогексану: (1) - плоска форма молекули; (2) - крісло;
(3) - ванна (“човен"}; (4) - твіст-конформація
60
1.3. Б удова біоорган іч н и х сполук . Ізомерія
Експериментально встановлено, що найбільш стабільною для цикло
гексану є конформація крісла, яка має найменшу потенційну енергію
серед інших конформерів. Саме така конформація притаманна молеку
лам біоорганічних сполук, що є похідними циклогексану або містять у
собі циклогексан як структурний фрагмент.
Стереохімія похідних циклогексану
В конформаційному стані крісла у звичайних умовах перебуває по
над 99 % всіх молекул циклогексану. Можливі два термодинамічно рів
ноцінні конформери крісла, між якими існує динамічна рівновага:
В конформації крісла розрізняють два типи зв’язків С— Н і відповід
но замісників, що сполучені з атомами вуглецю базової циклічної струк
тури, а саме:
- зв ’язки, що спрямовані паралельно осі симетрії циклогексану (шість
зв ’язків, направлених догори, та шість - вниз); ці зв’язки мають назву
аксіальних - а (лат. axis - вісь);
- зв’язки, що спрямовані перпендикулярно осі симетрії циклогекса
ну, тобто розташовані паралельно площині молекули - екваторіальні
зв’язки - е.
Просторову направленість аксіальних та екваторіальних зв’язків у
двох типах конформації крісла молекули циклогексану подано на рис. 1.15.
Як слідує з рисунка 1.15, в разі сполучення з кожним із атомів вугле
цю молекули циклогексану двох атомів водню (одного аксіального та
одного екваторіального) відбувається постійна осциляція структури мо
лекули від одного енергетично рівноцінного конформера до іншого. При
цьому відповідно змінюється і положення замісників при одному й тому
ж атомі вуглецю - аксіальні стають екваторіальними і навпаки.
Рис. 1.15. Аксіальні (а) та екваторіальні (є) замісники в конформерах циклогексану
6]
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
Введення в молекулу циклогексану відмінних від водню замісників,
між якими можуть виникати електростатичні взаємодії, призводить до
утворення додаткової торсійної напруги в молекулі. В цій ситуації конфор
мації' з аксіальним та екваторіальним розміїценням замісника (наприклад,
метальної групи) не є термодинамічно рівноцінними. Така ситуація вини
кає, наприклад, уже в молекулі 1-метилциклогеїссану. Внаслідок електро
статичної взаємодії аксіального метального радикала в положенні (1) з
воднями (3) та (5) такий сторичний ізомер буде значно менш стабільним,
ніж ізомер з екваторіальним метальним радикалом (рис. 1.16.):
Н
Рис. 1.16. Взаємовідношення стереоізомерів 1-метилциклогексану (В. П. Черних,
Б. С. Зіменковський, І. С. Гриценко, 1996)
Оскільки циклічні структури циклопентану та циклогексану є скла
довими молекул стероїдного ряду (похідних циклопеитанпергідрофенантрену), розглянуті закономірності мають суттєве значення для форму
вання тривимірної просторової структури природних фізіологічно актив
них сполук та ліків цього поширеного класу.
1.4. Реакції в біоорганічній хімії
1.4.1. Характ ерист ика типів реакцій
Реакція в біоорганічній хімії, як і хімічна реакція взагалі, - це процес, під
час якого змінюється атомний (елементний) стан певної молекули. Процес
хімічної реакції супроводжується зміною розподілу електронів на зовнішніх
(валентних) орбіталях реагуючих часточок (молекул, іонів, вільних радикалів)
з утворенням енергетично стабільніших атомних систем.
62
1.4. Р еак ц ії в біоорган іч ній хімії
Більшість органічних реакцій на шляху від вихідних субстратів (ча
сточок, що підлягають перетворенню) до кінцевих продуктів послідов
но проходить декілька стадій, опис яких називають механізмом реакції.
Звичайно механізм реакції включає в себе формування проміжного (або
активованого) комплексу, який являє собою тимчасове угруповання
атомів з найвищою хімічною енергією, що здатне до перетворення на
кінцеві продукти.
Виходячи з таких загальних уявлень, механізм перетворення речо
вини АВ (субстрату) за дії реагенту С на продукт АВС можна подати
такою схемою перетворень:
А - В + С -> А -В -С -» А - В - С
П ром іж ний комплекс
Перетворення субстрату (або субстратів) на продукт (продукти)
реакції через стадію проміжного комплексу вимагає певних енергетич
них затрат, необхідних для розриву існуючих хімічних зв’язків та утво
рення нових молекулярних орбіталей. Енергія, необхідна для такого пе
ретворення, дістала назву енергії акт ивації хімічної реакції (ДЕ). Ви
ходячи з цього, константа швидкості будь-якої органічної реакції (к) за
лежить від її енергії активації та температури, що виражається рівнян
ням Арреніуса в експоненційній формі:
к = А е-ш ет,
де експоненційний член рівняння с йт г(фактор Больцмана) позна
чає частку молекул у системі, які мають енергію, достатню для хіміч
ного перетворення.
В біохімічних системах, де реакції протікають, як правило, при ста
лих температурах (в клітинах вищих організмів при 36-37 °С), необхідна
для перетворення біомолекул швидкість реакції досягається за рахунок
застосування високоефективних біокаталізаторів - ферментів.
Реакції, що в них вступають біоорганічні сполуки, класифікують за
механізмом розщеплення хімічних зв’язків, спрямованістю процесу та
кінетичними критеріями.
63
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
І. Класифікація реакцій за механізмом
В основу класифікації органічних реакцій за механізмом покладено
способи розщеплення зв ’язків у молекулі субстрату, що підлягає пере
творенню. Згідно з цим принципом реакції поділяються на такі типи:
1) гетеролітичні, або іонні;
2) гемолітичні;
3) молекулярні.
1. Гетеролітичні, або іонні, реакції. Це - реакції, в ході яких пара
електронів, що уособлює хімічний зв’язок в молекулі, переходить до однієї
з часточок, які утворюються. Таким чином, в результаті гетеролітичної
реакції з молекули, що підлягала розщепленню, вивільняються два заряд
жених фрагменти - іони (позитивно заряджений катіон та негативно
заряджений аніон):
X
• У -> Х+ + у -
2. Гемолітичні, або радикальні, реакції. Цей тип реакцій полягає у
розщепленні хімічного зв’язку (електронної пари) з утворенням продук
тів, кожен з який має по одному з неспарених (неусуспільнених) елект
ронів, які утворювали пару (тобто були на одній молекулярній орбіталі).
Продукти реакції (атоми або групи атомів), на зовнішніх (атомних або
молекулярних) орбіталях яких знаходиться по одному неспареному елек
трону, називаються вільними радикалами. Загальна схема реакції:
Вільні радикали є хімічно надзвичайно активними і, як правило,
короткоживучими часточками. Гомолітичні реакції, що призводять до
утворення вільних радикалів, відбуваються при дії на молекулярні струк
тури опромінення, високої температури або під впливом деяких окислювально-відновлювальних ферментів, що здатні до каталізу одноелектронних реакцій (цитохромів). Внаслідок наявності у неспареного елек
трона певного спіну (тобто обернення навколо власної осі) вільні ради
кали мають нескомпснсований магнітний момент, що дозволяє виявля
ти (детектувати) їх наявність у хімічних та біохімічних системах за до
помогою методу електронного парамагнітного резонансу (ЕПР).
64
1.4. Р еак ц ії в біоорган іч ній хім ії
3.
Молекулярні, або синхронні, реакції, протягом яких відбуваєть
ся синхронний перерозподіл електронної густини між часточками, що
реагують, без розділення заряду.
II. Класифікація реакцій за спрямованістю перетворень
За спрямованістю та результатом хімічних перетворень органічні
реакції поділяють на такі класи:
1) реакції приєднання;
2) реакції відщеплення;
3) реакціїзаміщення;
4) реакції перегрупування;
5) реакції окислення та відновлення.
В свою чергу, реакції приєднання (А) та заміщення (S) можуть проті
кати за іонним - електрофільним (Е) чи нуклеофільним (N) або радикаль
ним (R) механізмом, що позначається такою знаковою системою:
А е - електрофільне приєднання;
A n- нуклеофільне приєднання;
A r - радикальне приєднання;
S - електрофільне заміщення;
SN- нуклеофільне заміщення;
SR- радикальне заміщення.
1. Реакції приєднання (позначаються символом А - addition (англ.)):
X=
Y
+ А— В
*- |С
f
А
В
Найбільш типовими серед реакцій приєднання є такі, що відбуваються
за механізмом АЕ (електрофільне приєднання). АЕ-реакції характерні
для иенасичених сполук, які містять кратні зв’язки, і супроводжуються їх
розривом. Найчастіше в АЕ-реакції вступають алкени, алкадієни (див.
наведене вище рівняння). Для органічних речовин, що містять полярні
подвійні зв’язки (зокрема, альдегідів та кетонів, у складі яких є карбоніль
на група) властиві реакції нуклеофільного приєднання (AN).
2. Реакції відщеплення (елімінування) - позначаються символом
Е (elimination —англ.):
65
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
|С —
|
А
В
*
X=
У
+ А— В
Такі реакції є властивими для галогенопохідних вуглеводнів, спиртів,
гідроксикислот, амінокислот. Реакціями елімінування (Е), характерними
для біохімічних систем, є відщеплення від відповідних метаболітів води
(дегідратація) або аміаку (дезамінування).
3. Реакції зам іщ ення (позначаються символом б - $иЬ5Іііи(іоп (англ)):
X
У
+
А
В
--------
X -- А
+
у ----- В
Це - поширений тип реакцій в біоорганічній хімії, що властиві різним
класам сполук. Зокрема, електрофільне заміщення (БЕ) властиве для
ароматичних сполук (аренів), нукпеофільне заміщення (8М) - для кар
бонових кислот (утворення амідів, ангідридів кислот тощо), спиртів, га
логенопохідних насичених вуглеводнів.
4. Р еакції перегрупування:
X
А
У
7.
— - X — У— г
А
Реакції перегрупування полягають у переході (міграції) окремих атомів
чи атомних груп у межах однієї молекули. Поширеним типом перегрупу
вання є кето-енольна таутомерія - реакція, що полягає у внутрішньомолекулярній міграції протона (іона водню) між атомами кисню та вуглецю.
5. Реакції окислення та відновлення
Цей тіш взаємно зворотних реакцій полягає у міжмолекулярному пе
реносі електронів, або міграції електронів до атома кисню, що приєдну
ється до субстрату (молекули, яка окиснюється). Формально окисновідновні реакції є різновидом реакцій приєднання та відщеплення, харак
терною їх ознакою є зміна ступеня окислення певного вуглецевого атома реакційного центру.
Сполука, що протягом окислювально-відновлювального процесу при
ймає електрони (або електрони разом з протонами - т. з. відновлювальні
66
1.4. Р еак ц ії в біоорган іч н ій хім ії
еквіваленти) має назву окисника, спряжена окиснику молекула, яка від
дає електрони, - відновника. Напрямок переносу електронів між двома
хімічними сполуками визначається значеннями окислювально-відновлювальних потенціалів (редокс-потенціалів) останніх. Окисно-відновні
реакції, що протікають в мембранних структурах біохімічних систем, є
джерелом хімічної енергії для всіх процесів життєдіяльності.
Ш. Класифікація за кількістю молекул, що беруть участь в реакції
У багатостадійному хімічному процесі (а практично кожна реакція
перетворення органічних речовин складається з декількох послідовних,
парціальних реакцій) сумарна швидкість накопичення продукту визна
чається швидкістю найповїльнішого етапу - реакції, яка в хімічній кінетиці
дістала назву “лімітуючої” . У свою чергу, константа швидкості лімітуючої
р еа кц ії залежить від кількості молекул субстрату та реагенту, що
взаємодіють. Відповідно до цього, розрізняють такі типи реакцій:
М ономолекулярні реакції - константа швидкості яких визначаєть
ся перетворенням однієї молекули речовини (А) за такими схемами:
А —» В + С (розщеплення вихідної сполуки на продукти реакції; в моно
молекулярних реакціях цього типу часто в якості реагенту
бере участь молекула води, але в більшості систем, що роз
глядаються в біоорганічній хімії, концентрація молекул води
значно перевищує концентрацію субстрату [А], що дозво
ляє приймати [Н ,0] за сталу величину);
А —> В
(реакції внутрішньомолекулярного перетворення, ізомеризації
молекули).
Б ім олекулярні р е а к ц ії - константа швидкості яких визначається
взаємодією двох молекул (субстрату та реагенту) за однією із схем:
А + В —> С
(реакція синтезу продукту з вихідних субстратів);
А + В —> С + О (реакція утворення нових сполук шляхом перегрупування).
До бімолекулярних належить більшість органічних реакцій та біохі
мічних перетворень у процесі обміну речовин.
Характеризуючи певну реакцію в біоорганічній хімії та біохімії, слід
враховувати її різні класифікаційні ознаки. Так, наприклад, виділяють
реакції мономолекулярного елімінування (НІ), мономолекулярного нуклеофільного заміщення (Б^І), бімолекулярного нуклеофільного заміщен
ня (3^2) тощо.
67
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
1.4.2. Реагенти та субстрати. Нуклеофіли, електрофіпи
На протязі органічної реакції в більшості випадків взаємодіють одна з
одною дві органічні сполуки. В цих умовах речовину, перетворення якої
вивчається (звичайно складнішу за своєю будовою молекулу) називають
субстратом реакції, а речовину, яка впливає на субстрат, спричиняючи
його хімічні зміни, -реагент ом (або “атакуючим реагентом”).
Залежно від будови часточок, що є інтермедіатами хімічного проце
су, та типу реакцій, в яких вони беруть участь, реагенти поділяють на:
- радикальні реагенти - такі, що спричиняють гомолітичні ради
кальні реакції;
- реагенти, які спричиняють гетеролітичні іонні реакції.
Реагенти, які беруть участь в іонних органічних реакціях, підрозділя
ються на нуклеофіли (нуклеофільні реагенти) - такі, що “тягнуться” до
“ядра” (позитивного заряду), та електрофіли (електрофільні реагенти) такі, що “тягнуться” до “електронів” (негативного заряду).
Нуклеофіли - це частинки (іони або молекули), що мають надли
шок електронів (взагалі електронної густини) та в ході органічної реакції
віддають електрони субстрату - епектрофілу (зазвичай атома вуглецю).
Позначення нуклеоф ілів- № г.
Електрофіли - частинки (іони або молекули), що мають нестачу
електронів (електронної густини) і схильні приєднуватися до нуклеофілу (звичайно, до позитивно зарядженого атома вуглецю). Позначення
електрофілів - Е+.
Нуклеофільними реагент ам и є:
1) негативно заряджені іони (аніони): ОН- , НаР(СТ, В г, Р , І ), СК ,
N 0 ,', 110 (алкоксид-іон), Н (гідрид-іон);
2) нейтральні молекули, в яких один атом має неподілену (вільну)
електронну пару: НОН, ЇЮН, ІІ(Ж , К -С О -О Н , № Н Д ;
3) ненасичені сполуки, що надають електрофілу свої р-електрони:
алкени, алкіни, арени та їх похідні.
Електрофільними реагент ам и є:
1) позитивно заряджені частинки: Н 1 (протон), N 0 / (катіон нітроїлу), Вг1(катіон брому), Ї13С+ (карбкатіон);
2) нейтральні молекули, що мають електронодефіцитний центр із дробним (частковим) позитивним зарядом, наприклад:
68
1.4. Р еакції в біоорган іч ній хімії
8+
5СЕЕЕЫ
5+
8ІЧ3С — —НаІ
3) вільні радикали: НаГ (С Г, Вг* т.і.), Я* (СН3* т.і.), КО*;
4) атоми в молекулах, що поляризуються в ході органічної реакції,
наприклад:
8+
Вг
8► Вг
Відповідні органічні реакції, в яких за реагент правлять нуклеофіли або
електрофіли, називають нукяеофиіьними або електрофільними реакціями.
1.4.3.
Кислоти та основи в біоорганічній хім ії
Загальні хімічні властивості сполук, що розглядаються в біоорганіч
ній хімії, значною мірою визначаються здатністю їх молекул виступати
в ролі органічних кислот або основ (лугів). За сучасними уявленнями,
кислотність та основність органічних сполук визначаються протояітичною (протонною) теорією Бренстеда - Лоурі (частіше позначається
як “теорія Бренстеда”) та електронною теорією Льюїса.
Кислотність та основність за теорією Бренстеда
Згідно з протолітичною теорією Бренстеда, кислотою є будь-яка ор
ганічна сполука, що є донором протонів, а основою - сполука, що є акцеп
тором протонів. За хімічною будовою:
кислоти за Бренстедом (протонні кислоти) - це нейтральні моле
кули або іони, що здатні відщеплювати протон в реакції з основою;
основи за Бренстедом - нейтральні молекули, що містять неподілену
пару електронів, або іони, що здатні приєднувати протон в реакції з кислотою.
Після втрати протона кислота перетворюється на спряжену осно
ву (тобто акцептор протонів), а основа в результаті приєднання протона
стає спряженою кислотою (тобто донором протонів).
Загальне рівняння взаємодії між кислотою та основою Бренстеда:
А-Н
К ислота
+
В
О снова
<-»
А-
+
С п ряж ен а основа
З урахуванням будови ковалентного хімічного зв ’язку як пари елек
тронів та наявності в складі основи неподіленої електронної пари, процес
переносу протона можна подати у вигляді таких рівнянь:
69
Розділ 1. ЗАГАЛЬНІ ПОЛОЖЕННЯ БІООРГАНІЧНОЇ ХІМІЇ
(а) А * * І Н
А_ + Н+
( б ) н * + : в н в +— н
Типи органічних кислот за Бренстедом
У складі органічних кислот атом водню може бути сполучений з
такими елементами (центрами кислотності), як кисень, сірка, азот та
вуглець. Відповідно до цього виділяють:
ОН-кислоти (карбонові кислоти, феноли, спирти);
ЭН-кислоти (тіоли);
ЫН-кислоти (аміди, аміни, іміди);
СН-кислоти (вуглеводні та їх похідні).
За викліочеїшям найбільш сильних карбонових кислот (найнижчі значен
ня рК), решта органічних сполук є слабкими кислотами; їх кислотність змен
шується в ряду: 5Н-кислоти >ОН-кислоти >ЇЧН-кислоти >СН-кислоти.
Типи органічних основ за Бренстедом
В якості негативно зарядженого центру, який взаємодіє з вільним прото
ном, основи Бренстеда містять неподілену електронну пару (т. з. “п-електрони”) або рухомі електрони я-зв'язку. Відповідно до цього, основи
Бренстеда розділяють на оніеві основи (п-основи) та я-основи:
Онієві основи (п-основи) - центрами основності в них є гетероатоми 14,
О, 8, які містять на зовнішніх АО неподілену пару електронів, що акцептує
протон. Онієві основи підрозділяють на амонієві, оксонієві та сульфонієві (з
атомами азоту, кисню та сірки як центрами основності, відповідно).
Прикладом функціонування онієвої основи може бути взаємодія з
водою метиламіну, що в результаті зв’язування протона спричиняє зсув
pH середовища в лужний бік:
СН3Ы:Н2 + Н20
СН3ЫН3+ + ОН~
п-основи - сполуки, в яких центрами основності є електрони я-зв’язку. Це - алкени, алкадієни, арени.
Кислотність та основність за теорією Льюїса
Згідно з теорією Льюїса, кислотами вважають часточки (атоми,
молекули, іони), що приєднують до себе електронну пару, а основами часточки, що віддають електронну пару акцептору (кислоті). Інакше
кажучи, кислоти Льюїса - це акцептори електронної пари, а основи
Л ью їса-д он ори електронів. За хімічною будовою:
70
1.4. Р еак ц ії в біоорган іч н ій хімії
кислотами Льюїса є хімічні часточки (атоми, молекули, катіони),
що мають незайняту парою електронів (вакантну) АО або МО. При
ймаючи електронну пару, ці речовини утворюють з донором електронів
ковалентний зв ’язок. Таким чином, в іонних реакціях кислоти Льюїса
виступають в якості епектрофільпих реагентів. Представниками ки
слот Льюїса є катіони металів, протони, галогеніди деяких металів (А1СЦ,
ВР3, БпС^, РеС13 тощо), що використовуються як каталізатори багатьох
важливих органічних реакцій;
основами Льюїса є хімічні часточки (атоми, молекули, аніони), що
містять на зовнішній орбіталі пару валентних електронів, які вони мо
жуть передавати акцептору з утворенням ковалентного зв ’язку. Таким
чином, основи Льюїса є нуклеофільними реагентами. Відповідно до
цього, до основ Льюїса належать сполуки, до складу яких входять гете
роатоми (14,0 , 8) з парою неподілених електронів (аміни, спирти, прості
ефіри, тіоли тощо). Крім того, слабкими основами Льюїса є речовини,
які містять рухомі я-електрони у складі р-зв’язку або системи спряже
них я-зв’язків (алкени, алкадієни, ароматичні сполуки).
71
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ
КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
2.1.
Вуглеводні та їх похідні
Вуглеводні - сполуки, молекули яких складаються з атомів вугле
цю та водню, - разом із своїми похідними утворюють один з найбільших
класів речовин, що вивчаються біоорганічною хімією.
Залежно від типу будови вуглецевих ланцюгів, вуглеводні підрозділя
ються на сполуки з відкритим (незамкненим) ланцюгом - ациклічні
(аліфатичні), та такі, що мають замкнений ланцюг, - циклічні вугле
водні. Циклічні вуглеводні мають ще назву гомоциклічних, або карбоциклічних (на відміну від гетероциклічних) речовин. В принципі, біль
шість інших органічних (та біоорганічних) сполук можна розглядати як
похідні вуглеводнів, утворені шляхом заміни атомів вуглецю нарізні ато
ми або групи атомів, що призводить до угворення інших класів речовин галогенопохідних, спиртів, альдегідів, органічних кислот, амінів тощо, для
яких молекули вуглеводнів є родоначальними структурами.
Як ациклічні, так і циклічні вуглеводні можуть мати у складі своїх вугле
цевих ланцюгів кратні (подвійні або потрійні) зв’язки, тобто бути насичени
ми або ненасиченими сполуками. Насичені аліфатичні вуглеводні звуться
штанами, такі, що вміщують один або два подвійних зв’язки - відповідно
алкенами та алкадієнами; сполуки з потрійним зв’язком - алкінами.
Карбоциклічні вуглеводні можуть мати цикли насиченого типу - аліциклічні вуглеводні або такі, що складаються з одного або декількох
бензольних кілець, які мають особливий (“ароматичний”) характер вуглець-вуглецевих зв’язків - ароматичні вуглеводні, або арени.
-►Алкани
-Ациклічні-
-Алкени, алкадієни
-Алкіни
Вуглеводні—
“Аліцикпічні
—►Циклічні —
-Ароматичні
72
2,1. В углеводні та їх похідні
2.1.1. Алкани
Ал кани - насичені аліфатичні вуглеводні, що мають лінійні нерозгалужені (н-алкани) або розгалужені (ізо- та нєоалкапи) вуглецеві лан
цюги, наприклад:
СН3- СНг- СН2- СН2- СН3
СН3- СН2- СН - С Н 3
н-П ен тан
|
сн3
Ізопентан
сн3
1
СН3—СН — СН3
Неопентан
І
сн3
Загальна молекулярна формула сполук гомологічного ряду алканів С Н 2п+2. Оскільки назви аліфатичних вуглеводнів є наріжним каменем
систематичної номенклатури всіх органічних сполук (розділ 1.1), наве
демо відповідну інформацію (таблиця 2.1). Перші члени ряду алканів гази, алкани С5- С |7 - рідини, вищі алкани - тверді речовини (парафін,
озокерит).
Таблиця 2.1.
Назви та молекулярні формули найбільш поширених алканів
Назва
алкану
Метан
Етан
Пропан
Бутан
Пентан
Г ексан
Г ептан
Октан
Нонан
Декан
Молекулярна
формула
СН4
с 2н 6
с 3н а
С4Н І0
с 5н 12
С*Н14
С 7Н І6
С 8Н 18
С 9Н 20
С 10Н 22
Назва алкану
Ундекан
Додекан
Тридекан
Т етрадекан
Ейкозан
Г енейкозан
Триаконтан
Т етраконтан
Пентаконтан
Г ектан
Молекулярна
формула
С:,Н 24
С 12Н 26
С 13Н 2В
С мН зо
С 20Н 42
с 2Ін 44
С 30н 62
С40н 82
С5оН|02
СіооНгм
73
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Хімічні властивості алканів
1. Реакції вшьнорадикального заміщення (8К)
Окремі атоми в молекулах алканів сполучені між собою простими
одинарними ст-зв’язками. Ці зв ’язки є неполярними (С -С -зв’язки) або
слабко полярними (С -Н -зв’язки). Тому міжатомні хімічні зв’язки в мо
лекулах алканів характеризуються значною міцністю порівняно з інши
ми зв’язками в молекулах органічних сполук і не схильними до гетеролітичного (іонного) розриву, виходячи з чого алкани являють собою хіміч
но інертні речовини.
Розщеплення внутрішньомолекулярних хімічних зв’язків у молеку
лах алканів відбувається за гемолітичним (вільнорадикальним) механіз
мом, який зазвичай реалізується за умов впливу високих температур
або дії на розчини вуглеводнів ультрафіолетового (УФ-) чи іонізуючого
випромінювання. За цих умов відбувається звичайно гемолітичний розрив
зв’язку С -Н з утворенням вільнорадикальних продуктів та заміщенням
водню на інший атом або групу атомів -р е а к ц ія вільнорадикального
■заміщення (5К). Більш жорсткі умови (подальше підвищення темпера
тури) призводять до гемолітичного розщеплення С -С -зв’язків (терміч
ний крекінг алканів).
1.1. Галогенування алканів
Взаємодія алканів з галогенами (хлором та бромом при третинному
атомі вуглецю) відбувається звичайно в умовах дії УФ-випромінювання
або високої температури і призводить до утворення моногалогеналканів,
які в подальшому можуть перетворюватися на полІгалогеналкани:
РМН + НаІ2 -> К-НаІ + Н-НаІ
Процес являє собою вільнорадикальну ланцюгову реакцію, що від
бувається в декілька стадій:
(1)
ініціювання ланцюга —здійснюється шляхом гемолітичного роз
риву неполярного ковалентного зв’язку в молекулі галогену з утворен
ням двох вільних радикалів:
СІ - 1 - С І
74
Ьу 0°)
> 2СГ
2.1. В углеводні та їх похідні
(2) зростання (подовження) ланцюга —реалізується в результаті
атаки радикалом галогену одного із зв’язків С—Н в молекулі алкану, який
піддається при цьому гемолітичному розриву; продуктами реакції є два нові
вільні радикали (алкану та водню), що підтягають подальшим перетворен
ням. В разі галогенування молекули метану (CH J процес має вигляд:
Н3С • • Н + С Г —» Н3С* (.метильний радикал) + НСІ
Радикал Н3С* атакує другу (нову в загальному випадку) молекулу
хлору, що призводить до утворення молекулярного продукту галогену
вання (хлорметану) та нового вільного радикала хлору:
Н 3С* + СІ • • СІ - > С Н 3СІ + СГ
Новий радикал хлору (СГ) може атакувати існуючі в реакційному
середовищі молекули алкану або хлоралкану, тобто виступати ініціато
ром подальшого утворення вільних радикалів і надання процесу харак
теру ланцюгового.
(3) обрив ланцюга - відбувається внаслідок зникнення або різкого
зменшення концентрації найбільш активних вільних радикалів. У випадку
реакцій галогенування це здійснюється в результаті взаємодії між со
бою (“рекомбінації”) радикалів хлору:
СГ + С Г
СІ • • СІ
або продуктів вільнорадикального окислення самих молекул алканів:
Н 3С -
+ Н 3С* -> с н 3 -
СН3
Обриву ланцюга сприяє також наявність у системі додаткових хіміч
них сполук - антиоксидантів, які, взаємодіючи з вільними радикала
ми, призводять до утворення малоактивних молекулярних продуктів.
Взаємодія вуглеводнів алканів та їх похідних з вільними радикалами має
важливе загальнобіологічне та медичне значення, оскільки за подібним
механізмом відбувається процес вільнорадикального перекисного окислен
ня поліненасичених жирних кислот біологічних структур (див. розділ 2.2).
75
Р озділ
г. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
1.2. Нітрування алканів
Нітрування алканів відбувається в умовах їх нагрівання з розбавле
ною азотною кислотою (реакція Коновалова):
Рі-Н + НКІ03 ->
к -и о 2 + н2о .
Реакція протікає також за механізмом вільнорадикального заміщен
ня (Бк) в результаті атаки молекули алкану вільним радикалом N 0 / .
2. Реакції окислення алканів
При повному окисленні (згорянні) алканів з утворенням кінцевих про
дуктів СО, та Н ,0 виділяється значна кількість енергії у вигляді тепла
та світла - 890 кДж/моль при згорянні одного моля метану, що дозволяє
використовувати ці сполуки як висококалорійне паливо. В умовах м ’яко
го окислення алканів у присутності каталізаторів (солей марганцю, хро
му) утворюється суміш кисеньвмісних похідних вуглеводнів - спиртів,
альдегідів, кетонів, карбонових кислот:
- Рі-СН 2ОН
R -C H 3-
R -C H =0
R-COOH
М едико-біологічне та фармацевтичне значення алканів та їх
похідних
Метан СН, (“болотний”, або “рудниковий” газ). Головна складова
(до 99%) природного паливного газу. В природних умовах утворюється
в результаті анаеробного бродіння целюлози, що входить до складу ви
копних рослинних залишків (“болотний газ”). Метан має слабку нарко
тичну дію, його присутність у вдихуваному повітрі в високих концентра
ціях призводить до розвитку тяжкої асфіксії.
Вазелінове масло медична - масляниста рідина, що являє собою
суміш алканів з довжиною вуглецевого ланцюга до С |5. Застосовується
як засіб з проносною дією (Oleum vaselini), розчинник при виготовленні
рідких лікарських форм та лініментів (Linimenta).
Вазелін - мазеподібна маса, що складається з рідких та твердих
алканів до С25. Використовується у фармацевтичній практиці як основа
для виготовлення мазей (Unguenta).
76
2.1. В углеводні та їх похідні
Парафій - суміш алканів С-1Й- С ,5, являє собого білу тверду масу.
Висока теплоємність парафіну сприяє його застосуванню як фізіотера
певтичного засобу.
Озокерит - природна суміш вищих алканів; тверда маса чорного
кольору Використовується у фізіотерапії за показаннями, близькими до
парафіну.
Хлороформ СНС13 (трихлорметан) - рідина із специфічним запахом,
що має наркотичну дію. Застосовується в медичній практиці як засіб
для інгаляційного наркозу (Chloroformium pro narcos і) в суміші з киснем
(0,5-1,5 об. %).
Йодоформ СНІ3 (трийодметан) - кристалічна речовина зеленува
то-жовтого кольору з характерним запахом. Використовується як анти
септичний засіб для зовнішнього використання (Iodoform іит) у вигляді
порошку або у складі мазей та паст.
Фторотан CF3CHBrCI (1,1,1-трифтор-2-хлор-2-бромстан) - летка
рідина, що має сильну наркотичну дію. Використовується як засіб для
інгаляційного наркозу {Phthorothanum).
Етилхлорид С,Н;С1 (хлоретил, хлоретан) - рідина з потужною нар
котичною дією. У зв’язку з високою леткістю хлоретил при взаємодії з
шкірою швидко випаровується, спричиняючи різке охолодження і знебо
люючу дію, що обумовлює його застосувашгя в медичній практиці як
засобу для місцевого знеболювання (Aethylii chloridum).
Нітроалкани. До нітроалканів належать нітрометан C H ,N 0 2та
нітропропани - 1-ніт ропропап CH 3CH 2CH 2N 0 2 та 2-ніт ропропаи
CH3CHNO,CH,. Ці речовини застосовуються як промислові розчинни
ки та компоненти ракетного палива і мають важливе токсикологічне
значення. При надходженні в організм нітроалкани спричиняють важкі
ураження центральної нервової системи та печінки.
2.1.2. А лкени. А лкадієни
Алкени - ненасичені вуглеводні, що мають один подвійний зв ’язок
(їх стара назва - “олефіни”). Загальна формула гомологічного ряду ал
кенів - С пН,п. Найпростішими представниками гомологічного ряду ал
кенів є:
77
Розділ
г. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
СН 2
СН2
Етеп
(ети л ен )
СН2 =
с н —с н 2— с н 3
Б у т е н -1
С Н 2 ------ СН
СНз
Пропен
(пропілен)
н3с - с н = с н — СНз
Бутен-2
Подвійний зв’язок є надзвичайно розповсюдженою структурою в біомолекулах, що надає останнім хімічних властивостей, характерних для
алкенів. Представниками метаболітів, що мають у своїй структурі один
подвійний зв’язок (моноєнові сполуки) є такі речовини, як мононенасичені
жирні кислоти (пальмітоолейюва, олеїнова, вакценова), інтермедіати
обміну вуглеводів (фумарова кислота), вітамін С (аскорбінова кислота)
тощо. Виходячи з цього, вивчення хімічних властивостей вуглеводнів,
що мають у структурі молекули подвійний зв ’язок, є надзвичайно важ
ливим для розуміння механізмів багатьох біохімічних реакцій.
Хімічні властивості алкенів
Як уже зазначено (розділ 1.3.1 ), атоми вуглецю в молекулі етилену
знаходяться в стані зр2-гібридизації. Це означає, що чотири валентності
вуглецю та відповідно чотири зв’язки, якими вуглець сполучається з інши-
Р ис.2,1. Перпендикулярні площини розміщення а- та л-зв’язків у молекулах вугле
воднів з подвійним зв'язком між атомами вуглецю
Л
78
2.1. В углеводні та їх похідні
ми атомами, неоднакові: три - а -зв ’язки, утворені гібридними зр2-орбіталями, та один л-зв’язок. Три сг-зв’язки (два зв’язки С -Н та один зв ’я
зок С -С ) розташовані в одній площині під кутом 120° один до одного,
я-зв’язок (другий зв ’язок С-С), який утворюється шляхом бічного пе
рекривання двох неспарених рг-електронів, розміщується в перпендику
лярній площині (рис. 2.1.); при цьому ділянки його максимальної електрон
ної густини скупчені по обидва боки (вище та нижче) лінії, що сполучає
ядра вуглецю - рис. 2.2.
Рис.2 .2 . Утворення іг-зв’язку в молекулах алкенів шляхом бічної взаємодії неспа
рених рг-електронів сусідніх атомів вуглецю
І. Реакиії електрофільного приєднання (А ^ до молекул алкенів
Два електрони я -зв’язку, що являють собою електронні хмари з мак
симумами щільності вище та нижче площини ог-зв’язку між атомами
вуглецю в молекулах алкенів, відрізняються за своїми властивостями
від електронів, які утворюють а -зв ’язок. Електрони я-зв’язку більш
рухомі і легко поляризуються під впливом електричного поля зовнішньої
е л е к тр о п о зи ти в н о ї ч асточки Е +. В иходячи з цього, най б ільш
характерними для алкенів є реакції електрофільного приєднання, що
відбуваються з розривом я-зв’язку.
Реакція відбувається за іонним (гетеролітичним) механізмом. Роз
глянемо взаємодію алкену з молекулою НА, що може дисоціювати на
іони Н+ та А“.
НА
►
Н+
+
А'
Сам процес електрофільного приєднання (АЕ) до молекули алкену
відбувається у дві стадії.
І стадія: Взаємодія електрофільної часточки (в даному випадку
протона Н+) з я-елекгронами подвійного зв ’язку. Результатом цього є
79
Розділ 2. ХАРАКТЕРИСТИКА О КРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
утворення проміжного я-комплексу, який шляхом перегрупування елек
тронів перетворюється на карбкатіон - комплекс, в якому два атоми
вуглецю сполучені ст-зв’язком (а-комплекс). Карбкатіон є часточкою,
що має позитивний заряд на атомі вуглецю:
Н
+ Н+
>
<
\
н+
а-комплекс
71-КОМПЛеКС
II стадія: Взаємодія карбкатіона з вільним аніоном А , що призво
дить до утворення з ним (аніоном) другого а -зв ’язку:
Н
-с-
+
A“
Н
А
-с-
-С-
За розглянутим механізмом відбувається взаємодія з подвійним
зв’язком алкенів галогеноводневих кислот (НСІ, НВг, НІ) та сірчаної
кислоти H ,S 0 4.
Приєднання до молекул алкенів води (гідратація) з утворенням спир
тів реалізується лише в присутності кислотних каталізаторів за схемою:
СН2 = С Н 2
+ Н 20
СН3 —
СН2ОН
Несиметричні алкени
Ш видкість взаємодії іонних сполук типу НА з алкенами суттєво за
лежить від ступеня поляризації подвійного зв’язку, що визначається індук
тивним та мезомерним ефектами сусідніх груп атомів. Зокрема, в мо
лекулах несиметричних алкенів (типу пропілену Н3С-СН =СН2) за раху
нок більшої елскгронегативності вуглецю порівняно з воднем відбува
ються електронні зсуви в напрямках Н—»С та Н3С—>СН. Це призво
дить до збільшення електронної густини на атомі вуглецю подвійного
зв’язку, що не сполучений з метальною групою, та утворенню на цьому
атомі часткового негативного заряду 5+:
80
2.1. В углеводні та їх похідні
н
1
6+
н — ►с — ► с н = с н 2
► СНз
$ -
СН=СН2
І
н
Внаслідок такого розподілу електричних зарядів зростає активність
приєднання протона Н* саме до атома вуглецю подвійного зв’язку, на яко
му зосереджений заряд 8' (більш гідрогенізованого атома вуглецю), а
аніона - до вуглецю, що має заряд 8+(мекш гідрогенізованого вуглецю).
Н+А’
с л
С Н 2 = с н — СН з
Такарегіоселективність взаємодії іонних сполук з подвійним зв’язком
молекул несиметричних алкенів дістала назву правила Марковмкова.
І. Г алогенування алкенів
При взаємодії алкенів з галогенами (хлором, бромом) відбувається
розрив подвійного зв’язку і утворення галогеноалкашв. Реакція також від
бувається за механізмом Аг В цьому випадку утворення електрофільної
часточки Е+відбувається шляхом поляризації ковалентного зв’язку в мо
лекулі галогену при дії п-електронної хмари алкену; в подальшому процес
відбувається двостадійно, як розглянуто для реакції гідрогалогенування.
У фармацевтичному аналізі в якості реакції на подвшнии зв’язок ви
користовується реакція взаємодії з алкеном брому, що супроводжує! ься
знебарвленням бурого кольору бромної води .
СН2 —
СН2 + Вт— *►Вг" — ► СН2Вг
СН2Вг
2 Р м к н ії відппвлешія молекул алкенів
^
>
Відновлення алкенів з приєднанням водню по місцю подвійного зв язку відбувається в присутності каталізаторів (нікелю, платини, паладію).
Результатом гідрування алкенів є утворення відповідних алканів:
81
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
І=і— с н = С Н — Н* + н2 — ► я — с н 2 — с н 2 — Я'
Процеси приєднання за місцем подвійного зв’язку широко розповсю
джені в біохімічних системах. Це переважно реакції гідратації та віднов
лення молекул а , |3-ненасичених карбонових кислот - низькомолекуляр
них метаболітів (фумарової, цис-аконіт ової, фосфоенолпіровиноградної', 5-дегідрош икімової кислот, транс-кротоніл-коензиму А) і
високомолекулярних (жирних) кислот, що протікають за участі біокаталізаторів - ферментів. Наявність карбоксильної групи призводить до
значної поляризації електронної густини впродовж вуглецевого ланцюга,
що призводить до суттєвих особливостей у протіканні реакцій приєднання
до біомолекул.
3. Окислення алкенів
Якісною реакцією на присутність подвійного вуглець-вуглецевого
зв’язку є також окислення алкенів перманганатом калію К М п04. В ре
зультаті реакції відбувається розрив я-зв’язку з утворенням двоатомного
спирту гліколю та випадання бурого осаду оксиду марганцю (IV):
ЗСН2=СН2 + 2КМ п0 4 + 4Н20 - > ЗСН2ОН-СН2ОН + 2КОН + 2Мп02
Алкадісіш
Вуглеводні, що містять два подвійні зв’язки, дістали назву алкадіснів
(дієнових вуглеводнів, або просто дієнів). Загальна формула гомологічно
го ряду алкадієнів - С Н . В залежності від локалізації подвійного зв’я
зку у вуглецевому ланцюгу дієнові вуглеводні розділяються на такі класи:
1) ізольовані алкадієни - алкадієни, в яких подвійні зв ’язки розді
лені впродовж ланцюга декількома метиленовими групами:
Р|2С =
с н — (СН2)П— СН =
СИ2
2) спряжені алкадієни - алкадієни, в яких два подвійні зв ’язки роз
ділені одним простим ст-зв’язком:
В2С = СН
СН = С И 2
Прикладами спряжених алкадієнів є:
1
2
СН2 = С Н
3
4
СН = С Н 2
СН2 = С Н ------С Н = С Н 2
СН3
Б у тад ієн -1,3
(ди вініл)
82
2 -м ети л б у тад ієн -1,3
(ізопрен)
2.1. В углеводні т а їх похідні
5
4
С Н 3—
СН =
3
2
СН —
1
СН = С Н 2
Пентадієн-1,3
3)
кумульовані алкадієни - такі, в яких два подвійні зв’язки розта
шовані біля одного атома вуглецю:
Я 2С = С
=
СИ2
Будова спряж ених алкадієнів
Алкадієни з ізольованими та кумульованими подвійними зв ’язками
за структурними особливостями та хімічною реактивністю майже не
відрізняються від звичайних дієнів. Найбільший інтерес становлять спря
жені алкадієни, що виявляють притаманні лише їм хімічні властивості;
до того ж спряжені подвійні зв’язки містяться у складі багатьох біомолекул ліпідної природи.
Найпростішим спряженим дієном € бутадієн-1,3, всі чотири атоми
вуглецю якого знаходяться в стані 5р2-гібридизації і лежать в одній пло
щині (рис. 2.3 -1).
Ііегібридизовані р^орбіталі цих атомів вуглецю перпендикулярні пло
щині а-скелета молекули і паралельні одна одній, що створює умови
взаємного перекривання р^-орбіталей не тільки між атомами С ,-С 2 та
С Г С4, а й між атомами С2 та С3 (рис. 2.3 -2). В результаті перекриван
ня чотирьох рг-орбіталей відбувається утворення спільної я-електронної хмари (рис. 2 .3 - 3 )., тобто спряж ення двох подвійних з в ’язків
(я-я-спряження).
Н1
2
с=с
( 1)
(2 )
(3)
Рис. 2 .3 . Електронна будова спряжених алкадієнів
У спряженій системі я-електрони не належать окремим вуглецьвуглецевим зв’язкам, в о н и —делокалізовані, тобто розподілені між трьо
ма зв’язками, що утворюють атоми С,,С2, С3т а С 4.
83
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Хімічні властивості спряжених дієнів
Подібно алкенам, для алкадієнів характерні реакції електрофільного
приєднання Ае. Проте, завдяки процесу спряження, в алкадієнах типу
бутадієну-1,3 при їх відновленні можуть утворюватися два продукти, що
є результатами 1,2- або 1,4-приєднання (рис. 2.4.):
Процес 1,4-приєднання є результатом спряження р-електронної хма
ри; в цьому випадку приєднання нових атомів відбувається на “кінцях”
системи, тобто до вуглеців С, та С4, що призводить до зсуву електронів
в напрямку “центру” і виникнення подвійного зв’язку між С2 та Сг
(1)
1
2
3
4
— *- с н 3 — с н 2 — с н = с н 2
1
2
сн2= с н
3
4
— СН = С И 2 —
1
(2 )
2
3
4
СН3 — С Н = С Н — СНз
Рис. 2 .4 . Відновлення спряжених алкадієнів: (1) 1,2-приєднання; (2) 1,4-приєднання
Біологічними представниками алкадієнів є ненасичені жирні кислоти
з двома подвійними зв’язками, що переважно входять до складу біоло
гічно важливих класів складних ліпідів, які утворюють основу біомембран та являють собою попередники в синтезі ендогенних фізіологічно
активних речовин. Біологічне та фармакологічне значення мають також
трісни та біомолекулн з декількома подвійними зв’язками - полієни:
поліненасичені жирні кислоти та відповідні ліпіди, похідні ізопренів, зокре
ма вітаміни А і Е та ейкозаноїди. Характерними для поліненасичених
жирних кислот є також реакції вільнорадикального перекисного окис
лення, що будуть окремо розглянуті в розділі 4.2.
2.1.3. Алкіни
А лкіни - ненасичені вуглеводні, що мають потрійний зв’язок; їх за
гальна формула С Н 2пГ Найпростішим представником гомологічного
ряду алкінів є ацетилен, тому ця група сполук має також назву аце
т иленових вуглеводнів.
НС ~ С Н
Е тин(ацетилен)
84
2.1. Вуглеводні та їх похідні
Назви алкінів за систематичною номенклатурою утворюються шля
хом заміни у відповідних алканах закінчення ~ан иа -ин(-ін), наприклад:
Н3С ~ С = С Н
П роп ін (м етил ацетил ен)
не
= с — с н 2 — СН3
Б у т и н -1
Хімічні властивості алкінів
Атоми вуглецю, що утворюють в молекулах алкінів потрійний зв’я
зок, знаходяться у стані зр-гібридизації, яка виникає в результаті ком
бінації однієї 25- та однієї 2р-орбіталі (рх). Гібридні зр-орбіталі двох ато
мів вуглецю, що розташовані лінійно, утворюють а -зв ’язки між собою
та з двома атомами водню.
Негібридизовані 2р-орбіталі (ру та рг), що шляхом бічного перекри
вання формують дві пари я-електронів, розміщені в площинах, перпен
дикулярних до площини а -зв ’язків (рис. 2.5. та 2.6.).
П лощ ина я - з в ’язку
Р ис.2 .5 . Розміщення а- та я-зв'язків у молекулах ацетиленових вуглеводнів у трьох
взаємно перпендикулярних площинах
Хімічні властивості алкінів зумовлюються наявністю в їх молекулах
потрійного зв’язку, тобто двох пар рухомих я-електронів. я-електронні
хмари алкенів є об’єктом атаки позитивно зарядженими електрофільними часточками Е+, тобто алкіни схильні до реакцій електрофільного при
єднання, подібно до алкенів. Проте алкіни менш активні в реакціях А Е,
що зумовлене більшою компактністю електронної густини в потрійному
зв’язку і, відповідно, меншою доступністю я-електронів для атаки електрофілами.
85
Розділ 2 . ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Р и с .2 .6 . Утворення тс-зв’язків у молекулах алкінів шляхом бічної взаємодії неспарених р2- та ру- електронів сусідніх атомів вуглецю
Ацетиленові вуглеводні в біологічних системах не зустрічаються,
проте деякі з них мають широке застосування в промисловості та фар
мацевтичному синтезі.
При надходженні в організм людини алкени діють як чужорідні хі
мічні сполуки (ксенобіотики), спричиняючи важкі отруєння.
2.2. А ром атичні вуглеводні (арени)
Аром атичні вуглеводні (арени) - сполуки, в молекулах яких міс
титься одне або декілька кілець бензолу (С6Н6) - циклічних структур,
що мають особливий характер зв’язків між атомами вуглецю.
2.2.1. Загальна характ еристика аромат ичних вуглеводнів
Загальновживана структурна формула бензолу (“формула Кекуле”):
Залежно від кількості бензольних кілець (циклів, ядер), що входять
до складу органічної молекули, арени підрозділяють на одноядєрні (моноциклічні) та багатоядерні (поліциклічні), до яких, у свою чергу, нале
жать арени з конденсованими (нафталін, антрацен, фенантрен тощо) та
ізольованими циклами.
Одноядєрні арени: будова, властивості, представники
До одноядерних (моноцгаогічних) аренів належить сам бензол та його
гомологи - похідні бензолу, в молекулах яких один або декілька аггомів вод
ню ароматичного кільця заміщені на вуглеводневі радикали, наприклад:
86
2 . 2 . А ром атичні вуглеводні (арени )
СоН
2П 5
М ети л бен зол (толуол )
Е тилбензол
Ізомери заміщених бензолів, що мають два замісники в положеннях
1,2; 1,3 та 1,4, називають орто- (о-), мета- (ш-) та пара- (р-) ізомерами
відповідно:
СН3
1,2-диметил бензол
(о-ксилол)
1,3-дим етилбензол
(.«-ксилол)
1,4-дим етил бензол
(л-ксилол)
Ароматичні радикали, що утворюються в разі відщеплення атома вод
ню від бензольного кільця або бічного вуглеводневого замісника, мають
назви “феніл” (фенільний радикал) та “бензил” (бензильний радикал):
\\ //
Ф еніл-
О -
с н 3—
Бензи л-
Похідні аренів, в яких атом водню заміщений на певну функціональну
групу (гідроксильну, карбоксильну, амінну тощо) розглядаються в межах
відповідних класів (гідроксисполук, карбонових кислот, амінів та ін.).
Медико-біологічне та фармацевтичне значення бензолу та його
похідних полягає в тому, що моноциклічні ароматичні структури входять до
складу багатьох біомолекул (зокрема, циклічних амінокислот та відповідно
пептидів і білків, вітамінів, коферментів, гормонів, нейромедіаторів) та
численних лікарських засобів природного і синтетичного походження.
Електронна будова ароматичних вуглеводнів
Наведені вище структурні формули бензолу, в яких чергуються оди
нарні та подвійні зв’язки, не зовсім адекватно характеризують його елек87
Р озділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
тронну будову та властивості. Насправді, всі вуглець-вуглецеві зв’язки в
молекулі бензолу та інших арєнів однакові ; до того ж бензол не виявляє
хімічних властивостей алкенів (наприклад, не знебарвлює “бромну воду”),
що свідчить про відсутність в його структурі справжніх подвійних
зв’язків.
В дійсності, всі шість С-атомів в молекулах аренів знаходяться в стані
5р2-гібридизації. Виходячи з цього, кожний атом вуглецю має три гібридні
орбіталі, які утворюють три ст-зв’язки (з двома сусідніми С-атомами та
одним атомом водню), що складають каркас молекули - правильний
шестикутник. Негібридизовані р-орбіталі (шість рг-АО) орієнтовані пер
пендикулярно площині шестикутника та паралельно одна одній (рис. 2.7 ):
Рис. 2 .7 . Електронна будова молекули бензолу. Показано ст-зв’язки, що формують
шестивуглецевий скелет молекули та негібридизовані рг-атомні орбі
талі
Бічна взаємодія зазначених шести рг-АО призводить до утворення трьох
я-зв’язків, або (користуючись уявленням про делокалізацію орбіталей, роз
глянутим на прикладі спряжених алкадієнів) - єдиної делокалізованої
я-системи. При цьому максимуми щільності я-електронної хмари розмі
щуються понад та під площиною ст-елеюронного скелета молекули:
я-електрокна
88
2 . 2 . А р ом ати ч н і вуглеводні (арени)
Рівномірний розподіл я-електронної щільності в межах циклічної сис
теми молекули бензолу дає можливість зображувати його будову у ви
гляді правильного шестикутника (кути між зв ’язками становлять 120°)
з кружечком всередині. Така модель будови бензолу відповідає повній
вирівняності довжини міжатомних С-С-зв’язків в його молекулі (0,139 нм,
що менше довжини одинарного зв’язку - 0,154 нм та більше подвійного 0,133 нм):
Замкнена спряжена система бензолу та його похідних характеризу
ється високою термодинамічною стійкістю молекули, оскільки при її
утворенні енергія електронної системи зменшується на 150 кДж/моль
(енергія спряж ення, або енергія делокалізацїі). Саме таку енергію
необхідно витратити для порушення розглянутої системи я-електронів у
молекулі бензолу, що пояснює високу хімічну стійкість цієї сполуки.
Критерії ароматичності органічних сполук
Розглянута електронна структура д істала назву “ароматичної
системи” і є характерною щонайперше для сполук бензольного ряду,
або таких, що містять у складі молекули ізольований бензольний цикл
(тобто моноцикл ічних арен ів та аренів з ізольованими циклами). Загальні
критерії ароматичності були сформульовані Е. Хюккелем (Е. Нііскеї).
Згідно з правилом Хюккеля, органічна сполука є ароматичною в
разі, якщо вона має плоский циклічний о-скелет та кількість спряжених
(усуспільнених) я-електронів (И), що дорівнює: N = 4п + 2, де п (число
циклів) = І, 2, 3 і т.д. Виходячи з цієї формули, кількість я-електронів в
ароматичних сполуках дорівнює 6 ( в молекулі бензолу), 10, 14 тощо.
Таким чином, ароматичними є також багатоядерні арени з конденсова
ними циклами (нафталін - 10 я-електронів, п=2; антрацен - 14 л-електронів, п=3; фенантрен - 14 я-електронів, п=3 тощо), а також деякі гете
роциклічні сполуки (групи піролу, піридину, фурану, тіофену тощо).
Сама назва “ароматичні сполуки” виникла історично для позначення
групи речовин природного походження, що були виділені на початку
XIX сторіччя з рослинних смол та бальзамів, які мали приємний запах.
89
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
2.2.2. Хімічні властивості ареиів
Характерною особливістю хімічних сполук із класу ареиів є їх здат
ність до реакцій заміщення, які не призводять до порушення ароматич
ності системи. Реакції приєднання, в результаті яких відбувається
порушення циклічної спряженої я-електронної системи, потребують
додаткових суттєвих енергетичних витрат і для ароматичних сполук
не властиві.
Оскільки в ході реакцій заміщення ареиів відбувається взаємодія з
я-електронною системою позитивно зарядженої (електрофільної) час
точки, такі реакції належать до електрофільного заміщення (8|;), що
може бути представлене у вигляді узагальненої схеми:
Н
Е
Загальний механізм реакцій 8Е, в які вступають арени, включає в
себе такі стадії:
1)
утворення електрофільної часточки Е+ за рахунок дисоціації або
гетеролітичного розщеплення сполуки ЕХ:
Е — X ----- Е+ + X
2)
утворення я-комплексу в результаті взаємодії електрофільного
реагенту Е+ (“електрофільної атаки”) з я-електронною хмарою арома
тичного кільця:
я-ком плекс
3) перетворення я-комплексу на 0-комплекс.
90
2.2. А р ом ати ч н і вуглеводні (арени )
Процес полягає в утворенні ковалентного зв’язку між електрофільною часточкою Е+та одним з атомів вуглецю ароматичного кільця. Зв’я
зок утворюється за рахунок двох електронів циклічної системи та при
зводить до переходу одного з С-атомів із зр3- у зр? - (тетраедричний)
стан, що позначається як формування а-комплексу. Решта (чотири)
я-електрони циклу розподіляються між п’ятьма іншими С-атомами, а
позитивний заряд, що вноситься Е'-часточкою, розподіляється в межах
всієї молекули, утворюючи карбкатіон:
-
V
V"
+
Н+
а-ком п лекс
(карб к атіон )
4) утворення кінцевого продукту слектрофільного заміщення.
Процес полягає у відщепленні від 0 -комплексу протона, сполученого
з атомом вуглецю, що зв’язаний з електрофільною часточкою. В ре
зультаті два л-електрони повертаються в циклічну систему, що призво
дить до відновлення ароматичної структури продукту реакції:
Реакції
в які вступають арени
До найбільш поширених 5^- реакцій, в які вступають арени, належать
нітрування, галогенування, сульфування, алкілування та ацилування.
І) Нітрування аренів
Нітрування аренів - це процес заміщення атома водню в бензольно
му ядрі на нітрогрупу - Ж ) г Реакція відбувається за умов дії на арени
91
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
так званої “нітруючої суміші”, що являє собою суміш концентрованих
азотної та сірчаної кислот:
нг3 0 4
с 6н6 + нм о3
■* с 6н5-м о г + н го
Електрофільиою часточкою в цій реакції є іон нітронію МО,+, що ви
никає при взаємодії азотної та сірчаної кислот. Перебіг реакції характер
ний для розглянутого вище загального механізму БЕ і може бути пред
ставлений такою послідовністю перетворень:
+
№
+ Н+
+ Ы02+
2) Галогенування аренів:
АІСІ3
+
СІ2
+
неї
Каталізатор реакції - хлорид алюмінію - являє собою кислоту Льюїса.
Він містить електронодефіцитний атом металу (алюмінію), під впливом
якого відбувається поляризація молекули галогену аж до утворення електрофільного катіона, що атакуєя-електронну систему бензолу:
§+
СГ + АІСІ4"
АІСк
СІ
С! +
3) Алкілування та аиилування аренів феакиія Фріделя - Крафтса):
Реакція полягає у введенні в бензольне ядро алкільного радикала, що
призводить до утворення гомологів бензолу -алкілбензолів:
,СН3
АІСІ3
+
92
СН3СІ
НСІ
2.2. А ром атичні вуглеводні (арени)
Продуктами реакції є метиловані, етиловані тощо похідні бензолу.
Процес відбувається за дії на ароматичні вуглеводні галогеноалканІв
в присутності каталізаторів-галогенідів алюмінію, які сприяють поля
ризації молекули галогеноалкану та утворенню електрофільної часточ
ки И.+, що атакує ароматичну систему (див, вище).
к - с і + А ісі3- ^ к +[А іс д Продуктом ацилування бензолу є ацетофенон (метилфенілкетон) сполука, що використовується в парфумерії та фармації як ароматичний
засіб.
Орієнтуючий вплив замісників у бензольному ядрі
Як було зазначено вище (розділ 1.3.3), різновид взаємного впливу ато
мів (замісників) в органічних молекулах, що передасться по системі спря
жених ті-зв’язків, має назву мезомерного ефекту. Молекула бензолу сама
по собі є спряженою системою з рівномірним розподілом щільності
ті-електронних хмар. Проте введення в ароматичне кільце замісника спри
чиняє перерозподіл електройної щільності, в результаті чого реакційна здатність молекули в реакціях електрофільиого заміщення змінюється.
Залежно від напрямку мезомерного ефекту, який вони спричиняють,
замісники в молекулі бензолу поділяються на два типи, а саме:
електронодонорні замісники (замісники І роду);
електроноакцепторні замісники (замісники II роду).
Електронодоиорні зам існики - такі, що збільшують електронну
щільність у спряженій ароматичній системі, зокрема за рахунок +М-ефекту.
До них належать, наприклад, гідроксильна група -О Н та амінна група
—МИ гетероатоми яких (кисень та азот) мають неподілені нари електро
нів, що вступають у спряжения з ті-електронною системою бензолу. В
результаті дії замісників І роду відбувається переважне зростання елект
ронної щільності в о- та ^-положеннях ароматичного кільця:
93
Р озділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
5-
6-
С
'Зі
Виходячи із зазначеного, введення в молекулу бензолу замісників
І роду активує протікання реакцій
в о- та р-положеннях (“орто- та
лорд-орієнтанти”)Електроноакцепторні замісники - такі, що зменшують електронну
щільність у спряженій ароматичній системі, тобто виявляють - М-ефект.
Електроноакцепторними замісниками є нітро- (-N 0 ,), сульфо- { - 5 0 3Н),
альдегідна (-С Н О ) та карбоксильна (-С О О Н ) групи. Ці замісники
відтягую ть на себе спряжену я-електронну хмару з ароматичної
системи. Введення в молекулу бензолу замісників II роду спричиняє
зростання електронної щільності в /«-положенні:
Результатом електрошіих ефектів замісників П роду є збільшення активносгі 8Е-реакцій в /я-положеніїі ароматичного кільця (“л/етя-орієнтанти”).
Реакції, іцо протікають за участю бічного ланцюга аренів
Реакції в бічному ланцюга гомологів бензолу відбуваються за загаль
ними закономірностями перетворень, властивих алкільним радикалам.
Зокрема, при дії сильних окислювачів відповідні ланцюги підлягають
окисленню. Кінцевим продуктом окислення бічного алкільпого радикала
є карбоксильна група, тому продуктами реакції є ароматичні карбонові
кислоти, наприклад:
94
2.2. А р ом атич ні вуглеводні (арени )
-сн .
ООН
Е тилбензол
М етилбензол
(толуол)
Подібні за загальною схемою перетворень реакції бічних ланцюгів
ароматичних вуглеводнів відбуваються в живих організмах у присут
ності специфічних каталізаторів - ферментів.
2.2.3. Багатоядерні арени: будова, представники
Багатоядерні арени, залежно від характеру сполучення в одній моле
кулі окремих бензольних циклів, підрозділяються на арени з конденсо
ваними циклами та з ізольованими бензольними циклами.
І)
Арени з конденсованими (анельованими) циклами, що мають
також назву “поліциклічних вуглеводнів”:
5
Н аф талін
4
5
10
А н трацен
4
1
Ю
Ф енантрен
Номенклатура похідних аренів з конденсованими циклами визначається
наведеною відповідно до правил ПО ПАК нумерацією вуглецевих атомів.
Хімічні властивості
Як зазначено вище, багатоядерні арени з конденсованими циклами
за своєю електронною структурою також належать до ароматичних
сполук. Тому їх хімічні властивості близькі до бензолу та його похідних.
Проте, оскільки в багатоядерних аренах відсутнє повне вирівнювання
л-електроиної щільності, ці сполуки є термодинамічно менш стійкими і
в цілому більш реакційноздатними, ніж бензольні сполуки. Зокрема,
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
8[:-рєакції у цих сполук протікають у м ’якших умовах, воші також більше
схильні до реакцій приєднання та окислення. З метою врахування не
рівнозначно сті С-атомів в молекулі нафталіну, положення 1,4,5,8 позна
чаються як а - , положення 2,3,6,7 - (ї. Це дає можливість розрізняти два
типи ізомерів нафталіну (а - та |3-), які є дещо відмінними за хімічними
властивостями - реакції електрофільного заміщення н молекулі нафта
ліну відбуваються переважно в а-положенні.
а
а
СН3
а -м е ти л н а ф та л іи
(З-метилнафталін
Медико-біологічне та фармацевтичне значення
Молекулярні структури поліциклічних конденсованих аренів є компо
нентами будови багатьох природних та синтетичних сполук, що мають
фізіологічну активність, у тому числі біомолекул та лікарських засобів.
Так, зокрема, похідна нафталіну -1,4-нафтохінон с основою будови
вітамінів К. Продукти гідрування фенантрену входять до складу природ
них фізіологічно активних сполук - похідних вуглеводню стерану - та
багатьох алкалоїдів — сполук рослинного походження, що мають лі
карські та токсичні властивості, в тому числі наркотичних засобів групи
морфіну. Широко поширені антибіотики класу тетрациклінів є похідними
поліциклічного ароматичного вуглеводню тетрацену:
Т етрац ен(н аф тацен )
Значний медико-біологічний інтерес представляють арени з конден
сованими циклами, що мають властивості хімічних канцерогенних спо
лук, які ушкоджують нормальну систему регуляції росту та поділу клі
тини. З них до найбільш активних сполук, що спричиняють злоякісну
трансформацію тваринних клітин, належать бенз(а)пірен, 3-мстилхолантрен, дибензантрацен - речовини, що містяться у кам’яновугільній смолі,
96
2.2. А ром атичні вуглеводні (арени)
тютюновому димі та викидах двигунів автотранспорту (т. з. “канцеро
генні вуглеводні”);
Бенз{а)пірен
3 -м етилхолантрен
7,12-дим етилбенз{а)антрацен
А зу ле н
Крім згаданих вище гетероциклів, представником небензоїдних аро
матичних сполук є азулен и - сполуки синьо-фіолегового кольору, моле
кула яких являє собою біциклічну конденсовану структуру. Структурни
ми компонентами азуленів є п’ятичлснне (циклопентадієнове) та шести
членне (циклогептатрієнове) кільця:
А зулен
(біцикло [ 3,5,0] декапентаєн)
Азулен та його алкілпохідні містяться в ефірних маслах деяких лі
карських рослин (ромашки, полину, евкаліпту тощо), які мають протиза
пальну активність.
2)
Арени з ізольованими циклами. В аренах з ізольованими цикла
ми бензольні ядра можуть буть сполучені або безпосередньо між собою,
або з участю аліфатичного ланцюга, наприклад:
Е іф екіл
Д и ф ен ілм етан
97
Р оїділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Хлоровані та бромовані похідні біфенілів (“полІхлороваиі біфеніли” ПХБ т а “полібромовані біфеніли” - ПББ) знайшли надзвичайно широке
застосування, зокрема в електротехнічній промисловості. Комплекси
ПХБ, починаючи з 20-х років XX сторіччя, на протязі багатьох десяти
річ вироблялись у великих кількостях у США (під комерційною назвою
“Ароклор”) та інших країнах, що призвело до масового забруднення цими
сполуками навколишнього середовища та надходження їх через повітря,
воду та продукти харчування в організм людини. Багато які з ПХБ та
ПББ мають токсичну та екоцидну дію, спричиняючи важкі захворювання
та генетичні порушення людей і тварин.
Похідним дифенілметану є активний пестицид (засіб для боротьби з
шкідливими комахами) ДЦТ (дихлордифенілтрихлоретан).
2.3. Гідроксисполуки (спирти, феноли). Тіоли
Гідроксильні (гідрокси-) сполуки - похідні вуглеводнів, у моле
кулах яких один або декілька атомів водню заміщені на гідроксильну
групу. Представниками гідроксисполук є спирти та феноли. Загальна
формула гідроксисполук - ROH, для фенолів використовується також
позначення АгОН.
Близькими за будовою та властивостями до гідроксипохідиих вуглевод
нів (спиртів та фенолів) є сполуки, в яких з атомом вуглецю зв’язанатіольна
(тіо-) група, - тіоли (меркаптани). Загальна формула тіолів - RSH.
За своїми хімічними властивостями спирти, феноли та меркаптани є
органічними протонними кислотами, кислотність яких зменшується в
ряду: АгОН > RSH > ROH.
2.3.1. Спирти (алкоголі): будова, властивості, представники
Спирти - похідні вуглеводнів, у молекулах яких гідроксильна група
сполучена з атомом вуглецю, що знаходиться в стані вр^-гібридизації.
Залежно від будови вуглеводневого радикала спирти поділяються на
аліфатичні, аліциклічні та ароматичні.
Аліфатичні спирти
Аліфатичні спирти складають гомологічний ряд, члени якого відрізня
ються на групу -С Н ,- Залежно від розміщення гідроксильної групи виді
ляють первинні (RCP^OH), вторинні (R-.CHOH) та третинні (R3COH) спир
ти. Наведемо приклади будови нижчих первинних аліфатичних спиртів:
98
2.2. А ром атичні вуглеводні (арени)
н 3с — с н 2— с н 2— о н
П р о п ан о л -1 (н-пропіловиіі спирт)
н 3с — с н 2—
с н 2— с н 2— ОН
Бутанол-1 (н-бутиловий спирт)
Аліциклічні спирти
Прикладом аліцшшічних спиртів є циклогексапол - речовина, у складі
якої гідроксильна група сполучена з одним із атомів вуглецю циклогексанового кільця;
Циклогсксанол
Аром ат ичні спирти
Ароматичні спирти (арилалканоли) —це похідні ароматичних вугле
воднів, в яких гідроксильна група сполучена з атомом вуглецю в бічно
му радикалі арену, наприклад:
Ф енілметанол (бензиловий спирт)
Сполуки, в яких гідроксильна група безпосередньо зв’язана з арома
тичним кільцем, складають клас фенолів.
Фізичні властивості спиртів
За фізичними властивостями нижчі аліфатичні спирти —рідини із спе
цифічним запахом. Вони широко застосовуються у фармації, лаборатор
99
Р озділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
ній практиці як жиророзчинники, компоненти сумішей для екстракції лі
підів з біологічних тканин.
Температури кипіння спиртів значно вищі, ніж у відповідних алканів.
Це пояснюється високою полярністю зв’язку -О Н , що спричиняє легкість
утворення водневих зв ’язків та формування асоціатів між окремими
молекулами спирту:
Я
Н
Полярність зв’язків -О Н пояснює також високу розчинність нижчих
спиртів у воді та їх застосування як розчинників для полярних речовин.
Ці властивості послаблюються із зростанням молекулярної маси та дов
жини вуглеводневого ланцюга аліфатичних спиртів.
Із збільшенням гідрофобності в ряду аліфатичних спиртів зроста
ють такі їх фізіологічні властивості, як наркотична дія та токсичність
(максимум у нерозгалужених спиртів з довжиною ланцюга С6- С 3).
Хімічні властивості спиртів
1. Кислотні властивості спиртів
Спирти мають властивості органічних ОН-кислот. Це зумовлено рух
ливістю атома водню гідроксильної групи, яка виникає внаслідок змі
щення електронної хмари від водню в напрямку електронегативного
атома кисню, що спричиняє поляризацію зв’язку - 0 - » Н і виникнення на
сусідніх атомах часткових зарядів:
5- 5 +
—о— Н
Внаслідок цього може відбуватися гетеролітичне розщеплення зв ’яз
ку О -Н , тобто полярна гідроксильна група є здатною до дисоціації. Така
реакція відбувається в присутності основ з утворенням алкоксид-аніона
та вільного протона Н+:
Я— ОН -4 Р — о - + Н +
100
2.3. Г ідрок си сполуки (спирти , ф ен оли). Тіоли
Спирти є слабкими протонними кислотами, зокрема рК а метанолу
та етанолу становить 16,0 та 18,0 відповідно, тоді як рКафенолу = 9,89, а
рКаоцтової кислоти = 4,76.
Кислотні реакції спиртів виявляються при їх реакціях з лужними ме
талами та кислотами:
а) взаємодія етанолу та інших нижчих спиртів з лужними металами
(натрієм, калієм) з утворенням алкоголятів:
2С2Н5-О Н + 2№ -> 2 С Д О - На* + Н2(г.)
Етанол
б) взаємодія етанолу з органічними та мінеральними кислотами з
утворенням складних ефірів (реакція етерифікації):
С2Н5-О Н + СН3-С О О Н -> С2Н5-0 - С 0 - С Н 3 + Н20;
Етилацетат
с 2н 5- о н + н о -г ю 2-> с 2н5- о - м о 2 + н 2о .
Етилнітрат
Механізм реакцій етерифікації буде розглянуто в розділі 2.5.
2. Пегідратаиія спиртів:
а)
внутрішньомолекулярна дегідратація з утворенням алкенів, що
протікає за механізмом елімінування (Е); реакція відбувається в умо
вах нагрівання спиртів з водовідні маючими речовинами при високих
температурах, наприклад:
н3с —сн— сн 2
_ _ і _____ І
І
і Н
1
Н23 0 4, 1 > 150°С
------------------
н3с —с н = с н 2
ГГропен (проп іл ен )
ОН {
П р о п ан о л -1
101
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
б)
міжмолекулярна дегідратація з утворенням простих ефірів; реак
ція протікає за механізмом нуклеофільного заміщення
при нижчих
температурах, наприклад:
г............. Н25 0 4, і <150°С
с 2н5— о н + но;— с 2н 5---------------------- - с 2н5— о — с 2н 5
Етанол
Е танол
Д іети л ов и й ефір
Реакція відбувається у дві стадії. Схема процесу:
1-ша стадія - протонування молекули спирту {за рахунок іона вод
ню мінеральної кислоти):
*
'
8.
+/ Н
я — с н 2— о — н + н+ — - я — с н 2— о ч
х н
2-га стадія - нуклеофільна атака оксонієвого катіона другою моле
кулою спирту; утворена перехідна структура втрачає молекулу води з
генерацією продукту реакції - простого ефіру; при цьому утворюється
оксонієвий катіон, в якому сусідній з киснем атом вуглецю має власти
вості електрофільного центру, з яким у подальшому може реагувати
нуклеофіл:
Н уклеоф ільний
центр
11
Е лектроф ільний
центр
X *
+ / 'н -н го
я — ОН
+ я — СН2— о х
—
*>
+
Я— О
- Н+
Я—
►
чн
—
-
я— о —
н
Н
3. Окислення спиртів
При окисленні спиртів утворюються карбонільні сполуки: при окис
ленні первинних спиртів - альдегіди (та в подальшому - карбонові кис
лоти), вторинних спиртів - кетони.
а) окислення первинних спиртів:
102
2.3. Гідрокси сполуки (спирти, феноли). Тіоли
я-сн2-он
[О]
* я-сн=о
[О]
» к-соон
б) окислення вторинних спиртів:
СН3-СН- С Н 3
О
Д и м ети л к етон (ацетон)
Окислення спиртів проводять за дії сильних окислювачів (хромової
суміші чи суміші перманганату калію з сірчаною кислотою) або дегід
руванням за участі металевих каталізаторів (міді, цинку, срібла). В біо
хімічних системах окислення спиртів відбувається шляхом дегідруван
ня переважно за умов дії специфічних ферментів - алкогольдегідрогеназ; таким шляхом в організмі людини етанол перетворюється на ацетальдегід, який в подальшому окислюється до двоокису вуглецю та води:
С Н 3— СН2— ОН
Е танол
-2Н
*
С Н — сн=о
А цетальдегід
Від накопичення в організмі ацетальдегіду - метаболіту, що може
утворювати ковалентні комплекси з білками мембранних структур ней
ронів головного мозку та клітин печінки, залежать церебро- та гепатотоксичні ефекти етилового спирту.
Ме.дико-біологічне та фармаиевтичне значення окремих спиртів
Метанол - безбарвна рідина (Ікиіі = 64,5 °С) з різким специфічним
запахом, близьким до етанолу, але відмінним від нього. Дуже токсична
сполука, при споживанні всередину може спричиняти ураження очних
нервів та сліпоту, у більших дозах (20-30 г) може давати летальний
ефект.
Етанол - безбарвна займиста рідина 0 ^ = 78 °С) із слабким запа
хом, легко змішується з водою. При споживанні людиною справляє спе
цифічний сп’яняючий ейфоризуючий психоневрологічний ефект, у вели
ких дозах проявляє наркотичну дію. Етанол широко використовується в
103
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
медичній практиці як знезаражуючий засіб для зовнішнього використан
ня, у фармації - як розчинник та екстрагуюча речовина при виготов
ленні рідких лікарських форм.
Широко розповсюдженим методом отримання етанолу є його фер
ментативний синтез, що використовується при виготовленні спиртних
напоїв. В основі методу лежить сукупність послідовних біохімічних реак
цій спиртового бродіння, які активно протікають за д ії на глюкозу
ферментів нижчих одноклітинних грибів - дріжджів Saccharomyces
cerevisiae:
С6Н1гО0 —> С гН5ОН + С 0 2 (г.)
Глюкоза
Е танол
Виходячи з цього, методом виготовлення вина (алкогольного налою,
що містить близько 12 % етанолу) є обробка комплексом дріжджових
ферментів (так званою “зимазою” - від “zyme" - дріжджі; грецьк.) ви
ноградного або інших фруктових соків, що містять значну кількість глю
кози. В разі використання для виготовлення спиртових напоїв крохмалю
картоплі або зерна злакових культур, полісахарид попередньо гідролізу ють з утворенням вільної глюкози. Видатний внесок у вивчення біохіміч
них механізмів та ферментів спиртового бродіння вніс визначний фран
цузький дослідник Луї Пастер (Louis Pasteur; 1822-1895).
Багатоатомні спирти
Спирти, до складу яких входить декілька гідроксильних груп, дістали
назву багатоатомних спиртів (поліолів). Розрізняють дво-, три- та
поліатомні спирти (зокрема, чотириатомні - еритрити, п ’ятиатомні пентити, шестиатомні - гексити).
Ациклічні багатоатомні спирти
Серед ациклічних багатоатомних спиртів значний інтерес становлять такі
сполуки, як двоатомний спирт етиленгліколь та триатомний - гліцерин.
он он
Етиленгліколь
(етан д іо л -1,2)
104
Гліцерин
(п р оп ан тріол-1,2,3)
2.3. Г ідрок си сп олукн (сп и р ти , ф еноли). Т іоли
Етиленгліколь - гігроскопічна в ’язка рідина, що застосовується як
антифриз. При споживанні per os в якості сурогата алкогольного напою
етиленгліколь виявляє значну токсичну дію з розвитком важкої нефро
патії, проте в мікромолярних кількостях утворюється внутрішньоклітин
но в процесі метаболізму.
Гліцерин (гліцерол) - безбарвна рідина з солодкуватим смаком. В
організмі людини і тварин гліцерин та його фосфорні ефіри є продуктами
метаболізму вуглеводів та ліпідів. У фармації гліцерин широко викорис
товується як основа для виготовлення рідких лікарських форм для зов
нішнього застосування.
Біологічно важливі ациклічні поліатомні спирти утворюються при
метаболізмі вуглеводів (сорбіт, ксиліт, маніт).
Циклічні багатоатомні спирти
Представником циклічних багатоатомних спиртів є інозит (інозитол; циклогексангексаол-1,2,3,4,5,6). Інозит є компонентом важливо
го класу мембранних ліпідів - інозитолфосфат идів. Фосфорильовані
похідні інозиту, зокрема інозитол-1,4,5-трифосфат, виконують роль внут
рішньоклітинних регуляторів транспорту та розподілу іонів кальцію та
активності Са2+-залежних біохімічних та фізіологічних функцій клітини.
Інозит
Інозитол-1,4,5-трифосфат
Альдегідо- та кетопохідними багатоатомних спиртів (ациклічних та
циклічних) є вуглеводи - група природних сполук, які у зв ’язку з їх розповсюдженістю та великим біологічним значенням виділені в окремий
клас сполук.
105
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
2.3.2.
Феноли: будова, властивості, представники
Феноли (ареноли) - похідні ароматичних вуглеводнів, молекули яких
містять одну або декілька гідроксильних груп, що сполучені з бензоль
ним кільцем. В молекулах фенолів група -О Н зв’язана з атомом вугле
цю, який знаходиться у стані зр2-гібридизації.
Залежно від кількості гідроксильних груп, розрізняють одноатомні (власне
ареноли), дво- та триатомні (арендіоли, арентріоли, відповідно) феноли:
Одноатомні феноли:
ОН
ОН
СНз
Ф енол
2-м етилф енол (о-крезол)
Двоат ом ні феноли:
он
,0 Н
он
1,2-д игід рокснб ензол
(пірокатехін)
НО
1,3-дигідроксибензол
(резорцин )
Триатомні феноли:
1,4-дигідроксибензол
(гідрохінон )
ОН
он
'ОН
он
1,2,3-тригідроксибензол
(пірогалол)
ОН
1,3,5 -тригі дрокси бензо л
(ф лороглю ц ин)
он
3,2,4-тригідроксибензол
(окси гідрохін он )
Фізичні властивості фенолів
Більшість фенолів є твердими речовинами з низькими температурами
плавлення (1пщщгі фенолу = 41 °С) або рідинами (деякі алкілфеноли) із спе
цифічним різким запахом “карболки”. У воді феноли малорозчинні.
