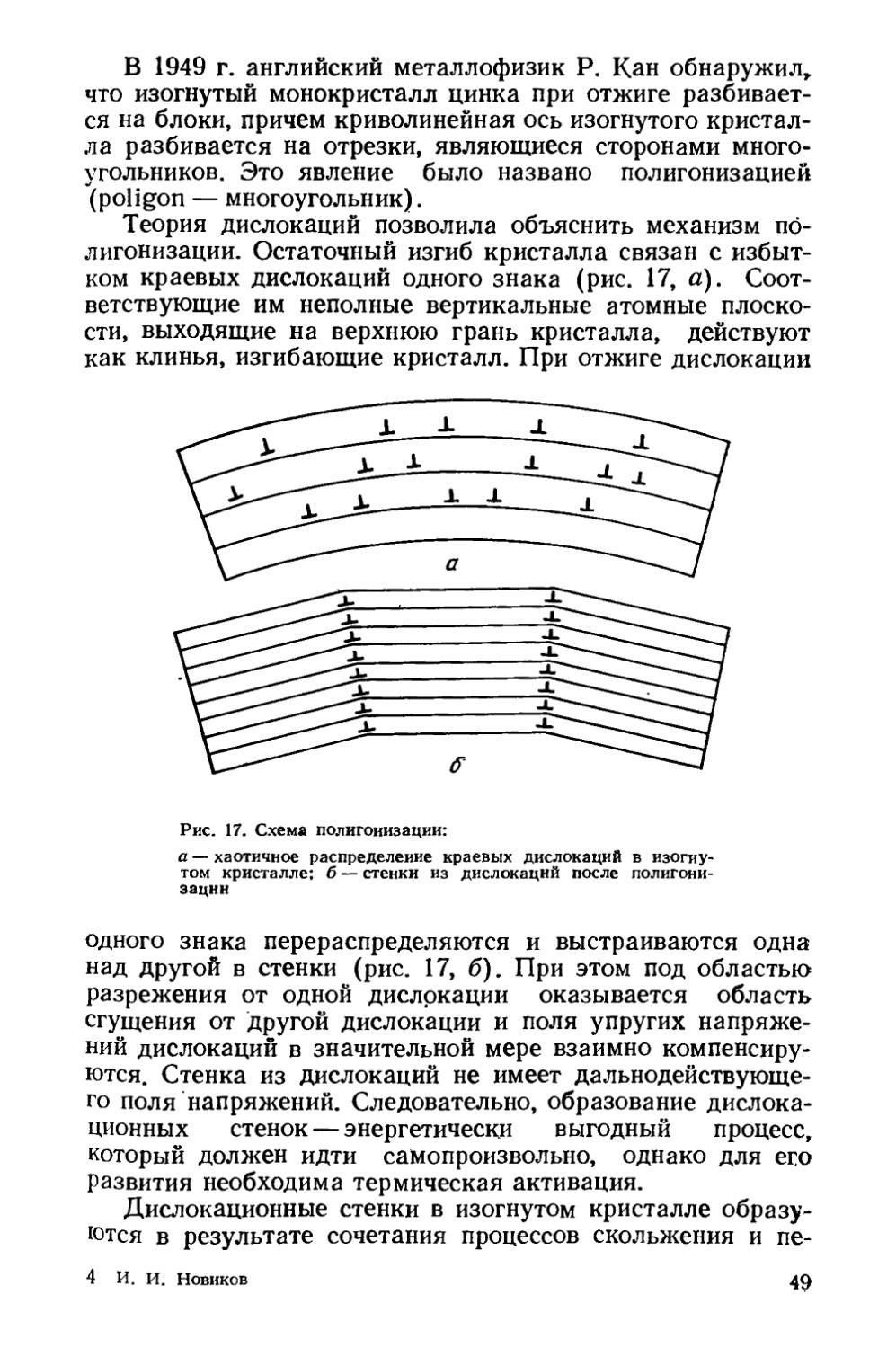
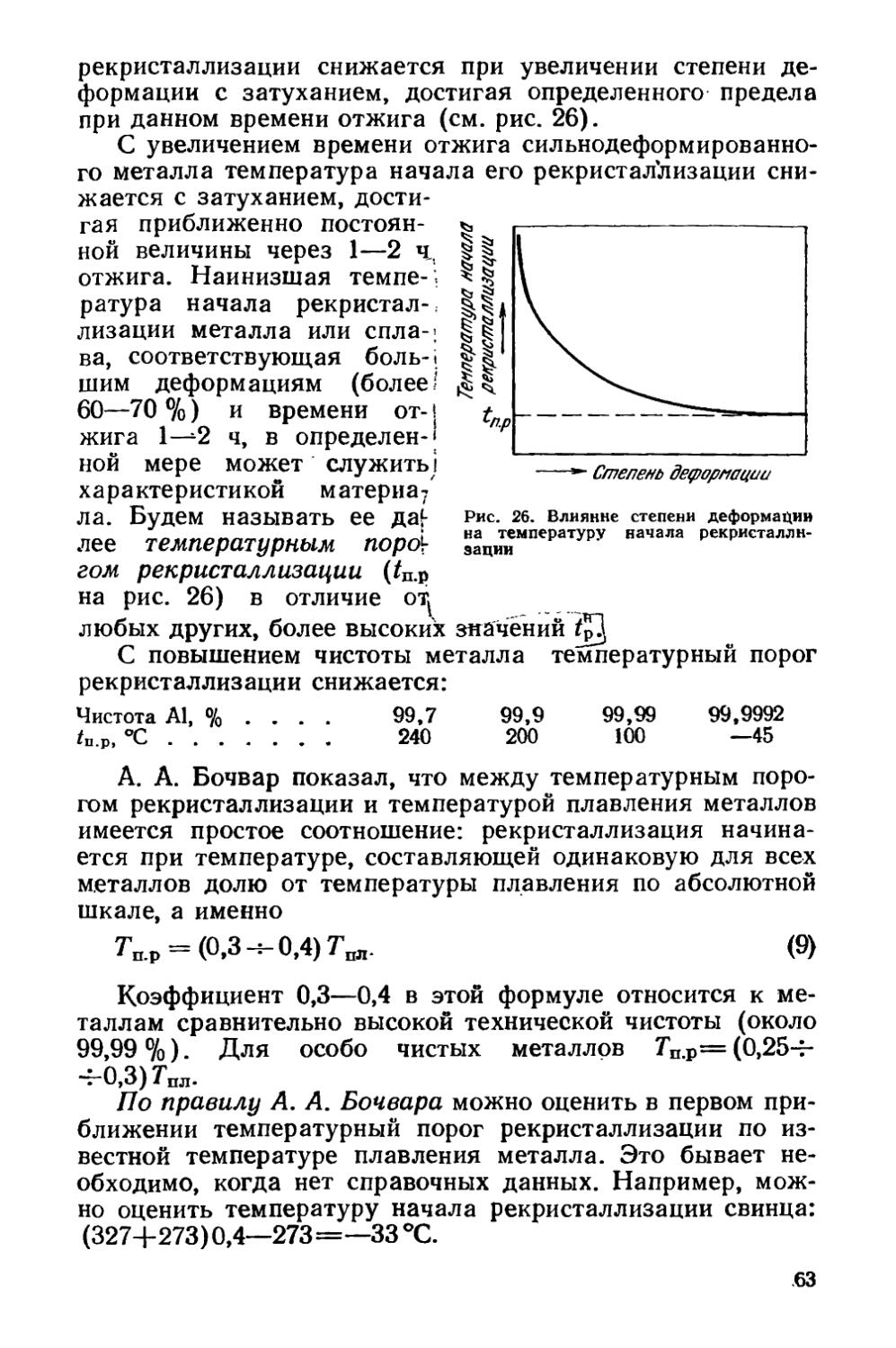
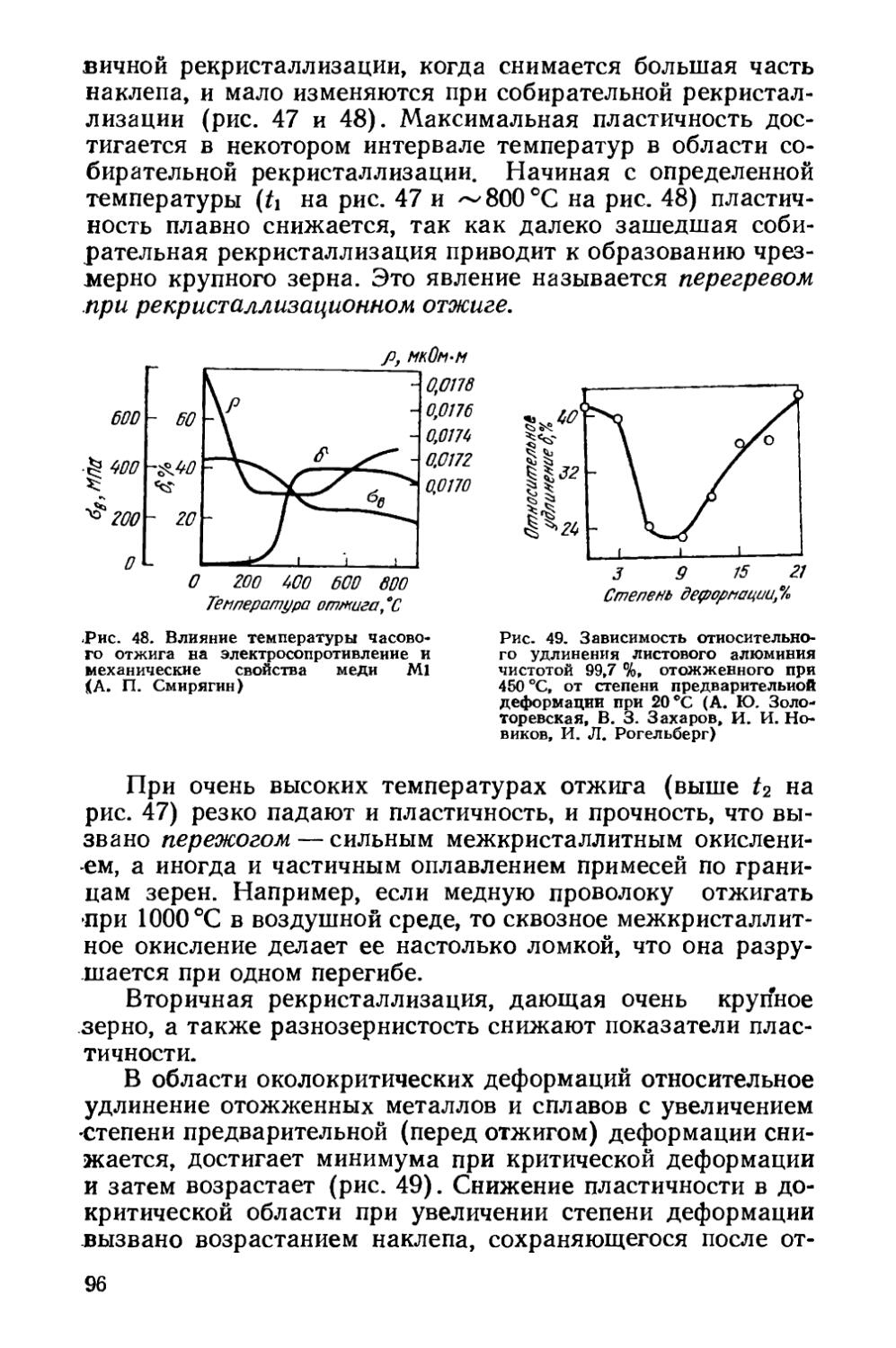
/









































































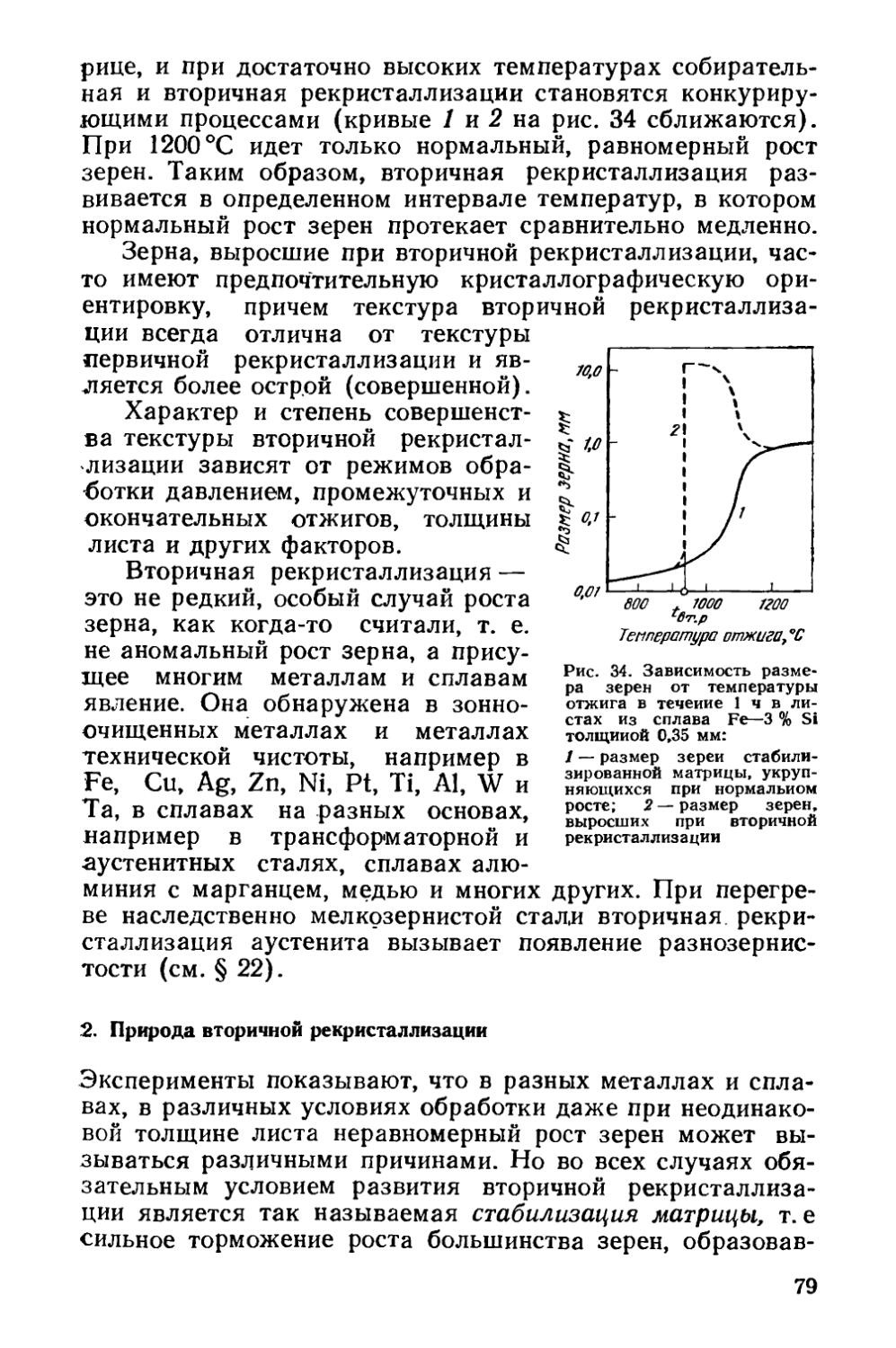



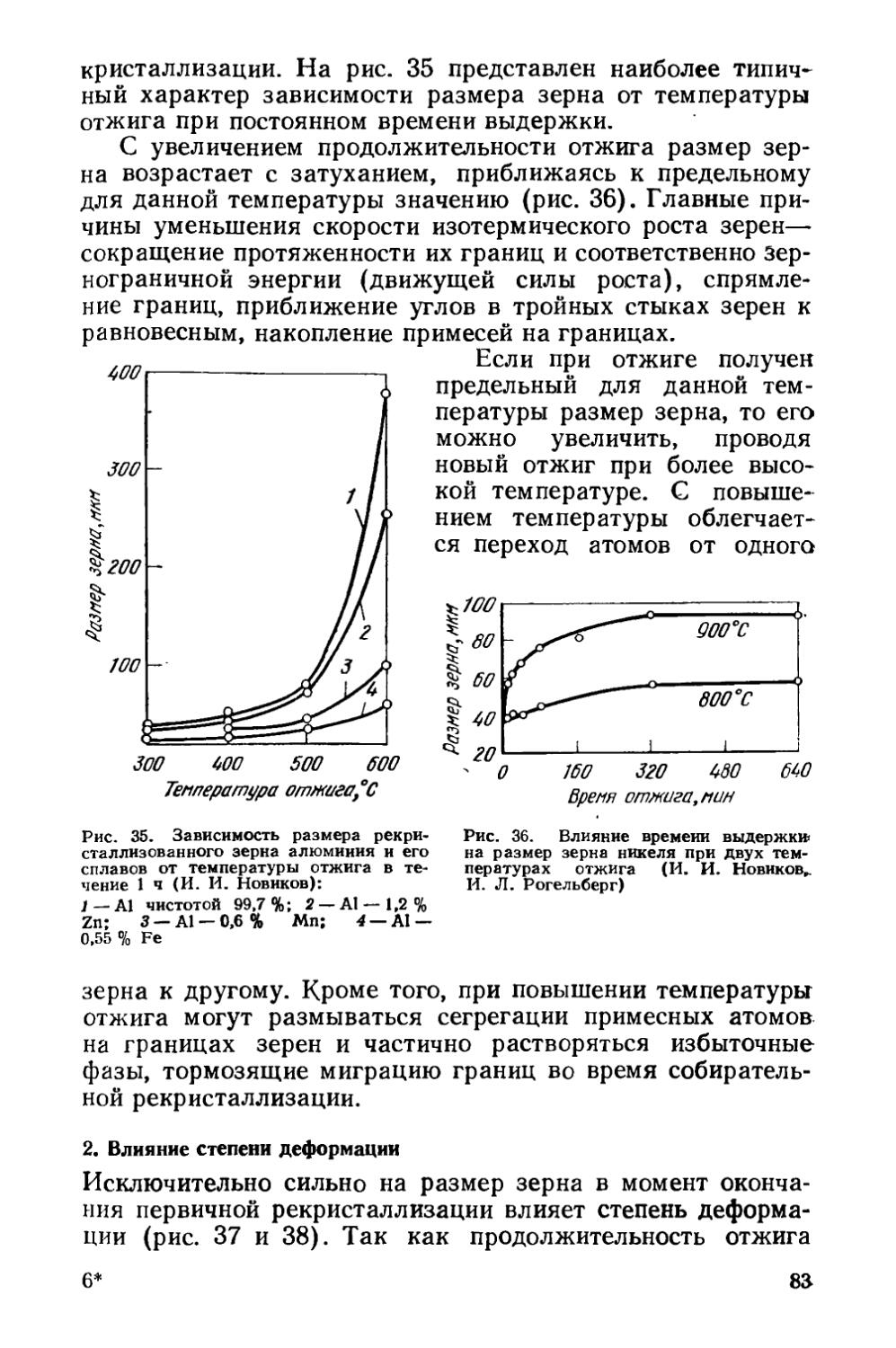

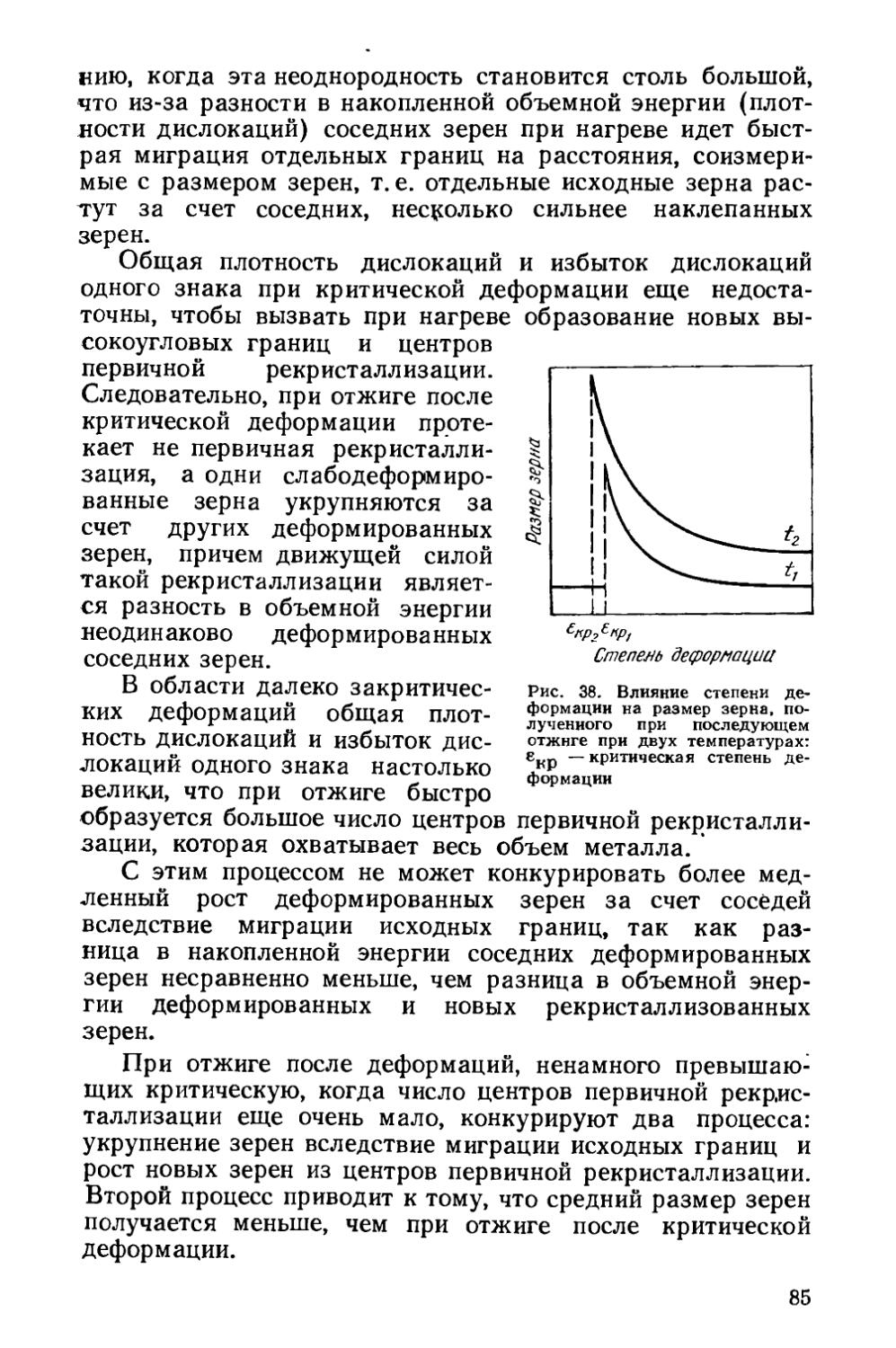

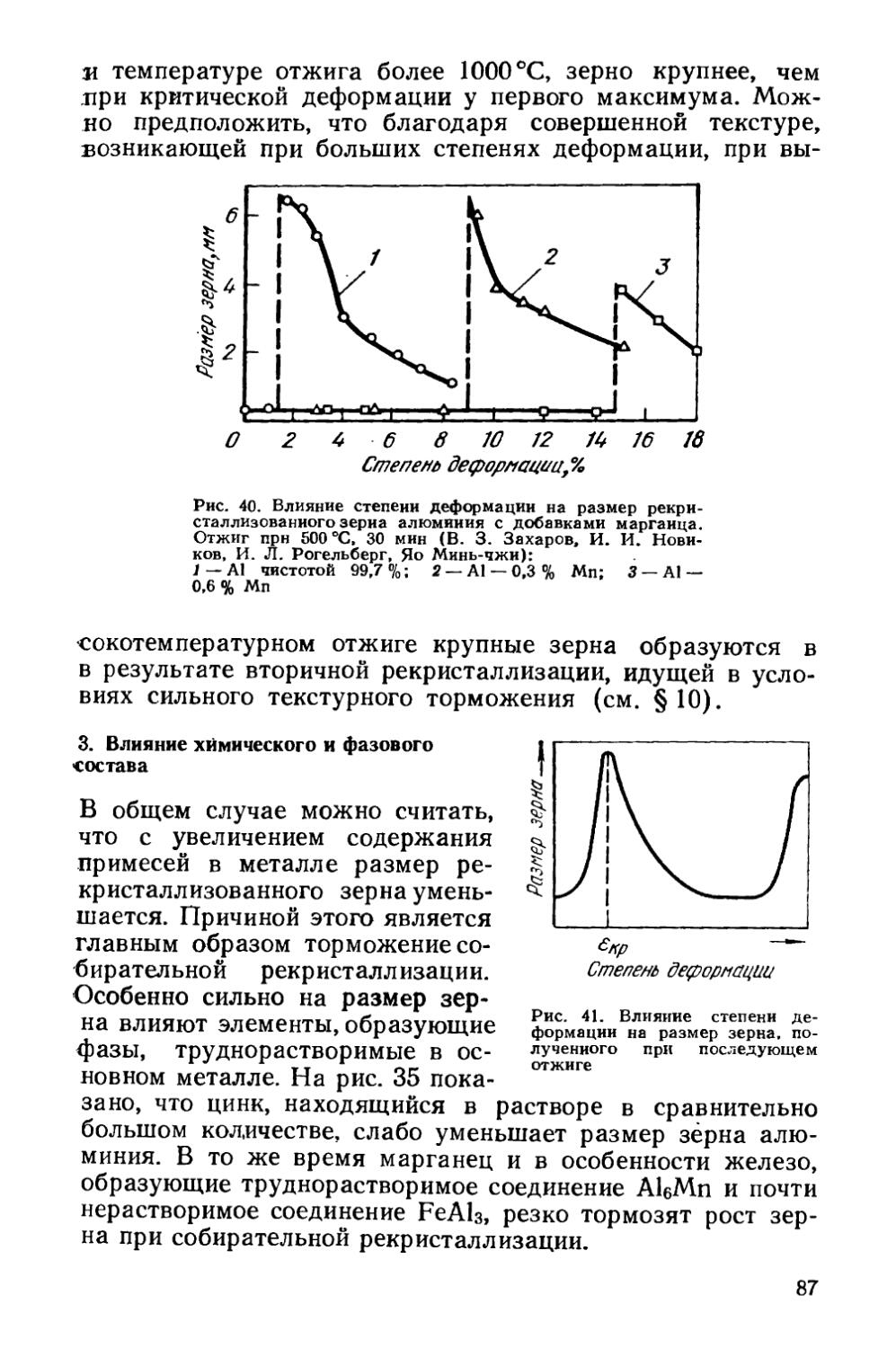

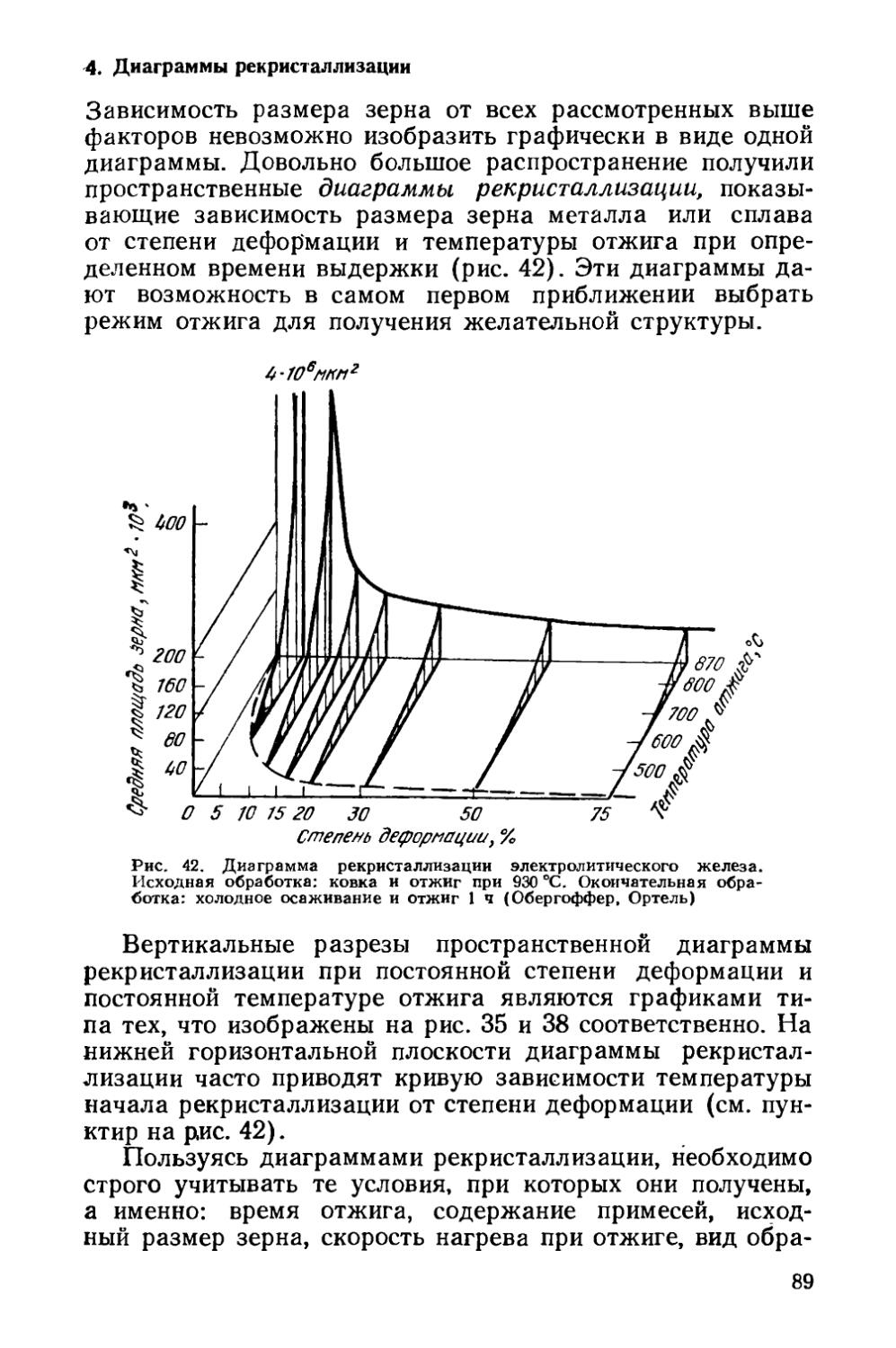



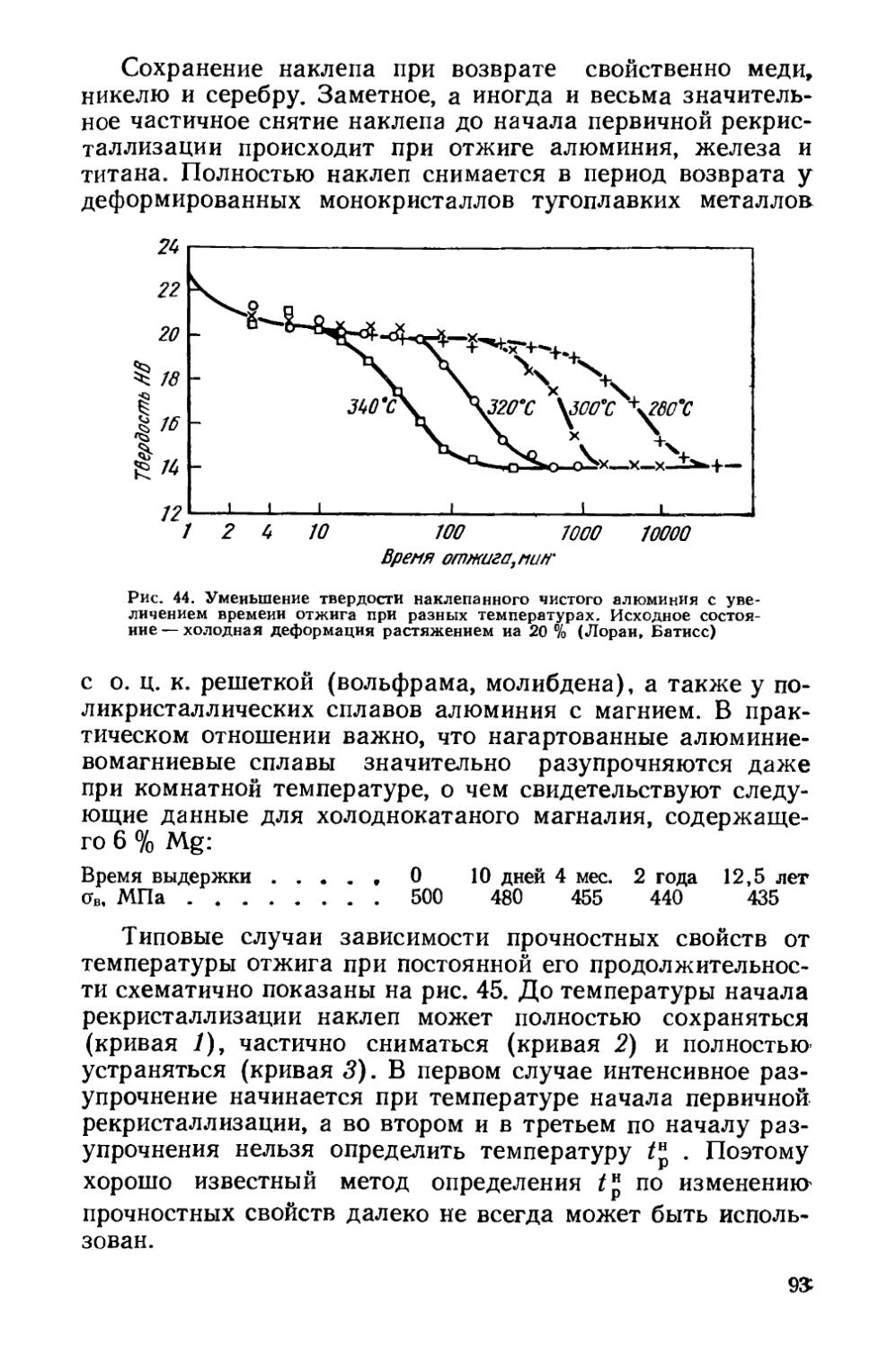

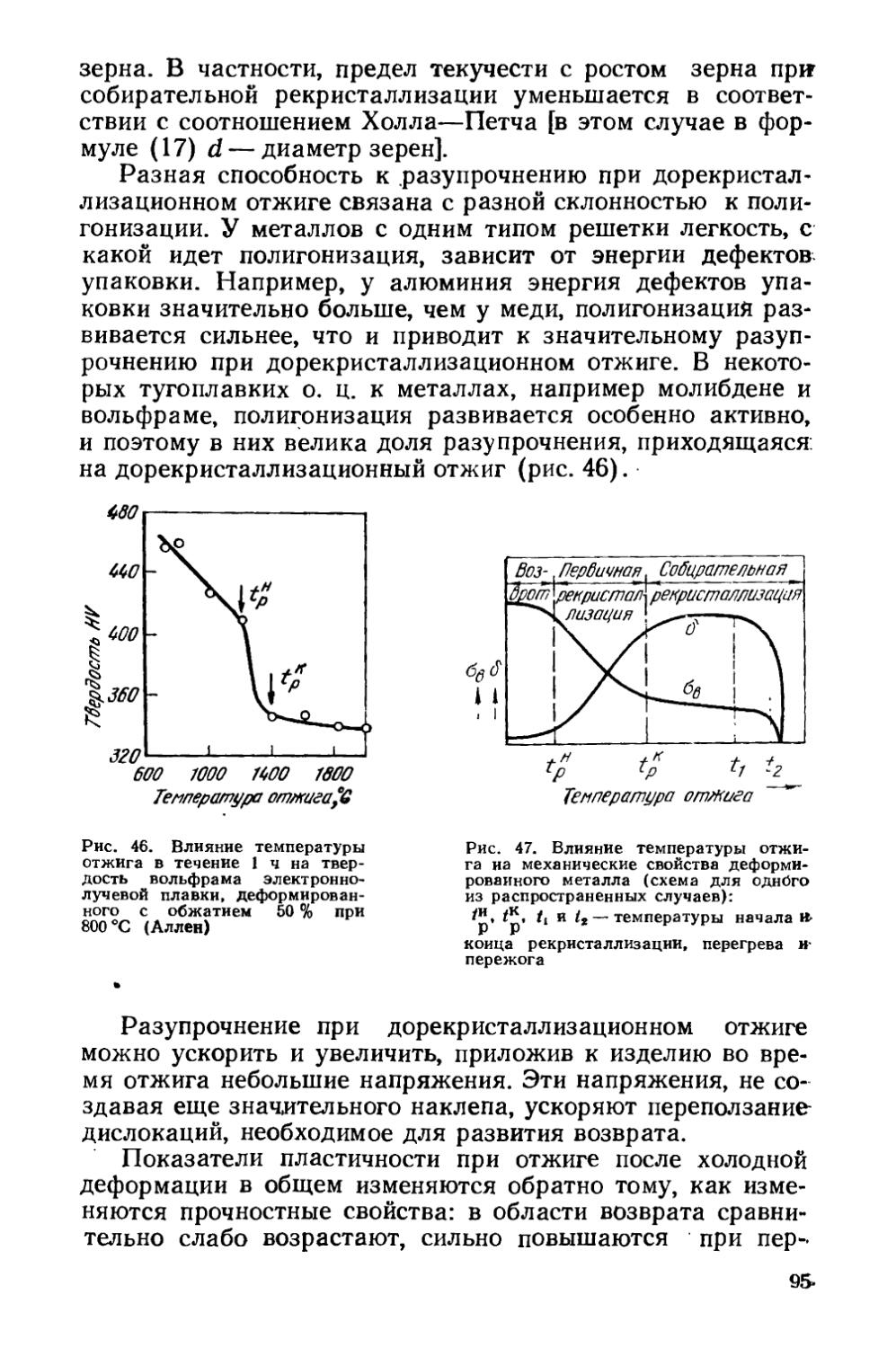
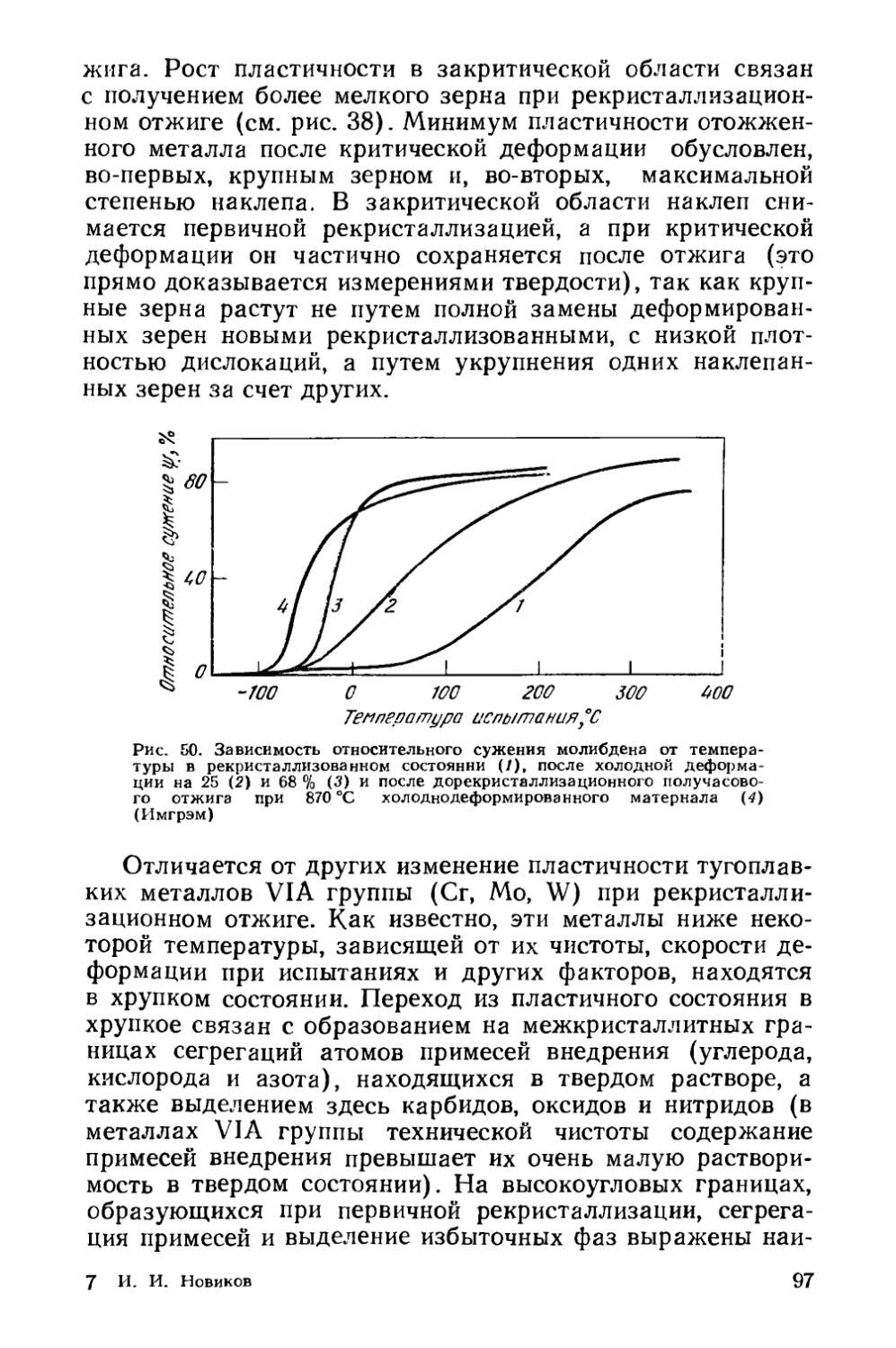

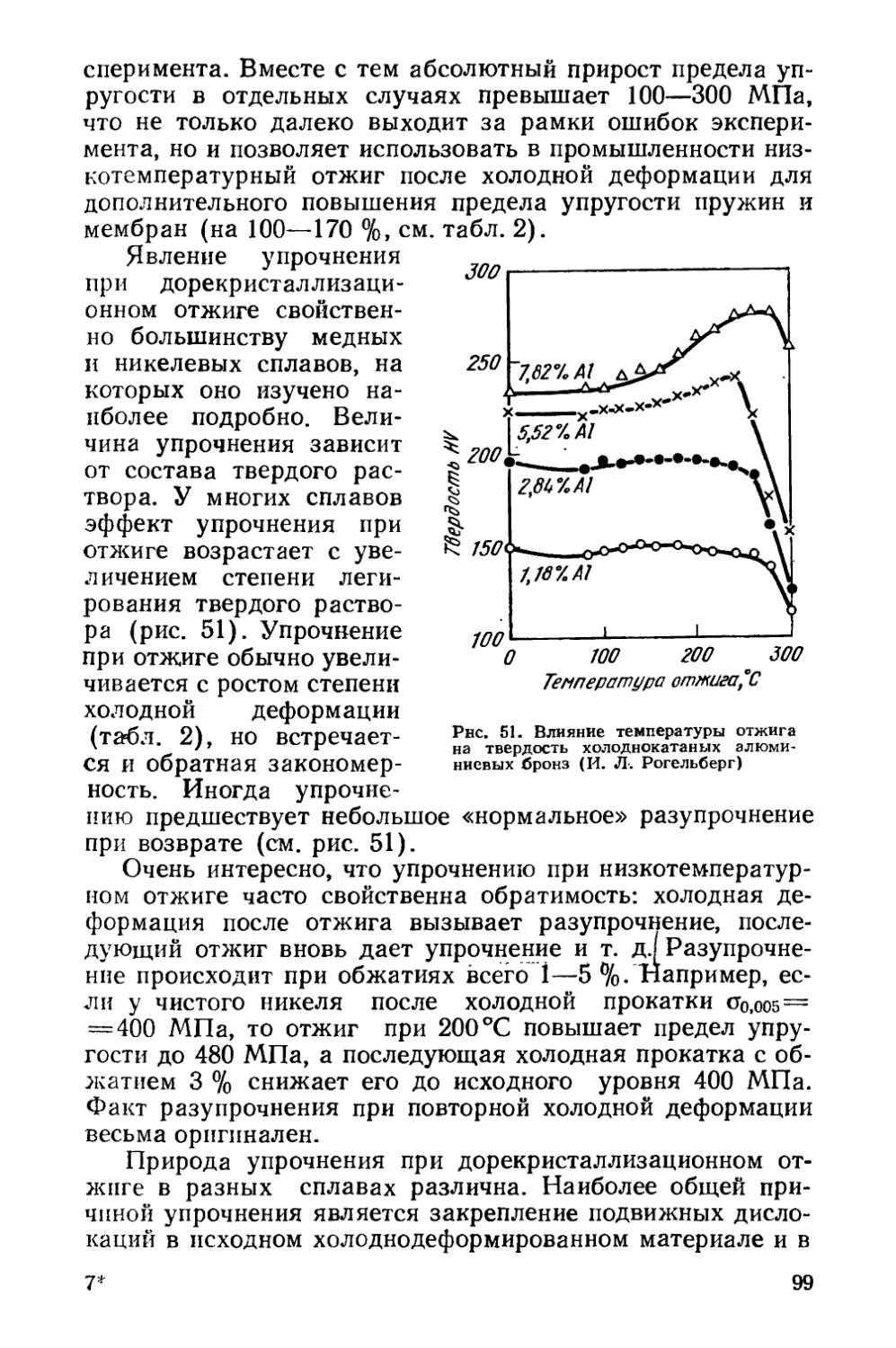


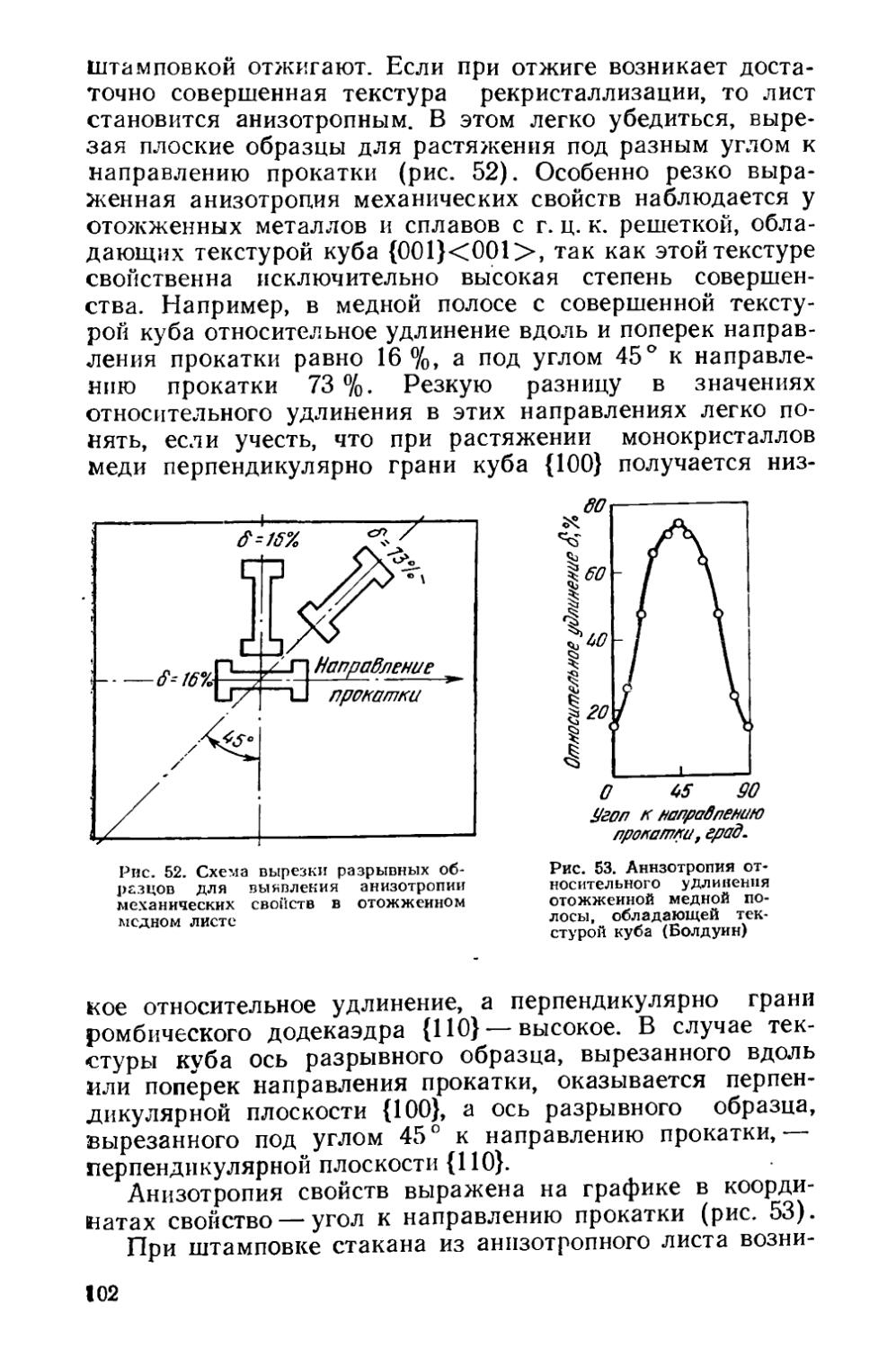






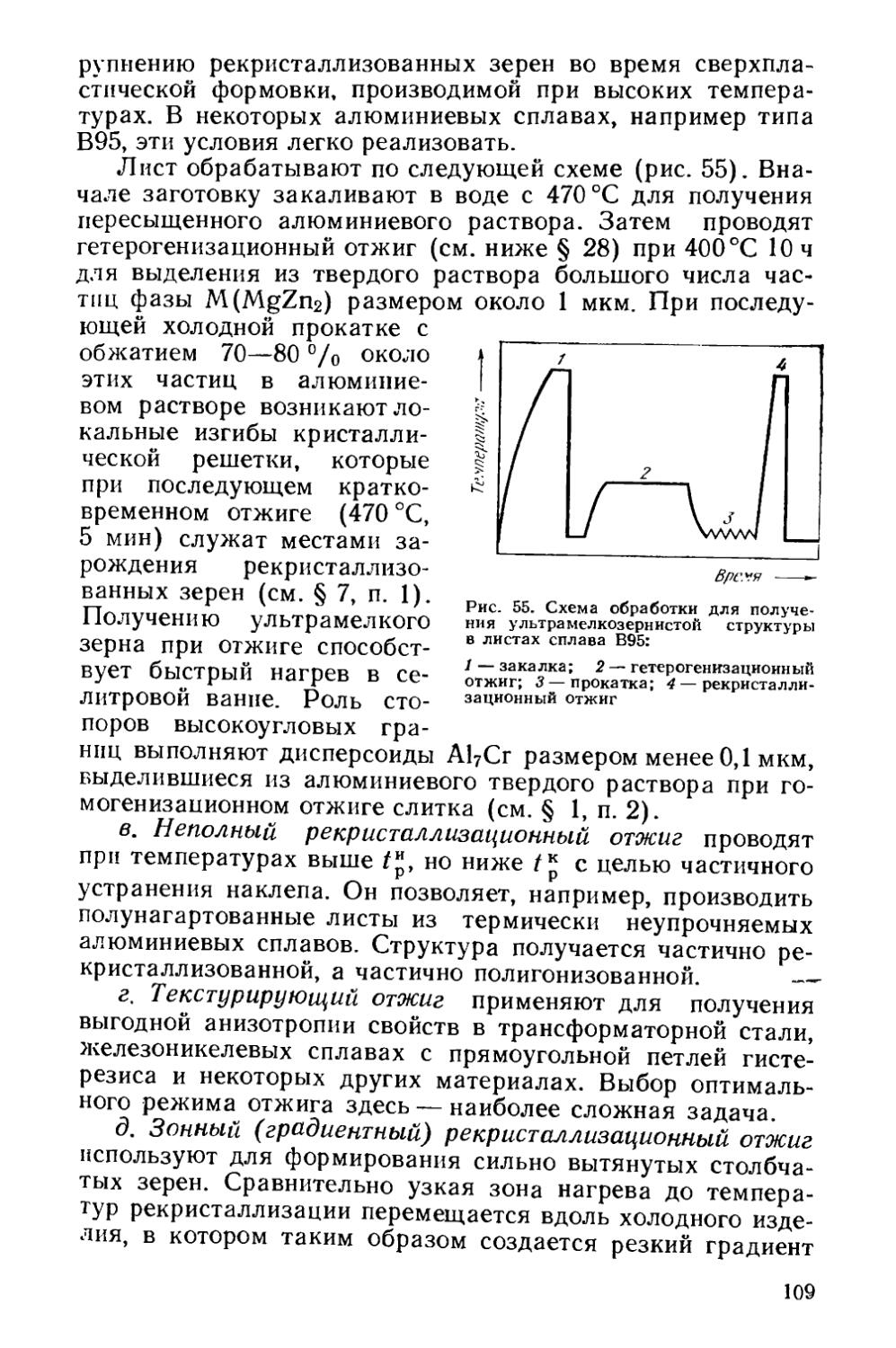
































































































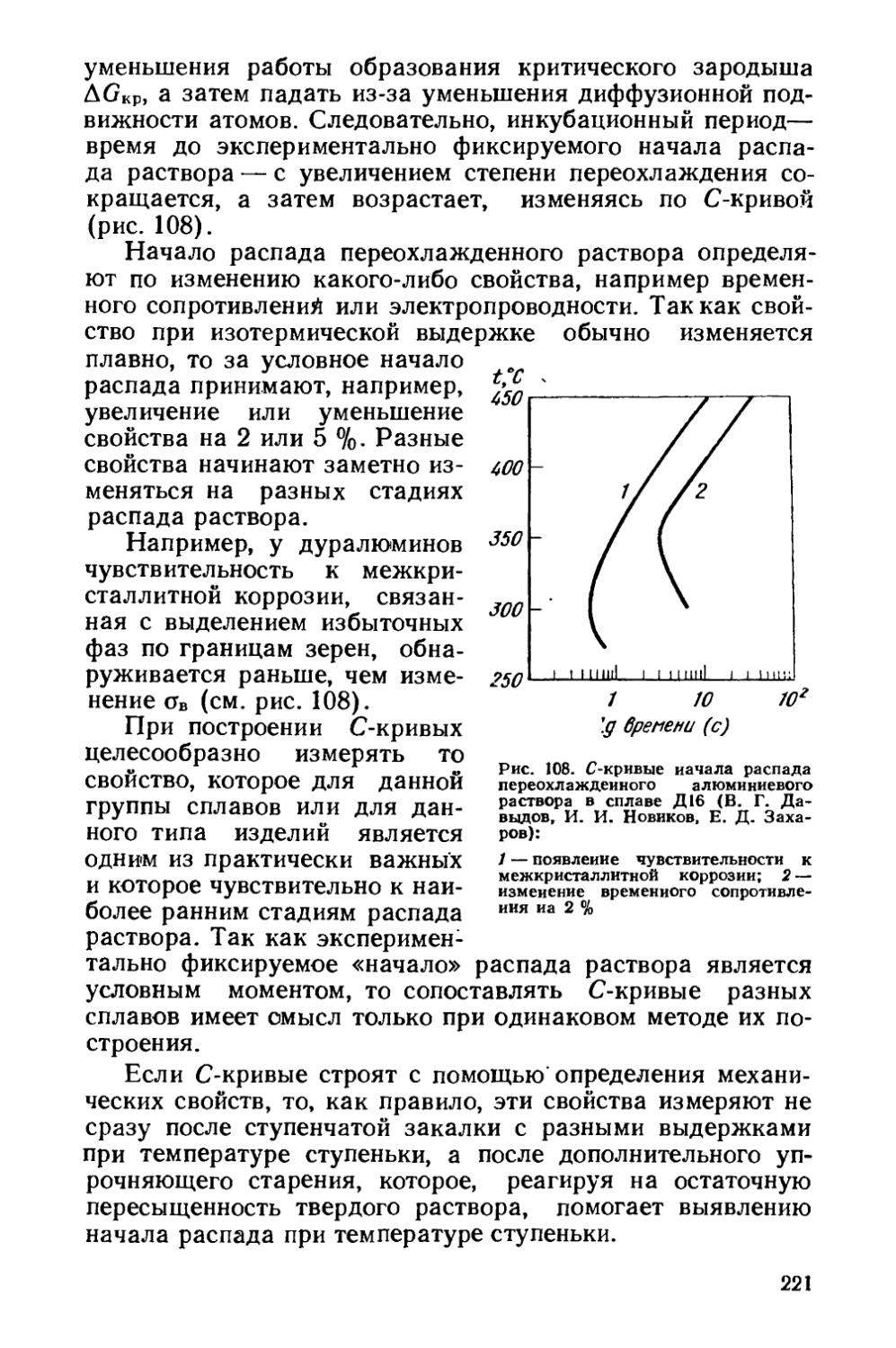


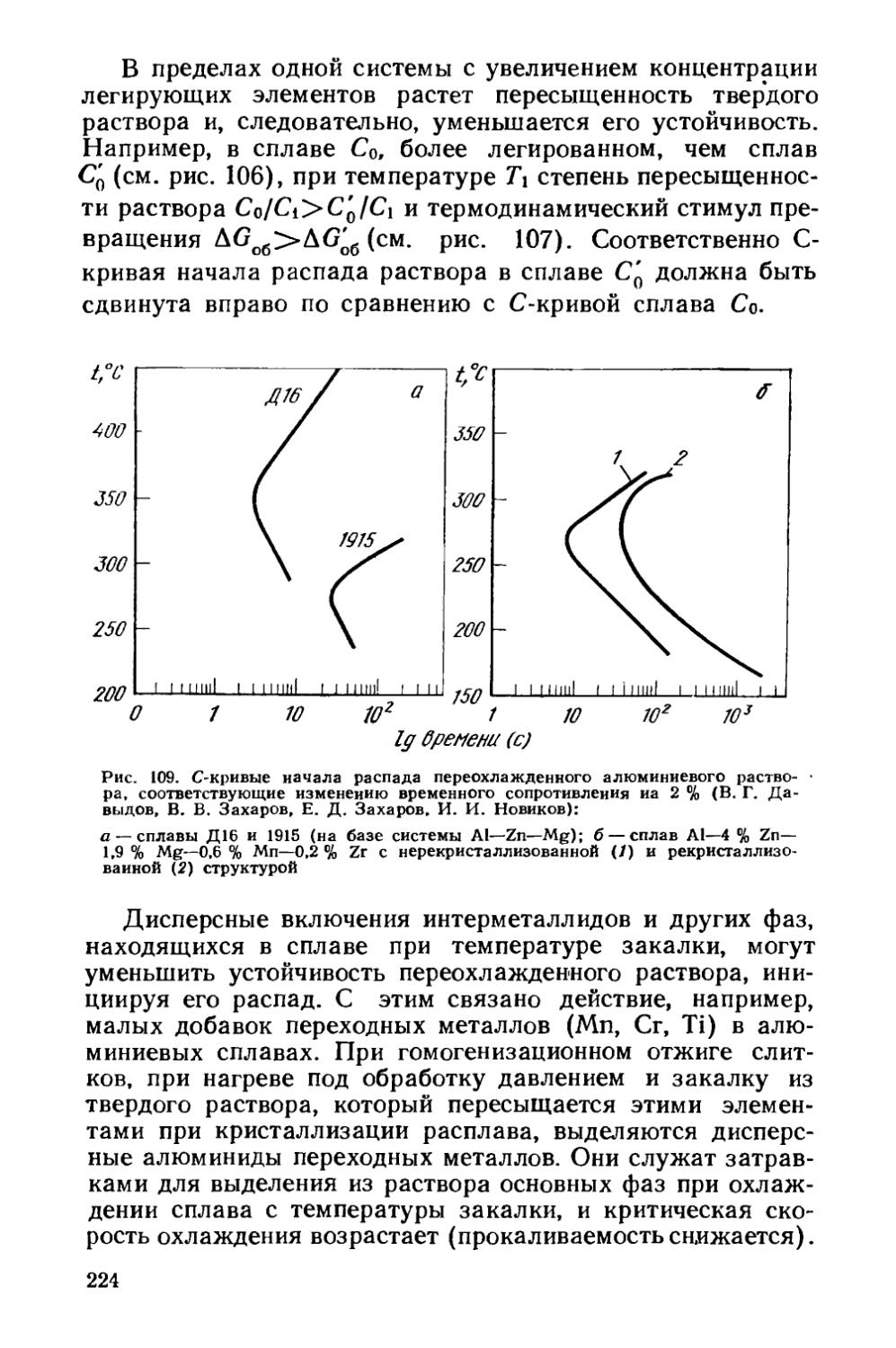

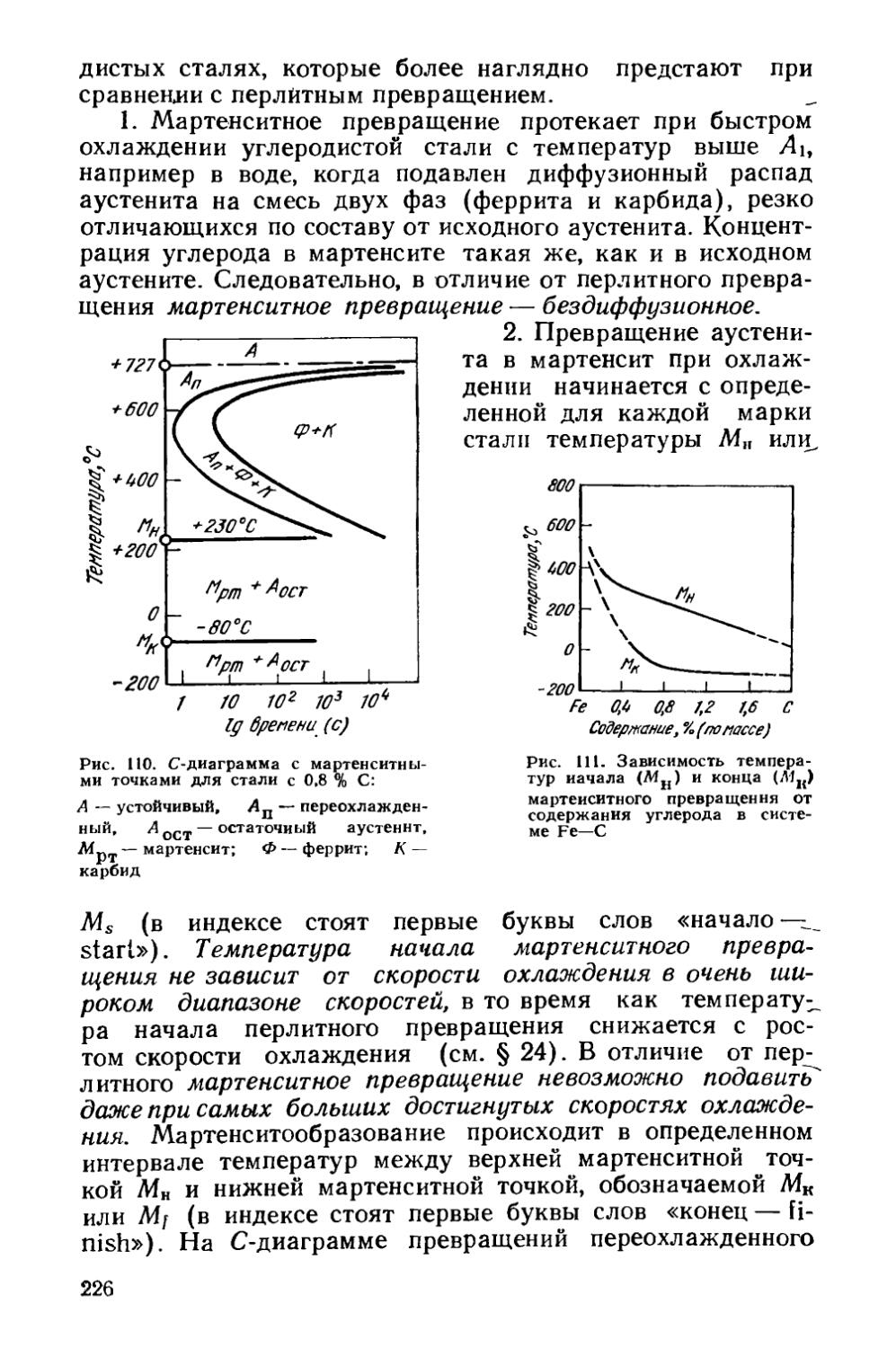






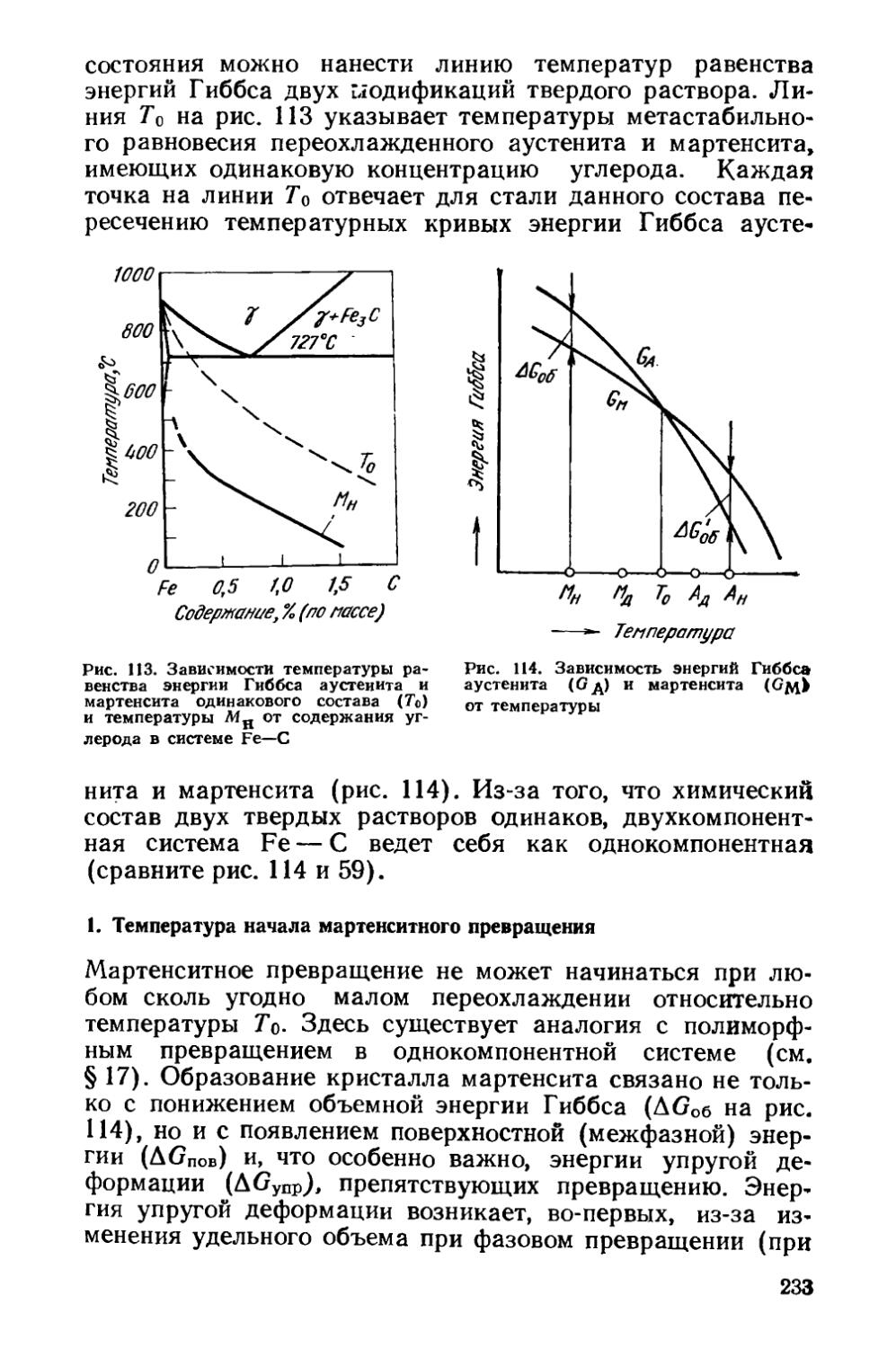































































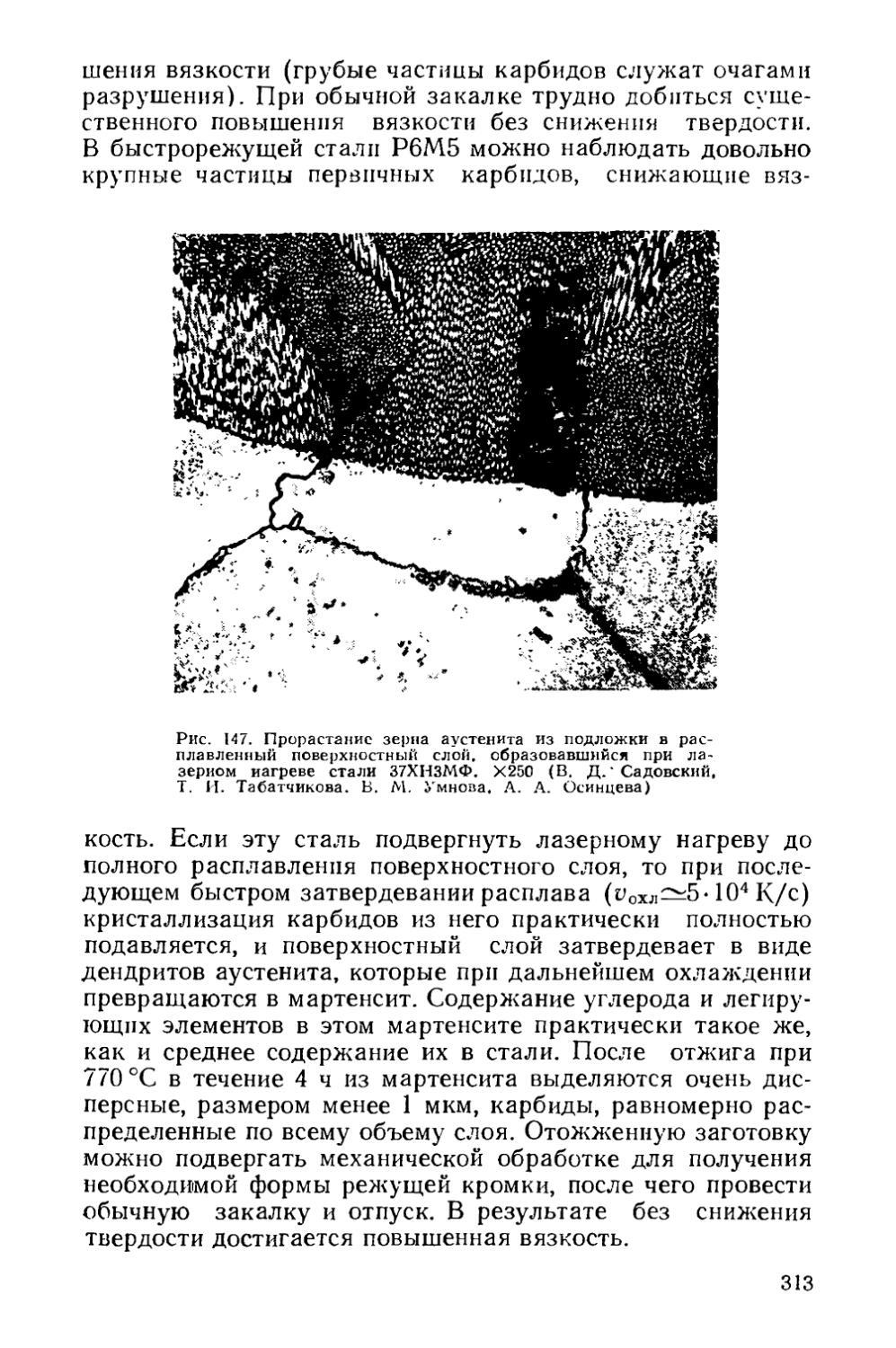









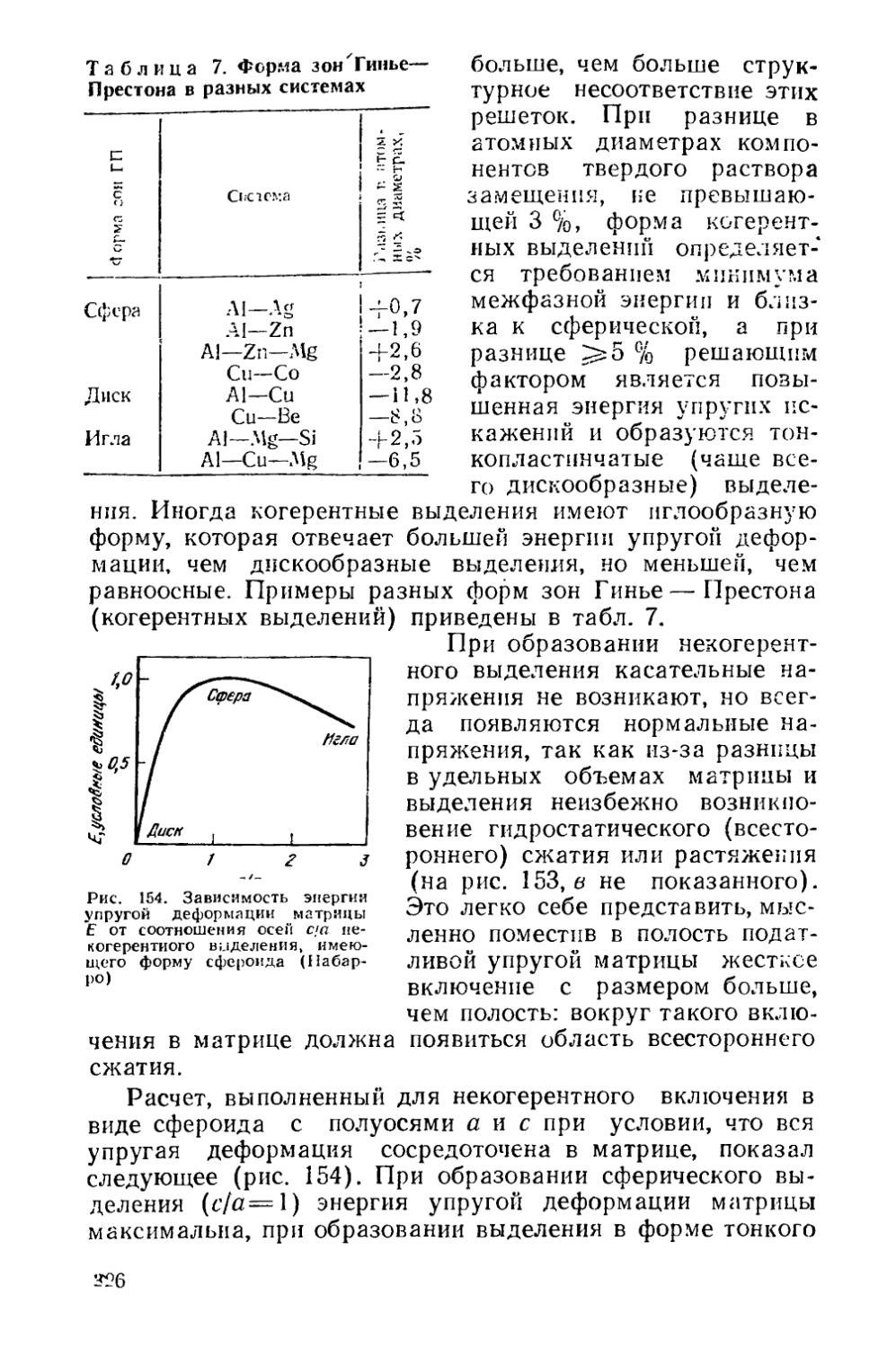







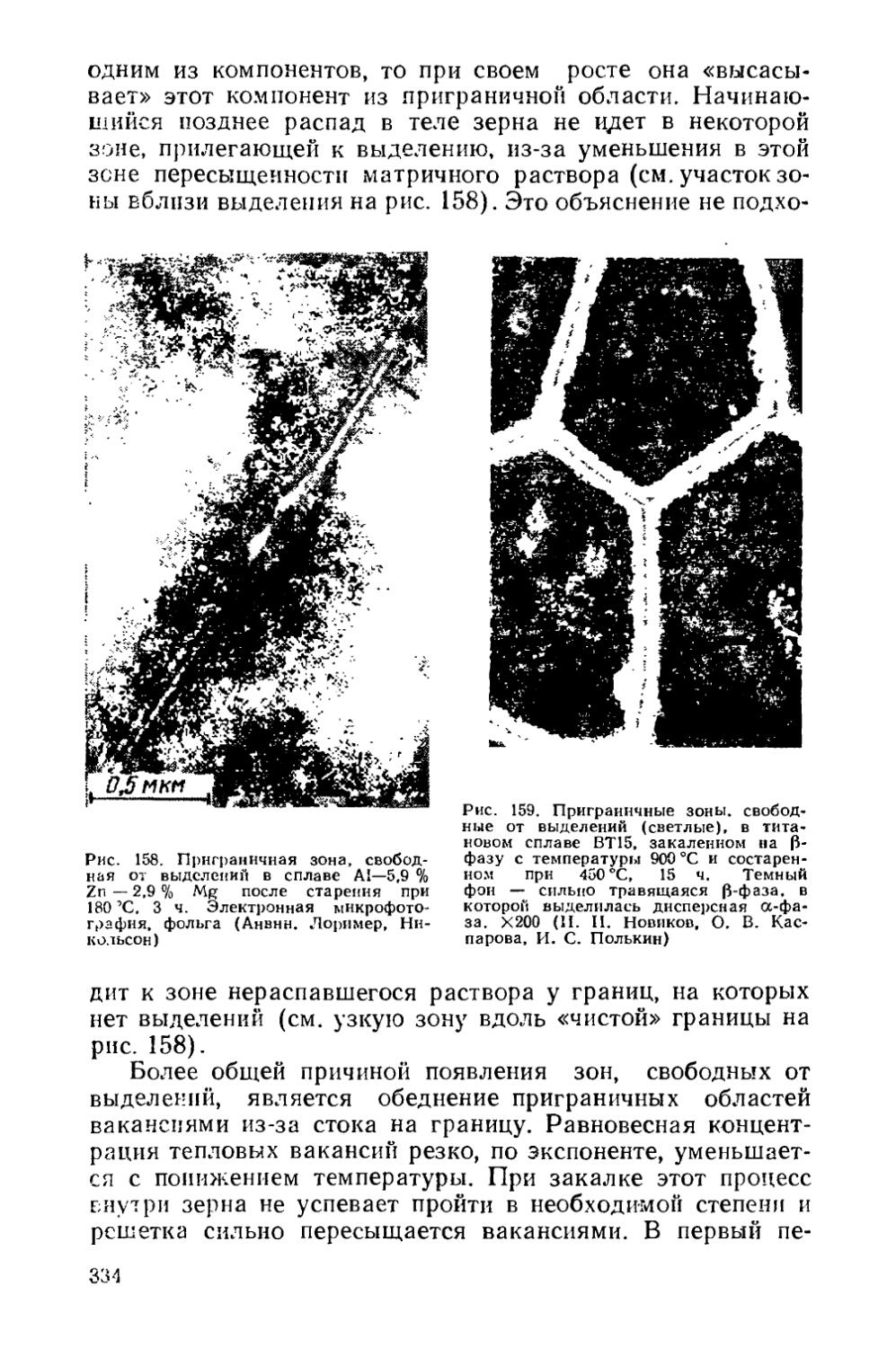
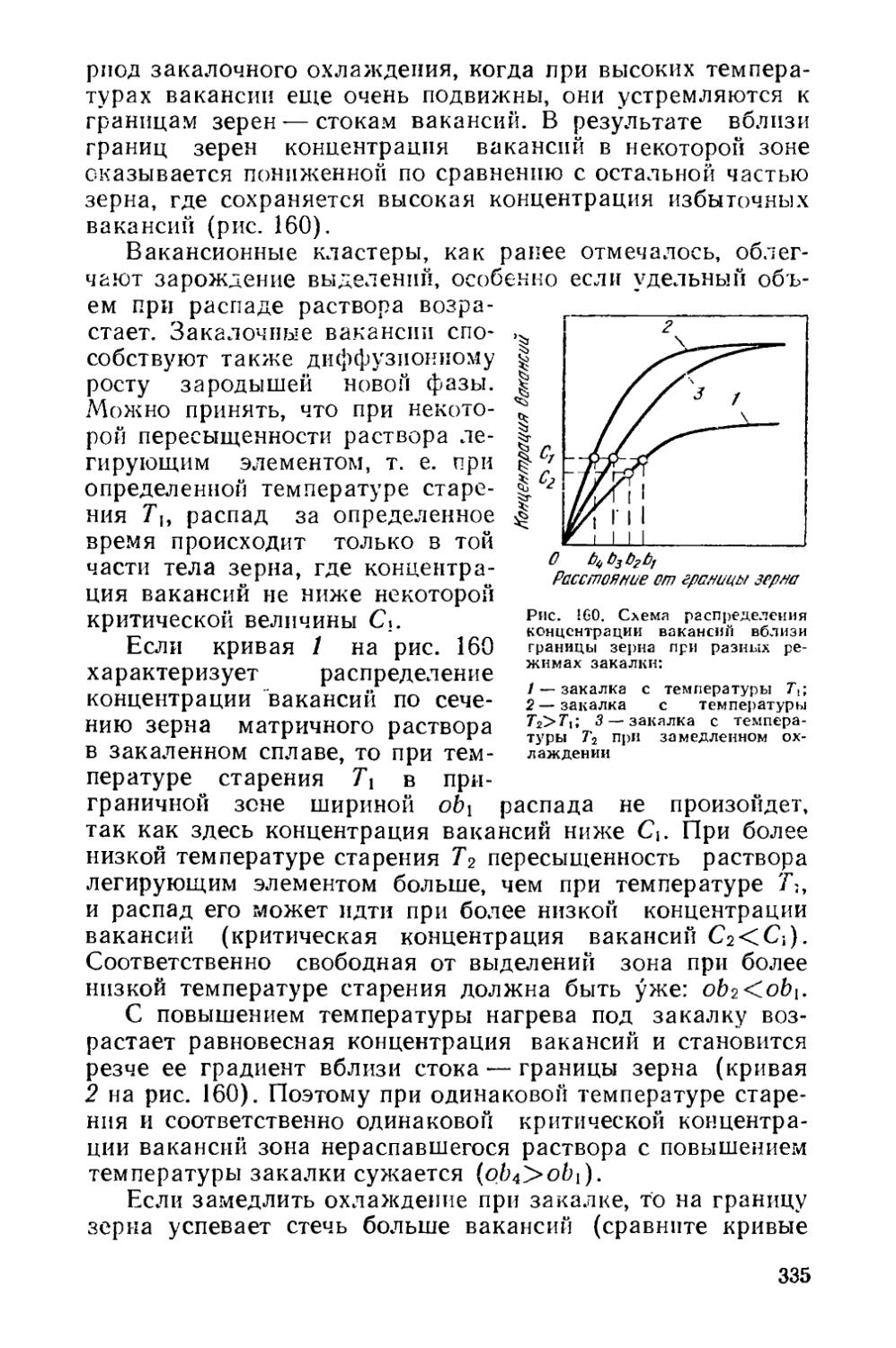





