/





















































































































Текст
Dietrich Braun,
Harald Cherdron,
Werner Kern
PRAKTIKUM DER
MAKROMOLEKULAREN
ORGANISCHEN
CHEMIE
2., VERBESSERTE UND ERWEITERTE
AUFLAGE MIT 35 ABBILDUNGEN
Dr. Alfred Hiithig Verlag Heidelberg
Д. Браун,
Г. Шердрон,
В. Керн
ПРАКТИЧЕСКОЕ
РУКОВОДСТВО
по синтезу
и исследованию
свойств
полимеров
Перевод с немецкого
Е. С. Гариной, М. Б. Лачинова,
Р. В. Тальрозе
Под редакцией В. П. ЗУБОВА
МОСКВА
ИЗДАТЕЛЬСТВО «ХИМИЯ»
1976
УДК 678.6/7:678-1 (076.5)
Браун Д., Шердрон Г., Керн В.
Практическое руководство по синтезу и исследованию
свойств полимеров. ФРГ, 1971. Пер. с нем. Под ред. до кт.
хим. наук В. П. Зубова. М., «Химия», 1976.
В книге рассмотрены основные методы синтеза, выделе-
ния, идентификации и исследования свойств полимеров. В на-
чале каждой главы приведено небольшое, но ценное теорети-
ческое введение, затем описываются хорошо подобранные
экспериментальные методики и даются практические советы.
Это руководство написано на высоком научном и методи-
ческом уровне широко известными учеными в области высо-
комолекулярных соединений и опытными педагогами.
Книга предназначена для студентов, изучающих химию
и физикохимию высокомолекулярных соединений. Она будет
полезна аспирантам, преподавателям и научным работникам,
а также работникам заводских лабораторий и отраслевых
научно-исследовательских институтов соответствующего про-
филя.
256 с.; 12 табл.; 35 рис.; список литературы 356 ссылок.
Б
31410-072
050(01)-76 72’7в
Перевод на русский язык.
© Издательство «Химия», 1976 г.
СОДЕРЖАНИЕ
От авторов............................................12
Предисловие...........................................13
1. Введение.......................................................15
1.1. Синтез высокомолекулярных соединений ........................ 15
1.2. Строение высокомолекулярных соединений........................17
1.3. Конформация макромолекул.................................... 32
1.4. Агрегатные состояния и свойства высокомолекулярных соединений 32
1.4.1. Твердые полимеры........................................32
1.4.2. Плавление полимеров.....................................36
1.4.3. Высокоэластпческое состояние полимеров..................39
1.4.4. Растворы полимеров......................................41
Литература.........................................................41
2. Общие методы химии высокомолекулярных соединений................. 44
2.1. Получение высокомолекулярных соединений..........................44
2.1.1. Работы, проводимые в отсутствие кислорода и влаги ... 44
2.1.2. Очистка и хранение мономеров...............................46
2.1.3. Реакционные сосуды для полиреакций.........................48
2.1.4. Температурный режим при полиреакциях.......................51
2.1.5. Проведение полиреакций.....................................51
2.1.5.1. Блочная полимеризация...............................51
2.1.5.2. Полиреакции в растворе ........ 52
2.1.5.3. Полимеризация в дисперсных системах.................55
2.1.5.3.1. Полиреакции в суспензии........................56
2.1.5.3.2. Полиреакции в эмульсии.........................56
2.1.5.4. Регулирование и прерывание полимеризации ... 58
2.1.6. Полимераналогичные превращения ............................59
2.1.6.1. Особенности полимераналогичных превращений . . 59
2.1.6.2. Выбор условий реакции..............................61
2.1.6.3. Аналитические методы, применяемые для характеристики
полимеров................................................. 63
2.2. Выделение и очистка полимеров....................................64
2.2.1. Выделение полимеров........................................64
2.2.2. Очистка и сушка полимеров..................................66
2.2.3. Стабилизация полимеров.....................................67
2.3. Характеристика полимеров.........................................67
2.3.1. Растворитель и растворимость...............................68
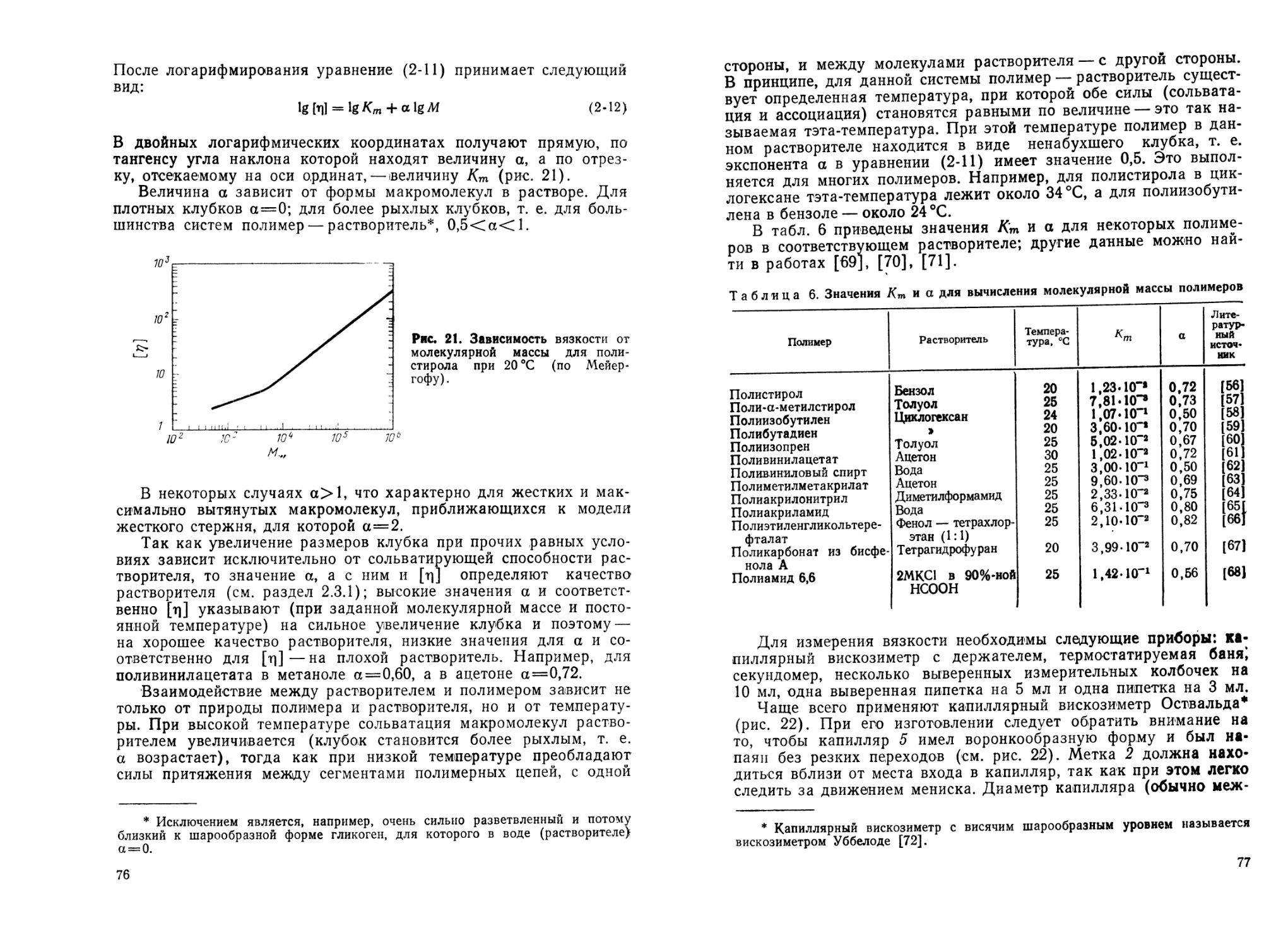
2.3.2. Определение молекулярной массы полимеров...................71
2.3.2.1. Определение вязкости растворов полимеров ... 73
2.3.2.2. Определение концевых групп полимеров .... 80
5
2.3.3. Фракционирование полимеров..................................81
2.3.4. Определение температуры стеклования, температуры размягче-
ния, интервала температур плавления и точки плавления кри-
сталлитов .........................................................87
2.3.4.1. Определение температуры стеклования..................87
2.3.4.2. Определение температуры размягчения..................87
2.3.4.3. Определение интервала температур плавления и точки
плавления кристаллитов.................................88
2.3.5. Определение вязкости расплавов полимеров...............89
2.3.6. Определение степени кристалличности полимеров .... 90
2.3.7. Определение плотности полимеров.............................99
2.3.8. Деструкция полимеров........................................91
2.3.8.1. Термическая деструкция полимеров............91
2.3.8.2. Химическая деструкция полимеров.............93
2.3.9. Оптические исследования полимеров..................93
2.3.10. Определение основных групп и элементов............94
2.3.11. Исследование сополимеров..........................95
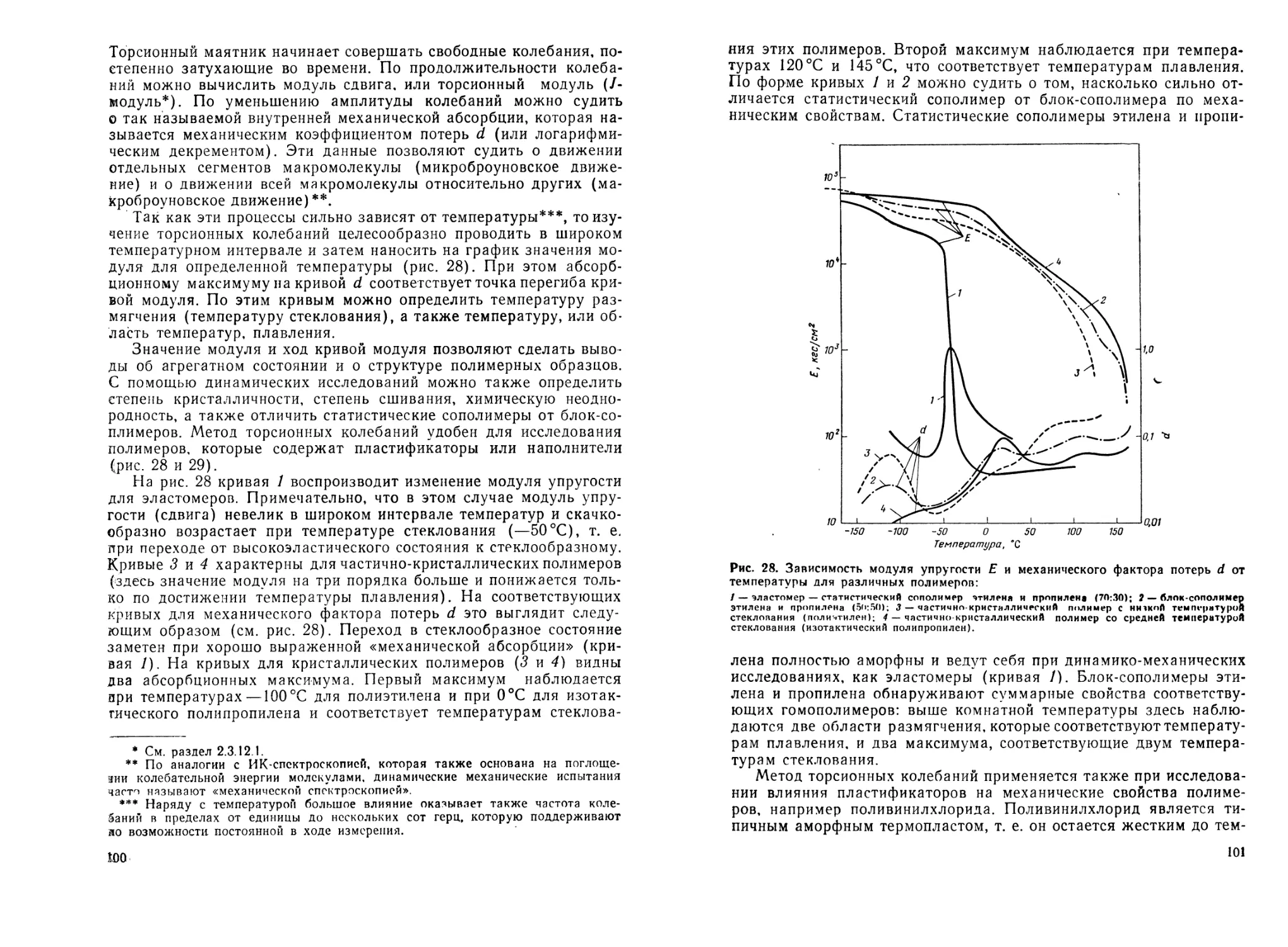
2.3.12. Механические методы исследования полимеров .... 96
2.3.12.1. Исследование деформации в зависимости от растяги-
вающего усилия.....................................97
2.3.12.2. Динамические механические испытания .... 99
2.3.12.3. Ударная вязкость..................................102
2.3.12.4. Определение твердости.............................103
2.4. Переработка полимеров...................................104
2.4.1. Измельчение полимеров......................................104
2.4.2. Переработка полимерных расплавов...........................104
2.4.2.1. Получение изделий прессованием......................105
2.4.2.2. Формование волокна из расплава......................105
2.4.3. Переработка растворов полимеров............................106
2.4.3.1. Получение пленок....................................106
2.4.3.2. Изготовление нитей из полимерных растворов («пря-
дение») ....................................................106
2.4.4. Получение вспененных полимеров (пенопластов) .... 107
Литература............................................................108
3. Синтез высокомолекулярных соединений методами полимеризации 112
3.1. Свободнорадикальная гомополимеризация................112
3.1.1. Полимеризация, инициированная перекисными соединениями 119
Опыт 3-01. Термическая полимеризация стирола в массе (влия-
ние температуры)....................121
Опыт 3-02. Полимеризация стирола в массе, инициированная
перекисью бензоила (влияние концентрации инициа-
тора) ......................................................122
Опыт 3-03. Эмульсионная полимеризация стирола, инициирован-
ная персульфатом калия......................................122
Опыт 3-04. Получение вспенивающегося полистирола и пено-
полистирола ................................................122
• Опыт 3-05. Полимеризация метилметакрилата в массе, иниции-
рованная перекисью бензоила..........................................123
Опыт 3-06. Полимеризация винилацетата в массе, инициирован-
ная перекисью бензоила .................................... 123
Опыт 3-07. Полимеризация винилацетата в водной дисперсии,
инициированная персульфатом аммония . . 124
Опыт 3-08. Полимеризация акрилонитрила в растворе, иниции-
рованная персульфатом аммония..............................124
Опыт 3-09. Суспензионная полимеризация винилацетата . . 125
Опыт 3-10. Полимеризация метакриловой кислоты в водном
растворе, инициированная персульфатом калия . 126
6
3.1.2. Полимеризация, инициированная азосоединениями . . . . 12 6
Опыт 3-11. Полимеризация стирола в массе, инициированная
азо-бис-изобутиронитрилом (влияние концентрации
инициатора)...................................................127
Опыт 3-12. Исследование дилатометрическим методом полиме-
ризации стирола в массе, инициированной азо-бис-
изобутиронитрилом ............................................127
Опыт 3-13. Полимеризация стирола в растворе, инициированная
азо-бис-изобутиронитрилом.....................................130
Опыт 3.14. Полимеризация метилметакрилата в массе, иниции-
рованная азо-бис-изобутиронитрилом .... *30
Опыт 3-15. Полимеризация винилацетата в различных раствори-
телях, инициированная азо-бис-изобутиронитрилом 131
Опыт 3-16. Определение молекулярно-массового распределения
поливинилацетата методом фракционной экстракции 131
3.1.3. Полимеризация, инициированная с помощью окислительно-вос-
становительных систем................................................133
Опыт 3-17. Полимеризация акриламида, инициированная окис-
лительно-восстановительной системой в водной среде 135
Опыт 3-18. Фракционирование полиакриламида методом гель-
фильтрационной хроматографии.............................135
Опыт 3-19. Полимеризация акриламида, инициированная окис-
лительно-восстановительной системой в воде (осади-
тельная полимеризация)...................................136
Опыт 3-20. Эмульсионная полимеризация изопрена, иницииро-
ванная окислительно-восстановительной системой 137
Опыт 3-21. Полимеризация стирола, инициированная окисли-
тельно-восстановительной системой в органическом
растворителе..................................................137
Опыт 3-22. Полимеризация винилхлорида, инициированная окис-
лительно-восстановительной системой в метаноле
(осадительная полимеризация)..................................138
3.2. Ионная полимеризация..............................................138
3.2.1. Ионная полимеризация по С = С-связи.139
3.2.1.1. Полимеризация, инициированная кислотами Льюиса 144
Опыт 3-23. Полимеризация изобутилена при низкой температу-
ре, инициированная газообразным трехфтористым
бором........................................................146
Опыт 3-24. Полимеризация винилизобутилового эфира, иниции-
рованная эфиратом трехфтористого бора при низкой
температуре.........................................146
Опыт 3-25. Димеризация стирола..........................147
Опыт 3-26. Катионная полимеризация а-метилстиролав растворе 147
3.2.1.2. Полимеризация, инициированная металлоорганическими
соединениями............................................148
Опыт 3-27. Полимеризация а-метилстирола, инициированная нат-
рийнафталиновым комплексом в растворе («живу-
щие цепи»).........................................148
Опыт 3-28. Стереоспецифическая полимеризация стирола, ини-
циированная н-амилнатрием....................................150
Опыт 3-29. Получение изотактического и синдиотактического
полиметилметакрилата, инициированного н-бутилли-
литием в растворе............................................152
Опыт 3-30. Стереоспецифическая полимеризация изопрена, ини-
циированная н-бутиллитием....................................152
3.2.1.3. Полимеризация на катализаторах Циглера — Натта . 154
Опыт 3-31. Полимеризация этилена на катализаторах Цигле-
ра — Натта . . . . ............155
Опыт 3-32. Стереоспецифическая полимеризация пропилена на
катализаторах Циглера — Натта ..... 156
7
Опыт 3-33. Стереоспецифическая полимеризация стирола на ка-
тализаторах Циглера — Натта...............................
Опыт 3-34. Стереоспецифическая полимеризация бутадиена на
катализаторах Циглера — Натта (получение цис-1,4-
полибута диена) ..........................................
3.2.2. Ионная полимеризация по С = О-связи.....................
Опыт 3-35. Анионная полимеризация формальдегида в растворе
(осадительная полимеризация)..............................
Опыт 3-36. Анионная полимеризация хлораля в растворе (оса-
дительная полимеризация)..................................
3.2.3. Ионная полимеризация по Ы=С-связи ......................
Опыт 3-37. Полимеризация w-бутилизоцианата в растворе, ини-
циированная цианистым натрием.............................
3.2.4. Полимеризация с раскрытием циклов.......................
3.2.4.1. Полимеризация простых эфиров с раскрытием цикла
Опыт 3-38. Полимеризация тетрагидрофурана в массе, иниции-
рованная пятихлористой сурьмой ...........................
3.2.4.2. Полимеризация циклических ацеталей с раскрытием
цикла ....................................................
Опыт 3-39. Полимеризация триоксана, инициированная эфира-
том трехфтористого бора...................................
3.2.4.3. Полимеризация лактонов (циклических сложных эфиров)
с раскрытием цикла........................................
Опыт 3-40. Полимеризация p-пропиолактона в массе, иницииро-
ванная алюминийорганическими соединениями
3.2.4.4. Полимеризация циклических амидов (лактамов) с рас-
крытием цикла ............................................
Опыт 3-41. Получение поли-е-капролактама в массе
3.3. Сополимеризация...............................................
3.3.1. Статистическая сополимеризация..........................
Опыт 3-42. Сополимеризация стирола с метилметакрилатом
Опыт 3-43. Свободнорадикальная сополимеризация стирола с
n-хлорстиролом (определение констант сополимери-
зации) ...................................................
Опыт 3-44. Катионная сополимеризация стирола с п-хлорстиро-
лом (определение констант сополимеризации)
Опыт 3-45. Свободнорадикальная сополимеризация стирола с
акрилонитрилом ...........................................
Опыт 3-46. Свободнорадикальная сополимеризация стирола с
бутадиеном в эмульсии.....................................
Опыт 3-47. Свободнорадикальная сополимеризация бутадиена с
акрилонитрилом в эмульсии.................................
Опыт 3-48. Свободнорадикальная сополимеризация винилхлори-
да с винилацетатом (внутренняя пластификация)
Опыт 3-49. Свободнорадикальная сополимеризация стирола с
дивинилбензолом в суспензии (трехмерная сополи-
меризация) ...............................................
Опыт 3-50. Катионная сополимеризация триоксана с 1,3-диокса-
ланом (сополимеризация с раскрытием цикла)
Опыт 3-51. Свободнорадикальная сополимеризация стирола с
малеиновым ангидридом («чередующаяся сополиме-
ризация») ................................................
Опыт 3-52. Свободнорадикальная сополимеризация циклогексе-
на с двуокисью серы («чередующаяся сополимериза-
ция») ....................................................
3.3.2. Блок- и привитая сополимеризация...................
157
158
159
160
161
161
162
162
162
164
164
165
166
166
167
168
169
169
175
176
177
178
179
179
180
181
181
181
182
182
8
Опыт 3-53. Получение блок-сополимера 4-винилпиридина со
стиролом методом анионной полимеризации . . 184
Опыт 3-54. Прививка стирола на полиэтилен .... 185
Литература.........................................................
4. Синтез высокомолекулярных соединений методами поликонденсации
и ступенчатой полимеризации.........................................188
4.1. Поликонденсация.................................................188
4.1.1. Сложные полиэфиры..........................................194
4.1.1.1. Полиэфиры на основе оксикарбоновых кислот . . . 194
4.1.1.2. Полиэфиры на основе диолов и дикарбоновых кислот 195
Опыт 4-01. Получение низкомолекулярных разветвленных поли-
эфиров поликоденсацией в расплаве . . . . 195
Опыт 4-02. Получение высокомолекулярных линейных полиэфи-
ров поликонденсацией в растворе ..... 196
4.1.1.3. Полиэфиры на основе диолов и дикарбоновых кислот 197
Опыт 4-03. Получение полиэфира из этиленгликоля и диметил-
терефталата поликонденсацией в расплаве . . . 198
Опыт 4-04. Получение поликарбоната из бисфенола А и фос-
гена поликонденсацией в растворе ......................... 198
4.1.1.4. Получение и сшивание (отверждение) ненасыщенных
полиэфиров.................................................199
Опыт 4-05. Получение ненасыщенного полиэфира и его отверж-
дение стиролом.............................................200
4.1.1.5. Получение и сшивание (отверждение) алкидных смол 201
Опыт 4-06. Получение и сшивание алкидных смол из глицерина
и фталевого ангидрида......................................202
Опыт 4-07. Получение ненасыщенной алкидной смолы («воздуш-
ной сушки»)......................................202
4.1.2. Полиамиды................................................ 203
4.1.2.1. Полиамиды на основе (о-аминокислот...........204
Опыт 4-08. Поликонденсация е-аминокапроновой кислоты в рас-
плаве .....................................................204
4.1.2.2. Полиамиды на основе диаминов и дикарбоновых кислот 205
Опыт 4.09. Получение найлона 6,6 из гексаметилендиаммоний-
адипата поликонденсацией в расплаве .... 205
4.1.2.3. Полиамиды на основе диаминов и производных ди-
кислот ...................................................206
Опыт 4-10. Получение найлона 6,10 из гексаметилендиамина
и хлорангидрида себациновой кислоты .... 206
4.1.3. Полиуретаны..........................................206
Опыт 4-11. Получение линейного полиуретана из этилен-бис-
хлорформиата и гексаметилендиамина поликонден-
сацией на границе раздела двух фаз .... 207
4.1.4. Фенолоформальдегидные олигомеры . ....................207
4.1.4.1. Поликонденсация фенола с формальдегидом в присутст-
вии кислотных катализаторов (новолаки) .... 208
Опыт 4-12. Поликонденсация фенола с формальдегидом, ката-
лизируемая кислотой........................................210
4.1.4.2. Поликонденсация фенола с формальдегидом в присут-
ствии щелочных катализаторов...............................210
Опыт 4-13. Поликонденсация фенола с формальдегидом, ката-
лизируемая щелочью.........................................211
9
4.1.5. Карбамидо- и меламиноформальдегидные олигомеры . . 211
4.1.5.1. Карбамидоформальдегидные олигомеры..................211
Опыт 4-14. Поликонденсация карбамида с формальдегидом . 213
4.1.5.2. Меламиноформальдегидные олигомеры...................213
Опыт 4-15. Поликонденсация меламина с формальдегидом . 214
4.1.6. Полиалкиленсульфиды ...................................... 215
Опыт 4-16. Получение полиалкиленсульфида из 1,2-дихлорэта-
на и тетрасульфида натрия.......................216
4.1.7. Полисилоксаны..............................................217
Опыт 4-17. Полимеризация с раскрытием никла олигосилоксана
с образованием линейного высокомолекулярного
полисилоксана с гидроксильными концевыми группа-
ми. Отверждение полимера...................................219
Опыт 4-18. Равновесная деструкция силиконового каучука с
образованием продукта с триметилсилильными кон-
цевыми группами (силиконового масла) . . . 220
4.1.8. Циклополиконденсация.......................................220
4.1.8.1. Комбинация полимеризации с поликонденсацией . . 220
4.1.8.2. Комбинация ступенчатой полимеризации с поликонден-
сацией .....................................................221
4.1.8.3. Комбинация поликонденсации с циклополиконденсацией 222
4.1.9. Дегидрирование ароматических соединений..................223
Опыт 4-19. Получение полидиметилфениленового эфира . . 224
4.2. Ступенчатая полимеризация......................................225
4.2.1. Полиуретаны..............................................225
4.2.1.1. Получение линейных полиуретанов...................226
Опыт 4.20. Получение линейного полиуретана из бутандиола-1,4
и гексаметилен-1,6-диизоцианата в расплаве . . 227
Опыт 4-21. Получение линейного полиуретана из бутандиола-1,4
и гексаметилен-1,6-диизоцианата в растворе (осади-
тельная ступенчатая полимеризация) .... 228
4.2.1.2. Получение разветвленных и сшитых полиуретанов . . . 228
Опыт 4-22. Получение эластичных пенополиуретанов . . . 230
Опыт 4-23. Получение жестких пенополиуретанов .... 231
4.2.2. Эпоксидные смолы...........................................231
Опыт 4.24. Получение эпоксидных смол из бисфенола А и эпи-
хлоргидрина в одну стадию..........................233
Опыт 4-25. Двухстадийный синтез эпоксидных смол из глицери-
на и эпихлоргидрина .......................... 235
Литература............................................................235
5. Реакция полимеров..................................................237
5.1. Химические превращения полимеров................................237
Опыт 5-01. Получение поливинилового спирта переэтерифика-
цией поливинилацетата; переацетилирование поли-
винилового спирта..........................................239
Опыт 5-02. Получение поливинилбутираля.......................239
Опыт 5-03. Омыление сополимера стирола и малеиновой кис-
лоты ......................................................240
Опыт 5-04. Этерификация полиметакриловой кислоты диазо-
метаном ................................................. 240
Опыт 5-05. Получение поли-п-винилацетофенона .... 240
Опыт 5-06. Ацетилирование целлюлозы.........................240
Опыт 5-07. Получение триметилцеллюлозы......................241
Опыт 5-08. Получение натриевой соли карбоксиметилцеллюлозы 241
Опыт 5-09. Ацетилирование полуацетальных групп полиоксиме-
тилена уксусным ангидридом.................................242
Опыт 5-10. Вулканизация бутадиен-стирольного сополимера , 242
10
5.2. Ионообменные полимеры (иониты)..................................243
Опыт 5-11. Получение катионита сульфированием сшитого поли-
стирола ..............................................245
Опыт 5-12. Получение катионита сульфированием фенолоформ-
альдегидного олигомера................................245
Опыт 5-13. Получение анионита из сшитого полистирола хлор-
метилированием и аминированием........................245
5.3. Деструкция полимеров........................................... 246
Опыт 5-14. Термическая деполимеризация поли-а-метилстирола
и полиметилметакрилата . 248
Опыт 5-15. Термическая деполимеризация полиоксиметилена . 248
Опыт 5-16. Термическая деполимеризация поли-п-винилацето-
фенона................................................249
Опыт 5-17. Термическое дегидрохлорирование поливинилхлорида 249
Опыт 5-18. Окислительная деструкция поливинилового спирта
под действием иодной кислоты..........................249
Опыт 5-19. Гидролитическая деструкция алифатического поли-
эфира ................................................250
Опыт 5-20. Гидролитическая деструкция целлюлозы и разделе-
ние продуктов гидролиза методом бумажной хрома-
тографии ..................................................250
Литература...........................................................252
Предметный указатель.................................................253
ОТ АВТОРОВ
За пятьдесят лет, прошедшие с тех пор, как Герман Штаудин-
гер заложил основы современной химии полимеров, она превра-
тилась в совершенно самостоятельную и обширную область зна-
ния. Число опубликованных работ по высокомолекулярной химии
особенно выросло в последние годы. Для того чтобы студенты мог-
ли овладеть основными понятиями химии полимеров, им необходи-
мо познакомиться не только с теоретическими аспектами этой
науки, но и приобрести некоторые практические навыки.
Хотя в 1961 г. У. Сёренсон и Т. Кемпбел уже опубликовали кни-
гу «Препаративные методы химии полимеров», мы подошли к рас-
смотрению этого вопроса с несколько иных позиций. В наши пла-
ны не входило дать исчерпывающий набор методик синтезов по-
лимеров. Мы стремились описать общие препаративные методы
и наиболее важные приемы, которые используются для синтеза и
исследования полимеров. Поэтому примеры, предназначенные для
иллюстрации аппаратурного оформления экспериментов, приведе-
ны также для химиков, не специализировавшихся ранее в об-
ласти высокомолекулярных соединений.
Первое немецкое издание «Практического руководства по хи-
мии макромолекулярных соединений» вышло в 1966 г. Позже оно
было переведено на испанский и японский языки. В начале 1971г.
появилось второе немецкое издание, в нем в некоторые главы был
внесен ряд изменений и дополнений. Число приведенных экспери-
ментальных методик близко к 100.
В 1972 г. второе издание книги было переведено на английский
язык. Мы приносим благодарность нашим коллегам и соавторам,
принимавшим участие в создании этой книги. Особую признатель-
ность мы выражаем доктору Р. Биндеру, осуществившему перевод
книги на английский язык, и мисс Р. Вейс за полезное корректи-
рование и составление указателя.
Д- Браун,
Г. Шердрон,
В. Керн
ПРЕДИСЛОВИЕ
Предлагаемое внимание читателей «Практическое руководство по
синтезу и исследованию свойств полимеров» написано группой не-
мецких ученых, активно работающих как в университете
(В. Керн), так и в промышленности пластических масс (Д. Браун,
Г. Шердрон). Эта книга представляет интерес для советского чи-
тателя в силу нескольких обстоятельств. Во-первых, выпущенное
у нас аналогичное руководство И. П. Лосева и О. Я. Федотовой*
уже в значительной степени устарело. Несколько позднее изданный
«Практикум по технологии полимеризационных пластических
масс»** посвящен более узкой области, и написан он с техноло-
гическим уклонам. Таким образом, очевидна необходимость изда-
ния современного исчерпывающего и доступного практикума по
химии полимеров. Во-вторых, в предлагаемой книге Д. Брауна и
др. наряду с описанием методик конкретных лабораторных задач
большое внимание уделено рассмотрению теоретических основ то-
го или иного метода синтеза или исследования свойств полимеров
и, что очень важно, есть специальные главы чисто методического
характера, в которых рассматриваются, например, такие вопросы:
как определять вязкость в растворе и расплаве, как оценивать
температуру стеклования и плавления, как человеку, не имеюще-
му специального опыта, ставить эксперимент по эмульсионной по-
лимеризации и т. д. Вот эти практические советы весьма полезны
и составляют необходимый элемент данного руководства. В-треть-
их, настоящее лабораторное руководство охватывает довольно ши-
рокий круг объектов и реакций, и читатель может найти здесь
примеры самых разнообразных синтезов, полезных как специалис-
там, так и тем, кто непорредственно не имеет дело с полимерами,
но у кого возникает утилитарная необходимость синтезировать
тот или иной полимерный препарат. Дополнительную ценность
имеет широко представленный раздел по химическим прев-
ращениям полимеров, включая полиреакции, сшивание, деструк-
цию. Наконец, достоинство данной книги заключается и в том,
* Лосев И. П., Федотова О. Я. Практикум по химии высокомолеку-
лярных соединений. Изд. 2-е. М., Госхимиздат, 1962. 174 с.
** Григорьев А. П. Практикум по технологии полимеризационных пла-
стических масс. Под ред. В. В. Коршака. М., «Высшая школа», 1964. 214 с.
13
что приводимые методики общедоступны и могут быть воспроиз-
ведены в рамках обычной химической лаборатории.
Таким образам, настоящее практическое руководство сущест-
венно дополняет известные советскому читателю книги У. Сёрен-
сона и Т. Кемпбела* и «Макромолекулярные синтезы»** и пред-
ставляет собой квалифицированное изложение большого числа
приемов, методов и конкретных работ, необходимых для знаком-
ства с химией высокомолекулярных соединений.
Не следует, однако, думать, что так называемая «теоретическая»
преамбула полностью решает задачу знакомства читателя с хи-
мией полимеров. Это всего лишь введение, и фундаментальные
вопросы химии и физики полимеров излагаются порой слишком
конспективно и упрощенно. Если читатель всерьез интересуется
тем или иным вопросом, ему надо обратиться к гораздо более
полным монографиям советских и зарубежных авторов. К сожале-
нию, в списке рекомендуемой литературы, составленном авторами,
отсутствуют многие ссылки на советские оригинальные работы.
Полагаю, что книга Д. Брауна, Г. Шердрона и В. Керна будет
полезна как учебное пособие к практикуму по общему курсу хи-
мии высокомолекулярных соединений для университетов и химико-
технологических институтов, а также как справочник для инжене-
ров-химиков при решении ими практических задач по синтезу и
химическим превращениям полимеров.
* Сёренсон У., Кемпбел Т. Препаративные методы химии полимеров.
М., Издатинлит, 1968. 564 с.
* * Макромолекулярные синтезы. М., «Мир», вып. I, 1966; вып. 2, 1969.
Чл.-корр. АН СССР Н. А. Платэ
1. ВВЕДЕНИЕ
Макромолекулярная органическая химия изучает соединения с до-
статочно высокими молекулярными массами. Однако нельзя про-
вести резкой границы между обычными малыми молекулами и
макромолекулами; скорее, здесь имеет место постепенный переход.
Поэтому условно макромолекулами называют такие молекулы,
которые состоят из нескольких сотен атомов. В настоящее время
к макромолекулярным соединениям относят вещества с молеку-
лярной массой не менее 1000.
В отличие от низкомолекулярных соединений, построенных из
молекул одинакового строения и одинакового размера, высокомо-
лекулярные соединения большей частью являются смесями макро-
молекул одинакового или сходного строения, но с различной моле-
кулярной массой. Высокая молекулярная масса и другие свойства
высокомолекулярных соединений (например, структурная изоме-
рия и стереоизомерия) обусловливают ряд особенностей поведе-
ния этих веществ, которые следует иметь в виду как при синтезе,
так и при анализе высокомолекулярных веществ.
1.1. СИНТЕЗ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИИ
При взаимодействии соединений с реакционноспособными группа-
ми, которые при выбранных условиях реагируют селективно, об-
разуются соединения определенного строения. Органическое соеди-
нение с одной реакционноспособной группой называется моно-
функциональным, с двумя группами — бифункциональным, с
тремя или более — олиго- или полифункционалыным. Функцио-
нальность соединения зависит от условий реакции. Так, например,
первичная аминогруппа при образовании амида кислоты является
монофункциональной, а при реакции с алкилгалогенидами— три-
функциональной. Соединения с одной непредельной группой,
эпоксиды и циклические эфиры при реакциях присоединения яв-
ляются монофункциональными, а при полимеризации — бифунк-
циональными.
Для образования макромолекул соединения должны иметь по
меньшей мере две функциональные группы. При взаимодействии
бифункциональных молекул образуются линейные макромолеку-
15
лы, в то время как при взаимодействии олигофункциональных мо-
лекул— сшитые полимеры. Монофункциональные соединения да-
же в реакциях с олиго-функциональными соединениями образуют
низкомолекулярные продукты реакции.
При поликонденсации или полицрисоединении образовавшие-
ся макромолекулы могут иметь функциональные группы, которые
вновь участвуют в реакции. Однако не все реакции бифункцио-
нальных мономеров приводят к получению высокомолекулярных
соединений. Так, окись этилена может димеризоваться в диоксан,
формальдегид тримеризоваться в триоксан, и при последующих
процессах поликонденсации и полиприсоединения наряду с линей-
ными макромолекулами будут образовываться низкомолекуляр-
ные циклические соединения. Таким образом, внутримолекуляр-
ная циклизация является побочной реакцией при межмолекуляр-
ном соединении; при соответствующих условиях эта реакция
может стать основной, например при синтезе многочисленных цик-
лов по принципу разбавления Руггли — Циглера.
Полиреакции, с помощью которых получаются высокомолеку-
лярные соединения, делятся на три группы: реакции полимериза-
ции, полиприсоединения и поликонденсации. Все эти реакции
очень чувствительны к различным примесям и загрязнениям ре-
агентов, поэтому следует обращать особое внимание на чистоту
исходных продуктов и на тщательное проведение самой реакции.
Полимеризацией (см. гл. 3) называется такая химическая ре-
акция, цри которой мономеры, содержащие реакционноспособные
двойные связи или мономеры циклического строения, путем после-
довательного присоединения образуют макромолекулы либо спон-
танно, либо под воздействием инициаторов или катализаторов.
Однако особенностью полимеризации являются не сами стадии
процесса присоединения, а, скорее, его кинетика: полимеризация
представляет собой цепную реакцию. Различают цепную ради-
кальную и цепную ионную полимеризацию; ионная полимеризация
может протекать по анионному и катионному механизмам. Про-
цесс образования сравнительно низкомолекулярных продуктов
называется олигомеризацией.
В табл. 1 сопоставлены типы инициаторов полимеризации по
механизму их действия. Механизм действия инициаторов известен
не для всех случаев. Особенно это относится к инициаторам, при-
меняемым в ионной полимеризации. Кроме того, в таблице не да-
ны сокатализаторы, которые очень важны при проведении неко-
торых реакций ионной полимеризации (см. гл. 3).
Способность мономеров участвовать в радикальной или ионной
полимеризации зависит от различных факторов [1], в частности
от поляризации двойной или других связей (например, в случае
циклических мономеров), числа заместителей, природы инициато-
ра и температуры. Если способность мономера к радикальной по-
лимеризации можно установить сравнительно легко, то в случае
ионной полимеризации, когда нужно получить полимеры с высокой
16
Таблица 1. Инициаторы полимеризации
Радикальные Катионные Анионные
1. Перекиси и другие пере- кисные соединения 1. Протонные кислоты 1. Щелочные металлы
2. Азосоединения 2. Кислоты Льюиса 2. Металлоорганические соединения*
3. Окислительно-восстанови- тельные системы 3. Вещества, образую- щие катионы, способ- ные к наращиванию цепи 3. Основания Льюиса
4. Световая энергия или другой вид излучения* 4. Частицы высокой энер- гии* 4. Частицы высокой энер- гии*
* Механизм полимеризации выяснен еще не для всех случаев.
молекулярной массой или особого строения, это сделать значи-
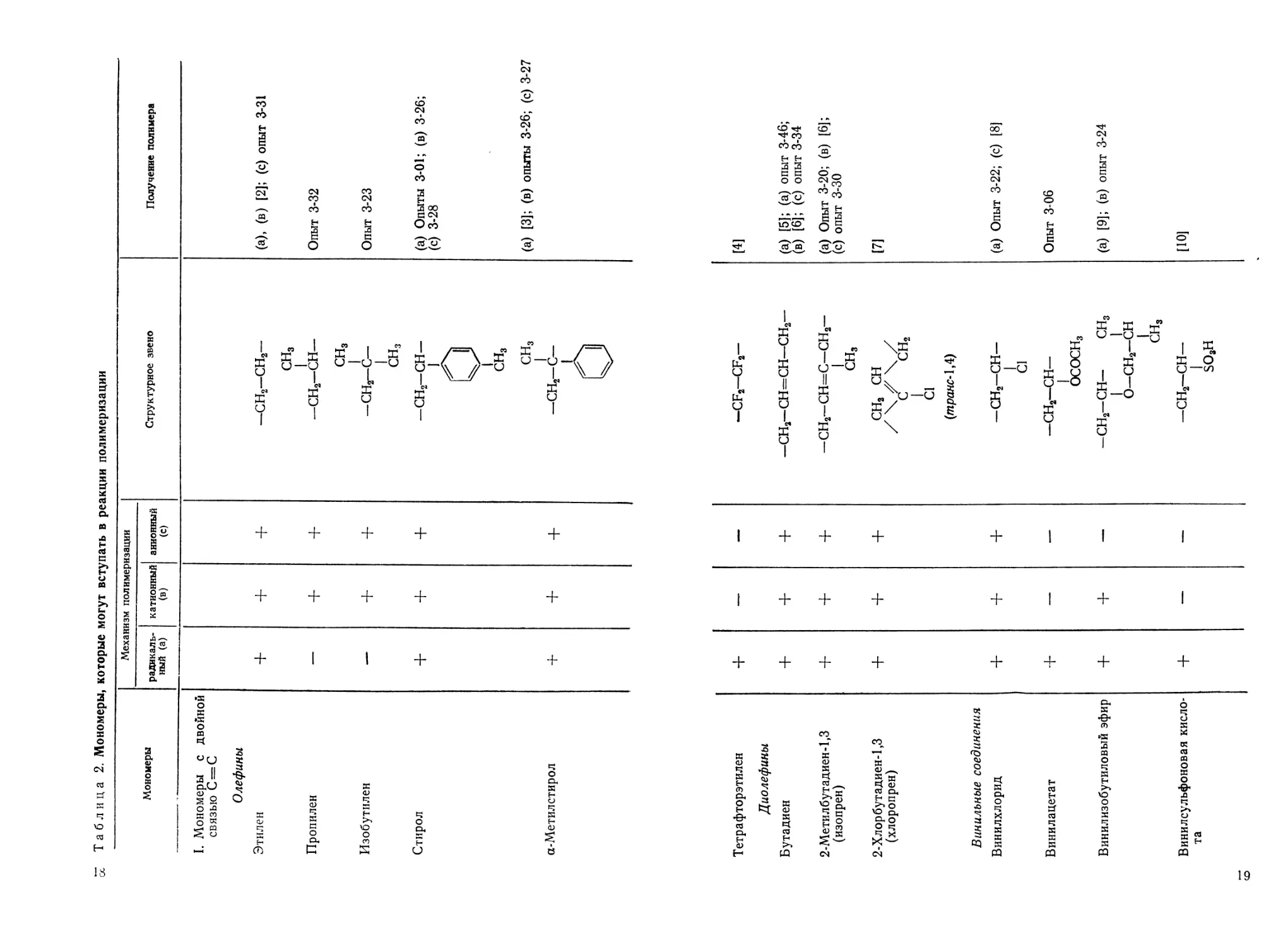
тельно труднее. В табл. 2 дан ряд различных мономеров и при-
ведена их способность к полимеризации. О мономерах, которые
в присутствии определенных инициаторов обладают пониженной
способностью к гомополимеризации, но хорошо сополимеризуются
(см. графу 4), будет упомянуто отдельно.
Поликонденсацией (см. раздел 4.1) называется такая химиче-
ская реакция, при которой образование макромолекул происходит
путем соединения би- или многофункциональных молекул, что со-
провождается выделением низкомолекулярных соединений (на-
пример, воды, спирта). Поликонденсация — типичная ступенчатая
реакция, и этим она также отличается от реакции полимеризации.
При полиприсоединении (см. раздел 4.2) би- или олигофунк-
циональные реагенты реагируют между собой без выделения низ-
комолекулярных соединений; обычно реакция протекает за счет
миграции водородного атома. Как и в случае поликонденсации,
при полиприсоединении макромолекулы (полиаддукты) образуют-
ся ступенчато. Этим полиприсоединение также отличается от поли-
меризации. Число известных пока реакций полиприсоединения
меньше, чем число полимеризационных или поликонденсациопных
реакций. Некоторые полиаддукты приведены в табл. 4.
Наконец, многие высокомолекулярные соединения получаются
по реакции обмена реакционноспособных полимеров с низкомоле-
кулярными соединениями (полимераналогичные реакции). Такие
реакции служат не только для модифицирования свойств уже из-
вестных полимеров, но иногда представляют единственный способ
получения полимеров (см. гл. 5).
1.2. СТРОЕНИЕ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
В состав макромолекул могут входить одно или несколько моно-
мерных звеньев (гомополимеры и сополимеры). По строению
основной цепи высокомолекулярные соединения делятся на карбо-
2—732
17
Таблица 2. Мономеры, которые могут вступать в реакции полимеризации
Мономеры Механизм полимеризации Структурное звено Получение полимера
радикаль- ный (а) катионный (в) анионный (с)
I. Мономеры с двойной связью С = С Олефины Этилен + + + -сн2-сн2- (а), (в) [2]; (с) опыт 3-31
Пропилен — + + сн3 —сн2—сн— Опыт 3-32
Изобутилен — + + сн3 -СНг-С— Опыт 3-23
Стирол + + + сн3 -сн2-сн- (а) Опыты 3-01; (в) 3-26;
а-Метил стирол + + + - 1 1^ сн3 сн3 —сн2—с— 1 (с) 3-28 (а) [3]; (в) опыты 3-26; (с) 3-27
Т етр а фторэти лен + — —
Диолефины
Бутадиен + + +
2-Метилбут адиен-1,3 (изопрен) + + +
2-Хлорбутадиен-1,3 (хлоропрен) + + +
Ванильные соединения Винилхлорид + + +
Винилацетат + — —
Винилизобутиловый эфир + + —
Винилсульфоновая кисло- та + — —;
—CF2 CF2’
—сн2-сн=сн—сн2-
-сн2-сн=с-сн2-
СН3
сн2 сн
/ \:н2
С1
(транс-1,4)
—СН2—СН—
С1
—СН2—СН—
ОСОСНз
-сн2—сн— сн3
I I
о-сн2-сн
I
сн3
-сн2-сн—
SO3H
[4]
(а) [5]; (а) опыт 3-46;
(в) [6]; (с) опыт 3-34
(а) Опыт 3-20; (в) [6];
(с) опыт 3-30
[7]
(а) Опыт 3-22; (с) [8]
Опыт 3-06
(а) [9]; (в) опыт 3-24
[Ю]
Продолжение
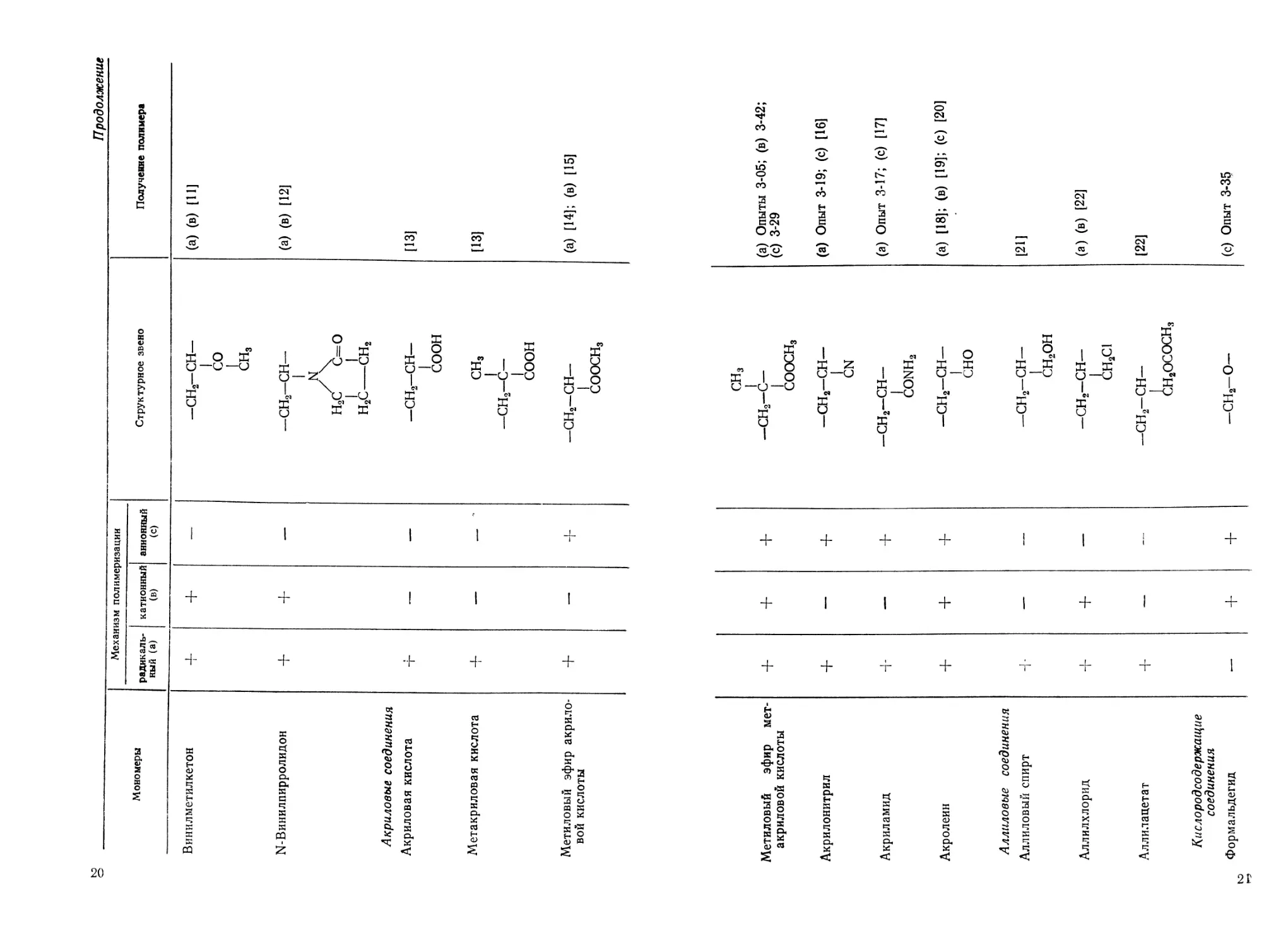
Мономеры Механизм полимеризации Структурное звено
радикаль- ный (а) катионный (в) анионный (с)
Винилметилкетон + — —СН2—СН— со СН3
N-Винилпирролидон Акриловые соединения + + — —СН2—СН— N н.с7 ^с=о Н2С—сн2
Акриловая кислота т- — — -СН2—СН— соон
Метакриловая кислота Д- — — СН3 1 —сн2-с— соон
Метиловый эфир акрило- вой кислоты + — + —сн2-сн— СООСНз
(а) (в) [11]
(а) (в) [12]
[13]
[13]
(а) [14]; (в) [15]
Получение полимера
Метиловый эфир мет- акриловой кислоты + ч- + сн3 —сн2-с— СООСНз (а) Опыты 3-05; (в) 3-42; (с) 3-29
Акрилонитрил 4- — 4- —сн2-сн— CN (а) Опыт 3-19; (с) [16]
Акриламид ч- — 4- —сн2-сн- conh2 (а) Опыт 3-17; (с) [17]
Акролеин 4- 4- 4- —СН2—СН— 1 сно (а) [18]; (в) [19]; (с) [20]
Аллиловые соединения
Аллиловый спирт ч- — — —сн2-сн— СН2ОН [21]
Аллилхлорид ч~ Ч- — -сн2-сн- СН2С1 (а) (в) [22]
Аллилацетат ч- — — —сн2-сн— С^Н2ОСОСН3 [22]
Кислородсодержащие соединения
Формальдегид — + 4- —СН2-О— (с) Опыт 3-3?
Продолжение
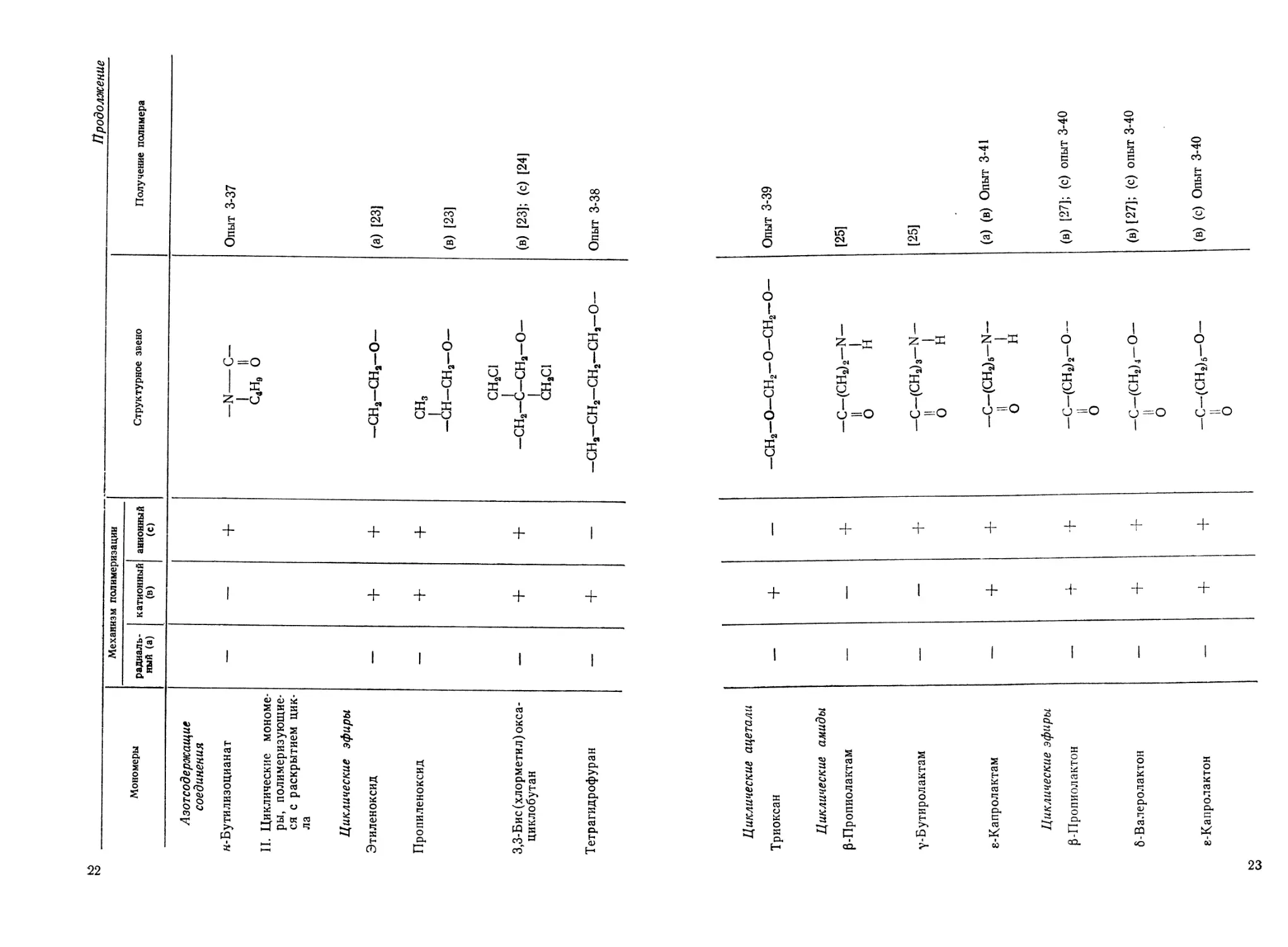
Мономеры Механизм полимеризации Структурное звено Получение полимера
радиаль- ный (а) катионный (в) анионный (с)
Азотсодержащие соединения н-Бутилизоцианат + —N С— Опыт 3-37
II. Циклические мономе- ры, полимеризующие- ся с раскрытием цик- ла Циклические эфиры Этиленоксид + + 1 II С4Нв О —сн2-сн2-о- (а) [23]
Пропиленоксид — + + сн3 1
3,3-Бис (хлорметил) окса- + + -СН-СН2—0- СН2С1 —сн2—<i—сн2—о— (в) [23] (в) [23]; (с) [24]
циклобутан Тетрагидрофуран — + — 1 СН2С1 —СН2—СН2—СН2—СН,—0— Опыт 3-38
Циклические ацетали Триоксан — + — —СН2—О—СН2—0—СН2—0— Опыт 3-39
Циклические амиды 0-Пропиолактам — — + 1 °- ? N> 1 [25]
у-Бутиролактам —- — + 1 Z—-X 1 со X У, 0=0 1 [25]
8-Капролактам — + + -C-fCHJs-N- 11 1 О н (а) (в) Опыт 3-41
Циклические эфиры р-Пропиолактон — + —С—(СН2)2—0— 11 0 (в) [27]; (с) опыт 3-40
б-Валеролактон — + + —С—(СН2)4—0— II 0 (в) [27]; (с) опыт 3-40
е-Капролактон — + + —С—(СН2)6—0— II 0 (в) (с) Опыт 3-40
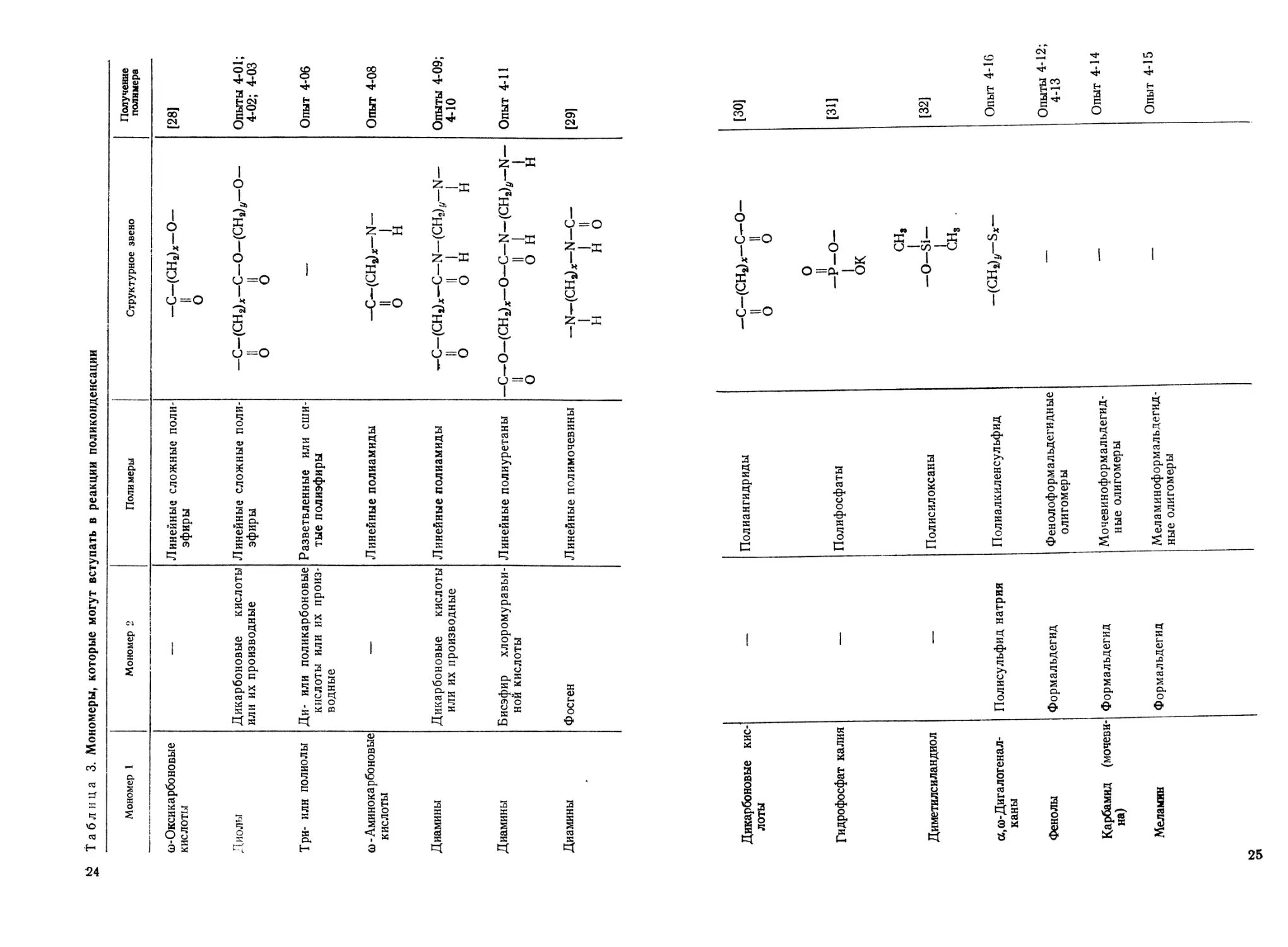
Таблица 3. Мономеры, которые могут вступать в реакции поликонденсации
Мономер 1 Мономер 2 Полимеры Структурное звено Получение полимера
со-Оксикарбоновые кислоты — Линейные сложные поли- эфиры -С-(СН2)х-О- II о [28]
Диолы Дикарбоновые кислоты или их производные Линейные сложные поли- эфиры -С-(СН2)х-С-О-(СН2)г/-О- II II О о Опыты 4-01; 4-02; 4-03
Три- илн полиолы Ди- или поликарбоновые кислоты или их произ- водные Разветвленные или сши- тые полиэфиры —- Опыт 4-06
со - Аминокарбоновые кислоты — Линейные полиамиды —С—(СНа)*—N— II 1 О н Опыт 4-08
Диамины Дикарбоновые кислоты или их производные Линейные полиамиды —С—(CHj)x—С—N—(CH2)V—N— II II 1 1 О он н Опыты 4-09; 4-10
Диамины Бисэфир хлоромуравьи- ной кислоты Линейные полиуретаны -С—О-(СНг)ж—О—С—N—(CH2)U—N— II II 1 1 О О н н Опыт 4-11
Диамины Фосген Линейные полимочевины -NHCHah-N-C- 1 1 II н но [29]
Дикарбоновые кис- лоты — Полиангидриды
Гидрофосфат калия — Полифосфаты
Диметилсиландиол — Полисилоксаны
ct, со-Дигалогена л- каны Полисульфид натрия Полиалкиленсульфид
Фенолы Формальдегид Ф ен ол оф о рм альдегидные олигомеры
Карбамид (мочеви- на) Формальдегид Мочевиноформальдегид- ные олигомеры
Меламин Формальдегид Меламиноформальдегид- ные олигомеры
к? сд J
ОК
СН8
—О—Si—
I
сн3 .
[30]
[31]
[32]
Опыт 4-16
Опыты 4-12;
4-13
Опыт 4-14
Опыт 4-15
Таблица 4. Мономеры, которые могут вступать в реакции полиприсоединения
Мономеры Мономеры Полимеры
Диолы Диизоцианаты Полиуретаны
Диамины Диизоцианаты Полимочевина
Ди- или полиэпоксиды Несопряженные диены Амины, ангидриды Бисмеркаптаны Эпоксидные смолы Политиоэфир
цепные полимеры, содержащие в главной цепи только углеродные
атомы (например, винильные полимеры), и гетероцепные полиме-
ры, содержащие помимо углерода другие атомы (например, кис-
лород в полиацеталях, азот в полиамидах) *.
Наименьшая часть макромолекул, из которых построена дан-
ная цепь, называется элементарным или основным звеном. Макро-
молекула может состоять из различных видов звеньев (сополиме-
ры) . Высокомолекулярные природные соединения также построе-
ны из основных звеньев. Эти звенья обычно определяются моно-
мерами, которые используются для синтеза соответствующих по-
лимеров. В случае полимеризации молекулярная масса основного
звена равна молекулярной массе мономера. В полимерах виниль-
ного ряда основное звено содержит два атома углерода; в основ-
ные звенья, однако, могут входить три или даже большее число
атомов. Существуют также звенья, содержащие только один
атом:
----СН2-СН2~СН.г~1СЩ------
основное звено с одним атомом (полиметилен из диазометана)
----СН2—СН2— ;СН2-СН^-----
основное звено с двумя атомами (полиэтилен)
—сн2—сн2—о— ^н2-сн2^о!—
основное звено с тремя атомами (полиэтиленоксид)
Помимо основного звена различают еще и структурную едини-
цу— ту наименьшую группировку атомов, которая периодически
повторяется в макромолекуле. Основное звено и структурная еди-
ница могут быть идентичны, как, например, в полимерах винило-
вого и акрилового рядов:
----СН2-СН-
I
X
сн2-сн
I
X
* По другой классификации все полимеры делятся на термопласты (пласто-
меры), каучуки (эластомеры) и реактопласты (дуропласты).
26
(ступенчатой полимеризации) 4
Структурное звено Получение полимеров
—С—N—(CH2)X—N—С—О—(СН2)(,—О— II 1 1 II он но Опыт 4-20
—С—N—(CHs)^—N—С—N—(СН2)„—N— II 1 1 II 1 1 ОН нон н [33]
— Опыт 4-24
-SHCHjh-S-CCHJj,- [34]
Структурная единица может содержать и несколько основных
звеньев, как, например, во многих поликонденсационных полиме-
рах, а также в сополимерах:
• • --------------------------------------------------
: I II! Hi I I II I
I H но о- H HO H
структурная единица из двух различных звеньев
В некоторых макромолекулах нельзя выделить структурную
единицу, хотя они построены из основных звеньев; таковы, напри-
мер, статистические сополимеры (стирола и метилметакрилата и
т. д.).
Макромолекулы, которые состоят из одинаковых основных
звеньев или из одинаковых структурных единиц, помимо вида кон-
цевых групп могут различаться также структурной изомерией и
стереоизомерией.
Линейные, разветвленные и сетчатые (сшитые) макромолеку-
лы, полученные из одних и тех же мономеров, являются структур-
ными изомерами. Разветвления образуются в тех случаях, когда
при синтезе применяются трифункциональные мономеры или при
полимеризации протекают побочные реакции, например реакции
передачи цепи (см. раздел 3.1) на полимер. Сшивки возникают в
тех случаях, когда в образовании полимера участвуют три- или
более функциональные исходные соединения (например, реакции
поликонденсации с триолами или реакции полимеризации с бута-
диенами, которые в этом случае могут реагировать как тетра-
функциональные. Ниже приведены примеры образования линей-
ных, разветвленных и сшитых полистиролов (см. опыт 3-49):
—сн2-сн-сн2~сн-сн2-сн-сн2-сн---
линейный полистирол
27
сшитый полистирол
Структурная изомерия может обусловливаться также (для ви-
нильных или винилиденовых соединений) различным способом
присоединения мономерного звена к растущей цепи:
-СН2— СН—СН2—СН--СН2-СН-
X X X
соединение по типу «голова—хвост»
---СН2-СН-:—СН—СН2~:-СН2~СН~
I И ; I
XX X
соединение по типу «голова—голова» или «хвост—хвост»
28
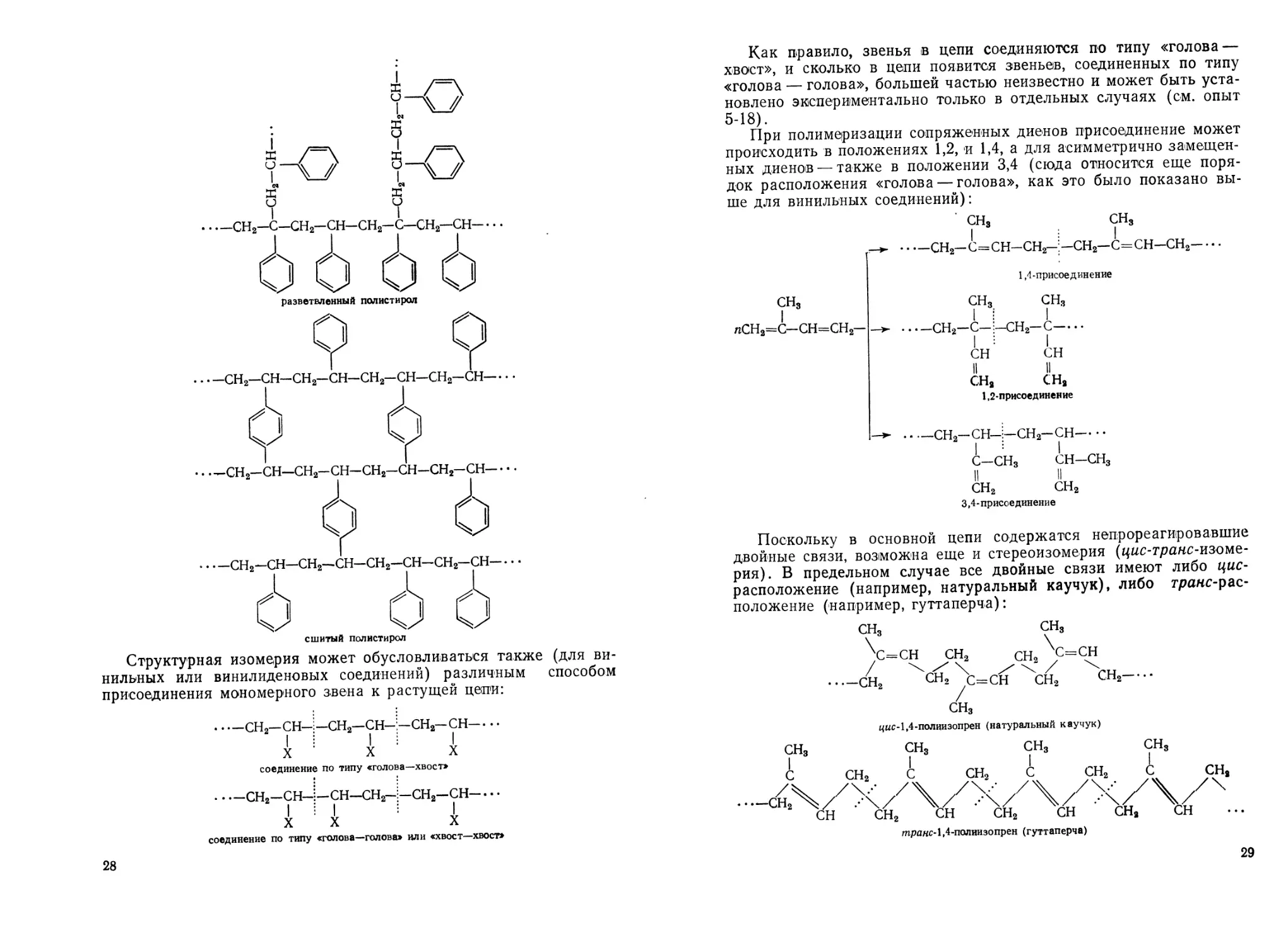
Как правило, звенья в цепи соединяются по типу «голова —
хвост», и сколько в цепи появится звеньев, соединенных по типу
«голова — голова», большей частью неизвестно и может быть уста-
новлено экспериментально только в отдельных случаях (см. опыт
5-18).
При полимеризации сопряженных диенов присоединение может
происходить в положениях 1,2, и 1,4, а для асимметрично замещен-
ных диенов — также в положении 3,4 (сюда относится еще поря-
док расположения «голова — голова»., как это было показано вы-
ше для винильных соединений):
CHS СН3
---СН2—С=СН—СН2—СН2—С=СН—СН2------
1,4-присоединение
сн3 сн3 сн3
I I i I
nCH2=C—СН=СН2— ---------------СН2—С— =-сн2— с--------
I I
СН СН
II II
СН3 СН,
1,2-присоединение
----сн2-сн-;:-сн2-сн--------
с—СН3 СН—сн3
II II
СН2 сн2
3,4- присоединение
Поскольку в основной цепи содержатся непрореагировавшие
двойные связи, возможна еще и стереоизомерия (цис-транс-изоме-
рия). В предельном случае все двойные связи имеют либо цис-
расположение (например, натуральный каучук), либо траис-рас-
положение (например, гуттаперча):
транс- 1,4-полиизопрен (гуттаперча)
29
Синтетические полидиены в большинстве случаев структурно
более неоднородны, чем природные полидиены. Однако при ис-
пользовании некоторых инициаторов и при тщательно контролиру-
емых условиях полимеризации (см. опыт 3-30) удается получить
цисЛ,4-полиизопрен («синтетический натуральный каучук») даже
в промышленном масштабе.
Для линейных макромолекул с боковыми заместителями, ко-
торые построены из основных звеньев типа —СН2—СНХ— (напри-
ш
Рис. 1. Плоскостное изображение макромолекул стереоизомерных полимеров
с основным структурным элементом —СН2—СНХ—:
I — изотактический полимер; II — синдиотактический полимер; III — атактический полимер,
мер, полимеры винильных соединений), наблюдаются еще два ви-
да изомерии: стереоизомерия (микротактичность) и оптическая
изомерия.
Стереоизомерия, названная Натта [35] микротактичностью,
обусловливается различным пространственным расположением за-
местителей X. Если представить себе линейную макромолекулу с
основным звеном —СН2—СНХ— вытянутой в форме плоского зиг-
зага (рис. 1), то заместители X могут быть расположены различ-
ным образом по обе стороны этой плоскости: по одну сторону (/),
по обе стороны (II) и, наконец, беспорядочно (III).
Полимеры первого типа называются изотактическими. Они по-
строены из закономерно повторяющихся основных звеньев с четвер-
тичными атомами углерода с одинаковой стерической конфигура-
цией. Полимеры, в которых каждый второй (замещенный) угле-
родный атом обладает противоположной стерической конфигура-
цией (полимеры второго типа), называются синдиотактическими.
30
Полимеры со статистическим распределением стерической конфи-
гурации называются атактическими полимерами.
До настоящего времени полимеры типа I и II получали из ви-
ниловых и акриловых мономеров, а также из сопряженных ди-
енов 1,2- и 3,4-. Возможна и стереоспецифическая полимеризация
эпоксидов и карбонильных соединений.
Оптическая изомерия наблюдается тогда, когда заместитель X
винилового мономера содержит асимметрический углеродный
атом. В этом случае полимеры оптически активны [36], причем
вращательная способность полимеров большей частью резко от-
личается от вращательной способности мономеров.
Для сополимеров принципиально возможны те же самые струк-
турная изомерия и стереоизомерия, что и для гомополимеров. Од-
нако для сополимеров существует еще структурная изомерия,
обусловленная различным распределением двух или более основ-
ных звеньев внутри главной цепи. В линейном сополимере, полу-
ченном из мономеров А и В, основные звенья могут либо строго
чередоваться, либо располагаться беспорядочно, т. е. статисти-
чески; чередоваться могут также блоки, состоящие из одинаковых
мономерных единиц. Соответственно различают чередующиеся,
статистические и блок-сополимеры. Кроме того, к основной це-
пи, которая состоит, например, из звеньев А, могут присоеди-
няться (прививаться) боковые цепи, например, из мономера В.
Такие разветвленные сополимеры называются привитыми:
—ABABABABABAB— чередующийся сополимер
—ААВАВВАВВАВА— статистический сополимер
—АААА—ВВВВ—АААА— блок-сополимер
—ААААААААААААА— привитой сополимер
В В
В в
в в
в в
в в
в
Аналогичным образом могут быть охарактеризованы продук-
ты сополиконденсации и сополиприсоединения.
Особый случай представляют так называемые стереоблок-сопо-
лимеры, в которых стереоизомерные звенья образуют длинные не-
прерывные последовательности.
Соединение звеньев в сополимере, т. е. возможность образова-
ния чередующихся или статистических сополимеров, зависит от
констант сополимеризации при данном составе исходной моно-
мерной смеси и способе инициирования (см. раздел 3.3), и поэто-
му их строение практически не может быть изменено. Наоборот,
блок- и привитые сополимеры во многих случаях могут быть по-
лучены любого строения (см. раздел 3.3.2).
31
1.3. КОНФОРМАЦИЯ МАКРОМОЛЕКУЛ
Макромолекулы в растворе обычно принимают наиболее ста-
тистически вероятную конформацию, которая приближается к
состоянию с максимально возможной энтропией. Согласно рас-
четам Куна [37] на моделях неразветвленных парафиновых
углеводородов эта наиболее вероятная конформация не яв-
ляется ни плотной шарообразной, ни вытянутой, а представляет
собой рыхлый статистический клубок. Конформация идеаль-
ного статистического клубка возможна для линейных нераз-
ветвленных макромолекул, но и то только тогда, когда их движе-
ние не ограничено никакими внешними силами. Такие идеальные
условия создаются в очень разбавленном растворе полимера в
инертном растворителе, когда дисперсионное взаимодействие меж-
ду индивидуальными макромолекулами незначительно и взаимо-
действие между сегментами, с одной стороны, и между сегмента-
ми и растворителем, с другой, одинаково. В этом случае размеры
статистического клубка могут быть определены с помощью так
называемой статистики случайных блужданий.
Отклонения от идеальности вызываются не только структурны-
ми изменениями в макромолекуле, например наличием разветвле-
ний или фрагментов, уменьшающих гибкость цепи (ароматические
кольца, гетероциклы), но и воздействием внешних сил, а также
агрегатным состоянием полимера (подробнее см. раздел 1.4).
1.4. АГРЕГАТНЫЕ СОСТОЯНИЯ И СВОЙСТВА
ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
1.4.1. Твердые полимеры
Полимеры могут находиться в аморфном, частично-кристалличе-
ском или высококристаллическом состоянии. Твердые аморфные
полимеры находятся обычно в стеклообразном состоянии, которое
характеризуется отсутствием дальнего порядка* в расположении
макромолекул, т. е. отсутствием кристаллических областей. При
охлаждении полимерного расплава может сохраниться определен-
ная ориентация макромолекул в потоке** («замороженные со-
стояния»). Ориентация молекулярного клубка может возникать
и тогда, когда полимер уже находится в твердом состоянии; это
достигается, например воздействием растягивающих сил. Нако-
нец, подобные ориентации могут возникать при получении пленок
из раствора, например при высушивании пленок (усадка) или при
снятии их с подложки. Эти ориентации вызывают анизотропию
различных физических свойств, которая отсутствует в полимере,
* Для кристаллизующихся низкомолекулярных соединений под дальним
порядком понимается закономерное расположение основных звеньев на рас-
стояниях больших, чем размеры молекулы. При ближнем порядке наблюдается
упорядоченность в размещении близко расположенных молекул.
** См. раздел 1.4.2.
32
состоящем из произвольно расположенных клубков. Подобные
ориентации можно наблюдать в прозрачном полимере (например,
в полистироле) в поляризованном свете. Ориентация также мо-
жет быть определена по характеру зависимости некоторых меха-
нических свойств полимера (см. раздел 2.3.12). Предотвратить
ориентационные явления довольно сложно: для этого полимер не-
которое время надо выдержать в расплавленном или размягчен-
ном состоянии и затем охладить его в таких условиях, при кото-
рых полимер не деформируется. В промышленности для получе-
ния определенных свойств, например улучшения разрывной проч-
Рис. 2. Статистический клубок линейной макромолекулы.
ности, ориентацию в пленках и волокнах создают искусственно
путем одно- или двухосного растяжения.
Макромолекулы кристаллизующихся полимеров [38], которые
в расплаве представляют собой беспорядочные, взаимно перепле-
тающиеся клубки, при медленном охлаждении ниже температу-
ры кристаллизации* образуют достаточно упорядоченные струк-
туры: в первую очередь возникают кристаллиты, которые затем
часто перерастают во вторичные агрегаты — сферолиты. Кристал-
литами называют наименьшие упорядоченные области в решетке
кристаллического полимера, обладающие протяженностью в не-
сколько сот ангстрем; их можно рассматривать как единичные
дефектные кристаллы. Образование кристаллитов часто объяс-
няют с помощью модели бахромчатой мицеллы или с помощью
модели складчатой пластины (рис. 2—4).
В некоторых областях бахромчатой мицеллы макромолекулы
укладываются параллельно друг другу (кристаллы с вытянутыми
цепями), т. е, создаются условия для частичной кристаллизации.
Эти высокоупорядоченные области могут прерываться областями
* Температура кристаллизации — это та температура, при которой про-
исходит кристаллизация из расплава. Она не совпадает с точкой плавления кри-
сталлов, а лежит существенно ниже вследствие ограниченной подвижности рас-
плавленных макромолекул.
3—732 ,33
е беспорядочным расположением макромолекул, т.е. аморфными об-
ластями. Так как длина макромолекул в несколько раз превышает
длину кристаллитов, то одна и та же полимерная цепочка участ-
вует в построении нескольких кристаллитов, т. е. она протягива-
ется через несколько кристаллических и аморфных областей. От-
дельные кристаллиты, таким образом, связаны между собой
аморфными областями. В соответствии с моделью бахромчатой
мицеллы предполагается, что макромолекулярные клубки уже до
начала кристаллизации разворачиваются. Такой процесс мало
вероятен, поэтому модель бахромчатой мицеллы непригодна для
Рис. 4. Складчатая пластина.
Рис. 3. Бахромчатая мицелла.
большинства синтетических полимеров, кристаллизующихся из
раствора или из расплава*. В то же время фибриллы многих
природных соединений, например целлюлозы и белков (коллаген,
шелк), состоят из пучков параллельно упорядоченных макромо-
лекул, что вполне согласуется с моделью бахромчатой мицеллы.
В большинстве случаев полимеры кристаллизуются вследствие
образования макромолекулами складок с постоянной длиной, что
приводит к пластинчатому строению кристаллита. Однако и в этом
случае дальний порядок в кристалле нарушается, так как петли,
образующиеся в местах перегиба цепей, могут приводить к обра-
зованию аморфных областей (см. рис. 4). Отсюда следует, что
полностью кристаллические полимеры, даже если они кристалли-
зуются в специальных условиях из очень разбавленных растворов,
получить нельзя. Представление о складчатости как о принципе
построения кристаллитов вытекает из рентгенографических и элек-
тронно-микроскопических исследований монокристаллов полиэти-
лена, полипропилена и других полимеров. Монокристаллы поли-
меров, которые можно получить путем медленного охлаждения
сильно разбавленных растворов (0,01—0,1%), возникают вслед-
ствие того, что складки образуются в одном направлении. У ра-
стущего кристаллита такого рода процессы образования складок
в разных областях могут протекать параллельно друг другу, что
* В особых условиях удается закристаллизовать полиэтилен в соответствии
с моделью бахромчатой мицеллы [39].
34
в конце концов должно приводить к образованию пластины или
нескольких пластин определенной толщины (см. рис. 4).
Поскольку в максимально растянутом состоянии макромолеку-
лы в соответствии с их молекулярной массой имеют длину от
100 до 10 000 А, а толщина пластины 100 А, то очевидно, что
пластина образована полимерными цепями, сложенными в склад-
ки и расположенными перпендикулярно ее поверх-
ности. В то же время толщина пластины мало зави-
сит от молекулярной массы полимера.
Конформации макромолекул в складчатой
пластине могут быть неодинаковыми для различ-
ных полимеров. Так, полиэтилен в кристаллическом
состоянии обнаруживает плоскую зигзагообразную
конформацию, в то время как некоторые другие по-
лимеры, в частности белки, имеют спиралевидную
конформацию (рис. 5).
Кристаллизация из расплавов по сравнению с
кристаллизацией из растворов — процесс более
сложный вследствие более плотной упаковки мак-
ромолекул; однако и здесь образование кристалли-
тов происходит по тому же принципу. При кристал-
лизации из расплавов, как правило, образуются не
монокристаллы, а сферолиты (рис. 6). Сфероли-
ты — морфологические единицы, которые могут до-
стигать в диаметре нескольких десятых долей мил-
лиметра и которые легко узнать под микроскопом в
поляризованном свете. Они состоят из пучка мель-
чайших кристаллических волоконец (фибрилл)}
расходящихся радиально из одного центра кристал-
лизации. Хотя фибриллы и обладают пластинчатым
строением, их следует рассматривать не как моно-
кристаллы, а как особую форму кристаллитов. Чис-
ло и размер сферолитов сильно зависят от условий
кристаллизации: температуры и числа зародышей.
С помощью искусственно добавленных зародышеоб-
разователей число сферолитов на единицу объема
может быть значительно увеличено и тем самым
О-сн3
Рис. 5. Модель
изотактического
полипропилена.
уменьшен их диаметр (гетерогенное зародышеобразование) [40}
(см. опыт 3-32). Это влияет на некоторые физические, в особен-
ности оптические, свойства полимеров, например на прозрачность
полимерного материала.
В заключение следует еще отметить, что один и тот же поли-
мер может кристаллизоваться в различных кристаллических ре-
шетках (полиморфизм [41]). Отдельные кристаллические формы
отличаются друг от друга некоторыми физическими свойствами,
в частности температурой плавления и плотностью.
Способность к кристаллизации различных полимеров зависит
3*
35
от ряда факторов, таких, как структура, число и длина боковых
цепочек, наличие разветвлений (например, для полиэтиленов вы-
сокого и низкого давления). Так, полиэтилен и полиоксиметилен—
высококристалличсские полимеры, изотактический полистирол мо-
жет быть и аморфным, и кристаллическим, полиэтилентерефталат
при быстром охлаждении ниже температуры кристаллизации ос-
тается аморфным, а при нагревании выше температуры стеклова-
Рис. 6. Сферолиты полипропилена.
ния рекристаллизуется. Некоторые полимеры кристаллизуются
только после предварительного нагревания или медленного ох-
лаждения расплава (см. изотактический полистирол, опыт 3-28).
1.4.2. Плавление полимеров [42]
Плавление кристаллических полимеров происходит не при одной
определенной температуре, а в значительном температурном ин-
тервале вследствие того, что существуют два вида теплового дви-
жения: движение сегментов в одной макромолекуле (микроброу-
новское движение), с одной стороны, и движение всей макромоле-
кулы в целом (макроброуновское движение), с другой стороны*.
Ниже определенной температуры, обычно называемой температу-
рой стеклования Tgt макроброуновское движение полностью пре-
кращается, а микроброуновское движение в значительной степени
* См. раздел 1.4.1.
36
замедляется («стеклообразное состояние»)*. Выше температуры
стеклования микроброуновское движение, т. е. подвижность ко-
ротких элементов цепочек (например, вращение вокруг С = С-свя-
зи), начинает усиливаться, что приводит к размягчению, при
котором возможно деформирование материала под воздействием
какой-либо силы. Это размягчение, как следствие микроброунов-
ского движения, наступает даже для сшитых полимеров. На прак-
тике вместо температуры стеклования Tg часто определяют темпе-
ратуру размягчения, которая для аморфных полимеров близка к
Tg, а для кристаллических, наоборот, значительно выше Tg. Тем-
пература, при которой наступает деформация под действием си-
лы, зависит от величины нагрузки**.
Выше температуры размягчения, а у кристаллических поли-
меров выше точки плавления деформация, вызванная внешней
силой***, уменьшается после прекращения действия этой силы, т. е.
полимер ведет себя как упругий каучукообразный материал. Эта
упругость не является следствием деформации валентных углов
или межатомных расстояний: она основана на том, что макромо-
лекулярные клубки выводятся из своей статистически наиболее
вероятной формы и вновь стремятся достичь этого состояния.
Выше температуры размягчения упругость полимеров не иде-
альна, так как упругое восстановление после деформации об-
разца не является полным («остаточная деформация»). Это про-
исходит потому, что внутренние напряжения внутри образца,
вызванные деформацией сегментов, при взаимном перемещении
макромолекул могут быть компенсированы, что, в свою очередь,
вызывает уменьшение восстанавливающей силы. Такого рода про-
цессы называются релаксационными. При более высоких темпе-
ратурах процессы релаксации протекают быстрее (усиление мак-
роброуновского движения), хотя сам полимер в расплавленном
состоянии еще остается упругим, так как макромолекулы нахо-
дятся в виде переплетенных клубков. Поэтому расплавы высоко-
молекулярных веществ называют также вязкоупругими жидкос-
тями. Вязкоупругие свойства отчетливо обнаруживаются только
в определенном температурном интервале: в непосредственной
близости от температуры размягчения полимеры являются на-
столько жесткими, что для их деформирования требуются значи-
тельные усилия и восстановление протекает весьма медленно.
Значительно выше температуры размягчения расплав легко де-
формируется, но на упругое восстановление накладывается тече-
ние вследствие усиления макроброуновского движения. Область
* При температуре стеклования такие физические свойства, как например,
показатель преломления, диэлектрическая проницаемость, теплоемкость, степень
растяжения, удельный объем, модуль упругости, изменяются скачкообразно.
Измерение температурной зависимости этих величин служит для определения
температуры стеклования (см. раздел 2.3.4.1).
** См. раздел 2.3.4.
♦** Предполагается, что деформирование происходит достаточно быстро.
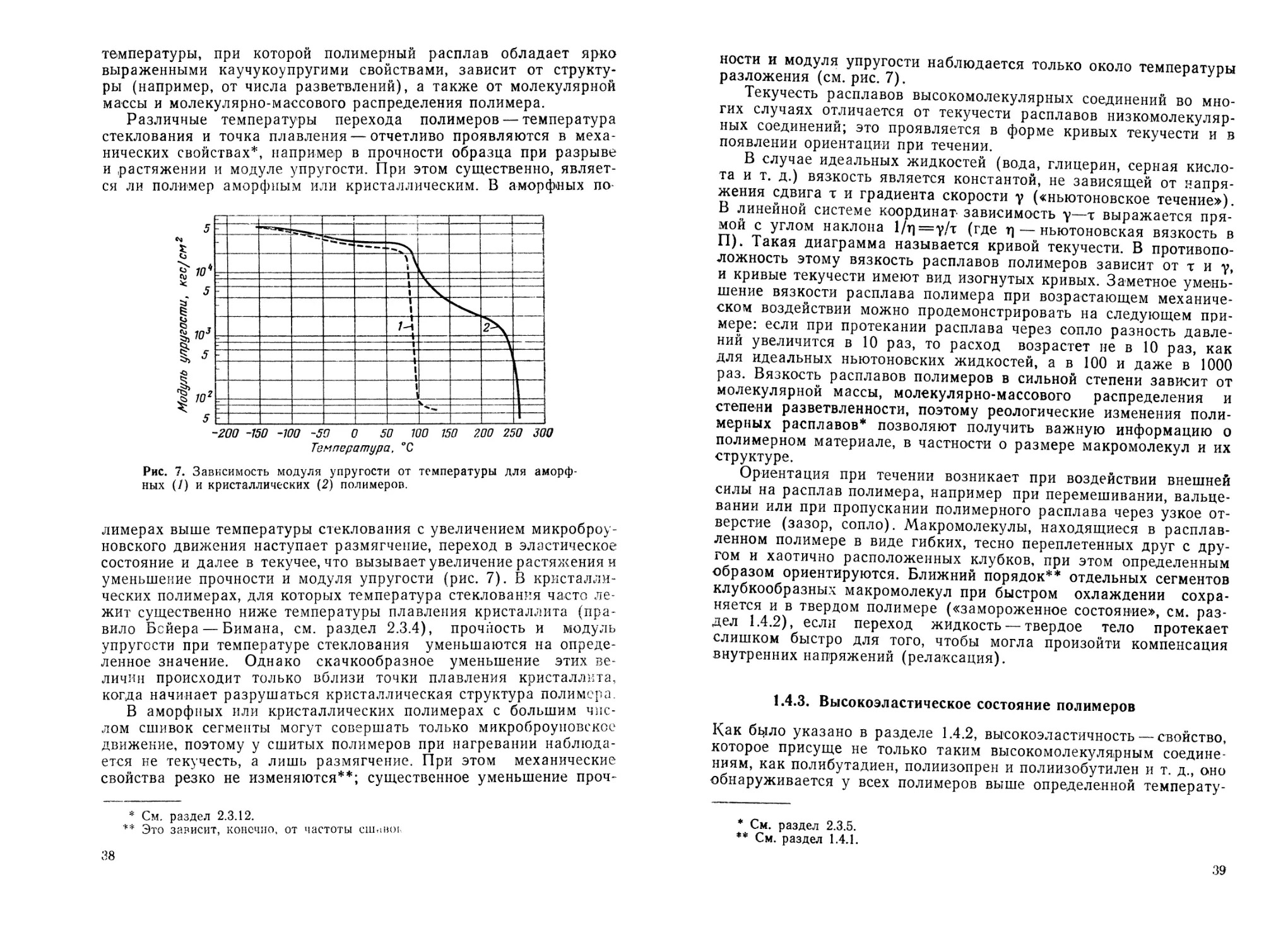
температуры, при которой полимерный расплав обладает ярко
выраженными каучукоупругими свойствами, зависит от структу-
ры (например, от числа разветвлений), а также от молекулярной
массы и молекулярно-массового распределения полимера.