106
2 3 . Г ід р о к с и с п о л у к и (с п и р т и , ф е н о л и ). Т іо л и
Х ім ічн і власт ивост і фенолів
Хімічні властивості фенолу С6Н5ОН та його похідних визначаються
наявністю гідроксильної групи та бензольного кільця, що взаємно впли
вають одне на одного. Ця взаємодія полягає у спряженні неподіленої
пари р-електронів атома кисню -ОН-групи з л-електронами ароматич
ного кільця (р, я-спряження), внаслідок чого відбуваються такі ефекти:
1) зменшення електронної густини на атомі кисню, що, у свою чергу,
супроводжується збільшенням поляризації гідроксильної групи та послаб
ленням зв ’язку О -Н ;
2) збільшення електронної густини в бензольному кільці з максиму
мами збільшення в о- та/> положеннях відносно гідроксильної групи.
Зазначені електронні ефекти визначають хімічні властивості фено
лів, а саме:
- їх кислотні властивості (внаслідок легкої дисоціації іона водню
поляризованої гідроксильної групи:
а
+М > -І
- здатність до реакцій електрофільного заміщення в о- та р-положеннях.
К ислот ні власт ивост і ф енолів. Феноли є слабкими кислотами,
проте їх кислотність вища, ніж у спиртів, зокрема рКа фенолу (карболо
вої кислоти) = 9,89. В якості кислот феноли здатні до взаємодії з лужни
ми металами та їх гідроксидами з утворенням відповідних феноляті в
(феноксидів):
а) взаємодія фенолу з лужними металами (натрієм, калієм):
2С6Н5-О Н + 2№ -> 2С6Н50 -
+ Н2 (г.)
б) взаємодія фенолу з гідроксидами лужних металів:
С6Н5-О Н + №ОН -> С6Н5а N3* + Н20.
107
Розділ 2. ХАРАКТЕРИСТИКА О КРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Реакції електрофільного заміщення. Як вже показано, гідроксиль
на група в молекулах фенолів є замісником (орієнтантом) І роду, тобто
вона збільшує електронну густину в ароматичному кільці в о- та р-попоженнях. При цьому відповідні атоми вуглецю набувають часткових нега
тивних зарядів і стають центрами, що здатні приєднувати електрофільні
часточки Е+, а на атомі кисню виникає частковий позитивний заряд:
Виходячи з цього, фенол та його похідні більше здатні до реакцій
електрофільного заміщення, ніж незаміщені арени. Прикладами зазна
чених реакцій є бромування та нітрування фенолу:
а) бромування фенолу (утворюється осад 2,4,6-трибромфенолу):
ОН
+
ЗНВг
ЗВг2
б)
нітрування ф енолу (утворю ється сполука жовтого кольору
2,4,6-тринітрофенол - пікринова кислота):
ОН
ОН
+
ЗНШ 3
зн2о
Наведені реакції поширені в лабораторній практиці як якісні реакції,
характерні для фенольного кільця (в тому числі у складі біомолекул та
фізіологічно активних сполук).
108
2.3. Пдроксисполукк (спирти, феноли). Тіоли
2.3.3. Тіоли (меркаптани)
Тіоли (тривіальна назва-м еркапт ани) - похідні вуглеводнів, у мо
лекулах яких один або декілька атомів водню заміщені на тіо- (меркапто-) групу -БН .
ТІовмісні аналоги спиртів мають назву тіоспиртів (К-8Н), фенолів тіофенолів (Аг-БН).
Представниками тіоспиртів є:
метантіол (метилмеркаптан)
СН3- БН
етантіол (етилмеркаптан)
С2н , - БН
пропантіол (пропілмеркаптан) С,Н 7- 8Н
Представниками тіофенолів є:
<^Узн
Т іоф ен ол (м еркаптобензол)
2-м ети лтіоф енол
Сульфіди та дисульфіди
Сірковмісні аналога простих ефірів та органічних пероксидів мають
назву тіоефірів - сульфідів (Я - Б - її) та дисульфідів (Я - Б - 8 - И):
СН3- 8 - С Н 3
Д и м ети л сул ьф ід
СН3- Б - Б - СН3
Д к м етш щ и сул ьф ід
Фізичні властивості тіолів
Тіоли є рідинами з неприємним запахом, що нагадує запах гниючої риби
(властивість використовується шляхом додавання етан- та пентантіолів
до побутового газу, що дозволяє органолептично виявити його витікання).
Внаслідок незначної схильності до утворення водневих зв’язків, темпера
тура кипіння тіолів значно нижча, ніж у відповідних спиртів.
Хімічні властивості тіолів
За своїми хімічними властивостями тіоли є значною мірою сірковміс
ними аналогами спиртів та фенолів і мають властивості органічних кислот.
І) Кислотні властивості тіолів
Тіоли є сильнішими кислотами, ніж відповідні спирти (рКа етантіолу = И , тоді як рКа етанолу = 18) і можуть утворювати з металами
солі —тіоляти (меркаптиди):
109
Роза і л 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
з лужними металами —розчинні в воді солі:
СН3СНг - ЗН + № О Н -» СН3СН2- 5 “Ма+ + Н20;
Е тантіол
Ет&нтіолят натрію
з іонами важких металів - нерозчинні у воді солі, наприклад, при вза
ємодії етантіолу з сулемою:
2СН3СН2- БН + НдС(2 -> (СН3СН2- Б)2Нд + 2 НСІ.
Е таитіолят ртуті
Утворення з іонами важких металів (ртуті, свинцю, цинку тощо) не
розчинних у воді тіолятів використовується в медицині шляхом застосу
вання тіолів (зокрема, унітіолу) як антидотів при отруєнні важкими ме
талами (ртуттю, свинцем, цинком тощо).
2) Окислення тіолів
При окисленні тіолів, яке відбувається по атому сірки, залежно від
умов процесу, утворюються різні групи продуктів:
а)
При окисленні однієї молекули тіолу послідовно утворюються такі
похідні як сульфенові, сульфінові тасульфонові кислоти (сульфокислоти Я - 8 0 3Н):
О
Я - 8 - ОН
Я -в-О Н
О
И - И - ОН
II
О
тіол
сульф енова
ки сл ота
сульф інова
ки сл ота
сульфонова
кислота
За такою схемою в організмі відбу вається окислення БН-вмІсної амі
нокислоти цистеїну (див. нижче).
Ароматичні сульфокислоти
Важливим прикладом ароматичної сульфокислоти (А г-8 0 3Н) є бен
зол сульфокислота С6Н5- 8 0 3Н, похідні якої —аміди сульфанілової кисло
ти (р-амінобензолсульфокислоти) є розповсюдженими антимікробними
засобами (сульфаніл):
110
2.3. Г ідрок си сп олуки (сп и р ти , ф еноли). Т іоли
О
II
— Б — ОН
О
Б єн зол су л ьфо к ис л ота
I
О
н 2м — \
о
Сульф аніл о в а кислота
(р-амі ноб ензолсульфоки слота)
і
О
Н2Ы
ЫН2
о
А м ід сульф анілової кислоти
(сульфаніламід; стрептоцид)
б)
М ’яке окислення двох молекул тіолів призводить до утворення
сульфідів (дисульфідів, алкілдисульфідів) за схемою:
СНз-Э
Н + 0 + Н Б - СН3
СН3- Б - Э - СН3 + Н20;
Ця реакція оборотна. Утворення дисульфідиих “місточків” - 8 - 8 - між
двома молекулами цистеїну у складі пептидних ланцюгів та їх зворотне
відновлення є розповсюдженим процесом у хімії та біохімії білків та
пептидів.
в)
Подальше окислення сульфідів дає такі продукти, як сульфоксиди
та сульфони (сульфонові кислоти, сульфокислоти):
111
Розділ
г. ХАРАКТЕРИСТИКА О КРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
[О ]
Я - Б - Р
[О ]
-------> Я - Э - И
||
> Я - Э - Р
о
О
Сульф он
Сульф оксид
Прикладом сульфоксидів є диметилсульфоксид (димексид) - ре
човина, що широко застосовується в фармакології та фармації.
СНз-Б-СНз
II
О
Д им етилеульф оксид
М едико-біологічне та фармацевтичне значення тіолів та су
льфідів
Цистеїн. Цистин. Глутатіон
Завдяки здатності до оборотного утворення дисульфідів, тіоли як біомолекули та фармацевтичні препарати приймають участь в регуляції
окисно-відновних реакцій у живих організмах. Таку функцію відіграє,
зокрема, 5Н-вмісна амінокислота цистеїн, яка шляхом окислення може
перетворюватися на дикарбоновий аналог дисульфід цистин:
NN 2
?Н
— сн;
СН2
Н о Ы -С Н -С О О Н
Ц и стеїн
СН
СООН
Ц истин
Тіогрупи цистеїну відіграють важливу роль у створенні та функціону
ванні активних центрів багатьох ферментів (дегідрогеназ тощо), а дисульфідні місточки - у формуванні тривимірної просторової конформації
багатьох білків з ферментною (рибонуклеаза) та гормональною (інсулін,
соматотропний гормон) активністю. Значне місце в контролі внутрішньо-
112
2.3. Гідрокснснолуки (спирти, феноли). Тіоли
клітинних окисно-відновних процесів посідає цистеїнвшсний трипептид
глутатіон.
Прикладом сполуки, що бере участь в реакціях біологічного окис
лення за рахунок зворотного переходу між тіоловою та дисульфідними
формами молекули, є біомолекула з вітамінними властивостями (кофер
мент) ліпоєва кислота:
СН2
Н2С|
о н — сн 2 — сн 2— сн 2— сн2— соон
5— Б
Л іп оєва кислота (6,8-дитіооктанова кислота)
Метилмеркаптан (СН^-БН) - газ, утворюється в товстому кишеч
нику людини та вищих тварин при розщепленні сірковмісних амінокис
лот продуктів харчування під впливом бактеріальної мікрофлори (“мік
робне гниття білків” у кишечнику).
Унітіол (2,3-димеркаптосульфонат натрію) - антидот для лікування
гострих та хронічних отруєнь сполуками важких металів (ртуті, миш’я
ку, хрому, вісмуту тощо), які за механізмом токсичної дії належать до
так званих “тіолових отрут”, тобто здатні в організмі людини і тварин
утворювати ковалентні зв ’язки із 8 Н - групами ферментних білків, що
призводить до втрати останніми каталітичних властивостей.
СН2 —
СН —
СН2
БН
БН
З 0 3№
Унітіол
сн2
БН
СН —
БН
СН2
ОН
БА Л
Унітіол близький за будовою та властивостями до так званого “бри
танського антилюїзиту (БАЛ)” - (2,3-димеркаптопропанолу) - фарма
кологічного засобу, що застосовується як антидот при отруєнні люїзи
том - бойовою отруйною речовиною, що містить миш’як.
Дішетилсульфоксид (димексид) - сполука, що є високоефективним
органічним розчинником; застосовується у фармацевтичній практиці як
зовнішній (поверх шкіри) засіб в якості розчинника для інших лікарських
речовин та ФАР із власними протизапальними властивостями.
113
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Іприт [сірчанистий іприт; Р, (У-дихлордіетилсульфід - (СІСЕ^СНДБ)] сполука, прийнята на озброєння арміями багатьох країн як БОР (“гірчич
ний газ”). За механізмом отруйного ефекту іприт належить до речовин
цитотоксичної дії із шкірнонаривним, імунотропним та (при високих
дозах) -загапьнотоксичним ефектом. Вперше був застосований у період
першої світової війни в 1917 р. збройними силами Німеччини поблизу
бельгійського містечка Іпр. Подібно до інших БОР, іприт належить до
отруйних речовин з надзвичайно високим ступенем токсичності; його
токсична концентрація в повітрі становить 0,0001 %.
2.4. Карбонільні сполуки (альдегіди, кетони)
До карбонільних сполук (оксосполук) належать альдегіди, ке
тони та карбонові кислоти - похідні вуглеводнів, що мають у своїй
структурі карбонільну (або оксо-) групу.
К арбон іл ьн а група
2.4.1.
Загальна характ ерист ика карбонільних сполук
Загальні формули представників класів, що належать до карбоніль
них сполук, мають вигляд:
я — с — Я'
II
о
Альдегіди
Кетони
Я = Н,Л1к,Лг
Я Д '= А1к,Аг
Карбонові кислоти
К = Н,Л Ік,А г
Незважаючи на близькість хімічної будови (наявність оксогрупи), ці
класи сполук (альдегіди і кетони, з одного боку, та карбонові кислоти з іншого) суттєво різняться між собою за хімічними властивостями, тому
карбонові кислоти будуть розглянуті окремо в розділі 2.5.
Номенклатура альдегідів та кетонів
Назви альдегідів за міжнародною номенклатурою угворюють шля
хом додавання суфікса -аль до назви відповідного вуглеводню, що має
найдовший вуглецевий ланцюг, який включає карбонільну групу (мета114
2.4. К арбон іл ь н і сполук и (ал ьдегіди , кетони)
наль, етаналь тощо). Тривіальні назви альдегідів мають в кореневій
основі назву карбонової кислоти, до якої окислюється цей альдегід, з
додаванням слова “альдегід”. Наприклад, перший представник ряду
альдегідів - метаналь - називають мурашиним альдегідом, або фор
мальдегідом, оскільки вій перетворюється на мурашину кислоту (Acidum
form icum - лат.)
Таблиця 2.2,
Найменування та скорочені структурні формули поширених
альдегідів
Назва альдегіду
Аліфатичні альдегіди
Метаналь (формальдегід,
мурашиний альдегід)
Етаналь (ацетальдегід, оцтовий
альдегід)
ГІропаналь (пропіоновий альдегід)
Пропеналь (акролеїн)
Бутаналь (бутиральдегід, масляний
альдегід)
Пентаналь (валеріановий альдегід)
Ароматичні альдегіди
Бензальдегід (бензойний альдегід)
2-Гідроксибензальдегід
(саліциловий альдегід)
Скорочена структурна
формула
н -сн о
СНз-СНО
СНг СН2-СНО
СН2 = с н - с н о
СН г СН2-СН2-СНО
С Н з-С Н гС Н гС Н гС Н О
С6Н5-СНО
С6Н5(ОН)-СНО
Назви кетонів за міжнародною номенклатурою утворюють шляхом
додавання до кореневого вуглеводню (з найдовшим вуглецевим лан
цюгом) суфікса -он (пропанон, бутанон тощо). Для найменування
кетонів, особливо ароматичних, використовується також радикало-функціональна номенклатура, згідно з якою в назві сполуки вказуються в
алфавітному порядку радикали, що сполучені з карбонільною групою, з
додаванням слова кетон. Багато кетонів мають також тривіальні на
зви, наприклад: диметилкетон —ацетон; дифеиілкетон - бензофенон.
115
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Б ен зал ьдегід
С аліци лови й
альдегід
В ан ілін (4-гідрокся3-м егоксн бензальдегід)
Таблиця 2.3.
Найменування та структурні формули поширених кетонів
Структурна
формула
Н азва кет ону
Аліфатичні кетони
СНз-С-СНз
II
0
Бутанон
СНз-С-СНз-СНз
(метилетилкетон)
II
0
3-М етил бутанон-2
СН з-С-СН-СН з
(метияізопропілкетон)
1 1
0 СН3
Пентанон-2
СН з-С -С Н 2-С Н 2-СНз
(метилпропілкетон)
і
0
Пентанон-3 (діетилкетон)
СНз-СН2-С -С Н 2-СНз
II
0
Пентсн-4-он-2 (метилалілкетон)
с н 3- с - с н 2- с н = с н 2
II
Пропанон
(диметилкетон, ацетон)
0
116
2 . 4 . К арбон ільні сполук и (ал ьдегіди , кетони )
Таблиця 2.3. (продовж ення)
Найменування та структурні формули поширених кетонів
Назва кет ону
Структурна
формула
Ароматичні кетони
С6Н ;-С -С 6Н5
II
0
С 6Н5-С-(СН2)2-С Н 3
Дифенілкетон (бензофенон)
Пропілфенілкетон
0
Будова карбонільної групи
Атоми вуглецю карбонільної групи С = 0 знаходяться у стані 5р2-гібридизації, тобто мають три гібридизовані орбіталі, що використовуються
для утворення трьох ст-зв’язків - двох із сусідніми С-атомами (радика
лами Я та К ' - рис. 2.8) та одного - з атомом кисню. Другий зв ’язок
між карбонільним вуглецем та киснем - це л-зв’язок, утворений за ра
хунок бічного перекривання двох нсспарених рх-електронів - одного від
атома вуглецю та одного - від кисню:
X
п
Рис. 2 .8 . Е л е к тр о н н а б у д о в а к ар б о н іл ьн о ї групи
Висока електронегативність атома кисню карбонільної групи сприяє
зміщенню до нього електронної густини хмари л-зв’язку. Внаслідок цього
на атомі кисню виникає частковий негативний заряд 8-, тобто утворюєть
ся електрофільний центр карбонільної групи:
117
Р озділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
2.4.2. Хімічні властивості альдегідів та кетонів
Характерними для альдегідів та кетонів є два типи реакцій:
1) реакції нуклеофільного приєднання (А^) до карбонільної групи; при
цьому альдегіди більш активні в реакціях
ніж кетони;
2) реакції відновлення та окислення.
Для карбонових кислот характерною реакцією є нуклеофільне замі
щення (Б Д що буде розглянуто в наступному розділі (2.5).
1. Нуклеофільне приєднання до карбонільних сполук
Здатність альдегідів та кетонів до реакцій нуклеофільного приєднан
ня А,,, визначається наявністю на атомі вуглецю карбонільної групи част
кового позитивного заряду, який виникає внаслідок зміщення електрон
ної густини в оксогрупі в бік кисню:
Завдяки такій електронній будові атом вуглецю оксогрупи є схиль
ним до атаки нуклеоф ілом, а атом кисню - електрофілом, що і виз
начає основний клас хімічних перетворень, до яких схильні альдегіди
(у більшій мірі) та кетони, а саме —реакції нуклеофільного приєд
нання А ы.
Загальна схема реакції нуклеофільного приєднання:
Е+
ІЧи
'Й
Розглянемо взаємодію карбонільної сполуки з реагентом {Х -У }, що
здатний до утворення іонів нуклеофільного (У-) та електрофільного (Х+)
характеру. Реакція Ам відбувається ступінчасто - у два етапи.
118
2.4. К арбон ільні сполук и (альдегіди, кетони)
1) На першому етапі (повільна, лімітуюча реакція!) відбувається вза
ємодія нуклеофільного реагенту з позитивно зарядженим вуглецем кар
бонільної групи. В результаті цієї “нуклеофІльної атаки” відбувається
гетеролітичне розщеплення зв’язку С = 0 І перехід пари електронів на
атом кисню з утворенням алкоксид-іона:
Я
Я
(1)
£.
) с = о
V
Я
V
--------
У “ ...... С = 0
ІЯ'
У— с — О*
І
Я'
2) Другий етап (відбувається швидко) полягає у приєднанні електрофілу (найчастіше - іона водню з середовища) до алкоксид-іона із стабілі
зацією останнього, тобто утворенням кінцевого продукту реакції.
Я
(2)
I
.
У— С — О
І
Я'
Я
Х+
|
► У— С — ОХ
І
Я'
Найбільш поширеними реакціями Ам, що їм підлягають карбонільні
сполуки, є взаємодія альдегідів з ціанідами, спиртами, водою, амінами,
бісульфітом.
1.1. Взаємодія з ціанідами металів
При взаємодії карбонільних сполук з солями ціановодневої кислоти
НСМ (ціанідам и) утворю ю ться ціангідрини (а-гідроксиніт рили).
Нуклеофільною часточкою в реакції є ціанід-іон СІЧ-:
Реакція важлива в органічному синтезі, оскільки гідроксинітрили є ви
хідними продуктами для отримання а-гідроксикислот та а-амінокислот.
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
1.2. Взаємодія із спиртами
При взаємодії карбонільних сполук (переважно альдегідів) з однією
або двома молекулами спирту утворюються напівацеталі та ацеталі
за механізмами нуклєофільного приєднання (А^) та нуклеофільного за
міщення (5,^) відповідно.
Н а п ів а ц е т а л і- похідні оксосполук, що містять при атомі вуглецю
гідроксильну та алкокешіьну групи.
А ц е т а л і- похідні оксосполук, що містять при атомі вуглецю дві алкоксильні групи.
он
ОЯ'
Н— С — О Я '
Н— с
ОЯ'
Н
Н
Н ап івац еталь
А цеталь
Нукпеофільною часточкою в цих реакціях є алкоксильна група спиртів.
Утворення напівацет алей (за механізмом А ^ :
ю н3
И—
но— сн3
г
с н — он
Утворення ацеталей (за механізмом 5у.ОСН3 _____
Я
СН— ОН
+
ОСНз
НО— СН3
я — с н — о с н 3 + н он
Реакції утворення ацеталей мають особливо велике значення в хімії
вуглеводів.
1.3. Взаємодія з водою
Альдегіди, як більш активні представники оксосполук, здатні до при
єднання молекули води з утворенням гідратів (діо.чів). Реакція оборотна,
і стабільні гідрати утворюються тільки з деякими активними альдегіда
ми, наприклад формальдегідом, трихлороцтовим альдегідом:
120
2.4. К арбон ільні сполук и (ал ьдегіди , кетони)
н ч / °н
н
/
с\
он
Г ід рат ф орм альдегіду
Рівновагу реакції взаємодії з водою формальдегіду суттєво зсуну
то праворуч; формальдегід у водних розчинах гідратований більш ніж
на 99,9 %.
При розчиненні у воді трихлороцтового альдегіду (хлоралю) утво
рюється стабільна сполука хлоралгідрат, що може бути виділена у
вигляді індивідуальної речовини. Хлоралгідрат має наркотичну актив
ність, що зумовило його застосування в медицині як снодійного засобу.
СІ
п
\
//
С І~ ^ 3 — с х
СІ
+ н 2о
Н
Чн
Х лорал ь
Х лоралгідрат
1.4. Взаємодія з амінами
Взаємодія карбонільних сполук з первинними амінами та іншими азотвмісними речовинами загального типу X -N H 2 відбувається за механіз
мом “приєднання-відщеплення” і складається з таких етапів:
1)
Реакція приєднання нуклеофільної часточки H2N -X (де X = -Н ,
-О Н , -N H j, -A lk, - А г ) до позитивно зарядженого вуглецю карбонільної
групи; при цьому подвійний зв’язок в групі С = 0 розривається:
Н?ІЇ — X
Н\
/ ° Н
/С ч
И'7
N4
X
2) Відщеплення від термодинамічно нестійкого продукту реакції Ам
молекули води (реакція елімінації - Е) з утворенням іміну (“основи
Шифа”):
121
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
в \
►
Я '/
\м [н ]— х
/С = м — X
р
Прикладами послідовного механізму “нуклеофільне приєднання елімінація” (Ак - Е) є реакції взаємодії альдегідів та кетонів з такими
похідними аміаку, як гідроксиламін, гідразин, фенілгідразин:
- взаємодія з гідроксиламіном:
Ич
- Н20
;с=о + н2м— он — -
«у
ус = и — он
Я '/
О ксим
- взаємодія з гідразином:
с=о +
Я '/
н2н—
ічн2
-н2о
►
п /
с— и—
рдн2
Гідразон
- взаємодія з фенілгідразином:
В\
С = 0 + Н2М-- МН
- Н?0
С6Н5 ---------- *► С = Н
І\ІН— С6Н5
Я '/
Фенілгідразок
Продукти реакцій - оксими, гідразони, фенілгідразони- є кристаліч
ними сполуками з визначеними температурами плавлення, що дозво
ляє використати розглянуті реакції для ідентифікації вихідних карбо
нільних сполук.
Основи Шифа є також інтермедіатами біохімічних реакцій проміжного
обміну амінокислот в організмі, зокрема процесів трансамінування та відновлювального амінування а-кетош елот до а-амінокнелот (розділ 2.7).
2. Реакції відновлення та окислення карбонільних сполук
Реакції відновлення
Реакції відновлення характерні як для альдегідів, так і для кетонів.
1) При відновленні альдегідів утворюються первинні спирти:
122
2.4. К арбон ільні сполук и (альдегіди, кетони)
И
н—
с—
он
2) При відновленні кетонів утворюються вторинні спирти:
С -0
+
В
Н2
СН2— ОН
Відновлення альдегідів та кетонів проводять за участю гідридів мета
лів (ЬіА1Н4, ЬіН, № Н), які вивільняють гідрид-іони Н , що є нуіслеофільними
часточками, які приєднуються до подвійного зв’язку С=0. Другий атом
водню (у вигляді іонаН4) постачається водою. В промисловості відновлен
ня карбонільних сполук до спиртів відбувається шляхом каталітичного
гідрування альдегідів та кетонів в присутності нікелю або паладію.
В біохімічних системах відновлення карбонільних сполук з утворен
ням первинних та вторинних спиртів каталізується ферментами дегідро
геназами, а донорами атомів водню є відновлені форми спеціальних коферментів НАД+ (нікотинамідаденіндинуклеотид) та НАДФ+ (нікотинамідаденіндинуклеотидфосфат). Реакції відбуваються за схемою:
НАД(Ф)Н + Н++ Р І-С Н = 0
НАД(Ф)+ + Р і-С Н 2-О Н .
Реакиії окислення
Реакції окислення характерні лише для альдегідів, які при цьому
перетворюються на відповідні карбонові кислоти:
[О]
Я — СООН
й — С Н = 0
Окислення альдегідів іонами металів {А§" та С и 1") широко викорис
товується в аналітичній практиці в біохімії та фармації для виявлення
альдегідів (наприклад, моносахаридів альдоз) та диференціювання аль
дегідів від кетонів:
]) Реакція “срібного дзеркала” - окислення альдегідів аміачним роз
чином оксиду срібла (реактивом Толенса) з виділенням металічного
срібла у вигляді блискучого шару на стінках пробірки:
Р - СН=0 + Ад20 -> И - СООН + 2Ад і .
Розділ 2. Х А Р А К Т Е Р И С Т И К А О К Р Е М И Х К Л А С ІВ Б ІО О РГА Н ІЧ Н И Х С П О Л У К
Механізм реакції полягає в окисленні альдегіду комплексною сполукою
[А £(№ і 3)2]ОН з утворенням солі амонію та відновленням оксиду срібла:
Р _С Н = 0 + 2[Ад(МН3)г]ОН -> Н-СООЫН4 + 2 А д і + ЗЫН3 + Н20 .
2)
Реакція відновлення альдегідами реактиву Фєлінга (комплексу
оксиду міді (II) з калій-натрієвою сіллю винної (виннокам’яної) кисло
ти). Для приготування реактиву змішують сульфат міді з лужним розчи
ном калій-натрій тартрату - сегнетовою сіллю, в результаті чого фор
мується тартратний комплекс міді:
СивОд
+
2 №ОН
► Си(ОН)2 +
СООЫа
СОО№
N8 2 8 0 4
СОО№
+Си(ОН)2
2
с н — ОН
Реактив Фєлінга легко відновлюється альдегідами з утворенням ок
сиду міді (І), що випадає у вигляді осаду червоно-цегляного кольору:
+ С и 20 |
Загальна схема реакції:
/Н
/О Н
^ С и 20 |
М едико-біологічне та фармацевтичне значення
Формальдегід (мурашиний альдегід, метаналь) СН20 - застосову
ється як дезінфікуючий та консервуючий засіб для анатомічних препа124
2 . 4 . Карбонільні сполуки (альдегіди, кетони)
ратів у вигляді 37-40 %-го водного розчину (“формалін”). Активована
за допомогою специфічних ферментів молекула формальдегіду в ком
плексі з вітаміном Вс (фолієвою кислотою) - формілт етрагідрофолат - бере участь у біосинтезі пуринового кільця нуклеотидів. Комплекс
форміату з амінокислотою метіоніном (И-формілметіоніи) є визначаль
ною біомолекулою в ініціації синтезу білка в рибосомах мікроорганізмів.
У незначних кількостях формальдегід утворюється в організмі людини
як продукт >ї-дезалкілування в геиатоцитах багатьох лікарських засобів.
Ацетон (диметилкетон) СН3-С О -С Н 3 - розповсюджений розчин
ник та речовина, що широко застосовується у фармацевтичному син
тезі. Ацетон також утворюється у значних кількостях в організмі людн
іш при розщепленні глюкози (гліколіз) у вільному стані (особливо при
цукровому діабеті) та у вигляді фосфорного ефіру діоксиацетонфосфату, який ферментативним шляхом зворотно перетворюється на свою
ізомерну форму гліцеральдегід-3-фосфат:
Д іоксиа цето нфо сфат
Гліцерал ьдегі д- 3 -фо сфат
Ацетальдегід (оцтовий альдегід) С К ІС Н О є одним із центральних
інтермедіатів проміжного метаболізму, який бере участь в енергетич
ному обміні в мітохондріях у вигляді тіоефіру з коферментом реакцій
ацетилування коензимом А (КоА-БН) - СН3-СО-8-КоА (ацетилкоензіш А). Утворюється при окисленні етилового спирту, в тому числі як
метаболіт ферментативного дегідрування етанолу в організмі.
Альдегіди та кетони є взагалі важливими проміжними продуктами
обміну речовин; вони утворюються в організмі людини як продукти ме
таболізму моносахаридів, жирних кислот, амінокислот. Карбонільні уг
руповання є функціональними групами багатьох лікарських засобів чис
ленних фармакологічних груп.
125
Р озділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
2.5. Карбонові кислоти та їх похідні
Карбонові кислоти - органічні сполуки, похідні вуглеводнів, що
мають у своєму складі карбоксильну групу (карбоксил) -СООН. Назва
класу походить від латинського найменування вугільної кислоти Acidum
carbonicutn.
— с — ОН
II
О
К арбокси л ьн а група
2.5.1. Загальна характ еристика карбонових кислот
Загальна формула карбонових кислот R-COOH. Залишок карбоно
вої кислоти без гідроксильної групи має загальну назву “ацил-” (напри
клад: “ацетил-“, “пропіоніл-“ , “бутирил-“ тощо) або “ацильна група”:
R— С —
II
О
А ц и л ьн а група
Карбонові кіслоти підрозділяються на такі типи:
за кількістю карбоксильних груп, що входять до їх складу, виділяють:
- м оно ка р б о но ві (одноосновні) кислот и (маю ть одну групу
-С О О Н );
- дикарбонові (двоосновні) кислоти (мають дві групи -СО О Н );
- трикарбонові (триосновні) (мають три групи -СО О Н );
за будовою вуглеводнового радикала виділяють:
- аліфатичні кислот и;
- ароматичні кислот и;
- аліциклічні кислоти.
За відсутністю або наявністю у складі вуглеводневого радикала по
двійного зв ’язку аліфатичні карбонові кислоти можуть бути насичени
ми та ненасиченими.
2.5.2. Монокарбонові аліфатичні кислоти
Насичені монокарбонові кислоти
Наведемо будову (скорочені та спрощені структурні формули) пер
ших членів гомологічного ряду насичених монокарбонових кислот (в
дужках - тривіальні назви кислот):
126
2.5. К арбон ові кислоти та їх похідні
О
н -с о о н
(
он
М етан ов а ки сл ота (м ураш и н а кислота)
О
н 3с - с о о н
-д
он
Е танова кислота (оцтова кислота)
О
н 3с - с н 2—С О О Н
З
.
1
\ ^ О
Н
П ропанова ки сл ота (пропіонова кислота)
О
Н3С — (СН2) 2
СООН
Бутанова кислота (н-м асляна кислота)
О
Н3С — (СН2)з
СООН
П ентанова кислота (н -валеріан ова кислота)
(СН3 )2СН—
с н 2----- СООН
3-м етилбутанова кислота (Ізовалеріанова кислота)
О
Н3с — (СН2)4
СООН
Г ексанова ки сл ота (н-капронова кислота)
127
Р озділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Вищі карбонові кислоти
Карбонові кислоти з великою кількістю вуглецевих атомів (С16, С !3,
С20 тощо) - пальмітинова С |;Н3,-СО О Н , стеаринова С |7Н35-С О О Н ,
додеканова С19Н39-СО О Н та їх ненасичені похідні дістали назву вищих
(жирних) карбонових кислот і будуть розглянуті при вивченні природ
них жирів (ліпідів).
Хімічні властивості карбонових кислот
Хімічні властивості карбонових кислот визначаються переважно бу
довою карбоксильної групи, в якій виділяють такі структурні елементи:
карбонільна група (карбоніл), гідроксильна група (гідроксил), водень
гідроксильної групи, що може дисоціювати, визначаючи головні хімічні
властивості класу в цілому На ступінь кислотних властивостей певних
карбонових кислот суттєво впливає хімічна природа радикала Л, що за
рахунок індуктивних ефектів замісників значно змінює ступінь дисоціації
гідроксильного водню.
Г—
карбоніл
гідроксил
П ---- С —
О Н -* — !
II
О
радикал
водень
кислоти
гідроксильної
групи
Відповідно до цього, хімічні властивості карбонових кислот можна
розділити на такі, що залежать від:
- наявності карбоксильної групи, які підрозділяються на:
а) такі, що зумовлені дисоціацією атома водню гідроксильної групи
карбоксилу (суто кислотні властивості);
б) властивості, спричинені впливом карбонільної групи (здатність до
нуклеофільного заміщенння в карбоксилі);
— хімічної природи раттикалакарбонової кислоти.
128
2.5. К арбон ові кислоти т а їх похідні
В цілому можна зауважити, що хімічні властивості, характерні для
карбоксильної групи аліфатичних насичених карбонових кислот, харак
терні І для інших підкласів карбонових кислот.
1. К ислот ні власт ивост і
Це властивості, що зумовлені здатністю атома водню гідроксильної
групи дисоціювати у вигляді протона Н+. Кислотні властивості карбоно
вих кислот визначаються зсувом електронної густини в карбоксильній
групі -С О О Н до карбонільного кисню = 0 внаслідок - І - та -М -ефектів
оксогруии С = 0 з виникненням на атомі вуглецю часткового позитивно
го заряду 6+:
Я—
Такий розподіл заряду в карбоксильній групі, у свою чергу, спричи
няє підвищену рухливість атома водню гідроксильної групи та можли
вість його дисоціації з утворенням ащиіат- (карбоксилат-) іона:
О
П—
ОН
С\
. +
Н+
О
Зсуву рівноваги реакції дисоціації вправо сприяє стабільність аніона,
що утворюється, яка обумовлена, у свою чергу, делокалізацієт заряду
(рівномірним розподілом негативного заряду) в ацилат-іоні:
-
1/2
-
1/2
ОН
Цей же ефект можна умовно зобразити через утворення в результаті
відщеплення (дисоціації) протона двох рівноцінних резонансних (гранич
них) структур:
129
Р озділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
&
я — С.
£ о -
О
я—
N.
я— о
СО'
ОН
Карбонові кислоти реагують з водою як із слабкого основою, в ре
зультаті чого при дисоціації карбонових кислот у водних, розчинах утво
рюються карбоксилатні аніони та іони гідроксонію:
Я
С О О Н + Н 20
Я
СОО"
+
Н
(Н 3З)
Гідрокс оній
Ступінь кислотності (сила карбонових кислот) залежить від будови
вуглеводневих радикалів та наявності у їх складі замісників з електронодонорними або електроноакцепторними властивостями. Наприклад,
значно збільшує силу кислоти наявність в радикалі атома хлору з вира
женим електроноакцепторним (негативним індуктивним, тобто -І-ефектом). Ароматичні кислоти, як такі, що, завдяки делокалізації заряду,
утворюють максимально стабільні ацилат-іони, мають сильніші кислотні
властивості, ніж незаміщені аліфатичні кислоти:
СІ
сн3—
С ООН С6 Н5
О ц това
кислота
р К = 4 ,7 6
С ООН СІ
Б ензой на
ки сл ота
р К = 4 ,1 9
-* -сн 2—
С ООН
Х лороц това
ки сл ота
рК = 2,85
СІ
і
1
СІ
с—
СООН
Т рихл ороц това
кислота
р К = 0 ,б б
Зростання кислотності
Солі карбонових килот. Мила
Кислотність карбонових кислот у хімічних та біохімічних системах
виявляється у реакціях утворення солей (реакція нейтралізації) при їх
взаємодії з сильними основами, наприклад:
130
2.5. К арбон ові кислоти та їх похідні
СН3
СООН + NaOH
О ц това кислота
С 1 7 Н3 5
СООН + NaOH
= =
СН3
COONa + Н20
А ц етат натрію
С 17н35
С теаринова кислота
COONa + Н20
С теарат натрію
На відміну від самих кислот, солі вищих карбонових насичених (жирних) кислот розчинні у воді завдяки дисоціації у водних розчинах за такою
реакцією:
Н20
с і 7 н зй— COONa — ► С-17Н35— СОО + Na+
Дисоційовані натрієві та калієві солі вищих карбонових (жирних) кис
лот мають амфіфільну природу, тобто виявляють як гідрофобні (за раху
нок довголанцюгового вуглеводневого радикала), так і гідрофільні (за
рахунок негативно зарядженої аніонної головки) властивості. Внаслідок
цього зазначеним солям притаманна поверхнева активність, тобто
вони знижують поверхневий натяг на межі поділу фаз (олія/вода) і їм
властива миюча дія. При цьому натрієві солі є твердими милами, а ка
лієві солі - рідкими. У медичній практиці застосовується рідке калієве
мило, яке дістало назву “мила медичного” (Sapo medicinalis). Рідке
мило, що має зелене забарвлення завдяки домішці метиленового синього,
називається “зеленим милом” (Sapo viridis).
2.
В ласт ивост і карбонільної групи (реакції нуклеоф іл ьного за
м іщ ення S y ).
Згідно з розглянутими вище закономірностями, атом вуглецю карбо
нільної групи карбоксилу має частковий позитивний заряд 5+, тобто являє
собою електрофільний центр. Як такий, він може атакуватися ігуклеофільними часточками N ir, в результаті чого здійснюється заміщення
гідроксильної групи ОН', тобто відбувається реакція нуклеофільного
заміщ ення SN.
М еханізм S^р е а к ц ій карбонових кислот'.
ОН
; ОН
Nu
-І
\м іі
131
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Характерними для карбонових кислот типами реакцій нуклеофільного заміщення з утворенням відповідних функціональних похідних є:
(1) утворення складних ефірів (реакції етерифікації); (2) утворення амі
дів кислот (реакції амідування); (3) утворення хлорангідридів або
(4) ангідридів кислот (реакції ангідридизації) - рис. 2.9.
<1)
с 2н5он
------
СНд—
н 2о
(2)
-М-----
Ж 3
х о — с 2н5
----- ►
- СНз
/О
Н20
о
мн2
с н 3он
(3)
РСІ5
-----►
/Р
Н20
С!
/О
(4)
СНз-СООН
н2о
Рис. 2 .9 . Схема
СН3
І- - <
>
СНз— С ч
х0
реакцій карбонових кислот на прикладі оцтової кислота
У біохімічних системах важливими З^.-реакціями в карбонових кис
лотах є заміщення-ОН-групи на спиртовий гідроксил (реакція етерифі
кації, що є ключовою при біосинтезі ліпідів), на аміногрупу (реакція амі
дування), фосфатну та пірофосфатну групи (утворення ацилфосфатів),
нуклеотид (утворення комплексів амінокислот із транспортними рибонук
леїновими кислотами тощо).
І) Реакиії етерифікації та їх біохімічне значення
Найбільш поширеною в хімічних та біологічних системах є реакція
взаємодії карбонових кислот із спиртами з утворенням складних ефірів
(реакція етерифікації). Реакцію звичайно проводять у присутності мі
неральних кислот як каталізаторів:
132
2.5. К арбон ові кислоти та їх похідні
О
я— с — ОН +
НО
1-ї
о
І!
и— с — о ^
Механізм реакції етерифікації включає в себе такі етапи:
1) протонування кислоти;
2) нуклеофільна атака утвореного катіона спиртом;
3) відщеплення молекули води;
4) депротонізація ефіру, що утворився.
Схема реакції (на прикладі утворення етилацетату):
Швидкість реакції етерифікації залежить від будови спирту, оскільки
можливе виникнення стеричних утруднень на етапі утворення проміжного
продукту приєднання (2 ). Відповідно до цього, первинні спирти етерифікуються значно легше, ніж вторинні та третинні {правило Меніиуткіна).
Реакція етерифікації є одним із найважливіших процесів органічної та
біоорганічної хімії і займає значне місце серед біохімічних реакцій мета
болізму в організмі людини, призводячи до утворення складних
ефірів, які є фізіологічно важливими біомолекулами (зокрема, численни
ми ліпідами, що є ефірами жирних кислот та таких спиртів, як гліцерол,
сфінгозин тощо). Етерифікація карбонових кислоту живих організмах
здійснюється шляхом введення у карбоксил замість -О Н активуючих
функціональних груп, основною з яких є похідна пантотенової кислоти
(вітаміну В5) - коензіш (кофермент) А (кофермент ацилування), що
містить тіолову (сульфгідрильну) групу -Б Н (скорочене позначення ко
ферменту Ш -К о А ).
При взаємодії карбонової кислоти з коензимом А утворюється тіосфір
типу Я -С О -Б -Я ', який, у свою чергу, виступає в ролі субстрату при
нуклеофільному заміщенні, зокрема в реакціях утворення складних ефірів:
133
Р озділ 2. ХАРАКТЕРИСТИКА О КРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
/Р
+
+ Н Б— КоА ----- ► К ~ с (
х ОН
х БКоА
в— с (
Н20
Реакція етерифікації в кислотному середовищі є оборотною, а в луж
ному середовищі відбувається необоротний гідроліз ефіру з утворенням
солі і спирту. Гідроліз складних ефірів у травному каналі та клітинах
різних тканин тваринного організму відбувається за участі спеціальних
ферментів з класу гідролаз - ліпаз (розділ 4.2 ).
2) Реакції амідування та і’у біохімічне значення
Аміди - функціональні похідні карбонових кислот, які утворюються
шляхом заміщення -ОН -групи карбоксилу на аміногрупу -КН.,.
Аміди утворюються шляхом взаємодії таких похідних карбонових
кислот, як галогеноангідриди, ангідриди, складні ефіри, з аміаком або
первинними чи вторинними амінами:
н— с;
Хх
+ NN 2— я' —
► я— а
+
я' х
Х ІЧН2
В реакції амідування в біохімічних системах вступають різні похідні
карбонових кислот, зокрема а-амінокислоти при біосинтезі пептидів та
білків, вільні дикарбоиові амінокислоти - Ь-аспарагінова та Ь-глутамінова при утворенні вільних амідів.
3) Реакції апгідридизаш ї та Ід~ біохімічне значення
В біохімічних процесах надзвичайне значення має утворення міша
них ангідридів карбонових та мінеральних кислот. Зокрема, в процесі
внутрішньоклітинного обміну речовин поширеним є заміщення ОН-груп
у карбоксилі природних карбонових кислот та їх похідних (гетерофункціональних сполук, зокрема, гідроксикислот, амінокислот) на фосфатну та
пірофосфатну групи. Ацилфосфати, що утворюються, мають таку будову:
РН3
<и°
/ОН
о — р==0
Х )Н
Ацетилфосфорна кислота
134
2.5. К арбонові к и сл оти та їх похідні
не— о н
1,3-диф осф огяіцеринова кислота
(зв ’язок у І -му полож енні - ф осф оеф ірний,
у 3-м у - ф осф оангідри дний )
Утворення ацилфосфатів є молекулярним механізмом метаболічної
активації біомолекул і сприяє подальшій участі цих сполук в біохімічних
перетвореннях. У вигляді фосфоангідридів ацильні залишки переносять
ся до гідроксильних, тіольних та аміногруп різних біологічних сполук:
о — 1^=0
\ /"VI і
НОРГ
О
І!
Я— С ~ О Я ”
НБЯ''
О
II
► Я— С —БЯ"
О
II
НгИЯ” -------► Я— - С - М Н Я "
Ацилпірофосфати утворюються в біохімічних системах при взаємодії
карбонових кислот та їх похідних з аденозинтрифосфорною кислотою
(АТФ). Так, наприклад, за механізмом утворення ацилпірофосфатів від
бувається активація протеїногенних а-Ь - амінокислот, що передує їх ви
користанню для синтезу поліпептидного ланцюга (розділ 4,3).
Н ен аси чекі м онокарбонові кислоти
Прикладами аліфатичних ненасичених монокарбонових кислот є:
СН2= С Н — СООН
П роп енова кислота (акрилова кислота)
135
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
с н 2= с — с о о н
І
СНз
2 -метил про пе нова кислота (метакрилова кислота)
Нч
/С О О Н
с=с
Н3СХ
\ Н
транс-бутен-2-оиа. кислота (к ротон ова кислота)
Похідними низькомолекулярних ненасичених монокарбонових кислот
є багато важливих інтермедіатів обміну речовин, що утворюються в
реакціях обміну ліпідів, вуглеводів та амінокислот. Структурними ком
понентами молекул природних ліпідів є моно- та поліненасичені жирні
кислоти (карбонові кислоти з С ]6 - пальмітоолеїнова, С 18 - олеїнова,
лінолева, ліноленова, С - арахідонова).
Хімічні та біохімічні властивості ненасичених карбонових кислот
Наявність у вуглеводневому радикалі ненасичених кислот подвійно
го зв’язку надає їм властивостей, характерних для алкенів, зокрема здат
ності до реакцій приєднання, окислення, полімеризації.
Процеси приєднання за місцем подвійного зв’язку широко розпо
всюджені в біохімічних системах. Це переважно реакції гідратації і від
новлення молекул а , Р-ненасичених карбонових кислот - низькомоле
кулярних метаболітів - та вищих (жирних) кислот, що відбуваються за
участю біокаталізаторів - ферментів. Характерною особливістю нена
сичених жирних кислот є також їх здатність до реакцій пероксидного
окислення у вуглеводневому радикалі. Ці реакції, що їм підлягають при
родні поліненасичені жирні кислоти, зокрема такі, що входять до складу
фосфоліпідів біологічних мембран, мають суттєве біологічне значення,
є розповсюдженими в біохімічних системах і будуть також розглянуті
при вивченні будови природних ліпідів.
Здатність ненасичених карбонових кислот та їх похідних до поліме
ризації використовується з метою створення важливих для медицини
пластичних мас. Наприклад, акрилова та метакрилова кислоти утворю
ють полімери - поліакрилати та поліметакрилаги, що застосовуються в
стоматології.
136
2.5. К арбонові кислоти та їх похідні
2.5.3. Дикарбонові та трикарбонові аліфатичні кислоти
Дмкарбонові кислоти
Дикарбоновими аліфатичними кислотами є такі похідні насичених та
ненасичсних вуглеводнів, у головний ланцюг яких включено дві карбок
сильні групи.
Насичені дикарбонові кислоти:
ноос— соон
Етандіова кислота (щ авлева кислота)
н о о с — СН2— СООН
П роп андіова кислота (м алон ова кислота)
НООС
(СН2)2
СООН
Бутан діова кислота {бурш тинова кислота)
НООС— (СН2)з—
соон
П ентандіова кислота (глутарова кислота)
НООС
(СН2)4
СООН
Г ександіова кислота (адипінова кислота)
Ненасичені дикарбонові кислоти:
цис-бутендіова кислота (малеїнова кислота)
Хімічні властивості. Біологічне та фармацевтичне значення
Хімічні властивості дикарбонових кислот близькі до монокарбонових. Нижчі члени ряду моиокарбонових кислот (щавлева, малонова)
мають більш виражені кислотні властивості, що пояснюється електроноакцепторною дією другої карбоксильної групи.
137
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Дисоціація карбоксильних груп у молекулах дикарбонових кислот
відбуваєтсья ступінчасто; вища кислотність дикарбонових кислот сто
сується лише дисоціації першої карбоксильної групи. Відрив протона від
другої карбоксильної групи відбувається значно важче, ніж від першої, і
за другим ступенем дисоціації кислотність дикарбонових кислот значно
менша:
СН3- СООН
РКа=4,76
НООС - СН2- СООН
р К ,= 2 , 8 6 ^ = 5 , 7 0
У хімічних та біохімічних системах дикарбонові кислоти утворюють
два типи солей - кислі (з одним еквівалентом основи) та середні (з дво
ма еквівалентами основи):
НООС - СООН + NaOH
->
НООС - COONa + Н20
Гідр оксалат натрію
(кисла сіль)
НООС - СООН + 2NaOH
NaOOC - COONa + Н20
О ксал ат натрію
(середн я сіль)
Як насичені, так і ненасичені дикарбонові кислоти (зокрема щавле
ва, бурштинова, глутарова, фумарова) належать до біомолекул, що міс
тяться в живих організмах, утворюючись як проміжні продукти внутріш
ньоклітинного обміну речовин. Солі та ефіри бурштинової кислоти (три
віальна назва - сукщшати; лат. —Acidum succinicum) використовують
також в синтезі лікарських засобів.
Важливими похідними бурштинової та тутарової кислот є гетерофункціональні сполуки (розділ 2.7) - а-амінокислоти: аспарагінова і глутамінова відповідно, що містяться у складі природних білків та виконують
важливі самостійні біохімічні функції. До біомолекул, що є гетерофункціональними сполуками - похідними дикарбонових кислот, належать та
кож гідроксикислоти (яблучна, винна) та оксокислоти (щавлевооцтова,
ct-кетоглутарова).
Т рн карб он ові ки сл оти
Біологічно важливими представниками трикарбонових кислот є їх гідроксипохідні лимонна (2-гідрокси-1,2,3-пропантрикарбонова) та ізопимонпа кислоти, а також ненасичена цис-аконітова кислота:
138
2.5. К арбон ові кислоти та їх п о ш п і
с н 2— с о о н
1у н 2— с о о н
но-
;г
т
-с о о н
с н 2— с о о н
Л и м он н а кислота
не— соон
но— с н — соон
Ізолим он на кислота
с н 2— с о о н
-с о о н
с н — соон
Цис-аконітова кислота
Лимонна кислота {Acidum citricum) є важливим інтермедіатом обміну
речовий у тваринних та рослинних організмах. Вона утворюється при кон
денсації біохімічно активованої форми оцтової кислоти - ацетилкоензиму А
та щавлевооцтової кислоти (оксалоацетату) в загальному метаболічно
му шляху внутрішньоклітинного перетворення вуглеводів, ліпідів та безазотистих залишків амінокислот-т. з. “цикли лимонної кислоти” (“цикли
трикарбонових кислот - ЦТК,” або “цикли Кребса”). Крім зазначених вище
трикарбонових кислот, метаболітами ЦТК є такі дикарбонові кислоти та їх
похідні, як фумарова, яблучна, щавлевооцтова та а-кетоглутарова.
Лимонна кислота у значній кількості міститься в плодах цитрусових
рослин. Вона використовується у харчовій промисловості як добавка до
фруктових сиропів, напоїв тощо. Цитрат натрію (тринатрієва сіль ли
монної кислоти) є фармацевтичним препаратом, що використовується
як консервант, оскільки протидіє позасудинному згортанню крові.
2.5.4. Ароматичні карбонові кислоти. Фенолокислоти
А ром ати чн і м онокарбонові кислоти
Аренмонокарбонові кислоти - це карбонові кислоти, в молекулах
яких карбоксильна група безпосередньо зв’язана з ароматичним цик
лом. До аренмонокарбонових кислот належать бензойна кислота та її
похідні, наприклад:
139
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
СООН
СООН
Б ензой на кислота
З - Еттшбе нзой на кислота
На хімічні властивості аренмонокарбонових кислот впливає наявність
ароматичного кільця, яке сприяє делокалізації заряду в молекулі і збіль
шенню стійкості аніона, що утворюється при дисоціації карбоксильної
групи. Тому кислотність ароматичних монокарбонових кислот переви
щує кислотність насичених та ненасичених кислот аліфатичного ряду.
Сполуки, в молекулах яких карбоксильна група розміщена в бічному
ланцюзі арену (наприклад - фенілоцтова кислота), формально нале
жать до карбонових кислот аліфатичного ряду.
с н 2— СООН
Ф енілоцтова кислота
Бензойна та фенілоцтова кислоти широко розповсюджені в клітинах
та біологічних рідинах живих організмів. Ці сполуки є продуктами нор
мального обміну речовин (метаболітами) або утворюються в організмі
людини та тварин в процесі біотрансформації чужорідних хімічних спо
лук - ксенобіотиків (лікарських засобів, промислових отрут тощо).
Бензойна кислота та її солі використовуються в медицині та фармації як
антисептичні препарати та вихідні речовини для синтезу багатьох лі
карських засобів.
А ром атичні д и карбон ові кислоти
Представниками арендикарбонових кислот є фталева кислота,
її ізомери - ізофталева, терефталева кислоти —та їх похідні.
140
2.5. К арбон ові к и сл оти та їх п охідні
соон
соон
соон
^
х соон
1,3-б ен зол д и к арб он ов а
1,2-бензолдикарбонова
кислота (о-ф снілендикарбонова,
(ізоф тал ева) кислота
ф талева кислота)
СООН
НООСГ
^
1 ,4-б ен золднкарбонова (тереф талева) кислота
Хімічні властивості арендикарбонових кислот суттєво не відрізня
ються від властивостей монокарбонових кислот. Фталева кислота при
нагріванні утворює фталевий ангідрид, який, у свою чергу, при взаємодії
з аміаком перетворюється на фталімід - сполуку, що широко викорис
товується в синтезі лікарських засобів, інсектицидів тощо:
О
О
О
О
О
Похідна фталевого ангідриду а , а-ди(4-оксифеніл)-фталід має вла
стивості кислотно-основного індикатора (фенолфталеїн) або ж у ви
гляді фармацевтичної форми застосовується в медицині як лікувальний
засіб послаблюючої дії (Пурген).
Ф енолокислоти
Ф еполокислот и - похідні ароматичних карбонових кислот, в моле
кулах яких один або декілька атомів водню заміщені гідроксилом. За
хімічною будовою фенолокислоти належать до гетерофункціоиальних
органічних сполук (розділ 2.7).
141
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Саліцилова кислота
Найпростішим представником фенолокислот, що має велике значення
в фармакології та фармації, є саліцилова (2-гідроксибензойна) кислота.
/^ /С О О Н
Саліцилова кислота має всі хімічні властивості, характерні як для
аренкарбонових кислот, так і для фенолу. Похідні саліцилової кислоти
використовують у медичній практиці як засоби, що мають протизапаль
ну, жарознижуючу та анальгетичну дію. Найбільш уживаними є ефіри та
солі саліцилової кислоти (саліцилати).
о
о
А ц ети л сал іц и л ова кислота
М етилсаліц илат
Антибактеріальну дію має феніловий ефір саліцилової кислоти (пре
парат Салол).
О
Ф енілсаліцилат
Синтез ацетилсаліцилової кислоти було здійснено у 1853 р. германсь
ким хіміком Герхардом, протизапальна і протиревматична активність
саліцилатів була виявлена в 1875 р., однак лише в 1893 р. Феліксом Гоф
маном у фірмі “Bayer” був уперше отриманий фармацевтичний препа
рат Аспірин. Незважаючи на визначний прогрес фармакології і впровад
ження нових лікарських засобів з протизапальною та жарознижуючою
дією (так званих “нсстероїдних протизапальних засобів” -Н П З З ), пре
142
2.6. Б іоор ган іч н і сп ол ук и азоту
парати ацетилсаліцилової кислоти залишаються одними з найбільш ужи
ваних. З’ясовано, що біохімічний механізм фармакологічної дії ацетил
саліцилової кислоти пов’язаний з пригніченням тканинного біосинтезу
простагландинів - фізіологічно активних похідних арахідонової кисло
ти (С20.4), що відіграють важливу роль у розвитку запалення та больово
го синдрому.
Галова кислота
Галова (3,4,5-тригідроксибензойна) кислота належить до фенолокислот природного походження. Галова кислота легко окислюється киснем
повітря, утворюючи хіноїдні похідні, що надає її водним розчинам тем
ного забарвлення. При декарбоксилуваині галової кислоти утворюється
триатомний фенол пірогалол (1,2,3-тригідроксибензол):
НО
Г алова кислота
Галову кислоту отримують при гідролізі танінів - комплексів гліко
зидних похідних галової кислоти, що, у свою чергу, є основними хімічни
ми компонентами т. з. дубильних речовин, які містяться в листі та корі
дуба, чайному листі тощо. У зв’язку з їх здатністю денатурувати білки,
препарати танінів застосовуються в медичній практиці як в ’яжучі, ду
бильні, антисептичні засоби місцевої дії при поверхневих запальних про
цесах, опіках.
2.6. Б іоорганічні сполуки азоту
Значне місце в біоорганічній хімії посідають азотвмісні похідні вуглевод
нів, зокрема нітросполуки, аміни та аміди. До азотвмісних сполук належать
як біомолекули, так і численні природні та синтетичні лікарські засоби.
2.6.1. Н іт росполуки
Н ітросполуки - похідні вуглеводнів, у молекулах яких один або де
кілька атомів водню заміщені на нітрогрупу - N 0 ,.
143
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Загальна формула нітросполук И-]ЧОг
Залежно від будови радикала розрізняють аліфатичні (насичені, або
нітроалкани, та ненасичені, або нітроалкени) та ароматичні нітро
сполуки (нітроарени), наприклад:
сн3
N 02
і ґ і
СН3
СН2
СН2
N 02
"N02
З-нітротолуол
Н ітробен зол
1-нітроп ропан
Електронна будова нітрогрупи
Хімічні властивості нітросполук визначаються будовою нітрогрупи, в якій
атом азоту та обидва атоми кисню знаходяться у стані 5р2-пбридизації.
Завдяки більшій слєктронегативності атомів кисню у складі нітро
групи відбувається зсув двох неспарених електронів (електронних хмар)
атома азоту до одного з атомів кисню. Відповідно до цього електронна
будова нітрогрупи являє собою резонансну структуру, граничні форми
якої можна подати таким чином:
О
/ °
<
\ з :
О
У зв’язку з рівноцінністю обох атомів кисню в групі - N 0 ,, електрон
ну будову нітрогрупи можна також подати із врахуванням рівномірного
розподілу двох часткових негативних зарядів, а саме:
я — й.
^ 0 - 1/2
Хімічні властивості нітросполук
Характерного реакцією нітросполук є відновлення їх нітрогрупи, в ре
зультаті чого утворюються первинні аміни:
+ 6[Н]
И
144
'ГМ02
ІЗ— ЫН2 + 2Н 20
2.6. Б іоор ган іч н і сполуки азоту
Відновлення нітросполук
Продуктами відновлення нітроалканів є алканаміни, наприклад:
С 2 Н5
N 02 + 6Н
► С 2 Н5
N 42 + 2Н20
В результаті відновлення ароматичних сполук (реакція Зініна) утво
рюються анілін та його похідні - сполуки, що становлять значний інте
рес у біоорганічній та фармацевтичній хімії (див. нижче). Проміжними
продуктами реакції є нітрозосполуки - речовини, що містять нітрозогрупу - N = 0 , та гідроксиламіни:
N 02
N = 0
МІН
ОН
МН2
+2Н
+2Н
ч ^
Н ітробен зол
+2Н
Ч ^
ч ^
Н ітрозобензол
Ф єн ілгідрокс ил ам і н
А м інобензол
(анілін)
Реакцію було відкрито російським хіміком M. М. Зініним (1812-1880),
який очолював кафедру загальної хімії Медико-хірургічної академії в
Санкт-Петербурзі.
2.6.2. А м іни
Аміни - похідні вуглеводнів, що містять одну або декілька аміно
груп - КН 2.
Аміни можна розглядати також як органічні похідні аміаку, в молеку
лах яких один, два або три атоми водню заміщені на вуглеводневі ради
кали. Відповідно до цього визначення, розрізняють первинні (К М і,),
вторинні (іу ^ Н ) та третинні ( їу ^ ) аміни.
Найбільше значення в біоорганічній хімії мають аліфатичні та аро
матичні аміни.
Аліфатичні аміни (алкіламіни):
Первинні
Вторинні
Третинні
c h 3- n h 2
GH3- N H - C H 3
CH , - N - C H ,
сн3
М етанамін (метиламін)
Д и м етилам ін
Т рим етилам ін
145
зділ 2. х а к а а і е г п '-. »
-
—
___________________________________
~3 азамїсниковою номенклатурою Д ^ ^ ^ ^ ^ д ^ ^ в о д ^ ^ д а д а НИХ амінів складають ^ ™
ванням суфіксу - « '» ■ Д .
”
0“ “ ш
вторинних і третинних амінів
^ іональну номенклатуру; при цьому на-
ГаТ
лівР переРахонУують в алфавітному порядку, наприклад:
л , т хтіт г н _ ГН - етилметиламін.
СН3- К Н - С Н 2
містять чотири радикали замість чоти
р
"
К
^
- м о н і й н ш групі мають назву
ам онієвих основ.
[Я 4 = N 1
ОН
Загальна будова четвертинних амонієвих основ
“ Бага'м ^ Г —
сполук,Г
=
—
Д 3 ^ . а_
Діаміни.
Г л а д і ДВІ аміногрупи.
М1^ ^ ^ ш Длу™ ^сч и н Т бутандіам ін-Ь |^та^^в£Р^л^С пен'гандіаш ^-
тяться в трупному матеріалі (лат. с а іт е )
тру
П утресцин
НгН - СНг- (СНг)г- С Н , - МНг
н 'м - с н ^ і с н ^ - с н ^ м н ,
Кадаверин
У складі хроматину клітинних ядер
д н к беручи участь
спермідт, що можуть утворювати іонні комплекси Д
, Р.
в контролі реплікації генетичного матеріалу.
Спермін
Н N - (СН 2) з - МИ - <СНг)4- МИ - (СНг)3- NN.
Нг2Ы- (СН2)4- NN - (СН2)4- м н 2
Спермідин
А р о м а т и ч н і аміни (ариламіни):
/С Н 3
О
Фсніламін
(анілін)
146
“"'
С і^ м Н г
Д и мети лфен іл амі н
(даметил анілін)
о-фенілендіамін
2.6. Б іоорган іч н і с п о л у к » азоту
Назви ароматичних амінів звичайне>будуть„"^(наприклад: мононазви родоночального представника ряду « о д и *
метиланілін, диметиланілін тощо).
Хімічні властивості амінів
1) 0£Ш 2Ш М й-гт ,вост і алцШ
- ттпп1. на атом; азоту аміногруЗ ар а х ^ іо к н а яв н о стів іл ьн о ^ е д а о н ^ ^
виражені у амінів
™ іфатичноторядапорівнягюз
З
З
Ї Ї ї д
а
- - -
к _ м н 2 + и х ^ [ Н - м н 3Гх
ДОвивільнення іона гідроксилу:
• ■ ^ ^ Хи+
о н - — ► ГИ
ІМН3Ґ + ОН'
Тому розчинення амінів у воді призводить до накопичення гідро
ксильних іонів, що поясиюелунші в л а с т ^ о с т ^ а ш ^ .
2) Е г т і і
ж ж т - „ а ш азоту аміногрупи надає цим спол у к а ^ ^ а с т и в о ^ т е ^ ^ и ^ ^ н у к л е о ф іл ів - В я к о с т ^ н у ^ о ф іл ^ а м і^ и
бонових кислот (2.2).
(2.1.)
(2.2.)
И— МН2 +
И— ИН2 +
Я*— СІ
Я'—
с“ .\ с (
■>
Я—
мн—
РГ + НС1
МН— с
о
ніліду):
я ' + неї
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
//
с 6н5— ын2 + с н 3— С \
Анілін
о
с 6н5- -ЫН-
-с ^
СІ
+ неї
СНз
Ацетанілід
Х лорангід рид
о ц тової кислоти
Продукт реакції - ацетанілід - є ключовим попередником у синтезі лі
карських засобів групи сульфаніламідів (див. нижче). Сам ацетанілід був
також одним із перших синтетичних жарознижуючих засобів, який дістав
назву Антифебрину. Жарознижуючим фармацевтичним препаратом, що є
похідним ацетаніліду, є Фенацетин (І-етокеи-4-ацетамінобензол):
Н3С — с о -
-и н -< 0
>— 1з- -с 2н5
Ф енацетин
3)
Заміщення в ароматичному кільиі амінів
Для ароматичних амінів характерні реакції електрофільного заміщення
в ароматичному циклі. Механізм реакції пояснюється електронодопор
ними властивостями аміногрупи, що є орієнтантом І роду, який сприяє
напрямку атаки електрофільних реагентів в о- та /^-положення арома
тичного кільця. Різновидом такого типу реакції є сульфування аніліну,
яке відбувається при нагріванні з концентрованою сірчаною кислотою:
Ш 2
Анілін
С ульф анілова кислота
А н ілін V синт езі лікарських засобів. Сульфаніламіди
Анілін та його гомологи є попередниками в цілому ряді синтезів лі
карських засобів та барвників. Так, зокрема, сульфанілова кислота, що
утворюється при сульфуванні аніліну, є структурним компонентом важ
ливої групи антимікробних лікарських засобів - сульфаніламідів.
148
2.6. Б іоорган іч н і сполук и азоту
Протимікробна дія т. з.“червоного стрептоциду”, що за хімічною
природою був азобарвником Пронтозилом, була вперше встановлена
в 1935 р. Г. Домагком. Пізніше було доведено, що фармакологічно актив
ним сульфаніламідом є “білий стрептоцид” (“стрептоцид”), що є продук
том перетворення пронтозилу в організмі людини та тварин.
Різні похідні сульфаніламідів е високоефективними хіміотерапевтич
ними препаратами, що відрізняються за спектром антибактеріальної
активності (рис, 2.10).
—
*■
h 2n —
( Г ^ Т ) — s o 2— NHZ
(2)
H2N—
— s o 2— NH— R
_ і/ 0СНз
H2W—
— SO, — NH—
<3)
>ОСНз
(1)
о
► H 2N—
— S O , — NH—
( 4)
Рис. 2 .1 0 . Сульфаніламіди з антибактеріальними властивостями:
(1) загальна структура сульфаніламідів; (2) стрептоцид; (3) сульфади
метоксин; (4) норсульфазол
Синтез стрептоциду з аніліну проходить через утворення ацетаніліду
(реакція ацилування) з метою захисту аміногрупи і включає стадії сульфохлорування та амінування бічної сульфогрупи:
-CH 3 COOH
А н іл ін
А цетанілід
+ n h 3 +h2o
Н3 ССО— NH— ( j ^ j ) — S 0 2CI ■
-HCI-CH3 COOH
П охідн а хлоран гідрид у сульф анілової кислоти
S
O
2 N
H
2
А м ід сульф анілової кислоти (стрептоцид)
149
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Біохімічний механізм антимікробного (бактеріостатичного) ефекту
сульфаніламідних препаратів полягає в їх антагонізмі щодо пара-амшобензойної кислоти (ПАБК). яку бактеріальні клітини використовують для
синтезу необхідного для їх життєдіяльності метаболіту - фолієвої кис
лоти (вітаміну В).
р-ам інобен зойна кислота
Завдяки структурній подібності сульфаніламідів до /ш/га-амінобеизойної кислоти, вони блокують дію ферментів, які взаємодіють з ПАБК
(конкурентне інгібування ферментів), що призводить до порушення ме
таболізму і затримки росту бактеріальних клітин.
Крім участі в утворенні важливого коферментного вітаміну - фоліє
вої кислоти, ПАБК є вихідною речовиною для синтезу деяких лікар
ських засобів, зокрема препаратів анестезуючої дії - Анестезину, Но
вокаїну, Лідокаїну, Тримекаїну тощо.
А нестезин (етиловий еф ір П А БК )
Н овокаїн ( (і-діетил амі п оети лови й ефір ПА БК)
Механізм місцєвоанестезуючої дії Анестезину, Новокаїну та їх струк
турних аналогів полягає у блокуванні К а+-каналів синаптичних мембран
аферентних нейронів, що робить тканини нечутливими до дії фізіологіч
них подразників.
150
2.6. Б іоорган іч н і сполук и азоту
2.6.3. Ам іди кислот
Аміди ки сл от-орган іч н і сполуки, що є продуктами заміщення гід
роксильної групи в карбоксилі карбонових кислот на аміногрупу -N1-1,.
Аміди можна також розглядати як продукти заміщення атома водшо в
молекулі аміаку на ацильну групу.
О
II
Загальна формула амідів кислот И - С - N Н2, наприклад:
сн.
,0
У/
сн 2— сн2— сн2-
ын2
,0
•//
•ч
НИ,
Бутанам ід (бутан оїлам ід)
Ацетамід
Б ензам ід
Схему реакції утворення амідів як функціональних похідних карбоно
вих кислот було наведено в розділі 2.5.
Важливе значення в біоорганічній хімії мають також И-заміщені амі
ди, які можна також розглядати як И-ацильні похідні первинних та вто
ринних амінів:
/°
сн3— с '
Х ^ С Н 3)2
>>ГДЧ-диметилацетамід
С6Н5— NN
/°
С^
СН3
N-(J)eнiлaцeтaмiл (ацетанілід)
Біохімічні реакції аміду вапни
В організмі людини і тварин в реакції амідування вступають біологіч
но важливі дикарбонові кислоти —Ь-глутамінова та Ь-аспарагінова, які
у значній кількості містяться у вільному стані в тканинах печінки та
головного мозку:
151
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
О
О
ОН
NH2
сн2
СН2
+
СН2
Нс
nh2
соон
L-гаутам ін ова кислота
NH3 — ►
СН2
+
Н20
НС— n h 2
соон
А м ід L-гл утам інової ки сл ота (гаутам ін)
Амідування дикарбонових кислот в організмі каталізується спеціаль
ними ферментами (відповідно глутамінсинтетазою та аспарагінст тстазою). Процес складається з двох стадій і включає в себе утворен
ня ацилфосфату (1) та подальше перетворення ацилфосфату на амід
карбонової кислоти (2):
О
(1)
R
С
+ ADP
Утворення ацилфосфату амінокислоти відбувається за рахунок фос
фатних зв ’язків аденозиитрифосфату (АТФ) (adenosintriphosphate ATP; англ.), що, втрачаючи один фосфатний залишок, перетворюється
на аденозиндифосфат (adenosinediphosphate -ADP).
Кислотоамідні (пептидні) угруповання є структурною основою
найбільш важливих для всіх процесів життєдіяльності біополімерів - білків
та пептидів. Механізми утворення пептидних зв’язків як реакцій нуклеофільного заміщення аміногрупою гідроксильних груп у молекулах при
родних L -a -амінокислот будуть розглянуті в розділі 4.3.
152
2,7. Г етер оф ун кц іон альн і сполуки
Фізіологічно важливим кінцевим продуктом перетворення в організмі
людини та вищих тварин азотвмісних білків, нуклеїнових кислот та низь
комолекулярних амінів є сечовина (urea - лат), що за хімічною будо
вою являє собою повний карбамід вугільної кислоти:
H2N— С —
nh2
О
С ечовина
У формі сечовини уреотелічні організми екскрєтують із сечею то
ксичний аміак, який постійно утворюється в живих тканинах при розще
пленні азотвмісних біомолекул. Хімічний синтез сечовини з ціанату амо
нію, який був здійснений у 1828 р. німецьким хіміком Ф, Вьолером, став
наріжним каменем розвитку органічної хімії, бо вперше підтвердив мо
жливість синтезу біомолекул поза живим організмом.
У фармацевтичному синтезі в якості реагентів застосовуються такі
аміди, як формамід H -C O -N H 2т а N .N -диметилформамщ Н-СО -(СН 3)г
2.7. Г етероф ун кц іон ал ьн і сполуки
До гетероф ункціональних сполук належать органічні сполуки, в
молекулах яких містяться різні функціональні групи: аміно-, гідрокси-,
оксо- та інші. До гетерофункціональних сполук належить більшість при
родних сполук та біомолекул тваринного і рослинного походження та
значна кількість лікарських засобів.
Гетерофункціональні сполуки можна подати у вигляді такої загальної
формули: X-R-Y.
Таблиця 2.4.
Ф у н кц іон альн і групи гетероф ункціональних
біоорганічних сполук
функціональна група
Y
X
-COOH
-nh2
-со о н
-ОН
-СООН
х= о
-nh2
-о н
Гетероф уткціональпі сполуки
Амінокислоти
Гідроксикислоти
Оксо-(кето-) кислоти
Ам іноспирти, амінофеноли
153
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
2.7.1. А м ін окислот и т а ід: власт ивост і
А мінокислоти - похідні карбонових кислот, у структурі вуглевод
невого радикала яких один або декілька атомів водню заміщені на амі
ногрупу -N11,. За будовою бічного радикала амінокислоти підрозділя
ються на аліфатичні, ароматичні та гетероциклічні.
Амінокислоти можуть містити у своєму складі одну або дві амінні
(моноаміно-, діаміно-) та одну чи дві карбоксильні (монокарбонові, дикарбонові) групи.
Ізомерія аліфатичних амінокислот зумовлена відмінностями у взаєм
ному розміщенні змінної та карбоксильної груп, виходячи з чого розріз
няють а -, Р-, у-амінокислоти тощо. До складу природних білків входять
а-амінокислоти, які позначаються звичайно тривіальними, історично
усталеними назвами (розділ 4.3 ). В систематичній номенклатурі аміно
кислот положення аміногрупи позначають цифровими локантами.
Приклади буцови аліфатичних, ароматичних та гетероциклічних аміно
кислот:
СН 3
СН
МН2
СООН
с н 2— с н 2— с о о н
ІЧН2
(і-аланін
а -а л а к ін
И - г -СН2----- С Н —
N
Н
Тирозин
соон
мн2
Гістидин
Біохімічне значення амінокислот полягає в тому, що певна група
цього класу біоорганічних сполук, а сам е-двадцять а-амінокислот сте
реохімічного Ь-ряду - є структурними компонентами природних білків
та пептидів (т. з. “протеїногенні амінокислоти”). Крім того, більшість
вільних, тобто не зв'язаних у иоліпептидні ланцюги, протеїногенних амі
нокислот мають велике самостійне фізіологічне значення, беручи участь
у реалізації життєво важливих функцій клітин та організму в цілому. Біо
логічне значення та біохімічні перетворення сс-Ь-амінокислот будуть
розглянуті в розділі 4.3.
154
2.7. Г етер оф уи кц іои альн і сп олук и
Хімічні властивості амінокислот
Хімічні властивості амінокислот визначаються наявністю в них карбо
ксильної груші (кислотного центру) та аміногрупи (основного центру), тобто
вони є амфотерними сполуками. Завдяки цьому амінокислоти у водних роз
чинах утворюють внутрішні солі, або біполярні Іони (цвіттер-іони):
И— СН
Н— С Н — С О О '
СООН
т2
мн3
За карбоксильною групою амінокислоти реагують як карбонові ки
слоти —вони утворюють солі, складні ефіри, аміди, галогеноангідриди:
1) утворення солей амінокислот:
о
О
Я— СН — С
+ № О Ни
ч
он
я— -с н — с ч
0№
ын2
[МН<
2) утворення складних ефірів амінокислот:
Я— СН — С
ч
О
о
он
ОИ'
ын2
І\ІН;
3) утворення амідів амінокислот:
о
О
Р'ІМН2
И— СН
я— с н -
он
МН2
ч
ІМИИ'
ж 2
1п аміногрупою амінокислоти реагують як первинні аміни - вступа
ють в реакції К-алюлування, N-ацилування, утворюють гідроксикислоти.
1) N-алкілування амінокислот:
155
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
о
Я— С Н — С \
+ В'НаІ
ОН
&
я — СН— с
ч
он
Ж И'
ЫН;
2) К-ацилування амінокислот:
/°
//
+Р'СОСІ
И— С Н — С
ч
ОН
т 2
о
я— сн — с
ч
ОН
МНСОРҐ
3) Утворення гідроіссикислот:
о
,0
Я— С Н — С
+ Н Ы 02
ч
ОН
//
и— СН— с
ч
ОН
Ін '
МН;
Реакції дегідратаиії та дезамінування амінокислот. Лактами
При нагріванні амінокислоти можуть підлягати реакціям дегідратації відщепленню молекули води - або дезамінування - відщепленню моле
кули аміаку. Ці реакції по-різному протікають з а , (і, у та 6-ам ін окис лотами:
а)
а-амінокислоти при нагріванні утворюють циклічні діаміди- дикетопіперазини. Ця реакція являє собою між молекулярну дегідра
тацію за рахунок взаємодії між собою функціональних груп двох моле
кул амінокислот:
Н
Ч
Н
-2Н20
н с Л о
а -а м ін о к и слоти
156
Хс
Г
.А А ,
Д и кетопіперази н
2.7. Г етер оф ун кц іон альн і сп олук и
б)
Р-амінокислоти при нагріванні підлягаютьвнутрішньамолекулярному дезамінуванню (реакція елімінування), тобто втрачають молеку
лу аміаку, перетворюючись на а -, |3-ненасичені карбонові кислоти:
Я
СН—
СН—
імн2
н
СООН
и— с = с —
н
СООН
н
в)
у-, 5- та є-амінокислоти при нагріванні підлягають внутрішньомолекулярній дегідратації а утворенням лактамів - циклічних п’ятичлен
них, шестичленних та семичленнпх амідів відповідно. У певних умовах
можливе формування і чотиричленного |3-лактаму при термічній циклі
зації р-амінопрогііонової кислоти. Утворення лактамів відбувається за
рахунок просторового зближення карбоксильної та аміногрупи в (3-, у-, 8- та
є-амінокислотах, що можуть перебувати у клешнеподібній конформації.
Лактами здатні гідролізуватися з утворенням вихідних амінокислот.
Прикладом реакції може бути утворення лактаму у-аміномасляноі
кислоти (у-бутиролактаму):
н
■у-амшомасляна кислота
у-бутиролактам
У процесі циклізації відбувається внутрІшньомодекулярне перегру
пування атомів, що підвищує кислотність кислотного центру амінокис
лоти. Лактамні цикли можуть існувати в лактамнін (а) та лактимній (в)
формах {пактам-лактішна таутомерія), які відрізняються кислотніс
тю та фізико-хімічними властивостями:
(а)
(Ь)
157
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Лактамні кільця присутні в структурі молекугї багатьох природних
біоорганічних сполук. Так, наприклад, чотиричленне Р-лактамне кільце
є компонентом будови молекул антибіотиків з класу пеніцилінів:
Пе н Іцилік V (фе нокси м етилпе ніцилін)
Гідроліз є-капролактаму (синтетичної сполуки, що використовується
для промислового отримання капрону) призводить до утворення є-амінокапронової (6-аміиогексанової) кислоти:
СН2— СНр
;=о
N— Н
^
Н2М
(СН2)5— СООН
+ н2°
сн— сн2
в -капролактам
е-амш о капронова кислота
є-амінокапронова кислота є потужним інгібітором процесів фібрино
лізу, у зв’язку з чим фармацевтичні препарати є-амінокапронату засто
совуються в медичній практиці як ефективні антигеморагічні (крово
спинні) засоби.
Лактам-лактимна таутомерія в молекулах азотистих основ пурино
вого та піримідинового ряду є важливим фактором у формуванні моле
кулярної організації нуклеїнових кислот ДНК та РНК, що, у свою чергу,
має ключове біологічне значення в процесах збереження та реалізації
генетичної інформації.
Реакції дезамінування протеїногенних а-Ь-амінокислот, що відбува
ються в біохімічних системах під дією ферментів, будуть розглянуті в
розділі 4.3.