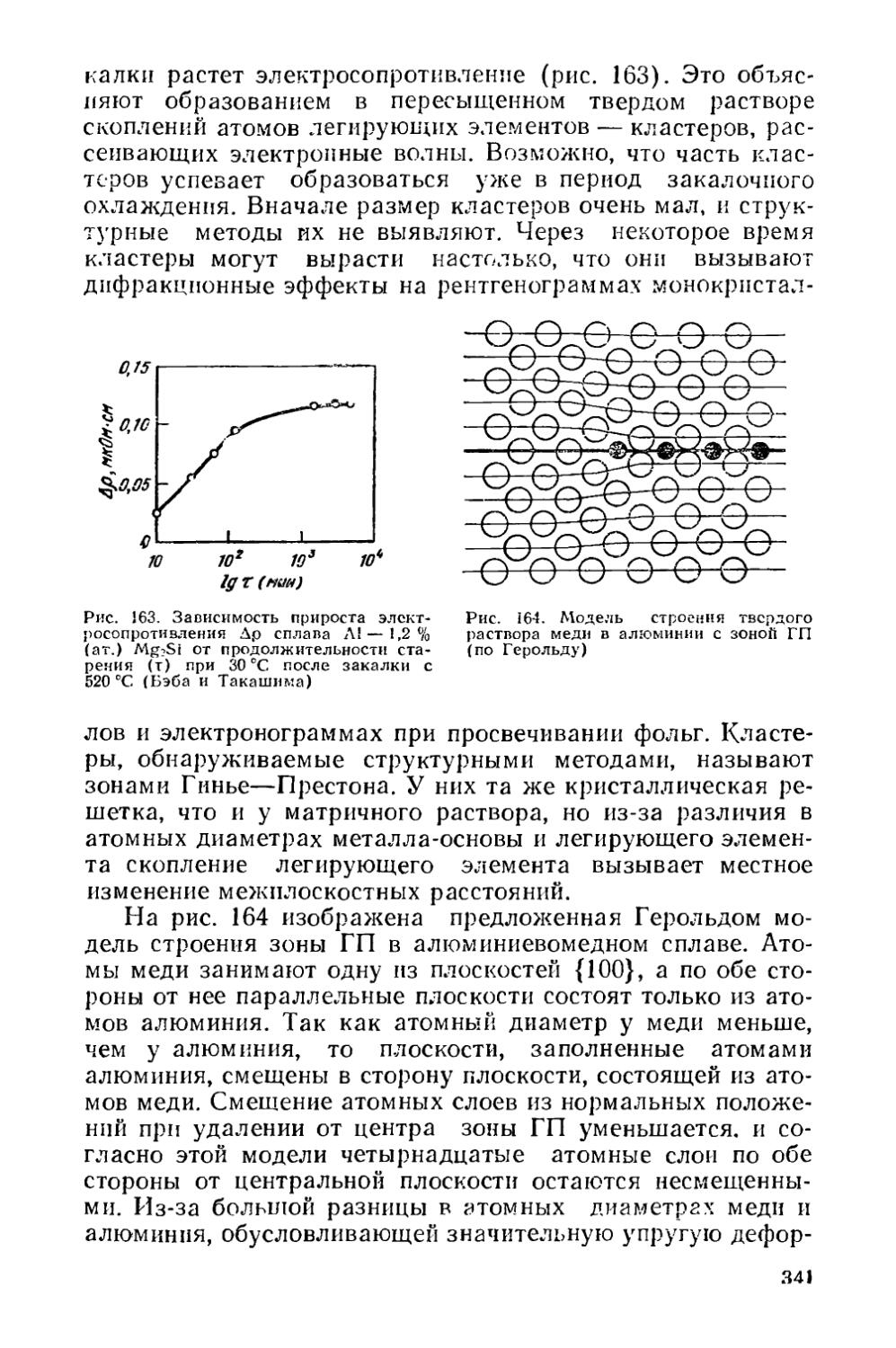



























































































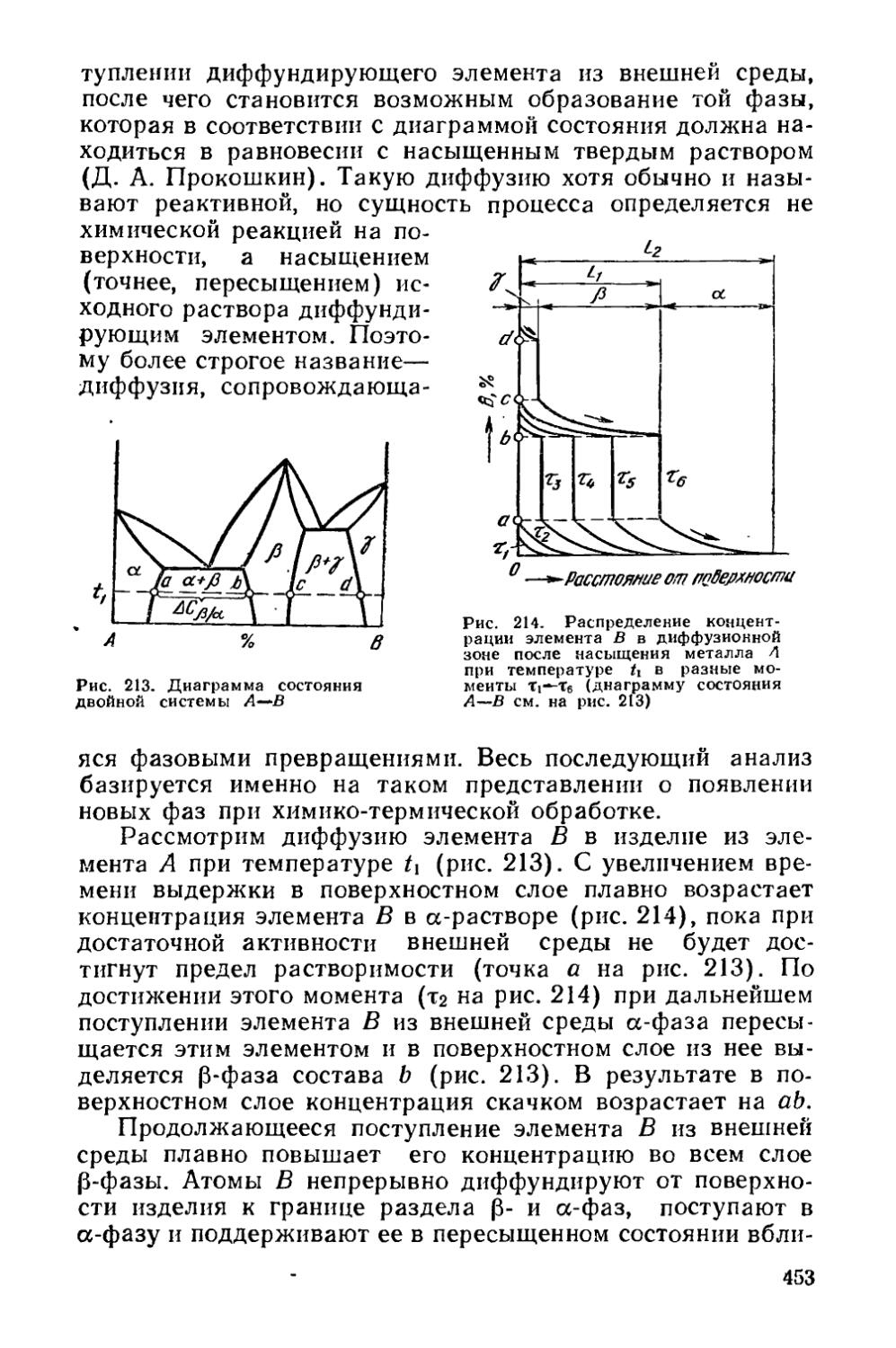


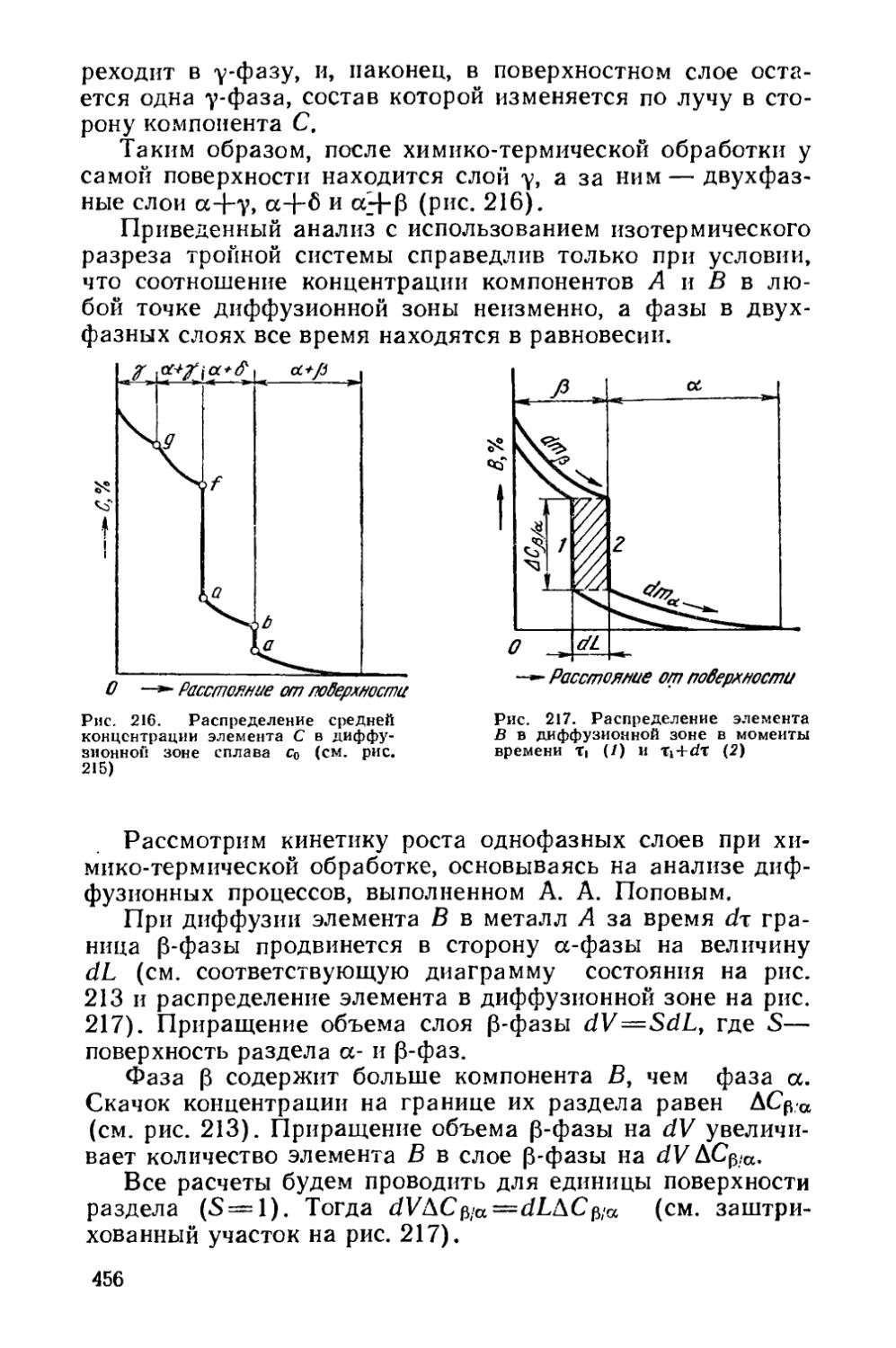

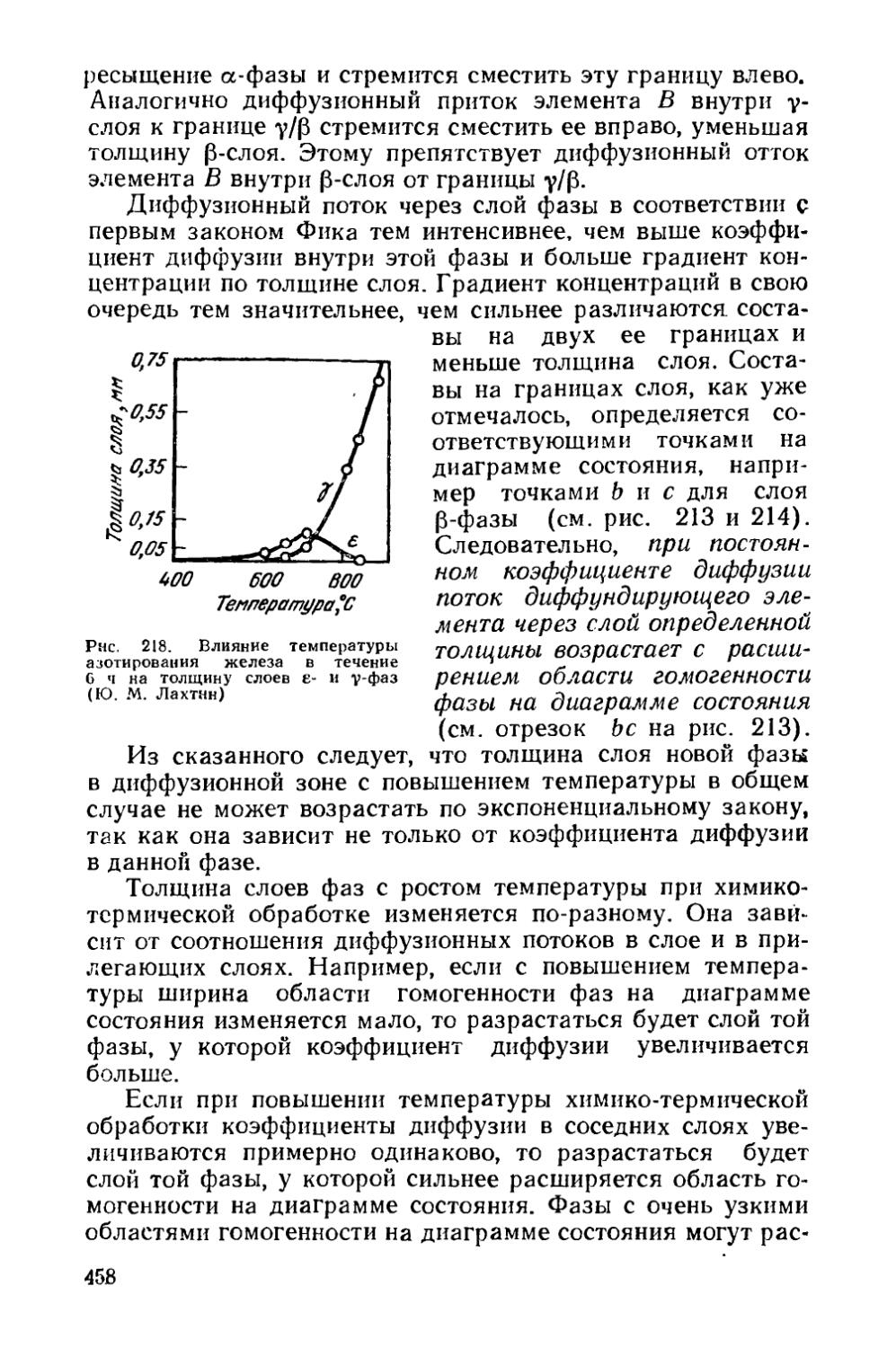







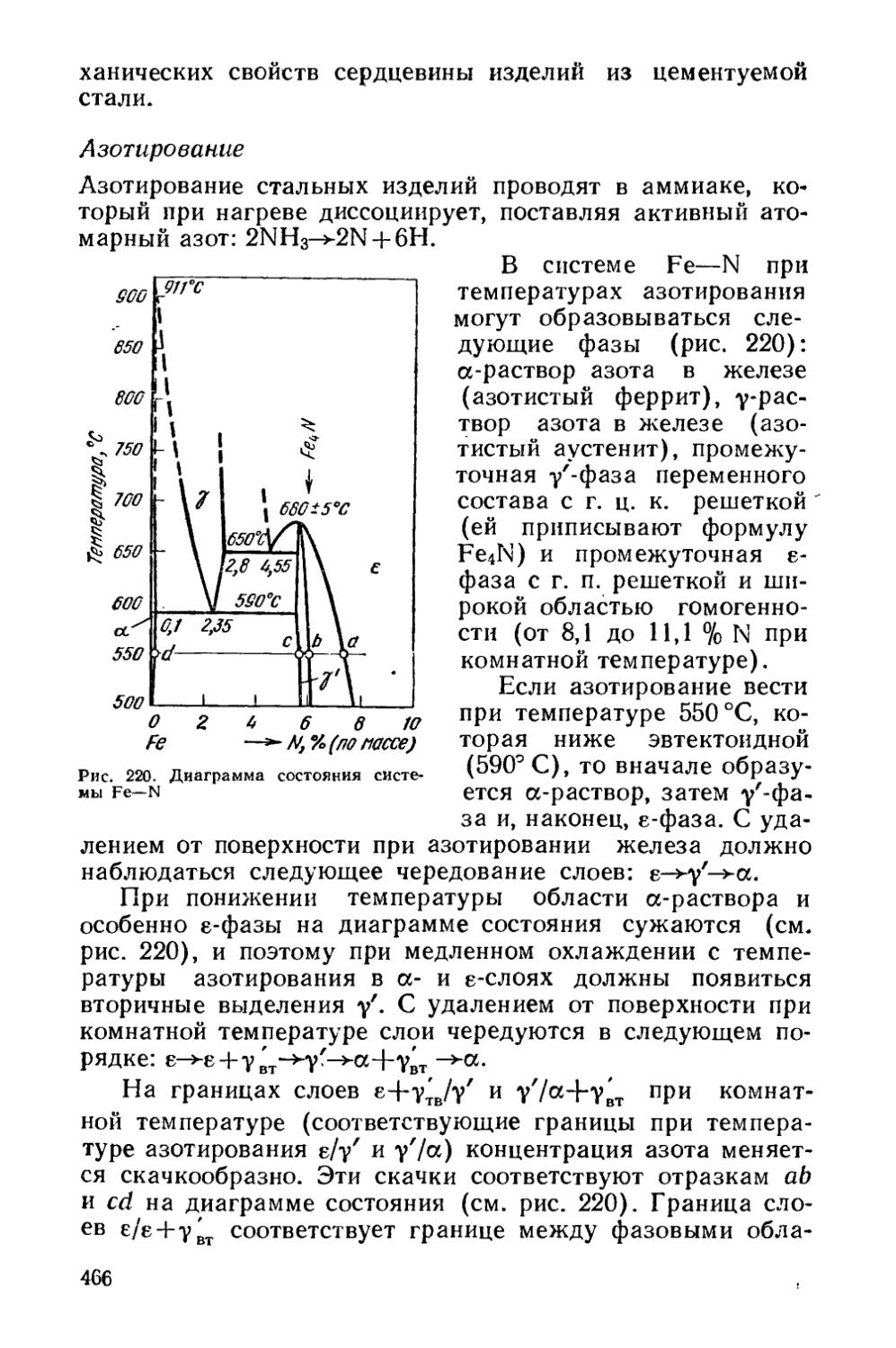















Текст
И. И. НОВИКОВ
И 4’1
ТЕОРИЯ ТЕРМИЧЕСКОЙ ОБРАБОТКИ МЕТАЛЛОВ
Издание 4-е, переработанное и дополненное
Допущено Министерством высшего и среднего специального образования СССР в качестве учебника для студентов вузов, обучающихся по специальности «Металловедение, оборудование и технология термической обработки металлов»
МОСКВА «МЕТАЛЛУРГИЯ» 1986
УДК 621.785'669.1/89
Рецензент: кафедра термообработки и физики металлов Уральского политехнического института им. С. М. Кирова
УДК 621.785'669.1/89
Теория термической обработки металлов: Учебник для вузов. 4-е изд., перераб. и доп.: Новиков И. И.: Металлургия, 1986. 480 с.
Изложена теория термической обработки сталей, чугунов, цветных металлов и сплавов. Проанализированы изменения структуры и свойств при закалке, отпуске, старении, отжиге с фазовой перекристаллизацией, рекристаллизационном и дорекристаллизационном отжиге, гомогенизации, отжиге для уменьшения напряжений, термомеханичёс-кой, химико-термической и других разновидностях термообработки.
Учебник рассчитан на студентов, специализирующихся по металловедению н термической обработке, а также студентов других металлургических специальностей и может быть полезен иц^кенерам — металловедам, термистам, литейщикам, сварщикам, специалистам по обработке металлов давлением и порошковой металлургии. Ил. 220. Табл. 11. Библиогр. список: 74 иазв.
УЧЕБНИК
Илья Изриэлович Новиков
ТЕОРИЯ ТЕРМИЧЕСКОЙ ОБРАБОТКИ МЕТАЛЛОВ
Редактор издателвства М. С. Архангельская
Художественный редактор Ю. И. Смурыгин
Технический редактор Г. Б. Ж а р о в а
Корректоры Г. Ф. Лобанова, Ю. И. Ко-ролева
И Б № 2982
Сдано в набор 11.04.86. Подписано в печать 23.06.86. Т-14670. Формат бумаги 84Х108’/з2. Бумага типографская № 1. Гарнитура литературная. Печать высокая. Усл. печ. л. 25,20. Усл. кр.-отт. 25,20. Уч.-изд. л. 29,06. Тираж 16 800 экз. Заказ 497. Цена 1 р. 30 к. Изд. № 1283
Ордена Трудового Красного Знамени издательство «Металлургия», 119857, ГСП, Москва, Г-34, 2-й Обыденский пер., д. Ц
Владимирская типография Союзполиграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли 600000, г. Владимир, Октябрьский проспект, д. 7
2704070000-—176
Н --------------- 73—86
040(01)—86
© Издательство «Металлургия», 1986
ОГЛАВЛЕНИЕ
Предисловие............................................ • • "
Введение................................................«... о
Раздел первый. ОТЖИГ ПЕРВОГО РОДА.............................15
Глава 1. Гомогенизациоиный отжиг..............................15
§ 1. Изменение структуры сплавов при гомогеиизационном отжиге ................................'.........................15
§ 2 Изменение свойств сплавов при гомогеиизационном отжиге 28
§ 3. Гомогенизация с иагревом выше температуры неравновесного солидуса....................................................31
Глава 11. Рекристаллизационный и дорекристаллизационный отжиги...................................................... 34
§ 4. Изменение структуры металла при холодной обработке давлением ....................................................♦ 34
§ 5. Изменение свойств металла при холодной обработке давлением ..........................................................42
§ 6. Изменение структуры при дорекристаллизациоином отжиге 45
§ 7. Первичная рекристаллизация (рекристаллизация обработки) 55
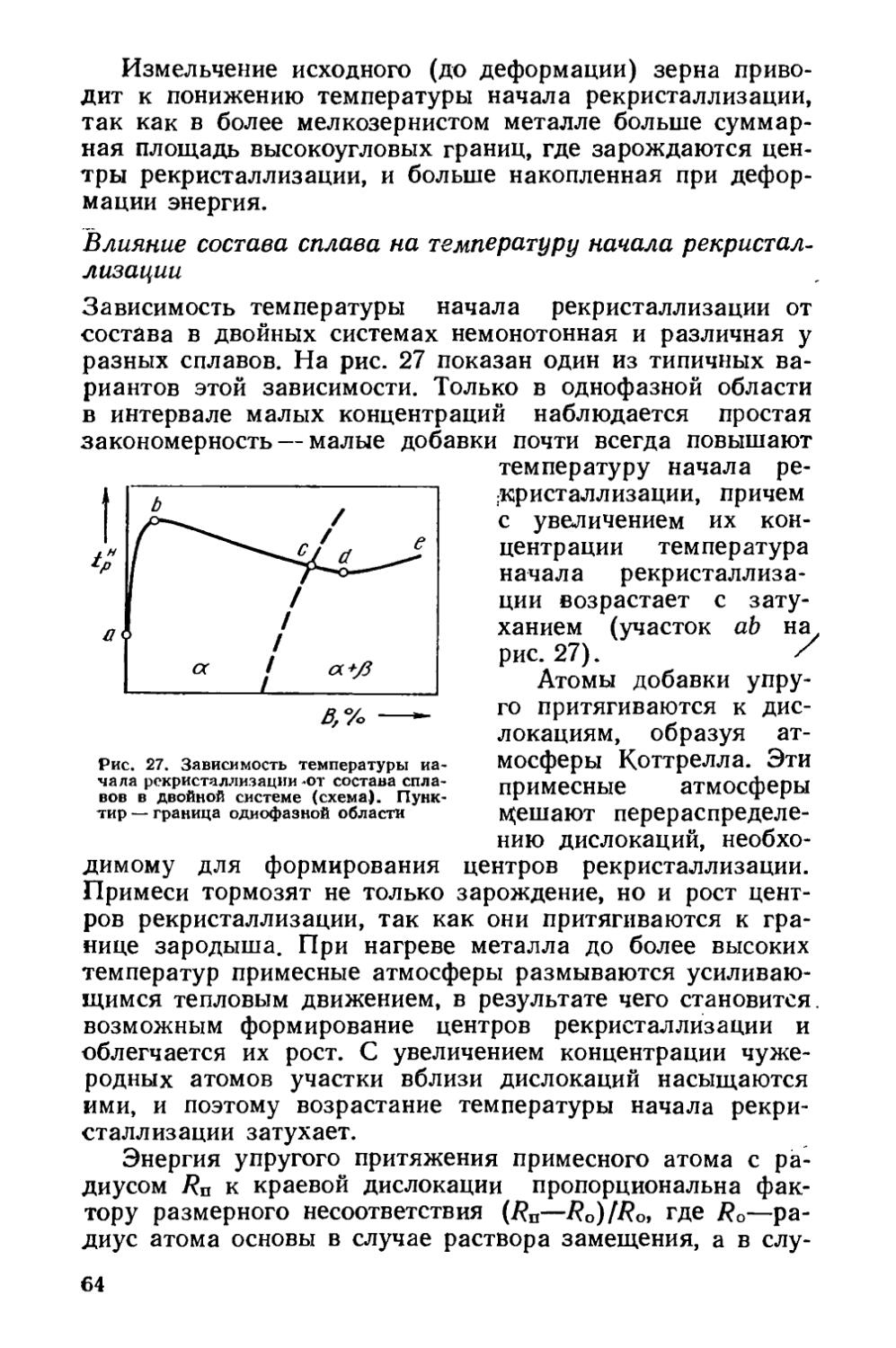
§ 8. Собирательная рекристаллизация 68
§ 9. Текстуры рекристаллизации 74
§ 10. Вторичная рекристаллизация ...««« 77
§ 11. Размер рекристаллизованного зерна . , 32
§ 12. Изменение свойств металла при дорекристаллизациоином и рекристаллизационном отжиге....................................91
§ 13. Анизотропия свойств отожженного металла .... 101
§ 14. Выбор режимов до рекристаллизационного и рекристаллизационного отжига.............................................. Ю5
Глава III. Отжиг, уменьшающий напряжения ..... НО
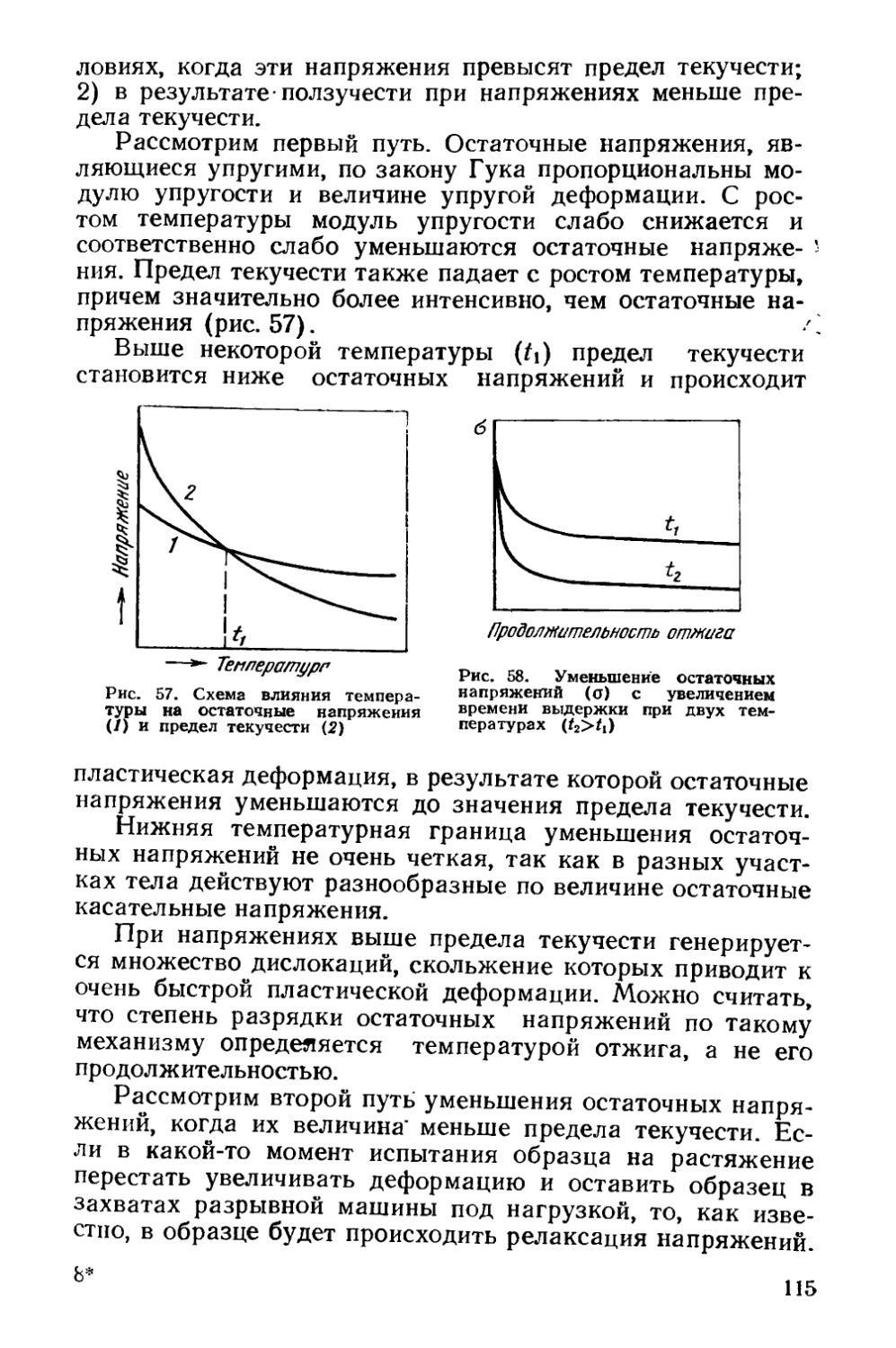
§ 15. Возникновение и роль остаточных напряжений . « . . НО § 16. Уменьшение остаточных напряжений при отжиге . . . 114
Раздел второй. ОТЖИГ ВТОРОГО РОДА 122
Глава IV. Общие закономерности фазовых превращений в твердом состоянии......................................... 123
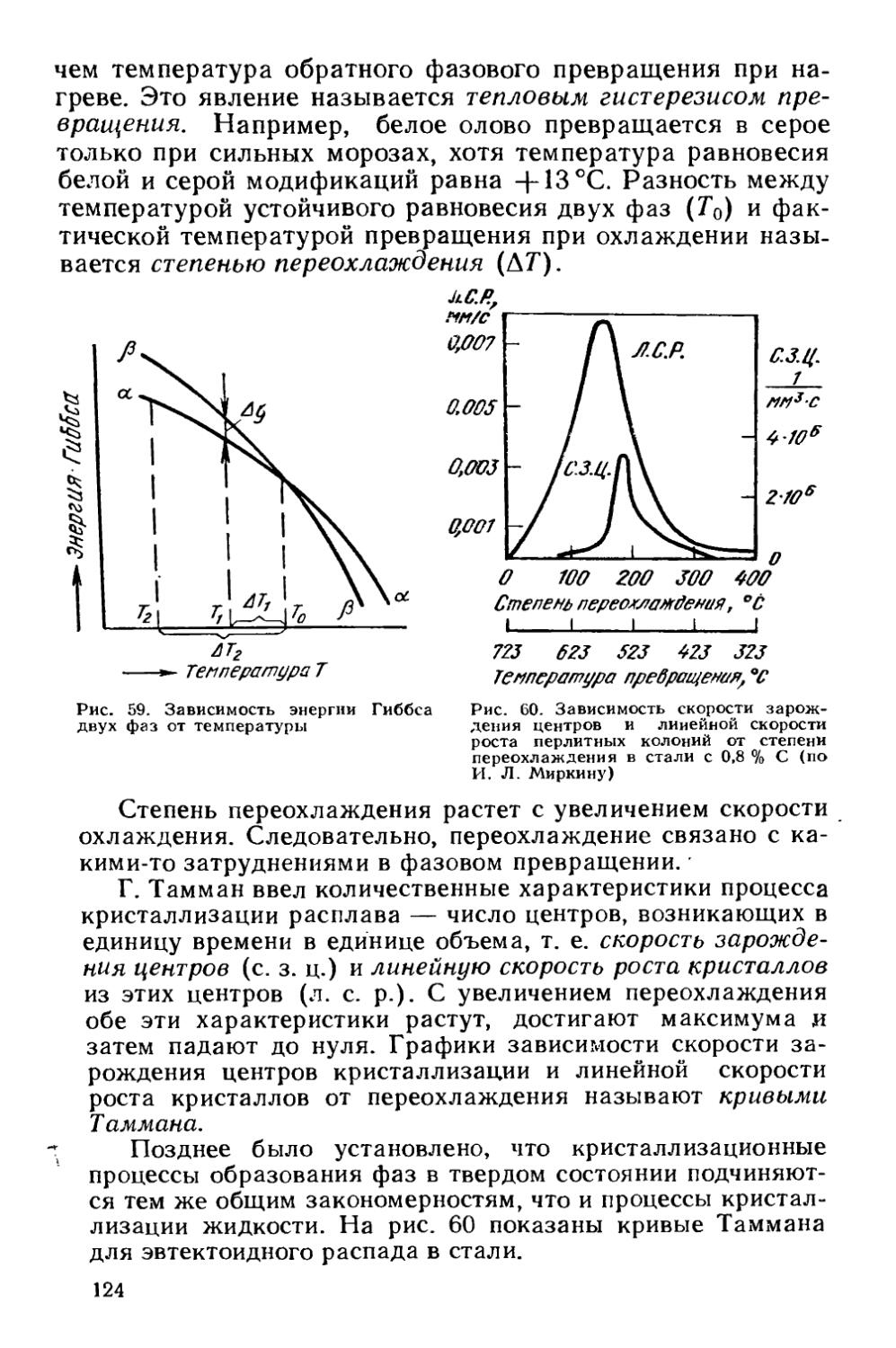
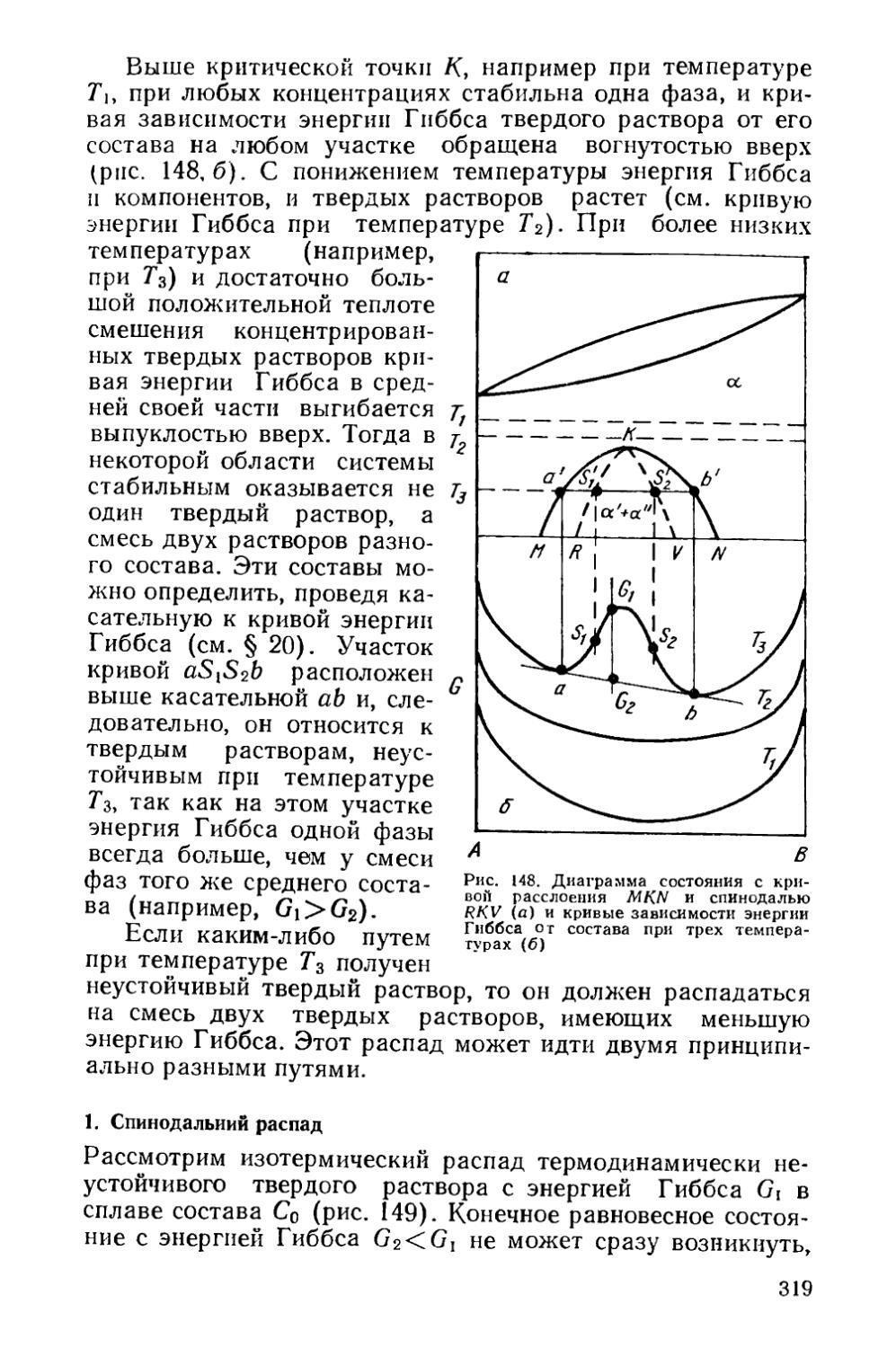
§ 17. Термодинамика фазовых превращений.....................123
§ 18. Роль строения межфазных границ при фазовых превращениях ........................................................132
§ 19. Гомогенное и гетерогенное зарождение фаз..............135
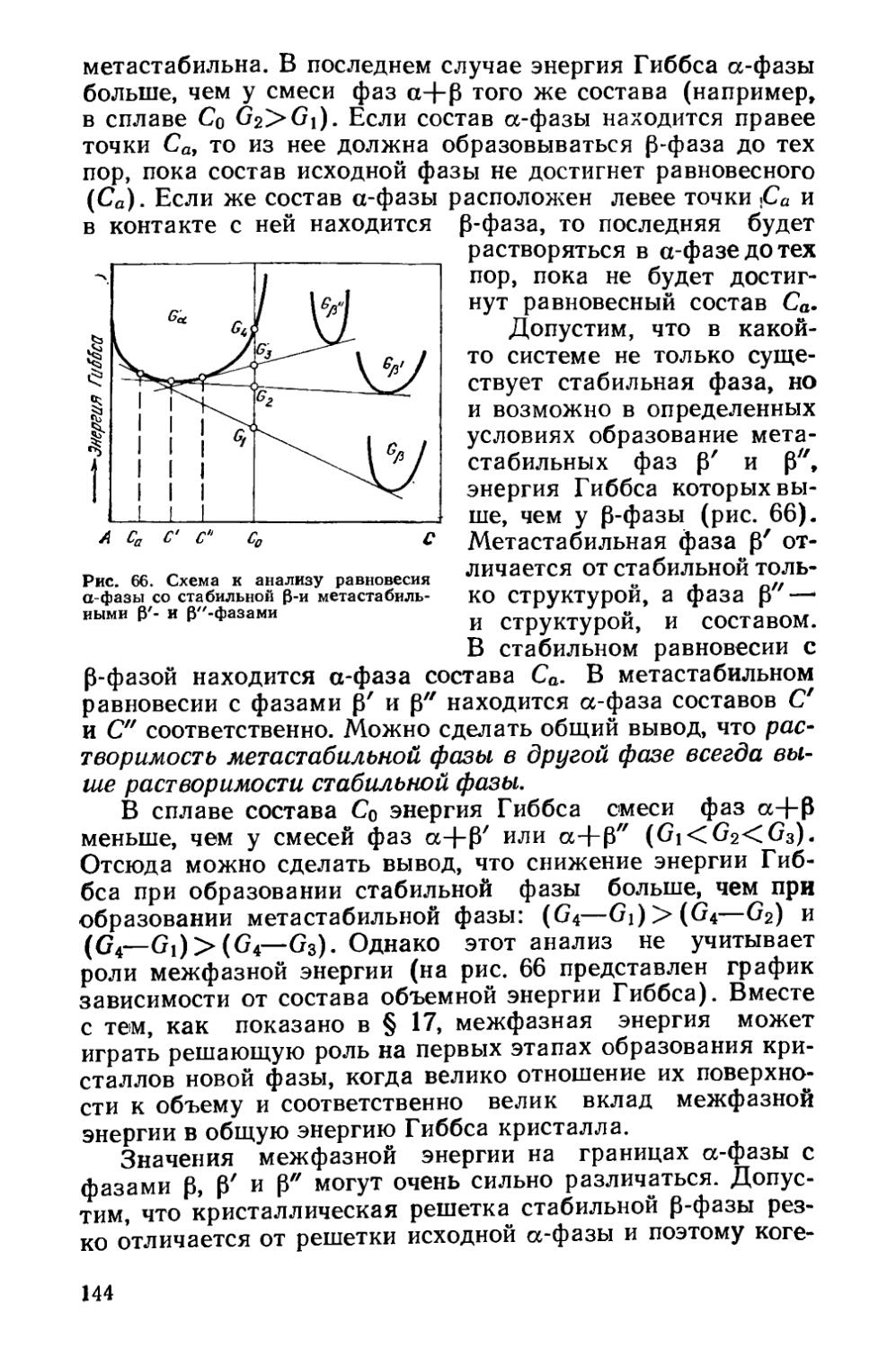
§ 20. Образование промежуточных метастабильиых фаз < . 143
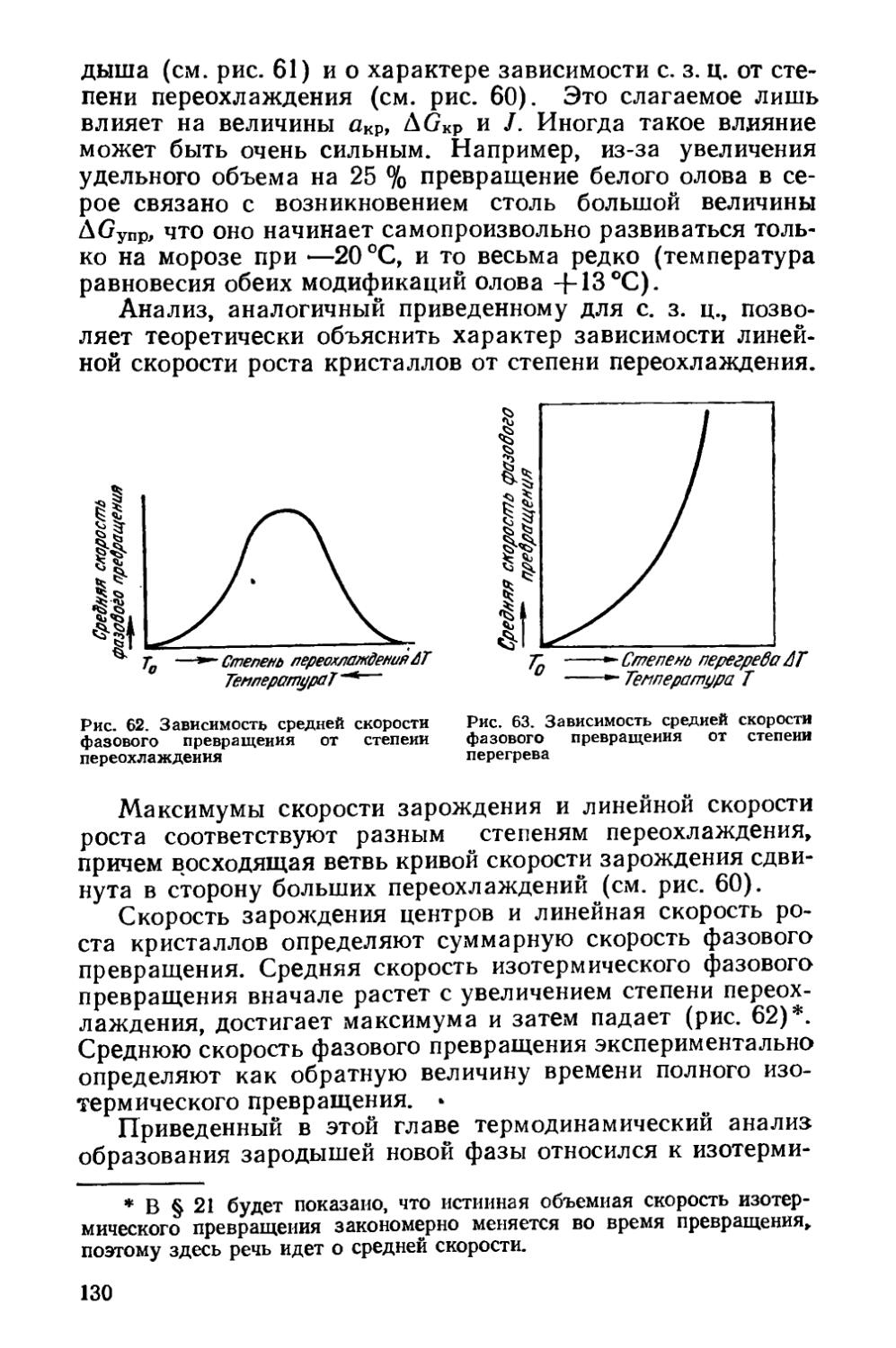
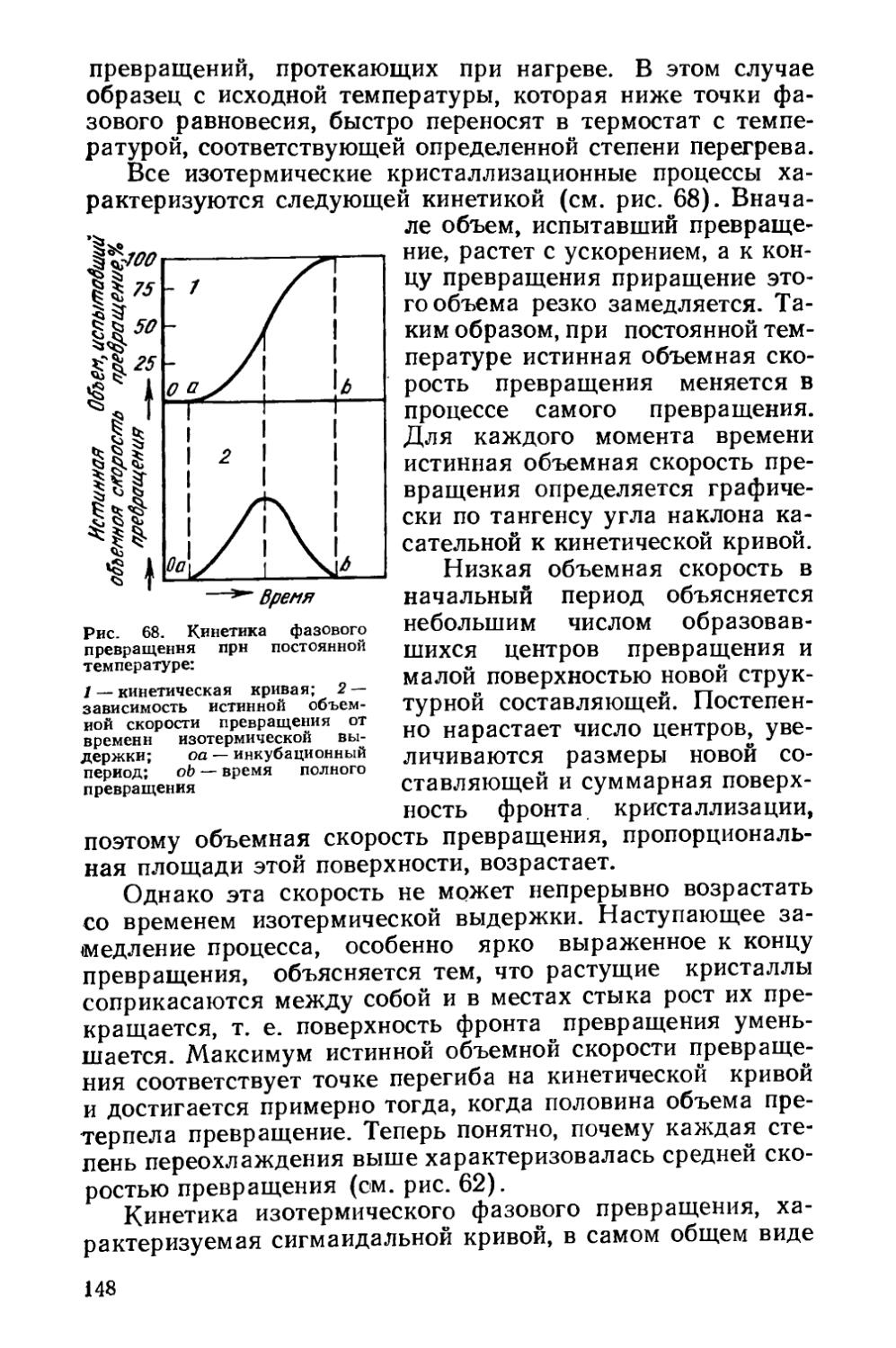
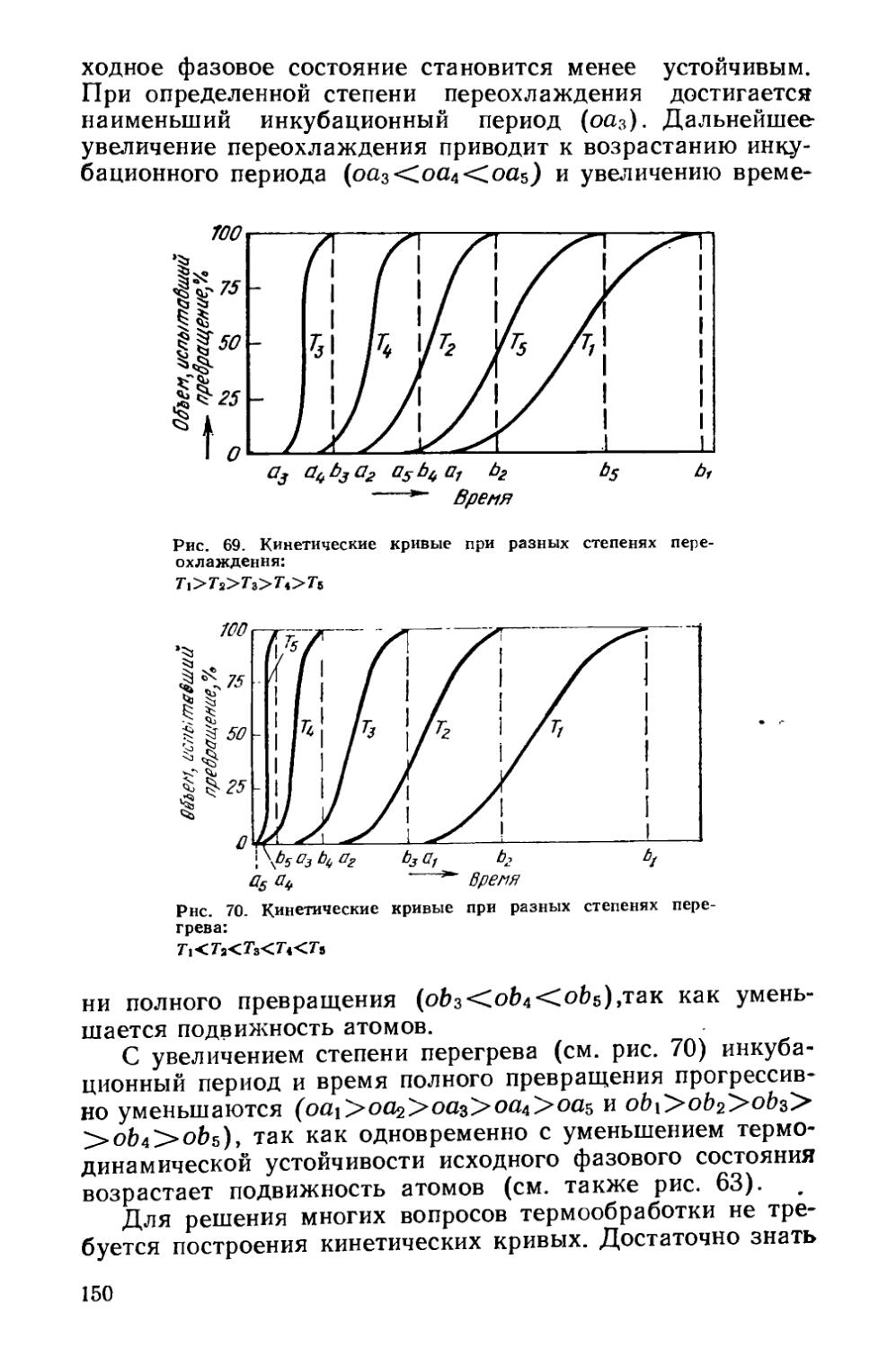
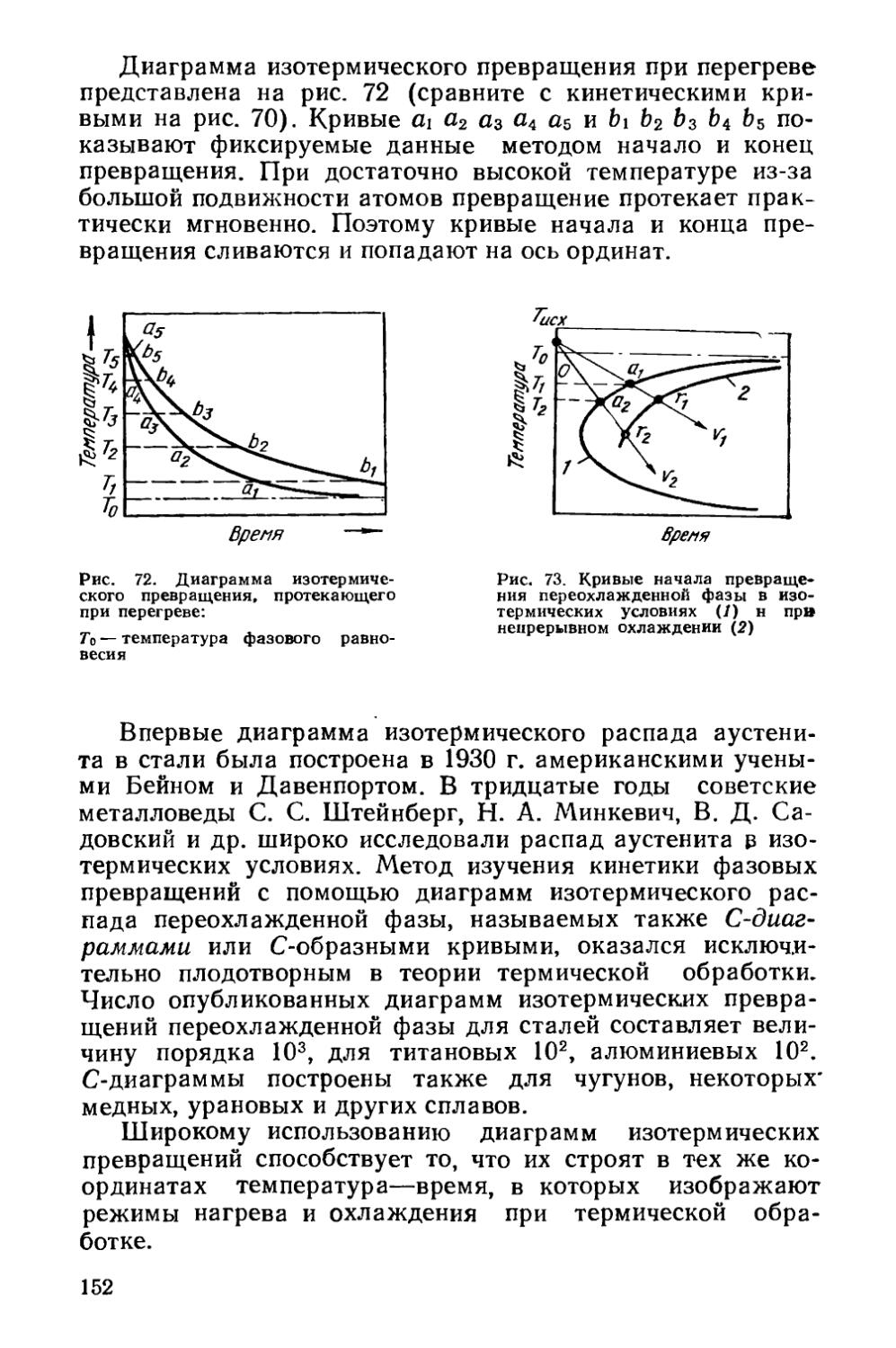
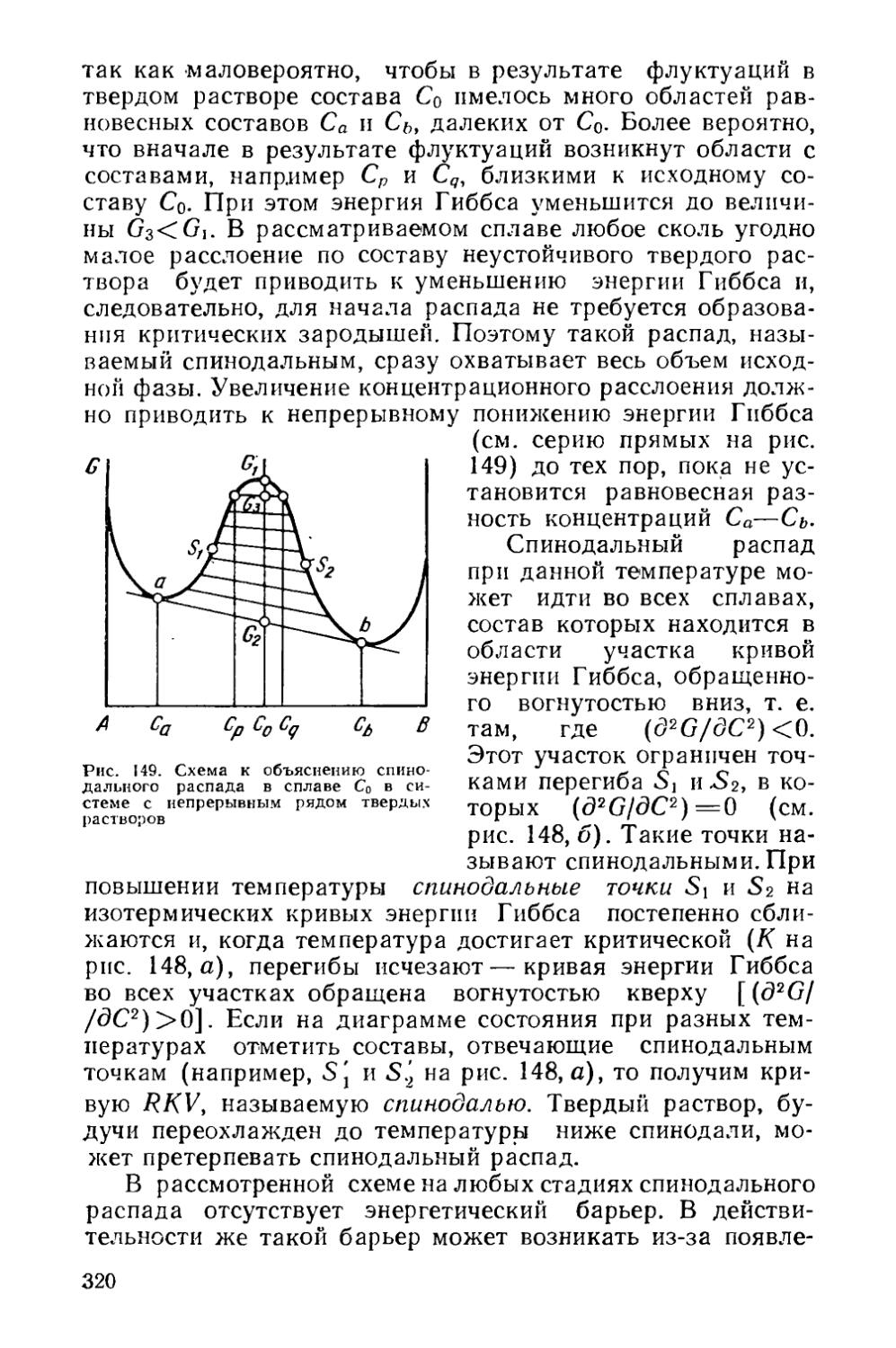
§ 21. Кинетика фазовых превращений..........................147
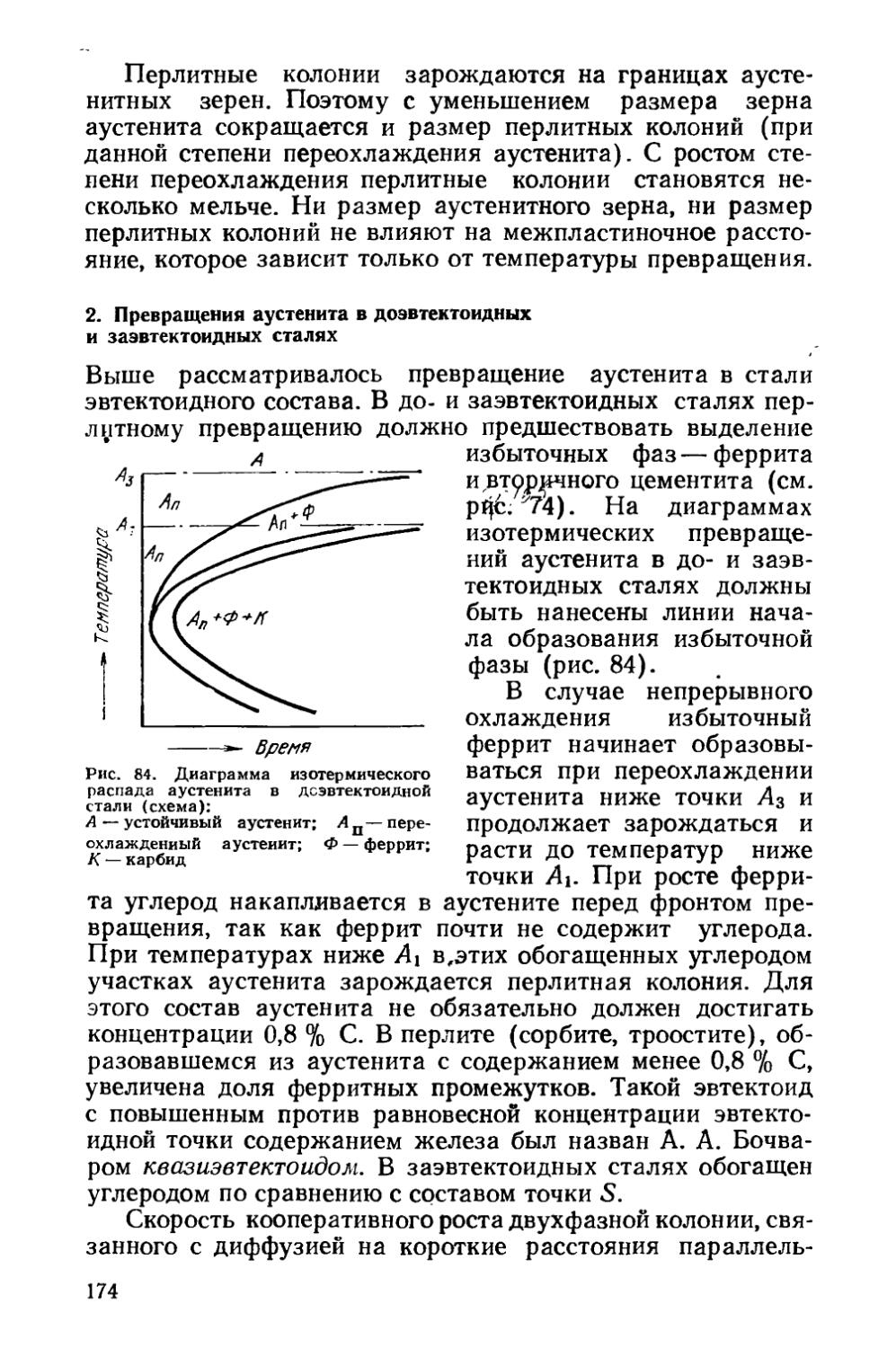
Глава V. Отжиг сталей..................................... 154
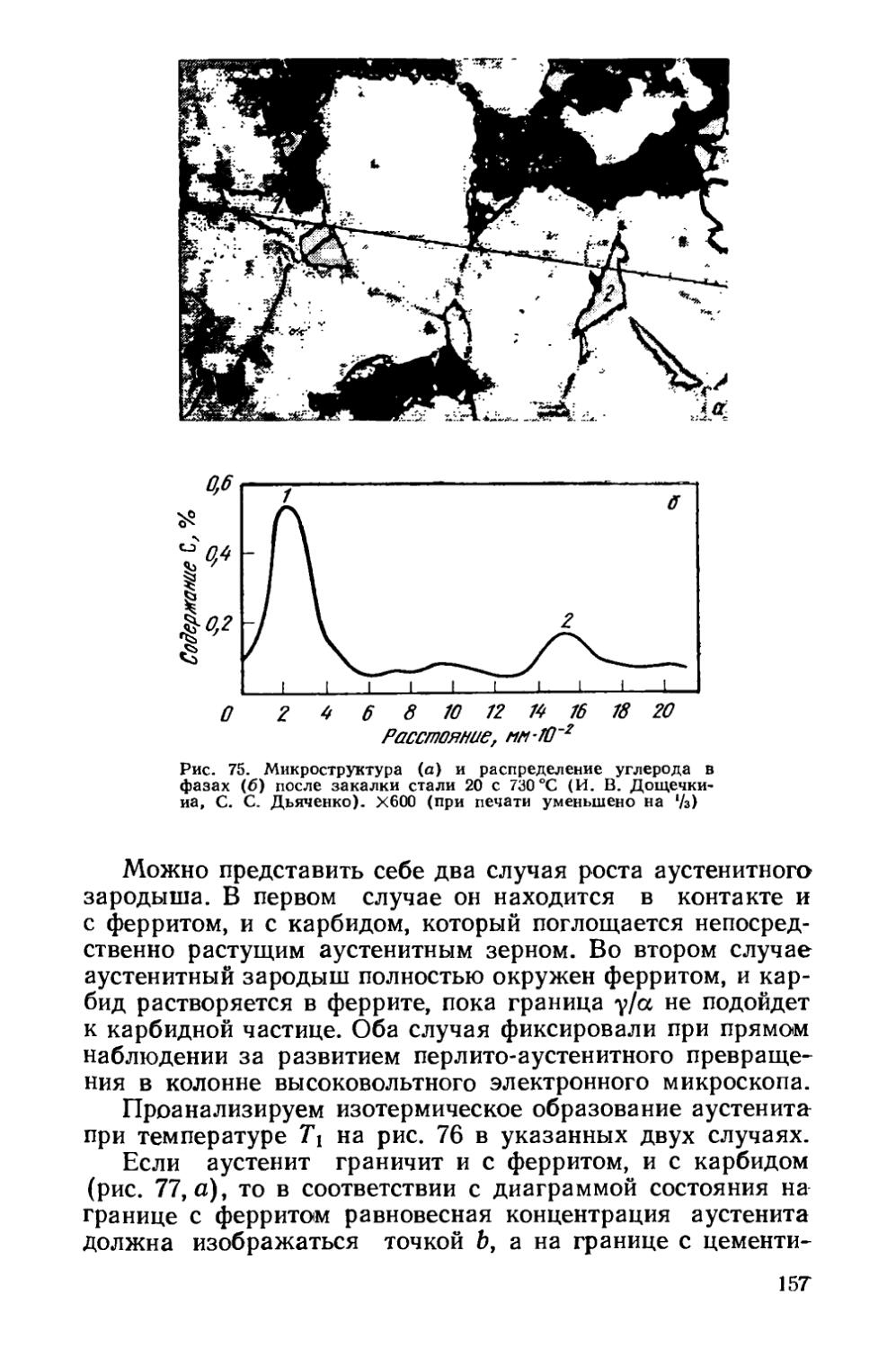
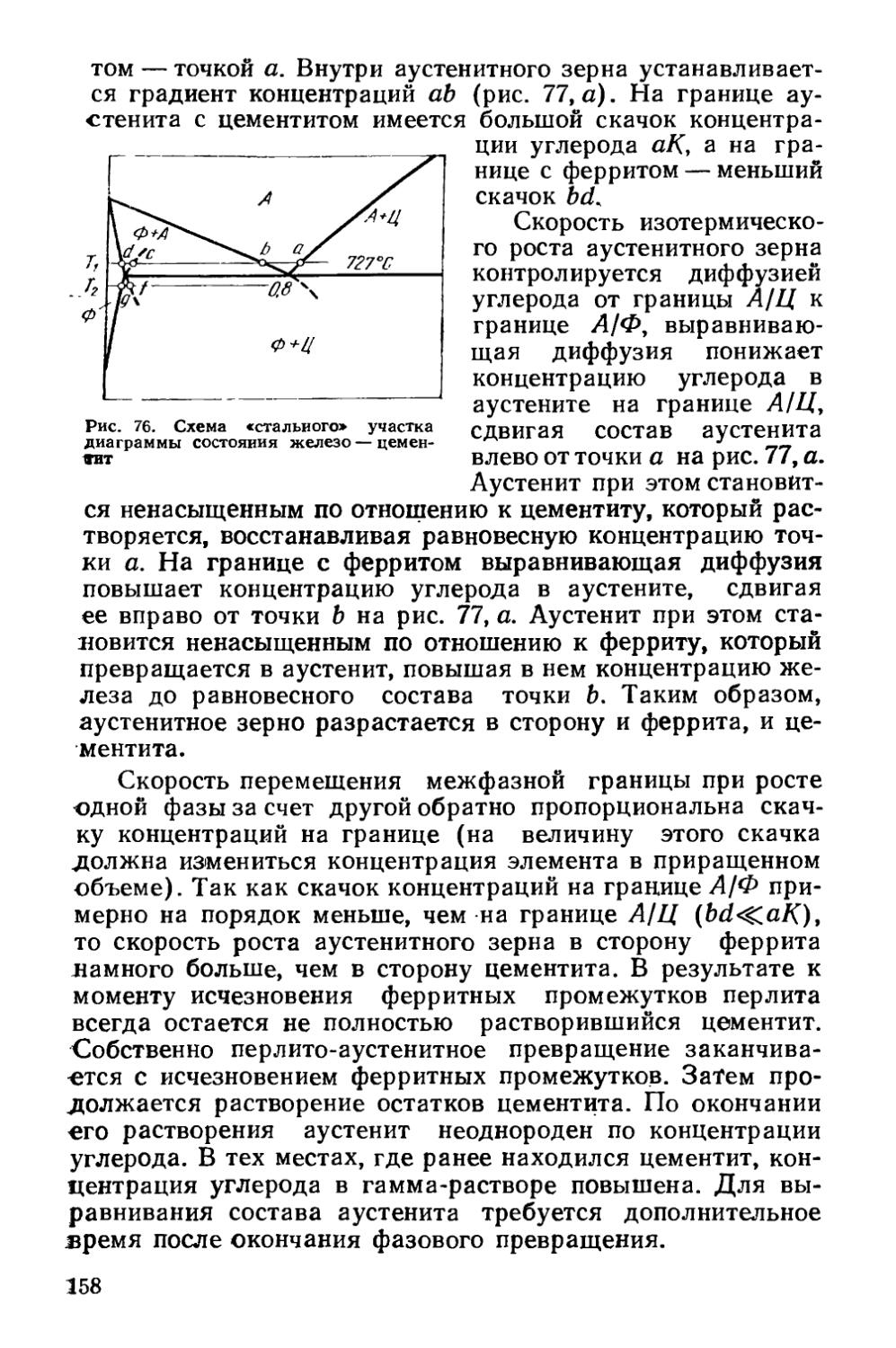
§ 22. Образование аустенита при нагреве % 155
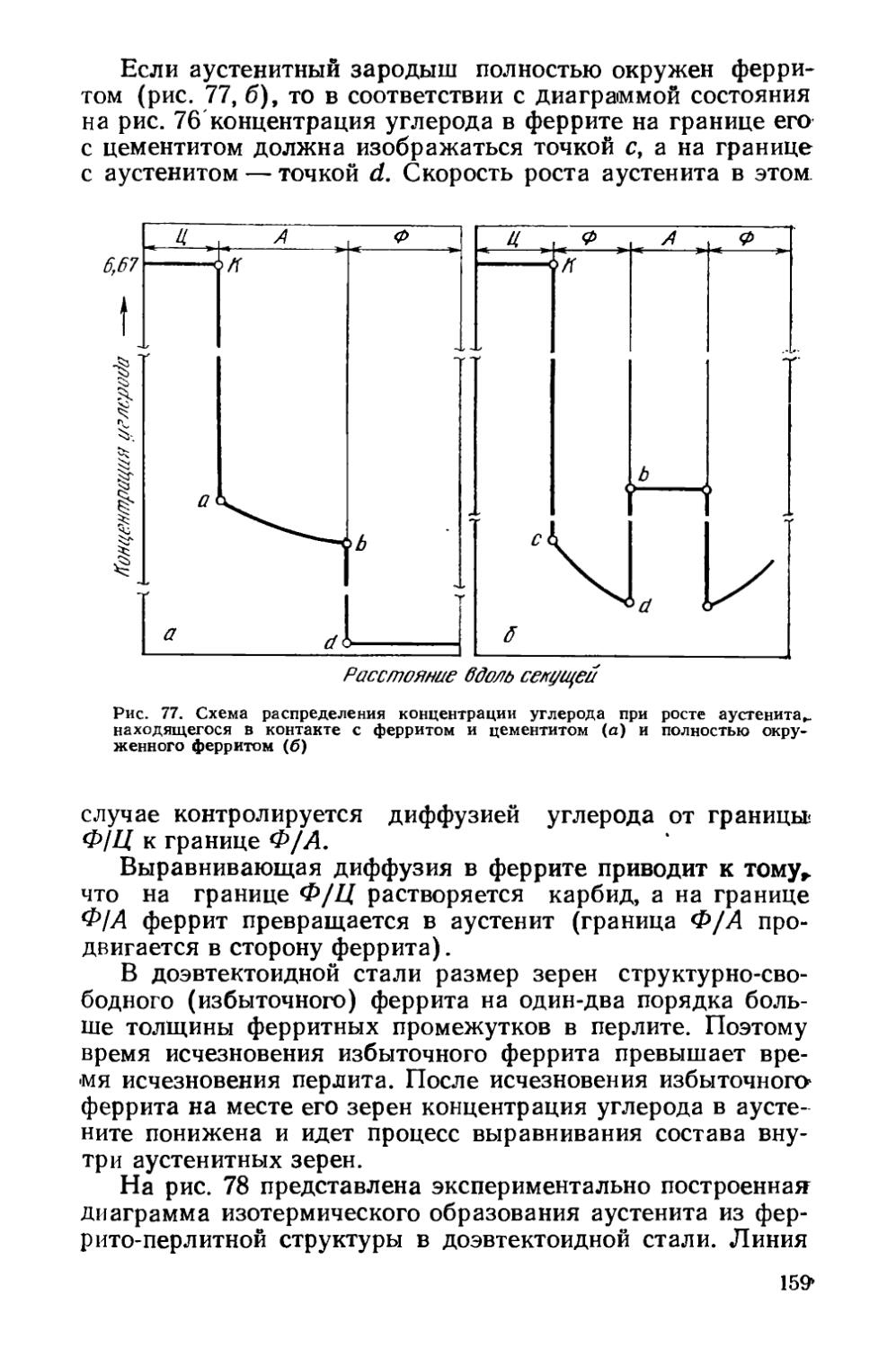
§ 23. Структурная наследственность и перекристаллизация аустенита ..................................................... 166
§ 24. Диффузионные превращения аустенита при охлаждении . 169
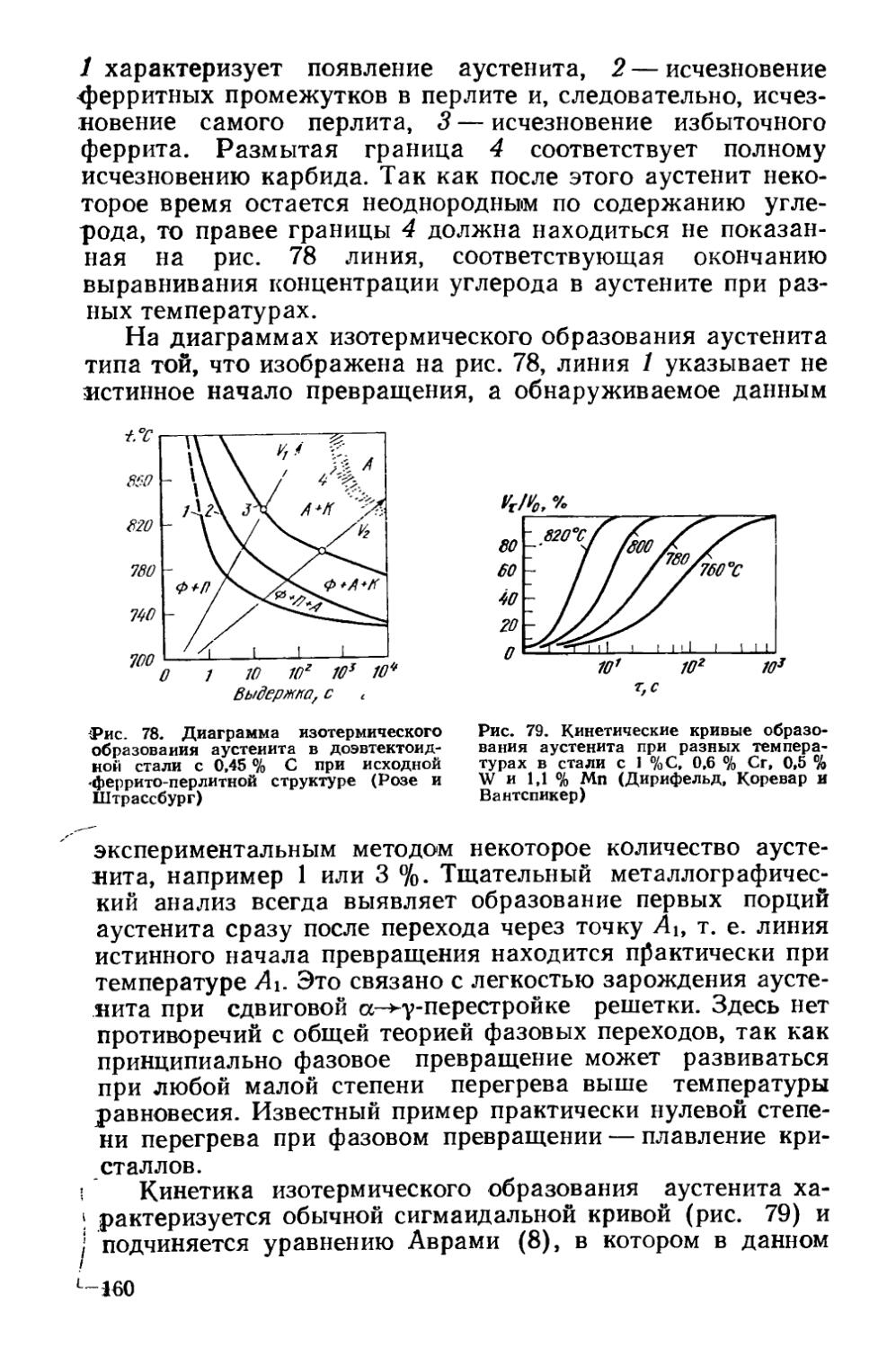
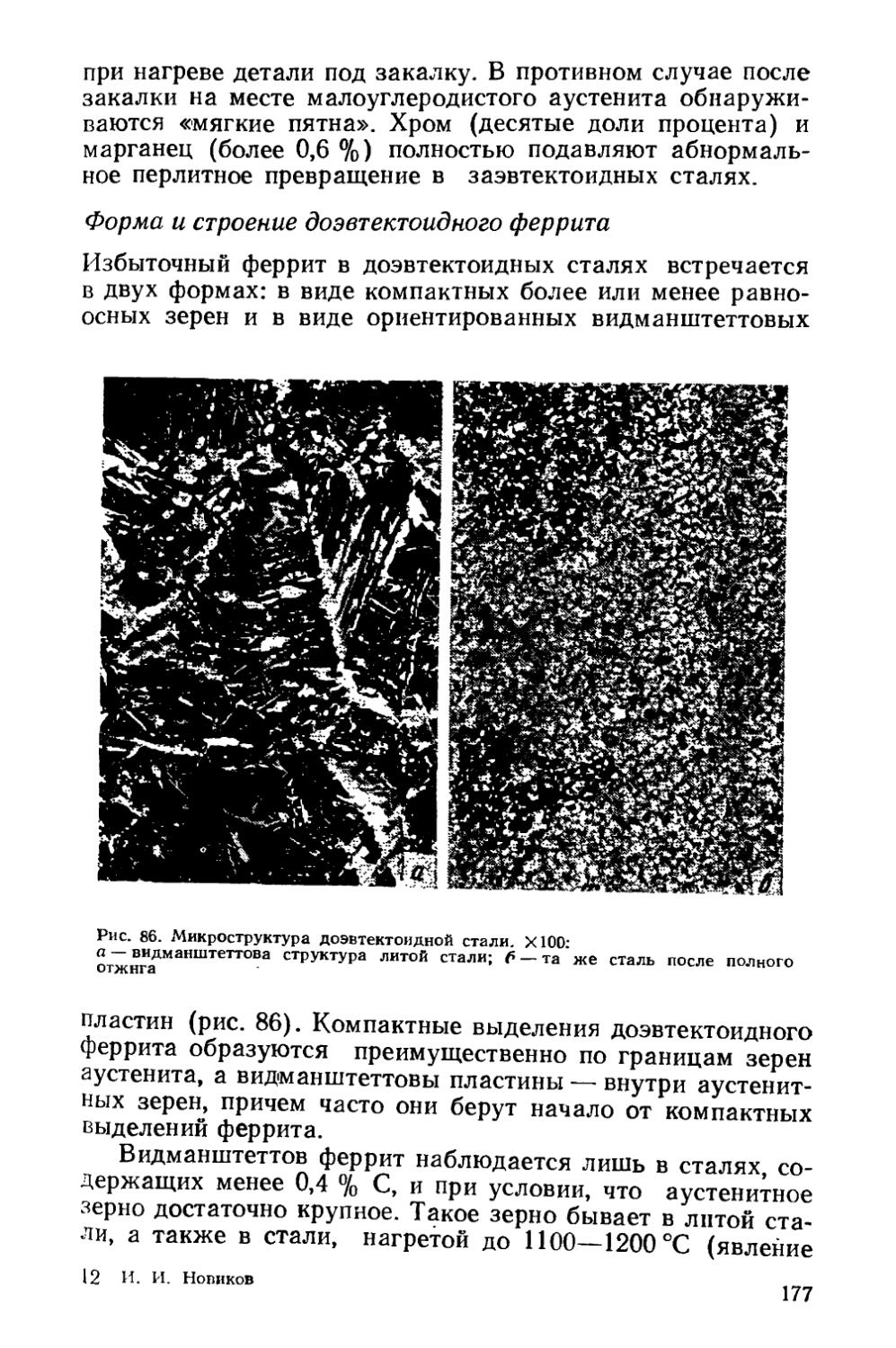
§ 25. Разновидности отжига сталей......................... 182
Глава VI. Отжиг чугунов.....................................193
§ 26. Графитизирующий отжиг чугуна....................... ’ 194
§ 27. Нормализация чугуна..........................* 202
Глава VII. Отжиг цветных металлов и сплавов . ’ * * 203
3
§ 28. Гетерогенизационпый отжиг...................• . . . 204
| 29. Отжиг с фазовой перекристаллизацией......................210
раздел третий. ЗАКАЛКА.........................................212
Глава VIII. Закалка без полиморфного превращения 213
§ 30. Изменение свойств при закалке без полиморфного превращения .......................................................214
§31. Нагрев и охлаждение при закалке без полиморфного превращения ....................................................217
Глава IX. Закалка с полиморфным превращением . . . 225
§ 32. Особенности _ мартенситного превращения в углеродистых сталях....................................................225
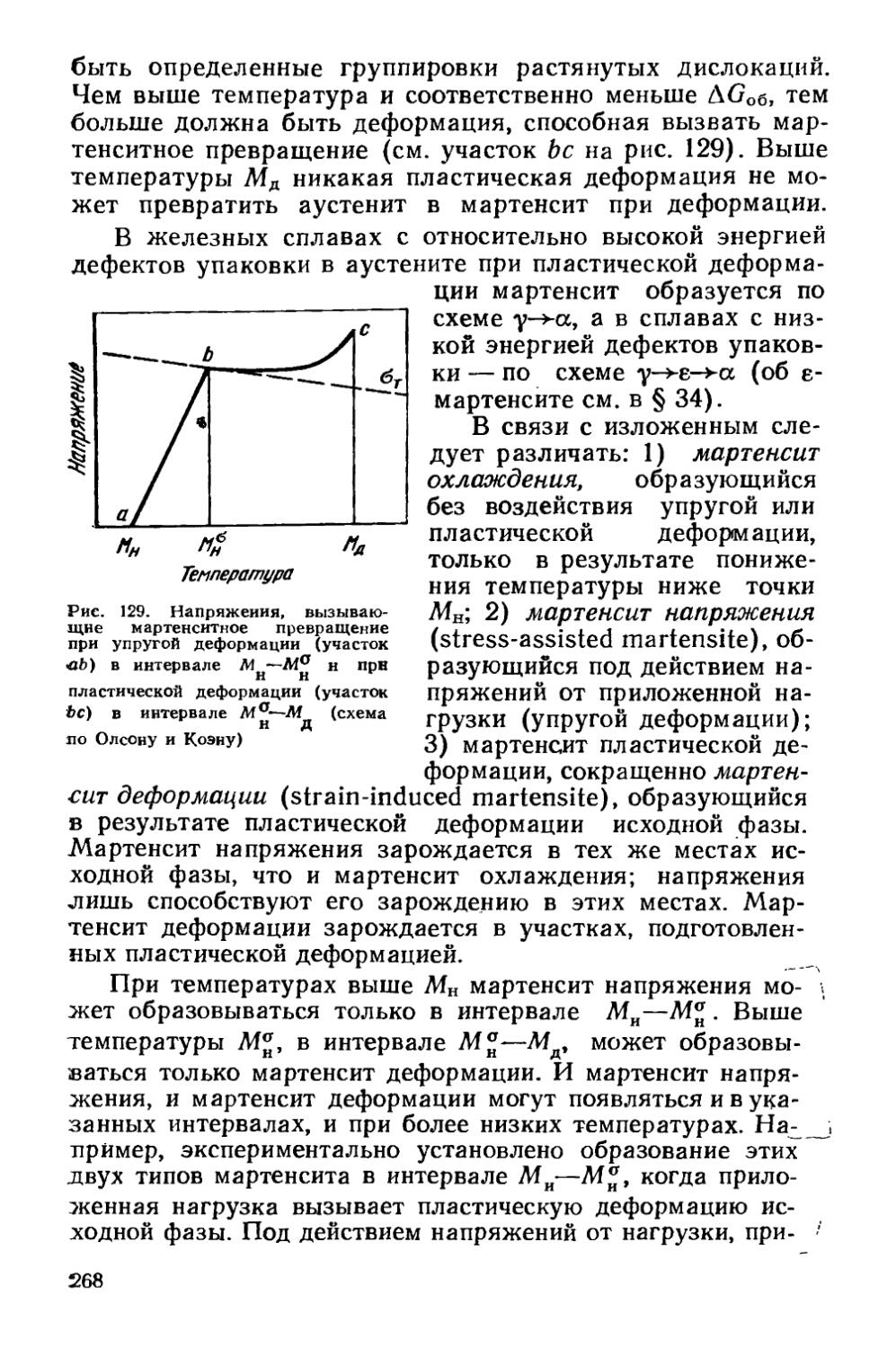
§ 33. Термодинамика мартенситных превращений.................228
§ 34. Механизм мартенситного превращения.....................236
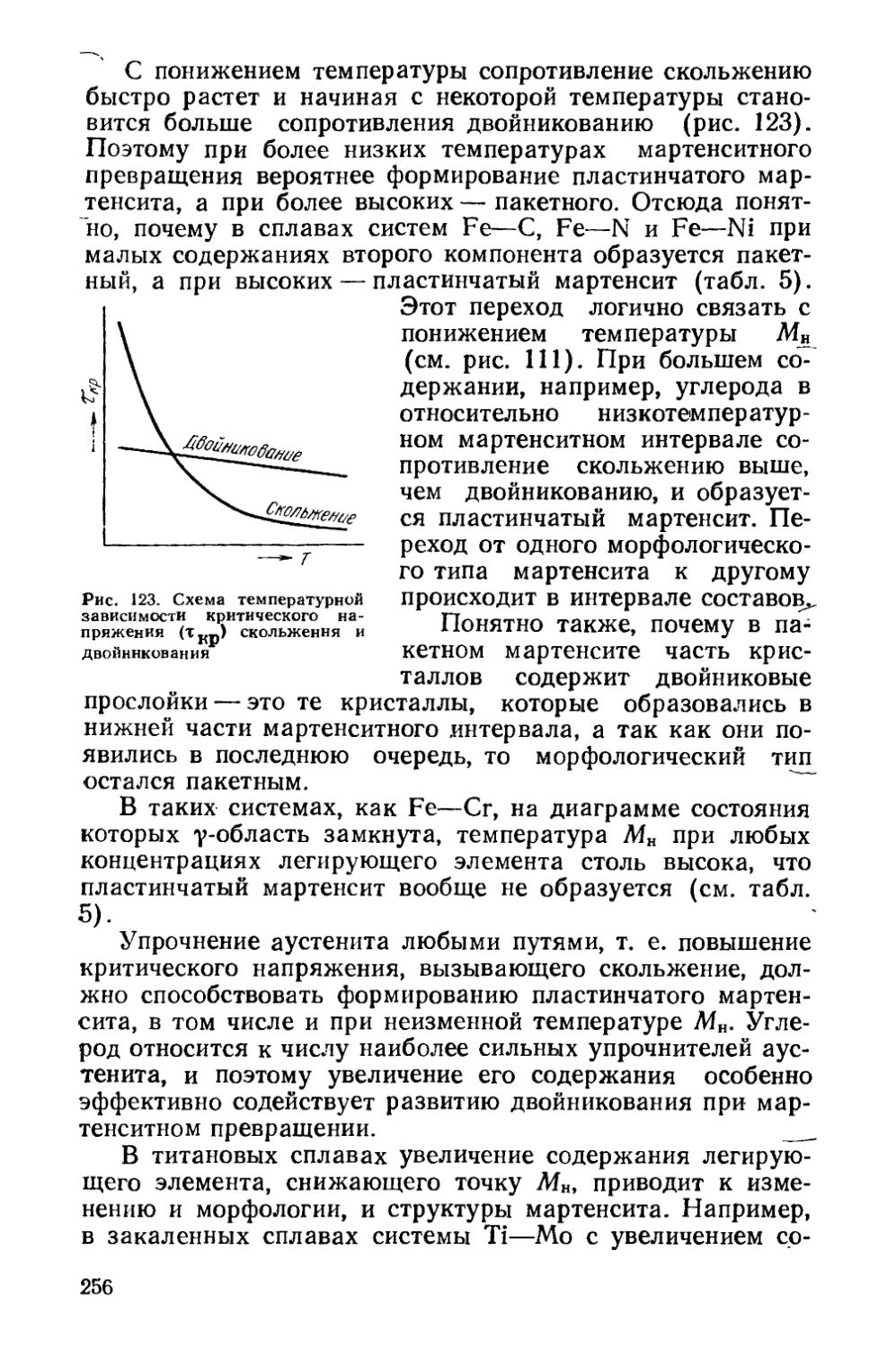
§ 35. Микроструктура и субструктура сплавов, закаленных на мартенсит.........................................249
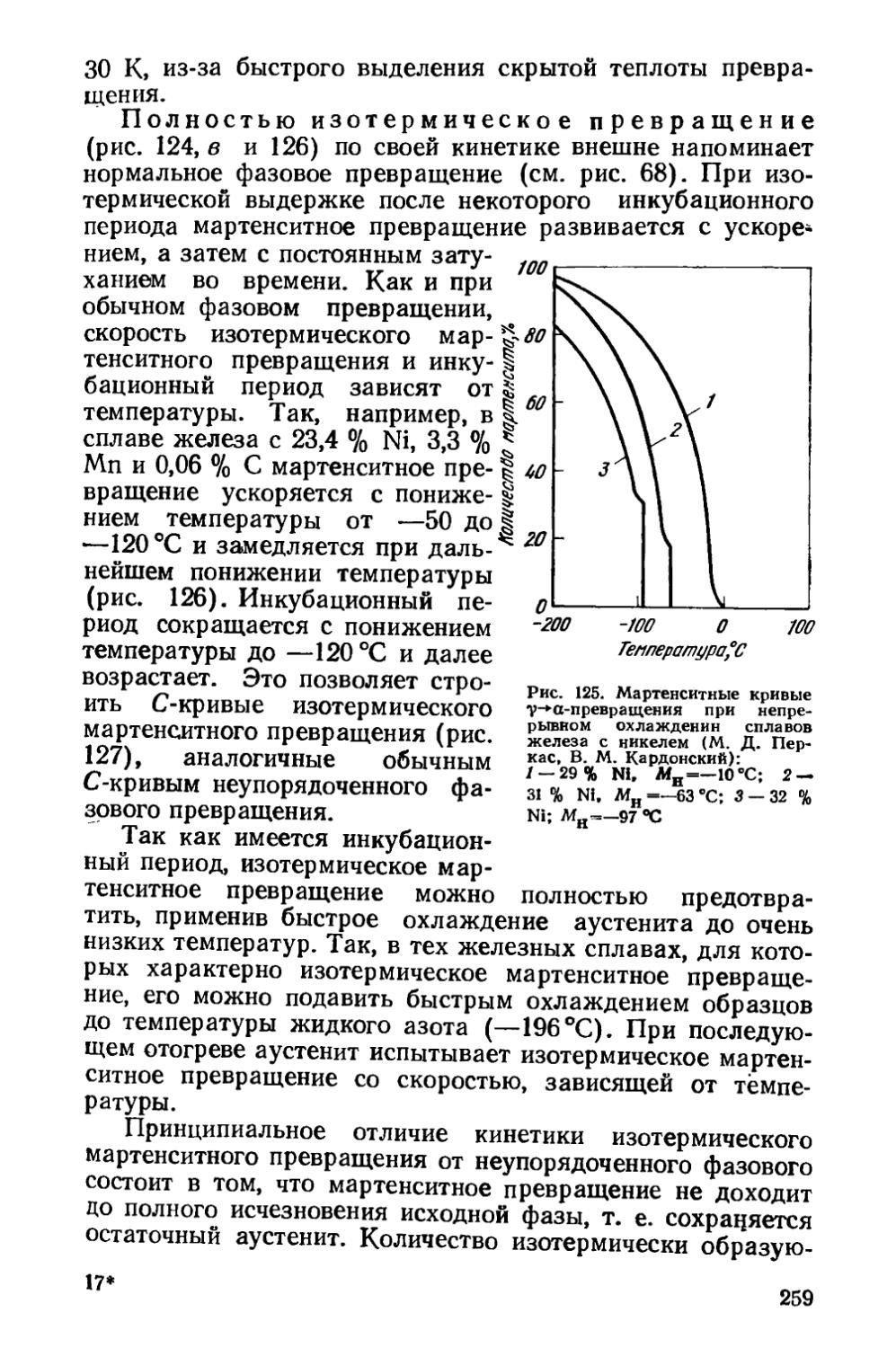
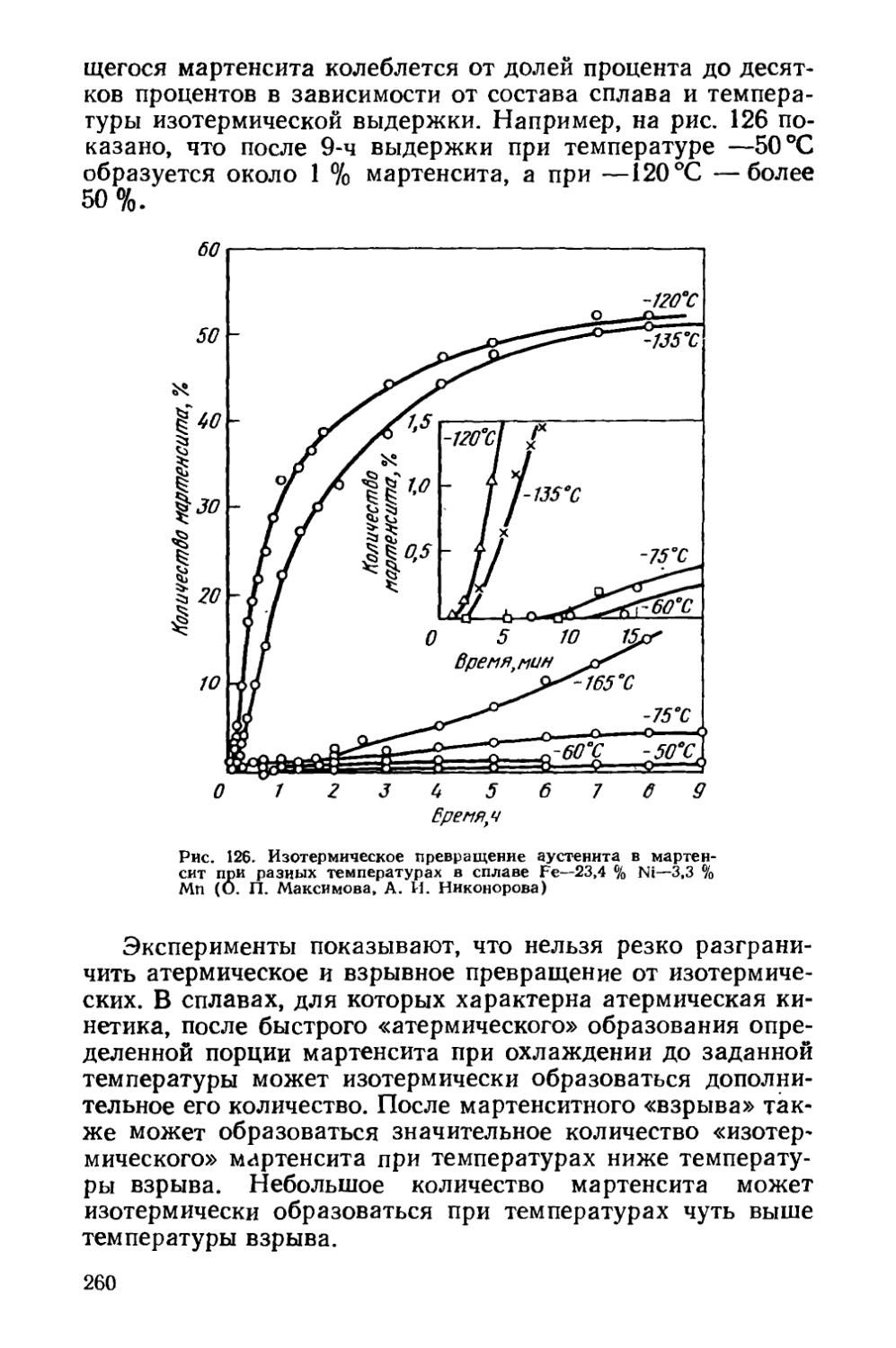
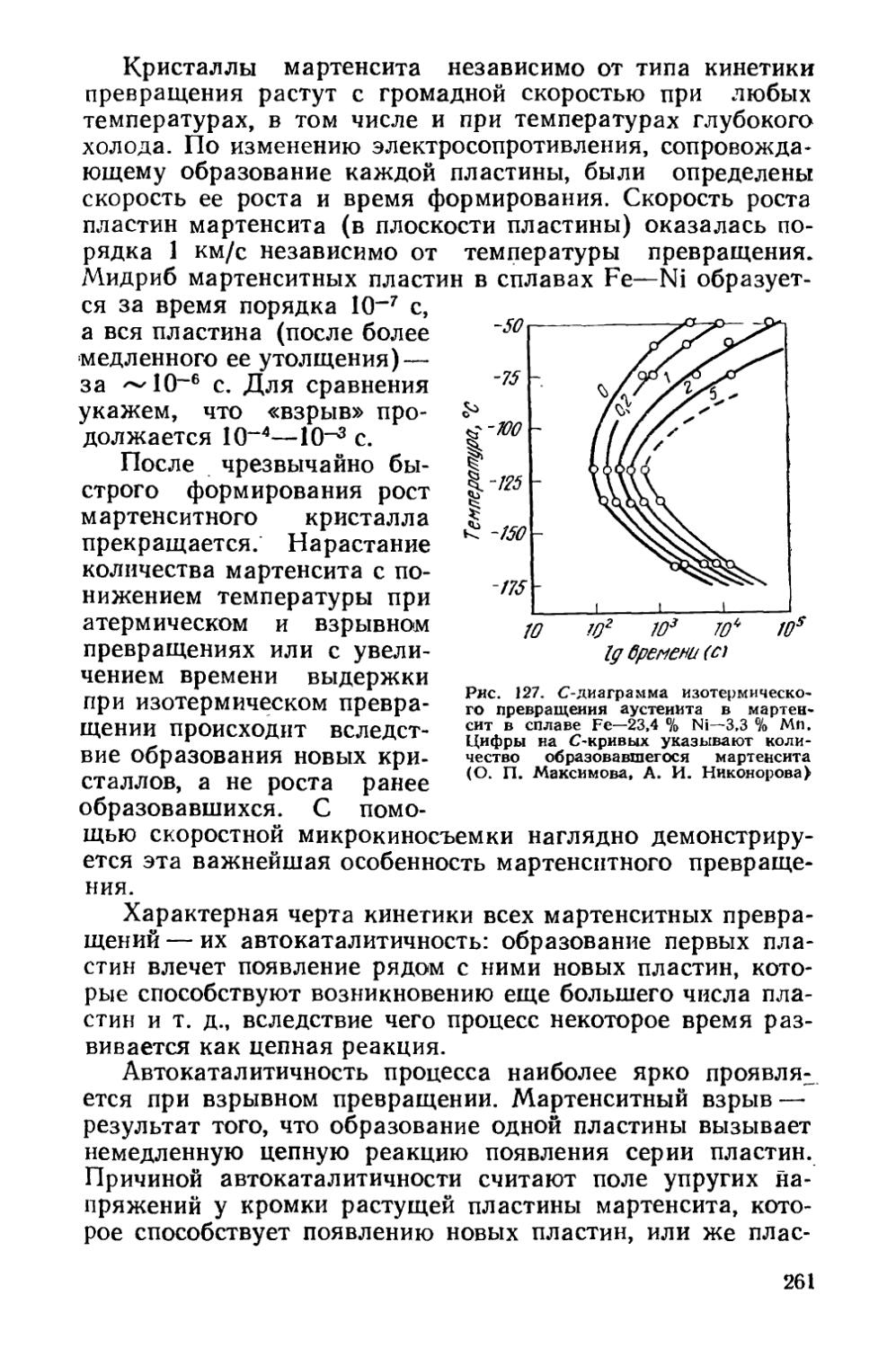
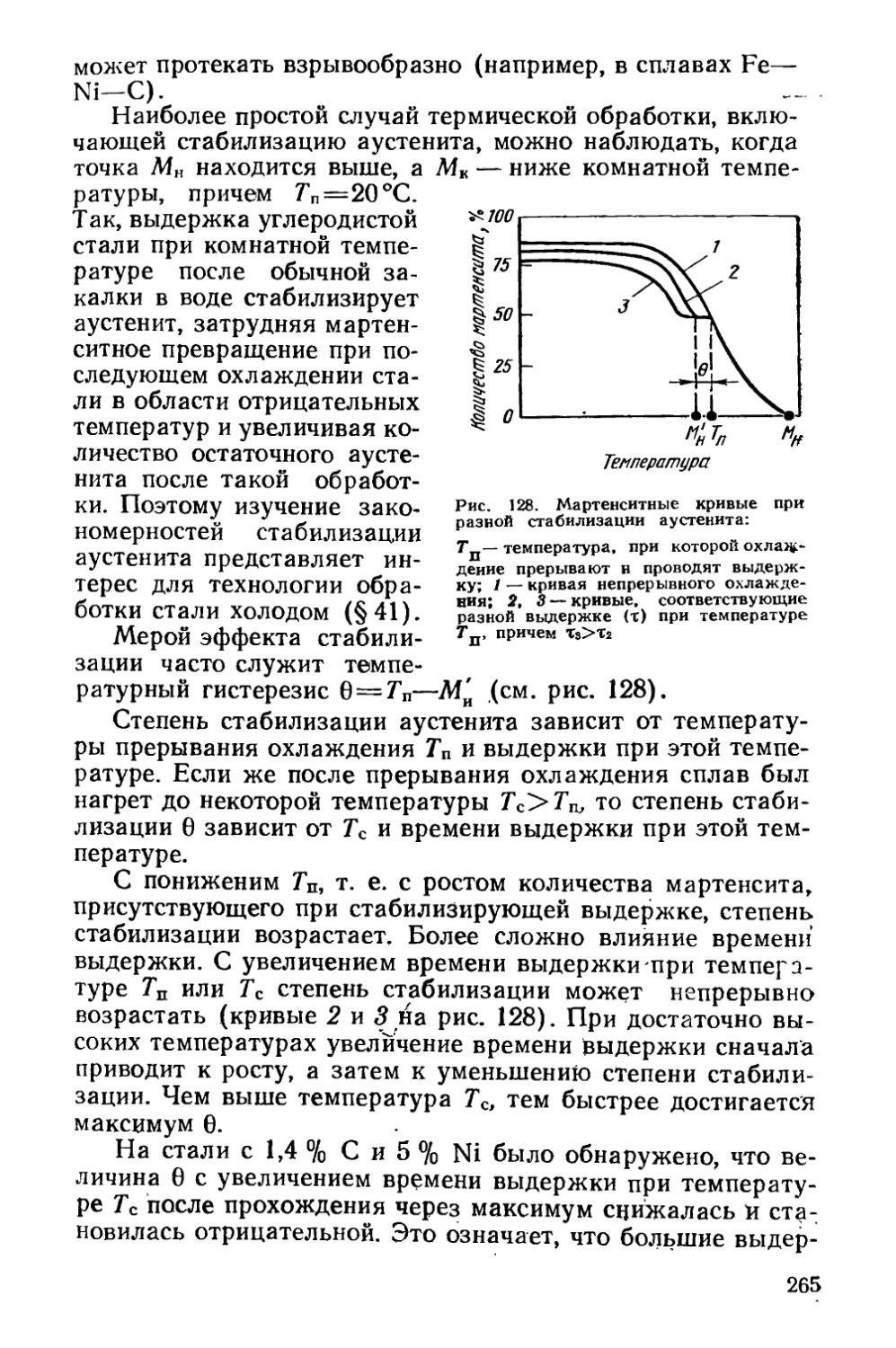
§ 36. Кинетика мартенситных превращений . . . * . . . 257
§ 37. Влияние деформации на мартенситное превращение . . 267
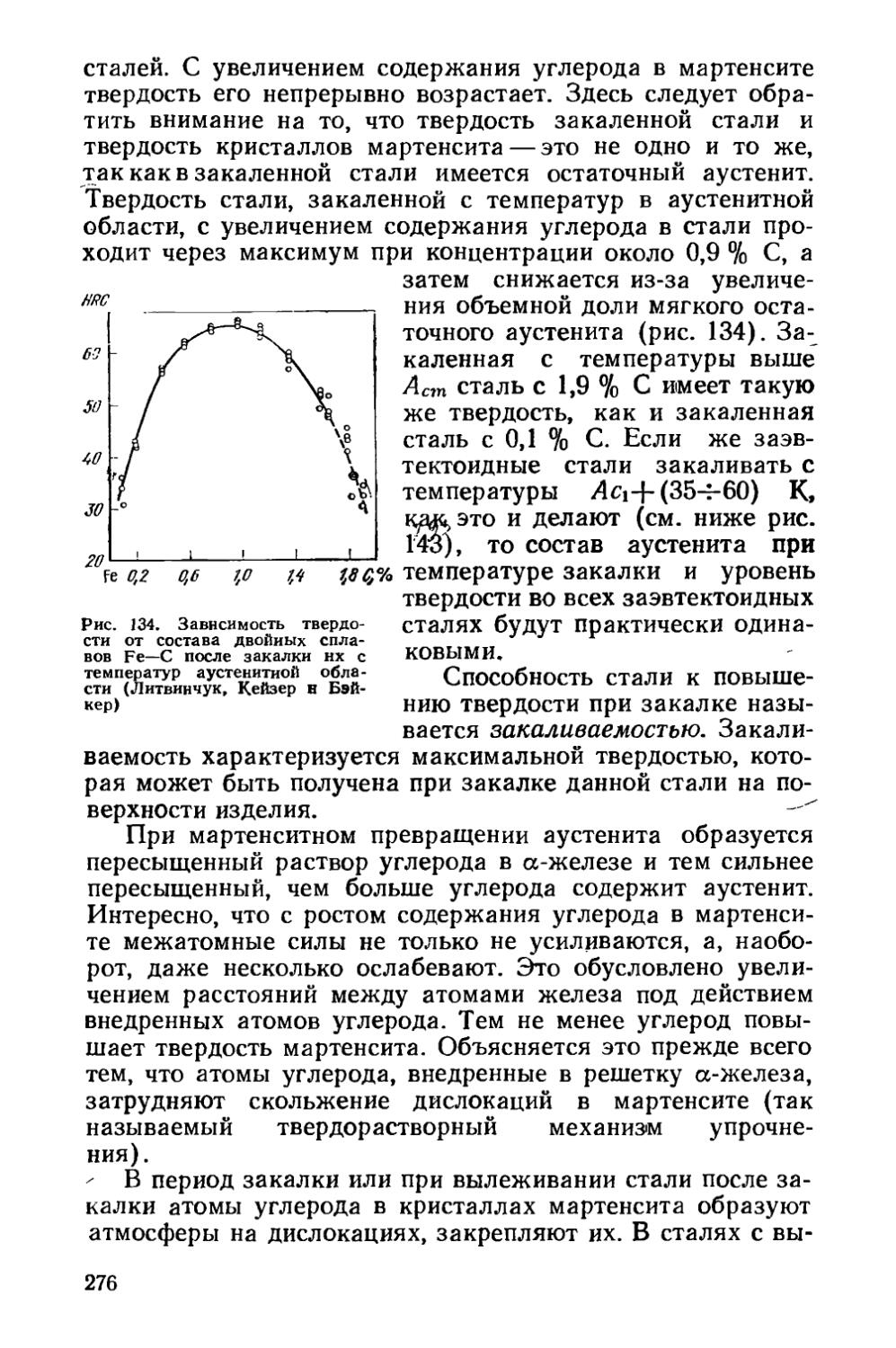
§ 38. Изменение свойств сплавов прн закалке на мартенсит' « . 274
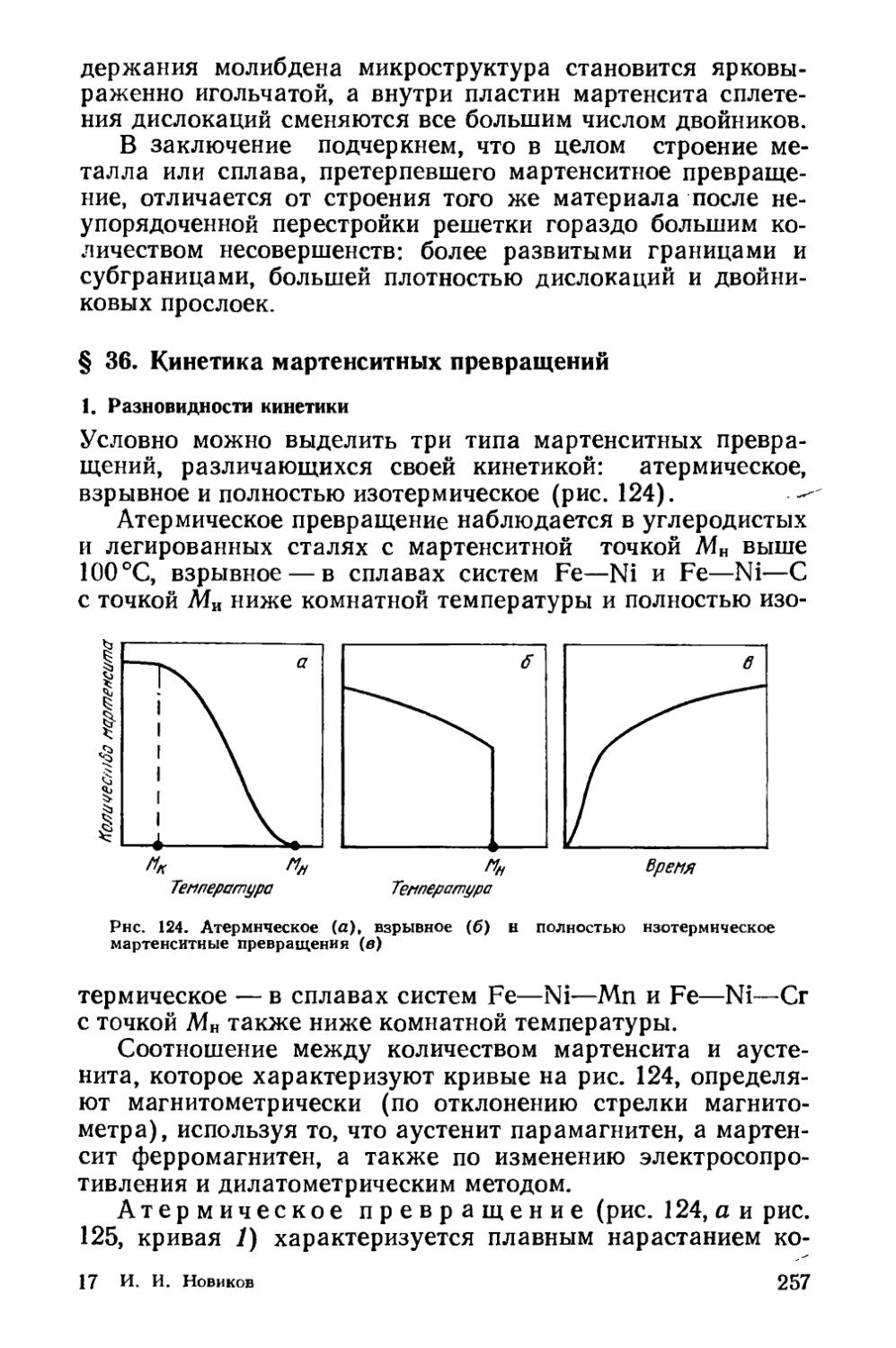
§ 39. Бейнитное превращение..................................279
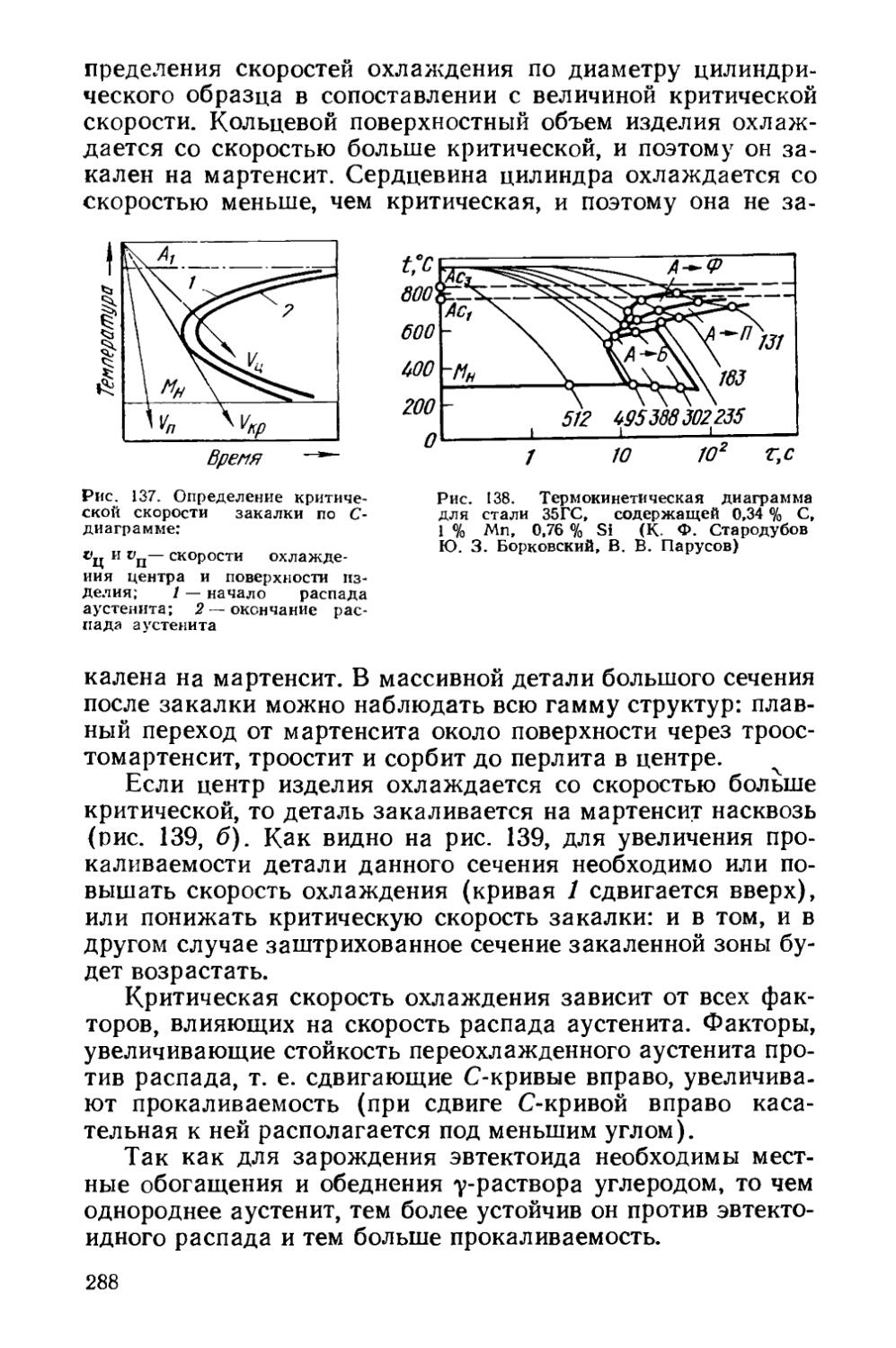
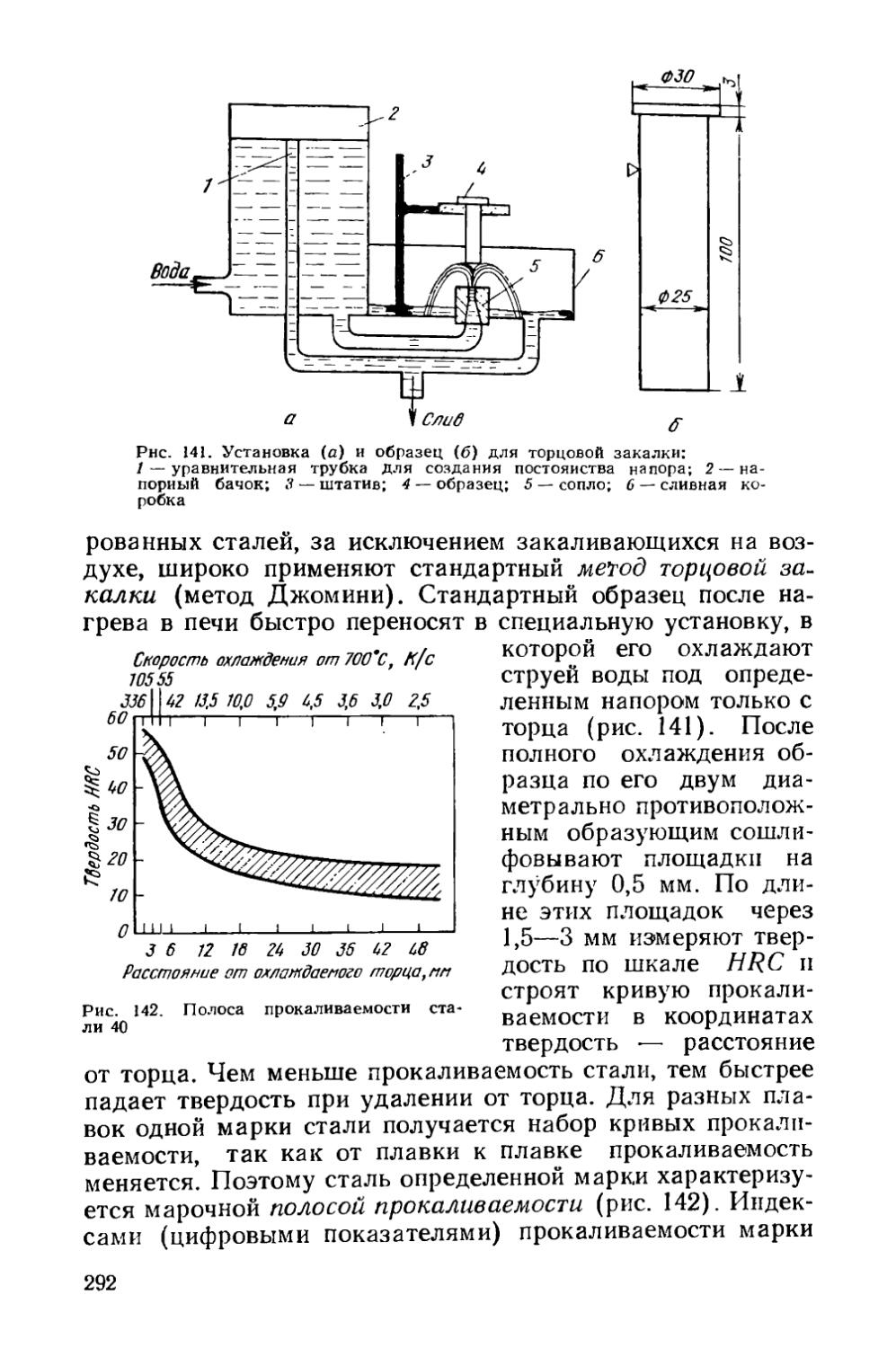
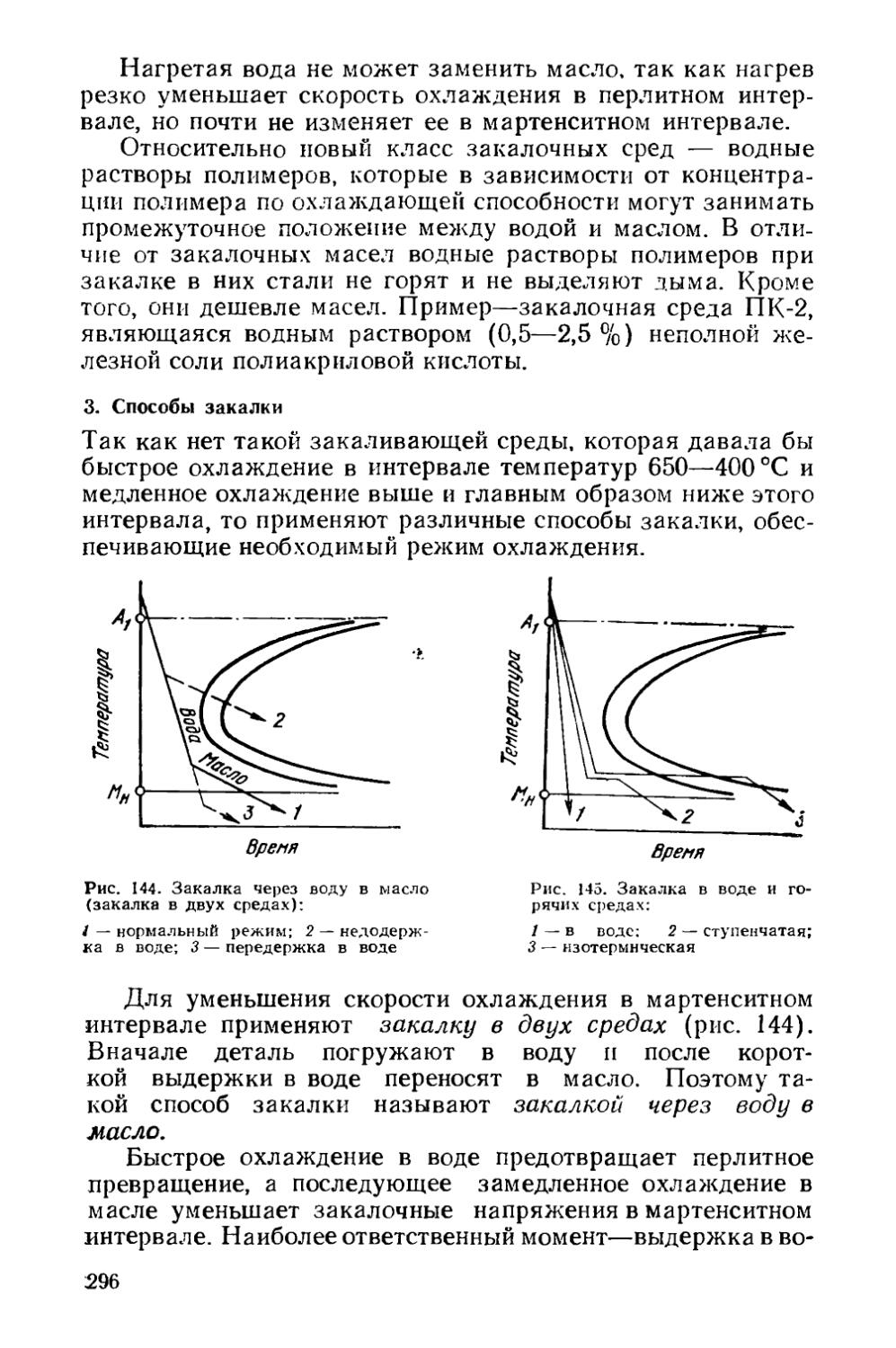
§ 40. Прокаливаемость сталей.................................287
§41. Объемная закалка сталей.....................293
§ 42. Поверхностная закалка сталей ........ 302
Глава X. Закалка с плавлением поверхности ..... 306
§ 43. Общие закономерности формирования структуры при сверхбыстром охлаждении расплава.......................307
§ 44. Изменение структуры и свойств при закалке с плавлением поверхности ,................................................310
Раздел четвертый. СТАРЕНИЕ И ОТПУСК ...... 315
Глава XI. Старение.............................................316
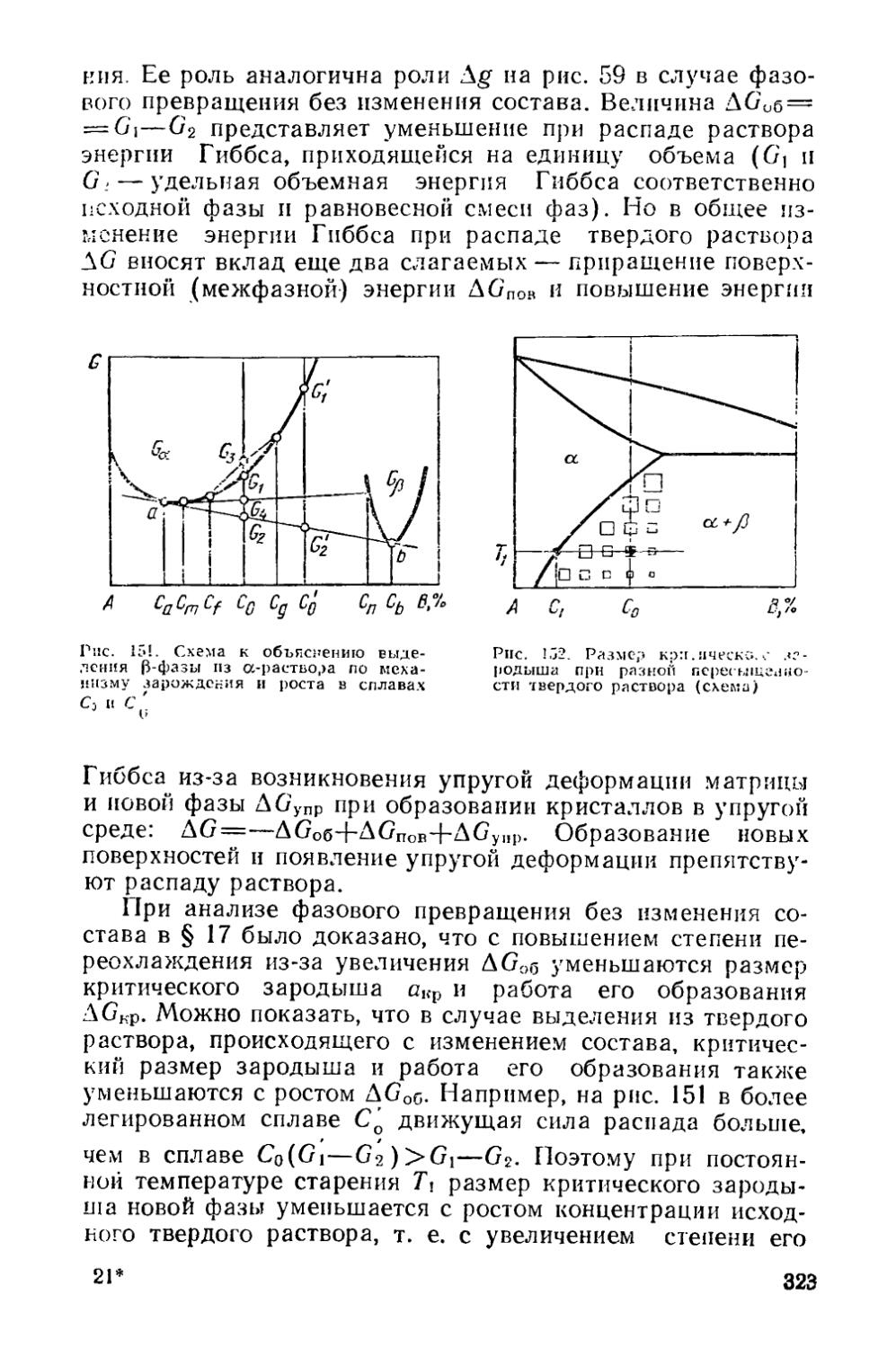
§ 45. Термодинамика процессов выделения из твердого раствора 318
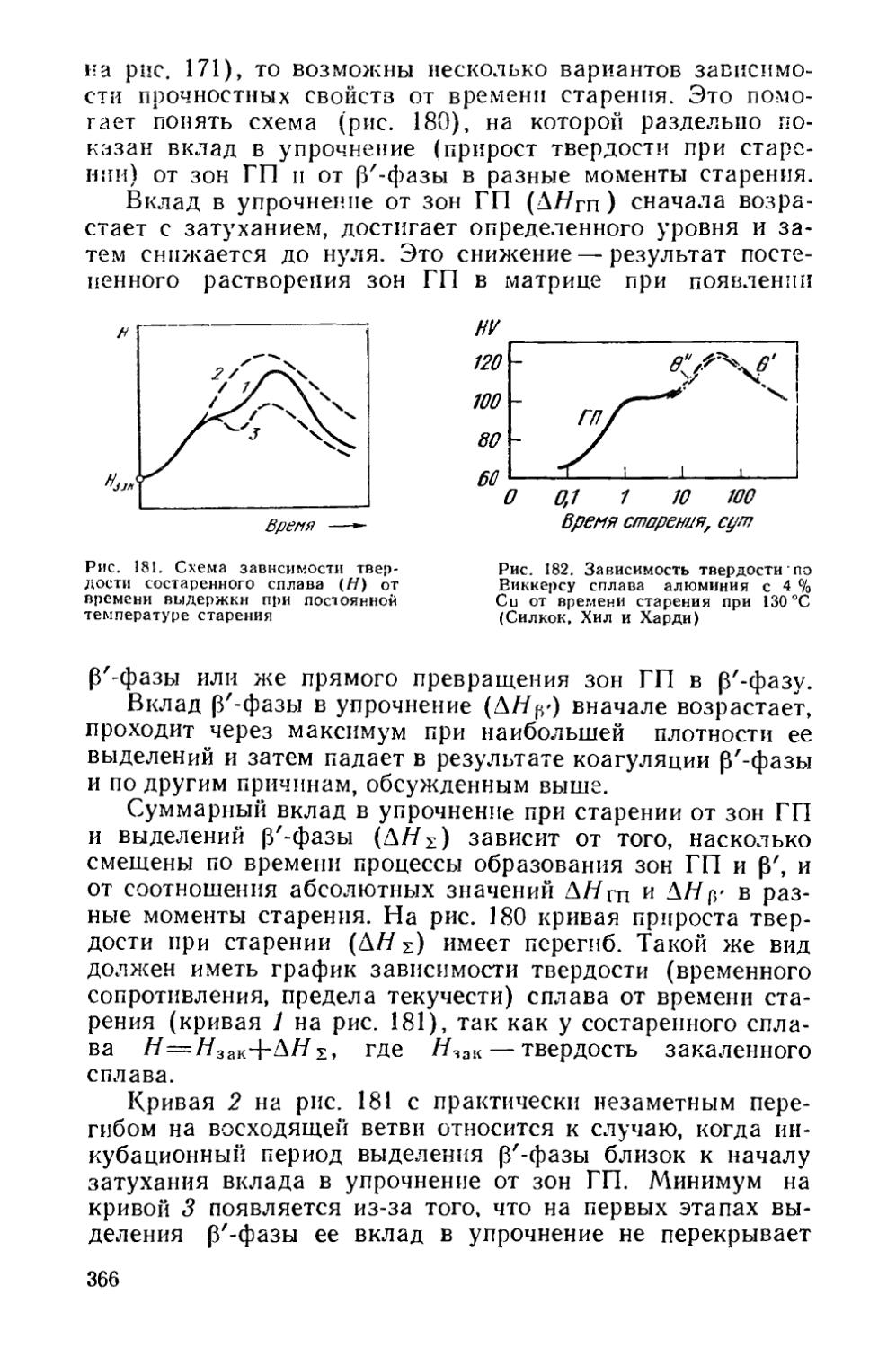
§ 46. Структурные изменения при старении..................... 324
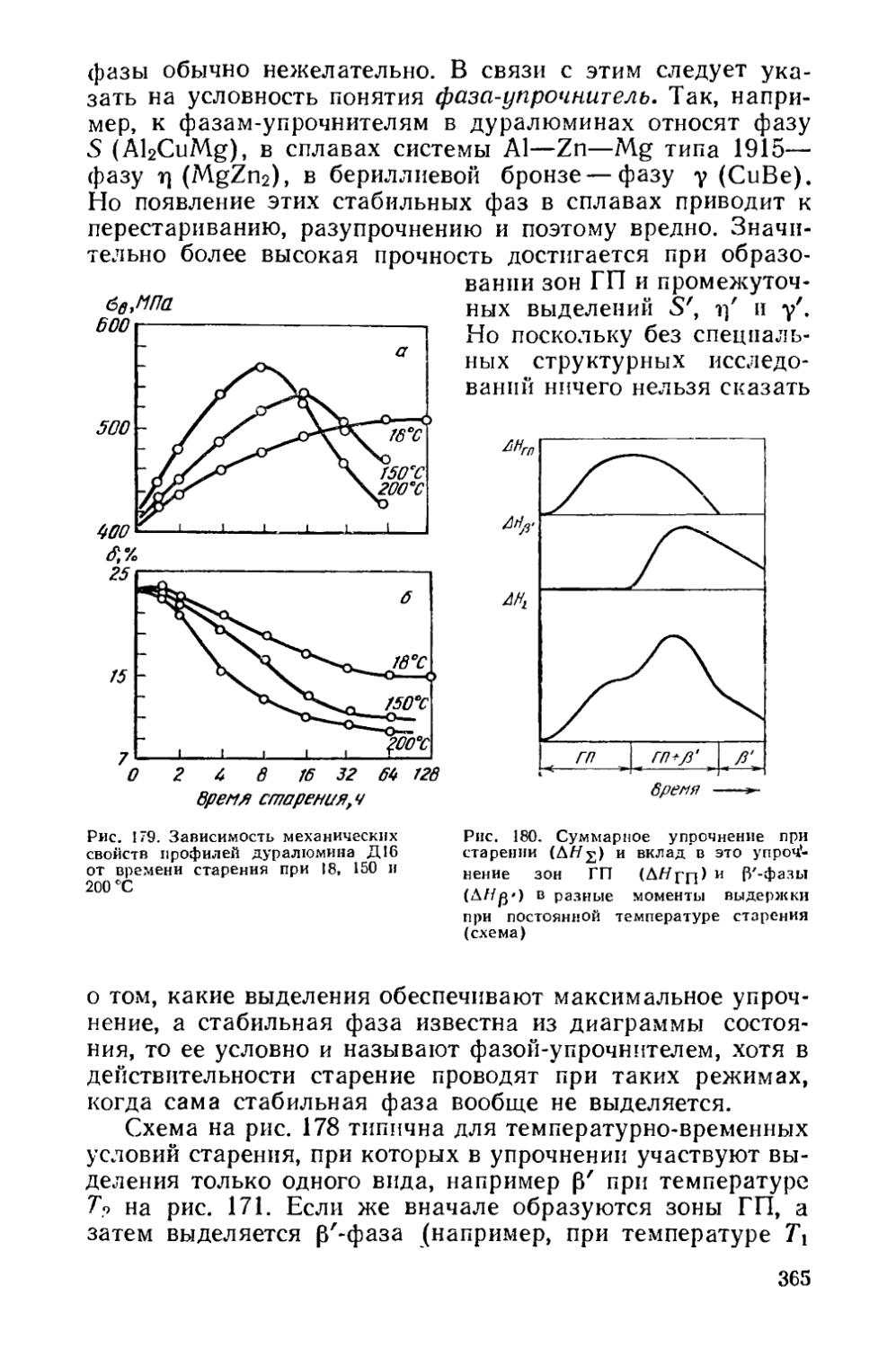
§ 47. Изменение свойств сплавов при старении...................358
§ 48. Влияние состава сплава на старение ...... 369
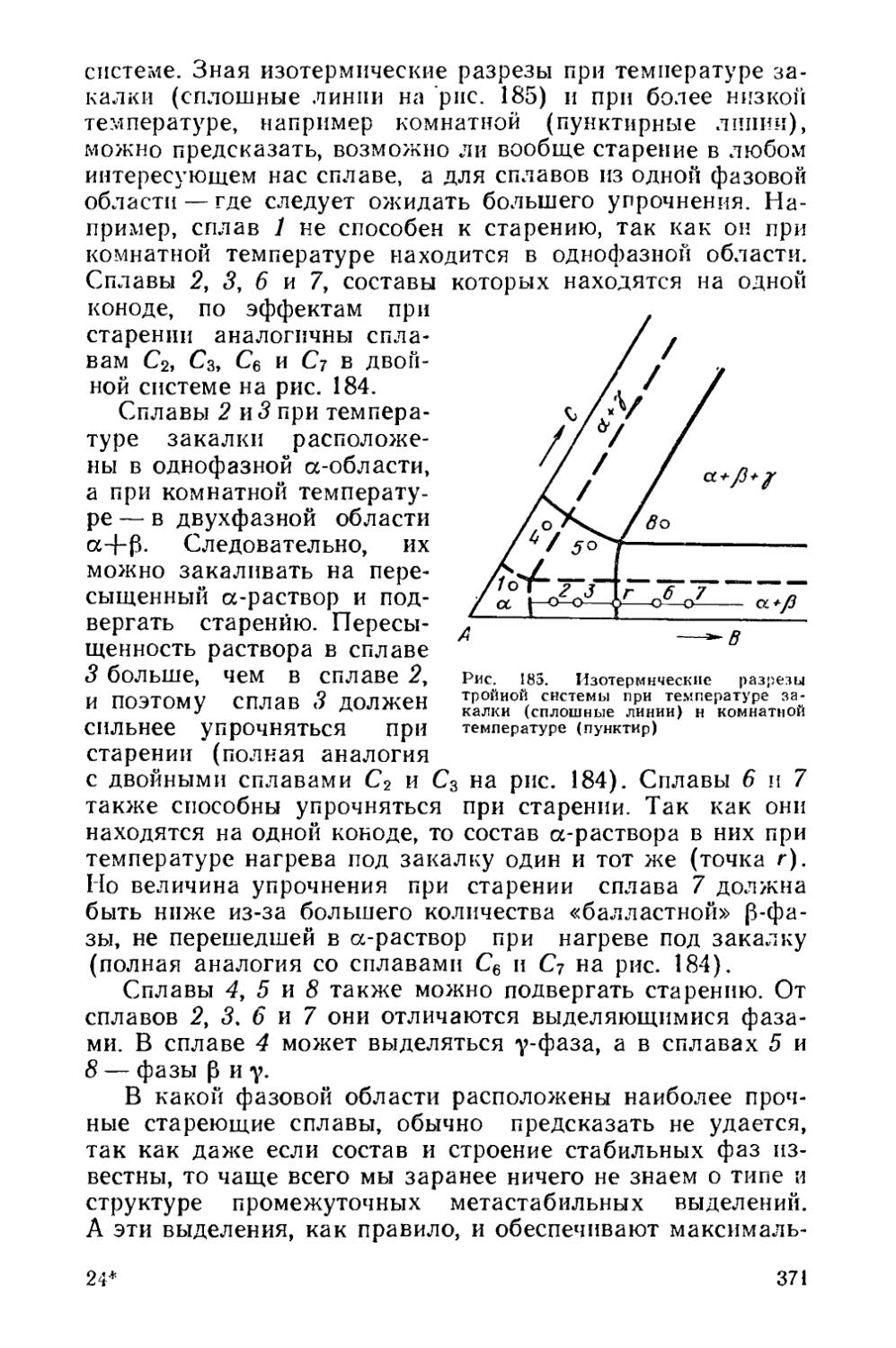
§ 49. Выбор режима старения....................................374
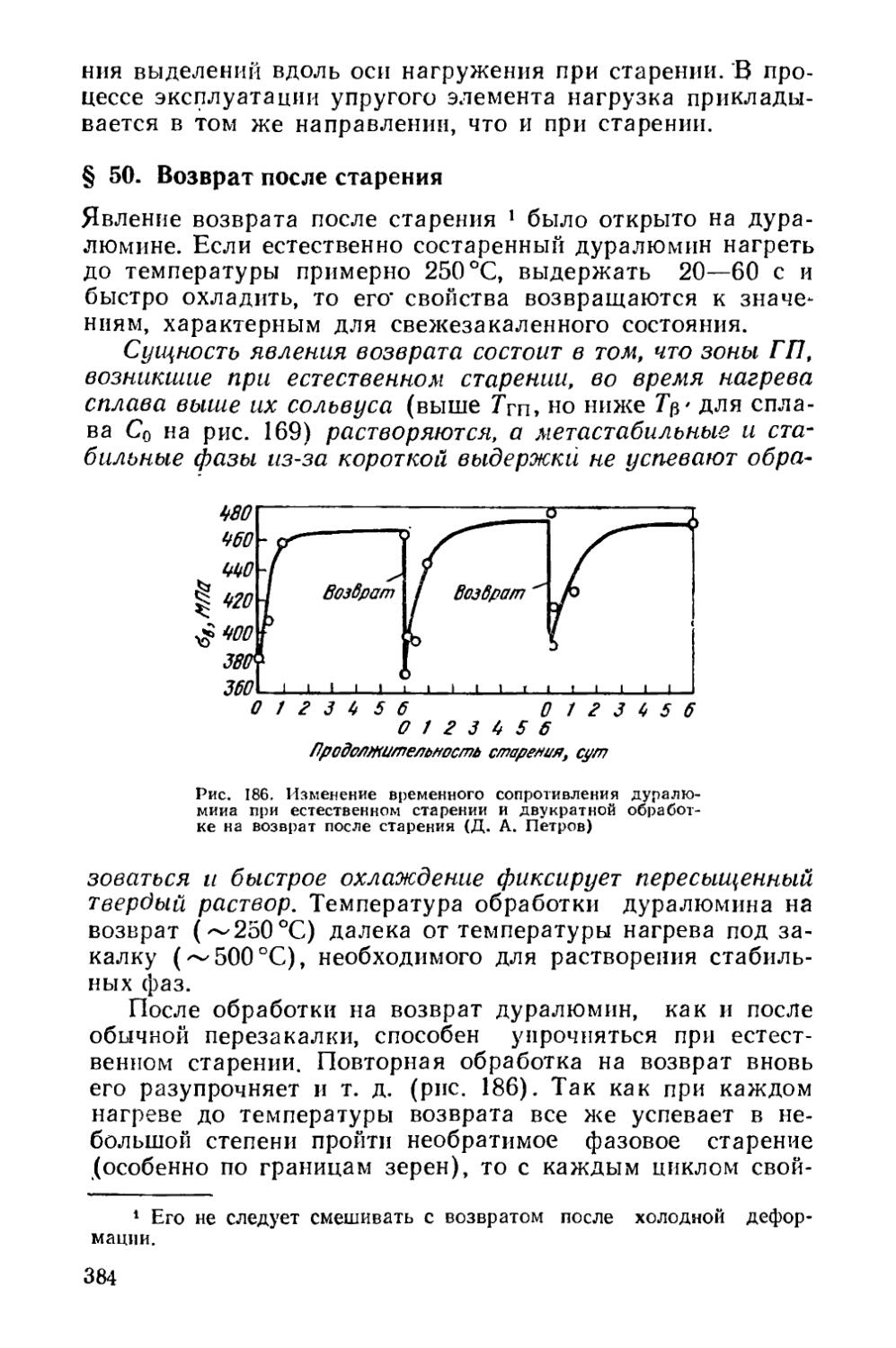
§ 50. Возврат после старения...................................384
Глава XII. Отпуск..............................................386
§ 51. Структурные изменения при отпуске сталей .... 387
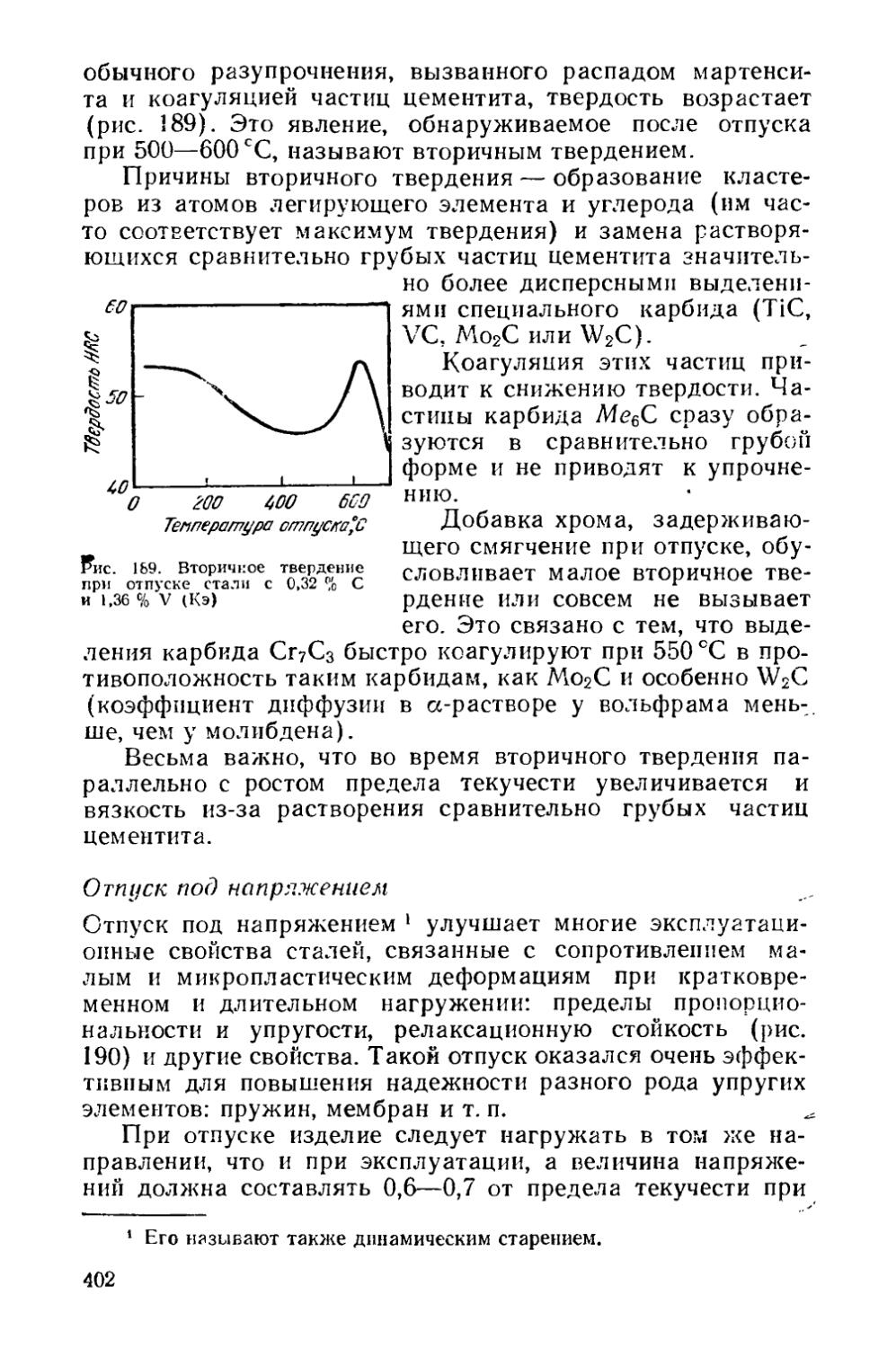
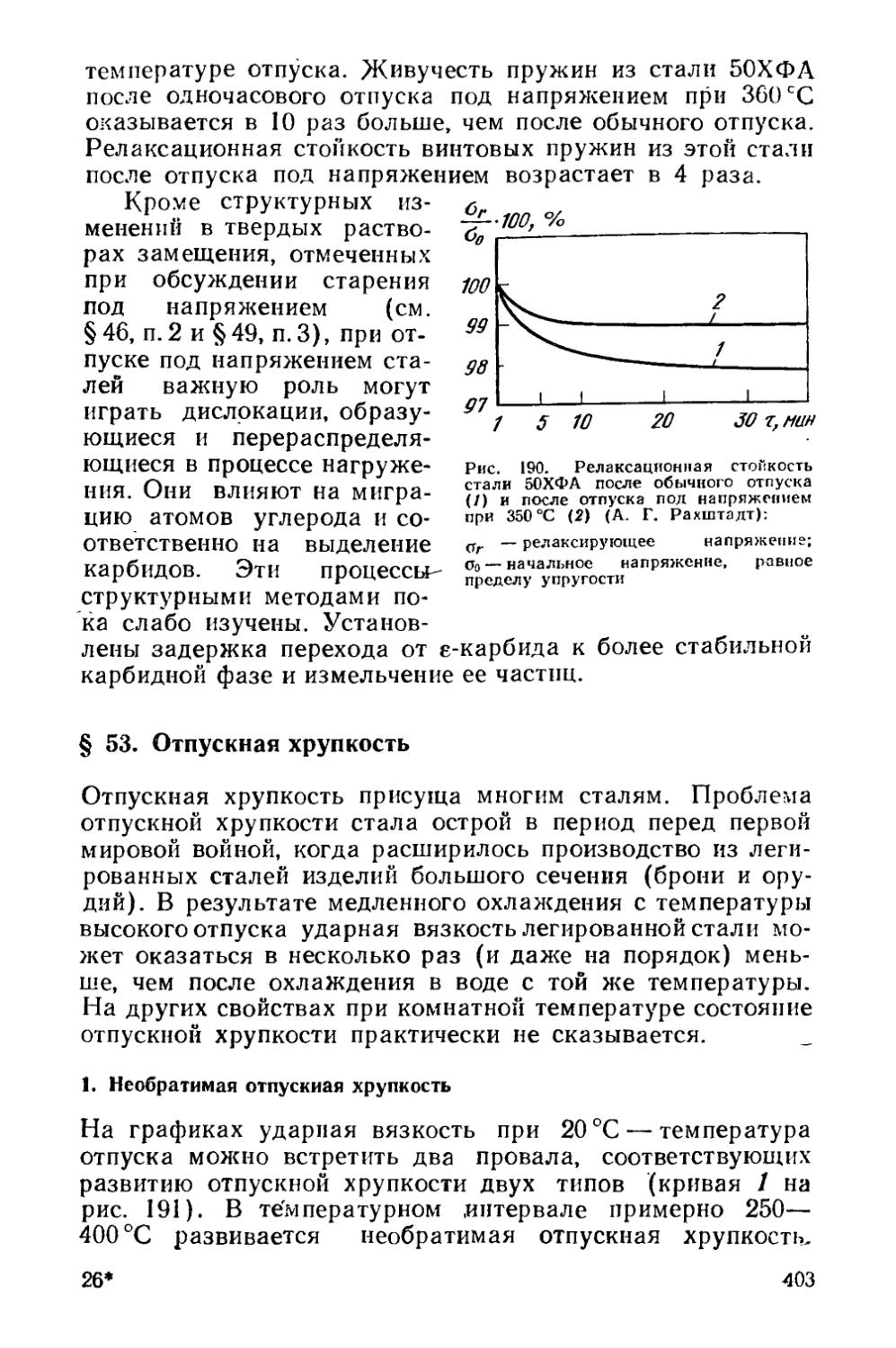
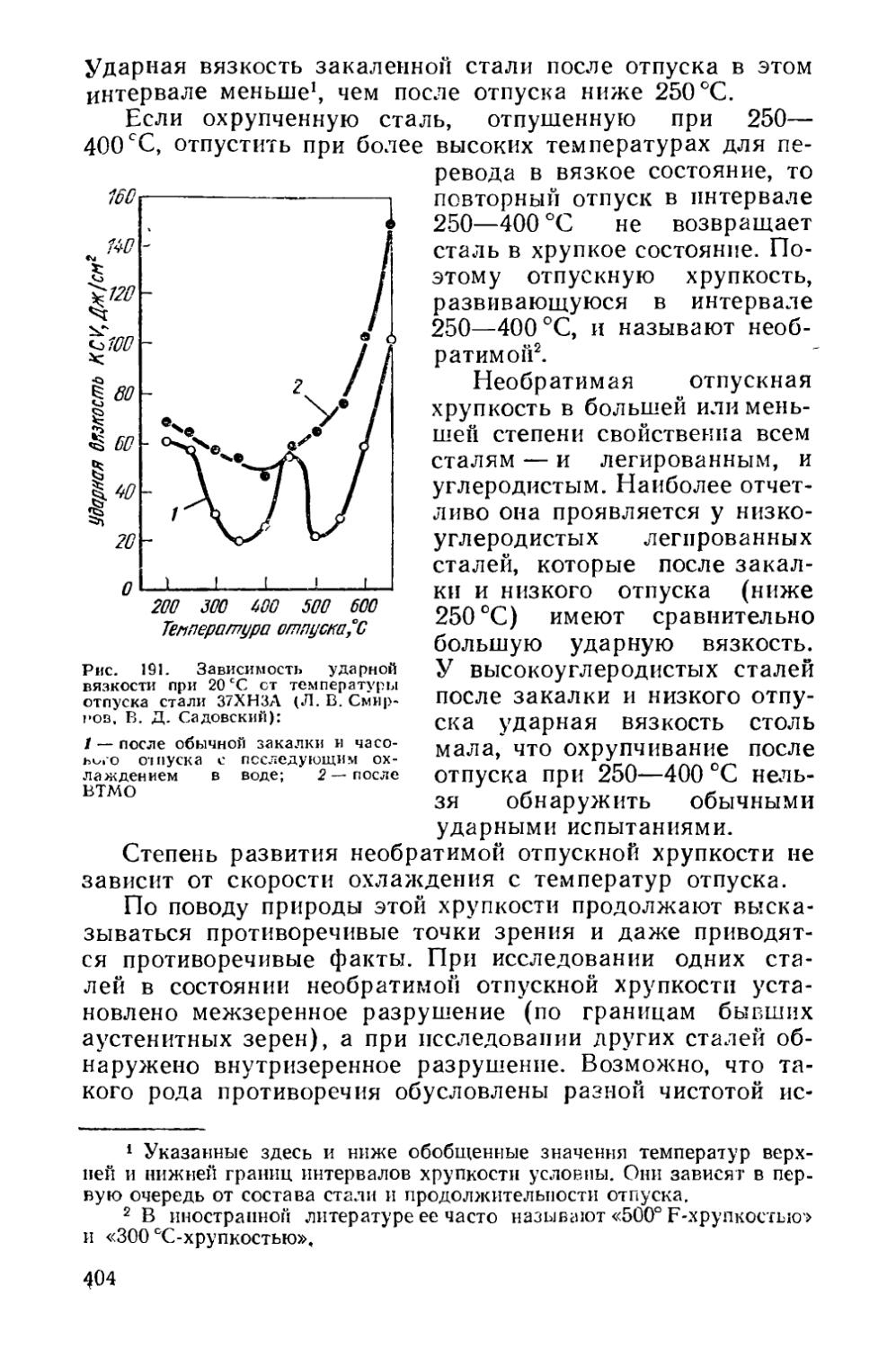
§ 52. Изменение механических свойств при отпуске сталей и выбор режима отпуска................................... , 397
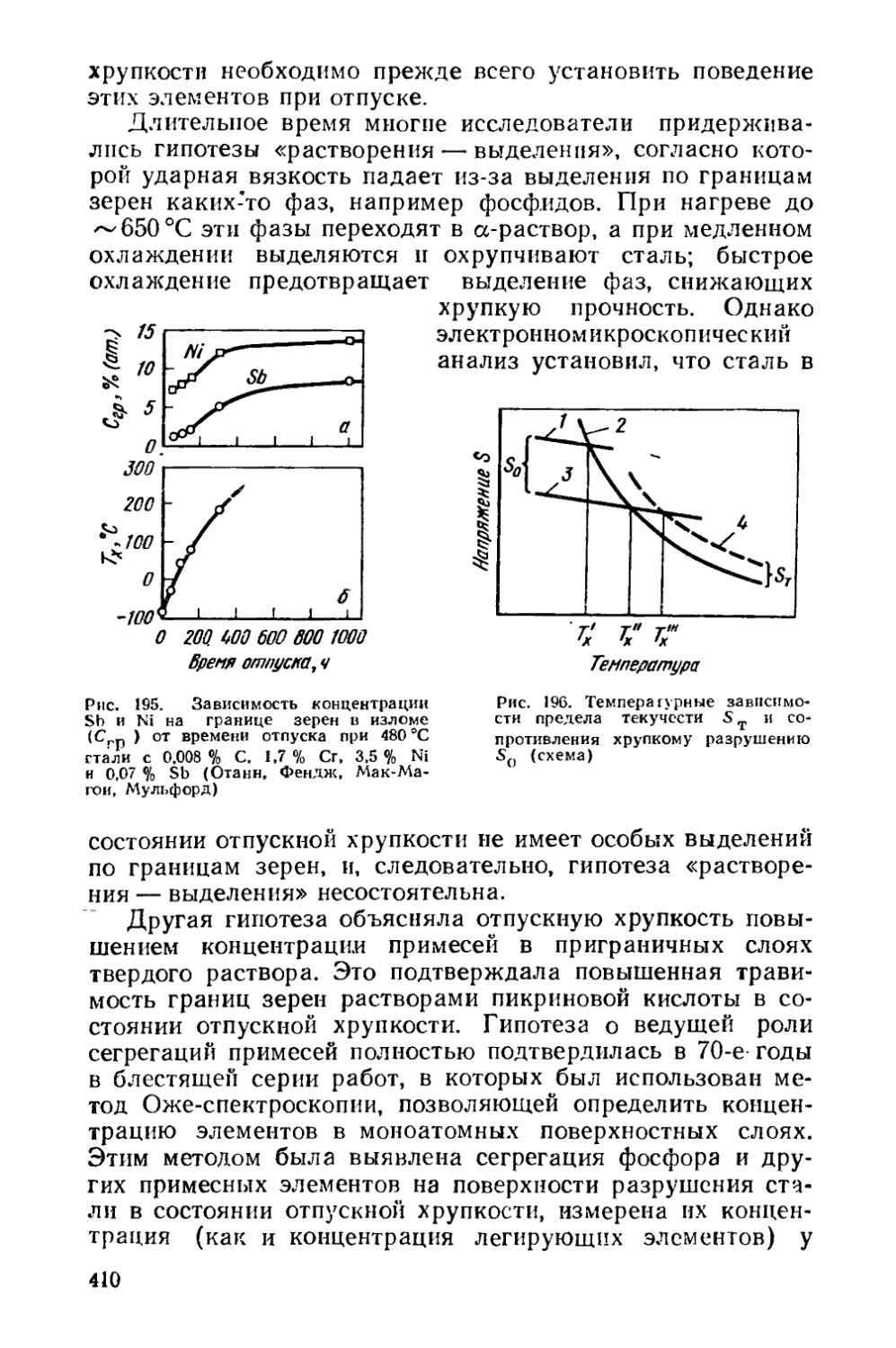
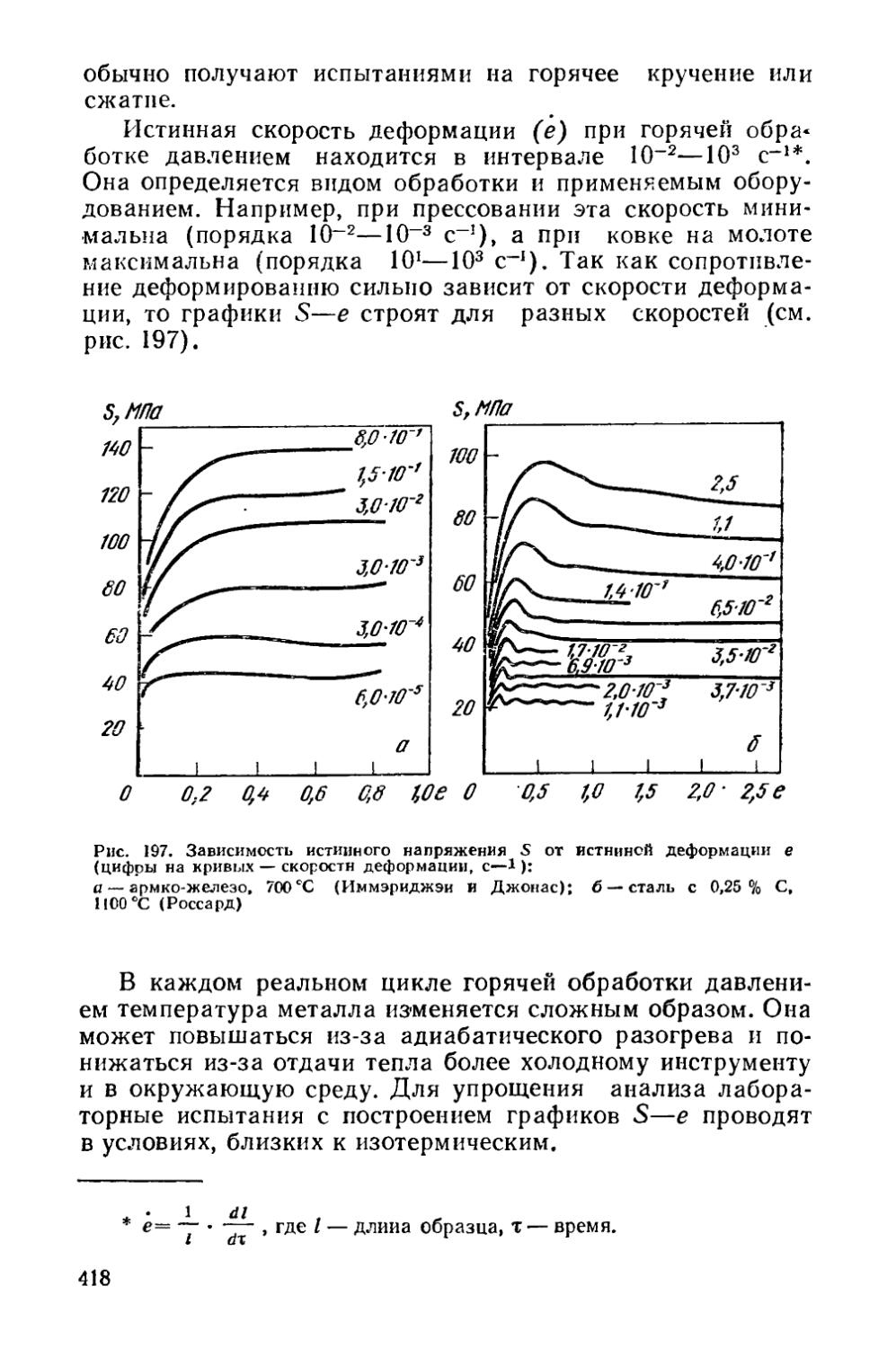
§ 53. Отпускная хрупкость............................... . 403
Раздел пятый. ТЕРМОМЕХАНИЧЕСКАЯ ОБРАБОТКА t . 416
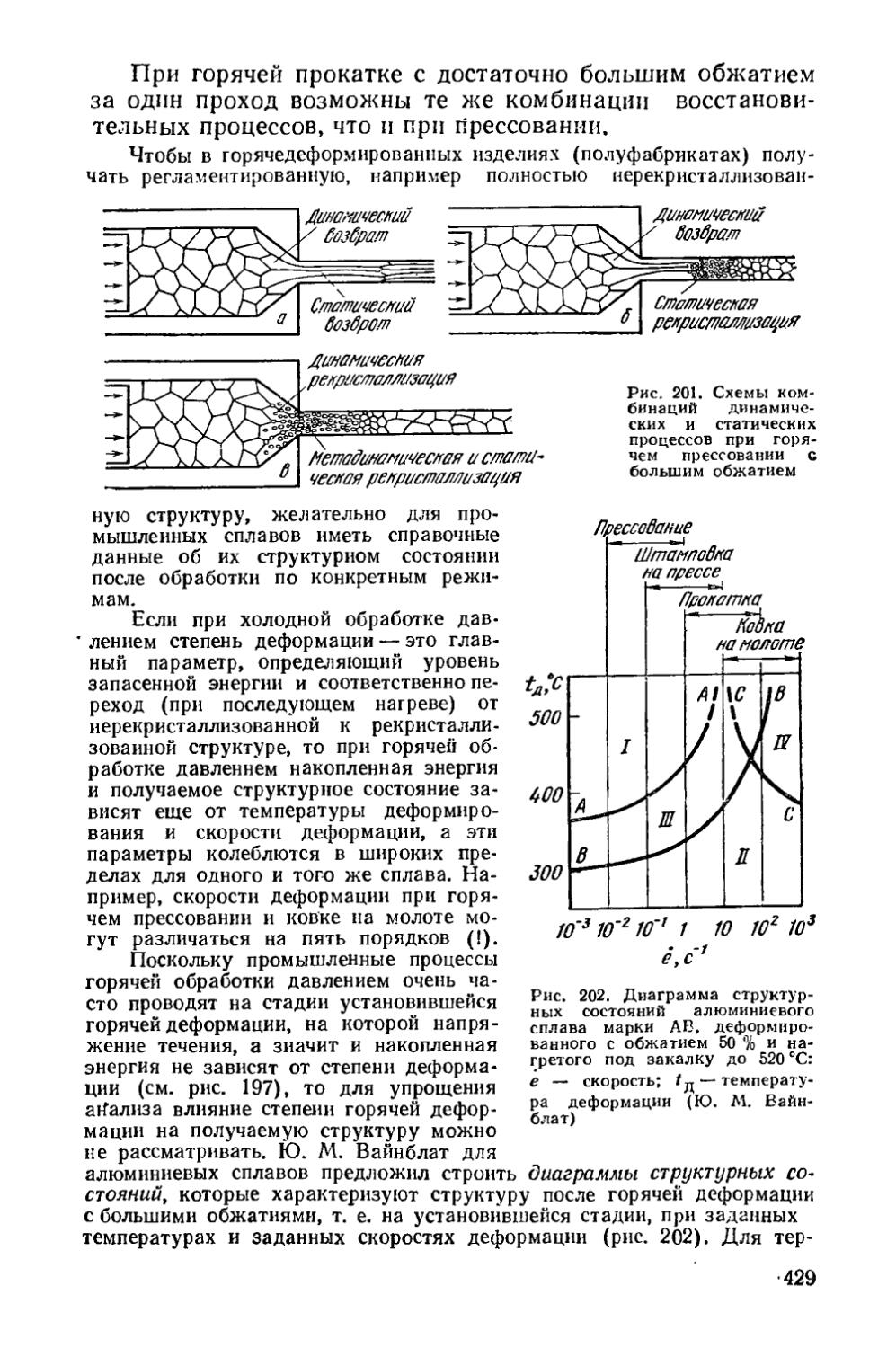
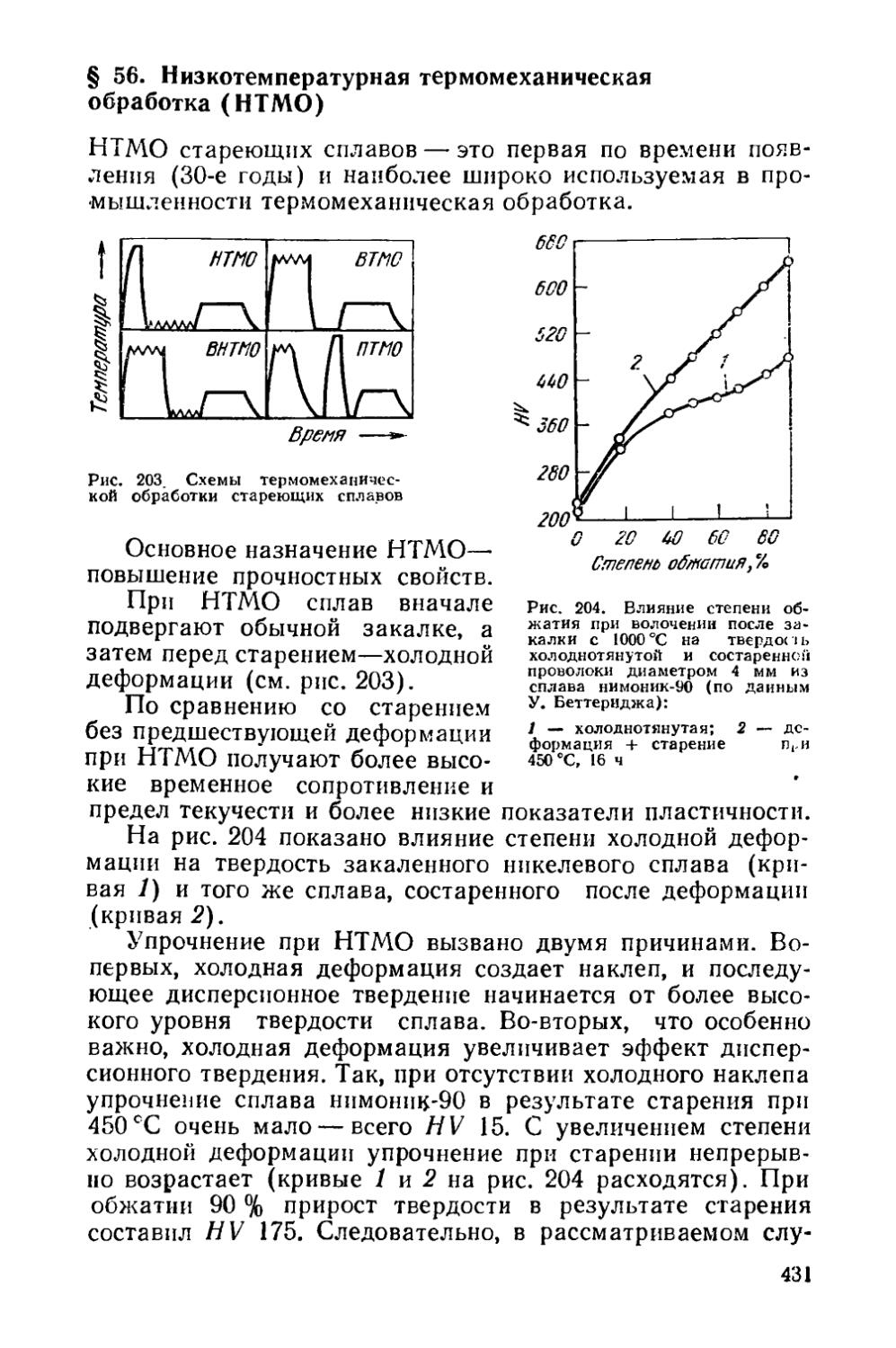
Глава XIII. Изменение структуры металла при горячей обработке давлением............................................ 417
§ 54. Структурные изменения во время горячей деформации . 419 § 55. Структурные изменения по окончании горячей деформации 425
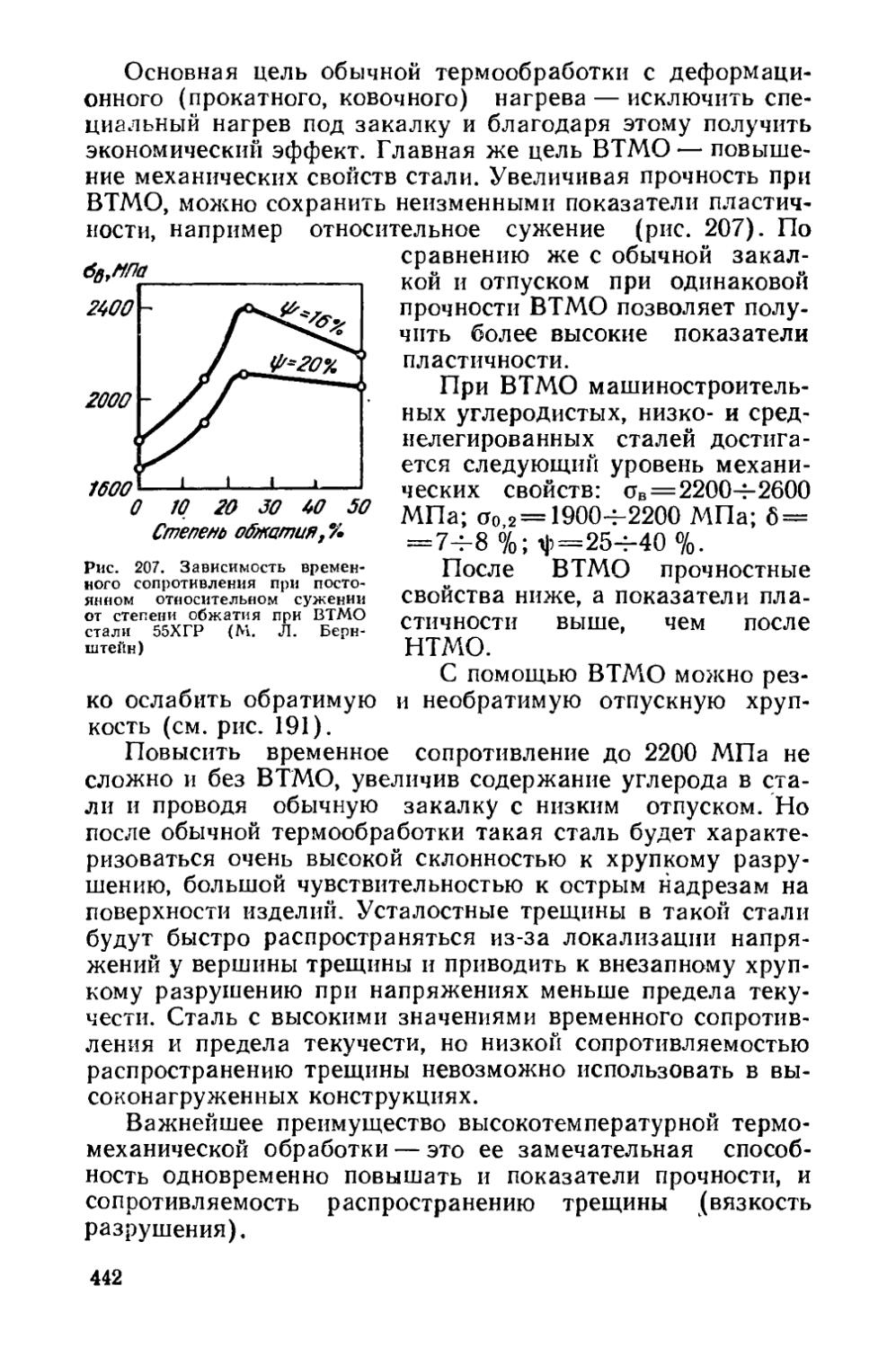
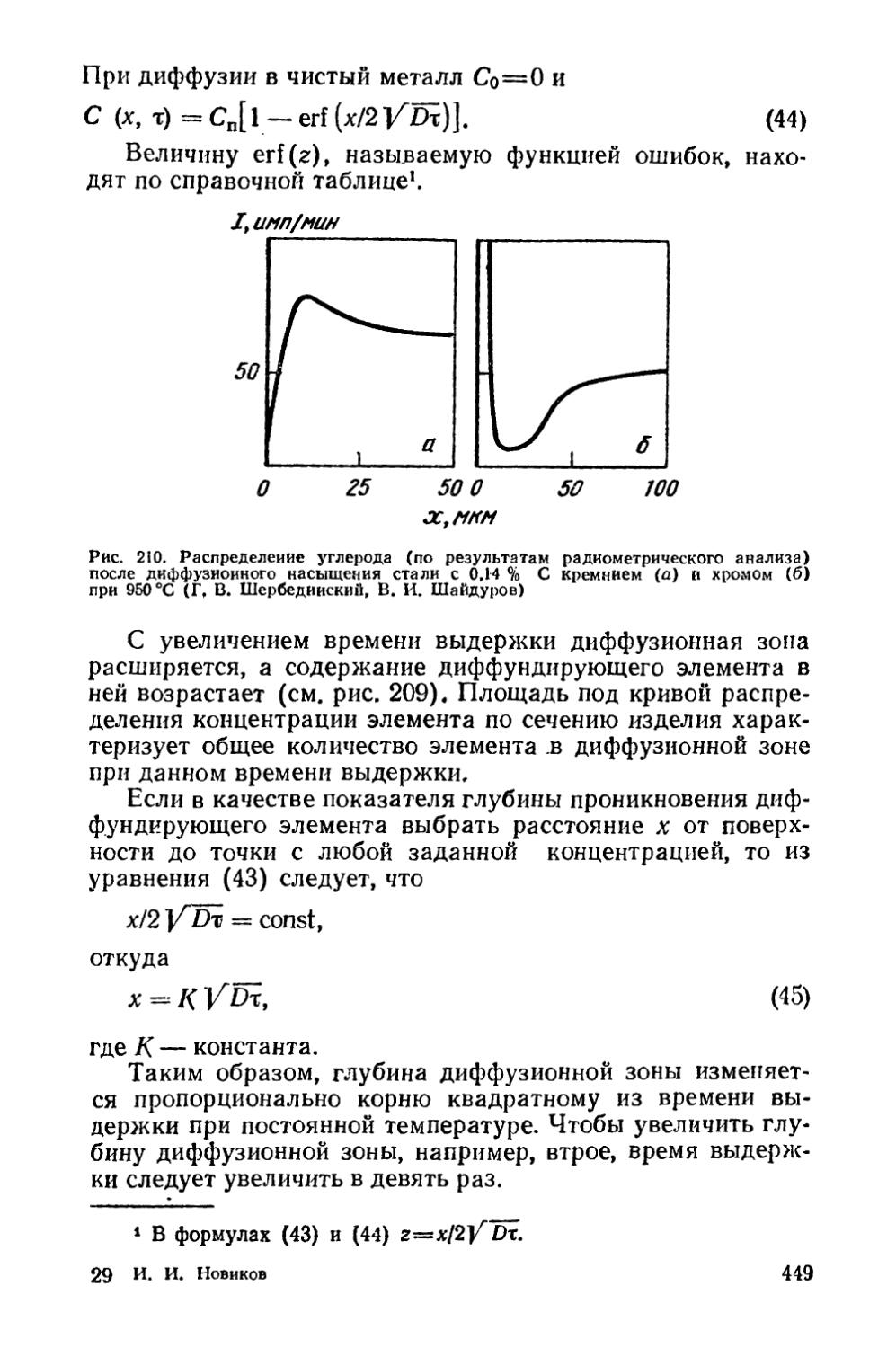
Глава XIV. Термомеханическая обработка стареющих сплавов 430
§ 56. Низкотемпературная термомеханическая обработка (НТМО) 431
4
§ 57. Высокотемпературная термомеханическая обработка (ВТМО) .....................................................436
§ 58. Предварительная термомеханическая обработка (ПТМО) , 438
Глава XV. Термомеханическая обработка сталей, закаливаемых на мартенсит.......................................... ... 440
§ 59. Низкотемпературная термомеханическая обработка (НТМО) 440
§ 60. Высокотемпературная термомеханическая обработка (ВТМО)......................................................441
§ 61. Термомеханическая обработка с деформацией во время перлитного превращения . . ...................444
§ 62. Контролируемая прокатка . .................... 445
§ 63. Предварительная термомеханическая обработка (ПТМО) . 446
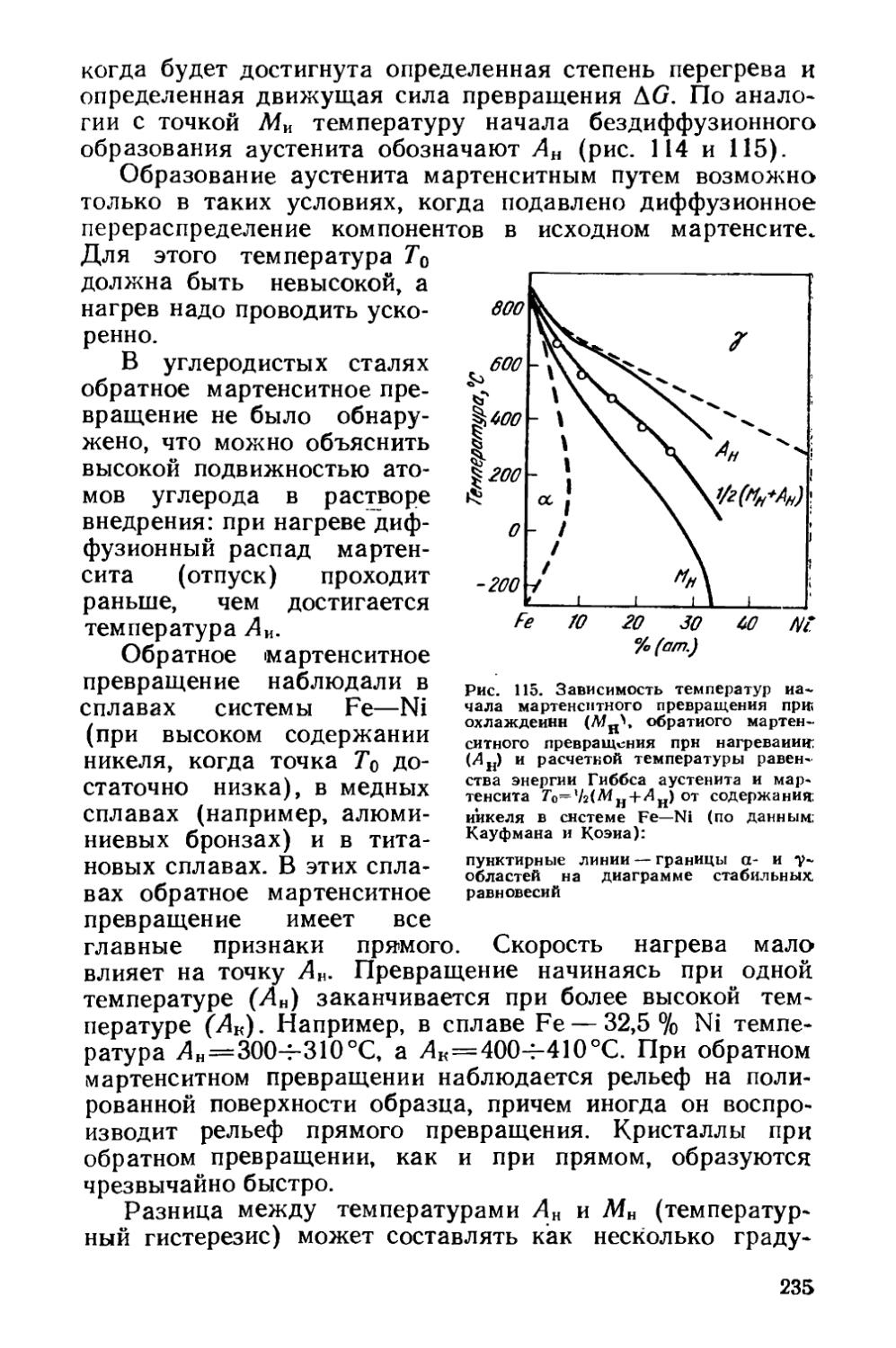
Раздел шестой. ХИМИКО-ТЕРМИЧЕСКАЯ ОБРАБОТКА . . 447
Глава XVI. Закономерности изменения состава и структуры при химико-термической обработке . . .............447
§ 64. Образование однофазной диффузионной зоны . . , ♦ 448
§ 65. Образование многофазной диффузионной зоны * 450
§ 66. Особенности строения диффузионной зоны............459
Глава XVII. Разновидности химико-термической обработки , 460
§ 67. Диффузионное насыщение неметаллами . 463
§ 68. Диффузионное насыщение металлами 468
§ 69. Диффузионное удаление элементов...................469
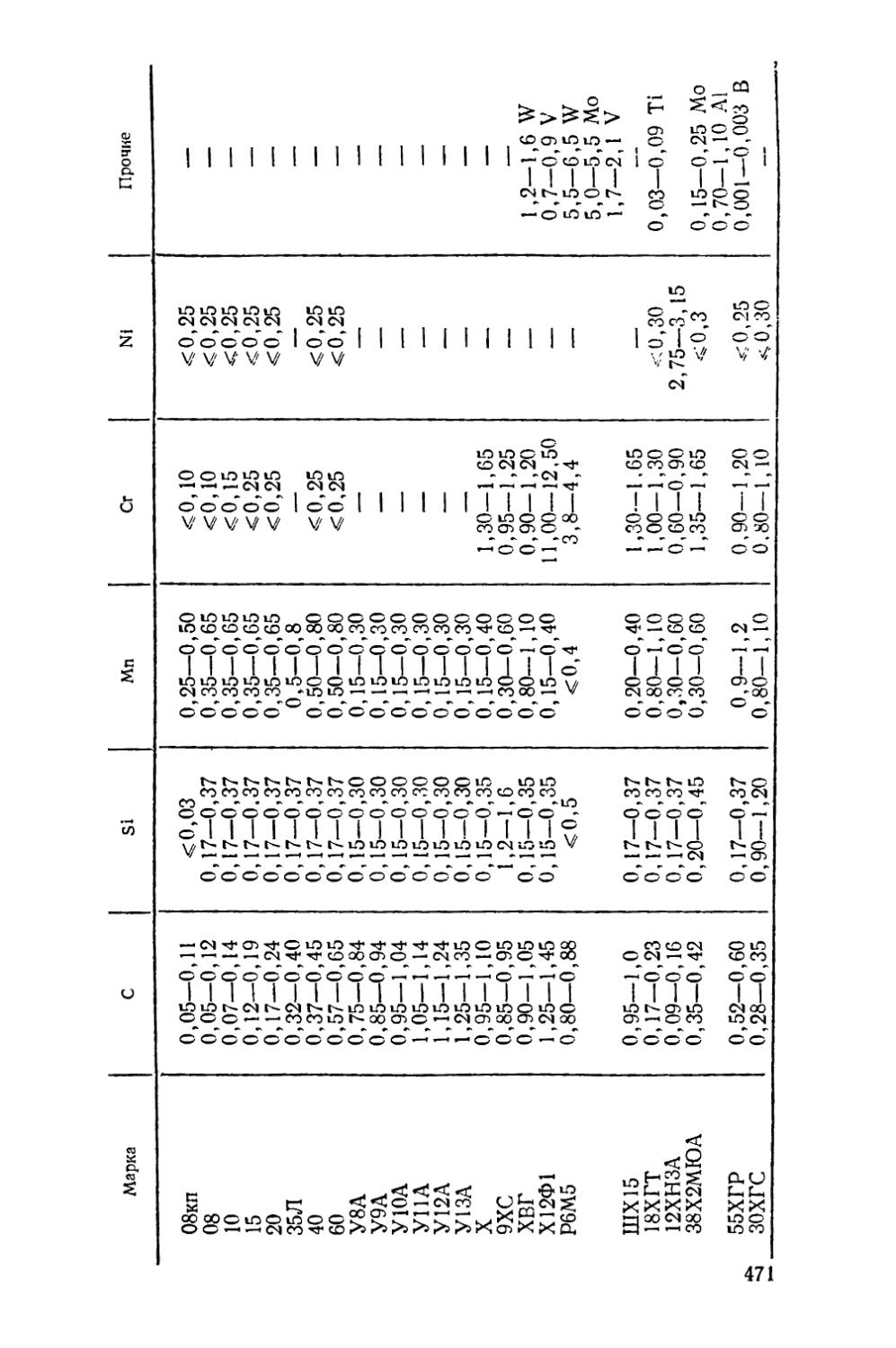
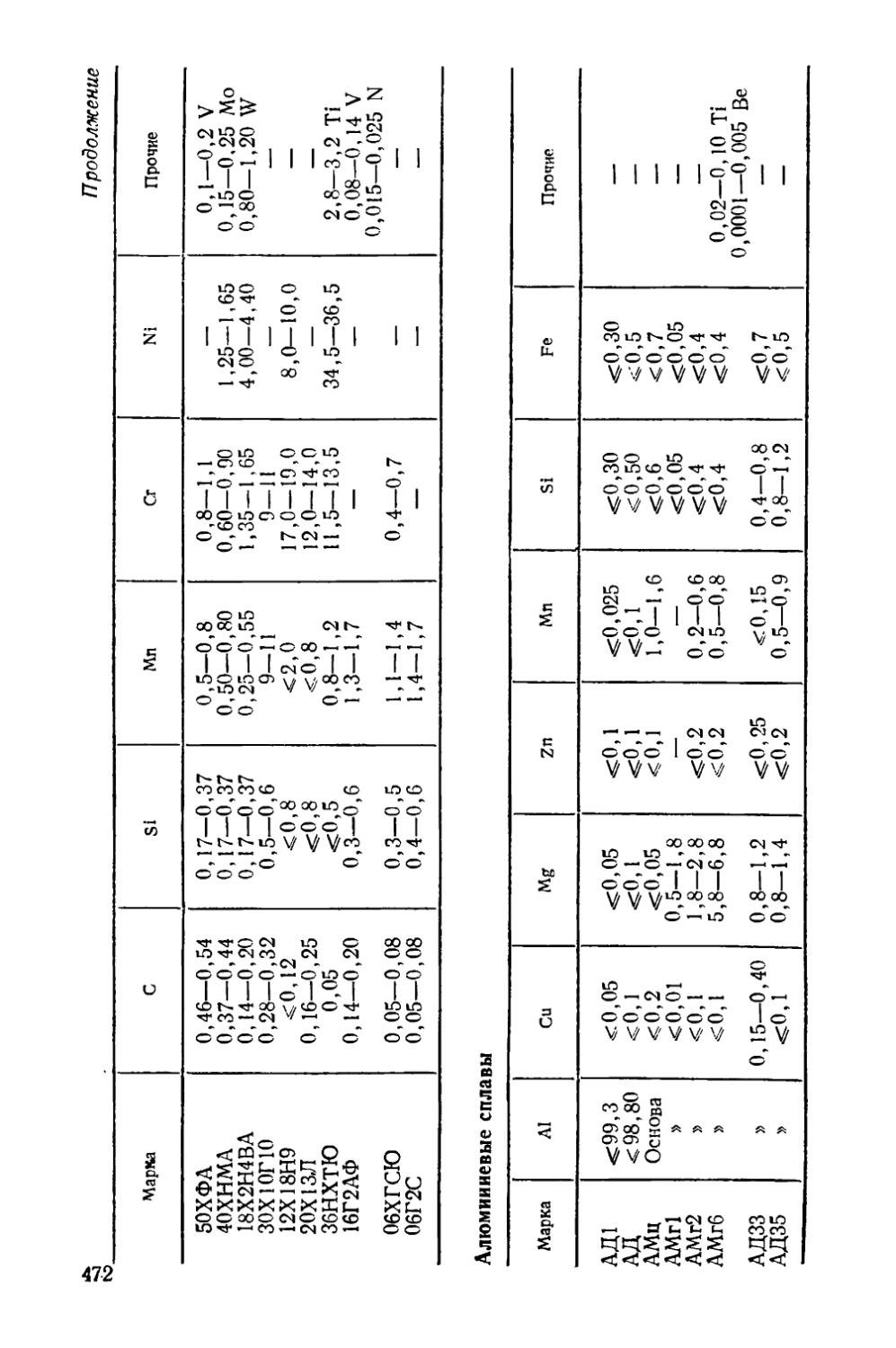
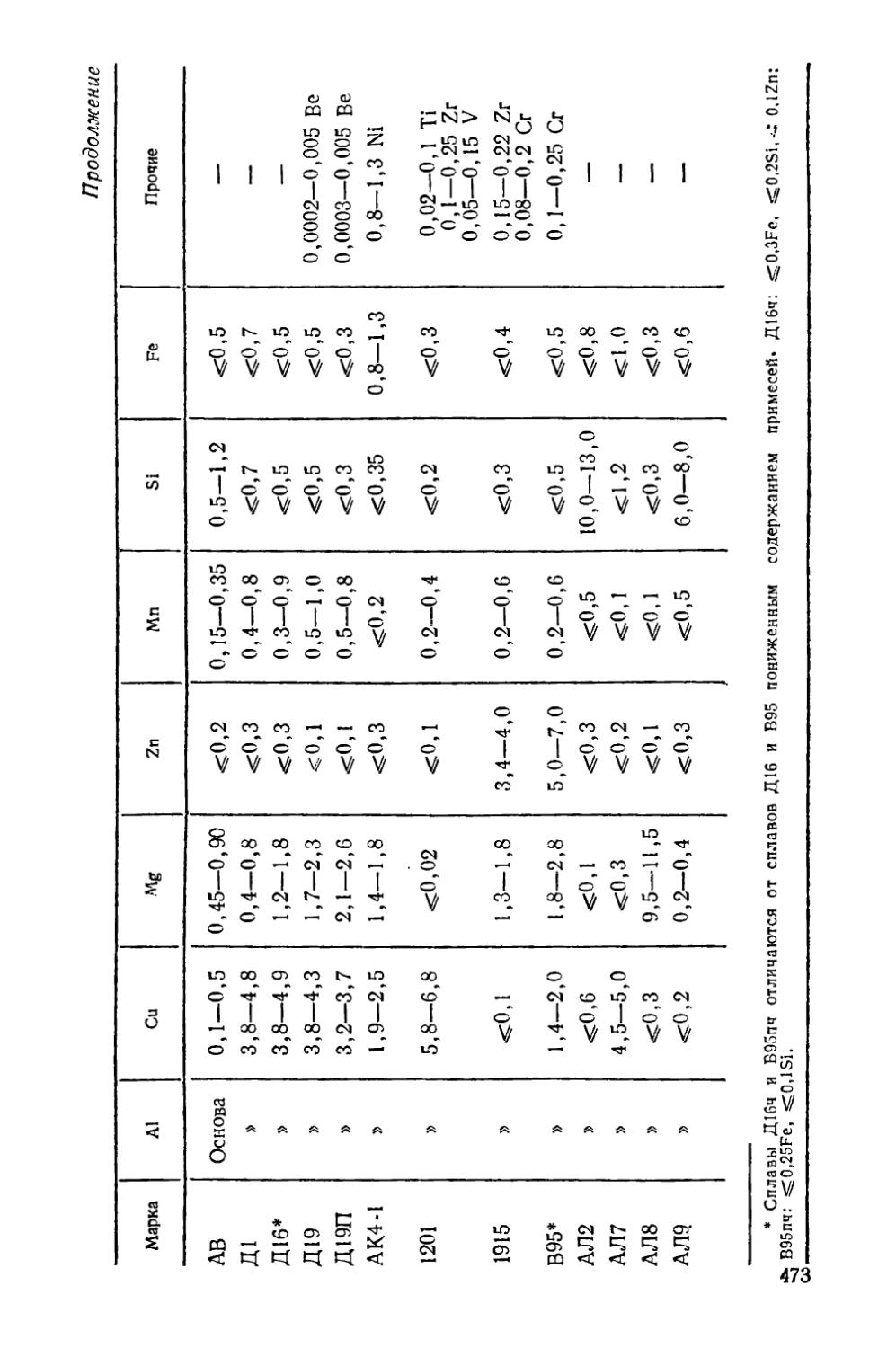
Приложение............................................. 471
Рекомендательный библиографический список..............
Предметный указатель . . * • ...................
Посвящаю моему учителю академику Андрею Анатольевичу Бочвару
ПРЕДИСЛОВИЕ
Книга написана в соответствии с учебной программой курса «Теория термической обработки», который автор на протяжении многих лет читает в Московском институте стали и сплавов.
Теория термической обработки является центральной дисциплиной в подготовке металловедов и термистов. Перед ее изучением студент должен освоить физическую химию, кристаллографию, металлографию, иметь представление о дефектах кристаллического строения, механических свойствах и методах испытания металлов. В свою очередь теория термической обработки — это база для изучения технологии термической обработки и таких разделов спецкурса металловедения, как «Легированные стали» и «Сплавы цветных металлов».
В последние десятилетия теория термической обработки развивалась очень быстрыми темпами. Для ее развития наиболее характерно все большее использование научных представлений и экспериментальных методов физики металлов, в особенности учения о дефектах кристаллической решетки, с целью более глубокого понимания природы, механизма и кинетики структурных изменений и закономерностей изменения свойств металлов н сплавов при тепловом воздействии. В результате весьма подробно изучены процессы термообработки давно используемых и новых металлических материалов. В одном учебнике уже невозможно изложить и общую теорию термической обработки, и особенности термообработки отдельных групп сплавов.
В книге дан анализ изменений структуры н свойств, вызываемых тепловым воздействием на металл. При этом большое внимание уделено таким структурным изменениям, как рекристаллизация, мартенситное превращение, распад твердого раствора, гомогенизация н др., которые особенно часто встречаются при термообработке разных групп металлических материалов.
Книга построена в соответствии с известной классификацией видов термообработки акад. А. А. Бочвара и рекомендациями по классификации и терминологии термической обработки Комиссии по стандарш-зации СЭВа.
6
Наиболее подробно представлены процессы отжига, закалки, отпуска, старения и термомеханической обработки. Менее детально рассмотрена химико-термическая обработка, так как она в неодинаковом объеме изучается студентами разных специализаций и для подробного изучения обычно выносится в спецкурс
При изложении общей теории термической обработки каждый вид термообработки проанализирован иа примере тех групп сплавов черных и цветных металлов, для которых соответствующие процессы исследованы подробнее и, как правило, имеют наибольшее практическое значение.
В приложении в конце книги приведен химический состав упомянутых в тексте сталей и сплавов цветных металлов.
В четвертое издание учебника внесены дополнения и исправления в соответствии с новой учебной программой по курсу, а также с уче-, том достижений в теории термической обработки за восемь лет, прошедших после предыдущего издания.
В учебник включена глава о новом виде термической обработки— закалке с плавлением поверхности (гл. X). Специалисты, знакомив* шпеся с рукописью учебника, высказали прямо противоположные точки зрения на целесообразность выделения закалки с плавлением поверхности как самостоятельного вида термической обработки. Автор убежден в правомочности и необходимости такого выделения, так как фазовые и структурные изменения при указанной обработке, определяющие ее главные преимущества, принципиально отличаются от фазовых и структурных изменений при других видах закалки.
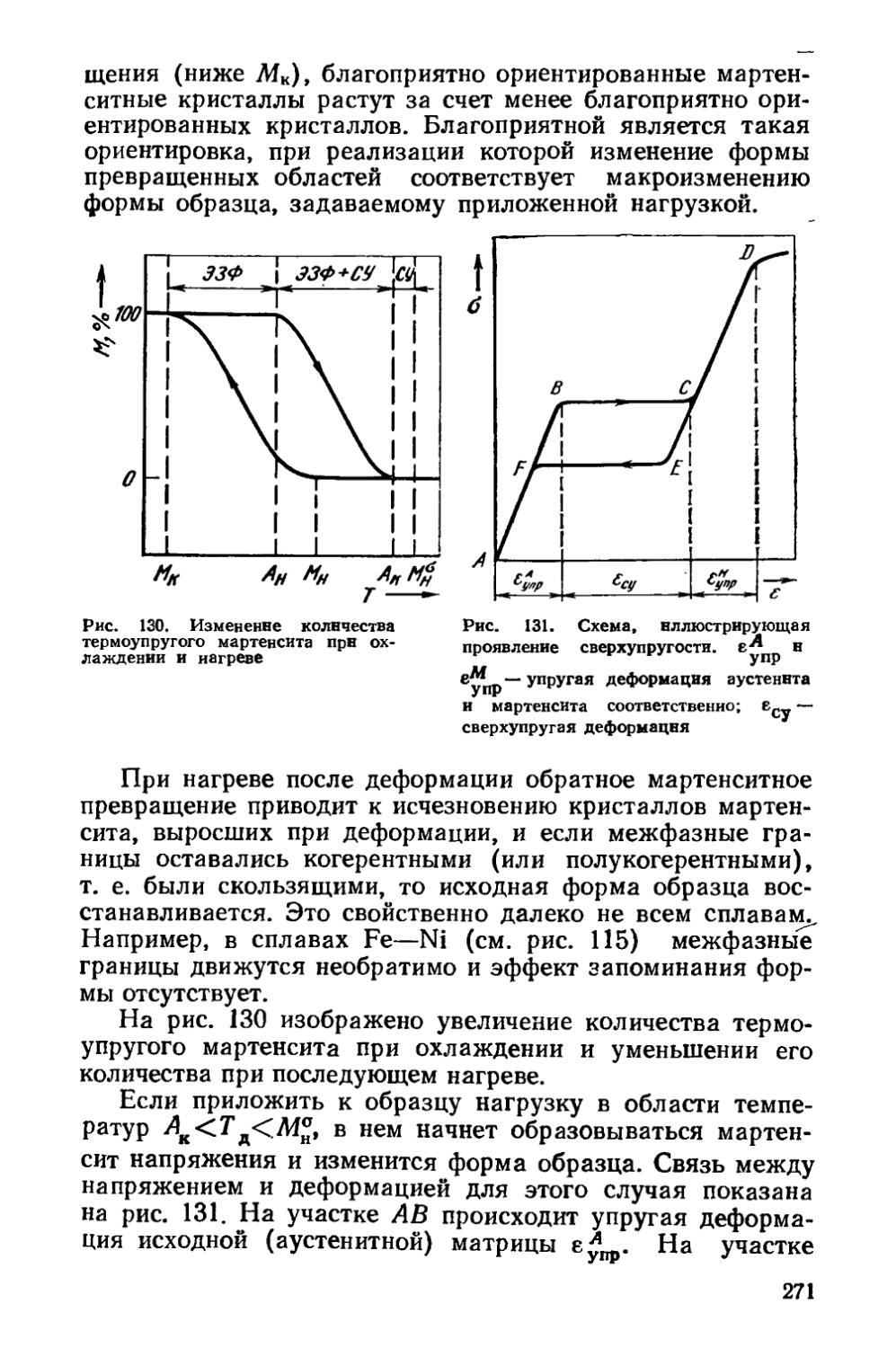
Новыми являются разделы, касающиеся абнормального эвтектоидного превращения в сталях (§ 24, п. 2), гетерогенизационного отжи-ia для улучшения деформируемости слитков (§ 28, п. 2), закалки сталей с температур межкритического интервала (§ 41, п. 5), поверхностной закалки на мартенсит с использованием лазерного нагрева (§ 42, и. 2), старения под напряжением (§ 46, п. 2 и § 49, п. 3), отпуска под напряжением (§ 52, п. 2), контролируемой прокатки (§ 62). Расширены сведения об образовании аустенита (§ 22), эффекте запоминания формы (§ 37, п. 2), кинетике старения (§ 46, п. 5) и о типах зависимости прочностных свойств от времени старения (§ 47, п. 3). Внесены также изменения и дополнения в другие разделы.
Автор благодарен коллективу кафедры термообработки и физики металлов Уральского политехнического института (зав. кафедрой проф. М. И. Гольдштейн) за подробный критический анализ рукописи. Автор признателен акад. В. Д. Садовскому, профессорам В. М. Глазову, С. С. Дьяченко, В. С. Золоторевскому, В. Ю. Новикову, А. Г. Рах-шгадту, Ю. А. Скакову, В. М. Счастливцеву, М. А. Штремелю и доц. А. И. Новикову за обсуждение отдельных разделов и замечания, которые помогли улучшить книгу.
7
ВВЕДЕНИЕ
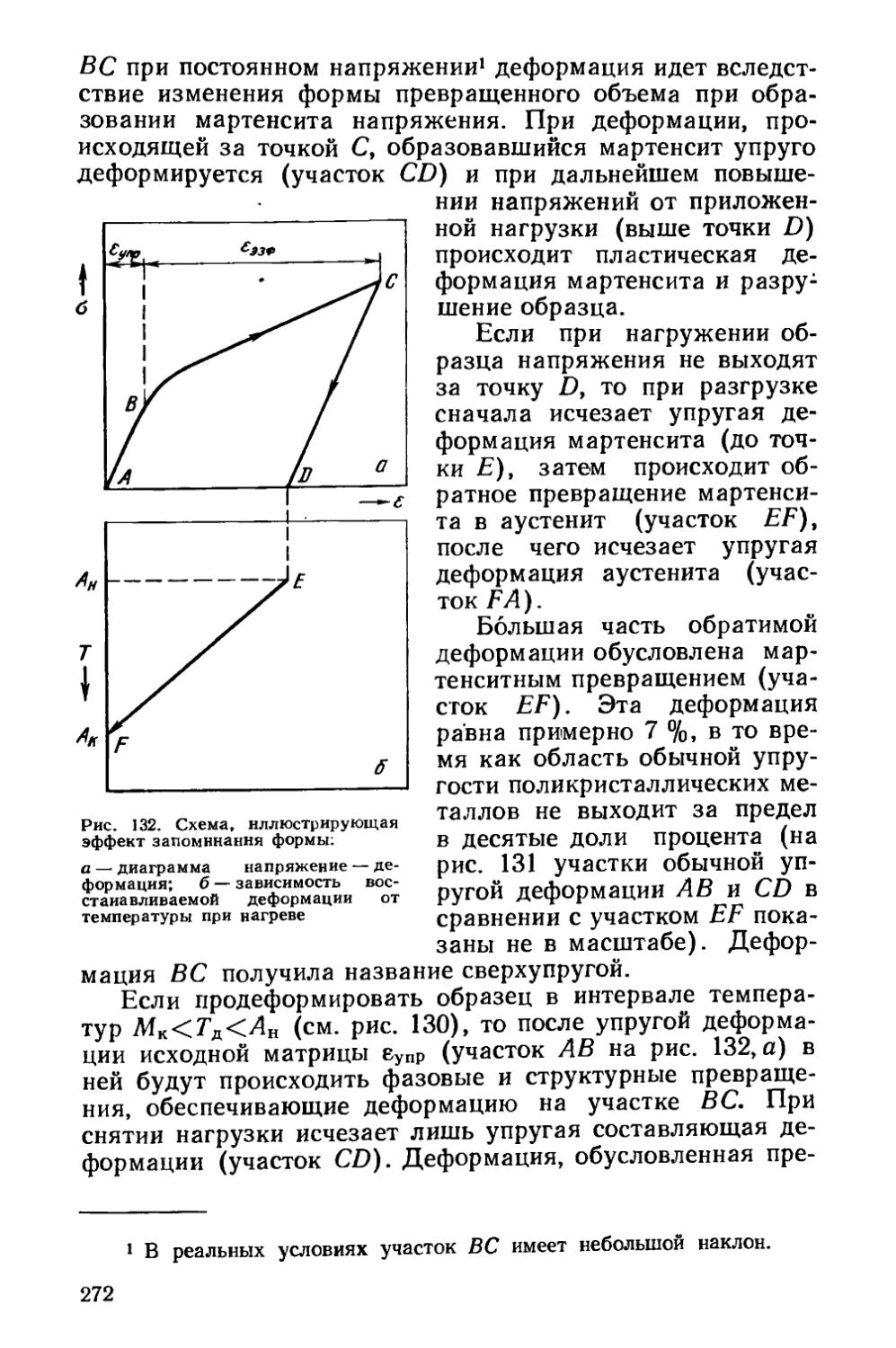
! Термической обработкой называют процесс обработки изделий из металлов и сплавов путем теплового воздействия с целью изменения их структуры и свойств в заданном на-_ правлении.
Это воздействие может сочетаться также с химическим, деформационным, магнитным и другими воздействиями.
Термическая обработка—самый распространенный в современной технике способ изменения свойств металлов и сплавов. На металлургических и машиностроительных за-"водах термическая обработка является одним из важнейших звеньев технологического процесса производства полуфабрикатов и деталей машин. Термообработку применяют как промежуточную операцию для улучшения технологических свойств (обрабатываемости давлением, резанием и др.) и как окончательную операцию для придания металлу или сплаву такого комплекса механических, физических и химических свойств, который обеспечивает необходимые эксплуатационные характеристики изделия. Чем ответственней конструкция, тем, как правило, больше в ней термически обработанных деталей.
Теория термической обработки является частью металловедения. Главное в металловедении — это учение о связи между строением и технически важными свойствами металлов и сплавов. При нагреве и охлаждении изменяется структура металлического материала, что обусловливает изменение механических, физических и химических свойств и влияет на его поведение при обработке и эксплуатации.
Теорию термической обработки составляет учение об изменениях строения и свойств металлов или сплавов при тепловом воздействии, не исчезающих после его прекращения.
По глубине и разнообразию структурных изменений, возникающих в результате термообработки, с ней не могут сравниться ни механические, ни какие-либо другие виды воздействия на металлы.
Краткий исторический очерк
Человек использует термообработку с древнейших времен. Анализ археологических находок позволяет сделать заключение о времени появления и характере операций термообработки. В переходный период от каменного века к бронзовому (в эпоху энеолита) появились первые металлические изделия, которые были получены ковкой с помощью каменного молота вначале самородных золота и меди, а затем и меди, выплавленной нз руды.
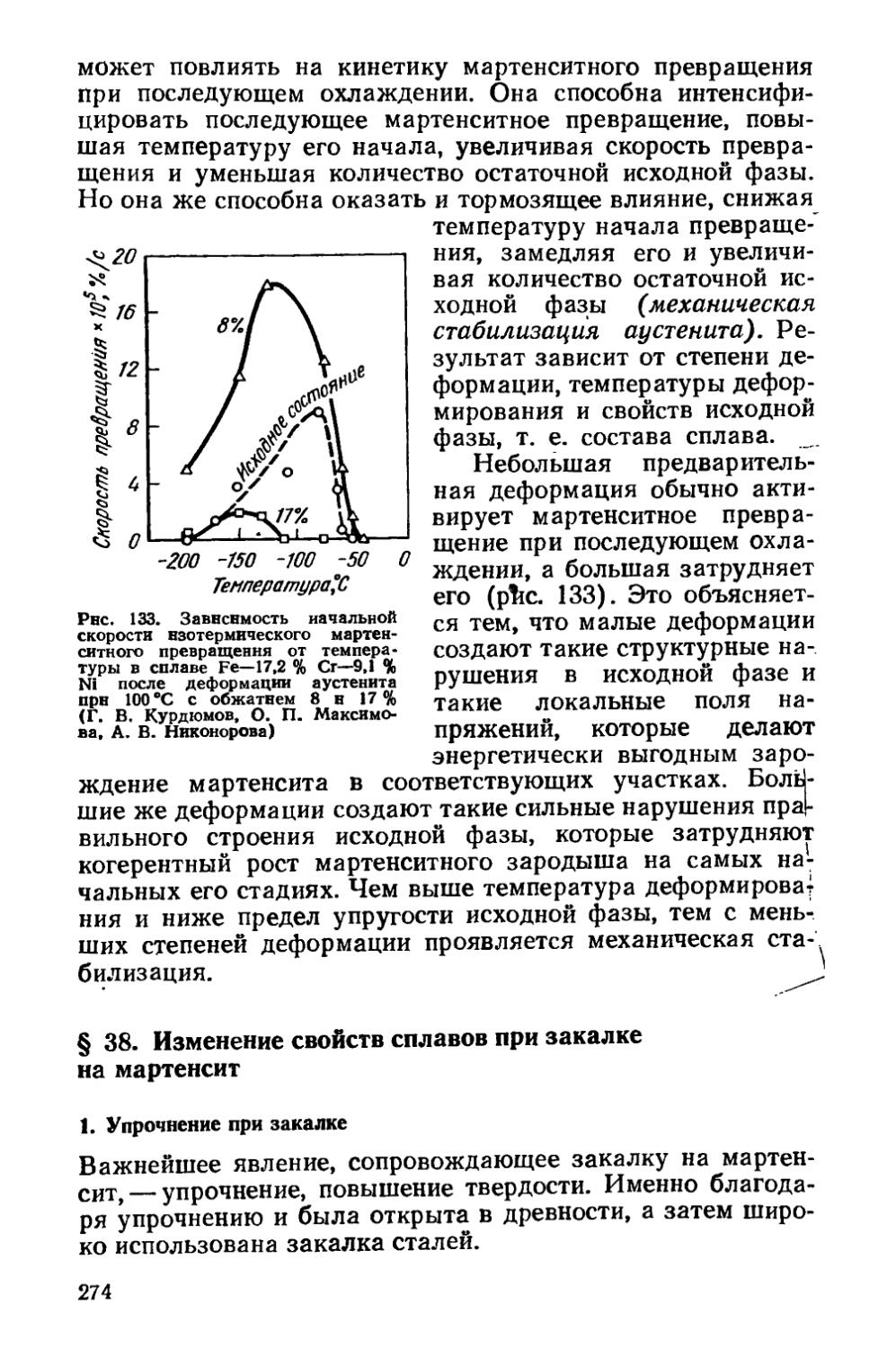
Применяя холодную ковку, первобытный человек столкнулся с яв-
8
лением наклепа, которое затрудняло изготовление изделий с тонкими лезвиями и острыми наконечниками, и для восстановления пластичности кузнец должен был нагревать холоднокованую медь в очаге. Наиболее ранние надежные свидетельства о применении такого рекристаллизационного отжига приходятся на конец V тысячелетия до н. э. Они получены при исследовании кованых изделий (например, ножей) из выплавленной меди, относящихся к южнотуркменской энеолитической культуре. Без промежуточного рекристаллизационного отжига для восстановления пластичности нельзя было обойтись н прн изготовлении методом ковки тонких медных, а позднее и бронзовых листов. Эти листы шли на выделку посуды, производившейся в значительных количествах во II тысячелетии до н. э. (в бронзовом веке).
Таким образом, рекристаллизационный отжиг по времени появления был первой операцией термической обработки металла и использовался уже начиная с V тысячелетия до н. э.
Закалка появилась значительно позднее отжига. Металлургическое производство железа началось с конца II тысячелетия до н. э. Железо получали тогда сыродутным способом непосредственно из железной руды. Из-за низкого содержания углерода оружие нз такого железа нельзя было упрочнить закалкой. В ранний период применения железа закалку проводили одновременно с цементацией. Нагревая заготовку для горячей ковки в древесноугольном горне, т. е. проводя науглероживание, и затем охлаждая ее в воде, кузнец столкнулся с резким улучшением качества оружия и орудий труда, сделанных из железа.
Открытие сыродутного способа производства железа с последующей его цементацией и закалкой было одним из важнейших достижений в истории человеческого , общества. Металлографическое изучение гальштатской кузнечной техники (Средняя Европа) показало, что цементация железных изделий (ножей, наконечников копий) и последующая их закалка в воде были известны уже в начале I тысячелетия до н. э. Однако эти виды упрочняющей термической обработки использовались в то время сравнительно редко.
В Древних Греции и Риме упрочняющая термообработка стали была хорошо известна, что подтверждается упоминаниями о закалке у античных авторов. В «Одиссее» Гомера (VIII—VII вв. до н. э.) в девятой песне есть такие строки: «Как погружает кузнец раскаленный топор иль секиру в воду холодную, и зашипит с клокотаньем железо — крепче железо бывает, в огне н воде закаляясь» (перевод П.вА. Шуйского). Аристотель (IV в. до н. э.) отмечал, что лучшую сталь получают нагревом железа в горне по нескольку раз. Такая обработка приводила к науглероживанию железа и обеспечивала после закалки высокую прочность и твердость изделий. Аристотель упоминал о закалке стали в масле. Плиний Старший (I в.) писал, что тонкие стальные изделия во избежание их коробления н растрескивания закаливают в масле.
Металлографический анализ европейских археологических находок показывает, что сталь (науглероженное железо) и закалка стальных изделий вошли во всеобщее употребление с V—IV вв. до н. э. Закалка медных сплавов была известна человеку также еще до и. э. Сравнительно недавние исследования литых этрусских зеркал из высокооловянной бронзы (Италия, V—IV вв. до н. э.) и сарматских зеркал (Поволжье, IV—II вв. до н. э.) показали, что эти изделия закаливали на мартенсит в воде скорее всего с целью улучшения зеркального блеска при полировке.
В средние века применяли самые разнообразные технологические операции: закалку в жидкости, закалку в воздушной струе, местную закалку лезвий, низкий, средний и высокий отпуск, цементацию, защиту
9
стали от обезуглероживания при иагреве, рекристаллизационный отжиг и др.
Микроструктурное изучение, рентгеновский анализ и измерение мик-ротвердостн многих сотен древнерусских находок из слоев X—XV в в. показали, что 9/ю исследованных стальных орудий труда и оружия находилось в термически обработанном состоянии, из них одна треть была закалена, а остальные закалены и отпущены. Цементацию в древесном угле или органическом веществе применяли к ножам, мечам, копьям, напильникам, резцам и другим инструментам. Искусство термообработки режущего и колющего оружия было высоко развито в средние века. Например, знаменитые клинки из булатной (дамасской) стали обладали прекрасными режущими и упругими свойствами благодаря сочетанию тонко разработанных способов плавки, ковки и термообработки.
Не зная сущности внутренних превращений в металле, средневековые мастера приписывали получение высоких свойств при термообработке проявлению сверхъестественных сил. Способы термообработки стали, особенно холодного оружия, детально описаны в средневековой литературе. Если из средневековых рецептов термообработки выбросить некоторые подробности и заклинания, то большая часть этих рецептов окажется оправданной с точки зрения современного термиста.
Термообработку использовали с древнейших времен как технологическую операцию, но развитие ее как науки стало возможным только с середины XIX столетия. До этого времени знания человека о термообработке представляли совокупность рецептов, выработанных на основе многовекового опыта. Эти рецепты, часто весьма ценные, передавали из поколения в поколение, секреты выполнения отдельных операции иногда терялись в веках и вновь познавались, но истинная природа процессов, происходящих в металле при термообработке, оставалась загадкой.
Развитие техники в XIX в. потребовало превращения термообработки из искусства в науку. В середине XIX столетия армия и флот стремились заменить бронзовые н чугунные пушки более прочными, а следовательно, и более мощными стальными орудиями. Начало широкого производства артиллерийских орудий из стали относится к 50-м годам прошлого века. В этот период проблема изготовления стальных орудийных стволов высокой и гарантированной прочности была чрезвычайно острой. Выдающиеся металлурги того времени, в том числе генерал П. М. Обухов, знали рецепты плавки и литья стали, но, несмотря на это, прн учебной стрельбе разрывы стальных орудий случались очень часто.
Много стальных крупповских орудий без видимых причин разорвалось в войну Пруссии с Австрией в 1866 г. Наступил кризис доверия к стали как к материалу для орудийных стволов, и начался возврат к бронзовым пушкам.
В 1866 г. на Обуховский сталелитейный завод в Петербурге был приглашен на должность техника молотового цеха Дмитрий Константинович Чернов (1839—1921 гг.). В 1868 г. в Русском техническом обществе Чернов делает знаменитый доклад «Критический обзор статей гг. Лаврова и Калакуцкого о стали и стальных орудиях и собственные Д. К. Чернова исследования по этому же предмету» *. В этом докладе он сообщает результаты работы по выяснению причин брака стальных поковок. Наблюдая под микроскопом шлифы, приготовленные из дул орудий, и изучая под лупой строение изломов в месте разрыва, Чернов пришел к выводу, что сталь тем прочнее, чем мельче ее структура. Тогда он «стал искать причину приобретения сталью мелкой структуры».
1 «Записки Русского технического общества», 1868, вып. 7, с. 399,
10
Сравнительные исследования стали после ковки при разных температурах показали, что «изменения в структуре стали нужно отнести к влиянию температуры, но не собственно механической обработки». После этого необходимо было для каждого сорта стали с определенным содержанием углерода найти температуры, при которых изменяется структура. Д. К. Чернов высказал гениальное предположение, что еле заметные поверхностные изменения, обнаруживаемые в темноте на охлаждающейся раскаленной поковке при двух температурах, связаны с глубокими внутренними изменениями структуры. Эти температуры Чернов определил на глаз и обозначил точками а и Ь. «Сталь, как бы тверда она ни была, будучи нагрета ниже точки а, не принимает закалки, как бы быстро ее ии охлаждали». Чтобы получить мелкозернистый излом, необходимо нагреть сталь немного выше точки Ь.
Таким образом, Д. К- Чернов в 1868 г. открыл внутренние структурные превращения в стали и связал с ними тепловой режим ковки и технологию термообработки. Тем самым великий русский металлург заложил научные основы термической обработки.
Основополагающий доклад Д. К. Чернова был переведен на иностранные языки, и предложенные им правила обработки стали вошлн в практику заводов разных стран. Работы Чернова в области металловедения и термической обработки получили мировое признание. Известный американский металловед А. Совер, обращаясь к Чернову, писал: «Вы создали теорию термической обработки стали рукою мастера, и Ваши ученики добавили сравнительно немногое к Вашим основным положениям». Яркую характеристику деятельности Чернова дал в некрологе французский металловед профессор А. Портевен: «Чернов был провозвестником и главой новой школы; его первые труды послужили фундаментом для последующего удивительного прогресса в области металлургии стали, для которой вторжение науки оказалось поистине революционным». «Столь прекрасная жизнь, получившая мировую оценку, делает великую честь России».
Выдающийся последователь Д. К. Чернова французский инженер Флорис Осмонд (1849—1912 гг.) применил в 1886 г. термопару Ле-Ша-телье для определения критических точек стали при термическом анализе. Работы Осмонда, подтвердившего и развившего выводы Чернова, привлекли внимание многих металлургов и химиков к проблеме структурных превращений в металлах и послужили дополнительным толчком для широких экспериментальных исследований в этой области.
В истории металловедения конец XIX и начало XX вв. характеризуются широким приложением термодинамического учения о гетерогенных равновесиях к металлическим системам. В разных странах были начаты систематические работы по построению диаграмм состояния. Эти диаграммы показывают, какие фазовые превращения возможны в сплавах, и, следовательно, дают исходные данные для анализа важнейших видов термической обработки.
Первые крупные исследования в области термообработки цветных сплавов были выполнены в начале XX в. В 1900 г. А. А. Байков (1870— 1946 гг.) на сплавах меди с сурьмой доказал, что способность к закалке присуща не только сталям, как это ранее считали, но и цветным сплавам. В 1903 г. в Германии был взят патент на «способ облагораживания алюминиевых сплавов нагреванием и закалкой»; было показано, что временное сопротивление литых сплавов алюминия с медью в результате закалкн возрастает в 1,5 раза.
В 1906 г. немецкий инженер А. Внльм (1869—1937 гг.) на изобретенном им дуралюмине открыл старение после закалки — один из основных способов упрочнения сплавов. В 1919 г. американский металловед
11
П. Мерика (1889—1957 гг.) вскрыл природу старения дуралюмипов, связав упрочнение при старении с образованием дисперсных выделений в пересыщенном твердом растворе. Это было одним из наиболее выдающихся достижений в теории термической обработки: по диаграммам состояния с переменной растворимостью компонентов в твердом состоянии стало возможным предсказывать области составов сплавов, способных к дисперсионному твердению.
Начиная с 20-х годов текущего столетия для развития теории термообработки характерно детальное изучение природы, механизма и кинетики структурных превращений в твердом состоянии с помощью разнообразных физических методов исследования и прежде всего рентгеновского анализа. В течение двух-трех десятилетий были накоплены обширные сведения о закономерностях изменений структуры и свойств металлов и сплавов при тепловом воздействии. С конца 50-х годов для выявления изменений субструктуры при термообработке все шире стали применять метод просвечивающей (дифракционной) электронной микроскопии.
На современном этапе для теории термообработки характерно широкое использование учения о дефектах кристаллической решетки металлов, так как эти дефекты оказывают сильное, а часто и решающее влияние на механизм и закономерности структурных изменений.
Одновременно с развитием теоретических представлений совершенствовались старые н разрабатывались новые способы термообработки, например, термомеханическая обработка, отпуск и старение под напряжением, закалка с плавлением поверхности и др.
Итогом многочисленных исследований явилась стройная теория термической обработки, которая позволяет научно обоснованно разрабатывать технологические процессы и получать сплавы с заданными свойствами.
Классификация видов термической обработки
Любой процесс термической обработки можно описать графиком, показывающим изменение температуры во времени. По такому графику можно определить температуру нагрева, время нагрева и охлаждения, средние и истинные скорости нагрева и охлаждения, время выдержки при температуре нагрева и общую продолжительность производственного цикла. Но по форме этого графика ничего нельзя сказать о том, с каким видом термообработки мы ! имеем дело. Вид термообработки определяется не характе-I ром изменения температуры во времени, а типом фазовых ; и структурных изменений в металле. Основываясь на по-~ следнем признаке, А. А. Бочвар разработал классификацию, охватывающую многочисленные разновидности термической обработки черных и цветных металлов и сплавов.
На основе классификации А. А. Бочвара Комиссией по стандартизации Совета Экономической Взаимопомощи Г были разработаны классификация видов и разновидностей I термической обработки сталей и цветных металлов и спла-{ вов, а также соответствующая терминология. На рис. 1 ^”12
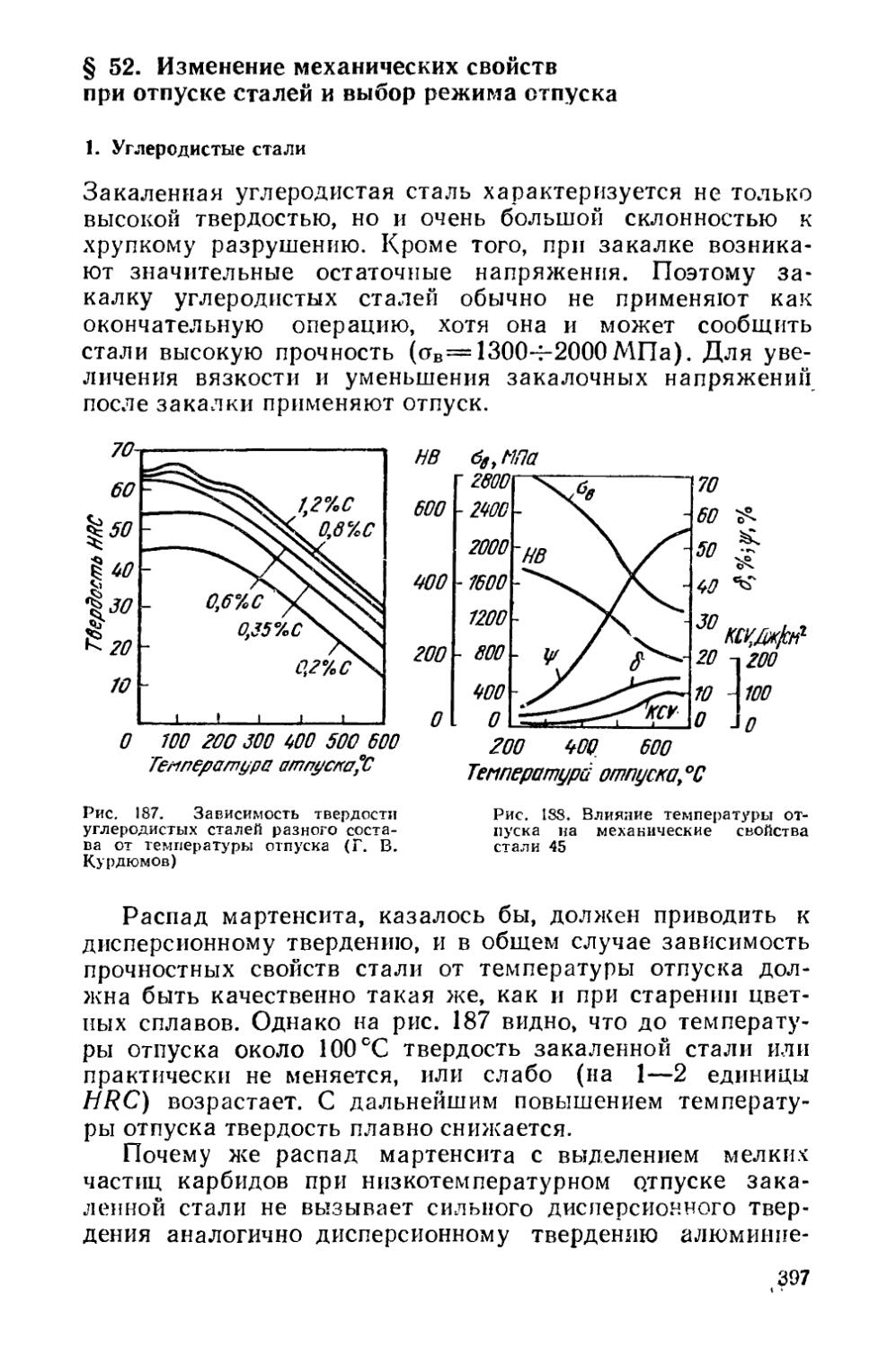
приведена схема классификации основных видов термической обработки металлов и сплавов 1.
Термическая обработка подразделяется на собственно термическую, термомеханическую и химико-термическую. Собственно термическая обработка заключается только в термическом воздействии на металл или сплав, термомеха-нпческая — в сочетании термического воздействия и пластической деформации, химико-термическая — в сочетании термического и химического воздействия.
Собственно термическая обработка включает следующие основные виды: отжиг 1-го рода, отжиг 2-го рода, закалку с полиморфным превращением, закалку без полиморфного превращения, закалку с плавлением поверхности, отпуск и старение. Эти виды термической обработки относятся и к сталям, и к цветным металлам и сплавам. Каждый из видов термообработки подразделяется на разновидности, специфические для сплавов на разных основах.
С отдельными видами термообработки приходится сталкиваться как с побочными процессами при горячей обработке давлением, литье, сварке и других технологических операциях. Например, частичная или полная закалка встречается при ускоренном охлаждении отливок после их затвердевания. При шлифовании деталей из-за разогрева
1 В дополнение к классификации Комиссии по стандартизации СЭВа автор включил в эту схему новый вид термической обработки, которому дано название «закалка с плавлением поверхности» (см. гл. X).
13-
поверхности может произойти отпуск. При сварке в зоне термического влияния сварного шва можно наблюдать рекристаллизационный отжиг и т.п. Побочные процессы термообработки бывают полезными, а могут вызывать и нежелательные изменения структуры и свойств изделий.
Производственные названия отдельных процессов термообработки складывались исторически и основывались не на характере внутренних превращений в металле или сплаве, а на чисто внешних признаках. Поэтому один и тот же термин иногда используют для обозначения разновидностей термообработки, совершенно различных по своей физической сущности. Например, нагрев с переходом за критическую точку, выдержку и охлаждение на воздухе обычно называют нормализацией. При такой обработке в углеродистой стали происходят процессы, которые относятся к отжигу 2-го рода, в высоколегированных сталях может образоваться мартенсит, т. е. происходит закалка с полиморфным превращением, а некоторые цветные сплавы подвергаются закалке без полиморфного превращения.
Примеров подобного рода можно привести множество. В связи с этим при употреблении некоторых производственных названий термической обработки иногда трудно понять, какова физическая сущность процессов, о которых идет речь. В таких случаях вместо устоявшихся производственных терминов или параллельно с ними необходимо использовать терминологию научной классификации раз-t новидностей термической обработки.
Раздел
первый ОТЖИГ ПЕРВОГО РОДА
Отжиг 1-го рода частично или полностью устраняет отклонения от равновесного состояния, возникшие при предыдущей обработке, причем его проведение не обусловлено фазовыми превращениями. Обработкой, предшествующей отжигу 1-го рода, могут быть литье, обработка давлением, сварка, термическая обработка и другие технологические процессы.
В зависимости от того, какие отклонения от равновесного состояния устраняются, различают следующие разновидности отжига 1-го рода: гомогенизационный, дорекри-сталлизационный, рекристаллизационный и уменьшающий напряжения отжиг. Процессы, устраняющие отклонения от равновесного состояния, идут самопроизвольно, и нагрев при отжиге 1-го рода проводят лишь для ускорения этих процессов. Основные параметры отжига 1-го рода — температуре нагрева и время выдержки. Скорости нагрева и охлаждения имеют подчиненное значение.
Глава 1
ГОМОГЕНИЗАЦИОННЫЙ ОТЖИГ
Гомогенизационный отжиг — это термическая обработка, при которой главным процессом является устранение последствий дендритной ликвации.
§ 1. Изменение структуры сплавов при гомогенизационном отжиге
В результате дендритной ликвации возникает химическая мпкронеоднородность внутри кристаллов твердого раствора — основы сплава и могут появиться неравновесные избыточные фазы. Например, в сплаве Х\ на рис. 2 при равновесной кристаллизации состав во всем объеме а-раствора изменяется по кривой bs, и кристаллизация заканчивается в точке равновесного солидуса s без образования эвтектики. При неравновесной кристаллизации состав позднее образующихся слоев а-раствора, обогащенных компонентом В, и состав ранее образовавшихся слоев не успевают выровняться, средний состав первичных кристаллов в сплаве X) изменяется по линии Ьс и кристаллизация его заканчивается при эвтектической температуре (точка т).
15
По окончании кристаллизации сплава в дендритныл ячейках — сечениях ветвей дендритов — концентрация ле'
тирующего элемента минимальна в середине, а в поверхностном слое соответствует точке предельной растворимости при эвтектической температуре (точка а на рис. 2). В этом сплаве, который в равновесных условиях должен быть однофазным, в результате дендритной ликвации кристаллизуется неравновесная эвтектика, количество которой равно ст/се. Очень часто неравновесная эвтектика вырождается в 0-фазу эвтектического происхождения, расположенную по границам дендритных ячеек (a-фаза состава
точки а из эвтектики кристаллизуется, как на подкладке,
на первичных а-кристаллах
Рис. 2. Неравновесный солидус A'ka в системе эвтектического типа и кривая изменения среднего состава твердого раствора Ьс при дендритной ликвации в сплаве Xi (схема)
и структурно не выявляется).
Точка k на диаграмме состояния (см. рис. 2)—это концентрационная граница появления неравновесной эвтектики в данных условиях. В диапазоне скоростей охлаждения при кристаллизации, который реализуется в производственных условиях, выравнивающая диффузия в твердом растворе сильно подавлена, и неравновесная избыточная фаза кристаллизуется при весьма малых концентрани-
качестве примера ниже
В
ях легирующего элемента.
приведены следующие дан
ные о концентрации второго компонента в точке предельной растворимости при эвтектической или перитектической температуре (а) и на концентрационной границе (k) появления неравновесной фазы в условиях литья в кокиль:
Система ..... f Al—Си Al—Mg Mg—Al Си—Sn
fl, % .......... 5,7 15,4 12,7 13,5
k, % t ..... . . 0,1 0,5 0,1 4,0
В результате отклонения от равновесия в процессе кристаллизации литой сплав имеет следующие основные недостатки:
1. Пластичность сплава обычно снижается, если в результате дендритной ликвации появляется избыточная хрупкая фаза. Особенно сильно снижается пластичность при образовании по границам дендритных ячеек сплошных
16
прослоек из грубых частиц хрупких соединений (интерме-таллидов, карбидов и др.).
2. Центральные участки дендритных ячеек и их границы, имеющие разный химический состав, образуют микро-гальванические пары. Поэтому внутрикристаллитная ликвация твердого раствора снижает стойкость против электрохимической коррозии. Появление неравновесной избыточной фазы в твердом растворе обычно также снижает стойкость против коррозии.
3. При обработке давлением, например прокатке и прессовании, микроучастки, имеющие разный химический состав, вытягиваются и может возникнуть строчечная структура. Такая структура обусловливает анизотропию свойств в изделии и повышенную склонность к межкристаллитному, а также шиферному излому. В поперечном направлении может наблюдаться сильное снижение относительного удлинения, сужения и ударной вязкости.
4. Дендритная ликвация понижает температуру солидуса сплава, что, как правило, нежелательно. Например, при быстром нагреве под закалку или обработку давлением изделие может частично оплавиться. Оплавляются участки, в которых находится неравновесно образовавшаяся эвтектика.
5. Структура и свойства литого сплава нестабильны во времени. В изделии, работающем при повышенных температурах, могут самопроизвольно постепенно выравниваться состав твердого раствора и рассасываться избыточные фазы. Эти процессы вызывают ускорение ползучести, а также постепенное изменение свойств, которое может выйти за допустимые пределы.
Для устранения недостатков литой структуры слиток или фасонную отливку подвергают отжигу-гомогенизации.
1. Основные структурные изменения
В однофазных сплавах, например в медноникелевом сплаве системы с непрерывным рядом твердых растворов, главный процесс при гомогенизационном отжиге — выравнивание состава зерен твердого раствора, т. е. устранение внутри-кристаллитной ликвации (рис. 3). В сплавах, содержащих неравновесную избыточную фазу, например в сплаве на_ рис. 2, при гомогенизационном отжиге протекают два основных процесса: выравнивание концентрации внутри зерен твердого раствора и растворение^неравновесных избы-, 2 И. И. Новиков П 4 ’ 17
точных фаз. В основе этих процессов лежит диффузия, и поэтому гомогенизационный отжиг называют также диффузионным.
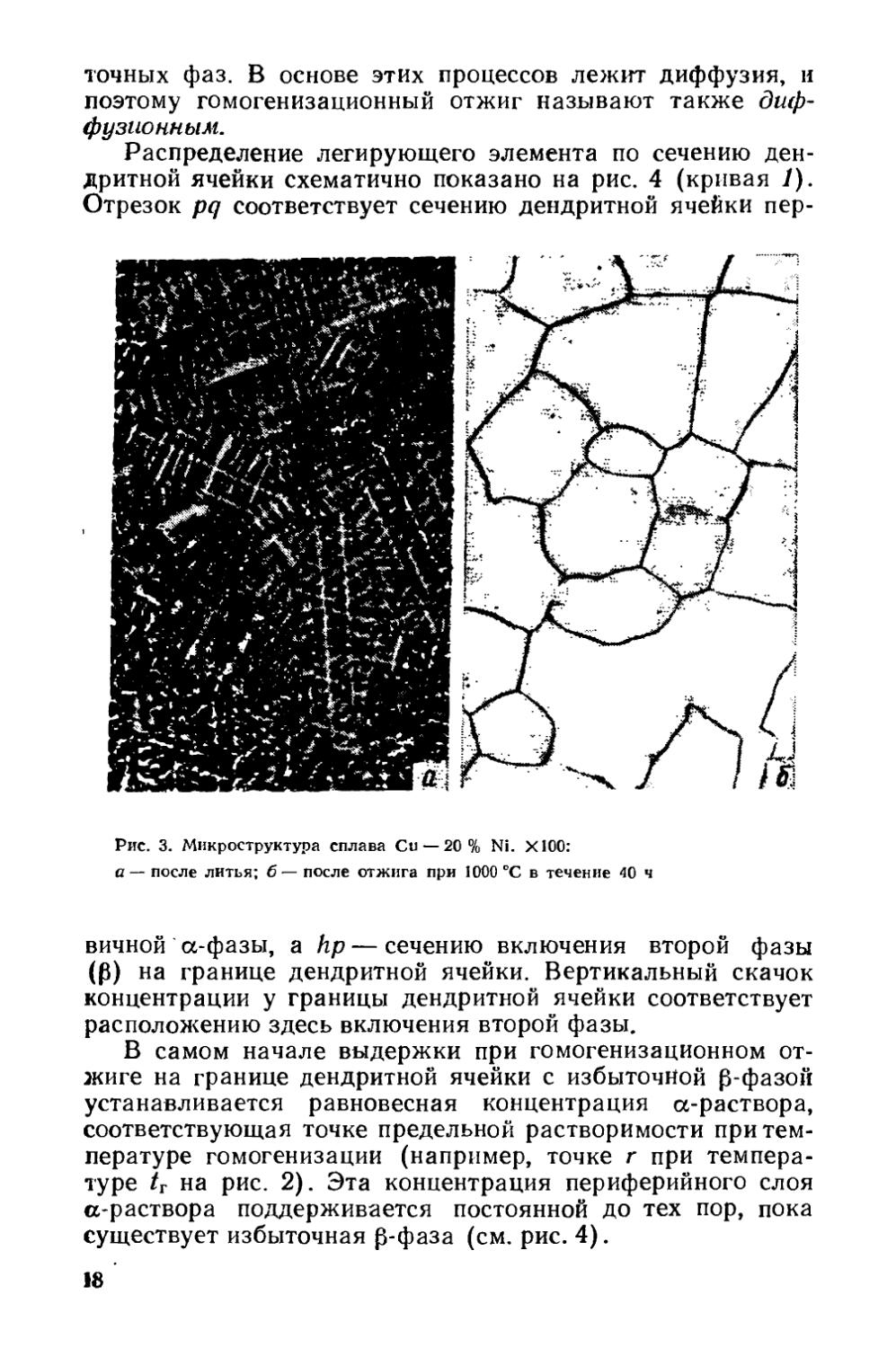
Распределение легирующего элемента по сечению дендритной ячейки схематично показано на рис. 4 (кривая 1). Отрезок pq соответствует сечению дендритной ячейки пер-
Рис. 3. Микроструктура сплава Си — 20 % Ni. Х100:
а— после литья; б— после отжига при 1000 °C в течение 40 ч
вичной a-фазы, a hp — сечению включения второй фазы (Р) на границе дендритной ячейки. Вертикальный скачок концентрации у границы дендритной ячейки соответствует расположению здесь включения второй фазы.
В самом начале выдержки при гомогенизационном отжиге на границе дендритной ячейки с избыточной р-фазой устанавливается равновесная концентрация а-раствора, соответствующая точке предельной растворимости при температуре гомогенизации (например, точке г при температуре 1Г на рис. 2). Эта концентрация периферийного слоя а-раствора поддерживается постоянной до тех пор, пока существует избыточная p-фаза (см. рис. 4).
18
Рис. 4; Схема распределения легирующего элемента по сечению дендритной ячейки а-раствора с P-фазой на ее границах в начальный (/), промежуточный (2) и конечный (3) моменты гомогенизации (точку г см. на рис. 2; состав сплава находится правее точки г
Выравнивание состава внутри дендритных ячеек является контролирующим звеном гомогенизации, определяющим скорость и время полного растворения избыточной фазы. Если условно отделить один от другого процессы выравнивания состава внутри a-фазы и растворения в ней избыточной 0-фазы, то можно схематично нарисовать такую картину. Выравнивание состава внутри а-раствора приводит к уменьшению его концентрации на границе с 0-фазой до значений ниже точки г на рис. 2 и 4, и периферийный слой дендритной ячейки становится ненасыщенным по отношению к 0-фазе. Поэтому 0-фаза растворяется, поднимая концентрацию в периферийном слое а-раствора до равновесной, и т.д. Постепенно граница а/0 перемещается в сторону растворяющейся 0-фазы, а состав a-фазы по сечению дендритной ячейки
выравнивается (см. пунктирные линии на рис. 4, соответствующие разным моментам гомогенизации).
Если избыточная 0-фаза должна полностью раствориться (в соответствии с диаграммой состояния), то после ее исчезновения через некоторое время завершается и выравнивание концентрации внутри а-раствора. Если же состав сплава таков, что он и в равновесных условиях не должен быть однофазным, то при гомогенизации растворяется только неравновесный избыток второй фазы (или нескольких фаз) и после отжига остается некоторое равновесное их количество (рис. 4, 5). Опыты с алюминиевыми сплавами показали, что время полного устранения внутрикристал-литной ликвации ненамного превышает время полного растворения (тР) неравновесного избытка фаз. Учитывая, что время тр можно легко определить, исследуя структуру с похмощью светового микроскопа, а для изучения кинетики выравнивания концентрации внутри твердого раствора необходимо использовать более дорогой и сложный микро
2* 19
рентгеноспектральный анализ, вполне допустимо в большинстве задач время окончания гомогенизации оценивать по времени окончания растворения неравновесного избытка фаз.
По первому закону Фика поток диффундирующего вещества /, т. е. количество вещества, проходящего в единицу времени через единицу площади сечения, пропорционален градиенту концентрации дс/дх вдоль направления х, перпендикулярного этому сечению:
I^ — Ddddx. (1)
Коэффициент диффузии D зависит от природы сплава, типа и состава твердого раствора, размера зерна и особенно резко от температуры:
£> = (2)
где R — газовая постоянная; Т — температура; Q — энергия активации диффузии; А — константа, практически не зависящая от температуры.
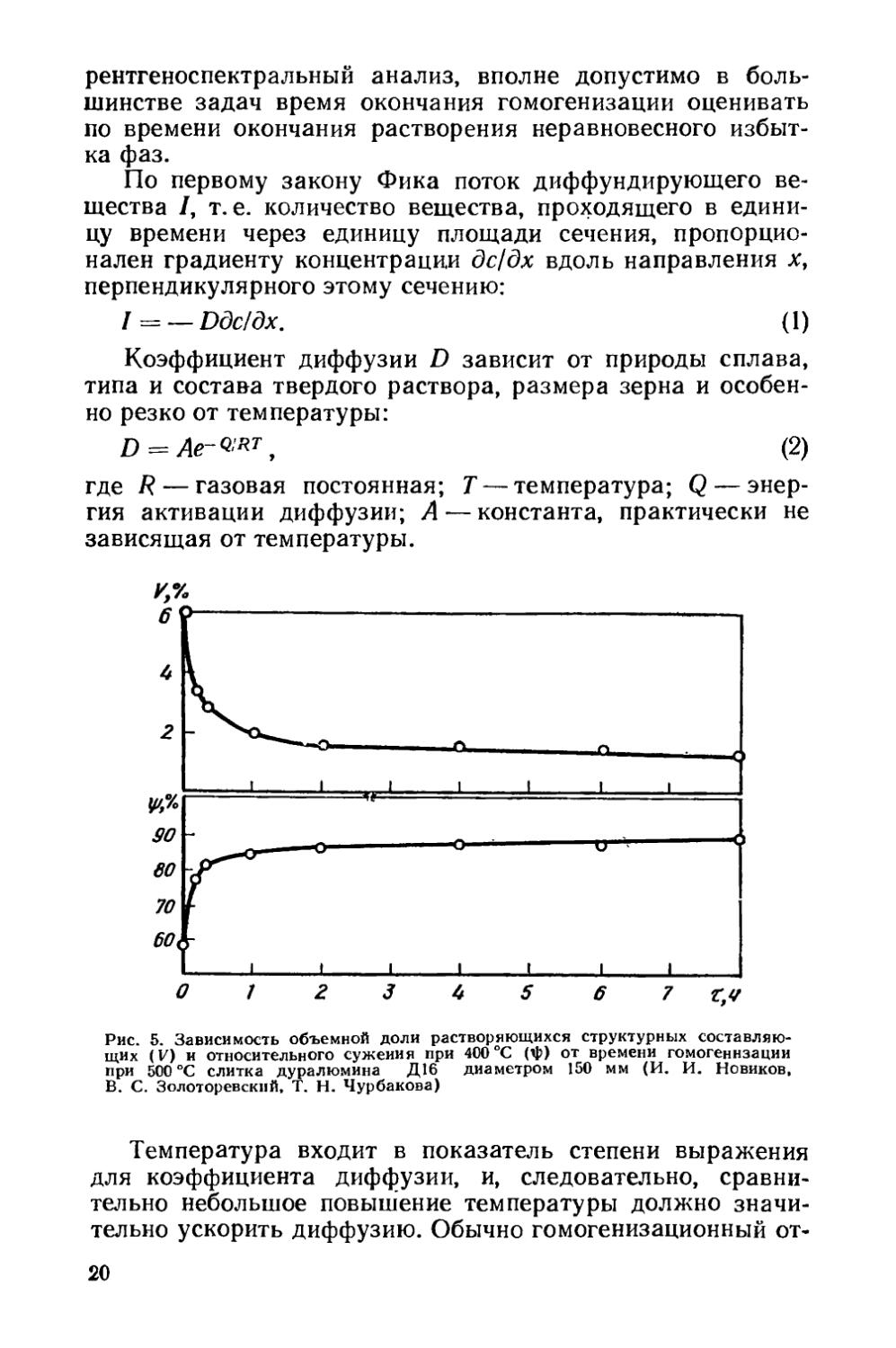
Рис. 5. Зависимость объемной доли растворяющихся структурных составляющих (V) и относительного сужения при 400 °C (ф) от времени гомогенизации при 500 °C слитка дуралюмина Д16 диаметром 150 мм (И. И. Новиков, В. С. Золоторевский, Т. Н. Чурбакова)
Температура входит в показатель степени выражения для коэффициента диффузии, и, следовательно, сравнительно небольшое повышение температуры должно значительно ускорить диффузию. Обычно гомогенизационный от-
20
жпг проводят при температурах выше 0,90—0,95 Тн.п (по абсолютной шкале), но ниже точки солидуса сплава. Иногда температура гомогенизации находится в интервале 0,8— 0,9 7ПЛ.
Энергия активации диффузии Q обеспечивает переход атомов из одного положения в решетке в другое. Необходимый для такого перехода избыток энергии приобретается атомом от его соседей благодаря тому, что атомы непрерывно обмениваются кинетической энергией. Так как величина энергии активации входит в показатель степени, то она очень сильно влияет на величину коэффициента диффузии.
Энергия активации диффузии элементов, растворенных по способу внедрения, меньше, чем у элементов, растворенных по способу замещения. Поэтому последние диффундируют медленнее. Например, легирующие элементы, растворенные в аустените по способу замещения, обладают значительно меньшей диффузионной подвижностью, чем углерод, растворенный в железе по способу внедрения. Энергия активации диффузии углерода в аустените среднеуглеродистой стали равна приближенно 130 кДж/моль, а энергия активации диффузии в аустените важнейших легирующих элементов обычно превышает 250 кДж/моль. Скорость диффузии углерода в аустените на несколько порядков больше скорости диффузии важнейших легирующих элементов.
Слитки из углеродистых сталей обычно не подвергают гомогенизационному отжигу, так как в них при нагреве под горячую обработку давлением из-за быстрой диффузии углерода в аустените дендритная ликвация успевает исчезнуть. Легированные стали для устранения внутрикристал-литной ликвации и растворения неравновесного избытка карбидов эвтектического происхождения приходится подвергать специальному нагреву — гомогенизационному отжигу при 1050—1250 °C.
Из закона Фика следует, что диффузионный поток должен уменьшаться по мере протекания гомогенизации, так как градиент концентрации дс)дх при гомогенизации уменьшается. Гомогенизация идет как бы с самоторможением. Отсюда следует важный практический вывод: наиболее интенсивно гомогенизация протекает в начальный период отжига. Этот вывод иллюстрируется графиком, приведенным на рис. 5, на котором уменьшение суммарного количества избыточных фаз при отжиге слитка дуралюми-
21
на Д16 в первые 30 мин выдержки значительно больше, чем в последующие 7 ч.
Чрезмерно большие выдержки при гомогенизации нецелесообразны, так как они малоэффективны и приводят к излишнему расходу электроэнергии (топлива) и к неоправданному уменьшению производительности. Повышение температуры отжига действует несравненно эффективнее, чем увеличение времени выдержки.
Время выдержки при гомогенизационном отжиге обычно колеблется в пределах от нескольких до десятков часов (не считая времени прогрева). Повышая температуру, можно сократить время выдержки при отжиге.
Время полной гомогенизации зависит не только от температуры отжига и природы сплава, определяющих диффузионную подвижность компонентов в твердом растворе. На время гомогенизации сильно влияет исходная мийро-структура^литого сплава. Скорость гомогенизации зависит от толщины частиц избыточной фазы и размера дендритной ячейки основной фазы.
Зависимость времени окончания растворения избыточной фазы тр от средней толщины частиц растворяющейся фазы т подчиняется уравнению
тр = ать , (3)
где а и b — константы для данного сплава и данной температуры гомогенизационного отжига. Показатель степени в этой формуле для алюминиевых сплавов находится в пределах 1,2—2,5. Естественно, что с уменьшением толщины включений время их растворения сокращается. В диапазоне практически встречающхся значений т связь тр с т близка к линейной.
С уменьшением размера дендритной ячейки возрастает средний градиент концентрации по ее сечению (при постоянной разности концентраций раствора на границе и в центре ячейки) и в соответствии с законом Фика должна ускоряться диффузия. Размер дендритной ячейки влияет па скорость гомогенизации и через толщину избыточной фазы, так как с уменьшением размера дендритной ячейки измельчается и избыточная фаза.
Можно использовать два пути ускорения гомогенизации, регулируя микроструктуру. Первый путь — увеличение скорости кристаллизации сплава. Чем выше скорость кристаллизации, тем меньше размер дендритных ячеек и тоньше частицы избыточных фаз, кристаллизующихся по их границам. Поэтому слитки и фасонные отливки, затвердевавшие
22
при большой скорости охлаждения, быстрее и полнее гомогенизируются, так как они отличаются более тонким строением. Например, слитки непрерывного литья малого сечения гомогенизируются быстрее, чем слитки большого сечения; кокильные отливки быстрее гомогенизируются, чем детали, отлитые в земляные формы.
Другой путь ускорения гомогенизации — измельчение структуры слитка обработкой давлением. Так, вместо длительного гомогенизационного отжига слитков легированной стали увеличивают продолжительность нагрева деформированной заготовки перед последним переделом.
Разные избыточные фазы могут характеризоваться существенно разными скоростями растворения. Так, в дур-алюмине Д16 по убыванию скорости растворения при гомо-генизационном отжиге избыточные фазы располагаются в следующий ряд: S(Al2CuMg), 6(CuAI2), Mg2Si.
Если элементы, входящие в состав соединений, имеют разную диффузионную подвижность в окружающем эти соединения твердом растворе, то в период растворения одна избыточная фаза может превращаться в другую. Например, в дуралюмине Д16 растворяющаяся S-фаза (Al2CuMg) из-за более быстрого по сравнению с медью ухода из нее магния в алюминиевый раствор превращается в 6-фазу (СиА12), которая затем сама растворяется.
Восходящая диффузия при отжиге
При гомогенизационном отжиге многокомпонентных сплавов диффузионное перераспределение отдельных компонентов в твердом растворе может быть значительно более сложным, чем это следует из классической теории диффузии, не учитывающей взаимодействия компонентов в растворе. Одно из проявлений этого взаимодействия — восходящая диффузия, т. е. поток компонента в сторону увеличения градиента концентрации.
Восходящая диффузия и другие факты сложного перераспределения компонентов становятся понятными, если воспользоваться представлениями термодинамики необратимых процессов, согласно которой в общем случае движущей силой диффузионного переноса является не градиент концентрации, а градиент химического потенциала р, которому пропорционален диффузионный поток:
I = — Мдр/дх. (4)
Закон же Фика (1) применим только к идеальному раствору, в котором диффузионные потоки разных компонентов не взаимодействуют.
23
Химический потенциал ьтого компонента в реальном растворе зависит от его термодинамической активности ар
= М£т + RT\nan
(5)
где pJT —химический потенциал в некотором стандартном состоянии, зависящий только от температуры.
Активность в реальном растворе определяет химический потенциал компонента так же, как определяет его концентрация в идеальном растворе. Если в соседних участках раствора активность f-того компонента одинакова, то он не будет перераспределяться. Градиент активности является движущей силой диффузии.
Следующий классический опыт Даркена демонстрирует роль градиента активности в появлении восходящей диффузии. Две плотно сложенные пластинки сплавов Fe — 0,49% С —3,8% Si и Fe —0,57% С —6,45% Мп отжигали в однофазном состоянии. После отжига на разном рас-
стоянии от границы раздела пластинок измеряли концентрацию углерода (рис. 6). Вместо «нормального» перехода
углерода от второго сплава, где его концентрация выше, к первому произошел обратный переход — от сплава с 0,49 % С к сплаву с 0,57 % С. Объясняется этот результат
Расстояние от границы, гт-
Рис. 6. Распределение углерода в составном образце после отжига при 1050 °C в течение 13 сут (Дар-кеи). Исходный левый образец содержал 0,49 % С и 3,8 % Si, правый 0,57 % С и 6,45 % Мп
тем, что, во-первых, кремний повышает, а марганец понижает активность углерода в аустените, и, во-вторых, тем, что диффузионная подвижность кремния и марганца па много порядков меньше, чем у углерода. Во время отжига марганец и кремний не успели перераспределиться, и углерод из раствора с кремнием, где его активность была выше, перехо-
дил в раствор с марганцем до тех пор, пока активности углерода по обе стороны от границы образцов не выровнялись (с ростом концентрации углерода в аустените его активность повышается). Если время отжига увеличить настолько, чтобы концентрация кремния и марганца в обоих образцах выравнялась, то и концентрация углерода также будет одинаковой. Таким образом, в рассмотренном случае при отжиге сначала идет восходящая диффузия углерода,
а затем выравнивающая.
24
Аналогичную картину можно наблюдать прн отжиге стали, в которой имеются микрообъемы с разной концентрацией кремния и других элементов из-за их внутрикри-сталлитной ликвации. При отжиге вначале быстрая восходящая диффузия углерода понижает концентрацию в микроучастках, обогащенных кремнием (здесь выше активность углерода), и только после длительного отжига, когда исчезнет градиент концентрации кремния, выравнивается и концентрация углерода по объему зерен аустенита. Если гомогенизация не была завершена, то в высококремнистых и одновременно малоуглеродистых участках аустенита при охлаждении формируется ферритная структура, а в малокремнистых — перлитная. Следовательно, диффузионный отжиг недостаточной продолжительности не только не устраняет, но даже может усилить концентрационную, а вместе с ней и структурную микронеоднородность в многокомпонентном сплаве.
Для качественного анализа возможных диффузионных перераспределений в стали полезно знать, что, как правило, карбидообразующие элементы понижают, а некарбидо-сбразующие повышают термодинамическую активность углерода в аустените.
2. Побочные структурные изменения
Одновременно с основными структурными изменениями, рассмотренными выше и составляющими сущность гомогенизации, могут протекать побочные изменения структуры, которые необходимо учитывать при выборе режима термической обработки.
Рост зерна. В сплаве, испытывающем полиморфное превращение, при гомогенизации в области высокотемпературной фазы может вырасти крупное зерно. Так, при гомогенизационном отжиге легированных сталей, который проводят при высоких температурах, вырастает крупное аустенитное зерно. В слитках это зерно измельчается при последующей обработке давлением, а фасонные отливки из легированной стали для устранения нежелательных последствий перегрева приходится после гомогенизационного отжига подвергать отжигу 2-го рода или нормализации для измельчения зерна (см. § 25).
Коагуляция избыточных фаз. Если сплав в равновесном состоянии не должен быть однофазным, избыточные фазы, которые не способны полностью раствориться при температуре гомогенизации, могут коагулиро
25
вать и округляться с увеличением длительности отжига. Например, в алюминиевых сплавах разветвленные кристаллы силицида Mg2Si, полностью не переходящего в твердый раствор, при достаточно высоких температурах отжига становятся более компактными. Подобным образом изменяется форма некоторых избыточных фаз в легированных сталях.
Гетерогенизация структуры. Для перевода неравновесного избытка фаз в твердый раствор выбирают такую температуру гомогенизационного отжига, чтобы в металле-основе растворимость компонентов, входящих в избыточные фазы, была высокой. В многокомпонентном сплаве при этой температуре может оказаться низкой растворимость компонентов, которые не входят в избыточные неравновесные фазы и находятся после кристаллизации в основном твердом растворе. Тогда при гомогенизационном отжиге будут одновременно протекать два процесса: растворение неравновесного избытка фаз в ненасыщенном по отношению к ним твердом растворе, т.е. собственно гомогенизация сплава, и выделение других фаз из пересыщенного по отношению к ним твердого раствора, т.е. гетерогенизация структуры. Такая* гетерогенизация играет важную роль при гомогенизационном отжиге многих алюминиевых сплавов.
Большинство алюминиевых сплавов содержит марганец, некоторые — цирконий и хром. Эти элементы при быстрой кристаллизации слитков непрерывного литья и тонкостенных отливок полностью или частично входят в твердый раствор на базе алюминия. Эвтектическая или перитектическая температура в соответствующих двойных системах близка к точке плавления алюминия (в системе Al — Мп 658°С; Al — Zr 660,5°С; AI — Сг 661,4°C). Температура гомогёйизационного отжига слитков алюминиевых сплавов (450—550 °C) значительно ниже указанных температур эвтектического и перитектического равновесия, при которых наблюдается предельная растворимость рассматриваемых элементов, и их равновесная растворимость при 450—550 °C намного меньше предельной. Так, в двойных системах растворимость при эвтектической или перитектической температуре и при температуре 500 °C соответственно составляет 1,4 и 0,34 % Мп; 0,28 и 0,05 % Zr; 0,72 и 0,19 % Сг. Поэтому при гомогенизационном отжиге слитков из пересыщенного марганцем, хромом и цирконием твердого раствора выделяются интерметаллиды, содержащие соответствующие элементы.
26
Гетерогенизация структуры при гомогенизационном отжиге может сильно влиять на поведение сплава при последующей обработке и на свойства изделий. Например, распад пересыщенного марганцем, хромом и цирконием алюминиевого раствора при гомогенизационном отжиге слитков может сказаться на величине пресс-эффекта (см. § 58). Дисперсные алюминиды этих переходных металлов повышают температуру начала рекристаллизации сплава. Если она окажется выше температуры нагрева под закалку, то деформированный полуфабрикат после полной термической обработки получается в перекристаллизованном состоянии и поэтому с повышенной прочностью. Высокая температура и большие выдержки при гомогенизационном отжиге слитков обусловливают коагуляцию продуктов распада пересыщенного раствора, и плотность выделений алкь. минидов уменьшается. В результате снижается температур ра начала рекристаллизации деформированных полуфаб-’ рикатов, и если она окажется ниже температуры нагрева под закалку, то пресс-эффект (повышенная прочность перекристаллизованного материала) не проявится. Таким образом, чтобы обеспечить рассмотренное полезное действие гетерогенизации структуры слитков, следует подобрать режим их отжига, обеспечивающий необходимую дисперсность и плотность выделений алюминидов переходных металлов.
Гетерогенизация структуры слитка может развиваться не только при изотермической выдержке, но и в период охлаждения с температуры отжига. Скорость охлаждения слитков при отжиге обычно не регламентируют. В производственных условиях садку охлаждают вместе с печью или выгружают из печи и охлаждают на воздухе. С понижением температуры уменьшается растворимость в алюминии основных легирующих элементов (меди, магния и др.). При очень медленном охлаждении слитков выделяются грубые частицы СиА12, S-фазы и других фаз. При нагреве под обработку давлением эти грубые выделения полностью не растворяются, вытягиваются в направлении главной деформации и снижают механические свойства, особенно показатели пластичности, в поперечном направлении. Для устранения этого и других нежелательных последствий гетерогенизации структуры слитки алюминиевых сплавов следует охлаждать с температуры гомогенизацион-ного отжига ускоренно (на воздухе).
Закалка. При охлаждении слитков легированных сталей с температуры гомогенизационного отжига на воз
21
духе может произойти полная или частичная закалка на мартенсит поверхностных слоев (например, в слитках из сталей 1Х2Н4ВА и 40ХН2МА). Если поверхностные дефекты слитка (приваренные брызги, плены, песочины и др.) не удаляются с окалиной и слитки подвергают обдирке, го необходимо после гомогенизационного отжига проводить смягчающий высокий отпуск.
Развитие вторичной пористости. С увеличением времени выдержки при отжиге литых алюминиевых сплавов иногда развивается пористость. Так, если в исходном литом образце дуралюмина Д16 объемная доля пор была равна 0,5 %, то после отжига при 490°C в течение 3 ч она составила 0,8 %.
Пористость, развивающуюся при нагревании сплава, называют вторичной в отличие от первичной, образующейся при кристаллизации. Чем выше температура отжига, тем больше вторичная пористость.
Одна из причин вторичной пористости — выделение водорода из пересыщенного им твердого раствора, образовавшегося при быстрой кристаллизации. Другой причиной может быть эффект Киркендалла — неравенство встречных диффузионных потоков атомов разных компонентов. При вакансионном механизме диффузии в тех участках твердого раствора, откуда уходят наиболее быстро диффундирующие атомы, появляются избыточные вакансии и возникает диффузионная пористость.
Увеличение пористости при обычном гомогенизационном отжиге по абсолютной величине весьма невелико и вряд ли значительно сказывается на свойствах изделий, особенно в тех случаях, когда поры завариваются при горячей пластической деформации. *
§ 2. Изменение свойств сплавов при гомогенизационном отжиге
1. Литые сплавы
Главное изменение свойств при гомогенизационном отжиге — повышение пластичности литого сплава. При выборе режима отжига слитка показатели пластичности следует измерять не при комнатной температуре, а при температуре первой операции горячей обработки давлением. Если, например, слитки сплава Д16 предназначены для прессования, то показатели пластичности следует определять при температуре прессования, равной 400 °C (см. рис. 5).
Механические свойства фасонной отливки следует из
28
мерять при температуре эксплуатации детали, например комнатной.
По мере растворения хрупких фаз пластичность растет и после окончания их растворения перестает изменяться (см. рис. 5). Относительное удлинение и сужение слитков алюминиевых сплавов при температурах горячей деформации возрастают в результате гомогенизации в 1,5—3 раза.
Если цель гомогенизационного отжига — повысить пластичность, то за оптимальное время гомогенизации можно принять время полного растворения неравновесного избытка фаз.
Повышение пластичности слитков легированных сталей, алюминиевых и других сплавов уменьшает брак по трещинам при первой операции горячей обработки давлением, позволяет увеличить степень деформации, особенно на первых обжатиях, повысить скорость деформирования, улучшает состояние кромки (уменьшает рванины горячекатаных полос).
Значение гомогенизации особенно велико для фасонных отливок, так как их не подвергают пластической деформации, измельчающей структуру. К фасонным отливкам из алюминиевых и магниевых сплавов гомогенизационный отжиг как самостоятельную операцию не применяют. Гомогенизация органически входит в операцию нагрева под закалку фасонных отливок. Этот нагрев проводят при таких высоких температурах и длительных выдержках, чтобы в твердый раствор перешло максимально возможное в производственных условиях количество избыточных фаз.
2. Деформированные сплавы
Хотя при горячей обработке давлением и происходит коренное изменение строения сплава, но оно все же недостаточно, чтобы полностью устранить влияние литой структуры на его технологичность при последующей холодной обработке давлением. «Наследственность» литой структуры с неустраненной дендритной ликвацией проявляется в снижении пластичности холоднодеформированного сплава. Объясняется это тем, что при горячей обработке давлением, несмотря на сильное измельчение и «перемешивание» структуры, полностью не устраняется микронеоднородность сплава, вызванная дендритной ликвацией. Гомогенизация слитка, повышая пластичность холоднодеформированного сплава, улучшает состояние кромки холоднокатаных полос, позволяет сократить промежуточные отжиги и увеличить
29
степень обжатия при холодной прокатке, улучшает штам-пуемость листов при глубокой вытяжке.
Наследственность литой структуры бывает весьма устойчивой и сказывается на служебных свойствах изделий, несмотря на то, что в технологическом цикле структура сплава испытывает такие мощные воздействия, как обработка давлением, закалка, отпуск и другие виды обработки. Так, в высокоуглеродистых сталях, легированных хромом и вольфрамом, в результате дендритной ликвации может появиться карбидная эвтектика. Это явление называют карбидной ликвацией. В изделиях, несмотря на горячую прокатку и закалку, сохраняются грубые скопления эвтектических карбидов. В этих местах выкрашиваются лезвие инструмента и трущаяся поверхность шарикоподшипника.
Гомогенизация слитка может не только улучшить, но и ухудшить некоторые свойства готовой продукции. Например, гомогенизация при 490 °C в течение суток слитка из дуралюмина марки Д16 повышает на несколько процентов относительное удлинение закаленных и состаренных листов, но одновременно снижает их временное сопротивление1 на 10—15 МПа. Причиной некоторого снижения прочности является особое поведение марганца при гомогенизации слитка. Как уже отмечалось, при температурах около 500 °C растворимость марганца в алюминии сравнительно невелика и пересыщенный марганцем раствор, образовавшийся при кристаллизации, распадается. Выход марганца из раствора при гомогенизации слитка и коагуляция выделившихся частиц марганцевого интерметаллида несколько снижают прочность рекристаллизованных листов дуралюмина, прошедших полную термическую обработку (закалку и старение).
Наиболее заметное и практически очень важное влияние гомогенизационный отжиг оказывает на показатели пластичности, ударную вязкость и усталостные характеристики изделий (прессованных полос, профилей, поковок и др.) поперек волокна, так как избыточные хрупкие фазы вытягиваются вдоль направления главной деформации.
Гомогенизационный отжиг сталей, требующий большого расхода топлива и сопровождающийся значительными потерями металла на окалину, применяют лишь к высококачественным легированным сталям ответственного назна
4 Устойчивое изменение временного сопротивления деформируемых легких сплавов на 10—15 МПа представляет практический интерес, в то время как для большинства сталей такое изменение можно не* принимать во внимание.
30
чения (температуру отжига выбирают в интервале 1050—-1250 СС). Из углеродистых сталей только автоматные подвергают гомогенизационному отжигу. Автоматные стали содержат повышенное количество серы, улучшающей обрабатываемость резанием (до 0,2—0,3 % вместо обычных 0,04—0,06 %). Сера сильно ликвирует к границам зерен при кристаллизации и вызывает красноломкость при прокатке. Гомогенизационный отжиг при 1150сС устраняет красноломкость автоматной стали.
Слитки большинства деформируемых алюминиевых сплавов подвергают гомогенизационному отжигу для улучшения обрабатываемости давлением и повышения механических свойств полуфабрикатов. Температуру отжига, обычно находящуюся в интервале 450—550 °C, выбирают в зависимости от марки сплава и вида полуфабрикатов.
Слитки деформируемых магниевых сплавов гомогенизируют при 390—405 °C с той же целью, что н слитки алюминиевых сплавов. Часто гомогенизацию совмещают с операцией нагрева слитков перед обработкой давлением, увеличивая выдержку в печи.
Если гомогенизационный отжиг позволяет существенно увеличить скорость обработки давлением, например прессования, то затраты на его проведение всегда с лихвой оку< паются, так как повышается производительность дорогих и сложных прессов.
§ 3. Гомогенизация с нагревом выше температуры неравновесного солидуса
В некоторых случаях степень гомогенизации, проводимой при температурах вблизи неравновесного солидуса, но ниже его, может оказаться недостаточной. Необходимая полнота гомогенизации при этом или вообще не достигается, или достигается при таких длительных выдержках, которые неприемлемы в производстве. Кроме того, всегда желательно сокращение времени гомогенизации. Поэтому весьма заманчива возможность проведения гомогенизационного отжига при температурах выше неравновесного солидуса. Такой отжиг с нагревом выше неравновесного, но ниже равновесного солидуса применяют в промышленности к слиткам некоторых алюминиевых сплавов.
Обычно отжиг промышленных слитков алюминиевых сплавов выше неравновесного солидуса вызывает опасения из-за возможности пережога (см. § 31).
В этой связи рассмотрим условия, в которых находится слиток во время гомогенизационного отжига при темпера
31
туре выше неравновесного, но ниже равновесного солидуса, например при температуре t\ на рис. 7. Твердый раствор на базе алюминия при температуре tx ненасыщен по отношению к неравновесным включениям расплава. Легирующий элемент из расплава (медь) будет диффундировать в алюминиевый раствор, и неравновесные включения расплава рассосутся. Таким образом, нагрев выше неравно-
весного, но ниже равновесного солидуса не вызывает пере-
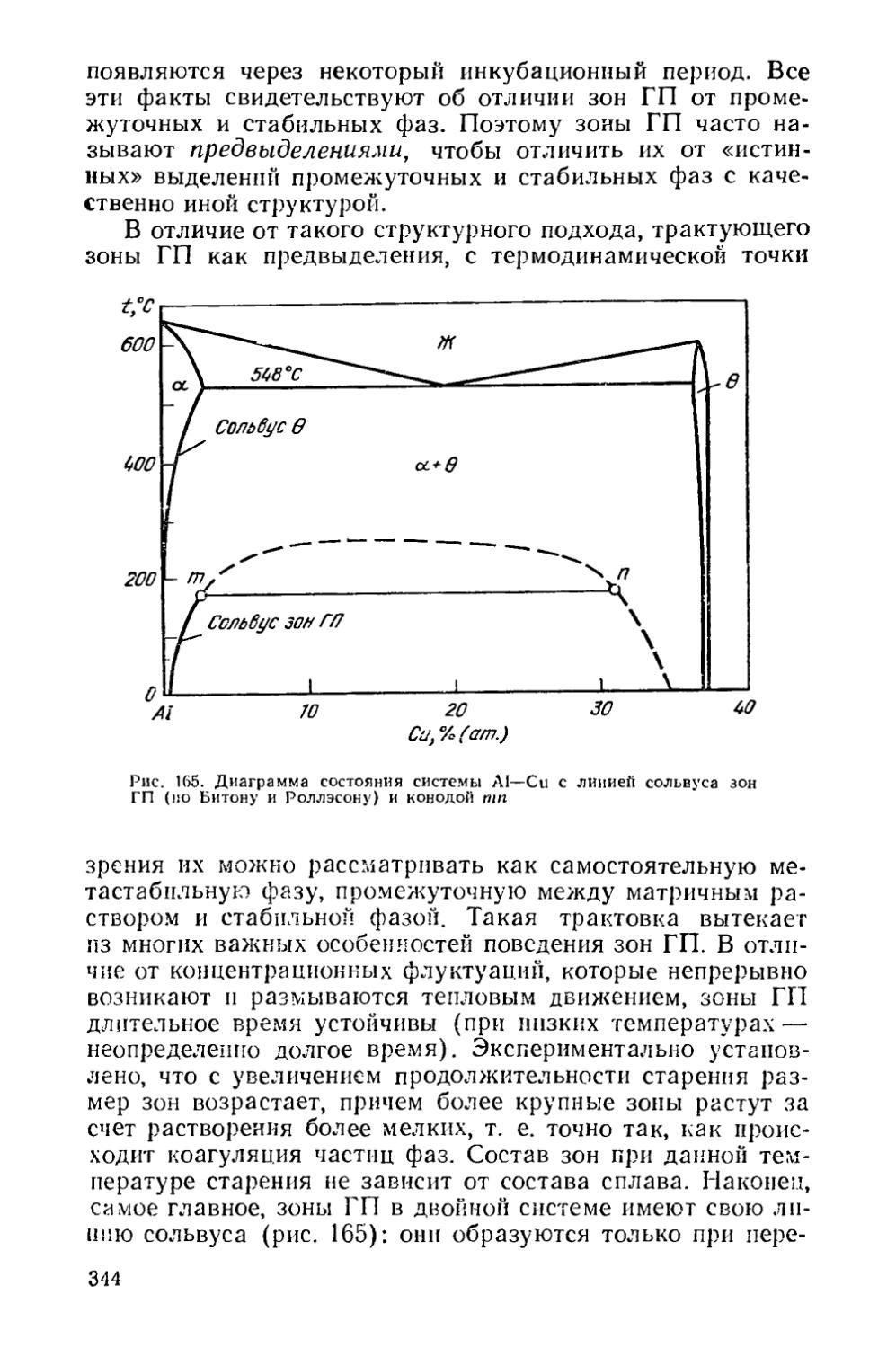
жога при оплавлении, так как участки расплава исчезают в процессе изотермической выдержки при гомогенизационном отжиге. У многих про
Рис. 7. Диаграмма состояния AI—Си: а — неравновесный солидус сплава AI — 2,3 % Си; b — равновесный солидус сплава AI—2,3 % Си; Л —температура гомогенизации
мышленных сплавов неравновесная эвтектика рассасывается уже в период нагрева слитка до температуры гомогенизации.
Пережог из-за межкристаллитного окисления не опасен потому, что, во-первых, на поверхности слитка алюминиевого сплава имеется плотная окисная пленка и, во-вторых, возможное межзеренное окисление в тонком поверхностном слое на свойствах слитка не скажется (в отличие от ли
стов, проволоки и других изделий тонкого сечения).
Процессы гомогенизации при отжиге с нагревом выше точки неравновесного солидуса идут гораздо быстрее, чем при обычном отжиге ниже солидуса. Например, у дуралю-мина Д16 температура неравновесного солидуса равна примерно 508 °C, а равновесного — около 530 °C. Время растворения неравновесного избытка фаз в промышленном слитке диаметром 150 мм при температуре отжига 515°С.в 2,5 раза меньше, чем при 480 °C.
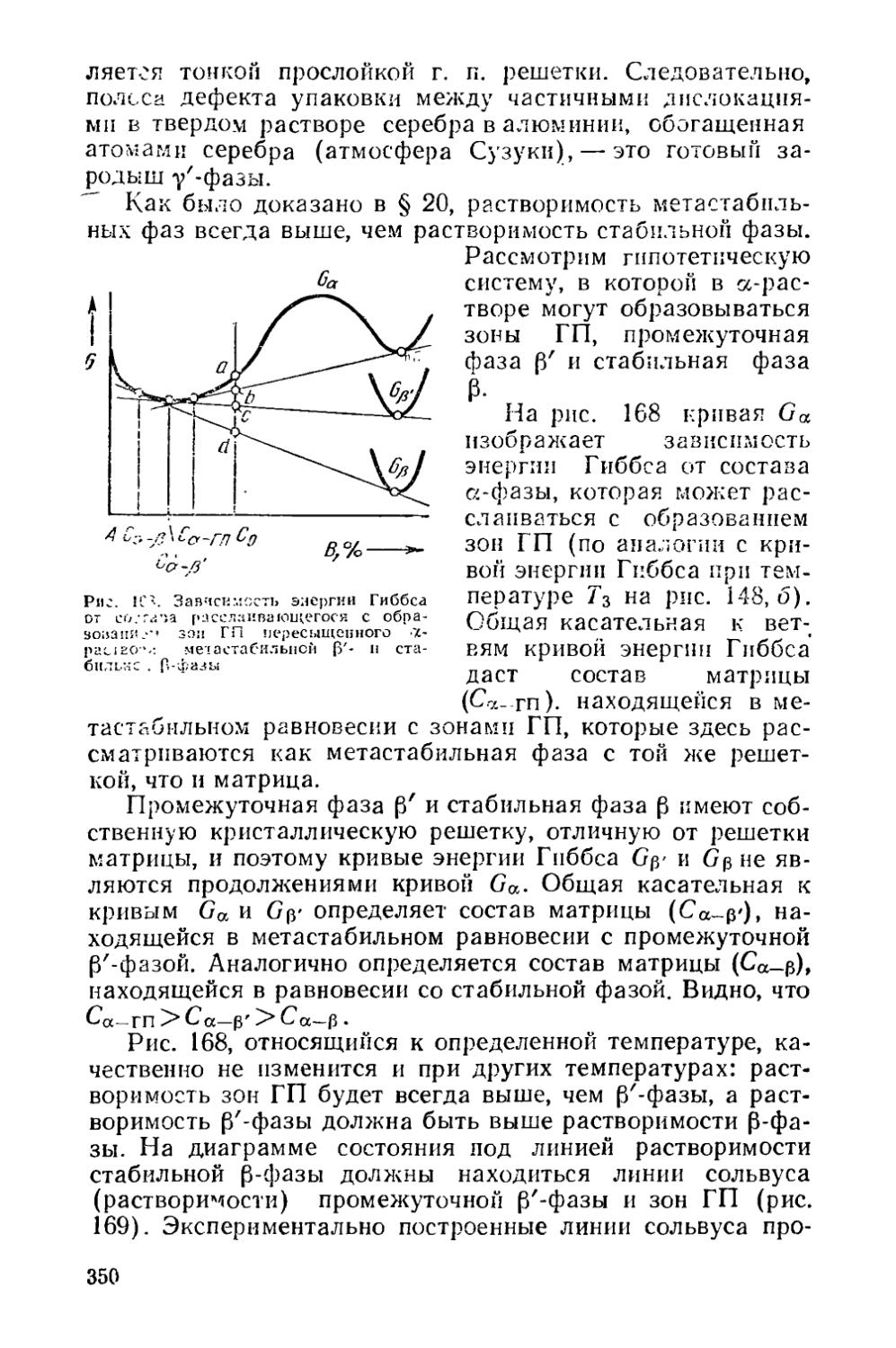
Причиной сильного ускорения гомогенизации при отжиге с нагревом до температур выше неравновесного солидуса является не присутствие жидкой фазы, а увеличение коэффициентов диффузии легирующих элементов с повышением температуры. Ведущим звеном процесса растворения фаз ниже и выше неравновесного солидуса является диффузионный отвод атомов легирующих элементов от межфазной границы в центральную зону дендритной ячейки.
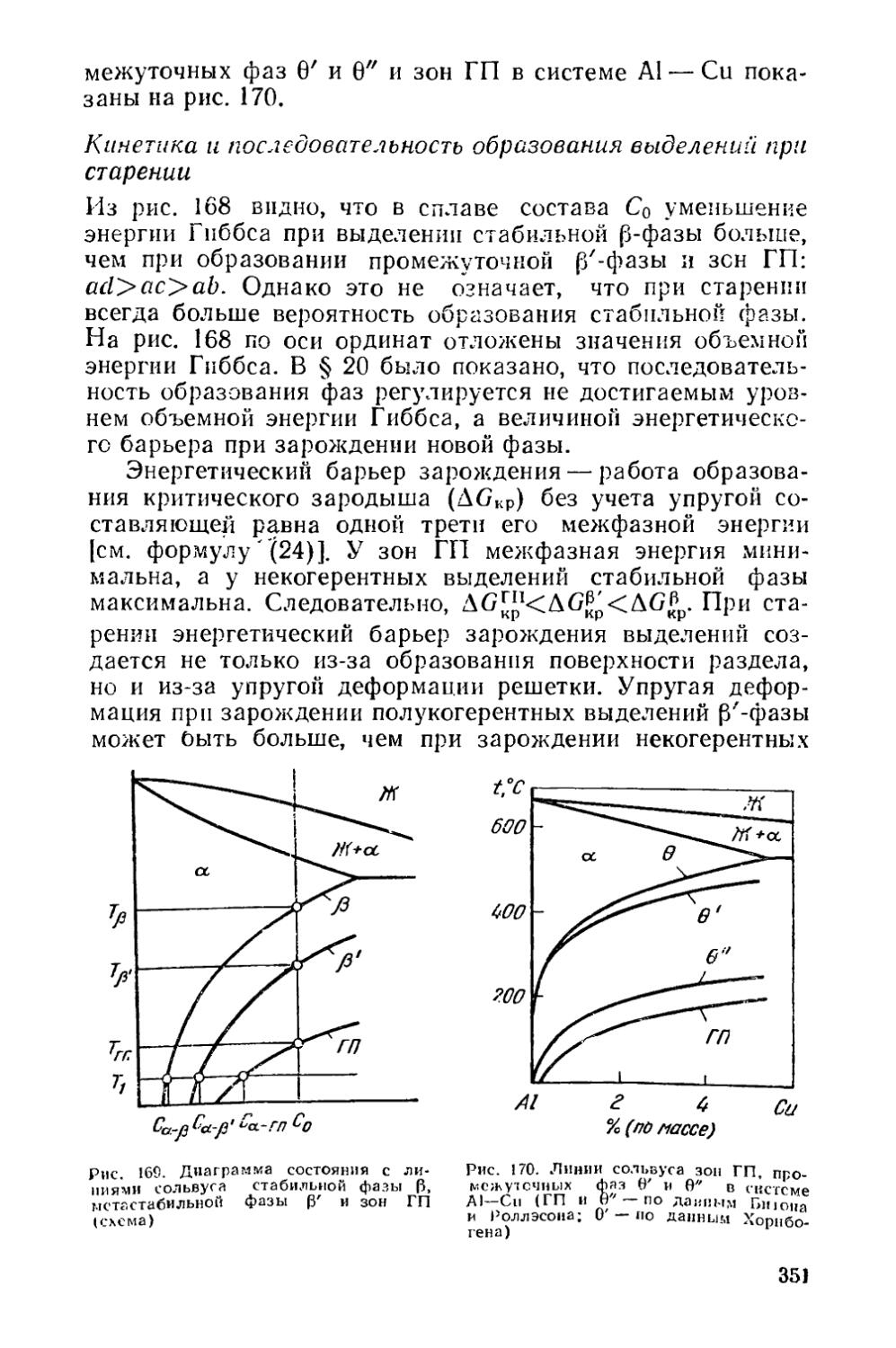
32
Сам по себе переход через точку солидуса не является критическим по отношению к процессу гомогенизации, но повышение температуры отжига даже на 20—30 К может резко, например в два раза, увеличить коэффициенты диффузии.
Гомогенизация при температурах выше неравновесного солидуса значительно сокращает продолжительность отжига, обеспечивающего необходимую технологическую пластичность при обработке давлением. Для некоторых сплавов возможно повышение обжатий и скорости обработки давлением, например скорости прессования. Кроме того, повышаются показатели пластичности деформированных полуфабрикатов, особенно поперек волокна.
Основной недостаток отжига с нагревом выше температуры неравновесного солидуса—значительно более быстрое развитие пористости (рис. 8). К причинам, вызывающим развитие пористости при обычном отжиге (см. § 1), здесь можно добавить еще и следующую (по Е. Д. Захарову). Твердый раствор на базе алюминия прл быстрой кристаллизации слитка пе-
ресыщается водородом. Растворимость водорода в расплаве намного больше, чем в кристаллах, и поэтому водород из пересыщенного им твердого раствора устремляется в жидкую фазу, появляющуюся при нагреве слитка выше точки неравновесного солидуса. Последующая сравнительно медленная изотермическая кристалли
Рис. 8. Зависимость объемной доли nop V от продолжительности отжига дуралюмииа Д16 при температурах ниже и выше неравновесного солидуса (508 °C) (И. И. Новиков, В. С. Зо-лоторевский, А. В. Курбатова)
зация . неравновесных
включений расплава приводит к выделению из него водорода, образующего газовые поры. Могут ли полностью завариваться эти поры при горячей деформации слитка и каково их влияние на эксплуатационные свойства изделий— еще точно не установлено. Поэтому гомогенизационный отжиг с нагревом выше температуры неравновесного солидуса, называемый в технологии алюминиевых сплавов «высокотемпературной» гомогенизацией, следует ис-использовать только после тщательного опробования, ис
ключив возможные нежелательные его последствия.
3 И. И. Новиков
33
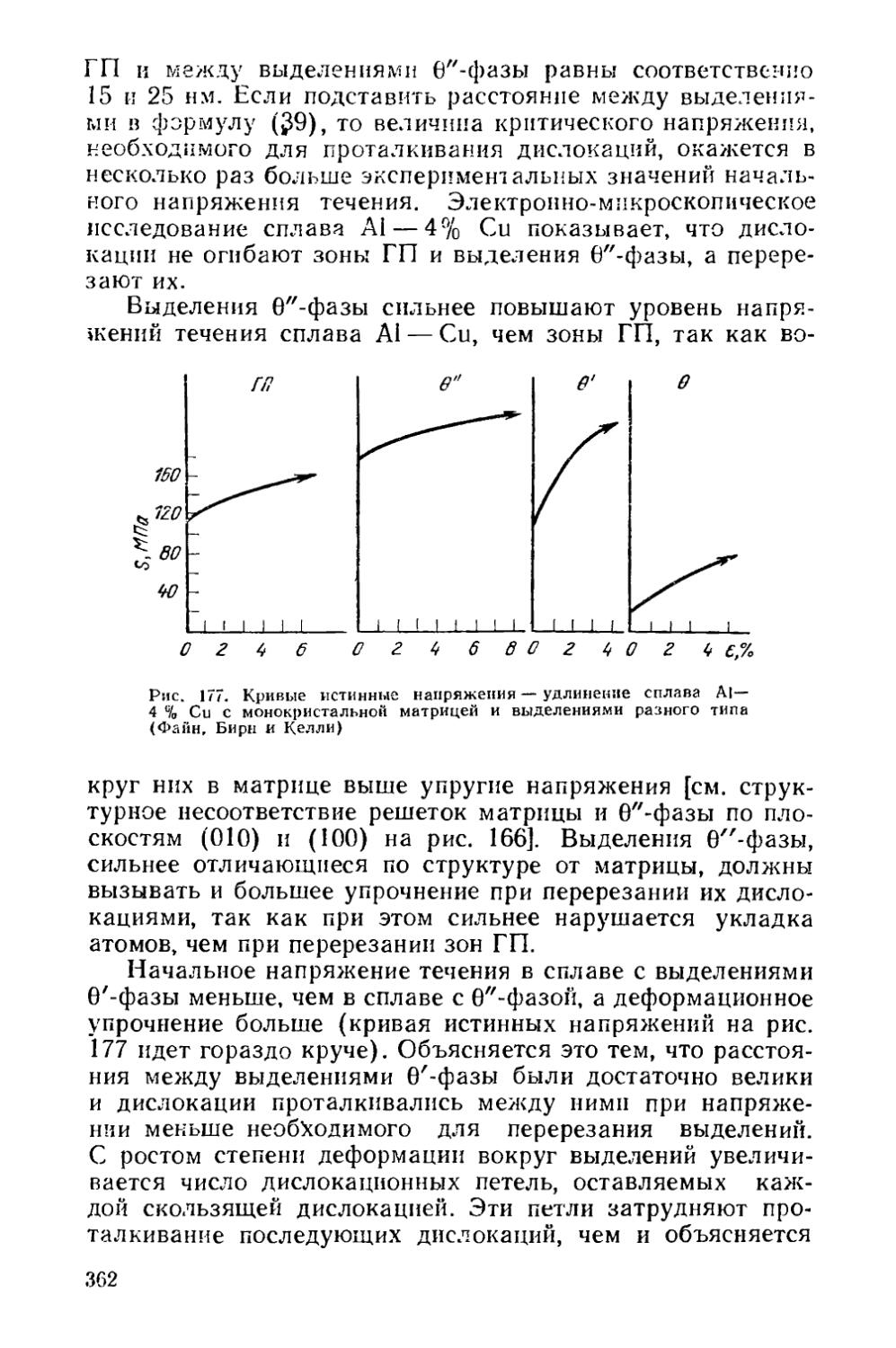
Глава II
РЕКРИСТАЛЛИЗАЦИОННЫЙ
И ДОРЕКРИСТАЛЛИЗАЦИОННЫЙ ОТЖИГ
Рекристаллизационный отжиг — это термическая обработка деформированного металла или сплава, при которой главным процессом является рекристаллизация.
Дорекристаллизационный отжиг — это термическая обработка деформированного металла или сплава, при которой главным процессом является возврат.
Обе разновидности термической обработки чаще применяют после холодной обработки давлением.
§ 4. Изменение структуры металла при холодной обработке давлением
Пластическая деформация вызывает в металле структурные изменения, которые включают изменение формы кристаллитов, их кристаллографической пространственной ориентировки и внутреннего строения каждого кристаллита.
1. Изменение формы зерен
и их кристаллографической ориентировки
Форма заготовки при обработке давлением изменяется под действием внешних сил вследствие пластической деформации каждого кристаллита в соответствии со схемой главных деформаций. Основное изменение формы кристаллитов состоит в том, что они вытягиваются в направлении главной деформации растяжения (например, в направлении прокатки или волочения). С повышением степени холодной деформации зерна все более вытягиваются и структура становится волокнистой.
При пластической деформации кристаллические решетки зерен приобретают преимущественную пространственную ориентировку-—возникает текстура деформации. Это одно из важнейших следствий кристаллографической направленности скольжения в каждом зерне по определенным плоскостям и направлениям пространственной решетки, которые закономерно поворачиваются по отношению к осям деформации изделия. Например, при растяжении монокристалла направление скольжения приближается к оси растяжения.
34
Характер текстуры деформации зависит от вида и условий обработки давлением (в основном от схемы главных деформаций), от типа кристаллической решетки и энергии дефектов упаковки.
Аксиальная текстура волочения характеризуется параллельным оси проволоки кристаллографическим направлением, вокруг которого решетка может быть повернута как угодно.
Тип решетки............. Г. ц. к. О. ц. к. Г. п.
Текстура..............<111>+<Ю0> < 110> <1010>
В металлах с г. ц. к. решеткой текстура двойная: в одних кристаллах параллельно оси проволоки устанавливается направление <111>, а в других <100>. Например, после волочения с обжатием 98 % ориентации < 111 > и <100> в медной проволоке имеют соответственно 64 и 36 % всех кристаллов, в алюминиевой 92 и 8 %.
Текстура прокатки характеризуется кристаллографическими плоскостью и направлением, параллельными плоскости и направлению прокатки соответственно (табл. 1). В металлах с г. ц. к. решеткой тип текстуры зависит от энергии дефектов упаковки. Материалы с низкой энергией дефектов упаковки (серебро, а-латунь с высоким содержанием цинка, аустенитная нержавеющая сталь) резко отличаются по текстурным компонентам от металлов со средней (медь) и высокой (алюминий) энергией дефектов упаковки.
Рентгеновский анализ фиксирует появление текстуры холодной деформации при обжатиях 10—20%. В дефор-
Таблица 1. Текстуры прокатки
р< Тип ешетки Материал Энергия дефектов упаковки, мДж/м* Текстура
основная дополнительная
г. ц. к. А1 135 {112} <111> {135} <112>
г. ц. к. Си 70 {135} <112> {110} <112>+ + {112} <111>
г. ц. к. Ag, Л68, 12Х18Н9 <30 {110} <112> {110} <100>
о. ц. к. a-Fe 140 {112} <1Ю>+ + {100} <110> {111} <110>
г. п. Mg — {0001}<1120> —
3*
35
мированном металле не все кристаллы имеют идеальную преимущественную ориентировку. В общем случае с увеличением степени деформации текстурные компоненты проявляются более четко.
2. Изменение внутреннего строения зерен
До 10 % работы, затраченной на холодную деформацию, поглощается металлом (остальная ее часть рассеивается в виде теплоты). Накопленная в металле энергия «задерживается» в виде энергии дефектов кристаллической решетки, образующихся при пластической деформации, а также в виде энергии упругой деформации при возникновении остаточных напряжений (о них см. в гл. III).
Важнейшее изменение внутреннего строения каждого кристаллита при холодной деформации — увеличение плотности дислокаций (отношения суммарной длины дислокаций к объему металла). У хорошо отожженного поликристалл ического металла плотность дислокаций равна 106— 108 см~2, при холодной деформации на несколько процентов она возрастает до 109—1010 см-2, а при сильной деформации (на 30—40 % и более)—до 10й—1012 см-2. Следовательно, плотность дислокаций при холодной обработке давлением может возрасти на пять — шесть порядков (!).
При пластической деформации возрастает и концентрация вакансий, генерируемых порогами скользящих винтовых дислокаций.
В процессах формирования структуры при отжиге холоднодеформированного металла исключительно важную роль играют разного типа и происхождения локальные изменения ориентировки кристаллической решетки, возникающие в результате пластической деформации. К настоящему времени накоплены весьма подробные сведения о таких изменениях ориентировки внутри зерен поликри-сталлического материала после обработки давлением со средними и большими обжатиями, представляющие большой практический интерес. Эти изменения ориентировки кристаллической решетки рассмотрены ниже.
Ячеистая структура
У большинства металлов уже при небольших степенях де-формациии (5—10%) начинает формироваться ячеистая структура: дислокации так перераспределяются в объеме зерна, что их сплетения образуют размытые стенки (границы), окружающие области, внутри которых плотность
36
Рис. 9. Ячеистая структура хромоцир-кониевой бронзы (0.38 % Сг, 0,07 % Zr) после растяжения на 5 %. Электронная микрофотография, фольга. X12 000 (В. М. Розенберг, А. И. Новиков)
дислокаций заметно меньше, чем в стенках (рис. 9). Средний размер таких ячеек составляет примерно 0,5—2 мкм, а толщина их стенок на порядок меньше. С увеличением, степени деформации в области средних и больших деформаций размер ячеек мало меняется, а плотность дислокаций в их размытых границах возрастает.
Из-за избытка дислокаций одного знака в стенке соседние ячейки разориентированы на углы до 1—2°. Направление разворота на границах между разными соседними ячейками разное, и поэтому в целом при ячеистой структуре не получается макроскопическое изменение ориентировки кристаллита.
Нет корреляции между ячеистой структурой в объеме зерна и линиями скольжения — следами выхода дислокаций на предварительно отполированную поверхность металла. Расстояние между линиями скольжения на порядок меньше размера ячеек дислокационной субструктуры. Этот и другие факты указывают на то, что ячеистая структура роз-никает в результате релаксационных процессов перераспределения дислокаций в объеме кристаллита в условиях, когда скольжение дислокаций по мере роста их плотности затрудняется. При образовании ячеистой структуры дислокации перераспределя
ются с обязательным участием поперечного скольжения. В сплавах с очень низкой энергией дефектов упаковки (а-латунь с 30 % Zn, нержавеющая аустенитная сталь и др.) поперечное скольжение столь затруднено, что ячеистая структура не возникает. В таких материалах растянутые дислокации образуют плоские скопления в своих плоскостях скольжения.
37
Деформационные и переходные полосы
В поликристаллическом материале каждое зерно деформируется под действием приложенных извне нагрузок при одновременном сложном взаимодействии с окружающими его зернами. При этом соседние участки одного зерна могут быть вынуждены поворачиваться по-разному (в противоположных направлениях или в разной степени в одном направлении). В результате соседние участки внутри одного зерна могут оказаться взаимно развернутыми на десятки градусов. Например, в поликристаллическом алюминии при обжатии 40 % в пределах одного зерна наблюдались разориентировки до 50°.
Участки, внутри которых ориентировка меняется незначительно, но которые сами развернуты на большие углы по отношению к соседним аналогичным участкам того же кристалла, называют деформационными полосами. В алю-
Рис. 10. Вытянутые вдоль направления прокатки деформационные полосы (ДП) и расположенные под углом 30—40° к направлению прокатки полосы сдвига (ПС) в сплаве алюминия с 10 % Mg. 0,1 % Ti и 0,1 % Zr после холодной прокатки с обжатием 70 %. Оксидированный шлиф, поляризованный свет (В. В. Истомин):
R — направление прокатки; /V — нормаль к плоскости прокатки
минии и алюминиевых сплавах деформационные полосы контрастно выявляются под световым микроскопом на оксидированных шлифах: в зависимости от кристаллографической ориентировки подложки прозрачная оксидная пленка в поляризованном свете имеет разную цветовую окраску. Деформационные полосы при средних и больших обжатиях вытянуты в направлении прокатки (рис. 10).
38
Если (более редкий случай) ориентировка решетки в кристалле одинакова по обе стороны от деформационной полосы, то ее называют полосой сброса (см. рис. 24, а).
Граница между соседними деформационными полосами сама является областью большей или меньшей ширины, в которой ориентировка изменяется от одной деформационной полосы до соседней полосы. Эту область называют переходной полосой.
Под световым микроскопом переходные полосы бывают видны в виде тонких линий или в виде границ областей разного контраста. Поэтому металлографически они могут и
не отличаться от границ вытянутых зерен. Вообще структурное различие между зернами и деформационными полосами из-за одинаково большой разориентировки по отношению соответственно к соседнему зерну и полосе состоит только в разном строении границы.
Просвечивание тонких фолы в электронном микроскопе выявило, что переходная полоса между деформационными полосами сама состоит из группы микрополос с почти парал-
Рис. 11. Микрополосы (отмечены буквами) в переходной полосе в меди после холодной прокатки с обжатием 90%. Электронная микрофотография, фольга (Ридха и Хачинсон):
R — направление прокатки; «V — нор маль к плоскости прокатки
лельными границами (рис. 11). Эти границы— две параллельные плоские стенки дислокаций— находятся в плоскости {111} в меди и в плоскости {110} в железе. Ш.и-
рина микрополос около 0,1—0,3 мкм, а длина может составлять десятки микрометров. Сама микрополоса по длине состоит из слегка разориентированных (не более 1—2°) фрагментов, разделенных пеперечными субграницами. Микрополосы вытянуты вдоль переходной полосы, т. е. вдоль направления прокатки при больших обжатиях.
В поперечном сечении переходной полосы размещается
39
от двух до десяти-пятнадцати микрополос. Соседние микрополосы разориентированы на углы не более 5°, причем очень важно, что разворот между ними в данной переходной полосе имеет одинаковый знак, т. е. разориентировка накапливается при переходе от одного края переходной полосы к другому. Так, например, в меди, прокатанной с обжатием 90 %, средний угол разориентировки соседних микрополос равен 4°, а группа из десяти таких микрополос обусловливает разориентировку между деформационными полосами величиной 40°.
Причины и механизм образования микрополос не установлены. Микрополосы — самостоятельный элемент субструктуры. Они не образуются в результате вытягивания ячеек по мере увеличения степени деформации, поэтому называть микрополосы вытянутыми ячейками, как это иногда делают, нецелесообразно. Отдельные микрополосы появляются на ранних стадиях деформации одновременно с формированием равноосных ячеек. Имеются свидетельства, что с микрополосами в объеме кристалла прямо связаны линии сдвига на его поверхности.
С увеличением степени деформации число микрополос растет. При больших деформациях они образуют группировки. Отдельные микрополосы и их группировки поворачиваются, становясь параллельными плоскости прокатки. Между микрополосами сохраняются мало меняющиеся равноосные ячейки. Между микрополосой и прилегающей к ней областью равноосных ячеек угол разориентировки не превышает нескольких градусов.
В твердых растворах с очень низкой энергией дефектов упаковки микрополосы, как и равноосные ячейки, при деформации не формируются, так как их образование, по всей видимости, связано с развитием поперечного скольжения.
Полосы сдвига
При обжатиях около 70 % в металлах с г. ц. к. и о. ц. к. решеткой в сечении, проходящем через направление прокатки и нормаль к плоскости прокатки, подобрав травитель, можно выявить систему параллельных полос, расположенных под углом 30—40° к направлению прокатки. Их называют полосами сдвига (рис. 12). В плоскости прокатки они перпендикулярны направлению прокатки. Часто можно наблюдать две системы полос сдвига, расположенных в виде «елочки» или взаимно пересекающихся. Полосы
40
сдвига в отличие от переходных полос могут пересекать границы зерен, а при очень больших обжатиях они проходят через всю толщину листа.
Полосы сдвига 1 названы так потому, что они являются участками сильной локализации сдвиговой деформации. При обжатиях более 90 % почти вся пластическая деформация сосредоточена в этих полосах.
Полосы сдвига имеют ширину порядка 1 мкм и состоят из микрополос, разделенных малоугловыми границами (рис. 13}.
Рис. 12. Полосы сдвига в меди после холодной прокатки с обжатием 90 % (Ридха и Хачинсон):
R — направление прокатки; N — нормаль к плоскости прокатки
Рис. 13. Микрополосы в полосе сдвига в сплаве алюминия с 10 % Mg. 0,1 % Ti и 0,1 % Zr после холодной прокатки с обжатием 70 %. Электронная микрофотография, фольга (В. В. Истомин):
R — направление прокатки; N — нормаль к плоскости прокатки
Таким образом, с увеличением степени холодной деформации возрастают плотность дислокаций и концентрация вакансий, появляются участки с локальной разориентиров-кой кристаллической решетки (деформационные и переходные полосы, полосы сдвига); при не слишком низкой энергии дефектов упаковки образуются микрополосы и формируется ячеистая структура.
1 Понятие «полоса сдвига» (shear band) не следует смешивать с понятием «полоса скольжения» (slip band)' Полосы скольжения выявляются в световом микроскопе после слабой деформации на полированной поверхности образца в виде линий, каждая из которых является следом пересечения этой поверхности группой активных соседних плоскостей скольжения.
4-1
На рис. 14 схематично изображены рассмотренные выше характерные особенности структуры сильно деформированного металла, обладающего средней или высокой энергией дефектов упаковки. Внутри деформационных полос показана ячеистая структура, а внутри переходных полос и полос сдвига изображены микрополосы.
Рис. 14. Схема строения зерна холоднокатаного металла с деформационными полоса мн (ДП), переходными полосами (ПП) и полосой сдвига (ПС):
Д — направление прокатки; N — нормаль к плоскости прокатки
§ 5. Изменение свойств металла при холодной обработке давлением
1. Наклеп
Обычно под наклепом понимают упрочнение при обработке давлением. В‘более широком понимании наклеп — это совокупность структурных изменений и связанных с ними изменений свойств при пластической деформации.
С увеличением степени холодной деформации показатели сопротивления деформированию (временное сопротивление, йредел текучести и твердость) возрастают, а показатели пластичности (относительное удлинение и сужение) падают (рис. 15). При деформировании металла со степенью деформации более 50—70 % временное сопротивле-
42
ние и твердость обычно увеличиваются в полтора — два, а иногда и в три раза в зависимости от природы металла и вида обработки давлением.
Небольшие деформации (до 10%), как правило, значительно сильнее влияют на предел текучести, чем на временное сопротивление. При больших степенях деформации
у некоторых сплавов предел текучести может возрасти в 5—8 раз и более.
Относительное удлинение резко уменьшается уже при сравнительно небольших деформациях (см. рис. 15). Сильная деформация, увеличивающая временное сопротивление и твердость в 1,5—2 раза, снижает относительное удлинение в 10—20, а иногда и в 30—40 раз и более.
Рост показателей сопро-
тивления деформированию и снижение показателей пластичности с увеличением степени предварительной холодной деформации происходят в результате повышения плотности дислокаций. В наклепанном металле из-за повышенной плотности дислокаций затруднено скольжение уже имеющихся, а также генерирование и скольжение «свежих» дислокаций.
Границы ячеек и субзерен служат барьерами для скользящих дислокаций. Уменьшение расстояний
Рис. 15. Зависимость механических свойств дуралюмииа Д1 от степени обжатия при холодной прокатке (А. В. Третьяков, К. М. Радченко)
между этими барьерами
(уменьшение размеров ячеек) способствует упрочнению при увеличении степени обжатия.
Электроны проводимости рассеиваются точечными дефектами и дислокациями. Поэтому увеличение числа дефектов решетки при холодной пластической деформации
вызывает рост электросопротивления. В то-время как механические свойства при больших деформациях изменяют-
ся в несколько или десятки раз, электросопротивление у чистых металлов обычно возрастает на 2—6 %,
У сплавов наблюдаются разнообразные по величине и даже разные по знаку изменения электросопротивления при холодной деформации.
43
У многих сплавов, содержащих переходные металлы, в том числе у широко используемых в промышленности сплавов нихром, хромель (Ni — Сг), алюмель (Ni — Сг — А1 — Мп — Si) и нейзильбер (Си — Ni — Zn), возникает так называемое /(-состояние, характеризующееся повышенным электросопротивлением по сравнению с электросопротивлением при статистически равномерном распределении атомов в твердом растворе.
Появление /(-состояния объясняют диффузионным перераспределением атомов, при котором число связей между атомами разного сорта становится больше, чем в статистически неупорядоченном твердом растворе. Наиболее характерным признаком /(-состояния является значительное уменьшение электросопротивления при холодной деформации. Это вызвано тем, что при холодной деформации нарушается свойственное /(-состоянию распределение атомов разного сорта и сплав переводится в состояние неупорядоченного твердого раствора.
2. Анизотропия свойств
Свойства холоднодеформированного металла по разным направлениям различны. Анизотропия свойств обусловлена двумя причинами: волокнистостью структуры и текстурой деформации.
По длине разрывного образца, вырезанного поперек волокна, число межзеренных границ значительно больше, чем в образце, вырезанном вдоль волокна. На межзеренных границах сосредоточены примеси и неметаллические включения, например оксидные плены. Естественно, что механические свойства металла вдоль и поперек волокна разные. Поэтому при контроле полуфабрикатов, полученных обработкой давлением, различают «долевые» и «поперечные» образцы и соответственно «долевые» и «поперечные» свойства. Обычно показатели пластичности и ударная вязкость на поперечных образцах ниже, чем на долевых.
Каждый кристаллит анизотропен, его свойства зависят ют кристаллографического направления. В металле с хаотичной ориентировкой кристаллов свойства по всем направлениям статистически усредняются. Такой металл ква-зиизотропен. В текстурованном металле с предпочтительной ориентировкой кристаллов имеются направления, вдоль которых одни свойства усилены, другие ослаблены. Поэтому текстура деформации обусловливает анизотропию «свойств.
44
§ 6. Изменение структуры при дорекристаллизационном отжиге
Холодная обработка давлением приводит металл в неравновесное состояние с повышенной энергией Гиббса. Наклепанный металл стремится самопроизвольно перейти в более равновесное состояние с меньшей энергией Гиббса. Восстановительные процессы сводятся в основном к уменьшению общего количества дефектов кристаллической решетки и перераспределению их в кристаллитах с образованием более равновесных конфигураций. Эти процессы совершаются путем перемещений атомов, и решающее влияние на них оказывает температура.
У большинства промышленных металлов и сплавов, исключая легкоплавкие, при комнатной температуре подвижность атомов недостаточна, чтобы обеспечить активное развитие восстановительных процессов, уменьшающих энергию Гиббса наклепанного материала. Чтобы частично или полностью устранить наклеп за практически приемлемое время, приходится проводить нагрев — отжиг после холодной обработки давлением.
В зависимости от температуры и продолжительности отжига в холоднодеформированном металле протекают различные структурные изменения, которые подразделяют на процессы возврата и процессы рекристаллизации.
После нагрева наклепанного металла при сравнительно низких гомологических1 температурах (для металлов обычной чистоты — ниже ~0,3 Тпл) под световым микроскопом не наблюдаются изменения формы и размеров деформированных зерен, не обнаруживаются новые, рекристаллизованные зерна. Однако такой дорекристаллизационный отжиг вызывает заметное изменение некоторых свойств металла, а с помощью рентгеноструктурного анализа, электронной микроскопии и других прямых и косвенных методов фиксируются изменения во внутреннем строении деформированных зерен.
Совокупность любых самопроизвольных процессов изменения плотности и распределения дефектов в деформированных кристаллах до начала рекристаллизации называют возвратом. Этот собирательный термин, относящийся к весьма разным по своему механизму явлениям, используют в связи с тем, что некоторые свойства наклепанного
1 Гомологической, или соответственной, температурой называют отношение данной температуры к температуре начала плавления по абсолютной шкале.
45
металла при дорекристаллизациоином отжиге частично или полностью возвращаются к значениям свойств перед холодной деформацией.
Если возврат протекает без образования и миграции субграниц внутри деформированных зерен, то его называют возвратом первого рода, или отдыхом. Если же при возврате внутри деформированных кристаллитов формируются и мигрируют малоугловые границы, то его называют возвратом второго рода, или полигонизацией.
1. Отдых
Наши знания о механизме отдыха базируются главным образом не на прямых структурных наблюдениях поведения дефектов кристаллов, а на результатах изучения кине-
тики изменения электросопротивления, выделения накопленной при наклепе энергии и дру-
гие. 16. Схема изменения физических свойств при отдыхе
гих косвенных данных.
Скорость отдыха максимальна в начальный момент и непрерывно уменьшается с увеличением времени изотермической выдержки (рис. 16). Характерная особенность отдыха — отсутствие инкубационного периода. Изменение свойств начинается с самого начала отжига.
Скорость убывания или приращения (ДХ) какого-то свойства
можно принять обратно пропорциональной времени отдыха (т):
dbXIdx = Кк,
(6)
где К — константа для данной температуры, зависящая от энергии активации процесса Q в соответствии с уравнением Аррениуса:
К = Лехр(-С//?Г). (7)
Отдых — самая низкотемпературная разновидность среди всех явлений самопроизвольного перехода наклепанного металла в более равновесное состояние. Эксперименты с медью и золотом, деформированными при температуре жидкого гелия, показали, что уменьшение электросопротивления начинается уже при температуре около —190°С. С повышением температуры на кривой изменения электросопротивления имеется несколько перегибов, свидетельствующих о разных стадиях отдыха, разных механизмах процессов уменьшения накопленной при деформации энергии.
46
К этим процессам относятся перераспределение точечных дефектов и уменьшение их концентрации, избыточной против равновесной для данной температуры.
Межузельные атомы аннигилируют на краевых дислокациях и прл встрече с вакансиями. Вакансии мигрируют к дислокациям и границам зерен и здесь аннигилируют.
Другие процессы при отдыхе — перегруппировка дислокаций и взаимная аннигиляция дислокаций разного знака. В деформированных зернах дислокации распределены неравномерно. При отжиге из-за термической активации простое и поперечное скольжение и переползание дислокаций на небольшие расстояния приводят к такой их перегруппировке, что энергетические пики сглаживаются. Во время перегруппировок дислокации разного знака, встречаясь, аннигилируют, и общая плотность дислокаций несколько снижается.
На стадии отдыха все перемещения дислокаций носят локальный характер.
Прямое наблюдение дислокационной структуры при комнатной температуре в алюминиевой фольге, деформированной непосредственно в электронном микроскопе, показало, что при отдыхе происходит лишь небольшое перераспределение дислокаций, а плотность дислокаций существенно не уменьшается. В технических металлах и сплавах- дислокации закреплены примесями, что дополнительно затрудняет их перемещение в интервале температур отдыха. .
По всей видимости, после холодной обработки давлением наиболее важным структурным изменением при отдыхе металлов и сплавов технической чистоты является уменьшение избыточной концентрации вакансий.
Скорость стока избыточных точечных дефектов зависит от температуры и энергии активации диффузии. Энергия активации диффузии Q у металлов растет линейно с повышением температуры плавления. Приняв Q=aTn^ получаем, что ехр (—Q//?T)=exp (—аТпл/#Т). Отсюда ве-величина ехр (—Q/RT) должна быть примерно одинаковой для всех металлов при температуре, составляющей одну и ту же долю от температуры павления. Следовательно, в соответствии с уравнениями (6) и (7) скорость отдыха при одинаковой гомологической температуре у разных металлов находится примерно на одинаковом уровне. Чем выше температура плавления, тем выше должна быть температура отжига для достижения одного и того же эффекта отдыха. Так, в холоднокатаных меди и никеле
47
большая часть избыточных вакансий, возникших при деформации, исчезает во время отдыха соответственно при 20 и 100 °C. Эти температуры соответствуют гомологической температуре 0,21 Тлл, Для алюминия комнатная температура— более высокая по гомологической шкале (0,31 Тпл). Поэтому после холодной деформации алюминия отдых при комнатной температуре приводит к практически полному исчезновению всех избыточных вакансий.
Атомы примесей и легирующих элементов могут стать ловушками точечных дефектов. Например, энергия искажений решетки вокруг примесного атома уменьшается, если в область искажений попадает вакансия. Выигрыш в энергии обеспечивает взаимное притяжение вакансии и примесного атома. Кроме упругого, между ними существует и электростатическое притяжение. Атомы примесей и легирующих элементов в твердом растворе, затрудняя перемещение вакансий, уменьшают скорость отдыха.
2. Полигонизация
Для лауэграммы деформированного монокристалла характерен астеризм — радиальная вытянутость рентгеновских пятен. Астеризм обусловлен тем, что ориентация решетки в деформированном, например изогнутом, кристалле непрерывно меняется, и соответствующие рентгеновские рефлексы оказываются размытыми. Отжиг в определенных условиях приводит к расщеплению размытого пятна на ряд пятен, причем общие очертания каждого исходного пятна астеризма сохраняются. Этот эффект, впервые обнаруженный С. Т. Конобеевским и И. И. Мирер в 1932 г. при отжиге изогнутых кристаллов каменной соли, можно трактовать как самопроизвольное разделение кристалла во время возврата на слегка разориентированные фрагменты (блоки), внутри которых кристаллографические плоскости выпрямлены. Каждый блок дает свой четкий рефлекс на лауэграмме. Малые расстояния между этими рефлексами и сохранение общих очертаний исходного пятна астеризма указывают на малую угловую разориентировку блоков.
Для характеристики дорекристаллизационного отжига, при котором зерна металла подразделяются на части, слегка различающиеся между собой по кристаллографической ориентировке, в 1933 г. Е. Ф. Бахметьев, А. А. Боч-вар, Г. С. Жданов и Я. С. Уманский предложили название «возврат второго рода» в отличие от возврата первого рода, не сопровождающегося образованием субзерен.
48
В 1949 г. английский металлофизик Р. Кан обнаружил, что изогнутый монокристалл цинка при отжиге разбивается на блоки, причем криволинейная ось изогнутого кристалла разбивается на отрезки, являющиеся сторонами многоугольников. Это явление было названо полигонизацией (poligon — многоугольник).
Теория дислокаций позволила объяснить механизм полигонизации. Остаточный изгиб кристалла связан с избытком краевых дислокаций одного знака (рис. 17, а). Соответствующие им неполные вертикальные атомные плоскости, выходящие на верхнюю грань кристалла, действуют как клинья, изгибающие кристалл. При отжиге дислокации
Рис. 17. Схема полигонизации:
а — хаотичное распределение краевых дислокаций в изогнутом кристалле; б — стенки из дислокаций после полигонизации
одного знака перераспределяются и выстраиваются одна над другой в стенки (рис. 17, б). При этом под областью разрежения от одной дислокации оказывается область сгущения от другой дислокации и поля упругих напряжений дислокаций в значительной мере взаимно компенсируются. Стенка из дислокаций не имеет дальнодействующе-го поля напряжений. Следовательно, образование дислокационных стенок — энергетически выгодный процесс, который должен идти самопроизвольно, однако для его развития необходима термическая активация.
Дислокационные стенки в изогнутом кристалле образуются в результате сочетания процессов скольжения и пе-
4 И. И. Новиков
49
реползания дислокаций. Из простого сопоставления рис. 17, а и б видно, что только скольжением в горизонтальных плоскостях дислокации не могут установиться одна над другой в виде вертикальной стенки. Для этого необходимо переползание, а оно состоит в достраивании или растворении кромок неполных атомных плоскостей и обеспечивается медленным диффузионным процессом. Скорость переползания — наиболее медленного процесса — определяет скорость выстраивания дислокаций в стенки.
Стенка из дислокаций одного знака является малоугловой границей, разделяющей соседние субзерна с небольшой разориентировкой решеток. Таким образом, при возврате субзерна возникают вследствие выстраивания дислокаций в стенки — малоугловые границы. Нагрев здесь необходим, чтобы активировать переползание большого числа дислокаций. Температура отжига для полигонизации должна быть выше, чем только для отдыха.
Простые границы наклона, состоящие из одних краевых дислокаций, и соответствующие им субзерна в виде параллельных пластинок, проходящих через весь кристалл, наблюдают обычно только при отжиге после деформации, когда действует одна система скольжения. В поликристал-лических металлах при средней и большой пластической деформации всегда скольжение идет по разным системам. Поэтому в них при отжиге образуются субзеренные границы, состоящие из смешанных дислокаций. Такие границы образуются в результате простого и поперечного скольжения и переползания дислокаций, причем самым медленным является переползание.
Угловая разориентировка соседних субзерен и в этом случае обусловлена избытком в субгранице дислокаций одного знака. Тело субзерен свободно или почти свободно от дислокаций. Субзеренная структура отличается от ячеистой тем, что ячейки окружены объемными, часто довольно размытыми стенками, состоящими из дислокационных сплетений (см. рис. 9), а субзерна окружены границами в виде плоских дислокационных сеток (рис. 18).
Если при пластической деформации возникла ячеистая структура (см. рис. 9), то полигонизация при отжиге состоит не в формировании субзерен из хаотично расположенных дислокаций, а в развитии имеющейся ячеистой структуры. Размытые, плохо оформленные ячейки полностью окружаются границами, объемные стенки ячеек становятся более узкими, плоскими, тело ячеек еще больше очищается
50
от дислокаций и ячейки постепенно превращаются в хорошо оформленные равноосные субзерна.
Подобрав травитель, можно металлографически выявить границы субзерен. Под микроскопом они обнаруживаются в виде цепочек ямок травления, каждая из которых соответствует месту выхода на поверхность шлифа дислокации внутри малоугловой границы. Чаще всего субзеренные границы видны на шлифах в виде сетки тонких линий внутри зерен, границы которых выявляются в виде более толстых линий.
Субзерна, образовавшиеся при полигонизации, с увеличением времени и повышением температуры отжига стремятся укрупниться. Экспериментально установлены два механизма этого укрупнения — миграция субграниц и коалесценция субзерен.
Рис. 18. Полигоиизоваиная структура сплава АМгб» отожженного прн 325 °C» 30 мнн после холодной прокатки с обжатием 45 %. Электронная микрофотография, фольга (В. В. Истомин)
На рис. *19 схематично изображен Y-образный стык трех субзереи, разделенных границами Р, Р' и Р". Две близко расположенные одна к другой дислокационные стенки Р' и Р" срастаются, продолжая стенку Р (тройной стык смещается вверх). При слиянии субграниц и перемещении тройного стыка два субзерна растут за счет третьего, а разориентировка субзерен около образующейся границы равна сумме разориентировок около исходных субгранцц.
4»
51
роль.
•Движущей силой этого процесса является стремление кристалла уменьшить энергию субграниц.
На рис. 19 стрелками показаны направления, по которым должны смещаться дислокации, участвующие в процессе слияния границ Р' и Р". Ясно, что это слияние наступает в результате сочетания скольжения и переползания. Одно скольжение не может привести к равномерному распределению дислокаций после слияния стенок. Следовательно, в укрупнении субзерен миграций субграниц, как и в их формировании на первых этапах полигонизации, переползание— самый медленный процесс — играет ведущую
Укрупнение субзерен путем коалесценции наблюдали при отжиге фольги непосредственно в колонне электронного микроскопа. Прямые наблюдения выявили постепенное размывание субграниц и исчезновение контраста между субзернами. А это значит, что субзерна приобретают одинаковую кристаллографическую ориентировку.
Стадии коалесценции двух субзерен показаны на рис. 20. Совершенно очевидно, что исчезновение субграницы должно сопровождаться некоторым поворотом решетки одного (рис. 20,6) или обоих субзерен, чтобы образовалась единая ориентация решетки в области, в которой ранее находились слегка разориентированные субзерна.
Граница между субзернами постепенно исчезает, так как дислокации уходят из нее в субграницы, окружающие эти субзерна. Поэтому иногда говорят, что граница «рассыпается». Предполагают, что в рассыпании субграницы главную роль играет переползание дислокаций.
Из схемы на рис. 20, б видно, что поворот субзерна при коалесценции возможен только в том случае, если атомы в окружающих его субзернах уйдут из заштрихованных участков. Следовательно, объемная диффузия является процессом, контролирующим коалесценцию. Скорость коалесценции пропорциональна коэффициенту самодиффузии и обратно пропорциональна кубу диаметра субзерен. Если субзерна крупные, то атомам необходимо диффундировать на большие расстояния. При очень больших или вытянутых субзернах механизм укрупнения их вследствие коалесценции может не срабатывать.
Л
Р
Рис. 19. Схема срастания соседних малоугловых границ, приводящего к укрупнению субзерен
52
Исчезновение субграницы, с которой всегда связан избыток энергии, — самопроизвольный процесс. Вместе с тем если при исчезновении субграниц возрастает угловая ра-зориентация у границ, окружающих сливающиеся субзерна, то энергия этих границ возрастает. Если у таких границ угловая разориентация больше, чем у исчезающей границы, то термодинамический стимул коалесценции сохраняется. Дело в том, что энергия границы Е с увеличением угла разориентации <р растет с затуханием и при одинаковом А<р в области больших углов ДЕ будет меньше, чем в области малых углов. Поэтому уменьшение энергии исчезающей малоугловой границы, происходящее в области малых углов, перекрывает повышение энергии границ с большим углом разориентации.
Рис. 20. Стадии коалесценции двух субзереи (схема Ли):
а — структура до коалесценции; б — поворот одного субзерна; в — структура сразу же после коалесценции; г — конечная структура после выпрямления субграниц вследствие их миграции
Наблюдение за структурой фольг, отжигаемых в колонне электронного микроскопа, выявило, что одновременно может рассыпаться несколько границ, разделяющих соседние субзерна, т. е. возможна не только парная, но и групповая коалесценция.
С увеличением выдержки и повышением температуры полигонизации субзерна могут вырасти до весьма больших размеров (~10 мкм). При этом их рост происходит в пре
53
делах кристаллографической ориентировки исходных деформированных зерен — очертания пятен астеризма на лауэграмме сохраняются. Такая далеко зашедшая полигонизация получила название рекристаллизации на месте (in situ — латинск.). Но рекристаллизация на месте не является разновидностью обычной первичной рекристаллизации, для которой характерно образование новых зерен, отделенных от матрицы высокоугдовыми границами. Рекристаллизация на месте по своей природе является не рекристаллизационным, а полигонизационным процессом.
Рост субзерен при полигонизации, связанный с увеличением избытка дислокаций одного знака в субграницах, как уже отмечалось, приводит к увеличению углов разориентировки соседних субзерен. Однако на стадии полигонизации, в том числе при рекристаллизации на месте, границы все время остаются малоугловыми, т. е. их строение описывается дислокационной схемой, например такой, как на рис. 17, б, а угол разориентировки соседних субзерен не превышает 10—15° (чаще всего соседние субзерна разориентированы на угол не более 1°).
Так как полигонизация состоит в постепенном формировании субзерен путем образования дислокационных стенок из отдельных дислокаций и в росте субзерен или же (при ячеистой структуре в деформированном металле) в развитии имеющихся ячеек и постепенном превращении их в субзерна, то температура начала полигонизации не является четко определенной физической константой, такой, например, как точка плавления.
Важнейшая характеристика металла — энергия дефектов упаковки — сильно влияет на склонность к полигониза-' ции. Чем меньше энергия дефектов упаковки, тем больше * ширина растянутых дислокаций и труднее проходят процессы переползания и поперечного скольжения, необходимые для полигонизации. Поэтому в алюминии, имеющем высокую энергию дефектов упаковки и, следовательно, слабо расщепленные дислокации, полигонизация идет сравни-. тельно легко. В меди она протекает труднее, а в а-латуни : с низкой энергией дефектов упаковки полигонизация г ^обычно не наблюдается.
Атомы примесей тормозят полигонизацию из-за образования атмосфер Коттрелла, затрудняющих перераспределение дислокаций скольжением и переползанием. При одинаковой температуре отжига более чистый металл полиго-низуется за более короткое время.
54
§ 7. Первичная рекристаллизация (рекристаллизация обработки)
Начиная с определенной температуры при отжиге холод-нодеформированного металла происходят сильные изменения микроструктуры, которые относятся к процессу, называемому рекристаллизацией. Наряду с вытянутыми деформированными зернами даже при небольших увеличениях светового микроскопа можно различить новые, более или менее равноосные рекристаллизованные зерна. По мере увеличения времени или. температуры отжига площадь шлифа, занятая новыми зернами, возрастает, а старые деформированные зерна постепенно исчезают. Рентгеновский анализ, а позднее электронная микроскопия фольг показали, что новые равноосные зерна отличаются от старых вытянутых зерен деформированной матрицы не только формой, но и, что гораздо важнее, более совершенным внутренним строением, резко пониженной плотностью дислокаций. Если плотность дислокаций в сильнодеформиро-ванном металле составляет 1011—1012 см~2, то после прохождения рекристаллизации она снижается до 106— 108 см~2.
В отличие от полигонизованной структуры, которая также более совершенна, чем деформированная матрица, рекристаллизованные зерна отделены от матрицы не малоугловыми, как субзерна, а высокоугловыми границами. Это различие имеет принципиальное значение.
Благодаря быстрой миграции высокоугловых границ рекристаллизованные зерна интенсивно «поедают» деформированную матрицу. Субзерна на стадии полигонизации имеют ориентацию деформированного кристаллита, а рост рекристаллизованного зерна, окруженного высокоугловой границей, может быть связан с сильной переориентацией кристаллической решетки.
Образование и рост зерен с более совершенной структурой, окруженных высокоугловыми границами, за счет исходных деформированных зерен той же фазы называют первичной рекристаллизацией или рекристаллизацией обработки.
Кинетика рекристаллизации похожа на кинетику фазового превращения — в обоих случаях в изотермических условиях она изображается кривой сигмаидального вида, характер которой объяснен в § 21 (сравните рис. 21 и 68).
При анализе кинетики первичной рекристаллизации оперируют теми же параметрами с. з. ц. и л. с. р., что и в
55
кинетике фазовых превращений. Скорость зарождения центров рекристаллизации (с. з. ц.) — это число центров рекристаллизованных зерен, возникающих в единицу времени в единице объема. Линейная скорость роста (л. с. р.) является скоростью перемещения границы зерна. С ростом температуры оба параметра экспоненциально увеличиваются.
Некоторое время новые зерна не обнаруживаются. Это время называют инкубационным периодом. С увеличением степени деформации и температуры отжига инкубационный период первичной рекристаллизации уменьшается.
Рис. 21. Развитие первичной рекристаллизации при разных температурах в алюминии чистотой 99,96 % после растяжения на 10 % (Андерсон И Мэйл)
Кинетика первичной рекристаллизации резко отличается от кинетики возврата. Если возврат не имеет инкубационного периода, скорость его максимальна в начальный период и непрерывно уменьшается во время изотермической выдержки (см. рис. 16), то рекристаллизация, наоборот, начинается после инкубационного периода, а скорость ее (приращение рекристаллизованного объема в единицу времени) нарастает от нуля до максимума, а затем снижается. Затухание рекристаллизации вызвано прекращением роста все большего числа новых зерен из-за их сопрокос-новения между собой.
Кинетика первичной рекристаллизации при постоянной температуре весьма удовлетворительно описывается уравнением Аврами:
VT/VO = 1-ехр (-Вт*), (8)
где Vt/Vo — доля объема, рекристаллизованного за время т; В — коэффициент, зависящий от с. з. ц. и л. с. р., т. е.
56
от температуры и степени деформации; k находится в интервале от 1 до 4,
Термодинамическим стимулом первичной рекристаллизации является накопленная при пластической деформации энергия, связанная с дислокациями, которые образуют хаотичные скопления, объемные стенки ячеек или же плоские субграницы. Уменьшение плотности дислокаций при первичной рекристаллизации приводит к высвобождению основной доли этой накопленной энергии, что обнаруживается при калориметрических исследованиях.
Уменьшение энергии в объеме кристаллов, происходящее в процессе повышения их структурного совершенства, перекрывает возрастание поверхностной энергии при любых, даже самых малых размерах рекристаллизованного зерна. Совсем не обязательно, чтобы зерна в рекристаллизованном металле были крупнее, чем в деформированном. К моменту окончания первичной рекристаллизации- суммарная поверхность равноосных зерен, выросших из множества центров, может быть больше суммарной поверхности вытянутых деформированных зерен. Несмотря на это, энергия Гиббса рекристаллизованного металла меньше, чем деформированного, из-за уменьшения общей плотности дислокаций внутри зерна.
1. Механизм зарождения центров рекристаллизации
Главным в механизме зарождения рекристаллизованных зерен в любых материалах и в любых условиях является формирование окруженного высокоугловыми границами участка с высоким структурным совершенством. В зависимости от особенностей строения деформированного материала и процессов возврата, протекающего на начальных стадиях отжига, конкретные места и механизмы зарождения центров рекристаллизации бывают весьма разными.
Центры рекристаллизации могут появляться: а) непосредственно на границах зерен деформированного материала и б) внутри зерен в участках сильного локального изменения ориентировки решетки (в переходных полосах, полосах сдвига, полосах сброса, около крупных включений).
а. Зародыши рекристаллизации непосредственно на границах деформированных зерен образуются при миграции небольших (размером порядка 1 мкм) участков готовой высокоугловой границы с образованием выступов, «языков» (рис. 22). Движущей силой такой миграции яв-
57
Рис. 22. Схема образования выступа иа границе путем ее миграции, вызванной наклепом (Бэк)
ляется разность в объемной энергии наклепанных участков по обе стороны от границы, которая продвигается в зерно с большей плотностью дислокаций, образующих хаотичные сплетения, плоские нагромождения, стенки ячеек или же субграницы в полигонизованной структуре (на рис. 22 разная суммарная плотность дислокаций и соответственно разная объемная энергия по обе стороны от высокоугловой границы обусловлена неодинаковой протяженностью границ ячеек). Продвигающаяся граница «выметает» на своем пути дефекты решетки в деформированном зерне. Увеличение поверхностной энергии при образовании «языка» перекрывается уменьшением накопленной при деформации энергии.
Участком, от которого начинается выгибание высокоугловой границы, может быть сравнительно крупное и совершенное субзерно. Образованию выступа может предшествовать также коалесценция субзерен вблизи высокоугловой границы (рис. 23), Микровыступ, «проникший» в соседнее зерно, отличаясь от него пониженной плотностью дислокаций и соответственно меньшей объемной энергией, способен к быстрому росту во все стороны путем миграции своей высокоугловой границы. На микроучастке размером порядка 1 мкм, откуда началось формирование рекристаллизованного зерна, его граница может остаться малоугловой или же здесь постепенно будет набрана высокоугловая разориентировка.
Механизм зарождения рекристаллизованных зерен путем вызванной градиентом наклепа миграции отдельных участков уже существующей высокоугловой границы наблюдали в алюминии, меди, серебре, никеле и железе после малых и средних деформаций.
б. Зарождение центров рекристаллизации внутри деформированного зерна связано с локальными изменениями кривизны решетки. Эту связь помогает понять схема на рис. 24, на которой процесс образования зародыша сведен к тому, что сравнительно плавное, хотя и сильное изменение кристаллографической ориентации (рис. 24, а), как бы сгущается, сосредоточиваясь в виде высокоугловых границ (рис. 24, б). Внутри же зародыша ориентация совершенной
58
решетки близка к средней ориентации в исходном участке с искривленной решеткой. Именно поэтому в таком участке и зародилось рекристаллизованное зерно, отличающееся по ориентации от деформированного.
Рассмотренная схема очень формализована, но она правильно отражает возникновение зародыша в местах с сильной локальной кривизной решетки деформированных зерен.
Рис. 23. Схема формирования выступа коалесценцией субзереи около высокоугловой границы ВГ:
а — до коалесценции; б — после коалесценции субзерен и миграции участка высокоугловой границы
При средних и особенно больших деформациях основным местом зарождения центров рекристаллизации являются переходные полосы и полосы сдвига (роль включений рассмотрена в § И, п. 3). Зародышами, способными превратиться в центры рекристаллизации, являются фрагменты микрополос, образовавшихся при холодной деформации (см. § 4, п. 2), или же наиболее крупные субзерна, сфор
мировавшиеся при полигонизации, которая прошла на
ранней стадии отжига.
Отдельные микрополосы растут за счет соседних микрополос, утолщаясь в направ-линии поперек переходной полосы. Это утолщение происходит путем выпучивания отдельных участков продольной границы микрополосы. Поскольку поперек переходной полосы существует сильное изменение ориентировки решетки в деформированном зерне, утол
а 6
Рис. 24. Схема образования зародыша рекристаллизации в полосе сброса (Шьюмон): а — изгиб решетки в полосе сброса до образования рекристаллизованного участка; б — зародыш рекристаллизации
щение микрополосы постепенно приводит к превращению малоугловой границы в высокоугловую, т. е. к формированию зародыша рекристаллизации. Этот зародыш, образовавшийся из длинной микрополосы, сам на начальной стадии имеет сильно вытянутую форму: его длина
59
Рнс. 25. Вытянутые рекристаллизованные зерна (светлые), образовавшиеся в переходных полосах и полосах сдвига на начальной стадии рекристаллизации меди, отожженной при 100 °C после холодной прокатки с обжатием 90 % (Ридха, Хачннсон);
R — направление прокатки; N — нормаль к плоскости прокатки
вдоль переходной полосы может составлять 20—50 мкм, а поперек — всего несколько микрометров. Последующая быстрая миграция высокоугловой границы в поперечном направлении делает новое зерно сравнительно равноосным.
На рис. 25 показаны вытянутые зерна, образовавшиеся на начальной стадий рекристаллизации в переходных полосах вдоль направления прокатки и в полосах сдвига в виде елочки.
С повышением степёни деформации растет число переходных полос и полос сдвига и соответственно увеличивается число центров рекристаллизации.
Другая возможность зарождения центров рекристаллизации — полигонизация в первый период отжига. Она может в результате ускоренной миграции некоторых субграниц или же групповой коалесценции субзерен приводить к появлению субзерна, отличающегося от соседнего существенно большим размером и большей разориентировкой. Такое субзерно получает преимущество в росте. Если оно находится на участке с повышенной кривизной решетки, т. е. с избытком дислокаций одного знака, то его ускоренный рост приводит к встраиванию в субграницу все большего числа конечном итоге, к превраще
нию субзерна в собственно центр рекристаллизации, окруженный высокоугловой границей.
При реализации любого из рассмотренных механизмов зародышеобразования ведущую роль играют диффузионные процессы, в частности диффузия, необходимая для миграции субграниц и коалесценции субзерен (см. § 6). Поэтому образование центров рекристаллизации — термически активируемый процесс, ускоряющийся с ростом температуры.
С. С. Горелик обратил внимание на то, что в процессе формирования центров рекристаллизации перераспределе
дислокаций одного знака и, в
60
ние и частичная аннигиляция дислокаций разного знака приводят к временному повышению концентрации вакансий, как бы «впрыскиванию» вакансий в металл.
Возврат, предшествующий рекристаллизации, влияет на зародышеообразование, причем влияние это двойственное.
Отдых всегда должен в той или иной степени затруднять зарождение центров рекристаллизации, так как уменьшение концентрации вакансий при отдыхе замедляет диффузионные процессы, контролирующие скорость формирования центров рекристаллизации.
Более сложно влияние полигонизации. Если в слабоде-формированном металле нет значительных локальных изменений кривизны решетки и по всему объему одновременно и с примерно одинаковой скоростью формируются и растут субзерна, то они могут достичь большого размера и высокого совершенства, а границы между ними остаются малоугловыми и малоподвижными. Такая полигонизация затрудняет рекристаллизацию из-за снижения накопленной энергии (термодинамического стимула рекристаллизации).
Далеко зашедшая полигонизация (рекристаллизация на месте) приводит к образованию столь совершенной субструктуры со стабильной сеткой малоугловых границ, окружающих крупные субзерна, что первичная рекристаллизация не наблюдается вплоть до точки плавления. Поэтому в данном случае, характерном для отжига после малых деформаций, можно говорить, что полигонизация — это процесс, конкурирующий с рекристаллизацией.
Если же в деформированном металле имеются участки с большой локальной кривизной решетки, то ускоренный рост субзерен в предпочтительных местах приводит к образованию крупных субзерен, которые растут за счет соседней, быстро «набирают» угловую разориентировку на своей границе и превращаются в центры рекристаллизации. Такая полигонизация является начальным этапом рекристаллизации, что характерно для отжига поликристаллов после средних и больших деформаций.
Полигонизацию, приводящую к формированию центров рекристаллизации, С. С. Горелик предложил называть предрекристаллизационной в отличие от стабилизирующей полигонизации, затрудняющей зарождение рекристаллизованных зерен.
2. Температура начала рекристаллизации
При разработке режимов термической обработки и для других целей необходимо уметь экспериментально фиксировать начало появления рекристаллизованных зерен сравнительно простым методом. При решении большинства прикладных задач условное начало рекристаллизации определяют с помощью световой микроскопии по появлению первых, обычно более светлых равноосных зерен на фоне сильнее травящейся деформированной матрицы или рентгеновским методом по появлению точечных пятен («уколов») на размытых интерференционных линиях рентгенограммы. Каждое такое пятно соответствует отражению рентгеновских лучей от рекристаллизованного зерна размером 2— 5 мкм. Световая микроскопия надежно выявляет рекристаллизованные зерна после достижения ими размера 10— 50 мкм. Иногда начало рекристаллизации определяют по началу интенсивного падения твердости или временного сопротивления. Но, как будет показано в § 12, этот метод .щшгоден не для всех материалов.
Температура появления рекристаллизованных зерен, т. е. температура начала рекристаллизации (/”), не явля? ется физической константой, такой как точка плавления.
В отличие от температуры фазового равновесия температура начала рекристаллизации зависит от времени отжига, что вытекает из анализа рис. 21. Если время отжига составляет 1000 мин, то при 325, 350 и 370 °C первичная рекристаллизация успевает полностью закончиться, а при 310 °C она еще только начинается. При времени отжига 10 мин начало рекристаллизации фиксируется при 370 °C, а при более низких температурах новые зерна не обнаруживаются. Следовательно, при длительности' отжига 1000 мин £”=310 °C, а при 10 мин t* =370 °C.
Другой фактор, сильно влияющий на температуру начала рекристаллизации, — степень деформации при обработке давлением. С увеличением степени деформации температура начала рекристаллизации снижается (рис. 26). Объясняется это тем, что с увеличением степени деформации растут плотность дислокаций и энергия, накопленная при деформации, т. е. возрастает термодинамический стимул рекристаллизации.
Так как плотность дислокаций и соответственно накопленная при деформации энергия с увеличением степени деформации растут с затуханием, то и температура начала
62
рекристаллизации снижается при увеличении степени деформации с затуханием, достигая определенного предела при данном времени отжига (см. рис. 26).
С увеличением времени отжига сильнодеформированно-го металла температура начала его рекристаллизации сни-
жается с затуханием, достигая приближенно постоянной величины через 1—2 ч. отжига. Наинизшая темпе-; ратура начала рекристал-, лизации металла или спла--; ва, соответствующая боль-5 шим деформациям (более-
Рис. 26. Влияние степени деформации на температуру начала рекристаллизации
60—70%) и времени от-1 жига 1-^2 ч, в определен-; ной мере может служить) характеристикой материа?' ла. Будем называть ее да!-лее температурным поро± гом рекристаллизации (/п.р на рис. 26) в отличие от^ любых других, более высоких значений
С повышением чистоты металла температурный порог
рекристаллизации снижается:
Чистота А1, % . . . . 99,7 99,9 99,99 99,9992
/п.р, °C................. 240 200 100 —45
А. А. Бочвар показал, что между температурным порогом рекристаллизации и температурой плавления металлов имеется простое соотношение: рекристаллизация начинается при температуре, составляющей одинаковую для всех металлов долю от температуры плавления по абсолютной шкале, а именно
Тп.р = (0,3-.-0,4)ТпЯ. (9)
Коэффициент 0,3—0,4 в этой формуле относится к металлам сравнительно высокой технической чистоты (около 99,99%). Для особо чистых металлов 7п.р= (0,25-4-<-0,3) Тпл.
По правилу А. А. Бочвара можно оценить в первом приближении температурный порог рекристаллизации по известной температуре плавления металла. Это бывает необходимо, когда нет справочных данных. Например, можно оценить температуру начала рекристаллизации свинца: (327+273)0,4—273=—33 °C.
63
Измельчение исходного (до деформации) зерна приводит к понижению температуры начала рекристаллизации, так как в более мелкозернистом металле больше суммарная площадь высокоугловых границ, где зарождаются центры рекристаллизации, и больше накопленная при деформации энергия.
Влияние состава сплава на температуру начала рекристаллизации
Зависимость температуры начала рекристаллизации от состава в двойных системах немонотонная и различная у разных сплавов. На рис. 27 показан один из типичных вариантов этой зависимости. Только в однофазной области в интервале малых концентраций наблюдается простая закономерность — малые добавки почти всегда повышают
Рис. 27. Зависимость температуры начала рекристаллизации-от состава сплавов в двойной системе (схема). Пунктир — граница однофазной области
димому для формирования Примеси тормозят не только
температуру начала рекристаллизации, причем с увеличением их концентр ации тем пер атура начала рекристаллизации возрастает с затуханием (участок ab на рис. 27). /
Атомы добавки упруго притягиваются к дислокациям, образуя атмосферы Коттрелла. Эти примесные атмосферы цешают перераспределению дислокаций, необхо-центров рекристаллизации, зарождение, но и рост цент-
ров рекристаллизации, так как они притягиваются к границе зародыша. При нагреве металла до более высоких температур примесные атмосферы размываются усиливающимся тепловым движением, в результате чего становится возможным формирование центров рекристаллизации и облегчается их рост. С увеличением концентрации чужеродных атомов участки вблизи дислокаций насыщаются ими, и поэтому возрастание температуры начала рекристаллизации затухает.
Энергия упругого притяжения примесного атома с радиусом /?п к краевой дислокации пропорциональна фактору размерного несоответствия (7?п—7?о)//?о, где /?о—радиус атома основы в случае раствора замещения, а в слу-
64
чае раствора внедрения — радиус такого жесткого шара, который, будучи внесен в то место решетки, где расположен примесный атом, не вызовет объемных искажений. Поэтому температура начала рекристаллизации растет тем сильнее, чем больше фактор размерного несоответствия атомов добавки.
Чем чище исходный металл, тем сильнее действие добавляемого элемента, так как в более грязном исходном металле на дислокациях уже имеются примесные атмосферы, затрудняющие рекристаллизацию.
В тех редких случаях, когда малая добавка понижает температуру начала рекристаллизации, это может быть результатом того, что исходный металл был загрязнен примесями, сильно повышающими температуру начала рекристаллизации, а добавляемый легирующий элемент связывает эти примеси в соединения и выводит их из твердого раствора.
Во многих системах по достижении максимума в области сравнительно небольших концентраций (десятые доли процента — проценты) температура начала рекристаллизации снижается. Это снижение можно объяснить влиянием легирующего элемента на структуру деформированного сплава. Если добавка уменьшает энергию дефектов упаковки твердого раствора, то дислокации сильно расщеплены, поперечное скольжение их затруднено и при пластической деформации образуются участки с большим избытком дислокаций одного знака. В таком твердом растворе температура начала рекристаллизации должна быть ниже, чем в менее легированном сплаве, где энергия дефектов упаковки более высокая.
Влияние состава на величину в области больших концентраций обусловлено главным образом изменением сил межатомной связи. Так, если при легировании температура плавления сильно понижается и силы межатомной связи ослабевают, то температура начала рекристаллизации в области больших концентраций твердого раствора непрерывно снижается (участок Ьс на рис. 27).
По сравнению с действием малых добавок на чистейший металл, когда сотые и десятые доли процента добавки могут повысить температуру начала рекристаллизации на сто градусов и более, увеличение содержания легирующих элементов в области больших концентраций на проценты и десятки процентов сравнительно слабо изменяет температуру начала рекристаллизации (максимум на десятки градусов). Соотношение температур начала рекристалли-
5 И. И. Новиков
зации и плавления у твердых растворов выше, чем у чистых металлов, причем в однофазных сплавах — растворах величина Тр/ТдЛ составляет максимум 0,6 (по сравнению с 0,25—0,4 у металлов).
При переходе через границу растворимости в двухфазную область температура начала рекристаллизации может несколько понизиться (участок cd на рис. 27). Это снижение обусловлено тем, что около частиц p-фазы, если они достаточно крупные, в a-матрице накапливаются дислокации и облегчается зарождение рекристаллизованных зерен (см. § 11, п. 3). При дальнейшем увеличении содержания второго компонента в сплаве возрастает объемная доля р-фазы и расстояние между ее кристаллами становится столь малым, что тормозятся формирование и рост зародышей рекристаллизации в a-фазе (участок de на рис. 27). __ Рассмотренный на рис. 27 вариант влияния состава сплава на температуру начала рекристаллизации предполагает, что сплавы перед холодной деформацией были с помощью отжига приведены в равновесное состояние. Если же в сплавах, состав которых находится правее границы растворимости на диаграмме состояния, закалкой (перед холодной деформацией) получить пересыщенный твердый раствор, тощего распад может сложным образом повлиять на температуру начала рекристаллизации. Выделения из пересыщенного раствора могут затруднять перераспределение дислокаций и миграцию малоугловых границ, т. е. мешать формированию центров рекристаллизации. Результат будет зависеть от того, проходит ли распад пересыщенного раствора до начала рекристаллизации или одновременно с ней, а также от типа и дисперсности выделений. Если распад раствора прошел до начала рекристаллизации, но при повышении температуры протекает коагуляция или растворение выделений, то их тормозящее влияние на рекристаллизацию ослабевает. В целом же у стареющих сплавов температура начала рекристаллизации выше, чем у близких к ним по составу однофазных сплавов, и находится в интервале (0,6—0,8) Гпл-
3. Температура конца рекристаллизации
Температуру полного исчезновения деформированной матрицы при данном времени отжига называют температурой конца рекристаллизации t*. Экспериментально ее определяют с помощью световой микроскопии — по исчезновению сильнее травящихся остатков деформированной матрицы
—66
и рентгенографически — по исчезновению на рентгенограмме размытых дебаевских колец. Кроме того, по рентгенограмме можно подсчитать число пятен — рефлексов от рекристаллизованных зерен. Максимальное число таких рефлексов соответствует концу первичной рекристаллизации, так как начинающаяся вслед за ней собирательная
рекристаллизация уменьшает число рекристаллизованных
зерен. —
При непрерывном наблюдении за первичной рекристаллизацией меди и алюминиевой бронзы с помощью фото-эмиссионного электронного микроскопа было обнаружено,
что в некотором интервале температур рекристаллизация не заканчивается полным исчезновением деформированных зерен: сигм аи дальние кривые аналогичны кривым на рис. 21, но они выходят на горизонталь, соответствующую большой доле объема перекристаллизованного металла. Следовательно, в определенном интервале температур первичная рекристаллизация достигает стадии «насыщения». Чем выше температура, тем больше макси-
$160 200 2^0^260 320 360
Тенпература°С
Рис. 28. Зависимость доли рекристаллизованного объема на стадии «насыщения» (QH) от температуры отжига меди, холоднотянутой с деформацией 48 % (Комт н Форм)
мальная доля рекристаллизо-
ванного объема, соответствующая этой стадии (рис. 28). Экстраполяция кривых на рис. 28 до значений QH=0% и Q.h=100% дает значения температур начала ($) и конца (/р) рекристаллизации. Выше температуры /р при достаточно длительной выдержке рекристаллизация доходит до конца.
На температуру конца рекристаллизации влияют все те факторы, которые изменяют температуру начала рекристаллизации. Чем выше /£, тем чаще всего (но не всегда) выше и /р.
4. Рост зерен при первичной рекристаллизации
Рекристаллизованные зерна растут путем быстрой миграции их границ в сторону деформированной матрицы. Движущей силой такой миграции является разность в упругой энергии кристаллов по обе стороны от границы из-за раз
5*
67
ной плотности дислокаций. В отличие от собирательной рекристаллизации (см. § 8) при первичной рекристаллизации граница зерна мигрирует в сторону от центра кривизны. Движущаяся высокоугловая граница «выметает» на своем пути дефекты решетки деформированной матрицы, увеличивая объем с более совершенной структурой, с меньшей плотностью дислокаций.
Средняя скорость миграции границ зерен при первичной рекристаллизации зависит от температуры:
v = v0e~^T9 (10>
где Q — энергия активации процесса миграции.
Миграцию высокоугловой границы при рекристаллиза
ции можно рассматривать как результат индивидуального
или группового неупорядоченного перехода атомов от деформированного зерна к рекристаллизованному. Атомы по обе стороны от границы при колебательном движении вре-
