/




















































































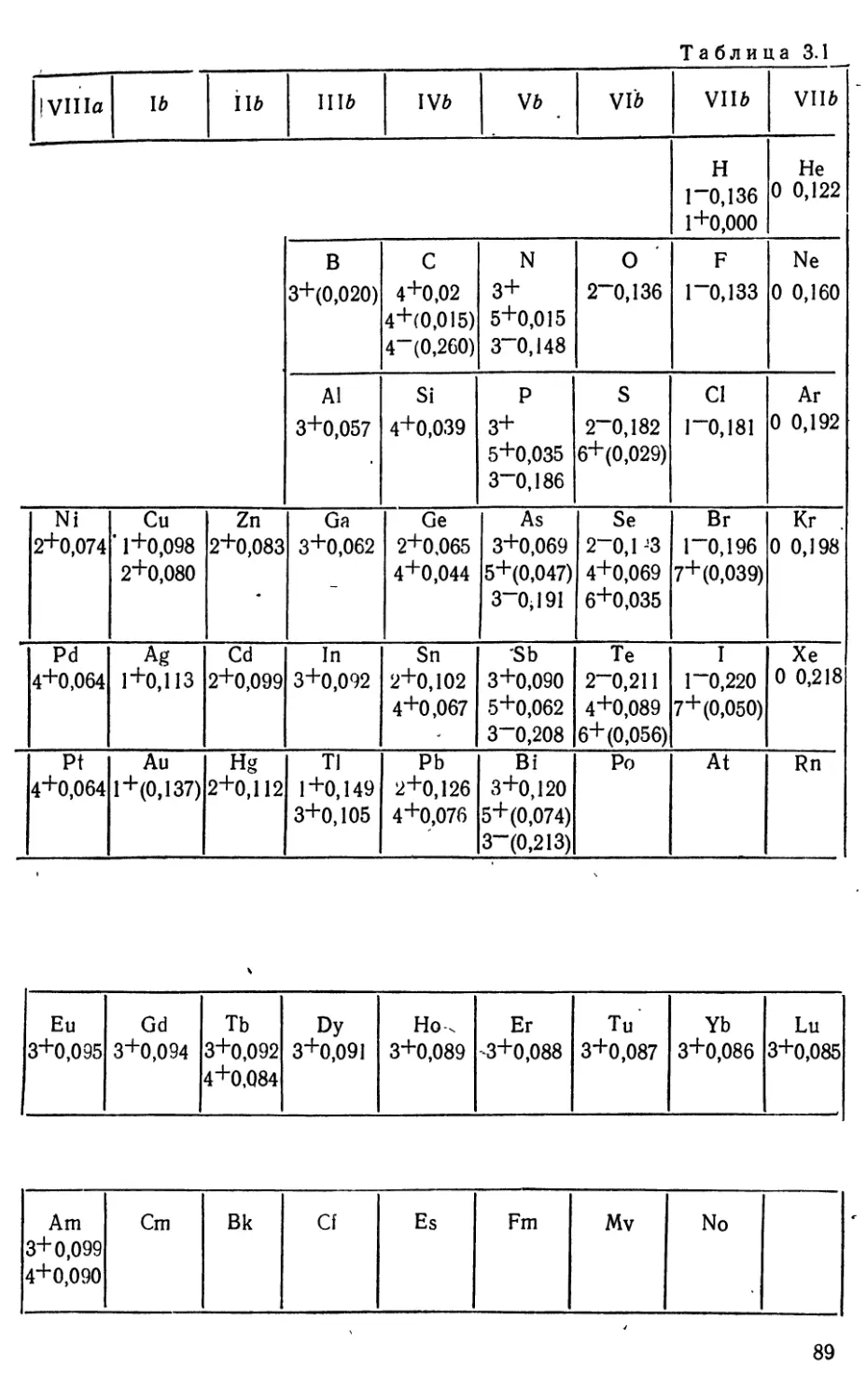










































































































Текст
3
щг метрология
Е П. Комарь
ГЕТЕРОГЕННЫЕ
ИОННЫЕ
РАВНОВЕСИЯ
УДК 543/545 ^ 541.122
Химическая метрология. Гетеро1енкые ионные равновесия.
Комарь Н. П.—Харьков: Вища школа. Изд-во при
Карьк. ун-те, 1984.—208 с.
Монография является второй из цикла книг Н. П. Кома-
ря, издаваемых под названием «Химическая метрология», в
которой теории ионных равновесий отведено важное место.
Она посвящена методам расчета гетерогенных ионных
равновесий в системах жидкая фаза — твердая фаза и
жидка я фаза—жидкая фаза. Рассмотрены процессы образования
твердой фазы из раствора, соосаждения, адсорбции,
промывания осадков. Особое внимание уделено вопросам
распределения веществ между жидкими фазами и
буферным системам. На большом числе конкретных примеров
расчета систем, используемых в химическом анализе,
показана необходимость последовательного метрологического
подхода к химико-аналитическим проблемам.
Для химиков-аналитиков, научных работников и
специалистов.
Нормативные материалы приведены по состоянию на
I января 1983 г.
Табл. 21. Ил. П. Библиогр.: 69 назв.
Научный редактор д-р хим. наук, проф. Л. П. Адамович
Рецензенты: чл.-кор. АН УССР, д-р хим. наук, проф.
Б. Л. НаваренкОу д-р хим. наук, проф. Л. Я. Бусев
Редакция естественнонаучной литературы
Зав. редакцией Е, П. Иваи^енко
^ М226(04)- 84
© Издательское
объединение
«Вища школа», 1984
Настоящая монография является непосредственным-
продолжением изданной ранее книги Н. П. Комаря «Химическая
метрология. Гомогенные ионные равновесия» [21]. В них изложены все
основные понятия, примеры и приложения теории ионных
равновесий. Монографии были задуманы автором как первые из
цикла книг, объединенных общим названием «Химическая метрология».
Название цикла отражает основную мысль Н. П. Комаря о том,
что аналитическая химия как наука об измерении химической
формы движения материи принадлежит и химии, и метрологии.
Поэтому она должна учитывать и использовать все
специфические законы химии, а также общие положения и приемы
метрологии. План этого- цикла изложен в предисловии к книге [21].-
К сожалению, Николай Петрович Комарь реализовал этот план
лишь частично, а завершенные рукописи не были опубликованы
п(.и жизни автора.
В редактировании и подготовке к печати настоящей
монографии приняли участие ученики Н. П. Комаря — сотрудники
кафедры химической метрологии Харьковского университета
и аналитической лаборатории ВНИИ монокристаллов (Харьков)
А. Б. Бланк, Т. В. Васильева, С. И. Вовк, Л. Л. Лукацкая,
Л. Е. Никишина, И. Г. Перьков, М. И. Рубцов.
Принципы редактирования этой книги были такими же, как
и монографии [21], и подробно изложены в предисловии
редактора к ней. Там же обсуждены применяемые единицы физических
и химических величин, а также некоторые специальные
обозначения.
В большинстве расчетных примеров, . приведенных в обеих
книгах по теории ионных равновесий, автор использовал
константы равновесий из учебных таблиц, составленных А. А. Буга-
евским [5]. Благодаря любезному согласию последнего, они
частично включены в настоящую монографию в виде приложения.
Все вопросы, замечания и пожелания, которые возникнут
у читателя при знакомстве с монографией, просим направлять
по адресу:
310077, Харьков-77, пл. Дзержинского, 4, университет,
химический факультет, кафедра химической метрологии.
Л. П. Адамович, д-р хим. наук
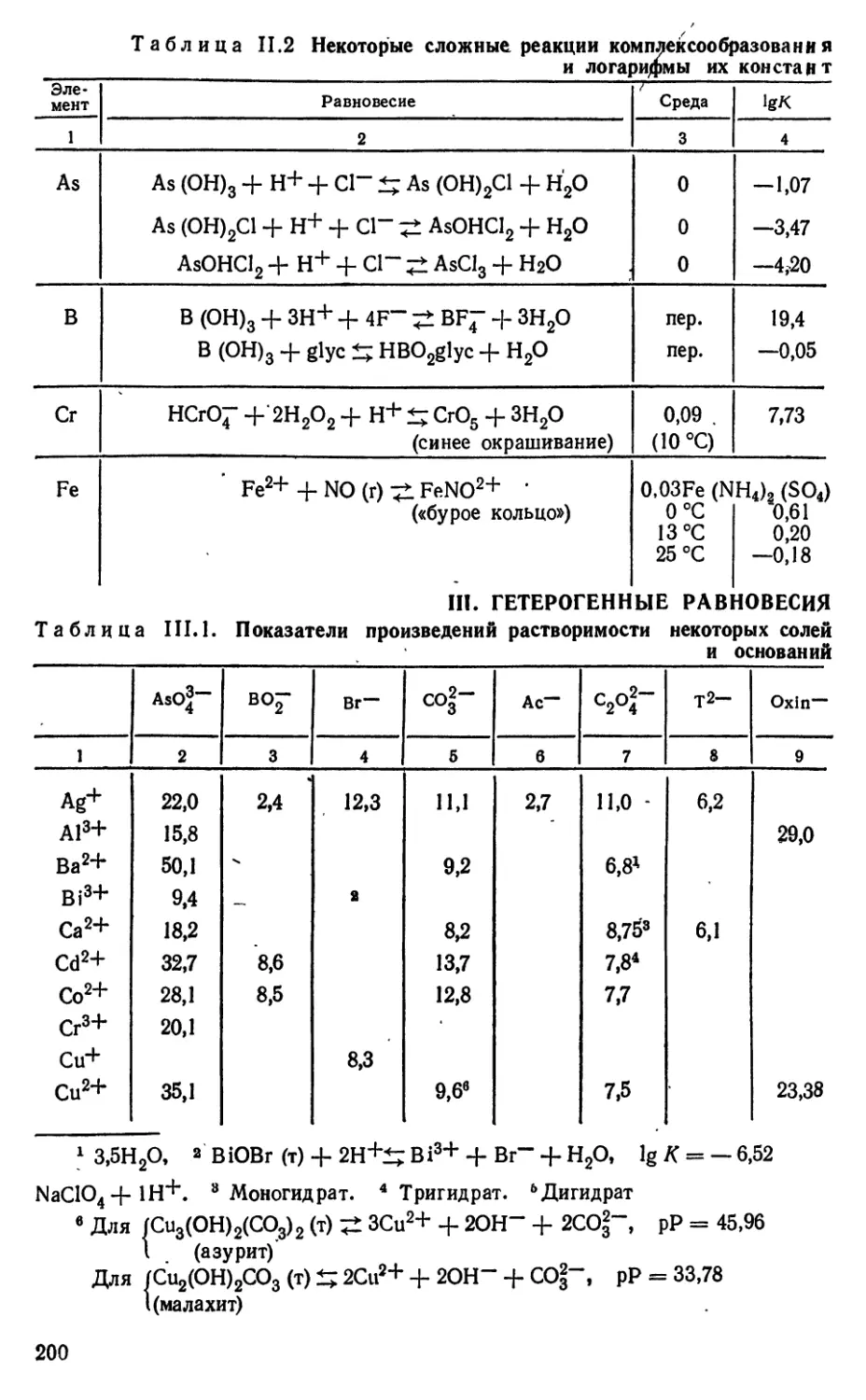
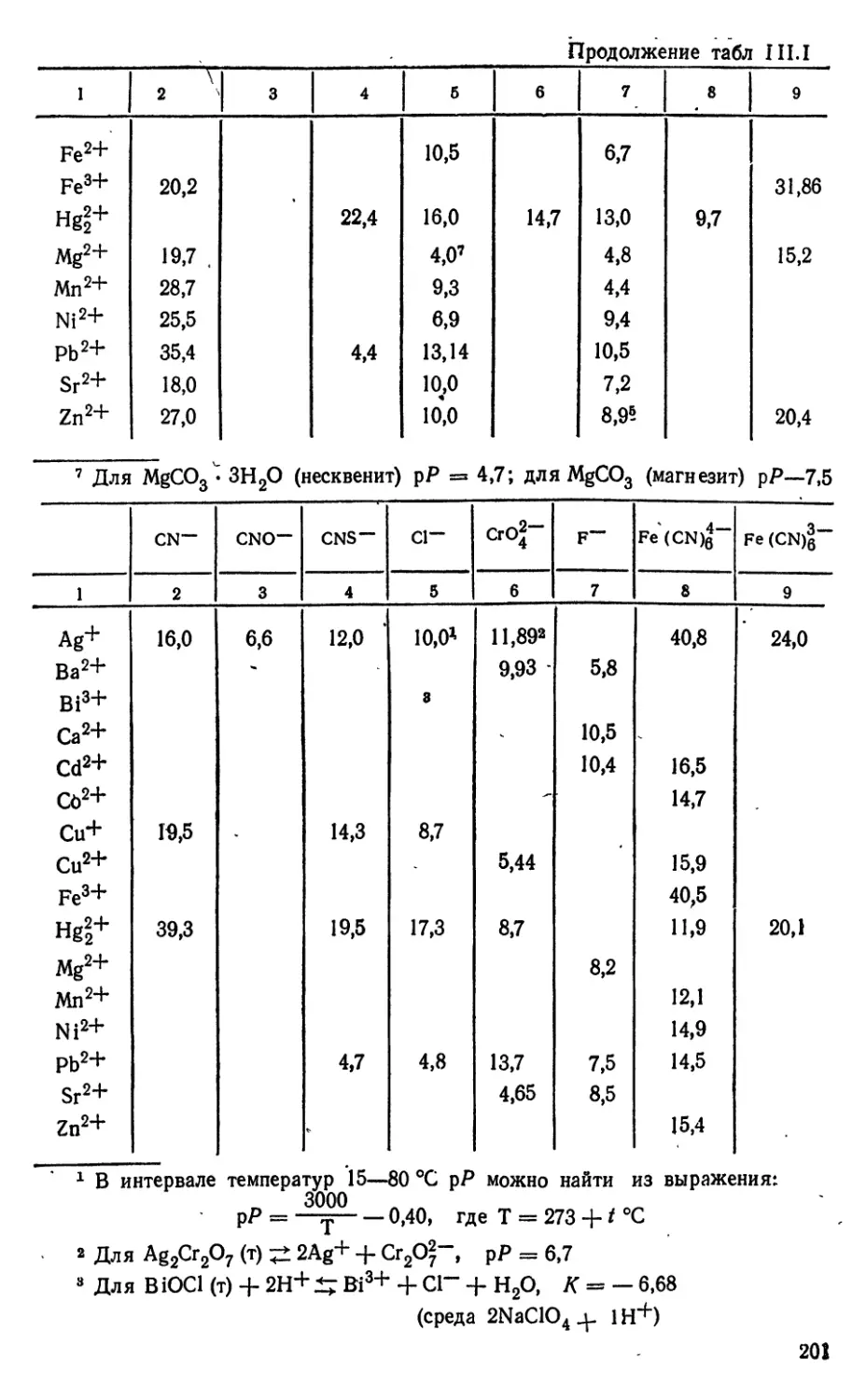
I РАСТВОРИМОСТЬ и ПРОИЗВЕДЕНИЕ
РАСТВОРИМОСТИ
1.1 Общие сведения о растворимости
При растворении малорастворимого вещества lAaA^, в воде ионы
МУ"^ и А^'", из которых состоит кристаллическая решетка, гидра-
тируются, т. е. превращаются в акво-комплексы и переходят в
раствор. По мере того как активности ионов М^^+ и А^'- в растворе
возрастают, все в большей степени начинает проявляться обратный
процесс дегидратации этих ионов и врастания их в решетку.
Равновесие между твердым веществом и его ионами,
находящимися в растворе, устанавливается, когда скорости перехода
твердого вещества в раствор и обратного
процесса—"кристаллизации станут одинаковыми. С этого момента
МаА,(т) + (ап^ + 1ЛП2) Н2О ^ аМ (0Н2)Й+ + [хА mfnT (1.1)
'и раствор считают насыщенным относительно твердой фазы МаА,^.
Концентрацию такого насыщенного раствора называют
растворимостью (L). Выражать ее можно в различных единицах, причем
всегда возможен переход к молярной концентрации.
В. Оствальд в 1897 г. установил, что растворимость зависит
от температуры и давления. При этом оказалось, что в большинстве
случаев растворение является процессом эндотермическим и
растворимость увеличивается с повышением температуры.
Соответствующие значения дают в виде таблиц и кривых растворимости (L, t).
Было найдено, что для полиморфных и аллотропных форм
одного и того же вещества L имеет разное значение. Так, Lz^s в
структуре цинковой обманки оказалась в десять раз ниже, чем для
того же ZnS в виде вюрцита с гексагональной структурой [71].
Малорастворимые вещества, полученные путем быстрого
осаждения, как отмечено в [52], часто имеют нарушенную решетку,
а в связи с этим считаются «активными», так как растворимость
их оказывается выше обычной. В некоторых случаях такие
вещества оказываются смесью гидратированных и дегидратированных,
активных и неактивных форм. Например, аморфный гидроксид
железа является сложной системой:
(аморфный, неактивный)
Fe (ОН)з ^ а — FeOOH
(активный) \^ (активный)
а — РегОз
Получить воспроизводимые значения (L, /) для такой системы,
конечно, нельзя.
Оказалось далее, что L может сильно возрастать для
кристаллов маленького размера и снижаться, стремясь к некоторому
предельному значению, по мере их роста. Одновременно была
установлена зависимость растворимости от величины поверхностного
натяжения а на границе раздела между кристаллом и насыщенным
раствором. Здесь а является мерой стремления данной
тонкораздробленной твердой фазы сократить площадь поверхности раздела
за счет перерастания в частицы
большего размера. В некоторые случаях
изменение а за счет веществ,
понижающих поверхностное натяжение,
может привести даже к изменению
кристаллической структуры твердой фазы.
Так, обнаружено, что при
кристаллизации NaCl из-за снижения а
получались только октаэдрические или
смесь октаэдрических и кубических
кристаллов [48].
Наконец, как это следует из
значений L, измеренных в присутствии
индифферентных солей,
растворимость может своеобразно и
значительно изменяться в зависимости от
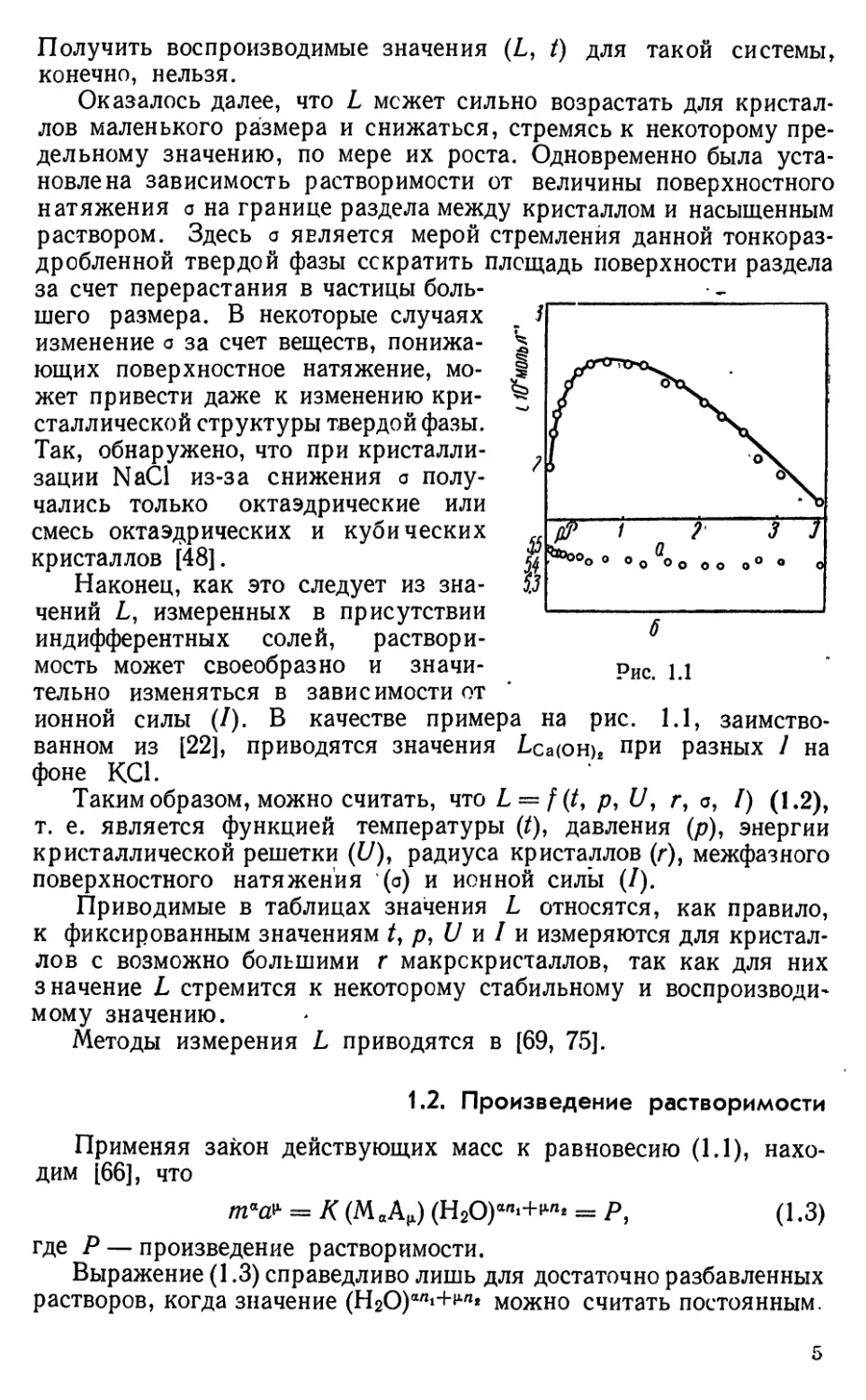
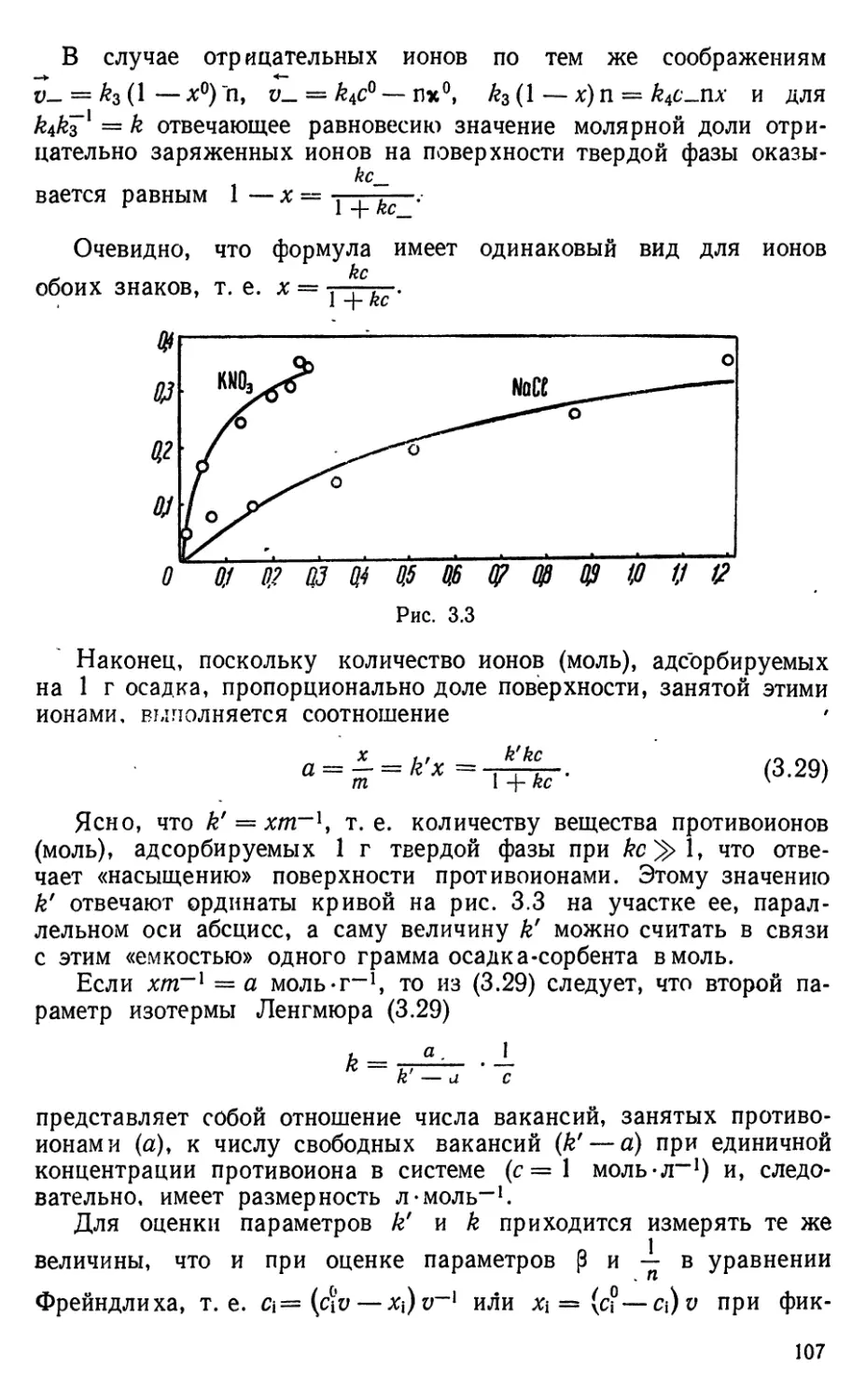
ионной силы (/). В качестве примера на рис. 1.1,
заимствованном из [22], приводятся значения Lca(OH)j при разных / на
фоне КС1.
Таким образом, можно считать, что L = f{t, р, (/, г, о, /) (1.2),
т. е. является функцией температуры {t), давления (р), энергии
кристаллической решетки ((/), радиуса кристаллов (г), межфазного
поверхностного натяжения (а) и ионной силы (/).
Приводимые в таблицах значения L относятся, как правило,
к фиксированным значениям U и I и измеряются для
кристаллов с возможно большими г макрокристаллов, так как для них
значение L стремится к некоторому стабильному и
воспроизводимому значению.
Методы измерения L приводятся в [69, 75].
1.2. Произведение растворимости
Применяя закон действующих масс к равновесию (1.1),
находим [66], что
т-а^ = К (МД,,) (H20)*'^i+f^«^ = Р, (1.3)
где Р — произведение растворимости.
Выражение (1.3) справедливо лишь для достаточно разбавленных
растворов, когда значение (H20)*'*i+»*'*« можно считать постоянным.
Активность твердой соли при атмосферном давлении считается
равной единице [56], так как предполагается, что равновесие (1.1),
* установившееся при данных t и не нарушается при возрастании
и уменьшении количества твердой фазы, и что величина (МаА,х)
не зависит от состава окружаюш.его раствора. Как следует из
раздела 1.1, предположения эти далеко не всегда выполняются,
а это сказывается, естественно, на воспроизводимости значений Р.
В частности, в связи с тем что твердая фаза может рключать
одновременно активные и неактивные формы вещества,
воспроизводимость значений Р, вычисляемых по результатам измерений
L (см. ниже), обычно тем хуже, чем меньше масса твердой фазы,
находящаяся в контакте с насыщенным раствором.
1.3. Связь между произведением растворимости
и растворимостью
Общие методы измерения Р приводятся в учебниках
физической химии и некоторых монографиях [52].
Значения Р можно рассчитать^ также по результатам
измерения L. Если растворимость МаЛц., найденная опытным путем при
температуре i, оказалась равной L, то согласно уравнению (1.1)
в насыщенном растворе, находящемся в контакте с твердым
веществом, См = aL, СА = [aL и матрица равновесий, если в системе
нет многоядерных частиц, имеет вид -
А«-
н+
к
0
1
0
0
0
I
—I
J
он-
м(он)г«
W
^ [OH~]=cp^Hte;;z-^
[M(OH)r"]=cp^,OH,,W-='^
[HjAi-*] = cpHjAajah^
Применяя закон сохранения начальной концентрации, получаем
еще два уравнения:
aL = m S cpм(OH),^кЛ-^
к=0
(1.4)
(1.5)
Определяя из них т, а и подставляя их значения в выражение
произведения раствсркмссти (1.3), находим, что
(1.6)
=0 / \j=0 V, /
Из (1.6) сразу находим, что в дбщем случае
/
(1.7)
т. е. зависит не только от а, [х и Р, но и от Л.,
Для вычисления h можно использовать выражение закона
сохранения электронейтральности, которое после подстановки
значений m и а из (1.4) и (1.5) принимает вид
к=0 -j-
-ft
к=0
(1.8)
S (J-«)?HjA^J^^
+ l^L^ = 0.
S ?H;A^j^^
Существенные упрощения возникают при расчетах по (1.6)
и (1.7) для очень малорастворимых соединений. В подобных случаях
даже сильно диссоциирующие аквометалло-ионы и очень легко
протонизующиеся основания не вызывают заметных изменений
рН. В результате можно заключить, что Л = 10-^ моль • л-^
В то же время ионная сила оказывается настолько близкой к нулю,
что все коэффициенты активности можно считать равными единице.
В условиях умеренной и значительной растворимости такие
предположения становятся незаконными, и значение h приходится
рассчитывать по (1.8), используя приемы, подробно
рассмотренные при исследовании кислотно-основного равновесия в водном
растворе хорошо растворимого соединения MaAjx.
Для малорастворимых солей XY (где X — катион сильного
основания, а Y —анион сильной кислоты) равновесие (1.1)
принимает вид XY(t)«XH- +Y- (1.9), формула (1.6) —(МО),
а формула (1.7) —L = j/p (1.11).
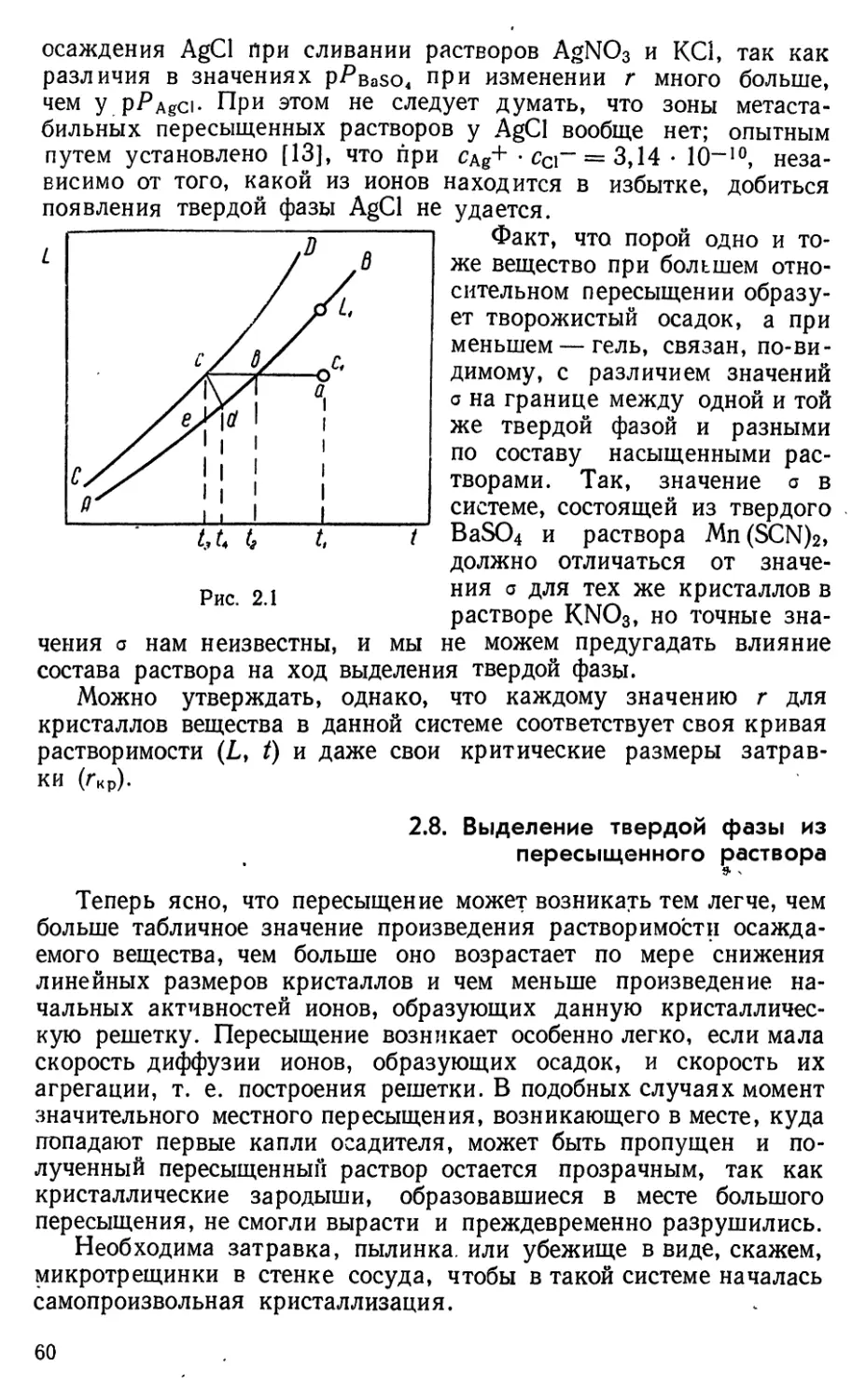
Само название «произведение растворимости» возникло,
по-видимому, в связи с тем, что долгое ^время при расчетах Р по
L кислотно-основные равновесия М?"^- и А'^'-ионов не учитывались.
1.4. Примеры расчета Р по измеренным L
Пример 1.1. Вычислить Pj^cio^» ^^^^ при 20 °С в 100 г воды растворяется
1,80 г этой соли и плотность раствора р = 1,011 г • см^. Поскольку молярная
масса М (KCIO4) = 138,55,
И ПО Девису
В результате Р'^^^Ю, = (10-°'««9 . Ю^^'^^^)^ « 10"^^^^^ = 9.6 • IQ-^.
Пример 1.2. Вычислить P^^^g, если при 20 ^Ag,S - 7,8 . Ю^^^ г
100 мл ВОДЫ.
Учитывая, что М (AggS) 247,80 г • моль""^ находим = 3,15 х
0,2 . 0,129 =-0,119 .
X 10""^^ мoль.л■"^ А = Cs « 3,15. 10""^^ и М
10~^4'2° моль.л~\ / « 9,35 . 10~*° моль • л~\ д. « J, рН - 7.
Матрица равновесий в э ом случае имеет следующий вид:
^Ag '•
6,30 . 10"" =
м
А
Ag+
S2-
н+
К
i=l
1=2
1 '
0
- 0
1
к
J
-11,7
^ 12,9
—23,8
19,95
При этом в формуле (1,6) а = 2, (х = 1, ^' = 2, а' = а = 2, ср. = 1 и,
следовательно,
у 2 \2 ? -1-.1
Лк=0 / j=0
Р = 22^3
Расчет Р по известной растворимости Ag2S дан в табл. 1.1.
(1.12)
Л
2к
10-7
10-42.904
1^—9,800
1,60 .10-^°
10-4.70
1
2,00-10""^
10^
\
1
1,00
t
1,00
10°
Пример 1.3. Вычислить /^рЬз(РО^)2, если в 1 л воды при 20 °С
растворяется 1,3 . 10-4 J, рЬз(Р04)2-
Здесь L = 1,3 . 10"~4 . (811,7)-^ = 1,6 • Ю^^ моль . л~^ М=3 • L=4,8 X
X 10-^ л=:2./, = з,2.10-^ /-^(з^ • ^^ + ^^ • =
8
=2,4.10-^ моль- л"~^ VI = 1,55.10-^ и Igcp3 - 0,0070 при расчете по Девису.
В этих условиях можно считать все ср^ = I.
Если пренебречь маловероятным при Мр^ =4,8 • 10—7 моль • л—1
образованием многоядерных гидроксоплюмбо-ионов, то матрица равновесий системы
примет вид:
3L
2L
РЬ2+
н+
i=l
i=«2
i=3
0
1
0
0
0
1
—1
—к
j
ОН-
РЬ (он)Г«
w
•riK
—14
-7,8
12,32
-17,2
19,53
" —28,0
21,68
В этом случае в формуле (1.6) а = 3, р. = 2, ja' = 3, а' = а = 3, ср^ = 1 и
г/ 3 \з / 3 \2"1-1
.Wo / \j«0 / .
Для первой оценки h предположим, что РО^^-ион протонизуется только
за счет Н"^-ионов, которые дает аквоплюмбо-ион. Тогда
РЬ2+ — Н+ PbOH+ IgYji = —7,8;
РО^- + Н+ НРО^- Iga « 12,32
РЬЧ +РО5-" :;±РЬОН+ +НР02-
с 4,8 . 10-7 3 2 . 10-7 _ _
П 1,6. 10-7+д; ^ 3,2. 10-7 3,2.10-7 —X
По закону действующих масс
(1,6 . \0-'^ + х)х
Ig/C - 4,62
(1.14)
(5.15)
-=l0-4'52 при
л:< 1,6. 10-7, х= 1,93. 10-^^
(3,2. 10-7
[РЬОН+] = [НР02-] = 3,2 . 10-7, [РЬ2+] -[РОЗ-] = 1,93.10-1^моль.л-^.
Таблица 1.1
^1
P
105.95
8,912.10^
105.9
1
7,943 • 10'
10°
i
1
JQ6.2269
t
1,686.10^
106.2269
7,4.10-^''
Теперь, применяя закон действующих масс к (1.15), находим
1,93 . 10-^
3,2- 10
,—7
;0-12.32^ /11 = 10-^'' моль.л-^, рН = 8,1,[0Н-]:
> == 1,26 . 10-6 . ^-1^
простое сравнение показывает, что равновесные концентрации всех частиц,
кроме [PO4—J, величины почти одного порядка. Тогда для расчета значения
Р, требующего повьш1енцой точности, следует использовать уравнения законов
сохранения:
3
cpb=3L= m S (1.16)
3
Таблица 1.2 ^Р04 = = а ^ . ^i, (1^7)
• j=o
>' w JL
^-Л+'^И (2-к).|к/1-^ +
к=0
3
+ aS^(j-3)aj/ii ==0. (1.18)
Из табл. 1.2, где
сопоставлены значения fi^^h"^ и a.Л^
следует, что при близком к
истине значении h= 10""^ мольх
л
§
а
%Л-2
7
(?
ч
10-7
10-7
10-3.2
10-0.8
10°
100.68- 1 105.53
J05.32
10°
Хл в процессе разворачивания
сумм в уравнении (1.18) и подстановке значений т и а из (1.16) и (1.17)
достаточно взять к = 0,1 и j = О, 1, 2. В результате после обычных преобразований
(1.18) переходит в
h Л + tii a^h^ + c^h+\
Таблица 1.3
h
33.22.L5
'2k
IQ—31,9462
jQ- 5,5408
2,88.10-6
10-2.2272
5,927.10-3
10-0.3136
0,4858
10°
1,0000
100.1737
1,4917
pP
10-0.7792
0,166
104.5572
36078
104.8336
68180
10°
1
los.oiso
t
104259
10l0,5571
42,50
3,1.10-43
a затем в
а/ + [aj + a, -f 4L}]/i4 + [y^^,^ _ a^tt^ + (2.i + .j^ag )L] /i^ _
— [(^1 +'^1^2)tt^ + '^l^l^ —^iJ^^ —"^1 (3L +aia;);i —Y|joy = 0. (1.20)
После подстановки численных значений с учетом безусловного неравенства
h > 10""^^'^^ получается расчетное уравнение
/1^ + 7,175 . 10-"7ft3 1324 . I0-lV_9 3i5 . io-22/j_9j72 . iQ-^o^ 0,
(1.21)
10
решив йоторое по Хорнеру, получим h = 3,262 • 10~^ моль • л~"^ и рН=7,4864.
Последующий расчет рРрь,(Р04)г ("Р^^^Р ^-3) приведен в табл. 1.3.
Он дает значение Р =3,1 • Ю^^з^ близкое к Р = 7,9 • \0~~^^,
приведенному в [27].
В тех случаях, когда металло-ион малорастворимой соли
координирует связанный с ним анион, приходится учитывать
процессы комплексообразования. При этом, во избежание возможной
неопределенности, необходимо точно указывать равновесие, для
которого рассчитывается рР. Согласно [1] само представление
о произведении растворимости становится неопределенным для
вещества типа Hgb, так как в насыщенном растворе таког^о
соединения одновременно находятся hg2+, Hgl"", Hgir, Hgl4 и I''—
ионы, a также комплексы Hgb, с преобладанием последних.
Значения рР для Hgb будут зависеть в таком случае от частиц,
избранных в качестве компонент гетерогенного равновесия.
Неопределенность становится в этом случае еще более ощутимой,
если учесть слоистый характер решетки Hgb [33, 37].
Поскольку ионы решетки не протонизуются и не
диссоциируют в растворе лишь в редких случаях и даже в растворах Hgb
всегда находится некоторое количество hg2+ и 1~-ионов,
очевидно, II в подобных случаях нет необходимости отказываться от
понятия «произведение растворимости».
Пример 1.4. Вычислить pPngi^' растворимость Hgig при 25
равна 1,3 . 10""^ моль/кг.
Плотность раствора Hglgi содержащего 0,0591 г этого соединения в 1000 г
воды, можно считать равной плотности воды. Тогда cfjgj^ = L = 1,3 . ю-"*»
:2L
^Hg = ^= 10-\
системы имеет вид (1—4 — значения /)
2,6 . 10 моль . л ^ и матрица равновесий
L
2L
1,3. ю-'^
2,6.10-^
Н+
Hg2+
1-
Ki
12 3 4
0
1
1
0
0
0
п
^ 0
-1
—к
0
j
0
0
0
I
он-
(ОН)^--''
w
\
h
—14
—3,96—6,52
12,87 23,82 27,60 29,83
Можно считать, что / < у (L . 2^ + 2L) = 3L = 3,9
10'
' ^ моль . л""^
Тогда по Девису Igcpj ;s:; 0,01, и, учитывая высокую закомплексованность, ве
дущую к снижению /, можно принять, что все = 1.
Последнюю строку матрицы учесть нельзя, так как протонизация Hgl^JjIp.
комплексов не изучалась.
Приближенную оценку" влияния комплексообразования -можно получить,
рассматривая только первую ступень координации.
11
в этом случае
Hg^++ l"-:;lHgI+lgpi = 12,87,
с1,3 . 10-^2,6 . 10-^
[ ]т 1,3.10-^+т, 1,3.10-^ —т.
По закону действующих масс (1,3- 10"^^ + т) m (1,3-10""*—m)=IO"~ ^^'^^
и если т< 1,3-10-^ то m=•10-*^'^^ моль-л'^ При столь малом m-[Hg^+]
можно считать pH,ie= 7 и исключить из рассмотрения первую и вторую строки
матрицы без учета последующей координации.
В этом случае
4
^^'^ S М"» (1.22)
п=0
4
2L = f/ + m 2 np„r/", (1.23)
n=0
2L=y + ^ • (1.24)
n=0
Уравнение (1.24) легко приводится к виду
hy' + (h + ^^h) у' + ih + ^h) / + Pi/ - (3^Pi - 1) ^/ -21 = 0.
(1,25)
После подстановки в (1.25) значений L и получаем
/ + 10-2. 2113 . у4 10-6.7687 . уЗ ^ ^^^16,96 . ^2 __ ю-^О.Збд . ^ _
— 10-33.4151 _ о, (1.26)
где 1/« 10-2'2113 „ 10-3.7687 „ возможно Г/» 10-^3.0461.
Если учесть эти неравенства, то (1.26) приобретает вид квадратного
уравнения
^ ^2^ 10-11.2013. 10-14.6103^ О,
из которого при и > 10-^*^'2°^3 j^Q^j^ . ^-1 находим г/= 10-7.3051 ^ 4 X
X 10-® моль . л-^
Подстановка этого значения в (1.26) подтверждает правильность решения.
Расчет равновесного состава системы (пример 1.4.) приведен в табл. 1.4.
Правильность расчетов подтверждает значение суммы [Hgl^—"] в последней
строке: 1,2997 . 10-^ вместо 1,3 . 10-^.
Теперь очевидно, что для гетерогенного равновесия Hgl2 (т) ;± Нб^"^ +
+ 21- рР « 27,7, а для Hgig (т) :;± Hgl+ + J-, рР 14,84.
Поскольку при расчетах рР соединений, дающих аквометалло-ионы и
гидроанионы, принимают во внимание схемы растворения, ведущие к образованию
простейших частиц, того же правила следует придерживаться и при наличии
комплексообразования. Тогда pP^gi^ = 2^,7 станет единственным значением.
12
Табл ица 1.4
hy
2
tn
|q0,6096
4
J05,6847
\
4,84-10^.
109,2098
\
1,621-109
105,5649
3,67.10^
10°
1
1
I09.2160
t
1,6218.10»
10-13,0961
[Hgir;
[HglJ
[Hgl+]
[Hg2+]
s
^ 10-12.4866
3,310-^3
10-7.4114
\
3,88-10-^
10- 3,8863
1,299.10-4
10-7.5312
\
2,94.10-^
10-13,0961
\
8.10-^4
1,2997 . 10-
1.5, Примеры расчета L no табличным значениям P
Для расчета L используется уравнение (1.7). Такое
вычисление растворимости по известному значению произведения
растворимости приходится^ производить гораздо реже, так как
большинство этих значений получено путем описанных расчетов по
экспериментально найденным значениям растворимости.
Существуют, однако, специальные методы определения произведений
растворимости, основанные на прямом измерении равновесных
концентраций (активностей) отдельных ионов, образующихся при
растворении вещества. Только в таких случаях и приходится
иногда искать растворимость вещества по величине произведения
растворимости. И здесь задача решается легко только в тех очень
редких случаях, когда ионы кристаллической решетки соединен
ния не подвергаются каким-либо превращениям, кроме
гидратации. В остальных случаях вычисления более сложны и ведутся,
как при расчетах Р по L.
Пример 1.5. Вычислить растворимость PbCOj при комнатной температуре,
Р^РЬсОз = 13,14. .
Если принять, что (РЬ2+) - (COf-) ~ /ю-^з.м ю-^.бт
_7 __1 , 1 . 7 -О . А
,, - J моль - Л \ то / ~
позволяет считать все ср^
' • 2 . 2,7
= 1.
10
=2,7Х
10""^- моль . л-^ Это
13
,-2
Используя равновесия ион изации аквоплюмбо-иона и протонизации СО"
иона и факт, что для РЬСОз начальные концентрации РЬ^"^ и СО^" ионов
совпадают с растворимостью, получаем матрицу равновесий системы;
L
L
IgKi
н+
i=2
i=3
0
1
0
4
3
6
1
0
0
1
0
0
0
1
—1
—к
j
—4
—4
-8
0
он-
РЬ (0Н)2-«
Н/С0^~2
Pb4 (OH)J+
РЬз (0H)^+
РЬб m)t^
РЬСОз (')
W
^44
•^34
Р
-14
-7,8 ^
10,33
-19,9
-23,4
-12,7
-13,14
-17,2
16,68
-28,0
Приводим систему уравнений, отвечающую матрице:
2
3
. (1.27)
L - m 5] -rj^fi-^ + 4Y)44m4/i-4 + З-^з^т^Л-^ ^ %r^^^m%-^, (1.28)
K=0
2 3
/t"-r + « S (/~2)a^/i^ + m (2-K)iri^/i-^ + 4^44m4/i-4 +
j=d K-o
+ 2Yi34m3/i-4 + 4Yie8m^/i-^ = 0, (1.29)
ma = P. (1.30)
В ней четыре неизвестных: Ь^йуЩ и h. Она линейна относительно 1,а,1л
решить ее можно только с помощью ЭВМ.
Для упрощения расчетов надо знать рН насыщенного раствора PbCOg, но
этих данных нет. Поскольку речь идет, однако, о соли, аквометалло-ион
которой являетс5![ донором, а анион — акцептором протонов, можно ожидать, что рН
системы^близок к 7. В то же время при рР = 13,14 можно ож'идать, что
tn^Yp^ 10-^'^^ моль . л-1.
2
Расчет значений 'П^^тЧ'"^ и Д] o.h\ при рН 6, 7 и 8, т = 10"^'^^ по-
к'1зал, что для данного интервала рН* уравнения (1.28). (1.29) принимают сле-
. ующий вид:
(1:31)
(1.32)
Исключая L из уравнении (1.27) и (1.31) и подставляя значение а из
(1.30), получаем формулу
14
т ■
f 2 ,
-8
V7
(1.33)
пригодную для расчета значений т при любом заданном рН, отвечающем
условию 6 < рН < 8. Правильность выбора рН контролируется путем подстановки
в (1.32) соответствующего значения Л, вычисленного для него по (1.33)
значения т и рассчитанного по (1.30) значения а.
Начало расчета приведено в верхней части табл. 1.5. Здесь, при первом
ориентировочном расчете заведомо небольшая разность h'—whr^ во внимание
не принималась. Фактические значения левой части уравнения (1.32), т. е. А,
даны в последней колонке таблицы. Из их сопоставления следует, что более
точное значение рН следует искать на интервале 6,5 < рН ^ 6,7 и при том
ближе к рН 6,7.
Таблица 1.5
рН
Л—и,/,—1
PS
т
6,5
6,6
6,64
6,66
6,65
1.792.10-7
,0-9,0774
jO-9,3495
10-9.2701
10-9,2970
10-9.2835
1040,0782
1041,6782
1041,1982
1041.3582
1041,2782
Продолж
10-7,0222
10-7,2897
10-7,2098
10-7,2365
10-7,2231
ение'табл^ L5
рН
а
^ 6,5
6,6
6,64
6,66
6,65
10-6,1178
10—5185 03
10-5,9302
10-5.9035
10-5,9169
10-2,2877
10-2,2202
10—2.2400
- 10-2,2333
10-2,2367
10-2,2311
10-2,2361
10-2,2367
10—2,2369
10-2.2365
+7,18. 10-4
—2,17 . 10-4
+4,4 . 10-5
—4,9 . 10-5
+3,18 . tO-6
Окончание оценки рН дано в нижних строках табл. 1.5, где сначала
вычисляются Д при рН 6,64 и 6,66, а затем для окончательно установленного
значения 6,65. В последнем случае принята во внимание и разность /i—а;Л~^ =
= 1,792. 10-^
Путем последующих приближений можно, конечно, получить-еще меньшее
значение Д, но этого делать не стоит, так как рассчитывать на очень высокую
надежность констант не приходится.
Здесь для вычисления L можно использовать только уравнение (1.27) или
(1.31). Оба они дают одно и тоже значение:
10-2.0604 _ 8^52 . ,0-3 моль .
Уравнение (1.7) в данном случае применить нельзя, так как оно пригодно
только для систем с одноядерными комплексами.
Пример 1.6. Вычислить растворимость BigSj при компактной
температуре, если рР = 97.
Сначала попробуем оценить растворимость ^Хс^-^^ пренебрегая ионизацией
аквависмути-ионов и протонизацией сульфид-ионов. В этом случае
BigSg (т) 2Bi3+ ^ 3S2-^ IgP _ ,_97,
= 2L и = 3L и ^ ^ 10~^^ моль . л""^
так как (2L)2 {Zlf ^ 10-^^
Очевидно, 410 в насыщенном растворе Ъ\с^^ ионная сила стремится к О,
У/=1 и рН-7.
15
Матрица равновесий системы имеет вид
2L
3L
н+
1
K, |i = l |i = 2
6
0
2
0
1
3
—12
j
0
BieOe^+
BigSg (T)
P
-1,58
12,9
-97
19,95
Для вычисления
неизвестных a, niyL при/1=10""
моль-л""^ получаем
уравнения: 2L = Gfim^h-^^ (1.34),
3L = а У ah^ (1.35),
j=0
т^а^^Р (1.36).
После исключения L из (1.34) и (1.35) и подстановки т из (1.36)
находим, что
,1=0
-1
(1.37)
Значения m и L получают после расчета а по (1.37). Растворимость,
вычисленная с учетом ионизации аквависмути-ионов и протонизации сульфид-
ионов, оказалась равной 2,3 • 10~^^ моль • л~"^ что на 4 порядка превышает
приближенную оценку (табл. 1.6).
Таблица 1.6
-12
n-1
п-1
L,
п-1
10
,-207,6259
105,95
Ф
'8,912-10^
105'9<
10°
Ф
1
10
-21,3852
10
-16,4222
10-15,636
1-16
106.2267
t
|7,943.10^| 1 11,6855.10^1 | 12,3-10
Пример 1.7. Вычислить растворимость MgNH^PO^ в воде при комнатнрй
температуре, если рРд^^Н4РО, = 12'^-
Грубая оценка L с помощью равновесия MgNH^PO^ (т) :^ Mg^+ + NH^+
+ РО^-» IgP = —12,6 дает L - 10-4.2 моль -л-!.
Здесь рН = 7, расчет по Девису дает оценки Igcpj =0,0092, lg<P2=0,0365,
lgcp3 =0,0824.
Сначала записываем матрицу равновесий системы, затем расчетные
уравнения законов сохранения и гетерогенного равновесия, возникающего в
системе. Здесь используются обозначения т = (Mg2+)\ (NHg), а= (РО^^):
L L
L
Mg2+
NH3
ро|-
н+
i = I
i=2
i==3
0
1
0
0
I
0
0
1
0
0
0
0
0
I
1
—1
—1
1
j
0
0
0
0
0
1
он-
MgOH+
NH+
HjPOi-3
MgNH4P04(T)
w
-n
a
p
—14
— 12,8
9,24
12,32
-12,6
19,53
21,68
16
L = m(cp2 + cpi7)/z 7,
' = r(cpia/i+ cpo),
J=0
= a (ТО^З'* + 9l^2^ + 92^1^ + 93)»
3
h^'Hr + m (292 + ^1^^""^) +^x^rh + aY (J - 3) ^j^a^j^^ = 0,
" J=0
(1.38)
(1.39)
(1.40)
(1.41)
pH
6
7
8
10
Таблица 1.7
eft
,0-6.8
,0-5.8
10-2.8
,03.24
,02.24
10-1.24
,0-0.76
,03,68
,00.68,
,0-2.32
,0-8,32
,07.53
,05.53
j03,53
,0-0.47
,06.32
,05,32
,04.32
102.32
mar/ia=P. (1.42)
Для сопоставления
значений отдельных слагаемых,
входящих в уравнения (1.38) —
(1.41), используем табл. 1.7.
Из табл. 1.7. следует, что
на интервале 6 < рН < 8cpi X
'Х < ср2, что при рН > 7
нельзя считать С 9i^^ и что
3
в сумме У на интервале 6 <
1=0
< рН < 8
во всех случаях сро^з'*^ " Ъ'^^х'^^ ^ ^2^1^^- ^ результате, считая cpQ = I,
преобразуем нашу систему и получаем:
L - cpgm, m = (1.43), /•=L(cpja/i + l)-"l (1.44),
Л2 {1 + cpjL [a (cpja/i + 1)-1 + «2 (cp,a2fi + cp2ai)-l]}
L [cp^a-iP (9,аЛ + 1) (cpjGg/l + cp2a,)]l/3.
Два последних уравнения обеспечивают возможность быстрой оценки рН
и L насыщенного раствора MgNH4P04. Для этого достаточно найти Л и L,
удовлетворяющие (1.46), где ш= 10""^"*. Простой подстановкой было найдено, что
рН= 10 подходит лучше рН = 9. Дальнейшие расчеты даны в табл. 1.8;, где
значки [ ] и { } отвечают выражениям в квадратных п фигурных скобках
уравнения (1.46).
Дальнейшее уточнение L = 10—3,1275 = 7^46 . 10—4 моль • л смысла не
имеет, так как использованные знaчeнияlg^<'^ нельзя считать абсолютно точными.
Таблица 1.8
(1.45)
(1.46)
(1.47)
рн
ср,ал + 1
£8
10
10,1
10,05
10-0,7508
10-0,8508
10—0,8008
100,0710
100.0573
100,0638
109.5392
109.4392
109.4892
1012,3579
1012,3572
1012,3572
10-9.3746
10-9.3890
10-9,3826
1моль-л—1
а
-<р1аЛ + 1
f J
10-3,1249
10-3,1297
10-3,1275
1,476 . 10^
1,523 . 10®
1,5004. 10®
1,486 . 10^
1,490 . 10^
1,488 . 10^
109.1735
109.1869
109,1805
10б,0578
1 об, 0664
106.0622
10—13,9422
10—14,1336
jO-14,0378
2 3-457
17
Из приведенных примеров видно^ что при вычислениях
произведений растворимости по данным о растворимости и наоборот
необходимо принимать во внимание взаимодействие ионов,
появляющихся в растворе, как с растворителем (т. е. ионизацию акво-
комплексов и протонизацию), так и друг с другом (т. е. комплексо-
образование). При этом, как следует из рассмотренных примеров,
начальные концентрации простых гидратированных ионов могут
снижаться за счет этих дополнительных процессов на много
порядков/так что недоучет этих равновесий может привести к грубейшим
ошибкам в оценках Put.
1.6. Растворение соединений, малорастворимых
в воде
Если соединение МаА,х практически не растворяется в воде»
то это значит, что концентрации М»^+-и А'*--ионов и
происходящих от них частиц в насыщенном водном растворе очень малы.
В этом случае для повышения растворимости МаАр. надо
сдвинуть равновесие (1.1) вправо, а добиться этого можно, лишь снижая
каким-нибудь способом концентрацию одного или обоих ионов —
продуктов гетерогенного равновесия (1.1). При этом, естественно,
произведение активностей ионов кристаллической решетки,
фактически находящихся в растворе, становится меньшэ произведения
растворимости; равновесие нарушается, и для его восстановления
в раствор переходит соответствующее количество твердого
вещества. Этот процесс может привести к полному растворению
«нерастворимого» вещества. Очевидно, что при этом ионный состав
«раствора» будет еще больше отличаться от ионного состава
кристаллической решетки, чем при растворении в чистой воде.
Существует три прямых метода снижения концентрации ионов
в растворе:
1) удаление иона за счет образования слабой кислоты или
основания;
2) перевод его в комплексный ион;
3) окисление или восстановление удаляемого иона.
Во всех этих случаях твердая фаза растворяется с помощью
надлежащим образом подобранного химического процесса.
Условия, обеспечивающие растворение осадка или
малорастворимого вещества, при использовании различных методов снижения
концентрации ионов проще всего разобрать на отдельных
конкретных примерах. При этом мы рассмотрим две основные задачи:
сколько молей малорастворимого вещества может раствориться
в 1 л раствора, который содержит заданное количество молей
растворяющего вещества; какую концентрацию растворяющего
вещества необходимо создать в 1 л раствора, чтобы обеспечить
растворение заданного количества малорастворимого соединения.
Пример 1.8. Вычислить ^саСаО* ^ растворе НС1, если Сдс!^^"^
= 0,1 моль • л""^ и в системе находится избыток СаС204.
18
Запишем матрицу равновесий системы:
Y
у
С1-
.Са2+
н+
i= 1
i=:2
0
1
0
1
0
0
1
1
— 1
— 1
j
0
он-
СаОН +
W
Р
— 14
— 12,6
4,25
— 8,75
5,52
Для грубой оценки / используем равновесия
СаС204 (т) :^ Ca^+ -{- CagO^"" Ig Р = - 8,75
Ср\- + 2Н+ 7^ Hf,p^ Ig = 5,52
СаС204(^)+ 2Н+ 7t Са2+ + Н2С2О4 Ig /С = - 3,23
^2^4(т)
с 0,1 —
[] 0,1 —2/п m
m2
m
т
10-3,23
(0,l-2m)2-^" ' 0,1-2m
- 10~^'^ m- 2,5 . 10~^
[H+] = 9,5 . 10-2, [Ca2+] = 2,5 • 10-"^ /= 0,1 моль . л-^
Расчет по формуле Девиса дает Ig«pi'i= 0,11, lg«p2= 0,44.