Различные температуры перехода полимеров — температура
стеклования и точка плавления — отчетливо проявляются в меха-
нических свойствах*, например в прочности образца при разрыве
и растяжении и модуле упругости. При этом существенно, являет-
ся ли полимер аморфным или кристаллическим. В аморфных по
Рис. 7. Зависимость модуля упругости от температуры для аморф-
ных (/) и кристаллических (2) полимеров.
лимерах выше температуры стеклования с увеличением микроброу-
новского движения наступает размягчение, переход в эластическое
состояние и далее в текучее, что вызывает увеличение растяжения и
уменьшение прочности и модуля упругости (рис. 7). В кристалли-
ческих полимерах, для которых температура стеклования часто ле-
жит существенно ниже температуры плавления кристаллита (пра-
вило Бейера — Бимана, см. раздел 2.3.4), прочность и модуль
упругости при температуре стеклования уменьшаются на опреде-
ленное значение. Однако скачкообразное уменьшение этих ве-
личин происходит только вблизи точки плавления кристаллита,
когда начинает разрушаться кристаллическая структура полимера.
В аморфных или кристаллических полимерах с большим чис-
лом сшивок сегменты могут совершать только микроброуновское
движение, поэтому у сшитых полимеров при нагревании наблюда-
ется не текучесть, а лишь размягчение. При этом механические
свойства резко не изменяются**; существенное уменьшение проч-
* См. раздел 2.3.12.
** Это зависит, конечно, от частоты сшиног
38
ности и модуля упругости наблюдается только около температуры
разложения (см. рис. 7).
Текучесть расплавов высокомолекулярных соединений во мно-
гих случаях отличается от текучести расплавов низкомолекуляр-
ных соединений; это проявляется в форме кривых текучести и в
появлении ориентации при течении.
В случае идеальных жидкостей (вода, глицерин, серная кисло-
та и т. д.) вязкость является константой, не зависящей от напря-
жения сдвига т и градиента скорости у («ньютоновское течение»).
В линейной системе координат- зависимость у—т выражается пря-
мой с углом наклона 1/т)=у/т (где ц— ньютоновская вязкость в
П). Такая диаграмма называется кривой текучести. В противопо-
ложность этому вязкость расплавов полимеров зависит от т и у,
и кривые текучести имеют вид изогнутых кривых. Заметное умень-
шение вязкости расплава полимера при возрастающем механиче-
ском воздействии можно продемонстрировать на следующем при-
мере: если при протекании расплава через сопло разность давле-
ний увеличится в 10 раз, то расход возрастет не в 10 раз, как
для идеальных ньютоновских жидкостей, а в 100 и даже в 1000
раз. Вязкость расплавов полимеров в сильной степени зависит от
молекулярной массы, молекулярно-массового распределения и
степени разветвленности, поэтому реологические изменения поли-
мерных расплавов* позволяют получить важную информацию о
полимерном материале, в частности о размере макромолекул и их
структуре.
Ориентация при течении возникает при воздействии внешней
силы на расплав полимера, например при перемешивании, вальце-
вании или при пропускании полимерного расплава через узкое от-
верстие (зазор, сопло). Макромолекулы, находящиеся в расплав-
ленном полимере в виде гибких, тесно переплетенных друг с дру-
гом и хаотично расположенных клубков, при этом определенным
образом ориентируются. Ближний порядок** отдельных сегментов
клубкообразных макромолекул при быстром охлаждении сохра-
няется и в твердом полимере («замороженное состояние», см. раз-
дел 1.4.2), если переход жидкость — твердое тело протекает
слишком быстро для того, чтобы могла произойти компенсация
внутренних напряжений (релаксация).
1.4.3. Высокоэластическое состояние полимеров
Как бь1ло указано в разделе 1.4.2, высокоэластичность — свойство,
которое присуще не только таким высокомолекулярным соедине-
ниям, как полибутадиен, полиизопрен и полиизобутилен и т. д., оно
обнаруживается у всех полимеров выше определенной температу-
* См. раздел 2.3.5.
** См. раздел 1.4.1.
39
ры, так называемой температуры стеклования («температуры пере-
хода второго рода»). Аморфные полимеры, температура стеклова-
ния которых лежит ниже комнатной температуры, называются
эластомерами; при комнатной температуре они обладают упруго-
стью. Однако их упругость при несшитом (невулканизованном) со-
стоянии при 20 °C похожа на упругость полимерных расплавов,
т. е. упругое восстановление после растяжения происходит тем
менее полно, чем больше было растяжение и чем дольше матери-
ал выдерживался в растянутом состоянии (остаточное удлинение).
Это объясняется уравновешиванием внутренних напряжений пу-
тем релаксации, т. е. взаимным перемещением деформированных
макромолекул. Если воспрепятствовать этим перемещениям путем
сшивания (вулканизация) и таким образом сделать невозможным
взаимные скольжения макромолекул друг относительно друга, то
можно получить эластомеры, которые после прекращения дейст-
вия силы даже при сильном и длительном растяжении возвраща-
ются вновь в исходное состояние. В технике такие сшитые эласто-
меры называются резинами. Резины отличаются весьма малыми
значениями модуля упругости в сочетании с высокой эластич-
ностью. Свойства резины существенно зависят от числа сшивок:
малое число сшивок дает высокую эластичность и низкий модуль
упругости, повышение числа сшивок уменьшает эластичность и
увеличивает модуль упругости; наконец, резины с большим чис-
лом сшивок в значительной степени теряют свои каучукоупругие
свойства (эбонит).
Как уже отмечалось выше, высокоэластичность полимерных
материалов является особым состоянием вещества, которое опре-
деляется тенденцией к увеличению энтропии, тогда как, например
у стали, возвращение к равновесию определяется тенденцией к
уменьшению внутренней энергии. Энтропийная природа упругости
объясняет тот факт, что напряжение растянутой резиновой ленты
возрастает с температурой, в то время как у стальной проволоки
оно снижается: при растяжении макромолекулы переходят из ста-
тистически наиболее вероятной формы клубка в статистически
наименее вероятное состояние растянутых цепочек. Чем выше тем-
пература, тем выше подвижность цепей и тем больше потеря эн-
тропии при переходе в растянутое состояние, при котором подвиж-
ность цепей сильно ограничена (ближний порядок, см. раздел
14.1). Таким образом, с повышением температуры увеличивается
стремление вернуться в исходное состояние с более высокой энтро-
пией.
В растянутом каучуке ориентации могут быть столь регуляр-
ными, что отдельные сегменты цепочек могут кристаллизоваться
(например, натуральный каучук). Такие образцы дают рентгено-
граммы, которые по виду напоминают рентгенограмму волокнис-
той структуры.
40
1.4.4. Растворы полимеров
В сильно разбавленных растворах в инертном растворителе мак-
ромолекулы находятся в виде почти идеальных статистических
клубков. В этих клубках макромолекулы сильно сольватированы,
т. е. клубки содержат значительные количества растворителя. Не-
смотря на то что растворитель находится благодаря диффузии
в постоянном обмене с окружающими клубок молекулами рас-
творителя, он все же настолько прочно связан с клубком, что дви-
жется вместе с ним, например,
трифуге. Поэтому макромоле-
кулярные клубки можно срав-
нить с малыми частицами геля,
которые состоят из каркаса
(макромолекулы клубка) и за-
ключенного в нем растворите-
ля. Эти представления схема-
тично изображены на рис. 8.
В зависимости от молеку-
лярной массы клубки в таких
растворах могут захватывать
растворитель в 20—1000-крат-
ном размере от объема цепи,
т. е. больше, чем 99%. Так как
диаметры таких гелевых клуб-
ков лежат между 100 и 1000 А,
эти высокомолекулярные рас-
творы относятся к коллоидным
системам. Но в противополож-
ность коллоидным частицам
седиментации в ультрацен-
1
Рис. 8. Схематическое изображение
структуры разбавленного раствора поли-
мера:
1 — «связанный» растворитель; 2 — «свобод-
ный» растворитель; 3 — объем клубка; 4 —
макромолекула.
обычных дисперсий в высокомоле-
кулярных растворах коллоидные частицы являются сольватиро-
ванными макромолекулярными клубками (молекулярные коллои-
ды).
Особое строение растворов полимеров выражается в некото-
рых свойствах, зависящих от молекулярной массы, природы рас-
творителя и температуры. Эти свойства используют при определе-
нии молекулярной массы полимеров с помощью метода измере-
ния вязкости разбавленных растворов, имеющего большое практи-
ческое значение. Этот метод будет рассмотрен в отдельном раз-
деле (2.3.2.1).
Литература
1. L й s s i Н., «Chimia», 1966, v. 20, р. 379.
2. Houben-Weyl, 1961, Bd. 14/1, S. 592.
3. Houben-Weyl, 1961, Bd. 14/1, S. 817.
4. Houben-Weyl, 1961, Bd. 14/1, S. 844.
5. Houben-Weyl, 1961, Bd. 14/1, S. 674.
<5. J. Polymer Sci., 1954, v. 13, p. 325.
41
7. Houben-Weyl, 1961, Bd. 14/1, S. 745.
8. J. Polymer Sci., 1964, v. 4, p. 299.
9. Houben-Weyl, 1961, Bd. 14/1, S. 956.
10. Houben-Weyl, 1961, Bd. 14/1, S. 1099.
11. Houben-Weyl, 1961, Bd. 14/1, S. 1093, 1095.
12. Houben-Weyl, 1961, 14/1, S. 1113.
13. Houben-Weyl, 1961, Bd. 14/1, S. 1019, 1023.
14. Sorenson-Campbell, 1968, p. 176.
15. Houben-Weyl, 1961, Bd. 14/1, S. 1065.
16. Houben-Weyl, 1961, Bd. 14/1, S. 977.
17. Sorenson-Campbell, 1968, p. 89.
18. Houben-Weyl, 1961, Bd. 14/1, S. 1084.
19. Makromol. Chem., 1955, Bd. 17, S. 62.
20. Makromol. Chem., 1963, Bd. 60, S. 139.
21. Houben-Weyl, 1961, Bd. 14/1, S. 1138.
22. Houben-Weyl, 1961, Bd. 14/1, S. 1139—1141.
23. Houben-Weyl, 1963, Bd. 14/2, S. 118.
24. Houben-Weyl, 1963, Bd. 14/2, S. 119.
25. Sorenson-Campbell, 1968, p. 241, 246.
26. Makromol. Chem., 1962, Bd. 53, S. 203.
27. Makromol. Chem., 1962, Bd. 56, S. 179.
28. Houben-Weyl, 1963, Bd. 14/2, S. 6.
29. Houben-Weyl, 1963, Bd. 14/2, S. 165.
30. Sorenson-Campbell, 1968, p. 138.
31. S о r e n s о n - C a m p b e 11, 1968, p. 141.
32. Sorenson-Campbell, 1968, p. 250.
33. S о r e n s о n - C a m p b e 11, 1968, p. 93.
34. Sorenson- Campbell, 1968, p. 126.
35. N a 11 a G., Angew. Chem., 1956, v. 68. p. 393; 1964, v. 76, p. 553: Farina M.,
P e r a 1 d о M., N a 11 a G., Angew. Chem., 1965, v. 77, p. 149.
36. Braun D., Houben-Weyl, 1961, Bd. 14/1, S. 119; S h u 1 z R. C., Advan-
ces Po’ymer Sci., 1965, v. 4, p. 236.
37. Kunn W., Kolloid-Z., 1934. p. 236.
38. Geil P. H., Polymer Singl Crystals, Polymer Reviews, 1963, v. 5;
Stuart H. A., Physik der Homopolimeren, 1955, Bd. 3; Zachmann H. G.,
Stuart H. A., Makromol. Chem., 1961, Bd. 44/46, S. 622.
39. Wunderlich B., Advances Polymer Sci., 1968, v. 5, p. 568.
40. Binsbergen F. L., Polymer, 1970, v. 11, p. 253; Binsbergen F. L.,
de Lange, Polymer, 1970, v. 11, p. 309.
41. D a n u s s о F., Polymer, 1967, v. 8, p. 281.
42. Philipp off W., Viskositat der Kolloide, Steinkopff-Verlag. 1942; Se-
vers E. T., Rheology of Polymers, Reinhold Publ., New York, 1962;
Meissner L, Kunststoffe, 1967, Bd. 57, S. 397; Scmjonov V., Advances
Polymer Sci., 1968, v. 5, p. 387.
Дополнительная литература
Учебные пособия
Batzer Н., «Einfiihrung in die makromolekulare Chemie», Htithig Verl., Heidel-
berg, 1958.
Бе м ф op д К., Барб У., Дженкинс А., О н ь о н П. Кинетика радикаль-
ной полимеризации виниловых соединений. Пер. с англ. Под ред. Ю. М. Ма-
линского. М., Издатинлит, 1961. 348 с.
Billmeyer F. W., Jr., Textbook of Polymer Science, New York — London,
1971.
Elias H. G., Makromolekiile, Hiitig Verlag, Heidelberg, im Druck.
Flory P. J., Principles of Polymer Chemistry, Cornell University Press, Ithaca,
New York, 1953.
42
He n r i c i - О 1 i v e G., Olive S., Polymerisation: Katalyse — Kinetik — Mecha-
nismen, Verlag Chemie, Weinheim, 1970.
Kern W. in: Karrer P., «Lehrbuch der organischen Chemie», Stuttgart, 1963,
S. 790—812.
К e 11 e у A. D., «The Stereochemistry of makromolecules, New York, 1967—68,
v. 1—3.
Kuehl er L., Polymerisationskinetik, Springer-Verlag, Berlin — Gottingen —
Heidelberg, 1951.
Лосев И. П., Федотова О. Я. Практикум по химии высокомолекулярных
соединений. Изд. 2-е. М., Госхимиздат, 1962. 174 с.
Pinner S. Н., A Practical Course in Polymer Chemistry, Oxford — London-
New York — Paris, 1961.
Сёренсон У., Кемпбел T. Препаративные методы химии полимеров. М.,
Издатинлит, 1968. 564 с.
Тобольский А. В. Свойства и структура полимеров. М., «Химия», 1964.
248 с.
X у в и н к Р., С т а в е р м а н А. Химия и технология полимеров. Л., «Химия»,
1965, т. I, 676 с.; 1966, т. И. 1124 с.
Staudinger Н., Organische Kolloidchemie, Vieweg, Braunschweig, 1950.
Stuart H. A., Die Physik der Hochpolymeren, Springer-Verlag, Berlin — Got-
tingen-Heidelberg, 1952—1956, Bd. 1, 2, 3, 4.
Vollmert B., Grundriss der makromolekularen Chemie. Berlin — Gottingen —
Heidelberg, 1962.
Справочники и монографии
Houben-Weyl-Muller, Methoden der organischen Chemie, 1961, Bd. 14;
1963, Bd. 14/2.
Hummel — Scholl, Atlas der Kunststoff-Analyse, Verlag Chemie, Munchen,
1968.
Brandrup J., I miner gut E. H., Polymer Handbook, New York, 1966. Poly-
mer Reviews, New York, 1966.
Plesch P. H., The Chemistry of Cationic Polymerisation, Oxford — London —
New York — Paris, 1963.
Staudinger H., Die Hochmolekularen organischen Verbindungen, Berlin — Got-
tingen— Heidelberg, 1960.
Staudinger H., Makromolekulare Chemie und Biologie, Basel, 1947.
Lap pert M. F., Leigh G. J., Developments in Inorganic Polymer Chemistry,
Amsterdam — London — New York, 1962.
Nitsche R., Wolf K- A., Kunststoffe, Physikalisches Verhalten und Prufung,
Springer-Verlag, Berlin — Gottingen — Heidelberg, 1961 —1962, Bd. 1—2.
2. ОБЩИЕ МЕТОДЫ ХИМИИ
ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
2.1. ПОЛУЧЕНИЕ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
2.1.1. Работы, проводимые в присутствии кислорода
и влаги [1], [2]
В большинстве случаев молекулярный кислород оказывает значи-
тельное влияние па процесс полимеризации, начиная или прекра-
щая рост цепи, дезактивируя или активируя ионные инициаторы
либо вызывая окислительную деструкцию уже полученных поли-
меров (особенно при поликонденсации). Так как это влияние ста-
новится заметным уже при очень малой концентрации кислорода,
то при синтезе высокомолекулярных соединений рекомендуется
работать в атмосфере азота или благородного газа. В лаборато-
рии азот высокой степени чистоты можно получить, пропуская
обычный азот из баллона над контактным катализатором, кото-
рый реагирует с имеющимся в азоте кислородом. Для этой цели
применяется катализатор «BTS-Kontakt», который благодаря
своей эффективности позволяет уменьшать содержание кислорода
при комнатной температуре до 10-4—10-5%. Этот катализатор
лучше применявшегося ранее медного катализатора Мейера —
Ронже [3]. Очистка азота может осуществляться с помощью рас-
творов [4] некоторых препаратов, таких, как пирогаллол, гидро-
сульфит натрия, кетилы металлов или алюминийорганические со-
единения. Однако такая очистка не имеет преимуществ по срав-
нению с очисткой с помощью контактных катализаторов.
Реакционный сосуд заполняется азотом путем многократного
введения и эвакуирования азота, так как обычно никогда нельзя
быть уверенным в полном вытеснении воздуха. Следует избегать
применения длинных резиновых трубок (включая и парафиниро-
ванные вакуумные трубки) и пользоваться вместо них поливинил-
хлоридными (PVC-трубки) или стеклянными трубопроводами.
На рис. 9 показано простое устройство для эвакуирования возду-
ха и наполнения аппаратуры азотом с одновременной очисткой и
осушкой азота.
Если реакция или перегонка проводится в токе азота, то необ-
ходимо предотвратить возможность диффузии кислорода из окру-
жающего воздуха. В простейшем случае достаточно применить
бунзеновский вентиль (резиновую трубку с продольными проре-
зями на стенках, закрытую на одном конце стеклянной палочкой)
либо ртутный или парафиновый затвор (см. рис. 9). При работе
с самовоспламеняющимися веществами (например, органическими
44
соединениями металлов) или при проведении реакций, сопровож-
дающихся быстрым изменением давления, применяют обратный
клапан (рис. 10). При создании разрежения жидкость (парафи-
новое масло) давит на пришлифованное полое тело в верхнем
конце камеры и таким образом отключает прибор от внешней
среды.
При ионной полимеризации реакционная смесь не должна со-
держать кислорода и воды. Обычные способы сушки при этом
большей частью неэффективны. В таких случаях стеклянную ап-
паратуру лучше всего высушивать при нагревании под высоким
вакуумом. Газы можно высушивать путем вымораживания или
при пропускании через колонку, заполненную подходящим осу-
шителем.
Рис. 9. Аппаратура для очистки и сушки азота при комнатной температуре:
/ — затвор; 2 — контактная колонка (100X5 см) с нагревательной спиралью (2000 Вт); 3 —
выпускной кран для водорода и водяного пара; 4 — счетчик пузырьков (парафиновое масло);
5 —осушающая колонка; 6 — трехходовой кран для эвакуирования или наполнения аппара-
туры азотом; 7 — ртутный манометр.
Рис. 10. Запорное устройство с пришлифованным обратным клапаном, заполнен-
ное жидкостью.
При высушивании охлаждением до —75 °C остаточная влаж-
ность составляет 0,001 мг Н2О/л газа (следует обратить внимание
на то, чтобы с газовым потоком из холодильника не уносились
кристаллы льда).
Ниже перечислены некоторые реагенты, применяемые для
осушки газов и жидкостей:
Остаточная
гласность
мг ЩО/л газа
Хлорид кальция......................От 0,3 до 1,2
Окись кальция................... 0,2
Силикагель...................... 0,006
45
Сиккон* . 0,005
Серная кислота, конц..... 0,003
КОН, расплавленный....... 0,002
Молекулярные сита**...... 0,001
Металлический натрий .... —
Гидрид кальция ........................... —
Перхлорат магния ...................... 0,0005
Фосфорный ангидрид*** .... 0,00002
♦ Полуобожженный гипс в виде гранул.
* * (мешанные с «линой цеолиты в форме гранул или палочек оп-
ределенного размера
* ** Лучше всею нанесенный на твердую подложку.
Жидкости могут быть обезвожены либо путем обработки со-
ответствующим осушителем при кипячении или перегонке, либо
азеотропной или экстрактивной дистилляцией [5]. При осушке
жидкостей следует использовать только нейтральные осушающие
средства, так как иначе могут протекать нежелательные побочные
реакции (например, стирол в присутствии концентрированной сер-
ной кислоты при комнатной температуре полимеризуется со взры-
вом) .
2.1.2. Очистка и хранение мономеров
При проведении реакций полимеризации особое значение приоб-
ретает очистка исходных веществ. Часто на течение реакций зна-
чительное влияние оказывают загрязнения, присутствующие даже
в очень небольших количествах [от 10”2 до 10—4% (масс.)]. В не-
насыщенных мономерах в основном присутствуют следующие при-
меси: побочные продукты реакции, образующиеся при получении
мономера (например, этилбензол и дивинилбензол в стироле,
ацетальдегид в винилацетате); добавленные стабилизаторы; ин-
гибиторы; продукты окисления и разложения мономеров (напри-
мер, перекиси в диенах, бензальдегид в стироле, синильная кисло-
та в акрилонитриле); примеси, которые попадают в мономер при
хранении (например, следы металлов или щелочей, остатки смаз-
ки кранов).
При выборе способа очистки необходимо руководствоваться
природой мономеров, ожидаемыми примесями и прежде всего ме-
ханизмом полимеризации (например, радикальная полимеризация
в водной эмульсии или ионная полимеризация с натрийнафтали-
новым комплексом). Так как универсальную схему очистки соз-
дать невозможно, то в каждом случае следует выбирать наиболее
действенный метод очистки. Для хорошей очистки необходима
максимальная чистота всех приборов (обработка горячей азотной
кислотой или хромовой смесью и многократное тщательное опо-
ласкивание дистиллированной водой).
Существуют следующие методы очистки: фракционная, азео-
тропная, или экстрактивная дистилляция в инертном газе [5, 6, 7,
8], кристаллизация, сублимация и хроматография на колонке.
Для жидких и нерастворимых в воде мономеров (например, сти-
46
рола, см. опыт 3-01) рекомендуется сначала удалить фенолы или
амины, добавленные в качестве стабилизаторов, путем встряхива-
ния с разбавленной щелочью или кислотой. Вследствие относи-
тельно высокой летучести подобных стабилизаторов полное их от-
деление путем перегонки часто бывает затруднительно. Для га-
зообразных мономеров (например, низших олефинов, бутадиена,
окиси этилена) применяют дополнительную очистку с помощью
Рис. 11. Градуированный приемник для мономеров и растворителей:
1 — насадка с капельницей; 2 — приемник с боковой насадкой для ртутного запора или для
подключения вакуумного насоса при дистилляции при пониженном давлении; 3 — ртутный
запор для дистилляции в струе азота при атмосферном давлении.
Рис. 12. Градуированный приемник для мономеров и растворителей:
1 — капельный наконечник; 2 — впускной кран с косым впуском (выпуск газа производится
путем вращения крана на 180°); 3 — кран.
Рис. 13. Приемник для мономеров и растворителя.
молекулярных сит для удаления воды или двуокиси углерода (см.
также раздел 2.1.1). Для особо тщательной очистки мономеров
применяют предварительную полимеризацию: уже очищенный мо-
номер путем нагревания, облучения или добавления инициаторов
полимеризуют примерно до 10—20%-ной конверсии. Затем моно-
мер отделяют от полимера фракционной перегонкой в атмосфере
азота. Таким образом удаляются примеси, которые влияют на
начало полимеризации (например, реакция с инициатором или
продуктами его распада), и примеси, которые реагируют с расту-
щими макромолекулами (обрыв, передача цепи).
Для определения степени чистоты мономера недостаточно из-
мерить обычные физические постоянные, например точку кипения
или показатель преломления, так как эти константы малочувстви-
тельны к небольшому содержанию примесей. Подходящими мето-
дами для этого являются газовая хроматография, инфракрасная
47
и ультрафиолетовая спектроскопия. Наиболее достоверные дан-
ные об отсутствии примесей получают с помощью самой реакции
полимеризации. Очистку повторяют до тех пор, пока поставлен-
ные при стандартных условиях полимеризационные опыты (проб-
ная полимеризация) по ходу реакции (кривая выход — время,
выход — вязкость полимеров) не совпадут друг с другом.
Хранение мономеров и растворителей часто требует особых ус-
ловий. Сосуды для хранения должны быть устроены так, чтобы
продукт мсжно было отдозировать в атмосфере инертного газа.
На рис. 11 и 12 показаны градуированные сборники, из которых
после перегонки мономер (или растворитель) непосредственно по-
дают в реакционный сосуд. Из сосуда для хранения (рис. 13)
проба отбирается пипеткой или медицинским шприцем, когда со-
суд закрыт самоуплотняющейся прокладкой (см. раздел 2.1.3).
Конструкции сосудов, показанные на рис. 12 и 13, обладают тем
достоинством, что их содержимое практически не соприкасается
со смазкой крапов.
Большинство мономеров сохраняется неизменным только ко-
роткое время (от нескольких часов до нескольких суток) даже в
атмосфере азота, в темноте и при низкой температуре. Для более
длительного хранения мономер необходимо стабилизировать. Эф-
фективными стабилизаторами (ингибиторами) радикальной поли-
меризации [9] являются хиноны, фенолы, амины, нитросоедине-
ния и соединения металлов. Для многих мономеров достаточная
стабилизация достигается путем добавления 0,1 — 1% гидрохино-
на или 4-трет-бутилпирокатехина.
Ингибирование ионной полимеризации может вызываться во-
дой, спиртами, эфирами или аминами. При этом, однако, следует
учитывать, что во многих случаях действие этих веществ в зави-
симости от их концентрации может быть различным. Например,
вода при полимеризации с кислотами Льюиса (BF3 для изобути-
лена) или металлоорганическими соединениями (алкилалюминия)
в малых концентрациях является сокатализатором (см. раздел
3.2), в больших же концентрациях, напротив, ингибитором (реак-
ция с инициатором и активными концами растущих макромоле-
кул).
2.1.3. Реакционные сосуды для полиреакций
В литературе [10] описывается большое число специальных экс-
периментальных установок, которые приспособлены для проведе-
ния реакций с соответствующими мономерами; все они являются
в основном вариантами описываемой ниже аппаратуры.
Радикальная полимеризация твердых и жидких мономеров, а
также ступенчатая полимеризация и поликонденсация обычно
проводятся в стеклянных ампулах или в трубчатых автоклавах.
Вследствие создающегося при реакции давления ампулы следует
изготавливать из толстостенного стекла и запаивать так, чтобы
48
а de
Рис. 14. Устройство для запол-
нения ампул твердым (а) или
жидким (б и в) веществом.
по возможности не возникало внутренних напряжений. Объем
ампулы не должен превышать 250 мл, а заполнение ни в коем
случае не должно превышать ее половины. При наполнении сосу-
да следует обращать внимание на то, чтобы в его верхней части
не осталось следов вещества, которое при нагревании может раз-
лагаться и заметно влиять на ход реакции. Это можно легко
предотвратить с помощью устройства, показанного на рис. 14. Уст-
ройство 14, а предназначено для работы с твердыми веществами.
Взвешенные количества компонен-
тов помещают с помощью длинной
воронки в ампулу, после чего ампу-.
лу эвакуируют и заполняют азотом.
Для жидкостей более предпочти-
тельным является устройство 14,6,
которое позволяет эвакуировать ам-
пулу перед заполнением и таким
образом избежать появления влаги
на внутренних стенках и потери лег-
колетучих веществ. Ампула с ини-
циатором с помощью резинового
шланга соединяется с насадкой, эва-
куируется и заполняется азотом
через боковой кран. После этого
ампула заполняется мономером (в
данном случае путем создания не-
большого вакуума при помощи бо-
кового крана). Наконец, ампулу от-
тягивают вниз настолько, чтобы ка-
пельница оказалась над местом от-
пайки, и производят отпаивание.
Для заполнения колб со шлифом или пробирок применяют уст-
ройство 14, в.
Полимеризацию в водной среде, так же как и некоторые реак-
ции ионной полимеризации в органических растворителях, прово-
дятся в многогорлых колбах (в круглодонных или с плоским
дном). Гетерогенную полимеризацию (полимеризацияс осадителем,
полимеризация в суспензиях и эмульсиях), при которой число час-
тиц и их размер играют решающую роль, рекомендуется прово-
дить при перемешивании. Для этого применяют мешалки со счет-
чиком числа оборотов. Для полимеризации, инициируемой сус-
пензиями (например, щелочными металлами), используют высоко-
скоростные мешалки. В этой связи следует отметить применение
в качестве реакторов для полиреакций высокоскоростных смеси-
телей (поликонденсация на границе раздела фаз).
В качестве реакционных сосудов для ионной полимеризации
прежде всего применяют колбы с самоуплотняющимся затвором
в сочетании с медицинскими шприцами. Это позволяет ввести ка-
тализатор или отобрать пробу при практически полном исключе-
4—732
49
нии воздуха и влаги и, наконец, отбирать пробы в ходе реакции
при определении зависимости время — превращение (см. опыт
3-13). Можно также применять обычные колбы со шлифом, кото-
рые закрывают, как показано на рис. 15. Наконец, можно снаб-
дить колбу самоуплотняющимся винтовым затвором; для этого в
крышке просверливают отверстие, помещают прокладку из рези-
ны толщиной примерно 2 мм, на нее кладут тонкую пластинку из
мягкого поливинилхлорида и затем завинчивают колбу. После
Рис, 15. Колба со шлифом.
Рис. 16. Дилатометр.
Рис. 17. Циркулирующее устройство для получения полиэфиров.
прокола уплотняющих прокладки и пластины для осторожности
отверстие в навинченной крышке можно заклеить кусочком лип-
кой ленты. Шприцы должны иметь стеклянный поршень (корро-
зия, чистка). Если полимеризация протекает под действием облу-
чения, то обычно применяют ампулы или кюветы заданных раз-
меров. Следует обратить особое внимание на точность юстировки
сосуда по отношению к источнику облучения. Детальное описание
установки приведено в литературе [11, 12]. Для кинетических ис-
следований гомогенных реакций полимеризации пользуются ря-
дом приборов и методов [12], из которых следует особо отметить
наиболее простой и широко применяемый дилатометрический ме-
тод [13, 14, 15] (рис. 16). Этот метод основан на измерении объ-
емного сжатия, вызванного различием в плотностях мономеров и
полимеров. Пересчет объемного сжатия раствора на глубину по-
лимеризации может быть выполнен с помощью градуировочной
50
кривой или путем расчета по удельным объемам (см. пример
3-12).
Реакции поликонденсации и ступенчатой полимеризации при
соблюдении особых мер (см. разделы 4.1 и 4.2) могут проводиться
в аппаратуре, обычно применяемой в органической химии. Для по-
лучения высокомолекулярных полимеров методом поликонденса-
ции в растворе применяют специальную рециркуляционную уста-
новку [16] (рис. 17).
2.1.4. Температурный режим при полиреакциях
Одним из важных условий при проведении полимеризации явля-
ется точное выдерживание определенной температуры, так как
скорость и степень полимеризации сильно зависят от температуры
реакции. Для точных работ применяют водяные или масляные тер-
мостаты. Реакции в ампулах или тугоплавких трубках проводятся
на паровых банях. Для этого запаянную с одного конца трубку
(диаметр 3—8 см) наполняют примерно на одну четверть соот-
ветствующей жидкостью и нагревают до кипения. Подлежащий
термостатированию сосуд подвешивают на несколько сантиметров
выше поверхности кипящей жидкости для наиболее полной кон-
денсации пара. Вещества, применяемые в качестве жидкостей для
паровых бань, описаны в литературе [17]. Для ряда исследований
полимеров при высоких температурах, например для исследова-
ния деструкции, применяют воздушные термостаты, в которых
можно поддерживать температуру с точностью до ±1°С.
Для поддержания температуры ниже комнатной применяют
обычные охлаждающие термостаты (примерно —80 °C) и охлаж-
дающие смеси.
2.1.5. Проведение полиреакций
Наряду с химической классификацией полиреакций существует
их классификация по способу проведения. Соответственно разли-
чают блочную полимеризацию, полимеризацию в растворе и в
дисперсии.
2.1.5.1. Блочная полимеризация
Блочная полимеризация в настоящее время применяется доволь-
но редко. Отвод теплоты реакции, нерастворимость полимера в
мономере, протекание побочных реакций в высоковязких систе-
мах (например, передача цепи на полимер, см. гл. 3) затрудняют
не только проведение полимеризаци, но часто влияют на свойст-
ва полученных полимеров. Поскольку эти недостатки часто сво-
дят на нет достоинства блочной полимеризации (большие молеку-
лярные массы при высоких скоростях полимеризации, очень чис-
тые полимеры), то для препаративных целей этот вид полимери-
4* 51
зации почти не применяется. В качестве примера блочной поли-
меризации можно назвать радикальную полимеризацию стирола
(опыт 3-02), винилацетата (опыт 3-08) и метилметакрилата (опыт
3-05), а также полимеризацию этилена при высоком давлении.
Блочная полимеризация очень удобна для кинетических исследо-
ваний, при которых ее проводят в основном до небольшой глуби-
ны превращения.
Большинство поликонденсатов получают блочной поликонден-
сацией при температуре выше точки плавления исходных или по-
лучаемых полимеров. Так как в ходе реакции молекулярная мас-
са и вязкость расплава возрастают, то удаление выделяющихся
легколетучих продуктов реакции (воды, спирта) из реакционной
смеси, даже в вакууме, все более затрудняется, что вызывает не-
обходимость повышения температуры реакции. В некоторых слу-
чаях для получения продуктов с высокой молекулярной массой к
концу реакции приходится повышать температуру до 250 СС и вы-
ше. Поэтому поликонденсацию в расплаве можно проводить толь-
ко тогда, когда и исходные компоненты, и получаемый поликон-
денсат термически стабильны; в противном случае начинают про-
текать побочные реакции, которые приводят к окрашиванию, сши-
ванию или снижению молекулярной массы. Этим способом нельзя
получать некоторые полиамиды с высокими температурами плав-
ления. Для их синтеза применяют поликонденсацию в растворе
или на границе раздела фаз между диаминами и хлорангидрида-
ми дикарбоновых кислот. Молекулярные массы, достигаемые
при поликонденсации в расплавах, обычно не превышают 50 000.
В некоторых случаях полимеризацию можно проводить не
только в жидкой, но и в твердой фазе, например полимеризацию
[18] акриламида или триоксана. Так называемая постконденса-
ция, например, полиэфиров (см. опыт 4-03) протекает в твердой
фазе. Наконец, в случае поликонденсатов с гетероциклами в глав-
ной цепи реакция замыкания кольца (например, при получении
полиамидов) также протекает в твердом состоянии.
2.1.5.2. Полиреакции в растворе
Наиболее распространенным способом получения высокомолеку-
лярных соединений является полимеризация в растворителе. Если
и мономер, и образующийся полимер растворяются в растворите-
ле, то такой процесс называют гомофазной полимеризацией; если
же полимер в процессе полимеризации выпадает в осадок, то го-
ворят о гетерофазной, или осадительной, полимеризации. При го-
мофазной полимеризации в растворе в инертных растворителях
при постоянной концентрации инициаторов с уменьшением кон-
центрации мономера понижается как скорость реакции, так и сте-
пень полимеризации*. При полимеризации в присутствии осадите-
* См. раздел 3.1.
52
ля часто наблюдаются отклонения от обычных кинетических за-
кономерностей.
Во многих случаях растворитель принимает участие в реакции,
что вызывает дополнительные отклонения от нормального проте-
кания полимеризации. Например, могут протекать реакции переда-
чи цепи на растворитель, благодаря чему средняя степень поли-
меризации при постоянной скорости полимеризации снижается.
В растворителях с высокими константами передачи цепи могут об-
разовываться только макромолекулы с низкой молекулярной мас-
сой, которые содержат осколки агента передачи цепи в качест-
ве концевых групп (теломеризация). При катионной полимериза-
ции влияние растворителя проявляется еще в большей степени
[19] и наряду с реакциями передачи цепи возможно протекание
реакций с инициатором (например, с кислотами Льюиса, с алкил-
галогенидами). Кроме того, при ионной полимеризации важную
роль играют диэлектрические свойства растворителя. В некоторых
случаях, например при анионной полимеризации, наблюдается так-
же влияние растворителя на стерическое расположение основных
звеньев (см. опыт 3-29). Следовательно, выбор растворителя дол-
жен проводиться очень тщательно с учетом конкретных условий,
в которых происходит полимеризация.
Реакции поликонденсации и ступенчатой полимеризации мо-
гут, хотя и реже, проводиться в растворе. Для получения поли-
эфиров, которые не могут быть получены путем поликонденсации
в расплаве, а также для получения полиэфиров с молекулярной
массой выше 30 000 применяют способ поликонденсации в раство-
ре при повышенных температурах (см. опыт 4-02).
Поликонденсацию проводят в 20 %-ном растворе в инертном
растворителе. Наиболее предпочтительными являются гидрофоб-
ные растворители, такие, как бензол, толуол, ксилол или хлорбен-
зол, которые образуют с выделяющейся водой азеотропную смесь
и препятствуют протеканию обратной реакции гидролиза эфирных
связей, образуя защитные сольватные слои. Благодаря более низ-
кой вязкости 20%-ного раствора по сравнению с расплавом выде-
ляющаяся вода удаляется значительно легче. Поэтому поликон-
денсацию в растворе можно проводить при более низкой темпе-
ратуре, которая определяется температурой кипения данного рас-
творителя. Однако для того чтобы и в этом случае этерификация
происходила с высокой скоростью, реакцию проводят на катали-
заторе (большей частью применяют кислотные соединения, как,
например, толуолсульфокислоту). Если один из исходных компо-
нентов (диол или дикарбоновая кислота) нерастворим в данном,
растворителе, то сначала проводят предварительную конденса-
цию в расплаве при 120—150 °C, а затем образовавшийся низко-
молекулярный полиэфир переводят в раствор и проводят дальней-
шую поликонденсацию.
Реакцию поликонденсации в растворе наиболее целесообразно
проводить в аппаратуре с рециркуляцией (см. рис. 17) [20]. При
53
этом воду отделяют путем азеотропной отгонки в разделителе Д,
а растворитель высушивают в сушильной трубке В.
Некоторые продукты поликонденсации, например полиамиды,
можно получить по реакции Шоттен — Баумана в растворе при
низкой температуре [21]. При этом исходные мономеры, напри-
мер диамин и хлорангидрид дикарбоновой кислоты, перемешива-
ют в инертном растворителе; выделившийся хлористый водород
улавливается поглотителем кислоты. Обычно работу проводят при
комнатной температуре с 10%-ными растворами в хлороформе,
дихлорэтане, бензоле или метилэтилкетоне, причем в качестве ак-
цептора кислоты применяют пиридин Этот метод обладает сле-
дующими достоинствами: поликонденсация осуществляется при
низких температурах (0—40°C), вследствие чего исключается воз-
можность протекания побочных реакций и, несмотря на низкие
температуры, ее скорость очень велика (поликонденсация обычно
заканчивается за несколько минут). Кроме того, исходные компо-
ненты могут вводиться не в строго эквимольных количествах, как
при других способах поликонденсации. Недостатками метода по-
ликонденсации в растворе являются необходимость иметь боль-
шие количества хорошо очищенных растворителей, а также обра-
зование в качестве побочных продуктов значительных количеств
солей. Однако несмотря на указанные недостатки, этот метод дает
возможность получать высокомолекулярные продукты поликонден-
сации без применения дорогостоящей аппаратуры и в течение до-
вольно короткого времени.
Реакция Шоттен — Баумана между хлорангидридами дикар-
боновой кислоты и диаминами может проводиться также путем
поликонденсации на границе раздела фаз при комнатной темпе-
ратуре. В этом случае оба исходных компонента растворяют в
двух различных растворителях, которые смешиваются либо час-
тично, либо не смешиваются совсем. Затем оба раствора осторож-
но сливают. При этом поликонденсация может происходить толь-
ко на границе раздела жидких фаз. Тонкая полиамидная пленка,
образующаяся практически мгновенно, препятствует дальнейшей
диффузии мономеров друг к другу. Поликонденсация может про-
должаться только после удаления этой пленки. Таким образом,
этот процесс возможно осуществлять непрерывным способом. По-
ликонденсация на границе раздела фаз проводится также и в дис-
персии. Для этого раствор хлорангидрида дикарбоновой кислоты
при энергичном перемешивании диспергируют в водном растворе
диамина в присутствии растворимого в воде стабилизатора дис-
персии. В этом случае поликонденсация происходит на поверхнос-
ти мелких капель. В качестве растворителя для диамина приме-
няют воду, а для хлорангидрида кислоты — хлорированные али-
фатические углеводороды.
Поликонденсация на границе раздела фаз может проводиться
как с алифатическими, так и с ароматическими хлорангидридами
дикарбоновой кислоты. Из диаминов используют только первич-
54
ные и вторичные алифатические диамины; ароматические диамины
по сравнению с ними реагируют слишком медленно. При этом
исходные компоненты могут вводиться не в строго эквимольных.
количествах. Если нужно получить продукты с высокими молеку-
лярными массами, то в каждом отдельном случае варьируют со-
отношение исходных компонентов и подбирают соответствующие
растворители. Большим достоинством поликонденсации на гра-
нице раздела фаз является высокая скорость реакции: во многих
случаях поликонденсация заканчивается за несколько минут.
Проведение полиреакций таким способом не требует примене-
ния сложной аппаратуры. Кроме того, полиреакции осуществляют
при низкой температуре, вследствие чего исключаются возмож-
ность протекания побочных реакций (например, переамидирова-
ние), а также окислительной и термической деструкций, которые
почти всегда сопровождают поликонденсацию в расплавах. С по-
мощью этого способа удается получать высокоплавкие полиамиды
с высокими молекулярными массами, что невозможно при исполь-
зовании обычных методов. Кроме того, в этом случае можно при-
менять исходные вещества, которые после окончания реакции еще
сохраняют реакционноспособные группы (например, гидроксиль-
ные группы, двойные или тройные С—С-связи). Наконец, преиму-
ществом этого метода по сравнению с поликонденсацией в раство-
ре при низкой температуре является то, что выделяющийся хло-
ристый водород не выпадает в виде соли, которую нужно спе-
циально отделять. Молекулярные массы полимеров, получаемых
межфазной поликонденсацией, обычно не ниже, чем при поли-
конденсации в расплаве (10 000—30 000), а во многих случаях зна-
чительно выше.
2.1.5.3. Полиреакции в дисперсных системах
Полиреакции, и в частности реакция полимеризации, могут про-
водиться в гетерофазной системе. В этом случае жидкий мономер
диспергируют в жидкости, в которой он или вовсе не растворяет-
ся, или же растворяется незначительно.
В ходе реакции агрегатное состояние дисперсной фазы изме-
няется, так как полимер получается в твердом виде, т. е. первона-
чальная эмульсия становится суспензией. Если полимер нерас-
творим в мономере, то этот переход происходит в самом начале-
реакции; если же полимер растворяется или набухает в мономе-
ре, то этот переход происходит только при больших степенях прев-
ращения.
Следовательно, полиреакции, известные под названием суспен-
зионной и эмульсионной полимеризации, происходят в химически
сходных коллоидных системах: в обоих случаях исходные системы
состоят из дисперсии жидкого мономера в жидкости (эмульсия),
который затем переходит в дисперсию твердого полимера в жид-
кости (суспензия). Понятие суспензионной полимеризации отно
55
сится к конечному состоянию системы, понятие эмульсионной по-
лимеризации, напротив, к начальному ее состоянию. Несмотря на
это формальное сходство, оба способа существенно различаются
размером образующихся полимерных частиц (0,1—0,5 мкм при
эмульсионной полимеризации, 0,5—2-Ю3 мкм при суспензионной
полимеризации) и кинетикой полиреакции (см. разделы 2.1.5.3.1
и 2.1.5.3.2).
Эти способы обладают существенными достоинствами: легкий
отвод теплоты полимеризации; низкая вязкость реакционной смеси
даже при больших степенях превращения, что существенно облег-
чает проведение реакции.
Реакции поликонденсации и полиприсоединения также могут
проводиться в дисперсиях (поликонденсация на границе раздела
фаз, см. раздел 4.1.2.3).
2.1.5.3.1. Полиреакции в суспензии
При полимеризации в суспензии [22] жидкий мономер, который
обычно содержит нерастворимый в воде инициатор (например, пе-
рекись бензоила, динитрил азо-бис-изомасляной кислоты), диспер-
гируется при интенсивном перемешивании в соответствующей сре-
де, в которой он либо совсем нерастворим, либо растворим час-
тично. Полимеризация происходит, таким образом, в капельках
мономера. При этом скорость полимеризации и средняя молеку-
лярная масса, а также свойства продуктов сравнимы с этими
параметрами при проведении блочной полимеризации. Дисперги-
рование мономера в воде может быть интенсифицировано добав-
лением небольших количеств (примерно 0,1%) защитных коллои-
дов (см. раздел 2.1.5.3.2) или мелкораздробленных неорганических
веществ (например, сернокислого бария или сернокислого маг-
ния), которые предотвращают соединение мономерных капелек
или коагулирование полимерных частиц в ходе дальнейшей реак-
ции.
В качестве диспергирующей среды (2—10-кратное количество
по отношению к мономеру) при радикальной полимеризации при-
меняют почти исключительно воду. Если мономер частично рас-
творим в воде или полимер нерастворим в мономере, полимер вы-
падает в осадок в виде частичек различной формы и размеров.
Если мономер и инициатор в воде нерастворимы, а полимер в мо-
номере растворим, то полимер образуется в виде шариков, диа-
метр которых по экспериментальным данным может составлять
от 0,5 мкм до нескольких миллиметров. Суспензионную полимери-
зацию такого типа называют бисерной полимеризацией.
2.1.5.3.2. Полиреакции в эмульсии
Как и при суспензионной полимеризации, при эмульсионной поли-
меризации мономер диспергируют в воде. Однако в отличие от
суспензионной полимеризации диспергирование мономера прово-
зе
дится в присутствии веществ, образующих мицеллы (эмульгато-
ры), в которых концентрируется мономер. Размер частиц при
этом существенно меньше, чем при суспензионной поли-
меризации (диаметр приблизительно 0,1 мкм). В каче-
стве инициаторов за редким исключением [23] применяют
соединения, растворимые в воде (персульфат калия,
окислительно-восстановительные системы). Полимеризация проте-
кает не в капельках мономера, а в мицеллах, поэтому скорость
полимеризации (при постоянной концентрации инициатора) зави-
сит от числа мицелл и, следовательно, от концентрации эмульга-
тора [24]. Эмульсионная полимеризация дает возможность полу-
чать полимеры с очень высокими молекулярными массами при
больших скоростях полимеризации. При применении для иниции-
рования окислительно-восстановительных систем реакцию прово-
дят при низких температурах, в некоторых случаях даже ниже
20 °C (см. опыт 3-22).
Процесс полимеризации в эмульсии очень сильно зависит от
эффективности эмульгатора. Так как эффективность эмульгатора
зависит от множества факторов (природа мономера, pH водной
фазы, температура), то в литературе дано описание большого чис-
ла эмульгаторов, отвечающих различным специальным требовани-
ям*. По характеру гидрофильных групп эмульгаторы можно разде-
лить на анионоактивные (щелочные соли жирных кислот, олеат
натрия, алкилсульфонаты), катионоактивные (соли аминов, чет-
вертичные аммониевые соли) и неионогенные эмульгаторы (ад-
дукты, окиси этилена в сочетании с алкилфенолом).
В сильно разбавленном водном растворе эмульгатор существу-
ет либо в ионизированном, либо в молекулярном состоянии. При
возрастающей концентрации эмульгатора внезапно наступает
скачкообразное изменение различных свойств раствора эмульга-
тора, например поверхностного натяжения, вязкости, осмотическо-
го давления и других. Это вызвано тем, что молекулы эмульгатора
собираются в молекулярные агрегаты (мицеллы), причем гидро-
фобные части молекул направлены внутрь мицелл, а гидрофиль-
ные— наружу, к водной фазе. Концентрация эмульгатора, при ко-
торой происходит молекулярная агрегация, называется критиче-
ской концентрацией мицеллообразования [27]; она характерна
для каждого эмульгатора. Концентрация эмульгатора при эмуль-
сионной полимеризации должна быть всегда выше критической
концентрации мицеллообразования; обычно она составляет 0,5—
5% (масс.) по отношению к мономеру. Количество воды в эмуль-
сии варьируется в пределах от половинного до учетверенного коли-
чества мономера.
В противоположность эмульгаторам защитные коллоиды не об-
разуют мицелл. Функция коллоидов состоит в том, чтобы предо-
* Данные об эмульгаторах для полимеризации в «обращенной эмульсии»
(вода — масло) приведены в работах [25] и [26].
57
хранить капельки мономера и частицы латекса от коагулирования.
В качестве защитных коллоидов применяют как растворимые в
воде полимеры (крахмал, пектин, альгинат, желатин), так и моди-
фицированные природные вещества и синтетические полимеры
(оксиэтилцеллюлоза, метилцеллюлоза, карбоксмметилцеллюлоза,
поливиниловый спирт, полиакриловая *кислота, поливинилпирро-
лидон).
2.1.5.4. Регулирование и прерывание полимеризации
Подбором подходящих условий полимеризации можно изменять
среднюю молекулярную массу и связанные с ней свойства поли-
меров. Так, при радикальной полимеризации повышение темпера-
туры реакции или содержания инициатора увеличивает число рас-
тущих радикалов. Так как скорость реакции цепи имеет первый
порядок по концентрации растущих радикалов, а скорость реакции
обрыва — второй порядок, то средняя молекулярная масса по-
нижается при повышении скорости полимеризации. Снижение
концентрации мономера также приводит к получению полимеров
с небольшой молекулярной массой; при этом скорость полимери-
зации тоже снижается. Вследствие возможности протекания по-
бочных реакций при высоких температурах и высоких концентра-
циях инициаторов молекулярную массу во многих случаях изме-
няют путем добавления регуляторов — веществ с высокими кон-
стантами передачи цепи (см. раздел 3.1 и опыт 3-14). Уже при
малых концентрациях эти вещества сильно снижают среднюю мо-
лекулярную массу. Скорость полимеризации при этом остается
практически неизменной. Осколки «регуляторов» входят в состав
молекул полимеров как концевые группы. Такими «регулятора-
ми» являются прежде всего меркаптаны (н-бутилмеркаптан, до-
децилмеркаптан) и другие серосодержащие органические соеди-
нения (например, диизопропилксантогенидсульфид), а также га-
логенсодержащие соединения, альдегиды и ацетали. В технике ре-
гуляторы играют важную роль при эмульсионной полимеризации
прежде всего при получении полимеров на основе бутадиена. Ре-
гулировать молекулярную массу можно и при ионной полимери-
зации [28].
Часто (например, при кинетических исследованиях, при опре-
делении констант сополимеризации или при получении неразвет-
влснных макромолекул) нежелательно доводить полимеризацию
до полного исчерпания мономера. Полимеризацию можно прек-
ратить различными способами. Иногда удается остановить реак-
цию одним лишь охлаждением реакционной смеси. В большин-
стве случаев полимеризацию можно прекратить смешиванием ре-
акционной смеси с определенным количеством осадителя. При
этом образовавшийся полимер выпадает в осадок, а мономер и
инициатор сильно разбавляются. Наиболее действенным способом
прекращения полимеризации является добавление ингибитора или
58
вещества, которое разлагает инициатор. При радикальной поли-
меризации применяют, например, гидрохинон или N-фенил-р-наф-
тиламин. Ионную полимеризацию прерывают добавлением воды,
кислот или оснований. Для прекращения ионной полимеризации,
особенно если ее проводят при низкой температуре, требуется раз-
ложить инициатор, так как при нагревании до комнатной темпе-
ратуры полимеризация может пойти с большей скоростью. Метал-
лоорганические инициаторы, а также катализаторы Циглера —
Натта могут быть разрушены водой или спиртом, кислоты Льюи-
са (BF3)—аминами.
Регулирование молекулярной массы в реакции поликонденса-
ции значительно проще: рост цепи происходит постепенно, и поли-
мер может быть выделен путем прекращения реакции на любой
нужной стадии. В принципе, можно направлять или прерывать
эти реакции, изменяя мольное соотношение бифункциональных
реагирующих компонентов или вводя соответствующие количества
монофункциональных соединений.
2.1.6. Полимераналогичные превращения
Реакции природных или синтетических полимеров отличаются от
химических превращений низкомолекулярных соединений (см. гл.
5); это также относится и к экспериментальным методам проведе-
ния реакций. Поэтому в следующих разделах поясняются некото-
рые детали, которые необходимо иметь в виду при проведении ре-
акций с полимерами.
2.1.6.1. Особенности полимераналогичных превращений
При реакции двух низкомолекулярных соединений А и В кроме
основного продукта С могут образоваться и побочные продук-
ты D:
АфВ-----► C4-D (2-1)
Кроме того, реакционная смесь всегда содержит некоторое ко-
личество исходных продуктов А и В.
При применении этой общей схемы к описанию химических
реакций полимеров следует учитывать, что реагирующие группы
А являются частью макромолекулы М, поэтому как группа С (ос-
новной продукт, возникающий при реакции с реагентом В), так
и группы D (побочный продукт) одинаково присоединены к мак-
ромолекуле М и разделить их невозможно:
М(А)Л 4- В -> М(А)Х (С)/у (D), (2-2)
(х+#+*=л)
В зависимости от состава и строения исходного полимера (или
сополимера) п может изменяться от единицы до значения степе-
ни полимеризации; в последнем случае реакционноспособные
59
группы входят в состав каждого мономерного звена. Количество
групп А, С и D в превращенном полимере описывается величина-
ми х, у и 2. Следует особо подчеркнуть, что не все макромолеку-
лы имеют одинаковое число этих групп; следовательно, х, у и z
представляют собой средние значения. Случай, когда х=г=0 и
у = п (превращение происходит полностью и без протекания побоч-
ных реакций), встречается крайне редко.
Важную роль при химических превращениях полимеров играют
стерический и статистический факторы. Подвижность реакцион-
носпособных групп сильно ограничена, так как они непосредст-
венно связаны с главной цепью макромолекулы. Кроме того, бо-
ковые реакционноспособные группы экранируются главной цепью
макромолекулы. Экранирование и возникающее вследствие этого
замедление реакции вызывается также и тем, что макромолекулы
в растворе представляют собой более или менее спутанные клуб-
ки, причем форма макромолекул может изменяться во время ре-
акции, облегчая или затрудняя превращения. При реакциях с
двойными связями у полидиенов (например, эпоксидирование)
можно использовать различия в положении двойных связей в 1,2-
или 1,4-звеньях. Наконец, при превращениях стереорегулярных
макромолекул (например, при омылении полиметакрилатов) сте-
реоизомерная структура макромолекулы влияет на ход реакции.
Степень превращения в полимераналогичных реакциях ограничи-
вается статистическими причинами и тогда, когда в реакции при-
нимают участие функциональные группы, принадлежащие различ-
ным звеньям макромолекулы. Например, при получении ацеталей
поливинилового спирта (см. опыт 5-02) две гидроксильные груп-
пы соседних звеньев реагируют с альдегидной молекулой. Соглас-
но расчетам Флори [29], максимальная степень превращения в
этом случае составляет 86,5% всех функциональных групп. Это
становится понятным, если представить себе, что каждая функ-
циональная группа, по статистическим причинам оставшаяся изо-
лированной, не может больше реагировать, так как необходимая
для реакции соседняя гидроксильная группа отсутствует:
---СН2-СН-СН2-СН—СН2— СН—СН2—СН—СН2—СН--------
I I I I I
он он он он он
+RCHO
сн2 сн2
---сн2—нс"/ СН-СН.-СН—СН2—Hff7 ^сн--
II I II
0 0 ОН 0 0
I
R
60
Если макромолекулы реагируют с бифункциональными соеди-
нениями, то реакция может протекать не только внутри молекулы,
но и между молекулами. При межмолекулярной полимеризации
образуются сшитые продукты, которые не растворяются и иногда
даже не плавятся. Такие реакции часто проводят специально (см.
пример 4-24), однако., они могут оказаться и нежелательными по-
бочными реакциями, сильно влияющими на растворимость реаги-
рующих продуктов, а также затрудняющими окончание работы.
Например, при реакциях с хлорангидридом полиакриловой кисло-
ты должна быть исключена вода, так как иначе возникают сво-
бодные карбоксильные группы, которые реагируют с хлорангид-
ридными группами другой макромолекулы с образованием анги-
дрида (т. е. сшитого продукта).
2.1.6.2. Выбор условий реакции
Перед проведением некоторых реакций с полимерами целесооб-
разно в каждом случае изучить соответствующую
реакцию на низкомолекулярном модельном веществе. В качестве
такой модели выбирают соединение, которое сходно с полимером
как в отношении реагирующей группы, так и по структуре. При
этом мономер, соответствующий изучаемому полимеру, неприго-
ден, так как он содержит двойную связь, которой нет в полимере.
Таким образом, в качестве модели для полистирола выбирают не
мономерный стирол, а кумол, для поливинилового эфира — соот-
ветствующий эфир изопропанола, для производных полиметакри-
ловой кислоты — соответствующее производное триметилуксусной
кислоты. Но так как далее приходится считаться с двусторон-
ним влиянием соседних реакционноспособных групп макромолеку-
лы, то выбирают такие модельные вещества, которые примерно
соответствуют димерам и тримерам, например пентадиол-2,4 как
модель для поливинилового спирта и производные глутаровой
кислоты, а-метилглутаровой кислоты или пентантрикарбоновой-
1,3,5-кислоты как модели для производных полиакриловой кисло-
ты. С такими модельными соединениями ставят предварительные
опыты, чтобы установить оптимальные условия реакции, а также
характер побочных продуктов. При этом одновременно получают
и модельные вещества для высокомолекулярных продуктов реак-
ции, на которых можно, например, провести исследования рас-
творов, а также аналитические исследования* (например, опреде-
ление функциональных групп, спектров в УФ- и ИК-областях, пи-
ролитическую газовую хроматографию). Данные, полученные та-
ким образом, не должны, однако, безоговорочно переноситься на
реакции с полимерами; это относится прежде всего к выбору рас-
творителя и температуры реакции, а также к процессам разделе-
ния смесей и их очистке.
* См. раздел 2.3.
61
В то время как реакции между низкомолекулярными соедине-
ниями можно проводить в газовой фазе, при работе с полимера-
ми этот способ исключается, так как полимеры не переходят в га-
зообразное состояние. Однако твердый или растворенный полимер
может реагировать с газообразным низкомолекулярным вещест-
вом [30]; например, при промышленном получении метилцеллюло-
зы метилирование проводят газообразным хлористым метилом.
Если реакцию приходится проводить с полимером в твердом
состоянии, например, когда он труднорастворим или вследствие
сшивания вообще нерастворим (как в случае ионного обмена, см.
опыты 5-11, 5-12, 5-13), или если реакция должна протекать толь-
ко на поверхности, целесообразно применять возможно более тон-
коизмельченный полимер (см. раздел 2.4.1). Затем полимер нуж-
но суспендировать в инертной среде, часто с добавлением ве-
ществ, в которых он набухает. Такой прием обеспечивает лучший
доступ низкомолекулярного реагента. Иногда сам низкомолеку-
лярный реагент вызывает набухание полимера, как, например,
при ацетилировании полуацетальных групп полиформальдегида
(см. опыт 5-09).
При проведении реакции в гомогенной фазе нужно учитывать
высокую вязкость полимерных растворов и возникающие вследст-
вие этого затруднения в теплообмене и доступе реагентов. В этом
случае для достижения хорошего перемешивания и предотвраще-
ния местных перегревов следует работать с мощными мешалками.
Если низкомолекулярное соединение при температуре реакции
находится в жидком состоянии, а полимер, подвергающийся прев-
ращению, или продукт реакции в нем растворяется, то, как пра-
вило, можно работать без дополнительного растворителя. В этих
случаях низкомолекулярный реагент берут в большом избытке и
затем обычным способом выделяют из раствора полимерный про-
дукт реакции (см. раздел 2.2). В качестве примера можно при-
вести превращение целлюлозы (см. опыты 5-06, 5-07, 5-08) и по-
лиакролеина [31]. Если, однако, это невозможно, то необходимо
применять растворитель. В этом случае подбирают растворитель,
общий для полимера, низкомолекулярного реагента и катализато-
ра. Следует, однако, иметь в виду, что растворимость полимера в
ходе реакции может существенно изменяться, даже если прореаги-
ровала лишь малая часть реакционноспособных групп. При этом
полимер может преждевременно выпасть в осадок, что затрудня-
ет дальнейшую работу. Если невозможно найти общий раствори-
тель или общую смесь растворителей для исходного полимера и
для конечного продукта, то реакцию следует проводить в такой
среде, которая являлась бы растворителем для продукта реак-
ции.
Особую осторожность следует соблюдать при выборе темпера-
туры реакции, которая заметно влияет на соотношение основных
и побочных продуктов. Полиреакцию рекомендуется проводить
при низких температурах, если возможно протекание реакций
62
сшивания, термической или термоокислительной деструкции (на-
пример, у полидиенов) или гидролиза (например, у целлюлозы),
которые могут вызывать значительное изменение молекулярной
массы, а тем самым и физико-механических свойств полимера. Это
необходимо иметь в виду, например, при работе с легкоокисляю-
щимися полимерами и проводить реакцию в атмосфере азота и да-
же в присутствии стабилизатора.
В большинстве случаев процессы разделения и очистки для мо-
дельных веществ и полимеров существенно различны. Для поли-
меров эти процессы ограничиваются соответственно отделением
высокомолекулярного продукта от низкомолекулярных побочных
продуктов (см. раздел 2.2).
2.1.6.3. Аналитические методы, применяемые
для характеристики полимеров
При определении степени превращения при реакциях полимеров
нельзя пользоваться критериями, применяемыми в химии низко-
молекулярных соединений. В разделе 2.3 подробно обсуждаются
методы, применяемые для характеристики полимеров.
Прежде всего необходимо качественно установить, протекает
ли нужная реакция, для чего используют УФ- и ИК-спектроско-
пию. Целью количественного анализа является получение данных
о долях компонентов А, С и D в уравнении (2-2). Для этого ис-
пользуют как обычные методы анализа, так и специальные мето-
ды (например, спектроскопию и газовую хроматографию продук-
тов пиролиза; см. раздел 2.3).
Результаты количественного анализа могут быть выражены в
массовых или мольных процентах. Результат, выраженный в мас-
совых процентах, характеризует число граммов найденного
повторяющегося звена в 100 г полимера. Однако это значение не
отражает долю прореагировавших звеньев, поэтому нагляднее
рассчитать мольную долю прореагировавших звеньев, т. е. пока-
зать, сколько молей повторяющегося звена прореагировало на
100 мономерных звеньев. Для этого нужно знать строение групп,
образующихся в результате побочных реакций, чтобы иметь воз-
можность вычислить молекулярные массы всех содержащихся
в полимере структурных элементов. Если принять для простоты,
что никаких побочных реакций не протекает, то можно рассмат-
ривать продукт реакции как двухкомпонентную систему, которая
состоит из исходных мономерных звеньев А и образующихся в
результате реакции звеньев С. Для пересчета массовых процен-
тов в мольные можно пользоваться следующими формулами:
100
а— 100—а / Л* V (2'3)
с — 100 —— а (2-4)
где а и с —содержание звеньев А и С соответственно, % (мол.); А* и би-
молекулярные массы звеньев А и С; а — содержание звеньев А, % (масс.).
63
Если необходимо выполнить ряд однотипных анализов, то це-
лесообразно построить диаграмму, на которой нанесены значения
аналитически найденных концентраций соответствующих групп
или соответствующих структурных элементов в зависимости от
состава, выраженного, с одной стороны, в массовых процентах, а
с другой — в мольных (рис. 18). Следует еще раз отметить, что,
как правило, найденные значения по уже указанным причинам
(см. раздел 2.1.6.1) меньше теоретических значений.
Степень превращения в мольных процентах удобнее предста-
вить в виде средней мольной степени превращения х:
— Число молей группы А в мономе ном звене X
х= 100
X Общая степень превращения в мол. %
10Q
Это рекомендуется делать в тех случаях, когда макромолекула
содержит более одной реакционноспособной группы, например
Рис. 18. Графическое определение сте-
пени превращения поливинилового спир-
та, ацилированного монохлоруксусной
кислотой.
три гидроксильные группы в
звене целлюлозы (см. опыты
5-06 и 5-07).
Приведенные выше расчеты
справедливы также и для тех
реакций, при которых образу-
ются три или большее число
различных групп: например
при проведении последователь-
ных реакций с полимером.
В этом случае из-за побочных
реакций число учитываемых
групп становится слишком
большим и проведение количе-
ственного анализа трудоемким.
Расчет степени превращения
при этом возможен только на
основании упрощающих допу-
щений, которые выбираются в
каждом конкретном случае.
Тогда ограничиваются анализом лишь тех групп, которые полу-
чаются в результате реакций, учитываемых при упрощении, и сте-
пень превращения вычисляется в массовых процентах с учетом
только этих реакций.
2.2. ВЫДЕЛЕНИЕ И ОЧИСТКА ПОЛИМЕРОВ
2.2.1. Выделение полимеров
Если в процессе образования полимеры выпадают в нераствори-
мый осадок (полимеризация с осаждением, суспензионная поли-
меризация, поликонденсация на границе раздела фаз), то выделе-
64
ние их из растворов не представляет никаких трудностей. Обра-
зовавшийся продукт отфильтровывают (водные растворы фильт-
руют с помощью бумажных фильтров, органические растворы —
с помощью фильтровального полотна или стеклянных фильтров)
или отделяют от растворителя центрифугированием. Если поли-
мер растворяется в реакционной смеси, то его можно выделить
из раствора двумя способами:
отогнать в вакууме находя-
щийся в избытке мономер, рас-
творитель и прочие летучие
компоненты или осадить поли-
мер с помощью осадителя.
Первый способ применяют
только в исключительных слу-
чаях, так как полимер обычно
выпадает в виде смолы или
плотного осадка и загрязнен
остатками инициатора, непро-
реагировавшего мономера и
растворителя. Второй способ
(выделение полимера с помо-
щью осадителей) является наи-
более удобным. Необходимо,
чтобы осадитель смешивался с
мономерами и с применяемыми
растворителями и растворял
все добавки (например, ини-
циатор), а также все образую-
щиеся побочные продукты (на-
пример, олигомеры). Кроме
Рис. 19. Сопла для распыления поли
мерных растворов в осадителе.
того, осадитель не должен рас-
творять полимер, а должен
осаждать его в виде хлопьев, а
не масла или смолы. Наконец,
осадитель должен легко отгоняться и по возможности мало адсор-
бироваться полимером.
Обычно реакционную смесь или раствор полимера при интен-
сивном перемешивании по каплям вливают в 4—10-кратный из-
быток осадителя. При этом концентрацию полимерного раствора
(как правило, не выше 10%) и количество осадителя подбирают
так, чтобы полимер выпадал в виде хлопьев. Часто выпавший по-
лимер остается в виде коллоида. В этом случае осаждение про-
водят при низкой температуре (внешнее охлаждение или последу-
ющее введение сухого льда) или добавляют электролиты (рас-
творы хлорида натрия или сульфата алюминия, разбавленную
соляную кислоту, уксусную кислоту или аммиак). Иногда поли-
мер выделяется в виде хлопьевидного осадка только после дли-
тельного интенсивного перемешивания или встряхивания. Полиме-
5—732
65
ры, трудно отделимые от растворителя и склонные к осмолению,
осаждают путем распыления в осадителе [32]. При этом полимер
осаждается в виде мелких хлопьев, которые вследствие их боль-
шой поверхности способствуют диффундированию непрореагиро-
вавшего мономера в осадитель. Прибор для распыления представ-
лен на рис. 19. Сопла должны быть не шире, чем показано на
рис. 19, так как иначе потребуется слишком сильный поток воз-
духа. Поток воздуха и образование тумана можно регулировать
с помощью крана Т-образной трубки воздухопровода. Среда, в ко-
торой происходит осаждение, должна интенсивно перемешиваться,
причем при объеме примерно до 300 мл можно пользоваться маг-
нитной мешалкой. Растворы, не стойкие на воздухе, можно распы-
лять азотом. В качестве растворителей применяют вещества с
температурой кипения не ниже 70 °C, так как иначе в устье сопла
произойдет слишком сильное охлаждение.
Из водных растворов полимеры, как правило, выделяют при
осаждении в спирт, ацетон или смесь метанол — эфир. Для выде-
ления полимера из водной эмульсии, которое часто бывает труд-
но осуществить, применяют следующие методы: разведение рас-
твора водой с одновременным добавлением электролита, вымора-
живание, а также впрыскивание раствора в спирт или ацетон.
2.2.2. Очистка и сушка полимеров
Тщательная очистка и сушка полимеров очень важны, прежде
всего для их анализа. Кроме того, различные примеси сильно
влияют на механические, электрические и оптические свойства по-
лимеров. Наконец, загрязнение может вызвать и ускорить процес-
сы деструкции и сшивания.
Обычные приемы очистки, используемые для низкомолекуляр-
ных соединений, такие, как перегонка, возгонка или кристаллиза-
ция, неприменимы для полимеров. Иногда загрязняющие примеси
удаляют путем холодной или горячей экстракции в подходящих
растворителях или путем перегонки с водяным паром. В случае
водорастворимых полимеров (например, полиакриловой кислоты,
поливинилового спирта, полиакриламида) низкомолекулярные
компоненты отделяют с помощью диализа [33] или электродиа-
лиза [33]. Однако чаще всего полимер очищают повторным осаж-
дением, при котором раствор полимера (не выше 5%-ной концен-
трации) вливают при перемешивании в 4—10-кратный избыток
осадителя. Эту операцию повторяют до тех пор, пока примеси не
перестанут обнаруживаться.
Сушка полимеров часто представляет большие трудности, так
как многие растворители или осадители прочно удерживаются по-
лимером. Очень часто даже в глубоком вакууме и при высокой
температуре продолжительность сушки составляет 2—Зсут. Перед
сушкой полимер необходимо измельчить (см. раздел 2.4). Особое
значение приобретает лиофильная сушка [34, 35]. В некоторых
66
случаях можно рекомендовать комбинацию лиофильной сушки с
осаждением путем распыления (см. раздел 2.2.1). Можно, напри-
мер, раствор полимера распылить в ступку, дно которой покрыто
кусками сухого льда размером примерно с лесной орех. Затем кус-
ки льда раздробить, поставить ступку в эксикатор и отвакууми-
ровать его с помощью масляного насоса. Можно также распылять
полимерный раствор в сильно охлажденную жидкость, которая не
смешивается с растворителем. Например, водный раствор полиме-
ра распыляют в охлажденный эфир. При этом полимер выпадает
в виде хлопьевидного осадка, после чего его отделяют деканта-
цией и отгоняют под вакуумом эфир.
2.2.3. Стабилизация полимеров [36]
Многие полицеры даже в отсутствие примесей претерпевают хими-
ческие изменения, которые влияют на их механические и физиче-
ские свойства (например, автоокисление под воздействием све-
та, гидролиз, ацидолиз, отщепление низкомолекулярных соедине-
ний). Для предотвращения подобных изменений проводят стаби-
лизацию полимера с помощью специальных добавок. В качестве
антиоксиданта для полидиенов и полиолефинов используют,
например, N-фенил-р-нафтиламин, который вводят в полимер в ко-
личестве 0,1—0,5%. Стабилизатор можно вводить в осадитель в хо-
де выделения полимера из раствора путем высаживания либо пу-
тем диспергирования измельченного полимера в эфирном раство-
ре стабилизатора при медленном испарении эфира. Примеры не-
которых других стабилизаторов приведены в работе [37]. Боль-
шие количества полимера рекомендуется перемешивать со стаби-
лизатором на вальцах.
2.3. ХАРАКТЕРИСТИКА ПОЛИМЕРОВ [38, 39]
Для характеристики низкомолекулярного соединения достаточно
указать его основные физические и химические свойства, такие,
как температура кипения, температура плавления, показатель
преломления и данные микроанализа. Если два низкомолекуляр-
ных соединения обладают одинаковыми характеристическими
свойствами, они идентичны.
Охарактеризовать высокомолекулярные вещества значительно
труднее. Так, у полимеров нет температуры кипения; точка плав-
ления кристаллических полимеров в большинстве случаев выраже-
на не резко, причем нередко наблюдается не плавление, а только
размягчение полимера, а иногда и его разложение. Поэтому на-
ряду с данными анализа необходимы дополнительные характери-
стики, такие, как растворимость, вязкость растворов, средняя мо-
лекулярная масса, молекулярно-массовое распределение, степень
кристалличности.
Основная трудность заключается в том, что полимеры не могут
быть получены с той степенью молекулярной однородности, как
5*
67
низкомолекулярные соединения [40]. Высокомолекулярные соеди-
нения одинакового аналитического состава могут различаться по
строению, структуре, стереорегулярности (см. раздел 1.2), а так-
же по ширине молекулярно-массового распределения. Иными сло-
вами, высокомолекулярные соединения — это смесь макромолекул
с различными характеристическими свойствами. Следовательно,
выражение «идентичный» к макромолекулам практически непри-
менимо. Так как до сих пор еще не существует способов получе-
ния высокомолекулярных соединений одинаковых строения и раз-
мера*, все физико-химические исследования полимеров могут дать
лишь средние значения измеряемой величины. Перечисленные осо-
бенности высокомолекулярных соединений вызывают необходи-
мость использования новых или видоизмененных методов иссле-
дования по сравнению с методами изучения низкомолекулярных
веществ.
Поскольку структура макромолекул, определяющая свойства
полимера, зависит от условий его получения (например, молеку-
лярной массы, степени кристалличности), наряду с методом опре-
деления структуры необходимо указать и способ получения поли-
мера (например, радикальная полимеризация при 80 °C, полиме-
ризация с определенным металлоорганическим смешанным ката-
лизатором при 20°C).
2.3.1. Растворитель и растворимость [41]
При изучении свойств полимера в первую очередь исследуют его
растворимость, поскольку это одна из наиболее важных характе-
ристик, по которой можно судить о степени сшивания (см. опы-
ты 3-49 и 4-06) и которая может быть использована для опреде-
ления изо-, синдио- и атактических структур (см. опыт 3-28), а
также сополимеров (см. опыт 3-42).
При определении растворимости следует иметь в виду, что по-
лимеры обычно либо полностью растворимы, либо практически
нерастворимы или только ограниченно набухают в растворителе.
Качество растворителя определяется не константой равновесия
между растворенным веществом и осадком, как для низкомолеку-
лярных соединений, а тем количеством осадителя, которое при до-
бавлении к раствору вызывает начало выпадения полимера. Ко-
нечно, более точным является сравнение значений второго вири-
ального коэффициента для различных систем полимер — раство-
ритель [42] или сравнение значений вязкости раствора полимера
в разных растворителях (см. раздел 2.3.2.1).
Ограниченная растворимость или набухание является типич-
ным признаком полимеров с очень высокой молекулярной массой.
* Даже полученные в специальных условиях полимеры с узким молекуляр-
но-массовым распределением (см. раздел 3.2.1) обладают определенной неодно-
родностью.
68
Таблица 5. Растворимость различных полимеров
Полимер Растворитель Осадитель
Полиэтилен, полибутен-1, изотактический поли- пропилен Атактический полипропи- лен Полиизобутилен ti-Ксилол*, трихлорбензол*, декан*, декалин* Углеводороды, изоамилаце- тат Гексан, бензол, четыреххло- ристый углерод, тетрагид- рофуран Алифатические и ароматиче- ские углеводороды Бензол, толуол, хлороформ, циклогексанон, бутилаце- тат, сероуглерод Тетрагидрофуран, циклогек- санон, метилэтилкетон, ди- метилформамид Циклогексанон, диметил- формамид Нерастворим Бензол, хлороформ, метанол, ацетон, бутилацетат Изолропанол, метилэтилке- тон, хлороформ, аромати- ческие углеводороды Ацетон, диоксан, хлороформ Хлороформ, ацетон, этил- ацетат, тетрагидрофуран, толуол Диметилформамид, диметил- сульфоксид, конц. серная кислота Вода Вода, разб. едкие щелочи, метанол, диоксан, диме- тилформамид Вода, метанол, диметил- сульфоксид Вода, диметилформамид*, диметилсульфоксид* Ацетон, диэтиловый эфир, низкие спирты Этилацетат, пропанол Ацетон, метанол, метил- ацетат
Полибутадиен, полиизо- прен Полистирол Поливинилхлорид Поливинилфторид Политетрафторэтилен Поливинилацетат Поливинилизобутиловый эфир Поливинилметилкетон Эфиры полиакриловой и полиметакриловой кис- лоты Полиакрилонитрил Полиакриламид Полиакриловая кислота Ацетон, диэтиловый эфир, низкие спирты Низкие спирты, диэтило- вый эфир, ацетон Метанол, ацетон, гептан Алифатические углеводо- роды, метанол Диэтиловый эфир, петро- лейный эфир, бутанол Метанол, ацетон Вода, алифатические уг- леводороды Метанол, диэтиловый эфир, петролейный эфир Спирты, диэтиловый эфир, вода, углеводо- роды Метанол, ацетон Углеводороды, метилаце- тат, ацетон
Поливинилсульфоновая кислота Поливиниловый спирт Углеводороды, ацетон Углеводороды, метанол, ацетон, диэтиловый эфир Ацетон, метанол Метанол, ацетон Метанол, диэтиловый эфир Этанол, диэтиловый эфир, петролейный эфир
Крахмал Целлюлоза Триацетат целлюлозы Вода, хлоральгидрат, ком- плекс этилендиамина с медью Водные растворы четвертич- ных аммониевых основа- ний, хлорида цинка, тио- цианата кальция Ацетон, хлороформ, диоксан
Триметиловый эфир цел- люлозы Хлороформ, бензол
Продолжение
Полимер Растворитель Осадитель
Карбоксиметилцеллю- лоза Вода Метанол
Алифатические полиэфи- Хлороформ, муравьиная кис- Метанол, диэтиловый
ры лота, бензол эфир, алифатические углеводороды
Полиэтилентерефталат м-Крезол, о-хлорфенол, нит- робензол, трихлоруксусная кислота Метанол, ацетон, алифа- тические углеводороды
Полиамид Муравьиная кислота, конц. серная кислота диметил- формамид, л<-крезол Метанол, диэтиловый эфир, углеводороды
Полиуретан Муравьиная кислота, у-бу- тиролактон, диметилформ- амид, лс-крезол Метанол, диэтиловый эфир, углеводороды
Полиоксиметилен у-Бутиролактон*, диметил- формамид*, бензиловый спирт* Метанол, диэтиловый эфир, алифатические углеводороды
Полиэтиленоксид Вода, бензол, диметилформ- амид Алифатические углеводо- роды, диэтиловый эфир
Политетрагидрофуран Бензол, метиленхлорид, тет- рагидрофуран Алифатические углеводо- роды, диэтиловый эфир
Пол ид иметилсил оксан Хлороформ, гептан, бензол, диэтиловый эфир Метанол, этанол
* Растворим при нагревании.
При набухании высокомолекулярные соединения поглощают боль-
шие количества растворителя и образуют гели (см. раздел 1.4.4).
Если этот процесс прекращается прежде, чем образуется гомо-
генный раствор, то имеет место ограниченное набухание, в про-
тивоположном случае — неограниченное набухание или раство-
рение. Степень набухания зависит от химической природы полиме-
ров и их молекулярной массы, а также от качества растворителя
и температуры. Для сшитых полимеров по степени набухания оп-
ределяют степень сшивания.
Несмотря на многочисленные исследования и термодинамиче-
ские расчеты [43], до сих пор нет единой теории растворимости
полимеров, в связи с чем приходится пользоваться эмпирически-
ми зависимостями [44]. В табл. 5 приведены примеры раствори-
телей и осадителей, применяемых для выделения полимеров. Ко-
нечно, не всякая комбинация растворителя с осадителем пригод-
на* для выделения полимера. Возможность применения каждой
из систем устанавливают предварительными опытами. Для ряда
полимеров подходящие комбинации растворитель — осадитель при-
ведены в гл. 3, 4, 5.
Для определения растворимости полимера поступают следую-
щим образом. В маленькие пробирки помещают 30—50 мг мелко-
* Растворители и нерастворители должны смешиваться между собой.
70
раздробленного полимера, заливают 1 мл растворителя и остав-
ляют на несколько часов. Содержимое пробирки периодически
перемешивают или встряхивают. Если при комнатной температуре
полимер не набухает, то смесь можно осторожно нагреть до тем-
пературы кипения растворителя. В некоторых случаях может на-
блюдаться изменение окраски раствора или выделение газов, что
указывает на разложение полимера (о стабилизаторах см. раздел
2.2.3). Если полимер набухает, но не растворяется, то опыт пов-
торяют с другими растворителями или смесями растворителей.
Если же и при повторных опытах происходит только набухание,
можно предположить, что полимер . имеет сетчатое строение
(сшивки).
Известны некоторые системы полимер — растворитель, которые
характеризуются своеобразными свойствами. Например, сущест-
вуют полимеры,4 растворимость которых в определенных раствори-
телях связана с отрицательным температурным коэффициентом.
Полиэтиленоксид растворяется в воде уже при комнатной темпера-
туре, а при нагревании снова выпадает в осадок. Подобным об-
разом ведут себя растворы поливинилметилового эфира в воде
(см. также [45].)
При применении смесей растворителей часто возникают неожи-
данные эффекты. Например, иногда смесь осадителей действует
как растворитель, и наоборот, смесь растворителей может дейст-
вовать как осадитель. Так, полиакрилонитрил как в нитрометане,
так и в воде полностью нерастворим, а в смеси этих осадителей
растворяется. Подобным же образом ведет себя полистирол при
растворении в смеси осадителей ацетон — гептан, а также поливи-
нилхлорид в смеси ацетон — сероуглерод. В качестве примера оса-
дителя, состоящего из смеси двух растворителей, можно привести
систему диметилформамид — динитрил малоновой кислоты для по-
лиакрилонитрила. Система поливинилацетат — формамид—ацето-
фенон является другим примером того же типа.
Наконец, следует отметить, что многие растворы полимеров в
одинаковых растворителях при смешении оказываются несовме-
стимыми [46]. Так, при смешении 10%-ных растворов полисти-
рола в бензоле и поливинилацетата в бензоле происходит разде-
ление фаз. Вначале смесь становится мутной, а затем полностью
расслаивается.
2.3.2. Определение молекулярной массы полимеров
В ряде случаев, например для кинетических, физико-химических
и технологических исследований, необходимо знать молекуляр-
ную массу полимерного продукта. При этом обычные методы оп-
ределения молекулярной массы, применяемые в химии низкомо-
лекулярных соединений, такие, как криоскопия и эбулиоскопия,
пригодны только для исследования полимеров с молекулярной
массой до 20000 [47]. Поэтому для полимеров используют осо-
71
бые методы, которые можно разделить на абсолютные и относи-
тельные. Возможность их применения определяется прежде всего
растворимостью данного полимера в соответствующем раствори-
теле.
Абсолютные методы дают непосредственно значение молеку-
лярной массы или степени полимеризации, причем в расчетное
уравнение, наряду с легко определяемыми константами, такими,
как плотность, показатель преломления и т. д., входят только уни-
версальные константы — газовая постоянная или число Авогадро.
В настоящее время наиболее важными абсолютными метода-
ми определения молекулярной массы являются [48] следующие:
1) осмометрический метод (определение осмотического давле-
ния);
2) метод ультрацентрифугирования (определение констант се-
диментации и диффузии);
3) методы светорассеяния (определение интенсивности тинда-
левского рассеяния в зависимости от длины волны падающего
света и угла наблюдения).
Эти методы требуют специального аппаратурного оформления
и экспериментальных навыков, поэтому они доступны далеко не
каждой лаборатории. В связи с этим по-прежнему широко исполь-
зуют химические методы (например, определение концевых групп) >
несмотря на некоторые ограничения (см. раздел 2.3.2.2).
С помощью относительных методов измеряется какое-либо
свойство полимера, которое однозначно зависит от его молекуляр-
ной массы, например степень растворимости в данном растворите-
ле, вязкость раствора. При этом для оценки молекулярной массы
необходимо иметь экспериментальную градуировочную кривую, по-
лученную путем сравнения с данными одного из абсолютных ме-
тодов. Одним из наиболее распространенных и широко применя-
емых относительных методов является измерение вязкости по
Штаудингеру [49]. Этот метод подробно рассмотрен в разделе
2.3.2.1.
Так как синтетические высокомолекулярные соединения пред-
ставляют собой смеси макромолекул с различными молекуляр-
ными массами, т. е. являются полидисперсными, можно опреде-
лить лишь среднее значение молекулярной массы.
Различные экспериментальные методы позволяют измерить
молекулярные массы разной степени усреднения. Так, средне-
массовая молекулярная масса Mw определяется по данным све-
торассеяния, ультрацентрифугирования и измерения вязкости*,
среднечисловая молекулярная масса Мп получается по данным из-
мерения осмотического давления или определения концевых групп.
Для монодисперсных полимеров Mw = Мп, для полидисперсных
Mw всегда больше Мп- Отношение среднемассовой и среднечисло-
* О более тонком “-различии между усредненными значениями молекулярной:
массы, полученной методами седиментации и вязкости, см. [50].
72
вой молекулярной массы дает величину, характеризующую поли-
дисперсность* соответствующего полимера:
U==MW/Mn (2-5)
В большинстве случаев это отношение больше 2. При полимери-
зации некоторых мономеров в присутствии специально подобран-
ных инициаторов и в особых условиях можно получить полимеры
с узким молекулярно-массовым распределением: U=ltl—1,3 (см.
раздел 3.2.1).
При определении молекулярной массы в растворе необходимо
предварительно убедиться в том, что. макромолекулы не ассоции-
рованы. Отсутствие ассоциатов может быть доказано с помощью
полимераналогичных превращений: если степень полимеризации
полимерного образца до реакции совпадает со степенью полимери-
зации прореагировавшего полимера, то ассоциация исключается.
Иногда для доказательства отсутствия ассоциации сравнивают
значения молекулярной массы, полученные в разных растворите-
лях: они должны совпадать.
2.3.2.1. Определение вязкости растворов полимеров [51]
Вискозиметрический метод определения молекулярной массы по
Штаудингеру основан на том, что линейные макромолекулы, на-
ходящиеся в растворителе, даже при относительно низких кон-
центрациях, значительно повышают его вязкость, причем повыше-
ние вязкости раствора пропорционально увеличению молекуляр-
ной массы. Этот метод применим только к линейным и мало раз-
ветвленным макромолекулам и не подходит для шарообразных
или сильно разветвленных макромолекул (глобулярные белки,
гликогены). Поскольку при определении молекулярных масс речь
идет не об абсолютной вязкости, а об относительном повышении
вязкости, то измерение заключается в определении вязкости рас-
твора полимера т) и чистого растворителя т]о и вычислений на осно-
ве этих измерений удельной вязкости т)уд:
Если измерения проводят в капиллярных вискозиметрах из-
вестных размеров (см. рис. 22) и при малых концентрациях (так
что плотность раствора приблизительно равна плотности раство-
рителя), то вместо вязкостей л и т]о в уравнение (2-6) можно
подставить непосредственно времена истечения раствора t и рас-
творителя /о. В этом случае уравнение упрощается:
Луд = (^ ^о)Ло (2-7)
Если разделить полученное значение на концентрацию полиме-
ра в растворе, то получают приведенную вязкость т]уд/С. Так как
* Для характеристики полидисперсности применяют также выражение
73
эта величина зависит от концентрации, то в качестве меры вяз-
костных свойств линейных макромолекул используют так назы-
ваемую характеристическую вязкость (или приведенное число
вязкости), которая определяется следующим образом:
[т]] = Ит (ЛМ (2-8)
С-»0 х
Поскольку т] —величина безразмерная, то [rj] имеет размерность
обратной концентрации (например, литр на грамм или 100 см3 на
грамм). В последнее время применяют также размерность см3/г
(показатель Штаудингера), поэтому при измерениях вязкости не-
обходимо указывать размерность концентрации.
Рис. 20. Графическое определение Пуд/С
по результатам измерения вязкости при
различных концентрациях:
1, 2, 3 — одинаковые количества полимера и рас-
творителя при возрастающей молекулярной массе;
4, 5 — полиэлектролит в воде до и после введения
низкомолекулярного электролита.
Измерение вязкости при бесконечно малой концентрации рас-
творов практически неосуществимо, поэтому измеряют вязкость
при возможно малой концентрации и полученные значения экст-
раполируют к нулевой концентрации. В этом случае используют
графические приемы, в которых для различных концентраций (на-
пример, 10; 5; 2,5 и 1,25 г/л) определяют значения г|уД/С и затем
наносят их против соответствующих концентраций. Экстраполя-
ция к нулевой концентрации дает на ординате отрезок, соответст-
вующий значению [т|]. Для полимеров с низкими или средними
молекулярными массами получают, как правило, прямые линии,
в то время как для очень высокомолекулярных образцов линии
часто искривлены кверху (рис. 20). В этом случае рекомендуется
проводить добавочные измерения при низких концентрациях (на-
пример, 0,6 г/л) и экстраполировать только линейную часть кри-
вой.
Совершенно другая зависимость вязкости от концентрации на-
блюдается для растворов полиэлектролитов [54] в полярных рас-
творителях (например, полимерные кислоты в воде, см. опыты
3-10 и 5-03). Правда, и здесь при уменьшении концентрации зна-
чения т]Уд/С снижаются, но затем круто поднимаются вверх (см.