Рнс. 29. Схема изменения энергии Гиббса прн переходе атома через границу между деформированным (Д) и рекристаллизованным (Р) зернами
мя от времени отрываются от одного зерна и присоединяются к другому. Такой механизм по своей природе — диффузионный (идет направленная самодиффузия атомов перпендикулярно границе).
На рис. 29 изображено изменение энергии Гиббса при переходе атома через границу между зернами. Видно, что благодаря разности в энергии Гиббса AG, связанной с различием в плотности дислокаций по обе стороны от границы, переход атомов от рекристаллизованного к деформированному зерну требует преодоления большего потенциального барьера (Q+AG), чем обратный переход (Q). Соответственно частота
перехода атомов в направлении от деформированного зерна к рекристаллизованному должна быть больше, чем в обратном направле-
нии, что и определяет «поедание» деформированного зерна рекристаллизованным.
§ 8. Собирательная рекристаллизация
По окончании первичной рекристаллизации, когда исчезают деформированные зерна, энергия Гиббса сильно понижена, однако структура остается нестабильной из-за раз-
68
витои поверхности границ рекристаллизованных зерен и неуравновешенности поверхностного натяжения на этих границах.
Рассмотрим зеренное строение металла в виде двухмерной модели. В стыке трех зерен (рис. 30, а) силы поверхностного натяжения должны быть уравновешены. В металле или однофазном сплаве поверхностное натяжение
на всех высокоугловых границах примем примерно одинаковым. Тогда силы поверхностного натяжения у, прило-
женные вдоль границ зерен, будут уравновешены при условии, что y=2ycos0/2 или cos6/2=V2 и 6/2=60°.
Следовательно, равновесной конфигурации отвечает встреча трех зерен с углами при вершине 120°.
В двухмерной модели зерна — правильные шестиугольники с прямыми сторонами, образующие гексагональную сетку границ, соответствуют равновесию сил поверхностного натяжения. Такая сетка границ механически стабильна.
Рис. 31. Плоская модель строения металла из зерен с разным числом границ (стрелки указывают направление миграции границ)
Рнс. 30. Схема границ на стыке трех зерен в разные моменты отжига:
а — до отжига; б — после установления равновесия снл поверхностного натяжения в тройной точке путем искривления
границ; в — после выпрямления границ, нарушившего равновесие в тройной точке
В реальном металле при первичной рекристаллизации зерна приходят в соприкосновение между собой в разные моменты времени в разных точках своей поверхности и по окончании первичной рекристаллизации они имеют неправильную форму, разные размеры и разное число граней.
69
В плоской модели такие зерна изображаются многоугольниками с разным числом сторон, например 3, 4, 6, 10, 50 и т. д. (рис. 31).
Для соблюдения указанного выше условия равновесия сил поверхностного натяжения в тройных стыках зерна с числом сторон меньше шести должны иметь границы, обра-х щенные вогнутостью в сторону зерна, а при числе сторон больше шести границы должны быть обращены вогнутостью в сторону соседей.
Границы искривляются миграцией с одновременным перемещением точки тройного стыка. На рис. 30, б сплошные линии изображают искривленные границы в равновесном тройном стыке после такой миграции, а пунктирные — исходное положение границ в неравновесном тройном стыке. Но искривленные границы нестабильны. Под действием поверхностного натяжения они стремятся уменьшить длину. Каждая граница выпрямляется миграцией в направлении центра ее кривизны. Эти направления на рис. 31 указаны стрелками. На рис. 30, в сплошные линии изображают спрямленные границы, а пунктирные —их исходное положение в равновесном стыке.
Миграция границы направлена к центру кривизны, потому что на вогнутой поверхности у атома больше связей с соседями из своего зерна, чем на выпуклой. Нескомпен-сированный переход атомов через границу и обусловливает рост зерен с вогнутыми границами за счет зерен с выпуклыми границами.
Спрямление границ нарушает достигнутое ранее равновесие в тройных стыках (на рис. 30, в угол при вершине зерна А стал меньше 120°). Для достижения равновесия в тройном стыке границы вновь изгибаются миграцией и т. д. Таким путем зерна Б и В на рис. 30 постепенно растут за счет зерна А.
В плоской модели зеренной структуры поликристалла зерна с числом сторон больше шести должны укрупняться, а с числом сторон меньше шести — «поедаться» соседями. В реальном металле зерно с одной стороны имеет вогнутую поверхность и растет за счет соседа, а с другого края оно может иметь выпуклую поверхность и само поглощаться соседом. В целом же средний размер зерен возрастает.
При изотермическом отжиге зерна укрупняются с затуханием, так как сетка границ приближается к равновесной и движущая сила миграции границ постепенно уменьшается. При анализе структур на шлифах следует иметь в
70
виду, что конфигурация трехмерной равновесной сетки границ зерен более сложная, чем двухмерной.
В пространстве равновесию сил поверхностного натяжения отвечает упаковка из зерен — четырнадцатигранни-ков с несколько изогнутыми границами. Даже если такая идеализированная равновесная конфигурация зерен и будет достигнута при отжиге, то в плоскости шлифа размеры произвольных сечений зерен получаются разными, а углы встречи трех границ будут отличаться от 120°.
Рост одних рекристаллизованных зерен за счет соседних рекристаллизованных зерен путем миграции высокоугловых границ называют собирательной рекристаллизацией L
Термодинамическим стимулом собирательной рекристаллизации является зернограничная энергия, а обязательным условием ее развития — неуравновешенность поверхностного натяжения, стремящегося выпрямить искривленные границы и создать равновесную конфигурацию границ в тройных стыках.
Скорость миграции границы должна быть тем больше, чем сильнее искривлена граница, т. е. чем меньше радиус ее кривизны. Если принять, что радиус кривизны границ равноосного зерна пропорционален его диаметру (г=р£>), то скорость роста зерен при собирательной рекристаллизации должна быть обратно пропорциональна диаметру зерен D и прямо пропорциональна поверхностному натяжению (зернограничной энергии) у:
dDIdx^KylpD, (11)
где К — константа, экспоненциально зависящая от температуры (K=Ae~QfRT).
Интегрируя, получаем
+ (12)
где Do — начальный диаметр рекристаллизованных зерен; т —* время отжига.
Если начальный диаметр зерен намного меньше текущего и Dq <^D2, to
D = (13)
Экспериментальные данные показывают, что- зависи-
1 В переводной и иностранной литературе первичную рекристаллизацию часто называют просто рекристаллизацией, а собирательную рекристаллизацию— ростом зерна (Grain growth — англ.). Последний термин нельзя признать удачным, так как и при первичной рекристаллизации растут новые зерна.
71
мость среднего размера зерен в металлах и однофазных сплавах от времени отжига подчиняется выражению
D = Вхп , (14)
где п=0,14-0,5, а В — константа, зависящая от температуры.
Отклонение показателя степени п от теоретической величины V2 в сторону меньших значений, т. е. менее интенсивный рост зерен, вызвано торможением границ из-за растворенных примесей и включений, а также другими
причинами.
Рис. 32. Выгибание границы зерна между частицами А12О3 при отжиге (1000 °C, 20 ч) после холодной деформации внутреинеокисленного сплава меди с 0,2 % Al. Х50 000 (Е. П. Данелия, М. Д. Теплицкий, В. М. Розенберг)
Атомы растворенных примесей упруго притягиваются к границе, и миграция ее связана с протаскиванием за собой примесных атомов. Мигрирующая граница встречает на своем пути примесные атомы, распределенные в теле «поедаемого» зерна, и примесь на границе накапливается, усиливая ее торможение.
Если мигрирующая граница встречает включения второй фазы, то она должна огибать эти включения и затем отрываться от них (рис. 32). Так как по мере развития собирательной рекристаллизации движущая сила ее уменьшается, то включения могут через некоторое время полностью затормозить рост зерна. Чем больше объемная доля включений f и меньше их
размер d, тем меньше конечный размер зерна DK, достигаемый при отжиге:
DK^d/f.
(15)
Следовательно, наибольшее препятствие росту зерен при собирательной рекристаллизации представляют высокодисперсные включения с большой объемной долей их в сплаве, а барьерный эффект от отдельных крупных включений невелик.
72
При достаточно большой движущей силе мигрирующая граница некоторое время может увлекать за собой включения второй фазы, а затем отрываться от них. Захватывая на своем пути включения в объеме соседнего зерна, мигрирующая граница оставляет за собой зону, свободную от этих включений. После отрыва границы от включений их цепочка остается внутри растущего зерна.
При отжиге тонких листов, когда размер зерен становится равным толщине листа, их рост замедляется, но зерна еще продолжают расти в плоскости листа. После того как размер зерен в плоскости листа становится в 2— 3 раза больше его толщины, рост совсем прекращается. Одной из причин тормозящего действия свободных поверхностей, называемого «.эффектом толщины», является образование канавок термического травления в местах выхода границ зерен на эту поверхность. Канавки образуются при отжиге из-за ускоренного испарения атомов с межзеренных границ и закрепляют границы, как бы «привязывая» их к соответствующим участкам поверхности.
Рост зерен при собирательной рекристаллизации может сильно замедлять текстура, возникающая при первичной рекристаллизации. «Текстурное торможение» обусловлено тем, что границы зерен с небольшой угловой разориенти-ровкой (а именно она характерна для совершенной текстуры) имеют пониженную поверхностную энергию и в соответствии с формулой (11) скорость роста зерна должна быть меньше.
Росту одних зерен за счет аналогичных зерен той же фазы необязательно должна предшествовать первичная рекристаллизация.
Часто считают, что в литых (недеформированных) металлах укрупнения зерен вследствие роста одних кристаллов за счет соседних не происходит. Это утверждение справедливо только по отношению к металлам невысокой чистоты, в которых примеси, концентрирующиеся по границам зерен в процессе кристаллизации, мешают миграции границ зерен при отжиге. В чистых литых металлах и однофазных сплавах при отжиге без предварительной деформации зерна могут быстро расти, т.е. фактически может протекать собирательная рекристаллизация.
§ 9. Текстуры рекристаллизации
1. Основные закономерности образования текстур рекристаллизации
Экспериментально установлены следующие три варианта взаимоотношения кристаллографических ориентировок в деформированном и рекристаллизованном металле. Во-первых, текстура деформации может перейти в тождественную ей текстуру рекристаллизации, т. е. рекристаллизованные зерна приобретают такую же предпочтительную ориентировку, какую имели исходные деформированные зерна. Во-вторых, текстура деформации может смениться отличной от нее текстурой рекристаллизации. Этот случай наиболее частый. В-третьих, рекристаллизованные зерна могут иметь хаотичную кристаллографическую ориентировку, в то время как исходный деформированный металл был текстурован.
Характер и степень совершенства текстуры рекристаллизации зависят от вида обработки давлением, степени и температуры последней деформации, температуры и времени последнего отжига, степени промежуточной деформации, температуры промежуточного отжига, содержания легирующих элементов, примесей, размера зерна перед холодной деформацией и других факторов. Следовательно, на текстуру рекристаллизации влияет гораздо большее число факторов, чем на текстуру деформации.
При отжиге ниже некоторой температуры текстура рекристаллизации качественно тождественна текстуре деформации. Различие может состоять в усилении одних и ослаблении других компонентов текстуры. Так, если холоднотянутая медная проволока имела двойную аксиальную текстуру <111 > и <100>, то после первичной рекристаллизации ее текстура такая же, но число зерен с ориентировкой <100> увеличено за счет зерен с ориентировкой < 111>.
Наиболее яркий и практически важный случай изменения текстуры при рекристаллизации — формирование весьма совершенной кубической текстуры {100} <100> при отжиге холоднокатаных г. ц. к. металлов со средней и высокой энергией дефектов упаковки. Текстура куба образуется при рекристаллизационном отжиге меди, никеля, алюминия и многих сплавов на их основе. Так, например, в меди текстура прокатки {135} <112> заменяется кубической текстурой: в рекристаллизованных зернах плоскость куба {100} оказывается параллельной плоскости прокат
74
ки полосы, а направление ребра куба <100> весьма точно ориентируется вдоль и поперек направления прокатки.
В г. ц. к. металлах и твердых растворах с очень низкой энергией дефектов упаковки (серебро, а-латунь с 30 % Zn, аустенитная нержавеющая сталь) текстура прокатки {110} <112> сменяется текстурой рекристаллизации {113} < 112>.
Как правило, чем совершеннее текстура деформации, тем более четкой получается соответствующая ей текстура рекристаллизации.
Исключительно сильно на характер и степень совершенства текстуры рекристаллизации влияют малые добавки и примеси, находящиеся в твердом растворе. Примеси могут предотвратить образование той, или иной текстуры рекристаллизации, а могут и усилить’ совершенство этой текстуры. Важно, что, во-первых, примеси и малые добавки, сильно изменяющие текстуру рекристаллизации, обычно не влияют на текстуру деформации. Во-вторых, результат их влияния на текстуру рекристаллизации зависит не только от концентрации, но и от степени деформации, температуры отжига и других технологических факторов. Простых закономерностей здесь нет. Противоречивость данных разных исследователей о зависимости характера и степени совершенства текстуры рекристаллизации от степени деформации, температуры и времени отжига для одних и тех же металлов и сплавов, особенно промышленной чистоты, может быть связана с разным содержанием примесей, в том числе и неконтролируемых.
2. Природа текстур рекристаллизации
Для объяснения текстур, формирующихся при первичной рекристаллизации, были предложены две гипотезы: ориентированного зарождения и ориентированного роста. Согласно первой в деформированной матрице образуются только такие зародыши рекристаллизованных зерен, кристаллографическая ориентировка которых соответствует получающейся текстуре рекристаллизации. Согласно второй гипотезе в деформированной матрице возникают по-разному ориентированные зародыши, но часть из них благодаря особой ориентировке относительно матрицы имеет наибольшую скорость роста. Эта гипотеза логично объясняет, почему в г. ц. к. металлах решетка рекристаллизованных зерен часто оказывается повернутой по отношению к ре-
75
шетке деформированных зерен вокруг общей оси < 111 > на угол 30—45°. Прямые измерения показали, что границь между кристаллами, развернутыми вокруг общей осг < 111 > на 40° в алюминии и на 30° в меди, обладают необычно высокой подвижностью.
Гипотеза избирательного роста не в состоянии объяснить сильное влияние локальных неоднородностей в структуре деформированного металла на формирование текстуры рекристаллизации. Эти неоднородности, как показанс в § 7, играют важную роль в образовании центров первичной рекристаллизации.
Трудно ожидать, что существует какой-то один механизм формирования текстуры рекристаллизации, действующий во всех металлах и при всех условиях обработки. Современные трактовки экспериментальных данных фактически исходят из объединенной гипотезы ориентированного зарождения и избирательного роста. Очень ценные сведения о механизмах формирования текстур рекристаллизации дают интенсивно проводимые в последние годы электронно-микроскопические исследования особенностей структуры металла после сильной холодной деформации и на ранних стадиях рекристаллизации с прямым измерением кристаллографических ориентировок в локальных областях. Рассмотрим на базе этих исследований, как формируется кубическая текстура рекристаллизации в силь-нодеформированной меди.
Микрополосы в переходных полосах, превращающиеся в центры рекристаллизации, имеют кубическую ориентировку {100} <100>. Внутри этих микрополос плотность дислокаций весьма низкая, что связано с быстрым протеканием в них динамического (во время деформации) или статического (при отжиге) возврата. Кристаллографический анализ показал, что именно в кристалле с кубической ориентировкой при прокатке существует уникальная ситуация — дислокации в действующих системах скольжения имеют взаимно ортогональные векторы Бюргерса и, следовательно, упруго не взаимодействуют, т. е. не могут образовывать устойчивых конфигураций. Таким образом, наличие в переходных полосах холоднокатаной меди длинных микрополос с кубической ориентацией и особая склонность к возврату внутри таких микрополос делают их зародышами формирования кубической текстуры рекристаллизации. Из-за того, что суммарный объем переходных полос с кубической ориентировкой составляет очень небольшую долю всего объема металла, рентгеновский ме-76
тод обычно не может зарегистрировать наличие компоненты {100}<100>* в текстуре прокатки (см. табл. 1).
В полосах сдвига образуются центры рекристаллизации с хаотичной ориентировкой, что мешает формированию совершенной кубической текстуры рекристаллизации. Кроме того, полосы сдвига, пересекая переходные полосы, делают микрополосы в них более короткими (см. рис. 14) и соответственно менее эффективными для зарождения рекристаллизованных зерен с кубической ориентировкой. Крупное исходное (перед прокаткой) зерно способствует развитию полос сдвига в меди, ослабляя тем самым кубическую текстуру рекристаллизации. Таким образом, воздействуя на развитие переходных полос и полос сдвига можно управлять текстурой рекристаллизации.
Сложное влияние примесей на текстуру рекристаллизации связано, по всей видимости, с их воздействием на подвижность малоугловых и высокоугловых границ при образовании центров рекристаллизации и росте зерен из этих центров.
Гипотеза ориентированного зарождения и избирательного роста позволяет понять, почему текстуры рекристаллизации (характер и степень совершенства) столь многообразны и зависят от большого числа факторов — гораздо большего, чем текстура деформации. Если на формирование текстур деформации главное влияние оказывают особенности скольжения дислокаций, то на формирование текстур рекристаллизации влияют исходная структура деформированного металла и скорость миграции границ разного типа, которые сами сильно зависят от многих факторов.
§10. Вторичная рекристаллизация
1. Закономерности вторичной рекристаллизации
При собирательной рекристаллизации зерна укрупняются более или менее равномерно и металл можно характеризовать одним средним значением размера зерна. Такой рост зерен, называемый нормальным, рассмотрен в§ 8.
В определенных условиях после отжига обнаруживается структура, состоящая из множества сравнительно мелких зерен примерно одинакового размера и гораздо меньшего числа очень крупых, иногда гигантских зерен, дости
77
гающих размера в несколько сантиметров (рис. 33). Такая структура возникает в результате неравномерного роста зерна, называемого вторичной рекристаллизацией: большинство зерен укрупняется очень медленно или практически вообще не растет, а отдельные зерна вырастают до больших размеров, «поедая» свое мелкозернистое окружение.
Вторичная рекристаллизация начинается с определенной температуры (на рис. 34 /ВТ.Р=925°С), ниже которой возможна только очень медленная собирательная рекристаллизация. При температуре 925 °C за время 1 ч, превы-
Рис. 33. Граница между мелкими зернами н крупным зерном, образовавшимся в цинке в результате вторичной рекристаллизации во время отжига при 200 °C. Х60 (Бурке)
шающее инкубационный период вторичной рекристаллизации, размер отдельных зерен увеличивается примерно в 50 раз, достигая величины 10 мм. Основная же масса зерен при 925 °C сохраняет размер около 0,02 мм.
Размер зерен получается максимальным при температуре /втр, а с ростом температуры выше /вт.р он уменьшается из-за большого числа центров вторичной рекристаллизации. Выше /вт.р ускоряется нормальный рост зерен в мат
78
рице, и при достаточно высоких температурах собирательная и вторичная рекристаллизации становятся конкурирующими процессами (кривые 1 и 2 на рис. 34 сближаются). При 1200 °C идет только нормальный, равномерный рост зерен. Таким образом, вторичная рекристаллизация раз-
вивается в определенном интервале температур, в котором нормальный рост зерен протекает сравнительно медленно.
Зерна, выросшие при вторичной рекристаллизации, часто имеют предпочтительную кристаллографическую ориентировку, причем текстура вторичной рекристаллиза-
ции всегда отлична от текстуры первичной рекристаллизации и является более острой (совершенной).
Характер и степень совершенства текстуры вторичной рекристаллизации зависят от режимов обработки давлением, промежуточных и окончательных отжигов, толщины листа и других факторов.
Вторичная рекристаллизация — это не редкий, особый случай роста зерна, как когда-то считали, т. е. не аномальный рост зерна, а присущее многим металлам и сплавам явление. Она обнаружена в зонно-очищенных металлах и металлах технической чистоты, например в Fe, Си, Ag, Zn, Ni, Pt, Ti, Al, W и Та, в сплавах на разных основах, например в трансформаторной и
Рис. 34. Зависимость размера зерен от температуры отжига в течение 1 ч в листах из сплава Fe—3 % Si толщиной 0,35 мм:
1 — размер зерен стабилизированной матрицы, укрупняющихся при нормальном росте; 2 — размер зерен, выросших при вторичной рекристаллизации
аустенитных сталях, сплавах алю-
миния с марганцем, медью и многих других. При перегреве наследственно мелкозернистой стали вторичная рекри-
сталлизация аустенита вызывает появление разнозернис-тости (см. § 22).
2. Природа вторичной рекристаллизации
Эксперименты показывают, что в разных металлах и сплавах, в различных условиях обработки даже при неодинаковой толщине листа неравномерный рост зерен может вызываться различными причинами. Но во всех случаях обязательным условием развития вторичной рекристаллизации является так называемая стабилизация матрицы, т. е сильное торможение роста большинства зерен, образовав
79
шихся при первичной кристаллизации. Если матрица в целом стабилизирована, то рост отдельных зерен, по каким-то причинам слабее заторможенный, и составляет сущность вторичной рекристаллизации. Следовательно, при анализе причин вторичной рекристаллизации в каждом конкретном случае необходимо установить, во-первых, почему стабилизирована матрица, и, во-вторых, почему отдельные зерна могут беспрепятственно расти, поедая стабилизированную матрицу.
Причинами стабилизации рекристаллизованной матрицы могут быть: а) дисперсные частицы или сегрегации примесей на границах; б) «текстурное торможение» и в) «эффект толщины». Все они уже были рассмотрены в § 8 при обсуждении причин торможения роста зерна во время собирательной рекристаллизации. Роль торможения дисперсными частицами, «текстурного торможения» и «эффекта толщины» доказана экспериментально в разных конкретных случаях вторичной рекристаллизации.
Более сложно выяснить, какие из рекристаллизованных зерен в стабилизированной матрице могут быстро расти, т. е. служат центрами вторичной рекристаллизации. При этом важно понять, почему они способны превзойти по размеру своих соседей на начальной стадии вторичной рекристаллизации. Последующий их преимущественный рост вполне понятен, так как всегда существует «движущая сила» роста более крупного зерна (размером £>к) за счет более мелких соседних зерен (размером £>м), пропорциональная разности этих размеров и зернограничной энергии у:
P^(DK-D^/(DKDM). (16)
Движущая сила Р — это уменьшение энергии Гиббса системы, отнесенное к единице объема. При росте крупного зерна за счет мелких движущей силой является уменьшение суммарной зернограничной энергии. Поскольку во время вторичной рекристаллизации размер зерен матрицы DM остается практически неизменным, то по мере укрупнения избранных зерен, т. е. с увеличением непрерывно возрастает движущая сила их роста.
Вернемся к вопросу, какие же зерна становятся центрами вторичной рекристаллизации.
После окончания первичной рекристаллизации всегда имеются зерна, размер которых больше среднего, и чем
80
крупнее такие зерна, тем меньше их количество. Если матрица стабилизирована дисперсными частицами второй фазы, то из-за неравномерности распределения этих частиц границы отдельных более крупных зерен могут быть значительно слабее заблокированы, чем границы большинства зерен, и такие зерна способны к избирательному росту. Тормозящее действие частиц зависит от их дисперсности, объемной доли [см. формулу (15)] и характера распределения, способности к коагуляции и растворению. Если тормозящее влияние слабое, то матрица недостаточно стабилизирована и происходит нормальный равномерный рост зерен. Если тормозящее действие частиц чрезмерно сильное, например объемная доля их очень большая, то матрица так «перестабилизирована», что не находится зерен, способных к росту, и вторичная рекристаллизация не идет.
В трансформаторной стали (Fe — 3 % Si) матрица стабилизирована частицами нитрида алюминия или сульфида марганца с объемной долей 0,01—0,1 % и размером 20— 100 нм. При 900—1000 °C эти частицы начинают растворяться или коагулировать, их тормозящее действие ослабевает и тогда наиболее крупные рекристаллизованные зерна матрицы начинают ускоренно расти за счет более мелких соседей. Поскольку при вторичной рекристаллизации трансформаторной стали формируется весьма совершенная ребровая текстура {110}<100>, то следует еще объяснить, почему именно зерна с такой ориентировкой способны к преимущественному росту. Возможно, это обусловлено повышенной подвижностью границ «ребровых зерен» в матрице, претерпевшей первичную рекристаллизацию.
Если причиной стабилизации матрицы служит совершенная текстура, возникшая при первичной рекристаллизации, то большинство зерен разделено границами с небольшим углом разориентировки и соответственно с низкой зернограничной энергией, вследствие чего эти границы малоподвижны. В условиях «текстурного торможения» к избирательному росту способны те немногие зерна, ориентировка которых сильно отличается от главной ориентировки стабилизированной матрицы и которые одновременно имеют больший размер, обеспечивающий движущую силу их роста за счет соседей.
В связи со сказанным можно понять, почему центром вторичной рекристаллизации становится лишь одно зерно из —105— 106 зерен матрицы.
6 И. И. Новиков
81
$ 1L Размер рекристаллизованного зерна
Размер рекристаллизованного зерна — одна из важнейших . характеристик отожженного металла. Время отжига, как правило, превышает продолжительность первичной рекристаллизации. Поэтому на размер зерна отожженного металла влияют все те факторы, которые сказываются и на первичной, и на собирательной рекристаллизации.
Размер зерна к моменту окончания первичной рекристаллизации зависит от соотношения скорости зарождения центров и линейной скорости роста. Чем больше с.з.ц. и меньше л. с. р., тем мельче получается зерно к моменту окончания первичной рекристаллизации, и наоборот. После окончания первичной рекристаллизации зерна укрупняются вследствие собирательной рекристаллизации. Поэтому на конечный размер зерна влияет также линейная скорость роста кристаллитов при собирательной рекристаллизации (вторичная рекристаллизация пока не рассматривается).
К основным факторам, влияющим на конечный размер рекристаллизованного зерна, относятся температура и время отжига, степень деформации, химический и фазовый составы сплава.
1. Влияние температуры и времени отжига
С повышением температуры отжига с. з.ц. и л. с. р. увеличиваются. Если оба параметра изменяются в зависимости ют температуры в одинаковой степени, то размер зерна при первичной рекристаллизации не должен зависеть от температуры отжига. Если же с. з. ц. увеличивается с повышением температуры интенсивнее, чем л.с. р., то размер зерна к моменту окончания первичной рекристаллизации должен быть тем меньше, чем выше температура отжига. Оба случая неоднократно наблюдались при отжиге алюминия, алюминиевых сплавов, латуни и других сплавов.
На первый взгляд может показаться, что эти факты не согласуются с обычными представлениями об укрупнении зерна при повышении температуры отжига. Кажущееся противоречие объясняется тем, что выше речь шла о размере зерна к моменту окончания первичной рекристаллизации, а обычно продолжительность отжига превышает длительность первичной рекристаллизации. В последнем случае укрупнение зерна при повышении температуры отжига объясняется интенсифицированием собирательной ре-
82
кристаллизации. На рис. 35 представлен наиболее типичный характер зависимости размера зерна от температуры
отжига при постоянном времени выдержки.
С увеличением продолжительности отжига размер зерна возрастает с затуханием, приближаясь к предельному для данной температуры значению (рис. 36). Главные при
чины уменьшения скорости изотермического роста зерен—
сокращение протяженности их границ и соответственно зернограничной энергии (движущей силы роста), спрямление границ, приближение углов в тройных стыках зерен к равновесным, накопление примесей на границах.
Рис. 35. Зависимость размера рекристаллизованного зерна алюминия и его сплавов от температуры отжига в течение 1 ч (И. И. Новиков):
1 — А1 чистотой 99,7 %; 2 — AI — 1,2 % Zn; 3 —Al —0,6% Мп; 4 — Al— 0,55 % Fe
Если при отжиге получен предельный для данной температуры размер зерна, то его можно увеличить, проводя новый отжиг при более высокой температуре. С повышением температуры облегчается переход атомов от одного
Рис. 36. Влияние времени выдержки на размер зерна никеля при двух температурах отжига (И. И. Новиков,. И. Л. Рогельберг)
зерна к другому. Кроме того, при повышении температуры отжига могут размываться сегрегации примесных атомов на границах зерен и частично растворяться избыточные фазы, тормозящие миграцию границ во время собирательной рекристаллизации.
2. Влияние степени деформации
Исключительно сильно на размер зерна в момент окончания первичной рекристаллизации влияет степень деформации (рис. 37 и 38). Так как продолжительность отжига
6*
83
обычно превышает длительность первичной рекристаллизации, то графики, подобные представленному на рис. 38, отображают влияние степени деформации на конечный размер зерна, полученный после собирательной рекристаллизации. Последняя лишь укрупняет зерно и качественно не меняет характера зависимости размера кристаллитов от-степени деформации в момент окончания первичной рекристаллизации.
При сравнительно небольшой критической деформации (обычно от 1 до 15%) при отжиге вырастает очень круп-
Рис. 37. Макроструктура пластинок чистого алюминия, отожженных при 500 °C после растяжения с разной степенью деформации (цифры на рисунке). Натуральная величина
ное зерно, иногда достигающее нескольких сантиметров. Механизм образования крупного зерна при критической деформации качественно отличен от механизма рекристаллизации в закритической области, т.е. при более высоких степенях деформации.
Структурные изменения при отжиге деформированного металла после критической деформации и близких к ней до- и закритических деформаций наиболее подробно экспериментально изучены и проанализированы С. С. Гореликом. Эти изменения состоят в следующем.
При отжиге после докритических деформаций происходит полигонизация, а также перемещение высокоугловых границ деформированных зерен на небольшие расстояния, составляющие всего лишь сотые — десятые доли размера зерен.
С увеличением степени деформации в докритической области возрастает неоднородность наклепа разных зерен. Критическая степень деформации соответствует состоя-
84
Степень деформации
Рис. 38. Влияние степени деформации на размер зерна, полученного при последующем отжиге при двух температурах: екр — критическая степень деформации
нию, когда эта неоднородность становится столь большой, что из-за разности в накопленной объемной энергии (плотности дислокаций) соседних зерен при нагреве идет быстрая миграция отдельных границ на расстояния, соизмеримые с размером зерен, т. е. отдельные исходные зерна растут за счет соседних, несколько сильнее наклепанных зерен.
Общая плотность дислокаций и избыток дислокаций одного знака при критической деформации еще недостаточны, чтобы вызвать при нагреве образование новых высокоугловых границ и центров первичной рекристаллизации. Следовательно, при отжиге после критической деформации протекает не первичная рекристаллизация, а одни слабодеформиро-ванные зерна укрупняются за счет других деформированных зерен, причем движущей силой такой рекристаллизации является разность в объемной энергии неодинаково деформированных соседних зерен.
В области далеко закритичес-ких деформаций общая плотность дислокаций и избыток дислокаций одного знака настолько велики, что при отжиге быстро
образуется большое число центров первичной рекристаллизации, которая охватывает весь объем металла.
С этим процессом не может конкурировать более медленный рост деформированных зерен за счет соседей вследствие миграции исходных границ, так как разница в накопленной энергии соседних деформированных зерен несравненно меньше, чем разница в объемной энергии деформированных и новых рекристаллизованных зерен.
При отжиге после деформаций, ненамного превышающих критическую, когда число центров первичной рекристаллизации еще очень мало, конкурируют два процесса: укрупнение зерен вследствие миграции исходных границ и рост новых зерен из центров первичной рекристаллизации. Второй процесс приводит к тому, что средний размер зерен получается меньше, чем при отжиге после критической деформации.
85
Чем выше температура отжига, тем меньше критическая деформация (см. рис. 38). Это вполне понятно, так как. чем выше температура отжига, тем при меньшей разности в накопленной энергии соседних зерен может начаться процесс роста зерен благодаря повышенной подвижности атомов.
С повышением температуры деформирования крити-
ческая деформация, выявляемая при последующем отжиге», возрастает (рис. 39). При более высоких температурах деформирования требуется большее обжатие, чтобы достичь необходимой неоднородности наклепа соседних зерен,.
так как уже во время самого деформирования происходит
возврат, частично устраняющий наклеп.
С повышением чистоты металла критическая деформа
ция уменьшается. Легирующие элементы по-разному вли-
яют на величину критической степени деформации. Одни элементы (например, марганец в алюминии, рис. 40) уже в небольших количествах резка увеличивают критическую степень деформации, а другие (например, цинк и медь в алюминии) даже в больших количествах оказывают слабое действие. Сильное влияние небольших количеств марганца обусловлено тем, что он в отличие от цинка и меди очень мало растворим в’ твердом алюминии и образует дисперсные частицы А16Мп, тормозя-
Рис. 39. Зависимость критической степени деформации алюминия от температуры деформирования. Отжиг при 450 °C, 30 мии (В. 3. Захаров, И. И. Новиков, И. Л. Ро-гельберг, Яо Минь-чжи)
щие миграцию границ исходных деформированных зерен.
На графиках размер рекристаллизованного зерна — степень деформации можно наблюдать практически скач-ковое (рис. 38 и 40) и плавное укрупнение зерна (рис. 41). В последнем случае восходящая ветвь (левее еКр) соответствует ускорению роста избранных исходных зерен за счет соседей из-за увеличения неоднородности в их наклепе с ростом степени деформации в докритической области.
При очень больших степенях деформации и высоких
температурах отжига у алюминия, меди и некоторых сплавов обнаружено появление второго максимума размера зерна (см. рис. 41). В отдельных случаях, например у электролитической меди при степени обжатия более 80 °/о
86
я температуре отжига более 1000 °C, зерно крупнее, чем при критической деформации у первого максимума. Можно предположить, что благодаря совершенной текстуре, возникающей при больших степенях деформации, при вы-
Рис. 40. Влияние степени деформации на размер рекристаллизованного зерна алюминия с добавками марганца. Отжиг прн 500 °C, 30 мин (В. 3. Захаров, И. И. Новиков, И. Л. Рогельберг, Яо Минь-чжи):
1 — А1 чистотой 99,7 %; 2 — А1 — 0,3 % Мп; 3 — А1 — 0,6 % Мп
сокотемпературном отжиге крупные зерна образуются в в результате вторичной рекристаллизации, идущей в условиях сильного текстурного торможения (см. § 10).
3. Влияние химического и фазового состава
В общем случае можно считать, что с увеличением содержания примесей в металле размер рекристаллизованного зерна уменьшается. Причиной этого является главным образом торможение собирательной рекристаллизации. Особенно сильно на размер зерна влияют элементы, образующие фазы, труднорастворимые в основном металле. На рис. 35 пока-
Степень деформации
Рис. 41. Влияние степени деформации на размер зерна, полученного при последующем отжиге
зано, что цинк, находящийся в растворе в сравнительно большом количестве, слабо уменьшает размер зёрна алюминия. В то же время марганец и в особенности железо, образующие труднорастворимое соединение AUMn и почти нерастворимое соединение FeAh, резко тормозят рост зерна при собирательной рекристаллизации.
87
Более подробный анализ показывает, что роль избыточных фаз в формировании рекристаллизованной структуры сильно зависит от размера их частиц и расстояния между частицами. Вторая фаза может и облегчать, и затруднять первичную рекристаллизацию по сравнению с однофазным сплавом (см. также рис. 27).
Если частицы второй фазы достаточно крупные (размером более 0,5—1 мкм), то при пластической деформации в матричной фазе непосредственно около них накапливаются дислокации одного знака и возникает локальное искривление кристаллической решетки. В этом участке матрицы уже во время пластической деформации или при последующем нагреве формируется субзерно с большой угловой разориентировкой по обе стороны от его субграницы.
Такое субзерно при нагреве способно к росту, при котором, набирая угловую разориентировку, субграница превращается в высокоугловую границу и формируется зерно— зародыш рекристаллизации (схему образования зародыша рекристаллизации в полосе изгиба решетки см. также на рис. 24).
Конечный размер рекристаллизованных зерен зависит от расстояния между крупными частицами. Если это расстояние большое, то рекристаллизованные зерна сравнительно свободно разрастаются. Если же объемная доля второй фазы высокая и соответственно расстояние между ее частицами маленькое, то эти частицы будут тормозить рост рекристаллизованных зерен, средний размер которых должен быть того же порядка,что и среднее межчастичное расстояние.
Около частиц малого размера (менее —0,1 мкм) при пластической деформации в матрице не возникает такого искривления решетки, которое способствовало бы зарождению центров рекристаллизации. Если очень дисперсные частицы равномерно распределены по объему на небольшом расстоянии одна от другой, то они затрудняют зарождение рекристаллизованных зерен, так как тормозят миграцию субграниц. Такие частицы могут затруднять и рост сформировавшихся зерен, являясь барьерами для миграции высокоугловых границ. Например, холоднокатаный сплав А1 — 6,5 % Mg — 0,2 % Sc с равномерно распределенными и мало склонными к коагуляции частицами AhSc размером около 0,03 мкм имеет температуру начала рекристаллизации на 200 К выше, чем сплав А1 — 6,5 % Mg.
88
4. Диаграммы рекристаллизации
Зависимость размера зерна от всех рассмотренных выше факторов невозможно изобразить графически в виде одной диаграммы. Довольно большое распространение получили пространственные диаграммы, рекристаллизации, показывающие зависимость размера зерна металла или сплава от степени деформации и температуры отжига при определенном времени выдержки (рис. 42). Эти диаграммы дают возможность в самом первом приближении выбрать режим отжига для получения желательной структуры.
Рис. 42. Диаграмма рекристаллизации электролитического железа. Исходная обработка: ковка и отжиг при 930 °C. Окончательная обработка: холодное осаживание и отжиг 1 ч (Обергоффер, Ортель)
Вертикальные разрезы пространственной диаграммы рекристаллизации при постоянной степени деформации и постоянной температуре отжига являются графиками типа тех, что изображены на рис. 35 и 38 соответственно. На нижней горизонтальной плоскости диаграммы рекристаллизации часто приводят кривую зависимости температуры начала рекристаллизации от степени деформации (см. пунктир на рис. 42).
Пользуясь диаграммами рекристаллизации, необходимо строго учитывать те условия, при которых они получены, а именно: время отжига, содержание примесей, исходный размер зерна, скорость нагрева при отжиге, вид обра
89
ботки давлением и др. Поэтому каждую диаграмму рекристаллизации следует сопровождать подробными сведениями об исходном состоянии и окончательной обработке* металла. Так как весьма небольшие колебания в содержании примесей, а также различные неучитываемые факторы существенно сказываются на размере зерна, то это в значительной мере снижает ценность диаграмм рекристаллизации как количественных характеристик металла. Эти диаграммы дают скорее полуколичественные данные, позволяющие ориентироваться в том, как степень деформации и температура отжига влияют на размер зерна металла или сплава.
5. Разнозернистые структуры
После рекристаллизационного отжига в разных участках изделия зерно может быть примерно одинаковым по размеру (например, только мелким или только крупным), но может наблюдаться и нежелательная разнозернистость. Основные ее формы:
1) равномерная разнозернистость — закономерное чередование по всему объему крупных и мелких зерен;
2) зональная разнозернистость — расположение крупных зерен в определенных зонах изделия;
3) строчечная разнозернистость — вытянутость крупных зерен обычно вдоль направления главной деформации;
4) островная разнозернистость — группы очень крупных или очень мелких зерен, незакономерно распределенные по объему изделия.
Причины и условия появления разнозернистости весьма многообразны и могут различаться даже в случае одного' типа разнозернистости в разных сплавах и изделиях.
Равномерная разнозернистость может возникнуть на. стадии незавершенной вторичной рекристаллизации.
Простейший случай зональной разнозернистости — возникновение ее при критической деформации металла в определенных зонах изделия. При сильной местной деформации степень деформации уменьшается от большой закри-тической, например у кромки пробитого отверстия, до нуля (вдали от этой кромки). В этом случае всегда какой-то* участок изделия оказывается деформированным на критическую степень и здесь при отжиге вырастает крупное зерно. Если технологически невозможно избежать критической деформ-ации, то предупредить разнозернистость можно, от
90
казавшись от рекристаллизационного отжига и применяя только дорекристаллизационный отжиг.
Строчечное расположение дисперсных частиц в дефор-мированном изделии может вызвать строчечную разнозер-нистость при рекристаллизационном отжиге.
Зональная ликвация в слитке приводит к макронеоднородности по химическому составу в деформированном изделии, и в участках разного состава с разной скоростью идут первичная, собирательная и вторичная рекристаллизации, вызывая зональную или островную разнозернис-тость.
Регулируя условия кристаллизации слитка, режим его гомогенизапионного отжига, условия обработки давлением, а также состав сплава, можно бороться с появлением разнозернистости. Из-за многообразия причин и форм проявления разнозернистости для конкретных изделий приходится разрабатывать свою технологию производства, обеспечивающую получение равномерной зернистой структуры.
В большинстве случаев желательно получить при отжиге не только равномерную, но мелкозернистую структуру. Если металл (сплав) не имеет полиморфных превращений, то крупнозернистость, полученную при рекристаллизационном отжиге, никакой последующей термообработкой не устранить, так как она соответствует состоянию с пониженной энергией Гиббса.
Рекристаллизационный отжиг используют и для намеренного получения крупнозернистой структуры, бикристаллов и монокристаллов, которые необходимы для исследовательских работ и специфических применений в технике. Монокристаллы и бикристаллы получают так называемым методом деформация — отжиг, применяя критическую деформацию и особые режимы отжига.
§ 12. Изменение свойств металла при дорекристаллизационном и рекристаллизационном отжиге
При отжиге наклепанного металла свойства изменяются обратно тому, как они изменялись при холодной деформации, а именно показатели сопротивления деформированию (временное сопротивление, предел текучести, твердость) уменьшаются, а показатели пластичности (относительное удлинение и сужение) возрастают.
91
1. Разупрочнение при отжиге
В зависимости от температуры и продолжительности отжига в металле с той или иной полнотой проходят разные структурные изменения и соответственно по-разному протекает разупрочнение.
На рис. 43 схематично показаны три типовых случая изменения прочностных свойств с увеличением продолжительности отжига при постоянной температуре. В инкубационный период до начала первичной рекристаллизации^ когда происходит только возврат, наклеп может практически совсем не уменьшаться (кривая /), частично уменьшаться (кривая 2) и полностью сниматься (кривая 3).
Характер изменения прочностных свойств до начала рекристаллизации полностью соответствует кинетике возврата (см. рис. 16): с увеличением времени возврата скорость снижения прочностных свойств уменьшается. Если по окончании возврата прочностные свойства еще не восстановились, то последующая первичная рекристаллизация полностью снимает наклеп. При этом с увеличением времени отжига скорость снижения прочностных свойств сначала возрастает, затем некоторое время остается неизменной и затем снижается в полном соответствии с сигмаидальной кривой нарастания рекристаллизованного объема (см. рис. 21).
На рис. 44 видно, как твердость наклепанного алюминия при изотермическом отжиге вначале снижается с затуханием (идет возврат), после чего сначала с ускорением, а затем с замедлением происходит интенсивное разупрочнение при первичной рекристаллизации.
Рис. 43. Схемы зависимостей прочностных свойств наклепанного металла от времени изотермического отжига
92
Сохранение наклепа при возврате свойственно меди, никелю и серебру. Заметное, а иногда и весьма значительное частичное снятие наклепа до начала первичной рекристаллизации происходит при отжиге алюминия, железа и титана. Полностью наклеп снимается в период возврата у деформированных монокристаллов тугоплавких металлов
Рис. 44. Уменьшение твердости наклепанного чистого алюминия с увеличением времени отжига при разных температурах. Исходное состояние — холодная деформация растяжением иа 20 % (Лораи, Батисс)
с о. ц. к. решеткой (вольфрама, молибдена), а также у по-ликристаллических сплавов алюминия с магнием. В практическом отношении важно, что нагартованные алюминие-вомагниевые сплавы значительно разупрочняются даже при комнатной температуре, о чем свидетельствуют следующие данные для холоднокатаного магналия, содержащего 6 % Mg:
Время выдержки............0 10 дней 4 мес. 2 года 12,5 лет
Ов, МПа................. 500 480 455 440 435
Типовые случаи зависимости прочностных свойств от температуры отжига при постоянной его продолжительности схематично показаны на рис. 45. До температуры начала рекристаллизации наклеп может полностью сохраняться (кривая 7), частично сниматься (кривая 2) и полностью устраняться (кривая 3). В первом случае интенсивное разупрочнение начинается при температуре начала первичной рекристаллизации, а во втором и в третьем по началу разупрочнения нельзя определить температуру t* . Поэтому хорошо известный метод определения по измененик> прочностных свойств далеко не всегда может быть использован.
9$
Если в интервале первичной рекристаллизации прочностные свойства сильно падают, то в интервале собирательной рекристаллизации они снижаются слабо.
Прочностные свойства зависят от дислокационной структуры и размера зерен. Если во время дорекристаллизацион-
ного отжига протекает толь-
Температура отжига
'Рис. 45. Схемы зависимости прочностных свойств наклепанного металла от температуры отжига
ко отдых и, следовательно, главные структурные изменения состоят в уменьшении концентрации точечных дефектов, а плотность дислокаций очень мало снижается, то упрочнение от деформации в основном сохраняется. Если же при до-рекристаллизационном отжиге развивается полигонизация, связанная с формированием и укрупнением субзерен и очищением их
объема от дислокаций, то прочностные свойства могут существенно снизиться.
Предел текучести <ут связан с диаметром субзерен d соотношением Холла—Петча:
<rT = cy0 + faW . (17)
В соответствии с этим соотношением рост субзерен при полигонизации приводит к снижению предела текучести при дорекристаллизационном отжиге.
Первичная рекристаллизация, резко снижая плотность дислокаций и «выметая» стенки субзерен, вызывает сильное разупрочнение, пропорциональное доле рекристаллизованного объема. С повышением температуры отжига между t* и / £ (см. рис. 45) или с увеличением времени отжига при постоянной температуре (правее на рис. 43) прочностные свойства интенсивно снижаются из-за первичной рекристаллизации. Кроме того, определенный вклад в разупрочнение вносит и продолжающийся возврат в тех деформированных зернах, которые еще не поглощены рекристаллизованными. На это указывает снижение микротвердости перекристаллизованных зерен.
После того как наклепанные зерна полностью заменились значительно более совершенными рекристаллизованными, собирательная рекристаллизация может лишь немного понизить прочностные свойства из-за укрупнения
ед
зерна. В частности, предел текучести с ростом зерна при собирательной рекристаллизации уменьшается в соответствии с соотношением Холла—Петча [в этом случае в формуле (17) d — диаметр зерен].
Разная способность к разупрочнению при дорекристал-лизационном отжиге связана с разной склонностью к полигонизации. У металлов с одним типом решетки легкость, с какой идет полигонизация, зависит от энергии дефектов упаковки. Например, у алюминия энергия дефектов упаковки значительно больше, чем у меди, полигонизация развивается сильнее, что и приводит к значительному разупрочнению при дорекристаллизационном отжиге. В некоторых тугоплавких о. ц. к металлах, например молибдене и вольфраме, полигонизация развивается особенно активно, и поэтому в них велика доля разупрочнения, приходящаяся, на дорекристаллизационный отжиг (рис. 46).
Рис. 46. Влияние температуры отжига в течение 1 ч на твердость вольфрама электроннолучевой плавки, деформированного с обжатием 50 % при 800 °C (Аллен)
Рис. 47. Влияние температуры отжига иа механические свойства деформированного металла (схема для однбго из распространенных случаев):
*Р* И ** ~ температуры начала » конца рекристаллизации, перегрева и пережога
Разупрочнение при дорекристаллизационном отжиге можно ускорить и увеличить, приложив к изделию во время отжига небольшие напряжения. Эти напряжения, не создавая еще значительного наклепа, ускоряют переползание-дислокаций, необходимое для развития возврата.
Показатели пластичности при отжиге после холодной деформации в общем изменяются обратно тому, как изменяются прочностные свойства: в области возврата сравнительно слабо возрастают, сильно повышаются при пер-
95.
вичной рекристаллизации, когда снимается большая часть наклепа, и мало изменяются при собирательной рекристаллизации (рис. 47 и 48). Максимальная пластичность достигается в некотором интервале температур в области собирательной рекристаллизации. Начиная с определенной температуры (6 на рис. 47 и ~ 800 °C на рис. 48) пластичность плавно снижается, так как далеко зашедшая собирательная рекристаллизация приводит к образованию чрезмерно крупного зерна. Это явление называется перегревом при рекристаллизационном отжиге.
Температура отжига, °C
Рис. 48. Влияние температуры часового отжига на электросопротивление и механические свойства меди Ml (А. П. Смирягин)
Рис. 49. Зависимость относительного удлинения листового алюминия чистотой 99,7 %, отожженного при 450 °C, от степени предварительной деформации при 20 °C (А. Ю. Золо-торевская, В. 3. Захаров, И. И. Новиков, И. Л. Рогельберг)
При очень высоких температурах отжига (выше #2 на рис. 47) резко падают и пластичность, и прочность, что вызвано пережогом — сильным межкристаллитным окислением, а иногда и частичным оплавлением примесей по границам зерен. Например, если медную проволоку отжигать ►при 1000 °C в воздушной среде, то сквозное межкристаллитное окисление делает ее настолько ломкой, что она разрушается при одном перегибе.
Вторичная рекристаллизация, дающая очень крупное зерно, а также разнозернистость снижают показатели пластичности.
В области околокритических деформаций относительное удлинение отожженных металлов и сплавов с увеличением -степени предварительной (перед отжигом) деформации снижается, достигает минимума при критической деформации и затем возрастает (рис. 49). Снижение пластичности в до-критической области при увеличении степени деформации вызвано возрастанием наклепа, сохраняющегося после от-
96
жига. Рост пластичности в закритической области связан с получением более мелкого зерна при рекристаллизационном отжиге (см. рис. 38). Минимум пластичности отожженного металла после критической деформации обусловлен, во-первых, крупным зерном и, во-вторых, максимальной степенью наклепа. В закритической области наклеп снимается первичной рекристаллизацией, а при критической деформации он частично сохраняется после отжига (это прямо доказывается измерениями твердости), так как крупные зерна растут не путем полной замены деформированных зерен новыми рекристаллизованными, с низкой плотностью дислокаций, а путем укрупнения одних наклепанных зерен за счет других.
Рис. 50. Зависимость относительного сужения молибдена от температуры в рекристаллизованном состоянии (/), после холодной деформации на 25 (2) и 68 % (3) и после дорекристаллизационного получасового отжига при 870 °C холоднодеформироваиного материала (4) (Имгрэм)
Отличается от других изменение пластичности тугоплавких металлов VIA группы (Сг, Mo, W) при рекристаллизационном отжиге. Как известно, эти металлы ниже некоторой температуры, зависящей от их чистоты, скорости деформации при испытаниях и других факторов, находятся в хрупком состоянии. Переход из пластичного состояния в хрупкое связан с образованием на межкристаллитных границах сегрегаций атомов примесей внедрения (углерода, кислорода и азота), находящихся в твердом растворе, а также выделением здесь карбидов, оксидов и нитридов (в металлах VIA группы технической чистоты содержание примесей внедрения превышает их очень малую растворимость в твердом состоянии). На высокоугловых границах, образующихся при первичной рекристаллизации, сегрегация примесей и выделение избыточных фаз выражены наи-
7 И. И. Новиков 97
более ярко. Здесь зарождаются хрупкие трещины, развивающиеся по границам или внутри зерен. Поэтому рекристаллизационный отжиг может резко (на 200—300 К) повысить температурный порог хрупкости хрома, молибдена и вольфрама по сравнению с деформированным состоянием (рис. 50). Эти металлы—оригинальный и практически важный пример того, как деформация, создающая наклеп, повышает пластичность (кривые 2 и 3), а рекристаллизация, снимающая наклеп, резко охрупчивает металл (кривая 7). Малоугловые границы субзерен в этом отношении ведут себя по-иному: полигонизация обычно снижает температурный порог хрупкости рассматриваемых металлов (кривая 4 на рис. 50).
2. Упрочнение при дорекристаллизационном отжиге
Давно было замечено, что при низкотемпературном отжиге некоторых металлов и сплавов до начала рекристаллизации значительно повышаются твердость, временное сопротивление и особенно пределы текучести и упругости (см. пунктир на рис. 45). У каждого из таких материалов имеется своя оптимальная температура отжига, при которой упрочнение максимально (табл. 2).
Упрочнение в температурной области возврата внешне противоречит самому понятию возврата, связанного с частичным снятием наклепа и возвращением свойств к их значениям до деформации. Поэтому упрочнение при дорек-ристаллизационном отжиге иногда объясняли ошибкой эк-
Таблица 2. Максимальный прирост предела упругости (Дсг/сг) после дорекристаллиэационного получасового отжига при оптимальной температуре (/опт) (Э. Н. Спектор, С. С. Горелик, А. Г. Рахштадт)
Материал е. % Да — 100% *опт’ °С *р. °C
Си (99.98 %-ная) . . . 75 10 150 200
Си—7% А1 7 35 280 450
90 170 280 350
Си—32 %Zn . . 60 100 200 250
Ni (99,99 %-ный) 60 40 200 350
Ni—20% Сг . 10 40 400 800
65 170 400 600
Nb (электронно-лучевой, по
<0,01 % С, N, О) 90 40 950 1100
Nb (0,02 % С; 0,03 %О; 0,02 %
N) 80 120 1000 1200
Примечание, g — степень обжатия при холодной прокатке перед отжигом.
98
сперимента. Вместе с тем абсолютный прирост предела упругости в отдельных случаях превышает 100—300 МПа, что не только далеко выходит за рамки ошибок экспери
мента, но и позволяет использовать в промышленности низкотемпературный отжиг после холодной деформации для
Влияние температуры отжига
Рис. 51. на твердость холоднокатаных алюминиевых бронз (И. Л-. Рогельберг)
дополнительного повышения предела упругости пружин и мембран (на 100—170 %, см. табл. 2).
Явление упрочнения при дорекристаллизаци-онном отжиге свойственно большинству медных и никелевых сплавов, на которых оно изучено наиболее подробно. Величина упрочнения зависит от состава твердого раствора. У многих сплавов эффект упрочнения при отжиге возрастает с увеличением степени легирования твердого раствора (рис. 51). Упрочнение при отжиге обычно увеличивается с ростом степени холодной деформации (табл. 2), но встречается и обратная закономерность. Иногда упрочне
нию предшествует небольшое «нормальное» разупрочнение при возврате (см. рис. 51).
Очень интересно, что упрочнению при низкотемпературном отжиге часто свойственна обратимость: холодная деформация после отжига вызывает разупрочнение, последующий отжиг вновь дает упрочнение и т. д.[ Разупрочнение происходит при обжатиях всего 1—5 %. Например, если у чистого никеля после холодной прокатки <Уо,оо5= = 400 МПа, то отжиг при 200 °C повышает предел упругости до 480 МПа, а последующая холодная прокатка с обжатием 3 % снижает его до исходного уровня 400 МПа. Факт разупрочнения при повторной холодной деформации
весьма оригинален.
Природа упрочнения при дорекристаллизациоином отжиге в разных сплавах различна. Наиболее общей причиной упрочнения является закрепление подвижных дислокаций в исходном холоднодеформированном материале и в
7*
99
дислокационных стенках, возникающих при полигонизации во время отжига, атомами растворенных примесей или легирующих элементов (например, алюминия в растворе на базе меди). С этим согласуется то, что у металлов высокой чистоты величина рассматриваемого упрочнения меньше.
В концентрированных растворах на базе меди и никеля еще одной причиной упрочнения при дорекристаллизационном отжиге является возникновение областей с ближним порядком.. Отрывом дислокаций от атмосфер и разрушением областей с ближним порядком объясняется разупрочнение при холодной деформации после дорекри-сталлизационного отжига.
При дорекристаллизационном упрочняющем отжиге предел упругости возрастает значительно сильнее, чем твердость или временное сопротивление. Это и понятно, так как на величину предела упругости, характеризующего сопротивление материала очень малым пластическим деформациям, сильно влияет исходная (до начала механических испытаний) дислокационная структура, мало изменяющаяся в процессе испытаний. В то же время при определениях твердости и временного сопротивления, характеризующих сопротивление материала большим пластическим деформациям, исходная дислокационная структура сильно изменяется в самом процессе механического испытания: растет плотность дислокаций, они перераспределяются и создается новая дислокационная структура. Поэтому закрепление дислокаций в исходной структуре при дорекристаллизационном отжиге несравненно сильнее влияет на предел упругости, чем на временное сопротивление. Даже рост предела текучести <70,2 может быть на порядок меньше роста предела упругости <70,02 того же сплава.
Повышение предела упругости при дорекристаллизационном отжиге — это весьма общее явление, свойственное металлам и сплавам с любым типом решетки, металлам обычной и высокой чистоты.
В целом же зависимость прочностных свойств от температуры отжига, изображенная на рис. 45 пунктиром, является одной из типичных и очень часто встречающихся.
3. Изменение электросопротивления при отжиге
Электросопротивление изменяется при отжиге сложным образом. У многих деформированных металлов электросо
100
противление в значительной степени восстанавливается при дорекристаллизационном отжиге (см. рис. 48).
Принято считать, что с повышением температуры рекристаллизационного отжига электросопротивление снижается (если при наклепе оно возрастало), но справочные данные не согласуются с этим представлением. У меди, никеля, серебра, алюминия, железа и многих сплавов с повышением температуры рекристаллизационного отжига после обычного снижения электросопротивление по невыясненным причинам аномально возрастает (см. рис. 48).
Если при холодной деформации электросопротивление уменьшалось, как у хромеля, алюмеля и нейзильбера, то естественно, что отжиг приводит к росту электросопротивления. Это обусловлено восстановлением при отжиге ^состояния, нарушенного предварительной деформацией.
В заключение отметим, что разные свойства начинают интенсивно изменяться при разных температурах отжига. Например, электросопротивление обычно начинает падать при отжиге раньше, чем начнется сильное уменьшение временного сопротивления (см. рис. 48), так как электросопротивление может значительно снижаться из-за уменьшения концентрации точечных дефектов при возврате, а интенсивное падение временного сопротивления становится возможным только тогда, когда резко уменьшается плотность дислокаций.
§13. Анизотропия свойств отожженного металла
В рекристаллизованном металле с хаотично ориентированными кристаллитами векторные свойства отдельных монокристаллов статистически усредняются по всем направлениям в макрообъеме поликристалла. При наличии текстуры рекристаллизации отожженный металл анизотропен. Его анизотропия проявляется тем сильнее, чем совершеннее текстура. Чаще всего анизотропия свойств вредна, но в отдельных случаях требуется получить изделие, в котором какое-то свойство должно быть усилено в определенном направлении. Получение отожженного изотропного металла, или наоборот, металла с ярко выраженной анизотропией свойств — важная научная и техническая задача.
1. Фестонистость
Наибольший вред текстура рекристаллизации приносит в том случае, когда листы или ленты предназначаются для глубокой вытяжки. Холоднокатаный лист или ленту перед
101
штамповкой отжигают. Если при отжиге возникает достаточно совершенная текстура рекристаллизации, то лист становится анизотропным. В этом легко убедиться, вырезая плоские образцы для растяжения под разным углом к направлению прокатки (рис. 52). Особенно резко выраженная анизотропия механических свойств наблюдается у отожженных металлов и сплавов с г. ц. к. решеткой, обладающих текстурой куба {001}<001>, так как этой текстуре свойственна исключительно высокая степень совершенства. Например, в медной полосе с совершенной текстурой куба относительное удлинение вдоль и поперек направления прокатки равно 16 %, а под углом 45° к направлению прокатки 73 %. Резкую разницу в значениях относительного удлинения в этих направлениях легко понять, если учесть, что при растяжении монокристаллов меди перпендикулярно грани куба {100} получается низ
Рнс. 52. Схема вырезки разрывных образцов для выявления анизотропии механических свойств в отожженном медном листе
Рис. 53. Анизотропия относительного удлинения отожженной медной полосы, обладающей текстурой куба (Болдуин)
кое относительное удлинение, а перпендикулярно грани ромбического додекаэдра {110} — высокое. В случае текстуры куба ось разрывного образца, вырезанного вдоль или поперек направления прокатки, оказывается перпендикулярной плоскости {100}, а ось разрывного образца, вырезанного под углом 45° к направлению прокатки,— перпендикулярной плоскости {ПО}.
Анизотропия свойств выражена на графике в координатах свойство — угол к направлению прокатки (рис. 53).
При штамповке стакана из анизотропного листа возни
102
кает фестонистостъ — волнистая кромка (рис. 54). Высота фестона при штамповке, например, алюминиевых бидонов достигает 15—25 % от высоты бидона. Возникновение фестонов, называемых также ушами или языками, является результатом разной вытяжки металла по разным направлениям с соответствующим разным утонением стенки стакана. В зависимости от типа текстуры встречаются четыре или шесть симметрично расположенных выступов кромки.
При массовом производстве штампованных стаканов фес-тонистость является серьезной проблемой: волнистую кромку приходится обрезать и направлять на переплав, в связи с чем повышается расход катанда заготовок. Разнотолщинность же стакана вообще неустранима.
Величина фестонов тем больше, чем большее число зерен имеет одинаковую кристаллографическую ориентировку. Поэтому понятно, что величина фестонов зависит от режима обработки. Можно подобрать такие оптимальные условия прокатки и отжига, при которых фестонистость не проявляется. Например, для получения изотропной ме-
ди И латуни часто рекомен- Рис 54 Штампованный алюминиевый ДУЮТСЯ НебОЛЬШОе послед- стакан с четырьмя фестонами нее обжатие и невысокая температура последнего отжига.
При благоприятном химическом составе бесфестонис-тые стаканы можно получать в широком диапазоне степеней деформации и температур отжига. Такой состав, а также режимы обработки пока подбирают чисто эмпирически.
2. Текстурованная трансформаторная сталь
* Важным примером промышленного материала, в котором текстура рекристаллизации полезна, может служить трансформаторная сталь. Стальной лист в сердечнике трансформатора непрерывно перемагничивается. Около 0,4 % обще
103
го расхода электроэнергии теряется на нагревание сердечников трансформаторов.
Высокий к. п. д. трансформаторов обеспечивается минимальными потерями на перемагничивание вдоль направления магнитопровода.
Трансформаторная сталь содержит 2,8—3,5 % Si и минимально возможное количество углерода. Магнитные свойства монокристалла кремнистого железа сильно анизотропны: магнитная проницаемость ртах вдоль направления <100> в 30 раз больше, чем вдоль направления <111>.
С середины 30-х годов начали разрабатывать технологию получения трансформаторной стали с резко выраженной текстурой рекристаллизации, которая обеспечивает наиболее высокую магнитную проницаемость в направлении прокатки и минимальные потери на гистерезис, при этом ухудшаются магнитные свойства в других направлениях. Магнитопровод из текстурованной стали изготовляют так, что направление прокатки листа совпадает с направлением магнитного потока.
В настоящее время промышленность все в больших масштабах производит холоднокатаную трансформаторную сталь с острой ребровой текстурой {110}< 100>, называемой текстурой Госса.
При ребровой текстуре ребро куба <100>, т. е. направление самого легкого намагничивания, параллельно направлению прокатки, а плоскость ромбического додекаэдра {110} параллельна плоскости проката. Направление трудного намагничивания <111> находится под углом 54,7° к направлению прокатки, а направление промежуточного намагничивания <110>— поперек прокатки.
Промышленный лист толщиной 0,2—0,5 мм с ребровой текстурой получают холодной прокаткой, отжигом при ~850 °C и высокотемпературным длительным отжигом при ~ 1100 °C в водороде.
На формирование острой ребровой текстуры {110}< 100> сильно влияет структура исходной горячекатаной полосы, в приповерхностном слое которой при горячей прокатке возникает ребровая ориентация. При первом отжиге холоднокатаной ленты после первичной рекристаллизации имеется некоторое количество ребровых кристаллов, причем также преимущественно в приповерхностных слоях. При заключительном высокотемпературном отжиге во время частичного растворения и коагуляции нитридов или сульфидов развивается вторичная рекристаллизация, создающая острую ребровую текстуру {110}< 100>. Ребровые зерна, име
104
ющие размерное преимущество, растут за счет зерен других текстурных компонент, причем вторичная рекристаллизация развивается от поверхности к центру сечения ленты. Химическое травление, удаляющее поверхностный слой с зародышами вторичной рекристаллизации, ослабляет ребровую текстуру в трансформаторной стали.
Холоднокатаные (и отожженные) трансформаторные стали с ребровой текстурой практически вытеснили изотропные горячекатаные в производстве сердечников силовых трансформаторов. Их применение позволяет уменьшить массу и габариты мощных трансформаторов на 20—25 % и сильно снижает потери электроэнергии в народном хозяйстве.
§ 14. Выбор режимов дорекристаллизационного и рекристаллизационного отжига
Основные параметры отжига наклепанных металлов и сплавов — температура и продолжительность. Они определяют характер и полноту структурных изменений при отжиге , а также свойства металла и сплава после отжига. В отдельных случаях, которые будут рассмотрены ниже, важную роль играют также скорость нагрева до температуры отжига и скорость охлаждения с этой температуры.
1. Дорекристаллизационный отжиг
Дорекристаллизационный отжиг бывает смягчающим и упрочняющим.
а. Дорекристаллизационный смягчающий отжиг используют для повышения пластичности при частичном co-f хранении деформационного упрочнения. Его применяют, когда необязательно или нежелательно полное смягчение, достигаемое рекристаллизационным отжигом. Смягчающий дорекристаллизационный отжиг чаще всего служит окончательной операцией, придающей изделию требуемое сочетание прочности и пластичности.
Алюминиевые листы марок АД, АД1 и др. в большом количестве выпускают после дорекристаллизационного смягчающего отжига при 150—300 °C (температура отжига зависит от содержания примесей в алюминии, влияющих на! ^р). Дорекристаллизационный смягчающий отжиг широко применяют к магналиям, чтобы обеспечить последующие операции гибки, отбортовки и др.
В § 12 уже отмечалась легкость полигонизации алюми-ниевомагниевых сплавов, которая обусловливает разупроч-
105
некие при дорекристаллизациоином отжиге. Такой отжиг сплавов АМг1 и АМг2 при 150—180 °C обеспечивает сочетание повышенной (по сравнению с рекристаллизованным состоянием) прочности и пластичности, достаточной для проведения последующих операций, включающих пластическую деформацию. Так как листы из алюминия и маг-налиев составляет большую часть листовой продукции, выпускаемой заводами по обработке легких сплавов, понятно, насколько широко п эффективно используется дорекристал-лизационный смягчающий отжиг.
Для тугоплавких металлов VIA группы (Мо и W) до-рекрпсталлизационный отжиг — единственный способ смягчения после обработки давлением, так как при рекристаллизации они сильно охрупчиваются. Дорекристаллизацион-ный отжиг этих металлов и сплавов на их основе не только помогает частично снимать деформационное упрочнение, но и снижает температуру перехода из пластичного состояния в хрупкое (см. рис. 50).
Дорекристаллизационный отжиг часто наряду с повышением пластичности преследует цель уменьшить остаточные напряжения, стабилизировать свойства и повысить стойкость против коррозии..
Для выбора режима дорекристаллизационного отжига необходимо знать t” (при данной степени деформации).
б, Дорекристаллизационный упрочняющий отжиг применяют для повышения упругих свойств пружин и мембран. Оптимальную температуру его (см. § 12) подбирают опытным путем.
При дорекристаллизациоином отжиге холоднодеформированных медныХ сплавов (алюминиевых и хромовых бронз, медноникелевых сплавов) можно встретиться с «огневой» хрупкостью из-за образования пор по границам зерен. Поры возникают при отжиге под действием остаточных напряжений и по аналогии с порами, образующимися при ползучести, растут вследствие конденсации вакансий. Для борьбы с огневой хрупкостью продолжительность отжига в соответствующем критическом температурном интервале должна быть минимальной.
2. Рекристаллизационный отжиг
По условиям проведения и по назначению можно выделить несколько разновидностей рекристаллизационного отжига.
а. Полный рекристаллизационный отжиг, обычно называемый просто рекристаллизационным, — одна из наиболее широко применяемых операций термообработки.
106
Рекристаллизационный отжиг используют в промыш- . ленности как первоначальную операцию перед холодной обработкой давлением (для придания материалу наибольшей пластичности), как промежуточный процесс между операциями холодного деформирования (для снятия наклепа) и как окончательную (выходную) термическую обработку (для придания полуфабрикату или изделию необходимых свойств). Рекристаллизационный отжиг сталей» цветных металлов и сплавов применяют после холодной прокатки листов, лент и фольги, холодного волочения труб» прутков и проволоки, холодной штамповки и других ВИДОВ/ холодной обработки давлением.
В производстве полуфабрикатов и изделий из цветных металлов и сплавов рекристаллизационный отжиг как самостоятельная операция термообработки распространен гораздо шире, чем в технологии производства стали. Объясняется это тем, что по сравнению со сталями холодной обработке давлением подвергают несравненно большую долю цветных металлов и сплавов.
Оптимальный режим отжига можно выбирать до гра-г фикам температурной зависимости свойств (см. *рйс. v47 и 48). Так, для восстановления пластичности меди можно рекомендовать часовой отжиг при 500—700 °C (см. рис. 48). Верхняя температурная граница отжига выбрана ниже температуры перегрева (~800°C), а нижняя—с некоторым превышением I* (~ 400 °C), так как при определенной «геометрической» степени деформации изделия отдельные его участки деформированы неравномерно. В участках с меньшей степенью деформации рекристаллизация заканчивается при более высоких температурах.
При выборе режима отжига можно использовать диаграммы рекристаллизации (типй ‘£иС. 42), избегая получения очень крупного зерна и разнозернистости. При этом следует хорошо представлять себе те ограничения, которые связаны с использованием диаграмм рекристаллизации.
О качестве отожженного материала не всегда можно судить только по механическим свойствам. Другим показателем в отдельных случаях служит размер рекристаллизованного зерна. Так, например, крупнозернистость является причиной появления апельсиновой корки — характерной шероховатости изделий после глубокой вытяжки, растяжки и т. п.
В крупнозернистом материале неоднородность пластической деформации разных зерен выражена особенно сильно. Неоднородность деформации крупных зерен на свобод
107
ной поверхности изделия и проявляется в виде апельсиновой корки. При мелком зерне такой шероховатости не видно.
Скорость нагрева до температуры отжига чаще всего не имеет значения. Но в отдельных случаях необходим ускоренный нагрев. Так, при медленном нагреве до температуры отжига холоднодеформированных полуфабрикатов из алюминиевого сплава марки АМц вырастают очень крупные зерна, обусловливающие шероховатость поверхности после правки и снижающие относительное удлинение. Крупные зерна вырастают из-за того, что в случае медленного нагрева первичная рекристаллизация начинается при сравнительно низких температурах и идет из малого числа центров. После быстрого нагрева до высокой температуры (например, при нагреве в селитровой ванне до 500 °C) сразу развивается интенсивная первичная рекристаллизация немногих центров и зерно получается мелким.
~~ Скорость охлаждения с температуры рекристаллизационного отжига металлов и однофазных сплавов не сказываемся на их свойствах. Полуфабрикаты из медных сплавов для лучшего отделения окалины иногда охлаждают в воде. Если же сплав способен упрочняться при закалке и старении, то скорость охлаждения с температуры рекристаллизационного отжига иногда приходится регламентировать. Так, в термически упрочняемом алюминиевом сплаве В95 при отжиге после холодной деформации, кроме основного процесса — рекристаллизации, может протекать также побочный процесс — частичная закалка (подкалка) с последующим старением. В результате при отжиге не достигается необходимое смягчение материала. Поэтому сплав В95 следует медленно охлаждать вместе с печью с температуры рекристаллизационного отжига (380—430 °C) до температуры 150 °C (со скоростью не более 30 К/ч).
б. Рекристаллизационный отжиг для получения ультра-мелкого зерна в последнее время стал приобретать все большее значение. Ультрамелкими называют зерна размером менее ~ 10 мкм. Такие мелкие зерна соизмеримы с субзернами и отличаются от них только высокоугловыми границами. Ультрамелкозернистую структуру должны иметь листы, предназначенные для сверхпластической формовки — выдувки под небольшим газовым давлением деталей сложной конфигурации. Отжиг для получения ультра мелкого зерна проводят при очень большом числе центров рекристаллизации, причем в сплаве должны также иметься какие-то стопоры, препятствующие быстрому ук
108
Рис. 55. Схема обработки для получения ультра мелкозернистой структуры в листах сплава B95:
1 — закалка; 2 — гетерогенизационный отжиг; 3 — прокатка; 4 — рекристаллизационный отжиг
рупнению рекристаллизованных зерен во время сверхпластической формовки, производимой при высоких температурах. В некоторых алюминиевых сплавах» например типа В95, эти условия легко реализовать.
Лист обрабатывают по следующей схеме (рис. 55). Вначале заготовку закаливают в воде с 470 °C для получения пересыщенного алюминиевого раствора. Затем проводят гетерогенизационный отжиг (см. ниже § 28) при 400°C 10 ч для выделения из твердого раствора большого числа частиц фазы M(MgZns) размером около 1 мкм. При последующей холодной прокатке с обжатием 70—80 °/о около этих частиц в алюминиевом растворе возникают локальные изгибы кристаллической решетки, которые при последующем кратковременном отжиге (470 °C, 5 мин) служат местами зарождения рекристаллизованных зерен (см. § 7, п. 1). Получению ультрамелкого зерна при отжиге способствует быстрый нагрев в селитровой ванне. Роль стопоров высокоугловых гра
ниц выполняют дисперсоиды А17Сг размером менее 0,1 мкм, выделившиеся из алюминиевого твердого раствора при гомогенизационном отжиге слитка (см. § 1, п. 2).
в. Неполный рекристаллизационный отжиг проводят при температурах выше /*, но ниже / £ с целью частичного устранения наклепа. Он позволяет, например, производить полунагартованные листы из термически неупрочняемых алюминиевых сплавов. Структура получается частично рекристаллизованной, а частично полигонизованной. —
г. Текстурирующий отжиг применяют для получения выгодной анизотропии свойств в трансформаторной стали, железоникелевых сплавах с прямоугольной петлей гистерезиса и некоторых других материалах. Выбор оптимального режима отжига здесь — наиболее сложная задача.
д. Зонный (градиентный) рекристаллизационный отжиг используют для формирования сильно вытянутых столбчатых зерен. Сравнительно узкая зона нагрева до температур рекристаллизации перемещается вдоль холодного изделия, в котором таким образом создается резкий градиент
109
температур. Кристаллы растут в направлении движения горячей зоны, «поглощая» на движущемся фронте рекристаллизации все новые зерна. В этом направлении в рекристаллизованном изделии будет резко уменьшено число поперечных межзеренных границ и могут улучшиться некоторые свойства, например жаропрочность.
Градиентный рекристаллизационный отжиг после критической деформации в лабораторной практике служит для выращивания крупных монокристаллов.
Глава III
ОТЖИГ, УМЕНЬШАЮЩИЙ НАПРЯЖЕНИЯ
При обработке давлением, литье, сварке, термообработке, шлифовании, обработке резанием и других технологических процессах в изделии могут возникать внутренние напряжения, которые взаимно уравновешиваются внутри тела без участия внешних нагрузок. В большинстве случаев внутренние напряжения полностью или частично сохраняются в металле после окончания технологического процесса и поэтому называются остаточными напряжениями.
В данной главе рассматриваются только напряжения, уравновешивающиеся в объеме всего тела или отдельных его макрочастей. Такие напряжения называют также зональными или напряжениями I рода, чтобы отличить их от напряжений II рода (микронапряжений), уравновешивающихся в объеме отдельных кристаллитов или их частей.
Отжиг, уменьшающий напряжения, — это термическая обработка, при которой главным процессом является полная или частичная релаксация зональных остаточных напряжений.
§ 15. Возникновение и роль остаточных напряжений
Причинами возникновения внутренних макронапряжений (напряжений I рода) являются неодинаковая пластическая деформация или разное изменение удельного объема в различных точках тела.
Следующий простой пример поясняет возникновение остаточных напряжений при неодинаковой пластической деформации в разных участках металла (рис. 56). Представим себе, что полоса металла прокатывается в бочкообразных валках, диаметр которых посередине значительно больше, чем по концам. Центральные слои полосы получают большее обжатие, чем крайние. Если бы заготовка была
ПО
составлена, например склеена, .из набора прутков, то каждый из этих прутков получил бы вытяжку в соответствии со своим обжатием: центральные прутки должны были бы вытянуться сильнее, чем крайние. Но полоса металла в действительности является монолитным телом, в котором центральные и крайние слои не могут изолированно одни от других вытягиваться на разную длину. Поэтому центральные слои, которые стремятся сильнее вытянуться, будут испытывать сдерживающее влияние крайних слоев и окажутся недовытянутыми. Иначе говоря, в центральных слоях возникнут сжимающие внутренние напряжения. Крайние слои, наоборот, будут под действием центральных слоев вытянуты на величину больше той, которая определяется их обжатием. Поэтому в крайних слоях возникнут растягивающие внутренние напряжения (см. рис. 56). Напря-
-----з— Направление пранатки
Рис. 56. Возникновение остаточных напряжений в полосе из-за разного обжатия центральных и крайних слоев прн прокатке в бочкообразных валках:
/ — полоса до прокатки; 2 — полоса после прокатки; 3 — центральный слой полосы в случае свободной вытяжкн (без взаимодействия с соседними слоями)
жения разного знака взаимно уравновешиваются внутри полосы и сохраняются в металле после окончания прокатки.
Удельный объем меняется при термическом сжатии и расширении, кристаллизации расплава, фазовых превращениях в твердом состоянии и изменении химического состава поверхностных слоев. Если бы термическое расширение или сжатие, кристаллизация расплава и фазовые превращения в твердом состоянии проходили одновременно и в одинаковой степени по всему объему тела, то внутренние напряжения не возникали бы. Но при нагреве и охлаждении всегда имеется градиент температур по сечению тела, и поэтому указанные выше изменения удельного объема в разных точках металла протекают неодинаково, в результате чего возникают внутренние напряжения.
111
Различают термические и фазовые (структурные) внутренние напряжения, которые возникают соответственно в результате термического сжатия или расширения и фазовых превращений в твердом состоянии. Внутренние напряжения могут возникнуть практически при любой обработке, причем одна технологическая операция может привести к созданию разных по своему происхождению остаточных напряжений: термических, фазовых и напряжений от неоднородной пластической деформации. Например, при -Форячей обработке давлением, кроме напряжений, образовавшихся из-за неоднородной пластической деформации, могут возникнуть термические, а также фазовые напряжения, если горячедеформированный сплав охлаждается ускоренно и в нем протекает фазовое превращение. При литье, сварке и закалке возникают термические и фазовые напряжения. Различные по своему происхождению остаточные напряжения аглебраически складываются и очень часто дают весьма сложные эпюры.
: В соответствии с названием технологического процесса
1 различают литейные, сварочные, закалочные, шлифовочные и другие остаточные напряжения.
Остаточные напряжения сказываются на поведении изделия при обработке, эксплуатации и даже при хранении на складе.
Остаточные напряжения, алгебраически складываясь с рабочими напряжениями, могут их усиливать или ослаблять.
Как правило, наиболее опасны растягивающие остаточные напряжения, так как они, складываясь с растягивающими напряжениями от внешних нагрузок, приводят к разрушению, хотя эти нагрузки могут быть и невелики.
Особенно опасны растягивающие напряжения при трехосном растяжении. Как известно, напряженное состояние при трехосном растяжении — наиболее «жесткое», так как касательные напряжения, вызывающие пластическое течение, чрезвычайно малы или равны нулю, вследствие чего создаются благоприятные условия для хрупкого разрушения. Остаточные напряжения особенно опасны также в изделиях из малопластичных сплавов и таких, которые становятся хрупкими при понижении температуры.
При больших остаточных напряжениях разрушение часто происходит от незначительных по величине нагрузок (особенно ударных). Так, например, трещины в стальных отливках могут возникать при очистке их пневматическим молотком и даже от сквозняка зимой (из-за добавления термических напряжений к остаточным). Крупные слитки полунепрерывного литья из малопластичных алюминиевых сплавов через не
112
которое время после окончания литья могут разрушаться от случайных небольших сотрясений или ударов; освобождающаяся при разрушении упругая энергия так велика, что одна часть слитка весом в сотни килограммов с сильным треском отрывается н отлетает на расстояние в несколько метров.
Остаточные растягивающие напряжения в сварных конструкциях иногда приводят к серьезным авариям. Разрушения сварных мостов н цельносварных судов часто связаны с проявлением больших остаточных напряжений, близких к разрушающим. Известны случаи, когда цельносварные суда нз-за остаточных растягивающих напряжений разрушались под воздействием незначительных внешних факторов, например от удара ломом прн очистке палубы от льда. Проблема остаточных напряжений в сварных конструкциях привлекла к себе особенно большое внимание после того, как несколько цельносварных судов разломилось пополам во время плавания нли стоянки.
Растягивающие остаточные напряжения в поверхностных слоях особенно вредны для деталей, работающих при знакопеременной нагрузке, так как такие напряжения способствуют усталостному разрушению (напомним, что усталостная трещина зарождается на поверхности изделия).
Вредное действие остаточных напряжений сказывается в повышении общей химической активности металла. Особенно вредно усиление межкристаллитной коррозии под действием растягивающих остаточных напряжений (сезонное растрескивание латуни).
В металле с остаточными напряжениями существуют области упругих деформаций разного знака. Если разрезать изделие или срезать (а также стравить) с него поверхностный слой, то становится возможным упругое снятие макронапряжений. На измерении возникающих при этом упругих деформаций основаны механические методы определения величины и знака остаточных напряжений (напряжения вычисляют по деформациям).
Остаточные напряжения могут вызвать искажение формы (коробление) и изменение размеров изделия во время его обработки, эксплуатации или хранения на складе. Коробление появляется в результате деформаций изгиба и кручения, возникающих при нарушении равновесия внутренних сил и моментов. Особенно частые и сильные коробления появляются при обработке резанием, так как удаление слоя металла нарушает равновесие остаточных напряжений. Например, если от полосы на рис. 56 отрезать с одной стороны слой, в котором действуют растягивающие напряжения, то полоса выгнется в плоскости чертежа из-за нарушения равновесия растягивающих и сжимающих остаточных напряжений.
Самопроизвольные изменения размеров и коробление при хранении деталей происходят из-за постепенного перераспределения остаточных напряжений при их релаксации. Скорость релаксации (уменьшения) напряжений зависит от их исходного уровня: чем он выше, тем быстрее
8 И. И. Новиков
113
идет релаксация. Так как в разных участках сечения изделия величина остаточных напряжений различна, то из-за неодинаковой скорости их релаксации при комнатной температуре нарушается исходное равновесие внутренних сил и ; моментов. При этом остаточные напряжения перераспределяются и устанавливается новое состояние равновесия. Величина коробления тем больше, чем больше различие в степени релаксации остаточных напряжений в разных участках сечения и чем меньше жесткость изделия при изгибе. Иногда после сборки станков появляются недопустимые зазоры или натяги в сопряженных деталях, ранее точно при-k гаанных одна к другой.
Создавая контролируемые остаточные напряжения, которые вычитаются из рабочих напряжений, можно повысить эксплуатационные свойства металла. Чаще всего в поверхностном слое намеренно создают сжимающие остаточные напряжения, которые уменьшают опасные растягивающие рабочие напряжения. С этой целью применяют ' Дробеструйный наклеп, азотирование (см. § 67) и другие виды поверхностной обработки металлов.
В подавляющем большинстве случаев величина, знак и распределение остаточных напряжений по объему изделия неизвестны. Для определения этих характеристик требуется нарушить целостность изделия.
Рентгенографический метод позволяет по относительному изменению меж плоскостного расстояния оценить остаточные макронапряжения только в поверхностном слое. Поэтому всегда можно ожидать, что неконтролируемые остаточные напряжения от обработки давлением, литейные, сварочные, закалочные и другие остаточные напряжения могут ухудшить свойства металла и оказаться опасными при его обработке и эксплуатации изделия. В общем случае желательно полностью или хотя бы частично снять неконтролируемые остаточные напряжения.
§ 16. Уменьшение остаточных напряжений при отжиге
Избыточная энергия в упругодеформированных областях может понизиться без нарушения целостности тела, если разрядка остаточных напряжений произойдет путем пластической деформации. Следовательно, чтобы добиться полного или частичного снятия остаточных макронапряжений в изделии, необходимо вызвать в нем пластическое течение.
Остаточные напряжения при отжиге уменьшаются двумя путями: 1) вследствие пластической деформации в ус
114
ловиях, когда эти напряжения превысят предел текучести; 2) в результате-ползучести при напряжениях меньше предела текучести.
Рассмотрим первый путь. Остаточные напряжения, являющиеся упругими, по закону Гука пропорциональны модулю упругости и величине упругой деформации. С ростом температуры модуль упругости слабо снижается и соответственно слабо уменьшаются остаточные напряже- ' ния. Предел текучести также падает с ростом температуры, причем значительно более интенсивно, чем остаточные напряжения (рис. 57).
Выше некоторой температуры (6) предел текучести становится ниже остаточных напряжений и происходит
Рис. 57. Схема влияния температуры на остаточные напряжения (/) и предел текучести (2)
Продолжительность отжига
Рис. 58. Уменьшение остаточных напряжений (о) с увеличением времени выдержки при двух температурах (/2>М
пластическая деформация, в результате которой остаточные напряжения уменьшаются до значения предела текучести.
Нижняя температурная граница уменьшения остаточных напряжений не очень четкая, так как в разных участках тела действуют разнообразные по величине остаточные касательные напряжения.
При напряжениях выше предела текучести генерируется множество дислокаций, скольжение которых приводит к очень быстрой пластической деформации. Можно считать, что степень разрядки остаточных напряжений по такому механизму определяется температурой отжига, а не его продолжительностью.
Рассмотрим второй путь уменьшения остаточных напряжений, когда их величина’ меньше предела текучести. Если в какой-то момент испытания образца на растяжение перестать увеличивать деформацию и оставить образец в захватах разрывной машины под нагрузкой, то, как известно, в образце будет происходить релаксация напряжений.
8*
115
Напряжения в этом опыте падают вначале быстро, затем все медленнее и медленнее, и, наконец, достигается некоторый практически неизменный уровень напряжений. Общая деформация образца складывается из упругой и пластической:
еобщ ~ еупр епл- (18)
Длина образца, находящегося в захватах машины, остается неизменной. Напряжения в нем снижаются потому, что упругая деформация уменьшается при одновременном и равном увеличении пластической деформации, развивающейся в условиях ползучести. Релаксация при заданной общей деформации развивается путем ползучести при падающем напряжении.
В детали, в которой остаточные напряжения меньше предела текучести, упругая деформация также может с течением времени переходить в пластическую.
•В области температур, где остаточные напряжения меньше предела текучести (ниже t\ на рис. 57), ползучесть является единственно возможным механизмом их уменьшения. При напряжениях меньше предела текучести не происходит массового размножения и массового скольжения дислокаций. Медленное пластическое течение осуществляется перемещением ограниченного числа легкоподвижных дислокаций. Встречаясь с препятствиями (дислокационными сплетениями, дисперсными частицами, границами зерен), скользящие дислокации тормозятся. Исчерпание легкоподвижных дислокаций приводит к затуханию ползучести, а уменьшение остаточных напряжений еще более усиливает это затухание.
Флуктуации тепловой энергии активируют скольжение. С увеличением продолжительности отжига они вовлекают в скольжение менее благоприятно расположенные дислокации, помогают дислокациям преодолевать и обходить препятствия. При температурах отжига выше 0,5 Гп.п препятствия обходятся дислокациями переползанием, как и при обычной высокотемпературной ползучести.
Остаточные напряжения в изделиях* несколько снижаются вследствие ползучести и при комнатной температуре. Чем выше температура, тем больше термические флуктуации и тем быстрее и полнее уменьшаются остаточные напряжения (рис. 58).
Если изделия нагревать до температур (выше t\ на рис. 57), при которых предел текучести становится меньше остаточных напряжений, то вначале происходит быстрая раз
116
рядка напряжений до величины предела текучести вследствие массового размножения и скольжения дислокаций, а затем этот механизм пластической деформации сменяется ползучестью, приводящей к постепенной и затухающей во времени релаксации напряжений.
Быстрая разрядка остаточных напряжений при повышенных температурах иногда бывает опасной, так как из-за неоднородного распределения напряжений по сечению и длине изделия она может привести к нарушению равновесия внутренних сил и моментов, сопровождающемуся короблением. Идеальна для отжига релаксация напряжений в ее классическом виде, когда медленная пластическая деформация нарастает при одновременном и равном уменьшении упругой деформации так, что линейные размеры в направлении действия сил не изменяются.
Чем выше уровень исходных напряжений, тем быстрее в первый период происходит их разрядка и тем больше опасность коробления.
Продолжительность отжига для уменьшения напряжений устанавливают опытным путем. Определенной температуре отжига в каждом конкретном изделии соответствует свой конечный уровень остаточных напряжений, по достижении которого увеличивать продолжительность отжига практически бесполезно (см. рис. 58).
При выборе производственного режима отжига следует прибегать к натурным испытаниям, так как каждая конкретная деталь характеризуется своим распределением остаточных напряжений до и после отжига.
Во многих случаях уменьшение остаточных напряжений является побочным процессом, совершающимся при разнообразных операциях термообработки одновременно с основными структурными и фазовыми изменениями. Например, литейные напряжения уменьшаются при гомогениза^ ционном отжиге. При высоком отпуске стали наряду с основным процессом превращения мартенсита в сорбит уменьшаются закалочные напряжения. Остаточные напряжения, возникшие в результате холодной обработки давлением, уменьшаются при рекристаллизационном отжиге, основной целью которого является снятие наклепа. _
Нагрев для уменьшения остаточных напряжений довольно часто приходится применять как самостоятельную операцию термообработки, которая в этом случае и называется отжигом для уменьшения напряжений.
Использование отжига для уменьшения напряжений лимитируется теми нежелательными структурными и фазовы
117
ми изменениями, которые могут произойти при нагреве. Например, чтобы достаточно полно снять закалочные напряжения в изделиях из тер’/пчески упрочняемых алюминиевых сплавов необходимо нагревать их до температур около 230—260°C. Но при таких температурах в алюминиевых сплавах происходит перестраивание, сопровождающееся снижением прочности. Для полною снятия остаточных макронапряжений поел? холодной обработки давлением желательно проводить о** жиг при таких темпе-турах, при которых протекает рекристаллизация. Но при этом снимается упрочнение от холодной обработки давлением, что во многих случаях недопустимо. Поэтому приходится либо мириться с недостаточно полным снятием остаточных напряжений при низких температурах, либо идти на компромисс, достигая более полного снятия напряжений при некотором ухудшении механических и других свойств.
Скорости нагрева и особенно охлаждения при отжиге должны быть небольшими, чтобы не возникли новые внутренние термические напряжения.
Отжиг для уменьшения остаточных напряжений широко применяют в технике. Он уменьшает вредные остаточные растягивающие напряжения, особенно опасные при объемном напряженном состоянии, позволяет повысить допустимые внешние нагрузки, повышает сопротивляемость усталости и ударным нагрузкам, снижает склонность к хрупкому разрушению, межкристаллитной коррозии и коррозионную усталость, стабилизирует размеры и предотвращает коробление и поводку изделий.
Если рабочие напряжения при эксплуатации изделий сравнительно небольшие (далеки от предела текучести) и основное требование к деталям — высокая стабильность размеров и формы, то для предотвращения коробления не обязательно добиваться значительного уменьшения остаточных напряжений. Примером могут служить малонагру-женные базовые детали точных станков и приборов, отливаемые из дешевого серого чугуна. Старинная практика предотвращения коробления таких отливок — вылеживание их на складе при температуре окружающей среды в течение нескольких месяцев и даже нескольких лет. Этот способ стабилизации размеров чугунных отливок называют естественным старением1. Как оказалось впоследствии, при
1 К широко известному виду термической обработки — естественному старению после закалки (см. § 49) — этот процесс никакого отношения не имеет.
118
длительном вылеживании чугунных отливок остаточные напряжения в них уменьшаются всего на 5—20 %, хотя такое вылеживание и предотвращает коробление деталей. Это можно объяснить тем, что коробление отливок из серого чугуна вызывается релаксацией главным образом пиковых напряжений в местах их концентрации около пластинок графита. При сравнительно невысоком общем уровне остаточных напряжений они около таких концентраторов, как графитовые включения, могут приближаться к пределу текучести и в этих местах быстрее релаксировать. Для доказательства образцы из стали, чугуна с шаровидным графитом и серого чугуна нагрузили до одинакового исходного напряжения. Затем в течение полугода следили за релаксацией напряжений. Оказалось, что медленнее всего напряжения релаксировали в образцах из стали, несколько быстрее — в образцах из чугуна с шаровидным графитом и наиболее быстро — в образцах из серого чугуна с грубыми пастинами графита. Можно предположить, что при комнатном вылеживании отливок из серого чугуна релаксируют главным образом пиковые напряжения около включений графита, что предотвращает коробление деталей, хотя и мало сказывается на измеряемых средних остаточных напряжениях*
Многомесячное вылеживание отливок из серого чугуна в условиях современного производства трудно использовать как основной способ предотвращения коробления. Для ускорения стабилизации размеров применяют отжиг чугунных отливок при 500—600 °C в течение 2—4 ч.
Стали отжигают для уменьшения напряжении при температурах обычно несколько ниже критической точки Ас19
Холоднокатаные листы и штамповки из меди, никеля, титана и деформируемых сплавов на их основе отжигают для уменьшения остаточных напряжений при температурах не выше точки начала рекристаллизации, чтобы сохранить высокие прочностные характеристики наклепанного металла. Отжиг для уменьшения напряжений широко применяют к латуням, содержащим более 20 % Zn, так как они характеризуются сильной склонностью к коррозии под напряжением («сезонная болезнь»).
Если слитки непрерывного литья из деформируемых алюминиевых сплавов не подвергались гомогенизационно-му отжигу, то перед разрезкой на мерные заготовки их следует отжечь при 300—350 °C для уменьшения остаточных напряжений во избежание опасного разрушения слитков при резке. Для уменьшения сварочных напряжений в осо
119
бо ответственных громоздких конструкциях строят специальные печи с большим рабочим пространством для помещения в них целиком всего изделия.
Временные перегрузки для уменьшения остаточных напряжений
Как уже отмечалось, разрядка остаточных напряжений при отжиге происходит только в результате развития пластической деформации. Если остаточные напряжения значительно меньше предела текучести, то релаксируют они медленно вследствие развития ползучести. Для более полного и более быстрого уменьшения остаточных напряжений используют различные временные перегрузки изделия, которые, суммируясь с остаточными напряжениями, вызывают пластическую деформацию во всем изделии или в отдельных его участках.
Самые старые способы, создания временных перегрузок— принудительная вибрация и остукивание со всех сторон чугунных отливок. Эти простые приемы весьма эффективно предотвращают коробление. Для стабилизации размеров чугунных отливок можно применять также временное статическое нагружение. Например, в длинных станинах создают с помощью домкратов изгибающие моменты.
Для снятия остаточных напряжений и одновременно для правки полуфабрикаты из алюминиевых сплавов (прутки, трубы и др.) растягивают на 1—3 % на специальных растяжных машинах.
Временную нагрузку создают и чисто термической обработкой, для чего используют два способа — термоциклирование и термоудары.
Термоциклирование наиболее целесообразно применять к сплавам, которые содержат фазы с резко различающимися термическими коэффициентами линейного расширения, например к силуминам. Термоциклированию подвергают детали из силуминов, к которым предъявляют особо жесткие требования по стабильности размеров во время хранения и эксплуатации высокоточных приборов. Детали из силу-мйнов типа АЛ2 и АЛ9 охлаждают до температур минус 40—минус 196 °C, затем отогревают до комнатной температуры и помещают в печь, нагретую до 150 °C (или же сразу переносят в печь). Затем детали охлаждают до комнатной температуры и вновь обрабатывают холодом. В течение трех циклов такой обработки (последней всегда должна быть операция нагрева) остаточные напряжения уменьшаются на 30—70 %. Обычный длительный отжиг при
120
верхней температуре термического цикла (150 °C) несравненно слабее уменьшает остаточные напряжения.
Из-за большой разницы в термических коэффициентах линейного расширения алюминиевой и кремниевой фаз (примерно в 6,5 раз) на межфазных границах возникают микронапряжения. При обработке холодом эти микронапряжения усиливаются и, складываясь с остаточными напряжениями, вызывают при нагреве значительные пластические деформации. Таким образом, чередование захолаживания до отрицательных температур и последующего нагрева усиливает пластическое течение в микроучастках п этим способствует более полному уменьшению остаточных напряжений.
Естественному термоциклированию (с незакономерными циклами изменения температуры под действием солнца, ветра и дождя) подвергаются чугунные отливки во время вылеживания под открытым небом. Возникающие при этом временные перегрузки способствуют большей стабилизации размеров, чем вылеживание тех же отливок в закрытом помещении.
Метод термоударов состоит в создании при быстром нагреве изделия термических напряжений, которые, вызывая временную перегрузку, способствуют релаксации остаточных напряжений. Самый простой способ — установка в разогретую печь чугунных отливок, имеющих большую разностей ность.
Чтобы снизить закалочные напряжения в плитах, штамповках и других деталях из алюминиевых сплавов, которые нельзя отжигать из-за недопустимости разупрочнения, предлагается применять обработку холодом с последующим термоударом. Закаленное изделие помещают в жидкий азот (—196°C), а затем быстро нагревают до невысоких температур в кипящей воде или струе пара. Предварительное захолаживание в жидком азоте позволяет увеличить термические напряжения во время термоудара при невысокой температуре нагрева.
Раздел
второй ОТЖИГ ВТОРОГО РОДА
Отжиг 2-го рода основан на использовании диффузионных (нормальных) фазовых превращений при охлаждении ме-таллов и сплавов1.
Принципиальную возможность применения к сплаву отжига 2-го рода можно установить по диаграмме состояния. В твердом состоянии протекают разнообразные фазовые превращения: полиморфное, эвтектоидное, растворение одной фазы в другой при нагреве и обратное выделение при охлаждении и др.
Отжиг 2-го рода можно проводить с полным изменением фазового состава, когда фазы, существовавшие при комнатной температуре, исчезают при нагреве, а фаза, стабильная при повышенной температуре, исчезает при охлаждении. Примером являются превращение перлита в аустенит и распад аустенита с образованием перлита (см. ниже рис. 74).
Если изменение фазового состава связано только с переменной растворимостью компонентов в твердом состоянии, то основная фаза, в которой растворяется избыточная •фаза, стабильна и при низких, и при высоких температурах. В сплавах этого типа при нагреве и охлаждении изменяется только количественное соотношение фаз (включая полное исчезновение одной из них при нагреве). Примером являются переход СиА12 в алюминиевый раствор при^ нагреве и выделение этого интерметаллида из твердого раствора- при охлаждении (см. рис. 7).
Практически целесообразность отжига 2-го рода определяется тем, насколько сильно структурные изменения влияют на свойства металла или сплава. Его целевое назначение весьма разнообразно и будет указано при рассмотрении конкретных разновидностей отжига.
Основные параметры отжига 2-го рода: температура нагрева, время выдержки при этой температуре и скорость охлаждения. Температура нагрева и время выдержки должны обеспечить необходимые структурные изменения, например полное растворение избыточной фазы. Скорость охлаждения должна быть достаточно мала, чтобы при понижении температуры успели пройти обратные фазовые
1 О механизме нормальных превращений см. в § 34.
122
превращения, в основе которых лежит диффузия (или са-модиффузия). Обычно при отжиге изделия охлаждают вместе с печью или на воздухе вне печи.
Так как отжиг 2-го рода'основан на использовании фазовых превращений в твердом состоянии, то вначале рассмотрим общие закономерности таких превращений.
Глава IV
ОБЩИЕ ЗАКОНОМЕРНОСТИ ФАЗОВЫХ ПРЕВРАЩЕНИИ В ТВЕРДОМ СОСТОЯНИИ
§17. Термодинамика фазовых превращений
Как известно, при постоянных температуре и давлении все самопроизвольные процессы в системе идут в сторону уменьшения энергии Гиббса. Равновесное состояние характеризуется минимальным значением энергии Гиббса. Энергия Гиббса (или изобарно-изотермический потенциал), являющаяся характеристической функцией системы, определяется следующим соотношением:
G^H—TS, (19)
где Н — энтальпия; S •— энтропия; Т — температура.
Из обобщенного уравнения первого и второго законов термодинамики следует, что (dG/dT)P=—S и (d2G/dT2)P = =—(d'S/dT)P<Of т. е. и первая, и вторая производные — отрицательные величины. Это означает, что с ростом температуры энергия Гиббса всегда уменьшается, а кривые, показывающие зависимость энергии Гиббса от температуры (рис. 59), должны быть обращены вогнутостью к оси температур (а не наоборот, как их часто неверно изображают).
Рассмотрим фазовое превращение в однокомпонентной системе, в частности полиморфное превращение в металле.
Если две фазы однокомпонентной системы могут находиться в равновесии, то кривые энергий Гиббса этих фаз должны пересекаться (см. рис. 59).
Точка пересечений кривых показывает равенство энер гий Гиббса двух фаз и соответствует температуре их устойчивого равновесия (Го).
Из графика на рис. 59 видно, что ниже температуры То a-фаза обладает меньшей энергией Гиббса, чем (3-фаза. Так как система стремится уменьшить свою энергию Гиббса, то при охлаждении (3-фаза должна при температурах ниже То превратиться в а-фазу.
Температура превращения при охлаждении лежит ниже,
123
чем температура обратного фазового превращения при нагреве. Это явление называется тепловым гистерезисом превращения. Например, белое олово превращается в серое только при сильных морозах, хотя температура равновесия белой и серой модификаций равна -J-13 °C. Разность между температурой устойчивого равновесия двух фаз (То) и фактической температурой превращения при охлаждении называется степенью переохлаждения (ДТ).
ЛСД, JWf/C
0,007
0005
0,003
0,001
СЗ.Ц. 1
210s
О
О 100 200 JOO 400
Степень переохлаждения, °C
I-------------------1__________________I__________________I___________________I
723 623 523 423 323
Температура превращена#, °C
Рис. 59. Зависимость энергии Гиббса двух фаз от температуры
Рис. 60. Зависимость скорости зарождения центров и линейной скорости роста перлитных колоний от степени переохлаждения в стали с 0,8 % С (по И. Л. Миркину)
Степень переохлаждения растет с увеличением скорости охлаждения. Следовательно, переохлаждение связано с какими-то затруднениями в фазовом превращении.'
Г. Тамман ввел количественные характеристики процесса кристаллизации расплава — число центров, возникающих в единицу времени в единице объема, т. е. скорость зарождения центров (с. з. ц.) и линейную скорость роста кристаллов из этих центров (л. с. р.). С увеличением переохлаждения обе эти характеристики растут, достигают максимума и затем падают до нуля. Графики зависимости скорости зарождения центров кристаллизации и линейной скорости роста кристаллов от переохлаждения называют кривыми Т аммана.
Позднее было установлено, что кристаллизационные процессы образования фаз в твердом состоянии подчиняются тем же общим закономерностям, что и процессы кристаллизации жидкости. На рис. 60 показаны кривые Таммана для эвтектоидного распада в стали.
124
Зависимости, полученные экспериментально, можно объяснить, используя представления термодинамики и статистической физики.
На первый взгляд кажется, что зародышем кристаллизации может явиться сколь угодно малая совокупность атомов, например одна элементарная ячейка новой фазы, которая растет вследствие постепенного присоединения атомов из материнской фазы. Более подробный анализ показывает, что это не так.
Рассмотрим изотермический процесс образования а-фа-зы в материнской p-фазе при некоторой температуре 1\ ниже температуры То (см. рис. 59).
При образовании частиц ос-фазы энергия Гиббса системы должна уменьшиться, так как при температуре 1\ новая фаза более устойчива и обладает меньшей энергией Гиббса, чем исходная p-фаза. Уменьшение энергии Гиббса при образовании одного кристаллика ос-фазы AGO6=^Ag, где V — объем этого кристаллика, Ag — разность удельных энергий Гиббса двух фаз при температуре (см. рис. 59)*.
С появлением нового кристалла связано возникновение поверхности раздела между новой фазой и исходной, на создание которой затрачивается работа. Поверхностная (точнее— межфазная) энергия кристалла АСПов=$у, где s — площадь поверхности кристалла, а у — межфазная энергия единицы поверхности (поверхностное натяжение).
Таким образом, при образовании частицы новой фазы внутри материнской энергия Гиббса системы, с одной стороны, должна уменьшиться вследствие перехода некоторого объема p-фазы в более устойчивую a-фазу, а с другой — увеличиться вследствие образования поверхности раздела с избыточной межфазной энергией. В итоге действительное изменение энергии Гиббса при образовании одного кристаллика выразится алгебраической суммой:
AG = — AGo6 4- AGnOB или AG — VAg + sy. (20)
Если принять, что форма кристаллика кубическая, то тогда AG=—a3Ag-|-6a2y, где а — длина ребра куба. Все последующие выводы могут быть получены и для кристаллов любой другой формы. Так как Ag и у постоянны для данной температуры Гь то AG является функцией размера кристалла (рис. 61). Физический смысл этой зависимости состоит в следующем: чем меньше частица, тем больше отношение ее поверхности к объему. Например, для куба если
* В данном случае под удельной энергией Гиббса понимают энергию Гиббса единицы объема.
125
а=10, то s/V= (6-102)/103=0,6; если а=1, то s/V= = (6-12)/13=6.
Следовательно , чем меньше частица, тем большая доля от общей энергии приходится на межфазную энергию. Если возникает частица с размером меньше некоторого aGf то приращение энергии Гиббса вследствие образования новой поверхности (+AGn0B) перекрывает уменьшение энергии Гиббса, происходящее в результате перехода части объема материнской фазы в более устойчивую модификацию (-—AGog) и в целом энергия Гиббса системы увеличивается (+AG). Чем больше размер частицы по сравнению с неко-
торым критическим размером акр, тем меньше сказывается межфазная энергия, а возникновение кристаллов с размером больше aG приводит к уменьшению энергии Гиббса системы (—AG) (см. рис. 61). <
Если появился кристаллик новой фазы с размером меньше критического, то дальнейший его
рост должен сопровождаться рис. 61. Зависимость изменения увеличением энергии Гиббса. Та-нии^^ристалла3 отП₽1его°^азм^а К0Й КрИСТЭЛЛИК ТерМОДИНЯМИЧе-(г2<Гь см. рис. 59) ски неустойчив, он рассасывает-
ся в исходной фазе. Если же каким-либо путем возникает кристаллик с размерами боль-'
ше акр» то он может самопроизвольно расти, так как его рост сопровождается уменьшением энергии Гиббса. Следовательно, центрами кристаллизации являются только способные к самопроизвольному росту зародыши новой фазы, имеющие размеры не меньше некоторой критической ве-
личины аКр.
Критический размер зародыша определяем, приравняв к нулю первую производную от AG по а, так как при а=акр функция проходит через максимум:
dAG/da = - 3^р Ag + 12акр у = О, откуда пкр = 4y/Ag. (21)
Так как при увеличении степени переохлаждения поверхностное натяжение у практически не изменяется, a Ag непрерывно растет (см. рис. 59), то критический размер зародыша уменьшается с ростом переохлаждения, что отображено на рис. 61, на котором пунктирная кривая соответствует большему переохлаждению.
126
Теперь остается выяснить, каким образом внутри материнской фазы сразу возникает частица с размерами не меньше определенного критического значения.
На образование критического зародыша затрачивается работа (+AGKp), так как образуется поверхность раздела фаз (см. рис. 61). Эту работу можно выразить через Ag и у:
ДОкР = -акрДёг + 6йкРТ. (22)
Подставляя сюда значение акр из выражения (21), получим
AGKP = — (4y/Ag)3 Ag + 6 (4y/Ag)2 у,
т. е. AGKP = 32y3/(Ag)2. (23)
Так как с увеличением степени переохлаждения величина у практически не изменяется, a Ag растет (см. рис. 59), то работа образования критического зародыша с ростом переохлаждения быстро уменьшается (обратно пропорционально квадрату Ag).
Работу образования критического зародыша при заданном переохлаждении можно выразить через поверхностную энергию. Из формулы (21) следует, что Ag=4y/aKp. Подставляя это значение в выражение (22), получим
AG = — а3 -^- + 6а2 6°KpY .
кр кр оКр кр 3
Так как 6a£p — это площадь поверхности зародыша s,
то АСкр = -1. sy. (24)
О
Таким образом, работа образования критического зародыша равна одной трети его межфазной энергии1. Остальные две трети межфазной энергии компенсируются уменьшением объемной энергии Гиббса.
Откуда же берется в замкнутой системе энергия, требуемая для образования критического зародыша?
В кристалле колеблющиеся атомы непрерывно обмениваются между собой кинетической энергией. Из-за хаотичности теплового движения энергия распределена между атомами неравномерно. В определенный момент энергия разных атомов различна, а для одного атома она меняется от
1 Так как поверхностное натяжение на разных гранях кристалла различно, то, строго говоря, в формуле для работы образования критического зародыша следует вместо sy поставить сумму S1Y1+S2Y2+ — -+s«Yn> гДе п— число граней.
127
одного момента времени к другому. Всегда имеются атомы и группы атомов, кинетическая энергия которых больше или меньше средней величины.
Отклонение энергии от среднего значения называется флуктуацией энергии. Флуктуационное повышение энергии в группе атомов материнской фазы может обеспечить работу образования критического зародыша. Критический зародыш образуется там, где участок материнской фазы размером не меньше критического обладает повышенной энергией не ниже определенного уровня.
Таким образом, хотя у решетки новой фазы запас энергии Гиббса меньше, чем у решетки материнской фазы, переход от одной фазы к другой при образовании центра кристаллизации происходит через промежуточное состояние с повышенной энергией Гиббса, большей, чем у исходной фазы.
Теперь можно объяснить зависимость скорости зарождения центров новой фазы от переохлаждения. С увеличением степени переохлаждения уменьшаются размер критического зародыша и работа его образования. Следовательно, чем больше степень переохлаждения, тем меньшая по уровню энергии и геометрическим размерам требуется флуктуация, на базе которой образуется критический зародыш, и-тем больше число таких флуктуаций. Поэтому с ростом степени переохлаждения увеличивается число критических зародышей, возникающих в единицу времени в единице объема в результате флуктуаций энергии. Их число должно быть пропорционально фактору e~^G^kT.
Критический зародыш становится центром кристаллизации только тогда, когда он присоединяет к себе один или несколько атомов от материнской фазы. Частота присоединения атомов к зародышу пропорциональна фактору e~Q/hT, где Q — энергия активации перехода атома через границу раздела фаз.
Следовательно, скорость зарождения центров кристаллизации, зависящую от вероятности образования определенных энергетических флуктуаций и вероятности присоединения атомов к критическим зародышам, можно представить следующим выражением:
j = Ae~^/kT е ~Q/kT . (25)
При температуре равновесия двух фаз Го, когда степень переохлаждения ДГ=0 и движущая сила превращения Ag=0 (см. рис. 59), в соответствии с выражением (21)
128
Окр = оо и превращение не может развиваться [сомножитель е~ЛС?кр/ЛТ в уравнении (25) равен нулю].
С увеличением степени переохлаждения быстро уменьшается работа образования критического зародыша AGKp и соответственно быстро нарастает число энергетических флуктуаций, на базе которых возникают критические зарсь. дыши. ' -
Однако одновременно с понижением температуры уменьшается вероятность присоединения атомов к критическому зародышу. Энергия активации Q не зависит от температуры, и сомножитель e~Q/hT в формуле (25) быстро уменьшается при снижении температуры, стремясь к нулю, когда температура приближается к абсолютному нулю.
Таким образом, с одной стороны, с увеличением степени переохлаждения возрастает разность объемных энергий Гиббса — движущая сила превращения, уменьшаются критический размер зародыша и работа его образования, что способствует увеличению с. з. ц. С другой стороны, с ростом степени переохлаждения уменьшается подвижность атомов, что затрудняет образование центров кристаллизации.
В результате совместного влияния двух противоположно действующих факторов кривая Таммана проходит через максимум. Важно, что с. з. ц. может стать ничтожно малой при температурах, далеких от абсолютного нуля (см. рис.. 60). Создавая достаточно большое переохлаждение, мы можем подавить диффузионное превращение в твердом состоя^ нии, на чем и основана закалка сплавов.
Во время превращения в твердом состоянии действует еще один фактор, затрудняющий зарождение центров превращения. Новая фаза всегда отличается от исходной структурой и удельным объемом. Так как превращение развивается в упругой кристаллической среде, то изменение удельного объема при этом вызывает появление энергии упругой деформации в одной или обеих фазах, что затрудняет превращение и повышает энергию Гиббса. Следовательно, в изменение энергии Гиббса при превращении в твердом состоянии вносит положительный вклад некоторая упругая составляющая AGynP, которую необходимо учесть в выражении (20):
ДО = - Д0об + ДОпов + Д6уПр. (26)
Слагаемое ДОупр, которое не было учтено при выводе формул (21), (23), (24) и (25), качественно не меняет заключения о характере зависимости изменения энергии Гиббса при изотермическом превращении ДО от размера заро-
9 И. И. Новиков
129
дыша (см. рис. 61) и о характере зависимости с. з. ц. от степени переохлаждения (см. рис. 60). Это слагаемое лишь влияет на величины аКр, AGKp и J. Иногда такое влияние может быть очень сильным. Например, из-за увеличения удельного объема на 25 % превращение белого олова в серое связано с возникновением столь большой величины AGynp, что оно начинает самопроизвольно развиваться только на морозе при •—20 °C, и то весьма редко (температура равновесия обеих модификаций олова + 13 °C).
Анализ, аналогичный приведенному для с. з. ц., позволяет теоретически объяснить характер зависимости линейной скорости роста кристаллов от степени переохлаждения.
Рис. 62. Зависимость средней скорости фазового превращения от степени переохлаждения
Рис. 63. Зависимость средней скорости фазового превращения от степени перегрева
Максимумы скорости зарождения и линейной скорости роста соответствуют разным степеням переохлаждения, причем восходящая ветвь кривой скорости зарождения сдвинута в сторону больших переохлаждений (см. рис. 60).
Скорость зарождения центров и линейная скорость роста кристаллов определяют суммарную скорость фазового превращения. Средняя скорость изотермического фазового превращения вначале растет с увеличением степени переохлаждения, достигает максимума и затем падает (рис. 62)*. Среднюю скорость фазового превращения экспериментально определяют как обратную величину времени полного изотермического превращения. *
Приведенный в этой главе термодинамический анализ образования зародышей новой фазы относился к изотерми
* В § 21 будет показано, что истинная объемная скорость изотермического превращения закономерно меняется во время превращения, поэтому здесь речь идет о средней скорости.
130
ческим условиям. Скорости зарождения и линейного роста участков новой фазы экспериментально также измеряют при определенных переохлаждениях. В практике же термической обработки наибольший удельный вес занимают процессы с непрерывным охлаждением. Это не обесценивает наши выводы, полученные для изотермического фазового превращения, так как при непрерывном охлаждении металл проходит через бесчисленное множество степеней переохлажде-_ ния. Чем быстрее охлаждается металл, тем меньше успеваГ ет образовываться центров кристаллизации новой фазы при каждой степени переохлаждения и тем сильнее переохлаждается материнская фаза.
До сих пор в этой главе рассматривалось фазовое превращение при охлаждении. Основные положения флуктуационной теории зарождения центров новой фазы справедливы и для процессов фазового превращения при нагреве. С увеличением степени перегрева выше TQ растет разность энергий Гиббса исходной и новой фаз (см. рис. 59) и уменьшаются размеры критического зародыша и работа его образования.
Основное отличие фазового превращения при нагреве от превращения при охлаждении — это нарастающее увеличение подвижности атомов при удалении от температуры равновесия двух фаз. Поэтому с увеличением степени перегрева, скорость превращения интенсивно растет вследствие умень/ шения размера критического зародыша и работы его обра; зования, а также увеличения подвижности атомов (рис. 63). Этим объясняется то, что при превращениях в твердом сог стоянии достигается степень перегрева обычно гораздр меньше, чем степень переохлаждения. -Д
При фазовых превращениях образующаяся фаза очень часто отличается по химическому составу от исходной фазы. Поэтому для образования критического зародыша недостаточно энергетической флуктуации, необходима еще флуктуация концентрации, В твердом растворе при определенном среднем химическом составе имеются участки с большим и меньшим содержанием того или иного компонента. Очевидно, чем больше отклонение от среднего состава и больше размер участка с таким отклонением, тем меньше вероятность концентрационной флуктуации в пределах исходной’ фазы. Например, чтобы в аустените эвтектоидного состава, (0,8 % С) образовался зародыш цементита, содержащего 6,67 % С, необходимо местное повышение концентрации углерода примерно в восемь раз. В аустените имеются участ^ ки, богатые и бедные углеродом. Расчет, основанный на 9* 13f
теории вероятностей, показывает, что в 1 см3 аустенита эвтектоидного состава имеется в 3,3 -1016 обогащенных угле-. родом участков размером в 6 элементарных ячеек с концентрацией углерода 6,67 % и всего лишь 1000 обогащенных углеродом участков с такой же концентрацией, но размером в 24 элементарные ячейки (всего в 1 см3 аустенита находится 2,14 -1022 элементарных ячеек).
Следует учитывать различие между флуктуациями состава внутри одной фазы и дисперсной смесью двух фаз. Флуктуации концентрации все время возникают и исчезают. Каждой температуре и среднему составу раствора соотвествует свое, не зависящее от времени распределение концентрационных флуктуаций по их величине. Такое распределение существует и в метастабильной, например переохлажденной, фазе до начала превращения. Двухфазная же система не может характеризоваться равновесным распределением частиц фаз по их размерам, так как эти частицы беспредельно стремятся к укрупнению — вначале растут за счет исходной фазы, а затем коагулируют соседние частицы, принадлежащие одной фазе.
Необходимость флуктуации концентрации в добавление к флуктуации энергии затрудняет образование центров новой фазы, особенно в тех случаях, когда составы исходной и новой фаз сильно различаются. Термодинамика процессов образования фазы, отличающейся по составу от исходной, более подробно изложена в § 45.
§ 18. Роль строения межфазных границ при фазовых превращениях
Межфазная энергия на границе зародыша с исходной фазой зависит от строения этой границы. Различают три типа межфазных границ: когерентные, полукогерентные и не когерентные.
На когерентной границе решетка одной фазы плавно переходит в решетку другой фазы (рис. 64, а): атомные плоскости не прерываются на такой границе, а лишь несколько изгибаются, как бы продолжаясь в другой фазе. Эта упругая деформация, называемая когерентной, обеспечивает плавную сопряженность решеток двух фаз, межатомные расстояния в которых всегда различны. Если новая фаза жестче исходной, то когерентная деформация сосредоточивается в основном в исходной фазе, и наоборот.
Если между кристаллами двух фаз имеется когерентная
132
граница или, коротко, когерентность1, то обе фазы (и их кристаллы) также называют когерентными. Если при фазовом превращении обеспечивается когерентность фаз, то это означает, что соседи любого атома в исходной фазе остаются соседями этого же атома в новой фазе (см. § 34).
Несоответствие двух решеток может быть скомпенсировано за счет когерентных деформаций только в том случае, когда разница в межатомных расстояниях сопрягающихся фаз достаточно мала.
На рис. 64, а видно, что при удалении от его центра к краям изгиб вертикальных атомных плоскостей нарастает.
Рис. 64. Схемы строения когерентной (а) и полукогерентной (6) границ между кристаллами фаз а и (3
Вполне очевидно, что с увеличением площади когерентной границы должна нарастать и упругая деформация на ней. Поэтому чисто когерентная межфазная граница возможна только на сравнительно небольшой поверхности раздела фаз, и тем меньшей, чем больше несоответствие их решеток.
При увеличении размера зародыша может быть достигнут момент, когда компенсация несоответствия решеток двух фаз становится энергетически более выгодной не в результате когерентной деформации по всей поверхности раздела, а частично за счет дислокаций (рис. 64, б). Эти дислокации называют структурными или дислокациями несоответствия. В промежутках между ними соблюдается когерентность решеток двух фаз. Межфазную границу, имеющую такое строение, называют полукогерентной. Если, например, степень несоответствия двух решеток е= (аа— —ар)/а0=0,01, а вектор Бюргерса структурных дислокаций равен одному межатомному расстоянию в плоскости сопряжения, то для компенсации указанного несоответствия ре
1 Слово «когерентность» произошло от англ, coherence, что означает связь, связность, согласованность. Поэтому следует признать весьма неудачным довольно распространенное словосочетание «когерентная связь».
133
шеток на полукогерентной границе дислокации должны отстоять одна от другой на 1/0,01 = 100 межатомных расстояний. Чем больше степень несоответствия решеток, тем выше плотность дислокаций на полукогерентной границе. При большой степени несоответствия решеток двух фаз расстояние между структурными дислокациями настолько уменьшается, что они теряют свою индивидуальность (их ядра сливаются). Такая межфазная граница называется некогерентной так же, как и сами фазы, разделяемые этой границей. Из сказанного следует, что прототипом некогерентной границы является высокоугловая граница зерен одной фазы, а прототипом полукогерентной — малоугловая граница.
Согласно оценочным расчетам, межфазная энергия когерентной границы не превышает 200 мДж/м2, у полукогерентной равна 200—500 мДж/м2, а у некогерентной составляет 500—1000 мДж/м2. Следовательно, при прочих равных условиях работа образования критического зародыша, имеющего когерентные границы, должна быть самой низкой, а скорость образования таких зародышей — наиболее высокой.
На рис. 64 видно, что решетка зародышей, отделенных от исходной фазы когерентной или полукогерентной границей, всегда* кристаллографически ориентирована определенным образом по отношению к решетке исходной фазы. Отсюда не следует делать обратного вывода, что две фазы с закономерной ваимной ориентацией их решеток обязательно когерентны. Во-первых, решетка некогерентной фазы может обладать закономерной ориентировкой по отношению к решетке исходной фазы, так как вначале зародыш новой фазы имел полукогерентную границу, затем при росте кристалла межфазная граница стала некогерентной, а ориентировка его сохранилась прежней. Во-вторых, если зародыш с самого начала имеет некогерентную границу, то также возможна закономерная ориентировка его решетки по отношению к решетке исходной фазы. Ориентированное превращение подчиняется общей закономерности, сущность которой была наиболее развернуто сформулирована П. Д. Данковым как принцип ориентационного и размерного соответствия: «Химическое превращение на поверхности твердого тела развивается таким образом, чтобы конфигурация атомов исходной твердой фазы сохранялась (или почти сохранялась) и в новой твердой фазе. Возникающая при указанном процессе кристаллическая решетка новой фазы сопрягается с кристаллической решеткой исходной фазы по
134
добными кристаллическими плоскостями, параметры которых отличаются друг от друга минимально». Причина закономерной ориентации двух фаз с термодинамической точки зрения состоит в том, что при кристаллизации в анизотропной среде «минимум поверхностной энергии обеспечивается при максимальном сходстве в расположении атомов на соприкасающихся гранях старой и новой фаз» (С. Т. Конобе-; евский).
Принцип ориентационного и размерного соответствия или, коротко, структурного соответствия, который также называют принципом Данкова—Конобеевского, наиболее ярко проявляется при образовании когерентной фазы, но он может определять ориентировку и некогерентного зародыша» так как и в этом случае закономерная ориентировка способна уменьшить энергию некогерентной границы.
Принцип структурного соответствия дает одно из объяснений появлению вытянутых, закономерно ориентированных кристаллов новой фазы. На шлифе они расположены параллельно один другому или составляют определенный угол. Такую структуру называют видманштеттовой по имени австрийского исследователя А. Видманштеттена, который одним из первых наблюдал в 1808 г. на железоникелевом метеорите характерный рисунок из переплетающихся полос.
§ 19. Гомогенное и гетерогенное зарождение фаз
Зарождение новой фазы, происходящее совершенно случайным образом в разных местах объема исходной фазы, называют гомогенным.
Одним из механизмов гомогенного зарождения является флуктуационное образование критических зародышей (см. § 17). Сами флуктуации энергии и концентрации являются результатом хаотичного теплового движения, и возникновение их в разных участках исходной фазы носит вероятностный характер. Поэтому и распределение по объему исходной фазы зародышей, возникших на базе таких флуктуаций, также случайно.
Классическая теория гомогенного зарождения была развита применительно к конденсации пара и кристаллизации жидкости. Но уже с самого начала было известно, что эти процессы происходят преимущественно на стенках сосуда или изложницы, а также на взвешенных частицах, т. е. на готовых поверхностях раздела.
В расплаве всегда присутствуют мельчайшие обычно неконтролируемые химическим анализом включения — окси
135
ды, нитриды, карбиды, сульфиды и др. Многочисленные «посторонние» включения служат затравками, и поэтому в обычных условиях мы имеем дело не с самопроизвольным гомогенным зарождением внутри объема чистой жидкости, а с зарождением на имеющихся поверхностях раздела с другими фазами. Поэтому такое зарождение было названо гетерогенным.
Гетерогенное зарождение не противоречит классической флуктуационной теории образования критических зародышей. Если межфазная энергия на границе зародыша с включением ниже, чем на границе с расплавом, то работа образования критического зародыша будет меньше1. Повышение скорости зарождения на готовых поверхностях раздела при росте переохлаждения объясняется, как и в случае гомогенной кристаллизации, уменьшением размеров критического зародыша и работы его образования.
В твердом состоянии условия для предпочтительного зарождения в определенных местах еще более благоприятны, так как в исходной фазе имеется множество мест с повышенной энергией Гиббса, которая способствует превращению. Такими местами являются границы зерен и субзерен исходной фазы, дисперсные включения других фаз, дислокации и дефекты упаковки.
Термин «гетерогенное зарождение» был распространен на все случаи предпочтительного зарождения в твердой фазе независимо от природы мест, в которых образуются зародыши.
При гетерогенном зарождении в твердом состоянии можно выделить две причины уменьшения работы образования критического зародыша: 1) пониженную межфазную энергию на готовой границе раздела зародыша и матрицы и 2) полное или частичное исчезновение в исходной фазе дефекта решетки и связанной с ним энергии Гиббса.
Рассмотрим раздельно гетерогенное зарождение в каждом из типовых мест предпочтительного образования зародышей.
1. Зарождение на границах зерен
Облегчение зарождения на границах зерен исходной фазы можно объяснить так же, как и предпочтительное зарождение кристаллов на включениях в расплаве. Это явление можно трактовать и несколько по-иному: при образовании зародыша новой фазы исчезает некоторая часть межзерен
1 Меньше числитель в формуле (23).
136
ной границы и высвобождающаяся при этом избыточная энергия межзеренной границы исходной фазы AGrp идет на образование зародыша новой фазы, т. е. на построение межфазной границы и компенсацию возникающей упругой энергии. Выражение (26) можно для этого случая записать в такой форме:
ДО = - Дбоб + Д0пов + ДОупр - Д6гр. (27)
Следовательно, работа образования критического зародыша из-за отрицательного вклада AGrp в общее изменение энергии Гиббса будет понижена.
Имеются и другие причины предпочтительного зарождения на границах зерен. Из курса физической химии известно, что те или иные компоненты жидкого раствора по-разному влияют на его поверхностное натяжение. Поверхностно-активные элементы, уменьшающие поверхностное натяжение раствора, должны самопроизвольно концентрироваться, адсорбироваться около его поверхности, так как при этом уменьшается энергия Гиббса системы. Поверхностная активность может проявляться не только в жидких, но и в твердых растворах. Растворенные элементы, снижающие энергию Гиббса межкристаллитной границы и обогащающие приграничную зону, называют горофильными. Самопроизвольное обогащение границ зерен растворенным элементом называют равновесной сегрегацией в отличие от неравновесного повышения концентрации твердого раствора вблизи межзеренных границ при дендр.итной ликвации. Если дендритная ликвация постепенно рассасывается при отжиге, то равновесная сегрегация, наоборот, усиливается с увеличением времени изотермической выдержки.
Одной из причин равновесной сегрегации является упругое взаимодействие атомов растворенного элемента с межзеренной границей. В растворах замещения из-за размерного несоответствия атомов компонентов вокруг атомов растворенного элемента решетка искажена. На межзеренной границе атомы расположены неправильно и имеются участки разрежения и сгущения. Атомы растворенного элемента с диаметром, большим, чем у основного металла, будут стремиться попасть в разреженные участки на границе зерен, а атомы растворенного элемента с меньшим диаметром, чем у растворителя, — в участки сгущения на межзеренной границе.
В растворах внедрения атомы растворенного элемента также создают искажения в объеме кристалла из-за того,
137
что их диаметр больше размера пустот решетки, в которых они размещаются. Этим атомам также энергетически более выгодно размещаться на межзеренной границе в разреженных участках.
