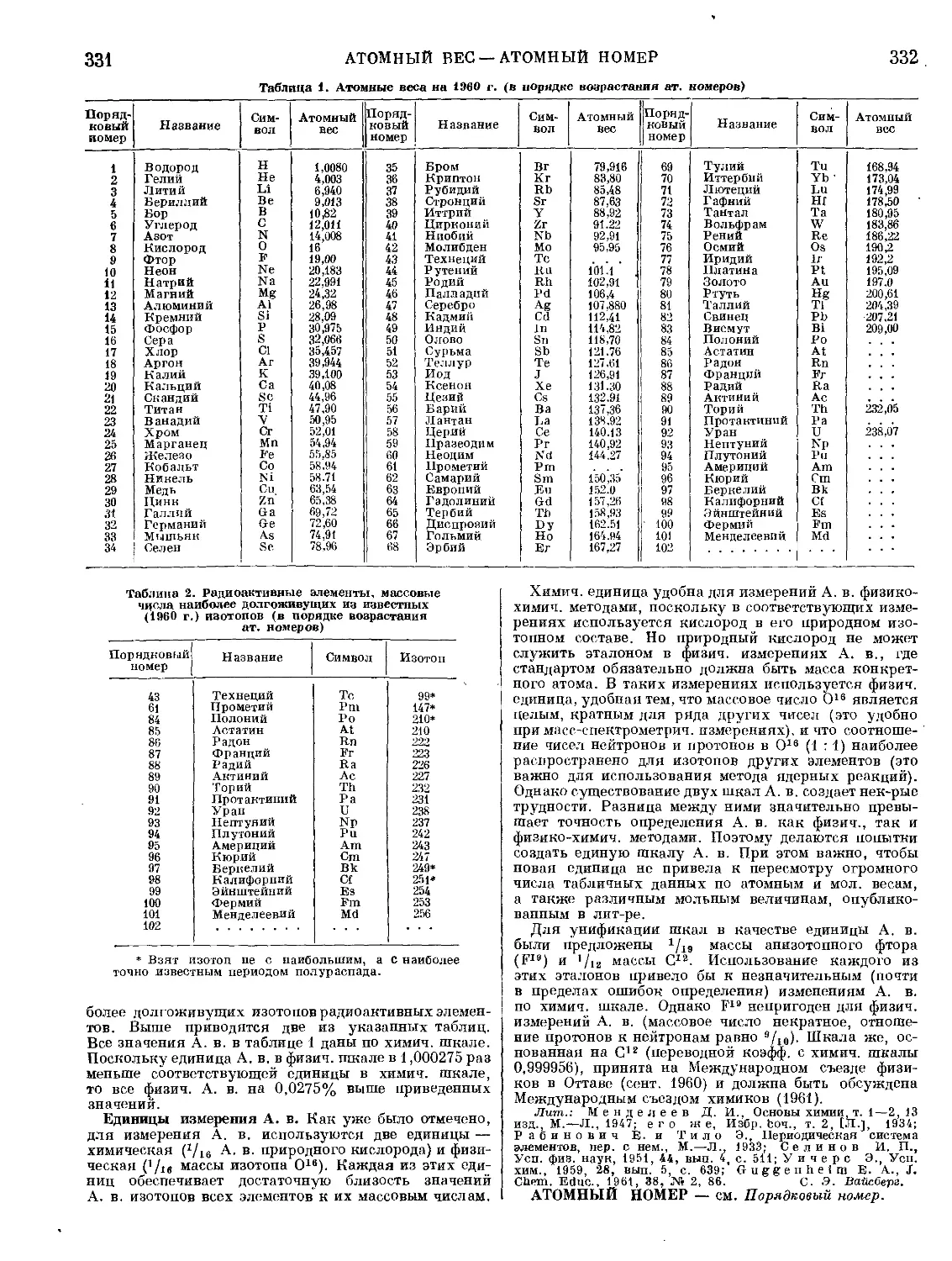
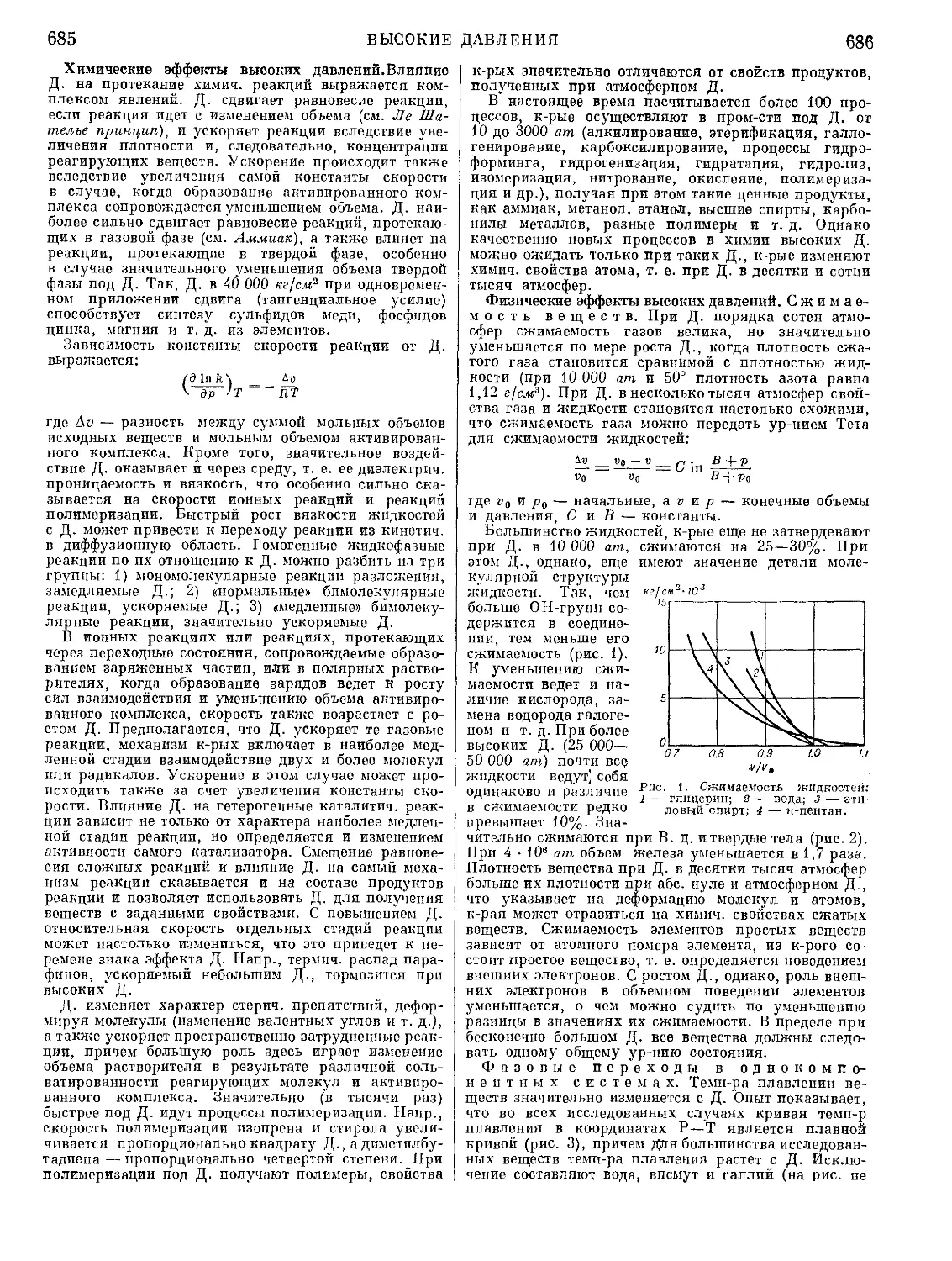
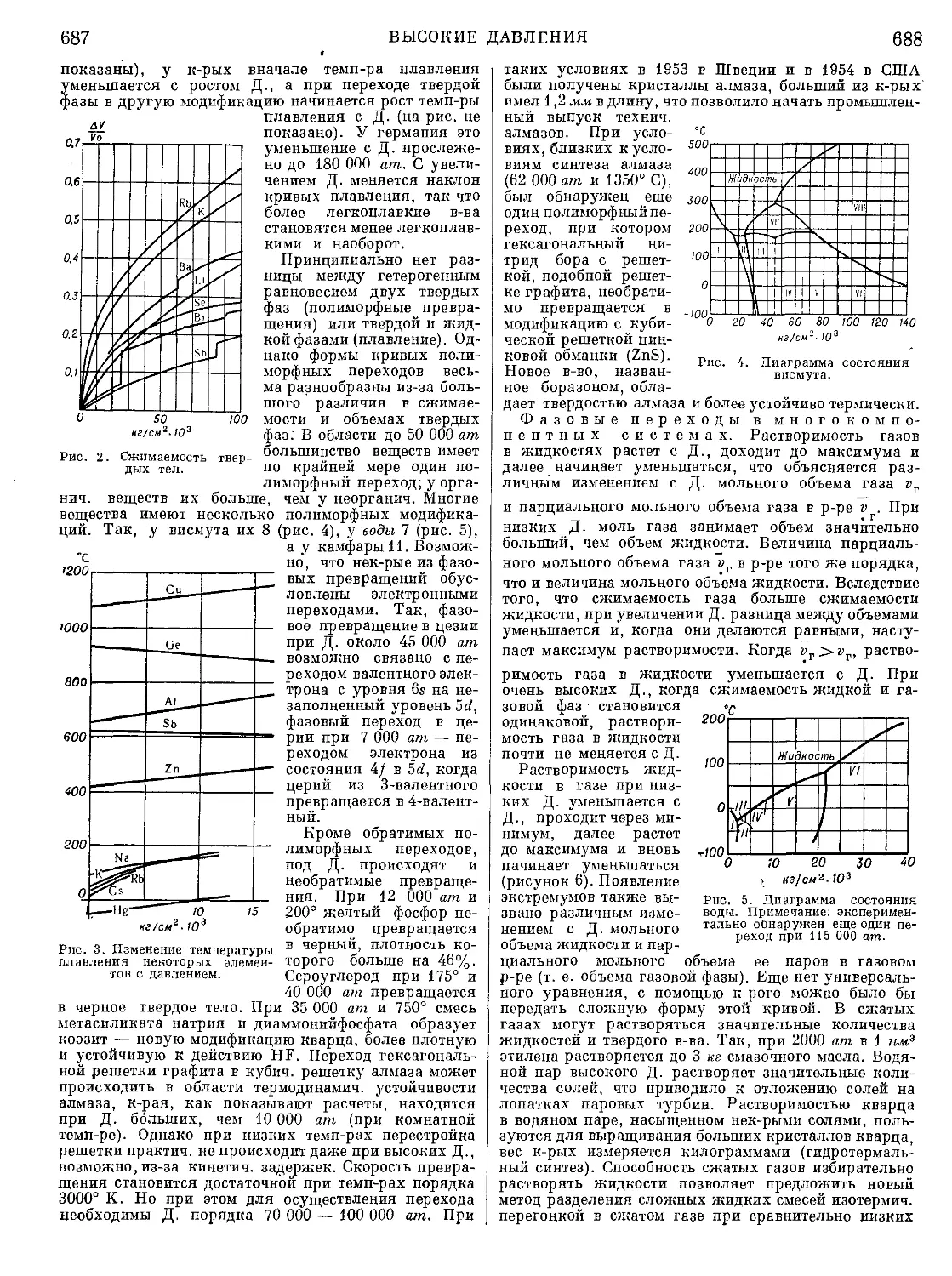
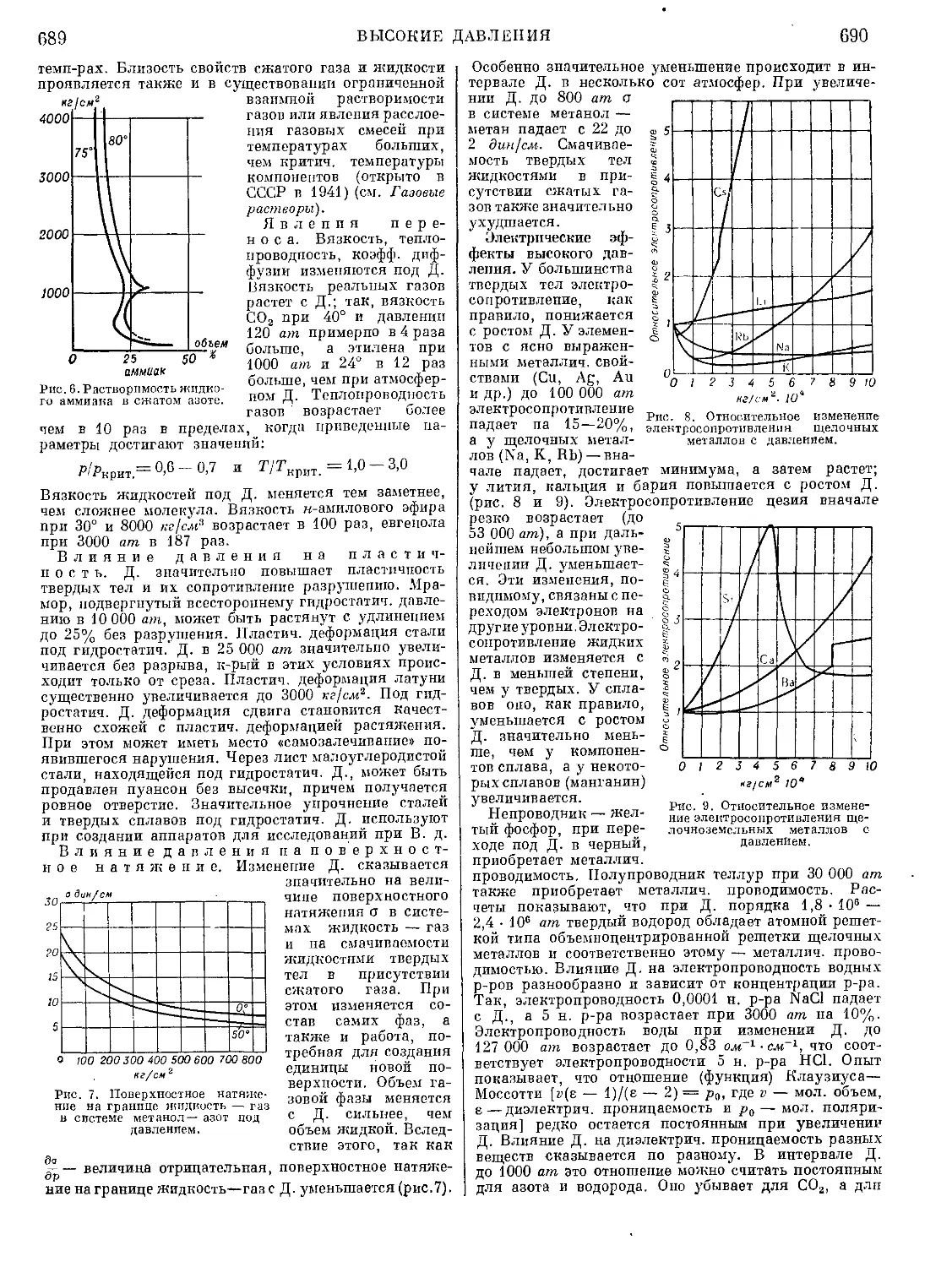
/