рис. 20, кривую 4). При добавлении нейтральной соли к водному
74
раствору полиэлектролита (например, 5% хлорида натрия) зави-
симость опять приобретает линейный характер (см. рис. 20, кри-
вую 5). Это вызвано тем, что для растворов полиэлектролитов
к факторам, влияющим на размеры клубка и его плотность, до-
бавляется еще один фактор: степень диссоциации. В диссоцииро-
ванном состоянии одинаково заряженные группы макромолекул
отталкиваются, что приводит к увеличению размеров клубка, а
это, в свою очередь, значительно увеличивает вязкость. Вязкость
водных растворов полиэлектролитов, например полиакриловой
кислоты (см. опыт 3-10), определяется конкуренцией двух про-
цессов. С одной стороны, вязкость уменьшается вследствие раз-
бавления, т. е. имеет место обычный ход кривых зависимости вяз-
кости от концентрации; с другой стороны, вязкость возрастает
вследствие ожСстчения макромолекул, связанного с увеличением
степени диссоциации карбоксильных групп. При добавлении к
раствору полиэлектролита хлорида натрия степень диссоциации
становится постоянной, и, таким образом, ожесточение цепей, т. е.
повышение вязкости при уменьшении концентрации полимера в
растворе, предотвращается.
Было предложено несколько эмпирических уравнений, с по-
мощью которых можно вычислить характеристическую вязкость
h] по одному измерению вязкости, например уравнение Шульца—
Блашке [55]:
(2-9)
гП]___W2—
М- 1+КпПуД
Поскольку измерения проводятся при низких концентрациях поли-
меров, то величина Ап обычно не зависит от растворителя и при-
роды полимера, т. е. является величиной постоянной (/<^ = 0,28).
Разумеется, это уравнение применимо не всегда, поэтому при ис-
следовании нового полимера целесообразно еще раз проверить,
совпадают ли вычисленное и графически найденное значения [ц].
Пока не существует удовлетворительного теоретического объ-
яснения концентрационной зависимости вязкости растворов поли-
меров. Так как характеристическая вязкость зависит не только от
размера макромолекул, но и от их формы, а также от свойств
применяемого растворителя, то до сих пор отсутствует простое
уравнение для непосредственного вычисления молекулярных масс
из измерений вязкости. Поэтому для каждой системы полимер—
растворитель при определенной температуре строят градуировоч-
ную кривую [ц]—молекулярная масса, причем молекулярную
массу определяют с помощью абсолютных методов. Известное
уравнение Штаудингера
(2-10)
применимо лишь в некоторых частных случаях. Более общим яв-
ляется уравнение, предложенное Куном
(2-И)
75
После логарифмирования уравнение (2-11) принимает следующий
вид:
lgfn] = lgK/n + algM
(2-12)
В двойных логарифмических координатах получают прямую, по
тангенсу угла наклона которой находят величину а, а по отрез-
ку, отсекаемому на оси ординат,—величину Кт (рис. 21).
Величина а зависит от формы макромолекул в растворе. Для
плотных клубков а=0; для более рыхлых клубков, т. е. для боль-
шинства систем полимер — растворитель*, 0,5<а<1.
Рис. 21. Зависимость вязкости от
молекулярной массы для поли-
стирола при 20 °C (по Мейер-
гофу).
В некоторых случаях <х>1, что характерно для жестких и мак-
симально вытянутых макромолекул, приближающихся к модели
жесткого стержня, для которой а=2.
Так как увеличение размеров клубка при прочих равных усло-
виях зависит исключительно от сольватирующей способности рас-
творителя, то значение а, а с ним и [т|] определяют качество
растворителя (см. раздел 2.3.1); высокие значения а и соответст-
венно [т]] указывают (при заданной молекулярной массе и посто-
янной температуре) на сильное увеличение клубка и поэтому —
на хорошее качество растворителя, низкие значения для а и со-
ответственно для [т|]—на плохой растворитель. Например, для
поливинилацетата в метаноле а=0,60, а в ацетоне а=0,72.
Взаимодействие между растворителем и полимером зависит не
только от природы полимера и растворителя, но и от температу-
ры. При высокой температуре сольватация макромолекул раство-
рителем увеличивается (клубок становится более рыхлым, т. е.
а возрастает), тогда как при низкой температуре преобладают
силы притяжения между сегментами полимерных цепей, с одной
* Исключением является, например, очень сильно разветвленный и потому
близкий к шарообразной форме гликоген, для которого в воде (растворителе)
а = 0.
76
стороны, и между молекулами растворителя — с другой стороны.
В принципе, для данной системы полимер — растворитель сущест-
вует определенная температура, при которой обе силы (сольвата-
ция и ассоциация) становятся равными по величине — это так на-
зываемая тэта-температура. При этой температуре полимер в дан-
ном растворителе находится в виде ненабухшего клубка, т. е.
экспонента а в уравнении (2-11) имеет значение 0,5. Это выпол-
няется для многих полимеров. Например, для полистирола в цик-
логексане тэта-температура лежит около 34 °C, а для полиизобути-
лена в бензоле — около 24 °C.
В табл. 6 приведены значения Кт и а для некоторых полиме-
ров в соответствующем растворителе; другие данные можно най-
ти в работах [69], [70], [71].
Таблица 6. Значения Кт и а для вычисления молекулярной массы полимеров
Полимер Растворитель Темпера- тура, °C а Лите- ратур- ный источ- ник
Полистирол Бензол 20 1,23.10"* 0,72 [56]
Поли-а-метилстирол Толуол 25 7,81 *10“* 0,73 [57]
Полиизобутилен Циклогексан 24 1,07'Ю"1 0,50 [58]
Полибутадиен » 20 3,60.10"* 0,70 [59
Полиизопрен Толуол 25 5,02.10"* 0,67 [60
Поливинилацетат Ацетон 30 1,02.10’2 0,72 61
Поливиниловый спирт Вода 25 3,00- Ю’1 0,50 62
Полиметилметакрилат Ацетон 25 9,60.10"3 0,69 63
Полиакрилонитрил Диметилформамид 25 2,33-10“2 0,75 64]
Полиакриламид Вода 25 6,31.10"3 0,80 651
Полиэтиленгликольтере- фталат Фенол — тетрахлор- этан (1:1) 25 2,10-IO’2 0,82 66
Поликарбонат из бисфе- нола А Тетрагидрофуран 20 3,99.10"2 0,70 [67]
Полиамид 6,6 2МКС1 в 90%-ной НСООН 25 1,42. кг1 0,56 [68]
Для измерения вязкости необходимы следующие приборы: ка-
пиллярный вискозиметр с держателем, термостатируемая баня;
секундомер, несколько выверенных измерительных колбочек на
10 мл, одна выверенная пипетка на 5 мл и одна пипетка на 3 мл.
Чаще всего применяют капиллярный вискозиметр Оствальда*
(рис. 22). При его изготовлении следует обратить внимание на
то, чтобы капилляр 5 имел воронкообразную форму и был на-
паян без резких переходов (см. рис. 22). Метка 2 должна нахо-
диться вблизи от места входа в капилляр, так как при этом легко
следить за движением мениска. Диаметр капилляра (обычно меж-
* Капиллярный вискозиметр с висячим шарообразным уровнем называется
вискозиметром Уббелоде [72].
77
ду 0,3 и 0,4 мм) подбирают так, чтобы время истечения раство-
рителя составляло примерно 60—150 с. В табл. 7 приведены вре-
мена истечения ряда растворителей (объем раствора 0,5 мл) при
20 °C; разумеется, оно должно быть установлено для каждого вис-
козиметра отдельно.
Поскольку вязкость раствора очень сильно зависит от темпе-
ратуры, необходимо обеспечить хорошее термостатирование (по-
Рис. 22. Капиллярный вискозиметр Оствальда (общая длина 25 см; длина ка-
пилляра 10 см; диаметр меньшего шарика 1,3 см; диаметр большего шарика
2,2 см; наполнение 2 мл; объем протекания 0,5 мл):
а, б, с — различные насадки со стеклянными фильтрами для фильтрования растворителя или
раствора полимера.
Рис. 23. Крепление и термостатирующая баня для вискозиметра Оствальда:
1 — резиновый шарик; 2 — осушающая трубка; 3 — трехходовой кран; 4 — контрольный тер-
мометр (1/10 °C); 5 — контактный термометр; 6 — мешалка; 7 — змеевик для охлаждения;
8 — погружной кипятильник.
стоянство температуры в пределах 0,05—0,1 °C). Постоянство тем-
пературы с точностью ±0,1 °C может быть осуществлено с по-
мощью прибора, изображенного на рис. 23 (в качестве сосуда
служит 10-литровый химический стакан или сосуд для фильтро-
вания), при правильно выбранном расстоянии между контактным
термометром и погруженным кипятильником или охлаждающим
змеевиком и при правильно проведенном охлаждении. Держа-
тель капиллярного вискозиметра должен быть установлен вер-
тикально (проверка с помощью отвеса) и не должен вибрировать
78
Таблица 7. Времена истечения некоторых растворителей
в вискозиметре Оствальда при 20 °C
(объем протекающей жидкости У=0,5 мл)
Растворитель Диа- метр капил- ляра, мм Время истечения с Растворитель Диа- метр капил- ляра, мм Время истечения ^0» с
Сероуглерод 0,3 58 Пиридин 0,4 72
Хлороформ 0,3 88 Вода 0,4 74
Ацетон 0,3 95 Уксусная кислота (ледя- 0,4 86
Гексан 0,3 114 ная)
Этилацетат 0,3 116 Диоксан 0,4 90
Четыреххлористый угле- 0,3 142 Циклогексан 0,4 92
род Муравьиная кислота 0,4 108
Толуол 0,3 160 Формамид 0,5 100
Метанол 0,3 171 Серная кислота 0,7 118
Бензол 0,3 172 и-Крезол 0,7 153
н-Бутилацетат 0,4 62
(двигатель для перемешивания и держатель закреплены на от-
дельных штативах; перемешивание не очень сильное).
Вязкость измеряют следующим образом: 100 мг хорошо высу-
шенного полимера взвешивают на аналитических весах в выверен-
ной колбочке на 10 мл и растворяют примерно в 10 мл раствори-
теля. После установления температуры раствора в 20 °C (подве-
шивание измерительной колбочки в бане вискозиметра) колбочку
наполняют до метки (концентрация полимера равна 10 г/л). За-
тем раствор полимера фильтруют через стеклянный фильтр (см.
с на рис. 22) в чистую и сухую пробирку (не ополаскивать). С
помощью пипетки наливают 3 мл приготовленного раствора (рас-
твор 1) в колено вискозиметра Оствальда. Вискозиметр подве-
шивают вертикально в бане, как показано на рис. 23. После вы-
равнивания температуры (примерно 5 мин при 20 °C) полимерный
раствор из колена 2 с помощью резиновой груши выдавливают
в колено 1 немного выше метки 1, снимают давление с помощью
трехходового крана и затем измеряют время, которое потребуется
для истечения раствора от метки 1 до метки. 2. За время истече-
ния раствора берут среднее значение из пяти измерений, которые
должны отличаться друг от друга не более чем на 0,2—0,4 с.
Аналогично определяют время истечения растворителя. Затем вис-
козиметр вынимают из бани и по возможности полностью выли-
вают раствор полимера через колено 2, После того как насаже-
ны насадки а и Ь, прибор несколько раз ополаскивают чистым
растворителем (на насадке а небольшой вакуум), затем чистым
ацетоном и в заключение высушивают (фильтр b целесоборазпо по-
крыть куском фильтровальной бумаги). После этого вискозиметр
пригоден для нового измерения. Таким же образом чистят и сушат
стеклянный фильтр с.
Данные, необходимые для графического определения характери-
стической вязкости h] при низких концентрациях, получают сле-
79
дующим образом. Градуированной' пипеткой наливают 5 мл рас-
твора 1 в 10-миллилитровую пробирку. Затем, как было указано
выше, раствор доливают до 10 ьй (концентрация полимера 5 г/л)
и после фильтрования (раствор 2) определяют время истечения.
После того как вискозиметр и фильтр вымыты и высушены, изме-
ряют время истечения растворов с концентрацией полимера 2 и
1,25 г/л, получаемых путем последовательного разбавления рас-
твора 2. Разумеется, эти растворы можно получить, приготовляя
для каждой концентрации отдельную навеску.
2.3.2.2. Определение концевых групп полимеров
Если макромолекулы содержат доступные анализу концевые груп-
пы, то кроме физико-химических методов определения молекуляр-
ной массы можно применять и химические методы. В полимеры,
которые получают радикальной полимеризацией, такие концевые
группы могут быть введены путем подбора соответствующего ини-
циатора (см. гл. 3). При этом существенно, по какому механизму
происходит обрыв цепи, так как от этого зависит число концевых
групп на одну макромолекулу (при рекомбинации — две концевых
группы, при диспропорционировании — одна группа). Следует учи-
тывать также возможность передачи цепи на мономер, так как
при этом число концевых групп на макромолекулу уменьшается,
что приводит к завышенному значению молекулярной массы. Для
определения числа концевых групп применяют специфические и
очень точные методы анализа, так как эти группы составляют
лишь малую долю макромолекулы (ниже 0,5% по молекулярной
массе). В качестве аналитических методов используют определе-
ние галогенов (например, при применении перекиси п-дибромбен-
зоила, определение 14С [74] при применении меченых перекисных
соединений [75] или азосоединений [76]) или спектроскопические
методы (при применении азосоединений с характеристическими
абсорбционными полосами [77]). Молекулярную массу полимеров,
синтезированных поликонденсацией или полиприсоединением,
также можно оценить путем определения концевых групп. Для
полиэфиров можно очень точно количественно определить число
гидроксильных или карбоксильных концевых групп с помощью
титрования или колориметрически [78]. Гидроксильные концевые
группы (например, в полиоксиметиленах) можно определить так-
же ацетилированием или метилированием [79]. Среднечисловую
молекулярную массу находят по уравнению
__ р
Af„ = 100Z— (2-13)
где Z — число концевых групп на макромолекулу; Е — молекулярная масса
концевой группы; е — экспериментально найденное содержание концевых
групп, %.
80
2.3.3. Фракционирование полимеров [80]
При синтезе высокомолекулярных соединений обычно получаются
смеси полимергомолов, содержащие макромолекулы с различны-
ми молекулярными массами. Экспериментальное исследование мо-
лекулярно-массовых распределений
проводят с помощью метода фракцио-
нирования. Такие исследования очень
важны, поскольку многие физические
свойства полимеров зависят не только
от средней молекулярной массы, но и
от молекулярно-массового распределе-
ния. В то же время анализ молекуляр-
но-массовых распределений позволяет
получить информацию о кинетических
особенностях процесса синтеза (о ме-
ханизме обрыва, передаче цепи ит. д.).
В настоящее время нет способа фрак-
ционирования, при котором из поли-
дисперсного образца получились бы
действительно однородные по молеку-
лярной массе фракции. В основном все
методы фракционирования основаны
на различной растворимости полиме-
ров разной молекулярной массы. Раз-
личают фракционирование осаждением
Ряс 21. Термэстэтируемые де-
лительные воронки для фрак-
ционирования осаждением (а)
а растворением (б).
и фракционирование растворением.
При фракционировании осаждением к раствору полимера (кон-
центрация полимера 0,1 — 1%) при постоянной температуре мед-
ленно добавляют осадитель до появления устойчивого помутне-
ния. Осажденный полимер (фракцию, содержащую наиболее вы-
сокомолекулярную часть) отделяют центрифугированием или де-
кантированием. Затем к оставшемуся раствору прибавляют сле-
дующую порцию осадителя и т. д. Фракционирование осаждени-
ем обычно проводят в колбах Эрленмейера, которые помещают
в термостат. Однако выгоднее использовать специальный сосуд
для фракционирования, показанный на рис. 24, а. Осадитель из бю-
ретки добавляют по каплям при перемешивании в термостатиро-
ванный раствор полимера до появления мути. После этого по-
вышают температуру до тех пор, пока раствор снова не станет
прозрачным, и, наконец, дают раствору остыть до первоначальной
температуры без перемешивания. При этом фракция отделяется
в виде маслянистой фазы, которую затем сливают через кран.
Недостаток фракционирования осаждением заключается в том,
что остающийся раствор становится все более разбавленным, и по-
этому отделение дальнейших фракций затрудняется. Кроме того,
этот метод требует довольно много времени, так как образование
геля происходит очень медленно.
6—732
81
Эти недостатки можно частично устранить при «треугольном»
фракционировании по Мейергофу [81]. К разбавленному поли-
мерному раствору прибавляют столько осадителя, чтобы выделить
примерно половину полимера. После отделения геля последний
снова растворяют, так что в результате получают два раствора,
в каждый из которых снова добавляют осадитель в количестве,
необходимом для выделения приблизительно половины полиме-
ра, и т. д. При этом удается избежать работы с большими объе-
мами раствора и, кроме того, получить существенную экономию
времени, так как несколько фракций можно выделять одновре-
менно.
Идея фракционирования заложена и в часто используемом для
анализа молекулярно-массовых распределений методе турбидимет-
рического титрования [82]. К разбавленному раствору полимера
добавляют осадитель и измеряют мутность раствора в зависимости
от количества прибавленного осадителя.
Для химически однородного полимера молекулярно-массовое
распределение можно определить по экспериментальной зависи-
мости простым расчетом. Турбидиметрическое титрование исполь-
зуют для ориентировочного установления условий фракциониро-
вания (например, выбор пары растворитель — осадитель, величи-
ны фракции и т. д.), которое предшествует собственно препара-
тивному фракционированию осаждением.
Фракционирование растворением не имеет недостатков, харак-
терных для фракционирования осаждением. В этом случае имеет-
ся набор смесей растворитель — осадитель с различной растворя-
ющей способностью. Растворение начинают со смеси, содержащей
максимальное количество осадителя, поэтому в противоположность
фракционированию осаждением первая фракция получается самой
низкомолекулярной, а последняя — высокомолекулярной. По Фук-
су [83], исследуемый полимер осаждают тонким слоем на очи-
щенную алюминиевую фольгу (толщина примерно 20—50 мкм при
общей поверхности 600—1000 см2) при погружении в 10%-ный рас-
твор полимера (0,5—1 г) в легколетучем растворителе. Фольгу
вынимают из раствора и дают растворителю медленно испарить-
ся. Нанесенное вещество должно составлять примерно 100 мг на
100 см2 фольги. Это значение может быть как угодно уменьшено,
но увеличено не более чем на 50%. После сушки пленки в ваку-
уме фольгу взвешивают и разрезают на полосы, которые поме-
щают в эрленмейеровскую колбу (250 мл) или в особый сосуд
для фракционирования (рис. 24, б), после чего последовательна
обрабатывают смесями растворитель — осадитель. Благодаря не-
большой толщине полимерной пленки (5—10 мкм) равновесие
достигается за 5—10 мин. По истечении этого времени раствор де-
кантируют (или сливают), а растворенный полимер получают вы-
париванием. Таким способом из 1 г полимера можно выделить
10—20 фракций за сравнительно короткое время (1—2 сут) (см.
опыт 3-16).
82
Особенно четкие фракции Получаются при фракционировании
на колонке. При этом полимер наносят на инертный носитель
(стеклянные шарики, песок или кизельгур) и в таком виде загру-
жают в вертикальную колонку. Затем колонку промывают спе-
циально подобранной смесью растворитель — осадитель, состав
которой непрерывно изменяется; вытекающую жидкость собира-
ют в фракционный приемник. Фракционирование на колонке про-
водят либо при постоянной температуре [84], либо в условиях гра-
диентов температуры и концентрации одновременно [85]. Фрак-
ционирование при постоянной температуре проще в аппаратурном
отношении, так как в качестве колонки можно использовать стек-
лянную трубку с рубашкой для термостатирующей жидкости.
Разновидностью метода фракционирования на колонке являет-
ся гель-хроматография [86]. В качестве разделительного вещества
применяют органические или неорганические вещества (например,
силикагель) пористой структуры с размером пор, зависящим от
плотности сшивок и условий получения. Для фракционирования
полимеров, растворимых в воде, чаще всего применяют набухший
в воде декстран с различной степенью сшивания (сефадекс). Для
растворов полимеров в органических растворителях применяют
сшитые полистиролы или сополимеры метилметакрилата с этилен-
гликольдиметакрилатом. Образец полимера растворяют, зали-
вают в колонку и элюируют, используя тот же самый раствори-
тель. Небольшие молекулы полимера свободно диффундируют
внутрь геля. Размеры некоторых молекул оказываются настолько
большими, что им не удается проникнуть внутрь пор, в результа-
те чего они первыми выходят из колонки при элюировании. Про-
должительность элюирования фракций возрастает с уменьшением
размера макромолекул. Существует критическое значение молеку-
лярной массы, ниже которого макромолекулы полимера могут
проникать в поры сетки и поэтому могут быть разделены. Моле-
кулы большего размера уже не могут быть разделены, так как
они не могут диффундировать в гель. Частота сетки геля и крити-
ческое значение молекулярной массы связаны между собой про-
стой зависимостью: чем чаще сетка, тем меньше критическое зна-
чение молекулярной массы.
Эффективность гель-хроматографического фракционирования
определяется не только природой геля, но и геометрией колонки.
Полный внутренний объем пор геля определяется количеством
сухой смолы и ее способностью к набуханию, которая, в свою оче-
редь, зависит от элюирующего растворителя. Общий объем геля
Vt складывается из собственного объема гелевого скелета Vg,
внутреннего объема пор геля Vi и свободного объема между
частицами геля. Для макромолекул с молекулярной массой выше
критической элюирующий объем Ve равен свободному объему Vo,
поскольку макромолекулы такого размера не проникают в поры
геля и проходят через колонку без задержки, что позволяет легко
определить Vo. Молекулы, размер которых настолько мал, что
они могут проникать во все поры, могут находиться как в любой
6*
83
части свободного объема Vo, так и в любой части объема пор
Vi, и поэтому для их элюирования необходимо пропустить через
колонку элюирующий объем:
Ve = V. + Vi (2-14)
Для молекул промежуточного размера доступна только доля
Kd(0^/<d^l) внутреннего объема пор V{ и весь свободный объем
Vo, поэтому элюирующий объем для них равен:
Ve = V0 + KdVi (2-15)
Константа Кл представляет собой коэффициент объемного распре-
деления между полным внутренним объемом К, и той его частью,
которая может быть занята молекулой растворенного вещества
KdVi. Числовое значение К<г зависит в первую очередь от размера
молекул вещества и в гораздо меньшей степени от их формы.
Концентрацию полимера в растворе, вытекающем из колонки,
определяют спектроскопически, рефрактометрически или посредст-
вом осаждения растворенного полимера. Для установления связи
между элюирующим объемом и молекулярной массой, зависящей
от природы полимера, характеристик геля и многих других фак-
торов, проводят калибровку колонки по образцам полимера с точ-
но известными молекулярными массами. Зависимость логарифма
молекулярной массы от элюирующего объема (как правило, почти
линейная) служит для калибровочной кривой при исследовании
молекулярно-массовых распределений.
После определения молекулярных масс отдельных фракций по-
лидисперсного полимера молекулярно-массовое распределение
удобно представить графически в виде кривой распределения по
массам
тР = Н (Р) (2-16)
где тР — масса макромолекул со степенью полимеризации Р, деленная на об-
щую массу полидисперсного образца.
Табл. 8 иллюстрирует обработку экспериментальных данных
по фракционированию образца полистирола со средней степенью
полимеризации 800.
На первый взгляд может показаться разумным представить
экспериментальные данные по фракционированию в виде ступен-
чатой зависимости массовой доли фракции со степенью полимери-
зации Р от средней степени полимеризации. Однако такая форма
представления информации корректна только в том случае, если
отклонения от средней степени полимеризации одинаковы для всех
фракций, т. е. если все ступеньки ступенчатой кривой имеют оди-
наковую ширину. На практике это выполняется очень редко. Из
второй графы табл. 8, например, видно, что сразу за максимумом
массовой доли (фракция 2) следует минимум (фракция 3). При-
чина такого различия не в том, что молекул со степенью поли-
меризации 1470 больше, чем со степенью полимеризации 1300, а
84
Таблица 8. Фракционирование полистирола со средней
степенью полимеризации 800
Номер фракции Количество фракции, % / (Р), % Wp-102, %* Р
8 3,4 1,7 1,10 169
7 3,7 5,25 3,45 363
6 7,3 10,75 4,35 433
5 16,8 22,8 6,95 680
4 24,9 43,7 7,55 900
3 9,9 61,6 5,30 1300
2 26,5 79,25 4,25 1470
1 7,5 96,25 1,40 2240
* Найдено графическим дифференцированием интегральной кривой распределения.
скорее в том, что фракция 2 охватывает гораздо больший интер-
вал молекулярных масс, чем фракция 3. Поэтому доли фракций
нужно представлять не вертикальными отрезками (высота сту-
пеней), а площадями, т. е. рассматривать функцию распределе-
ния масс [14] в интегральном виде
р
J(P) = ^H(P)dP (2-17)
1
Интегральную функцию распределения получают из эксперимен-
тальных данных следующим образом. Распределение масс внутри
одной фракции приблизительно симметрично. Следовательно, мож-
но предположить, что одна половина фракции содержит меньшие,
а другая половина большие степени полимеризации по сравнению
со средним значением Рт. Далее, фракция с полимерами от 1 до
(т—1) содержит макромолекулы со степенью полимеризации
меньшей, чем Рт, Если теперь суммировать массы всех фракций
от 1 до (т—1) и добавить половину массы m-й фракции, то полу-
чится массовая доля всех степеней полимеризации от 0 до Рт. Так,
массовая доля 1(Р) фракции 5 в табл. 8 получена суммирова-
нием процентных содержаний фракций 8, 7 и 6 (3,44-3,7 + 7,3 =
= 14,4) и половины доли фракции 5 (14,4+8,4 = 22,8). Зависимость
полученных таким образом величин от степени полимеризации и
представляет собой интегральную кривую распределения (рис.
25). Она указывает, например, на то, что 10,75% макромолекул
имеют степень полимеризации большую, чем 900 и т. д. Соответст-
венно этому полимер тем ближе приближается к монодисперсному,
чем круче интегральная кривая распределения.
Дифференциальная функция распределения масс имеет вид
dmP = H(P)dP (2-18)
где dmP— массовая доля макромолекул со степенью полимериза-
ции от Р до P+dP, которую получают обычно графическим диф-
ференцированием интегральной кривой [уравнение (2-17)]. Для
85
этого строят в возможно большем числе близко лежащих точек
касательные к интегральной кривой и определяют их наклон. По-
лученные таким образом значения Шр дают соответствующую диф-
ференциальную функцию распределения. Точка перегиба интеграль-
ной кривой соответствует максимуму дифференциальной функции
распределения. Чем уже фракция, т. е. чем больше число фракций,
тем более детальную информацию можно получить при фракцио-
нировании. Дифференциальная кривая распределения показыва-
Рис. 25. Функции распределения по степени полимеризации образца
полистирола:
/ — интегральная кривая; 2 — дифференциальная кривая.
ет, чему равна массовая доля макромолекул с той или иной сте-
пенью полимеризации Р в исследуемом образце. Обычно она име-
ет один максимум и по форме напоминает гауссову функцию.
Так как указанные методы фракционирования (кроме гель-
хроматографии) основаны на различии в растворимости, то фрак-
ционирование по молекулярным массам можно осуществить толь-
ко для химически однородных макромолекул. Для разветвленных
с различной степенью разветвления полимеров или для ’полиме-
ров, претерпевающих какие-либо химические превращения, а так-
же для статистических, привитых или блок-сополимров раствори-
мость зависит не только от молекулярной массы. Фракциониро-
вание в этом случае может привести к разделению макромолекул
полимера по их химическому составу [88], [89]. Результаты фрак-
ционирования можно, следовательно, использовать для расчета
молекулярно-массового распределения только тогда, когда уста-
новлена химическая идентичность фракций (элементный анализ,
ИК-анализ, пиролитическая газовая хроматография). Применение
различных растворителей и осадителей позволяет иногда провести
фракционирование как по молекулярным массам, так и по химиче-
скому составу.
86
2.3.4. Определение температуры стеклования,
температуры размягчения, интервала температур
плавления и точки плавления кристаллитов
Полимеры по отношению к нагреванию существенно отличаются
от низкомолекулярных соединений*. Только полимеры с высокой
степенью кристалличности имеют достаточно резко выраженную
точку плавления; аморфные полимеры, а также полимеры с низ-
кой степенью кристалличности размягчаются в довольно широком
температурном интервале. Наиболее важной характеристической
температурой для полимеров является температура стеклования.
2.3.4.1. Определение температуры стеклования
Ниже определенной температуры аморфный полимер может рас-
сматриваться как твердое стекло. Если его нагреть выше этой
температуры, то отдельные сегменты макромолекулы приобретают
большую подвижность, полимер становится мягким и, наконец, пе-
реходит в высокоэластическое состояние*. Температуру, при кото-
рой происходит это изменение, называют температурой стеклова-
ния Tg. Эта температура зависит от химической природы полиме-
ра, стереохимического строения его цепи, от степени разветвлен-
ности макромолекул. Для одного и того же образца Tg может
быть различной в зависимости от метода ее определения [90].
Температуру стеклования можно определить путем исследования
некоторых физических характеристик полимерного образца, таких,
как показатель преломления, модуль упругости, диэлектрическая
проницаемость, теплоемкость, коэффициент набухания, удельный
объем, в зависимости от температуры. При достижении темпера-
туры стеклования эти величины или их температурный ход резко
меняются. У аморфных полимеров температура размягчения часто
совпадает с температурой стеклования; у кристаллических поли-
меров точка плавления существенно выше, чем Tg. Температуру
стеклования кристаллических полимеров можно оценить по эмпи-
рическому правилу Бойера — Бимана: составляет примерно две
трети температуры плавления (в градусах Кельвина)**.
2.3.4.2. Определение температуры размягчения
Для определения температуры размягчения образец полимера
медленно нагревают под действием небольшой нагрузки до появ-
ления определенной деформации. Соответствующая температура
является температурой размягчения. Так как при определении
температуры размягчения условия испытания образца устанавли-
вают произвольно, то эта температура физически менее хорошо
* См. раздел 1.4.2.
** Сопоставление температур стеклования см. в работе [91].
87
обоснована, чем температура стеклования. Для определения тем-
пературы размягчения на практике применяют три метода [92].*
Наиболее распространенным является метод Вика, который
заключается в том, что на поверхность образца (площадью при-
мерно 1 см2 и толщиной 3—4 мм) вертикально устанавливают
тупую стальную иглу (площадь соприкосновения ее с образцом
1 мм2), которую нагружают определенным весом, например
б кгс. Образец нагревают в термостате со скоростью 50°С/ч и
определяют температуру, при которой игла вошла в образец на глу-
бину 1 мм; эта температура и является температурой размягчения
(температура размягчения по Вика).
По методу Мартенса образец в виде прутка укрепляется ниж-
ним концом в зажиме, а верхний нагружается. Образец нагрева-
ется с постоянной скоростью до тех пор, пока не появится заметный
изгиб. Установленная таким образом температура размягчения на-
зывается температурой размягчения по Мартенсу.
Наконец, по третьему методу пруток кладут обоими концами
на две опоры, нагружают в середине и медленно нагревают до
достижения определенного прогиба.
Эти методы определения температуры размягчения применяют
как для термопластов, так и реактопластов (сшитых полимеров).
Осуществить взаимные переводы температур размягчения, опре-
деленных этими методами, не удается.
2.3.4.3. Определение интервала температур плавления
и точки плавления кристаллитов
Полимеры редко имеют строго определенную температуру плав-
ления: они плавятся в достаточно широком температурном интер-
вале, который зависит от ряда факторов. Интервал плавления
кристаллических полимеров можно определить по изменению двой-
ного лучепреломления образца при нагревании. Сначала определя-
ют приблизительную температуру плавления [93]. Небольшое ко-
личество полимера расплавляют на предметном стекле при темпе-
ратуре, которая лежит немного выше области плавления, и за-
тем придавливают расплавленный полимер обычным покровным
стеклышком, нагретым примерно до той же температуры, так что
получается тонкая гомогенная пленка. Предметное стекло поме-
щают на столик микроскопа, нагретый примерно на 20 °C ниже
температуры плавления, и наблюдают образец в поляризованном
свете, одновременно медленно повышая его температуру. Фикси-
руют ту температурную область, в которой происходит изменение
двойного лучепреломления, до тех пор пока двойное лучепрелом-
ление не перестанет наблюдаться. За точку плавления принимают
среднее значение между первым и последним отсчетом. Многие
полимеры при охлаждении рекристаллизуются, поэтому возможно
повторное определение температуры плавления.
* В СССР широко применяют термомеханический метод (весы Каргина).
88
При определении области плавления этим методом надо иметь
в виду, что и некристаллический полимер может обнаружить двой-
ное лучепреломление, если его молекулы окажутся ориентирован-
ными под действием внешних сил. Ориентация может происходить
при придавливании расплавленного полимера покровным стек-
лышком или в ходе получения пленки, а также при разрезании
образца. В ориентированных полимерах исчезновение двойного
лучепреломления происходит не в области температуры плавле-
ния образца. Это можно исключить с помощью повторного опре-
деления, так как ориентация в расплавленном полимере не мо-
жет появиться без внешнего воздействи. Более точное определение
температуры плавления проводят с помощью дифференциального
термического анализа [94].
В случае аморфных полимеров температура и интервал плав-
ления* зависят от молекулярно-массового распределения и сте-
пени разветвленности исследуемого полимера. Зависимость темпе-
ратуры текучести от молекулярной массы можно наблюдать
для олигомеров в пределах одного гомологического ряда, напри-
мер для полиоксиметилендиметилового эфира [95], для олигомер-
ных полиамидов и полиэфиров [96], [97].
Сшитые полимеры не могут быть переведены в состояние рас-
плава и характеризуются только температурой размягчения. Су-
ществует также ряд несшитых полимеров, которые не имеют ни
температуры, ни области плавления (например, полиакрилони-
трил). Такие полимеры начинают разлагаться выше некоторой оп-
ределенной температуры.
2.3.5. Определение вязкости расплавов полимеров [98]
Для определения вязкости полимерного расплава** применяют
вискозиметры двух типов: ротационный вискозиметр (или виско-
зиметр с конусом и пластинкой) и капиллярный вискозиметр (или
капиллярный экструзиометр). Капиллярные вискозиметры относи-
тельно просты в обращении и, кроме того, их можно применять
при высоких напряжениях сдвига, которые часто встречаются на
практике. Для характеристики текучести полимеров при испыта-
ниях на капиллярных экструзиометрах определяют не вязкость
расплава т), а количество расплава, протекшее за определенный
промежуток времени (10 мин)—так называемый индекс расплава
i. Обычно указывают температуру измерения и напряжение сдви-
га или нагрузку, например (190 °C) =9,2 г/10 мин. Это означает,
что 9,2 г полимера протекло за 10 мин при 190 °C и нагрузке, рав-
ной 2 кгс.
Капиллярный экструзиометр для измерения индекса расплава
состоит из нагреваемого цилиндра, который нагружают определен-
* Имеется в виду температура перехода в вязкотекучее состояние.
** См. раздел 1.4.2.
89
ними грузами. Порошкообразный или гранулированный полимер
загружают в цилиндр, нагретый до нужной температуры, после
чего насаживают плунжер с грузом. Примерно через 5 мин арре-
тир плунжера освобождают, и полимерный расплав выдавливается
через сопло. Выдавленный столбик образца обрезается и взвеши-
вается. За индекс расплава принимают среднеарифметическое зна-
чение из 5 измерений.
2.3.6. Определение степени кристалличности полимеров
Для качественного определения степени кристалличности поли-
мера наблюдают двойное лучепреломление в поляризационном
микроскопе; при этом необходимо исключить влияние возможной
ориентации макромолекул, т. е. так называемое ориентационное
двойное лучепреломление*.
Количественную оценку степени кристалличности проводят с
помощью рентгеновских исследований, которые, кроме того, дают
возможность вычислить обычные кристаллографические данные.
В некоторых случаях кристаллические полимеры можно иссле-
довать методом ИК-спектроскопии, так как в ИК-спектрах появ-
ляются дополнительные полосы поглощения. Например, у поли-
этилена [99] дополнительная «кристаллическая» полоса наблю-
дается при 730 см'1, соответственно «аморфная» полоса — при
1300 см-"1, у полистирола [100] — при 982, 1318 и 1368 см-1. По ин-
тенсивности этих полос поглощения можно судить об изменении
степени кристалличности в зависимости от условий получения
полимера или при его нагревании.
Кроме того, степень кристалличности зависит от плотности по-
лимера, поэтому, 'определяя плотность образца, можно получить
относительную меру увеличения или уменьшения степени кристал-
личности.
2.3.7. Определение плотности полимеров
Определение плотности полимеров проводят пикнометрическим
или флотационным методом [101],
При пикнометрическом методе взвешиванием определяют объем
жидкости, вытесненной полимером. Большинство полимеров имеет
плотность больше единицы, поэтому при определении плотности
можно использовать воду. Вследствие сильно развитой поверхно-
сти полимерные порошки и пресс-материалы склонны к сорбции
воздуха, что может привести к существенным погрешностям при
измерении плотности. Этого можно избежать, если вакуумировать
пикнометр, содержащий полимер, перед заполнением жидкостью
или если понизить поверхностное натяжение воды добавлением
небольших количеств (0,1%) поверхностно-активных веществ.
* См. раздел 2.3.4.3.
’90
Для порошков часто используют другой метод, основанный на
установлении равновесия между массой частиц полимера и вытал-
кивающей силой в жидкости с плотностью меньшей, чем плотность
полимера, путем добавления более тяжелой жидкости. Так как в
состоянии равновесия плотность жидкой фазы и полимера одина-
кова, то задача сводится к определению плотности смеси жидко-
стей. Практически поступают следующим образом: порошкообраз-
ный полимер и определенное количество жидкости меньшей плотно-
сти помещают в термостатированный стакан. К этой смеси из бю-
ретки при слабом перемешивании добавляют жидкость с большей
плотностью до тех пор, пока не будет достигнуто состояние равно-
весия. Плотность жидкой смеси определяют пикнометрически или
получают ее с помощью калибровочной кривой. Для определения
плотностей полимеров меньших единицы (например, полиэтилен)
используют смеси этанол — вода, для плотностей больших едини-
цы— водно-солевые растворы (40%-ный СаС12,^4° = 1,40, 72%-ный
ZnClo, dl° = 1,95).
Следует специально отметить вариант метода, заключающийся
в измерении равновесной высоты в сосуде с градиентом плотности
[102].
2.3.8. Деструкция полимеров
Важную информацию о составе и строении полимеров, а также об
их стойкости часто можно получить при исследовании деструкции
полимеров [103]. Такие исследования выполняют как химическими
и колориметрическими методами (см. раздел 5.3), так и посредст-
вом облучения, ультразвуковых и механических воздействий.
В зависимости от способа экспериментального исследования де-
струкции получают существенно различающиеся сведения. Если
разложение полимера не доводить до конца, а летучие продукты
деструкции специально не исследовать, ограничивая исследование
определением потери массы и уменьшением молекулярной массы
(вязкость), то можно получить информацию о стабильности поли-
мера в условиях эксперимента. Если же исследовать состав и стро-
ение низкомолекулярных продуктов деструкции, то можно получить
также информацию о строении полимера, а в случае сополимера и
о содержании отдельных его компонентов*. В большинстве случаев
применяют термические или химичские методы деструкции, так как
их можно осуществить без применения сложного и дорогостоящего
оборудования.
2.3.8.1. Термическая деструкция полимеров
Термическая деструкция полимера (проводимая в инертной атмо-
сфере для исключения реакции с кислородом) приводит к образова-
* См. раздел 2.3.11.
91
нию различных низкомолекулярных продуктов в зависимости от
строения полимера и температуры деструкции*.
Для практического проведения таких исследований используют
прибор, показанный на рис. 26, который позволяет одновременно
проводить деструкцию нескольких образцов в токе инертного газа
или в вакууме. Для поддержания заданных температур используют
жидкостные (примерно до 250 °C) или воздушные (примерно до
400 °C) термостаты [105], Для более высоких температур применя-
ют бани с металлическими расплавами или электрические нагрева-
Рис. 26. Сосуд для термического разложе-
ния полимеров в атмосфере азота или
в вакууме:
1 — стеклянная палочка с резиновой пробкой для
извлечения пробирки с образцом из сосуда для
разложения полимера; 2 — пробирка для образца
(0,7X5 см); 3 —термометр со шлифом; 4—вы-
пускной кран для летучих продуктов распада.
тели. Пробирки для отдельных проб должны иметь одинаковый
внутренний диаметр (для навески 50—100 мг пригодны пробирки
диаметром 7 мм). Это необходимо для того, чтобы исключить вли-
яние толщины слоя на диффузию газообразных продуктов разло-
жения из расплава полимера. Навеска полимера должна оставать-
ся постоянной.
Если исследуют только термостойкость полимера, то через опре-
деленные промежутки времени путем взвешивания пробирок с про-
бами определяют потери полимера в массе (обычно в %); получен-
ные данные затем наносят на график в зависимости от времени
для различных температур деструкции. Для реакций первого поряд-
ка зависимость логарифма доли неразложившегося полимера от
времени линейна. Тангенс угла наклона прямой соответствует кон-
станте скорости деструкции при соответствующей температуре.
Оставшийся полимер можно извлечь из пробирок и использовать
для дополнительных исследований (вязкость, оптические или ана-
литические измерения). Для сравнения термостойкости полимеров
пользуются скоростью их разложения при 350 °C (в % за мин) или
температурой полураспада Гп, т. е. той температурой, при которой
полимер теряет половину своей массы при 30-минутном термоста-
тировании в вакууме [106].
Для того чтобы установить качественный и количественный со-
став продуктов разложения, их необходимо уловить и проанализи-
* См. раздел 5.3.
92
ровать. В простейшем случае для этого к крану 4 реакционного со-
суда (см. рис. 26) присоединяют холодильник и затем анализируют
собранный конденсат. При этом целесообразно использовать боль-
шие навески (примерно 1—5 г), чем при изучении термостойкости.
Для качественных и количественных исследований продуктов раз-
ложения полимеров применяют пиролитическую газовую хромато-
графию. Разложение (можно проводить с помощью нагретой током
проволоки непосредственно в газовом хроматографе [107] во внеш-
нем по отношению к хроматографу сосуде [108] (если для разло-
жения нужен катализатор [109]) или же в запаянных термостати-
рованных стеклянных трубках (с ампулами [109]). Применяемые
для этого количества вещества довольно малы: обычно не более
10 мг.
2.3.8.2. Химическая деструкция полимеров
Из химических методов деструкции для аналитических исследова-
ний полимеров наиболее пригодны ацидолиз и гидролиз. Окисли-
тельная деструкция (воздействие кислорода воздуха при повышен-
ной температуре) часто приводит к образованию большого числа
продуктов, анализ которых весьма сложен, поэтому однозначное за-
ключение о строении и составе полимера практически невозможно.
Эксперименты такого типа проводят с целью изучения стойкости
полимера и эффективности введенного стабилизатора.
Реакции химической деструкции полимеров протекают, как пра-
вило, по закону случая. Например, гидролиз полиэфира начинает-
ся не с конца цепочки и не развивается по механизму последова-
тельного отщепления мономерных единиц. Реакция начинается с
некоторой случайной группировки внутри цепи, поэтому образую-
щиеся продукты реакции имеют большую молекулярную массу.
Только в результате многократного повторения актов гидролити-
ческого расщепления полимер может быть разложен на фрагмен-
ты, соответствующие одной мономерной единице. Так как обычно
в условиях проведения гидролиза или ацидолиза даже низкомоле-
кулярные осколки полимера нелетучи, то за реакцией разложения
нельзя следить гравиметрически (в противоположность термическо-
му разложению). Часто в таких случаях реакцию контролируют,
применяя титрование для определения количества непрореагиро-
вавшего реагента. Весьма чувствительной пробой на деструкцию
является измерение вязкости раствора полимера по мере протека-
ния реакции (см. опыты 5-18 и 5-19).
2.3.9. Оптические исследования полимеров
Важнейшими оптическими методами исследования полимеров явля-
ются УФ- и ИК-спектроскопия. УФ-Спектроскопия [НО] может
быть использована только для исследования растворимых полиме-
ров. Ее с успехом применяют для количественных определений со-
93
става сополимеров (например, сополимеров стирола [111]), а так-
же для определения содержания незаполимеризовавшегося моно-
мера в полимере [П2].
Особенно большое значение имеет ИК-спектроскопия [ИЗ], так
как опа может быть применена для исследования нерастворимых
сшитых полимеров. Этот метод используется как в чисто аналити-
ческих целях, например для измерения количества функциональных
групп [114], [115], так и для определения строения [116] (напри-
мер, микроструктуры полидиенов, см. пример 3-30; разветвления
в полиэтилене [117]). Он является иногда самым надежным мето-
дом определения состава сополимеров (сополимеры этилена с про-
пиленом [118]). Определение степени кристалличности с помощью
ИК-спектроскопии рассматривалось в разделе 2.3.6.
При применении ИК-спектроскопии в химии полимеров исполь-
зуют обычные приемы работы. Если невозможно работать с рас-
творами, рекомендуется проводить измерения на пленках. В случае
легкоплавких полимеров пленку получают непосредственно плав-
лением на пластинах хлорида натрия или способом, описанным в
разделе 2.4. При использовании бромида калия или суспензий (в
парафиновом масле или перфторированных углеводородах) полимер
необходимо очень мелко измельчить (см. раздел 2.4.1). Для этого
готовую спрессованную таблетку бромида калия следует еще раз
хорошо растереть в ступке и потом спрессовать заново.
Вопрос о применении поляризационного микроскопа и рентге-
новских лучей уже обсуждался при рассмотрении измерения степе-
ни кристалличности (см. раздел 2.3.6). Сюда же следует отнести
метод электронной микроскопии, широко применяемый при иссле-
довании кристаллических структур [119], и метод ядерно-магнит-
иого резонанса, являющийся экспериментальной базой исследований
стереоизомерии полимеров [120].
2.3.10. Определение основных групп и элементов
Качественный и количественный анализ полимеров [121] прово-
дится обычными методами, применяемыми для низкомолекулярных
соединений [122], [123]. То же самое относится к определению лег-
ко идентифицируемых групп [123], например ацетильных, мет-
оксильных или амидных. Правда, при исследовании полимера ча-
сто возникают трудности, заставляющие изменять условия реакций
(например, продолжительность отдельных процессов). Специальные
методы, используемые при анализе концевых групп, описаны в раз-
деле 2.3.2.2.
Элементный анализ и анализ функциональных групп особенно
продуктивны для сополимеров или химически модифицированных
полимеров. Для гомополимеров, результаты элементного анализа
которых должны совпадать с соответствующими данными для
мономеров, отклонения от расчетных значений указывают на
94
протекание побочных реакций при полимеризации; однако эти
отклонения могут обусловливаться присутствием в полимере
остатков растворителей, осадителей, стабилизаторов и других до-
бавок. При подготовке образца к анализу на все это следует обра-
щать самое серьезное внимание (многократное переосаждение в
различных парах растворитель — осадитель, а также тщательная
сушка).
2.3.11. Исследование сополимеров [124]
При изучении сополимеров проводят качественное исследование,
при котором устанавливают, получен ли действительно сополимер
или только смесь гомополимеров, и количественное исследование,
при котором определяют массовую долю каждого из мономеров.
Качественное исследование сополимеров относительно просто,
если гомополимеры существенно различаются по растворимости;
например, если один сополимер растворяется в бензоле, а другой
нет. В этом случае одну пробу предполагаемого сополимера экст-
рагируют бензолом, а вторую пробу — растворителем второго гомо-
полимера. Если таким образом не удается проэкстрагировать чи-
стые гомополимеры, то исходный образец — истинный сополимер.
Разумеется, экстракция должна быть проведена очень тщательно и
повторена несколько раз, так как смеси полимеров обычно трудно
разделить экстрагированием [125]. Если соответствующие гомопо-
лимеры не различаются существенно по растворимости, то иногда
такое различие можно создать путем химических превращений, на-
пример омылением сополимеров винилацетата, акрилатов или мет-
акрилатов, эпоксидированием или гидроксилированием диенов. Ка-
чественное исследование сополимеров значительно осложняется,
если невозможно использовать различие в растворимости гомополи-
меров. В этом случае определяют другие физические константы
предполагаемых сополимеров (например, температуры размягче-
ния и плавления, плотность, степень кристалличности) и сравнива-
ют их с соответствующими значениями для смесей гомополимеров
разного состава. Часто сополимеры можно отличить от смесей го-
мополимеров, проводя качественный и количественный анализ про-
дуктов пиролиза (см. раздел 2.3.8).
Количественный анализ сополимеров относительно прост, если
один из мономеров содержит легко идентифицируемый элемент или
функциональную группу (см. опыты 3-43, 3-44, 3-45); С- и Н-ана-
лизы достаточно точны только при большом различии в содержании
углерода и водорода в реагирующих мономерах. Если посредством
элементного анализа или химическим путем не удается определить
состав сополимера, то его исследуют с помощью спектральных ме-
тодов (см. раздел 2.3.9), например УФ-спектроскопией (сополимеры
стирола), ИК-спектроскопией (сополимеры олефинов) или же с по-
мощью газовой хроматографии (см. раздел 2.3.8) после соответст-
вующего термического или химического разложения.
95
Состав сополимера может быть установлен также анализом со-
става непрореагировавшей смеси мономеров. Блок- и привитые со-
полимеры (см. раздел 3.3.2) принципиально могут быть охаракте-
ризованы теми же способами, однако при этом следует иметь в ви-
ду, что они могут содержать большие количества гомополимеров,
которые необходимо предварительно отделить.
К более точной характеристике статистического сополимера от-
носится определение констант сополимеризации (ri и г2) и вычи-
сление Q- и е- или q и е-значений (см. раздел 3.3 и опыты 3-43—
3-45).
2.3.12. Механические методы исследования полимеров
Изучение механических свойств полимера необходимо для опреде-
ления областей его применения. С первого взгляда такие исследо-
вания кажутся сугубо прикладными, однако некоторые из методов
позволяют узнать не только важные в практическом отношении
свойства полимера, но и дают возможность сделать выводы о его
строении, структуре и агрегатном состоянии. Следовательно, меха-
нические методы исследования могут дать ценную дополнительную
информацию о свойствах полимера (см. разделы 2.3.1—2.3.11). Для
характеристики полимера в твердом состоянии служат следующие
механические параметры: прочность и способность к растяжению,
упругость, хрупкость, ударная вязкость, твердость. Эти параметры
обсуждаются в разделах 2.3.12.1—2.3.12.4.
Многочисленные методы исследования механических свойств по-
лимеров в технике, а также специальные методы исследования кау-
чуков, пенопластов, лаков, пленок и волокон здесь не рассматрива-
ются [126].
Решающим фактором точности и воспроизводимости механиче-
ских исследований является соблюдение условий приготовления и
предварительной обработки полимерных образцов [127], [128].
Испытуемый образец должен быть, по возможности, изотропным,
т. е. во всех направлениях обнаруживать одинаковые свойства, и
должен быть свободным от внутренних напряжений. Кроме того,
температура испытания и влажность воздуха при всех измерениях
должны поддерживаться постоянными. Если последнее требование
выполнить относительно легко, то приготовить изотропные и пол-
ностью свободные от напряжений образцы довольно трудно (см.
раздел 1.4). Следует опасаться возникновения ориентации в испы-
туемых образцах, в особенности в термопластах, которая приводит
к тому, что значения предела прочности при растяжении или удар-
ной вязкости, измеренные в направлении ориентации, вдвое или
втрое больше, чем в перпендикулярном направлении. Возможности
снижения анизотропии многократно обсуждались в литературе [127,
128]. Образцы для механических исследований можно получать
либо литьем и прессованием под давлением, либо литьем в подхо-
дящих формах с последующей механической обработкой [127].
96
2.3.12.1. Исследование деформации в зависимости
от растягивающего усилия
При растяжении на исследуемый образец действует растягивающая
сила, которая в большей или в меньшей степени увеличивает его
длину. При этом поперечное сечение образца уменьшается, пока,
наконец, не наступает разрыв. В этих исследованиях применяют об-
разцы в виде лопаточек, разрыв которых происходит в месте наи-
меньшего поперечного сечения. Более широкие части образца слу-
жат для крепления в зажимные клеммы испытательной машины
Машина растягивает клеммы в противоположные стороны с посто-
янной скоростью. При этом к образцу прикладывается деформиру-
ющее усилие, которое вместе с соответствующим удлинением за-
писывается прибором в виде кривой. Кроме того, определяют мак-
симальное напряжение РМакс, возникающее в образце во время ис-
пытания, которое не всегда совпадает с растягивающим напряже-
нием в момент разрыва образца.
Частное от деления соответствующей силы растяжения на наи-
меньшее поперечное сечение So испытуемого образца до испытания
дает соответствующее растягивающее напряжение о; следователь-
но, оно представляет собой силу растяжения, отнесенную к единице
площади поперечного сечения (т. е. к 1 см2). Отношение макси-
мальной силы растяжения РМакс к начальному поперечному сече-
нию образца называется разрушающим напряжением (пределом
прочности) Ор = РМакс/^о. В общем случае под деформацией е пони-
мается удлинение растягиваемого бруска, отнесенное к его перво-
начальной длине. Относительное удлинение при разрыве ер пред-
ставляет собой растяжение Д/ = /—Zo при максимальной нагрузке
Лиакс, деленное на начальную длину
д/
/0:ев = -т— 100%
•о
Таким образом, по значениям предела прочности при растяже-
нии и соответствующего удлинения можно судить о способности
материала выдерживать определенную нагрузку. Для характери-
стики полимера обычно рассматривают не только разрушающее на-
пряжение при растяжении ор и относительное удлинение при раз-
рыве 8В; более информативными являются кривые нагрузка — уд-
линение. В зависимости от формы этой кривой материалы можно
разделить на три основные группы* (рис. 27).
К первой группе относятся полимерные материалы, для кото-
рых кривая нагрузка — удлинение имеет почти прямолинейную
* На практике между этими группами встречаются также и переходные
формы.
7-732 9J
форму и проходит очень круто*; при разрыве она изогнута едва
заметно (кривая 1 на рис. 27). Такие полимерные материалы, по-
добно металлам, даже при достаточно больших растягивающих
усилиях обнаруживают только небольшую деформацию. К ним от-
носятся все реактопласты и некоторые термопласты, такие, как
полистирол и полиметилметакрилат, т. е. полимеры, являющиеся
малоупругими и довольно хрупкими.
Деформация растяжения второй группы материалов описывает-
ся кривой 2 на рис. 27. Вначале эти материалы ведут себя, подобно
Рис. 27. Диаграмма нагрузка — удлинение по-
лимеров различных типов (пояснения см. в
тексте).
предыдущим, т. е. предел
пропорциональности здесь
невысок и деформация
при увеличении растяги-
вающего усилия невелика.
Потом внезапно происхо-
дит очень сильное измене-
ние длины, хотя дейст-
вующая сила остается по-
стоянной или даже умень-
шается; материал начина-
ет «течь»**. Точка <ув, при
которой начинается тече-
ние, называется пределом
текучести.
После того как макро-
молекулы благодаря про-
цессу деформации по-но-
вому сориентируются друг относительно друга, «текучесть» пре-
кращается и напряжение снова начинает возрастать***. Деформа-
ция при этом уменьшается, и наконец происходит разрыв. Напря-
жение пв в этой точке является разрушающим напряжением
(разрывной прочностью) при соответствующем удлинении 8Р. Ко
второй группе относятся многие термопласты, например полиоле-
фины, полиамид 6, полиамид 6,6 и непластифицированный поли-
винилхлорид.
Третью группу образуют материалы, которые уже при малом
растяжении обнаруживают относительно большие деформации.
Однако на кривой нагрузка — удлинение (кривая 3 на рис. 27) не
* Точка, в которой начинается отклонение от линейности на кривой нагруз-
ка — удлинение, называется пределом пропорциональности. Этот показатель
характеризует ту область деформации, где перестает выполняться закон Гука,
согласно которому изменение длины прямо пропорционально приложенной силе.
Положение предела пропорциональности зависит в значительной степени от
температуры, влияющей, как уже отмечалось выше, на механические свойства
полимеров (см. раздел 1.4).
О процессах, происходящих при течении полимеров, см. разделы 1.4.1—
*** Это явление имеет большое практическое значение для упрочнения пленок
и волокон.
93
наблюдается внезапного уменьшения напряжения, т. е. не сущест-
вует предела текучести. Здесь вследствие ориентации макромоле-
кул происходит постепенное ожестчение материала. Дальнейшее
увеличение растягивающего усилия приводит к разрыву. Соответ-
ствующее напряжение также называется разрушающим напряже-
нием Ор, а соответствующее удлинение — относительным разрывным
удлинением 8Р. К этой группе относятся термопласты, содержащие
пластификаторы (например, ПВХ), а также каучуки. По кривой
нагрузка — удлинение можно вычислить модуль упругости £, ко-
торый связан с жесткостью полимеров. По закону Гука между
напряжением при растяжении а и удлинением е существует ли-
нейная зависимость а = £е, т. е. модуль упругости легко вычислить
по тангенсу угла наклона этой кривой к оси абсцисс в линейной
области, т. е. ниже предела пропорциональности. Следовательно
Напряжение
Е Изменение длины
Таким образом, модуль упругости показывает, какая должна
быть нагрузка на единицу площади для того, чтобы образец поли-
мера в виде круглого стержня растянуть на его собственную длину.
Материалы с малым модулем упругости, например каучуки (£ =
= 10 кгс/см2), уже при небольших воздействиях обнаруживают зна-
чительные удлинения; материалы с большим модулем упругости,
например полиоксиметилен (£ « 35 000 кгс/см2) деформируются не-
значительно. При различных видах* нагрузки получают разные мо
дули упругости. При напряжениях растяжения, давления или изги-
ба говорят о модуле упругости (£-модуль), при напряжениях сдви-
га— о модуле сдвига, или торсионном модуле (G-модуль**).
2.3.12.2. Динамические механические испытания
При динамических механических испытаниях образец под действи-
ем приложенной нагрузки не разрушается. Такие испытания назы-
вают динамическими, поскольку механические свойства полимера
изучаются при колебательном воздействии на образец. Среди мно-
гочисленных измерительных устройств особенно хорошо зарекомен-
довал себя метод торсионных колебаний [129, 130]. При этом один
конец образца, имеющего форму прямоугольного параллелепипеда,
жестко укрепляется, а другой конец прикрепляется к колеблющему-
ся диску торсионного маятника. Образец находится в термостате.
* Кроме того, для полимеров больше, чем для металлов, имеет значение
длительность действия нагрузки. Если к полимерному материалу прикладывают
постоянное усилие, то материал обнаруживает деформацию е уже в момент на-
гружения, причем деформация возрастает во времени. Этот процесс называется
ползучестью. Если же образец растягивают на постоянную величину, то воз-
никает начальное напряжение о, которое постепенно убывает во времени. Этот
процесс называется релаксацией напряжения. Так как о и е являются функция-
ми времени, небезразлично, при каких условиях определяется модуль упру-
гости.
** См. раздел 2.3.12.2
7'
99
Торсионный маятник начинает совершать свободные колебания, по-
степенно затухающие во времени. По продолжительности колеба-
ний можно вычислить модуль сдвига, или торсионный модуль (/-
модуль*). По уменьшению амплитуды колебаний можно судить
о так называемой внутренней механической абсорбции, которая на-
зывается механическим коэффициентом потерь d (или логарифми-
ческим декрементом). Эти данные позволяют судить о движении
отдельных сегментов макромолекулы (микроброуновское движе-
ние) и о движении всей макромолекулы относительно других (ма-
кроброуновское движение)**.
Так как эти процессы сильно зависят от температуры***, то изу-
чение торсионных колебаний целесообразно проводить в широком
температурном интервале и затем наносить на график значения мо-
дуля для определенной температуры (рис. 28). При этом абсорб-
ционному максимуму на кривой d соответствует точка перегиба кри-
вой модуля. По этим кривым можно определить температуру раз-
мягчения (температуру стеклования), а также температуру, или об-
ласть температур, плавления.
Значение модуля и ход кривой модуля позволяют сделать выво-
ды об агрегатном состоянии и о структуре полимерных образцов.
С помощью динамических исследований можно также определить
степень кристалличности, степень сшивания, химическую неодно-
родность, а также отличить статистические сополимеры от блок-со-
плимеров. Метод торсионных колебаний удобен для исследования
полимеров, которые содержат пластификаторы или наполнители
(рис. 28 и 29).
На рис. 28 кривая 1 воспроизводит изменение модуля упругости
для эластомеров. Примечательно, что в этом случае модуль упру-
гости (сдвига) невелик в широком интервале температур и скачко-
образно возрастает при температуре стеклования (—50°C), т. е.
при переходе от высокоэластического состояния к стеклообразному.
Кривые 3 и 4 характерны для частично-кристаллических полимеров
(здесь значение модуля на три порядка больше и понижается толь-
ко по достижении температуры плавления). На соответствующих
кривых для механического фактора потерь d это выглядит следу-
ющим образом (см. рис. 28). Переход в стеклообразное состояние
заметен при хорошо выраженной «механической абсорбции» (кри-
вая /). На кривых для кристаллических полимеров (3 и 4) видны
два абсорбционных максимума. Первый максимум наблюдается
при температурах—100°C для полиэтилена и при 0°С для изотак-
тического полипропилена и соответствует температурам стеклова-
* См. раздел 2.3.12.1.
** По аналогии с ИК-спсктроскопией, которая также основана на поглоще-
нии колебательной энергии молекулами, динамические механические испытания
части называют «механической спектроскопией».
Наряду с температурой большое влияние оказывает также частота коле-
баний в пределах от единицы до нескольких сот герц, которую поддерживают
до возможности постоянной в ходе измерения.
Ю0
ния этих полимеров. Второй максимум наблюдается при темпера-
турах 120°C и 145°C, что соответствует температурам плавления.
По форме кривых / и 2 можно судить о том, насколько сильно от-
личается статистический сополимер от блок-сополимера по меха-
ническим свойствам. Статистические сополимеры этилена и пропи-
Рис. 28. Зависимость модуля упругости Е и механического фактора потерь d от
температуры для различных полимеров:
/ —эластомер — статистический сополимер этилена и пропилена (70:30); 2 — блок-сополимер
этилена и пропилена (5о:50); 3 — частично-кристаллический полимер с низкий температурой
стеклования (полиэтилен); 4 — частично-кристаллический полимер со средней температурой
стеклования (изотактический полипропилен).
лена полностью аморфны и ведут себя при динамико-механических
исследованиях, как эластомеры (кривая /). Блок-сополимеры эти-
лена и пропилена обнаруживают суммарные свойства соответству-
ющих гомополимеров: выше комнатной температуры здесь наблю-
даются две области размягчения, которые соответствуют температу-
рам плавления, и два максимума, соответствующие двум темпера-
турам стеклования.
Метод торсионных колебаний применяется также при исследова-
нии влияния пластификаторов на механические свойства полиме-
ров, например поливинилхлорида. Поливинилхлорид является ти-
пичным аморфным термопластом, т. е. он остается жестким до тем-
101
пературы стеклования, а затем жесткость его резко падает (рис.
29, кривая /). Путем добавления пластификаторов можно сдвинуть
температуру стеклования в область более низких температур. При
этом происходит уменьшение модуля в широком температурном ин-
тервале (кривая 2). Дальнейшее увеличение концентрации пласти-
фикаторов приводит к тому, что кривая по форме становится похо-
жей на соответствующую кривую для эластомеров (кривая 3). Мо-
жно провести параллель между этими тремя кривыми и кривыми
Рис. 29. Зависимость модуля упругости Е и механического фактора потерь d от
температуры для поливинилхлорида с различным содержанием пластифика-
торов:
/ — поливинилхлорид без пластификатора; 2 — поливинилхлорид с 20%-ным содержанием
пластификатора (диоктилфталат); 3 — поливинилхлорид с 40%-ным содержанием пластифика-
тора (диоктилфталат).
Рис. 30. Аппаратура для формования волокна из расплава полимера.
напряжение — удлинение, рассмотренными в разделе 2.3.12.1. Кри-
вая 1 характеризует полимер с высоким модулем упругости и ма-
лой способностью к растяжению, кривая 2—полимер с точкой те-
чения, содержащий небольшое количество пластификатора, кривая
3—систему полимер — пластификатор с низким модулем упругости
и высокой способностью к растяжению.
2.3.12.3. Ударная вязкость
Наряду с рассмотренными механическими методами, при которых
образец находится под действием нагрузки относительно длитель-
ное время, и поэтому скорость деформации мала (например, при
изучении зависимости напряжение — удлинение при измерении
твердости), большой интерес представляют и такие исследования,
102
при которых к материалу прикладывается мгновенная большая на-
грузка. Такие исследования могут дать информацию о хрупкости
и вязкости материала, в частности о так называемой ударной вяз-
кости. По этому методу измеряется энергия разрушения образца
маятниковым копром (кгс-см). При этом энергия разрушения от-
носится к единичной площади поперечного сечения образца, поэ-
тому ударная вязкость имеет размерность кгс-см/см2. В качестве
образцов применяют прямоугольные бруски как гладкие, так и с
надрезом; соответственно говорят о нормальной ударной вязкости ап
и об ударной вязкости с надрезом аь (в последнем случае на ис-
пытуемом образце перед исследованием делают V-образную на-
сечку, что способствует локализации напряжений при ударе в оп-
ределенном месте).
Определение ударной вязкости и ударной вязкости с надрезом
в основном проводится двумя методами, которые различаются меж-
ду собой только способами крепления образца. При испытании на
ударную вязкость при одновременном изгибе по Шарли образец
кладут горизонтально на две опоры и ударяют маятниковым коп-
ром посредине, в то время как по Изоду образец с закрепленным
нижним концом расположен вертикально, а по свободному верхне-
му концу ударяют маятниковым копром.
2.3.12.4. Определение твердости
Твердость — способность твердого тела противодействовать внед-
рению в него другого тела. Для оценки твердости измеряют силу,
необходимую для достижения определенной глубины внедрения.
Эта сила, или глубина внедрения, зависит от температуры и продол-
жительности измерения, а также от некоторых других факторов,
например от формы внедряемого тела. В настоящее время не су-
ществует единого метода измерения твердости полимерных мате-
риалов. Кроме того, существующие методы применимы не для всех
полимеров*. Имеются две группы методов изучения твердости: из-
мерение глубины внедрения по остаточной деформации без нагруз-
ки (метод Бринелля) и измерение глубины внедрения при полной
нагрузке**. Последний метод применяют для термопластов и реак-
топластов. Довольно часто используют следующий метод: стальной
шарик диаметром 5 мм вдавливают в образец в виде пластинки
* Твердость полимера можно определить также по модулю упругости:
высокий модуль упругости соответствует большей твердости. Преимущество
такого метода заключается в том, что значения модуля упругости можно по-
лучить одним и тем же методом: либо исследованием зависимости напряже-
ние — удлинение, либо изучением торсионных колебаний.
** Оценка твердости полимеров существенно зависит от используемого ме-
тода. Например, при определении твердости упругого материала (резины) по
методу Бринелля получается, что резина очень твердая (здесь измеряется толь-
ко остаточная деформация), в то время как при определении по методу вдавли-
вания шарика она оказывается мягкой, так как здесь одновременно учитывает-
ся и остаточная, и упругая деформации.
103
толщиной 4 мм с постоянной силой и через 10 и 60 с измеряют глу-
бину внедрения. Следует отметить, что полученные различными ме-
тодами значения твердости нельзя пересчитывать друг в друга.
2.4. ПЕРЕРАБОТКА ПОЛИМЕРОВ
Для ряда физических и химических исследований необходима
предварительная обработка полимеров. Для получения пленок, ни-
тей и других образцов для лабораторных испытаний методы, при-
меняемые в технике (литье под давлением, экструзия, каландрова-
ние), малодоступны. В лаборатории обычно используют методы,
описанные ниже.
2.4.1. Измельчение полимеров
Если в процессе синтеза, а также последующих операций осажде-
ния и сушки полимер получается недостаточно измельченным, то
его подвергают измельчению. Вследствие большой вязкости расти-
рание в ступке обычным способом редко приводит к желаемому ре-
зультату. Часто при измельчении образец сильно охлаждают, на-
пример жидким азотом, что увеличивает хрупкость. При растира-
нии полимера могут возникать электростатические заряды, которые
устраняют путем увлажнения образца небольшим количеством
эфира. Измельчение можно также проводить в мельницах, при этом
легкоплавящиеся полимеры начинают течь вследствие выделения
тепла. Для предотвращения этого добавляют небольшие количест-
ва сухого льда. Существуют также специальные мельницы с ох-
лаждением, которые очень удобны.
2.4.2. Переработка полимерных расплавов
При переработке полимерных расплавов предполагается, что при
высокой температуре переработки не происходит их заметного раз-
ложения. Полимеры, растворы которых трудно перерабатывать из-
за высокой вязкости или вследствие разложения при температуре
плавления, можно перевести в вязкотекучее состояние пластифи-
кацией и перерабатывать при более низкой температуре. В качест-
ве пластификаторов применяют высококипящие жидкости, совме-
щающиеся с полимерами, например эфиры фосфорной и фталевой
кислот (диоктилфталат), различные алифатические дикарбоновые
кислоты. Молекулы пластификатора располагаются между поли-
мерами цепочками, что приводит к уменьшению межмолекулярно-
го взаимодействия (внешняя пластификация)*. При этом подвиж-
ность полимерных цепочек возрастает, а температура стеклования
понижается. Пластифицированные полимеры являются более гибки-
ми и обладают меньшей твердостью по сравнению с непластифи-
цированными (см. опыт 3-48).
* Внутренняя пластификация описана в разделе 3.3.1.
104
2.4.2.1. Получение изделий прессованием
Для ряда физических исследований (микроскопия, ИК-спектроско-
пия, механические измерения) необходимы тонкие пленки полиме-
ров. Их получают в лаборатории следующим образом: на тонкую
(0,1 мм) алюминевую пластинку (15x15 см) насыпают нужное
количество порошка полимера, накрывают второй алюминиевой пла-
стинкой и помещают между плитами гидравлического пресса, наг-
ретыми до температуры плавления полимера. Образец прессуют в
течение примерно */г—1 мин, затем алюминиевые пластинки выни-
мают из пресса и охлаждают водой или двумя холодными метал-
лическими пластинами. Полученную полимерную пленку осторож-
но отделяют от алюминиевых пластинок. Если нужно получить плен-
ку заданной толщины, то между алюминиевыми пластинками по-
мещают шаблон (фольгу подходящей толщины). Оптимальные ус-
ловия изготовления пленок (количество полимера, температура,
давление и время прессования) подбирают эмпирически для каж-
дого отдельного случая. Если пленка получилась мутной, это озна-
чает, что температура прессования, по-видимому, была слишком
низкой; если она слишком тонка или содержит пузырьки газа
(разложение), то температура прессования была слишком высокой.
На свойства пленки может влиять и скорость охлаждения. Иногда
алюминиевые пластинки с трудом отделяются от полимера. Быст-
рое охлаждение водой, а также предварительное смазывание пла-
стин силиконовым маслом или водной дисперсией политетрафтор-
этилена помогает преодолеть эту трудность.
Если нет подходящего пресса, то можно поступить следующим
образом [133]. В двух нагревательных пластинах от электрическо-
го утюга просверливают отверстия для термопары, пластины со-
единяют параллельно и включают в сеть через регулировочный
трансформатор. Затем измеряют температуру при различных зна-
чениях напряжения и строят градуировочную кривую. Для получе-
ния пленки порошок полимера помещают между нагревательными
пластинами и сдавливают их в тисках в горизонтальном положе-
нии. Давление в этом случае измерить нельзя, но при некотором
навыке можно найти оптимальные условия прессования.
2.4.2.2. Формование волокна из расплава
Простейший способ формования волокна из расплава состоите том,
что полимер расплавляют в пробирке, опускают в расплав стеклян-
ную палочку и медленно ее вынимают. При этом обычно получают
короткие неодинаковые нити, которые для дальнейших исследова-
ний мало пригодны. Непрерывное волокно можно получить с по-
мощью прибора, представленного на рис. 30 [134]. Толстостенную
стеклянную трубку 1 диаметром примерно 3—4 см, вытянутую на
одном конце в короткий запаянный капилляр, опускают в сосуд с
двойными стенками 2 и наполовину заполняют полимером. Затем
систему эвакуируют и заполняют азотом через Т-образную труб-
105
ку 3. Полимер расплавляют, нагревая до кипения жидкость, нахо-
дящуюся в колбе 4 (см. раздел 2.1.4). Затем обрезают кончик ка-
пилляра 5 и давлением азота выдавливают расплав полимера в
виде тонкой нити. Полученную нить с помощью направляющего
ролика 6 наматывают на катушку 7. Температуру в капилляре ре-
гулируют, перемещая его (поднимая или опуская) относительно
сосуда 2.
2.4.3. Переработка растворов полимеров
При отсутствии оборудования для переработки расплава полимера,
или если полимер нельзя расплавить вообще, или если полимер при
плавлении разлагается, прибегают к переработке раствора поли-
мера. Однако перерабатывать растворы можно только в пленки и
волокна.
2.4.3.1. Получение пленок
Для получения пленок в лабораторных условиях раствор полиме-
ра наливают на стеклянную или металлическую пластинку, дают
растворителю испариться и осторожно снимают полимерную плен-
ку. При этом растворитель не должен испаряться слишком быстро,
так как образующаяся пленка будет морщиться и рваться. Этого
можно избежать, высушивая пленку в эксикаторе в атмосфере рас-
творителя. Температура кипения растворителя не должна быть
слишком высокой, так как это затрудняет его испарение. Концент-
рация полимера должна быть такой, чтобы раствор можно было
наливать, но чтобы он не стекал со стеклянной пластинки. Обычно
применяют примерно 20%-ные растворы. Чтобы получить пленки
одинаковой толщины, необходимо стеклянную пластинку устано-
вить горизонтально, а раствор полимера распределить равномерно
по поверхности пластинки. Это делают с помощью стеклянной па-
лочки, концы которой обмотаны липкой лентой или нитками. Для
более точной работы необходимо применять специальные приспо-
собления из металла, позволяющие регулировать толщину слоя.
Готовую пленку поддевают лезвием безопасной бритвы или ножом,
после чего осторожно стягивают ее со стеклянной пластинки. Вме-
сто стеклянной пластинки в качестве подложки можно использо-
вать ртуть, налитую в плоскую чашку.
2.4.3.2. Изготовление нитей из полимерных растворов («прядение»)
Различают два способа изготовления нитей: сухой и мокрый. При
получении нитей сухим способом полимерный раствор выдавлива-
ют через сопло в камеру, заполненную горячим воздухом или азо-
том; при этом растворитель испаряется, а полимер образует нить.
Этот процесс трудно провести без специального оборудования, по-
этому он малопригоден для лаборатории. При мокром способе
106
струя полимерного раствора впрыскивается в подходящий осади-
тель, при этом полимер коагулирует в форме нити. Процесс легко
осуществить в лабораторных условиях. Качество нитей в значитель-
ной степени зависит от условий осаждения (осадитель, температура
бани). Эти условия каждый раз нужно подбирать опытным путем.
Если в выбранном осадителе полимер коагулирует слишком быст-
ро, осадитель частично разбавляют растворителем (полиакрило-
нитрил, растворенный в диметилформамиде, осаждают смесью ди-
метилформамид — вода). Для впрыскивания полимерного раствора
в осадитель применяют медицинские шприцы, использование кото-
рых позволяет регулировать толщину нитей и скорость процесса
прядения (см. опыт 3-19).
2.4.4. Получение вспененных полимеров (пенопластов)
Вспененными материалами называют полимеры пористо-ячеистого
строения. Различают пенопласты с открытыми и замкнутами пора-
ми. Первые применяются в качестве губок, последние — в качестве
тепло- и звукоизоляционных материалов.
Пенопласты получают в основном двумя способами. По перво-
му способу пенопласт получают из готового полимера путем его
вспенивания. Этим способом изготовляют пенопласты из термопла-
стов, например пенополистиролы (см. опыт 3-04). Для получения
пенопласта из термопласта полимер, содержащий порообразователь,
нагревают выше температуры размягчения, при этом выделяющий-
ся газ вспенивает полимер. Затем следует быстрое охлаждение.
В качестве порообразователей применяют низкокипящие инертные
растворители (например, пентан или галогензамещенные алифа-
тические углеводороды) или такие соединения, которые при нагре-
вании разлагаются с образованием газа (бикарбонаты, азосоеди-
нения). При изготовлении пенистой резины используют способность
водных растворов, содержащих поверхностно-активные вещества,
сильно вспениваться при перемешивании, в особенности при введе-
нии воздуха. После вулканизации эмульгированного полимера по-
ристо-ячеистая структура фиксируется.
По второму способу вспенивание проводят одновременно с поли-
меризацией. Этим способом получают, например, вспененные фено-
лоформальдегидные полимеры, эпоксидные полимеры. Компонен-
ты, необходимые для образования реактопластов, смешивают с пе-
нообразователем (большей частью с низкокипящей жидкостью),
смесь наливают в форму и нагревают до температуры реакции.
Выделяющееся при реакции тепло испаряет пенообразователь, ко-
торый вспенивает смесь. Разновидностью этого метода является по-
лучение пенополиуретанов. Добавление специально вспенивающего
агента в этом случае не требуется, так как при конденсации диизо-
цианатов с диолами выделяется двуокись углерода, которая и вспе-
нивает материал (см. раздел 4.2.1.2).
107
Литература
1. Houben-Weyl, 1959, Bd. 1/2, S. 321.
2. Wei ssher ger A., ♦Technique of Organic Chemistry», New York — London;
1950, p. 605.
3. Meyer F. R., Ronge, Angew. Chem., 1939, Bd. 52, S. 637.
4. Houben-Weyl, 1959. Bd. 1/2, S. 321.
5. Houben-Weyl, 1958, Bd. 1/1, S. 866.
6. W e i s s b e r g e r A., «Technique of Organic Chemistry», New York, 1965,
p. 423.
7. В г e с к D. W., J. Am. Chem. See., 1956, v. 78, p. 5963.
8. Russian Chem. Rev., 1960, v. 9, p. 509.
9. Houben-Weyl, 1961, Bd. 14/1, S. 42, 746.
10. Houben-Weyl. 1961, Bd. 14/1. S. 45.
11. Ch a pi ro A., Radiation Chemistry of Polymers, New York — London, 1962.
12. Houben-Weyl. 1961, Bd. 14/1, S. 52.
13 Schulz G. V., Harbor th G., Angew. Chem., 1947, Bd. 59. S. 90.
14. Schulz G. V., Henri ci G.. Olive S., Z. Elektrochem., Ber. Bunsenges.
Physik. Chem., 1956. Bd. 60 S. 303.
15. Schuller H.. Angew. Makromol. Chem., 1968, Bd. 2, S. 64.
16. Houben-Weyl, 1963. Bd. 14/2, S. 18.
17. Сёрнсон У., Кемпбел T. Препаративные методы химии полимеров
М., Издатинлит, 1968. 564 с.
18. С h а р i г о A., J. Polymer Sci., 1964, v. С4, р. 1551.
19. Houben-Weyl, 1961. Bd. 14/1, S. 783; Kennedy J. P., Langer A. W.,
Advances Polymer Sci., 1964, v. 3, p. 508; Plesch P. H., «The Chemistry of
Cationic Polvmerisation». Oxford. 1963.
20. Houben-Weyl, 1963, Bd. 14/2, S. 18.
21. Morgan P. W., Kwoler S L., J. Polymer Sci., 1964, v. A2, p. 181, 209,
2693; Морган П. Поликонденсационные процессы синтеза полимеров. Л.,
«Химия», 1970. 426 с.
22. Houben-Weyl, 1961, Bd. 14/1, S. 133.
23. Breitenbach J. W., Edelhauser H., Makromol. Chem., 1961, Bd. 44/46,
S. 196.
24. В a г t h о 1 о m e E., G e r r i n s H., H e r b e с к R., Weitz H. M., Z. Elektro-
chem., 1956, Bd. 60, S. 334; Ger rens H., Advances Polymer Sci., 1959, v. 1,
p. 234.
25. Houben-Weyl, 1961, Bd. 14/1, S 190.
26. В a r 11 H., Bonin W., Makromol. Chem., 1962, Bd. 57, S. 74.
27. Wintgen R., Kolloid-Z., 1951, Bd. 124, S. 141.
28. J а а с к s V., В a a d e r H., Kern W., Makromol. Chem., 1965, Bd. 83, S. 56.
Hermann H. D., Fischer E., Weissermel K., Makromol. Chem.,
1966, Bd. 90, S. 1.
29. Flory P. J., J. Am. Chem. Soc., 1939, v. 61, p. 1518; 1942, v. 64. p. 177.
30. Kern W., Schulz R. C., Angew. Chem., 1957. Bd. 69, S. 168.
31. S c h u I z R. C., Angew. Chem.. 1964, Bd. 76. S. 357.
32. S c h u 1 z R. C., Sabel A., Makromol. Chem., 1954, Bd. 14, S. 115.
33. Ho u b e n - W e у 1, 1958, Bd. 1/1, S. 653.
34. Houben-Weyl, 1958, Bd. 1/1, S. 939.
35. Schulz R. C., Chem. Ing. Tech., 1956. Bd. 28, S. 296.
36. Фойгт И. Стабилизация синтетических полимеров против действия света и
тепла. Л., «Химия», 1972, 544 с.
37. Houben-Weyl, 1961, Bd. 14/1, S. 441.
38. Hoffman M., Schneider P., in Houben-Weyl, 1963, Bd. 14/2,
S. 917, 960.
39. N i t s c h e - W о 1 f, «Praktische Kunststoffprtifung», 1961.
40. Kern W., Angew. Chem., 1952, Bd 71, S. 585.
41. G n a m m, «Losungsmittel und Weichmachungsmittel», Wissenschaftliche Ver-
lagsanstalt, Stuttgart, 1972.
42. Schulz G. V., Angew. Chem., 1952, Bd. 64, S. 553.
108
43. Munster A., «Loslichkeit und Quellung» in: Physik der Homopolymeren,
. 1953, Bd. 2, S. 193.
44. Fuchs O., Kunststoffe, 1959, Bd. 43, S. 409; Makromol. Chem., 1956,
Bd. 18/19, S. 166.
45. Fuchs O., Farbe und Lack, 1965, Bd. 71, S. 104.
46. Fuchs O., Makromol. Chem., 1966, Bd. 90, S. 293; Angew. Makromol. Chem.,
1967, Bd. 1, S. 29.
47. S c h u 1 z G. V., Schon K. G., Z. Physik Chem., 1954, Bd. 2, S. 197.
48. Houben-Weyl, 1955, Bd. 3/1, S. 371; Stuart H. A., Physik der Hoch-
polymeren, 1953, Bd. 2, S. 373.
49. S t a u d i n g e г H., «Die Hochmolekularen Organischen Verbindungen»,' Ber-
lin, 1932. S. 52.
50. Stuart H. A., Physik der Hochpolymeren, 1953, Bd. 2, S. 280.
51. Houben-Weyl, 1955, Bd. 3/1, S. 431.
52 Houben-Weyl, 1961, Bd. 4/1, S. 83.
53. H i n к a m p P. E., Polymer, 1967, v. 8, p. 381.
54. Kern W., Z. Physik. Chem., 1938, Bd. 181. S. 249, 283: 1939. Bd. 184, S. 197,
302; Stuart H. A., Physik der Hochpolymeren, 1953, Bd. 2, S. 321, 680,
695.
55. Schulz G. V., Blaschke F., J. Prakt. Chem., 1941, Bd. 158, S. 130.
56. M e у e г h о f f G., Z. Physik. Chem., 1955, Bd. 4, S. 335.
57. M с С о r m i к H. W., J. Polymer Sci., 1959, v. 41, p. 327.
58. Krigbaum W. R., Flory P. J., J. Polymer Sci., 1953, v. 11, p. 37.
59. R i b e у г о 11 e s P., Guyot A., Benoit H., J. Chim. Phys., 1959, v. 56,
p. 377.
60. С a r t e r W. C., Scott R. L., Ma gat M., J. Am. Chem. Soc., 1946, v. 68r
p. 1480.
61. Matsumoto Ohyanagi Y., J. Polymer Sci., 1960, v. 46, p. 441.
62. D i a 1 e г K-, Vogler K., Pa tat F., Helv. Chim. Acta, 1952, v. 35, p. 869.
63. C h i n a i S. N., M a 11 a к J. D., R e s n i к A. L., Samuels R. J., J. Po-
lymer Sci., 1955, v. 17, p. 391.
64. Cleland R. L., S toe km a yer W. H., J. Polymer Sci., 1955, v. 17,
p. 473.
65. S c h о 11 a n W., Makromol. Chem., 1954, Bd. 14, S. 169.
66. С о n i x A., Makromol. Chem., 1958, Bd. 26, S. 226.
67. S c h u I z G. V., Horbach A., Makromol. Chem., 1959, Bd. 29, S. 23.
68. Elias H. G., Schumacher R., Makromol. Chem., 1964, Bd. 76, S. 23.
69. Stuart H. A., Physik der Hochpolymeren, 1953, Bd. 2, S. 304.
70. M e у e r h о f f G., Advances Polymer Sci., 1961, v. 3, p. 59.
71. Polymer Handbook, New York, 1966, Seite IV—I.
72. Houben-Weyl, 1955, Bd. 3/1, S. 435.
73. Kern W., Kammerer H., Makromol. Chem., 1948, Bd. 2, S. 127.
74. Bevington J. C., Advances Polymer Sci., 1960, v. 2, p. 1.
75. В e v i n g t о n J. C., J. Polymer Sci., 1956, v. 20, p. 133; 1956, v. 22, p. 257.
76. Bevington J. C., Trans. Faraday Soc., 1955, v. 51, p. 1392.
77. Kammerer H., Rocaboy F., Steinfort K.-G., Kern W., Makro-
mol. Chem., 1962, Bd. 53, S. 80.
78. К e r n W.. Munk R., S a b e 1 A., Schmidt К. H., Makromol. Chem.,
1955/1956, Bd. 17, S. 201, 219.
79. Koch T. A., Lindvig P. E., J. Appl. Polymer Sci., 1959, v. 1, p. 164.
80. Can tow M. J., «Polymer Fractionation», New York, 1967.
81. M e у e r h о f f G., Z. Elektrochem., 1957, Bd. 61, S. 325.
82. H e n g s t e n b e r g J., Z. Elektrochem., 1956, Bd. 60. S. 236.
83. Fuchs O., Makromol. Chem., 1951, Bd. 5, S. 245, Bd. 7, S. 259.
84. D e s r e u x V., Rec. Trav. Chim. Pays-Bas, 1949, v. 68, p. 789.
85. Baker C. A., Williams R. J. P., J. Chem. Soc., 1956, p. 2352.
109
86. Determann H., «Gelchromatographie», Berlin — Heidelberg — New York,
1967; Seidl J., Malinski J., Dusek K-, Heitz W., Advances Poly-
mer Sci., 1967, v. 5, p. 113; Can tow M. J. R., «Polymer Fractionation», New
York, 1967; Heitz W., Kern W., Angew. MakromoL Chem., 1967, Bd. 1,
S. 150.
87. Hou ben-We у 1, 1955, Bd. 3/1, S. 445; Stuart H. A., Physik der Hoch-
polymeren, 1953, Bd. 2, S. 355, 726.
88. Fuchs O., MakromoL Chem., 1962, Bd. 58, S. 65.
89. C a n t о w H. J., Fuchs O., MakromoL Chem., 1965, Bd. 83, S. 244.
90. Stuart H. A., Physik der Hochpolymeren, 1955, Bd. 3, S. 637.
91. Polymer-Handbook, 1966, New York, p. Ill—161.
92. Хувинк P., Ставерман А. Химия и технология полимеров. Л., «Хи-
мия», 1965, т. I, 676 с.; 1966, т. II. 1124 с.
93. Н о u b е n - W е у 1, 1953, Bd. 2, S. 788.
94. Polymer Reviews, 1964, v. 6, p. 347; High Polymers, 1962, v. 12/2, p. 159;
Murphy С. B., Analyt. Chem., 1962, Bd. 34, S. 298; Schultze R. D.,
Differentalthermoanalyse, Verlag Chemie, Weinheim, 1969.
95. Kern W., «Die hochmolekularen organischen Verbindungen», 1932, S. 224.
96. Rothe M., Polymer Handbook, p. 111-1, 1966.
97. Z a h n II., G I c i t s m a n n G. B., Angew. Chem., 1963, v. 75, p. 772.
98. Philippoff W., «Viskositat der KoTloide», Steinkopff — Verlag, 1942; Se-
vers E. T., «Rheology of Polymers», Rheinhold PubL, New York, 1962;
S e m j о n о w V., Kunststoffe, 1966, Bd. 56, S. 7.
99. Z a c h m a n n H. G., Stuart H. A., MakromoL Chem., 1961, Bd. 44/46,
S. 622; Stuart H. A., Physik der Hochpolymeren, 1955, Bd. 3, S. 244.
100. Braun D., Betz W., Kern W., Naturwissenschaften, 1959, Bd. 46,
S. 444.
101. Houben-Wey 1, 1955, Bd. 3/1, S. 188.
102. Weissberger A., «Technique of Organic Chemistry», 1959, Part 1.
p. 182.
103. Jellinek H. H. G., «Degradation of Vinyl Polymers», New York, 1955;
Грасси H. Химия процессов деструкции полимеров. М., Издатинлит, 1959.
214 с.; Hou ben-Weil, 1963, Bd. 14/2, S. 917.
104. Ch ap i ro A., «Radiation Chemistry of Polymeric Systems», High Polymers,
1962, v. 15.
105. Achhamer B. G., Tryon M., Kline G. M., Kunststoffe, 1959, Bd. 49,
S. 600.
106. Voigt J., Kunststoffe, 1961, Bd. 51, S. 18,314.
107. Braun D., Farbe und Lack, 1963, Bd. 69, S. 820.
108. Burg К. H., Fischer E., Weissermel K., MakromoL Chem., 1967,
Bd. 103, S. 268.
109. Ch er dr on H., Hohr L., Kern W., Angew. Chem., 1961, Bd. 73,
S. 215.
110. Schurz J., Stiibchen H., Z. Elektrochem., 1957, Bd. 61, S. 754.
111. К e r n W., Braun D., MakromoL Chem., 1958, Bd. 27, S. 23.
112. N e w e 11 I. E., Ana’yt. Chem., 1951, Bd. 23, S. 445.
113. Krimm S., Advances Polymer Sci., 1960, v. 2, p. 51.
114. H и m m e I - S c h о 1 1, «At as der Kunststoff-Analyse», Munchen, 1968.
115. Kammerer H., Rocaboy F., Stienfort K-G., Kern W., MakromoL
Chem., 1962, Bd. 53, S. 80.
H6. Schnell G., Ber. Bunsenges. Physik. Chem., 1966, Bd. 70, S. 297.
H7. Houben-Weyl, 1961, Bd. 14/1, S. 610.
H8. Gossl T., MakromoL Chem., 1960, Bd. 42, S. 1.