Применяя законы сохранения и учитывая гетерогенное равновесие, получаем
L = m(cp2 + cpiiri/i-^),
2
^ = а S ^j-2''j^^ = ^ (^2^2 + ^1^1^ + ^2)'
1=0.
2
У = ср^Л+ а 5] j . cpj«2^j^^ -TiW^-^ (1.50)
j=0
ma=P. (1.51)
Предварительный подсчет при Л = 10~^ моль . л""^ дает
(1.48)
(1.49)
4-2
,0-11,49
,03,52
,03,зб
,00,44
и приводит к неравенствам ^I'fih''^ < ср2» < даже при
т = 0,05 моль . л"^.
С их учетом и при = 1 получим:
(1.52)
19
2 *
L[\ + a^ (^2^+91^1)"^] = К- cpjfi = 0,1 - ср1л,
(1.53)
(1.54)
L=:[cp2P/2(a2^ + cp,a,)]2. (1.55)
Из (1.54) следует, ЧТО Л < 10"-^ • 10-°'^^ = Ю^^-^^ моль . д-^, а рН> 1,11.
Подстановка h< 10""^'^^ моль • л~^ в (1.55) дает соответствующие значения
Ly а подстановка того же Л и L в (1.54) обеспечивает правильность выбора h.
Ход решения дается в виде таблицы, как в разделе 1.5.
Результат: рН = 1,.138, L = [Са2+] = = 4,09 • 10~^ рСа == 2,8279,
РС2О4 = 5,9221, [СаОН+] = 1,4 • Ю""*^» [Щ^2^4] = 2,1 • 10-^ [HCgO^] =
= 1,995 . 10-^ [С202~] = 3,3 . 10-^ моль . л'К
Пример 1.9. Вычислить Z^Agci в 1 л раствора NH3, если Cj^^a =2 =
= А моль . л""* и в системе находится избыток AgCI.
Матрица равновесий в данном случае имеет вид
L
L ^
4
Ag+
С1~
NH3
н+
i= 1
1 = 2
1 = 3
1 = 4
0
1
I
V .
1
0
1
0 .
0
п
1
0
0
1
0
0
0
0
п
1
0
-1
-к
0
0
0
1
0
он-
Ag(OH)l-k
AgCli-"
AgvCP-^
Ag(NH3)+
NH+
AgCl (T)
w
\
Pin
Pv,
Pn
a
P
- 14
- 11,7
3,04
3,32
9.24
- ^i.75
-23,8
5.04
5,20
7,32
5,04
5,45
5,30
Пользуясь матрицей, легко записать уравнения законов сохранения:
к=0 n=l
v=2 n=l
4 3
n=0 v=2
2
n=0
2 4
h-j-fiV + m 2 0 - -Pi-k^k^"" + m S. С - ") ?i-nP щУ" +
(1.56)
(1.57)
(1.58)
k=0
3 2
v=2
n=l
20
Здесь, как и при решении других задач, очень важно оценить и сравнить
значения сумм, входящих в уравнения. Для этого необходимы первичные оценки
значении т, а, у, h и <р^..
Поскольку концентрания С^н, = ^ =2 "Л""^ очень велика, следует
начинать с оценки растворимости AgCl в NHg, считая, что образуется только
Ag(NH3)2'''-HOH:
AgCl (т) ' Ag+ + СГ Ig Р = - 9,75
Ag+ + 2NH3 ^ Ag (ЫНз)2+ к h = 7,23
AgCl (т) + 2NH3 ^ Q- + Ag (NH3)+ Ig /С == - 2,52
с 2 - . -
[] 2-2i/ у у
^2(1^1^)2. = у - lO""'' [NH3] - а ^ 1,8, т = Ру"' -
^ 10-8,75 мОЛЬ.Л-1.
Для последующей оценки h проще всего использовать равновесие NHg —
— Н2О:
NH3 + Н+ HN+ Ig а = 9,24
Н20:^Н+ + 0Н"' Iga; = —14
NH3 + Н2О :^ ЫН+ + ОН- Ig /С = - 4,76
с 1.8
f ] 1,8 —ш W W
f ^ 10-4.76^ Ш~ [NH+1^ [ОН-] = 10-2.25^ ^^jQ-lI,75 ^.^^.л-'.
1,8—ш ^ '
С достаточной точностью / системы определяется за счет [С1—] и [ Ag(NH3)J]
порознь равных ^ 0,1 моль • л-^ Тогда для / 0,1 моль • д-^ находим по
Девису: lg<pi = 0,11, Ig«p2 = (^»44, lgcp3 = 0,99 и считаем «ро = 1.
Последующие подсчеты дают значения сумм:
2 4
^ S - 5,3 « 10-9, m 2 ?i-nPini/" - 3.6 . 10-^'
к=0 n=i
3 2
v=2 П=4
Одновременно находим что ^^а ^ 10^*^® < P2^^ lO^'^t и cpjo^iЮ-^*^ С
« ?о = 1 •
С учетом всех этих неравенств записанная система уравнение чрезвычайно
упрощается: (1.56) переходит в L = (p^^2^a^ (1.60), вместо (1.57) принимаем
^=^9\У (1.61), и, поскольку L и у связаны лишь постоянным множителем,
сохраняем только (1.58). которое переходит в /1 = а + ^^s^^^^^c? (1.62), и ту = Р
(1.63). Из (1.60) и (1.62) а = Л—2L (1.64). Из (1.6J) и (ЬбЗ) m^^.PL"^
(1.65).
2)
"Подстановка значений а и т ъ (1.60) приводит к квадратному уравнению
относительно L: + 0,05058 L — 0,0253 = О (1.66).
В дальнейшем получаются следующие результаты:
L = = [С1~] = 0,1357 моль . л-^ Ig (/ = — 0.9773,
Ig m = - 8,7727, [Ag+] = 2,2 • 10-9,
a « JNH3] = 1,729, [ AgNH+] = 7,85 • 10-^
[Ag (ННз)^-] = L = 0,1367 моль . л^^
Пример 1.10. Вычислить растворимость CaCOj в 1 л раствора НАс, если
^НАс = 0»4 = Л моль . л""^
Запишем матрицу равновесий системы:
L
L
А
Са2+
С02-
Ас-
н+
Ki
1-1
1=2
0
I
0
0
I
0
0
1
0
I
0
0
0
I
0
-1
j
'I
0
он-
СаОН+
HjCOJ-2
НАс
СаСОз(„
W
'J
а
Р
— 14
- 12,60
10.33
4,76
-8,2
16,68
и уравнения законов сохранения:
L = m(<P2 + <f'il'«~').
J=0
л = а (<р, + 9о'Л).
Л-X + "? (2<Р2 + b-^h-^) + ^ Е (j - 2) <Pj_2«ift' -<Pie = О
" 1=0
Упрощенное решение задачи:
СаСОз(т) . :;±Са2++С0|- 1 lgP = -8.2
НАс :?iH+ + Ac- 2 _21ga = -9,52
С0|- + 2Н+ :^ + COj 1 . Ig «2 = »6,68
(1.67)
(1.68)
(1.69)
(1.70)
СаСОз(^)+2НАс ^ Са^++ 2Ас "+ Н2О + СО^ Ig Л" = - 1,04
■с- 0,4 — _ _
I ] 0,4 —2т т 2т m
4/п*
22
ДляpaвнoвecияC02;±C02(г),lgX = + 1,48.Это значит, что в насыщенном
растворе [COj] = 3,3 • 10~^ моль • л""' меньше найденного значения т =
= 0,1374 моль-л"' и задача должна быть пересчитана учетом этого
равновесия, считая (COj) = Рсо. = •^"i" (~ 0,1мПа):
СаСОз (т) + 2НАс :^Са^+гЛс" + ЩО + COj Ig /С = - 1,04,
СО,
iCOjfr)
lgX= 1,48
CaCO, (т) -f 2НАс 5± Са^+ 2Ас- + ЩО + COj (г) Ig АГ' = - 0,44
с
[]
0,4. - -
0,4 — 2т m 2m
4m^
lo"'". или m' - 0,363m2 + 0.1452m - 0.01452 = 0.
2^(0,2 —m) 2^
Расчет no Хорнеру дает
m ~ [Ca^+J = 0,1259 = 10-^'•^. 2m ~ (Ac"} = 0.2518 = IQ-O-Ssss^
tHAc] ~ 0.1482 = 10-0.829I моль • л"'.
Эти данные позволяют рассчитать [Н+]:
А ~ \0-*''^ • [НАс]. {Ас]-'= 10-'»-9902 „ ^ ^ JCO32-J = 10-7.3 .
Для оценки / достаточно учесть Са'^+ и Ас-.ионы. Тогда / ~ 1/2 (т • 22 +
+ 2т) ~ 0.3777 моль - л ' и по Девису: Ig 9, ~ 0.15 и Ig <f^ ~ 0,60. Принимаем:
'ill^iiirn'^^^^^^^^ входящих в уравнения
<p,o,ft
2?2
S
rS
,0-7.45
10-0.24
,05.48
,00.9
,05.48
,0-1.82
— 1 ( ^ \ к
Отсюда следует, что <p^fih * < cpg* — й"/ ^ ^Ь^* ^ ^1^ и 2^2 <
< (f>l<3^. Эти неравенства необходимо учесть при записи окончательной системы
уравнений. Уравнение (1.68) здесь использовать нельзя, так как трудно точно
учесть количество выделяющегося COg. Поскольку, однако, COg выделяется при
р Q = ! атм (-- 0,1 мПа), можно, используя одновременно равновесия
С02:;1С02(г) lgX=l,48
и
С0§"- + 2Н+ 7t ЩО + СО2 Ig ag,
записать вместо (1.68) зависимость г = (С0|") от h.
В результате система уравнений принимает вид: L = ^2^2 '^О = ^('^2^^)'"^
(1.72), а = Л (cpj + аЛ)-1 (1.73), 2^2^ — ср,о^г/1 — cpja = О (1.74), тг= Р (1.75).
Подстановка в уравнение (1.74) значений г, т, а, выраженных через
параметры и Л с помощью (1.72), (1.75), (1.73), приводит к уравнению
2cp2PX-la2h2-cp^Xaja7l/i-l -<f,Л(аЛ + <р,)-1 =0. (1.76)
23
После обычных пересчетов я преобразований получаем
2,454 . 10-^/1^-3,627 • 10-^^/1-5,875 • 10"^^ =0. (1.77)
Решая (1.77) по Хорнеру, находим /i = 10""^'^^°^ моль . л-^ Последующие
пересчеты приводят к значениям: г=10-^'^^9^, m = 10-'*^^^^ и искомой
L = 10—= 0,19 моль . л—^ Отметим, что первоначальная оценка L
0,1259 моль. л—^ составляет лишь 66% значения, полученного путем
точного расчета.
Пример 1.11. Вычислить растворимость РЬСгО^ в 1 л раствора NaOH, если
^NaOH — ^ ^^^^ ' Получаем матрицу равновесий системы, а затем
записываем уравнения законов сохранения, считая, что начальные концентрации
РЬ^"*"- и СгО^—-ионов одинаковы и равны искомой L, а Cq^^ = X = 1 моль • л—
Na+ РЬ2+
1=1
1=2
1=3
о
о
о
I
4
3
6
1
о
1
о
о
о
О
О
1
О
О
2
О
0.
О
О
О
он-
НСгО^
РЬ (0Н)2-^^
Pb4 (0Н)4+
РЬз (0Н)2+
РЬб(ОН)|+
РЬСгО* (т)
W
о
т
"^34
^68
Р
-14
6,50
1,64
—7,8
—19,9
—23,4
-12,7
-13,7
-28,0
Надо подумать, что при c^^qi^ = X = I моль . д-^ даже после
взаимодействия с РЬСг04 среда будет щелочной. Допустим, что w = (ОН) -
~ 10-^. моль . л—^* В этом случае /i < о), кроме того, для равновесия
CrOf- + Н+ НСГО4 Ig а = 6,50
в Z. . - -
~ [ ] (1 - a)Z. 10-^^ aL
= 10^»^° и а = 10-^'^^ т. е aL ^ 3 . 10-^ % от L
(1 —а) /. . 10-**
Поскольку образование Crpj"^ — процесс вторичный, можно считать
НСГО7] и [Сг202-]<[Сг02-].
Тогда система расчетных уравнений принимает вид
СгО^
,2-
(1.78)
к=0
-8
24
или после замены h"^ на ш""^ • «
3
к«0
+ 6<P4-ri68a'~^'«^W^ (1.79)
3
k=0
+ 4cp 2-^340'"" ^m^w^ + 8<P4Y;e8a''"^/nвa)^ (1.80)
rna = P, (1.81)
Даже предварительная оценка / в такой сложной системе связана с
трудностями и неопределенностью. Допустим для начала, что основную роль
играет образование одноядерных гидроксокомплексов: РЬ^^" + пОН"~
:^РЪ(ОН)^^. Используя значения у]^ и ш, находим, что Igpj = 10^'2,
Ig pg = 10,8 и Ig ?з = 14. Сопоставляя эти значения с Ig Р = —13,7, находим,
что заметной растворимости можно ожидать лишь за счет равновесий
РЬСг04(т) :^ РЬ2+ + Сг02- Ig Р = - 1з,7
РЬ2+ + ЗОН-^ РЬ (0Н)7 Jg |3 = 14,0
РЬСг04 (т) + 30H-:i± Сг02- + РЬ (ОН)^- ig /( « 0,~
с 1 - — ,
[ ] 1 — За а а
2
^^_^^^^з = 2, - 0,9815а2 + 0,3333а - 0,03704 = 0.
Решение по Хорнеру дает: а = [СгО|""] = 0,2336, [РЬ (ОН)^-] = 0,2336
и [ОН-] =0,3 мoль.л-^ а тогда с учетом [Na+] = 1 I • (I+0,3 +
+ 0,2336 + 0,2336 . 22) = 1,234 моль . л"* и по Девису: Ig cpj = 0,15, Ig =
= 0,60, Ig срз = 1,35, Ig cp4 = 2,4 и cpo = 1 •
Дальнейшее решение требует осторожного подхода, так как ОН—-ионы
распределяются между большим числом частиц и выбрать основные акцепторы
и доминирующие равновесия на основании одного рассмотрения матрицы
нельзя.
В этих условиях целесообразно провести оценку произведений известных
по величине параметров, входящих в отдельные слагаемые уравнений (1.79),
(1.80) после замены в них тна
После подстановки рассчитанных произведений в уравнения (1.79), (1.80)
шлуча ем
L =a-J (Ю^'^^Ч- 10^2,9^2^ 10-7'35^ + Ю-^З'О + 10-^5'^a-V +
+ 10-7.4^-3^4 1020'2782^-6^8^ ^,^2^
X = lO^'l^co + (10° 93^3 ^ 10-2.6^2 _^ 10-7.35^) _^
+ 10->5.7д-4^4 ^ jQ-7,3^-3^4 ^ 1020.4031^-6^8^
25
Поскольку а и О) не очень маленькие величины и входят в уравнения
(1.82), (1.83) в виде частных, можно предположить, что в уравнении (1.82)
основную роль играет последнее, а в (1.83) — последнее и первое слагаемые.
Тогда, отбрасывая остальные слагаемые и заменяя а на /gL, приходим
к системе = 1023.в782^« (1.84), (Х - Ю^.^М = 102^''''*(о» (1.85).
Умножая уравнение (1.84) на 4, (1.85)— на 3, вычитая и сокращая на
L^^O, находим, что L = j (X - Ю^'^^^^) (1.86).
^ В первом приближении при Ю^'^^о) < X =« 1, L = 0,75 моль . л""^ а тогда
из уравнения (1.84) о) = 10~3'°94* = 8,05 . 10"^ моль-д-^
Это первое приближение оказывается и последним, так как подотановка
в уравнения (1.79), (1.80) значений а =/^L = 10-0'7249^ m = 10-*2'9^5* ^
О) = 10""^'°^^^ моль.л""^ приводит к значениям: L х= Ю""^'*^^^ = 0,7495 и
Х= 1,00034 моль . л"^
Считая, что растворение РЬСгО^ в растворе NaOH приводит в основном
к появлению СгО!"" и РЬб (ОН)8"^-ионов, можно пересчитать /, принима
[Na+] = 1, [Cr02"J =0,75 и^[Pbg(OH)|+]= 0,125 моль-д-^ В этом случае
/ - 1/2 (1 + 0,75 . 2^ + 0,125 • 4^) = 2,5 моль . л""*. К сожалению, формула
Девиса при такой / для расчетов не годится. Нет смысла и в дальнейшем
уточнении значения L, так как все расчеты выполняются с помощью
параметров, значения которых нельзя считать окончательно установленными.
Пример 1.12, Вычислить растворимость CuS в растворе HNO3,
2 моль . л"
г-1
Запишем матрицу равновесий системы:
Cu2+
^2-
N0:
Н+
N0
К,
1 = 1
1 = 3
= 4
О
1
О
о
1
2
О
1
О
О
О
—к
—2
j
Си (0Н)2-к
Си(0Н)2+
HjSi-2
-^S.(T)
JNO
NO (г)
CuS (т)
■^22
^1
■ _±
К 2
К'
I
p
-8,0
-10,95
12,9
T -14,17
1.48,5
2,68
— 35,2
1-13,7
1*9,95
-27,3
—40,4
В первой' строке матрицы приведены значения четырех начальных
концентраций: С^ц, Cg, Cjqoj и Сн+, но использовать их для составления,
уравнений не так просто, поскольку в этом случае 52""-ионы образуют
нерастворимую серу, а NO~-HOH дает газообразный N0, который может полностью
остааься в растворе, если концентрация его мала или может частично
выделяться в атмосферу.
26
Начинать поэтому приходится с приближенного решения, считая сначала,
что N0 остается в растворе:
CuS(t)
S2- - 2е
NOf+ 4Н+ +3е
:Cu2+ + S2"-
:S
г NO + 2Н2О
3 (IgP =-35,2)3
3 (-Ig/C= 14,17)3
2 (Ig/C = 48,5)2
2N0J- + 3CuS (t) + 8H+ :^ 3Cu2+ + 3S (т) + 2N0 + 4H2O Ig К =
33,91
с 2
[ ] 1,5 +2x
8W(0Jb + xf ^ jQ«33,9i
8л: 3(0,25~;c) 2(0,25~;t)
и при X < 0,25 < 0,75 X = 10"^'^,
3^ .22 (0,25-л:)
a [NO] ^ 0,5 моль • л""^ что много больше (NO), отвечающей равновесию
N0 :;t N0 (г), IgX = 2,68, из которого при Pno=1 атм(;;^0,1 мПа) (N0) =
= 2,1 . 10-^ моль . л-*.
- Прибавляя два таких равновесия к записанному выше суммарному и
учитывая соответствующее изменение константы на 2 Ig X, получаем
2N0^ + 3CuS (т) + 8Н+ :^ 3Cu2+ + з5.(т) + 2N0 (г) + 4Н2О Ig К = 39,27.
В этом случае в расчетное уравнение вместо (N0) вводится pj^Q = 1 атм, а
остальные обозначения сохраняются. Тогда
8^^х^ .2(0,75 + а:)^
3З (0,25-xf
= 10-39.27
и при х«0,25 ;с= 10-5.9027^ [Н+] = 8х = 10-^'999б мoль.л-^pH = 5
[Cu2+] - L = 0,75,^ [NOfJ = 1,5 моль . л.
Здесь / = 1/2(0,75 . 22+ 1,5) = 2,25 моль . л""* расчет по приближенным
формулам вести нельзя — приходится считать, что все cpj = 1,
Теперь запишем систему уравнений:
4
^Си = ^ = ^ S ^к^""^ + 21^22^^^
к=«0
-2
(1.87)
(1.88)
так как из суммарного равновесия растворения следует, что на получение
трех Си2+-ионов расходуется два NO^~-HOHa;
4 2
c^=.Y =h^m Y к^к^""^-2^)22^2/1-2 + д ^ j^^/^j + 8/3L, (1.89)
к=0 j=0
поскольку из суммарного равновесия растворения следует, что на получение
трех Си2+ионов расходуется восемь Н+-ионов. Из матрицы видно, что
некоторое количество Н"^-ионов поступает в систему за счет ионизации акво-
купро-ионов, а часть расходуется на протонизацию 82~"-иона; та = Р (1.90)
^-2^, = 10-.-или
3lgm — 2lgy — 8]gh =
39,27.
(1.91)
27
Расчеты начнем с оценки отдельных слагаемых, входящих в уравнение
(1.87). Результаты при рН = 5, пример (1.12), приведены ниже:
^2Л-2
,0-20,4
10-12,3
10-3.7
0,0002
10-3.0
0,0010
10°
1,000
1,0012
10-0.649
0.224
Таким образом, уравнение (1.87) принимает вид
L = 1,0012/72 +-0,224/п2. (1.92)
Результаты такого же расчета для.уравнения (1.89) приведены ниже:
47)4Л~4
Зт1зЛ--3
2t)2/i-2
s
2а2Л2
,0-19.8
10-11.8
10-3'^
4 . 10""^
10-^
1 • 10-3
1,4 . 10-3
1010.251
1,782Х
107.9
8. 10^
1q10,253
1,79.10*0
Если учесть теперь, что h ^ 10 ^ моль * л *, m 0,75 й а = Р/п *
10—35 MQjjb . д—1^ то уравнение (1.89) можно записать так:
8
F = — 1,4 . 10-3 m —0,224 m^ + -Y^-
(1.93)
Подстановка L из уравнения (1.92) в (1.93) приводит, после обычных
преобразований и подстановок, к уравнению + 7,15т — 5,357 = О и
значениям m = [Cu2'^]= 0,684, L =0,7877 моль - л-*. Подстановка найденного
значения L в (1.88) дает у= 1,4749 моль • д-*.
Из уравнения (1.90) а ^ Ю-^^моль • д-*. Наконец, подстановка значений
т, г/, /I в уравнение (1.91) дает сумму 39,17 вместо 39,27, т. е. вполне
удовлетворительное совпадение. Поэтому можно считать, что искомая „растворимость
CuS в с (HNOg) = 2 моль • д—* равна L '^0,79 моль • д—*.
Пример 1.13. Вычислить = необходимую для растворения в 1 л
0,1 моль Саз(Р04)2.
Переход в раствор 0,1 моль Саз(Р04)2 приводит к появлению в растворе
0,3 моль Са^' и 0,2 моль РО^—-иона. Эти значения можно принять за
начальные концентрации М и А этих частиц.
Запишем матрицу равновесий системы:
Y
М А \ Y
?
0.3 1 0.2
?
CI- Са2+
РОЗ-
н+
•i = 1
i = 2
1 = 3
0
1
0
3
0
0
1
2
—1
—1
i
0
он-
Са (0Н)+
Н^РО^-з
Саз(Р04)2 (т)
W
0
Р
-14
—12,60
12,32
—26,0
19,53
21,68
28
Матрица приводит к следующей системе уравнений:
Af = m(cpjY|/i-"* +92).
3
j=o
3
J=o
n?a^ = P.
Начнем с предварительной оценки Ли/. Для суммарных равновесий имеем
(1.94)
(1.95)
(1.96)
(1.97)
Саз (Р04)2 (т) ЗСа2+ ^ gpof"
РОЗ- + jH+:;lHjPOV^
IgP
|21gaj
Саз (Р04)2 (т) + ]Н+ ЗСа2+ + 2рО^-з, Ig /(j= ig Р + 2 Ig
где Ig /Ci =-1,36, 1гА:2 - 13,06, Ig /Сз = 17,36.
Проводя расчеты для второго случая (] = 2), получаем:
' Саз (Р04)2 + 4Н+ ЗСа^+ + 2Н2РО7, Ig /С = 13,06
с у — —
[ ] ■ у-0,4 0,3 0,2
\ 3 ^ = 10~*з.0б^ Q^J^Удa ^ 0,4 + 10-^ h - 10-^ моль- л-^ рН - 4, / -
0,3 • 0,2
- 1/2 (0,3 . 22 + 0,2) = 0,7 моль • л""^ и по Девису: Ig cpj = 0,16, Ig cpg = 0,64,
lg?3 = l''^'^ и То=1-
Приводим результаты оценки значений отдельных слагаемых, входящих
в уравнения (1.94)—(1.96) при рН =4:
,0-8.44
,09.68
1011.69
108.96
101.44
,010.16
,011.99
,08.96
Из этих данных следует, что f jYj/i
—I
= 10
-8,44
« ср2 = 4,37.
Кроме того, в уравнении (1.95) можно пренебречь слагаемым с j = 0.
В результате уравнение (1.94) превращается в равенство
М = ср2т, m = /gM = 10-°'^^ . 0,3 = 1о-0'б«29 моль • л
—1
(1.98)
Из (1.97) сразу находим, что а = 10 "^^'9^^^ = 1,06 • 10-^2 моль • л"'•
Подстановка значения а в (1.95) и обычные преобразования приводят к
уравнению /1^+ 1,023 . 10-2/1^+ 1,905 . 10-9/1 — 3,950 • 10-^^ =0, решая которое
по Хорнеру, находим, что h = 6,185 . 10—^ = ю—^'^^^^ моль • л—^
Значение у у полученное путем уточненного расчета и равное 0,4008 моль-л—^
практически совпало со значением, найденным в первом приближении. Это
объясняется доминирующим характером величины cpic^fi^, очевидным из
последней таблицы.
Пример 1.14. Вычислить с^^у = К, достаточную для растворения в 1л
ее 0,2 моль свежеосаждеиного ZnS (вюрцита).
29
Из условия задачи ясно, что М = Л =0,2 моль • л""^ В приложении
находим все данные для составления матрицы равновесий системы. Образова*
нием хлорокомплексов можно пренебречь, так как значения очень малы.
Поскольку для H2S;^H2S ^(г) IgX = 0,99 и при P^^g = 1 атм (?^0,1 мПа),
^H,s ^^^^ ' ^ ^* вводим в матрицу это равновесие. В результате
С1-
М
0,2 0,2
Zn2+ S
г2-
HoS
1 = 1
I =2
О
1
2
О
О
1
О
о
о
1
о
1
-1
—1
—1
j
о
о
о
о
о
о
I
о
он-
'ZnOH+
Zn2(OH)3+
HjSl-2
ZnS (т)
w
X
р
—14
-8,96
-8,7
12,9
0,09
-21,6
19,95
Для суммарного равновесия
ZnS(T)i^Zn2++S2- lgP = -2,16
s2- + jH+i^'HjSl-2 Iga^
ZnS (t) + jH+^zi^2^
lg/fj=—8,7 и lg/C2 = —1»65. Это значит, что заметное растворение ZnS
возможно лишь при одновременном образовании эквивалентного количества
H2S. Но, как уже упоминалось, L^Is ^^^^ ' «"""^ и при Cj^^s = 0,2 мольХ
Хл—1 неизбежно выделение H2S. Тогда приходится рассматривать суммарное
равновесие: "
ZnS(T)+'2H+:;lZn2++H2S Ig/C2 = -I,65
HgSz^lHgSCr) lgX = 0,99
ZnS(T) + 2H+;±Zn2++H2S(r) lg/C = -0,66
с К - -
I ] У-0,4 0,2
0,2
Ph,s = 1 атм
10""°'^^, /1 = К —0,4- 0,956, У = 1,356.
(К-0,4)2 ■
Значение / в этой системе определяется равновесными концентрациями
tH+];==i 0,956, [CI-] ;^ 1,356 и [Zn^+l «0,2 моль-л ""Ч При этом /» 1,55
моль • л""^ и поскольку при такой / оценить Ig <Р| нельзя, приходится считать
все — 1.
Наконец, для системы, из которой выделяется HgS, нельзя записать
Уравнение закона сохранения = А, Поэтому система уравнений для нашей
30
задачи имеет вид: М = m (тг)Л * + l) + 2г^^^тЧ''^ (1.99), а = (Xagfi^)""* (1.100),
Г = Л—1Г)тЛ"-'—Y)2im2/i~^ + 2m+oiaft (1.101), та=Р (1.102).
При /I- 1 мoлb.л■~^ ф-^ 10"^'9^< 1. Тогда (1.99) переходит в М =
= m (1 + 2t]2im/i-0 ~ 'w. так как /п < 0,2, h 1 моль-л""' и ^fi^imh^^
10~9. Таким образом, из (1.99) получаемm = М = 0,2 = 10'~^'^99 моль.л'"*^,'
сразу же из (1.102) находим, что а= РтГ^ = 10~"2°'9 мoль.л-^ а из (1.100)
Л = 10-0.02 моль.д-».
Вычисление искомой ^нс! = ^ (1.101) (пример 1.14) приведено в
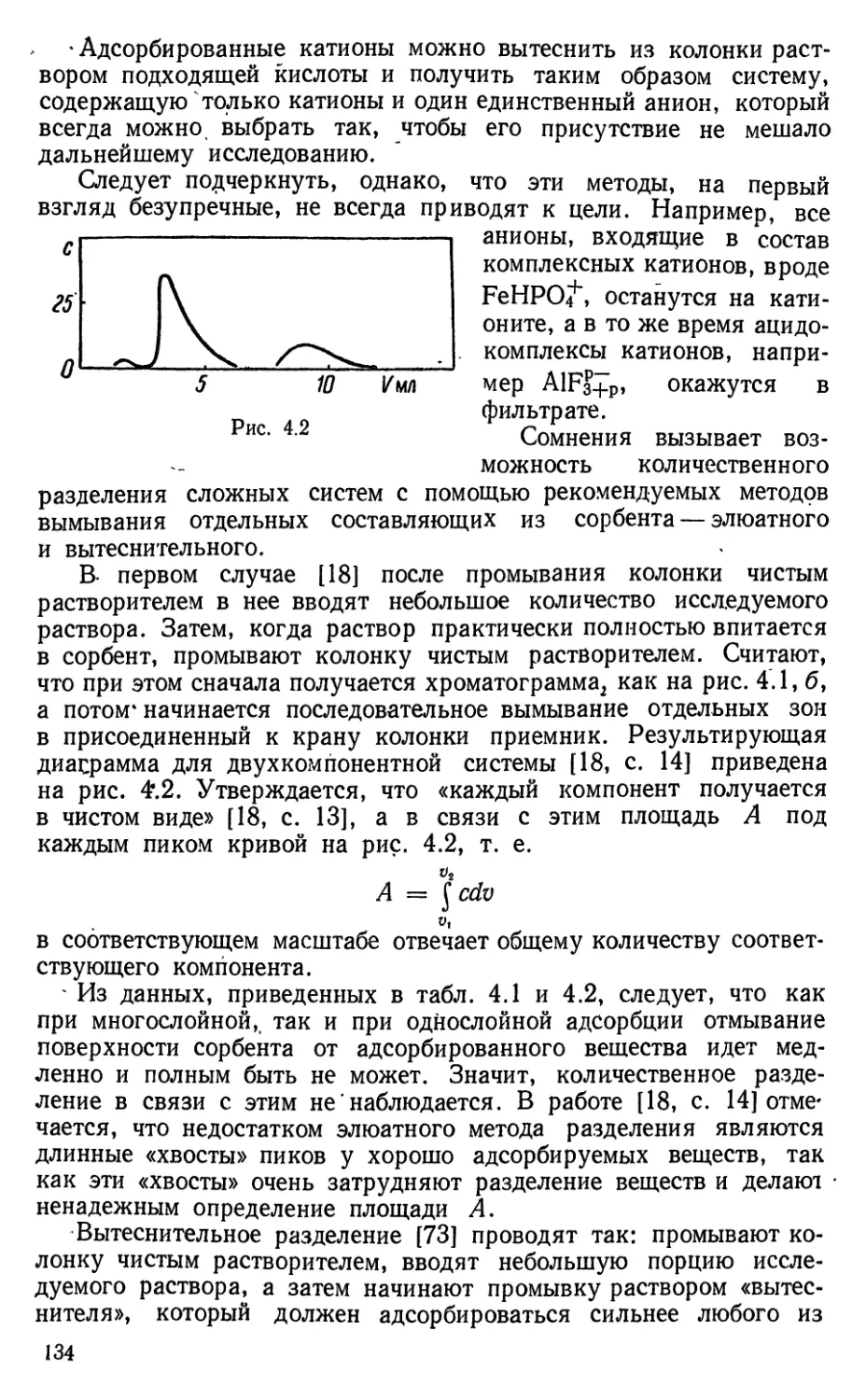
табл. 1.9.
Таблица 1.9
h, моль . л ^
ti2im2/i-l
2т
o\ah
моль-л—
10-0.02
Ф
0,955
10-9.639
lQ-10.078
0,4
10-8.02 :
1,355
Значение найденное уточненным способом, практически совпало со
значением, полученным при упрощенном расчете.
Пример 1.15. Вычислить сдсогн» достаточную для растворения в 1 л ее
О, 1 моля 5Гз(А804)2.
Введем обозначения: HCOgH = HR, H3ASO4 = Н3А. Запишем матрицу
равновесий системы:
R М А
? 0,3
0,2
R- Sr2+
AsO^- Н+
/Ci 1=1 1 1 = 2
1 = 3
0 0 0 -1
10 0 1
0 10—1
0 0 1 j
0 3 2 0
он-
HR
SrOH+
HjAsO{-3
5гз (As04)2 (т)
w
a
—14
3,75
—13,18
11,50 18,44 20,63
-18,1
Для суммарного равновесия
Srjf AsO^)2 (t) + 2jH+ :^ 3Sr2+ + 2HjAsOi-3,
Ig /С, = Ig P + 21g a,: Ig Ki = 4,9, Ig K2 = 18,78, Ig /С3 = 23,16.
Если источником Н+-ионов является HR, то при j = 2
5Гз(А804)2 (т) + 4Н+ :^ 3Sr2+ + 2H2ASO7, Ig К2 = 18,78
HR + R-. 41g g-^ = -15,00
Sr3(As04)2 (t) + 4HR :;i 3Sr2+ + 2H2ASO+ 4R- Ig A: = 3,78
31
с R , - -~
[ ] —0,4 0,3 0,2 0,4
0,4) ^ 10-3.78 д g первом приближении
0,33.0,2.0,4*
[HR] = —0,4 =8,23.10-3 и /?= 0,408 моль.л^^
Оценку /г получаем для равновесия HR Н++ R"" при [HR] = 8,2X
X 10-3 JJ [j^-] ^0,4 моль.д-^. Здесь /iW 10-3.75^
[HR] [R-]-^ = 10-5'43 моль.л-1.
Значение / определяется величинами lSr^+], [HgAsO^"] и [R""]; /
- 1/2.(0,3.22-1^0,2+0,4) = 0,9 мoль.л""^ а тогда по Девису; lgcpi = 0,15,
lgcp2 = 0,60 и lg'cp3= 1,35, далее полагаем, что <ро= 1.
Пользуясь матрицей, можно. записать расчетную систему уравнений:
M = m(cpi.)/i-i+T2) (^-103), i? = r(cpoa/i+9j) (1.104),
3
Л = fl ][] 9x_2P-^h^ (1.105), ^ Р (1.106),
j=0
3
cpj/i — cpit(y/i-^ + m (cpi-ri/i-i + 2CP2) — cpjr + a 5] (/ — 3) cpj_3aj/il. (1.107)
j=o
Значения слагаемых, входящих в систему уравнений, при рН=5,4
(пример 1.15) приведены ниже:
"РО'З*^
92»! А
¥3
2cp2»iA
3<рз
,0-7.63
10-1.65
104-«
107.79
106.70
,01.35
,07.00
101.83
Отсюда следует, что слагаемым cpjirj/i * можнсч пренебречь в (1,103)
и (1.107), а значения cpg ц Зсрз можно отбросить при разворачивании сумм
в (1.105) и (1.107).
Дальнейший расчет проводится так:
1) .из М = ср2т т= 10-0'^о.О,3= \{)-^^^'^^ моль-д-^; .
2) тогда из (1.106) а = ,(р/п-3)1/2 ^ 1q-7,3656 ^оль-д-^;
3) подстановка значения а в (1.105) приводит после обычных
преобразований к уравнению
/i3 + 9,120.10-3/i2 ^ 2,951. lO-^/i — 1,088- lO""^* = О, '
решение которого по Хорнеру дает
h = 9,423.10-7 _ 1Q-6.0258 моль.д-Ч
Вычисление ср^г по формуле
<р^г = cpj/ii- cpjoy/i-^ + 2cp2m — acp,a2/i2 — 2acp2^i^. (1.108)
32
полученной из (1.107) для рН = 5,4
приведено ниже
' 91^
292^^*
292^1ял
91Г
jQ-5,8758
10-7,8242
10-0,2219
0,5999
10-0,8272
-0,1488
10-0,9904
—0,1022
1Q—0,4574
t
0,3489
Подстановка значений г = 10 ^'^^^^ моль.л * и Л в (1,104) дает искомое
значение = 0,35 моль.д—Ч
Пример 1.16. Вычислить Cj^j = У, достаточную для растворения
0,1 моль HgO.
Матрица равновесий системы имеет вид:
Y
м
Y
? 0,1
?
Hg2+
1-
Н+
К,
i= 1 i = 2 i=3 i =4
0
1
1
1
0
0
п
0
—1
-к
0
—2
он-
Hg(0H)2-k
Hgir"
HgO(T)
w
Ik
к
p
-14
-3.96
12.87
—25.5
—6.52
23,82
-22.57
27,60
29,83
Составим расчетную систему уравнений:
3 4
к=0
n=I
n=0
mto^ = mw^hr^ = P, .
3
,f^h-^f^h-^w-\■Y + m V (2-k)^a-k'lk''"''+
k=0
(1.109J
(1.110)
(l.Ul)
+ m2 (2^n)92_„M"=0- (1-112)
n=i
Считая, что растворение HgO идет за счет комплексообразования,
находим:
HgO(T) + H20
Hg2+ + rtl-
tHg2+ + 20H-
tHgl2-°
IgPn
HgO (T) + H2O + nl- :j± 20H- + Hgl^-" Ig VC„ = Ig P + Ig fJ„
Ig/C, =—12,63, lg^tC2=—1,68, lgK3 = 2,l, lg/C4 = 4,33.
.8 3-457
33
Поскольку заметной растворимости можно ожидать лишь при п > 3,
предварительный расчет приходится вести для равновесия:
HgO (т) + Н2О + 31- 20Н- + Hgl Ig /Сз - 2,1
с Y — —
11 К-0,3 0,2 0,1
^^"""У = 10-2.', откуда = К-3 = 10-1">993 _3^2.10-2
0,2 «0,1
Y - 0,332, [ОН-] - 0,2 = 10-^'^^^, 1Н+] - Ю-^^.з^ ppj _ 3
Тогда из (1.111) 10-^*'^ моль-д-^
Величина / определяется здесь значениями: [К"^] = 0,332, [j-] = 3,2 X
X 10-2, [ОН-] =0,2 и [HgIJ-]=0,l моль-л-^ Для /- 0,332 моль-д-^
по Девису: Ig^j =0,15, lg^2 = ^»^^» а ?о=
Оценка произведений Тг-к'^к^""^ ^ Тг-пРп^'^» полученная с помощью
приведенных выше приближенных значений, дана в табл. 1.10.
Таблица 1.10
90^2^1-2
92
S
1017.18
1,514 . 10^7
1020,08
1,2023 . 102°
109,19
,00.б
1020,0805
t
1,2038 . 102°
10-4,0195
<Р2Р4И
90^2^2
9lPl£^
92
mS
1q24,4328
2,709 . 102*
^ jq23,2521
1,787 . 1023
jq20,8214
6,63 . 102^
ioii.52O7
ioo.6
1024,4607
t
2,8884 . 10^^
I00,3607
Из табл. I.IO следует, что суммами, включающими произведения
^2-к'^к'^~"^», можно во всех уравнениях пренебречь, а в суммах с индексом п
проводить суммирование от п = 2 до п = 4.
Теперь система принимает вид:
4 , 4
п=2 п«=2
4
■ = P(I.1II), -cpjCo + K + w^ (2-n)cp2_„p„r/'^ = 0 (1.115),
n=2
Дальнейшие вычисления следует вести так:
1) исключая Y из (1.114) и (1Л15), получаем
4
—^jo) + ^jr/ + 2w ^ ?2-nPn^" = О» •
n=2
(1.116)
2) замена суммы в (1.116) ее значением из (1.113) дает после обычного
преобразования значение
(1Л17)
34
3 последующее исключение т из (1.113) и (Г. 111) приводит к уравнению
9]Р (т2p4I/' + ^lPз?/' + ?oM^) --(2М + 9,г/)2М =0, (1.118)
переходящему в уравнение
/ + 2,089.10"V — 9,296. V — 3,328.10"^ — 2,355. Ю"» ^ о.
Расчет по Хорнеру дает l0-l•^^7° ^ 1,239-Ю"-^ моль.д'^
Дальнейший расчет по (1.1)3) и (1.П4) приводит к значениям m = io-23,8703 ^ К ==
= 0,4027 моль.л"^
Пример 1.17. Вычислить c^^^q^^Y, достаточную для растворения
0,03 моль AggS.
Из условия задачи следует, что = 2L == М = 0,06, а c^=zL ^
= 0,03 моль-л""Ч
Растворимость N0, которая должна получаться при взаимодействии HNO^
с AggS, равна 10~^*^ = 2'10~з моль.л"^ Следует ожидать поэтому, что в
нашем случае N0 будет выделяться из системы при р^^ = 1 атм (::^0,1 мПа).
Матрица равновесий системы имеет вид:
м
L \ Y \ Y
0,06 1 0,03
?
?
Ag+
S2- 1 NO3-
N0
1 1 = 1 1 i = 2
. 0
'Л
1
0
0
0
0
2
0
0
1
1/2
0
0
1
0
0
0
0
1/3
0
0
—1
-к
j
0
4/3
0
0 ^
0
0
0
0
0
1
0
6н-
1/2S(T)
1/3 NO
NO (г)
Ag2S(T)
w
X
p
-14
-11,7
12,9
1/2.14,17
1/3 48,5
2,68
—49,2
—23,8
19,95
Неизвестны Y, m a, y, h. Используем систему уравнений:
2
c^g=M = 0,06 моль.д-^ =/n ^ (Ы19)
k=0
^n03 = ^ = Ti^+2/3L, (1.120)
2 2
Ch - К = cpr'i - S k^i^kvji^^-k + a S j>j«2^j^^ + 8/3/:, (M21)
k=0 j=0
2
cpj/t —(piw/i~^—cp (/+m Y (l-^k)Ti-.k'^k^""^ +
k«o
3*
+ «S (j~2)9j^2^j/i^=0,
j=0
(Ы22)
(1.123)
35
Для предварительной оценки равновесных концентраций / и Ig<p| исполь-
ьУем приближенное решение:
AggSW :^2Ag++S2- 3
S2- -2e;±S(T) 3
NO 7 + 4H+ + 3e :^ NO + 2H2O 2
NO -l^ NO (r) 2
3lgP =-147,6
3Mg/C) = 42,5
2 Ig/С = 97,0
21gX = 5,4
3Ag2S (T) + 2n0^ + 8H+ :^ 6Ag+ + 3S (т) + 4H2O + 2n0 (г) Ig /С = -2,7
[) К-0,02 Г-0,08 0,06 J
(К—0,02)^.(К-0,08)^(0,06)""^ = 10^'^ Извлекая квадратный к'орень,
получаем {Y —0,02) (К —0,08)= 10"^'3154^ Допуская некоторое превышение
величины У, принимаем, что (К —0,08)^= 10~^.32. Хогда К =0,43, [NO^J =
= 0,41, [Н+]=0,35 и [Ag+] = 0,06 моль.л-!, / == 1/2 (0,41 + 0,35 + 0,06) =
= 0,41 моль.л""^ Далее принимаем <pq = 1 и находим по Девису: lg<Pi =
= 0,15 и Ig <Р2 = 0,60.
Результаты расчета отдельных произведений и сумм, входящих в
уравнения (1,119), (1.121), (1.122), приведены ниже:
■ 2
2<Ро02Л2
2
al
10-22.73
10-11.24
100.15
100.15
1019,331
1012,59
1019,331
10-27.7
Учитывая эти результаты, можно пренебречь выражениями a'^j^^_20.h^
в (1.121) и ^^(j—•2)(pj_2<^j^^ в (1.122). Далее, используя значения,
приведенные в первых трех столбцах таблицы, находим, что
2
^ S kcpi«kY)k^~^ = m(2cpji^2^"4TOi^--40.cpj|= 10-i2'46^^j.^
m ]2 (l~k)<pi_k^k^ ^ = '«(—'fi'^2^""^ + 0 + ?il = cpi'«
к=0
И приводим нашу систему к следующему виду:
М = 0,06 моль.д"-^ = <pjm, т = f^M,
r=^cpii/ + 0,02, t/ = /i(r-0,02),
r = (pj^+0,08, Л =/1 (К-0,08),
и дополняем ее уравнением
/Л«т-б=102.^
(1.124)
(1.125)
(1.126)
(1.127)
т. е. выражением закона действующих масс для суммарного процесса раство-
ч!1.ия AgSa в HNO3.
£6
Подстановка значений у, h а т ъ (1.127) приводит после обычных прэ
образований и извлечения квадратного корня к уравнению:
(У-.0,02) (У-0,08)^= \0^'^\fM^= 10-2'^^^^
1.128)
Значение Y моль.л""^ лежит на интервале 0,40 < У < 0,48. Границы
получаются, если один раз считать левую часть за К^, другой раз — за (К —
—- 0,08)^. Окончательное решение должно удовлетворять равенству
Ig (У - 0,02) + 4lg (У - 0,08) = —2,0154.
Его следует искать путем подстановки. Окончательный результат: У
= 0,464 моль.л~^
путем.
вместо К-=0,43 моль-л ^ полученного приближенным
1.7. Расчет оптимальных условий осаждения
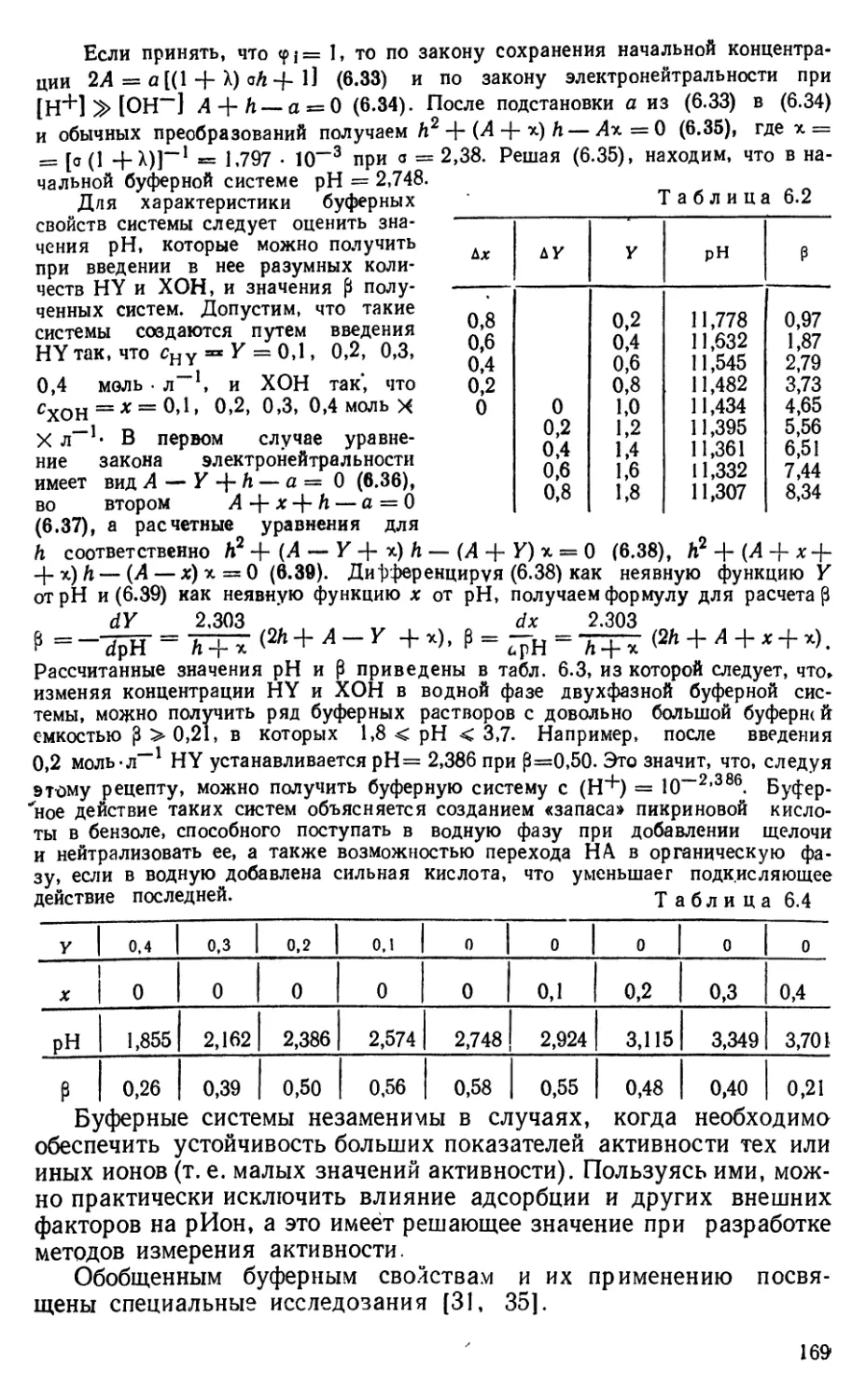
Растворимость любого осадка MaAjj. зависит от рН, так как
при росте рН снижается (Mi"+), за счет возрастания (M(OH)IJ'"''),
а при снижении рН уменьшается (А°'-) за счет роста (HjAJ-'*).
Ясно, что полнота осаждения MaA^^ должна обеспечиваться при
каком-то вполне определенном значении рН, обеспечивающем
оптимальные (А«-), (Mf^+).
Выбор соответствующего рН, отвечающего минимальной
растворимости малорастворимого вещества МаАр., чаще всего
осуществляют с помощью диаграмм Н. Бьер-рума [51], дающих
представление о доле данного иона {!А^+ или А°'-) в системе при разных
рН. Считают при этом, что оптимальные условия достигаются
при рН^ которому одновременно отвечают наибольшие доли
каждого из ионов решетки. Этот путь нельзя считать рациональным,
так как он дает лишь очень приближенную оценку рНопт»
Гораздо более точный аналитический подход сводится к
вычислению рН,, которому отвечает минимум растворимости вещества
Пусть ci^y^=iM, сх^А=А. Будем искать рН, которому отвечает
минимальная растворимость L, считая для простоты, что cpi=: 1.
Матрица равновесий системы, при отсутствии многоядер ности,
имеет вид
аА
М
А
Х+
Y~
н+
К,
0
1
0
а
0
0
1
—1
-к
j
0
0Н~
М (OH)iJ-«
МаА^ (т)
W
Для решения за ;ачи необходимы уравнения:
М = m Ц -»)кЛ-« + aS (1.129), А = a^ajhl +(л5, (1 '
37
где S —количество МаА{х(моль), выпадающее из 1 л системы
и т^а^ = Р (1.131).
Для единственного рНопт» который мы ищем, М, Л, S
—фиксированные величины, а разность M — aS или Л —|а5, в
зависимости от того, какой из ионов находится в избытке, является
растворимостью. Допустим, что в избытке взяты Л'*--ионы. Тогда
(1.129) можно записать в виде
L = M — aS^m Ц ^кЛ~«. (1.132)
к«=0
Дифференцирование всех уравнений по h (штрихами помечены
производные) и приравнивание L' нулю дают систему уравнений,
связывающую производные и переменные Л, m и а в экстремальной
точке:
L' = т' S ^h-^ — m S к-^к^-^-^ = О, (1.133)
к==0' к=0
а' а"'
0 = а' S <3ih^+a S jaJni-^ (1.134)
am«-im'a»^ + (xmW-^a' = 0, am'a + ]ima' = 0. (1.135)
Подстановка в (1.135) значений m' из (1.133) и а' из (1.134)
приводит после сокращения на та О к уравнению
о:"^^ = 0, (1.136)
, №0 j=0
позволяющему вычислить искомое значение Лопт.
Если уравнение (1.136) умножить на Л, то множитель при а
превратится в функцию Бьеррума пон = Л-н, выражающую среднее
число гидроксильных ионов, приходящихся на 1 моль частиц,
происходящих от катиона Ш"^ или П-н —среднее число оксоний-
ионов, отданных при ионизации гидратированного №+-иона. В то
же время множитель при (х представляет собой п+н —среднее
число протонов, входящих в образующиеся Н]А1~'*-ионы.
Уравнение (1.136) можно записать теперь так:
ал.н (М»^+) = (хл+н (А--). (1.137)
Таким образом, в рассмотренном случае, независимо от
начальных концентраций М и Л, растворимость МаАр. минимальна при
рН, при котором общее число протонов, получающихся при
образовании всех М (ОН)^""'^ из а моль М«^+-ионов, совпадает
с числом, идущим на образование всех HjA1~"-hohob, которые
дают и. молей аниона А*-.
38
2Л
Na+I
2M+R
cr
0
1
0
0
1
R+R JV1_
NHs
0
0
1
0
0
0
0
0
—1
j
1
—I
0
NH+
0H~
MgOH+
MgNH4P04
w
P
-14
12,32
9,24
-12,8
-12,6
1 = 2 i = 3
19,53
120,68
Пример 1.18. Вычислить рН, отвечающий минимуму растворимости
MgNH4P04, если с^а.нро, = А, Cд^gclз = с^ир = %н, = ^» ^ Mg^-t"
и ЫН|'-ионы находятся в избытке.
Считая все ср^ = I и используя матрицу равновесий системы, можно
записать систему уравнений:
3 3
А = а^ ^j^^ + 5или/, = Л — 5 = а^ a^hK (1.138)
j=o 1=0
R + R:=:r(ah+\) + S, (1.139)
M = m (1+^^-0 + 5, (1.140)
тагЛа = P или тлаЛ = Pa""^ .(1.141)
где L — растворимость; S —количество MgNH4P04 моль в 1 л системы и
r«(NHp).
Дифференцирование системы по h приводит к уравнениям
3 ' 3
L'=a' ajfil+a 2 jajfil-J, (1.142)
j-O j=0
0 = r'(a/i+ 1) + ar, (1.143)
0 = m' (1 + ^^""0 — ^m;i-2, (1.144)
mVa/i + mr'ah + тга'Л + mra = 0, (1.145)
так как при фиксированном Л, отвечающем минимуму L, S, также имеет фик,
сированное значение.
При L' =0 получаем значения а', г' и т' из (1.142)—(1.144) и
подставляем их в (1.145). После сокращения на тга и обычных преобразований
получаем
3
,~1
1
— и,
(1.146)
/=о
где первое слагаемое—молярная доля MgOH"^ в смеси Mg^+ ja MgOH+,
второе — молярная доля NHg в смеси NH"^ и NH3, а третье — равно п^ц{^0\'^^,
на 1 моль всех HjPO|f ^. Поскольку одновременно с образованием каждого
39
MgOH"^-HOHa в систему переходит один НдО^-ион и в том же соотношении
ионы получаются при образовании NH3 из NH+.ионов, физический смысл
уравнения (1.146) хорошо выражается равенством
«-н(Mg^'*- ) +"«-н'(^ + ^) - (РО'Г) = 0. . (1.147)
После обычных преобразований уравнение (1.146) переходит в уравнение
Saagfl^ + 2 (Tiaag + + ао^) +'^^^2 + ^^3 + <'2 + ^^l) ~
- h + ^l) + 1]^ - 27) = О, (1, ,48)
а после подстановки значений констант —в уравнение
;,5^ ,0-1,32б1^4_,_ ,о-8,833^3_ Ю'^^'^^^^^^ _ 10^42,8961 _ (1 ,49^
Анализ (1Л49) показывает, что при одновременном выполнении условий:
10-12,б232<<^^^ 10-7,5069 ^^10-1,3261 ^^^^^^^^ значения h ^ \(Г^^*'^^ ыолъ ^л^^
рН = 10,72.
Пример 1.19, Рассчитать условия наиболее полного Осаждения А1 (ОН)з,
если Сд|у =М=:0,1 моль.д""^ и А!^"*" не координирует У^-ионы.
Матрица равновесий системы, включающей осаждение мет'алло-иона с
помощью ХОН, имеет вид:
X
ZM
М
?
0,3
0,1
х+
Y-
0
1
I
2
1
—1
—1
—4
—2
—3
он-
А10Н2+
А1 (0Н)7
А1,(0Н)^+
А1 (ОН)з (т)
W
-Па
Р
-14
-4,3
-24,25
—14,56
- -32,4
Чтобы оценить /, будем считать, что для количественного осаждения
0,1 моль AIY3 понадобится 0,3 моль ХОН. Тогда в 1 л раствора оста!!ется
0,3 моль Х'^и 0,3 моль У'-ионов, что отвечает / = 0,3 моль.л~'- По Девису
Igcf, = 0,15, .lg<P2 = 0,60, Ig 93=1,35 и lgy4 = 2,4.
В этих условиях
/ =т (cp3 4-cp2Y;7i-l| + yj7)4mh-"^+y47i2m2fi-2 (1,150), m/i"-^ = .151),
где Z. — растворимость*
Дифференцирование по h дает:
L' = т' (уз Ч- 92^ЬГ^) — ?2^mft-2 + yj'ri4m'fi-^ — ^^'I'^A^f^'^^ +
+ 2(p4'r)2mm'/l~^ — 2^^г12тЧ-'^ = 0, (1 .Л 52)
m'/i~^ —3m/i-^=0. (1.153)
40
Полагая ^=0 и подставляя значение т' из (1.153) в (1.152), получаем после
обычных преобразований уравнение
(1.154)
4ср41Г)2Рш-3/1^ + Зсрз/1^ + 2cp2^fl^ ~ cpi7)4 = О
ИЛИ
+ 103'785;^4 ^ |0-1.44;^3 _ jo~22.142l ,55^
Безусловно, /i < 10^»^^^ моль.л""^ Тогда (1.155) можно записать в виде
3 Ig /I + Ig {h + 5,957. Ш-^) == —25,9271
и найти h путем пробных подстановок. С очень хорошим приближением
находим, что минимальной L отвечает рН= 6,903, а из (1.151)'получаем, что
при этом рА1 = 11,109 и т = lO'"^^•^°^ моль • л~^
Расчет L по уравнению (1.150) и при рН = 6,903 > дан в табл. 1.11.
Таблица 1.11
93
ср2'Г)Л--^
( )
m( )
L,
моль.л—^
S,
моль-л—^
iOl.35
2,2.10^
103.203
1,596-10^
1q3,2089
t
1,618-103
1^^7,9001
1,259.10-^
10-7,5970
2,530 X
xio-^
10-20,572
3,789 X
х10~^
0,1- '
3,789 X
хю""^
Из табл. 1.11 следует, что при правильном выборе рН в растворе
остается всего 3,8-10""^ % начального количества Al^+'nona. Расчет концентра-
лии ХОН, которую надо создать для достижения такого результата,
проводится по уравнению
. . . X = [А10Н2+] + 4 [А1 (0Н)7] + 2 [AI2 (0Н)^+] + 35. " (1.156)
Результаты расчета оптимальной концентрации ХОН при осаждении А13+
приведены ниже:
2ср41Г)2т2Л-2
35 моль-л ^
X моль.л ^
,0-7.906
1,242'. 10-^
10-6.9949
1,012 . 10"^
10-20,271
0,3-1,137-10-^
0,3—8.10--^ ^
Отсюда следует, что создать концентрацию X, точно отвечающую расчету,
п'рактически невозможно, особенно в тех случаях, когда ^aiyj неизвестна.