158
2.7. Гегероф у н ац іон ал ь н і сполуки
2.7.2. Гідроксикислоти та fcc властивості
Гідроксикислоти - похідні карбонових кислот, у структурі вугле
водневого радикала яких один або декілька атомів водню замішені на
гідроксигрупу -О Н .
Залежно від природи вуглеводневого радикала гідроксикислоти мо
жуть бути аліфатичними або ароматичними. Ароматичні карбонові
кислоти (фенолокислоти) розглянуті в розділі 2.5.
Аліфатичні гідроксикислоти
Структурна ізомерія аліфатичних гідроксикислот залежить від взаєм
ного розміщення карбоксильної та гідроксильної (гідроксильних) груп;
положення останніх, я к і в номенклатурі амінокислот, позначають циф
ровими локантами або буквами грецького алфавіту (а , Р, у тощо).
Розповсюдженими представниками природних аліфатичних гідрокси
кислот є такі сполуки;
Н2с^—1
с оон
он
он
Гліколева кислота
(гідроксиоцтова кислота)
HOOC— (j!H
сн 3—ун—соон
сн2— соон
ОН
Я бл уч н а кислота
(гідроксибурш тинова кислота)
М олочна кислота
(2-гід р о к си п р о п ан о вак и сл ота )
(а-гід р о к си проп іонова кислота)
HOOG— сн—
І
он
сн— соон
І
он
В инна кислота
(ос, а'-д и гід р о к си б у р ш ти ко в а кислота)
Більшість природних гідроксикислот є біомолекулами, що утворю
ються в організмі при перетворенні вуглеводів, амінокислот, жирних кис
лот, тобто є інтермедіатами внутрішньоклітинного обміну речовин. Зок
рема, біологічно важливими інтермедіатами обміну речовин є гідроксипохідні дикарбонових кислот —яблучна кислота —та трикарбонових
кислот —лимонна кислота (2-гідрокси-1,2,3-пропантрикарбонова) та
ізолимонна кислота.
159
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Проміжним метаболітом біосинтезу в організмі людини та тварин
холестерину та інших фізіологічно активних стероїдів с дифосфорний ефір
мевалонової кислоти — гідроксипохідної глутарової кислоти:
СН3
НООС— СНз— | — с н 2— СН2ОН
он
М с вал о нова кислота
Хімічні властивості гідроксикчслот. Біохімічне значення
Хімічні властивості гідроксикислот визначаються наявністю в їх мо
лекулах двох функціональних груп - карбоксильної та гідроксильної.
Карбоксильна група визначає кислотні властивості сполук, а гідроксиль
на - їх властивості як спиртів.
За карбоксильною групою гідроксикислоти дають реакції, харак
терні для карбонових кислот, тобто утворюють солі, складні ефіри, амі
ди, галогеноангідриди, наприклад:
1) утворення солей при взаємодії з лугами:
СНз— СН— СН2— СООН
+ МаОН1
ОН
СН3— ^ Н — СН2— С О О №
ОН
З-гід роксиб утанова кислота
3-гід рок си б утан оат
(З -гід р о кси б у ти р ат) натрію
2) утворення складних ефірів (реакція етерифікації) при взаємодії із
спиртами:
сн3— СН—
с (
±СНзОН
он
он
М ол оч н а кислота
С Н 5 - ЇН_ _ С^
|
он
ОСНз
М етиллактат
За гідроксильною групою гідроксикислоти утворюють алкоголяти,
прості та складні ефіри. При окисленні ОН-групи гідроксикислот утворю
ються оксокарбопові кислоти (альдегіде- та кетонокислоти). Наприклад:
160
2.7. Г стер оф ун кц іон альн і сполуки
1)
утворення складних ефірів при ацишованні гідроксикислот (вико
ристовуються галогеноангідриди або ангідриди кислот):
СН3
СН
СООН +- — 3— - 1
СН3 — С Н —
СООН
ОСОСНз
2-ацетокси пропанова кислота
2) окислення гідроксикислот з утворенням оксокарбонових кислот:
НО— '
он
Гліоксилова кислота
СНз— СН
СООН -
М » . сн3
СООН
II
о
ОН
М ол оч н а кислота
не
П іровиноградна кислота
Окислення гідроксикислот в біохімічних системах також відбуваєть
ся шляхом їх дегідрування за участю спеціальних ферментів дегідроге
наз. Акцептором атомів водню в цих реакціях є спеціальний кофермент
нуклеотидної природи, до складу якого входить піридинове кільце нікотинаміду (вітаміну РР) - никотинамідадсніндинуклсотид (НАД+) - розділ 5.3.
В ході окисно-відновної реакції до акцептора (НАДТ) приєднується протон
(іон водню) та два електрони, а другий протон, відщеплений від сполуки,
що окислю ється, залиш ається у вільном у стані, том у рівняння
ферментативного окислення молочної кислоти має вигляд:
СН3- СН - СООН + НАД+ - »
он
СН3- С - СООН + НАД+ + Н+
І!
о
161
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
Окислення в організмі гідроксипохідних довголанцюгових карбоно
вих кислот здійснюється шляхом дегідрування -ОН-груп, що знаходяться
у р-положенні, після переведення жирної кислоти в спеціальну активова
ну молекулярну форму —комплекс із коферментом ацилування —коензимом А,
Пегідратаиія гідроксикислот. Лактиди. Лактони
Подібно до амінокислот, гідроксикислоти при нагріванні вступають в
реакції елімінування, що проявляється внутрішньомолекулярною або
міжмолекулярною дегідратацією. При цьому можуть утворюватися цик
лічні структури - лактиди та лактони:
а)
а-гідроксикислоти при нагріванні підлягають міжмолекулярній
дегідратації з утворенням лактидів - циклічних складних ефірів, що
мають два складноефірні угруповання:
Л актид
а -п д р о к с и к и с л о т и
б)
р-гідроксикислоти при нагріванні зазнають внутрішньомолекулярної дегідратації] в результаті реакції утворюються а -, (ї-ненасичені карбонові кислоти:
Я
СН
СН
СООН
П----- С = С ------СООН
-Н20
он
н
н
н
в)
у та б-гідроксикислоти найлегше вступають в реакцію внутріишьомолекулярної дегідратації (вже при кімнатній температурі); при
цьому утворю ю ться складні ефіри - лактони. Реакція утворення
у-лактону з у-гідроксимасляної кислоти має вигляд:
162
2.7. Г етер оф ун кц іои альн і сп олук и
/
НгС\
\
он
*° )
А ь
‘ Нг0
он °
н* с
о
о
у-бутиролактон
у-гідроксим асляна кислота
Як і в разі утворення лактамів з амінокислот, формуванню лактонів
сприяє перебування молекул гідроксикислот у клешнеподібній конфор
мації. Лактони, як і лактиди, здатні до гідролітичного розщеплення з утво
ренням вихідних гідроксикислот.
Лактонні структури присутні в молекулах багатьох біоорганічних спо
лук. Утворення лактонів відбувається при відщепленні молекули води в
кислих розчинах альдонових кислот (розділ 4.1). Прикладом фізіологіч
но активного лактону є молекула Ь-аскорбінової кислоти (вітаміну С):
НО
ОН
ОН
Ь-а скорб і нова ки сл ота (у п л а то й 2,3-дегідро-Ь*гулон ової кислоти)
2.7.3. Оксокислоти та їх властивості
Оксокислоти (оксокарбонові кислоти) - похідні карбонових ки
слот, що містять у своєму складі, поряд з карбоксильною, також карбо
нільну групу. Залежно від типу карбонільної групи оксокислоти поділя
ються на сільдегідокис/іоти та кетокислоти.
Розміщення оксогрупи стосовно карбоксильної групи, як і для аміно
кислот та гідроксикислот, позначається цифровими локантами або від
повідними буквами грецького алфавіту.
Найбільш поширеними в біохімічних системах є такі оксокислоти:
163
Р озділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
° Ч
О
II
С— с(
не/
\
с н ^ с — соон
Гліокснлова кислота
(оксоеганова к ислота)
с н 3—
П іровиноград на кислота
(2-оксоп роп ан ова кислота)
с—
сн2—
соон
А ц етооцтова кислота (3-оксобутанова кислота)
ноос—
О
II
с—
сн2—
соон
Щ авлево оцтова ки сл ота (оксобутандіова кислота)
ноос—
О
II
с—
с н 2—
с н 2—
соон
сх-кетоглутарова ки сл ота (2-оксоп ентандіова кислота)
Хімічні властивості оксокислот
Відповідно до особливостей своєї будови, оксокислоти виявляють
хімічні властивості, характерні як для карбонових кислот, так і для аль
дегідів або кетонів.
За карбоксильною групою оксокислоти утворюють солі з основами
(1) та підлягають реакції етерифікації із спиртами з утворенням склад
них ефірів (2):
1) СН3-С О -С О О Н + ІМаОН - » СН 3-С О -С О О № + НгО;
2) СН3-С О -С О О Н + С2Н5-ОН - » СН3-С О -С О О -С 2Н5 + НгО.
В органічному синтезі, в тому числі синтезі лікарських засобів та
барвників, широко використовується етиловий ефір ацетооцтової кисло
ти —ацетооцтовий ефір.
164
2,7. Гетероф ум кц іон альн і сп олук и
О
II
с н 3— С
СН2
С
О
С 2 Н5
А ц етооцтови й еф ір
За карбонільною групою оксокислоти вступають в реаіщії нуклеофільного приєднання. Практичне значення мають реакції оксокислот з
гідроксиламінами (ИН2-О Н ) та гідразинами (МН2-МН2) з утворенням
специфічних оксимів та гідразонів, що використовуються в реакціях
ідентифікації вихідних оксосполук, наприклад, піровиноградної кислоти:
О
СНз— с — с о о н
+ Н2 п г - і^ п 2
V)
N— м н 2
( ! ) окси м пірови н оград н ої кислоти;
(2) гідразин пірови н оград н ої кислоти
Біохімічне значення та перетворення кетокислот
Піровиноградна кислота - кінцевий продукт внутрішньоклітинного
окислення глюкози (аеробного гліколізу). Концентрація піровиноград
ної кислоти у тканинах тваринних організмів перебуває в динамічній
рівновазі з продуктом її відновлення - гідроксикислотою лактатом (мо
лочною кислотою). Основний шлях розщеплення (катаболізму) пірови
ноградної кислоти в клітині полягає в її окислювальному декарбоксилуванні складною поліфермептпою системою з утворенням “активної форми
ацетату” - ацетилкоензиму А (ацетил-КоА), що підлягає подальшому
окисленню в циклі трикарбонових кислот до С 0 2 та Н20 :
165
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
сн3— с — с о о н
со
+НЗ-КоА
СН3— С — СОЭКоА
А ц ети л ю ен зи м А
Щавлевооцтовсі кислота та а-ке то глут арова кислота - інтермедіати загального біохімічного шляху внутрішньоклітинного перетво
рення вуглецевого скелету вуглеводів, жирних кислот та амінокислот
(циклу трикарбонових кислот , або циклу Кребса).
а -кетопохідні вищих карбонових кислот є проміжними продук
тами окислення в мітохондріях клітин жирних кислот - т. з. процесу
(3-окислення, що є головним біохімічним механізмом катаболізму жирів
з утворенням енергії у формі АТФ.
Ацетооцтова кислота - проміжний продукт [і-окислення жирних
кислот, що у тканинах тваринних організмів легко перетворюється на
(3-гідроксимасляну кислоту (1) та ацетон (2):
+2Н
ОН
*2Н с н 3— С Н — сн2— СООН
О
II
с н 3— С — СН2 — СООН-
( 1)
0р2
о
и
сн3— сс —
— СНз
(2 )
При деяких патологічних станах ацетооцтова кислота та ацетон мо
жуть накопичуватися в організмі людини у надзвичайно високих концен
траціях. Аномальне накопичення в плазмі крові цих сполук (“кетонових
166
2.7. Гстсрофу н ац іон ал ь н і сп олук и
тіл”), яке спостерігається при важких формах цукрового діабету, супро
воджується підвищеним виділенням цих сполук із сечею хворої людини
(іацетонурія, або кетонурія).
2.7.4. Аміноспирти. Амінофеноли
Аміноспирти та амінофеноли - похідні вуглеводнів, що мають у
своєму складі як амінну, так і гідроксильну групи. Похідними аліфатич
них вуглеводнів є аміноспирти, ароматичних - амінофеноли.
Хімічні властивості аміноспиртів та амінофенолів близькі між собою.
Для обох цих класів сполук властиві хімічні реакції, характерні для амі
нів. Проте аміноспирти вступають в реакції, притаманні спиртам, а амі
нофеноли - фенолам.
А. Аміноспирти
Хімічні властивості
РІаявність аміногрупи надає аміноспиртам виражених основних влас
тивостей. Як основи вони утворюють солі з мінеральними та сильними
органічними кислотами, а при взаємодії з водою утворюють гідроксиди:
НО— (СН2)П— ІМН2 + Н— ОН— Н Н О — (СН2) — ЙН3І0Н '
н о — {сн2)п— ж 2 + н — сі—► [но— {сн2)я— й н 3]с г
Медико-біологічне та фармацевтичне значення аміноспиртів
Важливими представниками аміноспиртів є т. з. біогенні аміни етаноламін (2-аміноетанол; коламін) та його ЇМ-ацильне похідне холін. Ці
сполуки використовуються в живих організмах як попередники для біо
синтезу інших складних біомолекул —ліпідів, нсйромедіаторів —та вико
ристовуються як лікарські засоби.
Н О — С Н 2— С Н 2 — Г\ІН2
К олам ін (етанолам ін)
Н О — С Н 2— С Н 2
N = ( С Н з ) з ОН"
Х олін (трим етил-2-гідрокси етилам оній)
Як і інші аміноспирти, коламін гідроксид завдяки своїм лужним влас
тивостям при взаємодії з кислотами утворює солі, а при розчиненні у
воді - четвертинні амонієві основи:
167
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
НО— СН^— СН2 — 1МН2+ Н— ОН— *~[НО— СН2— СН2— ЙНзІОН
У тканинах тваринних організмів коламін утворюється шляхом дєкарбоксилування амінокислоти Ь-серину, а подальше метилування коламіну призводить до утворення холіну:
Срг
но— с н 2— у н — С О О Н —
3-СНз
НО— СНг— сн2— мн2—
ІМНг
1-серии
К оламін
► НО
СН2
СН2
Й=
(С Н з )з
Холін
Ацет илхолін
Важливу фізіологічну роль в організмі відіграє сполука нейромедіаторної функції ацетилхолін - складний ефір холіну та оцтової кислоти. Аце
тилхолін утворюється шляхом ацетилування холіну за участі ацетилкоензиму А при каталітичній дії ферменту холінацетилтрансферази:
О
+
(С Н з)з= М
II
СН2 ----- СН2 — О Н +СН з— С — СОЗКоА-------►
Холін
А цетил-КоА
О
► (С Н з)з
—
С Н 2—
С Н 2—
О—
А ц ети л х о л ін
С — СНз + НЗ —
К оА
К о е н зи м А
Розщеплення ацетилхоліну здійснюється шляхом його гідролізу за
участю ферменту ацет ш холінест ерази:
О
+
(С Н 3)3 =
N
N
СН2
С Н 2 ------ О — С — С Н з + Н20 -------- ►
Ацетилхолін
(С Н 3)з =
N —
Холін
168
С Н 2 ------ С Н 2 —
О Н + С Н з ------- С О О Н
О цтова кислота
2.7. Гетерофуїї кц іок альн і сп олук и
Ацетилхолін є медіатором холінергічних нейронів центральної та пери
феричної нервової систем в організмі людини та вищих теплокровних
тварин; порушення його утворення або розщеплення призводить до важ
кої дисфункції в діяльності нервової та інших фізіологічних систем.
Б. Ам іноф еноли
Хімічні властивості
Подібно іншим ароматичним амінам, амінофеноли є слабшими
основами, ніж аміноспирти, але сильнішими від аніліну, що спричинено
електронодонорним впливом гідроксильної групи. Разом з тим, наявність
аміногрупи в ароматичному кільці дещо зменшує кислотні характерис
тики фенольного гідроксилу. Виходячи з цього, амінофеноли здатні реа
гувати як з мінеральними кислотами (основні властивості аміногрупи),
так і з лугами (кислотні властивості фенольного гідроксилу), тобто ма
ють властивості амфотерних сполук:
ІЧН3СІ
N 42
+НС1
+№ ОН
0 '№ +
р-амінофенолят натрію
ОН
р-амінофенол
ОН
р -гідрок си ан іл ін ів хлорид
Парацетамол. ПАСК
При М-ацетилуванні р-амінофенолу утворюється р-М-ацстамінофенол (Парацетамол), який нині є одним із найбільш поширених і потуж
них жарознижуючих, знеболюючих та протизапальних лікарських засобів.
+ (С Н 3С 0 ) 20
ІЧН2
р-ам ін оф ен ол
Ы НСОСНз
р-Ы -ацетам ш о ф енол
169
Розділ 2. ХАРАКТЕРИСТИКА ОКРЕМ ИХ КЛАСІВ БІООРГАНІЧНИХ СПОЛУК
До жарознижуючих препаратів з класу НПЗЗ належить також похідна
р-амінофенолу Фенацетин (1-етокси-4-ацетамінобензол).
Фармакологічно активною похідною т-амінофенолу є р-аміносаліцилова (4-аміно~2-гідро ксибензойна) кислота, що утворюється шляхом
карбоксилування т-амінофенолу:
т-амінофенол
р-аміносаліцилова кислота
р-Аміносаліцилова кислота (ПАСК) має високу бактеріостатичну
активність щодо мікобактерій туберкульозу і у вигляді натрієвої солі
(Natriumparaaminosalicilieum) застосовується як один з основних про
титуберкульозних засобів.
К ат ехолам іни
До фізіологічно активних сполук, які за хімічною будовою є аміно
спиртами, належать речовини, у складі молекул яких міститься зали
шок двоатомного фенолу пірокатехіну (1,2-дигідроксибензолу). Ці спо
луки мають назву пірокатехінамінів, або катехоламінів. До катехоламінів належать: дофамін, норадреналін та адреналін.
Катехоламіни належать до фізіологічно активних біогенних амінів і
утворюються при декарбоксилуванні гідроксипохідного L-фенілаланіну
амінокислоти 3,4-дигідроксифенілаланіну в результаті такої послідовності
ферментативних реакцій, що включають в себе процеси гідроксилування
та метилування:
H2N
ОН
но^уМ )
^ " " 'О Н
170
Т ирозин
2.7. Г етер оф ум кц іоп альн і сп ол ук и
НО
н
о
он
^ Н
3 , 4-ди гідроксиф енілалан ік
со
но
но-
NH2
ч
//
Дофамін
(^ о н
но
імн2
но- //
%
он
Н орадрсналін
СНз
/СН 3
НО
НО
—\
у
Г ~ын
(
А дреналін
ОН
Фізіологічні та біохімічні ефекти катехоламінів реалізуються шляхом
їх взаємодії із специфічними, високочутливими до цих сполук білковими
структурами на плазматичній мембрані клітин-мішеней, т. з. рецепт о
рами. Синтетичні біоорганічні сполуки, що за хімічною структурою
близькі до природних катехоламінів, застосовуються як лікарські засо
би нейротропної та психотропної дії.
171
Частина II
БІООРГАНІЧНІ СПОЛУКИ ЯК МЕТАБОЛІТИ
ТА ЛІКАРСЬКІ ЗАСОБИ
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Гетероциклічні сполуки - біоорганічні сполуки циклічної будови,
у складі циклів яких, крім атомів вуглецю, містяться також атоми інших
елементів (гетероатоми) - найчастіше азоту, кисню або сірки.
3.1. Загальні властивості гетероциклічних сполук
Гетероциклічні сполуки (гетероцикли) є надзвичайно різноманітним
класом речовин; деякі з них синтезуються в клітинах живих організмів,
решта є результатом штучного хімічного синтезу Гетероцикли та їх по
хідні особливо широко представлені серед природних сполук: як біомолекул - структурних компонентів клітини, так і низькомолекулярних фізіо
логічно активних речовин, зокрема рослинних алкалоїдів, синтетичних
лікарських засобів. В багатьох випадках молекули гетероциклів є поперед
никами (т. з. прекурсорами) в утворенні інших, складніших органічних
молекул з новими фізіологічними та фармакологічними властивостями,
що широко використовується у фармацевтичному синтезі.
Гетероцикли розрізняються між собою та класифікуються за природою
гетероатома (одного або двох), що входять до складу циклу, розміром цик
лу (звичайно зустрічаються п’яти- та шестичленні цикли, рідше - сеМичленні цикли), ступенем насиченості циклу. Серед природних сполук,
особливо таких, що мають фізіологічну активність, найчастіше зустрі
чаються ненасичені гетероцикли.
Номенклатура та класифікація гетероциклів
Гетероциклічні сполуки зазвичай мають тривіальні назви, які є до
пустимими відповідно до номенклатури ІЮПАК, в тому числі і в якості
назв родоначальних структур, які використовуються при побудові сис
тематичних назв їх похідних.
Нумерацію окремих атомів у структурі гетероциклічної сполуки по
чинають з гетероатома. Атоми вуглецю в молекулах гетероциклів мо
жуть позначатися також буквами грецького алфавіту (а, 3, у тощо), по
чинаючи від С-атома, розміщеного поряд з гетероатомом.
3.1. Загаль н і вл асти вості гетероц ик ліч н их сполук
Будова родоначальних предст авників окремих підкласів гет ер оииклів
Наведемо приклади будови найбільш поширених у біохімічних сис
темах гетероциклічних сполук з різною кількістю гетероатомів (в дуж
ках - назви гетероциклів за систематичною номенклатурою).
П ’ятичленні гетероциклы з одним гетероатомом;
Н
П ірол
Ф уран
{Азол)
(О ксол)
Т іоф ен
(Т іол)
П 'ят ичленні гетероцикли з двома гетероатомами:
н
Н
Ті азол
(1,3-тіазол)
П іразол
(1,2-діазол)
Ім ідазол
(1,3-діазол)
Ш естичленні гетероцикли з одним гетероатомом:
У
П іридин
(А зин)
У
у-піран
(4Н -піран)
173
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Ш естичленні гетероцикли з двома гетероатомами:
Ч ,
2кЧ
N
і
/
І
П ірим ідин
(1,3-діазин)
1
П іридазин
(1,2-діазин)
П іразин
(1,4-діазіш )
Конденсовані гет ероциклічні системи:
Гетероциклічні сполуки, молекули яких складаються з одного гетеро
циклічного та одного чи декількох бензольних або також гетероциклічних
ядер, мають назву конденсованих гетероциклічних систем, наприклад:
Індол
А кридин
N
N
\
/
N
Н
П урин
Аромат ичніст ь гетероциклів. Елект ронна конфігурація атома
азот у
Ряд найважливіших представників п ’яти- та шестичленних гетероциклів та побудовані на їх основі конденсовані гетероциклічні системи
мають ароматичні властивості (гетероароматичні сполуки). Такі гєтероцикли розповсюджені в біологічних об’єктах у вільному стані або
як компоненти складніших сполук - біомолекул тваринних організмів чи
рослинних алкалоїдів, що визначає необхідність вивчення особливостей
їх будови та реакційних властивостей.
174
3.1. Загал ь н і власти вості гетер оц и к л іч н и х сполук
Критерії ароматичності циклічних сполук були розглянуті в розділі 2.2.
Органічна сполука є ароматичною, якщо вона має плоский циклічний
скелет, створений о-зв’язками, та кількість спряжених я-електронів (N1),
яка дорівнює: N = 4п + 2, де п (число циклів) = 1, 2, 3 і т.д. (правило
Хюккеля). Відповідно до цього, для неконденсованих карбо- та гетероциклів (п = 1) ароматичними будуть структури, що містять 6 я-електронів.
Наявність гетероатома призводить до нерівномірності в розподілі елек
тронної густини в циклах. Маючи електронну конфігурацію 1522з22р3, атом
азоту у складі гетероциклічних сполук формує три гібридні зр2-орбіталі,
що утворюють, відповідно, три ковалентні о з в ’язки цього атома. При
цьому в залежності від розміру циклу (п’яти- чи шестичленний) та від
того, один чи два р-електрони (з двох неспарених електронів азоту) будуть
включені у спряжену я-систему, вплив гетероатомів азоту на утворення
ароматичного секстету буде різним. Згідно з цими особливостями,
розрізняють атоми азоту піридинового та пірольного типів.
У молекулі піридину атом азоту, що знаходиться в стані зр2-гібридизації, використовує для утворення ст-зв’язків дві з трьох яр2-гібридних орбіталсй. При цьому в ароматичний 6-членний цикл молекули постачаєть
ся один рг-електрон, що утворює циклічну систему я-зв’язків (рис. 3.1).
В ільна н еп од іл ен а пара ел ек трон ів, що м істи ться на третій
зр2-гібридній орбіталі IV-атома, визначає властивості піридину як основи
та нуклеофілу. Атом азоту з такою будовою зовнішнього електронного
(валентного) шару позначають як піридиновий азот. В результаті біль
шої електронегативності порівняно з атомами вуглецю піридиновий атом
азоту знижує електронну щільність на С-атомах ароматичного кільця.
Тому молекулярні системи з піридиновим азотом називають тс-недоспютніми ( п-електронодефіцитними).
У молекулі піролу в угворенні трьох а -зв ’язків атома азоту беруть
участь три електрони, що розміщені на трьох 5р2-гібридних орбіталях
К-атома. Для утворення ароматичного секстету 5-членного циклу
я-електронів використовуються два р^-електрони, розміщені на негібридизованій орбіталі (рис. 3.2 ).
Атом азоту в розглянутому електронному стані дістав назву пірольпого азоту. В молекулі піролу 6-електронна я-хмара локалізована на
5 атомах циклу, тому пірол являє собою п-надлишкову систему. До та
ких електронних систем належать також фуран і тіофен.
175
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Ф уран
Т іоф ен
N
••
П іридин
Рис. 3 .1 . Схема електронної будови піридинового атома азоту:
эрг-АО - три гібридизовані атомні орбіталі атома азоту; р,-електрон негібридизований електрон на р-орбіталі атома азоту, що використовується
для утворення ароматичного секстету іг-електронів молекули піридину
Рис. 3 .2 . Схема електронної будови пірольного атома азоту
Позначення - див. рис. 3.1.
176
3.1. Загальні властивості гетероциклічних сполук
Ароматичність гетероциклів з двома гетероатомами азоту
До гетероциклічних ароматичних сполук з двома атомами азоту на
лежать п ’ятичленні гетероцикли імідазол, піразол та шестичленна спо
лука — піримідин.
Два З а т о м и , що містяться в означених гетероциклах, мають різну
електронну будову і по-різному взаємодіють з циклічною структурою
відповідної молекули, а саме (рис. 3.3 ):
а) У шестиатомному піримідині обидва атоми азоту віддають у спря
жену циклічну систему по одному р-електрону та мають по одній парі
неподілених електронів на эр2-гібридизованій орбіталі. Отже, в піримідині
та інших шестичленних гетероциклах з двома М-атомами (діазинах) оби
два атоми азоту належать до піридинового типу;
б) У п ’ятичленних імідазол і та піразол і атоми азоту різні: один з них
віддає у спряжену систему два р-електрони - азот пірольного типу, а
другий К-атом - один р-електрон і є, відповідно, азотом піридинового типу.
Розглянуті особливості електронної будови гетероциклів з двома атоми
азоту визначають хімічні властивості відповідних біоорганічних сполук.
Атоми азоту піридинового типу
Атоми азоту пірольного типу
Рис. 3 .3 . Піридинові та пірольні атоми в гетероциклах з двома атомами азоту
177
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Гетероцикли як органічні кислоти та основи
Основні властивості зумовлені наявністю в молекулах гетероциклів
п ір и д и н о ви х атомів азоту Г\[-атоми піридинового типу мають на
їр^гібридизованій орбіталі неподілену пару електронів, що ке приймає
участі в ароматичному спряженні. За рахунок цієї пари електронів гетеро
циклічні сполуки з піридиновими атомами азоту являють собою основи
Льюїса, що можуть взаємодіяти з сильними кислотами, утворюючи солі:
НВг
а ?
*N‘
Вґ
“N
Н
П іридин
Ім ідазол
П іридин і н бром ід
Імідазш гій х л ори д
Кислотні властивості гетероциклів залежать від наявності в їх
структурі атомів азоту пірольного типу. У таких сполуках, як пірол,
піразол, імідазол та їх похідних, пірольні атоми азоту надають цим речо
винам властивостей слабких NH-кислот. У таких гетероциклах за дії
сильних основ від NH-груп може відщеплюватися атом водню із замі
щенням його на метал.
Амфотерні властивості, тобто здатність реагувати і як основи, і
як кислоти, мають п ’ятичлениі гетероцикли (імідазол, піразол), в яких
містяться як піридинові, так і пірольні атоми азоту.
178
3.2. П ’яти членні гетероцикли
3.2. П ’я тн ч л е н н і гетер о ц и кл н
3.2.1.
П ’ятичлснні гетероциклы з одним гетероатомом
До п ’ятичленних гетероциклів з одним гетероатомом (К, О та Б від
повідно) належать пірол, фуран та тіофен.
П ірол
Ф уран
Т іоф ен
Як уже зазначено, за своєю електронною будовою молекули цих спо
лук відповідають критеріям ароматичності, що визначає характер їх
реакці йноздатності.
Пірол - безбарвна рідина із запахом, що нагадує запах хлороформу.
Структура піролу та його похідних зустрічається у складі багатьох біомолекул, природних та синтетичних лікарських засобів.
Пірол має властивості основи (приєднує протон) та слабкої ІЧН-кислоти. Відповідно до ароматичного характера кільця, характерною влас
тивістю піролу, як і фурану та тіофену, є схильність до реакцій електрофільного заміщення (БЕ), що відбуваються переважно на а-атомах ву
глецю гетероцикла.
3.2.2. Хімічні властивості піролу
£ - р еакції піролу:
І) нітрування піролу (в реакції з ацетилнітратом):
Піроп
2-нітропірол
179
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
2) сульфування піролу (в реакції з піридинсульфотриоксидом):
П ірол
П ірол-2-сульф окислота
3) ацетилування піролу за Фріделем - Крафтсом (в реакції з ангі
дридами кислот):
N
N
П ірол
2-ацетилпірол
Відновлення піролу
Важливою для синтезу біоорганічних сполук, в тому числі створення
нових фармацевтичних засобів, є реакція відновлення піролу Відновлен
ня піролу відбувається в присутності металевих каталізаторів і призво
дить до утворення насиченого гетероциклу піролідину:
Пірол
180
2,5 - дигі дропіро л
(Піролін)
Т страгідропірол
(П ірол ідин)
3.2. П ’ятич ленні гегер оц н к л н
Піролідин є циклічним вторинним аміном, що має властивості сильної
основи. Структура піролідину входить до складу багатьох природних спо
лук з фізіологічною активністю, зокрема алкалоїдів (нікотину, атропіну, ко
каїну), амінокислоти проліну та синтетичних лікарських засобів.
Конденсовані гет ероцикли на основі піролу
Конденсованими гетероциклічними сполуками, що побудовані на ос
нові ядра піролу, є індол та його похідні.
Індол (бензо[Ь]пірол) - конденсований гетероцикл, що складається
з кілець піролу та бензолу.
Індол
У медицині та фармації мають важливе значення численні похідні
індолу, а саме: амінокислота Ь-триптофан та продукти її перетворення серотонін, триптамін, індоксил, р-індолілоцтова кислота, алкалоїди та
лікарські засоби, що містять у своїй молекулі структуру індолу.
Серотонін та триптамін - біогенні аміни, продукти біохімічного пере
творення (гідроксилування, декарбоксилування) триптофану в організмі
(рис. З.4.):
Рио. 3.4. Схема перетворення триптофану
(1) на 5-гідрокситриптофан (2), 5-гідрокситриптамін (серотонін) (3) та
триптамін (4)
181
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Серотонін - фізіологічно активна сполука, що має властивості
гормоната нейромедіатора, синтезується в серотонінергічних структурах
головного мозку, спеціальних клітинах сполучної тканини та кишечнику.
Серотонін збільшує артеріальний тиск, активує згортальну функцію крові,
є модулятором важливих психічних функцій людини, регулятором сну. З
порушенням обміну серотоніну пов’язують розвиток шизофренії, алко
голізму, ендогенної депресії.
Деякі індолвмІсні молекулярні структури з класу алкалоїдів мають
властивості психоактивних речовин, що порушують нормальну діяль
ність центральної нервової системи, спричиняючи важкі психічні розла
ди, виникнення галюцинацій. До рослинних галюциногенів, біохімічним
механізмом дії яких може бути порушення обміну серотоніну в головно
му мозку людини, належать псилоцибін та діетиламід лізергінової
кислоти (т. з. “сполука ЛСД”).
Кінцевими продуктами метаболізму серотоніну татриптаміну в ор
ганізмі є 5-окси-Р-індолілоцтова кислота та Р-індолілоцтова кислота, які
виділяються з сечею. В рослинних організмах Р-індолілоцтова кислота
виконує функцію гормону росту (гетероауксин) і з відповідною метою
застосовується в сільському господарстві.
5-окси-Р-індол іл оцтова кисл ота
(3- Індол Іл оц това кислота
Індоксил (3-оксиіндол) - похідна триптофану, що утворюється в тов
стому кишечнику в результаті перетворення (біотрансформації) амі
нокислоти ферментами мікроорганізмів (“гниття білків у кишечнику”).
В клітинах печінки Індоксил, що надходить з кишечнику, підлягає д е
токсикації шляхом утворення складного ефіру з сірчаною кислотою
(індоксилсульфату), який екскретується нирками у вигляді калієвої
солі - т . з. “тваринного індикану”. Таким чином, концентрація індикану
в сечі є біохімічним показником активності процесів гниття білків у
кишечнику та функціонального стану печінки:
182
3.2. П 'яти член ні гетероц ик ли
- С И — соон
I
ы н2 - ►
Триптофан
Індоксил
С катол
Індоксил сульфат
Індометацип. Синтетичним похідним Р-індолілоцтової кислоти є лі
карський засіб із протизапальною дією Індометацин НПЗЗ [2-метил-5метокси-1-(р-хлорбензоїл)індоліл-З-оцтова кислота]:
Індометацин
Тетрапірольні сполуки
Біологічно важливими похідними піролу є тетрапірольні сполуки похідні ароматичного макроциклу порфіну.
Порфіи - циклічна гстрапірольна структура, що побудована з піролінового (І), пірольного (III) та двох ізопірольних (II, IV) ядер, сполучених
між собою метановими групами = С Н -. Похідні піролу - порфірини в
комплексі з атомами заліза, міді або магнію - металопорфірини - є
компонентами (простетичнгши групами) складних білків, які беруть
участь у транспорті кисню та каталізі багатьох окислювально-відновлювальних реакцій в живих організмах.
183
Р озділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
П орф ін
Представником білків, що містять у своєму складі металопорфірини,
€ кисеньтранспортуючий білок еритроцитів людини гемоглобін. Порфіриновою структурою в цьому білку є т. з. протопорфірин III (за ста
рою номенклатурою Фішсра - протопорфірин IX, або 1,3,5,8-тетраметил-2,4-дивініл-6,7-дипропіоновокислий порфін), що в комплексі з іоном
заліза Бе (II) утворює гем - простетичну групу гемоглобіну. Гем є піг
ментом червоного кольору, що надає відповідного забарвлення еритро
цитам та крові людини в цілому.
Н3С ч
X
/С Н = С Н 2
р02+
н3С \
/С Н = С Н 2
і .
Н ЯС
Н— СНг
(СНг)г
184
снз
їоон
СООН
Протопорфірин IX
Гем
(Ре-протопорфірин IX)
3.2. П’яти членні гетероцнклн
Крім гемоглобіну, до металопорфіринових білків належать: міогло
бін (кисень-депонугочий білок м ’язів); дихальні ферменти мітохондрій
цитохроми; ферменти, що беруть участь в обміні перекису водню, каталаза та пероксидаза; хлорофіл - фотосинтезуючий білок рослин.
Модифікована макроциклічна тетрапірольна структура в комплексі з
іоном кобальту є також складовою молекули вітаміну В |2.
Синтез макроциклІчних тетрапірольних сполук в організмі здійсню
ється з відносно простих біомолекул-попередників - амінокислоти глі
цину та активної форми бурштинової кислоти -сукцинілу-КоА.
Тривалість життя зрілих еритроцитів людини складає 100-120 діб, пі
сля чого відбувається руйнування еритроцитів та окислювальний ката
болізм гемоглобіну. Розщеплення гему призводить до утворення ліній
них тетрапірольних структур, головна з яких - пігмент білірубін - вида
ляється з жовчю в кишечник.
СООН
СООН
Жовчний пігмент білірубін
Фуран - п’ятичленний гетероцикл, що містить атом кисню. За хіміч
ними властивостями фуран близький до піролу. Здатний до реакцій електрофільнош заміщення - нітрування, сульфування, ацилування.
Важливою похідною фурану є фурфурол (фуран-2-карбальдегід):
Ф уран
Ф урф урол
185
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Фурфурол являє собою маслянисту рідину із запахом свіжовипеченош житнього хліба (в якому він міститься). Продукт нітрування фурфуро
лу в 5-му положенні - 5-нітрофурфурол - при взаємодії з амінами утво
рює заміщені іміни, які є вихідною сировиною для синтезу важливого кла
су лікарських засобів з протимікробною (бактерицидною) активністю:
Ф урфурол
5-нітрофурфурол
СН=Ы Р
О
Імін 5-нітроф урф уролу
Поширеними в медичній практиці 5-нітрофуранами з антисептичною
дією щодо широкого класу патогенних мікроорганізмів є такі лікарські
сполуки, як Фурацилін та Фуразолідон (містить також відновлену ге
тероциклічну структуру оксазолу):
о
С Н = Ы — N14------ с
0 2м
ІЧН2
и
Ф урацилін (семікарбазон 5-нітрофурфуролу)
0 2Ы
■СН =
ІМ
N
.О
'О'
Ф уразолідон
{ 3-[М -(5-нітрофурф урііліден)аміно]оксазолідон-2)
186
3.2. П ’ятичленіїі гсгероцикли
Кільце фурану (фуранозний цикл) входить також до складу молекул
циклічних форм моносахаридів (розділ 4.1).
Тіофен - п’ятичленний гетероцикл, що містить у складі молекули
атом сірки.
Повністю гідроване кільце тіофену разом з гідрованим імідазолом є
структурною основою вітаміну Н (біотину). Біотин за біохімічним меха
нізмом дії належить до коферментних вітамінів, бере участь в реакціях
карбоксилування біомолекул ири синтезі жирних кислот, пуринових
нуклеотидів тощо.
Тіофен
Біотин
Тіофен та його похідні є фізіологічно активними складовими іхтіолової
мазі, що застосовується як засіб із місцевою протизапальною та антисеп
тичною дією. Деякі похідні тіофену мають антигельмінтну активність.
3.2.3. П ’я т и ч л е н н і гетероциклы з двома гетероатомами
П’ятичленні гетероцикли з двома гетероатомами, хоча б один з яких
є азотом, мають загальну назву азолів. Азоли підрозділяються на спо
луки з двома атомами азоту та двома різними гетероатомами.
П ’я т и ч ле н н і гет ероцикли, щ о м іст ят ь два ат оми азоту. До
цього підкласу належать речовини, які розрізняються за місцем локалі
зації азоту в циклі, а саме піразол та імідазол.
Як піразол, так і імідазол містять у своїй структурі атоми азоту пірольного та піридинового типів і проявляють властивості як органічних
основ (приєднують іон водню), так і N14-кислот (див. вище).
Піразол (1,2-діазол) - синтетична сполука, яку не виявлено в живій
природі:
187
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Н
Піразол
Піразол широко використовується у фармацевтичному синтезі. Прак
тично важливими хімічними властивостями піразолу є здатність до ре
акцій електрофільного заміщення, при цьому місце електрофільної атаки
залежить від природи реагенту та умов проведення процесу.
На основі похідного піразолу - піразолону-5 синтезується важлива
група лікарських засобів з анальгетичною та жарознижуючою дією Антипірин, Амідопірин, Анальгін:
Н
Піразол
І
н
Піразолон-5
С 6Н5
З-метал-1 -феніл-піразолок-5
188
3.2. П ’ятн'їленні гетероцикли
,сн.
,СН3 (СН3)2М
СН3 503№-СН2- і/
С6Н5
2,3 -дн метил-1-фен ілпіразолон-5
(Антипірин)
2,3-диметил-4диметил-аміно-1фенілпіразолон-5
(Амідопірин)
2,3-димети л-4-мети лам і но1-фенілпіразолон-5->і-метансульфонат натрію
(Анальгін)
Імідазол (1,3-діазол). Структура імідазолу входить до складу бага
тьох біомолекул та лікарських засобів.
Імідазол
Серед хімічних властивостей імідазолу в біохімічних системах ста
новлять найбільший інтерес його кислотно-основні властивості, проте
реакційна здатність щодо електрофільного заміщення порівняно з піразолом менш виражена.
Молекулі Імідазолу, як і піразолу, властива азольна таутомерія, як
різновид прототропної таутомерії. Це явище зумовлене рухомістю
атома водню кислотної МН-групи та можливістю його внутрішньомолекулярного переносу на центр основності, тобто піридиновий азот:
189
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
4
З
і1
4(5 )-метил і м ідаз ол
В результаті прототропної таутомерії похідні імідазолу, заміщені в
положеннях 4 та 5, є рівноцінними; виділити окремі таутомери (на
приклад, 4- та 5-метилімідазол, або відповідні таутомерні форми гісти
дину) неможливо. Для врахування цієї структурної особливості у назвах
відповідних сполук в дужках наводиться цифра, що позначає існування
двох таутомерів.
Похідні імідазолу
Гістидин. Пурин
Похідними імідазолу, що мають важливе біологічне значення, є протеїногенна амінокислота гістидин [а-аміно-|3-(імідазоліл-4)пропіонова
кислота] та конденсований гетероцикл азотиста основа пурин, що вхо
дить до складу багатьох вільних нуклеотидів та нуклеотидів як моно
мерів нуклеїнових кислот:
тг
СН2 — С Н — СООН
Н
А м інокислота гістидин (1) т а конденсований
гетер о ц и к л пурин (2) як похідн і ім ідазолу
190
3.2. П 'я ти ч л ен н і гетер оц и к л »
Імідазольний цикл, який е структурним компонентом молекули
протеїногенної амінокислоти Ь-гістидину, надає цій біомолекулі відповід
них амфотерних властивостей. Здатність різних атомів азоту гістидину
приєднувати протони (піридиновий атом - центр основності) та відда
вати протони (пІрольний атом - центр кислотності) визначає роль цієї
амінокислоти як центру кислотно-основного каталізу у складі активних
центрів багатьох ферментів гідролітичної дії (рис. 3.5 ).
А н іон
ім ід азол ію
Ім ідазол
К атіон
ім ідазолію
Р ис. 3 .5 . Схема участі Імідазолу у складі амінокислоти гістидину в кислотно-ос
новному каталізі ферментних білків
Гіст амін - фізіологічно активна сполука (біогенний амін гормональ
ної дії), що є продуктом декарбоксилування Ь-гістидину:
0р2
о
ГМ-*— СНг— СН— С О О Н
О
^ " Т “ СН2
СНІ2
ІЧЬІ2
N
Н
Г істи ди н
Гістам ін
Гістамін має властивості тканинного гормону, що розширює крово
носні судини; його концентрація в міжклітинних просторах різко зростає
в умовах розвитку запальних процесів, алергічних станах. Гістамін бере
також участь в регуляції секреції соляної кислоти в шлунку та виконує
функцію нейромедіатора в гістамінергічних структурах центральної
нервової системи. Сполуки, що протидіють зв ’язуванню гістаміну з чу
тливими рецепторами сполучної тканини, застосовуються як антиалер191
Розділ 3. ГЕТЕРОЦИКЛГЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
гічні засоби (Н, -гістаміноблокатори - Діше^рол, Діазолін), а фізіологіч
но активні речовини, які блокують взаємодію аміну з рецепторами сли
зової оболонки шлунку (антагоністи Н,-рецепторів—Ранітидин, Фамотидин), мають фармакологічні властивості антацидних засобів, що ви
користовуються при виразковій хворобі.
Бензімідазол - конденсована гетероциклічна система, що склада
ється з ядер бензолу та імідазолу:
Структура бензімідазолу є компонентом будови молекули вітаміну
ВІГ Похідним бензімідазолу є лікарський засіб Дибазол, що має гіпотен
зивну дію та адаптогенні властивості:
П 'я т и ч л е н н і гет ероцикли, що м іст ят ь два р Ь н и х гетероато
ми. До цього підкласу гетероциклічних сполук належать тісізол, окса
зол та ізоксазол:
Т іазол
( і ,3-тіазол)
О ксазол
(1,3-оксазол)
Ізоксазол
(1,2-оксазол)
Тіазол —кільце тіазолу разом з шестичленним гетероциклом піримідином (див. нижче) входить до складу вітаміну В! (тіаміну), що виконує
192
3.3. Ш естич лен ні гетероц ик ли
важливі каталітичні функції як кофермент складних ферментних систем
внутрішньоклітинного метаболізму.
Важливим похідним тіазолу є 2-амінотіазол - сполука, що викорис
товується в синтезі лікарських засобів, зокрема сульфаніламідних пре
паратів Норсульфазолу та Фталазолу.
м
Н2М
V
БОг
NN
Н о р су л ьф азо л
Оксазол та ізоксазол - кисеньвмісні аналога імідазолу та піразолу.
Кільце оксазолу міститься в структурі 5-нітрофураиового антисептику
фуразолідону (див. вище). Кільце ізоксазолу є структурним компонен
том молекул антибіотиків пеніцилінового ряду, зокрема Оксаципіну,
Диклоксаціиііну, повністю гідрованого ізоксазолу - протитуберкульоз
ного антибіотику Циклосерииу.
т 2
^с=о
н
Ц и кл осери н
{4-аміно-З - ізоксазолі динон)
3.3. Ш естичленні гетероцикли
3.3.1. Ш естичленні гетероцикли з одним гетероатомом
Представниками цього підкласу гетероциклів є сполуки, що містять
атом азоту - піридин - та кисню - піраті (а- та у-піран). В природних
системах містяться також конденсовані гетероциклічні системи, що
складаються з ядра піридину та сполученого з ним одного або двох
бензольних кілець - хінолін, ізохінолін, акридин.
Ш естичленні гетероцикли з атомом азоту
Піридин
Хімічні властивості піридин у
Хімічні властивості піридину та його похідних визначаються елект
ронною будовою гетероциклу, що була розглянута вище. За рахунок на193
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
явності на зр2-орбіталі піридинового атома азоту, яка не бере участі у
формуванні ароматичного секстету електронів, неподіленої електронної
пари, піридин має властивості основи та нуклеофілу.
Як основа піридин зв’язує вільний протон, що лежить в основі його
взаємодії з водою (зумовлюючи слаболужний характер розчинів) та
сильними кислотами (утворюючи піридинієві солі):
N
+ неї
+ НОН
СГ
+ОН'
N
N
н
н
Якнуклеофіл піридинієвий атом азоту атакує електрофільні центри в
молекулах алкілгалогенідів з утворенням солей алкілпіридинію:
8НаІ
НаГ
Б^-ре акції піридину
У зв’язку з більшою електронегативністю атома азоту порівняно з
вуглецем, електронну щільність на С-атомах молекули піридину зниже
но {р-недостатт система). При цьому найбільший ступінь зниження
електронної щільності в молекулі піридину спостерігається в положен
нях 2 ,4 та б, що зумовлює появу на 2-му, 4-му та 6-му С-атомах віднос
ного часткового позитивного заряду 5+. Виходячи з такої електронної
194
3.3. Ш ести ч л ен н і гетероц ик ли
структури гетероциклу, виникає можливість проходження в зазначених
положеннях молекули піридину реакцій нуклеофільного заміщення
N 1/
І
5+
8+
ІЧи
^М и'
N
До 8м-реакцій піридину належать:
(І)
амінування піридину при взаємодії з потужним нуклеофілом
амідом на'грію (реакція Чичибабіпа):
+
№ +МН2‘ - <
^ і Г ^ М Н г
2 -аміно піридин
Амінопіридини, які отримують в цій реакції, використовуються у фар
мацевтичному синтезі.
(2) гідроксилування піридину.
-
+ кон
*и
^
ч
&
2-гІдроксиш ридин
Відновлення та окислення піридину
(а)
відновлення ароматичного кільця піридину призводить до утво
рення насиченого гетероциклу піперидину (гексагідропіридину) :
+ ЗН2
~р Г
N
р?
N
Н
Піперидин
195
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Ядро піперидину входить до складу багатьох фізіологічно активних
речовин, зокрема ряду алкалоїдів (анабазину, лобеліну, коніїну), транквілізуючош (нейролептичного) засобу Галоперидолу, синтетичних замін
ників наркотичного анальгетику морфіну - Промедолу та Циклодолу.
С І— Н5С 6
X
он
о
СН2— СН2—
с н 2— С— с 6н5— ?
Г алоперидол
(4-(р-хлорф ен іл)-1-[3’-(р-фто рбе нзоїл)-п ропіл] -п Іперидино л-4)
П ром едол
(1,2,5-триметил -4-пропіоні локси -4-фенілпіпери ди н)
(б) окисленню підлягають звичайно бічні алкільні радикали гомоло
гів піридину, наприклад у складі метилпіридинів (лікопінів):
.с о о н
N
3-м етилпіридин
(Р-ПІКОЛІн)
196
Н ікотинова кислота
(р-п іри ди н карб он ова кислота)
3.3 . Ш естич лен ні гетероц ик ли
П охідні піридину
Гідроксипіридини (піридиноли) - група ізомерів, що розрізняються
положенням гідроксильної групи в циклі піридину відносно атома азоту
(а-, [З- та у-гідроксипіридини). Біологічно важливою похідною а-гідроксипіридинівє вітамін В ( -піридоксин, або піридоксол (3-гідрокси-4,5-ди
(гідроксиметил)-2-метилпіридин). Фосфорильовані похідні вітаміну В6
(піридоксаль-5-фосфат та піридоксамін-5-фосфат) виконують коферментні функції в реакціях обміну амінокислот.
Н2С'
.он
н2с "
он
Н2
с.
н2
^О Н
с.
р
1^
=0
^О Н
'О—
П ірид оксол-5-ф осф ат
Піридинкарбпнові кислоти. До піридинкарбонових кислот належать
три ізомери, що відрізняються положенням карбоксильної групи віднос
но атома азоту: а-(2-)піридинкарбонова(піколінова), Р-(3-)піридинкарбонова (нікотинова) тау-(4-)піридинкарбонова (ізонікотинова) кислоти:
соон
-соон
'
'І\І'
"-С О О Н
П іколінова кислота
І
N
Н ікоти нова кислота
Ізонікотинова кислота
Нікотинова кислота. Біологічно та фармакологічно активними похід
ними нікотинової кислоти є нікотинамід та ЇЧ^-діетиламід нікотинової
кіслоти (Кордіамін). Нікотинамід є вітаміном РР, що виконує в організм
людини і тварин важливі коферментні функції в реакціях біологічного окис197
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
лення. Кордіамін —лікарський засіб, який застосовується як потужний
стимулятор дихальної та серцево-судинної систем.
Синтез нікотинаміду та кордіаміну з нікотинової кислоти відбувається
через утворення хлорангідриду нікотинової кислоти за такою схемою:
(С2Н5)2МН
Рис. 3 .6 . Схема синтезу нікотинаміду (1) та кордіаміну (2) з нікотинової кислоти
їзонікотинова кислота. Фармакологічно важливими похідними ізонікотинової кислоти є сполуки, що складають найбільш поширену групу
лікарських засобів, які застосовуються при лікуванні туберкульозу Ц е гідразид ізонікотинової кислоти (Ізоніазид, ГІНК) та його похідні —пре
парати Фтивазид, Салюзид тощо:
О,У / М н — т 2
'Ы
Ізоніазид
(гідразид і зо ні коти нової
кислоти)
о
ЫН— М = С Н
Vі
ОСИ,
N
Ф тивазид
(3-метокси-4-оксибензил іден гід
разид ізоні коти н ової кислоти)
1,4-Дигідропіридини. Фармакологічно важливими синтетичними
похідними піридину є продукти його неповного відновлення - 1,4-дигідропіридини. 1,4-дигідропіридини є лікарськими засобами, що мають
властивості антагоністів (блокаторів) т, з. “повільних” кальцієвих кава198
3.3. Ш естичлен ні гетеро цикли
лів зовнішніх мембран збудливих клітин, особливо міоцитів. Виходячи з
цього, 1,4-дигідропіридини гальмують вхід іонів Са2<через канали Ь-типу
в клітини гладеньких м’язів, зокрема кровоносних судин, в тому числі
судин міокарда та головного мозку, що спричиняє вазодилатуючий та
гіпотензивний ефект.
1,4-дигідропіридин
1,4-дигідропіридини
Похідні 1,4-дигідропіридину знайшли широке застосування як одна з
найбільш ефективних фармакологічних груп при лікуванні гіпертонічної
хвороби та порушень мозкового кровообігу. Це - такі препарати, як Ніфедипін, Німодипін, Нікардипін, Ам лодипін:
Ніфедипін:
= Я ,= СН5; 1 ^ = Н; И4— N 0 ^
Німодипін-. 11,= СН(СН3)2; 11, = СН2СНг-0 -С Н ,. Я 3= Ш 2; И4=Н .
Нікардипін: II, = СН2СН,К(СН,)СН^С6Н5; = СН,; РЦ = Н;
N 0 ,.
Конденсовані гетероциклічні системи - похідні піридину
До конденсованих гетеро циклів - похідних піридину належать хіно
лін, ізохінолін, акридин:
199
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Х інолін
Хінолін. Ізохінолін
Хінолін та ізохінолін є ароматичними системами. їх хімічні власти
вості в цілому відповідають властивостям піридину та близькі між
собою.
Гетероатом азоту у складі хіноліну та ізохіноліну належить до піри
динового типу і надає цим сполукам основних властивостей. Для бензо
льного кільця хіноліну та ізохіноліну властиві реакції єлектрофільного
заміщення, а для піридинового - нуклеофільного заміщення.
П охідні хіноліну
3Е-реакції в бензольному кільці молекули хіноліну ідуть переважно в
положеннях 5 та 8; ц е -р е а к ц ії нітрування, сульфування. Б^-реакції, що
йдуть в піридиновому циклі, відбуваються переважно в положенні 2 з
утворенням гідрокси- таамінохінолінів.
Практично важливою похідною хіноліну є 8-гідроксихінолін, який вико
ристовується в аналітичній хімії як реагент на іони металів та у фармації
для створення деяких лікарських препаратів. Похідними 8-гідроксихіноліну з протимікробною активністю є Нітроксолін (5-НОК) (8-гідрокси-5нітрохінолін) та Ентеросептол (8-гідрокси-7-йод-5-хлорхінолін):
N 0?
СІ
Ы'
N
он
8-гідроксихінолін
он
Н ітроксолін
ОН
Е нтеросептол
3.3. Ш естичлен ні гетсроц ик ли
Клінічне застосування цих препаратів обумовлене особливостями їх
фармакокінетики в організмі людини. Нітроксолін екскретується у не
змінному вигляді нирками, супроводжуючись накопиченням у сечі, що є
підставою для застосування препарату при інфекціях сечовивідних шля
хів. Ентеросептол дуже повільно всмоктується поверхнею слизової обо
лонки шлунково-кишкового тракту, що обумовлює його переважну анти
мікробну активність при кишкових інфекціях.
Ядро хіноліну входить також до складу деяких алкалоїдів, що засто
совуються як лікарські засоби.
Похідні ізохіноліиу
Гетероциклічна структура ізохіноліну є компонентом важливого для
медицини класу алкалоїдів, що за хімічною будовою є похідними бензилізохіноліну (Папаверин, Наркотин) та фенантренізохіноліну (Морфін,
Кодеїн) - розділ 5.2.
А кридин
Акридин (дибензо[Ь,е] піридин) являє собою ароматичну систему із
слабкими основними властивостями. Його характерними хімічними ре
акціями є нуклеофільне заміщення в піридиновому циклі у положенні 9.
Важливим продуктом Б^реакцій акридину є 9-аміноакридин - поперед
ник у синтезі лікарських засобів з антисептичною (Етакридину Лак
тат; Риванол) та протималярійною (Акрихін) активністю.
N142
9-аміноакридин
N42
.ос2н5 СООН
• інон
Н2М
СНз
Етакридину лактат (6 ,9-діамшо-2-етоксиакридину лактат)
201
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПСКІДНІ
НдС^СН
(СН2)3- - ГМ(С2Н5)2
NN
Акрихін
(9-(4 '-діетиламіно-1 '-метилбутиламіко)-2-метокси-6-хлоракридин)
Важливим похідним акридйну є також поширений барвник акридФ
новий оранжевий —сполука, що використовується в молекулярній ге
нетиці як потужний індуктор штучних мутацій у мікроорганізмів. Меха
нізм мутагенної дії акридинового оранжевого полягає в здатності його
плоскої молекули вбудовуватися між окремими парами азотистих основ
ДН К(явищ е “інтеркаляції”), що призводить до порушення нормального
зчитування інформації з ланцюга нуклеїнової кислоти.
Акридиновий оранжевий
Ш естичленні гетероцикпи з атомом кисню
Піран. Шестичленними гетероциклами з атомом кисню є два ізоме
ри пїрану - а-піран та у-піран:
а-піран
(2Н-піран)
202
У"йіран
(4Н-піран)
3 .3. Ш ести ч л ен н і гетер оц и к л и
Ізомерні пірани не мають ароматичної будови молекул; до того ж ці
сполуки нестійкі, особливо а-піран, який існує тільки у формі похідних.
Важливими похідними а - та у-пірану є їх оксопохідні, що входять до
складу багатьох природних сполук - а-пірон та у-пірон:
О
о ^ \ 0-
О
у-пірон
а-пірон
Ядра а-пірону та у-пірону входять до складу багатьох природних фі
зіологічно активних сполук та синтетичних лікарських засобів. Важли
вими класами цих сполук є конденсовані гетероциклічні системи, що
складаються з ядер а-пірону чи у-пірону, відповідно, та бензольного кі
льця.
Конденсованими гетероциклами, які побудовані на основі а-нірону, є
1,2-бензопірон (кумарин) та його похідні - кумарини• До конденсова
них гетероциклів, побудованих на основі у-пірону, належать 1,4-бензопірон (у-хромон) та його похідні - флавоноїди.
Похідні а-пірону
Похідними а-пірону є кумарини (рослинні сполуки та їх синтетичні
Похідні) та деякі синтетичні речовини, що мають аналогічну фармаколо
гічну активність.
Кумарини - до цієї групи природних фізіологічно активних сполук
належить 1,2-бензопіран, власне Кумарин (бензо[Ь]пірон-2; лактон
о-оксикоричної кислоти) та його похідні. Фізіологічно активними речо
винами є 4-гідроксикумарини та деякі інші сполуки.
ОН
'О
Кумарин
4-гідроксикумарини
203
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
За своїми фізіологічними та фармакологічними властивостями кума
рини належать до природних антикоагулянтів. Надходження цих сполук
в організм супроводжується зменшенням здатності крові до згортання,
що протидіє внутрішньосудинному тромбоутворенню, яке спостерігаєть
ся при різних патологічних станах, зокрема атеросклерозі* гіпертонічній
хворобі. Представниками кумаринів з антикоагулянтною активністю є
Дикумарин, Неодикумарин, Фепромарон тощо.
Карбокромен. Синтетичним похідним а-пірану є конденсований гетероцикл з фармацевтичною назвою Карбокромен (Інтенкордин).
с2н5;
(СН2)2— N
'С2Н5
О
Н5С2
О—С— Н2СКарбокромен
(3-(сх-діетиламіноетил)-4-метил7-карбетокеиметокси-2-оксо -1,2-хромен)
За фармакологічною активністю Карбокромен є лікарським засобом,
що застосовується при ішемічній хворобі серця, розширюючи коронарні
судини таї сприяючи покращенню кровопостачанню міокарда.
П охідні у-пірону
Природними фізіологічно активними сполуками, що містять у-пірон,
є конденсовані гетероцикли флавоноїди, деякі рослинні органічні кисло
ти з лікарськими властивостями (хелідонова та м ет нова кислоти).
204
3.3. Шестичленні гетероцикли
Флавоноїди. Флавоноїди -чи сл ен н а група біоорганічних сполук, які
побудовані на основі 1,4-бензопірону або хромону (бензо[Ь]пірону-4) конденсованої гетероциклічної системи, що складається з ядер у-пірону
та бензолу. Різновидами флавоноїдів -»похідних хромону - є флавон та
ізофлавон.
О
Флавоноїди є широко розповсюдженими рослинними пігментами, що
постійно надходять з продуктами харчування в організм людини, часто у формі глікозидів. За своєю фізіологічною дією на організм лкрини фла
воноїди позначаються як вітамін Р —компйскс сполук, що зміцнюють
судинну стінку, зменшуючи її проникність, мають антигіпоксантні та ан
тиоксидантні властивості ІІредставником фізіологічно активних флаво*
ноїдів є кверцетин —гідроксипохідне флавону, якии є пігментом жовто
го кольору, що міститься в квітках багатьох рослин. Фармацевтичний
препарат кверцетину застосовується в медицині як лікарський засіб.
205
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Лікарський засіб “Вітамін Р” є фармацевтичною формою рут ину глікозиду кверцетину, вуглеводна частина якого являє собою дисахарид,
побудований із залишків Б-глюкози та Ь-рамнози. Природним джерелом рутину є зелена маса гречки, в якій концентрація флавоноїду стано
вить 1,5-6%.
Токофероли (вітаміни групи Е). До цієї групи ФАС належать похідні
хроману, що містять гідроване ядро у-пірану, конденсоване з кільцем
бензолу:
Хроман (бензо-у-дигідропіран)
Будова та біохімічні властивості токоферолів будуть розглянуті в роз
ділі 4.2. та 5,4.
3.3.2.
Ш ест и член н і гет ероцикли з двома гет ероат ом ам и
Ш ест и член н і гет ероцикли з двома атомами азоту
До шестичленних гетероциклов з двома гетероатомами азоту (діазинів) належать піридазин, щіримідии та піразин. Найбільш пошире
ними в біохімічних системах є молекули піримідину та його похідних.
Хімічні властивості діазинів —визначаються належністю їх моле
кул до спряжених ароматичних систем, що містять два атоми азоту
206
3.3. Шестичленні гетероцикли
піридинового типу. Наявність двох нсподілених пар р-електронів на кож
ному з електронегативних И-атомів супроводжується зменшенням елек
тронної густини на атомах вуглецю діазйну. Це, в свою чергу, призво
дить до значного послаблення здатності діазинів вступати в реакції електрофільиого заміщення і, навпаки, полегшує реакції нуклеофільного за
міщення. Крім того, наявність двох атомів азоту піридинового типу по
слаблює основні властивості цих сполук порівняно з піридином —незва
жаючи на наявність двох центрів основності, діазини реагують тільки з
одним еквівалентом кислоти з утворенням солей, наприклад:
|Й *І
+ неї
N
Піримідинійхлорид
Піримідин
ПІримідин (1,3-діазин). Похідними піримідину, що присутні в живих
організмах, є переважно гідрокси- та аміноПіримідини, які входять до складу
нуклеотидів нуклеїнових кислот ДНК та РНК, вітамінів та коферментів.
Компонентами нуклеотидів є т з . “азотисті основи” - похідні піримідину
урацил (2,4-дигідроксипіримідин), тимін (2,4-дигідрокси-5-метилпіримідин)
та цитозин (2-гідрокси-4-амінопіримідин), що можуть існувати в лактамних
та лакгимних таутомерних формах (детальніше - в розділі 4.4.).
ЫН2
NЯ
ь2
НО О
ТиміВ
6.
й
Цитозин
Заміщений піримідин, разом з п ’ятичленним гетероциклом тіазолом,
складає основу молекулярної будови вітаміну В, (тіаміну):
20 7
Розділ 3. ГЕТЕРОЦИКЛІЧНІ'СПОЛУКИ ТА ЇХ ПОХІДНІ
NN 2
— СНз
-СН2
N
о
Н3С
N
'Б
1
•сн2 — он
» *
Тіамін
Барбітурова кислота. Барбітурати. Серед лікарських засобів **
похідних піримідину ~ важливе місце займають препарати, що синтезу
ються на основі барбітурової кислоти {244,6-тріігщроксшірШЩШку}.
О
ОН
НІЧ'
НО
N
-
чОН
н
Барбітурова кислота (тригідрокси- та триоксоформи)
Барбітурова кислота у водних розчинах може існувати в декількох
таутомерних формах. Лактим-лактампа таутомерія молекули зумов
лена міграцією атомів водню між Ш - та карбонільними групами, кетоенольна таутомерія — міграцією водню між метиленовими —
карбонільними групами.
Барбітурати -г р у п а лікарських засобів, що є похідними барбітуро
вої кислоти, заміщеними в 5,5-положенні. За хімічними властивостями
5,5-алкілзаміщені барбітурової кислоти є слабкими органічними кисло
там и , що важко розч и н н і в воді; п ереваж аю чим и таутом ерам и
барбітуратів є оксоформи.
Фармакологічна активність барбітуратів проявляється в їх високій
снодійній, заспокійливій та протисудомній активності. Поширеними є такі
препарати, як Фенобарбітал (Люмінал) - 5-етил-5-фенілбарбітурова
кислота, Барбітал (Веронал) - 5,5-діетилбарбітурова кислота тощо.
208
Шя., Шестичленні гетероцикли
О
NN
ОТ
Ж
н
Барбітурати (5,5-похідні барбітурової кислоти)
Фенобарбітал: Я, ^ С2Н5;
Барбітал: Я, ~ С2Н3 .
С6Н5;
Піразин (1,4-діазин). За хімічними властивостями щразин є слаб
кою основою, як і інші діазини. Продукт повного відновлення піразину піперазин (гексагідропіразин) має більш виражені лужні властивості:
Н
Ж
З
+ 4Н
2
н
Піразин
Піперазин
Гетероциклічне ядро піперазину міститься в таких лікарських засо
бах, як Цинаризин, Флунаризин, що застосовую ться як препарати,
здатні нормалізувати кровообіг в судинах головного мозку.
о
Цинаризин
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
Сіль піперазину з адипіновою кислотою (Піперазину адипінат) ши*
роко застосовується в медичній та ветеринарній практиці як антигельмінтний засіб, ефективний при лікуванні аскаридозу, ентеробіозу.
Пурин. Пурин ~ конденсована система, що складається з ядер двох
гетероциклів - шестичленного піримідину та п ’ятичленного імідазолу.
Пурин
Аміно- та гідрокси-(оксо-)—похідні пурину є, поряд з відповідними похід
ними піримідину, основними структурними компонентами найважливішого
класу інформаційних біомакромолекул - нуклеїнових кислот, що буде
детально розглянуто у розділі 4.4. Крім того, під час ферментативного роз
щеплення нуклеотидів (структурних компонентів нуклеїнових кислот ДНК
та РНК) утворюються гідрокеипохідні пурину, що мають самостійне
медичне значення. Це такі продукти окислення пурину, як гіпоксантин,
ксантин та сечова кислотна (наведені оксоформи сполук).
Гіпоксантин
(6-оксипурин)
Ксантин
(2,6-діоксипурин)
Сечова кислота '
(2,6,8-триоксипурин)
Завдяки здатності молекул гіпоксантину, ксантину та сечової кисло
ти до лакгам-лактимноІ таутомерії» молекули цих речовин можуть існу
вати в оксо-(лактамній) та гідроксй-(лактимйіЙ) формах. У водних роз
чинах переважають оксоформи сполук.
2 10
3.3. Ш ести ч л ен н і гетероц и к ли
Сечова кислота
(оксоформа)
.а
. Сечова кислота
(гідроксиформа)
Сечова кислота являє собою кінцевий продукт розщеплення (ката
болізму) пуринових нуклеотидів у людини та інших ссавців -урикотелічних організмів (аеісіит игісит - сечова кислота; лат,). Всього в орга
нізмі людини при змішаному харчуванні утворюється за добу 0,5-1,0 г
сечової кислоти, яка у вигляді амонійних та натрієвих солей (уратів) екскретується нирками. Кислі натрієві та калієві солі сечової кислоти є ма
лорозчинними у водіД при підвищенні їх фізіологічних концентрацій в
сечі вони утворюють осади. Відкладення Натрієвих солей сечової кис
лоти в нирках є біохімічною основою сечокам ’яноїхвороби. Накопи
чення кристалів уратів у порожнині суйюбів, що спостерігається при
спадкових порушеннях обміну сечової кислоти - сечокислому діатезі призводить До виникнення важкої патології - подагри.
Рі-метильні похідні ксантину
Метильовані похідні ксантину (кофеїн, теофілін, теобромін) є фізіо
логічно активними речовинами, що належать до рослинних алкалоїдів.
Ці сполуки містяться в широко вживаних напоях та продуктах харчу
вання і завдяки їх фармакологічним властивостям використовуються як
лікарські засоби.
Кофеїн
Теофілін
Теобромін
Ж
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА К ПОХІДНІ
Кофеїн (1,3,7-триметилксантин) є алкалоїдом, що міститься в каво
вих зернах, листках чаю; стимулює діяльність серця, збуджує централь
ну нервову систему.
Теофілін (1,3-диметилксантин) є алкалоїдом, що знаходиться пере
важно в листі чаю; розширює кровоносні судини, має виражену сечогінну активність;
Теобромін (3,7-диметилксашин) міститься в бобах какао та в невеликш
кількості в чаї; стимулює діяльність серця, розширює кровоносні судини.
П т еридин. Птеридин (піразино[2,3-<1]піримідин) є конденсованою систе
мою, що складається з Ядер двох гетероциклів - піримідину та піразину.
Птеридин
Біциклічний гетероцикл птеридин лежить в основі структури розпо
всюдженого класу природних пігментів - т м и н ів . Вперше пі барвни
ки були виділені з пилку, що створює малюнок на крилах метеликів (звідси
назва: ріегоБ - крило; грецьк.).
Біологічно важливим похідним гетероциклічної структури птеридину
є необхідна для функціонування більшості ясцвих організмів сполука з
функціями коферменту Вітаміну Вс фолієва кислота (птероїлглутамінова кислота).
,
, ,
.
Крім того, ядро піридину входить до структури молекули рибофлавіну
(вітаміну В). У складі рибофлавіну двоядерниигетерощші піридин сполу
чений з кільцем бензолу, створюючи в цілому трициклічну конденсовану
систему - ізоалоксазин (флавін), який є структурною основою декількох
природних жовтих пігментів - флавінів (Даше - жовтий, золотавий; лат.).
о
Ізоалоксазин
212
3 .3 . Ш ести ч л ен н і гетероц и к л и
Завдяки наявності в молекулах флавінів системи спряжених зв’язків,
ці сполуки здатні до зворотного приєднання атомів водню. Ця власти»
вість лежить в основі коферментної функції вітаміну В2, що вхоДиТь до
складу окислювально-відновлювальних коферментів ФМН (флавінмононуклеотид) та ФАД (флавінаденіндинуклеотид).
Піридазин Щ^І-діазж), Хімічні властивості пірйдазину близькі До
таких піримідину. До праіктиЧно важливих похідних піридазину належать
сульфаніламідний препарат з високою антибактеріальною активністю
Сульфапіридазин та гербіцид Феназон.
Сульфаігіридазин
(З-м етокси-6-р-ам ш обензолсульф ам ідоп іри дазнн)
Ш ест ичленні гетероциклы з двома різним и гетероатомами
До цього класу біоорганічних сполук належать похідні гетероциклу
т іа зи н у - фенотіазини, що займають важливе місце в сучасній меди
цина, особливо фармакотерапії та лікуванні психічних захворювань.
Ф енотіазин. Фенотіазин - конденсована система, що складається
з шестичленного гетероциклу тіазину та щэнденсованих з ним двох
бензольних кілець.
Т іази н
(4 Н 4 ,4 -т іа зи н )
Ф енотіазин
(д и б с н зо -1,4-тіазии)
213
Розділ 3. ГЕТЕРОЦИКЛІЧНІ СПОЛУКИ ТА ЇХ ПОХІДНІ
фармакологічно активні похідні фенотіазину- це переважно сполуки,
що мають різні замісники в положеннях 2 та 10 гетероциклічної систе
ми. Більшість з таких речовин мають виражену,дію відносно централь
ної нервової с и с т е м и 4 застосовуються як препарати для терапй щ яж ірі
психічних захворювань, зокрема шизофренії, Особливості психотропної
дії фенотіазинів полягають в їх транквілізуючих ефектах (ІгапсщШНаз спокій, сумирність; дат.), здатності пригнічувати психічну діяльність
людини, нормалізуючи прояви порушених емоційних реакцій, зокрема
агресивності, тривоги, підвищеної збудженості. В цілому даний клас лі
карських засобів дістав назву антипсихотиків (“великих тражвілізат орів”, або нейролептиків).
;•
Найбільш поширеним нейролептиком фенотіазинового ряду є Аміна
зин (Хлорпромазин, Торазин), що був впроваджений у практику в 1952 р.»
докорінно змінивши обличчя психіатричних клінік.
/С Н з
А міназин
Залежно від будови
-
сполученого з азотом
серед OAQ
,
аліфатичні похідні (препарати Аміназин, Пропазин*, Левомтро-
-
мазин);
похідні піперазину (препарати Етаперазин, Трифтазин,Мете-
~
разин);
ПОХІДНІ піперидину (препарати Периціазин, Тіориот т ).
радикала,
фенотіазинового ряду розрізняють:
Вю
214
3.3. Ш ести ч л ен н і гетеро цикли
Лікарські засоби похідні фенотіазину:
Аміназин: 2-хлор-10-(3-диметиламійопропіл)-фенотіазин
Етаперазин: 2-хлор-10{3-[1-(в-оксиетил)-піперазиніл-4]пропіл}-фенотіазин
Периціазин: 2-ціан-10-[3-(4-оксипиперидино)-пропіл} - фенотіазин
Хімічна природа радикала при Щ | визначає особливості психотроп
ної фармакологічної дії фенотіазину; введення в положення С2галогену
або метоксигрупи збільшує проникність ФАС через ліпідний бішар
мембран нейронів. За механізмом фармако-біохімічної дії нейролепти
ки р похідні фенотіазину - є антагоністами нейронних структур, що
селективно збуджуються нейромедіатором дофаміном (антагоністи
Д2-рецепторів дофаміну).
Серед похідних фенотіазину є фізіологічно активні сполуки та відпо
відні лікарські засоби, що за особливостями дії на організм людини ви
ступають як антагоністи біогенного аміну тканинного гормону гістамі
ну (антагоністи Н,-рецепторів гістаміну). Такі препарати (Дипразин —
10-(2-диметиламінопропіл)-фенотіазин) є ефективними засобами при лі
куванні алергічних, анафілактичних станів.
Похідною фенотіазину є метиленовий синій - сполука, що широко
вживається як барвник у мікробіологічних дослідженнях, а також як
лікарський засіб антисептичної дії для поверхневого використання у ви
гляді спиртових розчинів.
3.4. С ем и ч л ен н і ге те р о ц и к л и
Семичленні гетероцикли з двома гетероатомами
Семичленні гетероцикли з двома атомами азоту - діазепіни у віль
ному стані в природних обєктах не існують. Значний інтерес в медицині
та фармації становлять похідні 1,4-діазепіну- конденсовані гетероцик
лічні системи 1,4-бензодіазепіни.
Н
1Н-1,4-діазепін
і ,4-бензодіазепіни
215
Похідні 1,4-бензодіазеніну є сполуками з фармакологічною активніс
тю, що проявляється заспокійливою дією, гальмуванням станів збудже
ності, тривоги, неспокою, Лікарські засоби, створені на основі 1,4-бензодіазепінів, знайшли застосування як найбільш ефективні анксіолітики
( a n x ie t y - т р и в о г а , неспокій, англ.) і ЇХ численні представники належать
до найбільш вживаних у світі класів ліків.
СН3
Хлодіазеиоксид. Еленіум
(2-метиламшо-4-оксо-5-феніл7-хлор-бензо-1,4-діазспін)
Діазепам. Сибазон
(1 -метил-2-оксо-5-феіші-7-хлорбензо-1,4-діазепін)
Біохімічні механізми психотропної дії бензодіазепінів полягають у їх вза
ємодії із спеціальними рецепторами на мембранах нейронів головного моз
ку, що є чутливими до у-аміномасляної кислоти (ГАМК)—гальмівного ме
діатора центральної нервової системи. Збільшуючи чутливість ГАМК-рецепторів до їх природного ліганда, фармакологічно активні бензодіазепіни
сприяють гальмівному процесу в структурах головного мозку.
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
4Л . Вуглеводи та їх похідні
4.1.1.
Загальна ха р а кт ер и ст и ка вуглеводів
Вуглеводи (шюциди, цукри) -біоорганічні сполуки, які за своєю хіміч
ною будовою є альдегіде- та кетопохідними багатоатомних спиртів,
або поліоксиальдегідами та птіоксикетонами. До класу вуглеводів на
лежать також численні похідні поліоксиальдегідівта поліоксикетонів.
Вуглеводи, що не підлягають гідролізу (тобто розщепленню водою
на більш прості сполуки)* є простими вуглеводами, або моносахари
дами. Вуглеводи, що гідролізуються до моносахаридів, мають назву
ст а д н и х вуглеводів •» олігосахаридів та полісахаридів.
Загальна емпірична формула вуглеводів Сп(Н20 ) т, що пояснює назву
класу, наприклад: С5Н 120 6 - глюкоза (а також ізомери цього монооахариду), С |2Н22О п - сахароза (а також лактоза, мальтоза).
Всі природні моносахариди є солодкими на смак, при цьому найсолодщою є Б-фруктоза. Якщо солодкість сахарози (що переважає вміст в
харчовому цукрі - буряковому чи тростинному) прийняти за 100, то
солодкість фруктози становить 173, глюкози - 74, лактози (молочного
ц у к р у ) - 16.
'
Природні дж ерела та біологічні функції. ,
Вуглеводи у вигляді глюкози утворюються в природі з диоксиду вуг
лецю та води за рахунок фотосинтезу, що протікає в рослинах за участі
ІУ^2+- протопорфіринвмісного пігментного біжи хлорофілу або відповід
них пігментів деяких мікроорганізмів:
6 с о 2 + 6 н2о Чі (С6н10с ц + 6 0 2.
Вуглеводи у вигляді рослинного полісахариду крохмалю, який є полі
мером глюкози, складають основну масу органічного вуглецю біосфе
ри; їх вміст у тканинах рослин становить біля 80 % маси. У свою чергу,
рослинні вуглеводи є'одним з основних джерел вуглецю для тваринних
організмів. У тваринних організмах вміст вуглеводів відносно невисокий в середньому 1—2 % сухої маси, в основному у вигляді резервного полі
сахариду глікогену Втім, в організмі людини і тварин вуглеводи відігра
ють життєво важливі енергетичні (моносахариди глюкоза, фруктоза,
гомополісахарид глікоген) та структурні (гетерополісахариди) функції.