Таким образом, движущей силой равновесной сегрегации является разность в энергии искажений решетки вокруг атома растворенного элемента в объеме кристалла и на межзеренной границе. Эта разность представляет собой энергию упругой связи атома растворенного элемента с границей зерен Е. От величины £ и от концентрации растворенного элемента в объеме зерна Со зависит концентрация его на границе:
г _ Сое^
ьгр “ 7777 '
Чем больше размерное несоответствие атомов, тем больше энергия упругого взаимодействия £ и, следовательно, выше СГр. В о. ц. к. решетке размер пустот, в которых размещаются атомы внедрения, меньше, чем в г. ц. к., и поэтому в ней атомы внедрения должны сильнее сегрегировать к межзеренной границе.
С ростом температуры равновесная сегрегация размывается тепловым движением и при достаточно высокой температуре практически полностью рассасывается.
Так как вокруг атомов растворенного элемента в объеме кристалла решетка всегда искажена, то, следовательно, всегда существует упругое взаимодействие этих атомов е межзеренной границей. Получается, что все растворенные элементы в той или иной степени должны быть горо-фильны. Однако имеются экспериментальные данные* о том, что некоторые элементы горофобны — их концентрация на границе зерен меньше, чем в объеме. Это значит, что, кроме упругого, существует еще и химическое взаимодействие атомов растворенного элемента с межзеренной границей, которое (в зависимости от особенностей электронного строения атомов) может способствовать равновесной сегрегации на границе, а может, перекрывая упругое притяжение, вызывать очищение приграничной зоны от атомов растворенного элемента.
Горофилен данный элемент или горофобен в конкретном растворителе, можно весьма надежно оценить, испЪльзуя представление об аналогии между строением расплава и неупорядоченной структурой на границе зерен. Из этой аналогии вытекает, что отношение концентраций растворенного элемента'в объеме зерна и на межзеренной грани
138
це должно быть равно отношению концентраций его в твердом (,СТ) и жидком (Сж) растворах, т. е. равно коэффициенту распределения:
С0/Сгр = Ст/Сж = k. (29)
Значение k легко определить по диаграмме состояния вблизи точки плавления основного компонента. Если добавка понижает точку ликвидуса, то А<1, и, следовательно, растворенный элемент должен быть горофилен: концентрация его в расплаве больше, чем в твердом растворе, а на границе зерен больше, чем в теле зерна. Если же добавка повышает точку ликвидуса твердого раствора, то А>1, и, следовательно, растворенный элемент горофобен: концентрация его в расплаве меньше, чем в твердом растворе, а на межзеренной границе меньше, чем в теле зерна. Однако надежных количественных предсказаний степени проявления горофильности или горофобности на основе указанного метода сделать нельзя хотя бы потому, что структура границ зерен не тождественна структуре жидкого раствора.
Равновесная сегрегация горофильного элемента или, наоборот, обеднение приграничной зоны горофобным элементом может сильно влиять на зарождение новой фазы по границам зерен, причем в принципе возможно и облегчение, и затруднение здесь фазового превращения. Особенно важно то, что при незначительном общем содержании горофильного элемента в сплаве концентрация его в растворе на межзеренной границе может быть достаточно велика, чтобы повлиять на фазовое превращение. Разные плавки сплава одной и той же марки в силу случайных обстоятельств содержат неодинаковое количество неконтролируемых обычными методами анализа горофильных примесей, и поэтому эти плавки могут существенно различаться по скорости зарождения новой фазы на границах зерен. Из-за равновесной сегрегации на фазовые превращения сильно влияют специально вводимые малые, «гомеопатические» добавки, например добавки бора, тысячные доли процента которого затрудняют распад аустенита по межзеренным границам.
Границы зерен влияют не только на зарождение, но и на скорость роста зародыша новой фазы. Если новая фаза отличается от исходной по химическому составу, то диффузионный рост ее зародышей по границам зерен идет быстрее, чем в теле зерна. Это объясняется тем, что, как известно, скорость диффузии по границам зерен, где строение
139
металла более «рыхлое», выше, чем в объеме зерен. Энергия активации зернограничной диффузии примерно вдвое меньше, чем у объемной диффузии. Так как величина энергии активации входит в показатель степени в формуле температурной зависимости коэффициента диффузии (D = =Ae~~QIRT)t то указанная разница в значениях энергии активации обусловливает большое различие в коэффициентах граничной и объемной диффузии. Например, у у-железа при 1000 °C коэффициент самодиффузии по границам зерен в 103 раза больше, чем внутри них.
Роль границ зерен в диффузионном превращении возрастает с понижением температуры в связи с тем, что диффузионная подвижность в объеме зерен становится весьма низкой. Границы зерен начинают заметно влиять на скорость диффузионных фазовых превращений при температурах ниже 0,75 Тпл.
2. Зарождение на дислокациях
Вокруг дислокации существует поле упругих напряжений. Например, в случае краевой дислокации под краем неполной атомной плоскости находится область растяжения, а над этим краем — область сжатия. Поэтому структурное несоответствие зародыша и исходной фазы может быть частично или полностью скомпенсировано дислокацией, что служит одной из причин предпочтительного образования на дислокациях зародышей с полукогерентными и некогерентными границами. При образовании такого зародыша упругая энергия решетки исходной фазы в некоторой области вблизи линии дислокации уничтожается. Это значит, что в формуле (26) слагаемое Д6упр имеет минус: упругая энергия дислокации способствует зарождению, суммируясь с движущей силой превращения AG06. По одной из оценок, в результате этого скорость зарождения на дислокациях в 1078 раз больше скорости гомогенного зарождения.
Другая причина предпочтительного зарождения на дислокациях — образование вдоль линий дислокаций атмосфер Коттрелла из атомов растворенного элемента. Если зародыш отличается от исходной фазы повышенным содержанием легирующего элемента, то естественно, что ему легче образоваться там, где уже имеется сегрегация этого элемента. Наконец, энергия активации диффузии вдоль краевых дислокаций примерно вдвое ниже, чем в объеме зерна вдали от дислокаций. Ускоренная диффузия по дислокационным «трубкам» облегчает диффузионный рост зароды
140
шей новой фазы, особенно при низких температурах, когда диффузия в объеме зерна исходной фазы становится очень медленной.
3. Зарождение на дефектах упаковки
Дефект упаковки, например в гранецентрированной кубической решетке, является прослойкой гексагональной плот-ноупакованной решетки, и наоборот. Если новая фаза имеет решетку того же типа, что и дефект упаковки, то он может служить готовым зародышем новой фазы. Так как растворимость легирующего элемента в общем случае должна быть разной в решетках разного типа, то атомы перераспределяются между дефектом упаковки и остальной решеткой исходной фазы, образуя атмосферы Сузуки, которые способствуют зарождению фазы, отличающейся по составу от исходной. По этим двум причинам растянутые дислокации, в которых дефект упаковки связывает частичные дислокации, являются местами предпочтительного зарождения новой фазы.
4. Зарождение на включениях
В исходной фазе часто имеются включения оксидов, карбидов, нитридов, интерметаллидов и других фаз. Включения могут облегчить зарождение новой фазы по той же причине, что и при гетерогенном зарождении в расплаве: работа образования критического зародыша уменьшается, если поверхностная энергия на границе зародыша с включением меньше, чем с материнской фазой. Это более вероятно в том случае, когда решетка включения изоморфна решетке зародыша.
Включения могут ускорять зарождение не только непосредственно, но и через образование дислокаций. Из-за разницы в термических коэффициентах расширения включений и матрицы при термической обработке вокруг включений могут возникнуть такие напряжения, что в материнской фазе генерируются дислокации, на которых может предпочтительно зарождаться новая фаза.
При затвердевании расплава образование центров кристаллизации на более тугоплавких включениях является основным механизмом гетерогенного зарождения. При превращениях же в твердом состоянии зарождение непосредственно на поверхности включений играет, как правило, второстепенную роль по сравнению с зарождением на разного
141
рода дефектах решетки (границах зерен, субграницах, дислокациях), число которых и протяженность обычно намного больше, чем включений.
5. Зарождение в микронесплошностях
На поверхности усадочных и газовых микропор, микротрещин и других несплошностей, возникающих по границам и внутри зерен при кристаллизации, пластической деформации и термической обработке, а также на открытой поверхности изделия, зарождение новой фазы может быть сильно облегчено, так как здесь отсутствует упругое сопротивление сплошной среды возникновению в ней кристаллов с другим удельным объемом. Эти места предпочтительного зарождения особенно эффёктивны, когда слагаемое AGynp в формуле (26) велико. Примерами являются зарождение графита в микронесплошностях аустенита и превращение белого олова в серое, начинающееся с открытой поверхности образца.
* * *
Дисперсность частиц новой фазы и характер их распределения по объему сплава при гетерогенном зарождении определяются числом и расположением мест предпочтительного зарождения. В отличие от гомогенного зарождения центры фазового превращения при гетерогенном зарождении могут быть резко неоднородно распределены по объему сплава, концентрируясь, например, на границах зерен. Очень часто кристаллы новой фазы, расположенные по границам зерен материнской фазы, дают четкие контуры этих границ. Но они могут и весьма равномерно распределяться по объему сплава, если места предпочтительного зарождения сами равномерно распределены по объему исходной фазы; это, например, возможно в случае зарождения на дислокациях, дефектах упаковки и включениях. Изменение числа и характера расположения мест гетерогенного зарождения под воздействием термической обработки, пластической деформации и другими способами является одним из самых эффективных путей регулирования структуры, получающейся при фазовых превращениях. Например, измельчение зерен исходной фазы (а значит, увеличение суммарной поверхности их границ) или повышение плотности дислокаций при пластической деформации увеличивает число центров фазового превращения.
142
§ 20. Образование промежуточных метастабильных фаз
Образование стабильной фазы приводит систему в состояние с абсолютным минимумом энергии Гиббса. Однако при определенных условиях зарождается и растет не абсолютно стабильная, а метастабильная фаза, образование которой
приводит систему в состояние с относительным минимумом энергии Гиббса.
Рассмотрим условия равновесия исходной фазы а с новой стабильной фазой 0, отличающейся от исходной структурой и составом. Используем для этого метод геометрической термодинамики, включающий построение графиков
энергия Гиббса — состав.
Как известно из курсов физической химии и металлографии, при постоянной температуре каждая из фаз характеризуется своей кривой зависимости энергии Гиббса от состава этой фазы (рис. 65). Если к кривым энергии Гиббса двух равновесных фаз провести общую касательную, то точки касания (а и Ь) укажут составы равновесных фаз при данной температуре. В общем случае эти составы не отвечают точкам минимума на рассматриваемых кривых, так как
Рис. 65. Диаграмма состояния (а) и зависимость энергии Гиббса от состава при температуре Л (б)
условием гетерогенного равновесия в системе, состоящей из более чем одного компонента,
является не достижение мини-
мума энергии Гиббса каждой из фаз, а равенство химических потенциалов компонента в соседствующих фазах.
Энергия Гиббса смеси фаз — всегда величина аддитивная и изображается точками на прямой, связывающей значения энергии Гиббса равновесных фаз. Следовательно, любая точка на отрезке abt например Gi (рис. 65,6), указывает энергию Гиббса смеси равновесных фаз с составами в точках а и b (поверхностная энергия межфазных границ здесь не учитывается).
Участок fa на кривой энергии Гиббса a-фазы относится к той области составов a-фазы (от А до Са), где она при данной температуре стабильна, а участок ag — где она
143
в контакте с ней находится
Рис. 66. Схема к анализу равновесия a-фазы со стабильной 0-и метастабиль-иыми 0'- и 0"-фазами
метастабильна. В последнем случае энергия Гиббса а-фазы больше, чем у смеси фаз а+р того же состава (например, в сплаве Со G2>Gi). Если состав a-фазы находится правее точки Са, то из нее должна образовываться p-фаза до тех пор, пока состав исходной фазы не достигнет равновесного (Са). Если же состав a-фазы расположен левее точки Са и p-фаза, то последняя будет растворяться в а-фазедотех пор, пока не будет достигнут равновесный состав Со. Допустим, что в какой-то системе не только существует стабильная фаза, но и возможно в определенных условиях образование мета-стабильных фаз р' и р", энергия Гиббса которых выше, чем у p-фазы (рис. 66). Метастабильная фаза р' отличается от стабильной только структурой, а фаза р"— и структурой, и составом. В стабильном равновесии с
р-фазой находится a-фаза состава Са. В метастабильном равновесии с фазами р' и р" находится a-фаза составов С' и С" соответственно. Можно сделать общий вывод, что растворимость метастабильной фазы в другой фазе всегда выше растворимости стабильной фазы.
В сплаве состава Со энергия Гиббса смеси фаз «+Р меньше, чем у смесей фаз сс+р7 или а+р" (Gi<G2<G3)e Отсюда можно сделать вывод, что снижение энергии Гиббса при образовании стабильной фазы больше, чем при образовании метастабильной фазы: (G4—G\)>(Gi—G2) и (G4—Gi)>(G4—G3). Однако этот анализ не учитывает роли межфазной энергии (на рис. 66 представлен график зависимости от состава объемной энергии Гиббса). Вместе с тем, как показано в § 17, межфазная энергия может играть решающую роль на первых этапах образования кристаллов новой фазы, когда велико отношение их поверхности к объему и соответственно велик вклад межфазной энергии в общую энергию Гиббса кристалла.
Значения межфазной энергии на границах a-фазы с фазами р, Р' и р" могут очень сильно различаться. Допустим, что кристаллическая решетка стабильной p-фазы резко отличается от решетки исходной сс-фазы и поэтому коге
144
рентная граница не может возникнуть. В то же время структура метастабильных фаз 0' и 0" ближе к структуре исходной a-фазы и межфазные границы а/0' и а/0" могут быть когерентными или полукогерентными. Межфазная энергия на таких границах намного ниже, чем на некогерентной границе а/0.
Если выигрыш в межфазной энергии существенно перекроет проигрыш в объемной энергии Гиббса, то тогда согласно формуле (26) работа образования критического зародыша Дбкр метастабильной фазы будет ниже, чем у стабильной, и скорость зарождения метастабильной фазы в соответствии с выражением (25) будет выше. В случае когерентной или полукогерентной границ метастабильных фаз для уменьшения AGKp выигрыш в межфазной энергии должен также перекрывать возрастание упругой энергии,, связанной с такими границами.
Если в системе могут существовать разные метастабиль-ные фазы, то при данной степени переохлаждения с увеличением времени выдержки вначале из-за большей скорости зарождения будет образовываться метастабильная фаза, у которой минимальна работа образования критического зародыша. Затем появятся метастабильные фазы с большей энергией образования критического зародыша й в последнюю очередь появится стабильная фаза, так как энергия активации ее образования самая высокая и, следовательно, скорость зарождения самая низкая.
Таким образом, последовательность образования фаз регулируется не достигаемым уровнем объемной энергии Гиббса, а величиной энергетического барьера при зарождении новой фазы, который сильно зависит от энергии межфазной границы. Чем больше указанный барьер, т. е. больше энергия активации образования критического зародыша, тем позднее образуется соответствующая фаза. Эта закономерность называется «правилом ступеней».
Когда после образования фазы 0" появляется относительно более стабильная фаза 0х, то около ее кристаллов в исходной a-фазе устанавливается равновесная концентрация С', в то время как на границе с 0"-фазой ранее установилась концентрация а-раствора С" (рис. 67). Диффузионное выравнивание состава в а-растворе стремится понизить его концентрацию на границе с 0"-фазой, что приводит к ее растворению в матрице. Благодаря такой «подпитке» вторым компонентом а-раствор на границе с 0"-фазой, пока она полностью не исчезнет, должен иметь концентрацию точки ,С". Если же условно разделить во
Ю И. И. Новиков
145
этои схеме
Рис. 67. Градиент концентрации компонента В в а-растворе на участке между выделениями Р" н Р' (С" и С' см. на рнс. 66)
нента В через матричный
времени процессы выравнивания состава в матрице и растворения в ней р"-фазы, то результат такого выравнивания состава можно схематично изобразить пунктирной линией на р.ис. 67.
перенос компонента В от границы а/(37' к границе а/07 повышает концентрацию а-раство-ра вблизи границы а/p7 и делает его здесь пересыщенным относительно р7-фазы. Вследствие этого выделяется р'-фаза на готовой поверхности р'-частицы и концентрация а-раствора снижается до равновесной (С'). Одновременно около границы а/p" матричный раствор становится ненасыщенным по отношению к р"-фаэе, которая растворяется в нем и поднимает концентрацию а-раствора до равновесной (С"). Эти процессы будут идти до тех пор, пока не растворится вся р"-фаза. Следовательно, более стабильная фаза р7 растет за счет растворения менее стабильной фазы р77 путем диффузионного переноса компо-а-раствор.
Аналогично при появлении стабильной фазы р должна растворяться фаза р7. Это соответствует общему правилу: образование более стабильной фазы приводит к растворению ранее образовавшейся менее стабильной фазы, которую обычно называют промежуточной. В конце концов система может прийти к состоянию, характеризующемуся абсолютным минимумом энергии Гиббса.
Образованию промежуточной фазы может способствовать также концентрационный фактор. Если стабильная фаза резко отличается по химическому составу от исходной, то для ее зарождения требуются большие флуктуации концентрации, а для ее роста необходим сравнительно большой диффузионный массоперенос. Если в этой же системе возможно существование 1метастабильной фазы, которая намного ближе по составу к исходной, то ее образование кинетически более выгодно, особенно при больших переохлаждениях, когда замедлены диффузионные процессы.
Классическим примером является образование из аусте
146
нита метастабильного цементита вместо стабильного графита. Состав аустенита (не более 2,14 % С) гораздо ближе к цементиту (6,67 % С), чем к графиту (100 % С). Вполне очевидно, что вероятность концентрационных флуктуаций, необходимых для зарождения цементита, несравненно больше вероятности концентрационных флуктуаций, необходимых для зарождения графита. Скорость диффузионного роста кристаллов цементита также выше, чем графита. Кроме этого концентрационного фактора, зарождению цементита способствует и структурный фактор: цементит ближе по структуре к аустениту, чем графит, и межфазная энергия на его границе с аустенитом должна быть ниже.
§ 21. Кинетика фазовых превращений
Кинетика кристаллизационных процессов в твердом состоянии определяется двумя параметрами — скоростью зарождения центров превращения и линейной скоростью роста из этих центров. Как было показано выше, оба параметра кристаллизации зависят от степени переохлаждения или перегрева.
Кинетика фазового превращения при определенном переохлаждении или перегреве изображается кинетической кривой, которая показывает нарастание количества новой " структурной составляющей во времени (рис. 68).
Кинетическую кривую для превращения, протекающего при охлаждении, строят следующим путем. Образец нагревают до температуры выше точки фазового равновесия. При этой температуре он может сколь угодно долго находиться в исходном фазовом состоянии. Затем образец быстро переносят в термостат с температурой ниже точки фазового равновесия, т. е. температура термостата соответствует определенной степени переохлаждения. Термостатом может служить соляная или масляная ванна, а также ванна из расплавленного металла.
Образец должен быть тонким, чтобы он мог быстро принять температуру термостата. За степенью превращения в образцах, находящихся в термостате, следят по изменению каких-либо свойств, например магнитных или электрических. Можно также закаливать образцы из термостата через определенные промежутки времени и измерять свойства или изучать микроструктуру закаленных образцов при комнатной температуре.
Аналогичным путем строят кинетические кривые для
10*
147
Рис. 68. Кинетика фазового превращения прн постоянной температуре:
1 — кинетическая кривая; 2 — зависимость истинной объемной скорости превращения от времени изотермической выдержки; оа — инкубационный период; ob — время полного превращения
превращений, протекающих при нагреве. В этом случае образец с исходной температуры, которая ниже точки фазового равновесия, быстро переносят в термостат с температурой, соответствующей определенной степени перегрева.
Все изотермические кристаллизационные процессы характеризуются следующей кинетикой (см. рис. 68). Вначале объем, испытавший превращение, растет с ускорением, а к концу превращения приращение этого объема резко замедляется. Таким образом, при постоянной температуре истинная объемная скорость превращения меняется в процессе самого превращения. Для каждого момента времени истинная объемная скорость превращения определяется графически по тангенсу угла наклона касательной к кинетической кривой.
Низкая объемная скорость в начальный период объясняется небольшим числом образовавшихся центров превращения и малой поверхностью новой структурной составляющей. Постепенно нарастает число центров, увеличиваются размеры новой составляющей и суммарная поверхность фронта кристаллизации,
поэтому объемная скорость превращения, пропорциональная площади этой поверхности, возрастает.
Однако эта скорость не может непрерывно возрастать со временем изотермической выдержки. Наступающее замедление процесса, особенно ярко выраженное к концу превращения, объясняется тем, что растущие кристаллы соприкасаются между собой и в местах стыка рост их прекращается, т. е. поверхность фронта превращения уменьшается. Максимум истинной объемной скорости превращения соответствует точке перегиба на кинетической кривой и достигается примерно тогда, когда половина объема претерпела превращение. Теперь понятно, почему каждая степень переохлаждения выше характеризовалась средней скоростью превращения (см. рис. 62).
Кинетика изотермического фазового превращения, характеризуемая сигмаидальной кривой, в самом общем виде
148
хорошо описывается уравнением Аврами (8), в котором для данного случая Vt/Vo — доля объема, испытавшего превращение за время т, а коэффициент В зависит от с. з. ц. и л. с. р.
А. Н. Колмогоров провел математический анализ кинетики фазового превращения с учетом роли постоянных с. з. ц. (м) и л. с. р. (с) при условии, что кристаллы новой фазы до момента соприкосновения одного с другим имеют сферическую форму:
Vt/V0= 1 — ехр[—— ПС3/). (30)
\ 3 /
Из этого уравнения следует, что изменение л. с. р., например при увеличении степени переохлаждения, должно влиять на долю превращенного объема значительно сильнее, чем изменение с. з. ц.
В соответствии с уравнением (30) в начальный период изотермической выдержки доля превращенного объема очень мала и очень медленно нарастает во времени. Поэтому начальный участок сигмаидальной кривой практически совпадает с осью абсцисс на участке оа (см. рис. 68).
Период оа называется инкубационным. В инкубационный период количество образовавшихся новых кристаллов настолько мало, что превращение не фиксируется обычными методами исследования. Конец инкубационного периода (точка а на рис. 68)—это фиксируемое данным методом начало превращения. Чем чувствительнее метод исследования, тем раньше обнаруживается появление новой структурной составляющей.
С практической точки зрения ничтожная степень превращения в инкубационный период не принимается во внимание, поэтому вполне целесообразно говорить о конце инкубационного периода как о начале превращения, всегда мысленно подразумевая, что к «началу» превращения уже образовалось некоторое количество новой структурной составляющей. Например, начало эвтектоидного превращения в стали фиксируется тогда, когда имеется 0,5—1 % перлита.
Кинетические кривые при различных степенях переохлаждения и перегрева представлены на рис. 69 и 70. В соответствии с закономерностями, графически изображенными на рис. 60 и 62, вначале с увеличением степени переохлаждения уменьшается инкубационный период (oai> >оа2>оа3 на рис. 69) и сокращается время полного превращения (о&1>о&2>о&з). Это объясняется тем, что ис-
149
ходное фазовое состояние становится менее устойчивым. При определенной степени переохлаждения достигается наименьший инкубационный период (оа3). Дальнейшее-увеличение переохлаждения приводит к возрастанию инкубационного периода (оаз<оа4<оа5) и увеличению време-
Рис. 69. Кинетические кривые при разных степенях переохлаждения:
7’1>7’2>7’з>7’4>7’5
Рнс. 70. Кинетические кривые при разных степенях перегрева: 7’1<7’а<Т3<7’4<7’9
ни полного превращения (оЬз<оЬ4<.оЬ5),так как уменьшается подвижность атомов.
С увеличением степени перегрева (см. рис. 70) инкубационный период и время полного превращения прогрессивно уменьшаются (оа1>оа%>оаь>оа4>оаь и ob\>ob2>obz> >ob4>ob^), так как одновременно с уменьшением термодинамической устойчивости исходного фазового состояния возрастает подвижность атомов (см. также рис. 63).
Для решения многих вопросов термообработки не требуется построения кинетических кривых. Достаточно знать 150
величину инкубационного периода и время полного превращения при разных температурах. Эти величины изображаются на диаграммах изотермического превращения. На рис. 71, а представлена диаграмма изотермического превращения, которое происходит при переохлаждении. Эта
Рис. 71. Диаграмма изотермического превращения, протекающего при переохлаждении (а), и зависимость средней скорости превращения от степени переохлаждения (б):
То — температура фазового равновесия
диаграмма построена по кинетическим кривым, показанным на рис. 69. Отрезки на рис. 71, а, отложенные при соответствующих температурах по горизонтали от ординаты до точек щ, а2, Ь2 и т. д., пропорциональны отрезкам оа^, obi, оа2, ob2 и т. д. на рис. 69.
Для построения диаграммы изотермического превращения ^совершенно не обязательно предварительно строить кинетические кривые. Достаточно опытным путем при каждой температуре определить время инкубационного периода и время полного превращения. Для этого образец, нагретый до температуры выше То, быстро переносят в термостат с температурой ниже То и фиксируют время появления новой структурной составляющей и время исчезновения остатков исходной структуры.
Левая кривая а\ а2 аз а$ является кривой начала превращения, показывающей зависимость величины инкубационного периода от степени переохлаждения. Правая кривая bi b2 Ъз bi bs является кривой конца превращения, показывающей зависимость времени полного превращения от степени переохлаждения. Обе кривые называются С-кривыми. Изгиб С-кривой соответствует максимальной скорости превращения (рис. 71, б).
151
Диаграмма изотермического превращения при перегреве представлена на рис. 72 (сравните с кинетическими кривыми на рис. 70). Кривые di а2 а3 ^4 и bi b2 Ь3 Ь4 Ь5 показывают фиксируемые данные методом начало и конец превращения. При достаточно высокой температуре из-за большой подвижности атомов превращение протекает практически мгновенно. Поэтому кривые начала и конца превращения сливаются и попадают на ось ординат.
Рис. 73. Кривые начала превращения переохлажденной фазы в изотермических условиях (/) н пр» непрерывном охлаждении (2)
Рис. 72. Диаграмма изотермического превращения, протекающего при перегреве:
То — температура фазового равновесия
Впервые диаграмма изотермического распада аустенита в стали была построена в 1930 г. американскими учеными Бейном и Давенпортом. В тридцатые годы советские металловеды С. С. Штейнберг, Н. А. Минкевич, В. Д. Садовский и др. широко исследовали распад аустенита в изотермических условиях. Метод изучения кинетики фазовых превращений с помощью диаграмм изотермического распада переохлажденной фазы, называемых также С-диаг-раммами или С-образными кривыми, оказался исключительно плодотворным в теории термической обработки. Число опубликованных диаграмм изотермических превращений переохлажденной фазы для сталей составляет величину порядка 103, для титановых 102, алюминиевых 102. С-диаграммы построены также для чугунов, некоторых' медных, урановых и других сплавов.
Широкому использованию диаграмм изотермических превращений способствует то, что их строят в тех же координатах температура—время, в которых изображают режимы нагрева и охлаждения при термической обработке.
152
С помощью С-диаграмм особенно удобно анализировать различные виды изотермических обработок, включающих ускоренное охлаждение и выдержку при постоянной температуре, например изотермический отжиг и изотермическую закалку (см. § 25 и 41). По С-диаграмме можно прямо определить время начала и конца превращения переохлажденной фазы при заданной температуре и в соответствии с поставленной задачей выбирать режим изотермической обработки.
Более сложно по диаграмме изотермических превращений анализировать процессы фазовых превращений, протекающие при непрерывном охлаждении, а именно такие процессы и преобладают в практике термической обработки. Некоторые качественные выводы при их анализе можно сделать по диаграмме изотермических превращений. Например, на рис. 73 кривая 1 указывает начало превращения переохлажденной фазы в изотермических условиях. Кривые v\ и и2 относятся к двум режимам непрерывного охлаждения. Если принять, что точки а,\ и а2 определяют начало превращения при непрерывном охлаждении, то рис. 73 наглядно показывает, что при более высокой скорости охлаждения превращение начинается при большем переохлаждении (То—Г2>Г0—Л).
Качественно этот вывод правилен. Количественно же нельзя определять начало превращения при непрерывном охлаждении по пересечению кривой охлаждения с С-кри-вой, построенной в изотермических условиях. Дело в том, что инкубационный период оах получен исследованием образца во время изотермической выдержки при температуре Ть Нас же сейчас интересует образец, непрерывно охлаждающийся по режиму vi. Этот образец в инкубационный период находился при переменных температурах, которые были выше температуры Т\. Но при более высоких температурах превращение в инкубационном периоде протекает более медленно, чем при температуре 1\. Следовательно, в непрерывно охлаждающемся образце по достижении температуры 7\ степень превращения должна быть меньше, чем в образце, который то же самое время находился в изотермических условиях при температуре 7ь Поэтому в образце, непрерывно охлаждаемом по режиму vlf температура фиксируемого начала превращения должна лежать ниже температуры Т\, которая определяется точкой пересечения кривой охлаждения и С-кривой, построенной в изотермических условиях.
Для количественного анализа превращений, протекаю
153
щих при непрерывном охлаждении, следует использовать диаграммы, которые получили название термокинетических.
Термокинетические диаграммы фазовых превращений строят, используя непрерывное охлаждение образцов, температура которых в период охлаждения записывается осциллографом. Можно измерять какую-либо характеристику образца в процессе его охлаждения (например, его длину при дилатометрическом методе) и по отклонению этой характеристики от плавного изменения определять начало превращения.
Другой способ состоит в охлаждении по одинаковому режиму серии образцов, которые в разные моменты времени закаливают в воде, а затем исследуют их структуру или свойства, определяя по ним начало и конец превращения (или определенную степень превращения) при одном режиме непрерывного охлаждения. Например, точки и и г? указывают начало превращения при скоростях охлаждения и съ (см. рис. 73). Соответствующая кривая 2 на рис. 73 указывает начало превращения при непрерывном охлаждении с разными скоростями. Эта кривая относится к термокинетической диаграмме, на которой можно аналогичным способом построить кривые конца превращений и кривые, соответствующие определенной степени превращения при непрерывном охлаждении с разной скоростью (см. рис. 138).
Расхождение в положении одинаковых по смыслу линий термокинетической диаграммы и диаграммы изотермических превращений может быть весьма существенным, как в случае сталей, и небольшим, как в случае дуралюминов. Заранее предсказать величину этого расхождения невозможно. Предложенные методы пересчета диаграмм изотермических превращений в термокинетические дают слишком грубые оценки. Поэтому оптимально построение для промышленных сплавов диаграмм превращений двух типов, хотя термокинетические диаграммы строить трудно.
Термокинетические диаграммы можно строить и для превращений, протекающих при нагреве.
Глава V
ОТЖИГ СТАЛЕЙ
Отжиг сталей (отжиг 2-го рода) — это термическая обработка, при которой главными процессами являются аустенитизация с последующим перлитным превращением.
154
§ 22. Образование аустенита при нагреве
1. Механизм и кинетика аустенитизации
Перед образованием аустенита исходной структурой чаще всего является смесь сс-фазы (о. ц. к.) и карбида. В углеродистых сталях аустенит при температуре несколько выше точки Ai содержит около 0,8 % С, в то время как феррит
в исходной структуре содержит сотые доли процента углерода (рис. 74). Каким образом возникает участок у-фазы с г. ц. к. решеткой и сравнительно высоким содержанием углерода? Этот вопрос давно дискутируется в литературе. Накопленные к настоящему времени экспериментальные данные указывают на двустадийное формирование аустенитных зародышей. Первая стадия — полиморфное а—>-
155
-^-превращение по сдвиговому механизму с образованием? метастабильного малоуглеродистого аустенита1. Вторая стадия — увеличение концентрации углерода в таком аустените, т. е. приближение его к равновесному составу, вследствие растворения в аустените частиц карбида.
На сдвиговый механизм «-^-перестройки решетки при образовании зародышей аустенита указывают следующие факты: игольчатая (точнее, пластинчатая) форма первых участков у-фазы, появление рельефа на полированной поверхности шлифа и «наследование» дислокационной субструктуры при «-^-превращении (см. § 60). При малой толщине плоского зародыша, образование которого связано с удвоением площади границ, выигрыш в объемной энергии Гиббса достаточен для развития превращения лишь в том случае, если новая граница когерентная (имеет низкую энергию), а такая граница характерна именно для сдвигового превращения (М. А. Штремель). По мере роста зародышей их граница из когерентной становится некогерентной и сдвиговый механизм сменяется нормальным механизмом роста аустенитных зерен. При этом игольчатость зародышей у-фазы исчезает и из них формируются обычные равноосные зерна.
Зародыши аустенита предпочтительно образуются на границах зерен феррита. В стали со структурой зернистого перлита (сферодита) аустенитные зародыши возникают около частиц карбида, расположенных на границах ферритных зерен.
В доэвтектоидных сталях первые участки аустенита предпочтительно появляются на границах зерен и субзерен в избыточном феррите и по границам перлитных колоний. На рис. 75, а виден участок 1 аустенита, появившийся около перлитной колонии, и участок 2 аустенита, образовавшийся в избыточном феррите стали 20. На рис. 75, б показано, по данным авторадиографии, распределение углерода вдоль прямой, отмеченной на рис. 75, а, в образце этой стали после нагрева до 730 °C с выдержкой 1 мин. На участке 1 содержание углерода доходит до 0,53 %, а на участке 2 — до 0,18 %, что вполне естественно, так как первый участок ближе к источнику углерода (цементиту в перлитной колонии). С увеличением выдержки состав аустенитных участков должен приближаться к равновесному, определяемому линией GS.
1 К этому вопросу необходимо вернуться после проработки § 34, в котором сопоставлены механизмы сдвигового и нормального превращений.
156
Расстояние, нн-Ю~2
Рис. 75. Микроструктура (а) и распределение углерода в фазах (6) после закалки стали 20 с 730 °C (И. В. Дощечки-иа, С. С. Дьяченко). Х600 (при печати уменьшено на */з)
Можно представить себе два случая роста аустенитного зародыша. В первом случае он находится в контакте и с ферритом, и с карбидом, который поглощается непосредственно растущим аустенитным зерном. Во втором случае аустенитный зародыш полностью окружен ферритом, и карбид растворяется в феррите, пока граница у/a не подойдет к карбидной частице. Оба случая фиксировали при прямом наблюдении за развитием перлито-аустенитного превращения в колонне высоковольтного электронного микроскопа.
Проанализируем изотермическое образование аустенита при температуре Tj на рис. 76 в указанных двух случаях.
Если аустенит граничит и с ферритом, и с карбидом (рис. 77, а), то в соответствии с диаграммой состояния на границе с ферритом равновесная концентрация аустенита должна изображаться точкой Ь, а на границе с цементи-
157
Рис. 76. Схема «стального* участка диаграммы состояния железо — цементах
том — точкой а. Внутри аустенитного зерна устанавливается градиент концентраций аЪ (рис. 77, а). На границе аустенита с цементитом имеется большой скачок концентрации углерода аК, а на границе с ферритом — меньший скачок bd.
Скорость изотермического роста аустенитного зерна контролируется диффузией углерода от границы А/Ц к границе А/Ф, выравнивающая диффузия понижает концентрацию углерода в аустените на границе А/Цу сдвигая состав аустенита влево от точки а на рис. 77, а. Аустенит при этом становит-
ся ненасыщенным по отношению к цементиту, который растворяется, восстанавливая равновесную концентрацию точки а. На границе с ферритом выравнивающая диффузия повышает концентрацию углерода в аустените, сдвигая ее вправо от точки b на рис. 77, а. Аустенит при этом становится ненасыщенным по отношению к ферриту, который превращается в аустенит, повышая в нем концентрацию железа до равновесного состава точки Ь. Таким образом, аустенитное зерно разрастается в сторону и феррита, и цементита.
Скорость перемещения межфазной границы при росте одной фазы за счет другой обратно пропорциональна скачку концентраций на границе (на величину этого скачка должна измениться концентрация элемента в приращенном объеме). Так как скачок концентраций на границе А/Ф примерно на порядок меньше, чем на границе А/Ц (bd<^aK), то скорость роста аустенитного зерна в сторону феррита намного больше, чем в сторону цементита. В результате к моменту исчезновения ферритных промежутков перлита всегда остается не полностью растворившийся цементит. Собственно перлито-аустенитное превращение заканчивается с исчезновением ферритных промежутков. Затем продолжается растворение остатков цементита. По окончании его растворения аустенит неоднороден по концентрации углерода. В тех местах, где ранее находился цементит, концентрация углерода в гамма-растворе повышена. Для выравнивания состава аустенита требуется дополнительное время после окончания фазового превращения.
158
Если аустенитный зародыш полностью окружен ферритом (рис. 77, б), то в соответствии с диаграммой состояния на рис. 76 концентрация углерода в феррите на границе его с цементитом должна изображаться точкой с, а на границе с аустенитом — точкой d. Скорость роста аустенита в этом
Рис. 77. Схема распределения концентрации углерода при росте аустенита^ находящегося в контакте с ферритом и цементитом (а) и полностью окруженного ферритом (б)
случае контролируется диффузией углерода от границы Ф/Ц к границе Ф/А.
Выравнивающая диффузия в феррите приводит к тому,, что на границе Ф/Ц растворяется карбид, а на границе Ф/А феррит превращается в аустенит (граница Ф/А продвигается в сторону феррита).
В доэвтектоидной стали размер зерен структурно-свободного (избыточного) феррита на один-два порядка больше толщины ферритных промежутков в перлите. Поэтому время исчезновения избыточного феррита превышает время исчезновения перлита. После исчезновения избыточного феррита на месте его зерен концентрация углерода в аустените понижена и идет процесс выравнивания состава внутри аустенитных зерен.
На рис. 78 представлена экспериментально построенная Диаграмма изотермического образования аустенита из феррито-перлитной структуры в доэвтектоидной стали. Линия
159*
1 характеризует появление аустенита, 2 — исчезновение ферритных промежутков в перлите и, следовательно, исчезновение самого перлита, 3 — исчезновение избыточного феррита. Размытая граница 4 соответствует полному исчезновению карбида. Так как после этого аустенит некоторое время остается неоднородным по содержанию углерода, то правее границы 4 должна находиться не показанная на рис. 78 линия, соответствующая окончанию выравнивания концентрации углерода в аустените при разных температурах.
На диаграммах изотермического образования аустенита типа той, что изображена на рис. 78, линия 1 указывает не истинное начало превращения, а обнаруживаемое данным
Рис. 78. Диаграмма изотермического образования аустенита в доэвтектоид-Hoii стали с 0,45 % С при исходной «феррито-перлитной структуре (Розе и Штрассбург)
Рис. 79. Кинетические кривые образования аустенита при разных температурах в стали с 1 % С, 0,6 % Сг, 0,5 % W и 1,1 % Мп (Дирифельд, Коревар и Вантспикер)
экспериментальным методом некоторое количество аустенита, например 1 или 3 %. Тщательный металлографический анализ всегда выявляет образование первых порций аустенита сразу после перехода через точку Аи т. е. линия истинного начала превращения находится практически при температуре Ai. Это связано с легкостью зарождения аустенита при сдвиговой а->у-пер ест ройке решетки. Здесь нет противоречий с общей теорией фазовых переходов, так как принципиально фазовое превращение может развиваться при любой малой степени перегрева выше температуры равновесия. Известный пример практически нулевой степени перегрева при фазовом превращении — плавление кристаллов.
Кинетика изотермического образования аустенита ха-i рактеризуется обычной сигмаидальной кривой (рис. 79) и I подчиняется уравнению Аврами (8), в котором в данном
L-1BO
случае Vt/V0—объемная доля образовавшегося аустенита, а т — время фазового превращения. Из рис. 79 видно, что сигмаидальные кривые образования аустенита при всех температурах выходят из точки с нулевой выдержкой и инкубационный период отсутствует.
На первый взгляд может показаться совершенно непонятным, почему в перлите превращение происходит при температурах, далеких от точки равновесия а- и у-мо-дифпкаций железа (911 °C). Дело в том, что в сталях феррит всегда содержит растворенный углерод. Линия GP на диаграмме состояния железо — цементит (см. рис. 74) показывает, что очень малые количества углерода в твердом растворе резко снижают температуру начала перехода сх->-у. Точка Р соответствует содержанию в феррите около 0,1 % (ат.) углерода. Следовательно, когда на 1000 атомов железа приходится лишь один атом углерода, граница устойчивости о. ц. к. решетки железа оказывается пониженной почти на 200 К.
Время окончания разных этапов образования аустенита уменьшается с ростом температуры (см. кривые 1—4 на рис. 78).
Если аустенитный зародыш находится в контакте и с ферритом, и с карбидом (см. рис. 77, а), то скорость процесса аустенитизации должна быть пропорциональна коэффициенту диффузии углерода в аустените и градиенту концентраций (Са—Сь)/Х, где X—сечение растущего зерна аустенита, Са и Сь — концентрации углерода в точках а и b на рис. 77, а. С повышением температуры экспоненциально растет коэффициент диффузии и увеличивается перепад концентраций Са—Съ в соответствии с расхождением линий GS и SE на диаграмме состояния Fe—С. Кроме того, с ростом температуры уменьшается скачок концентраций на границе А/Ф (укорачиваются коноды, соединяющие линии GP и GS), что также ускоряет рост аустенита за счет феррита.
С ростом скорости нагрева температура окончания всех этапов аустенитизации повышается, что видно из качественного сопоставления положения точек пересечения схематично проведенных лучей нагрева V\ и Кг, например, с линией 3 исчезновения феррита на диаграмме изотермических превращений на рис. 78. Для получения соответствующих количественных данных необходимо строить термокинетические диаграммы образования аустенита по результатам опытов с непрерывным нагревом образцов при разных скоростях. Практически важно, что при скоростном, например
Ц И. И. Новиков
161
индукционном или лазерном, нагреве до более высоких температур время полной аустенитизации меньше, чем при медленном печном нагреве до более низких температур.
С увеличением дисперсности исходной структуры время окончания всех этапов аустенитизации уменьшается.
Гомогенизация аустенита идет гораздо дольше в легированных сталях. Легирующие элементы неравномерно распределены между ферритом и карбидом. Некарбидообразующие элементы находятся в феррите, а карбидообразующие— преимущественно в карбиде. По окончании перлито-аустенитного превращения аустенит неоднороден. В участках аустенита, соответствующих исчезнувшим частицам феррита и карбида, различна концентрация не только углерода, но и легирующих элементов.
Хотя скорость диффузии углерода в аустените на несколько порядков больше, чем у легирующих элементов, его концентрация не может выравняться до тех пор, пока не исчезнет градиент концентраций легирующих элементов. Объяснение этому с учетом влияния легирующих элементов на термодинамическую активность углерода было дано в § 1.
Из всего изложенного о кинетике аустенитизации следует, что время выдержки при нагреве определяется тем, насколько полно должны пройти самые медленные процессы — растворение карбидов и гомогенизация аустенита. При нагреве доэвтектоидной стали с укороченными выдержками можно при температурах выше точки А3 получить аустенито-карбидную структуру.
2. Размер аустенитного зерна
Из-за очень большого числа центров образования аустенита к концу а—^-превращения формируется мелкое аустенитное зерно. Это зерно аустенита, называемое начальным, обычно имеет размер около 10—20 мкм.
— С повышением температуры с. з. ц. аустенита возрастает больше, чем л. с. р., и соответственно начальное зерно аустенита получается мельче. Следовательно, при быстром нагреве до высоких температур и коротких выдержках можно получать мелкое аустенитное зерно, что важно для термообработки со скоростным нагревом.
По окончании аустенитизации зерна аустенита способны к росту, движущей силой которого является зернограничная энергия. С повышением температуры рост зерна аустенита ускоряется. Зерно может расти равномерно, как
162
при собирательной рекристаллизации, но может наблюдаться и неравномерный рост, когда небольшое число крупных зерен аустенита растет за счет множества мелких, пока они не исчезнут. Этот процесс по существу является вторичной рекристаллизацией (см. § 10). Мелкие зерна аустенита стабилизированы против роста дисперсными частицами, в частности нитрида алюминия.
Хотя при охлаждении аустенит превращается в другие фазы, размер его зерна является важной характеристикой стали. Это объясняется тем, что все структурные составляющие при медленном и при быстром охлаждении стали формируются в пределах каждого аустенитного зерна. Чем мельче аустенитные зерна, тем мельче сетка избыточного феррита по их границам, меньше размер перлитных колоний (см. § 24) и меньше размер мартенситных кристаллов (См. § 35). Поэтому мелкому зерну в аустенитной области соответствует мелкокристаллический излом, а крупному аустенитному зерну — крупнокристаллический излом стали при тех температурах, где аустенита уже нет. К разному размеру зерна аустенита особенно чувствительна ударная вязкость, которая падает с его укрупнением. Температурный порог хладноломкости повышается с увеличением размера аустенитного зерна (см. § 53).
Размер зерна аустенита характеризуют условным номеров (баллом) стандартной шкалы микроструктур (ГОСТ 5639—65). Каждому номеру зерна N соответствует определенное среднее число зерен т на площади 1 мм2 шлифа и условный пространственный размер этих зерен D. 1
В основу построения стандартной шкалы микроструктур положена формула
m=2W+3. (31)
Стандартная шкала имеет 18 номеров зерна; —3, —2, —1,0, 1, 2,..., 14. Чем выше номер зерна, тем меньше его размер. Номеру зерна —3 соответствует структура с т—1 мм~2 и D=1 мм, номеру 6 — структура с /н=512 мм~2 и D— 40 мкм, номеру 14 — структура с т== 131072 мм~2 в D— 20 мкм. Из формулы (31) следует, что при возрастании номера зерна N на единицу удваивается величина т. Для быстрой оценки номера зерна применяют метод сравнения исследуемой структуры с эталонными структурами стандартной шкалы (при одинаковом увеличении) .
Повышение концентрации углерода в гамм а-растворе способствует росту зерна, что можно объяснить понижением солидуса сплава и соответствующим ростом гомологической температуры при постоянной температуре отжига. Повышение содержания углерода сверх предельной кон-11* - 163
центрации в аустените (линия ES диаграммы состояния Fe—РезС) затрудняет рост аустенитного зерна, что объясняется тормозящим действием частиц цементита.
Почти все легирующие элементы тормозят рост аустенитного зерна. Исключение составляет марганец, который усиливает рост зерна аустенита. Сильно тормозят рост аустенитного зерна V, Ti, Al, Zr, W, Mo и Сг и слабо действуют Ni и Si.
Основной причиной этого действия легирующих элементов считается образование труднорастворимых в аустените карбидов, нитридов и других фаз, которые являются барьерами для растущего зерна. Такие активные карбидообра-зователи, как Ti, Zr и V, сильнее тормозят рост зерна, чем Сг, W и Мо, так как карбиды первых более устойчивы и труднее растворяются в аустените.
Разные плавки стали одной марки могут сильно различаться по склонности к росту аустенитного зерна, так как они содержат разные количества дисперсных труднорастворимых частиц карбидов, нитридов и других фаз — барьеров для роста аустенитного зерна. Дисперсность и распределение этих частиц зависят не только от условий выплавки стали и горячей обработки давлением, но и от предварительной термообработки (отжига). Следовательно, склонность стали к росту зерна при нагреве зависит, помимо ее состава по основным компонентам, от металлургического качества, технологии производства, т. е. от ее истории, которая предшествует термообработке. В связи с этим различают наследственно крупнозернистые и наследственно мелкозернистые стали.
В наследственно крупнозернистой стали зерно интенсивно растет при относительно небольших превышениях температуры над точкой Ас3. В наследственно мелкозернистой стали мелкое аустенитное зерно получается в широком диапазоне температур: от точки Ас3 до 950—1100 °C. Переход через этот температурный порог приводит к перегреву наследственно мелкозернистой стали. Под перегревом здесь подразумевается интенсивное укрупнение зерна и связанное с этим падение ударной вязкости1. При столь высоких температурах зерно в наследственно мелкозернистой стали может оказаться даже более крупным, чем в наследственно крупнозернистой стали.
Алюминий особенно сильно влияет на рост зерна аусте
1 Не следует путать это понятие с перегревом в теории фазовых превращений.
164
нита при нагреве стали. Наследственно мелкозернистую сталь получают введением в ковш перед разливкой примерно 0,05 % А1. Мельчайшие частички нитридов и оксидов алюминия тормозят рост зерен аустенита. Интенсивный рост зерна в наследственно мелкозернистой стали выше 950—1100 °C объясняется растворением и, возможно, коагуляцией барьерных частиц. При этом из-за неодновременного в разных местах ослабления барьерного эффекта может возникнуть характерная для перегрева наследственно мелкозернистых сталей разнозернистость.
Для определения склонности стали к росту зерна пользуются стандартной технологической пробой, которая состоит в следующем. Доэвтектоидную сталь цементуют при 930 °C в течение 8 ч с последующим медленным охлаждением. Размер зерна определяют по карбидной сетке, окаймляющей границы аустенитных зерен. Заэвтектоидную сталь нагревают до 930 °C и после выдержки в течение 3 ч медленно охлаждают. Размер зерна определяют по сетке вторичных карбидов, выделяющихся по границам аустенитных кристаллов. Другой метод состоит в окислительном нагреве шлифа в течение 3 ч при 930 °C. Границы зерен аустенита выявляются сеткой оксидов.
При пробе на наследственное зерно температура нагрева 930 °C выбрана по следующим соображениям. Для большинства сталей температура нагрева при различных вилах термообработки не превышает 930 °C. Вместе с тем наследственно мелкозернистая сталь при 930 °C еще сопротивляется интенсивному росту зерна, а в наследственно крупнозернистой стали при этой температуре вырастает крупное зерно.
Стали с зерном аустенита № 6—14, т. е. с размером зерна менее примерно 40 мкм, считают мелкозернистыми, а с зерном от № 5 и ниже— крупнозернистыми.
Таким образом, различают наследственное зерно и действительное зерно аустенита. Наследственное зерно получается в стандартных условиях технологической пробы и характеризует склонность стали к росту зерна. Действительное зерно — это то зерно, которое получается в результате той или иной операции термообработки. Оно может быть больше или меньше наследственного зерна в зависимости от того, выше или ниже 930 °C температура нагрева стали при термообработке.
§ 23. Структурная наследственность и перекристаллизация аустенита
При охлаждении всех углеродистых и большинства легированных сталей аустенит претерпевает перлитное, ‘мартенситное или бейнитное превращение (о двух последних см. в гл. IX). При повторном нагреве, когда вновь образуется аустенит, в определенных условиях размер, форма и кристаллографическая ориентировка его зерен могут быть такими же, как и у исходного зерна аустенита перед первым перлитным, мартенситным или бейнитным превращением. Такое наследование размера, формы и ориентировки аустенитного зерна называют структурной наследственностью. Это явление наиболее подробно изучено в работах В. Д. Садовского, результаты исследований которого положены в основу приведенного описания.
Проявление структурной наследственности зависит от исходной (перед повторным нагревом) структуры стали и ее состава, от скорости этого нагрева и температуры аустенитизации. Наиболее ярко структурная наследственность проявляется, если перед повторным нагревом сталь имела мартенситную или бейнитную структуру. У стали с феррито-перлитной структурой исходное аустенитное зерно обычно не наследуется. Если ранее такая сталь была перегрета и имела крупное зерно аустенита, то при повторном нагреве до температур Ac3-f- (304-50) К аустенит, зарождаясь во многих местах, получается мелкозернистым. Крупное аустенитное зерно наследуется здесь только при исходной видманштеттовой структуре.
Для мартенситной, бейнитной, а также видманштеттовой структур общей чертой является кристаллографическая упорядоченность: в пределах объема зерна исчезнувшего аустенита пластины a-фазы имеют определенную кристаллографическую ориентировку. При нагреве стали зародыши у-фазы в свою очередь закономерно ориентированы относительно пластин a-фазы, в результате чего в объеме исходного аустенитного зерна при небольших перегревах выше точки А3 формируется точное такое же аустенитное зерно, называемое восстановленным.
Таким образом^’для восстановления аустенитного зерна при повторном нагреве стали необходимо, во-первых, проявление наследственности при охлаждении (образование кристаллографически упорядоченного комплекса a-кристаллов в пределах каждого исходного аустенитного зерна) и, во-вторых, проявление наследственности при последующем
166
нагреве стали (переход указанного комплекса a-кристаллов в одно восстановленное аустенитное зерно). _
Когда аустенит при охлаждении стали претерпевает перлитное превращение и не возникает видманштеттовой структуры, образующиеся зерна избыточного феррита и колонии перлита не имеют строго закономерной кристаллографической ориентировки по отношению к решетке исходного аустенитного зерна. В этом случае колонии перлита и зерна избыточного феррита не образуют единого кристаллографически упорядоченного комплекса в пределах объема исходного аустенитного зерна, внутри которого при последующем нагреве стали возникает много разориентиро-ванных один относительно другого зародышей аустенита, и ого зерно получается более мелким, чем исходное.
Если при нагреве стали выше точки Л3 вначале наследовалось крупное зерно аустенита, то при последующем повышении температуры, а иногда и при увеличении выдержки восстановленное зерно аустенита вместо того, чтобы укрупняться, измельчается. Такое измельчение зерна в температурной области, где нет никаких фазовых превращений, происходит в результате зарождения новых зерен аустенита в большом числе мест в пределах объема каждого восстановленного зерна. Этот процесс структурной перекристаллизации и по внешним признакам, и по физической сущности является рекристаллизационным. Только в отличие от обычной первичной рекристаллизации, когда необходимая для формирования ее зародышей повышенная плотность дислокаций создается деформацией металла внешними силами, в рассматриваемом случае повышенная плотность дислокаций возникает при внутреннем фазовом паклепе.
Фазовый наклеп — это повышение плотности дислока-^ цпй в новой фазе в ходе фазового превращения. Он может возникать при любом фазовом превращении из-за разности удельных объемов старой и новой фаз, а также при' мар-• енситном превращении, когда аккомодационная деформация обязательно сопровождает образование каждого кристалла (см. § 34). Повышенная плотность дефектов в новой । фазе может также возникнуть из-за «передачи» дислокан гшй от исходной фазы к новой.
Если в наследуемом крупном зерне аустенита имеется достаточно высокая плотность дислокаций, то это и обусловливает развитие первичной рекристаллизации, центры которой зарождаются одним из известных способов (см. § 7): а) «набором» растущим субзерном высокоугловой
167
разориентировки; б) выбрасыванием границей аустенитного зерна «языков». После окончания первичной рекристаллизации аустенита с увеличением времени выдержки или при повышении температуры идет обычный рост новых зерен.
Таким образом, имеются два этапа формирования мелкозернистой структуры в случае проявления структурной наследственности в стали. Первый этап — образование восстановленного зерна у-фазы с повышенной плотностью дислокаций из кристаллографически упорядоченного комплекса кристаллов a-фазы в пределах объема исходного аустенитного зерна. Второй этап — зарождение и рост более совершенных кристаллов у-фазы, т. е. рекристаллизация, приводящая к устранению структурной наследственности.
Окончание фазового превращения и первичная рекристаллизация аустенита могут быть разделены температурным интервалом в десятки и даже сотни градусов. Например, если отливки из углеродистой стали 35Л имеют крупнозернистую видманштеттову структуру, то при обычном нагреве до 850 °C восстанавливается исходное аустенитное зерно, а при нагреве до 900—960 °C, т. е. на 100— 150 К выше Ас3, измельчается зерно и ликвидируется наследственность исходной литой структуры. Чем больше видманштеттовых пластин феррита в стали, тем выше температура структурной перекристаллизации.
Для устранения структурной наследственности нагрев легированной стали обычно должен быть значительно выше точки Ас3, чем нагрев углеродистой. Например, для исправления крупного зерна отливок из стали 20X1ЗЛ их нагревают до 1100—1150 °C, т. е. выше Ас3 примерно на 300 К.
Исходное аустенитное зерно наследуется при небольших скоростях нагрева (градусы в минуту), характерных для обычного печного нагрева. С ускорением нагрева эффект наследования исходного зерна ослабевает. Если же закаленную и неотпущенную сталь нагревать очень быстро (сотни градусов в секунду), то снова проявится сильная структурная наследственность. Такое сложное влияние скорости нагрева пока трудно объяснить, так как нет надежных данных о механизме превращения сгри разных степенях перегрева.
В 1868 г. Д. К. Чернов установил, что для исправления крупнозернистости стали, определяемой по излому, необходим нагрев выше точки Ь. Обычно точку b Чернова отождествляют с критической точкой Ас3. Однако из работ
168
В. Д. Садовского следует, что точка 6, всегда соответствующая температуре структурной перекристаллизации, исправлению крупнозернистости, в зависимости от состава и исходной структуры стали может совпадать с а может находиться значительно, иногда на 200—300 К, выше Лс3. В последнем случае точка b соответствует температуре начала первичной рекристаллизации аустенита, а сама рекристаллизация является следствием фазового наклепа, возникающего при превращении.
§ 24. Диффузионные превращения аустенита при охлаждении
1. Перлитное превращение
Основное превращение, протекающее во время охлаждения при отжиге стали, — это эвтектоидный распад аустенита на смесь феррита с карбидом. Кинетика эвтектоидного превращения изображается С-образными кривыми на диаграмме изотермического превращения аустенита (рис. 80).
С-диаграмму строят следующим образом. Тонкие образцы стали нагревают до полной аустенитизации и затем быстро переносят в термостат (соляную ванну) с температурой ниже критической точки. Начало и конец распада аустенита при данной температуре можно определить несколькими методами. Микроструктурный анализ — самый простой, вместе с тем надежный, но и весьма трудоемкий метод. Образцы после различного времени выдержки в тер-: мостате закаливают в воде. Изотермически нераспавшийся аустенит превращается в мартенсит, который хорошо отли-( чается под микроскопом от продуктов распада аустенита?
При эвтектоидном превращении аустенита увеличивает7’ ся удельный объем, падает электросопротивление и сталь переходит из парамагнитного в ферромагнитное состояние. На исследовании изменений этих физических свойств основаны дилатометрический, электрический и магнитный методы определения времени начала и конца превращения. Магнитометрический метод позволяет количественно определять степень превращения аустенита в любой момент изотермической выдержки, так как интенсивность намагничивания образца пропорциональна количеству ферромагнитной альфа-фазы (аустенит парамагнитен).
При температуре 727 °C аустенит находится в термоди-^ намически устойчивом равновесии со смесью феррита и це
163
ментита. Чтобы начался распад аустенита, необходимо его переохладить ниже 727 °C.
Устойчивость переохлажденного аустенита характеризуется инкубационным периодом, т. е. отрезком времени (от оси ординат до левой С-кривой, рис. 80), в течение которого обычные методы не фиксируют появление продуктов распада.
' В эвтектоидной стали при температурах около 550 °C переохлажденный аустенит наименее устойчив. Как раз
/ 10 102 103 10^ ю5
1д времени (с'.
Рнс. 80. Диаграмма изотермического распада аустенита в эвтектоидной стали:
А — устойчивый аустенит; —переохлажденный аустенит; Ф—феррит: К — карбид
Рис. 81. Схема зарождения п роста двух перлитных колоний (Мэйл и Хэ-гель)
при этих же температурах обнаружены максимумььс^а^)-сти зарождения и скорости роста эвтектоида (см. рис. о0). ' В инкубационный период в переохлажденном аустените протекают подготовительные процессы перераспределения углерода.
Перлит растет из отдельных центров в виде колоний (рис. 81, 82). Зародышем перлитной колонии может быть или цементит (рис. 81, а), или феррит, зарождение которых облегчено на границе аустенитных зерен. При утолщении цементитной пластины вблизи нее аустенит обедняется углеродом и создаются условия для зарождения путем полиморфного у->а-превращения ферритных пластин, примыкающих к цементитной (рис. 81,6). При утолщении же ферритной пластины, практически не содержащей углерода, он оттесняется в аустенит, в результате чего создаются благоприятные условия для появления новых цементитных пластин, и т. д. (рис. 81, г, д). Такое многократное попере-170
-менное зарождение пластин феррита и цементита — не единственная возможность бокового роста эвтектоидных колоний. Другой экспериментально установленный механизм бокового роста — ветвление пластин обеих фаз, кото-
Рис. 82. Строение перлита около границы колонии (Вилел-ла)
рое может приводить к формированию как плоскопараллельных, так и веерных структур (рис. 83).
Если колония перлита зарождается из ферритного центра на границе двух аустенитных зерен yi/y2 и растет в зерне yi, то решетка феррита этой колонии сопрягается с решеткой «чужого» зерна у2, являющегося подкладкой для зародыша колонии. Поскольку зерна у! и у2 взаимно ориентированы произвольным образом, то в «своем» зерне феррит отделен от аустенита границей с высокой энергией и большой подвижностью. Ситуация аналогична и в случае зарождения колонии перлита из цементитного центра.
Внутри перлитной колонии пластины феррита и цементита сопрягаются гранями (101)ц и {112}ф или (001)ц и {125}ф. Соответствующие кристаллографические плоскости в решетках феррита и цементита имеют очень близкое строение.
Кроме бокового, идет торцовый рост пластин феррита и цементита (см. рис. 81, бив). Перед торцами пластин феррита и цементита концентрация углерода в аустените соответственно повышена и понижена, т. е. существуют градиенты концентрации перпендикулярно и параллельно фронту превращения (при росте одной фазы градиент концентра-
171
Рнс. 83. Ветвление цементита в перлитных колониях эвтектоидной стали. Растровая электронная микроскопия после вытравливания феррита на шлифе. Х540 (Ю. Н. Таран)
ративного роста колонии; 2) тает суммарная поверхность
ций только перпендикулярен фронту превращения). Рост перлитной'колонии контролируется диффузионным перераспределением углерода параллельно фронту превращения в объеме аустенита и прямо по границе перлитной колонии. Кооперативный рост двухфазной колонии путем диффузионного перераспределения компонентов — наиболее характерная особенность перлитного превращения.
Скорость роста колонии и межпластиночное расстояние (суммарная толщина пластин феррита и цементита или, что то же самое, расстояние между серединами ближайших одноименных пластин) постоянны при данной степени переохлаждения аустенита. Зинер предположил, что толщина пластин зависит от следующих факторов: 1) чем тоньше пластины обеих фаз, тем меньше пути диффузии углерода на фронте превращения и тем быстрее совершается его перераспределение, необходимое для коопе-с утонением пластин возрас-их раздела .и, следовательно,
уменьшается разность ДбОб—ДСПОв, являющаяся результирующей движущей силой превращения. При данной степени переохлаждения, т. е. при определенной величине разности энергий Гиббса аустенита и перлита (Д6Об), устанавливается такое межпластичное расстояние, при котором скорость роста, управляемая указанными факторами, максимальна.
С увеличением степени переохлаждения ДГ возрастает ЛОоб, что позволяет развиться большей поверхности Ф/Ц— межпластиночное расстояние в перлите уменьшается.
Межпластиночное расстояние в перлите — важнейшая структурная характеристика. С его уменьшением прочностные свойства стали возрастают.
В эвтектоидной стали при распаде аустенита в области температур от А{ до ~ 650 °C межпластиночное расстояние
172
в колониях равно 0,5—1 мкм, двухфазное строение колоний хорошо видно при средних увеличениях микроскопа. Такой эвтектоид называют перлитом. При распаде аустенита в интервале температур примерно 650—600 °C межпластиночное расстояние равно 0,4—0,2 мкм, двухфазное строение колоний выявляется лишь при больших увеличениях светового микроскопа (предельное разрешаемое расстояние светового микроскопа ~0,2 мкм). Такой эвтектоид называют сорбитом. Распад аустенита в интервале температур 600—500 °C дает очень тонкую эвтектоидную смесь с межпластиночным расстоянием около 0,1 мкм. Двухфазное строение такого эвтектоида, называемого тро-оститом, выявляется только под электронным микроскопом.
При непрерывном охлаждении указанные структуры образуются в углеродистой эвтектоидной стали в следующих условиях: перлит—при охлаждении стали из аустенитного состояния вместе с печью со скоростью несколько градусов в минуту, сорбит — при охлаждении на воздухе со скоростью несколько десятков градусов в минуту, троос-тит — при охлаждении в масле со скоростью несколько десятков градусов в секунду.
В стали с 0,8 % С твердость перлита, сорбита и троос-тита равна приближенно НВ 170—230, 230—330 и 330— 400 соответственно.
Весьма условное подразделение перлитных структур на собственно перлит, сорбит и троостит хотя и устарело, но продолжает использоваться, особенно в практике термической обработки стали.
Перлитное превращение в эвтектоидной стали протекает при диффузионном распаде аустенита в интервале от А\ до температур вблизи изгиба («носа») С-кривой (см. рис. 80). Ниже изгиба С-кривой в интервале примерно 500—250 °C происходит превращение дру'ого типа — бейнитное, которое является промежуточным между перлитным и мартенситным и поэтому будет обсуждено после рассмотрения мартенситного превращения (см. § 39).
Наряду с межпластиночным расстоянием внутри колоний перлита важной структурной характеристикой является размер колоний. Эти колонии при разрушении стали ведут себя как самостоятельные зерна. Размер фасеток в изломе в среднем равен размеру перлитных колоний. С уменьшением размера перлитных колоний, называемых также эвтектоидными зернами, ударная вязкость стали растет.
173
Перлитные колонии зарождаются на границах аустенитных зерен. Поэтому с уменьшением размера зерна аустенита сокращается и размер перлитных колоний (при данной степени переохлаждения аустенита). С ростом степени переохлаждения перлитные колонии становятся несколько мельче. Ни размер аустенитного зерна, ни размер перлитных колоний не влияют на межпластиночное расстояние, которое зависит только от температуры превращения.
2. Превращения аустенита в доэвтектоидных и заэвтектоидных сталях
Выше рассматривалось превращение аустенита в стали эвтектоидного состава. В до- и заэвтектоидных сталях пер-
лу тному превращению должно предшествовать выделение / избыточных фаз—феррита
и втэдцршого цементита (см. pfjc. 74). На диаграммах изотермических превращений аустенита в до- и заэвтектоидных сталях должны быть нанесены линии начала образования избыточной фазы (рис. 84).
В случае непрерывного охлаждения избыточный феррит начинает образовываться при переохлаждении аустенита ниже точки А3 и продолжает зарождаться и расти до температур ниже точки Ai. При росте ферри-
Время
Рис. 84. Диаграмма изотермического распада аустенита в дсэвтектоидной стали (схема):
А— устойчивый аустенит; Лп—переохлажденный аустенит; Ф — феррит; К — карбид
та углерод накапливается в аустените перед фронтом превращения, так как феррит почти не содержит углерода. При температурах ниже Ах в,этих обогащенных углеродом участках аустенита зарождается перлитная колония. Для этого состав аустенита не обязательно должен достигать концентрации 0,8 % С. В перлите (сорбите, троостите), образовавшемся из аустенита с содержанием менее 0,8 % С, увеличена доля ферритных промежутков. Такой эвтектоид с повышенным против равновесной концентрации эвтектоидной точки содержанием железа был назван А. А. Бочва-ром квазиэвтектоидом. В заэвтектоидных сталях обогащен
углеродом по сравнению с составом точки S.
Скорость кооперативного роста двухфазной колонии, связанного с диффузией на короткие расстояния параллель-
174
но фронту превращения (в отличие от диффузии на большие расстояния перпендикулярно фронту превращения при росте избыточного феррита), сравнительно высока, и перлитное превращение быстро охватывает весь объем аустенита. Поэтому после начала перлитного превращения образование доэвтектоидного феррита почти прекращается.
Относительное количество структурно-свободной избыточной фазы зависит от степени переохлаждения аустенита. С увеличением скорости охлаждения уменьшается количество успевающего обособленно вырасти избыточного феррита или вторичного цементита. При достаточно большом переохлаждении образование избыточной фазы в виде самостоятельной структурной составляющей полностью предотвращается и сталь неэвтектоидного состава приобретает чисто квазиэвтектоидную структуру.
Структурное вырождение перлита
В доэвтектоидной стали с малым количеством аустенита эвтектоидного состава, например при содержании в стали до 0,1 % С, при медленном охлаждении феррит, образующийся при эвтектоидном превращении, нарастает на зернах избыточного феррита, а эвтектоидный цементит остается в виде структурно-свободных прожилок по границам этих зерен. Здесь полная аналогия с вырождением эвтектики (а+₽) в p-прожилки по границам первичных a-кристаллов, на которых наросла a-фаза эвтектического происхождения (это происходит в том случае, когда в сплаве мало эвтектической жидкости и она кристаллизуется в узком пространстве между первичными кристаллами.
Вырождение перлита в протяжённые прослойки структурно-свободного цементита (их иногда путают с третичным цементитом, образующимся из феррита) снижает способность листовой стали к глубокой вытяжке, так как по этим прослойкам происходят разрывы. При рекристаллизационном отжиге холоднокатаного автолиста из стали 08кп нельзя превышать температуру точки Аь чтобы не появился аустенит, который при медленном охлаждении даст структурно-свободный цементит в нежелательной форме.
В заэвтектоидной стали эвтектоид также может структурно вырождаться. Цементит, образующийся в результате эвтектоидного распада, при очень малых переохлаждениях ниже (выше ~700 °C) наслаивается на вторичный цементит, оставляя рядом участки структурно-
175
Рис. 85. Заэвтектондная сталь с аб-нормальной структурой. ХЗОО (Ю. Н. Таран)
ницы с цементитом — точкой
свободного феррита (рис. 85). Такое эвтектоидное превращение, идущее с разделением фаз в пространстве, называют абнормальным в отличие от нормального эвтектоидного превращения, при котором происходит кооперативный рост феррита и цементита в виде колоний с регулярным чередованием двух фаз. При абнормальном превращении эвтектоид вырождается в грубый конгломерат феррита и цементита, не имеющий характерного эвтектоидного строения.
Во время абнормального эвтектоидного превращения, когда вторичный цементит окружается оболочкой из феррита, поступление углерода от распадающегося аустенита к цементиту, необходимое для его роста, происходит через эту оболочку. При температуре Т2 на рис. 76 концентрация углерода в феррите на границе с аустенитом изображается точкой f, а около гра-g. Градиент концентраций в
ферритной оболочке весьма небольшой, но подвижность углерода в о. ц. к. решетке феррита высокая и диффузионный поток углерода через феррит способен питать цементит, растущий при абнормальном эвтектоидном превращении.
В процессе эвтектоидного превращения механизм его может меняться от абнормального (рядом со вторичным цементитом) до нормального. При температурах ниже ~700 °C скорость нормального эвтектоидного превращения оказывается намного больше и абнормальное превращение подавляется. Поэтому при ускоренном охлаждении и соответственно большом переохлаждении аустенита абнормальное превращение может быть полностью подавлено.
Развитию абнормального превращения в заэвтектоид-ной стали способствует примесь кислорода, поэтому после цементации стали в смеси древесного угля с углекислым барием, содержащей много кислорода, в науглероженном слое часто образуется абнормальная структура. Она вредна, так как при грубых участках феррита требуется большее время или более высокая температура аустенитизации
176
при нагреве детали под закалку. В противном случае после закалки на месте малоуглеродистого аустенита обнаруживаются «мягкие пятна». Хром (десятые доли процента) и марганец (более 0,6 %) полностью подавляют абнормаль-ное перлитное превращение в заэвтектоидных сталях.
Форма и строение доэвтектоидного феррита
Избыточный феррит в доэвтектоидных сталях встречается в двух формах: в виде компактных более или менее равноосных зерен и в виде ориентированных видманштеттовых
Рис. 86. Микроструктура доэвтектоидной стали. Х100:
а — видманштеттова структура литой стали; б — та же сталь после полного отжига
пластин (рис. 86). Компактные выделения доэвтектоидного феррита образуются преимущественно по границам зерен аустенита, а видманштеттовы пластины — внутри аустенитных зерен, причем часто они берут начало от компактных выделений феррита.
Видманштеттов феррит наблюдается лишь в сталях, содержащих менее 0,4 % С, и при условии, что аустенитное зерно достаточно крупное. Такое зерно бывает в литой стали, а также в стали, нагретой до 1100—1200 °C (явление
12 И. И. Новиков 177
перегрева). С уменьшением размера аустенитного зерна доля видманштеттова феррита уменьшается, а доля феррита в форме равноосных зерен возрастает. Наиболее четко видманштеттова структура проявляется при ускоренном охлаждении стали.
Видманштеттов феррит образуется в интервале от Л3 минус 50 °C до 600—550 °C. С увеличением содержания углерода в стали доля видманштеттова феррита в структуре уменьшается.
Образование видманштеттова феррита кристаллографически упорядочено. Ориентация феррита подчиняется принципу структурного соответствия: {110}ф I | {111} А. Эти плоскости наиболее плотной упаковки соответственно в о. ц. к. и г. ц. к. решетках имеют очень близкое строение. Плоскость габитуса каждой ферритной пластины близка к {Н1} А-
Имеются две гипотезы о механизме образования видманштеттова феррита. Первая гипотеза связывает его образование со сдвиговой у->а-перестройкой решетки, характеризующейся упорядоченными взаимосвязанными перемещениями атомов (как при образовании мартенсита)1. Медленный рост кристаллов видманштеттова феррита по сравнению с ростом мартенсита предположительно объяснен тем, что в отличие от легко скользящей когерентной границы мартенсит — аустенит граница видманштеттов феррит — аустенит имеет такое строение, которое не может обеспечивать ее легкого скольжения (Р. И. Энтин). Мартенситоподобный механизм образования видманштеттовой пластины объясняет, почему в ней в отличие от равноосных а-зерен повышена на 1—2 порядка плотность дислокаций и появляется характерный рельеф на полированной поверхности шлифа.
Предполагается, что равноосные зерна феррита растут путем нормальной диффузионной перестройки решетки с неупорядоченным переходом атомов через границу у/а. Предпочтительное зарождение компактных выделений феррита на границах аустенитных зерен объясняют повышенной диффузионной подвижностью железа в этих местах, необходимой для реализации нормального механизма у->а-превращения. Чем крупнее зерно аустенита, тем меньше роль его границ в зарождении феррита нормальным путем и соответственно больше доля видманштеттова феррита.
1 К рассмотрению механизма образования видманштеттова феррита следует вернуться после проработки § 34.
178
Другая гипотеза исходит из механизма роста видманштеттовой пластины феррита путем миграции ступенек на ее широкой грани. Такую миграцию прямо наблюдали в электронном микроскопе (высота ступенек была порядка Ю1—Ю3 нм). Предполагается, что широкие плоские участки поверхности пластины (фасетки) являются полностью когерентными или частично когерентными границами и практически неподвижны (как когерентные границы двой
Гис. 87. Схема роста кристалла фер-г лта путем миграции ступенек вдоль его поверхности
Рис. 89. Схема зарождения и роста межфазных выделений карбида (Хо-и.чкомб)
Рис. 88. Цепочка межфазных выделений VC в феррите жаропрочной стали, содержащей 0,1 % С. 0,3 % Сг, 0,5 % Мо, 2,4 % V и 0,6 % Мп. Изотермическое превращение при 700 °C, 15 мии. Электронная микрофотография, фольга (Даилоп и Хоникомб)
ников), а ступеньки — высокоподвижными некогерентными границами, через которые может совершаться быстрый диффузионный переход атомов от аустенита к ферриту. Пробег одной ступеньки вдоль ферритной пластины (горизонтальные стрелки на рис. 87) означает утолщение этой пластины (вертикальная стрелка) на высоту ступеньки.
Во второй гипотезе учитывается, что рельеф на шлифе не обязательно вызывается сдвиговым превращением, а наблюдается у ряда сплавов и при нормальном превращении.
Схема роста кристаллов доэвтектоидного феррита путем Движения ступенек вдоль границы у/a представляет боль-
12*
179
той интерес, так как она позволяет понять важную особенность структуры феррита в некоторых сталях, легированных сильными карбидообразователями (Nb, V, Ti, Mo, W и др.). В этих сталях в определенных условиях обработки при формировании доэвтектоидного феррита из аустенита внутри a-кристаллов появляются плотно расположенные цепочки очень дисперсных частиц карбида NbC, VC, М02С и т.п. (рис. 88). Эти частицы часто имеют размер менее 10 нм, а расстояние между их цепочками колеблется от 5 до 50 нм. Такую плотную «начинку» доэвтектоидного феррита чрезвычайно дисперсными карбидными частицами можно увидеть только при достаточно больших разрешениях электронного микроскопа, а при меньших увеличениях ферритные зерна выглядят монокристаллами. Формирование такой структуры объясняют выделением карбида на межфазной границе у/а, причем частицы карбида зарождаются на плоских неподвижных фасетках и успевают лишь немного подрасти за время быстрой миграции ступеньки вдоль границы фасетки. На самой ступеньке из-за быстрого ее продвижения карбид не успевает выделиться (рис. 89). При пробеге каждой новой ступеньки образуется новая цепочка дисперсных частиц карбида. Расстояние между цепочками разное и равно высоте соответствующих ступенек.
Цепочки частиц карбида, образовавшиеся в период формирования доэвтектоидного феррита, получили название межфазных выделений. Эти выделения не только очень дисперсны и потому значительно упрочняют феррит, но и сильнее сопротивляются коагуляции при длительном нагреве по сравнению с частицами карбида, выделившимися из мартенсита при отпуске стали. Объясняется это тем, что в доэвтектоидном феррите плотность дислокаций и субграниц, ускоряющих диффузию и соответственно коагуляцию, меньше, чем в отпущенной стали. Повышенная стойкость дисперсных частиц против коагуляции важна для жаропрочных сталей.
Намеренное создание структуры с доэвтектоидным ферритом, пронизанным дисперсными межфазными выделениями карбидов, — это интересное направление упрочнения сталей. Для получения такой структуры сталь следует нагревать до растворения специального карбида в аустените и ускоренно охлаждать, чтобы избежать обычного выделения карбида прямо из аустенита до начала формирования доэвтектоидного феррита.
180
3. Влияние легирующих элементов на перлитное превращение
Легирующие элементы оказывают чрезвычайно важное для практики влияние на кинетику распада аустенита. За исключением кобальта, все широко используемые легирующие элементы, растворенные в аустените (Сг, Ni, Мп, W, Mo, V и др.), замедляют перлитное превращение, сдвигая верхнюю часть С-кривой вправо.
Природа увеличения устойчивости переохлажденного ахстенита под влиянием легирующих элементов довольно сложная. Если в углеродистых сталях перлитное превращение связано с у^а-перестройкой решетки и диффузионным перераспределением углерода, то в легированных сталях к этому могут добавиться образование специальных карбидов и диффузионное перераспределение легирующих элементов, по-разному растворенных в феррите и карбиде.
Растворенные в аустените легирующие элементы не только сами имеют малый коэффициент диффузии, иногда в десятки и сотни тысяч раз меньше, чем у углерода, но некоторые из них (Mo, W) замедляют диффузию углерода в у-решетке. Кроме того, некоторые элементы (Сг, Ni) замедляют у->а-перестройку, являющуюся составной частью перлитного превращения.
Таким образом, легирующие элементы могут замедлять перлитное превращение по следующим причинам: 1) из-за образования специальных карбидов и необходимости диффузионного перераспределения в аустените легирующих элементов, атомы которых несравненно менее подвижны, чем атомы углерода; 2) из-за замедления диффузии углерода; 3) из-за уменьшения скорости полиморфного у-нх-превращения. В зависимости от состава стали и степени переохлаждения аустенита решающим может оказаться то? иди иной из указанных факторов. Например, при добавке к эвтектоидной углеродистой стали 0,8 % Мо продолжительность полного распада аустенита при температуре вблизи изгиба С-кривой возрастает на четыре порядка (!), причиной чего является необходимость диффузионного перераспределения компонентов в аустените в связи с образованием карбида (Fe, Мо)2зС6.
Более сильный эффект оказывает легирование одновре-' менно несколькими элементами. Замедление распада аустенита под действием легирующих элементов широко используют для увеличения прокаливаемости стали (см. §40).
Здесь, в разделе об отжиге 2-го рода, важно отметить, что легирование приводит к большему переохлаждению
181
аустенита при неизменной скорости охлаждения (рис. 90)*. Так как с увеличением переохлаждения уменьшается межпластиночное расстояние, то при одинаковой скорости охлаждения легированная сталь после перлитного превращения оказывается прочнее.
Рис. 90. Схематические диаграммы изотермического распада аустенита в углеродистой (а) и легированной (б) сталях. При одинаковой скорости охлаждения степень переохлаждения AT2>ATi
§ 25. Разновидности отжига сталей
Основной фактор, от которого зависит микроструктура стали после отжига 2-го рода, — это степень переохлаждения аустенита. Разновидности отжига 2-го рода различаются главным образом способами охлаждения и степенью
переохлаждения аустенита, а
также положением температур нагрева относительно критических точек.
Необходимая степень переохлаждения аустенита достигается или при непрерывном охлаждении, или при изотермической обработке. На рис. 91 на примере доэвтектоидной стали схематична изображены режимы охлаждения, соответствующие основные разновидностям отжига 2-го ро-
Рис. 91. Основные разновидно- ДЭ2 ПОЛНЫЙ (?)> U3OTCpMU4CCKUU Йид^^йТталиГ° рода доэвтек' (2), нормализационный отжиг (нормализация) (3) и патентиро^ вание (4).
К заэвтектоидным сталям применяют сфероидизирую-
щий отжиг и нормализацию.
* Легированная сталь — многокомпонентная система, в которой в соответствии с правилом фаз вместо эвтектоидной температуры имеется эвтектоидный интервал температур, показанный на рис. 90 штриховкой.
182
I. Полный отжиг
Для проведения полного отжига доэвтектоидную сталь нагревают до температуры, которая на 20—40 К выше точки .4г3 (рис. 92).
Охлаждение при отжиге проводят с такой малой скоростью, чтобы аустенит распадался при небольшой степе-и переохлаждения. Так как в легированных сталях аусте-
Рис. 92. Температура нагрева сталей для отжига 2-го рода:
1 — полный отжиг; 2 — неполный отжиг; 3 — сфероидизирующий отжиг; 4 — нормализация
лит более склонен к переохлаждению (см. рис. 90), то их следует охлаждать при отжиге с меньшей скоростью, чем углеродистые. Если углеродистые стали можно охлаждать при отжиге со скоростью 200 К/ч, то легированные— со скоростью 100—30 К/ч.
Так как превращение аустенита при отжиге полностью завершается при температурах значительно выше изгиба кривых, то отжигаемые изделия можно выдавать из печи на спокойный воздух при 500—600 °C, если не опасны термические напряжения. Если же одной из целей отжига является уменьшение напряжений в детали сложной конфигурации, то следует проводить охлаждение с печью почти комнатной температуры.
Структура доэвтектоидной стали после отжига состоит в избыточного феррита и перлита.
Основные цели полного отжига следующие: устранение пороков структуры, возникших при предыдущей обработке металла (литье, горячей деформации, сварке и термообра
183
ботке), смягчение стали перед обработкой резанием и уменьшение напряжений.
В доэвтектоидной стали отжигом устраняют два порока структуры — крупнозернистость и видманштеттов феррит (см. рис. 86). Крупные зерна аустенита могут сформироваться при затвердевании отливок и сварных швов, из-за перегрева в околошовной зоне и в слитках, подвергнутых гомогенизации при 1100—1200°C. В этих же случаях может появиться грубая видманштеттова структура, сильно снижающая ударную вязкость стали.
При отжиге с нагревом до Лс3+ (204- 40) К образуется мелкое аустенитное зерно, из которого при охлаждении формируется равномерная мелкозернистая феррито-перлитная структура. Если в стали с видманштеттовым ферритом (а также с мартенситной или бейнитной структурой, образовавшейся из-за подкалки при ускоренном охлаждении на воздухе отливок, сварных швов, горячего проката и поковок из легированных сталей) при нагреве наследуется исходное крупное зерно аустенита, то для достижения структурной перекристаллизации следует проводить высокотемпературный отжиг. Например, отливки из стали 20Х13Л следует отжигать при 1100—1150 °C, что примерно на 300 К выше точки Лс3. При таком нагреве достигается точка b Чернова, соответствующая температуре первичной рекристаллизации аустенита.
Целям обычного полного отжига, измельчающего зерно, противоположна цель отжига, увеличивающего зерно. Отжиг на крупное зерно с нагревом до 950—1100 °C применяют к мягким низкоуглеродистым сталям для улучшения обрабатываемости резанием. Такие стали дают вязкую, трудно отделяемую стружку, способную привариваться к режущему инструменту, что делает поверхность шероховатой. Улучшению качества поверхности и большей ломкости стружки низкоуглеродистых сталей способствует структура с крупными колониями пластинчатого перлита, которую и получают при высокотемпературном отжиге, увеличивающем зерно.
В заключение подчеркнем, что если структуру перегрева стали можно устранить отжигом (или нормализацией), то пережог, связанный с окислением по границам аустенитных зерен, — это неисправимый брак.
2. Неполный отжиг
Неполный отжиг доэвтектоидной стали проводят при нагреве до температур выше Асх, но ниже Ас% (см. рис. 92).
184
Этот отжиг для доэвтектоидных сталей применяют ограниченно. При температуре неполного отжига избыточным феррит не исчезает. Следовательно, неполный отжиг не может устранить указанных выше пороков стали, которые связаны с нежелательными размерами и формой избыточного феррита.
Неполный отжиг доэвтектоидной стали используют для смягчения ее перед обработкой резанием, так как в результате эвтектоидного превращения при неполном отжиге образуется мягкий перлит. Неполный смягчающий отжиг позволяет сэкономить время и снизить стоимость обработки.
3. Сфероидизирующий отжиг
Для заэвтектоидных сталей полный отжиг с нагревом выше Асщ (линия ES) вообще не используют, так как при медленном охлаждении после такого нагрева образуется грубая сетка вторичного цементита, ухудшающая механические свойства. К заэвтектоидным углеродистым сталям широко применяют отжиг с нагревом до 740—780 °C и последующим медленным охлаждением. После такого нагрева в аустените остается большое число нерастворившихся включений цементита, которые служат центрами кристаллизации во время распада аустенита при охлаждении.
В результате образуется структура зернистого перлита (сферодита), почему этот отжиг и называют сфероидизирующим. Мелкие частицы цементита при температуре отжига в интервале А\ — АСт получаются в результате деления цементитных пластин. При делении пластины растворяются в наиболее тонких участках, а также в местах выхода на межфазную поверхность Ц/А субграниц в цементите или аустените.
В месте выхода субграницы в цементитной пластине на межфазную поверхность неуравновешенность сил поверхностного натяжения является стимулом для локального растворения цементита до такой конфигурации поверхности, чтобы эти силы уравновесились (рис. 93). Канавки растворения с выпуклыми в сторону аустенита стенками идут вдоль линий выхода субграниц на поверхность пластины.
Известно, что растворимость частицы зависит от радиуса кривизны ее поверхности, и эта зависимость описывается уравнением Томсона — Фрейндлиха:
lnCr/Coo = 2yV/kTr, (32)
где Сг—концентрация раствора около межфазной границы с радиусом г; Соо —концентрация раствора около плоской
185
Рис. 93. Канавки растворения в местах выхсда субграницы в цементите (показана пунктиром) на поверхность раздела цементит — аустенит
границы; у — поверхностная (межфазная) энергия на границе фаз; V — атомный объем.
Чем меньше радиус кривизны границы, тем больше около нее равновесная концентрация раствора. Следовательно, около выпуклых стенок канавок растворения в цементите (см. рис. 93) концентрация углерода в аустените будет больше, чем около остальной плоской поверхности пластины. Выравнивание состава в аустените снизит концентрацию углерода в нем около выпуклых стенок канавки, и так как аустенит здесь окажется ненасыщенным по отношению к цементиту, то цементит растворится и границы его спрямятся. Это в свою очередь вызовет нарушение равновесия сил поверхностного натяжения у выхода субграниц, по
следующее восстановление равновесия при углублении канавки и т. д. В результате пластина цементита будет разделена вследствие растворения по ее субгранице.
Деление цементитных пластин можно ускорить, применив холодную пластическую или теплую деформацию при температурах ниже А\. При этом цементитные пластины не разрушаются, как полагали ранее, а пластически деформируются. При нагреве после холодной и во время теплой деформации путем полигонизации образуются субграницы, способствующие делению цементитных пластин. Как показало электронно-микроскопическое просвечивание фолы, цементитные пластины делятся не только при их ускоренном растворении у выходов субграниц, но и в местах повышенной плотности дислокаций в цементите.
После деления пластин мелкие их частицы сфероидизируются. Около краев и вершин цементитных частиц с малым радиусом кривизны концентрация углерода в аустените повышена в соответствии с уравнением (32). Выравнивание состава внутри аустенита приводит к повышению его концентрации около участков границы с большим радиусом кривизны, где аустенит пересыщается и выделяет цементит. Параллельное снижение концентрации углерода в аустените около краев и вершин приводит к их растворению. В результате градиент концентраций в аустените восстанавливается и процесс растворения цементита в участках с меньшим радиусом кривизны границы и выделения его в
186
участках с большим радиусом кривизны приводит к округлению частиц.
Таким образом, сфероидизация частиц цементита идет путем переноса углерода через окружающий твердый раствор.
Рассмотренные механизмы деления пластин и сфероидизации частиц одной фазы, находящейся в равновесии с твердым раствором, — общие для сплавов на разных основах.
Для режима сфероидизирующего отжига заэвтектоид-ных сталей характерен узкий температурный «интервал огжигаемости». Нижняя его граница должна находиться выше точки Ль а верхняя граница не должна быть слишком высокой, так как иначе из-за растворения в аустените центров карбидного выделения при охлаждении образуется пластинчатый перлит. Так как точки и Ах сходятся при эвтектоидной концентрации, то у сталей, близких к эвтектоидному составу, «интервал отжигаемости» особенно узок. Например, для сталей У9А и У10А границы этого интервала 740—750°C, в то время как для сталей УНА, У12А и У13А они находятся в пределах 750—780 °C.
Конечная структура зависит от скорости охлаждения и температуры сфероидизирующего отжига. Чем меньше скорость охлаждения, тем до больших размеров вырастают глобули карбида при распаде аустенита. Регулируя скорость охлаждения, можно получать структуры глобулярного перлита от точечного до крупнозернистого. Более мелкозернистый перлит обладает повышенной твердостью.
Влияние режима сфероидизирующего отжига на твердость показано на рис. 94. С повышением температуры отжига до 800—820 °C твердость снижается из-за развития сфероидизации, а при дальнейшем увеличении температуры отжига твердость растет из-за появления все в большем количестве пластинчатого перлита.
Сфероидизирующему отжигу подвергают углеродистые и легированные инструментальные и шарикоподшипниковые стали. Сталь со структурой зернистого перлита обладает наименьшей твердостью, легче обрабатывается резанием, что особенно важно, например, для работы автоматических линий в условиях массового подшипникового производства. Кроме того, зернистый перлит является оптимальной исходной структурой перед закалкой. При исходной структуре зернистого перлита меньше склонность к росту аустенитного зерна, шире допустимый интервал закалочных температур, меньше склонность к растрескива
187
нию при закалке, выше прочность и вязкость закаленной стали (мелкие глобули равномерно распределены в мартенсите закаленной заэвтектоидной стали).
Поэтому металлургические заводы поставляют отожженную инструментальную сталь со структурой зернистого перлита. Поковки шарикоподшипниковой стали также отжигают на зернистый перлит.
Рис. 94. Зависимость твердости шарикоподшипниковой стали ШХ15 от температуры отжига при трех скоростях охлаждения (Я. Р. Раузии), К/ч:
i — 100; 2 — 30; 3 — 5
Рис. 95. Микроструктура заэвтектоидной стали У12 после маятникового отжига (зернистый цементит). Х300
Если при однократном отжиге не происходит полной сфероидизации цементита, то можно применить циклический или маятниковый отжиг, являющийся разновидностью сфероидизирующего. Например, углеродистую сталь несколько раз попеременно нагревают до 740°C и охлаждают до 680 °C. Пластина цементита при каждом нагреве частично растворяется в аустените. Растворение идет преимущественно с вершин и ребер пластин. При каждом охлаждении из аустенита выделяется цементит на нераст-ворившихся остатках цементитных пластин, причем выделение идет преимущественно вдали от вершин и ребер. Попеременно растворяясь и подрастая, цементитная пластина постепенно округляется (рис. 95).
188
К сожалению, в заводских условиях очень трудно создать контролируемые колебания темературы большой садки с амплитудой 40 К в заданном температурном интервале.
4. Изотермический отжиг
Малая степень переохлаждения аустенита, необходимая при отжиге, может быть получена не только при непрерывном охлаждении стали с печью. Другой путь — ступенчатое охлаждение с изотермической выдержкой в интервале перлитного превращения (см. рис. 91). Такая термообработка называется изотермическим отжигом. После нагрева до температуры выше Аз сталь ускоренно охлаждают до температуры изотермической выдержки, которая находится ниже точки Аь Затем проводят ускоренное охлаждение на воздухе; мелкие изделия простой конфигурации можно охладить в подогретой воде.
Время изотермической выдержки должно быть несколько больше времени полного изотермического превращения аустенита, определяемого по С-диаграмме.
Чем ближе температура изотермической выдержки к точке Ai, тем больше межпластиночное расстояние в перлите и мягче сталь, но больше и время превращения. Так как основное назначение изотермического отжига — смягчение стали, то практически выбирают такую температуру изотермической выдержки (на 30—100 К ниже температуры Ai), при которой получается достаточное смягчение стали за сравнительно небольшой промежуток времени.
Изотермический отжиг по сравнению с обычным отжигом имеет два преимущества. Прежде всего он может дать выигрыш во времени, если суммарное время ускоренного охлаждения, изотермической выдержки и последующего ускоренного охлаждения меньше времени медленного непрерывного охлаждения изделия вместе с печью.
Особенно большой выигрыш времени можно получить при изотермическом отжиге легированных сталей с устойчивым переохлажденным аустенитом.
Другое преимущество изотермического отжига — получение более однородной структуры, так как при изотермической выдержке температура по сечению изделия выравнивается и превращение во всем объеме стали происходит при одинаковой степени переохлаждения.
5. Нормализация
При нормализации сталь нагревают до температур на 30— 50 К выше линии GSE и охлаждают на воздухе (см. рис. 92). Ускоренное по сравнению с отжигом охлаждение обусловливает несколько большее переохлаждение аустенита (см. рис. 91). Поэтому при нормализации получается более тонкое строение эвтектоида (тонкий перлит или сорбит) и более мелкое эвтектоидное зерно. Кроме того, частично подавляется выделение избыточной фазы (феррита или вторичного цементита) и, следовательно, образуется квазиэвтектоид. Таким образом, прочность стали после нормализации должна быть больше, чем прочность после отжига.
Нормализацию применяют чаще всего как промежуточную операцию для устранения пороков строения и общего улучшения структуры перед закалкой, а также для смягчения стали перед обработкой резанием. Таким образом, назначение нормализации как промежуточной обработки аналогично назначению отжига. Так как нормализация выгоднее отжига (меньше задалживается печное оборудование), то ее всегда следует предпочесть отжигу, если оба эти вида обработки дают одинаковые результаты. Но нормализация не всегда может заменить отжиг как операцию смягчения стали. Это объясняется следующим. Склонность аустенита к переохлаждению растет с увеличением содержания в нем углерода и легирующих элементов, поэтому разница в свойствах после отжига и после нормализации зависит от состава стали. Например, твердость сталей, содержащих 0,2; 0,45 и 0,8 % С, после отжига равна соответственно НВ 120, 160 и 180, а после нормализации НВ 130, 190 и 240.
Нормализацию широко применяют вместо смягчающего отжига к низкоуглеродистым сталям, в которых аустенит слабо переохлаждается. Но она не может заменить смягчающий отжиг высокоуглеродистых сталей, которые весьма ощутимо упрочняются при охлаждении на воздухе из-за значительного переохлаждения аустенита. Что же касается средне- и высоколегированных сталей, то в них при охлаждении на воздухе может образоваться мартенсит, т. е. происходит воздушная закалка (см. § 40). Здесь следует уточнить понятие нормализации. Под нормализацией понимают такую термическую обработку стали, при которой охлаждение на воздухе приводит к распаду аустенита в температурном интервале перлитного превращения. Поэтому если охлаждение легированной стали на воздухе дает мартенсит, как в стали 18Х2Н4ВА, то такой процесс никакого отношения к нормализации не имеет.
190
Рис. 96. Строчечная структура доэвтектоидной стали (на фойе феррита видны вытянутые шлаковые включения). Х300
Нормализацию широко применяют взамен отжига для устранения пороков стали, возникших при горячей деформации и термической обработке: крупнозернистости, видманштеттовой структуры, строчечное™. Во многих случаях нормализация дает лучшие результаты, чем отжиг. Например, строчечность в стали (рис. 96) легче устранить нормализацией, так как при большем переохлаждении аустенита феррит выделяется не только на вытянутых шлаковых и сульфидных включениях, но и во всем объеме аустенитного зерна.
Если исходная структура стали была кристаллографически упорядоченной (например, видманштеттовой), то для обеспечения структурной перекристаллизации аустенита проводят высокотемпературную нормализацию с нагревом выше точки b Чернова (см. § 23). Например, отливки из стали 35Л следует нагревать под нормализацию до 950— 970 °C, т. е. примерно на 150 °C выше Ас$.
В заэвтектоидной стали нормализация устраняет грубую сетку вторичного цементита. При нагреве выше точки
Аст (линия ES) вторичный цементит растворяется, а при последующем ускоренном охлаждении на воздухе он не успевает образовать грубую сетку, понижающую свойства стали. Например, если после горячей прокатки в инструментальных сталях УН—У13 имеется грубая цементитная сетка, то перед сфероидизирующим отжигом ее устраняют нормализацией с нагревом до 850 °C и последующим охлаждением, ускоренным с помощью вентиляторов.
Очень часто нормализация служит для общего измельчения структуры перед закалкой. Если в стали перед закалкой имеются грубые выделения избыточного феррита, то при нагреве под закалку аустенит не успевает как следует гомогенизироваться. Участки аустенита, соответствующие местам залегания грубых включений феррита, будут
191
обеднены углеродом и после закалки не приобретут необходимую твердость. После предварительной нормализации измельчаются выделения избыточного феррита, эвтектоид становится более дисперсным и тем самым облегчается быстрое образование гомогенного аустенита при нагреве под закалку.
Нормализацию используют и как окончательную обработку средне- и высокоуглеродистых доэвтектоидных сталей, если требования к свойствам умеренные и необязательна закалка с высоким отпуском.
Как окончательную термообработку нормализацию применяют и к низкоуглеродистым низколегированным сталям, например к высокопрочной строительной стали с карбонитридным упрочнением марки 16Г2АФ. Нормализация этой стали обеспечивает сочетание высокой прочности и низкой температуры перехода из вязкого состояния в хрупкое.
В заключение отметим, что скорость охлаждения на воздухе зависит от массы изделия и отношения его поверхности к объему, вследствие чего эти факторы сказываются на получаемой структуре и свойствах нормализованной стали.
6. Патентирование
Для получения высокопрочной канатной, пружинной и рояльной проволоки применяют изотермическую обработку, которая известна с 70-х годов XIX в. и получила название патентирования. Проволоку из углеродистых сталей, содержащих от 0,45 до 0,85 % С, нагревают в проходной печи до температуры на 150—200 °C выше Ac3f пропускают через свинцовую или соляную ванну с температурой 450— 550 °C и наматывают на приводной барабан. Распад аустенита проходит около изгиба С-кривой вблизи нижней границы температурного интервала перлитного превращения (см. рис. 91). По выходе из ванны проволока имеет фер-рито-цементитную структуру с очень малым межпласти-" ночным расстоянием. Ее принято называть сорбитом патентирования и трооститом. Избыточный феррит или вторичный цементит не успевает образоваться, и вся структура является квазиэвтектоидной.
После патентирования проволоку подвергают многократному холодному волочению с большим суммарным обжатием. Обычно получают проволоку с временным сопротивлением, равным 1500—2000 МПа. На стали У9А, полу-
192
ченной вакуумно-дуговым переплавом для устранения вредного влияния неметаллических включений и примесей, после патентирования и холодной протяжки с суммарным обжатием 99 % был достигнут рекордный уровень временного сопротивления 5000 МПа.
В получении высокопрочного состояния патентирование играет двоякую роль. Во-первых, благодаря ему проволока способна выдерживать большие обжатия при холодной протяжке без обрывов. Это обеспечивается структурой тонко-пластинчатого перлита и отсутствием зерен избыточного феррита, вызывающего обрывы при сильном натяжении. Во-вторых, после холодной пластической деформации фер-рито-цементитная смесь, в которой межпластиночное расстояние еще меньше, чем после патентирования, обеспечивает сочетание высокой прочности с вязкостью при скручивании и изгибе. Границы между пластинами феррита и цементита представляют непроницаемые барьеры для дислокаций, и временное сопротивление патентированной проволоки подчиняется соотношению Холла — Петча (17), в котором d для данного случая — межпластиночное расстояние.
Высокая температура нагрева при патентировании (обычно 870—920 °C) необходима для гомогенизации аустенита. Скорость движения проволоки должна быть такой, чтобы время пребывания в ванне было несколько больше времени окончания перлитного превращения. В противном случае по выходе проволоки из ванны аустенит, не успевший претерпеть перлитный распад, превращается в нижний бейнит или мартенсит и пластические свойства проволоки резко снижаются.
Хотя патентирование и специфический процесс, но по главному классификационному признаку — типу фазовых превращений — он относится к отжигу 2-го рода, являясь одной из разновидностей изотермической обработки.
Глава VI
ОТЖИГ ЧУГУНОВ
Основные компоненты чугуна — железо, углерод и кремний. Кроме того, обычные чугуны содержат марганец, фосфор и другие элементы. Несмотря на сложность химического состава чугуна, важнейшие структурные изменения при его отжиге качественно можно проанализировать с ис
13 И. И. Новиков
193
пользованием диаграммы состояния двойной системы Fe — С. В этой системе, как известно, аустенит и феррит могут находиться в стабильном равновесии с графитом (пунктирные линии на рис. 74) и в метастабильном равновесии с цементитом (сплошные линии).
Графит по сравнению с цементитом труднее зарождается и труднее растет в металлической матрице. Для зарождения графита требуются гораздо большие флуктуации концентрации, так как графит — это практически 100 % С (растворимость железа в нем ничтожна), а цементит содержит только 6,67 % С. При росте графитного кристалла необходимо почти полное удаление атомов железа от фронта продвижения его границы в металлической матрице. Поэтому образование метастабильного цементита в определенных условиях, например при ускоренном охлаждении, кинетически более выгодно, чем образование стабильного графита. Однако метастабильное равновесие аустенита или феррита с цементитом соответствует относительному минимуму энергии Гиббса, а стабильное равновесие с графитом— абсолютному минимуму энергии Гиббса. Поэтому выдержка чугуна при повышенных температурах должна в конце концов привести к замене цементита графитом.
Кремний, никель, алюминий и другие элементы способствуют графитизации, а марганец, хром, ванадий, магний, церий, сера и другие элементы затрудняют ее.
Фазовые превращения при термической обработке чугунов включают все те основные процессы, которые встречаются в сталях, и дополнительно осложнены процессами, связанными с поведением графитной фазы.
Выдающийся вклад в изучение фазовых превращений в чугунах, в частности графитизации, внесли работы школы К. П. Бунина. В данной главе анализ процессов отжига чугунов базируется главным образом на обобщениях и результатах работ этой школы.
Основные разновидности отжига 2-го рода чугунов — графитизирующий отжиг и нормализация.
§ 26. Графитизирующий отжиг чугуна
Графитизирующий отжиг чугуна — это термическая обработка, при которой главным процессом является образование графита с одновременным частичным или полным исчезновением цементита.
Графитизирующему отжигу подвергают белые, серые и\ высокопрочные (модифицированные) чугуны.
194
1. Отжиг белого чугуна на ковкий
Белый чугун тверд и очень хрупок из-за большого количества эвтектического цементита в его структуре. Современный способ получения ковкого чугуна графитизирующим отжигом белого был изобретен в начале XIX в. В настоящее время ковкий чугун — это широко применяемый машино-
Вреня
Рис. 97. График отжига белого чугуна иа ковкий:
I и II — первая и вторая стадии графитизации
Рис. 98. Микроструктура ковкого чугуна на ферритной основе. Х120
строительный материал, сочетающий простоту и дешевизну получения отливкой фасонных деталей с высокими механическими свойствами.
Для производства ковкого чугуна используют отливки из доэвтектического белого чугуна, содержащего 2,2— 3,1 % С, 0,7—1,5 % Si, 0,3—1,0 % Мп и до 0,08 % Сг. Содержание в шихте кремния, облегчающего графитизацию, и марганца с хромом, затрудняющих ее, регулируют таким образом, чтобы подавить кристаллизацию графита из расплава и обеспечить возможно более быстрое прохождение графитизации при отжиге. Напомним, что при кристаллизации серого чугуна графит растет из расплава в неблагоприятной форме разветвленных крабовидных розеток, сечения которых на шлифе имеют вид изогнутых пластин. При отжиге белого чугуна (рис. 97) графит, называемый
13* 195
углеродом отжига, образуется в значительно более компактной, благоприятной для механических свойств форме (рис. 98). Хотя ковкий чугун и не куют, но относительное удлинение у него находится в пределах 2—20 % (в зависимости от структуры), в то время как у белого чугуна относительное удлинение не превышает 0,2 %, а у серого — не более 1,2 %.
Исходный фазовый состав белого чугуна такой же, как у стали: феррит и цементит, и поэтому механизм его аустенитизации аналогичен рассмотренному в § 22. При нагреве вначале происходит перлито-аустенитное превращение, затем растворение вторичного цементита и гомогенизация аустенита по С и Si.
Первая стадия графитизации
Во время выдержки около 10 ч ^ри 900—1050 °C проходит первая стадия графитизации (см. рис. 97), по окончании которой весь цементит эвтектического происхождения и остатки вторичного цементита заменяются графитом и структура из аустенито-цементитной превращается в аустенитографитную.
Предположение о разложении цементита с непосредственным выделением из него графита по реакции Fe3C-> ->3Fe+C не согласуется со многими фактами. В частности, форма углерода отжига в ковком чугуне не соответствует форме исходных кристаллов цементита.
Доказано, что графитизация белого чугуна на первой стадии состоит в зарождении графита на границе А/Ц и вдали от цементитных кристаллов и росте графита при одновременном растворении цементита в аустените путем переноса атомов углерода через аустенит от границы А/Ц к границе А/Г.
Удельный объем графита в несколько раз больше, чем у аустенита, и поэтому его гомогенное зарождение в плотной металлической матрице маловероятно — слишком велика упругая составляющая AGynp в формуле (26). Дислокации, субграницы и высокоугловые границы малоэффективны в качестве мест гетерогенного зарождения графита из-за большой величины AGynp.
Как известно, серое олово, удельный объем которого на одну четверть больше, чем белого, зарождается предпочтительно на открытой поверхности образца белого олова. Естественно, что при графитизации, когда удельный объем новой фазы еще более резко отличается от удельного объ-
196
ема исходной фазы, зародыши графита также преимущественно возникают на свободной поверхности аустенита.
В объеме отливки местами гетерогенного зарождения графита служат несплошности, скопления вакансий, усадочные и газовые микропустоты, микротрещины, разрывы на границе аустенита с неметаллическими включениями из-за разности их термического расширения. Местами за-
рождения графита могут быть диффузионные поры, возникающие при гомогенизации аустенита. Например, при выравнивании состава аустенита после ухода атомов кремния из обогащенных им участков остается избыток вакансий, образующих поры. Этим предположительно можно объяснить ускорение графитизации под действием кремния, которое происходит, несмотря на то, что кремний замедляет диффузию углерода в аустените.
После образования центров графитизации в аустените существует градиент концентрации углерода, так как предельная растворимость цементита в нем выше, чем графита (на диаграмме состояния рис. 74 линия ES находится правее линии E'S'). Например, если первая стадия графитизации проходит при температуре t* (рис. 99), то состав аустенита на границе с цементитом изображается точкой Ь, на границе с графитом — точкой а. Выравнивание концентрации углерода в аустените делает его ненасыщенным по отношению к цементиту (на границе А/Ц состав аустенита сдвигается влево от точки Ь) и пересыщенным по отношению к графиту (на границе А/Г состав сдвигается вправо от точки а). В результате непрерывно, вплоть до исчезновения, растворяется цементит и растет графит.
Кроме переноса атомов углерода через твердый раствор, для графитизации необходим еще один процесс — эвакуация атомов железа от поверхности растущего графита, чтобы освободить графиту «жизненное» пространст
Рис. 99. Участок диаграммы состояния системы Fe—С со сплошными линиями стабильного и пунктирными линиями метастабильного равновесия (схема)
ву
во. К. П. Бунин отметил, что именно этот диффузионный процесс, а не приток атомов углерода контролирует скорость роста графитных включений в аустените, так как диффузионная подвижность атомов железа намного меньше,, чем у углерода.
Форма графита зависит от температуры отжига и состава чугуна. Углерод отжига быстрее разрастается вдоль высокоугловых границ и субграниц, так как по ним быстрее отводятся атомы железа. Такое нежелательное разветвление графита усиливается с ростом температуры, и после отжига при температурах выше 1050—1070 °C механические свойства чугуна оказываются очень низкими. Этим определяется верхняя температурная граница первой стадии графитизации.
Добавки и примеси оказывают сложное влияние на рост углерода отжига. Например, малые добавки магния (~0,1 %) обеспечивают рост углерода отжига в компактной форме.
При охлаждении чугуна после окончания первой стадии графитизации состав аустенита изменяется по линии E'S' и из него выделяется вторичный графит. Эту стадию графитизации называют промежуточной. Вторичный графит наслаивается на включения углерода отжига и обычно самостоятельной структурной составляющей не дает.
Вторая стадия графитизации
Металлическая матрица ковкого чугуна формируется при эвтектоидном распаде аустенита.
Для получения чисто ферритной матрицы охлаждение в интервале температур эвтектоидного распада должно быть медленным (см. рис. 97). Здесь проходит вторая стадия графитизации — аустенит распадается по схеме А^Ф+Г.
Последовательность структурных изменений на второй стадии графитизации удобно проследить по диаграмме изотермических превращений аустенита в чугуне. На рис. 100 линия ОД — начало образования эвтектоидного феррита, BE — начало образования эвтектоидного цементита, РИ — окончание эвтектоидного превращения и БН — окончание графитизации перлитного цементита.
Рассмотрим превращения в аустените, переохлажденном до температуры t\. Вначале безынкубационно образуется графит на поверхности имеющихся включений углерода отжига. Начиная с момента, соответствующего точке 1 (линия ОД), Наряду с графитом образуется и эвтектоидный феррит, т. е. идет эвтектоидный распад по схеме А->Ф + Г. Он заканчивается в точке 2 полным исчезновением аустенита.
При эвтектоидном распаде графит наслаивается на уг: лерод отжига, образовавшийся в первую и промежуточную
198
стадии графитизации, и феррито-графитный эвтектоид как самостоятельная структурная составляющая в виде дисперсной смеси чередующихся фаз не образуется.
Эвтектоидный распад А—>-Ф+Г может идти нормальным и абнормальным путем. При нормальном распаде аустенит с образующимися из него
все время находится в контакте ферритом и графитом. На границе с графитом он имеет концентрацию точки g, а на границе с ферритом — точки h (см. рис. 99). Выравнивание состава в аустените приводит к пересыщению его углеродо!М на границе А/Г, выделению здесь графита и уменьшению концентрации углерода на границе А/Ф, вызывающему -^-«-перестройку — образование феррита.
Абнормалъное эвтектоидное превращение начинается в том случае, когда феррит полностью окружает графит и прерывается контакт аустенита с графитом. На границе с графи
Рис. 100. Диаграмма изотермических превращений аустенита в ковком чугуне, содержащем, %: 2,9 С; 0,88 Si; 0,36 Мп; 0,09 S (К. П. Бунин с сотр.)
том феррит имеет состав точки d, а на границе с аустенитом — состав точки /. Хотя градиент концентраций углерода и невелик, но диффузия элемента внедрения в о. ц. к. решетке феррита идет весьма быстро. От границы Ф/А углерод переносится к границе Ф/Г, где феррит пересыщается углеродом и выделяет графит, восстанавливая концентрацию, изображаемую точкой d. У границы Ф/А понижение концентрации углерода в феррите (сдвиг состава влево от точки f) делает его ненасыщенным по отношению к аустениту и происходит у-^а-превращение с образованием феррита состава точки f. Следовательно, при абнормальном эвтектоидном превращении графит выделяется не из аустенита, а из феррита.
При температурах выше точки В на рис. 100, т. е. при сравнительно небольших переохлаждениях аустенита (или медленном непрерывном охлаждении), полная ферритиза-ция матрицы происходит непосредственно при эвтектоидном распаде по схеме А—>-ФЦ-Г. При больших переохлаждени-, ях (ниже температуры точки В на рис. 100) становится возможным образование перлита (линия BE). Например,
199
при температуре /2 после безынкубационного выделения графита идет в период, обозначенный точками 3—4, эвтектоидный распад по схеме А->ФН-Г, а с момента, соответствующего точке 4, начинается перлитное превращение (А—>Ф-|-Ц). К моменту, соответствующему точке 5, оба эвтектоидных процесса (А-^Ф+Г и А-^Ф+Ц) приводят к полному исчезновению аустенита, и структура чугуна состоит из углерода отжига, феррита и перлита. Чем больше степень переохлаждения (или больше скорость охлаждения при непрерывном понижении температуры), тем большая доля аустенита распадается по схеме А->Ф+Ц. Если охлаждение отливки в интервале эвтектоидного превращения не замедлить, то получится чисто перлитный ковкий чугун. Его структура состоит из углерода отжига и перлитной матрицы.
Перлит примерно в 2,5 раза прочнее и тверже феррита, но менее пластичен. Поэтому с увеличением количества перлита в структуре ковкого чугуна прочностные свойства растут, а пластичность снижается. Например, у ферритного ковкого чугуна марки КЧ37-12 ав=370 МПа и S=12 %, а у перлитного чугуна ма’рки КЧ63-2 ов=630 МПа и 6 = = 2%.
Развитие второй стадии графитизации можно обеспечить не только замедлением охлаждения в эвтектоидном интервале температур, но и изотермической выдержкой после обычного, не замедленного охлаждения (см. пунктирную площадку на рис. 97). При непрерывном охлаждении идет перлитное превращение аустенита, а последующая длительная выдержка приводит к графитизации перлитного цементита. Механизм графитизации здесь принципиально такой же, как и на первой стадии; только графитизация идет путем переноса углерода не через аустенит, а через феррит от границы Ф/Ц к границе Ф/Г. Узким звеном процесса является эвакуация атомов железа в феррите от поверхности растущего графита.
На рис. 100 при температуре t2 перлитный цементит исчезает в момент, соответствующий точке 6. Чем ниже температура изотермической выдержки, тем большее время требуется для достижения этого момента (см. наклон кривой БН).
Одновременно с графитизацией перлитного цементита при изотермической выдержке происходят деление цементитных пластин и сфероидизация цементита. Механизм этих процессов аналогичен рассматриваемым в § 25 процессам получения сферодита в сталях.
200'
Поддерживая повышенное содержание марганца в шихте (до 1 %), можно резко замедлить графитизацию перлитного цементита, чтобы сохранить перлит и с помощью сфероидизации получить ковкий чугун с матрицей из сфе-родита.
Чугун с матрицей из зернистого перлита отличается сочетанием высокой прочности и повышенной пластичности, а также хорошей обрабатываемостью резанием и высокими антифрикционными свойствами. При ов=600 МПа относительное удлинение у такого чугуна достигает 10 %, и им можно заменить литую и кованую сталь. Из ковкого чугуна с основой из зернистого перлита изготавливают даже такие ответственные изделия, как коленчатые и кулачковые валы.
Отжиг белого чугуна на ковкий — длительный процесс, продолжающийся десятки часов. В производстве ковкого чугуна отжиг—наиболее дорогая операция, сильно влияющая на себестоимость литья. Поэтому разработаны разные способы ускорения отжига.
Весьма эффективна предварительная выдержка при температурах около 400 °C в течение примерно 6 ч, увеличивающая число центров графитизации на первой стадии.
Малые добавки модификаторов (0,02 % А1, 0,003 % В) также ускоряют графитизацию.
Предварительная закалка деталей из белого чугуна в масле резко ускоряет графитизацию благодаря образованию многочисленных закалочных субмикротрещин, заполняемых углеродом отжига на первой стадии графитизации. Но такая обработка применима только к мелким деталям простой формы.
2. Отжиг для устранения отбела
В тонких сечениях отливок из серого чугуна и высокопрочного чугуна с шаровидным графитом из-за ускоренного охлаждения кристаллизуется ледебурит, т. е. чугун получается белым. При литье в кокиль вся поверхность может получиться отбеленной. Для улучшения обрабатываемости резанием и повышения пластичности проводят графитизирующий отжиг, устраняющий отбел отливок.
Так как серый и высокопрочный чугуны содержат больше кремния, чем ковкий (до 3,3%), то графитизация в них развивается быстрее. Поэтому температура и время отжига для снятия отбела меньше, чем при отжиге белого чугуна на ковкий. Отливки для снятия отбела нагревают до
201
850—950 °C и после выдержки 0,5—5 ч охлаждают на воздухе. В зависимости от скорости охлаждения и состава чугуна матрица в бывших отбеленных участках получается перлитной или феррито-перлитной.
3. Низкотемпературный смягчающий отжиг
Этот отжиг применяют к отливкам из серого чугуна с перлитной или перлито-ферритной матрицей для снижения твердости, улучшения обрабатываемости резанием и повышения циклической вязкости. Отливки отжигают при 670— 750 °C в течение 1—4 ч для частичной графитизации перлитного цементита. Одновременно проходит частичная сфероидизация цементита. Механизм этих процессов такой же, как и во время изотермической выдержки на второй стадии графитизации ковкого чугуна.
В заключение отметим, что графитизирующий отжиг применяют также и к заэвтектоидным сталям, легированным кремнием (1—2%) или никелем (5—10%), способствующими образованию графита. Компактные включения графита в заэвтектоидной стали обеспечивают хорошие антифрикционные свойства и сопротивление изнашиванию. Прочность и предел выносливости у графитизированной стали выше, чем у ковкого чугуна.
§ 27. Нормализация чугуна
Упрочняющая термическая обработка серого чугуна не получила такого широкого распространения, как термообработка стали. Это объясняется тем, что пластинчатый графит, действуя как внутренние надрезы, сильно снижает прочность и пластичность металлической основы. Поэтому изменение ее строения при термической обработке не дает большого эффекта упрочнения и часто нерентабельно. Эффективнее термообработка серых чугунов с более благоприятной формой графита, в особенности высокопрочных чугунов' с шаровидным графитом. К такой термической обработке чугуна относится нормализация, повышающая прочность, твердость и износостойкость.
Нормализация чугуна — это термическая обработка, при которой главными процессами являются аустенитизация и последующее перлитное превращение.
Нормализации подвергают отливки главным образом с ферритной и феррито-перлитной матрицей и реже — с перлитной. Отливки нагревают до 850—950 °C и после вы
202
держки в течение 0,5—3 ч охлаждают на спокойном воздухе или в воздушной струе. Отливки сложной формы для уменьшения остаточных напряжений рекомендуется начиная с температуры около 500 °C охлаждать в печи.
При нагреве сначала проходит аустенитизация, включающая «-^-превращение, растворение в аустените графита и перлитного цементита (если матрица содержит перлит). После аустенитизации структура чугуна состоит из аустенита и графита. При дальнейшем нагреве графит частично растворяется в аустените в соответствии с ходом линии S'E' и количество связанного углерода возрастает.
В случае ускоренного охлаждения на воздухе идет перлитный распад аустенита и вся матрица приобретает перлитную или сорбитную структуру. Упрочнение при нормализации достигается благодаря двум факторам: устранению структурно-свободного феррита (повышению концентрации связанного в цементит углерода) и увеличению дисперсности перлита. Твердость чугуна возрастает от НВ 150 до НВ 200—250. Эффект упрочнения зависит от исходной структуры. Наибольшее упрочнение получается при нормализации ферритного чугуна, так как при этом мягкая ферритная основа заменяется более твердой перлитной. При нормализации перлитного чугуна лишь увеличивается дисперсность эвтектоида. Но даже в этом случае сопротивле* ние износу может возрасти в полтора раза.
Глава VII
ОТЖИГ ЦВЕТНЫХ МЕТАЛЛОВ И СПЛАВОВ
Отжиг 2-го рода в технологии цветных металлов и сплавов не используют так широко, как для сталей и чугунов.
Во многих промышленных цветных металлах и сплавах, например однофазных латунях, бронзах и электротехнических никелевых сплавах, отсутствуют фазовые превращения в твердом состоянии и к ним отжиг 2-го рода в принципе неприменим. Другие цветные сплавы претерпевают фазовые превращения в твердом состоянии, но отжиг 2-го рода вызывает такое изменение свойств, что применение его нецелесообразно.
В зависимости от типа фазовых превращений различают две разновидности отжига 2-го рода: гетерогенизацион-ный отжиг и отжиг с фазовой перекристаллизацией.
203
§ 28. Гетерогенизационный отжиг
Гетерогенизационный отжиг — это термическая обработка, при которой главным процессом является возможно более полное выделение из матричной фазы одной или нескольких фаз.
В подавляющем большинстве сплавов матричной фазой является твердый раствор на базе основного металла, а избыточной — соединение. К таким материалам относятся все термически упрочняемые сплавы на алюминиевой, магниевой, медной, никелевой и других основах, например дуралюмин, электрон, бериллиевая бронза, нимоник. Среди цветных сплавов эта группа самая многочисленная.
Относительное количество фазы, которая полностью переходит в твердый раствор при нагреве и выделяется при обратном медленном охлаждении, обычно не превышает 10—15 % от всего объема сплава. При таком фазовом превращении не происходит коренной ломки структуры по всему объему, как при отжиге углеродистых сталей с нагревом выше точки Дз. Тип кристаллической решетки матричной фазы при растворении в ней или выделении из нее другой фазы не меняется. Отжиг, включающий только процессы растворения и (или) выделения, приводит лишь к изменению концентрации компонентов в матричной фазе и к изменению количества, размера, а также формы частиц выделяющейся фазы. На этих изменениях и основано применение гетерогенизационного отжига. Цели его могут быть самыми разными. При обсуждении способа получения ультрамелкого зерна в листах из сплава В95 (см. рис. 55) уже рассматривался гетерогенизационный отжиг перед холодной прокаткой, проводимый для выделения частиц, которые способствуют увеличению числа центров рекристаллизации при заключительном рекристаллизационном отжиге (см. § 14, п. 2). Ниже рассмотрены другие случаи использования гетерогенизационного отжига цветных сплавов.
1. Смягчающий гетерогенизационный отжиг деформированных полуфабрикатов
Во многих сплавах гетерогенизация структуры при выделении избыточных фаз из матричного раствора, происходящая при медленном охлаждении с температуры отжига, приводит к разупрочнению, которое используют в разных целях.
204
Полный смягчающий отжиг применяют ко всем термически упрочняемым алюминиевым сплавам (типа дуралю-мин, авиаль и др.). Цель отжига — смягчить материал, сделать его пластичнее перед штамповкой, гибкой, отбортовкой и другими операциями холодной обработки давлением. Ускоренное охлаждение на воздухе горячекатаных рулонов
Рис. 101. Схема к объяснению выбора режима полного (Т п 0) н неполного (T’h.q) смягчающего гете-рогеннзацнонного отжига
Температура отжига°C
Рис. 102. Влияние температуры отжига после холодной прокатки на временное сопротивление дуралю-мнна Д16 при двух скоростях охлаждения (по данным В. А. Ливанова н С. М. Воронова):
/ — охлаждение с печью до 200 °C; 2 — охлаждение на воздухе
с температуры горячей прокатки приводит к частичной закалке (подкалке). Гетерогенизационный смягчающий отжиг рулонов, например дуралюмина, позволяет при последующей холодной прокатке повысить степень обжатия без промежуточных отжигов.
При полном смягчающем отжиге сплав нагревают до температур однофазной области, например до температуры Т’п.о на рис. 101, а затем очень медленно, с печью, охлаждают, чтобы в максимальной степени понизить концентрацию твердого раствора (в соответствии с линией сольвуса ас на рис. 101).
Чем ниже концентрация матричного раствора и больше расстояние между выделениями, тем больше разупрочнение при отжиге.
Большинство термически упрочняемых алюминиевых сплавов подвергают полному смягчающему отжигу при 380—420 °C в течение 10—60 мин с последующим охлаждением со скоростью не более 30 К/ч.
205
При полном отжиге, кроме основного процесса — гетерогенизации, может проходить и рекристаллизация, вносящая свой вклад в разупрочнение. Например, это происходит тогда, когда из-за низкой температуры окончания горячей прокатки лист был наклепан. Однако режим отжига для полного смягчения выбирают, исходя из требований гетерогенизации структуры, предусматривая медленное охлаждение, в то время как для чисто рекристаллизационного отжига однофазных сплавов скорость охлаждения не имеет значения.
Неполный (или сокращенный) смягчающий отжиг термически упрочняемых сплавов проводят при такой температуре ниже сольвуса, чтобы во время выдержки выделение второй фазы сильно снизило концентрацию матричного раствора (до точки b при температуре Тн.о на рис. 101).
Для многих систем характерно большое изменение растворимости в твердом состоянии в области высоких температур и очень слабое при более низких температурах (сравните наклон участков сольвуса ab Ьс на рис. 101). Поэтому охлаждение с температуры неполного отжига может быть быстрым (на воздухе или в воде)—сильного пересыщения матричного раствора не возникнет.
Из-за более низкой температуры сокращенного отжига время выдержки при этой температуре должно быть больше, чем при полном отжиге, однако общая продолжительность термической обработки уменьшается из-за последующего быстрого охлаждения.
На рис. 102 показано, что для максимального разупрочнения дуралюмина Д16 температура отжига в случае охлаждения с печью должна быть около 380—400 °C, а в случае охлаждения на воздухе — около 350—370 °C. При более высоких температурах нагрева S-фаза и СпА12 быстро переходят в алюминиевый раствор и при последующем охлаждении на воздухе происходит подкалка, повышающая прочность.
Сокращенный смягчающий отжиг большинства алюминиевых сплавов проводят при 350—370 °C с выдержкой 2—4 ч и охлаждением на воздухе или в воде.
Смягчающий гетерогенизационный ’отжиг применяют не только к алюминиевым сплавам. Если, например, (а+р)-латунь в результате ускоренного охлаждения из р-области после горячей обработки давлением имеет пониженную пластичность (из-за большого количества р'-фазы), то этот недостаток можно устранить отжигом с медленным охлаждением, во время которого из p-фазы более полно
206
выделяется пластичная a-фаза (см., например, сплав с 40 % Zn на рис. 103).
2. Гетерогенизационный отжиг .
для улучшения деформируемости слитков
При горячей обработке давлением алюминиевых сплавов наилучшая деформируемость обеспечивается тогда, когда концентрация легирующих элементов в твердом растворе на базе алюминия минимальна, а частицы избыточных фаз не слишком крупны и имеют компактную форму. В слитках, предназначенных для прессования, оптимальный размер частиц избыточных фаз находится в интервале 1—5 мкм. Частицы большего размера снижают пластичность, а меньшего слишком быстро растворяются при температуре прессования, и деформируемость ухудшается из-за повышения концентрации легирующих элементов в алюминиевом растворе.
Гомогенизационный отжиг, может не обеспечить оптимальную для деформируемости структуру слитка. При ускоренном охлаждении с температуры гомогенизации леги-рованность твердого раствора оказывается повышенной, а при замедленном охлаждении из твердого раствора могут выделиться частицы избыточной фазы размером более 10 мкм.
Если слитки после окончания гомогенизации охладить ниже сольвуса до температуры минимальной устойчивости переохлажденного твердого раствора и выдержать при этой температуре, то за сравнительно короткое время (например, за 3 ч при 350 °C в случае сплава Д16) концентрация
Таблица 3. Максимальное давление прессования (Ртах) и предельно допустимая скорость истечения (акр) при прессовании слитков алюминиевых сплавов (В. В. Захаров)
Сплав Режим отжига слитка* Р max’ МПа Гкр’ м/мин Сплав Режим отжига слитка* Р max’ МПа икр’ м/мин
АДЗЗ 1 105 40 Д16 1 180 4,0
2 95 ПО 2 160 6,0
АД35 1 105 35
2 95 170 В95 1 160 1,7
Д1 1 165 5,3 2 150 2,1
2 150 10,4
Режим 1 слитки охлаждены на воздухе с температуры гомогенизации: режим 2 после гомогенизации применен гетерогенизационный отжиг.
207
легирующих элементов в алюминиевом растворе сильно снизится, причем выделятся частицы избыточной фазы размером 1—5 мкм. В результате такого гетерогенизационного отжига, проводимого после обычной гомогенизации слитков, снижается давление прессования и в 1,5—3 раза возрастает предельно допустимая скорость истечения (скорость, еще не приводящая к появлению трещин). Это позволяет существенно увеличить производительность прессового оборудования. Наиболее сильно улучшается деформируемость при прессовании сплавов АДЗЗ и АД35 системы А1—Mg—Si (табл. 3).
3. Гетерогенизационный отжиг для повышения коррозионной стойкости
Если двухфазная структура с определенной степенью дисперсности и оптимальным распределением частиц избыточной фазы обеспечивает повышенную стойкость против коррозии, то такую структуру получают гетерогенизационным отжигом. Например, максимальную стойкость против кор
Рис. 103. Диаграмма состояния системы Си—Zn
розии под напряжением магналия марки АМгб обеспечивает двухфазная структура, в которой p-фаза (Al3Mg2) равномерно распределена в зернах алюминиевого раствора. Этого достигают отжигом магналия при 320 °C перед последней холодной нагартовкой.
4. Явление перегрева в литой ( а+р)-латуни
Границы зерен в технических литых сплавах весьма стабильны. В однофазном литом сплаве, например а-латуни, при нагреве зерно практически не укрупняется.
По-другому ведет себя (а-Ьр)-латунь (рис. 103), в слитках которой после перехода через границу а-Ьр/р возможен сильный рост зерна (явле-
ние перегрева), причем размер р-зе-рен может достигать нескольких сантиметров (рис. 104). Бурный рост зерна вызван фазовым наклепом из-за изменения удельного объема при растворении a-фазы в р-фазе (А. А. Бочвар), а также, возможно, из-за разности термических коэффициентов расширения этих фаз. В ре-
208
зультате естественной неравномерности распределения a-фазы по обе стороны от границы p-зерен местный наклеп здесь также неодинаков. Гранина p-зерна мигрирует в сторону более наклепанного участка, «выметая» на своем пути дислокации: одно p-зерно растет за счет соседнего. После того как отдельные зерна «вырвались вперед» по своему размеру, они растут за счет основной массы более мелких