Поскольку снижение 5 при X < ЗМ очевидно, остается выяснить вопрос об
изменении L в случае превышения оптимальной с^он*
Допустим, что ^хон "2 1 % превышает Х^^^ = 0,3. В этом случае X =
= 0)303, Ж = 0,1 мoль.л"■^ и можно записать, что M = L+S (1.157)^
где L выражается уравнением (1.150), и
X = cpja;/i-^ -.^^/i-j^^^r^mk-^ + 4^,714m/i"^ + 2cp47)2m^/i~"'^ + 35, (1.158)
где (pjfi эквивалентно доле [OH"~J за счет диссоциации воды.
и-1
41
Если (1.157) после подстановки в него значения L умножить/на 3,
вычесть его из (1.158) и заменить т, используя (1.151), то уравнение для
расчета h принимает вид
+ (X — ЗМ) л — (ср1'^4РйУ""3 — cpjay) = 0. (1.159)
или
_^ ,о4.3871д4 ^ 10-0,839^3 ,0-6.89^2 ,0-9.5629^ _ J0~20.8023 _ о.
(1.160)
При анализе этого уравнения, относящегося к щелочной области, можно
считать, что/1«10-^'°^1 < 10-5.2261 104,з871 мoль.л-^ тогда
^^ 10-11.2394 моль . л-1, рН= 11,24, т = 10-24'11»2 моль.д-^
Расчет L при рНг= 11,24 и m = Ю-^'*'^^®^ моль . д-^ (пример 1.18) дает
величину ;^ 5,5. ЮТ"* моль-д-^
Таким образом, возрастание X на 1 % ведет к увеличению L на 4
порядка, причем основную роль в росте L начинает играть второе слагаемое
правой части (1.150). Учитывая это, можно считать, что при избытке ХОН
и рН > рНоп^
Ig ^ = Ig ?i'^4^^""^ + рН = -1^,5 + рН. (1.161)
Значение h можно всегда рассчитать по (1.159) после подстановки в него
соответствующей разности X —3Af.
Проведенные расче1ы показали, что ^а1(0н)з можно свести к очень
малому значению, однако обеспечить соответствующие оптимальные урловия при
осаждении А1 (6Щ^ раствором ХОН практически нельзя. Рассмотрим в связи
с этим возможность замены ХОН каким-либо слабым основанием.
Поскольку слабые основания А и А"— являются такими же акцепторами
протонов, как ОН—-ионы, все предыдущие расчеты остаются в Силе и надо
установить лишь с^, или эквивалентную ранее вычисленному
Из сопоставления равновесий
A + H+:;tHA+ а
Нр :^Н+ +0Н- W
A + HjOz^tHA^+OH-
А- +Н+4:НА а
Н^О :^:н+ + ОН-ш
А- + HgO НА + ОН-, ZW
(второе из них отвечает А* при а = 1) следует, что к моменту, когда
расход ОН~-ионов достигнет значения X, в системе должно накопиться X моль
НА+ или НА.
Концентрации Ср^ = или с^д = ^2» обеспечивающие такое накопление,
определяются из соотношений
[НА+] = Л, = Х и НА = Л2 ^^/^ = (Ь162)
различающихся лишь значениями коэффициентов активности. Из них
следует, что
^на+^'^ ^на^^
42
Решающую роль в этих выражениях играет произведение ah. Так, при
^опт = I0""^*^ моль.д""* достаточно взять основание, для которого Iga =
= 6,9, чтобы и А2 стали примерно равными 2х, что заметно облегчает
регулировку рН до соответствующего изменения окраски подходящего
цветного индикатора.
Среди оснований можно выбрать и такие (для них Iga < рН^^х)» ДЛя
которых достижение опасной зоны, где [А1 (ОН)^] становится недопустимо
большой, вообще невозможно. ^
Пример 1.20. Выяснить путем расчетов условия, при которых можно
рассчитывать на возможно полное осаждение Hg^+.nona сероводородом.
Предположим, что раствор, содержащий Hg^+-иoны, насыщается HgS
при атмосферном давлении и избыток HgS уходит в атмосферу. Будем искать
рН, обеспечивающий минимальную растворимость осадка HgS, считая, что
Bcecpj=l. В этом случае при c^^gy^ = М моль-л~"^ матрица равновесий
системы имеет вид
2М
М
Hg2+
н+
Ki
i= 1
i = 2
1=3
1
0
0
1
1
0
1
0
2
1
—к
i
0
0
0
0
0
1^
0
0
Hg(0H)2--^
HiS^-2
HgS (г)
Hgs|-
HgS(T)
h
p
—3,96
12,9
0,99
—51,8
—6,52
19,95
54,7
-22,57
Растворимость выражается уравнением
3
L = m ^ fl^h-^ + pgmas.
k=0
(1.164)
Если насыщение сероводородом идет при 25 °С, то (HgS) = так как
PHsS = I ^™ 0,1 мПа). Но тогда (HgS) = Ogo/i^ = и а оказывается
фиксированной величиной: а = X^^a^^/i^^ (IM65).
Теперь растворимость и выражение произведения растворимости принимают
следующий вид:
к=0
m/i-2 = РХа.
(1.166)
-2. (1.167)
Дифференцируя два последних уравнения по Л и приравнивая L' нулю,
получаем условия экстремума:
3 3
^'==^'11 ^.к^"'^-'" II к^к^""^""^ - 4р2>^-2,-2д-5^ ^
к=0 к=0
(1.168)
43
m'/i-2 - 2т/г-з = О, или m'= 2m'i-^ (1.169 )
После подстановки значения т' в (1.168) и обычных преобразований
получаем расчетное уравнение
2X2a2/l4 + ^,Х2а|лЗ ~ y^^x2^2h - 2^2 = О
или
(1.170)
(1.171
д4 ^ ,0-4,261/^3 _ ,о-22.871д _ ,о12,82 _ ^
Здесь безусловно Л< 1035'^^ моль-л""^ и почти наверное /i>10-'*'^^^ моль-л^^
Тогда 10^^'®^ и ^опт °= 103'205 моль.л""^ что недостижимо.
В подобных случаях необходимо выяснить изменения, которые придется
внести в уравнение для расчета L при h < Л^^^, и провести -пробный расчет
при каком-нибудь доступном значении Л. Расчет растворимости осадка HgS
по (1.166) при /I = 1 приведен ниже, где из уравнения (t.167)
10-30.86 моль.л-1 и
= Р2Х-2а-2_1012.82^
1о
i, моль-л"'
,0-22.57
,0-6,62
,0-3,96
10'
10-30.86
,0-18.04
10-18.04
Отсюда следует, что даже при h, на три с лишним порядка меньшей
/Iqj^^, растворимость HgS достаточно мала, чтобы считать осаждение Hg2+-
иона практически полным. В то же время выясняется, чго при рН > рН^„^
основную роль в расчетной формуле (Цбб) играет последнее слагаемое.
Если, учитывая это, исключить из (1.166) первое слагаемое, заменить во
втором т, используя (1.167), и прологарифмировать полученное выражение, то
новая расчетная формула Ig L = 2рН — 18,04 (1.172) позволит быстро
находить растворимость при любом заданном рН.
Расчеты для систем, в которых . одновременно осуществляются кислотно-
основные равновесия, и комплексообразование, а тгкже идут окислительно-
восстановительные и гетерогенные реакции, оказались значительно сложнее
расчетов, " рассмотренных для одних кислотно-основных равновесий. В этом
случае нельзя установить общие критерии допустимости упрощений, а
приходится анализировать влияние отдельных блоков. Приведенные примеры
характеризуют общий подход к решению этих более сложных задач.
ВЫДЕЛЕНИЕ ТВЕРДОЙ ФАЗЫ
ИЗ ГОМОГЕННОЙ СИСТЕМЫ
2.1. Общие сведения
Если MaV(T)?=taMt^+ + txA«- Р (2Л), с^^ ^с^^^=^М,
% А ^А«- = ^ И ММ»" > Р (2.2), то можно ожидать
выделения твердой фазы МаА(х(т), а после достижения равновесия —
выполнения условия nV^ay-= Р (2.3).
Однако в некоторых случаях в системах, в которых условие
(2.2) выполняется, видимого выделения твердой фазы не
происходит. Осложнения этого рода возникают при пересыщении,
когда твердая фаза вообще не выделяется; при образовании
золя MaAjx, т. е. в тех случаях, когда твердая фаза выделяется
в настолько раздробленном (дисперсном) состоянии, что увидеть
частички ее в растворе невозможно и система сохраняет
кажущуюся гомогенность.
• Для того, чтобы разобраться в первом осложнении, надо
помнить, что значение произведения растворимости Р
характеризует устойчивость кристаллической решетки МаЛ^ в данной
гетерогенной системе.
Произведение растворимости зависит от кристаллической
структуры выделяющейся твердой фазы, от природы
растворителя, размеров выделяющихся криста;1лов, температуры (/) и
давления (/?):
Р = /((/, а, г, /, р), - (2.4)
где и — энергия кристаллической решетки; о — поверхностное
натяжение на границе между твердой фазой и раствором; г —
средний радиус кристалла.
Кристаллическая решетка подавляющего большинства
твердых солей имеет ионную структуру. Отдельные ионы,
образующие такую решетку, например Ка"^-ионы и С1~-ионы в случае
хлорида натрия, непрерывно колеблются вокруг определенных
положений равновесия — «узлов» кристаллической решетки, но
не могут далеко отходить от них. Если отдельные ионы
рассматривать как точечные заряды, то сила взаимодействия между
ними по закону Кулона равна
/ = С^. (2.5)
45
где zi и Z2—заряды ионов; / — среднее расстояние между ними,
в — относительная диэлектрическая проницаемость среды (в
вакууме е=1); С —константа.
Амплитуда колебаний ионов в решетке тем больше, чем выше
.температура. При некоторой, вполне определенной для каждого
данного кристаллического вещества температуре межионные силы
притяжения становятся неспособными противодействовать
хаотическому тепловому движению ионов — кристаллическая решетка
разрушается, вещество плавится.
Совокупность всех сил, удерживающих ионы в узлах
решетки, характеризуется энергией кристаллической решетки f/, т. е.
полной энергией, выделяющейся при образовании 1 моль
кристаллов данной соли из свободных газообразных ионов.
Согласно А. Ф. Капустинскому, значение V и свойства,
связанные с этой энергией, определяются количеством
структурных единиц кристалла Sn» т. е. общим числом ионов в
элементарной ячейке, их размерами, валентностями, а в ряде случаев
й поляризационными свойствами. Для расчета энергии
кристаллической решетки бинарных соединений в кДж-моль~^ можно
использовать формулу Капустинского:
и - 107,2 .^^^Т^, (2.6)
'к "Т" 'а
где гк, га — эффективные радиусы катиона и аниона по Гольд-
шмидту, выраженные в нм; шк, йуа —их валентности, 107,2 —
константа.
Например, в случае NaCl: йук=^==1, 2п= 2, rj^^4.= 0,098нм,
г^1-.= 0,181 нм, а значит II^^c\=T^^J кДж.мoль~•^ что
практически совпадает с экспериментальным значением.
Следовательно, при образовании 58,5 г кристаллов хлорида натрия из
газообразных ионов выделяется 768,7 кДж тепла.
Если вещество МаА^^ полиморфно, т. е. может существовать
в виде двух или нескольких кристаллических структур, то
структуре с наиболее плотной упаковкой будет отвечать
наибольшее значение U и наименьшее Р, Так, например, для ZnS
в структуре цинковой обманки (сфалерита), отвечающей наиболее
плотной кубической упаковке, рР = 23,8, а для того же ZnS
в более рыхлой структуре вюрцита рР = 21,6. В этом и в
других подобных случаях значения' рР всегда меняются
скачкообразно.
Отметим здесь же, что вероятность образования менее
упорядоченного и лучше растворимого соединения с меньшим рР всегда
выше. В результате, как правило, из раствора в первую очередь
выпадает соединение с большими Р, т. е. более растворимое,
хотя на первый взгляд можно ожидать обратного.
При погружении в растворитель ионы кристалла
подвергаются ударам его молекул. Одновременно уменьшается и сила
46
электростатического притяжения между противоположно
заряженными ионами, так как входящая в уравнение (2.5)
относительная диэлектрическая проницаемость е для любого
растворителя всегда больше единицы, а для водных растворов
достигает 80.
Оба эти обстоятельства способствуют растворению в тем
большей степени, чем меньше U и больше Р (например, для
ZnS в структуре вюрцита больше, чем для цинковой обманки).
2.2. Зависимость растворимости от температуры
Влияние температуры на растворимость характеризуется
обычно кривой растворимости данного вещества, выражающей
зависимость растворимости от температуры. Пример такого
графика дан на рис. 2.1. Каждая точка кривой АВ отображает
растворимость (L) макрокристаллов данного вещества*,
отвечающую данной температуре t.
Каждая точка плоскости, лежащая ниже кривой,
отображает в таком случае концентрацию вещества, меньшую, чем
растворимость L при той же температуре, т. е. соответствует
ненасыщенному раствору данного вещества. Любая точка выше
кривой показывает суммарное количество вещества в системе.
Часть этого количества, отвечающая значению L[t), находится
в растворенном состоянии, а все остальное в виде твердой фазы.
Рассмотрим теперь некоторые опытные факты. Допустим, что
взят ненасыщенный раствор, концентрация которого при
температуре ti выражается ординатой ati. Казалось бы при
постепенном снижении температуры концентрация должна сохранять
постоянное значение до температуры ^2, отвечающей точке й,
лежащей на кривой растворимости. При последующем же
снижении температуры, наоборот, можно ожидать снижения раство.
римости вдоль кривой растворимости АВ по направлению к А,
сопровождающегося выделением избытка вещества в виде
твердой фазы. Однако фактически для веществ, дающих
пересыщенные растворы, выделения твердой фазы при температуре /2 не
замечается. Снижение температуры системы можно продолжать
дальше, пока при какой-то температуре, скажем, /3, начнется
образование твердой фазы; для веществ, растворимость которых
возрастает с ростом температуры, .это сопровождается
выделением т^пла, затраченного ранее на растворение, а, значит,
и повышением температуры системы до /4.
При этом выделяется столько осадка, что концентрация
вещества в растворе становится равной нормальной
растворимости, изображаемой ординатой d^4. При дальнейшем снижении тем-
пературы концентрация вещества в растворе будет снижаться
соответственно кривой растворимости ВА.
* Макрокристаллами мы называем такие кржталлы, для которых
увеличение линейных размеров практически не влияет на L.
47
Многократно повторяя опыт при разных начальных
концентрациях ненасыщенного раствора, можно получить кривую CD,
ограничивающую область возможного переохлаждения растворов,
не сопровождающегося обязательным выделением твердой фазы.
Для каждой температуры, например hy все точки, лежащие
ниже точки отвечают ненасыщенным при этой температуре
растворам; точка е соответствует нормальной растворимости
макрокристаллов вещества при температуре ^з; точки между точками
е м с отвечают метастабильному пересыщению, так как в этой
области пересыщение иногда осуществить удается, а иногда нет.
Выше точки с лежит область концентраций, отвечающих
неустойчивому пересыщению, так как сохранить гомогенность при
этих концентрациях нельзя и все попытки этого рода
заканчиваются выделением избытка твёрдого вещества и нагреванием
раствора до температуры t^.
Для малорастворимых веществ, например BaS04, подобную
же диаграмму можно получить, рассматривая при постоянной
температуре системы, в которых' произведение сначала
меньше Р, затем становится равным Р, и, наконец, начинает
превосходить эту величину. Поскольку в этом случае изменение
температуры при выделении твердой фазы неощутимо, применяя
чувствительные методы анализа^ можно найти значения
концентраций, отвечающих точкам е \Ссу окаймляющим область
неустойчивого пересыщения.
В подобном виде явления пересыщения были впервые
представлены в 1897 году И. Оствальдом, который пришел к выводу,
что при постепенном повышении концентрации растворенного
вещества в системе она проходит через три состояния:
устойчивого ненасыщенного раствора, метастабильной зоны, где
кристаллизация невозможна без введения «затравки»; зоны лабильных
растворов с самопроизвольной (спонтанной) кристаллизацией.
Само явление пересыщения и его устранение с помощью
затравки были открыты русским академиком Т. Е. Ловицем в
1794 г. В те годы еще ничего не было известно о строении
кристаллической решетки, но многие знали, что кристаллы
раскалываются по плоскостям спайности и для данного вещества имеют
всегда определенную и такую же регулярную форму, как,
скажем, солдатский строй, который часто приходилось наблюдать
Ловицу в современном ему императорском Петербурге. Не даром
свои представления о возникновении кристалла в гомогенном
растворе Ловиц выразил в следующих словах: «Мне показалось,
что все находящиеся в растворе частички соли спорят из-за
первенства отделения от воды и что та часть соли, которой это
сначала удается, напоминает того полководца, который подает
другим сигнал следовать за собой». И далее: «Рассматривая
явление с этой точки зрения, я предположил, что, может быть,
одно простое вбрасывание кристалла соли во вполне охлажденный
и кристаллизующийся раствор может послужить подобным же
48
поводом к кристаллизации. Я произвел опыт и успех оправдал
мой ожидания» [44].
2.3. Кристаллизация чистых жидкостей
В дальнейшем первые наиболее интересные результаты
получены при изучении переохлажденных жидкостей. Оказалось, что
1) жидкость, профильтрованная через фильтр, диаметр пор
которого больше 1,5 мкм, кристаллизуется в процессе охлаждения
при обычной для нее температуре замерзания; 2) та же жидкость,
профильтрованная через тот же фильтр сразу после начала
кристаллизации, теряет способность кристаллизоваться при
повторном охлаждении, но сразу же кристаллизуется при введении
затравки; 3) расплавленная и выдержанная некоторое время при
температуре, превышающей точку плавления, она снова теряет
способность кристаллизироваться; 4) затравка вызывает
кристаллизацию переохлажденной жидкости только в том случае, если
размеры ее (эффективный радиус), превышают некоторую
критическую величину: г > Гкр.
Для объяснения всех этих явлений предложена теория
центров кристаллизации или кристаллических зародышей. Принято,
что кристаллизация переохлажденной жидкости мржет начаться,
если где-то в ее объеме возникнет первичный кристаллик,
достаточно прочный, чтобы противостоять столкновениям с
окружающими, молекулами.
Такой зародыш должен быть достаточно большим и должен
попасть в условия, способствующие его дальнейшему росту, так
ща^ кристаллические зародыши малого размера недостаточно
устойчивы и быстро исчезают, не успевая вызвать кристаллизацию
остальной жидкости.
Малая устойчивость очень маленьких зародышей объясняется
просто. Работа, которую необходимо затратить для отрыва от
кристалла частицы, находящейся где-то посередине одной из
его граней, значительно превыш^т величину работы отрыва
такой же частицы, находящейся • на ребре кристалла или на его
вершине. Первичный зародыш настолько мал, что большинство
частиц, его составляющих, можно считать расположенными как
раз на ребрах и вершинах; он и разрушается в связи с этим
гораздо легче больших кристаллов, для которых, наоборот, число
частиц, расположенных на гранях, во много раз превосходит
число более подвижных частиц, находящихся на ребрах и
вершинах.
Устойчивость зародыша тем меньше, чем больше его удельная
поверхность (отношение поверхности к объему), которая для
шара равна (47:^2^ ur^j = 3/г, для куба — {ЫЩ1^) = 6//, т. е.
всегда обратно пропорциональна линейным размерам
кристалла.
4 3-457 49
Самопроизвольную кристаллизацию чистой жидкости при
охлаждении объясняют снижением кинетической энергии ее
молекул. В результате становятся устойчивыми даже очень
маленькие кристаллические зародыши, легко «плавящиеся» при более
высоких температурах.
Для объяснения фактов легкой кристаллизации
нефильтрованных растворов предположили, что она связана с наличием
в первичной жидкости «пылинок». Выделившиеся на таких
пылинках первичные кристаллы оказываются защищенными от
молекулярных ударов, выживают и вызывают кристаллизацию
во всем объеме. При фильтровании жидкости, которая начала
кристаллизоваться, вместе с кристаллами удаляются все
пылинки, а лишенная их жидкость теряет способность
кристаллизоваться.
Существование пылинок в жидкостях, кристаллизующихся
при табличной температуре, доказано, а фильтрование
начинающих кристаллизоваться жидкостей используется в качестве
метода приготовления совершенно прозрачных — «оптически пустых»
жидкостей.
2.4. Распространение теории кристаллических
зародышей на растворы электролитов
Теория кристаллических зародышей была распространена на
растворы электролитов. Здесь процесс кристаллизации гораздо
сложнее, так как ионы, входящие в решетку, должны
предварительно дегидратироваться, а уж затем каким-либо образом
объединиться, образуя первичный кристаллический зародыш.
Комплексообразование и другие процессы, изменяющие начальный
состав системы, также приходится учитывать в этом случае.
Тем не менее, между кристаллизацией индивидуальных
жидких веществ и выделением твердой фазы из пересыщенных
растворов много общего. Так, например, специальные исследования
чрезвычайно разбавленных растворов солей радиоактивных
изотопов свинца и висмута [3, с. 82] показали, что выделение
твердой фазы из растворов этих веществ, приготовленных на воде,
освобожденной от примесей путем центрифугирования,
происходит гораздо труднее, чем из растворов той же концентрации,
приготовленных на обычной дистиллированной воде. Оказалось
далее, что чем меньше кристаллики Na2S04 в его пересыщенном
растворе, тем сильнее можно охладить раствор, не вызывая
кристаллизации. Таким образом, устойчивость пересыщенных
растворов электролитов также зависит от размера
кристаллических зародышей и от наличия посторонних примесей.
При образовании и растворении ионных кристаллов, по
представлениям В. Косселя (1928), основную роль играет
электростатическое притяжение между ионами. Согласно его расчетам
50
при кристаллизации веществ, в решетку которых входят только
однозарядные доложительные и отрицательные ионы, в первую
очередь должны возникать ионные цепочки, так как именно в
этом случае каждый заново присоединяющийся ион будет
испытывать наименьшее отталкивающее воздействие со стороны
одноименно заряженных ионов, уже включившихся в цепочку.
Взаимное соединение таких цепочек ведет к образованию
двухмерных ионных плоскостей, а настоящий трехмерный
кристалл получается из этих последних в результате взаимного
наложения их друг на друга.
Последующие теоретические исследования показали, однако,
что вероятность образования трехмерного кристалла за счет
наложения друг на друга двумерных листочков очень мала.
Оказалось, что появление и тем более сохранение на плоской поверх*-
ности такого листка только одного иона, начинающего второй
слой, настолько маловероятно, что может реализоваться лишь
при 50%-ном пересыщении. Возникло существенное расхождение
между теорией и практик^ой, так как согласно опытным данным
пересыщение, обеспечивающее образование и рост трехмерных
кристаллов, не превышает обычно 1 %.
Расхождение между расчетами и'результатами опытов удалось
объяснить лишь путем электронно-микроскопических
исследований. Франк [41] обнаружил, что поверхность граней кристаллов
никогда не бывает такой .совершенной, как представляли физики
и химики. Применение новой техники позволило выявить на
поверхности граней различные смещения, в частности винтовые.
При наличии таких смещений на поверхности грани всегда
имеются террасы, на которых рост может происходить и без
предварительного образования новых двухмерных зародышей. Если
существует одно такое смещение в центре грани, она может расти
спиральной лестницей как раз при тех небольших пересыщениях,
которые были обнаружены опытным путем.
Так был решен вопрос о причинах пересыщения и методах
его нарушения, наметилось решение вопроса относительно
причин возникновения метастаб ильной зоны и был выяснен до
известной степени механизм возникновения и роста ионных
кристаллов. Оставались неясными вопросы: почему одни вещества
дают пересыщенные растворы, а другие —золи, почему заметно
растут только кристаллы веществ, дающих пересыщенные
растворы, и прав ли Грэм, разделивший все вещества на
кристаллоиды, дающие только пересыщенные растворы, и коллоиды,
образующие твердую фазу толь{со в виде гелей и золей.
2.5. Теория относительного пересыщения
Первым приблизился к решению этой задачи П. П. Веймарн,
создавший так называемую теорию относительного пересыщения
(1910 г.^
4* 51
Согласно этой теории скорость образования центров
кристаллизации (ш) пропорциональна величине относительного
пересыщения и выражается формулой
w = K'-^, (2.7)
где К — фактор пропорциональности; с—полная концентрация
данного вещества в момент его осаждения; L — его табличная
' растворимость; с — L — абсолютное и (с— L)/L — относительное
пересыщение.
Если относительное пересыщение велико, то практически все
вещество, которое должно выпасть, выделяется сразу, образуя
множество центров кристаллизации или кристаллических
зародышей. Дальнейший рост этих зародышей практически
невозможен, так как избытка вещества в растворе нет. Этот процесс
ведет к образованию золя.
Если относительное пересыщение мало, т. е. L лишь
немного меньше с, то скорость образования кристаллических
зародышей мала, а число их 'невелико. В первый момент выделяется
лишь небольшое количество твердой фазы, остальная часть ее
выделяется постепенно на первичных центрах кристаллизации,
из которых при этом вырастают крупные кристаллы.
Таким образом, Веймарн, опираясь на формулу (2.7), отверг
предложенное Грэмом деление всех веществ на кристаллоиды
и коллоиды, и предположил, что любое твердое вещество может
в одних системах давать пересыщенные растворы и вести себя
как грэмовский кристаллоид, а в других — давать золи.
Это новое положение подтверждено блестящими и хорошо
воспроизводимыми опытами для растворов сульфата бария. В
данном случае L^YV^ 10-^ моль • л-^ и относительное
пересыщение равно (с—10-5)/10-^
Для наиболее концентрированного раствора,, который удалось
получить Веймарну путем смешения равных объемов
насыщенного раствора Ba(SCN)2 с концентрацией 3,5 моль • л~^ и
пересыщенного раствора MnS04 с такой же начальной концентрацией,
относительное пересыщение достигало огромной величины {с —
L)IL=\J5 ' 10^. В результате энергичного перемешивания сме- •
си она превращалась в студень, не выливавшийся из
перевернутого стакана.
При сниженных в 5 раз исходных концентрациях тех же солей,
т. е. при t: = Q,7 моль • л-^ и (с —L)/L = 3,5 . 10^ получен
творожистый осадок BaS04, не отличимый по внешнему виду от'
обычного осадка AgCl. При (с — L)/L = 2,6-10^ образовался
осадок, состоящий из перистых и звездоподобных кристаллов.
При (t:—L)/L = 250, т. е. при с =5,0 • 10-^ моль - л-!,
получились компактные кристаллы BaS04. Кристаллическими были
осадки и при более низких значениях относительного
пересыщения,
52
Опытным путем показана также возможность получения
в кристаллической форме таких веществ как А1 (ОН)з, ЗЬгЗз
и т. п., которые обычно выпадают из растворов в виде так
называемых аморфных осадков.
Практическое применение теории Веймарна и формулы
относительного пересыщения основывается на следующих
соображениях. Если необходимо получить данное вещество в виде
достаточно крупных кристаллов, следует уменьшить .значение (с—L)IL.
Для этого можно уменьшать с следующими способами: I)
разбавляя смешиваемые растворы, как это делал Веймарн в своих
опытах; 2) подавляя диссоциацию реагента, например, снижая рН,
если осадителем является А^'-'-ион, а его источником НаА; 3)
снижая исходные концентрации осаждаемого иона или иона-осадите-
ля путем комплексообразования; так получают, например,
кристаллы хлорида серебра при медленном испарении аммиака из
раствора, содержащего Ag+ и С1--ионы; 4) используя для
получения иона-осадителя медленно идущую реакцию, например,
омыление этил- или метилоксалата в случае осаждения оксалата кальция.
Можно, наоборот, увеличивать L, повышая температуру
системы или изменяя состав растворителя. В ряде случаев
комбинируют методы, одновременно снижающие значение с и
повышающие L.
Получение золей достигается увеличением с или снижением
L, а в случае необходимости путем одновременного использования
обоих путей.
Рассмотрим теперь пригодность формулы Веймарна для
предсказания хода выделения твердой фазы из двух растворов разного
состава при практически одинаковых начальных условиях.
Возьмем в качестве примера две системы. Пусть в первой
б^д^-ь = 0,01,6:(.j-=0,l моль • л~^ а во второй Cgg2-f=0,01 и c^q^- =
= 0,1 моль • л-Ч Поскольку в первой в избытке С1"--ионы, а во
второй S02-, можно считать, что для первой системы с=сА^а =
= 0,01, а для второй с = csaso^ = O7OI моль • л-^
Для определения значений L записываем:
Ag+ + С1~ ^ AgCl (т) Р = 10-^^ Ba2++SO|-^BaS04(t)P= 10-^^
с 10-2 0,1 10-2 0,1
[] X 9 • 10-2 +л: у 9 . 10-2+
X (9 . 10-2 + л:) = 10-10, у (9 - IO-2 + у) = lO-i^,
л: = 1,1 • iO-^ моль • л-^ у = 1,1 • 10-^ моль • л-»
Поскольку для обеих ^ селей при избытке аниона-осадр^теля
растворимость L определяется равновесной концентрацией
катиона, т.-е. величинами х и у, значение L для обеих систем
одинаково, равно 1,1 • 10-9, а тогда относительное пересыщение по
(2.8) для обеих систем также одно и то же:
10-2 _ и . 10-9
1,1 . 10-9
9 • W
5а
и выражается колоссальным числом, превышающим 1,75 • 10^,
при котором BaS04 (в несколько иных условиях) давал студень.
Следовало ожидать, что в обеих системах твердая фаза должна
присутствовать в виде золя.
Опыт показывает, однако, что золь образует только AgCl,
. да и то не студень, а творожистый осадок, тогда как в системе
BaS04 наблюдается пересыщение и образование кристаллического
осадка. Частицы золя AgCl почти не изменяют своих размеров
во времени. В то же время в осадке BaS04 происходят быстрые
изменения: мелкие кристаллы исчезают, а за их счет растут
кристаллы более крупные. Объяснить эти различия теория
Веймарна в ее обычной трактовке не в состоянии.
2.6. Зависимость растворимости от величины
поверхностного натяжения на границе
между твердой фазой и раствором
Более тонкие различия в свойствах гетерогенных систем
удалось установить, сопоставляя их поведение с явлениями,
наблюдавшимися в системах «насыщенный пар — капли жидкости».
Так, мелкие капельки ртути могут долго сохраняться под
колпаком, т. е. в контакте с воздухом, насыщенным парами ртути,
не меняя видимых размеров. Здесь не наблюдается заметный
рост крупных капель за счет мелких, как незаметен он и в золе
хлорида серебра. Совсем иная картина наблюдается для
изолированной системы, состоящей из капель какой-нибудь летучей
жидкости и воздуха, насыщенного ее парами. В такой системе
мелкие капли быстро испаряются, вещество их «перегоняется»
в соседние капли большего размера, растущие, таким образом, за
счет мелочи. При этом вокруг укрупняющихся капель образуются
свободные от мелких капель пространства — «дворики».
Подобным, же образом ведут себя кристаллы BaS04,
находящиеся в контакте с его насыщенным раствором.
Сходство в поведении позволило распространить на систему
«насыщенный раствор — твердая фаза» законы, установленные для
системы «насыщенный пар — жидкость».
В уравнении Гиббса-Томсона
'»^^(^-^).
выражающем зависимость между давлением пара и кривизной
поверхности капель жидкости, индексами 1 и 2 отмечены радиусы
двух капель и давление пара над ними р\ и р2, —плотность
жидкости, молярная масса, /? —универсальная газовая
постоянная, Г — термодинамическая температура, а —
поверхностное натяжение на границе жидкость-воздух, насыщенный ее
парами, которое является мерой стремления системы сократить
площадь поверхности раздела.
■54
Как следует из (2.8), давление пара обратно
пропорционально радиусу капель жидкости. Над мелкими каплями оно больше,
в результате чего жидкость из мелких капель перегоняется
в большие.
Формула (2.8) была использована Оствальдом для объяснения
зависимости растворимости от размеров частиц осадка. После
небольших поправок, введенных в нее Фрейндлихом, она
приняла вид
л _ 1 \
\ -
(2.9)
где Li — растворимость кристалла, радиус которого Г\\ — его
плотность; М — молярная масса; а — поверхностное натяжение на
границе между твердой фазой и насыщенным раствором; оно
является мерой стремления мелко раздробленной твердой фазы
сократить площадь поверхности раздела за счет перерастания
мелких частиц в частицы с большим радиусом. Эта тенденция
приводит одновременно к возрастанию растворимости более
мелких частиц.
Позже было показано [67], что для ионных кристаллов типа
МА, т. е. для случая |х = а, формула (2.9) принимает вид
" (2.10)
1 ^
" Pi ~ d^RT
где Pi — произведение растворимости кристаллов, радиус
которых равен ri. ' .
Считают, что для макрокристаллов Р имеет практически
такое же значение как для кристаллов, у которых г = оо. Тогда
_Р_ 2аМ 0,8686аМ
^оо ^ ^т^Тг Jg = d^RTr • - (2.11)
Если к тому же учесть, что для веществ с формулой МА P^L^,
то
, L qM . L 0,4343ауИ
In = kLZ== IJfT' (2.12)
Величины М, dj, R и Т известны, а значения и Роо
можно приравнять, с точностью до случайных погрешностей
измерения, табличным значениям этих величин. Это дает возможность
вычислять значения растворимости L и произведения
растворимости Р для кристаллов с заданным радиусом, если известны
значения а. К сожалению,, прямых методов измерения
поверхностного натяжения на границе между твердой фазой и
раствором пока не существует, величины а находят по результатам
измерения растворимости. Получаемые таким образом оценки а
•включают большие систематические погрешности. Так, по данным
разных авторов, оценки а для системы SrS04 — Н2О составляют
55
J,4 и 0,08, для системы BaS04 — HgO —1,25, 3 и 0,15 Дж ^-м^.
На границе раздела AgCl —НгО а 0,125 Дж • -м-.
Применять эти значения а для тЬчных расчетов нельзя, мы
их используем лишь для того, чтобы показать, что формулы
(2.11), (2.12> позволяют объяснить различия в характере систем,
образующихся при выделении AgCl и BaS04 из гомогенных
пересыщенных относительно этих веществ растворов.
Для дальнейших иллюстративных расчетов использовали
формулы (2.11), (2.12), записанные в виде
,gi..4,„^ = 4,,n^-E. (2.>3)
Расчет значений А, В и С, входящих в уравнения (2.13),
для AgCl и BaS04 при Г = 293 К приведен в табл. 2.1.
Та блица 2.1
Твердая
фаза
Мг МОЛЬ""^
d г • см—3
а . 105,
Дж.см~2
А см
в см
с, см
BaS04
AgCl
10->«
233,0
143,3
4,5
5,56
5,2
1,25
4,8-10-^
5,75.10-^
9,6.10-^
1,15.10-^
1,1 . 10~^
1,3 .10-^
Характер систем твердая фаза —раствор для AgCl и BaS04
может оказаться разным, если вероятность образования
кристаллов, ведущих себя как макрокдисталлы, для этих систем резко
различна.
Расчет числа ионов надо начинать с оценки радиуса
кристалла, растворимость которого практически не отличается от
табличной L, которую считают равной Loo- Пусть такая Lr =
= 1,01 • Loo, т. е. отличается от Loo всего на 1%, тогда lg(Lr/Loo)=
= Ig 1,01 = 0,0043, /'i=Ai/4,3 . 10~з и длина ребра кубического
кристалла / = 2/-. В то же время величина 2 {г, + гд)
представляет сумму диаметров катиона и аниона, входящих в решетку
данной соли; п = 1/2 (гк + г а) дает число пар ионов,
укладывающихся на длине ребра кристалла, 2a2—-число ионов на том же
ребре, а N = (2п)^ — общее число ионов, образующих
макрокристалл.
Результаты вычисления числа ионов, образующих кристаллы
AgCl и BaS04, растворимость которых практически не
отличается от табличной Loo, приведены в табл. 2.2.
Таблица 2.2
г. см
/. см
2<-К +
п
2п
N
BaS04
AgCl
1,Ы0"^
1,35.10-^
2,2.10-^
2,7.10-^
8,76.10-®
5,88.10"^
2,5.10^
4,6.10^
5,0 . 10^
9,2 . 10^
1,25 . 10^^
7,8 . 10®
56
Вычисленные N различаются больше, чем на два порядка, но
сами по себе они так велики,, что рассчитывать сразу на
образование макрокристаллов ни в том, ни в другом случае не
приходится. В связи с этим предположение, что в одной из систем
сразу выпадают макрокристаллы, исключается.
Поскольку все же разница в поведении систем является
безусловным фактом, стоит рассмотреть вопрос, не различается ли
заметно зависимость растворимости от размеров микрокристаллов
BaS04 и AgCl. Для решения этой задачи следует, очевидно,
проследить за изменением Рг от г для каждой из солей в
отдельности. В табл. 2.3 приведены ревультаты расчетов по одной
из формул (2.13), переписанной в виде
рРг = рРсо - В . == 10 - В . г-Ч (2.14)
где рР — показатель произведения растворимости; В — параметр,
взятый из табл. 2.1 (crrjrcrr тсй же таблР!це для обеих солей
рРоо = 10).
Та блица 2.3
п
n
BaS04
agcl
Л см 1 рР^
г, см 1 рРг
2
4
8
16.
32
64
128
256
512
1024
2048
64
512
4,1 . 103
3,3 . 104
2,6 . 105
2,1 .
1,7 ■
1,3 .
1,1
8,6.
6,9 .
106
107
108
109
109
10
10
8,76 .
1,75.
3,50 .
7,01 •
1,40 .
2,80 .
5,П .
1,12.
2,24 .
4,48.
8,96 .
-8
,—7
1-7
.—7
10
10
10'
10
10-6
10-6
10-6
10-5
10-5
10-5
10-5
-0,86
4,5
7,3
8,6
9,3
9,7
9,8
9,9
9,96
9,98
9,99
2,7 . 10^
562
22,4
5,0
2,2
1,4
1,26
1,12
1,05
1,02
1,01
5,90 •
1,18^
2,35 .
4,70-
9,40.
1,88 '
3,76.
7,52.
1,50 ■
3,00-
6,00 -
10-*
10
.—7
—7
10
10-^
10-^
10-^
10-^
10-^^
10-^
10-^
10""^
8,1
9,0
9,5
9,8
9,9
9,94
9,97
9,98
9,992
9,996
9,998
8,910-
3,162
1,678
1,259
1,122
1,072
1,035
1,023
1,010
1,005.
1,ооа
в первом столбце таблицы приведено число пар ионов п,
находящихся на ребре кристалла; во втором N = {2nf — число ионов
в кубе, на ребре которого находится п пар ионов. В столбцах
3 и 6 даны значения радиусов соответствующих кристаллов,
полученные по формуле г = п (Гк + г а) (гд^т-Ь = 0,113 нм, га -
= 0,181 нм, гва2+-= 0,145 нм, rso^"'^ 0,295 нм). В столбцах
4 и 7 приведены значения рРг, вычисленные по формуле (2.14),
а
а в столбцах 5 и 8 значения L^/Loo =/10-р^г/10-5 = Ю^^.
При сравнении значений рР;., приведенных в столбцах 4 и 7,.
видно, что значения эти для больших кристаллов AgCl и Ва304
57
почти одинаковы, но становятся резко различающимися для
кристаллов маленьких. Соответственно должны различаться
и растворимости. Так, например, если для кристаллов •BaS04
с двумя парами ионов на ребре действительно рРг = —0,86, то
Lr = 100'4з = 2,7 моль • л-1 = 629 г . л-\ т. €. примерно 63 г в
100 г воды. Это значит, что такие мелкие кристаллы BaS04
должны растворяться в два раза лучше макрокристаллов NaCl
и так же хорошо, как макрокристаллы СаСЬ. Более крупные
кристаллы BaS04 гораздо устойчивее, но вероятность их
возникновения очень мала. Отсюда пересыщение и появление широкой
зоны метастабильных растворов, которую теперь можно
представить как поверхность, покрытую рядом сливающихся одна
с другой кривых растворимости, относящихся к кристаллам
разного размера, т. е. кривым Lr, t,
В случае AgCl даже для кристаллов с п = 2 рР^ = 8,1. Это
значит, что самые мелкие кристаллы AgCl растворимы хуже
макрокристаллов PbS04.
В качестве подтверждения пригодности уравнения (2.14) для
таких расчетов ниже сопоставлены расчетные и опытные
результаты, полученные для кристаллов BaS04 разной величины.
Растворимость BaS04, моль • л""Ч
г, см Найдено экспериментально Вычислено
9 . 10-5 9,8 • 10-6 10 . 10-6
5.10-6 1,8-10-5 1,6-10-5.
2.7. Применение выражения относительного
пересыщения к системам, в которых
зависимость L от г различна
Попытаемся снова использовать формулу Веймарна для
случая BaS04 и AgCl. Допустим, что на грани устойчивости
находятся кристаллы, на ребре которых находится 4 пары ионов,
тогда pPr(AgCl) = 9 для г =1,18 нм и рР^ (BaS04) = 4,5 для
г = 1,75 нм. Повторяя расчет L для эгих значений рРг, получаем:
для AgCl для BaS04
Ag+ + Cl- ^AgCl(T) 109 Ba2+ + S02-:?:BaS04(T) lO^^s
€ 10-2 0,1 10-2 0,1
{] X 9 • 10-2 + x У 9 ' 10-2 + у
x(9 • 10-2 + x)= 10-9 у (9 . 10-2 + у) = 10-4.5
X = LAgci = 1,1 • 10-» у = LBaSO. = 3,5 . 10-4
= 9 . 10-5 ^ = 28
Найденные значения относительного пересыщения полностью
объясняют образование золя в случае AgCl и пересыщения для
58
BaS04. Правда, творожистый осадок в опытах Веймарна
получался для BaS04 при относительном пересыщении, равном 7 х
X 104, а в только что проведенном расчете для AgCl найдено
значение 9 • 10^, но если учесть ненадежность. значений а, то
качественное совпадение следует считать удовлетворительным.
Теперь можно объяснить факт сравнительно быстрого роста
крупных кристаллов BaS04 за счет мелких и практическое
отсутствие такого роста для золя AgCl. В 5-м и 8-м столбцах
табл. 2.3 приведены значения L^/Loo, из которых следует, что
растворимость самых маленьких кристаллов AgCl всего в — 9 раз
больше растворимости' макрокристаллов того же вещества, тогда
как для BaS04 эго отношение в 30 ООО раз больше. В результате
следует ожидать быстрого укрупнения мельчайших кристаллов
BaS04 с последующим замедлением этого роста для кристаллов
более крупных. Тенденция к росту кристаллов у AgCl
проявляется б незначительной степени.
К тем же результатам приводит формула, выражающая
относительное изменение растворимости в зависимости от радиуса
dL dr dL г
Кристалла, т. е. величина х ' 7"^ d7 ' 1*
Используя третью из формул (2.13), получаем
с с
L = Looe S ^ = ~ Looe ^ • С/-2 -LCr-^ а тогда
^: ± = _LCr-2 . rL-i = _ ^ и ^ = 4^ (2.15)
Знак «минус» в (2.15) показывает, что с ростом у
растворимость убывает. Прямо из формулы следует, что при
относительном увеличении радиуса у растворимость убывает в ^ раз
быстрее. Например, при увеличении кристалла AgCl, у которого
dL^ _ КЗ • 10"^
L 2,35 . 10~
растворимость снизится в 1,11 раза.
В то же время при уменьшении вдвое кристалла BaS04, у
которого г = 3,5 . 10-7 см, ^ = -iii:J2l!(_2) = 6,3; т. е. раство-
3,5 • 10
римость возрастает в 6,3 раза.
Становятся понятными и результаты опытов Оствальда,
.приведенные на рис. 2.1. Зона метастабильных пересыщенных
растворов представляет собой поверхность, покрытую множеством
кривых, выражающих растворимость кристаллов данного
вещества, имеющих разные радиусы и находящихся в контакте с
насыщенным раствором данного состава. Очевидно, что в случае,
скажем, осаждения BaS04 при сливании растворов нитрата бария
и сульфата калия эта зона будет много шире, чем в случае
г = 2,3а. 10-7 см, вдвое ^ = — 1'^'-2=-1,11, т. е.
59
Рис. 2.1
осаждения AgCl при сливании растворов AgNOa и КС1, так как
различия в значениях рЯва504 "ри изменении г много больше,
чем y.pPAgci. При этом не следует думать, что зоны
метастабильных пересыщенных растворов у AgCl вообще нет; опытным
путем установлено [13], что при CAg+ -Са-^З.И • 10-^^
независимо от того, какой из ионов находится в избытке, добиться
появления твердой фазы AgCl не удается.
^ Факт, что порой одно и то-
^1 ^ ' же вещество при большем
относительном пересыщении
образует творожистый осадок, а при
меньшем — гель, связан,
по-видимому, с различием значений
а на границе между одной и той
же твердой фазой и разными
по составу насыщенными
растворами. Так, значение а в
системе, состоящей из твердого
BaS04 и раствора Mn(SCN)2,
должно отличаться от
значения а для тех же кристаллов в
растворе KNO3, но точные
значения а нам неизвестны, и мы не можем предугадать влияние
состава раствора на ход выделения твердой фазы.
Можно утверждать, однако, что каждому значению г для
кристаллов вещества в данной системе соответствует своя кривая
растворимости (L, f) и даже свои критические размеры
затравки (гкр).
2.8. Выделение твердой фазы из
пересыщенного раствора
Теперь ясно, что пересыщение может возникать тем легче, чем
больше табличное значение произведения растворимости
осаждаемого вещества, чем больше оно возрастает по мере снижения
линейных размеров кристаллов и чем меньше произведение
начальных активностей ионов, образующих данную
кристаллическую решетку. Пересыщение возникает особенно легко, если мала
скорость диффузии ионов, образующих осадок, и скорость их
агрегации, т. е. построения решетки. В подобных случаях момент
значительного местного пересыщения, возникающего в месте, куда
попадают первые капли осадителя, может быть пропущен и
полученный пересыщенный раствор остается прозрачным, так как
кристаллические зародыши, образовавшиеся в месте большого
пересыщения, не смогли вырасти и преждевременно разрушились.
Необходима затравка, пылинка, или убежище в виде, скажем,
микротрещинки в стенке сосуда, чтобы в такой системе началась
самопроизвольная кристаллизация.
60
Допустим, например, что в гомогенном сильно пересыщенном
растворе при с^+==0,01 и с^о^-= 0,1 моль . л-^ находились
пылинки, послужившие, центрами кристаллизации зародышей,
отвечающих рРг = 4,5. Если таких пылинок очень много, то, как
следует из приведенного выше расчета, [Ba^+J == у = 3,5 • 10-^
моль . л^К Это значит, что в осадок перейдет (10-2 — 3,5.10-4)^
— 10-2 моль BaS04 в виде кристаллов, с / = 3,5 . 10-^ см.
Каждый такой кристалл весит 4,5 . (3,5 • Ю-^)^ = 1,9 . 10-^9 г, а всего
их должно выпасть из литра раствора 233 . 10-2/1,9 • 10-^9 =
= 1,2 . 10^9 штук. Если бы это число в дальнейшем не
изменялось и все они росли равномерно, то даже в случае полного
выделения оставшихся 3,5 • 10-^ моль BaS04 на их поверхности
размеры кристаллов остались бы субмикроскопическими (частицы
.с линейными размерами <0,16 . 10-4 см не видны в оптический
микроскоп).
Фактически в таких случаях сразу образуется белая «муть».
Это происходит, по-видимому, в связи с очень быстрым ростом
более крупных кристаллов за счет мельчайших, так как при
г --== 1,75 . 10-7 см Lr превышает Loo более чем в 500 раз.
Последующие визуальные наблюдения показывают, что выпадающий
hpH осаждении BaS04 осадок, состоящий из огромного числа
мельчайших частичек, вскоре становится крупнозернистым.
Снимки, полученные с помощью электронного микроскопа, дали
дополнительные сведения. Оказалось, что крохотные
кристаллические образования, размеры которых составляют лишь доли
микрометра, исчезают очень быстро, постепенно перерастая
в компактные кристаллы размерами около 4 . 10-"^ см. Дальней-
' ший рост этих образований, линейные размеры которых
практически такие же, как у макрокристаллов (табл. 2.2), уже не
заметен. На тех же снимках видно, что кристаллические сростки
BaS04 размером (6 — 8) . 10-4 находящиеся в контакте с
маточным раствором, не растут, а только перестраиваются,
становятся компактнее в полном согласии с данными о разной
«растворимости» граней, ребер и вершин одного и того же кристалла.
Приведенный цифровой материал нельзя считать безупречным,
так как:
1) формула Гиббса — Томсона (2.8) имеет в виду кристаллы
сферической формы, подобные каплям испаряющейся жидкости;
2) значения поверхностного натяжения а и даже плотности
кристаллов dr могут сами зависеть от радиуса г;
3) до сих пор не существует надежного метода определения
поверхностного натяжения на границе кристалл — насыщенный
раствор, поэтому любое из значений а, приведенных в
справочниках, вызывает сомнение.
Тем не менее, можно предполагать, что все вещества,
растворимость которых мало зависит от размеров кристаллов, должны
давать, подобно AgCl, золи или псевдорастворы. Наоборот, ве-
61
щества, растворимость которых сильно изменяется по мере
изменения величины кристаллов, должны давать пересыщенные
растворы.
Пересыщение устраняется сразу же после появления первых
кристаллов или введения затравки, а затем мелкие вначале
кристаллы этих веществ сравнительно бы:тро растут под слоем
маточного раствора.
Поскольку формула (2.10) дает возможность установить
зависимость произведения растворимости любого малорастворимого
вещества от размеров кристаллов, важно было бы разработать
достаточно точный и независимый метод измерения
поверхностного натяжения на границе между твердой фазой и ее
насыщенным раствором.
2.9. Образование золей
Вещества, растворимость которых почти не зависит от
размеров кристаллов, например хлорид серебра, устойчивых
пересыщенных растворов обычно не дают. Однако, если начальная
концентрация осаждаемого иона мала и анализируемый раствор
содержит мало посторонних ионов, то он может остаться
совершенно прозрачным на вид после прибавления
осаждающего' реагента. Если такой раствор не
выделяет осадка при внесении затравки и при
трении стеклянной палочкой о стенки сосуда,
но начинает мутнеть после прибавления
достаточного избытка электролита, то можно
утверждать, что вместо ожидаемого осадка образовался
коллоидный раствор или золь осаждаемого
вещества.
Рассмотрим на примере хлорида серебра про-,
цесс образования коллоидного раствора. Допустим,
что к 10 мл раствора, содержащего 2 • 10-^
моль • л-^ Ag+-HOHa, прибавлено 0,1 мл раствора,
хлорид-иона с концентрацией 2 • 10-2 моль • л-^
(рис. 2.2). Предположим теперь, что в первый
момент после добавления раствора хлорида хлорид-
ионы проникли лишь в верхний слой раствора,
содержащего Ag+-HOHbi, объем которого также равен 0,1 мл.
В этом случае объем зоны реакции равен 0,2 мл, са?+ = Ю"^,
сс1"" = 10-2 MQjib . а произведение CAg"*" • ^ci- = Ю-^.
Подстановка этого значения во второе равенство (2.13) при В = 1,15 х
X 10-7 см показывает, что в зоне реакции система пересыщена
относительно всех частиц, радиус которых г > 2,9 • 10-» см.
Допустим, что весь AgCl выделился в виде кристаллов сг =
== 1,18 • 10-7 см, в каждом из которых (см. табл. 2.3) N = 512
ионам. Для такой системы рР,- = 9,0, а тогда
Рис. 2.2
62
Ag+ +Cb«AgCl(T)
с 10-2
[] X 9,9. 10-3 +л; x= 1,1-10-7 моль .л-i
и можно утверждать, что из литра, такого раствора должно
выпасть (Ю-^ _ 1,1 . 10-7)_ 10-4 моль AgCl.
Это значит, что в слое объемом v = 0,2 мл выделяется 10-^ X
X 2 • 10-^ = 2 . 10-^ моль AgCl, каждый кристаллик которого
содержит d^t^M-i =5,56(2,36 . 10-7)з (143,3)-! _ 5 j . 10-22 ^оль
AgCl. Всего таких кристаллов в зоне смешения образуется 2 х
X 10-8/5,1 . 10-22 _ 3^9 . 1013 штук.
Допустим теперь, что при дальнейшем смешении реагирующих
растворов новых первичных кристаллов получаться не будет,
а весь излишек хлорида серебра будет выделяться вокруг
образовавшихся в первый момент центров кристаллизации. В этом
случае в нашем растворе по-прежнему будет всего 3,9 • Ю^з
кристаллов, и если все они будут расти равномерно, то даже после
полного выделения всего хлорида серебра, содержаш,егося в
растворе, т. е. 2 • 10-^ моль, каждый из кристаллов будет иметь
массу 143,3 . 2 • 10-6/3,9 • Ю^з/-! = 7,3 • Ю-^» г при длине ребра
3
1^У7,г . 10-^8/5^56^1 1^1 . 10-6_0,01 мкм.
Кристаллы такого размера нельзя увидеть даже в микроскоп,
так как разрешающая способность обычных оптических
микроскопов ограничена 0,16 мкм. Поэтому смесь, полученная в
результате реакции, будет казаться совершенно гомогенной, хотя
практически весь хлорид серебра выделился из нее в виде
твердой фазы.
В то же время общая поверхность всех частиц, AgCl в 10 мл
раствора S = 6 (1,1 • 10-^)2 . 3,9 • Ю^^з ^ 280 см2.
Таким образом, в 1 л полученной системы содержится 2 х
X 10-4 у1ол^ AgCI в виде 3,9 • 10^^ частиц с общей поверхностью
2,8 м2 при поперечнике частицы — 1 • 10-^ см и массе —7,3 X
X 10-^^ г. Эти ;з;анные можно считать общей характеристикой
системы. Ее можно дополнить значением «дисперсности», т. е.
отношением поверхности частицы к ее объему. Д/1я кубического
кристалла она.равна 6/2//з = 6/-^ и в нашем случае составляет
6(1,1 . 10-б)-1г=5,4 . 10? см-^
Возможно образование и гораздо более концентрированных
золей. Так, при осаждении AS2S3, нормальная растворимость
которого при 20° С L = 5,2 . 10-4 г . л-^ или 5,2 • Ю-^х(246,04)-^
= 2,1 . 10-е моль'. л-^ 'МОЖНО получить прозрачный в
проходящем свете раствор, содержащий до 70 г AS2S3 в 1 л. Если
принять, что rfAs,s, = 3,2 г • см-з, М (AS2S3) = 246 г • моль-^ и / =
=^ 10-6 см, т при дисперсности 6 • 10^ см-1, совпадающей с
дисперсностью для AgCl, эта система будет содержать в 1.л 70х
X (243,14)-^ = 0,28 моль AS2S3 в виде частиц с / 10-^ см, массой
m = 3,2 • (10-6)3 = 3,2 . 10"^^ г, при общем числе частиц в литре
63
раствора N = 70(3,2 • 10-^»)-^ = 2,2 • 10^9 с общей поверхностью
5 = 6(10-6)2 .'2,2 . 10^9= 1,32 . 108 см2= 13200 м2.
Такие системы называют псевдорастворами, коллоидными
растворами или золями.
2.10. Свойства золей
Размеры частиц. Все золи полностью проходят, подобно
обычным растворам, через фильтровальную бумагу, стеклянные,
фарфоровые и обычные мембранные фильтры и кажутся
прозрачными, но от настоящих растворов отличаются: 1) тем, что псевдо-
раство репное вещество может быть отделено на ультрафильтре,
т.. е. фильтре с очень мелкими порами; 2) наличием явления
Тиндаля, т. е. способностью рассеивать проходящий свет, подобно
тому, как рассеивают свет пылинки, взвешенные в воздухе;
3) присутствием множества частиц, обнаруживаемых с помощью
ультрамикроскопа, т. е. микроскопа,, в котором можно наблюдать
прозрачные среды на темном поле при очень сильном боковом
освещейии; 4) низкими значениями свойств, величина которых
пропорциональна числу частиц в единице объема: ничтожно
малым осмотическим давлением, понижением температуры
замерзания и повышением температуры кипения; 5) тем, что при
электролизе коллоидных систем все «растворенное» вещество
скапливается целиком у одного из электродов. Размеры пор
фильтрующих систем в мкм приведены в табл. 2.4 [51, с. 298].
Та бл и ца 2.4
Система
0i мкм
Система
0, мкм
Система
Система
Беззольная
бумага:
грубая
средняя
тонкая
7~7,'5
6-6,8
2,3-2,8
Стеклянные
фильтрующие тигли
ПОР 10Q
ПОР 40
ПОР 10
40—
100
16—40
3—10
Фарфоровые тигли
5—8
Мембранные фильтры
0,3—3
Понятно, что золи, в которых '/< 1 • 10-6 (^м^ должны
проходить, подобно обычным растворам, через все фильтрующие
системы, приведенные в табл. 2.4.
- Оптические свойства золей. Проходящий свет может
поглощаться и рассеиваться на частицах.
Белый свет, охватывающий интервал длин волн 4-10-5 <х<
< 7 . 10-5 cj^^ совсем не проходит, если расстояние между
частицами меньше любой X и коэффициент пропускания для всех
X видимой части спектра тх ^ О, или концентрация частиц так
64
велика, что для любой X оптическая плотность А\ очень велика,
хотя тх Ф 0. Именно так ведет себя обычная тушь.
Белый свет частично проходит через систему, если расстояния
между частицами меньше любой X на интервале 4- 10-"5<х<
< 7 • 10-^ см, но для любой длины волны тх Ф 0. Если тх
примерно одинаковы, то близки и Лх, в- силу чего система в
проходящем свете кажется «нейтрально» серой, как разбавленная тушь.
Если частицы пропускают только одну составляющую белого
света, а остальные не пропускают и расстояния между частицами
меньше длин волн видимой области спектра, то система окраши
вается в цвет, для которого А\ Ф 0.
Свет рассеивается, если расстояние между частицами одного
порядка с длинами волн падающего на систему света. Световой
поток рассеянного света определяется при этом формулой Релея:
Фх = ^|^Фох = А=^Фоь (2.16)
где N—-число частиц в единице объема раствора; t; — r^—объем
частицы; / — расстояние от рассматриваемого элемента объема
до поверхности, X—длина волны падающего света, световой
поток которого Фох, Uvd==c — концентрация частиц в единице объема
(формула-применима при г<30 нм = 3 • Ю-^ см).
Поскольку Фх обратно пропорционален Х^, короткие волны
рассеиваются сильнее длинных. Так, для Хф = 4 • 10~5 см и Хк=
= 7,5 • 10-5 см Фф . Фк^ = 7^ • 4-4 = 9,4. Это значит, что сине-
фиолетовая часть спектра рассеивается сильнее красной почти
в 10 раз, благодаря чему бесцветные золи, рассматриваемые при
боковом освещении, кажутся голубыми, а на просвет —
красноватыми.
Общий метод оценки расстояния между частицами проще
всего рассмотреть для конкретных случаев (см. раздел 2.9). Как
было подсчитано, в 1 л AgCl-золя находится 3 • 10^^ частиц.
Тогда на любом ребре куба длиной 10 см будет находиться
3
1/3,9 ' 10^5=:^ 1 Q . кристаллов при общей их длине 1,1.10-бх
X 1,6 • 10^ = 0,176 см; при этом длина свободного пространства
между ними составит (10— 0,176)9,8-см, а расстояние между
двумя частицами р = 9,8: 1,6- 10^ = 6,1 • 10-5 см. Значит, золь
AgCl должен рассеивать белый свет, так как 4 • 10-5 < р = 6,1 х
X 10-5 < 7 . 10-5 см.
В случае гораздо более концентрированного золя AS2S3 на
3
длине 10 см находится )/"2,2 • 10^9 = 2,8 • 10^ частиц, /=10-^ см,
а свободное расстояние между двумя такими частицами составит
(10 - Ю-б . 2,8 . 106) . (2,8 . 10б)-1 = 2,6 . 10-5 см. При таком
Р < 4 • 10-5 см можно ожидать или полного затемнения, или
появления собственной окраски. Поскольку золь AS2S3 в
проходящем свете кажется желтым, для него О и А» •> 0.
5 3 457 65
в обоих псевдорастворах параллельный пучок света кажется
расходящимся. На темном фоне камеры ультрамикроскопа
хорошо видны ярко светящиеся, т. е. рассеивающие свет частицы.