217
Розділ 4, БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
4.1.2. Моносахариди та їх похідні.
Будова та номенклатура
Залежно від кількості вуглецевих атомів моносахариди (монози) йоді*
ляються на тріози (С-3), тетрози (С-4), пентози (С-5), гексози (С-6),
гептози (С-7). Моносахариди з більшою довжиною вуглецевого ланцюга
в природі не знайдені» Найбільш поширеними в тваринних організмах
моносахаридами є гексози та пентози, які знаходяться в клітинах як
учасники обміну речовин —метаболіти у- та виконують певні структурні
функції, входячи до складу інших біомолекул.
^ Н 2ОН
С = 0
(Н-
С
ОН)п
С Н 2ОН
Відповідно до характеру оксогрупи моносахариди поділяються на
альдегідо- та кетоспирти —альдози та кетози відповідно, наприклад:
с = о
СН 2ОН
С Н 2ОН
Альдози (п * І - 5)
Кетози (п * 1 - 4)
При найменуванні моносахаридів використовують, як правило, триві
альні назви. Прикладом альдогексози є глюкоза (природний ізомер О-глюкоза), кетогексози - фруктоза (Б-фруктоза).
2 18
4.1. В углеводи т а їх п охідні
1СН2ОН
ЇМ Н ІЙ
но-?ь-н
н -^ о н
н --о н
6сн2он
2С = 0
ио--3!—н
н——он
н——он
6
О-глюкоза
Нумерацію атомів вуглецю в молекулах моносахаридів починають з
того атома, з яким сполучена оксогрупа (у альдоз) або з атома;'розта
шованого найближче до оксогрупи (у кетрз). Зірочками (*) позначаїоть
асиметричні атоми вуглецю. я
Стереохімія моносахаридів
Молекули моносахаридів мають декілька центрів хіральності (асиї?
метричних атомів вуглецю), тому вони існують у вигляді декількох про
сторових (стерео-) ізомерів.
Найпростіша за будовою апьдоза - гліцериновий альдегід (С-3) існує
у вигляді двох тріаз - стереоізомерів, що дістали назву О- та Ь-гліцеринових альдегідів:
С Н 2ОН
ІЗ- Гліцериновий альдегід
СН 2ОН
[^гліцериновий альдегід
Тетрози - моносахариди з С-4 мають 4 стереоізомери, пентози (С-5)
та гексози (С-б) - 8 та 16 відповідно.
В цілому кількість стереоізомерів для кожної альдози або кетози певної
довжини складає N = 2“ (де « - кількість асиметричних С-атомів),
Віднесення кожного моносахариду до Б - або Ь-стереохімічного ряду
проводять за співпаданням просторової будови (конфігурації) хірального
центру, найбільш віддаленого від оксогрупи (тобто сусіднього з кінцевою
групою СН2-О Н ), з конфігурацією Б - або Ь-гліцеринового альдегіду
відповідно. У пентоз таким центром буде С4, у гексоз - С,.
21 9
Розділ 4. БІОМОЛБКУЛИ ТА МЕТАБОЛІТИ
Виходячи із зазначеного, наведемо проекційні формули Фішера для
О- та Ь-стереоізомерів гаюкози:
№
“Ч и
н- -ОН
н о - -Н
н- -ОН
н- -о н
с н 2о н
;ОН
н
но
н
-Н
■он
•о н
н
с н 2о н
Ь-гаюкоза
О-глюкоза
Більшість природних моносахаридів (пентоз, гексоз) належить до
стереохімічного В-ряду (О-рибоза, О-глюкоза, О-галактоза, О-фруетоза). Ферменти організму людини та тварин завдяки хіральній структурі
своїх активних центрів не можуть метаболізувати Ь-моносахариди, тобто
використовувати ЇХ як енергетичні та пластичні субстрати.
Наведемо формули Фішера моносахаридів сімейства й-альдоз, що
містять від трьох до шести атомів вуглецю (рис. 4.1.),
ЄНО
н -|-о н
СН2ОН
3-гліцершювий альдегід
сно
н- -он
н- -ОН
СН2ОН ■
Б-еритроза
СНО
н - -о н
н - -о н
н - -о н
(: н 2о н
Б-рибоза
220
СНО
но— -н
н - он.
н - -о н
с н 2о н
0-арабіноза
сно
-н
нон- о н
СН2ОН
О-треоза
сно
СНО
н - -О Н
но— —Н
І Р -О Н
СН2ОН
Б-ксилоза
Н
НОНО-
н
н-
он
СН2ОН
0-ліксоза
4.1. Вуглеводи та їх похідні
сно
н- -он
н- -он
н- -он
н- -он
сн2он
О-алоза
СНО
Нн Мэн
н - -он*
но- -н
н - -он
СН2ОН
І)-гулоза
сно
но- Нй
н- -о н
н- г О Н
н- ~О Н
сн2он
О-альтроза
СНО
но- —н
н- -он
нон - н
н - -он
СН2ОН
О-ідоза
сно
ннонн-
сно
но-^-н
Н О - -н
, н - -о н
н - -он
сн2он
ОН
Н
ОН
он
СН2ОН
Р-глюкоза
Б-маноза
СНО
Н^
ОН
н
но-Н
нон- ОН
СН2ОН
О-галактоза
сно
НО
-Н
но
-Н.
но
н
■н
ОН
СН2ОН
Р-талоза
Рис, 4 ,1 . Моносахариди сімейства Р-альдоз (від С-3 до С-6)
Р ізновиди ст ереоізомерів моноз: енантіомери. діастереомери.
епім ери
Б* та Ь-стереоізомери певного моносахариду складають пару енантіомерів (дзеркальних ізомерів, або оптичних антиподів). Виходячи з
наведеного, зрозуміло, що існуіють 8 пар енантіомерів альдогексоз (тоб
то 8 сполук Б - та 8 - Ь-ряду), 4 пари енантіомерів альдопентоз та 2 пари
енантіомерів альдотетроз.
Стереоізомери моносахаридів, що розрізняються конфігурацією одно
го або декількох центрів хіральності, та не є енантіомерами (оптичними
антиподами), називаються діастереомерами (див, розділ 1.3.).'З наве
дених прикладів слідує, що діастереомерами є 8 стереоізомерів альдогек
соз, 4 стереоізомери альдопентоз та 2 стереоізомери альдотетроз.
Різновиди діастереомерів, що відрізняються один від одного конфі
гурацією тільки одного асиметричного атома вуглецю, мають назву
епімерів. Так, епімерамй відносно одне одного є О-глюкоза та П-галактоза, Б-глюкоза та Б-маноза, Б-рибоза та О-ксилоза (та відповідні
пари альдоз Ь-ряду).
221
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Оптична активність стереоізомерів моносахаридів
Природні моносахариди мають оптичну активність, тобто здат
ність обертати площину поляризації світла праворуч (+) або ліворуч (-).
Знак оптичної активності певного моносахариду визначається експери
ментально і не Співпадає з віднесенням сполуки До того чи іншого сте
реохімічного ряду. Наприклад, звичайна форма глюкози, що зустріча
ється в об’єктах живої природи, є правообертаючим ізомером гексози
(пйтоме обертання [а]“
а природна форма фруктози - лівообертаючим ([а]2р = -92,4°). :
;
У свіжовиготовлених водних розчинах моносахаридів відбувається
взаємна трансформація ізомерних форм молекул (лінійних та циклічних—
див нижче). Це супроводжується змінами оптичного обертання розчи
ну - явище, що дістало назву мутаротації.
Хім ічні властивості моносахаридів
Хімічні реакції, в які вступають моносахариди, можна розділити на
такі різновиди:
(а) за характером функціональних груп, що реагують:
- взаємодія карбонільної та спиртової груп;
- реакції карбонільної групи моносахариду;
- реакції гідроксильних (спиртових) груп моносахариду.
(б) за типом реакції, що відбувається:
«» утворення внутрішніх напівацеталей;
ж утворення глікозидів;
- окислення та відновлення;
- алкілування та ацилування;
- утворення амінопохідних;
- реакції дегідратації.
< ї
І|
Розглянемо найбільш важливі хімічні реакції моносахаридів.
1 . Утворення внутріш ніх напівацеталей.
Крім розглянутих вище відкритих ланцюгових форм моносахаридів,
останні існують у водних розчинах та природних системах також у ци
клічних (напівацетальних) формах.
Утворення внутрішніх напівацеталей в молекулах моносахаридів має
місце внаслідок перебування шести- та й’ятивуїлецевих ланцюгів у клеш-
222
4.1. В углеводи та їх п охідні
неподібній конформації, що призводить до просторового зближення атомів
С, з С4 або з С5. В результаті такого зближення відбувається внутрішньомолекулярна взаємодія відповідної карбонільної (альдегідної або кетонної)
та спиртової групи з формуванням напівацеталя циклічної будови- Реакція
відбувається за механізмом нуклеофільного приєднання Ак шляхом нуклеофільної атаки групи -О Н на атом вуглецю оксогрупи (розділ 2.4.).
При утворенні напівацеталей в молекулах альдогекроз в реакцію з
альдегідною групою при С1вступає переважно гідроксильна група біля
Сг При цьому атом водню гідроксильної групи переміщується до атома
кисню альдегідної групи, а між атомами С, та С5 виникає зв’язок через
атом киеню, що призводить до виникнення стійкого шестичленного
(піранозного) циклу:
Формування в альдогексозах п ’ятичленного (фуранозного) циклу
при взаємодії С, з С4 є термодинамічно менш вигідним процесом. Утво
рення піранозних циклів з лінійної відкритої форми О-ілюкози показано
на рис. 4.2,
Шч Ш
1
н-
...
.....
(
V
н
пн
1
1
м
п
н-
/оV н
АІ„|
им
н- 5
/оV - й
но
СН2ОН
а-Р-гаюкопіраноза
---!Л—
О-глюкоза (відкрита форма)
9
і 5
СН2ОН
(З-О-гаюкопіраноза
Р ис. 4 .2 . Утворення циклічних піранозних форм Э-глюкози (структури циклічних
форм моносахариду наведені у вигляді.формул Коллі - Толленса),
223
Розділ 4. БІОМОЛЕКУЯИ ТА МЕТАБОЛІТИ
Як слідує з будови циклічних форм глюкози, що наведені, форму*
вання внутрішнього напівацеталю призводить до виникнення в моле
кулі альдогексози додаткового центру хіральності - при С ь (аномерного центру). Завдяки цьому, виникають два нових стереоізомери «
аномери <х- та (3-, що відрізняються просторовим розташуванням двох
замісників - водню і гідроксильної групи, що зв ’язана з Щ (натвацетального гідроксилу). ,
У зв ’язку з особливостями молекулярної будови кетогексоз (напри
клад, фруктози) та альдопентоз (наприклад, рибози) ці моносахариди
утворюють внутрішні напівацеталі з формуванням стабільних п ’ятичлепних і (фуранозних) циклів (рис. 4.З.).
1 СН 2ОН
і
аС
~ 1
2 (^
ноннс н 2о н
а-Б-фруктофураноза
0 ^
-н
I
-о н
■И
*н
с н 2о н
Б-фруктоза
(лінійна форма)
с н 2о н
Р-Р-фруктофураноза
Р ис. 4 .3 . Утворення циклічних піранозних форм Э-фруктози (структури циклічни*
форм моносахариду наведені у вигляді формул Коллі - Толленса)
Ф орм ули Х еуорса
.
Наведені вище для зображення циклічних форм моносахаридів фор
мули Коллі - ТолЙенса (у проекції Фішера) недостатньо адекватно відо
бражують будову оксидних циклів, тому більш прийнятими в біоорганіч
ній хімії та біохімії для цих цілей є формуш Хеуорса.
У формулах Хеуорса піранозні та фуранозні цикли Подають у вигпяда
плоских багатокутників, що розміщені перпендикулярно площині рисун
ка. При цьому замісники розташовують вище та нижче площини циклу
При переході від циклічних формул Коллі - Толленса замісники, що у
проекціях Фішера знаходяться праворуч, у формулах ХеуОрса р о з м іЯ |^
ють під площиною циклу, а замісники, розташовані ліворуч - над пло
щиною циклу. При побудові формул Хеуорса у піранозних форм альдо224
4 .1 . В углеводи т а їх п охідн і
гексоз Б-ряду група СН2ОН (Св) розміщується над площиною циклу; у
фуранозних форм кетогексоз Б-ряду розміщення групи СН^ОН (С () буде
різним для а - та Р-аномерів (див. формули).
Наведемо структурні формули Хеуорса (Хт та |3-аномерів О-глюкопіранози та О-фруктофуранози:
6 СН2ОН
СН2ОН
О.
ОМ
он>-
я
1
И ОН
2
2
ОН
ОН
р -Р -гл ю и оп іран оза
а-О-глюкопіраноза
носн2
ОН
ОН
Р -Р -ф р у к то ф у р ан о за
а-О-фруктофураноза
Подамо у вигляді формул Хеуорса утворення циклічних фуранозних
форм а- та (3-аномерів альдопентози Б-рибози:
НОСН2
НОСН2
СН2ОИ
а -Б -р и б о за
(ц и кл ічн а ф орм а)
О -рибоза
(л ін ій н а ф орм а)
{Н )-рибоЗа
(циклічн а ф орм а)
225
Розділ 4, БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Таутомерія моносахаридів
Як вже зазначалося, у водних розчинах моносахаридів відбувається
зміна кута обертання площини поляризованого світла, що проходить через
цей розчин -? явище мутаротації. Хімічною основою мутаротації є здат
ність простих цукрів до т аут омерії—явища, що полягає у постійній
трансформації відкритої ланцюгової (лінійної) форми моносахариду в одну
з циклічних форм. Таке явище називається кільцево-ланцюговою, або
цикло-оксо-таутомерією.
Завдяки явищу Таутомерії, молекула будь-якої гексози, зокрЬма глю
кози, може існувати у вигляді п ’яти ізомерних форм, а саме: двох піранозних (Ос- та р-), двох фуранозних (а- та |3-) і лінійної нециклічнОЇ форми.
При цьому трансформація одна в одну як форм, що розрізняються роз
мірами циклу (піранозний або фуранозний), так і типом аномерії (а- або
(3-аномер) проходять через відкриту оксоформу;
а-О-глЮкопіраноза
а-Б-глюкофураноза
Б-глюкоза
(лінійна форма)
р-Б-гаюкопіраноза
(З-Б-глюкофураноза
Таутомерні перетворення окремих молекулярних форм 1)-піюкози
В стані рівноваги у водних розчинах глюкози кількісно переважають
найбільш стабільні її форми—о с - та (3-піранозні, а в найменшій кількості
міститься лінійна нециклічна форма.
Аналогічно для перетворень, розглянутих для глюкози, таутомерні
перетворення відбуваються у водних розчинах і з іншими моносахари
дам и ,^ також з більшістю дисахаридів. Так, наприклад, рівноважна су*
міш Найбільш поширеної в біологічних рідинах кстогексози Б-фруктози
також представлена п ’ятьма таутомерами, серед яких фуранозні форми
є переважаючими.
Конформаиії циклічних форм гексоз
У зв ’язку з наявністю значної кутової та торсійної напруги, похідні
циклогексану не можуть являти собою плоских молекулярних структур
226
4.1. Вуглеводи та їх похідні
та існують у термодинамічно найбільш вигідній конформації - конфор
м ації крісла (розділ 1.3.).
Н
Н
р-Р-глюконіраноза
Разом з тим дві кріслоподібні конформаціїа- та (3-аномерів В-гаюкопіранози відрізняються між собою за просторовим розташуванням напівацетального гідроксилу і, відповідно,-за енергетичними параметрами. В
а-аномері гідроксильна група при С, займає аксіальне, а в |3-аномері
екваторіальне положення, яке є енергетично менш напруженим і тому
термодинамічно преференційним. Виходячи з цього, в таутомерній суміші
В-глюкози, де значна частина моносахариду знаходиться у стані Б -т к м
копіранози, кількісно переважає саме (3-аномер (до 68 % складу).
2 . Утворення глікозидів.
Глікозиди-п охід н і циклічних форм моносахаридів, які утворюються
шляхом заміщення напівацетального гідроксилу на залишок спирту, фе
нолу, тіоепирту, аміну, гетероциклу тощо (О-, Б- та И -тікозиди відповід
но). Синтезують глікозиди конденсацією аглікону з активованою фор
мою вуглеводу.
Ацетальний атом вуглецю в модекулі моносахариду називається гліко
зидний центром (глікозидним атомом вуглецю), а залишок, що до нього
приєднаний - агліконом. Хімічний зв’язок між аномерним атомом вуглецю
(глікозидним центром) та агліконом дістав назву глікозидного зв 'язку.
Найменування глікозидів будують на основі назви відповідного моно
сахариду, замінюючи-суфікс -оза на -озид (глюкозид, галактозид, фруктозид, рибозид тощо). Залежно від розміру оксидного йикяу, глікозиди
поділяються на піранозиди та фуранозиди.
0-глікдзиди. Найбільш поширеним серед глікозидних зв’язків є О-глікозидний зв’язок, що сполучає циклічний моносахарид з іншою монозою (в
молекулах оліго- та полісахаридів) або з невупгеводним компонентом
(спиртом, фенолом). При цьому оліго- та полісахариди розглядають зви
227
Р озділ
4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
чайно як окремі класи вуглеводів, а під поняттям “глікозид*5 розуміють
структуру, в якій вихідний моносахарид сполучений глікозидним зв’язком
з невугаеводним компонентом, 3 метою термінологічного відокремлення
складних вуглеводів від глікозидів, що містять невуглеводний компонент,
для останніх іноді використовується термін гетеро глікозиди.
Загальна будова 0-гяікозиду (а-аномера) має такий вигляд:
СН2ОН
В процесі утворення О-ілікозидів незалежно від будови вихідного моно
сахариду утворюється суміш а - т а Р-аномерних форм молекули (рис. 4.4.).
D-гаюкопіраноза
Метил a -D -глюколіраиозид
#
Метил P-D-глюкопіранозид
Р ис. 4 .4 . Реакція утворення метилглікозиду. Хвиляста лінія при глікозидному атомі
вуглецю у вихідному моносахариді означає, що субстратом реакції може
бути я ка -, так і p-аномер, тобто незалежність стереохімічних властивостей
продуктів (утворення а- та (3-аномерів) від аномерної природи субстрату
Наведемо приклади О-глікозидів з фізіологічною активністю.
Рутин. Природною сполукою з властивостями вітаміну Р є О-глікозид рутин, дкий підтримує рівень проникності кровоносних судин
(permeability - проникність; англ.). Вуглеводна частина рутину є дисаха
ридом, що складається із залишків D-глюкрзи та L-рамнози; агліконом похідне флавону кверцетин (розділ 3.4.).
228
4.1. В угл еводи т а їх похідні
ц
не
он
Рутин
Ам игдалін сполука з токсичними якостями» що міститься в кісто
чках плодів багатьох рослин (слива, вишня, абрикос, персик, яблуня, ми
гдаль). Особливо багато амигдаліну в гіркому мигдалі. Вуглеводна«частина амигдаліну являє собою дисахарид гентіобіозу, побудований з двох
залишків Р-Б-глюкози. Аптікон складається із залишків бензойного аль
дегіду та синильної (ціановодневої) кислоти.
С 6Н5
ОН
ОН
Аміщадігі
При ферментативному гщролізі в кишечнику амигдаліну останній ви
вільняє синильну кислоту, яка є потужним інгібітором ланцюга біологіч
ного окислення в мембранах мітохондрій, спричиняючи феномен важкої
клітинної гіпоксії
До О-глікозидів належать також серцеві глікозиди —природні сполу
ки з кардіотонічною фармакологічною активністю, в молекулах яких
агліконами є стероїдні цикли (похідні циклопентанпергідрофенантрену).
Крім О-глікозидів, серед природних сполук поширеними є речовини з
К , Б, та С-глікозидними зв’язками.
N-глікозиди утворюються при взаємодії моносахаридів з ИН-вмісними сполуками (аліфатичними, ароматичними амінами, гетероциклами).
Зокрема, важливими для біологічних систем М-піікозидами є нуклеозиди структурні компоненти нуклеотидів нуклеїнових кислот (розділ 4,4.).
229
Розділ 4, БІОМОЛЕКУЛИ ТА МЕТАЮЛІТИ
О
h 2n
S-глікозиди - продукти взаємодії циклічних форм моносахаридів з тіоспиртами (тіоглікозиди). Тіоглікозиди представлені деякими природни
ми сполуками з фізіологічною активністю. Прикладом є тіоглікозид
сінггрин, що міститься в гірчиці (Sinapis alba)' при ферментативному
гідролізі сінігрину в теплому середовищі утворюється алілізотіоціанат
(гірчична ефірна олія), яка і справляє подразнюючу дію гірчичників.
0 S 0 20 K
сн—сн2
S-глікозид сінігрин
Н2С = сн— СН2— N = ^ C = S
Алілізотіоціанат
Гідроліз глікозидів. Характерною хімічною властивістю глікозидів є
їх здатність до кислотного гідролізу. В біохімічних системах глікозиди
підлягають ферментативному гідролізу за дії ферментів глікозидаз. Глікозидази є специфічними для а- або ^-глікозидних зв’язків, тому фермен
тативний гідроліз може бути використаний для встановлення конфігу
рації глікозидного атома вуглецю.
2 30
4.1. В углеводи та їх похідні
3. Окислення моносахаридів.
Моносахариди досить легко окислюються. Утворення різних проду
ктів окислення та участь в реакціях окремих структурних компонентів
молекули (карбонільної, спиртових груп) залежать від фізико-хімічних
умов середовища, дії певних каталізаторів та в живих організмах - при
сутності специфічних ферментів. Біологічний інтерес становить окис
лення карбонільних та первинноспиртової (при.С6) груп альдогексоз без
руйнування вуглецевого ланцюга молекули.
Реакції окислення альдогексоз широко використовуються також у
клінічній хімії як основа аналітичних методів виявлення “цукру” та кіль
кісного методу визначення глюкози в біологічних рідинах (сечі, крові).
Щ Утворення цукрових (глікарових) кислот.
За умов дії на альдози розбавленої азотної кислоти відбувається од
ночасне окислення альдегідної та первинноспиртової групи 3 утворен
ням глікарових (або цукрових) кислот:
СН2ОН
СО ОН
Глікарова кислота
А льдоза
Продуктом окислення в означених умовах глюкози є т ю т рова
кислота:
СООН
Н“ “ 0Н
Н О -т —н
н— он
н— он
СН2ОН
Б-глюкоза
[НМОзІ
н— он
н о — ї Нн
н— он
Н—р-ОН
СООН
В -глю карова кислота
231
Р озділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
(2) Утворення альдонових (гліконових) кислот.
При м ’якому окисленні альдоз бромною водою відбувається ізольо
ване окислення альдегідної групи до карбоксильної. Продукти реакції ■*
альдонові (гліконові) кислоти:
СО О Н
.
(Н -б -О Н )п
[Вг2/Н 20 ]
-■ - *
■
(Н -
сн2он
і
ОН)п
І
СН2ОН
А льдон ова кислота
А л ьд оза
Продуктом окислення бромною водою Б-глюкози є 0-ш ю конова
кислота. В кислих водщіх розчинах альдонові кислоти відщеплюють
молекулу води з формуванням лактонів.
Нч
Р
н- -он
н о - -н
н- -он
н- - о н
СН2ОН
D -глюкоза
«
[Вгг/НгО]
V
нНО-
нН-
-ОН
-Н
-ОН
-ОН
-Н20
сй2он
D -глю конова кислота
СН2ОН
Y-лактон D -ш ю ш н о в о ї
кислоти
•Інтермедіатом спеціалізованого (т. з. пентозофосфатного) шляху
окислення гаюкози в клітині є фосфорний ефір глюконової кислоти 6-фосфогаюконова кислота. Кальцієва сіль D-глюконової кислоти (Calcium
gluconicum) застосовується в медицині як протйалергічний та крово
спинний засіб.
(3) Утворення уронових (глікуроновіїх) кислот.
Ізольоване окислення в молекулах альдоз первинної спиртової групи
призводить до утворення біологічно важливих уронових (глікуронових)
кислот - глюкуронової та галактуронової. Такий тип реакції відбу-
4,1. Вуглеводи та їх похідні
вається в умовах хімічного захисту альдегідної групи, що може реалізу
ватися при дії окислювача на відповідний глікозид, наприклад:
соон
СН2ОН
2
но
он
Ешд-О-гаюиопіранозид
[Н20 ]
>~ос2н5- ^ ^ и
он
НО
ОН
Етил-О-глюкуронід
СООН
І
ОН
он
і р-глюкуронова кислота
Подібним шляхом утворюю ться інціі уронові кислоти, зокрема
О-галактуронова та Ь-ідуронова кислоти.
- <88он°>но
П
,
Ь-ідуронова кислота
Аналітичні реакції. В клінічній хімії для виявлення наявності глюкози
в біологічних рідинах (зокрема, в сечі людини) використовують здатність
альдогексоз відновлювати в лужному середовищі при нагріванні оксиди
металів - срібла Та міді. З цією метою застосЬвують такі реактиви:
1} реактив ТолліЬнса - аміачний розчин Ag20 ;
2) реактив Троммера - Си(ОН)2;
3) реактив Ф ел ін га- іон Си2%стабілізований аніоном винної кислоти
(тартрат-аніоном). *
Окйслювально-відновлювальні реакції відбуваються за такою схемою:
233
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
н ч jO
х с
+ Ч)Ад20
+2) 2Cu(OH ) 2
COOH
+ 1)2Ag
(H *lC -O H )4 + 2,3)2 C u (O H )
CH2OH
Cu20 + H20
COOK
В першій реакції (“реакція срібного дзеркала”) оксид срібла віднов«
люється до металічного срібла з утворенням тонкого блискучого шару
срібла на поверхні пробірки, В другій* та третій реакціях двовалентна
мідь Си(ІІ) відновлюється до одновалентної Си(І) з утворенням осаду
цегляно-бурого кольору Си20 . Усі реакції відбуваються при нагріванні.
4.
Відновлення моносахаридів.
в Відновлення моносахаридів відбуваються за їх карбонільними група
ми (альдегідною чи кетонною) з утворенням багатоатомних спиртів,
що мають назву цукрових спиртів. В якості донорів відновлювальНих
еквівалентів використовують боргідрид натрію № В Н 2 або алюмогідрид
літію А1ЬіН4, каталізаторів - метали (нікель, паладій).
Біологічно важливими багатоатомними спиртами є продукти віднов
лення альдогексоз а гексити:
СН2ОН
(н "с —он)4
СН2ОН
Альдогексоза
СН2ОН
Гексит
При відновленні глюкози утворюється спирт сорбіт, галактози - д у льцит, манози - маніт. Продуктами відновлення фруктози є суміш сор
біту та маніту, що є результатом(появи нового хірапьного центру при
І утворення двох діастереомерів.
234
4,1. Вуглеводи та їх похідні
СН2ОН
н— он
но—
Н --О Н
к -|-о н
сн2он
В-пнокоза
н
н— -он
н -]-о н
СН 2ОН
И-сорбіт
Цукрові спирти с кристалічними сполуками, розчинними у воді, Вони
є солодкими на смак, і такі їх представники, як сорбіт та ксиліт, вико
ристовуються в якоетї замінників харчового цуіфу при цукровому діа
беті. Гіпертонічний розчияманіту (манітол) використовується в якості
осмотичного діуретику; введення маніту в судинне русло спричиняє збіль
шення осмотичного тиску плазми крові та перехід значної кількості води
з тканин (позаклітинних просторів) в кров з подальшим зростанням діурезу.
5. Алкілування та ацилювання моносахаридів.
Гідроксильні групи моносахаридів (як спиртові, так і напівацетальна)
здатні утворювати прості ефіри в реакціях алкілування. Алкілування
моносахаридів відбувається при їх взаємодії з алкілгалогенідами та ал
кілсульфатами.
Біологічно значущі продукти утворюються при ацилуванні моноса
харидів. Зокрема важливими інтермедіатами обміну речовин є складні
ефіри моносахаридів та фосфорної кислоти, такі як глюкозо-фосфати,
фруктозо-фосфати, пентозо-фосфати тощо (див. нижче). Фосфати мо
носахаридів утворюються переважно при фосфорилюванні цукрів аденозинтрифосфорною кислотою (АТФ), наприклад:
О-глюкоза + АТФ
й-глюкозо-б-фосфат + АДФ.
Ефіри сірчаної кислоти у вигляді сульфатів моносахаридів (зокрема
сульфати К-ацетил-О-галактозаміну) є структурними компонентами гетерополісахаридів (глікозамінгліканів) сполучної тканини хондроїтин-
235
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
_________________________________
сульфатів, кератансульфатів, гепарансульфатів та антикоагулянту
гепарину (див. нижче).
6. Утворення амінопохідних моносахаридів.
В живих тканинах структурні та регуляторні функції виконують чис
ленні амінопохідні моносахаридів (аміноцукри). У складі аміноцукрів
аміногрупа -1ЧН2 звичайно заміщує гідроксильну групу при С2. Аміно
група в аміноцукрах є в більшості випадків ацетильованою (ЇЧ-ацетиламіни) або іноді - сульфагованою.
Утворення аміноцукрів (найбільш поширеними з яких є Р-глюкозамін та Ь-галактозамін) відбувається шляхом ферментативного аміну
вання моносахариду в реакції, де донором аміногрупи виступає амід
Ь-піутамінової кислоти глутамін. Біологічно важливі аміноцукри роз
глянуті нижче.
7. Д егідрат ація м оносахаридів.
При нагріванні монбсахаридів з мінеральними кислотами відбувається
внутрішньомолекулярна дегідратація молекули з відщепленням трьох
молекул води. В цих умовах альдопентози утворюють фурфурол, а альдогексози та кетОгеКсози - 5-гідроксиметилфурфурол.
О-фруктоза
5-гідроксиметилфурфурол
фурфурол дає червоне забарвлення прй реакції з аніліном, що є якісною
реакцією на пентози, а 5 -гідроксйметилфурфурол - червоне забарвлення
при взаємодії з резорцином (якісна реакція Селівтова на фруктозу)*
Характерист ика окремих представників моносахаридів та їх
похідних.
Гексози - підрозділяються на:
а)
альдогексози, до яких належ ать а-Э -глю коза і її епімери $-0 -галакгпоза, а-О^мшноза т а інші.
236
4.1. Вуглеводи та їх дохідні
—
ОН.
к
он
се-О-тюкоза
сн2он
сн2он
СН2ОН
о
он
он
/ о н
он
а-Р-галактоза
он>і З
г . он *
вШ-маноза
б) кетогексози, представником яких є О-фруктоза
а-Б-фруктоза
И-глюкоза (виноградний цукор, декстроза) - моносахарид, що широко
розповсюджений в природі: у вільному стані гаюкоза знакодиться в росли
нах, крові людини (4 ,4 4 - 6 ,6 6 ммоль/л) та інших біологічних рідинах.
Глюкоза є структурним компонентом дисахаридів сахарози, лактози та гомополісахаридів крохмалю, клітковини та глікогену. Рос
линний полісахарид Крохмаль є основним джерелом надходження глю
кози в організм людини.
Глюкоза та інші прості вуйїеводи;(зокрема фруктоза та галактоза)
після всмоктування в кишковому тракті та проникнення всередину клі
тин підлягають метаболічним перетворенням, які становлять підґрунтя
їх біоенергетичної функції. Безпосередній і найбільший внесок в отри
мання хімічної енергії у формі АТФ дає окислення глюкози до кінцевих
продуктів катаболізму —діоксиду вуглецю та води.
С6Н12 0 6 +
602 -> 6 С 0 2 + 6 н 2о .
В організмі люДини за рахунок аеробного окислення глюкози вивіль
няється в середньому 60-70% хімічної енергії, необхідної для енергоза
безпечення процесів життєдіяльності.
Інтермедіатами глюкози, що утворюються в процесі її внутрішньо
клітинного перетворення (гліколіз, біосинтез та розщеплення глікогену),
є фосфорні ефіри глюкозо-6 -фосфат та глюкозо- 1 -фосфат.
237
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАЮЛІТИ
\У о —
ОН
І
Глюкозо-6-фоефат
о
н
ОН
/ он
р==0
х он
: Глюкозо-1ч
Глюкозо-1-фосфат
Ь-фруктоза (левульоза) - кето гексоза, що міститься у вільному ви
гляді в плодах та нектарі квіток (“плодовий цукор”). Розчини Б-фруктози
обертають шющину поляризованого світла ліворуч (-)г звідки походить
ще одна назва моносахариду —“левульоза”. Фруктоза разом з глюкозою є
структурним компонентом дисахариду сахарози, а також мономером
деяких рослинних полісахаридів (поліфруктозидів), зокрема інуліну, До
складу полісахаридів фруктоза входить у вигляді Б-фруктофуранози.
Енергетична функція фруктози в організмі людини реалізується че
рез її внутрішньоклітинне перетворення на глюкозу. Фосфорними ефіра
ми фруктози, що утворюються при її ферментативних перетвореннях, є
фруктозо- 6 -фосфат та фруктозо- 1 ,6 -дифосфат.
НО\
О
Р— О — О Но
_
СН2ОН
/О Н
о — Р==0
х он
Р-галакт вза - входить до складу дисахариду лактози, який міс
титься в молоці, а також в гетерополісахаридах (иіікозамінгліканах, або
238
4.1. Вуглеводи та їх похідні
мукополісахаридах) тваринних тканин» Головним джерелом галактози
для організму людини є дисахарид лактоза, яка після гідролізу звіль
нює галактозу, здатну перетворюватися на глюкозу.
И -м аноза - є структурним компонентом полісахариду манану, щ о є
компонентом рослиннихта бактеріальних ілікопротеїнів; у вільному стані
міститься в шкірці цитрусових. У тваринному організмі входить до складу
олігосахаридної частини шіколшідів та ілікопротеїнів мембран та біоло
гічних рідин. Як уже вказано, продукт відновлення манози - шестиатом
ний спирт маніт застосовують в медицині як осмотичний діуретик та
як замінник сахарози в дієті хворих на цукровий діабет.
П ент ози - підрозділяються на альдопентози та кегпопентот, Біо
логічно важливими представниками пентоз є:
- альдопентози; О-рибоза, 2-дезокси-В-рибоза\
НОСН2
носн2
-О
"Ч -
он
он
н І
Ч
о
ОН
он
н
н
2-дезокси-Р-рибоза
а-Р-рибоза
О-рибоза - аяьдопентоза, що в Р-фуранозній формі входить до скла
ду нувдеотидів рибонуклеїнових кислот (РНК) та вільних рибонуклеотидів, ряду коферментів (НАД, НАДФ, ФАД, ФМН),
2-деіокси-В-рибоза (дезоксирибоза) - пентоза, що відрізняється від
Р-рибози відсутністю атома кисню в 2-му положенні* Входить до скла
ду нуклеотидів дезоксирибонуклеїнових кислот (ДНК).
- кето пентози: О-рибулоза, О-ксилулоза.
СН2ОН
СН2ОН
© =0,
Н-
ОН
НОч)
-н
11
1І
СН2ОН
(
-ОН
V
і
СН2ОН
Р-рибулоза
О-ксилулоза
Н-
-ОН
239
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
р-рибулоза, О-кснлулоза — кетопентози, які у вигляді фосфорних
ефірів (певтозо-5-фоефатів) утворюються в організмі людини і тварин
як метаболіти пентозофосфатного шляху обміну глюкози.
Амінопохідні моносахаридів (аміноцукри)
До найбільш поширених аміноцукрів належать 2-амінопохідні гексоз
Б-глю кози та ©-галактози щ гексозаміни — ІУ-глюкозамін (хітозамін)
та О-галактозамін (хондрозамін) .
С Н 2О Н
С Н 2О Н
° \ і
Ч ? н
ОНЧ
'
У *
- г ОН
N 42
В-рнокозамнг
° у
—
К о н
І
° \ і
>
г он
ічн2
ми
О-галактозамін
И-ацетильовані похідні гексозамінів найчастіше зустрічаються у
складі гетерополісахаридів —глікозамінгліканів (компонентів білково-вуглеводних комплексів протеогліканів), олігосахаридних ланцюгів глікопротеїнів та гліколіпідів.
сн2он
ЧОН
онЧ
сн2он
й он
о
МН— <х '
X СНс
К-ацетилглюкозамін
ІЧ-ацетилгалактозамін
Біологічно важливими амінопохідними моносахаридів є похідне дев’ятивугаецевого цукру нонулози нейрамінова кислота та її
та О-ацильні
похідні (сіалові кислоти). Нейрамінова та сіалові кислоти є структурни
ми компонентами тк в л іш д ів біомембран (гангліозидів), глікопротеїнів та
протеогліканів біологічних рідин, слизів, сполучної тканини, виконуючи важ
ливі механічні та імунохімічні функції.
240
4.1. В углеводи та їх п охідн і
СНоОН
сн2он
І
И2Н
(СНОИ )2
— о
соон
соон
/ о н
он
Нейрамінова кислота
(3,5-дидезокси-5-амінононулонова кислота)
М-ацетилнейрамінова кислота
ж (Сіалова кислота)
Глікарові (цукрові) кислоти
Вище було зазначено, що при окисленні моносахаридів утворюються
глікарові (цукрові) кислоти, які поділяються на окремі хімічні групи
залежно від того, яка функціональна група (альдегідна чи первинноспиртова) підлягає окисленню.
Альдонові кислоти - продукти окислення альдоз за альдегідним
атомом вуглецю. Прикладом альдонових кислот є глюконова кислота,
яка у вигляді фосфорного ефіру (б-фосфогяюконовоїкислоти) утворю
ється при окисленні глюкози в організмі (в реакціях пентозо-фосфатного
шляху окислення).
Уронові кислоти - продукти окислення первинної гідроксильної групи
альдоз. Важливе біологічне значення мають уронові кислоти, що утворю
ються при окисленні ілюкози та галактози - О-глюкуронова та О-галактуронова кислоти та похідне гексози Ь-ідози Ь-ідуронова кислота.
Уронові кислоти (О-глюкуронова, О-галактуронова та Ь-ідуронова) ра
зом з аміноцукрами є компонентами гетерополісахаридів сполучної ткани
ни * гіалуронової, хондроїтпнсірчаної кислот, гепарансудьфатів, дерматансульфатів та природного антикоагулянту гепарину (див. нижче).
Крім того, О-глюкуронова кислота у вигляді комплексу з нуклеозиддифосфатом УДФ (уридиндифосфоглюкуронова кислота) бере участь
в реакціях детоксикації в печінці чужорідних хімічних сполук (ліків, от
руйних речовин) та деяких ендогенних токсичних метаболітів (продук
тів катаболізму гормонів, жовчного пігменту білірубіну) шляхом фор
мування водорозчинних глюкуронідів, що екскретуються з сечею:
241
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
соон
соон
тон
< °н ~ Р >
Г
ноч ,]1
он
1—
- н 2о
но
Токсичний
метаболіт
Б -гл ю ку р о н о ва
ки сл ота
о.
он
О“
в
П родукт гаю к у р о н у в а я я я
(глю курон ід)
Ь-аскорбінова кислота
Ь-аскорбінова кислота (вітамін С) є похідним гексоз і структурно
близька до моносахаридів. Хімічний синтез Ь-аскорбінової кислоти здій
снюють з Б-глюкози через Ь-сорбозу.
За своєю будовою Ь-аскорбінова кислота є у-лактоном 5-кето-Ь-гулонової кислоти:
СООН
І ■
Ь
с=о
НОІг
-н
Н20
-он
н о - -н
сн2он
н о \.4
со
5= 0
и
п
0
н
но-— — н
сн2он
Кн
нно-
-н
сн2он
Т аутом ерні п е р етв о р ен ая :
у-лактону 2 -к е т о Л -їу а о я о в о ї кисаоуи
2-кето-Ь -гулон ова
ки сл ота
снон—сн2он
О —'
' Ь -асгорб ін ов а кислота
(ци кл ічн а ф орм а за Х еуорсом )
242
о
4.1. Вуглеводи та їх похідні
Ь-аскорбінова кислота є водорозчинним вітаміном (вітамін С); вона
не синтезується в організмі людини, який залежить від постійного над
ходження цієї сполуки з продуктами харчування рослинного походження
(50-75 мг/добу). В рослинних організмах та у більшості тварин присут
ня повна ферментна система біосинтезу Ь-аскорбінової кислоти з альдонової кислоти - Ь-гулонової кислоти, яка може утворюватися при від
новленні тю куронової кислоти.
Ь-аскорбінова кислота є потужним відновлювачєм. При окисленні
шляхом дегідрування вона перетворюється на Ь-дегідроаскорбінову кис
лоту (оборотна реакція!), що має властивості вітаміну С. Подальше
окислення Ь-дегідроаскорбінової кислоти з розривом лактонного. кільця,
яке відбувається у водних розчинах (особливо в присутності іонів мета
лів) призводить до утворення продуктів без вітамінної активності.
СО
1
1
НО-
І — ОН
- 2Н
11
сЫ ОН
+ 2Н-1
шо
с) —
0
І1
п
СН2ОН
Ь -аскорбінова
кислота
соон
І
СО
но-
п
СН2ОН
Ь-дегідроаскорбінова
кислота
+ н 2о
с= о
С=0
нЧ — он
но- — Н
СН2ОН
2,3-Дикето-ьгулонова кислота
Біохімічна функція Ь-аскорбінової кислоти полягає в її участі у ряді
важливих окислювал^но-відновлювальних реакцій - процесах окислю
вального гідроксилювання, регуляції перекисного окислення ліпідів. Не
стача в раціоні харчування людини вітаміну С призводить до розвитку
важкого авітамінозу - цинги (скорбуту), характерними проявами якого
є порушення синтезу колагену - структурного білка сполучної тканини внаслідок неспроможності ферментних систем гідроксилювати аміно
кислоту Ь-пролін до 4-гідроксипроліну.
243
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАЮЛІТИ
4.1.3. О лігоєдхариди. Гомополісахариди
С клад н і вуглеводи (глікани) - продукти конденсації моносахари
дів та їх похідних, що містять у своєму складі від двох, трьох або чоти
рьох ірлігосахариди) до багатьох тисяч (полісахариди) мономерних
залишків цукрів.
А. Олігосахариди. Дисахариди
Д и сахари д и
Серед природних олігосахаридів найважливіше значення в біохімії і
фізіології людини мають дисахариди лактоза, сахароза, мальтоза. У
складі рослинних продуктів харчування до шлунково-кишкового тракту
людини потрапляють трисахарид рафіноза з цукрового буряку та тетрасахарид стахіоза.
Для запису назв вільних олігосахаридів та олігосахаридних радикалів
глікопротеїнів та гаіколіпідів використовують міжнародну систему ско
рочених позначень моносахаридних залишків, що входять до їх складу:
Glc - глюкоза
Gal - галактоза
M an - маноза
Fra - фруктоза
Fuc - фукоза
Rib - рибоза
Ага - арабіноза
GlcNAc - N -ацетилглюкозамін
GalNAc - N-ацетилгалатозамін
ManNAc - N-ацетилманозамін
GlcA - глюкуронова кислота
GalA *»галактуронова кислота
NeuNAc - N-ацетилнейрамінова кислота
Позначення моносахаридних залишків за наведеною номенклатурою
відносяться до піранозних форм молекул; для позначення фуранозних
форм на кінці скорочень додають літеру f, наприклад:
P-D-Fruf - P-D-фруктофураноза.
Л акт оза (Р-0-галактозидо-1,4-а-0-глю коза) - молочний цукор,
складається з залишків P-D-галактози та a -D -глюкози, сполучених
1,4-шікозидним зв’язком.
Структурна формула лактози:
244
4.1. В углеводи т а їх п охідні
Скорочене позначення лактози:
- > 4 )-с х - 0 - 6 Іс .
Лактоза є важливим компонентом харчування людини, особливо в
дитячому віці, входячи до складу жіночого (6-7% ) та коров’ячого (біля
5%) молока. Перетравлення лактози в тонкому кишечнику людини здій
снюється за дії ферменту лактази ф-галактозидази).
Сахароза (а-Б-глю козил-1,2-(3-В-фруктозид) - один з найбільш розіповсюджених в природі та практично важливих дисахаридів» що міс*
титься в стеблах, коренях, бульбах та плодах рослин. Молекула сахаро
зи побудована із залишків а-О-глюшзи (а-Б-глюкопіранози) та (З-Б-фруктози ((З-Б-фруктофуранози); в утворенні глікозидного зв’язку беруть
участь аномерні гідроксильні групи обох моносахаридів, тобто між за»
лишками глюкози та фруктози утворюється 1 —> 2 - глікозидний зв ’язок.
Виходячи із означеного, наводимо структурну формулу молекули са*
харози:
245
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Скорочена назва сахарози має вигляд:
сс-0-0Іс-(1
2)-0-О-РгиГ.
Оскільки обидва аномерні гідроксили в молекулі сахарози сполучені
між собою глікозидним зв’язком, сахароза не має відновлюючих влас
тивостей, як, наприклад, мальтоза та лактоза, зокрема не дає позитивної
реакції з реактивами Толленса та Фелінга (“нередукуючий дисахарид”).
В харчуванні людини сахароза має найбільше значення як компонент
Харчового цукру, джерелами якого є цукровий буряк та цукрова трости
на, В порожнині тонкого кишечнику сахароза розщеплюється гідроліти
чним шляхом ферментом сахаразою ф-фруктофуранозидазою); вільні
глюкоза та фруктоза всмоктуються в кров’яне русло.
Мальтоза Оі УИііп її НИЙ їді ШІІіІ..||ІІІІІІШІ солодовий цукор - ди
сахарид, що складається з залишків двох молекул глюкози.
6СН2ОИ
6СН2ОН
^
он
Мальтоза
Скорочена назва мальтози:
ос-0-0іс-(1
—>
4)-сс-0-СІс.
Мальтоза утворюється в травному каналі людини під дією на харчо
вий полісахарид крохмаль ферменту / 2-амілази.
Б. П олісахариди. Гомополісахариди
П олісахари д и
Полісахариди - складні вуглеводи, які за хімічною структурою є
полімерами, побудованими із залишків десятків або сотень тисяч моле
кул моносахаридів та їх похідних, об’єднаних за допомогою реакції полі
конденсації За особливостями хімічної будови ці сцолуки поділяються
на гомопт ісахариди т а 2бщерошліс 0щ риди.
246
4.1. В углеводи т а їх похідні
Гомополісахариди -ск л а д н і вуглеводи, мономерами яких є залиш
ки однакових моносахаридів (найчастіше глюкози) або їх похідних, Гомополісахариди підрозділяються на вуглеводи тваринного (глікоген, хі
тин), рослинного (крохмаль, клітковина, інулін, пектини) та мікробного
(декстран) походження.
Гетерополісахариди — складні вуглеводи, утворені 3 різних за
хімічною структурою мономерів - похідних гексоз.
Гомополісахариди
Крохмаль - рослинний гомополісахарид, що надходить в організм
людини з рослинними продуктами харчування. Крохмаль складається з
двох фракцій — амілози та амілопектину, які становлять 15-20% та
80-85 % загальної маси крохмалю, відповідно.
Амілоза - лінійний полісахарид, молекули якого містять від 200 до
1000 мономерів (залишків глюкози); м.м. амілози-4 0 -1 6 0 кД. В складі
амілози мономери сполучені а-1,4-глікозидними зв’язками. Гомополімери амілози формують спіральні структури, кожен виток яких включає
шість молекул глюкози.
Амілопектин - розгалужений полісахарид з м.м. від 1 до 6 мли. Д.
Головний ланцюг амілопектину утворений а-1,4-ілікозидними зв’язками;
розгалуження формуються а-1,6-глікозидними зв’язками. Між точками
розгалужень містяться. £ 0 —З 0 глюкозиднцх. мономерів. ,
Глікоген - гомополісахарид тваринного походження з м.м. близько
100 млн. За хімічною структурою глікоген близький до амілопектину
крохмалю (“тваринний крохмаль”), але має значно більш розгалужені
молекули. Лінійні' відрізки основного ланцюга гайкогену вміщають 6 -1 2
залишків молекул глюкози, об’єднаних а-1,4-глікозиДними зв ’язками;
розгалуження формуються за рахунок (х-1 ,6 -глікозидних зв’язків.
Глікоген утворює внутрішньоклітинні грайули - лабільне депо мета
болічної енергії, в яких резервується надлишок глюкози, що надходить з
їжею. Найбільша кількість глікогену в організмі людини міститься в пе
чінці (2-5 %) та хребцевих м ’язах (0,5-2 %).
Целюлоза (кліт ковина) - гомополісахарид, який є головним струк
турним компонентом клітинних стінок рослин. До складу целюлози вхо
дить понад 50% всього органічного вуглецю біосфери; деревина Скла
дається з целюлози приблизно наполовину, а бавовна являє собою май
же чисту целюлозу.
247
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Молекули целюлози являють собою нерозгалужені ланцюги, що скла*
даються з залишків молекул глюкози, сполучених а-1,4-глікозидними
зв ’язками. Макромолекулярний ланцюг целюлози утворюється з 250012 000 молекул глюкози, м.м. - 1-2 млн. У травному каналі людини
целюлоза не розщеплюється.
Будова фрагменту молекули гомополісахаридів крохмалю та глікогену
Крім целфлозй, з їжею до травного каналу лкзйини потрапляють і інші
рослинні гомо- і ґетерополісахарнди, що формують харчові волокна
кишечнику. До цих полісахаридів належать геміцелюлоза та пектини.
Декспіран - гомополісахарид мікробного походження, що складається із залишків а-О-глюкози. Являє собою розгалужений полімер, що
синтезується в клітинах дріжджів та деяких бактерій. Лінійні ланцюги
молекули декстрану утворюються за рахунок а-(1 —» 6)-гаікозидного
зв’язку між молекулами глюкози. Розгалуження формуються зв’язками
типів а-(1 —>2), а-(1 —> 3) або а-(1 —> 4)У зв ’язку з високою гідрофільністю та осмотичною активністю пре
парати частково гідролізованого декстрану застосовують у фармації як
плазмо- та кровозамінники (препарати Поліглюкін, Реополіглюкін). У
вигляді гелів декстрани застосовують в біохімічній практиці як основу
для колонкової хроматографії (гель-хроматографія).
Ін у лін - гомополісахарид, щ о міститься в бульбах деяких рослин.
За хімічною будовою інулін є поліфруктозидом, побудованим із залиш
ків (З-О-фруктофуранози. Молекули О-фруктози сполучені в лінійних
248
4.1. Вуглеводи та їх похідні
ланцюгах інуліну (2 —» 1)-глікозидними зв ’язками. Окремі ланцюги іну
ліну (довж иною близько ЗО м оном ерів) закінчую ться залиш ком
а-О-глюкопіранози.
Інулін використовують як джерело для отримання Е)-фруктози, а та
кож у фізіологічних дослідженнях при визначенні швидкості клубочкової
фільтрації в нирках.
4.1.4. Гет ерополісахариди
Гетерополісахариди - біополімери, що побудовані із значної кількості
(десятки тисяч мономерів) різних моносахаридів та їх похідних. Найбільше
значення в біохімії та фізіології організму людини має клас гетерополісахаридів, що має назву глікозамінглікани (застаріла —
• “мукополісахариди”).
Глікозамінглікани побудовані з дисахаридних залишків, що багато
разів повторюються. МоноСахаридними компонентами цих дйсахаридних повторів є найчастіше гексуронові кислоти (Б-глюкуронова, іноді Ь-ідуронова) та И-ацетилпохідні гексозамінів.
Глікозамінглікани є Структурними біополімерами, що у комплексі з біл
ками (протеоглікани) утворюють міжклітинний матрикс сполучної ткани
ни, є основою слйзів (лїуцинів)}які виконують функції фізіологічного масти
ла в порожнині суглобів. В цілому глікозаміншікани забезпечують механіч
ну міцністьта пружність органів і тканин, еластичність їх сполучень.
Представниками глікозамінгліканів є такі сполуки, як гіалуронова
кислот а, хондроїт инсуяьф ат и, дерматансульфати, кератанСуяьфати, гепарансульфати, гетерополісахарид з антикоагулянтними вла
стивостями гепарин.
Завдяки значній кількості полярних радикалів, що входять до їх скла
ду (гідроксильні, карбоксильні, сульфатні, амінні, ацетиламінні групи),
глікозамінглікани є гідрофільними структурами, що забезпечує їх здат
ність утримувати в біологічних тканинах значну кількість води.
Гіалуронова кислота. Гіалуронова кислота є лінійним полісахари
дом з м.м. 105—107Д, у якому дисахаридні фрагменти побудовані із залиш
ків О-ілюкуронової кислоти та И-ацетил-Р-ілюкозаміну, сполучених між
собою |И -4 3)-глікозидним зв’язком. Окремі дисахаридні фрагменти
об’єднані в біопблімер (3-(1 —> 4)-глікозидними зв ’язками.
24 9
Розділ 4. БЮМШЕКУДИ ТА МЕТАБОЛІТИ
А
А
Будова ланцюга молекули гіалуронової кислоти:
А - дисахаридиі фрагменти; (а) - О- глюкуронова кислота;
(Ь) - К-айетил-О-глюкозамін.
Хондроїт инсульф ат и. Молекули хондроїтинсульфатів у комплексі
з відповідними тканинними білками (протеоглікани) є структурними
компонентами хрящової тканини.
Дисахаридні фрагменти хондроїтинсульфатів складаються із залиш
ків О-глюкуронової кислоти та сульфатованого М-ацетил-О-галактозаміну, сполучених між собою Р-(1 —>3)-глікозидним зв’язком. Сульфатна
група присутня в 4-му або 6 -му положеннях ІЯ-ацетил-Б-галактозаміну
(хондроїтин-4- та хондрогага-6 -сульфати). Окремі дисахаридні фрагменти
об’єднані між собою а-(1 —> 4)-щікозидними зв’язками.
Будова ланцюга м #л е*^ й хондроїтш-б-суйьфаіу:
А -дисахаридні фрагменти;
( а ) - Р - гаюкуронова кислота; (Ь) - К-ацетил-б-сульфо-О-галакгозамін.
Білково-полісахаридні комплекси, що формують надмолекулярні струк
тури тканинних протеогліканів (у складі білків та шікозамінгліканів - гіа
луронової та хондроїтинсірчаної кислот), утворюю ться за рахунок
О-глікозидних зв’язків між гідроксильними групами моносахаридів (олігосахаридних ланцюгів) та ОН-групами бічних ланцюгів амінокислот
серину або треоніну відповідних поліпептидів.
2 50
4.2. Л іп ід и . С тероїди . Т ерп ен и
Гепарин. Гетерополісахарид гепарин є гаікозамінгліканом, що син
тезується опасистими клітинами сполучної ткатШ (гепариноцитами).
Гепарин виконує функцію природного тканинного антикоагулянту,
механізм дії якого полягає в активації антитромбіну III - фактора антизгортальнси системи, що протидіє внутрішньосудинному згортанню крові.
Дисахаридні одиниці в ланцюгах молекули гепарину складаються із
залишків №■ або О-сульфатованого В-глюкозаміну (чи ІчТ-ацеТил-В-глюкозаміну), сполучених а-(1 —>4)-глікозидним зв’язком із залишком уронової кислоти (Ь-ідуронової або О-глюкуронової). При цьому Ь-ідуронова кислота кількісно значно переважає О -тю куронову, становлячи до
90 % загального вмісту уронових кислот в молекулі гепарину.
Окремі дисахаридні фрагменти пов’язані між собоюМ ІИр4)-глікозидними зв’язками, що утворені між уроновою Кислотою одного дисахаридн&го
повтору та шюкозаміном ~ іншого. М.м, гепарину складає 16-20 кД.
А
в
Будова фрагменту Молекули гепарині',
А , В - дисахаридні фрагменти складу:
( а . ) - О-, И-сульфатобаний глюкозамін;
(а,) - О-сульфатований И-аиетил глюкозамін;
(Ь) Ц О-сул ьфатована Ь-ідуронова кислота.
Медичне застосування препаратів гепарину спрямоване на протидію
патологічно активованому при ряді захворювань згортанню крові, утво
ренню внутрішньосудинних тромбів при ішемічній хворобі серця, пору
шеннях мозкового кровообігу.
4.2. Л іпіди. С тероїди. Т ерпени
4.2.1.
Ліпіди: будова, властивості. Ж ирні кислоти
Ліпіди - клас біоорганічних сполук, характерною ознакою яких є нероз
чинність у воді та інших полярних розчинниках і здатність до розчинення в
неполярних (гідрофобних) рідщіах. Неполярні розчинники (діетиловий ефір,
251
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
дихлоретан, хлороформ тощо) використовують для екстрагування ліпідів з
біологічних об’єктів (крові, тканин, продуктів харчування та ін,).
За хімічною структурою ліпіди є складними ефірами вищих карбоно
вих (жирних) кислот та спиртів (гліцеролу, сфінгозину, холестеролу тощо).
До складу багатьох класів ліпідів (складних ліпідів) входять також залиш
ки фосфорної кислоти, азотистих спиртів (коламіну, холіну), вуглеводів.
Біологічні функції ліпідів:
X. Енергетична функиія. Цю функцію виконують переважно нейт
ральні жири, що за хімічною будовою є триацилгліцеролами (тригліцеридам и).
2. Структурні функиії - виконують полярні ліпіди, що складають
основу будови біологічних мембран. Це переважно - складні ліпіди, що
містять залишок фосфорної кислоти та є ефірами гліцерину і сфінгозину ф осф огліцериди та сф інголіпіди.
3. Регуляторні функиії - багато ліпідів мають властивості фізіоло
гічно активних сполук, в тому числі гормонів та клітинних біорегуляторів. Ці функції виконують ліпіди таких класів:
- ліпіди стероїдної будови (стероїдні гормони - чоловічі та жіночі
статеві гормони; гормони кори наднирників —■глюткортитіди
та мінералокортикоїдщ похідні вітаміну /3 - регулятори гомео
стазу кальцію; ж овчні кислоти - фізіологічні емульгатори);
- похідні арахідонової кислоти (ейкозаноїди) - біорегулятори із
властивостями гормонів та клітинних медіаторів - простаглан
дини, простацикліни, тромбоксани, лейкотрієни.
Ж и р н і ки сл оти
Найважливішою ознакою, що визначає фізико-хімічні та біологічні
властивості ліпідів, є їх жирнокиспотний склад. Кількість вуглецевих
атомів та відповідно довжина вуглеводневого ланцюга, ступінь насиче
ності жирних кислот, що входять до складу природних ліпідів (нейтраль
них жирів, фосфоліпідів, сфінголіпідів тощо) визначають їх консистенцію
(рідкі, тверді) та поверхневу активність, зокрема, здатність до комплексоутворення з білками і, відповідно, утворення міцел, біщарів, транспорт
них ліпопротеїнів, ліпідного матриксу біологічних мембран.
До складу ліпідів організму людини і вищих тварин входять жирні
кислоти з парним числом вуглецевих атомів, що містять від 1 2 до
24 атомів вуглецю, переважно від С 16 до С 20 (вищі жирні кислоти).
2 52
4.2. Л іп іди . С тер оїди . Т ерп ен и
Як приклади природних жирних кислот можна навести такі сполуки,
найбільш розповсюджені в ліпідах тканин організму людини:
Н а си ч ен і к и сло т и (СяН 2п+1СООН):
Пальмітинова (н-гексадеканова) кислота - С)5Н3,СООН (СІ60)
Стеаринова (н-октадеканова) кислота - С 17Н35СООН (С180)
Арахінова (н-ейкозанова) кислота - С 19Н ,9СООН (С20..)
Н енасичен і к и сло т и |
м оноєнові кислот и (СпН2п ,СООН):
Пальмітоолеїнова (г^мс-гєксадецен-10-ова) кислота - С15Н,9СООН (С 6 )
Олеїнова (г^мс-октадецен-9-ова) кислота - С 18,. (С І7Н33СООН)
полієнові к и сл о т и (СпН2п3(5 СООН):
Лінолева (і^мс-,і/г<с-октадекадієн-9,12-ова) кислота - С17Н3]СООН (С182)
Арахідонова (і/мс-,і/мс-,г/ис-і#ис-ейкозатетраєн-5,8,11,14-ова) кислота - С 19Н31СООН (С20:4)
Будову вищих жирних кислот часто зображують за допомогою спро
щених формул (с. 15), які більш наглядно відображують їх молекулярну
організацію, зокрема довжину вуглеводневого ланцюга та валентні кути
між атомами вуглецю:
О
16
но
СН;'з
.
П альм іти нова кислота
О
18
НО г
СНз
С теари н ова ки сл ота
О
'з
НО 1
Олеїнова кислота
О
Ш
. Ш 19
но
18
'з
НО
Л ін ол ева кислота
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
В організмі людини жирні кислоти знаходяться частково у вільному,
тобто неетерифікованому стаді (неетерифіковані жирні кислоти М*
НЕЖК) - головним чином в плазмі крові - та переважно входять до
складу тригліцеридів жирової тканини як резерв енергетичного матеріал
лу та полярних структурних ліпідів біомембран (таблиця 4.1).
Таблиця 4 .1.
Вміст жирнйх кислот в тригліцеридах жирової
тканини людини (Ю. І. Губський, 2000)
Насичені жирні кислоти
3
20
5
Ненасичені жирні кислоти
ПальМітоодеїнова
5
55-60
Олеїнова
Лінолева
10
Арахідонова
0 ,2
Температура
плавлення, °С
Г Ц И
*
+ 62,8
+ 69,6
+ 1 ,0
+ І3 ,0 о
М іристинова
Пальмітинова
Стеаринова
Вміст, %
1
Ж ирні кислоти
-т 49,5
Серед насичених жирних кислот в жирах людини переважає пальмі
тинова кислот а ( С 15Н 3,С О О Н ), сер ед н е н аси ч ен и х - олеїнова
(С 17Н 33СООН), яка становить близько 60 % від загальної Кількості жир
них кислот, що входять до складу тригліцеридів жирової тканини. Наяв*
ність в ліпідах значної кількості олеїнової кислоти з низькою температу
рою плавлення обумовлює рідкий стан жирів тіла людини, температура
плавлення яких становить в середньому 10-15 °<Х Так, зокрема, Ліпіди*
депоновані в адипоцитах жирової тканини, являють собою сферичну
ліпідну краплину, Що складається переважно з тригліцеридів; ліпіди
плазми крові являють собою ліпідні міцели, стабілізовані білками у ви?
гляді ліпопротеїнів.
С труктура т а ф ункції окрем их кл асів ліпідів
Залежно від хімічного складу компонентів, що вивільняються за умов
їх повного гідролізу, ліпіди поділяються на такі класи:
254
4.2. Ліпіди. Стероїди. Терпени
1.
Прості ліпіди.
1.1. Ацилгаіцериди.
1 .2 . Цериди (воски).
Д. Складні Шгпіди.
2.1. Фосфоліігіди
2.1.1. Гліцерофосфоліпіди.
2.1.2. Сфінгофосфоліпіди.
2.2. Гліколіпіди.
2.2.1, Глііш илтіцерм ю .
2.2.2, Глікосфінголіпіди.
Означені кваси ліпідних сполук відносять дощ з. омилюваних ліпідів,
тобто сполук, які здатні до кислотного або лужного гідролізу з утворен
ням спиртів та карбонових кислот • прості ліпіди та додаткових речовин
(фосфорної кислоти, азотистих сполук, моно- та олігосахаридів) - складні
ліпіди.
За фізико-хімічними властивостями до ліпідів відносять також різно
манітні “неомилювані ліпіди” - численні стероїди та компоненти ефір
них олій рослин - тертий (дйв. нижче).
1, Прості ліпіди - ліпіди» що при гідролізі утворюють спирт та жирні
кислоти.
'
,
1.1.
Ацилгаіцериди (ацилгліцероли) - ліпіди, що є складними ефіра^
ми гліцеролу (гліцерину) та вищгіх жирних кислот. Триацилгліцероли
(тригяіцериди) мають емпіричну назву нейтральні жири.
Триацилгяідерол (тригліцерид)
В разі якщо тригліцериди містять залишки однакових жирних кислот,
їх називають простими, якщо різних - змішаними. Природні тригліце255
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАЮЛІТИ
риди є змішаними ліпідами, тобто до їх складу входять залишки різних
жирних кислот.
Тригліцериди, до складу яких входять залишки насичених жирних
кислот, за своєю консистенцією при звичайній температурі оточуючого
середовища є твердими ~ їх називають звичайно жирами; тригліцериди, які містять залишки ненасичених кислот, є рідкими, - їх називають
оліями, наприклад:
0
Н2С ^ О -
1
' 1 7 НЗЗ
О
не— о
- 4 І— С 1 7 Н33
о
II
Н2С - “ - О — С——С і 7 Н3 3
Тристеароїлгаіцерин
(тристеарин); Iе плавл. = + 71 °С,
Триоленоїлгціцерин
(триолеїл); § плавл. = - 17 °С.
Прикладами твердих жирів є овечий та яловичий жири. З підвищенням
вмісту ненасичених жирних кислот температура плавлення тваринних жирів
знижується, вони стають більш легкоплавкими (свиняче сало, вершкове
масло). Рослинні жири, що містять залишки переважно ненасичених жирних
кислот, найчастіше бувають рідкими (оліями) - соняшникова, льняна та
інші олії. Проте деякі тваринні жири мають рідку консистенцію (риб’ячий
жир), відомі також тверді рослинні жири (наприклад, кокосове масло).
Ступінь ненасиченості різних жирів оцінюють йодним числом, яке
дорівнює масі йоду (в г),.що приєднується до 1 0 0 г досліджуваного жиру.
П ри взаєм о д ії з тр и гл іц ери дам и м олекули йоду взаєм одію ть з
ненасиченими зв’язками залишків жирних кислот, тому зростання зна
чення йодного числа відповідає більшому ступеню ненасиченості жир*
них кислот, що входять до складу даного тригліцериду (таблиця 4.2.).
В організмі людини нейтральні жири є основною складовою части
ною ліпідів адипоцитів жирової тканини, являючи собою молекулярну
форму зберігання вищих жирних кислот-найбільш енергоємного мета
болічного палива. Нейтральні жири адипоцитів є мішаними тришіцери256
4.2. Ліпіди. Стероїди. Теряени
дами; завдяки великій кількості олеїнової кислоти при температурі тіла
(близько 37 °С) вони знаходяться в рідкому стані.
За рахунок окислення саме жирних кислот тригліцеридів організм
людини отримує значну кількість енергії, необхідної для життєдіяльності.
Добова потреба в жирах дорослої людини —близько 70 г, що забезпечує
орієнтовно ЗО % добової калорійності їжі (для порівняння - людина отри
мує за добу близько 500 г вуглеводів, їх калорійність становить близько
70 % від добової потреби).
Таблиця 4.2.
Ж и р н о ки сл о тн и й скл ад д еяки х природних ж и рів
т а олій (за Н. А. Тюкавкіною, 1989, із змінами)
Ж ир, олія
Ж ирні
Температура
кислоти, %
насит
нена- плавлення, °С
сичені
чені
Тваринні жири
%
Йодне
число
Свинячий жир
48
52
22-32
53-75
Вершкове масло
60
40
18-23
24-32
Трісковий жир
16
Щ15-30
84
РоЄдиннг олії
150-175
-1 6 -1 8
119-140
Соняшникова олія
8 -10
90-92
Оливкова олія
8-14
86-92
Льняна олія
8 -10
90-92
-
2 -6
-1 8 -2 7
80-88
174-184
Тригліцериди, що виконують енергетичну функцію, містять у складі
своїх молекул багато атомів водню, тому при їх окисленні шляхом дегі
дрування вивільняється приблизно в 2,5 рази більше хімічної енергіі
(9,3 ккал/г, або 38,94 кДж/г), ніж при окисленні вуглеводів (4,1 ккал /г, або
17,17 кДж/Г). Біологічною перевагою жирів як енергетичних джерел є
також їх здатність утворювати значні енергетичні резерви у вигляді жи
рових включень в адипоцитах жирової Тканини, що створює умови три
валого енергозабезпечення організму в умовах недостатнього надход
ження енергетичних поживних продуктів з їжею.
257
ш
а
й
з
а
в
»
які * сюадн“
'Ф 'Р“™ вищих
5
п т ?
високомолекулярних спиртів, зокрема нетипового
^ іб зз°Н ) та мірицилового (С Н ОШ Ло »««гі» „
Ч а т о в о го
5
.
^
а
* , І | * П-упу складних ліпіді» с к л а д а ю т ь С
р
О
и
д
а
* — »
«2
и
НгС О—С—5 ,
І
о /О »
ні <б р - о - ^
НО—{і—о—сна
* "ш
і
д
і д
в
в
а
НаС
0(І
І
НО— Р— О—-СІ
ін
он
Фосфатиднакислота
„„„®0Сф0ДІЄфІРН" *
І Й В а Р Я І И І Х0" » У Й
гТ
а
М вдрофрсфолшідів утворений гід
М
в
Ш Я Ш К Щ Ш Ш Ш або 2
« « Р З Д п Д ет»5
| або серину £
ньому ( 1 -му, а -) положенні задники насичених, а у ц етрал ьн ом у ( ї м ,
Р-) положенні - ненасичсних жирних КИСЛОТ.
258
,
4 .2 . Л іп іди . С тероїди . Т ерп ен и
о
н2с —о —с— &
о
не— о —Л— и2
І
о
_ +
и
І
(СНз)з = N"1042)2— О— Р ^ 0 — СН2
ОН
Фосфатидилхолін (загальна формула)
о
НгС— о —с —
о
н е — О - Л — и2
о
Н2И —(СН 2 )2— О — Р — О — СН 2
ін
Фосфатидилетаноламін (загальна формула)
О
о
(С Н з )з = іФ —
сн2—сн2—о-
Ан
Фосфатидилхолін
{1-пільмітоїд-2чшмй(&-фосфатвдш[Х9лін)
Як слідує з наведених структурних формул, у молекулах гліцерофосфолійідів є кислотний (фосфорна кислота) та основний (аміногрупи)
центри, що обумовлює їх амфіфільні (дифільні) фізико-хімічні власти
вості та здатність до утворення внутрішніх солей. Біологічно важливою
259
г
Розділ 4. БІОМОДЕКУДИ ТА МЕТАБОЛІТИ
Д
И
” * ^ е р и с т и к о ю гліцерофосфоліпідів е наявність у складі
ку Гідрофобних, неполярних (радикали вищих жирних кислот) та
п ч р о ф ™ * . полярних (залишки фосфорної кислоти та а 2 Г и Ирті2
угрупувань. Такий Я ракгер молекул гліцерофосфоліпідів
н Г Г ш ш а п о Г СТЬ Д° Ф° РМУВаННЯ 8 П0ЛЯРНИХ г а д о в и щ а х ламеляр!
них та бішарових структур, що є основою ролі гліцерофосфоліпідів як
с л ї ртл Т
~
Л Ш ,Д Н 0Г0 матриксу біологічних мембран
Сфгнгофосфолгпгди - складні ефіри багатоатомного аміш кпирту
а
г
а
жирних кислот’ щ° м°жуть так° * міс™™ залиш-
( Н - а ^ ^ о з Г в ) ° : УТВОРЄНМ П° ПЄрЄДНИКІВ ^ Ф ™ п і дів ^
Н О -г -С Н 2
н і ---- ІЧН5
н о — с н — с н = с н — (СН2)12— онз
Сфінтозйн
(звичайна структурна та спрощена структурна, формули)
н о — СНс
о
не — N4
С-
— ОН— С Н = С Н — (СН2)12"
И-ацилсфінгозин (церамід)
260
-СН ,
4.2. Ліпіди. Стероїди. Терпени
Сфінгафосфоліпіди є фосфорними ефірами церамідів та аміноспиртів «£
холіну, етаноламіну, серину. В нервовій тканині людини присутні переважно
холінвмісні сф інгоф осф оліпіди «** сфінгомієліни Щ -ацилсф інгенілфосфохоліни):
О
(СЬЩШтЩ
1—0(0 Н2)2 — г>—
О
Р
ОН
но
_сн2
о
н е— N4— (5 -іN
—(Ін-**1С Н = С Н — (СН2 )і 2 ^ С Н
3
Сфінгомієлін
Наявність в молекулах сфіншфосфоліпідів двох гідрофобних вугле
водневих радикалів (спирту сфінгеніну та ацилу жирної кислоти) поряд з
гідрофільною голівкою (фосфорна кислота, етерифікована аміноспиртом)
робить цей клас ліпідів також ідеально пристосованим для утворення
ламелярних структур, що лежать в основі біомембран.
2.2.
Гліктіпіди “ сполуки, в яких ліпідна частина ковалентно зв ’яза
на з вуглеводною. Ліпідна частина у складі гаіколіпіду є складним ефі
ром гліцерину або сфінгозину та вищих жирних кислот; вуглеводний
компонент — залишок моносахариду (глюкози, галактози та їх похід
них) або олігосахаридна група.
Гліколїшди, іцо є ефірами гліцерину, мають назву глікозилгліцеридів,
гліколіпіди, що єефірами М-ацилсфінгозинів (церамідів)—гпікосфінголіпідів.
СН2ОН
НО
—О-—СНг
і
" ° \ |
N 2!! \У
ОН
НС— О
1
С — ІЇ2
О
Н 2 С — О — С— ІЧ3
Глікозилгаіцерид(моногалакгозШідіацшшіінорин)
Глікосфінголіпіди є різноманітним за будовою підкласом сфінголіпідів, до яких належать такі ліпіди нервової тканини, як цереброзиди, ганг261
Розділ 4. БІОМОЛЕКУЛИуТА МЕТАБОЛІТИ
зюзиди, глобозиди та ін. Вони мають особливо важливе біологічне зна
чення у зв ’язку з їх структурною роллю в якості компонентів мембран
нейронів; до того ж існує ряд спадкових захворювань з важкими нерво
во-психічними порушеннями, що пов’язані з генетичними дефектами
метаболізму шікосфіншліпідів.
Наведемо приклади будови глікосфінголіпідів, які мають в якості ву
глеводної Частини залишок гексози (цереброзид) та олігосахариду лак
този (глобозид):
СН 2ОН
Цереброзид (галактозилцерамід)
НО— СН— С Н ==С Н — (СН2)12— СН3
Глобозид (лактозилцерамід)
Фізико-хімічні властивості ліпідів
За своєю молекулярною будовою молекули нейтральних жирів є непо
лярними сполуками, тобто мають гідрофобні властивості. Завдяки
цьому жири та олії йе розчиняються в воді або водних розчинах, і прй
намаганнях змішування з водою або іншими полярними розчинниками
утворюють двофазні системи з поверхнею розділу. Лише за певних умов
ліпіди при змішуванні з полярними розчинами можуть утворювати дис
персні системи типу емульсій “олія у воді” чи “вода в олії”; такими
умовами є наявність поверхнево активних речовин (ПАР).
262
4.2. Ліпід». Стероїди. Терпени
До ПАР, що функціонують в біологічних системах, належать жовчні
кислоти (див. нижче) та деякі білки. Саме завдяки дії цих фізіологічних
ПАР, природні жири присутні в біологічних розчинах (плазмі крові, цитозолі клітин) в емульгованому стані - у вигляді мікрокраплин, стабілізо
ваних зазначеними фізіологічними ПАР; процес емульгування є також
необхідною умовою для розщеплення (гідролізу) жирів харчових продук
тів у шлунково-кишковому тракті людини.
На відміну від нейтральних жирів, складні ліпіди (гліцерофосфоліпіди,
сфінгофосфоліпіди, шіколіпіди) є амфіфільними сполуками, що зумовле
не наявністю в структурі їх молекул ях гідрофобних жирнокислотних
“хвостиків”, так і полярних “голівок” (залишків фосфату, аміногруп, монотаолігосахаридних радикалів). Такі особливості будови та фізико-хімічних властивостей складних ліпідів є визначальними для їх участі в гГобудові біологічних мембран.
Л іпопротеїни. Роль ліпідів у побудові біом ембран
Ліпопротеїни «*комплекси ліпідів різної хімічної будови з білками. В їх
утворенні беруть участь нековалентні зв’язки та фізико-хімічш взаємодії
(водневі, іонні, ван-дер-ваальсові, гідрофобні).
Найбільше фізіологічне значення мають ліпопротеїни, в складі яких
ліпіди знаходяться в плазмі крові людини, та ліпопротеїни, що є інтеграль
ними компонентами біологічних мембран.
Ліпопрот еїни плазм и крові є молекулярними комплексами різних
класів ліпідів (тригаіцеридів, холестерину, фосфогліцєридів) з білкамИ,;ВДО
утворюють міцелярні структури, в складі яких ліпіди розчинені (суспен
довані) в плазмі, яка за своїми фізико-хімічними властивостями являє
собою водний розчин. Фізіологічною функцією цих ліпопротеїнів є міжорганний транспорт ліпідів - транспортні ліпопротеїни,
Ліпопрот еїни біологічних м ем бран —комплекси ліпідів з білками,
що складають основу структури (матриксу) біологічних мембран різно
го типу - супрамолекулярних структур, що відокремлюють клітину від
навколишнього середовища та розділяють внутрішньоклітинний простір
на певні тмпартменти (органели, субклітинні структури).
Всі біом ем брани- зовнішня клітинна мембрана (плазматична мем
брана) та внутрішньоклітинні мембрани (ядерна мембрана, мембрани
мітохондрій, ендоплазматичного ретикулуму, лізосом тощо) - являють
263
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
собою тришарові структури, що утворені внутрішнім ліпідним шаром,
вкритим ззовні та з середини білковими молекулами. Встановлено, що
8 0 - 9 0 0/о загального вмісту мембранних ліпідів складаютьрізні підкласи
шіцерофосфоліпідів (фосфатидилхоліни, фосфатидилетаноламіни, фосфатидилсерини) та сфінгомієліни. Найбільша концентрація фосфоліпідів при
сутня в мієлінових мембранах, що оточують відростки нейронів, форму
ючи нервові стовбури центральної та периферичної нервової системи.
Для хімічного складу зовнішніх клітинних (плазматичних) мембран ха
рактерним є також високий вміст гаіколіпідів та холестерину (див. ниж
че), який бере участь у стабілізації, зменшенні плинності фосфоліпідного бішару.
Хім ічні властивості ліпідів
Наведемо характеристику хімічних властивостей нейтральних жи
рів та інших пхіцероліпідів, найбільш цікавих з точки зору медико-біологічних властивостей та їх застосування в фармації.
А Гідроліз тригліиеридів та фосфпгліиеридіа
• Прості та складні ліпіди як складні ефіри спирту та жирних кислот
здатні до гідролізу, тобто розщеплення за участі молекул води. Гідроліз
ліпідів з розривом складноефірних зв ’язків є зворотним процесом до ре
акції етерифікації, відбувається в присутності мінеральних кислот або
лугів і протікає за механізмом нуклеофільного заміщення
(розділ 2 .5 .).