Рнс. 104. Зерна-гиганты, образовавшиеся при перегреве слитка (аЧ-р)-латунн. Уменьшено в 2,5 раза
зерен (на этой стадии процесс аналогичен вторичной рекристаллизации) .
Чем меньше a-фазы в исходной структуре, тем слабее должен быть фазовый наклеп при нагреве литой (а+р)-латуни и меньше склонность к росту 0-зерна. Например, в литой латуни с 40 % Zn объемная доля a-фазы при комнатной температуре составляет 80 %, а в латуни с 45 % Zn — около 5 % (см. рис. 103). Соответственно рассмотренный процесс бурного роста 0-зерен наблюдается только в первом сплаве. При многократном изменении температуры (20^820 °C)
14 И. И. Новиков
209
с медленным охлаждением для более полного выделения a-фазы и быстрым нагревом для обеспечения сильного фазового наклепа небольшой слиток латуни с 40 % Zn можно постепенно превратить в монокристалл 0-фазы (после последнего нагрева проводят закалку).
Из диаграммы состояния Си — Zn (см. рис. 103) вытекает, что количество a-фазы в («+0) -латунях при уменьшении содержания цинка возрастает от 0 до 100 %. В термически упрочняемых алюминиевых, магниевых и медных сплавах количество вторых фаз относительно мало, и объемные изменения при растворении этих фаз не приводят к достаточно сильному фазовому наклепу. Поэтому в этих сплавах в литом состоянии нагрев при переходе через границу растворимости в однофазную область, за редкими исключениями, не приводит к сильному росту зерна основного твердого раствора.
§ 29. Отжиг с фазовой перекристаллизацией
Отжиг с фазовой перекристаллизацией — это термическая обработка, при которой главным процессом является нормальное полиморфное или эвтектоидное превращение. Такой отжиг приводит к коренной перестройке структуры по всему объему сплава.
Полиморфное превращение в металлах можно использовать для устранения текстуры и изменения размера зерна. В этом отношении интересным примером является термическая обработка урана (так называемая 0-термообработка) .
Уран ниже 668 °C имеет ромбическую «-решетку, а от 668 до 774 °C — тетрагональную 0-решетку. В начальный период развития атомной техники было установлено, что если изделие из поликристаллического урана подвергается многократным нагревам и охлаждениям в температурном интервале «фазы, то оно меняет свои размеры. Обработанное давлением изделие сильно удлиняется, «растет» в одном направлении, сокращаясь в другом (рис. 105).
Наиболее важным фактором, определяющим величину «роста» урана, является текстура, которая возникает при обработке давлением в a-области. Преимущественная ориентировка кристаллов в урановом изделии приводит к сильной анизотропии термического расширения, из-за которой при термических циклах возникают напряжения и направленное изменение размеров изделия. Для предотвращения вредного явления «роста» урана необходимо устранить
210
текстуру, которая создается при обработке давлением в а-области.
Если урановый стержень с резко выраженной текстурой нагреть до температуры p-области (выше 668 °C) и затем охладить, то произойдет фазовая перекристаллизация, причем a-кристаллы, образующиеся при полиморфном превращении р-^а, не получают резко выраженной предпочтитель-
Рис. 105. «Рост» уранового прутка, прокатанного при 300 °C (Чизунк и Кель-ман):
вверху — пруток до воздействия теплосмен; внизу — тот же пруток после 3000 теплосмен в интервале 50—550 ®С
ной ориентировки. Следовательно, фазовая перекристаллизация устраняет текстуру, в результате чего во всех направлениях коэффициент термического расширения становится одинаковым. После такой термообработки скорость «роста» уранового стержня при воздействии теплосмен резко снижается.
Чем больше размер зерна, тем ярче выражена шероховатость поверхности уранового изделия, возникающая при воздействии многократных термических циклов. Зерна урана можно измельчить при фазовой перекристаллизации. Размер a-зерен уменьшается с ускорением охлаждения из Р-состояния.
По аналогии с ураном полиморфное превращение в других цветных металлах и твердых растворах может быть использовано для устранения текстуры и измельчения зерна.
Эвтектоидное превращение в цветных сплавах пока редко используют как основу отжига 2-го рода.
В заключение заметим, что при полиморфном и эвтектоидном превращении нагрев выше критической точки может привести к сильному росту зерна деформированного, а также литого металла или сплава.
14*
Раздел третий ЗА КАЛ КА
Закалка — это термическая обработка, при которой главным процессом является формирование неравновесной структуры во время ускоренного охлаждения.
Существуют три принципиально отличных один от другого вида закалки: закалка с полиморфным превращением, закалка без полиморфного превращения и закалка с плавлением поверхности. Из самого названия этих видов термической обработки видно, что они различаются типом фазовых превращений.
Закалку с полиморфным превращением (или закалку на мартенсит) человек начал использовать задолго до нашей эры. На протяжении веков это был основной способ упрочнения сталей. Теперь закалку на мартенсит применяют не только к сталям, но и к сплавам цветных металлов.
Закалка без полиморфного превращения была открыта на рубеже XX в. Ее промышленное использование началось одновременно с применением дуралюмина в авиастроении. С 20—30-х годов закалка без полиморфного превращения в сочетании со старением становится основным способом упрочнения сплавов на основе цветных металлов.
Закалка с плавлением поверхности — это новый вид термической обработки, имеющий значительно более узкое применение по сравнению с двумя первыми видами закалки. Она появилась в 70-е годы, когда в промышленности начали использовать лазерный нагрев изделий.
Основные параметры закалки любого вида — температура нагрева, время выдержки и скорость охлаждения.
Температура нагрева и время выдержки должны быть такими, чтобы произошли необходимые фазовые изменения, например образование высокотемпературной фазы из одной или нескольких низкотемпературных фаз, растворение избыточной фазы в матричной и др. В этом отношении закалка аналогична отжигу 2-го рода.
Скорость охлаждения при закалке должна быть достаточно большой, чтобы при понижении температуры были подавлены диффузионные фазовые превращения и образовывалась метастабильная структура. Этим закалка отличается от отжига 2-го рода.
212
Глава VIII
ЗАКАЛКА БЕЗ ПОЛИМОРФНОГО ПРЕВРАЩЕНИЯ
Закалка без полиморфного превращения — это термическая обработка, фиксирующая при более низкой температуре состояние сплава, свойственное ему при более высокой температуре.
Закалка без полиморфного превращения применима к любым сплавам, в которых одна фаза полностью или частично растворяется в другой. Например, в сплаве Со на рис. 106 при нагреве до Тзак 0-фаза растворяется в матричной a-фазе. При обратном (медленном охлаждении 0-фаза выделяется из a-фазы, в которой концентрация компонента В уменьшается в соответствии с ходом сольвуса nb*.
Так как составы а- и 0-фаз различны, то выделение 0-фазы связано с диффузионным перераспределением компонентов. При достаточно быстром охлаждении
диффузионное перераспределение, необходимое для зарождения и роста кристаллов 0-фазы, не успевает пройти и 0-фаза не выделяется из а-раствора. После такой термообработки (закалки) сплав при комнатной температуре состоит из одной a-фазы, как и при температуре нагрева под закалку.
При температуре нагрева под закалку Тзак в сплаве Со твердый раствор ненасыщенный. После закалки а-раствор при комнатной температуре имеет такой же состав, как и при температуре закалки, но он уже пересыщенный, так как состав насыщенного раствора соответствует точке Ь. z Таким образом, при закалке без полиморфного превращения образуется пересыщенный твердый раствор. Такая закалка к чистым металлам принципиально неприменима. Рассмотренную на примере сплава Со закалку без полиморфного превращения широко применяют к алюминие
* Линией сольвуса называют кривую ограниченной растворимости в твердом состоянии.
213
вым, магниевым, никелевым, медным и другим сплавам, а также к некоторым легированным сталям.
5^' Закалкой далеко не всегда фиксируют однофазное состояние. Например, в сплаве С2 (см. рис. 106) при любых температурах, вплоть до эвтектической, содержится 0-фаза. При температуре закалки Т3ак в этом сплаве находятся насыщенный твердый раствор состава точки т и нераст-воренный избыток 0-фазы. При достаточно медленном охлаждении из-за выделения 0-фазы из а-раствора его состав должен изменяться по линии mb. При быстром охлаждении происходит закалка: 0-фаза не успевает выделиться из а-раствора и состав его при комнатной температуре так же, как и при температуре закалки, определяется точкой т. Следовательно, закаленный сплав С2 содержит пересыщенный а-раствор состава точки т и избыточную 0-фазу, не растворившуюся при нагреве под закалку. Закалкой сплава С2 зафиксировано состояние, которое было стабильным при температуре нагрева под закалку.
Начинающие изучать термическую обработку иногда ошибочно считают, что закалка применима лишь к такому сплаву, ордината которого на диаграмме состояния пересекает линию фазового равновесия в твердом состоянии, например к сплаву Со на рис. 106. Но, как было показано на примере сплава С2, это совсем не обязательно. Принципиально закалка без полиморфного превращения возможна всегда, когда в равновесном состоянии при разных температурах имеется различие в химическом составе фаз.
§ 30. Изменение свойств при закалке без полиморфного превращения
Изменение свойств при закалке зависит от фазового состава и особенностей структуры сплава в исходном и закаленном состояниях, от условий закалки, предыдущей обработки и других факторов. Направление и величина изменения свойств в разных сплавах весьма различны.
Встречается ошибочное утверждение, что закалка всегда приводит к упрочнению, причем термины «закалка» и «упрочнение» часто неверно считают синонимами. В действительности же закалка может и упрочнять, и разупроч-нять сплав. У одних сплавов закалка повышает прочность, но снижает пластичность, у других, наоборот, снижает прочностные характеристики и повышает показатели пластичности, а у третьих повышает и прочность, и пластичность. Наконец, у очень многих сплавов, к которым в прин-
214
Таблица 4. Механические свойства сплавов в литом, отожженном и закаленном состояниях
Сплав МПа б. % . I Сплав ов. МПа б. %
ОТЖИГ закалка отжиг закалка литье закалка литье закалка
Д16 200 300 25 23 АЛ8 150 300 1 12
БрБ2 550 510 22 46 АЛ9 МЛ5 160 160 200 250 2 3 6 9
ципе можно применить закалку, она практически не изменяет свойств.
Сильное упрочнение с одновременным резким снижением пластичности в промышленных сплавах, подвергаемых закалке без полиморфного превращения, не наблюдается.
При закалке без полиморфного превращения деформируемых сплавов наиболее частый случай — повышение прочности при сохранении высокой пластичности, которая может мало отличаться от пластичности отожженного сплава. Типичный пример —дуралюмин Д16 (табл. 4).
Реже встречаются сплавы, у которых при закалке снижается прочность и сильно возрастает пластичность по сравнению с отожженным состоянием. Типичный пример— бериллиевая бронза БрБ2 (табл. 4). У нержавеющей хромоникелевой стали 12Х18Н9 относительное удлинение при закалке после горячей прокатки возрастает с 20 до 45%. Полуфабрикаты из таких сплавов, как бериллиевая бронза и сталь 12Х18Н9, для повышения пластичности перед холодной деформацией не отжигают, а закаливают.
Повышение или снижение прочности при закалке зависит от следующего. С увеличением концентрации легирующего элемента в твердом растворе прочность его возрастает. Поэтому пересыщенный раствор в закаленном сплаве прочнее менее легированного раствора в отожженном сплаве. Прочность отожженного сплава определяется прочностью матричного раствора, а также размером и структурой частиц избыточной фазы и расстояниями между этими частицами. Если в отожженном сплаве торможение дислокаций избыточной фазой не вносит большого вклада в прочность двухфазной смеси (из-за большого расстояния между ее частицами), то при закалке упрочнение раствора благодаря увеличению его легированности перекомпенсирует разупрочнение, связанное с растворением избыточной фа
215
зы, и прочность сплава возрастает. Если же растворение избыточной фазы приводит к сильному разупрочнению, перекрывающему рост прочности, связанный с повышением концентрации матричного раствора, то сплав при закалке разупрочняется. На суммарном эффекте сказываются величина изменения растворимости при нагреве под закалку и прирост прочности матричного раствора, приходящийся на каждый процент растворяющегося элемента.
При закалке литейных сплавов прочность и пластичность обычно растут по сравнению с литым состоянием (см. сплавы АЛ8, АЛ9 и МЛ5 в табл. 4). В структуре литейных промышленных сплавов избыточные фазы обычно находятся в форме сравнительно крупных частиц с большим межчастичным расстоянием. После их растворения прочность сплава становится выше из-за большей легированности
' матричного раствора.
< По грубым и хрупким включениям избыточной фазы, ‘например интерметаллида, происходят отрыв и скол в ли-;том сплаве. Поэтому пластичность сплава после закалки •оказывается повышенной. Особенно сильно она возрастает при полном растворении избыточной фазы, как в алюми-Чшевом сплаве АЛ8 (растворяется Al3Mg2) и магниевом сплаве МЛ5 (растворяется Mgi7Ali2), которые после закалки практически однофазны (табл. 4). В силумине АЛ9 при закалке пластичность возрастает благодаря частичному растворению и коагуляции кремния и полному растворению силицида Mg2Si.
Основное назначение закалки без полиморфного превращения— подготовка сплава к старению. Закалку некоторых сплавов (БрБ2, 12Х18Н9) используют и как промежуточную смягчающую операцию (вместо отжига) перед холодной деформацией. Наконец, закалка служит окончательной термообработкой для придания изделию необходимого комплекса свойств. Однофазный закаленный сплав может обладать значительно большей пластичностью и более высокой стойкостью против коррозии, чем состаренный. Литейный алюминиевый сплав АЛ8 применяют только в закаленном состоянии именно по этим причинам.
§ 31. Нагрев и охлаждение при закалке без полиморфного превращения
1. Нагрев при закалке
Основное исходное положение при выборе температуры закалки— возможно более полное растворение избыточных фаз в матричной фазе.
Если сплав расположен в той области диаграммы состояния, где он способен при нагреве полностью перейти в однофазное состояние (сплав Со на рис. 106), температура закалки должна находиться выше линии сольвуса двойной системы. При закалке сплава Со с температуры ниже То сохраняется нерастворившаяся p-фаза и матричный а-ра-створ оказывается менее легированным, чем при закалке с температур выше То.
Верхнюю границу интервала закалочных температур во, избежание пережога выбирают ниже точки солидуса сплава с учетом возможного перепада температур в садке и, точности теплового контроля. При оплавлении границ связь,; между зернами нарушается и под действием закалочных, напряжений возникают межкристаллитные трещины. Мел-, кие трещины можно не обнаружить, но они вызывают сни-; жение пластичности, ударной вязкости, циклической долт говечности. При пережоге могут появляться усадочные поры, главным образом, в тройных стыках зерен. Механи-i ческие свойства снижаются при пережоге из-за образова-i ния в процессе закалки межкристаллитных прослоек хруп-! кой фазы в результате неравновесной кристаллизации! оплавленных участков, обогащенных легирующими эле-: ментами. Кроме того, причиной пережога служит быстро^! проникновение по оплавленным участкам компонентов атмосферы печи, приводящее к образованию оксидов и газовых пузырей. На шлифе небольшой пережог выявляется в виде утолщений границ зерен. Пережог — это неиспраД вимый и самый опасный брак.
Возможная ширина интервала закалочных температур в двойной системе определяется «вилкой» между точками солидуса и сольвуса. Из рис. 106 видно, что у сплава возможный интервал закалочных температур шире, чем у сплава Со.
При закалке литейных сплавов, которые в отличие от деформируемых предварительно не подвергают гомогени-зационному отжигу и технологическому нагреву для горячей обработки давлением, необходимо учитывать плавление
217
неравновесной эвтектики (см. пунктир ka на рис. 2). Если для более полного растворения избыточных фаз литейный сплав необходимо нагревать до температуры выше точки ’ неравновесного солидуса (например, выше точки т у сплава на рис. 2), то проводят ступенчатый нагрев: при температуре первой ступени, которая должна быть ниже точки неравновесного солидуса, растворяется легкоплавкая составляющая и окончательную температуру закалки можно поднять, не опасаясь пережога. Например, отливки с массивным сечением из сплава МЛ5 перед нагревом под закалку до 415 °C следует выдерживать 3 ч при температуре 375 °C.
Если содержание легирующего элемента превышает ! предел растворимости и сплав нельзя перевести в однофаз-j ное состояние, например Сг на рис. 106, то температуру нагрева под закалку выбирают возможно ближе к эвтектической (перитектической) температуре с учетом технических возможностей избежать пережога.
В зависимости от системы легирования, содержания легирующих элементов, от того, насколько сильно на свойства сплава влияет неполнота растворения избыточных фаз, интервал закалочных температур составляет градусы и десятки градусов (редко более 100°C). Например, у ду-ралюминов разных марок интервал закалочных температур колеблется от 5 до 15 °C. У сплава Д1 его пределы 495—510 °C, у сплава Д16 490—500 °C и у высокомагние-вого дуралюмина Д19П 502—508 °C. У дуралюминов верхняя граница интервала закалочных температур может отличаться от точки солидуса всего на несколько градусов. Поддержание температуры нагрева под закалку в очень узком интервале возможно только в селитряных ваннах и печах с принудительной циркуляцией воздуха.
Противоположный пример — деформируемые и литейные алюминиевые сплавы на базе системы А1—Zn—Mg, у которых возможный интервал закалочных температур на порядок (!) больше, чем у дуралюминов, и составляет около 150 °C. Сплавы этой системы можно закаливать с температур 350—500 °C. Ясно, что такие сплавы несравненно проще нагревать под закалку, не опасаясь пережога или недогрева.
/ Если возможный интервал закалочных температур дос-' таточно широк, то температуру нагрева внутри него выби-! рают более высокой, чтобы сократить продолжительность i растворения избыточных фаз. Однако предельную темпе-। ратуру закалки может лимитировать не солидус, а рост
218
зерна в деформированном сплаве. Кроме того, в некоторых полуфабрикатах требуется сохранить после обработки давлением нерекристаллизованную структуру, и поэтому температура нагрева под закалку не должна превышать температуру начала рекристаллизации (см. § 58).
Время выдержки при температуре нагрева под закалку выбирают так, чтобы завершились процессы растворения избыточных фаз. Чем дисперснее избыточные фазы, тем быстрее они растворяются. Деформированные полуфабрикаты часто (но не всегда) выдерживают при температуре закалки меньше, чем отливки, так как в них при гомогенизационном отжиге слитков растворялись грубые частицы избыточных фаз, а обработка давлением измельчила структуру. Например, листы из алюминиевых сплавов выдерживают при температуре нагрева под закалку 10— 40 мин, а фасонные отливки 2—6 ч (иногда—более 10 ч).
В тонких (0,5—1 мм) холоднокатаных лентах из алюминиевых сплавов с хороша проработанной структурой, не содержащих грубых частиц избыточных фаз, выдержка при нагреве под закалку составляет всего 1— 3 мин. Это позволяет с большой скоростью (до 50 м/мин) перемещать ленту через агрегат непрерывной закалки. '
Время растворения избыточных фаз при нагреве под закалку литейных сплавов связано с толщиной их частиц соотношением (3). Детали, отлитые в земляную форму, следует дольше нагревать под закалку, чем детали, отлитые в кокиль, из-за более грубой литой структуры. Чем толще сечение отливки и -соответственно меньше скорость охлаждения при литье, тем толще частицы избыточных фаз и больше должно быть время нагрева под закалку. Поэтому при закалке отливок время выдержки колеблется от десятков минут до 10—20 ч.
2. Охлаждение при закалке
С понятием «закалка» обычно ассоциируется представление о быстром охлаждении. Действительно, многие изделия закаливают в воде. Однако/при закалке необязательно быстрое охлаждение. Важно литпь^-дтобы при ох и а ж пении-не успел произойти распад матричного растврра.^В-дависимо-стрГ от скорости этого распада скорость охлаждения„при закалке может быть различной. Для одних сплавов обязательна закалка. в холодной воде, а другиеЦв которых ласт-вор распадается медленно, можно закаливать .с^лхлал^де^
219
нием на воздухеД Существует много промышленных сплавов на основе железа, никеля, алюминия и магния, закаливаемых на воздухе.
Кинетика распада переохлажденного раствора
Если тонкий образец сплава Со (см. рис. 106), находящегося при температуре Г3ак в состоянии ненасыщенного твердого раствора, быстро перенести в термостат с температурой 71 <TQl то а-раствор окажется пересыщенным, так как при температуре Т\ насыщенным является раствор со-
Рис. 107. Зависимость энергии Гиббса а- и (З-фаз и двухфазной смеси а+р от состава в двойной системе при температуре Т\ (см. рис. 106)
става точки г. Степень пере-сыщенности можно характеризовать отношением Со/С1. -
При температуре 7\ пересыщенный а-раствор состава Со одновременно является переохлажденным относительно температуры То, выше которой стабильна одна a-фаза, а ниже — двухфазная смесь а+р. Степень переохлаждения А7= = 7о—7'ь Чем ниже темпе
ратура термостата, в koto-i рый мы переносим образец, нагретый до температуры 7зак, тем больше степень переохлаждения и степень пересыщен-ности твердого раствора.
Термодинамическим стимулом распада переохлажденного раствора является разность AGoc между его энергией Гиббса Gi и энергией Гиббса двухфазной смеси G2 (рис. 107).
Скорость образования центров выделения новой фазы в переохлажденном растворе, как и в случае фазового превращения вл чистом металле, определяется выражением (25);-У
В случае распада твердого раствора Q представляет энергию активации диффузии наиболее медленно диффундирующего элемента. Такая диффузия необходима для образования центров выделения новой фазы, отличающейся
по составу от матричного раствора.
В соответствии с формулой (25) скорость образования центров избыточной фазы в сплаве Со на рис. 106 при увеличении степени переохлаждения ниже То сначала должна возрастать из-за увеличения AGoc и соответствующего
220
\д времени (с)
Рис. 108. С-кривые начала распада переохлажденного алюминиевого раствора в сплаве Д16 (В. Г. Давыдов, И. И. Новиков, Е. Д. Захаров):
1 — появление чувствительности к межкристаллитной коррозии; 2 — изменение временного сопротивления на 2 %
уменьшения работы образования критического зародыша AGKP, а затем падать из-за уменьшения диффузионной подвижности атомов. Следовательно, инкубационный период— время до экспериментально фиксируемого начала распада раствора — с увеличением степени переохлаждения сокращается, а затем возрастает, изменяясь по С-кривой (рис. 108).
Начало распада переохлажденного раствора определяют по изменению какого-либо свойства, например временного сопротивлений или электропроводности. Так как свойство при изотермической выдержке обычно изменяется плавно, то за условное начало распада принимают, например, увеличение или уменьшение свойства на 2 или 5 %. Разные свойства начинают заметно изменяться на разных стадиях распада раствора.
Например, у дуралюминов чувствительность к межкристаллитной коррозии, связанная с выделением избыточных фаз по границам зерен, обнаруживается раньше, чем изменение Ов (см. рис. 108).
При построении С-кривых целесообразно измерять то свойство, которое для данной группы сплавов или для данного типа изделий является одним из практически важных и которое чувствительно к наиболее ранним стадиям распада раствора. Так как экспериментально фиксируемое «начало»
условным моментом, то сопоставлять С-кривые разных сплавов имеет смысл только при одинаковом методе их построения.
Если С-кривые строят с помощью определения механических свойств, то, как правило, эти свойства измеряют не сразу после ступенчатой закалки с разными выдержками при температуре ступеньки, а после дополнительного упрочняющего старения, которое, реагируя на остаточную лересыщенность твердого раствора, помогает выявлению начала распада при температуре ступеньки.
является
221
Критическая скорость охлаждения при закалке и прокали-ваемость
С Количественным критерием устойчивости переохлажденного твердого раствора является критическая скорость охлаждения (v«p) — наименьшая скорость непрерывного охлаждения, позволяющего избежать распада раствора, или, то-; чнее, избежать изменения на определенную величину вы-; бранного свойства. Если скорость охлаждения в центре ^сечения изделия больше t>Kp, то изделие прокаливается насквозь. Критическую скорость охлаждения определяют, проводя из точки, соответствующей температуре нагрева под закалку, касательную к С-кривой условного начала распада переохлажденного раствора.
р" Величина цкр зависит от чувствительности метода, который был выбран для построения С-кривой начала распа-/да. Например, для дуралюмина марки Д16 в случае построения С-кривой по появлению чувствительности к межкристаллитной коррозии цкр=550 К/с, а при построении С-кривой по изменению на 2 % предела прочности состаренного сплава цКр=90 К/с. Если же за критерий начала распада принять изменение прочности на 5%, то vKp= =20 К/с.
В § 21 было показано, что количественные оценки при непрерывном охлаждении следует делать не по С-кривым изотермического распада, а по термокинетическим диаграммам. Но такие диаграммы для сплавов, закаливаемых без полиморфного превращения, пока насчитываются единицами, и поэтому приходится пользоваться С*кривыми изотермического распада.
Так как кривая начала превращения исходной фазы на термокинетической диаграмме сдвинута вправо и вниз по сравиению^с. С-жривой, построенной в изотермических условиях (смТрис. /3), то расчет с использованием касательной к такой С-кривой даст завышенную против истинной величину иКр. В отдельных случаях это завышение невелико.
Прокаливаемость при данной скорости охлаждения тем больше, чем меньше -икр, т. е. чем правее находится С-кри-_вая. При качественном сравнении прокаливаемости разных сплавов можно сопоставлять положение их С-кривых, не рассчитывая величину икр.
; Количественной характеристикой является глубина про-I каливаемости в данном охладителе — расстояние от охла-/ ждаемой поверхности до слоя, у которого заданное свой-
222
ство (например, временное сопротивление, предел текучести или твердость) после старения изделия отличается не более чем на 5 % от соответствующего свойства поверхностного слоя. —-
Для сравнения прокаливаемости разных сплавов удобно использовать критический диаметр, т. е. диаметр максимального сечения прутка, который в данном охладителе. прокаливается насквозь. Ниже в качестве примера привб^ дены значения критического диаметра (dKp) при закалке алюминиевых сплавов в холодной воде:
Сплав . . . В95 Д1 Д16 Д19 1201 АВ АК4-1
с?кР, мм . .... 120 150 150 150 150 150 250
Факторы, влияющие на устойчивость переохлажденного раствора
Устойчивость переохлажденного твердого раствора и соответственно прокаливаемость зависят от содержания легирующих элементов и структуры сплава перед закалкой.
В сплавах на разной основе и с разными легирующими элементами при одной основе различна диффузионная подвижность атомов [величина Q в формуле (25)]. Работа образования критического зародыша зависит от межфазной энергии на границе матрицы и выделения, а также от энергии упругой деформации, возникающей из-за различия в удельных объемах фаз. Поэтому скорость зарождения выделяющейся фазы [см. формулу (25)] в разных системах различна. Так, сплавы на базе системы алюминий — медь—магний (дуралюмины) выделяются среди алюминиевых весьма низкой устойчивостью переохлажденного твердого раствора, а сплавы на базе системы А1—Zn—Mg (типа 1915 и 1925)—очень высокой (рис. 109,а). Разница в устойчивости переохлажденного раствора в сплавах на базе этих двух систем предопределяет резкое различие в технологии их термообработки: если сплавы типа «дуралю-мин» необходимо закаливать в воде, то сплавы на основе системы А1—Zn—Mg можно закаливать с охлаждением на спокойном воздухе. Прессованные полуфабрикаты из сплавов 1915 и 1925 вообще не подвергают операции закалки — они самозакаливаются при охлаждении профилей и труб на воздухе с температуры прессования.
Закалку с охлаждением на воздухе широко применяют к жаропрочным аустенитным стареющим сталям с интер-металлидным упрочнением, жаропрочным никелевым сплавам «нимоник», магниевым сплавам типа «электрон» и др.
223
В пределах одной системы с увеличением концентрации легирующих элементов растет пересыщенность твердого раствора и, следовательно, уменьшается его устойчивость. Например, в сплаве Со, более легированном, чем сплав Ср (см. рис. 106), при температуре Л степень пересыщеннос-ти раствора C0/Ci>C'/Ci и термодинамический стимул превращения AGo6>>AG'6 (см. рис. 107). Соответственно С-кривая начала распада раствора в сплаве С' должна быть сдвинута вправо по сравнению с С-кривой сплава Со.
Zy бремени (с)
Рис. 109. С-кривые начала распада переохлажденного алюминиевого раство- • ра, соответствующие изменению временного сопротивления иа 2 % (В. Г. Давыдов, В. В. Захаров, Е. Д. Захаров. И. И. Новиков):
а — сплавы Д16 и 1915 (на базе системы Al—Zn—Mg); б — сплав А1—4 % Zn— 1,9 % Mg—0,6 % Мп—0,2 % Zr с перекристаллизованной (/) и рекристаллизованной (2) структурой
Дисперсные включения интерметаллидов и других фаз, находящихся в сплаве при температуре закалки, могут уменьшить устойчивость переохлажденного раствора, инициируя его распад. С этим связано действие, например, малых добавок переходных металлов (Мп, Сг, Ti) в алюминиевых сплавах. При гомогенизационном отжиге слитков, при нагреве под обработку давлением и закалку из твердого раствора, который пересыщается этими элементами при кристаллизации расплава, выделяются дисперсные алюминиды переходных металлов. Они служат затравками для выделения из раствора основных фаз при охлаждении сплава с температуры закалки, и критическая скорость охлаждения возрастает (прокаливаемостьснижается).
224
С повышением чистоты алюминиевого сплава уменьшается число частиц нерастворимых фаз, содержащих железо и кремний, благодаря чему повышается устойчивость переохлажденного твердого раствора. Поэтому сплавы повышенной чистоты Д16ч и В95лч обладают большей прока л иваемостью, чем сплавы Д16 и В95.
Температура закалки может оказаться ниже температуры начала рекристаллизации. Сплав с перекристаллизованной, лолигонизованной структурой обладает меньшей устойчивостью переохлажденного твердого раствора (рис. 109,6) из-за облегченного зарождения избыточных фаз на дислокациях и субграницах.
Увеличение размера рекристаллизованного зерна может существенно уменьшить критическую скорость охлаждения при закалке. Например, с увеличением размера зерна дуралюмина Д16 с 15 до 50 мкм в 1,5 раза уменьшилась
При закалке в воде возникают большие остаточные напряжения и коробление изделий. Поэтому там, где это возможно, скорость охлаждения уменьшают, используя закалку в подогретой и кипящей воде. В последнее время для закалки алюминиевых сплавов стали применять в качестве более мягкой закалочной среды водные растворы полимеров. Некоторые жаропрочные аустенитные стали закаливают в масле.
Глава IX
ЗАКАЛКА С ПОЛИМОРФНЫМ ПРЕВРАЩЕНИЕМ
Закалка с полиморфным превращением — это термическая обработка металла или сплава, при которой главным является мартенситное превращение высокотемпературной фазы. Поэтому такую термообработку обычно называют закалкой на мартенсит. Эта закалка в принципе применима к любым металлам и сплавам, в которых при охлаждении перестраивается кристаллическая решетка.
§ 32. Особенности мартенситного превращения в углеродистых сталях
Мартенситное превращение было открыто при изучении закалки сталей. Ниже кратко рассмотрены наиболее характерные особенности мартенситного превращения в углеро-
15 И. И. Новиков
225
дистых сталях, которые более наглядно предстают при
сравнении с перлитным превращением.
1. Мартенситное превращение протекает при быстром охлаждении углеродистой стали с температур выше например в воде, когда подавлен диффузионный распад аустенита на смесь двух фаз (феррита и карбида), резко отличающихся по составу от исходного аустенита. Концент
рация углерода в мартенсите такая же, как и в исходном
аустените. Следовательно, в отличие от перлитного превра-
щения мартенситное превращение — без диффузионное.
Рис. 110. С-диаграмма с мартенситными точками для стали с 0,8 % С:
А — устойчивый, Дп — переохлажденный, А ост — остаточный аустенит, 2Ирт ~~ мартенсит; Ф — феррит; К — карбид
2. Превращение аустенита в мартенсит при охлаждении начинается с определенной для каждой марки стали температуры МИ или.
Содержание, (по пассе)
Рис. 111. Зависимость температур начала (2ИН) и конца (Л1К) мартенситного превращения от содержания углерода в системе Fe—С
Ms (в индексе стоят первые буквы слов «начало — start»). Температура начала мартенситного превращения не зависит от скорости охлаждения в очень широком диапазоне скоростей, в то время как температур ра начала перлитного превращения снижается с ростом скорости охлаждения (см. § 24). В отличие от перр литного мартенситное превращение невозможно подавит^ даже при самых больших достигнутых скоростях охлаждения. Мартенситообразование происходит в определенном интервале температур между верхней мартенситной точкой AfH и нижней мартенситной точкой, обозначаемой Мк или Mf (в индексе стоят первые буквы слов «конец—finish»). На С-диаграмме превращений переохлажденного
226
аустенита при температурах Л4Н и Л4К проходят горизонтали (рис. ПО), указывающие на начало и конец мартенситного превращения. Зависимость температур начала и конца мартенситного превращения от содержания углерода показана на. рис. 111. •
3. При температуре Л4Н превращение только начинается, появляются первые кристаллы мартенсита. Чтобы мартенситное превращение развивалось, необходимо непрерывно охлаждать углеродистую сталь в мартенситном интервале Мн — МИ. Если охлаждение приостановить и выдерживать углеродистую сталь при постоянной температуре внутри этого интервала, то образование мартенсита почти сразу же прекращается. Эта особенность наиболее ярко~ отличает кинетику мартенситного превращения от перлитного, которое, как показывает С-диаграмма, всегда доходит до конца при постоянной температуре ниже точки Д1, т.е. оканчивается полным исчезновением аустенита, если время изотермической выдержки достаточно велико. После мартенситного превращения даже при охлаждении cTq/fi практически «мгновенно», образуется некоторое количество, остаточного аустенита.
4. В отличие от перлитного мартенситное превращение в углеродистой стали не имеет инкубационного периода. Длина горизонтали М„ на рис. ПО никакого физического7 смысла как отрезок времени, в течение которого идет мартенситное превращение, не имеет. Горизонталь Мн соответствует температуре, ниже которой чрезвычайно быстро, практически «мгновенно», образуется некоторое количество мартенсита.
5. Мартенсит образуется в форме пластин, растущих с громадной скоростью (порядка 1 км/с) при любых температурах, в том числе и ниже О °C. После «мгновенного», образования мартенситная пластина не растет. Количество мартенсита при охлаждении ниже точки Мн увеличивается не вследствие подрастания уже образовавшихся пластин, а в результате «мгновенного» возникновения все новых и новых пластин. Эта особенность также резко отличает мартенситное превращение от перлитного. В процессе развИг тия перлитного превращения не только образуются новые, но и растут ранее образовавшиеся колонии (см. рис. 81).
6. Между решетками кристаллов мартенсита и исхоД^ ного аустенита имеется определенное ориентационное соотношение, закономерная ориентировка решетки мартенсита по отношению к решетке аустенита, в то время как. при перлитном превращении решетки фаз, входящих в эв
15*
227
тектоидную смесь, могут быть и произвольно ориентированы по отношению к решетке исходного аустенитного зернах 7. При мартенситном превращении в углеродистых сталях на плоской полированной поверхности образца образуется характерный рельеф, свидетельствующий об изменении формы превращенного объема аустенита. При перлиту ном превращении такой рельеф не возникает. Характерный рельеф на исходной плоской поверхности образца может служить главным внешним признаком мартенситного превращения в стали.
Мартенситное превращение, открытое при изучении закалки углеродистых и легированных сталей, как выяснилось впоследствии, является одним из фундаментальных способов перестройки кристаллической решетки, свойственным самым разным классам кристаллических веществ: чистым металлам, безуглеродистым сплавам на основе железа, сплавам цветных металлов, полупроводниковым соединениям и др.
Для теории термической обработки наиболее важны исследования мартенситных превращений в системах Fe — С (см. рис. 74) и Fe — Ni (см. ниже рис. 115).
Обе системы представляют исключительно большой практический интерес: Fe — С как основа сталей, a Fe — Ni как основа сравнительно молодой группы высокопрочных мартенситно-стареющих сплавов.
Ниже на примере этих систем рассмотрены основные закономерности мартенситных превращений.
§ 33. Термодинамика мартенситных превращений
Начнем термодиначеский анализ с простейшего случая (рис. 112)—система характеризуется полиморфизмом твердого раствора и отсутствием трехфазных равновесий (эвтектоидных, перитектоидных). При достаточно медленном охлаждении сплава Со (рис. 112, а) в интевале температур от точки 1 до точки 2 протекает полиморфное превращение Р Г—2'
При любой температуре а- и р- растворы на базе модификаций компонента А характеризуются своими кривыми энергия Гиббса — состав (рис. 112, б). При температуре То кривая энергии Гиббса p-фазы расположена ниже соответствующей кривой a-фазы, т. е. p-раствор при любой его концентрации стабильнее а-раствора (Gp<Ga). Кривые Ga и Gp сходятся в точке равновесия двух модификаций чистого компонента А (см. соответствующую температуру Го на рис. 59).
228
С понижением температуры до Тъ Т2 и Г3 энергия Гиббса а- и ₽-фаз возрастает, причем так, что кривые Ga и Gp пересекаются. Общая касательная к двум кривым энергии Гиббса определяет концентрации фаз, находящихся в равновесии при данной температуре (см. § 20). Например, касательная ab на рис. 112, б характеризует равновесие а-фазы состава а' и £-фазы состава Ъ' при температуре (см. рис. 112, а).
Рис. 112. Участок диаграммы состояния системы с полиморфизмом твердого раствора (а) и кривые энергия Гиббса— состав для а- и p-фаз при разных температурах (б):
Т^Г^— линия температур равенства энергий Гиббса а- и р-фаз одинакового состава
229
Стабильное равновесие ctQz+pd, достигается при очень медленном охлаждении, когда полиморфное превращение сопровождается диффузионным перераспределением компонентов между исходной и новой фазами. При ускоренном охлаждении сплава из p-области такое перераспределение компонентов может быть полностью подавлено.
Сопоставим при разных температурах значения энергии Гиббса а- и p-фаз одинакового состава, отвечающего составу сплава Со.
При температуре T1Gp<Ga (точки d и f на рис. 112, б), при Т2 Gp=Ga (точка g) и при Г3 Gp>Ga (точки m и п). Следовательно, энергия Гиббса исходной p-фазы при охлаждении возрастает интенсивнее, чем энергия Гиббса a-фазы того же состава, и ниже некоторой температуры (Т2 для сплава Со) фазы меняются местами по величине энергии Гиббса: исходная фаза обладает большей энергией Гиббса, чем новая. Кривые температурной зависимости энергии Гиббса двух модификаций твердого раствора одинакового состава идут, пересекаясь, аналогично соответствующим кривым двух модификаций чистого компонента (см. рис. 59). Принципиальное различие состоит в том, что в случае чистого металла точка пересечения кривых энергии Гиббса отвечает температуре стабильного равновесия двух его модификаций, а в случае сплава — температуре метастабильного равновесия переохлажденного р-раствора с метастабильным а-раствором того же состава. Если бы успело произойти диффузионное перераспределение компонентов, то при рассматриваемой температуре метастабильного равновесия Т2 в сплаве Со образовалась бы равновесная смесь фаз разного состава ctQ, +рд,.
Для любого сплава рассматриваемой системы можно указанным способом отметить на диаграмме состояния температуру равенства энергий Гиббса метастабильных а-и p-фаз одинакового состава. Геометрическим местом точек таких температур является линия TGT'Q на рис. 112, а.
В любом сплаве при переохлаждении исходной р-фазы до температур ниже линии ТоТ'^ ее энергия Гиббса становится больше, чем у a-фазы того же состава, т.е. появляется термодинамический стимул AG = Gp—Ga полиморфного превращения р->а без изменения состава. Например, в сплаве Со при превращении p-фазы состава Со в а-фазу того же состава этот стимул («движущая сила» превращения) при температуре Т3 равен отрезку тп.
230
Превращения без перераспределения компонентов, называемые бездиффузионными 1 или безызбирательнымщ возможны и при кристаллизации жидких растворов. Но если при кристаллизации металлического расплава обычно трудно достичь таких переохлаждений, которые бы обеспечили прохождение бездиффузионной кристаллизации жидкого раствора, то в твердом состоянии из-за гораздо меньшей диффузионной подвижности атомов очень часто достигаются переохлаждения ниже линии Т-Т'^, когда становится термодинамически возможным полностью бездиф-фузионное полиморфное превращение твердого раствора. Кинетически такое превращение в условиях низкой подвижности атомов возможно, так как оно совершается путем особого, мартенситного механизма перестройки решетки (см. §34). Образующаяся при этом фаза называется мартенситом.
При бездиффузионном мартенситном и при диффузионном «нормальном» превращении в сплаве Со образуется фаза, которая при комнатной температуре является ненасыщенным твердым раствором компонента В в низкотемпературной модификации компонента А (а-раствор насыщен в точке г).
В более легированном сплаве состава (см. рис. 112, а) в результате бездиффузионного превращения образуется мартенситная a-фаза, которая при комнатной температуре является пересыщенным твердым раствором компонента В в низкотемпературной модификации компонента Л, так как в этом сплаве состав мартенсита (С^) находится правее точки насыщения a-фазы в равновесных ( условиях (г). Следовательно, мартенсит в сплавах может\ быть и пересыщенным, и ненасыщенным твердым раство- ( ром легирующего элемента в низкотемпературной модифи- i кации базового компонента. )
Рассмотрим теперь на примере сплавов Fe — С превращения в системе эвтектоидного типа.
Распад аустенита на феррито-карбидную смесь — это связанный с перераспределением углерода диффузионный процесс с перемещением атомов на расстояния, намного превышающие период решетки аустенита.
При быстром охлаждении углеродистой стали (закалка в воде — сотни градусов в секунду) аустенит успевает сильно переохладиться, не претерпев диффузионного рас
1 О трактовке этого термина см. в § 34.
23!
пада на смесь двух фаз. Но аустенит не может полностью сохраниться при любом сколь угодно быстром охлаждении до комнатной температуры, так как в углеродистой стали его энергия Гиббса начиная с некоторой температуры Го, оказывается выше энергии Гиббса пересыщенного твердого раствора углерода в a-железе (мартенсита), имеющего состав, одинаковый с составом исходного аустенита, и отличающегося от него типом кристаллической решетки.
Мартенсит — метастабильная фаза; она отсутствует на диаграмме состояния Fe — С (см. рис. 74), так как в равновесных условиях, когда система обладает абсолютным минимумом энергии Гиббса, структура сталей ниже точки Ах (727 °C) состоит из смеси двух стабильных фаз — феррита и цементита. Мартенсит при любых температурах обладает большей энергией Гиббса, чем эта смесь, но ниже температуры TQ он отличается меньшей энергией Гиббса, чем переохлажденный аустенит. Когда в условиях большого переохлаждения не может произойти диффузионный распад аустенита на феррито-карбидную смесь, приводящий систему к абсолютному минимуму энергии Гиббса, то аустенит бездиффузионным способом превращается в мартенсит, что приводит систему к относительному минимуму энергии Гиббса. У аустенита гранецентрированная кубическая решетка у-железа, а у мартенсита — тетрагональная, близкая к объемноцентрированной кубической решетке сс-железа. Ниже точки Ах решетка у-железа стабильно существовать не может. При больших переохлаждениях в условиях малой подвижности атомов полиморфное у-кх-превращение происходит, но углерод не успевает выделиться из раствора в виде карбидной фазы.
В a-железе при комнатной температуре равновесная растворимость углерода ничтожна (см. рис. 74). Поэтому мартенсит, образующийся в результате бездиффузионного полиморфного превращения аустенита в углеродистой стали, всегда является пересыщенным раствором внедрения углерода в a-железе. Чем больше углерода в исходном аустените, тем больше его и в мартенсите. С увеличением пересыщенности мартенсита углеродом растет тетрагональное искажение его решетки: отношение осей с/а= = 1Ч-0,046р, где р — концентрация углерода, % (по массе). У чистого a-железа с/а=1 (кубическая решетка), а у мартенсита, содержащего 2% углерода, с/а=1,09 (природа тетрагонального искажения решетки мартенсита объяснена в § 34).
В системе с эвтектоидным превращением на диаграмму
232
состояния можно нанести линию температур равенства энергий Гиббса двух модификаций твердого раствора. Линия То на рис. ИЗ указывает температуры метастабильно-го равновесия переохлажденного аустенита и мартенсита» имеющих одинаковую концентрацию углерода. Каждая точка на линии TQ отвечает для стали данного состава пересечению температурных кривых энергии Гиббса аусте
Рис. 113. Зависимости температуры равенства энергии Гиббса аустенита и мартенсита одинакового состава (То) и температуры Мн от содержания углерода в системе Fe—С
Рис. 114. Зависимость энергий Гиббса аустенита (G д) и мартенсита (бзд) от температуры
нита и мартенсита (рис. 114). Из-за того, что химический состав двух твердых растворов одинаков, двухкомпонентная система Fe — С ведет себя как однокомпонентная (сравните рис. 114 и 59).
1. Температура начала мартенситного превращения
Мартенситное превращение не может начинаться при любом сколь угодно малом переохлаждении относительно температуры То. Здесь существует аналогия с полиморфным превращением в однокомпонентной системе (см. § 17). Образование кристалла мартенсита связано не только с понижением объемной энергии Гиббса (AGoe на рис. 114), но ис появлением поверхностной (межфазной) энергии (Дбпов) и, что особенно важно, энергии упругой деформации (AGynJ, препятствующих превращению. Энергия упругой деформации возникает, во-первых, из-за изменения удельного объема при фазовом превращении (при
233
у-^а-превращении в сплавах на основе железа удельный объем возрастает на 2—3 %), и, во-вторых, из-за когерентности решеток мартенсита и исходной фазы (об этом подробнее см. в § 34).
Результирующее уменьшение энергии Гиббса системы AG =—Дбоб+ЛСпов-FAGynp. Для начала мартенситного превращения необходимо настолько переохладить аустенит ниже температуры метастабильного равновесия его с мартенситом, чтобы термодинамический стимул превращения (AG об) достиг необходимой величины. Поэтому температура начала мартенситного превращения (мартенситная точка Мн) всегда находится ниже температуры То. В системе Fe — С разность температур TG — Л1Н~ 2004-250 К (см. рис. 113).
Температура Л4Н может служить характеристикой сплава определенного состава (при данном режиме предварительной обработки). Горизонталь при температуре Мн на рис. ПО указывает, что мартенситное превращение в данной стали начинается при одной и той же температуре независимо от скорости охлаждения.
Состав сплава обычно сильно влияет на температуру Начала мартенситного превращения. С повышением содер- \ жания углерода в стали точка Мк сильно снижается: bJ -сталях, содержащих менее 1 % С, она находится выше 200 °C, а при содержании углерода около 2 % близка к комнатной температуре (см. рис. 111).
Содержание углерода в стали и в аустените не всегда одинаковое, так как углерод входит в состав карбидов. Карбиды, сосуществующие с аустенитом, сами по себе не влияют на точку AfH. При повышении температуры закалкиТ? когда карбиды растворяются в аустените и концентрация углерода в нем возрастает, точка Мн снижается.
Изменение состава влияет на температуру начала мартенситного превращения, так как меняется температура То, а также степень переохлаждения TG — Мн. В частности, добавки, увеличивающие модуль упругости матрицы (аустенита), затрудняют образование мартенсита: AGytTp растет и соответственно степень переохлаждения TG — Мн увеличивается.
2. Обратимость мартенситного превращения
Из схемы на рис. 114 вытекает, что сплав, закаленный на мартенсит, при нагреве до температур выше То может претерпеть обратное мартенсито-аустенитное превращение,
234
когда будет достигнута определенная степень перегрева и определенная движущая сила превращения ДО. По аналогии с точкой 7ИН температуру начала бездиффузионного образования аустенита обозначают Ан (рис. 114 и 115).
Образование аустенита мартенситным путем возможно только в таких условиях, когда подавлено диффузионное
перераспределение компонентов в исходном мартенсите.
Для этого температура То должна быть невысокой, а нагрев надо проводить ускоренно.
В углеродистых сталях обратное мартенситное превращение не было обнаружено, что можно объяснить высокой подвижностью атомов углерода в растворе внедрения: при нагреве диффузионный распад мартенсита (отпуск) проходит раньше, чем достигается температура Лн.
Обратное мартенситное превращение наблюдали в сплавах системы Fe—Ni (при высоком содержании никеля, когда точка То достаточно низка), в медных сплавах (например, алюминиевых бронзах) и в титановых сплавах. В этих сплавах обратное мартенситное
Рис. 115. Зависимость температур начала мартенситного превращения при? охлаждении (AfH\ обратного мартенситного превращения при нагревании; (Лн) и расчетной температуры равенства энергии Гиббса аустенита и мар« тенсита 7о=’/2(Мн + Лн) от содержания; никеля в системе Fe—Ni (по данным; Кауфмана и Коэна):
пунктирные линии — границы а- и V-областей на диаграмме стабильных, равновесий
превращение имеет все
главные признаки прямого. Скорость нагрева мало влияет на точку А„. Превращение начинаясь при одной температуре (Лн) заканчивается при более высокой температуре (Лк). Например, в сплаве Fe — 32,5 % Ni температура Лн=300-=-310°С, а Лк=4004-410 °C. При обратном мартенситном превращении наблюдается рельеф на полированной поверхности образца, причем иногда он воспроизводит рельеф прямого превращения. Кристаллы при обратном превращении, как и при прямом, образуются чрезвычайно быстро.
Разница между температурами Лн и AfH (температурный гистерезис) может составлять как несколько граду-
235
сов, так и сотни градусов. Большой гистерезис обусловлен высокой энергией упругой деформации матрицы (AGynp), препятствующей образованию в ней кристаллов новой фазы.
§ 34. Механизм мартенситного превращения
1. Сдвиговый (мартенситный) и нормальный механизмы перестройки решетки
Изучение мартенситных превращений привело к представлению о том, что,, в зависимости от условий могут реализоваться два механизма перестройки решетки — сдвиговый (мартенситный) и так называемый «нормальный».
у"- Нормальное превращение. При нормальном полиморфном превращении кристаллы новой фазы растут путем неупорядоченных, взаимно не связанных переходов атомов через границу. Отрываясь в результате термической активации от решетки «исходной фазы, например у-же-леза, атомы поодиночке (или небольшими группами) присоединяются к решетке новой фазы, например сс-железа, и в результате граница кристалла а мигрирует в сторону кристалла у: новая фаза «поедает» материнскую фазу. Такое независимое одно от другого неупорядоченное перемещение атомов через границу раздела фаз похоже на само-диффузионный процесс роста кристаллов при первичной рекристаллизации и отличается от него лишь происхождением термодинамического стимула. В случае первичной рекристаллизации разность энергий Гиббса исходных и новых зерен является следствием большей дефектности строения исходных (деформированных) зерен той же фазы, а при полиморфном превращении металла термодинамическим стимулом является разность энергий Гиббса двух фаз с разной кристаллической решеткой. Как и рекристаллизация, неупорядоченная перестройка решетки (нормальное превращение) может идти только при высоких температурах.
Сдвиговое (мартенситное) превращение. Механизм перестройки решетки при мартенситном лревра-кцении описан в классических работах Г. В. Курдюмова. ^Сдвиговый (мартенситный) механизм фазового превращения отличается упорядоченным,, кооперативным, взаимосвязанным характером перемещений атомов на расстояния меньше межатомных без обмена атомов местами так, что соседи любого атома в исходной фазе остаются его соседями в новой, мартенситной фазе. В этом состоит глав-236
ная особенность сдвиговой перестройки решетки, позволяющая понять многие характерные черты мартенситных превращений, прежде всего когерентность на границе растущего мартенситного кристалла.
Действительно, если при превращении соседи любого атома в исходной фазе остаются его же соседями в новой фазе, на границе фаз окружение из старых соседей вокруг каждого атома также должно сохраняться, т.е. границу должна быть когерентной (см. рис. 64).
Когерентность, упругое сопряжение двух решеток на границе мартенсита и исходной фазы обусловливают возможность чрезвычайно быстрого движения границы в сторону матрицы даже при очень низких температурах, так как для такого «скользящего» движения не требуется диффузии с миграцией атомов на расстояния, превышающие межатомные. На границе происходит лишь кооперативное перемещение атомов на расстояния меньше межатомного, результатом которого и является перемещение самой границы в сторону исходной фазы, т.е. рост мартенситного кристалла/
С ростом мартенситного кристалла на когерентной границе накапливается упругая деформация, пока, наконец, не достигается предел текучести и наступает разрядка упругих напряжений вследствие нарушения когерентности.' Теперь уже, когда на границе кристалла мартенсита с материнской фазой возникает неупорядоченное расположение атомов, «скользящее» движение границы становит-^ ся невозможным и быстрый рост кристалла по мартенситному механизму прекращается. Дальнейший рост кристалла мартенсита возможен только путем неупорядоченного перехода атомов через границу, а так как мартенситное превращение протекает в области температур, где само-диффузия идет крайне медленно, то и подрастание мартенситного кристалла после разрыва когерентности может, практически не наблюдаться.
Таким образом, рассмотренный механизм сдвиговой перестройки решетки объясняет две важные особенности мартенситного превращения: громадную скорость роста кристаллов мартенсита в условиях малой подвижности атомов (вплоть до температур, близких к абсолютному нулю) и быстрое прекращение их роста.
Кооперативное, одинаково направленное перемещение атомов при сдвиговом превращении вызывает макроскопическое изменение формы превращенного объема, а в месте выхода мартенситного кристалла на открытую по
237
верхность исходной фазы — наклон соответствующего участка поверхности. Этим объясняется обязательное образование характерного рельефа при мартенситном превращении на исходной плоской поверхности образца.
Для полиморфного превращения твердых растворов по мартенситному механизму характерна еще одна важная особенность — отсутствие диффузионного перераспределения компонентов. Действительно, если атомы во время превращения не обмениваются местами и каждый атом не меняет своего окружения, то концентрация образующегося твердого раствора должна быть в точности такой же, как и в исходной фазе (например, концентрация углерода в мартенсите такая же, как и в аустените).
Отсутствие диффузионного перераспределения атомов разного сорта само по себе не является определяющим признаком мартенситного превращения. Во-первых, в чистом металле, например железе, состоящем из атомов одного сорта, при полиморфном превращении вообще не может быть речи о таком перераспределении. В то же время и в чистом металле в определенных условиях проходит мартенситное полиморфное превращение, отличающееся, от нормального превращения в том же металле. Во-вторых, в сплавах возможно образование новой фазы того же состава, что и исходная, путем нормальной перестройки решетки. Такое превращение называют массивным
Массивное превращение, являющееся разновидностью нормального, происходит вследствие перемещения некогерентной межфазной границы в сторону исходной фазы в результате неупорядоченных переходов атомов на короткие расстояния в районе этой границы. Диффузии на дальние расстояния, необходимой для диффузионного перераспределения компонентов, здесь не требуется, в связи с чем скорость роста массивной фазы сравнительно большая (достигает значений около 1 см/с)* * и превращение может успеть произойти даже в период закалочного охлаждения.
Массивные кристаллы (в отличие от тонких мартенситных пластин) имеют искривленные границы неправильной формы.
1 Термин предложили, исходя из названия продукта превращения — «массивная фаза» (по аналогии с названиями других превращений — перлитного и мартенситного).
* Тем не менее это на пять порядков меньше скорости роста мартенситного кристалла.
238
Массивное превращение происходит в граничных твердых растворах на базе металла [например, в сплавах Fe — Ni, содержащих до 6 % (ат.) Ni] и в промежуточных фазах [например, p-xx-превращение в латуни с 37—38 % (ат.) Zn].
Если p-фаза в сплавах системы на рис. 112 переохлаждена до температур ниже линии TGr (границы а-области), но выше линии температур начала мартенситного превращения Мн (не показанной на рис. 112), то становится возможным нормальное превращение р~>а без диффузионного перераспределения компонентов, т. е. массивное превращение. При переохлаждении p-фазы до температур двухфазной области ниже линии но выше линии Тог (и одновременно выше температур 7ИН) в принципе возможны два типа нормальных превращений: массивное и с диффузионным перераспределением компонентов в соответствии с положением линий фазового равновесия на диаграмме состояния. В этом случае массивное превращение вероятнее встретить при температурах ближе к линии Тог.
Нормальные полиморфные превращения в чистых металлах по существу также являются массивными, хотя использование этого понятия для чистых металлов не имеет особых преимуществ, так как других нормальных превращений в чистых металлах нет.
Мартенситное превращение часто называют бездиффу-зионным. Этот термин следует понимать не как отсутствие диффузионного перераспределения компонентов, что свойственно и нормальному массивному превращению в сплавах, а как отсутствие неупорядоченных перемещений атомов, характерных для самодиффузии.
2. Термоупругое равновесие фаз
Представление о когерентном росте позволило Г. В. Курдюмову предсказать явление термоупругого равновесия кристаллов мартенсита и исходной фазы, позднее обнаруженное в алюминиевых бронзах, латунях и других сплавах. Сущность этого явления состоит в следующем.
При когерентном росте кристалла мартенсита результирующее изменение энергии Гиббса Д0 =—Д00бЧ-Дбпов + + ДбУ1тр. Накопление энергии упругой деформации решетки может привести к тому, что рост кристалла мартенсита при понижении температуры прекращается еще до разрыва когерентности: увеличение составляющей Дбупр идет быстрее, чем увеличение «движущей силы» ДбОб, и резуль
239
тирующая «движущая сила» превращения AG становится равной нулю. Тогда устанавливается термоупругое равновесие между мартенситом и матрицей. Это равновесие смещается в ту или иную сторону с изменением температуры: при понижении температуры Д0Об возрастает и кристалл растет, пока не установится новое термоупругое равновесие (или не нарушится когерентность), а при повышении температуры А0Об уменьшается и кристалл будет сокращаться в размерах (если при охлаждении граница не превратилась в некогерентную).
Межфазные границы при повышении температуры перемещаются по путям, обратным тем, которые они проходили при охлаждении. Важно подчеркнуть, что в случае термоупругого равновесия обратное мартенситное превращение начинается не выше То (см. рис. 114), а ниже температуры начала прямого мартенситного превращения, т. е. Дн<Мн.
Основные условия реализации термоупругого равновесия: 1) накопление большой упругой энергии, которая может скомпенсировать рост AGo6 при охлаждении и реализоваться в виде движущей силы обратного мартенситного превращения при нагреве; 2) отсутствие релаксационных процессов, которые могут нарушить когерентность на межфазной границе и термоупругое равновесие. Если когерентность полностью нарушается, то обратное мартенситное превращение сможет начаться только при перегреве мартенсита выше То (Ац>Т0).
Обнаружение «термоупругих» кристаллов мартенсита служило блестящим подтверждением правильности представлений о когерентности на границе мартенсита с исходной фазой и о ведущей роли соотношения AGoe и AGynp в термодинамике мартенситных превращений. Позднее оказалось, что термоупругое равновесие представляет очень большой практический интерес в связи с разработкой сплавов, способных «запоминать» форму (см. ниже §37).
3. Условии реализации нормального и мартенситного превращений
Основные условия, необходимые для проявления того или иного механизма превращения высокотемпературной модификации в низкотемпературную, проанализированы Г. В. Курдюмовым.
Если температура стабильного равновесия двух модификаций чистого металла достаточно высока (например, 911 °C у железа, 882,5 °C у титана, 865 °C у циркония и
240
660°C у урана), то могут реализоваться оба механизма перестройки решетки. При сравнительно малых переохлаждениях, когда подвижность атомов достаточно высока, идет нормальное полиморфное превращение с неупорядоченной перестройкой решетки. Мартенситное превращение при малых переохлаждениях идти не может и поэтому не составляет конкуренции нормальному превращению. Объясняется это тем, что при неупорядоченной перестройке решетки упругая деформация кристаллов исходной фазы обусловлена только изменением удельного объема, а при мартенситном превращении — также и когерентностью решеток исходной фазы и мартенситного кристалла. Большая величина AGynp при мартенситном превращении требует большого термодинамического стимула (Д0Об) для развития превращения и, следовательно, большего переохлаждения высокотемпературной модификации, чем это необходимо для развития нормального превращения.
С ростом степени переохлаждения скорость неупорядоченной перестройки решетки возрастает, достигает максимума и затем падает в соответствии с С-кривой (см. рис. 71). При сравнительно больших переохлаждениях, когда разность энергий Гиббса двух модификаций становится достаточной, чтобы шло мартенситное превращение, оно происходит намного раньше, чем успеет начаться очень вялое при низкой температуре нормальное превращение Например, у чистого железа (при содержании 0,0015% С) температура Л1н равна 750 °C, что соответствует степени переохлаждения около 160 °C. При переохлаждении у-железа до температур, находящихся в интервале 911— 750 °C, протекает нормальное у-т-превращение, а ниже 750 °C — мартенситное у->а-превращение.
У металлов с высокой температурой равновесия модификаций не всегда легко получить такие большие степени переохлаждения, которые необходимы для начала мартенситного превращения. Так, например, для реализации мартенситного механизма полиморфного превращения в железе образцы следует сильно перегревать в у-области и очень быстро охлаждать, чтобы подавить развитие нормального превращения при меньших степенях переохлаждения.
Если у чистого металла температура равновесия двух модификаций сравнительно низкая (например, 400 °C у кобальта и —196°C у лития), то из-за малой подвижности атомов реализуется только мартенситное превращение, а неупорядоченная перестройка решетки вообще не наблюдается.
16 И. И. Новиков
241
Из всего изложенного следует, что определение «нор-мильное» превращение — полностью условное, так как мартенситное превращение является не менее обычным для фазовых переходов в твердом состоянии.
В твердых растворах на базе полиморфных металлов реализация того или иного механизма превращения зависит от того, как легирующий элемент изменяет температуру равновесия двух модификаций. Например, при легировании железа никелем и марганцем можно так сильно понизить температуру равновесия двух фаз, что нормальное у->а-превращение становится невозможным и наблюдается только мартенситная перестройка решетки.
Противоположный случай — легирование кобальта элементами, которые так повышают температуру равновесия двух модификаций, что, кроме мартенситного превращения, идущего при достаточно большом переохладении, становится возможным и нормальное превращение (при малых степенях переохлаждения). %
4. Кристаллогеометрия превращения аустенита в мартенсит
Для понимания строения мартенсита в сталях необходимо знать кристаллогеометрию перестройки г. ц. к. решетки аустенита в объемноцентрированную тетрагональную решетку мартенсита, близкую к о. ц. к. решетке а-железа. Следующее простое изменение формы элементарной ячей^ ки аустенита, известное в литературе как деформация Бейна, позволяет составить представление о характере кристаллогеометрии мартенситного превращения в стали^. (рис. 116).
На рис. 116, а показаны две соседние кубические элементарные ячейки г. ц. к. решетки аустенита. Атомы железа, отмеченные кружками, расположены в вершинах кубов и центрах их граней. Атомы углерода, растворенного в у-железе по способу внедрения, статистически равномерно занимают часть октаэдрических пустот г. ц. к. решетки1. Октаэдрические пустоты, помеченные крестиками, находятся на серединах ребер и в центре объемов кубических ячеек (рис. 116, а). Все эти пустоты структурно эквива-^ лентны, неразличимы. —
В г. ц. к. решетке аустенита можно мысленно выделить не только кубическую, но и тетрагональную элементарную ячейку (на рис. 116, а она показана жирными линиями).
1 Для упрощения указаны не все положения атомов железа и углерода.
242
В этой тетрагональной ячейке аустенита отношение периодов cla— 1 2, в то время как у решетки мартенсита экспериментально установленная степень тетрагональности (с/а) < 1,09. Деформация Бейна, простейшим способом превращающая решетку аустенита (см. рис. 116, а) в решетку мартенсита (рис. 116,6), состоит в сжатии тетрагональной ячейки аустенита вдоль ее оси с и одновременном увеличении размеров вдоль осей а.
В г. ц. к. решетке аустенита атомы углерода статистически равномерно распределены по октаэдрическим, пусто-
Рис. 116. Деформация Бейна° превращающая решетку аустенита (а) в решетку мартенсита (б)
там вдоль направлений трех ребер куба [100], [010] и [001J (см. крестики на рис. 116.а). Но в той же самой решетке аустенита атомы углерода избирательно расположены по-отношению к направлениям ребер тетрагональной ячейки: они находятся только на середине ребер вдоль оси [001] и В‘центре горизонтальных граней тетрагональной ячейки* т.е. тоже вдоль направления [001]. По окончании превращения внедренные атомы углерода продолжают располагаться в решетке мартенсита в октаэдрических пустотах только вдоль направления [001], не занимая пустот в направлениях [1Q0] и [010] (см. рис. 116, б). Находясь между атомами железа в рядах, параллельных оси [001], атомы углерода не позволяют деформации Бейна превратить г. ц. к. решетку в о. ц. к. с отношением периодов, равным единице. Степень тетрагонального искажения решетки мартенсита с!а растет прямо пропорционально концентрации в нем углерода.
Подвижность атомов углерода даже при комнатной температуре достаточна, чтобы по окончании превращения
16* 243
они смогли перераспределиться и занять ближайшие свободные октаэдрические пустоты вдоль направлений [100] и [010] с одновременным исчезновением тетрагональности. Для этого достаточно диффузионых перемещений на очень малые расстояния — в пределах одной элементарной ячейки. Однако в действительности решетка мартенсита сохраняет тетрагональность при комнатной температуре. Теоретический анализ показал, что между атомами углерода в мартенсите стали существует такое деформационное взаимодействие, которое делает термодинамически выгодным их упорядоченное распределение с предпочтительным расположением вдоль одной из кристаллографических осей. Таким образом, тетрагональное искажение решетки мартенсита отвечает минимуму энергии Гиббса благодаря минимизации энергии упругой деформации решетки, связанной с внедренными атомами углерода, при их упорядоченном расположении.
С повышением температуры дальний порядок в расположении атомов углерода должен был бы исчезнуть. Но этому препятствует упруго-напряженное состояние кристалла мартенсита в матрице остаточного аустенита. Благодаря ему дальний порядок в пересыщенном твердом растворе углерода в a-железе (мартенсите) сохраняется при всех температурах, при которых мартенсит еще не претерпевает распада.
Важную роль в развитии представлений о механизме мартенситного превращения сыграло установление рентгеновским методом ориентационных соотношений решеток исходной и мартенситной фаз. Для сплавов железа известны три главных ориентационных соотношения решеток аустенита и мартенсита: Курдюмова — Закса, Нншиямы и Гренингера — Трояно.
Ориентационное соотношение Курдюмова — Закса (пример— углеродистые стали с 0,5—1,4% С) можно записать в следующей форме: *
(Ш)а || (101)м и [1Т0]А||[11Т]м.
Такого рода взаимная ориентация решеток легко объяснима: плоскость плотнейшей упаковки {111} в г. ц. к. решетке наиболее близка по атомному строению к плоскости плотнейшей упаковки {110} в о. ц. к. решетке, а* направление плотнейшей упаковки <110> в г. ц. к. решетке наиболее близко по атомному строению к направлению плотнейшей упаковки < 111 > в о. ц. к. решетке. Подобная взаимная ориентация решеток наиболее полно удовлетворяет
244
принципу структурного соответствия. Так как в г. ц. к. решетке аустенита имеются четыре кристаллографически эквивалентные плоскости типа {111}, а именно (111), (111), (111), (111), и шесть кристаллографически эквивалентных направлений типа <110>, то относительно одного положения кристалла аустенита возможны 24 ориентации кристаллов мартенсита, удовлетворяющие соотношению Курдюмова — Закса.
Ориентационное соотношение Нишиямы (пример — сплавы железа с 27—34 % Ni) можно записать в форме
(111д II (101)м и [121]а II [101]м.
Соотношение Гренингера — Трояно (пример — сплав Fe—22 % Ni — 0,8 % С) является промежуточным между соотношениями Курдюмова — Закса и Нишиямы.
Деформация Бейна, наглядно пояснившая, как с помощью кратчайших атомных смещений г. ц. к. решетка аустенита может превратиться в объемноцентрированную тетрагональную решетку мартенсита, одна не в состоянии привести, например, к 24 ориентациям Курдюмова — Закса, так как ребра элементарной ячейки мартенсита остаются параллельными ребрам исходной тетрагональной ячейки аустенита (см. рис. 116). Для получения ориентационного соотношения Курдюмова — Закса необходимы более сложные траектории атомных перемещений, чем при деформации Бейна. Истинные траектории движения атомов при мартенситном превращении неизвестны. Формально все экспериментально обнаруженные ориентационные соотношения решеток аустенита и мартенсита можно получить, дополнив деформацию Бейна поворотом решетки мартенсита, при котором становятся параллельными соответствующие плоскости и соответствующие направления в решетках исходной и мартенситной фаз.
5. Инвариантность габитусной плоскости мартенсита и аккомодационная деформация при мартенситном превращении
Вслед за установлением ориентационных соотношений исключительно важную роль в понимании кристаллогеомет-рии мартенситных превращений и внутреннего строения мартенситной фазы сыграло количественное изучение рельефа на плоской полированной поверхности образца. Появление такого рельефа всегда сопровождает мартенситное
245
превращение из-за изменения формы превращенного объема.
На рис. 117 плоская поверхность FGHE относится к исходному аустенитному состоянию образца. При образовании мартенситной пластины ABCDLMNO поверхность образца на участке ABCD наклоняется по отношению к ее исходной плоскости. Как показали эксперименты, линия АВ остается неповернутой, а риски типа STT'S', которые до превращения были прямолинейными, остаются прямыми на наклонном участке поверхности ABCD и непрерывными на всей своей длине. Так как это справедливо по отноше-
Рис. 117. Рельеф на исходной плоской поверхности FCjEH образца при образовании в нем мартенситной пластины ABCDLMNO (по Бнлбн н Кристиану):
Stt'S' — ломаная риска, бывшая до превращения прямой
Рис. 118. Виды деформации при мартенситном • превращении (по Билбн н Кристиану)
нию к любой выбранной исходной поверхности образца, то делается вывод, что в процессе мартенситного превращения габитусная плоскость мартенситного кристалла ABML (плоскость раздела мартенситной и аустенитной фаз} макроскопически' приблизительно инвариантна — она не искажается и не вращается (любые отрезки на этой плоскости остаются неизменными по длине и направлению).
Инвариантность габитусной плоскости обеспечивает минимум упругих деформаций при превращении. Это особенно легко себе представить в случае превращения с одной поверхностью раздела между исходным и мартенситным кристаллом: Инвариантность этой поверхности предотвращает образование упругих макродеформаций в исходной и мартенситной фазах.
246
Возникает вопрос, как может реализоваться инвариантность плоскости раздела фаз, если при мартенситном превращении изменяется элементарная ячейка, т. е. деформируется сама кристаллическая решетка. Схема, показанная на рис. 118, помогает ответить на этот вопрос.
На рис. 118, а представлен участок исходной фазы до превращения, а на рис. 118, б тот же участок после превращения, состоящего в изменении типа решетки. Хорошо видно, как изменение формы и размеров элементарных ячеек, т. е. однородная деформация решетки, приводит к макроизменению формы превращенного участка.
Макроскопическое изменение формы участка исходной фазы можно получить принципиально иным путем, без изменения элементарных ячеек—пластической деформацией скольжением (рис. 118, в), а также двойникованием.
Комбинация деформации решетки (изменения элементарной ячейки) и пластической деформации, например, скольжением (рис. 118, г) может сохранить неизменной исходную форму макроучастка, несмотря на прохождение фазового превращения (тип решетки на рис.-118, г отличается от исходного на рис. 118, а). Естественно, что боковая поверхность габитуса кристалла новой фазы на рис. 118, г является инвариантной только в макромасштабе: из-за развития скольжения на ней образовались микроскопические ступеньки.
Таким образом, важнейшее следствие из экспериментально обнаруженного факта инвариантности плоскости габитуса мартенситного кристалла состоит в том, что его образование должно заключаться не только в изменении типа кристаллической решетки, но и в одновременной пластической деформации, возникающей вследствие скольжения или двойникования. Такая дополнительная (аккомодационная) деформация, являющаяся неотъемлемой частью механизма мартенситного превращения, обеспечивает минимум энергии упругих искажений на инвариантной поверхности раздела фаз. Этот вывод особенно важен для понимания субструктуры мартенсита (см. ниже § 35).
Экспериментально установлены разные габитусные плоскости кристаллов мартенсита. Для сплавов на основе железа наиболее характерны плоскости габитуса {225}д, {259)д, {3, 10, 15}ди {111} а- Буква «А» здесь указывает, что индексы плоскости раздела двух фаз даны в кристаллографических осях решетки аустенита.
Тип габитуса связан с видом аккомодационной деформации решетки при мартенситном превращении. Например,
247
теория предсказывает, что если такая деформация идет путем двойникования по плоскости {112}м, то габитус мартенсита будет {3, 10, 15}д.
6. Зародыши мартенсита
Вопрос о том, что представляют собой зародыши мартенсита, наиболее трудный во всей проблеме мартенситных превращений. Гипотеза гомогенного зарождения, связанного с флуктуационным образованием зародыша критического размера, большинством исследователей отвергается, так как из-за высокой энергии упругих искажений работа гомогенного образования критического зародыша столь велика, что вероятность его флуктуационного появления ничтожна.
Большинство гипотез предполагает гетерогенное зарождение мартенсита, привязывая центры превращения к особым субмикроучасткам в исходной фазе, причем в разных металлах и сплавах природа и строение таких участков зарождения могут быть разными. Из рассмотрения исключаются границы и субграницы, так как эксперимент показывает, что они не являются местами предпочтительного образования мартенситных кристаллов.
Прямым доказательством гетерогенного зарождения считаются результаты опытов по исследованию мартенситного превращения в дисперсных частицах сплава Fe— 30,2 % Ni. Около 7го общего числа частиц диаметром ~40 мкм и менее остались в аустенитном состоянии, не испытав мартенситного превращения, вплоть до —196 °C, в то время как у обычного массивного образца Мп——20 °C. Это объясняется тем, что гомогенно мартенсит не зарождается, а вероятность нахождения участков гетерогенного зарождения в частице аустенита снижается с уменьшением ее размера.
Наиболее просто природа мест гетерогенного зарождения мартенсита трактуется в случаях, когда из фазы с г. ц. к. решеткой образуется мартенсит с гексагональной плотноупакованной (г. п.) решеткой. Дефекты упаковки в г. ц. к. фазе, возникшие при расщеплении дислокаций, являются тонкими прослойками г. п. решетки и поэтому представляют собой как бы готовые двухмерные зародыши гексагонального мартенсита. Такая ситуация имеется в случае превращения ₽г.ц.к.-няг.п в кобальте.
В сталях с высоким содержанием марганца (более 10 %), в также в нержавеющих хромоникелевых сталях об
248
разуется так называемый е-мартенсит, имеющий г. п. решетку, а из него может образоваться обычный а-мартенсит с объемноцентрированной кубической или тетрагональной решеткой. Готовыми зародышами е-мартенсита являются дефекты упаковки в аустените. В стали с повышенным содержанием марганца и аустените хромоникелевых нержавеющих сталей из-за очень низкой энергии дефектов упаковки дислокации сильно расщеплены и, следовательно, имеется множество зародышей гексагонального е-мартенсита.
Прямыми наблюдениями в высоковольтном электронном микроскопе обнаружено, что в нержавеющих сталях зародыши Е-мартенсита действительно появляются на дефектах упаковки аустенита. Вместе с тем а-мартенсит зарождается в местах скопления дислокаций.
Роль дислокаций в гетерогенном зарождении а-мартен-сита бесспорна, но в чем конкретно состоит эта роль, пока не ясно. По одним гипотезам, группы из нескольких дислокаций в г. ц. к. решетке образуют такую конфигурацию, которая превращает соответствующий микроучасток в зародыш с о. ц. к. решеткой. Другой подход рассматривает дислокации как источник поля внутренних напряжений, уменьшающего работу образования критического зародыша (вплоть до безбарьерного зарождения).
Зарождение мартенсита рассматривают также как результат потери механической устойчивости решетки аустенита в предмартенситном состоянии. В подтверждение этого приводят факты падения модулей упругости аустенита при приближении к точке Л1н у некоторых сплавов.
§ 35. Микроструктура и субструктура сплавов, закаленных на мартенсит
1. Микроструктура
При исследовании структуры закаленных углеродистых сталей и безуглеродистых сплавов на железной основе выявлены два главных морфологических типа мартенсита: пластинчатый и пакетный.
Эти два типа мартенсита различаются формой и взаимным расположением кристаллов, субструктурой, а также габитусной плоскостью. --
Пластинчатый мартенсит (который называют также игольчатым, низкотемпературным и двойникованным)— это хорошо известный «классический» тип мартенсита, наиболее ярко выраженный в закаленных высокоуг-
249
Рнс. 119. Фермообразное расположение пластин мартенсита и остаточный аустенит (светлый фон) в закаленной стали с 1,Ь6 % С. В пластинах виден мидрнб. Х55О (Краус и Мадер)
леродистых сталях и в безуглеродистых железных сплавах с высокой концентрацией второго компонента, например в. сплавах Fe—Ni при содержании более 28 % Ni. Кристаллы мартенсита имеют форму тонких линзообразных пластин (рис. 119). Такая форма пластин мартенсита соответствует минимуму энергии упругих искажений при образовании его в аустенитной матрице п аналогична форме механических двойников. Попадание пластины своей большой поверхностью в плоскость шлифа — крайне редкий случай (рис. 120). Произвольные сечения мартенситных пластин плоскостью шлифа при небольших увеличениях микроскопа создают ложное впечатление об игольчатой форме кристаллов. Однако исторически сложившиеся термины «крупноигольчатый» и «мелкоигольчатып» мартенсит широко распространены.
Г абитусом пластинчатого мартенсита в зависимости от состава сплава могут быть плоскости {225}д и {259}4 в системе Fe—С и {3, 10, 15}а в системе Fe—Ni.
параллельны одна другой и час
то образуют фермоподобные ансамбли (см. рис. 119). Ввиду интерференции дальнодействующих полей упругих напряжений от каждой пластины такое закономерное их расположение в матричной фазе обеспечивает минимум упругой энергии суммарного поля всего ансамбля из пластинчатых кристаллов.
Пластины, возникающие в первую очередь (вблизи точки Мн), проходят через все аустенитное зерно, расчленяя его на отсеки. Через границу зерна матричной фазы мартенситная пластина, как следует из механизма ее образования, пройти не может, и поэтому максимальный размер мартенситных пластин ограничен размером аустенитных, зерен.
В отсеках аустенита при понижении температуры образуются новые мартенситные пластины, размер которых уже
Соседние пластины не
250
ограничен размерами отсека матрицы (см. рис. 119 и схему на рис. 121). По мере развития превращения аустенитное зерно расчленяется на все более мелкие отсеки, в которых образуются все более мелкие мартенситные пластины. При мелком аустенитном зерне, например при малых
Рис. 120. Редкий случай расположения мартенситной пластины в плоскости шлифа. Х750 (А. П. Гуляев, Е. В. Пету-ни.-а)
Рнс. 121. Схема образования мартенситных пластин разной
длины в одном аустенитном зерне
перегревах стали выше Ас3, пластины мартенсита столь мелки, что «игольчатое» строение на шлифе не видно и мартенсит называют бесструктурным. Такой мартенсит наиболее желателен.
После закалки между пластинами мартенсита при комнатной температуре сохраняется остаточный, аустенит (см. рис. 119). ___
251
В местах столкновения пластин мартенсита, растущих под углом одна к другой, могут возникать механические двойники и микротрещины. С укрупнением аустенитного зерна укрупняются пластины мартенсита и образование микротрещин в местах их столкновения усиливается.
Пакетный мартенсит (который называют также реечным, массивным, высокотемпературным и недвойнико-
Рис. 122. Пакетный мартенсит в закаленном сплаве Fe—1,94 % Mo. X 200 (Краус н Мадер)
ванным) — это широко распространенный морфологический тип, который можно наблюдать в закаленных низкоуглеродистых и сред-неулеродистых сталях, большинстве конструкционных легированных сталей, сравнительно низколегированных безуглеродистых железных сплавах, например в сплавах Fe—Ni при концентрации менее 28 % Ni. Кристаллы этого мартенсита имеют форму примерно одинаково ориентированных тонких пластин, припасованных одна к другой и образующих более или менее равноосный пакет. Габитус-
ная плоскость пластин —
«реек» близка к плоскости Пластины реечного мартенсита разделены малоугловыми границами.
Ширина пластин в пределах пакета примерно одинакова и находится в диапазоне от нескольких микрон до долей микрона (обычно 0,1—0,2 мкм), т. е. может находиться на пределе разрешающей способности светового микроскопа и даже за этим пределом. Поэтому «реечные» кристаллы под световым микроскопом или совсем не видны, или же выявляются как тонкая структура пакетов. В связи с этим в качестве основного структурного элемента шлифа выступает пакет из пластин (рис. 122), а не отдельные очень тонкие кристаллы. Поэтому мартенсит с такой структурой и был назван пакетным, или массивным, в отличие от игольчатого.
В конструкционных легированных сталях внутри мар
252
тенситных пакетов между кристаллами мартенсита находятся прослойки остаточного аустенита толщиной 20— 50 нм. В одном аустенитном зерне может образоваться несколько мартенситных пакетов. От зерен феррита, образующихся при нормальном превращении, пакеты мартенсита отличаются не только указанным внутренним строением, которое может не выявляться под световым микроскопом, но и изрезанными вытянутыми контурами.
Образованию пакетного мартенсита свойственны все основные отличительные признаки мартенситного превращения, в том числе и возникновение рельефа на полированной поверхности, соответствующего внутреннему реечному строению.
2. Субструктура
Просвечивающая электронная микроскопия выявила во многих сплавах весьма сложную тонкую структуру мартенситных кристаллов с большим количеством дислокаций и двойников. Такая субструктура может возникнуть двумя принципиально разными путями: во-первых, при аккомодационной пластической деформации (скольжением или двойникованием), которая, как показано в § 34, является неотъемлемой составной частью механизма мартенситной перестройки решетки, и, во-вторых, при пластической деформации после образования мартенсита из-за воздействия на мартенситный кристалл окружающей упругой среды. В первом случае можно говорить о первичной субструктуре превращения, а во втором — о вторичной субструктуре деформации. Соответственно различают понятия о двойниках превращения и деформационных (механических) двойниках. Различить же происхождение субструктуры экспериментально не всегда удается. Обсуждаемые ниже факты рассматриваются в предположении, что мы имеем дело с субструктурой превращения. __
Субструктура пластинчатого мартенсита характеризуется тем, что уже под световым микроскопом в сечении пластин можно увидеть среднюю линию или, точнее, среднюю зону повышенной травимости, называемую также мидрибом* (см. рис. 119).
Электронная микроскопия показала, что мидриб — это область с плотным расположением параллельных тонких двойниковых прослоек. Плоскость двойникования в мартенсите сплавов на железной основе чаще всего {112}м- Тол-
1 От анг. midrib — сокращенное midale ribbon (средняя полоска).
253
щина двойниковых прослоек в зависимости от состава сплава- и условий образования мартенсита колеблется от нескольких десятых до десятков нанометров.
По обе стороны от средней двойникованной зоны находятся периферийные области мартенситной пластины, в которых видны дислокации с относительно небольшой плотностью (Ю9—1010 см-2).
Ширина двойникованной зоны в пластине мартенсита зависит от состава сплава. Так, в сплавах системы Fe—Ni с увеличением содержания никеля ширина этой зоны возрастает, пока все сечение мартенситной пластины не становится занятым двойниками превращения.
Поскольку субструктура и габитус кристалла зависят от характера аккомодационной деформации при мартенситном превращении, то естественно, что форма кристаллов мартенсита, их плоскость габитуса и субструктура взаимосвязаны. Так, в сплавах системы Fe—Ni отклонение габитуса от плоскости {3, 10, 15)д, наблюдаемое при снижении содержания никеля от 33 до 30 %, сопровождается уменьшением плотности двойников превращения {112}м в пластинах мартенсита и ширины зоны этих двойников, а сами пластины становятся менее линзообразными и более зазубренными. Усложнение субструктуры пластин при отклонении габитуса от плоскости {3, 10, 15}д указывает на усложнение характера аккомодационной деформации при мартенситном превращении.
В связи с тем что мартенситная пластина в общем случае состоит из средней зоны двойникования и периферийных зон скольжения, была выдвинута гипотеза о двух стадиях формирования мартенситной пластины. Вначале в бразовании пластины (будущего мидриба) главную роль играет аккомодационная деформация двойникованием. Из-за громадной скорости роста пластины теплота превращения не успевает отводиться и температура на поверхности пластины сильно возрастает, что приводит к смене механизма аккомодационной деформации: переходу от двойникования к дислокационному скольжению (хорошо известно, что с понижением температуры в металлах с о. ц. к. решеткой при быстрой пластической деформации скольжение заменяется двойникованием),
Субструктура пакетного мартенсита качественно отличается от субструктуры пластинчатого мартенсита отсутствием зоны из тонких двойниковых прослоек. Субструктура пакетного мартенсита имеет сложное дислокационное строение, для которого характерны дислокаци
254
онные сплетения при высокой плотности дислокаций — порядка 1011—1012см-2, т. с. такой же, как и в металле после сильной холодной деформации. Рейки часто состоят из вытянутых слегка разориентированных субзерен. Двойниковые прослойки могут встречаться в пакетном мартенсите, но плотность их несравненно меньше, чем в мидрибе пластинчатого мартенсита, а многие рейки вообще не содержат двойников.
Субструктура остаточного аустенита от-личается от субструктуры исходного аустенита большей плотностью несовершенств, возникающих при локальной пластической деформации аустенита под действием мартенситных кристаллов. В аустените вблизи мартенситных кристаллов наблюдаются плоские скопления дислокаций, дислокационные сплетения и дефекты упаковки.
3. Переход от одного морфологического типа мартенсита к другому
Два основных типа мартенсита различаются микро- и субструктурой, а также габитусной плоскостью. Вполне логично сделать вывод, что ведущую роль в образовании пластинчатого мартенсита, по крайней мере на первой стадии, играет аккомодационная деформация двойникованием, а в образовании пакетного мартенсита — аккомодационная деформация скольжением.
Какой тип мартенсита формируется, зависит от соотношения в аустените критических касательных напряжений, вызывающих скольжение и двойникование. Если для начала скольжения требуется большее касательное напряжение, чем для начала двойникования, то образуется пластинчатый мартенсит, а в противоположном случае — пакетный. Основываясь на этом положении, легко объяснить многие закономерности перехода от одного типа мартен^ сита к другому и прежде всего влияние состава сплава на морфологию мартенсита (табл. 5).
Таблица 5. Границы образования пакетного и пластинчатого мартенсита (Краус и Мадер)
Система Содержание второго компонента в сплаве, %
пакетный мартенсит пластинчатый мартенсит
Fe—С <1,0 0,6—2,0
Fe—N <0,7 0,7—2,5
Fe—Ni <29 29—34
Fe—Сг <10 —
255
Рис. 123. Схема температурной зависимости критического напряжения (тКр) скольжения и двойникования
С понижением температуры сопротивление скольжению быстро растет и начиная с некоторой температуры становится больше сопротивления двойникованию (рис. 123). Поэтому при более низких температурах мартенситного превращения вероятнее формирование пластинчатого мартенсита, а при более высоких — пакетного. Отсюда понятно, почему в сплавах систем Fe—С, Fe—N и Fe—Ni при малых содержаниях второго компонента образуется пакетный, а при высоких — пластинчатый мартенсит (табл. 5).
Этот переход логично связать с понижением температуры Мн (см. рис. 111). При большем содержании, например, углерода в относительно низкотемпературном мартенситном интервале сопротивление скольжению выше, чем двойникованию, и образуется пластинчатый мартенсит. Переход от одного морфологического типа мартенсита к другому происходит в интервале составов^ Понятно также, почему в пакетном мартенсите часть кристаллов содержит двойниковые
прослойки — это те кристаллы, которые образовались в нижней части мартенситного интервала, а так как они появились в последнюю очередь, то морфологический тип остался пакетным.
В таких системах, как Fe—Сг, на диаграмме состояния которых у-область замкнута, температура Мп при любых концентрациях легирующего элемента столь высока, что пластинчатый мартенсит вообще не образуется (см. табл. 5).
Упрочнение аустенита любыми путями, т. е. повышение критического напряжения, вызывающего скольжение, должно способствовать формированию пластинчатого мартенсита, в том числе и при неизменной температуре Мн. Углерод относится к числу наиболее сильных упрочнителей аустенита, и поэтому увеличение его содержания особенно эффективно содействует развитию двойникования при мартенситном превращении. ___
В титановых сплавах увеличение содержания легирующего элемента, снижающего точку ЛГН, приводит к изменению и морфологии, и структуры мартенсита. Например, в закаленных сплавах системы Ti—Мо с увеличением со-
256
держания молибдена микроструктура становится ярковы-раженно игольчатой, а внутри пластин мартенсита сплетения дислокаций сменяются все большим числом двойников.
В заключение подчеркнем, что в целом строение металла или сплава, претерпевшего мартенситное превращение, отличается от строения того же материала после неупорядоченной перестройки решетки гораздо большим количеством несовершенств: более развитыми границами и субграницами, большей плотностью дислокаций и двойниковых прослоек.
§ 36. Кинетика мартенситных превращений
1. Разновидности кинетики
Условно можно выделить три типа мартенситных превращений, различающихся своей кинетикой: атермическое, взрывное и полностью изотермическое (рис. 124).
Атермическое превращение наблюдается в углеродистых и легированных сталях с мартенситной точкой Мн выше 100 °C, взрывное — в сплавах систем Fe—Ni и Fe—Ni—С с точкой Ми ниже комнатной температуры и полностью изо
Рнс. 124. Атермическое (а), взрывное (б) н полностью изотермическое мартенситные превращения (в)
термическое — в сплавах систем Fe—Ni—Мп и Fe—Ni—Сг с точкой Мн также ниже комнатной температуры.
Соотношение между количеством мартенсита и аустенита, которое характеризуют кривые на рис. 124, определяют магнитометрически (по отклонению стрелки магнитометра), используя то, что аустенит парамагнитен, а мартенсит ферромагнитен, а также по изменению электросопротивления и дилатометрическим методом.
Атермическое превращение (рис. 124, а и рис. 125, кривая 1) характеризуется плавным нарастанием ко
17 И. И. Новиков 257
личества мартенсита при непрерывном понижении температуры в мартенситном интервале Мн—Мк. Остановка охлаждения приводит к практически полному прекращению превращения. Когда образец в аустенитном состоянии перебрасывают в ванну с температурой ниже Л1н, превращение идет с очень большой скоростью за те несколько секунд» в течение которых температура образца выравнивается и становится одинаковой с температурой ванны. Во время последующей изотермической выдержки мартенсит не образуется, несмотря на большое количество исходной фазы (аустенита), или, точнее, скорость изотермического превращения аустенита ничтожно мала (отсюда и название — «атермическое» превращение).
Количество остаточного аустенита после закалки с охлаждением до комнатной температуры зависит от состава исходного аустенита и фактически связано с положением верхней мартенситной точки. С увеличением содержания углерода точка Ми снижается (см. рис. 111) и количество остаточного аустенита при комнатной температуре возрастает. Например, при закалке из аустенитного состояния в сталях с 0,6 и 1,2 % С это количество равно соответственно 2 и 20 %. В закаленных высокоуглеродистых легированных сталях может содержаться 60 % остаточного аустенита.
Остаточный аустенит сохраняется и при температурах ниже точки конца мартенситного превращения Мк. По достижении этой точки мартенситная кривая становится горизонталью, соответствующей некоторому практически неизменному соотношению количеств мартенсита и остаточного аустенита (см. рис. 124, а).
Атермическое превращение представляет наибольший практический интерес, так как в отличие от взрывного и полностью изотермического, характерных главным образом для области отрицательных температур, оно протекает при температурах выше комнатной, т. е. в обычных условиях закалки промышленных сплавов.
- Взрывное превращение характеризуется скачкообразным (взрывообразным) появлением определенного количества мартенсита при температуре точки 7ИН или несколько ниже (рис. 124,6 и рис. 125, кривые 2, 3). Количество мартенсита, образовавшегося взрывообразно, зависит от состава сплава и положения точки Мн, колеблясь от нескольких процентов до примерно 70 %. Взрыв сопровождается звуковым эффектом (отчетливо слышимым щелчком) и временным повышением температуры, иногда на
258
30 К, из-за быстрого выделения скрытой теплоты превра
щения.
Полностью изотермическое превращение (рис. 124, в и 126) по своей кинетике внешне напоминает нормальное фазовое превращение (см. рис. 68). При изотермической выдержке после некоторого инкубационного периода мартенситное превращение развивается с ускоре-
нием, а затем с постоянным зату- 1дд ханием во времени. Как и при обычном фазовом превращении, скорость изотермического мар-*Л? тенситного превращения и инку-1 бационный период зависят от | бд температуры. Так, например, в |.0 сплаве железа с 23,4 % Ni. 3,3 % £ Мп и 0,06 % С мартенситное пре-1 со вращение ускоряется с пониже-нием температуры от —50 до | —120 °C и замедляется при даль-4 20 нейшем понижении температуры (рис. 126). Инкубационный пе- о
риод сокращается с понижением температуры до —120 °C и далее
-200 -юо о юо
Теппература°С
возрастает. Это позволяет строить С-кривые изотермического мартенситного превращения (рис. 127), аналогичные обычным С-кривым неупорядоченного фазового превращения.
Так как имеется инкубационный период, изотермическое мар-
Рис. 125. Мартенситные кривые ^-►а-превращения при непрерывном охлаждении сплавов железа с никелем (М. Д. Пер-кас, В. М. Кардовский): / — 29% Ni, -------10 °C; 2 —
31 % Ni, Мн----63 °C; 3 — 32 %
Ni; Мн=—97 °C
тенситное превращение можно полностью предотвра-тить, применив быстрое охлаждение аустенита до очень низких температур. Так, в тех железных сплавах, для кото-
рых характерно изотермическое мартенситное превращение, его можно подавить быстрым охлаждением образцов до температуры жидкого азота (—196°C). При последующем отогреве аустенит испытывает изотермическое мартенситное превращение со скоростью, зависящей от температуры.
Принципиальное отличие кинетики изотермического мартенситного превращения от неупорядоченного фазового состоит в том, что мартенситное превращение не доходит до полного исчезновения исходной фазы, т. е. сохраняется остаточный аустенит. Количество изотермически образую-
17*
259
щегося мартенсита колеблется от долей процента до десятков процентов в зависимости от состава сплава и температуры изотермической выдержки. Например, на рис. 126 показано, что после 9-ч выдержки при температуре —50 °C образуется около 1 % мартенсита, а при —120 °C —более 50 %.
Рис. 126. Изотермическое превращение аустенита в мартенсит при разных температурах в сплаве Fe—23,4 % Ni—3,3 % Мп (О. П. Максимова, А. И. Никонорова)
Эксперименты показывают, что нельзя резко разграничить атермическое и взрывное превращение от изотермических. В сплавах, для которых характерна атермическая кинетика, после быстрого «атермического» образования определенной порции мартенсита при охлаждении до заданной температуры может изотермически образоваться дополнительное его количество. После мартенситного «взрыва» также может образоваться значительное количество «изотермического» мартенсита при температурах ниже температуры взрыва. Небольшое количество мартенсита может изотермически образоваться при температурах чуть выше температуры взрыва.
260
Кристаллы мартенсита независимо от типа кинетики превращения растут с громадной скоростью при любых температурах, в том числе и при температурах глубокого холода. По изменению электросопротивления, сопровождающему образование каждой пластины, были определены скорость ее роста и время формирования. Скорость роста пластин мартенсита (в плоскости пластины) оказалась порядка 1 км/с независимо от температуры превращения. Мидриб мартенситных пластин в сплавах Fe—Ni образует
ся за время порядка 10-7 с, а вся пластина (после более медленного ее утолщения) — за ~10~6 с. Для сравнения укажем, что «взрыв» продолжается 10“4—10""3 с.
После чрезвычайно быстрого формирования рост мартенситного кристалла прекращается. Нарастание количества мартенсита с понижением температуры при атермическом и взрывном превращениях или с увели-
рие. 127. С-диаграмма изотермического превращения аустенита в мартенсит в сплаве Fe—23,4 % Ni—3,3 % Мп. Цифры на С-кривых указывают количество образовавшегося мартенсита (О. П. Максимова, А. И. Никонорова>
чением времени выдержки при изотермическом превращении происходит вследствие образования новых кристаллов, а не роста ранее образовавшихся. С помо-
щью скоростной микрокиносъемки наглядно демонстрируется эта важнейшая особенность мартенситного превраще-
ния.
Характерная черта кинетики всех мартенситных превращений — их автокаталитичность: образование первых пластин влечет появление рядом с ними новых пластин, которые способствуют возникновению еще большего числа пластин и т. д., вследствие чего процесс некоторое время развивается как цепная реакция.
Автокаталитичность процесса наиболее ярко проявляется при взрывном превращении. Мартенситный взрыв — результат того, что образование одной пластины вызывает немедленную цепную реакцию появления серии пластин. Причиной автокаталитичности считают поле упругих напряжений у кромки растущей пластины мартенсита, которое способствует появлению новых пластин, или же плас
261
тическую деформацию аустенита, сопровождающую рост мартенситного кристалла.
Ярко выраженный взрыв происходит тогда, когда пластины имеют габитус {259}. Показано, что именно при таком габитусе у кромки пластины мартенсита возникают высокие напряжения в аустените, стимулирующие появление новых пластин. При образовании мартенситных пластин с габитусом {225} взрыва не наблюдается. Поэтому мартенсит с габитусом пластин {259} и характерным двойникованным мидрибом иногда называют взрывным мартенситом.
Резкой разницы между взрывным и обычным атермическим превращением не существует. При изменении состава сплава происходит постепенный переход от атермического к ярко выраженному взрывному превращению. Например, в системе Fe—Ni при содержании 29 % Ni превращение характеризуется типично атермической кинетикой, а при содержании 31 и 32 % Ni — взрывной, причем величина взрыва у сплава с 32 % Ni больше, чем у сплава с 31 % Ni (см. рис. 125).
Взрывное превращение можно рассматривать как разновидность атермического, когда автокаталитичность процесса особенно ярко выражена. В свою очередь атермическое превращение принципиально не отличается от изотермического. Обе эти разновидности кинетики мартенситных превращений, как показано Г. В. Курдюмовым, следует рассматривать на основе единых представлений о температурной зависимости скорости зарождения центров превращения.
Поскольку скорость роста кристаллов мартенсита независимо от температуры громадная, то наблюдаемое с понижением температуры сначала увеличение, а затем уменьшение скорости изотермического превращения (см. рис. 126 и 127) обусловлено только температурной зависимостью скорости зарождения кристаллов мартенсита.
Скорость зарождения мартенсита, как и при любом фа-зовом превращении, J=Ae~&G*^kTe~Q/kT [см. формулу (25)].
В некоторой промежуточной области еще не слишком низких температур уменьшение работы образования зародышей может привести к столь быстрому превращению, что ни инкубационный период, ни изотермичность превращения (его развитие во времени) не удается экспериментально зафиксировать.
262
В зависимости от состава сплава и абсолютного значения Tq можно встретить разные экспериментально наблюдаемые случаи. Например, если точка То низка, то при любых температурах обнаруживается изотермичность превращения (см. рис. 126 и 127), так как при любых температурах в мартенситном интервале скорость превращения сравнительно невелика. Если же температура То значительно выше комнатной, то изотермическое превращение можно наблюдать только в области температур вблизи точки Л4Ц и при сравнительно низких температурах (обычно отрицательных, реже — комнатной). Между этими интервалами температур, где экспериментально наблюдается изотермическое превращение, может находиться область температур, в которой изотермическое превращение протекает с громадной скоростью и поэтому создается впечатление об отсутствии изотермичности. С этих позиций не исключено, что собственно атермического превращения вообще не существует, так как оно является обычным изотермическим превращением, протекающим с громадной скоростью при каждой температуре в период закалочного охлаждения. Именно поэтому в сплавах, для которых в целом характерна «атермическая» кинетика, часто в той или иной степени обнаруживается изотермическое образование мартенсита при остановке охлаждения.
Обнаружение Г. В. Курдюмовым и О. П. Максимовой полностью изотермического мартенситного превращения явилось прямым доказательством того, что зарождение кристаллов мартенсита — это зависящий от температуры, термически активируемый процесс, как и любой процесс зарождения кристаллов новой фазы (см. § 17). В связи с этим само понятие «атермическое превращение» противоречит термической природе образования зародышей. Более подходяще, но менее распространено другое название— «неизотермическое превращение», указывающее лишь на то, что мартенсит образуется не при одной температуре, а при разных температурах во время охлаждения.
Важнейшие особенности кинетики мартенситных превращений — прекращение образования мартенсита при наличии неизрасходованной исходной фазы (аустенита), необходимость понижения температуры для возобновления превращения. Так как мартенситное превращение развивается вследствие образования новых, а не роста ранее образовавшихся кристаллов, то прекращение превращения означает прекращение образования зародышей мартенсита. В связи с тем что механизм зарождения мартенсита надеж
263
но не установлен, рассматриваемая важнейшая особенность кинетики мартенситных превращений имеет только предположительное объяснение.
Современные трактовки исходят из гипотезы гетерогенного зарождения мартенсита в заранее подготовленных участках аустенита с особым строением (см. § 34). Эти особые участки в аустените отличаются один от другого размерами. Когда при охлаждении термодинамический стимул превращения— разность энергий Гиббса аустенита и мартенсита—достигает определенной величины, указанные особые участки в аустените превращаются в активные, способные к росту зародыша мартенсита. Вблизи точки Мн в виде центров превращения реализуются только те участки аустенита, в которых работа образования зародышей мартенсита минимальна. По исчерпании таких участков превращение останавливается. Для его возобновления необходимо понизить температуру, чтобы увеличить разность энергий Гиббса аустенита и мартенсита.
Затуханию мартенситного превращения при изотермической выдержке способствует также прогрессирующее разбиение аустенитного зерна на все более мелкие отсеки (см. рис. 121). С сокращением размера таких отсеков уменьшается размер новых мартенситных кристаллов, и поэтому объемная доля образующегося мартенсита, приходящаяся на каждый его зародыш, становится меньше.
2. Термическая стабилизация аустенита
Под термической стабилизацией исходной фазы понимают затруднение ее превращения в мартенсит в результате теплового воздействия.
Термическая стабилизация аустенита, которую обычно называют просто стабилизацией, наблюдается при временной остановке охлаждения железного сплава в мартенситном интервале атермического превращения. Если прервать охлаждение при температуре 7П<МН (но выше Мк) и сделать здесь выдержку, то аустенит стабилизируется. Стабилизация проявляется в том, что по возобновлении охлаждения превращение начинается не сразу при температуре Гп, а после переохлаждения аустенита (гистерезиса) до некоторой температуры М' (рис. 128). При этом мартенсита часто образуется меньше, чем при непрерывном охлаждении (мартенситная кривая 2 на рис. 128 идет ниже кривой /), и количество остаточного аустенита возрастает. Возобновляющееся при температуре М* мартенситное превращение
264
может протекать взрывообразно (например, в сплавах Fe— Ni—С).
Наиболее простой случай термической обработки, включающей стабилизацию аустенита, можно наблюдать, когда точка Мн находится выше, а Мк — ниже комнатной темпе
ратуры, причем 7^=20 °C. Так, выдержка углеродистой стали при комнатной температуре после обычной закалки в воде стабилизирует аустенит, затрудняя мартенситное превращение при последующем охлаждении стали в области отрицательных температур и увеличивая количество остаточного аустенита после такой обработки, Поэтому изучение закономерностей стабилизации аустенита представляет интерес для технологии обработки стали холодом (§41).
Мерой эффекта стабили-
Рис. 128. Мартенситные кривые при разной стабилизации аустенита:
Тп— температура, при которой охлаждение прерывают и проводят выдержку; 1 — кривая непрерывного охлаждения; 2t 3 — кривые, соответствующие разной выдержке (т) при температуре 7 , причем TS>T2
зации часто служит темпе-
ратурный гистерезис 6 = ГП—М'и (см. рис. 128).
Степень стабилизации аустенита зависит от температуры прерывания охлаждения Тп и выдержки при этой температуре. Если же после прерывания охлаждения сплав был нагрет до некоторой температуры Тс>Тп, то степень стабилизации 6 зависит от Тс и времени выдержки при этой температуре.
С пониженим Тп, т. е. с ростом количества мартенсита, присутствующего при стабилизирующей выдержке, степень стабилизации возрастает. Более сложно влияние времени выдержки. С увеличением времени выдержки при температуре Тп или Тс степень стабилизации может непрерывно возрастать (кривые 2 и 3 йа рис. 128). При достаточно высоких температурах увеличение времени Выдержки сначала приводит к росту, а затем к уменьшению степени стабилизации. Чем выше температура Тс, тем быстрее достигается максимум 0.
На стали с 1,4 % С и 5 % Ni было обнаружено, что величина 6 с увеличением времени выдержки при температуре Тс после прохождения через максимум снижалась И становилась отрицательной. Это означает, что большие выдер
265
жки после прерывания охлаждения приводили не к стабилизации (М'к<Тп), а к противоположному эффекту— активированию мартенситного превращения (М'>ГП).
Термическая стабилизация аустенита — сложный процесс. Вполне возможно, что в разных случаях, например в разных температурных интервалах, действуют разные механизмы стабилизации.
В термическую стабилизацию при температурах ниже точки Мн определенный вклад может внести релаксация упругих напряжений в аустенитной матрице вокруг мартенситных пластин. Эти напряжения, как уже отмечалось, вызывают автокаталитический эффект, свойственный всем мартенситным превращениям. Уменьшение упругих напряжений вокруг мартенситных кристаллов хорошо объясняет повышение степени стабилизации с увеличением времени выдержки, но не может объяснить снижения 0 при больших выдержках.
Для понимания природы стабилизации аустенита весьма важно, что термическая стабилизация наблюдается только в тех сплавах на основе железа, которые содержат, хотя бы и в сравнительно небольшом количестве, элементы внедрения: углерод и азот. Поэтому почти все современные гипотезы механизма термической стабилизации аустенита исходят из предположения о решающей роли сегрегации атомов углерода или азота.
Атомы элементов внедрения во время стабилизирующей выдержки могут сегрегировать в потенциальные участки зарождения в аустените, препятствуя превращению этих участков в зародыши мартенсита. Они могут сегрегировать на межфазную границу матрицы и зародыша, препятствуя его росту.
Весьма убедительна гипотеза, связывающая термическую стабилизацию с деформационным старением — образованием сегрегаций из внедренных атомов на дислокациях в аустените. Так как образование мартенситного кристалла вызывает пластическую деформацию аустенитной матрицы, то упрочнение ее при деформационном старении затрудняет мартенситное превращение. Развитие деформационного старения аустенита с увеличением времени выдержки при температуре Тс объясняет рост 6. Уменьшение эффекта стабилизации с дальнейшим увеличением времени’выдержки легко объяснить перестариванием (см. § 49), с которым связано падение предела текучести аустенита. Достижение отрицательных значений 0 можно связать с далеко зашедшим перестариванием, которое, во-пер
266
вых, сильно разупрочняет аустенит и, во-вторых, может привести к обеднению аустенита углеродом вблизи выделений карбида. Такие обедненные углеродом участки имеют повышенную температуру Мн. С повышением температуры Тс деформационное старение ускоряется и соответственно ускоряется достижение максимума 0.
При комнатной и более высоких температурах могут действовать оба механизма стабилизации: образование сегрегаций углерода и релаксация напряжений.
§ 37. Влияние деформации на мартенситное превращение 1. Мартенсит напряжения и мартенсит деформации
Деформация исходной фазы оказывает сложна влияние на мартенситное превращение.
Деформация выше точки Ми вызывает мартенситное превращение в интервале между температурой Ми и некоторой температурой ЛТД, которая находится где-то ниже точки То, иногда очень близко от нее. Аналогично деформация мартенсита вызывает обратное мартенситное превращение в области температ^-^цже точки Ан вплоть до некоторой температуры Ад (см. рисГтг4).
Мартенситное превращение выше 7Ии вызывается и упругой, и пластической деформацией (рис. 129). Выше точки Ми термодинамический стимул превращения ДбОб не может вызвать превращения. Выше Мп в некотором интервале AfH—упругие напряжения способствуют зарождению кристаллов мартенсита в тех же участках исходной фазы, где они появляются ниже Мп без участия этих напряжений. Энергия напряжений от приложенной нагрузки как бы добавляется к термодинамическому стимулу превращения Дбоб, и мартенситное превращение становится возможным при меньших степенях переохлаждения по отношению к То, т. е. при более высоких температурах. Чем больше удалена температура от Мн (в сторону 70)> тем меньше ДбОб и соответственно больше должно быть напряжение упругой деформации, способное вызвать мартенситное превращение (см. участок ab на рис. 129). С повышением температуры деформации достигается точка при которой указанное напряжение становится равным пределу текучести. Выше температуры пластическая деформация обусловливает иной механизм стимулирования мартенситного превращения, а именно образование под действием деформации новых мест зарождения мартенсита. Это и понятно, если учесть, что зародышами мартенсита могут
267
Рис. щне при ab)
Мн Мд
Температура
129. Напряжения, вызываю-мартенситное превращение упругой деформации (участок в интервале Л4н—AfJJ н при пластической деформации (участок Ьс) в интервале М*^—Мд (схема но Олсону и Коэну)
быть определенные группировки растянутых дислокаций. Чем выше температура и соответственно меньше ЛбОб, тем больше должна быть деформация, способная вызвать мартенситное превращение (см. участок Ьс на рис. 129). Выше температуры Мд никакая пластическая деформация не может превратить аустенит в мартенсит при деформации.
В железных сплавах с относительно высокой энергией дефектов упаковки в аустените при пластической деформации мартенсит образуется по схеме а в сплавах с низкой энергией дефектов упаковки — по схеме у->-е->сс (об е-мартенсите см. в § 34).
В связи с изложенным следует различать: 1) мартенсит охлаждения, обр а зующи йся
без воздействия упругой или пластической деформации, только в результате понижения температуры ниже точки 7МН; 2) мартенсит напряжения (stress-assisted martensite), образующийся под действием напряжений от приложенной нагрузки (упругой деформации); 3) мартенсит пластической деформации, сокращенно мартен
сит деформации (strain-induced martensite), образующийся в результате пластической деформации исходной фазы. Мартенсит напряжения зарождается в тех же местах исходной фазы, что и мартенсит охлаждения; напряжения лишь способствуют его зарождению в этих местах. Мартенсит деформации зарождается в участках, подготовленных пластической деформацией.
При температурах выше Мн мартенсит напряжения мо- \ жет образовываться только в интервале Л4И—М£ . Выше температуры Л4£, в интервале М°—Мд9 может образовываться только мартенсит деформации. И мартенсит напряжения, и мартенсит деформации могут появляться и в указанных интервалах, и при более низких температурах. На-____,
пример, экспериментально установлено образование этих двух типов мартенсита в интервале МИ—Мкогда приложенная нагрузка вызывает пластическую деформацию исходной фазы. Под действием напряжений от нагрузки, при- ’
268
ложенной при температурах ниже М& мартенсит напряжения образуется в дополнение к мартенситу охлаждения.
Автоката л итичность, характерная для любого мартенситного превращения (см. § 36), по существу является результатом инициирующего влияния напряжения. Только в этом случае напряжения возникают в исходной фазе при появлении каждого нового кристалла мартенсита.
Эффект мартенситообразования под действием деформации нашел разнообразное практическое применение.
Высокая кавитационная стойкость сталей с механически нестабильным аустенитом (например, стали 30Х10Г10, точка 7ИН=—40 °C) объясняется образованием под действием гидравлических микроударов мартенсита в поверхностном слое изделия, сердцевина которого осхается аустенитной.
В 60-е годы появился новый класс высокопрочных сталей — так называемые трип-стали или ПНП-стали1. Эти стали, легированные хромом, никелем, молибденом, марганцем и другими элементами, имеют точки Мн и Мд ниже комнатной температуры. В аустенитном состоянии при 400— 600°C трип-стали наклепывают для повышения прочности. При этом на дислокациях выделяются карбиды, аустенит обедняется углеродом и точка Мд поднимается до температуры выше комнатной. Такая сталь обладает ценным сочетанием повышенной прочности (благодаря наклепу и деформационному старению аустенита) и очень высокой пластичности. Когда во время растяжения при комнатной температуре разрывного образца начинается локализация деформации, то под ее действием образуется мартенсит, упрочняющий образец в этом месте, и деформация распространяется на соседние участки аустенита. Таким образом, образование мартенсита под действием пластической деформации предотвращает развитие локализации деформации (образование шейки), чем обеспечивается высокое относительное удлинение (до 100—150 %). Недостаток трип-сталей — трудность получения больших обжатий при сравнительно низких температурах, когда велико сопротивление деформированию и требуется мощное оборудование.
2. Эффект запоминания формы и сверхупругость
Интерес к закономерностям влияния деформации на мартенситное превращение усилился в 70-е годы в связи с промышленным использованием эффекта запоминания формы и сверхупругости.
1 ПНП — пластичность, наведенная превращением (англ. — Transformation Induced Plasticity, или сокращенно TRIP).
269
Эффект запоминания формы (ЭЗФ) проявляется в том, что после придания образцу определенной формы при повышенной температуре ему придают новую форму при более низкой температуре, а после нагрева исходная форма образца восстанавливается.
ЭЗФ внешне проявляется весьма необычно и эффектно. Например, если проволоку, закрученную при повышенной температуре в спираль причудливой формы, выпрямить при комнатной температуре, то при последующем нагреве проволока сама закручивается в спираль точно такой формы, как исходная.
Сверхупругость проявляется в том, что образец претерпевает обратимые деформации, которые на 1—2 порядка (1) больше, чем деформация металлических материалов до условного предела упругости. Металлический сплав, обладающий сверхупругостью, ведет себя подобно резине.
ЭЗФ и сверхупругость связаны с одним и тем же явлением — обратимостью макродеформации образца, обусловленной движением скользящих границ между кристаллами при сдвиговом превращении. Термин «скользящая» применительно к границе используется в том же смысле, что и для дислокации. При скользящем движении атомы смещаются по отношению к своим соседям на расстояния меньше межатомного, нет потока вакансий и других точечных дефектов, энергия активации существенно меньше энергии активации самодиффузии.
В разных сплавах и даже в одном сплаве в неодинаковых условиях могут действовать разные механизмы восстановления формы. Среди них для промышленного использования наиболее важны те, что связаны с обратным мартенситным превращением после деформации.
Ранее уже отмечалось, что мартенситное превращение обязательно сопровождается изменением формы превращенного объема и что в случае термоупругого мартенсита при обратном превращении межфазные границы проходят пути, обратные тем, что они проходили при прямом превращении.
Для сплавов, обладающих ЭЗФ, наиболее важны следующие два воздействия деформации. Во-первых, под действием деформации выше Л4К зарождаются кристаллы мартенсита с преимущественной ориентировкой или предпочтительно растут за счет исходной фазы ранее образовавшиеся кристаллы, имеющие эту ориентировку. Во-вторых, если деформацию проводят по окончании прямого превра
270
щения (ниже Л1К), благоприятно ориентированные мартенситные кристаллы растут за счет менее благоприятно ориентированных кристаллов. Благоприятной является такая ориентировка, при реализации которой изменение формы превращенных областей соответствует макроизменению формы образца, задаваемому приложенной нагрузкой.
Рис. 131. Схема, иллюстрирующая проявление сверхупругости. Супр н Супр~ УпРУгая Деформация аустенита и мартенсита соответственно; ЕСу — сверхупругая деформация
Рис. 130. Изменение количества термоупругого мартенсита при охлаждении и нагреве
При нагреве после деформации обратное мартенситное превращение приводит к исчезновению кристаллов мартенсита, выросших при деформации, и если межфазные границы оставались когерентными (или полукогерентными), т. е. были скользящими, то исходная форма образца восстанавливается. Это свойственно далеко не всем сплавам^ Например, в сплавах Fe—Ni (см. рис. 115) межфазные границы движутся необратимо и эффект запоминания формы отсутствует.
На рис. 130 изображено увеличение количества термоупругого мартенсита при охлаждении и уменьшении его количества при последующем нагреве.
Если приложить к образцу нагрузку в области температур ИК<ТД<Л1^ в нем начнет образовываться мартенсит напряжения и изменится форма образца. Связь между напряжением и деформацией для этого случая показана на рис. 131. На участке АВ происходит упругая деформация исходной (аустенитной) матрицы . На участке
271
Рис. 132. Схема, иллюстрирующая эффект запоминания формы:
а — диаграмма напряжение — деформация; б — зависимость стаиа вливаемой деформации температуры при нагреве
ВС при постоянном напряжении1 деформация идет вследст-ствие изменения формы превращенного объема при образовании мартенсита напряжения. При деформации, происходящей за точкой С, образовавшийся мартенсит упруго деформируется (участок CD) и при дальнейшем повышении напряжений от приложенной нагрузки (выше точки D) происходит пластическая деформация мартенсита и разрушение образца.
Если при нагружении образца напряжения не выходят за точку D, то при разгрузке сначала исчезает упругая деформация мартенсита (до точки £), затем происходит обратное превращение мартенсита в аустенит (участок EF), после чего исчезает упругая деформация аустенита (участок FA).
Большая часть обратимой деформации обусловлена мартенситным превращением (участок EF). Эта деформация равна примерно 7 %, в то время как область обычной упругости поликристаллических металлов не выходит за предел в десятые доли процента (на рис. 131 участки обычной упругой деформации АВ и CD в сравнении с участком EF показаны не в масштабе). Дефор
мация ВС получила название сверхупругой.
Если продеформировать образец в интервале температур АТк<Гд<Лн (см. рис. 130), то после упругой деформации исходной матрицы еупр (участок АВ на рис. 132, а) в ней будут происходить фазовые и структурные превращения, обеспечивающие деформацию на участке ВС. При снятии нагрузки исчезает лишь упругая составляющая деформации (участок CD). Деформация, обусловленная пре
вос-от
1 В реальных условиях участок ВС имеет небольшой наклон.
272
вращением, не начнет исчезать до тех пор, пока температура не превысит Дн, когда станет возможным протекание обратных превращений (участок EF на рис. 132,6). При нагреве выше точки Дк деформация, обусловленная превращением, полностью исчезнет и восстановится первоначальная форма образца, т.е. проявляется эффект запоминания формы.
В температурном интервале ЛН<ГД<ЛК восстанавливаемая деформация является суммой деформаций есу+
ЭЗф . Температурные интервалы деформации, приводящей к раздельному и совместному проявлению эффекта запоминания формы и сверхупругости, указаны на рис. 130.
Первые промышленные сплавы, обладающие эффектом запоминания формы, — это сплавы нитинол на базе интер-металлида NiTi, у которого высокотемпературная модификация претерпевает мартенситное превращение.
Из сплавов нитинол с точкой Мн выше комнатной температуры [пример — сплав с 50 % (ат.) Ni и 50 % (ат.) Ti] изготовляют саморазвертывающиеся устройства, в частности антенны. Изделию £фи повышенной температуре придают требуемую для эксплуатации форму, при которой оно занимает большой объем или имеет большую длину. Затем изделие свертывают (деформируют) при комнатной температуре, чтобы транспортировать его в компактном виде. После доставки к месту назначения изделие нагревают для восстановления формы и оно само развертывается. Такие развертывающиеся устройства представляют большой интерес для космической техники.
Сплавы нитинол с точкой Мк вблизи —150 °C [например, сплав с 50 % (ат.) Ti, 3—4 % (ат.) Fe, ост. Ni] применяют для изготовления соединительных муфт и уплотнений трубопроводов. Деформацию такой детали, например для увеличения внутреннего диаметра муфты, проводят в жидком азоте. После установки муфты на стык труб она отогревается до комнатной температуры и, восстанавливая свою первоначальную (до деформации) форму, обеспечивает плотное и прочное соединение труб.
Разработано множество других предложений практического использования ЭЗФ и сверхупругости. Создаются более дешевые, чем нитинол, медные сплавы, обладающие этими свойствами.
3. Механическая стабилизация аустенита
Пластическая деформация; не вызывающая образования мартенсита непосредственно в период деформирования,
18 И. И. Новиков 974
-200 -150 -ЮО -50 О
Темпера тура°С
Рис. 133. Зависимость начальной скорости изотермического мартенситного превращения от температуры в сплаве Fe—17,2 % Сг—9,1 % Ni после деформации аустенита при 100 РС с обжатием 8 и 17 % (Г. В. Курдюмов, О. П. Максимова, А. В. Никонорова)
может повлиять на кинетику мартенситного превращения при последующем охлаждении. Она способна интенсифицировать последующее мартенситное превращение, повышая температуру его начала, увеличивая скорость превращения и уменьшая количество остаточной исходной фазы. Но она же способна оказать и тормозящее влияние, снижая температуру начала превраще-' ния, замедляя его и увеличивая количество остаточной исходной фазы (механическая стабилизация аустенита). Результат зависит от степени деформации, температуры деформирования и свойств исходной фазы, т. е. состава сплава.
Небольшая предварительная деформация обычно активирует мартенситное превращение при последующем охлаждении, а большая затрудняет его (рЪс. 133). Это объясняется тем, что малые деформации создают такие структурные нарушения в исходной фазе и такие локальные поля напряжений, которые делают энергетически выгодным заро-
ждение мартенсита в соответствующих участках. Большие же деформации создают такие сильные нарушения пра|-вильного строения исходной фазы, которые затрудняют когерентный рост мартенситного зародыша на самых начальных его стадиях. Чем выше температура деформирова? ния и ниже предел упругости исходной фазы, тем с меньших степеней деформации проявляется механическая стабилизация. J
§ 38. Изменение свойств сплавов при закалке на мартенсит
1. Упрочнение при закалке
Важнейшее явление, сопровождающее закалку на мартенсит,— упрочнение, повышение твердости. Именно благодаря упрочнению и была открыта в древности, а затем широко использована закалка сталей.
274
Природа сильного упрочнения при закалке сталей представляла загадку на протяжении многих веков. Сейчас кажутся необычайно наивными средневековые представления о сверхъестественных силах, вызывающих упрочнение при погружении раскаленного клинка в шипящую воду, а также попытки даже на рубеже XX столетия связать высокую твердость закаленной стали с переходом ее углерода в алмаз.
В познании механизма упрочнения при закалке решающую роль сыграли выяснение природы самого мартенситного превращения и изучение структуры мартенсита. В общих чертах механизм упрочнения при закалке на мартенсит теперь вполне понятен, но многие важные его детали еще требуют выяснения.
Попытки объяснить большую твердость закаленной стали какой-либо одной причиной экспериментально не подтвердились. Можно уверенно утверждать, что упрочнение при закалке стали на мартенсит является результатом действия нескольких механизмов торможения дислокаций.
Важнейшая роль во всех теориях упрочнения при закалке сталей справедливо отводится углероду. Однако необходимо иметь в виду, что мартенситное превращение в чистом железе и в безуглеродистых сплавах способно привести к повышению прочностных свойств в 3—4 раза по сравнению с отожженным состоянием. Так, по сравнению с обычной ферритной структурой твердость железа в результате мартенситного превращения возрастает с HV 60 до HV 200, а временное сопротивление — с 200 до 900 МПа. У отожженного сплава железа с 8 % Сг и 0,45 % Ni предел текучести равен 220 МПа, а у закаленного с 1000 °C он составляет 800 МПа.
Мартенсит в отличие от фазы того же химического состава, но образовавшейся при медленном охлаждении вследствие неупорядоченной перестройки решетки, характеризуется повышенной плотностью дефектов: двойниковых прослоек и дислокаций (см. § 35). Плотность дислокаций в мартенсите доходит до 1010—1012 см~2, т. е. по порядку величины такая же, как и в холоднодеформированном металле. Границы двойников и сплетения дислокаций служат барьером для скользящих дислокаций, т. е. упрочняют мартенсит. Фазовый наклеп, возникающий при мартенситном превращении, в той или иной степени вносит вклад в упрочнение всех металлов и сплавов, закаливаемых на мартенсит.
Рассмотрим роль углерода в упрочнении при закалке
18*
275
сталей. С увеличением содержания углерода в мартенсите твердость его непрерывно возрастает. Здесь следует обратить внимание на то, что твердость закаленной стали и твердость кристаллов мартенсита — это не одно и то же, так как в закаленной стали имеется остаточный аустенит. Твердость стали, закаленной с температур в аустенитной области, с увеличением содержания углерода в стали проходит через максимум при концентрации около 0,9 % С, а
h'RC
20 fe
60
50
АО
50
затем снижается из-за увеличения объемной доли мягкого остаточного аустенита (рис. 134). Закаленная с температуры выше Асчп сталь с 1,9 % С имеет такую же твердость, как и закаленная сталь с 0,1 % С. Если же заэв-тектоидные стали закаливать с температуры Лй+ (354-60) К, кда>это и делают (см. ниже рис. 143), то состав аустенита при
о,г о,6 1,0 w температуре закалки и уровень
Рис. 134. Зависимость твердости от состава двойных сплавов Fe—С после закалки их с температур аустенитной области (Литвинчук, Кейзер н Бэйкер)
твердости во всех заэвтектоидных сталях будут практически одинаковыми.
Способность стали к повышению твердости при закалке называется закаливаемостью. Закали-
ваемость характеризуется максимальной твердостью, которая может быть получена при закалке данной стали на поверхности изделия.
При мартенситном превращении аустенита образуется пересыщенный раствор углерода в a-железе и тем сильнее пересыщенный, чем больше углерода содержит аустенит. Интересно, что с ростом содержания углерода в мартенсите межатомные силы не только не усиливаются, а, наоборот, даже несколько ослабевают. Это обусловлено увеличением расстояний между атомами железа под действием внедренных атомов углерода. Тем не менее углерод повышает твердость мартенсита. Объясняется это прежде всего тем, что атомы углерода, внедренные в решетку а-железа, затрудняют скольжение дислокаций в мартенсите (так называемый твердорастворный механизм упрочнения).
' В период закалки или при вылеживании стали после закалки атомы углерода в кристаллах мартенсита образуют атмосферы на дислокациях, закрепляют их. В сталях с вы-
276
сокой точкой ЛТН, например в углеродистых, содержащих менее 0,5 % С (MH>300°C, см. рис. 111), в период закалочного охлаждения в мартенситном интервале создаются наиболее благоприятные условия для частичного распада мартенсита с выделением дисперсных частиц карбидов, т. е. самоотпуска (см. § 52). Кроме того, в любых сталях углерод при обычных скоростях закалки успевает образовывать сегрегации на дефектах решетки аустенита в период охлаждения стали выше точки Мн. Сегрегации углерода в аустените наследуются мартенситом, а поскольку он и так пересыщен углеродом, то эти сегрегации становятся местами зарождения частиц карбида. С этим согласуется то, что при очень больших скоростях охлаждения твердость кристаллов мартенсита оказывается почти в полтора раза ниже, чем после обычной закалки.
Из-за действия указанных выше механизмов упрочнения углерод оказывает столь сильное упрочняющее влияние на мартенсит, что твердость закаленной стали практически не зависит от содержания легирующих элементов, растворенных в мартенсите по способу замещения, а определяется только концентрацией углерода.
Суммируя, можно сделать заключение, что сильное упрочнение сталей при закалке на мартенсит обусловлено образованием пересыщенного углеродом а-раствора, появлением большого числа двойниковых прослоек и повышением плотности дислокаций при мартенситном превращении, образованием на дислокациях атмосфер из атомов углерода и выделением из а-раствора дисперсных частиц карбида. Вклад каждого из этих факторов в общее упрочнение при закалке окончательно не установлен.
Начинающие изучать термическую обработку часто рассматривают сильное упрочнение при закалке сталей как явление, исключительное по своему эффекту, по приросту твердости. Обратимся к фактам. Твердость эвтектоидной стали в отожженном состоянии составляет НВ 180, а в закаленном НВ 650. Следовательно, закалка повышает ее твердость в 3,5 раза.
Упрочнения металлов и сплавов достигают также другими путями. При холодной пластической деформации твердость легко увеличить вдвое и даже втрое. Увеличивая концентрацию твердого раствора при легировании металла, особенно раствора внедрения, можно повысить твердость в несколько раз. Например, твердость раствора титана с 0,7 % О в три раза больше твердости чистого титана (атомы кислорода внедрены в решетку а-титана). Сильное упроч
277
нение достигается при дисперсионном твердении стареющих сплавов. Например, старение бериллиевой бронзы при 350 °C приводит к увеличению твердости в четыре раза (I) по сравнению с закаленным состоянием. При сопоставлении с приведенными примерами эффект упрочнения при закалке сталей не выглядит чем-то выдающимся, особенно если учесть одновременное действие нескольких механизмов упрочнения.
В сплавах других систем (не на основе железа) упрочнение при закалке на мартенсит может быть вызвано действием тех же механизмов, что и при закалке сталей и без-углеродистых железных сплавов. Наиболее общим является механизм фазового наклепа, свойственного всем мартенситным превращениям в металлах и сплавах и состоящего в увеличении плотности дефектов кристаллической решетки.
2. Изменение пластичности при закалке
Закалка углеродистых сталей приводит к резкому снижению всех показателей пластичности. Относительное удлинение и сужение закаленных высокоуглеродистых сталей равны нулю. В то время, когда считалось, что мартенситное превращение протекает только в сталях, обычно подчеркивалось, что мартенсит тверд, но хрупок. Когда же обнаружили мартенситное превращение в безуглеродистых железных сплавах и сплавах цветных металлов, то оказалось, что хрупкость не является общим свойством, присудим любому мартенситу. Теперь установлено, что низкое сопротивление хрупкому разрушению мартенсита в сплавах железа связано с присутствием в о. ц. к. решетке элементов внедрения — углерода и азота.
Основная причина резкого охрупчивания при закалку углеродистых сталей — малая подвижность дислокаций в мартенсите, содержащем углерод. Это вызвано, во-первых, повышенным сопротивлением решетки раствора внедрения скольжению дислокаций и, во-вторых, закреплением дислокаций атмосферами из атамов углерода. Из-за низкой подвижности дислокаций не происходит релаксации напряжений у вершины хрупкой трещины вследствие микропласти^ ческой деформации, чем и объясняется низкое сопротивлений мартенсита распространению трещины. '
— Зарождению хрупких трещин способствует локальная концентрация микронапряжений около скоплений дислокаций в мартенсите. Следовательно, мартенсит углеродистых; сталей обладает пониженным сопротивлением зарождений особенно низким сопротивлением развитию хрупких треЗ
278
щин. В высокоуглеродистых сталях микротрещины можнс увидеть сразу же после закалки, еще до приложения внешней нагрузки.