Осмотические свойства золей. Все осмотические свойства
определяются истинным числом отдельных частиц, находящихся в
растворе.
Известно, что молярная депрессия для Н2О равна 1,86 °С на
1 моль • л-Ч т. е. когда в 1 л системы находится 6,06 • Ю^з
отдельных частиц.
Так, в 1 л раствора КС1 с концентрацией с = 2 • 10-^ моль • л-^
общее количество вещества с учетом полной диссоциации /г = 4 х
X 10-^ моль, а депрессия Akci = 1,86.4 • 10.-^ = 7,5 • 10-^ К -моль-^.
Золь AgCl той же концентрации замерзает практически при той же
температуре, что и чистая вода, так как C(Agci, золь) =3,9 X
X 10^5 (6,06 • 1023)--1 = 6,4 . 10-9моль . л-^ а тогда депрессия
AAgci, золь = 1,86 .6,4 . 10-9= 1,2 . 10-8 К*.
Картина ещё разительнее для концентрированных золей. Так,
при кажущейся ^(As^Ss) = 0,28моль • л-^, с учетом полной
диссоциации на 5 ионов, можно было ожидать снижение точки
замерзания на 1,86 . 5 • 0,25 = 2,6 К. Фактически и этот золь замерзает
при той же температуре, что и чистая вода. Это понятно, так как
истинная C(As,s„ЗОЛЬ) = 2,2 . 10^9(6,06.1023)-! =3,6-10-5 моль . л-1,
а тогда Aas^s,. золь = 1,86 • 3,6 • 10-^ = 7- IQ-^ К.
Поведение золей в гравитационном поле. Сравнение масс
отдельных частиц золей mAgci. золь = 7,3 • 10-^^ г и /tzas^s,, золь ==
= 3,2 • 10-!® г с массами отдельных молекул этих^ веществ 2,5 х
X 10-22 р для AgCl и 4 . 10-22 г для AS2S3 показывает, что
псевдорастворы должны походить на суспензии или взвеси, которые
можно получить путем встряхивания мелко измельченного
твердого вещества с жидкостью, в которой . оно практически не
растворяется.
Однако непосредственное сравнение настоящих коллоидных
растворов с обычными механическими смесями очень тонко
измельченных твердых веществ с жидкостями показывает, что
между этими системами очень мало общего. Особенно существенны
для нас следующие различия.
Механические смеси, полученные посредством встряхивания
очень тонко измельченных малорастворимых веществ с водой,
очень нестойки. Частицы твердой фазы такой суспензии пребывают
в состоянии непрерывного броуновского движения, интенсивность
которого тем выше, чем мельче частицы. В то же время все эти
частицы падают под влиянием силы тяжести и при том тем
быстрее, чем больше их радиусы. Взаимные столкновения отдельных
частиц взвеси, происходящие в процессе броуновского движения,
* К —Кельвин, единица термодинамической температуры, численно
равная градусу Цельсия.
66
сопровождаются их слипанием — агрегацией. В результате этого
процесса по мере «старения» такой механической смеси число мелких
частиц уменьшается, а крупных — растет. Это приводит к
дальнейшему ускорению оседания твердой фазы вплоть до полного
отделения ее от жидкости.
В коллоидных растворах наблюдается такое же энергичное
броуновское движение, как и в обычных механических смесях
твердых порошков с жидкостями, но оно не приводит к слипанию
сталкивающихся частиц. Частицы коллоидного раствора если
и укрупняются при его хранении, то лишь очень медленно, и
твердая фаза из золя не выпадает. Таким образом, настоящие
коллоидные растворы отличаются от простых смесей повышенной
устойчивостью.
Простой расчет показывает при этом, что объяснить
стабильность настоящих золей только ничтожными размерами отдельных
частиц нельзя. В самом деле, допустим, что частицы золя
шарообразны, тогда сила тяжести, под влиянием которой они должны
оседать из раствора, выразится формулой
F'^jT.r^di-do)g, (2.17)
где г — радиус частицы; rfi —плотность вещества частицы; do —
плотность вещества среды; g = 9,81 м • с-^ — ускорение силы
тяжести.
Для случая, когда размеры частицы таковы, что их оседание
можно считать движением равномерным, сила тяжести
уравновешивается^ силой трения. Эта сила для частицы шарообразной
формы вычисляется по формуле Стокса: /==6:итд/'с; (1.18), где
г —радиус частицы; t; —скорость; iq— динамическая вязкость
среды.
В случае равновесия F = f послб приравнивания правых частей
формул (2.17) и (2.18) общее выражение для скорости падения
шарообразной частицы, находящейся в жидкой среде, в поле
земного тяготения принимает вид
„=-.,<^-. ,2.,9,
В случае осаждения AgCl, которое было рассмотрено в разделе
JQ—б
2.9, di = dAgci = 5,56 г • cм-^ do = dn.o = 1 г • см-з, г = -j- =
= 5 . 10-7 см и ^ = 0,1 Па • с для воды при 20 °С. Значит,
у = 2,5 . 10-8 см . с-1.
Вычисленная скорость падения очень мала. Однако и при
такой ничтожной скорости все частицы радиуса 5 • 10-7 должны
полностью осесть из слоя раствора толщиной 2 мм за время, равное
0,2/2,5 . 10-8 = 8 . 10^0 = 8 . 106/60 • 60 . 24 = 93 сут, т.е.
примерно за три месяца, даже в том случае, если исключено слипание.
При наличии агрегации частиц оседание их должно ускоряться
67
пропорционально квадрату радиуса частиц, как это видно из
формулы (2.19). Но как показали специальные наблюдения,
частицы настоящих золей не проявляют никакого стремления
к взаимному слипанию, и коллоидные растворы могут
сохраняться, не выделяя заметного осадка, очень долгое время.
В 1909 г. А. В. Думанский впервые применил для исследования
коллоидных растворов центрифугу. Ему удалось показать, что
после центрифугирования коллоидных растворов Ре(ОН)з и ЗЬг^з
электропроводность в верхней и нижней части раствора становится
резко различной. Несколько позже для оценки результатов
центрифугирования были использованы радиоактивные изотопы.
В случае центрифугирования коллоидные частицы
подвергаются воздействию нормального или центростремительного ускорения
a = V^/Ry где У —линейная скорость, i? —радиус центрифуги.
Поскольку V = 27ri?a), где ш частота вращения, а = 4'ic2/?a)2
(2.20).
Например, в случае использования центрифуги, у которой
= 10 см, О) = 100 с-\ а = 47г2 . 10 . 1002 ^ 3^95 . юе см/с2,
откуда видно, что для такой центрифуги а = 3,95 • 10^/981 =4 • Wg
и частицы коллоидного раствора подвергаются в ней воздействию
ускорения, которое в четыре тысячи раз превышает ускорение
силы тяжести.
В таких условиях скорость оседания части коллоидного
раствора AgCl составит у = 2,5 . 10~^ • 4 • 10^ =х 10-^ см/с, а время,
необходимое для оседания всех частиц радиусом 5 • 10-^ см из '
слоя толщиной 0,2 см, равно 0,2/10-^' = 2 • 10^ с = 33,5 мин.
Еще быстрее должно происходить такое оседание при
использовании современных ультрацентрифуг. Так, для стандартных
приборов этого типа, для которых расстояние центра кюветы от
оси вращения {R) равно 6,5 см, а частота вращения а) = '1,16 х
X 103 с~1, а = 47г2;?а)2 = 4 . 9,8696 . 6,5(1,16 • 103)2 = 3,454 х
X 10^см/с2=3,5- 10^^. Работая на такой установке, можно ожидать,
что частицы AgCl осядут из слоя толщиной 2 мм за время
в 3,5 • 10V4 • 10^^88 раз меньшее, чем в поле обычной
центрифуги, т. е. примерно за 23 с.
Такие смещения дисперсной фазы в поле центробежных сил
действительно удается наблюдать с помощью специального
оптического устройства во время вращения центрифуги. Но смещения
эти имеют такой же временный характер, как и изменения
электропроводности, которые наблюдал Думанский, и дисперсная '
фаза снова распределяется по всему объему системы после
окончания вращения ротора центрифуги.
Электрические свойства золей. Противодействие уплотнению
дисперсной фазы и агрегации ее .частиц объясняется тем, чго
частицы золей несут на себе электрические заряды. В этом можно
убедиться, сопоставляя поведение в электрическом поле обычных
механических суспензий и настоящих коллоидных растворов.
68
Механические взвеси, полученные при энергичном встряхивании
тонко измельченного малорастворимого твердого вещества с какой-
нибудь жидкостью, например водой, имеют электропроводность
этой последней. Частицы твердой фазы в этом случае не
испытывают заметного воздействия со стороны введенных в раствор
заряженных электродов, а это может быть в том случае, если
они сами не заряжены.
Коллоидные растворы проводят электрический ток. При этом
все коллоидно растворенное вещество передвигается к какому-
нибудь одному из электродов. Например, лри электролизе золя
трисульфида мышьяка весь AS2S3 всегда передвигается в сторону
анода и у него выделяется. При электролизе золя хлорида серебра,
полученного приливанием какой-нибудь соли Ag+ к раствору,
содержащему избыток С1--конов, весь коллоидно растворенный
AgCl также передвигается к аноду. Наоборот, в случае золя
хлорида серебра, полученного посредством приливания раствора,
содержащего С1--ионы, к раствору, содержащему избыток Ag+-
ионов, частицы AgCl сосредоточиваются у катода.
Согласно современным представлениям, частички золя
адсорбируют из окружающего их маточного раствора ионы
кристаллической решетки, находящиеся в нем в избытке. Эти ионы
дегидратируются и врастают в поверхность кристаллов, образуя потен-
циалопределяющий слой. Для таких веществ, как галогениды
серебра, гидроксид алюминия и им подобные, которые могут
выпадать как при избытке катиона, так и при избытке аниона,
пстенциалопределяющий слой, а значит, и знак заряда, будет
зависеть от условий, в которых выделяется твердая фаза. Для
золей типа трисульфида мышьяка потенциалопределяющий слой
может образоваться лишь за счет сульфид-ионов, так как
содержание простых As3+-H0H0B даже в кислых растворах H3ASO3
ничтожно. В литературе есть указания, что в некоторых случаях
потенциалопределяющий слой может формироваться из ионов,
не входящих в кристаллическую решетку осадка.
Вокруг каждой такой частицы концентрируются ионы, несущие
заряд противопсложнсто знака. Часть этих противоионов образует
адсорбционный слой. Суммарная толщина потенциалопределяюшего
и адсорбционного слоев порядка поперечника молекулы. На этом
более или менее фиксированном в данных условиях расстоянии
потенциал грани компенсируется тем сильнее, чем - больше
противоионов находится в адсорбционном слое. Остальные противо-
ионы, вперемежку с молекулами-растворителями и ионами, знак
заряда которых совпадает со знаком заряда ионов, образовавших
потенциалопределяющий слой, образуют вторую часть двойного
электрического слоя, диффузную из-за теплового движения.
Согласно правилу Панета-Фаянса-Хана в адсорбционный слой
преимущественно втягиваются противоионы, образующие с ионами
потенциалопределяющего слоя наименее диссоциированные или
наименее растворимые соединения, а также ионы, легче других
6^^
поляризуемые в поле потенциалопределяющего слоя. Так, из
раствора, содержащего К+, Na+ и Н+-ионы, потенциалопределяю-
щий слой, состоящий из S2--H0H0B, будет извлекать
преимущественно Н+-И0НЫ; потенциалопределяющие Ag+-HOHbi из раствора,
содержащего N07» Ас~ и С10Г"-ионы, будут извлекать Ас"--ионы,
а у поверхности частиц, покрытых АР+-ионами, будут
концентрироваться не NOr-ионы, а легко поляризующиеся анионы конго-
красного, что используется в качественной реакции на А1з+-ионы.
хсм
кем
хсм
Рис. 2.3
Распределение зарядов в двойном электрическом слое
схематически представлено на рис. 2.3 для одной грани микрокристалла
с отрицательно заряженным потенциалопределяющим слоем. На
рис. 2.3 пространство влево от оси ординат — твердая фаза,
расстояние от оси ординат до пунктирной линии — толщина
закрепленной части двойного слоя (она дана здесь в очень большом
масштабе). В пределах закрепленной части потенциал cpi снижается
до значения ср2. Дальнейшее снижение потенциала до срво,
отвечающего значению потенциала в глубине раствора, вызывается
избытком противоионов в диффузной части двойного слоя. Разность
92 —фоо = С называют электрокинетическим или дзета-потенциалом.
Этот потенциал равен разности потенциалов между закрепленной
и подвижной частями двойного слоя и может быть вычислен из
скорости электрофореза.
70
Количество противоположных зарядов в закрепленном и
диффузном слоях одинаково. Вся такая частдчка-мицелла может
считаться поэтому электронейтральной, а это исключает
возможность какого-либо взаимодействия между зарядами обкладок
частицы и малым зарядом, находящимся вне частицы.
В электрическом поле такие частицы поляризуются. Диффузная
часть двойного слоя смещается по направлению к электроду,
несущему заряд противоположного знака. Сами частицы с
закрепленной на них частью двойного слоя направляются к
противоположно заряженному электроду. Так, частичка, представляемая
формулой {MaAjx}nM^"^.Rp"", где {MaAiA,}n— твердая фаза, будет
двигаться к аноду, так как она несет избыточный положительный
потенциал за счет М»*'+-ионов потенциалопределяющего слой,
а {MaAj,}nA«-, ВР+_к катоду.
В маточном растворе при отсутствии ориентирующего электрг-
ческого поля мицеллы пребывают в состоянии непрерывного
броуновского движения, не испытывая ни взаимного притяжения,
ни отталкивания, так как заряды потенциалопределяющего слоя
полностью экранируются зарядами адсорбционного и диффузного
слоев.
Электростатическое взаимодействие между частичками
образовавшегося золя возникает лишь при столкновении,
сопровождающемся взаимным перекрыванием диффузных слоев. Для достаточно
устойчивых золей оно всегда ведет к взаимному отталкиванию
частиц и обеспечивает стабильность системы.
2.11. Коагуляция золей
Твердое вещество, суспендированное в жидкости, оседает
самопроизвольно. Золь остается стабильным, но в нем можно
вызвать коагуляцию диспергированой фазы путем введения
достаточного количества электролита.
Процесс коагуляции. Коагуляцией называют уменьшение
дисперсности, сопровождающееся слипанием частиц дисперсной фазы,
т. е. превращением их в частицы с гораздо большей массой,
и ведущее к седиментации (осаждению) агрегатов. . Происходит
флоккуляция — образование осадков, в виде хлопьев.
Установлено, что коагуляцию вызывает любой электролит,
вернее один из его ^ ионов, противоположный по знаку заряду
коллоида. Коагуляция происходит, если концентрация противоиона
с достигла некоторого значения, называемого порогом коагуляции.
Коагулят, т. е. осевшая в виде хлопьев дисперсная фаза золя,
всегда уносит с собой часть противоионов. При этом и побочные
ионы увлекаются в эквивалентных количествах.
Порог коагуляции тем ниже, чем выше валентность
противоиона. Обратная ему величина, коагулирующая способность,
определяется правилом Шульце-Харди. Приводим значения относитель-
71
ного порога коагуляции: CyCi ^ и относительной коагулирующей
способности: ^z^T^ для противоионов разной валентности z:
Z 1 1 2 1 3 1 4 6
I
1
0,02
50
1,75 . 10-3
570
5,9 .10-^
1700
7,1 . 10-6
14 000
Правило Шульце-Харди выполняется лишь приближенно. В
качестве примера в табл. 2.5 проводится сопоставление опытных
данных для некоторых золей [25, с. 176] с теоретическими
значениями YzTi ^»
Таблица 2.5
Золь
Знак г
2 = 2
2=3
2=4
AS2S3
Au
^AgJ
AlA
Теоретическое
±
77
62
59
83
62
588
3333
'2000
667
769
588
25000
10 000
1000
4 167
Значения различаются тем сильнее,
зарядов несет противоион.
Некоторую ясность в наши представления о поведении частиц
золя, находящихся в состоянии теплового (броуновского) движения,
■ внесли исследования Б. В. Дерягина. Непосредственными
наблюдениями он установил: 1) частицы золя, содержащего невысокую
концентрацию противоионов, могут свободно и без всякой затраты
внешней энергии сближаться на расстояния порядка Ю-^ см,
2) дальнейшее сближение (по мнению Дерягина) требуег затраты
энергии на выталкивание гидратированных ионов и молекул
из пространства между сближающимися частицами, причем
затрата эта быстро растет по мере сближения, 3) пройдя для расстояния
около 2 нм через максимум, затрата энергии падает до нуля, когда
частицы сближаются на расстояние 1 нм. В этот момент (по Деря-
гину) прослойка между частицами самойроизвол1?нр раскрывается
и частицы слипаются.
Теория коагуляции додана объяснить все наблюдаемые при
коагуляции явления, в частности, различия коагулирующей
способности для противоионов, несущих разное число зарядов, т. е
правило Шульце-Харди.
В курсах коллоидной химии подробно излагаются
разновременно предлагавшиеся теории коагуляции. В большинстве из них
коагуляцию при введении электролита (концентрационную
коагуляцию) объясняли резким возрастанием количества противоионов
в адсорбционном слое, соответствующим снижением (практически
чем большее число
72
до нуля) электрокинетического потенциала и протяженности
двойного электрического слоя, т.е. фактическим превращением золя
в обычную нестойкую суспензию. О силах, вызывающих слипание
частиц при этом, не думали.
Протяженность двойного электрического слоя можно рассчитать
по формуле
где D — диэлектрическая проницаемость среды, ^ —постоянна
Больцмана, е — заряд электрона, с\ — концентрация и Zi — число
зарядов i-ro иона.
Для водных растворов при 25 °С
8о = 4,31 . 10-8
см.
(2.22)
Как следует из (2.21), значение 85 снижается с температурой
и ростом и й.
Та бл и ца 2.6
Валентный тип
электролита
Концентрация
электролита,
моль . л~"^
0,1
0,01 0,001
1 — 1
1—2 и 2—1
2—2
1—3 и 3—1
0,96
0,56
0,48
0,39
3,1
1,9
1,5
1,4
9,6
5,6
4,8
3,9
Приведенные в табл. 2.6
оценки 8о-.при 25 °С не
образуют ничего сходного с рядом
Шульце-Харди и имеют
размерность длины, а не силы, а
это значит, что уравнение (2.21)
имеет для теории коагуляции
лишь второстепенное значение.
В частности из сопоставления
результатов опытов Б. В. Де-
рягина и данных табл. 2.6
следует, что ожидать образования стойких золей в системах,
содержащих больше 0,1 моль • л~^ любого электролита, не
приходится.
В стабильном золе заряды потенциалопределяющего слоя
отдельных частиц полностью экранируются зарядами
адсорбционного и диффузного слоев. В результате никакого
электростатического взаимодействия между частицами золя не возникает и его
можно рассматривать как совокупность молекул, ионов и мицелл
в состоянии теплового движения. Если толщина двойного слоя
частиц такого золя заметно превосходит критические размеры,
. установленные Б. В. Дерягиным, то при случайном перекрывании
ионных оболочек, которое может возникнуть в результате
столкновений, сразу же возникает взаимное отталкивание и
стабильность золя сохраняется. Наоборот, если толщина ионных оболочек
по каким-либо причинам окажется ниже критической, каждое
соударение, сопровождающееся перекрыванием оболочек, будет
приводить к слипанию частиц и потере устойчивости коагуляции.
73:
Критерии устойчивости золей. В случае интересующей цас
концентрациснной коагуляции основную роль играет не природа
сил отталкивания и притяжения, а условия, обеспечивающие
снижение толщины двойного электрического слоя, окружающего
частицы, до размеров, допускающих преимущественное проявление
сил сцепления.
Эти условия — критерии устойчивости, обычно выражают в
единицах коагулирующей способности противоионов, вызывающих
это сокращение. Выводят соответствующие формулы чаще всего
эмпирическим путем. Проверяют их пригодность по сходимости
с правилом Шульце-Харди.
Естественно, например, предложить, что значения ^гтГ^
следует связать с концентрациями и числом зарядов противоиона,
снижающего толщину 8i двойного электрического слоя частиц
устойчивого золя до толщины 82» при которой начинается
самопроизвольная коагуляция. Пусть согласно (2.22) при 25°С Ь\=±'
= 4,31 • 10 ^(S^iZ^) ^ и для снижения Ъ\ до 82 используется соль
MYz, содержащая противоион М^+. Если для достижения 82
в системе надо создать неизвестную ^my^ = ^z, то по (2.22) заданная
82= ' (2.23)
(Sqzf+C,z2 + ZC,)/
Тогда
I
z(l + z)
|4,31 . 10-у
(2.24)
причем выражение в квадратных скобках имеет фиксированное
значение и первоначальный ионный состав, т. е. С{ и zi не имеют
значения, если не возникает взаимодействия с добавляемым
электролитом MYz. Очевидно, что = [ ] • 2-^ С2 = f ] • 6-4 ...,
■^z = [ ][z(l+z)]-^ ... и, в свою очередь, Yi = 2 [ . ..,
72=z(l+z)[ Соответствующий разным z ряд значений
ТгтГ^ определяется формулой
TzTr' = l/2-z(l + z) (2.25)
имеет следующий вид:
1
6
10 21
т с рядом Шульце-Харди не совпадает.
Поскольку и другие варианты увязки влияния Cz на 8 с
правилом Шульце-Харди благоприятных результатов не дали, для
74
оценки зависимости yz от Cz был использован сам ряд Шульце-
Харди, согласно которому
-(jj-^ I 50 570 1700 14 000
Положим, что 72ТГ^ = Z" и \g{^z^V^) = nlgz. Тогда
Z
12 3 4 6
0 1,7 2,76 3,23 4,15
0 0,3 0,48 0,6 0,78
n
5,7 ' 5,87 5,38 5,32
Из сопоставления значений п в последней строке следует, что
5 < п < 6. Расчеты для п (5, 6) дают:
п =5
Z
12 3 4 6
1 32 243 1024 7776
п =■ 6
1 64 72 9 4096 46 656
Хотя и в этом случае полного соответствия между расчетными
и опытными значениями ^гтГ^ не наблюдается, в [И, 12] при
выводе критерия устойчивости было принято, что TzT"^—z^.
Критерий Дерягина—Ландау [12] опирается на опытные данные
Б. В. Дерягина, теоретические соображения, правило Шульце-
Харди и представляется в виде формулы
СП,
(2.26)
где с—молярная концентрация электролита; П]—число
противоионов в молекуле электролита; р = 2227* — отношение валентностей
побочного иона и противоиона; А — постоянная Ван-дер-Ваальса;
е —заряд электрона; zi — валентность противоиона: D
—диэлектрическая проницаемость; k — постоянная Больцмана; с = 2Dr^2 X
X (^Тр2)-^— безразмерная постоянная, в выражении которой г —
радиус, ф —потенциал, /^(Р) — функция асимметрии электролита,
в первом приближении равная 1 + Р.
Пригодность своего критерия Дерягин и Ландау
продемонстрировали с помощью табл. 2.7, в которой сопоставлены значения
7zTr\ вычисленные по (2.26), с ^ найденными опытным путем.
75
Как следует из табл. 2.7, резкие скачки возникают только
при изменении электровалентности противоиона (zi). Валентность
побочного иона (Z2) в.^ияет гораздо слабее.
Критерий (2.26) был получен на основании некоторых
электродинамических соображений, а доводился и пpoвepяJ:cя с учетом
правила Шульце-Харди. Тот же подход можно использовать для
Таблица 2.7
Тип электролита
вычислено
опытные
данные
1,5
2,0
2,5
48
64
486
608
2560
27 216
1
1,65
2.5
2,8
51
47
573
938
1720
14 050
1
соответствующего
исправления и превращения в
критерий устойчивости золей
выражения (2.25). Учтем для
этого, что в (2.26) принято во
внимание только снижение
толщины двойного
электрического слоя, и запишем
полное выражение критерия в
виде
—2- z".
(2.27)
Для расчета п используем
выражение n=[lg(7zTr%x-
— Ig [^z(z + l)J(lgz)-^ где (TzTf^hux—значения из правила
Шульце-Харди. Расчеты дают
4.07
4.13
3.70
3,63
И Приводят к очень простому критерию
ТгТГ' .z-(z+ l)z4. (2.28)
Расчет значений относительной коагулирующей способности
при разных z с помощью (2.28) приводит к следующим значениям
48
486
2560
27 217
Т.е. к тем же значениям, которые дает критерий (2.26). Надо
подчеркнуть, что в обоих случаях вычисленные значения угтГ^
тем сильнее превышают найденные опытным путем, чем выше
электровалентность противоиона.
Основные причины, вызывающие, расхождения между
вычисленными и опытными значениями О неточности правила ШyльцeJ
Харди писал еще Р. Зигмонди (1933 г.), хорошей иллюстрацией
этого могут служить данные табл. 2.7. Основной причиной рас-
73
хождений считают недоучет специфичности вызывающего
коагуляцию соединения. Например, для КАс [54, с. 575] 71 в два
с лишним раза меньше, чем для КС1; наоборот, массивные
органические противоионы проявляют гораздо большую
коагулирующую способность, чем несущие столько-же зарядов неоргани-
чесгше ионы. Так, однозарядный неофуксин имеет fi в—'495 раз
больший, чем К+-ион, проявляя коагулирующую способность,
совпадающую с тзтГ^ из правила Шульце-Харди.
Не следует забывать и о том, что аквометалло-ионы являются
слабыми кислотами, а многозарядные анионы — слабыми основа-
1*\:!и11. Ионизация первых и протонизация вторых неизбежно
ведет к снижению числа частиц, несущих заданное высокое число
Japядoв. В результате среднее число зарядов на 1 моль частиц,
выражаемое функцией Н. Бьеррума (Пг), снижается при С =
= 0,1 моль . л""^ для Ре"^^-иона до 2,75, а для Р04'"-иона до 2,55.
Совместное воздействие всех этих и, вероятно, многих других
факторов приводит к огромным расхождениям между опытными
данными, правилом Шульце-Харди и теоретическими построениями.
В этих условиях сам выбор правила Шульце-Харди в качестве
критерия для оценки пригодность теоретических разработок
вопроса кажется сомнительным.
2.12. Некоторые практические выводы из
теории коагуляции
При химических измерениях осаждение проводится обычно
в растворах, содержащих значительные концентрации
электролитов. В связи с этим золи образуются сравнительно редко и почти
всегда не очень устойчивы. Для их коагуляции обычно используют
аммонийные соли, если в качестве противоионов нужны катионы,
и уксусную кислоту, когда необходимы противоионы — анионы.
Последующее кипячение раствора повышает кинетическую энергию
частиц, облегчает возможность их сближения, слипания и
оседания.
Выпавший при коагуляции осадок обратно в раствор обычно
не переходит. Однако в тех случаях, когда осадок содержит
большое количество маточного раствора, возможна пептизация,.
т. е. обратное превращение коагулята в золь.
Пептизация начинается чаще всего при тщательной промывке^
осадка чистой водой. При'этом избыток электролита, вызвавший
коагз^ляцию, вымывается; ионные атмосферы отдельных мицелл
расширяются и радиусы их возрастают до критического значения,
при котором силы отталкивания уже превышают силы притяжения.
В результате снова образуется коллоидный раствор и осажденное
вещество может быть потеряно.
Пептизация нарушает нормальный ход исследования и
измерений и может повести к большим ошибкам. Предогвратить ее мож ю,
77
во-первых, всеми способами, снижающими расстояние между
частицами коагулята, в частности, отсасыванием осадка на фильтре
или его центрифугированием; во-вторых, мерами,
предотвращающими возможность расширения ионных атмосфер отдельных частиц
коагулята при промывании осадка. Например, для промывки
используют не чистую воду, а раствор подходящего электролита.
Если пептизация произошла, то приходится заново добиваться
коагуляции образовавшегося золя. Поскольку, однако,
устойчивость такого золя мала ^2 ^iZ? в нем достаточно велика j, очень
часто для его коагуляции достаточно простого кипячения,
повышающего кинетическую энергию частиц и возможность их
агрегации.
Коагулят всегда увлекает с собой противоионы, попавшие
в адсорбционный слой. Иногда это может повести к потере
некоторых ионов и ошибкам. Освободить ионы, захваченные осадком,
промыванием последнего очень трудно, так как поверхности
частиц, покрытые адсорбированными ионами и пленкой маточного
раствора, слипаются и становятся недоступными для промывной
жидкости.
В подобных случаях лучше всего переосадить осадок, т. е.
заново растворить его в подходящем растворе и снова осадить
в условиях, исключающих возможность адсорбции противоионов.
Теорию промывания и переосаждения осадков рассмотрим позже.
2.13. Последовательное выделение осадков
из гомогенного раствора
В тех случаях, когда система включает два или несколько
ионов, дающих с реагентом малорастворимоэ соэдинение, а
образование золя и пересыщение практически невозможны, возникает
вопрос, в какой последовательности будут выпадать
соответствующие осадки.
Рассмотрим сначала простейший случай. Допустим, что система
включает две соли XY(i„ XY(2), причем cxY(1) == ^Yfj] = У]. cxy^2) =
= Cyj2) = ^2- Спрашивается, в какой последовательности будут
выпадать осадки MY(i){x и MY(2)p. при постепенном введении
в систему раствора MY^,, если ^my^^^, = Pi и Рт^2)^ = ^2-
При решении этой и других таких же задач надо помнить,
что использовать законы, касающиеся гетерогенных равновесий,
можно только для систем, в которых твердая фаза находится
в равновесии с насыщенным раствором. На первый взгляд поэтому
кажется, что к вопросу о последовательности выпадения осадков
из раствора применять эти законы нельзя. Однако простой прием
делает такое использование вполне законным.
78
Допустим, что оба осадка выпали и между ними и раствором
установилось равновесие. Тогда
MY(,),(t)^M^+ + (.Yu), (2.29)
■ _ MY(2)^t)5:M'^++!xYH), (2.30)
и по закону действующих масс
mt/i = Pu ту^2 = Р2. (2.31)
Исключая т из уравнений (2.31), находим, что
У 2
I
Это очень важное соотношение показывает, что после выделения
обоих осадков отношение активностей осаждаемых ионов должно
сохранять постоянное отношение, характерное для этого двойного
гетерогенного равновесия. Если осадки MYd)^^ и МУ(2)ц, не могут
образовать твердый раствор, то, зная значение константы /С, можно
решить вопрос о том, какой из двух осадков будет выпадать
первым. Для этого находят отношение начальных концентраций:
^ = 9 (2.33)
и Сравнивают его со значением К из (2.32). При этом возможны
три случая: д = К, д <К, д > К.
Если д = Kj то отношение У1/У2 в системе совпадает с самого
начала с тем, которое должно установиться при наличии двойного
гетерогенного равновесия. В этом случае оба осадка начинают
выпадать одновременно и выпадают в таком соотношении, что
отношение r/i/r/2 остается постоянным^ равным д.
Если д< Ку т. е. Y1/Y2 < У\/у2^ устанавливающегося при
равновесии, то первым должен начать выпадать осадок MY(2),x.
При этом Yi остается неизменным, а Y2 стремится к такому
значению г/2, при котором будет достигнуто соотношение
^ = /(. (2.34)
Очевидно, ^2==^^l^C""^ после достижения этого значения оба
"осадка начнут выпадать одновременно и притом в количествах,
обеспечивающих выполнение условия (2.32).
Если д > Ку т. е. Y1/Y2 > У\1у2. устанавливающегося при
равновесии, то первым начнет осаждаться осадок MYd)^^ и будет
осаждаться до тех пор, пока установится соотношение y\IY2 = К.
В этот момент ух =^ KY2y а в дальнейшем будут выпадать оба
осадка.
Систем, в которых последовательное осаждение не связано
с дополнительными процессами, почти нет, и подобрать безупреч-
79
ный пример очень трудно. Пожалуй, ближе всего подходит, если
пренебречь образованием хлорокомплексов, осаждение хлоридов
Ag+ и Hg2+-H0H0B.
Допустим, что к раствору, в котором CAguo^ = 0,1 = Mi
и CHg,(No,h = 2 • 10-2 iQ-ij моль . л-^ подкисленному
HNO3, постепенно приливают разбавленный раствор НС1.
Попытаемся выяснить, в какой последовательности будут выпадать
осадки.
Поскольку закон действующих масс можно применять лишь
к установившимся гетерогенным равновесиям, допустим, что оба
осадка выпали. Тогда
AgCl (т) ^ Ag+ + С1-, miy = 10-9.75^
Hg2Cl2 (т)« Hgl+ + 2С1-, т2у' - 10~^''^ ^-
и можно утверждать, что в системе, в которой выпали оба осадка,
при дальнейшем добавлении НС1 оба осадка будут выпадать
одновременно в таких количествах, что отношение тЬ^2 будет
оставаться постоянным и равным Ю-^-^ при t = 25° С.
Поскольку соответствующее отношение начальных
концентраций M?Mr^ = (10~^f (10""^'^Г^=Л0""°'^==0,5 больше величины
10-2'2, К которой должна стремиться равновесная система, в первую
очередь будет выпадать AgCl, а М2='10'-^''^молъ -л-^ будет
оставаться неизменной (если считать, что объ^м системы не меняется) до
момента, когда т\МГ^ станет равным 10~^•^. Это произойдет,
очевидно, когда концентрация Ag+-HOHa снизится до т\ =
= l/lO-^'^. 10-2.2 10-1.95 ^ 1,1 . 10-2 моль • л-^ с этого
момента начнется совместное осаждение обоих хлоридов.
,Расчеты усложняются, если осаждаемые ионы участвуют в пэ-
бочных процессах, снижающих их активность.
Пусть, например, в систему, где CmV^, = М, св\^ == В, с,^^ = 31,
вводится реагент ХаА, образующий малорастворимые соединения
MaAj^, BaAv. Спрашивается, какой из осадков начнет выпадать
первым, если M(0H2)n^ и В (0H2)n" диссоциируют как кислоты,
а В*+-ион, кроме того, координирует основание 3t.
Матрица равновесий системы MY^ — ВYv — 91 — ХаА — Н2О
представлена в табл. 2.8.
Для системы, в которой уже выпали оба осадка, выполняются
равенства т«а»^ = Pi и =; (2.35), приводящие к важному
условию
80
= /С, (2.36)
1
Таблица 2.8
К—
м
0
1
О
о
о
о
а
О
О
о
1
о
о
1
о
а
О
о
о
I
о
п
О
о
о
о
о
о
1
о
-1
-к
-к
1
i
о
о
о
в (ОН)^-'*
Ног^+
вог;;+
К\ (Т)
BaAv (Т)
W
а
'\
Pi
Р2
определяющему, какой осадок будет выпадать первым, если
известны активности т, b осаждаемых ионов в начальной системе.
Применяя к системе, не содержащей осаждающего реагента
ХаА, закон сохранения начальной концентрации, получаем систему
уравнений:
М = m S cpM(ON)k'^ik/^"^^, (2.37)
к=0
V 1^
N
(2.38)
(2.39)
Если рН задан или измерен, что всегда возможно, то из (2.38)
и (2.39) можно исключить Ь, а затем решить полученное уравнение
относительно а и, зная h и а, рассчитать т и b с помощью (2.37)
и (2.38).
Если рН не измерялся, а также в присутствии многоядерных
гидроксокомплексов, расчеты возможны лишь на ЭВМ.
Рассмотрим в качестве примера пocлeдoвafeльнocть, в которой
должны выпадать осадки Са(10з)2 и Cd(I03)2 4i3 системы, в которой
Сса(С1о.ь = М = 0,2, Ccd(ci04b = В = 0,1, = 31=0,5 моль • л-^1
при введении в нее КЮз.
/ Поскольку pPcadOeb = 6,15 и pPcddOe), = 7,6,. отношение
тЬ"^ = 10^'^^, а = I0°"^ можно ожидать, что первым будет
выпадать Сс1(10з)2, так как возрастание отношения Ю^^'З до 3^^.
чения Ю^'^**^ возможно, на первый взгляд, только за счет снижения
В == Ccd(Ci04),.
Однако такой вывод в данном случае нельзя считать
окончательным, поскольку последовательность выпадения определяется
6 3-457
81
00
ю
Таблица 2.9
2(М В)
f
м в ^
0.6
0.2
0.1
0.5
С10~
Са2+
Cd2+
NH3
Н +
1 = 1 i = 2 1 i = 3 \ i = 4
.0
1
0
0
0
0
1
Q
0
0
1
0
0
1
0
1
0
0
0
1
0
n
0
0
0
0
0
0
1
0
2
2
—1
— 1
—к
1
1
0
0
0
0H~
, Ca(OH)+
Cd(0H)2-k
NH+
HIO3
Cd(NH3)2+
Ca (103)2 (T)
С(1(10з)2(т)
^k
Pi
P2
— 14
-12,6
—9,30
9,24
0,78
2,51
—6,15
-7,6
-19,17
4,47
—32,48
5,77
не отношением начальных концентраций Са2+ и Сс12+-ионов.
а отношением их активностей пг и b в начальной системе. Для
оценки этого отношения используем матрицу равновесий системы,
приведенную в виде табл. 2.9 в той части, которая отвечает си-
стеме до введения в нее ЮГ-ионов.
Применение закона сохранения начальной концентрации
приводит к системе уравнений:
5=Ь
М^т (срсаОн^зЛ-^ + срса)» ' (2.40)
(2.41)
• 3 4
S TCd(OH)k'i[lk^-^ + S TCd(NHs)nPna"
к=0 п= 1
о ^
St = а (срын.аЛ + cpNH.) + 6 S ncpcd(NH3)„Pna". (2.42)
n=l
Используя значения начальных концентраций, находим, что
/ = 0,9 моль • л-^ а затем получаем по Девису Igcpi = 0,14 и lgcp2 =
= 0.56.
Измерение дало рН == И. Уравнение сохранения
электронейтральности оказалось ненужным, а для расчета а путем исключения
b из (2.41) и (2.42) было получено уравнение
4
Щ = а (ср1аЛ + Фо) + В -3 ^ . (2.43)
к=0 п=1
Подстановка в (2.43) численных значений и обычные
преобразования приводят к уравнению + 10-1'^893^4_ 10-Ьб278^3 —
— 10-2.6401^2 10-4.6510^ _ 10-3.9522 =3 о и значению а = 0,1932
моль . л-1. Из (2.40) т = 0,0545 = 10-Ь2б32 ^оль • л-i. Из (2.41)
b = 2,58 • 10-6 ^ 10-5.5883 моль л-К Значит, тЬ"^ = Ю^.згб! >р
> lO^'^s и первым должен выпадать Са(10з)2.
Этот пример показывает, что недоучет одновременно
возникающих равновесий может привести к совершенно неправильным
выводам относительно последовательности осаждения.
Расчет равновесия в системе, описанной в табл. 2.8 в тот
момент, когда одновременно с осадком МаА^ начинает выпадать
BaAv, связан с гораздо большими трудностями. В самом деле,
если первым выпадает MaAj^, то здесь уже нельзя считать, что
активность (В^Н-) остается равной Ь. Возможность измерения рН
исключается, так как просто неизвестно, в какой момент это
из мерение надо производить. Появляются новые неизвестные:
А г=сх^Ау а = (А*-) и Si— количество вещества МаА^ (моль),
83
выпадающего к моменту начала совместного выпадения обоих
осадков.
В этом случае приходится использовать систему уравнений:
= Д срм(он)к^1к/1-'' + a5i, . (2.44)
(2.45)
5 = & ^ cpB(0H)k'^2k/i ^ + S фвот Рпбе"
к=0 п==1 "^^п
N
31 = а (срнщ + + щ) + b ncpegj^^Pna", (2.46)
а'
>l = aS^<PHjA8j/i4-t*Sb (2.47)
^vj,-^ ^ (2.36), /„«а^^ = Pi, (2.35)
p.'
(рнЛ ~ сроншЛ-' — ((аЛ1 — vB) + аЛ + — к) (pM(OH)kiSik/iIi;'' +
v' N
+ Д — к) (рв(он)к^2кЛ-''+ <pB5i^i' РпЛ" +
a
+ срнщ ааЛ + а 2 (J — а) cpHjAajW,= 0. (2.48)
В этой системе семь неизвестных: /п, h, S\, 6, а, Л, а.
Расчеты даже в случае отсутствия многоядерных комплексов
связаны с трудностями из-за одновременной нелинейности
уравнений относительно нескольких неизвестных, и для их проведения
приходится привлекать ЭВМ.
3. СООСАЖДЕНИЕ
При выделении твёрдой фазы из гомогенного раствора
попадание посторонних веществ в нее обусловлено несколькими
причине ми, в частности, частичным замещением основных ионов
решетки другими ионами; притяжением посторонних ионов из
маточного раствора ионами решетки, находящимися на поверхности
выпадающих кристаллов; включениями маточного раствора и
адсорбированных ионов в неправильно срастающиеся кристаллы
и т. л.
В первом случае говорят о загрязнении твердой фазы из-за
образования твердого раствора, во втором —адсорбции, в
третьем — окклюзии.
3.1. Твердые растворы
Термин «твердый раствор» введен профессором Харьковского
университета Павлом Эйнбродтом (1851 г.). Принятое
определение этого понятия: «твердый раствор — однородный комплекс
нескольких тел, соотношения между которыми могут меняться при
сохранении однородности», — принадлежит Вант-Гоффу (1890 г.).
Огромный фактический материал и ряд вгжных обобщетй по
этому вопросу даны в диссертациях Д. И. Менделеева.
Безусловно, работа над первой «Изоморфизм в связи с другими
отношениями кристаллической формы к составу» (1856 г.) стала
началом будущих великих открытий.
Еще совсем недавно твердыми растворами интересовались
главным сбразом радксхимики [42], использовавшие образование
твердых растворов для извлечения радиоактивных веществ из сверх-
разб^вленных систем. Даже в самых полных • монографиях
по теории аналитической химии им уделялись считанные строки.
Од1 ако по мере осложнения исследуемых систем старые прописи
все чаще приводили к серьезным систематическим ошибкам,
причиной возникновения которых оказалось образование твердых
растворов. Это обстоятельство вызвало появление ряда
фундаментальных работ, выполненных Кольтгофом с сотрудниками [19] и
другими авторами, сводка работ которых дана а [26].
Поскольку ошибки за счет образования твердых растворов
могут вызывать серьезные осложнения при качественных и
количественных определениях и образование твердых растворов ис-
85
пользуется для разделения и концентрирования определяемых
компонентов, необходимо выяснить следующее:
1) как предугадать (обеспечить) образование твердого
раствора; 2) как рассчитать равновесие в системе, где образуется
твердый раствор, в частности, как оценить возникающую за счет
образования твердого раствора ошибку; 3) как предотвратить
образование твердого раствора, если оно нежелательно, или как
обеспечить образование твердого раствора, если оно полезно.
В гетерогенных системах раствор-осадок твердые растворы
получаются за счет замещения ионов основной решетки (в
процессе ее образования) другими ионами по законам изоморфизма,
играющим огромную роль в геохимии [38]. Под изоморфизмом
понимают при этом способность элементов замещать друг лруга
в кристаллической решетке при условии близости размеров
составляющих кристалл единиц (атомов, ионов и т. п.), тождества
знака заряда (но не числа зарядов) и относительно близких
величин'их поляризации.
Геохимики установили ряд важных правил изоморфного
замещения.
1. Возможность изовалентного изоморфизма определяется в
первую очередь близостью радиусов ионов, которые должны
различаться не более, чем на 15%. При этом решетки соединений
MaAjx и BaAj,,, где В«^+— внедряющийся ион, должны иметь
одинаковую структуру.
Так, С1--И0Н может заместить Вг--ион в твердом AgBr, так
как Г(>|_ = 0,181 нм, т.е. всего на 8,3% отличается от rg^.-=
= 0,196 нм, и-AgCl, AgBr кристаллизуются в структуре
каменной соли. В то же время К^-ион не может внедриться в решетку
Т1С1, хотя rj^+= 0,133 нм не выходит за пределы интервала
10,127—0,171] нм, охватывающего все радиусы катионов,
способных замещать Т1+-ионы (/-71 = 0,149 нм), так как KCI
кристаллизуется в решетке типа. NaCl, а НО —типа CsCl. В случае
NH4CI образование твердых растворов с Т1С1 возможно, так как
структуры их одинаковы и г^^^4. = 0,143 нм.
2. По гетеровалентной линии изоморфное замещение также
возможно; радиус иона, несущего большее число зарядов, может
быть при этом равен или превышать не более чем на 10 % радиус
замещаемого иона; радиус же иона с меньшим числом зарядов
может быть меньше, но не более чем на 10 %, или равен
замещаемому. В обоих случаях должна происходить замена
противоиона, обеспечивающая сохранение электронейтральности решетки.
Так, при замене в системе СаЗЮз иона Са^+ на Na+-HOH (Гсагч- =
= 0,104 нм, Tj^g^. = 0.098 нм) в нее одновременно должны войти
F- или ОН--ИОНЫ, а при осаждении BaS04 из растворов, содер-
л<ащих КМп04, выпадает осадок, окрашенный в фиолетовый цвет.
8G
;В этом случае rj^+ = 0,133 нм. Гз^г-ь = 0,138, /-^^„7+= 0,046,
Vs6+ = 0,034 нм.
3. Уже очень давно Д. И. Менделеев ввел представление
о «компенсационном изоморфизме», когда оказываются
соизмеримыми не отдельные ионы, а суммы ионов, что и обеспечивает
возможность взаимного замещения. Так, сравнивая формулы
полевого шпата (KAlSisQs) и Лабрадора (CaAl2Si208), Менделеев
указывал, что Са и А1 играют в Лабрадоре почти ту же роль,
что К и Si в полевом шпате, что здесь не просто А1 изоморфно
замещает Si, а сумма А1+Са замещает сумму K + SIb порядке
компенсации свойств. Лишь через несколько десятилетий, когда
уже стали известны размеры ионов, представления о
компенсационном изоморфизме были сведены к обобщенной формулировке:
в сложных структурах атомы группируются по величине
радиусов с одновременной компенсацией зарядов. Для полевого
шпата Sz = 1 + 3 + 4-3= 16, а Lr = 0,133 + 0,057 + 0,039 • 3 =
= 0,-307, а для Лабрадора Sz = 2 + 3-2 + 4-2 = 16 и Sr =
= 0,104 + 0,057.2 + 0,039-2 = 0,296.
А. Е. Ферсман [38, с. 141] предложил характеризовать
возможности изоморфного замещения путем построения звезд
изоморфизма, в центре которых находится замещаемый ион, по
вертикали располагаются изовалентные заместители, а по
горизонтали — гетеровалентные. В [38] указано, что равные (или
почти равные) радиусы имеют ионы элементов, расположенных
по направленным слева направо (вниз) диагоналям в длинной
форме таблицы Менделеева (см. табл. 3.1), например: г^^^ =
= 0,068, Гд,^2+ = 0,074, rs^3+ = 0,083, г^^,+ = 0,087, = 0,098,
г^^2+ = 0,104 нм. Это правило облегчает составление звезде
помощью матрицы ионных размеров, которой является табл. 3.1.
В качестве примера построим звезду изоморфизма для Ва2+-
иона. Поскольку Гз^2+== 0,138 нм, на изовалентный интервал
попадают М2+-ионы, для которых 0,117 < г< 0,159, М+ и
ионы, для которых 0,i24<r<0,38 и МЗ+-ионы, для которых
0,138 <Гд^з+< 0,152 нм. В результате отбора получаем первый
вариант звезды в виде
Sr2+(0,120)
РЬ2+ (0,126)
/Vu+(0,137)K+ (0,133) Ва2+(0,138)
Ra2+(0,144)
M3+-H0HQB, радиусы которых удовлетворяют условию замещения
Ва2+-иона, нет.
Из рассмотрения звезды следует, что важнейшими ионами,
которые могут давать твердые растворы с солями Ва2+-нона,
87
la
Па
Ша
IV а
Va
Via
Vila
Villa
Li
11+0,078
Be
|2+0,034
Na
11+0,0981
Mg
2+0,074!
К
1+0,133
Ca
2+0,104
Sc
13+0,083
Ti.
2+0,078
3+0.069
4+0.C64
V
2+0,072
3+0,067
4+0,061
5+0,04
Cr
2+0,083
3+0,064
6+0,035
Mn
2+0,091
3+0,070
4+0,052
7+0.046
Fe
2+O.O80I
3+0.067
Co
2+0,078
3+0,064
Rb
11+0,1491
Sr
5+0,120
Y
|з+0,097|
Zr
4+0,092
Nb
4-<-0,067
5+0.066
Mo
4+0,068
|б+0,0б5
Тс
Ru
14+0,062
Rh
3+0,075
4+0,065
Cs
)+0,165|
Ba
i+0,I38|
La
3+0,106
4+0,090
Hf
4+(0,078)
Та
|5+(0,06B)
W
|4+0,068
6+0,065
Re
6+0,052j
Os
|4+0,065|
Ir
|4+0,065
Fr
Ra
7+0,144
Ac
3+0,111
Ce
3+0.103
4+0,0j2
Pr
|з+0,101
4+0,0 0|
N'J
3+0 099
Pm
3+(0,098)
Sm
3+0.09б|
Th
3+0,108
4+0,0:)5
Pa
3+0,106
4+0,036
и
3+0,103
4+0,093
Np
3+0,101
4+0,092
Pu
3+0,100
4+0,086
Таблица 3.1
IVlIIfl
lb
116
U\b
Vb
Vlb
WUb
WUb
H
1-0,136
1+0,000
He
0 0,122
В
3+(0,020)
с
4+0,02
4+(0,015)
4-(0,260)
N
3+
5+0,015
3~0,148
0
2-0,136
F
1-0,133
Ne
0 0,160
A!
3+0,057
Si
4+0,039
P
3+
5+0,035
3-0,186
S
2-0,182
6+(0,029)
CI
1-0,181
Ar
0 0,192
Ni
2+0,074
Cu
' 1+0,098
2+0,080
Zn
2+0,083
Ga
3+0,062
Ge
2+0,065
4+0,044
As
3+0,069
5+(0,047)
3-0J9I
Se
2-0,1-'3
4+0,069
6+0,035
Br
1-0,196
7+(0,039)
Kr .
0 0,198
Pd
4+0,064
Ag
1+0,113
Cd
2+0,099
In
3+0,092
Sn
2+0,102
4+0,067
'Sb
3+0,090
5+0,062
3^0,208
Те
2-0,211
4+0,089
6+(0,056)
I
1-0,220
7+(0,050)
Xe
0 0,218
Pt
4+0,064
Au
1+(0,137)
Hg
2+0,112
Tl
1+0,149
3+0,105
Pb
2+0,126
4+0,076
Bi
3+0,120
5+(0,074)
3-(0,213)
Po
At
Rn
Eu
3+0,095
Gd
3+0,094
Tb
3+0,092
4+0,084
Dy
3+0,091
Ho.
3+0,089
Er
3+0,088
Tu
3+0,087
Yb
3+0,086
Lu
3+0,085
Am
3+0,099
4+0,090
Cm
Bk
Cf
Es
Fm
Mv
No
89
следует считать изовалентные Sr2+, РЬ2+, Ra2+; гетеровалентные
К+ и Аи+/
При замене Ва2+ на К+ и Аи+ необходима одновременная
замена 504""-иона на удовлетворяющий требованиям
изоморфного замещения одновалентный анион.
Для первичной проверки правильности построенной звезды
можно использовать сведения о химическом составе минералов,
приводимые в справочниках по описательной минералогии. Так,
например, в [10, с. 322] указано, что барит (BaS04), целестин
(SrS04) и англезит (PbS04) изоморфны и что обычными примесями
к бариту являются эти минералы и, что несколько неожиданно,
CaS04.
Дополнительные сведения о ионах, способных замещать Ва^+-
ион, можно найти в специальной литературе, главным образом,
в работах, посвященных исследованию аналитических прописей,
измерению атомных масс и радиохимии. Так, например, радио-
химики указывают, что Ва2+ и Ra2+ всегда дают истинные
смешанные кристаллы, т.е. твердые растворы. В специальной
литературе подтверждается возможность замещения Ва2+-ионов
Канонами. Относительно Аи+-ионов указаний нет, но зато отмечается
возможность образования твердых растворов, включающих BaS04
и NH4HSO4 или NaHS04, LiHS04 и LiHS04-2H20, НМПО4 и Н2О.
Поскольку Na+ и Ы+-ионы не могут непосредственно
заменять Ва2+-ионы, так как г^^^ = 0,098 нм и г^^^ = 0,068 нм не
попадают в интервал [0,124—0,138] нм, отвечающий гетерова-
лентному лучу звезды Ва2+.иона для М+-ионов, надо думать, что
фактически вместо Ва^''"-ионов внедряются Li (ОН2)2'-, Li(0H2)t-
и Na(0H2)+-H0Hbi, так как, согласно данным, приведенным в [45,
^- 37], /-^^^^^^^+=0,115 нм, /-^,^^^^^+ = 0,137 нм, а тогда
^Na(oH2)+^^''^ НМ, как МОЖНО заключить из величины/-^^^^^ ^^.=:
= 0,146 нм и изменений радиусов гидратированных Li+ и Са2+.
ионов на один моль воды.
Если теперь учес1ь, что /'н^о = ^н,о+^''^8 нм и данные,
приведенные в [45], то можно построить новую более подробную
звезду изоморфии для Ва2+-иона;
Sr2+ (0,120)
РЬ2+(0,126)
Са(ОН2)2+ (0,127)
Na(0H2)+ (0,120) К+ (0,133) Аи+, Ва2+ (0,138)
Li(0H2)^ (0,137)НзО+(0,138)
Ra2+(0,144)
Вода в звезду не включена, так как она внедряется в решетку
BaS04 вместе с НМПО4, т. е. в виде Н3ОМПО4, подобно КМПО4.
190
Звезду для Ва2+-иона следовало, конечно,' пополнить гидро-
ксоионами, например, FeOH2+, внедрением которых объясняется
обнаруженное присутствие железа в осадке BaS04. К сожалению,
сведений о радиусах комплексных ионов очень мало, а это
ограничивает возможность предсказания систематических ошибок за
счет образования твердых растворов.
3.1.1. ЭКСПЕРИМЕНТАЛЬНАЯ ПРОВЕРКА ЧИСТОТЫ ОСАДКОВ
Сь].еделением примесей, увлекаемых осадками, занимались
специалисты по измерению етомных масс и химики, проверявшие
аналитические Прописи. Исследовали и высушенные, и
прокаленные осадки. Использовались очень сложные и трудоемкие
методы. Так, для определения примесей в осадке BaS04 проводили
следующие операции.
Для измерения примеси H2SO4 высушенный при 105° осадок
помещали в платиновую лодочку и прокаливали в токе чистого
сухого воздуха в трубке для прокаливания. Выделяющуюся
H2SO4 помещали в 3%-ный раствор перекиси водорода, а затем
осаждали в виде BaS04 и взвешивали.
Для оценки содержания НС1 прокаленный осадок BaS04
растворяли в горячей концентрированной H2SO4, отгоняли НС1 в
раствор AgNOs и взвешивали образующийся осадок AgCl.
Прокаленный осадок BaS04 растворяли в горячей
концентрированной H2SO4, охлаждали полученный раствор, вливали при
помешивании в холодную воду, фильтровали, выпаривали
фильтрат до полного удаления H2SO4. Остаток растворяли в воде,
раствор выпаривали в платиновом тигле. Привес после
прокаливания при красном калении учитывался как примесь сульфатов
щелочных металлов.
В связи с трудоемкостью всех этих операций и измерений
большинство исследователей ограничивалось определением
прироста *или убыли массы осадка, образующегося в той или иной
системе по сравнению с ожидаемым теоретическим выходом. Так
поступают даже современные исследователи [53], хотя
использование прямых методов определения микропримесей в
образующихся осадках могло бы дать более точные и исчерпывающие
данные по этому вопросу.
3.1,2. РАВНОВЕСИЯ В СИСТЕМЕ ТВЕРДЫЙ РАСТВОР — ЖИДКАЯ ФАЗА
Твердые растворы, больше всего нас интересующие,
образуются при выделении ионных кристаллов из гомогенного
раствора в результате внедрения в решетку посторонних ионов по
описанным выше правилам изоморфного замещения. Такие
кристаллы должны быть электронейтральными, что позволяет
приписывать им формулы соответствующих молекулярных летучих
веществ, например, безводному ферри-хлориду— формулу FeCb.
91
Надо только помнить, что каждый такой кристалл, по существу,
является макроскопичесьсй молекулой, молярная масса которой
тем больше, чем больше кристалл. Формулу такого кристалла
можно записать в виде {{МаАр,)п}.
Изоморфное замещение в ней части Mf^+-ионов на
изовалентные В1^+-ионы приведет к образованию ряда электронейтральных
соединений с формулами вида {(M«(i-v)BavAtx)n}» где v может
изменяться от О до 1, если ВаА(д. и МаА^, ^•cгyт смешиваться во
всех отношениях и от О до какого-то предельного значения,
лежащего между О и 1, в случае ограниченной растворимости.
Получающиеся соединения следует считать неопределенными, так
как их состав и молярные массы изменяются го мере замещения
М''+-ионов В1^+-ионами непрерывно.
Различают гомогенные и гетерогенные твердые растворы.
В первом случае твердая фаза состоит из кристаллов, в
которых содержание посторонних ионов сохраняет неизменное
значение в любом элементе объема, включая центр кристалла и его
края. Гомогенные твердые растворы образуются в тех случаях,
когда в процессе выделения твердой фазы весь образовавшийся
осадок постоянно находится в равновесии со всем раствором.
Во втором случае в каждый момент времени в равновесии с
раствором находится только последний (поверхностный) слой
кристаллов, состав которых в дальнейшем не меняется. Первичные
кристаллы будут состоять при этом из наименее растворимого
вещества, допустим, МаА(д., которое должно выделяться первым.
Последующий микрослой, выделяющийся на поверхности
первичных кристаллов из раствора, в котором концентрация М'^+-ионов
уже понизилась, будет содержать небольшую примесь BaA^,.
В микрослоях, выделяющихся вслед за первым, содержание
ВаА^ будет постепенно возрастать, и в результате получится
твердая фаза, состоящая из отдельных кристаллов слоистого
строения. В этом случае каждый микрослой является твердым
раствором, которому отвечает свой, возникший в данных
условиях осаждения состав.
Надо думать, что в чистом виде ни один из случаев не
реализуется. Любые расчеты в связи с этим могут дать лишь при- •
ближенные результаты, для получения которых можно,
естественно, воспользоваться простейшими моделями и методами.
3.1 3. РАВНОВЕСИЯ в СИСТЕМЕ ЧИСТАЯ ТВЕРДАЯ ФАЗА — РАСТВОР,
СОДЕРЖАЩИЙ ЗАМЕЩАЮЩИЙ ИОН
Допустим, ЧТО равновесие между твердой фазой и раствором
устанавливается практически мгновенно и перекристаллизация
твердой фазы, ведущая к образованию гомогенного твердого
раствора, осуществляется с такой же высокой скоростью. Тогда
вместо изучения процесса постройки решетки из двух одновре-
92
менно встраивающихся в нее в соответствующем соотношении
изоморфных соединений, можно рассматривать взаимодействие
чистой твердой фазы, которая должна выпасть первой, с
раствором, содержащим интересующую нас начальную концентрацию
замещающего иона. Такой подход сводит задачу в простейшем
случае к расчету гетерогенного равновесия
МА(т) + В«ВА(т) + М, К (3.1)
эквивалентному сумме гетерогенных равновесий: МА (T)y:i=tМ + А,
которому отвечает произведение растворимости Рь и B + A5=i'
5=tBA(T), которому отвечает константа равновесия PJ^ (где Р2—
произведение растворимости ВА).
.Полагая неизвестные коэффициенты активности М, В, А-ионов
в растворе и тдердой фазе равными единице и применяя к
равновесию (3.1) закон действующих масс, находим, что
[MJ пва_. ^1
(3.2)
ма
mr
AgBr
BaCr04
RaS04
BaS04
Pb (103)2
p1p2 ^
-3
где Hi—молярная доля i-й частицы в твердой фазе.