Кислотний гідроліз призводить до утворення гліцерину та вйщих
жирних кислот. При гідролізі природних жирів звичайно отримують су
міш насичених та ненасичених жирних кислот, кількісні взаємовідношення
між якйми залежать від характеру жиру
Лужний гідроліз (в середовищі МаОН або КОН) дає гліцерин та
натрієві або кйлієві солі жир.них кислот (мила) —тверде та рідке мило
відповідно, які є поверхнево активними сполуками і мають миючу дію,
звідки походить Історично усталена назва процесу - “омилення ліпідів”,
В живих організмах реалізується кислотний гідроліз ліпідів за участі
ферментів ліпаз, що розщеплюють нейтральні жири, або фосфоліпаз,
що розщеплюють фосфошіцериДи.
Загальна схема кислотного ліполізу трипхіцеридів:
264
4.2. Ліпіди. Стероїди. Терпени
О
Н2С — О — С —
н 2С — о н
л ,— с о о н
І
II
+ Н 20 ( Н +)
І
н е — о — с — н 2 — - — ► н е - — о н + р 2— СООН
о
[
Н2С-— О-— й — В 3
Н2С — О Н
Яз
соон
2. Гідрогенізаиія ж ирів
Під гідрогенізацією жирів розуміють відновлення залишків ненасиченйх жирних кислот у складі тригліцерйдів рослинник олій та деяких
тваринних легкоплавких жирів. Гідрогенізація відбувається за дії водніб
в присутності металічних каталізаторів (№ , Рі):
О
Н2 с — О — С — С 1 7 Н3 3
О
О
Н2 С — О — С — С 1 7 Н3 5
І
о
н е — о — й — Сі7Н29 + н?.(г% . н е — О — С — ■С
( 1 7 Н3 5
О
І
о
Н2С — О — б — Сі 7Н33
1,3-олеїноїл-2-лінолеїл-гліцерин
Н2С — О— (Я— -С17Н35
Тристеароїлгліцерин
(тристеарин)
■
Гідрогенізацію тришіцеридів у складі природних жирів використову
ють в промисловості для отримання насичених жирних кислот. Напри
клад, шляхом гідрування та подальшого гідролізу бавовняної, соняшни
кової та коріандрової олій або тваринних жирів отримують суміш стеа
ринової та пальмітинової кислот - т. з. “стеарин”, який поряд з парафі
ном використовують у фармації для виготовлення свічок.
3. Окислення ліпідів
Окислювальні реакції, що їм підлягають різні класи ліпідів, є надзви
чайно різноманітними. При цьому найбільш поширеними є хімічні про
цеси, які полягають у взаємодії кисню з ацильними радикалами, хцо вхо
дять до складу різних класів ліпідів. В результаті таких реакцій най
265
Розділ 4. БЮМОЛЕКГУЛИ ТА МЕТАБОЛІТИ
більш інтенсивно руйнуються залишки поліненасичених жирних кислот,
особливо у складі природних тригліцеридів та фосфогяіцеридів, що по
значається як “гіркнення” жирів.
В результаті процесів окислювального гіркнення жирнокислотні ради
кали перетворюються на коротколанцюгові альдегіди, які обумовлюють
специфічний неприємний запах жирів, що згіркли. Подібні за механізмами
реакцій процеси відбуваються в живих організмах* спричиняючи процеси
перекисного (пероксидного) окислення ліпідів біологічних мембран,
Реакції перекисного окислення ліпідів
Органічні пероксиди за хімічною будовою можна розглядати як
органічні похідні пероксиду водню НО-ОН, в молекулі якого один (в
гідропероксидах) або два атоми водню (в алкілпероксидах) заміщені на
алкільні радикали:
Я О -О Н
Р О -С Ж
Утворення органічних пероксидів алканів, алкенів та їх похідних від
бувається в хімічїшх та біологічних системах за умов наявності вільних
радикалів, тобто часточок, що мають на зовнішніх атомних або моле
кулярних орбіталях неспарені електрони. Для біоорганічної хімії найбільше
значення мають вільнорадикальні реакції утворення пероксидів поліне
насичених жирних кислот у складі ліпідних молекул, які утворюють біо
логічні мембрани, ліпопротеїнові міцели плазми крові, нуклео-протеноліпідні комплекси ядерного хроматину.
Існують такі етапи генерації вільних радикалів в означених біологіч
них системах, ініціювання та подальшого ланцюгового розвитку процесу
ліпопероксидації(Ю . І. Губський, 1997):
1)
Ініиіювання проиесу - відбувається шляхом генерації вільного
радикала Я* вуглеводню (або поліненасйченої жирної кислоти - перева«
жно С,8.2, С 20,4 або С226) та, після його взаємодії з киснем, утворення
перекисного радикала ЇЮ *:
1 а )К -Н -> К * ;
16) ЯГ + 0 2-> Щ ф
Механізми генерації вихідного радикала Л* можуть бути складними і
різнитися в окремих біохімічних системах - процес може бути ініційова
ним хімічно активними формами кисню (аніон-радикалом 0 2*-, синглет266
4.2. Л іп іди . С т ер оїд и . Т ерп ен н
ним киснем ‘О , гідроксильним радикалом ОН*), іонами Fe2/\ продуктам
ми радіолізу води, вільнорадикальними метаболітами ксенобіотиківтощо.
Сполуки, що сприяють активації вільнораДикального переш еного окис
лення ліпідів (Іони металів, ксенобіотики) дістали назву прооксидантів.
2) подовження ланиюга # полягає у взаємодії перекисного ради®
кала з іншою молекулою жирної кислоти, з утворенням гідроперекису
R-О О Н та нового радикала:
R 0 2* + R -H -> R-О О Н + R*.
У свою чергу, гідроперекиси жирних кислот Можуть перетворюватися
на стабільні молекулярні продукти- альдегіди, кислоти, спирти, кількісне
визначення одного з яких - малонового діальдегіду (Н О С -С Н 2-СО Н )
використовується як метод оцінки активності процесу ліпопероксидації.
Пероксидація ліпідів біологічних структур призводить до руйнування
життєво важлйвйх біомолекул та є одним з фундаментальних молекуляр
них механізмів пошкодження клітин при дії іонізуючої радіації, ігіїхіксикаціях, в процесі старіння особини.
3) обрив ланиюга - відбувається внаслідок рекомбінації вільних
радикалів (За) або їх взаємодії з інгібітором (36), а саме:
За) R* + R* -> R -R ;
36) R* ЩІп-Н -> R -H * Щ г Вільнорадикальйі форми інгібітору Ін* є кінетично малоактивними спо
луками, вони можуть взаємодіяти між собою з утворенням молекуляр
них продуктів, що призводить до гальмування процесу Ліпопероксидації:
Зв) In* +-ІП* —> In-In.
Органічні речовини, що зупиняють реакції ліпопероксидації в біологі
чних системах шляхом взаємодії з хімічно активними вільними ради
калами (виступаючи в ролі гасників, або “пасток” вільних радикалів),
називаються антиоксидантами.
Вітамін Е
Найбільш активгіим та поширеним у тваринних тканинах антиокси
дантом є жиророзчинний вітамін Е (а-токоферол).
267
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Як видно з наведеної формули, молекула а-токоферолу складається
з конденсованого і-етероциклу хроману (бензо-у-дигідропірану) з гідро
ксильною групою в 6 -му положенні ;щ ізопреноїдного бічного ланцюга.
Молекулярна основа антиоксидантної активності вітаміну Е поля
гає в його спроможності виступа-щ інгібітором вільцорадикальних реакцій
за розглянутим вище механізмом;
Іп-Н + ЯГ ф !п’ + Р -Н .
Здатність молекули а-токоферолу до взаємодії з вільними радикала
ми обумовлена активністю рухомого атома водню у складі гідроксиль
ної ірупи хроманового кільця:
Радикали а-токоферолу, що утворилися, рекомбінують між собою
подібно до інших інгібіторів типу їй*.
4.2.2. Стероїди та їх похідні
С тероїд и -вели ки й клас біоорганічних сполук, що є похідними циклічного вуглеводню сщерану (циюіопентанпергідрофенантрецу), Скелет
268
Ль.
4.2. Ліпіди. Стероїди. Терпени
стерану складається з повністю гідрованих трьох нелінійно конденсова
них кілець циклогексану (А, В, С) та ядра циклопентану (Б).
Стеран
(циклоиектагоергідрофенактрен)
Незважаючи на специфічність хімічної будови, за фізико-хімічними вла
стивостями, зокрема розчинністю в неполярних гідрофобних середовищах,
стероїди відносять до ліпідів. Разом з терпенами (розділ 4.2.3.) стероїди
складають групу т. з.”неомилюваних ліпідів”, тобто таких жироподібних
єполук, що, на відміну від розшянутих вище гаіцероліпідів, сфінголіпідів та
восків, не підлягають гідролізу в кислому або лужному середовищах.
За біологічними, властивостями стероїди належать до низькомо
лекулярних біорегуляторів метаболічних процесів та фізіологічних функ
цій організму. Це - вітаміни Трупи
та В 3), стероїдні гормойи кори
наднирників, чоловічі та жіночі статеві гормони, жовчні кислоти.
Хімічна будова більшості природних стероїдів характеризується на
явністю кисеньвмісного замісника при С, (-О Н ; -(Ж ), метильних ради
калів С , 8 і С)9.та алкільного замісника при С 1Г,
Будова загального скелету стероїдів
Залежно від будови замісник? ПРИ С І7 стероїди диференціюються
на декілька груп, що розрізняються також за ступенем ненасиченостт
складових стеранового скелету, біологічною активністю та фізіологіч
ними функціями (таблиця 4.З.).
2 69
Розділ 4„ БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Таблиця 4.3.
Групи стероїдів залеж но від будови
вуглеводневого р а д и к ал а п ри С ]7
Радикал при Сп
Назва
вугле
водню
Будова радикала .
■0 івСІЇ!з
К-ть
С-атомів
Стерини
«СНП “ с н 3
Холестан
Назва групи
стероїдів
8
21СН3
' -л -
27СН3
■Ш ^Ш Й ;ійСНїО'й СНа.й І4СН,
Холан
5
Жовчні
кислоти
- , гіС И і .
Прегнан
Естран
Андростан
О 20СН2О 2>СН3
<_ - ЩІК (Сі9 відсутні).
а н
Жіночі статеві
гормони (гей- *‘2
тагсни) та кор- |
тикостероїди
Жіночі статеві
0 ’ . гормони
(естрогени)
Чоловічіста0
теві гормони
(андрогени)
Стереохімія стероїдів
Всі циклогексанові кільця; що складають молекулу стероїдів, знахо
дяться в конформації крісла. При цьому в природних стероїдах кільця А
і В можуть бути як в транс-, так і в умс-зчленуванні, тоді як кільця В і
С завжди, а С і О - майже завжди мають транс-зчленування. Загальна
структура молекул стероїдів є жорсткою, мало схильною до конформаційних перетворень.
Конформація стерану в умовах транс-зчленування кілець А/В» В/С та СЯ)
2 70
4.2. Ліпіди. Стероїди. Терпени
Н
Конформація стерану в умовах цис-зчленування кілець А/В
та транс-зчленування кілець B/Ç і C/D
При зображенні молекул стероїдів на папері їх звичайно подають у
вигляді плоских циклічних структур. Положення атомів водню та заміс
ників (-О Н , -C H j тощо) позначається літерою ос, якщо вони знаходять
ся нижче від умовної площини кільця (тобото за площиною проекції),
або Р, якщо вони розташовані вище від площини кільця (перед площи
ною проекції). Для зображення різної просторової орієнтації зазначених
замісників застосовують спеціальні позначення типу, жирного або заштри
хованого клина (розділ 1.3.):
У більшості випадків при напиеанйі структурних формул стероїдів
просторову конфігурацію замісників не враховують, тобто зображують
ЇХ такими, що знаходяться в площині циклічної структури,
С терини
Найбільш розповсюдженими стероїдами, що присутні в мембранних
структурах всіх тваринних та рослинних клітинах, є стериніі (зоостерипи та фітостерини відповідно). За хімічною будовою стерини є
Похідними вуглеводню холестану (див. таблйцю 4гЗ-.), що містять в 3-му
271
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
положенні стеранового циклу гідроксильну групу, тобто являють собою
вторинні спирти - стероли.
Стерини (стероли) здатні утворювати складні ефіри з жирними кис
лотами І> стериди.
Холест ерин
Із стеринів тваринного організму найбільше значення має холесте
рин (холестерол), !■ похідне холестану, особливостями » ’мінної будови
якого є наявність гідроксильної в Р-конфігуращїта подвійного зв’язку в
кільці В між 5-м та 6 -м атомами вуглецю:
Холестерин міститься в більшості тканинних ліпідів, входячи як струк
турний ліпід до складу плазматичних мембран. Крім того, холестерин
присутній; у вільному або етерифікованому жирними кислотами (холестериди) стані в пЛазмі крові та жовчі. В плазмі крові людини холесте
рин міститься у вигляді комплексів з білками (транспортних ліпопро-
2 71
4.2. Ліпіди^ С т ер оїд и . Т ерп ен и
теїнах плазми крові - див. нижче), переважно у складі т. з. ліпопротеїнів
низької щільності - ЛНП. Фізіологічні функції холестерину полягають в
його участі у структуруванні біологічних мембран та ролі як попередни
ка в синтезі багатьох інших стероїдів, зокрема вітаміну Б ^та жовчних
кислот. Організм людини отримує близько половини необхідного для ЙОШ
життєдіяльнос*гі холестерину з продуктами харчування тваринного
походження (в середньому 1,0-1,5 г/добу); крім того, ферментні системи
більшості тканин (особливо печінки) здатні до біосинтезу холестерину з
активної форми оцтової кислоти — ацетил-КоА. Порушення обміну
холестерину проявляються здебільшого накопиченням аномально великої
к іл ьк о с ті стер и н у в крові (гіперхолест еринем ія) та тк ан и н ах з
відкладанням його кристалів в стінках судин (атеросклероз), ‘що при•
•
зводить до глибоких порушень у функціонуванні серцево-судинної сис
теми та кровопостачанні головного мозку.
Віт амін
В тканині печінки частина холестерину окислюється до 7-дегідрохолестерцну, який надходять у жирову тканину та шкіру, де під впливом
ультрафіолетового опромінення (складової сонячного світла) перетво
рюється на вітамін /),:
Вітамін БДхолекальциферол) належить До жиророзчинних вітамінів,
іо г о фізіологічна роль полягає в регуляції обміну іонів кальцію та фосфа
тів, необхідних дл/побудови кісткової тканини. Нестача в організмі ди
тини вітаміну Б 3 призводить до важкого захворювання - рахіту. Близь*
ким за будовою та біологічними властивостями до вітаміну 0 3 іе віта
мін Ь , (ергокальциферол) - сполука рослинного походження, біологіч
ним попередником якої є ергостерин рослинних олій.
273
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАЮЛІТИ
Жовчні кислоти і
Жовчні кислоти —похідні вуглеводню холану, в бічному ланцюзі яко
го дистальний метил (при С24) є окисленим до карбоксильної групи (холанова кислота)'.
Жовчні кислоти є гідроксшюхідними холанової кислоти. До них нале- жать такі сполуки, як холева (3,7,12-тригідрокеихоланова), дезоксихолева (3,12-дигідроксихоланова), літохолева (3-гідроксихоланова),;сенодезоксихолева (3,7-дигідроКсихолаиова) кислоти. 70-80 % жовчних ки
слот, що містяться в жовчі людини, становлять холева та дезоксихолева кислоти. Розміщення гідроксильних груп в жовчних кислотах має
а-конфігурацнр:
Холева кислота
Дезоксихолева кислота
Жовчні кислоти здатні утворювати аміди з амінокислотою шіцином
та таурином - т. з. “парні” жовчні кислоти - глікохшеву та таурохолеву кислоти.
274
4.2. Ліпіди. Стероїди. Терпени
Завдяки присутності у складі молекул гідрофобної стеранової струк
тури Та гідрофільних гідроксильних Та карбоксильної груп, жовчні кис
лоти є поверхнево активними речовинами, необхідними для .емульгу
вання харчових ліпідів (переважно тригліцеридів) в порожнині тонкого
кишечника людини. Натрієві солі глікохолевої та таурохолевої кислот є
найбільш активними ПАР, сприяючи утворенню емульсії тригліцеридів
та створюючи оптимальні умови для дії кишечної ліпази.
Стероїдні гормони
До стероїдів належить також група фізіологічно важливих гормонів,
що у людини та вищих тварин синтезуються в ендокринних залозах—
корі надннрників та єтдтевих залозах - стероїднихгормонів. Це - кор
тикостероїди - похідні прегнану (С21), чоловічі статеві гормони (андро
гени) - похідні андростану (СІ9), жіночі статеві гормони -п о х ід н і ест
рону (С 18) та прегнану (С21).
18
27 5
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Будова та властивості окремих представників стероїдних гормонів
будуть розглянуті в розділі 5.5.
Стероїдні аглікони серцевих глікозидів
Серед лікарських засобів, що містять у своїх молекулах стероїдні
структури, важливе значення для медичної практики мають т.з “серцеві
глікозиди *—речовини, що виділені з різних видів наперстянки (Напер
стянки пурпурової- Digitalis purpurea; Наперстянки шерстистої - Digitalis
lanata) та строфанту (Строфанту гладкого - Strophanthus gratus, Стро
фанту Комбе—Strophanthus Kombe). Серцеві глікозиди використовуються
як кардіотонічні засоби для стимуляції скоротливої функції міокарда при
серцевій недостатності.
За хімічною будовою молекули серцевих глікозидів складаються з двох
компонентів - аглікону (невуглеводного фрагменту) та гпікону, в ролі якою
виступає один з цукрів—D-дйгітоксоза, D-ілюкоза, D-цимароза, D-рамноза
тощо. А ш ію н являє собою похідну стерану, яка сполучена з іліноновою
частиною за рахунок гідроксилу при С3 (О-глікозидним зв’язком).
Стероїдними агліконами найбільш поишреної групи серцевих глікози
дів - карденолідів є дигМокєигенін та строфантидин, Які £ гідроксипохідниМи стерану сполученими в 1?-йу положенні з п ’яТИчЛЄннйй лактонним кільцем:
Дигітоксигенін є структурним компонентом серцевого глікозиду
Дигіт оксину- препарату з Digitalis purpurea та Digitalis lanata; гліконовою частиною дигітоксину є трис&харид, що складається з трьох залишків
D-дигітоксози.
276
4.2. ЛІПІДИ; Стероїди. Терпенй
Серцевий тптзт&Дигітоксіф з В%йаІі$ рцгригеі
Строфантидин входить до складу серцевого глікозиду Строфан
тину (препарату із Б^орЬапЛиз КоггіЬе), глікон якого є дисахаридом, що
м і с т и т ь залишиш Б-цимарози та Г>-тюк6зи.
4.2.3. Терпени: будова, представники
Близьким за фізико-Хімічними властивостями до ліпідів та стероїдів
є клас терпенів, до якого входить значна кількість сполук тваринного
та рослинного походження.
Терпени - це клас біоорганічних сполук, що є ненасиченими вугле
воднями, в основі будови яких лежатьзалишки молекули ізопрену (С5Н8),
що Повторюються (ізопреноїди).
фщ
ш СИ2 = С
СН = С Н
2
Ізопрен (2-метилбутадієн-1,3)
Загальна емпірична формула терпенів (терпенових вуглеводнів) (С5Н8)п>де п становить переважно від 2 до 8 або більшої кількості зали
шків ізопрену (поліізопреноїди). Перші представники класу терпенів
277
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
були виділені із скипидару - продукту перегонки рідкої смоли хвойних
дерев. Давня назва скипидару - “терпентинова олія” (ОНеш ІегеЬіШЬтае),
звідки похоДить назва класу. У зв’язку з антисептичною дією компонен
тів терпентинної олії, останню використовували в Стародавньому Єгипті
як бальзамуючий засіб. В подальшому багато терпенів знайшли засто
сування в медицині та парфумерії.
До терпенів іноді відносять також близькі до них за будовою каро
тиноїди (див. нижче) та стероїди, бірсіртез яких відбувається за ме
ханізмом конденсації ізопренових залишків.
Терпено'щи. Кисеньвмісні похідні терпенових вуглеводнів (спирти,
альдегіди, кетони, ефіри) дістали назву терпеноїдів. Природними дже- •
релами різноманітних терпеноїдів та терпенових вуглеводнів є ефірні олії,
що надають специфічного унікального запаху рослинам. Біагатр терпенів
та їх похідних мають властивості бірлогіщ р активних сполук і деякі з
них застосовуються в медицині та фармації (ментол, валідол, камфора).
КпасисЬікація терпенів.
За кількістю ізопреноїдних фрагментів терпени поділяються в а щ р
нотерпени (містять два ізопреноїдних залишки; ЗзАб)> ^т ерпени (мі
стять чотири ізопреноїдних фрагменти —С20), тритерпени (С3(>), тетратерпени (С40) та політерпет - сполуки, які містять більше восьми
Ізопреноїдних ОДИНИЦЬ.
. Г
: ?
/
За особливостями будови вуглеводневого ланцюга виділяють ацик
лічні та циклічні терпени.
Аииклічні тевпени ,
‘
В ациклічних терпенах ізопреноїдні залищкиспряучені між собою за
типом “голова” до “хвоста”, що може бути продемонстровано на,при
кладі найпростішого ациклічного дитерпену - оцимену, що міститься в
олії хмелю:
СНз
СНд
І
1'
Н2 С = С — - с н 2 — с н 2 —
1
2
3
4
Оцймен (С10Н|6)
278
2'
З-
4'
н с = с |— с н = с н
2
4.2. Ліпіди. Стероїди. Терпени
Важливим ациклічним політерпеном €
де п > 500, який широко використовуєш » в ринил галузях промисло
вості Природний каучук добувають у формі колоїдного розчину Т _
латексу з виділень рослии-каучуконосів (Hevea brasilicnsis), и о після
S
9
S
S
- т ~— промиетову
сировину.
Циклічні терпени
ж
Циклічні терпени за кількістю циклів поділяються на моноциклічш
біциклічні.
S
&
щ°лежитьв°сновімоноцикл™х^
ненів є вуглеводень ментан, який можна розглядати як продукт кон
денсації двох молекул ізопрену, сполучених вуглецевими містками між
С _ е 4 та С 5- С 6 карбоциклічного ядра. Похідним ментану, що застосо
вується в медичній практиці Жментол:
М ентан
(р-метилізопропілциклогексан)
—
и
(меитанол-3)
Ментол - сполука з м ’ятним запахом, є головним компонентом ефір
ної олії м ’яти перцевої. В медицині ментол використовується як засіб,
що має антисептичну та слабку заспокійливу і мішевоанестезуючу дію
у вільному стані та як компонент багатьох складних лікарських препа
ратів Ш ироке застосування ментол має як складова Валідолу - фар
мацевтичного засобу, що розширює кровоносні судини та застосовуєть
ся при синдромі стенокардії.
279
Розділ 4. БІОМОЛЕКУЛИ: ТА МЕТАБОЛІТИ
Л ім он ен (м ен тад ієн - 1 , 8 ) є також моноциклічним ненасиченим
терпеном. Лімонен існує в природі у вигляді двох дзеркальних ізомерів —
(+) ~ та (-) - лімонену, вдо містяться в ефірних оліях лимону, апельсину»
кмину та хвойних дерев і широко застосовується в парфумерії. При
гідратації лімонену в кислому середовищі утворюється двоатомний спирт
терпін (ментандіол *1 , 8 ):
СНЧ
н3с
Лімонен
Терпін
Кристалогідрат терпіну (фармацевтична назва —Терпінгідрат) за
стосовується при лікуванні захворювань верхніх дихальних шляхів як
відхаркуючий засіб.
Біииклічні терпени
В основі будови біциклічних терпенів Та терпеноїдів лежить шести
членний вуглецевий цикл, що конденсований із Циклами менших розмі
рів. Родоначальниками цього підкласу терпенів є вуглеводні пінан та
борнан (камфан).
Пінан
Борнан (Камфан)
а-пінен
сс-пінен похідне пінану —, є головним компонентом скипидарів, які
добувають з хвойних дерев, переважно Сосни приморської (Pinus pinaster)
та Сосни лісової (Pinus silvestris). В медицині скипидар застосовують як
місцево-подразнюючий засіб при простудних захворюваннях, міалгіях
та артралгіях шляхом зовнішніх розтирань або скипидарних ванн.
2 80
4.2. Ліпіди. Стероїди. Терпени
Камфора (борнанон-2) є важливим похідним борнана. Камфора існує
у вигляді двох енантіомерів, що розрізняються за оптичною активніс
тю - (+) - та (-) - камфора, але мають однакову фізіологічну дію як
ефективний кардіотонічний засіб.
СН 3
р » і'' Камфора
СН 3
( - ) - Камфора
Природну правообертаючу (+) - камфору виділяють з коричника
(лавру) камфорного (С іш іато п и т СатрЬога), що росте у країнах ГОвдеігао-Східної Азії (т. з. “японська” камфора), лівообертаючу (^ -к а м
фору отримують з борнеолу, який міститься в ефірних оліях ялиці сибір
ської, щавлії камфорної, полину тощо. Рацемічна камфора (суміш двох
оптичних стереоізомерів) може бути отримана хімічним шляхом з
а-пінену.
Фармацевтичним препаратом камфори є Камфорне масло (20 %
олійний розчин порощку камфори), яке при підшкірному введенні стиму
лює судинно-руховий та дихальний центри, поліпшує метаболічні проце
си в міокарді. Препарати камфори для зовнішнього застосування вико
ристовують при артритах, ревматизмі, м ’язових болях. Похідне камфо
ри ~ Бромкамфору - застосовують як препарат заспокійливої дії при
збудженні центральної нервової системи.
Біологічно важ ливі похідні терпенів. Каротиноїди
До біологічно важливих похідних терпенів належать: каротиноїди (ка
ротини та вітамін А, вітамін Е - похідні дитерпенів), вуглеводень сквален
(С30Н50) - попередник в біосинтезі з ізопреноїдних фрагментів холестерину.
Каротини - група рослинних пігментів (а-, (3- та у-каротини), що
надають жовтого забарвлення багатьом овочам (моркві, томатам, ку
курудзі) та фруктам (від саіхла —морква, лат.). Найбільше значення для
організму людини має (З-каротин, який є біологічним попередником віта
міну А (“провітаміном” вітаміну А). За хімічною природою (З-каротин є
281
/
Р озділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
полієновим вуглеводнем - октамером ізопрену (С49), що містить на двох
протилеж них кінцях п оліненасиченого вуглеводневого ланцю га
шестичленні цикли (3-іонону.
В печінці тваринних організмів (3-каротин розщеплюється гідролітич
ним шляхом за дії ферменту каротинази з утворенням двох фізіологіч
но активних мОлекул вітаміну А 1 {ретинолу):
Вітамін А, являє собою жовту кристалічну речовину, що за фізикохімічними властивостями належить до жиророзчинних вітамінів. Він
надходить в організм людини переважно з тваринними продуктами харчування йі жовтком яєць, вершковим маслом, риб’ячим \^иром - або
утворюється з рослинних каротинів. Функції вітаміну А в організмі людини
полягають в регуляції зорового аналізатора шляхом утворення разом з
білком опст ом світлочутливого пігменту ф&ківкй ока - родопсину]
крім того, вітамін А необхідний для нормального росту організму та
утворення складних білків шікопротеїнів, що складають основу біологічних слизів (муцинів, мукопротеїдів)*
Будова та біохімічні функції вітаміну Е були розглянуті вище. Як вже
відмічеНо, похідними терпенів е також стероїди, біосинтез яких в живих організмах відбувається на основі реакцій поліконденсації ізопреноїдних фрагментів.
282
|
1
1
|
І
|
]
)
З
|
]
4.3. Білки, пептиди. Амінокислоти
4.3. Білки, пептиди. Амінокислоти
4.3.1. Білки: визначення, загальна характ ерист ика
Б ІЛ К И "* біоорганічні високомолекулярні сполуки; молекули яких є
гетерополімерамй, побудовані із залишків а-Ь-амінокислот, об’єднаних
кислото-амгдними ( пепт идніїми) зв ’язками (-С О -И Н -). П епт иди
(поліпептиди) - біомолекули, що відрізняються від власне білків мен
шою молекулярною масою та фізико-хімічними характеристиками.
Білки та пептиди є найбільш розповсюдженими з усіх класів біомолекул; вони входять до складу’всіх клітинних компонентів мікроорганізмів,
рослин» тварин (ядра, біомембран, цитоплазми) та міжклітинних струк
тур. Білковий склад живих клітин залежить від ступеня складності гено
му та етапу еволюційного розвитку організму. Кількість різних білків в
прокаріотичнїй клітині Б.Соїі близько 3 000, в організмі людини - біля
5 000 000, всього в різних видах організмів, що складають біосферу Землі, ж
1 0 хо—1 0 12 різних білків.
М олекулярна м аса білків
Білки є високомолекулярними сполуками (біополімерами); їх моле?
кулярна маса коливається в межах від декількох тисяч до декількох
мільйонів. Наприклад, м,м. білкового гормону інсуліну - 5,7 кД, фермен
ту рибонуклеази -1 2 ,6 кД, ферменту пепсрну 35,5 кД, кисень-транспортуючого білка гемоглобіну - 64,5 кД.
Молекули білків можуть складатися з одного або декількох окремих
поліпептидних ланцюгів, що об’єднані ковалентними (дисульфідними) або
нековалентними зв ’язками. Білки, які мають молекулярну масу від 5-6
до ~50 кД, складаються звичайно з однієї субодиниці (протамеру).
Білки з більшою М;М. (в сотні тисяч кілодальтонів) складаються, як пра
вило, з декількох субодиниць (протомерів), ідо утворюють складні між
молекулярні агрегати; це - олігомерні білки. Це, як правило, складні
білки ферменти; наприклад молекулярна маса ферменту піруваткінази - 240 кД, фермеНту глікогенфосфорилази - 495 кД, ферментного
комплексу синтетазц жирних кислот 2.300 кД.
Пептиди (олігопептидй, поліпептиди) відрізняються від власне біл
ків молекулярною масою (звичайно меншою 5-6 кД) та відповідними
фізико-хімічними властивостями.
283
Р озділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Біологічні функції' білків та пептидів,
Кі Ферментативна (каталітична) функція. Всі ферменти (біокаталізатори) являються за своєю хімічною природою білками або комплек
сами білків з небілковими сполуками (коферментами, кофакторами),
2. Структурна функція. Білки входять до структури біомембран»
складають основу цитоскелету (мікротрабекулярна сітка, мікрофіламенти), міжклітинного матриксу (колаген, еластин) та певних спеціалізова
них тканин (кератини).
3. Енергетична функція - за рахунок окислення вуглецевого скеле
ту амінокислот білків їжі або тканинних білків організм людини отримує
до 1 0 % хімічної енергії, необхідної для його життєдіяльності.
4. Регуляторна та рецепторна функція. Білкову та пептйдну при
роду мають численні біорегулятори: гормони, медіатори та модулято-і
ри, що продукуються в ендокринній, нервовій та імунній системі. Білками
є також мембранні та цитозольнірецептори - ультраструктурні утворення,
що сприймають хімічний сигнал від певного біорегулятора.
5. Транспортна функція. Спеціалізовані білки зв’язують та здійсню
ють міжклітинний та внутрішньооклітинний (трансмембранний, цито
плазматичний) транспорт різних лігандів - біомолекул, кисню, іонів металів*|
лікарських засобів (гемоглобін, трансферин, сироваткові альбуміни тощо):1'
6 . Скорочувальна функцШ. Білки є молекулярними структурами, які
реалізують Скорочувальну функцію м ’язів (акты«, міозин), клітинних
джгутиків та війочок (динеїн, тубулін).
•7. Захисна функція. Спеціалізовані білки здійснюють функцію імун- ]
ного захисту (імуноглобуліни, інтерлейкіни, інтерферони тощо). Біл- *
ки згортальної, антизгортальної та фібринолітичної систем крові проти*
діють кровотечі та тромбоутворенню.
Фактично, взаємодія та/або перетворення певних білкових молекул є
основою реалізації будь-якої фізіологічної функції на всіх рівнях струк
турної організації живих організмів. Тому біоорганічна хімія білків протеаміка - є одним з найбільш розвинених та перспективних напрямів
сучасної біомедицини.
4.3.2. Амінокислотний склад білків та пептидів
Йри гідролізі природних білків та пептидів вивільняється близько 2 0
а-Ь-амінокислот, розміщення кожної з яких в поліпептидному ланцюга
кодується триплетом нуклеотидів в ДНК геному, що кодує даний білок*
284
4.3. Б іл к и , п еп ти ди . А м ін ок и сл оти
Будова протеїногенних амінокислот
Амінокислоти, що входять, до складу природних білків та пептидів
(протеїногенні амінокислоти) мають хімічну будову, що представле
на загальною структурною формулою:
Я
и
н
с' «—
щ,
соон
Сх-Ь-амінокисяота
Як слідує з наведеної формули, протеїногенні амінокислоти мають
такі структурні особливості:
1 ) а-атом вуглецю всіх протеїногенних амінокислот, за виключен
ням гліцину, зв’язаний з чотирма різними функціональними групами (аси
метричний атом), і є хіральним центром молекули. Виходячи з цього,
протеїногенні амінокислоти є оптично активними сполуками,.тобто
здатні до обертання площини поляризованого світла. За своєю стереохі
мічною конфігурацією протеїногенні амінокислоти є стереоізомерами
Ь-ряду. Б-амінокислоти до складу природних білків не входять; Б-амінокислоти зустрічаються в бактеріальних та рослинних об’єктах, входять
до складу деяких антибіотиків (граміцидин, актиноміцин Б ). Фер**
ментні системи організму людини, як правило, не сприймають Б-амінокислоти. Цікаво при цьому, що амінокислоти Ь- та П-ряду можуть ди
ференціюватися за смаком.
2) Природні (протеїногенні) амінокислоти Я (Х-амінокислотами, тоб
то в них аміногрупа сп олучен і з атомом вуглецю, що міститься в
а-положенні по відношенню до карбоксильної групи. Деякі протеїногенні
амінокислоти (лізин, аргінін) та природні амінокислоти, що не входять до
складу білків (орнітИн, цитрулін) мають додаткову аміногрупу, що розта
шована в кінцевому ( о ) положенні радикала Я.
Класифікація прот еїногенних амінокислот
Залежно від хімічної будови бічного радикала природні амінокислоти
поділяють на такі класи:
285
/
Розділ 4. БІОМОЛЕКУЛИ ТА-МЕТАБОЛІТИ
1) Ациклічні амінокислоти:
а) моноаміномонокарбонові амінокислоти;
б) моноамінодикарбонові амінокислоти;
в) діаміномонокарбонові амінокислоти;
г) діамінодикарбонові амінокислоти.
2) Циклічні амінокислоти:
а) карбоциклічні (ароматичні) амінокислоти;
б) гетероциклічні амінокислоти:
- з первинною аміногрупою в бічному ланцюзі;
— імінокислоти.
Наведемо формули основних представників а-Ь-амінокислот, що
входять до складу природних білків І пептидів (в дужках - їх скорочені
назви за міжнародною номенклатурою).
М оноаміномонокарбонові амінокислот и;
НІ
!
СН3
V
И #—
Н2М Р ^ * # с н ^ С О О Н
с н — со о н
А л ан ін (А іа)
(ос-ам інопропіонова кислота)
Гліцин (С Іу)
(ам інооц това ки сл ота)
СН
н2іч
сн —соон
В алін (Уаі) (а-ам ін о ізо в ал ер іан о в а кислота)
Н3с
н3с.
сн
СН2
н 2м— с н -с о о н
Лейцин (Ьеи) (а-ам іноізокапронова кислота)
2 86
\
СН 2 ^ с н 3
сн
4.3. Б іл к и , п еп ти ди . А м ін ок и сл оти
ОН
І
СНа
ОН
І
С Н -С Н з
І
Н2М— с н -с о о н
і у | — С Н -С О О Н
аряМ М
Т реонін (ТИг)
(а -а м ін о -0 -о к си м а с л я н а
кислота)
(а -а м ін о -Р -о к с и п р о л іо н о в а
кислота)
СН3
,
Б
г
сн 2
І сн 2
І
— с н -с о о н
ЄН
І
сн 2
І
■оьсоон
Ц истеїн {Су«)
(<х-аміно-Р-тіоирогаонова
кислота)
М етіонін (М еі)
(а -ам іи о -у -м ети л тіо м асл ян а
кислота)
М оноамінодикарбонові амінокислоти:
СО О Н
сн 2
СООН
І
сн 2
*
;
.
сн 2
І
і
Н з^ С Н -С О О Н
нлч- -С Н “ СООН
А спарагінова ки сл ота (Авр) ,
(а -а м ін о б у р ш ти н о в а кислота)
Г лутам ін ова ки сл ота (О іи)
(а-ам ін о гл у т ар о ва кислота)
287
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Діаміномонакарбонові амінокислоти:
ІЧН2
КІН2
с= м н
І
сн 2
І
сн 2
сн 2
сн 2
сн 2
ж
г
Ег
сн 2
сн 2
н2іч— с н -с о о н
н2м— с н -с о о н
Л ізш і (Ьув)
(а,є-д іа м ін о к ап р о н о в а кислота)
Аргінін (Аг§)
(а-аміно-5-гуанідиновалеріанова кислота)
Аромат ичні амінокислоти
ОН
СН 2
НгМ— с н —соон
Фенілаланін (РЬе)
(а-аміно-р-фенілпропіонова
кислота)
288
сн 2
н2м— сн *^ со о н
Тирозин (Туг)
(а-аміно-Р-р-оксифенілпропіонова
кислота)
4.3. Білки, пептиди. Амінокислоти
Гетероциклічні амінокислоти та імінокисноти
*сн 2
н ^ — с н — соон
— с н — соон
Триптофан (Тгр)
(а-аміно-Р-індолілпропіонова
кислота)
Гістидин (Нів)
(а-аміно-р-імідазолілпропіойова
кислота)
*
СООН
Пролін (Рго)
(піролідин-а-карбонова
кислота)
До основних (“канонічних”) амінокислот, що утворюють природні біл
ки, належать також аміди аспарагінової,та ш утамінової кислот - аспаг
рагін та глутамін відповідно. Крім того, у складі деяких білків виявле
но похідні цих амінокислот, зокрема 4-гідроксипролін, 5-гідроксилізин,
К-метиллізин, 3-метилгістидин, фосфосерин, фосфотреонін, дийодтирозин. Хімічна модифікація (гідроксилювання, метилювання, фосфорилювання, йодування) амінокислот відбувається вже після рибосомального
синтезу поліпептидних ланцюгів,
Ф ізико-хімічні властивості протеїногенних амінокислот
За фізико-хімічниМи властивостями (полярності та знака заряду біч
ного радикала И.) амінокислоти розділяють на такі к л а с и :.
*•« амінокислоти 3 неполярними (гідрофобними) Я-групами {аланін,
валін, лейцин, ізолейцин, метіонін, пролін, триптофан, фенілаланін);
- амінокислоти з полярними (гідрофільними) незарядженими Я-групами {гліцин, серин, треонін, цистеїн, тирозин, аспарагін, глутамін);
289
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
«■ амінокислоти з негативно зарядженими. И-групами (кислі аміно
кислоти) (аспарагінова кислота, глутамінова кислота)',
- амінокислоти в позитивно зарядженими ІІ-групамщ: (основні амі
нокислоти) (аргінін, лізин, гістидин).
Кислот но-основні властивості амінокислот
Амінокислоти є амфотерними електролітами, що можуть дисоцію
вати з утворенням іонних форм —аніона або катіона. У водному середо
вищі амінокислоти Ішуіоть у вигляді рівноважної суміші, що складаєть
ся з внутрішньої солі' (біполярного іона, цвіттер-іона% аніонної та каті
онної форм, присутність яких залежать від рНсередовища:
+ Н+^ R— СН—-С ^ °
н+
І
"ОН
NHq+
R— CH— f f — ► R — CH— C t° ~
I
ОН
І
О*
Н"
°
-О Н '
R— CH— С
І
0‘
nh2
Наведена схема відповідає механізму кислотно-основної дисоціації
моноамінОмонокарбонових амінокислот, що мають По одній а-амінній
та ot-карбоксильній групі. В цьому найпростішому випадку рівновага
між: позитивно Та негативно зарядженими іонами може теоретично
досягатися в нейтральних- розчинах, тобто при рН=7.
Разом з тим, деякі амінокислоти Мають бокові ланцюги R, що міс
тять додаткові функціональнітрупи, здатні до дисоціації:
Ä кислотні групи Asp, Glu;
--** Основні групи Lys, Arg, His.
Вйходячиз цього, сумарний заряд молекул амщокиелот (та відповідно
білків і пептидів, до складу яких вони входять) визначається:
а) взаємовідношенням між кількістю вільних кислотних та Основних
груп радикалів амінокислот;
б)’ ступенем дисоціації (рКа) амінокислотних радикалів;
в) pH середовища, в Якому розчинена амінокислота чи білок (полі*
пептид).
290
4,3. Білки, пептиди. Амінокислоти
В кислих розчинах переважає катіонна форма амінокислот (молекул
ли заряджени позитивно), в лужних розчинах - аніонна (амінокислоти
'шряджені негативно). Ці фізико-хімічні властивості аміиокислог визнача
ють їх здатність до електрофорезу » розділенню в високовольтному по
стійному електричному полі. При рівновазі позитивних та негативних за
рядів молекула амінокислоти перебуває в ізоепектричнаму стані. Харак
терне для кожної амінокислоти значення рН, при якому амінокислота має
еумарний нульовий заряд, називається р Н ізоелектричної точки (рі).
Розчинність ам інокислот т а їх взаємодія з р ізн и м и лігандам и
Залежно від будови бічних радикалів Б., амінокислоти в більшій або
меншій мірі взаємодіють з диполями води, тобто проявляють гідрофільні
цбо гідрофобні властивості. Розчинними у воді, водних розчинахда інших
полярних розчинниках є амінокислоти з полярними незарядженимм, не
гативно та позитивно зарядженими Я-групами. Амінокислоти з неполяр
ними (гідрофобними) бічними ІІ-грунами, навпаки, здатні до взаємодії з
неполярними розчинниками, зокрема сполуками ліпідної природи, напри
клад у складі біомембран, ліпопротеїнів.
Полярність функціональних груп амінокислот разом з їх кислотноосновними властивостями визначають особливості структури, більшість
фізико-хімічнихта відповідно біологічних властивостей білків, які син
тезуються з цих амінокислот.
Х ім ічні в л а с ти в о ст і п ротеїй оген н н х ам ін о ки сл о т
Характеристику хімічних властивостей амінокислот як гетерофункціональних сполук було наведейо в розділі 2.7. Нижче розглянемо здат
ність амінокислот до утвореная кислотоамідних зв’язків, що лежить в
основі формування пептидів і білків, біохімічні пертворення амінокислот
в живих організмах та деякі хімічні реакції амінокислот, що використа
ну ються в аналітйчшй біохімії та фармації для ШіДеігШфЙМХйГта кількіспого визначення.
/. Утворення кислотоамідних (пептидних) з в ’язків.
Характерною хімічною властивістю протеїногенних амінокислот є
здатність їх а-карбоксильних груп взаємодіяти з а-аміногрупами інших
амінокислот з видіДШням елементів молекули водй та утворенням кис
лотоамідних і^шнтидщш) зв’язків. Продукти, іЦо формуються (аміди
амінокислот), дістали назву пепт идів (дипепт идів* т рипепт идів
... олігопептидів) та поліпептидів (при кількості реагуючих молекул
291
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАЮЛІТИ
амінокислот більше десяти). Наведемо реакцію утворення дипептиду
при взаємодії а-карбоксильної групи амінокислоти аланіну з ос-аміногрупою амінокислоти цистеїну:
р-™-'---,
СНз
Нг^*- С Н -С~і_ОНг--й^’М
Д
°
ЭН
сн2
СН3
н
ЗН
сн2
СН~С_0Н — ^ Н2М — СН-С- N— СН-С-ОН
А.
і
о
0
0»
Утворений пептидного зв’язку в дипептиді аланіл-цистеїн
Реакція утворення пептидних зв’язків йде за механізмом нуклеофільного заміщення
(розділ 2.5.). При цьому нуюіеофільний реагент
(нуклеофільний центр амінокислоти, що вступає в реакцію - в наведено
му прикладі —їШ 2-група а-цистеїну), взаємодіє з позитивно зарядже
ним ( 8 +) атомом вуглецю амінокислоти —субстрату реакції (в розгляну
тому випадку —сс-аланіну), В результаті цієї нуклеофільної атаки сто
совно електрофільного центру а-аланіну відбувається заміщення гіді
роксильної групи на залишок амінокислоти з вільною а-аміногрупою (в
наведеному прикладі ос-цистеїну) та утворюється дипептид, в молекулі
якого залишки двох амінокислот сполучені кислотоамідним (пептидним)
зв’язком:
Ri *
L
H 2N -C H -C '
И
Л
R2
І І 1
- ШО
+ и >Ч-СН-СООН
h 2n
ин п
Ri
-
<
c h
-
c
Ö
-
Н R2
і '
m - c h - c o o h
Синтез пептидів
Хімічний синтез пептидів
У зв ’язку з широким застосуванням білково-пептидних препаратів у
медицині з метою діагностики та лікування, сільському господарстві,
харчовій промисловості, проблема штучного синтезу пептидів та білків
має велике наукове та практичне значення. Вперше пептидний синтез
був здійснений видатним німецьким хімішм-органіком Емілем Фішером
(E. Fischer; 1852-1919), якийу 1901 р. отримав дипептид гліцил-гліцин, а
в 1903 р. запропонував хлорангідридний метод утворення пептидів.
2 92
4.3. Білки, пептиди. Амінокислоти
При штучному хімічному синтезі природних пептидів застосовують
такі загальні методичні підходи:
1) метод послідовного нарощування амінокислотного ланцюга, що
дає змогу отримана невеликі (п » 9 - 1 0 ) пєптидні молекули; ,
2) метод блочного синтезу, який полягає у з ’єднанні в одну молекулу
кількох Малих пептидів (п = 9-10), що були отримані послідовним наро<щуванням пептидного ланцюга.
З метою створення оптимальних умов для формування кислотоамідних зв’язків між окремими амінокислотами усі методи хімічного синтеї
зу пептидів та білків грунтуються на послідовному здійсненні таких трьох
стадій (Ю. А. Овчинников, 1987):
а)
блокуванні (“захисті**) функціональних груп амінокислоти (аміно
кислот), або пептиду (пептидів), що не беруть участі в реакції утверенпя пептидного зв’я зк у ;'
б) конденсації активованої Карбоксильної групи одного реакційного
компонента з вільною аміногрупою іншЬго компонента;
в)
видаленні захисних груп для отримання вільного пептиду (кінче
ного продукту) або продовження синтезу.
Активацію' карбоксшіьної групи в амінокислотах та пептидах здій
снюють за рахунок утворення хлорангідридів, азидів, арильних ефірів,
їм ішаних ангідридів при взаємодії карбоксилу з похідними вугільної кис
лоти, е^илхлорформіатом.
Захист аміногрупи здійснюють шляхом взаємодії амінокислоти або пе
птиду з бензилоксикарбоніпхлоридом аботирега-бутипокснкарбонілхлоридом.
Захист карбоксильної групи здійснюють за допомогою бензилброміду, ізобутену тощо.
Наведемо схему хлорангідридного методу синтезу пептидів в умопах попереднього захисту аміногрупи першої амінокислоти (2 ,), карбок
сильної групи другої амінокислоти
та активації карбоксильної групи
першої амінокислоти:
О
293
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Твердофазний синтез пепт идів
Важливим внеском у проблему штучного синтезу пептидів став за
пропонований Р. Мерифілдом (її. МеггШеИ) метод твердофазного син
тезу. Головна ідея цього методу полягає в закріпленні поліпептидного
ланцюга, що утворюється, На полімерному нерозчинному носії, поверх
ня якого містить хлорметияьні “якірні” групи. За допомогою розробле
ного методу Р. Мерифілд здійснив синтез таких природних пептидів, як
брадикінін (нанопептид), ангіотензин (декапептид) та перший штуч
ний синтез ферментного білка - рибонуклеази, що містить 124 аміно
кислотних залишки.
В методі рідшфазного синтезу, запропонованому в 1965 р. академіком
М, М. Шемякіним, вперше був застосований розчинний носій полістирол,
що дало змогу суттєво збільшити швидкість реакцій пептидного синтезу.
Біохімічні р е а к ц ії синтезу пептидів
Синтез білків та пептидів в живих організмах здіснюється за участі
складної мультимолекулярної субклітинної системи, що включає струк
турний аппарат (білки, рибонуклеїнові кислоти)рибосом, комплекс при
родних а-Ь-амінокислот та сукупність специфічних ферментів - процес
трансляції. Визначальною особливістю рибоеомального синтезу білків
є його матричний характер, тобто включення кожного амінокислот
ного залишку в певне місце поліпептидного ланцюга кодується трипле
том нуклеотидів відповідної матричної (інформаційної) РНК, що, у свою
чергу, визначається комплементарною послідовністю нуклеотидів у ге
нетичній ДНК клітини.
Безпосередня взаємодія окремих а-£^амінокислотз рибосомальним
апаратом трансляції відбувається у вигляді комплексів амінокислот із
спеціальними транспортними молекулами РНК (тРНЕС ~ розділ 4.4,), що
утворюються заучасті аденозинтрифосфорної кислоти (АТФ). Цей про
цес дістав назви активації амінокислот в ході біосинтезу білка і скла
дається з таких етапів:
а) взаємодія амінокислоти з ДТФ з утворенням аміноациладенілату.
?"
нум— С Н -С ^
о
ОН
Г
+ АТФ “ ^ Н2М—с н —с х
+ ФФп
ІО — АМФ
4.3. Білки, пептиди. Амінокислоти
б)
перенос амінокислотного з а л и ш к у з аміноациладенілату натРН К
з утворенням комплексу амінокислоти з тРНК ^ аміноацил-тРНК'.
Вп
,0
1
^Р
н2іч—сн-сг
н2іч—сн-с*
о - ЧВМАП
О — АМЯ
В подальшому аміноацил-тРНК взаємодіє з рибосомою, до якої вже
прикріплений комплекс пептиду, що синтезується, з іншою тРН К - пептидил-тРНК довжиною (п-1) амінокислотний залишок. В ході цієї взає
модії пептидний фрагмент довжиною (п-1) амінокислотний залишок пе
реноситься на комплекс нової —п-ої амінокислоти з тРНКп(амііюацилптРНК ) з утворенням наступного пептидного зв ’язку. В результаті*розглянутого процесу відбувається подовження поліпептидного ланцюга, що
синтезується на рибосомі, на один амінокислотний залишок —реакція
елонгації (подовження - анга.) - рис. 4.5.
(п -1 )
1
.0
Н2М-
-сн— с^
Г
І
І °І
І
+ Н2И- - с н —с -нІ м н — с н —с г 'О-^—
о- іЗДЧАп.і
о — іеша„
?(2-п)
Н2Ы—
І
сн— с -и№ — сн— с;V
І
V
° [г
+ АКАП
’О- *- ®№АП
□
л г
(и)
Рис. 4 .5 . Схема процесу подовження пептидного ланцюга
{елонгації) в рибосомі
Детальніше процеси біосинтезу природних білків та пептидів розгля
ди ються в курсах біологічної хімії та молекулярної біології.
295
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
■2. Біохімічні перетворення амінокислот
Головним джерелом природних а-Ь-амінокислот для організму люг
дини є білки харчових продуктів. Проте частина природних амінокислот
може синтезуватися в організмі (“замінні” амінокислоти), тоді як інші
(валін, лейцин, ізолейцин, треонін, метіонін, лізин, фенілаланін) не синте
зуються (“незамінні’’ амінокислоти); деякі амінокислоти (аргінін, гісти
дин) синтезуються лише частково (“умовно незамінні” амінокислоти).
Вільні амінокислоти за участі специфічних ферментів використову
ються для внутрішньоклітинного біосинтезу білків та пептидів або всту
пають у біохімічні реакції, що ведуть до утворення різних необхідних;
для метаболізму інтермедіатів —переважно беазотистих продуктів обміну речовин (карбонових кислот, гідрокси- та кетокислот, спиртів тощо).
Біохімічні перетворення амінокислот в організмі поділяються на такі,
що є загальними майже для всіх амінокислот, та специфічні для окре
мих амінокислот щдяхй перетворення. До загальних шляхів метаболіз
му амінокислот належать реакції дезамінування, трансамінування та
декарбоксш ю вання.
Д езамінування амінокислот
В біологічних об’єктах можливі такі типи реакцій дезамінування амі
нокислот (відщеплення аміногрупи з утворенням аміаку):
а)
відновне дезамінування:
В — С Н -С О О Н
+ 2Н +
------- ►
И — С Н 2 — С О О Н + |\ІН 3
Ж 2
Карбонова кислота
б)
гідролітичне дезамінування:
+ Н20
И— «С Н -С О О Н
і
ІЧН2
------ ►
ІЗ— С Н — СООН + ІМНз
і
ОН
І’ідроксикислота
2 96
4.3. Б іл к и , пеп ти ди . А м ін ок и сл оти
в) внут ріш ньом олекулярне дезам інування
-Ж 3
я—сн—с о о н — і* Я—с н = с н + ічн3
І
ІЧНо
Ненасичена карбонова кислота
г) окислю вальне дезам інування:
+ 1/2 02
я — С Н - С О О Н --- *- в — С — С О О Н + N N 3
І
І!
N42
О
ое-кетокислота
- ,
Для тваринних організмів; переважним типом дезам інування ам іно
кислот є окисне, хоча виявляються І ін ш і типи дезам інування (Напри
клад, гістидин у печінці та шкірі м ож е зазнавати внутріш ньомолекуляриого дезамінування). Р ізн и х типів дезам інування зазнають ам інокисло
ти за д ії ферментів мікроорганізмів, щ о містяться в товстом у кишечни
ку людини (“м ікробне гниття білків” у кишечнику).
М еханізм окисного дезамінування ам інокислот .
Окисне дезамінування амінокислот здійсню ється в два етапи:
1) дегідрування а-ам інокислоти з утворенням імінокислоти (фермен*
тнтивна реакція):
-2 Н
Я— СН— СООН
—
►
и
ЬІНг
а-амінокисяйта
| — СООН
ИН
Іміяекиеяота
2) гідролітичне розщ еплення ім іну з утворенням (^кетокислоти (неферментативна стадія реакції окисного дезамінування):
р — ц — -СО О Н
N4
Імінокислота
+ Н20
------- ІМкг Я— С
СООН + N43
О
а-кетокислота
297
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Найбільш поширеною реакцію окислювального дезамінування є де
замінування L-глутамінової кислоти з утворенням ос-кетогаутаровоїки
слоти —важливого інтермедіату окислювального метаболізму в мітохондріях. Реакція каталізується ферментом h-глутаматдегідрогеназою,
коферментом якого є нуклеотадний кофермент НАД+(нікотинамідаденіндинуклеотид; NAD):
со
он
1
СО О Н
СНо
NAD
1
сн2
О
NADH 2
*
1
соон
Н20
сн2
Q
сн2
І
не— nh2
І
соон
NH3 сн2
сн2
І
с=о
C=N H
1
соон
соон
■гпутамінова
кислота
Іміноглутарова
кислота
а-кстоглутарова
кислота
Р еакц ії т рансам інування
Трансамінування - реакція міжмолекулярного перенесення аміно
групи —Ї*Ш2 від амінокислоти на ос-кетокислоту з утворенням нової ке
токислоти та нової амінокислоти:
Ri
Н С — NH 2 #
I
соон
R2
^1
С =*0
0 = 0
I
соон
CO OH
fp2
+
Н С — NH 2
I
I
'
CO O H
Процес трансамінування є оборотним і найбільш активно відбувається
за участю хоча б одній дикарбонової зміно- чи кетокислоти, наприклад:
298
4.3. Білки, пептиди. Амінокислоти
СООН
соон
ї
сн2
І
І
СН 2
СНз
н о — -ЫН2 + с
еІо о н
а-аланін
І
сн2
СНз
СН2
с = о
о
+
І
соон
Іс о_о н
І
соон
Піровиноградна
кислота
а-кетогаутарова
кислота
н е — мн2
^іяут&мінова
кислота
Реакції трансамІнування каталізуються ферментами амінотрансфе
разами (трансаміназами), коферментом яких є фосфорильований піридоксаль (альдегідна форма вітаміну В6 - піридоксину). Під час ката
літичного циклу відбувається поперемінне перетворення піридоксальфосфату на пірйдоксамінфосфат (коферментні форми вітаміну В6).
V
НОч ^ к ^ С Н г О -
СН2ЫН2
/О Н
,0Н
=0
с н 2о -
но
-р % 0
^ о н
Ч0Н
Н3с
Н зС ^ы "
N
у
Пірйдоксамінфосфат
Піридоксальфосфат
Д екарбоксилю вання амінокислот
Декарбоксилювання амінокислот з виділенням діоксиду вуглецю С 0 2
відбувається шляхом розщеплення С -С -зв’язку між сс-вуглецем та кар
боксильною групою. Реакції декарбоксилювавння с незворотними:
и — с н —соо н
І
ын2
а-амщокислота
— ►
СН2— МН2 +
С02
Біогенний амін
299
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Біогенні аміни - продукти декарбоксилювання протеїногенних аміно
кислот в організмі, належать до фізіологічно активних сполук—гормонів,
нейромедіаторів. Наприклад, придекарбоксилюванні похідних ароматич
них амінокислот утворюються катехоламійи адреналін та норадреналін, при декарбоксилюванні Ь-глутамінової кислоти т у-аміномасляна
кислота (ГАМК) - нейромедіатор з гальмівною активністю, при декарбоксилюванні Ь-гістидину—тканинний гормон та медіатор гістамін:
^-гістидин-'
<
г
І
Гістамін
Ферменти, що каталізують реакції декарбоксилювання амінокислот декарбоксилази; їх коферментом-є фосфорильована альдегідна форма
вітаміну В6 -піридоксальфосфат.
Шмічні реакції, що використовуються для аналізу амінокислот
Завдяки різноманітності своїх функціональних груп, молекули сс-амінокислоТ можуть вступати в хімічні реакції, що застосовуються в ана
літичній та клінічній біохімії для ідентифікації і кількісного визначення
окремих амінокислот. Ці реакції (так звані “кольорові реакції”) викорис
товуються як для визначення вільних амінокислот, що містяться в
біологічних об єктах (плазмі крові, сечі та ін.), так і при аналізі аміно
кислотного складу білків та пептидів, що може використовуватися як в
біохімії, так і в фармацевтичній хімії.
1) Н інгідринова реакція
Н інгідрин (трикетогідринденгідрат) є високочутливим реактивом на
а-амінокислоти, що при нагріванні утворює комплекс синьо-фіолетового
забарвлення з максимумом поглинання при
—570 нм. За допомогою
нінгідринової реакції можна детектувати 1 нмоль амінокислоти.
2) Ф луорескамінова реакція.
Високочутливим реагентом на а-амінокислоти є також флуорест мін, який утворює з амінокислотами флуоресціюючі комплекси. Флуо-
4.3. Білки, пептиди. Амінокислоти
рескамінова реакція є чутливішою, ніж нінгідринова, і дозволяє визнача
ти амінокислоти в кількостях 10-50 пмолей.
О
Нінгідрин
Флуорес камін
Спектрофотометричне або спектрофлуорометричне вимірювання
комплексів амінокислот з нінгідрином або флуорескаміиом дозволяє кіль
кісно визначати амінокислоти не тільки як вільні метаболіти, а і у складі
білкових гідролізатів після їх хроматографічного розділення, що викори
стовується в аналізі первинної структури білків та пептидів.
Крім зазначених, в клінічній біохімії застосовують такі кольорові ре
акції амійокислот, як:
— ксант опрот еінова реакція
характернаедЛя бензольного ядра
циклічних амінокислот (фенілаланіну, тирозіну,триптофану),яке нітрусться при дії концентрованої азотної кислоти з утворенням нітросполук
жовтого кольору;
- реакція М ілона - специфічна реакція на тирозин (амінокислоту,
що містить фенольний гідроксил), В умовах нагрівання фенолів Та їх
похідних з реактивом Мілону (суміш нітратів ртуті (І) та (II) утворю
ються ртутні похідні цегляно-червоного кольору;
■ реакція Сакагучі - реакція, щ о застосовується для ідентифікації
гуанідинової групи аргініну. При взаємодії гуанідину з й-нафтолом та
гіпохлоритом натрію в лужних умовах утворюються сполуки з черво
ним забарвленням;
■ реакція Е рліха - застосовується для виявлення індольного кіль
ця триптофану, яке при реакції з р-диметиламінобензальдегідом. в кис
лому середовищі дає сполуки з фіолетовим забарвленням;
301
Р озділ 4. БІОМОЛЕКУЛЙ ТА МЕТАБОЛІТИ
- реакція Фоля - реакція, характерна для сірковмісних амінокислот.
При кип’ятінні розчину білка або відповідних амінокислот а лугом в прису
тності плюмбіту нагрію утворюється чорно-бурий осад сульфіду свинцю.
ЙЗкИ Ст рукт урна організація білків т а пептидів
Всі білки та пептиди мають унікальну тривимірну просторову органі
зацію (конформацію), яка сосновою виконання білком його специфічних
біологічних функцій.
Високовпорядковані конформації білкових молекул стабілізуються за
рахунок утворення між амінокислотними залишками певних ділянок поліпептидних ланцюгів міцних ковалентних зв’язків та слабких фізико-хімічних зв’язків та взаємодій.
Типи зв язків у молекулах білків та пептидів.
1. К овалент ні зв'язки.
1. 1 . Пептидні з в ’язки— утворюються внаслідок взаємодії між «-кар
боксильними та а-аміногрупами амінокислот, що утворюють пептидний ланцюг.
1 .2.
Дисульфіди} зв язки (—8 —8 —) ~ утворюються між залишками мо
лекул цистеїну, що входять до одного або різних нептидних ланцюгів.
2. Н ековалент нізв’язки т а слабкі взаємодії—фізико-хімічні зв’яз
ки, що приймають участі» у взаємодії як певних частин одного, пептид
ного ланцюга, так і різних, близько розташованих- ланцюгів, утворюючи
вищі рівні конформації білкових молекул.
2 .1 .
Водневі зв язки — виникають між двома електронегативними
атомами за рахунок атома водню, ковалентно зв’язаного з даним із елек
тронегативних атомів. В молекулах білків водневі зв’язки найчастіше
утворюються між воднем» що входить до складу груп - Ш , -О Н , - 8 Н
та сусіднім атомом кисню. Водневі зв’язки можуть сполучати між собою
ділянки одного й того ж поліпептидного ланцюга чи різні поліпептиди.
Водневі зв язкй:мають особливо велике значення для утворення вищих
рівнів структурної організації білкових молекул.
О — н —о =
с
Схема утворення водневих зв’язків у молекулах білків та неитиада
302
4.3. Білки, пептиди. Амінокислоти
2.2. Іонні (сольові) зв’язки - сполучають між собою іонізовані амінні
та карбоксильні групи (головним чином, бічних радикалів діаміно- та
дикарбонових амінокислот).
2.3. Дипольні зв ’язки {або Ван-дер-ВашіьсОві сили взаємодії) електростатичні взаємодії постійних чи індукованих диполів, які можуть
утворюватися між радикалами полярних амінокислот (серину, треоніну,
цистеїну, тирозину та ін.), що входять до складу білковйх м о л е н а .
2.4. Гідрофобні взаємодії - слабкі взаємодії, що виникаючііміж Ііч11 ими радикалами неполярних амінокислот за рахунок ЇХ виштовхуван
ня” з полярної (зазвичай, водної) фази. Гідрофобні зв'язки виникають
здебільшого між радикалами таких амінокислорт, як валін, лейцин, ізо
лейцин, фенілаланін та ін.
Рівні структурної о рган ізац ії білків
1. П ервинна структ ура білків.
Під п е р в и н н о ю структурою білків розуміють лінійну будову пепти
дного (поліпептидногр) ланцюга, що складається із залишків сх-Ь-амінокислот. В поняття первинної структури білка або пептиду входять його
якісний, кількісний амінокислотний склад та порядок чергування (послі
довність) окремих амінокислотних залишків.
Загальнийвигаяд п е р в и н н о ї структуриліоліпептид ного ланцюга,
/"щ о складається з п-амінокислотних залишків
303
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Н3С
/С Н з
сн
єн
Первинна структура тетрапептиду аланіл-цистеїніл-лейцил-гліцин
При утворенні назв пептидів, що визначають їх первинну струїсгуру
першим вказується залишок амінокислоти, що має вільну а-аміногрупу
(так званщ М -инцевої амінокислоти”; у назвах всіх амінокислот; що
еруть участь в утворенні пептидного зв ’язку сс-карбоксильною гру
пою, закінчення "-ин (-ін)” змінюється на “ -ил Щ
амінокислота, яка
має вільну «-карбоксильну групу (“С-кінцева амінокислота”) свого за
кінчення (-И Я , -ін) не змінює.
Користуючись системою міжнародних скорочених символів, наве
демо приклади первинної структури деяких коротколанцюгових пептидів
що мають певну біологічну активність.
Агд-Рго-Ргр-РЬе-Зег-Рго-РІїе-Агд
Брадикінін (окгапептид із судиішодилатуючою активністю)
Туг-<ЗІу-ОІу-Р(іе-МеІ
Туг-в1у-6Іу-РІіе-І_еи
Ендорфіни(мет~енкефаліншлей-енкефалінш •
пептиди головного мозку з оніатоподібною активністю)
1
Скорочені записи пептидів мають особливе значення для позначення
білків, до первинної структури яких входять багато сотень амінокислот
них залишків.
304
4.3. Б іл к и , п еп т и ди . А м ін ок и сл оти
Будова пєпт идної групи
Чотири атоми угрупування -СО~ІЧН— складають пептидну групу,
особливостями структури якої є такі:
— всі чотири атоми пєптидної групи -С О -Ы Н - розміщені в одній
геометричній площині (компланарні);
чя групи С = 0 та N -11 знаходяться в транс-пояожтні;
- довжина зв’язку між атомами С і N (0,132 нм) є проміжною між
такою для одинарного С—Ы (0,147 нм) та подвійного С=Ы (0,127 нм)
зв’язків; це означає, що реальною формою цього зв’язку є резонансна
структура:
Н
О
Резонансна будова пептидного зв’язку
- вільне обертання в поліпептидному ланцюгу з утворенням різних
конформацій білкової молекули можливе лише навкруги груп -С Н Я
сусідніх амінокислотних залишків.
Виходячи з розглянутих особливостей будови пєптидної групи, наве
демо модель будови фрагменту первинної структури пептидного ланцюга
з урахуванням транс-положення груп СО-ЬІН та валентних кутів між
окремими атомами:
305
Розділ 4. БЮМОЛЕКУЛИ ТА МЙТАГОЛГТИ
У зв’язку із здатністю окремих функціональних груп пептидного
ланцюга до міжмолекулярних взаємодій, пептидний ланцюг набуває
певної просторової структури. Розрізняю ть два рівні Просторової
організації пептидного ланцюга (та відповідно, білкової молекули, що
складається на його основі) ** вторинна та третинна структури'.
Вт оринну, т рет инну Та четвертинму (див. ниж че) стр у кту р и
позначаю ть разом, як вищі рівн і сяпруктурної організації білкових
молекул,
2. Вт оринна структура білків.
Вторинна структура білків —це ряд упорядкованих конформадай*
утворення яких зумовлено водневими зв’язками між окремими ділянка
ми (переважно, дептидними групами) пептидного ланцюга або різними
пептидними ланцюгами.
Рис. 4 .6 . Модель вторинної структури поліпептидного ланцюга у вигляді а-спіралі
(за Л. Полінгом та Р. Корі)
306
4.3, Б іл к и , и еи ти ди . А м ін ок и сл оти
Розрізняють такі основні типи впорядкованої вторинної структури
природних білків: а-спіраль, спіраль колагену та ß -структуру.
а-спіраль - конформація, яка утворюється при просторовому скру
чуванні поліпептидного ланцюга за рахунок водневих з в ’язків, що вини
кають між С = 0 - та N H - групами поліпептидного ланцюга, які відда
лені одна від одної на чотири амінокислотних залишки. Водневі зв ’язки
її а-спіралі направлені паралельно осі молекули.
а-спіраль можна уявити собі у вигляді лінії, що йде по боковій по
верхні уявного циліндру. Напрямок обертання поліпептидного ланцюга в
природних білках - правий (“права” а-спіраль). Геометричні параметри
а-спіралі: радіус - 0,25 нм; крок спіралі - 0,54 нм; на один оберт а-спіралі
припадає 3,6 амінокислотних залишків (рис. 4.6.).
Спіраль колагену,
Формування а-спіралі залежить від просторових (стеричних) взає
мовідносин між амінокислотними залишками, щ о утворюють поліпептидний ланцюг. Окремі амінокислоти (Pro, Gly, Glu, Asp, Arg та ін.) про
тидіють утворенню а-спіралі або дестабілізують її.
У зв’язку із зазначеним можливе утворення спіральних структур,
що за своїми геометричними параметрами відрізняються від агспірані.
І Ірикладом t спіраль білка колагену “ головного білкового компоненту
сполучної тканини, особливістю первинної структури якого є постійний
повтор мотиву Gly-Pro:
-Gly-Pro-Met-Gly-Pro-Ser-.
Всього у складі колагену міститься близько 33 % гаіцину і 21 % про
ліну та гідроксипроліну.
ß-структура - структура типу складчастого шару, утворюється з
декількох зигзагоподібно розгорнутих пояіпептидних ланцюгів,,що роз
ташовані поряд (двох або більшої кількості).
ß-структури формуються: переважно за рахунок міжланцюгових водпепих зв’язків між групами -С = 0 та -N H сусідніх поліпептидів (рис. 4.7).
307
Р озділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Р и с. 4 .7 . р-конформація поліпептидного ланцюга. Схематично зображені три па
ралельні ланцюги, що утворюють структуру типу складчастого шару.
Прикладом білків Ц ^-конформацією є ^-кератини, що складаються
із зигзагоподібних антипаралельно орієнтованих поліпептидних ланцю
гів.- Представником Р-кератинів є фіброїн т фібрилярний нерозчинний
білок шовку та павутиння.
Крім впорядкових типів конформації (а-спіралі, спіралі колагену та
|3-структури) вторинна структура білка може являти собою нерегулярну,
невпорядковану (хаотичну) конформацію.
В багатьох природних білках на протязі одного поліпептидного
ланцюга присутні як а-спіралізовані ділянки, так І зони, що являють собою
складчастий шар ф-структури) або мають нерегулярну конформацію.!
Наприклад, в молекулі хімотрипсину до 14 % загальної кількості а м н
нокислотних залишків входить до складу а-спіралей, 45 % - (З-структур,
61 % —ділянок з невпорядкованою структурою.
С упервт оринні ст рукт ури
Декілька білкових молекул з вторинною структурою у вигляді спіра
лей можуть взаємодіяти одна з одною, утворюючи міжмолекулярні ком
плекси, що являють собою суперспіралізовані (“супервторинні”) струк
тури. Таким шляхом відбувається формування багатьох біологічно
308
4.3. Б іл к и , п еп т и ди . А м ін ок и сл оти
нижливих фібрилярних білків, наприклад мікрофібрия тропоколагену—
структурних одиниць колагенових фібрил сполучної тканини, молекул
Р-кератинів - основного типу фібрилярних білків, з яких побудовано
зовнішні покривні тканини (захисні покриття) хребетних тварин, зокрема
епідерміс, волосся, нігті людини.
3. Трет инна ст рукт ура білків.
Третинна структура білків являє собою спосіб укладання у тривимірно
му просторі поліпептидного ланцюга з певною вторинною структурою.
1) утворенні та стабілізації третинної Структури приймають участь
нодневі, іонні, гідрофобні зв’язки та взаємодії.
Залежно від форми та особливостей тривимірної просторовоїорганітц ії виділяють глобулярні та фібрилярні білки. ®
Глобулярні білки, —білки, що мають окрушу (кулеподібну або еліпсо
подібну) форму. Глобулярні білки можуть бути добудованими з одного або
декількох зв’язаних дисульфідними місточками поліпептидних ланцюгів,
що згорнуті у щільні кулеподібні форми. Це -альбум ін сироватки крові*
міоглобін м ’язів, гемоглобін еритроцитів, більшість фермейтйих білків.
Модель третинної структури білка міоглобіну, створену на підставі
рснтгеноструктурного аналізу білкових кристалів,'буЛ о вперше запропо
новано в 1958 р. англійським дослідником Дж. Кендр’ю (ГКепсІгеш)
(рис. 4.8).
Стабілізація компактної глобули реалізується за рахунок водневих
(переважно), іонних та дипольних зв’язків між бічними радикалами амі
нокислотних залишків, які фіксують відносно один одного певні частини
поліпептидного ланцюга (або декількох ланцюгів, сполучених З-Б -зв’язкими). Характер розташування полярних та неполярних амінокислотних
радикалів (йа поверхні або всередині білкової глобули) визначає фізико*
хімічні та біологічні властивості білків із третинною структурою, зокре
ма їх розчинність у воді, здатність до комплексування з ліпідами, вуглем
водами або іншимилїігандами,
Ф ібрилярні білки - білки, структурною особливістю яких є витяг
нута (подовжена) форма молекул. Такі білки схильні до утворення мультимолекулярних ниткоподібних комплексів - фібрил, що складаються з
док іл ькох паралельних поліпептидних ланцюгів.
309
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Р ис. 4 .8 . Просторова конформація міоглобіну із зображенням характеру втфИ№
ної ,структури поліпептидного ланцюга
Фібрилярні білки є зазвичай структурними компонентами сполучної або
інших опорних тканин організму. Прикладами структурних фібрилярних
білків є: колаген - найбільш розповсюджений білок організму лщцини, що
посідає до ЗО % загальної кількості тканинних білків; еластин сполучної
тканини; кератини покривних тканин (епідермісу, волосся та ін.).
Д оменні білки
Домени - структурні ділянки білкових молекул, що являть собою
глобулярні утворення в середині білків з третинною структурою. Окремі
домени є функціонально відносно автономними утвореннями у складі
білкових молекул, і доменні білки у цьому відношенні подібні до олігомерних білків. Але, на відміну від олігамерів (білків з четвертинною струк
турою - див. нижче), окремі Глобули в доменних білках утворюються
тим самим поліпептидаим ланцюгом. Ці доменні ділянки сполучені між
собою фрагментами єдиного поліпептидного ланцюга *»»так званими!
шарнірними ділянками, і тому зв' язки між доменами можна розіцеиитй
тільки за допомогою протеолітичних ферментів. Прикладами доменних
білків є ферменти гаіколітичного циклу окислення глюкози.
310
4.3. Б іл к и , п еп ти ди . А Ш нок ислотя
4. Чет верт инна ст рукт ура білків.
Білки з молекулярною масою більше 50 кД (в сотні тисяч кілодальтонів і більше), як правило, складаються з декількох субодиниць (протомерів), тобто являють собою складні міжмолекулярні агрегати - так
звані олігамерні білки. Такі білки називаються білками з четвертинпою структурою. Олігомернї білки мають зазвичай парну кількість
протомерів (2 ,4 ,6 ,8 ,1 0 ,1 2 ).
Окремі протомери (субодиниці) в білках з четвертинною структу
рою зв’язані нековалентними зв’язками; це спричиняє можливість їх
дисоціації при змінах фізико-хімічних умов середовища (зміні pH, іонної
сили розчинів, додаванні: сечовини). Разом з там , така дисоціація при
зводить до втрати специфічної для даного білка біологічної активності,
яка притаманна лише цілісному олігомерному утворенню, тобто білка з
четвертинною структурою.
Типовим представником білків, які мають четвертинну структуру, є
гемоглобін (НЬ) еритроцитів, що виконує функцію транспортера кисню
в організмі людини та вйщйх тварин. Молекулу гемоглобіну (м.м. = 68 кД)
побудовано з чотирьох попарно однакових субодиниць - двох а- та
двох (3-поліпептидних ланцюгів. Кожен з цих ланцюгів сполучений з небілковою сполукою гемам —порфіриновим похідним, що зв’язує моле
кулу кисню. Умовна формула гемоглобіну: НЬ = а 2(32 (рис. 4.9).
Прикладами ферментних білків з четвертинною структурою є такі
олігомерні білки: алкогольдегідрогеназа (2 протомери); піруваткінача (4 протомери); альдолаза (4 протомери), гдутамінсинтетаза (8 про
томерів). Протомери, що утворюють олігомерний комплекс (білок з че
твертинною структурою) можуть бути однаковими за будовою та біохі
мічними властивостями або різними. Наприклад, молекула ферменту
лакт ат дегідрогенази; ЛДГ, що каталізує зворотнє перетворення
піровиноградної кислоти на молочну, складається з чотирьох протомерів,
які містять два типи ПоліпейТидних ланцюгій; Н - серцевийтип (від англ.
heart - серце) та М - м ’язовий тип (від англ. muscle - м ’яз). Завдяки
різним сполученням цих субодиниць можливе існування п ’яти форм фер
менту - НННН (Н4), НННМ (Н3М), ННММ (Н2М2), РМММ (НМ3) та
ММММ (МД. Ці молекулярні форми ЛДГ, що відрізняються будовою та
каталітичною активністю, містяться переважно в різних органах і тка311
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
нинах; такі множинні форми одного й того ж ферменту називають ізо
ферментами. Визначення в сироватці крові людини окремих ізоферме
нтів ЛДГ має діагностичне значення у зв’язку з можливістю визначення
локалізації ураження патологічним процесом того чи іншого органа.
Рис. 4 .9 . Ч е т в е р т и н н а с тр у к т у р а м о л ек у л и г ем о гл о б ін у
Ф ізико-хімічні властивості білків
Кислот но-основні властивості білків
Завдяки наявності значної кількості іоногенних груд (а-амінні та
(Х-карбоксильні кінцеві групи, бічні радикали кислих та основних аміно
кислот) білкові молекули є амфотерними електролітами. Відповідно
до цього, білки у водних розчинах утворюють амфіонц, знак та заряд
яких залежить від їх амінокислотного складу та рН середовища. Подіб
но до вільних амінокислот, в кислому середовищі переважають катіонці
форми білгових молекул, в лужних - аніонні.
Змінюючи рН, можливо перевести біяок у стан, при якому сумарний
електричний заряд білкової молекули дорівнює нулю (ізоелектричний
стан). Відповідне значення рН дістало назву р Н ізоелектричноїточ
ки білка (рі). В складі білків плазми крові людини кількість аніоногенних амінокислотних залишків перевищує кількість катіоногенних залипі312
4 .3. Б іл к и , п еп ти ди . А м ін ок и сл оти
ків, тому для цих білків рі знаходиться в кислому середовищі, і при ней
тральних або слаболужниХ значеннях рН вони знаходяться у формі ані
онів. Лужними білками € ядерні білки гістони, що містять у своєму
складі значну кількість залишків позитивно заряджених амінокислот
аргініну та лізину.