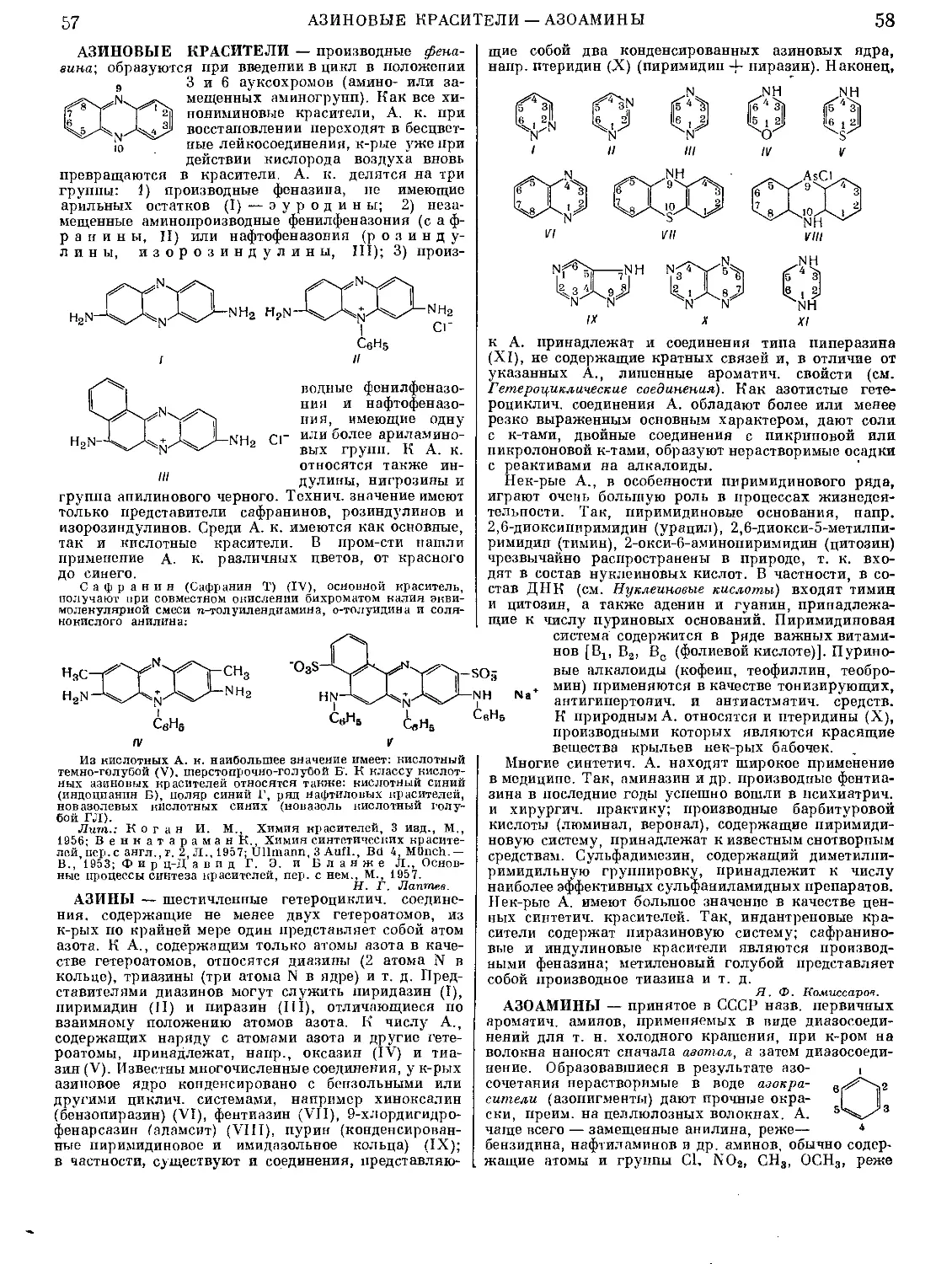

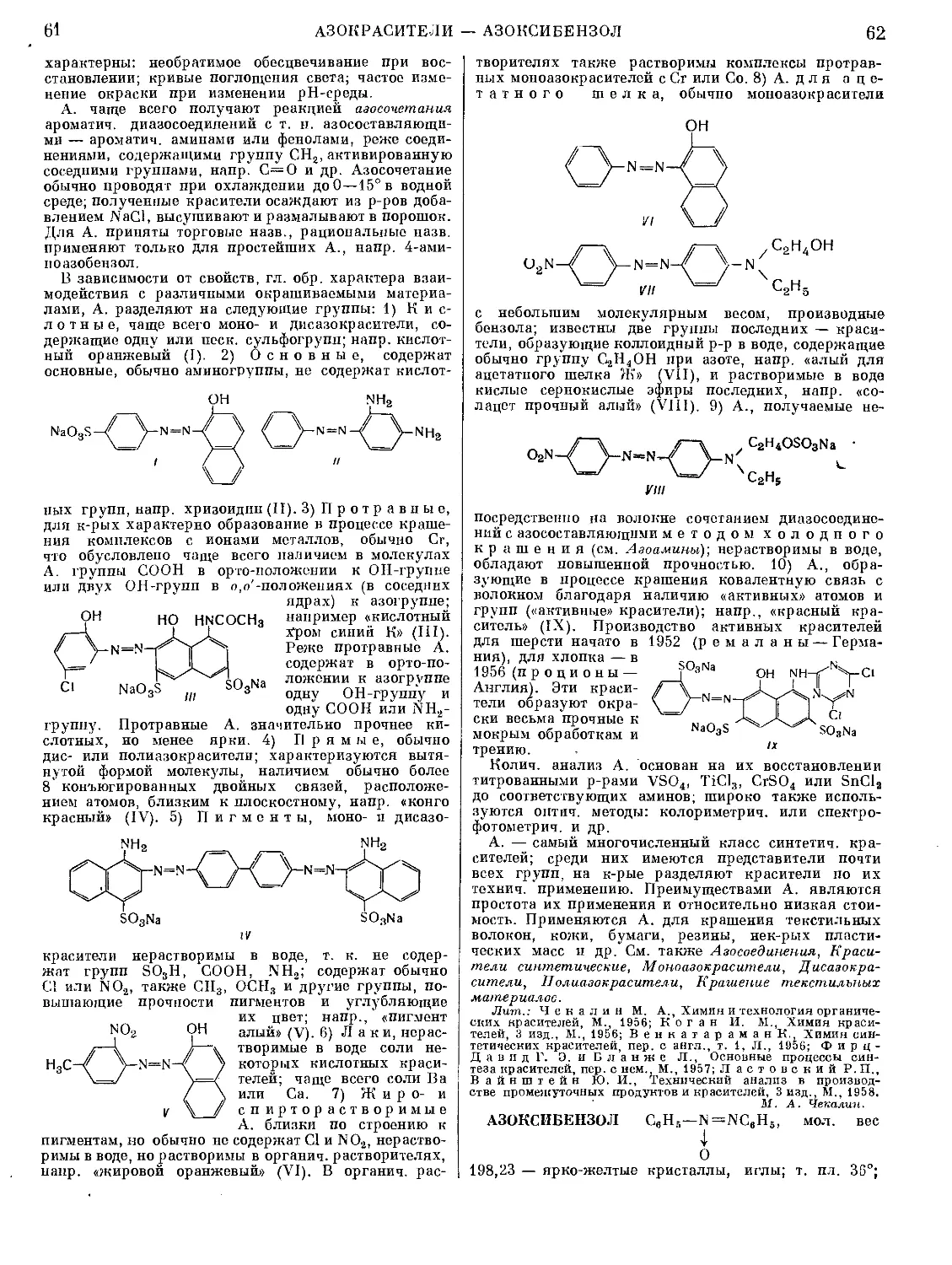









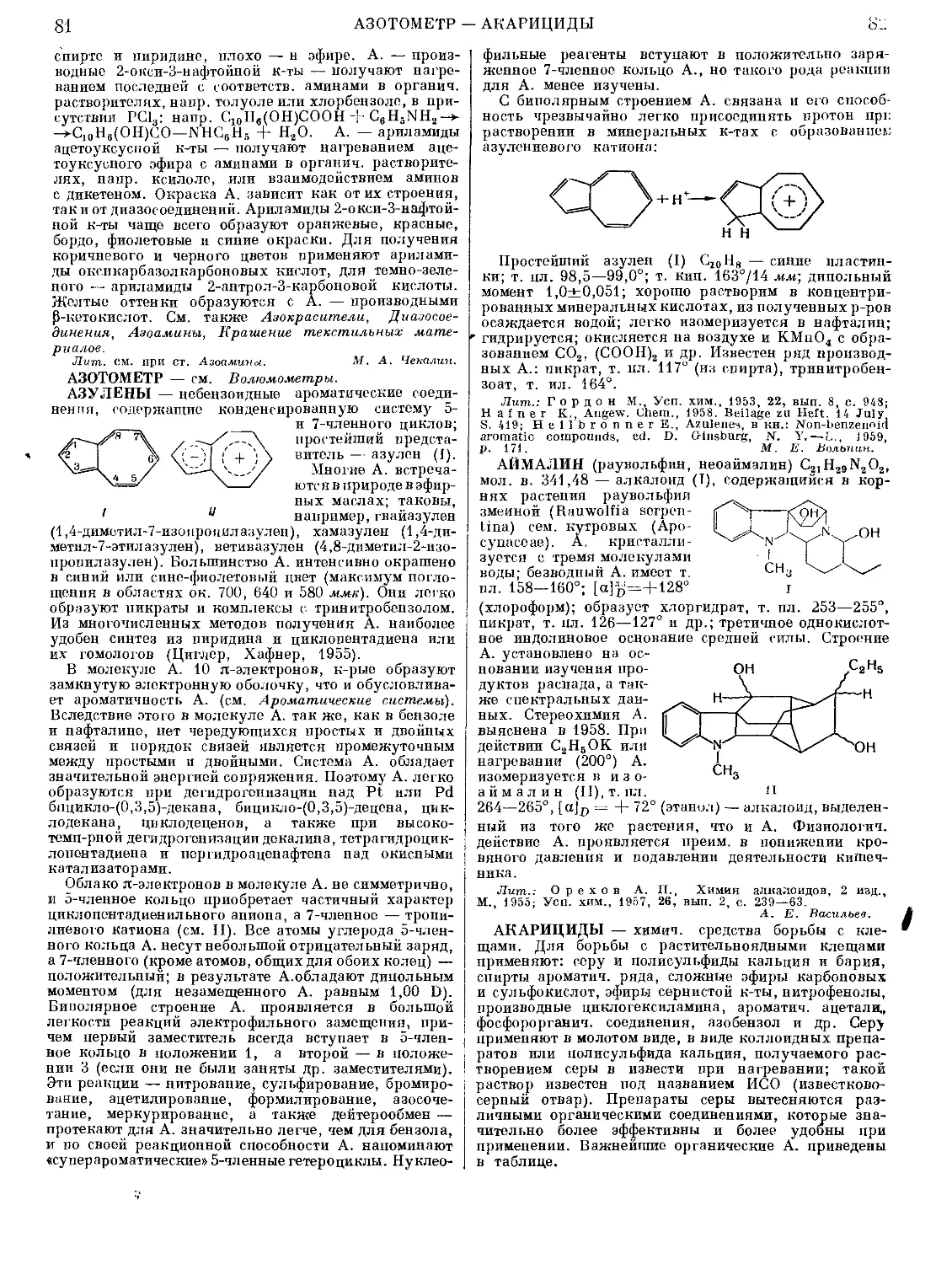
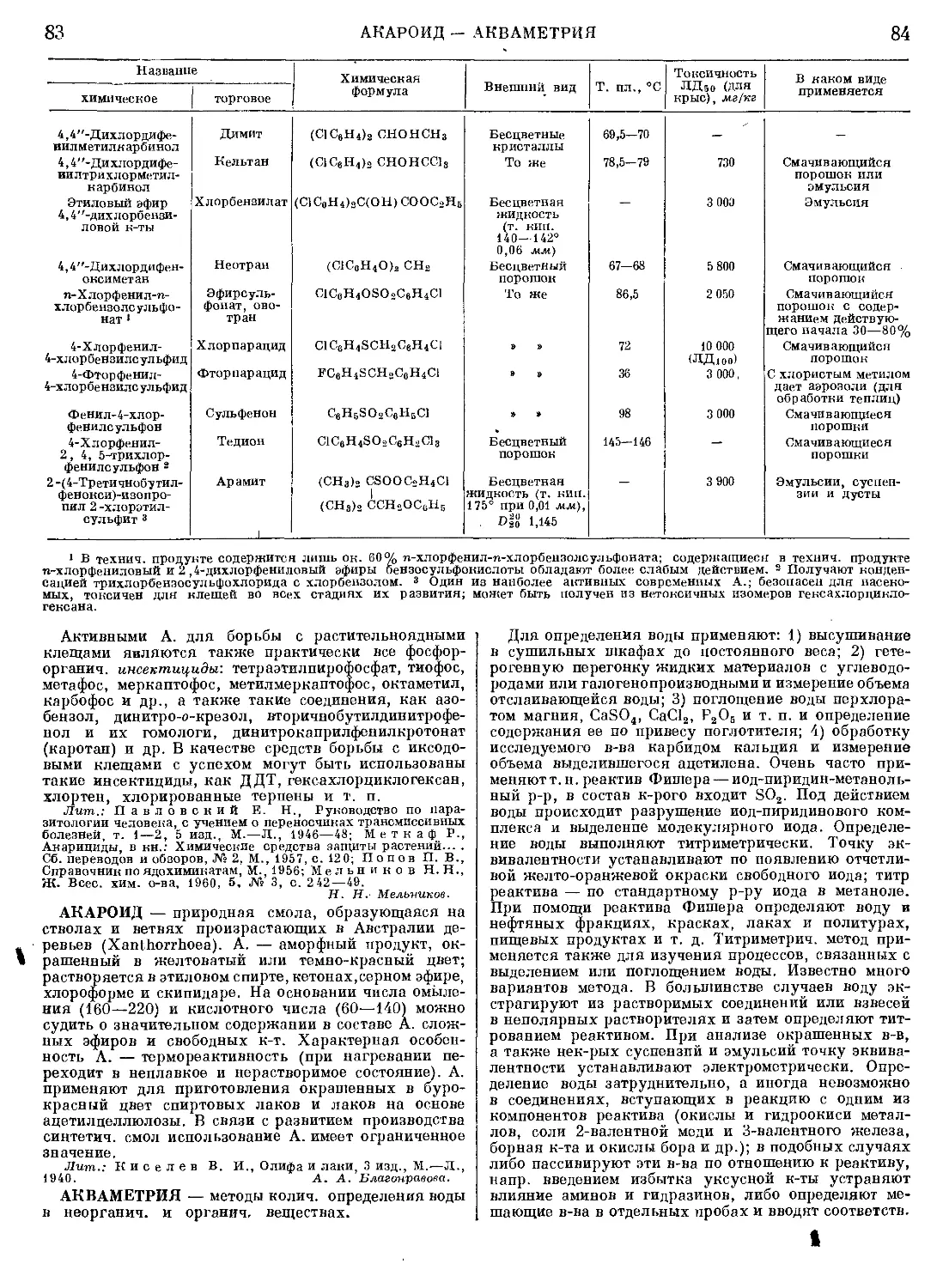








































