Картина резко меняется, если углерод вывести из соста. ва сплава. Так называемые мартенситно-стареющие стали, являющиеся безуглеродистыми сплавами железа с никелем, кобальтом, молибденом и титаном, после закалки на мартенсит отличаются высокими показателями пластичности (6=14—20%, ф=70-е-80 %). Эти сплавы в закаленном состоянии можно обрабатывать при комнатной температуре давлением с высокими обжатиями, в то время как закаленную высокоуглеродистую сталь из-за хрупкости вообще невозможно обработать давлением при комнатной температуре.
Высокие показатели пластичности безуглеродистых железных сплавов после закалки на мартенсит объясняются следующим. В мартенсите закаленных мартенситно-старе-ющих сталей, содержащих не более 0,03 % С, имеются незакрепленные дислокации. Никель и кобальт, содержащиеся в большом количестве в этих сталях (например, сталь с 18 % Ni и 8 % Со), увеличивают подвижность дислокаций в мартенсите, так как они понижают сопротивление решетки мартенсита скольжению дислокаций и уменьшают степень закрепления дислокаций атомами примесей углерода и азота. Высокая подвижность дислокаций в железоникелевом мартенсите обеспечивает разрядку напряжений у вершины трещины, повышая сопротивление хрупкому разрушению.
Если отвлечься от проблемы хрупкости твердых растворов с о. ц. к. решеткой, содержащих элементы внедрения, то следует заметить, что закаленные на мартенсит металлы и сплавы в общем могут иметь довольно высокие показатели пластичности, хотя после мартенситного превращения из-за повышенной плотности дефектов решетки показатели пластичности должны быть ниже, чем после полиморфного превращения с неупорядоченной перестройкой решетки.
§ 39. Бейнитное превращение
t. Строение бейнита
В углеродистых сталях ниже изгиба С-кривой, в интервале примерно 500—250 °C, происходит бейнитное превращение. Оно называется также промежуточным превращением — промежуточным между перлитным и мартенситным. Кинетика этого превращения и получающиеся структуры имеют
279
черты кинетики и структур, наблюдаемых при диффузионном перлитном и бездиффузионном мартенситном превращениях.
В результате бейнитного превращения образуется смесь a-фазы (феррита) и карбида, которая называется бейнитом. Карбид в бейните не имеет пластинчатого строения, свойственного перлиту. Карбидные частицы в бейните очень дисперсны, их можно обнаружить только под электронным микроскопом.
Различают верхний и нижний бейнит, образующийся соответственно в верхней и нижней части промежуточного интервала температур (условная граница между ними 350°C). Верхний бейнит имеет перистое строение, а нижний— игольчатое, мартенситоподобное. Нижний бейнит по виду микроструктуры бывает трудно отличить от отпущенного мартенсита.
Электронно-микроскопический анализ показал, что субструктура a-фазы верхнего бейнита аналогична субструктуре реечного мартенсита низкоуглеродистых сталей, а строение онфазы нижнего бейнита ближе к строению мартенсита сталей с более высоким содержанием углерода. В верхнем бейните a-фаза состоит из реек шириной 5— 10 мкм и толщиной менее 1 мкм, а карбидные частицы расположены между рейками или/и по границам, и внутри реек. Следовательно, в верхнем бейните карбид выделяется преимущественно из аустенита. В нижнем бейните частицы карбида находятся внутри пластин a-фазы, т. е. карбид образуется при ее распаде. Внутри a-фазы и в верхнем, и в нижнем бейните повышена плотность дислокаций.
Продукты изотермического превращения переохлажденного аустенита в промежуточном интервале температур по своим физическим свойствам, составу и структуре фаз близки к продуктам отпуска мартенсита закаленной стали, если температуры бейнитного превращения и отпуска одинаковы1. Ферритная фаза в бейните является пересыщенным раствором углерода в а-железе.
Карбидная фаза в верхнем бейните — цементит, а в нижнем бейните — е-карбид, который заменяется цементитом с увеличением времени выдержки (как и при отпуске стали).
Специальные карбиды в легированных сталях, образующиеся в перлитном интервале, при бейнитном превращении не появляются. Содержание легирующих элементов в
1 Поэтому к изучению § 39 следует вернуться после проработки гл. ХП. г
280
е-карбиде и .цементите бейнита такое же, как и в исходном аустените. Здесь полная аналогия с отпуском легированных сталей (см. § 51, п. 2). Следовательно, легирующие элементы при бейнитном превращении не перераспределяются.
В сталях, легированных кремнием, карбид может вообще не образовываться и бейнит состоит из a-фазы и остаточного аустенита.
2. Кинетика бейиитного превращения
В бейнитном интервале температур, так же как и в перлит-' ном, переохлажденный аустенит начинает распадаться после, некоторого инкубационного периода. У сталей, легирован^ ных карбидоообразующими элементами (Сг, W, Мо и др.), под С-кривыми перлитного распада находятся С-кривые начала и конца бейиитного превращения (рис. 135). Области
Рис. 135. Диаграммы изотермического распада аустенита легированных сталей с обособленными С-кривыми перлитного и бейиитного превращений (Розе и Петер):
а — сталь с 0,43 % С, 1,22 % Сг, 0,82 % Мп н 0,11 % V: 1 — начало образования феррита; 2— начало образования перлита; 3 — конец образования перлита; 4—начало образования бейнита; 5 — конец образования бейннта; б — сталь с 0,43 % С и 3,52 % Сг: 1 — начало образования перлита; 2 — конец образования перлита; 3 — начало образования бейиита; 4 —конец образования бейиита
перлитного и бейиитного превращений в этих сталях могут частично перекрываться. Например, на рис. 135, а показано, что при 500 °C через 100 с начинается бейнитное превращение, которое примерно через 60 мин от начала изотермической выдержки переходит в перлитное.
У высоколегированных сталей С-кривые перлитного и бейиитного превращения могут быть разделены температурным интервалом высокой устойчивости переохлажденного аустенита, в котором перлитный распад не происходит в течение многих часов, а для бейиитного превращения переохлаждение еще недостаточно велико (рис. 135, б). Максимальную температуру на С-кривой начала бейиитного превращения обозначают точкой Бп (или Bs).
281
Кинетика бейнитного превращения похожа на кинетику перлитного не только наличием инкубационного периода, но и характером нарастания объема во время изотермической выдержки: вначале доля превращенного объема аустенита нарастает с ускорением, а затем с замедлением (см. кинетическую кривую на рис. 68). Вместе с тем во многих легированных сталях бейнитное превращение имеет характернейшую особенность мартенситного превращения — оно не доходит до полного исчезновения аустенита. Каждая точка на С-кривой конца бейнитного превращения (например, кривой 4 на рис. 135, б) соответствует определенному (и разному) количеству остаточного аустенита. С удалением температуры превращения от точки Бн количество бейни-та возрастает (от нуля при температуре Бк), а количество остаточного аустенита уменьшается (здесь аналогия с уменьшением количества остаточного аустенита при понижении температуры мартенситного превращения, см. § 36).
В большинстве сталей при бейнитном превращении существенно возрастает содержание углерода (и период решетки) в аустените. В некоторых сталях при увеличении изотермической выдержки концентрация углерода в аустените начинает снижаться, а в высокоуглеродистых сталях она снижается с самого начала бейнитного превращения, что связано с выделением карбида из аустенита.
Аустенит, оставшийся непревращенным в бейнитном интервале, при охлаждении стали до комнатной температуры частично превращается в мартенсит, причем точка Л1Н оказывается ниже, чем у той же стали при образовании мартенсита из исходного аустенита. Аустенит после бейнитного превращения неоднороден по содержанию углерода, и мартенсит образуется в участках, обогащенных углеродом.
Бейнитное превращение, как и перлитное, может совершаться не только при изотермической выдержке, но и при непрерывном охлаждении (см. ниже на рис. 138 пересечение кривой охлаждения с кривой начала бейнитного превращения на термокинетической диаграмме).
Размер аустенитного зерна практически не сказывается на кинетике бейнитного превращения.
У углеродистых сталей бейнитное превращение не обособлено от перлитного. Например, в эвтектоидной стали перлит и бейнит образуются в интервале температур —400—* 600 °C. При более высоких температурах в структуре доминирует продукт перлитного превращения, а небольшие участки верхнего бейнита трудно выявить. При более низких температурах в структуре доминирует бейнит.
282
Тщательно поставленные эксперименты показали, что начало распада аустенита при перлитном и бейнитном превращениях в углеродистых сталях характеризуется раздельными С-кривыми, но так как инкубационные периоды у этих превращений при одной и той же температуре очень близки, то распад аустенита углеродистых сталей изображают единой С-кривой (см. рис. 80).
>. Механизм бейнитного превращения
Бейнитное превращение включает следующие основные процессы: у->а-перестройку решетки, перераспределение углерода, выделение карбида. Механизм бейнитного превращения все еще остается дискуссионным.
Спорными являются следующие важные вопросы: 1) в какой последовательности совершаются процессы перестройки, перераспределения углерода и выделения карбидов; 2) каков механизм образования феррита; 3) из какой фазы (аустенита или феррита) выделяется карбид.
Большинство исследователей считают, что во всем температурном интервале бейнитного превращения феррит образуется из аустенита по мартенситному механизму. В пользу этого представления говорят следующие факты: образование рельефа на плоской полированной поверхности образца (при перлитном превращении рельеф не возникает), присутствие в легированных сталях остаточного аустенита после окончания бейнитного превращения (перлитное превращение всегда доходит до исчезновения аустенита), сходство микроструктур нижнего бейнита и отпущенного мартенсита, близость кристаллографии феррита в бейните и мартенсита, сходство субструктур верхнего бейнита и малоуглеродистого мартенсита.
Механизм бейнитного превращения, своеобразие его кинетики, близость и к перлитному, и к мартенситному превращениям можно объяснить следующим. Бейнитное превращение происходит при температурах ниже 500—450 °C, т. е. ниже порога рекристаллизации железа. Это означает, что в бейнитном интервале практически полностью подавлено диффузионное перемещение атомов основного компонента— железа. Поэтому становится невозможным образование феррита путем неупорядоченной у—>а-перестройки, т. е. подавляется перлитный распад. Но выше 200—250 °C еще достаточно активно идет диффузия углерода, которая делает возможным выделение карбида из аустенита и феррита.
Имеются доказательства того, что в инкубационный период в исходном аустените углерод перераспределяется пе-/
283
ред бейнитным превращением. Участки аустенита с низкой концентрацией углерода претерпевают мартенситное у-+а-превращение.
Кристаллы а-фазы образуются в промежуточном интервале температур путем когерентного роста при упругой связи с исходной у-фазой, т. е. точно так, как растут пластины мартенсита при превращении ниже мартенситной точки Л4Н. Но в отличие от чисто мартенситного превращения, для которого характерно «мгновенное» образование пластин мартенсита, при бейнитном превращении пластинки а-фазы растут сравнительно медленно. Это объясняется тем, что в промежуточном интервале температур a-фаза может образоваться только из у-фазы, обедненной углеродом, и, следовательно, скорость роста кристаллов a-фазы контролируется скоростью диффузионного отвода атомов углерода. При отводе этих атомов точка _Л4Н в аустените непосредственно у границы с a-фазой повышается и мартенситная перестройка у->а идет при температуре, которая выше точки Мн, характерной для данной стали.
В момент мартенситного превращения концентрация углерода не меняется, а изменяется только решетка и образуется пересыщенный а-раствор. Поэтому сейчас же после у->а-превращения начинается выделение карбида (цементита или е-карбида) из а-раствора, т. е. фактически происходит отпуск (см. § 51).
В участках аустенита, которые перед у—н%-превращени-ем обогатились углеродом, может выделиться карбид.
Легирующие элементы, растворенные по способу замещения, не успевают перераспределиться при бейнитном превращении, и содержание их в феррите и аустените одинаково.
Разницу между строением верхнего и нижнего бейнита связывают с разной подвижностью углерода в верхней и нижней части температурного интервала бейиитного превращения.
При высоких температурах бейиитного превращения а-фаза содержит мало углерода, успевающего отводиться в аустенит от продвигающейся границы a/у, и карбид выделяется прямо из аустенита между пластинами а-фазы — формируется верхний бейнит. При низких температурах превращения из-за меньшей подвижности углерода а-фаза пересыщена им и частицы карбида выделяются внутри а-пластин — формируется нижний бейнит.
Термин «бейнитное превращение», первоначально предложенный при изучении изотермических превращений в ста
284
лях, впоследствии был распространен на некоторые цветные сплавы: р-латуни, алюминиевые и оловянные бронзы, титановые сплавы и др.
В латуни из p-фазы при охлаждении может выделяться сс-фаза (см. рис. 103). При небольших переохлаждениях а-фаза выделяется обычным диффузионным путем. При больших переохлаждениях, например ниже 350 °C в сплаве с 41 % Zn, a-фаза образуется в виде тонких пластин, давая рельеф на полированной поверхности образца. Пластины a-фазы состоят из регулярных рядов дефектов упаковки, как и мартенсит, который в р-латуни образуется при температурах ниже 0 °C. Ориентационное соотношение решеток а-и p-фазы подобно ориентационному соотношению в латуни, испытавшей мартенситное превращение. Вместе с тем от чисто мартенситного превращения рассматриваемое выделение пластин a-фазы качественно отличается тем, что происходит диффузионное перераспределение цинка: а-фаза содержит меньше цинка, чем исходная p-фаза. Указанные особенности позволили отнести образование пластинчатой а-фазы в переохлажденной р-латуни к бейнитным превращениям.
При бейнитном превращении в р-латуни сначала происходит бездиффузионная мартенситная перестройка решетки р->а, а затем диффузионное перераспределение компонентов между а- и p-фазами вслед за медленно продвигающейся границей а/p. Бейнитное превращение в латуни, как и в стали, развивается в том температурном интервале, где подвижность атомов основного компонента (меди) уже очень мала, а у другого (цинка) еще достаточна, чтобы осуществить его отвод от продвигающейся межфазной границы мартенситной фазы.
4. Механические свойства стали с бей нит ной структурой
Бейнит прочнее перлита, причем его прочностные свойства растут с понижением температуры изотермического превращения (рис. 136).
Повышенная прочность бейнита обусловлена малым размером ферритных кристаллов, дисперсными выделениями карбида, повышенной плотностью дислокаций, закрепленных атомами углерода, и искажением решетки феррита из-за пересыщенности его углеродом и легирующими элементами.
Предел текучести бейнита связан с размером ферритных кристаллов соотношением Холла—Пётча (17), так как границы a-кристаллов служат эффективными барьерами для
285
дислокаций. С понижением температуры изотермического
превращения в бейнитном интервале из-за более медленно-
го отвода в аустенит атомов углерода от границы a/у кристаллы феррита получаются мельче, и это является одной из главных причин повышения прочностных свойств бейнита.
Частицы карбидов, находящиеся внутри a-фазы, тормозят движение дислокаций тем эффективнее, чем больше их
в единице объема и чем они равномернее распределены. В
ТЕмпЕратрра пребращения'С
Рис. 136. Зависимость механических свойств эвтектоидной стали от температуры превращения аустенита (Гензамер)
верхнем бейните карбидные частицы расположены главным образом по границам ферритных кристаллов и поэтому не вносят существенного вклада в упрочнение. С понижением температуры превращения дисперсность карбидов возрастает и они располагаются в основном внутри феррита, повышая прочность бейнита.
Феррит в бейните имеет большую плотность дислокаций, чем избыточный феррит в доэвтек-тоидной стали. Дислокации в бейнитном феррите закреплены коттрелловскими атмосферами из
атомов углерода и вносят значительный вклад в упрочнение. Твердорастворный механизм упрочнения элементами,1
растворенными по способу замещения, по-видимому, несколько повышает упрочнение бейнита.
На рис. 136 показано, что пластичность при переходе из перлитной области в бейнитную падает, а затем с понижением температуры вновь возрастает. Снижение пластичности связано с тем, что строение верхнего бейнита сравнительно грубое. Частицы карбида, расположенные по границам ферритных кристаллов, понижают пластичность бейнита. В нижнем же бейните частицы карбидов находятся внутри a-фазы, и поэтому пластичность у него более высо-
кая.
Стали со структурой нижнего бейнита, как правило, обладают большей вязкостью, чем после закалки на мартенсит и отпуска на равную твердость и прочность. Вязкость особенно высока у сталей, легированных кремнием, в которых бейнит состоит из a-фазы и обогащенного углеродом аустенита, а карбидной фазы нет.
На получении бейнитной структуры основана бейнитная закалка (см. § 41). Кроме того, в так называемых бейнит-
286
ных сталях бейнитная структура формируется при охлаждении на воздухе с температуры горячей прокатки или при простой термообработке с нагревом до аустенитного состояния и охлаждением на воздухе, В таких сталях С-кривая бейиитного превращения должна быть сильно сдвинута к оси ординат, а выделение избыточного феррита, наоборот, должно быть медленным, чтобы он не выделился при непрерывном охлаждении до начала бейиитного превращения. Этим требованиям удовлетворяет, например, малоуглеродистая сталь, легированная 0,5 % Мо и бором.
§ 40. Прокаливаемость сталей
1. Прокаливаемость и критическая скорость охлаждения
При закалке на мартенсит сталь должна охлаждаться с закалочной температуры так, чтобы аустенит, не успев претерпеть распад на феррито-карбидную смесь, переохладился ниже точки AfH. Для этого скорость охлаждения изделия должна быть выше критической. Критическая скорость охлаждения — это минимальная скорость, при которой аустенит еще не распадается на феррито-карбидную смесь.
В первом приближении критическая скорость охлаждения при закалке определяется наклоном касательной к С-кривой начала распада аустенита (t^ на рис. 137). При таком определении получается величина, примерно в 1,5 раза превышающая истинную критическую скорость. Истинную скорость икр можно получить при использовании термокинетических диаграмм. На рис. 138 представлена такая диаграмма для стали 35ГС. Пересечения кривых охлаждения с линиями термокинетической диаграммы показывают температуры начала и конца соответствующего превращения, а цифры у кривых — значения твердости по Бринеллю по окончании охлаждения. Минимальная скорость охлаждения при закалке на мартенсит стали 35ГС должна быть такой, чтобы не началось бейнитное превращение (А->Б).
Скорость охлаждения на поверхности изделия может быть больше критической, а в центре меньше. В этом случае аустенит в поверхностных слоях превратится в мартенсит, а в центре изделия испытывает перлитное превращение, т. е. деталь не прокалится насквозь. Прокаливаемость — одна из важнейших характеристик стали. Под прокаливаемостью понимают глубину проникновения закаленной зоны.
Прокаливаемость зависит прежде всего от критической скорости охлаждения. На рис. 139 изображена кривая рас-
287
пределения скоростей охлаждения по диаметру цилиндрического образца в сопоставлении с величиной критической скорости. Кольцевой поверхностный объем изделия охлаждается со скоростью больше критической, и поэтому он закален на мартенсит. Сердцевина цилиндра охлаждается со скоростью меньше, чем критическая, и поэтому она не за
Рис. 137. Определение критической скорости закалки по С-диаграмме:
и гп— скорости охлаждения центра и поверхности изделия; 1 — начало распада аустенита; 2 — окончание распада аустенита
Рис. 138. Термокинетическая диаграмма для стали 35ГС, содержащей 0,34 % С, 1 % Мп, 0,76 % Si (К. Ф. Стародубов Ю. 3. Борковский, В. В. Парусов)
калена на мартенсит. В массивной детали большого сечения после закалки можно наблюдать всю гамму структур: плавный переход от мартенсита около поверхности через троос-томартенсит, троостит и сорбит до перлита в центре.
Если центр изделия охлаждается со скоростью больше критической, то деталь закаливается на мартенсит насквозь (рис. 139, б). Как видно на рис. 139, для увеличения про-каливаемости детали данного сечения необходимо или повышать скорость охлаждения (кривая 1 сдвигается вверх), или понижать критическую скорость закалки: и в том, и в другом случае заштрихованное сечение закаленной зоны будет возрастать.
Критическая скорость охлаждения зависит от всех факторов, влияющих на скорость распада аустенита. Факторы, увеличивающие стойкость переохлажденного аустенита против распада, т. е. сдвигающие С-кривые вправо, увеличивают прокаливаемость (при сдвиге С-кривой вправо касательная к ней располагается под меньшим углом).
Так как для зарождения эвтектоида необходимы местные обогащения и обеднения у-раствора углеродом, то чем однороднее аустенит, тем более устойчив он против эвтектоидного распада и тем больше прокаливаемость.
288
С укрупнением действительного аустенитного зерна уменьшается суммарная межзеренная поверхность, на которой предпочтительно начинается распад, и прокаливаемость
увеличивается.
Увеличение температуры нагрева и времени выдержки перед закалкой приводит к выравниванию концентрации у-раствора и к росту аустенитного зерна, т. е. повышает устой-
a
Рис. 139. Прокаливаемость цилиндра:
а — несквозная прокаливаемость; б — сквозная прокаливаемость; I — кривая распределения скоростей охлаждения по диаметру цилиндра; 2— критическая скорость охлаждения (заштрихован слой, закаленный на мартенсит)
чивость переохлажденного аустенита. Поэтому с ростом температуры нагрева и выдержки перед закалкой прокаливаемость стали увеличивается, причем первый фактор более эффективен.
Очень сильно на прокаливаемость влияет химический состав аустенита. С повышением концентрации углерода в аустените он делается устойчивее и критическая скорость закалки уменьшается. Наименьшей критической скоростью, т. е. наилучшей прокаливаемостью, обладают стали, состав которых близок к эвтектоидному. Более высокая критическая
скорость у заэвтектоидных сталей объясняется тем, что они закаливаются не из аустенитной области, а с температур выше Л1, но ниже АСт (см. ниже.рис. 14’3). С увеличением содержания углерода в заэвтектоидной стали концентрация его в аустените при нормальной температуре закалки (ДЦ-ЗБ-^60) К не повышается, а количество цементита растет. Частицы цементита, являясь затравкой для перлитного превращения, уменьшают устойчивость переохлажденного аустенита. Поэтому с повышением содержания углерода в заэвтектоидной стали критическая скорость закалки возрастает. Если заэвтектоидные стали закаливать с температур выше Аст (из аустенитной области), то критическая скорость охлаждения будет непрерывно уменьшаться с увеличением содержания углерода в стали, так как при этом повышается концентрация углерода в аустените.
Прокаливаемость углеродистых и низколегированных сталей значительно возрастает при введении в них бора в количестве тысячных долей процента, что нашло практическое применение.
19 И. И. Новиков
289
Увеличение прокаливаемости под влиянием очень малых количеств бора объясняется тем, что этот элемент в аустените поверхностно-активен. Он сегрегирует на границы зерен аустенита, что доказано экспериментально, и затрудняет здесь зарождение избыточного феррита. Предполагается, в самом общем виде, что бор, сегрегируя на границу, неким образом изменяет строение тех микроучастков границы, которые являются потенциальными местами зарождения избыточного феррита.
Сталь одной марки, но разных плавок обладает различной прокаливаемостью, что объясняется различием в размере аустенитного зерна, влиянием неконтролируемых количеств растворенных примесей и включений оксидов, нитридов, сульфидов и др.
За исключением кобальта, все легирующие элементы, растворенные в аустените, затрудняют его распад, уменьшают критическую скорость закалки и улучшают прокаливае-мость. Для этой цели широко используют добавки марганца, никеля, хрома и молибдена. Особенно эффективно комплексное легирование, при котором полезное влияние отдельных элементов на прокаливаемое™ взаимно усиливается. Например, для стали с 0,4 % С и 3,5 % Ni критическая скорость закалки равна 150 К/с, а добавление 0,75 % Мо снижает эту скорость примерно до 4 К/с.
Увеличение прокаливаемое™ при легировании используют в двух направлениях. Во-первых, применение легированной стали обеспечивает сквозную прокаливаемое™ в таких больших сечениях, которые невозможно прокалить насквозь, если использовать углеродистую сталь. Например, при закалке в воде стали 45 критический диаметр равен 20 мм, в то время как изделия из стали 40ХН2МА диаметром 120 мм прокаливаются насквозь при охлаждении в масле. Во-вторых, для изделий небольшого сечения замена углеродистой стали легированной позволяет перейти к менее резкому закалочному охлаждению. Применяя углеродистую сталь, можно прокалить насквозь изделие небольшого сечения при закалке в воде. Но при этом могут возникнуть недопустимо большие остаточные напряжения, а также коробление и трещины, особенно в изделиях сложной формы. С использованием легированной стали закалку в воде можно заменить более мягкой закалкой в эмульсии, масле или даже на воздухе.
2. Характеристики прокаливаемое™
Простейшей характеристикой является глубина прокаливае-мости изделия в определенном охладителе. Глубину прокаливаемое™ можно определить методом пробной закалки по излому, макрошлифу и распределению твердости в сечении изделия. Закаленная на мартенсит сталь хрупка; в закаленной зоне изделие имеет ровный мелкозернистый, матово-серый, часто фарфоровидный излом. Непрокаленная
сердцевина—более вязкая; у нее неровный, шероховатый, слегка волокнистый излом. Граница между этими двумя зонами очень хорошо видна в изломе.
На макрошлифе закаленная и непрокаленная зоны травятся по-разному и поэтому различаются довольно четко.
Распределение твердости по сечению закаленного цилиндра показано на рис. 140. Характерно интенсивное изменение твердости на определенном расстоянии от поверхности изделия, причем это
Рнс. 140. Распределение твердости по диаметру образца из стали 60, закаленного с 815 °C: 1 — закалка в воде; 2 — закалка в масле; HRC пм — твердость
расстояние соответствует границе между закаленной и непрокален-ной зонами, выявляемой по изло-
стали с полумартенситной структурой
му и макрошлифу.
Глубиной прокаливаемости условно считают расстояние от охлаждаемой поверхности до слоя с полумартенситной структурой (50 % троостита и 50 % мартенсита). В районе этого слоя на твердость очень сильно влияет расстояние от поверхности изделия. Поэтому глубину прокаливаемости как расстояние от охлаждаемой поверхности до слоя с полумартенситной структурой можно весьма точно определить, измеряя твердость изделия и пользуясь справочными данными о твердости сталей разного состава с полу мартенситной структурой (HRCnM на рис. 140). Пруток диаметром 38 мм из стали 60 при закалке в воде имеет глубину прокаливаемости 7 мм, а при закалке в масле 0,4 мм.
Характеристикой прокаливаемости может, служить также критический диаметр, т. е. диаметр максимального сечения, которое в данном охладителе прокаливается насквозь.
Для определения прокаливаемости углеродистых и леги-
19*
291
Рнс. 141. Установка (а) и образец (б) для торцовой закалки:
/ — уравнительная трубка для создания постоянства напора; 2 — напорный бачок; 3 — штатив; 4 — образец; 5 — сопло; 6 — сливная коробка
рованных сталей, за исключением закаливающихся на воз-
духе, широко применяют стандартный метод торцовой закалки (метод Джомини). Стандартный образец после на-
грева в печи быстро переносят в
Скорость охлаждения от700*С, К/с 10555
JJ6II4Z 13.5 10,0 5,9 7,5 3,6 3,0 2,5
ббгттп----1---1—“Г—1-----1---1--1—
/7LLLLI--1---1---1-----1-1----1---1-----
3 6 12 16 2k 30 36 4Z 46
Расстояние от охлаждаемого торца, мм
Рис. 142. Полоса прокаливаемости стали 40
специальную установку, в которой его охлаждают струей воды под определенным напором только с торца (рис. 141). После полного охлаждения образца по его двум диаметрально противоположным образующим сошли-фовывают площадки на глубину 0,5 мм. По длине этих площадок через 1,5—3 мм измеряют твердость по шкале HRC п строят кривую прокаливаемости в координатах
твердость •— расстояние
от торца. Чем меньше прокаливаемость стали, тем быстрее падает твердость при удалении от торца. Для разных плавок одной марки стали получается набор кривых прокали-
ваемости, так как от плавки к плавке прокаливаемость меняется. Поэтому сталь определенной марки характеризуется марочной полосой прокаливаемости (рис. 142). Индексами (цифровыми показателями) прокаливаемости марки
292
могут служить максимальная и минимальная твердость на определенном расстоянии от охлаждаемого торца, или максимальное и минимальное расстояние от торца до точек с заданной твердостью (например, «полумартенситной»).
§41. Объемная закалка сталей
При объемной закалке изделие нагревают до заданной температуры, одинаковой по всему его объему.
Рис. 143. Интервал температур на-гпева под закалку углеродистых сталей
1. Нагрев под закалку
Превращения в стали при нагреве описаны в § 22. Температуры нагрева под закалку углеродистых сталей можно выбрать по диаграмме состояния (рис. 143). Доэвтектоидные стали закаливают с температур, превышающих точку Лз на 30—50 К. Наследственно мелкозернистая сталь допускает более высокий нагрев. При перегреве наследственно крупнозернистой стали закалка дает структуру крупноигольчатого мартенсита. При закалке с температур ниже точки Д3 (неполная закалка) наряду с мартенситом остается избыточный феррит, который снижает твердость закаленной стали и ухудшает механические свойства после отпуска.
Заэвтектоидные стали закаливают с температур, превышающих точку Л1 на 35—60 К. При закалке с температур в интервале А}—Аст в заэвтекто-
идных сталях наряду с мартенситом имеется вторичный цементит, который повышает износостойкость инструмента. Нагрев до температур выше АСт вреден, так как твердость закаленной стали получается ниже из-за растворения твердых цементитных частиц и повышения количества остаточного аустенита (см. рис. 134), укрупняется аустенитное зерно, усиливается обезуглероживание поверхности и растут закалочные напряжения.
Температура нагрева под закалку большинства конструкционных и инструментальных легированных сталей находится в пределах 800—880 °C (для каждой марки стали экспериментально подбирают свой более узкий интервал закалочных температур). Стали с большим содержанием карбидообразующих элементов (W, Mo, V, Сг) закаливают с
2ча
более высоких температур, иногда близких к солидусу, чтобы перевести в аустенит большое количество труднорастворимых карбидов. Например, быстрорежущую сталь Р6М5 закаливают с температур 1200—1230 °C, а штамповую Х12Ф1 — с температур 1030—1050 °C.
Продолжительность выдержки при температуре нагрева должна быть такой, чтобы прошла гомогенизация аустенита. Скорость нагрева желательно иметь максимальной для повышения производительности. При закалке с высоких температур некоторых высоколегированных, например быстрорежущих, сталей для уменьшения термических напряжений используют ступенчатый нагрев.
2. Охлаждение при закалке
Режим охлаждения при закалке должен прежде всего обеспечить необходимую глубину прокаливаемости. В то же время режим охлаждения должен быть таким, чтобы не возникали сильные закалочные напряжения, приводящие к короблению изделия и образованию закалочных трещин.
Закалочные напряжения складываются из термических и структурных напряжений. При закалке всегда возникает перепад температур по сечению изделия. Разная величина термического сжатия наружных и внутренних слоев в период охлаждения обусловливает возникновение термических напряжений.
Мартенситное превращение связано с увеличением объема на несколько процентов. Поверхностные слои раньше достигают мартенситной точки, чем сердцевина изделия. Мартенситное превращение и связанное с ним увеличение объема происходят в разных точках сечения изделия не одновременно, что приводит к возникновению структурных напряжений.
Суммарные закалочные напряжения растут с увеличением температуры нагрева под закалку и с повышением скорости охлаждения, так как в обоих этих случаях возрастает перепад температур по сечению изделия. Увеличение перепада температур приводит к росту термических и структур, ных напряжений.
Для сталей наиболее вероятно возникновение закалочных трещин в интервале температур ниже мартенситной точки, когда появляются структурные напряжения и обру-зуется хрупкая фаза — мартенсит. Выше мартенситной точки возникают только термические напряжения, причем сталь находится в аустенитном состоянии, а аустенит пластичен.
294
Как показывает С-диаграмма, быстрое охлаждение необходимо в районе наименьшей устойчивости переохлажденного аустенита. Для большинства сталей этот район находится в интервале 650—400 СС. Выше и ниже этого интервала температур аустенит гораздо более устойчив против распада, чем около изгиба С-кривой, изделие можно охлаждать относительно медленно. Медленное охлаждение особенно важно проводить начиная с температур 300—200 СС, ниже которых в большинстве сталей образуется мартенсит. При замедленном охлаждении выше изгиба С-кривой уменьшаются только термические напряжения, а в мартенситном интервале снижаются и термические, и структурные напряжения.
В качестве закалочных сред наиболее широко используют холодную воду, 10 %-ный водный раствор NaOH или ХаС1 и масла.
В табл. 6 приведены значения скорости охлаждения небольших стальных образцов в двух температурных интервалах для различных сред. Пока не найдено такой закалочной жидкости, которая быстро охлаждала бы в перлитном интервале температур и медленно в мартенситном.
Холодная вода — самый дешевый и весьма энергичный охладитель. Он быстро охлаждает и в перлитном, и в мартенситном интервалах температур. Добавки соли или щелочи увеличивают охлаждающую способность воды в перлитном интервале. Главный недостаток воды — большая скорость охлаждения в мартенситном интервале.
Минеральное масло медленно охлаждает в мартенситном интервале (это его главное преимущество), но оно медленно охлаждает и в перлитном интервале (это его основной недостаток). Поэтому масло применяют для закалки сталей с хорошей прокаливаемостью.
Таблица 6. Скорость охлаждения стали в различных средах
Закалочная среда Скорость охлаждения, К/с, в интервалах, °C
650-—550 | | ,W—
Вода при 18 °C .... 600 270
Вода при 74 °C 30 200
10 %-ный раствор NaOH в воде при 18 °C ... 1200 300
Минеральное масло . . , 100—150 20—50
295
Нагретая вода не может заменить масло, так как нагрев резко уменьшает скорость охлаждения в перлитном интервале, но почти не изменяет ее в мартенситном интервале.
Относительно новый класс закалочных сред — водные растворы полимеров, которые в зависимости от концентрации полимера по охлаждающей способности могут занимать промежуточное положение между водой и маслом. В отличие от закалочных масел водные растворы полимеров при закалке в них стали не горят и не выделяют дыма. Кроме того, они дешевле масел. Пример—закалочная среда ПК-2, являющаяся водным раствором (0,5—2,5%) неполной железной соли полиакриловой кислоты.
3. Способы закалки
Так как нет такой закаливающей среды, которая давала бы быстрое охлаждение в интервале температур 650—400 °C и медленное охлаждение выше и главным образом ниже этого интервала, то применяют различные способы закалки, обеспечивающие необходимый режим охлаждения.
Рис. 144. Закалка через воду в масло (закалка в двух средах):
/ — нормальный режим; 2 — недодержка в воде; 3 — передержка в воде
Рис. 145. Закалка в воде и горячих средах;
1 — в воде; 2 — ступенчатая; 3 - - изотермическая
Для уменьшения скорости охлаждения в мартенситном интервале применяют закалку в двух средах (рис. 144). Вначале деталь погружают в воду п после короткой выдержки в воде переносят в масло. Поэтому такой способ закалки называют закалкой через воду в масло.
Быстрое охлаждение в воде предотвращает перлитное превращение, а последующее замедленное охлаждение в масле уменьшает закалочные напряжения в мартенситном интервале. Наиболее ответственный момент—выдержка в во
296
де, продолжительность которой устанавливают для каждого конкретного изделия. И передержка, и недодержка в воде могут привести к браку. При недодержке в воде происходит частичный или полный распад аустенита и получается заниженная твердость, а при передержке возникают более сильные закалочные напряжения, которые могут привести к короблению и образованию трещин. Несмотря на указанные недостатки, закалку через воду в масло широко применяют в производстве режущего инструмента из углеродистой стали (углеродистая сталь обладает небольшой прокаливае-мостью, и режущий инструмент из нее нельзя закаливать в масле).
Другой способ уменьшения скорости охлаждения в мартенситном интервале — ступенчатая закалка (рис. 145). Нагретое до температуры закалки изделие быстро погружают в ванну с горячей средой, а затем после некоторой выдержки выдают на воздух или погружают в холодное масло. Впервые закалка в горячей среде была описана Д. К. Черновым. В 1885 г. в известном докладе «О приготовлении стальных бронепробивающих снарядов» Д. К. Чернов сообщил, что снарядная сталь при закалке в расплаве свинца с оловом приобретает такую же твердость, как и при закалке в холодной воде.
При выборе режима ступенчатой закалки помогает С-диаграмма. Температуру горячей среды (температуру «ступеньки») выбирают вблизи мартенситной точки (на 20— 30 К выше нее) в области высокой устойчивости переохлажденного аустенита. Время выдержки в горячей среде (длина «ступеньки») должно быть меньше инкубационного периода при соответствующей температуре. Ступенчатая закалка более проста в исполнении, чем закалка через воду в масло, и дает более стабильные результаты. Другое важное преимущество ступенчатой закалки — выравнивание температуры по сечению изделия при выдержке в горячей среде. Мартенситное превращение после этой выдержки происходит при медленном охлаждении и одновременно по всему объему, в результате чего уменьшаются закалочные напряжения. Наконец, весьма сушественное преимущество — то, что сталь при температуре «ступеньки» находится в аустенитном состоянии. После извлечения нз горячей среды изделие некоторое время пластично и его можно править для устранения коробления. Это особенно ценно для тонких и длинных изделий, при закалке которых даже в горячей среде неизбежно коробление. Часто используют правку вручную, но наилучшие результаты дает правка под прессом.
29Т
Правка может продолжаться при охлаждении ниже точки так как непосредственно в момент мартенситного превращения (но не после его окончания) пластичность повы-шена.
Основной недостаток ступенчатой закалки — малая скорость охлаждения в горячей среде (сравните кривые 1 и 2 на рис. 145). Поэтому применение ступенчатой закалки к углеродистым сталям ограничено изделиями небольшого сечения (до 8—10 мм толщиной). Изделие большого сечения охлаждается в горячей среде медленно, и аустенит успевает претерпеть эвтектоидный распад. Изделия из легированных сталей, у которых меньше критическая скорость охлаждения, проще подвергать ступенчатой закалке. Так, например, ступенчатой закалкой широко пользуются при обработке инструментов и деталей машин из хромистой стали (ШХ15, ХВГиЭХС).
Разновидностью ступенчатой закалки является закалка в горячей среде, температура которой несколько ниже мартенситной точки. Более низкая температура «ступеньки» обеспечивает большую прокаливаемость, а так как количество мартенсита еще невелико, то основные преимущества ступенчатой закалки сохраняются. Но править изделие при этом уже нельзя.
При ступенчатой закалке используют три группы горячих сред: минеральные масла, расплавы селитр и расплавы щелочей.
Если длина «ступеньки», находящейся в температурном интервале бейиитного превращения, больше времени изотермического распада аустенита, то операцию термообработки называют изотермической или бейнитной закалкой {кривая 3 на рис. 145).
Резкое уменьшение закалочных напряжений и коробления — важное преимущество изотермической закалки. Кроме того, у изотермической закалки есть и другое преимущество. При бейнитном превращении в некоторых легированных сталях сохраняется большое количество остаточного аустенита, который не превращается в мартенсит при охлаждении после изотермической выдержки. Изотермическая закалка таких сталей обеспечивает высокую ударную вязкость, резко уменьшает чувствительность к надрезу по сравнению с закаленной на мартенсит и отпущенной сталью. Следовательно, изотермическая закалка позволяет повысить конструктивную прочность стали. Ниже сравниваются «свойства стали ЗОХГС после обычной закалки с отпуском и изотермической закалки:
298
МПа G_ о. МПа в 0’2 Дж/см2
Закалка с 880 °C в воде + отпуск 520 СС........................... Н00 850 45
Изотермическая закалка с 880 °C в селитре при 300 °C . . . 1650 1300 60
4. Закалка с обработкой холодом
Во многих сталях мартенситный интервал (Мп—Мк) простирается до отрицательных температур (см. рис. 111). В этом случае в закаленной стали содержится остаточный аустенит, который можно дополнительно превратить в мартенсит, охлаждая изделие до температур ниже комнатной. По существу такая обработка холодом (предложена в 1937 г. А. П. Гуляевым) продолжает закалочное охлаждение, прерванное при комнатной температуре, которая не является критической для металла.
Обработку холодом можно проводить всегда, когда точка Мн лежит ниже нуля. Эффект обработки холодом зависит от количества остаточного аустенита при комнатной температуре. С увеличением содержания углерода в стали мартенситный интервал снижается в область более низких температур и увеличивается количество остаточного аустенита, который превращается в мартенсит при охлаждении закаленной стали до температуры нижней мартенситной точки Мк. Охлаждение стали ниже точки Мк не имеет смысла, так как оно не приводит к дополнительному мартенситному превращению.
Основное назначение обработки холодом — стабилизация размеров изделий. Структура закаленной стали с большим количеством остаточного аустенита не стабильна. Уже при комнатной температуре, а тем более при небольшом климатическом понижении температуры остаточный аустенит постепенно превращается в мартенсит. Это превращение сопровождается увеличением объема, и размеры изделия меняются. У таких изделий, как шарико- и роликоподшипники, калибры и другой измерительный инструмент, размеры должны выдерживаться с точностью до микрометра. Эти изделия для стабилизации размеров обрабатывают холодом. Во многих случаях даже не требуется глубокого охлаждения ниже нуля, а достаточно охладить закаленную деталь в воде с температурой от +5 до» + 10 °C.
Другое назначение обработки холодом •— повышение твердости и износостойкости режущего инструмента, штампов и измерительного инструмента.
299
Обработка холодом повышает твердость и износостойкость и устраняет шлифовочные трещины в деталях из легированных конструкционных сталей, подвергнутых цементации. В высокоуглеродистом цементованном слое после закалки содержится значительное количество аустенита, который уменьшает твердость стали и вследствие распада которого во время шлифования появляются трещины.
Обработкой холодом можно повысить магнитные свойства постоянных магнитов в результате дополнительного перехода парамагнитного аустенита в ферромагнитный мартенсит.
При обработке холодом следует учитывать явление стабилизации аустенита (см. § 36). Разрыв во времени между операцией закалки и обработкой холодом приводит в некоторых сталях к сильной стабилизации аустенита при комнатной температуре, а стабилизация уменьшает эффект обработки холодом. Поэтому предельно допустимый разрыв регламентируют. Так, для измерительных плиток из стали X он не должен превышать 30 мин.
Обычно для обработки холодом требуются температуры не ниже —80 °C.
5. Закалка с температур межкритического интервала
Длительное время считалось, что недогрев до температуры Ас% при закалке должен приводить к браку. Действительно, при такой неполной закалке среднеуглеродистых сталей из-за присутствия в их структуре мягкого феррита прочность и твердость получаются ниже, чем после полной закалки из аустенитной области.
С конца 70-х годов закалку с температур межкритического интервала Асх—Ас3 начали подробно изучать в связи е разработкой и все более широким применением, особенно в автомобилестроении, так называемых двухфазных феррито-мартенситных сталей (сокращенно ДФМС). Эти низкоуглеродистые и низколегированные стали (типичные марки 06ХГСЮ, 06Г2СЮ) закаливают с температур а+у-области для получения мелкозернистой ферритной матрицы с равномерно распределенными в ней островками мартенсита, объемная доля которого составляет 15—25 %. За рубежом такие материалы называют сталями с «дуальной структурой».
ДФМС отличаются высокой технологической пластичностью при холодной штамповке листовых деталей и холодной высадке крепежных деталей. Хорошая штампуемость ДФМС обусловлена тем, что благодаря специфике состава 300
и структуры у них низкий предел текучести, отсутствует площадка текучести на кривой растяжения, низкое отношение о02/ов и соответственно большое равномерное удлинение. Большой коэффициент деформационного упрочнения обеспечивает достаточно высокую прочность после изготовления детали холодной штамповкой или высадкой.
Закалка доэвтектоидных сталей с температур межкритического интервала характеризуется следующими особенностями, прямо вытекающими из диаграммы состояния. В стали заданного состава с повышением температуры закалки в межкритическом интервале возрастает объемная доля аустенита и уменьшается содержание в нем углерода; соответственно увеличивается объемная доля мартенсита, уменьшается концентрация в нем углерода, повышаются критическая скорость охлаждения при закалке и температура начала мартенситного превращения.
Для уменьшения предела текучести ДФМС желательно феррит исходной структуры частично заменить на перекристаллизованный феррит, получающийся при охлаждении аустенита с температуры нагрева в межкритическом интервале и содержащий меньше кремния и фосфора, которые снижают пластичность. Увеличение доли перекристаллизованного феррита достигается повышением температуры нагрева в межкритическом интервале и проведением охлаждения в две стадии: сначала замедленно, со скоростью 5— 30 К/с, и затем в воде. В первую стадию охлаждения из аустенита выделяется «новый» феррит, а во вторую образуется мартенсит. При этом возникает эффект саморегулирования структуры: чем выше температура нагрева, тем больше аустенита в стали, меньше содержание в нем углерода и меньше устойчивость такого аустенита, т. е. больше получается феррита на стадии замедленного охлаждения. В результате если в момент начала охлаждения объемная доля аустенита была равна 50—65 %, то после закалки получается около 25 % мартенсита.
Конкретный режим термообработки (температуру в межкритическом интервале и скорость замедленного охлаждения) выбирают для каждой стали с учетом требуемого комплекса технологических и эксплуатационных свойств. Например, при количестве мартенсита 10—18 %ов^450 МПа, а при количестве мартенсита 20—28 % ав^550 МПа, но равномерное удлинение снижается с ростом объемной доли мартенсита.
§ 42. Поверхностная закалка сталей
Многие изделия должны иметь высокую поверхностную твердость, высокую прочность поверхностного слоя и вязкую сердцевину. Такое сочетание свойств на поверхности и внутри изделия достигается поверхностной закалкой. Для поверхностной закалки стального изделия необходимо нагреть до температур аустенитной области только поверхностный слой заданной толщины. Этот нагрев должен совершаться быстро, чтобы сердцевина вследствие теплопроводности также не прогрелась до закалочных температур. Разновидности поверхностной закалки различаются способами нагрева.
1. Закалка с индукционным нагревом
Как известно, с увеличением частоты переменного тока возрастает неравномерность распределения его по сечению проводника. При высоких частотах можно считать, что ток практически идет лишь в тонком поверхностном слое. На использовании этого поверхностного эффекта основан индукционный высокочастотный нагрев для поверхностной закалки, предложенный в 1985 г. В. П. Вологдиным. Изделие помещают в индуктор с током высокой частоты. Для конструкционных сталей можно принять, что выше точки Кюри глубина проникновения тока, мм
6~500///, (33)
где f — частота тока, Гц. В слое этой толщины выделяется примерно 87 % всей тепловой энергии. Следовательно, глубина проникновения тока уменьшается с увеличением его частоты. Так, например, для стали 45 при 850 °C эта зависимость иллюстрируется следующими данными:
/, Гц . . . 50 10000 70000 400000
6, мм . . . 80 5,5 2,1 0,9
После нагрева в индукторе на разогретую поверхность из спрейера выбрызгивается закалочная жидкость (спрейе-ром может служить сам индуктор в виде полой трубки с отверстиями).
Скорость высокочастотного индукционного нагрева может доходить до 300—500 К/с, что на 2—4 порядка (!) больше скорости обычного печного нагрева. Высокая скорость индукционного нагрева обусловливает важные особенности фазовых превращений и получающейся структуры.
На рис. 78 видно, что с повышением скорости нагрева фазовые превращения смещаются в область более высоких 302
температур. К этому следует добавить, что в доэвтектоид-ных сталях повышение температуры при индукционном нагреве как бы обгоняет диффузию углерода, в результате чего избыточный феррит превращается в малоуглеродистый аустенит.
Следовательно, температура высокочастотной закалки должна быть выше температуры закалки при обычном печном нагреве, и тем выше, чем больше скорость нагрева и гпгбее выделения избыточного феррита. Например, сталь 40 при печном нагреве закаливают с температур 840—• 860 °C, при индукционном нагреве со скоростью 250 К/с — с 880—920 °C, а при скорости нагрева 400 К/с — с 930— 980 °C.
Высокочастотной закалке желательно подвергать сталь с измельченной исходной структурой, для чего можно использовать нормализацию, а в отдельных случаях и улучшение, т. е. обычную сквозную закалку с высоким отпуском на сорбит.
С увеличением степени перегрева скорость зарождения центров аустенита растет быстрее линейной скорости их роста. Поэтому в условиях высокочастотного нагрева, отличающихся сильным перегревом выше точек и Лз и отсутствием выдержки при максимальной температуре, образуется очень мелкое аустенитное зерно. Его размер может находиться в интервале 2—7 мкм (до № 14 по ГОСТу), в то время как при печном нагреве под закалку обычно образуются аустенитные зерна размером более 10 мкм (№ 7— 10).
Из-за большой скорости нагрева и отсутствия выдержки при максимальной температуре цикла диффузионные процессы оказываются незавершенными, и углерод распределен неоднородно в микрообъемах аустенита. Например, в стали 40 могут образоваться микроучастки аустенита, содержащие 1,7 % С.
Очень мелкое зерно, т. е. большая протяженность межзеренных границ, и неоднородность в распределении углерода делают аустенит менее устойчивым при перлитном превращении. Поэтому после индукционного нагрева требуется интенсивное закалочное охлаждение.
В микрообъемах аустенита с разной концентрацией углерода образуются кристаллы мартенсита, также различающиеся по составу. В участках аустенита с низкой концентрацией углерода точка Л4Н высокая (см. рис. 113) и могут выделяться дисперсные карбиды при охлаждении мартенситных кристаллов в интервале от Мп до комнатной
303
температуры. Аустенит с очень мелким зерном превращается в очень мелкие кристаллы мартенсита.
Указанные выше особенности строения мартенсита в стали после индукционного нагрева обусловливают повышенное сопротивление хрупкому разрушению и даже несколько большую твердость (на 4—6 ед. HRC выше, чем при закалке с печным нагревом).
Закалку с высокочастотным индукционным нагревом, отличающуюся большой производительностью, широко применяют для повышения твердости поверхности изделий, увеличения сопротивления изнашиванию и усталости. Повышению предела выносливости способствуют остаточные сжимающие напряжения в поверхностных слоях. Так как глубина проникновения тока обычно не превышает глубины прокаливаемости углеродистых сталей, то не требуется использовать легированные стали с повышенной прокали-ваемостью. Высокочастотную закалку широко применяют к деталям из дешевых углеродистых сталей, содержащих 0,4 % С и более.
Для каждой конфигурации детали необходимо изготовить свой индуктор и подобрать свой режим обработки. Поэтому поверхностная закалка с индукционным нагревом рентабельна при серийном производстве однотипных деталей, как, например, в автомобильной промышленности.
2. Закалка с лазерным нагревом
Технологические лазеры начали сравнительно недавно использовать в промышленности для термической обработки металлов. Источником излучения, как правило, служат газовые СО2-лазеры непрерывного действия.
Особенностью лазерной закалки является очень высокая концентрация энергии в тонком (обычно до 1 мм) поверхностном слое. Этот слой за чрезвычайно короткое время (сотые доли секунды и менее) нагревается до высоких закалочных температур, а остальной объем изделия остается холодным. Когда нагретый участок выходит из-под пучка (например, в результате перемещения детали), он очень быстро охлаждается из-за теплоотвода в соседние холодные слои изделия.
Лазерную закалку проводят с плавлением и без плавления поверхностного слоя. Первый случай, принципиально отличающийся от всех остальных видов термической обработки, является самостоятельным видом закалки и анализируется в гл. X. Ниже кратко рассмотрена лазерная закалка без плавления поверхности. Она включает те же
304
фазовые превращения, что и обычная закалка на мартенсит. Основное отличие лазерной закалки от поверхностной индукционной закалки состоит в гораздо более быстром (на 2—4 порядка) нагреве до более высоких температур (благодаря очень высокой концентрации энергии в тонком слое) и в более быстром охлаждении (благодаря соседству холодных слоев). Скорость лазерного нагрева составляет 104—106 К/с, скорость охлаждения 103—106 К/с.
Такой тепловой режим обусловливает следующие структурные особенности: чрезвычайно мелкое аустенитное зерно, большую степень неоднородности распределения углерода в микрообъемах аустенита и соответственно мартенсита, повышенное количество остаточного аустенита, не успевшие раствориться дисперсные частицы карбидов. Дисперсная структура определяет повышенную прочность, износостойкость и сопротивление усталости поверхностного слоя.
Важные преимущества лазерной закалки — еще меньшая, чем при высокочастотной закалке, окисляемость поверхности (ничтожно время пребывания металла при высокой температуре), практически полное отсутствие коробления, возможность обработки труднодоступных участков поверхности, требующих упрочнения.
3. Закалка с нагревом пламенем горелки
При сгорании в смеси с кислородом газообразного горючего, например ацетилена, образуется пламя с высокой температурой. Газокислородное пламя с температурой 2000— 3000 °C от закалочного наконечника горелки направляют на изделие, оно быстро разогревает участок его поверхности на глубину 2—4 мм до температур аустенитной области. Затем на разогретый участок через отверстия в охлаждающей части закалочного наконечника выбрызгивают под давлением воду.
Закалку с нагревом пламенем горелки целесообразно применять тогда, когда высокочастотная и лазерная закалка нерентабельны или практически неприменимы: в условиях ремонтного хозяйства (в том числе полевых), при поверхностной закалке крупногабаритных изделий (например, прокатных валков, крупных шестерен). Недостатки этого простого способа поверхностного упрочнения — неточная регулировка толщины и температуры закаливаемого слоя, возможность сильного перегрева вплоть до оплавления.
20 И. И. Новиков
Глава X
ЗАКАЛКА С ПЛАВЛЕНИЕМ ПОВЕРХНОСТИ
Закалка с плавлением поверхности — это термическая обработка, при которой главным процессом является быстрое затвердевание поверхностного слоя с образованием мета-стабильной структуры.
При высокой концентрации энергии в тонком поверхностном слое изделия этот слой может разогреться до таких температур, что полностью перейдет в жидкое состояние, а после прекращения нагрева затвердевает с очень большой скоростью охлаждения из-за быстрого отвода теплоты внутрь холодного объема. При такой самозакалке тонкого слоя расплава (без использования специальных закалочных сред) у поверхности изделия может сформироваться структура, улучшающая его эксплуатационные свойства.
Оплавление при термической обработке всегда считалось браком, как правило, неисправимым. Рассмотренный в § 3 гомогенизационный отжиг с переходом через точку неравновесного солидуса (тоже нетрадиционная разновидность термической обработки) включает появление мпкроучастков расплава, распределенных по всему объему, слитка. Обсуждаемый здесь вид закалки связан с появлением непрерывного слоя расплава на макроучастке поверхности детали. Этот слой имеет такую небольшую толщину (обычно порядка 10-1 мм), что при его появлении и последующем затвердевании форма детали не меняется, т. е. мы имеем дело с термической обработкой изделия, а не с формированием изделия из расплава, что относилось бы к литейному производству.
Получить расплавленный слой заданной небольшой толщины на заданном участке поверхности детали можно разными способами, применяя для нагрева высокоэнергетические направленные потоки, такие как лазерное излучение и электронный пучок, которые все шире используют для поверхностного упрочнения деталей. Особенно перспективен для этой цели нагрев с помощью технологических газовых лазеров непрерывного действия (например, СО2-лазе-ров), которые в отличие от электронно-лучевого нагрева не требуют вакуума. Ценное качество лазерного нагрева при закалке с плавлением поверхности — возможность обработки строго очерченных участков, в том числе в труднодоступных местах поверхности. Ввиду высоких капитальных
306
затрат наибольший экономический эффект от применения лазерного нагрева достигается при обработке небольших ответственных участков поверхности деталей массового производства (например, из серого чугуна).
При закалке с плавлением на исходной гладкой поверхности детали могут появляться мелкие неровности—результат усадочных процессов и газовыделения при затвердевании. Так, например, после шлифования детали из серого чугуна неровности на ее поверхности имели размер 0,5 мкм, а после лазерной закалки с плавлением эти неровности достигли высоты 80 мкм. В случае необходимости для устранения подобного рода дефектов после закалки применяют хонингование.
§ 43. Общие закономерности формирования структуры при сверхбыстром охлаждении расплава
В традиционной литейной технологии скорости охлаждения при затвердевании отливок даже с очень тонким сечением не превышают 103 К/с. Охлаждение расплава со скоростями более 103—104 К/с называют сверхбыстрым или же закалкой из жидкого состояния.
Сверхбыстрое затвердевание расплава реализуется в металлургических процессах получения порошков (гранул, чешуек), в литейных процессах получения порошков (гранул, чешуек), в литейных процессах производства тонкой литой леиты и литой проволоки. Существует множество способов создания высоких и сверхвысоких скоростей охлаждения расплава в лабораторных и промышленных условиях: распыление расплава на мелкие частицы, катапультирование расплава на быстровращающийся медный цилиндр с размазыванием его в тонкие пленки па поверхности этого цилиндра, подача тонкой струи расплава на вращающийся металлический диск с непрерывным формированием тонкой ленты, вытягивание из расплава тонких нитей и др.
При закалке с плавлением поверхности деталей скорость охлаждения во время затвердевания расплавленного слоя благодаря его малой толщине и быстрому теплоотводу внутрь холодной детали обычно имеет величину порядка 104—106 К/с и может достигать при использовании специальных лазеров 1010 К/с. Поэтому закалку с плавлением поверхности, относящуюся к технологии термической обработки деталей, можно рассматривать как частный случай закалки из жидкого состояния иа собственной теплоотводящей подложке.
Чтобы понять изменения структуры и свойств в результате закалки с плавлением поверхности, необходимо предварительно изложить кратко общие сведения о формировании структуры при сверхбыстром охлаждении расплавов.
1. Микроструктура
Первичные кристаллы твердого раствора иа базе металлов при обычных скоростях охлаждения растут в форме дендритов. С увеличением скорости охлаждения (ц) усиливается разветвление дендритов, уменьшается толщина их ветвей и соответственно уменьшается размер дендритных ячеек (d) —сечений ветвей дендритов плоскостью шлифа:
d = Av~n> (34)
20*
307
где А и п — константы, мало различающиеся у разных сплавов на одной основе. Это позволяет использовать формулу (34) для оценки скорости охлаждения при кристаллизации тонких слоев расплава по измеренным значениям d.
Размер дендритов в отличие от толщины их ветвей неоднозиачио связан со скоростью охлаждения, так как сильно зависит от присутствия в расплаве зародышей кристаллизации, направления теплоотвода и других факторов. Например, при высоких скоростях охлаждения с направленным теплоотводом могут формироваться длинные (в направлении теплоотвода) и тонкие дендриты.
По достижении определенных скоростей охлаждения (~106 К/с для алюминиевых сплавов) дендритная форма роста первичных кристаллов сменяется на иедендритную: из множества центров формируются не успевшие разветвиться компактные мелкие зерна («субдендритиые зерна»). Их размер (менее 10 мкм) уменьшается с увеличением скорости охлаждения по тому же закону (34), что и размер дендритных ячеек.
Эвтектика при обычных скоростях охлаждения растет в виде колоний. Каждая эвтектическая колония — это двухфазный бикристалл, сросток двух разветвленных кристаллов разных фаз. Кристалл каждой из фаз в этом стростке имеет множество пластинчатых, стержневых и другой формы ответвлений, причем в пространстве ответвления двух фаз перемежаются и дают на шлифе, т. е. в произвольном плоском сечении, картину из чередующихся мелких участков обеих фаз.
Одна нз фаз эвтектики-—ведущая: ее ответвления растут с некоторым опережением, а между ними в расплаве формируются ответвления второй фазы. С ускорением охлаждения усиливается разветвление ведущей, а следовательно, и второй фазы и толщина ответвлений обеих фаз в эвтектической колонии уменьшается — происходит общее диспергирование структуры внутри колоний.
При достижении достаточно больших скоростей охлаждения (порядка 106 К/с) колониальное строение эвтектик исчезает. Вместо него появляется структура очень тонкого конгломерата фаз, образование которой можно объяснить зарождением при больших переохлаждениях множества центров кристаллизации обеих фаз эвтектики.
По достижении определенной скорости охлаждения выделение первичных кристаллов в виде самостоятельной структурной составляющей может быть полностью подавлено и вся структура становится квазиэв-тектической (см. аналогичное явление при образовании квазиэвтектоида, § 24). Например, в доэвтектическом сплаве Fe — 3,5 % С отсутствует первичный аустенит и виден только ледебурит, хотя согласно диаграмме состояния содержание углерода в эвтектике 4,3 %. Чем больше скорость охлаждения, тем шире область составов сплавов с квазиэвтекти-ческой структурой. Квазиэвтектика имеет колониальное строение, а при очень больших" скоростях охлаждения представляет собой структуру тонкого конгломерата фаз, кристаллы которых росли из множества центров.
2. Фазовый состав
При закалке из жидкого состояния наблюдаются разнообразные изменения фазового состава (по сравнению с равновесным), различные для разных систем, интервалов скоростей охлаждения и разных сплавов одной системы.
Вплоть до весьма больших скоростей охлаждения в первичных кристаллах твердого раствора может развиваться виутрикристаллитная ликвация: составы ранее и позднее образующихся участков твердого раствора могут резко различаться, как и при обычной кристаллизации.
308
С увеличением скорости охлаждения ветви дендритов первично кристализующегося твердого раствора становятся тоньше, а число их больше, и возрастает суммарная доля приграничных объемов в этих ветвях, обогащенных из-за внутрикристаллитной ликвации легирующими элементами (например, медью и магнием в алюминиевых сплавах). В результате суммарное содержание легирующих элементов в первичных кристаллах возрастает и соответственно уменьшается объемная доля соединений, в состав которых входят эти элементы (например. СиА12 и Mg2Si в алюминиевых сплавах).
При чрезвычайно больших скоростях охлаждения становится возможной бездиффузионная (безызбирательная) кристаллизация: состав образующегося твердого раствора такой же, как исходный состав жидкого раствора. Термодинамическая возможность кристаллизации без пе
рераспределения компонентов появляется тогда, когда расплав переохлаждается ниже температуры равенства свободных энергий жидкого и твердого растворов одинакового состава. Для анализа здесь можно воспользоваться схемой на рис. 112, заменив обозначения (3 на Ж (жидкость) и на G
Для закалки с плавлением поверхности большой интерес представляет расширение области твердых растворов при сверхбыстрой крис-
таллизации. Можно выделить три случая этого расширения, которые рассмотрим на примере эвтектических систем.
Первый случай — кристаллизация происходит без изменения числа и типа образующихся фаз (рис. 146). Так как при быстром охлаждении эвтектическая кристаллизация может начинаться при температуре значительно ниже точки эвтектического равновесия, .то состав периферийных слоев первичных a-кристаллов продолжает изменяться по пунктирному продолжению линии солидуса, заходя за точку предельной растворимости а. Состав a-фазы в эвтектике (или в квазиэвтектике) также будет иахо-
Рис. 146. Расширение области a-раствора при сверхбыстром охлаждении расплава
литься на пунктирном продолжении линии солидуса. Пример — сплавы А1—Si, в которых при самых высоких достигнутых скоростях охлаждения не найдено никаких фаз, кроме a-раствора кремния в алюминии и кремния. При скорости охлаждения порядка 10е К/с концентрация кремния в a-фазе достигает 2—3 % (в условиях равновесия предельная растворимость кремния 1,65 %).
Второй случай расширения области твердых растворов — наличие в системе метастабильного эвтектического равновесия с участием метаста-бильного соединения, например цементита (вместо графита) в системе Fc С. При метастабильном равновесии концентрация граничного раствора всегда выше, чем при стабильном (сравните точки Е и Е' на рис. 74), и поэтому кристаллизация в соответствии с метастабильиой диаграммой состояния приводит к образованию более концентрированного твердого раствора (как в виде первичных кристаллов, так и в эвтектике). В случае системы Fe—С различие в концентрациях точек Е и Е' очень маленькое, а в других системах оно может быть весьма большим. Например, в системе А1—Мп в стабильном эвтектическом равновесии с соединением АЦМп участвует a-раствор с 1,8 % Мп, а в метастабильном равновесии при более низкой температуре (с участием фазы А14Мп) находится a-раствор с ~4 % Мп.
309
Третий случай расширения области твердых растворов — бездиффу-зноиная кристаллизация, когда полностью подавлено образование второй фазы и составы твердого раствора и исходного расплава одинаковы.
Для закалки с плавлением поверхности очень важно образование новых метастабильных промежуточных фаз при увеличении скорости охлаждения. Хрестоматийный пример — образование цементитной эвтектики вместо графитной в системе Fe—С, происходящее при ускорении охлаждения в диапазоне обычных скоростей.
При высоких и сверхвысоких скоростях охлаждения из расплава могут кристаллизоваться метастабильные фазы, которых вообще нет в данной системе, или такие фазы, которые в условиях равновесия не должны встречаться в данном сплаве, но существуют в других сплавах той же системы. Число обнаруженных таких фаз при закалке из жидкого состояния очень велико, прогнозировать же их появление весьма сложно.
Практически очень важно, что при быстром затвердевании может быть полностью подавлена кристаллизация отдельных промежуточных фаз, которые ухудшают свойства изделия.
Предельный случай изменения фазового состава при быстром затвердевании — получение сплава в аморфном состоянии, в виде металлического стекла, когда подавлен процесс кристаллизации. Составы сплавов, в которых при скоростях охлаждения до 10е—108 К/с получается аморфное состояние, весьма далеки от составов стандартных конструкционных и инструментальных сплавов, поэтому проблема образования металлических стекол в связи с применением закалки с плавлением поверхностного слоя пока не представляет большого практического интереса.
§ 44. Изменение структуры и свойств при закалке с плавлением поверхности
При закалке с плавлением поверхности на стадии затвердевания происходят такие фазовые и структурные изменения, которые вообще невозможны при нагреве без расплавления. Вместе с тем во время быстрого охлаждения полностью затвердевшего поверхностного слоя протекают процессы, характерные для закалок на мартенсит и без полиморфного превращения. Благодаря сверхвысоким скоростям охлаждения при закалке с плавлением поверхности CTpyicrypa по своей морфологии и фазовому составу качественно отличается от того, что получается, если ту же деталь производить методами литья. Таким образом, в результате закалки с плавлением поверхности возникает структура, которую невозможно получить ни при закалке с нагревом в твердом состоянии, ни при литье деталей.
Номенклатура сплавов и деталей, подвергаемых закалке с плавлением поверхности, а также области применения этого нового вида термической обработки непрерывно расширяются. В настоящее время закалку с плавлением поверхности используют в основном для увеличения сопро
310
тивления разным видам износа, повышения твердости и стойкости против коррозии.
1. Закалка чугунов
Множество деталей нз серого чугуна с пластинчатым графитом, высокопрочного чугуна с шаровидным графитом и ковкого чугуна выходит из строя в результате изнашивания рабочих поверхностей. Учитывая, что в машино- и станкостроении чугун — самый дешевый литейный материал в массовом производстве, очень большой интерес представляет повышение износостойкости чугунных деталей с помощью закалки с плавлением поверхности.
При лазерном нагреве графит полностью или частично растворяется в образующемся на поверхности жидком слое, который при последующем быстром охлаждении затвердевает в виде белого чугуна. Возможны два случая формирования структуры при кристаллизации.
Первый случай — вначале образуются дендриты первичного аустенита, а затем кристаллизуется ледебурит. Эвтектика может вырождаться: ее аустенит нарастает на дендриты первичного аустенита, между которыми располагается почти непрерывная сетка из эвтектического цементита.
Второй случай — расплав при определенном режиме нагрева сильно перегревается, вследствие чего и больше переохлаждается. В результате подавляется образование первичного аустенита и весь объем расплавленного слоя затвердевает в виде аустенито-карбидной эвтектики перистого (тонкопластинчатого) строения.
В первом случае твердость поверхностного слоя HV 600—800, во втором она доходит до HV 1200. Реализовать в производстве второй случай обычно трудно из-за образования трещин.
Концентрация углерода в аустените эвтектического происхождения, а также в периферийных слоях первичных кристаллов аустенита должна быть близкой к точке предельной растворимости в системе Fe—С—Si для случая равновесия с участием карбида (~1,6°/о С). При такой концентрации углерода в аустените двойных сплавов Fe—С мартенситная точка Мп близка к комнатной температуре (см. рис. 111). В чугунах высокоуглеродистый аустенит содержит марганец, хотя и другие элементы, понижающие точку Мн до температур ниже комнатной. В результате в зоне полного расплавления после охлаждения до комнатной температуры содержится очень много аустенита (более половины объема всего слоя).
311
Участки первичного аустенита, образовавшиеся при более высоких температурах, должны содержать меньше углерода и соответственно иметь более высокую точку Ми. В этих участках происходит мартенситное превращение. Скорость охлаждения с понижением температуры сильно уменьшается, и возможен самоотпуск свежеобразованного мартенсита с появлением феррита и дисперсных частиц карбида. Таким образом, поверхностный слой после закалки состоит из аустенита, карбида эвтектического происхождения и образовавшегося при самоотпуске и а-фазы, которая может представлять собой малоуглеродистый мартенсит или феррит. Аустенит в этом слое метастабилен и так как его точка Л4Н находится вблизи комнатной температуры, то при механическом воздействии он может превратиться в мартенсит напряжения или мартенсит деформации (см. § 37).
Повышение твердости в 3—5 раз и резкое измельчение структуры поверхностного слоя серого, ковкого и высокопрочного чугунов обусловливают значительное увеличение сопротивления изнашиванию рабочей поверхности в трущихся парах. Отсутствие частиц графита, являющихся центрами эрозии в потоке со взвесью абразивных частиц, обусловливает повышение стойкости против этого вида износа.
При закалке чугунов с плавлением поверхности обычно образуются три зоны: 1) рассмотренная выше зона полного расплавления; 2) переходная зона частичного оплавления с большим количеством графита и 3) зона нагрева до температур выше Ас3 без оплавления, где при охлаждении прошла закалка на мартенсит. В третьей зоне вблизи границы со второй очень много остаточного аустенита, так как концентрация углерода в нем должна быть близка к предельной при эвтектической температуре.
Глубина зоны полного расплавления в зависимости от режима обработки (удельной мощности облучения и скорости перемещения детали относительно луча) колеблется в пределах от 0,05 до 1 мм.
Чугун •— это первый объект широкого промышленного использования закалки с плавлением поверхности.
2. Закалка сталей
Преимущества закалки сталей с плавлением поверхности по сравнению с поверхностной закалкой на мартенсит (без оплавления) менее очевидны, чем в случае чугунов. Одно из возможных направлений ее использования — измельчение первичных карбидов в легированных сталях для повы
312
шения вязкости (грубые частицы карбидов служат очагами разрушения). При обычной закалке трудно добиться существенного повышения вязкости без снижения твердости. В быстрорежущей стали Р6М5 можно наблюдать довольно крупные частицы первичных карбидов, снижающие вяз-
Рис. 147. Прорастание зерна аустенита из подложки в расплавленный поверхностный слой, образовавшийся при лазерном иагреве стали 37ХНЗМФ. Х250 (В. Д.* Садовский, Т. И. Табатчикова. В. М. Умнова. А. А. Осинцева)
кость. Если эту сталь подвергнуть лазерному нагреву до полного расплавления поверхностного слоя, то при последующем быстром затвердевании расплава (y0XJI~5-104 К/с) кристаллизация карбидов из него практически полностью подавляется, и поверхностный слой затвердевает в виде дендритов аустенита, которые прп дальнейшем охлаждении превращаются в мартенсит. Содержание углерода и легирующих элементов в этом мартенсите практически такое же, как и среднее содержание их в стали. После отжига при 770 °C в течение 4 ч из мартенсита выделяются очень дисперсные, размером менее 1 мкм, карбиды, равномерно распределенные по всему объему слоя. Отожженную заготовку можно подвергать механической обработке для получения необходимой формы режущей кромки, после чего провести обычную закалку и отпуск. В результате без снижения твердости достигается повышенная вязкость.
313
При лазерном нагреве до оплавления поверхности сталей в определенных условиях можно наблюдать эффект кристаллизации на подкладке: формирующееся из расплава зерно аустенита является продолжением зерна из пограничной нерасплавленной зоны (рис. 147).
3. Закалка сплавов цветных металлов
Закалку с плавлением поверхности можно использовать для увеличения сопротивления изнашиванию детален из силуминов. При быстром затвердевании расплавленного слоя резко измельчается микроструктура: становятся мельче кристаллы а-раствора на базе алюминия, кремния и других фаз, в том числе железосодержащих, снижающих пластичность. Кроме того, пересыщенность а-раствора оказывается увеличенной по сравнению с обычной закалкой; это — результат расширения области а-раствора при быстром затвердевании с одновременным уменьшением объемной доли избыточных фаз кристаллизационного происхождения (Si, CuAI2, Mg2Si и др.). Более высокая пересыщенность твердого раствора обусловливает больший прирост твердости при старении. Все эти факторы и определяют увеличение износостойкости силум.ипа. Твердость некоторых силуминов после такой обработки возрастает в 1,5 раза.
При закалке с плавлением поверхности многокомпонентных алюминиевых бронз -можно получить гомогенную однофазную структуру. Так, например, бронза марки Бр.АЖ9-4 в обычном состоянии состоит из а-раствора на базе меди, эвтектоида а+у2 и железосодержащей фазы. Эта фаза при закалке с плавлением поверхности не успевает образоваться из расплава (уохл^Ю6 К/с), который кристаллизуется в виде одной фазы (вероятно (3), не претерпевающий в твердом состоянии распада с образованием а- и у2-фаз. Однофазный слой имеет повышенную стойкость против коррозии и кавитационного воздействия.
Закалка с плавлением поверхности еще слабо изучена. По мере углубления наших знаний о ней, а также с увеличением доступности технологических лазерных и электронно-лучевых установок для ее проведения в промышленности применение этого нового вида термообработки, несомненно, будет расширяться. Поиск областей использования этой обработки в первую очередь следует вести применительно к сплавам, в которых при быстром затвердевании образуются новые метастабильные фазы (включая аморфные), существенно расширяется область твердого раствора, а также подавляется кристаллизация вредных фаз.
314
Раздел
четвертый
СТАРЕНИЕ И ОТПУСК
Закаленный сплав находится в метастабильном состоянии и обладает повышенной энергией Гиббса. При закалке без полиморфного превращения и в подавляющем большинстве случаев при закалке с полиморфным превращением образуется пересыщенный твердый раствор и закаленный сплав стремится понизить свою энергию Гиббса, в результате чего твердый раствор распадается. Уже при комнатной температуре могут образовываться выделения из пересыщенного раствора, однако в большинстве сплавов диффузионная подвижность атомов при комнатной температуре недостаточна, чтобы распад раствора прошел в необходимой степени за приемлемое время. Поэтому для изменения структуры и свойств закаленного сплава его нагревают — подвергают старению или отпуску.
Исторически так сложилось, что для одних сплавов, например алюминевых, использовали преимущественно термин «старение», для других, например углеродистых сталей, — «отпуск», а для третьих, например бронз и титановых сплавов, оба эти термина использовали на равных правах. Сравнительно недавно было предложено термин «отпуск» применять к тем сплавам, которые были подвергнуты закалке с полиморфным превращением, а термин «старение»— в случае закалки без полиморфного превращения. Такое классификационное деление использовано в книге.
Главным процессов при старении и отпуске большинства закаленных промышленных сплавов является распад мета-стабильного твердого раствора. При этом сплав переходит в более стабильное состояние, хотя обычно и далекое от истинного равновесия, для которого характерен абсолютный минимум энергии Гиббса. Процессы распада пересыщенного раствора в закаленном сплаве протекают самопроизвольно с выделением теплоты превращения.
Факт нагрева сплава не противоречит представлению о самопроизвольности процессов, происходящих в сплаве при старении и отпуске, так как нагрев необходим лишь для ускорения диффузии, лежащей в основе всех структурных изменений при распаде пересыщенных растворов.
315
Основные параметры старения п отпуска — температура и время выдержки. Скорости нагрева и охлаждения обычно играют подчиненную роль. Исключение составляет специфическое явление отпускной хрупкости легированных сталей при замедленном охлаждении с температуры отпуска (см. § 53).
Глава XI
СТАРЕНИЕ
Старение — это термическая обработка, при которой в сплаве, подвергнутом закалке без полиморфного превращения, главным процессом является распад пересыщенного твердого раствора.
В 1906 г. немецкий инженер Альфред В ильм открыл явление естественного старения, обнаружив, что вылеживание закаленного сплава алюминия с медью и магнием (дуралюмина) при комнатной температуре приводит к повышению твердости.
Вильм получил патент на способ облагораживания дуралюмипов, заключающийся в закалке сплавов с последующим естественным старением, в результате которого повышаются твердость, временное сопротивление и предел текучести.
В 1919 г. американские исследователи Мерика, Вальтенберг и Скотт опубликовали знаменитую статью, в которой впервые был дан анализ природы старения дуралюмина. Мерика выдвинул гипотезу, согласно которой старение дуралюмина связано с переменной растворимостью соединения СиА12 в алюминии. При нагреве сплава соединение СиА12 переходит в твердый раствор и не успевает обратно выделиться во время быстрого охлаждения (закалки), а последующее вылеживание сплава при комнатной температуре приводит к постепенному выделению из пересыщенного раствора очень дисперсных и потому невидимых под микроскопом частиц СиА12, которые и вызывают упрочнение. Эта гипотеза просто и убедительно объяснила имевшиеся к тому времени экспериментальные данные.
Отсутствие упрочнения во время выдержки закаленного образца при —180 °C было объяснено низкой скоростью образования выделений, а появление упрочнения с повышением температуры вылеживания до комнатной и до 100 °C связывалось с тем, что подвижность атомов становилась достаточной для выделения СиА12 в дисперсной форме. Рост твердости во время старения при 200 °C, достижение максимума упрочнения и дальнейшее снижение твердости были объяснены образованием частиц СиА12 с определенной степенью дисперсности, соответствующей максимальной твердости, и последующей коагуляцией этих частиц.
Увеличение эффекта упрочнения при старении в результате повышения температуры нагрева под закалку легко было связать с более полным растворением СиА12 при нагреве, образованием более пересыщенного раствора при закалке и соответственно появлением при старении большего числа дисперсных выделений из такого раствора.
Джеффрис и Арчер в 1921 г. развили гипотезу дисперсионного твердения 'Дерика и предложили общую теорию упрочнения сплавов дисперс-316
пымн частицами любого происхождения. Согласно этой теории твердые дисперсные частицы, действуя как «шипы», «заклинивают» плоскости скольжения и вызывают упрочнение сплава. При коагуляции частиц, когда суммарный их объем не изменяется, а лишь уменьшается их число, многие плоскости скольжения освобождаются от «шипов» и происходит разупрочнение. Максимуму твердости и прочности соответствует некоторая критическая степень дисперсности твердых частиц. Такая степень дисперсности легко достигается при распаде пересыщенных твердых растворов.
Первоначальная теория, объяснявшая упрочнение дуралюмина при комнатном старении выделением из пересыщенного твердого раствора частиц СиА12, уже в 20-е годы столкнулась с рядом противоречий. Выделение СиА12 и соответственно обеднение твердого раствора медью должны были бы привести к снижению электросопротивления, а в действительности электросопротивление при естественном старении возрастало. Далее с выделением СиА12 из твердого раствора должен был бы увеличиваться период его решетки из-за меньшего (чем у алюминия) атомного диаметра меди, растворенной по способу замещения. Однако рентгеновские исследования не обнаружили изменения периода решетки при достижении сплавом максимальной твердости, и только после перестаривания при повышенных температурах, когда происходило разупрочнение, период решетки возрастал, что прямо указывало на выделение СиА12 из пересыщенного раствора.
Учитывая эти противоречия и большое число других новых экспериментальных факторов, Мерика в 1932 г. предложил следующее объяснение упрочнения при старении, явившееся развитием его первоначальной гипотезы. Упрочнение при любых температурах старения происходит во времени. Выдержка необходима для развития диффузионных процессов, лежащих в основе распада пересыщенного твердого раствора. При повышенных температурах диффузия атомов меди обеспечивает собственно выделение частиц СиА12, «заклинивающих» плоскости скольжения. При комнатной температуре диффузионные процессы лишь подготавливают твердый раствор к образованию кристаллов СиА12, сами же частицы СпА12 не выделяются. Мерика высказал предположение, что вследствие диффузии в пересыщенном твердом растворе образуются скопления, сегрегаты атомов меди, не обособленные от решетки матрицы поверхностями раздела. Строение этих областей в алюминиевом твердом растворе получается таким, какое необходимо для последующего возникновения здесь частиц СиА12 или же некоего промежуточного ме-тастабильного соединения. Из-за скоплений атомов меди должно происходить местное сжатие решетки, и такие сильно искаженные участки твердого раствора затрудняют скольжение при пластической деформации, вызывая упрочнение.
Эти предсказания блестяще оправдались. В 1935 г. немецкие исследователи Вассерман и Вирте на стадии максимального упрочнения сплавов А1—Си рентгенографически обнаружили образование в пересыщенном растворе промежуточной фазы (которую теперь обозначают как 6' в отличие от равновесной фазы 0-СиА12), а в 1938 г. Гинье во Франции и Престон в Англии независимо друг от друга истолковали эффекты диффузного рассеяния на лауэграммах естественно состаренных монокристаллов А1—Си как результат образования в пересыщенном растворе малых областей, обогащенных медью. Позднее эти области были названы зонами Гинье—Престона (сокращенно — зоны ГП).
Таким образом, исходное положение теории Мерика о том, что старение сплавов связано с переменной растворимостью избыточной фазы, а упрочнение при старении происходит в результате образования дис
317
персных выделений при распаде пересыщенного твердого раствора, в основе своей оказалось правильным и исключительно плодотворным, хотя это первоначальное положение со временем претерпело ряд существенных уточнений и было позднее дополнено представлениями о роли пред-выделений и промежуточных фаз.
Вся история возникновения и развития представлений о старении сплавов весьма поучительна. Открытие В ильмом старения дуралюмииа трудно переоценить, но оно само по себе не могло стать базой для разработки новых стареющих сплавов, так как совершенно не ясно было, какова природа этого загадочного для того времени явления и в каких сплавах его следовало ожидать.
Создание основ теории старения — это яркий пример того, как научная гипотеза, в общих чертах правильно отразившая природу нового важного явления, послужила фундаментом для последующего удивительно быстрого прогресса в области теоретического и прикладного металловедения. Если в период после открытия Вильмом старения дуралюмнна и до появления теории Мерика не было найдено нн одного стареющего сплава, то после опубликования этой теории уже в 20-е годы старение было предсказано и обнаружено в десятках сплавов алюминия, железа, никеля и других металлов, были разработаны новые промышленные сплавы, упрочняемые старением. Начиная с 30-х годов быстро нарастал поток информации о структурных изменениях при старении в разных группах сплавов. В последние три десятилетия наиболее важные результаты были получены с помощью метода электронной микроскопии, позволившего «увидеть» дисперсные образования.
Создание новых стареющих сплавов с высокими механическими и особыми физическими свойствами, разработка оптимальных режимов их термической обработки, продолжающиеся обширные исследования механизма и закономерностей старения в разных группах сплавов составили одно из центральных направлений в развитии современного металловедения, причем по-прежнему теоретические и прикладные работы в этой области базируются на общих исходных положениях теории старения, предложенной Мерика.
§ 45. Термодинамика процессов выделения из твердого раствора
В общем случае из пересыщенного твердого раствора выделяется фаза, отличающаяся от матрицы и химическим составом, и структурой. Рассмотрим вначале наиболее простой случай, когда выделяющаяся фаза отличается от матрицы только составом.
На рис. 148, а изображена диаграмма состояния с непрерывным рядом твердых растворов при высоких температурах. Линия MKN, часто называемая кривой расслоения (по аналогии с соответствующей кривой для жидких растворов), является границей растворимости в твердом состоянии. При охлаждении сплава до температуры ниже этой линии из исходного твердого раствора выделяется другой твердый раствор с той же кристаллической решеткой, но иным химическим составом.
318
Рис. 148. Диаграмма состояния с кривой расслоения MKN и спинодалью RKV (а) и кривые зависимости энергии Гиббса от состава при трех температурах (б)
Выше критической точки К, например при температуре Т], при любых концентрациях стабильна одна фаза, и кривая зависимости энергии Гиббса твердого раствора от его состава на любом участке обращена вогнутостью вверх (рис. 148,6). С понижением температуры энергия Гиббса п компонентов, и твердых растворов растет (см. кривую энергии Гиббса при температуре Г2). При более низких температурах (например, при Г3) и достаточно большой положительной теплоте смешен ия концентр иров а н-ных твердых растворов кривая энергии Гиббса в средней своей части выгибается выпуклостью вверх. Тогда в некоторой области системы стабильным оказывается не один твердый раствор, а смесь двух растворов разного состава. Эти составы можно определить, проведя касательную к кривой энергии Гиббса (см. § 20). Участок кривой aSiSzb расположен выше касательной ab и, следовательно, он относится к твердым растворам, неустойчивым при температуре Тз, так как на этом участке энергия Гиббса одной фазы всегда больше, чем у смеси фаз того же среднего состава (например, Gi>G2).
Если каким-либо путем при температуре Т3 получен
неустойчивый твердый раствор, то он должен распадаться на смесь двух твердых растворов, имеющих меньшую энергию Гиббса. Этот распад может идти двумя принципиально разными путями.
1. Спинодальиий распад
Рассмотрим изотермический распад термодинамически неустойчивого твердого раствора с энергией Гиббса G[ в сплаве состава Со (рис. 149). Конечное равновесное состояние с энергией Гиббса G2<Gj не может сразу возникнуть,
319
так как маловероятно, чтобы в результате флуктуаций в твердом растворе состава Со имелось много областей равновесных составов Са и СЬу далеких от Со. Более вероятно, что вначале в результате флуктуаций возникнут области с составами, например Ср и Cq, близкими к исходному составу Со. При этом энергия Гиббса уменьшится до величины G3<Gb В рассматриваемом сплаве любое сколь угодно малое расслоение по составу неустойчивого твердого раствора будет приводить к уменьшению энергии Гиббса и, следовательно, для начала распада не требуется образования критических зародышей. Поэтому такой распад, называемый спинодальным, сразу охватывает весь объем исход
ной фазы. Увеличение концентрационного расслоения должно приводить к непрерывному понижению энергии Гиббса
Рис. 149. Схема к объяснению спино-далыюго распада в сплаве Со в системе с непрерывным рядом твердых растворов
(см. серию прямых на рис. 149) до тех пор, пока не установится равновесная разность концентраций Са—Сь.
Спинодальный распад при данной температуре может идти во всех сплавах, состав которых находится в области участка кривой энергии Гиббса, обращенного вогнутостью вниз, т. е. там, где (<32G/c/C2) <0. Этот участок ограничен точками перегиба Si Иа$2, в которых (d2G/dC2)=0 (см. рис. 148, б). Такие точки называют спинодальными. При
повышении температуры спинодальные точки Si и S2 на изотермических кривых энергии Гиббса постепенно сближаются и, когда температура достигает критической (К на рис. 148, а), перегибы исчезают—кривая энергии Гиббса во всех участках обращена вогнутостью кверху [ (d2G/ /дС2)>0]. Если на диаграмме состояния при разных тем-
пературах отметить составы, отвечающие спинодальным точкам (например, Sj и S’, на рис. 148, а), то получим кривую RKV, называемую спинодалью. Твердый раствор, бу-
дучи переохлажден до температуры ниже спинодали, может претерпевать спинодальный распад.
В рассмотренной схеме на любых стадиях спинодального распада отсутствует энергетический барьер. В действительности же такой барьер может возникать из-за появле-
320
нпя энергии упругой деформации решетки. Участки твердого раствора с разной концентрацией, хотя и характеризуются однотипным строением, но отличаются удельными объемами. Так как граница между этими участками когерентная, то с ее появлением связана упругая деформация сопряжения участков с разным периодом решетки.
Возникающая при спинодальном распаде упругая энергия вносит положительный вклад в энергию Гиббса, что не учитывалось схемой на рис. 148, б. Этот факт может обусловить необходимость для начала спннодального распада дополнительного переохлаждения исходного твердого раствора на десятки и сотни градусов против положения «химической спинодали» RKV иа рис. 148, а, В отличие от нее расположенную ниже линию температур начала спп-нодального распада, рассчитанных с учетом упругой деформации на когерентных границах фаз, называют «когерентной» спинодалъю.
Первоначальная идея о спинодальном распаде, выдвинутая на примере жидких растворов Гиббсом в XIX в., длительное время рассматривалась в классических курсах термодинамики. Затем, когда с середины 20-х годов быстро распространилась теория кристаллизации путем образования и роста зародышей новой фазы, спинодальный распад был почти забыт. В последнее время интерес к нему вновь возник, в частности в связи с возможностью получения при термообработке дисперсных продуктов распада, равномерно распределенных по объему сплава (см. § 46).
2. Распад по механизму образования и роста зародышей
Проанализируем изотермический распад твердого раствора, состав которого Со не лежит в спинодальной области (рис. 150). Его энергия Гиббса Gi больше энергии Гиббса равновесной двухфазной смеси G2. Если в результате флуктуаций на ранних стадиях распада образуются две фазы с составами Cf и CQ, близкими к Со, то энергия Гиббса такой двухфазной смеси будет выше, чем у исходного раствора Со (Gs>Gi). Это неизбежно, так как кривая энергии Гиббса вне спннодального интервала обращена вогнутостью вверх. Только при появлении большой разницы по составу, например Сп,—Сп, энергия Гиббса понижается (G4<Gij.
Таким образом, в отличие от спннодального распада, при котором энергия Гиббса непрерывно снижается, в рассматриваемом случае превращение сопровождается вначале повышением, а затем снижением энергии Гиббса, т. е.
21 И. И. Новиков
321
Рис. 1.50. Схема к объяснению распада по механизму зарождения и роста в сплаве Q в системе с непрерывным рядом твердых растворов'
существует термодинамический барьер образования достаточно больших участков новой фазы даже без учета роли межфазной энергии и энергии упругой деформации решетки. Следовательно, в рассматриваемом случае для того, чтобы распад раствора начался и самопроизвольно протекал с уменьшением энергии Гиббса, необходимы зародыши.
Распад такого типа не имеет особого краткого наименования, так как является обычным, наиболее распространенным в металлических сплавах. Иногда его называют распадом по механизму образования и роста зародышей, чтобы отличить от спинодального распада. В системах с расслоением в твердом состоянии он протекает в области между кривой расслоения MKN и спи-
нодалыо RKV (см. рис. 148). В таких системах, в которых для спинодального распада требуется переохлаждение ниже когерентной спинодали, обычный распад может протекать и при температурах ниже химической спинодали.
В то время как спянодальный распад более вероятно встретить в системах с расслоением, где решетка новой фазы такая же, как и у исходной, обычный распад происходит в любых системах с переменной растворимостью компонентов в твердом состоянии. Наибольшее практическое значение для разработки стареющих сплавов имеют системы с промежуточными фазами (соединениями). Выделяющаяся в них фаза отличается от исходного твердого раствора не только составом, ио и типом кристаллической решетки. Все, что сказано по поводу схемы обычного, не спинодального распада в системе с расслоением (см. рис. 150) относится и к системе с промежуточной фазой (рис. 151). Обозначения на обоих графиках одинаковы. Различие между ними состоит лишь в том, что на рис. 151 кривая энергии Гиббса второй фазы Gp — самостоятельная и не продолжает соответствующую кривую матричного раствора а.
Выше никак не учитывалась роль межфазной энергии и энергии упругой деформации в образовании новой фазы по механизму зарождения и роста. На рис. 150 и 151 разность Gi—G2 является термодинамическим стимулом превраще’
?-22
вия. Ее роль аналогична роли Sg на рис. 59 в случае фазового превращения без изменения состава. Величина AGO6= = Gi—G2 представляет уменьшение при распаде раствора энергии Гиббса, приходящейся на единицу объема (Gj и Gj —удельная объемная энергия Гиббса соответственно исходной фазы и равновесной смеси фаз). Но в общее изменение энергии Гиббса при распаде твердого раствора AG вносят вклад еще два слагаемых — приращение поверхностной (межфазной) энергии ДОПов и повышение энергии
Гис. 151. Схема к объяснению выделения P-фазы из <Х’раство,)а по меха-
низму зарождения и роста в сплавах G и С '
с*
Рис, 132, Размер кр:г. лческз, с зародыша при разной иересышенно-сти твердого раствора (схема)
Гиббса из-за возникновения упругой деформации матрицы и новой фазы AGynp при образовании кристаллов в упругой среде: AG =—Д6об+АОпов+АбуПр. Образование новых поверхностей и появление упругой деформации препятствуют распаду раствора.
При анализе фазового превращения без изменения состава в § 17 было доказано, что с повышением степени переохлаждения из-за увеличения AGo6 уменьшаются размер критического зародыша акр и работа его образования AGKP. Можно показать, что в случае выделения из твердого раствора, происходящего с изменением состава, критический размер зародыша и работа его образования также уменьшаются с ростом AGog. Например, на рис. 151 в более легированном сплаве С'о движущая сила распада больше, чем в сплаве C0(Gi—G2)>Gi—G2. Поэтому при постоянной температуре старения Ti размер критического зародыша новой фазы уменьшается с ростом концентрации исходного твердого раствора, т. е. с увеличением степени его
21* 323
пересыщения C0/Ci (рис. 152). При постоянной концентра’ дни исходного твердого раствора, например в сплаве Со (см. рис. 152), размер критического зародыша возрастает с повышением температуры старения, так как при этом уменьшается пересышенность раствора.
§ 46. Структурные изменения при старении
Основные структурные изменения при старении сводятся к разным этапам распада пересыщенного твердого раствора, полученного в результате закалки сплава. Так как распад пересыщенного раствора является диффузионным процессом, то степень распада, тип выделений из раствора, их дисперсность, форма и другие структурные характеристики зависят от температуры и продолжительности старения и, конечно, от природы сплава, его химического состава по основным компонентам. Кроме того, па структуру состаренного сплава влияют примеси, температура нагрева и скорость охлаждения при закалке, пластическая деформация после закалки (перед старением), продолжительность вылеживания закаленного сплава при комнатной температуре перед искусственным старением и другие факторы.
Зависимость структуры состаренного сплава от большого числа факторов и многостадийность процесса распада пересыщенного твердого раствора в сочетании с высокой дисперсностью выделений, особенно на начальных этапах распада, весьма осложняют изучение структурных изменений при старении. Основные методы изучения строения состаренных сплавов — электронная микроскопия и рентгеноструктурный анализ.
Кроме того, полезные данные получают, изучая изменение механических и физических свойств, особенно электросопротивления, при старении. Это изучение свойств позволяет делать предположения, а в отдельных случаях и выводы о характере и последовательности структурных изменений, прежде всего на ранних стадиях' распада, когда возможности прямых структурных методов ограниченны.
1. Типы выделений
В зависимости от строения поверхности раздела между выделением и матрицей различают три типа выделений: полностью когерентные, частично когерентные и некогерентные, У полностью когерентного выделения вся поверхность раздела с матрицей когерентная и решетка матрицы вокруг
324
выделения упруго искажена (рис. 153, а). У частично когерентного выделения хотя бы одна из границ с матрицей когерентная, а остальные могут быть полукогерентными (рис. 153, б) или даже некогерентными. Некогерентное выделение не имеет ни одной когерентной границы с матрицей
Рис. 153. Схема строения матрицы с полностью когерентным (а), частично когерентным (б) и некогерентным (в) выделениями
(рис. 153, в). В стареющих сплавах А1—Си примером полностью когерентных выделений являются зоны ГП и 0"-фа-за, частично когерентных б'-фаза и некогерентных СиА12 (их структура рассмотрена ниже, см. рис. 166).
2. Форма и пространственное расположение выделений
Форма выделении
В стареющих сплавах выделения из твердого раствора встречаются в следующих основных формах: тонкопластинчатой (обычно дискообразной), равноосной (обычно сферической или кубической) и игольчатой.
Форма выделений определяется двумя конкурирующими факторами: поверхностной (межфазной) энергией и энергией упругой деформации, стремящихся к минимуму. Требование минимума межфазной энергии обусловливает стремление к равноосной форме выделений и к появлению граненых форм с наименьшим поверхностным натяжением на всех гранях. Энергия упругих искажений минимальна для выделений в форме тонких пластин. В зависимости от того, какой из указанных двух факторов преобладает, форма выделений ближе к равноосной или тонкопластинчатой.
У полностью и частично когерентных выделений упругая деформация, обеспечивающая плавную сопряженность решеток на когерентной границе, распространяется от нее в глубь матрицы и выделения (рис. 153, а, б). Энергия упругой деформации решеток матрицы и выделений тем
325
Таблица 7. Форма зон'Гинье—
Престона в разных системах
□д ног ииЛор Система ; Газьицэ г. атомных диаметрах, (M /0
Сфера Диск Игла Al—Ag 1 Al—Zn Al—Zn—Mg Cu—Co Al—Cu Cu—Be Al—Mg—Si Al—Cu—Mg 1 -4-0,7 — 1,9 -4-2,6 —2,8 — 11,8 —8,8 + 2,5 —6,5
ния. Иногда когерентные выделения имеют
больше, чем больше структурное несоответствие этих решеток. При разнице в атомных диаметрах компонентов твердого раствора замещения, не превышающей 3 %, форма когерентны х в ы де л ен и и он р ед ел яе т-ся требованием минимума межфазной энергии и близка к сферической, а при разнице ^5 % решающим фактором является повышенная энергия упругих искажений и образуются тонкопластинчатые (чаще всего дискообразные) выделе-иглообразную
форму, которая отвечает большей энергии упругой деформации, чем дискообразные выделения, но меньшей, чем
равноосные. Примеры разных форм зон Гинье — Престона (когерентных выделений) приведены в табл. 7.
Рис. 154. Зависимость энергии упругой деформации матрицы Е от соотношения осей с;а не-когерентиого выделения, имеющего форму сфероида (Набар-ро)
При образовании некогерентного выделения касательные напряжения не возникают, но всегда появляются нормальные напряжения, так как из-за разницы в удельных объемах матрицы и выделения неизбежно возникновение гидростатического (всестороннего) сжатия или растяжения (на рис. 153,н не показанного). Это легко себе представить, мысленно поместив в полость податливой упругой матрицы жесткое включение с размером больше,
чения в матрице должна
чем полость: вокруг такого вклю-появиться область всестороннего
сжатия.
Расчет, выполненный для некогерентного включения в виде сфероида с полуосями а и с при условии, что вся упругая деформация сосредоточена в матрице, показал следующее (рис. 154). При образовании сферического выделения (с/а=\) энергия упругой деформации матрицы максимальна, при образовании выделения в форме тонкого
326
диска (с7а<С1) она минимальна, а при игольчатой форме (с/а^ 1) имеет промежуточное значение.
Модулированные структуры
Стремление к минимуму энергии упругих искажений влияет не только на форму, но и на взаимное расположение выделений. Требование минимума упругой энергии при распаде твердого раствора обусловливает образование в ряде сплавов так называемых модулированных (или периодических) структур, для которых характерно законо
Рис. 155. Модулированная структура в сплаве Си — 33,5 % Ni — 15 % Fe с ?.=25,4 нм после старения при 775 °C в течение 15 мин. Электронная микро-фотография, фольга (Батлер и Томас)
Рис. 156. Модулированная структура в сплаве Ni 6,7 % Ai, состаренном при 750 °C в течение 96 ч. Электронная микрофотография, фольга (Эрделл и Ннкольсон). Выделения -/(NisAI) выстроены вдоль направлений <100>
мерное пространственное расположение когерентных выделений на определенном расстоянии одно от другого, называемом периодом модуляции структуры X (рис. 155 и 156).
Так как период модуляции обычно не превышает нескольких сотен межатомных расстояний, то под световым микроскопом модулированная структура не выявляется. Наилучший метод ее изучения — электронно-микроскопический анализ тонких фольг.
В одних сплавах модулированная структура возникает на самых ранних стадиях распада, например при спино-дальном распаде, в других она появляется через некоторое время после начала распада.
327
Модулированные структуры в разных сплавах и в зависимости от режима термической обработки отличаются разной морфологией. В одних случаях видны тонкие пластины выделений, расположенные на одинаковом расстоянии одна от другой и параллельные определенной кристаллографической плоскости, в других — периодически расположенные стержиеобразные выделения, которые могут образовывать трехмерную сетку взаимноперекрывающихся в пространстве стержней, параллельных определенным направлениям в матрице, например «упругомягкому» направлению <100> в кристаллах с кубической решеткой. Такая сетка выделений часто выявляется в виде характерного узора «корзиночного плетения» (см. рис. 155). Одна из типичных разновидностей модулированных структур представляет собой ряды выделений кубической формы, параллельные направлениям <100> кубической решетки матрицы (см. рис. 156).
Модулированная структура возникает при старении таких сплавов, в которых когерентные выделения создают вокруг себя сравнительно сильные поля упругих напряжений, например из-за большой разницы в удельных объемах исходной и новых фаз. Классическим примером являются дисперсионнотвердеющие сплавы для постоянных магнитов типа кунифе и кунико, относящиеся к системам Си—Ni—Fe и Си—Ni—Со. Закалка фиксирует в этих сплавах пересыщенный твердый раствор с г. ц. к. решеткой, а старение приводит к его распаду на два раствора, которые в равновесном состоянии также имеют г. ц. к. решетку, отличаясь один от другого и от матрицы составом, а следовательно, периодом решетки и удельным объемом (аналогичная двойная система описывается диаграммой состояния на рис. 148, а).
Другим примером сплавов с детально изученной модулированной структурой являются стареющие никелевые сплавы, в частности типа нимоников, в которых при распаде пересыщенного раствора с г. ц. к. решеткой выделяется когерентная у'-фаза, также имеющая г. ц. к. решетку. При изучении двойных сплавов Ni—Al показано, что вначале при старении образуются хаотично расположенные выделения у' кубической формы, которые по мере увеличения времени старения постепенно выстраиваются в ряды, параллельные направлениям <100> матрицы (см. рис. 156). Это выстраивание происходит не в процессе самого распада пересыщенного раствора, а в период медленной коагуляции, когда одни частицы растворяются, а другие растут. Осо
328
бенностью рассматриваемого процесса коагуляции является то, что из-за упругого взаимодействия выделений у'-фазы селективно растут благоприятно расположенные частицы и растворяются другие частицы, вследствие чего и развивается модулированная структура.
Влияние напряжений от внешней нагрузки
Напряжения ниже макроскопического предела текучести, действующие в период старения, изменяют форму выделений, их ориентацию и взаимное расположение в матрице. Поле напряжений от внешней нагрузки взаимодействует с полями напряжений от выделений, которые образуются в такой форме, с такой ориентировкой и взаимным расположением, чтобы обеспечивался минимум упругой энергии всей системы. В частности, энергетически выгодно образование таких выделений, у которых уменьшено несоответствие решетки выделений и матрицы. Определенное влияние должно оказывать соотношение модулей упругости матрицы и выделений.
Морфология выделений зависит от кристаллографического направления приложения внешней нагрузки растяжения или сжатия. Например, при старении (170 °C) монокристалла сплава А1 •— 3,7 % Си под растягивающей нагрузкой, приложенной вдоль оси [001], почти все дискообразные выделения б'-фазы оказываются расположенными своей плоскостью габитуса в кристаллографических плоскостях (100) и (010), а при сжатии вдоль той же оси почти все диски О'-фазы расположены в плоскости (001). Следовательно, диски О'-фазы ориентированы параллельно оси растяжения и перпендикулярно оси сжатия.
В рассмотренном примере не сама O'-фаза реагирует на приложенную во время ее выделения нагрузку. Приложенная нагрузка заставляет ориентированно образовываться зоны ГП, которые с увеличением выдержки преобразуются в О'-фазу.
Под действием приложенной нагрузки растет упорядоченность в пространственном расположении выделений. Например, в сплаве никеля с 15 % Сг и 10 % А1 при обычном старении образуются сферические статистически неупорядоченно распределенные в матрице выделения у'-фазы, а под действием внешней нагрузки при старении выделяются эллипсовидные частицы у', выстроенные в виде цепочек по определенным кристалографическим направлениям.
а
Расстояние
Л s с а Г
Ряс. 157. Схема эвол.’оцпн распределения лспг'л ющсто элемента в пересы-твердом растворе на разных схлпя.х спинодального распа-
да (и?) и распада по механизму зарождения и роста (б) (Дж. Каи). К жцептрации Си, СQ н С^ см. на рис. 149 и 151
3. Структурные изменения при спинодальном распаде
В начальной стадии спинодального распада возникает вы-сокодпсперсная смесь фаз, решетки которых когерентны — плавно переходят одна в другую, и межфазные границы не резкие, а сильно размытые. Чем же отличается эта смесь фаз от флуктуаций состава, всегда имеющихся в твердом растворе, в том числе и выше спинодальной кривой RKV на рис. 148, а? При флуктаци-ях состава соседние участки твердого раствора также характеризуются разной концентрацией и соответственно разным периодом решетки. Как уже отмечалось в § 17, флуктуации состава непрерывно возникают и исчезают. В области спинодального распада участки твердого раствора с повышенной и пониженной концентрацией флуктуационного происхождения становят
ся устойчивыми, они не только не исчезают, но, наоборот, растут.
На рис. 157, а схематически показана последовательность изменений случайной флуктуации состава по мере развития спинодального распада. В первый момент (/ на рис. 157, а) в твердом растворе образовался кластер — устойчивый участок с повышенной концентрацией растворенного элемента (выше средней концентрации Со), окруженный зоной с пониженной концентрацией. Предпочтительное взаимное притяжение атомов одного сорта приводит в следующие моменты (например, II на рис. 157, а) к еще большему повышению концентрации в кластере и дальнейшему обеднению соответствующим компонентом прилегающей зоны. Этот процесс обеспечивается восходящей диффузией (на рис. 157, а указана стрелками), что соответствует отрицательному коэффициенту диффузии. Последнее обусловлено тем, что в формулу для коэффициента диффузии D одним из сомножителей входит вторая производная от энергии Гиббса по концентрации, а так как в области спинодального распада (d2G/dC2) <0 (см. § 45), то D<0. Это важнейший признак,, отличающий спинодальный распад от обычного распада по механизму зарождения и роста, который будет рассмотрен позже.
330
Атомы одного сорта в исходной матрице, расположенные по внешнему краю обедненной зоны, также испытывают предпочтительное взаимное притяжение. Так как силы . их взаимного притяжения короткодействующие, то указанные атомы не «чувствуют» существования готового кластера, а испытывают только влияние непосредственно граничащей с ними обедненной зоны. Поэтому они удаляются от обедненной зоны и образуют новый кластер, также окруженный обедненной зоной. Таким образом, образование одного кластера приводит к образованию соседнего и так далее: этот процесс в виде концентрационной волны быстро распространяется по решетке матрицы. На одинаковом расстоянии один от другого, называемом длиной концентрационной волны, последовательно возникают все новые и новые кластеры.
Наиболее подробно структурные изменения при спино“ дальном распаде изучены в сплавах системы Си—Ni—Fe, находящихся по составу в центре области расслоения на диаграмме состояния. На электронно-микроскопических снимках, полученных методом просвечивания тонких фольг, светлые участки относятся к областям, обогащенным медью, а темные — обогащенным железом и никелем (см. рис. 155). В твердом растворе Си—Ni—Fe, характеризующемся, как и многие другие кристаллы с кубической решеткой, значительной анизотропией модуля упругости, спино-дальный распад идет вдоль каждого из трех «упруго-мягких» направлений <100>. Поэтому первоначально при спинодальном распаде в сплавах Си—Ni—Fe образуется модулированная структура, состоящая из стержнеобразных областей, разделенных размытыми границами («корзиночное плетение»). По мере увеличения времени старения растут амплитуда концентраций и длина концентрационной волны, модулированная структура грубеет, а границы между когерентными выделениями становятся менее размытыми. Постепенно расслоение по составу достигает максимума, соответствующего равновесию двух фаз с г. ц. к. решеткой. Когерентность теряется (из-за роста упругих напряжений), причем на межфазных границах образуются структурные дислокации. Потеря когерентности сопровождается исчезновением модулированной структуры и сильным огрублением выделений в результате коагуляции.
Особых морфологических признаков, которые были бы характерны только для структур, полученных при спинодальном распаде, нет. Спинодальный распад не обязательно дает модулированную структуру, а модулированная
331
структура не обязательно связана со спинодальным распадом, как это считали раньше. Вместе с тем в упругоанизо-троппых кристаллах весьма вероятно образование при спи-нодальном распаде модулированной структуры корзиночного плетения.
Термодинамика и механизм спинодального распада предопределяют его гомогенность: предпочтительного образования выделений на границах зерен или на дислокациях при спинодальном распаде не наблюдалось. Для практики весьма важно и то, что для спинодального распада характерна высокодисперсная структура, равномерная по всему объему зерен исходной фазы.
Из-за отсутствия специфических структурных признаков не всегда легко установить, спинодальный ли распад в данном сплаве. К промышленным сплавам, в которых при старении действительно протекает спинодальный распад, можно отнести магнитно-твердые сплавы типа кунифе и кунико, легированные куниали для изготовления пружин и высокопрочные сплавы системы Си—Ni—Sn.
4. Непрерывный и прерывистый распад
Непрерывный распад
При непрерывном распаде в исходном пересыщенном растворе образуются и растут отдельные выделения избыточной фазы. Так как выделения обогащены одним из компонентов, то матричная фаза обеднена этим компонентом и в ней существует градиент концентраций.
Кристаллы избыточной фазы растут вследствие обычной нисходящей диффузии: поток атомов (см. стрелки на рис. 157,6) направлен в сторону понижения концентрации, и коэффициент диффузии D положителен. Последнее обусловлено тем, что вне области спинодального распада вторая производная от энергии Гиббса по концентрации d2G/dC2>Q (сравните со спинодальным распадом путем восходящей диффузии при отрицательных значениях D).
Растущие выделения при непрерывном распаде постепенно «высасывают» легирующий элемент из матричной фазы, обедняя ее по всему объему до равновесной концентрации С, (см. рис. 151 и 157, б).
Размер выделений г при непрерывном распаде с увеличением продолжительности старения т возрастает приближенно по параболическому закону:
Г==(£)т)12. (35)
332
Скорость роста выделений контролируется коэффициентом объемной диффузии D в решетке матрицы.
Характерная особенность рассматриваемого процесса — непрерывное по всему объему исходных зерен уменьшение концентрации легирующего элемента. Поэтому распад и называют непрерывным. Кристаллографическая ориентировка зерен исходной фазы при непрерывном выделении не изменяется.
По микроструктурным признакам непрерывный распад раствора при старении подразделяют на равномерный (или общий) и локализованный.
При равномерном распаде выделения однородно распределены по объему зерна. Зарождение при равномерном распаде может быть гомогенным или гетерогенным (см. § 19). В последнем случае места предпочтительного зарождения (дислокации, скопления вакансий и др.) распределены равномерно по телу зерна.
При локализованном распаде выделения неравномерно распределены по телу зерна. Продукты распада обнаруживаются у границ зерен и субзерен, в полосах скольжения и других местах. Зарождение при локализованном распаде — всегда гетерогенное.
Зоны, свободные от выделений
При старении некоторых сплавов (алюминиевых, титановых, железных, никелевых и др.) вблизи границ зерен матричного твердого раствора распада не происходит и отчетливо видны зоны, свободные от выделений (рис. 158 и 159). В алюминиевых сплавах ширина таких зон составляет обычно сотые — десятые доли .микрометра, и они видны только под электронным микроскопом. В титановых р-спла-вах после старения зоны, свободные от выделений, имеют ширину порядка нескольких микрометров п хорошо видны в световой микроскоп.
Существование приграничных зон, свободных от продуктов распада раствора, не противоречит положению о том, что границы зерен облегчают зарождение выделений новой фазы. Речь идет не о самой границе, а о примыкающей к ней приграничной области. Часто можно наблюдать выделения непосредственно на границе зерна и рядом с ними приграничную зону, свободную от выделений (см. рис. 158) <
Появление приграничных зон, свободных от выделений, можно объяснить двумя причинами. Если распад начинается с границ и здесь зарождается фаза, обогащенная
333
одним из компонентов, то при своем росте она «высасывает» этот компонент из приграничной области. Начинающийся позднее распад в теле зерна не идет в некоторой зоне, прилегающей к выделению, из-за уменьшения в этой зоне пересыщеиности матричного раствора (см. участок зоны вблизи выделения на рис. 158). Это объяснение не подхо
Рис. 158. Приграничная зона, свободная от выделений в сплаве AI—5,9 % Zn — 2,9 % Mg после старения при 180 ’С, 3 ч. Электронная микрофотография, фольга (Анвнн. Лоример, Нн-кольсон)
Рис. 159. Приграничные зоны, свободные от выделений (светлые), в титановом сплаве ВТ15, закаленном на 13-фазу с температуры 900 °C и состаренном при 450 °C, 15 ч. Темный
фон — сильно травящаяся p-фаза, в которой выделилась дисперсная а-фа-за. Х200 (И. И. Новиков, О, В. Каспарова, И. С. Полькин)
дит к зоне нераспавшегося раствора у границ, на которых нет выделений (см. узкую зону вдоль «чистой» границы на рис. 158).
Более общей причиной появления зон, свободных от выделений, является обеднение приграничных областей вакансиями из-за стока на границу. Равновесная концентрация тепловых вакансий резко, по экспоненте, уменьшается с понижением температуры. При закалке этот процесс внутри зерна не успевает пройти в необходимой степени и решетка сильно пересыщается вакансиями. В первый пе-
334
Расстояние от границы зерна
рпод закалочного охлаждения, когда при высоких температурах вакансии еще очень подвижны, они устремляются к границам зерен — стокам вакансий. В результате вблизи границ зерен концентрация вакансий в некоторой зоне сказывается пониженной по сравнению с остальной частью зерна, где сохраняется высокая концентрация избыточных вакансий (рис. 160).
Вакансионные кластеры, как ранее отмечалось, облегчают зарождение выделений, особенно если удельный объем при распаде раствора возрастает. Закалочные вакансии способствуют также диффузионному росту зародышей новой фазы. Можно принять, что при некоторой пересыщенности раствора легирующим элементом, т. е. при определенной температуре старения распад за определенное время происходит только в той части тела зерна, где концентрация вакансий не ниже некоторой критической величины Ct.
Если кривая 1 на рис. 160 характеризует распределение концентрации вакансий по сечению зерна матричного раствора в закаленном сплаве, то при температуре старения 1\ в приграничной зоне шириной оЬ{
так как здесь концентрация вакансий ниже Сь При более низкой температуре старения Т2 пересыщенность раствора легирующим элементом больше, чем при температуре 'Л, и распад его может идти при более низкой концентрации вакансий (критическая концентрация вакансий С2<С5). Соответственно свободная от выделений зона при более низкой температуре старения должна быть уже: ob2<ob[.
С повышением температуры нагрева под закалку возрастает равновесная концентрация вакансий и становится резче ее градиент вблизи стока — границы зерна (кривая 2 на рис. 160). Поэтому при одинаковой температуре старения и соответственно одинаковой критической концентрации вакансий зона нераспавшегося раствора с повышением температуры закалки сужается (ob4>ob[).
Если замедлить охлаждение при закалке, то на границу зерна успевает стечь больше вакансий (сравните кривые
Рис. 160. Схема распределения концентрации вакансий вблизи границы зерна при разных режимах закалки:
1 — закалка с температуры Д; 2 — закалка с температуры T2>Ti; 3 — закалка с температуры 7’2 при замедленном охлаждении
не
335
2 и 3) и свободная от выделений зона оказывается шире (оЬ3>оЬ4).
Таким образом, для сужения приграничных зон, свободных от выделений, следует повышать температуру закалки, ускорять закалочное охлаждение и понижать температуру старения. Пластическая деформация закаленного сплава перед старением, способствуя распаду пересыщенного рас-тзора, может полностью предотвратить появление этих зон.
Возможны случаи, когда непосредственно у межзеренной границы находится узкая свободная от выделений зона, обедненная легирующими элементами, которые сегрегировали на границу, а по обе сторон1 т к ней примыкает зона, свободная от выделений из-за стока вакансий на границу. В этой зоне раствор имеет исходную концентрацию легирующих элементов.
Рассмотренные закономерности влияния разных факторов на ширину зон, свободных от выделений, неоднократно устанавливались при изучении алюминиевых и титановых сплавов. Роль этих зон при эксплуатации состаренных сплавов во многих случаях окончательно не ясна.
Чаще всего считают, что зоны, свободные от выделений, вредны, так как из-за меньшего предела текучести в них локализуется пластическая деформация, приводящая к межзеренному разрушению. Согласно одной из гипотез внутри «слабой» зоны легко генерируются дислокации, которые скользят вдоль границы зерна и образуют плоские скопления вблизи тройных стыков, где и зарождаются меж.зерен-пые трещины.
Пластически деформированная зона, свободная от выделений, является анодом по отношению к остальнохМу зерну и служит причиной ускоренного межзеренного разрушения при коррозии под напряжением.
Однако имеется и другая точка зрения, согласно которой рассматриваемые зоны полезны, так как в них полнее релаксируют напряжения, которые концентрируются в месте остановки полосы скольжения границей зерен. Чем шире мягкая зона, тем полнее проходит эта релаксация и соответственно труднее зарождается и растет трещина.
Получить прямые доказательства роли зон, свободных от выделений, не просто, так как с изменением их ширины при варьировании режима термообработки одновременно изменяются и другие структурные характеристики, влияющие па свойства сплава.
336
Прерывистый (ячеистый распад)
При прерывистом распаде в зернах исходного пересыщенного раствора ап зарождаются и растут ячейки (колонии) двухфазной смеси «i+р, имеющие перлитообразное строение (рис. 161). У оо-фазы внутри ячеек — та же решетка, что и у исходной фазы ап, но состав ее является равновесным при данной температуре распада или промежуточным между исходным и равновесным. Рассматриваемое превращение можно записать в следующей форме:
4-₽. (36)
Средний состав двухфазной смеси оо+р внутри ячейки такой же, как состав исходного раствора ап.
Распад развивается при продвижении фронта ячейки в исходный раствор вследствие кооперативного роста а!- и p-фаз аналогично росту перлитной колонии.
Во время превращения концентрация исходного раствора остается все время неизменной, пока этот раствор совсем не исчезнет. На границе ячейки и исходного раствора в узкой зоне происходит резкий скачок концентрации — от исходной в растворе ап до концентрации раствора ai внутри ячейки. Поэтому распад и называют прерывистым в отличие от непрерывного, при котором в исходном растворе концентрация легирующего элемента плавно снижается, так как он посте
пенно «высасывается» в результате роста избыточной р-фазы.
Рентгенографически прерывистый распад впервые был обнаружен Н. В. Агеевым, М. Хансеном и Г. Заксом в 1930 г. на сплаве серебра с медью. При непрерывном распаде период решетки матричного раствора плавно измени-
Рнс. 161. Колонии прерывистого распада в сплаве Ni — 20 % Cr-9% Nb. Старение при 850 °C. 2 ч после закалки с 1180'С. Х1350 (А. Г. Рахштадг,
О. М. Ховова, Ц. Н. Гевелннг)
22 И. И. Новиков
337
ется из-за постепенного уменьшения его концентрации по всему объему и линии рентгенограммы соответственно смешаются и размываются. При прерывистом распаде на рент-1енограмме отмечаются две системы линий: одна соответствует исходному раствору ап с определенным периодом решетки, а другая — раствору czj с конечной концентрацией и своим периодом решетки. С развитием прерывистого распада линии рентгенограммы не смещаются, т. е. периоды решеток а-раствора двух составов не изменяются. Постепенно ослабляются интерференции от раствора с исходной концентрацией, так как количество его уменьшается, и усиливаются отражения от раствора с конечной концентрат* ией.
Прерывистый распад начинается от границ зерен. При малом межпластиночном расстоянии в ячейках пли сильной травпмостп превращенной области она выявляется под световым микроскопом в виде темных участков, обычно резко отличающихся от светлых зерен исходного пересыщенного раствора.
Кристаллографическая ориентация агфазы внутри ячейки отличается от исходной ориентации аи-фазы в том зерне, в котором растет эта ячейка. Вместе с тем ориентация агфазы внутри ячейки такая же, как в соседнем зерне по другую сторону от границы, где начался прерывистый распад. Таким образом, продвижение фронта ячейки прерывистого распада в сторону одного зерна сопровождается переориентацией кристаллической решетки матричной фазы и его можно трактовать как продвижение межзеренной границы в сторону «поедаемого» зерна. Это внешне похоже на образование выступов, «языков» при первичной рекристаллизации, когда отдельные участки высокоугловой границы выгибаются и продвигаются в сторону одного из зерен (см. рис. 22).
При прерывистом распаде избыточная фаза выделяется из матрицы позади межзеренной границы, продвигающейся в сторону соседнего зерна. Термодинамическим стимулом рассматриваемого продвижения межзеренной границы является разность объемных энергий Гиббса исходного пересыщенного раствора и двухфазной смеси внутри ячейки (Gj—G2 на рис. 151). Если состав фаз внутри ячейки и не достигает равновесных значений, то все равно образование этой смеси приводит к уменьшению объемной энергии Гиббса, хотя и не предельно возможному.
Механизм зарождения ячейки прерывистого распада точно пе выявлен. Одно из предположений, базирующееся
338
Рис. IG2. Схема формирования ячейки прерывистого распада у мигрирующей границы (Фурнелле и Кларк)
на строении фронта превращения, сводится к следующему. Граница зерен мигрирует, растворенный элемент сегрегирует около нее и выделяется в виде частиц, локально закрепляющих границу. Продолжая мигрировать, граница выгибается между выделениями, а они удлиняются при росте, следуя за продвигающейся границей (рис. 162). Так формируется чередование участков аг и p-фаз, т. е. зарождается перлитообразная ячейка позади мигрирующей межзеренной границы. Предложены и другие гипотезы зарождения ячейки, в частности учитывающие повторное зарождение пластин 13-фазы на мигрирующей границе а-зерна.
Механизм роста сформировавшейся ячейки более ясен. Перед торцами пластин или стержней аг и 13-фаз концентрация легирующего элемента В в матрице соответственно повышена и понижена (имеется в виду,
что p-фаза обогащена компонентом В, а агфаза км обеднена). Как и при росте перлитной колонии в аустените, при кооперативном росте двухфазной ячейки прерывистого рас-пала компоненты диффузионно перераспределяются вдоль межфазной границы матрицы с ячейкой. При непрерывном же распаде рост выделения контролируется объемной диффузией перпендикулярно поверхности выделения. Скорость диффузии вдоль межфазной границы матрицы с ячейкой намного больше, чем объемной, а пути диффузии очень короткие, так как межпластиночное расстояние в ячейке небольшое. Поэтому прерывистый распад способен быстро протекать при относительно низких температурах, в том числе и при таких, когда рост изолированных выделений P-фазы цо механизму непрерывного распада идет с очень малой скоростью или практически полностью подавлен. Межпластиночное расстояние внутри ячеек уменьшается с понижением температуры старения.
Во время роста колонии внутри нее поддерживается примерно постоянное, характерное для данной температуры старения, межпластиночное расстояние. Это обеспечивается увеличением числа пластин в колонии в результате ветвления пластин, а не повторного их зарождения.
При более низких температурах прерывистого распада/
22*
339
соседние ячейки растут от одной границы по обе ее стороны, т. е. в прямо противоположных направлениях.
Пластическая деформация исходного пересыщенного раствора, увеличивающая в нем плотность дислокаций, может и ускорять, и замедлять прерывистый распад. Практически очень важно, что небольшая добавка третьего элемента к двойному сплаву также может ускорять или замедлять прерывистый распад. Одной из причин замедления прерывистого распада, вплоть до полного его подавления, является затруднение миграции границ в результате сегрегации на них добавки третьего элемента.
Среди промышленных сплавов прерывистый распад в заводской практике встречается в бериллиевой бронзе (например, марки БрБ2), магниевых сплавах на базе системы Mg—Al—Zn (например, марки МЛ5), аустенитном железном сплаве марки 36НХТЮ.
При старении обычно стараются избежать прерывистого распада, так как двухфазная структура с некогерентными выделениями после прерывистого распада получается более грубой и соответственно менее прочной, чем после обычного дисперсионного твердения, когда образуются дисперсные когерентные или полукогерентные выделения. Кроме того, некогерентные пластинчатые выделения избыточной фазы на границах зерен охрупчивают сплав.
От вредного для бериллиевых бронз прерывистого распада можно полностью избавиться, применив сильную холодную деформацию (более 90 %) или же вводя небольшие добавки, например 0,2 % Со пли 0,1 % Mg.
В магниевых сплавах замедление охлаждения при закалке увеличивает объем, претерпевший при старении прерывистый распад. В отливках, закаленных с охлаждением на воздухе, границы зерен на шлифе сильно растравливаются в результате прерывистого распада, успевшего пройти здесь во время медленного закалочного охлаждения.
Иногда прерывистый распад полезен. Если при прерывистом распаде выделяется фаза, когерентная матрице, каж-у'-фаза (типа Ni3Al) в сплаве 36НХТЮ, то после старения получается дисперсная структура и механические свойства повышаются.
5. Стадии распада раствора при дисперсионном твердении
Образование зон Гинъе—Престона
У дисперсионнотвердеющих алюминиевых сплавов и бериллиевых бронз при комнатной температуре сразу после за-
340
кадки растет электросопротивление (рис. 163). Это обт^яс-пяют образованием в пересыщенном твердом растворе скоплений атомов легирующих элементов — кластеров, рассеивающих электронные волны. Возможно, что часть кластеров успевает образоваться уже в период закалочного охлаждения. Вначале размер кластеров очень мал, и структурные методы их не выявляют. Через некоторое время кластеры могут вырасти настолько, что они вызывают дифракционные эффекты на рентгенограммах монокристал-
лов и электронограммах при просвечивании фольг. Кластеры, обнаруживаемые структурными методами, называют зонами Гинье—Престона. У них та же кристаллическая решетка, что и у матричного раствора, но из-за различия в атомных диаметрах металла-основы и легирующего элемента скопление легирующего элемента вызывает местное изменение межплоскостных расстояний.
На рис. 164 изображена предложенная Герольдом модель строения зоны ГП в алюминиевомедном сплаве. Атомы меди занимают одну из плоскостей {100}, а по обе стороны от нее параллельные плоскости состоят только из атомов алюминия. Так как атомный диаметр у меди меньше, чем у алюминия, то плоскости, заполненные атомами алюминия, смещены в сторону плоскости, состоящей из атомов меди. Смещение атомных слоев из нормальных положений при удалении от центра зоны ГП уменьшается, и согласно этой модели четырнадцатые атомные слои по обе стороны от центральной плоскости остаются несмещенными. Из-за большой разницы в атомных диаметрах меди и алюминия, обусловливающей значительную упругую дефор-
341
мацню решетки, форма зон 1 П в алюминиевомедных сплавах тонкопластинчатая, дискообразная.
В сплавах А1—Ag и А1—Zn разница в размерах атомов добавки и алюминия мала, упругая деформация решетки невелика и форма зон ГП сферическая (см. табл. 7). Атомы серебра и цинка образуют сферические кластеры, вокруг которых раствор обеднен легирующим элементом.
Размер зон ГП зависит от состава сплава, температуры и продолжительности старения н составляет величину порядка i —10 им. Так, например, дискообразные зоны ГП в сплавах А1—Си имеют диаметр порядка 10 нм.
Образование кластеров — диффузионный процесс, связанный с перемещением атомов легирующего элемента в решетке твердого раствора. Важнейшей особенностью кинетики образования кластеров является необычайно высекая диффузионная подвижность атомов растворенных элементов. Кластеры образуются даже при отрицательных температурах (в сплавах А1—Zn при минус 100°C).
Коэффициент диффузии легирующих элементов в алюминиевом растворе при комнатной и более низких температурах можно рассчитать по скорости роста кластеров, например по формуле (35). Такие расчетные значения оказались на много порядков больше коэффициента диффузии, полученного экстраполяцией значений, экспериментально определенных при высоких температурах в обычных диффузионных опытах. Эта разница для раствора меди в алюминии при комнатной температуре составляет 10s (!).
Аномально высокая скорость диффузии при образовании кластеров во время старения обусловлена пересыщением твердого раствора вакансиями при закалке. Равновесная концентрация вакансий при температуре закалки на много порядков больше, чем при температуре старения. Во время закалки значительная часть вакансий не успевает аннигилировать в стоках и твердый раствор оказывается пересыщенным не только легирующим элементом, но и вакансиями. Так как механизм диффузии в растворах замещения вакансионный, то закалочные вакансии резко ускоряют -миграцию атомов легирующего элемента, чем и обусловлена очень высокая скорость образования кластеров при сравнительно низких температурах.
С повышением температуры нагрева под закалку возрастает пересыщение твердого раствора вакансиями и ускоряется образование кластеров. Противоположный эффект дает замеледнпе охлаждения при закалке, так как больше вакансий успевает стечь в стоки (на дислокации, границы
зерен и свободную поверхность образца) в период закалочного охлаждения.
После быстрого роста электросопротивления в начальный период старения («быстрая реакция»), продолжающийся обычно несколько десятков минут, оно медленно возрастает длительное время (см. рис. 163). В период этого медленного роста электросопротивления («медленная реакция») скорость диффузии, обеспечивающей образование зон, остается все еще аномально высокой. Основная теоретическая трудность состоит в том, чтобы объяснить длительное (в течение многих часов и суток) сохранение значительного избытка закалочных вакансий, несмотря па существование большого числа стоков для них (дислокаций, границ зерен).
Одна из гипотез, выдвинутых для преодоления этой трудности, сводится к следующему.