Наявність заряду у молекул білків визначає ЇХ здатність до електро
форезу - руху в постійному електричному полі. Електрофоретична ру
хомість молекул білків залежить від їх заряду та молекулярної маси, що
дозволяє застосовувати метод електрофорезу для фракціонування склад
них білкових сумішей. Розчинніст ь білків
Розчинність окремих білків у різних фізико-хімічних сербдовищах
залежить від їх заряду та переважання в їх складі полярних або неполяр
них амінокислотних залишків.
Більшість глобулярних білків (зокрема, білків сироватки крові та інших
біологічних рідин) містять на своїй поверхні гідрофільні залишки поляр
них незаряджених або заряджених амінокислот, які взаємодіють з дипо
льними молекулами води, утворюючи навколо білкових молекул гідратні
оболонки, і тому вони легко розчинні в воді або слабких сольових розчинах
солей лужних металів.
Збільшення в розчинах білків вмісту катіонів металів та амонію су
проводжується нейтралізацією поверхневого заряду та дегідратацією
білкових молекул і осадженням певних білків (метод висолювання). З
цією метою найчастіш е використовуються концентровані розчини
сульфату амонію, сульфату натрію, хлоридів натрію та калію.
Д енат урація білків
Під денатурацією розуміють втрату білковою молекулою прита
манної їй просторової структури {нашивної конформації) та порушення
хнрактерних фізико-хімічних властивостей. Денатурація супроводжується
іииженням або втратою специфічної для даного білка біологічної актив
ності (ферментативної, гормональної та ін.).
Денатурація відбувається внаслідок дії на білкові розчини та білки,
що знаходяться в біологічних середовищах, жорстких хімічних, фізикохімічних та фізичних факторів. Денатурацію спричиняють: дія кислот,
лугів, органічних розчинників, нагрівання білків до 60-80 °С, дія високих
доз ультрафіолетового та іонізуючого випромінювання.
313
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Механізм впливу денатуруючих агентів полягає в руйнуванні слаб
ких зв’язків (водневих, іонних, дипольних, гідрофобних), що стабілізу
ють впорядковані типи просторової організації білкових молекул (вто
ринну та третинну структуру):
С кладні білки
Існують класи білків, які постійно зв’язані з певними небілковими
сполуками, що являються інтегральними структурними компонентами
цих білків. Такі білки, до складу яких входять приєднані ковалентними
або нековалентними зв’язками інші біомолекули або іони металів, дістав
ли назву скла д н и х білків. Білки, до складу яких входять лише залишки
амінокислот; сполучені в поліпептидні ланцюги, - це прост і білки.
Сполуки неамінокислотної (непептидної) природи, що входять до скла
ду складних білків - це вуглеводи, ліпіди, нуклеїнові кислоти, нуклеотиди, порфірини, іони металів, залишки фосфорної кислоти. Небілкова час
тина складного білка може бути зв’язана з білковою частиною (апо~
протеїном) як ковалентними, так і нековалентними (водневими, іонними^
гідрофобними) зв’язками. Небілкова частина, що сполучена з апопротеїном ковалентним зв ’язком, дістала назву простетичної групи.
В залежності від хімічної природи небілкової частини (вуглевод, ліпід-,
нуклеїнова кислота, пігментна сполука, залишок фосфорної кислоти)
складні білки поділяються на гяікопротеіни, ліпопротеїни, нуклеопрот еїни, хрвмопрот еїни, мет алопрот еїни, фосфопротеїни.
4.3.4, М ет оди виділення т а аналізу білків і пепт идів
В иділення т а ф ракціонування білків
Виділення індивідуальних білків з тканин, клітин та біологічних рідин
живих організмів (тварин, рослин, бактерій, сироватки крові людини) є
розповсюдженою біохімічною та біотехнологічною процедурою, що
широко застосовується з метою отримання лікарських засобів (гормо
нів, ферментів, імуношобулінів, інтерферонів, факторів росту тощо).
В иділення білків
При виділенні білків з біологічних об’єктів першим етапом процеду
ри є їх екстрагування за допомогою різних розчинників, вибір яких за
лежить від фізико-хімічних властивостей білка або групи білків, яку по
трібно отримати. В разі необхідності одержання білків, які локалізовані
в певних субклітинних органелах та зв’язані з біоструктурами, гіроцеду314
4.3. Білки« пеп ти ди . А м ін ок и сл от и
ра виділення білка включає в себе руйнування тканинних та клітинних
структур, диференційне центрифугування тканевих гомогенатів з метою
отримання ізольованих фракцій клітини (ядер, мітохондрій, мембран єн*'
доплазматичного ретикулуму, лізосом та ін.), переведення білків субклі
тинних фракцій в розчинний етан обробкою біоструктур детергентами
або сольовими розчинниками, осадження білків шляхом висолювання
або застосування таких дегідратуючих реагентів, як ацетон, етанол.
Ф ракціонування білків
Подальшим етапом виділення індивідуальних білків £ фракціонуван
ня білкових сумішей, яке здійснюється на основі відмінностей у фізико»
хімічнйх властивостях індивідуальних білків (молекулярній масі, заряді,
розчинності, хімічній та біохімічній активності), 3 цією метою застосо
вуються: метод ультрацентрифугування (осадження, або сєдименгГіації
окремих білків у швидкісних ультрацентрифугах), хроматографічні ме
тоди —гель-фільт рації (гел ь-хромат ографіі), іонообмінної хрома
тографії, хроматографії за спорідненістю білків до певних лігандів
(“афінна” хроматографія), електрофорез білкових сумішей.
Детально методи виділення та фракціонування білків Та пептидів
розглядаються в курсі біологічної хімії.
Визначення амінокислот ного складу і первинної
ст рукт ури білків т а пепт идів
А напа амінокислот ного складу білків
Аналіз амінокислотного складу (ідентифікації окремих амінокислот
них залишків та визначення їх кількісного складу) є першим етапом у
визначенні первинної структури білка чи пептиду.
Етапи аналізу амінокислотного складу білків (пептидів):
а) гідроліз пепт идного ланцю га білка чи пепт иду.
Гідроліз пептидного ланцюга здійснюється шляхом кислотного роз
щеплення пептиднихзв’язків. Стандартною процедурою єкип’ятіння білка
в НС1 (5,7 моль/л) при 105-110 °С протягом 24 год.
б) розділення амінокислот гідролізат у.
Здійснюється шляхом іонообмінної хроматографії компонентів гідролі
зату на сульфополістиролових смолах, що несуть на своїй поверхні каті
онні групіровки К - 8 0 3'Ка+. При пропусканні через хроматографічну ко
лонку, що заповнена сульфополістиролом, суміші амінокислот останні
315
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
сорбуються на певних рівнях колонки. Подальше промивання колонки !
буферними розчинами з різними значеннями рН приводить до вимивана і
ня (елюції) певних амінокислот.
в)
ідентифікація т а кількісне визначення окремих амінокислот
Здійснюється за допомогою реакції з нінгідрином або фпуорескамі-1
нам, що дають при реакції з амінокислотами відповідні кольорові або
флуоресціюючі комплекси (див. вище). Найбільш часто з цією метою застососвується нінгідрин, який при нагріванні з а-амінокислотами спричиняє |
їх декарбоксилювання з утворенням >Ш3/ С 0 2 та альдегіду ^продукту!
окислювального декарбоксилювання амінокислоти. В подальшому аміак*
що вивільнився, реагує з відновленим нінгідрином; комплекс, що утворю
ється, має максимум поглинання при 570 нм, що1дозволяє кількісно визнай І
чати а-амінокислоти, застосовуючи метод спектрофотометрії:
+ R -C H O + С 0 2
Иінгідринова реакція визначення а-амінокислот
Метод кількісного аналізу амінокислотного складу білків та пептидів
був вперше запропонований в 1958 р. У. Стейном (W. Stein) та С. Муром
(S. Moore) і в наш час реалізується за допомогою автоматичних аміно
кислотних аналізаторів.
Розш ифровка первинної ст рукт ури білків т а пепт идів
Визначення послідовності амінокислотних залишків в молекулах білків
та пептидів (розшифровка первинної структури) складається з таких етапів;
316
4.3 . Б іл к и , п еп т и ди . А м ін ок и сл оти
І) ідент ифікація N - т а С-кіпцевих амінокислот .
Оскільки в поліпептидному ланцюзі, що формує первинну структуру
будь-якого білка, на одному кінці розташована вільна a-N H 2-rpyna, а на
другому - вільна ос-карбоксильна група, визначення N- та С-кінцевих
амінокислотних залишків дозволяє оцінити кількість поліпептидних лан
цюгів, що складають дану білкову молекулу.
Ідент ифікація N -кінцевих амінокислот — здійснюється шляхом
використання хімічних реагентів, що ковалентно зв’язуються з кінцем
пою а-амінною групою пептидного ланцюга. Для взаємодії з N-кінцевими амінокислотами в білках та пептидах застосовують такі реагенти:
- 2,4-дипітрофторбензол (ДНФБ) - реагент, який, взаємодіючи з
а-аміногрупою кінцевої амінокислоти, утворює ДНФ-похідне білка чи
пептиду. Подальший кислотний гідроліз продукту арилювання пептид
ного ланцюга призводить до вивільнення ДНФ-похідного N-кінцевої
амінокислоти та суміші вільних амінокислот, які ідентифікують хрома
тографічно - метод Ф. Сенгера (F. Sanger);
—
М 02
^2-п
Н Ri
^ TN- Chf r *- {COOH+H2 N—С Н —СООН)(пи}
Схема утворення ДНФ-похідних N-кінцевих амінокислот за Сенгером
- дансил-хлорид (1-диметиламінонафталін-5-сульфохлорид, ДНС) реагент, що в результаті взаємодії з N-кінцевою амінокислотою утво317
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
рює ДНС-похідні білків та пептидів («дансилування пептидів»). Після
подальшого кислотного гідролізу ‘-міченого” пептиду модифікований К
термінальний амінокислотний залишок ідентифікується завдяки специі
фічній флуоресценції продуктів дансилування.
СІ
Дансилхлорид
(димєтиламінонафталінсульфонійхлорид)
Ідентифікація С-кінцевих амінокислот
здійснюється шляхом
ферментативного гідролізу білків та пептидів карбоксипептидазами
або методом гідразинолізу за Акаборі.
Карбоксипептидазнийметод використовує застосування протеолітичних
ферментів, які специфічно відщеплюють від поліпептидного ланцюга С-кінцеву
амінокислоту »подальшою її хроматографічною ідентифікацією.
Гідразиноліз за Акаборі (АкаЬоґі). Згідно з цим методом досліджу
ваний поліпептид обробляють гідразином ІШЬШШй що веде до розщеп
лення пептидних зв’язків і утворення гідразидів усіх амінокислот, крім
С-кінцевої. С-кінцева амінокислота лишається, не підлягає хімічній мо
дифікації іможе бути ідентифікованою в реакційній суміші.
2) розділення білкових м олекул на окремі поліпептидні ланцю
ги - шляхом відновлення або окислення дисульфідних ( в-Б )-зв’язків.
Відновлення Б -З -зв’язкІв здійснюють шляхом відновлення їх меркаптоетанолом або дитїотреїтолом до цистеїну, окислення **' обробкою надмурашиною кислотою з утворенням залишків цистеїнової кислоти.
3) фрагмент ація поліпептидного ланцю га та розділення пеп
т идних фрагментів.
Розщеплення досліджуваного білка чи пептиду на короткі фрагменти
здійснюється звичайно методом ферментативного гідролізу із заетосу318
4.3. К іл ки, п еп ти ди . А м ін ок и сл оти
м и ш і я м протеолітичних ферментів (трипсину, хім от рш єцму та ін.).
Утворені пептидні фрагменти складаються з 10-20 амінокислотних
І В Л и ш к і в і можуть бути розділені за допомогою хроматографічного
методу (зокрема, методу високоефективної р ідинної хромат ограф ії
■ ВЕРХ) або методу електрофорезу.
4)
визначення амінокислот них П ослідовностей в окрем их пеп*
пик)и их фрагмент ах.
Основним сучасним методом розшифровки амінокислотних повлідониостей є метод ступінчастої деградації поліпептидних ланцюгів фенШ зо-тіоціанатом (ФІТЦ). Метод був запропонований шведським
біохіміком П. Едманом (Р. Есітап, 1950—1956 рр).
Унікальною особливістю методу Едмана є послідовне Скорочення
досл іджуваного пептиду з ІЧ-кінця на один мономер без руйнування иепТидпих зв’язків між іншими амінокислотними залишками. Таким чином
стік: можливим повторення зазначеної послідовності операцій та аналіз
вього пептиду шляхом поступового відщеплення та ідентифікації його
^кінцевих амінокислотних залишків:
Вп
/ \ А , СООН
п
Фон ілізотіоціанат
Пептид (п амінокислотних залишків)
ОН'
Вп
1
/ \ А . < ■СООН
І4 Н ^ -М Н -С Н ^ —
о
н+
319
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Фецілтіогідшшгїнова
похідна N-кінцевої
амінокислоти
Залишок пептиду
( ( n - І) амінокислотних залишків)
Автоматизація методу Едмана, що реалізована в спеціальному приладі“сіквенаторі” (англ. “sequence” - послідовність) дозволяє аналізувати про-]
дукги білкового гідролізу і природні пептвди з високою ефективністю. ...
4.4.
Н уклеотиди. Нуклеїнові кислоти
Нуклеїнові кислоти - це полінуклеотиди, що складаються з моно-|
мерних ланок - нуклеотидів (мононуклеотидів). Вони поділяються на j
два класи - дезоксирибонуклеїнові кислоти (ДНК) та рибонуклеїнові ]
кислоти (РНК).
Відкриттям нуклеїнових кислот людство зобов’язане швейцарсько- і
му лікарю та досліднику Фрідріху Мішеру (F. Miescher, 1844-1895), який!
вперше виявив у клітинних ядрах (nucleus) фосфатвмісні сполуки кислого!
характеру (1869 p.).
Нуклеїнові кислоти є Високомолекулярними сполуками з молекулярні
ною масою від декількох тисяч (транспортні Р Н К ) до кількох мільйонів J
дальтон (ДНК еукаріотів). Це - біоподімери, які разом з білками нале- j
жать до класу інформаційних біомакромолекул. Нуклеїнові кислоти '
виконують ряд унікальних біологічних функцій, не властивих іншим біо- і
полімерам: забезпечують зберігання і передавання нащадкам спадко
вої інформації, беруть безпосередню участь у механізмах реалізації цієї 1
інформації шляхом програмування матричного синтезу всіх білків інди- і
відуального організму.
Сукупність біологічних функцій нуклеїнових кислот та механізмів їх реалі-1
зації складають потік генетичної інформації в живих організмах'.
ДНК —> РНК
320
білок
4.4. НуклеотИди. Нуклеїнові кислоти
Иуклеотиди є структурними компонентами (мономерними ланка
ми) молекул нуклеїнових кислот ДНК та РНК. За хімічною будовою
Нйііи є трикомпонентними сполуками, що складаються 3 азотистої осноїїи пуринового чи піримідинового ряду, залишків пентоз (рибози або
дезоксирибози) та фосфату.
4.4.1. Будова, власт ивост і та ф ункц ії нуклеот идів
Ст рукт ура нуклеот идів
За умов повного гідролізу нуклеїнових кислот (кислотного або луж
ного) вивільняються пуринові та піримідинові азотисті основи, пентози
(0-рибоза або 2’-дезокси-0-рибоза) та фосфорна кислота.
В основі структури азотистих основ нуклеотидів лежать гетероциклічні
вполуки пурин та піримідин.
Н
Пурин
Піримідин
П уринові основи нуклеїнових кислот
І Іуриновими основами, що входять до складу нуклеотидів н
них кислот, є: аденін (А) та гуанін (Г), що мають таку будову:
О
Аденін (6-амінопурин)
Гуанін (2-аміно-6-оксипурин)
П іримідинові основи нуклеїнових кислот
До складу нуклеотидів нуклеїнових кислот входять три головні ріриМіди нові основи: цит озин (Ц), урацил (У),т и м ін (Т):
321
Розділ 4. БЮМОЛЕКУЛИГ ТА МЕТАБОЛІТИ
О
NN
О
HN '
HN'
і/*
н
Цитозин
УрацйЯ
(2,4-діоксипіримідин)
(2-окси-4-амінопіримідин)
А
Р ;.
Тшін
(5-метил-2,4-дюксипІримідин)
Оксипохідні пурину та ліримідину, залежно від рН середовища, мо
жуть перебувати в двох таутомерних формах —лактамних (оксоформах) і лактимних:
О
ОН
N
НО
HN
N
Н
N
Л акти м н а ф о р м а
Л актам н а ф орм а
г _____
Урацил
У складі нуклеотидів нуклеїнових кислот всі оксипохідні пурину та пірим ідину перебуваю ть у лактамній ф ормі, що сприяє утворенню
міжмолекулярних водневих зв ’язків між пуринами та піримідинами
окремих ланцю гів у дволанцю говій структурі молекул ДНК та в
одноланцюговйх РНК.
Н уклеозиди - двокомпонентні біоорганічні молекули, що складають
ся з азотистої основи пуринового чи піримідинового ряду та пентози
(Р-рибози або 2 -дезокси-Б-рибози) рибонутеозиди та дезоксирибонуклеозиди відповідно:
322
4.4. Н ук л еоти дн . Н у к л еїн ов і кислоти
Загальні формули рибонукяеотидів (1) та дезоксирибокуклеотидів (2). В - азотиста основа {base - анга.)
„
Зи своєю хімічною будовою нуклеозиди є N-піікозидами рибози або
дезоксирибози та азотистої основи. В утворенні відповідних N -гдікозидНИХ зв’язків в піримідинових нуклеозвдах бере участь N-1 піримідину та
Б> 1 пентози, а в пуринових - N-9 пурину та С-1 пентози.
Рибонуклеозиди:
О
NH;
-N
N
N'
HN
N
%
H 2N ^ ^ N
Гуанозин
323
Розділ 4. БІОМШЕКУЛИ ТА МЕТАБОЛІТИ
О
ОН
он
ОН
он
Уридин
Цитидин
Дезоксирибонуклеозиди:
О
NH;
N
N
If
Дезоксиаденозин
О
Декзоксицитидии
324
Дезокситимідин
4.4. Н ук л еоти д и . Н ук л еїн ов і к и сл оти
У зв’язку з можливістю вільного обертання навколо ^глікозидного
іи'й'іку, нуклеозиди (та нуклеотиди, що їм відповідають - див. нижче)
Можу ть знаходитися у анти- та сни-конформаціях:
Вільні пуринові нуклеозиди (та нуклеотиди), що знаходяться в розчині,
Можугь набувати як син-, так і анти-конформащї, тоді як для вільних піриміДИнових нуклеозидів (та нуклеотидів) переважаючою є шши-конформація. В
ТОЙже час у складі нуклеїнових кислот (особливо у двоспіральних молекулах
ДНК та РНК) просторове взаєморозташування азотистих основ та пентоз
відповідає антм-конформації, що є необхідною стеричною умовою для
утворення водневих зв’язків між комплементарними нуклеотидами.
Нуклеотиди. Фосфорилювання (ацилювання фосфорною кислотою)
Певного гідроксилу в пентозі, що входить до складу нуклеозиду, призводить
до утворення нуклеотиду (нуклеозидфосфату). До складу нуклеотидів (та
нуклеозидів) Д Н К входить 2 ’-дезокси-Б-рибоза, РН К -О-рибоза:
ОН
И
(-Н, -ОН)
Схема будови нуклеотиду
(рибонуклеозид-, дЄзоксирибонуклеозид-5’-монофосфату)
325
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Залежно від місця фосфорилювання пентозного гідроксилу розрізня
ють три типи нуклеотидів (нуклеозидфосфатів):
В результаті гідролізу нуклеїнових кислот, залежно від місця розщеп
лення фосфодієфірних зв’язків в молекулах полінуклеотидів, утворюються
нуклеозид-5'-фосфати та нуклеозид-3-фосфати (первинну структуру ДНК
та РНК див. нижче).
Крім різниці в пентозах, нуклеотиди РНК та ДНК розрізняються та
кож за складом піримідинових основ (в РИК?-*цитозин, урацил, в ДНКЛ
цитозин, тимін). Номенклатуру нуклеозидів та нуклеотидів (нуклеозид5'-моиофосфатів) ДНК і РНК подано в таблиці.
М індрні нуклеотиди
Крім зазначених вище основних п ’яти азотистих основ (двох пурино
вих та трьох піримідинових), до складу деяких нуклеїнових кислот вхо
дять у відносно незначних кількостях додаткові (мінорні) азотисті осйови та відповідні їм мінорні нуклеотиди.
Найбільша кількість мінорних нуклеотидів зустрічається в молеку
лах транспортних РНК (тРНК) —до 5% нуклеотидного складу. До мінор
них нуклеотидів належать метильовані дохідні звичайних азотистих основ,
наприклад 1-метиладенін, 1-метилгуанін, 3-метилцитозин. ДНК людини
містять значну кількість 5 -метилцитозину.
Нуклеотидами незвичайної структури в складі тРНК є дигідроуридин та псевдоуридин, в якому рибоза приєднана до урацилу в 5-му по
ложенні, тобто не азот-вуглецевим, а вуглець-вуглецевим зв ’язком:
326
1
№
і
я
1
в
о
в
а
о
№
3
3
1
%
о.
'б
Г уанш
Цитидий
Уридин
Дезоксиаденозин
Дезокси1 гуаиозин
Дезоксицитцдин
Тимідин
(А; А) ;
О
Тимін
( 1
Ш
Цитозин
«
о
■
8
К
‘І
я
&
в
]
Щ
(Г ;0 )
В
ЦМФ
УМФ
Цитидилова кислота (цнтидин-5’-фосфат)
Уридилова кислота (уридин-5’-фосфат)
Дезоксицитидилова кислота
(дезоксицитидин-5 ’-фосфат):
Тимідилова кислота (тимідин-5 ’-фосфат)
= Дезоксиаденілова кислота
і (дезоксиаденозин-5’-фосфат)
Дезоксигуанілова кислота
(дезоксигуанозин-5’-фосфат
ТМФ
дЦМ Ф
дГМФ
дАМФ
АМФ
ГМФ
позначення
нуклеотидів
Аденілова кислота (аденозии-5’-фосфат
Гуанілова кислота (гуанозин-5 ’фосфат)
РН К
Е4
Я
14
Пуринові:
Аденін
Аденозин
Гуаиозин
(А; А)
и 3
О
Цитозин
Урацил
Й
о
X
и
0
X
Пуринові:
Аденін
Гуанін
Н уклеотиди
а)
О
5
а
і
О
Скорочені
укр.; англ.
Нуклеози
ди
Номенклатура нуклеозндів 1 нуклеотмлів РНК та ДНК
4.4. Нуклеотиди. Нуклеїнові кислоти
327
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
5-метилцитозин
Дигідроуридин
Псевдоуридин
Біологічні функції мінорних нуклеотидів до кінця не з ’ясовані.
Деякі вільні рибонуклеотиди та їх похідні, що не входять до складу
нуклеїнових кислот, виконують функції коферментів, кофакторів, алостеричних ефекторів різних ферментних систем.
Б іохім ічні ф у н к ц ії віл ьн и х нуклеотидів:
1) участь в реакціях біосинтезу ДНК та РНК як попередників - дезокси-рибонуклеозидтрифоефати (дНТФ) дАТФ, дГТФ, дТТФ, дЦТФ
та нуклеозидтрифосфати (НТФ) АТФ, ГТФ, УТФ, ЦТФ, відповідно^
2 ) участь в енергетичному обміні - дифосфорильовані та трифосфорильовані нуклеотиди т. з. аденілової системи: аденозинтрифосфорна
кислота (АТФ) та аденозиндифосфорна кислота (АДФ).
МН2
ї ї )
О
О
а
-О — Р — ОН
ін
АДФ
АТФ
328
О
Ан
Ін
4.4. Нуклеотиди. Нуклеїнові кислоти
Утворення АТФ з АДФ та неорганічного фосфату (Фи) є централь
ною реакцією окисного фосфорилювання, що відбувається в мембраних мітохондрій і є головним процесом запасання (акумуляції) хімічної
енергії, яка вивільняється в результаті реакцій біологічного окислення:
АДФ + Ф„ - » АТФ.
Особливістю фосфоангідридного зв’язку між залишками фосфату в
(}- та у-положеннях молекули АТФ є висока стандартна вільна енергія
(ДО0) гідролізу або переносу у-фосфатної групи на інший акцептор:
АТФ -» АДФ + Фн
ДЄ° = - 7,3 ккал/м оль (- 30,5 кдж/моль).
Такі хімічні зв ’язки та біомолекули, що їх містять, дістали в біохімії
назву макроергічних сполук. Хімічна енергія, що вивільняється при
ферментативному розщепленні макроергічних зв’язків в молекулах АТФ,
є головним джерелом протікання інших ендергонічних реакцій та фізіо
логічних процесів у живих організмах.
Нуклеотиди АТФ, АДФ та АМФ можуть також виступати в роді алостеричних модуляторів певних регуляторних ферментів,
3) Участь в метаболічних реакціях в ролі коферментів:
- НАД, НАДФ, ФАД, ФМН - в реакціях біологічного окислення;
- УТФ, УДФ - в реакціях біосинтезу глікогену;
- ЦТФ, ЦДФ - в біосинтезі гліцерофосфоліпідів.
Синтетичні аналоги азотистих основ та нуклеотидів як антиметабо
літи та лікарські Засоби.
Деякі похідні азотистих основ пуринового та піримідиновго ряду при
їх надходженні всередину клітин можуть заміщати за механізмом кон
курентного зв’язування нормальні азотисті основи у складі нуклеїнових
кислот, що синтезуються. Таке конкурентне заміщення природних ме
таболітів їх синтетичними аналогами призводить до порушення норма
льної течії біохімічних процесів, зокрема функцій ДНК і РНК. Виходячи
з цього, антиметаболіти - аналоги пуринів та піримідинів - порушують
процеси нормального біосинтезу нуклеїнових кислот та білків, що
призводить до гальмування мітозу, особливо в клітинах, які здатні до
активного клітинного поділу. Такі сполуки ( 6 -меркаптопурин, 6 -тіогуа-
329
Розділ 4. БЮМОЛЕКУЛИ ТА МЕТАБОЛІТИ
нін, 5-фторурацил) застосовуються в медичній практиці як хіміотерапев
тичні препарати при лікуванні деякйх форм онкологічних захворювань»
6-меркаптопурин
,
6-тіогуаніи
5-фторурацил
4.4.2.
П ервинна ст рукт ура нуклеїнових кислот ; полярніст ь;
полінуклеот идів
П ервинна ст рукт ура нуклеїнових кислот
ВдІ класи нуклеїнових кислот - ДНК та РНК - є високомолекулярнимй
сполуками, основою первинноїструктури яких є полінуклеогидний ланцюг.
Окремі нуклеотиди зв’язані між собою в полінуклеотидний ланцюг
за рахунок фосфодіефірних зв ’язків, що утворюються між 3'- та 5'- гід
роксильними групами пентоз (рибоз або дезоксирибоз) сусідніх Нуклеотидів (рис. 4.10.).
А
Т
\
5*
5‘
Кінчена
3‘-ОН-група
3 30
\
Ф
\
5‘
Ц
3‘
3‘
3‘
ф
л
Г
А
5‘
3‘
3'
\
•ф
\
ф
\
Т
\
5‘
ф
\
л
5‘
Г
3‘
чф
ф
\
Ц
3‘
\
\
5‘
3‘
\
он
ф
\
5‘
5‘
3‘
Кінцевий
5 ‘-фосфат
4.4. Нукдеотмди. Нуклеїнові кислоти
З'
Рис. 4 .1 0 . Первинна структура полідезоксирибонуклеотидного ланцюга ДНК
При схематичному зображенні полінуклеотидних ланцюгів ДН К та
РНК пентози подають вертикальними лініям^ 3',5'-фосфодіефірні зв’яз
ки - похилими лініями з літерою Ф (або) посередині;
Цей же ланцюг можна подати такою системою скорочених Позначень:
фАфТфГфЦ...
Полярність полінуклеотидів.
В полінуклеотидному ланцюгу ДНК або РНК виділяють два кінці:
5 '-кінець, тобто той,-що містить вільний (не зв’язаний з черговим нукле0Т И Д 0 М )5 '-Г ІД Р 0 К С И Л пентози, та 3'-кінець-той, що містить вільний {не
зв’язаний 3 нуклеотидом) 3,'-гідроксил пентози. В природних нуклеїно
331
Розділ 4. БІОМОЛЕКУЛМ ТА МЕТАБОЛІТИ
вих кислотах 5'-кінець (5'-гідроксил кінцевої рибози або дезоксирибози)
звичайно фосфорильований, 3'-кінець — містить вільну ОН-групу.
Прийнято вважати, що така нуклеїнова кислота полярна і має напрямок
ланцюга 5' —» З'.
Відмінності в первинній структурі Д Н К та РНК:
1.
У складі нуклеотидів ДНК міститься цукор 2'-дезоксирибоза,
міеть рибози в складі нуклеотидів РНК.
2„ Нуклеотиди ДНК та РНК відрізняються за складом піримідинових основ:
- в ДНК міститься піримідин тимін (5-метилурацил)§
в РНК міститься піримідин урацил (заміси» тиміну).
3. Первинна структура ДНК та РНК різниться за наявністю деяким
мінорних нуклеотидів.
4, Певні класи ДНК та РНК мають специфічні для них послідову
ності нуклеотидів, що визначають їх біологічні функції.
Таблиця 4.5 і
Особливості первинної структури ДНК та РНК
Н уклеїнові
кислоти
ДНК
РНК
Азот ист і основи
пурини
піримідини
Аденін
Цитозин
Г уанін
Тимін
Аденін
Цитозин
Гуанін
Урацил
Пентоза
Дезоксирибоза
Рибоза
4.4.3. Будова, властивості т а ф ункції Д Н К
Біологічні функції Д Н К
Біологічними функціями ДНК, які вони виконують у всіх живих орга
нізмах, є:
1. Збереження генетичної (спадкової) інформації»
2. Передавання генетичної інформації нащадкам.
Генетична роль ДНК (тобто її провідна роль у збереженні та реалі-*
зації генетичної інформації) була доведена в результаті вивчення такого'
біологічного явища, як феномен трансформації пневмококів Streptococcus
pneumoniae (Ф. Гріффіт (P. Griffith), 1928; О. Евері, К. Мак-Леодом та;
М. Мак-Карті (О. Avery, C. McLeod, M. M cCarthy), 1944) та механізму!
332
4,4> Н ук л еот и д и . Н у к л еїн о в і к и сл оти
Піредивання генетичного матеріалу бактеріофагу Т2 (Альфред Д. Херші
(А, Hershey), Марта Чейз (M. Chase), 1952).
М олекулярна м аса т а розм іри м олекул Д Н К
Молекулярна маса дезоксирибонуклеїнових кислот суттєво варіює у різних
І Ішюгічних об’єктів: вірусів, прокаріотичних та еукаріогичних клітин.
Точному визначенню молекулярної маси різних зразків ДНК пере
шкоджає гідродинамічна ламкість гігантських молекул нуклеїнових кисЛОТ, особливо у вищих організмів, які за спроб виділити їх в інтактному
втпиі руйнуються на більш короткі фрагменти. Втім, застосування су
часних фізико-хімічних методів дослідження та електронноїмікроскопії
Дозволяло встановити, що молекулярна маса ДНК (при розрахунку на
ОДИН полінуклеотидний ланцюг) складає в середньому діапазон від 106
до 10" дальтон (Д).
Д Н К вірусів
Найменшу молекулярну масу та довжину молекули мають ДНК найПростіших живих утворень-вірусів, зокрема вірусів бактерій (бактеріофигіп) - в середньому 1Q6 - 1.08 Д.
14
М М ^ НК вІРУСУ поліоми ** 3 ' 106 Д. бактеріофага
Д Н К прокаріот ів
І)
прокаріотичних клітинах мікроорганізмів кількість ДНК та її моле
кулярна організація значно вищі, ніж у вірусів. Зокрема, ДНК кишкової
Пнлички E. Coli являє собою ковалентно замкнене дволанцюгове кільце
4M,м . 1 ,9 1 09Д. Відповідно до зростання складності біологічної органі
ч н ії при переході від вірусів до прокаріотів (а далі - еукаріотів) зростає
й кількість нуклеотидних пар в дволанцюгОвих молекулах ДНК.
Д Н К еукаріот ичних клітин
У клітинах без’ядерних прокаріотів міститься одна молекула ДНК,
ЯКИ розташована в спеціальній зоні цитоплазми - нутеоїді, ДНК ядер
них (еукаріотичних) клітин розміщена в ядрі і, поза фазами клітинного
ПМІлу, входить до складу аморфного нуклеопротеїнового утворення Яде/того хроматину. В період підготовки до мітозу - фазі S клітинного
Циклу - відбувається подвоєння ДНК з подальшою конденсацією хромитину 1утворенням цитологічних структур—хромосом, в яких сконцен
трований ядерний генетичний матеріал клітини.
Кількість хромосом в еукаріотичних клітинах специфічна для біоло
гічного виду; соматичні клітйни людини мають 46 хромосом (23 пари).
333
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
Молекулярна маса ДНК з хромосом людського організму дорівнюєпр
1
близно 1 6 • Р дальтон, що відповідає 2,4 1 09 пар азотистих основ. Ф ь 1
зична довжина розгорнутих молекул ДНК з клітин еукарютів досягає 1
декількох сантиметрів.
В и в ^ н м н у ^ еоти д н ого складу молекул ДНК з різних біологічних і
об’єктів показало, що, незалежно від джерела походження(бактеріальну 1
рослинні, тваринні організми), всі ДНК мають певні кшькісш взаємовідносини між вмістом пуринових та шримідинових нуклєотидт.
Згідно з цими закономірностями - правилами Чаргафа (Е. Chargatt), |
У С^ ; Г п у р и н о в и х основ дорівнює сумі піриміданових основ, тобто.
А + Г = Т + Ці
або (А + Ґ)/(Т ♦. Ц) ** 1;
2)
к і л ь к і с т ь
НТ » ”
6 -аміногруп
"
в
и
дорівнює кількості 6 -кетогруп (за хімічною
Юе вмісту тиміиу, а « і » гуаніну дорівнює
вмісту цитозину {правило еквівалентності).
А -Т , Г »Ц .
Зазначені кількісні взаємовідношення між азотистими основами а
т а м ж результати вивчення будови молекул ДНК методом рентгенеструктурного аналізу, здійснені М. Уілкінсом (М. Wilkins), дозвмили аме
риканському біохіміку Джеймсу Уотсону та англійському фізику Френ
сису Кріку, (J. Watson, Fr. Crick), що працювали в Кембриджському у н т
верситеті, запропонувати просторову модель структури молекули.ДНК
(1953 p.). Ця просторова модель являла собою подвійну спір
звана В-форма молекули ДНК за Уотсоном і Кріком).
Згідно з моделлю Уотсона - Крика, молекула ДНК складається з двох
т т ^ ралеп ьн и х полінуклеотидних ланцюгів. Ці ланцюги сполучені м ш
S
B
зв’язками, що утворюються між протилежно розташо
ваними так званими комплементарними (додатковими, англ.) азота
тими основами (аденіном ітиміном та гуаніном і цитозином, вщповідн ),
що пояснює зазначені вище емпіричні правила Чаргафа (рис. 4.11.).
Дваангипарапеиьні полінуклеотидні
утворюють стабілізо
вану водневими зв’язками правообертаючу спіраль, в якій обидва пошл а н ц ю
334
г и
4.4. Н ук л еот и д и . Н у к л еїн о в і к и сл оти
Ііуклеотидні ланцюги закручені навколо центральної осі. Структурні осо
бливості подвійної спіралі: діаметр спіралі - 2 нм; відстань між азотис
тими основами впродовж осі спіралі 0,34 нм; спіральна структура повто
рюється з інтервалом в 3,4 нм, тобто через 10 нуклеотидних пар.
Рис. 4 .1 1 . Схема утворення водневих зв’язків між аденіном і тиміном та гуаніном
і цитозином
Крім водневих зв’язків, стабільність молекули ДНК підтримується
також за рахунок взаємодій між р-електронними хмарами гетероциклів
азотистих основ, що розміщені один під одним впродовж осі спіралі —гак звані “стекінг-взаємодіР’.
Зазначені структурні особливості стосуються запропонованої Уотсо
ном і Кріком В-форми молекули ДНК (рис. 4.12; 4.13; 4.14).
335
Розділ 4. БЇОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
\
5'
Т 3' ч
В ІТ р ^
Т ЗС З. р
' р 5'
А З—А
«
рЗ
Р
3
5 ' ни
р З
р З
5'
Т з*
>3
—X
‘р З
чр з
-А .
5'<
Р ис. 4 .1 2 . Схематична модель двоспіральної молекули ДНК за Уотсоном і
Кріком
т
Р
5’
С З- ч _
ШНІ \ — 5
в 3' “ 5'
Т 3і А ,
т
А ЗЛ ,
---- V “ !
Рис. 4 .1 3 . Антиларалельність п о л ін у Н
леотидних ланцюгів в молекул. ДНК
Крім В-форми, молекула ДНК при взаємодії з різною кількістю мо
лекул води та катіонами набуває інших структурних форм: А, С та Ъ, які
можуть спостерігатися в різних фізіологічних умовах, зокрема при ком
плексуванні з білками ядерного хроматину (С-форма).
Третинна структ ура Д Н К
В живій клітині подвійна спіраль, що становить вторинну структуру
ДНК, не має вигляду розгорнутої молекули, а додатково згорнута в про
сторі, утворюючи певні третинні структури - єуперспіралі.
В суперспіралізованому стані молекули ДНК в комплексі з певними
клітинними білками входять до складу нуклеоїду прокаріотів та утво
рює нуклеосоми ядерного хроматину еукаріотів. Завдяки суперспіралізації довгі молекули ДНК формують компактні утворення, зокрема
хромосоми ядра. В результаті компактизації ядерна молекула ДНК клі
тин організму людини, що становить біля 8 см, вміщується в хромосомі
довжиною 5 нм.
Біосинтез Д Н К
Біосинтез ДНК здійснюється за механізмом елонгації, тобто подов
ження вже існуючих ланцюгів шляхом приєднання нових нуклеотидів до
336
4 .4. Н ук л еоти ди . Н ук л еїн ов і к и сл оти
вільних 3 ^-кінців одного з ланцю
гів наявного тюлінуклеотиду:
(дНМФ)п + дНТФ —>
(дНМФ)в+І + ФФЙ.
Механізм елонгації ДНК поля-«
гає в утворенні нових 3‘- 5‘-фосфодіефірних зв’язків, що здійснюєть
ся в напрямку 5‘ —> 3 ‘. Фосфодіефірний ЗВ ‘яЗОК формується в ре«і
зультаті нуклеофільної атаки з боку
вільної 3 ‘-ОН-групи пентози кінце
вого нуклеотиду на а -ф о с ф а т
дНТФ, що вступає в реакцію (див.
рис. 4.15).
Рис. 4 . 1 4 . В-форма молекули ДНК
Б іосинтез ДН К є складним
молекулярнобіологічним проце
сом, що каталізується сукупністю ферментів, головними з яких еД Н К полімерази з різною специфічністю та ефективністю дії.
Ф ізико-хімічні власт ивост і Д Н К
1) Хімічні властивості ДНК.
Всі полінуклеотиди, і ДНК зокрема, є сильними багатоосновними
кислотами з низьким значенням рК. Кислотність ДНК обумовлена вто«
ринними фосфатними групами, що при фізіологічних значеннях pH повні
стю іонізовані.
Завдяки кислотним властивостям і наявності на своїй поверхні нега
тивних зарядів молекули ДНК при фізіологічних значеннях pH активно
реагують і утворюють комплекси з катіонами:
- основними білками (гістонами, протамінами);
т катіонами металів (Са2+, М§2+, Ре2*);
*ш поліамінами (спермідином, сперміном).
2) В ’язкість розчинів ДНК.
Висока молекулярна маса і значна довжина молекул ДНК зумовлю
ють високу в ’язкість навіть дуже розбавлених їх розчинів. В ’язкість мо
лекул ДНК в розчині залеж ну від їх конформації і суттєво змінюється за
33 7
но—Р
он .
О — р —-О— Р— о
он
он
Рис. 4 .1 5 . Схема елонгації полінуклеотидного ланцюга ДНК.
Н І Г ' н
он н
в ) Поглинання в УФ-області.
Азотисті основи (та відповідні нуклеотиди), що входять до складу
нуклешових кислот ДНК і РНК, м а т ь вяастивість поглинати у л ь т р а Г
олетове світло з максимумом поглинання при 260 нм
по: Г ; Г : ОЛІНУКЛЄОТВДІВСуПр0 В0ДЖУється певним зниженням УФ.
ошинання порівняноз сумішшю вільних нуклеотидів. Поглинання пои
260 нм нативної молекули ДН К в середньому на 40 % нижче відповідного поглинання суми азотистих основ, що входять до складу полінукле
т Г ^ ф Т р З Г л н к ^ ” ” ' ПР" П° РУШЄННІ в" ° Р * » о ї двоспіральн и конформації ДНК та структурних взаємовідносин між азотистими
основами спостерігається гіперсроннии ефект, то6то зросташ я „о
338
І
4.4. Н ук л еот и ди . Н у к л еїн о в і к и сл оти
4) Денат урація.
* Денатурація Д Н К N* це порушення нативної двоспіральної конфорМйЦІЇ молекул ДНК та їх впорядкованого просторового розташування з
УТІорснняМ невпорядкованих одноланцюгових клубків. Ренатурація відновлення нативної вторинної Конформації ДН К, що спостерігається
ю цепних умов.
Денатурація спричиняється:
різкими змінами pH в кислий Ябо лужний бік;
1 нагрівання розчинів ДНК до певних температур.
ІК умов денатурації ковалентні зв’язки в ДН К зберігаються, проте
В и б у в ає т ь с я розкручування подвійної спіралі з втратою специфічних
Ііирмодій між азотистими основами. Молекулярною основою денатурМЦІЇ молекул ДНК є руйнування водневих зв ’язків між комплементар
ними азотистими основами А - Т т а Г - Ц . Денатурація ДНК супрово
джується гіперхромним еф ект т та зменшенням в ’язкості її розчинів.
Нуклеїнові кислоти, що зазнали денатурації, втрачають свої біолоI Ічні властивості.
Термічна денатурація ДНК дістала назву' процесу плавлення (melting Ш м е н н я ; англ.). Для кожного типу молекул ДНК (тобто молекул, що
ММІОТЬ певний нуклеотидний склад) характерна відповідна температура
^вІНїіурпціі, що позначається як температура (точка) плавлення - Т .
Оскільки між гуаніном та цитозином в складі двоспіральної молеку
ли ДНК утворюється три водневих зв’язки (на відміну від двох - між
ШИШОМ та тиміном), термічна дисоціація дари G-С потребує більших
ІІТриг енергії, тобто відбувається при більш високих температурах, ніж
руйнування пари А-Т. Виходячи з цього, температура плавлення (Т )
Молекул Д Н К прямо пропорційна вмісту в них G -C-nap, що дозволяє
Використати визначення Тт як показника нуклеотидного складу ДНК.
4, 4. 4 .
Чудова, власт ивост і т а ф ун к ц ії Р Н К
Рибонуклеїнові кислоти (РНК) - за характером структури та біологіч
них функцій підрозділяються на такі класи: інформаційні (матричні
мшнджерні) РНК (мРНК), транспортні РНК (тРНК), рибоеомні РНК
(рІЧІК) (табл. 4.6). Кожен з цих класів РНК виконує свої специфічні
функції в переносі та реалізації генетичної інформації в клітині.
Первинна структура Р Н К - за первинною структурою РНК є полі рНЙ(ЩШ<ютидами (див. вище). На відміну від дволанцюгових ДНК,
339
Розділ 4. БІОМОЛЕКУЛИ ТА МЕТАБОЛІТИ
м о л еку л и Р Н К ви щ и х орган ізм ів є од н олан ц ю гови м и пол ін уклеотидам и. Разом з тим, одноланцюгові РНК за рахунок внутрішньомолекулярних взаємодій набувають конформацій, що позначаються як
вторинні та третинні структури.
Таблиця 4.6.
В л асти вості основних к л а с ів Р Н К в б ак тер іал ьн ій к л іти н і
Е.С оїі Цін РО Д ІМ ІЙ'і^ із змінами)
Тип РНК
Відносна
к іл ьк іс ть,
%
Рибосомальна РНК (рРНК)
80
1,2 х Ю3
0,55 х 103
3,6 х Ю!
2,5 x 1 0 і
Гетеро
генний
15
Транспортна РНК (тРНК)
Інформаційна (матрична)
РНК (мРНК)
М аса, кД
і
К іл ькість
нуклеотидів
3700
п оо
120
75
склад
Біосинтез окремих типів РНК каталізується РНК-пдлімеразами різ
ної клітинної локалізації та специфічності.
/ч
ям
НЬ
І І
О»
гсцТ
XX
У
Х
А
р
А
V
4/
ь
т
>
Vа г Чш іНІз
г
Ц І г А~у 1 І І . І
г
у
Р ис. 4 .1 6 , Механізм утворення “шпильок" у вторинній структурі РНК.
340
_
[-
4.4. Нуклеотиди. Нуклеїнові кислоти
Вторинна структура одноланцюгових полірибонуклеотидів еукаріотів характеризується наявністю ділянок, що мають двоспіральну стру
ктуру. Ці ділянки молекул РНК - так звані “шпильки” - утворюються за
рахунок згинів полірибонуклеотндного ланцюга та взаємодії між собою
комплементарних азотистих основ (А - Y та Г - Ц) в межах одного
ланцюга. Такі еніралізовані ділянки включають в себе 20 - ЗО нуклеотидних пар і чергуються з неепіралізованими фрагментами РНК (рис. 4.16.).
Фізико-хімічні властивості РНК.
Подібно до ДНК, полінуклеотиди РНК характеризуються максиму
мом поглинання при 260 нм, обумовленим азотистими основами, мають
гіпохромний ефект, підлягають денатурації при дії жорстких фізико-хімічцих факторів.
V.
Інформаційні (матричні) РН К
Це - клас РНК, що складають 2-5% загальної кількості клітинної
РНК. МРНК виконують функцію переносників генетичної інформації від
генома (ядерної ДНК) до білоксинтезуючої системи клітинй (рибосом).
Для мРНК властиві найбільша гетерогенність молекулярної маси
та розмірів молекул ш від 25103 до 1 -2 1 0 6 з константами седиментації
6-25Щ- та метаболічна нестабільність. Широкий спектр молекул мРНК
відповідає кількості білків організму, носіями генетичної інформації для
синтезу яких вони являються.
Первинна структура мРНК. За своїм нуклеотидним складом мРНК
в і д п о в і д е ® (з урахуванням принципу комплементарності) нуклеотидній
послідовності фрагменту одного з ланцюгів ядерної ДНК, транскршшда
якого вона (мРНК) являється. Виходячи з цього, мРНК і інформаційни
ми матрицями, які визначають амінокислотні послідовності в поліпепти
дах, що синтезуються в рибосомах.
Особливістю первйн'нОЇ структури мРНК є наявність на 5' та З’-кінцях
молекули характерних для цього класу РНК нуклеотидних послідовностей.
5 '-кінець всіх молекул мРНК еукаріот в якості першого нуклеотиду
містить 7-метилгуанозин, який через трифосфатвий залишок сполуче
ний з 5'-гідроксилом сусіднього (другого) нуклеотиду. Нуклеотид, з яким
зв’язаний 5'-кінцевий 7-метилгуанозин, маєметильовану по С-2'-рибозу.
Модифікований 7-метилгуанозином 5'-кінець мРНК носить назву
“кепа” (cap - шапочка; аніл.). До складу кепу можуть входити 1-3 залишки
7-метил гуанозину.
341
Розділ 4. БІОМОЛЕКУЙЙ ТА МЕТАБОЛІТИ
З '-кінець багатьох мРНК еукаріотів містить відносно довгі поліаденілатні (ро1у(А)) послідовності. До складу роІу(А)-”хвостів” мРНК
входять 20—250 нуклеотидів.
Вважають, що 5'-кепу^ання та З'-поліаденілування стабілізує молекули
мРНК, запобігаючи дії нуклеаз, та має значення для зв’язування мРНК
з рибосомами в процесі трансляцій у
4
*
Вторинна структура мРНК характеризується численними внутрішньоланцюговими двоспіральними ділянками — шпильками , до складу
яких входить до 40—50% нуклеотидногоскладу полірибонуклеогиду.
Транспорт ні РН К
На долю тРНК припадає 10-20 % клітинної РНК. Молекули тРНК
являють собою полірибонуклеотидні ланцюги довжиною в 70-90 нукле
отидів. Молекулярна маса тРНК —23-28 кД, константа седиментації *»
4з. Всього в клітинах знаходиться не менше 20 типів тРНК, що відпові
дає кількості Природних Ь-амінокислот, з якими тРНК взаємодіють в
ході трансляції.
Первинна структура тРНК відзначається великою кількістю м і
норних нуклеотидів 2 метильованих, псевдоуридилових та дигідроуридилових залишків.
Дтпринна структура молекул тРНК в двомірному просторі має
конформацію “листка конюшини” , що утворюється за рахунок специфіч
ної взаємодії комплементарних азотистих основ на протязі полірибонуклеотидного ланцюга. Неспарені нуклеотидні послідовності формуюсь спе
цифічні для будови тРНК структурні елементи:
- акцепторну гьї^у (стебло) - З'-кінедь молекули, який містить тер
мінальну послідовність нуклеотидів ЦЦА. кінцевий аденозин через
З'-гідроксильну групу рибози акцептує амінокислоту в процесі трансляції,
* антикодонову пет:ію - містить групу з трьох нуклеотидів {антикодон), які комплементарні триплету нуклеотидів (кодону) в складі
мРНК. Антикодонова петля відповідає за взаємодію тРНК з певними
нуклеотидами мРНК в процесі трансляції,
, .
і* дигідроуридилову петлю ■“ складається з І Н 'ї і нуклеотидів,
містить в собі 1—4 дигідроуридилові залишки,
- псевдоуридилову петлю - ділянку тРНК, яка містить у своєму
складі нуклеотид псевдвуридин та послідовність 5*-'Я0ЦСі-3 , вважають*
що псевдоуридилова петля необхідна для взаємодії тРНК з рибосомою.
342
4.4. Н ук л еоти ди . Н ук л еїн ові кислоти
додаткову гілку - структуру, за
кількістю нуклеотидних залишків у якій
тРНК поділяються на два класи: тРНК
класу І містить 3-5 нуклеотидів та
тРН К класу Ш (13-21 нуклеотид).
Схему будови тРНК - див. рис, 4.17.
Притаманні тРНК структури типу
“листка конюшини” можуть утворю
вати більш компактні просторові кон
формації-третинні структури. Зада
ними рентгеноструктурного аналізу,
третинна структура молекул тРНК
нагадує велику латинську літеру Ь.
Рибосомні Р Н К
Рибосомні РНК (рРНК) - клас
Рис. 12. Узагальнена схема вторин
клітинних РНК, що входять до скла
ної структури молекули тРНК
ду рибосом прокаріотичних та еукатипу “листка конюшини1’
ріотичних клітин. На долю рРНК прииадає до 90 % загальної кількості клітинних РНК.
рРНК разом із специфічними білками складають основу структури
та функції рибосом - рибонуклеопротеїнових часточок, в яких відбува
ється процес трансляції - біосинтез поліпептидних ланцюгів на основі
коду, що поставляється мРНК. Рибонуклеїнові кислоти цього типу є
метаболічно стабільними мрлекулами; взаємодіючи з рибосомними біл
ками, вони виконують функцію структурного каркасу для організації
системи білкового синтезу.
Модифікованих (мінорних) основ у складі рРНК значно менше, ніж в
тРНК. Проте рибосомні РНК є також високометильованими полірибоиуклеотидамк, в яких метильні групи зв’язані або з азотистими основа
ми, або з 2'-гідроксильними групами рибози.
Вторинна структура рРНК представлена значною кількістю корот
ких двоспіральних ділянок» що мають вишяд “шпильок” або паличок.
Крім зазначених кланів РНК, в ядрах Клітин ссавців містяться рибо
нуклеїнові кислоти різної молекулярної маси, так звані гетерогенні ядерні
141К (гяР Н К ). М олекулярна м аса гяРН К може бути значною і
перевищувати 107Д. ГяРНК являють собою продукти транскрипції ге
мін, що не зазнали посттранскрипційної модифікації-процесингу.
З'
343
Розділ 5. Ф ІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
5.1.
З а га л ь н а х а р а к те р и с ти к а ф ізіологічно ак ти в н и х с п о л у к и
Ф ізіологічно (біологічно) а к ти в н і сполуки - речовини, що приі
взаємодії з живим організмом (людини, тварин, рослин, мікроорганізмі^*
спричиняють специфічні для дії даного класу ФАС зміни у перебігу біо-1
хімічних та фізіологічних процесів. Вплив фізіологічно активних сполуга
на організми відбувається в більшості випадків у дозах і концентраціях!
що не перевищують частки грама або міліграма.
Багато біологічно активних сполук у низьких дозах впливають на ор-і
ганізм людини як лікарські засоби (тобто мають фармакологічну дію), у ]
високих дозах — діють як отрути (мають токсичну дію), тобто призвО*|
дять до порушення нормального функціонування біохімічних та анатомо-1
функціональних систем і можуть спричинити загибель цілісного організм}«
В основі впливу фізіологічно активних сполук на організм людини лежати
такі фундаментальніклітинні та біохімічні механізми (Ю. І. Губський, 1994Я
1.
Рецепт орна дія т а вп ли в на ф ункціонування р е гу ля т о р н и х '
сист ем кліт ини.
Рецепторні ефекти лежать в основі фармакологічної активності меді-а
аторів нервової системи (норадреналіну, дофаміну, серотоніну, ацетилхоі
ліну у-аміномасляної кислоти, нейропептидів), гормонів (адреналіну, білково-пептидних гормонів, гормонів щитовидної залози), численних лікар
ських засобів та токсичних сполук, що виступають як агоністи чи
антагоністи нейромедіаторів та гормонів.
В основі рецепторного механізму лежить взаємодія ФАС з білковимй
рецепторами (К), що містяться в плазматичних мембранах клітини і
здатні до специфічного зв ’язування ендогенних та екзогенних лігандівЗ
Взаємодія молекули ліганда (ФАС) з білковим рецептором відбувається
за участю водневих, іонних, диполь-дипойьних, гідрофобних зв’язків.
Після взаємодії з фізіологічно активними сполуками - лігандами хі
мічний сигнал передається з рецепторного білка (Я) на каталітичну субодиницю ферменту аденілатциклази за допомогою білка-трансдукто^
ра (О-білка). Внаслідок цього відбувається активація аденілатциклази
та посилене утворення з аденозин-триф осфату (АТФ) циклічного
аденозинмонофосфату ( 3 \ 5 ’-АМФ; цАМФ) - “вторинного посеред
ника”, “месенджера”, який, у свою чергу, є активатором багатьох внут
рішньоклітинних ферментів.
344
5.1. Загальна характеристика фізіологічно активних сполук
АТФ - » 3’,5 ’-АМФ + ФФН
ОН
Циклічний АМФ (3’,5’-АМФ )
Розглянута схема дістала назву “каскадного механізму” трансфор
мації зовнішнього хімічного сигналу з боку гормону або іншої ФАС в
(Кти націю специфічної біохімічної та фізіологічної функції клітини.
2 . М ет аболіт на т а ант имет аболіт на дія.
Цей тип дії ФАС на клітину характеризується безпосереднім вили
тім ми перебіг метаболічних процесів таких біомолекул, як амінокисло
ти, вуглеводи, нуклеотиди, вітаміни* коферменте. Крім того, багато ФАС
виступають як антиметаболіти, тобто речовини, що конкурують із
впрпвжніми метаболітами за активні ц ен тр і певних ключових фермен
тів, що каталізують реакції біохімічного перетворення сполук. Такий тип
ДІЇ мають насамперед антибіотики - препарати, що порушують ріст
ТВ розмноження бактеріальних клітин або клітин злоякісних пухлин,
ИНТИнітаміни, антиметаболіти, які є структурними аналогами амінокис
лот, пуклеотидів тощо.
Аитиметаболітну дію проявляють також синтетичні лікарські засо
би Ті токсичні сполуки, що пригнічують активність деяких ферментів за
рихунок хімічної модифікації молекул останніх. Наприклад, цей тип акі иипості лежить в основі протизапальних ефектів саліцилатів (аспірину
Ті Інших похідних ацетилсаліцилової кислоти), що спричиняють хімічну
Модифікацію та гальмування активності циклооксигенази - ферменту
Ш іісіу простагландинів - сполук, що беруть участь у перебігу процесу
345
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
запалення. За принципово подібним механізмом реалізується негативним 1
вплив на організм людини та багатьох тварин деяких токсинів, наприклад і
фосфорорганічних сполук, що незворотно пригнічують активність фермен- і
ту ацетилхоліиеетерази, необхідного для нормального перебігу еинап- 1
тичних процесів у головному мозку та парасимпатичнім нервовій системі. І
3. М ембранот ропна дія.
Мембранотропні ефекти властиві ФАС* шо за своєю фізико-хімічною |
природою здатні безпосередньо взаємодіяти з ліпідним випаром біодй- |
гічних мембран, порушуючи функціонування мембранозв’язаних фер- ;
ментів, іонних каналів, систем активного транспорту іонів. Таку мем- |
бранотропну (мембранотоксичну) дію мають численні гідрофобні про
мислові отрути (тетрахлорметан, дихлоретан, інші похідні алканів, бензол, вищі спирти тощо), хлорорганічні пестициди.
4. Вплив на систему біосинтезу білка.
Цей тип дії близький до антимєтаболітного і характеризується здат
ністю певних ФАС активних сполук втручатися в перебіг рибосомного
синтезу. Деякі антибіотики (тетрацикліни, стрептоміцин, хлорамфенікол, пураміцин) виступають у ролі конкурентних інгібіторів окремих
стадій трансляції (тобто зчитування інформації з мРНК з утворенням
поліпептидного ланцюга), що лежить в основі їх антибактеріальної або
антипухлинної дії.
5. Взаємодія з компонентами генетичного апарату клітини.
Деякі біоорганічні сполуки здатні безпосередньо зв’язуватися з ДНК
або білками нуклеосомних комплексів ядерного хроматину, порушуючи
нормальну, реплікацію ДНК або транскрипцію мРНК. Класичними
представниками цього класу ФАСє протипухлинні антибіотики тину ак
тиноміцину О, що розміщується між двома ланцюгами молекул ДНК
(“інтеркаляція”), протидіючи перебігу ДНК-полімеразних реакцій; алкілуючі агенти (похідні хлоретиламінів, етиленіміну, алкан-та аренсульфонових кислот), що призводить до ковалентної модифікацй (алкілуван
ня) азотистих основ у молекулах ДНК.
5.2. А лкалоїди я к л ік ар с ь к і засоби
азотовм ісіі сполуки, що мають хімічні властивості ос
нов та впливають на організм людини й тварин як фізіологічно активні
сполуки. Згідно із сучасною класифікацією біоорганічних сполук алка
А л к а л о їд и -
346
5.2« А л к ал оїд и я к л ік ар сь к і засоби
лоїди є природними похідними N-вмісних гетероцикл ів. У великих дозах
більшість алкалоїдів характеризується токсичною Дією, в малих - деякі
з них використовуються як важливі лікарські засоби.
За хімічними властивостями алкалоїди є біоорганічними сполу
ками основного характеру, обов’язковим компонентом структури яких є
третинний атом азоту: N as. Цей третинний азот надає молекулам алі*
калоїдів лужних властивостей, тобто здатності відщеплювати іон водню
Н+ подібно до того, як це притаманне амонійним солям:
Н+Х'
+
:N
N
+
Х‘
Н
Алкалоїди синтезуються в рослинах, вищих та нижчих грибах, водо**
ростях. Вміст алкалоїдів у рослинах, де вони знаходяться переважно у
вигляді органічних або мінеральних солей, невеликий &* 1 - 2 %.
Головними попередниками в процесі біосинтезу алкалоїдів є аміноки
слоти орнітин, лізин, аспарагінова кислота, фенілаланін, тирозин, трипто
фан. Більшість алкалоїдівЩце кристалічні речовини, оптично активні.
На сьогоднішній день відомо понад 5000 різних алкалоїдів, які за хіміч
ною природою є похідними гетероциклічних сполук.
Класифікація алкалоїдів
Алкалоїди класифікують за хімічною природою гетероциклів, що вхо
дять до їх складу (похідні піридину, піперидину, хіноліну, тропану, індолу
тощо) або за характером фізіологічної та фармакологічної дії на тва
ринні організми (нейротропні, кардіотропні, спазмолітичні, анальгетичні
засоби тощо). За механізмом фармакологічних ефектів алкалоїди налеі*
жать до ФАС із рецепторною Дією.
Реакції ідент ифікації алкалоїдів
Важливим завданням фармацевтичної хімії та токсикологічної (судо
вої) хімії є розробка методів виявлення (ідентифікації) алкалоїдів як у
складі лікарських засобів, так і в якості компонентів невідомих речовин
та їх сумішей, а також біологічних об’єктів (крові, сечі людини тощо).
Реакції ідентифікації, що застосовуються для виявлення алкалоїдів,
поділяються на загальні (групові), тобто такі, що є властивими для всієї
групи алкалоїдів, та. специфічні, які притаманні лише певному алкалоїду.
347
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
В якості групових реакцій на алкалоїди найчастіше застосовують;
досить численні реакції осадження, в якості специфічних більш вжич
ваними є кольорові реакції (реакції забарвлення), що зумовлені осо-:
бливостями хімічної будови алкалоїду та наявністю в структурі його
молекули певних функціональних груп.
Реакції осадження ґрунтуються на здатності алкалоїдів з певними ре^
активами утворювати прості та комплексні солі, що є нерозчинними у воді’.
До групових реактивів, що спричиняють осадження алкалоїдів, належать:*
- реактиви Вагнера, Лю гом, Бушарда (розчини йоду в йодиді калію);
- реакт ив Майєра (розчин йодиду ртуті в йодиді калію);
- реакт ив Драгендорфа (розчин йодиду вісмуту в йодиді калію);
- реакт ив Зонненштейна (фосфорномолібденова кислота);
«* реакт ив Ш ейблера (фосфорновольфрамова кислота)!
* розчини таніну та пікринової кислоти.
Реакції забарвлення є специфічними для окремих алкалоїдів або їх
груп. Більшість з цих реакцій грунтуються на дегідратації або окисленні
алкалоїду чи його конденсації з альдегідами. В якості таких застосову
ються: концентровані сірчана та азотна кислоти; суміш сірчаної та азотної
кислот (реактив Ердмана)', суміш концентрованої сірчаної кислоти та
триоксиду молібдену (реактив Фреде% суміш концентрованої сірчаної
кислоти та формальдегіду (реактив М аркі), що є специфічним для
алкалоїдів опію (морфіну, кодеїну, папаверину) і є вживаним у судовою
хімічній практиці.
А лкалоїди групи піридину т а піперидину
Н ікот ин {3-[2 ’-(>>Г-метилтролщил)]тридин}. До алкалоїдів групи
піридину належить нікотин - алкалоїд, молекула якого складається з
циклів піридину та ЇЯ-метилпіролідину:
Нікотин
348
5.2. А л к ал оїди я к л ік а р сь к і засоби
Нікотин є токсичним алкалоїдом, летальна доза якого для лю д и ш
становить близько 40 мг. Вміст нікотину в листках тютюну (Nicotiana
tabacum) досягає 8 %.
За фармакологічною дією нікотин є головним представником класу
н-холіноміметиків *- фізіологічно активних сполук, що стимулюють
центри вегетативної нервової системи, нейрони якої містять специфічні
иікотинові (н-) рецептори. Нікотин стимулює діяльність центральної нер
вової системи, звужує кровоносні судини, впливає на гладенькі м ’язи та
інші анатомо-функціональні системи організму. Тютюнокуріння розгля
дається в наш час як різновид наркоманії.
Похідними піперидину є алкалоїди лобелін та коніїн.
Лобелін [Ь-(-)2-бензоїлметил-6-(2 ’-гідрокси - 2 ’-фенілетил)-1-метил5*
піперидин] - алкалоїд, похідне піперидину, що міститься в рослині Lobelia
inflata. Лобелін використовується в медичній практиці як засіб, що сти
мулює дихальний центр головного мозку (аналептична дія). Коніїн високотоксичний алкалоїд, що є діючою речовиною рослинної отрути з
болиголову, або цикути (Conium maculatum).
Н
Н
Коніїн
Лобелін
Алкалоїдом, молекула якого складається з циклів піридину та піпери
дину, є анабазин.
Анабазин [Р -(а’-піперидил)піридин] - алкалоїд, що добувають з рос
лини іжника безлистого Anabasis aphylla; його застосовують в медицині як
засіб проти куріння та в сільському господарстві як потужний інсектицид.
N
Анабазин
349
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
Хінуклідин (1,4-етиленліперидин; 1-азабіцикло-[2,2 42]-октан)-алка
лоїд, який можна розшядати як похідну піперидину, в молекулі якої атом
азоту і атом вуглецю, що знаходиться відносно нього в «ара-положенні,
сполучені містком - СН 2с« СН 2 - :
4
1
Хінуклідин
Залишок хінуклідину ■ складовою частиною молекули лікарських
препаратів протималярійного засобу Хініну (див. нижче) та сполуки з
антиаритмічною дією Аймаліну Крім того, структура хінуклідину вхо*'
дить до складу молекули синтетичної нейротропної ФАС ацеклідину
(3-ацетоксихінуклідину) з холіноміметичною дією, що стимулює рецептори^
чутливі до ацетилхоліну та мускарину (так званих м-холінорецептори). ї
Похідна хінуклідину - хінуклідил-3-бензилат є високотоксичною
отрутою з психо- та нейротропною дією, що застосовується як компо
нент хімічної зброї (В2; Бі-Зет). Вже при вдиханні повітря, що містить
ЕХ в концентрації 0 , 1 мг/л, у отруєної людини наступають дискоординація рухівта важкі розлади психічної діяльності.
О
II
Х>
С
СН3
N
Ацеклідин
Хінуклідил-3 -бензилат
При отруєнні ацеклідином та хінуКлідил-3-бензилатом як антидот
застосовують холінолітичний засіб Атропін (див. нижче).
Алкалоїди групи хіноліну т а ізохінолІну
З природних сполук, що є похідними хіноліну, важливе значення в
фармакології має алкалоїд хінін.
350
5.2. Алкалоїди як лікарські засоби
Хінін [(6’-метоксихіноліл-4’)-(5-вінілхінуклідил-2)-карбінол] - алка
лоїд з кори хінного дерева (СЬіпсІюпа). До складу молекули хініну вхо
дять залишки хіноліну та хінуклідину:
Хінолін
Хінін являє собою безбарвну кристалічну речовину, надзвичайно гір
ку на смак, яка застосовується в медичній практиці як протималярійний
засіб. З фармакологічної точки зору хінін є протипаразитарним засобом,
ефективним щодо збудників малярії Plasmodium vivax та Plasmodium
malariae. В якості протималярійного засобу кора хінного дерева була
відомою з XVIII сторіччя, її алкалоїдний склад був вивчений професо
ром Харківського університету Ф. І. Гізе в 1814 р. та французькими
дослідниками П. Пелтье та Ж. Кавенту в 1820 р.
До групи алкалоїдів »» похідних ізохіноліну належать фізіологічно
активні сполуки, що входять до складу опію - соку, який міститься в
незрілих голівках снодійного маку (Papaver somniferum): морфін, геро
їн, кодеїн, папаверин. За характером фармакологічної дії ці алкалоїди
належать до класу наркотичних анальгетиків; крім того, вони спричиня
ють складний психоемоційний стан внутріЩнього задоволення - ейфорії
та при повторному вживанні, можуть спричиняти розвиток синдрому
хворобливої залежності - наркоманії. Сама назва головного представ
ника алкалоїдів цієї4групи походить від імені давногрецького бога Мор«
фея - бога сну та сновидінь,
Морфін - алкалощ, за хімічною будовою є N-метилпохідною фенантренізо-хіноліну (морфінану) - М-метил-3,6-діокси-7,8-дидегідро-4,5-епоксиморфінан. Всього в наш час відомо більше 40 морфінанових алкалоїдів.
351
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
не
о
"Х Н з
но
Морфін
Кодеїн. ГероШ О-метильна похідна морфіну - кодеїн - є вживаним)
у медицині Іфармакопейнйм) протикашлевим засобом. О-діацетильна
похідна морфіну - героїн - належить до найбільш небезпечних наркоти
чних речовин, т. з. “важких наркотиків” :
О
II
Кодеїн
Героїн
За механізмом дії морфін та подібні до нього речовини (опіоїдні нарко
тики) є сполуками, що взаємодіють з тими ж рецепторними структурам^
в головному мозку, що й природні нейромедіаторкпептидної природи, так
звані опіоїдні нейропептиди (енкефаліни, ендорфіни). Всі ці сполуки (яій
природні, так і синтетичні) регулюють складні фізіологічні, психологічні та
емоційні процеси, що пов’язані з відчуттями болю, задоволення та спокою.
Вперше морфін був виділений з опію в 1806 р. германським хіміком та
фармацевтом Ф. Сергюрнером, його хімічна структура була встановлена
в 1925-1927 рр. британським хіміком-органіком Р. Робінсоном, повний
хімічний синтез здійснений М. Гейтсом у 1952 р. і лише в 1958 р. за
допомогою рентгеноструктурного аналізу було завершено з ’ясування
стереохімічної будови алкалоїду СД- Ходаскін).
352
5.2. Алкалоїди як лікарські засоби
У наш час проблема наркоманії набула надзвичайно важливого соці
ального значення. Тому вельми актуальною проблемою медичної хімії
та фармакології є пошук такого анальгетику, що, маючи знеболювальні
властивості морфіну, не виявляв би його негативних наслідків, а саме
розвитку психічної та фізичної залежності. Англійським хіміком К. Бентлі
було синтезовано таку похідну морфіну, яка за знеболювальною та За
спокійливою активністю в 10 600 разів (?) активніша за морфін. В станом
влено, що 1 мг цієї речовини (“сполуки Бентлі”) може заспокоїти розлю
ченого дикого слона, проте, на жаль, цей засіб виявився токсичним (при<*
гнічує дихальний центр) та, знову ж таки, здатним Спричиняти хвороб
ливу пристрасть до нього:
Папаверин [6,7-диметокси-1-(3 ’,4’-диметоксибензил)ізохінолін] алкалоїд, що є похідною бензилізохіноліну.
Папаверин
353
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
Папаверин має здатність розслаблювати гладенькі м ’язи внутрішніх«
органів та кровоносних судин і використовується в клінічній медицині які
спазмолітичний засіб.
Алкалоїди групи тропану
Тропан є конденсованою біциклічною сполукою, до складу якої bxo J
дять п ’ятичленне піролідинове та шестичленне Піперидинове кільця!
Найбільш відомими тропановими алкалоїдами є сполуки холінолітичної
дії атропін, гіосціфіШ та скополамін,
Атропін [тропіновий ефір (±)~тропової кислоти] похідна тропану, що
міститься в рослинах родини пасльонових, зокрема в красавці (беладонні)]
Atropa belladonna, дурмані D a to a stramomutn), блекоті (Hyoscyamus niger)J
Атропін
Тропан
Фактично алкалоїд атропін є рацематом двох ізомерів - ефірів аміно
спирту тропіну та тропової(Р-гідроксі-а-фенілвропіонової) кислоти; лі- 1
вообертаючий ізомер дістав назву Гіосціаміну. Вважають, що в рослині
міститься переважно гіосціамін, який В процесі екстрагування за раху-1
нок наявності в троповій кислоті асиметричного атома вуглецю пере- ?
творюється на суміш двох стереоізомерів - раЦемат атропін.
Скополамін [скойдаовий ефір (^-троіКіваї; кислоти] - ййкалоїд, що ]
також міститься в пасльонових рослинах красавці, дурмані тощо. За х і-|
мічною будовою скополамін є близьким до атропіну та гіосціаміну, явля* І
ючи собою складний ефір тропової кислоти та аміноспирту скопіну.
о
Скополамін
3 54
>— о н
5.2. Алкалоїди як лікарські засоби
За механізмом дії атропін, гіосціамін та скополамін є антагоністами
м-холіиорецепторів, які блокують передавання нервових імпульсів в
холінергічних центрах головного мозку та парасимпатичної нервової
системи. Застосування цих сполук в медичній практиці ґрунтується на
їх холінолітичній дії. Зокрема, атропін використовується як антидот при
отруєнні фосфорорганічними сполуками, що діють як інгібітори ацетипхолінестерази.
Кокаїн (метиловий ефір бензоїлекшніну). Похідною Трепану є також
алкалоїд кокаі'щ який добувають з листів південноамериканської росли
ни - чагарника кока (ЕгуШгохуіоп еоса).
О
№
Екгоиіи
(тропін-2-карбоиова кислота)
Кокаїн
Кокаїн є активним місцевоанестезуючим та наркотичним засобом.
Діючи на мембрани нервових закінчень, алкалоїд блокує мембранний
транспорт іощв Иа*, що пригнічує проведення імпульсів по аферентних
чутливих нейронах. У зв’язку з можливістю викликати хворобливу за
лежність (кокаїнізм), в наш час кокаїн має обмежене застосування в
медичній практиці переважно для місцевої анестезії слизових оболонок,
кон’юнктиви ока тощо.
Алкалоїди групи індолу
Ядро індолу входить до складу багатьох алкалоїдів. Найбільш за
стосовуваними в фармакології алкалоїдами групи індолу є резерпін, фізостигмін, стрихнін.
Резерпін - алкалоїд, що міститься в коренях індійської рослини ра
увольфії зміїної Ш ш т . зегрепйпа). За хімічною структурою резер
пін є похідною конденсованої гетероциклічної сшстемиаллоіохімбану (11,17-диметокси-16 -карб-метокси-18-(3’,4 ’,5 ’-триметоксибензоїл)оксиаллоіохімбан):
355
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
Резерпін має гіпотензивні та седативні властивості, що визначає його
застосування в якості лікарського засобу при гіпертонічній хворобі, під-.вищеній збудливості центральної нервової системи.
Фізостигмін (езерин) - алкалоїд, який виділяють з плодів отруйної І
африканської рослини РЬувовйдша уепепозит, відомої також як “калані
барські боби” (РаЬа саІаЬагіа).
п*
СНз
Н3С— NН— С—Оч
.
N
І
А
N
1
Р
СНз СНз
Фізостигмін
356
5.3. А л к ал оїд и я к л ік ар сь к і засоби
За механізмом фармакологічної дії фізостигмін є зворотним інгібітором
ацетипхолінестерази - ферменту, що розщеплює нейромедіатор аце
тилхолін. Виходячи з цього, при надходженні в організм фізостигміву від
бувається накопичення значної кількості ацетилхоліну, що і пояснює фізіо
логічні та біохімічні ефекти цього алкалоїду та показання дО його викори
стання в медичній практиці як стимулятора холінергічних процесів.
Стрихнін - алкалоїд, що міститься в насінні блювотного горіху (чилі?
бухи) (Strychnos nux vomica). За хімічною будовою стрихнін являє собою
складну багатоядерну конденсовану систему, що містить залишок-індолу:
Стрихнін являє собою токсичну сполуку, що діє збудливим чином по
відношенню до центральної нервової системи; алкалоїд спричиняє важкі
судоми шляхом стимуляції відповідних центрів спинного мозку. За біохіь
мічним механізмом дії стрихнін є антагоністом гальмівних ефектів нейромедіаторної амінокислоти гліцину. Може застосовуватися в медицині
як засіб тонізуючої дії.
5.3.
А н ти б іоти ки я к л ік а р с ь к і засоби
А нтибіотики ЯГфізіологічно активні сполуки, що протидіють росту
та розмноженню клітин як прокаріотів (мікроорганізмів, бактеріальних
клітин), так і еукаріотів (клітин тваринних організмів). Антибіотики, що
гальмують біосивгетичні процеси у прокаріотів, дістали поширення як
протимікробні, проти інфекційні лікарські засоби; антибіотики, які діють
на еукаріотичні клітини, використовуються як протипухлинні препарати.
За механізмами біохімічної дії більшість антибіотиків виступають як
антиметаболіти', вони блокують біосинтез білка та нуклеїнових кис
357
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
лот, як правило, витісняючи з клітинних місць зв ’язування (рибосоми,ДНК, ядерний хроматин) природні метаболіти (амінокислоти, пурини^
піримідини, нуклеотида, фолієву кислоту).
Пеніциліни. Антибіотична дія пеніциліну була вперше помічена в 1929 р.
англійським бактеріологом О. Флемінгом, який спостерігав антимікроб
ний вплив (гальмування розвитку стафілококів) активного фактора, що/
виділявся зеленою пліснявою з виду РепісіПіит поСаШт. Натрієва сіль
пеніциліну з високою антистафілококовою активністю була виділена в
І940 р. X. Флорі та Е. Чейном. Пізніше був виділений та охарактеризо^
ваний з хімічної та біологічної точки зору цілий клас антибіотиків - пені
цилінів, що є похідними пепіциланової кислоти — конденсованої сис
теми, що складається з п ’ятичленного тіазолідинового кільця (А) та
(3-лактамного (В),
Загальна структура пеніцилінів, як похідних 6 -аміноценіциланової ки
слоти, ма є такий вигляд:
О
6-амінопеніциланова кислота
Пеніциліни
Залежно від хімічної природи радикала Я розрізняють численні Пені-4
циліни, які дещо відрізняються за своїми хімічними властивостями та
антимікробною активністю:
Р ад и кал И:
Н азв а пеніциліну:
С6Н ,-С Н „ Бензилпеніцилін (пеніцилін Ж
с6н5-о -с н 2Феноксиметилпеніцилін (пеніцилін V)
С 6Н5-СН Ы Н 2Ампіцилін
О
Оксацилін
358
3.3. Алкалоїди як лікарські засоби
Біохімічний механізм антимікробної дії пеніцилінів полягає в вс здатності
інгібувати фермент транспептидазу, необхідний для синтезу складного
вуглеводного каркасу (пептидоглікану муреїну) клітинної стінки бактерій.
Деякі так звані “грамнегативні” мікроорганізми виявляють нечутли
вість до пеніцилінів, що пов’язано з їх здатністю виробляти спеціальний
фермент пеніциліназу ((3-лактамазу), який розщеплює (3-лактамне кіль
це в молекулі пеніциланової кислоти з утворенням біологічно неактивної
пеніцилоїнової кислоти:
сн3
ІЗ'»
Г ^Н 3
+н2о
£ ]
-М-
Я —
//
о ' Ч' п ін
^ с о о н
О
Пеніцилін
СН3
'СО ОН
Пеніциярїнова кислота
Цефалоспорини. Цефалоспорин С - антибіотик, що був уперше ви
ділений в 1961р. з екстракту мікроскопічного гриба СерЬаІоврогіпшп
асгетоп іи т. За хімічною будовою цефалоспорини є п о х і д н и м и 7-амі~
ноцефалоспоранової кислоти (7-АЦК) та 7-амінодезацетоксицефалоспоранової кислоти (7-АДЦК) і близькі до пеніцилінів, оскільки містять
(3-лактамне кільце, сполучене з кільцем частково гідрованого 1,3-тіазину:
(В)Н2МЧ
(Р О Н г^
В
о
N.