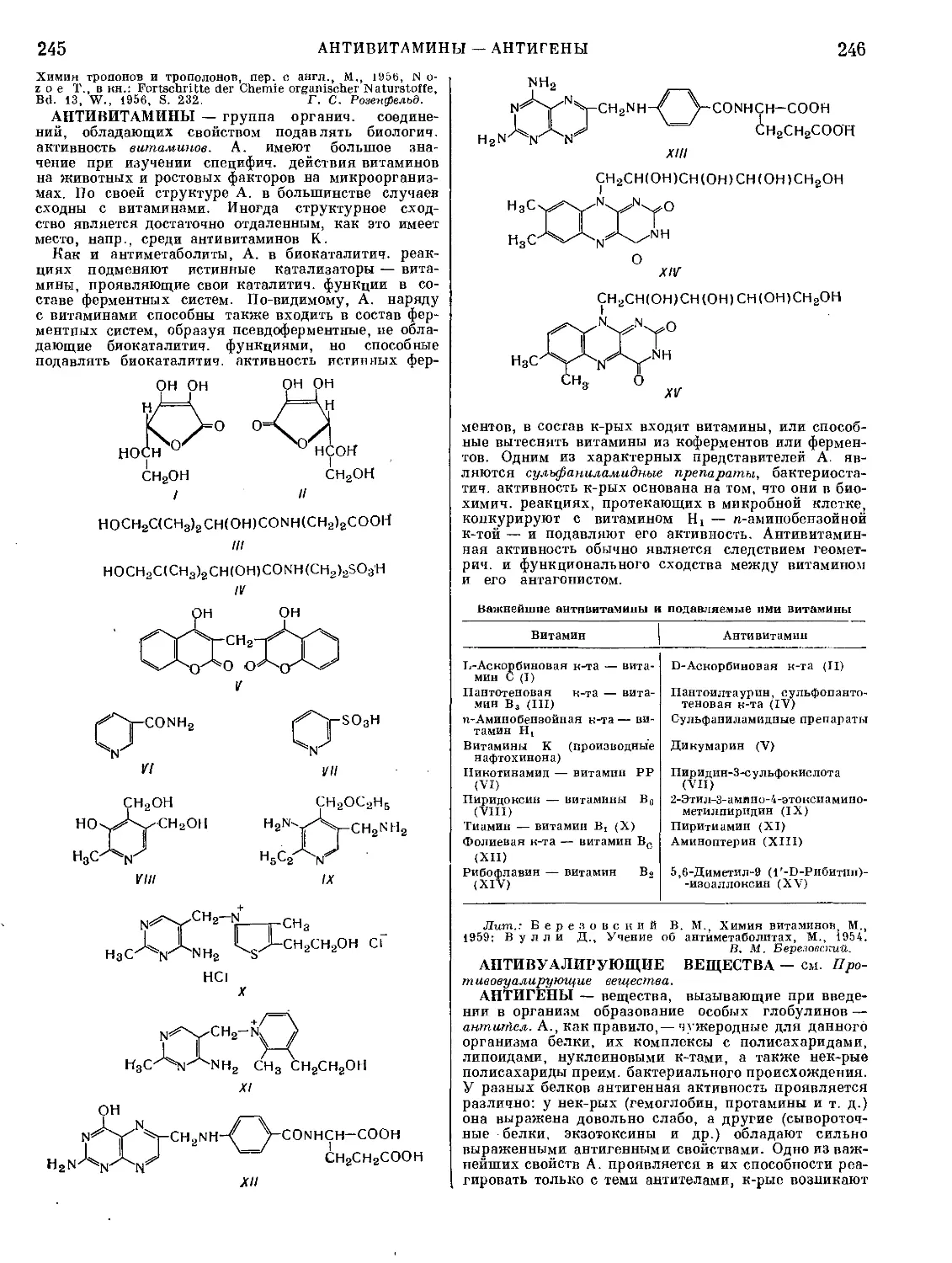






























































































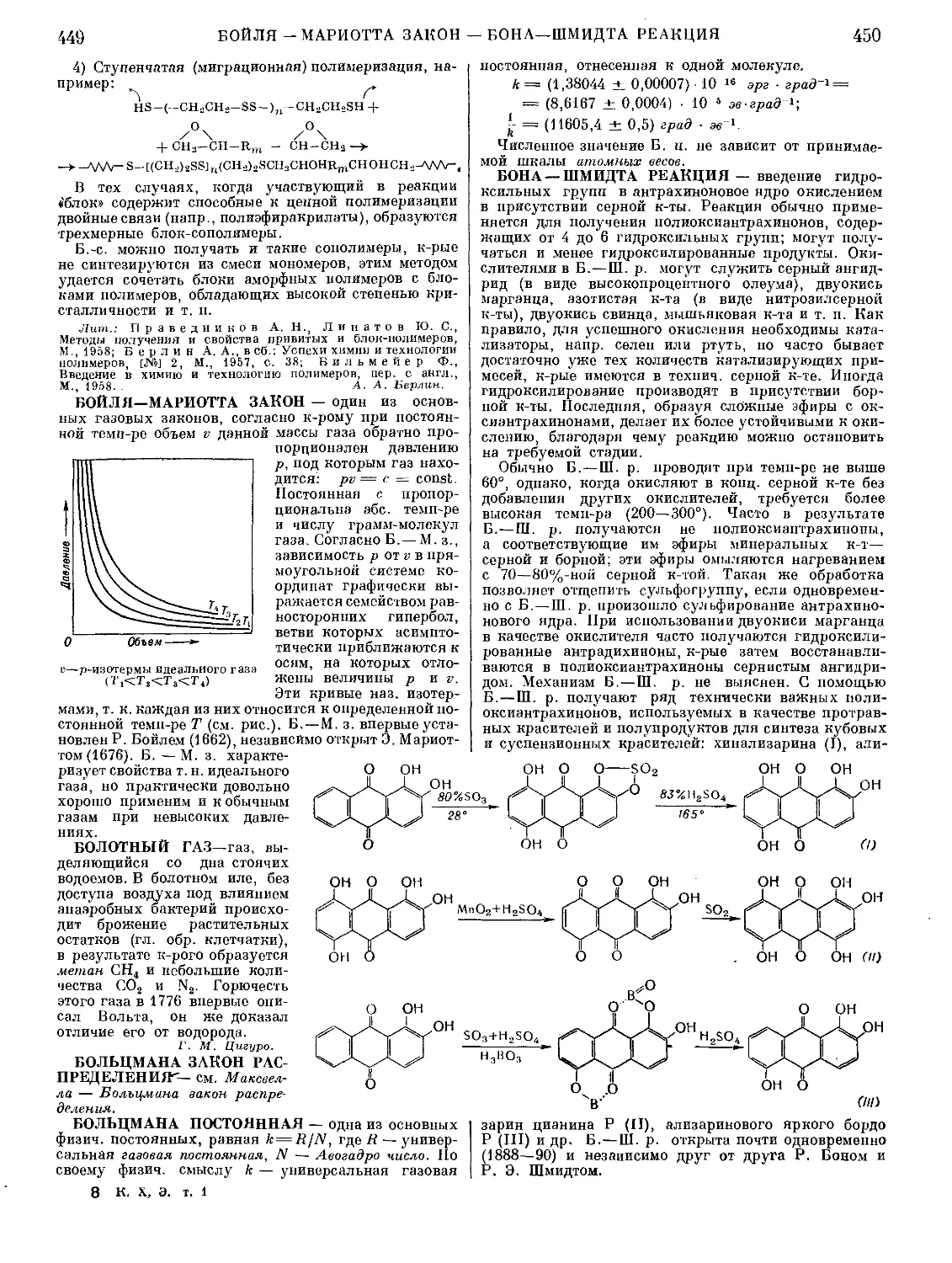





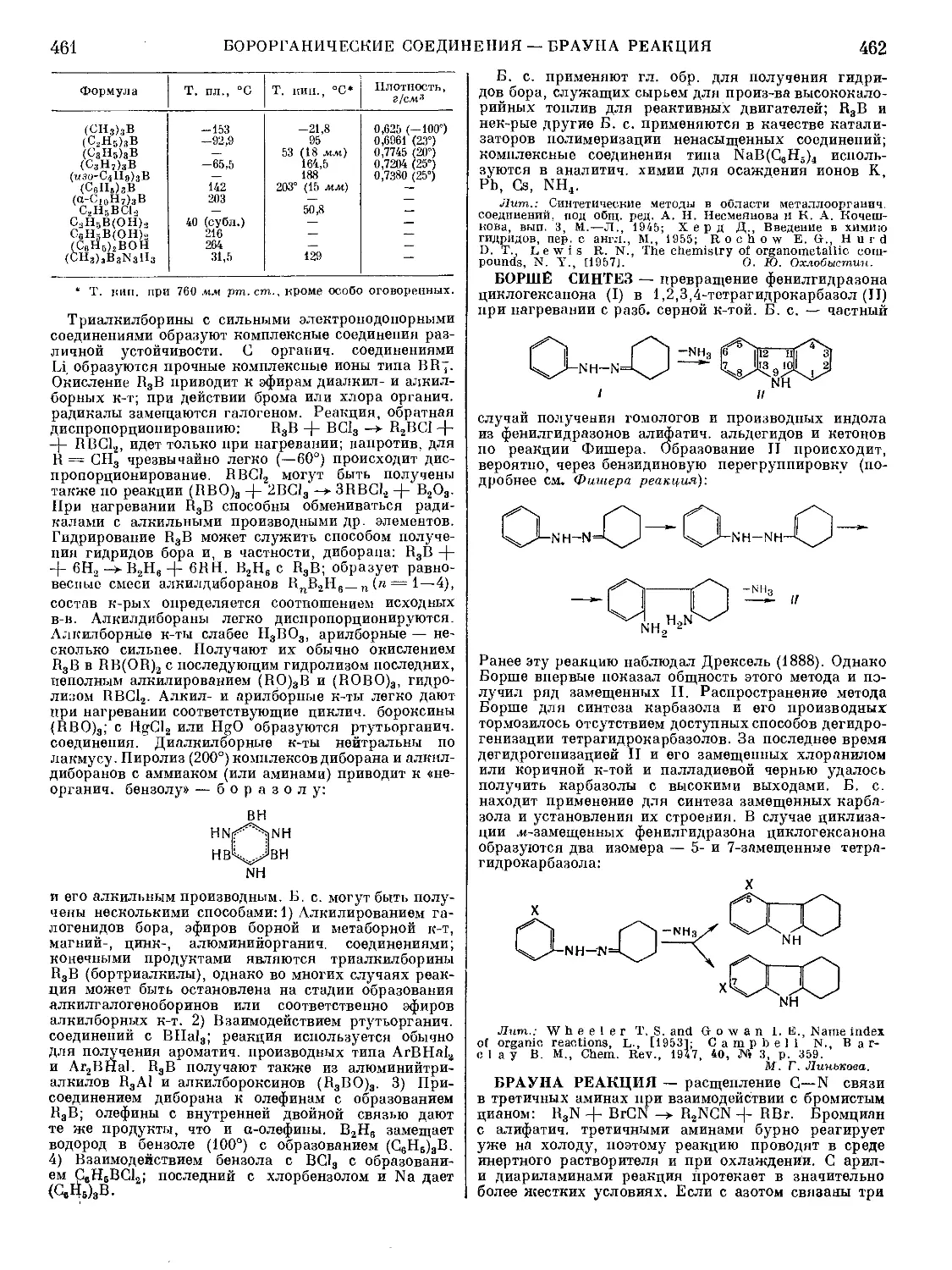



