Вакансии связаны с атомами растворенного элемента в комплексы, и рост зон обеспечивается миграцией этих комплексов. Приблизившись к зоне, комплекс диссоциирует на вакансию и атом легирующего элемента, который присоединяется к зоне. Повышение концентрации свободных вакансий у границы зоны создает направленную их миграцию в матрицу, где они встречаются с атомами растворенного элемента и образуют новые комплексы. Так как около границы зоны концентрация комплексов меньше, чем вдали ст нее, то комплексы мигрируют к зоне, диссоциируют, отдавая ей атом легирующего элемента, и т. д. Зона действует как помпа, выкачивающая атомы растворенного элемента из матрицы и посылающая вакансии в матрицу. Часть свободных вакансий аннигилирует в стоках, и поэтому скорость диффузии, обеспечивающей образование и рост зон, падает.
При обсуждении природы зон ГП обычно их сопоставляют с местабильными промежуточными фазами. При этом часто подчеркивают, что зона ГП — это не новая фаза, а участок исходного твердого раствора, обогащенный растворенным элементом. В отличие от промежуточных фаз, характеризующихся собственной решеткой, зона ГП имеет ту же решетку, что и матричный раствор, только она деформирована из-за различия в атомных диаметрах растворимого и растворителя. Между зоной и окружающим раствором нет четкой границы раздела. В некоторых сплавах зоны ГП (кластеры) образуются безынкубационно, сразу же после закалки или даже в период закалочного охлаждения, в то время как промежуточные и стабильные фазы
343
появляются через некоторый инкубационный период. Все эти факты свидетельствуют об отличии зон ГП от промежуточных и стабильных фаз. Поэтому зоны ГП часто называют предвыделениями, чтобы отличить их от «истинных» выделений промежуточных и стабильных фаз с качественно иной структурой.
В отличие от такого структурного подхода, трактующего зоны ГП как предвыделения, с термодинамической точки
Рис. 165. Диаграмма состояния системы AI—Си с линией сольвуса зон ГП (во Битону и Роллэсону) и конодой тп
зрения их можно рассматривать как самостоятельную ме-тастабпльную фазу, промежуточную между матричным раствором и стабильной фазой. Такая трактовка вытекает из многих важных особенностей поведения зон ГП. В отличие от концентрационных флуктуаций, которые непрерывно возникают и размываются тепловым движением, зоны ГП длительное время устойчивы (при низких температурах—• неопределенно долгое время). Экспериментально установлено, что с увеличением продолжительности старения размер зон возрастает, причем более крупные зоны растут за счет растворения более мелких, т. е. точно так, как происходит коагуляция частиц фаз. Состав зон при данной температуре старения не зависит от состава сплава. Наконец, самое главное, зоны ГП в двойной системе имеют свою линию сольвуса (рис. 165): они образуются только при пере
344
охлаждении матричного раствора ниже этой линии. Если же в сплаве имеются зоны ГП, то они растворяются в матрице при нагреве до температуры выше линии их сольвуса. Здесь полная аналогия с выделением и растворением стабильной фазы.
Таким образом, зоны ГП можно рассматривать как вторую фазу, находящуюся в метастабильном равновесии с матричным раствором. То, что зоны не отличаются по типу решетки от матрицы, не противоречит этому утверждению, так как, например, в системе с непрерывным рядом твердых растворов (см. рис. 148, а) выделяющаяся под куполом расслоения стабильная фаза имеет ту же решетку, что и матрица. По аналогии с кривой расслоения, относящейся к стабильному равновесию двух фаз с одинаковой решеткой (см. рис. 148,а), на диаграмме состояния можно провести куполообразную кривую метастабильного равновесия матричного раствора и зон ГП (см. пунктир на рис. 165). При любой температуре конода, например mn, должна соединять точки состава зон ГП (на правой ветви куполообразной кривой) и обедненного матричного раствора (на левой ветви). Здесь обедненный легирующим элементом матричный раствор и зоны ГП рассматриваются как метастабиль-ные фазы, образующиеся из исходного однородного твердого раствора.
От других промежуточных фаз зоны ГП отличаются строением границы раздела с матрицей. Зоны — полностью когерентные выделения, и поэтому граница их раздела с матрицей размыта.
Из сказанного видно, что термин «предвыделение» применительно к зонам ГП весьма условен, так как их можно считать когерентными выделениями.
Механизм зарождения зон ГП слабо изучен. Плотность дислокаций в рекристаллизованном закаленном сплаве равна 10' —108 см-2, и в случае гетерогенного зарождения на дислокациях число зон в единице сечения должно было бы характеризоваться этой величиной. В действительности же плотность выделений зон ГП (число зон в единице объема) измеряется величиной порядка 1018 см-3, а в сечении-— порядка 10'2 см~2. Считают, что для кластеров характерно главным образом гомогенное зарождение на концентрационных флуктуациях.
Если рассматривать зону ГП как фазу, то при ее зарождении изменение энергии Гиббса сплава AG = —AGc6-r +А6ПОв4-А6упр. Из-за когерентности зоны и матрицы составляющей АСпов можно пренебречь. У зоны малого раз-
345
мера (на начальной стадии ее формирования) составляющая \Gynp тоже очень мала. Тогда при достаточно большом Пересыщении энергетический барьер для зарождения зоны ГП должен быть очень мал, чем и объясняется появление кластеров сразу после закалки или даже во время закалочного охлаждения.
Большое практическое значение имеет то, что зоны ГП легко зарождаются по всему объему матричного твердого раствора и дают структуру равномерного распада с высокой плотностью выделений (см. § 47).
Выделение промежуточных и стабильных фаз
В большинстве стареющих промышленных сплавов из пересыщенного раствора может выделяться метастабильная фаза, структура которой является промежуточной ,мв>КДУ матричным раствором п стабильной фазой (см. Tat) л. 8).
Рис. 166. Элементарные ячейки стабильной (0) и метастабильных промежуточных фаз (О' и 0"), которые могут выделяться из алюминиевого раствора при старении сплавов AI—Си (Хорнбоген)
В некоторых системах выделяются две промежуточные ме-тастабильные фазы.
При образовании зародыша любого выделения общее изменение энергии Гиббса складывается из трех составляющих : A G=—A GO6+A GDoe4-A Gyпр.
Энергия Гиббса метастабильной фазы выше, чем у стабильной, и образование ее зародыша в матрице вызывает меньшее снижение объемной энергии Гиббса. Причина, обусловливающая выделение промежуточной фазы вместо стабильной, — значительно меньшая межфазная энергия на границе с матрицей и соответственно меньший прирост AGnoB при образовании зародыша (см. § 20). Поэтому наиболее важной общей чертой структуры всех * промежуточных фаз в стареющих сплавах является то, что она обес-346
лечивает минимум одну когерентную границу между выделением и матрицей. Выделения промежуточных фаз—• обычно частично, а иногда и полностью когерентные.
Полностью п частично когерентные выделения промежуточных фаз подробно изучены в стареющих сплавах А1 — Си. На рис. 166 изображена элементарная ячейка стабильной фазы 0 (СиА12) и метастабпльных промежуточных фаз 0х и 0". Здесь же приведена элементарная ячейка алюминия, приближенно характеризующая строение матрицы (без учета атомов меди в матричном ct-растворе, несколько уменьшающих период решетки).
Решетка стабильной 0-фазы—тетрагональная с периодами а=0,607 нм и с=0,487 нм. Строение 0-фазы в плоскостях (001), (010) и (100) (и любых других плоскостях) сильно отличается от строения матрицы, и поэтому выделения 0-фазы полностью некогерентные.
Решетка промежуточной фазы 6" — тетрагональная, состав фазы соответствует СиА12. В структуре 0ХХ часть плоскостей занята только атомами алюминия, а часть — только атомами меди. Выделения 0" — полностью когерентные, причем по плоскости (001) сопряжение с алюминиевой матрицей идеальное (у 0", как и у алюминия, период решетки а = 0,404 нм). По плоскостям же (010) и (100) когерентность обеспечивается большой упругой деформацией, так как около слоев, занятых атомами меди, межплоскостное расстояние уменьшено (0,182 нм вместо 0,202 нм).
Максимальная толщина выделений бхх составляет 10 им, а диаметр — до 150 нм. Их называют также зонами ГП2 в отличие от зон ГП1, имеющих решетку матрицы и обозначавшихся выше как зоны ГП. В настоящее время все чаше используют обозначение 0", а не ГП2, так как решетка этих выделений отличается от решетки матрицы.
Решетка промежуточной фазы 0х — тетрагональная с периодами а=0,404 нм, с=0,580 нм, состав отвечает соединению СиА12. По плоскости (001) пластинчатое выделение 0х имеет с матрицей когерентную границу с идеальным сопряжением решеток. По плоскостям (010) и (100) несоответствие строения 0х и матрицы значительно, и межфазная граница полукогерентна. Таким образом, выделения О'-фазы — частично когерентные, и поле упругих напряжений вокруг них меньше, чем вокруг когерентных выделений 0х,-фазы и зон ГП. Форма выделений О'-фазы хорошо видна на рис. 167, где ее пластинки расположены параллельно и перпендикулярно плоскости фольги.
Промежуточные и стабильные фазы зарождаются гете-
347
рэгенно в отличие от зон ГП, для которых характерно гомогенное зарождение. Исключение составляет стабильная фаза Al3Sc в алюминиевых сплавах, зарождающаяся гомогенно, равномерно по всему объему матрицы. Это объясняется тем, что соединение Al3Sc— единственная в алюминиевых сплавах стабильная фаза, выделения которой благодаря очень хорошему структурному соответствию полностью когерентны матрице.
Рис. 167. Пластинки б'-фазы, перпендикулярные (/) и параллельные (2) плоскости фольги (001). и зоны ГП (3) в сплаве Л! — 3% Си, состаренном при 130 °C в течение 18 дней. Электронная микрофотография, фольга (Филипс)
Местами предпочтительного зарождения промежуточных фаз (при старении) служат отдельные дислокации, малоугловые границы (стенки дислокаций), дефекты упаковки и предположительно вакансионные кластеры. В зонах ГП также может зарождаться промежуточная фаза.
Некогерентные выделения стабильной фазы предпочтительно зарождаются на высокоугловых границах и вакан-сионных кластерах. Кроме того, стабильные фазы могут зарождаться па ранее появившихся выделениях промежуточных фаз.
Зарождение выделений промежуточных фаз на дислокациях облегчено главным образом потому, что структур
348
ное несоответствие зародыша и матричного раствора частично или полностью компенсируется разрежением или сгущением около края экстраплоскости. Пластинки промежуточной фазы зарождаются с такой ориентировкой на краевых дислокациях, что поля напряжений от этих пластинок и дислокаций частично гасят одно другое. В формуле (26) слагаемое AGynp, затрудняющее зарождение, при выделении полукогерентной частицы на дислокации мало и даже может быть отрицательным — упругая энергия дислокации способствует зарождению.
При зарождении некогерентного выделения стабильной фазы с высокой поверхностной (межфазной) энергией в формуле (26) определяющую роль играет составляющая AGnOB, а не AGynp. При образовании некогерентного выделения на межзеренной границе, являющейся готовой поверхностью раздела, составляющая AGDob, затрудняющая зарождение, оказывается уменьшенной, т. е. зарождение некогерентного выделения на высокоугловой границе облегчено.
У когерентного и полукогерентного выделений межфазная энергия значительно меньше, чем у полностью некогерентного. Поэтому при их зарождении определяющую роль в формуле (26) играет составляющая AGynp, а не AGnOB.
Дислокации и границы зерен служат местами предпочтительного зарождения еще и потому, что на них образуются сегрегации атомов растворенного элемента — атмосферы Коттрелла на дислокациях и равновесная сегрегация’ на высокоугловых границах. Так как промежуточные и стабильные фазы характеризуются повышенной концентрацией легирующего элемента, то им легче образоваться в участках матрицы, уже обогащенных этим элементом.
На границах зерен выделения стабильной фазы могут появиться даже в период закалочного охлаждения. Это происходит, в частности, при закалке некоторых алюминиевых сплавов в кипящей воде.
Если у промежуточных и стабильных фаз удельный объем больше, чем у матрицы, то естественно, что зарождение их облегчено на вакаисионных кластерах, которые быстро образуются в матрице из-за пересыщения ее закалочными вакансиями.
Дефекты упаковки служат местами гетерогенного зарождения лишь в тех случаях, когда структура выделения тождественна структуре дефекта упаковки. Например, в сплавах А1 — Ag промежуточная фаза у' имеет г. и. решетку, а дефект упаковки в г. ц. к. решетке, как известно, яв-
349
Рис. !Сч Зависимость энергии Гиббса от со/газа расслаивающегося с обра-воза ни?*’ зон Г И пересыщенного X-рас 1£О^: метастабильной (У- п ста-бнльпС . П-фазы
ляетгя тонкой прослойкой г. п. решетки. Следовательно, полоса дефекта упаковки между частичными дислокациями в твердом растворе серебра в алюминии, обогащенная атомами серебра (атмосфера Сузуки), — это готовый за-£одыш /-фазы.
Как было доказано в § 20, растворимость метастабиль-ных фаз всегда выше, чем растворимость стабильной фазы. Рассмотрим гипотетическую систему, в которой в a-растворе могут образовываться зоны ГП, промежуточная фаза р' и стабильная фаза ₽•
На рис. 168 кривая Ga изображает зависимость энергии Гиббса от состава a-фазы, которая может расслаиваться с образованием зон ГП (по аналогии с кривой энергии Гиббса при температуре Г3 на рис. 148, б). Общая касательная к ветвям кривой энергии Гиббса даст состав матрицы (Cv-m). находящейся в ме-
тастабильном равновесии с зонами ГП, которые здесь рассматриваются как метастабильная фаза с той же решеткой, что и матрица.
Промежуточная фаза р' и стабильная фаза р имеют соб-
ственную кристаллическую решетку, отличную от решетки матрицы, и поэтому кривые энергии Гиббса Gp' и GpHe являются продолжениями кривой Ga. Общая касательная к кривым Ga и Gp' определяет состав матрицы (Са-р')> находящейся в метастабильном равновесии с промежуточной Р'-фазой. Аналогично определяется состав матрицы (Са-р)> находящейся в равновесии со стабильной фазой. Видно, что Ga rn > Са—0' > Са-0 .
Рис. 168, относящийся к определенной температуре, качественно не изменится и при других температурах: растворимость зон ГП будет всегда выше, чем р'-фазы, а растворимость р'-фазы должна быть выше растворимости р-фа-зы. На диаграмме состояния под линией растворимости стабильной p-фазы должны находиться линии сольвуса (растворимости) промежуточной р'-фазы и зон ГП (рис. 169). Экспериментально построенные линии сольвуса про
350
межуточных фаз 6' и 6" и зон ГП в системе А1 — Си показаны на рис. 170.
Кинетика и последовательность образования выделении при старении
Из рис. 168 видно, что в сплаве состава Со уменьшение энергии Гиббса при выделении стабильной p-фазы больше, чем при образовании промежуточной р7-фазы и зон ГП: аеГ>ас>аЬ. Однако это не означает, что при старении всегда больше вероятность образования стабильной фазы. На рис. 168 по оси ординат отложены значения объемной энергии Гиббса. В § 20 было показано, что последовательность образования фаз регулируется не достигаемым уровнем объемной энергии Гиббса, а величиной энергетического барьера при зарождении новой фазы.
Энергетический барьер зарождения — работа образования критического зародыша (AGKp) без учета упругой составляющей равна одной трети его межфазной энергии [см. формулу ' (24)]. У зон ГП межфазная энергия минимальна, а у некогерентных выделений стабильной фазы максимальна. Следовательно, AG^<AG^<AG£p. При старении энергетический барьер зарождения выделений создается не только из-за образования поверхности раздела, но и из-за упругой деформации решетки. Упругая деформация при зарождении полукогерентных выделений р'-фазы может быть больше, чем при зарождении некогерентных
Рис. 169. Диаграмма состояния с линиями сольвуса стабильной фазы р, метастабильной фазы (У и зон ГП (схема)
Рис. 170. Линии сольвуса зон ГП, про-уточных фаз О' и 0" в системе А!—Си (ГП и v" — по данным Биюпа и Роллэсона; О' — по данным Хорнбо-гена)
351
выделений p-фазы. Тогда неравенство AGj?p<AG£p выпол-няется только в том случае, если выигрыш в межфазной энергии перекрывает возможный проигрыш в энергии упругой деформации. Скорость зарождения выделений в стареющем сплаве определяется формулой (25), в которой Q— энергия активации диффузии наиболее медленно диффун
дирующего элемента
Чем больше скорость зарождения, тем меньше инкубационный период—время выдержки до фиксируемого дан-
Рис. 171. C-кривые образования зон ГП, промежуточной фазы 0' и стабильной фазы Р при распаде пересыщенного раствора в стареющем сплаве (схема):
ТУп* ^0' и температуры сольвуса выделений в сплаве Со на рис. 169
ным методом начала выделения. С повышением температуры старения инкубационный период вначале сокращается из-за увеличения диффузионной подвижности атомов, а затем возрастает из-за уменьшения пересы-щенности твердого раствора по отношению к данной фазе.
Зоны ГП, промежуточная и стабильная фазы ха-
рактеризуются своими С-кривыми (рис. 171). Верхние ветви С-кривых асимптотически приближаются к соответствующим температурам сольвуса Ггп, и ,
При заданной температуре старения, т. е. при неизменной диффузионной подвижности атомов, скорость зарождения выделений в соответствии с формулой (25) определяется только величиной AGKp. Первыми появляются выде
ления, для которых работа образования критического
зародыша минимальна, а затем выделяется фаза с большей величиной AGKp. Например, при температуре старения Тх через период времени тгп появляются зоны ГП, затем
по достижении момента тр, появляется промежуточная Р'-фаза и, наконец, после выдержки тр стабильная р-фаза. Такую последовательность образования выделений часто записывают в виде схемы ос->ТП->р'->р.
При температуре старения Г2, превышающей температуру сольвуса зон ГП, из пересыщенного раствора вначале выделяется р'-фаза, а затем p-фаза, т. е. а->р'-^р. При температуре старения Гз, превышающей температуру соль-
352
Таблица 8. Стадии распада пересыщенного раствора в промышленных сплавах
Система Сглав Стадии распада
А1—Си AT—Mg Al—Си—Mg Al—Си—Mg—Fe—Ni Al—Mg—Si Al—Zn—Mg Al—Zn—Mg—Си Си—Be АЛ7 АЛ8 Д16 AK4-1 АД35 1915 B95 БрБ2 Зоны ГП-^0"-*0'->0 (CuA12) Зоны ГП—>Р'—>Р (А!зМ§2) Зоны rn->S'->S (AkCuMg) Зоны ГП->8'->5 (Al2CuMg) Зоны ГП->Р'->Р (Mg2Si) Зоны (MgZn2)-> ->Т (AI2Mg3Zn3) Зоны ГП-^М'—М [Mg(CuZn)2] Зоны ГП->у/~*'¥ (СнВе)
вуса [З'-фазы, из пересыщенного раствора может выделяться только стабильная (3-фаза. Это соответствует общему правилу: чем меньше степень пересыщенности твердого раствора по отношению к стабильной фазе, тем меньше число промежуточных превращений. Выше это правило было продемонстрировано на примере одного сплава: степень пересыщенности раствора уменьшалась с повышением температуры старения, оно справедливо и для сплавов разного состава при постоянной температуре старения. Например, если взять сравнительно малолегированный сплав с составом левее точки Са-гпна рис. 169, то при температуре старения Т{ зоны ГП в нем не могут образоваться.
В табл. 8 приведены примеры последовательности появления выделений в сплавах разных систем с ростом продолжительности старения или температуры старения (при постоянной выдержке). Таблица демонстрирует не последовательность выделений при старении по промышленным режимам, а возможное для данного сплава число стадий выделений в широком диапазоне температур и времен выдержки при старении, в том числе и при режимах, не используемых в промышленности. Например, для большинства сплавов режим старения подбирают так, чтобы не выделялась стабильная фаза (см. § 47 и 48).
Для теории и практики старения (см. § 49) очень важно знать, как зарождаются и растут более стабильные выделения, если имеются ранее образовавшиеся менее стабильные выделения. Запись процесса распада пересыщенного раствора в виде схемы ос->ГП->pz-указывает лишь на временную (при постоянной температуре) или температурную (при постоянной выдержке) последовательность появления разного типа выделений. Эту запись не следует трак-
23 И. И. Новиков О Г-О
товать так, что зоны ГП всегда превращаются вследствие перестройки решетки в р'-фазу, а р'-фаза — в р-фазу.
Возможны три способа образования более стабильных выделений. Первый способ — указанное прямое превращение менее стабильных выделений в более стабильные, т.е. изменение типа решетки, аллотропический переход в пределах объема выделения без участия матрицы (/ на рис. 172). Этот путь может реализоваться только при небольшой разнице в структуре выделений и поэтому встречается редко. При исследовании стареющих сплавов А1 — Сн
Рис. !72. Три варианта зарождения в a-матрице более стабильной фазы р при наличии ранее образовавшейся менее стабильной фазы 0'
получены экспериментальные данные, которые можно трактовать как прямую перестройку зон ГП в выделения (/'-фазы, которые поэтому и были названы зонами ГП2.
Второй способ — зарождение Р'-фазы на зонах ГП и рост выделений Р' в матрице и аналогично зарождение p-фазы на выделениях р' и рост частиц р в матрице (2 на рис. 172).
Третий способ — независимое
зарождение более стабильной фазы в матрице вдали от выделений менее стабильной фазы и зон ГП (3, рис. 172). Выше температуры сольвуса промежуточной фазы стабильная p-фаза может выделяться только из матрицы. Каковы будут места ее предпочтительного зарождения при более низких температурах старения, когда ранее уже вы-
делилась промежуточная фаза, заранее предсказать пока нельзя. Например, в сплавах А1 — Си 6-фаза (СиА12) на
ранних стадиях распада зарождается на границах зерен матрицы, а на поздних — на границах раздела б'-фазы с алюминиевым раствором. В этих же сплавах б'-фаза на
ранних стадиях распада выделяется на дислокациях, а на более поздних зарождается на выделениях 6"-фазы. Некоторые исследования указывают, что 6"-фаза образуется не перестройкой и развитием зон ГП, а прямо выделяется из
хматрины.
Многочисленные данные показывают, что в сплавах системы Л1 — Zn — Mg выделения if-фазы при искусственном старении зарождаются на зонах ГП, образовавшихся при естественном старении. Это явление используют при выборе режимов старения (см. § 49). В бериллиевой бронзе у'-фаза также зарождается на зонах ГП.
354
Если образующаяся позднее стабильная фаза зарождается на ранее появившихся выделениях метастабильной р'-фазы, то при температурах ниже точки пересечения С-кривых этих фаз инкубационный период образования p-фазы может быть существенно меньше, чем при независимом ее выделении из матрицы. Соответственно вместо плавного продолжения С-кривой (участок оа, рис. 173) должна находиться смещенная влево ветвь ob.
Рис. 173. С-кривые выделения из пересыщенного раствора стабильной (|3) и метастабильной ((У) фаз (схема для двух вариантов С-кривой Р-фазы)
своеобразный случай
Если р'-фаза не оказывает инициирующего воздействия на зарождение p-фазы, то в принципе возможно и некоторое смещение кривой оа вправо, в сторону больших выдержек, из-за уменьшения пересыщенности матричного раствора при образовании р'-фазы.
При температруах, близких к сольвусу р7-фазы, инкубационный период ее образования может быть больше, чем у стабильной p-фазы, и тогда в последовательности появления при старении фазы меняются местами. Этот
должен реализовываться в очень узком интервале температур старения от Г) до Тг/, где низка пересыщенность матричного раствора по отношению к р'-фазе (см. рис. 172).
Метастабильная фаза может зарождаться позже стабильной и по другим причинам. Например, на границах зерен стареющего сплава* можно увидеть некогерентные выделения стабильной фазы раньше, чем в объеме зерен появятся частично когерентные выделения метастабильной фазы. Этому способствует отсутствие в объеме зерен дислокаций— мест гетерогенного зарождения метастабильной фазы. Еще один случай выделения стабильной фазы раньше, чем метастабильный, рассмотрен в § 56.
В § 20 было доказано, что образование более стабильной фазы должно приводить к растворению менее стабильной фазы. При старении сплавов это правило играет важную роль. Около зон ГП концентрация матричного раствора равна Са-гп (см. рис. 168 и 169). После образования промежуточной р'-фазы на границе с ней устанавливается концентрация раствора Са-$'. Следовательно, в матрице возникает градиент концентраций Са-гп—Ca-iv. Выравнивающая диффузия в матрице в направлении этого гра-
23*
355
диента создает пересыщение раствора относительно р'-фа-зы и делает его ненасыщенным по отношению к зонам ГП. В результате выделения р'-фазы будут расти вследствие одновременного растворения зон ГП.
Аналогично после образования стабильной p-фазы концентрация раствора на границе с ней <Са-$' и выделения р будут расти вследствие одновременного растворения р'-фазы.
Рис. 174. Структура сплава AI— 15 % Ag, закаленного с 520 “С и состаренного при 160 °C, 10 дней. Электронная микрофотография, фольга. X 22500 (Кларк)
На рис. 174 видны серые области с ранее образовавшимися сферическими зонами ГП и позднее выделившиеся иглы у'-фазы, около которых светлые поля — это участки матрицы, где зоны ГП растворились при одновременном подрастании игл
Коагуляция выделений
В процессе непрерывного распада твердого раствора суммарный объем выделений увеличивается, а концентрация легирующего элемента в растворе снижается. Когда состав матричного раствора становится близким к равновесному при температуре старения, суммарный объем выделений перестает изменяться, но структура состаренного сплава нестабильна — дисперсные выделения склонны
к укрупнению, коагуляции.
Выделения в состаренном сплаве, отделенные одно от другого решеткой матрицы, не могут укрупняться слиянием так, как сливаются капли ртути под действием сил поверх-
ностного натяжения.
Движущей силой коагуляции является разность энергий Тиббса более мелких и более крупных частиц. В состаренном сплаве из-за разных локальных условий роста размеры выделений разные. Чем мельче выделение, тем больше доля атомов, расположенных на его поверхности
356
(по отношению ко всем атомам выделения), и тем, следовательно, выше средняя энергия Гиббса, приходящаяся на 1 моль выделения.
На рис. 175 кривая энергии Гиббса мелких частиц р-фа-зы (G“) расположена выше кривой энергии Гиббса крупных частиц этой же фазы (Gp. Из схемы на рис. 175 видно, что концентрация а-раствора, находящегося в равновесии с мелкими выделениями p-фазы (См), должна быть
Рис. 175. Зависимость от состава энергии Гиббса а-раствора (Ga), крупных (ср) и мелких (G j*) выделений (3-фазы
Рис. 176. Градиент концентрации легирующего элемента в а-растворе между мелким и крупным выделениями (3-фазы (Ски См см. на рис. 175)
выше, чем при равновесии с крупными выделениями (Ск). Этот же вывод следует из уравнения (32).
Таким образом, в матричном растворе существует градиент концентраций легирующего элемента между выделениями разного размера (рис. 176). Этот градиент непосредственно и вызывает коагуляцию. Выравнивающая диффузия понижает концентрацию раствора на его границе с мелким выделением, и оно растворяется, поддерживая равновесную концентрацию раствора на своей границе. Та же диффузия повышает концентрацию раствора на его границе с крупным выделением, раствор здесь пересыщается и выделяет p-фазу, поддерживая равновесную концентрацию Ск. Фаза р выделяется на готовой поверхности крупной частицы, которая таким путем растет при одновременном растворении мелкого выделения вплоть до его полного исчезновения. Следовательно, коагуляция выделений во время старения происходит вследствие переноса вещества через матричный раствор (из-за градиента концентраций) при растворении более мелких и росте более крупных выделений.
Средний радиус частиц г с увеличением времени старе
357
ния при коагуляции изменяется в соответствии с уравнением Лифшица — Слезова:
г3 = го + 5т, (37)
где го — средний начальный радиус выделений перед коагуляцией.
В = 8DyCx VWkT. (38)
Здесь D — коэффициент диффузии; у—межфазная энергия на границе выделения с матрицей; Соо — равновесная концентрация матричного раствора у плоской поверхности раздела с избыточной фазой; V — объем выделения, приходящийся на один атом растворенного элемента.
Скорость коагуляции увеличивается с ростом D и у. Коэффициент диффузии с повышением температуры возрастает по экспоненте, и поэтому коагуляция сильно ускоряется с ростом температуры старения. Этому способствует также увеличение Соа с ростом температуры.
Коагуляция является единственным структурным изменением стареющего сплава после образования выделений стабильной фазы. Но это не значит, что коагулируют только выделения стабильной фазы. Аналогично могут коагулировать и выделения промежуточных фаз и зоны Гпнье— Престона, так как концентрация раствора, находящегося в метастабильном равновесии с ними, зависит от размера соответствующих выделений (в том числе и зон ГП). Поэтому коагуляцию можно наблюдать на разных стадиях распада раствора. Но особенно большой практический интерес она представляет как заключительная стадия распада.
Для некогерентных выделений стабильных фаз характерна большая межфазная энергия у, способствующая коа-гуляциии выделений. Стабильная фаза AhSc, отличающаяся когерентностью выделений в алюминиевых сплавах, из-за малой величины у трудно коагулирует.
§ 47. Изменение свойств сплавов при старении
1. Природа упрочнения при старении
Упрочнение при старении — результат торможения дислокаций теми выделениями, которые образовались при распаде пересыщенного твердого раствора. Можно указать три главные причины упрочнения: 1) торможение дислокаций полем упругих напряжений в матрице вокруг выделе-
358
iniii; 2) упрочнение при перерезании выделении дислокациями; 3) упрочнение при огибании частиц дислокациями Ч
П о л е у п р у г и х напряжений неизбежно возникает в матрице при образовании когерентных и полукоге-рентных выделений, так как когерентность решеток обеспечивается упругой деформацией их около границы раздела (см. рис. 153, а, б). Величина упругих напряжений тем больше, чем больше размерное несоответствие структуры матрицы и выделения, выше модуль упругости матрицы и больше площадь когерентной границы. Для продвижения дислокаций через упругодеформированную матрицу требуется напряжение, превышающее среднее напряжение поля упругих деформаций вокруг выделений. Соответствующее упрочнение является результатом дальнодействующего влияния выделений на дислокации.
Упрочнение при перерезании частиц — результат ближнего взаимодействия дислокаций и выделений, когда дислокации проходят через выделения и они деформируются вместе с матрицей.
Решетка выделения не идентична решетке матрицы, даже если речь идет о полностью когерентном выделении. Поэтому дислокация, входящая со своим вектором Бюргерса в выделение, нарушает укладку атомов вдоль плоскости скольжения. Чем больше отличается строение выделения в плоскости перерезания от строения матрицы в этой же плоскости, тем сильнее нарушение укладки атомов внутри выделения и тем выше требуется напряжение для перерезания выделений дислокациями. В случае когерентного выделения (зоны ГП) поверхностная энергия на плоскости его «среза» составляет величину порядка 102 мДж/м2, а при перерезании некогерентного выделения — порядка 103 мДж/м2 (как на высокоугловой границе).
Модуль сдвига выделения обычно больше, чем у матрицы. Чем жестче выделение, тем труднее дислокации его перерезать.
Еще одна причина торможения дислокаций — образование выступов на перерезанном выделении и соответственно увеличение его поверхности, с которой связан избыток энергии.
Упрочнение при огибании частиц дислокациям и возникает тогда, когда дислокации не перерезают 'выделения. В этом случае необходимо повысить
1 Торможение дислокаций дисперсными частицами подробнее рассматривается в курсах «Кристаллография н дефекты кристаллической
решетки металлов» н «Механические свойства металлов».
359
напряжение, чтобы выгнуть дислокацию между выделениями.
Критическое напряжение сдвига обратно пропорционально расстоянию I между выделениями:
ткр - Gb/tt (39)
где G — модуль сдвига матрицы; b — вектор Бюргерса дислокации.
2. Величина упрочнения при образовании выделений разного типа
Величина упрочнения зависит от типа выделений, их строения, свойств, размера, формы, характера и плотности распределения, степени несоответствия решеток матрицы и выделения, а также от температуры испытания.
Благодаря гомогенному зарождению плотность, распределения зон ГП весьма большая, и расстояние между ними обычно настолько мало (порядка 10 нм), что для огибания зон ГП дислокациями требуются большие напряжения, чем для перерезания зон. Если разница в атомных диаметрах растворимого и растворителя небольшая, то энергия упругих деформаций матрицы мала и упрочнение при перерезании зон ГП является единственной причиной повышения прочности при старении (пример — сплавы А1 — Ag и А1 — Zn). При большой разнице в атомных диаметрах, например в сплавах AI — Си и Си — Be, вокруг зон ГП создается поле значительных упругих напряжений, которое вносит свой вклад в торможение дислокаций зонами и в упрочнение при старении.
Спинодальный распад, на ранних стадиях которого структура отличается очень высокой дисперсностью когерентных областей с разной концентрацией компонентов и равномерностью их распределения по всему объему зерна, может обеспечить большое увеличение предела текучести. Это увеличение пропорционально квадрату амплитуды концентрационной волны (см. рис. 157, а) и, например, у сплава меди с 9 % Ni и 6 % Sn доходит до 500 МПа. Поэтому изыскание сплавов, в которых возможен спинодальный распад, следует рассматривать как один из перспективных пу, тей создания пружинных и других материалов с высоким пределом текучести.
Появившиеся недавно промышленные сплавы системы Си —Ni — Sn, названные спинодальными, после закалки, холодной деформации и старения по уровню прочностных свойств близки к бериллиевым бронзам.
360
Полукогерентные выделения промежуточной фазы могут оказывать сильное упрочняющее влияние, если расстояние между ними мало. При прочих равных условиях поле напряжений вокруг полукогерентных выделений слабее, чем вокруг когерентных, и, следовательно, соответствующая составляющая упрочнения при старении должна быть меньше. Однако выделения промежуточной фазы сильнее, чем зоны, отличаются по структуре от матрицы, и поэтому при их перерезании дислокациями создается большое нарушение укладки атомов. Следовательно, каждое выделение промежуточной фазы способно вызвать более сильное упрочнение при перерезании дислокациями, чем зона ГП, что при достаточно высокой плотности выделений может привести к получению большей прочности состаренного сплава по сравнению с зонной стадией распада. Если же плотность выделений промежуточной фазы значительно ниже, чем зон ГП, то напряжения, способные выгнуть дислокации между выделениями, могут оказаться ниже напряжений, требуемых для перерезания зон ГП. В этом случае прочность сплава на стадии выделений промежуточной фазы ниже, чем на зонной.
Выделения стабильной фазы обычно некогерентны матрице, вокруг них нет полей упругих напряжений, а расстояния между выделениями достаточно велики, и дислокации под действием сравнительно небольших напряжений могут их огибать. Поэтому выделения стабильных фаз обычно вызывают значительно более слабое упрочнение при старении, чем зоны ГП и выделения метастабильных фаз.
Проследим роль типа выделений на примере сплавов А1 — Си. На рис. 177 показаны кривые нарастания истинных напряжений течения при деформирован™ кристаллов пересыщенного твердого раствора AI — 4 % Си. в котором предварительным старением были получены выделения разного типа: зоны ГП, О"-, 0х- или 0-фаза. Рис. 177 позволяет сравнить значения начального (критического) напряжения течения и способность к деформационному упрочнению — нарастанию напряжения течения с ростом степени деформации (по наклону кривых).
Сплав с зонами ГП и когерентными выделениями ©"-фазы отличается высоким начальным напряжением течения и малой величиной деформационного упрочнения. Наклон кривых истинных напряжений сплава с зонами ГП и ©"-фазой небольшой.
В монокристаллах, результаты испытания которых представлены на рис. 177, расстояния между центрами зон
361
ГП и между выделениями б^-фазы равны соответственно 15 и 25 нм. Если подставить расстояние между выделениями в формулу (39), то величина критического напряжения, необходимого для проталкивания дислокаций, окажется в несколько раз больше экспериментальных значений начального напряжения течения. Электронно-микроскопическое исследование сплава А1 — 4% Си показывает, что дислокации не огибают зоны ГП и выделения 0"-фазы, а перерезают их.
Выделения 0"-фазы сильнее повышают уровень напряжений течения сплава А1 — Си, чем зоны ГП, так как во-
Рис. 177. Кривые истинные напряжения — удлинение сплава Al— 4 % Си с монокристальной матрицей и выделениями разного типа (Файн, Бирн и Келли)
круг них в матрице выше упругие напряжения [см. структурное несоответствие решеток матрицы и 0"-фазы по плоскостям (010) и (100) на рис. 166]. Выделения 0"-фазы, сильнее отличающиеся по структуре от матрицы, должны вызывать и большее упрочнение при перерезании их дислокациями, так как при этом сильнее нарушается укладка атомов, чем при перерезании зон ГП.
Начальное напряжение течения в сплаве с выделениями О'-фазы меньше, чем в сплаве с 0"-фазой, а деформационное упрочнение больше (кривая истинных напряжений на рис. 177 идет гораздо круче). Объясняется это тем, что расстояния между выделениями 0'-фазы были достаточно велики и дислокации проталкивались между ними при напряжении меньше необходимого для перерезания выделений. С ростом степени деформации вокруг выделений увеличивается число дислокационных петель, оставляемых каждой скользящей дислокацией. Эти петли затрудняют проталкивание последующих дислокаций, чем и объясняется
362
интенсивный рост сопротивления деформированию с уве^ личением степени деформации.
Аналогичная картина наблюдается в сплаве с выделениями стабильной 6-фазы. Так как эти выделения полностью некогерентны матрице (см. рис. 166), а расстояние между ними еще больше (порядка 1 мкм), то начальное напряжение течения в сплаве со стабильной 6-фазой (СнА12) значительно ниже, чем в сплаве'с зонами ГП и выделениями (У'- и O'-фаз (см. рис. 177). Коэффициент же деформационного упрочнения (наклон кривых) у сплава с 6-фазой больше, чем у сплава с зонами ГП или 6"-фазой, из-за накопления дислокационных петель вокруг выделений.
3. Влияние продолжительности и температуры старения
на механические свойства сплавов
Временное сопротивление, предел текучести и твердость сплава с увеличением продолжительности старения возрастают, достигают максимума и затем снижаются (см. кривые Т2 и Тз на рис. 178).
Старение до достижения максимума прочностных свойств (восходящая ветвь кривых) называют упрочняю-
Bfertfi старения '
Рис. 178. Схе.ма зависимости прочностных свойств от продолжительности старения при разных температурах (Л< <Л<7Д
щим, а правее максимума (нисходящая ветвь) —разупроч-няющим старением или перестариванием. При этом подразумевается разупрочнение по сравнению со сплавом, который подвергался старению более короткое время. По сравнению же с исходным закаленным сплавом (начальная точка на оси ординат) перестаренный сплав может быть значительно прочнее.
3G3
Упрочнение с увеличением времени старения на восходящей ветви кривых может быть вызвано разными причинами. Во-первых, возможны случаи, когда на стадии упрочняющего старения плотность выделений столь велика, что дислокации не могут огибать выделения и перерезают их (при достаточно большом приложенном напряжении). Рост прочностных свойств с увеличением продолжительности старения в этих случаях обусловлен укрупнением выделений (в частности, зон ГП), увеличением плотности их распределения в матрице и появлением трудно перерезаемых частиц более стабильной фазы (например, 6' в дополнение к 6" в сплавах А1 — Си). Во-вторых, возможны случаи, когда на стадии упрочняющего старения дислокации огибают выделения. В этих случаях рост прочностных свойств с увеличением продолжительности старения обусловлен увеличением плотности выделений при развитии распада и соответственно ростом критического напряжения сдвига [см. формулу (39)].
В перестаренном сплаве дислокации не перерезают выделения, а только огибают их при напряжениях меньше тех, которые необходимы для перерезания. Снижение прочностных свойств при переходе от упрочняющего старения к перестариванию может быть вызвано несколькими причинами. Одна причина — увеличение расстояний между ранее образовавшимися выделениями из-за их коагуляции. Другая причина — замена менее стабильных выделений более стабильными, характеризующимися меньшим числом частиц в единице объема матрицы. Третья возможная причина перестаривания — уменьшение или исчезновение поля упругих напряжений в матрице при замене когерентных выделений сначала полукогерентными, а затем и некогерентными.
Если температура старения достаточно низка, то перестаривание не достигается и сплав упрочняется из-за повышения плотности зон ГП и их укрупнения, причем эти процессы и соответствующее упрочнение развиваются с затуханием (см. кривую на рис. 178). Так ведет себя, например. дуралюмин при комнатном старении (рис. 179,а).
С повышением температуры старения достигается стадия перестаривания и тем раньше, чем выше температура (см. рис. 178 и 179, а). Это и понятно, так как все процессы развития распада раствора — диффузионные.
Стабильная фаза обычно выделяется на стадии перестаривания. Если сплав должен эксплуатироваться в максимально упрочненном состоянии, то появление стабильной
364
фазы обычно нежелательно. В связи с этим следует указать на условность понятия фаза-упрочнителъ. Так, например, к фазам-упрочнителям в дуралюминах относят фазу S (Al2CuMg), в сплавах системы А1—Zn—Mg типа 1915— фазу т] (MgZn2), в бериллиевой бронзе — фазу у (СиВе). Но появление этих стабильных фаз в сплавах приводит к перестариванию, разупрочнению и поэтому вредно. Значительно более высокая прочность достигается при образовании зон ГП и промежуточных выделений S', т/ и у'. Но поскольку без специальных структурных исследований ничего нельзя сказать
Рис. 179. Зависимость механических свойств профилей дуралюмина Д!6 от времени старения при 18, 150 и 200 °C
Рис. 180. Суммарное упрочнение при старении (Д/7у) и вклад в это упрочнение зон ГП (Atfrpp и (У-фазы (Д//^*) в разные моменты выдержки при постоянной температуре старения (схема)
о том, какие выделения обеспечивают максимальное упрочнение, а стабильная фаза известна из диаграммы состояния, то ее условно и называют фазой-упрочнителем, хотя в действительности старение проводят при таких режимах, когда сама стабильная фаза вообще не выделяется.
Схема на рис. 178 типична для температурно-временных условий старения, при которых в упрочнении участвуют выделения только одного вида, например р' при температуре То на рис. 171. Если же вначале образуются зоны ГП, а затем выделяется р'-фаза (например, при температуре Л
365
на рис. 171), то возможны несколько вариантов зависимости прочностных свойств от времени старения. Это помогает понять схема (рис. 180), на которой раздельно показан вклад в упрочнение (прирост твердости при старении) от зон ГП и от р'-фазы в разные моменты старения.
Вклад в упрочнение от зон ГП (Л7/гп) сначала возрастает с затуханием, достигает определенного уровня и затем снижается до нуля. Это снижение — результат постепенного растворения зон ГП в матрице при появлении
Рис. 181. Схема зависимости твердости состаренного сплава (Н) от времени выдержки при постоянной температуре старения
Время старения, сдт
Рис. 182. Зависимость твердости по Виккерсу сплава алюминия с 4 % Си от времени старения при 130 °C (Силкок, Хил и Харди)
Р'-фазы или же прямого превращения зон ГП в р'-фазу.
Вклад р'-фазы в упрочнение (А#|у) вначале возрастает, проходит через максимум при наибольшей плотности ее выделений и затем падает в результате коагуляции р'-фазы и по другим причинам, обсужденным выше.
Суммарный вклад в упрочнение при старении от зон ГП и выделений р'-фазы (ДА/^) зависит от того, насколько смещены по времени процессы образования зон ГП и р', и от соотношения абсолютных значений АА/гп и Д7/ф' в разные моменты старения. На рис. 180 кривая прироста твердости при старении (Д# х) имеет перегиб. Такой же вид должен иметь график зависимости твердости (временного сопротивления, предела текучести) сплава от времени старения (кривая 1 на рис. 181), так как у состаренного сплава 7/=//зак+Д7/ у, где Н зак — твердость закаленного сплава.
Кривая 2 на рис. 181 с практически незаметным перегибом на восходящей ветви относится к случаю, когда инкубационный период выделения р'-фазы близок к началу затухания вклада в упрочнение от зон ГП. Минимум на кривой 3 появляется из-за того, что на первых этапах выделения р'-фазы ее вклад в упрочнение не перекрывает
366
уменьшения вклада в упрочнение от растворяющихся зон ГП. Все три типа кривых зависимости прочностных свойств от времени старения наблюдали у сплавов на разных основах, в которых старение было многостадийным. У некоторых сплавов перегиб переходит в плато. На рис. 182 для сплава А1 — 4 % Си показана зависимость твердости от времени старения, в котором участвуют выделения трех типов: зоны ГП, 6" и 6'.
Если перегиб, плато или минимум находятся в интервале таких сравнительно небольших выдержек, при которых по каким-либо причинам старение не изучалось, то экспериментально установленная зависимость прочностных свойств от времени старения описывается простейшей кривой с одним максимумом (см. рис. 179, а).
В разных сплавах и при разных температурах старения одного сплава максимум упрочнения соответствует разным структурным состояниям. Без экспериментов нельзя предсказать, какова должна быть в данном сплаве конкретная структура, обеспечивающая максимальное упрочнение. Ответ зависит от того, какие стадии распада возможны в этом сплаве при данной температуре старения, какова структура выделений, плотность выделений каждого типа, и от других факторов. Можно лишь указать, что чаще всего максимальное упрочнение достигается в сплаве, в котором внутри зерен пересыщенного раствора образовались зоны ГП и выделения промежуточной метастабильной фазы или только выделения этой фазы (при высокой плотности их распределения). Например, у дуралюмина Д16 максимальное упрочнение при искусственном старении (190 °C) достигается на стадии образования частично когерентных выделений промежуточной S'-фазы. У сплава В95 максимальное упрочнение при старении (125 °C) достигается также на стадии выделения промежуточной ЛГ-фазы (см. табл. 8).
В сплаве алюминия с 4 % Си при температуре старения 130 °C максимуму упрочнения соответствует структура с выделениями 0" и O'-фаз (см. рис. 182). Когерентные выделения 6" создают более сильные упругие деформации в матрице, чем полукогерентные выделения О'-фазы. Однако выделения О' значительно труднее перерезать дислокациями. Поэтому сочетание выделений 0" и 6' в определенном соотношении обусловливает максимальную прочность состаренного сплава. Перестаривание сплава алюминия с 4 % Си при 130 СС можно связать, во-первых, с уменьшением плотности выделений 8", которые постепенно заменяются выделениями 8', в результате чего ослабляются поля
367
нить, что соседние точки ветви могут относиться
----*- Температура старения
Рис. 183. Схема зависимости прочности от температуры старения при постоянной выдержке
упругих напряжений в матрице и растет среднее расстояние между частицами, и, во-вторых, с коагуляцией выделений 6'.
Выбирая практически температурный режим старения, часто строят графики в координатах прочность — температура старения (рис. 183). На таком графике всегда имеются участки повышения и снижения прочности с ростом температуры старения. Анализируя такие кривые, следует пом-на восходящей (или нисходящей) к разным стадиям старения при разных температурах. Например, при выдержке п с ростом температуры старения от 7\ до Т3 прочность непрерывно возрастает (см. рис. 178). Вместе с тем точки а и b соответствуют стадии упрочняющего старения при температурах Л и Т2у а точка с — стадии перестаривания при температуре Т3. Если время выдержки равно т2> то с ростом температуры старения от до Г3 прочность
падает (точки d, е и f), причем при температуре Т\ сплав находится на стадии упрочняющего старения, а при температурах Т2 и Т3— на стадии перестаривания..
М. В. Захаров, обобщив данные для сплавов на разных основах, установил, что температура старения на максимальную прочность и твердость составляет определенную долю от температуры солидуса (по абсолютной шкале): Гстар^^н-О.б)^. (40)
Это эмпирическое соотношение нельзя рассматривать как формулу, позволяющую по точкам плавления точно рассчитывать значения температур старения на максимальную прочность. Оно позволяет оценивать лишь ориентировочный уровень таких температур, если сравнивать сплавы с сильно различающимися точками солидуса и, следовательно, с резко разной диффузионной подвижностью компонентов при одинаковой температуре (например, сплавы на базе разных металлов).
При повышении температуры старения прочность сплава может оказаться ниже, чем в исходном закаленном состоянии (см. рис. 183). Такое сильное перестаривание вызвано далеко зашедшей коагуляцией выделений и сильным уменьшением легированное™ матрицы. Соответствующую
368
термообработку иногда неточно называют отжигом, хотя сущность процессов здесь та же, что и при обычном старении: распад раствора и коагуляция выделений.
Относительное удлинение при упрочняющем старении существенно снижается (см. рис. 179,6), а при развитии перестаривания чаще всего меняется незначительно, продолжая слабо снижаться, или же слабо возрастает.
§ 48. Влияние состава сплава на старение
1. Влияние состава в двойных системах
Рис. 184. Схема зависимости максимально возможного прироста твердости при старении от состава сплавов в двойной системе:
А/7 — разность значений твердости состаренного и закаленного сплава
На рис. 184 линия Атпр схематично показывает, как влияет содержание легирующего элемента в двойном сплаве на прирост твердости при старении по режиму, обеспечивающему максимальное упрочнение1. Подобный график может характеризовать влияние состава и на прирост временного сопротивления или предела текучести при старении.
В сплавах с концентрацией компонента В ниже Ci старение невозможно, так как в них нельзя получить пересыщенный твердый раствор (закалка невозможна). Во всех сплавах с концентрацией легирующего элемента больше С! при закалке фиксируется пересыщенный твердый раствор и старение возможно. Если эти сплавы подвергать старению по оптимальному для каждого из них режиму, обеспечивающему максимальное упрочнение, то можно ожидать, что с увеличением концентрации
второго компонента прирост твердости при старении будет возрастать (участок тп), достигать максимума и затем постепенно снижаться (участок пр).
В сплаве С3 при прочих равных условиях можно получить более высокую плотность выделений, чем в сплаве С2, из-за большего пересыщения твердого раствора. Сле
1 Влияние температуры и времени старения на максимальную прочность у разных сплавов одной системы может существенно различаться.
24 И. И. Новиков 369
довательно, у сплава Сз можно получить большее упрочнение, чем у сплава С2.
Теоретически эффект старения должен быть максимален у сплава состав которого отвечает точке предельной растворимости при эвтектической температуре. Практически же невозможно получить а-раствор состава С5, так как для этого потребовалось бы закаливать сплав точно с температуры плавления эвтектики. Так как температуру закалки во избежание пережога выбирают ниже температуры солидуса, то максимальная пересыщенность раствора и максимальное упрочнение при старении достигаются при концентрации легирующего элемента в сплаве несколько левее точки предельной растворимости, например в сплаве состава С4.
Уменьшение упрочнения на участке пр объясняется следующим. В сплавах С6 и С7, закаленных с одной температуры, а-раствор имеет одинаковый состав (точка г). Следовательно, в этих сплавах плотность выделений в a-фазе после старения при одинаковой температуре будет одной и той же и прирост твердости а-раствора в обоих сплавах должен быть одинаковым. Но в закаленных сплавах С6 и С7, кроме первичных a-кристаллов, находится еще и избыточная p-фаза из эвтектики. В сплаве С7 ее больше, а a-фазы меньше, чем в сплаве Cq (rs>rq). Так как упрочнение при старении происходит в результате распада а-раствора, то из-за меньшего его количества прирост твердости сплава С7 должен быть ниже, чем у сплава С6. Иными словами, при одинаковом приросте микротвердости первичных a-кристаллов твердость всего сплава С7 при старении растет слабее из-за большего количества «балластной» p-фазы, не участвующей в старении.
Прочность состаренного сплава зависит от исходного уровня — прочности закаленного сплава. Так как прочность а-раствора возрастает с увеличением в нем концентрации легирующего элемента, то сплавы, близкие по составу к точке предельной растворимости при эвтектической температуре, обладают высокой прочностью в закаленном состоянии и большим упрочнением при старении. Отсюда следует вывод, что составы наиболее прочных стареющих сплавов находятся на диаграммах состояния вблизи точек предельной растворимости.
2. Влияние состава в тройных системах
Закономерности влияния состава на старение сплавов тройной системы качественно такие же, как и в двойной
370
в однофазной области, которых находятся на одной
Рис. 185. Изотермические разрезы тройной системы при температуре закалки (сплошные линии) н комнатной температуре (пунктир)
системе. Зная изотермические разрезы при температуре закалки (сплошные линии на рис. 185) и при более низкой температуре, например комнатной (пунктирные липни), можно предсказать, возможно ли вообще старение в любом интересующем нас сплаве, а для сплавов из одной фазовой области — где следует ожидать большего упрочнения. Например, сплав 1 не способен к старению, так как он при комнатной температуре нахе Сплавы 2, 3, 6 и 7, составы коноде, по эффектам при старении аналогичны сплавам С2, С3, С6 и С? в двойной системе на рис. 184.
Сплавы 2 и 3 при температуре закалки расположены в однофазной «-области, а при комнатной температуре — в двухфазной области «+Р- Следовательно, их можно закаливать на пересыщенный «-раствор и подвергать старению. Пересы-щенность раствора в сплаве 3 больше, чем в сплаве 2, и поэтому сплав 3 должен сильнее упрочняться при старении (полная аналогия
с двойными сплавами С2 и С3 на рис. 184). Сплавы 6 и 7 также способны упрочняться при старении. Так как они находятся на одной коноде, то состав «-раствора в них при температуре нагрева под закалку один и тот же (точка г). Но величина упрочнения при старении сплава 7 должна быть ниже из-за большего количества «балластной» р-фа-зы, не перешедшей в «-раствор при нагреве под закалку (полная аналогия со сплавами С6 и С7 на рис. 184).
Сплавы 4, 5 и 8 также можно подвергать старению. От сплавов 2, 3. 6 и 7 они отличаются выделяющимися фазами. В сплаве 4 может выделяться -у-фаза, а в сплавах 5 и 8 — фазы р и -у.
В какой фазовой области расположены наиболее прочные стареющие сплавы, обычно предсказать не удается, так как даже если состав и строение стабильных фаз известны, то чаще всего мы заранее ничего не знаем о типе и структуре промежуточных метастабильных выделений. А эти выделения, как правило, и обеспечивают максималь
24*
371
ное упрочнение при старении. Наглядным примером могут служить промышленные сплавы на базе системы А1—Zn— Mg (типа 1915). В этих сплавах согласно диаграмме состояния в стабильном равновесии с алюминиевым а-раст-вором находится тройное соединение Т (Al2Mg3Zn3), а при старении, обеспечивающем высокую прочность, выделяется т)'-фаза, промежуточная между а-раствором и фазой т] (MgZn2). Предсказать подобные случаи пока невозможно.
Несмотря на определенные ограничения, диаграмма состояния все же необыкновенно ценна при выборе состава и режима термической обработки стареющего сплава. Она указывает, в области каких составов следует искать стареющие сплавы, позволяет выбрать интервал закалочных температур (см. § 31), температурный уровень старения на максимальную прочность [см. соотношение (40)] и выявить для экспериментального опробования составы сплавов вблизи границы растворимости при температуре закалки. Чем больше разница в предельной растворимости при эвтектической (перитектической) и комнатной температурах (С3— Ci на рис. 184), тем большее упрочнение следует ожидать при закалке и старении сплавов, так как при закалке можно получить большую пересыщенность твердого раствора.
Если уже известно, что в тройной системе А—В—С сплавы из двухфазной области а+р сильно упрочняются при старении, то в другой системе А—В—D термически упрочняемые сплавы следует прежде всего искать в области составов, где из а-раствора может выделяться p-фаза. Такой подход существенно облегчает поиск высокопрочных стареющих сплавов, ориентируя исследователя на определенные фазовые области диаграммы состояния.
3. Влияние малых добавок и примесей
Специальные добавки и случайные примеси, содержащиеся в сплаве в тысячных — десятых долях процента, иногда сильно влияют на кинетику распада раствора, структуру л свойства состаренного сплава.
Влияние добавок, прямо связанное с образованием новой фазы, ниже не рассматривается, так как такая добав* ка действует как обычный компонент сплава. Механизмы влияния малых добавок, не вызывающих качественного изменения фазового состава, могут быть разными. Добавка может характеризоваться высокой энергией связи с вакансиями, и ее атомы в твердом растворе действуют как ло
372
в ушки для вакансий. Такое действие оказывают добавки Cd, In, Sn и Be в алюминиево-медном растворе.
Захват вакансий примесными атомами приводит к уменьшению их участия в транспортировке атомов основного легирующего элемента (меди в сплавах А1—Си) к зонам ГП. Малые добавки Cd, In, Sn и Be задерживают образование и укрупнение зон ГП в сплавах А1—Си и упрочнение при старении замедляется.
Добавка может оказаться горофильной и сегрегировать на границу раздела матрицы с выделением, уменьшая здесь межфазную энергию. Так, в сплаве А1—4 % Си на границе О'-фазы с алюминиевым раствором у=1530 мДж/м2, а в том же сплаве, но с добавкой 0,1 % Cd у=250 мДж/м2. При снижении межфазной энергии уменьшается в соответствии с формулой (24) работа образования критического зародыша, т. е. растет число выделений в единице объема. Кроме того, уменьшение энергетического барьера при зарождении выделений под действием горофильной добавки может сократить инкубационный период. Так, в сплаве А1—3,5 % Си при 160 °C инкубационный период образования О'-фазы составляет ~3ч, а добавка 0,15% Cd уменьшает его до ч.
В соответствии с формулами (37) и (38) при уменьшении межфазной энергии замедляется коагуляция выделений. Например, малая добавка кадмия в пять раз снижает скорость коагуляции выделений О'-фазы в сплаве Al-Си. Торможение коагуляции особенно ценно для стареющих жаропрочных сплавов, в которых таким путем затрудняется разупрочнение во время эксплуатации изделия при повышенных температурах.
Еще один практически важный эффект от малой добавки кадмия — снижение температуры сольвуса 0"-фазы в сплавах А1—Си. Так, в сплаве А1—5 % Си добавка 0,15 % Cd снижает сольвус 0"-фазы, с ~210 до ~ 170°C и одновременно ускоряет образование O'-фазы. В результате в сплаве с кадмием при 170 °C вместо 0"-фазы выделяется O'-фаза, являющаяся более сильным упроч-нителем (при достаточной плотности ее выделений). Именно этим обусловлено полезное влияние малой добавки кадмия к некоторым промышленным сплавам на базе системы А1—Си, для которых температура промышленного режима старения как раз и составляет 170—180 °C.
Таким образом, одна и та же малая добавка может по-разному влиять на процессы старения в разных температурных интервалах. Добавка 0,15—0,20 % Cd к сплаву А1—5 % Си, задерживая образование зон ГП, замедляет
373
и уменьшает упрочнение при естественном старении. Она же повышает плотность выделений б'-фазы при 220 СС, усиливая упрочнение. Наконец, эта же добавка при температуре старения 170°C приводит к замене менее эффективного упрочнителя (0") на более эффективный (О').
Еще один механизм влияния малой добавки связан с вхождением ее в состав зон ГП и стабилизацией зон. Примесь 0.25 % Si в сплаве АК4-1 (система А1—Сн—Mg—ге— Ni) увеличивает плотность выделений S'-фазы и измельчает этИ' выделения при 190 СС, повышая тем самым прочность сплава. Предполагается, что атомы кремния входят в состав зон ГП, делают зоны стабильными до более высоких температур так, что на них, а не на дислокациях зарождается S'-фаза при 190 СС. Так как плотность распределения зон ГП, зарождающихся гомогенно, очень высокая, то плотность выделений S'-фазы оказывается повышенной.
Концентрирование атомов малой добавки в выделяющейся фазе может так понизить ее объемную энергию Гиббса, что уменьшится работа образования критического зародыша и возрастет плотность выделений (при смещении вниз кривой Gp на рис. 151 увеличивается разность объемных энергий Гиббса G{—G^ являющаяся термодинамическим стимулом превращения). Возможно, что так действует малая добавка серебра в сплавах А1—Zn—Mg. Под ее влиянием в этих сплавах измельчаются выделения т^-фазы.
Введение малых добавок и регулирование содержания примесей является одним из наиболее эффективных путей управления процессами старения.
§ 49. Выбор режима старения
1. Выбор температуры и продолжительности старения
После предварительной оперен температурного уровня старения по соотношению (46) "или по аналогии с другими сплавами на базе того же металла экспериментально отрабатывают режим старения, строя графики, подобные рис. 178 и 183.
Как известно, старение подразделяют на естественное, происходящее при комнатной температуре, и искусственное, требующее нагрева до определенной температуры.
В большинстве стареющих сплавов вылеживание при комнатной температуре после закалки не дает такого изменения свойств, которое можно было бы практически использовать. Механические свойства закаленных медных, 374
никелевых и многих других сплавов вообще не изменяются при комнатной температуре, так как она слишком низка для достаточного развития в них диффузионных процессов.
В алюминиевых сплавах (дуралюминах и др.) образование зон ГП при естественном старении приводит к сильному упрочнению, что широко используют в промышлен^ ности.
Понятия «естественное» и «искусственное» старение характеризуют условия его проведения, но однозначно не определяют характер структурных изменений в пересыщенном твердом растворе. Если исключить из рассмотрения легкоплавкие сплавы, у которых естественное старение протекает при высокой гомологической температуре (для свинцовых сплавов — около 0,5 Тгл) и приводит к далеко зашедшему распаду, то можно считать, что у большинства сплавов при естественном старении образуются только кластеры. В то же время при искусственном старении в зависимости от его температуры и продолжительности распад раствора останавливается или на зонной стадии, или на стадии выделения промежуточных фаз либо доходит до коагуляции выделений стабильной фазы.
И. Н. Фридляндер предложил параллельно с понятием естественное и искусственное старение использовать понятия зонное и фазовое старение. Зонное старение алюминиевых сплавов может быть естественным и искусственным и заканчивается на стадии образования зон ГП. Фазовое старение алюминиевых сплавов, как правило, бывает искусственным. Исключение составляют многолетние выдержки при комнатной температуре сплавов на базе системы Al—Zn—Mg. Практически важно, что сплав после зонного и фазового старения характеризуется разным комплексом свойств.
Для зонного старения алюминиевых сплавов характерны большое относительное удлинение (6> 10-^15 %), значительная разница, между временным сопротивлением и пределом текучести (оо,г/оъ—0,7-:-0,8), высокое сопротивление удару и стойкость против коррозии под напряжением. Зонное старение бывает только упрочняющим. _
Фазовое старение может быть упрочняющим и разупроч-няющим (перестаривание)1. Для упрочняющего фазового старения характерны пониженное относительное удлинение, малая разница между временным сопротивлением и пре
1 Перестаривание на стадии коагуляции выделений можно назвать коагуляционным старением.
373
делом текучести (oro>2/orB = 0,8ч-0,95), пониженная ударная вязкость и пониженная стойкость против коррозии под напряжением.
Подразделение старения на зонное и фазовое в значительной мере условно. Во-первых, условно само подразделение выделений на зоны ГП и промежуточные фазы (см. § 46), что нашло отражение в двойственном названии зоны ГП2 — фаза 6". Во-вторых, при распаде раствора зоны ГП заменяются выделениями промежуточных фаз постепенно и могут сосуществовать с ними длительное время (см. рис. 182). В-третьих, моменты появления промежуточной фазы или полного исчезновения зон ГП могут не отразиться на кривых изменения свойств.
Без прямых структурных исследований, измеряя только свойства, нельзя однозначно сказать, с каким структурным состоянием мы имеем дело. Однако подразделение старения на зонное и фазовое, несмотря на его условность, полезно, так как позволяет ориентироваться в выборе режима старения для получения определенного комплекса свойств.
При выборе оптимального режима старения часто исходят из требования достичь максимальной прочности. Но для многих изделий критерием оптимальности режима старения служит не максимальная прочность, а сочетание разных свойств.
В зависимости от режима, структурных изменений и получаемого комплекса свойств искусственное старение можно подразделить на полное, неполное, перестаривание и стабилизирующее старение (соответствующие режимы и свойства приведены в табл. 9 для литейного алюминиевого сплава АЛ9).
Полное искусственное старение проводят при такой температуре и продолжительности, которые обеспечивают достижение максимальной прочности.
Таблица 9. Режимы старения и свойства сплава АЛ9
Разповиднссть старения Обозначение режима Температура нагрева, сС Выдержка, ч ав’ МПа ао.2 МПа f>. %
Без предваритель- ной закалки Т1 170—180 5—17 170 140 2
Неполное Т5 145—155 1—3 230 170 4
Полное Тб 195—205 2—5 240 210 2
Стабилизирующее Т7 215—230 3—6 190 140 4,5
376
Неполное искусственное старение — это старение с более короткой выдержкой или при более низкой температуре, чем полное, с целью повысить прочность при сохранении достаточной пластичности. Режимы неполного,старения соответствуют восходящим ветвям кривых на рис. 178 и 183. Некоторая потеря возможного прироста прочности компенсируется меньшим снижением пластичности.
Перестаривание — это старение при более высокой температуре или большей выдержке, чем полное, с целью получить сочетание повышенных прочности, пластичности, коррозионной стойкости, электропроводности и других свойств. Режимы перестаривания соответствуют нисходящим ветвям кривых на рис. 178 и 183. По сравнению с неполным старением перестаривание при той же прочности обеспечивает большую степень распада твердого раствора и коагуляцию выделений, что часто позволяет добиться требуемого комплекса разнообразных свойств.
Стабилизирующее старение — это разновидность перестаривания, целью которого является стабилизация свойств и размеров изделия.
Жаропрочные сплавы, предназначенные для длительной службы, обычно подвергают старению при температуре выше рабочей. В противном случае при эксплуатации изделия в нем будут активно протекать структурные изменения, приводящие к разупрочнению и нестабильности свойств изделия. Очень часто термическую обработку жаропрочных сплавов проводят в режиме перестаривания.
Режим старения следует выбирать с учетом условий закалки. С повышением температуры нагрева под закалку нз однофазной области (выше То в сплаве Со на рис. 106) старение ускоряется из-за повышения концентрации закалочных вакансий, которая входит в предэкспоненциальный множитель А в выражении (25) для скорости зарождения новой фазы. Таким образом, С-кривые распада раствора на рис. 171 с повышением температуры закалки сдвигаются влево, причем этот сдвиг больше в низкотемпературной области, где роль закалочных вакансий особенно велика.
Некоторые сплавы подвергают старению без специального нагрева под закалку. В таких случаях пересыщение раствора достигается ускоренным охлаждение с температуры конца затвердевания отливки или горячей обработки давлением. Упрочнение здесь не достигает максимально возможного для данного сплава из-за меньшей пересы-щенности твердого раствора1, но экономическая эффектив-
1 См. режим Т1 в табл. 9.
377
пость (исключение операции закалки) делает указанное старение целесообразным для ряда деталей. Для отдельных сплавов, например для сплава МЛ 12 системы Mg— Zn—Zr, старение отливок без специального нагрева под закалку является основным способом термической обработки.
Скорость охлаждения после старения не влияет на свойства сплава. Обычно с температуры старения изделия охлаждают на воздухе.
2. Ступенчатое старение
Старение с выдержкой вначале при одной, а затем при другой температуре называют ступенчатым. Как правило, температуру первой степени выбирают ниже, чем второй. Основная цель двухступенчатого (двойного) старения—создать большее число центров выделений на низкотемпературной ступени, когда пересыщенность твердого раствора велика (на рис. 152 степень пересыщенности C0/Ci растет с понижением температуры 7i), а затем на высокотемпературной ступени получить необходимую степень распада раствора и оптимальный размер выделений. В результате достигаются более высокая плотность и однородность распределения выделений, чем это возможно при одноступенчатом старении при повышенной температуре.
Допустим, что по условиям эксплуатации необходимо иметь искусственно состаренный сплав с выделениями промежуточной фазы. Если эта фаза при оптимальной температуре старения зарождается гетерогенно на дислокациях, границах зерен и субзерен, то плотность ее выделений сравнительно низкая. Если при этом она способна зарождаться на зонах ГП, то предварительное низкотемпературное старение, в том числе и естественное, может резко увеличить плотность выделений промежуточной фазы и измельчить ее на высокотемпературной ступени старения. Примером являются сплавы на базе системы А1—Zn—Mg (типа 1915), старение которых при 150—175°C обеспечивает повышенную коррозионную стойкость. В этих сплавах зоны ГП, образующиеся на низкотемпературной ступени (100°C), увеличивают плотность выделений т)'-фазы (см. табл. 8) на высокотемпературной ступени (175 °C), и в результате двойного старения достигается сочетание повышенной прочности и сопротивления коррозии под напряжением. Роль низкотемпературной ступени здесь может выполнять и естественное старение в течение одного месяца, но более эф
378
фективно проводить первое старение при 100СС в течение 10—20 ч.
Другой пример — двухступенчатое старение сплава В95пч. Если этот сплав подвергнуть одноступенчатому зонному старению на максимальную прочность по режиму Т1 (120 СС, 24 ч), то коррозионная стойкость окажется пониженной. Двухступенчатое же старение по режиму ТЗ (115 СС, 5 ч+165°С, 25 ч) при несколько меньшем временном сопротивлении обеспечивает высокое сопротивление коррозионному растрескиванию и расслаивающей коррозии и одновременно высокую вязкость разрушения. На первой ступени образуются зоны ГП, которые на второй ступени способствуют зарождению большего числа выделений М'-фазы. Кроме того, выделяется стабильная М-фаза (Mg, Си) Zn2 и частично проходит коагуляция выделений.
В производственных условиях изделия не всегда сразу после закалки можно загрузить в печи для искусственного старения, т. е. между закалкой и искусственным старением неизбежен перерыв, иногда весьма длительный. Следовательно, естественное старение, даже если оно специально не планируется, обычно предшествует искусственному, которое фактически является высокотемпературной ступенью двойного старения (непланируемое естественное старение называют вылеживанием после закалки). Поэтому закономерности ступенчатого старения представляют более широкий интерес, чем может показаться на первый взгляд. Эти закономерности бывают весьма сложными и часто не соответствуют нарисованной выше упрощенной схеме благотворного влияния предстарения.
Рассмотрим практически важный случай сложной роли естественного старения на примере сплавов системы А1— Mg—Si, находящихся на разрезе А1—Mg2Si или недалеко от него (сплавы типа авиаль). В этих сплавах при естественном старении образуются игольчатые зоны ГП, обогащенные магнием и кремнием, а при искусственном (170°С) — метастибильная р'-фаза (см. табл. 8). Макси-нальное упрочнение достигается при искусственном старении.
Давно было известно, что перерыв между закалкой и искусственным старением сплавов типа авиаль снижает их прочность в искусственно состаренном состоянии. Электронная микроскопия показала, что это обусловлено огрублением структуры. Одно из объяснений вредного влияния естественного старения сводится к следующему. При образовании зон ГП во время естественного старения матрпч-
379
нг<й раствор вокруг них обедняется легирующими элементами. При нагреве до температуры искусственного старения степень пересыщенности раствора еще больше снижается, так как возрастает равновесная растворимость (С\ на рис. 152). Каждой степени пересыщенности соответствует свои минимально возможный размер выделений (в том числе и зон ГП). С уменьшением пересыщенности этот размер возрастает. При нагреве естественно состаренного сплава до температуры искусственного старения мелкие зоны ГП растворяются и только более крупные служат центрами зарождения р'-фазы — структура получается грубой. Если же после закалки сплав сразу подвергнуть искусственному старению, то в начальный его период пересыщен-ность раствора будет высокой и в нем гомогенно зарождается большое число мелких выделений р'-фазы.
Предложено несколько способов предотвратить снижение свойств искусственно состаренных сплавов, обусловленное вылеживанием после закалки. Первый способ, самый простой, но не всегда выполнимый — ограничение времени вылеживания или хранение изделий после закалки при пониженной температуре (в холодильнике). Другой способ— кратковременный (1—3 мин) промежуточный нагрев изделия из сплавов типа авиаль до ~250°C перед окончательным старением при 160—170°C. Во время такого нагрева зоны ГП, образовавшиеся при естественном старении, полностью растворяются, и сплав оказывается в состоянии, аналогичном свежезакаленному (см. обработку на возврат, § 50). Самый эффективный способ — введение малых добавок, задерживающих естественное старение.
Снижение свойств из-за вылеживания после закалки характерно не только для сплавов А1—Mg—Si. Оно наблюдалось и в стареющих сплавах других систем. В самой же системе А1—Mg—Si вредное влияние вылеживания свойственно сплавам, содержащим не менее ~1°/о Mg2Si. В сплаве с 0,8 % Mg2Si комнатное предстарение не снижает, а повышает прочность в искусственно состаренном состоянии. Объясняется это тем, что из-за меньшей леги-рованности магнием и кремнием и соответственно меньшей пересыщенности твердого раствора в свежезакаленном сплаве с 0,8 % Mg2Si р'-фаза зарождается при 170 °C толь-го гетерогенно, на дислокациях, и структура получается грубой. Комнатное предстарение, создавая зоны ГП, являющиеся центрами зарождения р'-фазы, измельчает структуру искусственно состаренного сплава с 0,8 %
380
MgzSi и делает более равномерным распределение выделений этой фазы.
Таким образом, ступенчатое старение в зависимости от состава сплава и температуры ступеней может быть полезным или вредным.
Если зоны ГП не влияют на характер выделения метастабильной фазы, то зонное старение не сказывается на последующем фазовом старении. Например, свойства дуралюмина Д16 после искусственного (фазового) старения не зависят от длительности перерыва между закалкой и этим старением, несмотря на то, что вылеживание при комнатной температуре приводит к развитию зонного старения.
С ролью предстарения тесно связан вопрос о роли скорости нагрева при одноступенчатом старении. Обычно на скорость нагрева до температуры старения не обращают внимания. Однако начальные стадии распада при замедленном нагреве могут влиять на свойства состаренного сплава. Так, например,, замедленный нагрев до температуры старения некоторых алюминиевых сплавов позволяет несколько повысить их прочность.
Выше обсуждалось ступенчатое старение, у которого первая ступень — низкотемпературная. Несравненно реже используют старение вначале при повышенной, а затем при более низкой температуре. Интересный пример такой термообработки— старение нимоника ЭИ437Б (система Ni— Сг—Al—Ti). При старении этого сплава из матричного никелевого раствора с г. ц. к. решеткой выделяется когерентная у'-фаза состава Ni3(Al, Ti), имеющая также г. ц. к. решетку. Важная особенность сплава состоит в том, что упрочнение при старении растет с увеличением объемного содержания у'-фазы и практически не зависит от размера ее выделений в широком диапазоне размеров. В связи с этим для достижения максимального упрочнения необходимо добиваться выделения возможно большего количества у'-фа-зы. Чем ниже температура старения, тем меньше предельная растворимость легирующих элементов в никелевом растворе и, следовательно, больше должно выделиться у'-фа-зы. Так, при 700 °C можно получить около 15 % (объемн). у'-фазы, а при 850°C — только 9 %. Однако для получения при 700 °C максимального упрочнения, соответствующего ~15% (объемн.) у'-фазы, необходимы выдержки продолжительностью в несколько сотен часов. Если же нимоник вначале выдержать 2 ч при 850 °C, то последующее старение при 700 °C в течение всего лишь 10 ч обеспечит выделение ~15% (объемн.) у'-фазы. Таким образом, двойное
381
старение 850+700 °C с суммарной выдержкой около 12 ч дает такой же эффект, как одинарное старение при 700 °C в течение нескольких сотен часов.
Роль предварительной высокотемпературной выдержки заключается в следующем. При 700 °C выделяются дисперсные частицы у'-фазы (размером в нанометры), которые коагулируют очень медленно. В соответствии с уравнением Томсона — Фрейндлиха (32) концентрация раствора, находящегося в равновесии с частицами малого радиуса, выше концентрации раствора на границе с более крупны ми частицами. Во время предварительного старения при 850 СС выделяются частицы у'-фазы сравнительно большого размера (~20 нм) и при последующем старении при 700 СС дораспад идет за счет роста этих частиц, на границе с которыми концентрация раствора ниже и соответственно пересыщенность его выше, чем в случае выделения очень дисперсных частиц при одинарном старении. Этим обеспечивается большая полнота до распада при 700 °C за время, которое на порядок (!) меньше времени достижения того же объемного количества выделения у'-фазы при одинарном старении. Рассмотренный способ ускорения старения неприменим к тем сплавам, для которых режим старения выбирают в расчете на получение предельно высокой дисперсности и плотности выделений.
О величине прироста прочностные свойств при старении промышленных сплавов можно судить по данным табл. 10. В ней разность значений свойств состаренного и закаленного сплава отнесена к значению свойства исходного закаленного сплава. Данные приведены для режимов старения на максимальную прочность.
Предел текучести при старении повышается сильнее, чем временное сопротивление, и отношение Оо>2/ов возрас-
та б л п н а 10. Прирост временного сопротивления н предела текучести промышленных сплавов в результате полного старения
Металлов нова Марка сплава 4%. % Д<т0 2. % °0.2/ап
после закалки после старения
Ti ВТ22 72 80 0,93 0,98
Си БрБ2 160 — — 0,94
А1 Д16 50 80 0,73 0,88
1915 70 170 0,50 0,78
АЛО 20 90 0,55 0,87
МЛ5 2 40 0,34 0,47
382
тает. Это — типичная картина при полном искусственном старении.
Бериллиевая бронза и алюминиевый сплав 1915 на базе системы А1—Zn—Mg сильно упрочняются при старении, а магниевый сплав МЛ5—слабо упрочняется (поэтому сплав МЛ5 чаще используют в закаленном состоянии).
Сплавы, упрочняющиеся при старении, называют термически упрочняемыми, дисперсионнотвердеющими или облагораживаемыми. Разработка термически упрочняемых сплавов — одна из главных задач металловедения.
3. Старение под напряжением 1
В последние годы было показано, что нагрузка, приложенная в период старения, может существенно улучшить некоторые эксплуатационные свойства сплавов, главным образом характеристики сопротивления малым и микропласти-ческим деформациям (пределы пропорциональности и упругости, релаксационную стойкость) при практически неизменных временном сопротивлении и относительном удлинении. Указанное улучшение свойств используют при изготовлении упругих элементов, например мембран из бериллиевой бронзы.
С ростом приложенной нагрузки достигаемый эффект возрастает, однако из-за развития ползучести в период старения может возникнуть нежелательное изменение конфигурации изделия. Чтобы избежать этого и одновременно достичь максимального эффекта улучшения свойств, можно применить программированное ступенчатое нагружение по мере упрочнения сплава при старении. Например, у бериллиевой бронзы БрБНТ1,9 Мг после обычного старения при 340 СС, 4 ч предел упругости оОСЮ5 = 730 МПа, а после старения со ступенчатым, через каждый час, увеличением напряжения (с поддержанием его на уровне 0,65 002)00005 = =950 МПа.
Разновидностями рассматриваемой термической обработки являются достаривание под напряжением после старения без приложенной нагрузки и двухступенчатое старение с приложением нагрузки на одной из ступеней.
Улучшение свойств бериллиевой бронзы в результате старения под напряжением можно объяснить тем, что выделяются главным образом такие частииы у'-фазы (см. табл. 8), габптусная плоскость которых параллельна оси приложения нагрузки или составляет с ней малый угол (см. § 46, п. 2). При этом увеличивается плотность распрсделе-
1 Его называют также динамическим старением.
383
ния выделений вдоль оси нагружения при старении. В процессе эксплуатации упругого элемента нагрузка прикладывается в том же направлении, что и при старении.
§ 50. Возврат после старения
Явление возврата после старения 1 было открыто на дура-люмине. Если естественно состаренный дуралюмин нагреть до температуры примерно 250 °C, выдержать 20—60 с и быстро охладить, то его* свойства возвращаются к значениям, характерным для свежезакаленного состояния.
Сущность явления возврата состоит в том, что зоны ГП, возникшие при естественном старении, во время нагрева сплава выше их сольвуса (выше Тгп, но ниже Т$' для сплава Со на рис. 169) растворяются, а метастабильные и стабильные фазы из-за короткой выдержки не успевают обра-
Рис. 186. Изменение временного сопротивления дуралю-мииа при естественном старении и двукратной обработке на возврат после старения (Д. А. Петров)
зоваться и быстрое охлаждение фиксирует пересыщенный твердый раствор. Температура обработки дуралюмина на возврат (~250°С) далека от температуры нагрева под закалку (~500°C), необходимого для растворения стабильных фаз.
После обработки на возврат дуралюмин, как и после обычной перезакалки, способен упрочняться при естественном старении. Повторная обработка на возврат вновь его разупрочняет и т. д. (рис. 186). Так как при каждом нагреве до температуры возврата все же успевает в небольшой степени пройти необратимое фазовое старение (особенно по границам зерен), то с каждым циклом свой-
1 Его не следует смешивать с возвратом после холодной деформации.
384
ства несколько отклоняются от свойств свежезакаленного сплава. В частности, усиливается склонность дуралюмина к межкристаллитной коррозии.
Обработку на возврат можно применить, когда требуется восстановить пластичность дуралюмина перед гибкой, отбортовкой и т. п., а перезакалка нежелательна из-за коробления. Основной технологический недостаток обработки на возврат — необходимость строгого (с точностью до 10 с) регулирования времени выдержки изделий в селитряной ванне. Если эта выдержка окажется короче оптимальной, то не все зоны ГП растворятся и разупрочнение будет неполным, а при передержке упрочнение произойдет из-за начавшегося фазового старения. По этой причине, а также из-за снижения коррозионной стойкости возврат после старения дуралюминов не нашел применения.
В теории старения явление возврата играет большую роль, так как позволяет оценить стабильность зон ГП, полученных в разных условиях старения, и определить температуру их растворения.
Температура нагрева для полного возврата свойств (температура растворения зон ГП) в общем случае не является константой для данного сплава. Она зависит от стабильности зон, а значит от температуры и времени их образования. Чем больше продолжительность низкотемпературного старения, тем крупнее и стабильнее зоны ГП и тем выше должна быть температура нагрева для их полного растворения. Начиная с некоторой выдержки при старении температура полного возврата не зависит от времени старения. Такая критическая температура возврата характерна для сплава, состаренного при определенной температуре, и она растет с повышением температуры старения из-за увеличения стабильности зон ГП. Максимально возможная, предельная температура, до которой наиболее стабильные зоны ГП могут существовать в данном сплаве, отвечает точке кривой сольвуса зон на диаграмме метастабиль-иых равновесий (см. рис. 169).
Из сказаного следует, что нельзя определять температуру сольвуса зон ГП, нагревая, например, естественно состаренный сплав. В таких опытах определяют не максимально возможную, предельную температуру существования зон ГП в данном сплаве, а какую-то более низкую температуру растворения зон, образовавшихся за определенное время при комнатной температуре. Линия сольвуса зон ГП, показанная на рис. 170, фактически сольвусом не является, так как она указывает температуры растворения зон, об-
25 И. И. Новиков
385
разевавшихся в течение 10 дней комнатного старения. Линия истинного сольвуса зон ГП в системе А1—Си должна проходить выше, чем это изображено на рис. 170. Например, у сплава А1—4 % Си максимальная температура существования зон ГП равна не 180 °C, а ~275 °C.
Возврат можно наблюдать не только после старения с образованием зон ГП и не только в алюминиевых сплавах. Обработкой на возврат в принципе можно растворять и зоны ГП, и выделения метастабильных фаз в сплавах на разной основе. Для каждого сплава и режима старения необходимо подбирать свою температуру (/в) и время выдержки (тв) при обработке на возврат. Например, для естественно состаренных дуралюминов рекомендуются следующие режимы:
Д1 Д16 Д19
сС......... 240—250 265—275 270—280
тп с........... 20—45 15—30 10—15
Глава XII
ОТПУСК
Отпуск — это термическая обработка закаленного на мартенсит сплава (или металла), при которой главными процессами являются распад и (или) возврат и рекристаллизация мартенсита.
Ниже рассмотрены процессы отпуска только сталей, так как они составляют подавляющее большинство промышленных сплавов, закаливаемых на мартенсит. Несмотря на многочисленность исследований отпуска с широким привлечении рентгеноструктурного анализа, электронной микроскопии, измерений разнообразных физических свойств, до сих пор трактовки отдельных важных процессов, происходящих при отпуске сталей, спорны. Причины этого — многообразие параллельно идущих структурных изменений, высокая дисперсность выделений на ранних стадиях отпуска и трудность расшифровки структуры выделений.
Структура закаленной стали метастабильна. При нагреве после закалки вследствие увеличивающейся подвижности атомов создаются условия для процессов, изменяющих структуру стали в направлении к более равновесному состоянию. Характер этих процессов определяется тремя важнейшими особенностями строения закаленной стали: сильной пересыщенностью твердого раствора — мартенсита, повышенной плотностью в нем дефектов кристаллической решетки — дислокаций, малоугловых и высокоугло-386
вых границ, двойниковых прослоек и присутствием во многих сталях значительных количеств остаточного аустенита.
Распад мартенсита с выделением карбидов — главный процесс при отпуске сталей. Закономерности распада мартенсита во многом сходны с закономерностями распада пересыщенного раствора при старении сплавов, подвергающихся закалке без полиморфного превращения. Распад мартенсита в зависимости от температуры и продолжительности отпуска проходит через стадии предвыделения, выделения промежуточных метастабильных карбидов, выделения цементиту и коагуляции.
Структурные изменения при отпуске могут осложняться распадом остаточного аустенита.
Повышенная плотность дислокаций из-за аккомодационной деформации во время мартенситной перестройки решетки делает субструктуру мартенсита похожей на субструктуру наклепанного металла (см. § 35). В результате при отпуске создается стимул к развитию полигонизации и рекристаллизации.
К процессам отпуска целесообразно относить совокупность любых структурных изменений при нагреве после закалки на мартенсит, а не только распад пересыщенного раствора. Тогда нагрев после закалки чистых полиморфных металлов, в которых прошло мартенситное превращение, и сплавов, в которых мартенсит не является пересыщенным раствором (например, закаленный сплав Со на рис. 112), тоже следует относить к отпуску. Во всех таких материалах при мартенситном превращении возникает повышенная плотность дислокаций, в результате чего оказывается возможным проводить отпуск, при котором структурные изменения состоят только из полигонизационных и рекристаллизационных процессов. Этим отпуск принципиально отличается от старения, которое применимо лишь к сплавам, содержащим пересыщенный твердый раствор1.
§ 51. Структурные изменения при отпуске сталей
L Отпуск углеродистых сталей
Характер структурных изменений при отпуске углеродистых сталей зависит от температуры и продолжительности
1 Предлагаемый подход к понятию отпуска чистых металлов и ненасыщенных твердых растворов со структурой мартенсита условен. Соответствующую термообработку в принципе можно относить и к отжигу. Однако обязательность предварительной закалки и специфика субструктуры мартенсита позволяют предпочесть в этом случае термин «отпуск».
25*
387
отпуска и содержания углерода в стали. С повышением содержания углерода в аустените возрастает пересышенность сс-раствора, снижается температура Мк, происходит переход от пакетного мартенсита к пластинчатому и увеличивается количество остаточного аустенита. Все это сказывается на процессах отпуска.
Сегрегация углерода в кристаллах мартенсита является первым структурным изменением при отпуске углеродистых сталей. Возможны два разных по природе процесса сегрегации углерода: образование примесных атмосфер на дефектах решетки мартенсита и возникновение кластеров.
Дефекты кристаллической решетки — энергетически более выгодные места для атомов углерода, чем нормальные позиции этих атомов в решетке мартенсита. Атомы углерода упруго притягиваются к дислокациям и дислокационным стенкам. Такие сегрегации углерода успевают образовываться на структурных несовершенствах в мартенсите уже в период закалочного охлаждения (если точка Л4Н высокая) и при комнатной температуре сразу после закалки.
Другой процесс сегрегации не связан с притяжением атомов углерода к структурным несовершенствам. При температурах отпуска до ~ 100 °C в мартенсите высокоуглеродистых сталей электронографически обнаружены кластеры — плоские скопления атомов углерода. Естественно, что с образованием таких скоплений связано значительное смещение атомов железа, т. е. упругое искажение решетки мартенсита. С повышением температуры отпуска эти кластеры укрупняются. Образование кластеров углерода можно трактовать как зонную стадию распада раствора, аналогичную концентрационному расслоению при старении твердых растворов замещения.
В свежезакаленном мартенсите перераспределение углерода заметно уже при —40 °C. Вначале на каждый кластер приходится лишь несколько атомов углерода. Кластеры постепенно укрупняются, а содержание углерода в них становится соответствующим составу, который можно описать формулой Fe4C. Этот процесс контролируется диффузней углерода.
Выделение промежуточных карбидов из мартенсита — следующая после сегрегации углерода стадия структурных изменений при отпуске. Начиная примерно с температуры 100 °C экспериментально обнаруживается ме-тастабильный е-карбид (Fe2C), отличающийся от цементи-388
та типом решетки (гексагональная у е-карбида, ромбическая у цементита).
Промежуточный карбид образуется прямо из кластеров углерода путем небольшой перестройки их решетки с одновременным увеличением соотношения концентраций углерода и железа. Он может также выделяться непосредственно из а-раствора независимо от кластеров углерода. В сталях, содержащих менее 0,2 % С, е-карбид не образуется.
По отношению к матрице е-карбид ориентирован так, что (ООО 1) е II (011) м, чем обеспечивается когерентность па границе раздела фаз. При низких температурах е-карбид выделяется в виде очень дисперсных (10—100 нм) пластин или стержней. 'С повышением температуры или увеличением продолжительности отпуска частицы е-карбида укрупняются.
Предпочтительное образование промежуточного карбида вместо более стабильного цементита Fe3C можно объяснить тем, что на границе мартенсита с е-карбидом сопряжение решеток лучше и, следовательно, поверхностная энергия ниже, чем на границе матрицы с цементитом.
Согласно некоторым электронно-микроскопическим исследованиям первой фазой, выделяющейся из мартенсита, является не гексагональный е-карбид, а т]-карбид (Fe2C) с ромбической решеткой, которая также хорошо сопрягается с решеткой мартенсита. Этот карбид обнаружен при температурах отпуска 100—200 °C. В высокоуглеродистых сталях после отпуска при 250—300°C преобладают выделения ромбического %-карбида (Fe5C2), сменяющие выделения т]-карбида.
В сталях с высокой точкой Л4Н, т. е. во многих конструкционных сталях, частичный распад мартенсита с выделением промежуточного карбида успевает пройти во время закалочного охлаждения в мартенситном интервале. Следовательно, в период закалки таких сталей происходит самоотпуск.
Образование цементита Fe3C со структурой, одинаковой или близкой к структуре цементита отожженной стали, происходит при температурах выше 250°C, причем наиболее активно в интервале 300—400 °C.
Цементит Fe3C — более стабильная фаза, обладающая меньшей объемной энергией Гиббса, чем промежуточный карбид.
Установлены два механизма зарождения цементита.
389
Во-первых, цементит выделяется прямо из пересыщенного а-раствора, причем рост частиц Fe3C сопровождается растворением выделений ранее образовавшегося менее стабильного карбида (см. § 20). Во-вторых, цементит образуется перестройкой решетки промежуточного карбида в решетку Fe3C (в пределах объема частиц промежуточного карбида).
При повышении температуры и (или) увеличении продолжительности отпуска возрастает отношение Fe/C в карбиде: от ~2 (е и ц) до ~2,5 (х) и 3 (цементит).
Коагуляция и сфероидизация цементита— завершающая стадия процессов карбидообразования при отпуске. При сравнительно низких температурах цементит растет в виде дисперсных пластин, полукогерентных матрице. Размер цементитных пластин различен. Концентрация углерода в а-растворе около относительно мелких частиц выше, чем около более крупных [см. формулу (32)]. Эта разность концентраций обеспечивает диффузию углерода в а-растворе от более мелких цементитных частиц к более крупным. В результате выравнивающей диффузии а-раствор становится ненасыщенным около мелких частиц и пересыщенным около крупных. Более мелкие цементитные частицы растворяются, а более крупные подрастают. Цементит выделяется из а-раствора на крупных частицах вдали от их вершин и ребер, и форма крупной частицы приближается к сферической. Таким образом, переносом вещества через раствор осуществляются коагуляция и сфероидизация цементита при отпуске стали. Ниже 350 °C эти процессы развиты очень слабо. По-настояшему интенсивная коагуляция и сфероидизация начинаются с 350— 400*эС. Выше 600 СС все частицы цементита сферические и идет только их коагуляция. При изотермической выдержке коагуляция цементита интенсивно развивается в течение короткого времени (первого часа) и затем затухает. Средний размер цементитных частиц растет с повышением температуры отпуска.
Распад остаточного аустенита играет существенную роль в процессах отпуска высокоуглеродистых сталей, где он находится в значительном количестве. Распад аустенита активно протекает в интервале температур примерно 200—300 ЭС. Остаточный аустенит при отпуске превращается в нижний бейнит.
Главное внимание выше было уделено карбидообразо-ванию при отпуске. Не менее важны изменения состояния матричной фазы, включающие уменьшение концентрации
390
углерода в а-растворе, и изменения субструктуры и микроструктуры а-фазы.
Уменьшение концентрации углерода в а-растворе идет во всем температурном интервале выделения из него карбидной фазы. По изменению дифракционной картины на рентгенограмме Г. В. Курдюмов подразделил распад мартенсита на две стадии.
" Первая стадия распада мартенсита—так называемый «двухфазный» распад (ниже 150°C).
Подвижность атомов углерода при температурах ниже 150 ЭС еше слишком мала. Эта подвижность вполне достаточна, чтобы обеспечить образование карбидных пластин за счет углерода из ближайшего мартенситного окружения. Но она недостаточна, чтобы обеспечить рост выделившихся частиц карбида за счет диффузии углерода из участков мартенсита, еще не охваченных распадом и имеющих исходную высокую концентрацию углерода. В результате такого распада мартенсит становится неоднородным с различным содержанием углерода в разных участках. В тех участках, где выделился карбид, концентрация углерода и, следовательно, степень тетрагональности меньше, чем в участках, не затронутых распадом. Два твердых раствора с разной концентрацией углерода сосуществуют длительное время из-за низкой скорости диффузии, и поэтому распад называется «двухфазным». «Двухфазный» распад мартенсита развивается не вследствие роста карбидных частиц из окружающих их участков обедненного углеродом раствора, а в результате выделения новых частиц карбида в участках мартенсита с исходной концентрацией углерода.
Так как выделившиеся при низких температурах карбидные частицы чрезвычайно мелки, то они находятся в метастабильном коллоидном равновесии с окружающим их раствором, содержащим избыток углерода [см. формулу (32)]. Для дальнейшего обеднения углеродом участков раствора, окружающих карбид, необходимо укрупнение карбидных частиц.
Вторая стадия распада мартенсита — обеднение углеродом а-раствора при одновременном росте карбидных частиц (150—300 °C). Скорость диффузии углерода при температурах выше 150°C достаточна, чтобы обеспечить рост карбидных частиц при переносе атомов через а-рас-твор. Поэтому выше 150 °C одновременно с «двухфазным» распадом происходит обычный диффузионный рост карбидных частиц. Но все же до 300 °C кристаллы карбида рас-
391
тут медленно и остаются чрезвычайно мелкими. Кинетика такого распада характеризуется тем, что а-раствор быстро обедняется углеродом в сравнительно короткий промежуток времени, зависящий от температуры, а затем обеднение раствора углеродом быстро затухает. При 300 ЭС в а-рас-творе остается около 0,1 % С. Выше этой температуры обычный рентгеноструктурный анализ не обнаруживает разницы между решетками а-раствора и a-железа. Ниже 300 ЭС степень тетрагональности (с/а>1) еще измерима. При 400 гС или при несколько более высокой температуре а-раствор полностью освобождается от избытка углерода и переход мартенсита в феррит заканчивается.
Возврат и рекристаллизация в a-фазе происходят в широком интервале температур отпуска. Развитие этих процессов сдерживается частицами карбидных выделений, закрепляющих отдельные дислокации, дислокационные стенки и высокоугловые границы. Закрепление слабее выражено в низкоуглеродистых сталях, где соответствующие процессы изучены подробнее.
Нижнюю температурную границу возврата при отпуске трудно указать. Изменения дислокационной структуры a-фазы, отчетливо различимые при электронно-микроскопическом анализе, начинаются с температур около 400°С. Протяженность малоугловых границ, существовавших в реечном мартенсите, при температурах отпуска выше 400 °C в доли секунды резко падает. Одним из механизмов этого может быть «рассыпание» дислокационных стенок, о котором упоминалось при рассмотрении коалесценции субзереи во время отжига холоднодеформированного металла (см. § 6). В первые моменты отпуска карбидные выделения еще малочисленны и поэтому не являются эффективными стопорами для малоугловых границ и отдельных дислокаций. Затем выделение большого числа карбидиых частиц стабилизирует структуру матрицы. Вытянутость реек а-фазы в низкоуглеродистых сталях сохраняется до высоких температур отпуска.
Формирование центров рекристаллизации a-фазы и развитие их в рекристаллизованные зерна, подобно тому, как это происходит при первичной рекристаллизации холоднодеформированного металла, при отпуске сталей не наблюдались, несмотря на высокую плотность дислокаций в мартенсите.
После достаточно длительного отпуска при высоки?; температурах (выше ~ 600 °C), когда в результате коагуляции цементитных частиц закрепление границ ослабевает, 392
происходит рекристаллизационный рост зерен миграцией исходных высокоугловых границ. Микроструктура при этом теряет характерные морфологические признаки реечного мартенсита.
В высокоуглеродистых сталях из-за сильного торможения миграции границ частицами цементита рекристаллизационный рост зерен а-фазы идет еще труднее и «игольчатый» характер структуры сохраняется до температур отпуска около 650 °C.
Из сказанного видно, что при отпуске закаленной углеродистой стали протекают разнообразные процессы, которые по времени и температурному интервалу своего развития накладываются один на другой.
Все указанные выше температурные границы структурных изменений разного типа весьма условны. Они снижаются при увеличении продолжительности отпуска и смещаются вверх или вниз с изменением содержания углерода в стали.
Традиционно принято выделять три температурных интервала и соответствующие им три «превращения» при отпуске углеродистых сталей. Это подразделение, основанием для которого в свое время послужил анализ объемных изменений при отпуске, весьма условно, но как первое приближение его можно принять.
Первое «превращение» при отпуске относят к интервалу температур 100—200°С. При этих температурах закаленный образец укорачивается. Так как из всех структурных составляющих стали наибольший удельный объем у мартенсита, то первое «превращение» связывают с его распадом.
Второе «превращение» при отпуске относят к интервалу температур 200—300 °C. При выдержке в этом интервале Длина закаленных образцов средне- и высокоуглеродистых сталей увеличивается и тем больше, чем выше содержание углерода в стали. Так как удельный объем аустенита наименьший и количество остаточного аустенита растет с увеличением содержания углерода в стали, то второе «превращение» связывают с его распадом. При этом, конечно, следует иметь в виду, что в температурном интервале второго «превращения» продолжается распад мартенсита.
Третье «превращение» при отпуске относят к интервалу температур 300—400 °C. В этом температурном интервале длина образцов сокращается. Природа третьего «превращения» остается дискуссионной. В интервале этого
393
«превращения» исчезают промежуточные карбиды и заканчивается выделение Fe3C из мартенсита.
Структуру, получающуюся после отпуска стали при температурах ниже 300 СС, называют отпущенным мартенситом. Под микроскопом он отличается от мартенсита закалки большей травимостью из-за выделений карбидов. После отпуска при 300—450 °C обнаруживается особенно сильно травящаяся игольчатая структура, которую называют трооститом отпуска. В интервале температур 450— 650 °C получается сорбит отпуска. Его двухфазное строение отчетливо выявляется при больших увеличениях светового микроскопа. Высокие температуры отпуска приводят к потере игольчатого вида сорбита, который приобретает явно точечное строение.
2. Влияние легирующих элементов
Диффузионная подвижность атомов легирующих элементов, растворенных в a-железе по способу замещения, на - много порядков ниже, чем диффузионная подвижность атомов углерода, который растворен в железе по способу внедрения. При температурах отпуска ниже примерно 400°C в матрице не происходит диффузионного перераспределения легирующих элементов; из а-раствора вначале выделяется е-карбид, а затем цементит, в которых концентрация легирующих элементов такая же, как в мартенсите. Атомы легирующих элементов в решетке промежуточного карбида и цементита, образующихся при температурах ниже ~400°C, частично замещают атомы железа: (Ее, Сг)3С, (Fe, V)3C и т. д.
Цементит выделяется в виде топких пленок и грубых пластинок па высокоугловых границах, а также в виде ориентированных пластинок внутри а-матрицы.
На первую стадию распада мартенсита, т. е. на «двухфазный» распад при температурах ниже 150 °C, легирующие элементы не оказывают какого-либо существенного для практики влияния. Это согласуется с тем, что на первой стадии главный процесс — зарождение карбидных частиц,— зависит в основном от пересыщенности а-раствора углеродом, а диффузионный рост карбидных выделений и в углеродистых сталях развит очень слабо.
На вторую стадию распада мартенсита многие легирующие элементы влияют очень сильно, замедляя рост карбидных частиц и сохраняя пересыщенность а-раствора углеродом, т. е. сохраняя состояние отпущенного мартенсита до температур 450—500 °C. Так действуют, например, до
394
бавки Сг, W, Mo, V, Со и Si. Задержка распада мартенсита объясняется двумя причинами.
Во-первых, такие легирующие элементы, как ванадии, молибден и хром, снижают скорость диффузии углерода в в а-растворе. Этим нельзя объяснить задерживающее влияние кобальта и кремния, которые не уменьшают коэффициента диффузии углерода в железе. Другая причина — повышение прочности межатомных связей в решетке а-раствора под влиянием таких элементов, как Со, Si, Сг, Мо и W, при котором затрудняется переход атомов через границу а-раствор — карбид и, следовательно, затрудняется распад мартенсита.
На карбидные превращения при отпуске легирующие элементы сильно влияют при температурах выше ~450?С, когда становится возможным их диффузионное перераспределение. В результате этого влияния образуются специальные карбиды. Возможны два механизма их появления. Во-первых, концентрация карбидообразующего легирующего элехмепта в результате его диффузионного перераспределения между а-раствором и цементитом возрастает до такой величины в цементите, что он превращается в специальный карбид. Например, легированный цементит (Fe, Сг)3С так превращается в карбид хрома (Сг, Ре)?Сз. Во-вторых, специальный карбид может зародиться прямо в пересыщенном легирующим элементом а-растворе. Вначале вблизи дислокаций образуются кластеры из атомов легирующего элемента и углерода размером 3—5 им. При повышении температуры до ~550 °C они преобразуются в частицы специального карбида, например VC или ЛАозС.
Очень важно (см. § 52), что выделения таких карбидов, как VC, TiC, ЛА02С и W2C, намного мельче растворяющихся частиц цементита. Одна из причин этого — малая диффузионная подвижность атомов легирующих элементов.
С повышением температуры или увеличением длительности отпуска одни промежуточные метастабильные карбиды могут заменяться другими, более стабильными. Например, в молибденовых и вольфрамовых сталях первым специальным карбидом, образующимся при отпуске вслед за цементитом, является карбид Ме2С (Мо2С или \У2С). Одновременно с ним или несколько позже появляется карбид Ме23С6, а еще позже — карбид Ме6С.
Последовательность карбидообразования в указанных сталях можно записать в виде схемы:
Fe3C^ Ме2С + TWe2Sq. -> Л1е6С.
393
Легирующие элементы влияют на скорость коагуляции карбидных частиц. Никель ускоряет коагуляцию, а хром, молибден, вольфрам, ванадий и некоторые другие элементы затрудняют ее. Элементы, усиливающие межатомную связь в решетке а-раствора и карбида (в последнем случае сильные карбидообразователи) и уменьшающие скорость диффузии углерода в а-растворе. затрудняют переход атомов через границу карбид — а-раствор »и перенос углерода через раствор. Такие элементы задерживают растворение мелких и рост крупных частиц при коагуляции.
Легирующие элементы, входящие в состав специальных карбидов, сами должны диффундировать в а-растворе от растворяющихся мелких частиц к растущим более крупным частицам карбида. Из-за малой скорости диффузии легирующих элементов коагуляция специальных карбидов идет медленно. Даже легированный цементит (Fe, Сг)С3в углеродистой стали с добавкой лишь 0,1 % Сг коагулирует намного медленнее, чем Fe3C.
Полигонизационные и рекристаллизационные процессы при отпуске могут задерживаться под действием добавок легирующих элементов, во-первых, из-за замедления диффузионных процессов переползания дислокаций и, во-вторых, в результате закрепления дислокаций, малоугловых и высокоугловых границ трудно коагулирующими дисперсными частицами специальных карбидов. Особенно эффективны в этом отношении высокодисперсные карбиды Nb и Ti, образующиеся при небольшом (не более 0,1 %) содержании этих добавок в стали.
Большинство легирующих элементов повышает температурный интервал распада остаточного аустенита. Если при отпуске углеродистой стали остаточный аустенит распадается в интервале 200—300 °C, то в легированной стали он сохраняется до 500—600 °C В закаленной высоколегированной высокоуглеродистой стали, например в быстрорежущей, содержится большое количество остаточного аустенита. Если такую сталь отпустить при 500—600 °C, то остаточный аустенит приобретает способность к мартенситному превращению при охлаждении с температуры отпуска. Причиной этого является выделение карбидов из остаточного аустенита и обеднение его углеродом и легирующими элементами при высокотемпературном отпуске. В результате мартенситная точка А4Н повышается и остаточный аустенит становится способен к мартенситному превращению при охлаждении с температуры отпуска.
396
§ 52. Изменение механических свойств при отпуске сталей и выбор режима отпуска
1. Углеродистые стали
Закаленная углеродистая сталь характеризуется не только высокой твердостью, но и очень большой склонностью к хрупкому разрушению. Кроме того, при закалке возникают значительные остаточные напряжения. Поэтому закалку углеродистых сталей обычно не применяют как окончательную операцию, хотя она и может сообщить стали высокую прочность (ов= 13004-2000МПа). Для увеличения вязкости и уменьшения закалочных напряжений после закалки применяют отпуск.
Рис. 187. Зависимость твердости углеродистых сталей разного состава от температуры отпуска (Г. В. Курдюмов)
НВ вв.НПа
Рис. 188. Влияние температуры отпуска на механические свойства стали 45
Распад мартенсита, казалось бы, должен приводить к дисперсионному твердению, и в общем случае зависимость прочностных свойств стали от температуры отпуска должна быть качественно такая же, как и при старении цветных сплавов. Однако на рис. 187 видно, что до температуры отпуска около 100сС твердость закаленной стали или практически не меняется, или слабо (на 1—2 единицы HRC) возрастает. С дальнейшим повышением температуры отпуска твердость плавно снижается.
Почему же распад мартенсита с выделением мелких частиц карбидов при низкотемпературном одпуске закаленной стали не вызывает сильного дисперсионного твердения аналогично дисперсионному твердению алюмшше-
397
вых и других стареющих сплавов? Объясняется это тем, что из-за высокой подвижности атомов углерода они успевают образовывать сегрегаты на дислокациях уже в период закалочного охлаждения, причем дисперсионное твердение может дойти до стадии максимального упрочнения^ Поскольку углерод, растворенный в a-железе, вносит большой вклад в упрочнение мартенсита (см, §38), то обеднение раствора углеродом при выделении промежуточных карбидов (например, е-карбида) уже при низких температурах отпуска вызывает разупрочнение.
С ростом температуры отпуска разупрочнение усиливается из-за следующих причин: 1) уменьшения концентрации углерода в а-растворе; 2) нарушения когерентности на границе карбид—матрица и снятия упругих микронапряжений; 3) коагуляции карбидов и увеличения межчастичного расстояния; 4) развития возврата и рекристаллизации. В разных температурных интервалах преобладает действие разных факторов разупрочнения в соответствии с интенсивностью развития тех или иных структурных изменений (см. § 51). В высокоуглеродистых сталях, содержащих значительное количество остаточного аустенита, распад его с выделением карбида задерживает падение твердости, а в интервале температур 200—250 °C даже несколько увеличивает ее (см. рис. 187).
Так как упрочняющий отпуск закаленной углеродистой стали не имеет практического значения, то часто с отпуском любых сталей связывают представление об обязательном смягчении, хотя, как будет показано ниже, это представление ошибочно.
Прочностные характеристики углеродистой стали (временное сопротивление, предел текучести и твердость) не прерывно уменьшаются с ростом температуры отпуска выше 300 °C, а показатели пластичности (относительное удлинение и сужение) непрерывно повышаются (рис. 188).-Ударная вязкость начинает интенсивно возрастать при отпуске выше 300 °C. Максимальной ударной вязкостью обладает сталь с сорбитной структурой, отпущенная при 600 °C. Некоторое снижение ударной вязкости при температурах отпуска выше 600 °C можно объяснить тем, что частицы цементита по границам ферритных зерен, растущие вследствие растворения частиц внутри а-фазы, становятся слишком грубыми.
По температуре нагрева различают низкий, средний и высокий отпуск.
Низкий отпуск на отпущенный мартенсит (120—250 °C)
398
широко применяют после закалки инструментов, цементованных и планированных изделий и после поверхностной закалки. Цель низкого отпуска — уменьшение остаточных закалочных напряжений; температуру низкого отпуска выбирают такой, чтобы твердость и износостойкость не снизились или слабо снизились. Выдержка при температуре низкого отпуска обычно не превышает 1—3 ч; с дальнейшим увеличением выдержки остаточные напряжения очень слабо уменьшаются.
Разновидность низкого отпуска — стабилизирующий отпуск. В закаленной стали даже при комнатной температуре, а тем более в результате климатических колебаний температуры происходят медленные (в течение многих Лет) процессы распада мартенсита, перехода остаточного аустенита в мартенсит и снятия напряжений. Все эти явления ведут к постепенному изменению размеров изделия. Для таких изделий, как мерительный инструмент высокого класса точности и прецизионные подшипники, недопустимы изменения размеров даже на несколько микрометров. Поэтому размеры изделий необходимо стабилизировать. Вредное влияние остаточного аустенита устраняют, уменьшая его количество при обработке холодом (см. §41). Стабилизации мартенсита и напряженного состояния достигают низким (стабилизирующим) отпуском при 100—180 °C с выдержкой до 30, а иногда и до 150 ч.
Средний отпуск на троостит (350—450 °C) — сравнительно редкая операция. Ее используют тогда, когда необходимо сочетание высокой прочности, упругости и вместе с тем достаточной вязкости. Среднему отпуску подвергают пружины и рессоры.
Высокий отпуск на сорбит (450—650 СС) широко применяют в машиностроении к изделениям из конструкционной стали, которые должны характеризоваться не только достаточной прочностью, но и хорошей сопротивляемостью ударным нагрузкам. Выдержку при высоком отпуске (обычно несколько часов) подбирают опытным путем для получения заданного комплекса свойств.
Квазиэвтектоидную сорбитную структуру можно получить нормализацией непосредственно из аустенита при охлаждении стали, причем достигают твердости, равной твердости стали после высокого отпуска. Однако при одинаковой твердости относительное сужение и ударная вязкость будут значительно выше у отпущенной стали. Объясняется это тем, что твердость зависит главным образом от дисперсности феррито-цементитной смеси, а на 01 носитель-
399
ное сужение и ударную вязкость сильно влияет форма цементита. В сорбите, полученном при распаде аустенита, цементит имеет форму длинных пластин, а в сорбите отпуска — форму коротких пластинок с округлыми краями или сфероидальную форму, обеспечивающую более высокую вязкость стали.
Двойная операция получения сорбита — закалка с высоким отпуском — называется улучшением. Эту операцию применяют к соеднеуглеродистым сталям, содержащим от 0,35 до 0,6 % С.
Такие стали называют улучшаемыми в отличие от низкоуглеродистых цементуемых.
Качество закалки сильно сказывается на свойствах стали после высокого отпуска. Деталь при улучшении должна прокаливаться насквозь. В противном случае после высокого отпуска внутренние слои при одинаковой твердости будут иметь меньшую ударную вязкость, чем внешние слои, так как первые будут содержать пластинчатый цементит, а вторые — точечный или зернистый. Если при охлаждении не было полностью подавлено выделение избыточного феррита во внутренних слоях, то понижается не только ударная вязкость, но и временное сопротивление, твердость и особенно усталостная прочность.
Например, заготовка из стали 45 диаметром 15 мм прокаливается в воде насквозь, и в центре ее после высокого отпуска получается структура зернистого сорбита, характеризующаяся следующими свойствами: ов = 800 МПа, по,2=650 МПа, 6 = 16 % и KCV—100 Дж/см2. Центральные слои заготовки диаметром 100 мм охлаждаются со скоростью значительно меньше критической скорости закалки. При этом в центральных слоях получается структура пластинчатого сорбита с избыточным ферритом, которая характеризуется пониженными показателями прочности и пластичности по сравнению со структурой зернистого сорбита: ов = 700 МПа, о0 2=450 МПа, 6 = 13% и ЯСУ = 50 Дж/см2.
Скорость охлаждения с температуры отпуска не влияет на механические свойства углеродистых сталей, и если не опасны термические напряжения, то можно проводить ускоренное охлаждение.
Иногда закалку и отпуск совмещают в одной операции, которую называют закалкой с самоотпуском. Изделие кратковременно погружают в воду или обрызгивают водой. Поверхностный слой закаливается па мартенсит и затем отпускается за счет тепла внутренних слоев изде
400
лия. Таким способом проводят сорбитизацию поверхностного слоя головки рельса.
В низкоуглеродистых сталях, у которых точка конца мартенситного превращения Л1К находится выше 300 °C, структурные изменения, характерные для отпуска, успевают протекать во время закалочного охлаждения в мартенситном интервале Мн—Мк и ниже температуры Л1К.
Явление такого самоотпуска было использовано К. Ф. Стародубовым в предоложенном им процессе термического упрочения проката. Этот процесс состоит в том, что в условиях непрерывного потока продукции прокатного стана прокат интенсивно охлаждается водой непосредственно на выходе из стана. Изделия из низкоуглеродистых сталей (арматура железобетона, листы и др.) после достижения температуры Л4К охлаждаются на воздухе. При таком прерванном охлаждении происходит закалка с самоотпус-ком. Исключение из технологического процесса самостоятельной операции отпуска позволило на крупных металлургических заводах избежать строительства большого числа отпускных печей. Дополнительное по сравнению с обычной закалкой упрочнение проката происходит благодаря тому, что в условиях интенсивного теплоотвода сразу по выходе из прокатного стана затруднено развитие рекристаллизации деформированного аустенита (см. §60 об упрочении при ВТМО).
2. Легированные стали
Легирующие элементы, затрудняющие распад мартенсита и коагуляцию карбидов (см. §51), смещают температурную границу начала интенсивного разупрочнения при отпуске с 200—300 до 450—550 °C. Повышение красностойкости закаленной стали, т.е. способности ее сопротивляться смягчению при нагреве, — одна из основных целей легирования в производстве инструмента.
Для конструкционных легированных сталей весьма важно, что специальные карбиды выделяются при высоком отпуске в более дисперсной форме, чем цементит. Это обеспечивает повышенную вязкость, так как микропустоты (очаги разрушения) зарождаются около мелких частиц специального карбида труднее, чем около более крупных частиц цементита.
Вторичное твердение
В сталях с добавками титана, молибдена, ванадия или вольфрама при повышении температуры отпуска после
26 И. И. Новиков
401
обычного разупрочнения, вызванного распадом мартенсита и коагуляцией частиц цементита, твердость возрастает (рис. 189). Это явление, обнаруживаемое после отпуска при 500—600СС, называют вторичным твердением.
Причины вторичного твердения — образование кластеров из атомов легирующего элемента и углерода (ши час-
то соответствует максимум твердения) и замена растворяющихся сравнительно грубых частиц цементита значитель-
Рис. 189. Вторичное твердение при отпуске стали с 0,32 % С и 1,36 % V (Кэ)
но более дисперсными выделениями специального карбида (TiC, VC, Мо2С или W2C).
Коагуляция этих частиц приводит к снижению твердости. Частины карбида Ме6С сразу образуются в сравнительно грубой форме и не приводят к упрочнению.
Добавка хрома, задерживающего смягчение при отпуске, обусловливает малое вторичное твердение или совсем не вызывает его. Это связано с тем, что выде-
ления карбида Сг?Сз быстро коагулируют при 550 СС в противоположность таким карбидам, как Мо2С и особенно W2C (коэффициент диффузии в а-растворе у вольфрама меньше, чем у молибдена).
Весьма важно, что во время вторичного твердения па
раллельно с ростом предела текучести увеличивается и вязкость из-за растворения сравнительно грубых частиц
цементита.
Отпуск под напряжением
Отпуск под напряжением 1 улучшает многие эксплуатационные свойства сталей, связанные с сопротивлением малым и микропластическим деформациям при кратковременном и длительном нагружении: пределы пропорциональности и упругости, релаксационную стойкость (рис. 190) и другие свойства. Такой отпуск оказался очень эффективным для повышения надежности разного рода упругих элементов: пружин, мембран и т. п.
При отпуске изделие следует нагружать в том же направлении, что и при эксплуатации, а величина напряжений должна составлять 0,6—0,7 от предела текучести при
1 Его называют также динамическим старением.
402
температуре отпуска. Живучесть пружин из стали 50ХФА после одночасового отпуска под напряжением при 360 СС оказывается в 10 раз больше, чем после обычного отпуска. Релаксационная стойкость винтовых пружин из этой стали после отпуска под напряжением возрастает в 4 раза.
Кроме структурных изменений в твердых растворах замещения, отмеченных при обсуждении старения под напряжением (см. § 46, п. 2 и § 49, п. 3), при отпуске под напряжением сталей важную роль могут играть дислокации, образующиеся и перераспределяющиеся в процессе нагружения. Они влияют на миграцию атомов углерода и соответственно на выделение
Рис. 190. Релаксационная стойкость стали 50ХФА после обычного отпуска </> и после отпуска под напряжением при 350 °C (2) (А. Г. Рахштадт):
— релаксирующее напряжение;
По —начальное напряжение, равное пределу упругости
карбидов. Эти процессы структурными методами пока слабо изучены. Установлены задержка перехода от Е-карбида к более стабильной карбидной фазе и измельчение ее частиц.
§ 53. Отпускная хрупкость
Отпускная хрупкость присуща многим сталям. Проблема отпускной хрупкости стала острой в период перед первой мировой войной, когда расширилось производство из легированных сталей изделий большого сечения (брони и орудий). В результате медленного охлаждения с температуры высокого отпуска ударная вязкость легированной стали может оказаться в несколько раз (и даже на порядок) меньше, чем после охлаждения в воде с той же температуры. На других свойствах при комнатной температуре состояние отпускной хрупкости практически не сказывается.
1. Необратимая отпускная хрупкость
На графиках ударная вязкость при 20 °C — температура отпуска можно встретить два провала, соответствующих развитию отпускной хрупкости двух типов (кривая 1 на рис. 191). В температурном интервале примерно 250— 400 °C развивается необратимая отпускная хрупкость.
26*
403
Ударная вязкость закаленной стали после отпуска в этом интервале меньше1 2, чем после отпуска ниже 250 °C.
Если охрупченную сталь, отпущенную при 250— 400СС, отпустить при более высоких температурах для пе-
Теппература огппуска'С
Рис. 191. Зависимость ударной вязкости при 20 СС ст температуры отпуска стали 37XH3A (Л. В. Смирнов, В. Д. Садовский):
/ — после обычной закалки и часового c i пуска с последующим охлаждением в воде; 2 — после ВТМО
ревода в вязкое состояние, то повторный отпуск в интервале 250—400 °C не возвращает сталь в хрупкое состояние. Поэтому отпускную хрупкость, развивающуюся в интервале 250—400 °C, и называют необратимой2.
Необратимая отпускная хрупкость в большей или меньшей степени свойственна всем сталям — и легированным, и углеродистым. Наиболее отчетливо она проявляется у низкоуглеродистых легированных сталей, которые после закалки и низкого отпуска (ниже 250 °C) имеют сравнительно большую ударную вязкость. У высокоуглеродистых сталей после закалки и низкого отпуска ударная вязкость столь мала, что охрупчивание после отпуска при 250—400 °C нельзя обнаружить обычными
ударными испытаниями.
Степень развития необратимой отпускной хрупкости не
зависит от скорости охлаждения с температур отпуска. По поводу природы этой хрупкости продолжают выска-
зываться противоречивые точки зрения и даже приводятся противоречивые факты. При исследовании одних сталей в состоянии необратимой отпускной хрупкости установлено межзеренное разрушение (по границам бывших аустенитных зерен), а при исследовании других сталей обнаружено внутризеренное разрушение. Возможно, что такого рода противоречия обусловлены разной чистотой ис
1 Указанные здесь и ниже обобщенные значения температур верхней и нижней границ интервалов хрупкости условны. Они зависят в первую очередь от состава стали и продолжительности отпуска.
2 В иностранной литературе ее часто называют «500° F-хрупкостыо'» и «300 сС-хрупкостью».
404
следованных материалов. Так, например, межзеренный характер разрушения не проявляется в стали вакуумной лабораторной плавки с очень низким содержанием фосфора. В сталях промышленной чистоты установлена сегрегация фосфора по границам бывших аустенитных зерен, развивающаяся при температурах у-области. Такая сегрегация фосфора может служить причиной межзеренного разрушения при характерном для интервала необратимой отпускной хрупкости структурном состоянии стали.
В высокочистых углеродистых и легированных сталях после отпуска при тех же температурах разрушение внут-ризеренное. В этом случае просвечивающая электронная микроскопия выявила связь необратимой отпускной хрупкости с распадом остаточного аустенита. В реечном мартенсите между рейками находятся тонкие прослойки, пленки остаточного аустенита (объемная доля 1—5%), повышающие ударную вязкость закаленной стали. При отпуске этот аустенит распадается как раз в области температуры 300°C, образуя между рейками а-фазы пленки карбида, которые способствуют зарождению и развитию трещин внутризеренного (по отношению к границам исходного аустенитного зерна) разрушения. Такая структура отпущенной стали похожа на верхний бейнит. Легирующие элементы, затрудняющие выделение карбида из остаточного аустенита (Si, Al), сдвигают интервал необратимой отпускной хрупкости к более высоким температурам.
Поскольку необратимую отпускную хрупкость не научились устранять, средний отпуск, как правило, не используют.
2. Обратимая отпускная хрупкость
Второй провал на кривой ударной вязкости приходится на интервал температур отпуска примерно 450—600°C (кривая 1 на рис. 191). Для нижней и верхней границ этого интервала часто указывают также значения 375 и 575 СС соответственно.
На рис. 191 два провала ударной вязкости ярко выражены, так как разделены узким интервалом температур отпуска (вблизи 450 °C) в котором отпускная хрупкость не проявляется. У многих сталей этот горб на кривой ударной вязкости может совсем отсутствовать, и тогда обнаруживается одна широкая область температур отпуска, после которого ударная вязкость понижена.
Охрупчивание при высоком отпуске может возникать в результате нагрева до 450—600 °C (независимо от ско-
405
Рис. 192. Зависимость ударной вязкости стали с 0,42% С. 0,77% Сг. 0,76 7о Мп и -0,036% Р от температуры испытаний посл'е часового отпуска при 620 “С с охлаждением в воде (/) и со скоростями охлаждения 660 (2), 100 (3) и 9 К/ч (О
Тенпераггура исг!ытания°С
рости последующего охлаждения) и в результате отпуска при температурах выше 600 СС с последующим медленным охлаждением в интервале 600—450°C. Быстрое охлаждение с температур отпуска выше 600°C, например в воде, предотвращает развитие отпускной хрупкости. Быстрое охлаждение после отпуска при 450—600 °C не предотвращает отпускной хрупкости. Таким образом, попадание в опасный интервал температур «снизу» (при нагреве и выдержке) или «сверху» (при медленном охлаждении) приводит к качественно одинаковому результату.
Важнейшая особенность охрупчивания при высоком отпуске состоит в его обратимости. Если сталь, охрупченную в результате отпуска выше 600 °C с последующим медленным охлаждением или отпуска при 450—600° С (с любой скоростью охлаждения), вновь нагреть до температур выше 600°C и быстро охладить, то ударная вязкость восстанавливается. Если после этого сталь вновь попадает в опасный интервал температур отпуска, то она повторно охрупчивается. Новый нагрев выше 600 °C с быстрым охлаждением устраняет хрупкость и т. д. Поэтому рассматриваемое явление называют обратимой отпускной хрупкостью. Так как высокий отпуск широко применяют к разнообразным конструкционным сталям, то обратимую отпускную хрупкость особенно подробно изучали на протяжении многцх десятилетий. Когда говорят об отпускной хрупкости легированной стали, то обычно имеют в виду обратимую отпускную хрупкость.
Количественная оценка склонности стали к отпускной хрупкости
По величине ударной вязкости, измеренной при одной, обычно комнатной температуре, нельзя оценивать склонность стали к отпускной хрупкости. Необходимо измерять ударную вязкость при разных, в том числе отрицательных,
406
от
Температура испытания
Рис. 193. Схема зависимости ударной вязкости от температурь» испытания стали, не склонной к отпускной хрупкости (/), и стали в состоянии отпускной хрупкости (2)
температурах, т. е. строить так называемые сериальные кривые.
Если сталь и не находится в состоянии отпускной хрупкости, то у нее все равно при понижении температуры наступает известный для хладноломких металлов переход от вязкого разрушения к хрупкому, сопровождающийся рез-ким сниженим ударной вязкости в узком интервале температур (кривая 1 на рис. 192). Развитие отпускной хрупкости смещает интервал перехода разрушению в область более высоких температур (кривые 2—4).
Если склонность стали к отпускной хрупкости оценивать по результатам испытаний только при одной температуре, то можно встретиться со следующими случаями (рис. 193). При температуре испытания Т\ значения ударной вязкости в двух состояниях сильно различаются (именно этому случаю соответствуют условия испытаний на рис. 191). При температуре же испытания Л, которая может быть и ком-
натной, по величине ударной вязкости вообще нельзя выявить склонность к отпускной хрупкости, хотя в действительности эта хрупкость имеется (кривая 2 на рис. 193 смешена вправо по сравнению с кривой /). Нельзя выявить отпускную хрупкость и испытаниями при низкой температуре 7V Таким образом, хотя приводимые в справочной литературе графики зависимости ударной вязкости при 20°C от температуры отпуска и представляют значительный интерес, отсутствие провалов ударной вязкости на таких графиках не может свидетельствовать об отсутствии склонности стали к отпускной хрупкости.
Для оценки условной температуры перехода из вязкого состояния в хрупкое (Тх), называемой также критической температурой хрупкости и порогом хладноломкости, используют разные критерии: температуру достижения определенного уровня ударной вязкости (например, 30 Дж/см2), верхнюю границу или температуру середины переходного интервала, температуру, при которой 50 % излома занято вязкой (волокнистой) составляющей, и др.
Повышение Тк в результате отпуска, т. е. величина Д7\, служит надежной количественной мерой развития отпускной хрупкости.
407
Закономерности развития отпускной хрупкости
Ниже рассмотрены основные закономерности развития обратимой отпускной хрупкости:
1. Углеродистые стали, содержащие менее 0,5 % Мп, не склонны к обратимой отпускной хрупкости. Эта хрупкость появляется только у легированных сталей. Легирующие элементы по-разному влияют на склонность к отпускной хрупкости. К сожалению, именно наиболее широко используемые легирующие элементы — хром, никель и марганец — способствуют развитию отпускной хрупкости. Раздельно эти элементы действуют слабее, чем при совместном введении в сталь. Наиболее сильно отпускная хрупкость проявляется у хромоникелевых и хромомарганцевых сталей. Небольшие добавки молибдена (0,2—0,3%) ослабляют отпускную хрупкость, а при большем его содержании отпускная хрупкость усиливается. С уменьшением содержания углерода в стали отпускная хрупкость ослабевает, но не исчезает полностью.
2. Важнейшим является тот факт, что легированные стали очень высокой чистоты совершенно не склонны к отпускной хрупкости, которая вызывается присутствием в промышленных сталях разных примесей, прежде всего фосфора, олова, сурьмы и мышьяка. Если в очень чистую сталь раздельно добавлять эти примеси, то по убыванию силы воздействия на склонность к отпускной хрупкости (ДГХ) в расчете на 0,01 % они располагаются в следующий ряд: Sb, Р, Sn, As. С увеличением содержания каждой вредной примеси Д7\ возрастает.
3. Скорость и степень развития отпускной хрупкости зависят от температуры и времени выдержки стали в опасном температурном интервале (450—600°C). Эта зависимость изображается С-кривыми достижения определенных значений Тх (рис. 194) *. При некоторой температуре отпуска ( — 550 °C на рис. 194) начальные стадии охрупчивания наступают значительно быстрее, чем при более высоких или низких температурах.
При температурах несколько выше «носа» С-кривой у многих сталей с увеличением времени выдержки степень охрупчивания проходит через максимум.
4. Независимо от степени развития отпускной хрупкости кратковременный (несколько минут) повторный нагрев
* С-кривые охрупчивания ничего общего, кроме формы, не имеют с известными С-кривыми фазовых превращений.
408
до температур выше 600 °C с последующим быстрым охлаждением может полностью ее устранить.
Рис. 194. С-кривые достижения заданных значений температурного порога хрупкости (Тх) при отпуске стали с 0,39 % С, 0,77 % Сг, 1,26 % Ni, 0,79 % Мп и 0,15 % Г (по данным Джаффи и Баффума). Цифры на кривых — температура Тх,
5. Сталь в состоянии отпускной хрупкости имеет структуру, состоящую из феррита и карбида, а разрушение при ударных испытаниях проходит преимущественно по границам исходных аустенитных зерен. Чем крупнее исходное аустенитное зерно, тем при прочих равных условиях больше Д7\ в результате развития отпускной хрупкости.
6. Охрупчивание в интервале температур 450—600°C свойственно не только закаленным на мартенсит сталям. Это охрупчивание — довольно общее явление. Оно, хотя и в меньшей степени, проявляется у сталей с бейнитной структурой и еще слабее — у сталей с перлитной структурой, причем во всех случаях охрупчивание обратимо. Стали с высокой склонностью к отпускной хрупкости рекомендуется й при отжиге ускоренно охлаждать в опасном интервале температур.
Теория отпускной хрупкости должна объяснить все эти факты.
Роль примесей
Поскольку отпускная хрупкость в очень чистой стали не возникает и непременным условием ее развития является присутствие вредных примесей: Р, Sb, Sn, As, то для понимания природы и основных закономерностей развития
409
хрупкости необходимо прежде всего установить поведение
этих элементов при отпуске.
Длительное время многие исследователи придержива
лись гипотезы «растворения — выделения», согласно которой ударная вязкость падает из-за выделения по границам зерен каких-то фаз, например фосфидов. При нагреве до ~ 650 °C эти фазы переходят в а-раствор, а при медленном охлаждении выделяются и охрупчивают сталь; быстрое
охлаждение предотвращает
выделение фаз, снижающих хрупкую прочность. Однако электронномикроскопический анализ установил, что сталь в
Вренр отпуска, ч
Рис. 195. Зависимость концентрации Sb и Ni на границе зерен в изломе (Срр ) от времени отпуска при 480 °C стали с 0,008 % С, 1,7 % Сг, 3,5 % Ni и 0,07 % Sb (Отанн, Фендж, Мак-Ма-гои, Мульфорд)
Рис. 196. Температурные зависимости предела текучести S т и сопротивления хрупкому разрушению S(J (схема)
состоянии отпускной хрупкости не имеет особых выделений по границам зерен, и, следовательно, гипотеза «растворения — выделения» несостоятельна.
Другая гипотеза объясняла отпускную хрупкость повышением концентрации примесей в приграничных слоях твердого раствора. Это подтверждала повышенная трави-мость границ зерен растворами пикриновой кислоты в состоянии отпускной хрупкости. Гипотеза о ведущей роли сегрегаций примесей полностью подтвердилась в 70-е годы в блестящей серии работ, в которых был использован метод Оже-спектроскопии, позволяющей определить концентрацию элементов в моноатомных поверхностных слоях. Этим методом была выявлена сегрегация фосфора и других примесных элементов на поверхности разрушения стали в состоянии отпускной хрупкости, измерена их концентрация (как и концентрация легирующих элементов) у
410
поверхности излома и показано, что развитие отпускной хрупкости (увеличение Тх) прямо связано с ростом концентрации примесей вблизи границ бывших аустенитных зерен (рис. 195).
В результате равновесной сегрегации концентрация вредных примесей на поверхности излома может в десятки и сотни раз превышать их среднюю концентрацию в стали. Обычно концентрация вредных примесей в стали промышленной чистоты составляет тысячные — сотые доли процента, а на поверхности излома она измеряется целыми процентами. Толщина приграничного слоя сегрегации — несколько атомных диаметров.