Для измерения X осаждают рассчитанное количество твердой
фазы МА из раствора, содержащего известную начальную
концентрацию замещающего иона В. Последующие измерения
равновесных концентраций обоих ионов М и В в растворе, позволяют
рассчитать молярные доли МА и ВА в твердой фазе, а затем и X
по (3.2). В табл. 3.2 сопоставлены измеренные значения X и
соответствующие значения
PiPJ^ для некоторых рав- Таблица 3.2
новесий вида
МА(т)+ R^MR (т) + А.
Сопоставление
приводит к заключению, что,
наряду с
удовлетворительным согласием X и "PiPJ"^
эти значения могут
различаться на несколько
порядков.
В некоторых
специальных случаях количество
твердой фазы МА бывает относительно очень большим, [М]
определяется растворимостью МА и остается практически постоянной, а съ
настолько мала, что практически полное внедрение В-иона в
решетку МА не может изменить значение Пмд. В этих условиях, с
которыми столкнулись В. Г. Хлопни и его сотрудники [43] при изучении
образования твердых растворов в системах
BaCl2 (т) + Ra^+ RaC^ (т) + Ва-^-ь,
AgCl
BaS04
BaS04
PbS04
Ba (10з)2
2,2 . 10-3
3,8 . I0~3
14,7 . 10-3
0,91
0,56
0,12
4 . 10-2
5 . 10
2
0,43
5 . 10-3
2,5... lO-'^
93
Ba(N03)2 (т) -1- Ra2+?=i Ка(КОз)2(т) + Ва2+
уравнение (3.2) имеет вид
В (3.3) т<^1ва\, и m<AZBaY2, где L—растворимость чистого
BaY2, т—ничтожный вклад в [Ba^+J за счет Ва2+-ионов,
вытесняемых из решетки Ва¥2-ионами Ra2+. Тогда
'^RaY^ ^BaY, ^
и в общем случае
-щу = X, (3.4)
где X—константа распределения-В-иона между твердой и жидкой
фазами.
Рассмотрим некоторые случаи применения равновесия (3.1)
и уравнения (3.2).
Допустим, что в 1 л раствора, в котором исходные
концентрации 'М и В-ионов равны = и Св== ^2, введено такое коли-^
честно А-иона, что б'А = и^ь Если по расчету первым должен
выпадать осадок МА и и > 1, то можно для начала предложить,
что М-ионы будут осаждены полностью, а остаток осадителя
(и—1)^1 будет полностью израсходован на осаждение В-иона.
При точном выполнении этого предположения суммарное количество
МА и ВА в осадке составит u^i моль, , начальные концентрации
М и В-ионов будут соответственно равны: = 0; = ^2 —
— (и—1)^1, а молярные доли МА, ВА в твердом растворе
определятся выражениями
о , о (4 — 1)^1
ntAA = Ci/UCu ПвА = —г-;; .
Поскольку предположение, чтосм=0, лишено смысла,
допустим, что aci моль М-иона остается в растворе, а такое же
количество В-иона дополнительно внедряется в твердую фазу.
Тогда равновесные концентрации этих ионов в растворе и
соответствующие значения щ в осадке будут определяться
выражениями, приведенными в следующей схеме:
А, МА(т) + В - 7^ ВА(т) +М, X
Со UCj ^2 Ci
с О —(U — l)Cj О
по
[] ^2 —(U—1) Cj — ас, ас^
С,(1-а) (u-l)c,+aci
94
Подстановка этих значений в (3.2) приводит после обычных
преобразований к квадратному уравнению
[q-(U-l+a)J(l-a)
где q = СгсГ', и а — молярная доля М-иона, замещенного В-ионами
в твердой фазе.
Решение этого уравнения приводит, к расчетной формуле
„ 2X[q-(u-l)]
[(u-l)(l-X) + (H-q)M +
(3.6)
+ >/[(u-l)(l-X) + (l+q>Xp + 4X(l-X) (q -(u-l)
при u = 1 уравнение (3.5) переходит в
а расчетная формула (3.6) принимает вид
^ ^ 2qx ^3
(1 + q) X + + q)2X^ + 4qX (1 X)'
Для u < 1 путем приведенных рассуждений находим, что [М] =
(и — а) С, аСх
^=={1 —u)ci +(хси [В] = С2 — аси АгмА = ———,АгвА = —.
Подстановка этих значений в (3.2) приводит к уравнению
(l-,U+a)a ,OQ4
и значению
2uqX
а =
(1 - U) + (U + q) X +/[(1 ~ U) + (U+ q) ХР + 4uqX (1 - X)*
(3.10)
Очевидно, что при и=1 (3.9) переходит в (3.7), а (3.10) —
в (3.8).
С помощью приведенных выше формул можно вычислять
значения а, т. е. степень загрязнения первой твердой фазы второй
при разных X и начальных условиях, и, наоборот, значения X,
обеспечивающие заданную степень чистоты.
Так, используя для системы Вг-, С1~, Ag+ при q = 1 фор-
мулу-(3.8) и считая Х = 2,54-10-3, находим, ^ло а = 4,8-10-2.
Это значит, что при добавлении к раствору, содержащему Вг-
и С1- в эквимолярном соотношении, Ag+-HOHOB в количестве,
достаточном для осаждения только AgBr, получить чистый AgBr
нельзя, так как молярная доля оставшегося в растворе Вг"*-иона
95
составит 4,8 % и такой же будет молярная доля AgCl,
внедрившегося в осадок AgBr.
В качестве второго примера в табл. 3.3 приведены
результаты расчета по формуле (3.7) значений X, обеспечивающих
заданные значения а при и = 1 для разных q = С2сТ^.
Из табл. 3.3 следует, например, что при q = 10 рассчитывать
на то, что загрязнение осадка МА ионом В не превысит
а = 10""^ = 0,1 %, можно лишь
при условии, что Яма на семь
порядков меньше Рва, так
как X — Pi -РТ\ Поскольку
далее pPAgBr = 12,3; рРд^с! = 9,75
и X 10~2'55^ при U = 1 и q = 1
рассчитывать на получение
осадка AgBr, практически не
содержащего AgCl, не
приходится.
Формулы (3.5)—(3.10),
полученные по (3.2) в расчете на
образование гомогенного твердого раствора, дают представление
о возможном максимальном загрязнении основного осадка более
растворимым компонентом. При этом надо учесть, что близость
радиусов и однотипность решеток могут привести к образованию
концентрированных твердых растворов только при относительно
больших значениях X. При малых X погрешности так малы, что
ими часто можно пренебречь.
' Таблица 3.3
а
\
10"-^
10-2
0,1
0,5
Ю-'
10-^
1,ыо-з
5,3. 10-2
10-^
10-^-
1,2. 10-2
1
10"-^
1,1 . 10-3
3.1.4. РАВНОВЕСИЯ ОБРАЗОВАНИЯ ГОМОГЕННОГО ТВЕРДОГО РАСТВОРА
ПРИ ОДНОВРЕМЕННОЙ ЗАСТРОЙКЕ РЕШЕТКИ М, В и А-ИОНАМИ
Если равновесие в системе, содержащей М, В, А-ионы,
устанавливается практически мгновенно, то загрязнение твердой фазы
МА-ионами В можно рассматривать также и в процессе
одновременной застройки решетки М, В и А-ионами. Тогда в какой-то
момент осаждения можно найти долю а-конкурирующих ионов В,
внедрившихся в решетку, когда доля /W-иона в ней равна и.
Если с^ = С\, св = С2 и А-ионы расходуются только на
образование твердой фазы, то схема, ведущая к дальнейшим выводам,
принимает вид:
МА (т) +
ВА (т)
+ М, X
I ]
так как суммарное количество МА и ВА в твердой фазе равно
U^l + а^2.
По закону действующих масс
(1 — Ц) . ^2 _ ^
откуда
« = П=7гЬх). (3.11)
Например, в случае осаждения Вг--ионов Ag+-HOHaMH в
присутствии С1--И0Н0В Х = 2,54-10-3. Если и = 0,952, как в ранее
рассмотренном примере, то
а = 0,952-2,54-10-з ^
1 -0,952(1 -2,54-10-3)
ЧТО Практически совпадает со значением а = 0,048, рассчитанным
по формуле (3.8).
3.1.5. РАВНОВЕСИЯ ПРИ ОБРАЗОВАНИИ ГЕТЕРОГЕННОГО
ТВЕРДОГО РАСТВОРА
Пусть в систему, содержащую М и В-ионы, вводится А-ион,
образующий с ними малорастворимые соединения МА и ВА.
Если МА и ВА могут образовать только гетерогенный твердый
раствор и при начальных условиях первым должен выпадать
осадок МА, .то первичные кристаллы его будут состоять только
из М и А-ионов, поскольку См в растворе велика. В дальнейшем
по мере убывания с^. последующие слои, нарастающие на
поверхности первичных кристаллов, будут включать постепенно
возрастающие количества В-ионов. При этом, поскольку твердая
фаза перекристаллизации практически не подвергается, соотно;
шение между молярными долями МА и ВА в последовательно
выделяющихся на кристаллах слоях остается неизменным.
Вывод уравнения для расчета равновесий, ведущих к
образованию гетерогенных твердых растворов, дан в [49].
Пусть б:м = М, св = В и после добавления некоторого
количества ионов А в осадок ушло т моль иона М и 6 моль иона В.
Это изменение в системе можно представить в виде схемы
МА + В5:ВА + М
со В М
с В — Ь М — т
Допустим теперь, что в следующий микрослой перейдет dm
моль МА и db моль ВА. Этому переходу отвечают равновесие
и схема
7 3-457 97
МА + В ±: ВА + М, X
[] В — Ь — АЬ M — m — dm
dm дЬ
П
где дифференциально малые величины dm и db много меньше,
чем М — т и В — Ь.
Тогда по закону действующих масс
После разделения переменных и интегрирования в пределах
от О до. /72 и от О до получаем
~1п {В — Ь)\о = —X In {М — т) \^
и расчетную формулу в виде
. В —Ь . М — т . [В] ^ 1 [М] /о 1 оч
Из формулы (3.13) следует, что при расчетах равновесий,
ведущих к образованию гетерогенных твердых растворов, состав
твердой фазы непосредственно не учитывается.
В общем случае надо иметь в виду два варианта: с избытком
и недостатком осаждающего иона, при разных соотношениях
между начальными концентрациями М, В-ионов.
Пусть, например, = Л1, с^в = 5 = qM и = иМ, где и > 1,
а q принимает любое значение. Тогда при и > 1 в первый момент
== О, с^м = О и св= 'qM — (и — 1) М, а затем в условиях
равновесия [MJ = т, [ВJ = qM — (u — 1)Af —ти расчетная
формула принимает вид
Ы ^^-^''"'"^^'^Ж- (3.14)
или
так как
g(l-^-|) = Xlga, (3.15)
т
= а . (3.16)
— доля В-иона в гетерогенном твердом растворе.
Формулы, отвечающие и = 1 и q = 1, получаются без труда.
В случае и < 1 выражения для Ci сохраняются. Значения
начальных концентраций имеют вид: сд = О, св = q^f и см =
= (1 —и)М. Для равновесных концентраций находим значения:
98
[В] = qM — m и [М] = (1 — u) М + m. В этом случае расчетная
формула имеет вид
= ^-Ig ^^-Т""" или Ig (1 - = X Ig (1 - U + а).
(3.17)
Очевидно, что при и = 1 и q = 1 уравнение (3.17) будет иметь
тот же вид, что (3.15) при тех же условиях.
Логарифмические уравнения (3.15), (3.17) и их варианты
приходится решать путем пробных подстановок.
Пусть в систему, где свг = = 0,1 и cci = 5 = 0,5 мoль•л■"^
введено CAg = ^ = 0,098 моль-л-^ Здесь q = 5, и и = 0,98 < 1,
формула (3.17) принимает вид Ig ^1 — -^j = X Ig (0,02 + а), где
а =и Х = 2,54-10-3, расчетную формулу можно представить
м
виде
R (а) = Ig (0,02 + a)/lg (1 -1) = 393.
Решение методом постепенного приближения дано ниже.
Подстановка начата с а = 0,05, что соответствует 5% внедрения С1--
иона в гетерогенный твердый раствор:
0,02 + а Ig(0,02 +а) а/5 1 _ Ig (l _^] Ig(а) (а)
5
а
5
0,05 0,07 —1,1549 0,01 0,99 —0,0044 2,4190 262
0,03 0,05 -1,3010 0,006 0,994 -0,0026 2,6992 500
0,04 0,06 —1,2218 0,008 0,992 —0,0035 2,5429 349
0,035* 0,055 -1,2596 0,007 0,993 -0,0031 2,6088 406
Дальнейшее уточнение значения а смысла не имеет, а
окончательные результаты можно представить в следующем виде:
m = аМ = 3,5.10-3, [СЬ] = qM — m = 0,5 — 3,5 • Ю-з,
[Br-j (1 — u) М + m = 5,5.10-3 моль• л-^,
^Agci = аМ/0,98М = 0,036, /ZAgBr = 0,964.
В шяние условий осаждения на гомогенность выпадающего
твердого раствора.
В работе [59] описаны результаты осаждения эквимолярной
смеси Вг-, С1--И0Н0В раствором AgNOa в отсутствие и в
присутствии А13+-И0Н0В. Авторы считали, что в первом случае
образуется мелкодисперсный золь и что это должно облегчать
перекристаллизацию и способствовать гомогенности выпадающей
в таком виде твердой фазы. Во втором случае, т. е. в условиях
быстрой коагуляции и образования гораздо более крупных
частиц, полную перекристаллизацию они считали невозможной и
рассчитывали получить неоднородные твердые растворы. В табл. 3.4
полученные в [59] опытные данные сопоставлены с результатами
расчетов. Авторы названной работы проводили расчеты по
формулам, аналогичным приведенным выше, используя полученное
в результате тех же измерений значение Х = 2,54-10-з.
7* 99
Таблица 3.4
u
Остаток Вг~-иона в растворе, молярная доля в %
Al^-H-HOHbi не вводились
в присутствии Al^-b-ионов
вычислен
по (З.б) и (3.10)
измерен
вычислен
по (3.15) и (3.17)
измерен
через 3 мин 40 с
0,98
0,99
1,00
1,01
1,02 .
5,90
5,33
4,80
4,33
3,90
5,82
5,30
4,81
4,26
3,87
1,99
1,13
0,39
0,03
2,18
1,52
1,19
Из данных табл. 3.4 следует, что состав твердого раствора
безусловно зависит от условий, в которых он образуется.
Данные, приведенные в двух последних столбцах, показывают, что
авторам не удалось получить путем введения коагулянта
идеального гетерогенного твердого раствора AgCl в AgBr. Если учесть
далее разброс значений X в табл. 3.2, принять во внимание, что
хорошее согласие между значениями, приведенными во втором
и третьем столбцах табл. 3.4, связано с тем, что использованное
значение Х = 2,54«10--з рассчитано по данным третьего столбца,
то придется отметить, что предельные случаи вряд ли когда-
нибудь реализуются.
3.1.6. ПРЕДОТВРАЩЕНИЕ ОБРАЗОВАНИЯ ТВЕРДЫХ РАСТВОРОВ
Считают [42 с. 105], что в случае образования твердого
раствора добиться очистки даже путем перекристаллизации
невозможно, поэтому особое значение приобретают приемы,
рассчитанные на предотвращение образования смешанных кристаллов.
В случае изовалентного и гетеровалентного замещения для
этого используют: методы, ведущие к превращению замещающего
В-иона в частицы, заметно отличающиеся от замещаемого М-иона
размерами, числом зарядов, знаком заряда; условия
осаждения, ведущие к образованию твердой фазы, включающей не-
М-ионы, а комплексы этих ионов, не замещаемые простыми В-
ионами; изменения среды, снижающие до безопасных пределов
концентрацию ионов, необходимых при внедрении в решетку МА
гетеровалентных В-ионов по компенсационному типу.
Так, захват РЬ2+-ионов осадком SrS04 практически полностью
предотвращается в присутствии галогенидов щелочных металлов
(ХГ), превращающих РЬ2+-ионы в РЬГп"""-комплексы [58]. При
осаждении ВаЗОц можно практически полностью исключить
внедрение Ре^+-ионов в решетку BaS04, переводя их в комплекс
с этилендиаминтетраацетат ионом при рН = 1 [53]. Из системы,
содержащей Са^-ь, СгО^'^-ионы, при комнатной температуре
осаждается смесь СаС204-2Н20 и СаС204.3Н20, а при ЮО^С —моно-
iOO
гидрат [63]. Поскольку радиусы три-, ди- и моноаквокальций
ионов различны, можно, меняя температуру, предотвращать
образование нежелательных твердых растворов, если известны
остальные ионы системы и их радиусы. В [53J приведены данные,
показывающие, что при рН > 5 замещение Ва2+-иона на К+-ион
в системе, содержащей Ва^"*", К"^, CP, 504"'-ионы, становится
невозможным из-за резкого снижения концентрации Н507"ИОнов.
Наконец, в [42, с. 150] отмечено, /что малорастворимые вещества
неполярной природы типа Hgl2, Hg2Cl2 редко включают
посторонние вещества. Возможно, что осадки, получаемые с помощью
органических осадителей, обладают той же полезной особенностью.
. Однако следует заметить, что систематических исследований,
касающихся образования твердых растворов, явно недостаточно,,
хотя вопрос этот имеет большое и общее значение.
3.2. Адсорбция
Под адсорбцией понимают процессы, ведущие к тому, что
активности растворенных веществ в непосредственной близости
к твердой фазе оказываются существенно иными, чем на
достаточном -расстоянии от нее.
В химико-аналитической практике существенное значение
имеют три вида адсорбции: адсорбция потенциалопределяющих
ионов; молекулярная адсорбция или адсорбция ионных пар;
однослойная ионная адсорбция.
3.2.1. АДСОРБЦИЯ ПОТЕНЦИАЛОПРЕДЕЛЯЮЩИХ ИОНОВ
Как было показано ранее, стойкость золей объясняется тем,
что частицы их оказываются одноименно заряженными за счет
адсорбции на их поверхности тех ионов решетки, которые в
данный момент находятся в избытке в окружающем растворе.
В работе [64| установлено, что во многих случаях адсорбция
цотенциалопределяющих ионов в каждый момент пропорцисг-
нальна логарифму концентрации адсорбируемого иона:
^ = ^dlnci. (3.18)
Интегрирование этого уравнения в пределах от О до л; и от с\
до Ci приводит к уравнению
= k\n\ (3.19)
\ где л; — количество адсорбированного иона (^oль); т —масса
.осадка; Ci — фактическая концентрация i-ro иона в решетке;
'С? — изоэлектрическая концентрация i-ro иона; k — константа.
10)
Из уравнения (3.19) следует, что] при Ci<Ci i-й ион вообще
не может адсорбироваться осадком. В'этом случае
преимущественное право на внедрение в потенциалопределяющий слой
получает второй, противоположно заряженный, ион той же решетки,
если его с > с^. Это ведет к перемене знака заряда твердой фазы
на противоположны и j
На рис. 3.1 представлена зависимость потенциала на частице
AgCl (условная шкала) от p^Ag. Значения потенциала при pcAg <
< pCAg положительны. Они сначала круто падают, а затем, по
мере приближения к pcAg = 4, асимптотически приближаются к
нулю; на интервале p^Ag от»'4 до 4,3 сохраняют нулевое
значение потенциала; при p^Ag > 4,3 (где
рсс\ = p^ci) принимают сначала
медленно, а потом быстрее растущие
отрицательные значения за счет
входящих в потенциалопределяющий
слой С1--И0Н0В.
Значения констант входящих
в (3.18) и (3.19), в работе [64J
отсутствуют. Авторы [64] лишь утверждают,
что при CAg— lO^Ag каждые 100Г--ио-
нов, находящихся на поверхности
кристалла AgF, привлекают в потенциалопределяющий слой 4—5
Ag+-HOHOB. Для Agl при CAg, превышающей соответствующую
Сац примерно в 3000 раз, они ожидают адсорбции 23 Ag"^-HOHOB
на каждые 100 1--ионов, находящихся на поверхности кристаллов.
Пусть, например, из 1 л раствора выпадает 0,01 моль AgCl
при избыточной [Ag+] = 10-^ мoль•л"-^ т.е. в условиях, когда
каждые 100 С1"~-ионов привлекают в потенциалопределяющий
слой 5 Ag+-HOHOB. Предположим, что выпадают кристаллы AgCl,
на ребре которых размещается 20Ag+ и 20 С1—-ионов. Тогда,
используя табличные данные:
M(Ag) = 107,9, ЛГ(AgCl) = 143,3 г.мoль-^ rfAgci = 5,56 г-см-^,
/'Ag+ = 1,13 и Га- = 1,8Ь 10-8 см, Z = 2 (rAg+ + Га-), L = 201
и число Авогадро Л/'= 6,02 • 102^ мoль-^ можно рассчитать
возрастание массы осадка А/тг за счет Ag+-HOHOB
потенциалопределяющего слоя. Расчет ведется по формуле
I ^2
Рис. 3.1
Ат= l[0,01M(AgCl).(L3rf)--i.6L2]2
10--2>5>Al(Ag).Ar--i=Q-QQ^^(^f)'^(^^\
(3.20)
где 0,0Ш (AgCl) {ЬЫ)-^ = п — число кристаллов в системе; п • 6L2=
= 5 —общая поверхность всех кристаллов; 5 2 -—длина стороны
102
соответствующего квадрата; S ^ /-^ — число СН (и в то же время
Ag+) ионов на стороне квадрата; SM —число С1--ионов на всей
поверхности золя; 0,0551-2 —число Ag+-ионов в потенциалопре-
деляющем слое; Ьт — их масса. В данном случае она равна
0,0034 г = 3,4 мг. К сожалению, прямые измерения Am связаны
с огромными трудностями и до сих пор не проводились.
Как следует из работы [64] и рис. 3.1, при избыточных
концентрациях ионов решетки, близких к изоэлектрическим,
образование потенциалопределяющего слоя за счет этих ионов
становится практически невозможным. В подобных случаях
возникает, по-видимому, возможность образования
потенциалопределяющего слоя за счет других ионов, находящихся в системе
(Кольтгоф с сотр., 1933—1947 гг.). Показано, что анионы
красителя «Понсо 4R» способны вытеснять с поверхности PbS04
эквивалентное количество 804""-ионов. Описаны результаты
встряхивания осадка СаС204, несущего потенциалопределяющий слой
из С204""-ионов, с растворами, содержащими ЮГ» SO^"" или 0Н~-
ионы, показавшие возможность замены С204""-ионов этими
последними. Обнаружено, что цитрат-ионы успешно конкурируют
с бО^^-ионами в создании на осадке BaS04 отрицательного
заряда.
Простейшее гетерогенное равновесие этого типа можно
представить в виде схемы
{(МА)„А} + R {(MA)„R} + А, К, (3.21)
Тогда по закону действующих масс
(А)
(R)
раст
(R)
(А)
по
= /(. (3.22)
К сожалению, методов измерения активностей ионов в потен-
циалопределяющем слое пока нет, значений К в связи с этим
также не суш.ествует.
3.2.2. МОЛЕКУЛЯРНАЯ АДСОРБЦИЯ
Молекулярной адсорбцией считают адсорбцию на поверхности
твердой фазы полярных молекул. Поскольку на поверхности
первого адсорбированного слоя может нарастать второй, а на
втором — третий и т.д., эту адсорбцию называют еще
многослойной.! Отличительным признаком гетерогенных систем, в
которых осуществляется молекулярная адсорбция, считают поэтому
монотонное возрастание количества вещества, адсорбируемого на
1 г твердой фазы, по мере возрастания концентрации
адсорбируемых молекул в растворе. Типичный график изотермы такой
адсорбции, т.е. зависимости хт-^ (л: — количество
адсорбированного вещества, мо/.ь; т — масса твердой фазы) от концент-
103
рации при постоянной температуре дан на рис. 3.2 (кривая /).
Молекулярной адсорбции противопоставляется ионная или
однослойная адсорбция. График изотермы такой адсорбции (см. рис. 3.2,
кривая 2) характеризуется тем, что, начиная с некоторой
концентрации, дальнейшее возрастание этой величины не вызывает
роста A:/n'~^ так как адсорбционный слой оказывается
насыщенным противоионами.
\,Поскольку в случае адсорбции ряда электролитов насыщения
не наблюдалось, возникла мысль, что в подобных случаях катион
и анион адсорбируются в
эквивалентных количествах, одновременно
занимая на поверхности осадка соседние
места. Этот случай получил название
адсорбции ионных пар. Считают, что
именно таким образом адсорбируется
КВгОз на осадке BaS04. .
Уравнением, наиболее
подходящим для выражения зависимости
хт-^^ от концентрации, считаютГизо-
терму Фрейндлиха [54, с. 240, 1225]
Рис. 3.2
(3.23)
где р, Аг — постоянные величины; с—равновесная концентрация
адсорбируемого вещества. Размерности х, т, с, которым
отвечают используемые значения констант, должны быть точно
указаны. Константы р, п не относятся к числу фундаментальных
величин, так как сохраняют постоянство лишь при условиях
выделения твердой фазы, в которых они получены. Их
приходится измерять для каждого конкретного случая, поскольку в
справочниках они отсутствуют.
Для измерения р, п ставят ряд" опытов, осаждая обычно
одно и то же количество осадка т(г) в одном и том же объеме
раствора v (л), при разных начальных концентрациях
адсорбируемого вещества с? (моль-л~^). Путем последующих измерений
находят либо х\у т. е. полное количество вещества (моль),
адсорбированного на m г осадка, либо равновесную концентрацию
того же вещества в растворе с\ (моль-л-^). В первом случае,
зная ATi, вычисляют с\ = {с% ~ х\) v'~'\ во втором •->-а:1= (с? —^j)t;.
Если проведено v таких измерений, то получается система v
условных уравнений
1
Xi/n-i = р [{cfv — Xi) v-'Y . (3.24)
Их линеаризуют путем логарифмирования и получают новую
систему:
Ig^ + I Ig [{ciV ~Xi) V-'] = Ig(хт-^). (3.25)
104
в ней неизвестными константами являются lgP = Kb = V^2»
коэффициентами при них Ац = 1, Ai2= lg[{ciV — Xi)v^^]y и,
наконец, Oi == Igixifn-^),
Свертывание системы по методу наименьших квадратов дает
V V V
an = Ti^ii = v; «12 = «21 == 2^11^12= IIlg[Kt; —A:i)tr-i];
i=l i=l i=l
^22= S Л?2= S { \g\(c^-xdv-A\ 91 - 1]ЛпФ1 =
i=l i=l I J i=i .
i = l
* 92= i]/li20i= i,{\g[(c?v-Xi)v-^]}\g(Xim-^).
i=l i=l
Последующее решение системы нормальных уравнений
апГ1+а.2Г2 = срь ^3 26>
^2ll^l + ^221^2 = Т2
дает наивероятнейшие значения y?=lgp, F2 =-~> отвечающие
условиям осаждения, в которых происходило выделение твердой
фазы.
Располагая значениями Igp, , можно решать любые
задачи, связанные с расчетами адсорбции в данной системе.
Пусть, например, для данной системы lgp= 1,313 и -~ =
== 0,25; необходимо рассчитать Xi, й, если т = 0,5 г, v =0,2 л,
с? = 420 мкмоль-л-' и константы отвечают Х[ в мкмоль и ci в:
мкмольл-^ Расчетное уравнение имеет вид
lg^= 1,313 + 0,25 Ig(420 .0,2-^0 (0,2)-^
Оно легко преобразуется в уравнение
^.6.10.. (3.2Г,.
которое дает Х[ = 39, а затем ci = (420-0,2 — xi) (0,2)-^ =
= 225 мкмоль-л-Ч
Физический смысл константы р легко устанавливается с
помощью уравнения (3.23), так как при с=1 мoль•л~^ m = 1 г,
Р = л:, т. е количеству вещества (моль), адсорбированному на
поверхности 1 г твердой фазы.
Смысл второй константы, входящей в уравнение (3.23),
становится ясным при решении задачи об отношении относительного
105^
изменения адсорбции к относительному изменению концентрации,
адсорбируемого вещества в растворе. Пользуясь уравнением (3.23^
в виде х = т^с^у находим, что
. ^ = ^ . ± = 1 т^Л -^ ^ = 1 . т?с'^- = 1,
x ' с dc x п ^ x п —-— п
x
откуда
x '~ п с'
(3.28)
Это значит, что параметр — является фактором
пропорциональности, определяющим зависимость между относительным
изменением адсорбции вещества на 1 г твердой фазы и относительным
изменением его равновесной концентрации в растворе.
Например, при -^=0,25 возрастание концентрации на 1% ведет к
возрастанию адсорбции на 0,25%.
3.2.3. ОДНОСЛОЙНАЯ (ИОННАЯ) АДСОРБЦИЯ
)
в тех случаях,! когда адсорбируемые противоионы могут
занимать на поверхности осадка только ограниченное число пози-_
ций и образование нескольких молекулярных слоев невозможно, ^
' для выражения равновесия между адсорбированным веществом'
*в растворе и на поверхности осадка приходится пользоваться
каким-либо из вариантов изотермы адсорбции Ленгмюра.
Для вывода этого уравнения предположим, что па
поверхности твердой фазы находится в данный момент всего п ионов,
из них пл:°—положительных, п (1—л:°) ~ отрицательных. Пусть
далее в тот же момент концен'1:рация положительных ионов в
растворе равна ^4. и отрицательно заряженных —cL. Тогда
скорость ухода положительных ионов в раствор будет равна
vj^ = кхпх^, а скорость выхода их из раствора на поверхность vj^ =
= ^2C+(1 — л:°)п, так как положительные противоионы могут
адсорбироваться лишь на отрицательно заряженных участках
поверхности. ]
По мере приближения к состоянию равновесия скорости
и vj^ выравниваются, а и стремятся к значениям и л:,
отвечающим состоянию динамического равновесия. 1 После его
достижения kixix = k2C^ (1 — л:) п, и, если k2kT^ = Л, то
106
в случае отрицательных ионов по тем же соображениям
tL. = ^3 (1 — х^) п, = k^c^ — nx^ *з (1 — а:) n = *4б_пл- и для
kikT^ = k отвечающее равновесию значение молярной доли
отрицательно заряженных ионов на поверхности твердой фазы оказы-
вается равным 1 — х= ^ _^ .•
Очевидно, что формула имеет одинаковый вид для ионов
kc
обоих знаков, т. е. х = j^fj^'
0.1 О,? ИЗ G4 0.5 0.6 (ff
Рис. 3.3
Наконец, поскольку количество ионов (моль), адсорбируемых
на 1 г осадка, пропорционально доле поверхности, занятой этими
ионами, Б1лг1олняется соотношение
a = ± = k'x =■
k'kc
\ + kc '
(3.29)
Ясно, что k' = xm-^, т.е. количеству вещества противоионов
(моль), адсорбируемых 1 г твердой фазы при ^с>1, что
отвечает «насыщению» поверхности противоионами. Этому значению
отвечают ординаты кривой на рис. 3.3 на участке ее,
параллельном оси абсцисс, а саму величину k' можно считать в связи
с этим «емкостью» одного грамма осадка-сорбента в моль.
Если хт-^ = а моль-г-^, то из (3.29) следует, что второй
параметр изотермы Ленгмюра (3.29)
k' — а
представляет собой отношение числа вакансий, занятых
противоионами (а), к числу свободных вакансий (k' — a) при единичной
концентрации противоиона в системе (с=1 моль-л"*^) и,
следовательно, имеет размерность л-моль-^
Для оценки параметров к' w k приходится измерять те же
величины, что и при оценке параметров р и -~ в уравнении
Фрейндлиха, т.е. с\= {c\v — х)или Xi=^[c\—c)v при фик-
107
сированных гПу V и с\. Линеаризацию системы уравнений (3.29)
можно осуществить различными способами. В частности, после
освобождения от знаменателя, деления всех уравнений на d и
подстановки значений си система имеет вид
k'k-^k = —^ (3.30)
В ней Ли = 1, Г] = k'ky А\2 = —Xitn-K Y2 = k,
Oi = XiV [m {c% —
Свертывание системы (3.30) проводится по формулам, данным
для системы (3.25). Решение системы нормальных уравнений,
аналогичной системе (3.26), дает значения = k'k и У\=
откуда /^'-r?(r^)"■^
В разделе 3.2.2 отмечалось, что]^в ионных системах с
отчетливо выраженным насыщением (рис. 3.2, кривая 2)
адсорбируются отдельные ионы, а в системах, где насыщение не
достигается (рис. 3.2, кривая I), идет адсорбция ионных пар.'
В частности, по этой причине пришли к выводу, что при
осаждении BaS04 из раствора, содержащего К"^ и ВгОГ-ионы, на
поверхности кристаллов адсорбируются не отдельные
противоионы, а ионные пары {К"^, ВгОз"}. Если подойти с этой точки
зрения к результатам измерения адсорбции KNO3 и NaCl
выраженным в ммоль-г""^и нанесенным в виде отдельных точек на
рис. 3.3 в зависимости от с\ моль-л-^ в растворе, то придется
признать, что К"^ и ЫОз"-ионы адсорбируются по законам
молекулярной адсорбции, а Na"^ и СР-ионы по ионному типу.
Трудно предположить, чтобы в водных растворах, где для солей
типа KNO3, NaCI и им подобных двойники обнаружить не
удалось [68], адсорбция происходила по молекулярной схеме.
Особенно трудно допустить такое толкование в случае адсорбции
на кристаллах с ионной решеткой, практически всегда несущих
потенциалопределяющий слой. Вероятнее, что во всех подобных
случаях адсорбция выражается изотермой Ленгмюра и идет в
порядке заполнения противоионами адсорбционного слоя, но в
условиях, при которых насыщение еще не достигается.
Расчеты, проведенные для двух последних случаев,
подтверждают сделанное предположение. Так, для системы
KNO3 — BaS04 k'k = 5,68, k' = 0,42, k = 13,4;
NaCl — BaS04 k'k = 0,79, k' = 0,46, = 1,7.
Ясно, что при c^^Q =0,3 моль-л-* условие kc^l не
достигается, так как kc = 4,02, и что для NaCl даже при с^ао
= 1,2 моль-л-' ^с = 2,04, чем и объясняется очень медленное
стремление к насыщению.
108
Учитывая результаты, приведенные в работе [68], и наши
расчеты, можно считать, что практически всегда на поверхности
кристаллов ионного типа сначала адсорбируются потенциалопре-
деляющие ионы, а затем противоионы. При этом адсорбции
противоионов благоприятствуют следующие условия: высокая
концентрация адсорбируемого противоиона; наличие на нем большого
числа зарядов, особенно, если радиус иона невелик;
незначительная растворимость соединения, образуемого потенциалопре-
деляющим ионом и противоионом; слабая диссоциация
соединения, которое противоион образует с потенциалопределяющим
ионом; наличие в двойном слое сильно поляризующего катиона
и легко поляризуемого аниона.
Применение этого комплекса условий, называемого обычно
правилами адсорбции Панета-Фаянса-Хана, можно
проиллюстрировать примерами. Так, при осаждении Ва^'^-ионов избытком
504""-ионов в присутствии Са^"^ и Mg^"^ в адсорбционный слой
преимущественно попадают Са^'^-ионы, так как растворимость
MgS04 во много раз превосходит растворимость CaS04. Мицеллы
{CuSjnS^-, образующиеся при осаждении Си^'^-ионов
сероводородом в присутствии Н"^, К"^ и Na'^-HOHOB, адсорбируют в
основном Н'^-ионы, дающие с 5^~-ионами слабо диссоциирующие
Н5""-ионы и HgS. Профильтрованный раствор Ьа(ОН)з,
полученный встряхиванием с водой отмытого и осажденного на холоду
при избытке ЬаЗ"^-ионов гидроксида лантана, остается бесцветным
при добавке раствора тимолфталеина, так как в таком растворе
рН^9. Осадок же (La (ОН)з} La^"^ окрашивается в таком
растворе в синий цвет за счет попадающих в адсорбционный слой
легко поляризуемых Ьа^'^-ионами анионов индикатора.
Задача об отношении относительного изменения адсорбции к
относительному изменению концентрации адсорбируемого
противоиона в растворе решается точно так же, как в случае
молекулярной адсорбции. Исходя из (3.29), находим, что
Г k'k k'k^c
с I
откуда
da ^ dc __da с_
'а * T~"dc ' а \ \ + kc ~ (T-M^J ^
_ 1 dc
а \ +kc ' с'
(3.31)
Из (3.31) следует, что по мере возрастания с, т.е.
концентрации адсорбируемого противоиона в растворе, одинаковым
приращениям величины будут отвечать все меньшие и меньшие
Приращения величины —.
109
3.2.4. СНИЖЕНИЕ АДСОРБЦИИ
] Соосаждение за счет адсорбции является одной из причин,
вызывающих загрязнение осадков, используемых в химическом
анализе для разделения сложных систем на менее сложны^,
вплоть до выделения малорастворимого соединения какого-лйёо
одного элемента, количество которого подлежит измерению.
Поскольку [одновременно соосаждаются адсо]рбируемые противоионы,
что порой ведет к большим ошибкам при измерении их
концентрации в растворе, из которого выпал осадок] адсорбция
оказывается явлением крайне нежелательным при таких
разделениях и измерениях.
Самым общим \методом снижения адсорбции следует считать
уменьшение величины относительного пересыщения и тем самым
дисперсности твердой фазы и ее удельной поверхности^
^ Снизить загрязнение твердой фазы за счет молекулярной
адсорбции можно.;лишь, как это следует из (3.23),' путем
уменьшения концентрации адсорбируемого вещества.,'
У В случае ионной адсорбции можно добиваться снижения со-
осаждения двумя путями т--г препятствуя накоплению на
поверхности твердой фазы /большого числа потенциалопределяющих
ионов и снижая актив^юсть противоионов.\
Для выполнения первого условия используют различные
методы. Для осаждения "^пользуются слабыми электролитами,
например, 'осаждают гидроксиды раствором аммиака, в котором
концентрация свободных ОН~-ионов мала. Применяют реактивы»
генерирующие осаждающие ионы в процессе осаждения, напри*
мер, осаждают СаС204 путем нагревания раствора, содержащего
Са^"^-ионы и (С2Н5)2С204, при медленном* омыле1ии которого
образуются НС2ОГ и С204~-ионы. Следует, по-видимому, отдавать
предпочтение 1 методам осаждения, дающим осадки с решеткой
неионного типа, в частности, осаждению с помощью
органических реактивов.
' " Поскольку при наличии потенциалопределяющего слоя
адсорбция противоионов неизбежна,' возникает вопрос об
избирательном заполнении адсорбционного слоя ионами,? наиболее
подходящими в данных условиях. Так, используя комплексоэбразователи,
можно замаскировать и практически полностью сохранить в
растворе все ионы, содержание которых подлежит определению!
В некоторых случаях осаждение ведут при высоких
концентрациях специально введенных противоионов в расчете на замену
ими тех, которые должны остаться в фильтрате. Очень, часто
при этом, конкурирующий ион подбирают так, чтобы вместе с
ионами потенциалопределяющего слоя он давал летучее или
термически нестойкое соединение. Этот подход рассчитан на
получение чистой твердой фазы при последующем ее прокаливании.
ПО
Например, таким образом получаются осадки ^A<?CI\iCl , Н"^и
{BaS04}nS0r, NH^.
3.2.5. АДСОРБЦИЯ НА КОЛЛЕКТОРЕ
Итак, адсорбция является одной из основных причин
загрязнения осадков, а значит многих качественных и количественных
ошибок в химической метрологии и в связи с этим явлением
нежелательным. Вместе с тем изучение законов адсорбции
показало, что, рационально используя это явление, можно создать
некоторые' принципиально новые методы разделения сложных
систем.
Одним из этих методов является применение так называемых
«коллекторов». Он применяется в тех случаях, когда
концентрация изучаемого иона так мала, что даже обнаружить его с
помощью обычных реактивов и приемов невозможно, т. е. в
случаях, когда дело идет о следах того или иного элемента. В такой
раствор нарочно вводят небольшое количество постороннего иона
и осаждают его подходящим реактивом. При этом
микроколичества определяемого элемента адсорбируются на выпадающем
осадке, который называется коллектором (собирателем), так как
увлекает с собой ничтожные количества вещества, осаждение
которых обычными способами невозможно.
Для оценки коэффициента полезного действия коллекторов
молекулярного и ионного типа надо преобразовать уравнения
(3.24) и (3.29) для случая, когда т=1 г и у = 1 л, а затем
выделить из каждого отношение х{с^)-'.
В первом случае получаем выражение
во втором
X kk' ~ kx
^0 x^kk'-^ kx
(3.33)
и в том и в другом случае при с° О стремится к нулю и
Ху так как по самому смыслу этих величин всегда х < с^. Тогда
в первом случае
lim ^0=1. (3.34)
а во втором
Й^=ГТТГ- (3.35)
Это значит, что в случае молекулярной адсорбции по мере
снижения с° коллектор адсорбирует следы все полнее и полнее.
В случае ионной адсорбции рассчитывать на полный захват всего
111
''li, мин
T2i, мин
Примечание
3
4
3
3
3
О
10
20
60
30
0,373
0,892
0,915
0,945
0,001
^HgCU
нали1|ного количества ионов можно лишь в тех случаях, когда
А'А>1. В остальных случаях при с°->0 >^^у+Ш^^'
житель при например, для захвата KNO3 осадком BaS04
составит 5,68(1 +5,68)-1 = 0,85, а для NaCl всего 0,79(1 +
+ 0,79)-1 = 0,44.
3.3. Последующее осаждение
Авторы первого сообщения о последующем осаждении [61]
считают, что в этом случае первичный осадок выпадает чистым
и загрязняется лишь за
Таблица 3.5 счет последующего
выпадения второго осадка в тех
случаях, когда маточный
раствор пересыщен по
отношению ко второму
веществу и оно
кристаллизуется из него очень медленно.
При этом ионы,
образующие второй осадок, сначала
входят в качестве
противоионов в адсорбционный
слой, а затем, если произведение активностей в двойном слое
превысит произведение растворимости, дают второй осадок, который
может образовать с основной твердой фазой твердый раствор.
В качестве примеров Кольтгоф и соавт. (1933—1938 гг.)
приводят последующее осаждение MgC204-2H20 на смеси осадков
СаС204'2Н20 и СаС204-ЗН20; ZnS на HgS; ZnS на В128з; NiS
на CuS, HgS и ZnS; FeS на CuS.
Рассмотрим результаты, описанные в [62]. В табл. 3.5
показано возрастание последующего осаждения ZnS с увеличением
промежутка времени между осаждением и фильтрованием. В
систему, содержавшую 25 мл раствора HgCb с концентрацией
0,05 молЬ'л-^ и 25 мл раствора ZnS04 такой же концентрации,
приготовленных на растворе H2SO4, с -i-'^2S04^= 0,35 моль-л~1
Tl мин пропускали H2S. Затем закрывали колбу с раствором
пробкой, через которую проходила трубка, присоединенная к
аппарату с выделяющимся H2S. Система, находившаяся под
давлением H2S, встряхивалась еще Т2 мин, после чего в ней
измерялась доля а начальной концентрации Zn^"^-HOHOB, перешедших в .
осадок.
Из табл. 3.5 видно, что из раствора, не содержащего Hg^"^-
ионов, выделяется лишь ничтожная доля Zn^"^-иoнoв, тогда как
в присутствии осадка HgS осаждение ZnS проходит почти
количественно. При этом оказалось, что осадок HgS не содержит
сколько-нибудь заметных количеств ZnS, пока в растворе оста-
112
ются неосажденные Попытки извлечь Zn "^-ионы из
осадка путем обработки раствором НС1 не обеспечили полноты
растворения даже при C(hci)= 3 моль • л-^, что привело к мысли
об образовании твердого раствора. Поскольку в одной из работ,
предшествовавших введению представления о последующем
осаждении, показано, что присутствие тонкого инертного порошка
облегчает выпадение осадка ZnS из пересыщенного раствора,
но вызываемое действие оказалось незначительным, результаты,
приведенные в табл. 3.5, безусловно следует приписать только
специфическому влиянию осадка HgS.
В разделе 2.1 указано, что, как правило, из раствора в
первую очередь выпадают более растворимые структуры. Это
значит, что в нашем случае первым, как это и наблюдается,
должен выпадать черный меркуросульфид, кристаллизующийся в
кубической структуре, и ZnS(p), дающий гексагональные
кристаллы. В приложении находим для Hg^'^-иoнa p-qi = 3,96, а
для Zn^'^-иона = 8,96. Поскольку обычный подсчет
показывает, что при с^^=0,35 моль'л""^ даже аквомеркуро-ион
диссоциирует всего на —0,03%, при оценке последовательности
осаждения HgS (ч) и ZnS (Р) можно считать, что до начала
пропускания HgS в системе находятся только Hg^"^ и Zn^■^-иoны.
Используя приемы, описанные в разделе 2.12, и значения
p/^HgS(4) = 51,8 и pPznS(p) 21,6 из приложения, находим, что
в системе, содержащей оба осадка
Es!i)-ioz!!i'-_ 10-30.2
(Zn^+) ~ 10-21'^
Тогда очевидно, что первым должен выпадать черный меркуро-
сульфид, так как с^^ : = ^ ^ 10-зо.2^ причем о какой-либо
примеси к нему ZnS(P) говорить не приходится, так как к
началу осаждения ZnS активность Hg^'''-иона в растворе должна
снизиться до (Hg^+)б'2п • 10""^^*^~ 10'"^^'\ отвечающей одному
Hg^+-HOHy в -^4-107 л раствора.
Таким образом, указание авторов [62], что осадок HgS
выпадает чистым, не расходится с теоретическими соображениями.
Для проверки второго предположения [62], что последующее
осаждение происходит за счет избытка реактива, если маточный
раствор пересыщен по отношению ко второму веществу,
рассмотрим систему
H2S(r) ^HgS -IgX =-0,94
HgS e2H++S' -lga2 = -19,95
Zn^-^ + S'-- ;:^ZnS(t) — lgP= 21,6
Zn^-^+ HgS (t) 5=^ 2H++ ZnS (t) Ig /С = 0,71
с 0,025 0,35
[ ] m 1 0,4 —2m
8 3-457 113
По закону действующих масс (0,4 —2т)2= 10^'^^ т,
т2 1,6825/п + 0,04 = О и m == 0,0243 моль-л-Ч
Это значит, что за счет избытка осадителя может выпасть
всего (0,025 — 0,0243) • (10-2 • 0,025)-i ;^ 3 % Zn^+-иoнoв, на-
ходившихся в первоначальном растворе. Говорить о
пересыщении здесь рисковано.
В работе [60] высказывается предположение о возможности
образования твердого раствора ZnS в jHgS, причем полностью
извлечь Zn^"^ из этого раствора не удается даже путем
длительной обработки раствором НС1 с концентрацией 3 моль .л-^
Предположение об образовании твердого раствора не
выдерживает критики по следующим причинам: 1) г^^24- = 0,112 нм >
> 1,15г2„2+= 1,15 .0,083 = 0,096 нм; 2) HgS(4) HZnS(p) кри-
сталлизуются в разных структурах; 3) пользуясь схемой
HgS (т) + Zn^+ « ZnS (т) + Hg2+, X ^ 10-'''2
и уравнением (3.7) при q= 1, находим, что степень загрязнения
HgS растворенным ZnS а—10-^^'^ это отвечает растворимости
ZnS в HgS порядка 10-^^%.
Поскольку пересыщение в растворе-относительно ZnS и
образование концентрированного твердого раствора ZnS в HgS в
условиях работы [62] оказываются недостижимыми, остается
рассмотреть последнее предположение авторов [62], согласно
которому осадок ZnS образуется в двойном слое. Само по себе это
предположение, если учесть практически полное осаждение ZnS
при равенстве с^^ и с^^, следует считать маловероятным.
Оно несостоятельно еще и потому, что не объясняет
неполноту извлечения ZnS из осадка даже путем длительной
обработки его раствором НС! при c^j^j = 3 моль-л-^.
В самом деле, для схемы
' ZnS(p)(T) + 2H+«Zn2+ + H2S(r) lg/(= -0,71
с 3 0 0
[ ] 3 — 2m m 1 атм
находим, что m2 —4,282m-f 2,2.5 = О, m = 0,61 мoль•л-^ с
использованием НС1 на 2т.100 %/3--41 %, что совершенно не
совмещается с установленной опытным путем [60] неполнотой
извлечения цинка.
Из работ [60, 621, в частности, из даннь|х табл. 3.5 следует,
что в системе Hg^"^—Zn^"^ —H2SO4 сначала количественно
осаждается HgS(4), а затем практически полностью ZnS, причем
выпадающий в этих условиях ZnS ведет себя совершенно иначе,
чем ZnS, осаждаемый Н25из растворов, не содержащих Hg^"*"-HOHOB.
Анализ объяснений странного поведения ZnS, приведенных в
работах [60, 62], показал их несостоятельность.
114
Все недоумения устраняются, если предположить, что в
присутствии HgS осаждается не ZnS(P), а ZnS (а), для которого
рР = 23,8. Последовательность осаждения в этом случае не
меняется, а возможность образования твердого раствора ZnS (а)
в HgS также исключается, но зато в этом случае
Zn'++H2S(r)«2H+ + ZnS(t), !g/( = 2,91
с 0,026 0,35
[ ] m 1 атм 0,4 — 2m
m2 —203,6/n + 0,04 = 0, m=l,96.10-4 моль.л-^ или --0,8%
от начальной концентрации Zn^"^-HOHa. Растворимость ZnS (а) в
НС1 при =3 моль-Л"^ по схеме
ZnS (а) (т) + 2H+:г=tZn^+ + HgS, \gK = -2,91
с 3 0 0
[ ] 3 —2m m m,
определяемая по закону действующих масс, оказывается равной
т —3,7-10-3 моль-л~^ что согласуется с наблюдениями в
работе [60].
Осаждение ZnS (а) вместо ZnS"(P) объясняется здесь тем,
что HgS(4) кристаллизуется в кубической системе цинковой
обманки, т.е. в той же системе, что ZnS (а), и играет в этом
случае роль затравки.
Утверждать, что и в других случаях последующее осаждение
возникает таким же образом, конечно,^ нельзя, но для
полиморфных веществ типа NiS, CoS, ZnS и им подобных такую
возможность следует учитывать.
Таким образом, в этой главе мы рассмотрели правила и закономерности,
позволяющие предугадать и рассчитать возможность загрязнения
выпадающей из раствора твердой фазы за счет различных видов соосаждения.
Приведены уравнения, включающие параметры, которые не являются
фундаментальными постоянными, и поэтому должны измеряться для каждой конкретной
системы. Результаты расчетов с помощью этих уравнений должны
контролироваться опытным путем.
Соосаждение ведет порой к полной «потере» отдельных компонентов системы
при ее исследовании, серьезным количественным и даже качественным
погрешностям, недопустимым при химических измерениях* Контроль за соосаждением,
меры по его учету и ослаблению — одна из важнейших обязанностей химика-
метролога.
Аналогичных источников ошибок и трудностей в физико-механической
метрологии не встречается.
115
4. ПЮМЫВАНИЕ ОСАДКОВ
4.1. Предварительные замечания
Первоначально считали, что осадки выпадают химически
чистыми и загрязняются лишь пленкой приставшего к ним маточ-
ного раствора. Исходя из этих соображений, Р. Бунзен
разработал теорию промывания осадков для случая использования п
порций растворителя равного объема. Равенство объемов в его
выводе обосновано не было.
Прямые опыты показали, однако, что отмывание осадков от
загрязнений растворителем не подчиняется теории Бунзена, что
примеси удаляются во много раз медленнее, чем предсказывают
формулы, и что, более того^! удалить их полностью путем
промывания растворителем вообще нельзя.
Специалисты, занимавшиеся измерением атомных масс,
разработали специальные методы исследования осадков, пользуясь
.которыми им удалось количественно оценить загрязнения,^
остающиеся в осадках после их промывки.
Все это привело к мысли, что примеси, попавшие в осадок
или на егЬ поверхность за счет различных видов соосаждения,
отмываются растворителем гораздо хуже, чем маточный раствор.
Появились экспериментальные исследования, посвященные этому
вопросу [46, 50].
Высказывались надежды на самоочистку осадков в процессе
их перекристаллизации в контакте с маточным раствором.
Начали применять переосаждение осадков и промывание их
специально подобранными растворами электролитов.
Теория всех этих методов и приемов не разрабатывалась, а
при любых метрологически обоснованных операциях очень важно
иметь предварительные оценки как погрешностей, так и
достижимой степени их снижения.
4.2. Отмывание осадка от маточного раствора
Полный объем растворителя, используемого для промывания,
выбирают так, чтобы потерей части промываемой твердой фазы
из-за растворимости можно было пренебречь. Допустим, что
такой объем равен Ул.
Учтем еще, что при любом методе отделения промывной
жидкости от осадка, например, с помощью центрифугирования
или фильтрования, на нем за счет капиллярных сил
удерживается примерно один и тот же объем раствора, равный w л.
т
Допустим теперь, что вся промывная жидкость разделена
на п порций разного объема, так что
У= ft;!, (4.1)
i=i
И попытаемся найти, какими должны быть эти объемы чтобы
концентрация в пленке после, п промывок имела минимальное
значение.
"Если концентрация отмываемого вещества в маточном
растворе равна со, то при описанных условиях общее количество
примеси, остающееся в пленке, приставшей к осадку, равно
cqw, После добавления первой порции vi растворителя
концентрация примеси в растворе, окружающем осадок^ снизится до Ci =
^ cow(w+ vi)'^K После удаления избытка раствора общее
содержание примеси станет равным c\wy а концентрация в системе
после введения второй порции растворителя упадет до значения
. _ _^ '0'^ (л оч
Теперь видно, что после к-й промывки
а после последней п-й
Сп = „ . (4.4)
П (ш + vi)
i=i
Наша задача —найти объемы vi, обеспечивающие достижение
минимального значения ^п- При ее решении надо учесть, что
cot(y"—фиксированная величина и что любое из значений vi
можно выразить через V и сумму остальных v с помощью (4.1).
Пользуясь первым обстоятельством, определение значений vi^
обеспечивающих минимальное значение Сп, заменяем поиском
тех же значений, при которых знаменатель уравнения (4.4)
принимает максимальное значение. С учетом второго условия этот
знаменатель принимает вид
w+V-'^y ViVniw + vi), (4.5)
i=l /1=1
Z =
если принять, что
t;n = V-"2^i. (4.6)
Поскольку Z —функция от п — 1 переменных, условия
экстремума находятся в общем случае путем решения системы из
117
n-1 / n-1 \k-l n-I
^^-n {w + vO + {w + V- ^vi] n{w+Vi) П (w+Vi),
уравнений, получающихся tiyreM приравнивания нулю п —I
частных производных от Z по каждой из переменных.
Общий вид этой системы в нашем случае
dz dz dz
W^-o ШГ^ = о,..., 1^;;^; = о- ^4.7)
Частная производная от z по имеет вид
n-I
+Vi) П (w+v
(4.8)
dz
Приравнивая ^ нулю, сокращая полученное уравнение на
к-1 ' п-1 п-1
Yl{w + Vi) П {w + Vi) ФО и подставляя вместо V — ^vi значе-
ние этой разности из (4.6), находим, что
^ = -t^k + t;n = 0. (4.9)
Поскольку все остальные уравнения системы (4.7) будут иметь
тот же вид, что (4.9);
— t;i-f Уп = 0,—==0,..., —1/п-1+Уп=0,
(4.10)
приходим к выводу, что Z достигает максимального, а Сп
минимального значения при условии
t/i = ... = t;i = . .. = t;k = . .. = t/n = t;, (4.11)
т. е. в том случае, когда объем растворителя V разбивается на
п равных порций, каждая объемом v:^V = nv (4.12).
При этом (4.4) переходит в формулу Бунзена
^п=-^. (4.13)
Если y=quy, qw^wH q>l (как это всегда бывает, так как
ш —небольшой объем жидкости, пропитывающей осадок или
центрифугат, а у — прибавляемый объем растворителя), то
^п^^ = Q = (1 + q)-" и IgQ = -п lg(l + q) -п Igq. (4.14)
Тогда число промывок п, необходимых для получения
заданного Q при данном q, равно
„=-!f. ,4..5,
Так, если мы хотим добиться Q == 10"^, т. е. снижения Со
в 10^ раз, а q = 10, то п == — ^ = 6.
К сожалению, опытные данные, как упоминалось в разделе
4.1, не подтверждают этого оптимистического прогноза. Поэтому
118
в дальнейшем исследуется процесс отмывания растворителем
примесей, адсорбированных только на поверхности твердой фазы.
Возможность удаления таким путем примесей, образующих с
основным осадком твердые растворы, совсем не учитывается, так
как в подобных случаях промывание растворителем никакой
пользы не приносит [42]. Использование растворителя (воды) для
отмывания примесей, выпавших при последующем осаждении,
также не имеет смысла, поскольку относительное содержание
таких веществ в осадке может быть большим, а растворимость
их в растворителе, как правило, очень мала.
4.3. Отмывание примеси, попавшей на поверхность
осадка за счет молекулярной адсорбции
Отмывание примесей, остающихся на поверхности осадка,из-за
молекулярной адсорбции, имеет существенное значение при
выделении из раствора малорастворимых органических веществ,
комплексных соединений металло-ионов с органическими лиганда-
ми и, возможно, комплексных неорганических соединений с
кристаллическими решетками неионного типа.
Допустим, что начальная концентрация адсорбируемого
вещества в маточном растворе со, объем раствора t^o л и масса осадка
т г. Тогда распределение адсорбируемого вещества между
раствором и поверхностью осадка описывается с помощью изотермы
Фрейндлиха:
f^o^^^j,,,=.m?{co~'^^n. (4.16)
В результате хо моль вещества фиксируется на осадке, (cqVo—xo)
моль остается в растворе, а равновесная концентрация в маточном
растворе оказывается равной ^о —~.
Если после удаления основной массы жидкой фазы в осадке
остается w л маточного раствора, то полное число молей примеси
в системе окажется равным
Хо +
w=Qo, (4.17,N
и новая начальная концентрация будет равна
Q,
.1=^, (4.18)
если к осадку, смоченному w л маточного раствора, добавлено
(v — w) л растворителя.
119
в новой системе распределение описывается с помрщью той
же изотермы (4.16) с заменой индекса О при х п с на/1, а Vona
V, с учетом уравнения (4.18) получаем
X, = (с, - = тр (^5 - (4.19)
Если опять на осадке останется w л раствора, а затем снова
объем будет доведен до t; л за счет добавки растворителя, то
общее количество остающегося в системе вещества будет равно
Xi +
W моль, а С2 =
■ /Qo 11
Снова используя (4.16) с заменой индекса О при хи с на 2,
а V0 на V, находим, что распределение выражается формулой
,п.
(4.20
При этом общее количество остающегося в системе вещества
/Qn
L\t^ V/ V * V V
. l\V VI V ^ V V
хз = (сг — ^)n,
w.
w
Хг
= mp({.
vl
V * V V
(4.21)
Если взять теперь за скобку v"^ и принять, что wv^ = q, то
после обычных преобразований уравнение (4.21) переходит в
выражение
XS = m^v " [(Qo - xi) q2 + (x, - ^^2) q + {X2 - X3)]", (4.22)
a тогда изотерма, определяющая равновесие при i-й операции
промывки, имеет вид
XI = ^ [(Qo - xi) q>~^ + (J^i - X2) q^-2 + . . . + (^i~i - Xi) q^\\
(4.23)
Для заданных co, vo, m, v и q и при известных p и п расчет
ведется в следующем порядке: 1) используя уравнение (4.16),
переписанное в виде
4
(тр)"
(4.24)
12(3
находят w, 2) с помощью уравнений (4.17), (4.18) вычисляют Qo
и ^i; 3) находят х\ из (4.19), представленного в виде
. (тр)".