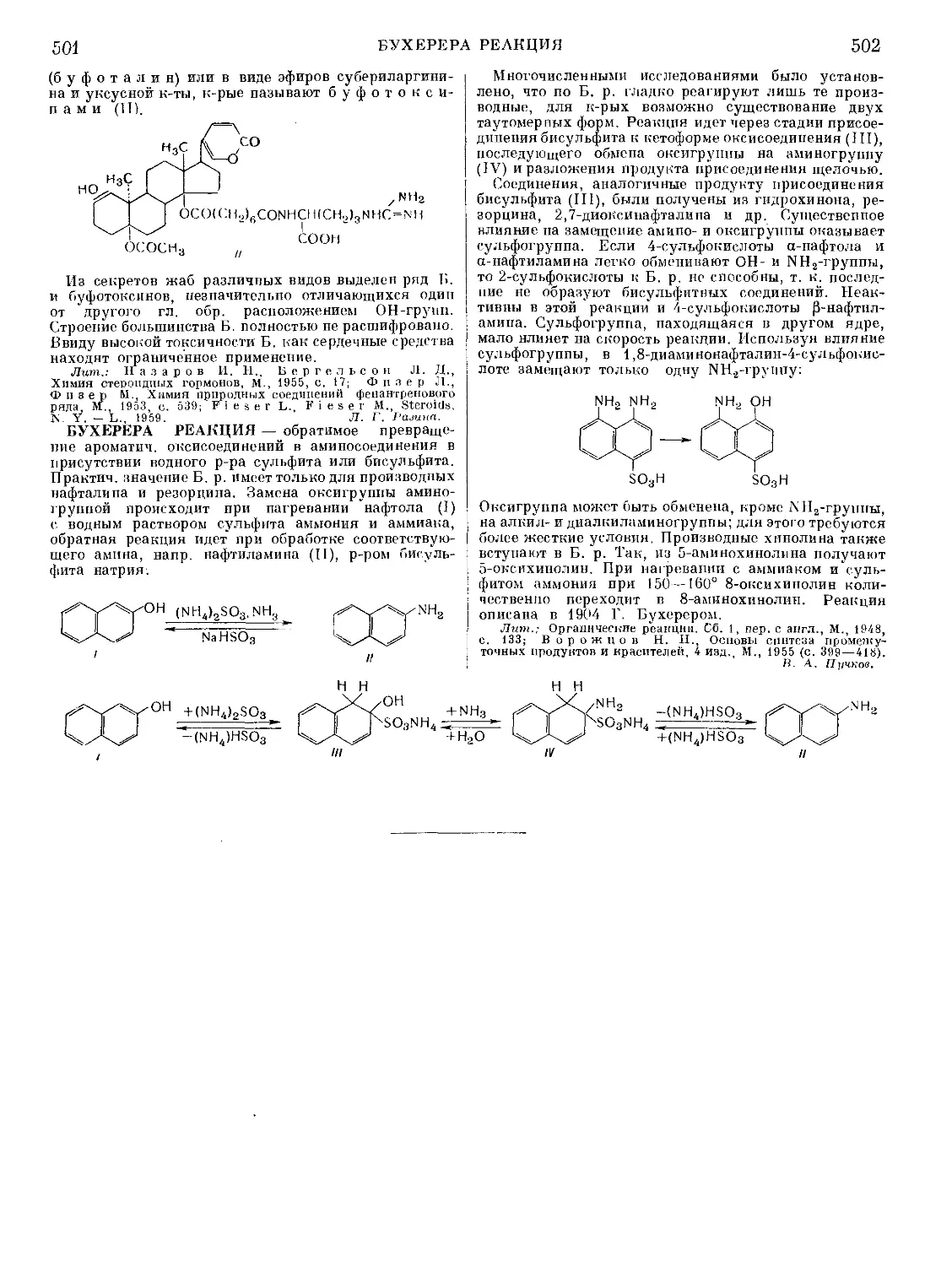









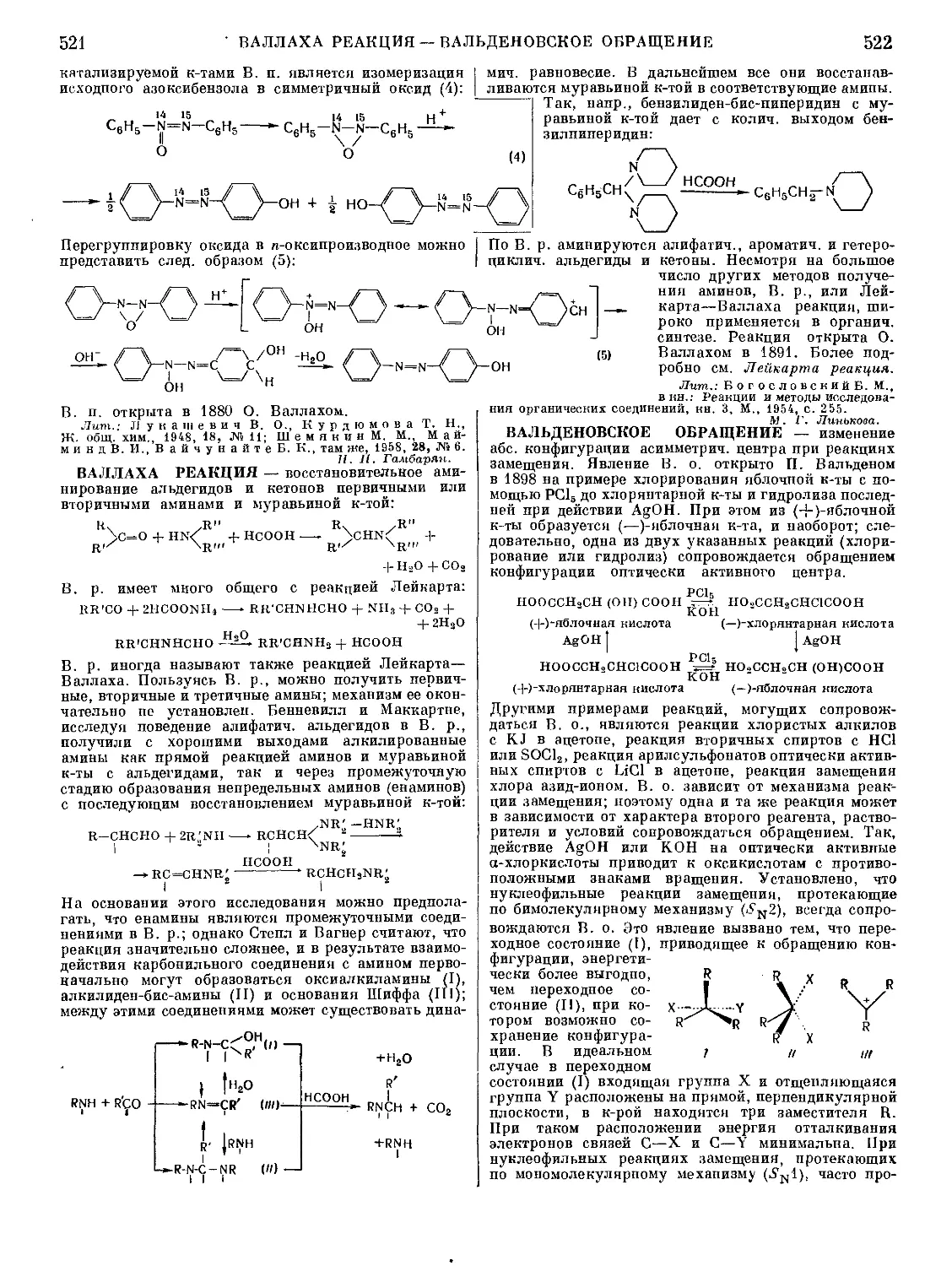




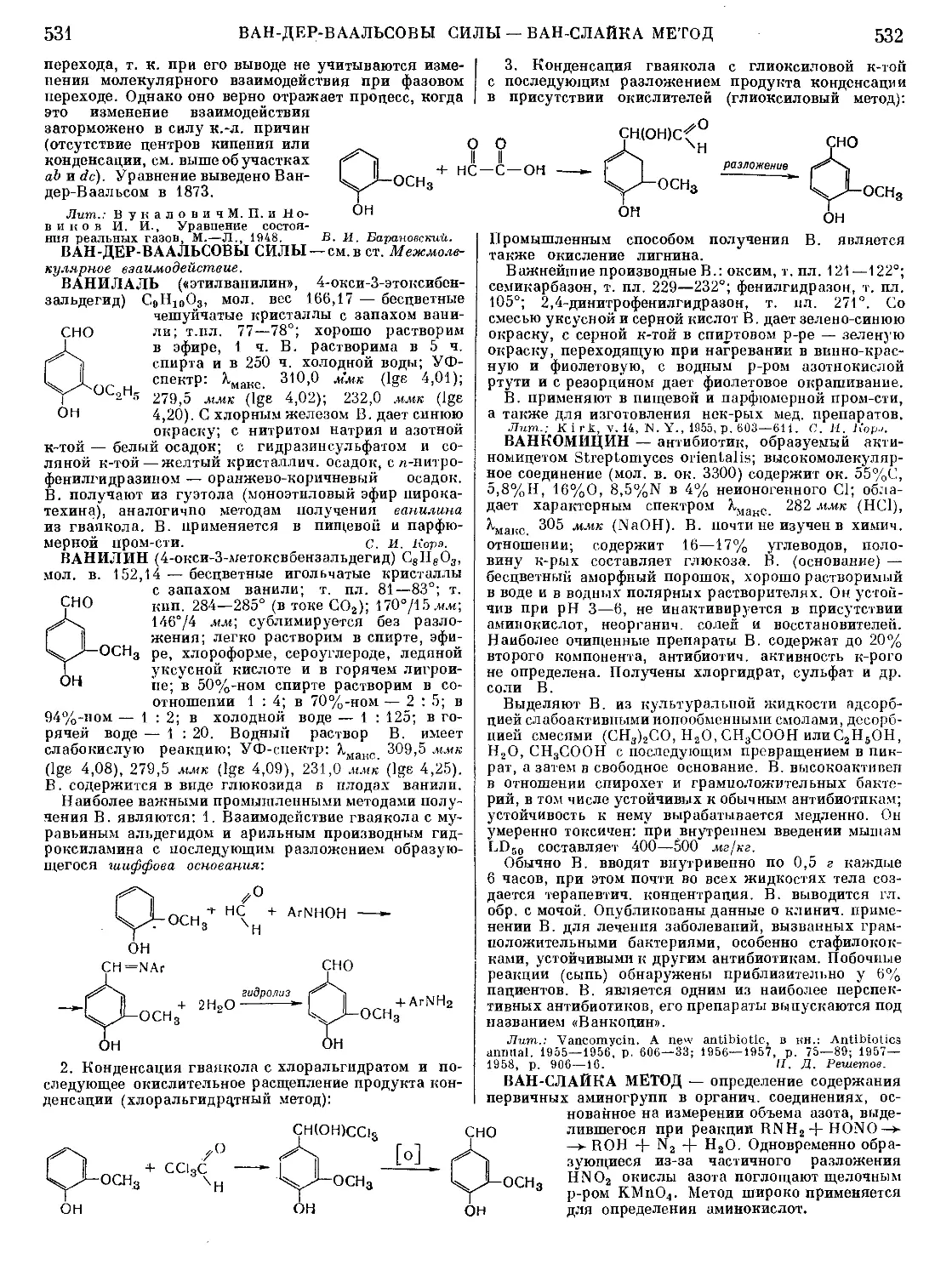






































































































