7-АЦК
— СН2— О —СОСН3
7-АДЦК
Окремі представники цефалоспоринів розрізняються за хімічною при?*
родою радикала Я {цефалоспорин С, цефазолін, цефацетрил, цефаклор, цефаміцин Сі цефакситин). Аналогічно пеніциліну цефалоспо
рини пригнічують активність транспептиЬази, що бере участь у син*
тезі муреїну - основи клітинної стінки чутливих до антибіотика бакте
рій. Пеніциліни та цефалоспорини - найбільш поширені в наш час анти
біотики, їх виробництво становить Д о 60-65 % вартості усіх антибіоти
ків на міжнародному фармацевтичному ринку.
359
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
С трептом іцини. Стрептоміцин -антибіотик, що продукується ниж
чим грибом актиноміцетом Actinomyces globisporus streptomycini. Нале
жить до класу аміноглікозидних антибіотиків, які за своєю хімічною
будовою є похідними вуглеводів, зокрема глюкозаміну, сполучених між
собою глікозидними зв ’язками. Молекула стрептоміцину складається з
дисахаридного фрагменту (И-метил-Ь-глюкозамін (А), зв ’язаного із
L-стрептозою (В)), та аглікону - стрептидину (С):
ОН
Стрептоміцин
За механізмом дії стрептоміцин належить до фізіологічно активних
сполук, щ о гальмують біосинтез білка в рибосомах. Зв’язуючись з пев
ним рибосомним бійком ЗОБ-субодиниць прокаріотів, аміногдікозидні
антибіотики протидіють включенню амінокислот в поліпептидні ланцю
ги, що синтезуються. Стрептоміцин використовується переважно як
антибактеріальний засіб ііри лікуванні туберкульозу. Крім того, препа
рат є ефективним при лікуванні туляремії, чуми, бруцельозу та деяких
інших важких інфекційних захворювань.
Тетрацикліни. До класу тетрациклінів належать поліоксикарбонільні
сполуки природного походження, основою хімічної структури яких є ча-стково гідрований цикл тетрацену (нафталіну). В медичній практиці за
стосовують препарати Тетрациклін (4-диметиламіно- 1,4,4а,5а,6,11,12аокгагідро-3,6,1ОД 2,1 2 а-пентаок-си-6 -метил- 1 ,1І -дикетонафтацен- 2 -карбоксамід), Окситетрациклін, Метациклін, Морфоциклін, Доксициклін.
360
5.4. В ітам ін и . К оф ер м ен ти
Тетрацен
Тетрациклін
Для отримання антибіотиків тетрациклінового ряду застосовують
мікроорганізми ^ ге р іо ту се в аигео£асіеп8 та БігерЮтусев гіто вш . Тет
рацикліни належать до антибіотиків широкого спектру дії, ефективних
щодо багатьох патогенних для людини мікроорганізмів.
5.4. В іта м ін и . К оф ерм енти
В ітам іни - біоорганічні сполуки, що є життєво необхідними компо
нентами обміну речовин; на відміну від інших біомолекул, вітаміни, як
правило, не синтезуються в організмі людини, а надходять з продукта
ми харчування. ГІроте, як Встановлено новітніми дослідженнями, деякі
вітаміни повністю (вітамін В 3) або частково (вітамін А, вітаміни РР, Вй,
біотин, фолієва кислота) синтезуються в тканинах організму людини або
мікрофлорою 1кишечника за умов достатнього надходження з їжею їх
метаболічних попередників.
Класифікація вітамінів
Враховуючи, що відкриття перших препаратів вітамінів значно пе
редувало розшифровці їх хімічної структури, історично склалися по*
значення вітамінів, що містять велику літеру латинського алфавіту з
цифровим індексом та, в деяких випадках*—основний біологічний ефект
з префіксом “анти-”. В сучасних назвах вітамінів вказують також їх
хімічну природу.
Залежно від фізико-хімічних властивостей (розчинності в воді або в
ліпідах) вітаміни традиційно поділяють на дві великі групи: водорозчинні
та жиророзчинні.
До водорозчинних вітамінів належать: вітаміни В,, В,, В 6 В.», віта?»
мін РР (В5), пантотенова кислота (вітамін В3), фолієва кислота (вітамін
Вс), біотин (вітамін Н), вітакін С (аскорбінова кислота).
361
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
До ж иророзчинних віт амінів належать вітаміни А, Е, К, О.
М еханізми д ії віт ам інів
За біохімічними механізмами дії більшість водорозчинних вітамінів
(т. з. “вітаміни групи В”) є коферментами, тобто небілковими компонен
тами складних білків-ферментів або попередниками в синтезі певних
коферментів: Жиророзчинні вітаміни входять до складу біомембран як
антиоксиданти і виконують певні регуляторні функції на рівні окремих
клітинних структур та цілого організму.
Патологічні стани, що розвиваються внаслідок зменшення (або від
сутності) певного вітаміну в організмі, дістали назву гіповіт амінозів та
авіт амінозів. Причиною розвитку цих патологічних процесів є недо
статність певних вітамінів в продуктах харчування або неспроможність
організму до їх використання, наприклад порушення всмоктування пев
них жиророзчинників вітамінів у кишечнику.
Гіпо- та авітамінози призводять до глибоких порушень метаболічних
процесів та клітинних функцій, в яких беруть участь певні вітаміни як
специфічні біомолекули.
Коферментні вітаміни: будова, біохімічні функції
До цього класу належать вітаміни, що с кристалічними сполуками,!
добре розчинними в воді та інших полярних розчинниках. Механізми
біхійічИОЇ дії більшості водорозчинних вітамінів (за виключенням віта-:
Мїйу С) полягають у Їх використанні в побудові коферментів, які каталі
зують у складі ферментних білків важливі метаболічні реакції - кофер
м ент ні ф ункції віт амінів.
Вітамін В , (тіамін). За хімічною будовою тіамін є продуктом конден
сації двох гетероциклічних сполук - похідного піримідину (2-метил-5-гідроксиметил-6 -амінопіримідину) та тіазолу (4-метил-5-гідроксіетилтіазолу):
Вітамій В, був першим вітаміном, щодо якого було доведено значен
ня як необхідного фактора харчування. Вітамін В, був отриманий в
362
5.4. В ітам ін и . К о ф ер м ен т е
1911 р. польським дослідником К. Функом (1884-1967) з рисових висівок.
Сполука, виділена К. Функом, попереджала розвиток хвороби бері-бері
(специфічного поліневриту, спричиненого тривалим споживанням поліг
рованого рису) і містила в своїй структурі аміногрупу, що і стало підста
вою запропонованого для всіх додаткових факторів харчування терміну
“вітаміни” ( у і ї а т т и т - амін ж и т т я ; лат.).
Біологічна активність вітаміну В, полягає в участі в енергетичному,
зокрема вуглеводному, обміні. Біохімічній механізм дії вітаміну зумов
лений його коферментною формою —тіаміндифосфатом (ТДФ), який
утворюється в результаті фосфорилювання вільного тіаміну за участю
ферменту тіамінфосфокінази:
Тіамін + АТФ -> Тіаміндифосфат + АМФ.
ТДФ є коферментом, що бере участь у функціонуванні складних поліферментних комплексів піруватдегідрогенази та б-кетоглутаратдегідрогенази, які каталізують реакції окислювального декарбоксилювання
кетокислот - проміжних інтермедіатів вуглеводного обм іну-піровиног
радної та а-кетоглутарової.
Вітамін В г За хімічною будовою вітамін В 2 {рибофлавін) є похід
ним трициклічної сполуки ізоалоксазину та спирту рибітолу —6,7-диметил-9-рибітилізо-алоксазин:
Рибофлавін
Біохімічні функції вітаміну В 2 полягають в його участі в окислювально-відновлювальних реакціях. Коферментнгши формами рибофлавіну
363
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
Є флавінаденіндинуклеотид (ФАД) та флавінмононуклеотид (ФМН) *
простетичні групи багатьох анаеробних та аеробних дегідрогеназ та
оксидаз - ферментів, що каталізують окисленні численних інтермедіатів вуглеводного, ліпідного та амінокислотного обміну.
Ж 2
А З
>
П О Р -О Н
ОН ОН
І
0
СН 2 0 Р 0 3 Н2
о=р— он
(СНОН)з
1
СНд— О
(Ін о н ь
сн2
срн2
г т т
о
-Ж
т
Ні
о
ФАД
ФМН
В іт ам ін Р Р (вітамін В5). Біологічні властивості вітаміну РР (ніацину) мають нікотинова кислота (піридин-3-карбонова кислота) та її амід,
які є в організмі взаємно перетворюваними молекулярними формами:
ІЧН2
ОН
'О
N
Нікотинова кислота
364
N
Нікотинамід
5.4. В ітам ін и . К оф ер м ен ти
Коферментними формами вітаміну РР є коферменти анаеробних
дегід роген аз Н А Д + (н іко ти н ам ід ад ен ін д и н у кл ео ти д ) та Н А Д Ф +
(нікотинамідаденіндинуклеотидфосфат), до складу яких входить амід ні
котинової кислоти. НАД* та НАДФ+ є коферментами дегідрогеназ - фе*
рментів, що каталізують біохімічні реакції окислення численних субстра
тів вуглеводного, ліпідного, амінокислотного та інших видів метаболізму.
NH2
Коферменти НАД* (R W -Н) та НАДФ+ (R
-Р 0 3Н2)
Недостатність вітаміну РР —пелагра (pelle agra шершава шкіра;
італ.); проявляється характерними патологічними змінами шкіри та (при
тривалому перебігу патології) важкими нейропсихічними розладами.
В іт ам ін В 6 (піридоксин). Терміном піридоксш о б ’єднують три
близькі за дією та взаємно перетворювані в біологічних тканинах по
хідні піридину (розділ 3.4.): піридоксш (2-метил-3-окси-4,5-діоксиметилпіридин), піридоксаль Та піридоксамін\
365
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
,0
но
он
■ОН
НО
он
м н 2
но
он
Біохімічні функції вітрйіну Вв пов’язані з участю в реакціях айіиоіййслотного обміну. Його коферменмними формами є піридоксаль-5-фос
фат (ПАЛФ) та піридоксамін-5-фосфат (І1АМФ), що є коферментами т рансам іназ та дека р б о кси ла з, які беруть участь в к атал ізі
відповідних реакцій біотамічного перетворення амінокислот.
Г1іридоксаль-5-фосфат
Піридоксамін-5-фосфат
Вітамін
(кобаламін). Вітамін В12 (кобаламін) за хімічною будовою
належить до класу кориноїдів; йош молекула складається з двух частин В
кобапьтвмісної порфіриноподібноїта нуклеогидної структур. Атом Со", що
міститься в центрі ядра хромофорної структури, в комерційних препаратах
вітаміну В12 сполучений з ціанідною трупою (ціанакобаламін).
Коферментними формами вітаміну В (2, що присутні в організмі, єг
метилкобаламін та 5-дезоксіаденозилкобаламін.
366
5.4. Вітаміни. К оф ерм ент
н о н 2с
п
Іф огн
о
Н О - Р Н |- 0
ОН
н3с - і н
С Н г ^ Н -С О -С Н г
М Н г-С О ^С Н гГ 1.
Вітамін В 12(кобаламін)
Біохімічна функції коферментних ф орм ивітам іну В 12 полягають в
участі в каталізі біохімічних реакції обміну амінокислот та нуклеотидів.
Найбільша кількість цього вітаміну в тваринних організмах міститься в
печінці. Недостатність вітаміну В,2 проявляється у вигляді т. з, макроцитарйої (“злоякісної’^перніціозної) анемії ^хвороба Адисона - Бірмера.
Біохімічною основою розвитку злоякісної вітамін В 12-залежної анемії є
порушення біосинтезу нукл еїнових кислот та білків у процесі дозрівання
клітин крові.
Фолієва кислот а (вітамін Вс). Фолієва кислота за хімічною приро
дою є похідною птеринів — птероїлгяутпаміновою кислотою, що міс
тить у складі молекули сполучені з похідним птерину фрагменти
п-амінобензойної кислоти (ПАБК) та Ь-глутамату:
367
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНГ СПОЛУКИ
ОН
соон
СООН
Фолієва кислота
Коферментною формою фолієвої кислоти є й гідроване похідне
5Д |7, 8-тетрагідрофол ієва кислота (ТГФК). Коферментні функції ТГФК
полягають в міжмолекулярному переносі одновуглецевих фрагментів
(метильного, метиленового, метенільного, оксиметильного, формільного, форміміно-), що використовуються в багатьох реакціях обміну амі-»
нокислог, синтезі нуклеотидів ДНК та РНК, фізіологічно активних сполук!
Проявом авітамінозу ФК є захворювання “спру”, яке характеризуй
ється макроцитарною анемією та пінистими проносами (§рги\у - піна;
голландськ.); захворювання розвивається внаслідок споживання раціо
ну, збідненого білками, що призводить до порушення синтезу мікроорганізмами кишечника фолієвої кислоти.
Віт амін Н (біотин). За будовою молекули вітамін Н (2-кето-3,4-імідазолідо-^тетрагідротіофенвалер’янова кислота) є продуктом конден-|
сації сечовини татіофенвалер’янової кислоти:
О
СООН
т г
Біотин
Коферментна функція біотину полягає в його участі в реакціях карбоксилювання. Акцентуючи С 02 з утворенням М рбоксибіот ш у, віта-'
368
5.4. В ітам ін и . К оф ер м ен ти
мін Н бере участь в біохімічних реакціях, пов‘язаних з ускладненням
структури біомолекул при утворенні жирних кислот синтезі глюкози (гаюконеогенезі), пуринових нуклеотидів. Прояви вітамінної недостатності
можуть розвиватися за умов порушення функціонування нормальної мі
крофлори кишечника (нераціональне використання сульфаніламідів та
антибіотиків) І проявляються патологічними змінами шкіри за типом
себореї, що дістало відображення в назві вітаміну (haut - шкіра; нім.).
Пантотенова- кислота (вітамін В3). Молекула пантотенової кис
лоти являє собою сполучення (3-аланіну з похідним масляної кислоти —
а,у-дигідрокси-(3,(3‘-диметилбутиріл-Р-аланіном:
Павтотенова кислота
В організмі людини пантотенова кислота використовується для син
тезу коензиму А (КоА-БН) - коферменту ацилювання, що є одним з
ключових коферментів в реакціях метаболізму вуглеводів, окислення та
синтезу жирних кислот, обміну амінокислот та ін.
Віт амін С (аскорбінова кислота)
За хімічною будовою вітамін С (Ь-аскорбінова кислота єу-лакгоном
2,3-ди-гідро-Ь-гулонової кислоти:
ОН
СН2'
| ІІ-аскорбінова кислота
У водних розчинах Ь-аскорбінова кислота (Ь-АК) зворотно перетво
рюється на дегідроформу - Ь-дегідроаскорбінову кислоту (Ь-ДАК) - роз
діл 4.1, яка повністю зберігає біологічні властивості вітаміну С; подальші
369
Розділ 5, ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
окислювальні перетворення L-ДАК є незворотними і призводять до утво
рення похідних, що не мають вітамінних властивостей. Подібних ж©
перетворень L-AK зазнає і в організмі (іп vivo).
Біологічні властивості та механізми dit
Вперше L-асюрбінова була виділена у чистому вигляді видатним біохі
міком угорського походження А. Семт-Дьєрдьїзнаднирників бика (1928 p.),
а потім з червоного перцю та лимонного соку. L-аскорбінова кислота
синтезується в більшості рослинних та тваринних організмів і не синте
зується (тобто І вітаміном) у людини, Морських свинок, деяких прима
тів та летючих мишей, що пов’язують з мутаціями певних генів на ета
пах прадавньої еволюції тваринних організмів*
Емпірична назва вітаміну С —аскорбінова кислота вказує на його
профілактичну дію по відношенню до цинги, або екорбушу (scorbut;
scurvy; англ.) - специфічного патологічного процесу, спричиненого екзо
генною недостатністю вітаміну при недостатності в раціоні харчування
свіжих продуктів переважно рослинного походження (овочів, фруктів).
Найбільш виражені симптоми ЦгШги стосуються системи сполучної
тканини і проявляються ураженням судинних стінок і Опорних тканин,
що містять структурний білок колаген: у хворих відмічається-збільшен
ня проникностістінок кровоносних судан з розвитком підвищеної крово
точивості; (особливо ясен), виникненням “петехій” (дрібнокрашшнних
крововиливів) на шкірі і слизових оболонках , випадіння зубів, порушення
структури і функції суглобів.
Біохімічні функції вітаміну С в організмі людини полягають в участі
L-аскорбінової ксилоти в реакціях Ре3+-аскорбатзалежного гідроксилю- j
ваннії деяких важливих біомолекул, а саме: гідроксилюваниі залишків
амінокислот проліну та лізину до відповідних гідроксиамінокисдот в
процесі синтезу зрілого білка колагену; гідроксилюванні в процесі си н И
тезу біогенних амінів (дофаміну, норадреналіну, адреналіну, серотоніну)
та стероїдних гормонів.
Ж иророзчинні вітам іни
Вітаміни, що входять до цього класу, являють собою олієподібні ре- І
човини, які добре взаємодіють з гідрофобними розчинниками. Завдяки j
наявності в структурі молекул довгих вуглеводневих (переважно - ізо - 1
преноїдних) радикалів, більшість з цих вітамінів є струкгурнйми iÉR)Я
понентами біомембран, у складі яких вони виконують специфічні біологічні і
370
5.4. В ітам ін и . К оф ер м ен ти
функції, зокрема є потужними біоантиотидантами (вітаміни В, А, К).
Порушення всмоктування жиророзчинних вітамінів в тонкому кишечнику»
яке залежить від наявності поверхнево-активних молекул жовчних кис
лот, може супроводжуватися розвитком вітамінної недостатності.
Вітамін А (ретинол), Сполуки, щ о мають біологічні властивості
вітаміну А, е похідними Р-іОЩону. Дві молекулярні форми вітаміну А
(вітамери) - А. та А, - є циклічними ненасиченими спиртами (трансізомери), щ о ш ю т ь в якості бічного радикала гідрофобну диізопреноїдну групу, завдяки якій ці речовини розчиняються в ліпідному біщарі
мембран. Найбільша кількість вітаміну А міститься в продуктах тва
ринного походження (вершковому маслі, молоці, печінці, ясчному жовт
ку, риб’ячому жирі):
Н3 С С Н
3
Н3 С С Н
3
В
Вітамін А, -ренянв»(А) та а і ш ш
дегідроретивод (В)
У рослинних організмах містяться провітаміни (біологічні поперед
ники) вітаміну А ,— жовті пігменти а, Р та у-каротини (розділ 4,2.).
Активними молекулярними формами вітаміну А: спирт (ретинол), аль
дегід (ретиналь) та трансретиноєва кислота.
Біологічні властивості
Біологічна активність вітаміну А полягає в регуляції таких функцій
організму: процесів темпового (нічного) зору - недостатність вітамі
ну А супроводжується порушенням темнового зору і розвитком “куря
371
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
чої сліпоти” (гемералопії), процесів рост у і диференціювання клітин
(“вітамін росту”) та утворення біологічних слизів шляхом шікозилювання білкових молекул.
Роль вітаміну А в процесах темнового зору грунтується на участі
альдегідної форми вітаміну - 11-цис-ретиналю в Побудові фоторецепторного білка родопсину, що міститься у спеціалізованих світлочутли
вих клітинах сітківки - паличках. Участь вітаміну А в процесах росту
та диференціювання клітин реалізується /п/?аке-ретиноєвою кислотою,
яка утворюється з альдегідної форми. Значення вітаміну А для глікозилювання білків полягає в коферментної функції ретинолу у складі глікозштрансфераз як ліпідного переносника олігосахаридних залишків через
лілопротеїнові мембрани до місць глікозилювання пептидної частини
гаішпротешів, що утворюють біологічні слизі.
Вітамін К. Властивості вітаміну К має група вітамерів - похідних
2-метил-1,4-нафтохінону. Розрізняють вітамін К, (філохінон) та вітамін
К , (фарнохінон).
Вітамін К, є похідним 2-метил-1,4-нафтохінону, бічним вуглеводне
вим радикалом в якому є похідне ізопрену - фітил (2-метил-3-фітил-1,4нафтохінон). Цей вітамер був вперше виділений з люцерни і є біологічно
найбільш активною формою вітаміну К.
СНс
СН3
С Н = С — СН2— (СН2— СН2— СН— СН2)3Н
О
Вітамін" К,
Вітамін К, має довший бічний ізопреноїдний ланцюг, являючи собою
за хімічною будовою 2-метил-3-фарнезил-1,4-нафтохінон; вітамер був
вперше виділений з рибного борошна.
372
5.4. Вітаміни. Коферменті!
О
Вітамін Kj
Біологічні властивості
Біологічна дія вітаміну К в організмі людини і тварин полягає в його
впливі на згортання крові, як кофактора реакцій коагуляції. Вітамін К є
необхідним компонентом для утворення факторів коагуляції крові II, VII,
IX, X (“антигеморагічний” вітамін). При недостатності вітаміну К розви
ваються важкі кровотечі внаслідок порушення здатності крові до коагуляції.
Віт амін Е. Властивості вітаміну Е має група похідних токолу
(2-метил-2(4',8’,12'-триметилтридецил)-6-хроманолу - а , (3 та у-токо
фероли, що були вперше виділені з рослинних олій. Найбільшу біологічну
активність має а-токоферол (5,7,8-триметилтокол):
СН3
ÎHg— С Н 2— С Н — с н 2)3н
з
Вітамін Ê (а-токоферол)
Біологічні властивості та механізми д ії
Вітамін Е має широкий спектр біологічної активності - його недостат
ність супроводжується численними змінами обмінних процесів та фізіо
логічних функцій організму. Найбільш характерними для Е-авітамінозу є
глибокі порушення репродуктивної функції як у чоловіків (аномальний
сперматогенез) так і жінок (неспроможність запліднення та виношування
вагітності), м ’язові дистрофії, некрозодистрофічні процеси в печінці.
373
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
Згідно з сучасними уявленнями, основні молекулярні механізми фізіо-|
логічної активності сс-токоферолу полягають у його антиоксидантній д ії
як інгібітора реакцій вшьнораДикального окислення поліненасичениХі
жирних кислот (розділ 4.2,}.
Вітамін Щ До вітамінів групи Щ належать вітамери: вітамін 0 3
(холекальциферол, вітамін О тваринного походження) та вітамін В 2(ер
гокальциферол), вітамін О рослинного походження:
374
U . Гормони. Нейромедіатори
Біологічні властивості та механізм д ії
На відміну від розглянутих вище жиророзчинних вітамінів, вітамін D3
повністю синтезується в організмі людини з похідної холестерину —
7 -дегідрохолестерину при УФ-опроміненні шкіри (розділ 4.2.). Біологіч
ною функцією вітамінів групи D є регуляція гомеостазу кальцію та фос
фатів. Зокрема, вітамін D 3 є попередником фактору гормонального типу
д іїкальцитріолу (l,25(O H )2D|J, якии стимулює утворення в кишечнику
Ca-зв’язуючих білків ентероцитів - регуляторів всмоктування в кишеч
нику іонів Са2+та фосфатів, необхідних для кісткоутворення та контролю
багатьох Са-залежних біохімічних процесів. Недостатність вітамінів D
у дітей супроводжується порушенням кальцієво-фосфорного обміну,
остеомаляцією і розвитком рахіту (rhachis М. хребет; хребетний стовп).
Особливо багатим джерелом вітаміну D 3 є риб’ячий жир, що широко
використовується для профілактики і лікування рахіту.
5.5. Г орм они, Н ей ром едіатори
Гормони - фізіологічно активні сполуки, що продукуються залозами
внутрішньої секреції (ендокринними залозами) або іншими спеціалізова
ними клітинами і діють як біорегулятори, контролюючи перебіг біохіміч
них реакцій та фізіологічних функцій в організмів Н ейром едіатори (нейротрансміттери) ~ біомолекуяи, що забез
печують передавання хімічних сигналів (нервових імпульсів) у нервовій
системі з одного нейрона на інший або з нейрона на ефекторний орган.
За своїми фізіологічними функціями як гормони» так і нейромедіато
ри є сигнальними молекулами, що реалізують процеси міжклітинної
комунікації в складних багатоклітинних організмах.
За хімічною будовою гормони та нейромедіатори поділяються на
такі класи:
1) білки та пептиди;
2 ) аміни та їх похідні; вільні амінокислоту;
3) стероїди;
4 ) ейкозаноїди (похідні арахідонової кислоти).
Гормони та н ейром едіатори б іл кової т а п еп ти д н ої природи
До цього класу належить численна група ФАС із гормональними та
нейромедіаторними властивостями, що продукуються в різних органах і
тканинах та за своєю хімічною природою є поліпептидами (простими
375
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
білками, низькомолекулярними пептидами) або глікопротеїнами. Крім
“справжніх” гормонів, що синтезуються в залозах внутрішньої секреції
(інсуліну, глюкагону, окситоцину, вазопресину, паратгормону, кальц
цитоніщу, гормонів гіпофіза тагіпоталамуса тощо), найбільше фізіологіч-і
не значення мають опіоїдні пептиди головного мозку та інші нейропеп«
тиди (енкефаліни, ендорфінщ сполука Р), пептиди гастро-інтестинальної системи (гастрин, холецистокіиін), компоненти кінінової (бра^
дикінін, каллідин) та ренін-ангіотензинової системи, гормони та медіан
тори імунної системи (імун оглобул іни, інтерферони, інтерлейкіни) та*
функціонально близькі до них пептидні фактори росту, натрійуретичніі
пептиди серця та мозку тощо. Детально будова та властивості цих*
біорегуляторів розглядаються в курсах біохімії та спеціальних медико -,1
біологічних дисциплін.
Аміни, амінокислоти та їх похідні я к гормони та нейра/иедіаторил
До низькомолекулярних сигнальних молекул, що виконують функцій
гормонів або нейромедіаторів, належать ацетилхолін, тиреоїдні гормони - |
похідні Ь-тирозину, біогенні аміни (катехоламіни, індоламіни, імідазола-:
міни), нейромедіаторні амінокислоти (Ь-глутамат та Ь-аспартат - збу
джувальні кислі амінокислоти; у-аміномасляна кислота, гліцин, а-ала-і
нін, таурин - гальмівні нейтральні амінокислоти).
Ацетилхолін - складний ефір метильованого аміноспирту холіну та
оцтової кислоти - є найбільш поширеним медіатором центральної та;
периферичної нервової системи.
СН
СН 3
Ацетилхолін
Біосинтез та розщеплення ацетилхоліну відбуваються в хрлінергічних центрах (ядрах, нейронах) нервової системи. Сполуки, що вплива
ють на активність розщеплення ацетилхоліну в організмі (інгібітори
ферменту ацетилхолінестерази) та його взаємодію із специфічними
холінорецепторами (холінолітики) знайшли широке застосування в
клінічній медицині.
376
5.5. Гормони. Иейромедіатори
Тиреоїдні гормони: тироксин. трийодтиронін
Тироксин. Трийодтиронін. Тироксин (3,5,3 ‘,5 ‘-тетрайолтиронін; Т4)
та трийодтиронін (3,5 53 ‘-трийодтиронін; Т ^ - гормони щитовидної зало
зи, що є йодованими похідними амінокислоти Ь-тирозину.
5'
6'
Тироксин (Т )
Трийодтиронін ( ТЛ
Біосинтез цих гормонів відбувається у фолікулярних клітинах щито
видної залози шляхом включення молекулярного йоду в структуру білка
тиреоглобуліну, який в подальшому гідролізується з утворенням віль
них молекул гормонів Т4 та Тг Порушення біосинтезу тиреоїдних гор
монів, що спостерігається найчастіше за умов ендемічної недостатності
солей йоду в питній воді та продуктах харчування, проявляється різними
формами функціональної недостатності щитовидної залози (гіпотиреоз,
“ендемічний зоб”).
Катехдяаміни. До катехоламінів (пірокатехінамінів) належать по
хідні амінокислоти Ь-тирозину: адреналін, норадреналін, дофамін.
Адреналін. Норадреналін. Дафамін. Адреналін, норадреналін та
дофамін є біогенними амінами, що синтезуються з Ь-тирозину в ре
зультаті послідовних реакцій декарбоксилювання діоксифеиілаланіну
(ДОФА) та подальш их реакцій гідрокеилю вання та метидю вання
(розділ 2.7.). Норадреналін (норепінефрин) відрізняється від адрена
ліну (епінефрину) відсутністю метильного радикала в бічному ланцюзі
‘ЧЧ опЬе НасЦкаІ”
без радикала” - нім.).
377
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
Адреналін
Норадреналін
Дофамін
Катехоламіни мають властивості як гормонів, так і медіаторів централь
ної і периферичної нервової системи з широким спектром фізіологічної
активності.
Вони синтезуються в хромафінних клітинах мозкового шару наднир*
никових залоз (переважно адреналін), гангліях симпатичної нервової си
стеми, адренергічних (норадреналін) та дофамінергічних (дофамін)
структурах центральної нервової системи*
Індолам іни
Серотонін. Фізіологічно активним представником індоламінів є
індолалкіламін серотонін ( 5 -гідрокситриптамін)—продукт послідовного
гідроксшдавання та декарбоксилювання амінокислоти Ь-триптофану.
Н
Серотонін
Серотонін належить до ФАС, що мають властивості як тканинного
гормону, так і медіатора серотонінергічних структур центральної та
периферичної нервової системи. Фізіологічні функції серотоніну в голо- ,
вному мозку людини полягають в регуляції разом із специфічними нейи
ропептидами таких психоемоційних реакційна станів, як тривога, неспо-|
кій, агресивність, Імпульсивні потяги, процеси с н у Порушення обміну]
серотоніну та функцій серотонінових рецепторів є біохімічною ОСНОВОЮ ]
патогенезу депресивних станів, потягів до самогубства, розвитку!
шизофренії, алкоголізм^ наркоманій.
Порушення базових нейропсихічних функцій головного мозку епрйй
чиняють галюциногени (дизлептики), у складі молекул яких знаходиться
залишок індолу Це, зокрема, такі речовини, як діет мамід лізергінової
378
5.5. Горм они . Н ей ром едіатор и
кислоти (“сполука ЛСД”) Та рослинні алкалоїди - пстоцин та пеилоцибін, що містяться в мексиканському грибі РзіїосуЬе техісапа.
О
<р
О
С Н 2С Н г М + ( С Н з ) з
\
н
Псилоцибін
їм ід аз ол ам іни
Гістамін Щбіогенний амін (імідазоламін), що є продуктом декарбоксилювання амінокислоти Ь-гістидину.
о
н
Гістамін
379
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
Найбільша кількість гістаміну синтезується в спеціалізованих клітиі
нах сполучної тканини —тканинних базофілах та Мстамінергічних
структурах головного мозку, У зв’язку з різноманітністю фізіологічних]
ефектів гістаміну, широке застосування в медичній практиці знайш ли
лікарські засоби, що протидіють взаємодії цієї ФАС із специфічними кліч
тинними рецепторами. Зокрема, блокаториН^рецепторів гістаміну знай-І
шли широке застосування Як антиалергічні засоби (Димедрол, Діазолін,]
Кетотіфен), блокатори Н 2-рецепторів (Ранітідин, Ф ам отід и н )- як
антисекреторні препарати при Лікуванні виразкової хвороби.
С тероїд н і горм он и
Стероїдні гормони є похідними вуглеводнів прегнану, естрану та]
андростану (розділ 4.2.), За-фізіологічними та біохімічними ефектамш
які вони спричиняють в організмі, стероїдні гормони підрозділяються на]
такі класи:
~ стероїдні гормони кори наднирників (“кортикостероїди”) — по-|
хідні прегнану С2 ,-стероїди, до яких належать:
глюкокорптюїди (гормони, що регулюють переважно обмін ш ю ш зиУ
мінералокортшоїди (гормони, що є регуляторами обміну м іне-|
ральних елементів, переважно Иа+ та ІС+);
- стероїдні гормони статевих залоз, до яких належать:
чоловічі статеві гормони (андрогени) *■ похідні андростану - 1
С 19-етероїди;
ж іночі ст ат еві гормони - п о х ід н і ест рану (ест рогени) - і
С 1&-стероїди та прегнану (гестагени) — С2,-стероїди.
Найбільш важливими представниками гормонів означених класів у люди-І
ни є кортизол, альдостерон, тестостерон, естрадіол та прогестерон. 1
Кортизол - головний представник глюкортикоїдів. Як і альдостерон-І
належить до С21-стероїдів. Основним біологічним ефектом кортйзолу Є ]
регуляція вуглеводного обміну, що виражається стимуляцією синтезу!
глюкози в печінці з безазотистих продуктів катаболізму амінокислоті
(процесу глюконеогенезу).
Альдост ерон - найбільш активний представник мінералокортикої-1
дів. Гормональна дія альдостерону полягає в затримці, в організмі іонів
Иа+ і стимуляції екскреції іонів К+та водню Н+. Біохімічні ефекти альдо-1
стерону реалізуються через його вплив на транспорт іонів в клітинах!
ниркових канальців. Збільшене утримання в організмі натрію супрово-1
380
5.5. Горм они. Н ей р ом ед іатор и
джується також затримкою в тканинах води, тобто головний фізіологіч
ний ефект альдостерону полягає в зменшенні сечоутворення (діурезу).
с н 2о н
СН 2ОН
Л ,
І
СН 3 С = 0
и он
Кортизол
сУ/
с=о
Альдостерон
Тестостерон - основний представник чоловічих статевих гормонів
(андрогенів), що належить до представників С, 9-стероїдів. Гормон
синтезується в клітинах Лейдіга сім’яників та контролює розвиток і функ
цію статевих залоз та експресію вторинних чоловічих статевих ознак.
Естрадіол (17-Р-естрадіол) - основний, найбільш активний предста
вник естрогенів - жіночих статевих гормонів (С 18-стероїдів), що син
тезуються у фолікулах яєчників, а в період вагітності також у плаценті.
Разом з прогестероном є регулятором менструального циклу, стимулю
ючи проліферацію клітин ендометрію, а також контролює розвиток жіно
чих вторинних статевих ознак.
Прогестерон - головний представник гестагенів —жіночих стате“
вих гормонів, що, як і кортикостероїди, належить до С2|-стероїдів - ПОХІД
НИХ прегнану. Синтезується переважно в другу фазу менструального
циклу, в жовтому тілі яєчників, а також у плаценті (“гормон вагітності”).
381
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
Е йкозаноїди я к м іж к л іти н н і біорегулятори
Ейкозаноїди - клас фізіологічно активних похідних арахідонової кис
лоти (ейкозатетраєн-5,8,11,14-ової кислоти; С20;4). Залежно від особли
востей хімічної структури та біологічних властивостей, ейкозаноїди пси'
діляються на простаноїди (простагландини та простацикліни), тромбов
ксани та лейкотрієни.
Ейкозаноїди синтезуються в багатьох клітинах тваринного організму
і розглядаються як тканинні гормони місцевої дії. Порушення обміну
цих ФАС євавжливою лайкою патогенезу багатьох хвороб, що протіка
ють з явищами запалення; алергічних та анафілактичних процесів, дис
функції судинного тонусу, скорочення гладеньких м ’язів тощо. У зв’язку
з високою фармакологічною активністю препарати ейкозаноїдів знай
шли застосування в сучасній фармакології. Як приклад наведемо будову
простагландину Е,, лікарські форми якого (препарати Мізопростол,
Цитотек) використовуються при лікуванні виразкової хвороби.
О
382
5.6. П ести ц и ди та ін ш і біоц и дн і ксен обіоти к и
5.6. П естициди та іяш і біоцидні ксенобіотики
П естициди т сполуки, що застосовуються в сільському господарстві
як засоби захисту культурних рослин від різних шкідників. За хімічною
будовою структура пестицидів надзвичайно різноманітна. їх класифіку
ють залежно від типу шкідників, на який вони чинять токсичну (біоцидну) дію. Так* розрізняють: інсектициди (засоби боротьби з комахами),
гербіциди (діють проти бур’янів), акарициди (засоби проти кліщів) тощо.
За механізмом дії пестициди виступають як інгібітори ферментів (фос
форорганічні сполуки), засоби, що ушкоджують біомембрани,системи
синтезу білків і ДНК (більшість хлорорганічних сполук).
Фосфорорганічні пестициди. Фосфорорганічні пестициди (фосфор
органічні сполуки; ФОС)) є незворотними інгібіторами ацетиЛхолінестерази - ферменту (Е), що в синаптичній щілині каталізує гідроліз аце
тилхоліну - одного з головних медіаторів центральної та периферичної
нервової системи:
О
Е
+
( С Н з ) з ^ N— СН2 — СН2 — О— С
СН3 + Н20 -------►
+
-► ( С Н з ) з ^ N—
СН2
СН2
ОН + НООС —
СН3
Накопичення ацетилхоліну супроводжується різким збудженням не
рвової системи з розвитком судом, порушенням функцій серцево-судинноїі Гастроінтестинальної та інших систем організму Препарати фосфо
рорганічних сполук є високотоксичними отрутами не лише для комах, а
й для теплокровних тварин і людини, при цьому загибель настає від
паралічу дихального центру.
Хімічна будова всіх фосфорорганічних сполук', що пригнічують акти*
вність щєтшхсшінестерази, може бути виражена такою загальною
формулою (О. І, Черкес, 1964):
383
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
де Я, і
- різні або однакові алкіли, алкоксил- чи алкіламіни; X залишок неорганічної або органічної кислоти (найчастіше — фтор або
інші галогени, СІЧ), а також залишок нітрофенолу, інших гідроксилзамі«
щених ароматичних чи гетероциклічних сполук.
Механізм токсичного ефекту ФОС полягає в їх здатності сполучати^
ся ковалентними зв ’язками з гідроксильною групою серину в активна-!
му центрі ацетилхолінестерази, блокуючи таким чином зв ’язування
ферменту з субстратом - ацетилхоліном. Цю реакцію можна проілюст
рувати на прикладі дії модельного ФОС диізопропілфторфосфату (ДФФ):
^С Н 3
СН2-;-О Н
:
і+ / \
нсх
»
/ СНз
N3— сне'
АХЕаза
{База
ДФ Ф
О
3
/ 'С Н з
Фосфорильована Д Ф Ф АХЕаза
Розроблено спеціальну групу лікарських засобів - антидотів при от
руєннях фосфорорганічними сполуками, які за механізмом молекуляр
ної дії € реактиваторами АХЕази, тобто сприяють вивільненню акти
вного центру ферменту від ковалентного зв’язку з молекулою отрути
(Алоксим, Дипіроксим).
Представниками фосфорорганічних пестицидів, які широко викорис
товуються в сільському господарстві й побуті, є такі сполуки:
н3с— о
\
Дихлорф ос
[0,0-ди м ети л -0-(2,2-ди хл ор він іл )ф осф ат; Д Д В Ф ]
384
5.6. П ес т и ц и д и та ін ш і біоц и дн і к сен обіоти к и
н3с— о
н3с— ох
о— с6н4мо2
Метафос (метилнаратіон)
Н3С— о
\ р^
Н3С——о
8
в— снсоос2н5
сн2соос2н5
Карбофос (малатіон)
Направлений синтез фосфорорганічних сполук, що є високотоксич
ними для організму теплокровних тварин, сприяв отриманню нервовопаралітичних отрут, які були прийняті на озброєння як бойові отруйні ре
човини (БОР). Найбільш відомими БОР з групи фосфорорганічних спо
лук є такі речовини:
Табун
Зарин
сн3
Зоман
385
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
Великі запаси фосфорорганічних БОР, що були накопичені в 70-х-80-х
роках XX сторіччя у ряді країн світу, зробили актуальною технічною,
еш цігічною та медичною проблемою розробку методів їх знешкодження, безпечних для здоров’я людей та стану навколишнього середовища.
Х лорорганічні пестициди. Найбільш поширеним хлорорганічним
пестицидом протягом багатьох років був диХлордифенілтрихлоретан
(ДДТ), токсичність якого для комах була вперше встановлена в 1939 р.
II. Мюллером:
ССІз
сін О - ':н—С ^ сі
ДДТ
Застосування ДДТ в роки другої світової війни дало можливість при
пинити поширення епідемії висипного тифу, а пізніте —ліквідації осеред
ків малярії та сонної хвороби.
Прикладом високоактивного інсектициду є також у-стереоізомер гекеахлорциклогексану (гексахлорану), що дістав комерційну назву “Ліндан”:
СІ
•Гекеахлорцикяогексан
Піретроїди. Хлорорганічні пестициди досить токсичні для організ
му людини та сільськогосподарських тварин, до того ж, у зв’язку з ліо
фільною природою, вони схильні до акумуляції в жировій тканині. Забруд
нення біосфери цими пестицидами спонукало до пошуку нових, менш
токсичних речовин, що призвело до виявлення піретроїдів - сполук, які
практично не токсичні для теплокровних організмів і менш стабільні в
навколишньому середовищі.
386
5.6 . П ест и ц и д и та ін ш і біо ц и д к і к сен обіоти к и
Піретроїди належать до фізіологічно активних сполук природного (ро
слинного) походження, один з перших активних препаратів {піретрин 1 )
був виділений з багатокомпонентного екстракту ромашки “піретруму” :
Піретрин І
Пізніше були синтезовані штучні аналоги піретроїдів (декаметрин,
перметрин, циперметрин), що значно перевершують піретрин 1 за інсе
ктицидною активністю та є малотоксичними для організмів людини і
тварин. На сьогодні кількість синтетичних піретроїдів, що застосову
ються в сільському господарстві промислово розвинутих країн, налічує
сотні назв. Встановлено, що комахами та кліщами знищується до 20 %
світової сільськогосподарської продукції, у зв’язку з чим проблема син
тезу високоефективних та екологічно безпечних пестицидів має надзви
чайно велике соціальне значення.
Хлоралкани. До хлоралканів з токсичною дією щодо організму
людини та тварин належать т. з. “промислові отрути” - хлоровані вугле
водні тетрахлорметан СС14 та 1,2-дихлоретан СН 2С1—С Н 2СІ. Ці
сполуки широко використовуються в промисловості як розчинники та
реагенти в багатьох хімшьтехнологічних процесах, а при надходженні в
організм вони викликають важкі пошкодження клітин печінки, нирок,
міокарда, головного мозку,
В основі молекулярних механізмів цитотоксичної дії ксенобіотиків з
класу хлоралканів лежить утворення з цих сполук за участі мікросомальних монооксигеназ печінки (цитохрому Р-450) вільнорадикальних
продуктів метаболізму типу R* та R 0 2*. Класичним прикладом утво
рення в біохімічній системі вільного радикала ксенобіотика є тетрахлор-
Розділ 5. ФІЗІОЛОГІЧНО АКТИВНІ СПОЛУКИ
метан, молекула якого зазнає гемолітичного розщеплення за такою схе
мою (Ю. І. Губський, 1989):
ССІ4 —> ССІд* + СІ* ;
СГ + 0 2-> СЮ 2*,
Радикали СГ та С10 2 можуть вступати у взаємодію з молекулами
поліненасичених жирних кислот, білків та азотистих основ ДНК, призводячи до вільнорадикального ушкодження цих біомолекул (розділ 4.2) та
глибоких Порушень у функціонуванні ферментних систем біомембран
та генетичного апарату клітини.
388
ДОДАТОК
Міністерство охорони здоров’я України
Центральний методичний кабінет з вищої медичної освіти
Б ІО О Р Г А Н ІЧ Н А Х ІМ ІЯ
Програма
дпя студентів медичних та стоматологічних факультетів
вищих медичних закладів освіти
ІЕИ У рівнів акредитації
Київ
2000
ДОДАТОК
П огодж еної
Директор
Центрального методичного
кабінету з вищ ої медичної освіти
І. С. Вітенко
4. 05.2000 р.
Зат вердж ено:
Начальник
Управління освіта та медичної
науки МОЗ України
Ю. В. Вороненко
5.05,2000 р.
Програму розроблено
кафедрою біоорганічно% біологічної та фармацевтичної дгшй" 1
Національного медичного університету імені О. О. Богомольця зав. кафедри —член-кор. АМН України, заслужений діяч науки і
техніки України, професор Ю . І. Губський).
Рецензенти:
— завідувач кафедри Загальної хімії Національного медичного універ-ситету ім. О. О. Богомольця проф. В. О. Калібабчук;
— завідувач кафедри біологічної хімії Української медичної стомато
логічної академії проф. Л. М. Тарасенко, доценти Р. Я. Юхновець,!
В. К. Григоренко, О. І. Цебржинський, М. Я. Червиць;
— Завідувач кафедри біоорганічної, фізколоїдної та біонеорганічноїхімії,
Харківського державного медичного університету доц. Л. Г. Шаповал2
П О Я С Н Ю В А Л Ь Н А ЗА П И С К А
Біоорганічна хімія вивчає будову та реакційну здатність органічних
речовин, що входять до складу живих організмів, - низькомолекулярних
біомолекуд, біополімерів (білків, нуклеїнових кислот, полісахаридів),
природних та синтетичних фізіологічно активних сполук (гормонів, віта
мінів, лікарських засобів, токсичних речовин тощо). Завдання біооргані
чної хімії полягають у визначенні структури біомолекул, природнихі син
тетичних біорегуляторів, виявленні залежностей між їх молекулярною,
електронною будовою та фізіологічними, зокрема фармакологічними
ефектами, вивченні закономірностей щ перетворень.
У вищих медичних закладах освіти біоорганічна хімія викладається
на першому курсі і передує вивченню біомедичних дисциплінцбіологіч3 90
ДОДАТОК
ної хІмЙ, фармакології, токсикології, імунології та ін., теоретичною осно
вою яких вона К В основу навчальної Програми, що подається, покладе
но багаторічний Досвід навчального процесу на кафедрі біоорганічної,
біологічної та фармацевтичноїхімії Національного медичного універси
тету ім. О. О. Богомольця, інших провідних медичних вузів України.
При розробці програми використано системний підхід до структури
курсу на єдиній теоретичній основі—електронній та просторовій будові
органічних сполук з урахуванням механізмів їх хімічних перетворень.
Викладення навчального матеріалу здійснюється поступово - від фун
даментальних загально-теоретичних положень до вивчення окремих
класів органічних сполук з урахуванням їх реакційної здатності.
Матеріал курсу з біоорганічної хімії представлений двома основними
розділами:
І. Теоретичні основи біоорганічної Хімії.
Д. Будова та властивості окремих класів біоорганічних сполук.
У І розділі представлено: будову молекул, природу хімічного зв’язку
в біоорганічних сполуках, взаємний вплив атбмів в молекулах, види ізо
мерії, типи та механізми реакцій, яким підлягають біоорганічні сцолуки.
У II розділі розглядаються гідроксюївміснІ та карбонільні сполуки,
аміни, гетерофункціональні біоорг анічні сполуки (багато з яких є проміж
ними продуктами розщеплення або синтезу природних органічних речо
вин: аміноспирти та їх йдхідні, біогенні аміни, гідроксикислоти, оксокислоти та ін.), гетероциклічні сполуки та їх похідні —важливі складові
біомолекул та фізіологічно активних речовин, У цьому розділі також пред
ставлені біополімери (вуглеводи, білки, нуклеїнові кислоти), біорегулятори
та і нігті ФАС (вітаміни, гормони, антибіотики, алкалоїди, пестициди).
Матеріал програми інтегрований з вивченням навчальних дисциплін
медико-біологічного циклу. Зокрема, в перелік реакційної здатності ор
ганічних сполук включено переважно реакції, які відбуваються в біосистемах; охарактеризовано структуру і властивості білків “ рівні їх струк
турної організації,^методи осадження та денатурації, будову ферментів,
нуклеїнових кислот, деяких природних фізіологічно активних сполук та
біорегуляторів. Розглянуто багато біоорганічних речовин, які мають вла
стивості лікарських засобів: антибіотиків, сульфаніламідів, протизапаль
них, нейро- та психотропних препаратів т а ін.
391
ДОДАТОК
Під час викладання курсу біоорганічної хімії застосовуються традив
ційні форми навчання —лекції проблемної спрямованості та лабораторій
заняття, на яких виконуються експериментальні роботи, що формують
у студентів певні вміння і практичні навички. Контроль за засвоєння^
знань та вмінь проводиться у вигляді контрольних робіт. Крім того, на
більшості занять використовуються поточний тестовий контроль та
контроль за виконанням лабораторних робіт.
Кінцева мета вивчення курсу біоорганічної хімії:
1. Навчити студентів загальним принципам .оцінки хімічних властш
востей та перетворень біоорганічних речовин в процесі життєдіяльності ■
організму.
2, Розкрити практичні аспекти біоорганічної хімії, її значення для ви
вчення фундаментальних медичних дисциплін (біохімії, фармакології ]
тощо) та використання здобутих знань в клінічній практиці і профілактичнім
медицині.
Після вивчення курсу біоорганічної хімії студенти повинні вміти:
1. Юіасифі кувати біоорганічні сполуки за будовою вуглецевого лан?,!
цюга і природою функціональноїтрупи.
2 . Користуватись правилами систематичної номенклатури для н а -1
писання структурних формул за назвою і знати зображені структурні
формули сполук найбільш важливих класів біоорганічних речовин.
3. Складати рівняння реакцій перетворень найбільш поширених к л а-1
сів біоорганічних сполук.
4. Проводити якісні реакції на виявлення ненасиченості, на доведен- І
ня відновних властивостей сполук (зокрема, моносахаридів), наявнісТ^ і
пептидної групи, на деякі амінокислоти, що входять до складу білків.
5. Користуватися хімічним посудом та оволодіти навичками прове-1
дення хімічного експерименту, техніки безпеки роботи в хімічній лабо - 1
раторії, реєструвати та аналізувати експериментальні результати, оформ - 1
ляти протоколи експериментальних досліджень.
У програмі наведено також орієнтовні тематичні плани лекцій, лабораторно-практичних занять, самостійної позааудйторної роботи студен- |
тів, розраховані на такий розподіл навчальних годин: всього годин - 60, в
тому числі: лекцій - 1 6 , лабораторно-практичних та контрольних занять - І
24, сам остійної позааудйторної роботи студентів - 2 0 . Кінцевим
контролем набутих знань є залік або іспит. Зміни в зазначеній кількості І
ї
і
392
ДОДАТОК
годин, відведених на вивчення біоорганічної хімії в медичних універси
тетах, інститутах, академіях та на медичних факультетах загальноосві
тніх університетів, можуть бути проведені Окремими навчальними за
кладами за погодженням з МОЗ України.
1, ВСТУП. БІООРГАНІЧНА ХІМ ІЯ ЯК НАУКА
Біоорганічна хім ія як наука, що вивчає хімічну структуру і власти
вості органічних сполук, які входять до складу живих організмів та впли
вають на процеси життєдіяльності
Предмет, завдання та розділи біоорганічної хімії як науки; та навчаль
ної дисципліни. Органічна хімія як фундаментальна основа біоорганічної
хімії. Будова та властивостіфізіологічно активних сполук як роздідбіоорганічної хімії, Методи дослідження в біоорганічній хімії,
Значення вивчення біоорганічної хімії в системі вищої медичної осві
ти, її необхідність для оволодіння іншими медико-біологічними дисцип
лінами (біологічною хімією, молекулярною біологією, загальною та клі
нічною фармакологією, токсикологією, імунологією тощо).
2. ТЕОРЕТИ ЧНІ ОСНОВИ БІООРГАНІЧНОЇ ХІМ ІЇ
2.1. Класифікація та номенклатура органічних сполук
2.1.1. Класифікація органічних сполук. Класифікаційні ознаки: будог
ва вуглецевого ланцюга, природа функціональних груп. Класи органічних
сполук за будовою вуглецевого ланцюга. Характеристика найважливі
ших класів органічних (біоорганічних) сполук за природою функціональ
них грушспиртів, фенолів, тіолів, альдегідів, кетонів* карбонових кислот
та їх похідних, складних ефірів, амідів, нітросполук, амінів;
2.1.2. Номенклатура органічних сполук. Номенклатурні системи:
тривіальна, раціональна, міжнародна (номенклатура ІЮПАК). Принци
пи утворення назв органічних сполук за номенклатурою ІЮ ПАК: замісниковий, радикало-функціональний.
2.2. Будова біоорганічних сполук
2.2.1.
Теорія будови біоорганічних сполук. Хімічна будова моле
кул органічних сполук; поняття про структурні ізомери (ізомери будови).
Природа хімічного зв’язку в органічних сполуках: гібридизація орбіталей атома вуглецю, електронна будова сполук вуглецю (валентні ста
ни атома вуглецю).
393
ДОДАТОК
Делокалізація електронів та спряжені системи в органічних сполу
ках: р, я - та 71, я-спряження. Спряжені системи з відкритим ланцюгом;
електронна будова 1,3-дієнів (бутадієну, ізопрену); особливості хімічної
поведінки 1,3-дієнів. Спряжені системи із замкненим ланцюгом: елект*
ронна будова бензолу; ароматичність в ряду одно- та багатоядерних
аренів, гетероциклічних сполук.
Взаємний вплив атомів в органічних молекулах: поляризація зв’язків;
індуктивний (І-) та мезомерний (М-) ефекти; Електронодонорні та електроноакцепторні замісники, їх вплив на реакційну здатність молекулі
Орієнтуючий вплив замісників І та II роду в реакціях аренів.
2.2.2.
П рост орова будова м олекул біоорганічних сполук. П рос
торова будова сполук вуглецю; стереохімічні формули; конфігурація та
конформація органічних сполук. Просторові ізомери (стереоізомери^
геометричні, онтичні, поворотні (конформери).
Геометрична ізомерія: приклади біоорганічних сполук, що мають цей
тин ізомерії (заміщені алкени, циклоалкани, ненасичені вищі жирні кис-*
Лоти, дикарбонові кислоти). Цис~, транс- та E/Z-системи позначень
геометричних ізомерів.
Оптична ізомерія; асиметричний (хіральний) атом вуглецю; хіральність
молекул органічних сполук. D/L- та R/S- стереохімічні системи. Проекційні '
формули Фішера. Приклади біоорганічних сполук - оптичних антиподів
(енантіомерів) та діастереоізомерів (гліцериновий альдегід, молочна
кислота, «-ам інокислоти, винні кислоти, м оносахариди). З в ’язок
просторової будови з біологічною активністю сполук. Рацемічні суміші
біоорганічних сполук; асиметричний синтез в біохімічних системах.
Поворотна (конформаційна) ізомерія. Проекційні формули Ньюмена.
Енергетичні характеристики конформаційних ізомерів сполук з відкри
тим ланцюгом в син-, анти- та гош-конформаціях. Конформації цикліч
них сполук; стереохімія циклогексану, аксіальні та екваторіальні зв ’яз
ки. Значення шнформаційної ізомерії для утворення просторової струк
тури біомолекул (білків, вищих жирних кислот, стероїдів).
2.3. Т ипи р еакц ій біоорганічних сполук
Загальна характеристика реакцій біоорганічних сполук. Типи реакцій
в органічній хімії: приєднання (А), замінність (S), відщеплення (еліміну
вання) (Е), перегрупування, окислення і відновлення. Класифікація реак
цій за механізмом: гемолітичні (радикальні), гетеролітичні (іонні) реакцій ;
394
ДОДАТОК
Електрофільні та нуклеофільні реагенти в гетеролітичних органічних
реакціях. Приклади перетворень біоорганічних сполук за зазначеними
типами та механізмами:
Б. (реакції радикального заміщення в алканах та циклоалканах);
А е (реакції електрофільного приєднання в неНасичених сполуках алкенах, алкадієнах);
Бе (реакції електрофільного заміщення в аренах І гетероциклічних
сполуках);
(реакції нуклеофільного приєднання в оксосполуках);
(реакції нуклеофільного заміщення в галогеналканах, спиртах, кар
бонових кислотах і гетероциклах);
Е (реакції елімінування на прикладі спиртів та гетерофункціональних
сполук).
Окислювально-відновні реакції біоорганічних сполук: дегідрування,
міжмолекулярного транспорту електронів, приєднання кисню. Вільнорадикальйі реакції утворення біоорганічних пероксидних сполук, їх біомедичне значення в нормі та за умой патології клітини,
Кислотні та основні властивості біоорганічних сполук: протонна тео
рія Бренстеда; теорія кислот і основ Льюїса.
3. БУДОВА І В Л А С ТИ В О С ТІ О К РЕ М И Х КЛ А С ІВ БІО О РГА
Н ІЧ Н И Х С П О Л У К
З .ї. Г ідроксилвм існі сполуки т а тіоли в біоорганічній хімії
Спирти: будова, властивості та перетворення одно- і багатоатомних
спиртів. Біомедична характеристика найбільш поширених одноатомних
(метанол, етанол) та багатоатомних (етиленгліколь, гліцерин, ксиліт, сорбіт) спйртів. Прості ефіри: будова, властивості.
Феноли: будова, властивості, біомедичне значення. Одноатомні (фе
нол, крезоли) та двоатомні (пірокатехін, резорцин, гідрохінон) феноли.
Тіоли (меркаптани) та їх похідні; сульфіди, дисульфіди—їх біомедич
не значення.
3.2. К арбонільн і сполуки в біоорганічній хімії
3.2.1.
Альдегіди та кетони. Будова та реакційна здатність карбоніль
ної групи. Властивості альдегідів та кетонів. Біомедичне значення
найбільш поширених представників.
395
ДОДАТОК
3.2.2.
Карбонові кислоти. Стадні ефіри. Будова і хімічні властим
вості карбонових кислот. Функціональні похідні карбонових кислот: соліа
ангідриди, складні ефіри, аміди. Реакції декарбоксилювання карбонових
кислот залежно від кількості і взаємного розташування карбоксильних груці
Дикарбонові кислоти: щавлева, малонова, бурштинова, піутарова, фу
марова. Вугільна кислота та ЇЇ похідні: уретани, уреїди кислот, сечовинам
Складні ефіри карбонових кислот: номенклатура, способи добування!
властивості.
3.3. Аміни в біоорганічній хім ії
Аміни: номенклатура, хімічці властивості, біомедичне значення.
Аліфатичні аміни. Біогенні аміни (адреналін, норадреналін, дофаміні
триптамін, Серотонін, гістамін) як гормони та нейромедіатори. Поліамід
ни (путресцин, спермідин, спермін, кадаверин).
Ароматичні аміни. Анілін як попередник в синтезі лікарських засо-|
бів - сульфаніламідів, фенацетину, анестезину, новокаїну.
3.4. Г етероф ун кц іон ал ьн і біоорганічні сполуки
3.4.1. Аміноспирти. Будова, властивості, біомедичне значення амҐ-І
носпиртів: етаноламіну (шламіну), холіну, ацетилхоліну.
3.4.2. Гідроксикислоти та амінокислоти. Властивості та специ- ]
фічні хімічні реакції аліфатичних а-, (3-, у-гідроксикислот та а-, 3-, у-аміно-!
кислот. М онокарбонові (молочна, (3-гідроксимасляна), дикарбонові
(яблучна, винні) і трикарбонові (лимонна, ^мс-аконітова) гідроксикисло-!
ти. Реакції біохімічних перетворень амінокислот: дезамінування, тран с-*
амінування, декарбоксйлування.
3.4.3. Оксокислоти. Будова та властивості найбільш поширених в
біооб’єктах оксокисЛот: піровиноградної, ацетооцтової, ЩавлевооцтовоО
а-кетошутарової. РеакцйГ утворення з ацетооцтової кислоти ацетону та'
Р-гідроксибутирату (кетонових тіл).
3.4.4.Фенолокислоти. Саліцйлова кислота: х ім й н і властивості як
біфуйкціональної сполуки; похідні саліцилової кислоти як нестероїдні про
тизапальні (ацетилсаліцилова кислота - аспірин, метилсаліцилат, салі
цилат натрію) та протимікробні (фенілсаліцилат) засоби. Галова кислота.
3.5. Г етероциклічні сполуки та їх похідні
3.5:1. П ’я тичленні гетероцикли з однім гет ероат ом і». Пірол, ■
фуран, тіофен - будова, властивості. Біомедичне значення гетрапірольних сполук; порфінів, порфіринів, Гема. БейзпіроЛгі: індол та його похідні
396
ДОДАТОК
(індоксил, скатол, скатоксил), їх утворення в процесах гниття білків у
кишечнику; триптофан і реакції утворення триптаміну та серотоніну. Тіофен як структурний компонент молекули біотину (вітаміну Н).
3.5.2. П ’ятичленні гетероциклы з двома гетероатомами. П ’яти*
членні гетероцикли з двома гетероатомами азоту. Піразол, ціразолон;
похідні піразолону - 5 як лікарські засоби (антипірин, амідопірин, аналь
гін). Імідазол та його похідні: гістидин, гістамін,
П’ятичленні гетероцикли з двома різними гетероатомами: тіазол, оксазол. Тіазол як структурний компонент молекули тіаміну (вітаміну В,).
3.5.3. Шестичленні гетероцикли з одним гетероатомом. Ш е с
тичленні гетероцикли з атомом азоту. Піридин, його ароматичність, Хі
мічні властивості, похідні; Нікотинова кислота! її амід (вітамін РР) як
складові частини окисно-відновних коферментів НАД (нікотинамідаденіндинуклеотиду) та НАДФ (нікотинамідаденіндинуклеотидфосфату).
ПіридоксИн (вітамін В6), його молекулярні форми (піридоксол, піридоксаль, піридоксамін) та коферментні функції.
Ш естичленні гетероцикли з атомом кисню, ос-піран та (3-піран.
Участь у-пірану в утворенні флавонів (вітаміну Р).
3.5.4. Шестичленні гетероцикли з двома гетероатомами. Ш ес
тичленні гетероцикли з двома атомами азоту. Діазини: піримідин, піразин, піридазин. Піримідин та його похідні (урацил, цитозин, тимін); тауто
мерні форми піримідинових основ. Похідні піримідину як лікарські засо
би: 5-фторурацил, оротат калію, барбітурати. Фенобарбітал, веронал як
снодійні та протиепілептичні засоби. Піперазин (гексагідропіразин) як
антигельмінтний засіб.
Шестичленні гетероцикли з різними гетероатомами. Фенотіазини (амі
назин та ін.), як антипсихотичні (нейролептичні) засоби.
3.5.5. Семичленні гетероцикли з двома гетероатомами. Діазепіни.
Бензо-*1,4-діазепіни як найбільш поширені транквілізатори та анксіоЛітики.
3.5.6. Конденсовані системи гетероцитів. Пурин та його п охідн і
Амінопохідні пурину (аденін, гуанін), їх таутомерні форми, біохімічне
значення. Гідроксийохідні пурину (гіпоксантин, ксантин, сечова кисло
та, їх таутомерні форми)< Метильовані похідні ксантину (кофеїн, теофі
лін, теобромін) як фізіологічно активні сполуки, що діють на центральну
нервову систему, серце, судини.
Алоксазин. Ізоалоксазин як структурний компонент молекули рибо
флавіну (вітаміну В2).
397
ДОДАТОК
3.6. Вуглеводи т а їх похідні
3 6.1. Моносахариди. Визначення, класифікація (альдози і кетози;тріозй, тетрози, пентози, гексози, гептози), біомедйчне значення окре
мих представників. Пентози: рибоза, 2-дезоксирибоза, ксилоза, рибулоза, ксилулоза. Гексози: глюкоза, галактоза, маноза, фруктоза.
Стереоізомерія моносахаридів (О- і Ь-форми). Відкриті та циклічні
форми моносахаридів; циклооксотаутомерія ( формули Фішера, Коллі Толленса, Хеуорса). Фуранозні та піранозйі цикли; а - та (3-аномери.
Хімічні властивості моносахаридів: реакції за участю оксо- Та гідро
ксильних груп; якісні реакції на моносахариди. Окислення моносахари
дів* утворення альдонових та уронових кислот. Ь-аскорбінова кислота
(вітамін С). Глікозидний гідроксил, його властивості; О- і М-гаікозиди.
Амінопохідні моносахаридів: глюкозамін, галактозамін. Будова серце
вих глікозидів. Відновлення моносахаридів; утворення сорбіту, маніту.
3.6.2. Олігосахариди: Будова. Дисахариди.(сахароза, лактоза, маль
тоза): структура, біомедйчне значення.
3.6.3. Полісахариди. Гомополісахариди. Полісахариди: класифіка
ція, структура. Гомополісахариди: крохмаль, глікоген, целюлоза (клітко
вина), декстрани: будова, гідроліз, біомедйчне значення. Якісна реакція
на крохмаль.
3.6.4. Гтеропачісахариди. Визначення, структура. Будова та біомедичне значення глікозаміногліканів (мукополіеахаридів) - гіалуронової кислоти, хондроїтинсульфатів, гепарину.
3.7. Л іп ід и
3.7.1.
Прості Ліпіди. Триацилгліцероли (тригліцериди, нейтральні
жири): будова, хімічні властивості, фізіологічне значення. Вищі жирні
кислоти: пальмітинова, стеаринова, олеїнова, лінолева, ліноленова, арахі
донова. Залежність фізико-хімічних властивостей природних жирів від їх
жирнокислотпого складу. Аналітична характеристика жирів (йодне чис
ло). Гідроліз жирів (кислотний, лужний, ферментативний). Мила: будова,
фізикохімічні та біологічні властивості як поверхнево-активних сполук.
З.Я,2- Складні ліпіди. Класифікація та будова основншс класів скла
дних.ліпідів,- Фоефоліпіди: фосфатидна кислота, фосфатидилетаноламін,
фосфатидилхолін, фосфатидкисерин. Сфінголіпіди. Гліколіпіди. Роль скла
дних ліпідів у побудові біомембран.
398
ДОДАТОК
3.7.3.
Ізопреноі'ди(терпени, стероїди)■. Терпени (терпенові вугле
водні): будова; біомедичне значення як лікарських засобів (камфора,
ментол, ефірні олії) та компонентів будови жиророзчинних вітамінів (ві
тамінів К та Е). Стероїди як похідні циклопснтанпергідрофенантрену
(стерану). Будова біологічно важливих представників стероїдів, холес
терину, вітаміну О, жовчних кислот, кортикостероїдів (кортизолу, альдо
стерону), статевих гормонів (тестостерону, естрадіолу, прогестерону).
3 .8 . П р о теїн о ген н і ам ін о к и с л о ти , п еп ти д и , б іл ки
3.8.1. Амінокислот ний склад білків та пептидів. П ри р о д н і
І ,-а-амінокислоти: класифікація зй структурою радикала Я і кількістю
амінних і карбоксильних груп та за полярністю і зарядом радикала Я.
Фізико-хімічні властивості протеїногенних амінокислот як амфотерних
електролітів. Реакції, застосовувані для кількісного визначення вільних
аміно- та карбоксильних груп (реакції Ван-Слайка, Серенсена). Хімічні
реакції, що використовуються для аналізу амінокислот в біосередовищах
та гідролізатах білків і пептидів: нІнгідринова, флуорескамінова, ксантопротеїнова, реакції Мілона, Фоля. Хроматографічне розділення амінокислот.
3.8.2. Будова і властивості білків та пептидів. Молекулярна маса
та форма білків. Пептидний зв’язок: утворення, структура;. біуретова
реакція на пептидну Групу Типи зв’язків між амінокислотними залишка
ми у білкових молекулах: ковалентні (гіептидні, $йсульфідні), нековалейші,
слабкі взаємодії (водневі, іонні, дипольні зв’язки, гідрофобні взаємодії).
Рівні структурної організації білкових молекул: первинна, вторинна,
третинна та четвертинна. Олігомерні білки. Фізико-хімічні властивості
білків. Методи осадження. Денатурація. Методи фракціонування та ана
лізу білків і пептидів (седиментація, хроматографія, електрофорез). Ана
ліз первинної структури білків і пептидів: методи С е т е р а (застосування
2,4-дииітрофторбєнзолу) та Едмана (застосування фені лізотіоціанату).
Ферменти як біологічні каталізатори білкової природи. Принципи но
менклатури та класйфікації ферментів. Поняття про механізми фермен
тативного каталізу.
3.9. Н у к л ео ти д и , н у к л еїн о в і к и с л о ти
3.9.1.
Нуклеозиди та нуклеотиди. АзотйСТІ основи пуринового і
піримідинового ряду як компоненти нуклеозидів та нуклеотидів: будова,
хімічні властивості, таутомерія. Мінорні азотисті основи.
399
ДОДАТОК
Нуклеозвди. Нуклеотиди як фосфорильовані похідні нуклеозидів (нуклеозид-монофосфати, ди- і трифосфати АМФ, ГМФ, УМФ, ЦМФ, ТМФ); вза
ємодія як структурних компонентів нуклеїнових кислот. Вільні нуклеотиди;
нуклеотиди - коферменте; циклічні нуклеотиди З \ 5 ’-АМФ, 3 ’,,5’-ГМФ.
3.9.2.
Нуклеїнові кислоти: дезоксирибонуклеїнові рибонуклеїнові
як полінуклеотиди. Полярність полінуклеотидних ланцюгів.
..Характеристика первинної, вторинної, третинної структур нуклеїно
вих кислот; зв’язки, що їх формують. Генетична роль ДНК. Типи РНК
та їх роль в біосинтезі білка.
ЗДО. Ф ізіологічно а к ти в н і сполуки. Б іорегулятори
Поняття про фізіологічно активні сполуки (ФАС). Рецепторні та метаболітні механізми дії ФАС на функціонування клітин.
» Вітаміни; загальна характеристика; поняття про коферментну дію
вітамінів. Будова вітамінів В(, В 2, В6, РР, С.
О рієнтовний тем ати ч н и й п л ан лекц ій з біоорганічної хім ії
N
1.
Т ем а л екц ії
Біоорганічна хімія як наука; її місце в медичній
освіті. Класифікація, будова та реакційна
здатність органічних сполук
2.
Гідрокси- та оксосполуки: будова та реакційна
здатність
3. Карбонові кислоти та їх функціональні похідні.
Ліпіди; будова, властивості
4. Амінокислоти, пептиди, білки: будова,
властивості, біологічні функції
5. Вуглеводи: класифікація; будова і властивості
моносахаридів та їх похідних
6.
Олігосахариди та полісахариди: будова,
властивості
7. Гетероциклічні сполуки. Нуклеотиди.
Нуклеїнові кислоти: будова, біологічні функції
8.
Фізіологічно активні сполуки. Поняття про
вітаміни, гормони, алкалоїди, антибіотики
400
К іл ькість
годин
2
2
2
2
2
2
’ 2
2
•
ДОДАТОК
Гормони: поняття про гормони як біорегулятори. Загальна характе
ристика гормонів білково-пептидної групи, похідних амінокислот, стеро
їдів та ейкозаноїдів.
Алкалоїди: загальне визначення, значення алкалоїдів як діючих речо
вин лікарських засобів (класів піридину та піперидину, хіноліну та ізохіноліну, тропану, індолу).
*
Антибіотики: поняття про антибіотики; характеристика антибіотиків
класів пеніцилінів, цефалоспорйнів, стрептоміцинів.
Пестициди: загальне визначення; фосфори та хлорорганічні пести
циди * застосування в медицині» сільському господарстві, токсиколо
гічне значення.
О рієн товн и й те м а ти ч н и й п л ан л а б о р ато р н о -п р ак ти ч н и х
за н я ть з біоорганічної хімії
N
1.
2.
' 3,
4.
5.
6.
7.
8.
9.
10.
.
12.
11
Т ем а зан яття
Кількість
годин
Принципи класифікації та номенклатури
органічних сполук. Ізомерія
Природа хімічного зв’язку в органічних
сполуках. Взаємний вплив атомів; електронні
ефекти замісників
Реакційна здатність органічних сполук
2
Гідрокси- та оксосполуки
Карбонові кислоти та їх похідні.Ліпіди
2
Гетерофункціональні сполуки. Контрольна
робота N 1
Ь-б-амінокислоти, пептиди, білки
Вуглеводи; будова і властивості моносахаридів
2
Вуглеводи: ди- і полісахариди, Контрольна
робота N2
Гетероциклічні сполуки
Нуклеотиди, нуклеїнові кислоти
Фізіологічно активні сполуки. Вітаміни,
гормони, атибіотищ
2
2
2
2
2
2
2
2
2
40І
ДОДАТОК
Теми позааудиторної самостійної роботи студентів
з біоорганічної хімії
N
! і.
2,
3.
4.
5.
6.
7.
Тема заняття
Поняття про конфігурацію молекул біоорга
нічних сполук. Конформація сполук з відкри
тим та закритим вуглецевим ланцюгом.
Стереохімія біоорганічних сполук Цнс-*
трансізомерія; О/Ь та К/Б-ізомери. Зв’язок між
просторовою будовою та біологічною
активністю
Характеристика ароматичного зв’язку-. Типи
спряження: р, р та р, р. Відкриті та закриті
спряжені системи. Ароматичність аренів,
гетероциклічних сполук. Особливості ХІМІЧНИХ
властивостей спряжених систем при взаємодії
з галогенами, воднем, галогеноводнями (на
прикладі реакцій бутадієну-^ 1,3)
Хімічні властивості гаЛОгеналканів, спиртів,
фенолів. Механізм цих реакцій
Кислотність та основність біоорганічних
сполук за Бренстедом та Льюїсом
Хімічні властивості аліфатичних амінів, якісні
реакції виявлення первинних, вторинних,
третинних амінів. Ароматичні аміни, методи
добування, особливості хімічних реакцій.
Адреналін, норадренаЛіни
Аміди вугільної кислоти (карбамінова кислота
та сечовина). Одержання біурету. Біуретова
реакція.
Гетероцикли в біологічних системах та
лікарських препаратах. Алкалоїди як лікарські
засоби
К іл ькість
годин
4
4
4
2
3
2
3
ДОДАТОК
П Е РЕ Л ІК
к он трольн и х п и тан ь до зал іку (іспиту)
з біоорганічної хімК
1. Біоорганічна хімія як наука: визначення, предмет і завдання, розді
ли, методи дослідження. Значення в системі вищої медичної освіти.
2. Класифікація органічних сполук за будовою вуглецевого радикала
та природою функціональних груп.
3. Будова найважливіших класів біоорганічних сполук за природою
функціональних груп: спиртів, фенолів, тіолів, альдегідів, кетонів, карбо
нових кислот, складних ефірів, амідів, нітросполук, амінів.
4. Номенклатура органічних сполукі тривіальна, раціональна, між
народна. Принципи утворення назв органічних сполук за номенклату
рою П0ПАК: замісниковий, радикало-функціональний,
5. Теорія будови органічних сполук. Хімічна будова молекул; по
няття про структурні ізомери.
6.
Природа хімічного зв’язку в органічних сполуках: гібридизація
орбіталей, електронна будова сполук вуглецю.
7. Делокалізація електронів та спряжені системи в органічних спо
луках. Спряжені системи з відкритим ланцюгом: електронна будова та
хімічні властивості 1,3-дієнів.
8.
Спряжені системи із замкненим ланцюгом: електронна будова
бензолу; ароматичність в ряду одно-та багатоядерних аренів, гетероцик
лічних сполук.
9. Взаємний вплив атомів в органічних молекулах: поляризація зв’яз
ків; індуктивний (І-) та мезомерний (М-) ефекти. Вплив електронодонорних та елекгроноакцепторних замісників на реакційну здатність молекул .
10. Просторова будова біоорганічних сполук: стереохімічні форму
ли; конфігурація та конформація. Стереоізомери: геометричні, оптичні,
поворотні (конформери).
1 1 . Геометрична ізомерія в заміщених алкенах, циклоалканах, ненасичених вищих жирних кислотах, дикарбонових кислотах. Цис-, т рат
та E/Z-номенклатурні ейстемй.
12. Оптична ізомерія; хіральність молекул органічних сполук. D/L- та
R/S- стереохімічні номенклатури. Енантіомери та діастереоізомери біоор
ганічних сполук. Зв’язок просторової будови з фізіологічною активністю.
403
ДОДАТОК
13. Поворотні (конформаційні) ізомери; проекційні формули Ньюме
на. Енергетичні характеристики конформаційних ізомерів вуглеводнів в
син-, анти- та гош-конформаціях.
14. Конформаційні ізомери циклічних вуглеводнів; аксіальні та еква
торіальні зв’язки в молекулі циклогексану. Значення конформаційної ізо
мерії для утворення просторової структури біомолекул.
15. Типи реакцій в біоорганічній хімії: класифікація за результатом
(спрямованістю) та механізмом реакції.
16. Характеристика та приклади окремих типів реакцій в біоорганіч
ній хімії: приєднання, заміщення, відщеплення (елімінування), перегрупу
вання, окислення і відновлення.
17. Характеристика та приклади гемолітичних (радикальних) та ге-’
теролітичних (іонних) реакцій в біоорганічній хімії. Електрофшьні та нуклеофільні реагенти.
1В. Окислювально-відновні реакції в біоорганічній хімії. ВільгіорадикальнІ реакції утворення біоорганічних пероксидних сполук, їх біомедичне
значення в нормі та за умов патології клітини,
19. Кислотні та основні властивості біоорганічних сполук: протонна
теорія Бренстеда; теорія кислот і основ Льюїса.
20. Гідроксилвмісні сполуки (спирти) та тіоли в біоорганічній хімії:
будова, властивості, біомедичне значення окремих представників.
21. Феноли: будова, властивості, біомедичне значення. Характерис
тика представників одноатомних (фенол; крезоли) та двоатомних (піро
катехін, резорцин, гідрохінон) фенолів.
22. Тіоли (меркаптани), сульфіди та дисульфіди в біоорганічній хімій
будова, властивості.
23. Карбонільні сполуки в біоорганічній хімії. Хімічні властивості та
біомедичне значення альдегідів і кетонів.
24. Карбонові кислоти в біоорганічній хімії: будова і хімічні власти
вості; функціональні похідні карбонових кислот (ангідриди, аміди, складні
ефірй). Реакції декарбоксилювання.
25. Будова і властивості дикарбонових кислот: щавлевої, малонової,;
бурштинової, глутарової, фумарової.
26. Будова І властивості вугільної кислоти та її похідних. Уретани*;
уреїди кислот, сечовина.
27. Складні ефіри карбонових кислот: номенклатура, утворення, вла?
стивості.
404
ДОДАТОК
28. Аміни: номенклатура, властивості. Біомедичне значення біоген
них амінів (адреналіну, норадреналіну, дофаміну, триптаміну, серотоніну,
гістаміну) та поліамінів (спермідину, сперміну, путресцину, кадаверину).
29. Ароматичні аміни: будова, властивості. Анілін як попередник в
синтезі лікарських засобів - сульфаніламідів, фенацетину, анестезину,
новокаїну.
30. Аміноспирти: будова, властивості. Біомедичне значення етаноламіну (коламіну), холіну, ацетилхоліну.
3 1. Гідроксикислоти в біоорганічній хімії: будова і властивості монокарбонових (молочної, (3-гідроксимасляної), дикарбонових (яблучної,
винних) та трикарбонових (лимонної, ^ис-акоиітової) гідроксикислот.