Физическая природа (механизм) охрупчивания из-за сегрегации Р, Sb, Sn и As не выяснена. Согласно одному предположению примесные элементы снижают поверхностную энергию и тем самым уменьшают работу образования межзеренной трещины. Прямых подтверждений этого пет. Согласно другому предположению вредные примесные элементы, замещающие атомы железа, из-за большего, чем у железа, атомного лизиметра увеличивают среднее расстояние между поверхностными атомными слоями по обе стороны от границы. В результате сцепление между соседними зернами ослабевает. Чем больше атомный диаметр примеси превышает атомный диаметр железа, тем сильнее ее охрупчивающее действие. Этим можно объяснить, почему сурьма намного сильнее охрупчивает сталь, чем фосфор, если концентрация этих элементов (число атомов на межзеренной поверхности) одинакова.
Для дальнейшего анализа воспользуемся классической схемой акад. А. Ф. Иоффе (рис. 196), объясняющей переход материала из пластичного состояния в хрупкое тем, что с понижением температуры предел текучести (ST) растет интенсивно, а сопротивление отрыву (So) мало изменяется. При температуре перехода Гх' кривые ST и So пересекаются. Ниже Г* материал разрушается хрупко, так как сопротивление хрупкому разрушению (кривая Г) достигается раньше, чем может быть достигнут предел текучести (кривая 2). Если в состоянии отпускной хрупкости сцепление зерен, т. е. сопротивление межзеренному разрушению, понижено (кривая 3), то при том же пределе текучести (кривая 2) температура перехода окажется выше (Т">Т*). Повышение предела текучести (кривая 4) при неизменной прочности границ зерен (кривая 3) может усилить отпускную хрупкость
411
Используя эту схему и экспериментальные данные о сегрегации примесей, можно объяснить основные факты проявления отпускной хрупкости.
Экспериментально показано, что температура перехода Л растет с повышением концентрации вредной примеси на границах зерен. Сегрегация контролируется диффузионным подводом атомов элемента к границе зерна, причем концентрация примеси в приграничном слое растет с затуханием (см. рис. 195, а), стремясь к предельной величине, характерной для данной температуры [см. формулу (28)]. Одновременно растет с затуханием и Т* (см. рис. 195,6).
У стали, загрязненной фосфором, при достаточно высокой температуре отпуска величина проходит через максимум после достижения равновесной концентрации фосфора на межзеренной границе. Это обусловлено тем, что длительный отпуск приводит к разупрочнению тела зерен при неизменном сопротивлении межзеренному разрушению, а снижение предела текучести, как следует из схемы на рис. 196, приводит к понижению Гх.
В стали, загрязненной сурьмой, с увеличением времени отпуска Тх не проходит через максимум, так как сурьма, диффундирующая намного медленнее фосфора, после разупрочнения при отпуске еще продолжает сегрегировать на границы, снижая здесь хрупкую прочность.
С ростом температуры диффузионный процесс образования зернограничной сегрегации ускоряется и одновременно уменьшается абсолютная величина равновесной сегрегации, размываемой тепловым движением. Действие этих факторов обусловливает С-образный характер кривых времени достижения определенной Тх (см. рис. 194).
Расчеты показывают, что зависимость времени достижения заданной степени равновесной сегрегации фосфора от* температуры изображается С-кривыми, аналогичными С-кривым Тх. Чем меньше общее содержание фосфора в стали, тем правее расположены С-кривые определенной степени его сегрегации и соответственно меньше склонность стали к обратимой отпускной хрупкости.
При температурах выше 600—650 °C сегрегация примеси или полностью исчезает (Sb), или находится на очень низком уровне (Р). При последующем охлаждении стали в воде сегрегации не успевают восстанавливаться.
Конструкционная электросталь открытой дуговой плавки обычно содержит 0,01—0,02 % Р, 0,005—0,01 % Sn, 0,002—0;005 % Sb и 0,005—0,01 % As. В стали, полученной кислородно-конвертерным и мартеновским способами, ти-.412
личное содержание фосфора еще больше: 0,02—0,03 и 0,025—0,035 % соответственно. Чем больше примеси в стали, тем выше ее концентрация на границе зерна [см. формулу (28)]. Поэтому хотя сурьма значительно сильнее повышает Тх, чем фосфор (при равной концентрации примеси на границе), но так как в промышленных сталях фосфор содержится в больших количествах (сотые доли процента), чем сурьма (тысячные доли процента), он дает больший вклад в повышение Тх при отпуске. Кроме того, по сравнению с сурьмой фосфор значительно быстрее сегрегирует на границы зерен. С учетом всего этого фосфор следует считать наиболее вредной примесью и для уменьшения отпускной хрупкости от него необходимо очищать сталь в первую очередь.
Роль легирующих элементов
В развитии отпускной хрупкости не меньшую роль, чем примеси, играют легирующие элементы. Сплавы Fe—С, содержащие те же количества вредных примесей, что и легированные стали, не склонны к обратимой отпускной хрупкости. В железоуглеродистых сплавах сегрегация вредных примесей столь мала, что не вызывает отпускной хрупкости. В присутствии легирующих элементов (Ni, Сг и Мп) сегрегация вредных примесей резко увеличена. При этом сами легирующие элементы, не образующие равновесных сегрегаций в сталях высокой чистоты, в присутствии вредных примесей сегрегируют к границам зерен (см. рис. 195, а). Следовательно, можно говорить о взаимодействия легирующего элемента и примеси в а-растворе, способствующем их совместной сегрегации. Предполагается, что если атомы примеси и легирующего элемента сильнее взаимно притягиваются, чем атомы этой же примеси и железа, то сегрегация примеси и легирующего элемента будет взаимно усилена. Именно так ведут себя Р и Ni, Р и Сг, Sb и Ni, Sb и Мп, а также другие пары примесь — легирующий элемент. Второй легирующий элемент может дополнительно усилить сегрегацию примеси. Например, никель и хром вместе вызывают большую сегрегацию сурьмы, чем это следует из простого сложения эффектов их раздельного влияния.
Повышение концентрации вредных примесей в приграничных слоях твердого раствора под действием легирующих добавок ослабляет межзеренную связь (So на рис. 196) и является одной из причин высокой склонности к &т-
£13
пускной хрупкости легированных сталей, содержащих Ni, Сг и Мп.
Выделение карбида при отпуске может повлиять на кинетику сегрегации примеси. Рассмотрим это влияние на примере сплава Ее—С—Ni—Sb. Из-за того, что растворимость никеля в карбиде очень мала по сравнению с его растворимостью в a-фазе, атомы никеля при выделении карбида оттесняются на границу карбид—феррит, где образуют узкий слой а-раствора с неравновесным избытком никеля, который медленно рассасывается.
Если предположить, что химический потенциал сурьмы в этом слое понижен по сравнению с в объеме a-фазы, то атомы сурьмы устремляются к границе карбид—феррит, одновремеино понижая здесь химический потенциал никеля и стабилизируя его сегрегацию. Сурьма будет до тех пор сегрегировать на границу с карбидом, пока ее химический потенциал здесь ие станет равным химическому потенциалу в объеме а-фазы.
Если выделения карбида отсутствуют, первичным процессом является сегрегация атомов сурьмы на границу зерна. Снижение химического потенциала иикеля в обогащенном сурьмой слое обусловливает подвод атомов никеля к границе, который, снижая здесь химический потенциал сурьмы, усиливает ее сегрегацию. Совместная сегрегация сурьмы и никеля будет продолжаться до тех пор, пока ие установятся равенства химических потенциалов = Нм = Нм •
Граница зерен, заранее обогащенная никелем, вызывает более быструю сегрегацию сурьмы, и поэтому выделение карбида, приводящее к обогащению никелем, может ускорить развитие отпускной хрупкости, по крайней мере, на начальных стадиях.
Легирующие элементы могут повышать Гх, не только усиливая сегрегацию вредных примесей и ослабляя тем самым сцепление зерен So, но и повышая предел текучести ST (см. схему на рис. 196). Такова, например, роль во многих сталях хрома, задерживающего разупрочнение при отпуске. Хромоникелевая сталь, загрязненная фосфором, обладает значительно большей склонностью к обратимой отпускной хрупкости, чем никелевая, главным образом из-за большего предела текучести и в меньшей мере из-за повышенной сегрегации фосфора. С увеличением содержания углерода в хромоникелевой стали отпускная хрупкость развивается сильнее вследствие упрочнения тела зерен (на сегрегации фосфора содержание углерода не сказывается).
Природа ослабления обратимой отпускной хрупкости под действием небольших добавок молибдена и вольфрама не установлена. Одно из предположений — резкое снижение предельной растворимости фосфора в a-фазе под действием молибдена и соответственно уменьшение зернограничной сегрегации фосфора в твердом растворе.
Роль границ аустенитных зерен
Предпочтительное разрушение стали, находящейся в состоянии отпускной хрупкости, по границам исходных аусте-414
нитных зерен связано с сегрегацией вредных примесей на этих границах.
Следует подчеркнуть, что после высокого отпуска в стали нет аустенитных зерен как таковых. По исходным границам аустенитных зерен в отпущенной стали стыкуются кристаллы а-фазы из соседних зерен и находятся выделения карбида. Разрушение происходит по границам а/а и а/карбид.
Сильная сегрегация вредных примесей происходит лишь на высокоугловых границах. В отпущенной стали все границы а/а вдоль исходных границ аустенитных зерен высокоугловые в отличие от внутренних границ между реечными кристаллами а-фазы, образовавшимися при мартенситном превращении. Предпочтительное выделение при отпуске более крупных кристаллов карбида по исходным аустенитным границам и соответственно усиленная сегрегация на границе а/карбид также могут быть причиной локализации здесь разрушения.
Чем крупнее исходные зерна аустенита, тем выше Тх в состоянии отпускной хрупкости, так как при крупном зерне развивающаяся межкристаллитная трещина реже меняет свое направление.
Меры борьбы с обратимой отпускной хрупкостью
Основные меры борьбы с отпускной хрупкостью стали следующие: 1) уменьшение содержания вредных примесей; 2) ускоренное охлаждение с температуры высокого отпуска (выше 600°C); 3) введение небольших добавок молибдена (0,2—0,3%); 4) использование высокотемпературной термомеханической обработки (об этом см. ниже § 60).
При разработке высокопрочных сталей с большим пределом текучести целесообразно во избежание усиления отпускной хрупкости снижать допустимый предел содержания вредных примесей. К сожалению, в процессе выплавки стали не удается достичь очистки от основной вредной примеси — фосфора. Поэтому большое значение здесь имеет использование более чистой исходной шихты.
Быстрое охлаждение с температур отпуска выше 600 °C — самый дешевый и простой способ борьбы с хладноломкостью. Однако, во-первых, он не позволяет устранить отпускную хрупкость в центре массивных изделий, и, во-вторых, при охлаждении в воде изделий с резкими переходами и отверстиями могут образоваться трещины (особенно в сталях с повышенным содержанием углерода).
415
Раздел пятый
ТЕРМОМЕХАНИЧЕСКАЯ ОБРАБОТКА
Пластическая деформация изменяет характер распределения и увеличивает плотность несовершенств кристаллического строения — дислокаций, вакансий, дефектов упаковки, мало- и высокоугловых границ. Так как дефекты кристаллической решетки сильно влияют на формирование структуры сплавов при фазовых превращениях, то пластическую деформацию перед фазовыми превращениями или в период их развития можно использовать для создания оптимальной структуры термически обработанного сплава.
Термомеханическая обработка (ТМО)—это термическая обработка, включающая пластическую деформацию, которая благодаря повышенной плотности дефектов влияет на формирование структуры при фазовых превращениях, происходящих во время термического воздействия.
Следовательно, к ТМО* нельзя относить любое сочетание операций деформирования, нагрева и охлаждения. Например, если пластическая деформация проводится после всех операций термообработки, то мы имеем дело не с ТМО, а с обычной термообработкой и последующей обработкой давлением. Такая пластическая деформация, например холодная прокатка после старения, может создать наклеп, повысить прочностные свойства, но она не влияет на формирование структуры при фазовых превращениях, так как эти превращения прошли до деформации.
Если пластическая деформация была проведена до термообработки, но ие оказала определяющего влияния на формирование окончательной структуры сплава при фазовых превращениях, то такое сочетание пластической деформации и последующей термообработки также нельзя относить к ТМО. Например, холодная прокатка с последующим нагревом под закалку, при котором проходит рекристаллизация, не являются составными частями ТЛЮ, так как ре-
* Точнее, ее следовало бы назвать деформационно-термической обработкой.
416
кристаллизованная структура характеризуется низкой плотностью несовершенств кристаллического строения.
Процессы пластической деформации и термической обработки при ТМО могут быть совмещены в одной технологической операции, но могут проводиться и в разное время, например с разрывом в несколько суток. Важно лишь, чтобы при этом фазовые превращения проходили в условиях повышенной плотности дефектов решетки, созданных пластической деформацией.
В настоящее время в промышленности используют и опробуют разнообразные схемы ТМО, включающие горячую и (или) холодную пластическую деформацию, которая оказывает определяющее влияние на формирование структуры сплава при старении, при перлитных, бейнитных и мартенситных превращениях.
Прежде чем приступать к анализу изменений структуры и свойств металла при ТМО, необходимо рассмотреть, как формируется структура при горячей обработке давлением (структурные изменения при холодной деформации рассмотрены в § 4).
Глава XIII
ИЗМЕНЕНИЕ СТРУКТУРЫ МЕТАЛЛА ПРИ ГОРЯЧЕЙ ОБРАБОТКЕ ДАВЛЕНИЕМ
На протяжении десятилетий принято было считать, что в процессе горячей обработки давлением всегда протекает рекристаллизация. Именно в этом видели основное отличие горячей обработки от холодной. Начатые в 60-х годах подробные исследования механизма горячен пластической деформации выявили ошибочность указанного утверждения. Оказалось, что истинная структура, формирующаяся непосредственно в процессе горячей деформации, часто сильно искажена рекристаллизацией, проходящей во время охлаждения металла с температуры окончания горячей деформации. Очень быстрое охлаждение образца в момент прерывания горячей деформации помогает выявить структуру, сформировавшуюся к этому моменту. Так как сопротивление деформированию реагирует на структурные изменения, то ценную информацию о сущности процессов, происходящих при горячей деформации, можно также получить, анализируя форму кривых истинное напряжение 6 — истинная деформация е (рис. 197)*. Такие кривые
* e=ln(Z/Zo), где /0 — начальная длина образца, а / — его длина в заданный момент деформации,
27 И. И. Новиков 417
обычно получают испытаниями на горячее кручение или сжатие.
Истинная скорость деформации (е) при горячей обра< ботке давлением находится в интервале 10-2—103 с-1*. Она определяется видом обработки и применяемым оборудованием. Например, при прессовании эта скорость минимальна (порядка 10“2—10-3 с-1), а при ковке на молоте максимальна (порядка 101—103 г1). Так как сопротивление деформированию сильно зависит от скорости деформации, то графики S—е строят для разных скоростей (см. рис. 197).
Рис. 197. Зависимость истинного напряжения S от истинной деформации е (цифры на кривых — скорости деформации, с—1):
а — армко-железо, 700 СС (Иммэриджэи и Джонас); б — сталь с 0,25 % С, 1100 °C (Росса рд)
В каждом реальном цикле горячей обработки давлением температура металла изменяется сложным образом. Она может повышаться из-за адиабатического разогрева и понижаться из-за отдачи тепла более холодному инструменту и в окружающую среду. Для упрощения анализа лабораторные испытания с построением графиков S—е проводят в условиях, близких к изотермическим.
* • 1 at .
* у. — э Где I — длина образца, т — время.
418
§ 54. Структурные изменения во время горячей деформации
На начальных этапах горячей деформации всегда происходит деформационное упрочнение, связанное с повышением плотности дислокаций. Восстановительными, разупрочняю-щпми процессами, уменьшающими плотность дислокаций во время горячей деформации, могут быть только возврат или возврат и рекристаллизация.
Процессы разупрочнения во время горячей деформации аналогичны процессам разупрочнения при отжиге после холодной деформации: при возврате плотность дислокаций уменьшается в результате переползания и поперечного скольжения с выстраиванием дислокаций в стенки (полигонизация), а при рекристаллизации — в результате «выметания» дислокаций мигрирующими высокоугловыми границами. Восстановительные процессы, идущие во время деформации, имеют и свои особенности, рассмотренные ниже. В связи с этим были введены термины динамический возврат (в том числе динамическая полигонизация) и динамическая рекристаллизация в отличие от статического возврата и статической рекристаллизации, которые идут при отжиге после холодной деформации или по окончании горячей деформации (во время последеформационной выдержки и охлаждения).
1. Динамическим возврат
Если динамический возврат — это единственный восстановительный процесс при горячей деформации, то кривые напряжение — деформация имеют наиболее простой вид: рост напряжения течения плавно затухает до достижения определенной величины, не зависящей далее от степени деформации (см. рис. 197, а). Если исходный металл был отожжен и имел рекристаллизованную структуру с плотностью дислокаций порядка 106—108 см~2, то в период деформационного упрочнения плотность дислокаций плавно возрастает, достигая на стадии установившегося течения величины порядка 1010 см“2.
На стадии деформационного упрочнения (горячего наклепа) вначале образуются дислокационные клубки, а затем ячеистая структура. Постепенно формируется субзеренная структура, т. е. происходит динамическая полигонизация. На стадии установившегося течения средний размер субзереп, ’их разориентировка и средняя плотность дислокаций внутри субзерен остаются постоянными.
27*
419
С ростом степени деформации скорость генерирования дислокаций мало меняется, а скорость их аннигиляции из-за роста плотности дислокаций возрастает до тех пор, пока не сравняется со скоростью генерирования. Это и соответствует достижению стадии установившегося течения. С понижением температуры или повышением скорости деформации восстановительные процессы возврата в расчете на одну и ту же величину деформации успевают проходить в меньшей степени и поэтому стадия установившегося течения наступает позднее. Истинная деформация, соответствующая достижению стадии установившегося течения, находится в интервале от 0,1 до 0,5 в зависимости от температуры и скорости деформирования.
На установившейся стадии субзерна остаются равноосными вплоть до очень больших деформаций, в то время как зерна сильно вытягиваются в направлении течения. Сохранение равноосности субзерен можно объяснить процессом реполигонизации — многократной повторной полигонизации, состоящей в рассыпании субзеренных границ и новом их формировании в таких местах, что средний размер субзерен остается неизменным.
Чем выше температура и ниже скорость деформации, тем меньше напряжение течения на стадии установившегося течения, меньше общая плотность дислокаций и крупнее субзерна.
Динамический возврат может развиваться и при холодной деформации, однако здесь он не приводит к стадии установившегося течения: деформационное упрочнение идет непрерывно, а динамический возврат проявляется только в уменьшении коэффициента деформационного упрочнения с ростом деформации. Плотность дислокаций в стенках ячеек при холодной деформации возрастает, в то время как на установившейся стадии горячей деформации общая плотность дислокаций остается постоянной. Указанные различия в поведении металла при горячей и холодной деформациях обусловлены различиями в механизмах динамического возврата. При холодной деформации единственный механизм динамического возврата — поперечное скольжение дислокаций, которое позволяет дислокациям обходить барьеры и тем самым уменьшает коэффициент деформационного упрочнения. При горячей деформации динамический возврат, кроме поперечного скольжения, включает еще и переползание дислокаций, в связи с чем становится возможной реполигонизация и поддерживается постоянной общая плотность дислокаций, т. е. достигается
420
стадия установившегося течения. Переползание дислокаций как наиболее медленный процесс является контролирующим механизмом динамического возврата при горячей деформации. Нижней границей температурной области горячей деформации можно условно считать гомологическую температуру 0,5 ТПл, выше которой активно идет переползание дислокаций.
Напряжение о на стадии установившегося течения при горячей деформации не зависит от степени деформации, а определяется температурой Т и скоростью деформации е:
& - Xeexp(WT), (41)
где Q — энергия активации пластической деформации; А и п— константы (я=34-6).
Произведение еехр (Q/RT) —Z называют параметром Зинера—Холомона. Этот параметр характеризует совместное влияние скорости и температуры деформации на напряжение течения.
Поскольку поперечное скольжение и переползание дислокаций являются главными элементарными процессами динамического возврата при горячей деформации, а с увеличением энергии дефектов упаковки и соответственно с уменьшением ширины растянутых дислокаций протекание этих процессов облегчается, то динамический возврат металлов с более высокой энергией дефектов упаковки идет интенсивнее и приводит к образованию более совершенной субструктуры с более крупными субзернами, чем в металле с низкой энергией дефектов упаковки (при одинаковой гомологической температуре). Металлами с активно развивающимся динамическим возвратом при горячей деформации являются, например, Al, a-Fe, Mo, W, a-Zr, Be и Zn. Введение добавок, входящих в твердый раствор и уменьшающих энергию дефектов упаковки, затрудняет динамический возврат. Такое легирование может привести к росту напряжения течения при горячей деформации на порядок.
Во время горячей деформации могут протекать процессы, усложняющие вид кривых S—е. Напряжения течения после достижения стадии установившегося течения может плавно снижаться вследствие адиабатического разогрева деформируемого металла. Снижение этого напряжения может также вызываться коагуляцией выделений, которой способствует ускоренная диффузия растворенного элемента по дислокационным стенкам. Если при горячей деформации раствор распадается с образованием дисперсных выделений, то напряжение течения возрастает.
421
2. Динамическая рекристаллизация
На кривых напряжение — деформация динамическая рекристаллизация проявляется в падении напряжения течения. На стадии установившегося течения возможны два типа поведения металла (см. рис. 197,6). При более высоких скоростях деформации напряжение течения после спада остается неизменным1, а при более низких скоростях напряжение течения осциллирует около некоторого среднего уровня, который тем ниже, чем меньше скорость деформации. С ростом степени деформации амплитуда колебаний напряжения постепенно уменьшается и кривая S—е сглаживается.
Исследование резко охлажденных образцов, деформацию которых прерывали на разных стадиях, позволило составить следующую картину структурных изменений. Вначале происходит деформационное упрочнение с параллельно идущим динамическим возвратом (участок подъема кривой истинных напряжений). Если динамический возврат слабо развит, то с повышением плотности дислокаций создаются условия для формирования центров динамической рекристаллизации. При малых скоростях деформации они возникают по механизму локального выгибания существующих высокоугловых границ (см. рис. 22), а при повышенных скоростях стенки ячеек аккумулируют все больше дислокаций и превращаются в высокоугловые границы зародыша динамической рекристаллизации.
Пока рекристаллизованные зерна с пониженной прочностью занимают небольшую часть объема металла, еще продолжается рост напряжения течения. С увеличением суммарного объема рекристаллизованных участков разупрочнение перекрывает деформационное упрочнение и напряжение течения падает. Следовательно, критическая степень деформации, соответствующая началу динамической рекристаллизации, несколько меньше деформации, соответствующей пику истинного напряжения течения.
Динамическая рекристаллизация отличается от статической тем, что появившиеся рекристаллизованные зерна с низкой плотностью дислокаций во время своего роста постепенно наклепываются из-за продолжающейся деформации — в них повышается плотность дислокаций. Участки,
1 При достаточно больших скоростях из-за адиабатического разогрева образца может наблюдаться плавный спад напряжения на стадии установившегося течения (см. верхнюю кривую на рис. 197,6),
422
рекристаллизовавшиеся в первую очередь, начинают наклепываться раньше и в них быстрее достигается критическая плотность дислокаций, необходимая для зарождения новых рекристаллизованных зерен, которые затем наклепываются, и т. д. Многократно чередующиеся циклы динамической рекристаллизации и наклепа рекристаллизованных зерен соответствуют установившейся стадии с неизменным средним размером зерна.
Наклеп рекристаллизованных зерен уменьшает разность в плотности дислокаций по обе стороны от мигрирующей границы, а так как эта разность является движущей силой миграции, то скорость роста рекристаллизованных зерен уменьшается. Чем выше температура и ниже скорость деформации, тем крупнее и совершеннее рекристаллизованные зерна.
При малой скорости деформации цикл рекристаллиза- • ции успевает закончиться раньше, чем в участках, рекристаллизованных в первую очередь, будет достигнута критическая плотность дислокаций и начнется новый цикл рекристаллизации. Поэтому при малой скорости деформации вначале наблюдается четкое чередование спадов (рекристаллизационное разупрочнение) и подъемов (деформационное упрочнение) напряжения течения. Так как эти процессы в разных участках макрообъема образца в силу разных, в том числе случайных причин перестают совпадать по фазе, то осцилляция напряжения течения уменьша’ ется и кривая S—е сглаживается (см. рис. 197,6).
При больших скоростях деформации скорость увеличения плотности дислокаций больше скорости рекристаллизации, и до окончания первого цикла рекристаллизации достигается критическая плотность дислокаций, т. е. начинается второй цикл рекристаллизации. В результате такого перекрытия циклов рекристаллизации напряжение течения не осциллирует: вслед за достижением пикового напряжения оно падает до определенного уровня, промежуточного между сопротивлением деформированию рекристаллизованных и перекристаллизованных участков.
Характерные особенности структуры металла на стадии динамической рекристаллизации следующие: а) неоднородность субструктуры по объему металла и внутри отдельных зерен, связанная с тем, что одни участки только что рекристаллизовались, а ранее рекристаллизованные участки подверглись наклепу и динамическому возврату; б) неровность, зубчатость границ зерен, вызванная выбрасыванием «языков» при зарождении новых зерен; в) появление коло
423
ний новых зерен преимущественно около границ исходных кристаллов; г) равноосность зерен (в отличие от вытянутых зерен на стадии динамического возврата).
При сильном развитии динамического возврата критическая плотность дислокаций, необходимая для зарождения центров рекристаллизации, может быть не достигнута вплоть до самых больших степеней деформации, и тогда динамическая рекристаллизация вообще не начнется. Именно такая ситуация наблюдается при прессовании полуфабрикатов из промышленных алюминиевых сплавов. В металлах со сравнительно низкой энергией дефектов упаковки (Си, Ni, Со, у-Fe, Ag, Au, Pb) поперечное скольжение и переползание дислокаций, а соответственно и динамический возврат затруднены, и при достаточно большой горячей деформации, например при прессовании, может быть достигнута критическая плотность дислокаций, необходимая для зарождения центров динамической рекристаллизации.
Если степень деформации меньше критической, например при горячей прокатке, то и в этих металлах мы имеем дело только с динамическим возвратом.
Легирующие элементы, уменьшающие энергию дефектов упаковки, затрудняют динамический возврат и тем самым облегчают достижение критической плотности дислокаций, необходимой для динамической рекристаллизации. При оценке роли легирующих добавок следует учитывать возможное затруднение динамической рекристаллизации из-за торможения дисперсными частицами миграции высокоугловых границ.
К промышленным сплавам, в которых при достаточно большой горячей деформации возникает динамическая рекристаллизация, относятся углеродистые и легированные стали в аустенитном состоянии, жаропрочные никелевые сплавы и латуни. Критическая степень деформации, необходимая для начала динамической рекристаллизации, уменьшается с ростом скорости деформации и с понижением температуры.
Таким образом, в зависимости от типа металла, состава сплава, температуры, степени и скорости деформации <мы можем иметь дело при горячей обработке давлением только с динамическим возвратом или с ним и с динамической рекристаллизацией. В любом случае для горячей деформации в отличие от холодной характерно наступление после деформационного упрочнения стадии установившегося течения.
§ 55. Структурные изменения по окончании горячей деформации
На любой стадии горячей деформации металл имеет повышенную плотность дислокаций, которая служит термодинамическим стимулом для восстановительных процессов после окончания деформации. Установлены три типа самопроизвольных восстановительных процессов, идущих по окончании горячей деформации: статический возврат, статическая рекристаллизация и метадинамическая рекристаллизация.
sstna
Рис. 198. Кривая течения стали с 0,68 % С, деформированной сжатием при 780 °C со скоростью 1,3-10—Зс —1 (о), и зави-
симость степени разупрочнения этой стали от последеформационноЙ выдерж*» ки при 780 °C (б) (Джейк и Джонас):
1—4 — выдержки после истинных деформаций 0,055; 0,098: 0,24 и 0.41 соответственно
Первые два процесса имеют аналоги при отжиге после холодной деформации, а метадинамическая рекристаллизация может идти только после горячей деформации.
Тип восстановительных процессов, их кинетика и вклад в разупрочнение зависят от того, на какой стадии была прервана горячая деформация. На рис. 198, а показана кривая течения углеродистой стали в аустенитном состоянии при 780 СС, а на рис. 198,6 — кинетика разупрочнения 1 во время последеформационноЙ изотермической выдержки после прерывания горячего сжатия при разных степенях деформации. Кривая 1 на рис. 198,6 относится к прерыванию горячего сжатия на стадии начального деформационного упрочнения (см. соответствующую точку 1 на кривой течения, рис. 198,а). В этом случае наклеп к моменту окончания деформации еще слишком мал, чтобы при последе-формациоиной выдержке стала возможной статическая рекристаллизация, и последеформационное разупрочнение идет только за счет статического возврата. Этот возврат начинается сразу по окончании горя-
1 Степень разупрочнения — разность между напряжением течения перед разгрузкой и пределом текучести повторно нагруженного образца, отнесенная к разности между этим же напряжением и начальным пределом текучести.
425
чей деформации (без инкубационного периода) и идет с затуханием, завершаясь примерно за 100 с (см. выход кривой 1 на горизонталь, рис. 192, б).
Кривая 2 относится к прерыванию горячего сжатия тоже на стадии деформационного упрочнения, ио степень деформации больше (см. соответствующую точку 2 на рис. 198, а) и наклеп достаточен для развития статической рекристаллизации при последеформационной выдержке. Эта рекристаллизация начинается примерно через 104 с (инкуба* циоиный период) от начала выдержки и приводит к полному разупрочнению (см. второй подъем иа кривой 2). С повышением скорости горячей деформации до заданной степени растет плотность дислокаций и в результате ускоряется статическая рекристаллизация при последеформационной выдержке (скорость рекристаллизации при отжиге после колодной деформации малочувствительна к скорости холодной деформации).
В инкубационный период проходит только статический возврат, который развивается несколько быстрее и приводит к большему разупрочнению по сравнению с предыдущим случаем (плато на кривой 2 выше плато на кривой /).
Особенно интересна кривая 5, характеризующая разупрочнение после деформации, прерванной на стадии начала динамической рекристаллизации, но еще до достижения установившейся стадии (см. точку 3 иа спаде после пикового напряжения, рис. 198, а). Разупрочнение в этом случае проходит в три этапа. Первый и третий подъемы на кривой 3 аналогичны подъемам иа кривой 2 и соответствуют статическому возврату и статической рекристаллизации, начинающейся после инкубационного периода. Новым является промежуточный подъем на кривой 3, соответствующий метадинамической (послединамической) рекристаллизации, которая в отлнчие от классической статической рекристаллизации протекает без инкубационного периода.
Когда деформация прервана на стадии динамической рекристаллизации, имеется множество свежих, не успевших подвергнуться наклепу зародышей рекристаллизованных зерен, способных к росту в статических условиях сразу после прекращения горячей деформации. Многие высокоугловые границы рекристаллизованных зерен, по обе стороны которых резко различна плотность дислокаций, мигрировали в сторону более наклепанных зерен при горячей деформации и могут продолжать такую миграцию сразу по окончании деформации. Этим обусловлена важнейшая для практики особенность метадинамической рекристаллизации— отсутствие инкубационного периода, рекристаллизационный рост зерен за короткое время охлаждения металла с температуры деформации.
Первая задержка в разупрочнении (см. маленькое плато на рис. 198, б)—это результат того, что разупрочнение при быстром статическом возврате завершается раньше, чем станет заметным разупрочнение от развивающейся метадинамической рекристаллизации.
Кривая 4 на рис. 198,6 соответствует разупрочнению после прекращения горячей деформации на стадии установившегося течения (точка 4 на рис. 198,а). Остановки разупрочнения по окончании статического возврата здесь нет, так как до этого момента уже сильно 426
развито разупрочнение от метадипамической рекристаллизации. Новым является то, что метадннамическая рекристаллизация приводит к полному разупрочнению за время меньше инкубационного периода статической рекристаллизации, которая поэтому не идет.
Чем больше степень горячей деформации до начала стадии установившегося течения, техМ быстрее проходит разупрочнение при последеформационной выдержке и мельче рекристаллизованное зерно. Увеличение степени деформации на стадии установившегося течения не влияет ни на скорость разупрочнения, ни на размер зерна при последеформационной выдержке. Скорость последеформационногс разупрочнения возрастает с повышением температуры горячей деформации.
Зерна, сформировавшиеся при последеформационной выдержке, отличаются от зерен, возникших на стадии динамической рекристаллизации, отсутствием субструктуры и менее извилистыми границами.
Вклад трех рассмотренных восстановительных процессов в после-деформационное разупрочнение зависит от степени деформации, что схематично изображено на рис. 199. На этой схеме ес — критическая степень горячей деформации, необходимой для начала статической рекристаллизации, ея — деформация в момент начала динамической рекристаллизации, еу — деформация, соответствующая началу стадии установившегося течения. Например, при деформации степень разупрочнения от статического возврата характеризуется отрезком ab> от мета-динамической рекристаллизации — отрезком Ьс и от статической (классической) рекристаллизации — отрезком cdt сумма которых составляет 100 %-
Если деформация е<ес> то разупрочнение является результатом только статического возврата и никогда не бывает полным (заштрихо-
ванная область—нереализуемые степени разупрочнения). Если ес<е< <ед, то статический* возврат и позднее идущая статическая рекристаллизация приводят к полному разупрочнению. Если ед<е<еу, то метадинамическая рекристаллизация также дает вклад в разупрочнение, доля которого нарастает с увеличением степени горячей деформации. При е>£у, т. е. в случае установившегося течения, все последеформаци-онное разупрочнение обеспечивается статическим возвратом и метадн-намической рекристаллизацией (без участия статической рекристаллизации). Вклад статического возврата в последеформационное разупрочнение обычно не превышает 50 %.
<700
§150
Статиче-ская(клас сическал)
Статический возврат
рекрисъ
изацил
\Иетадинамическая с рекристаллизация
Степень деформации е
199. Схема влияния степени
Рис, горячей деформации на долю пос-ледеформационного разупрочнения металла под действием статического возврата, классической статической рекристаллизации и ме-тадинамической рекристаллизации (Джейк и Джонас)
В заключение рассмотрим возможные комбинации дина
427
мических и статических восстановительных процессов при горячей обработке давлением в промышленных условиях (рис. 200 и 201).
При горячей прокатке степень деформации за один проход обычно сравнительно небольшая и в зоне деформации (между валками) протекает только динамический возврат, не изменяющий формы зерен. По выходе из валков в охлаждающемся металле может протекать только статический возврат и зерна остаются вытянутыми (рис. 200, а) или
Рис. 200. Схемы комбинаций динамических и статических процессов при горячей прокатке с небольшим обжатием
возможна также статическая рекристаллизация, в результате которой образуются равноосные зерна (рис. 200,6). Первый случай характерен для металлов с высокой, а второй — с низкой энергией дефектов упаковки.
Если между проходами статическая рекристаллизация не идет и наклеп накапливается, то, несмотря на сравнительно небольшую степень деформации за каждый проход, начиная с определенного суммарного обжатия при горячей прокатке в зоне деформации становится возможной динамическая рекристаллизация1.
При горячем прессовании часто достигаются очень большие степени деформации. Если в зоне деформирования протекал только динамический возврат, то при охлаждении прессованного полуфабриката может в зависимости от степени, скорости и температуры деформации, а также от энергии дефектов упаковки протекать один статический возврат (рис. 201, а) или возможна также классическая рекристаллизация (рис. 201,6). Если же во время горячей деформации развивается динамическая рекристаллизация, то по выходе из пресса в полуфабрикате идет метадинами-ческая и, возможно, статическая (классическая) рекристаллизация (рис. 201, в).
1 Соответствующие структурные изменения см. па схеме рис. 201, в.
428
При горячей прокатке с достаточно большим обжатием за один проход возможны те же комбинации восстановительных процессов, что и при прессовании.
Чтобы в горячедеформированных изделиях (полуфабрикатах) получать регламентированную, например полностью иерекрнсталлизован-
динамическая декристаллизация
Рис. 201. Схемы комбинаций динамических и статических процессов при горячем прессовании с большим обжатием
Метадияаяическая астати
ческая рекристаллизация
ную структуру, желательно для промышленных сплавов иметь справочные данные об их структурном состоянии после обработки по конкретным режимам.
Если при холодной обработке дав-леннем степень деформации — это главный параметр, определяющий уровень запасенной энергии и соответственно переход (при последующем нагреве) от перекристаллизованной к рекристаллизованной структуре, то при горячей обработке давлением накопленная энергия и получаемое структурное состояние зависят еще от температуры деформирования и скорости деформации, а эти параметры колеблются в широких пределах для одного и того же сплава. Например, скорости деформации при горячем прессовании и ковке на молоте могут различаться на пять порядков (!).
Поскольку промышленные процессы горячей обработки давлением очень часто проводят на стадии установившейся горячей деформации, на которой напряжение течения, а значит и накопленная энергия не зависят от степени деформации (см. рис. 197), то для упрощения анализа влияние степени горячей деформации на получаемую структуру можно не рассматривать. Ю. М. Вайнблат для
Рис. 202. Диаграмма структурных состояний алюминиевого сплава марки АВ, деформированного с обжатием 50 % и нагретого под закалку до 520 СС: е — скорость; /д — температура деформации (Ю. М. Вайнблат)
алюминиевых сплавов предложил строить диаграммы структурных состояний, которые характеризуют структуру после горячей деформации с большими обжатиями, т. е. на установившейся стадии, при заданных температурах и заданных скоростях деформации (рис. 202). Для тер-
429
мическн упрочняемых алюминиевых сплавов важно знать структуру не столько в свежедеформированном состоянии, сколько после нагрева до температуры закалки, которая может быть как выше, так и ниже температуры начала рекристаллизации (так называемое структурное упрочнение обусловлено сохранением при закалке перекристаллизованной структуры, см. § 57). В связи с этим диаграмму структурных состояний промышленных алюминиевых сплавов целесообразно строить для стандартных условий нагрева под закалку (520 °C для сплава АВ на рис. 202).
На диаграмме структурных состояний линия АА отделяет область горячей деформации I, после которой при нагреве под закалку рекристаллизация не идет. Линия АА характеризует взаимосвязь двух критических параметров горячей деформации — критической скорости деформации и критической температуры деформирования. При скорости ниже критической и при температуре выше критической рекристаллизация при последующей стандартной термообработке не идет. Например, если сплав АВ (см. рис. 202) деформировать со скоростью около 10-1 г1, то перекристаллизованную структуру в закаленном изделии можно получить, проводя деформирование при температурах выше 400 °C. С другой стороны, если деформировать сплав при 400 °C, то перекристаллизованную структуру после закалкн можно получить только при скоростях деформации меньше 10-1 с"1. При горячей обработке давлением критическая скорость деформации как бы выполняет роль, которую играет критическая степень деформации при холодной обработке давлением.
Линия А А, отделяющая область I докритических состояний, где рекристаллизация не идет, от области закритических состояний, называется линией критических состояний. Любая точка на этой линии соответствует одной и той же температуре начала рекристаллизации (после деформации на установившейся стадии), равной температуре нагрева под закалку: (^р)уСТ=^эак. Следовательно, можно сделать вывод, что любая точка на линии критических состояний АА соответствует одинаковому уровню накопленной энергии. Естественно, что чем выше температура деформирования, тем больше должна быть критическая скорость деформации, чтобы накопить энергию, достаточную для начала рекристаллизации.
Линия ВВ на рис. 202 — это температурно-скоростная граница области II, где рекристаллизация при нагреве под закалку проходит полностью, т.е. на этой линии (^р)уст = ^зак Между линиями АА и ВВ находится область III частичной рекристаллизации. Наконец, линия СС отделяет область IV, в которой рекристаллизация проходит сразу после окончания горячей деформации, без дополнительного нагрева (см. выше метадинамическую рекристаллизацию).
Глава XIV
ТЕРМОМЕХАНИЧЕСКАЯ ОБРАБОТКА СТАРЕЮЩИХ СПЛАВОВ
На рис. 203 приведены основные схемы ТМО стареющих сплавов. Зубчатыми линиями обозначена пластическая деформация.
430
§ 56. Низкотемпературная термомеханическая обработка (НТМО)
НТМО стареющих сплавов — это первая по времени появления (30-е годы) и наиболее широко используемая в промышленности термомеханическая обработка.
Степень обматия, 7*
Рис. 204. Влияние степени обжатия при волочении после закалки с 1000 °C на твердое зь холоднотянутой и состаренном проволоки диаметром 4 мм из сплава нимоник-90 (по данным У. Беттериджа):
1 — холоднотянутая; 2 — деформация + старение пги 450 °C, 16 ч
Время —*
Рис. 203. Схемы термомеханической обработки стареющих сплавов
показатели пластичности.
Основное назначение НТМО— повышение прочностных свойств.
При НТМО сплав вначале подвергают обычной закалке, а затем перед старением—холодной деформации (см. рис. 203).
По сравнению со старением без предшествующей деформации при НТМО получают более высокие временное сопротивление и предел текучести и более низкие
На рис. 204 показано влияние степени холодной деформации на твердость закаленного никелевого сплава (кривая 7) и того же сплава, состаренного после деформации (кривая 2).
Упрочнение при НТМО вызвано двумя причинами. Во-первых, холодная деформация создает наклеп, и последующее дисперсионное твердение начинается от более высокого уровня твердости сплава. Во-вторых, что особенно важно, холодная деформация увеличивает эффект дисперсионного твердения. Так, при отсутствии холодного наклепа упрочнение сплава нимоник-90 в результате старения при 450 СС очень мало — всего //V 15. С увеличением степени холодной деформации упрочнение при старении непрерывно возрастает (кривые 1 и 2 на рис. 204 расходятся). При обжатии 90 % прирост твердости в результате старения составил HV 175. Следовательно, в рассматриваемом слу
431
чае холодны» наклеп увеличил упрочнение при старении на порядок (!). Такой сильный эффект упрочнения от НТМО по сравнению с упрочнением при термической обработке по обычной схеме (закалка+старение) — сравнительно редкое явление. Обусловлен он тем, что температура старения 450 °C слишком низка для нимоника, и при отсутствии холодного наклепа распад пересыщенного раствора при этой температуре развивается очень вяло. Если после закалки проводить старение при температуре, оптимальной для максимального упрочнения (около 700 °C), то эффект от введения холодного наклепа будет значительно меньше.
В самом первом приближении можно утверждать, что холодный наклеп, повышая плотность несовершенств в кристаллах пересыщенного раствора, делает его термодинамически менее стабильным и ускоряет старение. Однако экспериментальные факты и более детальный анализ показывают, что влияние наклепа на старение может быть весьма сложным. Характер этого влияния зависит от режимов закалки, деформации и старения, от природы сплава, а для одного сплава — от типа выделений при старении.
Рассмотрим вначале влияние холодной деформации на зонное старение. Казалось бы, что деформация, увеличивая плотность дислокаций и концентрацию вакансий, должна ускорять зонное старение. Но, во-первых, зоны зарождаются гомогенно, а не на дислокациях, и, во-вторых, дислокации являются эффективными местами стока вакансий. Очень сильная пластическая деформация повышает концентрацию вакансий (отношение числа вакансий к числу атомов) всего на 10~6, в то время как закалка создает значительно более сильное пересыщение решетки вакансиями: концентрация закалочных вакансий достигает величины порядка 10-4.
«Выметание» большого числа закалочных вакансий скользящими дислокациями при пластической деформации закаленного сплава может полностью перекрыть сравнительно небольшое поступление в кристалл вакансий, генерируемых порогами скользящих дислокаций. В результате пластическая деформация закаленного сплава замедляет зонное старение, так как избыточные вакансии играют важную роль в диффузионном переносе атомов растворенного элемента к растущим зонам (см. § 46).
В сплавах с небольшой концентрацией растворенного элемента холодный наклеп может замедлить образование зон при старении из-за притяжения к дислокациям атомов
432
этого элемента и соответствующего обеднения ими матрицы.
Замедление роста зон в наклепанном сплаве проявляется в замедлении упрочнения п уменьшении прироста прочности при старении. Такой результат получается, например, при холодной деформации свежезакаленного дур-алюмина. Поэтому НТМО с зонным старением как заключительной операцией обычно не используют.
Если при закалке изделие охлаждалось сравнительно медленно, например при закалке в воздушной среде, и решетка сильно не пересыщалась закалочными вакансиями, то замедление зонного старения под действием наклепа может и не проявиться. Более того, наклеп в этом случае способен даже ускорить зонное старение из-за повышения концентрации подвижных вакансий в результате разрушения дислокациями вакансионных скоплений, успевших образоваться при замедленном закалочном охлаждении. Кроме того, в этих условиях ускорение зонного старения может быть также результатом генерирования вакансий порогами скользящих дислокаций. В таких случаях НТМО, включающую зонное старение-, можно использовать для дополнительного упрочнения сплава.
Несравненно больший интерес представляет НТМО, включающая фазовое старение. Дислокации облегчают за = рождение выделений фаз. Поэтому в наклепанном сплаве с повышенной плотностью дислокаций фазовое старение протекает быстрее, а плотность выделений и соответственно упрочнение оказываются более высокими, чем при старении ненаклепанного сплава. Чем больше степень холодной деформации, тем выше плотность дислокаций и соответственно выше плотность выделений и упрочнение при фазовом старении (см. рис. 204).
При нагреве под старение после холодной деформации рекристаллизация, как правило, не протекает, а развиваются процессы отдыха и полигонизации, несколько уменьшающие упрочнение от НТМО. Следует иметь в виду взаимное влияние этих процесов и распада раствора: выделения тормозят полигонизацию, а полигонизация, если она успела пройти, изменяет плотность и характер распределения выделений.
Холодный наклеп не только ускоряет распад раствора и увеличивает плотность выделений. Он может вызвать появление фазы, которая без наклепа при том же режиме старения не выделяется, и даже способен изменить последовательность появления фаз при старении.
28 И. И. Новиков
433
Например, в ненаклепанном сплаве А1—4 % Си во время старения при 150 °C (/-фаза рентгенографически обнаруживается через 15 дней, а 0-фаза (СиА12) вообще не появляется. После холодной прокатки с обжатием 90 % во время старения при той же температуре сначала (через 5 мин) обнаруживается 0-фаза, а позднее (через 30 мин) O'-фаза. Появление метастибильной фазы после выделения стабильной вызвано следующим. После холодной деформации с большим обжатием по всему объему кристаллов твердого раствора решетка искажена столь сильно, что полукогерентные выделения (/-фазы зарождаться не могут. Вместе с тем в сильно искаженных участках облегчено зарождение некогерентных выделений б-фазы. Через некоторое время отдых и полигонизация, протекающие при температуре старения, делают решетку в отдельных участках кристаллитов настолько совершенной, что здесь уже могут зарождаться полукогерентные выделения О'-фазы (при условии, что в этих участках раствор еще не распался с образованием 9-фазы).
Если твердый раствор претерпел частичный распад перед холодной деформацией, то это сказывается на кинетике окончательного старения и свойствах сплава. Например, в случае холодной деформации свежезакаленного сплава А1—4 % Си максимум твердости во время старения при 160 °C достигается через 20—30 ч, а если ту же деформацию проводить после естественного старения, то максимальное упрочнение во время старения при 160 °C достигается через 8—10 ч. Это ускорение искусственного старения можно объяснить тем, что при пластической деформации естественно состаренного сплава дислокации останавливаются зонами ГП и при последующем старении вблизи дислокаций уже имеются обогащенные медью участки, в которых облегчено зарождение б'-фазы. Следовательно, в данном случае для ускорения искусственного старения выгодно деформировать сплав после естественного старения.
Предстарение (перед пластической деформацией) может не только ускорять заключительное старение, но и усиливать упрочнение от него.
В связи со сказанным для естественно стареющих алюминиевых сплавов необходимо регламентировать перерыв между закалкой и холодной деформацией при НТМО.
Предстарение может быть не только естественным, но и искусственным. Для сплавов на разных основах следует шире опробовать усложненные схемы НТМО типа закал-ка->старение->холодная деформация-устарение.
434
Повышенную прочность можно получить, применяя обычную холодную деформацию после старения (без НТМО). По сравнению с такой обработкой НТМО обеспечивает при равной прочности более высокую пластичность, меньшие остаточные напряжения и термически более стабильную структуру.
В настоящее время НТМО широко применяют в технологии производства полуфабрикатов и изделий из стареющих медных, алюминиевых и аустенитных сплавов.
В результате холодной прокатки перед старением бериллиевой бронзы БрБ2 предел текучести дополнительно возрастает на 20 %, При НТМО пружинных аустенитных сплавов типа 36НХТЮ с обжатием 50 % предел упругости ао,о02 оказывается на 20—30 % выше, чем после закалки и старения.
Иногда НТМО используют для получения небольшого по абсолютной величине, но практически важного повышения прочностных свойств. Например, применяемая в промышленности НТМО листов из алюминиевого сплава 1201, включающая холодную прокатку с обжатием 7—10 % перед старением при 1.75 °C, повышает 0^,2 на 30 МПа по сравнению с обычной обработкой (старением после правки закаленных листов).
Целью НТМО может быть не только упрочнение. Алюминиевые сплавы типа АМгб рекомендуется деформировать вхолодную на 30— 40 % с последующим старением при 220 °C в течение нескольких часоз. В первые 20 мин нагрева после деформации проходит полигонизация, а затем равномерное выделение ₽-фазы (Al3Mg2) по субграннцам. Такая структура обеспечивает повышенную стойкость против коррозии под напряжением.
Полуфабрикаты из стареющих алюминиевых сплавов (профили, панели, трубы, листы) после закалки обязательно правят растяжением или прогладкой. Степень деформации при правке невелика — обычно не более 3 %. Но даже небольшая холодная деформация со степенью 1—3 % может сильно увеличивать упрочнение при последующем искусственном старении. Например, предел текучести дуралюмина Д16, состаренного при 190 °C, в результате применения перед старением растяжки на 1,5 % возрастает с 400 до 455 МПа. Технологический процесс по схеме закалка~>правка~>старение обычно не называют термомеханической обработкой, но фактически он является типичной НТМО.
Благодаря простоте технологии и эффективности процесса число стареющих сплавов на разных основах и номенклатура изделий, подвергаемых НТМО, будет расширяться. Но, назначая НТМО, всегда следует учитывать и возможные отрицательные последствия — снижение пластичности, характерное для большинства сплавов, уменьшение сопротивления ползучести некоторых алюминиевых сплавов, анизотропию свойств и др.
28*
435
§ 57. Высокотемпературная термомеханическая обработка (ВТМО)
При высокотемпературной термомеханической обработке проводят горячую деформацию, закалку с деформационного нагрева и старение (см. рис. 203).
Сущность ВТМО состоит в том» что после горячей деформации и закалки получается пересыщенный твердый раствор с перекристаллизованной структурой, т. е. с новы* шенной плотностью несовершенств (границ субзерен, свободных дислокаций). В результате старения сплава с такой структурой возникают повышенные механические свойства. В большинстве случаев оптимальной является полигонизо-ванная матрица закаленного сплава.
При проведении ВТМО должны выполняться минимум три условия: 1) получение к концу горячей деформаци^Гне-рекристаллизованной структуры; 2) предотвращение возможной рекристаллизации после окончания горячей деформации; 3) достижение необходимой для старения степени пересыщенности твердого раствора.
Если два первых условия не выполнены и закаленный сплав полностью рекристаллизован, то мы имеем дело не с ВТМО, а с закалкой, проведенной с температуры деформационного нагрева. Такое совмещение операций горячей деформации и нагрева под закалку экономически выгодно, но оно не приводит к улучшению свойств по сравнению с обычной термообработкой, включающей специальный нагрев под закалку.
Применение ВТМО ограничивают следующие факторы.
Сплав может отличаться столь узким интервалом температур нагрева под закалку, что поддерживать температуру горячей обработки давлением в таких узких пределах практически невозможно (например, в пределах ±5 К для дуралюмина Д16).
Оптимальный температурный интервал горячей деформации может находиться значительно ниже интервала температур нагрева под закалку. Например, при прессовании алюминиевых сплавов скорость деформирования без прояви ления поверхностных трещин тем ниже, чем выше температура. Если требуется значительно повысить температуру горячей деформации до температур нагрева под закалку, то приходится снижать скорость деформирования, и производительность оборудования падает.
В сплаве с нерекристаллизованной структурой распад переохлажденного раствора может идти значительно быст
436
рее, чем в рекристаллизованном сплаве (см. рис. 109,6). Если такой сплав обладает невысокой прокаливаемостыо, то в условиях ВТМО она оказывается еще ниже и может не обеспечить сквозную закалку.
При высокой склонности сплава к рекристаллизации трудно создать к моменту окончания деформации перекристаллизованную структуру.
Рис. 205. Микроструктура титанового [З сплава (ВТ15) после обычной закалки (а) и после ВТМО (б). Х300 (С. Г. Глазунов, И. С. Полькин)
ВМТО используют для повышения прочностных свойств сплава по сравнению со свойствами того же сплава с рекристаллизованной структурой. Прочность при ВТМО растет в результате общего повышения плотности несовершенств уже в закаленном состоянии и более равномерного распада пересыщенного раствора по телу зерен при старении (упрочняющая фаза выделяется по границам субрезен и на одиночных дислокациях внутри них).
Более равномерный распад раствора и характерная для многих сплавов измельчепность зерен и искривленность их границ (рис. 205) обусловливают высокий уровень пластичности после ВТМО. В отличие от НТМО, заметно снижающей пластичность, дополнительное упрочнение от
437
ВТМО достигается при практически неизменном уровне пластичности. У алюминиевых сплавов ВТМО может даже повысить пластичность и ударную вязкость.
Упрочнение от ВТМО сохраняется до более высоких температур, чем после НТМО. Повышенная жаропрочность сплавов после ВТМО связана с зубчатостью границ зерен, затрудняющей межзеренное разрушение, и наличием субзеренных границ, закрепленных частицами выделений.
Из алюминиевых сплавов только сплавы на базе систем Al—Mg—Si (АД35) и Al—Zn—Mg (1915) широко подвергают ВТМО. Алюминий характеризуется высокой энергией дефектов упаковки, и полигонизация в нем проходит очень легко. При прессовании алюминиевых сплавов, как правило, формируется весьма стабильная полигонизованная структура, и поэтому никаких специальных мер для ее получения и сохранения при ВТМО принимать не приходится.
Указанные алюминевые сплавы характеризуются широким интервалом температур нагрева под закалку (от 350 до 500 СС для сплавов на базе системы А1—Zn—Mg) и обладают очень высокой прокаливаемостью (см. рис. 109, а). Все это предопределяет простоту технологии ВТМО. При прессовании тонкостенных профилей (толщиной до 12 мм) охлаждение на воздухе обеспечивает сквозную прокаливаемость. При большей толщине профилей из сплавов АД35 и 1915 их охлаждают водой прямо на прессе. ВТМО стареющих сплавов в промышленности применяют значительно реже, чем НТМО, из-за указанных выше технологических ограничений и меньшего эффекта упрочнения.
Для освоения в производстве рекомендован процесс, включающий комбинацию ВТМО и НТМО, — так называе* мая высоконизкотемпературная термомеханическая обработка (ВНТМО). При ВНТМО вначале проводят закалку с деформационного нагрева, затем холодную деформацию и старение (см. рис. 203). После ВНТМО прочность получается выше, а пластичность ниже, чем после ВТМО.
§ 58. Предварительная термомеханическая обработка (ПТМО)
Сущность ПТМО заключается в том, что полуфабрикат, полученный после горячей деформации в перекристаллизованном состоянии, сохраняет перекристаллизованную структуру и при нагреве под закалку. ПТМО отличается от ВТМО тем, что операции горячей деформации и нагрева под закалку разделены (см. рис. 203).
438
ПТМО широко применяют в технологии производства полуфабрикатов из алюминиевых сплавов. Давно было известно, что прессованные полуфабрикаты из сплавов типа «дуралюмин», «авиаль» и др. отличаются значительно более высокой прочностью, чем катаные и кованые. Это явление было названо пресс-эффектом. Разница в прочности обусловлена тем, что прессованные полуфабрикаты после закалки имели нерекристаллизованную структуру, а катаные и кованые — рекристаллизованную. Позже оказалось, что горячекатаные листы и штамповки из ряда сплавов после закалки также находятся в перекристаллизованном состоянии и характеризуются повышенной прочностью.
Вместо терминов пресс-эффект, прокат-эффект и т. п. В. И. Добаткпн предложил обобщающий термин структурное упрочнение, под которым понимается повышение прочности термически обработанных полуфабрикатов, обусловленное сохранением после закалки перекристаллизованной структуры. Свойства сплава в состоянии структурного упрочнения обычно сравнивают со свойствами того же сплава в рекристаллизованном состоянии. Прирост временного сопротивления и предела текучести вследствие сохранения перекристаллизованной структуры у стареющих алюминиевых сплавов составляет от 10 до 40%. Этот прирост проявляется уже в свежезакаленном состоянии из-за повышенной плотности несовершенств. Старение перекристаллизованного сплава дает большое упрочнение, увеличивая разницу в свойствах рекристаллизованного и перекристаллизованного полуфабриката. Пластичность же выше у сплавов в рекристаллизованном состоянии. Поэтому полуфабрикаты выпускают с рекристаллизованной структурой и повышенной пластичностью, а также с полигонизо-ванной структурой и повышенной прочностью. Например, у дуралюминовых прутков Д1Ро ^380 МПа и 6^14 %, а у прутков Д1ПП* ов^430 МПа и 10 %.
В алюминиевых сплавах, как уже отмечалось, интенсивно развивается полигонизация, создающая стабильную сетку субграниц, и поэтому в них легко получить нерекристаллизованную структуру, особенно при прессовании.
Добавки марганца, циркония и других антирекристаллизаторов, образующих дисперсные алюминиды (А16Мп, AhZr и др.), повышают температуру начала рекристаллизации и способствуют получению перекристаллизованной
* Р и ПП — это тестированные обозначения прутков с рекристаллизованной структурой и повышенной прочностью соответственно.
439
структуры при горячей деформации. Особенно важно, что эти добавки, содержащиеся во многих алюминиевых сплавах, способствуют сохранению перекристаллизованной структуры при последующем нагреве под закалку.
Структурное упрочнение алюминиевых сплавов (ПТМО) давно и широко используется в крупнотоннажном массовом производстве полуфабрикатов и является примером эффективного промышленного применения термомеханической обработки.
Глава XV
ТЕРМОМЕХАНИЧЕСКАЯ ОБРАБОТКА СТАЛЕЙ, ЗАКАЛИВАЕМЫХ НА МАРТЕНСИТ
Процессы ТМО сталей начали интенсивно изучать с середины 50-х годов в связи с изысканием новых путей повышения конструктивной прочности.
§ 59. Низкотемпературная термомеханическая обработка (НТМО)
При НТМО переохлажденный аустенит деформируется в области его повышенной устойчивости, но обязательно ниже температуры начала рекристаллизации и затем превращается в мартенсит (рис. 206). После этого проводят низкий отпуск (на рис. 206 не показан).
Сильное упрочнение в результате пластической деформации переохлажденного аустенита с последующей закал: кой с температуры деформирования было открыто в 1954 г. Этот процесс, названный аусформингом, позволил повьь сить временное сопротивление конструкционных легированных сталей до 2800—3300 МПа при 6=54-7 %. Показатели пластичности и ударной вязкости получались не ниже, а в некоторых случаях даже выше, чем после обычной термообработки, обеспечивающей ов= 18004-2200 МПа. Понятно, что достижение «сверхпрочности» сталей методом аусфор-минга вызвало громадный интерес.
Причина упрочнения стали при НТМО — наследование мартенситом дислокационной структуры деформированного аустенита.
Холодной деформацией нельзя сильно упрочнить сталь с мартенситной структурой, так как мартенсит, содержащий углерод, хрупок и не поддается большим обжатиям. Аустенит же при температурах ниже температуры начала
140
рекристаллизации можно деформировать с большими обжатиями, при которых сильно возрастает общая плотность дислокаций.
При мартенситном превращении соседи любого атома
в аустените остаются соседями этого же атома в мартенсите. Поэтому дислокации при у->а-превращении по мар-
тенситному механизму не исчезают, а «передаются» от исходной фазы к новой, т. е. мартенсит наследует субструктуру деформированного аустенита. Очень высокая плотность
дислокаций в мартенсите, закрепленных атомами углерода и карбидными выделениями, обусловливает получение рекордных значений прочности после НТМО.
Измельченностью кристаллов мартенсита объясняется приемлемый уровень показателей пластичности стали, находящейся в высокопрочном состоянии.
НТМО применима только к легированным сталям, обладающим значительной устойчивостью переохлажденного аустенита.
Рис. 206. Схема высокотемпературной (ВТМО) и низкотемпературной (НТМО) термомеханической обработки легированной стали, закаливаемой на мартенсит
Внедрение НТМО в производство сильно затруднено необходимостью использования мощного оборудования для обработки
давлением, так как для получе-
ния высокой прочности сталь должна подвергаться большим обжатиям (не менее 50%) при таких температурах, при которых сопротивление деформированию очень высо-
кое.
Другой существенный недостаток НТМО — невысокая
сопротивляемость хрупкому разрушению сильно упрочненг ной стали.
§ 60. Высокотемпературная термомеханическая обработка (ВТМО)
При ВТМО аустенит деформируют в области его термодинамической стабильности и затем проводят закалку на мартенсит (см. рис. 206), После закалки следует низкий отпуск.
441
Основная цель обычной термообработки с деформационного (прокатного, ковочного) нагрева — исключить специальный нагрев под закалку и благодаря этому получить экономический эффект. Главная же цель ВТМО — повыше-
ние механических свойств стали. Увеличивая прочность при
ВТМО, можно сохранить неизменными показатели пластичности, например относительное сужение (рис. 207). По
сравнению же с обычной закалкой и отпуском при одинаковой прочности ВТМО позволяет получить более высокие показатели пластичности.
При ВТМО машиностроительных углеродистых, низко- и среднелегированных сталей достигается следующий уровень механических свойств: ов=22004-2600 МПа; оо 2 = 19004-2200 МПа; б = = 74-8 %; ^=254-40 %.
После ВТМО прочностные свойства ниже, а показатели пластичности выше, чем после НТМО.
1600^----1---1---1---1---
О 10 20 30 40 50
Степень о&катия, %
Рис. 207. Зависимость временного сопротивления при постоянном относительном сужении от степени обжатия при ВТМО стали 55ХГР (М. Л. Бернштейн)
С помощью ВТМО можно резко ослабить обратимую и необратимую отпускную хрупкость (см. рис. 191).
Повысить временное сопротивление до 2200 МПа не сложно и без ВТМО, увеличив содержание углерода в стали и проводя обычную закалку с низким отпуском. Но после обычной термообработки такая сталь будет характеризоваться очень высокой склонностью к хрупкому разрушению, большой чувствительностью к острым надрезам на поверхности изделий. Усталостные трещины в такой стали будут быстро распространяться из-за локализации напряжений у вершины трещины и приводить к внезапному хруп
кому разрушению при напряжениях меньше предела текучести. Сталь с высокими значениями временного сопротивления и предела текучести, но низкой сопротивляемостью
распространению трещины невозможно использовать в вы-соконагруженных конструкциях.
Важнейшее преимущество высокотемпературной термомеханической обработки — это ее замечательная способность одновременно повышать и показатели прочности, и сопротивляемость распространению трещины (вязкость разрушения).
442
Наилучший комплекс механических свойств стали после ВТМО достигается, когда мартенсит образуется из аусте-нита с хорошо развитой полнгоннзованнон структурой. В таком аустените получаются значительно мельче пакеты и кристаллы мартенсита. Размер многих мелких кристаллов мартенсита не выходит за пределы размеров субзерен аустенита» В сталях со смешанной морфологией мартенсита ВТМО с полигонизацией аустенита увеличивает долю пакетного мартенсита за счет пластинчатого. Мартенситные кристаллы наследуют дислокационные субграницы горяче-деформнрованного аустенита. Эти и другие особенности структуры сталей, ннзкоотпущенных после ВТМО, обусловливают получение указанного выше комплекса механических свойств.
Режим ВТМО выбирают так, чтобы к началу мартенситного превращения аустенит имел развитую полигонизо-ванную структуру. Степень горячей деформации при ВТМО не должна быть слишком большой, иначе развивается рекристаллизация, снижающая упрочнение (см. рис. 207). Применение дробной деформации с суммарным обжатием, равным большому обжатию за один проход, облегчает деформирование н способствует развитию динамической полигонизации.
После окончания деформирования углеродистых и низколегированных сталей необходима немедленная закалка, чтобы предотвратить статическую рекристаллизацию и сохранить полнгоннзованную структуру к началу мартенситного превращения.
М. Л. Бернштейн и А. Г. Рахштадт обнаружили интересное явление наследования упрочнения от ВТМО при повторной термической обработке. Оказалось, что упрочнение от ВТМО сохраняется, если сталь перезакалить с кратковременной выдержкой при температуре нагрева под закалку или если упрочненную ВТМО сталь вначале подвергнуть высокому отпуску, а затем перезакалить. Структура, созданная при горячей деформации аустенита, один раз наследовалась при у->а-превращении во время ВТМО, в определенной мере сохранялась при высоком отпуске и еще дважды наследовалась во время перезакалкн при ос->у- и у-^ос-превращениях. «Передачу» дислокаций от а- к у-фазе легко представить, если принять сдвиговый механизм ос->у-превращения. Короткая выдержка при повторной закалке предотвращает развитие рекристаллизации аустенита, которая . уничтожила бы полнгоннзованную структуру и упрочнение от предшествующей ВТМО.
443
ВТМО, несмотря на менее сильное упрочнение, имеет неоспоримые преимущества перед НТМО, К ним относятся одновременное повышение прочности и вязкости разрушения, высокая технологичность (для деформирования не требуется специализированного мощного оборудования),
Рес. 208. Схемы термомеханической обработки с деформацией в аустенитной области и последующим бейнитным превращением (/) и с деформацией во время перлитного превращения (2)
применимость не только к легированным сталям с повышенной устойчивостью переохлажденного аустенита, но и к углеродистым и низколегированным сталям.
Классическая схема ВТЛ4О предусматривает обязательную закалку на мартенсит. Разновидностью ВТМО являются горячая деформация стали в аустенитном состоянии и ускоренное охлаждение с последующим бейнитным превращением (рис. 208, кривая /). Повышение комплекса механических свойств при такой обработке обусловлено уменьшением размеров пакетов бейнит-ных кристаллов. Горячая де-
формация аустенита замедляет бейнитное превращение, т. е. бейнитная прокаливаемость при ВТМО увеличивается,
§ 61. Термомеханическая обработка
с деформацией во время перлитного превращения
При обработке, известной под названием «изоформинг», сталь после аустенитизации охлаждают до температур перлитного превращения и деформируют во время этого превращения (рис. 208, кривая 2). Такая обработка приводит к формированию тонкой субзеренной структуры в феррите (с размером субзерен менее 1 мкм) и замене карбидных пластин в перлите на дисперсные сферические равномерно распределенные частицы диаметром порядка 10~2— 10-1 мкм. По сравнению со сталью с феррито-перлитной структурой, полученной после обычной горячей обработки давлением, сталь после изоформинга обладает более высоким пределом текучести и, что особенно ценно, резко увеличенной вязкостью. Сопротивление распространению трещины может возрасти на порядок (!).
444
Совершенная субзеренная структура феррита формируется в результате полигонизации во время пластической деформации и в паузах между обжатиями. Дисперсные сферические частицы карбида могут зарождаться на дислокациях в аустените, а могут также появляться в результате фрагментации карбидных пластин и их сфероидизации, ускоренной деформацией. Существует оптимальная температура пластической деформации, выше которой субзерна и карбидные частицы получаются крупнее, а ниже не успевают сформироваться совершенные субзеренные границы, что отрицательно сказывается на вязкости стали. Суммарная степень обжатия для получения оптимальной структуры должна быть не менее 70 %.
Пластическая деформация аустенита уменьшает инкубационный период перлитного превращения. Это прямо противоположно влиянию ее на бейнитное превращение.
Основной недостаток изоформинга — высокое сопротивление стали деформированию при пониженных температурах обработки давлением.
§ 62. Контролируемая прокатка
Контролируемая прокатка — это горячая прокатка по регламентированному режиму, включающему запрограммированные температуры начала и окончания деформации, обжатия и скорость охлаждения.
Контролируемой прокатке подвергают ннзкоуглероднс-тые стали с микродобавками таких карбидо- и ннтрндооб-разователей, как ванадий, ниобий и титан. Пример — сталь 09Г2ФБ, содержащая 0,06 %V, 0,04 % Nb и до 0,015 % N.
Цель контролируемой прокатки — сформировать структуру с мелким зерном полнгоннзованного феррита и дисперсными выделениями карбонитрндов, обеспечивающую повышение (на 100—150 МПа) предела текучести, снижение температуры хрупко-вязкого перехода и улучшение свариваемости.
При контролируемой прокатке во время деформации и в паузах между обжатиями проходят следующие процессы: наклеп и рекристаллизация аустенита, превращение его в феррит, выделение карбидов и нитридов ванадия, ниобия и титана из аустенита и феррита, полигонизация феррита.
Начинают прокатку при температурах выше ~950°С. Пластическая деформация при понижающейся температуре и выделения карбидов из аустенита способствуют формированию аустенитных зерен с развитой сеткой субграниц и
445
полосами деформации. Из таких аустенитных зерен образуется большое число мелких ферритных зерен. Дисперсные карбонитриды препятствуют укрупнению зерен феррита. Образовавшийся феррит полигонизуется, а внутри него выделяются еще более дисперсные (размером до 5 нм) частицы карбонитридов, обусловливающих дисперсионное упрочнение стали.
Указанные структурные изменения позволяют рассматривать контролируемую прокатку как разновидность ТМО. После контролируемой прокатки сталь не подврегают тер* мической обработке.
Низкие температуры окончания контролируемой прокатки (700—800°C) и значительные (до 20 %) обжатия при таких температурах обусловливают большие усилия прокатки и необходимость в специализированных станах. Несмотря на это, контролируемую прокатку используют в крупнотоннажном производстве наиболее прочных и сравнительно дешевых сталей для промышленного строительства, мостостроения, газопроводов и других целей.
§ 63. Предварительная термомеханическая обработка (ПТМО)
ПТМО проводят по схеме холодная пластическая деформа* ция->дорекристаллизационный нагрев->закалка со скоростным нагревом и короткой выдержкой->отпуск.
Перед деформированием исходной является структура феррито-карбидной смеси. Холодная деформация повышает плотность дислокаций, перестройка которых при доре-кристаллизационном нагреве создает полигонизованную структуру феррита. При последующей закалке со скоростным нагревом и короткой выдержкой субструктура наследуется при и у—^-превращениях и кристаллы мартенсита получаются фрагментированными. Следовательно, механизм упрочнения при ПТМО тот же, что и в случае получения повышенной прочности при повторной закалке после ВТМО.
Предварительная термомеханическая обработка привлекает простотой технологии. Перерыв между холодной деформацией и нагревом никак не регламентирован. Специализированного оборудования для деформации не требуется. ПТМО с применением кратковременного нагрева под закалку в соляных ваннах или токами высокой частоты является эффективным способом упрочнения холоднокатаных листов и тонкостенных труб.
446
Раздел шестой
ХИМИКО-ТЕРМИЧЕСКАЯ ОБРАБОТКА
Глава XVI
ЗАКОНОМЕРНОСТИ ИЗМЕНЕНИЯ СОСТАВА И СТРУКТУРЫ ПРИ ХИМИКО-ТЕРМИЧЕСКОЙ ОБРАБОТКЕ
Химико-термическая обработка — это термическая обработка, сочетающая тепловое воздействие с химическим, в результате чего изменяются состав и структура в поверхностных слоях, а иногда и по всему объему изделия.
Для изменения химического состава изделие нагревают в активной среде. Во время выдержки изделие диффузионно обогащается элементами из внешней среды. Кроме того, химико-термическую обработку можно использовать для диффузионного удаления из изделия примесей, а в отдельных случаях и основных компонентов.
Подавляющее большинство промышленных процессов химико-термической обработки включает диффузионное обогащение поверхностных слоев изделий неметаллами или металлами из внешней активной среды, для чего используют газовые, жидкие и твердые среды. Самый известный из этих процессов — цементация (науглероживание стальных изделий в древесном угле при высоких температурах) относится к числу древнейших операций термической обработки, использовавшихся задолго до н. э.
Можно выделить три одновременно идущих процесса, обеспечивающих обогащение изделия элементами из внешней среды.
Первый процесс — образование химического элемента в активированном атомарном состоянии (in statu nascendi) в результате разнообразных химических реакций (см. гл. XVII), а также в результате испарения. В отдельных случаях, например при поступлении атомов металла непосредственно из расплава, эта стадия отсутствует.
Второй процесс — адсорбция атомов поверхностью изделия. Адсорбционный процесс может включать простую физическую адсорбцию, при которой моно- или полиатомный
адсорбционный слой на всей поверхности изделия или в ее активных участках образуется благодаря действию ван-дер-ваальсовых сил притяжения. Одновременно возможна и химическая адсорбция (хемосорбция) с возникновением сильных химических связей между адсорбируемыми атомами и атомами металлической поверхности. Адсорбция — всегда экзотермический процесс, приводящий к уменьшению энергии Гиббса.
Третий процесс при химико-термической обработке — диффузия адсорбированных атомов от поверхности в глубь изделия. Адсорбция протекает очень быстро, а диффузия идет медленно. Так как глубина зоны измененного состава (диффузионной зоны) и распределение концентрации внутри нее зависят главным образом от развития диффузии, то при анализе химико-термической обработки основное внимание уделяется закономерностям диффузии.
§ 64. Образование однофазной диффузионной зоны
Рассмотрим процесс диффузионного проникновения элемента с поверхности в глубь изделия. В любой точке диффузионной зоны на расстоянии х от поверхности изделия концентрация раствора С изменяется во времени т в соответствии со вторым законом Фика:
дС/дх = Dd2C/dx*. (42)
км
§ сл
I
§ ^0,5
В- С
5? ''О
<8
Х0,5
—Расстояние от поверхности ос
Рис. 209. Распределение концентрации в однофазной зоне диффузионно» о обогащения в разные моменты времени при постоянной концентрации на поверхности С
Здесь и ниже для упрощения принимаем, что коэффициент диффузии D не зависит от концентрации.
Уравнение диффузии (42) решают, выбрав определенные граничные условия. В нашем случае можно
использовать решение для диффузии в полубесконечное пространство с постоянной концентрацией на поверхности (рис, 209). В точке на расстоянии х от поверхности изделия концентрация С(х, т) зависит от концентрации на этой поверхности Сп, исходной концентрации в объеме Со, коэффициента диффузии D и
времени выдержки т от начала процесса:
С (х, т) = Сп — (Cn - Со) erf (х/2 /А).
(43)
448
При диффузии в чистый металл Со=О и
С (х, т) = Cn[ 1 — erf (х/2 УТ*)]. (44)
Величину erf (г), называемую функцией ошибок, находят по справочной таблице1.
Рис. 210. Распределение углерода (по результатам радиометрического анализа) после диффузионного насыщения стали с 0,14 % С кремнием (а) и хромом (б) при 950 °C (Г. В. Шербедииский, В. И. Шайдуров)
С увеличением времени выдержки диффузионная зона расширяется, а содержание диффундирующего элемента в ней возрастает (см. рис. 209), Площадь под кривой распределения концентрации элемента по сечению изделия характеризует общее количество элемента в диффузионной зоне при данном времени выдержки.
Если в качестве показателя глубины проникновения диффундирующего элемента выбрать расстояние х от поверхности до точки с любой заданной концентрацией, то из уравнения (43) следует, что
х/2 = const,
откуда
(45)
где К — константа.
Таким образом, глубина диффузионной зоны изменяется пропорционально корню квадратному из времени выдержки при постоянной температуре. Чтобы увеличить глубину диффузионной зоны, например, втрое, время выдержки следует увеличить в девять раз.
1 В формулах (43) и (44) г=х/2рЛ Dx.
29 И. И. Новиков 449
В реальных процессах химико-термической обработки параболический закон роста диффузионной зоны, описываемый формулой (45), может нарушаться из-за непостоянства концентрации на поверхности изделия (например, при большой активности окружающей среды концентрация Сп может возрастать).
Зависимость глубины однофазной диффузионной зоны от температуры, как показывают опыты, является экспоненциальной или близкой к экспоненциальной. Этого и следовало ожидать, учитывая, что коэффициент диффузии, входящий в формулу (45), сам экспоненциально растет с повышением температуры [см. формулу (2)].
Рассмотренные выше простые закономерности образования диффузионной зоны относятся к двухкомпонентному твердому раствору. Закономерности формирования диффузионной зоны в многокомпонентном растворе значительно сложнее, так как изменение концентрации одного элемента влияет на термодинамическую активность других элементов и на пх распределение1. Например, при силицировании— диффузионном насыщении стали кремнием — активность углерода вблизи поверхности изделия, где кремния больше, повышена и углерод как бы оттесняется от поверхности (рис. 210, а). При диффузионном хромировании картина прямо противоположная. Хром понижает активность углерода в аустените и в приповерхностных слоях, где хрома больше, концентрация углерода повышена, а прилегающий более глубокий слой обеднен углеродом (рис. 210, б). Такое необычное на первый взгляд перераспределение углерода (восходящая диффузия) в обоих случаях идет в сторону уменьшения градиента его активности. В общем при насыщении стали карбидообразующими элементами, снижающими активность углерода, можно ожидать обогащения углеродом приповерхностного слоя и обеднения им следующего слоя, а при насыщении некарбидообразующими элементами, повышающими активность углерода, следует ожидать оттеснения его от поверхности.
§ 65. Образование многофазной диффузионной зоны
В однофазной диффузионной зоне при охлаждении изделия с температуры химико-термической обработки могут протекать фазовые превращения: полиморфное превращение
1 О роли термодинамической активности элементов в диффузионных процессах см. в § 1,
450
твердого раствора, эвтектоидное превращение, выделение избыточной фазы и др.
Допустим, что во время науглероживания железа при температуре t\ (выше 911 °C) концентрация углерода на поверхности изделия возрастает от точки а (чистое железо) до точки Ь, не достигая границы аустенитной области '(рис. 211,/). При температуре химико-термической обработки диффузионная зона в любой момент времени имеет аустенитную структуру с переменной концентрацией углерода, уменьшающейся при удалении от поверхности в глубь изделия (см. кривую на рис. 211, //).
При медленном охлаждении с температуры науглероживания в аустените происходят фазовые превращения в соответствии с диаграммой состояния Fe—С. На участке диффузионной зоны, примыкающем к поверхности и имеющем концентрацию углерода выше 0,8 %, при комнатной температуре находятся вторичный цементит и перлит, а на участке с концентрацией углерода менее 0,8 % — избыточный феррит и перлит. При удалении от поверхности из-за непрерывного уменьшения концентрации углерода постепенно исчезает сетка вторичного цементита и структура становится чисто перлитной, а затем постепенно уменьшается количество перлита и структура становится ферритной (рис. 212).
Таким образом, в рассмотренном случае при химикотермической обработке диффузионная зона — однофазная, а в результате превращения при охлаждении она становится по всему своему сечению двухфазной (феррит+цемен-тит).
Новые фазы могут появиться непосредственно в процессе диффузионного изменения состава при температуре химико-термической обработки. Диффузию, сопровождающуюся появлением новых фаз, называют реактивной (или реакционной).
Принципиальную возможность протекания реактивной диффузии устанавливают по диаграмме состояния. В системе с непрерывным рядом твердых растворов реактивная диффузия невозможна, а в любой системе с ограниченной растворимостью компонентов в твердом состоянии она может протекать.
Предложены два объяснения механизма образования новых фаз при реактивной диффузии.
По одному из них, непосредственно на поверхности раздела металла с окружающей средой протекает химическая реакция образования соединения. Такая реакция рассмат
29*
451
ривается как первичный процесс реактивной диффузии, определяющий ее сущность (В. 3. Бугаков). Новая фаза об-
разуется в результате проявления сил химического взаимо-
действия реагирующих элементов. Таким способом может образоваться лишь очень тонкий (порядка атомных размеров) слой соединения, так как, возникнув, он разобщает атомы реагирующих элементов. Дальнейший рост новой фазы обеспечивается диффузией одного или обоих реагирующих элементов через образовавшийся слой.
Другой механизм реактивной диффузии включает обязательное достижение предельной растворимости при пос
Рис. 212. Микроструктура диффузионной зоны после медленного охлаждения с температуры науглероживания железа. Х200
Рис. 211. Участок диаграммы состояния системы Fe—С (/) и кривая распределения углерода по глубине диффузионной зоны (II) после науглероживания железа при температуре /1 (схема)
452
туплении диффундирующего элемента из внешней среды, после чего становится возможным образование той фазы, которая в соответствии с диаграммой состояния должна находиться в равновесии с насыщенным твердым раствором (Д. А. Прокошкин). Такую диффузию хотя обычно и называют реактивной, но сущность процесса определяется не
химическом реакцией на поверхности, а насыщением (точнее, пересыщением) исходного раствора диффундирующим элементом. Поэтому более строгое название— диффузия, сопровождающа-
Рис. 214. Распределение концентрации элемента В в диффузионной зоне после насыщения металла Л при температуре ti в разные моменты T^T6 (диаграмму состояния Л—В см. на рис. 213)
Рис. 213. Диаграмма состояния двойной системы Л—В
яся фазовыми превращениями. Весь последующий анализ базируется именно на таком представлении о появлении новых фаз при химико-термической обработке.
Рассмотрим диффузию элемента В в изделие из элемента А при температуре t\ (рис. 213). С увеличением времени выдержки в поверхностном слое плавно возрастает концентрация элемента В в а-растворе (рис. 214), пока при достаточной активности внешней среды не будет достигнут предел растворимости (точка а на рис. 213). По достижении этого момента (тг на рис. 214) при дальнейшем поступлении элемента В из внешней среды a-фаза пересыщается этим элементом и в поверхностном слое из нее выделяется p-фаза состава b (рис. 213). В результате в поверхностном слое концентрация скачком возрастает на ab.
Продолжающееся поступление элемента В из внешней среды плавно повышает его концентрацию во всем слое р-фазы. Атомы В непрерывно диффундируют от поверхности изделия к границе раздела р- и a-фаз, поступают в a-фазу и поддерживают ее в пересыщенном состоянии вбли
453
зи границы p/а. Поэтому при непрерывном поступлении элемента В из внешней среды на границе p/а из пересыщенного раствора непрерывно выделяется p-фаза и эта граница перемещается в глубь изделия (см. положения границ слоев в моменты т3 и т4 на рис. 214).
Когда в поверхностном слое достигается предельная для данной температуры концентрация элемента В в р-фазе (точка с на рис. 213) и затем p-фаза становится Пересы-щенной, в этом слое образуется у-фаза состава точки d. При дальнейшем поступлении компонента В концентрация его в у-фазе плавно возрастает, приводя к смещению границы у/p в глубь изделия.
Можно отметить следующие характерные особенности образования многофазной диффузионной зоны.
Последовательность образования фаз при диффузионном изменении состава соответствует последовательности их расположения на диаграмме состояния (а->р->у на рис. 213). В таком чередовании фазы появляются при повышении концентрации диффундирующего элемента в поверхностном слое изделия с увеличением времени выдержки и в таком же порядке они располагаются в диффузионной зоне по направлению от сердцевины к поверхности (см. мо-» мент времени Тб на рис. 214). Если в диффузионной зоне какая-либо из фаз не обнаруживается экспериментально обычно под световым микроскопом или рентгеновским методом), то это не значит, что она была «пропущена» при формировании зоны. Толщина ее из-за малой скорости диффузионного роста такова, что при данной продолжительности химико-термической обработки экспериментально эта фаза не выявляется. При большем времени выдержки «пропущенная» фаза обычно обнаруживается.
По толщине каждого растущего слоя фазы имеется градиент концентрации. Без такого градиента диффузионное проникновение элемента в глубь изделия, т. е. формирование диффузионной зоны, было бы невозможно.
В диффузионной зоне составы фаз на границе раздела соседних слоев определяются составами фаз, находящихся в равновесии на диаграмме состояния [см., например, точки а и b или с и d на рис. 213). Это следует из того, что новая фаза образуется только после достижения предела насыщения исходной фазы.
Двухфазные области в диффузионной зоне при насыщении одного элемента другим отсутствуют, т. е. граничат всегда однофазные слои (см. рис. 214). Это объясняется тем, что в двухфазной смеси составы каждой из фаз при
454
любом их количественном соотношении неизменны и опре-
деляются соответствующими точками на диаграмме состо-
яния (например, а и b на рис. 213). В то же время для диффузионного роста слоя необходим градиент концентраций в фазе переменного состава. Поэтому рост двухфазного слоя, по сечению которого состав каждой из фаз постоянен,
к вообще диффузионный перенос элемента от поверхности в глубь изделия через такой слой невозможны.
Двухфазные слои могут расти при химико-термической обработке не чистого металла, а сплава или же при диф-
фузионном насыщении металла сразу двумя элементами. В этих случаях в диффузионной зоне мы имеем дело с тройной системой, в которой составы равновесных фаз в двухфазной области переменны.
Допустим, что в двухкомпонентный сплав состава cQ диффундирует элемент С (рис. 215). При неизменном соотношении компонентов Л и В в диффузионной зоне ее состав изменяется по
Рис. 215. Схема изотермического разреза тройной системы А— В—С, поясняющая последовательность образования слоев при диффузии элемента С в сплав Со (по А. П. Гуляеву)
лучу с0С. Элемент С растворяется в фазах аир и, когда средний состав двухфазного слоя а+р изменяется по линии с^а,
состав а-фазы изменяется по
линии hk, р-фазы — по линии тп. Трехфазный слой образоваться не может, так как в соответствии с изотерми-
ческим разрезом составы трех фаз а*, Рл и бР, находящихся в равновесии, постоянны, и через трехфазный слой невозможна диффузия в глубь изделия. Поэтому, когда средний состав двухфазного слоя а+р достигает точки а, он изменяется скачком до точки Ь: в результате замены фазы рл на фазу 6Р образуется двухфазный слой а*+бР.
При изменении среднего состава слоя а+б по линии bd составы а- и б-фаз изменяются по линиям kq и рг. При дальнейшем поступлении компонента С в диффузионную зону ее средний состав на поверхности скачком изменяется от точки d до точки f в результате замены фазы бг на фазу у$. Далее средний состав двухфазного слоя изменяется по линии [g, а составы а- и у-фаз — по линиям qg' и sg (пунктиром gg' на.рис. 215 изображена конода, указывающая составы равновесных фаз а и у). При этом а-фаза пе
455
реходит в у-фазу, и, наконец, в поверхностном слое остается одна у-фаза, состав которой изменяется по лучу в сторону компонента С.
Таким образом, после химико-термической обработки у самой поверхности находится слой у, а за ним — двухфазные слои а-Ьу, а 4-6 и а^-р (рис. 216).
Приведенный анализ с использованием изотермического разреза тройной системы справедлив только при условии, что соотношение концентрации компонентов Л и В в любой точке диффузионной зоны неизменно, а фазы в двухфазных слоях все время находятся в равновесии.
Рис. 216. Распределение средней концентрации элемента С в диффузионной зоне сплава с0 (см. рис.
215)
Рис. 217. Распределение элемента В в диффузионной зоне в моменты времени Т| (/) и Ti+dT (2)
Рассмотрим кинетику роста однофазных слоев при химико-термической обработке, основываясь на анализе диффузионных процессов, выполненном А. А. Поповым.
При диффузии элемента В в металл А за время dx граница p-фазы продвинется в сторону а-фазы на величину dL (см. соответствующую диаграмму состояния на рис. 213 и распределение элемента в диффузионной зоне на рис. 217). Приращение объема слоя p-фазы dV=SdL, где S— поверхность раздела а- и р-фаз.
Фаза р содержит больше компонента В, чем фаза а. Скачок концентрации на границе их раздела равен ДСра (см. рис. 213). Приращение объема p-фазы на dV увеличивает количество элемента В в слое p-фазы на dV ДСр/а.
Все расчеты будем проводить для единицы поверхности раздела (5=1). Тогда dVACp/a=dLACp;a (см. заштрихованный участок на рис. 217).
456
Диффузия в p-фазе подводит атомы В к поверхности раздела p/а, а диффузия в a-фазе отводит атомы В от этой поверхности. Количество элемента В, подведенного к границе p/а через p-фазу, обозначим через dm$ , а количество элемента В, отведенного от этой границы через а-фазу, обозначим через dm^. Разность dm$—dma равна увеличению общего содержания элемента В в слое p-фазы при продвижении ее границы на dL.
Следовательно, dUsC^a— din$—dma.
Учитывая первый закон Фика [см. формулу (1)], можем записать (в расчете на единицу площади поверхности раздела фаз):
dLACfl/a — (дСр /дх) dr — Da (дСа /дх) dx, где т — время.
Отсюда скорость продвижения границы p-слоя в сторону а-фазы
д^/а
Решение этого уравнения при условии постоянства концентрации на поверхности изделия показывает, что толщина слоя новой фазы (L) растет с увеличением времени выдержки (т) по параболическому закону:
L = KJ/T. (47)
Эта формула аналогична уравнению (45) для роста диффузионной зоны при химико-термической обработке без образования новых фаз, но коэффициент пропорциональности К зависит от значительно большего числа факторов.
Из уравнения (46) следует, что толщина слоя новой фазы тем больше, чем интенсивнее диффузия в нем, медленнее диффузия в слое соседней фазы и меньше скачок концентраций на границе фаз.
Если диффузионная зона состоит из слоев нескольких фаз, то толщина промежуточного слоя (р на рис. 214) зависит от соотношения скоростей продвижения его границ и, следовательно, от развития диффузионных процессов в каждой из фаз.
Направление диффузионных потоков в слоях показано стрелками на рис. 214. Диффузионный приток элемента В внутри p-слоя к границе p/а, поддерживая около нее пересыщение a-фазы, стремится сместить эту границу вправо и увеличить толщину p-слоя. Диффузионный отток элемента В внутри a-слоя от границы p/а уменьшает около нее пе
457
ресыщение a-фазы и стремится сместить эту границу влево. Аналогично диффузионный приток элемента В внутри у-слоя к границе у/p стремится сместить ее вправо, уменьшая толщину p-слоя. Этому препятствует диффузионный отток элемента В внутри p-слоя от границы у/р.
Диффузионный поток через слой фазы в соответствии с первым законом Фика тем интенсивнее, чем выше коэффициент диффузии внутри этой фазы и больше градиент концентрации по толщине слоя. Градиент концентраций в свою
очередь тем значительнее, чем сильнее различаются, соста-
Рнс. 218. Влияние температуры азотирования железа в течение 6 ч на толщину слоев е- и у-фаз (Ю. М. Лахтнн)
вы на двух ее границах и меньше толщина слоя. Составы на границах слоя, как уже отмечалось, определяется соответствующими точками на диаграмме состояния, например точками Ь и с для слоя p-фазы (см. рис. 213 и 214). Следовательно, при постоянном коэффициенте диффузии поток диффундирующего элемента через слой определенной толщины возрастает с расширением области гомогенности фазы на диаграмме состояния (см. отрезок Ьс на рис. 213).
Из сказанного следует, что толщина слоя новой фазы в диффузионной зоне с повышением температуры в общем
случае не может возрастать по экспоненциальному закону, так как она зависит не только от коэффициента диффузии в данной фазе.
Толщина слоев фаз с ростом температуры при химико-термической обработке изменяется по-разному. Она зависит от соотношения диффузионных потоков в слое и в прилегающих слоях. Например, если с повышением температуры ширина области гомогенности фаз на диаграмме состояния изменяется мало, то разрастаться будет слой той фазы, у которой коэффициент диффузии увеличивается больше.
Если при повышении температуры химико-термической обработки коэффициенты диффузии в соседних слоях увеличиваются примерно одинаково, то разрастаться будет слой той фазы, у которой сильнее расширяется область гомогенности на диаграмме состояния. Фазы с очень узкими областями гомогенности на диаграмме состояния могут рас-
458
ти так медленно, что их слои внутри диффузионной зоны экспериментально не будут обнаружены.
Из-за одновременного и разного изменения коэффициентов диффузии, градиентов концентраций внутри фаз и скачков концентрации на их границах скорость роста слоя каждой фазы, определяемая уравнением (46), может изменяться с повышением температуры химико-термической обработки сложным образом. Например, при азотировании (см. § 67) толщина слоя е-фазы растет с повышением температуры процесса примерно до 700 °C, а затем уменьшается (рис. 218). Вместе с тем весьма часто зависимость толщины слоя фазы от температуры бывает близкой к экспоненциальной (см. кривую для у-фазы на рис. 218),
§ 66. Особенности строения диффузионной зоны
Диффузионную зону на шлифе можно выявить травлением благодаря измененному химическому составу поверхностного слоя. В однофазной зоне концентрация плавно изменяется от поверхности в глубь изделия (см. рис. 209), и поэтому под микроскопом граница такой зоны размыта или чаще вообще не выявляется.
Если диффузия сопровождается фазовыми превращениями, то строение диффузионной зоны резко отличается от структуры глубинных слоев. Различие в типе решеток и скачок концентраций на границе новой и исходной фаз обусловливают резкую разницу в травимости по обе стороны от этой границы или способствуют вытравливанию самой границы в виде так называемой диффузионной линии (рис. 219).
Расстояние от поверхности изделия до границы с исходным раствором (Li на рис. 214) обычно принимают за условную глубину диффузионной зоны (фактически глубина ее £2 больше на величину слоя исходного твердого раствора измененного состава).
Кристаллы новой фазы, образующейся в диффузионной зоне, имеют столбчатую форму (см. рис. 219). Объясняется это тем, что кристаллы новой фазы растут только у границы слоев разных фаз, где из-за диффузионного притока элемента поддерживается пересыщенность исходной фазы. По мере продвижения границы слоев в глубь изделия растут в этом направлении и кристаллы новой фазы.
Если при охлаждении с температуры химико-термической обработки происходит полиморфное превращение, то столбчатые кристаллы фазы, образовавшейся при диффузи
459
онном насыщении, могут заменяться равноосными кристаллами фазы, появляющейся при охлаждении.
Диффузия по границам зерен идет быстрее, чем в их теле, и поэтому при химико-термической обработке иногда можно наблюдать более глубокое проникновение диффундирующего элемента по границам зерен исходной фазы и
Рис. 219. Микроструктура диффузионной зоны после насыщения железа молибденом при 1200 °C в течение 12 ч. Х200 (Г. H. Дубинин)
выделение здесь в виде сетки кристаллов новой фазы. Однако при сравнительно высоких температурах, при которых обычно и проводят химико-термическую обработку, разница в скоростях диффузии по границам и телу зерен нивелируется.
Глава XVII
РАЗНОВИДНОСТИ ХИМИКО-ТЕРМИЧЕСКОЙ ОБРАБОТКИ
В промышленности применяют множество способов химико-термической обработки, различающихся диффундирующими элементами, типом и составом внешней среды, химизмом процессов в ней, техникой исполнения и другими признаками.
В зависимости от агрегатного состояния внешней среды, в которую помещают обрабатываемое изделие, различают химико-термическую обработку в твердой, жидкой и газовой средах.
460
Атомы диффундирующего элемента поступают из твердого вещества в местах прямого контакта его с поверхностью изделия. Этот процесс малоэффективен, и применяют его редко. Обычно твердую среду используют для создания активной газовой или паровой фазы, из которой атомы поступают в изделие. Например, при цементации в твердом карбюризаторе (древесном угле) атомы углерода, диффундирующие в сталь, образуются из оксида углерода (2СО->С+СО2), а древесный уголь необходим лишь для образования газовой фазы. Другой пример— диффузионное хромирование в «твердой» среде, когда изделие упаковано с порошком хрома или феррохрома. При нагреве хром испаряется и его атомы поступают в изделие главным образом из паровой фазы, а не в местах прямого контакта порошка с поверхностью изделия.
При химико-термической обработке в жидкой среде атомы элемента, диффундирующего в изделие, образуются в результате химических реакций в расплавленной соли (например, в NaCN при цианировании стали) или поступают непосредственно из расплавленного металла (например, из расплава алюминия при диффузионном алюминировании стали).
При химико-термической обработке в газовой среде диффундирующий элемент образуется в результате реакций диссоциации (СН4->С+2Н2), диспропорционирования (2СО->С+СО2), обмена (CrCU+Fe—xCrJ^FeCh) и восстановления (VC12+H2^V+2HC1).
Газовая среда и активная газовая фаза, образующаяся при нагреве изделий в твердой среде, в промышленных процессах служат самыми распространенными поставщиками атомов элементов, которыми обогащаются поверхностные слои изделия.
Для химико-термической обработки наиболее удобна чисто газовая среда: ее состав легко регулировать, она быстро прогревается до заданной температуры, позволяет полностью механизировать и автоматизировать процесс химико-термической обработки и сразу проводить закалку (без повторного нагрева).
Приведенные примеры показывают, что классификация методов химико-термической обработки по агрегатному состоянию среды, в которую помещено изделие, не всегда согласуется с физико-химической сущностью процесса обработки.
Основываясь на физико-химической характеристике активной фазы, поставляющей диффундирующей элемент,
461
Таблица 11. Разнввздыости химико-термической обработки
Диффузионное насыщение неметаллами Диффузионное насыщение металлами Диффузионное удаление элементов
Науглероживание (цементация) Азотирование Цианирование Нитроцементация Борирование Силицирование Сульфидирование Сульфоцианирование Насыщение кислородом* Внутреннее окисление* Алитирование Хромирование диффузионное Хромоалитирование Цинкование диффузионное Меднение диффузионное Титанирование Бериллизация Ванадироваиие Обезводороживание Обескислороживание Обезуглероживание Комплексное удаление примесей
* Насыщение кислородом — поверхностное, а внутреннее окисление — это насыщение всего объема изделия кислородом с целью образования дисперсных оксидов.
Г. Н. Дубинин предложил следующую классификацию методов химико-термической обработки: насыщение из твердой фазы, насыщение из паровой фазы, насыщение из газовой фазы и насыщение из жидкой фазы. Согласно этой классификации цементацию стали в твердом карбюризаторе следует относить к методу насыщения из газовой фазы, а диффузионное хромирование в порошке хрома — к методу насыщения из паровой фазы.
По характеру изменения химического состава обрабатываемого изделия все разновидности химико-термической обработки можно разделить на три группы: диффузионнное насыщение неметаллами, диффузионное насыщение металлами и диффузионное удаление элементов. Эти группы включают разнообразные промышленные процессы химикотермической обработки (табл. 11).
Каждый процес химико-термической обработки может осуществляться разными методами (насыщением из газовой, паровой, жидкой или твердой фазы) и в самом разнообразном техническом исполнении (например, с получением активной газовой фазы в рабочем пространстве печи пли в отдельном генераторе).
Ниже в качестве примеров кратко рассмотрены некоторые типичные разновидности химико-термической обработки. Эти примеры относятся главным образом к сталям,
462
так как химико-термическую обработку чугунов и цветных металлов и сплавов в промышленности применяют несравненно реже.
§ 67. Диффузионное насыщение неметаллами
Поверхностное насыщение стали углеродом и азотом или совместно этими элементами — наиболее широко используемые процессы химико-термической обработки.
Углерод и азот растворяются в железе по способу внедрения и поэтому могут быстро диффундировать на значительную глубину. Активные среды, содержащие эти элементы, дешевы, а фазы, образующиеся с участием углерода и азота в процессе насыщения или при последующей термообработке, резко изменяют механические и физико-химические свойства стали.
Науглероживание (цементация)
Цементации подвергают изделия из сталей с низким содержанием углерода (обычно до 0,25 %).
При цементации в твердом карбюризаторе изделия укладывают в ящики, куда засыпают древесный уголь, смешанный с 20—25 % ВаСОз. При нагреве углерод древесного угля, соединяясь с кислородом воздуха, находящегося в цементационном ящике между частицами карбюризатора, образует оксид углерода:
2С (уголь) + О2 2СО.
В контакте с железом оксид углерода дает атомарный углерод:
2СО СО2 + С (атомарн.).
Этот активный углерод (in statu nascendi) поглощается аустенитом и диффундирует в глубь изделия.
Добавка ВаСОз сильно интенсифицирует процесс цементации, поставляя дополнительное количество оксида углерода и соответственно активного углерода:
ВаСОз+С (уголь)->ВаО+2СО
и 2СО СО2 4- С (атомарн.).
Для газовой цементации в качестве карбюризатора используют природный газ (состоит в основном из СН4), контролируемые атмосферы, получаемые в специальных генераторах, а также жидкие углеводороды (керосин, бензол и др.), каплями подаваемые в герметичное рабочее
463
пространство печи, где они образуют активную газовую фазу.
Основной поставщик углерода в газообразных карбюризаторах— метан: СН4->2Н2+С.
В искусственно полученной контролируемой атмосфере таким поставщиком является оксид углерода.
В зависимости от состава газовой смеси и состава стали атмосфера в печи может быть науглероживающей, обезуглероживающей и нейтральной. В последнем случае концентрация углерода в стали является равновесной для данного состава газовой смеси, и такую концентрацию (а более строго — соответствующую термодинамическую активность углерода) называют углеродным потенциалом данной атмосферы. Если концентрация углерода в стали ниже той, которая соответствует равновесию с атмосферой, то будет происходить науглероживание.
Газовая цементация — основной процесс при массовом производстве, а цементацию в твердом карбюризаторе используют в мелкосерийном производстве.
Глубина цементации в зависимости от назначения изделия и состава стали обычно находится в пределах 0,5— 2,0 мм.
Цементацию проводят при 910—930 °C, а иногда для ускорения при 1000—1050 °C. С повышением температуры уменьшается время достижения заданной глубины цементации. Так, при газовой цементации науглероженный слой толщиной 1,0—1,3 мм получают при 920 °C за 15 ч, а при 1000 °C — за 8 ч. Чтобы предотвратить сильный перегрев (рост аустенитного зерна), высокотемпературной цементации подвергают наследственно мелкозернистые стали.
Концентрация углерода в поверхностном слое изделия обычно составляет 0,8—1,0% и не достигает предела растворимости при температуре цементации. Следовательно, сетка ГезС при температуре цементации не образуется, и поверхностный слой, как и сердцевина, находится в аустенитном состоянии. После медленного охлаждения цементованный слой с переменной концентрацией углерода состоит из феррита и цементита и характеризуется гаммой структур, типичных для заэвтектоидной, эвтектоидной и доэвтектоидной стали (см. рис. 212).
Цементация является промежуточной операцией, цель которой — обогащение поверхностного слоя углеродом. Требуемое упрочнение поверхностного слоя изделия достигается закалкой после цементации. Закалка должна не только упрочнить поверхностный слой, но и исправить
464
структуру перегрева, возникающую из-за многочасовой выдержки стали при температуре цементации.
После цементации в твердом карбюризаторе ответственные изделия подвергают двойной закалке, так как содержание углерода в сердцевине и на поверхности изделия разное, а оптимальная температура нагрева под закалку зависит от содержания углерода в стали (см. рис. 143).
Первую закалку проводят с нагревом до 850—900 °C (выше точки А3 сердцевины изделия), чтобы произошла полная перекристаллизация с измельчением аустенитного зерна в доэвтектоидной стали. В углеродистой стали из-за малой глубины прокаливаемости сердцевина изделия после первой закалки состоит из феррита и перлита. Вместо первой закалки к углеродистой стали можно применять нормализацию. В прокаливающейся насквозь легированной стали сердцевина изделия состоит из низкоуглеродистого мартенсита. Такая структура обеспечивает повышенную прочность и достаточную вязкость сердцевины.
После первой закалки цементованный слой оказывается перегретым и содержащим повышенное количество остаточного аустенита. Поэтому применяют вторую закалку с температуры 700—780 °C, оптимальной для заэвтектоид-ных сталей. После второй закалки поверхностный слой состоит из мелкоигольчатого высокоуглеродистого мартенсита и глобулярных включений вторичного карбида.
При газовой цементации чаще всего применяют одну закалку с цементационного нагрева после подстуживания изделия до 840—860°C. После закалки цементованные изделия всегда нагревают до 160—180 °C для уменьшения закалочных напряжений.
Цементацию широко применяют в машиностроении для повышения твердости и износостойкости изделий с сохранением высокой вязкости их сердцевины. Удельный объем закаленного науглероженного слоя больше, чем сердцевины, и поэтому в нем возникают значительные сжимающие напряжения. Остаточные напряжения сжатия в поверхностном слое, достигающие 400—500 МПа, повышают предел выносливости изделия.
Низкое содержание углерода (0,08—0,25%) обеспечивает высокую вязкость сердцевины. Цементации подвергают качественные стали 08, 10, 15 и 20 и легированные стали 12ХНЗА, 18ХГТ и др. Основное назначение легирования здесь — повышение прокаливаемости и соответственно ме-
зо И. И. Новиков
465
ханических свойств сердцевины изделий из цементуемой стали.
Азотирование
Азотирование стальных изделий проводят в аммиаке, ко* торый при нагреве диссоциирует, поставляя активный ато
В системе Fe—N при температурах азотирования могут образовываться следующие фазы (рис. 220): а-раствор азота в железе (азотистый феррит), у-рас-твор азота в железе (азотистый аустенит), промежуточная у'-фаза переменного состава с г. ц. к. решеткой" (ей приписывают формулу Fe4N) и промежуточная е-фаза с г. п. решеткой и широкой областью гомогенности (от 8,1 до 11,1 % N при комнатной температуре).
Если азотирование вести при температуре 550 °C, которая ниже эвтектоидной (590° С), то вначале образуется а-раствор, затем у'-фа-за и, наконец, е-фаза. С уда-
марный азот: 2NH3^2N + 6H.
Рис. 220. Диаграмма состояния системы Fe—N
лением от поверхности при азотировании железа должно наблюдаться следующее чередование слоев: е->у'->а.
При понижении температуры области а-раствора и особенно е-фазы на диаграмме состояния сужаются (см. рис. 220), и поэтому при медленном охлаждении с температуры азотирования в а- и е-слоях должны появиться вторичные выделения у'. С удалением от поверхности при комнатной температуре слои чередуются в следующем порядке: е^е + у'т->у'-к*+у'т -+а.
На границах слоев е+у^/у' и у7а+у'т ПРИ комнатной температуре (соответствующие границы при температуре азотирования е/у' и у7«) концентрация азота меняется скачкообразно. Эти скачки соответствуют отразкам ab и cd на диаграмме состояния (см. рис. 220). Граница слоев е/е + увТ соответствует границе между фазовыми обла
466
стями е и е-Ь/ на диаграмме состояния (8,1 % N при комнатной температуре). Скачка концентрации азота на границе слоев е/е+у'т не должно быть.
При температуре азотирования 600 °C с удалением от поверхности изделия должно наблюдаться чередование слоев При медленном охлаждении аустенит
распадается при температурах ниже 590 °C, давая эвтектоидную смесь «+/, а в а- и 8-слоях появляются вторичные выделения -/-фазы. При комнатной температуре после медленного охлаждения должно быть следующее чередование слоев от поверхности в глубь изделия: е->е+-/т -* ->/->эвтектоид («+/)-хх+Твт-^а.
В легированных сталях азот образует с алюминием, хромом, молибденом и другими элементами нитриды в очень дисперсной форе, вследствие чего азотированный слой приобретает твердость, намного превышающую твердость цементованных сталей.
Образование азотированного слоя, сопровождается увеличением удельного объема, и у поверхности возникают остаточные сжимающие напряжения (до 800 МПа). Остаточные напряжения сжатия повышают предел выносливости азотированных изделий.
Тонкий слой 8-фазы (0,01—0,03 мм) хорошо защищает сталь от коррозии во влажной атмосфере и других средах. Азотированием повышают твердость, износостойкость, предел выносливости, а также коррозионную стойкость сталей.
Азотирование с целью повысить твердость и износостойкость применяют к деталям из сталей типа 38Х2МЮА. Перед азотированием изделие подвергают закалке и высокому отпуску для повышения прочности и вязкости сердцевины. Азотирование проводят при 500—520 °C. Из-за низкой температуры и, следовательно, низкой подвижности атомов азота процесс длительный (24—90 ч). Толщина азотированного слоя составляет 0,3—0,6 мм. Можно сократить продолжительность азотирования, повышая температуру, но при этом сильно падает твердость из-за коагуляции нитридов легирующих элементов. Длительный процесс азотирования для повышения твердости и износостойкости целесообразно применять только к изделиям ответственного назначения.
Ускорение азотирования достигают, используя тлеющий разряд между анодом и деталью — катодом (деталь
30*
467
бомбардируют ионами азота, образовавшимися в плазме разряда).
Для повышения коррозионной стойкости азотируют детали из разных сталей (главным образом, из углеродистых). Так как большая твердость здесь не требуется, то температуру процесса выбирают высокой (600—700°C); продолжительность такого процесса от 15 мин до 10 ч.
Некоторое применение нашло азотирование деталей из высокопрочных чугунов и титановых сплавов.
Азотирование титановых сплавов — один из немногих примеров промышленного использования химико-термической обработки сплавов цветных металлов. После азотирования повышается износостойкость и уменьшается схватывание деталей при работе в условиях трения.
Цианирование и нитроцементация
Насыщение поверхности изделий одновременно углеродом и азотом в расплавленной цианистой соли называют цианированием, а в газовой среде — нитроцементацией. Соотношение углерода и азота в диффузионной зоне можно регулировать, изменяя состав среды и температуру процесса.
Цианирование сталей проводят в ванне, содержащей NaCN, при 820—960 °C в течение 30—90 мин. При окислении цианистого натрия образуются атомарный азот и оксид углерода.
Преимущества цианирования по сравнению с цементацией — значительно меньшая продолжительность процесса и более высокая износостойкость и коррозионная стойкость (благодаря азоту в поверхностном слое). Недостаток процесса — использование ядовитых цианистых солей.
Нитроцементацию сталей, называемую также газовым цианированием, проводят при 850—870°С в течение 2—10 ч в среде, содержащей аммиак и науглероживающий газ. По сравнению с газовой цементацией нитроцементация имеет следующие преимущества: ниже температура процесса и, следовательно, меньше рост зерна, выше износостойкость, меньше коробление деталей.
§ 68. Диффузионное насыщение металлами
Металлы растворяются в железе и других металлах по способу замещения и поэтому медленнее, чем неметаллы, диффундируют в изделие. Как правило, диффузионное насыщение металлами проводят при более высоких темпера
468
турах, чем насыщение неметаллами. Типичные примеры-— алитирование и хромирование.
Алитирование применяют для повышения окалино-стойкости сталей и реже чугунов. Алитируют также литые лопатки газотурбинных двигателей из жаропрочных никелевых сплавов. При нагреве алитированного изделия в окислительной среде на его поверхности образуется тонкая и прочная пленка А12Оз, предохраняющая изделий от дальнейшего окисления. Глубина алитирования в зависимости от метода и режима составляет 0,2—0,8 мм.
Наибольшее распространение получило алитирование стальных изделий в порошках с насыщением из газовой фазы. Порошкообразная смесь состоит из ферроалюминия, хлористого аммония и окиси алюминия. В присутствии NH4C1 образуется газообразный хлорид алюминия А1С13, являющийся поставщиком активных атомов алюминия. Окись алюминия предотвращает спекание частиц ферроалюминия. Алитирование проводят при 950—1050 °C в в течение 3—12 ч.
Диффузионное хромирование позволяет повысить коррозионную стойкость, окалиностойкость и износостойкость стальных деталей.
Наибольшее применение получило хромирование в порошкообразных смесях феррохрома (или хрома), хлористого аммония и оксида алюминия при 1000—1050 °C с выдержкой 6—12 ч. Образующийся газообразный хлорид СгС12 является поставщиком активных атомов хрома. Используют также хромирование в вакууме при 1000— 1050 °C в течение нескольких часов с насыщением из паровой фазы, которая получается при испарении порошка хрома.
Для повышения коррозионной стойкости и окалино-стойкости поверхностный слой должен иметь структуру пластичного твердого раствора хрома в a-железе. Если одна из целей хромирования — повышение твердости, то в поверхностном слое должны образоваться карбиды хрома (Сг23С6, Сг7С3). Для этого выбирают сталь, содержащую более 0,4 % С. Толщина хромированного слоя обычно не превышает 0,2 мм,
§ 69. Диффузионное удаление элементов
Удаление вредных примесей при нагреве в вакууме и других средах — важная для ряда изделий разновидность химико-термической обработки. В основе ее лежат диффузи
469
онный процесс перемещения атомов из сердцевины к поверхности изделия и удаление элемента с поверхности. Как правило, требуется сквозное удаление вредных примесей (по всему объему, а не только в поверхностных слоях). Примером является обезводороживание титановых сплавов при нагреве в вакууме для предотвращения водородной хрупкости и повышения ударной вязкости. Обезводороживание проводят при 670—700 °C в течение 2—6 ч и давлении не более 10-4 гПа.
Для комплексного удаления примесей внедрения из тугоплавких металлов их нагревают в вакууме.
В промышленности давно применяют сквозное обезуглероживание трансформаторной стали отжигом листов в водороде.
Иногда для изменения свойств поверхностного слоя используют несквозное удаление одного из основных компонентов сплава.
Оригинальной разновидностью химико-термической обработки является термовакуумная, обработка с сублимацией одного или нескольких легирующих элементов (С. 3. Бокштейн и М. Б. Бронфин). Например, выдержка при 500 °C, 9 ч в вакууме 10~3 Па образцов из сплава А1 — 4,5 % Zn — 2 % Mg приводит к сублимации из поверхностного слоя цинка и магния, благодаря чему значительно возрастает сопротивление коррозионному растрескиванию (высокое суммарное содержание этих элементов в объеме сплава обеспечивает необходимый уровень прочности).
Процессы химико-термической обработки благодаря неисчерпаемому разнообразию химически активных сред и богатым возможностям изменения свойств поверхностных слоев и всего объема изделий широко используют в промышленности. Они быстро развиваются, завоевывая новые области применения.
Марка с Si
08кп 0,05—0,11 <0,03
08 0,05—0,12 0,17—0,37
10 0,07—0,14 0,17—0,37
15 0,12—0,19 0,17—0.37
20 0,17—0,24 0,17—0,37
35Л 0,32—0,40 0,17—0,37
40 0,37—0,45 0,17—0,37
60 0,57—0,65 0,17—0,37
У8А 0,75—0,84 0,15—0,30
У9А 0,85—0,94 0,15—0,30
У10А 0,95—1,04 0,15—0,30
У11А 1,05—1,14 0,15—0,30
У12А 1,15—1,24 0,15—0,30
У13А 1,25-1,35 0,15—0,30
X 0,95-1,10 0,15—0,35
9ХС 0,85-0,95 1,2-1,6
ХВГ 0,90—1,05 0,15—0,35
Х12Ф1 1,25—1,45 0,15-0,35
Р6М5 0,80—0,88 <0,5
ШХ15 0,95—1,0 0,17—0,37
18ХГТ 0,17—0,23 0,17—0,37
12ХНЗА 0,09—0,16 0,17—0,37
38Х2МЮА 0,35-0,42 0,20—0,45
55ХГР 0,52—0,60 0,17—0,37
ЗОХГС 0,28—0,35 0,90—1,20
Мп | ° Ni Прочие
0,25—0,50 <0,10 <0,25 —
0,35—0,65 <0,10 <0,25 —
0,35-0,65 <0,15 <0,25 —
0,35—0,65 <0,25 <0,25 —
0,35—0,65 <0,25 <0,25
0,5—0,8 — —
0,50—0,80 <0,25 <0,25 —
0,50—0,80 <0,25 <0,25 —
0,15—0,30 — — —
0,15—0,30 — —
0,15—0,30 — — —
0,15—0,30 — —
0,15—0,30 —
0,15—0,30 — —— —
0,15—0,40 1,30—1,65 — —
0,30—0,60 0,95—1,25 —
0,80—1,10 0,90—1,20 — 1,2—1,6 W
0,15—0,40 11,00—12,50 0,7—0,9 V
<0,4 3,8—4,4 — 5,5-6,5 W 5,0—5,5 Мо 1,7—2,1 V
0,20—0,40 1,30—1.65 — .—
0,80—1,10 1,00—1,30 <0,30 0,03—0,09 Ti
0,30-0,60 0,60—0,90 2,75—3,15
0,30—0,60 1,35—1,65 <0,3 0,15—0,25 Мо 0,70—1,10 А1
0,9-1,2 0.90—1,20 <0,25 0,001-0,003 в
0,80—1,10 0.80—1,10 <0,30 —
Марка С Si
50ХФА 0,46—0,54 0,17—0,37
40ХНМА 0,37—0,44 0,17—0,37
18Х2Н4ВА 0,14—0,20 0,17—0,37
30Х10Г10 0,28-0,32 0,5—0,6
12Х18Н9 <0,12 <0,8
20X1ЗЛ 0,16—0,25 <0,8
36НХТЮ 0,05 <0,5
16Г2АФ 0,14—0,20 0,3—0,6
06ХГСЮ 0,05-0,08 0,3-0,5
06Г2С 0,05—0,08 0,4-0,6
Алюминиевые сплавы
Марка А1 Си Mg Z
АД1 <99,3 <0,05 <0,05 <0
АД <98,80 <0,1 <0,1 <0
АМц Основа <0,2 <0,05 <0
АМг1 » <0,01 0,5—1,8 —
АМг2 » <0,1 1,8-2,8 <0
АМгб » <0,1 5,8—6,8 <0
АДЗЗ » 0,15—0,40 0,8—1,2 <0
АД35 » <0,1 0,8—1,4 <0
Продолжение
Мп Сг Ni Прочие
0,5—0,8 0,8—1,1 — 0,1—0,2 V
0,50—0,80 0,60—0,90 1,25—1,65 0,15—0.25 Мо
0,25-0,55 1,35-1,65 4,00—4,40 0,80—1,20 W
9-11 9—11 — —
<2,0 17,0-19,0 8,0—10,0 —
<0,8 12,0—14,0 — —
0,8-1,2 11,5-13,5 34,5—36,5 2,8—3,2 Ti
1,3—1,7 — -— 0,08—0,14 V
0,015—0,025 N
1,1-1,4 0,4—0,7 — —
1,4—1,7 — — —
п Мп Si Fe Прочие
,1 <0,025 <0,30 <0,30
,1 <0,1 <0,50 <0,5 —
,1 1,0—1,6 <0,6 <0,7 —
- —— <0,05 <0,05 —
,2 0,2—0,6 <0,4 <0,4 —
,2 0,5—0,8 <0,4 <0,4 0,02—0,10 Ti 0,0001—0,005 Be
,25 <0,15 0,4—0,8 <0,7
,2 0,5—0,9 0,8-1,2 <0,5 —
Продолжение
Марка А1 Си Mg Zn Мп Si Fe Прочие
АВ Основа 0,1—0,5 0,45—0,90 <0,2 0,15—0,35 0,5-1,2 <0,5 —
Д1 » 3,8—4,8 0,4—0,8 <0,3 0,4—0,8 <0,7 <0,7 ——
Д16* » 3,8—4,9 1,2—1,8 <0,3 0,3—0,9 <0,5 <0,5 —
Д19 » 3,8—4,3 1,7—2,3 <0,1 0,5—1,0 <0,5 <0,5 0,0002—0,005 Be
Д19П » 3,2—3,7 2,1—2,6 <0,1 0,5—0,8 <0,3 <0,3 0,0003—0,005 Be
АК4-1 » 1,9—2,5 1,4—1,8 <0,3 <0,2 <0,35 0,8-1,3 0,8—1,3 Ni
1201 » 5,8-6,8 <0,02 <0,1 0,2—0,4 <0,2 <0,3 0,02-0,1 Ti 0,1—0,25 Zr 0,05—0,15 V
1915 » <0,1 1,3—1,8 3,4—4,0 0,2-0,6 <0,3 <0,4 0,15—0,22 Zr 0,08—0,2 Cr
В95* » 1,4—2,0 1,8—2,8 5,0—7,0 0,2—0,6 <0,5 <0,5 0,1—0,25 Cr
АЛ2 » <0,6 <0,1 <0,3 <0,5 10,0—13,0 <0,8 —
АЛ7 » 4,5—5,0 <0,3 <0,2 <0,1 <1,2 <1,0 ——
АЛ8 » <0,3 9,5—11,5 <0,1 <0,1 <0,3 <0,3 —
АЛ9 » <0,2 0,2-0,4 <0,3 <0,5 6,0—8,0 <0,6 —
* Сплавы Д16ч и В95пч отличаются от сплавов Д16 и В95 пониженным содержанием примесей» Д 16ч: <0,3Fe, <0,25'1,^ O.lZn: В95пч: <0,25Fe, <0,1 Si.
оо —-------- --------_ ... , ,, , , , , , ________________________ .
Магниевые сплавы
Марка Al Zn Mn
МЛ5 МЛ 12 7,5—9,0 0,2—0,8 4,0—5,0 0,15—0,5 0,6—1,1 Zr
Титановые сплавы
3 Марка Al Mo Cr Прочие
BT8 5,8—7,0 2,8—3,8 0,20—0,35 Si
BT15 2,3—3,5 6,5—7,5 9,5—11,5 .—
BT22 4,5—5,9 4,0—5,5 0,5—2,0 4,0—5,5 V 0,5—1,5 Fe
Никелевые сплавы
Марка Сг Ti Al Прочие
ХН77ТЮР 19—22 2,4—2,8 0,60—1,0 <0,01 в
(ЭИ437Б) Нимоник-90 18—21 1,8—3,0 0,8—2,0 15-21 Co
Медиые сплавы
Марка
Л 70
БрБ2
БрБНТ1, 9Мг
БрАЖ9-4
Содержание элементов
69,0—72,0 Си, ост. Zn 1,8—2,1 Be, 0,2—0,5 Ni 1,85—2,1 Be. 0.2—0,4 Ni 0,1—0,25 Ti, 0,07—0,13 Mg 8—10 Al, 2—4 Fe
РЕКОМЕНДАТЕЛЬНЫЙ БИБЛИОГРАФИЧЕСКИЙ СПИСОК
К введению
Д. К. Чернов и наука о металлах/Под ред. Н. Т. Гудцова. М.: Ме-таллургиздат, 1950. 563 с.
Бочвар А. А. Основы термической обработки сплавов. М.: Метал-лургиздат, 1940. 298 с.
К главе I
Новиков И. И., Золоторевский В. С. Дендритная ликвация в сплавах. М.: Наука, 1966. 155 с.
Голиков И. Н., Масленков С. Б. Дендритная ликвация в сталях и сплавах. М.: Металлургия, 1977. 223 с.
Золоторевский В. С. Структура и прочность литых алюминиевых сплавов. М.: Металлургия, 1981. 191 с.
К главе II
Горелик С. С. Рекристаллизация металлов и сплавов. Мл Металлургия, 1978. 565 с
Рекристаллизация металлических материалов/Под ред. Ф. Хессмера: Пер. с аигл. М.: Металлургия, 1982. 352 с.
Лариков И. Н. Залечивание дефектов в металлах. Киев: Наукова думка, 1980. 278 с.
Тофпенец Р. Л. Разупрочняющие процессы в стареющих сплавах. Минск: Наука и техника, 1979. 184 с.
Бородкина М. М., Спектор Э. Н. Рентгенографический анализ текстуры металлов и сплавов. М.: Металлургия, 1981. 271 с.
а К главе III
Кудрявцев П. И. — В кн.: Материалы в машиностроении. Т. 2. М.: Машиностроение, 1967, с. 210—227.
Коцюбинский О. Ю. Стабилизация размеров чугунных отливок. М.: Машиностроение, 1974. 296 с.
Хенкин М. Л., Локшин И. X. Размерная стабильность металлов и сплавов в точном машиностроении и приборостроении. М.: Машиностроение, 1974. 255 с.
Технология термической обработки стали. Разд. 1.3 и 3.2.3: Пер. с нем./Под ред. М. Л. Бернштейна. М.: Металлургия, 1981. 607 с.
Колачев Б. А., Габидулин Р. М.л Пигузов Ю. В. Технология термической обработки цветных металлов и сплавов. Гл. 1. М.: Металлургия, 1980. 279 с.
К главе IV
Физическое металловедение/Под ред. Р. Кана. Вып. II., гл. V: Пер. с англ. М.: Мир. 1968. 490 с.
Баранов А. А. Фазовые превращения и термоциклирование металлов. Киев: Наукова думка, 1974. 230 с.
Попов А, А., Попова Л. Е. Изотермические и термокинетические диаграммы распада переохлажденного аустенита: Справочник термиста. М.: Металлургия, 1965. 495 с.
К главе V
Курдюмов Г. В., Утевский Л. М.л Энтин Р. И. Превращения в железе и стали. М.: Наука, 1977. 236 с.
475
Гуляев А. П. Термическая обработка стали. Гл. II., III и VIII. М.: Машгиз, 1960. 496 с.
Садовский В. Д. Структурная наследственность в стали. Мл Металлургия, 1973. 208 с.
Дьяченко С, С. Образование аустенита в железоуглеродистых сплавах. Мл Металлургия, 1982, 128 с.
Долженков И. Е., Долженков И, И. Сфероидизация карбидов в стали. М.; Металлургия, 1984. 142 с.
Гольдштейн М. И., Грачев С. В., Векслер Ю. Г. Специальные стали, Гл. VII и VIII. Мл Металлургия, 1985. 408 с.
Металловедение и термическая обработка стали: Справочник, т.
II, разд. 19, 23, 24, 25.4 и 25.5. Мл Металлургия, 1983. 367 с.
К главе VI
Бунин К. П., Таран Ю. Н. Строение чугуна. Гл. IV. Мл Металлургия, 1972. 160 с.
Материалы в машиностроении. Т. 4: Чугун. Гл. 2 и 4. М.: Машиностроение, 1969. 248 с.
К главе VII
Бочвар А. Л. Основы термической обработки сплавов. Гл. IV. Мл Металлургиздат, 1940. 298 с.
Колачев Б. А., Ливанов В. А., Елагин В. И. Металловедение и термическая обработка цветных металлов и сплавов. Мл Металлургия, 1981. 414 с,
К главе VIII
Колачев Б. А., Ливанов В. А., Елагин В. И. Металловедение и термическая обработка цветных металлов и сплавов. М.: Металлургия, 1981. 414 с.
Диаграммы изотермическою распада раствора в алюминиевых сплава х/В. Г. Давыдов, В. В. Захаров, Е. Д. Захаров, И. И. Новиков, Мл Металлургия, 1973. 152 с.
Металловедение алюминия и его сплавов: Справочник. Гл. 7. Мл Металлургия, 1983. 279 с.
К главе IX
Курдюмов Г. В. Утевский Л. M.t Энтин Р, И. Превращения в железе и стали. /М.: Наука, 1977. 238 с.
Гуляев А. П. Термическая обработка стали. Гл. IV, VII и IX. Мл Машгиз, 1960. 496 с.
Попов А. А., Попова Л. Е. Изотермические и термокинетические диаграммы распада переохлажденного аустенита: Справочник термиста. Мл Металлургия, 1965. 495 с.
Кидин И, И. Физические основы электротермической обработки металлов и сплавов. Мл Металлургия, 1969. 375 с.
Шепеляковский К. 3. Упрочнение деталей машин поверхностной закалкой при индукционом нагреве. Мл Машиностроение, 1972. 287 с.
Петров Ю. Н. Дефекты и без диффузионное превращение в стали. Киев: Наукова думка, 1978. 262 с.
Винтайкин Е. 3. — В кн.: Итоги науки и техники. Мл ВИНИТИ, 1983, с. 3-63.
Варлимокт X., Дилей Л. Мартенситные превращения в сплавах на основе меди, серебра и золота: Пер. с англ. Мл Металлургия, 1980. 205 с.
Эффект памяти формы в сплавах: Пер. с англ./Под ред. В. А. Зай-мовского. Мл Металлуртя, 1979. 468 с.
476
Металловедение и термическая обработка стали: Справочник. Т. II гл. 25. М.: Металлургия, 1983. 367 с.
Термическая обработка в машиностроении: Справочник. Гл. 7, 9 и 10. М.: Машиностроение, 1980. 783 с.
К главе X
Мирошниченко И, С. Закалка из жидкого состояния. М.: Металлургия, 1982. 168 с.
С каков Ю. А., Крапошин В. С. — В кн.: Итоги науки и техники. Металловедение и термическая обработка. Т. 13. М.: ВИНИТИ, 1980, с. 3—78.
К главе XI
Хачатурян А. Г. Теория фазовых превращений и структура твердых растворов. М.: Наука, 1974. 384 с.
Келли А., Никлсон Р. Дисперсионное твердение: Пер. с англ. М.: Металлургия, 1966. 300 с.
Чуистов К. В. Модулированные структуры в стареющих сплавах, Киев: Наукова думка, 1975. 229 с.
Лариков Л. Н., Шматко О. А. Ячеистый распад пересыщенных твердых растворов. Киев: Наукова думка, 1976. 223 с.
Металловедение алюминия и его сплавов: Справочник. Гл. 6 и 7. М.: Металлургия, 1983. 279 с.
Мартин Дж. У. Микромеханнзмы дисперсионного твердения сплавов: Пер. с англ. М.: Металлургия, 1983. 166 с.
Суховаров В. Ф. Прерывистое выделение фаз в сплавах. Новосибирск: Наука, 1983. 167 с.
Пастухова Ж- П., Рахштадт А. Г., Каплун Ю. А. Динамическое старение сплавов. М.: Металлургия, 1985. 221 с.
К главе XII
Курдюмов Г. В., Утевский Л. М., Энтин Р. И. Превращения в железе и стали. М.: Наука, 1977. 238 с.
Белоус М. В., Черепин В. Т., Васильев М. А. Превращения при отпуске стали. М.: Металлургия, 1973. 231 с.
Гольдштейн М. И„ Фарбер В. М. Дисперсионное упрочнение стали. М.: Металлургия, 1979. 208 с.
Металловедение и термическая обработка стали: Справочник. Т. II, разд. 25.7. М.: Металлургия, 1983. 367 с.
К главе XIII
Бернштейн М. Л., Займовский В. Л., Капут кина Л, М. Термомеханическая обработка стали. Гл. I, II. М.: Металлургия, 1983. 480 с.
Металловедение и термическая обработка стали: Справочник. Т. II, разд. 27, 28.3. М.: Металлургия, 1983. 367 с.
Структура и свойства полуфабрикатов из алюминиевых сплавов: Справочник. Гл. П. М.: Металлургия, 1984. 408 с.
К главе XIV
Рабинович М. X. Термомехаиическая обработка алюминиевых сплавов. М.: Машиностроение, 1972. 160 с.
Елагин В. И., Захаров В. В., Дриц А. М. Структура и свойства сплавов системы Al—Zn—Mg. Гл. VIII. М.: Металлургия, 1982. 222 с.
К главе XV
Бернштейн М. Л. Термомеханическая обработка металлов и сплавов. М.: Металлургия, 1968. 1171 с.
477
Бернштейн М. Л.» Займовский В. А., Капуткина Л. М. Термомеханическая обработка стали. М.: Металлургия, 1983. 480 с.
Контролируемая прокатка / Погоржельский В. И., Литвиненко Д. А., Матросов Ю, И., Иваницкий А. В. М.: Металлургия, 1979. 184 с.
Металловедение и термическая обработка стали: Справочник. Т. III, разделы 34.6.3 и 37. М.: Металлургия, 1983. 215 с.
К главе XVI
Лахтин Ю. М., Арзамасов Б. И. Химико-термическая обработка металлов. Гл. I. М.: Металлургия, 1985. 256 с.
Попов А. А. Теоретические основы химико-термической обработки стали. Свердловск: Металлургиздат, 1962. 120 с.
К главе XVII
Лахтин Ю. М., Арзамасов Б. Н. Химико-термическая обработка металлов. Мл Металлургия, 1985. 256 с.
Минкевич А. Н. Химико-термическая обработка металлов и сплавов. М.: Металлургия, 1965. 491 с.
Лахтин Ю. М., Коган Я- Д- Азотирование стали. М.: Машиностроение, 1976. 254 с.
Дубинин Г, Н. Диффузионное хромирование сплавов. М.: Машиностроение, 1964. 451 с.
Арзамасов Б. Н. Химико-термическая обработка металлов в активизированных средах. М.: Металлургия, 1979. 224 с.
Химико-термическая обработка металлов и сплавов: Справочник. М.: Металлургия, 1981. 424 с.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
А
Азотирование 466
Алитирование 469
Аустенит остаточный 227
Аустенитизация 155
Аусформинг 440
Б
Бейна деформация 242
Бейнит верхний 280
— нижний 280
Бочвара А. А. правило 63
Р-термообработка урана 210
В
Возврат:
второго рода 46
динамический 419
первого рода 46
после старения 384
статический 419
Выделения:
когерентные 324
межфазные 180
некогерентиые 324
частично когерентные 324
Г
Гетерогенизация при отжиге 26. 204
Гинье — Престона зона 340, 341
Гистерезис тепловой 124
Глубина прокаливаемости 222, 291
Гомогенизация высокотемпературная 33
Графитизация 196, 198
Д
Данкова — Конобеевского принцип 135
Деформация:
аккомодационная 245
критическая 84
Джомини метод 292
Диаграмма:
изотермического превращения 151
рекристаллизации 89
термокииетическая 154
Диаметр критический 223, 291
3
Закаливаемость 276
Закалка:
без полиморфного превращения 213
бейнитная 298
в двух средах 296
высокочастотная 303
из жидкого состояния 307
изотермическая 298
и а мартенсит 225
неполная 293
объемная 293
с индукционным нагревом 302
— лазерным нагревом 304
— нагревом пламенем горелки 305
— плавлением поверхности 306
— полиморфным превращением 225
— самоотпуском 400
478
ступенчатая 297
через воду в масло 296 Зарождение:
гетерогенное 136 гомогенное 135 Зерно: восстановленное 166 действительное 165 наследственное 165 начальное 162 ультра мелкое 108 эвтектоидное 173
И
Изоформинг 444
Интервал мартенситный 227 — отжига емости 187
К
Карбюризатор 463 Квазиэвтектоид 174 Кластер 341 Коагуляция выделений 356 Коалесценция субзерен 53 Колония перлита 170 — прерывистого распада 337 «Корка апельсиновая» 107 Красностойкость 401 Курдюмова — Закса соотношение 244
М
Мартецсит: бесструктурный 251 взрывной 262 высокотемпературный 252 гексагональный 249 двойникованный 249 деформации 268 игольчатый 249 массивный 252 напряжения 268 медвойникованный 252 низкотемпературный 249 отпущенный 394 охлаждения 268 пакетный 252 пластинчатый 249 реечный 252
Мартенситное превращение: атермическое 257 взрывное 258 изотермическое 259
Метод пробной закалки 291 — торцовой закалки 292 Мидриб 253
Н
Наклеп: горячий 419 при обработке давлением 42 фазовый 167
Напряжения: зональные НО структурные 294 фазовые 112
Напряжения остаточные 112 Наследственность структурная 166 Нитроцементация 468 Нормализация 190
О
Облагораживание 383
Обработка термовакуумная 470
— термомеханическая (ТМО) 416
— —высокотемпературная (ВТМО) 436, 441
---низкотемпературная (НТМО) 431, 440
---предварительная (ПТМО) 438» 446
— химико-термическая 447
Отдых 46
Отжиг:
l-ro рода 15
2-го рода 122
гетерогенизационный 204
гомогенизационный 15
градиентный 109
графитизирующий 194
диффузионный 18
для уменьшения напряжений 110
— устранения отбела 201
дорекристаллизационный 34, 105
зонный 109
изотермический 189
маятниковый 188
' на крупное зерно 184
неполный 184
нормализационный 182
полный 183
рекристаллизационный 34, 106, 108
с фазовой перекристаллизацией 210
сфероидизирующий 185
текстурирующий 109
циклический 188
Отпуск:
высокий 399
низкий 398
под напряжением 402
средний 399
стабилизирующий 399
упрочняющий 398
П
Патентирование 192
Перегрев:
при отжиге 96
латуни 208
стали 164
Пережог 96, 217
Перестаривание 363, 377
Перлит:
глобулярный 187
пластинчатый 173
Полигонизация 46, 48
— динамическая 419
— предрекристаллизационная 61
— стабилизирующая 61
Полоса:
деформационная 38
переходная 39
прокаливаемости 292
сброса 39
сдвига 40
скольжения 41
Потенциал углеродный 464
Правило ступеней 145
Превращение:
бейнитное 279
бездиффузионное 231
безызбирательное 231
массивное 238
нормальное 236
перлитное 169
промежуточное 279
сдвиговое 236
эвтектоидное абнормальное 176, 199
Предвыделение 344
Пресс-эффект 439
Прокатка контролируемая 445
479
Принцип структурного соответствия 135
Прокаливаемость 287
Р
Равновесие термоупругое 238
Распад:
двухфазный 391
локализованный 333
непрерывный 332
общий 333
прерывистый 337
равномерный 333
спинодальный 319
Рекриста л л иза ция: вторичная 77 динамическая 419 in situ 54 метадинамическая 425 на месте 54 обработки 55 первичная 55 собирательная 68
Реполигоинзация 420
Рост зерен: неравномерный 78 нормальный 77
С
Самоотпуск 277, 400
С-диаграмма 152
С-кривая 151
Скорость:
зарождения центров 56, 124
охлаждения критическая 222, 287
роста линейная 56, 124
Сорбит:
отпуска 394
патентирования 192
Спи иода ль: когерентная 321 химическая 321
Стабилизация аустенита: механическая 273 термическая 264
Старение:
без нагрева под закалку 377
двойное 378
двухступенчатое 378 динамическое 383 естественное 374 зонное 375 искусственное 374 коагуляционное 375 под напряжением 383 разупрочияющее 363 стабилизирующее 377 ступенчатое 378
упрочняющее 363
фазовое 375
Степень перегрева 131
— переохлаждения 124
Структура:
видманштеттова 135
дуальная 300
модулированная 327
периодическая 327
разнозернистая 90 ячеистая 36
Т
Таммана кривая 124
Твердение вторичное 401
— дисперсионное 383
Текстура:
деформации 34
рекристаллизации 74
Температура:
гомологическая 45
конца рекристаллизации 66
начала рекристаллизации 62
Термоудар 121
Термоцнклирование 120
Томсона — Фрейндлиха уравнение 185
Торможение текстурное 73
Точка мартенситная верхняя 226 — — нижняя 226 — спинодальная 320
Троостит 173
— отпуска 394
У
Углерод отжига 196
Улучшение стали 400
Упрочнение структурное 439
Ферритизация 199 /
Фестонистость 101
X
Хрупкость огневая 106
Хрупкость отпускная необратимая 403
---обратимая 405
Ц
Цементация 463
Цианирование 468
Ч
Чернова Д. К. точка 168
Э
Эффект запоминания формы (ЭЗФ) 269, 270 — толщины 73