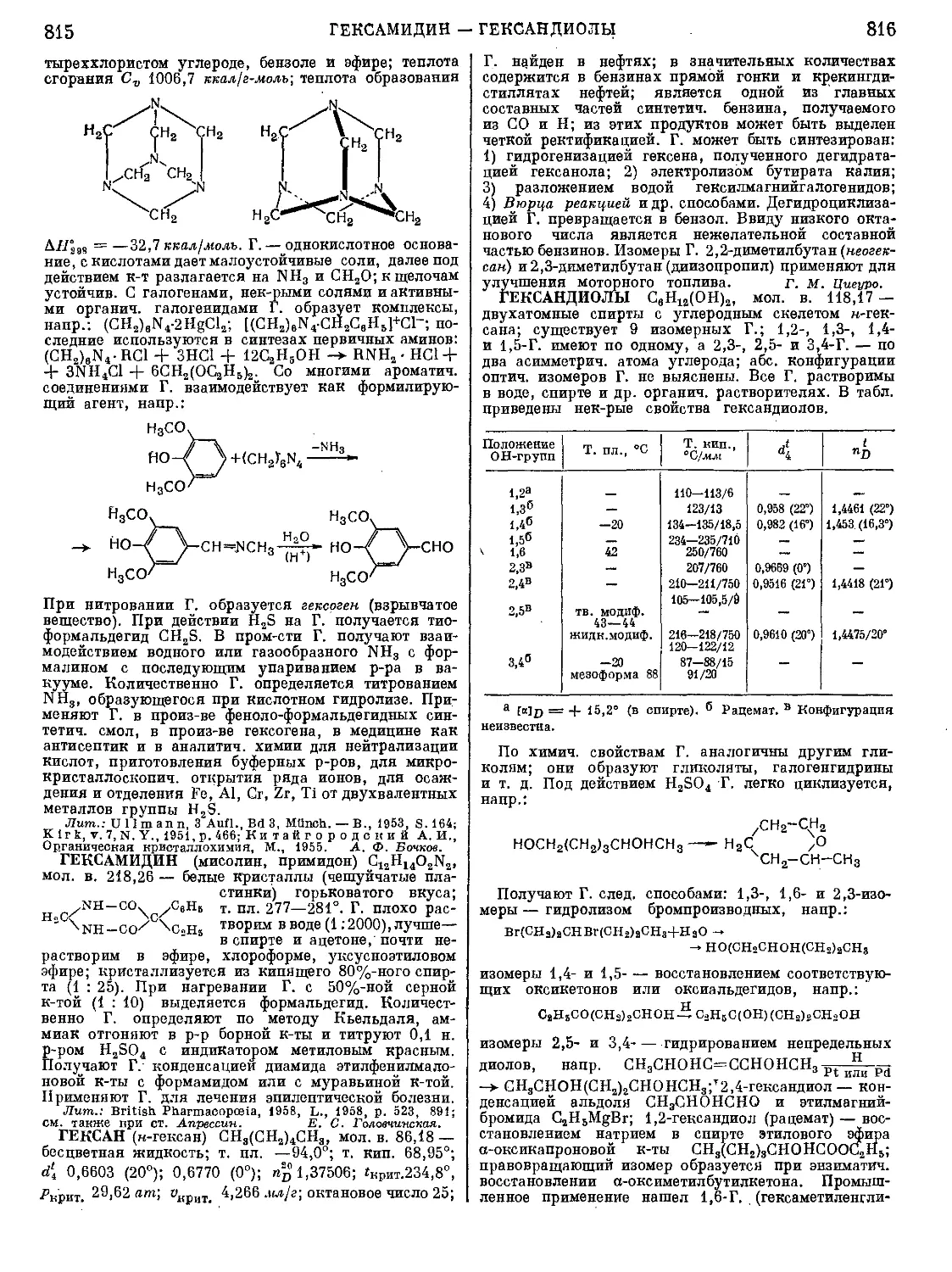


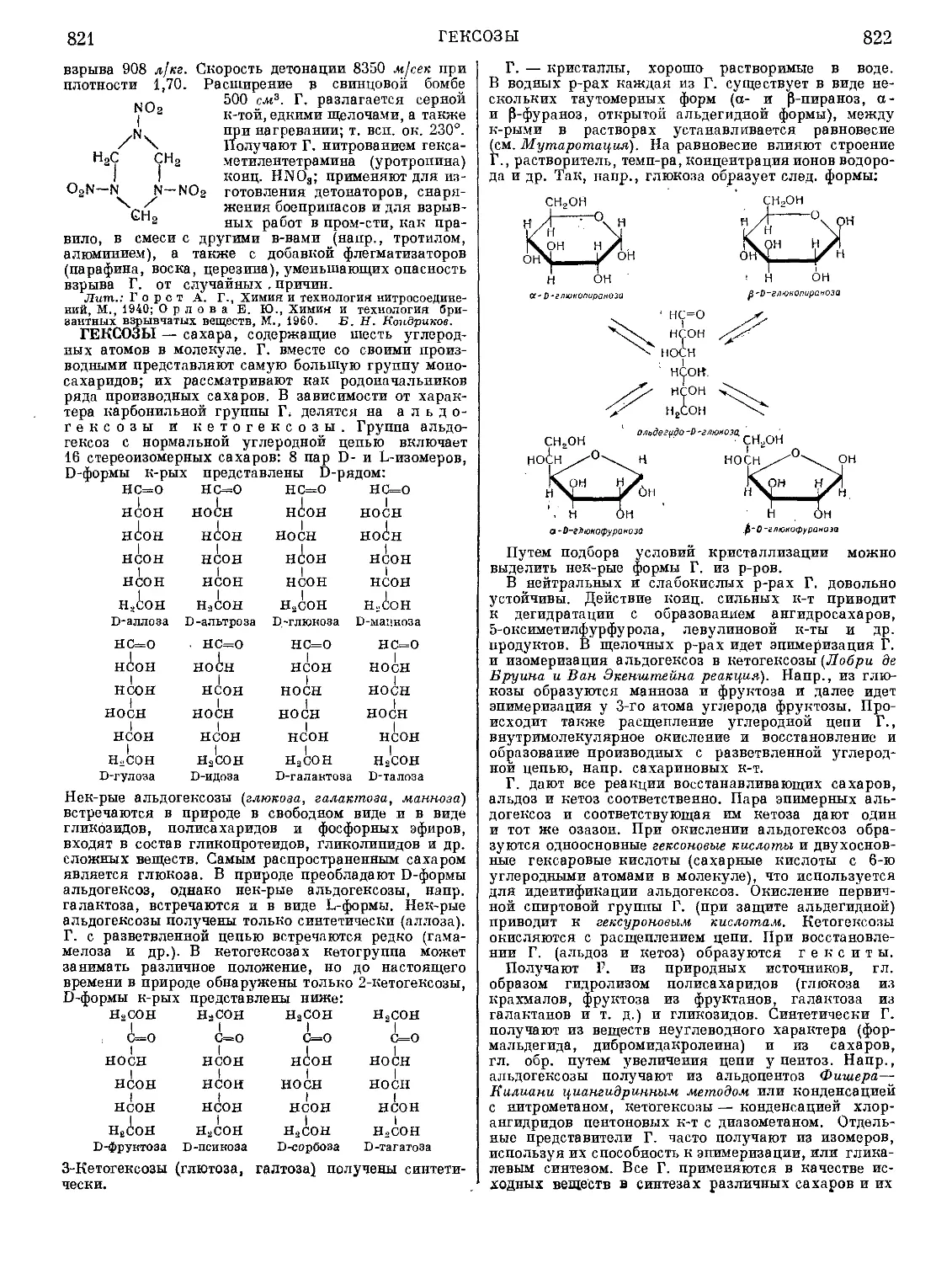




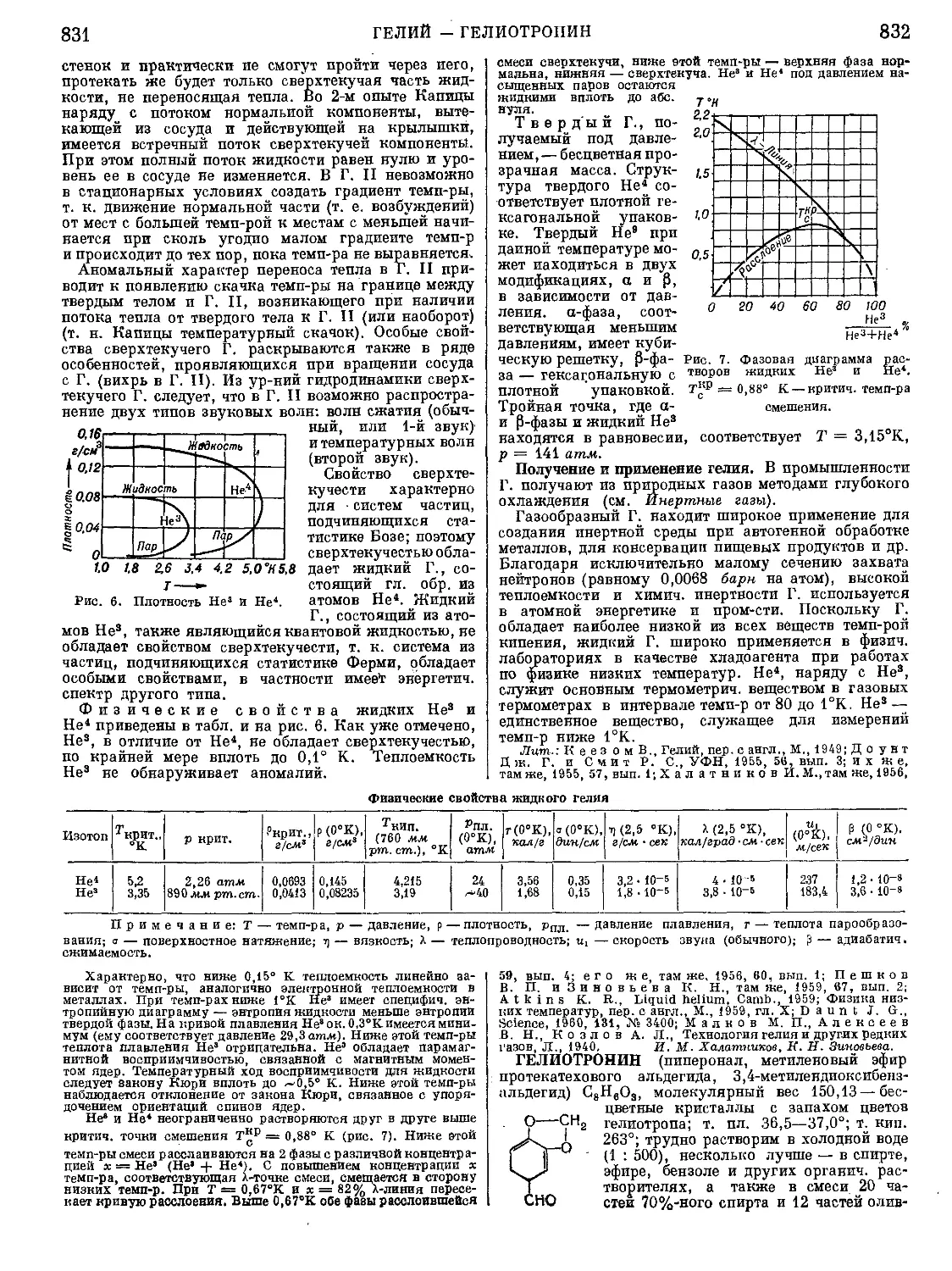
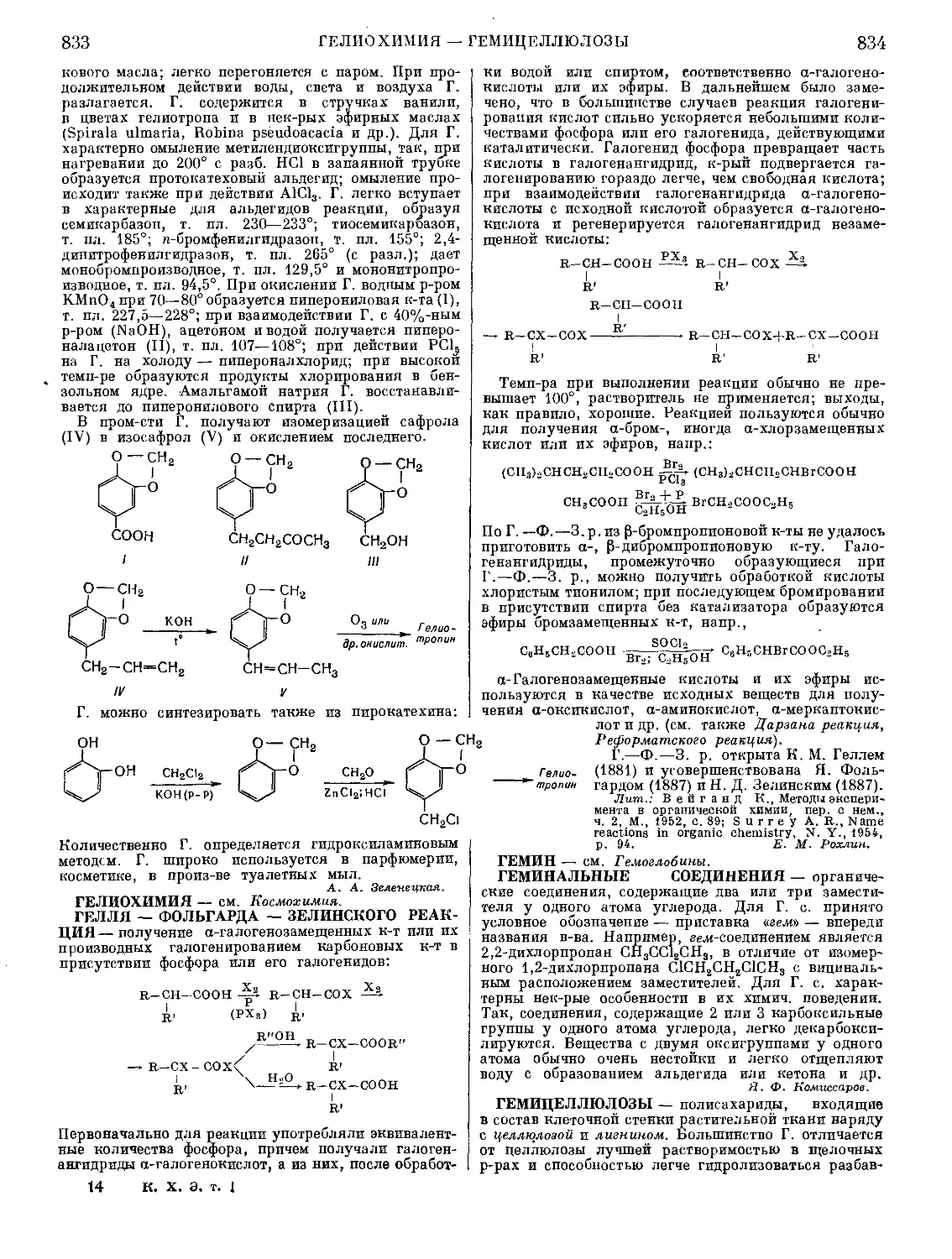




























