32. Амінокислоти: будова, стереоізомерія, хімічні властивості. Біо
медичне значення Ь-а-амінокислот. Реакції біохімічних перетворень
амінокислот: дезамінування, трансамінування, декарбоксилування.
33. Будова і властивості найбільш поширених в біооб’єктах оксокисдбт: піровиноградної, ацетооцтової, щавлевооцтової, а-кетоглутарової.
Поняття про кетонові тіла.
34. Фенолокислоти. Саліцилова кислота та її похідні як протизапальні
(ацетилсаліцилова кислота, метилсаліцилат, саліцилат натрію) та проти
мікробні (фенілсаліцилат) засоби.
35. П ’ятичленні гетероцикли з одним гетероатомом (пірол, фуран,
тіофен). Біомедичне значення тетрапірольних сполук: порфінів, порфіри
нів, гема.
36. Індол та його похідні триптофан і реакції утворення триптаміну
та серотоніну; індоксил, скатол, скатоксил - значення в процесах гниття
білків у кишечнику. .
37. П ’ятичленні гетероцикли з двома гетероатомами азоту. Піразол, піразолон; похідні піразолону-5 як лікарські засоби (антипірин, амі
допірин, анальгін). Імідазол та його похідні: гістидин, гістамін.
38. ІГятичленні гетероцикли з двома різними гетероатомами: тіазол,
оксазол. Тіазол як структурний компонент молекули тіаміну (вітаміну В.).
39. Шестичленні гетероцикли з атомом азоту; піридин. Нікотинамід
(вітамін РР) як складова частина окисно-відновних піридинових Коферментів. Шридоксин та молекулярні форми вітаміну В6.
40. Шестичленні гетероцикли з двома атомами азоту. Діазини: піримідин, піразин, піридазин. Азотисті основи- похідні піримідину (урацил,
цитозин, Тимін).
405
ДОДАТОК
41. Похідні піримідину як лікарські засоби: 5 -фторурацил, оротат
калію. Барбітурова кислота; барбітурати як снодійні та протиепілептичні
засоби (фенобарбітал, веронал).
42. Шестичленні гетероцикли з різними гетероатомами. Фенотіазини (аміназин та ін.) як антипсИхотичні (нейролептичні) засоби.
43. Семйчленні гетероцикли з двома гетероатомами. Діазепіни; бензо- 1 ,4 -діазепіни як найбільш поширені транквілізатори та анксіолітики.
44. Пурин та його похідні. Амінопохідні пурину (аденін, гуанін), їх таутомерні форми; біохімічне значення в утворенні нуклеотидів та коферментів.
45. Гідроксипохідні пурину: гіпоксантин, ксантин, сечова кислота. Метильовані похідні ксантину (кофеїн, теофілін, теобромін) як фізіологічно ак
тивні сполуки з дією на центральну нервову та серцево-судинну системи.
46. Вуглеводи: визначення, класифікація. Моносахариди (альдози |
кетози; тріози, тетрози, пентози, гексози, гентози), біомедичне значення
окремих представників.
47. Моносахариди: пентози (рибоза, 2-дезоксирибоза, ксилоза), гекч
сози (глюкоза, галактоза, маноза, фруктоза) —будова, властивості. Якісні
реакції на глюкозу.
48. Будова та властивості похідних моносахаридів. Амінопохідні:
глюкозамін, галактозамін. Урокові кислоти. Ь-аскорбінова кислота (ві
тамін С). Продукти відновлення Моносахаридів: сорбіт, маніт.
49. Олігосахариди: будова, властивості. Дисахариди (сахароза, лак
тоза, мальтоза), їх біомедичне значення.
50. Полісахариди. Гомополісахариди: крохмаль, глікоген, целюлоза, декстрани —будова, гідроліз, біомедичне значення. Якісна реакція на крохмаль.
51. Гетерополісахариди: визначення, структура. Будова та біомеди
чне значення іл ікозаміногліканів (мукополісахаридів)—гіалуронової кис
лоти, хондроїтин-сульфатів, гепарину.
52. Ліпіди: визначення, класифікація. Вищі жирні кислоти: пальміти
нова, стеаринова, олеїнова, лінолева, ліноленова, арахідонова. Прості лі
піди. Триацил-пхіцероли (нейтральні жири): будова, фізіологічне значені:
ня, гідроліз.
53. Складні ліпіди. Фосфоліпіди: фосфатидна кислота, фосфатидилетаноламін, фосфатидилхолін, фосфатидилсерин. Сфінголіпіди. Гліголіпіди. Роль складних ліпідів у побудові біомембран.
4 06
ДОДАТОК
54. Стероїди як похідні циклопентднпергідрофенантрену (стерану).
Будова біологічно важливих представників стероїдів: холестерину, віта
міну О, жовчних кислот, кортикостероїдів, статевих гормонів.
55. Амінокислотний склад білків та пептидів; класифікація природ
них Ь-а-амінокислот. Хімічні та фізико-хімічні властивості протеїногенних амінокислот. Нінгідринова реакція, її значення в аналізі амінокислот.
56. Білки та пеггщци: визначення, класифікація, біологічні функції. Типи
зв’язків між амінокислотними залишками в білкових молекулах, Пептидний зв’язок: утворення, структура; біуретова реакція.
57. Рівні структурної організації білків: первинна, вторинна, третинна
та четвертинна структури. Олігомерні білки.
58. Фізико-хімічні властивості білків; молекулярна маса, заряд. М е
тоди осадження. Денатурація білків.
59. Методи фракціонування Та аналізу білків і пептидів (седимента
ція, хроматографія, електрофорез). Аналіз первинної структури білків І
Пептидів: методи Сенгера та Едмана60. Ферменти як біологічні каталізатори білкової природи. Принципи
номенклатури та класифікації ферментів.
61. Нуклеозиди, нуклеотида. Азотисті основи пуринового і піримідинового ряду, що входять до складу природних нуклеотидів. Мінорні азо
тисті основи.
62. Нуклеозиди. Нуклеотиди як фосфорильовані похідні нуклеозидів
(нуклеозидмоно-, да- і трифосфати). Номенклатура нуклеозидів і нуклеотидів як компонентів РНК та ДНК.
,63. Будова та біохімічні функції вільних нуклеотидів: нуклеотидні
коферменти; циклічні нуклеотиди 3’,5’-АМФ, 3’,5,-ГМФ.
64. Нуклеїнові кислоти (дезоксирибонуклеїнові,рибонуклеїнові) як
полінуклеотиди. Полярність полінуклеотидних ланцюгів ДНК та РНК.
65. Будова та властивості ДНК; нуклеотидний склад, комплементарність азотистих основ. Первинна, вторинна та третинна структура ДНК.
6 6 . РНК: будова, тиц|і РНК та їх роль в біосинтезі білків,
67. Біоорганічні сполуки з фізіологічно активною дією (ФАС): визна
чення, класифікація, рецепторні та метаболітні ефекти ФАС..
6 8 . Вітаміни: загальна характеристика; поняття про коферментну дію
вітамінів. Будова та властивості вітамінів В р В2, В6, РР.
407
ДОДАТОК
69. Гормони: поняття про гормони як біорегулятори. Загальна хара
ктеристика гормонів білково-пептидної групи, похідних амінокислот, сте
роїдів та ейкозаноїдів.
70. Алкалоїди: визначення; значенні алкалоїдів як діючих речовин
лікарських засобів (класів піридину та піперидину, хіноліну та ізохіноліну
тропану, індолу).
71. Антибіотики: загальне поняття; характеристика антибіотиків
класів пеніцилінів, цефалоспоринів, стрептоміцинів. :
72. Пестициди: визначення; найбільш поширені фосфор- та хлорор
ганічні пестициди» їх токсикологічне значення.
П е р е л ік
Практичних навичок, якими повинні оволодіти студенти
під час вивчення біоорганічної хімії
Проведення йодоформної проби на ацетон.
Проведення реакції Тромера з формальдегідом.
Проведення реакції Вагнера з олеїновою кислотою.
Доведення ш насиченості жиру (реакція з К М п04).
Доведення наявності вільного фенольного гідроксилу в саліциловій кислоті.
Доведення наявності вільного фенольного гідроксилу в салолі.
Одержання реактиву Фелінга.
Проведення реакції Фелінга з глюкозою, лактозою, мальтозою.
Проведення якісної реакції на крохмаль.
Одержання глюконату міді.
Проведення нінгідринової реакції з сх-амінокислотами, пептидами, білками;
Проведення ксантопротеїнової реакції з ароматичними амінокислотами,
пептидами, білками.
Проведення реакції Фоля із сірковмісними амінокислотами, пептидами,
білками.
Проведення біуретовогреакції з пептидами та білками.
Осадження білків трихлороцтовою кислотою.
Осадження білків сульфосаліциловою кислотою.
408
Виявлення в гідролізаті нуклеопротеїну пурину.
Виявлення в гідролізаті нуклеопротеїну рибози.
Виявлення в гідролізаті нуклеопротеїну фосфорної кислоти.
Виявлення ферменту амілази в слині.
Виявлення гормонів білкової та пептидної природи біуретовою реакцією.
Виявлення адреналіну реакцією з хлоридом заліза.
РЕ К О М Е Н Д О В А Н А
Л ІТЕРА ТУ РА
1.
Альберт Э. Избирательная токсичность. - М.: Мир, 1971. - 432 с.
2. Беккер Г. Введение в электронную теорию органических реак
ц и й ,—М.: Мир, 1965. —576 с.
3. Беликов В. Г, Фармацевтическая хймйя. В 2 ч. — Пятигорск,
1996. - Ч. 1 . 4 3 2 с.; Ч. 2. - 608 с.
4. Боєчко Ф. Ф., Боєчко Л. О. Основні біохімічні поняття, визначен
ня і терміни: Навч. посібник. - К.: Вища шк.. І993. - 528 с.
5. Губськйй Ю. І. Біологічна хімія, -Київ-Тернопіль: Укрмедкнига,
2000. - 508 с.
6.
Губский Ю. И. Коррекция химического поражения печени. - К .:
Здоров'я, 1989. я£_ 168 с.
7*, Г убськ й й Ю . Г., Х м ел ев ськи й Ю . В.» й уд ари кова Я . Г.,
Усатенко О. К. Біоорганічна хімія. - К.: Вища школа, 1997. - 286 о.
8.
Губский Ю. И ., Ш аповалова В. А.* Кутько И. И., Ш апова
лов В. В. Л екарственны е средства в психофармакологии. ** Киев:
Здоров’я; Харьков: Торсинг, 1997. - 288 с.
9. Гудман М.,’ Мор хауз Ф. Органические молекулы в действии. М.: Мир, 1 9 7 7 .-3 3 5 с.
10. Дрюк В. Г., Малиновский М. С. Курс органической химии. - К.:
Вища шк. Головне видавництво, 1987. —400 с.
409
РЕКОМЕНДОВАНА ЛІТЕРАТУРА
И . Кан Р., Дермер О. Введение в химическую номенклатуру. - М.:
Химия, 1983. - 224 с.
12. Косовер Э. Молекулярная биохимия. :- М.: Мир, 1964, —336 с.
13. Крам Д., Хэммонд Дж. Органическая химия. - М.: Мир, 1964. Ч
714 с. *
14. Кузнецова М. А. Лекарственное растительное сырье й препараты; Справочное пособие. ^ М.: Высш. шк., 1987. - 191 с.
15. М арри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия человека
(в 2 т . ) . - М.; Мир, 1993. —Т. Ъ ~ 384 с.; Т. 2. - 415 с.
16. Мецлер Д. Биохимия (в 3-х т. ), - М.; Мир, 1980. - Т. 1. - 408 с.;
Т. 2, - 606 с.; Т. 3 .- 4 8 8 с.
17. М ещишев I. Ф,, Пішак В. П., Григор’єва Н. П. Біомолекули: структура та функції. -’•Чернівці: Медик, 1999. - 149 с.
18. Овчинников Ю. А. Биоорганическая химия. - М .: Просвещение,
1 9 8 7 .-8 1 5 с.
19. Пивоваренко В. Г,Основи біоорганічної хімії. -К иїв: Освіта, 1998. 176 с.
20. Пацак И. Органическая химия. - М.: Мир, 1986, - 366 с.
1 |* Руководство к лабораторным занятиям по биоорганичеекой хи
мии /Под ред. Н. А. Тюкавкиной. - М.: Медицина, 1985. - 256 с.
22» Сергеев П. В., Шимановский Н. Л. Рецепторы физиологически
активных веществ. - М.: Медицина, 1987. -г 400 с.
23. Степаненко Б .Н . Курс органической химии (в 2-х частях). - М.:
Высшая шк., 1976. - ч. 1 - 448 с.; ч. 2 - 302 с,
24. Сгеценко О. В., Виноградова Р. П. Біоорганічна хімія: Навч. посібник. - К.: Вища шк., 1992. - 327 с.
25. Тюкавкина Ж, А., Бауков Ю. И. Биоорганическая химия. - М.:
Медицина, 1 9 8 5 .-4 8 0 с.
26. Черних В. П., Зіменковський Б. С., Гриценко I. С. Органічна хімія:
у 3 кн. - Харків: Основа, 1997, - Кн. 1. - 145 С4 Кн. 2. - 480 с.; Кн. 3.
- 256 с.
27. Шаповалов В. В., Шаповалова В. А., Губский Ю. И,, Минкр А. И.»
Кузьминов В. Н , 8 Петренко С. Л. Лекарственные средства в наркопсихофармакологйи. Харьков: Прапор, 2002. - 592 с.
1
1
1
3
1
I
1
1
1
|
|
I
1
1
а
1
П р ед м етн и й п о к а ж ч и к
А
Ацеклідин (3-ацетоксихінуклідин) 350
Авітамінози 243,362,368,373
Аглікон 227-229,276,360
Аденілатциклаза 344
Аденілова кислота (АМФ) ????? ІД б
Адешн 321,334-335,339
Аденозин 323,325,342,344
Аденозиндифосфорна кислота (АДФ) 328-329
Аденозинтрифосфорна кислота (АТФ) 135,152,235,328,344
Адипінова кислота 137
Адреналін 54» 170-171,300,344,370,377-378
0-(-)-адреналш 54
Ь-(+)-адреналін 54
Азин 173
Азол 173
Азоли 187
Азольна таутомерія 189
Азулени 97
Аймалін 350
Акридин 174,193, І9 9 ,201-202
Акридйновий оранжевий 202
Акрилова кислота 135-136
Акрихін 201-202
АкроЛеїн 42
Аксіальні замісники 61
Аксіальні зв’язки 61
Активація амінокислот 135,294
АлайІн 286,289,292.
а-аланін 154,292*299,376
Р-аланія 154,369
Алілізотіоціанат 230
Аліфатичні аміни 145
Аліфатичні вуглеводні 72-73,167
ПРЕДМЕТНИЙ ПОКАЖЧИК
Аліфатичні гідроксикислоти 159
Аліфатичні кетони
Аліфатичні спирти 98-100
Алідщслічні спирти 99
Алкадієни 6 5 ,7 0 -7 2 ,7 7 ,8 2 -8 3
Алкалоїди 22,181,346-350,354—3.55
Алканаміни 145
Апкани 23,7 2 -7 4
н-алкани 73
Алкени 3 6 ,6 5 ,6 8 ,7 0 ,7 2 ,7 7 ,'8 0 ,8 6
Алкіламіни 145,384
Алкілбензоли 92
Алкілдиеульфіди 111
Алкілпероксиди 266
Алкілтіольна група
Алкільні групи 23 і
Алкільні радикали 23 ,4 1 ,9 2 ,9 4 ,2 6 6
Алкіни 36 ,6 8 ,7 2 ,8 4 -8 5
Алкоголі 98
Алкогольдегідрогенази 103,311
Алкоксид-іон 68,119
Алиокєикарбонільна група 120
Алкокєильна група
Аллоіохімбан 355—356
D-алоза 221
Алоксим 384
Альбумін 284,309
Альдегіди 102*114,118,120*124,266,278
АльдегідОкислоти 163
Альдогексози 218,224,236
Альдози 218-219,131
Альдонові кислоти 232,241
Альдостерон 380-381
D-альтроза 221
4 12
ПРЕДМЕТНИЙ ПОКАЖЧИК
Амігдалін 229
Аміди 70,110,,134,143,151*153,155-160,274,289,291
А м і д й кислот 151
Амідна група
Амідопірин 188-189
ß -амілаза 246
Амілоза 247
Амілопектин 247
Аміназин 214-215
Аміни 7 0 -7 1 ,8 7 ,1 2 1 ,1 3 4 ,443-147,151,153,155,167,170,181,186,192,
227,363,370,375-і376
Амінна група 20 ,3 8 ,4 2 ,8 7 ,9 3 ,132,134-135,145,147,154,236,263,298
п-амінобензолсульфокислота Ш
2-аміно-З-фенілпропіонова кислота 27
2-аміно-6-оксипурин 321
а-аміно-в-фенілпропіонова кислота 28,288
Аміноациладеніпати 294
Амшоацил-тРНК 295
Амінобензол 145
6 -аміногексанова кислота 158
Аміноглішзидні антибіотики 360
а-аміноглутарова кислота 28,287
Аміногрупа 42,236,284,291*302
7-амійодезацетоксицефалоспоранова кислота (7-АДЦК) 359
є-амінокапронова кислота 158,288
Амінокислоти 22,28,51-52,55,134,138^ 154—157, 168,283,285,290-291,
293,296
D-амінокислоти 285
а - L-амі нокислоти 154,158,284,286,2 9 4 ,296,303
а-амінокислоти 285
f -аміномасляна кислота 376
6 -амінопеніциланова кислота 358
а-амінопентандіова кислота 28
2-амінопентандіова кислота 27
413
ПРЕДМЕТНИЙ ПОКАЖЧИК
2-амінопіридин 195
Амінопохідні моносахаридів 236,240
6 -амінопурвд 321
р-аміносаліцилова кислота 170
Аміноспирти 22,167,169,258
Амінотрансферази (трансамінази) 299
р-амінофенол 169
Амінофеноли 167, ,169
7-аміноцефалоспоранова кислота (7-АЦК) 359
Аміноцукри 236,240
Амлодипін 199
Амонієві основи 167
Ампіцилін 358
Анабазин 196,349
Анальгін 188-189
Ангідриди 134,161
Андрогени 239,380
Андростан 275,380
Анестезин 150
Анілін 42,145-149,169,236
Аномери 224
Аномерний центр 224
Антибіотики 96,345-346,357,359-360
Анти-конформація 325
Антиметаболіти 329,345,357
Антиоксиданти 362
Антипірин 188-189
Антйфебрин 148
Антрацен 86,89,95
D-арабіноза 220
Арахідонова кислота 136,143, 252-253,382
Арахінова кислота 25 З
Аргінін 285,288,290,296,301,313
Арени 6 8 *7 0 ,7 2 ,8 6 ,8 9 -9 1 , 94-97,108
414
ПРЕДМЕТНИЙ ПОКАЖЧИК
Арени з ізольованими цйВЗйми 97
Аренкарбонові кислоти
Аренмонокарбонові кислоти 139
Ареноли 106
Арилалканоли 99
Ариламіни 146
Ароклор 98
Ароматичні альдегіди
Ароматичні аміни 145-146
Ароматичні амінокислоти 288
Ароматичні вуглеводні 72,86,93
Ароматичні дикарбонові кислоти 140
Ароматичні карбонові кислоти 94,139,159
Ароматичні кетони
Ароматичні монокарбонові кислоти 139
Ароматичні спирти 99
Ароматичні сульфокислоти 110
Ароматичність органічних сполук
Асиметричний атом вуглецю 50
Ь-аскорбінова кислота 162,242-243,363,369-370
Аспарагін 289
Аспарагінова кислота 287,290,347
Атомна орбіталь 2 8 -3 0 ,3 2 ,3 4 -3 5 ,8 8 , 176
Атропін 22,350,354-355
Афінна хроматографія 53
р-І^-ацетамінофенол 169
Ацеталі 120
Ацетальдегід 44, 103,125
Ацетамід 151
Ацетанілід 148-149,151
Ацетат натрію 131
М-ацетил-В-глюкозамій 250
И-ацетилталактозамін 240,244
М-ацетилглюкозамін 240,244,250
415
ПРЕДМЕТНИЙ ПОКАЖЧИК
Ацетилен 84
Ацетиленові вуглеводні 8 6
Ацетилкоензим А 125
И-ацетилнейрамінова кислота 241,243
Ацетилсаліцилова кислота 142
Ацетилфосфорна кислота 134
Ацетилхолін 168-169,357,376
Ацетилхолінестераза 168,346,355,357,383
Ацетилювання за Фриделем-Крафтсом 180
2 -ацетоксипропанова кислота 161
А цетоні 03,115,125,166,315
Ацетооцтова кирлота 164,166
Ацетооцтовий ефір 164
Ацетофенон 93
Ациклічні амінокислоти 286
Ациклічні вуглеводні 59
Ациклічні терпени 278
Ацилат-іон 129-130
Ацилгаіцериди 255
Ацилпііцероли 255
Ацилпірофосфати 135
Н-ацилсфінгозини 260-261
Ацилфосфатй 134
Ацильнагрупа 126
Б
Багатоатомні спирти 104—105
Багатоядерні (цоліциклічні) арени 89,95
Барбітал 208
Барбітурати 208-209
Барбітурова кислота 20,208
Бенз(а)пірен 96
Бензальдегід 116
Бензамід 151
416
ПРЕДМЕТНИЙ ПОКАЖЧИК
Бензиловий спирт 98
Бензилпеніцилін (пеніцилін G) 358
Бензильний радикал 87
Бензімідазол 192
Бензо[Ь]пірол 181
Бензо[Ь]пірон-4 205
Бензо-г-дигідропіран 206,268
1,4-бензодіазепіни 215
Бензойна кислота 139
Бензойний альдегід
Бензол 1 7 ,2 0 ,8 6 -8 8 ,3 4 6
Бензолсульфокислота Д10
1,2-бензопіран 203
1,,4-бензопірон 203
Бензофенон 115
Бери-бери 363
Білий стрептоцид 149
Білірубін 185
Білки 143,263,283-284,289,294,296,302,309-314,375
Бімолекулярні реакції 67
Біогенні аміни 167,181,300,376
Біокаталізатори 284
Біосинтез ДНК 336
Біотин 187,361,368
Біфеніл 96
Бойові отруйні речовини (БОР) 385
Борнан (Камфан) 280
Брадикінін 294,304
Британський антилюїзит (БАЛ) 113
г-2-бромо-3-хлоробутен-2 56
1-бромпропан44
2-бромпропан 44
Бурштинова кислота 137
Бутадієн-1,3 82
417
ПРЕДМЕТНИЙ ПОКАЖЧИК
Бутан 17,22
н-бутан 43, 58
н-бутиловий спирт 99
Бутаналь
Бутанамід 151
Бутандіамін-1,4146
Бутандіова кислота І37
Бутанова кислота 12?
Бутаноїламід 151
Бутанол 26
Бутанол-1 99
Бутанон 115
Бутен-1 44, 78
Бутен-2 44 ,7 8
Бутин-1 85
Бутиральдегід
у-бутиролактам 157
у-бутиролактон 163
В
Вазелін 76
Вазелінове масло 76
Валентні стани атома вуглецю 31-33
Валентність атома 32
Валеріановий альдегід
н-Валеріанова кислота 127
Валін 286,289,296,303
Ван-дер-Ваальсівські радіуси 47
Ванілін 116
Веронал 208
Винна кислота 159
Виннокам’яна кислота
Вищі жирні кислоти 252
Вищі карбонові кислоти 128
418
ПРЕДМЕТНІЙ ПОКАЖЧИК
Вільні радикали 64,69,75
Вітамери 371,374
Вітамін О 374
Вітамін Б 2 (ергокальциферол) 374
Вітамін Б , (холекальциферол) 374
Вітамін А 281-282,361,371
Вітамін А, (ретинол) 371
Вітамін А 2 (дегідроретюгол) 371
Вітамін В, 371-363
Вітамін В 12 361,366
Вітамін В 2 361,363
ВіхаМдЛї В 3 361,369
Вітамін В 5 361,364
Вітамін В 6 197,361,365-367
Вітамін Вс 361,367
Вітамін Е 267,281,373
Вітамін К 372-373
Вітамін К, (філохінон) 372
Вітамін К 2 (фарнохінон) 373
Вітамін Н 361,368
Вітамін Р 205
Вітамін РР 361,364
Вітамін С 78,242,361,369
Вітаміни 84,206,269,345,361,362-363,370
Водорозчинні вітаміни 243,361
Вторинна структура білків 306
Вторинна структура ДНК 332
Вторинна структура молекул тРНК 342
Вторинна структура мРНК 342
Вторинний атом вуглецю 44
Вторинні аміни 134,145,151,181
Вугільна кислота 126,153,293
Вуїяеводи 22,105,217,237,244,246,258,314,345
Вуглеводневий радикал 23
.
419
ПРЕДМЕТНИЙ ПОКАЖЧИК
Вуглеводні 1 9 ,2 3 -2 4 ,5 9 ,7 0 ,7 2 -7 3 ,7 7 ,8 2 ,8 6 ,9 3 ,9 7 ,2 8 0 ,3 8 7
В-форма молекули ДН К 334, 337
Г
D-галактоза 220-221,236,238
D-галактозамін 236,240,250
Галактозид 227
ß-галактозидаза 245
Галактозилцерамід 262
D-галактуронова кислота 233,241
Галова кислота 143
Г алогеноалкани 81
Галогеноангідриди 134,155,160
Г алогенова група 22
Галогенопохідні вуглеводнів 56
Галоперидол 196
Гангліозиди 261
Гексагідропіридин 195
н-гексадеканова кислота 253
Гексан 18
н-гексан 18
Гександіова кислота 137
Гексанова кислота 127
Гексахлоран 386
Гексахлорциклогексан 386
Гексити 104,234
Гексозаміни 240
Гексози 5 4,218-220,226,236,241-242,247
Гектан
Гем 184
Гемералопія
Геміцелюлоза 248
Гемоглобін 184,284,309,311
Генейкозан
420
ПРЕДМЕТНИЙ ПОКАЖЧИК
Гентіобіоза 229
Геометричні ізомери 47,54-55
Гепаран сульфати 249
Гепарин 249,251
Гептан
Гептози 218
Героїн 351
ГестагеНи 380
Гетерогенні ядерні РНК (гяРНК) 343
Гетероглікозиди 228
Гетеролітичні реакції 54,58,79
Гетерополісахариди 217,246,248-249
Гетерофункціональні сполуки 138,153
Гетероцикли 20,172-174,177-178,181, L87,192-193,202,204,206,213,
215
Гетероциклічні амінокислоти 286,289
Гетероциклічні сполуки 20,89,172,178,321
Гіалуронова кислота 249
Гібридизація атомних орбіталей 32-34
Гібридні орбіталі 32,86
Гідразин 122,165
Гідразиноліз за Акаборі 318
Гідразон 122,165
Гідриди металів 123
Гідрид-ю н 6 8
Гідрогенізація жирів 265
Гідроксалат натрію 138
2-гідрокси-1,2,3 -пропантрикарбонова кислота 138,159
4-гідрокси-З-метоксибензальдегід 116
2 -гідроксибензальдегід
3 -гідроксибутанова кислота 26,160
Гідроксикислоти 51,53, 138,155,159-160,162
4-гідроксикумарини 203
Гідроксил 128,132,301,384
421
ПРЕДМЕТНИЙ ПОКАЖЧИК
Гідроксиламін 122
Гідрокеиламіни 145
Гідроксильнагрупа 93,98-100,108,120,128,223,227
Гідроксильні (гідрокси-) сполуки 98
р-гідроксимасляна кислота 166
у-гідроксимасляна кислота 162
5-гідрокєиметил фурфурол 236
Гідроксинітрили 119
2-гідроксипіридин 195
Гідроксипірвдини 197
2 -гідроксипропанова кислота 159
5-гідрокситриптамін 181,378
5-гідрокситриптофан 181
8 -гідроксихінолін 2 0 0
Гідропероксиди 266
Гідрофобні взаємодії 303
Гідрохінон 106
ГШ К 198
Гіосціамін 354-355
Гіперхромний ефект 338
Гіповітамінози 362
Гіпоксантин 210
Ппохромний ефект 338,341
Гістамін 190*30,379
Гістидин 154, Г90-191 ^2§9-290,296
Гістони 3 13
Глікани 244
Глікарові кислоти 231,241
Глікоген 217,247
Глікозамінілікани 249
Ш юозидази 230
Глікозиди 227,230,276
О-ппкозиди 227
]Ч-глікозиди 227,229
422
ПРЕДМЕТНИЙ ПОКАЖЧИК
8-гаікозиди 227,230
Глікозидний зв’язок 227,229-230,245,249,360
Глікозидний центр 227
Глікозиліліцерид 261
Глікозшшііцериди 261
Глікозилтрансферази
Гліколева кислота 159,161
Гліколіз 125,237
Гліколіпіди 255,261,263
Глігонові кислоти 232
Глшзпротеїни 314
Глікосфіншліпіди 255,261
Глікохолева кислота 275
Глікуронові кислоти 232
Гліоксилова кислота 161,164
ГліцеральдегіД-З-фосфат 125
Гліцерин 104,264
Б-гліцериновий альдегід 50,219
Ь-гліцериновий альдегід 50,219
Гліцерол (гаіцерин) 105,133,258,264
Гліцерофосфоліпіди 255,258,263
Гліцин 286,289,292,304,376
Глобозиди 262
Глобулярні білки 309
Ь-глутаматдегідрогеназа 298
Глутамін І 52,236,289
Глутамінова кислота 27 -2 8 ,1 5 2 ,2 8 7 ,2 9 0
Глутарова кислота 137
Глутатіон 112
О-глюкарова кислота 231
О-глюкоза 218,220-221,223,224,231,235-237,244,246,276
Ь-глюкоза 220,236,240,250
П-шюкозамін 236,240,250
Глюкозиди 227
423
ПРЕДМЕТНИЙ ПОКАЖЧИК
Глюкозо-1-фосфат 237
Глюкозо-6 -фосфат 235,237
0 -глюконова кислота 232
а-О-шюкопіраноза 223,225-226,245,249
Р-О-глюкопіраноза 223,225-226
Глюкокортикоїди 380
Глюкуроніди 241-242
О-глюїіуронова кислота 233,241
Глюциди217
Головний ланцюг 23-25,137,247
Гомолітичні реакції 64
Гомополісахариди 244,246-246
Гомоциклічні вуглеводні 72
Гормони 252,269,275,284,375-377,380,382
Гош-конформація 58-59
Гуанілова кислота (ГМФ)
Гуанін 321,335
Гуанозин323
О-гулоза 2 2 1
д
Дансилхлорид (ДНС) 317-318
Двовалентні радикали 23
Ь-дегідроаскорбінова кислота
Дегідрогенази 112, 160-161,364-365
7-дегідрохолестерин 273
Дезамінування амінокислот 156,296-297
2-дезокси-Б-рибоза 239
Дезоксиаденілова кислота (дАМФ)
Дезоксигуанілова кислота (дГМФ)
Дезоксирибоза 239,332
Дезоксирибонуклеїнові кислоти (ДНК) 146,158,202,207,210,239,284,
294, 320, 322, 325-326, 329, 331-339, ;341, 346, 358, 368, 383,
388
424
ПРЕДМЕТНИЙ ПОКАЖЧИК
Дезоксирибонуклеозиди 322,324
Дезоксихолева кислота 274
Дезоксицитидилова кислота (дЦМФ) 366
5-дезоксиаденозилкобаламін 324
Дезоксиаденбзин 324
Дезоксигуанозин 324
Дезокситимідин 324
Декан
Декарбоксилази 300
Декарбоксилування амінокислот 298
Декзоксицитидин 324
Декстран 247-248
Делокалізація заряду 129-130,140
Денатурація білків 313
Денатурація ДНК 339
Дерматансульфати 249
Дибазол 192
Дибензо[Ь,е]пІридин 201
1 .2 -дйгідроксибензол 106
1,4-дигідропіридини 199
Дигідроуридин 326
Дигітоксигенін 276
Дигітоксин 277
D-дигітоксоза 276
Диізопропілфторфосфат (ДФФ) 384
Дикарбонові кислоти 137-140,151
2 .3 -дикето-Ь-гулонова кислота 243
Дикетопіперазини 156
Диклоксацилін 193 ,
Дикумарин 204
Димедрол 192,380
Димексид 112-113
2.3-димеркаптопропанол 113
2.3-димеркаптосульфонат натрію 113
425
ПРЕДМЕТНИЙ ПОКАЖЧИК
3,6-диметил-гептадієн-2,5-аль 27
Диметиламін 145
Диметиланілін 146
NjN-диметилацетамід 151
7,12-диметилбенз(а)антрацен 97
Диметилбензол 87
Диметилдисульфід 109
Диметилкетон 103,115* 125
Диметиловий ефір 44
Диметилсульфід 109
Диметилсульфоксид 112-113
Диметилфеніламін 146
М,ї«ї-диметилформамід 153
1.2-диметилциклопропан 55
Динамічна поляризація 39
2,4-динітрофторбензол (ДНФБ) 317
Дипептиди 291-292
Дипіроксим 384
Дипразин 215
Дисахариди 244
Дисульфіди 109
Днтерпени 278
6 , 8 -дитіооктанова кислота 113
Дифенілкетон 115
Дифенілметан 97
1.3-дифосфогліцеринова кислота 135
Дихлордифенілтрихлоретан (ДДТ) 386
Р, р ‘-дихлордіетилсульфід 114
1.2-дихлоретан 387
Дихлофос (ДДВФ) 384
Діазепам 216
Діазепіни215
1 .2 -діазин 174,213
Діазини 207,209
426
ПРЕДМЕТНИЙ ПОКАЖЧИК
Діазолін 192,380
Діаміни 146
Діамійодикарбонові амінокислоти 286
Діаміномонокарбонові амінокислоти 286,288
Діастереомери 47-49 ,5 4 ,2 2 1
Діетиламід лізергінової кислоти І 82,378
5,5-діетилбарбітурова кислота 208
Діетилкетон
2,4-діоксипіримідин 322
ДіоксиацеТонфосфаТ 125
Діоли 120
Додекан
Додеканова кислота
Домени 310
Доменні білки 310
Донорно-акцепторні зв ’язки 31,37,3 8-39
Дофамін 170,377
Дульцит234
Е
Е-2-бромо-3-хлоробутен-2 56
Ейкозан
Ейкозаноїди 84,252,375,382
н-ейкозанова кислота 253
Екваторіальні замісники 61
Екваторіальні зв ’язки 61
Екгонін 355
Еластин 284,310
Електронеґативність атома 117
Електронегативності шкала 39
Електронна конфігурація ЗО, 32,37,174
Електронна структура атома вуглецю 2$, 89
Електронні формули ЗО
Електроноакцепторні замісники 93
427
ПРЕДМЕТНИЙ ПОКАЖЧИК
Електронодонорні замісники 93
68-69
Електрофільне заміщення 65-66
Електрофільне приєднання. 65
Електрофорез 315
Еленіум 216
Енантіомери 47-48,52,221
Ендорфінй 304,352,376
Енергія активації 63
Енергія делокалізації 89
Енергія спряження 89
Ентеросептол 200
Епімери 221,236
Еритрити 104
D-еритроза 220
Естрад іол 380-381
17-р-Естрадіол 381
Естран 275
Естрогени 380
Етакридину лактат 201
Етан 16,22
Етаналь 115
Етандіова кислота 137
Етанова кислота 127
Етанол 17,28,99,101—104^315
Етаноламін 167
Етантіол 109-110
Етаперазин 214
Етен 78
4-Етил-2-метилгептан 19
5-Етил-5-фенілбарбітурова кислота 208
Етилацетат 101
З-Етилбензойна кислота 140
Етилбензол 87,95
Етилен 23,78
і Електрофіли
428
ПРЕДМЕТНИЙ ПОКАЖЧИК
Етиленгліколь 104
Етилнітрат 101
Етиловий спирт 16,28,44,99,103* І50
Етилхлорид 77
Етин 85
Ж
Жирні кислоти 78,84,136,251-252,254—255
Жиророзчинні вітаміни 362,370
Жіночі статеві гормони 252,269,275,380
Жовчні кислоти 252,263,269,274-275
З
Загальмована конформація 57-58
Замінні амінокислоти
Замісник 2 3 -2 6 ,2 8 ,4 0 ,4 2 ,5 2 ,5 4 -5 5 ,6 0 -6 1 ,9 3 ,1 2 8 ,1 3 0 ,2 2 4 ,2 7 0
Замісники І роду 93-94
Замісники П роду 93-94
Зарин 385
Заслонена конформація 57
я -зв ’язки 3 5 -3 6 ,4 1 ,7 1 ,7 8 -7 9 , 82,88,93
ст-зв’язки 3 5 -3 6 ,4 0 ,5 5 ,5 7 ,7 4 ,7 8 -7 9 ,8 8 ,1 1 7 ,1 7 5
Зв’язки водневі 31,39,302,306-307,325
З в’язки електровалентні 38
Зв’язки іонні 31,38,303,314
Зв’язки неполярні ковалентні 36,74
Зв’язки полярні ковалентні 36,40
Зв’язки сольові 31,38,303
Зв’язок семіполярний (координаційний) 31,38
Зв’язок фосфоангідридний 135,329
Зв’язок фосфоефірний 135
Зелене мило 13 1
Зигзагоподібна конформація 59
Зоман 385
429
ПРЕДМЕТНИЙ ПОКАЖЧИК
Зоостерини 271
І
Ідентифікація N-кінцевих амінокислот 317
Ідентифікація С-кінцевих амінокислот 317-31В
D-ідоза 221
L-ідоза 233
L-ідуронова кислота 233,241,251
Ізоалкани212
Ізоалоксазин 2 1 2 х
Ізобутан
їзо й л ер іан о ва кислота
Ізоелектрична точка
Ізоелектричний стан
ІзоксазОл 192
Ізолейцин 286
Ізолимонна кислота 139
Ізольовані алкадієни
Е-, Z-ізомери 56
Ізомери ланцюга 43
Ізомери положення 43
Ізомери функціональних груп 44
Ізомерія 28,4 2 -4 3 ,1 5 4
Ізомерія будови 43
Ізоникотинова кислота
Ізоніазид 198
Ізопентан 73
Ізопрен (2-метилбутадієн-1,3) 277
Ізопрено'їди
Ізоферменти
Ізофлавон205
Ізофталева кислота
Ізохінолін 2 0 0
Імідазол 173,177-178,188,191
430
ПРЕДМЕТНИЙ ПОКАЖЧИК
Імідазоламіни 379
Іміни І 8 6
Іміноглутарова кислота 298
їмшокислоти 297
Індоксил 182-183
Індоксилсульфат 183
Індол 174,181
Індоламіни 378
р-індолілоцтова кислота 181-183
Індометацин 183
Індуктивний ефект 40-41
Інозит (інозитол) 105
Інозитол-1,4,5-трифосфат 105
Інозитолфосфатиди / • '
Інтенкордин 204
Ійуяія248
Інформаційна (м а т р и ц а ) РНК (мРНК) 339, 341-342, 346
Іонні реакції 54
Р-іонон
Іцрит 114
Й
й од н е число 256
Йодоформ 77
К
Кадаверин 146
Кальцитріол (1,25(0Н )20 3)
Камфора 278,281
є-капролактам 158 *
н-капронова кислота 127
Карбкатіон 6 8 , 80,91
Карбокромен 204
Карбоксамідна група
431
ПРЕДМЕТНИЙ ПОКАЖЧИК
Карбоксибіотин 368
Карбоксил 126, 128,133-134*151
Карбоксилат-іон 38,129
Карбоксильна група 94,126,139,160,317
Карбоксипептидазний метод 318
Карбоніл 128
Карбонільна група 65,114,128
Карбонільні сполуки 102,114,119
Карбонові кислоти 7 0 ,9 4 ,1 0 2 ,1 1 4 ,1 2 3 , 1 2 8 , 1 3 0 , 1 3 6 ,
162
Карбофос (малатіон) 385
Карбоциклічні амінокислоти 286
Карбоциклічні вуглеводні 72
(3-каротин 281-282
Каротини 281,371
Каротиноїди 278,281
Каталаза 185
Катехоламіни 170,300,376
К аучук279
Квантове число головне 29
Квантове число магнітне 29
Квантове число орбітальне (побічне) 29
Квантове число спінове 30
Квантові числа 28
Кверцетин 205
Кератансульфати 249
а-кератини 310
Р-кератини 308-309
Кетогексози 218,226,236
а-кетоглутаратдегідрогеназа 363
а-кетопхутарова кислота 138-139,164,166,298,363
Кетози 218-219
Кетокислоти 163,297-298
а-кетокиелоти 122,297
432
13 9 , 15 5 , 1 5 7 ,1 5 9
ПРЕДМЕТНИЙ ПОКАЖЧИК
Кетон 28
Кетони 65,76,102,114-116,122-123,125,164,278
Кетонові тіла 166
Кефаліни 258
Кислота бурштинова 137—138,185
Кислота яблучна 22, 138-139, 159
Кислоти за Бренстедом 69
Кислоти Льюїса 70
Кислотоамідні зв’язки 291-293
Кільцево-ланцюгова таутомерія 226
Класи органічних сполук 20
Класифікація органічних сполук 18
Клешнеподібна конформація 59
Клітковина 247
Кобаламін 366
Ковалентні зв’язки 31
Кодеїн 201,351
Коензим А
Кокаїн 355
Колаген 284,310,370
Коламін І67-168
ті-комплекс 80,90
ст-комплекс 80, 90
Комплементарні азотисті основи 334,341-342
Конденсовані (анельовані) цикли 8 6,89,95-96
Конденсовані гетероцикли 181,204
Коніїн 349
Конфігураційні ізомери 47
Конфігурація 30 -32,37,52,56, 174
Конформаційні ізомери 47,56
Конформація 5 6,58-60,270,307-308,310,325
Конформація ванни 60
Конформація крісла 61
Конформери 57-58,61
433
ПРЕДМЕТНИЙ ПОКАЖЧИК
Кордіамін 197
Кориноїди 366
Кортизол 380
Кортикостероїди 275,380-381
Кофеїн 210
Коферменте 345,361* 365
Коферментні вітаміни 362
о-крезол 106
Кротонова кислота 136
Крохмаль 2 37,246-247
Ксантин 210
Ксантопротеїнова реакція 301
Ксенобіотики 86,267,383
І>кбилоза 2 2 0 - 2 2 1
Ксилол 87
Б-ксилупоза 239
Кумарини 203
Кумульовані алкадієни 83
Кутова (байєрівська) напруга 59
Л
Лактаза 245
Лактами 156
Лактам-лактимна таутомерія 157-157
Лактат 201
Лактатдегідрогеназа (ЛДГ) ЗІ І
Лактиди 162-163
Лактоза 217» 139,244-246
Лактозилцерамід 262
у-яактон О-ілюконової кислоти 232
Лактони 162
Левомепромазин 214
Левульоза 238
Лей-енкефалін 304
43 4
ПРЕДМЕТНИЙ ПОКАЖЧИК
Лейкотрієни 252,382
Лейцин 286,289,296,303
Лецитини 258
Лимонна кислота 139,159
Лідокаїн 150
Лізин 285,288,290,296,313,347,370
О-ліксоза 2 2 0
Лімітуюча реакція Н І
Лімонен 280
*
Ліндан 386
Лінолева кислота 253
Ліноленова кислота 136,253
Ліпази 264,275
Ліпіди 84,100,105* 128,132, 136,139,1 6 7 ,2 4 3 ,2 5 1 -2 5 2 ,2 5 4 -2 5 6 , 258,
261-267,269,272,274* 277,291,309,314
Ліпоєва кислота 113
Ліпопероксидація 266-267
Ліпопротеїни 263,314
з в іі*
Дітохолева кислота 274
'Н е
Лобелін 349
Л о к ан т2 3 ,2 6 ,28,154,159,163
ЛСД 182,379
Люїзит 113
Люмінал 208
М
М алеїнова кислота 55,137
М алонова кислота 137
Малоновий діальдегід 267
Мальтоза 217,244,246 *
Маніт 105,234,239
Б-маноза 221,23 6,23 9
Масляний альдегід
н-масляна кислота 127
435
ПРЕДМЕТНИЙ ПОКАЖЧИК
Мевалонова кислота 160
Мезомерний ефект 40-41
Меконова кислота 204
Ментан 279
Ментол 278-279
Меркаптани 98,109
Меркаптобензол 109
Меркаптогрупа
6 -меркаптопурин 329
Метакрилова кислота 136
Металопорфірини 183
Метан 2 2 ,3 2 ,7 5 -7 6
М етаналь 115,124
Метанамін 145
М етанова кислота 127
М етанол 99,101,103
М ета-орієнтанти 94
Метафос (метилпаратіон) 385
Мет-енкефалін 304
М етеразин 214
2-метил-1,4-нафтохінон 372
5 -метил- 2 ,4 -діоксипіримідин 322
Метил- P-D-глюкопіранозид 228
М етил-а-О-шюкопіранозид 228
Метил алілкетон
Метиламін 70,145
М етилацеїилен 85
Метилбензол 85,93
3 -метилбутанова кислота 127
З-метилбутанон-2
7-метилгуанозин 341
Метиленовий синій 215
Метилетиламін 146
Метил етилкетон
436
ПРЕДМЕТНИЙ ПОКАЖЧИК
Метилізопропілкетон
Метилкобаламін 366
Метиллактат 160
Метилмеркаптан 109,113
а-метилнафталін 96
Р-метилнафталін 96
Метиловий спирт 99
Меяилпіридини 196-197
2 -метилпропенова кислота 136
Метилпропілкетон
М етилсаліцилат 142
2 -метилтіофенол 109
Метилфенілкетон 93
2-метилфенол 106
3-метилхоЛантрен 96-97
1-метилциклогексан 62
5-метилцитозин 326,328
Метіонін 125,287,289,296
М етод Едмана 321-322
Метод Сенгера 317
Мила 130-131,264
Мило медичне 131
Мізопростол 382
Мінералокоргикоїди 252,380
Мінорні нуклеотиди 326
Міоглобін 185,309
Мірициловий спирт 258
Моделі Драйдинга 45
Моделі кульострижневі 46
Моделі масштабні 46
Моделі молекулярні 45
Моделі напівсферичні 46-47
Моделі скелетні 45-46
Моделі Стюарта - Бріглеба 46
'
43 7
Пр е д м е т н и й
по каж чик
Молекулярна асиметрія 48
Молекулярні реакції 65
М олочна кислота 51,159—161
Моноамінодикарбонові амінокислоти 286-287
Моноаміномонокарбонові амінокислоти 286
Моногалактозилдіацилгаіцерин 261
Моногалогеналкани 74
Моноєнові кислоти 253
Монози218
Монокарбонові аліфатичні кислоти 126
Монометиланілін 147
Мономолекулярні реакції 67
Мононуклеотиди 320
Моносахариди 51,217-224,231
Монотерпени 278
Моноциклічні терпени 279
Морфін 96,196,201,348,351-353
Мурашина кислота 127
Мурашиний альдегід 124
Муреїн 359
Мутаротація 222,226
Муцини 249,282
Н
НАД* 123,161,298,365
НАДФ+ 123,365
Напівацеталі 120,224
Напівацетальний гідроксил 224,227
Насичені дикарбонові кислоти 137
Насичені кислоти 253
Насичені монокарбонові кислоти 126
Нафталін 8 6 ,89,95-96,360
НафтацеН 96
1,4-нафтохінон 96
438
ПРЕДМЕТНИЙ ПОКАЖЧИК
Негативний (-1) індуктивний ефект 40
Негативний (-М) мезомерний ефект 41
Неетерифіковані жирні кислоти (НЕЖК) 254
Незамінні амінокислоти 240
Нейрамінова кислота 240
НейроМедіатори 352» 375-376
Нейропептиди 352,376
Нейтральні жири 252,255-256,264
Ненасичені кислоти 253
Ненасичені монокарбонові кислоти 135
Неоалкани 73
Неодикумарин 204
Неомшповані ліпіди 255
Неопентан 73
Неподілена електронна пара 37
Нерегулярна конформація 59,308
Нестероїдні протизапальні засоби (НПЗЗ) 142,170,183
Шацин364
Нікардипін 199
Нікотин 181,348
Нікотинамід 161» 197,364
Нікотинамідаденіндинуклеотид 123,298,365
Нікотинамідаденіндинуклеотидфосфат 123
Нікотинова кислота 196-197,364
Німодипін 199
Нінгідрин 300-301,316
Нінгідринова реакція 300,316
Нітрили
Нітрильна (ціано-)група
Йітроалкани 77,144
Нітроалкени 144
Нітроарени 144
Нітробензол 144-145
Нітрогрупа 42
439
ПРЕДМЕТНИЙ ПОКАЖЧИК
Нітрозобензол 145
Нітрозогрупа 145
Нітрозосполуки 145
№ троксолін 2 0 0
Нітрометан 77
2-нпропірол 179
1-нітропропан 77,144
Нітропропани 77
Нітросполуки 143
3-нітротолуол 144
5-нітрофурани 186
5-нітрофурфурол 186
ЗЙіфедипін 199
Новокаїн 22,150
5-НОК 200
Номенклатура ІЮПАК 22-27
Номенклатура міжнародна 2 2
Номенклатура органічних сполук 18,2 2
Номенклатура радикало-функціональна 27
Номенклатура систематична 22
Номенклатура тривіальна 2=2
Нонан
Норадреналін 170-171,300,344,370,377-378
Норсульфазол 149,193
Нуклеїнові кислоти 294,314» 320,339
Нуклеозиди 229,322,325
Нуклеозидфосфати 325
Нуклеотиди 314,320,325-329,332,338,345,358
Нуклеофіли 68-69
Нуклеофільна атака 102,133
Нуклеофільне заміщення 65-66,118,201
Нуклеофільне приєднання 65,118,122
Нуклеофільні реагенти 6 8
51
{
я
. І
440
ПРЕДМЕТНИЙ ПОКАЖЧИК
О
Одновалентні радикали 23
Одноядерні (моноциклічні) арени 86
Озокерит 73,77
Окисне дезамінування амінокислот 297
Оксазол 186,192-193
Оксалат натрію 138
Океалоацетат 139
Океацилін 193,358
2-окси-4-амінопіримідин 322
Оксим 122,165
Оксо-(кето-)кислоти 22,163
Оксогрупа 219
Оксокарбонові кислоти 160,163
Оксокислоти 22,138,163-164
Оксол 172
Оксонієві основи 70
Оксосполуки 114,120,165
Октан 350
н-октадеканова кислота 253
Олеїнова кислота 253
Олефіни 77
Олігомерні білки 283,311
Олігопептиди 283
Олігосахариди 244
Онієві основи (п-основи) 70
Оптична активність 48,5 3 ,2 2 2
Оптична ізомерія 48
Оптичні антиподи 47,52
Оптичні ізомери 47-50
Органічний радикал 23
Органічні кислоти 178,204
Органічні основи 147
Ориітин 285,347
V
441
ПРЕДМЕТНИЙ ПОКАЖЧИК
Орто-орієнтанти 94
Основи за Бренстедом 69
Основи Льюїса 70,178
Основи Шифа 121
Олеїнова кислота 122
Оцнмен 278
Оцтова кислота 127» 131,168
Оцтовий альдегід 125
П
Паличкова модель 46
Пальмітинова кислота 17,22,253
Пальмітоолеїнова кислота 361
Пантотенова кислота 361,369
Папаверин 22,201,351,353
Пара-амінобензойна кислота (ПАБК) 150, 367
Пара-орієнтанти 94
Парафін 73,77
Парацетамол 169
ПАСК 169
Пектюпс 247-248
Пелагра 365
Пеніциланова кислота 358-359
Пеніцилін Y 158,358
Пеніциліназа (ß-лактамаза) 359
Пеніциліни 158,358-359
Пеніцилоїнова кислота 359
Пентаконтан
Пентан 17,73
н-пентан 73
Пентаналь
Пентандіамін-1,5 146
Пентандіова кислота 137
Пентанова кислота 127
442
ПРЕДМЕТНИЙ ПОКАЖЧИК
ГІентанон-2
Пентанон'3
Пентен-1 26
Пентен-2 26
Пентен-4-он-2
Пентити 104
Пентози 218-219,236,239,321-322,331
Пептиди 283,295,302,304,320,375
Пептидил-тРНК 295
Пептидна група 305
Дептидні зв’язки 152,283,291,295,302,304
Первинна структура білків 303
Первинна структура мРНК 341
Первинна структура нуклеїнових кислот 330
Первинна структура тРНК 342
Первинний атом вуглецю 44
Первинні аміни 144,155
Перекисне окислення ліпідів 243,266-267
Периціазин 214-215
Пероксидаза 185
Перспективні формули 45,56
Пестициди 346,383
Піколіни 196
Піш лінова кислота 197
Пікринова кислота 108
Пінан 280
а-пінен 280
Піперазин 209,214
Піперазину адипінат 2 1 0
Піперидин 195,214,347-350
Піразин 174,206,209,212
Піразол 173,177-178,187-188,193
Піразолон-5 188
Піран 173,193,202-203,206
443
ПРЕДМЕТНИЙ ПОКАЖЧИК
4Н-піран 173,202
у-піран 202,173
Піранозиди 227
Шранозний цикл 223-224
Піретрин 1 387
Піретроїди 386
Піридазин 174,206,213
Піридин 173,175,178,193-195,347-349,365
ß-піридинкарбонова кислота 196
Піридинкарбонові кислоти 197
Піридиновий азот 175,189
Піридиноли 197
Піридоксаль-5-фосфат 197,366
Піридоксамін-5-фосфат 197,366
Піридоксин 197,299,365
Піридоксол 197,365
Піридоксол-5-фосфат 197
Піримідин 1 74,177,192,206-208,210f 212-213
Піримідинові основи 321
Піровиноградна кислота 161,164-165
Пірогалол 106,143
Пірокатехін 106,170
Пірокатехійаміни 170,377
Пірол 8 9 ,1 7 3 ,17 5 ,178-181,183,185
Пірол-2-сульфокислота 180
Шролідин 180
Ціролін 180
Пірольний азот 175-177
а-пірон 203
у-пірон 203-204
Піруватдегідрогеназа 363
Поворотні ізомери 47
Позитивний (+1) індуктивний ефект 40
Позитивний (+М) мезомерний ефект 42
444
ПРЕДМЕТНИЙ ПОКАЖЧИК
Полібромовані біфеніли” (ПББ) 98
Полігалогеналкани 74
ПолідезоксирибонуклеотидниЙ ланцюг 331
Полієнові кислоти 253
Поліізопреноїди 277
Пошненасичені жирні кислоти 84,136
Полінуклеотиди 320,326,330,336-337,341
Полінуклеотидний ланцюг 330-341,333-334,336
Поліоксикетони 217
Поліоксиальдегіди 217
Поліоли 104
Поліпептиди 250,283-284,290-291,302
Поліпептидний ланцюг 135, 153, 283-284, 289,294-295, 302-303, 305,
307-311,314
Полісахариди 104,217,227,237,239,244,246,248
Політерпени 278
Поліхлоровані біфеніли (ПХБ) 98
Поліциклічні вуглеводні
Поляризація електричного заряду 41
Полярність зв’язків 331
Полярність полінуклеотидів 330
Порфін 183
ч
Порфірини 183,314
Правила Чаргафа 334
Правило Марковнікова 81
Правило Меншуткіна 133
Правило Хюккеля 89,175
Прегнан 275,380-381
Провітаміни 281,371
Прогестерон 380-382
Проекційні формули Ньюмена 45,56-57
Проекційні формули Фішера 45,50,220
Пролін 181,289,370
Промедол 196
445
ПРЕДМЕТНИЙ ПОКАЖЧИК
Пронтозил 149
Прооксиданти 267
Пропазин 214
Пропан 16,22
Пропаналь
Пропандіова кйслота 137
Пропанова кислота 127
Пропанол-1 99,101
Пропанон 115
Пропен 23,78
Пропеналь
Пропенова кислота 135
Пропілен 78,80,101
н-Пропіловий спирт 99
Пропіл фенілкетон
Пропін85
Пропіонова кислота 127
Пропіоновцй альдегід
Простагландин Щ, 382
Простагландини 143,252,345
Простаноїди 3 8 t
Простацикліни 252,382
Простетична група
Прості білки 314
Прості вуглеводи 237
Прості ефіри 71» 235
Прості ліпіди 255,258
Просторова ізомерія 42,45
Протаміни 337
Протеїногенні амінокислоти 154,285
Протеоглікани 240,249-250
Протонні кислоти 69
Протопорфірин I I I 184
Протопорфірин ЕХ 184
446
ПРЕДМЕТНИЙ ПОКАЖЧИК
Прототропна таутомерія 189
Псевдоуридин 326,342
Ш илоцибін 182,379
/
Псилоцин 379
Птерйдий212
Птерини 212,367
Птероїлгаутамінова кислота 212
Пурген 141
Пурин 17,174,190,210,321-323,329,358,369
Пуринові основи 321
Путресцин 146
j
Р
Радикальне заміщення 65
Радикальне приєднання 65
Радикальні реакції 64,68
D-рамноза 276
L-рамноза 206,228
Ранітидин 192,380
Рафіноза 244
Рахіт 273,375
Рацемати 52-53
Рацемічної форми 53
Реагенти 68, 317,387
Реактив Драгендорфа 348
Реактив Зонценштейна 348
Реактив Майєра 348
Реактив Толецса 122,233,246
Реактив Троммера 233
Реактив Фелінга 124,233,246
Реактив Ш ейблера 348
Реактиви Вагнера, Люголя, Бушарда 348
Реакції алкілування 147
Реакції амідування 132,134,151
44 7
ПРЕДМЕТНИЙ ПОКАЖЧИК
Реакції ангідридизації 132,134
Реакції відщеплення 65
Реакції вільнорадикального заміщення 74
Реакції елімінування 65» 162
Реакції етерифікації 10J, 132-133
Рещсщї заміщення 65-66
Реакції ідентифікації алкалоїдів 347
Реакції карбоксилювання 187,368
Реакції окислення та відновлення 65-66
Реакції перегрупування 65-66
Реакції приєднання 65
Реакції трансамінування 298
Реакція “срібного дзеркала” 123
Реакція ЕрлІха 301
Реакція Зініна 145
Реакція Коновалова 76
Реакція Мілона 301
Реакція Сакагучі 301
Реакція Селіванова 236
Реакція Фоля 302
Реакція Фріделя - Крафтса 94
Реакція Чичибабіна 195
Резерпін 355
Резорцин 106,236
Ретиналь 371
Ретинол 282
Рибітол 363
D-рибоза 220-221,225,239
a -D -рибоза 225,239
ß-D-рибоза 225
Рибозид 227
Рибонуклеїнові кислоти (РНК) 158,207,239,294,320-322,325,328,332,
339-345,368
Рибонуклеозиди 322
4 48
ПРЕДМЕТНИЙ ПОКАЖЧИК
Рибосомальна РНК (рРНК) 339, 343
Рибофлавін 16,212,363
В-рибулоза 239
Риванол 201
Рівняння Арреніуса 63
Родоначальна структура 23-24
Рутин 206,228
С
Саліцилати 142,345
Саліцилова кислота 142
Саліциловий альдегід 116
Салол 142
Салюзид 198
Сахараза 246
Сахароза 217,237-239,244-245
Сегнетова сіль 124
Серин 1 6 8 ,250,258,261,287,289
Серотонін 181-182,344,371,379
Серцеві глікозиди 229,276
Сечова кислота 210-211
Сечовина 153
Сибазон216
Синігрин 230
Синільна кислота
Синтез пептидів 292,294
Синхронні реакції 65
ВД>система 50
іШ -система 51
Сіалові кислоти 240
Сірчаністий іприт 114
Сквален281
Складні білки 283,314
Складні вуглеводи 244
^
44 9
ПРЕДМЕТНИЙ ПОКАЖЧИК
Складні ефіри 134,155,160,162,235,258,260,264,272
Складні ліпіди 252,255,258,263-264
Скопін 355
Скополамін 354-355
Б -сорбіт 235
Ь-сорбоза 242
Спермідин 146,337
Спермін 146,337
Спирти 2 8 ,6 6 ,7 1 -7 2 ,9 8 -1 0 5 ,1 0 9 ,1 6 0 ,2 1 7 ,2 3 4 ,2 5 2 ,2 5 4 ,2 7 2 ,2 7 8 ,3 4 6 ,
372
а-спіраль 307
Спіраль типу колагену 307
Сполука Бентлі 353
Сполука поліфункціональна 22
Сполуки аліфатичні 18,72,144,229
Сполуки аліфатичні циклічні 19
С полуш аліцгослічні 18
Сполуки ароматичні 18
Сполуки ациклічні 18,72
Сполуки гетерофуикціональні 22,134,138,141,146,153
Сполуки гетероциклічні 20,24,89,154,172,174
Сполуки жирного ряду 18
Сполуки карбоциклічні 19
Спряжена кислота 69
Спряжена основа 69
СпряжейІ алкадієни 82-83,88
Спряжені си стем и 41,83,93,175
Спряження 41,83
Старшинство замісників 52
Старшинство функціональних груп 23
Статична поляризація 39
Стахіоза 244
Стеарат натрію 131
Стеаринова кислота 131,253
'
*
450
ПРЕДМЕТНИЙ ПОКАЖЧИК
Стсран 96,268-269,276
Стереоізомери 4 5 ,4 7 ,4 9 ,6 2 ,2 1 9 ,2 2 1
&і ~ t
Стереоізомерія 43,45
Стереоселективні реакції 53
Стереоспецифічні ферменти 53
Стереоформули 45
Стереохімічні формули 45,49
Стериди 272
Стерини 271,273
5Р-стероїд 271
5 а-стер о їд 271
Стероїди 160,229,251,255,268-271,273,275,277,282
Стероїдні гормони 275,371,377,381
Стероли 272
Стрептидин 360
L-стрептоза 360
Стрептоміцини 346,360
Стрептоцид 111,149
Стрихнін 356-357
Строфантидин 276
Строфантин 277
Р-структура 307-308
Структурна ізомерія 45,161
Субстрати 63, 68, 220
Сукцинати 138
Сукциніл-КоА 185
Сульфадиметоксин 149
Сульфаніламід Ш , 148-149,193.
Сульфанілова кислота 111,148
Сульфапіридазин 213
Сульфгідрильна група 133
Сульфенові кислоти 110
Сульфіди 109,111-112
Сульфідна група
«
і gm
451
ПРЕДМЕТНИЙ ПОКАЖЧИК
Сульфінові кислоти ІДО
Сульфокислоти 110-111
Сульфоксиди 111-112
Сульфони 111-112
Сульфонієві основи 70
Сульфонова (сульфо-)група 94
Сульфонові кислоти 110
Супервторинні структури 308
• Сфінгозин 133» 252,258,260-261
Сфінгоміеліни
Сфінгофосфоліпіди 255,258,260-261,263
Т
Д аб у н 387
D-Ііл о за 22Д
Т аніни!43,348
Таурохолева кислота 274
Таутомери 44,190
Таутомерія 44,66,157,189,, 208,210,226
Твіст-конформація 60
Теобромін 211
Теофілін 211
Терефталева кислота 140
Терпени 251,255,269,277-282
Терпенові вуглеводні 277
Терпенрїди 278,280
Терпін 280
Терпінгідрат 280
Тестостерон 382-383
Тетрагідропірш 180
5,6,7,8-тетрагідрофолієва кислота (ТГФК) 369
Тетрадекан
Тетраконтан f
Тетрапірольні сполуки 183
4 52
ПРЕДМЕТНИЙ ПОКАЖЧИК
Тетратерпени 278
Тетрахлорметан 346,387
Тетрацен 96, 360
Тетрацикліни 96,346,360-361
Тетрози218-219
Тимідилова кислота (ТМФ)
-ЗА~(Тимін 207,321,326,332,335
Тирозин 154,170,288-289,301,347
Тироксин 16,377
Тіазин 213,359
Тіазол 1 7 3 ^ 9 2 ,2 0 7
Тіамін 20,192,208,362-363
Тіаміндифосфат (ТДФ) 363
Тіошікозиди 230
6-тіогуанін 329
Тіоефіри 109,125,133
Тіоли 70-71,98,109-112
Тіолова група 133
Тіольна група 98
Тіоридазин 214
Тіоспирти 109,227,230
Тіофен 20,89,173,175-176,179,187
Тіофеноли 109
а-токоферол 267-268,373
Токофероли 206
Толуол 87,95
Тореійна напруга 60
транс-бутен-2-ова кислота 136
транс-бутендіова кислота 137
Транспептидаза359
Транспортан РНК (тРНК) 294-295, 326, 339, 342-343
Транс-ретиноєва кислота 371
В-треоза 220
Треонін 250,287,289,303
453
ПРЕДМЕТНИЙ ПОКАЖЧИК
Третинна структура білків 309
Третинна структура ДНК 336
Третинний атом вуглецю 44,74
Третинні аміни 145
Триаконтан
Триацилшіцероли 252,255
3.4.5-тригідроксибензойна кислота
2.4.6-тригідроксишримідин 208
Трипхіцериди 252,254-256,264,275
Тридекан
Трийодметан 77
Трийодтиронін 377
Трикарбонові кислоти 138
Тримекаїн 150
Триметиламін 145
2.4.6-тркнітрофенол 108
2.4.6-трйоксипіримідин 2 0
2 ,6 ,8 -триоксипурин 2 1 0
ТриоленоїЛгаіцерин 256
Трипептиди 291
Триптамін 181-182
Триптофан 1 8 1 ^ 1 ^ 2 ^ 9 ,301,347
1яаш §8
Р® -«Ш І ш т і І
Тристеарин256,265
ТристеароШліцерин 256,265
Тритерпениі278
Трифтазин214
Трихлорметан 77
Трихлороцтова кислота 130
Тріози 218-219
Тромбоксани 252,382
Тропан 347,334
Тропін 354 І
Тропова кислота 354
Тропоколаген 309
454
ПРЕДМЕТНИЙ ПОКАЖЧИК
У
Ундекан
Унітіол 110,113
Урацил 207,321-322,326,332
Уридилова кислота (УМФ)
Уридин 230,324
Уридиидифосфоглюкуроиова кислота (УДФ) 241,329
Уронові кислоти 233,241
Ф
ФАД 213,239,329,364
Фактор Больцмана 63
Фамотидин 192,380
Феназон213
Фенантрен 2 0 ,8 6 ,8 9 ,9 5
Фенантренізохіноліну (морфінан) 201
Фенацетин 148,170
Феніл амін 146
N -фенілацетамід 151
Фенілгідразин 122
Фенілгідразон 122
Фенілгідроксиламін 145
о-фенілендіамін 146
Фенілізотіоціанат 319
Фенілметанол 99
Фенілоцтова кислота 140
Фенілсаліцилат 142
Фенільний радикал 87
Фенобарбітал 208
Феноксиди 107
Феноксиметилпеніцилін 158
Феноксиметилпеніцилін (пеніцилін V) 158,358
Феноли 99,106-109,301
455
ПРЕДМЕТНИЙ ПОКАЖЧИК
Фенолокислоти 141-143
Фенолфталеїні 41
Феноляти 107
Фенотіазини 213
Фепромарон 204
Ферменти 53-54,62-64,82,95,103-104, 125*134;1 3 6 ,150,158,161,230,
264,294,296,310,314 , 318,329,337
Фібрилярні білки 308-310
Фіброїн 308
Фізіологічно активні сполуки (ФАС) 39,54, 6 0 ,2 0 6 ,2 1 4 ,3 4 4 ,3 4 6 -3 4 7
350,375,382
Фізостигмін (езерин) 355-357
Фітостерини 271
Флавінаденіндинуклеотид 213
Флавіни 212
Флавінмононуклеотид 213,364
Флавон 205,228
Флавоноїди 203-206
Флороглюцин 106
Флуорескамін 301,316
Флуорескамінова реакція 300,316
Ф М Н 213,239
Фолієва кислота 212,361,367
Формалін 125
Формальдегід 115,120,124,348
Формамід 153
N-формілметіонін 125
Формула емпірична 16
Формула Кекуле 86
Формула молекулярна 16
Формула структурна 16
Формуйа структурна скорочена 17
Формула структурна спрощена 17
456
ПРЕДМЕТНИЙ ПОКАЖЧИК
Формули Коллі “ Толленса 224
формули Хеуорса 224
Фосфатидилетаноламіни 258,264
Фосфатидилсерини 258,264
Фосфагадилхоліни 258,264
Фосфатидна кислота 258
Фосфотяіцериди 252,258,263-264
6-фосфогаюконова кислота 232,242
Фосфоліпази 264
Фосфоліпіди 136,252,255,258,264
Фосфорорганічні пестициди 383
Фракціонування білків 314—315
D-фруктоза 217-218,224,236-238
Фруктозид 227
Фруктозо-1,6-дифосфат 238
Фруктозо-6-фосфат 238
a -D -фруктофураноза 224—225
ß-D-фруктофураноза 214-225,244-245,248
ß -фруктофуранозидаза 246
Фталазол 193
Фталева кислота 140
Фталевий ангідрид 141
Фтивазид 198
Фторотан 77
5-фторурацил 330
Фумарова кислота 55,7 8 ,1 3 7
Функціональні ірупи 21,23-24,27,44,153,241,290
Фуразолідон 186,193
Фуран 89,173,175-176,179,185,187
Фуранозиди 227
Фуранозний цикл 187
Фурацилін 186
Фурфурол 20,185-186,236
ПРЕДМЕТНИЙ ПОКАЖЧИК
X
Характеристична група 23-24
Харчові волокна 248
Хвороба Адисона щ Бірмера 367
Хелідонова кислота 204
Хенодезоксихолева кислота 274
Хімічні зв’язки 17,31,37
Хімотрипсин 308,319
Хінін 350-351
Хінолін 1943,199-201,347,350
Хінуклідил-З-бензилат 350
Хінуклідин Зж)—351
Хіральний атом 48-49,52
Хіральні сполуки 53-54
Хіральні центри 51-52,219,234,285
Хіральність 48 _
Хітозамін 240
Хлодіазепоксид 216
Хлоралкани 75,387
Хлораль 121
Хлоральгідрат 121
Хлоретан 77
Хлоретил 77
Хлорорганічні пестициди 346,386
Хлорофіл 185,217
Хлороформ 77,179,252
Хлороцтова кислота 130
Холан 274
Холанова кислота 274
Холева кислота 274
Холестан271
Холестериди 272
Холестерин 160,263,272,281,375
Холестерол 252,272
458
ПРЕДМЕТНИЙ ПОКАЖЧИК
Холін 167-168,252
Холінацетилтрансераза 168
Холінолітики 376
н-холіноміметики 349
Хондрозамін 240
Хондроїтинсірчана кислота 250
Хондроїтинсульфати 235,249-250
Хроман 206,268
Хроматографія 53,248,315
Хромон 203,205
ц
Цвіттер-іони 155,290
Целюлоза 76,247
Цераміди 260-261
Цереброзиди 261-262
Цериди (воски)255,258
Цетиловий спирт 258
Цефалоспорин С 359
Цефалоспорини 359
Цикл Кребса 139, 166
Цикл лимонної кислоти 139
Цикл трикарбонових кислот (ЦТК) 139
Циклічний АМФ (3‘,5‘-АМФ; цАМФ) 345 •
Циклічні амінокислоти 286
Циклічні вуглеводні 59,72
Циклічні терпени 278-279
Циклогексан 19,60-61,226,269
Цикпогексанол 99
Цикло-оксо-таутомерія 226
Циклопентан 19,60,62,269
Циклопентанпергідрофенантрен 62,229,268
Циклопропан 19
Циклосерин 193
459
ПРЕДМЕТНИЙ ПОКАЖЧИК
В-цимароза 276
Цинаризин209
Цинга (скорбут) 243,370
Цис-, транс-ізомери 54
Цис-, транс-номенклатура 55
цис-аконітова кислота 138
цис-бутендіова кислота 137
Цистеїн 110-112,287,292,302,318
Цистин 112
ЦшшДшіова кислота (ЦМФ)
Цитидин 324
Цитозин 207,321,326,334-335,339
Цитотек 382
Цитохром Р-450 387
Ціангідрини 1 2 1
Ціаніди 119
Ціановоднева кислота 119,229
Ціанокобаламін 366
Цукри 22,217,226,235,244
Цукрові кислоти 231,241
Цукрові спирти 234
Ч
Частковий електричний заряд 40-42
Червоний стрептоцид 149
Четвертинна структура білків 311
Четвертинні амонієві основи 167
Чоловічі статеві гормони 275,380
Щ авлева кислота 137
Щ авлевооцтова кислота 164,166
Я
Яблучна кислота 159
46 0
Нові книги від “НОВА КНИГА»
М едичні: науки
В ийш ли з друку:
Бардов В. Г., Сергета І, В. та ін. Загальна гігієна та екологія
людини (навчальний посібник для стоматологічних факультетів ВМНЗ
III - IV рівнів акредитації)
Вітент І. С„ Вітенко Т. І Основи психології (підручник для ВМНЗ ІІГ- IV
рівнів акредитації)
Громовик Б. П. Організація роботи аптек (навчальний посібник для
фармацевтичних факультетів ВМНЗ Ш - IV рівнів акредитації)
Середюк Н. М. та ін. Діагностика та лікування невідкладних станів та
загострень терапевтичних захворювань (навчальний посібник для ВМНЗ
III - I V рівнів акредитації)
Терещук С, І. та ін. Система бухгалтерського обліку в аптеках
^навчальний посібник дйя фармацевтичних факультетів ВМНЗ III - IV
рівнів акредитації)
Левітін Є. Я. та ін. Загальна та неорганічна хімія (підручник для
фармацевтичних факультетів ВМНЗ III - IV рівнів акредитації)
Левітін Є. Я. та ін. Загальна та неорганічна хімія (практикум для
фармацевтичних факультетів ВМНЗ III - IV рівнів акредитації)
Тарасюк В. С. та ін.,Медична сестра в інфекційному контролі лікарні
(навчальний посібник для ВМНЗ І - II рівнів акредитації)
М. Туркевич, О. Владимирська, Р. Лесик. Фармацевтична хімія
(підручник для фармацевтичних факультетів ВМНЗ III—Ш рівнів
акредитації)
Головко Н. В. Ортодонтія (навчальний посібник для студентів
стоматологічних факультетів ВМНЗ І - її рівнів акредитації)
46 І
Готуються до видання:
Пішак В. П. та ін. Медична біологія (підручник для ВМНЗ III - IV
рівнів акредитації)
Сербія А. Г, та ін. Медична ботаніка (навчальний посібник для
фармацевтичних факультетів ВМНЗ III - IV рівнів акредитації).
Сєркова В. К. та ін. Факультетська терапія (підручник для с т у д е н т ів
ВМНЗ III - IV рівнів акредитації)
Губський Ю. І. Біоорганічна хімія (підручник для студентів
фармацевтичних факультетів ВМНЗ III - IV рівнів акредитації)
Митченок В. І. та ін. Пропедевтика хірургічної стоматології
(підручник для студентів стоматологічних факультетів ВМНЗ III - І щ |
рівнів акредитації)
Якубисяк М., Чоп ’я к В. В. Імунологія (підручник для студентів
ВМНЗ III - IV рівнів акредитації)
Пухлик Б. М. Алергологія (навчальний посібник для студентів В М Н Зя
III - IV рівнів акредитації)
Маленький В. П., М аєш Н. П. Клінічна електрокардіографія
(навчальний посібник Для студентів ВМНЗ III - IV рівнів акредитації) Щ
Маленький В. 77. та ін. Професійні хвороби (підручник для студентів
ВМНЗ III - IV рівнів акредитації)
Маленький В. П. та ін. Госпітальна терапія (навчальний посібник
для студентів ВМНЗ Ш - IV рівнів акредитації)
Шапринський В. О. та ін. Факультетська хірургія (підручник для
студентів ВМНЗ ПІ IV рівнів акредитації)
Біктіміров В. В. та ін. Біопсійно-секційний курс (навчальний
посібник для студентів ВМНЗ III - IV рівнів акредитації)
Мороз В. М. та іиі Охорона праці у медичній галузі (навчальний
посібник для студентів ВМНЗ III - IV рівнів акредитації)
Мороз В. М. та ін. Нормальна фізіологія (практикум для студентів
ВМНЗ ІЇІ - IV рівнів акредитації),^
Мороз В. М. та ін. Нормальна фізіологія (атлас для студентів ВМНЗ
III —IV рівнів акредитації)
Мороз В. М. та ін. Нормальна фізіологія (курс лекцій Для студентів
ВМНЗ I I I - IV рівнів акредитації)
Ш евчук В. Г. та ін. Нормальна фізіологія (підручник для ВМНЗ III IV рівнів акредитації).
Петренко В. І. Фтизіатрія, (підручник для студентів ВМНЗ III - IV
рівнів акредитації)
Ш лопов В. Г. та ін. Патологічна анатомія (підручник для студентів
ВМНЗ III - IV рівнів акредитації)
Ш лопов В. Г. та ін. Патологічна анатомія (атлас для студентів $
ВМНЗ I I I - IV рівнів акредитації)
Громовик Б. П. Маркетинг у фармації (навчальний посібник для
фармацевтичних факультетів ВМНЗ III - IV рівнів акредитації)
Громовик Б. П., Терещук С. І. Практикум з організаціх та економіки
фармації (навчальний посібник для фармацевтичних факультетів
ВМНЗ III - IV рівнів акредитації)
Глушко Л. В., Волошинський О. В. Довідник з інтенсивної терапії
невідкладних станів та медицини катастроф (довідник для практичних
лікарів)
М ороз А. С. Біофізична та колоїдна хімія (підручник для
фармацевтичних факультетів ВМНЗ III —IV рівнів акредитації)
«В&пшцьюий л чржаївтідй
ІМіейїгчиад унщг -'осит'зТ
БІаЛІОГЕКА
\|
Б ільш дегттвтгвна інф орм ація
www.novaknyha.com.ua
Навчальне видання
Юрій Іванович ГУБСЬКИЙ
БІООРГАНІЧНА ХІМІЯ
Підручник для студентів вищих фармацевтичних закладів освіти
та фармацевтичних факультетів вищих медичних закладів освіти
Щ—IV рівнів акредитації
Коректор Л. Я. Шутова
Комп’ютерна верстка: А. М. Райфурщ, Щ Ж. Слободянюк.
СвідоцтвоДК№ ї 03
г
;
Підписано до друку 03.03.2004 р. Формат 60890/16. Гарнітура Тип Тайме.
Папір офсетний. Друк Офсетний. Ум. друк. арк. 26,97.
Облі-вид. арк.27. Тираж2 000 прим. Зам. Да 42.
Видавництво "Нова книга"
21100, м. Вінниця, вуй. Квятека, 20
тел. (0432)52-34-80,52-34-81,52-34-82
E-mail: newbookl@vinnitsa.com
www.novaknyha.com.ua
Віддруковано з готових діапозитивів
наЩ1 Короновський I.A.
2ІА00, м. Вінниця, вул. Гоголя, 19 офіс 114.
ІЩЦЧіІІ Ції ПІЧИІІ».....
я б Ш Й Ш з
д іи я ш т о т в а