119. Stuart H. A., Physik der Hochpolymeren, 1955, Bd. 3; Zachmann H. G.,
Advances Polymer Sci., 1964, v. 70, p. 581.
120. Bovey F. A., Tiers G. V. D., Advances Polymer Sci., 1963, v. 3, p. 139;
Johnson LL, Ber. Bunsenges. Physik. Chem., 1966, Bd. 70, S. 320.
121. Houben-Weyl, 1963, Bd. 14/2, S. 918.
t’22. S t a и d i n ger - К e r n - К a m m e r er, «Anleitung zur organischen qualita-
tiven Analyse», Berlin — Heidelberg — New York, 1968.
HO
123. Gattermann-Wieland, «Die Praxis des organischen Chemikers», Ber-
lin, 1960.
124. Heinze D., MakromoL Chem., 1967, Bd. 101, S. 166.
125. Dexheimer H., Fuchs O., MakromoL Chem., 1966, Bd. 96, S. 172.
126. N i t s c h e - W о I f, «Praktische Kunststoffpriifung», 1961; Carlowitz B.,
«Tabellarische Ubersicht uber die Priifung von Kunststoffen», Frankfurt (Main),
1966; Хувинк P., Ставерман А. Химия и технология полимеров. Л.,
«Химия», 1965, т. I. 676 с.; 1966, т. II. 1124 с.
127. Nitsche-Wolf, «Praktische Kunststoffpriifung», 1961, S. 24.
128. Dasch J., Kunststoffe, 1967, Bd. 57, S. 117; Winter ger st S., Kunststof-
fe, 1967, Bd. 57, S. 188.
129. Nitsche-Wolf, «Praktische Kunststoffpriifung», 1961, S. 148.
130. Schmieder K., Wolf K., Kolloid-Z., 1952, Bd. 127, S. 65; 1953, Bd. 134,
S. 149.
131. Bohn L., Kolloid-Z., 1966, Bd. 213, S. 55.
132. Nitsche-Wolf, «Praktische Kunststoffpriifung», 1961.
133. Сёрнсон У., Кемпбел T. Препаративные методы химии полимеров.
М., Издатинлит, 1968. 564 с.
134. Сёрнсон У., Кемпбел Т. Препаративные методы химии полимеров.
М., Издатинлит, 1968. 564 с.; Houben-Weyl, 1963, Bd. 14/2, S. 138.
3. СИНТЕЗ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
МЕТОДАМИ ПОЛИМЕРИЗАЦИИ
3.1. СВОБОДНОРАДИКАЛЬНАЯ ГОМОПОЛИМЕРИЗАЦИЯ
Начальной стадией свободнорадикальной полимеризации является
стадия инициирования, в результате которой образуются свободные
радикалы. Свободные радикалы могут быть получены при нагрева-
нии самого мономера, однако это достоверно установлено только
для стирола (опыт 3-01). Обычно радикалы получают при терми-
ческом разложении инициаторов, иногда называемых также ката-
лизаторами. Свободные радикалы реагируют с мономером, возни-
кающие реакционноспособные частицы последовательно присоеди-
няют молекулы мономера, что приводит к образованию макроради-
калов. Этот процесс продолжается до тех пор, пока реакция роста
не прекратится. Обрыв растущих цепей происходит в результате
рекомбинации или диспропорционирования двух макрорадикалов.
Таким образом, свбодные радикалы, которые получаются при раз-
ложении инициатора, входят в состав макромолекул в качестве
концевых групп. Образующиеся при этом макромолекулы содержат
один или два концевых фрагмента инициатора, что зависит от ме-
ханизма реакции обрыва.
Если в системе возможно протекание реакции передачи цепи,
то рост макрорадикалов ограничивается их взаимодействием с мо-
лекулой агента передачи ZH. В результате этой реакции обрузует-
ея свободный радикал Z*, способный начать новую цепь. В этом
случае один первичный радикал инициатора может привести к об-
разованию нескольких полимерных цепей, которые получаются в
результате роста одной кинетической цепи. Таким образом, длина
кинетической цепи (т. е. среднее количество молекул мономера,
заполимеризовавшихся под действием одного первичного радика-
ла) может быть много больше степени полимеризации полученных
макромолекул. Кинетическую схему радикальной полимеризации
можно представить следующим образом.
Инициатор—(первичный акт)
Рост цепи:
R. +СН2=СН --> R—СН2— СН
X X
R—СН2—СН + лСН2=СН > R—Г—СН2—СН—СН2—СН
1Я2
Передача цепи:
R—Г—СН2—СН—1—СН2—СН + ZH
R—Г—СН2—СН—“l—СН2—СН2 4- Z
X Jn X
Обрыв рекомбинацией:
R-[--CH2-CH---СН2—СН + R—f-CHa—СН— ]-СН2-СН
X
X
X
X
R—Г—СН2—СН—1—R
т
. п
X
п+т+2
Обрыв диспропорционированием:
R—Г—СН2—СН—]—СН2—СН + R—
-СН2-СН-]-СН2-СН ---->
I I
X \т X
-СН2-СН-]-СН2-СН2
I I
X J,„ X
Таким образом, процесс свободнорадикальной гомополимериза-
ции состоит из трех стадий: инициирования, роста и обрыва поли-
мерных цепей (реакция передачи цепи пока не учитывается). Ско-
рость инициирования при разложении инциатора I описывается сле-
дующим уравнением:
^ин = [I] (3-1)
Константа скорости инициирования Дин содержит поправочный
фактор, учитывающий эффективность инициирования, поскольку
не все свободные радикалы, которые образуются при разложении
инициатора, вызывают полимеризацию. Некоторые из них могут
рекомбинировать или участвовать в побочных реакциях. Эффектив-
ность инициирования определяется отношением числа молекул ини-
циатора, начавших рост полимерных цепей, к общему числу рас-
павшихся в условиях полимеризации молекул инициатора. Для
большинства инициаторов это отношение лежит в пределах 0,6—0,9,
что зависит также от природы исходного мономера.
Скорость второй стадии — роста цепи выражается следующей
формулой:
d ГМ]
fP-= (3-2)
где [R*] — суммарная концентрация радикалов в системе.
Это уравнение основано на допущении о равенстве скоростей всех
последовательных актов роста цепи между собой, поскольку актив-
ность радикалов предположительно не зависит от длины цепи.
Скорость реакции обрыва цепи описывается уравнением
^об = IR-12 (3-3)
Согласно принципу Боденштейна скорости образования и гибели
радикалов равны друг другу для стационарных цепных реакций
8—732
ИЗ
(условие стационарности). Это справедливо для большинства поли-
меризационных процессов, по крайней мере при невысоких степе-
нях превращения. При выполнении этого условия иин и иОб можно
приравнять:
^ин = Уоб или £ин [I] = £Об [R• I2 (3-4)
Отсюда следует, что общая концентрация радикалов в системе
равна
[R-] = ()’/2 [1]1/2 (3-5)
Подставляя это выражение для [R-] в уравнение, определяющее
скорость реакции роста (3-2), получим:
d [М] / £ин \V2 1Z
(3-6)
Здесь ир идентична общей скорости полимеризации, поскольку
при достаточно большой длине цепей мономер расходуется только
в реакции роста. Таким образом, скорость полимеризации пропор-
циональна концентрации мономера в первой степени и корню квад-
ратному из концентрации инициатора. При полимеризации в массе,
когда [М] практически постоянна на начальных стадиях превра-
щения
vp ~ [1]1/а (3-7)
Если полимеризацию инициируют УФ-лучами или облучением
частицами высокой энергии, концентрацию инициатора в рассмат-
риваемых кинетических уравнениях заменяют энергией излучения.
Повышение температуры свободнорадикальной полимеризации
приводит к увеличению суммарной скорости полимеризации, так
как изменение температуры в первую очередь сказывается на ско-
рости разложения инициатора, а следовательно, и на числе ради-
калов, генерируемых в единицу времени. В то же время средняя
степень полимеризации образующегося полимера уменьшается, так
как согласно уравнению (3-3) скорость обрыва цепи также зависит
от концентрации радикалов в системе (см. опыт 3-02). Кроме того,
повышение температуры благоприятствует побочным реакциям, на-
пример реакциям передачи и разветвления. При полимеризации ди-
енов температура реакции оказывает влияние на микроструктуру
повторяющихся звеньев в цепи полимера.
Несмотря на то что приведенные уравнения несколько упроще-
ны, они достаточно точно описывают кинетические закономерности
многих реакций полимеризации [1—4]. Положение изменяется, од-
нако, если полимеризация протекает в гетерогенных условиях, на-
пример суспензионная или эмульсионная полимеризация (см. ли-
тературные ссылки, приведенные в разделе 2.1.5.3).
При свободнорадикальной полимеризации в массе наблюдаются
особые закономерности, если степень превращения превышает неко-
114
торое значение. Вследствие повышения концентрации полимера в
системе вязкость реакционной смеси увеличивается, приводя к
уменьшению подвижности растущих макрорадикалов. В результа-
те скорость обрыва существенно снижается, в то же время скорость
реакции роста остается практически постоянной, так что скорость
полимеризации и степень полимеризации образующегося полимера
одновременно возрастают. Система более не находится в стационар-
ных условиях, и концентрация свободных радикалов возрастает.
Увеличение скорости реакции ухудшает отвод теплоты полимериза-
ции, что вызывает повышение температуры реакционной системы.
При этом реакция автоускоряется, что может привести к термиче-
скому взрыву. Это явление автоускорения, известное под названием
гель-эффекта или эффекта Троммсдорфа, особенна отчетливо выра-
жено при полимеризации метилметакрилата (см. опыт 3-14А). Ана-
логичный эффект наблюдается и при полимеризации некоторых мо-
номеров в плохих растворителях, в которых макромолекулы сверну-
ты более плотно, чем в хороших растворителях; в последних поли-
мерные клубки набухают в большей степени (о качестве раствори-
телей см. раздел 2.3.1).
Скорости реакций образования радикалов, их роста и обрыва
определяют суммарную скорость полимеризации и в отсутствие ре-
акции передачи цепи — степень полимеризации образующегося по-
лимера. В результате реакций передачи цепи образуется насыщен-
ная полимерная цепь, одновременно получается новый радикал,
способный начать рост цепи, а следовательно, и продолжить рост
кинетической цепи. Длина кинетической цепи определяется числом
молекул мономера, заполимеризовавшихся в результате одного акта
инициирования. Поскольку эффективность большинства инициато-
ров точно неизвестна, длину кинетической цепи обычно измеряют
отношением скоростей роста и инициирования или обрыва. В от-
сутствие реакции передачи цепи и при условии обрыва по механиз-
му диспропорционирования длина кинетической цепи v идентична
средней степени полимеризации:
as V = Vp/UHH = Vp/Uo6 (3-8)
Если, однако, обрыв осуществляется таким путем рекомбина-
ции двух радикалов, то средняя степень полимеризации равна уд-
военной длине кинетической цепи:
Рп = 2v = 2рр/уин = 2vp/vo6 (3-9)
Если в системе возможно протекание реакции передачи цепи
(R-+ZH = RH + Z-) и ее скорость определяется уравнением
^пер == ^пер [R*l [ZH] (3-10)
то можно получить следующие соотношения для средней степени
полимеризации образующегося продукта.
Для диспропорционирования:
8*
115
Для рекомбинации:
Если теперь перейти к обратной величине Рп и подставить значе-
ния ир, иин и иПер из уравнений (3-1), (3-6) и (3-10), то
1____**ин . ?пер________^ин ГЛ_____ , ^пср [7Н1
?„-<№₽+ Vp - + *plMJ
где а равна 1 или 2 для обрыва цепей, протекающего по механизму диспропор-
ционирования или рекомбинации соответственно.
Поскольку мономер, так же как и инициатор, растворитель L,
введенный агент передачи цепи (регулятор ZH) и образовавшиеся
полимерные цепи, может участвовать в реакции передачи цепи, то
уравнение (3-13) примет вид
ь ш1'2 , г , r IB , r Tlj" [ZH]
Рп [М] +См+С« [MJ + Се[мГ+С2Н~ТмГ+"‘ (3’,4)
где константы передачи С соответствующих передающих агентов
определяются отношениями йцер/&р (см. опыт 3-14). Константа пе-
редачи цепи через большинство инициаторов приблизительно рав-
на 0, поэтому из уравнения (3-14) следует, что при невысоких кон-
версиях обратная средняя степень полимеризации пропорциональна
корню квадратному из концентрации инициатора. В свою очередь,
скорость полимеризации пропорциональна [I]0,5, поэтому средняя
степень полимеризации уменьшается с ростом скорости полимери-
зации.
Особое значение имеют реакции передачи цепи на растворитель
и на соединения, получившие название регуляторов, ввиду
их большого влияния на молекулярные массы образующихся поли-
меров. В то время как для большинства растворителей константы
передачи невелики (например, константа передачи цепи радикала-
ми роста полистирола на бензол при 60 °C имеет порядок 10~5), су-
ществует ряд растворителей, имеющих относительно высокие кон-
станты передачи цепи. Например, при полимеризации стирола в
четыреххлористом углероде константа передачи цепи полистироль-
ными радикалами на этот растворитель равна примерно 10-2, поэ-
тому образующийся полистирол имеет очень низкую степень поли-
меризации и состоит из смеси олигомеров. Реакции передачи цепи
можно описать следующими уравнениями:
R. + СС1, -> R-C1 + .СС13 .СС13 + М -> СС13-М.
Образующиеся макромолекулы содержат на концах атом хло-
ра или трихлорметильную группу. Аналогичным образом при поли-
меризации этилена в четыреххлористом углероде образуются оли-
гомеры следующего строения: С1—(СН2—СН2)П—СС13 (где
п = 2, 3 и 4), которые при реакции гидролиза могут быть превра-
щены в соответствующие со-карбоновые кислоты. Реакции образова-
на
ния олигомеров при полимеризации получили название' реакйяв
теломеризации [5—6].
Возможность получения низкомолекулярных полимеров с по-
мощью регуляторов имеет важное значение, так как управление мо-
лекулярной массой полимера путем изменения температуры реак-
ции либо концентрации мономера и инициатора возможно лишь в
узких пределах. Поэтому регуляторы молекулярной массы, пред-
ставляющие собой агенты передачи цепи, широко используются в
производственной практике. Среди них чаще всего применяют мер-
каптаны, которые необходимо добавлять в очень малых количест-
вах (около 0,1% по отношению к мономеру) из-за их высокой ак-
тивности в реакции передачи цепи (например, при полимеризации
стирола Сп=19 для н-додецилмеркаптана). Константу передачи це-
пи на регуляторы обычно определяют по уравнению (3-14) (для
полимеризации при различной концентрации регулятора и при по-
стоянных концентрациях мономера и инициатора).
В отсутствие регулятора ZH
1 ь |1|1/2 л. г л. г [П Д.Г 14 И 1-П
р„,о “*W+ m+ c,W + Cl W (3 5)
В присутствии регулятора
1 1 [ZHJ
Зависимость обратной степени полимеризации \/Рп (среднечисло-
вой молекулярной массы) полимеров, полученных в присутствии
регулятора с различной концентрацией, относительно величины
[ZH]/[M] представляет собой прямую линию, которая отсекает
на ординате отрезок, равный 1/7\,о; тангенс угла наклона прямой
позволяет определить константу передачи цепи Czh регулятора
(см. раздел 2.1.5.4. и опыт 3-14).
Наконец, имеется большое число соединений, оказывающих ин-
гибирующее влияние на полимеризацию. Такими соединениями яв-
ляются молекулярный кислород, окись азота, фенолы — гидрохи-
нон, трет-бутилкатехин, некоторые ароматические амины, например
N-фенил-р-нафтиламин, нитросоединения и ряд серосодержащих
соединений. Механизм действия большинства ингибиторов точно
не установлен. Ингибитор может реагировать либо с инициирую-
щим радикалом, либо с растущей цепью с образованием продуктов,
не способных к дальнейшему присоединению молекул мономера.
Стабильные свободные радикалы, как, например, дифенилпикрил-
117
также являются ингибиторами. Поскольку этот радикал ярко ок-
рашен, за его исчезновением во время ингибирования удобно сле-
дить фотометрически [7]; в этом случае, однако, реакция ингиби-
рования, по-видимому, не является простой рекомбинацией стабиль-
ного свободного радикала с активным концом растущей цепи.
Ингибиторы часто используют для быстрого прекращения по-
лимеризации, например при кинетических исследованиях. Другим
важным применением ингибиторов является их использование для
стабилизации мономеров во время хранения. Под действием кисло-
рода воздуха в результате автоокисления могут образоваться пе-
рекиси, которые медленно разлагаются даже при низких темпера-
турах, генерируя свободные радикалы и инициируя полимериза-
цию. Для подавления преждевременной полимеризации в мономер
вводят ингибитор в качестве стабилизатора (см. раздел 2.1.5.4),
который, конечно, необходимо удалять перед полимеризацией мо-
номера (см., например, опыт 3-01). Эффективность действия инги-
биторов прежде всего зависит от их строения. Поскольку ингиби-
тор расходуется в реакции с растущими радикалами, продолжи-
тельность действия ингибитора, т. е. индукционный период, зави-
сит от концентрации ингибитора (см. опыт 3-19).
Молекулярный кислород играет особую роль при полимериза-
ции. Как известно, кислород довольно быстро реагирует с углево-
дородными радикалами с образованием перекисных радикалов:
---R- 4- О2 ->----R—О—О-
Поэтому, естественно, что кислород необходимо удалять при сво-
боднорадикальной полимеризации. Перекисные радикалы значи-
тельно менее активны, чем свободные углеводородные радикалы,
тем не менее они способны к дальнейшему присоединению моле-
кул мономера с повторным образованием углеводородных радика-
лов. Последние вновь могут реагировать с кислородом. Таким обра-
зом, полимеризация в присутствии кислорода значительно замед-
ляется по сравнению с реакцией в отсутствие кислорода. В основ-
ном происходит чередующееся присоединение молекул мономера и
кислорода, в результате которого образуются полимерные перекиси
(сополимеризация с молекулярным кислородом):
---СН2—СН—О—-О—СН2—СН—О—О—• • •
I I
X X
Только после полного исчерпания молекулярного кислорода начи-
нается обычная полимеризация. Однако дополнительное термичес-
кое разложение присутствующих перекисей на активные радикалы
увеличивает скорость реакции. Следовательно, молекулярный кис-
лород вначале ингибирует полимеризацию, а затем после его ис-
черпания увеличивает скорость реакции.
В отличие от ионной полимеризации свободнорадикальная поли-
меризация осуществляется только для ненасыщенных соединений.
118
Эта реакция может быть инициирована термически, УФглучами
или другим более жестким излучением. Обычно используют пере-
кисные инициаторы или азосоединения, а также окислительно-
восстановительные системы.
3.1.1. Полимеризация, инициированная перекисными
соединениями
Органические и неорганичесие перекисные соединения широко ис-
пользуются в качестве инициаторов свободнорадикальной полиме-
ризации. Наибольшее распространение получили следующие орга-
нические перекисные соединения: гидроперекиси, диалкильные и
диацильные перекиси, а также некоторые перэфиры. Поскольку эти
соединения растворимы не только в органических растворителях,
но и в большинстве мономеров, они могут использоваться при про-
ведении полимеризации в растворах, в массе, а также при суспен-
зионной полимеризации. Разложение этих соединений на свободные
радикалы может осуществляться либо нагреванием, либо облуче-
нием, либо с помощью окислительно-восстановительной реакции
(см. раздел 3.1.3). Скорость разложения органических перекисных
соединений зависит от их строения и температуры. Гидроперекиси
обычно менее стойки, чем диацильные или диалкильные перекиси.
Полимеризация, инициированная термическим разложением орга-
нических персоединений, как правило, протекает с обычными ско-
ростями лишь при температурах выше 50 °C. Исключение состав-
ляют некоторые перэфиры (например, диэтилпероксидикарбонат),
быстро распадающиеся даже при комнатных температурах, вслед-
ствие чего их следует использовать только в разбавленных рас-
творах.
Одной из наиболее широко применяемых перекисей является пе-
рекись бензоила, которую вводят в реакционеную смесь в количестве
0,1—1,0% (масс.) (см. опыты 3-02, 3-05 и 3-06). В интервале тем-
ператур 50—80 °C перекись бензоила в растворах распадается в
основном с получением бензоатных радикалов, от которых при бо-
лее высоких температурах отщепляется двуокись углерода, образуя
фенильные радикалы:
Таким образом, концевыми группами образующихся макромо-
лекул являются либо способные гидролизоваться бензоатные груп-
пы, либо неспособные гидролизоваться фенильные группы.
119
Хлор- или бромзамещенная перекись бензоила распадается зна-
чительно быстрее незамещенной перекиси. Замещенные перекиси
используются также для введения в макромолекулы аналитиче-
ски определяемых галогенсодержащих концевых групп с целью
облегчения измерения молекулярной массы полученного полимера
(см. раздел 2.3.2.2). При использовании перекиси бензоила и боль-
шинства ее замещенных производных выполняются соотношения
(3-6) и (3-8), согласно которым скорость полимеризации возра-
стает, а степень полимеризации уменьшается с увеличением кон-
центрации инициатора.
Органические перекисные соединения в основном применяются
при полимеризации в массе или в органических растворителях, в
то время как неорганические перекисные соединения преимущест-
венно используются для инициирования полимеризации в водных
растворах, в эмульсиях или в суспензиях. Перекись водорода, как
правило, используют при окислительно-восстановительном иници-
ировании (см. опыт 3-22). Персульфаты калия и аммония часто
употребляют без восстановителей, поскольку они распадаются уже
при температурах около 30 °C с образованием свободных радика-
лов, инициирующих полимеризацию:
О О
II II
-о—s—о—o-s—о- —> 2.so;
Il II
о о
2.so; 4- 2Н2О -> 2HSO7 4- 2НО-
Персульфат аммония растворяется в воде лучше, чем персульфат
калия; кроме того, он растворим в некоторых полярных органиче-
ских растворителях (например, в диметилформамиде), поэтому он
используется иногда для инициирования полимеризации в органи-
ческих средах. Поскольку при полимеризации, инициированной пер-
сульфатами, реакционная среда становится кислой, обычно в си-
стему добавляют буфер (см. опыт 3-20).
При полимеризации в органических растворителях, иницииро-
ванной перекисными соединениями, необходимо учитывать проте-
кание реакций передачи цепи на растворитель, которые приводят
к уменьшению степени полимеризации. Растворителями с малой
константой передачи цепи являются бензол и циклогексан. Соглас-
но уравнениям (3-6) и (3-14) как скорость полимеризации, так и
степень полимеризации образующегося полимера понижаются при
уменьшении концентрации мономера при постоянной концентрации
инициатора.
В табл. 9 приведены некоторые наиболее распространенные
перекисные инициаторы.
Необходимо отметить, что любой мономер, способный полимери-
зоваться по радикальному механизму, может быть заполимеризо-
ван с помощью любого перекисного или азосоединения, распада-
ющегося на свободные радикалы, т. е. инициатор в основном опре-
120
Таблица 9. Перекисные соединения, используемые
для инициирования свободнорадикальной полимеризации
Перекисные соединения Формула Температура применения, С
Перекись водорода н—О—о-н О О II II 30-80
Персульфат калия КО — S — О—О — S — ОК II II О о 30—80
Перекись бензоила С,Н&С(О)ООС(О)С,Н4 40—100
Гидроперекись кумола СеН5С(СН3)2ООН 50-120
Ди- грет-бутилперекись С(СНз)зООС(СН^8 80—150
деляет скорость и степень полимеризации, а также тип концевых
групп и степень разветвленности. Однако инициатор не влияет на
способность мономера к полимеризации. Это положение не выпол-
няется для окислительно-восстановительных систем, тем более в
случае ионных инициаторов. Таким образом, для выяснения спо-
собности данного ненасыщеного соединения (тщательно очищенно-
го) к радикальной полимеризации его необходимо нагревать в те-
чение некоторого времени в присутствии 1% (масс.) перекиси бен-
зола (или персульфата калия при работе с водным раствором) при
температуре 50—120 °C в атмосфере азота.
Опыт 3-01. Термическая полимеризация стирола в массе (влияние темпера-
туры)
Технический стирол отмывают от фенольного ингибитора, встряхивая его
2 раза с 10%-ным раствором едкого натра, а затем мономер 3 раза промывают
дистиллированной водой. После этого мономер сушат над прокаленным хлори-
дом кальция или силикагелем. Высушенный мономер перегоняют при понижен-
ном давлении (т. кип. 82 °C при 100 мм рт. ст. или 46 °C при 20 мм рт. ст.).
Перегнанный мономер до использования хранят в холодильнике.
В 5 ампул объемом по 15—20 мл помещают по 4 г (38,4 ммоля) очищен-
ного стирола. Ампулы через переходник (см. раздел 2.1.3) поисоединяют к водо-
струйному насосу и помещают в охлаждающую смесь из хлористого метилена
и сухого льда (т. пл. стирола —30,6 °C).
После того как мономер замерзнет, ампулу откачивают на водоструйном
насосе, содержимое размораживают и в ампулу подают азот. Эту операцию
повторяют 2 раза и ампулу запаивают под азотом. Приготовленные образцы
полимеризуют при 80, 100, 110, 120 и 180 °C, для чего ампулы погружают в тер-
мостат или баню с соответствующей температурой (меры предосторожности:
ввиду возможного взрыва ампул их нагревание проводят за экраном). Через 6 ч
ампулы быстро охлаждают, опуская их в холодную воду (надеть защитные
очки) и вскрывают. Содержимое каждой ампулы растворяют в 20—30 мл бен-
зола и к раствору из капельной воронки постепенно добавляют 200—300 мл
метилового спирта для осаждения образовавшегося полистирола. Осадок отфиль-
тровывают и высушивают до постоянной массы в вакуумном сушильном шкафу
при 50 °C. Строят график зависимости выхода полимера (в %) от температуры
полимеризации. С помощью вискозиметра Оствальда (диаметр капилляра
0,3 мм) определяют характеристические вязкости полученных образцов в бен-
зольном растворе при 20 °C (см. раздел 2.3.2.1), рассчитывают средние степени
полимеризации и строят график их зависимости от температуры полимеризации,
121
Опыт 3-02. Полимеризация стирола в массе, инициированная перекисью бен-
зоила (влияние концентрации инициатора)
В 4 толстостенные ампулы со стандартными шлифами, с помощью которых
ампулы можно присоединять к вакуумной линии, помещают 4,7-10“3 г
(0,019 ммоля); 9,3-10-3 г (0,038 ммоля); 46,5* 10~3 г (0,19 ммоля) и 93*10~3 г
(0,38, ммоля) перекиси бензоила и по 4 г (38,4 ммоля) очищенного стирола
(см. опыт 3-01); в пятую ампулу наливают 4 г стирола без инициатора. Встря-
хиванием растворяют инициатор в мономере. К ампулам присоединяют краны,
закрепляют их с помощью пружинок, реакционные смеси вакуумируют и ампу-
лы заполняют азотом, как описано в предыдущем опыте 3-01. Образцы опус-
кают в термостат при 60 °C на 4 ч, после чего их быстро охлаждают в ледяной
воде и из реакционной смеси выделяют полимер (см. опыт 3-01). На основании
полученных результатов строят зависимость скорости полимеризации (в % пре-
вращения в ч) от корня квадратного из концентрации перекиси бензоила
{в % (мол.)] и зависимость степени полимеризации полимера от обратного
корня квадратного из концентрации инициатора. Полученные данные сопостав-
ляют с данными термической полимеризации стирола и с результатами поли-
меризации, инициированной азо-бис-изобутиронитрилом (см. опыт 3-11).
Опыт 3-03. Эмульсионная полимеризация стирола, инициированная персуль-
фатом калия
Трехгорлую колбу, емкостью 250 мл, снабженную мешалкой, термометром
и вводом для азота, откачивают и запопняют азотом (операцию повторяют
3 раза). Затем в токе азота ib колбу вводят 0,122 г (0,45 ммоля) персульфата
калия и 0,05 г NaH2PO4, 1 г олеата или лаурилсульфата натрия и 100 мл про-
кипяченной в токе азота воды. После растворения в колбу при постоянном
перемешивании вводят 50 мл очищенного от ингибитора стирола. Образовав-
шуюся эмульсию перемешивают с постоянной скоростью в слабом токе азота
в течение 6 ч при 60 °C. Затем реакционную смесь охлаждают, пипеткой отби-
рают 30 мл полистирольного латекса и переносят в химический стакан. Добав-
лением равного объема концентрированного раствора сульфата алюминия осаж-
дают полимер (если необходимо, смесь кипятят). Вторую пробу (также 30 мл)
осаждают добавлением 300 мл метилового спирта. Латекс, оставшийся в колбе,
коагулируют добавлением к нему концентрированной соляной кислоты. Полу-
ченные образцы полимеров промывают водой и метанолом, отфильтровывают
на стеклянном фильтре и высушивают до постоянной массы в вакуумном су-
шильном шкафу при 50 °C. Определяют суммарный выход полимера и харак-
теристическую вязкость (степень полимеризации) одного из образцов. Получен-
ные результаты сопоставляют с данными полимеризации в массе (см. опыты 3-01
и 3-02) и в растворе (опыт 3-13).
Опыт 3-04. Получение вспенивающегося полистирола и пенополистирола
Навеску 4 г полистирола, полученного, как это описано, например, в опы-
те 3-02 или 3-03, с 0,1% (мол.) инициатора тщательно перемешивают с раство-
ром, содержащим 6 г стирола, 0,6 г пентана и 0,08 г перекиси бензоила. Полу-
ченную массу переносят в ампулу (реакционная смесь не должна заполнять
более ’/4 объема ампулы). Содержимое ампулы замораживают при —76 °C
смесью хлористого метилена и сухого льда, ампулу запаивают в токе азота
и затем выдерживают в водяной бане при 30 °C в течение 8 суток. По истече-
нии этого срока температуру повышают до 85 °C и ампулу оставляют на 6 ч.
После охлаждения ампулу осторожно вскрывают (внутри ампулы избыточное
давление: необходимо соблюдать меры предосторожности). Способный вспени-
ваться полистирол, содержащий окклюдированный пентан, получается в виде
прозрачного стекла**.
* NaH2PO4 добавляют для создания слабощелочной среды, в которой сти-
рол л^гчше полимеризуется.
** В промышленности вспенивающийся полистирол получают аналогичным
способом для суспензионной полимеризации стирола. Гранулы полистирола,
содержащие вспенивающее вещество, нагревают паром выше температуры раз-
мягчения, в результате чего выделяется газ и материал вспенивается.
122
Для получения пенополистирола поступают следующим образом. Навеску
полимеризата около 1 г помещают в химический стакан с кипящей водой емко-
стью 500 мл. Полистирол размягчается и вспенивается под действием газообраз-
ного пентана. Образец удерживают под водой в течение 5 мин с помощью спе-
циально согнутой вилкообразной проволоки, после чего его вынимают из ки-
пятка.
Полученный вспененный материал погружают в градуированный цилиндр
с метанолом для грубой оценки объема пенопласта. После высушивания образ-
ца в эксикаторе его взвешивают и вычисляют кажущуюся плотность, которая
составляет обычно менее 0,1 г/см3 (кажущаяся плотность пробки 0,2 г/см3).
Опыт 3-05. Полимеризация метилметакрилата в массе, инициированная пере-
кисью бензоила
Этот опыт демонстрирует простой способ получения полиметилметакрилата
(органического стекла) с использованием замкнутой формы в качестве полиме-
Рис. 31. Полимеризационная форма
для получения оргстекла из полиме-
гилметакрилата:
I — метилметакрилат; 2 — стеклянные
пластинки; 3 — липкая лента; 4 — тефло-
новые бруски.
ризационного сосуда. Между двумя чистыми стеклянными пластинками (разме-
ром 10 X 10 X 0,5 см), высушенными при 80 °C, помещают в четырех углах не-
большие кусочки тефлона или поливинилхлорида толщиной примерно в 3 мм,
затем форму по торцу дважды обертывают липкой лентой. В ленте прокалывают
небольшое отверстие и готовую форму помещают в эксикатор.
Навеску 50 мг перекиси бензоила растворяют в смеси 5 г дибутилфталата
и 70 г метилметакрилата (предварительно очищенного от ингибитора и перегнан-
ного при пониженном давлении в атмосфере азота), полученный раствор филь-
труют в круглодонную колбу емкостью 250 мл. К колбе присоединяют обрат-
ный холодильник и нагревают на водяной бане до слабого кипения раствора.
Спустя 25—30 мин (продолжительность нагрева нельзя увеличивать, так как
реакционная смесь становится слишком вязкой) реакционную смесь перели-
вают в вертикально расположенную форму следующим образом: в верхнее
отверстие формы вводят капилляр (рис. 31) и с помощью водоструйного насоса
в форме создают разрежение. Одновременно форполимеризат вводят в форму
через нижнее отверстие с помощью медицинского шприца (лучше без иглы вви-
ду большой вязкости раствора). При заполнении формы необходимо избегать
образования пузырей. Отверстие в заполненной форме заклеивают и форму
помещают в печь при 45 °C на 24 ч. Для завершения полимеризации темпера-
туру печи ступенчато, через каждые 2 ч, повышают до 60, 80 и 120 °C. Затем
форму погружают в воду при 80 °C и постепенно охлаждают до комнатной
температуры. Стеклянные пластинки легко отлипают от полученного блока поли-
метилметакрилата, после того как с формы будет снята липкая лента.
Опыт 3-06. Полимеризация винилацетата в массе, инициированная пере-
кисью бензоила
Навеску 0,14 г (0,58 ммоля) перекиси бензоила растворяют в 20 г (0,23 мо-
ля) винилацетата (предварительно очищенного от ингибитора перегонкой в токе
азота). Полученный раствор хранят в холодильнике. Гриньяровскую колбу
емкостью 100 мл, снабженную обратным холодильником и капельной ворон-
кой, откачивают и заполняют азотом (операцию повторяют 3 раза). Поскольку
образующийся полимер сильно прилипает к стенкам, во избежание поломки
123
нужно использовать толстостенную колбу. Колбу нагревают на водяной бане
до 80 °C и приготовленный раствор мономера вводят в колбу через капельную
воронку с такой скоростью, чтоб раствор кипел в колбе. Одновременно в ап-
парат пропускают слабый ток азота (обратный холодильник снабжен краном,
см., например, раздел 2.1.1). После того как весь мономер введен в реакцион-
ную колбу, смесь выдерживают при 80 °C в течение 30 мин, затем температуру
повышают до 90 °C и при этой температуре колбу выдерживают еще 1 ч. Не-
заполимеризовавшийся винилацетат отсасывают в течение 10 мин при
90 °C. Затем смесь нагревают до 170 °C и вязкий расплав поливинилацетата
переносят из колбы шпателем. Оставшийся на стенках колбы полимер вымы-
вают метанолом, который добавляют порциями по 20 мл. Около 1 г полимера
растворяют в 20 мл метанола, затем полимер высаживают добавлением 6—8-
кратного количества воды. Отфильтрованный полимер высушивают в вакуумном
шкафу при 50 °C. Поливинилацетат растворим в бензоле, толуоле, ацетоне, ме-
таноле, этилаиетате и хлористом метилене. Определяют характеристическую
вязкость полученного полимера в растворе ацетона при 30 °C и рассчитывают
среднюю молекулярную массу поливинилацетата (см. раздел 2.3.2.1).
Синтезированный полимер можно использовать для получения поливинило-
вого спирта (см. опыт 5-01).
Опыт 3-07. Полимеризация винилацетата в водной дисперсии, инициирован-
ная персульфатом аммония
Трехгорлую колбу емкостью 500 мл с механической мешалкой, капельной
воронкой, обратным холодильником, термометром и вводом азота откачивают
и заполняют азотом. В колбу вносят 5 г поливинилового спирта (ем. опыт 5-01)
и растворяют при перемешивании в 100 мл дистиллированной воды при 60 °C.
К полученному раствору добавляют 2,2 г этоксилированного нонилфенола. 0.4 г
(1,8-10~3 моля) персульфата аммония и 0,46 г ацетата натрия в качестве бу-
фера для предотвращения гидролиза мономера — винилацетата. Колбу поме-
шают на водяную баню, раствор нагревают до 72 °C и, медленно по каплям,
вводят 25 г (0.29 моля) винилацетата (свежеперегнанного в токе азота) После
этого температуру бани повышают до 80 °C. Как только температура реакцион-
ной смеси достигнет 75 °C, в колбу постепенно вводят следующую порцию
винилацетата (75 г, или 0,87 моля). Скорость добавления мономера регули-
руют так, чтобы температура реакционной смеси находилась в пределах 79 —
83 °C при умеренной флегме. Введение второй порции мономера занимает в
среднем около 20 мин. Затем в колбу вводят 0,1 г (0.44 ммоля) персульфата
аммония, растворенного в 1 мл дистиллированной воды. Вскоре дефлегмация
уменьшается, и температура повышается примерно до 86 °C. Реакционную
смесь оставляют полимеризоваться еще на 30 мин при температуре бани, равной
80 °C. После охлаждения получают дисперсию кремообразной консистенции, со-
держащую менее 1 % мономера (содержание твердого вещества в полученной
массе составляет около 50%). Полимер можно высадить добавлением трехкрат-
ного количества ненасыщенного раствора хлористого натрия. Поливинилацетат-
ную дисперсию можно нанести тонким слоем на поверхность стеклянной пла-
стинки. При высыхании на воздухе полимерные частицы слипаются, образуя
гомогенную водостойкую пленку, которая крепко схватывается с поверхностью
пластины. Поскольку подобные дисперсии очень стабильны и весьма нечувст-
вительны к добавлению пигментов, электролитов, а также к умеренному изме-
нению температуры, они широко используются в качестве красителей и покры-
тий для дерева и штукатурки (дисперсионные краски), для склеивания дере-
вянных поверхностей, а также для пропитки кожи, бумаги и текстильных мате-
риалов.
Опыт 3-08. Полимеризация акрилонитрила в растворе, инициированная пер-
сульфатом аммония
А. В диметилформамиде
Навеску 0,025 г (1,1 -10~4 моля) персульфата аммония вносят в колбу
емкостью 250 мл, из которой откачивают воздух (см раздел 2 1.3). и запол-
няют азотом. В колбу заливают 90 мл диметилформамида (перегнанного в токе
азота) и встряхиванием растворяют персульфат. К полученному раствору до-
бавляют 125 мл (0,19 моля) акрилонитрила, очищенного от ингибитора перегон-
124
кой в токе азота. После этого колбу закрывают и нагревают в термостате до
50 °C. Через 48 ч бледно-желтую вязкую жидкость доиавляют по каплям при
постоянном перемешивании к 2 л воды, в результате чего полимер выпадает
в осадок в виде мелких хлопьев. Полимер фильтруют, промывают метанолом
и высушивают в вакуумном сушильном шкафу при 50 °C. Полученный белый
порошок полиакрилонитрила растворяется в очень ограниченном числе раство-
рителей (например, в диметилформамиде) и не плавится. Определяют выход
полимера и характеристическую вязкость полиакрилонитрила в растворе диме-
тилформамида при 20 °C.
Б. В 60%-ном водном растворе хлористого цинка
Четыре круглодонные колбы емкостью 50 мл откачивают, как описано
выше, и в атмосфере азота заполняют следующими компонентами;
15 мл раствора 100 г безводного ZnCh в 55 мл перегнанной в азоте воды;
5 мл (7,64-10“2 моль) акрилонитрила, перегнанного в атмосфере азота,
0,2 мл раствора 5 г персульфата калия в 75 мл перегнанной в токе азота
воды (5Ю“4 моль Кг^гОв);
в четвертую колбу дополнительно ничего не вносят.
Затем во вторую колбу добавляют 0,1 мл конц. соляной кислоты, в третью
колбу — 0,2 мл 20%-ного водного раствора бисульфита натрия (2.1-Ю-4 моль
Na2S20s), а в четвертую колбу — 0.1 мл конц. соляной кислоты и 0,2 мл
20%-ного водного раствора Na2S2O5- Все 4 колбы закрывают стеклянными проб-
ками и, встряхивая, тщательно перемешивают. Первые две колбы оставляют
при 50 °C на ночь. Две другие, которые содержат бисульфит натрия в качестве
восстанавливающего агента (см. окислительно-восстановительную полимеризат
цию, раздел 3.1.3), оставляют при комнатной температуре. В этих колбах реак3
ция начинается сразу же после перемешивания. При этом выделяется тепло, и
смесь становится вязкой; реакционная смесь, содержащая соляную кислоту,
остается бесцветной, а смесь без соляной кислоты желтеет Реакционные смеси,
оставленные полимеризоваться на ночь, содержат захваченные пузырьки, кото-
рые медленно перемещаются при наклоне колбы из-за высокой вязкости. К утру
смеси приобретают желтоватый оттенок.
Вязкие растворы полимера извлекают из колб шпателем и переносят в ста-
кан с водой. Большие куски полимера разрезают ножницами на мелкие и про-
мывают большим количеством воды (4—5 раз), пока промывные воды не пере-
станут давать положительную реакцию на хлор-ион. Затем образцы отфильтро-
вывают на стеклянном фильтре и высушивают до постоянной массы при 50 °G
в сушильном вакуумном шкафу. Выход полимера составляет практически
для реакций, проводимых при 50 °C, и около 80% для окислительно-восстанови-
тельного инициирования. Измеряют характеристические вязкости полученных об-»
разцов в растворе диметилформамида и сопоставляют с характеристической вяз*
костью полимера, полученного в предыдущем опыте. Если полимеры нераствори-
мы полностью, определяют фракцию полимера, экстрагируемую диметилформ-
амидом, взвешивают, высушивают нерастворимую фракцию. Растворимую фрак-
цию разбавляют чистым растворителем до концентраций, необходимых для
измерений характеристической вязкости. Если полимер растворим полностью, его
осаждают водой, высушивают до постоянной массы и готовят из него раствор
для вискозиметрических измерений.
Опыт 3-09. Суспензионная полимеризация винилацетата*
Навеску 0,15 г сополимера стирола с малеиновой кислотой (см. опыт 5-03)
растворяют в 150 мл горячей дистиллированной воды. Полученный 0,1%-ный
раствор нейтрализуют несколькими каплями аммиака и переносят в трехгорлую
колбу емкостью 500 мл, снабженную механической мешалкой, обратным холо-
дильником. термометром и капельной воронкой, укрепленной наверху обратного
холодильника. Раствор нагревают примерно до 70 °C на водяной бане, нагретой
до 80 °C. Затем растворяют 0.6 г (2.5 10“3 моля) перекиси бензоила в 100 г
(1,16 моля) свежеперегнанного винилацетата и полученный раствор вносят
в колбу через капельную воронку при сильном перемешивании в течение 30 мин.
Температуру водяной бани поддерживают на уровне 80 °C, винилацетат добав-
* Этот опыт не обязательно проводить в атмосфере азота.
125
ляют с такой скоростью, чтобы происходила умеренная дефлегмация. После
того как весь мономер введен в колбу, температура смеси повышается пример-
но до 80 °C (дефлегмация прекращается за несколько минут до достижения
этой температуры).
Для отгонки непрореагировавшего винилацетата через смесь в течение
30 мин пропускают пар. Затем реакционный сосуд охлаждают до комнатной
температуры и содержимое его доводят до 500 мл добавлением холодной воды.
Только после этого прекращают перемешивание и после седиментации частиц
полимера сливают водный слой. Полученный полимер промывают холодной во-
дой, пока промывные воды не перестанут пениться. Влажный полимер наносят
тонким слоем на стекло и высушивают при комнатной температуре в вакууме.
Измеряют характеристическую вязкость полученного полимера в растворе аце-
тона при 30 °C и рассчитывают среднюю молекулярную массу (см. раз-
дел 2.3.2.1).
Опыт 3-10. Полимеризация метакриловой кислоты в водном растворе, ини-
циированная персульфатом калия*
В трехгорлой колбе емкостью 50 мл, снабженной обратным холодильником
и двумя капельными воронками, нагревают 11 мл дистиллированной воды до
80 °C. По достижении этой температуры в колбу при слабом перемешиваний
магнитной мешалкой вводят по каплям 6 г (0,07 моля) метакриловой кислоты,
предварительно перегнанной под вакуумом в токе азота, и 0,18 г (6,6-10~4 мо-
ля) персульфата калия, растворенного в 4 мл воды. Метакриловая кислота
сразу же начинает полимеризоваться, что заметно по возрастанию вязкости
раствора. После того как все реагенты введены в колбу, реакционную смесь вы-
держивают при 80 °C в течение 1 ч.
Полученный довольно вязкий раствор разбавляют смесью 70 мл ацетона и
20 мл воды, фильтруют и затем добавляют по каплям к 1 л смеси 4 ч. ацетона
и 1 ч. петролейного эфира (т. кип. 50—70 °C). При этом полимер выпадает в
виде мелких частиц, которые затем слипаются друг с другом. После осаждения
отстой декантируют и добавляют 500 мл новой порции осаждающей смеси.
Полимер фильтруют, при необходимости дробят на более мелкие кусочки и за-
тем экстрагируют в аппарате Сокслета петролейным эфиром в течение 5 ч. По-
лученный продукт высушивают в вакуумном шкафу при 50 °C. Выход полимера
практически количественный.
Полиметакриловая кислота растворима в воде, а также в металоне, диокса-
не и в диметилформамиде. Измеряют вязкость полученного полимера в водном
растворе при 20 °C, используя следующие концентрации: 0,5; 0,7; 1,0; 1,5; 2,0;
2,5; 3,0; 3,5 и 4,0 г/л (измерение проводят с помощью вискозиметра Оствальда,
диаметр капилляра 0,3 мм). При построении графика цуд/С от С получают
типичную кривую для полиэлектролитов (см. рис. 20).
Если же измерения проводят с теми же концентрациями полимера в 1 н.
растворе NaCl, то вязкость изменяется так же, как и в случае неионогенных
полимеров. С этой целью поступают следующим образом: навеску 30 г NaC!
растворяют в 100 мл воды в мерной колбе (при этом получают 5,13 н. раствор).
Для приготовления растворов полимера указанных концентраций необходимое
количество полиметакриловой кислоты взвешивают в мерных колбах емкостью
10 мл и растворяют добавлением примерно по 5 мл воды. Затем в каждую
колбу приливают по 2 мл 5,13 н. раствора NaCl и доводят до метки дистилли-
рованной водой. Приготовленные растворы имеют концентрацию 1,03 моль/л.
3.1.2. Полимеризация, инициированная азосоединениями
В качестве инициаторов свободнорадикальной полимеризации ши-
роко используют азосоединения, в которых азогруппа с обеих сто-
рон связана с четвертичным углеродным атомом, связанным, в свою
очередь, с нитрильной или эфирной группой и алкильными ради-
* Этот опыт не обязательно проводить в атмосфере азота.
126
калами. Эти соединения достаточно стабильны при комнатных тем-
пературах, но разлагаются при нагревании (выше 40 °C) или под
действием УФ-лучей (даже ниже 40 °C) с образованием свободных
углеродных радикалов и выделением азота. Азот может вызывать
ряд нежелательных побочных эффектов, таких, как, например, за-
купоривание капилляра в ходе дилатометрических измерений, а
при полимеризации в блоке образующийся полимер часто содержит
окклюдированные пузырьки газа.
Разложение азосоединений обычно не зависит от природы ис-
пользуемого растворителя и в большинстве случаев подчиняется
закономерностям реакций первого порядка, в связи с чем такие
инициаторы широко используются при кинетических исследованиях
(см. раздел 3.1 и опыт 3-11). Наиболее распространенным из этих
инициаторов является азо-бис-изобутиронитрил (АИБН), который
распадается по уравнению
СН3 СН3 СН3
I I I
СН3—-С—N=N—-С—-СН3 —2СН3—-С* 4- N2
I I I
сн3 сн3 сн3
Однако выход инициирующих свободных радикалов часто ока-
зывается гораздо ниже, это следует из приведенного уравнения
Это обусловлено тем, что кроме цианизопропильных радикалов в
результате их рекомбинации или диспропорционирования могут
образоваться также динитрилтетраметил янтарной кислоты или
метакрилонитрил и изобутиронитрил. Азосоединения широко ис-
пользуют для инициирования полимеризации в массе и в органиче-
ских растворителях.
Опыт 3-11. Полимеризация стирола в массе, инициированная азо-бис-изобу-
тиронитрилом (влияние концентрации инициатора)
В 4 ампулы с вакуумными кранами загружают по 4 г (3,84-10~2 моля)
перегнанного стирола (см. опыт 3-01) и следующие навески инициатора:
3,2 мг (1,9 -10-6 моля), 6,3 мг (3,8-10—5 моля), 31,5 (1,9-10~4 моля) и 6,3 мг
(3,8-10-5 моля) АИБН*. В пятую ампулу заливают только стирол. Далее работу
проводят в соответствии с описанием опыта 3-02. По полученным данным строят
зависимости скорости полимеризации (в % превращения в ч) от корня квадрат-
ного из концентрации инициатора [уравнение (3-6)] и степени полимеризации
от обратного корня квадратного из концентрации инициатора [уравнение (3-8)].
Результаты сопоставляют с данными термической полимеризации и полимериза-
ции, инициированной перекисью бензоила (см. опыт 3-02).
Опыт 3-12. Исследование дилатометрическим методом полимеризации стиро-
ла в массе, инициированной азо-бис-изобутиронитрилом
Степень превращения при полимеризации можно легко определить с доста-
точной точностью по уменьшению объема реакционной смеси в дилатометре.
Такое уменьшение объема обусловлено значительной разностью плотностей мо-
номера и полимера. Зная исходный объем реакционной смеси V и контракцию
AV, можно вычислить степень превращения Q (в %) в отсутствие раствори-
телей:
= 100AV/V (3-17)
где — константа, запис^щая от удельных объемов мономера и полимера при
соответствующих температурах.
* Синтез АИБН описан в работе [8].
127
Взаимосвязь между Q и AV может быть установлена также эмпирически.
Калибровка дилатометров
Дилатометр представляет собой стеклянную ампулу со шлифом, выше ко-
торого находится карман для запирающей жидкости (см. рис. 16), и с капил-
лярной трубкой длиной около 30 см (0,2 мл объема), градуированной с ценой
деления 0,001 мл. Конец капиллярной трубки пришлифован к шлифу ампулы
дилатометра.
Для определения объема дилатометр, наполненный ртутью до верхней
кромки шлифа, термостатируют при 20 °C. При вставлении капилляра в ампулу
часть ртути попадает в капиллярную трубку, а избыток ртути вытесняется в за-
пасной карман. Затем капилляр с помощью пружинок закрепляют на ампуле
дилатометра, используя специально припаянные усики, после чего, переворачи-
вая дилатометр, избыток ртути выливают из кармана, закрыв предварительно
верхний конец капиллярной трубки. Дилатометр вновь помещают в термостат до
достижения температурного равновесия. Отмечают уровень ртути в капилляре,
капилляр осторожно вынимают (необходимо проверить, вся ли ртуть вылилась
из него). Ртуть переливают в предварительно взвешенный стаканчик и опреде-
ляют ее массу с точностью до 10 мг. Зная массу и плотность ртути при 20 °C
(р= 13,5457 г/см3), определяют ее объем. Для точного определения истинного
объема дилатометра необходимо учесть объем ртути, заполнявшей капилляр.
Каждый дилатометр калибруют таким образом несколько раз и определяют
среднее значение, которое и используется в дальнейших исследованиях.
Для прецизионных измерений объем дилатометра необходимо определять при
температуре полимеризации (в данном случае при 60 °C). Однако в общем
случае изменением объема дилатометра с температурой можно пренебречь, по-
скольку термический коэффициент линейного расширения стекла меньше коэффи-
циента объемного расширения жидкостей в 100 с лишним раз.
Определение термического коэффициента расшире-
ния стирола
Значение термического коэффициента расширения стирола необходимо для
дальнейших дилатометрических опытов с целью определения количества стирола
в дилатометре по объему, измеренному при температуре полимеризации. Опре-
деление термического коэффициента расширения а проводят в дилатометре. Для
этого дилатометр заполняют стиролом, стабилизированным 0,1% гидрохинона.
Вставляют капилляр и в качестве затворной жидкости заливают немного ртути.
Заполнение дилатометра проводят при температуре примерно на 10 °C выше,
чем наименьшая температура измерений, так что мениск в капилляре будет
опускаться при помещении дилатометра в термостат при начальной температуре
измерений. После достижения температурного равновесия фиксируют уровень
мениска, затем повышают температуру термостата и повторяют измерение.
Строят диаграмму зависимости объема (объем Vo минус показания дилатомет-
ра) от температуры. По углу наклона полученной прямой определяют среднее
значение термического коэффициента расширения в измеренном интервале тем-
ператур:
__ К<он ^нач
уначдт
(3-18)
где Укпн — объем при максимальной температуре опыта, мл; УНяч — объем
при минимальной температуре опыта, мл; ДТ — интервал температур между
максимальной и минимальной температурой опыта; а — термический коэффи-
циент расширения.
Термический коэффициент расширения а измеряют в интервале от 20 до
30 °C и от 50 до 60 °C, среднее из двух значений используют в дальнейшем во
всем интервале от 20 до 60 °C.
Дилатометрические измерения
В 4 калиброванные мерные колбы емкостью 25 мл вносят 35, НО, 180
и 250 мг (0,21, 0,67, 1,10 и 1,52 ммоля) азо-бис-изобутиронитрила, в колбы
наливают при 20 °C до метки очищенный от ингибитора стирол и взвешивают
128
для определения количества стирола. Можно вычислить плотность стирола при
20 °C (пренебрегая объемом растворенного инициатора).
Растворы стирола заливают в 4 дилатометра, вставляют капилляры и за-
твор заполняют ртутью. Дилатометры помещают в большой водяной термостат
при 60 °С±0,05 °C так, чтобы заполненная часть капилляра тоже была погруже-
на. Термостат легко собрать из следующего оборудования: большой стеклянной
банки, погруженной в термостатирующую жидкость нагревателя, хорошей ме-
ханической мешалки, ртутно-толуольного контактного терморегулятора и реле.
Для крепления дилатометров в термостате должны быть предусмотрены специ-
альные держатели. После погружения дилатометров в термостат уровень сти-
рола в капилляре повышается за счет термостатирования мономера (если необ-
ходимо, избыток стирола можно удалить шприцем с длинной иглой либо с
помощью тонких стеклянных капилляров).
Мениск некоторое время остается на постоянном уровне (индукционный
период, обусловленный растворенным кислородом), а затем начинает снижаться,
что свидетельствует о начале полимериза'ции. Уровень мениска фиксируют каж-
дую минуту после погружения дилатометров в термостат и показания наносят
на графике в зависимости от времени. Когда снижение уровня приобретает ста-
ционарный характер, измерения можно проводить через каждые 5 мин. Время
начала реакции определяют по пересечению горизонтальной и наклонной пря-
мых графика. После того как объем смеси уменьшится на 0,1—0,2 мл, реакцию
прекращают, вынув дилатометр из термостата (предварительно нужно отметить
время и уровень мениска) и быстро охладив его ледяной водой.
Полученный полимер извлекают из дилатометра следующим образом. После
охлаждения до 10 °C дилатометр осторожно переворачивают и выливают из за-
твора ртуть в стаканчик. Карман затвора промывают метанолом (ацетоном),
затем бензолом и высушивают, продувая его воздухом. Вынимают капилляр и
раствор полимера выливают в маленький стаканчик. Капилляр и ампулу дила-
тометра промывают несколько раз небольшими порциями бензола, которые до-
бавляют к основной массе раствора полимера. Полученный раствор приливают
при перемешивании к 8—10-кратному количеству метанола. Полимер фильтруют
и сушат до постоянной массы.
Для вычисления степени превращения необходимо знать начальное количе-
ство стирола. Для этого измеренный при 20 °C объем пересчитывают на темпе-
ратуру 20 °C по формуле
^о = 7Г^4оГ (3’19)
Умножив Кго на плотность стирола при 20 °C, получают исходное количество сти-
рола, после чего легко вычисляют степень превращения.
Обработка дилатометрических данных
Изменение объема ДУ определяют по дилатометрическим измерениям с мо-
мента начала реакции до ее прекращения по графику зависимости уровня мени-
ска от продолжительности реакции.
Для каждого эксперимента по формуле (3-17) вычисляют константу К и по-
лученные результаты усредняют. По разбросу данных определяют статистиче-
скую ошибку (без использования формулы Гаусса). Затем строят зависимость
скорости полимеризации (% конверсии в ч) от корня квадратного из концент-
рации инициатора [% (мол.)] — см. уравнения (3-6) и (3-7) раздела 3.1. Кроме
этого, измеряют вискозиметрическим методом степень полимеризации получен-
ных полимеров и строят зависимость степени полимеризации от обратной вели-
чины корня квадратного из концентрации инициатора [см. уравнение (3-8)]. Со-
поставляют полученные данные с данными опыта 3-02.
Статистический разброс значений К может и не быть связан с точностью
дилатометрического метода, поскольку основные ошибки индивидуальных опы-
тов тождественны. Поэтому необходимо определить точность метода, вычисляя
интервал разброса данных, и отметить основные источники ошибок и их при-
мерную величину.
9—732
129
Опыт 3-13. Полимеризация стирола в растворе, инициированная
азо-бис-изобутиронитрилом
А. Кривая конверсия — время
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, обратным хо-
лодильником и трубкой для ввода азота, помещают 100 мг (0,61 ммоля) азо-
бис-изобутиронитрила, после чего колбу откачивают и заполняют азотом 3 раза.
Затем в колбу заливают 100 мл чистого толуола, перегнанного в токе азота,
10 мл (0,09 моля) очищенного от ингибитора стирола и колбу помещают на
водяную баню. Через реакционную смесь пропускают слабый ток азота (газо-
вый вывод не должен пропускать внутрь прибора кислород — см. раздел 2.11)
и баню нагревают до кипения. Реакционную смесь, вязкость которой увеличи-
вается во времени, слабо перемешивают. С интервалом в 1 ч из колбы отби-
рают с помощью пипетки пробы по 10 мл (во время отбора пробы ток азота
увеличивают). Отобранную пробу сразу же приливают по каплям при переме-
шивании к 100 мл метанола. После 6 ч нагрева полимеризацию прекращают
путем охлаждения колбы. Содержимое колбы через капельную воронку прили-
вают к 50 мл метанола при перемешивании. Выпавший в осадок полимер
фильтруют и сушат в вакуумном сушильном шкафу при 50 °C до постоянной
массы. Измеряют конверсию и характеристическую вязкость образцов (см.
опыт 3-01). Строят зависимости конверсии и степени полимеризации образую-
щегося полимера от продолжительности реакции.
Б. Влияние концентрации мономера
В 7 ампул со шлифами вносят по 23 мг (0,14 моля) азо-бис-изобутирони-
трила, ампулы присоединяют к вакуумной линии, последовательно откачивают и
заполняют азотом несколько раз. Затем в токе азота в ампулы вводят 0,5; 1,0;
1,5; 2,0; 2,5 и 3,0 мл (4,36; 8,72; 13,07; 17,42; 21,78 и 26,13 ммоля) очищенного
стирола и смесь доводят в каждой ампуле до 15 мл добавлением чистого то-
луола, перегнанного в токе азота. В последнюю ампулу вносят 15 мл стирола
(полимеризация в массе). Ампулы отсоединяют от вакуумной линии и немедлен-
но закрывают пришлифованными пробками, закрепляя их пружинками. Затем
ампулы помещают в кипящую водяную баню. Через 6 ч ампулы вынимают из
бани, охлаждают и из них выделяют полимер, как описано в предыдущем опы-
те (при необходимости содержимое ампул разбавляют толуолом).
Постройте зависимости конверсии и степени полимеризации полимера от
концентрации мономера в исходной реакционной смеси. Отметьте, в чем различие
между данным образцом и образцом, полученным полимеризацией в массе.
Опыт 3-14. Полимеризация метилметакрилата в массе, инициированная азо-
бис-изобутиронитрилом
А. Автоускорение (гель-эффект, эффект Троммсдорфа)
В токе азота перегоняют 100 мл метилметакрилата в градуированный
приемник или в капельную воронку с уравнивающей давление трубкой (см.
раздел 2.1.2), которые содержат 100 мг (0,61 ммоля) заранее введенного азо-
бис-изобутиронитрила. Затем 10 ампул со шлифами откачивают, заполняют азо-
том и в каждую вводят по 10 мл (0,936 моля) полученного, как указано выше,
метилметакрилата. Ампулы отсоединяют от заливочного устройства при неболь-
шом избыточном давлении азота и быстро закрывают пришлифованными проб-
ками, закрепляя их пружинками, после чего ампулы охлаждают и до полиме-
ризации помещают в охлаждающую смесь сухого льда с ацетоном.
Перед началом опыта все ампулы размораживают и доводят до комнат-
ной температуры, затем их одновременно погружают в термостат при 50° С.
С интервалом в 1 ч ампулы вынимают из термостата и быстро охлаждают в
смеси сухого льда с ацетоном. Содержимое ампул разбавляют 50 мл хлорофор-
ма, полученный раствор вводят по каплям при перемешивании в 500 мл н-геп-
тана или петролейного эфира. После фильтрования и высушивания полимера
при 50 °C до постоянной массы определяют характеристическую вязкость полу-
ченных образцов в растворе хлороформа при 20 °C. Строят зависимость конвер-
сии и степени полимеризации от продолжительности реакции.
Конверсию можно также определять рефрактометрически, поскольку изме-
нение показателя преломления реакционной смеси прямо пропорционально сте-
130
пени превращения. Измерения можно проводить на рефрактометре Аббе на об-
разцах, полученных до гель-стадии. Конверсию в этом случае определяют по
калибровочной кривой, которую строят следующим образом: откладывают по-
казатель преломления чистого мономера для нулевой конверсии п^= 1,4140 и
показатель преломления полиметилметакрилата и 1,4915 для 100%-ной кон-
версии, эти две точки соединяют прямой линией.
Б. Регулирование молекулярной массы; определение
константы передачи цепи на н-Додецилмеркаптан
В 5 ампул со шлифами заливают, как описано выше, по 10 мл (93,6 ммоля)
метилметакрилата, содержащего 0,1% азо-бис-изобутиронитрила. Затем в 4 ам-
пулы добавляют 0,1; 0,5; 1 и 2% (мол.) н-додецилмеркаптана (лаурилмеркап-
тана) в качестве регулятора (пятая ампула без агента передачи используется
для сравнения), ампулы закрывают и до полимеризации помещают в охлаж-
дающую смесь сухого льда с ацетоном. Перед началом полимеризации ампу-
лы нагревают сначала до комнатной температуры, затем их одновременно по-
мещают в термостат при 50 °C. Через 2 ч (конверсия не должна превышать
10%, иначе отношение [ZH]/[M] нельзя считать постоянным в ходе полимери-
зации) ампулы вынимают из термостата, содержимое ампул разбавляют 30 мл
хлороформа, полученные растворы осторожно приливают при перемешивании
к 300 мл петролейного эфира или н-гептана. Осадок фильтруют и высушивают,
затем образцы повторно растворяют в хлороформе и вновь осаждают, фильтру-
ют и сушат до постоянной массы в вакуумном сушильном шкафу при 50 °C.
Определяют характеристические вязкости переосажденных образцов в растворе
хлороформа при 20 °C и рассчитывают их степени полимеризации (см. раз-
дел 2.3.2.1). Для определения константы передачи цепи [см. раздел 3.1, формула
(3-16)] строят зависимость обратной степени полимеризации полученных образ-
цов от мольного отношения н-додецилмеркаптана к мети л метаакрил ату:
[ZH]/{*M]. Полученная прямая отсекает на оси ординат отрезок 1/Рп.о, соот-
ветствующий опыту без регулятора, а по тангенсу угла наклона прямой рассчи-
тывают константы передачи цепи Czn. Полученное значение, однако, не является
точной величиной Czh, поскольку используемое уравнение строго справедливо
только для среднечисловой молекулярной массы образцов. На втором графике
строят зависимость конверсии от величины [ZH]/[M]; реакция передачи цепи
не влияет на скорость полимеризации.
Опыт 3-15. Полимеризация винилацетата в различных растворителях, иниции-
рованная азо-бис-изобутиронитрилом
В 4 ампулы емкостью по 50 мл вносят по 0,03 г (0,18 ммоля) азо-бис-изо-
бутиронитрила и по 20 г одного из следующих растворителей: толуола, хлори-
стого метилена, метанола, трет-бутилового спирта, перегнанных под азотом, и
по 10 г (0,12 моля) винилацетата, очищенного от ингибитора перегонкой в токе
азота. Воздух из ампул удаляют повторным откачиванием и заполняют азотохм
3 раза; откачивание ампул продолжают до появления первого пузыря в рас-
творе, после чего ампулу заполняют азотом. Ампулу запаивают и опускают в
термостат при 70 °C.
Через 4 ч ампулы вскрывают и их содержимое выливают в чашки Петри;
полимеризацию прекращают добавлением в каждый образец по 0,5 мл кротоно-
вого альдегида. Растворители и непрореагировавший мономер откачивают под
вакуумом при 60 °C. После взвешивания около 1 г каждого из образцов рас-
творяют в 20-кратном количестве метанола и высаживают в воду. После вы-
сушивания в вакуумном сушильном шкафу при 50 °C измеряют характеристиче-
ские вязкости образцов в растворе ацетона при 30 °C (диаметр капилляра вис-
козиметра 0,3 мм) и рассчитывают молекулярные массы (см. раздел 2.3.2.1). За-
полняют таблицу, в которой растворители располагаются согласно их влия-
нию на конверсию и средневязкостную молекулярную массу полученных
образцов.
Опыт 3-16. Определение молекулярно-массового распределения поливинил-
ацетата методом фракционной экстракции
Фракционная экстракция поливинилацетата производится согласно методу
Фукса (см. раздел 2.3.3). Для этого приготавливают 5—10%-ные растворы 1 г
9* 131
полимера* в этилацетате. Концентрацию растворов выбирают в зависимости от
молекулярной массы фракционируемого образца: чем выше молекулярная мас-
са полимера, тем ниже должна быть концентрация раствора. Нужную концент-
рацию легко определить в нескольких предварительных опытах, для чего куски
алюминиевой фольги погружают в растворы различной концентрации, затем
образцы вынимают, высушивают и взвешивают. Количество полимера, прилип-
шего к поверхности, должно быть около 100 мг на 100 см2 (это количество
может быть несколько меньше, но не более 50%). Вырезают 4 куска алюминие-
вой фольги (толщиной около 20—50 мкм) размером 7X16 см и взвешивают
их (общая площадь всех кусков составляет около 1000 см2). Куски фольги по-
гружают в раствор полимера, после чего их высушивают горячим воздухом и
оставляют на ночь в вакуумном шкафу при 40 °C. Количество полимера, прилип-
шего к фольге, определяют взвешиванием, после чего фольгу разрезают на по-
лоски размером 1X7 см. Для предотвращения слипания фольги при последую-
щей экстракции полоски сгибают гармошкой с помощью пинцета (высота склад-
ки 0,5—1 см). Экстракцию предпочтительно осуществлять в специальном приборе
(см. раздел 2.3.3) смесью очищенного метилового эфира уксусной кислоты и
петролейного эфира (последний не должен содержать фракций, кипящих при
температуре выше 60 °C, во избежание окклюзии при концентрировании и высу-
шивании фракций полимера). Полоски алюминиевой фольги встряхивают в
смеси в течение 10 мин, после чего раствор полимера сливают во взвешанные
колбы Эрленмейера емкостью 100 мл. Для полного удаления раствора со
складок фольги прибор для фракционирования встряхивают несколько раз. По-
следовательно, изменяя состав экстрагирующей смеси, выделяют 10 фракций
полимера. Полученные растворы осторожно концентрируют и затем высушивают
при 50 °C сначала на воздухе, потом в вакуумном шкафу.
Состав экстрагирующей смеси для индивидуальных фракций зависит от
молекулярной массы поливинилацетата, которую необходимо подбирать так,
чтобы массы отдельных фракций заметно не различались. Экспериментальные
данные, приведенные ниже, получены на образце поливинилацетата с т]уд/С=
= 0,12 (измеренной в растворе ацетона при 30°C при С = 10 г/л). В таблице
приведено только количество метилового эфира уксусной кислоты в смеси, по-
скольку суммарный объем постоянен и равен 100 мл. Количество петролейного
эфира определяют по разности. Если для исследуемого образца величина Луд/^
превосходит указанное значение более чем на 5%, концентрацию метилового
эфира уксусной кислоты в смеси необходимо увеличить; для образцов с меньшей
молекулярной массой следует увеличить содержание петролейного эфира в
смеси.
После определения количества сухого полимера в каждой фракции в кол-
бах Эрленмейера приготавливают растворы полученных образцов в ацетоне
концентрацией 10 г/л. При этом предполагается, что объем образующегося рас-
твора равен объему добавляемого ацетона. Колбы плотно закрывают пришлифо-
ванными пробками (нанести немного смазки на верхнюю часть шлифа) и поме-
щают на несколько часов на качалку до полного растворения полимера. С по-
мощью вискозиметра Оствальда (диаметр капилляра 0,3 мм) при 30 °C опреде-
ляют удельную и характеристические вязкости выделенных фракций, затем по
уравнению Шульца — Блашке (К=0,27) рассчитывают молекулярные массы
фракций (см. раздел 2.3.2.1).
Разультаты фракционирования сводят в таблицу. Ранее в разделе 2.3.3 было
показано, что данные, приведенные в 5-й графе, получены суммированием всех
предыдущих фракций из 4-й графы плюс половина данной фракции. Интеграль-
ную кривую распределения получают построением зависимости суммы массы
фракций от степени их полимеризации J (Р). Графическим дифференцированием
(см. раздел 2.3.3) этой зависимости получают величины Шр (6-я графа), а кри-
вуЮ-Дифференциальиого распределения получают построением зависимости тр
от Р.
* Типичная кривая распределения получена для образца поливинилацетата,
синтезированного полимеризацией в растворе трет-бутилового спирта (см.
опыт 3-15).
132
Фракционирование поливинилацетата
{общее количество полимера 1016 мг)
№ опыта Метиловый эфир уксусной кислоты, мл Полимер 1{Р), % mp-10‘3, % Чуд/С; опытные данные Гп1; расчетные данные р
мг %
1 52,0 43 4,2 k 2,1 5,8 0,020 0,019 400
2 54,0 84 8,3 8,4 11,9 0,041 0,037 1080
3 56,0 146 14,4 19,7 15,6 0,071 0,060 2020
4 54,0 152 15,0 34,4 16,6 0,096 0,076 2790
5 57,5 201 19,8 51,8 11,8 0,142 0,103 4220
6 58,0 220 21,6 72,5 7,4 0,214 0,136 6250
7 58,5 128 12,6 89,6 4,1 0,338 0,177 9030
8 59,0 18 18 96,8 — — — —-
9 60,0 9 0,9 98,2 — — — —
10 62,0 7 0,7 99,0 — —. —. —
11 100,0 5 0,5 99,8 — — — —
3.1.3. Полимеризация, инициированная с помощью
окислительно-восстановительных систем
При окислительно-восстановительных реакциях в системе возни-
кают свободные радикалы, которые могут инициировать полимери-
зацию. Чаще всего в качестве окислительных агентов используют
органические или неорганические перекисные соединения, а в ка-
честве восстановительных агентов ионы металлов, находящихся в
низшем валентном состоянии, либо неметаллические, легко окисля-
емые соединения (например, некоторые серосодержащие соедине-
ния). Можно использовать также системы из трех компонентов,
а именно: перекисного соединения, иона металла (например, Fe2+)
и другого восстановителя типа кислого сульфита. В последнем слу-
чае ионы трехвалентного железа, получаемые в результате окис-
лительно-восстановительной реакции между Fe2+ и перекисным со-
единением, вновь восстанавливаются кислым сульфитом до Fe2+,
поэтому для реакции достаточно очень малого количества ионов
Fe в системе.
В двухкомпонентной системе, состоящей, например, из перекис-
ного соединения и ионов Fe2+, свободные радикалы образуются в
результате переноса электрона с ионов двухвалентного железа к
перекиси, которая при этом распадается на анион и свободный ра-
дикал; последние затем могут реагировать с мономером:
R—О—О—R + Fe2+ --> R—О" + Fe3+ 4- R—О*
R—О* +СН2=СН ----> R—О—СН2—СН
Ниже приведены некоторые типичные окислители и восстанови-
тели, применяемые для окислительно-восстановительного иници-
ирования свободнорадикальной полимеризации
133
Окисляющие агенты Восстанавливающие агенты
Перекись водорода Ag+, Fe2+, Fe3+
Пероксидисульфат Сульфиты, кислые сульфиты
Ацильные перекиси Тиосульфаты, меркаптаны, сульфиновые
кислоты, амины (например, М,М-диме-
тиланилин)
Эндиолы (сахар)
Бензоин/Fe2*
Кислый сульфит/Ре2+
Необходимо отметить, что окислительно-восстановительные си-
стемы в отличие от перекисных инициаторов или азосоединений не
всегда способны инициировать полимеризацию ненасыщенных мо-
номеров. Поэтому при исследовании полимеризации новых соедине-
ний целесообразно всегда начинать с полимеризации, инициирован-
ной не окислительно-восстановительной системой, а, например, пе-
рекисью бензоила (см. раздел 3.1.1).
Эффективность окислительно-восстановительных систем зависит
от ряда факторов, поэтому для создания оптимальных условий
проведения полимеризации необходимо тщательно сбалансировать
окислительно-восстановительные компоненты. Так, наиболее бла-
гоприятные условия не всегда достигаются при эквимольном соот-
ношении компонентов инициирующей системы. Обычно справедливо
правило, что при постоянном отношении окислителя и восстанови-
теля скорость полимеризации возрастает с увеличением концент-
рации инициатора, а степень полимеризации образующегося поли-
мера при этом уменьшается (см. опыт 3-19).
Порядок введения реагентов в систему также может играть су-
щественную роль. И если обычно первым к мономеру добавляют
восстановитель (для удаления возможно присутствующего кисло-
рода) с последующим постепенным добавлением окислителя, в не-
которых случаях используют обратный порядок введения компо-
нентов инициирующей системы. Наконец, величина pH также су-
щественна при полимеризации в водных средах. Если реакцию
проводят в щелочном растворе, необходимо помнить, что соли же-
леза следует использовать в совокупности с комплексообразующи-
ми соединениями, такими, например, как пирофосфат натрия
Na4P2O7.
Окислительно-восстановительную полимеризацию обычно про-
водят в водных средах, в суспензиях или эмульсиях и реже в сре-
де органических растворителей (см. опыт 3-21). Основной особен-
ностью этих инициаторов является возможность проведения поли-
меризации с высокими скоростями при относительно низких тем-
пературах, при этом образуется полимер с высокой молекулярной
массой. Реакции передачи и разветвления цепи в этом случае не
играют заметной роли. В промышленности окислительно-восстано-
вительное инициирование впервые было использовано при получе-
нии синтетического каучука из стирола и бутадиена (буна S) при
5 °C (см. опыт 3-46).
134
Опыт 3-17, Полимеризация акриламида, инициированная окислительно-вос-
становительной системой в водной среде
В колбе емкостью 1 л, снабженной мешалкой (с вводом и выводом для
инертного газа), растворяют 5 г перекристаллизованного из бензола акрил-
амида в 500 мл дистиллированной воды, перегнанной в токе азота. К получен-
ному раствору добавляют 25 мл 0,1 М водного раствора (МН^гРе^О^г-бНгО
и 25 мл 0,1 М раствора перекиси водорода. Через раствор в течение 5 мин про-
пускают азот для удаления кислорода. Полимеризацию проводят при комнат-
ной температуре (около 20 °C). Спустя 30 мин полученный вязкий раствор вво-
дят по каплям при быстром перемешивании к 4 л метанола, подкисленного со-
ляной кислотой. Осадок, имеющий коричневый цвет, обусловленный присутствием
гидроокиси трехвалентного железа, вновь растворяют в 50 мл воды. К раствору
добавляют избыток аммиака для полного осаждения железа в виде гидроокиси,
которую отфильтровывают. Полимер повторно высаживают в 500 мл метанола,
осадок фильтруют и сушат при помощи водоструйного насоса, а затем в вакум-
ном сушильном шкафу при 20°C. Выход полимера составляет около 50%.
Полученный полиакриламид растворим в воде, но не плавится. Определяют ха-
рактеристическую вязкость полученного полимера в водном растворе при 25 °C
(диаметр капилляра 0,35 мм).
Опыт 3-18. Фракционирование полиакриламида методом гель-фильтрацион-
ной хроматографии*
Навеску 10 г сефадекса G 100 выдерживают в течение 2 сут в 400 мл
воды в химическом стакане. На дно двухметровой колонки диаметром 1,5—2 см
у выходного крана помещают несколько стеклянных бусинок и слой стекловаты.
Колонку частично заливают водой, после чего в нее переносят набухший сефа-
декс. При заполнении колонки необходимо избегать образования пустот и пу-
зырей. После того как частицы геля осядут, отстой осторожно сливают. Гель
промывают свежей водой, до тех пор пока промывная вода, добавленная к ме-
танолу, не перестанет вызывать муть (продажный сефадекс часто содержит во-
дорастворимые соединения, неблагоприятно влияющие на фракционирование).
Далее сливают отстойную воду, оставляя над поверхностью геля небольшой
слой толщиной в несколько миллиметров.
В колонку заливают раствор 250 мг полиакриламида (полученного в опы-
те 3-17) в 25 мл дистиллированной воды, открывают кран колонки и собирают
фракции вытекающей жидкости по 10 мл. При правильной набивке колонки
время элюирования одной фракции должно составлять около 1 ч. Когда мениск
раствора полиакриламида достигнет поверхности геля, в колонку в качестве
элюента добавляют дистиллированную воду.
Каждую из собранных фракций добавляют к 100 мл метанола, подкислен-
ного 2 каплями конц. соляной кислоты. При добавлении фракции с 6-й по 20-ю
образуется муть или осадок. Выпавшие фракции полимера фильтруют, промы-
вают метанолом и высушивают до постоянной массы в вакуумном сушильном
шкафу при 20 °C. Измеряют характеристические вязкости полученных образцов
в воде при 25°C (диаметр капилляра вискозиметра 0,35 мм). Для уменьшения
ошибки измерений раствор для вязкостных измерений готовят достаточно кон-
центрированными— 10 г/л. Затем по уравнению Шульца — Блашке (см. раз-
дел 2.3.2.1) рассчитывают молекулярные массы фракций. Соседние фракции,
количества которых недостаточны для вискозиметрических измерений, могут
быть объединены.
Строят зависимость количества выделенных индивидуальных фракций от
продолжительности элюации. Сопоставляют суммарную массу всех фракций с
количеством введенного в колонку полимера для определения потерь полимера
при Фракционировании; эти потери не должны превышать 3%.
Строят интегральную и дифференциальные кривые распределения по моле-
кулярным массам, как было описано ранее в разделе 2.3.3 и в опыте 3-16. При
суммировании фракций (см. опыт 3-16) отсчет начинают с последней фракции,
имеющей наименьшую молекулярную массу, и переходят к первым фракциям
по возрастанию молекулярных масс.
* См. раздел 2.3.3.
135
Опыт 3-19. Полимеризация акриламида, инициированная окислительно-вос-
становительной системой в воде (осадительная полимеризация)
А. Влияние отношения окислителя к восстановителю
Приготавливают следующие растворы в дистиллированной воде:
5%-ный раствор Na2S2O5;
5%-ный раствор персульфата калия K2S2O8;
10 мг FeSO4-7H2O в 100 мл воды +2 мл конц. H2SO4.
Четыре круглодонные колбы емкостью по 250 мл откачивают и заполняют
азотом* (см. раздел 2.1.3). Затем в колбы вносят реагенты, указанные в при-
веденной ниже таблице; при этом раствор персульфата калия вводят во все
колбы одновременно и в последнюю очередь. Содержимое встряхивают и остав-»
ляют при 20 °C. Отмечают время, когда раствор в колбах начинает мутнеть.
№ колбы Н2О*. мл Акрило- нитрил**, мл Na2S2O5 FeSO4 K2S2O8
мл ммоль мл ммоль мл ммоль
1 175 15 0,5 0,13 2,5 9-Ю-4 2,5 0,46
2 173 15 2,5 0,66 2,5 9-Ю-4 2,5 0,46
3 170 15 5,0 1,32 2,5 9-Ю-4 2,5 0,46
4 165 15 10,0 2,63 2,5 9-Ю-4 2,5 0,46
* Перегнанная под азотом.
'♦ Очищенный от ингибитора перегонкой в токе азота.
Через 20 мин реакцию во всех колбах прекращают и фильтруют осадок. Осадок
промывают на фильтре водой, потом метанолом и оставляют на ночь в вакуум-
ном сушильном шкафу при 50 °C. Строят зависимость конверсии, скорости поли-
меризации и характеристических вязкостей полученных образцов от соотноше-
ния окислителя к восстановителю в исходной реакционной смеси (вязкости по-
лимера измеряют в растворе диметилформамида при 25°C).
Б. Влияние концентрации инициатора при постоянном
отношении окислителя к восстановителю
В 4 круглодонные колбы емкостью по 250 мл загружают, как описано выше,
следующие реагенты:
№ колбы н2о*. мл Акрило- нитрил** , мл Na2S2O5 FeSO4 K2S2O8
мл ммоль мл ммоль мл ммоль
1 181 15 0,5 0,13 0,5 1,8.10"4 0,5 0,09
2 175 15 2,5 0,66 2,5 9,0.10’4 2,5 0,46
3 167 15 5,0 1,32 5,0 1,8.10"3 5,0 0,93
4 152 15 10,0 2,63 10,0 3,6.10’3 10,0 1,85
* Перегнанная под азотом.
♦♦ Очищенный от ингибитора перегонкой в токе азота.
После смешения компонентов колбы оставляют на 20 мин при 20 °C, а за-
тем выделяют полимер, как описано выше. Строят зависимость скорости поли-
* Этот опыт не обязательно проводить в атмосфере азота.
136
меризации (в % конверсии в 1 мин) от корня квадратного из концентрации
инициатора (К2$2О8 моль/л), а также зависимость характеристических вязко-
стей или молекулярных масс полученных образцов от обратной величины кор-
ня квадратного из концентрации инициатора.
В. Ингибирование полимеризации
В 4 круглодонные колбы емкостью по 250 мл вносят 5, 20, 100 и 200 мг
гидрохинона, колбы откачивают и заполняют азотом. Затем в колбы заливают
по 15 мл акрилонитрила, очищенного от ингибитора перегонкой в токе азота,
165 мл воды, также перегнанной под азотом, 10 мл 5%-ного раствора ЫагБгОб
и 2,5 мл раствора FeSO4 той же концентрации, что и в опыте А. Затем в каж-
дую из четырех колб добавляют по 2,5 мл 5%-ного раствора персульфата ка-
лия. Реакцию проводят при 20 °C, отмечают время индукционного периода по
появлению мути в растворах. Сопоставьте времена индукционных периодов,
включая четвертый образец из опыта А.
Г. Мокрое прядение полиакрилонитрила
Навеску 3,5 г одного из полученных образцов полиакрилонитрила раство-
ряют в 25 мл диметилформамида. Вязкий раствор заливают в оттянутую с ниж-
него конца стеклянную трубку диаметром 1 см. Оттянутый конец трубки опуска-
ют в лоток с холодной водой. Под действием собственного веса раствор поли-
мера медленно вытекает из трубки в воду, при этом полимер выпадает в оса-
док в виде тонкой бесконечной нити, которую, пропустив через ванну с водой,
можно наматывать на вращающийся барабан. Вместо оттянутой с одного кон-
ца трубки можно использовать шприц с иглой, при этом скорость прядения и
толщину нити легко варьировать.
Опыт 3-20. Эмульсионная полимеризация изопрена, инициированная окис-
лительно-восстановительной системой
В большинстве случаев эмульсионную полимеризацию проводят в присут-
ствии водорастворимых инициаторов; однако в описываемом опыте один из ком-
понентов инициирующей окислительно-восстановительной системы (перекись
бензоила) нерастворим в воде.
Трехгорлую колбу емкостью 100 мл, снабженную мешалкой и вводом для
азота, откачивают и заполняют азотом 3 раза. Приготавливают следующие рас-
творы: а) 500 мг олеата натрия (или лаурилсульфата натрия) в 16 мл деаэри-
рованной воды; б) 125 мг (0,32 ммоль) Fe(NH4)2(SO4)2 и 125 мг пирофосфата
натрия в 4 мл деаэрированной воды (для создания буфера). Этот раствор
встряхивают в течение 15 мин при 60—70 °C и затем выливают в колбу вместе
с раствором, указанным в пункте «а». После охлаждения до комнатной тем-
пературы в колбу вносят 20 мл (0,2 ммоль) изопрена, перегнанного в атмосфе-
ре азота и содержащего 50 мг (0,21 ммоль) перекиси бензоила. Сильное пере-
мешивание способствует образованию стабильной эмульсии, вязкость которой
возрастает во времени. После 6-часовой выдержки при комнатной температуре
изопрен почти полностью полимеризуется. Полимер высаживается в виде хлопьев
из латекса при добавлении эмульсии по каплям к 500 мл метанола, в котором
содержится 500 мг N-фенил-Р-нафтиламина, необходимого для стабилизации
полиизопрена; образование осадка можно усилить добавлением в осадитель не-
скольких капель соляной кислоты. После фильтрования с отсасыванием и про-
мывки метанолом прочный эластичный образец высушивают в вакуумном су-
шильном шкафу при 50 °C. Определяют растворимость полученного полимера
в различных растворителях, измеряют характеристическую вязкость в растворе
толуола при 25 °C, содержание 1,2- и 1,4-звеньев в цепи, а также соотношение
цис- и грянс-структур (см. опыт 3-30). Сопоставьте полученные данные с ре-
зультатами полимеризации изопрена под действием бутиллития (опыт 3-30).
Опыт 3-21. Полимеризация стирола, инициированная окислительно-восстано-
вительной системой в органическом растворителе
Три круглодонные колбы емкостью по 100 мл со специальными приспособле-
ниями для заполнения (см. раздел 2.1.3) откачивают и заполняют азотом 3 ра-
за. В одну из колб вносят 3 мл бензоата железа (II), в другую 3 мг ацетил-
ацетоната железа (II). Затем в каждую из этих колб вносят раствор 121 мг
(5-10-4 моль) перекиси бензоила в 5 мл абсолютированного бензола, перегнанно-
го в токе азота, и раствор 106 мг бензоина (5-10~4 моль) в 15 мл бензола.
137
В третью колбу вносят раствор 121 мг перекиси бензоила в 5 мл абсолютиро-
ванного бензола. Затем в токе азота в каждую колбу .вводят пипеткой 11 мл
(0,1 моль) высушенного и очищенного от ингибитора стирола. Содержимое
колб доводят до 50 мл добавлением бензола. При небольшом избыточном дав-
лении азота вынимают приспособления для заполнения и колбы закрывают
пришлифованными пробками, закрепляя их пружинками. Колбы выдерживают
48 ч при 50 °C, после чего полимеризацию прекращают прибавлением по каплям
реакционных смесей при перемешивании к 500 мл метанола. Выпавшие в оса-
док полимерные образцы фильтруют, высушивают до постоянной массы в ва-
куумном сушильном шкафу при 50 °C, определяют конверсию, характеристиче-
скую вязкость полимеров (в растворе бензола при 20 °C), рассчитывают молеку-
лярные массы образцов (см. раздел 2.3.2.1).
Опыт 3-22. Полимеризация винилхлорида, инициированная окислительно-вос-
становительной системой в метаноле (осадительная полимеризация).
Трехгорлую колбу емкостью 250 мл, снабженную мешалкой, термометром и
вводом для азота (с краном), охлаждают смесью сухого льда с метанолом или
в охлаждающем термостате до —30 °C, вакуумируют и заполняют азотом 3 ра-
за. Затем в колбу наливают 50 мл метанола, перегнанного в токе азота, и ох-
лаждают ее. После охлаждения в колбу из специального градуированного при-
емника (см. раздел 2.1.2) вводят 30 мл (0,47 моля) винилхлорида. В приемнике
мономер находится в конденсированном состоянии (во время этой операции
азотная трубка соединяется через трехходовой кран с приемником). Далее в
колбу при перемешивании вводят 0,135 г (0,77 ммоля) аскорбиновой кислоты,
0,40 мл (3,53 ммоля) 30%-ной Н2О2 и 0,15 мл 1%-ного водного раствора
FeSO4-7H2O (5,4-10”6 моля). Закрывают кран на выходе и вводную трубку
для азота заменяют резиновой камерой, заполненной азотом (вновь открыть
кран на выходе из системы).
Через 15 мин реакционная смесь мутнеет, и наконец полимер выпадает в
осадок. После 5 ч полимеризации при —30 °C образовавшийся поливинилхлорид
быстро фильтруют на стеклянном фильтре, промывают осадок метанолом и вы-
сушивают в вакуумном сушильном шкафу при 50 °C. В результате реакции
получают около 6 г порошкообразного полимера, растворимого в циклогексане
или тетрагидрофуране. Определяют характеристическую вязкость полученного
образца в растворе тетрагидрофурана при 20 °C. Возможность термического де-
гидрохлорирования поливинилхлорида можно продемонстрировать на простом
опыте (см. опыт 5-17).
3.2. ИОННАЯ ПОЛИМЕРИЗАЦИЯ
Как и свободнорадикальная, ионная полимеризация является цеп-
ной реакцией; кинетика ионной полимеризации, однако, существен-
но отличается от кинетики свободнорадикальной полимеризации.
Скорости ионной полимеризации слабо зависят от температуры,
поскольку энергии активации стадий инициирования и роста цепи
малы (существуют исключения из этого положения, например
при инициировании на катализаторах Циглера—Натта). Поэтому
в отличие от радикальной полимеризации ионная полимеризация
протекает с высокими скоростями при низких температурах. Так,
полиизобутилен в промышленности получают в присутствии трех-
фтористого бора при —100 °C в жидком пропане (см. опыт 3-23).
Однако температура полимеризации оказывает решающее значение
на структуру образующегося полимера.
При ионной полимеризации бимолекулярный обрыв цепей не-
возможен, поскольку активные концы растущих цепей взаимно
138
отталкиваются вследствие того, что они несут одноименные элек-
тростатические заряды. Обрыв цепи может происходить только на
соединениях, которые реагируют с активными центрами, например
на воде, спиртах, кислотах и аминах. При определенных условиях
ионной полимеризации активные центры сохраняют свою актив-
ность в течение продолжительного времени (так называемые «жи-
вущие» полимеры, см. раздел 3.2.1).
В зависимости от того, является ли активный центр катионом
или анионом, различают катионную и анионную полимеризацию.
В соответствии с этим соединения, инициирующие процессы ион-
ной полимеризации, классифицируют как катионные и анионные
инициаторы. Существуют ионные инициаторы, механизм действия
которых пока полностью не выяснен. Особенностью некоторых
ионных инициаторов является то обстоятельство, что на них воз-
можно осуществление стереоспецифической полимеризации, т. е.
получение стереорегулярных полимеров.
Известные до сих пор инициаторы свободнорадикальной полиме-
ризации позволяют проводить реакции только по двойной С = С-
связи. Число мономеров, способных полимеризоваться по ионному
механизму, гораздо больше и включает соединения, содержащие
С = О- и С = Ы-группы, а также ряд гетероциклических соединений.