^1 _
4) Х2 рассчитывают из уравнения
(4.20), записанного
(Qo-^i)q + (^i-^2)
5) любое Xi находят из уравнения
(4.25)
в виде
(4.26)
-I
(Qo ~ ^l) Я'^' + (^i - ^2)'Я'-'' + . . . + (Xc^i - X,)
соответствующую
концентрацию
адсорбируемого вещества в растворе
вычисляют по формуле
Ci^m,-x{)q'-'' +
+ (^i-^2)q'*~^+.:.+
(Xi^x-Xi) qO]-i.
(4.28)
(mp)"
= —, (4.27)
Таблица 4.1
110.2
98.7
89,4
81,7
74.6
462
620
527
438
399
0,92
1,24
1.05
0.88
0.80
Qi
QiQo ^
111.1
99.9
90.4
82.6
75,4
0.93
0.84
0,76
0.69
0,63
В табл. 4.1 приведены результаты отмывания вещества,
адсорбированного на поверхности осадка в виде отдельных молекул
чистым растворителем. Расчеты приводились по уравнениям (4.23),
(4.27), (4.28) для / = ТЗ, t;o = 0,l л; йу = 0,002 л; t; = 0,02 л,
q = 0,1, m = 0,25 г, = 2150 мкмоль • л-^ Использовались
значения р == 20,6 и п = 2,2, полученные для расчета xi в мкмоль х
X г~^ если а выражаются в мкмоль • л~Ч
В этом случае уравнение (4М6) приводится к *
у"
(тр)"
ИЛИ
у2,2
^0
215 —лго
= 368,2.
Из последнего находим, что л:о= 117,5 мкмоль • г"-^ Тогда из
уравнения (4.17) Qo = 119,45 мкмоль. Как следует из последней
КОЛОНКИ табл. 4.3, отмывание осадка от адсорбированного
вещества происходит очень медленно. После пятой промывки система
содержит еще 63 % от начального содержания примеси. При
этом, как следует из сопоставления значений Xi и {ci — XiV-'^)Wy
главная масса примеси находится на поверхности твердой фазы.
Результаты не совсем точно характеризуют процесс
промывания, так как все расчеты выполняются для равновесных условий,,
а при промывании трудно рассчитывать на строгое выполнение
этого предположения. Скачок для значений d — Xiv^ w {ci —
— Xt""^)^ при переходе от/= 1 к / = 2 (табл. 4.3) объясняется
121
тем, что все значения, отвечающие t = 1, соответствуют t;o== 0,1 л,
а для всех последующих / v = 0,020 л.
4.4. Отмывание примеси, попавшей на поверхность
осадка за счет ионной адсорбции
В разделе 3.2.1 показано, что при осаждении плохо
растворимых веидеств в виде кристаллов с ионной решеткой на их
поверхности образуется потенциалопределяющий слой за счет того
из ионов решетки, который находится в растворе в избытке.
Образование этого слоя определяется уравнением (3.19).
Адсорбционный слой заполняется при этом противоионами в соответствии
с правилом Панета — ФаянсаХана (раздел 3.2.3) и изотермой
Ленгмюра (3.29).
При рассмотрении процесса промывания такого осадка чистым
растворителем целесообразно учитывать одновременное отмывание
маточного раствора по формуле Бунзена (4.13), а также
постепенное удаление противоионов вследствие снижения j^x
концентрации в диффузном слое и потенциалопределяющих ионов из-за
приближения их концентрации в растворе к изоэлектрической.
Поскольку отмывание адсорбируемого вещества учитывает
одновременное разбавление остающегося маточного раствора,
необходимость в применении формулы Бунзена отпадает. Точное
решение вопроса об одновременном воздействии разбавления
жидкой фазы системы растворителем на концентрации потенциал-
определяющих ионов и противоионов на поверхности осадка не
рассматривалось. Поэтому приходится ограничиться уравнением
(3.29), хотя рассчитывать на постоянство «емкости» поверхности
твердой фазы, положенное в основу вывода уравнения (3.29), при
промывании осадка нельзя.
Пусть начальная концентрация противоиона в маточном
растворе Со, объем раствора —- vo, масса осадка — т, константы к'
и к, входящие в уравнение Ленгмюра, измерены заранее. Если
т г осадка адсорбировали в этих условиях хо моль противоиона,
то равновесная концентрация противоиона в маточном растворе
равна {cqVo — л:o)^'~^ а уравнение Ленгмюра выглядит так:
fo _ ^'^ (^0^0-^о) ,4 29)
Из (4.29) следует, что
^0 у [(vok-^ + к'т + Wo) — YXvok-^ + к'т + CoVof — ^k'mcoVo],
(4.30)
причем минус перед корнем поставлен потому, что всегда хо <
< CoVo.
122
После удаления маточного раствора в системе остается
хо + (^0 ~ ^) ^ = Qo моль (4.31)
противоиона, если объем маточного раствора, задерживающегося
на m г осадка, равен w л. Если для каждой операции
промывания используется (v — w) л растворителя, то начальную
концентрацию противоиона при первой промывке надо считать равной
d z= Qov^^ (4.32). Значение х\ в новой системе рассчитывается
по формуле (4.30) с заменой vq на v и остальных индексов О на
1. Тогда
Х\ = ^ [{vk-^ + k'm + civ) —
— У {vk-^ + k'm + cxvY — Ak'mcxvl (4.33)
В уравнении. (4.33) согласно (4.32) c^v = Qo, a vk-^ + k'm и 4k'm —
величины фиксированные. Полагая vk-^ + k'm == a и 4k'm =
=1 b (4.34), получаем расчетную формулу
Xi^J [(a + Qo) - Vr^ + Qo)'-bQo]. (4.35)
Поскольку при принятых условиях промывания осадка
Q^^xi + [cx-^)w, (4.36)
а значения а и b сохраняются,
д:2 = Т [(а + Qi) - l/(a + Qi)2-bQi], (4.37)
и вообще
xt+i = т + Qi) - Vi^+Qiy-bQtl (4.38)
Комбинируя уравнения (4.36) и (4.32), находим, что
Qi=^i + (Qo-Xi)f (4.39)
Пусть далее wv-^ == q, q2 = а:2 + {С2 — Х2^^) ш и ^2 = Q\v-K
Тогда, учитывая (4.39), q2 = а:2 + (Qo — xi) q^ + (xi — X2) q, и
вообще Qi = Xi + (Ci —XiV^) Wy a также
Qi = Xi + (Qo — Xi) q^ + (xi — X2) q^-^ + . . . + (Xi-i — Xi) q.
(4.40)
Пример применения этого метода расчета приведен в табл. 4.2
для промывания осадка BaSO^, полученного при осаждении S0|-
иона раствором ВаСЬ в присутствии КАс. В табл. 4.2 приняты
следующие начальные условия: /п=0,5г, ио==0,25 л, Co^l • Ю-^моль •
. л-1, W = 0,001 л, и = 0,020 л, q = 0,05, coVo 2,5 • lO-^ моль.
В результате измерений k'k = 2,01, k = 0,88, k' = 2,29. Тогда 4 —
123
— 1,429 Хо + 2,8625 • 10-^ = О, где, безусловно, хо < 2,5 • Ю-^ <^
« 1,429, а значит 1,429л:о= 2,8625-10-4, хо- 2,003-10-^ моль. Со—
-xov-^' = 1,988 - 10-4, (co — xoV-')w= 1,988 • 10-^ и Qp-2,005X'
X 10-4.
Таблица'4.2
,-1
(q — x\v
QiQo ^
i,965 . 10-4
1,928 . 10"4
1,892 . 10-4
1,856 . 10-^
1,821 . 10-4
1,787 . 10-4
2,000
1,950
1,900
1,900
1,850
1,800
10-^
10-4
10-^
10-^
10-4
10-^
2,00
1,95 «
1,90
1,90
1,85.
1,80 .
10'
,—7
1-7
i—7
1-7
i-7
,—7
1,967 . 10-4
1,930 . 10-^
1,894 . 10-^
i,858 ' 10-4
1,823 . 10-4
1,789 . 10"^
0,981
0,963
0,945
0,927
0,909
0,892
В дальнейших расчетах a == 1,168, k'm = 1,145, b = 4,58.
Поскольку, однако, оказалось, что во всех квадратных уравнениях
л:] —(а + Qi--i)A:o + /j'mQi-i = О, < а + Qi_i и они сводятся
к формулам Х[ = k'mQ\^\ (а + Qi-i)-i, все значения х\,
приведенные в табл. 4.2, рассчитывали по этим формулам.
Значения QiQ^^ характеризуюш;ие снижение общего содер"-
жания примеси в зависимости от числа операций промывания,
показывают, что отмывание с помощью растворителя дает в
рассматриваемой системе, т. е. для отвечающих ей значений k'' и
очень плохие результаты. Фактические результаты должны быть
еще хуже, так как расчеты предусматривают в каждом случае.
до(Тижение равновесия, а в гетерогенной системе надеяться на
это не приходится. В связи с этим данные табл. 4.2 следует
считать предельными. Реализоваться такие данные могут лишь
при переосаждении осадков, когда .последующие операции
сводятся: 1) к отделению твердой фазы от основного маточного
раствора; 2) полному растворению твердой фазы в подходящем
растворителе; 3) повторному осаждению твердой фазы; 4) отделению
ее от раствора и дальнейшему повторению этих операций.
Результаты расчетов, приведенные в табл. 4.2, показывают,
что, только измерив заранее k'' и k для данной конкретной
системы, можно правильно решить вопрос об эффективности
промывания чистым растворителем соответствующей твердой фазы.
4.5. Очистка осадков за счет «старения»
при дигерировании
При дигерировании, т. е. более или менее длительном
пребывании осадка в контакте с маточным раствором, содержащим
избыток иона-осадителя, образующего потенциалопределяющий
124
слой, выпавшая твердая фаза может подвергаться ряду превра-
ш.ений.
В главе 2 показано, что для осадков, растворимость которых
сильно зависит от радиуса частиц, маленькие кристаллы должны
исчезать, а крупные расти. Обш,ая поверхность твердой фазы
должна за этот счет сокращаться, а это должно вести к
самоочищению осадка за счет «старения». В табл. 4.3 приводятся
результаты, дающие представление о постепенном снижении количества
Na2S04 на поверхности осадка BaS04, полученного при
осаждении Ва2+-иона избытком 502~-иона. В третьем столбце таблицы
приведены значения Xh равные массе Na2S04, обнаруженного на
осадке после старения за времят^. Как видно из табл. 4.3, даже для
осадка BaS04, растворимость которого сильно изменяется номере
изменения размеров частиц, особых надежд на самоочищение за
интервалы времени, имеющие практическое значение, возлагать
не приходится.
Таблица 4.3
i
XI, мг
XI Xi, мг
Х\ -Xi
xi.io-3
10-2.2'^/»
1
2
3
4
5
6
7
8
15 мин
3 ч
18 ч
2 суг
4 сут
48 сут
150 сут
210 сут
22,6
19,0
17,4
14,0
12,2
10,4
8.2
7,6
3,6
5,2
8,6
10,4
12,2
14,4
15,0
16
23
38
46
54
64
66
1,13
0,95
0,87
0,70
0,61
•0,52
0,41
0,38
Считают, что в некоторых случаях существенного улучшения
природы и чистоты твердой фазы можно добиться за счет
перехода ее при дигерировании в другую кристаллическую
модификацию или даже в другое, менее растворимое соединение. Так,
Винклер (1918 г.) показал, что после тщательной подгонки рН до
величины, обеспечивающей достаточную растворимость желатино-
образного осадка Mga (Р04)2, он преврашается в менее
растворимый MgNH4P04 • 6Н2О, иглы которого достигают длины 1—2 мм.
Сокращение поверхности ведет в этом случае к снижению ошибки
за счет адсорбции. Таким образом, старение осадков и их
промывание чистым растворителем не могут обеспечить практически
полное освобождение твердой фазы от адсорбированных примесей.
4.6. Промывание осадков растворами электролитов
Промывание осадков растворами электролитов проводят для
замены противоионов, попавших в адсорбционный слой
коагулята в процессеосаждения, другими специально подобранными
ионами. Ионы выбирают так, чтобы соединение, которое они дают
125
с ионами потенциалопределяющего слоя, как можно больше
отличалось по своим свойствам от основной твердой фазы. Если это
возможно, то достаточно полную очистку осадка удается
осуществить путем последующей обработки. Этот подход используют
в технологии. Он же имеет большое значение при решении многих
задач химической метрологии. Например, при промывании осадка
{BaS04}nS0r^» Na+, образующегося при осаждении Ва2+-ионов
избытком 504"-ионов в присутствии Na+-HOHOB, раствором NH4CI,
стремятся получить твердую фазу {BaS04};, SO^"", NH^, в расчете
на последующую очистку Ва50"4 путем прокаливания, так как •
(NH4)2S04 разлагается и улетучивается при температуре,
значительно более низкой, чем BaS04.
Вытеснение противоионов, попавших в адсорбционный слой
коагулята, другими ионами происходит по законам ионного обмена.
При выводе всех уравнений ионного обмена исходят из
представления об эквивалентности обмела. Принимают, что равновесия
ионного обмена подчиняются закону действующих масс при огра*
ничейной емкости адсорбента. Это единство подхода приводит,,
естественно, к тому, что различные уравнения ионного обмена
оказываются разными лишь по форме. Поэтому дальше приведен
вывод лишь одного из них, наиболее приспособленного к
интересующим нас расчетам [7, 8].
Пусть R—доля мицеллы, несущая один 4- или одрш —
заряд, тогда равновесие ионного обмена представляется схемой
RM(,), + |М(^2|^ RM(2)j + ^ M^it, К, (4.41)
п v
причем максимальное число вакансий на поверхности
фиксированной массы твердой фазы, которые могут занять однозарядные
ионы, считается равным Гоо.
Допустим, что начальная концентрация М"+-иона в системе
равна СМ(1),. а иона М'+ —СМ(2)- Предположим, что в начальный
момент число вакансий, занятых на поверхности твердой фазы
о о
М"+-ионами, равно Гоо — Г, а М'+-ионами — Г. Тогда
начальная скорость выхода М"+-ионов на поверхность осадка
i 1 о
c;i= Wa)/u)A (4.42)
а М'+-иона
i 1 о
V2 = hcm) fh) (Г 00 — Г), (4.43)
так как М"+-ионы могут выxoдиtь на поверхность осадка только
вытесняя М''+-ионы и наоборот. В равновесной системе vi = V29
о
Г переходит в Г, а начальные концентрации в равновесные.
126
Приравнивая правые части уравнений (4.42), (4.43), после
обычных преобразований получаем
: 7 -rz^~k:=^ (4.44)
или, считая неизвестные коэффициенты активности = 1,
Wit У"
(4.45)
Сопоставляя равновесие (4.41) и уравнение (4.45) для
начальных и равновесных условий, находим, что переход к равновесию
следует учитывать для твердой фазы (ТФ) и жидкой фазы (ЖФ)
отдельно:
Начальные
условия
ТФ
ЖФ
о
т
о
Г
^М(г)
^M(i)
Равновесные
условия
ТФ
ЖФ
о
т
т
В этой схеме Гоо, Г н Г определяются числом однозарядных
вакансий на т2 твердой фазы. Поскольку число вакансий
пропорционально количеству вещества эквивалента любого иона,
а в уравнение (4.45) входит отношение Г(Гоо —Г)~^ в
дальнейшем все значения Г выражены количеством вещества эквивалента
(в молях).
Путь к измерению констант /( и Гоо, входящих в уравнение
(4.45), становится ясным после приведения этого уравнения к виду
* 1 t W Ч1_1_ -■ 1 /ч,
(4.46)
Для измерения значений [M(\|li, WT)]i и Г\ входящих в i-e
уравнение (4.46), осаждают в i-м опыте при v = const и прочих
фиксированных условиях то же, что и в других опытах,
количество осадка (т) в присутствии М^^ и М(2) начальные
концентрации которых соответственно равны Сщ^)1 и Сщ^)1. В аликвотах
равновесного раствора измеряют [Mfitli и [M(^li, а Г[
вычисляют по формуле Л = V (cM^2)i — W^]u (4-47).
Полученную таким образом систему условных уравнений
(4.46), где An = -1, Xi = /(, Ai2 - ГГ\ Х2 == /СГео, Фг = [M?it]"
127
W^] " свертывают по методу наименьших квадратов и
находят наивероятнейшие значения К и /СГоо. а затем Гоо.
Применяя уравнение (4.45) надо помнить, что Гоо выражается
количеством вещества эквивалента (в молях), отнесенным к
фиксированной массе твердой фазы в контакте с 1 л раствора,
причем начальная концентрация второго противоиона в этом растворе
может иметь любые значения. Если фактическая масса осадка
отличается от той, для которой были найдены Гоо и /С в q раз,
то и число вакансий в данном конкретном случае изменяется
в q раз. Если при этом объем раствора равен не I л, а у л, то
число вакансий изменяется в раз, начальная же
концентрация второго противоиона сохраняется. Это значит, что при
изменении массы осадка и объема раствора значение /С, получен--
ное для уравнения (4.45) сохраняется, а значение Гоо изменится
в qy-^ раз.
При промывании осадка можно считать, что масса его
остается практически неизменной. Объем раствора соли, расходуемый
на каждую операцию промывки, также можно считать
фиксированным. Если учесть далее, что общая масса загрязнений,
остающихся в пленке раствора на поверхности осадка, значительно
меньше адсорбированной части, как это видно из сопоставления
значений {Ci—XiV-^)w и ATi, приведенных в табл. 4.2 и 4.3, то
загрязнениями в пленке можно пренебречь. Тогда достаточно
один раз пересчитать значение емкости для данной массы осадка
т и объема промывной жидкости v п в дальнейшем использовать
полученное значение Гоо на всех стадиях промывки.
К началу первой стадии промывки будем считать, что в
адсорбционном слое мицелл находятся только M(V]"-HOHbi. Тогда
рассчетная схема принимает вид:
RM(iM + 1 M(^2j « RM(2)^ + ^ M?i|, К, Гоо
о
г Г.. О
v
Г Г«, — пх\ nxi
По закону действующих масс
1_ 1_
= /С, или
1 (r„-nxi) 1
- (4.48)
128
На второй стадии, учитывая изменения в адсорбционном слое,
имеем
RM(oi + 1 Mbt ^ RM(2)_i + \Kit^ к. Гоо
п v
О
Г Гоо —пл:1 пх\
П с—--^2 ^2
V
г Гоо — п (л;1 + Х2) п (л;1 + л;2)
Отсюда расчетное уравнение выглядит так:
j
4(^1 + ^2)
= (^)1К. (4.49)
Теперь очевидно, что рассчитать лгт, т. е. число молей Mff}"-
иона, появляющееся в литре раствора после m промывок,
можно с помощью уравнения
= К. (4.50)
Количество же вещества противоиона Мо"]" моль остающееся на
поверхности осадка равно
^ m
Хш = п-1ГооXI. (4.51)
i=i
8 уравнениях (4.50), (4.51) vn^-'c, п-^Гоо и ("vj"^ имеют
фиксированные значения. По мере возрастания i сумма I,Xi растет,
а п~^Гоо —уменьшается, что ведет к постепенному убыванию
значений Xi, т. е. снижению эффективности ионного обмена в
адсор^бционном слое.
В качестве примера рассмотрим результаты ионного обмена
на осадке {Fe (ОН)з}пОН-, Ca^^^RCai при промывании массы
1 2
его, равной 0,2 г, отдельными порциями раствора NH4CI
объемом V = 0,015 л каждая, если cnh.ci == 1 моль • л~^ К = 0,456
и пересчитанное на приведенные выше условия значение Гоо =
= 0,037 моль.
9 3-457 129
Из схемы RCa, + NH+5=tRNH4 + yCa2+ следует, что п
2
= 2, V = 1, тогда
1 m-l
= 2К, или
(0,5с-^^)
/ т—1
i=l
1 m—1
i=l
(0^5 ~^m)
m—1 \
= 0,912.
0,0185— у
В уравнении для первой стадии промывания
1
1^1
-0,912 и 0<;ci < 0,0185.
(0,5 —(0,0185
Пользуясь методом последовательных приближений, находим, что
Xi = 0,01475 моль . л-1, 0,5Гоо — a:i = 3,75 • Ю-з моль.
Тогда для второй стадии можно сразу записать расчетное
уравнение
0,912, где 0<а:2<3,75.10-з<^0,5моль.
4(0.01475 H-ATg)
(0,5 —ATg) (3,75 . Ю-^-^х^)
Пользуясь последним неравенством, упрощаем расчетное уравнение
0,456
3,75 . 10-3
и находим, что л:2 = 2,05 • 10-^, Х\ +Х2 = 0,0168 и 0,5Гоо —{xi +
+ л:2) = 1,7 • 10-3 моль.
Тогда после третьей промывки
_^(.^Ш + ,з) ii£l!|±5), 0,456,
(0,5-;сз) (1,7 . 10-3-л:з) 1,7.10-3-л:з
так как О < ;сз < 1,7 • Ю-з <^ 0,5 моль. Решение дает: хг=^7х
X 10-4, л:1 + л:2 + л:з = 0,0175 и 0,5Гоо — (a:i + л:^ + лгз) == 1, Ох
X 10-3 моль. Таким образом,
. после No промывок 12 3
осталось на осадке Сг^+ 20,3 9,2 5,4 %
заменено на NH^ 79,7 90,8 94,6 %
130
при последующем прокаливании осадка всего 5,4 % Са2+'
иона останется на нем в виде СаО.
Следует помнить, что и в этом случае фактические
результаты будут приближаться к расчетным лишь при достижении
равновесия. Промывание следует вести поэтому путем декантации,
т. е. вливать раствор соли в стакан с осадком, хорошо
перемешивать, давать отстояться, сливать через фильтр и повторять
эту операцию несколько раз.
4.7. Разделения на сорбентах различной природы
4.7.1. ОБЩИЕ СВЕДЕНИЯ О ХРОМАТОГРАФИИ
В 1903 Г. М. с. Цвет показал, что при фильтровании раствора
хлорофилла через стеклянную' трубку, заполненную порошком
карбоната кальция, отдельные компоненты раствора
задерживаются не одновременно и образуют разноокрашенные зоны. При
последующем промывании колонки тем или иным растворителем
зоны разделяются и передвигаются вниз, каждая с присущей
данному веществу и в данных условиях скоростью.
Расцвеченный столбик сорбента Цвет назвал хроматограммой,
а метод — хроматографическим. Появление нескольких
окрашенных зон указывало на сложность первичной системы и -
возможность ее разделения на данном сорбенте. Для идентификации
отдельных составляющих хроматограмму можно разделить на
окрашенные цилиндрики, извлечь из каждого адсорбированное
вещество и установить его состав и количество. Другой путь
основан на постепенном вымывании зон из колонки
растворителем, собирании соответствующих растворов в отдельные приемники
и последующем исследовании полученных таким образом фракций.
Часто разделение на сорбентах называют хроматографическим
анализом. Следует четко различать операции- разделения и
последующего исследования фракций, для чего можно использовать
самые разнообразные методы измерения.
В качестве простейшего примера рассмотрим разделение Си^"*"
и Со2+-ионов на оксиде алюминия. Для приготовления колонки
можно использовать стеклянную трубку длиной 120 мм и внутренним
диаметром 10 мм, с краном, как у бюретки. В нижнюю часть
трубки вкладывают тампон из стеклянной ваты, а затем
заполняют трубку до половины жидкой кащицей из мелко
измельченного оксида алюминия и воды. Адсорбент должен равномерно
заполнять всю нижнюю часть трубки, а над ним обязательно
должен оставаться слой воды. Если в верхнюю часть такой
колонки влить смесь растворов CUSO4, C0SO4 и открыть кран, то
из него начнет вытекать вода, а в верхнюю часть сорбента начнет
просачиваться раствор, содержащий .Cu2+> Со2+-ионы. Кран
следует закрыть, когда над поверхностью сорбента останется
небольшой слой исходного раствора. Колонка сорбента окрашива-
9* 131
Синял
зона
Синяя
Розовая
ется (см. рис. ,4.1, а): вверху образуется синяя зона, ниже —
фиолетовая и еще ниже — розовая. Это значит, что Си^^-ионы
адсорбируются на поверхности АЬОз лучше Со2+-ионов. При
последующем промывании колонки водой происходит разделение
зон и получается иная хроматограмма. (рис. 4.1, б). На этой
стадии от колонки можно отделить два цилиндра: один розовый,
содержащий Со^^-ионы, и второй голубой, окрашенный Си2+-ио-
нами. После извлечения Со2+, Си^+-ионов можно измерить массу
каждого из них тем или иным
способом и завершить таким образом
исследование состава первоначальной
системы.
Можно добиться разделения и
несколько иным путем: продолжать
отмывание водой до момента, когда
розовая зона приблизится вплотную
к крану. С этого момента начинают
\Роэо6ая собирать вытекающий розовый
раствор (его называют элюатом) в
отдельный приемник. Заканчивают
операцию, когда из колонки выйдет вся
розовая зона. Подобным же образом
собирают элюат, содержащий Си^^-
ионы. В элюатах раздельно измеряют
содержание Со2+, Си^^-ионов. В
рассмотренном случае адсорбция описывается уравнением Ленгмюра.
При отмывании существенна та же изотерма, но в гораздо более
сложных условиях.
Когда сорбент состоит не из ионов, а из атомов или молекул,
подобно активированному углю, и разделяется смесь веществ,
не являющихся электролитами, результаты адсорбции и
промывания растворителем определяются изотермой Фрейндлиха.
Было показано (см. табл. 4.1 и 4.2), что растворитель
вымывает адсорбированные вещества с поверхности твердой фазы очень
медленно. Гораздо эффективнее проходит процесс замещения одних
адсорбированных противоионов другими, основанный на законах
ионного обмена.
Различные' варианты этих адсорбционных методов разделения
широко применяются в науке и практике. Их изучение имеет
огромное значение при решении многих биологических проблем,
в медицине, сельском хозяйстве и т. д.
Рис. 4.1
4.7.2. ИОНООБМЕННИКИ И ИХ ПРИМЕНЕНИЕ
Уже давно отмечали, что некоторые алюмосиликаты — цеолиты
обладают рыхлой кристаллической структурой и способны
обменивать содержащуюся в них воду на другие жидкости (спирт,
аммиак и т. п.), а катионы, входящие в их состав, — на различ-
132
ные другие катионы. Состав цеолитов выражается схематической
формулой МхЭуОгу • пНгО, где М металло-ион, обычно Са или
Na и гораздо реже Ва, Sr или К, Э обозначает Si или А1,
которые входят в переменных отношениях.
Основой цеолита является прочный алюмосиликатный скелет,
представляющий собой высокомолекулярный анион, который
несет на себе относительно слабо связанные одно- и двухзарядные
катионы и молекулы волы. При фильтровании через слой цеолита
раствора, содержащего какие-нибудь катионы, металло-ионы,
входившие в состав исходного материала, вымываются и
замещаются ' эквивалентным количеством ионов, входивших в состав
раствора. Это свойство цеолитов и использовано для удаления
Са2+, Mg2+, Fe^+, Мп2+-ионов из природных жестких вод.
Природные цеолиты были первыми ионитами, сознательно
использованными для смягчения воды. Иониты или ионообмен-
ниКи, основой которых является прочный анионный скелет,
получили название катионитов, так как на поверхности такого
каркаса могут задерживаться и обмениваться только катионы.
Позднее начали применять искусственные цеолиты, так
называемые пермутиты-обменники, получающиеся при сплавлении
кварца с каолином и содой. После измельчения сплава и
обработки его водой получаетсй желтовато-белая пористая масса
пермутита натрия Na2 lH4Al2Si20io] = Ма2П, способного к
обратимым ионообменн1?1м процессам. Например,
Са2+ + Na2n 2Na+ + СаП
СаП + 2Na+ ^Са+ NagH.
В настоящее время промышленность выпускает различные
типы синтетических катионитов и анионитов. Эти вещества
представляют собой чрезвычайно малорастворимые пластмасды, в
состав которых для получения катионитов вводят различные кислые
группы:—SO3H,— СООН, —ОН, —SH и т. п. Для получения
анионитов, наоборот, вводят основные группы: —NH2, —NHR,
NR2 и другие.
Особо чистые и малорастворимые иониты используют для
различных разделений при химических исследованиях.
Иониты, предназначаемые для таких разделений,
предварительно подвергают соответствующей обработке непосредственно
в той колонке, в которой они находятся. Так, например, для
удаления катионов, которые могут мешать исследованию аниоп
него состава, через катионит пропускают раствор НС1. Это
приводит к насыщению поверхности катионита НзО+-ионами, т. е.
превращению его в Н-катионит. Если теперь через
подготовленную таким образом ионообменную колонку» профильтровать
исследуемый раствор, то находящиеся в нем катионы задержатся на
сорбенте, а в элюат перейдут анионы и эквивалентное, вытеснеи-
i:o2 катионами, количество Н3О+-ИОНОВ.
133
' * Адсорбированные катионы можно вытеснить из колонки
раствором подходящей кислоты и получить таким образом систему,
содержащую только катионы и один единственный анион, который
всегда можно, выбрать так, чтобы его присутствие не мешало
дальнейшему исследованию.
Следует подчеркнуть, однако, что эти методы, на первый
взгляд безупречные, не всегда приводят к цели. Например, все
анионы, входящие в состав
комплексных катионов, вроде
FeHP04", останутся на кати-
оните, а в то же время ацидо-
комплексы катионов,
например AIF3+P, окажутся в
фильтрате.
Сомнения вызывает
возможность количественного
разделения сложных систем с помощью рекомендуемых методов
вымывания отдельных составляющих из сорбента — элюатного
и вытеснительного.
В- первом случае [18] после промывания колонки чистым
растворителем в нее вводят небольшое количество исследуемого
раствора. Затем, когда раствор практически полностью впитается
в сорбент, промывают колонку чистым растворителем. Считают,
что при этом сначала получается хроматограмма^ как на рис. 4.1, б,
а потом* начинается последовательное вымывание отдельных зон
в присоединенный к крану колонки приемник. Результирующая
диаграмма для двухкорлпонентной системы [18, с. 14] приведена
на рис. 4'.2. Утверждается, что «каждый компонент получается
в чистом виде» [18, с. 13], а в связи с этим площадь А под
каждым пиком кривой на рис. 4.2, т. е.
А = ^cdv
в соответствующем масштабе отвечает общему количеству
соответствующего компонента.
^ Из данных, приведенных в табл. 4.1 и 4.2, следует, что как
при многослойной,, так и при однослойной адсорбции отмывание
поверхности сорбента от адсорбированного вещества идет
медленно и полным быть не может. Значит, количественное
разделение в связи с этим не наблюдается. В работе [18, с. 14]
отмечается, что недостатком элюатного метода разделения являются
длинные «хвосты» пиков у хорошо адсорбируемых веществ, так
как эти «хвосты» очень затрудняют разделение веществ и делают •
ненадежным определение площади А,
Вытеснительное разделение [73] проводят так: промывают
колонку чистым растворителем, вводят небольшую порцию
исследуемого раствора, а затем начинают промывку раствором
«вытеснителя», который должен адсорбироваться сильнее любого из
134
компонентов исследуемой системы. Предполагается, что при
просачивании раствора вытеснителя через адсорбент смешанная зона
адсорбированной системы разделяется на отдельные зоны,
продвигающиеся вниз к спускному крану колонки. При этом впереди
других движется зона * компонента, вытесняемого легче других,
в последней находится наиболее сильно адсорбируемое вещество,
а по мере продвижения этой последней зоны вниз на поверхности
сорбента остается только вытеснитель. В работе [18, с. 75]
отмечается, как большое преимущество метода, что при этом «каждое
вещество находится лишь в одной из ступенек и нигде более».
К сожалению, из данных, приведенных в разделе 4.6, где
рассматривается один из вариантов вытеснительного метода —
ионный обмен, видно, что абсолютного разделения, когда
«каждое вещество находится лишь в одной из ступенек», и здесь
ожидать нельзя. Это следует, например, из выражения гг^^Гоо —
т
— S^Tt, входящего в формулу (4.50), поскольку в нуль эта
разность обратиться не может. Опытные данные подтверждают
этот вывод. Так, в работе [18, с. 15, 74, 79] указывается, что
вытеснительный метод не всегда применим и что при наличии
осложнений «возможность вытеснительного проявления в каждом
отдельном случае следует устанавливать опытным путем».
4.8. Несколько общих замечаний об использовании
гетерогенных систем, состоящих из твердой
и жидкой фаз
Со времен Лавуазье большинство методов измерения состава
основывается, на выделении соответствующей составляющей из
Ж11Дкой системы (раствора) в виде малорастворимого соединения.
Появление этой твердой фазы в данных конкретных условиях
подтверждает присутствие в изучаемой системе вполне
определенной составляющей. Масса ее, выраженная в граммах,
используется в дальнейшем для расчетов, ведущих к оценкам любых
проявлений химической формы движения материи (ХФДМ).
Исследование процесса образования твердой фазы показало,
чта фактическая масса ее может уменьшаться вследствие
растворимости и изменяться из-за различных видов соосаждения.
Рассмотрены современные теоретические положения,
используемые для объяснения этих изменений массы, математические
выражения и результаты примерных расчетов. Оказалось, что
рассчитывать на^ получение теоретического выхода абсолютно
чистой твердой фазы нельзя.
При точных физических измерениях массы объекта, например
металлического цил^^нд^а, также приходится учитывать ряд
факторов, влияющих на результат взвешивания, например, массу-
вытесняемого. объектом воздуха. Но для оценки этой массы
достаточно найти в таблицах плотность материала, из которого из
готовлен объект, коэффициент его объемного расширения, плотность
135
воздуха и измерить температуру, при которой производилось
взвешивание. Этих данных достаточно, чтобы однозначно оценить
массу вытесненного воздуха и ввести соответствующую
аэростатическую поправку в результате измерения.
В уравнения, с помощью которых можно было бы рассчитать
поправки, учитывающие растворимость и загрязнение выпадающей
твердой фазы, входят либо параметры еще не известные,
например, коэффициенты активности, либо параметры, имеющие особое
значение для каждой конкретной системы, вроде констант в
уравнениях изотерм адсорбции и ионного обмена, а такие константы
не табулируют.
В связи с этим при химических исследованиях, связанных
с измерением массы твердой фазы, выпадающей из раствора, вы-
бирают из таблиц все параметры, которые могут пригодиться при
измерениях и расчетах, например, плотность твердой фазы,
произведение ее растворимости, константы протонизации аниона
и ионизации аквокатиона,- радиусы ионов, находящихся в
системе, и дополнительно измеряют параметры равновесий
соосаждения в условиях, точно отвечающих принятой прописи осаждения.
Располагая всеми перечисленными, данными, можно получить
приближенные оценки растворимости твердой фазы,' состава
адсорбируемого вещества и изменения массы вследствие всех
возможных видов соосаждения. Имея эти оценки, можно планировать
исследование по-разному, в зависимости от требуемой точности.
Если рассматривать грубые измерения, и рассчитанная общая
погрешность окажется допустимой, то считают выход твердой
фазы количественным и используют фактически найденную ее
массу для оценки всех заданных проявлений ХФДМ.
В случаях, когда рассчитанная погрешность превышает
допустимую (измерения не претендуют на особую точность),
рассматривают все возможные меры по устранению причин, ведущих
к снижению массы твердой фазы и ее загрязнению за счет
соосаждения, например, путем создания рН, обеспечивающего
минимальную растворимость, введения реагентов, маскирующих
сопутствующие ионы и т. п. Если последующий расчет покажет, что
исправленная таким образом пропись пригодна для получения
результатов с ошибкой, не превышающей допустимую, то ею
пользуются так, как будто она дает идеальные результаты.
Для получения особо точных результатов каждое измерение
массы выпавшей твердой фазы должно сопровождаться рядом
дополнительных измерений: содержания осаждавшегося иона
в маточном растворе, т. е. определением потери за счет
растворимости; обнаружением и количественным измерением всех
примесей к твердой фазе; расчетом массы твердой фазы с учетом
всех возможных поправок. Осложнения, возникающие при
выделении изучаемого объекта из раствора в виде твердой фазы,
связаны с неизбежным образованием огрОхМной поверхности
раздела между фазами.
136
5.
РАСПРЕДЕЛЕНИЕ ВЕЩЕСТВА МЕЖДУ
ДВУМЯ ЖИДКИМИ ФАЗАМИ
5.1 .Общие сведения о процессе распределения
Как мы показали, выделение твердой фазы из гомогенного
раствора всегда сопровождается соосаждением посторонних
веществ, исключить которое вообще нельзя и даже снизить в
необходимой степени очень трудно. В связи с этим за последнее
время резко возрос интерес к разделениям путем экстракции,
так как поверхность раздела между двумя разделяемыми жидкими
фазами можно снизить до долей квадратного сантиметра, т. е.
сделать в сотни и тысячи раз меньше поверхности раздела между
мелкодисперсной твердой фазой и маточным раствором.
Распределение между двумя жидкими фазами используют
в аналитической и препаративной химии, технологии атомного
горючего, гидрометаллургии, биохимии. Оно будет играть
существенную роль при решении многих геотехнологических задач. Все
это повышает интерес к теории этого метода разделения.
В дальнейшем рассматривается распределение в системах:
водный раствор — жидкое органическое соединение или смесь
органических соединений, водный раствор — ртуть или амальгама.
Неводную фазу, будем называть экстрагентом.
5.2. Краткие сведения о применяемых экстрагентах
Достаточно полные перечни органических растворителей,
применяемых при химических измерениях, даны в работах [30, с. 270;
28, с. 10; 32, с. 207]. Сведения об экстрагентах, имеющих
существенное значение в технологии, приведены в [39, с. 154].
К органическим растворителям [30, с. 270] относятся:
углеводороды, относительная диэлектрическая проницаемость которых*
(s) составляет от 1,9 до 2,6; замещенные углеводороды, для
большинства которых е = 2,2-f-10,4; только для нитроэтана е =
= 28,1 и нитрометана s = 35,9; одноатомные спирты s = 8,2-^
32,6; многоатомные спирты s > 32; эфиры е = 2,2-ь-4,3; кетоны
6 = 11,9-7-25,7; сложные эфиры е = 5,0-4-8,1; амины с е = 2,9-г-
12,3. Многие из этих растворителей сами по себе применяться
в качестве экстрагентов не могут, так как очень хорошо или
относительно хорошо растворимы в воде. Их используют в
качестве компонентов сложных органических фаз.
В работе [39, с. 154] кроме растворителей, приведенных
в [30, с. 270], даны важнейшие кислоты и основания, образующие
137
с металло-ионами или их анионокомплексами растворимые в
органических растворителях соли или внутрикомплексные соединения.
5.3. Механизм распределения
Случаев чисто «физического» распределения, когда вещество,
частично переходящее в другую фазу, совершенно с ней не
взаимодействует, совсем нет или они очень редки. Сюда относят
распределение ОеСЦ между водой и CCl4[30;34], меркурогалоге-
нидов между водой и органическими растворителями, например,
этиловым эфиром [24]. Вероятно, физическим можно считать
распределение иода между водными растворами, содержащими
1Г-И0Н, и CCI4.
В большинстве остальных случаев следует учитывать
различного вида химические взаимодействия, предшествующие,
сопровождающие и способствующие распределению. При этом в водной
фазе приходится принимать во внимание возможность
возникновения всех ранее рассмотренных равновесий, а также
дополнительные взаимодействия между частицами, находящимися в
водной фазе, и частицами органической фазы, попадающими в
водный раствор из-за взаимного растворения. Для органической
фазы особенно важную роль играет значение относительной
диэлектрической проницаемости s, поскольку в первом приближении
можно считать [47], что константа любого процесса диссоциации,
равная в водном растворе /С, в органической фазе становится
хменьше в —^SOe—^ раз, а константы ассоциации в такой же степени
возрастают. Это приводит обычно к тому, что диссоциацией даже
электролитов, переходящих в органическую фазу, можно
пренебречь, а возможность образования ассоциатов следует всегда иметь
в виду. Важно помнить при этом, что значения s могут сильно
изменяться за счет взаимной растворимости воды в органической
фазе и органической фазы в воде.
В результате распределения возникает равновесие между двумя
фазами. Оно характеризуется одновременным равенством
химических потенциалов в обеих фазах для всех способных
распределяться, т. е. незаряженных частиц. Это значит, что, если для i-й
распределяемой частицы \11^ы+ЯТ\па[ в водной фазе и =
IS SI
^ Ы = RTlmi в органической фазе, после распределения, т. е.
IS
после достижения фазового равновесия, fxf = fxi и
IS ^ol
А = е = Xi (Г = const), (5.1)
где Xi — константа распределения i-й частицы между водой и
соответствующей органической фазой. Такие же равенства надо
138
записать для всех остальных незаряженных частиц. Их
единовременное выполнение отвечает установившемуся межфазному
динамическому равновесию, при котором в единицу времени через
поверхность раздела переходит в противоположных направлениях
одно и то же число частиц i-ro сорта. Так, например, в
двухфазной системе ССЦ —раствор Ь в водном растворе KI должны
учитываться распределения четыреххлористого углерода, воды
и Ь между раствором U CCI4 и водным раствором,
содержащим К+, I"", CCI4.
Попытки классификации экстракционных равновесий ввиду их
сложности не привели к однозначным результатам [30, с. 10; 39,
с. 9; 34, с. 18].
Гораздо важнее сведения о составе распределяющихся незг-
ряженных частиц, без которых нельзя обойтись при составлении
матриц равновесий и уравнений законов сохранения. К
сожалению, и эти сведения зачастую не вполне однозначны. Так, при
распределении FeCb между водным раствором, содержащим НС1,
и кислородсодержащим растворителем (например, диэтиловым
эфиром) [16, с. 135], по данным -различных авторов в
органическом слое присутствуют ионные ассоциаты, образованные из соль-
ватированных ионов. {Solv . Н+, Реа4 . wSolv-} [30, с. 12],
ионные ассоциаты, которые не только сольватируются, но и уносят
с собой часть воды [40], соединения, в которых вода и
органический растворитель связаны только с катионной частью ассоци-
ата {mSolv • ПН2О • Н+, FeCU""} [55].
По данным [65], сольват HFeCU • 2ДПЭ извлекается в диизо-
пропиловый эфир (ДПЭ) только в сопровождении "пяти молей
Н2О на каждый- ЕеЗ+-ион.
В трибутилфосфат (ТБФ) одновременно извлекаются HFeCU X
X 2ТБФ и FeCla • ЗТБФ [70]. Совместная экстракция Н1пВг4
и 1пВгз рассмотрена в работе [57]. Сведения о гидратации в [70]
сольватации и гидратации в [57] не приводятся.
Взаимная растворимость жидких фаз также связана* с
образованием гидратов и сольватов [29, с. 248, 334]. Их образуют
даже сильные кислоты. Так, в [24] отмечается образование
(С2Н5)20 • HCI, а в [39, с. 46]—экстракция HCIO4 в
трибутилфосфат в виде 2HCIO4 • 4ТБФ.
В ряде случаев при использовании смешанных растворителей
наблюдается значительное изменение константы распределения
экстрагируемого вещества. Это явление называется синэнергети*
ческим эффектом. Его приписывают образованию смешанных соль-
ватокомплексов. Теория вопроса рассматривается в работах [39,
с. 84; 34, с. 59]. В частности, вводится представление о диспро-
порционировании двух обычных сольватов в смешанный, идущем
без изменения координационного числа, МА^. . 2Si -f MAj,. . 2S2 5=t
<=t2MAj,SiS2.
139
5.4. Расчеты равновесий в двухфазных жидких системах
Пример 5.1. Расчет равновесия в системе, полученной путем встряхивания
У = 0,1 л раствора Ig в CCI4, в котором Cj^ == У = 0,08 моль - л * с с» = 0,5 л
воды, если X =: 85,5 = 10^*^^^ и CCI4 предварительно насыщен водой,
а вода —CCI4.
В работе [23, с. 192] находим, что в 100 мл СС14 растворяется 0,01 г НдО,
а в 100 мл HgO — 0,08 г CCI4. Считая d^^Q^l г • см""^ и d^^^^ == 1,595 г .cм-^
находим, что Гн^о = 55.46, ^^01, = * 1^"^' "^^н.о = ^'^ * ^0""^ ^CCU =
= 10,37 моль • л-^ .
Поскольку далее Ig-—неэлектролит, ионная сила в обеих фазах очень мала,
и все cpj можно считать равными 1, а равновесные концентрациисовпадаю-
и'ими с активностями. Если попустить, что I2 не координирует CCI4 и HgO,
то матрица равновесий системы имеет вид:
?
55,46
5,2 . 10-3
10,37
5,6. 10-3
0,08
Н2О
CCI4
CCI4
Н2О
I2
К
lg/<
1
1
X
1,932
Если объемы v и v после достижения равновесия сохраняют начальное
значение, то, применяя закон сохранения количества вещества (начального числа
молей), находим, что
Yv= [h]v + [\2]v = lll,]v + [l,{v.
Тогда [Ig] = 8,85 • 10-^ моль • л-^ [У = 7,567 • IQ-^ моль . л"^ [laJ-O.S
= 4,42.10-'* моль, [Ig] • 0,1 = 7,567 • 10-3 моль, и задача о распределении
I2 между двумя фазами решена.
Пример 5.2. Расчет равновесия в системе, полученной путем встряхивания
у = 0,8 л водного раствора пикриновой кислоты (НА), в котором = Л =
= 3 . 10-3 j^Q^j^ . ^-1 ^ Q 4 ^ бензола, если X = 232 = Ю^'Зббб^ ^ ^ 2,4 =
_ Iq0,3802 ^ Qpj^ предварительно насыщен водой, а вода — бензолом.
В работе [23, с. 191] указано, что в 100 мл СвНе растворяется 0,054 г Н3О,
а в 100 мл воды 0,082 г СвНв. Считая d^^^Q^ 1 г • см-з и cf^^H.^ ^'^^^ г . см"3,
находим, что ^н^О = 55»46, с^^^^ = 0,0105, ^н^о = ^,0 • 10 и с^^и,=
11,44 моль . л—^
Путем обычного расчета находим, что пикриновая кислота при А =
= 3 . 10-3 дает [Н3О+] = [А"-] = 2,98 • Ю^з моль • л-\ т. е. диссоциируег
практически полностью. Тогда ионная сила / = 3 . 10—з моль • л—* и Ig^i
0,0257, если расчет вести по Дэвису. В литературе нет сведений о
гидратации пикриновой кислоты и о сольватах ее с бензолом. Нет сведений и об обр^-
140
зова НИИ ассоциатов, хотя при е^-.^^, = 2,3 этого можно было ожидать.
Диссоциацией НА в бензольном растворе в первом приближении можно пренебречь,
80
так как ее константа- снижается примерно в 23 =35 раз.
Учитывая замечания и отсутствие информации по ряду вопросов, махри[1у
равновесий системы можно представить в следующем виде:
А
3,0.10-^
11,44
55,46
0,0105
3,0.10-3
Н20
СбНв
Н,0
СвНо
Н+
НА
НА
НА
К
1
0
1
0
0
1
а
X
0,380
2,336
[НА] = ^wppah
[НА] = ^iiploah
По закону сохранения количества вещества (начального чпсла молей)
0 Q 0
Av = а (<рд + ^'liA^h) V + ^^ploahv. Полагая ср^^д = ср^д = = подставляя
v=-0,Sv и сокращая на у, находим А= аЕсрд-f а/г(1 + 0.5Х)] (5.2).
Учет электронейтральности водной фазы приводит к выражениям:
(р„/1~срда=0, a = — h = bh==h.
Подстановка этого значения а в уравнение (5.2) приводит к а (1 -f- 0.5Х) -f
+ срд/i — Л = О и /i2 + 3,779 . 10-^/1 — 1,068 . 10"^ = О (5.3). Из уравнения
(5.3) /1 = а = 1,885 - 10"^ [H+J = [А"] = 2,001 • Ю'^, [НА] = 8,54 . Ю"^
и [НА] = 1,98-10 ^ моль - л . Эю значит, что в 0,4 л неводного раствора
перешло 7,92 • 10"""* моль НА, а в 0,8 л водного раствора осталось 6,832 . 10^^
моль НА и 1,6008. 10""3 моль А".иона, что в сумме почти точно дает
Av = 2,40 • 10"^ моль. Поскольку при расчетах не были учтены все
возможные осложнения, полученные результаты следует считать условными и
представить их в округленном виде: [Н+] = [А"] = 2 • 10"^ [НА] = 8,5 . 10"^
И [НА] =2.10 моль . л . Подсчитанная по новым данным ионная сила
/ = 2 . 10"^ моль . л""^, но пересчитывать задачу не следует, поскольку
полной информацией о системе мы не располагаем.
Пример 5.3. Расчет равновесия в системе, полученной путем встряхивания
О •
уд = 0,050 л водного раствора, в котором cpecls = ^ ^ ^нс1~^» ^ равным
объемом Vq этилового эфира (R), если водный раствор предварительно
насыщен эфиром, а эфир водой.
Используя данные, приведенные в работе [23, с. 191], считая с^НвО^
ж 1 г . см rfj^ =0,719 г . см находим, что в неводной фазе cHtO ~
: 0,588 и = 10,45, а в «водной» — Cpj^Q = 49,96 и = 1,06 моль • л"
,^1
141
Матрица равновесий сисгемы FeCIg — HCI — Hfi — (С^0^)20, основанной
на фактических данных, приведенных в [34; 39; 71; 72], представлена в табл. 5.L
Образование гидроксо- и гидроксоферри-комплексов не учитывается, так как
лри высоких значениях Y их" равновесные концентрации-> О и должны
приниматься во внимание только в выражениях закона действующих масс.
Принято, что все первичные равновесия возникают в водной фазе, а затем уже
устанавливаются межфазные равновесия. Возможность распределения РеС1з
не рассматривалась. Никаких серьезных изменений в матрице введение этого
равновесия не вызовет.
Для расчета равновесия надо знать фактические объемы водной и эфир-
ной фаз равновесной системы, поскольку при высоких концентрациях
распределяющихся веществ первоначальные объемы могут заметно измениться,
Вычислить у и у, не зная равновесного состава системы, нельзя. Для.их
измерения надо тщательно разделить фазы, взвесить их порознь и измерить
плотность.
Используя матрицу, вырази^! через «pj константы равновесий и активности
простейших частиц (HgO) ==/, (/?) = /-, (Fe+3) = ;п, (Н+) = fi, (СП = г/
активности всех сложных частиц:
[РеС1„3~п] _ срз_„3„т^/", [НС1] = cp^a/i^, [HR+] = ср^а^Аг, ^ _
[fecl4 • 2R-] = <PiPi2P4/n^V. [QI == Ъ^l^2h2hhtnrY^
[HCI . R J = cpoa/ir^/, [Q (HaO)^] = сроа^а^рхаРз^Р^Д/^тгУ,
- q q q s si is
[H2O] = cpoXi/, [R] = cpoXg/", [HCI . R] = cpoXgo/ir^/,
[Q (H20)n] = 9ovia2pi2p3np4/i/"mry.
Из дальнейшего просмотра матрицы следует, что неизвестны и подлежат
измерению в первую очередь стехиометрический коэффициент п в формуле
Q(H20)„ и константы; pig, ^21^ Рз^, р4, ^i.. ^2» >^i» К h и х4. Необходимо
установить также рациональный подход к оценке <т для НС1 и других сильных
ки:лот. Поскольку при высокой / рассчитать cpi трудно, нyж^6 заранее измерить
параметры уравнений, связывающих с / логарифмы: «Ppecin~"» ^нс1» thr"^,
0
ТреС!.. 2R-T{hr+.fecl4 • 2R-}, ^HCl • r' cp{hr+.FeCU- 2R-} • (h,0)n» <Рн о'
0 si is *
tr» thci.r' t{hr+, Feci. . 2r-} (H,0)n.
При наличии всех данных решение задачи сводится к вычислению при
заданных начальных условиях активное гей /i, /, m, г, (/, а также дальнейшему
расчету всех равновесных концентраций. Здесь проще всего применить закон
сохранения количества вещества(нача;1ьного числа молей) Н2О, R,Fe3+, Q"",
Н-^: W = (0,588+49,96) Уо, Е = (10,45+ 1,03) Уо. М\ = ^> (ЗМ + К) v, = Y
и Куо = я.
Пользуясь матрицей, получаем систему уравнений:
W = (сроУ + ^oXiyl / + п.3123зп?4^1^2 (?оУ + hl^'mr^y'^; (5.4)
q q is is
Е = (срру + сроХгУ) г + cpioiy/ir + ^21^ ('fo^ + hry + 2^^^i^^^vmrY +
^^h2h^i^z^hntrY + Зр12рзпр4<^1<^2 + П'^Л /i/nmrV; (5.5)
142
Таблица 5.1
0Х
а:
о
X
X
FeCl4-2R—
x
Q(H20)n
о
о
О
О
О
О
О
О
О
О
О
1 '
О
О
О
О
О
О
О
О
о
о
о
о
о
1
о
о
о
о
о
о
о
о
о
о
о
I
о
о
о
о
о
о
о
о
о
1
о
о
о
о
о
о
о
о
о
о
1
о
о
о
о
о
о
о
о
о
о
о
о
1
о
о
о
о
о
о
о
о
о
о
о
о
о
1
о
о
о
о
о
о
о
о
о
о
о
1
FeCl
3—п
п
НС1
HR +
FeCl4 . 2R-
Q
HQ . R
Q(H20)„
H^O
s
R
la
HCl • R
Q(H20)„
hi
hn
1,48
—5
2,13
1,13
Q= {HR+, FeCU'- 2R-}
n=0
+ Pl2p3nP4^1^2 (^0^ + W) ^"'"^V; (5.6)
iS Q 4 -
n==0
Q
-\- ^nh2h^fnrY + 4cpoPi2p4^i^at;/zm/-y + 4р12рзпр4<^1^2 (?ot^ + ^h^WnirYl (5 7)
+ 9ohv) hry + ?t2p3rtP4<Ji^2 (?ot^ + ?o>4C') hl^'mrY' (5-^)
В системе уравнений (5.4) — (5.8) индексы при ср указывают для сокращения
записей только число зарядов, когорое несет частица. Следует помнить, однако,
что для каждой частицы Igf имеег индивидуальное значение. Система линейна
только относительно h и т. Это значит, что, даже располагая значениями всех
параметров, расчеты следует вести на ЭВМ. 'Расчеты упрощаются при наличии
результатов прямых измерений h и у, которые можно осуществить с помощью
рН- и рС1-метрических установок, / и г по измерениям их в газовой фазе.
Попытка расчета распределения ИОз^^-иона между водной и какой-либо
н '-'.одной фазой привела бы к подобным результатам.
5.5. Некоторые выводы, связанные.с расчетами
равновесий распределения
Прежде всего следует отметить значительные пробелы в
сведениях о параметрах, характеризующих процессы сольватации и
гидратации распределяющихся частиц, параметрах, входящих в
уравнения для расчета коэффициентов активности, и константах
распределения. Отсутствуют сведения и об очень важных при
учете синергезиса константах образования тройных комплексов
и константах распределения этих сложных частиц.
Не существует общепринятых и хорошо отработанных
методов измерения активностей растворителей, а без этих методов
нельзя вести измерения отсутствующих значений параметров и
проводить дополнительные измерения, ведущие к упрощению и
уточнению расчетов равновесий.
В примере 5.1 найдено, что в результате встряхивания с
водой раствора h в ССЦ, только 4,42-10-4 (10-2-7,567-10-з)-1 =
= 5,84 % иода оказалось в водном слое. В подобных случаях
может возникнуть вопрос, сколько же повторных экстракций
нужно сделать, чтобы количественно, скажем на 99,99 %,
извлечь экстрагируемое вещество.
В примере 5.2 установлено, что при однократном встряхивании
в бензол должно перейти 7,92-10"''^ моль, а в воде остаться
всего 6,83-10-6 моль пикриновой кислоты, т. е. меньше 1 %.
Такой результат на первый взгляд кажется хорошим. Если
учесть, однако, что наряду с пикриновой кислотой в водной
144
фазе должно остаться 1,6-Ю-з моль пикрат-иона, то станет
ясно, что и в этом и во многих других случаях возникнет
необходимость в расчетах условий, обеспечивающих заданную
полноту разделения.
Из раздела 5.4 видно, какую важную роль играют при
расчетах константы распределения Х,-, однако служить мерой
разделения они могут лишь в частных случаях. Для практической
оценки любой операции распределения важен коэффициент
распределения i-й частицы Д, равный отношению произведения
аналитической концентрации i-частицы в неводной фазе на объем
этой фазы к аналогичному произведению для водной фазы:,
00
^'■=V (5.9)
Так, в случае пикриновой кислоты
0 0
Г) _ t^^i^ 1,98. 10-3-Од
^на - ([НА] + [А-]) v - (8,54 . 10-« + 2,001 • Ю"») о,8
Подобным же образом коэффициент распределения для эфира в
примере 5.3 можно было бы рассчитать по формуле
00
([R] + [HC1.R] + 3[Q {Н^0)^])%
([RI + [HR+] + 2 [FeCl^ .2R ]+ 3 [Q] + [HCI-R] + 3 [Q (HgO)^]) v
где Q^{HR+ FeCU -гЕ-}.
/ 5.6. Теория экстракции для случая, когда
экстрагируемое вещество ни в первой, ни
во второй жидкой фазе никаким превращениям
не подвергается
Пусть объем первой фазы w == const и для экстрагирования
намечено использовать V второго растворителя так, что V =
п
= ^ t;i. Спрашивается, по какому закону должны изменяться
i=l
объемы VU чтобы после намеченных п операций экстрагирования
исходная конц-ентрация экстрагируемого вещества Са= А
изменилась до минимального значения.
Допустим, что растворители друг в друге практически не
растворяются,и вещество ни с одним из них не взаимодействует.
0'
Тогда следует учесть только распределение A<=iA. Если кон-
10 3-457 • 145
0 0 0
сганта распределение X, (А) = а, (А) = а, /1 = 1, а значит [А] =
= а и [А] = а, то а == Ха (5.10).
Первая операция распределения приводит к следующему
выражению закона сохранения количества вещества А в системе;
о 0
Следовательно, после достижения равновесия
lA].=(A),=a,^^.
При втором распределении подобным же образом
0
aiw = a2W + a2V2 == a2W + \v2a2 =^a2(w + Х^г);
^2 = ш + Хг;2 (ta;+Xt;i)(^ + Xt,2),
a после достижения равновесия в п-м распределении
о
AxsP const
an =
П (te^+X^i) П(ш + Ху^)
i=l i=l
(5.11)
(5.12)
(5.13)
(5.14)
(5.15)
Из уравнения (5.15) следует, что an-^-min, когда z=s П (^+
п
-fXuO-^max. Поскольку Vvx^V, можно принять, что
•t;n=:V^ (5.16)
Тогда
z =
/ n-l
йУ + Х У — V У1
п—1
П (tc^ + ^t;i)=/(t;,, t;n-i) (5.17)
i=l
И Принимает максимальное значение, когда одновременно
дг
О —-О -А- = о
(5.18)
-x"n(t£; + Xt;i)+X
at;,
Рассмотрим подробнее (dz/dvw) = 0:
г / п-1 Nik—1
L V
+ X^iVn (re; + Xt;i) = 0. (5.19)
/k+I
k-l n-l
После сокращения на 1 n{w + Ivi) П (гг^ + Щ ФО и замены
i=l l=k+l
n—1
V — ^ Vl на Vn находим, что оптимальный эффект получается
1=1
146
при условии vu = Vn = Vi = v (5.20), т. е. последовательном п-
кратном экстрагировании равными объемами экстрагента.
Тогда при фиксированном v = qw (5.21)
ап =
Aw""
(5.22)
(а^+Ь)" (1+Xq)" •
Если^ X и q известны, то для снижения А до значения а =
'=QA необходимо провести п повторных экстракций, причем
0 0 0
Пусть в t; = 0,l л исходного раствора U в CCU ci^ = y =
= 0,08 мoль•л-^ Сколько операций экстракции (п) надо
провести, используя для каждого распределения у = 0,5 л воды,
0
чтобы снизить Y в 10^ раз, если для равновесия
Ь (Н2О) 5=t I2 (CCI4) IgX = 1,932.