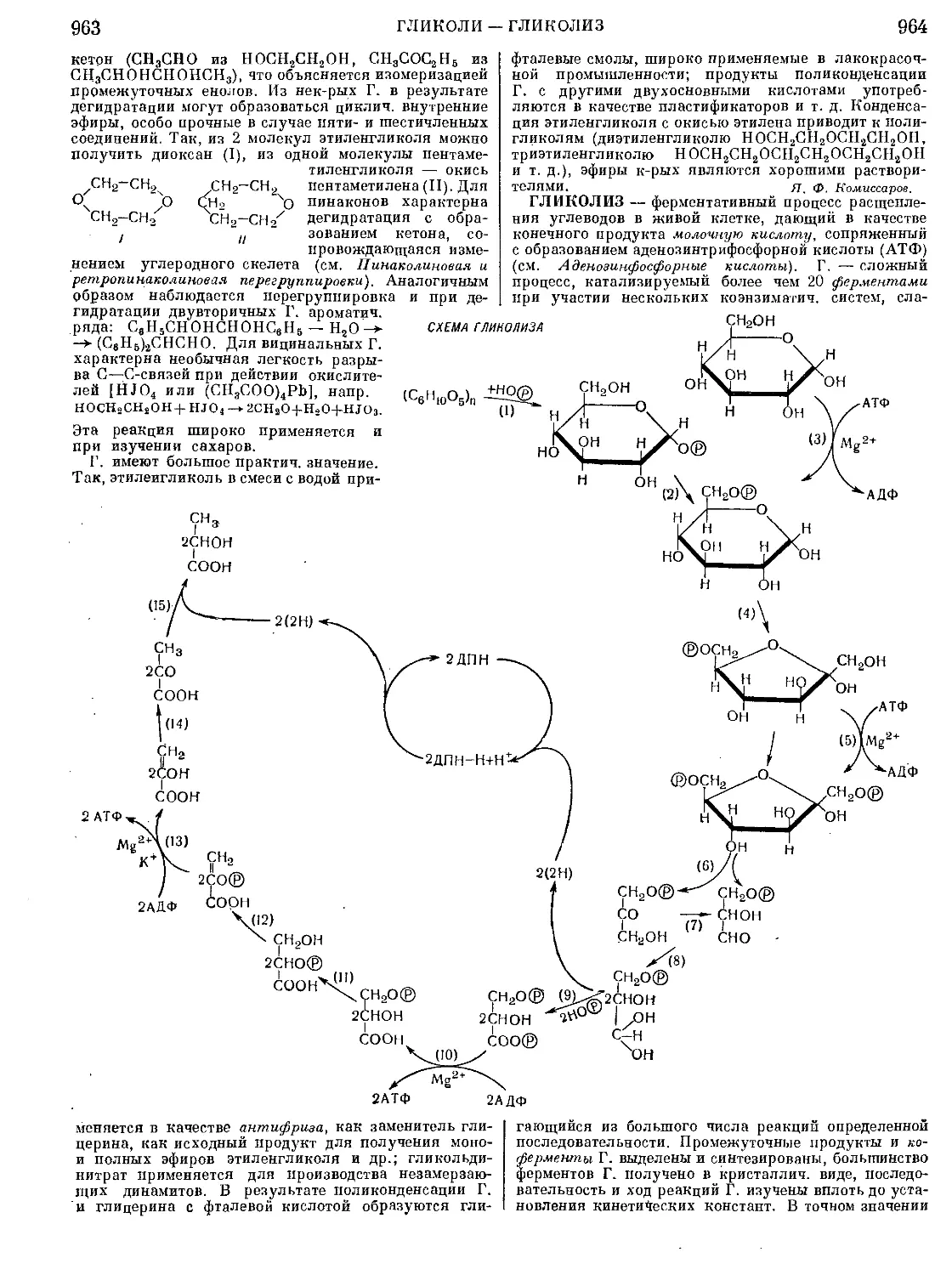










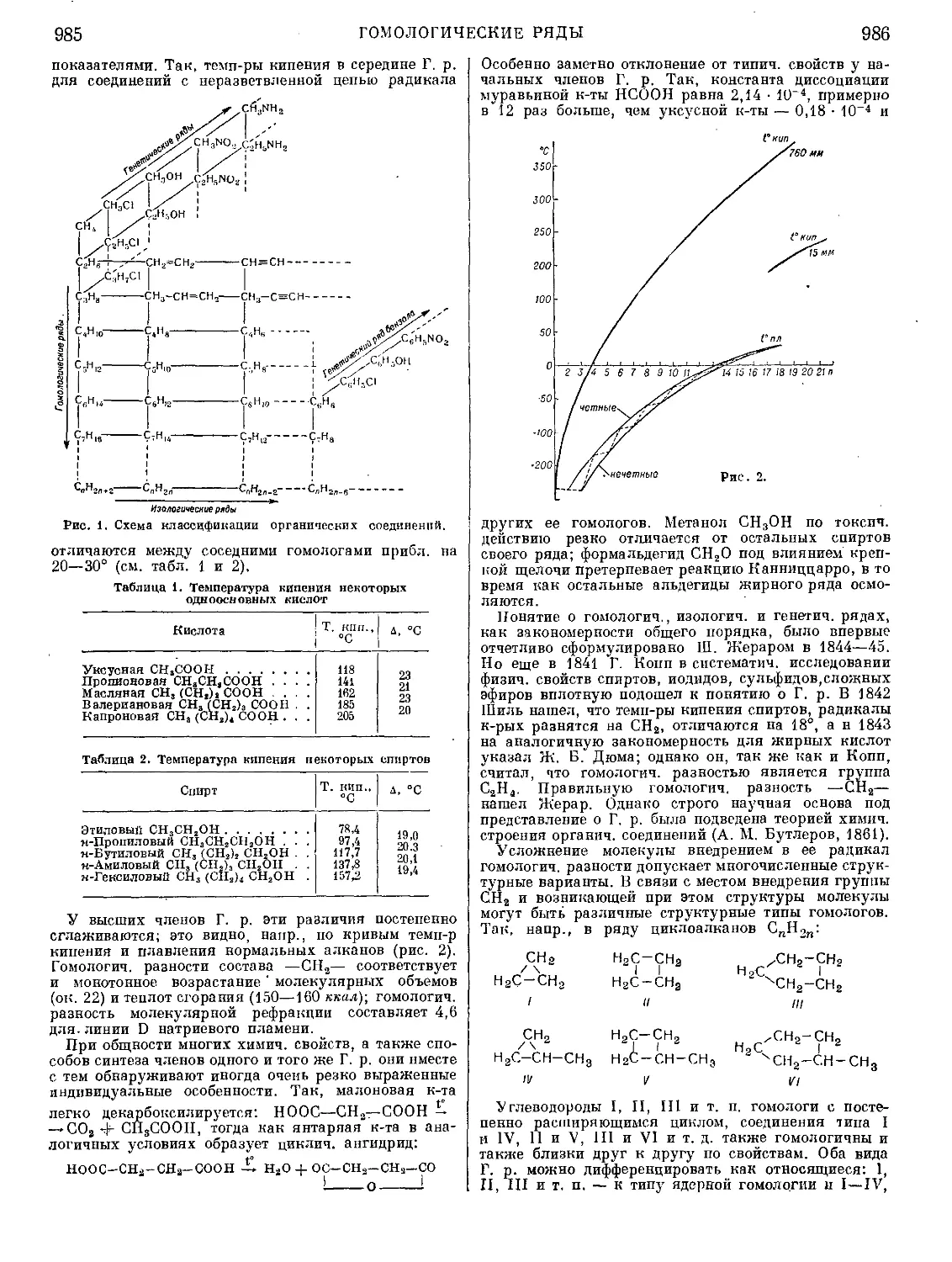












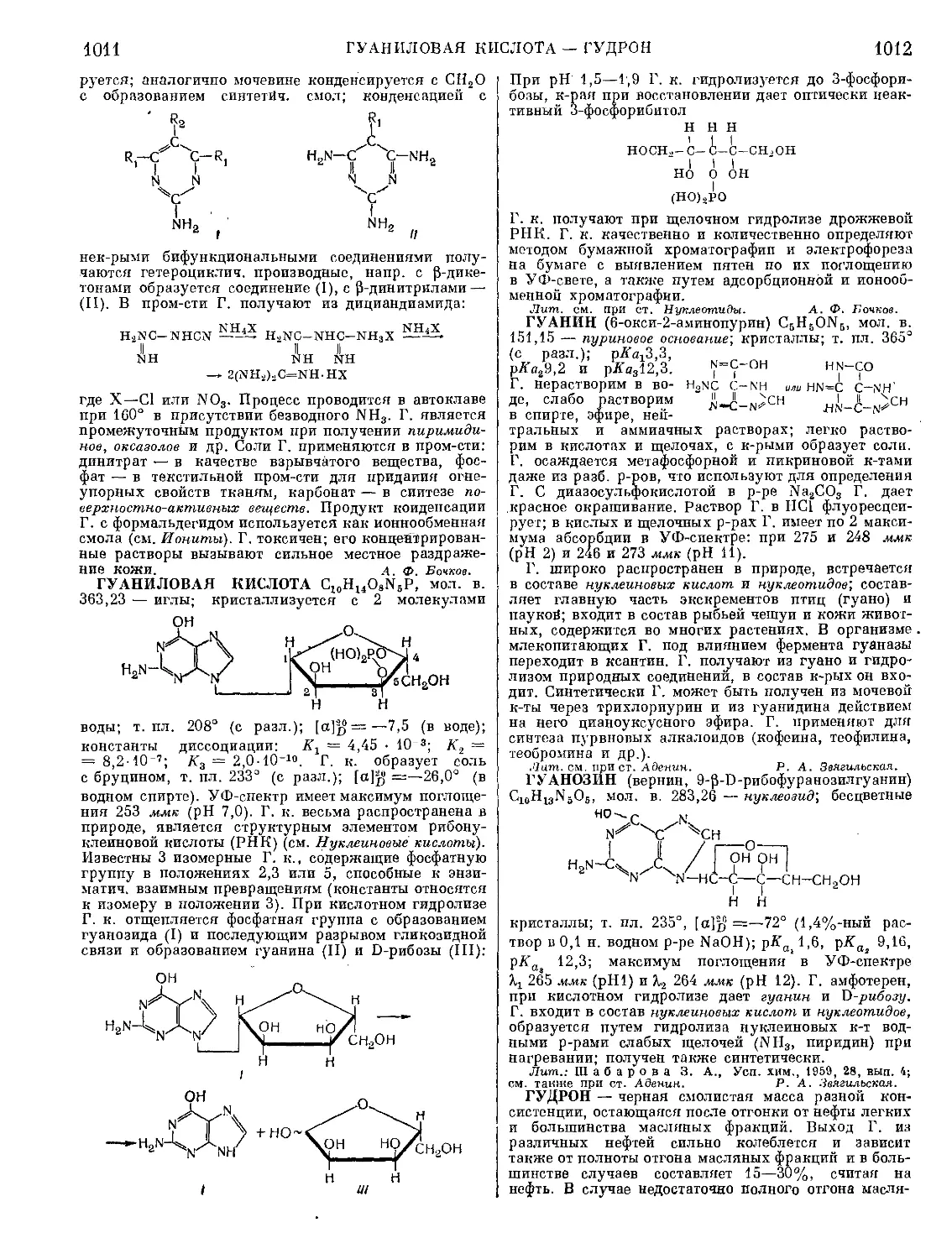




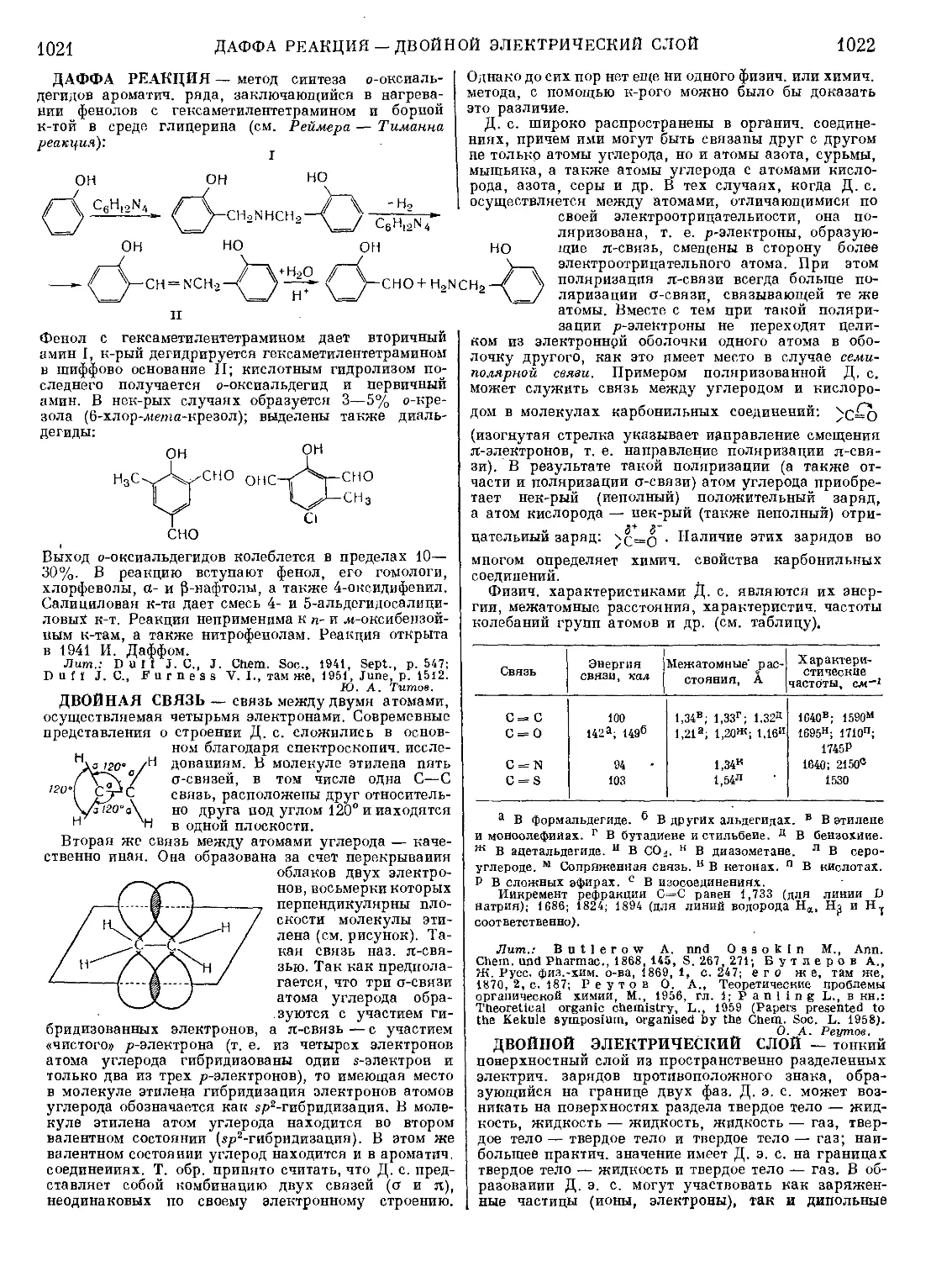







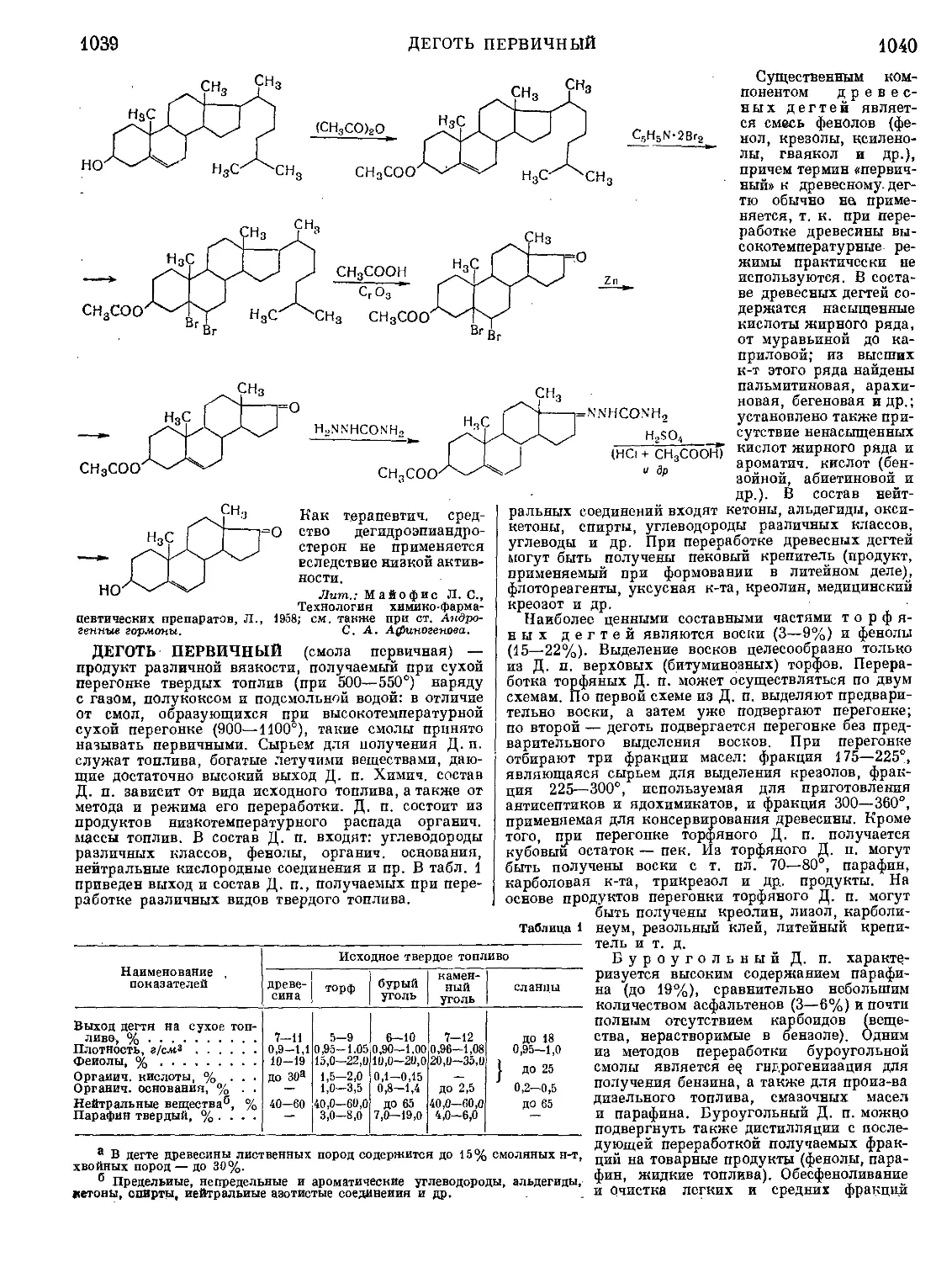


































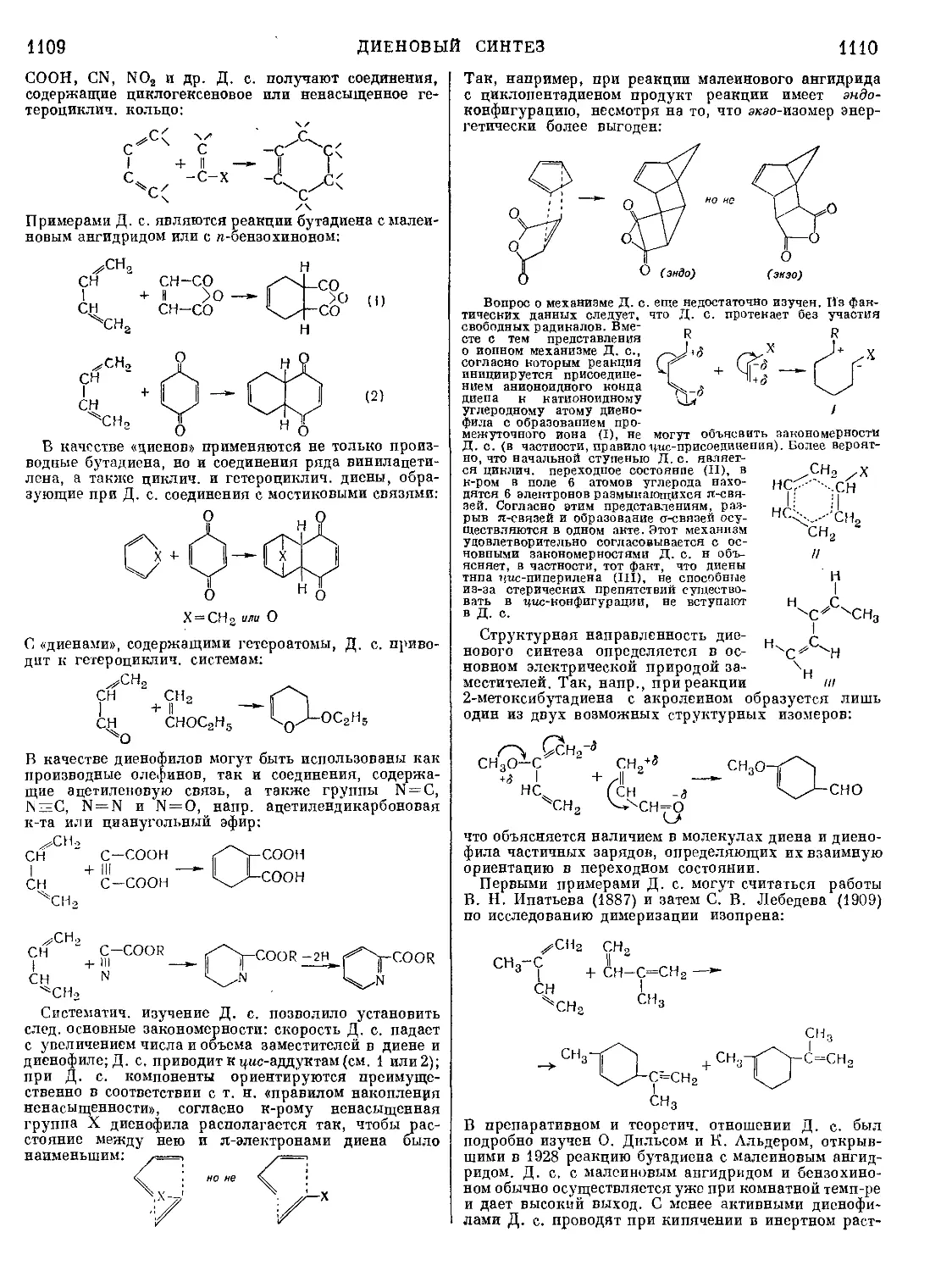