3.2.1. Ионная полимеризация по С=С-связи
Способность ненасыщенных соединений к катионной или анионной
полимеризации зависит от типа заместителя при двойной связи,
по которой происходит полимеризация. Известны мономеры, поли-
меризующиеся только по катионному или только по анионному
механизму; существуют и такие мономеры, которые способны поли-
меризоваться по обоим механизмам (см. раздел 1.1, табл. 2).
Электронодонорные группы (алкильные, фенильные, алкокси-
группы) поляризуют двойную связь мономера, при которой неза-
мещенный углеродный атом ненасыщенной группы приобретает
частичный отрицательный заряд. К этому незамещенному углерод-
ному атому могут присоединяться протоны или другие катионы,
при этом замещенный углеродный атом приобретает способность
к участию в реакциях роста, т. е. последовательному присоедине-
нию молекул мономера [10, 11]. При катионной полимеризации
рост цепи может прекратиться только при присоединении соответ-
ствующего аниона, поскольку рекомбинация двух макрокатионов
невозможна. Возможна также реакция передачи цепи за счет
отрыва протона от предпоследнего углеродного атома растущей
цепи:
Инициирование:
6- 6+ +
R+ + CH2=:CH -> R—СН3-СН
139
Рост цепи:
4- 'д— б+
R—СНа—СН 4- СН2=СН
X X
—> r-ch2-ch-ch2-ch
I I
X X
Обрыв цепи:
---СН2-СН + в- —> —сн2-сн-в
I I
X X
Передача цепи:
---СНа—СН --------СН=СН + Нь
X X
Такие нуклеофильные агенты, как вода, спирты, простые и слож-
ные эфиры, ацетали, могут участвовать в реакции передачи цепи,
присоединяясь к макрокатиону и образуя новый катион. Послед-
ний вызывает рост новой цепи. Таким образом, в результате этой
реакции кинетическая цепь не обрывается:
---СН2—СН + НОН -> ---сн2—сн—он + н+
Л— Л 4- 4-
Н+4-СН2—СН СН3—СН
Катионную полимеризацию инициируют протонными кислотами
(серной кислотой, хлорной кислотой, трифторуксусной кислотой),
кислотами Льюиса (см. раздел 3.2.1.1), а также соединениями,
образующими катионы (иод, ацетилперхлорат). Кроме того, неко-
торые мономеры полимеризуются по катионному механизму под
действием облучения частицами высокой энергии.
Выбором соответствующих условий в некоторых случаях удает-
ся остановить реакцию роста на стадии димера. Например, в слу-
чае стирола образуется ненасыщенный линейный или циклический
димер (см. опыт 3-25):
СвН.-,-С11--СН2
Н+ 4- СН2=СН -> СН3-СН
140
—> сн3— сн— сн=сн + н+
Ненасыщенные соединения, содержащие электроноакцепторные
заместители (карбоксиалкильные, нитро-, нитрильную, винильную
группы), могут полимеризоваться под действием таких анионов,
как карбанион ОН" или NH~ [12]. Присоединение аниона в этом
случае происходит к ненасыщенному углеродному атому, несущему
частичный положительный заряд. Поскольку активный конец рас-
тущей цепи представляет собой истинный анион, обрыв может про-
исходить при взаимодействии с катионом. Как и при катионной
полимеризации, бимолекулярный обрыв невозможен; однако воз-
можна реакция передачи цепи с участием электрофильных соеди-
нений.
Инициирование:
64- д- -
R- + СН2=СН ---> R—СН2—СН
Рост цепи:
— 6+ 6- _
R—СН2—СН + СН2=СН > R—СН2—СН—СН2—СН
II II
XX XX
Обрыв цепи:
R—СН2- СН + Нь
I
X
r-ch2-ch2
X
Передача цепи:
r-ch2-ch + hoh-----> R-CH2-CH2 + но-
I I
X X
б+ 6— -
но- + сн2=сн —> но-сн2—сн
I I
X X
Даже водные растворы щелочей могут вызвать анионную поли-
меризацию некоторых мономеров (например, эфиров а-циансорби-
новой кислоты СНз—СН = СН—CH = C(CN)—COOR или нитроэти-
лена СН2 = СН—NO2). Другими анионными инициаторами явля-
141
ются основания Льюиса (третичные амины или фосфины), щелоч-
ные металлы (в виде тонкой суспензии в высококипящих угле-
водородах) и металлоорганические соединения (см. раздел 3.2.1.2).
Эффективность действия перечисленных инициаторов сущест-
венно различается, поскольку поляризуемость ненасыщенных со-
единений значительно зависит от типа заместителя и природы ис-
пользуемого растворителя.
Непосредственный перенос электрона представляет еще один
путь инициирования анионной полимеризации [13]. Так, при взаи-
модействии металлического натрия с нафталином происходит пе-
ренос одного электрона от атома натрия к молекуле нафталина
без замещения водородного атома. Этот электрон может быть пе-
ренесен к молекуле стирола или а-метилстирола, образуя ион-
радикал:
Ион-радикалы очень быстро димеризуются с образованием ди-
анионов, которые могут присоединять молекулы мономера с обоих
концов:
2«СН2—СН СН—СН2—СН2—СН
I I I
XX X
Если реакционная смесь достаточно чиста, так что дианионы
не могут реагировать с загрязнениями (например, с водой), то
рост полимерных цепей продолжается до тех пор, пока весь моно-
мер не исчерпается. При добавлении новых порций стирола к сме-
си эти «живые» цепи будут продолжать расти до тех пор, пока не
произойдет обрыв, например, на доноре протонов с образованием
«мертвого» полимера.
«Живущие» полимеры могут быть использованы для получения
блок-сополимеров путем добавления второго мономера в реакцион-
ную смесь после того, как первый мономер израсходуется. При
этом рост продолжается с обоих концов первоначально получен-
ного блока. При обрыве живых цепей на электрофильных соеди-
нениях в макромолекулы могут быть введены определенные кон-
цевые группы. Так, при взаимодействии «живущего» полистирола
с двуокисью углерода получают полистирол, содержащий карб-
оксильные концевые группы.
В случае идеальной анионной полимеризации два ион-радика-
ла инициируют рост одной полимерной цепи, так что при равно-
мерном распределении молекул мономера среди активных центров
степень полимеризации будет равна удвоенному отношению моль-
ных концентраций мономера и инициатора:
142
Полидисперсность полимеров, полученных анионной полимери-
зацией’ обычно достаточо низка. Например, отношение Mw/Mn
для поли-а-метистирола, полученного по описанному методу, рав-
но 1,05. Однако получение таких полимеров возможно только при
тщательнейшей очистке реагентов и при мгновенном и полном сме-
шении мономера с раствором инициатора (см. опыт 3-27).
При анионной полимеризации а-метилстирола устанавливается
(можно наблюдать) термодинамическое равновесие (зависящее от
температуры) между мономером и полимером. Интенсивно-зеленая
окраска раствора инициатора при добавлении мономера переходит
в красную за счет образования карбанионов. При низкой темпера-
туре (между —40 и —70°C) образуются «живущие» цепи, и рас-
твор становится вязким. При нагревании раствора полимер депо-
лимеризуется, а при охлаждении вновь полимеризуется. Температу-
ра, при которой равновесие сдвинуто полностью в сторону моно-
мера, называется предельной температурой [14], для а-метилсти-
рола она составляет 60 °C, в то время как для большинства осталь-
ных мономеров с двойной С = С-связью предельная температура
лежит выше 250 °C. Предельная температура некоторых мономеров,
полимеризующихся по С = О-связи, и ряда циклических мономеров
также относительна низка; например, для формальдегида или
триоксана она равна 126 °C, для тетрагидрофурана 85 °C. Несмот-
ря на свою термодинамическую неустойчивость поли-а-метилсти-
рол может быть выделен после обрыва «живущих» цепей, по-
скольку блокирование концов цепей обрывом на молекулах воды
или двуокиси углерода кинетически предотвращает деполимериза-
цию. Только при температурах выше 200 °C термическое разложе-
ние полимера протекает с высокими скоростями (см. опыт 5-14),
Металлоорганические катализаторы, открытые Циглером с сотр.
[15, 16], также относятся к ионным инициаторам. На этих ини-
циаторах можно полимеризовать этилен при атмосферном давле-
нии. И хотя относительно механизма полимеризации в присутствии
этих инициаторах существуют различные концепции [17, 18], со-
вершенно точно установлено, что они не являются свободноради-
кальными инициаторами. Как было показано Натта с сотр. [19],
при полимеризации многих виниловых мономеров эти инициаторы
осуществляют стереоспецифический катализ, т. е. позволяют полу-
чить стереорегулярные полимеры. Последние вследствие своей ре-
гулярной структуры отличаются по многим свойствам (см. раз-
дел 1.2) от атактических полимеров, расположение заместителей
в цепи которых имеет беспорядочный характер.
С тех пор было открыто много других инициирующих систем,
позволяющих проводить стереоспецифическую полимеризацию. Так,
при полимеризации стирола под действием металлоорганических
соединений щелочных металлов можно получить стереорегулярный
полистирол [20, 21] (см. опыт 3-28). При катионной и даже при
свободнорадикальной полимеризации некоторых мономеров осуще-
ствляется определенный стереоконтроль реакции роста.
143
Стереоспецифическая полимеризация имеет особое значение при
получении стереорегулярных полидиенов. При свободнорадикаль-
ной полимеризации бутадиена и изопрена молекулы мономера
присоединяются в 1,4-положение (как в цис-, так и в транс-положе-
ние), а также в 1,2- й 3,4-положения в соотношении, зависящем
от условий реакции. Использование специальных инициирующих
систем (см. опыт 3-34) позволяет получить синтетический нату-
ральный каучук, содержащий в цепи более 90% звеньев цисЛД-
изопрена.
В табл. 10 приведены некоторые инициаторы и сложные ини-
циирующие системы для полимеризации диенов, а также структура
образующихся при этом полидиенов.
Таблица 10. Содержание различных структур в полидиенах,
полученных в присутствии разных инициаторов (в %)
Бутадиен Изопрен
1,4-цис 1,4-транс сч о 3 3" 1,4-транс со
Инициатор
Перекиси, применяемые при эмульсионной по- 28 51 21 22 65 6 7
лимеризации 90
А1С13 —— — — — — —
Li в гептане — — —- 93 — —. —
Li в диэтиловом эфире — — — 4 21 6 63
Na в гептане 14 23 61 — 46 9 45
К в гептане — — — — 55 9 36
AlRs/TiCU 49 49 о 97 — — 3
AIR3/VCI4 — 99 — — 99 — —
AlRs/ацетилацетонат Сг 90 (изотакти- ческий)
Как видно из приведенной таблицы, растворители и катализа-
торы значительно влияют на структуру образующихся полидиенов.
Так, строение цепи полиизопрена, полученного на различных ще-
лочных металлах в одном и том же растворителе, значительно
различается. При полимеризации изопрена и бутадиена в присут-
ствии одного и того же инициатора реализуются различные типы
присоединения, что отчетливо демонстрируется в опыте с типич-
ным катализатором Циглера—Натта — системе триалкилалюми-
ния с четыреххлористым титаном.
3.2.1.1. Полимеризация, инициированная кислотами Льюиса
Катионную полимеризацию можно инициировать с помощью сле-
дующих кислот Льюиса: BF3, А1С13, TiCl4, SnCl4, SnCl2, FeCl3 и др.
При этом значительную роль играют так называемые сокатализа-
торы. Сокатализаторами являются протонно-донорные соединения
144
(протонные кислоты, вода, спирты), а также такие соединения, как
галогеналкилы, образующие с кислотами Льюиса ионные комп-
лексы, которые диссоциируют с выделением катиона, инициирую-
щего полимеризацию. При использовании в качестве сокатализа-
торов воды или спиртов истинным инициатором является протон,
образующийся по схеме
BF3 ROH-------->
ч=> BF3-OR F Н+
А при использовании галогеналкилов — алкилкатион:
SnCl4 + RC1 ----------> [RSnCl6] SnC17 + R+
При катионной полимеризации концентрация инициатора ниже,
чем при свободнорадикальной; очень часто концентрации инициато-
ра порядка 1 • 10~3—1 -10~3 моль на моль мономера достаточно для
достижения высоких скоростей полимеризации. При катионной по-
лимеризации не существует единой зависимости скорости и степе-
ни полимеризации от концентрации инициатора; в каждом случае
она определяется как природой мономера, так и рядом других
факторов.
Тип и количество сокатализатора, необходимого для получения
оптимальных условий проведения полимеризации, следует оп-
ределять в каждом отдельном случае; в общем можно лишь ут-
верждать, что количество сокатализатора меньшее, чем эквимоль-
ное по отношению к катализатору, является достаточным.
Полимеризация ненасыщенных мономеров с кислотами Льюиса
проводится при низких температурах около —100 °C и ниже (см.
опыт 3-23). Однако полимеризация циклических мономеров часто
проводится при более высоких температурах.
Катионная полимеризация ненасыщенных соединений практи-
чески всегда осуществляется в растворе. При свободнорадикаль-
ной полимеризации разбавление мономера растворителем приво-
дит к уменьшению скорости полимеризации и молекулярной мас-
сы. При катионной полимеризации разбавление подходящим рас-
творителем в 1—4 раза по отношению к мономеру во многих слу-
чаях приводит к значительному росту молекулярной массы обра-
зующегося полимера, а иногда к увеличению скорости полимериза-
ции. Это объясняется в основном лучшей теплопроводностью,
влиянием полярности и иногда сокаталитическим эффектом рас-
творителя. Последние два обстоятельства играют существенную
роль при подборе растворителей. Как правило, скорость полимери-
зации и молекулярная масса возрастают с увеличением полярности
растворителя и его диэлектрической проницаемости. Кроме того,
ряд растворителей образует комплексы с кислотами Льюиса, ко-
торые затем инициируют катионную полимеризацию. Растворите-
ли могут также влиять на катионную полимеризацию, участвуя в
реакции передачи цепи. Принимая во внимание эти ограничения,
10—732
145
для проведения катионной полимеризации можно рекомендовать
следующие растворители: бензол, циклогексан, хлористый метилен,
четыреххлористый углерод, дихлорэтан, трихлорэтан, хлорбензол,
нитробензол и жидкую двуокись серы.
При полимеризации, инициированной кислотами Льюиса в рас-
творах, реакция часто не начинается сразу же после введения
инициатора; обычно наблюдаются индукционные периоды, которых
не удается избежать даже при самой тщательной очистке реаген-
тов и растворителей. В то же время некоторые реакции протекают
так быстро (флеш-полимеризация), что даже при разбавлении пол-
ная конверсия достигается за минуты (например, для изобутиле-
на, см. опыт 3-23).
Реакции катионной полимеризации крайне чувствительны к по-
сторонним примесям, которые могут либо ускорять (за счет сока-
тализа), либо замедлять реакцию (например, третичные амины).
Кроме того, они могут приводить к реакциям передачи или обрыва
цепи, уменьшающим степень полимеризации получаемого полиме-
ра. Поскольку даже очень малые количества примесей могут при-
водить к подобным эффектам [10“3 % (мол.) и меньше], необхо-
дима тщательнейшая очистка и сушка всех реакционных сосудов.
Опыт 3-23. Полимеризация изобутилена при низкой температуре, иницииро-
ванная газообразным трехфтористым бором
Мономер — изобутилен пропускают через осушительную колонку, заполнен-’
ную твердым КОН, и конденсируют в сухой охлаждаемой ловушке.
Трехгорлую колбу емкостью 100 мл, снабженную мешалкой, притертой проб-
кой и термометром, охлаждают смесью сухого льда с метанолом до —80 °C,
затем при перемешивании в колбу вносят 10 мл изобутилена и 5 г сухого льда,
взятого из центра большой глыбы и содержащего минимально возможное ко-
личество влаги. В сухую пипетку объемом 10 мл набирают газообразный трех-
фтористый бор (при заполнении использовать сухую промежуточную емкость),
затем BFa вводят в жидкий мономер. Полимеризация начинается мгновенно с
образованием каучукоподобного эластичного продукта. Через 45 мин охлаждаю-
щий сосуд убирают, так что непрореагировавший мономер может медленно уле-
тучиваться при нагревании до комнатной температуры. Полученный полиизобу-
тилен растворим в алифатических, циклоалифатических растворителях и в хло-
рированных углеводородах. В растворе циклогексана при 24 °C определяют ха-
рактеристическую вязкость полимера, рассчитывают молекулярную массу (см.
раздел 2.3.2.1).
Опыт 3-24. Полимеризация винилизобутилового эфира, инициированная эфи-
ратом трехфтористого бора при низкой температуре
Пробу 35 мл винилизобутилового эфира промывают 5 раз дистиллированной
водой, сушат над A4gSC>4, перегоняют в присутствии твердого NaOH в токе
азота (т. кип. 80 °C) в сухой приемник (см. раздел 2.1.2). Около 80 мл пропана
конденсируют в сухую охлаждаемую ловушку, предварительно пропустив газ
через колонку, заполненную твердым КОН.
Сухую трехгорлую колбу емкостью 500 мл, снабженную мешалкой, термо-
метром и вводом для азота, откачивают и нагревают пламенем горелки для
удаления следов влаги. Колбу заполняют азотом. Для предотвращения обратной
диффузии азота внутрь прибора (см. раздел 2.1.1) должна быть предусмотрена
ловушка, которую охлаждают смесью сухого льда с ацетоном до —70 °C. В кол-
бу заливают конденсированный пропан и 20 мл (0,15 моля) винилизобутилового
эфира. При сильном перемешивании при —70 °C в колбу вводят по каплям
0,3 мл (0,39 ммоля) свежеперегнанного эфирата трехфтористого бора №о =
= 1,125), через 30 мин в смесь приливают еще 0,3 мл эфирата и содержимое
146
колбы перемешивают еще 30 мин при —70 °C. Все время через прибор пропу-
скают медленный ток азота; полимеризация осуществляется на поверхности
капель инициатора. Для прекращения реакции в колбу добавляют избыток цик-
логексиламина, смесь постепенно нагревают до комнатной температуры, пропан
при этом улетучивается. Полимер в виде небольших кусочков высушивают в ва-
куумном шкафу при 50 °C; перед высушиванием кусочек образца растворяют
в бензоле и высаживают в петролейный эфир. Переосажденный образец исполь-
зуют для определения характеристической вязкости в растворе бензола при
20 °C и измерения температуры плавления полимера (90 °C; см. раздел 2.3, 4.3).
Опыт 3-25. Димеризация стирола
Смесь 52 г (0,5 моля) очищенного от ингибитора стирола (см. опыт 3-01) и
раствора 5 мл конц. H2SO4 в 7,5 мл воды интенсивно перемешивают в трехгор-
лой колбе, емкостью 250 мл, снабженной мешалкой, термометром и обратным
холодильником, после чего колбу нагревают на масляной бане до 120 °C. Внача-
ле образуется молочно-белая эмульсия, которая примерно через 10 мин окра-
шивается в слабо-желтый цвет. Температуру смеси поддерживают равной
120 °C и через каждые 15 мин измеряют показатель преломления органической
фазы (при 20°C), по которому судят о завершении димеризации. Для отбора
пробы перемешивание прекращают и оставляют смесь для расслоения фаз; из
верхней органической фазы, содержащей мономер и димер, отбирают каплю.
После того как несколько измерений дадут постоянный показатель преломле-
ния (что занимает около 4 ч), опыт прекращают, поскольку димеризация за-
вершилась. Строят зависимость уменьшения показателя преломления от про-
должительности реакции (для мономерного стирола — 1,5472; для димерного
стирола п2^ = 1,5877).
Закрытую колбу выдерживают 1 ч в водяной бане при 50 °C; затем в дели-
тельной воронке отделяют органическую фазу от воды. Органическую фазу раз-
бавляют 15 мл бензола, трижды промывают 10%-ным раствором хлористого
натрия и 2 раза насыщенным раствором соды (разделение фаз происходит мед-
ленно) и в заключение насыщенным раствором СаС12. После отстаивания и от-
деления от водной фазы органическую фазу сушат над СаС12 в течение несколь-
ких часов, затем перегоняют при пониженном давлении (0,5 мм рт. ст., форва-
куумный насос). Отбирают фракцию, кипящую при 120—123°C; показатель пре-
ломления фракции должен быть близок к 1,5877, но может быть и меньше за
счет присутствия (около 3%) циклического димера стирола. Выход димерного
стирола 50% от теоретического.
Опыт 3-26. Катионная полимеризация а-метилстирола в растворе
Мономер а-метилстирол очищают от ингибитора (фенола) двукратным про-
мыванием 10%-ным раствором NaOH затем мономер трижды промывают дистил-
лированной водой для удаления NaOH, после чего сушат над гидридом каль-
ция. Перед использованием мономер дополнительно перегоняют над гидридом
кальция в вакууме в токе азота (т. кип. 54 °C при 12 мм рт. ст.). Хлористый ме-
тилен кипятят над Р2О5 (лучше гранулированным или нанесенным на силикагель)
в течение 1 ч и затем перегоняют в сухой приемник.
Тщательно высушенную трехгорлую колбу емкостью 100 мл, снабженную
мешалкой, термометром (с вводом для азота), откачивают и заполняют азотом.
В колбу вносят 5,9 г (0,05 моля) а-метилстирола и 50 мл хлористого метилена.
Полученный раствор охлаждают до —75 °C смесью сухого льда с метанолом.
Затем при помощи пипетки вносят 0,040 мл (0,75 ммоля) конц. H2SO4. Неболь-
шой разогрев смеси свидетельствует о начале полимеризации. После 3 ч реак-
ции полученный вязкий раствор приливают по каплям к 500 мл метанола, оса-
док фильтруют и промывают метанолом. Повторно переосаждают часть получен-
ного образца из 2%-ного бензольного раствора в метанол. Оба образца высуши-
вают до постоянной массы в вакуумном сушильном шкафу при 50 °C; конверсия
составляет около 70%. Определяют температуру стеклования переосажденного
образца и его характеристическую вязкость в растворе толуола при 25 °C (моле-
кулярная масса около 100 000).
Этот опыт можно провести также со стиролом. Кроме того, в качестве ини-
циатора можно использовать эфират трехфтористого бора или SnCl4.
10* ' 147
З.2.1.2. Полимеризация, инициированная
металлоорганическими соединениями.
Многие металлоорганические соединения способны иницииро-
вать полимеризацию ненасыщенных соединений. Из них особое
значение имеют металлоорганические соединения щелочных метал-
лов, цинкорганические и кадмийорганические соединения (напри-
мер, диэтилцинк, диизобутилцинк) и магнийорганические соедине-
ния. Полимеризацию под действием металлоорганических соедине-
ний обычно проводят в растворах. Наиболее распространенными
растворителями являются алифатические и ароматические углево-
дороды (гексан, гептан, декалин, бензол, толуол).
При полимеризации ненасыщенных соединений под действием
металлоорганических соединений обычно используют концентрации
инициатора порядка НО"1—1-10~4 молей на моль мономера; со-
катализаторы, как правило, не нужны. Температура полимериза-
ции ниже комнатной обычно достаточна для получения разумных
скоростей полимеризации.
Все перечисленные выше инициаторы крайне чувствительны к
соединениям, содержащим активный водород. Следовательно, при
проведении опыта необходимо полностью исключить присутствие
кислот, воды, спиртов, меркаптанов, аминов и производных ацети-
лена. Кислород, двуокись углерода, окись углерода, карбонильные
соединения и галогеналкилы также должны отсутствовать, по-
скольку они реагируют с катализаторами. Необходимы тщательная
очистка и высушивание всех исходных реагентов и используемых
приборов, особенно при синтезе «живущих цепей» (см. опыт 3-27).
Опыт 3-27. Полимеризация а-метилстирола*, инициированная натрийнафта-
линовым комплексом в растворе («живущиеэ цепи)
Для проведения этого опыта необходимо, чтобы все операции осуществля-
лись при полном отсутствии воздуха и влаги.
Очистка мономера и растворителей
Мономер а-метилстирол очищают от ингибитора и высушивают по методи-
ке опыта 3-26. Чистый тетрагидрофуран кипятят над металлическим калием
в атмосфере чистого азота по крайней мере 1 сут. Затем в колбу добавляют
немного бензофенона. Если тетрагидрофуран достаточно чист, то содержимое
колбы окрашивается в голубой цвет за счет образования кетилов металла
(если изменение цвета не происходит, кипячение над калием следует продол-
жать). Необходимое для опыта количество тетрагидрофурана отгоняют от рас-
твора кетила в атмосфере азота непосредственно перед использованием.
Приготовление раствора инициатора
Перед началом опыта тщательно моют все части прибора (рис. 32), соби-
рают его, откачивают масляным насосом и высушивают, нагревая на открытом
пламени; затем прибор заполняют очищенным сухим азотом* **.
Тонкодисперсный высокоактивный натрий, относительно безопасный в обра-
щении, получают в атмосфере азота смешением 50 г нейтральной окиси алюми-
ния и 5 г металлического натрия при интенсивном перемешивании в интервале
температур от 150 до 170 °C. Полученный тонкий порошок легко дозировать с
помощью бюретки, изображенной на рис. 32.
Для приготовления раствора инициатора 1,28 г (10 ммоля) особо чистого
нафталина помещают в сосуд Шленка 1 и через вводную трубку 6 в сосуд про-
В этом опыте в качестве мономера можно использовать стирол.
** Азот сушат, пропуская его либо через охлажденную при —180 °C ловуш-
ку, либо над Р2О5 и далее через раствор кетила.
148
Рис. 32. Прибор для получения
натрийнафталинового комп-
лекса:
1 — сосуд Шленка; 2 — сосуд; 3, 4 —
измерительные бюретки; 5 — ЭПР-
ампула; 6 — вводная трубка; 7 —
стеклянный фильтр; 8, 9, 10 — со-
единительные шлифы и пробки.
пускают азот. Затем вносят 100 мл абсолютированного тетрагидрофурана через
ввод 8, не допуская при этом попадания в систему воздуха, и включают магнит-
ную мешалку. Бюретку 3, содержащую порошок натрия и окиси алюминия,
вставляют в пришлифованное отверстие 8 (диаметр отверстия крана, закрываю-
щего бюретку, 4 мм, объем бюретки 10 мл). Прекращают подачу азота и за-
крывают все краны прибора. В сосуд 1 при перемешивании вводят 5 мл порош-
ка натрия. При этом раствор почти мгновенно приобретает интенсивно-зеленую
окраску. После этого бюретку можно заменить пришлифованной пробкой. Для
завершения взаимодействия натрия с нафталином раствор перемешивают еще
в течение 30 мин. Затем прибор переворачи-
вают так, чтобы раствор инициатора фильтро-
вался через стеклянный фильтр 7 в сосуд 2,
а непрореагировавший натрий оставался на
фильтре. Если в лаборатории имеется спектро-
метр ЭПР, можно зарегистрировать ЭПР-
спектр раствора инициатора, свидетельствую-
щий о наличии в нем анион-радикалов. Для
этого раствор инициатора заливают в ампу-
лу 5, охлаждают до —76 °C, отпаивают ампу-
лу и помещают ее в резонатор спектрометра
ЭПР для регистрации спектра. Для определе-
ния содержания натрийнафталинового комп-
лекса в растворе инициатора бюреткой 4 от-
бирают 10 мл раствора и добавляют пробу к
небольшому количеству дистиллированной во-
ды в колбе Эрленмейера. Продукт гидролиза
титруют 0,1 н. HCI; раствор должен содер-
жать около 100 ммоль/л инициатора.
Полимеризация
В сосуд Шленка, высушенный нагревани-
ем под ваккумом и заполненный азотом, вно-
сят 50 мл абсолютированного тетрагидрофу-
рана. Пропуская через сосуд слабый ток азо-
та, его присоединяют к бюретке 4 прибора
(см. рис. 32) с помощью шлифа 10. Для уда-
ления возможных следов примесей в сосуд
приливают несколько капель раствора инициа-
тора. Если зеленая окраска раствора сохра-
няется, из бюретки 4 добавляют 1 ммоль ини-
циатора (около 10 мл; точно рассчитать по
результатам титрования). Сосуд Шленка еще
раз продувают азотом, закрывают пробкой и
помощью сухого медицинского шприца при интенсивном перемешивании содер-
жимого сосуда в сосуд вводят 5,90 г (0,05 моля, что соответствует примерно
5,5 мл) а-метилстирола (за один прием). Раствор мгновенно окрашивается в
красный цвет, что обусловлено образованием карбанионов.
Реакционную смесь выдерживают в течение 1 ч при —78 °C, затем из со-
суда в токе азота отбирают определенный объем раствора с помощью сухого
медицинского шприца, заполненного азотом. Отобранную пробу по каплям при-
бавляют к 10-кратному объему метанола, в результате чего поли-а-метилстирол
выпадает в осадок. К оставшейся в сосуде реакционной смеси добавляют новую
порцию (5,9 г) а-метилстирола; при этом красный цвет раствора не должен
исчезать. . Через 1 ч из смеси вновь отбирают пробу и в сосуд опять добавляют
свежую порцию мономера. Еще через 1 ч опыт прекращают добавлением мета-
нола к реакционной смеси, и полимер выпадает в осадок. Полученные образцы
переосаждают из раствора тетрагидрофурана в метанол, фильтруют с отсасы-
ванием и высушивают в вакуумном сушильном шкафу при 50 °C.
Определение молекулярной массы п о л и-а-м ет и л ст и р о л а
Если полимеризация а-метилстирола под действием патрийнафталинового
комплекса протекает согласно описанному механизму без обрыва, то степень
охлаждают до —78 °C. Затем с
149
полимеризации полученного полимера должна подчиняться следующему просто-
му соотношению:
Р = 2
где концентрация мономера и инициатора выражены в молях.
Следовательно, если к 0,05 моля а-метилстирола добавить 0,001 моля ини-
циатора, степень полимеризации полученного полимера будет составлять 100
(это соответствует молекулярной массе 11 800).
Определите характеристическую вязкость полученных образцов полимера
в растворе толуола при 30 °C и рассчитайте их молекулярные массы. Сопоставь-
те полученные значения с предсказываемыми теоретически.
Опыт 3-28. Стереоспецифическая полимеризация стирола, инициированная
«-амилнатрием
Получение «-а милнатрия*
н-Гептан и чистый петролейный эфир (т. кип. 110 °C) раздельно кипятят
в атмосфере азота над металлическим натрием в течение 4 ч, перегоняют и хра-
нят над натрием.
«-Амилнатрий получают в сухой трехгорлой колбе емкостью 250 мл, имею-
щей вмятины с четырех сторон для увеличения турбулентности при перемеши-
вании, впаянный ввод для газа, высокоскоростную мешалку (частота вращения
около 20 000 об/мин), переходник для капельной воронки и обратный холодиль-
ник; третье горло колбы закрывают пришлифованной пробкой. Обратный холо-
дильник имеет затворный кран и соединен с промывалкой или затвором для га-
за (см. раздел 2.1.1). В целях безопасности в качестве охлаждающей жидко-
сти вместо воды используют толуол. Перед началом работы необходимо убе-
диться, что весь прибор собран без напряжения, которое может привести к по-
ломке из-за вибрации при перемешивании. Все краны и пришлифованные соеди-
нения должны быть закреплены предохранительными пружинами.
После того как прибор откачан и 3 раза заполнен азотом, в него в токе
азота вносят 100 мл абсолютированного петролейного эфира и 4,6 г натрия,
тщательно очищенного с поверхности под слоем петролейного эфира. Петролей-
ный эфир нагревают до кипения на закрытой электрической плитке (открытое
пламя недопустимо) и после того, как натрий расплавится, мешалку постепенно
(примерно в течение 2 мин) переводят на максимальную скорость. Через 5 мин
перемешивание прекращают и сразу же убирают нагреватель, так что получен-
ная суспензия натрия (диаметр частиц 5—20 мкм) может остывать до комнат-
ной температуры. Петролейный эфир заменяют «-гептаном: для этого обычную
пришлифованную пробку заменяют специальной пробкой с резиновой проклад-
кой, которую прокалывают шприцем, и отсасывают петролейный эфир. Затем в
реакционный сосуд осторожно приливают при интенсивном перемешивании через
капельную воронку 100 мл абсолютированного «-гептана. После того как натрий
осядет, «-гептан удаляют; эту операцию повторяют дважды. Пробку с проклад-
кой вновь заменяют обычной и через капельную воронку вводят 100 мл абсо-
лютированного «-гептана. Затем при интенсивном перемешивании в реакцион-
ную смесь приливают по каплям раствор 10,66 г (0,1 моля) хлористого «-амила
(перегнанного в атмосфере азота) в 20 мл абсолютированного «-гептана. При
добавлении первых капель раствор окрашивается в темный цвет, и колбу охлаж-
дают смесью сухого льда с метанолом до —25 °C. Остальной раствор хлористого
«-амила вводят в течение 30 мин, поддерживая температуру при —25 °C (реак-
ционная смесь не должна нагреваться выше —20 °C, при необходимости следует
добавить сухой лед). Темно-синюю суспензию инициатора перемешивают еще
30 мин при —25 °C и затем переносят шприцем в специальный сухой приемник;
при —20 °C инициатор сохраняет активность в течение длительного времени.
Перед употреблением определяют содержание «-амилнатрия в суспензии
следующим образом [23].
* Работу с н-амилнатрием обязательно вести в защитных очках] Все опе-
рации необходимо проводить в условиях, исключающих попадание влаги и воз^
духа в реакционный сосуд.
150
Определение суммарного содержания щелочи. Пробу 2 мл
суспензии инициатора разлагают добавлением к 50 мл перегнанного метанола.
Затем добавляют 20 мл воды и образец титруют 0,1 н. НС1 в присутствии фе-
нолфталеина в качестве индикатора (Vi).
Определение содержания свободной щ е л о ч и. Пробу 5 мл
суспензии инициатора добавляют к 15 мл перегнанного бензилхлорида. После
добавления 20 мл перегнанного метанола и 30 мл воды раствор титруют 0,1 н.
НС1 в присутствии того же индикатора (V2) . Содержание я-амилнатрия полу-
чают по разности этих определений, как показано на следующем примере.
Суммарное содержание щелочи: Vt = 14,53 мл 0,1 н. НС1 со-
ответствует содержанию 0,7260 ммоля Na в 1 мл суспензии инициатора.
Содержание свободной щелочи: V2 = 7,33 мл 0,1 н. НС1 соот-
ветствует содержанию 0,1465 ммоля Na в 1 мл суспензии инициатора.
Содержание амилнатрия: 0,7260—0,1465 = 0,5795 ммоля Na на
1 мл суспензии инициатора (79,8% щелочи титруется), что соответствует
0,5795 моля амилнатрия в 1 л.
Полимеризация
В трехгорлую колбу емкостью 500 мл (с вводом и выводом для азота),
снабженную мешалкой и термометром, наливают 250 мл абсолютированного
я-гептана и 26 г (0,25 моля) сухого стирола, перегнанного в атмосфере азота.
После охлаждения до —20 °C из колбы откачивают воздух и заполняют ее азо-
том, повторяя эту операцию 3 раза. Вместо термометра вставляют специальную
пробку с резиновой прокладкой, которую прокалывают шприцем, и в колбу
вводят 0,05 моля (0,2 моля на моль стирола) я-амилиатрия, что соответствует
в рассмотренном примере 86,4 мл суспензии инициатора. При непрерывном
перемешивании реакционной смеси в колбу вновь вставляют термометр и,
пропуская слабый ток азота, реакционную смесь выдерживают при —20±1 °C
еще 6 ч. Полимеризацию прекращают добавлением в колбу около 10 мл мета-
нола. Затем реакционную смесь при перемешивании приливают к 2,5 л метанола,
выпавший осадок фильтруют с отсасыванием, промывают метанолом и сушат
в вакуумном сушильном шкафу в течение ночи при 50 °C. Для удаления остат-
ков неорганических соединений сухой полимер растворяют в бензоле и центри-
фугируют 30 мин с частотой вращения мешалки 4000 об/мин. Полистирол выса-
живают из чистого раствора, используя в качестве осадителя метанол. Полимер
фильтруют, промывают метанолом и сушат при 50 °C в вакуумном сушильном
шкафу до постоянной массы (около 20 ч). Выход составляет 25%.
Кристаллизация и определение характеристических
свойств изотактического полистирола
Полученный полистирол не должен содержать атактическую фракцию и,
следовательно, должен кристаллизоваться. Для этого 4 г полимера кипятят
в 200 мл чистого я-гептана в течение 4 ч. В горячем виде раствор фильтруют
для отделения нерастворимой изотактической фракции полимера (для сравнения
рекомендуется проделать эти же операции с атактическим полистиролом умерен-
ной молекулярной массы, полученным свободнорадикальной полимеризацией).
Атактическую фракцию полистирола, оставшуюся в растворе я-гептана, вы-
саживают в метанол и фильтруют; оба образца сушат в вакуумном шкафу при
50 °C и взвешивают. Затем проверяют растворимость обеих фракций в метил-
этилкетоне, определяют плотности образцов (см. раздел 2.3.7), их характеристи-
ческие вязкости в растворе бензола при 20 °C; сопоставляют рентгенограммы
и инфракрасные спектры (см. разделы 2.3.6 и 2.3.9). Сопоставьте полученные
результаты с характеристиками полистирола, полученного свободнорадикальной
полимеризацией.
С помощью микроскопа, имеющего предметный столик с нагревом, опреде-
ляют интервал температур плавления изотактического кристаллического образ-
ца. При плавлении наблюдаются следующие стадии: размывание краев, начало
агломерации (спекания), начало плавления, начало течения расплава, появление
чистого расплава.
Температурный интервал между 4-й и 5-й стадиями принимается за ин-
тервал плавления; для полимера, полученного в данном опыте, он находится
между 205 и 215 °C.
151
Опыт 3-29. Получение изотактического и синдиотактического полйметилмет-
акрилата, инициированного н-бутиллитием в растворе
Получение раствора инициатора
В тщательно высушенную трехгорлую колбу емкостью 250 мл, заполненную
азотом и снабженную мешалкой и вводом для азота, вносят 50 мл абсолютиро-
ванного бензола, около 0,5 г (0,072 моля) мелко нарезанного лития и 4,64 г
(0,050 моля) свежеперегнанного н-бутиллития (необходимо исключить возмож-
ность попадания воздуха и влаги во время этих операций). На колбе укрепляют
обратный холодильник и при перемешивании колбу осторожно нагревают до
80 °C. Реакционную смесь выдерживают при этой температуре в течение 4 ч,
после чего выключают нагрев и перемешивание прекращают. Полученный рас-
твор н-бутиллития имеет примерно 1 М концентрацию. Для точного определения
концентрации инициатора из раствора отбирают пробу 2 мл с помощью шприца
(при отборе пробы необходимо пропускать ток азота), добавляют ее к 20 мл
чистого метанола и титруют 0,1 н. НС1, используя в качестве индикатора мети-
ловый красный.
Полимеризация
Метилметакрилат, бензол, толуол, 1,2-диметоксиэтан ректифицируют и пе-
регоняют над гидридом кальция в токе сухого азота в специальный приемник
(см. раздел 2.1.2).
Две трехгорлые колбы емкостью 250 мл (с вводом для азота) высушивают,
нагревая в пламени горелки при откачке воздуха, и затем несколько раз запол-
няют сухим азотом. Каждую колбу снабжают мешалкой и специальной пробкой
с резиновой, самозатягивающейся прокладкой (см. раздел 2.1.3). В первую кол-
бу заливают 100 мл толуола, во вторую — 100 мл 1,2-диметоксиэтана и в обе
колбы добавляют по 0,006 моля н-бутиллития (примерно 6 мл 1 7И раствора
инициатора). Колбы охлаждают до — 78 °C, затем в каждую из них с помощью
шприца вводят по 10 мл (0,6 моля) метилметакрилата. Через 30 мин полиме-
ризацию прекращают добавлением в реакционную смесь 10 мл метанола и каж-
дый образец высаживают в 1,5 л низкокипящего петролейного эфира. После
фильтрования с отсасыванием влажные образцы полимера растворяют в бензо-
ле и центрифугируют около 30 мин при частоте вращения мешалки 4000 об/мин
для отделения от нерастворимых продуктов (сшитого полимера и неорганиче-
ских продуктов гидролиза). Образцы полимера переосаждают из бензольного
раствора в петролейный эфир (15-кратнос количество), фильтруют и сушат в
вакуумном шкафу при 40 °C. Выход изотактического полимера, полученного в
растворе толуола, составляет 60—70%, а выход синдиотактического полиметил-
метакрилата, полученного полимеризацией в растворе 1,2-диметоксиэтана, соот-
ветственно равен 20—30%. Определяют характеристические вязкости получен-
ных образцов в растворе ацетона при 25 °C (см. раздел 2.3.2.1), записывают
ПК-спектры полимеров между пластинами из КВг (см. раздел 2.3.9). Количество
изо- и синдиоструктур в образцах полимера можно определить качественно и
количественно по ПК-спектрам [24].
Опыт 3-30. Стереоспецифическая полимеризация изопрена, инициированная
я-бутиллитием
А. Полимеризация в массе (получение ф^с-1,4-п о л и изо-
прена)
Изопрен очищают от ингибитора и сушат по методике, описанной в опыте
3-01. Перед использованием мономер в атмосфере азота пропускают через колон-
ку высотой 30 см, наполненную нейтральной окисью алюминия.
Сухую двухгорлую колбу емкостью 100 мл, снабженную магнитной мешал-
кой, термометром и (вводом для азота), нагревают на открытом пламени при
откачивании и затем заполняют азотом, пропущенным над Р2О5. После этого
в колбу в токе азота заливают 38 г (0,56 моля) сухого изопренш и 0,5 ммоля
^-бутиллития (2,5 мл 0,2 М раствора в бензоле; см. опыт 3-29). Раствор ини-
циатора и мономер осторожно перемешивают и колбу нагревают на водяной
бане до 30 °C. Как только начнется полимеризация, о чем свидетельствует заки-
пание изопрена (т. кип. 34 °C), водяную баню убирают. Если полимеризация не
начинается, то это значит, что в колбу попала влага или другие загрязнения,
разрушающие инициатор. В этом случае в колбу вносят еще одну порцию ипи-
152
циатора и повторяют описанную операцию. Через 7 ч реакция заканчивается
практически с количественным выходом. Полученный высокоэластичный прозрач-
ный продукт охарактеризовывают, как описано в пункте Б. Для этого около
1 г полимера очищают растворением в 50 мл бензола и затем высаживают
в 500 мл этанола. Полученный полимер растворим в бензоле, толуоле, декали-
не, сероуглероде. Определяют характеристическую вязкость полимера в растворе
толуола при 25 °C и рассчитывают его молекулярную массу (см. раздел 2.3.2.1).
Б. Полимеризация в растворе (получение 3,4-п о л и изо-
прена)
Изопрен очищают, как описано в пункте А. Циклогексан и 1,2-димет-
оксиэтан кипятят в течение 6 ч над металлическим натрием и перегоняют в атмо-
сфере азота.
Тщательно высушенную двухгорлую колбу емкостью 250 мл, снабженную
термометром, вводом для азота и магнитной мешалкой, повторно откачивают
и заполняют высушенным над Р2О5 азотом. В токе азота в колбу наливают
20 мл 1,2-диметоксиэтана, 100 мл циклогексана и 6,8 г (0,1 моля) изопрена.
Включают магнитную мешалку и при комнатной температуре в раствор добав-
ляют 0,1 ммоля н-бутиллития (0,5 мл 0,2 М раствора в бензоле); при этом
раствор окрашивается в желтый цвет и разогревается, что свидетельствует о на-
чавшемся процессе полимеризации. Если реакция не начинается (из-за присутст-
вия загрязнений), в реакционную смесь повторно вносят раствор инициатора,
пока не получится устойчивой желтой окраски смеси. Через 3 ч реакция завер-
шается, и к полученному продукту добавляют 50 мг N-фенил-Р-нафтиламина
в качестве антиоксиданта. Полимер высаживают в этанол, фильтруют и сушат
в вакуумном сушильном шкафу при 50 °C. Выход составляет 90—95%. Для
определения характеристических свойств полимера (см. пункт В) около 1 г
полученного продукта переосаждают из 2%-ного бензольного раствора в 10-крат-
ное количество этанола.
В. Исследование микроструктуры полидиенов с по-
мощью ПК-спектроскопии
Качественная и количественная оценка строения цепи полидиенов может
быть получена с помощью ПК-спектров полимера*. Для этого на пластины из
NaCl наносят тонкий слой 2%-ного раствора полимера в сероуглероде (спек-
трально-чистом) и после испарения растворителя на пластине получают тонкую
пленку полимера. Затем пластины с нанесенным полимером помещают в спектро-
метр и регистрируют ПК-спектр. Возможные типы соединения звеньев в цепи
полидиенов и соответствующие этим звеньям характеристические частоты при-
ведены в табл. 11.
В первом приближении достаточно провести качественный анализ полученных
спектров. Полиизопрен, полученный при полимеризации в растворе, имеет интен-
Таблица 11. Различные типы присоединения звеньев
в цепи полидиенов и их характеристические частоты
в ИК-спектрах
Тип звена Длина волны, мкм Волновое число, СМ-1 1 Тип звена Длина волны, мкм Волновое число, см—1
Полибутадиен 1,2- .... 11,0 909 1 Полиизопрен 1,2- .... 11,0 909
транс-1,4- . . 74 1355 3,4- .... 11,27 888
10,3 981 транс-1,4-. . 7,55 1325
zpc-1,4 . . . 7,62 1311 8,71 1148
13,5—13,8 741—725 цис-\,4 . . . 7,60 1315
8,87 1127
* Звенья, соответствующие 1,2- и 1,4-присоединению, можно различить по
взаимодействию полимера с гидроперекисью бензоила [25].
153
сивную полосу 888 см-1, которая свидетельствует о большой вероятности при-
соединения в положение 3,4. Для образца, полученного при полимеризации в
массе, интенсивность этой полосы гораздо меньше по сравнению с полосами при
1127 и 1315 см"1; в этом образце преобладает присоединение звеньев в
цис- 1,4-положение. В полимере, полученном при свободнорадикальной полиме-
ризации в эмульсии (опыт 3-20), присутствуют все возможные типы присоеди-
нения; содержание звеньев, соответствующих цис- 1,4-присоединению, невелико.
3.2.1.3. Полимеризация на катализаторах Циглера—Натта
Точный механизм полимеризации на металлоорганических катали-
заторах, открытых Циглером [15, 16], до конца еще не выяснен.
В основном эти катализаторы состоят из алюминийорганического
соединения и соединения переходного металла, например титана.
На этих катализаторах впервые удалось осуществить нерадикаль-
ную полимеризацию этилена и получить высокомолекулярный про-
дукт при атмосферном давлении и невысокой температуре. До это-
го открытия полиэтилен получали только по механизму свободно-
радикальной полимеризации при высоких давлении и температуре.
Кроме того, применение катализаторов Циглера—Натта позволи-
ло провести полимеризацию высших а-олефинов и ряда других мо-
номеров; во многих случаях были получены стереорегулярные по-
лимеры [19, 26—28].
Полимеризация на катализаторах Циглера—Натта является
сложной реакцией, которая зависит от многих факторов и не всег-
да хорошо воспроизводится.
На эффективность действия этих металлоорганических катали-
заторов влияет также тип алюминийорганического соединения; для
этих целей применяют триэтилалюминий, диэтилалюминийхлорид,
этилалюминийдихлорид, триизобутилалюминий, этилалюминийхло-
рид. Существенную роль играют также тип соединения переходного
металла и его кристаллическая модификация. При использовании
хлоридов титана в качестве второго компонента катализатора при-
меняют либо твердый кристаллический TiCl3, либо жидкий TiCl4.
TiCl3 добавляют к алюминийорганическому соединению в виде
суспензии в алифатических углеводородах; смесь этих компонентов
образует активный катализатор. Жидкий TiCl4 сразу же восста-
навливается алюминийорганическим соединением (см. опыт 3-31).
Поскольку для этой реакции требуется определенное время, макси-
мум активности катализатора достигается не сразу. В этом случае
перед добавлением мономера целесообразно выждать некоторое
время для завершения взаимодействия компонентов катализатора
(т. е. «вызревания» катализатора [29]). Отношение Al/Ti также
является важным фактором, поскольку оно влияет как на скорость
полимеризации, так и на молекулярную массу получаемого про-
дукта. Реакции этого типа часто являются гетерогенными, поэтому
размер частиц твердого соединения переходного металла, а также
процессы диффузии (скорость перемешивания) могут играть за-
метную роль. Кроме того, полимеризация на катализаторах Цигле-
ра—Натта очень чувствительна к таким примесям, как кислород,
154
вода, спирты, кислоты, основания. Полимеризацию, как правило,
проводят в алифатических или ароматических углеводородах в ин-
тервале температур 20—100 °C.
Опыт 3-31. Полимеризация этилена на катализаторах Циглера—Натта
При работе с алюминийорганическими соединениями необходимо проявлять
особую осторожность, поскольку они легко воспламеняются при контакте с воз-
духом и водой. Все операции следует выполнять так, чтобы исключить попада-
ние влаги и воздуха. Кроме того, при попадании на кожу этих соединений об-
разуются труднозаживающие раны. Работу с этими соединениями надо прово-
дить в защитных очках.
Рис. 33. Прибор для полимеризации на катализаторах Циглера — Натта:
1 — промывалка; 2 — сосуд для сброса избыточного давления этилена; 3 реакционный со-
суд; 4 — затвор.
Прибор для проведения полимеризации (рис. 33) состоит из четырехгорлой
колбы, емкостью 1 л, снабженной мешалкой, термометром и вводом и выводом
для азота (с кранами на обеих трубах). Вводная трубка соединена с сосудом
(2), позволяющим стравливать избыточное давление, и промывалкой (7),
с обеих сторон которой имеются краны. На выводной трубке также имеется
затвор 4, в качестве которого можно использовать промывалку (см. раз-
дел 2.1.1). Из собранного прибора удаляют воздух повторным откачиванием и
наполнением азотом (вакуумная линия и трубка, через которую подают азот,
соединены с прибором через трехходовой кран Z?i). При пропускании через при-
бор азота обе промывалки 1, 4 и сосуд 2 заливают на треть сухим петролейным
эфиром (т. кип. 100—140 °C), предварительно перегнанным над натрием в атмо-
сфере азота, а в колбу 3 наливают 500 мл петролейного эфира. Для очистки
этилена в промывалку 1 заливают 5 мл триэтилалюминия*. От тройного крана
(£1) отсоединяют вакуумную линию и присоединяют баллон с этиленом. Вклю-
чают мешалку и через реакционный сосуд начинают пропускать этилен, одно-
временно колбу 3 нагревают на бане до 50 °C. Через 15 мин раствор в колбе
насыщается этиленом. Кран на выходной трубке закрывают, так что утечка
этилена может происходить только через затвор 2. Затем вынимают термометр
и через это горло при перемешивании в колбу быстро с помощью шприца вно-
сят 2 мл (15 ммоля) триэтилалюминия** и 1 мл (9,1 ммоля) TiCl4; воздух при
этом в колбу не попадает, поскольку в нее все время подается этилен.
* Пипетку из-под триэтилалюминия следует сразу же промыть несколько
раз сухим гептаном, затем ацетоном и высушить.
** Вместо триэтилалюминия можно использовать диэтилалюминийхлорид
А1(С2Н5)2С1 либо этилалюминийсесквихлорид А12(С2Н5)3С13.
155
После введения катализатора реакционная смесь окрашивается в коричне-
вый цвет; в это время может происходить усиленное поглощение этилена. Реак-
ционную смесь перемешивают в течение 30 мин с частотой вращения мешалки
700 об/мин, а затем из-за возрастания вязкости среды частоту вращения увели-
чивают до 900 об/мин. Скорость подачи этилена регулируют так, чтоб через
затвор 2 происходила минимальная утечка мономера (любое изменение скоро-
сти полимеризации легко обнаруживается по изменению уровня жидкости в за-
творе 2). Если этилен подают из газометра, то за ходом полимеризации следят
по расходу газа через каждые 5 мин и строят график зависимости расхода эти-
лена от времени.
С началом полимеризации температура реакционной смеси повышается до
55—60 °C, и прозрачный раствор становится мутным. По мере протекания
реакции скорость полимеризации уменьшается, и система охлаждается до 50 °C.
За 45—55 мин содержимое колбы превращается в коричневую кашицеобразную
массу. Через 60 мин после начала реакции полимеризацию прекращают добав-
лением 30 мл н-бутанола, в результате чего реакционная смесь сразу же стано-
вится белой. Примерно через 10 мин в колбу добавляют 150 мл смеси метано-
ла и соляной кислоты (2:1) и перемешивание продолжают еще 10 мин. Поли-
мер фильтруют с отсасыванием, тщательно промывают метанолом, затем аце-
тоном и сушат в вакуумном шкафу при 50 °C; выход составляет около 50 г.
В зависимости от молекулярной массы полиэтилен может быть мягким вос-
кообразным либо твердым, кристаллическим. В данном опыте образуется доста-
точно высокомолекулярный продукт, плавящийся при температуре около 130 °C.
При комнатной температуре он нерастворим, однако при повышенной темпера-
туре (100—150 °C) растворяется в алифатических и ароматических углеводоро-
дах. Измерение вязкости можно проводить в ксилоле, тетралине или декалине
при 135 °C, во избежание окислительной деструкции к полимеру добавляют
около 0,2% антиоксиданта — М-фенил-0-нафтиламина. Полиэтилен легко пере-
рабатывается под давлением. При нагревании полиэтилена между металлически-
ми пластинками до 180—190 °C из него можно получать тонкую пленку (см.
раздел 2.4.2.1). Полученную пленку охлаждают водой и отделяют от пластин.
Пленку можно использовать для регистрации ПК-спектра полимера для опреде-
ления степени его кристалличности (см. раздел 2.3.6) и степени разветвленности
(см. раздел 2.3.9).
Опыт 3-32. Стереоспецифическая полимеризация пропилена на катализато-
рах Циглера—Натта
А. Получение изотактического полипропилена
Пропилен можно полимеризовать на том же самом катализаторе и в тех
же условиях, что и этилен (см. опыт 3-31); выход полимера 3—4 г.
Полученный таким образом полипропилен содержит низкомолекулярную
и аморфную фракции, представляющие собой липкую или каучукообразную
эластичную массу. Они могут быть извлечены из полученного продукта экст-
ракцией диэтиловым эфиром, а затем н-гептаном в аппарате Сокслета в тече-
ние 24 ч в атмосфере азота. Из 4 г исходного продукта около 0,2 г растворимы
в диэтиловом эфире и 1,2 г в н-гептане; растворенные фракции могут быть вы-
делены выпариванием растворителя либо высаживанием в метаноле.
Изотактический полипропилен обладает такой же растворимостью, как и по-
лиэтилен; плавится он, однако, при более высокой температуре (интервал тем-
пературы кристиллизации 160—170 °C).
Б. Влияние гетерогенных зарод ышеобразователей на
кристаллизацию изотактического полипропилена
При кристаллизации изотактического полипропилена [30—32] из расплава
введение в него специальных зародышей может оказывать влияние на число и
размер образующихся сферолитов (а следовательно, и на скорость кристаллиза-
ции). Образование мелких сферолитов увеличивает прозрачность полипропилено-
вой пленки и в некоторых случаях влияет па механические свойства образца
[33].
Влияние гетерогенного зародышеобразования на кристаллизацию изотакти-
ческого полипропилена из расплава можно продемонстрировать следующим обра-
зом. Небольшое количество порошка полимера растирают в ступке с 0,1% бен-
156
зоата натрия*. Небольшую порцию этой смеси плавят при 250 °C на пластинке,
затем накрывают покровным стеклом и прижимают его корковой пробкой, что-
бы образующийся слой полимера имел минимальную толщину. Образец выдер-
живают несколько минут при 220—250 °C и затем кристаллизуют изотермически
при 130 °C на нагреваемом предметном столике поляризационного микроскопа.
Полипропилен без добавок кристаллизуют аналогичным образом**. При наблю-
дении в микроскопе за кристаллизацией обоих образцов легко обнаружить раз-
личие в размерах образующихся сферолитов. Можно использовать и обычный
микроскоп с подогревом предметного столика, но в этом случае необходимо
иметь два поляризатора, которые помещают перед конденсером и перед окуля-
ром. Затем необходимо добиться максимального затемнения поля при вращении
одного поляризатора относительно второго.
Опыт 3-33. Стереоспецифическая полимеризация стирола на катализаторах
Циглера-Натта
Сухую трехгорлую колбу емкостью 1 л, снабженную мешалкой, термомет-
ром, вводом и выводом для газа и капельной воронкой с трубкой для вырав-
нивания давления, откачивают и заполняют азотом несколько раз. Пропуская
ток азота через колбу, в нее вносят с помощью шприца 0,9 мл (8,2 ммоля)
TiCU. Затем из капельной воронки вводят в течение 20 мин при перемешивании
смесь 3,3 мл (24 ммоля) триэтилалюминия*** (или триизобутилалюминия) с 5 мл
абсолютированного н-гептана. Поскольку реакция компонентов катализатора
вначале протекает с выделением большого количества теплоты, реакционную
смесь необходимо охлаждать на бане с температурой около 0 °C. Во избежание
воспламенения при поломке колбы охлаждающая жидкость не должна содер-
жать воды {триэтилалюминий реагирует с водой со взрывом), поэтому рекомен-
дуется использовать, например, смесь сухого льда с 1,2-диметоксиэтанолом. По-
сле введения всего катализатора в колбу реакционную смесь перемешивают
еще в течение 30 мин при комнатной температуре. Затем из другой капельной
воронки быстро вводят 400 мл (3,5 моля) тщательно высушенного стирола (см.
опыт 3-01). Увеличивают скорость перемешивания и нагревают реакционную
смесь до 50 °C на масляной бане. Через 1—2 ч содержимое колбы становится
вязким и наконец гелеобразным (через 3—6 ч). Убирают баню и из капельной
воронки при интенсивном перемешивании постепенно добавляют 50 мл метанола
(в течение 10 мин). Добавлять метанол следует очень осторожно при тщатель-
ном перемешивании. После разложения катализатора в систему быстро добав-
ляют еще 350 мл метанола при интенсивном перемешивании. В результате про-
исходит осаждение полистирола в виде мелких хлопьев. Систему перемешивают
еще 10 мин, осадок фильтруют, отсасывают и промывают метанолом. Для пол-
ного удаления катализатора полимер в течение 1 ч перемешивают с 500 мл ме-
танола, подкисленного 5 мл конц. соляной кислоты. После фильтрования и про-
мывки метанолом образец высушивают в вакуумном сушильном шкафу при
60 °C; выход полимера 5—30%.
Для отделения аморфной фракции сухой полимер перемешивают в течение
3 ч со смесью 500 мл ацетона и 2 мл конц. НС1; оставшийся нерастворимый про-
дукт фильтруют и сушат в вакуумном сушильном шкафу при 60 °C; выход со-
ставляет 85—95% полистирола, способного кристаллизоваться.
Нерастворимый в ацетоне полистирол кипятят 2 ч в свежеперегнанном ме-
тилэтилкстоне. Затем смесь выдерживают в течение ночи при комнатной темпе-
* Достаточно поместить небольшую порцию порошка полипропилена на
нагретую пластину и добавить несколько крошек бензоата натрия. При кристал-
лизации можно наблюдать, что сферолиты, образующиеся вблизи бензоата нат-
рия, имеют меньший размер, чем в остальной массе образца.
** Для количественных исследований (которые -требуют одинаковой терми-
ческой предыстории образцов) предпочтительно оба образца оставлять остывать
до комнатной температуры на выключенной электрической плитке.
♦** При работе с алкилалюминием необходимо полностью исключить возмож-
ность его контакта с влагой и воздухом.
157
ратуре, полимер фильтруют и сушат в вакуумном сушильном шкафу при 60 °C.
Выход кристаллизующегося изотактического полистирола 95—100% от нерас-
творимой в ацетоне фракции. Определяют температуру (интервал) кристаллиза-
ции (см. опыт 3-28), плотность образца (см. раздел 2.3.7) и характеристическую
вязкость в растворе бензола при 20 °C.
Опыт 3-34. Стереоспецифическая полимеризация бутадиена на катализаторах
Циглера—Натта (получение zvrzc-l,4-пол и бутадиена)
Ловушку с трехходовым краном сушат в течение 1 ч при 120 °C, затем от-
качивают и заполняют чистым сухим азотом. После этого ловушку через кран
и осушительную колонку, заполненную фосфорным ангидридом, соединяют с
Рис. 34. Прибор для стереоспецифи-
ческой полимеризации бутадиена на
катализаторах Циглера — Натта:
/ — ловушка с бутадиеном; 2 — реакцион-
ный сосуд; 3 — капельная воронка со спе-
циальной пробкой с самозатягивающейся
прокладкой; 4 — затвор.
баллоном, наполненным бутадиеном. Воздух из соединительных трубок и кра-
нов вытесняют бутадиеном. При охлаждении смесью сухого льда с метанолом
в ловушку конденсируют 20 г (33 мл) бутадиена.
Бензол кипятят над калием в течение дня, затем в токе азота перегоняют
200 мл абсолютированного таким образом бензола в сухую капельную воронку
с трубкой, позволяющей выравнивать давление. В сухую ампулу с краном за-
гружают 300 мг (0,84 ммоля) ацетилацетоната кобальта(III). Ампулу откачи-
вают и заполняют азотом, затем с помощью шприца через отверстие в пробке
крана в ампулу вводят 10 мл сухого бензола. Закрытую ампулу встряхивают
до полного растворения ацетилацетоната кобальта.
Полимеризацию проводят в четырехгорлой колбе, емкостью 500 мл, снаб-
женной мешалкой, термометром, переходной трубкой с краном и вводом и вы-
водом для азота (рис. 34). Все части прибора сушат, прогревая при 120 °C,
и в горячем виде быстро собирают. Собранный прибор повторно откачивают и
заполняют высушенным над Р2О5 азотом. Затем укрепляют капельную воронку
с 200 мл абсолютированного бензола и закрывают ее специальной пробкой
с самозатягивающейся прокладкой из резины. Далее с помощью шприца (пред-
варительно высушенного при 120 °C и продутого азотом) в капельную воронку с
бензолом через прокладку вводят 0,5 мл (2,3 ммоля) А12(С2Н5)3С13. На иглу
шприца наносят немного парафина во избежание поломки иглы при прокалыва-
нии прокладки; сразу же после использования шприц промывают смесью изо-
пропанола с декалином (1 : 1). Полученный раствор этилалюминийсесквихлори-
да* вводят в колбу, температуру в которой поддерживают на уровне 20—25 °C.
Затем в капельную воронку аналогичным образом вводят 10 мл раствора соли
кобальта и смешивают оба компонента катализатора. Через 10 мин после смеше-
* При работе с алюминийсесквихлоридом исключить возможность его кон-
такта с воздухом или влагой.
158
ния компонентов катализатора ловушку с бутадиеном размораживают и 20 г
(0,37 моля) бутадиена постепенно испаряют в колбу (примерно в течение 1 ч)
при непрерывном перемешивании реакционной смеси» Реакционную смесь выдер-
живают еще 1 ч при 20 °C, после чего полимер высаживают добавлением полу-
ченного вязкого раствора к 5-кратному количеству метанола при перемешива-
нии. После отстаивания осадка верхний слой жидкости декантируют, полимер
размельчают и промывают свежей порцией метанола в течение нескольких ми-
нут; эту операцию повторяют 2 раза. Полимер фильтруют с отсасыванием и
сушат в вакуумном сушильном шкафу при 40 °C, выход больше 90%. Около
1 г полученного полибутадиена повторно переосаждают из бензольного раствора
в метанол. Определяют характеристическую вязкость полимера в растворе цикло-
гексана при 20 °C, рассчитывают молекулярную массу и получают другие харак-
теристики полимера (см. опыт 3-30).
3.2.2. Ионная полимеризация по С=О-связи
Полимеризацию мономеров по С = О-связи (например, формальде-
гида) изучают довольно давно [34]. Согласно электронной (резо-
нансной) структуре формальдегид может полимеризоваться как по
анионному, так и по катионному механизму [35, 36]:
СН2=О -<—> СН2— О:-
Анионную полимеризацию можно инициировать третичными
фосфинами или аминами, металлоорганическими соединениями или
алкоголятами. Инициирование происходит при нуклеофильной ата-
ке карбонильного углеродного атома:
R—Li + СН2—6:- --> R-CH2--O:-Li+
R-CH2-O:-Li+4-CH2-O:- ---> R-CH2—O-CH2-O:-Li+
Катионную полимеризацию формальдегида, которую необходи-
мо проводить в абсолютно безводных условиях во избежание про-
текания реакций передачи цепи, можно инициировать протонными
кислотами, кислотами Льюиса (см. раздел 3.2.1.1) или другими
соединениями, способными давать катионы (например, иод, аце-
тилперхлорат) :
Н+ + -:Ь—СН2---> НО-СН2
но—сн2+7 О—СН2-----но—сн2—о—сн2
Полимеры формальдегида с концевыми полуацетальными груп-
пами термически нестойки; они начинают отщеплять мономерный
формальдегид уже при температуре 150 °C. При ацетилировании
концевых гидроксильных групп термическая стойкость возрастает
до 220. °C. Алкилированный полиформальдегид становится стойким
к действию щелочей; однако он нестоек к действию кислот, по-
скольку последние расщепляют ацетальную связь (см. опыты 5-09
и 5-15).
159
Другие карбонилсодержащие соединения, например ацетальде-
гид и пропионовый альдегид, могут быть заполимеризованы с об-
разованием высокомолекулярных продуктов; однако они менее
стойки, чем полиоксиметилены с защищенными концевыми группа-
ми [37—39].
Опыт 3-35. Анионная полимеризация формальдегида в растворе (осадитель-
ная полимеризация)
Получение раствора мономерного формальдегида
Прибор для получения мономерного формальдегида состоит из двухгорлой
колбы А емкостью 1 л, одно горло которой неплотно закрыто пробкой, легко
открывающейся при избыточном давлении. Во второе горло колбы вставлена
согнутая под прямым углом двухметровая стеклянная трубка диаметром не
менее 2 см, второй конец трубки введен в двухгорлую колбу Б емкостью 500 мл,
заполненную кольцами Рашига, другое горло этой колбы используется для вы-
вода газа и соединено с охлаждаемой ловушкой и объемОхМ 1 л. Выходное от-
верстие из ловушки должно быть соединено с затвором (см. раздел 2.1.1) для
предотвращения попадания влаги и двуокиси углерода из воздуха в ловушку
(СОз является сильным ингибитором полимеризации). В колбу А загружают
100 г полиоксиметилена (параформа) и 100 г сухого жидкого парафина. Оба
компонента смешивают и нагревают на водяной бане до 130 °C. Образующийся
газообразный формальдегид пропускают не в прибор, а выпускают под тягу, что
необходимо для обезвоживания полиоксиметилена.
Одновременно остальные части прибора обжигают пламенем горелки при
откачивании масляным насосом, а затем заполняют сухим азотом. В ловушку
наливают 500 мл абсолютированного над натрием диэтилового эфира и охлаж-
дают до —78 °C. Двухгорлую колбу А с помощью двухметровой трубки присо-
единяют к колбе Б. Газообразный формальдегид, получаемый в результате пиро-
лиза а-полиоксиметилена, частично полимеризуется в стеклянной трубке и в кол-
бе Б с образованием низкомолекулярного полиоксиметилена, содержащего на
концах цепи гидроксильные группы (очистка путем форполимеризации). Необхо-
димо следить, чтобы осаждающийся полимер не забил соединительную трубку,
в этом случае прекращают пиролиз и очищают трубку. Во избежание заедания
шлифов и кранов их обильно смазывают вакуумной смазкой. Пиролиз проводят
быстро (в течение 1 ч), поскольку в противном случае теряется много мономе-
ра при форполимеризации; температура масляной бани должна быть около
200 °C. Полученный эфирный раствор содержит примерно 70 г (4 моль/л) фор-
мальдегида. Очень чистый мономерный формальдегид можно получить при раз-
ложении триоксана в газовой фазе при 220 °C на фосфорнокислотнохМ контакт-
ном катализаторе [40, 41].
Полимеризация
В предварительно обожженную трехгорлую колбу емкостью 250 мл, охлаж-
денную до —78 °C, переносят 150 мл полученного раствора формальдегида в
диэтиловом эфире так, чтобы не попала влага. Колба снабжена эффективной
мешалкой, пробкой с самозатягивающейся прокладкой и уравнивателем давле-
ния с трубкой, заполненной едкихМ натром. При интенсивном перемешивании
в охлажденный раствор мономера постепенно вводят в течение 15 мин раствор
1 мг пиридина в 5 мл абсолютированного диэтилового эфира. Через 1 ч до-
стигается конверсия, превышающая 90% (запах формальдегида практически
исчезает). Если скорость полимеризации вследствие присутствия примесей мала,
в систему дополнительно вводят инициатор. Образующийся полиоксиметилен
фильтруют с отсасыванием, промывают диэтиловым эфиром и сушат в вакуум-
ном сушильном шкафу при комнатной температуре. Полимер плавится в интер-
вале 176—178 °C. Измеряют характеристическую вязкость образца в 1%-ном
растворе диметилформамида при 140*С (т)Уд/С«0,08 л/г, что соответствует мо-
лекулярной массе около 80 000). Определяют термическую стойкость полимера
до и после превращения концевых гидроксильных групп (см. опыты 5-09 и 5-15).
Полиоксиметилен, полученный при полимеризации формальдегида или триок-
сана, представляет собой высококристаллический продукт, нерастворимый при
160
комнатной температуре в большинстве растворителей (за исключением гексафтор-
ацетонгидрата); при повышенных температурах он растворим в некоторых по-
лярных растворителях (например, при idO’C в диметилформамиде и диметил-
сульфоксиде). При блокировке нестабильных полуацетальных концевых групп
(см. опыт 5-03) полиоксиметилен можно перераоатывать в термопластичном
состоянии без разложения при повышенных температурах в присутствии ста-
билизаторов.
Опыт 3-36. Анионная полимеризация хлораля в растворе (осадительная по
лимеризация)
Хлораль (трихлоруксусный альдегид) может полимеризоваться как по ка-
тионному механизму (под действием А1С43 или А1Вгз), так и по анионному
[42]. В последнем случае инициаторами могут служить металлоорганические
соединения и третичные амины, .например триэтиламин или пиридин. Из-за низ-
кой предельной температуры полимеризации реакцию необходимо вести также
при низкой температуре.
Хлоральгидрат, используемый для получения хлораля, перемешивают
с 4-кратным количеством подогретой конц. серной кислоты. Образующийся слой
хлораля отделяют и перегоняют (т. кип. 98 °C, т. пл. —57 °C).
В открытую двухгорлую колбу емкостью 100 мл, снабженную термометром
и магнитной мешалкой, вносят 30 г хлораля и 30 г сухого тетрагидрофурана
(см. опыт 3-27). После охлаждения содержимого колбы до —70 °C охлаждаю-
щую смесь убирают и в колбу вводят 2 мл пиридина (предварительно перег-
нанного над твердым КОН). Мгновенно образуется белый осадок полихлораля,
и температура смеси повышается. Колбу вновь опускают в охлаждающую
смесь и выдерживают при —70 °C в течение 15 мин. Полимер быстро фильтру-
ют при возможно более низкой температуре. Осадок выдерживают в вакуумном
эксикаторе несколько часов. Выход составляет 30—40%.
Полученный полимер нестоек даже при комнатной температуре и медленно
деполимеризуется с образованием мономерного хлораля. При повышенных тем-
пературах деполимеризация протекает очень быстро, так что за процессом можно
наблюдать, например, с помощью микроскопа с нагреваемым предметным столи-
ком при 150 °C. Деполимеризацию можно подавить, как и в случае полиметиле-
на, ацетилированием полуацетальных и гидроксильных концевых групп. Поли-
хлораль с блокированными концевыми группами стоек даже при 255 °C; он не-
растворим и не размягчается до 400 °C.
Анионная полимеризация хлораля является относительно простым и нагляд-
ным опытом определения предельной температуры. Для этого в несколько ампул
загружают по 2 мл эквимольной смеси мономера и тетрагидрофурана, ампулы
охлаждают до различных температур в интервале от 0 до —50 °C. В каждую
ампулу добавляют по капле пиридина и выдерживают при соответствующей
температуре. Максимальная температура, при которой в ампуле образуется
муть, свидетельствующая о протекании полимеризации, является примерной оцен
кой критической температуры.
3.2.3. Ионная полимеризация по М = С-связи
Димеризация и тримеризация изоцианатов с образованием цикли-
ческих соединений известны уже давно; моноизоцианаты могут,
однако, полимеризоваться с образованием и линейных макромоле-
кул [44, 451:
R О R О
I II I II
nN=C ----->-----N-C------
Полимеризацию инициируют соединения основного характера,
например цианистый натрий в растворе диметилформамида. При
этом могут быть получены продукты высокой молекулярной мас-
сы, которые не содержат в цепи NH-групп. В отличие от обычных
11—732
161
полиамидов они не способны образовывать водородных мостиков.
Полимерные изоцианаты термически относительно нестойки; при
нагревании происходит деполимеризация, а в присутствии основ-
ных катализаторов деструкция протекает даже при комнатной
температуре.
Опыт 3-37. Полимеризация н-бутилизоцианата в растворе, инициированная
цианистым натрием
Двухгорлую колбу емкостью 100 мл высушивают в пламени горелки при от-
качивании и заполняют сухим азотом. В колбу вносят 1 г тонкодисперсного циа-
нистого натрия, предварительно высушенного над Р2О5 в течение 24 ч. В токе
азота заливают 50 мл диметилформамида, предварительно высушенного над
СаС12 и трижды перегнанного над Р2О5 в атмосфере азота. Колбу закрывают
и встряхивают, через час образуется 1%-ный раствор цианида в диметилформ-
амиде.
Полимеризация
Сухую четырехгорлую колбу емкостью 250 мл, снабженную мешалкой, тер-
мометром, вводом и выводом для азота, пробкой с самозатягивающейся про-
кладкой (см раздел 2.1.3), обжигают пламенем горелки при откачивании и за-
полняют сухим азотом; газоотводная трубка при этом соединена с обратным
затвором для предотвращения попадания воздуха в прибор (см. раздел 2.1.1).
В токе азота в колбу вносят 100 мл сухого диметилформамида и 10 мл (0,09 мо-
ля) н-бутилизоцианата (трижды предварительно перегнанного при пониженном
давлении в токе азота) и полученную смесь охлаждают до —60 °C. Затем с по-
мощью шприца в колбу по каплям медленно (в течение 10 мин) вводят 5 мл
раствора инициатора (что соответствует примерно 1 ммолю NaCN). Полимериза-
ция начинается и протекает с выделением тепла. Реакцию продолжают при
—60 °C еще 45 мин, образующийся полимер медленно осаждается. Полимериза-
цию прекращают введением в реакционную смесь 100 мл метанола; при этом
весь полимер выпадает в осадок. Полимер фильтруют с отсасыванием, промыва-
ют несколько раз метанолом и сушат в вакуумном сушильном шкафу при
50 °C; выход составляет около 6 г. Синтезированный полимер плохо растворим
в бензоле или тетрагидрофурано вследствие очень большой молекулярной мас-
сы. Из разбавленных растворов полимера может быть получена прозрачная плен-
ка. Температура размягчения полимера лежит около 180 °C, температура плав-
ления — около 209 °C. При длительном нагревании при 200 °C образец деполи-
меризуется. Определяют характеристическую вязкость полимера в бензольном
растворе при 20 °C.
3.2.4. Полимеризация с раскрытием циклов [46]
Многие гетероциклические соединения под действием ионных ини-
циаторов могут полимеризоваться с раскрытием цикла, образуя
линейные макромолекулы. К таким соединениям относятся прос-
тые циклические эфиры, циклические ацетали, циклические слож-
ные эфиры (лактоны), циклические амиды (лактамы) и цикличе-
ске амины. Полимеризацию с раскрытием цикла проводят в таких
же условиях и часто в присутствии тех же инициаторов, что и ион-
ную полимеризацию ненасыщенных мономеров (см. раздел 3.2.1);
следовательно, эти реакции чувствительны к тем же примесям.
З.2.4.1. Полимеризация простых эфиров
с раскрытием цикла
При полимеризации трех-, четырех- и пятичленных циклических
простых эфиров (например, эпоксидов, оксоциклобутана, тетрагид-
рофурана) образуются полиэфиры [38, 47, 48], однако шестичлен-
Г62
ные циклические простые эфиры (диоксан) до настоящего времени
заполимеризовать не удалось.
Эпоксиды [49], например окись этилена, которые не полиме-
ризуются по радикальному механизму, способны полимеризоваться
под действием катионных инициаторов (в частности, кислот Льюи-
са) и анионных инициаторов (например, алкоголятов, металлоор-
ганических соединений):
R+ 4- Н2С-^СНа-> Н2С—СН2--► R-O-CH2-CH2
V ¥
R—O-CHj-CHa -F H2C—CHa -> R—О—СН2—СН2—О—СН2—СН2
RO" 4- Н2С—СН2-> RO—( Н2-СН2-О-
\/
RO—СН2~СН2-О-4-Н2С—сн2 —> ro-ch2-ch2-o-ch2—сн2-о-
При полимеризации окиси этилена под действием тщательно
очищенных карбонатов щелочноземельных металлов (например,
карбоната стронция) образуются высокомолекулярные продукты
[50]. Полиэтиленоксиды с молекулярными массами до 600 пред-
ставляют собой вязкие жидкости; полимеры с большими молеку-
лярными массами — воскообразные или твердые, кристаллические
продукты, хорошо растворимые в воде и некоторых органических
растворителях, в частности в бензоле или в хлороформе. Произ-
водные окиси этилена (например, окись пропилена) в общем слу-
чае полимеризуются с образованием атактических аморфных, а
также изотактических кристаллических полимеров [51, 52]. При
полимеризации /-окиси пропилена может быть получен оптически
активный полипропиленгликоль [53].
Полимеризация четырехчленных циклических простых эфиров
может протекать как под действием катионных инициаторов (на-
пример, кислот Льюиса), так и под действием металлоорганиче-
ских соединений. Полимер 3,3-бис-хлорметилоксациклобутана от-
личается очень высокой температурой размягчения, а также хими-
ческой стойкостью [54].
Тетрагидрофуран может полимеризоваться только под дейст-
вием катионных инициаторов, например трехфтористого бора или
пятихлористой сурьмы [55, 56]. Стадия инициирования заключа-
ется в образовании циклического оксониевого иона; при последую-
щей атаке одной из двух активированных метиленовых групп в
a-положении молекулой мономера происходит раскрытие цикла.
11*
163
Рост цепи осуществляет третичный ион оксония, а не свободный
ион карбония:
R+4-
Н2С-СН2
Н2С-СН2
Н2С^ /СН2
о
СН2—(СН2)3—О—R
Опыт 3-38. Полимеризация тетрагидрофурана в массе, инициированная пя-
тихлористой сурьмой
Тетрагидрофуран очищают, как описано в опыте 3-27, и перегоняют перед
использованием в сухой приемник (см. раздел 2.1.2).
Сухую круглодонную колбу емкостью 250 мл, снабженную соединительным
устройством (см. раздел 2.1.3), обжигают открытым пламенем горелки при
откачивании и заполняют азотом. Затем, укрепив приемник на переходнике,
в колбу наливают 15 г (0,21 моля) тетрагидрофурана, создавая небольшое раз-
режение через переходник. Одновременно пропускают азот в прибор через трех-
ходовой кран, припаянный к боковой стенке приемника. Колбу охлаждают до
—30 °C и в токе азота в колбу вводят 0,6 г (2,0 ммоля) свежеперегнанной пяти-
хлористой сурьмы. При небольшом избыточном давлении азота переходник от-
соединяют от колбы, которую сразу же закрывают пришлифованной пробкой,
закрепляя ее с помощью пружинок. Содержимое колбы перемешивают и остав-
ляют при комнатной температуре на 24 ч. К образовавшейся вязкой смеси до-
бавляют 2 мл воды и 150 мл тстрагидрофурана. Полученную смесь кипятят
около 30 мин до получения гомогенного раствора. К вязкому раствору добавля-
ют 100 мл тетрагидрофурана, затем фильтруют при отсасывании для удаления
продуктов гидролиза инициатора, после чего полимер высаживают приливанием
полученного раствора при интенсивном перемешивании к 3 л воды. Осадок
фильтруют и сушат в вакуумном сушильном шкафу при 20 °C. В зависимости
от молекулярной массы политетрагидрофураны могут быть вязкими маслами,
воскообразными или твердыми, кристаллическими продуктами (интервал плав-
ления около 55 °C). Полиэфир, полученный по настоящей прописи, представляет
собой немного липкий твердый продукт, плавящийся при температуре около
40 °C. Он растворим в бензоле, четыреххлористом углероде, хлорбензоле, тетра-
гидрофуране, диоксане, этилацетате и нерастворим в воде, метаноле и ацетоне.
Определяют характеристическую вязкость полученного полимера в бензоле при
20 °C.
3.2.4.2. Полимеризация циклических ацеталей
с раскрытием цикла
Циклические формали (1,3-диоксалан, триоксан и др.) и тетра-
гидрофуран полимеризуются только под действием катионных ини-
циаторов. При полимеризации с раскрытием цикла триоксана [35,
36. 57] (циклического тримера формальдегида) получается поли-
оксиметилен (см. опыт 3-39), обладающий таким же строением
цепи, как и полиформальдегид (см. опыт 3-35). Эти полимеры
термически нестойки, если их концевые полуацетальные гидро-
№4
ксильные группы полностью не блокированы (см. опыт 5-09). Как
и циклические простые эфиры, триоксан полимеризуется путем
присоединения инициирующего катиона к одному из кислородных
атомов цикла с образованием оксониевого иона, который затем
при раскрытии цикла превращается в карбониевый ион; далее
рост цепи происходит за счет присоединения молекул триоксана:
СН2 СН2
o' ^0 R—О +
R+ + I | ---* | I ------► R^O-(GH2-O)2-CH3
HaC^^CHt HtC^ GHt
^CHf ^CHa
R—O—(CH2—O)2—CH2 + ° ° ---> ° ° -
H2C CH2 H2C CH2
R-O-(CH2—O)2-CH,
--► R—O—(CHj—O)&—CH2
Поскольку полиоксиметилен растворим очень плохо, полиме-
ризация с раскрытием цикла триоксана в массе (расплаве), а так-
же в растворе является гетерофазным процессом. В твердом со-
стоянии триоксан может полимеризоваться под действием облуче-
ния частицами высокой энергии [58, 59].
Опыт 3-39. Полимеризация триоксана, инициированная эфиратом трехфтори-
стого бора
А. Полимеризация в массе
Около 150 г триоксана кипятят (с воздушным холодильником) над 9 г нат-
рия или калия в атмосфере сухого азота 48 ч (т. кип. 115 °C)*. Навеску 20 г
очищенного таким образом мономера перегоняют в сухую колбу емкостью 50 мл,
обожженную пламенем горелки (при откачивании), снабженную впаянной в стек-
ло магнитной мешалкой. Колбу закрывают пробкой с самозатягивающейся про-
кладкой (см. раздел 2.1.3), нагревают до 70 °C и с помощью шприца вводят
0,05 мл (4-10-4 моля) эфирата трехфтористого бора (d2o = 1,125) в 10%-ном
растворе нитробензола. Неооходимо следить за тщательностью перемешивания
расплава триоксана при введении инициатора, за его быстрым и гомогенным
распределением в реакционной смеси. Сразу после введения инициатора обра-
зуется полиоксиметилен, выпадающий в осадок из расплава мономера; примерно
через 10 с вся реакционная смесь затвердевает. Полимеризацию прекращают
добавлением ацетона и после тщательного перемешивания (при необходимости
дробления) полимер фильтруют на стеклянном фильтре. Затем образец дважды
кипятят в 100 мл ацетона по 20 мин, фильтруют и сушат в вакуумном шкафу
при комнатной температуре. Выход полимера составляет около 50%; интервал
плавления 177—180 °C. Определяют характеристическую вязкость полученного
образца в 1%-ном растворе диметилформамида при 140°C (т]уд/С«0,06 л/г
примерно соответствует молекулярной массе 60 000). Исследуют термическую
* Мономер чистый для полимеризации можно получить также кипячением
триоксана в течение 12 ч в присутствии 5% 1,5-нафтилендиизоцианата и
0,2 бис-н-бутиллаурата олова, триэтилендиамина или нитрата висмута.
16S
деструкцию полимера до и после блокирования концевых гидроксильных групп
(см. опыты 5-09 и 5-15).
Б. Полимеризация в растворе (осадительная полиме-
ризация)
Около 90 г триоксана, очищенного по методике, описанной в пункте А,
перегоняют в колбу емкостью 500 мл (отожженную пламенем при откачивании),
содержащую 200 мл предварительно высушенного над Р2О5 дихлорэтана. Колбу
закрывают пробкой с самозатягивающейся прокладкой (см. раздел 2.1.3). При
перемешивании в колбу вводят 0,06 мл (0,5 ммоля) эфирата трехфтористого
бора, растворенного в 7 мл дихлорэтана, и содержимое колбы нагревают до
45 °C. После небольшого индукционного периода, составляющего примерно 1 мин,
полиоксиметилен начинает выпадать из раствора в осадок, а затем ‘и все со-
держимое колбы затвердевает. Через 1 ч полученный продукт переносят в 200 мл
ацетона, фильтруют, кипятят, как описано в пункте А, и сушат. Для удаления
окклюдированного инициатора полимер кипятят в 1 л диэтилового эфира, содер-
жащего 2% (масс.) трибутиламина. Полимер фильтруют и сушат в вакуумном
сушильном шкафу при комнатной температуре. Конверсия составляет 90—95%,
интервал плавления полученного образца 176 —178 °C. Определяют характери-
стическую вязкость полимера в растворе диметилформамида (молекулярная мас-
са порядка 60 000) и термическую стойкость полимера (см. пункт А).
3.2.4.3. Полимеризация лактонов (циклических
сложных эфиров) с раскрытием цикла
Циклические эфиры w-оксикарбоновых кислот могут полимеризо-
ваться с раскрытием цикла и образованием линейных алифатиче-
ских полиэфиров. Способность к полимеризации зависит от числа
членов в цикле: p-пропиолактон, 6-валеролактон и 8-капролактон
могут полимеризоваться под действием катионных и анионных ини-
циаторов [60].
В зависимости от типа инициатора и природы мономера поли-
меризация протекает с расщеплением связи алкил—кислород(I)
либо связи ацил—кислород(П):
Н2С--С=О +
R3N + | | > R3N—СН2—СН2—С—О~
н2с—!—-о о
Н2С-С=О1 НоС—С=о +
R++ | | ---> I -I— —> R~O-CH2-CH2-C
Н2С-0 Н2С-О+ |
I О
Способность к полимеризации лактонов зависит не только о г
числа членов в цикле, но также от числа, размера и положения
заместителей.
Опыт 3-40. Полимеризация p-пропиолактона в массе, инициированная алю-
минийорганическими соединениями
Мономерный p-пропиолактон перегоняют при пониженном давлении в азоте
(т. кип. 62 °C при 20 мм рт. ст.). Для удаления следов воды и других примесей
мономер повторно перегоняют в токе азота в присутствии 2% толуолдиизоциа-
ната и хранят в тщательно высушенном приемнике (см. раздел 2.1.2).
Круглодонную колбу емкостью 50 мл повторно откачивают и заполняют азо-
том с помощью переходника (см. раздел 2.1.3). Затем в колбу помещают 10 мл
166
(0,16 моля) p-пропиолактона и 0,2 мл (1,6 ммоля) диэтилалюминийхлорида*
в 10%-ном бензольном растворе. При небольшом избыточном давлении азота
переходник убирают и на его место быстро вставляют пробку, закрепляя ее
пружинками. Колбу нагревают до 50 °C, выдерживают при этой температуре
20 ч, затем реакционную смесь разбавляют хлороформом и полимер высаживают
приливанием смеси к диэтиловому эфиру. Осадок фильтруют и сушат в вакуум-
ном сушильном шкафу при комнатной температуре. Выход составляет 10—15%.
Получается алифатический кристаллический полиэфир с выходом 10—15%. Он
растворим в хлороформе, диоксане и муравьиной кислоте. Определяют характе-
ристическую вязкость полимера в растворе хлороформа при 20 °C.
Поли-Р-пропиолактон количественно деструктирует с выделением акриловой
кислоты при нагревании до 200—250 °C.
д-Валеролактон также может быть заполимеризован по описанной методике.
Поли-б'Валеролактои отличается от поли-Р-пропиллактона температурой плавле-
ния (50—60*С) и растворимостью (растворим в бензоле и толуоле). При нагре-
вании до 200—250 ЪС он деполимеризуется с образованием б-валеролактона.
3.2.4.4. Полимеризация циклических амидов (лактамов)
с раскрытием цикла
Полимеризация лактамов [61, 62], протекающая с раскрытием
цикла, осуществляется под действием ионных инициаторов. В ре-
зультате полимеризации образуются линейные полиамиды. Как
и в случае лактонов, способность мономеров к полимеризации су-
щественно зависит от числа членов в цикле, от числа и располо-
жения заместителей [63]. Пятичленный лактам (у-бутиролактам)
полимеризуется по анионному механизму при низких температу-
рах; однако образующийся полиамид вновь деполимеризуется в
присутствии инициаторов при 60—80°C с образованием мономера
[64]. Соответствующий шестичленный лактам (6-валеролактам)
также способен полимеризоваться [63]. Семичленный лактам
(е-капролактам) может полимеризоваться по катионному, а так-
же по анионному механизмам с образованием высокомолекуляр-
ных полиамидов.
Полимеры, образующиеся при анионной полимеризации 8-кап-
ролактама при высокой температуре, имеют вначале высокие мо-
лекулярные массы, которые при длительном нагревании реакцион-
ной смеси уменьшаются и наконец достигают равновесного значе-
ния. Это изменение молекулярно-массового распределения обус-
ловлено реакцией переамидирования между растущими и «мертвы-
ми» цепями полиамида.
Полимеризация Е-капролактама с анионными инициаторами
является истинной цепной реакцией и описывается следующими
уравнениями:
СО
R- 4- (СН2)5
R—С—(СН2)5-~NH
NH
* В качестве инициаторов можно использовать также А1(С2Н5)з и А1С2Н5С12;
при работе с алюминийорганическими соединениями необходимо полностью ис-
ключить их контакт с влагой и воздухом.
167
co
R—С- (СН2)6— NH + (СН2)5
II \
о \
R-C-fCHjJj-NH-C-fCHiJs-NH
II II
О о
NH
В то же время полимеризация 8-капролактама в присутствии ка-
талитических количеств воды, 2-аминокапроновой кислоты или бен-
зойной кислоты представляет собой типичную ступенчатую реак-
цию полиприсоединения:
СО
H2o 4- (СН2)б
NH2-(CH2)5-C-OH
NH
О
СО
NH2-(GH2)5-C-OH + (СН2)5
о
NH
NH2—(СН2)5—С—NH—(СН2)5—С—ОН
О О
С увеличением конверсии возрастают молекулярные массы.
Путем добавления небольших количеств соединений, способных к
реакции переамидирования с цепями полиамидов (например, бен-
зойной кислоты), можно регулировать молекулярную массу поли-
меров. Реакции переамидирования и гидролиза амидной связи при-
водят к установлению равновесного распределения по молекуляр-
ным массам (см. раздел 4.1).