Здесь первой фазой является раствор в ССЦ, значит q = 5,
l^(qX) = — 1,233, 1 + qX = 1,0585, Q = Ю-з и по (5.23)
5.7. Теория экстракции для случая, когда
экстрагируемое вещество может взаимодействовать
с обоими растворителями, но не дает
многоядерных комплексов
В качестве несложного примера рассмотрим результаты
распределения НаА при п-кратной экстракции незаряженных частиц
НаА из водного раствора при фиксированном vh^o л. Будем
о п
считать си^А== Ау суммарный объем экстрагента V = V vi л и
допустим, что в неводной фазе находятся только мономерные
частицы НаЛ. Тогда приходится учитывать только протонизацию
А*--иона и распределение. НаА, и матрица равновесий системы
принимает вид:
А
н+
К
1 j
0 0
0
1
Н,А'-«
0
Н.А
"i
0 0
10*
147
После первой экстракции по закону сохранения начального
количества вещества НаА в системе
[На
0 / ^
+ У1Срн«АЬа/1*
Aw
Aw
\i=Q J /
-1
0
a, SI?H,A^i^^ + 4^HaA^a^
j=0
^ CL
(5.24)
•. (5.25)
J=0
Аналитическая концентрация НаА в водной фазе после первой
экстракции с учетом значения щ из (5.25) равна
Л, = ш X4>H,A«ift' % — = 5^^.- (5.26)
j=0
^ и
ТН^А^а^*
j=0 ^
где X'—фиксированная величина, если в системе поддерживаются
рН = const и / = const.
После введения второй порции vl л, несмешивающегося с
водой растворителя, встряхивания и достижения равновесия
/а' \ 0 / Ci' \
Л,ш = ^ [HjAi-«] + [НаА]2У2 == аз ш У TH.AajA^ + ^gXcaA ,
\J=0 /2 \ j=0 /
(5.27)
«2 = -5J+T;7(S ^H^^ajA^ ' (5.28)
и, учитывая уравнение (5.26),
An-
Aw""
i=l
(5.30)
Теперь из сопоставления уравнений (5.30) и (5.15) очевидно,
что и в случае взаимодействия экстрагируемой частицы с
растворителями равенство v\^=^Vn=^Vi =v (5.20) обеспечивает опти-
148
мальный результат экстракции, если взаимодействие не приводит
к образованию многоядерных комплексов.
Если многоядерность возникает, например, в неводной фазе
0 0
за счет равновесия рНаА (НаА)р с константой тс появляются
частицы'(НаА)р, то выражение (5.24) усл( живется:
а' 0 0
= Ш ;2 ?HjAC^jai/l* + t^lf H^Abaaift* + /7^1^(Н«А)р (kCauihy. (5.31)
j=0
В нем уже нельзя взять общим множителем аь и вопрос об
оптимальном подборе объемов экстрагента vi остается нерешенным.
5.8. Множитель №аА^а^* S^H:A^j^'^ при IVi
в уравнении (5.26), его роль и использование
Умножение числителя и знаменателя множителя
0 /а' ' \-1
(5.32)
на а дает выражение
0 „ 0 0
УНдА^а^^ - "^^«А [Н^А] ^ [Н^А]
1=0 ^ j=:0
ИЗ которого следует, что этот множитель почти точно равен доле
экстрагируемой частицы в водном растворе в тех условиях, при
которых происходит распределение. Если ' экстрагируемое
вещество не взаимодействует с растворителями, он равен единице,
В этом случае для расчетов используются формулы (5.22), (5.23).
При наличии такого взаимодействия множитель (5.32) обычно
меньше единицы, а X' соответственно меньше X.
В выражении (5.32) значение множителя зависит от рН.
В случае распределения комплексных незаряженных частиц это
значение может зависеть и от показателя адденда и рН. Это
дает возможность экспериментатору^ меняя рН или pAd, или оба
показателя одновременно, изменять коэффициент распределения
и величину Qn по своему усмотрению.
Так, в случае однопротонной кислоты НА при а' = а =^= U
умножая числитель и знаменатель выражения (5.32) на /а,
приводим его путем последовательных преобразований к виду:
^НА ?НА/А^^ ТИА 1 _
Уна Уна/а^Л + 1 Уна (^на/а^^)""^ + *
0
^НА 1 ^ , _ >
= анА ^ ;
Тна ioP"~^«^-^6mAfA ^1 - jO^H-ig о ^ J
(5.33)
149
Тогда для однопротонной кислоты НА
Qn
(ш»+Х1>Она)п
W + .
(5.34)
10PH-lR» + 1 j
Например, для пикриновой кислоты в системе бензол —вода
X = 232, !go = 0,38. Тогда при п = 4, ш = 0,5, v = 0,03 л и
разных рН найдем
о, = Л.- -
^* А ~ (0,5 + 232 . 0,03(10Р"-"-38+
X (10Р"-"-38 + I)-']-* = (1 + Ы)-*.
[-1 + 13,92 X
рН
-0,5
0
1
2
рН—0,38
jQpH-0.38
,0РН-0.38 ^ ,
а
1+и
Q4
—0,88
0,13
1,13
12.3
13,3
3,2- 10-5
-0,38
0,42
If
foie
7,35 . 10-5
0,62
4,17
5,17
-2,69
3.69
5,4 • 10-3
1,62
41.7
42.7
0,33
1,33
■0,32
Ясно, что рН можно использовать в качестве своеобразного
регулятора при распределении пикриновой и других кислот
между водной и неводной фазами. В частности, это относится
и к комплексным кислотам, например, НРеСЦ, особенности
распределения которой между эфироводной и водноэфирной фазами
рассматривались в примере 5.3. Рациональный выбор условий
распределения возможен во всех этих случаях лишь при наличии
достоверных знаний всех парамегров, необходимых для
предварительных расчетов.
5.9. Оптимизация распределения
Если путем предварительного исследования установлено, что
в системе, которая может содержать несколько
электронейтральных соединений данного элемента, например, MY^, HMY^+i, ...
. .., HpMY^+p, ..., Hn-jxAIYn, лучше всех экстрагируется
данным растворителем какая-то одна из них, то возникает вопрос
о мерах, ведущих к повышению равновесной концентрации этой
частицы в водной фазе.
Если такой частицей является MY^, то рН надо подобрать
гак, чтобы свести к минимуму ионизацию М(ОН2)ы*'-ионов. В то
же время нельзя сильно снижать рН, так как за счет протони-
':»ации анионных комплексов MYjJi^^p возможны нежелательные
сдвиги равновесий в сторону их образования.
150
Рассмотрим случай, когда с^^^^^^^ c^^=.Y м c^^^^^=^U,
считая, что М**"^ не координируют ионы и что в системе не
образуется заметных количеств многоядерных частиц.
В этом случае матрица равновесий системы MY —HY —
—ХУ-^НгО имеет вид:
Y
|хЛ1 +Н
м
Y
и
х+
y—
н+
К
0
1
1
0
0
0
п
0
-I
-к
0
j
0
0
0
1
он-
W
Ik
Pn
Значения щ позволяют рассчитать chy(1)= достаточную
для подавления ионизации М (OHg)^^ на первой ступени. Если
найденное значение Н не очень велико, а рН не очень низок,
то образованием W^TAY^^^ можно пренебречь и учитывать только
образование МУ1;"""-комплексов.
В этом случае
п=0
п=0 "
(5.35)
(5.36)
г^{т^)=^^ту^ (5.37)
и выход комплекса при фиксированном М зависит только от Y.
Дифференцируя (5.35)- и (5.37) по у, находим:
О = ^' ii УмУпРпУ" + m ^ П9му„М"-^, (5.38)
п=0 п=0
z' = pj, (m V + ту^-^). (5.39)
В экстремуме z' = О (5.40) уравнение (5.39) после
сокращения на ^^у^"^^ Ф О переходит в пСу + р-т = О (5.41), а
подстановка в (5.41) значения т! из (5.38) после обычных преобразований
приводит к уравнению
i;(n-[^)9MYnM". (5.42)
п=0
дающему значение у, при котором [МУ^х] достигает максимальной
величины.
151
Подстановка найденного значения у в уравнение (5.35)
позволяет найти т, отвечающее М, а последующая подстановка т
Vi у в (5.36) дает искомое значение = ^•
К сожалению, констант Oj в справочниках почти нет,
коэффициенты активности не измерялись, и возможность даже
предварительных, приближенных расчетов оказывается осуществимой
в очень редких случаях.
Если преимущественно экстрагирующейся частицей является
HuMYjx+u, где u<N —(X, или какой-нибудь тройной комплекс,
обеспечивающий синергетический эффект^, решение экстремальной
задачи в общем виде связано с громоздкими выкладками. Здесь
проще искать частные решения для конкретных случаев,
используя ЭВМ. К сожалению, как следует хотя бы из табл. 5.1,
параметров, необходимых для расчетов процессов распределения,
явад не хватает.
/ В качестве ^примера предварительной оптимизации состава
водной фазы рассмотрим экстракцию Hg^"*■-иoнa, если
преимущественно экстрагируются частицы HgCl2.
Пусть CHg(NOsb == Л1 = 0,1 и Chno,== Н. Будем искать Скс1= К,
при котором [HgCb]-> max. Поскольку сведений а^значениях ср
нет, будем считать <pi= 1.
Пользуясьматрицей (с. 151), вполне отвечающей данному случаю,
и значением pigi = 3,96, путем элементарного расчета находим
величину Н, достаточную для практически полного подавления
ионизации Hg(0H2)n^-H0Ha:
Hg^+ т=± HgOH+ + Н+ р^, = 3,96
с 0,1 — Н
[ ] 0,1 10-4 н+10-4
^^"'^ = 10-3,96^ Н 10-0.96 = 0,11.
Значений aj для HgCir и HgCl^"" в литературе нет. Принимаем
в связи с этим, что эти анионы не протонизуются заметным
образом при рН = 0,96, и используем для расчета г/ —(С1-),
обеспечивающей максимальный выход HgCb, уравнение (5.42), в
котором N = 4, (X = 2 и ^MYn== ^•
2 (n-2)P„i/n = 0,
n»=0
И, соответственно, lgP4= 14,92, lgp3= 13,92, lgp2== 12,78 и
IgPi =5,27. Решая полученное уравнение
у^ + 0,05^3 _ 1,12 . 10-iOr/— 1,208 • Ш^^^ _ о
по способу Хорнера, находим, что у, при котором^HgCb]
достигает максимального значения, равен 5,2 • 1G~^ моль-л-*.
Расчет m, [HgCl^"""] и ^/ дан в табл. 5.2.
152
у
P.P-
m
J 0—4.2840
10-2.216
6,08 • 10-^
101.066
11,64
104.212
16294
,100.986
9,68
10°
1.00
104.2127
16316
10-5,2127
[HgCl2-]
[HgCljJ
[HgCl^}
[HgCl+]
Pi my
[Hg2+]
m
2
10-7,4287
3,7 . 10-8
10-4,2061
6,22 . 10-5
10-1,0007
9,985. 10-2
10—4,2267
5,93 • 10-5
20—5,2127
6,1 .10-^
9,998 . 10-2
ЗРзУ^
hy ■
—
Y
5,2 . 10-^
JO—1,6139
2,43 • 10-2
,0l.543I
34,92
.J04,5130
32580
100.986
9,68
—
104.5135
32624,6
10-0,6992
0,1999
0,2
. Из табл. 5.2 следует, что при вычисленном оптимальном
значении Cj^^^ = 0,2 моль - л
1
99,85 % Hg^"^-HOHa будет находиться
в водной^ фазе в виде HgCb. Результат следует считать
приближенным, так как не были учтены и возможность
протонизации HgCl?+p ионов.
5.10. Экстракционные системы. Соэкстракция
Как видно из последней главы [30], где описаны избранные
методы экстракции 66 элементов, в большинстве случаев
экстрагируются комплексные соединения с 30 органическими и 12
неорганическими лигандами. Среди указанных [30] 26 различных
неводных растворителей чаще других применяются бензол,
пиридин (Ру), хлороформ, четыреххлористый углерод, этилацетат
(Et-Ac) и диэтиловый эфир >(Et20). В табл. 5.3 приводятся
сведения о 23-х комплексообразующих реагентах: и 63 элементах,
для практически количественной экстракции которых из водной
фазы можно использовать указанные растворители.
Таблица 5.3
Реагент
СвНв
Ру
CHCla
ecu
Et-Ac
Ацетилаце-
тон
1
AI, Be, Со,
Cr, Fe, Ga,
In, Mo, V,
Zr
Бензоат-ион
AI, Fe,
Ga, In, Sc
Вг
In
Бутират-ион
Си 1 Be
НА
Cr
Дибензоил-
метан
'u
Диметил-
глиоксим
Ni, Pd
Дитизон
Bi,Cd, In,
Pb
Ag, Co,
Cu, Hg,
Zn
Диэтил-
дитиокарба-
минат-ион
-
As
Ag, Bi, Cd,
Co, Cu, Hg,
In, Pb, Pd,
Sb, Se, Те,
TI, Zn
154
продолжение табл. 5.3
Реагент
QH.
Py
CHCl,
CCI4
Et - Ac
I
Sb
In, Hg, Sn
Купферон
Cu, Fe, Sb
Fe
Fe, и
Неокуп рои н
' Cu
и
Am, Np
1-нитрозо-
2-нафтол
Co
8-Оксихино-
лин
Al, Ca, Co,
Cu„Fe, Mn,
Mo, Nb, Ni,
Sn, Th, Ti,
U, V, W
Os
Mn, Re,
Rh, Tc
SCN-
Cd
Hf, Nb,
Sc, W
Теноилтри-
фторацетон
Ас, Am,
Be, Cm, Hf,
Np, Pa, Po,
Sc, Rh, Th,
Tl
Тетрафенил-
арсоний
хлорид
B, Co, Mn,
Re, Tc
Тетрафенил-
стибоний
сульфат
F
Тио-
карбанилид
Os
СГ
As
Ge Sb
Ga, Sc, Tl
1
P
Sn
Эфедрин
гидрохлорид
Os
155
Из табл. 5.3 следует, что лишь немногие элементы могут
экстрагироваться без введения комплексообразователей. Так
экстрагируется Os в виде Os04 в CCI4 и Р в виде Н3РО4 в Et20. Пиридин
извлекает из водных растворов металлы (М): Мп, Re и Тс в виде
пиридиниевых солей РуН-М04. В остальных случаях между
фазами распределяются комплексные соединения. В прописях,
приведенных в [30], даны подробные указания относительно
условий проведения . экстракционных разделений. В [16, гл. 6|
приведены избранные методики анализа, в которых групповое
экстракционное концентрирование элементов сочетается с их
различными инструментальными методами определения в экстрактах.
В соответствии с законом распределения Нернста каждое из
экстрагируемых веществ должно распределяться соответственно
присущей ему константе распределения независимо от
присутствия других веществ. Однако в некоторых случаях эта
закономерность не соблюдается. Указанное явление, называемое соэкст-
ракцией [14, с. 177], чаще всего объясняется взаимодействием
экстрагируемых веществ друг с другом. Например, соэкстракция
кальция и скандия, по-видимому, связана с распределением
между фазами соединения Са (8004)2 [14, с 179]. К сожалению,
возникающие при соэкстракции дополнительные равновесия почти
не изучены, что делает невозможным их количественный учет.
5.11. Выводы и задачи
Как показано ранее, выделение любой твердбй фазы из гомогенной жидкой
системы всегда сопровождается потерями за счет растворимости и загрязнением,
связанным с различными видами соосаждения. Оказалось, что обычно
рекомендуемые приемы очистки поверхности твердой фазы хороших результатов дать
не могут, так как поверхность раздела между фазами очень велика и заметно
сократиться не может.
В случае экстрагирования межфазная поверхность в сотни раз меньше,
однако возможны загрязнения за счет соэкстракции. К сожалению, изучению
- процессов распределения уделялось мало внимания. Не хватает сведений
о составе экстрагируемых частиц, почти нет достоверных значений констант
распределения, совсем нет параметров для расчета коэффициентов активности,
внушают большие сомнения значения констант комплексообразования,
практически не изучалась протонизация комплексных бренстедовских оснований.
В результате теряется возможность теоретического подхода к повышению
селективности экстракционных равновесий.
По-видимому, уже теперь метод распределения должен гораздо шире
использоваться при наиболее точных химических измерениях в простейших
системах, где соэкстракция невозможна. Одновременно следует- выделить
системы, планомерное исследование которых желательно начать в первую очередь,
и обеспечить соответствующие измерения полным набором параметров,
необходимых для рациональной разработки аналитических прописей, расчета
возможных погрешностей и путей их устранения.
156
6. БУФЕРНЫЕ СИСТЕМЫ
6.1. Предварительные сведения
Буферностью или буферным действием следует считать
способность любой замкнутой равновесной системы противодействовать
нарушению установившегося в ней равновесия при любых
внешних воздействиях.
Такими внешними воздействиями могут быть: изменение
температуры системы, давления, под которым она находится,
разбавление всей системы, изменение аналитических концентраций
одного или нескольких компонентов системы, введение в нее
новых веществ.
Мерой буферности может служить величина, на которую
должен измениться воздействующий фактор, чтобы вызвать
изменение какого-либо интенсивного свойства, связанного с
равновесным состоянием системы, на определенную величину. Обычно
в качестве свойства выбирают рН, а мерой буферности служит
буферная емкость
величина, обратная скорости изменения рН при вводе в систему
сильного основания или выводе из нее сильной кислоты. Чем
больше р, т. е. чем больше нужно изменить аналитические
концентрации ХОН или HY для изменения рН на заданную
величину,, тем выше буферная емкость системы и наоборот*.
Для открытых систем типа живых организмов падение
буферности приводит к потере жизнеспособности, болезням и гибели.
Контролировать нарушение буферности в этом случае можно
путем измерения температуры организма, показателей разл ичных
ионов (рИЬн) в плазме и соках и т. п. Не даром классическое
определение буферной емкости возникло в биологии [74], и
можно ожидать, что буферная емкость, определяющая
жизнеспособность, станет одной из важнейших величин биологической
метрологии.
Система может проявлять буферность лишь при наличии в
ней резерва, за счет которого может восстанавливаться в той
или иной мере нарушенное равновесие. Таким резервом, как
♦ Иногда буферной емкостью называют максимальное изменение
аналитической концентрации ХОН или НУ, при котором изменением рН еще
можно пренебречь.
157
показано в [20], может быть: слабая кислота, поставляющая
Н3О+-ИОНЫ по мере их расходования; твердый гидроксид металла
или соль слабой кислоты, снижающие активность НзО"^-ионов,
вводимых в систему; комплексное соединение, реакция
образование которого пригодна для регулировки активности
координирующей группы и лиганда; дополнительная жидкая фаза,
содержащая резервное количество вещества, обеспечивающего
буферность водного раствора; кристаллосольват, поддерживающий
постоянство парциального давления пара соответствующего
растворителя и т. п. Буферное действие таких систем ограничено
запасом соответствующего вещества-регулятора ь системе.
В открытых системах энергия и вещества, необходимые для
сохранения равновесия, восполняются .за счет обмена с
окружающей средой. Буферное действие таких систем ограничивается
только наличием этой среды и возможностью ее использования.
6.2. Буферность гомогенных кислотно-основных систем
6.2.1. БУФЕРНОСТЬ СИСТЕМЫ: СИЛЬНАЯ КИСЛОТА И ЕЕ СОЛЬ
Допустим, что объем системы v фиксирован, что Chy = К =
= с(1 — а) и Cj^y = ^ = ас или с^^ = 7 = с, а c^qj^ == X = ас,
как при титровании HY раствором ХОН. Тогда в общем случае
[Y""] == Су [Х"^] == ас и О < а < 1. При а = О в растворе нет XY,
а chy = У * При а = 1 в растворе нет HY, и с^у = с. При О <
< а < 1 по закону электронейтральности
Н+]-[ОН-~] + [Х+] —[Y~] ^^н'^ —^оншЛ"^ + ас—с = Аг+
.+ ас—с = 0. (6.2)
Здес^ь для разности [Н*^] —[0Н~], входящей в уравнения
электронейтральности для всех систем, введено обозначение, данное
в [6]: ^н^~ ^он ^'^""^ =(6.3). Тогда из (6.2) получаем ас =
= с^п (6.4).
d Л!'
При выводе формулы для расчета р = учтем, что по
определению рН = — \gh, т. е. Л = 10""^", и, следовательно,
^ = — 2,303 10-Р" == — 2,303Л. (6.5)
Кроме того, в дальнейшем нам будет полезно выражение для
производной Получим его, дифференцируя (6.3)
З^Л = ?н + f он = (Тн'» + ?он (6.6)
158
Согласно [6] введем обозначение + ^он ^'^""^^ (^•'^)» ^^^^^
(6.6) можно переписать в виде
^ = Л-п2. -• (6.8)
Дифференцируя (6.4) по рН как сложную функцию, с учетом
(6.5), получаем
аЖ~"ТГ Т?Н==-'' Аг2(-2,303Л).
Поскольку ас = X, последнее выражение дает формулу для
расчета р.-
Из (6.9) следует, что при а=1, т. е. при Cj^y = О и с^^ =с
буферная емкость р = 2,303 (Ю-^ + ю-^) 4/506 • 10~7 моль,
а при а = О, когда [Н+] = с > [ОН-], буферная емкость р =
= 2,303 с, т. е. определяется с^у = Y.
6.2.2. БУФЕРНОСТЬ СИСТЕМЫ: СИЛЬНОЕ ОСНОВАНИЕ И ЕГО СОЛЬ
Уравнение электронейтральности в этом случае имеет вид
[Н+] — [ОН-] + [Х+] — [Y-] = 0. При начальных условиях с^^ =
— 7 = ас и с^он = X =={\ — а)с это уравнение можно
переписать в виде
<РнЛ — cpoHtC'A-i + ас + (I —о()с = п + с—ас =0,
откуда ас = Аг+с (6.10). Дифференцирование ас по рН с учетом
(6.5) и (6.8) дает
. ^ = (—2,303/1) = — 2,303п2
и
р = - ^ = - = 2,303^2 = 2,303 ([Н+] + [ОН-]), (6.11)
т. е. буферная емкость определяется той же суммой равновесных
концентраций ионов Н+ и ОЙ-, которая определяет Р для
системы HY + XY (раздел 6.2.1).
6.2.3. ЭКСТРЕМАЛЬНОЕ ЗНАЧЕНИЕ Р НА КРИВОЙ ф. рН) ДЛЯ
СИСТЕМ ИУ —XY И XOH — XY
В предыдущих разделах было установлено, что буферная
емкость таких систем описывается одним и тем же уравнением
(6.9) или (6.11). Для определения координат точки экстремума
159
на кривой (|3, рН) приравняем нулю первую производную р по
рН, полученную дифференцированием (6.11). Для этого предва-
рительно получим выражение -jj из (6.7):
^ == ?н — ?он г<уЛ-2 == /1-1 (срнй _ срон шЛ-1) = Л-^л.'(6.12)
Теперь дифференцируем (6.11) с учетом (6.5) и (6.12):
- — 2,3032л.
О 0,2 0,4 0.6 0,3 (О 0,8
Рис. 6.1
Условием экстремума будет п =^ ^Hh — (foHwh-^ = О, т. е.
h = Vfn^OHW ^ 10-7.
Подстановка значения п =^0 в (6.4) и (6.10) дает а = 1. Это
значит, что для обеих систем HY + XY и ХОН + XY
минимальное значение буферной емкости^ соответствует раствору
нейтральной соли и равно р = 2,303 • 2 . 10-^ = 4,606 • 10-^ моль,
поскольку [Н+] ;:;:^[0Н~] 10-7 молЬ'Л""^ Других,
экстремальных точек на кривой нет.
6.2.4. КРИВЫЕ (Igp, а) ДЛЯ СИСТЕМ HY — XY И ХОН —XY
На рис. 6.1 приведены графики (lg|3, а) для систем HY — XY
и ХОН — XY при с= 1 моль • л-^ и с = lO-^ моль • л^К Для
расчета отдельных точек при заданных с и а находим по (6.4)
160
или (6.10) n, затем h, п'^ и по формуле (6.11)— значение р.
Для а = 1 можно воспользоваться значением р, полученным в
предыдущем разделе. Графики для систем HY — XY и ХОН —
XY при одном и том же с симметричны. Уменьшение с
приводит к снижению буферной емкости. По мере возрастания а,
т.е. содержания соли XY в системе, IgP монотонно убывает.
Это значит, что для снижения активности Н3О+ на одну и ту же
величину нужно добавить в систему ХОН при высоких а меньше,
чем при низких.
6.2.5. БУФЕРНОСТЬ СИСТЕМЫ: СЛАБАЯ КИСЛОТА И ЕЕ СОЛЬ
Выражение закона электронейтральности для такой системы
имеет вид [Н+] — [ОН-] + [Х+] — [А-] = 0.'
При начальных условиях с^д = Л" = ас, Сна =; Л = (1 — а) с
и фиксированном значении с общая концентрация Сд частиц,
содержащих А, равна с, а [Л*+] = ас. По закону сохранения
начальной концентрации с = а (^на^Л + срд), где а — активность
А-, а—константа протонизации А-. Доли суммарной
концентрации с частиц А- и НА выражаются дробями 1/(<рна/а<зЙ+1)
и срнА/А<'й/(?нА/АаЛ + 1) соответственно.
Если ввести обозначение [4] л = рН — Iga — IgcpHA^A (6.13),
то выражения для долей частиц А- и НА можно переписать
в виде
1/(д^)=: 1/(10-^+ 1) и у{-х)^ 1/(10^+1). (6.14)
Как показано в [4], эти функции оказываются полезными при
исследовании буферных свойств таких систем. Заменив [А-] на
су{х) и [Х+] на ас, перепишем выражение закона
электронейтральности п + ас — су (х) = 0. Отсюда ас = су (х) — п (6.15).
Чтобы вывести формулу для расчета буферной емкости р =
= = ЧЩ*' необходимо продифференцировать это
выражение по рН. Предварительно найдем
^У{х) ' d
(6.16)
Теперь с учетом этого выражения и формул (6.5) и (6.8)
получим выражение, определяющее р
Р = ^ [<?i/ W - «J • = 2,303 [су (х) у(-х) + п% (6.17)
Из (6.17) следует, что р — аддитивная величина, включающая
буферную емкость чистого растворителя и растворов сильных
И 3-457 1 61
кислот, оснований и их солей (л^) и буферную емкость раствора
слабой кислоты и ее соли. Очевидно, что буферная емкость
системы, включающей п слабых кислот с константами протониза-
ции аниона выразится расчетным уравнением
п
Р = 2,303
i = l
6.2.6. ЭКСТРЕМАЛЬНЫЕ ЗНАЧЕНИЯ р ДЛЯ СИСТЕМЫ НА —ХА
Экстремальным значениям р будут соответствовать Л, при
которых j-^ = 0. Используя уравнение (6.16) и полученное
аналогичным путем ^ ^^'^ = h-^y (х) •у (— л:), а также (6.12),
найдем выражение для производной Р по Л, дифференцируя (6.17):
i| = 2,303/1-1 {с [у {xWy {-х)-с [у (- х)]^у (х) + п}: •
После приравнивания нулю и сокращения на 2,303 h"^ Ф О
уравнение для расчета Л, отвечающих экстремальным
значениям р, принимает вид п — с - у (х) • у {—х)[у (— х) — у {х)] = 0.
Подстановка в него у{х) и у{—х) после алгебраических
преобразований приводит к' уравнению
+ З/на^аКА^ — {ЫЫа9аКс — З^иаЛК^ + /нсронш) +
+ ihfWAK'c + ^иаЛК' - 3fH90HfHA9AKw} А2 - '
— З/нСрОн/нАТа^^ Wh —fn^OHfuA^AK^W = О,
где/с = 0-1. в нем три перемены знака. Это дает право
рассчитывать на получение трех положительных значений Л,
которые можно вычислить на ЭВМ после подстановки значений
коэффициентов активностей и /С. К сожалению, в большинстве
случаев <р| неизвестны, поэтому интересны ;сотя бы приближенные
расчеты, проведенные для 9i=l. Такие расчеты показывают,
что функция, описывающая зависимость р от А (или а), имеет
два минимума —при а = 0 (раствор кислоты НА) и a='l
(раствор соли ХА) — и один максимум при промежуточном
значении а. Оценим h в точках экстремума.
Для раствора слабой кислоты (а = 0), описываемого
равновесием НА ^ Н+ + А-, /С, h оценивается равенством h = У'Кс,
справедливым при А<с. Так, при /С = 10-^ и с=1 моль-л-*
А= 10-^.5 моль-л-!.
Равновесие в растворе соли ХА (а = 1) можно представить
в виде
А- + Н+ ^ НА, а
Н20^Н+ + 0Н-, W
А-+Н20ч:^НА +0Н-, aW
162 '
Для него справедливо h = YKw/c, если h<^K. Для прежних
значений= 10-^ис= 1 моль-л-* получаем Л = 10-^'^ моль-л-Ч
Прежде чем приступить к поиску Л, соответствующего
максимуму р, заметим, что буферная емкость должна возрастать с
ростом обоих слагаемых, входящих в (6.17).
Продифференцировав по h функцию 2 = у{х) ' у{—х) и приравняв нулю
производную, получим условие максимума для. первого слагаемого:
срна/аоЛ=1; при максимуме буферной емкости h будет
выражаться равенством h = [на'РаК тем точнее, чем строже
выполняется неравенство <^с ' у{х) ' у{—х). Для /С =10-5 ц с =
= 1 моль • л-1 10-5 моль • л~Ч
6.2.7. КРИВЫЕ (Ig а) ДЛЯ СИСТЕМ HY - XY И НА —ХА,
ОТНОСЯЩИЕСЯ к ОДНОЙ ОБЛАСТИ рН
Как показано в предыдущем разделе, для системы НА--ХА
со значением /(=10-^ и с=1 моль • л~^ максимум буферной
емкости соответствует рН = 5. Для создания такого же рН в
системе HY —XY надо использова- ь сну == Ю-^ моль • л~^ Для
таких систем (НА — ХА с /С = 10- , с = 1 моль.л~^ и HY — XY
с с = 10~5 моль-л"-^) на рис. 6.2
приведены кривые (IgP, а).
В основу расчета точек кривых
для системы НА — ХА было
положено вычисление значений h при
заданных а с помощью уравнения
(6.15), а затем «2, С'у{х) -у{—х)
и р. Результаты приведены в табл.
6.1. Отметим, что значения 2.303X
^У {х)у (— х) заметно отличаются от р
лишь при малых а. Значит, для
буферных систем НА — ХА при р/С — 5
не-
0.2 0.4 0.0 0.8
Рис. 6.2
вклад в значение р за счет
значителен.
Из рис. 6.2 видно, что значение р для системы НА — ХА
близко к максимальному в широком интервале а — от 0,2 до 0,8.
Сопоставление кривых (Igp, а) показывает, что буферная
емкость системы HY — XY на 4—5 порядков ниже буферной
емкости системы НА —ХА. Большая буферная емкость
последней системы позволяет поддерживать значение рН вблизи
заданной величины при введении в систему относительно больших
количеств щелочи и кислоты. Так, при введении в систему
НА — ХА с с = 1 мoль•л-"^ а = 0,5, для которой рН = 5, 0,1 моль
сильной кислоты получается система с а 0,4 и рН 4,82, как
163
видно из табл. 6.1. Введение же в нее 0,1 моль щелочи
приводит а;^0,6 и рН;5^5,18, т.е. отклонения рН от заданного
невелики. Такие же добавки сильной кислоты и щелочи к системе
HY —XY с рН = 5 (С= 10-5 моль.л-1) приведут к рН;^1 и
рН;^13 соответственно, т.е. сдвигу заданного значения рН на
несколько порядков.
Таблица 6.1
а
рН
2,Шу (x) X
X у{—х)
Р
а
рН
2,303// (а;) X
X у{-х)
Р
0
0,05
0,1
0,2
0,3
0,4
0,5
2,60
3,72
4,04
4,40
4,63
4,82
5,00
0,0091
0,1095 •
0,2072
0,3683
0,4824
0,5513
0,5756
0,0149
0,1139
0,2093
0,3694
0,4830
0,5517
0,5758
0,6
0.7
0,8
0,9
0,95
1,0
5,18
5,37
5,60
5,96
6,28
9,40
0,5513
0,4824
0,3683
0,2070
0,1093
0,0001
0,5513
0,4824
0,3683
0,2070
0,1093
0,00016
Высокая буферная емкость системы HY — XY, очевидная из
рис. 6.1, объясняется очень большой концентрацией ионов Н3О+,
испытывающей лишь незначительные изменения при небольших
добавках HY и ХОН.
6.3. Ионобуферные гомогенные системы
Свойством буферности обладают и системы, включающие
комплексное соединение и продукты его диссоциации. Такие системы
можно назвать металлобуферными. В общем случае, если система
способна поддерживать постоянной концентрацию какого-либо
идна, о ней говорят как об ионобуферной системе.
В связи с современным распространением ионоселективных
электродов следует ожидать широкого признания буферных
свойств, характеризующих устойчивость концентраций металлоинов
и лигандов. Поэтому возникла необходимость такого обобщения
понятия буферной емкости, чтобы оно охватывало взаимосвязь
между изменением показателя активности любой частицы системы
и изменением ее начального состава.
Подход к расчетам такой буферной емкости проще всего
рассмотреть на примерах. /
Пример 6.1. Оценка буферной емкости р
'dPBa
для системы, в которой
^ВаССЮ.ь ^ М =-0,5 моль - л ^ и с^^р^^ принимает значения Л, равные 0,5; 0,6;
0,7; 0,8; 0,9; 1,0 моль . л""'.
В^связи с отсутствием необходимых данных о коэффициентах активности
ионов примем, что cpj = Ь
164
Ион Ва2+ с ацетат-ионом образует комплекс ВаАс"^ (константа образования
р = 2,57). По -Закону сохранения начальной концентрации
М = [Ва2+] + [ВаАс+]
А = [Ас-] + [ВаАс+] + [НАс].
Оценим величину последнего слагаемого, учитывая, равновесия
Ас- + Н+ 7^ НАс, 10*'''*
:^н+ + он-, 10-^*
Ас- + Н20^НАс + 0Н-, 10-^'2*
с 1 — О О
[ ] 1—0) — О) (О
Из выражения закона действующих масс a)2(l —(о)—^ = 10"^'^^ следует, что
[НАс] ^ 10"~*'^ моль • л—^ Более точное значение [НАс], полученное с учетом
образования ВаАс+, будет еще меньше, так как общая концентрация протони
зующегося иона Ас— станет меньше 1 моль . л—^ Тогда, пренебрегая
последними слагаемыми в выражениях закона сохранения начальной концентрации,
получим М = m + рта (6,18), Л = а-f рта (6.19). Исключая а, получаем
уравнение
рта+т[р(Л+ 1]~.М=0, (6.20)
а из него расчетную формулу, для т:
т = ^^Г' (е.2,)
Л — + р~1 + у {А — М + р-^^ + 4Мр-1
Для вывода расчетной формулы буфеоной емкости продифференцируем (6.20)
как неявную функцию Л по т:
(!Л 2рт + Р(Л--Л^)+ 1 2т+ Л —Л^ + р-^
dm mp tn
dm
Аналогично (6.5) -^j^ = — 2,303 m, поэтому
P = d?^ = • d£ = 2.303 (2m+A-M + Г'). (6.22)
Результаты расчетов p^^ = — Igm и P при М = 0,аи /С""^ =0,389 приведены,
ниже.
Л, моль . л-^ 0,5 0,6 0,7 0,8 0,9 1,0
р^^ 0,54 0,59 0,63 0,67 0,70 0,74
р 2,22 2,31 2,43 2,57 2,74 2,89
Ш
Пример 6.2. Оценка буферной емкости р = для систем, в которых
^ХС1 = ^= 0,2МОЛЬ.Л-^ и ^HgCClO.b ^ =0,15МОЛЬ.Л-^ или Ccd(Cl04)3 = =
= 0,15 моль . л—^
Буферная емкость относительно СГ" для этих систем создается благодаря '
способности Cd^+ и Hg^"^ образовывать хлорокомплексы. Существование
равновесия между ионами металлов и их хлорокомплексами позволяет поддерживать
165
"равновесную концентрацию CI приблизительно постоянной при внешних
воздействиях на систему. Если таким воздействием будет изменение начальной
концентрации иона металла, то буферной емкостью можно считать р = -j^.
Логарифмы общих констант комплексообразования р„ для хлорокомплексов
Cd^+ и Hg^'^ имеют следующие значения:
12 3 4
HgCl2-" 5 27 12,78 13,92 14,92
CdCl^-" 1,95 2,49 2,34 1,64
Для обеих систем при cpj = 1 законы сохранения начальных концентраций
в общем виде записываются как *
М^т ?у (6.23), К = ^ + m ^ пру. (6.24)
п=0 п==0
Образование гидроксокомплексов Cd^'^ и Hg^"^ не учитываем, так как оценочные
расчеты для уравнений
Hg2+ + Н2О HgOH+ + Н+ 10-3.96
Cd2+ + Н2ОCdOH+ 4- Н+ 10"^'^^
показывают, что [HgOH+] < 4 • 10"^ моль . л^^, а [CdOH+] < Ю-^моль х
X л'"^ Подстановка т из (6.23) в (6.24) дает уравнение
Y = y+M"^ .
которое удобнее переписать в виде
4 4
S Рп^""^^ + 2 ("^ - ^) Рп^" = О' (6.25)
П==0 п=0
Дифференцируя (6.25) как неявную функцию М от у, получаем
^ (п + 1) p„t/" + ^ п (пМ - К)
дМ п=о п=о
dy - 4
п=0
ду
С учетом соотношения д^ = —2,3031/ легко получить выражение для
буферам ой емкости
2,303
4 4 (6.26)
n=0 n=0
n=0
Чтобы рассчитать p, необходимо знать у = [С1~] — корень уравнения (6:25).
После подстановки в него М =0,15 моль • л""^ Y = 0,2моль • л""^ и конкрет-
166
ных значений р^^, а также обычных преобразований, получим для раствора
соли Hg2+«/5 4- 0,5001 И+ 3,224. ?.0~2|/^ + 7,224 . Ю-У—1,119. 10-^^ —
—2,404 . 10"*-^^ = О (6.27), и для раствора соли Cd^^ / + 5,410//* + 8,331^/3 +
+ 2,750^/2 — 0,7917^ — 4,581 • lO^^ = О (6.28).
Можно ожидать, что значение [С1~"] для раствора соли Hg^'^ невелико,
так как константы образования хлорокомплексов значительны. Предположив,
что 0,5001 моль . л""^ преобразуем уравнение (6.27) в квадратное —
— 1,545 • 10~"^у — 2,323 • 10""^^ = О, из которого находим у = 5,837' 10""'^ мольХ '
Хл""^ рС1 = 6,2338. Расчет по (6.26) дает р = ^^"^ = 0,22 моЯь.
dpCl
Для раствора Cd (С104)2 сделать предположение < 5,41 моль-л""^ рискован--
но, так как константы образования хлорокомплексов в этом случае не так велики.
Нужно находить корень полного уравнения (6.28). Это можно сделать
приближенными методами, например, по схеме Хорнера или по формуле Ньютона.
Получим 1/= 0,05218 моль . л"" ^ рС1 = 1,28. Расчет р по (6.26) дает то же
dC
значение р = = 0,22 моль, что и для системы Hg (С104)2— XCI.
dpCl
Сопоставление результатов расчета для двух систем показывает, что при
одинаковой буферной емкости с помощью системы Hg^^—*С\~~ можно создать
[С1""] на 5 порядков ниже, чем с помощью хистемы Cd^^ —CI"". Поскольку
pCl около 1—3 можно обеспечить и с помощью растворов HCI и ХС1, можно
утверждать, что системы с IgPn около 2—3, как Cd^"^ — С1"", применения не.
найдут. Система же Hg^"^ — CI"" и ей подобные, поддерживающие рС1 > 4,
могут оказаться полезными во многих случаях. Для расчета рС1 подобных
систем необходим, конечно, полный набор параметров равновесий ионизации
ионов М (OHg)^"^, образования комплексов MCIjj—" и протонизации анионов
MClJljIp. Полного набора таких параметров, т. е. констант равновесий и
коэффициентов активности, нет пока ни для одной системы. В некоторых случаях,
в частности для С1~", рИон той или иной буферной системы может быть
измерен с помощью соответствующей прокалиброванной рИон-метрической
установки. Такая буферная система может рассматриваться и использоваться
в дальнейшем как рабочая мера данного рИон.
6.4. Ионобуферные гетерогенные системы
В гетерогенных ионобуферных системах запас вещества,
поддерживающего стабильность показателя активности
соответствующего иона в системе, сосредоточивается в твердой или второй
жидкой фазе, соприкасающейся с основным раствором.
Рассмотрим примеры подхода к подобным системам.
Пример 6.3. Оценка буферной емкости систем, насыщенных Са (0Н)2
По данным [2] в качестве дополнительного стандарта используют
насыщенный при 25 °С и отфильтрованный раствор Са (0Н)2 с ра^ = 12,45.
Оценим способность гетерогенной системы, содержащей избыток Са(0Н)2.
поддерживать постоянным значение рН. Для нее при ср^ = 1.
Са (0Н)2 (т) :г Са2+ + 20Н-, Ig Р = —5,43
[ ] - т 2т
167
4т /0-5.43 ^ ^ рса = 2,01
рОН = —Ig (2m) = 1,71 и рН = 12,29.
Если в 1 л такой системы ввести Y моль НС1, т. е. создать c^^j = К, то
при наличии твердой фазы т = Pto—2 (6.29).
Уравнение закона электронейтральности [Н"^! — [ОН"] + 2 [Са^"^] — [Cl""] =
= О с учетом того, что в насыщенном относительно Са (ОН)., растворе [Н"^] <
< [ОН—], можно переписать в виде —а) + 2т-—К=0 (6.30), а с учетом
(6.29) —в виде (0^+Уа)2 —2Р = 0 (6.31). Расчет равновесных концентраций
по (6.31) и (6.29) при F=0,1 моль . л-^ дает рОН = 2,08, рН = 11,92.
рСа=1,2б.
Если в 1 л насыщенного раствора Са (0Н)2 ввести х моль ХОН, то при
наличии твердой фазы т{Х + 2т)^ = Р.
Например, при X = 0,1 моль • л"^ рСа = 3,43, рОН = 1,00, рН = 13,00.
Результаты расчетов приведены ниже, из которых следует, что, несмогря на
присутствие твердой фазы, насыщенный раствор Са (OHJg не обладает
надежными буферными свойствами.
Система
Са(ОНЬ
Ca(OHb+HY(0.1 моль.л-1)
Са(ОНЫ-ХОН (0,1 моль.л-1)
рН
12,29
11,92
13,00
: 2,303 (APw-^^ + wh-^) = 2,303 (6Pw-^h^ - У).
Как видно из уравнения (6.31), величиной, с помощью которой можно
повлиять на (о (и рН), является значение F, т. е. концентрация HY, введенной
в систему. Это наводит на мысль выбрать в качестве исходной буферную систему,
в которой при избытке твердой Са(0Н)2 уже находится HY с концентрацией
CpjY = У®. Будем в такую систему вводить дополнительно HY так, чтобы
общая концентрация НУ становилась У = (1 + а) или ХОН так, чтобы
\^ = (1 — а) уо. Тогда расчет ы при разных а должен проводиться по уравнению
(6.31) с соответственно измененным значением У.
Уравнение для расчета буферной емкости р получается из уравнения (6.30)
после замены в нем (о на суД—^ и mm Pw'^^h^: —шЛ-^+2Рш—V—У==о (6.32.
Дифференцирование Y из (6.32) по рН с учетом (6.6) дает
dY
P = ~t/pH
В табл. 6.2 приведены результаты расчета значений рН и р, соответствующих
введению в начальную буферную систему с1 моль * л-1 различных С^он =
= Да; и Сну ^Y,
Из табл. 6.2 следует, что исходная система с 1= моль . л""^ является
надежной буферной системой, так как ее рН меняется несущественно. Даже при
добавлении 0,8 моль . л""' сильной кислоты или щелочи нельзя получить таких
изменений рН, как для насыщенного раствора Са (0Н)2 при добавлении
0,1 моль . л-^ НУ или ХОН.
Пример 6.4. Оценка буферной емкости системы бензол — вода, содержащей
пикриновую кислоту (НА) и ее соль (ХА).
В воде растворимость пикриновой кислоты около 0,06 моль . л""^ в
бензоле она значительно выше, так как коэффициент распределения X = 232,6.
Поэтому начальное условие для общих концентраций tJ^д = с^д = Л =
= 0,5 моль . л""' при ^HjO^^CeHe реально. Из него следует, что =
= 2Л = i ^оль • л"
1
А = 0,5 моль . л"
168
Да:
рН
0,8
0,6
0,4
0,2
О
Если принять, что «pi= I, то по закону сохранения начальной
концентрации 2Л = + X) аЛ+1] (6.33) и по закону электронейтральности при
[Н+1>[0Н-1 Л + Л —а = 0 (6.34). После подстановки а из (6.33) в (6.34)
и обычных преобразований получаем + (А + г) h — Ау. (6.35), где у. =
= [о (1 +Х)]""^ = 1,797 • 10""^ при а = 2,38. Решая (6.35), находим, что в
начальной буферной системе рН = 2,748.
Для характеристики буферных Т а б л и ц а 6.2
свойств системы следует оценить
значения рН. которые можно получить
при введении в нее разумных
количеств HY и ХОН, и значения р
полученных систем. Допустим, что такие
системы создаются путем введения
НУ так, что Сну = 1^ = 0.1, 0,2, 0,3,
0,4 моль . л""^ и ХОН так, что
^ХОН = ^ = ^'^' 0,2, 0,3, 0,4 мольх
X л"~^- В первом случае
уравнение закона электронейтральности
имеет видЛ — К + Л — а = О (6.36),
во втором А + х + h — a = Q
(6.37), а расчетные уравнения для
h соответственно + (А---У + х) h — {А + Y) % =^0 (6.38), + (Л + д: +
+ х) /I — (Л — л;) 7. = О (6.39). Ди1зференцируя (6.38) как неявную функцию Y
отрН и (6.39) как неявную функцию х от рН. получаем формулу для расчета р
dY 2,303 „ dx 2.303
Рассчитанные значения рН и р приведены в табл. 6.3, из которой следует, что»
изменяя концентрации HY и ХОН в водной фазе двухфазной буферной
системы, можно получить ряд буферных растворов с довольно большой буферн( й
емкостью р > 0,21, в которых 1.8 < рН < 3,7. Например, после введения
0,2 моль.л"^ HY устанавливается рН= 2,386 при р=0,50. Это значит, что, следуя
этому рецепту, можно получить буферную систему с (Н"^) = 10"^'^^^ Буфер-
"ное действие таких систем объясняется созданием «запаса» пикриновой
кислоты в бензоле, способного поступать в водную фазу при добавлении щелочи
и нейтрализовать ее, а также возможностью перехода НА в органическую
фазу, если в водную добавлена сильная кислота, что уменьшаег подкисляющее
действие последней. Таблица 6.4
О
0,2
0.4
0,6
0,8
0,2
0,4
0,6
0,8
1.0
1,2
1,4
1,6
1,8
11,778
11.632
11,545
11,482
11,434
11,395
11,361
11,332
11,307
0,97
1,87
2,79
3.73
4,65
5,56
6,51
7,44
8.34
у 1 0.4
0,3
0,2 1 0.1 1 0 0 1 0 1 0
0
x
0
0
0
0
0 0,1
0,2
0,3
0,4
рН
1,855
2,162
2,386
2,574
2,748 2,924
3,115
3,349
3,701
Р
0,26
0,39
0,50
0,56
0,58 0,55
0,48
0,40
0,21
Буферные системы незаменимы в случаях, когда необходимо
обеспечить устойчивость больших показателей активности тех или
иных ионов (т. е. малых значений активности). Пользуясь ими,
можно практически исключить влияние адсорбции и других внешних
факторов на рИон, а это имеет решающее значение при разработке
методов измерения активности.
Обобщенным буферным сво.й[ства.м и их применению
посвящены специальные исследозания [31, 35].
169
ЗАКЛЮЧЕНИЕ
Измерение химической формы движения материи связано с рядом
специфических операций, проводимых в гомогенных и гетерогенных ионных системах.
Полнота осуществляемых разделений и превращений, выбор оптимальных
условий для проведения операций, ошибки за счет адсорбции и других
воздействий окружающей среды должны учитываться точно так же, как при
измерениях массы учитывают аэростатическое воздействие, неравноплечесть весов
и другие факторы.
Как было показано ранее и в работе [21], опираясь на закон
действующих масс, законы сохранения и уравнения, связывающие активности и
равновесные концентрации, можно при строгом соблюдении- законов размерности
оценить возникающие погрешности и найти пути их полного или частичного
устранения в большинстве случаев.
Введение матриц равновесий систем, блоков, объединяющих совокупности
родственных частиц, единообразной системы обозначений и широкое
использование функции Н. Бьеррума позволило унифицировать и упростить составление
систем уравнений, характеризующих равновесные системы.
Одновременно вскрыты пробелы в существующих табличных данных и в
ряде случаев приведены примеры оценки достоверности этих данных.
Особое внимание уделено анализу получившихся уравнений,
определяющему возможность их законного упрощения. Всюду, где это возможно,
отмечались пути упрощений за счет дополнительных измерений.
Применения теории ионных равновесий, рассмотренные в этой монографии
и книге [21] имеют непосредственное значение для всех разделов химии и хи«
шческой технологии.
список ЛИТЕРАТУРЫ
1. Бабко А. К. Растворимость осадков в присутствии общих и
посторонних ионов.—Журн. аналит. химии, 1952, 7, вып. 1, с. 3—13.
2. Бейтс Р. Определение рН. Теория и практика / Пер. с аШл. Изд. 2-е.—
* Л.: Химия, 1972.—400 с.
3. Б р ее л ер С. Е. Радиоактивные элементы.—М.; Л.: Гос.' изд. техн.-
теорет. лит., 1949.—308 с.
4. Бугаевский А. А. Исследование уравнений, описывающих системы
со ступенчатыми равновесиями: Автореф. дне. ... канд. хим. наук.—
Харьков, 1965.—13 с.
5. Бугаевский А. А. Таблицы констант важнейших равновесий,
имеющих значение в аналитической химии.— X.: Изд-во при Харьк. ун-те,
1967,-67 с.
6. Бугаевский А. А., Комарь Н. П. Буферные свойства кислотно-
основных систем.—Уч. зап. Харьк. ун-та, 133, Тр. хим. фак-та и НИИ
химии, 1963, 19, с. 123—134.
7. Г а п о н Е. Н., Гапон Т. Б. Хроматографический анализ М. С.
Цвета и ионный обмен.—В кн.: Хроматографический анализ. Разделение
ионов. М.: Изд-во иностр. лит., 1949, с 9—14.
8. Г а п о н Е. Н., Г а п о и Т. Б., Ж у п а х и н а Е. С. Теория
ионообменной хроматографии.— В кн.: Исследования в области хроматографии.
Тр. Всесоюз. совещания по хроматографии. Москва 21—24 ноября
1960 г.- М.: Изд-во АН СССР, 1952, с. 5—28.
9. Глесстон С. Введение в электрохимию /Пер. с англ. Под. ред.
Б. Н. Кабанова.—М.: Изд-во иностр. лит., 1951.—768 с.
10. Д а н а Э. С. Описательная минералогия (Справочник) / Пер. с англ.
под общ. ред. акад. А, Е. Ферсмана.—М.; Л.: Изд-во глав. ред. геолого-
развед. и геодезич. лит-ры, 1937.-423 с.
11. Дер яги н Б. В., Ландау Л. Д. Теория устойчивости сильно
заряженных лиофобных золей и слипания сильно заряженных частиц в
растворах электролитов.—В кн.: Ландау Л. Д. Собр. трудов. /Под ред.
Е. М. Лифшица. М.: Наука, 1969, 1, с. 388—411.
12. Л а н д а у Л., Д е р я ги н Б. Теория устойчивости сильно заряженных
лиофобных золей и слипания сильно заряженных частиц в растворах
электролитов.—Журн. экспер. и теорет. физики, 1945, 15, вып. 2, с. 663—
682.
13. Да вис К., Джонс А. Осаждение хлористого серебра из водных
раствогов.— В кн.: Новые исследования по кристаллографии и
кристаллохимии. 1. Рост кристаллов. М.: Изд-во иностр. лит., 1952, с. 80—84.
14. Золотое Ю. А. Экстракция внутрикомплексных соединений.—М.:
Наука, 1968.-314 с.
15. Золотое Ю. А., И о ф а Б. 3„ Ч у г а л и н Л. А. Экстракция галогенид-
'пых ьомплексов металлов.—М.: Наука, 1973.-580 с.
16. Золотов Ю. А., Кузьмин Н. М. Экстракционное концентрирование.—
М.. Химия, 1971.—272 с.
17. Киргницев А. Н., Т р у ш н и к о в а Л. И., Л а в р е н т ь е в а В. Г.
Растворимость неорганических веществ в воде*. Справочник.—Л.: Химия,
1972.-246 с.
171
18. к л асе он С. Адсорбционный анализ смесей /Пер. с англ.—М.; Л.:
Госхимиздат, 1950.—152 с.
19. Кольтгоф И. М., Стенгер В. А. Объемный анализ. 1. / Пер. с
англ.—М.; Л.: Госхимиздат, 1950.—376 с.
20. Комарь И. П. Основы качественного химического анализа.—X.: Изд-
во Харьк. ун-та, 1955—448 с.
21. Комарь И. П. Химическая метрология. Гомогенные ионные
равновесия.—Харьков: Вища школа, 1983.-208 с.
22. К о м а р ь Н. П., В о в к С. И., С и н ю т а Т. И. Измерение
коэффициентов активности Са2+-ионов и произведения растворимости Са(0Н)2.—
Журн. физ. химии, 1976. 50, с. 2403—2404.
23. Корен ман И. М. Экстракция в анализе органических веществ.—
М.: Химия, 1977.—200 с.
24. Кузнецов В. И. Химические теоретические основы экстракционного
извлечения элементов.—Усп. химии, 1954, 23, № 6, с. 654—671.
25. Л а й т и н 6 н Г. А. Химический анализ /Пер. с англ.— М.: Химия,
1966.—656 с.
26. Л а й т и н е н Г. А., X а р р и с В. Е. Химический анализ /Пер с англ.—
М.: Химия, 1979.—624 с.
27. Лурье Ю. Ю. Справочник по аналитической химии.—М.: Химия,
1979.-480 с.
28. Мазуренко Е. А. Справочник по экстракции.—Киев: Техшка, 1972.—
448 с.
29. Менделеев Д. И. Избр. соч. Т. 3.—Л.: Госхимиздат, 1934.-467 с.
30. Моррисон Дж., Фрейзер Г. Экстракция в аналитической химии.—
Л:: Госхимиздат, I960.—312 с.
31. Мухина Т. П., Рудная Л. Е., Б у г а е в с к и й А. А. Расчет
равновесий в сложных системах. Сообщ. 4. Оценка буферных свойств
системы.—Жури, аналит. химии. 1970, 25, № 4. с. 640—645.
32. Николотова 3. И. Карташова Н. А. Экстракция
нейтральными органическими соединениями. Справочник по экстракции. Т. 1.—М.:
Атомиздат, 1976.-598 с.
33. Ормонт Б. Ф. Структура неорганических соединений.—М.; Л.: Гос.'
изд. тёхн.-теорет. лит., 1950.-968 с.
34. Р о 3 е н А. М. Физическая химия экстракционных равновесий.— В кн.:
Экстракция. Теория, применение, аппаратура. Вып. 1. М.: Атомиздат,
1962, с. 6-87.
35. Р у д н а я Л. Е. Обобщенные буферные свойства и некоторые их
применения: Автореф. дне. канд. хим. наук.— Харьков, 1975.-24 с.
36. Справочник по растворимости / Под ред. В. В. Кафарова. т. 1. Би-
' парные системы.—М.; Л.: ВИНИТИ АН СССР. Кн. 1, 1961.-960 с.
37. У э л л с А. Строение неорганических веществ. (Структурная
неорганическая химиям /Пер. с англ —М.: Изд-во иностр. лит., 1948.-691 с.
38. Ферсман А. Е. Избранные труды. Т. 3.—М.: Изд-во АН СССР,
1955.-798 с.
39. Фомин В. В. Химия экстракционных процессов.—М.: Атомиздат,
.1960.-166 с.
40. Ф о м и н В. В., М о р г у н о в а А. Ф. Экстракция гидратов хлорного
железа эфирами.—Журн. неорг. химии, 1960, 5, № 6, с. 1385—1389.
41. Франк Ф. Влияние смещений на рост кристаллов.—В кн.: Новые
исследования по кристаллографии и кристаллохимии. Сб. 1. Рост кристаллов.
М,: Изд-во иностр. лит., 1950, с. 41—46.
42. Хан О. Прикладная радиохимия.— Л.; М.: Госхимиздат, 1947.-276 с.
43. Хлопни В. Г. Избранные труды. Т. 1.—М.; Л.: Изд-во АН СССР,
1957.—372 с.
44. Я Ц и м и р с к и й К. Б. Из .истории аналитической химии в России.—
Усп. химии, 1949, 18, № 5, с. 623—628.
45. Я ц и м и р с к и й К. Б. Термохимия комплексных соединений.— М.:
Изд-во АН СССР, 1951.-252 с.
172
46. Bala rev D. Disperse structure of solid systems and its thermodinamic
foundation. IV.—koUoid. Z., 1941, 96, S. 19-23.
47.Bjerrum N. Untersuchungen iiber lonenassoziation lonen bei mit-
tleren Assoziationsaraden (Det. Kgl. Danske Vide.nskabernes Selskab, Mathe-
matisefysiske meddelelser, 1926, B. 7,9, S. 1—48.
48. В u n n C. W. Adsorption, Oriented Overgrowth and Mixed Crystal
Formation.—Proc. Roy Soc, Ser. A, 1933, 141, N A 845, p. 567—593.
49. Doerner H. A., Hoskins W. Co-precipitation of radium and barium
sulfates.—Journ. of Amer. Chem. Soc, 1925, 47, p. 662—675.
50. Enustun B. v., J u r ke v ic h. The variation of solubility of
strontium sulphate with particle size. Radioisotopes in the physic, sciense and
indystrie (Vienna), 1962, 3, p. 531—548.
51. Erdey L. Theorie und Praxic der gravimetrischen Analyse, Budapest,
Akad. Kiado, 1960, b. 1.-340 S.
52. Fe itknec ht, P. Schindler. Solubility constants of metal oxides,
metal hydroxides and metal hydroxide salts in aqueous solution — Pure
Appl. Chem., 1963, 6, N 2, p. 130—199.
53. Fischer R. В , R h i n e h a m m e r T. B. Rapid precipitation of barium
sulfate —Anal. Chem,, 1953, 25, N 10, p. 1544—1548.
54. Freundlleh H. Кар illarchemie.—Leipzig Akad. Verlagsgesel. M. B. H.,
1923.— 1225 с
55. Friedman H. L. The Visible and Ultraviolet Absoption, Spectrum of
the Tetrachloroferrate (III) Ion In Various Media.— Journ. Amer. Chem.
Soc, 1952, 74, N 5, p. 5—10.
56. H 1 a ss t one S. Thermodynamics for chemists. 3-d Ed. N, York — Toronto—
London D. van Mostrand Co., inc., 1948.-522 p.
57. I r v i n g H., R о ss о 11 i F. J. C. The Extraction of Indium.at Tracer
Concentrations from Acid. Bromide Solutions into iso Butyl Methyl ketone.—Journ.
Chem. Soc, 1955, N 6, p. 1927—1966,
58. Kading Uber den Einbau kleiner Mengen von Blei in Alkalihalo-
^ |enide.—Z. Physlk. C^hem., 1932, A J_62^ S. 174—186.
59. Kolthoff I. M., Eggertsen F T. Studies on Ading and Copre-
cipitation. 24. The Coprecipitation of Silver Chloride with Colloidal and
Flocculated Silver Bromide.—Journ. Amer. Chem. Soc, 1939, 61, p. 1036—1040.