Полимеры, полученные при достижении равновесия при темпе-
ратурах 250—270еС, содержат около 10—12% циклического моно-
мера и олигомеры (частично циклические), которые могут быть
выделены экстракцией водой или низшими спиртами. Олигомеры
могут быть выделены и идентифицированы хроматографически
(см. опыт 4-08).
Опыт 3-41. Получение поли-е-капролактама в массе
А. Полимеризация, инициированная е-а ми иокап р о новой
кислотой
е-Капролактам дважды перекристаллизовывают из циклогексана и сушат
в вакуумном эксикаторе над Р2О5 в течение 48 ч при комнатной температуре
(т. пл. 68—69 °C).
Трехгорлую колбу емкостью 250 мл, снабженную мешалкой, термометром,
с вводом и выводом для азота и обратным холодильником, откачивают и за-
полняют азотом. Затем 90 г (0,8 моля) очищенного s-капролактама и 10 г
(76,2 ммоля) е-аминокапроновой кислоты загружают в колбу и нагревают в ат-
мосфере азота на песочной бане. После того как смесь расплавится, в нее по-
гружают термометр. По достижении 140 °C начинают перемешивание смеси.
168
одновременно нагревая ее до 250 °C. Через 5 ч образуется почти бесцветный,
очень вязкий расплав, из которого с помощью стеклянной палочки можно вы-
тянуть нить. Расплав быстро переносят в стакан (при необходимости колбу
дополнительно нагревают горелкой), где он и застывает. Полученный полиамид
(найлон 6) плавится при температуре около 216 °C. Он содержит циклические
олигомеры, которые можно экстрагировать водой (см. опыт 4-08). Определяют
характеристическую вязкость полученного образца в растворе л£-крезола или
конц. серной кислоты.
Б. Полимеризация, инициированная анионным инициа-
тором (флеш-полимеризация)
На колбу с коническим дном емкостью 50 мл надевают переходник, отка-
чивают и заполняют азотом. В колбу загружают 25 г (0,22 моля) чистого
е-капролактама (об очистке см. пункт А) и нагревают до 80—100 °C. Зате.м
к расплавленному е-капролактаму добавляют 0,04-0,08% металлического натрия,
диспергированного в ксилоле*. (Полученная смесь е-капролактама и его натрие-
ВОЙ СОЯМ стабильна при 80—100°C в течение нескольких часов). В колбу до
ДИВ Опускают капилляр, через который медленно пропускают ток азота, продол-
жая нагревать колбу на песочной бане до 255—265 °C. Спонтанно начинающаяся
полимеризация завершается в течение 5 мин; за полимеризацией можно следить,
фиксируя время подъема пузырьков пропускаемого азота. Расплав полиамида
быстро переливают в стакан. Определяют характеристическую вязкость полиами-
да в растворе л£-крезола или конц. серной кислоты, сопоставляют ее с характе-
ристической вязкостью полимера, полученного в предыдущем опыте. Если рас-
плав полимера выдерживать при 255—265 °C более 6 мин, становится заметной
деструкция полимера, соответственно уменьшается характеристическая вязкость
полимера.
3.3. СОПОЛИМЕРИЗАЦИЯ
3.3.1. Статистическая сополимеризация
Совместная полимеризация двух или нескольких мономеров с об-
разованием макромолекул, содержащих в основной цепи звенья
различных исходных мономеров, называется сополимеризацией
[65—69]. Необходимо отметить, что не все мономеры, способные
гомополимеризоваться, вступают в реакцию совместной полимери-
зации. Так, при радикальной сополимеризации стирола с винилаце-
татом лишь очень небольшое число звеньев винилацетата включа-
ется в цепь сополимера (табл. 12). В то же время некоторые моно-
меры, не способные гомополимеризоваться, могут вступать в реак-
цию сополимеризации.
Сополимеризация двух мономеров исследована очень подробно;
изучение сополимеризации трех или более мономеров [65, 70, 71]
несколько затруднено, что обусловлено большим числом перемен-
ных параметров, хотя ряд терполимеров (сополимеров трех моно-
меров) производится в промышленном масштабе.
Ниже рассматривается простейший случай сополимеризации
двух мономеров. При этом возможно протекание четырех элемен-
тарных реакций роста: молекула мономера Mi может присоеди-
* Вместо дисперсии металлического натрия можно использовать едкий натр,
гидрид натрия или литийалюмогидрид.
169
няться к активному центру (радикальному или ионному) расту-
щей цепи, и концевым звеном может быть либо Мь либо М2; то
же относится и к присоединению мономера М2:
^11
----+ М, ----->-----Mi — MJ
—mj + m2 —Mi-mj
^21
----mj + Mi —-------м2 — m;
—mj + м2 —м2 — mj
где Ли, &i2, k2\ и k22 — константы скоростей роста; первый индекс соответствует
типу конца активного центра, второй индекс — типу присоединяющейся моле-
кулы мономера.
Если длина кинетической цепи достаточно велика, скорости
реакций роста становятся лимитирующими; в этом случае, как и
при гомополимеризации, выполняется условие квазистационарно-
сти. При малых конверсиях (<10%) и выполнении условия ква-
зистационарности можно показать, что отношение скоростей расхо-
да двух мономеров, т. е. отношение концентраций мономерных
звеньев в цепи сополимера описывается уравнением
d [MJ __ гиг __ JMJ Z1 [MJ 4JM2L zo
~d[M2] - ~ |M2]~ [Mj + r2[M2]
где ri = ^u/^i2 и r2 = k22lk2\ — так называемые константы сополимеризации;
[MJ и [М2] — мольные концентрации мономеров в исходной реакционной смеси.
Состав образующегося сополимера зависит, следовательно, от
соотношения концентраций мономеров в исходной смеси. Посколь-
ку параметры г4 и г2 являются отношением констант роста, они
отражают тенденцию растущих цепей к присоединению одного из
мономеров. Если значение г близко к 1, это значит, что присоеди-
нение Mi и М2 происходит статистически равновероятно; если
г>1, то это значит, что активный центр предпочтительно при-
соединяет «свой» мономер. Константы сополимеризации слабо за-
висят от температуры реакции, поскольку они являются отноше-
нием двух констант роста. Однако, строго говоря, они относятся
только к температуре реакции. Поэтому, приводя значения кон-
стант сополимеризации, необходимо указывать температуру, при
которой проводилась реакция.
Если константы сополимеризации и г2 известны, то для полу-
чения сополимера заданного состава (f = mi/m2) необходимо взять
мономеры в соотношении, определяемом из преобразованного
уравнения (3-20):
4Й-==_27Г 1(/~ ° + [(/ ~ °’+ 4fir^|I/2} (3-21)
Зависимость состава сополимера от соотношения концентраций
мономеров в исходной смеси отражается так называемой сополи-
170
меризационной диаграммой (рис. 35). Такую диаграмму получают
построением зависимости мольной доли одного из двух сополиме-
ров в цепи образующегося полимера от мольной доли того же
мономера в исходной реакционной смеси (первую величину полу-
чают по данным анализа полимера или остаточной мономерной
смеси). Состав сополимера может быть вычислен по уравнению
(3-20) [72].
Из уравнения (3-20) и из сополимеризационной диаграммы сле-
дует существование осрбых случаев сополимеризации. Если соста-
вы мономерной смеси и образующегося полимера идентичны, сопо-
* "n yft-n '-ртг- :
Рис. 35. Диаграмма сополимеризации
стирола (Mi) с метилметакрила-
том (М2):
/ — радикальная сополимеризация (60 °C;
Г1=0,52; г2=0.46); 2 — катионная сополи-
меризация (25 °C; инициатор SnBr4; fi =
= 10,5; r2=0,l); 3 — анионная сополимери-
зация (—50 °C: инициатор Na в жидком
аммиаке; п=0,12; rj—6,4).
лимеризацию называют азеотропной [73]; уравнение (3-20) прини-
мает вид
mY__ __ |Mt]______r2— 1
~ [М21 ~ 7*1— f
(3-22)
В этом случае сополимеризация протекает до глубоких конвер-
сий без изменения состава сополимера (см. опыт 3-45). Азеотроп-
ный состав соответствует точке пересечения кривой составов с
диагональной линией на диаграмме сополимеризации. В соответ-
ствии с рис. 35, характеризующим свободнорадикальную сополи-
меризацию стирола с метилметакрилатом, азеотропный состав мо-
номерной смеси содержит 53% (мол.) стирола.
Если Гг = О и ^22 = 0, то мономер М2 не присоединяется к концу
растущей цепи, оканчивающемуся звеном М2, т. е. мономер М2
не способен полимеризоваться, и уравнение (3-20) имеет вид
При этом даже при высокой концентрации М2 в исходной мо-
номерной смеси сополимер максимально может содержать 50%
(мол.) М2. Это справедливо, например, для сополимеризации с
участием малеинового ангидрида, а также таких «мономеров», как
молекулярный кислород и двуокись серы. Для последних двух не-
171
зависимо от природы второго сополимера образуются чередую-
щиеся сополимеры, содержащие примерно эквимольные количества
обоих мономеров.
Наконец, известны случаи, когда оба сополимеризующихся мо-
номера не способны гомополимеризоваться (йц = &22 = 0), при этом
образуются строго чередующиеся сополимеры (см. опыт 3-52).
Необходимо отметить, что способность мономера к полимериза-
ции и сополимеризации в очень большой степени зависит от при-
роды растущего конца цепи. При радикальной сополимеризации
инициатор не влияет на образующийся сополимер. В общем слу-
чае, однако, существует различие в составе сополимеров, получен-
ных под действием катионных и анионных инициаторов. Даже при
использовании различных катионных либо различных анионных
инициаторов состав получаемого сополимера может зависеть от
природы инициатора и в особенности от типа противоиона.
При свободнорадикальной сополимеризации стирола с метил-
метакрилатом вероятности вхождения в цепь мономеров близки.
Однако при сополимеризации эквимольной смеси этих мономеров
под действием анионных инициаторов предпочтительнее включение
в цепь звеньев метилметакрилата. Напротив, цепь сополимера, по-
лученного в результате катионной сополимеризации этой же сме-
си мономеров, почти полностью состоит из звеньев стирола (см.
опыт 3-42). Такие мономерные системы, как стирол—метилмет-
акрилат, полимеризационное поведение которых изучено достаточ-
но подробно, могут быть использованы для изучения механизма
инициирования новых, недостаточно изученных инициирующих си-
стем.
Поскольку состав сополимера зависит от способности мономера
полимеризоваться по данному механизму, существует большое
число исследований, посвященных установлению взаимосвязи кон-
стант сополимеризации со строением мономера. Установлен ряд
полуэмпирических зависимостей между резонансной стабилиза-
цией растущего конца цепи, электронной плотностью двойной свя-
зи ненасыщенного соединения и константами сополимеризации
[68, 74—76]. Здесь эти зависимости не рассматриваются.
В большинстве случаев сополимеризации один из мономеров
чаще включается в цепь сополимера. Состав мономерной смеси
при этом меняется с глубиной конверсии, следовательно, в ходе
реакции изменяется и состав образующегося сополимера. Поэтому
при определении констант сополимеризации важно заканчивать
сополимеризацию при малых степенях превращения, когда состав
мономерной смеси мало отличается от исходного. Если же реакцию
в препаративных целях необходимо вести до глубоких степеней
превращения, то систему надо постоянно подпитывать более реак-
ционноспособным мономером.
Для определения констант сополимеризации [77] необходимо
знать составы сополимеров, полученных при сополимеризации раз-
личных составов мономерных смесей (существует ряд аналити-
ческих методов определения состава сополимеров; см. раз-
172
дел 2.3.11). Константы g и г2 можно вычислить по уравнению
(3-20), если известны по крайней мере составы двух сополимеров,
полученных путем сополимеризации различных смесей мономеров
Mi и М2. Чаще, однако, определяют составы сополимеров, полу-
ченных из нескольких мономерных смесей, затем для каждого опы-
та определяют величину г2 из преобразованного уравнения соста-
ва сополимера при некоторых произвольно выбранных значе-
ниях Гь
JMJ / m, / , JMJ \
“ |М*Г ( «Н 1М,1
(3-24)
/ЛИ ТОСТро«И111 зависимостей г2 ОТ для нескольких опытов
МраЬечеиие прямых в точке, соответствующей искомым
КОКСТантам сополимеризации. Обычно же линии не пересекаются
в одной точке, а ограничивают некоторую область наиболее ве-
роятных значений и и г2, по которой можно вычислить средние ве-
личины и ошибку опыта.
Простой метод определения констант сополимеризации был
предложен Файнманом и Россом [78]. Уравнение состава сополи-
мера (3-20) преобразуется к виду
О) (/-!) = r^lf ~г2 (3-25)
или
(f- -rJ/F^r, (3-26)
где f = F = [MJ [М2]
Построением зависимости (Т7//) (f—1) от отношения F2/f полу-
чают прямую в координатах уравнения (3-25), по тангенсу угла
наклона которой определяют величину гь а по отрезку, отсекаемо-
му на оси ординат, — г2. Аналогичным образом результаты сопо-
лимеризации можно обработать и по уравнению (3-26). Ошибки
в определении констант и г2 при этом могут быть вычислены
методом наименьших квадратов по разбросу точек от прямой ли-
нии. Подробные таблицы констант сополимеризации большинства
известных мономеров можно найти в литературе [65]; некоторые
наиболее характерные константы приведены в табл. 12.
Сополимеры в большинстве случаев существенно отличаются
по своим физическим свойствам от соответствующих гомополиме-
ров. Например, при включении небольшого количества винилацета-
та в поливинилхлорид достигается внутренняя пластификация (см.
раздел 1.4). Окрашиваемость синтетических волокон может быть
улучшена включением малого количества специально подбираемо-
го сомономера. Кроме того, в общем случае существует большое
различие в растворимости сополимеров и соответствующих им го-
мополимеров (см. опыт 3-42). Свойства сополимеров, содержащих
эквимольные количества звеньев обоих типов, распределенных ста-
тистически, часто значительно отличаются от свойств соответст-
вующих им гомополимеров. Так, полиэтилен и изотактический по-
липропилен представляют собой кристаллические полимеры, имею-
173
Таблица 12. Константы сополимеризации
Ml М2 п Г2 Инициирование Темпе- ратура, Ю
Стирол Метилметак- 1 илат 0,52±0,03 0,46 + 0,03 Свободнорадикальное 60
10,5 0,12 Катионное (SnBr4) 25
0,12 6,4 Анионное (Na в жидком -50
Стирол аммиаке)
Бутадиен 0,78+0,01 1,39 + 0,03 Свободнорадикальное 60
Стирол Винилаиетат 55 4- Ю 0,01 + 0,01 То же 60
Стирол 2,5-Дихлор- 0,32+0,06 0,08+0,05 » 65
Изобутилен стирол 14,8f0,2 0,34+0,15 Катионное (А1С1з) 0
Бутадиен 0,01+0,01 115+15 То же — 103
Малеиновый Стильбен 0,03 <0,03 0,03+0,03 Свободнорадикальное 60
ангидрид «-Метилсти- 4-Хлорстирол 28±2 0,12±0,03 Катионное (SnCl4) —78
рол «-Метилсти- Акрилонит- 0,1+0,02 0,06+0,02 Свободнорадикальное 75
рол рил
щие свойства, характерные для термопластов. Сополимер же эти-
лена с пропиленом состава 70:30 является аморфным эластичным
каучукоподобным продуктом.
Большое практическое значение имеет трехмерная сополимери-
зация, при которой используются мономеры, содержащие две или
более двойных связей. Эти мономеры могут участвовать в процес-
се полимеризации, приводя к образованию сшитых продуктов,
имеющих трехмерную сетку. В качестве примера можно привести
синтез сшитого полистирола, полученного сополимеризацией сти-
рола с небольшими количествами дивинилбензола (опыт 3-49).
Подобные сшитые сополимеры не растворяются и не плавятся, они
способны к ограниченному набуханию в органических растворите-
лях. Это их свойство используется для получения ионообменных
смол (см. раздел 5.2). В результате сополимеризации ненасыщен-
ных эфиров малеиновой и фумаровой кислот со стиролом также
получаются трехмерные сополимеры (см. опыт 4-05).
Кинетические закономерности свободнорадикальной сополиме-
ризации (скорость реакции и молекулярные массы образующихся
продуктов) в основном аналогичны закономерностям свободнора-
дикальной гомополимеризации (см. раздел 3.1). Так, с увеличе-
нием концентрации инициатора скорость сополимеризации возрас-
тает, а молекулярные массы уменьшаются; повышение температу-
ры приводит к такому же эффекту.
Свободнорадикальную сополимеризацию, как и свободноради-
кальную гомополимеризацию, проводят в массе, в растворе или в
дисперсиях. При этом необходимо соблюдать те же предосторож-
ности, о которых говорилось ранее при обсуждении гомополиме-
ризации (см. разделы 2.1.5 и 3.1). Состав сополимера, полученного
в эмульсии или суспензии, может, однако, отличаться от состава
174
сополимера при сополимеризации в массе, если один из мономеров
больше растворим в воде. В этом случае исходный состав мономе-
ров не соответствует составу мономеров в органической фазе, в ко-
торой осуществляется сополимеризация.
Методика эксперимента ионной сополимеризации не отличается
существенно от методики ионной гомополимеризации (см. раз-
дел 3.2). В то же время тип инициатора и природа растворителя
оказывают заметное воздействие на процесс ионной сополимериза-
ции и на состав образующегося сополимера; таким образом, опти-
мальные условия проведения ионной сополимеризации необходимо
подбирать отдельно для каждой мономерной пары.
В большинстве случаев состав получаемого сополимера изме-
няется с глубиной превращения. Поэтому при проведении реакции
ДО глубоких конверсий с целью получения большого количества
продукта в ходе реакции в реакционную смесь необходимо вводить
более реакционноспособный мономер, чтобы состав мономерной
смеси все время оставался постоянным [79]. Это достигается сле-
дующим образом: состав смеси, необходимый для получения за-
данного сополимера, определяют по диаграмме сополимеризации
(или рассчитывают по константам сополимеризации). Затем стро-
ят кривую конверсия — время и находят состав сополимера через
определенные промежутки времени. Таким образом определяют
количество мономера, которое необходимо добавить в систему.
Для качественного и количественного анализа сополимеров тре-
буются специальные методы, которые рассмотрены в разделе 2.3.11.
Опыт 3-42. Сополимеризация стирола с метилметакрилатом
Стирол и метилметакрилат очищают от ингибитора и высушивают по мето-
дикам, описанным в опытах 3-01 и 3-29. Приготавливают смесь 8,00 г (76,8 ммо-
ля) стирола и 7,68 г (76,8 ммоля) метилметакрилата в сухом сосуде в атмосфе-
ре азота. До начала опыта смесь мономеров хранят в холодильнике.
А. Свободнорадикальная сополимеризация
В колбу емкостью 50 мл помещают 16 мг (0,1 ммоля) азо-бис-изобутиро-
нитрила, присоединяют переходник, откачивают вакуумным насосом и заполняют
азотом. После этого в колбу с помощью пипетки быстро вносят 5 мл смеси моно-
меров. При небольшом избыточном давлении азота переходник заменяют при-
шлифованной пробкой, которую закрепляют с помощью пружинок, и колбу по-
мещают в термостат при 60 °C. Через 4 ч колбу охлаждают ледяной водой и
реакционную смесь разбавляют 25 мл бензола. Для выделения сополимера по-
лученный раствор приливают при перемешивании к 200—250 мл петролейного
эфира. Сополимер выпадает в осадок в виде мелких белых хлопьев, склонных
к взаимному слипанию (использование в качестве осадителя метанола по этой
причине менее желательно). Полученный осадок фильтруют через стеклянный
фильтр, промывают свежей порцией петролейного эфира и сушат в вакуумном
сушильном шкафу при 50 °C до постоянной массы; выход сополимера составляет
10—20%.
Б. Анионная сополимеризация
Получение раствора инициатора. Фенилмагнийбромид получа-
ют смешением 12 г (76.5 ммоля) бромбензола и 1.86 г порошка магния в 30 мл
абсолютированного тетрагидрофурана (см. опыт 3-27). Прибор, в котором про-
водят реакцию, предварительно тщательно высушивают. Реакцию можно вести
в присутствии кислорода воздуха. Для завершения реакции смесь немного нагре-
вают с тем, чтобы добиться максимального превращения магния.
175
Проведение сополимеризации. В колбу емкостью 50 мл, высу-
шенную пламенем горелки при откачивании и заполненную сухим азотом, зали-
вают 5 мл приготовленной ранее смеси мономеров. Колбу закрывают пробкой
с самозатягивающейся резиновой прокладкой (см. раздел 2.1.3) и охлаждают
до —50 °C; затем с помощью шприца в колбу через прокладку вводят 2 мл све-
жеперегнанного раствора инициатора. Реакционную смесь выдерживают 90 мин
при —50 °C, после чего в колбу добавляют 25 мл бензола. Затем сополимер вы-
саживают приливанием его раствора при перемешивании к 200 мл метанола,
содержащим 10 мл 2 н. НС1 или H2SO4 Далее полученный сополимер выделяют
по методике, описанной в пункте А. Выход сополимера составляет примерно
10—20%. Реакцию анионной сополимеризации необходимо проводить только при
низкой температуре, так как при более высоких температурах возможно взаимо-
действие магнийорганического соединения со сложноэфирной группой метилмет-
акрилата.
В. Катионная сополимеризация
Колбу емкостью 100 мл, снабженную переходником (см. раздел 2.1.3), высу-
шивают пламенем горелки при откачивании вакуумным насосом, а затем запол-
няют сухим азотом. В колбу наливают 5 мл приготовленной ранее смеси моно-
меров и 40 мл раствора инициатора [последний готовят растворением 300 мл
(2,25 ммоля) безводного хлористого алюминия в 50 мл чистого сухого нитро-
бензола — см. опыт 3-50]. При небольшом избыточном давлении азота заменч
ют переходник пришлифованной пробкой, содержимое колбы перемешивают и
оставляют при комнатной температуре на 1 ч. Для выделения продукта реак-
ционную смесь приливают по каплям к 200 мл метанола, подкисленного 2 н. НС1,
далее сополимер выделяют по методике, описанной в предыдущих пунктах А и
Б. Выход сополимера составляет 40—50%.
При данных условиях катионная сополимеризация протекает так быстро,
что и при более низкой концентрации инициатора примерно половина мономер-
ной смеси заполимеризовывается в течение 1 ч. При 50%-ном превращении реак-
ция прекращается, поскольку при катионном инициировании только стирол вхо-
дит в полимерные цепи.
Г. Идентификация сополимеров
Осаждение из растворов сополимеров, полученных тремя различными мето-
дами из идентичной смеси мономеров, свидетельствует о различии этих продук-
тов. Проверяют растворимость полученных сополимеров. С этой целью отбирают
по 50 мг каждого из полученных сополимеров, а также смеси полистирола и
полиметилметакрилата(III) и нагревают в 5 мл следующих растворителей:
1) смеси ацетона с метанолом (2:1), 2) ацетонитриле, 3) смеси циклогексана
с бензолом (4:1).
Полученные растворы отделяют от осадка (если он остался) и выливают в
метанол, в котором все полимерные продукты выпадают в осадок.
Первый и второй растворители являются хорошими растворителями для
полиметилметакрилата; полистирол хорошо растворяется в третьем раствори-
теле.
Опыты по растворимости свидетельствуют о том, что образец, полученный
свободнорадикальной полимеризацией, нерастворим ни в одном из взятых рас-
творителей, т. е. его растворимость отличается от растворимости полиметилмета-
крилата и полистирола.
Продукт, полученный анионной полимеризацией, растворим в ацетонитриле
и при нагревании в смеси ацетона с метанолом, однако он не растворяется
в смеси циклогексана с бензолом. Таким образом, растворимость этого образца
аналогична растворимости полиметилметакрилата.
И наконец, продукт, полученный катионной полимеризацией, немного рас-
творим в смеси ацетона с метанолом, практически нерастворим в ацетонитриле,
но хорошо растворим в смеси циклогексана с бензолом. Следовательно, по рас-
творимости он соответствует гомополистиролу.
Опыт 3-43. Свободнорадикальная сополимеризация стирола с п-хлорстиро-
лом (определение констант сополимеризации)
В 9 ампул со шлифами загружают по 20 мг (0,12 ммоля) азо-бис-изобути-
ронитрила и очищенные стирол и ц-хлорстирол (см. опыт 3-44) в количествах,
176
указанных в следующей таблице (взвешивать с точностью до I мг). Если образ-
цы не полимеризуют в тот же день, их следует хранить в холодильнике.
№ ампулы Стирол л-Хлорстирол № , ампулы Стирол л-Хлорстирол
г ммоли г 1 ммоли r 1 ммоли г ммоли
1 з.о 29 0 0 6 0,7 7 2,3 17
2 2,6 25 0,4 3 7 0,3 3 2,7 19
3 2,3 22 0,7 5 8 0,1 1 2,9 21
4 1,6 15 1.4 10 9 . 0 0 3,0 22
5 1Л 11 1,9 14
Ампулы присоединяют через переходник (см. раздел 2.1.3) к вакуумной
линии, при повторном замораживании (в смеси сухого льда с метанолом) и раз-
мораживании вакуумируют и наполняют азотом. При небольшом избыточном
давлении азота ампулы отсоединяют от вакуумной линии и закрывают пришли-
фованными пробками, которые закрепляют с помощью пружинок. После этого
ампулы выдерживают в термостате при 50 °C в течение 8 ч. По истечении этого
времени ампулы охлаждают, их содержимое разбавляют 5 мл бензола. Для вы-
деления сополимеров полученные смеси приливают при перемешивании к 80 мл
метанола. Осадок фильтруют с отсасыванием, дважды переосаждают и сушат
в вакуумном сушильном шкафу при 50 °C до постоянной массы. Выход в каж-
дой ампуле составляет около 300 мг (10%).
В высушенных образцах определяют содержание хлора (например, грави-
метрически методом по Вюрцшмиту [80]). По данным анализа определяют со-
став сополимеров, рассчитывают константы сополимеризации, строят диаграмму
сополимеризации. Сопоставьте полученные данные с результатами следующего
опыта по катионной сополимеризации этих же мономеров (см. опыт 3-44).
Опыт 3-44. Катионная сополимеризация стирола с n-хлорстиролом (опреде-
ление констант сополимеризации)
Стирол и ц-хлорстирол очищают от ингибитора двукратной экстракцией 2 н.
раствором NaOH, потом мономеры промывают дистиллированной водой до ней-
трального pH сливаемой воды, сушат над безводным сульфатом магния и, на-
конец, перегоняют при пониженном давлении в токе азота (т. кип. п-хлорстирола
81 °C при 21 мм рт. ст.).
Подготовка прибора для проведения полимеризации
Сополимеризацию проводят в колбах емкостью 50 мл или других подходя-
щих сосудах, которые могут закрываться пробкой с самозатягивающейся про-
кладкой (см. раздел 2.1.3). Сосуды должны быть хорошо высушены. Для этого
их тщательно моют, споласкивают чистым ацетоном и выдерживают под ва-
куумом при НО °C в течение 1 ч. После этого еще горячими их помещают в ва-
куумный эксикатор на 2 ч. Через 2 ч в эксикатор впускают сухой азот и, откры-
вая его в токе азота, вынимают чистые и сухие сосуды и быстро закрывают их
пробками с самозатягивающимися прокладками.
Получение раствора инициатора
В одно горло двугорлой колбы вставляют ввод для азота, на второе горло
надевают небольшой обратный холодильник, к верхнему концу которого через
переходник присоединяют прямой холодильник. На прямой холодильник наде-
вают капельную воронку (приемник) с боковой трубкой, позволяющей уравно-
вешивать давление (см. раздел 2.1.1). Прибор помещают под тягу. В собранном
таким образом приборе кипятят около 20 г SnCl4 над Р2О5 в слабом токе азота.
Через 1 ч выключают охлаждение в обратном холодильнике и SnCI4 перегоняют
в приемник (первую фракцию SnCl4 сливают через кран капельной воронки).
После окончания перегонки увеличивают ток азота, капельную воронку снимают
и быстро закрывают резиновой пробкой. С помощью сухого шприца, заполнен-
ного азотом, из воронки отбирают 1,36 мл (11,5 ммоля) SnCl4 (р = 2.23 г/см3)
и быстро вводят через самозатягивающуюся прокладку в заранее приготовлен-
ную колбу. Затем в ту же колбу наливают предварительно перегнанный под
Р2О5 четыреххлористый углерод в таком количестве, чтобы суммарный объем
12—732
177
раствора составил 125 мл (полученный раствор содержит 24 мг SnCl4 в 1 мл).
Проведение полимеризации
В реакционные ампулы шприцем вводят тройное количество (по сравнению
с предыдущим опытом) стирола и я-хлорстирола. Кроме того, в каждую ампулу
вводят по 12 мл сухого четыреххлористого углерода. Растворы охлаждают до
О °C и ниже, помещая ампулы в охлаждающую смесь снега с солью, после чего
в каждую ампулу шприцем вводят по 18 мл приготовленного раствора ини-
циатора (это соответствует 1,7 ммоля SnCl4). Содержимое ампул тщательно пе-
ремешивают, затем ампулы помещают в сосуд Дьюара (при О °C). Через 6 ч
ампулы вскрывают и их содержимое разбавляют 50 мл четыреххлористого угле-
рода. Сополимеры высаживают добавлением полученных растворов к 10-кратно-
му количеству метанола. Осадок фильтруют с отсасыванием и дважды переосаж-
дают из бензола или метилэтилкетона в метанол. После высушивания образцов
до постоянной массы в вакуумном сушильном шкафу при 60 °C определяют со-
держание хлора в сополимерах. Далее расчет ведут в согласии с описанием
предыдущего опыта 3-43.
Опыт 3-45. Свободнорадикальная сополимеризация стирола с акрилонит-
рилом
А. Определение констант сополимеризации
В 6 ампул со шлифами помещают перекись бензоила [0,1% (мол.)], сти-
рол и акрилонитрил в количествах, указанных в приведенной ниже таблице (об
очистке акрилонитрила и стирола см. опыты 3-01 и 3-08).
№ ампулы Перекись бен-оила, мг Стирол Акрилонитрил Время полимеризации, мин
г ммоли г ммоли
1 24,2 1,0 10 4,7 90 105
2 24,2 3,1 30 3,7 70 120
3 12,1 2,0 20 1,5 30 135
4 12,1 3,1 30 1,0 20 180
5 12,1 4,1 40 0,5 10 270
6 9,7 3,7 36 0,2 4 360
Ампулы присоединяют к вакуумной линии и откачивают при повторном за-
мораживании (в смеси сухого льда с метанолом) — размораживании и запол-
няют азотом. Затем ампулы погружают в термостат при 60 °C на время, ука-
занное в таблице. После истечения этого времени ампулы извлекают из термо-
стата, вскрывают и добавляют в каждую около 40 мл диметилформамида; сопо-
лимеры высаживают добавлением полученных растворов к 10-кратному количе-
ству метанола. Осадок фильтруют, полученные образцы дважды переосаждают
из диметилформамида в метанол, переосажденные сополимеры сушат до по-,
стоянной массы в вакуумном сушильном шкафу при 60 °C. Степень превращения'
в каждой из ампул не должна заметно превышать 10%, иначе при определении
констант сополимеризации нельзя считать постоянным отношение концентраций
мономеров в полимеризующейся смеси.
Состав сополимеров определяют по содержанию азота методом Кьельдаля
[81], используя полиакрилонитрил в качестве стандарта, или спектральными ме-
тодами (см. раздел 2.3.9). На основании полученных данных рассчитывают кон-
станты сополимеризации. Определяют растворимость полученных сополимеров,
температуры их размягчения (например, на термомеханических весах), получен-
ные результаты сопоставляют с характеристиками гомополимеров.
Б. Азеотропная сополимеризация стирола с акрилони-
трилом
Описанный ниже опыт демонстрирует независимость состава сополимера
от степени превращения при сополимеризации азеотропной смеси. В 6 ампул
со шлифами помещают по 3,23 г (31 ммоль) стирола, 1,01 г (19,0 ммоля) акри-
лонитрила и 12,1 мг (0,05 ммоля) перекиси бензоила. Ампулы откачивают, за-
полняют азотом и помещают в термостат при 60 °C. Через 2, 4, 6, 8, 12 и 20 ч
178
ампулы по очереди вынимают из термостата и быстро замораживают в охлаж-
дающей смеси, затем содержимое ампул растворяют в диметилформамиде.
В образцы, которые за время полимеризации не успели затвердеть, добавляют
по 50 мл диметилформамида, содержащего немного гидрохинона. Те же образ-
цы, которые успели затвердеть ко времени окончания полимеризации, растворяют
в 100 мл диметилформамида, также содержащего немного гидрохинона. Из по-
лученных растворов сополимеры высаживают добавлением к 10-кратному количе-
ству метанола, осадки фильтруют и высушивают. Строят график зависимости
выход полимера — время полимеризации (обсудить отклонение от линейности).
Сополимеры дважды переосаждают из диметилформамида в метанол, тщатель-
но промывают метанолом и сушат до постоянной массы. Состав сополимеров
определяют по содержанию азота методом Кьельдаля.
Опыт 3-46. Свободнорадикальная сополимеризация стирола с бутадиеном
8 эмульсии t
Стирол очищают от ингибитора (см. опыт 3-01) и перегоняют в токе азота
в специальный приемник (см. раздел 2.1.2). Бутадиен конденсируют из баллона
в охлаждаемую ловушку, заполненную азотом, и помещают в смесь сухого льда
с метанолом. Полимеризацию проводят в специальном сосуде емкостью 500 мл,
испытанном на давление 25 атм. Сосуд заполняют азотом, затем в него наливают
раствор 5 г олеата натрия (или лаурилсульфата натрия) в 200 мл кипяченой
воды, 0,5 г додецилмеркаптана (используемого в качестве регулятора молеку-
лярной массы) и 0,25 г (0,93 ммоля) персульфата калия. Содержимое перемеши-
вают встряхиванием сосуда до полного растворения всех компонентов. Доводят
pH раствора до 10—10,5 добавлением разбавленного раствора NaOH. В сосуд
под азотом заливают 30 г (0,29 моля) стирола и 70 г (1,30 моля) бутадиена и
плотно закрывают. Бутадиен переливают в полимеризационный сосуд следующим
образом. Сосуд, погруженный в охлаждающую баню со смесью сухого льда
с метанолом, ставят на весы (под тягой) и из ловушки быстро наливают бута-
диен. Избыток бутадиена испаряют. Закрытый сосуд помещают за экран и нагре-
вают до комнатной температуры. Сосуд заворачивают в ткань и интенсивно
встряхивают для получения эмульсии. Полимеризацию проводят при 50 °C. Для
этого сосуд ставят на термостатируемую переворачивающую качалку, а если
ее нет, то его интенсивно встряхивают примерно через каждый час. Продолжи-
тельность реакции 15 ч (обязательно использовать защитный экран). Затем со-
суд охлаждают вначале до комнатной температуры, а затем до 0 °C (в ледяной
воде). Сосуд повторно взвешивают для проверки утечки бутадиена. Полученный
латекс под тягой медленно выливают при перемешивании в 500 мл этилового
спирта, содержащего 2 г N-фенил-Р-нафтиламина для стабилизации полученно-
го сополимера против окисления. Непрореагировавший бутадиен испаряется; со-
полимер выпадает в виде слабо слипающихся хлопьев. Осадок фильтруют и
сушат в вакуумном сушильном шкафу при 50—70 °C в течение 1—2 сут. Со-
став сополимера можно определить аналитически по содержанию двойных свя-
зей либо спектроскопически по содержанию стирола (см. раздел 2.3.9); конфигу-
рацию звеньев бутадиена в цепи сополимера определяют по ИК-спектрам (см.
опыт 3-30). Сополимер можно превратить в нерастворимый высокоэластичный
продукт вулканизацией (см. опыт 5-10).
Опыт 3-47. Свободнорадикальная сополимеризация бутадиена с акрилони-
трилом в эмульсии
Акрилонитрил перегоняют в атмосфере азота в специальный приемник (см.
раздел 2.1.2). Бутадиен конденсируют из баллона в охлаждаемую смесью сухо-
го льда с метанолом ловушку, заполненную азотом. По методике, описанной в
предыдущем опыте, проводят полимеризацию в смеси, содержащей 10 г (0,19 мо-
ля) акрилонитрила, 25 г (0,46 моля) бутадиена, 0,1 г додецилмеркаптана (для
регулирования молекулярной массы), 0,25 г (0,93 ммоля) персульфата калия
(инициатора) и 50 мл 5%-ного водного раствора олеата натрия (или лау-
рилсульфата натрия). Через 18 ч полимеризационный сосуд охлаждают до ком-
натной температуры, а затем до 0 °C (льдом). Сосуд взвешивают для определе-
ния утечки бутадиена во время полимеризации. Латекс выливают в химический
стакан, добавляют при перемешивании 0,5 г N-фенил-р-нафтиламина, используе-
мого в качестве антиоксиданта. Затем для осаждения сополимера в стакан при
12*
179
перемешивании приливают 5%-ный раствор NaCl в 2%-ном растворе H2SO4. Оса-
док фильтруют, промывают несколько раз водой и сушат до постоянной массы
в вакуумном сушильном шкафу при 65—70 °C. Определяют содержание азота
или двойных связей в сополимере (см. опыт 3-45), рассчитывают состав сопо-
лимера. Методом ИК-спектроскопии определяют конфигурацию звеньев бутадие-
на в сополимере (см. опыт 3-30).
Полученный сополимер бутадиена с акрилонитрилом (буна N) нерастворим
в алифатических углеводородах и поэтому является маслостойким; однако эти
сополимеры растворимы в ароматических или хлорированных углеводородах.
Опыт 3-48. Свободнорадикальная сополимеризация винилхлорида с винилаце-
татом (внутренняя пластификация)
А. Получение сополимеров
Винилхлорид пропускают через промывную склянку с 50%-ным раствором
КОН и трубку длиной 40 см, заполненную хлористым кальцием, после чего
винилхлорид конденсируют в охлажденной до —70 °C ловушке. Винилацетат
очищают от ингибитора перегонкой.
В 4 толстостенные ампулы емкостью 30 мл помещают по 0,5 г олеата нат-
рия (лаурилсульфата натрия), 25 мг (0,1 ммоля) персульфата аммония и 10 мг
(0,1 ммоля) кислого сернистокислого натрия. Ампулы через специальный пере-
ходник (см. раздел 2.1.3) несколько раз откачивают и заполняют азотом; затем
в каждую из градуированной капельной воронки добавляют по 10 мл дистилли-
рованной воды. При небольшом избыточном давлении азота ампулы отсоединя-
ют, быстро закрывают резиновыми пробками и охлаждают до —70 °C в невос-
пламеняющейся охлаждающей смеси сухого льда с хлористым метиленом. Рези-
новые пробки вынимают и добавляют необходимое количество охлажденного
винилхлорида и винилацетата. Избыток винилхлорида испаряют на весах.
№ ампулы Винилаиетат Винилхлорид
г ммоли г ммоли
1 0 0 5,0 80
2 0,5 5,8 4,5 2,0
3 1,0 11,6 4,0 64,0
4 1,5 17,4 3,5 56,0
Заполненные охлажденные (предпочтительно жидким азотом) ампулы быст-
ро запаивают острым пламенем газовой горелки. При запаивании ампул нужно
следить за тем, чтобы мономерная смесь не разморозилась, иначе легколетучий
винилхлорид попадет в зону разогрева и ампулу не удастся герметично запаять.
Запаянные ампулы помещают в металлические гильзы, закрепляют в гори-
зонтальном положении на качалке в термостатируемом устройстве при 60 °C
(использовать защитный экран). Через 3 ч ампулы замораживают до —10 °C и
осторожно вскрывают, содержимое ампул размораживают, опуская ампулы в во-,
ду. Полученную эмульсию (около 50 мл в каждой ампуле) выливают в хими-
ческие стаканы емкостью 250 мл и медленно нагревают до 80 °C. Для осажде-
ния сополимеров в каждый стакан при перемешивании добавляют по 50 мл
10%-ного раствора сульфата алюминия. Полученные осадки фильтруют и не-
сколько раз тщательно промывают холодной водой, затем образцы сушат до
постоянной массы в вакуумном сушильном шкафу при 50 °C.
Б. Получение пленки из раствора
Эффект внутренней пластификации можно продемонстрировать на примере
получения эластичной пленки. С этой целью по 2 г каждого сополимера и со-
ответствующего гомополимера растворяют в 30 мл тетрагидрофурана. Получен-
ные растворы выливают на горизонтально установленные стеклянные пластинки,
которые обрамлены металлическими рамками (0,5ХЮХЮ см). Медленно испа-
ряют растворитель под тягой и через несколько часов пленку можно отделить
от стекла. Пленки высушивают на воздухе в течение двух суток. Эластичность
и жесткость пленок определяют сгибанием и разгибанием, а также процарапы-
ванием ногтем. Для проявления эффекта внешней пластификации пленку полу-
чают из раствора 1,6 г гомополивинилхлорида и 0,4 г диоктилфталата в 30 мл
180
тетрагидрофурана. Пленку получают по описанной выше методике. Пленки, мо-
дифицированные пластификатором (диоктилфталатом) либо полученные сопо-
лимеризацией с винилацетатом, более эластичны и гибки, чем пленки непласти-
фицированного ПВХ.
Опыт 3-49. Свободнорадикальная сополимеризация стирола с дивинилбензо-
лом в суспензии (трехмерная сополимеризация)
Стирол и дивинилбензол (последний используют в виде 50—60%-ного рас-
твора в этилбензоле) очищают от ингибитора и перегоняют по методике, описан-
ной в опыте 3-01.
Трехгорлую колбу емкостью 500 мл, снабженную мешалкой (желательно
с указателем числа оборотов), термометром, обратным холодильником и вводом
для азота, повторно откачивают и заполняют азотом. В колбу вносят 250 мг
поливинилового спирта (см. опыт 5-01) и растворяют при 50 °C в 150 мл воды,
предварительно переданной в атмосфере азота. К полученному раствору при
постепенном перемешивании добавляют свежеприготовленный раствор 0,25 г
(1,03 ммоля) перекиси бензоила в 25 мл (0,22 ммоля) стирола и 2 мл (7 ммолей)
дивинилбензола. В результате перемешивания смеси образуется эмульсия мелких
капелек мономера в воде. Через реакционную смесь пропускают слабый ток
азота и при постоянном перемешивании колбу нагревают на водяной бане до
90 °C. Через 1 ч примерно при 5%-ном превращении происходит образование ге-
ля. Эмульсию перемешивают еще в течение 7 ч при 90 °C, затем колбу охлаж-
дают до комнатной температуры. Перемешивание прекращают и отстоявшуюся
жидкость декантируют. Гранулы полимера несколько раз промывают метанолом
и в заключение выдерживают 2 ч в метаноле. Полимер фильтруют и сушат
в течение ночи в вакуумном сушильном шкафу при 50 °C. Выход практически
количественный. Полученный сшитый сополимер стирола с дивинилбензэлом
можно использовать для приготовления ионообменной смолы (см. опыты 5-11
и 5-13).
Для определения степени набухания 1 г сухого сополимера выдерживают в
закрытой колбе в толуоле в течение 3 сут при комнатной температуре. На-
бухшие гранулы фильтруют с отсасыванием на стеклянном фильтре в течение
5 мин и быстро взвешивают на фильтре. Под микроскопом сопоставляют раз-
мер набухших гранул с размером исходных гранул.
Опыт 3-50. Катионная сополимеризация триоксана с 1,3-диоксаланом (сопо*
димеризация с раскрытием цикла)
Триоксан и 1,3-диоксалан очищают по методике, описанной в опыте 3-39:
кипячением в течение дня над гидридом кальция и последующей перегонкой.
Нитробензол кипятят над Р2О5 и перегоняют.
Колбу емкостью 500 мл высушивают в пламени горелки при откачивании и
заполняют сухим азотом или воздухом. В колбу вносят 90 г (1 моль) триоксана
и 9 г (0,12 моля) 1,3-диоксалана в 300 мл нитробензола, колбу закрывают
пробкой с самозатягивающейся прокладкой. Затем шприцем в колбу вводят
0,18 мл (1,4-10—3 моля) эфирата трехфтористого бора в 10 мл нитробензола.
Реакционную смесь нагревают до 45 °C, и через несколько минут сополимер на-
чинает выпадать в осадок. Через 2 ч полимер промывают ацетоном, фильтруют
и вновь промывают ацетоном. Для удаления инициатора и нитробензола сопо-
лимер кипятят 30 мин в 1 л этанола, содержащего 1 % (масс.) трибутиламина;
сополимер фильтруют, промывают ацетоном и высушивают. Выход сополимера
состав ^рт ™пло 90 г. Его макромолекула содержит примерно одно звено
—О—СН2—СН2— на тридцать звеньев —О—СН2—*. Полученный продукт пла-
вптгп пои температуре около 156—159 °C и имеет молекулярную массу около
30 000 (что соответствует т)уд/С«0.04 л/г в растворе диметилформамида при
140°C). Сопоставьте термическую стойкость сополимера при 190°C (см. опыт
5-15) сп стойкостью гомополимера триоксана (см. опыт 3-39).
Опыт 3-51. Свободнорадикальная сополимеризация стирола с малеиновым
ангидридом («чередующаяся сополимеризация»)**
* Сополимеры триоксана предпочтительно анализировать методом пиролити-
ческий газовой хроматографии [82].
** Опыт не обязательно проводить в отсутствие воздуха.
181
В трехгорлую колбу емкостью 500 мл, снабженную мешалкой, термометром
и обратным холодильником, загружают 300 мл перегнанного бензола, 10,4 г
(0,1 моля) очищенного от ингибитора стирола (см. опыт 3-01), 9,8 г (0,1 моля)
чистого малеинового ангидрида* и 0,1 г (0,4 ммоля) перекиси бензоила.
Смесь перемешивают при комнатной температуре до получения прозрачного
раствора. Содержимое колбы при постоянном перемешивании нагревают до ки-
пения на водяной бане. Постепенно начинает образовываться осадок сополимера.
Через 1 ч смесь охлаждают, полимер фильтруют и сушат в вакуумном сушиль-
ном шкафу при 60 °C. Выход полимера составляет 19—20 г.
Полученный сополимер содержит строго чередующиеся звенья стирола и ма-
леинового ангидрида; омылением его можно получить кислоту (см. опыт 5-03).
Сополимер нарастворим в четыреххлористом углероде, хлороформе, бензоле, то-
луоле и метаноле, однако он растворим в тетрагидрофуране, диоксане и диме-
тилформамиде.
Опыт 3-52. Свободнорадикальная сополимеризация циклогексена с двуокисью
серы («чередующаяся сополимеризация»)
Циклогексен перегоняют в атмосфере азота в специальный приемник (см.
раздел 2.1.2). Двуокись серы конденсируют в охлажденную до —30 °C ловушку,
которую затем заполняют азотом.
Две бутылки, выдерживающие избыточное давление 3—5 атм, моют, запол-
няют азотом и охлаждают до —20-i—30 °C. Бутыли загружают следующим об-
разом.
Первая бутыль: 30 мл (0,3 моля) циклогексена, 20 мл (0,45 моля) жидкой
двуокиси серы, 5 мл перегнанного под азотом этилового спирта и 3 мл 2%-ного
водного раствора Н2О2 (1,76 ммоля).
Вторая бутыль: 10 мл (0,1 моля) циклогексена, 40 мл (0,89 моля) жидкой
двуокиси серы, 5 мл перегнанного под азотом этилового спирта и 3 мл 2%-ного
водного раствора Н2О2 (1,76 ммоля).
Бутыли закупоривают и выдерживают при комнатной температуре под тя-
гой за защитным экраном в течение 7 ч. Затем бутыли осторожно** переносят в
кастрюлю, наполненную метанолом или ацетоном. Добавлением сухого льда бу-
тыли охлаждают до —20 °C. Через несколько минут бутыли открывают и после
добавления в каждую бутыль по 50 мл хлороформа их вынимают из охлаждаю-
щей бани. Бутыли нагреваются до комнатной температуры и их содержимое пе-
реливают в стаканы емкостью 500 мл. При интенсивном перемешивании в стаканы
быстро добавляют по 300 мл диэтилового эфира, в результате чего сополимеры
выпадают в осадок. Осадок фильтруют и переосаждают, растворяя в 50 мл
хлороформа и высаживая в диэтиловый эфир, содержащий около 10% метанола
(приливанием раствора в осадитель при непрерывном перемешивании). Пере-
осажденчые образцы фильтруют и сушат в вакуумном сушильном шкафу
при 50 °C.
Сополимеры двуокиси серы с циклогексеном растворимы в хлороформе и
четыреххлористом углероде, они также растворимы в конц. серной кислоте и
могут быть высажены из этого раствора добавлением воды (без деструкции);
сополимеры разрушаются под действием горячих щелочей. Они термически стой-
ки вплоть до 200 °C.
Содержание серы в сополимере определяют методом Шенигера и по дан-
ным анализа рассчитывают состав сополимеров. Измеряют характеристическую
вязкость в растворе хлороформа при 20 °C.
3.3.2. Блок- и привитая сополимеризация
В результате обычной сополимеризации получают статистические,
или регулярные (чередующиеся), сополимеры. Разработаны так-
* Если используемый ангидрид недостаточно чист, образуется мутный рас-
твор.
** Всю работу обязательно проводить за защитным экраном; работать сле-
дует в перчатках во избежание порезов при возможном разрыве бутылей (внут-
реннее давление до 3 атм).
182
же специальные методы, позволяющие получать блок- и привитые
сополимеры, которые весьма интенсивно исследуются в последнее
время [83—87].
В состав макромолекул блок-сополимеров входят участки це-
пи, построенные из различных последовательностей звеньев типа
А и В:
—ААААААААААА—ВВВВВВВВВВ-АЛААААЛААА-
Для синтеза блок-сополимера можно исходить из готовых бло-
ков, содержащих на концах реакционноспособные группы, которые
могут инициировать радикальную или ионную полимеризацию вто-
рого мономера, в результате чего образуется блок звеньев второго
типа. Наиболее перспективны для получения блок-сополимеров
«живущие» полимеры [88], (см. раздел 3.2.1.2 и опыт 3-52), спо-
собные инициировать анионную полимеризацию добавленного мо-
номера.
Блок-сополимеры состоят из линейных макромолекул, в то
время как привитые сополимеры состоят из разветвленных макро-
молекул. Основной цепью привитого сополимера обычно является
гомополимер или статистический сополимер, к которому привиты
боковые цепи второго полимера, например
АААААААААААААААААААААА
В В
В В
В В
В В
В В
В В
В
В
Для синтеза привитых сополимеров разработан ряд специаль-
ных методов. Можно, например, получать активные центры (ради-
калы) в основной цепи, которые затем инициируют полимеризацию
второго мономера. Активные центры можно получить облучением
УФ-лучами (см. опыт 3-54), у-излучением или автоокислением,
последнее приводит к образованию гидроперекисных групп в цепи,
которые, в свою очередь, при термическом разложении образуют
свободные радикалы. Для осуществления привитой сополимериза-
ции можно использовать реакции передачи цепи; однако в этом
случае обычно образуется также и гомополимер второго, прививае-
мого мономера.
Больший интерес представляет ионная привитая сополимериза-
ция, так как в этом случае рост привитых цепей происходит толь-
ко на основной цепи без образования гомополимеров.
Исследование свойств блок- и привитых сополимеров в общем
случае намного сложнее исследования обычных статистических со-
183
полимеров (см. раздел 2.3.11). Свойства таких сополимеров в зна-
чительной степени зависят от числа и размеров блоков или развет-
влений; кроме того, их свойства зависят от структуры последова-
тельностей и мольного соотношения концентраций мономеров в
цепях сополимеров. Обычно свойства блок- и привитых сополиме-
ров аддитивны свойствам соответствующих гомополимеров, в то
время как свойства статистических сополимеров в общем случае
являются средними между свойствами соответствующих гомополи-
меров. Таким образом, можно получать сополимеры с комплексом
требуемых свойств. Это невозможно достичь простым смешением
соответствующих гомополимеров, поскольку химически различные
полимеры весьма редко совместимы друг с другом.
В заключение необходимо добавить, что привитые и блок-со-
полимеры могут быть получены также с использованием реакций
поликонденсации и ступенчатой полимеризации.
Опыт 3-53. Получение блок-сополимера 4-винилпиридина со стиролом мето-
дом анионной полимеризации
Как и в опыте 3-27, кислород и воздух должны быть полностью исключены
при получении блок-сополимера.
Стирол очищают от ингиоитора по методике, описанной в опыте 3-01, и су-
шат над СаСЬ. Затем мономер выдерживают 24 ч над СаНг и перегоняют при
пониженном давлении в токе азота в тщательно осушенный сосуд Шленка.
Чистый 4-винилпиридин дважды перегоняют над КОН при пониженном давле-
нии, затем мономер перегоняют на ректификационной колонке при пониженном
давлении в токе азота (т. кип. 62 °C при 12 мм рт. ст.) в тщательно высушенный
сосуд Шленка. Колбы закрывают и мономеры замораживают (до применения).
Методика получения раствора инициатора (натрийнафталинового комплекса)
описана в опыте 3-27.
Полимеризацию проводят следующим образом.
В сосуд Шленка, высушенный пламенем горелки и заполненный азотом, вно-
сят 50 мл тетрагидрофурана и 1 ммоль натрийнафталинового комплекса (см.
опыт 3-27). К этому раствору при перемешивании с помощью шприца, запол-
ненного азотом, добавляют 4,6 мл (40 ммолей) стирола при комнатной темпера-
туре; через сосуд одновременно пропускают ток азота. Сосуд закрывают и вы-
держивают при комнатной температуре 15 мин. Реакционная смесь при полиме-
ризации окрашивается в красный цвет.
Через 15 мин при интенсивном встряхивании в реакционный сосуд к раство-
ру «живущего» полистирола добавляют 5 мл раствора 5,3 г 4-винилпиридина
в 50 мл тетрагидрофурана (что соответствует 5 ммолям 4-винилпиридина). Еще
через 15 мин в сосуд вводят 40 ммолей стирола и через следующие 15 мин
вводят новую порцию (5 ммолей) 4-винилпиридина. Эти операции повторяют
еще раз с интервалом в 15 мин. Через 15 мин после добавления последней пор-
ции мономера блок-сополимер осаждают в смеси 300 мл диэтилового эфира и
300 мл петролейного эфира. Полимер фильтруют, промывают диэтиловым эфи-
ром и сушат в вакуумном сушильном шкафу при комнатной температуре.
Блоки 4-винилпиридина не должны быть длинными, иначе сополимер выпа-
дает в осадок из тетрагидрофурана. Это легко продемонстрировать введением
в «живущий» полимер вместо 5 ммолей 4-винилпиридина 20 ммолей. Раствор
вскоре мутнеет, однако при повторном добавлении стирола муть исчезает.
Для сопоставления получают гомополимеры стирола и 4-винилпиридина
анионной полимеризацией на натрийнафталиновом комплексе. Поли-4-винилпи-
ридин осаждается из раствора тетрагидрофурана. Осадок переносят в 200 мл
диэтилового эфира и фильтруют. Сополимер переосаждают из пиридина в
10-кратное количество диэтилового эфира. Затем осадок сушат в вакуумном су-
шильном шкафу.
184
Записывают ИК-спектры трех полимеров и сопоставляют их между собой.
Состав блок-сополимера можно определить по анализу на азот.
Проверьте растворимость полимеров. Поли-4-винилпиридин растворим в пи-
ридине, метаноле, хлороформе, он нерастворим в бензоле и диэтиловом эфире;
в воде поли-4-винилпиридин значительно набухает. Как и полистирол, блок-со-
полимер растворим в пиридине, хлороформе, бензоле; в отличие от полистиро-
ла он хорошо набухает в метаноле.
Опыт 3-54. Прививка стирола на полиэтилен
Сухой взвешенный кусок полиэтиленовой пленки (длиной 50 мм, шириной
25 мм, толщиной 0,1—0,2 мм) помещают в ампулу емкостью 70 мм со шлифом.
В ампулу добавляют несколько кристалликов оензофенона (в качестве сенсиби-
лизатора), в шлиф вставляют кран и открытую ампулу выдерживают 1 ч на
водяной бане при 60 °C. Ампулу вынимают, закрывают кран и облучают 15 мин
ртутной лампой, основная часть излучения которой лежит в области 2530—2540 А.
Во время облучения пленки во вторую ампулу помещают 50 мл очищенного
от ингибитора стирола (см. опыт 3-01) и 0,1 мл тетраэтиленпентамина. Затем
эту ампулу откачивают и заполняют азотом; 40 мл полученного раствора с по-
мощью шприца заливают в первую ампулу с пленкой. Ампулу откачивают, за-
полняют азотом и ставят в термостат при 60 °C на 2 ч. Полученную привитую
пленку экстрагируют в аппарате Сокслета этилацетатом в течение 1 ч. Затем
пленку сушат в вакуумном шкафу при 50 °C и взвешивают. Экстракт по каплям
добавляют к 400 мл метанола, выпавший в осадок полистирол фильтруют, сушат
при 50 °C в вакуумном сушильном шкафу и взвешивают.
Количество привитого стирола определяют по увеличению массы пленки;
ее можно найти также по плотностям полиэтилена dh полистирола d2 и получен-
ного привитого сополимера d$ по формуле
100— х х __100_
di d2 d3
где х — количество привитого полистирола в % (масс.) по отношению к приви-
той пленке. Плотность полистирола d2=l,05; плотность полиэтилена d\ и при-
витого сополимера da определяют на небольших образцах (0,2—0,5 см2) суспен-
зионным методом в смеси этанола с водой (см. раздел 2.3.7). Определите соотно-
шение привитого и гомополистирола.
Литература
1. Kuehl er L., Polymerisationskinetik, Springer-Verlag, Berlin — Gottin-
gen— Heidelberg, 1951.
2. Бемфорд К., Барб У., Дженкинс А., О н ь о н П. Кинетика ради-
кальной полимеризации виниловых соединений. Пер. с англ. Под ред.
Ю. М. Малинского. М., Издатинлит, 1961. 348 с.
3. Н е n г i с i - О 1 i v ё G., Olive S., Advances Polymer Sci., 1961, v. 2,
p. 496.
4. Henrici-Olive G., Olive S., Polymerisation : Katalyse— Kinetik —Me-
chanismen, Verlag Chemie, Weinheim, 1970.
5. В г e i t e n b a c h I. W., Maschin A., Z. Physik Chem., 1940, v. A187,
p. 175.
6. F r e i d 1 i n a R. К h., Karapetyan S b. A., Telomerisation and New Synthe-
tic Materials, Pergamon Press, London, 1961.
7. Bartlett P. D., К wart H., J. Am. Chem. See., 1950, v. 72, p. 1051.
8. T h i e 1 e I., Heuser K-, Liebigs Ann. Chem., 1896, v. 290, p. 30.
9. Kern W., Angew. Chem., 1949, v. 61, p. 471.
10. Плеш П. Катионная полимеризация. Пер. с англ. Под ред. В. А. Кабанова..
М., «Мир», 1971. 524 с.
11. Kennedy I. Р., Langer A. W., Advances Polymer Sci., 1961, v. 3,
p. 508.
12. О v e r b e г g e г C. G.s Schiller A. M., Advances Polymer Sci., 1961, v. 3,
p. 106.
185
13. Шварц М. Анионная полимеризация. Пер. с англ. Под ред. Н. С. Ениколо-
пяна. М., «Мир», 1971. 670 с.
14. С о о k R. Е., Dainton F. S., Ivin К- L. J- Polymer Sci., 1958, v. 29,
p. 549.
15. Ziegler K., Holzkamp E., Breil H., Martin H., Angew. Chem.,
1955, v. 67, p. 541.
16. Z i e g I e г K-, Angew. Chem., 1964, v. 76, p. 545.
17. Cooper W., Stereospecific Polymerization in: Progress in High Polymers,
v. 1, Heywood and Co. Ltd., London, 1961.
18. В awn С. E. H., Led with A., Quart. Rev. (London), 1962, v. 16, p. 361.
19. N a 11 a G., Angew Chem., 1956, v. 68, p. 393.
20. Braun D., Kern W., J. Polymer Sci., 1964, v. C4, p. 197.
21. Kern W., Braun D., Chemiker-Ztg./Chem. Apparatus 1963, v. 87, p. 799.
22. Houben-Weyl, 1961, v. 14/1, p. 635.
23. Houben-Weyl, 1961, v. 14/1, p. 798.
24. В a u m a n n U., Schreiber H., Tessmar K-, Makromol. Chem., 1960,
v. 36, p. 81.
25. Houben-Weyl, 1961, v. 14/1, p. 692.
26. N a 11 a G., Danusso F., Stereoregular Polymres and Stereospecific Poly-
merizations, v. 1 and 2, Pergamon Press, Oxford, 1967.
27. R a f f R. A., Doak K. W., Crystalline Olefin Polymers, High Polymers,
1964—65, v. 20.
28. Kunststoff-Handbuch, Hanser Verlag, Miinchen.
29. Schnee ко H. W„ Rein mo 11 er M., Lintz W., Weirauch K-,
Kern W., Makromol. Chem., 1965, v. 84, p. 156.
30. Kuhre C. L, Wales M., Doybe M. E., SPE-Journal, 1964, v. 20, p. 1113.
31. Beck H. N., J. Appl. Polymer Sci., 1967, v. 11, p. 673.
32. В i n s b e r g e n F. L., Polymer, 1970, v. 11, p. 253, 309.
33. Leu ger ing H. I., Makrcmol. Chem., 1967, v. 109, p. 204.
34. Kern W., in: H. Staudinger, Die hochrnolekularen organischen Verbindungen,
1932, p. 286.
35. Kern W., Cherdron H., Jaacks V., Angew. Chem., 1961, v. 73, p. 177.
36. Kern W., Chemiker-Ztg./Chem. Apparatus 1964, v. 88, p. 623.
37. V о g 1 O., J. Polymer Sci., 1964, v. A2, p. 4591.
38. Фурукава Дж., Саегуса T. Полимеризация альдегидов и окисей. Пер.
с англ. Под ред. Н. С. Ениколопяна и В. А. Кабанова. М., «Мир», 1965.
480 с.
39. J. Makromol. Sci., 1967. v. Al, p. 201.
40. G i e f e r A., Kern W., Makromol. Chem., 1964, v. 74, p. 39.
41. Jaacks V., Kern W., Makromol. Chem., 1964, v. 74, p. 46.
42. Rosen I., J. Makromol. Sci., 1967, v. Al, p. 267.
43. Rosen I., Hud gen D. E.. Sturm C. L., McCain G. H., W i 1 h-
j e i m R. M., J. Polymer Sci., 1965, v. A3, p. 1535.
44. Sh ash ova V. E., Sweeny W., Tietz R. F., J. Am. Chem. Soc., 1960,
v. 82, p. 866.
45. N a 11 a G., di Pietro J., Cambini M., Makromol. Chem., 1961, v. 56,
p. 200.
46. F r i s c h К. C., R e i g e n S. L., Ring-opening Polymerization, Dekker, New
York. 1969.
47. Houben-Weyl, 1963, v. 14/2, p. 425.
48. Easth am A. M., Advances Polymer Sci., 1960, v. 2, p. 18.
49. Gurgiolo A. E., Reviews Macromol. Chem., 1966, v. 1, p. 39.
50. H i 11 F. N., В a i 1 e у F. E., F i t z p a t r i c k J. T., Ind. Eng Chem., 1958,
v. 50, p. 5.
51. О s g a n M., Price С. C., J. Polymer Sci., 1959, v. 34. p. 153.
52. Furukawa J. and coworkers, Makromol. Chem., 1959, v. 32, p. 90.
53. Price С. C. and coworkers, J. Am. Chem. Soc., 1956, v. 78, p. 690, 4787.
54. T а у 1 о r G. M., Wenger E. C., Ind. Eng. Chem., 1961, v. 53, p. 46.
55. Meer we in H., Delfs D., Morschel H., Angew. Chem., 1960, v. 73,
p. 927.
186
56. Drey fuss P.. Drey fuss M. P., Fortschr. Hochpolymeren-Forsch., 1967,
v. 4, p. 528.
57. Weisermel K-, Fischer E., Gutweiler K-, Hermann H. D.,
Cher dron H., Angew. Chem., 1967, v. 79, p. 512.
58. C h a p i г о A., J. Polymer Sci., 1964, v. C4, p. 1551.
59. Fadner T. A., Mor a we tz H., J. Polymer Sci., 1960, v. 45, p. 475.
60. Ch er dron H., Ohse H., Korte F., Makromol. Chem., 1962, v. 56, p. 179,
187.
61. Rinke H., Istel E., Houben-Weyl, 1963, v. 14/2, p. 111.
62. W i c h t e r 1 e O., S e b e n d a J., К r a 1 i с e к I., Advances Polymer Sci., 1961,
v. 2, p. 578.
63. C u b b on R. С. P., Makromol. Chem., 1964,- v. 80, p. 44.
64. H a 11 H. K. j r., J. Am. Chem. Soc., 1958, v. 80, p. 6404.
65. Хэм Д. Сополимеризация. Пер. с англ. Под ред. В. А. Кабанова. М., «Хи-
мия», 1971. 616 с.
66. Mark Н., Angew. Chem., 1949, v. 61, р. 313; 1951, v. 63, р. 341.
67. Kuehl er К., Polymerisationkinetik, 1951, р. 160.
68. V о I I m е г t В., Grundriss der makromo'ekuJaren Chemie, 1962, p. 72.
69. Ring W., Makromol. Chem., 1967, v. 101, p. 145.
70. V a 1 v a s s о r i A., Sartori G., Advances Polymer Sci., 1967, v. 5, p. 28.
71. Ring W., European Polymer J., 1968, v. 4, p. 413.
72. Izu M., O’Driscoll K. F., J. Appl. Polymer Sci., 1970, v. 14, p. 1515.
73. Wit tm er P., Hafner F., Ger rens H., Makromol. Chem., 1967, v. 104,
p. 101.
74. A 1 f г e у T., Price С. C., J. Polymer Sci., 1947, v. 2, p. 101.
75. Price С. C., J. Polymer Sci., 1948, v. 3, p. 772.
76. S c h w a n T. C., Price С. C., J. Polymer Sci., 1959, v. 40, p. 457.
77. Tidwell P. W., Mortimer G. A., J. Macromol. Sci., 1970, v. C4, p. 28L
78. F i n e m a n M., Ross S. D., J. Polymer Sci., 1950, v. 5, p. 259.
79. Ring W., Makromol. Chem., 1964, v. 75, p. 203.
80. Houben-Weyl, 1953, v. 2, p. 41.
81. Houben-Weyl, 1953, v. 2, p. 191.
82. В u г g К. H., Fischer E., Wissermel K-, Makromol. Chem., 1967, v. 103,
p. 268.
83. Ко л e с н и к о в Г. С., Я ралдов Л. К., Усп. хим., 1965, т. 34, с. 195.
84. Цереза Р. Блок- и привитые сополимеры. Пер. с англ. Под ред. С. Р. Ра-
фикова. М., «Мир», 1964. 288 с.
85. S m е t s G., Hart A., Advances Polymer Sci., 1960, v. 2, p. 173.
86. Mark H., Angew. Chem., 1955, v. 67, p. 53.
87. Greber G., Makromol. Chem., 1967, v. 101, p. 104.
88. S z w a г с M., Makromol. Chem., 1960, v. 35, p. 151.
4. СИНТЕЗ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
МЕТОДАМИ ПОЛИКОНДЕНСАЦИИ
И СТУПЕНЧАТОЙ ПОЛИМЕРИЗАЦИИ
4.1. ПОЛИКОНДЕНСАЦИЯ
Процессы поликонденсации представляют собой ряд последова-
тельных реакций между, по крайней мере, бифункциональными
компонентами. При этом выделяются побочные продукты, такие,
как вода или спирт. Существуют два основных способа получения
линейных, продуктов поликонденсации из бифункциональных ком-
понентов*: либо из мономера, содержащего две различные функ-
циональные группы, способные к конденсации (реакции типа I),
либо из двух различных мономеров, содержащих по две одинако-
вые реакционноспособные группы (реакции типа II).
Примером реакций первого типа является поликонденсация
оксикислот:
I. nHO-(CHJx--COOH -----> НО-Г-(СН2)х-С-О-1—(СН2)Х-СООН
—(Zi-LH2O Д
С) Jn-i
Примером реакций второго типа может быть поликонденсация
диолов с дикарбоновыми кислотами:
II. пНО—(СН2)Х—ОН 4-пНООС—(СН2)^—СООН ———>
I ZrZ”—“ 1
+ Н—Г-О—(СН2)Х-О-С—(СН2)7—С-1—он
Поликонденсация является ступенчатым процессом. Например,
при поликонденсации оксикислоты на первой стадии образуется
димер, содержащий те же концевые группы, что и молекулы исход-
ного мономера:
2НО—(СН2)х—СООН ---> НО-(СН2)х-С—О-(СН2)Х—СООН
-Н2о ||
О
На следующей стадии концевые группы этого димера могут реаги-
ровать с мономером или с другой димерной молекулой и т. д.
В отличие от свободнорадикальной полимеризации, в данном слу-
чае молекулярная масса образующихся макромолекул увеличива-
* Это характерно и для полифункциональных соединений, способных к об-
разованию разветвленных или сшитых полимеров.
188
ется в процессе реакции. Промежуточные продукты, получающиеся
на каждой отдельной стадии, представляют собой олигомерные
или полимерные молекулы, содержащие те же функциональные
группы, что и исходный мономер. В принципе, они могут быть вы-
делены, при этом они сохраняют способность к дальнейшему росту.
Таким образом протекают все реакции типа
Mi 4- Mj -► Mi+j при 1 /, / < х
где Mt и Mj — соответственно олигомерный и полимерные продукты, содержа-
щие I и / мономерных звена.
Система уравнений, описывающая множество возможных реак-
ций между /-мерами и /-мерами с различными константами ско-
рости кц и соответствующими концентрациями Mi и Mj, была бы
весьма сложной. Однако кинетическая обработка процесса поли-
конденсации значительно упрощается, если предположить, что все
функциональные группы реагируют идентично и с одинаковой ско-
ростью независимо от молекулярной массы макромолекул, в со-
став которых они входят. «Принцип равной реакционной способно-
сти» применим как к процессам поликонденсации, так и к ступен-
чатой полимеризации. Это означает, что нет различия между реак-
ционной способностью концевых групп мономера, димера, тримера
и т. д., так что в течение всего времени скорость реакции не зави-
сит от степени полимеризации.
На основании этого была разработана кинетика поликонденса-
ции [1, 2], причем особый интерес представляют следующие во-
просы:
зависимость средней молекулярной массы от конверсии;
зависимость средней молекулярной массы от мольного соотно-
шения функциональных групп;
зависимость конверсии и средней молекулярной массы от равно-
весного состояния процесса конденсации;
обменные реакции, такие, как переэтерификация и переамиди-
рование.
За протеканием процесса поликонденсации можно легко про-
следить, анализируя количество непрореагировавших функцио-
нальных групп. Если реакционноспособные группы присутствуют
в эквимольном соотношении, как это обычно требуется (см. ниже),
достаточно определить количество одной из двух групп. Примером
может служить анализ карбоксильных групп в реакциях этери-
фикации. Если Nq — число исходных карбоксильных групп, N — их
число через определенное время t, то степень полимеризации опре-
деляется долей прореагировавших за это время функциональных
групп:
(умножая полученное значение на 100, получаем степень конвер-
сии в %).
189
__ В реакциях поликонденсации средняя степень полимеризации
Р определяется отношением исходного числа молекул мономера
к полному числу различных молекул на соответствующей стадии
реакции (N включает и все непрореагировавшие молекулы моно-
мера). Принимая во внимание уравнение (4-1), получаем:
Следовательно, Рп зависит от глубины превращения р.
Зависимость средней степени полимеризации Рп и молекуляр-
ной массы Мп от конверсии в реакциях поликонденсации и сту-
пенчатой полимеризации:
Степень конверсии Л Степень конверсии Рп Мп
50 2 200 99.5 200 20 000
75 4 400 99.7 300 30 000
90 10 1000 99,95 2 000 200 000
95 20 2 000 99,97 3 000 300 000
99 100 10 000
Молекулярная масса звена считается равной 100.
Из приведенных данных видно, что для получения полимера,,
представляющего промышленный интерес, т. е. полимера с моле-
кулярной массой 20 000—30 000, нужно проводить реакцию до
99 %-ной конверсии.
Помимо конверсии на степень полимеризации влияет также
мольное соотношение реагирующих групп А и В. Если N&—Nb—
число исходных функциональных групп А и В и r — NA/N-Q — их со-
отношение, то степень полимеризации равна
— 1 + г
Рп = 2r (1 — р) -f- 1 — г (4’3^
Если две функциональные группы А и В присутствуют в экви-
.мольном соотношении, т. е. г=1, уравнение (4-3) превращается
в уравнение (4-2).
Если все группы А прореагировали, т. е. р= 1, то уравнение (3-3)
превращается в
^=4=7 (4'4)
В общем случае влияние избытка одного из компонентов тем
сильнее, чем выше степень конверсии: при 99 %-ной конверсии мо-
лекулярная масса уменьшается вдвое при избытке, равном 2%
(мол.), в то время как при 99,5%-ной конверсии избыток, равный
1% (мол.), приводит к тому же результату. При 100%-ной кон-
версии вещества А влияние неэквивалентного количества компо-
нентов на степень полимеризации (связь между степенью полиме-
ризации Рп и соотношением числа функциональных групп А и В
190
в реакциях поликонденсации и ступенчатой полимеризации) можно
проиллюстрировать приведенными ниже данными:
Избыток компонента В (МОЛ.) Рп
10 0.9 19
1 0.99 199
0J 0,999 1 999
0,01 0,9999 19 99J
Совершенно очевидно, что важно применять эквимольные ко-
личества функциональных групп в реакциях поликонденсации, так
как даже при избытке одного из компонентов, составляющем 1%
(мол.), максимально достижимая степень полимеризации оказы-
вается меньше 200. В полиреакциях типа I (с окси- или аминокис-
лотами) точные эквимольные соотношения заданы априори, так
как мономер содержит обе реакционноспособные группы. В то же
время в полиреакциях типа II (диол и дикарбоновая кислота) да-
же небольшой избыток одного из компонентов быстро прекраща-
ет реакцию, так как остается только одно соединение, взятое в из-
бытке, концевые группы которого не могут реагировать друг с
другом.
Даже если две функциональные группы присутствуют вначале
в эквимольных количествах, их соотношение может изменяться
в ходе реакции в результате испарения, сублимации или побочных
реакций одного из реагентов.
Присутствие монофункционального соединения, которое может
реагировать с одним из бифункциональных компонентов, оказы-
вает то же действие, что и избыток бифункционального компонен-
та. Следовательно, для получения высокомолекулярного продукта
поликонденсации необходимо использовать особо чистые мономе-
ры. Кроме того, если требуется точный контроль за средней моле-
кулярной массой, нужно брать определенный избыток одного из
реагентов или добавлять соответствующее количество монофунк-
ционального соединения.
Помимо соотношений, представленных уравнениями (4-1) —
(4-4), описывающих как процессы поликонденсации, так и процес-
сы ступенчатой полимеризации, имеются два дополнительных фак-
тора, которые необходимо принимать во внимание при рассмотре-
нии поликонденсации. Во-первых, существует поликонденсацион-
ное равновесие, которое лимитирует степень конверсии, а следо-
вательно, и среднюю молекулярную массу. Как и реакции между
монофункциональными соединениями, реакции поликонденсации
являются равновесными процессами*, подчиняющимися закону
действующих масс. Например, в случае полиэтерификации, реак-
ция 1 моля гидроксильных групп (1/2 моля диола) с 1 молем карб-
* По существу, условия равновесия реализуются и в реакциях ступенчатой
полимеризации (например, обратимая реакция образования гидроксильных и
изоцианатных групп из уретановых), что, однако, возможно только при повы-
шенных температурах.
191
оксильных групп (1/2 моля дикарбоновой кислоты) описывается
уравнением (4-5), отвечающим закону действующих масс [1, 2]:
(1—р)2 (4’5)
где в соответствии с уравнением (4-1) р=(М0—N)/N — доля прореагировавших
функциональных групп (например, доля эфирных групп в данный момент време-
ни; No и N — число молей исходных и непрореагировавших функциональных
групп соответственно); (1—р) — мольная доля свободных гидроксильных или
карбоксильных групп; nw — стационарная концентрация воды в реакционной
смеси.
Решая уравнение относительно р, получаем верхний предел
значений конверсии как функцию отношения р = К//ги.
р = с + 4₽) 1 <4-б)
Так как среднечисловая степень полимеризации Рп обратно про-
порциональна доле непрореагировавших функциональных групп
(1—р), т. е.
= (4-7)
то предельная степень полимеризации в условиях равновесия опи-
сывается уравнением
Так как в большинстве случаев желательна максимальная конвер-
сия (т. е. р^1), то уравнения (4-6) и (4-8) можно упростить:
/“Т/р" (4-9)
рп^ (4-ю)
Образование высокомолекулярного продукта поликонденсации
прекращается при выделении 10% воды от ее полного количества,
получающегося в процессе реакции (если константа равновесия
имеет величину порядка 1—10). Поэтому очень важно удалять
побочные продукты поликонденсации настолько полно, насколько
это возможно.
И, наконец, следует рассмотреть обменные реакции, которые
могут протекать, например, между свободными концевыми группа-
ми и группами, связанными в цепь, например между гидроксиль-
ными концевыми группами и эфирными группами полиэфира (пере-
этерификация) :
О О
II II
• О-(СН2)Х-О-С-(СН2)^С-...
НО-(СН2)х-О—с-----
II
о
192
о о
II II
С-(СН2),-С----
—>-------о-(СН2)л-он+ |
О—(СН2)х—О—с-----
II
о
Хотя такие реакции не влияют ни на число свободных функ-
циональных групп, ни на число молекул (среднечисловая степень
полимеризации остается постоянной), они могут заметно изменять
среднемассовую степень полимеризации, а следовательно, и моле-
кулярно-массовое распределение. Например, две макромолекулы
одного размера могут взаимодействовать друг с другом с обра-
зованием одной очень длинной и одной очень короткой макромо-
лекулы; и наоборот, две различные макромолекулы могут реаги-
ровать, давая две макромолекулы одной длины. Независимо от
исходного распределения в таких обменных реакциях в каждом
случае устанавливается состояние равновесия, в условиях которо-
го скорости образования и разложения равны. Это приводит к
равновесному молекулярно-массовому распределению, которое
формально согласуется с распределением, получаемым в результа-
те случайной поликонденсации. Поэтому при обычной поликон-
денсации обменные реакции не влияют на молекулярно-массовое
распределение. Однако при смешении высоко- и низкомолекуляр-
ных полиэфиров в расплавленном состоянии вскоре достигается
равновесное молекулярно-массовое распределение: вместо двух
различных максимумов вначале появляется один. Обменные реак-
ции такого типа наблюдали также на полиамидах, полисилокса-
нах и полиангидридах.
Хотя методы синтеза продуктов поликонденсации во многих
отношениях идентичны методам конденсации монофункциональных
соединений, необходимо учитывать ряд дополнительных факторов,
особенно если ставится цель получения высокомолекулярных про-
дуктов. Эти факторы следуют из уравнений (4-1)—(4-5), (4-9),
(4-10). Прежде всего, реакция поликонденсации должна быть
специфичной и проходить с высоким выходом. Кроме того, реаги-
рующие между собой группы должны присутствовать в строго
эквимольных количествах в течение всей реакции. Равновесие ре-
акции должно быть сдвинуто в сторону образования продуктов
поликонденсации настолько, насколько это возможно. Равновесие
можно сдвинуть либо удалением выделяющегося побочного про-
дукта (например, воды), либо вакуумированием или азеотропной
разгонкой. Для усиления диффузии продуктов реакции из очень
вязкой смеси смесь полезно перемешивать либо механическим пу-
тем, либо пропускать через нее инертный газ. Исходные вещества
в реакциях поликонденсации должны быть очень чистыми. Прежде
всего, их необходимо очистить от монофункциональных примесей,
способных блокировать концевые группы растущих макромолекул
13—732 193
и, следовательно, прекращать реакцию. При синтезе линейных
продуктов поликонденсации нежелательно присутствие полифунк-
циональных соединений, так как они вызывают разветвление и
сшивание. И, наконец, поликонденсацию следует проводить в от-
сутствие кислорода во избежание окислительной деструкции, по-
скольку реакции часто протекают при повышенных температурах.
4.1.1. Сложные полиэфиры
Полиэфиры представляют собой полимеры, макромолекулы кото-
рых содержат сложноэфирные группы:
НО—(СН^х—О—Г—С—(СН2)^—С—О—(СН2)Х—О—"I—С—(СН2)^—СООН
Линейные полиэфиры обычно растворимы в хлороформе, ди-
хлорбензоле и муравьиной кислоте. Свойства полиэфиров в зна-
чительной степени определяются их составом. Алифатические по-
лиэфиры плавятся обычно при температурах ниже 100 °C (темпера-
тура плавления возрастает с увеличением числа метиленовых
групп между эфирными группами) и легко омыляются. В то же
время полиэфиры ароматических или циклоалифатических дикар-
боновых кислот и диолов являются высокоплавкими продуктами,
которые гидролизуются с трудом (например, полиэфиры терефта-
левой кислоты и этиленгликоля или 1,4-бис-оксиметилциклогекса-
на). Такие полиэфиры используются в производстве волокон и
пленок. Полиэфиры получают обычно следующими методами [3,
4]:
поликонденсацией оксикислот;
поликонденсацией диолов с дикарбоновыми кислотами;
поликонденсацией диолов с производными дикарбоновых кис-
лот (например, ангидридами или эфирами дикарбоновых кислот);
полимеризацией лактонов с раскрытием циклов (см. раз-
дел 3.2.4.3).
Для ускорения этих реакций поликонденсации во многих слу-
чаях используют кислые или щелочные катализаторы [5], а имен-
но сильные кислоты, такие, как /г-толуолсульфокислота, окислы
металлов (двуокись свинца, трехокись сурьмы), соли щелочных и
щелочноземельных металлов слабых кислот (ацетаты, бензоаты)
или алкоголяты.
4.1.1.1. Полиэфиры на основе оксикарбоновых кислот [6]
Поликонденсацию алифатических оксикарбоновых кислот прово-
дят обычно в расплаве и получают высокомолекулярные полиэфи-
ры, используя в качестве исходных веществ гликолевую, а-окси-
пропионовую или рицинолевую кислоты. Однако в этой реакции
часто образуются циклические эфиры (лактоны). Способность к
194
циклизации определяется числом метиленовых звеньев между карб-
оксильными и гидроксильными группами; в наибольшей степени
она проявляется для у-оксимасляной, д-оксйкапроновой и е-окси-
валериановой кислот.
Ароматические карбоновые кислоты также могут вступать в
реакции поликонденсации с образованием высокомолекулярных
полиэфиров.
4.1.1.2. Полиэфиры на основе диолов и дикарбоновых кислот
Поликонденсацию диолов с дикарбоновыми кислотами часто про-
водят в расплаве. Однако в этом случае не всегда получаются
высокомолекулярные полиэфиры, так как исходные соединения
или образующиеся полимеры оказываются нестабильными при вы-
соких температурах. Более того, скорости реакций между диолами
и дикарбоновыми кислотами часто очень малы. В таких случаях
полезно использовать производные дикарбоновых кислот (ангидри-
ды или эфиры) вместо самих кислот. Для получения полиэфиров
с очень большими молекулярными массами используют реакции
поликонденсации в растворе при повышенных температурах (см.
также раздел 2.1.5.2).
Опыт 4-01. Получение низкомолекулярных разветвленных полиэфиров поли-
конденсацией в расплаве
А. Синтез слаборазветвленных полиэфиров
Прибор для получения разветвленных полиэфиров состоит из трехгорлой
колбы емкостью 500 мл, снабженной вводом для азота, мешалкой, термометром,
касающимся дна колбы, дефлегматором длиной 20 см (в верхнюю часть дефлег-
матора вставлен термометр) и холодильником с «пауком» и приемником. В кол-
бу помещают 146 г (1 моль) адипиновой кислоты, 110,5 г (1,04 моля) дигликоля
(или ди-Р-оксиэтилового эфира) и 9,0 г (0,067 моля) безводного 1,1,1-тримети-
лолпропана. Смесь постепенно нагревают, пропуская через нее небольшой ток
азота. При 80—100 °C содержимое колбы становится настолько жидким, что
можно включить мешалку. При 130—140 °C начинается реакция поликонден-
сации, о чем свидетельствует появление воды. После этого температуру начина-
ют повышать до 200 °C с такой скоростью, чтобы температура в верхней части
дефлегматора не превышала 100 °C. За это время отгоняется основное количест-
во воды (Зо г), поэтому перемешивание нельзя прерывать, так как в противном
случае жидкость может вспениться.
Как только температура в верхней части дефлегматора опустится ниже
100 °C, систему откачивают до 12—14 мм рт. ст. На этой стадии следует обра-
тить внимание на возможность переброса дигликоля и по мере необходимости
откачивать более медленно. Поддерживая постоянными температуру (200 °C) и
давление реакционной смеси (12—14 мм рт. ст.), через каждые 5 ч отбирают
пробу для определения кислотного числа. Для отбора пробы систему заполня-
ют азотом, а затем снова откачивают. Реакцию прекращают, когда кислотное
число становится меньше двух (полное время реакции составляет 35—40 ч).
После охлаждения в атмосфере азота слаборазветвленный полиэфир представ-
ляет собой вязкую, светло-желтую массу. Число гидроксильных групп* равно
примерно 60, и полиэфир может быть непосредственно использован для полу-
чения эластичной полиуретановой пены (см. раздел 4-22).
Б. Синтез сильноразветвленных полиэфиров
Навески 16,9 г (0,12 моля) адипиновой кислоты, 42,5 г (0,15 моля) олеино-
вой кислоты, 44,4 г (0,30 моля) фталевого ангидрида и 105 г (0,78 моля)
* Смотри пункт Г.
13*
195
1,1,1-триметилолпропана помещают в прибор, аналогичный описанному выше.
Систему вакуумируют и заполняют азотом, а затем постепенно нагревают в токе
азота. При 120 °C содержимое колбы плавится, и смесь начинают перемешивать.
Температуру реакционной смеси повышают до 190 °C в течение 2 ч. Как только
температура в верхней части дефлегматора упадет ниже 70 °C, систему медлен-
но откачивают до 30 мм рт. ст. в течение 2 ч. При этом давлении и постоянно
поддерживаемой температуре 190 °C смесь перемешивают еще 8 ч. Затем колбу
заполняют азотом и охлаждают. Получившаяся вязкая жидкость и есть развет-
вленный полиэфир, кислотное число которого равно двум, а гидроксильное чис-
ло — 350; полимер можно использовать для получения жесткой полиуретановой
пены (см. опыт 4-23).
В. Определение кислотного числа
Навеску 1—2 г полиэфира растворяют в ацетоне при нагревании и после
охлаждения быстро титруют 0,1 н. спиртовым раствором КОН с фенолфталеи-
ном до появления красной окраски, сохраняющейся в течение нескольких секунд.
В холостом опыте определяют щелочной вклад растворителя. Кислотное число
определяется количеством КОН, приходящимся на 1 г образца
к. ч. = 5,6If . —
где А — количество КОН, израсходованное на титрование образца, мл; В — ко-
личество КОН, израсходованное на титрование растворителя, мл; Е — навеска
образца, г; f — титр спиртового раствора КОН.
Г. Определение гидроксильного числа [7]
Для ацетилирования свободных гидроксильных групп полиэфира готовят
раствор 150 г свежеперегнанного ангидрида (т. кип. 140 °C) в 350 г свеже-
перегнанного безводного пиридина (т. кип. 114,5 °C). Раствор, немного желтею-
щий при стоянии, помещают в темную бутыль.
В каждую из трех колб Эрленмейера емкостью 250 мл с коническим дном
помещают по 2,5 г полиэфира (точность взвешивания ±0,001 г). Образцы рас-
творяют в 10 мл пиридина при слабом нагревании. После охлаждения в каждую
колбу добавляют по 10 мл ацетилирующего раствора. Для холостых опытов две
колбы Эрленмейера заполняют 10 мл пиридина и 10 мл ацетилирующего рас-
твора. Пять колб закрывают стеклянными, плотно притертыми пробками и по-
мещают в сушильный шкаф при температуре ПО °C на 70 мин (вынимая колбы
из шкафа, будьте осторожны: наденьте защитные очки и асбестовые перчатки).
После охлаждения к каждому образцу добавляют несколько капель 0,1%-ного
водного раствора «хлорида нильского синего»* и растворы титруют 1 н. раство-
ром КОН до изменения красного цвета на фиолетовый**. Гидроксильное число
определяется количеством КОН в мг, эквивалентным количеству свободных гид-
роксильных групп в 1 г полимера:
Р В —Л
г. ч. = 5,61/-—g—
Где л — количество КОН, израсходованное на титрование образца, мл; В —
количество КОН, израсходованное на титрование растворителя; Е — навеска
образца, г; f — титр спиртового раствора КОН.
Опыт 4-02. Получение высокомолекулярных линейных полиэфиров поликон-
денсацией в растворе
В колбу (см. прибор для перегонки, рис. 17) помещают 59,05 г (0,5 моля)
перегнанного гександиола-1,6, 59,09 г (0,5 моля) перекристаллизованной янтар-
ной кислоты, 200 мл безводного толуола и 1,5 г чистой п-толуолсульфокислоты;
сифон заполняют толуолом таким образом, чтобы циркуляция растворителя на-
чалась немедленно. Насыпав в колбу кипятильники, смесь разогревают до кипе-
* Фирма «Мерк», Дармштадт, ФРГ.
** Гидроксильное число можно также определить титрованием по фенолфта-
леину [8].
196
ния на масляной или воздушной бане; толуол должен циркулировать через труб-
ку с осушителем и возвращаться в колбу. Известь в осушительной трубке сле-
дует заменить спустя несколько часов, когда 3Д от теоретического количества
воды соберется в сепараторе. Еще через несколько часов осушитель вновь за-
меняют.
Вязкость реакционной смеси постепенно возрастает с повышением темпера-
туры в колбе. Как только температура смеси достигнет 130 °C, в колбу добав-
ляют 50 мл абсолютированного толуола для поддержания интенсивной пере-
гонки.
Через 25 ч содержимое колбы охлаждают до комнатной температуры, а за-
тем смесь по каплям приливают в 10-кратный избыток метанола. Осадок от-
фильтровывают и сушат под вакуумом при 40 °C. Выход полимера составляет
около 90%.
Синтезированный в растворе линейный полиэфир имеет большую молеку-
лярную массу, чем полимер, полученный в расплаве. Он растворим в бензоле,
толуоле, хлороформе, муравьиной кислоте и легко гидролизуется (см.
опыт 5—19).
Температура плавления полиэфира около 100 °C. За увеличением молеку-
лярной массы в ходе реакции можно проследить, отбирая пробы по мере ее
протекания, выделяя и высушивая полимер. Молекулярную массу полимера
определяют по вязкости либо по концевым карбоксильным группам (см. раз-
дел 2.3.2.2).