60. Kolthoff I. M., Griffith F. S. Pos.tprecipitation of nickel sulfide
with copper sulfide, mercuric sulfide and zinc sulfide.—Journ. Phys.
Chem., 1938, 42, p. 541-545.
61. Kolthoff I. M., Moltza u D. R. Induced' precipitations and pro-'
perties of metal sulfides, 1935, 17, N 3, p. 293—325.
62. Kolthoff I. M., Mol tzau D. R. Jhe postprecipitation of zinc sulfide.—
Journ. Phys. Chem., 1936, 40, p. 779—798.
63. Kolthoff I. M., Sandell E. B. The Coprecipitation of Alkali Ions
with Calcium Oxalate and the Adsorbent Properties of the Latter, Journ.
Phys. Chem., 1933. 37, p. 443—458.
64. Lange E., Berger R. IJber die Messung potential Bestimmender lonen-
adsorption am Agl mittls potentiometrischer. Fallungstitration.— Zeitschrift
fur Electrochemie und ingewandte physikalische Chemie, 1930, B. 36, N 3,
S. 171—179.
65. L a u r e n с A. A., С a m p b e 11 D. E., W i b e r 1 e у S. E., С 1 a r к A. M.
The Extraction of Ferric Chloride b у Isopropyl Ether. Part 1. The signifi-
cance of water in the Extrated Iron Complex,—Journ. Phys. Chem., 1956,
60, N 7, p. 901—904.
66. Lew in S. The solubility product principle: an introduction to its uses
and limitations.—London: Pitman, I960.—113 p.
67. May D. R., Kolthoff I. M. Studies on the Aging of Precipitates
and Coprecipitation. XL. The solubility of Lead. Chromate as' a Function
of the Particle Size.—Journ. phys. and Colloid Chem., 1948, 52, N 5,
p. 836 — 853.
68. Pur lee Е. L., Grunwald E. The Potentiometric Measurement of
Ion-pair Dissociation Constants HCl, NaCI and KCl in 10% Dioxane —30%
Wates,—Journ. of Amer. Chem. Soc, 1957, 79, N 6, p. 1366—1372.
69. Seidell A. Solubilities of inorganic and methal organic compounds of
quantitative solubifity data from the periodical literature, 3-d ed., vol. 1-—
N. York, D. Van Nostrand Company, Inc., 1940.—1698 p.
70. Specker H., CremerM. Dber Eisen (III) — Verbindungen in chlorid-
haltigen verteilungs systemen.—2. andL. Chem., 1959, 167, N 2, S. 110—115.
71. Stability constants of metal — ion complexes. Special publication N 17.
CompI, by Sillen L. G. Martell A. E. —London.: Chem. Soc., 1964.—754 p.
72. Stability constants of metal — ion complexes. Suppl. N 1, Special
publication N 25 (Compl. by Sillen L. G:, Martell A. E. —London: The Chem.
Soc, 1971.—865 p.
73. Tiselius A. Studied uber Adsorptionsandlyse. I. —KoHoid Z., 1943,
B. 105, H 2. S. 101-109.
74. Van Slyke D. D. The measurement of buffer values and the
relationship of buffer value to the dissociation Constant of the buffer and the
concentration and the reaction of the buffer solution.—Journ. Biol Chem.,
1922, 52, p. 525—570.
75. Zimmermann H. K. The experimental determination of solubilities.—
Chem. Revs, 1952, 52, N 1, p. 25—65.
ПРИЛОЖЕНИЕ
Таблицы констант важнейших равновесий, имеющих
значение в аналитической химии (5)
Сокращения, принятые для некоторых органических веществ
Сокращение
Формула
Название
Ас-
cit^-
Edta^-
Oxin~
Py
Sal2-
Suc2-
T2-
^ CH3COO-
СзН4(СОО)|-
НзС—C=:N=0
HsC-C«NOH
(OOCCH3)2N (CH2)2N (CHaCOO),
<3-n.n-/y^
OM
CH2OH—CHOH—CH2OH
>—\.. ; c,HeON-
N
\_
>
CeH40C002-
\СЩ)2 (C00)2-
(CH0H)2 (C00)|-
ацетат
цитрат
диметилглиоксимат
этилендиаминтетраацетат
(трилонат)
эриохромат (черный Т)
глицерин
8-гидроксохинолат (окси-
нат)
пиридин
салицилат
сукцинат
тгртрат
I. КИСЛОТНО-ОСНОВНЫЕ РАВНОВЕСИЯ
Таблица 1.1 Показатели ионного произведения воды,
выраженного в (моль/л)^ при различных температурах
9q
ра;
«С
«С
рш
PC
pie;
0
5
во
15
14,89
14,68
14,44
14,24
20
22
25
30
14,07
14,00
13,90
13,72
35
40
50
60
13,57
13,42
13,25
12,90
70
80
90
100
12,68
12,47
12,28
12,13
Для различной среды при < = 25 °С: гЫаСЮ, — 14,08, SNaClO*—14,22,
4NaCI04- 14,80.
176
Таблица 1.2. Важнейшие кислоты, значения логарифмов общих констант
образования (протонизации) (а) и показателей констант ступенчатой
ионизации (К)
jA— +;iH+5=iHiA<J'"-')-,
Н,А(«-')-г±Н++ Hi_iA<"-'-»-,
(HiAP"-')-)
(A«-)J(H+)'
(H+) (Hi-iA^'-'-n-)
(H,A<''-"-)
Кислота
формула
H3ASO4
HgAsO-
HAs02-
HAsOg
HAsSa
мышьяковая
дигидроарсенат
моногидроарсенат
мышьяковистая
тиомышь яковиста я
Н3ВО3 I борная
ЗВО-+2Н++Н20^ВзОз(ОН)~
4В07 + ЗН+ + H205=iB404(OH)f
НВО2 • glyc I глицеринборная
НВгОз
НВгО
НВг
бромноватая
бромноватистая
бромистоводородная
Среда
Ig J
пер.
пер.
пер.
О
пер.
20,63
18,44
11,50
9,29
3,7
О
О
О
пер.
9,24
35.17
43,81
6,5-
сильная
I 8,6
сильная
РК
2,19
6,94
11,50
9,29
3,7
9,24
0,5
8,0
СО2+Н2О
НСО3-
HCN
HCNO
HCNS
НСООН
НАс
CH2CICOOH
CHCI2COOH
CClgCOOH
Н2С2О4
HCgO-
СН3СНОНСОООН
H2SUC
HSuc-
Н2Т
HT-
HDm
CeHgOH
угольная
гидрокарбонат
цианистоводородная
циановая
р од анистоводородна я
муравьиная
уксусная
монохлоруксусная
дихлоруксусная
трихлоруксусная
щавелевая
гидрооксалат
молочная
янтарная
гидросукцинат
винная
гидротартрат
диметилглиоксим
фенол
- О
О
пер.
пер.
О
О
О
О
О
О
О
О
О
О
О
О
О
0,01
16,68
10,33
9,35
3,66
0,85
3,75
4,76
2,85
1,23
0,66
Ь,52
4,25
3,85
9,85
5,63
7,41
4,37
11,1
9,99
6.35
10,33
9,35
3,66
0,85
3,75
> 4,76
2,85
1,23
0,66
1,27
4,25
3,85
4,22
5,63
3,04
4,37
11,1
9.99
176
Продолжение табл. 1.2
HaCit-
HCit^-
CeHgCOOH
HgSal
HSal-
CeHa (N02)3COOH
CeH4 (C00H)2
CeH4C00HC00~
HaOxin^
HOxin
H4Edta
HgEdta-
HgEdta^-
HEdta^-
НзЕг
HgEr-
HEr^-
пикриновая
лимонная
дигидроцитрат
моногидроцитрат
бензойная
салициловая
гидросалицилат
тринитробензойная
0-фталевая
гидро-о-фталат
8-гидроксохинолиний
8-гидроксохинолин
этилендиаминтетрауксус-
ная кислота
тригид рютрилонат
дигидротрилонат
моногидротрилонат
эриохром черный Т
дигидроэриохромат
моногидроэриохромат
О
О
О
о
о
о
о
о
о
о
0,1
0,1
0,1КС1
0,1КС1
0,1КС1
0,1КС1
0,02
0,02
0,02
0,32
13,30
10,16
5,39
4,20
16,37
13,4
0,65
8,35
5,41
15,0
9,90
21,09
19,09
16,42
10,26
17,85
11,55
HCIO4
НСЮз
НСЮз
нею
НС1
хлорная
хлорноватая
хлористая
хлорноватистая
соляная
О
О
сильная
сильная
1,96
7,53
сильная
Н2СГО4
НСг07
хромовая
гидрохромат
1ЫСЮ4
О
2Сг02~ + 2Н+5=^Сг202-+Н20
6,50
14,64
HF I фтористоводородная
2F~ + H+*^HF~
О
О
3,17
3,76
h4Fe(CN)e
H3Fe(CN)j-
HgFe (CN)|-
HFe{CN)3-
гексацианотетрагидррже-
лезо
тригидрогексацианофер-
роат
дигидрогексацианоферро-
ат
моногид рог ексациа нофер-
роат
О
О
сильная
сильная
7,30
4130
H5lO«
Н41ОГ
НЮв
ортоиодная
тетрагидроортоиодат
йодноватая
О
О
О
9,82
8,27
0,78
12 3 457
177
Продолжение табл. 1.2
1+ + Н2О (вНЮ + Н+)
НЮ
HI
иодноватистая
иодистоводородна я
0,1КС1
-О
16,8
12,3
сильная
НМПО4
марганцовая
сильная
HNOs
HNO2
HN3
NH3NH2+
NHgNH^
NH3OH+
NH+
CH3NH+
(СНз)2НН^
(СНз)зЫН+
C2H5NH+
C6H5NH+
(ЫНз)2(СН2)|+
NH2(CH2)2NH+
C6H5NH+
CH3C5H5NH+
азотная
азотистая
азотистоводо родная
дигидрогидразиний
гидразиний
гидроксиламмониЛ
аммоний
метила ммоний
ди метила ммоний
три мети ламмоний
этиламмоний
фениламмоний
этилендиаммоний
этилена минаммоний
пиридиний
пиколиний
0,001
О
О
О
О
О
О
О
О
О
О
О
О
О
О
сильная
3,29
4,72
8,21
7,94
5,98
9,24
10,60
10,87
9,87
10,75
4.61
18,17
10,65
5,20
5,97
Н2О2
перекись водорода
о
11,8
Н3РО4
Н2РОГ
НР02-
Н4Р20,
НЗР207
Н2Р20Г
НР2О2Г
Н2РО3Н
НРОзН""
НРО2Н2
фосфорная
дигидрофосфат
моногидрофрсфат
пирофосфорная
тригидропирофосфат
ди гид ропи рофосфат
моногидропирофосфат
фосфористая
гидрофосфит
фосфорноватистая
О
О
О
О
О
О
О
пер. 18^
пер. 18°
пер. 18*^
21,68
19,53
12,32
19,73
18.21
15,85
9,25
8,58
6,58
1,07
178
продолжение табл> 1.2
HS07
H2S203
HSjOl"
SO2 + HjO
Hsog-
Hsr •
HS|-
HS7
hs-
При температуре "С:
серная
гидросульфат
тиосерная
гидротиосульфат
гидрогипосульфит
сернистая
гидросульфит
гидропентасульфнд
гидротетрасульфид
гидротрисульфид
гидродисульфид
сероводородная
гидросульфид
О
О
О
~0
-о
~0
?
?
?
?
о
о
сильная
1,99
2.32
1,72
2.45
8,97
7,21
8.4
11.6
12,3
14,0
19,95
12,9
lgej= 12,9-0,031 (< —25)
lga,= 19.95 - 0,0435 (<-25)
1,99
0,60
1,72
2,45
1,76
7,21
8,4
11.6
12,3
14,0
7.05
12,9
Si
si(oh;4
SiO (OH)J-
SiO(OH)J-
4Si02(OH)^-+6H+
4Si02(OH)|- + 4H +
кремневая
гидросиликат
кремневая
гидросиликат
^Si^O^ (ОН)*-+4Н20
0,6NaClO4
0.5NaClO4
3NaC104
3NaC104
|0.6NaClO4
3NaC104
22,02
12.56
22,14
12,71
100,65
121.04
9.46
12,56
9,43
12,71
12»
Таблица 1.3. Аквокомплексы, показатели общих констант ионизации (yj).
Гидрокомплексы, показатели. ступенчатых констант основной ионизации (Kqch)
Дквок омплеке ы
ш'++И,о««,,он,«'-»++,н-ь, ,„ „,.(«,'°«)""-"-Ь(Н+>'
Гидрокомплексы
М (OH)j'*-'>+ ;=tM (OH)<,i^J+l>+ + ОН-. Коси i ■■
(М (ОН)'|^'+')+)(ОН-)
(M(OH)<f-i)+)
Элемент
Уравнение иониза1;ии
Среда
P^och
I
2
3
4
5
Ag
Ag+ + ЩО ^ AgOH ^ Н+
Ag+ + 2Ир-^ Ag (0Н)7 + 2Н+
Ag (0Н)7 5=> AgOH (т) + ОН-
0
0
0
11,7
23,8
2,3
1,9
—3,5
Al
А12+ + ЩО ^ А1(0Н)2+ + Н+
2А13+ _^ 2Щ0 ^ Al2(0H)^+ + 2Н+
Al3+ + 4Н2О ^ Al (0Н)7 + 4Н+
А1 (OH)f:^ Al (ОН)з (т) + ОН-
0
0
пер
пер
4,3
14,56
24,25 '
9,7
0,68
Ва
Ba^t + HgO 5=± ВаОН+ + Н+
0
13,36 .
0,64
Be
Ве2+ + Н2О <=± ВеОН+ + Н+
2Ве2+ + Н2О « BegOH^-*" -К Н+
lNaC104
lNaC104
6,52
3,51
—
Bi
Bi3+ + Н20 ^ BiOH2+ + Н+
бВ|2++6Н20;г=гВ1еО|++ 12Н+
3NaC104
3NaC104
1,58
0,33
12,64
Са
Са2+ + Н2О ^ СаОН+ + Н+
0
12,60
1,40
Cd
Cd2+ + Н2О « CdOH+ + Н+
2Cd2+ + Н2О CdgOH^^ + Н+
3LiC104
3LiCI04
10,2
9,10
—
Со
СоЗ+ + Н2О ^ СоОН2+ + Н+
Со2++ HgO ^ СоОН+ + Н+
lNaC104
0
1,78
11,20
2,80
Cr
СгЗ+ + Н2О :!=t СгОН2+ + Н+
СгЗ+ + 2Н2О « Сг (0Н)+ + 2Н+
СгЗ+ + 4Н2О <=i Сг (OH)J- + 4Н+
Сг (0Н)7 <=t Сг (ОН)з(т) + ОН-
-0
-0
пер.
пер.
3,8
10,0
26
10,2
7,8
-0,4
ISO
Продолжение табл. 1.3
Cu2+ + Н2О <=± CuOH+ + Н+
Cu2++ 2Н20^ Cu(0H)2 + 2Н+
2Cu2+ + 2Н2О ^ Cu2 (0Н)2+ + 2Н+
Cu2+ + ЗН О « Си (ОЫ)^ + ЗН+
Cu2+ + 4Н2О 5=± Си (ОН)!" + 4Н+
Си (0Н)7 5=t Си (0Н)2 (т) + 0Н~
8,0
6,8
10,9
27,2
40,3
Fe^H- + Н2О ^ РеОН2+ + Н+
2Fe3+ + 2Н2О 5=t Feg (0Н)^+ + 2Н+
Fe2+ + Н2О <=t FeOH++ H+
0
0
0
2,17
2,85
5,92
Hg2+ -i- H2O HgOH+ + H+
Hg2+ + 2H2O « Hg (0H)2 (T) + 2H+
Hg2+ + ЗН2О 5=± Hg (OH)f + 3H+
Hg2+ + H2O z=± Hg20H+ + H+
0
0
0
0,5NaClO4
3,65
7,72
22,57
5,0
Mg2+ + H2O <=t MgOH"^ i- H+
0,1
12,8
Mn2+ + HgO ^ MnOH+ + H+
Mn (OH)^ ^ Mn(0H)2 (T) + OH-
10,6
Ni2+ + HgO ^ NiOH+ + H+
8,94
Pb2+ + H2O ^ PbOH+ + H+
Pb2+ + 2H2O <=t Pb (0H)2 + 2H^
Pb2+ + ЗН2О ^ Pb (OH)- + 3H+
4Pb2+ + 4H2O 5=i Pb4(0H)J+ + 4H+
3Pb2+ + 4H20.=t Pbg (0H)2-b + 4H+
6Pb2+ + 8H2O 5=i Pbg (0H)^+ + 8H+
0,3NaClO4
0,3NaClO4
0,3NaCIO4
0,3NaClO4
0,3NaClO4
0,3NaClO4
7,8
17,2
28,0.
19,9
23,4
24,7
SbO+ + 2H2O <=t Sb (ОН)з + H+
SbO+ + ЗН2О 5=± Sb (OH)^- + 2H+
0
0
0,87
11,87
181
Продолжение табл. 1.3
Sn2+ + Н2О 5=t SnOH+ + Н+
Sn2+ + 2H2O 5=s Sn (0H)2 (T) + гн+
Sn2+ + SHgOsrt Sn (OH)J- + 3H+
Sn2+ + H20.=t SnOH+ + H+
2Sn2+ + 2H2O 5=t Snj (0H)|+ + 2H+
3Sn2+ + 4H2O *=t Snj (0H)2+ 4. 4H+
Sn (OH)g- s=s Sn (0H)2 (T) + OH-
Sn
Sr
0
0
0
3NaCI04
3NaC104
3NaC104
0
Sr2+ + H2O e 8ЮН+ + H+
Zn2+ + HjOszt Zn 0H+ + H+
Zn2+ + ЗН2О ^ Zn (OH)J- + 3H+
2Zn2+ + HjO Zn^0H3+ + H+
2,07
7.06
16,61
3.9
4,45
6,77
13,18
11,93
9,01
4,45
10,3
-0,8
0,82
Zn
0,lNaClO4
3LiC104
8,96
28,4
8,7
4.94
182
Таблица 1.4 Кислотно-основные индикаторы; показатели констант
кислотной ионизации, области 1Герехода и окраска (при комнатной
температуре)
Индикатор
РК
Область
перехода
Окраска в среде
кислой
щелочной
Тимоловый синий
Пентаметокси — красный
Метиловый оранжевый
Р-динитрофенол (1-окси-
2,6-динитробензол)
а-ди нитрофенол (1-окси-
2,4-д ннитробензол)
Бромфеноловый синий
Бромкрезоловый зеленый
Метиловый красный
7-динитрофенол (1-окси-
2,5- динитробензол)
Бромкрезоловый
пурпурный
П-нитрофенол
Бромтиловый синий
Феноловый красный
М-нитрофенол
0-крезолфталеин-
Тимоловый синий
Фенолфталеин
Три нитробензол (I, 3, 5)
1,51
1,86
3,52'
3,69
4,06
4,23
4,92
5,04
5,2
6,3
7,1
7,24
8,08
8,35
/8,8
19,4
8,9
/8,94
19,55
13,0
1,2^2,8
1,2-3,2
3,0-4,5
2,2-4,0
2,8—4,5
3,0-4,6
3,8-5,4
4,4-6,2
4,0—5,5
5,2—6,8
5,6—7,6
6,0—7.6
6,8-8,4
6.7—8,4
8,0-
-10,0
а,0—9,6
8,U
—10,2
12—14
красный
красно-фиолетовый
красный
бесцветный
бесцветный
желтый
желтый
красный
бесцветный
желтый
бесцветный
желтый
желтый
бесцветный
бесцветный
желтый
бесцветный
бесцветный *|
желтый
бесцветный
желтый
желтый
желтый
синий
синий
желтый
желтый
пурпурный
желтый
синий *
красный
желтый
красный
синий
красный
оранжевый
183
II. РАВНОВЕСИЯ КОМПЛЕКСООБРАЗОВАНИЯ
Таблица ПЛ. Комплексные соединения, логарифмы общих (р)
и ступенчатых (k) констант образования
MAj^i . А .-.MAi , (MA[Lr^~^^^l+).(A«-)
Координирующий ион
Адденд (А)
формула
комплекса
Среда
1
2
3
1
4
5
6
Ag+
Ag^
Вг-
CN-
CNS-
ci-
F-
I-
NH,
AgAf-
AgA2-
AgA7
AgA
Ag2A+
AgA^-
AgA|-
AgAf
AgA^-
AgA|-
AgAJ-
AgAf-
AgAf-
AgA^
AgA
Ag2A+
Ag3A^+
AgA
AgA3-
AgA|-
AgA+
AgA+
0
0
0
0
пер.
0
0
0.3
2.2.
2.2
2,2
0
0
0
0
пер.
пер.
О
1,6
1.6
-О (За'С)
-О (30 "С)
8.73
8.00
7.34
4.38
9,70
20.68
21,8
21.1
10.08
9,08
7.57
5.30
5.04
5,04
3.04
5,20
5.45
0.36
13,75
13.95
7,23
3.32
0,73
0.66
2.96
4.38
-1,13
0.70
1.0
2.51
0.26
0.00
2.00
3.04
0.36
-0,20
3.92
3,32
Расчетная формула для других ионных сил (в NH,NOs) и температур:
lg*a = lg *2-0'010J -0,017 (t-30)
184
Продолжение табл. П.1
4.
Ag+
N0^
NO7
so?-
Ac~
' Ac-
Edta*-
HEdta^-
Py
AgA
AgA^
AgA|-
AgA-
AgA3-
AgA-
AgA-
AgA7
AgA
AgA-
AgA2-
AgA+
AgA+
0
пер.
2NaNOs
2NaN08
0
0
0
0,001
0,lKNO3
O.IKNO3
0
0
—0,18
2,83
7,8
5,4
13,46
8,82
1.3
0,64
0,73
~0
7.32
3,07
4,35
1,97
—0,18
2,4
5,4
4,64
8,82
1,3
—0,09
0,73
~0
7,32
3,07
4,35
1.97
A13+
A13+
A10H2+ .
A13+
Edta*-
HEdta^-
Edta"-
Sal2-
AIA|-
A1A|-
AIA7
AIA3
A1A+
A1A2+
A1A»-
AIA7
AlA-
AlA
A10HA2-
A1A+
0,53
0,53
0,53
0,53
0.53
0,53
0
0
0,1KNO«
0,lKNO3
0,1
19,84
19,37
17,75
15,00
11,15
6,13
16,3
13.0
16.13
8,40
22.70
14,0
0.47
1,62
2.75
3.85
5.02
6,13
3.3
16,13
8.40
22,70
14.0
Ba2+
P2O7-
s,ol-
Ac-
BaA+
BaA+
BaA2-
BaA
BaA+
0
0
пер.
0
0
<0.45
0,92
4,64
2,33
0,41
<0,45
0,92
4.64
2.33
0,41
185
продолжение табл. П.1
Ва2+
Т2-
нт-
Cit^-
Edta*-
HEdta*-
Oxin-
HSal-
BaA
BaA
BaA+
BaA-
BaA2-
BaA-
BaA+
BaA+
0
0.2
0.2
O.lSNaCl
0
P.1
0
0
2.31
1.62
0,88
2,98
7,78
2,07
2,07
0,21
Bi3+
Br-
Br-
CNS-
ci-
NO3-
HT-
OH-
X2-
BiA^
BiAg
BiAf
BiA2+
BiAi-
BiA^
BiA^
BiA2+
BiA|-
BiAf-
BiA7
BiAj
BiA+
BiA2+
BiA|-
BiA2-
BiA:f
BiA2+
BiA7
В1(ОН)зТ2-
3,0=
(2NaC104 -I-
+ 1H+)
0,4HClO4
0,4HClO4
0,4HCIO4
O.4HCIO4
4,0 =
=(3LiCI04+
-f 1H+)
3,0 =
=(2NaC104+
+ 1H+)
пер.
7,80
6,18
4,26
2,26
4,23
3,41
2,26
1,15
7,36
7.3
6,75
5.8
3,5
2.2
18.9
16.80
14.95
1.26
8.30
31,0
Ca2+
HCO3-
F-
NO5-
CaA+
CaA+
CaA+
0,15NaCl
0
0
0,81
<1,04
0,28
186
Продолжение табл. II. I
3
6
HPOf-
Р2О7-
S2or
so|-
Ac-
HT-
Cit3-
HCit2-
HjCit-
Edta-*-
HEdta^-
Ег»-
Oxin-
HSal-
CaA
CaA2-
CaA
CaA
CaA+
CaA
CaA
CaA+
CaA-
CaA
CaA
CaA2-
CaA-
CaA-
CaA+
CaA+
nsp.
0
0
0,20
0
0,2
0,2
0
0
0
{?..
0,1
0,02
0
0
2,40
5,00
1,91
2,31
0,53
~3
1,80
1,11
4.90
3,05
1.15
/11,0
I 10,7
3,51
5,4
3,27
0,55
2.40
5,00
1.91
2.31
0.53
~3
1,80
1.11
1.85
1.90
1.15
/11.0
tl0,7
3,51
5,4
3.27
0,55
BO7
Br-
CN-
CNS-
CNS-
CdA2-
CdA2-
CdAf
CdAg
CdA+
CdA|-
CdAf
CdAj
"cdA+
CdA-
CdAj
CdA+
CdA2-
CdAj-
CdAj
CdA+
CdAa
пер.
0
0
0
0
3,0
3,0
3,0
3,0
3,0
3,0
3,0
0
0
0
0
INaClOi
10,6 (?)
2,9
2.83
3,00
2.23
18.85
15.30
10,60
5.54
2.55
1,95
1.36.
1.64
2,34
2,49
1.95
0,53
0,10
—0,17
0,77
2,23
3,55
4,70
5.06
5.54
0,60
0,59
1,36
—0,70
—0,15
0,54
1,95
0.07
187
ci-
Продолжение табл.
F-
1-
CdA+
CdA2-
CdA-
CdAj
CdA+
CdA|+
CdA|+
CdA|+
CdA^+
INaCIOi
0
0
0
0
-0(30 °C)
-0 (30 °C)
-0 (30 °C)
'0 (30 °C)
0,46
6,10
5.0
3,92
2,28
6,56
5,77
4,47
2,51
Расчетная формула для других ионных сил и температур
Ig = Ig *° + 0.070/ - 0.008 {t - 30)
no:
N07
P2O7-
S02-
spl-
S02-
Ac-
Cit3-
0H-, cit^-
Edta*-
HE dta^-
Py
Oxin-
CdAf-
CdA7
CdAj
CdA+
CdA+
CdA2-
CdA2-
CdA
CdA
CdAj
CdA+
CdA2-
CdA
CdA-
CdOHCit^-
CdA2-
CdA-
CdA2+
CdA2+
CdAj
CdA+
CdA+
3NaC104
SNaCld^
3NaC104
3Naci04
0
INaNOg
0
0
0
0
0,1
0.1
O.IKNO,
0,1 KNO,
0.5
0.5
0.01
0.01
0
3,11
3,81
3,01
1,80
0,40
5.57
4,19
3.94
2,29
2,77
1.77
5,29
3,52
4,22
5.05
16.46
9,10
2.1
1.27
13.4
7.2
7.78
0,46
1.10
1.08
1,64
2,28
0,79
1,30
1.96
2.51
0.7
0.80
"1.21
1.80
0.40
5.57
3.94
2.29
1.00
1.77
1.77
3.52
4.22
16,46
9.10
0.8
1.27
5,2
7.2
7.78
188
Продолжение табл. II.l
I
2
3
4
5
б
ci-
CI2
ВГ2
ICl
IBr
I2
CI3-
С1ВГ2
IC17
ClBrl-
CII7
0
пер.
пер.
пер.
0
—0,72
0,08
2,22
1,64
0,09
-0,72
0,08
2,22
1,64
0,09
Со2+
Вг-
CNS-
СГ
СоА+
СоА+
СоА2-
СоА+
CoAf-
СоА+
СоА2-
СоА+
CoAf-
СоА+
2NaC104
О
40%-ный
ацетон
50%-ный
' ацетон
60%-ный
ацетон
100%-ный
ацетон
I,5NaC104
—0,12
1,32
4,11
1,28
5,38
1,55
6,59
1,78
13,72
-0,4
F —комплексы не обнаружены даже в 10 м раствора KF
NH,
СоА|+
СоА§+
СоА|+
СоА|+
СоА|+
СоА2+
О (30 °С)
О (30 °С)
О (30
О (30 °С)
О (30 °С)
о (30 "О
■ 4,39
5,13
5,07
4,43
3,50
1,99
Расчетная формула для других ионных сил и температур
■lg*a
S20i-
sof-
Edta-"
HEdta^-
Py
Ig *2 + 0.062/ — 0,005 (t — 30)
CoA
CoA
CoA
CdA2-
CoA-
CoA|+
0
0
. 0
0,1 KCl
0,lKNO3
0,5
2,05
2,47
4,7 •
16.21
9,15
1,54
-0,12
1,32
1,28
1.55
1.78
—0.4
—0,74
0.06
0.64
0,93
1,51
1,99
2,05
2,47
4,7
16,21
9,15
0,40
189
Продолжение табл.
1
2
3
4
5
6
Со2+
Oxin—
СоА2+
CoAj
СоА+
0.5
0,01
0.01
1.14
17,2
9.1
1,14
8,1
9.1
СоЗ+
NH.
Edta*-
CoAi+
СоА|+
CoAl+
СОАЗ+
СоАЗ+
СоАЗ+
СоА-
2NaN0,
(30 °С)
35,16
30.75
25,7
20.1
14.0
7,3
36,0
4.41
5.05
5.6
6.1
6.7
7.3
36.0
CNS-
С1-
СгА+
СгА2+
СгА+
СгА2+
СгАз
СгА+
СгА2+
1.0
1,0
О
О
0,5
0,5
0,5
2,98
1.87
-0,11
0,60
10.29
7.81
4.41
1.11
1,87
—0.71
0.60
2,48
3,40
4.41
CU+
Вг-
CN-
CNS-
С1-
1-
NH,
Р2О7-
so§-.
CuA^
CuAj-
CuA|-
CuA7
CuA^-
CuA^
CuA^
CuA^
CuA+
CuA+
CuA^-
CuA^-
CuA3-
6
0
0
0
4,2
3.09
0
0
2NH4NO8
2NH4NOS
пер.
5.92-
30.29
28,59
24,0
5,18
12,11
—4.94
8,85
10,86
5.93
£6.72
8.5
11,69
1.7
4,59
-6,93
/4,93
5,93
190
Поодолжение табл. II. 1
Вг-
CN-
CNS-
ci-
NH,
CuA^
CuA+
CuA2-
CuA|-
CuA^
CuAf-
CuAJ-
CuAj
CuA+
CuA+
CuA+
CuA2+
CuA|+
CuA2+
CuA2+
пер.
0
0,7Mg(NOs)2
0,7Mg(NO3)3
пер.
пер.
пер.
пер.
О
О
~0 (30 »С)
~0 (30 °С)
~0 (30 "С)
~0 (30 »С)
—4.40
—0.03
25(?)
6,52
5,19
5,62
4,89
4,40
2,80
1,23
0,82
12,03
10.06
7,33
3,99
CuNH|+
Расчетная формула для других ионных сил и температур
Ig Afl = 'g а2+0'080 / — 0,013 (t — 30)
INaClO* 1,48
ШаСЮ« 1,23
INaClO^ 1,0
пер. 10,30
пер.' 5,20
пер. 12,29
О 2,37
О 3,30
О 2,24
0,1 8,5
— 3,03
- 3.3
~0 12,44
О 19,14
>0,5 14,21
N07
P2OI-
soj-
Ао-
нт-
0Н-, т2-
CuAj
CuA+
CUNH3A+
CuA|-
CuA2-
CuA2-
CuA
CuAj
CuA+
CuA|-
CuA
CuAg
CuOHT-
Cu(OH)2T2-
CuA-
—0,03
1,33
0,73
0,49
1,60
2.80
1,23
0.82
1.97
2.73
3,34
3,99'
0,25
1,23
1.0
5,10
5,20
2,37
1,06
2,24
3,03
14,21
191
Продолжение табл. IIЛ
Cu2+
3—
|0Н- Cif
HCit2-
0Н-, Cit^-
Edta<-,
HEdta^-
Py
Oxin-
Oxin-
Sal2-
Cu (OH)2Cit3-
CuHCitHjCit
CuOHCit^-
CuA2-
CuA-
CuA2+
CuA|+ .
CuA2+
CuA2+
CuAg
CuA+
CuA2-
CuA
>0.5
0,1 KCI
O.lKNOs
0,5
0,5
0,5
0,5
0.01
0,01
19.30
4.0
16,35
18.79
11.54
6.03
5.43
4.29
2.41
23,4
12.2
16.9
10.6 '
Fe2+
Fe3+
CN-
CNS-
NO(r)
spl-
so|-
C201-
Cit3-
HCit2-
Edta"-
HEdta^-
Py
Oxin-
Br-
CN-
FeA|-
FeAj
• FeA+
FeA+
(CM. табл. 11.2)
FeA
FeA
FeA2-
FeA
FeA-
FeA
FeA2-
FeA-
FeA|+
FeA2+
FeAj
FeA+'
FeA2+
FeA|-
0
пер.
пер.
пер.
О (6.1 °С)
l,lNaC104'
>1
О
ШаС104
lNaC104'
0,1 KCI
0.1 KCI
0.5
0.5
0.01
0.01
О
О
35
0.07
0,95"
<1.5 ;
2,17
0.04
>7,75
>4.7
3.08
2.12
14.33
6.86 .
6.7 (?)
0.71
15,0
8.0
0,49
42
0.71
7,0
8.0
0,49
192
Продолжение табл. IIЛ
Fe3+
FeCN2+
Fe3+
Fe(CN)jN02-
(нитропрус-
сид)
Fe3+
FeOH2+
Fe
,3+
CNS-
ci-
F-
NO7
HPOf
S2-
S02-
' sof-
HS07
HClt2-
Edta*-
HEdta^-
Edta''-
Oxin-
Sal«-
FeA|-'
FeA,-
FeAg
FeA^
FeA2+
РеАз
FeA+
FeA2+
FeA3
■ FeA+
FeA2+
FeCNA+
FeA2+
FeA+
Fe(CN)5N0Ae-
Fe (CN)5N0A*-
Fe(CN)5N0Ae-
Fe(CN)5N0A*-
FeA+
FeA+
FeA2+
FeA3-
FeA^
FeA+
FeA+
FeA
FeA+
FeA-
FeA
FeOHA^-
FeAj
FeA+
F*A2+
FeA|-
6
пер
пер
пер
пер
пер
О
О
О
0,5
0,5
0.5
0.5
О
0,665
~0
~0
0.4
0.4
0.47.
0.15
0.15
,0 .
О
О
пэр.
INaClOi
INaClOi
0.1 KCI
0.1 KCI
0.1 KCI
0.01
0.01
0.01
7.19
7.17
6.37
4.97
3,03
1.13
2.13
1.48
12,06
9.30
5.28
3.92
1.0
9.35
5.3
5.0
0.40
1.17
2.10
2.36
1.78
20.2
16.2
9.4
7.49
11.85
6.3
25.1
16.2
20.1
33.9
23.6
12,3
'33,53
13 3.457
193
Продолжение табл. ПЛ
Fe3+
Hg|+
Sal
2—
NOi"
Br-
CN-
CNS-
ci-
P-
HgO
I-
I-
NH,
FeA7
FeA+
Hg2A+
HgjAj
Hg2A+
HgA|-
HgA7
HgA2
HgA+
HgA^-
HgAr
HgAj
HgA+
HgA2-
HgAj
HgA|-
HgAr
HgAa
HgA+
HgA+
HgA2+ = Hg|+
HgA^
HgA7
HgAj
HgA+
HgA|+
HgA2+
HgA|+
HgA2+
0
3NaC104
3NaC104
27,85
16,4
<0,6
—0,30
0,02
0,5
0,5
0,5
0,5
o,r
0,1
0,1
0,1
0
0,35
0,5
0,5
0,5
0,5
0,5
0
0,5
0,5
0,5
0,5
2NH4NO,
2NH4NO3
2NH4NOS
2NH4NO8
21,00
19,74
17,32
9,05
41,51
38,53
34,70
18,00
21,89
17,47
14,92
13,92
12,78
5,27
1,03
-8,26
29,83
27,60
23,82
12,87
19,28
18,50
17,50
8,8
» Hg (раствор в B0Ae):;lHg; lgK = — 6,5
194
Продолжение табл. ИЛ
HgOH-+
Hg2+
NOr
S2-
S02-
Ac-
Edta*-
HEdta^-
Edta*-
Py
HgA2
HgA+
HgA|-
HgA2-
HgAf-
HgA|-
HgA2-
HgA
HgAj
HgA2-
HgA-
HgOHA^-
HgA|+
HgAi+
HgA2+
3NaC104
3NaC104
пер.
3,0
0
0
0
0,33NaClO4
0,lKNOs
0,lKNOs
0,IKNOs
.0,5
0.5
0,5
0,01
0,11
54,7
22.85
33.61
Э2.26
29.86
1.42
8.43
21.80
14.6
15.4
10.4
10.0
5.1
Mg2+
HCO3-
NH,
I3-
MgA+
MgA+
.MgA2+
0 (25 »C)
пер.
0
~0
2.87
. 3.7
1.82
-0,1
HPO|-
P2O7-
Ac-
HT-
Cit^-
Edta*-
MgA
MgA2-
MgA
MgA
MgA+
MgA
MgA
MgA+
MgA-
MgA2-
0
0,02KNOs
0
0
0
0,2
0,2
0.2
0.16
0
0,1 KCI
2,5
5,70
1,79
2,29
0.82
3,43
1.36
0,92
3.2
9; 12
8,69
* hlr)i^h; /C= r-2,88(25»C)
13»
195
продолжение таб^> II.I
HEdta^-
Oxin"
MgA-
MgA-
MgA+
0,1 KCl
0,08
0
2,28
7,0
4,74
HCO3-
ci-
NH,
sot
Ac~
Edta^-
Oxin-
MnA+
MnA+
MnA2+
MnA2+
MnA
MnA
MnA+
MnA^Z
MnA
МпА2-'
MnAg
MnA+
0
0
nep.
пер.
0
0
0
0
0
0,1 KCl
0,01
0,01
1.8
-0
1,3
0.8
1.95
2,28
1.2
5,25
3,82
13.58
12,6
6,8
Br-
CN-
CNS-
NHo
NiA2-
NiAg
NiA2-
NiAg-
NiAg
NiA+
NiA2+
NiA|+
NiA2+
NiA2+
NiA2+
NiA2+
0
INaClO^
INaCIO^
INaClOa
-0(30 °C)
-0(30 °C)
-0 (30 °C)
-0(30 °C)
-0(30 °C)
-0 (30 °C)
—8.12
—3,24
22
1.81
1.64
1.18
8,01
8.10
7.47
6,40
4.79
2,67
Расчетная формула для других ионных сил и температур
Ig ==lgkl + 0.0611 — 0.0075 (t — 30)
196
продолжение > табл. 11.1
6
S02-"
Ас-
С2ОГ
Edta^-
Ру
Oxin"
NiA|-"
NiA2-"
NiA
NiA
NiAg
NiA+
NiAf-
~NiA2-
NiA2+
NiA2+
NiA2+
NiA2
NiA+
пер.
пер.
О
О
1,0
1,0
о
0,1 КС!
0,5
0,5
0,5
0,01
0,01
7; 19
5,82
2,06
2,40
1,26
0,67
6,51
18,56
3,14
2,83
1,78
18,7
9,9
1,37
5,82
2,00
2,40
0,59
0,67
18,56
0,31
1,05
1,78
8,8
9,9
Вг-
CNS-
С1-
I-
N07
Р^О^-
S2OI-
Ас-
С20Г
нт-
он-, т2-
Cit^-
Edta^-
Oxin-
PbA+
РЬАз
PbA+
PbA+
PbA+
PbA2-
РЬАз-.
PbA+
PbA+
PbA^-
PbA^-
PbA2- .
PbA+
PbA2-
PbA-
Pb (0H)2T2-
PbA-
PbA2-
. PbA-b
0
пер.
пер.
О
О
О
О
О
О
пер.
пер.
пер.
О
О
О
0,1 КС1
О
1,77
2,52
1,09
1,42
<0,8
3,85
4,65
1,46
1,19
5,32
6,35
5,13
2,52
6,54
4,7
14,1
6,50
18;з
9,02
1,77
1,43
1,09
1,42
<0,8
-0,8
1,46
1,19
1,22
2,52
6,50
18,3
9,02
197
Продолжение табл. ИЛ
Sn2+
SnOH+
Sn2+
SnOH+
Sn<+
ВГ
ci-
ci-
ci-
SnAj
SnAz
SnA+
SnOHA
SnA2-
SnAj
SnAj
SnA+
SnOHA
SnA+-
SnA2-
SnA2-
0
0
0
•^SNaClO.
0
0
0
0
3NaC104
0
nep.
nep.
1,46
1,81
1.11
0.70
1.48
2,03
2,24
1,51
1.04
4.85
-14
0,82
Sr2+
, NOJ-
Р2ОГ
Ac-
С2ОГ
HT-
Edta<-
HEdta^-
Oxin~
SrA+
SrA2-
SrA
SrA+
Sr4
SrA
SrA+
SrA-
SrA2-
SrA-
SrA+
nep.
0
0
0
0,2
0,2
0,15
0
0,1 KCl
0,1 KCl
0
0,82
4,66
2.04
0,44
2,54
1,65
0,91
2,90
8.80
8,63
2,30
2.56
Zn2+
Br-
CN-
CNS-
ci-
ZnA^-
ZnAj-
ZnAj
ZnA+
ZnA2-
7nA+
ZnA2-
ZnAj-
4,5
4.5
4.5
4,5
nep.
0.1
0
0
-1.26
-1,70
-0.97
-0.60
12.60
1.68
—1.0
0.0
198
Продолжение табл. П.1
Zn2+
NH
3
ZnAg
ZnA+
ZnA+
ZnA+
ZnA|+
ZnA|+
ZnA|+
ZnA2+
0
0
0
3,0
-0 (30 °C)
-0 (30 ^C)
-0(30 ^C)
-0 (30 °C)
-1,0
—0,5
1,26
-1,3
8,70
6,74
4,43
2,18
Расчетная формула для других ионных сил и температур:
Ig = lgkl + 0,095/ — 0,008(/ — 30)
ZnA|-
.4-
Р2О7
S20I-
sof-
Ас-
Т2-
0Н-, т2-
Edta'«-
РУ
©xin-
ZnA
ZnA
ZnA+
ZnA2-
ZnA
ZnA
ZnOHT-
ZnA2-
ZnA-
ZnA2+
ZnA2+
ZnAj
ZnA+
пер. .
пер.
О ■
0,1
О
О
0.2
0,1 KCI
0,5
0,5
О
0,3NaClO4
7.24
-2,3
2.28
1,70
7.04
4,68
2.68
7,62
16.26
13,0
1.45
0.95
17.56
9,34
-0.5
—0.5
1.26
-1.3
1.96
2.31
2.25
2.18
-2.3
2.28
1.70
2.36
4.68
, 2,68
16.26
13.0
0.5
0.95
8.22
9.34
Таблица II.2 Некоторые сложные реакции комплексообразования
Элемент
Равновесие
1
Среда
1
2
3
4
As
As (ОН)з + Н+ + С1- ±; As (O^gCl + Н^О
As (0Н)2С1 + Н+ + С1" 7^ AsOHCIg + ЩО
AsOHClg + Н+ + С1~ :^ AsCIg + Н2О
0
0
0
-1,07
—3,47
—4,20
В
В (ОН)з + ЗН+ + 4F~ 7^ BF- + ЗЩО
В (ОН)з + glyc и HBOgglyc + HgO
пер.
пер.
19,4
—0,05
Сг
НСГО7 H-^HgOg + Н+1; CrOg + SHgO
(синее окрашивание)
0,09 .
(10 X)
7,73
Fe
Fe2++ NO (г) 71 FeN02+ '
(«бурое кольцо»)
0.03Fe (N
0°С
13 °С
25 X
Н4)2 (SO4)
"0,61
0,20
—0,18
ПК ГЕТЕРОГЕННЫЕ РАВНОВЕСИЯ
Таблица III. 1. Показатели произведений растворимости некоторых солей
и оснований
AsO|-"
ВО^
Вг—
со|-
Ас-
Cg02~
Т2-.
Oxin—
1
2
3
4
б
б
7
8
9
Ag+
Ва2+
В,-з+
Са2+
Cd2+
Со2+
СгЗ+
CU+
Cu2+
22.0
15.8
50.1
9.4
18,2
32,7
28,1
20,1
35.1
2.4
8,5
12,3
11.1
9,2
8.2
13,7
12,8
9,6»
2,7
11.0 -
6,8*
8,75'
7.8*
7,7
7,5
6,2
6,1
29,0
23,38
1 3,5Н20. а ВЮВг (т) + 2H+S: Bi^ + Вг" + ЩО, lgK = — 6,52
NaC104+lH+. 'Моногидрат. * Тригидрат. 'Дигидрат
• Для |Сиз(ОН)2(СОз)2 (т) :^ ЗСи^+ + гОН" + 2С02-, рР = 45.96
I (азурит)
Для |Си2(ОН)2СОз (т) t; 2Cv?+ + 20Н- + С0§-, рР = 33,78
I (малахит)
200
Продолжение табл ИМ
9
Fe2+
РеЗ+
Hg|+
Mg2+
Мп2+
№2+
РЬ2+
Sr2+
Zn2+
20,2
19,7
28,7
25,5
35,4
18,0
27,0
22,4
4,4
10,5
16,0
4.0'
9,3
6,9
13,14
iq_,o
10,0
14,7
6,7
13.0
4.8
4.4
9.4
10,5
7.2
8,95
9,7
31,86
15.2
20.4
' Для MgCOg • ЗН2О (несквенит) рР = 4.7; для MgCOj (магнезит) рР—7.5
CN—
CNO-
CNS—
С1-
СГ02-
6
Fe(CN)|-
Fe(CN)e'
Ag+
Ва2+
81»+
Са2+
Cd2+
0)2+
Cu+
Cu2+
РеЗ+
Hg|+
Mg2+
Mn2+
N12+
Pb2+
Sr2+
Zn2+
16.0
6.6
19,5
39.3
12,0
14.3
19,5
4,7
10,0*
11,89*
9,93
5,8
10,5
10,4
8,7
17,3
4,8
5,44
8,7
13,7
4,65
8.2
7.5
8.5
40.8
16.5
14.7
15.9
40,5
11,9
12,1
14,9
14.5
15.4
24,0
20.1
^ В интервале температур 15—80 °C pP можно найти из выражения:
3000
рР = —J—— 0.40, гдеТ = 273 + /°С
* Для Ag2Cr207(T):-:2Ag+ + Cr202- рР = 6.7
» Для В iOCl (т) + 2Н+ ^ Bi^+ + СГ + ЩО, К = - 6.68
(среда 2NaC104+ Ш+)
201
Ag+
Ва2+
Ы'+
Cd2+
Со2+
CU+
Cu2+
Fe2+
Hg2+
Mg2+
Mn2+
Ni2+
Sr2+
Zn2+
Hg{CNS)|
2—
5^2
6,54
7.48
I—
4-
7,51
16,0»
18,1
12,0
7,86*
7,5
8,8
6,15»
7,6»
7.12
28,3
34,0
12,8
6,48
5,4
15,1
13,7
18,7
12.8
7,7
32,4
30,4
5.43
13,55 (акт); 14,20 (стар)
14,2 (синий)
14,8 (роз., свеж.); 15,7 (роз.,
стар.)
29,8-30,7
14.0
19,8
16,1
37 аморф.
42.5 (0,5FeA — «)
4'4 (FeOOH - а)
неустойчив
25.5
9,2 (акт.); 10,9
12,6
14,7 (свеж.); 14,2 (стар.)
14,9; 15,1 (желт); 15,3 (краен.)
26,2
16,7 (т); 16,9 (е); 16,89 (ZnO)
15,68— 15,95 (аморф.)
1 Данные для Sbj08(T) см. в табл. III.2.
I В интервале температур 12—87 °С рР = 5090/Т — 1,52, где Т=273+/ "6.
» Гексагидрат.
* При 65 -С рР = 6,5Р
202
Ва2+
Bi3+
Ca2+
Cd2+
Co2+
Cf3+
Cu+
Cu2+
Fe2+
Hg2+
Mn2+
Ni2+
Pb2+
Sn2+
Sr2+
Zn2+
P03-
Hpo:
19,9
18,2
22,5
22,9
26,0
32,6
34,7
22,62
(зел)
17,0
(фиол)
7,0
7,0
6,7
so
.2—
4,8
9,963
5,04^
.2—
•2^3
(2)
4,8
27,2«
30,3
41,2
27,4
32.0
9,9
6,2
47,6 -26,6 + 0,096
I I I I I (^ — 25)
36,9 II I I I 35,2 -14,2 + 0,054
(/-.25)
17,2 3,8-0,01 (/ — 25)
21,9 I 88?
'2»4 ^ 6,2 выпадают HgS (т) +
+Hg(T)
51,8 (черн.)—30,8+0,121
(/-25)
52,4 (крас.)-31,4+0,121
(/-25)
9,6 (роз) 11,4—0,023
(^-25)
12,6 (зел) 8,4 — 0,023
(/-25)
18,5(а); 24,0(3); 25,7(7)
7,66^ I 6,4 I I 26,6 -5,6+0,044
(/-25)
25,0 -4,0 + 0,027
6,46 вюрцит 21,6 (Р)—0,6+0,010
(/-25)
шпалерит 1 23,8 (а) —2,8 + 0,018
(/-25)
^ Здесь приведены температурные зависимости Ig/С для реакций типа»
MiS (т) + 2Н+ iM(2/^)+ + HgS (г).
, 2 NaAgSgOg . HgO (т) U Na"*" + Ag"*- + Spl^^ ЩО; pP = 12,64(4NaC104).
8 При 95°e= pP = 9,59.
^ При 20 °C pP = 4,99; при 100 °C pP = 5,9.
^ Для MgNH4P04 (T) Mg2+ + NH+ + P0^~; pP = 12,6.
« При 5 °C pP == 7.85; при 50°C pP = 7,44.
13,9
7,1
s2~(i')
49,2
97
26,1 -5,1 + 0,030
(/-25)
20,4 (a) 0,6-0,01
(/-^25)
24,7 (P)-3,7-0,01
(/-25)
203
Таблица III.la. Показатели произведений растворимости некоторых солей
реактивов-осадителей, не включенных в табл. П1.1
Элемент
Равновесие
рР
Cs
Hg
к
NH+
N1
AgBrOg (т) 7^ Ag+ + Br07
AgN02(t)i5Ag+ + NO+
AggFe (CN)5 NO (т) ±; 2Ag+ + Fe (CN)5N02-
Ag2Mo04 (t) :^ 2Ag+ + MoOf-
CsC104(t)1;Cs+ + C10J-
HgFe (CN)5 NO (т) :^ Hg2+ + Fe (CN)5N02-
KHT (t)±5:K++ HT~
KCIO4 (t)±;K+ + CI07
KgPdCIg (t) :^ 2K+ + PdCl|-
KgPtCleCT):^ 2K+ + PtCl|-
(NH4)2 NaCo (NOg)^ (т) 2NH+ + Na+ + CO (NOgj
NiNi (CN)4 (t) Ni2+ + Ni (CN)2~
Ni(Dm)2 7^Ni2+ + 2Dm"-
RbC104l;Rb+ + C10-
3-
4,28
3,8
12,1
10,5
2,4
8,82
' 3,58
3,67
5,2
4,17
13
8,77
24,64
2,6
Таблица II 1.2. Логарифмы констант некоторых сложных равновесий
с участием твердой фазы
Элемент
Равновесие
Среда
As
2As (0Н)з + SHgS 1; AsgSg (т) + 6H2O
AS2S3 (t) + S2+^2AsS7
AS2S3 (т) + 20Н~ ^ AsS~ + AsS (0Н)7
8Ь20з (т) (кубич) + 2Н+ ;± 2Sb0+ + Н2О
5Ь20з (т) (орторомбич) + 2Н+ :^ 2SbO+ + ЩО
ЗЬзЗз (т) + 2Н2О + 2Н+ ±; 2SbO+ + 3H2S
Sb2S3(t)+S2--Sb2Sf-
SnS2 (t) + HS-:;lHSnSj-
irep.
nep.
nep. (20" C)'
0
0
0
nep.
nep.
25,2
2,0
2,15
—6,10
—4,64
-27,8
2,08
—0,7
204
Т а блица III.3. Логарифмы постоянных закона Генри, X,
для равновесий типа R i;R (г); X = P(r)/(R) между газами и водными
растворами
Гаэ»С
20
25
30
40
60
80
90
N0 .
ЩО
S02
С1,
3.02
1.Г2
3.00
2.48
2.66
0.69
-0.62
0,68
3,10
1,42
3,17
2.68
1.57
2.85
0.94
-0.18
1.00
3.12
1.48
3.21
2.72
1.62
2.92
0.99
-0.10
1.05
3.14
1.54
3.24
2.77
2.96
1.05
—0.02
1,09
3.16
1,66
3.30
2.82
3.02
1.14
0.12
1,19
3.24
1.89
3.43
2,96
3.15
1.27
0.31
1,34
3.41
3,62
3.18
3.37
3.64
3.85
3.42
3.60
(1,44 при
100 "О
0,48
1.51
0.55
1.74
Таблица III.4. Константы и их логарифмы для равновесий распределения
некоторых веществ между водой и органическими растворителями
KURiopr), X = (R(opr))/(R)
Вещество
Органический растворитель
X
h
h
CgHgNHj (анилин)
CCI4
CCI4
CHCI3
CgHe (бензол)
27
89.9
250
10
1.43
1.954
2.40
1.00
205
ОГЛАВЛЕНИЕ
1. Растворимость и произведение растворимости 4
1.1. Общие сведения о растворимости , 4
1.2. Произведение растворимости 5
1.3. Связь между произведенией растворимости и растворимостью б
1.4. Примеры расчета Р по измеренным L 8
1.5. Примеры расчета L по табличным значениям Р 13
1.6. Растворение соединений, малорастворимых в воде ..... 18
1.7. Расчет оптимальных условий осаждения . 37
2. Выделение твердой фазы из гомогенной системы 45
2.1. Общие сведения 45
2.2. Зависимость растворимости от температуры 47
2.3. Кристаллизация чистых жидкостей 49
2.4. Распространение теории кристаллических зародышей на
растворы электролитов 50
2.5. Теория относительного пересыщения 51
2.6. Зависимость растворимости от величины поверхностного
натяжения на границе между твердой фазой и раствором .... 54
2.7. Применение выражения относительного пересыщения к
системам, в которых зависимость L от r различна 58
2.8. Выделение твердой фазы из пересыщенного раствора .... 60
2.9. Образование золей , 62
2.10. Свойства золей 64
2.11. Коагуляция золей 71
2.12. Некоторые практические выводы из теории коагуляции ... 77
2.13. Последовательное выделение осадков из гомогенного раствора 78
3. Соосаждение 85
3.1. Твердые растворы . 85
3.1.1. Экспериментальная проверка чистоты осадков .... 91
3.1.2. Равновесия в системе твердый раствор — жидкая фаза 91
3.1.3. Равновесия в системе чистая твердая фаза — раствор,
содержащий замещающий ион 92
3.1.4. Равновесия образования гомогенного твердого раствора
при одновременной застройке решетки М, В и А-ионами 96
3.1.5. Равновесия при образовании гетерогенного твердого
раствора 97
3.1.6. Предотвращение образованная твердых растворов ... 100
3.2. Адсорбция 101
3.2.1. Адсорбция потенциалопределяющих ионов 101
3.2.2. Молекулярная адсорбция 103
3.2.3. Однослойная (ионная) адсорбция . 106
3.2.4. Снижение адсорбции 110
3.2.5.. Адсорбция на коллекторе 111
3.3. Последующее осаждение . 112
4. Промывание осадков 116
4.1. Предварительные замечания 116
4.2. Отмывание осадка от маточного раствора 116
4.3. Отмывание примеси, попавшей на поверхность осадка за счет
молекулярной адсорбции . . . 119
206.
4.4. Отмывание примеси, попавшей на поверхность осадка за счет
ионной адсорбции . . . . 122
4 5. Очистка осадков за счет «старения» при дигерировании ... 124
4.*6. Промывание осадков растворами электролитов 125
4.7. Разделения на сорбентах различной природы 131
4.7.1. Общие сведения о хроматографии 131
4.7.2. Ионообменники и их применение 132
4.8. Несколько общих замечаний об использовании гетерогенных
систем, состоящих из твердой и жидкой фазы ....... 135
5. Распределение веществ между двумя жидкими фазами ... 137
5.1. Общие сведения о процессе распределения . 137
5.2. Краткие сведения о применяемых экстрагентах ...... 137
5.3. Механизм распределения 138
5.4. Расчеты равновесий в двухфазных жидких системах 140
5.5. Некоторые выводы, связанные с расчетами равновесий
распределения . , 144
5.6. Теория экстракции для случая, когда экстрагируемое вещество
ни в первой, ни во второй жидкой фазе никаким превращениям
не подвергается 145
5.7. Теория экстракции для случая, когда экстрагируемое вещество
может взаимодействовать с обоими растворителями, но не дает
многоядерных комплексов . 147
5.8. Множитель <p^^.^^ c^h I ^ ^н\к'^\^^ в уравнении (5.26), его роль и использование , . 149
5.9. Оптимизация распределения 150
5.10. Экстракционные системы. Соэкстракция ........ 154
5.11. Выводы и задачи . 156
6. Буферные системы 157
6.1. Предварительные сведения 157
6.2. Буферность гомогенных кислотно-основных систем ..... 158
6.2.1. Буферность системы: сильная кислота и ее соль .... 158
6.2.2. Буферность системы: сильное основание и ее соль ... 159
6.2.3. Экстремальные значения р на кривой (Р, рН) для
систем HY-XY, XOH-XY , 159
6.2.4. Кривые (Ig р; а) для систем HY-XY и XOH-XY 160
6.2.5. Буферность системы: слабая кислота и ее соль .... 161
6.2.6. Экстремальные значения р для системы НА — ХА . . . 162
6.2.7. Кривые (lgp, а) для систем HY — XY и НА — ХА, относящиеся к одной области рН 163
6.3. Ионобуферные гомогенные системы 164
6.4. Ионобуферные гетерогенные системы 167
Заключение 170
Список литературы 171
Приложение , , 175
НИКОЛАЙ ПЕТРОВИЧ
КОМАРЬ
Химическая Метрология
ГЕТЕРОГЕННЫЕ ИОННЫЕ
РАВНОВЕСИЯ
Редактор Л. Ф. Кизилова, переплет художника В. Б. Мартыняка,
художественный редактор В. Е. Петренко, технический редактор
Г. П. Александрова, Koppeктopы Л. А. Марченко, Г. В. Всеволожская.
ИБ № 8184
Сдано в набор 12,12.83. Подп. в Ре*1ать Oo.va.oi. ЬЦ 09149.
Формат 60Х90/и. Бумага типогр. №^3, Лнт. гарн. Выс. печать.
13 печ. л. 13,26 кр.-отт. 16 уч.-изд. л. Тираж 1260 экз.
Изд. № 1190. Зак. 3-467. Цена 2 р. 40 к.
Издательство при Харьковском государственном университете
издательского объединения «Внща школа», 310003, Харьков-3,
ул. Университетская, 16
Харьковская книжная фабрика «Коммунист», 310012,
Харьков-12, ул. Энгельса, 11
химическая
метрология
Н. П. Комарь
ГЕТЕРОГЕННЫЕ ИОННЫЕ
1ЭДВН0ВЕСИЯ
ХАРЬКОВ
ИЗДАТЕЛЬСТВО ПРИ ХАРЬКОВСКОМ
ГОСУДАРСТВЕННОМ УНИВЕРСИТЕТЕ
ИЗДАТЕЛЬСКОГО ОБЪЕДИНЕНИЯ
«ВИЩА ШКОЛА»
1984