4.1.1.3. Полиэфиры на основе диолов и дикарбоновых кислот
Если при поликонденсации диолов и дикарбоновых кислот заме-
нить кислоту ее производным (например, диэфиром), то поликон-
денсацию в расплаве можно проводить при более низких темпе-
ратурах. Переэтерификация протекает под действием кислотных
или щелочных катализаторов и сопровождается выделением легко-
летучего спирта (например, метанола). Вместо диэфиров и ди-
карбоновых кислот можно использовать диангидриды (см.
опыт 4-06).
Реакцию поликонденсации диолов с хлорангидридами карбо-
новых кислот используют главным образом для синтеза поликар-
бонатов [9, 10]. Наиболее важные в промышленном отношении
поликарбонаты получают из бисфенолов и фосгена. В отличие от
продуктов взаимодействия алифатических диолов с фосгеном эти
поликарбонаты имеют более высокие температуры размягчения и
стеклования. Они растворяются в ряде органических растворите-
лей (в хлорированных углеводородах и циклических эфирах);
обладают высокой стойкостью к воде и водным минеральным кис-
лотам, но деструктируются под действием щелочей, аммиака и
аминов. Реакции между диоксипроизводными и фосгеном
СН8
пНО——он + пСОС12
СН3
197
протекают даже при низких температурах, например при
комнатной температуре, и могут ускоряться при добавлении чет-
вертичных аммониевых солей. Обычно реакция идет в безводных
органических растворителях (например, пиридине) или в смесях
метиленхлорида или бензола с водным раствором едкого натра.
Таким методом получают поликарбонаты с молекулярной массой
от 25 000 до 75 000 и выше.
Опыт 4-03. Получение полиэфира из этиленгликоля и диметилтерефталата
поликонденсацией в расплаве
Этиленгликоль в присутствии 2% металлического натрия кипятят с обратным
холодильником в течение 1 ч и затем перегоняют. Диметилтерефталат перекри-
сталлизовывают из метанола и тщательно сушат в вакууме (т. пл. 141—142 °C).
В круглодонную колбу емкостью 50 мл помещают 9,7 г (0,05 моля) диме-
тилтерефталата, 7,1 г (0,115 моля) и этиленгликоля, 0,015 г чистого безводного
ацетата кальция и 0,04 г трехокиси серы. Колбу соединяют с дефлегматором,
воздушным холодильником, «пауком» и приемником. Систему откачивают и за-
полняют азотом, а содержимое расплавляют на масляной или металлической
бане при 170 °C. Через длинный капилляр, опущенный практически до дна кол-
бы, пропускают ток азота. Переэтерификация происходит моментально. Метанол
отгоняют и собирают в приемник для определения степени конверсии. Как толь-
ко прекращается выделение спирта (через 1 ч), температуру повышают до 220 °C
и поддерживают ее в течение 2 ч для того, чтобы отогнать остатки метанола.
Избыток этиленгликоля удаляют, повысив температуру до 220 °C на 15 мин, а
затем до 280 °C. Еще через 15 мин приемник заменяют круглодонной колбой
и систему откачивают до 0,5 мм рт. ст., поддерживая постоянную температуру
на уровне 280 °C. Через 3 ч реакция поликонденсации заканчивается. Пропуская
ток азота в систему, колбу охлаждают, а затем осторожно разбивают молотком
и извлекают твердый полиэтиленгликольтерефталат. Полиэфир растворим в
//-крезоле и может быть переосажден эфиром или метанолом. Определите вяз-
кость полимера в //-крезоле или в смеси фенола с тетрахлорэтаном (1:1) (см.
раздел 2.3.2.1) и температурный интервал размягчения полимера. Волокна, по-
лученные из расплава, можно растягивать руками. Для синтеза полиэфира мож-
но использовать прибор, описанный в опыте 4-08.
Опыт 4-04. Получение поликарбоната из бисфенола А и фосгена поликонден-
сацией в растворе
Под тягой собирают прибор, состоящий из 3 промывалок, соединенных с
баллоном с фосгеном. Две крайние промывалки оставляют пустыми и соединя-
ют между собой обратимо, навстречу друг другу, а среднюю заполняют жидким
парафином (глицерином нельзя). Последнюю промывалку соединяют с трехгор-
лой колбой, снабженной мешалкой, термометром и трубкой для ввода и вывода
газа. Термометр вставляется через Т-образную газовую трубку, служащую одно-
временно для вывода газа, и опускается глубоко в реакционную смесь. Трубка
для ввода газа диаметром не менее 5 мм тоже должна быть погружена в реак-
ционную смесь, причем конец ее устанавливают чуть выше лопасти мешалки
для предотвращения забивания ее осаждающимся пиридинийгидрохлоридом. Ко-
лено для вывода газа соединяют с пустой промывалкой, которая, в свою очередь,
связана с промывалкой, содержащей жидкий парафин.
В колбу насыпают 22,5 г чистого бисфенола (т. пл. 155—157 °C) и прибав-
ляют 230 мл перегнанного пиридина (осторожно: бисфенол А — сильный лакри-
матор). Реакционную смесь энергично перемешивают, пропуская в раствор фос-
ген с такой высокой скоростью, что пузырьки газа едва различимы. (При откры-
вании крана баллона с фосгеном и при разборке прибора следует надевать про-
тивогаз) .
Реакция начинается мгновенно, и содержимое колбы постепенно мутнеет
в результате выпадания осадка гидрохлорида пиридиния. Температуру реакции
следует поддерживать около 25 °C, и если она повысится до 30 °C, надо охладить
колбу на водяной бане. После завершения реакции смесь становится очень вяз-
кой (через 45—60 мин). Фосген пропускают в колбу до тех пор, пока он не
198
окажется в небольшом избытке (спустя 5 мин); в этом случае образуется комп-
лекс фосгена с НС1 желтого цвета. Затем прибор разбирают (следует надеть
противогаз).
Газовую трубку заменяют капельной воронкой и выделяют полимер, при-
бавляя к реакционной смеси при сильном перемешивании 250 мл метанола в те-
чение 10 мин. Смесь продолжают перемешивать в течение нескольких минут,
продукт фильтруют и сушат в вакууме при 50 °C, переосаждают из раствора в
метиленхлориде метанолом и снова сушат в вакууме при 50 °C. Поликарбонат
растворим в метиленхлориде, хлороформе, пиридине, диоксане, тетрагидрофура-
не. Температура плавления полимера лежит в интервале от 225 до 250 °C. Опре-
делите вязкость поликарбоната в тетрагидрофуране и рассчитайте молекуляр-
ную массу (см. раздел 2.3.2.1).
4.1.1.4. Получение и сшивание (отверждение)
ненасыщенных полиэфиров
При полимеризации диолов или дикарбоновых кислот, содержа-
щих двойные связи, образуются ненасыщенные полиэфиры. В ка-
честве исходных мономеров для их получения удобно использовать
малеиновую и фумаровую кислоты (или ангидрид малеиновой кис-
лоты) или бутен-2-диол-1,4. Количество двойных связей в полиэфи-
ре можно варьировать, используя смеси насыщенных и ненасы-
щенных дикарбоновых кислот и диолов (сополиконденсация).
Обычно ненасыщенные полиэфиры получают поликонденсацией в
расплаве. Конечные продукты, как правило, представляют собой
вязкие или воскоподобные вещества относительно небольшой моле-
кулярной массы.
Двойные связи вводят в макромолекулы полиэфиров для облег-
чения реакций сшивания, которые протекают по свободнорадикаль-
ному или ионному механизму.
Наибольший интерес представляют ионные реакции сшивания
по двойным связям. Однако радикальные процессы сшивания яв-
ляются практически более важными. Инициирование таких реак-
ций может происходить под действием кислорода или света, осо-
бенно в присутствии соответствующих катализаторов, например
соединений двухвалентного кобальта («воздушная сушка»). Боль-
щие возможности в этом плане представляет сополимеризация.
Для этого ненасыщенный полиэфир растворяют в мономере, спо-
собном к радикальной сополимеризации, и добавляют соответст-
вующий инициатор. Выбранный инициатор определяет темпера-
туру полимеризации. При использовании перекисей, таких, как
перекись бензоила, перекись циклогексанона или гидроперекиси,
полимеризацию проводят при 70—100°C («горячее отверждение»),
в присутствии окислительно-восстановительных систем — при ком-
натной температуре («холодное отверждение»). Наиболее распро-
страненными окислительно-восстановительными системами явля-
ются смеси перекиси и восстановителя, растворимого в органиче-
ской среде (например, нафтенат или октоат кобальта или меди и
третичный амин, такой, как М,1\[-диметиланилин). В качестве сши-
вающего агента обычно используют стирол. В результате реакции
образуются прозрачные нерастворимые термостойкие продукты с
199
хорошими механическими свойствами, которые применяются в ка-
честве связующего при получении стеклопластиков. Строение
ненасыщенных полиэфиров можно представить следующим обра-
зом:
I i
•. —ОСО-СН-СН—СОО—СН2—СН2—ОСО -сн— сн—соо—сн2—сн2—о-
---осо—сн—сн— соо—сн2—сн2—осо—сн—сн—соо—сн2—сн2— о---
сн2 сн2
Реакции сшивания проводят по следующей методике. Поли-
эфир растворяют при перемешивании в виниловом мономере (7:3)
и смесь нагревают до 130 °C для более интенсивного растворения.
Для предотвращения преждевременной полимеризации в систему
добавляют ингибитор (0,1—0,5% гидрохинона или 4-трет-бутилка-
техина). После того как смесь гомогенизируется, раствор охлажда-
ют до температуры полимеризации и добавляют инициатор (1—
3% от общего количества реагентов). Образование геля в реак-
ционной смеси служит доказательством начала полимеризации.
В зависимости от условий реакции отверждение полимера проте-
кает приблизительно 24 ч. Полученный сшитый полимер часто
содержит некоторое количество растворимых продуктов (раство-
римый виниловый гомополимер), которое можно точно определить
путем экстракции подходящим растворителем (например, бензо-
лом, если в качестве винилового компонента взят стирол). Коли-
чество растворенного вещества служит мерой степени сшивания
и прививки.
Опыт 4-05. Получение ненасыщенного полиэфира и его отверждение стиро-
лом
Синтез ненасыщенного полиэфира
В трехгорлую колбу емкостью 500 мл, снабженную мешалкой, термометром,
трубкой для ввода азота и холодильником, помещают 80 г пропиленгликоля-1,2
(1 моль+5°/0-ный избыток), 49 г (0,5 моля) малеинового ангидрида, 47 г
200
(0,5 моля) фталевого ангидрида и 37 г (0,02%) гидрохинона (ингибитор по-
лимеризации). Систему откачивают и заполняют азотом, а затем постепенно на-
гревают в токе азота. При 80—90 °C реакционная смесь плавится и мешалка
начинает вращаться. При 180—190 °C начинается этерификация. Доказатель-
ством начала реакции служит выделение воды, которую отгоняют. Температуру
реакционной смеси поддерживают постоянной до тех пор, пока кислотное число
не понизится до 50, что занимает приблизительно 5—6 ч. Расплав охлаждают
до 140 °C и быстро (в течение 1 мин) добавляют 100 г перегнанного стирола
(при перемешивании), содержащего 0,03% гидрохинона. После этого смесь мед-
ленно охлаждают до комнатной температуры (на водяной бане) для предотвра-
щения полимеризации. Получается вязкий светло-желтый раствор полиэфира
в стироле.
Отверждение ненасыщенного полиэфира стиролом
Для сшивания полиэфира при комнатной температуре (холодное отвержде-
ние) 10 г раствора полиэфира помещают в химический стакан или в плоскую
оловянную чашку и при перемешивании последовательно добавляют компоненты
окислительно-восстановительной системы (одновременно не добавлять). В каче-
стве инициаторов можно использовать, например, 0,06 мл 10%-ного раствора
нафтената кобальта(II) в стироле (можно октоат кобальта); 0,2 мл перекиси
циклогексанона или перекиси метилэтилкетона (в виде 50%-ного раствора в
дибутилфталате) или 200 мг перекиси бензоила и 0,05 мл чистого диметил-
анилина.
Через 30—40 мин смесь разогревается, что свидетельствует о начале от-
верждения. Спустя 1 ч большая часть стирола заполимеризовывается, и образец
отверждается.
Для сшивания при высоких температурах (горячее отверждение) 0,1 г пере-
киси бензоила растворяют в 10 г раствора полиэфира. Полимеризация начи-
нается через несколько минут после достижения 80 °C (образуется гель) и в ос-
новном заканчивается через 15 мин. Полученные образцы заполимеризованы
еще не полностью, и для достижения оптимальной жесткости отверждение же-
лательно продолжать еще 1—2 ч при 70—100 °C. По 1 г каждого из образцов
(холодное, горячее и постотверждение) тщательно растирают и обрабатывают
10 мл бензола в течение 30 мин. После фильтрования и промывания бензолом
образцы сушат в вакууме при 60 °C для определения потери массы. Для осаж-
дения и выделения полистирола бензольные растворы выливают в метанол.
Определите способность к набуханию отвержденных образцов в органических
растворителях.
Для получения стеклопластиков стеклянное волокно пропитывают полиэфир-
ным раствором, содержащим радикальный инициатор, и отверждают при опре-
деленной температуре.
4.1.1.5. Получение и сшивание (отверждение)
алкидных смол
Алкидные смолы [11] представляют собой разветвленные или сши-
тые полиэфиры, получаемые поликонденсацией, например, дикар-
боновых кислот с олигофункциональными спиртами. Реакции при-
вивки и сшивания протекают гладко и легко контролируются. Так,
при поликонденсации фталевой кислоты (или ангидрида) с гли-
церином образуется полиэфир, который остается растворимым и
плавким, если прекратить реакцию на стадии превращения карб-
оксильных или гидроксильных групп менее чем на 75%. Если сте-
пень конверсии достигает 75% (мол.), полиэфиры превращаются
в полностью нерастворимые продукты. В отличие от реакций сши-
вания методом привитой сополимеризации ненасыщенных поли-
эфиров синтез сшитых алкидных смол следует проводить при
201
200 °C, так как сшивание происходит в результате поликонденса-
ции (отверждение лаков). Однако, если карбоновые кислоты со-
держат двойные связи, можно приготовить алкидные смолы, сши-
вающиеся при низких температурах (например, модифицированные
маслом алкидные смолы, см. опыт 4-07). При этом механизмы
реакций сшивания могут быть различными. Обычно используют
фталевую кислоту, ее ангидрид или их смесь с адипиновой или не-
насыщенной кислотой. Кроме глицерина в качестве полигидрок-
сильного компонента часто используют пентаэритрит и гексантри-
ол. Синтез алкидных смол, так же как и их сшивание, обычно
проводят в расплаве (за исключением ряда ненасыщенных алкид-
ных смол) по методу, описанному в разделе 4.1.
Опыт 4-06. Получение и сшивание алкидных смол из глицерина и фталевого
ангидрида
В трехгорлую колбу емкостью 250 мл, снабженную отводом Клайзена, ме
шалкой, холодильником Либиха и трубкой для ввода азота, помещают 74 г
(0,5 моля) фталевого ангидрида и 30,5 г (0,33 моля) глицерина. Смесь нагрева-
ют до 200 °C в токе азота. Через десятиминутные интервалы отбирают пробы
расплава с помощью стеклянного капилляра и тотчас же выливают на алюми-
ниевую фольгу. После охлаждения образцов определяют кислотное число (см.
опыт 4-01). Дополнительно проверяют растворимость смол в смеси толуола с
бутилацетатом (1 : 1). Для этого 1 г порошка смолы смешивают с 5 мл раство-
рителя в ампуле и оставляют на 1 ч при комнатной температуре. Результат
может быть следующим: полимер полностью растворим, частично растворим или
нерастворим. Реакцию поликонденсации ведут до тех пор, пока кислотное число
не достигнет значения, равного 127—132 (приблизительно через 1,5 ч). На этой
стадии реакции разветвленный полимер еще растворим и плавок. За постконден-
сацией, которая приводит к полному сшиванию, наблюдают, нагревая 10 про-
бирок, содержащих по 1 г смолы, до 200 °C и определяя растворимость через
каждые 5 мин. Данные о времени поликонденсации, кислотном числе и раство-
римости впишите в таблицу.
Опыт 4-07. Получение ненасыщенной алкидной смолы («воздушной сушки»)
Высыхающие на воздухе лаки, сшивающиеся под действием кислорода воз-
духа при комнатной температуре (например, их используют в производстве мас-
ляных красок), представляют собой так называемые модифицированные маслами
алкидные смолы. Для сшивания смолы в макромолекулу полимера в процессе
поликонденсации вводят двойные связи, добавляя определенное количество не-
насыщенной карбоновой кислоты.
Многогорлую колбу емкостью 0,5 л с мешалкой, термометром, капельной
воронкой, обратным холодильником и трубкой для ввода азота заполняют азо-
том и наливают 118 г льняного масла. Масло нагревают до 235 °C в атмосфере
азота. К маслу добавляют 0,5 г хорошо измельченной окиси свинца, а затем
медленно (в течение 20 мин) вводят из капельной воронки 26 г (0,28 моля)
глицерина. Через 30 мин образуется гомогенная смесь, в которую вводят сразу
5 г (0,36 моля) ангидрида фталевой кислоты, предварительно расплавленного
для предотвращения попадания кислорода в систему. Капельную воронку заме-
няют сепаратором для отделения воды, образующейся в процессе поликонден-
сации.
Смесь перемешивают при 250 °C до тех пор, пока се кислотное число не
станет меньше 5 (об определении кислотного числа см. с. 196). Во избежание
попадания кислорода в реакционный сосуд через колбу непрерывно пропускают
медленный ток азота. Через 6 ч поликонденсацию заканчивают и охлаждают
смесь до комнатной температуры. Полученная алкидная смола представляет со-
бой вязкую жидкость, растворимую в ароматических углеводородах, бутилаце-
тате и ацетоне.
202
Если 50%-ный раствор ненасыщенной алкидной смолы в бензоле, содержа-
щий 1,5% (по отношению к количеству смолы) нафтената кобальта в качестве
катализатора, налить на металлическую поверхность и дать «просохнуть» при
комнатной температуре на воздухе, то он образует гладкую прозрачную лако-
вую пленку.
Белый лак можно приготовить, тщательно перемешав в ступке 50 г раство-
ра полученной смолы, 75 г литопона, 25 г окиси цинка, 27 г скипидара и 1 г
нафтената кобальта. Эта смесь образует лаковую пленку в тех же условиях,
что и неокрашенный лак.
4.1.2. Полиамиды
Полиамиды представляют собой полимеры, макромолекулы кото-
рых содержат амидные группы:
H2N~(CH2)x-N-r—C-(CH2)Z/—C-N—(СН2)Х—N-l-С—(СН2)Д,~СООН
I II II I I II
H L О ОН н Jrt о
Полиамиды принято классифицировать в соответствии с числом
атомов углерода в диамине (первая цифра) и дикарбоновой кис-
лоте (вторая цифра). Так, продукт поликонденсации гексамети-
лендиамина и адипиновой кислоты называют полиамидом 6,6 (най-
лон 6,6), а продукт поликонденсации гексаметилендиамина и се-
бациновой кислоты — полиамидом 6,10 (найлон 6,10). Полиамиды,
полученные в результате конденсации аминокислот или полимери-
зации лактамов с раскрытием цикла, обозначают одной цифрой:
полиамид 6 (найлон 6) — это полиамид, полученный из е-амино-
капроновой кислоты или g-капролактама.
Многие свойства полиамидов обусловлены образованием во-
дородных связей между NH-группами и СО-группами соседних
макромолекул. Это доказывается их растворимостью в серной и
муравьиной кислотах и в л/-крезоле, высокими температурами
плавления (даже если они состоят только из алифатических ком-
понентов) и стойкостью к омылению.
Температуры плавления алифатических полиамидов зависят от
числа алифатических групп исходных мономеров: с увеличением
числа атомов углерода между амидными группами температуры
плавления соответствующих полиамидов понижаются; полиамиды,
содержащие мономерные звенья с равной длиной алифатических
частей, плавятся при более высоких температурах, чем те, которые
состоят из сомономеров разной длины. Боковые алкидные группы
(N-алкилированные полиамиды) понижают температуру плавле-
ния и улучшают растворимость полиамида. Полиамиды в боль-
шинстве своем являются исходными материалами для производст-
ва волокон и других материалов, которые должны выдерживать
большие механические нагрузки. При проведении синтеза поли-
амидов [12, 13] следует просмотреть разделы 2.1.5.1, 2.1.5.2 и 4.1.
Полиамиды получают следующим способом:
поликонденсацией со-аминокислот;
поликонденсацией диаминов с дикарбоновыми кислотами;
203
поликонденсацией диаминов с производными дикарбоновых
кислот (например, с хлорангидридами);
полимеризацией лактамов с раскрытием цикла (см. раз-
дел 3.2.4.4).
Достижение равновесия при поликонденсации можно ускорить
использованием катализатора, однако в отличие от полиэтерифи-
кации нет необходимости проводить указанные реакции в равно-
весных условиях. Первые две реакции получения полиамидов про-
водят обычно в расплаве; поликонденсацию в растворе (например,
в ксилоле или п-трет-бутилфеноле) используют значительно реже
из-за плохой растворимости полиамидов. В то же время поликон-
денсацию диаминов с дихлорангидридами проводят либо в рас-
творе, либо на границе раздела фаз.
4.1.2.1. Полиамиды на основе со-аминокислот
Синтез полиамидов путем поликонденсации аминокислот при по-
вышенных температурах (опыт 4-08) возможен лишь в том случае,
если аминокислоты содержат более четырех метиленовых групп
между амино- и карбоксильной группами. Реакция сопровождается
выделением воды; а и б-аминокислоты в условиях дегидратации
обычно циклизуются (глицин—>*2,5-дикетопиперазин, у-аминомас-
ляная кислота —>у-бутиролактам).
Опыт 4-08. Поликонденсация 8-аминокапроновой кислоты в расплаве
Реакцию можно проводить в приборе, описанном в опыте 4-03. Для получе-
ния небольших количеств найлона 6 используют простой прибор, позволяющий
одновременно определять количество выделившейся воды. Навеску 15 г хорошо
растертой е-аминокапроновой кислоты помещают в пробирку на 50 мл. Пробирку
соединяют с маленьким дефлегматором, который через стеклянный переход
связан с U-образной трубкой, содержащей определенное количество безводного
хлорида кальция. Чтобы вода не конденсировалась в приборе до U-образной
трубки, дефлегматор и стеклянный переход заполняют алюминиевой стружкой
и асбестом. Дефлегматор снабжен трубкой для ввода азота. Медленный ток
газа обеспечивает перенос выделяющейся воды в U-образную трубку и предот-
вращает попадание кислорода в реакционный сосуд.
Аминокислоту плавят прямо в пробирке, которую помещают в масляную
или металлическую баню при 220 °C. Температуру быстро повышают до 260 °C
и поддерживают в течение 15 мин. Если в процессе поликонденсации вода все-
таки конденсируется в приборе, ее выдувают горячим воздухом, а затем рас-
плав охлаждают в токе азота. Полиамид извлекают из пробирки, хлоркальцие-
вую трубку взвешивают для определения количества выделившейся воды. Опыт
повторяют дважды, увеличив время реакции до 30—60 мин. Определите вязкость
трех образцов полиамида в конц. H2SO4 при 30 °C (С =10 г/л) в вискозиметре
Оствальда (диаметр капилляра 0,6 мм). Возрастание т\уд/С с увеличением
продолжительности реакции является мерой степени поликонденсации. Получен-
ный найлон 6 имеет температуру плавления, равную 215 °C; из его расплава
можно тянуть нити. Полиамид содержит примеси линейных и циклических олиго-
меров, которые можно экстрагировать из хорошо растертого образца метанолом
в аппарате Сокслета (12 ч). Экстракт содержит циклические и линейные олиго-
меры вплоть до пентамера, количество которых можно определить после удале-
ния метанола в вакууме. е-Капролактам удаляют промыванием остатка безвод-
ным эфиром. Остаток вновь растворяют в метаноле (1%-ный раствор) и про-
пускают раствор через катионит [14], промытый метанолом; линейные олиго-
меры задерживаются в колонке. Количество циклических олигомеров определяют
204
взвешиванием осадка, полученного после удаления растворителя из элюата. Их
можно разделить хроматографией на бумаге [15] в смеси тетрагидрофурана,
циклогексана и воды (186 : 14 : 10); значения R/ возрастают с уменьшением чис-
ла членов в 'цикле.
4.1.2.2. Полиамиды на основе диаминов и дикарбоновых кислот
Поликонденсация диаминов с дикарбоновыми кислотами может
быть осуществлена просто при плавлении смеси хорошо очищен-
ных компонентов при 180—300 °C в атмосфере азота. В этом слу-
чае значительное количество диамина может быть потеряно в про-
цессе отгонки воды (особенно в конце реакции при работе в ва-
кууме), и реагенты могут оказаться не в эквимольном соотноше-
нии. Так как эквимольное соотношение компонентов является усло-
вием получения полимеров с высокими молекулярными массами,
диамин следует добавлять в определенном избытке, который уста-
навливается экспериментально для каждого конкретного случая.
Изящной модификацией описанного метода является конденса-
ция соли карбоновой кислоты и диамина (см. опыт 4-09). Получе-
ние соли и саму поликонденсацию проводят в две стадии. Соли по-
лучают в кристаллической форме путем растворения эквимольных
количеств диамина и дикислоты в растворителе, в котором соль
не растворяется (например, в спирте). Для получения высокомо-
лекулярных продуктов исходные соли должны быть нейтральными
(эквимольное соотношение диамина и дикислоты) и очень чистыми
(например, перекристаллизованными из смеси этанола с водой).
Опыт 4-09. Получение найлона 6,6 из гексаметилендиаммонийадипата поли-
конденсацией в расплаве
Получение диаммониевой соли
Навеску 36 г (0,245 моля) чистой адипиновой кислоты (т. пл. 152 °C) рас-
творяют в 300 мл 95%-ного этанола, а 29 г (0,25 моля) гексаметилендиамина
растворяют в смеси 80 мл этанола и 30 мл воды. Оба раствора фильтруют (если
он непрозрачен).
Раствор адипиновой кислоты наливают в стакан емкостью 500 мл и в те-
чение 8 мин к нему при перемешивании приливают раствор диамина. Обра-
зование соли сопровождается разогревом раствора до 40—45 °C. При дополни-
тельном перемешивании в течение 30 мин соль кристаллизуется из раствора при
охлаждении; затем ее фильтруют на воронке Бюхнера, дважды промывают
95%-ным метанолом и сушат в вакууме. Т. пл. 183°C (в отсутствие воды);
pH 9,5%-ного раствора равен 7,62. Для очистки соль можно перекристаллизовать
из смеси метанола с водой (3: 1).
Поликонденсация диаммониевой соли
Колбу с коническим дном емкостью 50 мл, снабженную дефлегматором, воз-
душным холодильником, «пауком» и приемником, на % заполняют диаммоние-
вой солью, вакуумируют и заполняют азотом. Соль нагревают в атмосфере азо-
та на металлической бане при 220 °C в течение 1 ч и при 260—270 °C — бо-
лее 3 ч.
После охлаждения колбу разбивают. Полученный полиамид (найлон 6,6)
плавится при 265 °C. Его можно тянуть из расплава, а образовавшиеся волокна
подвергать холодной вытяжке. Определите вязкость полиамида в jn-крезоле,
конц. H2SO4 иля в 2М растворе КС1 в 90 %-ной муравьиной кислоте (см. раз-
дел 2.3.2.1).
205
4.1.2.3. Полиамиды на основе диаминов
и производных дикислот
Как и при синтезе полиэфиров, реакцию получения полиамидов
можно проводить при более низких температурах, если вместо
самих дикарбоновых кислот использовать их производные. Особен-
но эффективными являются хлорангидриды кислот, которые реаги-
руют с диаминами по реакции Шоттен—Баумана даже при ком-
натной температуре (см. ниже, раздел 2.1.5.2). Поликонденсацию
можно проводить в растворе или на границе раздела фаз.
Опыт 4-10. Получение найлона 6, 10 из гексаметилендиамина и хлорангидри-
да себациновой кислоты
А. Поликонденсация в растворе при комнатной тем-
пературе (осадительная поликонденсация)
Для проведения реакции необходимо подготовить очищенный от спирта
хлороформ, который получают, пропуская 200 мл хлороформа через колонку
с окисью алюминия.
Хлорангидрид себациновой кислоты готовят следующим образом. Навеску
60 г себациновой кислоты и 150 г тиопилхлорида кипятят на водяной бане в
течение 2 ч. Затем отгоняют избыток тионилхлорида, а хлорангидрид дикарбо-
новой кислоты перегоняют и сушат в вакууме (т. кип. 142°C при 2 мм рт. ст.).
В трехгорлой колбе емкостью 500 мл, снабженной мешалкой и обратным
холодильником, растворяют 5,3 г (50 ммолей) гексаметилендиамина и 15,3 мл
триэтиламина в 100 мл хлороформа, очищенного от спирта. Быстро (за 10 с)
приливают 10,7 мл (50 ммолей) дихлорангидрида себациновой кислоты в 40 мл
хлороформа. При этом раствор сильно перемешивают при комнатной температу-
ре, и перемешивание продолжают еще 5 мин после добавления хлорангидрида.
Поликонденсация происходит очень быстро с интенсивным выделением тепла
и осаждением полимера. После охлаждения смеси продукт фильтруют, тщатель-
но промывают хлороформом, петролейным эфиром, 1 н. раствором НО, водой
и 50%-ным ацетоном и, наконец, сушат в вакууме при 50 °C. Выход продукта
составляет около 70%, низкомолекулярные фракции могут быть отделены обра-
боткой фильтрата петролейным эфиром.
Б. Поликонденсация на границе раздела фаз
Раствор 3 мл (14 ммолей) свежеперегнанного дихлорангидрида себациновой
кислоты (синтез см. в пункте А) в 100 мл четыреххлористого углерода помеща-
ют в стакан емкостью 250 мл. Раствор 4,4 г (38 ммолей) гексаметилендиамина
в 50 мл воды осторожно наливают на раствор дихлорида (для того чтобы луч-
ше видеть границу раздела, в водную фазу добавляют несколько капель фенол-
фталеина). Пленку полиамида, которая немедленно образуется на поверхности
раздела, зажимают пинцетом и вынимают. Постепенно образующуюся узкую
нить наматывают на катушку, вращающуюся от электродвигателя. Поликонден-
сация прекращается, как только перестает наматываться нить, и сразу же после
первого поворота катушки начинается вновь, даже если прошло уже несколько
часов.
Нить полиамида тщательно промывают 50%-ным этанолом или ацетоном с
водой, а затем сушат при 30 °C в вакууме. Полученный таким методом найлон
6,10 имеет те же характеристики, что и образцы, полученные конденсацией в
расплаве или растворе. Он плавится при 228 °C. Из расплава можно формо-
вать волокна (например, с помощью стеклянной палочки), которые в дальней-
шем могут быть подвергнуты холодной вытяжке.
4.1.3. Полиуретаны
Линейные полиуретаны обычно получают при взаимодействии дио-
лов с диизоцианатами. Их можно также синтезировать, конден-
сируя дихлорпроизводные с диаминами в присутствии соединений,
связывающих свободную НС1:
206
NaOH
C1-C-O-(CH2)X-O-C-C1 + H2N-(CH2).-NH2 ----->
II II
о о
--->-----С—О—(СН2)Х—О—С—NH—(CH2)n--NH------
II II
о о
Так как эта реакция может протекать в водной среде, ее вы-
годно вести на границе раздела фаз (см. раздел 2.1.5.2), особенно
в тех случаях, когда необходимо получить полимеры с высокими
молекулярными массами при синтезе нетермостойких полиурета-
нов. Из вторичных диаминов получают N-алкилированные поли-
уретаны, стойкие к гидролизу и обладающие особыми свойствами,
обусловленными отсутствием водородных связей. Такие полимеры
нельзя получить реакцией диолов с диизоцианатами.
Опыт 4-11. Получение линейного полиуретана из этилен-бис-хлорформиата и
гексаметилендиамина поликонденсацией на границе раздела двух фаз
Получение эфира
Под тягой баллон с фосгеном соединяют с 3 последовательно соединенными
промывалками. Центральную промывалку заполняют жидким углеводородом
для проверки тока газа, а две других оставляют пустыми. Последняя промывал-
ка на выходе соединена с охладительной трубкой, погруженной в баню с сухим
льдом и метанолом. Эта трубка соединена с другой пустой промывалкой, кото-
рая, в свою очередь, связана с промывалкой, заполненной концентрированным
раствором аммиака. (При открывании крана баллона и при разборке прибора
следует надеть противогаз').
После конденсации 55—60 мл (около 80 г) фосгена в охладительной трубке
его осторожно переливают в двугорлую колбу емкостью 100 мл, охлаждаемую
смесью поваренной соли со льдом. В одно горло колбы вставляют термометр,
опущенный непосредственно в фосген, а во второе — отвод Клайзена с термо-
метром, капельной воронкой и холодильником Либиха. На холодильник надева-
ют алонж с круглодонной колбой в качестве приемника. Около 10 г безводного
этиленгликоля добавляют по каплям в фосген таким образом, чтобы темпера-
тура смеси не превышала 5 °C. После этого нагревают реакционную смесь до
комнатной температуры. Газообразный фосген, попадающий в холодильник, про-
ходит через пустую промывалку в охлаждаемую трубку, с которой соединен ряд
пустых промывалок и одна, содержащая аммиак; охлаждаемая трубка погру-
жена в баню со смесью сухого льда с метанолом (фосген, конденсирующийся в
охлаждаемой трубке, впоследствии разлагают аммиаком). В колбу вставляют
стеклянный капилляр и нагревают смесь до 40—50 °C, откачивая систему водо-
струйным насосом. Бисхлорформиат перегоняют в вакууме (т. кип. 81 °C при
0,8 мм рт. ст.).
Получение полиуретана поликонденсацией па грани-
це раздела фаз
Раствор 5,8 г (0,05 моля) чистого гексаметилендиамина 10,6 г (0,1 моля)
карбоната натрия и 1,5 г эмульгатора (например, лаурилсульфата натрия) в
150 мл воды охлаждают до 5 rfC. К этому раствору при сильном перемешивании
(лучше с помощью мешалки) приливают раствор 9,35 г (0,05 моля) этилен-бис-
хлорформиата в 125 мг бензола; охлажденный до 10 °C. Через несколько минут
полиуретан отфильтровывают, промывают водой и сушат в вакууме. Хлопьевид-
ный полимер (выход 70%) имеет т. пл. 180 °C. Определите его вязкость в
jw-крезоле.
4.1.4. Фенолоформальдегидные олигомеры
Продукты поликонденсации фенола с карбонилсодержащими сое-
динениями [16] (особенно с формальдегидом) называют обычно
207
фенольными смолами. Они представляют собой смеси легкораство-
римых и плавких олигомерных соединений, которые могут сши-
ваться (отверждаться) в соответствующих условиях. В зависимо-
сти от природы катализатора (кислый или щелочной) получают
различные продукты. Мольное соотношение фенола и формальде-
гида также влияет на свойства поликонденсата. Конденсация в
кислой среде приводит к образованию растворимых и плавких
фенольных олигомеров (новолаков), которые в основном соедине-
ны через метиленовые группы в орто- и пара-положениях феноль-
ного ядра. Средняя молекулярная масса олигомеров достигает
600—1500. Длительное нагревание таких продуктов не вызывает
дополнительной конденсации. Однако эти соединения можно под-
вергнуть сшиванию взаимодействием с подходящими олигофунк-
циональными компонентами (например, с формальдегидом). В то
же время в щелочной среде получают растворимые и плавкие ме-
тилолфенолы (резолы), содержащие одно или два бензольных
ядра; их средняя молекулярная масса 300—700. В отличие от
новолаков резолы при нагревании превращаются в нерастворимые,
неплавкие продукты (резиты).
Наиболее важными фенольными смолами являются продукты
конденсации фенола с формальдегидом. Они обладают высокой
прочностью, хорошими электрическими и механическими свойства-
ми и высокой химической стойкостью.
4.1.4.1. Поликонденсация фенола с формальдегидом
в присутствии кислотных катализаторов (новолаки)
При экзотермической реакции поликонденсации фенола с формаль-
дегидом в кислых средах (теплота реакции 23 ккал/моль) обра-
зуются растворимые фенольные смолы. В зависимости от моле-
кулярной массы (от 600 до 1500) смол температура плавления
изменяется от 100 до 140 °C. Соединение фенольных ядер может
происходить как в орто-, так и в пара-положении, поэтому воз-
можно большое разнообразие этих структур [18, 17]. Строение
новолаков можно представить схематически следующим образом:
СН2
ОН
208
Поликонденсацию проводят обычно в водном растворе, медлен-
но приливая 30%-ный раствор формальдегида к кислому раствору
фенола либо смешивая компоненты при комнатной температуре,
а затем нагревая смесь. Во избежание преждевременного сшива-
ния соотношение фенола с формальдегидом в смеси не должно
превышать 1 :0,8. После добавления формальдегида смесь нагре-
вают до исчезновения запаха формальдегида. Водный слой слива-
ют и продукт промывают горячей водой для удаления кислоты.
Отфильтрованный полимер нагревают в вакууме для удаления во-
ды и непрореагировавшего фенола. Получающиеся смолы обычно
растворимы в спиртах, низших эфирах, кетонах и в разбавленной
щелочи. При выборе кислоты необходимо принимать во внимание
возможность ее удаления (промыванием, перегонкой). Наиболее
пригодны соляная кислота, щавелевая кислота или их смесь. Ща-
велевую кислоту можно удалить и промыванием и нагреванием
продукта до 180 °C.
Так как новолаки не содержат функциональных групп, способ-
ных к эффективному сшиванию, то их можно сшивать, добавляя
соответствующий полифункциональный компонент, реагирующий с
фенолами. Такими соединениями, например, являются формальде-
гид, метилолсодержащие соединения, аминобензиловые спирты,
оксибензиламины, аминобензиламины и диоксидибензиловые эфи-
ры. Наиболее часто используемым сшивающим агентом является
гексаметилентетрамин. Сшивание происходит при смешивании ком-
понентов и нагревании смеси до 150—220 °C в течение нескольких
минут. Строение образующейся фенолоформальдегидной смолы
очень сложно. Если сшивающим агентом является гексаметилен-
тетрамин, сшивки преимущественно состоят из дибензиламинных
и третичных аминных мостиков:
14—732
209
Опыт 4-12. Поликонденсация фенола с формальдегидом, катализируемая кис-
лотой
В трехгорлую колбу с мешалкой и обратным холодильником вводят 130 г
(1,38 моля) фенола, 92 г 37%-ного водного раствора формальдегида (1,13 мо-
лей), 15 мл воды, 2 г дигидрата щавелевой кислоты. Смесь кипятят в течение
1,5 ч. После добавления 300 мл воды смесь быстро перемешивают и охлаждают.
Водный слой сливают, а остаток воды удаляют, медленно повышая температу-
ру до 150 °C и откачивая систему до 50—100 мм рт. ст. Эта температура под-
держивается постоянно (около 1 ч) до тех пор, пока образцы, извлеченные из
расплава, не затвердеют при охлаждении. При выливании еще теплой смолы из
колбы она отверждается в виде бесцветной прозрачной массы, растворимой
в спирте.
Полученный новолак используют для получения формовочной массы из но-
волака, гексаметилендиамина и опилок. Для этого 50 г хорошо измельченного
новолака, 50 г сухих опилок, 7 г гексаметилендиамина, 2 г окиси кальция (для
связывания остатка кислоты) и 1 г стеарата кальция (в качестве смазки) тща-
тельно перемешивают (лучше в шаровой или аналитической мельнице) и смесь
нагревают в форме в течение 5 мин при 160 °C и давлении 140 атм. Получив-
шийся сформованный материал не плавится и не растворяется.
4.1.4.2. Поликонденсация фенола с формальдегидом
в присутствии щелочных катализаторов
При поликонденсации фенола с избытком формальдегида в ще-
лочной среде образуются смолы (резолы) с молекулярной массой
от 300 до 700. Такие смолы обычно растворимы в воде и спирте.
Как и новолаки, резолы состоят в основном из фенольных ядер,
связанных метиленовыми группами. Однако они отличаются от но-
волаков присутствием в них гидроксиметиленовых групп, которые
определяют способность резолов к ряду химических реакций (на-
пример, этерификации и образованию эфира). Связывание фе-
нольных групп и введение метилольных остатков происходит в ор-
то- и пара-положениях, причем в щелочной среде ни одно из поло-
жений не является предпочтительным.
Схематически строение резолов можно представить следующим
образом:
Резолы получают нагреванием водного раствора фенола с из-
бытком формальдегида (в соотношениях от 1:1,1 до 1 : 1,5) в ще-
лочной среде. Кроме гидроокисей щелочных и щелочноземельных
металлов подходящими катализаторами являются аммиак, первич-
ные и вторичные амины. Амины могут включаться в состав фе-
210
нольной смолы. Температуру реакции необходимо поддерживать
ниже 70 °C, так как в противном случае уменьшается раствори-
мость смолы в воде. Продолжительность реакции составляет обыч-
но 2—5 ч. В отличие от новолаков резолы можно перевести в не-
растворимые и неплавкие продукты путем удаления воды из мети-
лольных групп при относительно мягких условиях реакции. В этом
случае образуются эфирные мостики [19], т. е. сшивание может
происходить и в отсутствие сшивающего агента. Реакции сшива-
ния с изменением строения продукта (удаление формальдегида)
можно индуцировать нагреванием продуктов конденсации до 150—
200 °C. Время, требуемое для полного сшивания, составляет от
нескольких минут до нескольких часов в зависимости от строения
рассматриваемого резола.
Опыт 4-13. Поликонденсация фенола с формальдегидом, катализируемая ще-
лочью
В трехгорлую колбу емкостью 500 мл, снабженную мешалкой и термомет-
ром, помещают 94 г фенола (1,0 моля), 125 г 37%-ного водного раствора фор-
мальдегида (1,54 моля) и 4,7 г Ва(ОН)2-8Н2О; смесь нагревают при 70 °C в те-
чение 2 ч. После этого реакционную смесь подкисляют 10%-ным раствором
серной кислоты до pH 6—7. Измерение pH проводят после выключения мешал-
ки и расслоения смеси на фазы. Обратный холодильник заменяют холодильни-
ком Либиха и отгоняют воду при 30—50 мм рт. ст.; температура может не
превышать 70 °C. При отгонке через каждые 20 мин отбирают пробы. Если
образец затвердевает с образованием нелипкой массы, реакцию заканчивают
(как только смола станет вязкой, пробы отбирают каждые 10 мин; после пре-
кращения реакции продукт спустя 20 ч должен быть довольно вязким при 70 °C,
но еще хорошо текучим). При выливании реакционной смеси из колбы она от-
верждается с образованием плавкой и растворимой массы (резола).
Если смолу продолжать нагревать, конденсация продолжается и образуются
нерастворимые, но набухающие и плавящиеся продукты, которые называют рези-
толами.
Продолжительное нагревание приводит к образованию сильносшитых нерас-
творимых и неплавких резитов. Нагревайте образец смолы в пробирке до 100 °C
до тех пор, пока не образуется очень твердый продукт, который извлекают, раз-
бив пробирку. Сравните растворимость резита и резола в спирте. При добавле-
нии таких наполнителей, как опилки, известь, красители и т. д., резол можно
приготовить в виде формовочного материала, который можно прессовать под
давлением.
4.1.5. Карбамиде- и меламиноформальдегидные
олигомеры [20]
4.1.5.1. Карбамидоформальдегидные олигомеры
В зависимости от условий реакции (pH среды, температуры, про-
должительности реакции, мольного соотношения компонентов) при
поликонденсации карбамида с формальдегидом образуются продук-
ты различного строения с разными свойствами. При поликонденса-
ции в щелочной среде образуются в основном соединения, содер-
жащие метилольные группы. Кроме того, появляются эфирные
группы, особенно при нагревании. Было доказано, что основными
14*
211
продуктами такой поликонденсации является моно- и диметилол-
мочевина:
он-
NH2—С—NH2 4- СН2О -> NH2-C~NHCH2OH и HOCH2NH-C-NHCH2OH
Присутствие три- и тетраметилолмочевины до сих пор окончатель-
но не доказано. Эти полупродукты растворимы в воде и спирте.
При дальнейшей поликонденсации и при удалении воды они пре-
вращаются в легкорастворимые высокомолекулярные соединения,
которые при сшивании переходят в нерастворимое состояние.
Строение сшитых (отвержденных) карбамидоформальдегидных
смол еще полностью не установлено.
Растворимые метилолсодержащие соединения перед отвержде-
нием (сшиванием) могут быть химически модифицированы путем
взаимодействия с монофункциональными соединениями (напри-
мер, с целью введения простых или сложноэфирных групп). Тем
самым оказывается возможным существенно изменить свойства не
только конечных сшитых продуктов, но и исходных материалов.
Например, при частичной этерификации метилольных соединений
бутанолом образуются продукты, растворимые в неполярных рас-
творителях (например, в толуоле), но сохраняющие способность
к сшиванию в отсутствие сшивающих агентов.
Обычно карбамидоформальдегидные смолы синтезируют поли-
конденсацией в водных и щелочных средах. В зависимости от ко-
нечного применения продукта используют 1,5—2-кратный избыток
формальдегида. В качестве катализаторов можно применять все
соединения основного характера при условии их достаточной рас-
творимости в воде. Наиболее широко используются щелочи. Од-
нако pH реакционной смеси не должен превышать 8—9 во избе-
жание протекания реакции Канниццаро для формальдегида. Так
как pH раствора уменьшается в процессе реакции, его необходимо
поддерживать неизменным, либо используя буферный раствор,
либо добавляя щелочь. При этих условиях продолжительность
реакции составляет 10—20 мин при 50—60 °C. После завершения
реакции необходимо оттитровать непрореагировавший формальде-
гид с гидросульфитом натрия [21] или гидрохлоридом гидроксил-
амина. Титрование необходимо проводить очень быстро и при низ-
ких температурах (10—15°С), так как иначе расщепление мети-
лольных соединений с образованием формальдегида приводит к
ошибке в анализе. Из-за этой обратимости реакции выделение
растворимых продуктов конденсации оказывается возможным лишь
при осторожном выпаривании воды в слабощелочной среде в ва-
кууме при температуре ниже 60 °C. Дальнейшую поликонденсацию
с целью получения сшитых продуктов обычно проводят в исходном
водном растворе либо нагреванием нейтрального раствора до 120—
140°C (10—60 мин), либо проводя кислотный катализ при низких
212
температурах. Каталитическое сшивание (кислотное отверждение)
проводят в присутствии свободных кислот (например, фосфорной
кислоты) или соединений, кислотные свойства которых проявля-
ются при нагревании (скрытые отвердители). Для этого применяют
натриевые соли галогенкарбоновых кислот, эфиры фосфорной кис-
лоты, хлорид аммония или гидрохлорид пиридина. Так, при до-
бавлении больших количеств фосфорной кислоты (pH ^2) сши-
вание происходит уже при комнатной температуре (клей, засты-
вающий на холоду). Все реакции сшивания карбамидоформальде-
гидных смол протекают вследствие дальнейшей поликонденсации
гидроксимстильных соединений, сопровождающейся выделением
воды. При отверждении больших формованных кусков удаление
воды может вызвать появление гетерогенностей и разложение.
Эти осложнения можно устранить введением наполнителей, погло-
щающих воду, таких, как целлюлоза или другие полиспирты (см.
опыт 4-14).
Опыт 4-14. Поликонденсация карбамида с формальдегидом
В трехгорлой колбе емкостью 500 мл, снабженной мешалкой, термометром
и обратным холодильником, нагревают 243 г 37%-ного водного раствора форм-
альдегида (3 моля) до 50 °C, а затем растворяют в нем 120 г (2 моля) карб-
амида. После добавления 10 мл конц. раствора аммиака раствор нагревают до
85 °C. Через 20 мин раствор мутнеет, и вязкость возрастает, в то время как pH
понижается до 5. Через 1 ч нагревание прекращают и отбирают небольшой обра-
зец для проверки растворимости продукта поликонденсации в воде.
К охлажденному раствору при перемешивании добавляют порошок целлю-
лозы до тех пор, пока масса еще поддается перемешиванию. Затем продукт
помещают в большой стакан и добавляют столько целлюлозного порошка, что-
бы общее количество порошка составляло 140 г. Смесь перемешивают рукой
{наденьте резиновые перчатки).
Гранулированный продукт сушат в вакууме при 50 °C в течение 24 ч. Сухое
вещество растирают в ступке и обрабатывают 1%-ными растворами хлорида
аммония и стеарата цинка (смазка). Образец сшивают (отверждают) в пресс-
форме при 160 °C под давлением 300—400 атм в течение 10 мин. Получающая-
ся тонкая прозрачная пластинка больше не растворяется и не поддается воз-
действию воды. Проверьте это, выдерживая маленький кусочек сформованного
образца в стакане с водой в течение ночи.
Если пресс-форма (пресс) недоступна или ее использование невозможно,
поступают следующим образом: две плоские стальные пластины толщиной 2 см
нагревают в печи до 240 °C, а затем извлекают из нее. Одну пластину помеща-
ют на нижнюю часть горизонтально укрепленных тисков (положите асбестовую
прокладку между тисками и пластиной) и дают остыть до такого состояния, что-
бы гранулы полимера не разлагались на стальной пластине. Далее несколько
граммов образца помещают на пластину и сверху кладут другую пластину; изо-
лировав пластину от верхней части тисков куском асбеста, тиски зажимают.
4.1.5.2. Меламиноформальдегидные олигомеры
Формальдегидные смолы с улучшенными водо- и термостойкими
свойствами получают при частичной (сополиконденсация) или пол-
ной замене карбамида меламином. Такие реакции проводят глав-
ным образом в щелочных средах, в результате образуются раство-
римые полупродукты. Они состоят в основном из три- и гексаме-
тилольных производных меламина:
213
NH-CH2-OH
зсн2о
---->
0Н“
С C-NH~CH2OH
зсн2о
6СН2О
---->-
ОН“
HOH2C-N-CH2OH
С
НОН2С\ N N /СН2ОН
)n-c c-n(
Н0Н2(7 \ / 42H2OH
N
Эти соединения наиболее стойки при pH 8—9. При последую-
щей поликонденсации (преимущественно при удалении воды из
ЛЛметилольных групп и остатка NH-групп) они превращаются в
ограниченно растворимые, а затем в нерастворимые сшитые про-
дукты. Растворимые полупродукты могут быть также модифициро-
ваны введением, например, сложно- или простых эфирных групп.
Практически синтез меламиноформальдегидных смол проводят
при тех же условиях, что и получение карбамидоформальдегидных
смол. Меламин, первоначально нерастворимый в водной реакцион-
ной смеси, полностью растворяется в процессе поликонденсации.
За ходом реакции можно легко проследить, титруя непрореагиро-
вавший формальдегид гидросульфитом натрия (см. раздел 4.1.5.1),
так как метилоламины значительно стабильнее, чем соответствую-
щие производные карбамида.
Сшивание (отверждение) полупродуктов можно проводить тем
же методом, что и отверждение карбамидоформальдегидных смол;
лучше всего это делать при pH от 3,5 до 5. Меламиноформальде-
гидные смолы, полученные при 2,8—3-кратном избытке формальде-
гида, сшиваются быстрее.
Карбамиде- и меламиноформальдегидные смолы применяют в
качестве формовочных материалов, лаков, клеев (для дерева) и
составных частей тканей (для несминаемости) и бумаги (для улуч-
шения прочности во влажном состоянии).
Опыт 4-15. Поликонденсация меламина с формальдегидом
Прибор для синтеза состоит из трехгорлой колбы емкостью 250 мл, снаб-
женной мешалкой, двугорлым отводом с обратным холодильником и термомет-
ром, и резиновой пробки с отверстием, через которое вводят стеклянную палоч-
ку. В колбу насыпают 63 г меламина (0,5 моля) и наливают 150 г 40%-ного
водного раствора формальдегида (2 моля). Смесь перемешивают и добавляют
несколько капель 20%-ного едкого натра до pH 8,5. Продолжая перемешивание,
214
смесь нагревают до 80 °C на водяной бане в течение 5—10 мин. При 70—80 °C
образуется гомогенный раствор. Во время нагревания необходимо поддерживать
pH 8,5, добавляя по каплям 20%-ный раствор едкого натра. Смесь непрерывно
перемешивают при 80 °C и постоянном pH 8,5 до тех пор, пока коэффициент
осаждения не станет равным 2:2 (см. ниже). После охлаждения раствор филь-
труют для удаления небольших количеств нерастворимого продукта.
Меламин растворяется в водном растворе формальдегида при нагревании
с образованием метилольных соединений, которые представляют собой кристал-
лические вещества, растворимые в горячей и ограниченно растворимые в холод-
ной воде (следовательно, при охлаждении раствора, полученного сразу же после
полного растворения меламина, ограниченно растворимые метилольные соедине-
ния осаждаются).
При дальнейшем нагревании получаются продукты поликонденсации, еще
растворимые в воде при всех концентрациях. Они содержат довольно большое
количество мономерных метилольных соединений, поэтому при охлаждении рас-
твор мутнеет. Водные растворы остаются прозрачными и при продолжительном
нагревании, однако на этой стадии растворы сохраняют прозрачность только
при высоком содержании полимера; в разбавленных растворах полимер выса-
живается. С увеличением продолжительности реакции сродство к воде умень-
шается таким образом, что в конце концов меламиноформальдегидная смола
выпадает в осадок.
Определение коэффициента осаждения
При добавлении небольшого количества реакционной смеси в ледяную воду
вода мутнеет, если с момента начала реакции прошло около 50—60 мин. После
этого отбирают по 2 мл образцов смеси. К охлажденным до 20 °C образцам до-
бавляют по каплям дистиллированную воду и смесь перемешивают. Опыт за-
канчивают, если при добавлении 2 мл воды соответствующий образец мутнеет.
Пропитывание бумаги -
Приготовленный раствор смолы переносят в фарфоровую чашку и погру-
жают в него 10—20 круглых фильтровальных бумажек. Фильтры выдерживают
в растворе 1—2 мин, затем их вынимают пинцетом и дают стечь избытку раство-
ра. Пропитанную бумагу укрепляют на растянутом шнуре и сушат в течение
ночи.
Производство слоистого пластика
Десять листов пропитанной фильтровальной бумаги складывают стопкой и
помещают между двумя кусками алюминиевой фольги (15X15 см). Стопку
сжимают гидравлическим прессом до давления 40—100 атм при 135 °C в тече-
ние 15 мин. Образец вынимают из-под пресса прежде, чем он успеет остыть.
Для получения прозрачных слоистых пластиков формование следует прово-
дить при давлениях, указанных выше. Все обогреваемые лабораторные гидрав-
лические прессы пригодны для этой цели. Если пресса в лаборатории нет, можно
использовать тиски (см. раздел 2.4.2.1) с парой стальных пластин (никелирован-
ных), которые нагревают до 140 °C. Проверьте стойкость отвержденного мела-
миноформальдегидного пластика по отношению к растворителям и другим хими-
ческим реагентам.
4.1.6. Полиалкиленсульфиды
При обработке соответствующих галогенпроизводных алифатиче-
ских углеводородов полисульфидами щелочных или щелочнозе-
мельных металлов образуются линейные резино- или смолоподоб-
ные полиалкиленсульфиды [22]:
nCl— R—С1 4- nNa2Sx -> [-—R—Sx—-]n 4- 2nNaCl
Наиболее широкое применение находят 1,2-дихлорэтан, бис-2-хлор-
этиловый эфир и бис-2-хлорэтилформиат. Если, кроме того, ис-
пользуют олигогалогенпроизводные (например, 2% 1,2,3-трихлор-
215
пропана), образуются разветвленные или сшитые продукты. В ка-
честве полисульфидного компонента чаще всего используют тетра-
сульфид натрия Na2S4, так как он содержит очень мало моносуль-
фида, взаимодействие которого с дигалогеном приводит к образо-
ванию побочных циклических продуктов с неприятным запахом.
Серу можно удалить из полиалкиленсульфида с помощью со-
единений, связывающих свободную серу (например, обработкой
водной дисперсии сульфидом натрия, NaOH или Na2SO3 при 30—
100°C). При этом строение макромолекулы не изменяется до тех
пор, пока по крайней мере 2 атома серы приходятся на одно мо-
номерное звено:
[—R—S4—]„ + 2nS2~ >- [—R—S2—]„ + 2/iSi-
Последующее десульфирование вызывает деструкцию полиал-
киленсульфидов с образованием низкомолекулярных продуктов.
При вулканизации полиалкиленсульфидов могут образовываться
-сшитые, эластичные полимеры, имеющие важное промышленное
значение, обусловленное их химической стойкостью по отношению
к растворителям и маслам. Более того, они значительно менее
чувствительны к кислороду и свету, чем большинство синтетиче-
ских каучуков. Технологические свойства этих материалов можно
модифицировать, прежде всего, варьируя содержание серы.
Полиалкиленсульфиды получают, приливая дигалоген к уме-
ренно концентрированному водному раствору полисульфида (избы-
ток 10—20%) при сильном перемешивании. Используемый дигало-
ген определяет температуру и продолжительность реакции: с 1,2-
дихлорэтаном реакция протекает при 50—80 °C в течение примерно
5 ч, а с бис-2-хлорэтиловым эфиром — при 100 °C в течение 20—
30 ч. Дальнейшее протекание реакции и промывание продукта ос-
ложняется тем, что нерастворимые в воде полиалкиленсульфиды
склонны к слипанию. Поэтому поликонденсацию полезно прово-
дить в присутствии диспергирующего агента (например, 2—5%
гидроокиси магния). Добавлять эмульгаторы не рекомендуется,
так как они усложняют последующую очистку. После завершения
реакции хлорид натрия и непрореагировавший полисульфид нат-
рия отделяют многократным промыванием полимера водой. До-
бавление соляной кислоты вызывает коагуляцию самого полиалки-
ленсульфида.
Опыт 4-16. Получение полиалкиленсульфида из 1,2-дихлорэтана и тетрасуль-
фида натрия
Приготовление раствора тетрасульфида натрия
Тетрасульфид натрия можно приготовить, растворяя серу в сульфиде нат-
рия или в растворе едкого натра
Na2S + 3S --> Na2S4
6NaOH 4- 10S -* Na2S2O3 + 2Na2S4 -h 3H2O
Образующийся тиосульфат натрия не мешает протеканию реакции конденсации.
Приготовление раствора из сульфида натрия и серы.
Навеску 240 г (1,0 моля) кристаллического сульфида натрия Na2S-9H2O (вы-
пускаемого в промышленности), 96 г (3,0 г-атома) порошковой серы и 50 мл
216
воды нагревают при перемешивании на водяной бане в отсутствие воздуха да
растворения серы (30—60 мин). Процесс растворения серы можно ускорить до-
бавлением нескольких миллилитров спирта или следов осушающего агента. Да-
лее смесь нагревают еще 30 мин при 90—95 °C, а затем, не давая ей остыть,
фильтруют на воронке Бюхнера для удаления следов шлама сульфида железа.
Раствор разбавляют 190 мл воды.
Приготовление раствора из едкого натра и серы.
В смеси 300 г 40%-ного раствора NaOH (3,0 моль) и 120 мл воды растворяют
при перемешивании 160 г порошковой серы (5 г-атомов) при 90—95 °C.
Получение полиалкиленсульфида. В трехгорлую колбу емко-
стью 500 мл, снабженную мощной мешалкой, термометром, обратным холодиль-
ником и капельной воронкой, наливают 290 г одного из описанных выше раство-
ров тетрасульфида натрия (0,5 моля). К этому раствору при перемешивании
одновременно добавляют раствор 3 г кристаллического хлорида магния в 10 мл
воды и 7,5 мл 2 н. едкого натра. Поддерживая температуру реакционной смеси
на уровне 50—60 °C, в течение 2 ч по каплям добавляют 40 г (0,40 моля)
1,2-дихлорэтана. После этого смесь нагревают еще 2 ч при 70—80 °C, охлаж-
дают и получившуюся дисперсию выливают в воду (1 л). Полиалкиленсульфид
отделяют и очищают от хлорида натрия и непрореагировавшего тетрасульфида
натрия многократным промыванием водой, центрифугированием и декантацией.
Дисперсию, полученную после очистки, подкисляют 5 мл конц. НС1, которая
вызывает коагуляцию и осаждение полимера в виде пористой массы. После
фильтрования на воронке Бюхнера желто-белый полимер с неприятным запахом
сушат в вакууме над Р2О5. Сухой продукт частично растворим в сероуглероде
и набухает в четыреххлористом углероде и бензоле. Он размягчается при 130 °C.
4.1.7. Полисилоксаны [23, 24]
Полисилоксаны*, или кремнийорганические полимеры содержат
силоксановую связь —Si—О—Si—. В общем виде строение линей-
ного полисилоксана можно представить следующим образом:
R R R R
---Si—О—Si—О—Si—О—Si—О— • •
1111
R R R R
где R — алкил или арил
По своим свойствам полисилоксаны занимают промежуточное
положение между чисто органическими полимерами и неорганиче-
скими силикатами. Изменяя природу заместителей, свойства поли-
силоксанов можно смещать в ту или иную сторону. В промышлен-
ности наиболее широко применяются полисилоксаны с метильными
заместителями. Для получения полисилоксанов используют различ-
ные силаны (например, R2SiC12 или RSiCls). Сначала силаны гид-
ролизуются до соответствующих силанолов, которые очень неста-
бильны и легко конденсируются с выделением воды и образовани-
ем линейных полимеров:
R R
I н2о |
nCl—Si—С1 ---> п НО—Si—ОН
—(п—1)Н2О
* Обычно в промышленности используют термин «силиконы» (силиконовые
масла, силиконовые каучуки и т. д.).
217
Линейные полисилоксаны, образованные в результате гидролиза
дихлорсиланов, имеют относительно низкие молекулярные массы,
однако при дополнительном нагревании они могут конденсировать-
ся дальше за счет взаимодействия концевых ОН-групп.
Так как Si(R)2—О-группы склонны к образованию циклических
соединений, то кроме линейных силоксанов продуктами гидролиза
дихлорсиланов являются также циклические олигосилоксаны, со-
держащие в кольце от 3 до 9 Si—О-групп. В соответствующих
условиях реакции получаются только циклические соединения (см.
раздел 4-17). Циклические силоксаны могут превращаться в вы-
сокомолекулярные линейные продукты либо при полимеризации
с раскрытием цикла, либо при катионной (в присутствии кислот
Льюиса), либо при анионной (в присутствии щелочей) полимери-
зации:
Н3С СН3
НзС\ / \ СН3 сн3 сн3 сн3
,Si О сн кон 1111
.ur/l I / --->---Si-O—Si—О—Si—О—Si—О--
пн3С о si/ 1111
\ / \сн СН3 СНз СНз СН3
Si—О 3
НзС^ ^СН3
Наиболее удобны в этом отношении тримерные и тетрамерные ди-
метилсилоксаны (см. опыт 4-17).
Полисилоксаны, полученные анионной полимеризацией, харак-
теризуются более высокой молекулярной массой, чем синтезирован-
ные катионной полимеризацией. Полимеризация с раскрытием
цикла в присутствии катионных инициаторов позволяет вводить
стабильные концевые группы (если ее проводят с соответствующи-
ми агентами передачи цепи). Таким образом, полисилоксаны с три-
метилсилильными концевыми группами получают катионной поли-
меризацией октаметилциклотетрасилоксана в присутствии гекса-
метилдисилоксана в качестве агента передачи цепи:
Н3С
Si—О
СН3
СН,
Н3С
сн3
сн3 сн3
I I н4-
+ СН3—Si— О-Si—СН3 ----->
I I
СН3 СНз
“ СН3 " СН3
I I
—Si-О---------Si—СН3
I I
_ СН3 _ mi сн3
сн3
I
CH3—Si— О—
I
СН3
218
Низкомолекулярные полисилоксаны с триметилсилильными кон-
цевыми группами можно также получить гидролитической деструк-
цией высокомолекулярных полисилоксанов с концевыми ОН-груп-
пами, катализируемой кислотой в присутствии гексаметилдиси-
локсана (равновесная реакция; см. опыт 4-18). Полисилоксаны, не
имеющие концевых ОН-групп, не способны к дальнейшей поликон-
денсации при нагревании, поэтому их молекулярные массы не из-
меняются при повышенных температурах, что очень важно для
практического применения силиконовых масел.
Низкомолекулярные полисилоксаны представляют собой масло-
или воскоподобные вещества, в то время как высокомолекулярные
продукты очень эластичны: путем сшивания их можно превратить
в силиконовый каучук. На практике силиконы сшивают перекися-
ми (см. опыт 4-17), которые, по-видимому, отрывают атомы водо-
рода от метильных групп, образуя углеводородные радикалы вдоль
макромолекулы полисилоксана. При рекомбинации углеводород-
ных радикалов различных макромолекул происходит сшивание за
счет образования С—С-связей.
Опыт 4-17. Полимеризация с раскрытием цикла олигосилоксана с образова-
нием линейного высокомолекулярного полисилоксана с гидроксильными концевы-
ми группами. Отверждение полимера
А. Получение октаметилциклотетрасилоксана
В капельную воронку, снабженную хлоркальциевой трубкой, помещают
200 г диметилдихлорсилоксана и медленно при перемешивании приливают его
к 600 мл воды. После добавления 200 мл эфира органический слой отделяют
от водного, дважды промывают дистиллированной водой и сушат над сульфатом
магния. Эфир отделяют вакуумной перегонкой. Маслянистый остаток, состоящий
преимущественно из циклических олигосилоксанов, фракционируют перегонкой.
После отгонки некоторого количества (около 0,5%) гексаметилциклотрисилокса-
на (т. кип. 134 °C, т. пл. 64 °C) отгоняется октаметилтетрасилоксан (т. кип.
175 °C, 74 °C при 20 мм рт. ст., т. пл. 17,5 °C).
Выход составляет около 40%. При температурах около 200 °C и выше от-
гоняется пентамер (т. кип. 101 °C при 20 мм рт. ст.) и гексамер (т. кип. 128 °C
при 20 мм рт. ст.).
Остаток представляет собой вязкое масло, состоящее из более высокомоле-
кулярных продуктов. Нагреванием этого масла до 400 °C можно выделить дру-
гую группу циклических олигомеров, в основном тримеры и тетрамеры. Для это-
го колбу перегонного аппарата нагревают до 400—500 °C, пропуская через систе-
му слабый ток азота. Полученный дистиллят фракционируют так, как описано
выше.
Б. Полимеризация олигосилоксана
Навеску 60 г предварительно перегнанного октаметил'циклотетрасилоксана
смешивают с 30 мг хорошо растертого едкого кали в колбе Эрленмейера емко-
стью 250 мл. Колбу нагревают на масляной бане до 140 °C. Возрастание вязко-
сти можно наблюдать при медленном перемешивании. Через 20—30 мин поло-
вину сиропообразной жидкости отбирают и обрабатывают в соответствии с пунк-
тами В и Г. Остаток реакционной смеси нагревают еще 2—3 ч для превращения
ее в пластичную, похожую на замазку массу. При охлаждении получается кау-
чукоподобный полимер; его выход составляет 90—95%.
В. Горячее отверждение каучукоподобного полимера
Приготовленный полисилоксан растворим в бензоле и толуоле. Горячим
отверждением его можно перевести в нерастворимый продукт — силиконовый
каучук. Для этого смешивают в небольшом смесителе 10 г полимера с 10 г по-
рошка кварца (или 7,5 г растолченного кизельгура) и 0,6 г перекиси 2,4-дихлор-
219
бензоила (50%-ный раствор в силиконовом масле). Без смесителя перемешива-
ние полимера с другими ингредиентами оказывается неудовлетворительным и
длительным процессом. Смесь нагревают в печи при НО °C в течение 10 мин.
Проверьте растворимость исходной смеси и конечного продукта в бензоле.
Г. Холодное отверждение сиропообразного полимера
при комнатной температуре
Навеску 10 г сиропообразного полисилоксана, полученного по методике, опи-
санной в пункте Б, растирают в ступке с 5 г порошкообразного кварца или
кизельгура; вязкий сироп переливают в стакан и смешивают с 0,3 г тетраэтилси-
ликата (сшивающий агент) [25] и 0,3 г дибутилоловодилаурата (катализатор).
Масса отверждается в течение 1—2 ч с образованием эластичного силиконового
каучука. Такой каучук сохраняет свою эластичность в необычайно широком тем-
пературном интервале (от —90 до +300°C); он обладает высокой стойкостью
по отношению к атмосферным воздействиям.
Опыт 4-18. Равновесная деструкция силиконового каучука с образованием
продукта с триметилсилильными концевыми группами (силиконового масла)
Навеску 10 г каучукоподобного полимера, полученного в опыте 4-17
(пункт Б), растворяют в 20 г толуола; к вязкому раствору прибавляют 0,5 г
96 %-ной серной кислоты и 0,2 г гексаметилдисилоксана [26]*. Смесь перемеши-
вают до йолного растворения. После добавления 0,3 мл воды смесь перемеши-
вают еще 2 ч. Раствор промывают водой в делительной воронке до нейтральной
реакции промывных вод. После отгонки толуола остается прозрачное жидкое
масло, выход которого составляет 70%.
4.1.8. Циклополиконденсация
При циклополиконденсации процесс протекает в две стадии. При
этом получаются макромолекулы, содержащие карбо- или гетеро-
циклические основные цепи [27—29]. Примером циклополикон-
денсации является получение лестничных полимеров. На первой
стадии полимер получается из тетрафункциональных мономеров по
соответствующей полиреакции, в которой принимают участие две
из четырех функциональных групп. На второй стадии в результат
конденсации двух оставшихся функциональных групп происходит
циклизация, приводящая к образованию лестничных полимеров.
На первой стадии можно использовать любую полиреакцию.
4.1.8.1. Комбинация полимеризации с поликонденсацией
Примером такого процесса является свободнорадикальная поли-
меризация метилвинилового кетона с последующей циклической
конденсацией полиметилвинилового кетона, которая приводит к об-
разованию лестничного полимера с карбоциклической основной
цепью. Первую реакцию — свободнорадикальную полимеризацию
проводят по стандартной методике при температурах ниже 80 °C;
вторую реакцию — циклическую поликонденсацию — при 300°C:
* Гексаметилдисилоксан получают гидролизом 5 г диметилхлорсилана в ки-
пящей воде. Через несколько минут органическую фазу отделяют и перегоняют
над Р2О5 (т. кип. 100 °C; выход 80%).
220
Первая стадия (полимеризация)
~80°С
СН2=СН-С-СН3----► • • •
300 сс
---->
—лН2О
со со со со
сн3сн3<!н3сн3
Вторая стадия (циклическая поликонденсация)
Получающийся полимер окрашен в красный цвет, не растворяется
в растворителях, но еще плавится.
4.1.8.2. Комбинация ступенчатой полимеризации
с поликонденсацией
Синтез полиамидов является типичным примером ступенчатой по-
лимеризации. Первая стадия реакции заключается во взаимодей-
ствии бисангидрида с ароматическим диамином, которое протекает
очень быстро при 20—40 °C в сильнополярных растворителях (ди-
метилформамиде, диметилацетамиде, N-метилпирролидоне). Обра-
зующийся сильновязкий раствор полимерной аминокислоты нали-
вают тонким слоем и нагревают до 150—250 °C; растворитель уле-
тучивается, а при выделении воды происходит циклизация:
Первая стадия (ступенчатая полимеризация)
О о
221
Полиамиды, полученные из пиромеллитового диангидрида и 4,4'-
диаминодифенилового эфира, отличаются хорошей термостойкостью
(температура размягчения лежит выше 800°C).
4.1.8.3. Комбинация поликонденсации с циклополиконденсацией
Типичными представителями полимеров, полученными этим мето-
дом, являются полибензимидазолы, синтезированные Марвелом и
сотр. На первой стадии ароматический тетрамин конденсируют с
дифениловым эфиром ароматической дикарбоновой кислоты при
220—260 °C с образованием полимерного аминоамида и выделени-
ем фенола. На второй стадии идет циклизация при 400 °C и выде-
ляется вода:
Первая стадия (поликонденсация)
Вторая стадия (циклополиконденсация)
В атмосфере азота полиимидазолы стабильны до 600 °C и теря-
ют только около 30% массы за 1 ч при 900 °C.
Хотя лестничные полимеры не растворяются и практически не
плавятся, двухстадийные процессы, описанные выше, позволяют
222
формовать детали из этих полимеров на месте применения. Раство-
римые и плавкие линейные полимеры получают в требуемом виде
(например, пленки или волокна), синтезируя на первой стадии
реакции, а затем на второй стадии их переводят в лестничные по-
лимеры циклополиконденсацией.
Лестничные полимеры относятся к термостойким полимерам,
которые обычно разлагаются только при температурах, заметно
превышающих 400 °C; кроме того, они сохраняют механические
свойства в широком температурном интервале.
4.1.9. Дегидрирование ароматических соединений
Особым типом реакции поликонденсации является дегидрирование
ароматических соединений, при котором соединение низкомолеку-
лярных молекул происходит в результате межмолекулярного де-
гидрирования. В отличие от стандартных реакций поликонденсации
дегидрогенизационная поликонденсация возможна лишь в присут-
ствии катализатора. Катализатор образует комплекс с мономером,
и только этот комплекс способен реагировать в требуемом направ-
лении.
Примерами дегидрогенизационной поликонденсации являются
синтез n-полифенилена из бензола в присутствии кислот Льюиса
(катализаторы [30]):
ОАЮз/Ог
--------
—н2
и получение полифениленовых эфиров из замещенных фенолов
Мономер, предназначенный для получения полифениленовых
эфиров, должен отвечать следующим требованиям. При получении
в основном линейных продуктов заместители в фенольном ядре
должны располагаться в положениях 2 и 6. Атомы галогенов не
замедляют процесса, однако в ходе полиреакции они могут от-
щепляться и вызывать разветвление. Фенол должен легко окис-
ляться; заместители, понижающие его окислительный потенциал,
замедляют или ингибируют реакцию дегидрогенизации.
Таким образом, из 2,6-дихлорфенола можно получить только
низкомолекулярные полифениленовые эфиры, а 2,6-динитрофенол
223
вовсе не способен к реакции. Заместители в положениях 2 и 6 не
должны быть больших размеров, так как в противном случае бу-
дет происходить присоединение по типу «голова к голове», приво-
дящее к образованию дифенилхинона:
В случае фенолов, содержащих относительно небольшие заме-
стители, хинонов образуется очень мало, однако их выход заметно
увеличивается с повышением температуры. Желтоватый оттенок
поли-2,4-диметилфениленового эфира, по-видимому, также обус-
ловлен присутствием хинонов.
Подходящими катализаторами являются соли меди (например,
хлорид, бромид и сульфат) в сочетании с аминами; однако высо-
комолекулярные полифениленовые эфиры получают только с неко-
торыми аминами, например с пиридином. Более того, если соотно-
шение амина и соли меди в смеси недостаточно высоко, увеличива-
ется доля образующегося дифенилхинона, а молекулярная масса
полимерного продукта соответственно уменьшается.
Опыт 4-19. Получение полидиметилфениленового эфира
Поли-2,6-диметил-п-фениленовый эфир можно получить при комнатной тем-
пературе дегидрогенизацией 2,6-диметилфенола кислородом в присутствии хло-
рида меди и пиридина в качестве катализатора. Реакция протекает по ступенча-
тому механизму и включает образование фенолятного комплекса, который впо-
следствии дегидрогенизируется [32, 33]:
Опыт проводят в трехгорлой колбе емкостью 500 мл, снабженной мешалкой,
капилляром для газа и термометром. В колбу помещают 0,4 г хлорида меди
в смеси со 100 мл хлороформа и 20 мл пиридина. Кислород пропускают через
раствор при непрерывном перемешивании. Через 10—20 мин образуется прозрач-
ный темно-зеленый раствор. К этому раствору добавляют 10 г (0,082 моля)
чистого 2,6-диметилфенола и непрерывно перемешивают, продолжая пропускать
кислород через раствор. В процессе добавления диметилфенола температура
раствора медленно повышается до 40 °C, и цвет смеси изменяется до желто-ко-
ричневого. Спустя 30 мин вязкий раствор наливают в 1 л метанола, содержаще-
224
го 10 мл конц. НС1 для разрушения медного комплекса. Полимер промывают
метанолом и переосаждают из хлороформа (100 мл) метанолом (1 л +10 мл
конц. НС1). Выход количественен. При введении больших количеств реагентов
температуру можно поддерживать постоянной, используя внешнее охлаждение.
Для получения высокомолекулярного продукта реакцию следует проводить при
температуре около 30 °C. Молекулярная масса полимера возрастает с увеличе-
нием концентрации диметилфенола.
Температура размягчения полидиметилфенилового эфира лежит около 250 °C,
он растворим в хлорированных углеводородах (например, хлороформе, четырех-
хлористом углероде и тетрахлорэтане), а также в нитробензоле и толуоле. Опре-
делите характеристическую вязкость полимера в хлороформе при 25 °C.
Для приготовления пленки спрессуйте 2 г полимера в течение 5 мин между
двумя металлическими пластинками, нагретыми до 320 °C (см. раздел 2.4.2.1).
После охлаждения металлических пластин пленку можно вынуть.
4.2. СТУПЕНЧАТАЯ ПОЛИМЕРИЗАЦИЯ
В противоположность поликонденсации ступенчатая полимериза-
ция протекает без выделения побочного продукта; связывание мо-
номерных звеньев сопровождается в этом случае передачей атома
водорода (см. уравнение реакции образования полиуретана из дио-
ла и диизоцианата в разделе 4.2.1). Как и поликонденсация, сту-
пенчатая полимеризация представляет собой ступенчатый процесс,
состоящий из отдельных независимых стадий. Поэтому средняя
молекулярная масса образующегося полимера непрерывно возрас-
тает в процессе реакции. Олигомерные и полимерные продукты, об-
разующиеся на каждой стадии, содержат те же функциональные
группы, что и исходный мономер. Эти продукты можно выделить,
причем их реакционная способность не уменьшается (в отличие от
продуктов полимеризации). Ступенчатая полимеризация подчиня-
ется тем же кинетическим закономерностям, что и равновесная по-
ликонденсация [см. уравнения (4.1) — (4.4) в разделе 4.1]. Аппа-
ратурное оформление в основном то же самое.
4.2.1. Полиуретаны [34—37]
Полиуретаны представляют собой полимеры, макромолекулы кото-
рых содержат уретановые группы. Их получают преимущественно
реакцией би- или полифункциональных оксисоединений с би- или
полифункциональными изоцианатами*:
nHO-(CH2)x-OH + nO=C=N—(СН2)^—N=C=O
НО~(СН2)х--Ю—|’--C--N“(CH2)z/r-N--C—(^(CHaJj^-O—"1---C~NH-(CH2)^-№C-0
ОН НО
п-1 О
Эта реакция протекает легко и с количественным выходом. Она
может сопровождаться побочными реакциями [38], в результате
которых образуются также амидные, карбамидные, биуретановые,
аллофонатные и изоцианатные группы. Иногда такие побочные
реакции оказываются желательными (см. раздел 4.2.1.2).
* См. опыт 4-11 — получение полиуретанов по реакции поликонденсации.
15—732 225
Линейные полиуретаны, полученные из короткоцепных диолов
и диизоцианатов, представляют собой высокоплавкие кристалличе-
ские термопласты, по свойствам напоминающие полиамиды, что
обусловлено сходным строением их основных цепей. Однако обычно
полиуретаны плавятся при более низких температурах, а их раство-
римость оказывается выше, чем полиамидов (например, в хлори-
рованных углеводородах). Термическая стабильность полиурета-
нов ниже: в зависимости от структуры полимера уже при 150—
200 °C начинается заметная диссоциация уретановых групп до ис-
ходных функциональных групп; расщепление аллофонатных групп
начинается даже при 100 °C. Полиуретаны используются для про-
изводства волокон. Сшитые полиуретаны применяются в качестве
лаков, клеев, покрытий (для тканей и бумаги), эластомеров и пе-
нопластов.
Для получения полиуретанов очень важна реакционная способ-
ность диизоцианатов [39]. Ароматические диизоцианаты более ре-
акционноспособны, чем алифатические; диизоцианатные группы
при первичном атоме углерода реагируют быстрее, чем группы
при вторичном или третичном атоме. Наиболее распространенны-
ми в промышленности и легкодоступными диизоцианатами явля-
ются гексаметилен-1,6-диизоцианат, дифенилметан-4,4'-диизоциа-
нат, нафтилен-1,5-диизоцианат и смесь толуол-2,4-диизоцианата и
толуол-2,6-диизоцианата (80 :20).
Как уже упоминалось, уретаны разлагаются при нагревании с
образованием изоцианатов и оксисоединений, причем температура
разложения зависит от состава уретана. Эту особенность уретанов
используют в том случае, когда полимерные оксисоединения реаги-
руют с «защищенными» диизоцианатами, такими, как диуретаны
ароматических диизоцианатов и фенолов, которые разлагаются с
выделением исходных компонентов при низких температурах:
150—180 °C. Такие диуретаны можно смешивать с полимерными
оксисоединениями при комнатной температуре (они хранятся без
изменения). При нагревании диизоцианат выделяется и реагирует
с гидроксильными группами с образованием уретановых групп
большей стабильности, тогда как фенол отгоняется.
Взаимодействие изоцианатов с оксисоединениями ингибируется
кислотами (например, НС1 или м-толуолсульфокислотой), а уско-
ряется основными соединениями (например, четвертичными амина-
ми, пиридином, 1\Г,М-диметилбензиламином, и прежде всего N,N-
эндоэтиленпиперазином), солями некоторых металлов и металло-
органическими соединениями (например, нитрат висмута, ацетил-
ацетонат цинка, дибутилоловодилаурат). Эти катализаторы часто
эффективны в количествах, меньших 1%.
4.2.1.1. Получение линейных полиуретанов
Реакция между диоксисоединениями и диизоцианатами протекает
при смешении и слабом нагревании компонентов. Линейные поли-
уретаны с молекулярными массами до 15 000 получают именно та-
226
ким методом при определенных условиях (см. раздел 4.2.1). Как
и в случае полиамидов и полиэфиров, температуры размягчения
алифатических полиуретанов зависят от числа атомов углерода
между функциональными группами. Полиуретан, полученный из
бутандиол а-1,4 и гексаметилен- 1,6-диизоцианата, имеет промыш-
ленное значение, обусловленное его высокими показателями
свойств, например высокой температурой плавления (около 184 °C)
и большей, чем у найлона 6,6, стойкостью к гидролизу. Реакцию
можно проводить как в расплаве, так и в растворе.
При проведении реакции в расплаве к тщательно очищенному
и высушенному гликолю по каплям при сильном перемешивании
добавляют диизоцианат. Для поддержания постоянной температу-
ры следует тщательно контролировать количество добавляемого
диизоцианата и, если необходимо, охлаждать систему снаружи
(теплота реакции 33,6 ккал/моль уретановых групп). Так как реак-
ция в расплаве проводится при относительно высоких температу-
рах и, более того, в результате разложения образующегося поли-
уретана под действием диизоцианата могут легко протекать по-
бочные процессы (см. раздел 4.2.1), то линейные высокомолеку-
лярные полиуретаны лучше получать в растворе. Наиболее при-
годны для этой цели такие инертные растворители, как толуол,
ксилол, хлорбензол и о-дихлорбензол. Обычно реакцию проводят,
медленно добавляя диизоцианат к раствору гликоля, при опреде-
ленной температуре (точку кипения растворителя выгодно выби-
рать в качестве температуры реакции). Образующийся полимер
часто выпадает в осадок из реакционной смеси, т. е. в этом случае
он менее склонен к побочным реакциям, чем в расплаве.
Линейные полиуретаны можно также получать поликонденса-
цией диаминов с бисхлорпроизводными (см. раздел 4.1.3).
Опыт 4-20. Получение линейного полиуретана из бутан диол а-1,4 и гексаме-
тилен-1,6-диизоцианата в расплаве
В сухую трехгорлую колбу емкостью 250 мл, снабженную стеклянной или
металлической мешалкой, капельной воронкой с трубкой для выравнивания дав-
ления, хлоркальциевой трубкой и капилляром для продувки азота, помещают
22,5 г (0,25 моля) чистого бутандиола (т. пл. 19,7 °C, т. кип. 120 °C при
10 мм рт. ст.). Систему вакуумируют и заполняют азотом. В токе азота содер-
жимое колбы нагревают до 55 °C и при сильном перемешивании вводят по кап-
лям 42 г (0,25 моля) хорошо очищенного гексаметилендиизоцианата (т. кип.
122—125 °C при 10 мм рт. ст.) в течение 60 мин. При добавлении диизоцианата
лучше руководствоваться следующей системой (реакционную смесь нагревают до
нужных температур на масляной бане):
Объем добавляемого диизоцианата, мл Продолжительность добавления, мин т, °с
5 12 95
10 24 125
20 35 145
25 45 165
30 50 190
42 55 200
15*
227
После введения всего гексаметилендиизоцианата температура реакционной
смеси немного понижается. Реакцию ведут еще в течение 15 мин при 195 °C,
а затем расплав полиуретана переливают в стакан или в фарфоровую чашку,
где он отверждается. Полиуретан, полученный на основе бутандиола и гексаме-
тилендиизоцианата, плавится при 181—183 °C и растворяется в ле-крезоле и
формамиде. Сравните характеристическую вязкость и цвет полученного полимера
с полимером, синтезированным методом осадительной ступенчатой полимериза-
ции.
Опыт 4-21. Получение линейного полиуретана из бутандиол а-1,4 и гексаме-
тилен-1,6-диизоцианата в растворе (осадительная ступенчатая полимеризация)
В сухую трехгорлую колбу емкостью 500 мл, снабженную мешалкой, термо-
метром, обратным холодильником с осушителем и капилляром для пропускания
азота, помещают 225 г (0,25 моля) чистого бутандиола, 42 г (0,25 моля) чи-
стого гексаметилендиизоцианата и 125 мл безводного хлорбензола.
Колбу вакуумируют и насыщают азотом, затем осторожно нагревают на мас-
ляной бане при пропускании слабого тока азота. При температуре около 95 °C
реакционная смесь, вначале мутная, становится прозрачной. Начиная с этого
момента, температура повышается достаточно быстро и может превысить тем-
пературу масляной бани. Через 15 мин после начала кипения (132 °C) раствор
постепенно становится опять мутным (о чем свидетельствует голубой оттенок
поверхности реакционной смеси и стенок сосуда), пока, наконец, высокомолеку-
лярный полиуретан не осядет в виде порошка. Для полноты реакции нагревание
продолжают еще 15 мин. После охлаждения порошок полиуретана фильтруют
на воронке Бюхнера (если раствор фильтруется при 100 °C, около 1—3% низко-
молекулярного полимера остается в растворе и может быть выделено при осаж-
дении метанолом). Полимер отделяется от адсорбированного хлорбензола пере-
гонкой с водяным паром.
При этом получается 62 г (96%) белого порошка с т. пл. 181—183 °C.
Сравните вязкость и цвет продукта с полиуретаном, полученным в распла-
ве (опыт 4-20).
4.2.1.2. Получение разветвленных и сшитых полиуретанов
Наиболее распространенными методами синтеза разветвленных и
(или) сшитых полиуретанов являются следующие.
1. Взаимодействие диизоцианатов с соединениями, содержащи-
ми более двух гидроксильных групп в молекуле.
Степень сшивания в этом случае зависит в основном от строе-
ния и природы полиоксисоединений. Этот способ обычно применя-
ется при приготовлении лаков (реакция диизоцианатов при низкой
температуре в безводных растворителях, таких, как бутилацетат)
или формовочных деталей (обычно при применении «защищенных»
диизоцианатов при повышенных температурах).
2. Взаимодействие линейных полиуретанов, содержащих либо
гидроксильную, либо изоцианатную концевые группы, с соответ-
ствующими бифункциональными соединениями.
Этот метод впервые был применен при синтезе полиуретановых
эластомеров [40]. Он состоит из двух стадий. На первой стадии
тщательно очищенный, относительно низкомолекулярный простой
или сложный алифатический полиэфир, содержащий концевые гидр-
оксильные группы, реагирует с избытком диизоцианата. При этом
около двух-трех линейных молекул диола сшиваются друг с дру-
гом, начиная «рост цепи» с образованием линейных полимерных
цепей, которые содержат некоторое количество уретановых групп
228
и концевые изоцианатные группы. Удобными исходными соедине-
ниями являются, например, полиэфиры из этиленгликоля и ади-
пиновой кислоты или политетрагидрофуран с молекулярной массой
около 2000. Концевые гидроксильные группы этих соединений ре-
агируют затем с реакционноспособными диизоцианатами, такими,
как нафтилен-1,5-диизоцианат, фенилен-1,4-диизоцианат или ди-
фенил метан-4-4'-диизоцианат.
На второй стадии полученные линейные полиэфируретаны мо-
гут сшиваться водой, гликолем или диамином. Если в качестве
сшивающего агента использовать воду, то две полимерные цепи
с изоцианатными концевыми группами связываются вместе, обра-
зуя линейные карбамидные мостики (с выделением двуокиси уг-
лерода):
OCN---------NCO OCN-------------NCO OCN-------------NCO
н2о н2о
| —2СОо
НН НН
OCN---------N-C-N-------- • --N-C-N------ —NCO
U II
О о
По-видимому, сшивание происходит за счет взаимодействия
водородных атомов карбамидных мостиков с концевыми изоциа-
натными группами исходного полимера с образованием биуретано-
вых групп:
О ОН
II II I
---N—С—N---- ----N—С—N------
A i i-0
NCO NH
I I
Развитие цепи и сшивание с гликолями или диаминами проте-
кает по такой же схеме (но без выделения двуокиси углерода).
При сшивании гликолем рост цепи происходит вначале за счет
образования уретановых мостиков, которые в реакции сшивания
взаимодействуют с концевыми изоцианатными группами с образо-
229
ванием аллофонатных групп. Использование диамина в качестве
сшивающего агента приводит к росту полимерных цепей через
карбамидные мостики, а последующее сшивание протекает точ-
но так же, как это было показано для реакции сшивания с по-
мощью воды.
Гликоли и диамины играют важную роль в качестве сшиваю-
щих агентов в производстве полиуретановых эластомеров. Выде-
ляющаяся двуокись углерода используется как вспениватель.
Свойства полиуретанов, полученных этим методом, зависят как
от природы исходных соединений, так и от условий реакции.
Эластичные пенопласты получаются при небольшой степени
сшивания. В этом случае удобными диоловыми компонентами яв-
ляются линейные простые или сложные полиэфиры с относительно
высокой молекулярной массой и низким гидроксильным числом
(40—60).
Жесткие пенопласты получаются при увеличенной степени сши-
вания (за счет использования короткоцепных или разветвленных
полифункциональных спиртов, таких, как продукты присоединения
окиси пропилена к полифункциональным спиртам с п^роксиль-
ными числами от 300 до 600). Удобным диизоцианатн^.м компо-
нентом является смесь 2,4- и 2,6-изомеров толуолдиизоцианата
(80:20), которая широко используется в технике. Если пенообра-
зование должно происходить при комнатной температуре и осо-
бенно если используют соединения со вторичными ОН-группами
(полипропиленгликоли), то обычно применяют катализатор. На
однородность и размер закрытых и открытых пор пенопластов мо-
жет влиять введение таких добавок, как эмульгаторы и стабилиза-
торы. Эмульгаторы (например, натриевые, кальциевые и цинковые
соли длинноцепочечных жирных кислот) обеспечивают гомогенное
вспенивание за счет равномерного распределения воды внутри ре-
акционной смеси. Стабилизаторы (силиконовые масла) предотвра-
щают разрушение ячеистой структуры на начальной стадии реак-
ции.
В лабораторных опытах вполне достаточно тщательно переме-
шивать отдельные компоненты. Для получения гомогенных пено-
пластов с оптимальными свойствами реагенты и добавки необхо-
димо тщательно взвешивать. На практике смешивание проводят
с помощью специального дозатора.
Опыт 4-22. Получение эластичных пенополиуретанов
В стакане емкостью 600 мл деревянной палочкой в течение 1 мин тщатель-
но перемешивают 100 г полиэфира с гидроксильным числом 60 (см. опыт 4-01)
и 35 г смеси 2,4- и 2,6-изомеров толуолдиизоцианата (65:35). После этого при
сильном перемешивании добавляют смесь следующих веществ: 1 г М,Ь1-диметил-
бензиламина, 2 г 50%-ного водного раствора неионного эмульгатора, 1 г
50%-ного водного раствора лаурилсульфата натрия, 0,25 г полидиметилсилокса-
на и 1 г воды. После перемешивания в течение 20—30 с уже вспенившуюся
смесь быстро переливают в сосуд объемом не менее 2 л, содержащий бумажную
прокладку. Через 1 мин образование пены практически заканчивается. Через
20—30 мин (в зависимости от температуры) поверхность полиуретановой пены
230
перестает быть липкой. Эластичную пену (полный объем около 1500 см3) можно
отделить от бумажной прокладки и подвергнуть механической обработке.
Опыт 4-23. Получение жестких пенополиуретанов
В стакане емкостью 600 мл деревянной палочкой перемешивают 110 г силь-
норазветвленного полиэфира с гидроксильным числом около 350 (см. опыт 4-016)
и 85 г смеси 2,4- и 2,6-изомеров толуолдиизоцаната (65:35). Затем добавляют
смесь 3 г неионного поверхностно-активного вещества и 2 г лаурилсульфата
натрия в воде. Смесь перемешивают до появления пены. Сразу после этого ее
переливают в бумажный пакет с плоским дном (минимальный объем 1 л). Чёрез
1 ч из бумажного пакета можно извлечь жесткую хрупкую пену (полный объем
пены около 1 л).
4.2.2. Эпоксидные смолы [41]
Под эпоксидными смолами обычно понимают продукты реакции
полифункциональных гидроксилсодержащих соединений с эпихлор-
гидрином в щелочной среде. В простейшем случае 2 моля эпихлор-
гидрина реагируют с 1 молем бисфенола А, например:
+ Н2С----СН-СН2С1 ---->
За счет взаимодействия диэпоксида (I) с бисфенолом образу-
ются продукты с большими молекулярными массами:
При этом могут протекать побочные реакции. Так, молекула
бисфенола может связываться только с одной эпоксигруппой либо
некоторые эпоксигруппы могут гидролизоваться таким образом,
что число гидроксильных групп не будет соответствовать форму-
ле II.
231
Первая и вторая реакции могут протекать либо параллельно,
либо последовательно. Если с самого начала реакцию проводить
в щелочной среде (например, добавляя требуемое количество эпи-
хлоргидрина к смеси гидроксилсодержащего компонента и экви-
мольное количество водной щелочи при 50—100°C), первая и вто-
рая реакции идут параллельно (опыт 4-24). Если гидроксилсодер-
жащее соединение реагирует с эпихлоргидрином в безводной среде
в присутствии кислого катализатора, то образуется только хлор-
гидрин, который затем на второй стадии при добавлении экви-
мольного количества щелочи переходит в эпоксисоединения
(опыт 4-25).
Условия реакции заметно влияют на строение и молекулярную
массу образующейся эпоксидной смолы. Большой избыток эпихлор-
гидрина (около 5 молей на 1 гидроксильную группу фенола) вы-
зывает предпочтительное образование концевых эпоксигрупп; одна-
ко молекулярная масса, а с ней и температура размягчения пол. w
мера уменьшаются с увеличением избытка эпихлоргидрина. Кроме
того, важны соотношение реагентов и температура реакции: высо-
кие температуры способствуют вторичным процессам, таким, как
гидролитическое расщепление эпоксигрупп, что приводит к появле-
нию дополнительного количества гидроксильных групп.
Наиболее важные в промышленном отношении эпоксидные смо-
лы получают из 2,2-бис (n-оксифенил) пропана (бисфенол А) и эпи-
хлоргидрина. Их молекулярные массы колеблются от 450 до 4000
(что соответствует изменению п в формуле II от 1 до 12), а тем-
пературы размягчения лежат между 30 и 155 °C. Такие эпоксид-
ные смолы еще растворимы, однако их можно перевести в нерас-
творимое и неплавкое состояние путем последующего сшивания
(отверждения).
Эту реакцию отверждения проводят обычно с би- или олиго-
функциональными соединениями, способными к присоединению
к эпоксидной группе*. Более того, в присутствии каталитических
количеств третичного амина, кислотных соединений, таких, как
серная кислота или катализаторов Фриделя—Крафтса (в основном
в форме их аддуктов к эфирам или спиртам), может происходить
самосшивание смол. Эта реакция идет даже при низких темпера-
турах, но протекает неравномерно. Сшивание эпоксидных смол
представляет собой экзотермический процесс с теплотой реакции
22—26 ккал/моль эпоксигрупп. В большинстве случаев сшиваю-
щий агент берется в эквимольных количествах (по отношению к
аналитически определяемому количеству эпоксидных групп). От-
верждение обычно проводят в блоке, а иногда и в растворе (ла-
ки). Для проведения реакций сшивания в растворе иногда ис-
пользуют реакционноспособные разбавители, т. е. вещества, обла-
дающие меньшей вязкостью, чем эпоксидные смолы; такими ве-
* Часто гидроксильные группы также участвуют в реакциях сшивание
эпоксидных смол.
232
ществами являются, например, маловязкие моно- и диэпоксиды
или алилглициновый эфир.
Большинство реакций сшивания начинается только при повы-
шенных температурах, поэтому смеси эпоксидной смолы и сши-
вающего агента можно хранить при комнатной температуре. На
практике в качестве сшивающих агентов чаще всего использу-
ются многоосновные карбоновые кислоты, их ангидриды и амины.
Если карбоновые кислоты реагируют достаточно быстро только
при температурах около 180 °C, то ангидриды кислот (например,
ангидрид фталевой кислоты) эффективно сшивают уже при 100 °C,
особенно в присутствии каталитических количеств третичного ами-
йа. Оптимальное сшивание происходит при соотношении, пример-
но равном V2 моля ангидрида на моль эпоксидных групп.
Реакция сшивания с аминами происходит либо каталитически
с третичными аминами (например, с 1Ч,М-диметилбензиламином),
либо при повышенных температурах при взаимодействии экви-
мольных количеств эпоксидов с первичными или вторичными оли-
гоаминами. Такие реакции используются очень широко. В качест-
ве катализаторов применяются вещества, способные к образова-
нию водородных связей (вода, спирты, фенолы, карбоновые кис-
лоты и т. д.). Наиболее предпочтительное соотношение амина и
эпоксида устанавливается эмпирически для каждого конкретного
случая.
В сшитом состоянии эпоксидные смолы стойки к действию хи-
мических реагентов и растворителей и обладают хорошими элект-
рическими свойствами. Благодаря этому они используются в ка-
честве лаков и покрытий. Эпоксидные смолы являются также
прекрасными клеями для большинства пластмасс, дерева и ме-
таллов («двухкомпонентные адгезивы»).
Опыт 4-24. Получение эпоксидных смол из бисфенола А и эпихлоргидрина
в одну стадию
А. Приготовление эпоксидной смолы с молекулярной
массой 900
Трехгорлую колбу емкостью 250 мл, снабженную мощной мешалкой (эф-
фективное перемешивание необходимо, так как система становится гетероген-
ной в ходе реакции), термометром и обратным холодильником, помещают под
тягу*. В колбе смешивается 45,6 г (0,2 моля) чистого бисфенола А (перекристал-
лизованного из разбавленной уксусной кислоты) с раствором 15 г NaOH
{0,375 моля) в 150 мл воды. Реакционную смесь нагревают до 50 °C на
масляной бане при сильном перемешивании в течение 10 мин. Затем 29,0 г
(0,314 моля) свежеперегнанного эпихлоргидрина приливают к исходной смеси
(мольное соотношение эпихлоргидрина и бисфенола А составляет 1,57), темпера-
туру повышают до 95 °C в течение 20 мин и поддерживают ее постоянной еще
40 мин. Температура не должна превышать 95 °C, так как при более высоких
температурах возможность протекания побочных реакций увеличивается. После
* Порошок бисфенола А и эпихлоргидрин вызывают воспаление слизистой
оболочки глаз и дыхательной системы. Кроме того, адсорбируясь на коже,
эпихлоргидрин вызывает отравление. Поэтому необходимо избегать прямого
контакта с этим веществом; при попадании на кожу эпихлоргидрин следует
смыть большим количеством воды. (Необходимо надеть защитные очки и пер-
чатки.)
233
этого холодильник убирают, а мешалку выключают, чтобы смола могла осесть.
Верхний прозрачный водный слой осторожно сливают и смолу тщательно переме-
шивают с дистиллированной водой при 90 °C при температуре бани 80—95 °C.
После осаждения водный слой опять сливают.
Операцию промывки проделывают несколько раз пока на 100 мл промывной
воды не будет расходоваться при титровании менее 0,15 мл 0,1 н. НС1 (инди-
кат'ча — метиловый красный). Для удаления окклюдированной воды промытую
су Ду нагревают при умеренном перемешивании при 150 °C в течение 30 мин (в
процессе такой обработки она становится прозрачной). Затем смолу помещают
в фарфоровую чашку, где она затвердевает.
Твердая эпоксидная смола с молекулярной массой около 900 (ц-2 в форму-
ле II) размягчается при 70 °C. Эпоксидное число* равно приблизительно 0,2.
Соответственно смола содержит 1,8 эпоксидной группы в молекуле (эквивалент-
ная масса 500). Она растворяется, например, в ароматических углеводородах,
тетрагидрофуране, хлороформе.
Б. Приготовление эпоксидной смолы с молекулярной
массой 1400
Навеску 45,6 г (0,2 моля) бисфенола А, 11,1 г (0,28 моля) (растворенного
в 112 мл воды) и 22,6 г (0,245 моля) эпихлоргидрина смешивают в тех же
условиях, что и в описанном выше опыте А.
Так как мольное соотношение эпихлоргидрина и бисфенола А, равное 1,22
меньше, чем в опыте А, то образующаяся эпоксидная смола имеет более высо-
кую молекулярную массу, чем в опыте А. Эпоксидное число равно примерно 0,1,
что соответствует 1,44 эпоксидной группы в молекуле (эквивалентная масса
около 970). Смола с молекулярной массой около 1400 (п = 3,7 в формуле II)
размягчается при 97—103 °C.
Определение эпоксидного числа основано на реакции при-
соединения галогенводорода к эпоксигруппам (1 моль галогенводорода эквива-
лентен 1 молю эпоксидных групп). Навеску 0,5—1 г смолы кипятят с обратным
холодильником с избытком (50 мл) раствора солянокислого пиридина (16 мл
чистой конц. НС1 разбавляется в отношении 1 : 1 чистым пиридином). После ох-
лаждения смесь титруется 0,1 н. NaOH по фенолфталеину.
_ (В — A) N [“Число грамм-эквивалентов эпоксидного кислорода
Эпоксидное „число = —— -----------------------100~г-------------------:
где А — объем 0,1 н. NaOH, израсходованный на титрование образца; В —
объем 0,1 н. NaOH, израсходованный на титрование гидрохлорида пиридина;
/V — нормальность растворов NaOH; Е — масса смолы, г. Эквивалентная мас-
са — масса смолы, содержащей 1 эквивалент эпоксидных групп; он равен 100
на эпоксидное число.
С. Сшивание (отверждение) эпоксидных смол
Отверждение аминами. После определения эпоксидного числа и эквивалент-
ной массы смолы, полученной по методике, описанной в пункте Б, небольшой
образец плавят при 150 °C и хорошо перемешивают с эквивалентным количест-
вом (0,25 моля на 1 моль эпоксидных групп) мелкорастертого 4,4-диаминоди-
фенилметана в течение 0,5 мин. При нагревании смеси в течение 1 ч при 150 °C
образец переходит в нерастворимое и неплавкое твердое состояние.
Отверждение ангидридами. В стакане расплавляют 5 г смолы
при 120 °C, приготовленной по методике, описанной в пункте А, и расплав пе-
ремешивают с 1,5 г фталевого ангидрида (0,6—0,8 эквивалентов на 1 эквивалент
эпоксидных групп). Сначала смесь выдерживают при 120 °C (после этой обра-
ботки смола еще растворима в ацетоне и хлороформе), а затем в течение 1—2 ч
отверждают при 170—180 °C.
Сшивание эпоксидных смол ангидридами карбоновых кислот катализируется
третичными аминами. В рассмотренном выше опыте отверждение проходит за
1 ч при 120 °C (в присутствии 50 мг ^Кт-диметиланилина).
* Эпоксидное число означает число грамм-эквивалентов эпоксидного кисло-
рода, приходящееся на 100 г смол.
234
Опыт 4-25. Двухстадийный синтез эпоксидных смол из глицерина и эпихлор-
гидрина
Получение хлоргидрина
В трехгорлую колбу емкостью 1 л, снабженную мешалкой, термометром и
обратным холодильником, помещают 46 г (0,5 моля) перегнанного глицерина и
0,25 мл эфирата BF3 и нагревают до 40 °C на водяной бане. В течение 45 мин
по каплям прибавляют 140 г (1,5 моля) свежеперегнанного эпихлоргидрина, хо-
рошо перемешивая смесь. Спонтанная реакция протекает с выделением тепла.
При добавлении эпихлоргидрина температуру реакции поддерживают равной
44—45 °C, используя внешнее охлаждение. Охлаждение прекращают и смесь
продолжают перемешивать еще 2 ч. Эпоксидное число (определение см. в опыте
4-24Б) должно быть равным нулю.
Получение эпоксидной смолы
- Полученный хлоргидрин растворяют в 100 мл смеси бутанола и толуола
(1:1) и охлаждают до —5 °C. Поддерживая температуру от 5 до 0 °C к этой
смеси в течение 30 мин добавляют 135 мл 45%-ного раствора NaOH, а затем
оставляют при этой температуре на 3 ч. Смесь отмывают от NaCl 60 мл воды;
водный слой (pH 7—8) отделяют в делительной воронке. Смесь растворителей
осторожно отгоняют в вакууме, а оставшуюся сильновязкую смолу фильтруют
на воронке Бюхнера после добавления некоторого количества кизельгура (для
облегчения процесса фильтрования). Прозрачный сильновязкий раствор, получен-
ный с 80%-ным выходом, характеризуется эпоксидным числом, равным 0,6, и
молекулярной массой около 300. Его можно сшивать так, как описано в опы-
те 5-24, получая твердый нерастворимый продукт.
Литература
1. Flory Р. J., Principles of Polymer Chemistry, Ithaca, N. Y., 1953.
2. Schulz G. V., Z. Physik. Chem., 1938, Bd. A182, S. 127.
3. W i 1 f о n g R. E., J. Polymer Sci., 1961, v. 84, p. 385.
4. Muller E., Houben-Weyl, 1963, v. 14/2, p. 1.
5. Korshak V. V., Vinogradova S. V. Polyesters, Pergamon Press, Ox-
ford, 1965.
6. Muller E., Houben-Weyl, 1963, v. 14/2, p. 6.
7. Houben-Weyl, 1963, v. 14/2, p. 17.
8. Сёренсон У., Кемпбел T. Препаративные методы химии полимеров.
М. Издатинлит, 1968. 564 с.
9. Schneel Н., Angew. Chem., 1956, Bd. 68, S. 633; Polymer Reviews, 1964, v. 9,
p. 103.
10. К r i m m H., Houben-Weyl, 1963, v. 14/2, p. 196.
11. Houben-Weyl, 1963, v. 14/2, p. 40.
12. R i n k e H., I s t e 1 E., Houben-Weyl, 1963, v. 14/2, p. 99.
13. Корша к В. В., Фрунзе Т. М. Синтетические гетероцепные полиамиды.
М., «Наука», 1964. 496 с.
14. Van Velden Р. F., Van der Want G. M., К г u i s s i n k Ch. A., Her-
mans P. H., Stevcrman A. J., Rec. Trav. Chim., Pays-Bas, 1955, v. 74,
p. 1376.
15. Rothe M., J. Polymer Sci., 1958, v. 30, p. 227.
16. W e g 1 e r R., H e r 1 i n g e r H., Houben-Weyl, v. 14/2, p. 193.
17. Kammerer H., Schweikert H., MakromoL Chem., 1963, Bd. 60, S. 155;
1965, Bd. 83, S. 188.
18. Kammerer H., Angew. Chem., 1965, Bd. 77, S. 965.
19. Kammerer H., Grossman M., Um son st G., MakromoL Chem., 1960,
Bd. 39, S. 39.
20. W e g 1 e r R., Houben-Weyl, 1963, v. 14/2, p. 319.
21. Lem me G., Chemiker-Ztg., 1903, Bd. 27, S. 896.
22. Spielberger G., Houben-Weyl, 1963, v. 14/2, p. 591.
23. Ro chow E. G., An Introduction to the Chemistry of Silicones, 2nd ed., John
Wiley and Sons, N. Y., 1951.
235
24. Noll W., Chemie und Technologie der Silicone, 2nd ed. Verlag Chemie, Wein-
cheim, 1968.
25. Dearing A. W., Reid E. E., J. Am. Chem. Soc., 1928, v. 50, p. 3058.
26. Sauer R. O., J. Am. Chem. Soc., 1944, v. 66, p. 1707.
27. Wallenberg F. T., Angew. Chem., 1964, Bd. 76, S. 484.
28. V о 11 m e r t B., Kunststoffe, 1966, Bd. 56, S. 680.
29. Segal B., J. Macromol. Sci., 1967, v. Al, p. 1.
30. Kovacic P., Feldman M. B., Kovac ic J. P., Lando J. B., J. AppL
Polymer Sci., 1968, v. 12, p. 1735.
31. Hay A. S., J. Polymer Sci., 1962, v. 58, p. 581.
32. Enders G. F., Hay A. S., Eu stance J. W., J. Org. Chem., 1963, v. 28,
p. }300.
33. H у A. S., Advances Polymer Sci., 1967, v. 4, p. 496.
34. B*yer O., Angew. Chem., 1947, Bd. 59, S. 257.
35. Muller E., Houben-Weyl, 1963, v. 14/2, p. 57.
36. Саундерс Дж. X., Фриш К. К. Химия полиуретанов. М., «Химия»,
1968. 470 с.
37. Wind emu th Е., Kunststoffe, 1967, Bd. 57, S. 337.
38. Merten E., Lauer er D., Braun G., Dahm M., Macromol. Chem., 1967.
Bd. 101, S. 337.
39. S i e f к e n W., Liebigs Ann. Chem., 1949, Bd. 562, S. 75.
40. Bayer O., Muller E., Angew. Chem., 1960, Bd. 72, S. 934.
41. Wegler R., Schultz-Josten R., Houben-Weyl, 1963, v. 14/2, p. 462
5. РЕАКЦИИ ПОЛИМЕРОВ
5.1. ХИМИЧЕСКИЕ ПРЕВРАЩЕНИЯ ПОЛИМЕРОВ [1-5]
Синтез полимеров основан на способности низкомолекулярных
полифункциональных соединений к реакциям полимеризации, по-
ликонденсации и ступенчатой полимеризации. Однако сами по се-
бе макромолекулы также способны к химическим превращениям
(см. раздел 2.1.6), что часто используется в практических целях.
В химии высокомолекулярных соединений различают реакции
функциональных групп, протекающие без изменения строения ос-
новной цепи, а следовательно, и степени полимеризации, и реак-
ции, протекающие с деструкцией макромолекулярных цепей. Во
многих случаях обе эти реакции протекают одновременно. Кроме
того, возможны реакции сшивания и прививки, которые могут
быть использованы для получения блок- и привитых сополимеров
(см. раздел 3.3.2).
В реакциях низкомолекулярных соединений образующиеся ве-
щества можно отделить от непрореагировавших исходных и по-
бочных продуктов, хотя иногда это связано с экспериментальны-
ми трудностями. Более того, продукты реакции дают информацию
о строении исследуемых соединений.
В химических реакциях высокомолекулярных соединений все
усложняется тем, что одна и та же макромолекула принимает
участие как в основных, так и в побочных реакциях. Например,
если только 80 из 100 мономерных звеньев участвуют в данной
химической реакции, а остальные либо претерпевают превращение
в побочном процессе, либо не реагируют вовсе, то две эти группы
звеньев невозможно изолировать друг от друга, так как они вхо-
дят в состав одной макромолекулы. Таким образом получается по-
лидисперсный продукт реакции (см. раздел 2.1.6).
Если в реакции макромолекул все звенья реагируют одинако-
вым образом без разрывов основной цепи и без участия в побоч-
ных реакциях, молекулярная масса полимера изменяется, а сте-
пень полимеризации остается постоянной. Например, при омылении
неразветвленного поливинилацетата с образованием поливинило-
вого спирта молекулярная масса полимера уменьшается, а степень
полимеризации не изменяется. Такие реакции, в которых основная
цепь макромолекулы сохраняется, называются полимераналогич-
ными превращениями. Они играют важную роль в структурных ис-
237
следованиях высокомолекулярных соединений. Полимераналогич-
ные превращения были использованы для подтверждения того
факта, что полимеры действительно состоят из длинных макромо-
лекул, а не являются агрегатами ассоциированных низкомолеку-
лярных веществ. Так как молекулярные массы мономерных звень-
ев исходного полимера, побочных продуктов и основного продукта
реакции, как правило, различаются, выход и степень конверсии
должны определяться по отдельности.
В упомянутом примере полимераналогичного омыления поли-
винилацетата свойства исходного и конечного продукта различны,
однако степени полимеризации обоих полимеров одинаковы. Полу-
ченный омылением поливиниловый спирт можно этерифицировать
с образованием поливинилацетата исходной молекулярной массы,
обладающего теми же свойствами. В то же время, если в процес-
се этих превращений происходит разрыв цепи, то это определя-
ется по характеристической вязкости переацеталированного полиме-
ра (опыт 5-01). Кроме таких реакций, в которых должны участ-
вовать все мономерные звенья полимерной цепи, протекают реак-
ции с участием двух соседних звеньев. Например, практически
важной является реакция между соседними гидроксильными груп-
пами, которая протекает при ацеталировании поливинилового
спирта с образованием цикла, содержащего карбонильную альде-
гидную группу. В силу статистических условий в лучшем случае
только 86,5% всех функциональных групп могут претерпевать пре-
вращения в таких реакциях (см. раздел 2.1.6.1).
Здесь невозможно рассмотреть все огромное многообразие хи-
мических превращений макромолекул [5]. Такие реакции позволя-
ют иногда синтезировать полимеры, мономеры которых не извест-
ны (например, поливиниловый спирт) или трудно синтезируемы,
не способны к полимеризации или полимеризуются плохо. Это от-
носится к винилгидрохинону, который, как и сам гидрохинон, яв-
ляется ингибитором полимеризации. Проведя «защиту» гидроксиль-
ных групп фенола путем ацетилирования, получают продукты, спо-
собные к полимеризации, с которых после полимеризации снимают
«защиту».
При получении ионообменных смол функциональные группы
часто вводят в (обычно сшитые) макромолекулы после полимери-
зации (см. раздел 5).
К числу наиболее важных химических превращений макромоле-
кул относятся различные реакции целлюлозы [6], в мономерном
звене которой все или часть гидроксильных групп можно превра-
тить в простую или сложноэфирную группу. В зависимости от чис-
ла ацетилированных гидроксильных групп в мономерном звене
получают триацетат целлюлозы, вторичный ацетат целлюлозы
и т. д. Другой важной реакцией является превращение целлюлозы
в ксантогенаты. Водные растворы натриевых солей ксантогенатов
целлюлозы, известные как «вискоза», пропускают через ванны,
содержащие минеральные кислоты, для регенерации целлюлозы и
238
коагуляции полимера; полученные волокна называются вискозным
или искусственным шелком.
Реакции концевых групп макромолекул, протекающие без из-
менения основной цепи, также относятся к полимераналогичным
превращениям (например, реакции концевых гидроксильных групп
полиэфиров с ангидридом фталевой кислоты и карбоксильных
групп с диазометаном). Такие превращения блокируют концевые
группы, т. е. придают им устойчивость. Так, полиоксиметилены
(полученные из формальдегида или триоксана) термически неста-
бильны ввиду присутствия полуацетальных групп. Однако конце-
вые группы можно стабилизировать ацеталированием. В соответ-
ствующих условиях эту реакцию можно проводить без заметного
разрушения основной цепи (опыт 5-09) [7].
Аналогичные превращения протекают в реакциях свободных
аминогрупп концевых аминокислот в пептидах и белках, напри-
мер, с 2,4-динитрофторбензолом; при гидролизе пептидных связей
[8] образующиеся динитрофениловые кислоты можно идентифи-
цировать. О методах определения концевых групп см. раз-
дел 2.3.2.2.
Опыт 5-01. Получение поливинилового спирта переэтерификацией поливинил-
ацетата; переацетилирование поливинилового спирта
А. Получение поливинилового спирта
В трехгорлую колбу емкостью, 500 мл, снабженную мешалкой, обратным
холодильником и капельной воронкой, помещают 50 мл 1%-ного метанольного
раствора NaOH и нагревают на водяной бане до 50 °C. В течение 30 мин по кап-
лям при сильном перемешивании добавляют раствор 15 г поливинилацетата
(см. опыт 3-06) в 100 мл метанола. Переэтерификация начинается немедленно,
о чем свидетельствует осаждение поливинилового спирта. После введения поли-
винилацетата перемешивание продолжают еще 30 мин. Порошкообразный осадок
фильтруют, отмывают от щелочи метанолом и сушат в вакууме при 30—40 °C.
Поливиниловый спирт растворяется или набухает только в небольшом числе
органических растворителей (например, в теплом диметилформамиде), однако он
хорошо растворим в теплой воде.
Б. Переацетилирование поливинилового спирта
В колбу емкостью 100 мл помещают 5 г полученного поливинилового спирта
и 75 мл ацетилирующего агента (состоящего из смеси пиридина, ацетангидрида
и уксусной кислоты в соотношении 1 : 10: 10 по объему). Содержимое колбы
нагревают с обратным холодильником до 100 °C. Избыток ацетилирующего аген-
та упаривают в вакууме при комнатной температуре и полимер переосаждают
водой из метанола. Определите содержание ацетильных групп [9] и раствори-
мость полимера и сравните ее с растворимостью поливинилового спирта и исход-
ного поливинилацетата. Кроме того, определите характеристическую вязкость
исходного и полученного образцов поливинилацетата. Опыт покажет, что в про-
цессе омыления и переацетилирования полимер частично разлагается, так как
величина цуд/С переацетилированного образца ниже, чем исходного. За измене-
нием растворимости поливинилового спирта с изменением степени ацетилирова-
ния легко проследить по растворимости в воде, ацетоне и метаноле проб, отоб-
ранных в различное время. При степени ацетилирования выше 20% (мол.) рас-
творимость в воде уменьшается, а в органических растворителях возрастает.
Опыт 5-02. Получение поливинилбутираля
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, обратным
холодильником и капельной воронкой, помещают 3,3 г перегнанного бутираль-
альдегида, к которому добавляют при перемешивании раствор (нагретый до
65 °C) 5 г поливинилового спирта (опыт 5-01) в 50 мл воды и 0,3 г конц.
H2SO4. Раствор следует добавлять в течение нескольких минут при интенсив-
239
ном перемешивании, отрегулированном так, чтобы оно происходило без разбрыз-
гивания. Поливинилбутираль сразу же выпадает в осадок. После добавления
1 г серной кислоты (средней концентрации) реакционную смесь выдерживают
при 50—55 °C в течение 1 ч. После охлаждения до комнатной температуры по-
лимер фильтруют при отсасывании и промывают водой до нейтральной реакции.
Полимер затем переосаждается водой из раствора метанола и сушится в
вакууме при 40 °C; его выход составляет около 6 г. Проверьте растворимость
поливинилбутираля и сравните ее с растворимостью поливинилового спирта.
Растворимость может сильно меняться при изменении степени ацеталирования.
Опыт 5-03. Омыление сополимера стирола и малеиновой кислоты
Навеску 2 г сополимера стирола и малеиновой кислоты (см. опыт 3-51) и
50 мл 2 н. NaOH нагревают в круглодонной колбе емкостью 100 мл, снабжен-
ной обратным холодильником.
Полимер растворяется за несколько минут. После нагревания в течение
нескольких минут смесь охлаждают и помещают в 500 мл 2 н. НС1. Выпавшую
в осадок полимерную кислоту фильтруют на воронке Бюхнера. Если осадок
отделяется плохо, то подкисленную дисперсию встряхивают со 100 мл эфира
в делительной воронке. При этом полимер осаждается в виде эластичной клей-
кой массы на границе раздела двух жидкостей. Сополимер фильтруют и промы-
вают холодной водой (сополимер растворим в воде) и сушат на воздухе. Для
очистки полимерную кислоту растворяют в 50 мл тетрагидрофурана или диокса-
на и высаживают в 500 мл бензола. Через несколько часов сополимер осаж-
дается, так что его можно отфильтровать и высушить в вакууме при 50 °C.
В отличие от исходного продукта сополимер стирола и малеиновой кисло-
ты растворяется в горячей воде. Водный раствор поликислоты дает кислую реак-
цию.
Опыт 5-04. Этерификация полиметакриловой кислоты диазометаном
Навеску 1,2 г полиметакриловой кислоты (см. опыт 3-10) растворяют в
10 мл метанола и раствор охлаждают на ледяной бане. К холодному раствору
по каплям при перемешивании прибавляют эфирный раствор диазометана* [10]
до появления желтой окраски. В процессе добавления диазометана полностью
осаждается этерифицированная полиметакриловая кислота. Ее фильтруют и су-
шат в вакууме при 50 °C. Определите характеристическую вязкость этерифициро-
ванного полимера при 25 °C в ацетоне и рассчитайте молекулярную массу
(раздел 2.3.2.1).
Опыт 5-05. Получение поли-п-винилацетофенона
В трехгорлую колбу емкостью 500 мл, снабженную мешалкой, обратным
холодильником и капельной воронкой, помещают 100 мл сероуглерода и 27 г
безводного порошкообразного хлорида алюминия. После введения 12 г (0,15 мо-
ля) свежеперегнанного ацетилхлорида по каплям, не прекращая перемешивания,
прибавляют раствор 10,4 г (0,1 моля) полистирола** в 10 мл сероуглерода. По-
следующая энергичная реакция сопровождается выделением НС1. Реакционную
смесь кипятят в течение 1 ч. После этого заменяют обратный холодильник от-
водом с холодильником Либиха и приемником и отгоняют сероуглерод. Остаток
отделяют и тщательно обрабатывают ледяной разбавленной соляной кислотой
для разложения хлорида алюминия. После декантации остаток снова обрабаты-
вают ледяной НС1 и многократно промывают водой. Полимер отфильтровывают,
промывают метанолом и сушат в вакууме при 50 °C. Частично ацетилированный
полистирол растворим в свежеперегнанном ацетофеноне, горячем диоксане, аце-
тоне и в ледяной уксусной кислоте; Ьн нерастворим в метаноле. Его можно
деполимеризовать с выделением мономера n-винилацетофенона (см. опыт 5-16).
Опыт 5-06. Ацетилирование целлюлозы
Навеску 10 г целлюлозы в виде ваты или кусков фильтровальной бумаги***
помещают в широкогорлую бутыль емкостью 25 мл и заливают раствором 0,5 г
* Соблюдайте осторожность при получении диазометана.
** Лучше использовать низкомолекулярный полистирол, так как в противном
случае потребуется очень много растворителя для получения реакционной смеси
низкой вязкости.
*** От вида исходного материала зависит скорость реакции.
240
конц. H2SO4 в 5 мл уксусной кислоты. Смесь перемешивают стеклянной палоч-
кой для равномерного увлажнения 'целлюлозы. Бутыль закупоривают и оставля-
ют на 1 ч при комнатной температуре. После этого к содержимому бутыли
добавляют смесь 50 мл 95%-ного ацетангидрида и 20 мл ледяной уксусной кис-
лоты и закрытую бутыль оставляют на водяной бане при 50 °C. Через 15 мин
целлюлоза растворяется, и через 30 мин реакция завершается. Исходный раствор
делят на две части, которые используют для получения триацетата и вторичного
ацетата целлюлозы.
А. Получение триацетата целлюлозы
К одной части полученного раствора, осторожно помешивая, добавляют
25 мл 80%-ной уксусной кислоты (60 °C) для разложения избытка уксусного
ангидрида. Необходимо проследить, чтобы ацетат целлюлозы при этом не высаж-
дался. После нагревания раствора до 60 °C в течение 15 мин наливают в стакан
емкостью 600 мл и осторожно добавляют 25 мл воды, перемешивая содержимое
стакана. После прибавления еще 200 мл воды триацетат целлюлозы осаждается
в виде рыхлого белого порошка, который легко поддается промывке. Продукт
фильтруют, а затем обрабатывают 300 мл воды, которую декантируют через
15 мин. Промывание повторяют до нейтральной реакции промывных вод. Поли-
мер отделяют от воды, насколько это возможно фильтрованием или центрифуги-
рованием, а затем сушат при 105 °C. Выход составляет около 7 г триацетата
целлюлозы. Продукт растворим в смеси метиленхлорида и метанола (9:1) и
практически нерастворим в ацетоне или в кипящей смеси бензола и метанола
(1:1).
Б. Получение вторичного ацетата целлюлозы
Для частичного омыления целлюлозы 50 мл 70%-ной уксусной кислоты,
нагретой до 60 °C, и 0,14 мл конц. H2SO4 медленно добавляют при перемеши-
вании к остатку раствора, полученного в опыте А. Реакционную смесь в за-
крытом сосуде выдерживают в течение 3 ч на водяной бане при 80 °C, а затем
обрабатывают так, как указано в пункте А. Выход составляет 6—6,5 г. Вторич-
ный ацетат целлюлозы (содержание ацетильных групп 40%) растворим в ацето-
не, смеси метиленхлорида с метанолом (9:1) ив кипящей смеси метанол—бен-
зол (1 : 1).
Проверьте по растворимости, прошло ли частичное омыление так, как это
было необходимо. Для осаждения ацетата целлюлозы к 1 мл раствора добав-
ляют воду. Осадок быстро отделяют от кислоты промыванием водой и сушат
между двумя фильтровальными бумажками. Несколько еще влажных волокон
нагревают в пробирке с 15—20 мл смеси бензол—этанол (1:1) на водяной
бане до кипения растворителя. Если волокна растворились, то реакция прошла
так, как описано выше.
Опыт 5-07. Получение триметилцеллюлозы
В трехгорлую колбу емкостью 1 л, снабженную магнитной мешалкой, обрат-
ным холодильником и двумя капельными воронками (прибор держите под кол-
паком), помещают 10 г растворимого в ацетоне вторичного ацетата целлюлозы
(содержание ацетатных групп 40%, см. опыт 50-06Б) и растворяют его в 200 мл
ацетона при 55 °C. К этому раствору при перемешивании равномерно (в течение
1,5 ч) приливают из двух воронок 120 мл диметилсульфата (осторожно) и
320 мл 30%-ного раствора NaOH. После этого ацетон отгоняют, а остаток от-
фильтровывают в горячем состоянии и многократно промывают большим коли-
чеством кипящей воды для удаления щелочи. Для очистки триметилцеллюлозу
обрабатывают ацетоном и эфиром, а затем сушат в вакууме при комнатной тем-
пературе.
Триметилцеллюлоза растворима в хлороформе, бензоле и горячем циклогек-
саноне и нерастворима в этаноле, эфире и петролейном эфире.
Опыт 5-08. Получение натриевой соли карбоксиметилцеллюлозы
В колбу емкостью 1 л, снабженную мешалкой, капельной воронкой и труб-
кой для ввода газа, помещают 15 г целлюлозы* и 400 мл изопропанола. Воз-
дух вытесняют из сосуда, заполняя его азотом. Через 15 мин добавляют 50 г
* Кусочки фильтровальной бумаги смачивают водой и образующуюся каши-
цу фильтруют и сушат.
16—732
241
30%-ного раствора NaOH при сильном перемешивании. Перемешивание продол-
жают еще 30 мин, после чего в течение 30 мин приливают раствор 17,5 г хлор-
уксусной кислоты в 50 мл изопропанола. Раствор, нагретый на водяной бане
до 60 °C, перемешивают еще 4 ч. Реакционную смесь нейтрализуют несколькими
каплями ледяной уксусной кислоты (индикатор — фенолфталеин) и фильтруют
в горячем состоянии. Волокна натриевой соли карбоксиметилцеллюлозы смачи-
вают 40 мл 80%-ного водно-метанольного раствора при 60 °C, фильтруют и про-
мывают водным метанолом. Очистку проводят несколько раз до полного удале-
ния хлорида натрия. После промывки метанолом полимер сушат при 80 °C. Вы-
ход составляет 20—25 г. Проверьте растворимость сухой натриевой соли карб-
оксиметилцеллюлозы в воде.
Опыт 5-09. Ацетилирование полуацетальных групп полиоксиметилена
уксусным ангидридом
Полиоксиметилен получают полимеризацией безводного формальдегида
(опыт 3-35) или триоксана (опыт 3-39); продукт полимеризации параформальде-
гида нельзя ацетилировать в гетерогенной среде. Уксусный ангидрид и Ы,М-ди-
метилциклогексиламин тщательно фракционируют, а ацетат натрия обезвожива-
ют при плавлении.
А. Ацетилирование в гетерогенной среде
В колбе емкостью 100 мл, снабженной воздушным холодильником и хлор-
кальциевой трубкой, в течение 2 ч кипятят при перемешивании 3 г хорошо рас-
тертого полиоксиметилена (139 °C) с 30 мл уксусного ангидрида и 30 мг без-
водного ацетата натрия. Полимер фильтруют на стеклянном фильтре и тщатель-
но промывают (5 раз) горячей дистиллированной водой (80 °C), к которой до-
бавлено немного метанола. После этого полимер кипятят в ацетоне при переме-
шивании в течение 1 ч и отфильтровывают (хранят полимер над СаС12 и NaOH
в эксикаторе). Выход составляет 9% (масс.) от исходного полимера. Интервал
плавления* полимера 174—177 °C. Термически стабильная часть** 88%.
Б. Ацетилирование в расплаве
В полимерную пробирку помещают 3 г полиоксиметилена, 6 мл уксусного
ангидрида и 2 мл Н,М-диметилциклогексиламина (для предотвращения ацидо-
лиза) и нагревают до 200 °C в течение 30 мин. Продукт реакции тщательно
промывают этанолом на стеклянном фильтре. Затем полимер кипятят в ацето-
не в течение 1 ч, перемешивая раствор, фильтруют и сушат сначала на стеклян-
ном фильтре до исчезновения запаха, а затем в эксикаторе над СаС12 и грану-
лированным NaOH. Выход составляет 90% (масс.). Интервал плавления***
ацетилированного полимера 170—174 °C; термически стабильная часть****
97%. Если ацетилирование проводить в тех же условиях, но без И,И-диметил-
циклогексиламина, выход составляет 72% (масс.); температурный интервал плав-
ления 169—171 °C, а термически стабильная часть 98%.
Опыт 5-10. Вулканизация бутадиен-стирольного сополимера
В промышленности вулканиза'цию дисковых гомополимеров и сополимеров
проводят элементарной серой при высоких температурах: 100—140 °C (горячая
вулканизация). В лабораторных условиях этот процесс обычно не проводят вви-
ду сложного аппаратурного оформления. Однако принцип вулканизации можно
продемонстрировать на примере сшивания бутадиен-стирольного сополимера
буна S хлористой серой S2C12 при комнатной температуре (холодная вулканиза-
ция).
Поместите в пробирку кусок сополимера бутадиена со стиролом (опыт 3-46)
и S2C12. После заполнения пробирки азотом5* закройте ее и оставьте стоять на
1 ч. Декантируйте S2C12 и добавьте бензола. Сравните растворимость и (или) на-
бухание образца с растворимостью сополимера, не обработанного S2C12.
* Интервал плавления определяли в тонких стеклянных капиллярах.
** Через 10 ч при 190 °C в азоте (опыт 5-15).
*** Интервал плавления определяли в тонких стеклянных капиллярах.
**** Через 10 ч при 190 °C в азоте (опыт 5-15).
5* Так как опыт носит качественный характер, работать в атмосфере азота
нс обязательно.
242
Развитие реакции сшивания можно легко продемонстрировать с помощью
следующего эксперимента. В колбе Эрленмейера с коническим дном (250 мл)
растворяют 2 г сополимера бутадиена со стиролом в 100 мл бензола в токе
азота*. Половину раствора смешивают с 1 мл S2CI2, сильно встряхивая смесь,
а вторую половину хранят в атмосфере азота, но без S2C12. Через 5 мин раствор,
обработанный S2CI2, становится заметно более вязким, чем необработанный, а
спустя 10 мин он превращается в гель. Через 20 мин образуется плотная желс-
подобная масса. Еще через несколько часов степень сшивания уже настолько
велика, что происходит разделение фаз с выделением растворителя в результа-
те синерезиса.
5.2. ИОНООБМЕННЫЕ ПОЛИМЕРЫ (ИОНИТЫ)
Ионообменные полимеры представляют собой полиэлектролиты,
которые состоят из твердого сшитого и, следовательно, нераство-
римого полимера, содержащего кислые или щелочные группы. Дав-
но известные ионообменные материалы такие, как природные не-
органические минералы (например, цеолиты) и некоторые синте-
тические материалы (например, пермутиты), в настоящее время
играют второстепенную роль. Им на смену пришли ионообменные
полимеры, получаемые путем введения ионогенных групп в уже
сшитые продукты полимеризации или поликонденсации, синтезиро-
ванные из соответствующих мономеров [11—17].
Наиболее распространенными ионообменными полимерами яв-
ляются поликислоты или полиоснования, способные к обмену ионов
Н+ или ОН- на катионы (катиониты) или анионы (аниониты) со-
ответствующих низкомолекулярных солей. Этот принцип использу-
ется, например, для очистки воды:
Ро1~Н+ 4- Na+ Pol~Na+ + Н+
Pol+OH- + Cl- «=> Pol+Cl- + OH-
Pol~H+ + Pol+OH- + NaCl ч=> Pol~Na+ 4- Pol+Cl" 4- H2O
где Pol“ и Pol+ — ионогенные группы нерастворимого сшитого полимера, кото-
рые связывают обменивающиеся ионы.
Так как процессы ионного обмена представляют собой равно-
весные реакции, то отработанный катионит или анионит можно ре-
генерировать обработкой соответственно кислотой либо щелочью.
Промышленные ионообменные полимеры часто выпускают в соле-
вой форме, поэтому перед использованием их необходимо переве-
сти в кислую или основную форму. Однако многие катионы метал-
лов могут непосредственно обмениваться друг с другом.
Катиониты содержат обычно такие активные кислотные груп-
пы, как сульфо- и карбоксильные группы; кислотные группы фос-
форной кислоты используют значительно реже. Аниониты прежде
всего содержат первичные, вторичные и третичные аминогруппы,
однако в них могут входить и четвертичные полимерные аммоний-
ные основания. Обычно иониты получают из полимерных материа-
* Так как опыт носит качественный характер, работать в атмосфере азота
не обязательно.
16* 243
лов, главным образом из сополимеров стирола и дивинилбензола
(см. опыт 3-49), которые лучше всего использовать в виде гранул
диаметром 0,1—2 мм. Концентрация дивинилбензола [в % (масс.)]
в мономерной смеси определяет степень сшивания. Чем выше сте-
пень сшивания, тем ниже степень набухания полимера; однако по-
следнее зависит также от типа противоиона и ряда других фак-
торов.
Функциональные ионогенные группы последовательно вводят
в полимер путем полимераналогичных превращений. Можно ис-
пользовать и мономеры, уже содержащие необходимые функцио-
нальные группы, так что ионит образуется прямо в процессе по-
лимеризации. Таким методом получают слабые (катиониты из мет-
акриловой кислоты.
По другому способу в качестве исходного материала исполь-
зуют нерастворимый продукт поликонденсации (например, феноло-
формальдегидной смолы), в который вводят ионогенные группы,
если только функциональные группы уже не вошли в процессе
поликонденсации например, при использовании фенолсульфокис-
лоты.
Для научных целей иониты получают также из целлюлозы. Их
готовят из щелочной целлюлозы с хлоруксусной кислотой
(опыт 5-08). При обработке |3-хлорэтилдиамином получают анио-
нит, называемый DEAE-целлюлозой.
Кроме размеров гранул, степени сшивания и набухания важ-
ной количественной характеристикой ионитов является их емкость.
Она представляет собой число эквивалентов обменивающихся про-
тивоионов в полимерной сетке, приходящихся на единицу массы
или объема сухого либо набухшего ионита. В лабораторной прак-
тике емкость обычно выражается в мэкв/г; емкость ионита для
смягчения жесткой воды часто дается в граммах СаО на 1 л
ионита.
Существуют различные способы проведения реакции обмена.
Реакция может проводиться в статических условиях, при кото-
рых ионит находится в контакте с раствором, содержащим обмени-
вающиеся ионы, до установления равновесия. В таких реакциях
обмена равновесие сдвинуто в сторону образования целевого про-
дукта, что достигается введением избытка ионита. Обычно ионо-
обменные реакции проводят в колонках в динамическом режиме.
Как и в хроматографической колонке, исходный раствор пропус-
кают через колонку сверху вйиз. Ионит, используемый в качестве
наполнителя, помещают в колонку уже в набухшем состоянии, так
как давление набухания может разрушить колонку.
Некоторые иониты можно использовать как в промышленности
(например, для смягчения жесткой воды, извлечения металлов из
отработанных вод, рафинирования сырого сахара), как и в лабо-
раторных целях. С их помощью можно легко очищать водные рас-
творы (очистка муравьиной кислоты от растворов формальдегида),
готовить кислоты или основания высокой степени чистоты (напри-
244
мер, тиоциановую кислоту из тиоцианата алюминия). Иониты при-
меняются также для катализа химических реакций, например в
реакциях этерификации и гидролиза белков. В литературе описа-
ны и другие примеры применения ионообменных смол.
Опыт 5-11. Получение катионита сульфированием сшитого полистирола
А. Сульфирование сшитого полистирола
Из полистирола, сшитого дивинилбензолом, сульфированием легко получает-
ся ионит. Для этого смесь 0,2 г сульфата серебра и 150 мл конц. H2SO4 нагре-
вают до 80—90 °C в трехгорлой колбе емкостью 500 мл, снабженной мешалкой,
обратным холодильником и термометром. К этой смеси при перемешивании до-
бавляют 20 г гранулированного сополимера стирола с дивинилбензолом
4опыт 3-49), что вызывает спонтанное повышение температуры до 100—105 °C.
Реакционную смесь выдерживают при 100 °C в течение 3 ч, после чего охлаж-
дают до комнатной температуры и оставляют стоять на несколько часов. Затем
смесь выливают в колбу Эрленмейера (на 1 л) с 500 мл 50%-ного раствора
серной кислоты. После охлаждения смесь разбавляют дистиллированной водой,
желто-коричневые гранулы отфильтровывают и промывают большим количеством
зоды.
Б. Определение обменной емкости
Для определения обменной емкости сульфированный полимер помещают
в стеклянную трубку с краном на конце, в который кладется стеклянная вата
для предотвращения засорения крана. Через колонку пропускают 100 мл 2 н.
раствора NaCl, а затем 100 мл 2 н. НС1. Колонку непрерывно промывают ди-
стиллированной водой и из промывных вод отбирают пробы по 10 мл, которые
титруют 0,1 н. NaOH по фенолфталеину. Промывание заканчивают, когда нор-
мальность воды становится ниже 0,001. Смолу сушат, еще во влажном состоя-
нии перекладывают в стакан и 3 образца по 2 г немедленно взвешивают в кол-
бах Эрленмейера емкостью по 100 мл. Одну колбу помещают в сушильный шкаф
и сушат смолу до постоянной массы для определения влажности образца. В две
оставшиеся колбы наливают по 50 мл 0,1 н. NaOH и смесь перемешивают, силь-
но встряхивая. Через 30 мин образцы фильтруют, промывают водой и оттитро-
вывают 0,1 н. НС1. Обменную емкость выражают в мэкв/г сухой смолы.
Опыт 5-12. Получение катионита сульфированием фенолоформальдегидного
олигомера
В круглодонной колбе емкостью 250 мл нагревают с обратным холодильни-
ком смесь 40 г феполоформальдегидной смолы и 120 г 95%-ной серной кисло-
ты до 140 °C. Как только смола полностью растворится, раствор охлаждают до
комнатной температуры, переливают в металлическую емкость и быстро переме-
шивают с 30 мл 37%-ного водного раствора формальдегида. Металлическую
чашу помещают на масляную баню при НО °C и ее содержимое отверждают
в течение 2 ч. После охлаждения продукт промывают водой, разбивают молот-
ком и растирают в ступке до получения кусочков размером 1—3 мм. Смолу
промывают водой до тех пор, пока промывные воды не становятся прозрачны-
ми. Определите обменную емкость так, как описано в опыте 5-11.
Опыт 5-13. Получение анионита из сшитого полистирола хлорметилированием
и аминированием
А. Хлорметилировапие сшитого полистирола
В трехгорлую колбу емкостью 250 мл, снабженную мешалкой, обратным хо-
лодильником с хлоркальциевой трубкой (третье горло плотно закрыто пробкой),
помещают 20 г сшитого гранулированного стирола (например, можно использо-
вать сополимер стирола с дивинилбензолом, полученным в опыте 3-49), и рас-
твор 50 г дихлорметилового эфира* в 40 мл тетрахлорэтилена. Смесь перемеши-
вают в течение 30 мин при комнатной температуре, что вызывает некоторое на-
бухание полимера. Через 1 ч прибавляют 10 г безводного хлорида цинка, непре-
рывно перемешивая смесь при 40—60 °C, затем смесь перемешивают еще 2 ч. Не-
прореагировавший дихлорметиловый эфир разрушают, осторожно прибавляя
* Работайте под тягой. Дихлорметиловый эфир сильнотоксичен.
245
воду, и, наконец, полимер неоднократно промывают водой и сушат в вакууме
при 50 °C. Содержание хлора составляет около 15% (масс.).
Б. Аминирование хлорметилированного полистирола
В трехгорлой колбе емкостью 100 мл, снабженной мешалкой, обратным хо-
лодильником, термометром и трубкой для ввода газа, кипятят смесь 20 г хлор-
метилированного полистирола с 50 л бензола в течение 30 мин. После охлажде-
ния до 30—35 °C газообразный безводный триметиламин пропускают в раствор,
постепенно повышая температуру до 50—55 °C. (Газообразный триметиламин по-
лучают, приливая его водный раствор к раствору едкого натра; перед пропуска-
нием через реакционную колбу газ проходит через трубку, наполненную твер-
дым NaOH.) Через 4 ч прекращают пропускать газ и реакционную смесь вы-
держивают при комнатной температуре около 3 ч. Гранулы фильтруют, отсасы-
вая, промывают несколько раз бензолом и сушат в вакууме при 50 °C. После
этого сухие гранулы выдерживают в 100 мл 5%-ного раствора соляной кислоты
в течение 2 ч, а затем промывают водой до нейтральной реакции (по метило-
вому оранжевому).
В. Определение обменной емкости
Гранулы, полученные по методике, описанной в пункте Б, тщательно филь-
труют и 3 еще влажных образца массой по 2 г быстро взвешивают в 3 колбах
Эрленмейера емкостью по 100 мл. Одну колбу помещают в сушильный шкаф
при НО °C для определения влажности образца. В каждую из двух оставшихся
колб добавляют по 10 мл 15%-ного раствора едкого натра и смесь перемеши-
вают магнитной мешалкой в течение 30 мин; в результате образуется четвертич-
ное аммониевое основание. Избыток щелочи удаляют водой (определяют по
фенолфталеину) и продукт фильтруют. Еще влажные гранулы количественно
переносят в колбы Эрленмейера и в каждую добавляют по 50 мл 0,1 н. НО.
Смесь изредка встряхивают, через 30 мин фильтруют, промывают водой и вы-
сушенную кислоту оттитровывают 0,1 н. NaOH. Обменная емкость выражается
в мгэкв/г сухой смолы (в форме хлорида).
5.3. ДЕСТРУКЦИЯ ПОЛИМЕРОВ [18, 19]
Разрыв связей в основной молекулярной цепи может осуществлять-
ся как химическим, так и физическим путем. При расщеплении
макромолекул сначала образуются макромолекулы, имеющие
строение исходной основной цепи, но меньшую степень полимери-
зации. С увеличением степени деструкции могут образовываться
низкомолекулярные фрагменты (например, олигомеры и мономе-
ры), свойства которых позволяют получить информацию о структу-
ре полимера в целом. Особенно важными являются термическая и
химическая деструкция; однако механодеструкция и деструкция
под действием радиационного ^излучения также играют существен-
ную роль в теории и практике.
Термическая деструкция полимеров обычно представляет собой
свободнорадикальный процесс. Разрыв цепи может происходить
либо по закону случая, либо по слабым местам (например, вблизи
разветвлений или структурных нерегулярностей), либо на концах
цепи. Термическая деструкция ряда полимеров (например, полиэти-
лена, полипропилена, эфиров полиакриловой кислоты, полиакри-
лонитрила, полибутадиена) не приводит к образованию мономера;
в этих случаях протекает собственно деструкция. Если основным
продуктом деструкции полимера является мономер (например, по-
246
листирол, поли-а-метилстирол, полиизопрен, полиметилметакрилат,
полиформальдегид), то протекает деполимеризация. Если термиче-
ская деструкция начинается на подвижных концевых группах, то
стабильность полимера можно значительно повысить, защитив кон-
цевые группы (см. опыт 5-15, термическая деполимеризация поли-
формальдегида).
Тип осколков, образующихся в процессе деструкции, зависит
преимущественно от строения полимера и температуры его разло-
жения. Так, при термической деструкции полиакрилатов мономер
практически не образуется, в то время как он является основным
продуктом деструкции полиметилметакрилата в тех же условиях.
До 250 °C при деструкции полистирола получаются олигомерные
осколки, такие, как димер, тример и более высокомолекулярные
гомологи, однако при повышении температуры до 350 °C образует-
ся уже заметное количество мономерного стирола.
В ряде случаев нагревание инициирует реакции, сопровождаю-
щиеся выделением низкомолекулярных продуктов без заметной
деструкции цепи. Так, при нагревании поливинилхлорида выделя-
ется хлористый водород, что очень нежелательно из-за его агрес-
сивности и последующего изменения цвета полимера. Этот процесс
деструкции можно затормозить на некоторое время, добавив ста-
билизатор [20]. Аналогичным образом при нагревании поливинил-
ацетата выделяется уксусная кислота.
Продуктами деструкции поли-трет-бутилметакрилата при тем-
пературе около 250 °C являются изобутилен, вода и ангидрид поли-
метакриловой кислоты.
При термической деструкции полимеров на воздухе реакции
окисления превалируют над реакциями деструкции, поэтому часто
совершенно неясно, чем вызвана реакция.
Деструкция может быть вызвана окислением, автоокислением
или гидролизом. Как и низкомолекулярные соединения, полимеры
подвержены автоокислению в соответствующих условиях, особенно
при повышенных температурах ['21, 22]. Образующиеся в процессе
окисления гидроперекиси обычно нестабильны при высоких темпе-
ратурах и разлагаются на свободные радикалы, т. е. реакция мо-
жет стать автокаталитической; вторичные продукты могут иниции-
ровать последующие реакции. Это в основном реакции деструкции
и сшивания цепей. В промышленности эти процессы подавляют,
добавляя антиоксиданты, такие, как фенолы и амины; поэтому не-
которые полимеры «стабилизуют» сразу после синтеза. Это очень
важно для полидиенов, в,которых двойные связи особенно склонны
вызывать автоокисление полимера.
Кроме молекулярного кислорода окислительную деструкцию
могут вызывать и другие окислители. Окислением поливинилового
спирта иодной кислотой можно определить количество звеньев,
соединенных «голова к голове», так как расщепляются только
структуры 1,2-диола; поэтому вискозиметрические измерения дают
информацию о количестве таких структурных нерегулярностей
247
(опыт 5-18). Другим примером химической деструкции является
озонолиз натурального каучука с образованием у-кетовалерианово-
го альдегида и у-кетовалериановой кислоты.
Гидролитическая деструкция имеет важное значение для таких
полимеров, как полиэфиры, которые можно омылять, выделяя из
них исходные мономерные продукты. Ацетильные связи в синтети-
ческих (полиформальдегид) и природных (целлюлоза) полимерах
могут гидролизоваться под действием кислот. Стойкость полимера
по отношению к гидролизу зависит от строения полимера. Напри-
мер, омылять эфиры терефталевой кислоты нелегко, тогда как али-
фатические полиэфиры обычно гидролизуются очень просто.
Полиамиды проявляют большую, чем полиэфиры, стойкость к
гидролизу; их можно расщеплять, используя методы расщепления
полипептидов и белков.
Многие полимеры подвергаются механодеструкции в процессе
размола или вальцевания. При так называемой мастикации нату-
рального каучука происходит механически инициируемая, авто-
окислительная деструкция, которая приводит к снижению молеку-
лярной массы, что делает более удобным его переработку [23].
Опыт 5-14. Термическая деполимеризация поли-а-метилстирола и полиметил-
метакрилата
В круглодонную колбу емкостью 100 мл помещают 5 г полимера (точная
навеска). Колбу с помощью изогнутой под прямым углом стеклянной трубки
соединяют с двумя ловушками (—78°C), которые охлаждаются смесью сухого
льда с метанолом. Систему откачивают до остаточного давления 0,1 мм рт. ст.
и затем нагревают на металлической бане, температуру которой можно регулиро-
вать горелкой Бунзена с помощью ртутно-толуольного регулятора и газопрово-
да с точностью до ±3 °C. Прежде чем опустить колбу*, металлическую баню на-
гревают до 100 °C для плавления металла. После этого металлическую баню бы-
стро нагревают (в течение 6—8 мин) до требуемой температуры деполимериза-
ции. Температура деполимеризации поли-а-метилстирола 280 °C, а полиметилмет-
акрилата 330 °C. В зависимости от скорости разложения температуру поддер-
живают в течение 1—2 ч, непрерывно откачивая систему масляным насосом.
Опыт заканчивают, прекратив нагревание и охладив баню.
Как только мономер, затвердевший в охлаждающей ловушке, переходит
в жидкое состояние, в систему пускают воздух. Взвешивая остаток и (или) соб-
ранный мономер, определяют глубину деполимеризации. Идентифицируйте моно-
меры по их показателям преломления (для а-метилстирола п2£ = 1,5386, для
метилметакрилата п^~ 1,4140). Уменьшение молекулярной массы в результате
деполимеризации определяют по характеристической вязкости исходного и остав-
шегося полимеров в бензоле при 20 °C. Для сравнения проведите деструкцию
полистирола, при которой в этих условиях образуется очень мало мономера.
Опыт 5-15. Термическая деполимеризация полиоксиметилена [24]
Разложение полиоксиметилена можно проводить в приборе, описанном в
разделе 2.3.8.1. Каждый из указанных ниже образцов массой 100 мг помещают
в пробирки одинакового диаметра:
полиоксиметилен с гидроксильными концевыми группами (из опыта 3-35;
полимер из опыта 3-39 менее пригоден);
полиоксиметилен с ацетатными концевыми группами (из опытов 5-09А
и 5-09Б).
* Рекомендуется закоптить дно колбы на пламени горелки Бунзена перед
тем, как поместить ее в металлическую баню, во избежание растрескивания
стекла при извлечении колбы из бани.
248
Пробирки помещают в сосуд, который многократно откачивают и заполняют
чистым азотом. Сосуд нагревают до 190 °C на масляной бане или с помощью
воздушного термостата, пропуская через сосуд медленный ток азота. Пробирки
вынимают последовательно через каждый час и взвешивают. Строят график за-
висимости количества оставшегося вещества в % (масс.) от продолжительно-
сти термической обработки.
Термическое разложение полиоксиметилена при 190 °C начинается по кон-
цевым группам, тогда как окислительная и химическая (ацидолиз) деструкция
протекают по случайным местам цепи. Следовательно, при разложении полиок-
симетилена на воздухе или в присутствии сильных кислот (например, /г-толуо-
сульфокислоты или фосфорной кислоты) образцы с блокированными концевыми
группами также подвергаются разложению.
Опыт 5-16. Термическая деполимиризация поли-п-винилацетофенона
Круглодонную колбу емкостью 100 мл, соединенную с двумя охлаждаемыми
ловушками с помощью изогнутой под прямым углом стеклянной трубки, запол-
няют 5 г поли-п-винилацетофенона (опыт 5-05) и откачивают до остаточного
давления 1 мм рт. ст. Вторую ловушку охлаждают до —50 °C при помощи ох-
лаждающей смеси сухого льда с ацетоном. Сосуд нагревают по меньшей мере
до 300 °C на металлической бане. Нагревание продолжают до тех пор, пока не
прекратят отгоняться продукты пиролиза (около 30 мин). Продукты пиролиза
растворяют в небольшом количестве эфира и сушат над хлоридом кальция
около 1 ч. После фильтрования в раствор добавляют 1 % гидрохинона (относи-
тельно количества продуктов пиролиза) и эфир отгоняют с помощью водоструй-
ного насоса. Продукт перегоняют при 0,1—0,5 мм рт. ст. Отбирают промежуточ-
ную фракцию в виде прозрачного масла (т. кип. 75 °C), при охлаждении которо-
го образуются бесцветные кристаллы (т. пл. 33 °C). Выход составляет около 3 г
п-винилацетофенона.
Для идентификации продукта готовят его оксим. С этой целью смесь 0,5 г
свежеперегнанного п-винилацетофенона, 0,5 г солянокислого гидроксиламина,
5 мл воды, 10 мл этанола и 6 гранул твердого едкого натра (смесь должна да-
вать щелочную реакцию) кипятят в течение 15 мин. Потом добавляют 20 мм
50%-ного водного раствора спирта и смесь подкисляют разбавленной серной кис-
лотой до кислой реакции, что вызывает помутнение раствора. Кристаллизацию
оксима п-винилацетофенона проводят в холодильнике. После перекристаллиза-
ции из раствора спирта в воде температура плавления оксима равна 116—118 °C.
Небольшое количество п-винилацетофенона полимеризуют в атмосфере азо-
та в полимерной пробирке в присутствии 0,1% (мол.) азо-бис-изобутиронитрила
при 60 °C. Полимеризацию прекращают на низких степенях конверсии (спустя
6 ч), так как этот мономер проявляет склонность к побочным реакциям с обра-
зованием сшитых продуктов (реакции передачи). Для прекращения реакции
содержимое пробирки растворяют в 10-кратном количестве ацетона и раствор
декантируют. Полимер выделяют, прибавляя раствор к 10-кратному количеству
метанола, фильтруют и сушат в вакууме при 50 °C. Проверьте его раствори-
мость в растворителях, перечисленных в опыте 5-05.
Опыт 5-17. Термическое дегидрохлорирование поливинилхлорида
В 2 пробирки насыпают по 1 г поливинилхлорида (см. опыт 3-22). В одну
из пробирок добавляют 100 мг стеарата свинца в качестве стабилизатора {хоро-
шо перемешайте в ступке). Пробирки неплотно закрывают корковыми пробками,
к нижним концам которых прикреплена полоска влажной индикаторной бумаги
длиной 4 см. Пробирки нагревают до 170—175 °C в течение 10 мин в стакане
с бесцветным силиконовым маслом. Если индикаторная бумага, помещенная над
полимером, стабилизированным стеаратом свинца, практически не изменяется, то
над нестабилизированным образцом она обесцвечивается вследствие выделения
НС1 из полимера. Более того, стабилизированный образец только немного тем-
неет, тогда как нестабилизированный полимер превращается из розовато-красно-
го в коричневый. Выделение НС1 можно также наблюдать, пропуская пары над
стаканом с аммиаком или пропуская их в подкисленный раствор нитрата се-
ребра.
Опыт 5-18. Окислительная деструкция поливинилового спирта под действием
иодной кислоты
249
Навеску 2 г поливинилового спирта (опыт 5-01) растворяют в 70 мл дистил-
лированной воды в стакане емкостью 250 мл; для ускорения растворения смесь
перемешивают стеклянной палочкой и немного подогревают. Как только обра-
зуется гомогенный раствор, его охлаждают до комнатной температуры и филь-
труют на стеклянном фильтре в мерную колбу на 10 мл для отделения твердых
частиц. Стакан ополаскивают водой и промывную воду фильтруют в мерную
колбу. После термостатирования колбы при 25 °C раствор разбавляют водой до
метки. Готовят раствор 1,7 г иодной кислоты (Н1О4-2Н2О = Н5Юб) в 45 мл ди-
стиллированной воды, фильтруют его в мерную колбу емкостью 50 мл, а затем
при 25 °C, разбавляют водой до метки.
После окислительной деструкции определяют вязкость в вискозиметре
Оствальда (диаметр капилляра 0,4 мм) при 25 °C. Ниже перечислен порядок
проведения всех необходимых измерений.
1. В мерной колбе на 10 мл готовят раствор 5 мл иодной кислоты в 5 мл
воды, фильтруют и измеряют время истечения этого раствора to при 25 °C.
2. Определяют время истечения 5 мл раствора поливинилового спирта
с 5 мл дистиллированной воды /.
3. Смешивают 5 мл раствора поливинилового спирта с 5 мл иодной кисло-
ты и определяют время истечения полученного раствора ta сразу же после при-
готовления. Затем через короткие промежутки времени повторяют измерения ta
до получения постоянного значения.
Если предположить, что иодная кислота не влияет на вязкость раствора
полимера, удельная вязкость неразложившегося полимера определяется как
t-tQ
- ta
а удельная вязкость разложившегося полимера
Рассчитайте характеристическую вязкость и средние молекулярные массы
полимеров по уравнению Шульца — Блашке (k = 0,27; см. раздел 2.3.2.1). На
основании полученных результатов определите содержание 1,2-диоловых групп
в исходном полимере.
Опыт 5-19. Гидролитическая деструкция алифатического полиэфира
В мерной колбе емкостью 100 мл растворяют 1 г линейного алифатического
полиэфира (например, полиэфира янтарной кислоты и гександиола, опыт 4-02)
в безводном тетрагидрофуроне и раствор разбавляют до метки при 30 °C. К по-
ловине этого раствора прибавляют 2 мл 30%-ной серной кислоты и оба раство-
ра помещают на водяную баню при 30 °C. Вязкость растворов измеряют сразу
же после приготовления (измерения повторяют несколько раз с интервалами
в 1 ч) в вискозиметре Оствальда (диаметр капилляра 0,3 мм) при 30 °C; для
этого в вискозиметр пипеткой наливают пробы раствора по 3 мл. Колбы хра-
нят при 30 °C в течение ночи и измерения продолжают на следующий день до
тех пор, пока время истечения не перестанет изменяться (как правило, через
20 ч). Вязкость образца, не содержащего серной кислоты, остается неизменной
в течение всего опыта.
Строят графики зависимости удельной вязкости (или времени истечения) от
времени. Время истечения растворителя to измеряют для тетрагидрофуранового
раствора серной кислоты (50 мл тетрагидрофурана +2 мл 30%-ной H2SO4), так
как вязкость тетрагидрофурана значительно увеличивается при добавлении
H2SO4. После вискозиметрических измерений по 20 мл растворов гидролизован-
ного и негидролизованного полимеров добавляют к 200 мл метанола. Если
гидролиз прошел полностью, полимер не высаживается.
Опыт 5-20. Гидролитическая деструкция целлюлозы и разделение продуктов
гидролиза методом бумажной хроматографии
Навеску 100 мг -целлюлозы (например, в виде кусочков фильтровальной
бумаги) хорошо перемешивают с 1 мл охлажденной 72 %-ной серной кислоты
250
с помощью стеклянной палочки. Полученную смесь хранят при О °C в течение
ночи. Массу разбавляют 1 мл 25%-ной серной кислоты и выдерживают при 50 °C
в течение 2 ч. После охлаждения добавляют 40 мл ледяной воды и кипятят
1 ч.
Для нейтрализации* продукт гидролиза пропускают через колонку (дли-
на 15 см, а ширина 1 см) с анионитом (например, можно использовать анионит
из опыта 5-13). После этого колонку промывают 50 мл воды. Последние капли
промывных вод должны давать отрицательную реакцию на сахар при нанесении
на хроматографическую бумагу и последующем проявлении анилинфтал атом
после высыхания бумаги. В этом случае раствор концентрируют, отгоняя раство-
ритель в вакууме, до объема 5 мл.
Разделение продуктов гидролиза проводят хроматографически на бумаге
[25]. Для того чтобы доказать селективность метода, бумагу пропитывают рас-
твором кислого фосфорнокислого натрия. С этой целью готовят раствор 15,6 г
(0,1 моля) NaH2PO4-2H2O в 1 л воды. В мелкую чашку наливают 100 мл этого
раствора и медленно пропитывают им бумагу (для каждого куска бумаги
нужно брать по 100 мл свежего раствора, так как раствор быстро впитывается).
Листы бумаги развешивают в линию для сушки на воздухе. Сухие листы режут
на куски длиной 60 см и шириной 10 см. Базисную линию слегка наносят каран-
дашом на уровне 8 см от верхней части длинного листа (необходимо учитывать
направление потока, указанное на листах). С помощью пипетки по 5 мм3 рас-
твора наносят на базовую линию (желательно удалить растворитель горячим
воздухом сразу после нанесения образцов на бумагу для предотвращения не-
правильного распространения пятен):
1) 5 мм3 продукта гидролиза (относительно 0,1 мг гидролизованной целлю-
лозы) ;
2) 5 мм3 раствора UOO мг глюкозы в 5 мл воды (относительно 0,1 мг
глюкозы);
3) 5 мм3 раствора 5 мг ксилозы в 5 мл воды (относительно 0,005 мг
ксилозы);
4) 5 мм3 раствора 5 мг маннозы в 5 мл воды (относительно 0,005 мг ман-
нозы).
Разделение продуктов гидролиза проводят методом распределительной хро-
матографии со смесью бутанола, ацетона и воды (40 : 50 : 10). Для этого в стек-
лянный сосуд наливают растворитель так, чтобы закрыть дно. После того как
желоб, закрепленный в верхней части сосуда, заполнится наполовину, закрытый
сосуд оставляют на несколько часов для насыщения воздуха внутри сосуда па-
рами растворителя. Только после этого лист бумаги помещают в сосуд, погрузив
верхний край листа в желоб и придерживая его тяжелой стеклянной палочкой
так, чтобы базисная линия находилась на 3—5 см ниже края желоба. Сосуд за-
крывают крышкой, и спустя 2—3 ч фронт растворителя должен пройти четверть
бумажной полосы. (В зависимости от конструкции сосуда и желоба хромато-
граммы будут несколько различаться, поэтому оптимальное время распростране-
ния фронта растворителя можно установить, только повторяя опыт.) Бумагу вы-
нимают и сушат на воздухе. Затем обрабатывают анилинфталатом и после
сушки на воздухе в течение 10 мин ее помещают в сушильный шкаф при 100 °C
еще на 10 мин для проявления сахарида в виде фиолетово-коричневых пятен.
Этот простой метод дает лишь качественную характеристику состава целлю-
лозного гидролизата. Однако по размеру и интенсивности пятен можно устано-
вить, что используемая целлюлоза содержит более 90% звеньев глюкозы и что
другим важным копмонентом является ксилоза.
Количественные данные с помощью указанного метода можно получить в
других условиях с помощью фотометрического анализа бумажных хроматограмм
в УФ-лучах [26].
* Раствор нельзя нейтрализовать карбонатами щелочноземельных металлов
во избежание превращения глюкозы в маннозу, которое возможно вследствие
образования комплексов ионов щелочноземельных металлов с маннозой.
251
Литература
1. Kern W., Schulz R. C., Angew. Chem., 1957, Bd. 69, S. 153.
2. Kern W., Chemiker-Ztg., 1958, Bd. 82, S. 71.
3. Kern W., Schulz R. C., Braun D., Chemiker-Ztg., Apparatur, 1960,
Bd. 84, S. 385.
4. S m e t s G., Macromol. Chem., 1959, Bd. 34, S. 190.
5. Schneider P., Houben-Weyl, v. 14/2, 1963, v. 14/2, p. 661.
6. Husemann E., Werner R., Houben-Weyl, 1963, v. 14/2, p. 862.
7. Kern W., Cherdron H., Jaacks V., Angew. Chem., 1963, Bd. 73,
S. 177.
8. В r a u n i t z e r G., Angew. Chem., 1957, Bd. 69, S. 189.
9. Gattermann-Wieland, Die Praxis des organischen Chemikers, De Gruy-
ter und Co., Berlin.
10. De Boer Th. J., Backer H. J., Org. Slynth. Coll., 1963, Bd. 4, S. 250.
11. Griessbach R., Austauschadsorption in Theorie und Praxis, Akademie Ver-
lag, Berlin.
12. Heffrich F., lonenaustauscher, 1959, Bd. I, Verlag Chemie GmbH, Wein-
heim/Bergstrafie.
13. Dor fn er K-, lonenaustauscher, 1970, eed. Walter de Gruyter und Co., Ber-
lin.
14. Helfrich F., Angew. Chem., 1954, Bd. 66, S. 241.
15. Inezedy J., Analytische Anwendungen von lonenaustauschern, 1964, Verlag
der Ungarischen Akademie der Wissenschaften, Budapest.
16. Inezedy J., Kun in R., Ion Exchange Resins, 1958, John Wiley and Sons,
N. Y.
17. Osborn G. H., Synthetic Ion Exchangers, 1961, Chapman and Hall, Ltd.,
London.
18. Грасс и H. Химия процессов деструкции полимеров. М., Издатинлит, 1959.
214 с.
19. М a do г sky S. L., Thermal Degradation of Organic Polymers, 1964, Polymer
Preprints, p. 7.
20. J a s c h i n g W., Kunststoffe, 1962, Bd. 52, S. 458.
21. Rieche A., Kunststoffe, 1964, Bd. 54, S. 428.
22. D u 1 о g L., Radlmann E., Kern W., Makromol. Chem., 1963, Bd. 60,
S. 1; 1964, Bd. 80, S. 67.
23. Watson W. F., Makromol. Chem., 1959, Bd. 34, S. 240.
24. Kern W., Cherdron H., Makromol. Chem., 1960, Bd. 40, S. 101.
25. G r a m e r F., Papierchromatographie, 1962, 5th ed., Verlag Chemie, Weinheim,
196.
26. J ay me G., Knoll e H., Z. Anal. Chem., 1960/1961, Bd. 178, S. 84.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автоокисление 247
Автоускорение 130
Агрегатные состояния полимеров 32
Азеотропная очистка 46
Азеотропная сополимеризация 171, 178
Алкидные смолы 201, 202
Аморфное состояние 32
Аморфные полимеры 38, 89
Аниониты 243, 245
Анионная полимеризация 16, 18—23. 159—
161, 218
Анионная сополимеризация 175
Анионоактивные эмульгаторы 57
Антиоксиданты 247
Аппаратура
для очистки азота 45
для полимеризации 155, 158
для полиреакций 48 сл.
для разложения полимеров 92
для сушки азота 45
для формования волокна 102
Ассоциация 77
Атактические полимеры 30, 31
Ацетаты целлюлозы 69
Гетероцепные полимеры 26
Гидроксильное число 196
Гидролитическая деструкция 248, 250
«Голова — хвост» 28
Гомофазная полимеризация 52, 112 сл.
Гуттаперча 29
Дальний порядок 32, 34
Дегидрогенизационная поликонденсация
223 сл.
Деполимеризация 247—249
Деструкция полимеров 91, 246 сл.
Деформации 97 сл.
Дилатометр 128
Динамические механические испытания
99 сл.
Диспропорционирование 112, 113, 115
Дифференциальная кривая распределения
85, 86
Дуропласты 26
«Живущие» полимеры 139, 142, 148, 183
Бахромчатые мицеллы 33, 34
Бисерная полимеризация 56
Ближний порядок 32, 39, 40
Блок-сополимеризация 182 сл.
Блок-сополимеры 31, 101, 183, 184
Блочная поликонденсация 52
Блочная полимеризация 51
Боденштейна принцип 113
Бойера—Бимана правило 38, 87
Вика метод 88
Вискозиметрия 73 сл.
Вискозиметры 89
Оствальда 77, 78
Внутренняя пластификация 173, 180
Вспененные полимеры 107 сл.
Вулканизация 40, 442
Выделение полимеров 64 сл.
Высококристаллическое состояние 32
Высокомолекулярные соединения
синтез 15 сл., 44 сл.
строение 17 сл.
поликонденсация 188 сл.
полимеризация 112 сл.
Высокоэластические свойства 39, 40
расплавов полимеров 89 сл.
Вязкость 39, 73, 103
Вязкоупругие жидкости 37
Вязкоупругие свойства 37
Гель-фильтрационная хроматография 135
Гель-эффект 115, 130
Гетерогенная полимеризация 49
Гетерофазная полимеризация 52
«Замороженное состояние» 32, 39
Зародышеобразование 35
Защитные коллоиды 57
Звенья 26
Измельчение полимеров 104
Изотактические полимеры 30, 151, 152, 156
Изоцианаты 161
Ингибирование 137
Ингибиторы 48, 117
Индекс расплава 89
Инициаторы 16, 57, 139
Инициирование 112, 139, 141
азосоединениями 126 сл.
катализаторами Циглера—Натта 154
кислотами Льюиса 144 сл.
металлоорганическими соединениями
148
окислительно-восстановительными сис-
темами 133 сл.
перекисными соединениями 119 сл.
Интегральная кривая распределения 85, 86
Иониты 243 сл.
Ионная полимеризация 16, 49, 138 сл.
по С=С-связи 139 сл.
по С=О-связи 159 сл.
по Ы=С-связи 161 сл.
Ионная сополимеризация 175
Ионообменные полимеры 238, 243 сл.
Капиллярный вискозиметр 89
Карбамидоформальдегидные олигомеры 25,
253
Карбоцепные полимеры 17
Катализаторы 112, 143, 154
Катиониты 243, 245
Катионная полимеризация 16, 18—23, 147,
159, 218
Катионная сополимеризация 176, 177
Катионоактивные эмульгаторы 57
Кислотное число 196
Константы сополимеризации 170, 174, 176,
177
Конформации макромолекул 32 сл., 35
Концевые группы 80 сл.
Коэффициент
механических потерь 100
объемного распределения 84
осаждения 215
термического расширения 128
Крахмал 69
Кремнийорганические полимеры 217 сл.
Кривые
нагрузка — удлинение 97, 98
текучести 39
Кристаллизация 46
Кристаллиты 33
Кристаллические полимеры 38, 100
Критическая концентрация мицеллообразо-
вания 57
Куна уравнение 75
Линейные полимеры 27, 220, 222
Логарифмический декремент 100
Лучепреломление двойное 88
Макроброуновское движение 36, 100
Макромолекулы 15
Мартенса метод 88
Мастикация 248
Меламиноформальдегидные олигомеры 25,
211, 213
Метод
Бринелля 103
«Механическая абсорбция» 100
«Механическая спектроскопия» 100
Механические методы 96 сл.
Механические свойства 38
Механодеструкция 248
Микроброуновское движение 36, 100
Мицеллы 57
Модельные соединения 61
Модуль
сдвига 99, 100
упругости 38, 40, 99
Молекулярная масса 55, 57, 58, 71 сл., 149,
189
определение 73, 80
регулирование 131
Молекулярно-массовое распределение 81,
84, 131
Молекулярные коллоиды 41
Монокристаллы 35
Мономеры
очистка 46
для поликонденсации 24 сл.
для полимеризации 18 сл.
хранение 46
Набухание 68
Найлон 203—206
Натуральный каучук 29
Неионогенные эмульгаторы 57
Ненасыщенные полиэфиры 199, 200
Новолаки 208
Ньютоновские жидкости 39
Обезвоживание 46
Обменная емкость 244, 246
Обменные реакции 189, 193
Обрыв цепи 112, 113, 140, 141
Окислительная деструкция 93, 247, 249
Окислительно-восстановительное иницииро-
вание 120, 133 сл.
Олигомеризация 16
Оптическая изомерия 30, 31
Оптические методы 93
Ориентационное лучепреломление 90
Ориентация 32, 39, 89
Осадители 69
Осадительная полимеризация 52, 160, 161,
206
Осаждение 65, 81
Осмометрия 72
Остаточная деформация 37, 40
Оствальда вискозиметр 77, 78
Отверждение 199—202, 212, 214, 219, 220
Отгонка 65
Очистка
мономеров 46 сл.
полимеров 64 сл., 66 сл.
Пенопласты 107
Пенополистирол 122
Передача цепи 53, ИЗ, 115, 116, 120, 131
139—141
Перекисные соединения 119
Переработка 104 сл.
расплавов 104
растворов 106
Пикнометрический метод 90
Плавление полимеров 36 сл.
Пластификаторы 101, 102, 104
Пластификация 173
внешняя 104
внутренняя 180
Пленки 106
Плотность полимеров 90
Ползучесть 99
Полиаддукты 17
Полиакриламид 69, 135, 136
Полиакрилаты 69
Полиакриловая кислота 69
Полиакрилонитрил 69, 77, 125, 137
Полиалкиленсульфиды 25, 215—217
Полиамиды 24, 70, 77, 203—206, 221
Полиангидриды 25
Полиацетали 60
Полибензимидазолы 222
Полибутадиен 69, 77, 158
Полибутен-1 69
Поливинилацетат 69, 77, 124, 126, 131, 132
Поливинилбутираль 239
Поливинилизобутиловый эфир 69, 146
Поливинилметилкетон 69
Поливиниловый спирт 69, 77, 239, 249
Поливинилсульфоновая кислота 69
Поливинилфторид 69
Поливинилхлорид 102, 138, 249
Полидиены 30, 144, 153
Полидисперсность 73, 143
Полиизобутилен 69, 77, 146
Полиизопрен 69, 77, 137, 152
Поликапроамид 167, 168, 204
Поликарбонат 77, 197, 198
Поликонденсационное равновесие 191
Поликонденсация 16, 17, 49, 51, 188 сл.
блочная 52
на границе раздела фаз 54, 206, 207
в дисперсии 54, 56
в расплаве 52
в растворе 53
дегидрогенизационная 223 сл.
карбамида с формальдегидом 213
меламина с формальдегидом 214
регулирование 59
фенола с формальдегидом 208, 210, 211
и циклополиконденсация 222
254
Прлимераналогичные превращения 17, 59,
237
Полимеризация 16
акриламида 135, 136
акрилонитрила 124
ацеталей 164 сл.
блочная 51
бутадиена 158
н-бутилизоцианата 162
в водной среде 49
винилацетата 53, 123—125
винилизобутилового эфира 146
винилхлорида 138
изобутилена 146
изопрена 137, 152
ионная см. Ионная полимеризация
на катализаторах Циглера—Натта 154
лактамов 167
„ лактонов 166
метакриловой кислоты 126
метилметакрилата 52, 123, 130, 152
а-метилстирола 147, 148
олигосилоксана 219
и поликонденсация 220
пропилена 156
простых эфиров 162
с раскрытием цикла 162 сл., 219
в растворе 52
регулирование 58
стереоспецифическая 139
стирола 52, 121, 122, 127, 130, 137, 150,
157
в суспензии 56
в твердой фазе 52
тетрагидрофурана 164
триоксана 165
формальдегида 159, 160
этилена 52, 155
Полиметакрилаты 69
Полиметакриловая кислота 126, 240
Полиметилметакрилат 77, 123, 130, 152, 248
Поли-а-метилстирол 77, 147—149, 248
Полиморфизм 35
Полимочевины 24, 26
Полиоксиметилен 36, 70, 99, 161, 242, 248
Полиприсоединение 16, 17
в дисперсиях 56
Полипропилен 36, 69, 101, 156
Полиреакции
в дисперсных системах 55
в растворе 52 сл.
в суспензиях 56 сл.
температурный режим 51
в эмульсии 56 сл.
Полисилоксаны 25, 70, 217
Полистирол 27, 28, 36. 69, 77, 85, 121, 122,
127, 130, 137, 150, 151, 157, 245, 246
Политетрагидрофуран 70, 164
Политетрафторэтилен 69
Политиоэфиры 26
Полиуретаны 24, 26, 70, 206, 207, 225
Полифениленовые эфиры 223, 224
Полифосфаты 25
Поли-3,3-бис-хлорметилоксациклобутан 163
Полиэтилен 36, 69, 101, 155, 185
Полиэтиленоксид 70, 71, 163
Полиэтилентерефталат 36, 70, 77
Полиэфиры 24, 70, 195—198, 250
диолов и дикарбоновых кислот 195, 197
оксикарбоновых кислот 194
Порообразователи 107
Постконденсация 52
Правило Бойера—Бимана 38, 87
Предел текучести 98
Предельная температура 143
Прерывание полимеризации 58 сл.
Прессование 105
Приведенная вязкость 73
Приведенное число вязкости 74
Привитые сополимеры 31, 183
Примеси 46, 146
Принцип Боденштейна 113
«Прядение» 106, 137
Радикальная полимеризация 16, 18—23, 49
Разветвленные полимеры 27
Разрушающее напряжение 97, 99
Расплавы полимеров 37
Растворение 68 сл., 81, 82
Растворители 6, 53, 68—70, 77, 79
Растворы полимеров 41 сл.
Реактопласты 26, 98
Регулирование полимеризации 58 сл.
Регулярные сополимеры 182
Регуляторы 58, 116
Резины 40
Резиты 208
Резолы 208, 210
Рекомбинация 112, 113, 115, 116
Релаксация 37, 39, 40, 99
Рост цепи 112, 113, 140, 141
Ротационный вискозиметр 89
Светорассеяние 72
Свободнорадикальная гомополимеризация
112 сл.
Свободнорадикальная сополимеризация
175, 176, 178—182
Сетчатые полимеры 27
Силиконы 217, 220
Синдиотактические полимеры 30, 152
Складчатые пластины 33, 34
Сложные полиэфиры 194 сл.
Слоистый пластик 215
Сокатализаторы 48, 144
Сольватация 77
Сополиконденсация 31
Сополимеризация 169 сл.
бутадиена с акрилонитрилом 179
винилхлорида с винилацетатом 180
стирола с акрилонитрилом 178
----бутадиеном 179, 242
------- дивинилбензолом 181
----малеиновым ангидридом 181
----метилметакрилатом 175
— — а-хлорстиролом 176, 177
Сополимеры 31, 95, 101, 442
Сополиприсоединение 31
Спектроскопические методы 93, 153
Стабилизаторы 48, 67
Статистическая сополимеризация 169 сл.
Статистические сополимеры 31
Стеклообразное состояние 37
Степень
диссоциации 75
кристалличности 90
полимеризации 115, 142, 189, 190
превращения 63, 64
Стереоблок-сополимеры 31
Стереоизомерия 27, 29
Стереорегулярные полимеры 143
Стереоспецифическая полимеризация 139,
143, 150, 152, 156—158
Структурная изомерия 27, 28, 31
Структурные единицы 27
Ступенчатая поликонденсация 53
Ступенчатая полимеризация 17, 49, 51, 168,
225 сл.
и поликонденсация 221
Сублимация 46
Суспензионная полимеризация 55, 125
Сушка 45
полимеров 66 сл.
Сферолиты 33, 34
Сшивание 27, 40, 199—220, 209, 212, 214, 219
Твердость 103
Твердые полимеры 32
255
Текучесть 38, 39, 98
Теломеризация 53, 117
Температура
кристаллизации 33
плавления 88, 100
предельная 143
размягчения 37, 87, 100
стеклования 36, 40, 87, 100
Термическая деструкция полимеров 91, 246,
Термомеханический метод 88
Термопласты 26, 98, 99
Торсионный модуль 99, 100
Трехмерная сополимеризация 174, 181
Троммсдорфа эффект 115, 130
Тэта-температура 77
Ударная вязкость 102
по Изоду 103
по Шарпи 103
Удельная вязкость 73
Ультрацентрифугирование 72
Упругость полимеров 37, 40
Уравнение
Куна 75
состава сополимера 173
Штаудингера 75
Шульца—Блашке 75
Фракционирование полимеров 81 сл., 135
осаждением 81
растворением 82
Фракционная очистка 46
Фракционная экстракция 131
Функциональность 15
Характеристическая вязкость 74
«Хвост — хвост» 28
Химическая деструкция полимеров 93
Хранение мономеров 46
Хроматография 46, 93, 135
Хрупкость 103
Целлюлоза и ее производные 69, 238, 240,
241, 244, 250
Центрифугирование 65
Циклополиконденсация 220 сл.
Частично-кристаллические полимеры 32,
100, 101
Чередующиеся сополимеры 31, 172, 181, 182
Шоттен—Баумана реакция 54
Штаудингера уравнение 75
Шульца—Блашке уравнение 75
Фенолоформальдегидные олигомеры 25,
207, 245
Фенольные смолы 208
Фибриллы 34, 35
Фильтрование 65
Флеш-полимеризация 146, 169
Флотационный метод 90
Формование из расплава 105
Эбонит 40
Экстрактивная дистилляция 46
Эластомеры 26, 40
Эмульгаторы 57
Эмульсионная полимеризация 55, 122, 137
Эпоксидные смолы 26
Эфиры целлюлозы 69
Эффект Троммсдорфа 115, 130
Эффективность инициирования 113, 134
ДИТРИХ БРАУН,
ГАРОЛЬД ШЕРДРОН,
ВЕРНЕР КЕРН
ПРАКТИЧЕСКОЕ РУКОВОДСТВО
ПО СИНТЕЗУ И ИССЛЕДОВАНИЮ
СВОЙСТВ ПОЛИМЕРОВ
Редактор Рогайлина А. А.
Художник Михайлов А. Я. -
Художественный редактор Носов Н. В.
Технический редактор Скитина В. М.
Корректор Голубева О. И.
Сдано в наб. 21/VI 1976 г. Подп. в печ. 20/VIII 1976 г. Формат
бумаги 60X90V16. Бумага тип. № 2. Усл. печ. л. 16.
Уч.-изд. л. 20,58. Тираж 4800 экз. Зак. 732. Изд. № 935.
Цена 1 р. 19 к.
Издательство «Химия». 107076, Москва, Стромынка, 13.
Московская типография № 11 Союзполиграфпрома при Госу-
дарственном комитете Совета Министров СССР по делам изда-
тельств, полиграфии и книжной торговли. Москва, 113105, Нага-
тинская ул., д. 1.
256