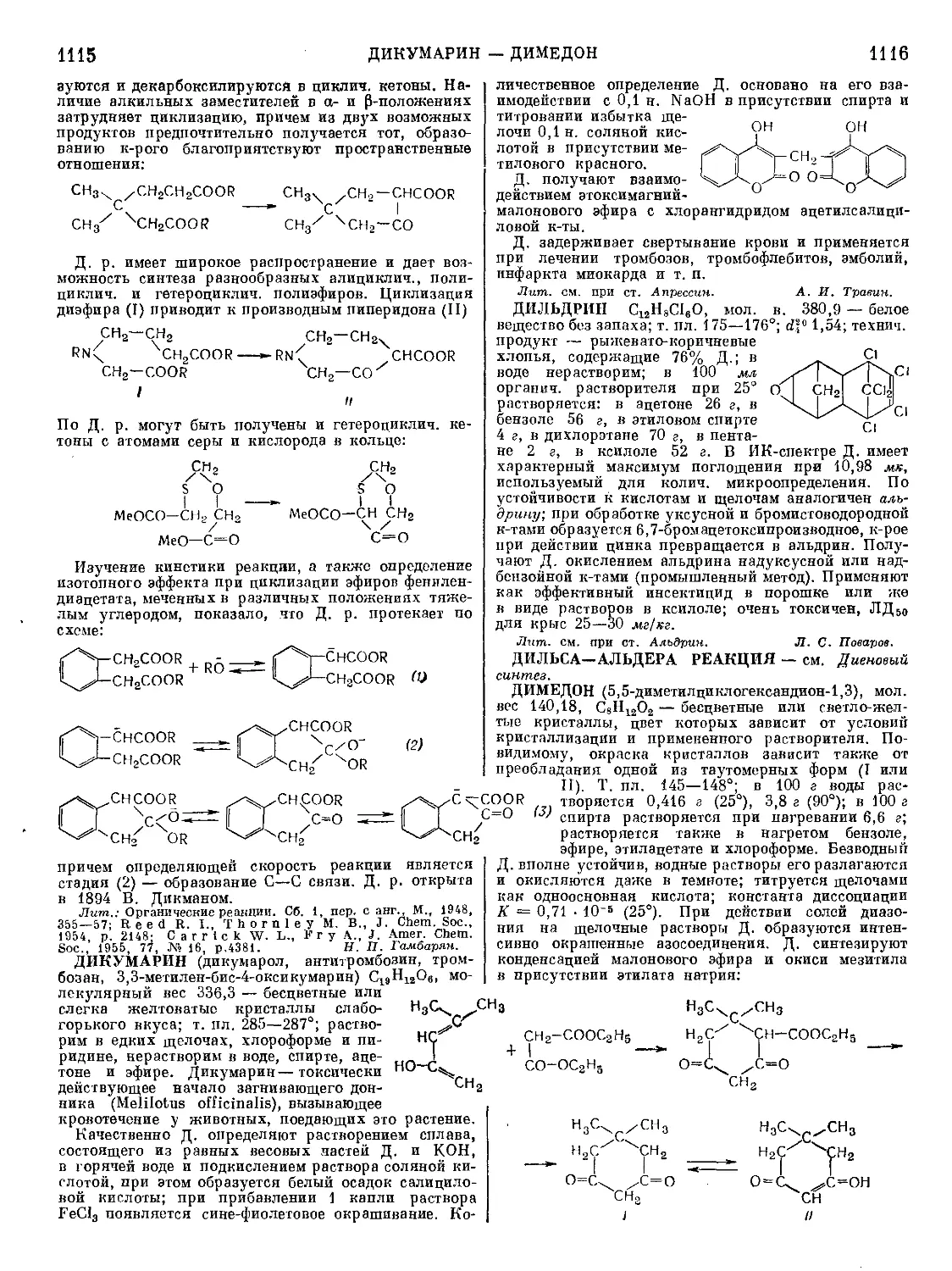









































































Текст
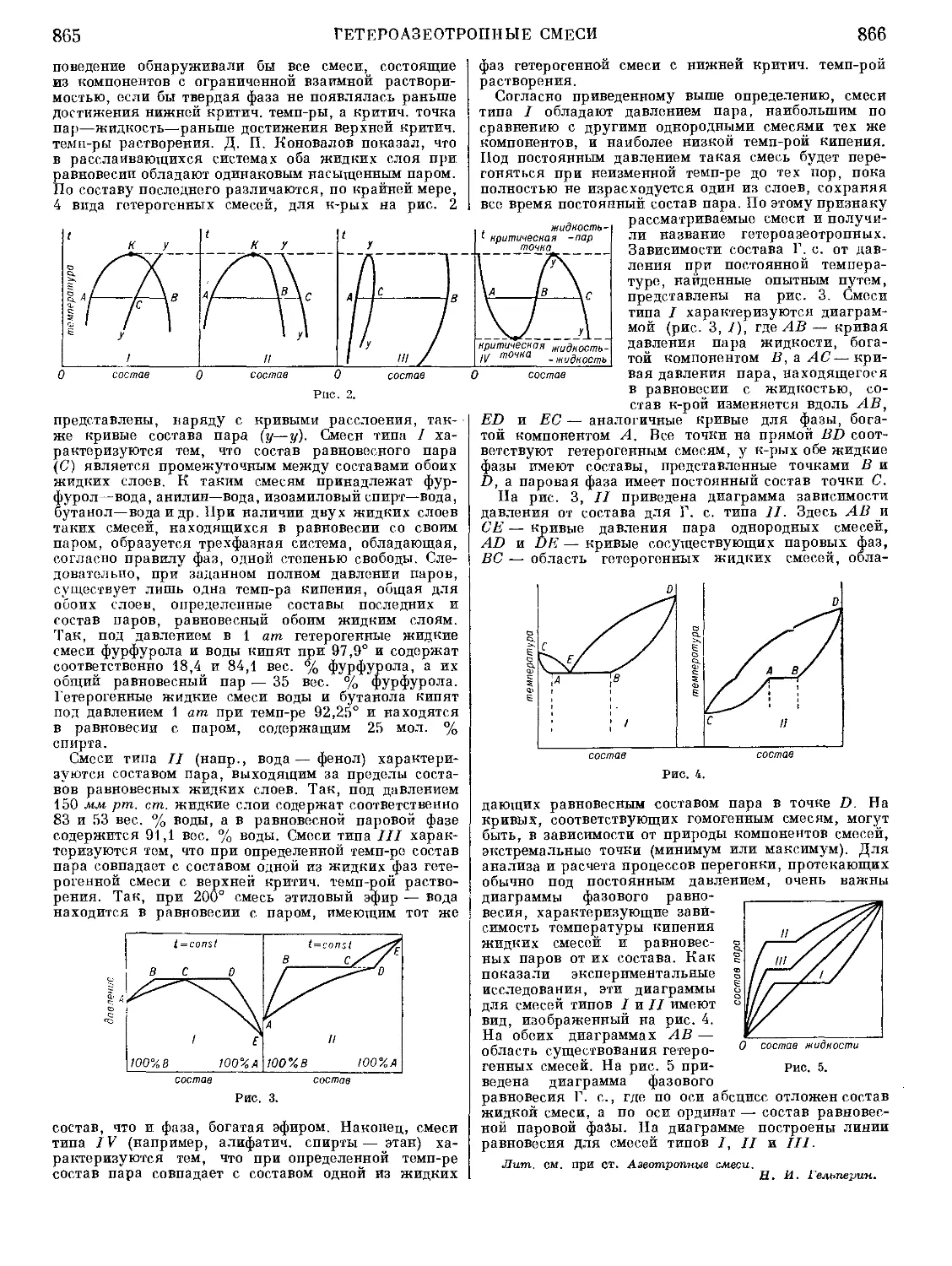
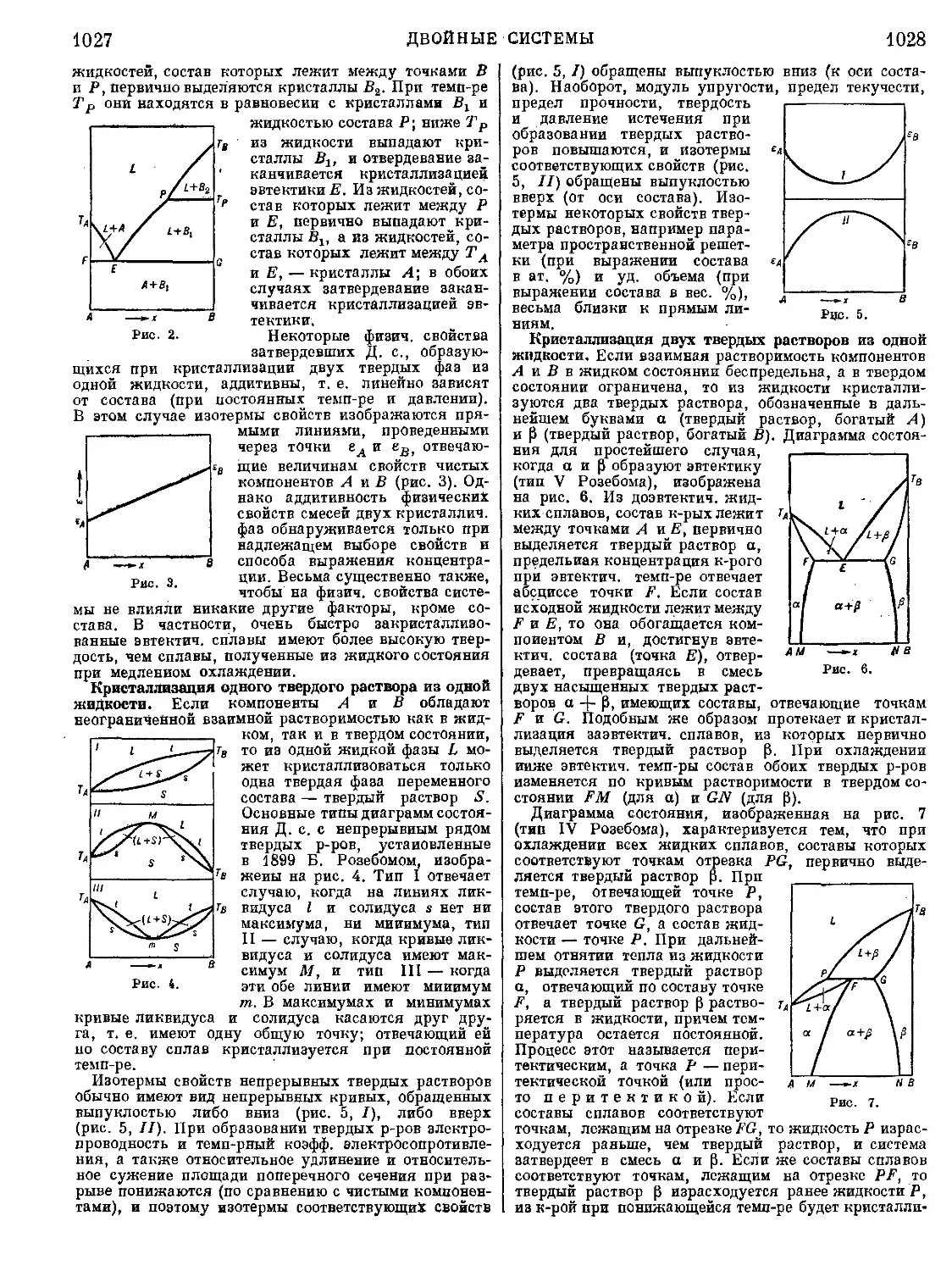
।
КРАТКАЯ
ХИМИЧЕСКАЯ
ЭНЦИКЛОПЕДИЯ
РЕДАКЦИОННАЯ КОЛЛЕГИЯ
И. Л. КНУНЯНЦ (главный редактор), Г. Я. БАХАРОВСКИЙ (зам. глав-
ного редактора), А. И. БУСЕВ, Я. М. ВАРШАВСКИЙ, II. И. ГЕЛЬПЕ-
РИН, П. И. ДОЛИН, В. А. КИРЕЕВ, Г. А. МЕЕРСОИ, А. И. МУРИН,
С. А. ПОГОДИН, П. А. РЕБИНДЕР, Г. Л. СЛОНИМСКИЙ, Б. Н. СТЕ-
ПАНЕНКО, Д. А. ЭПШТЕЙН
1
ГОСУДАРСТВЕННОЕ НАУЧНОЕ ИЗДАТЕЛЬСТВО
«СОВЕТСКАЯ ЭНЦИКЛОПЕДИЯ»
ГОСУДАРСТВЕННОЕ НАУЧНОЕ ИЗДАТЕЛЬСТВО
«СОВЕТСКАЯ ЭНЦИКЛОПЕДИЯ»
ЭНЦИКЛОПЕДИИ
СЛОВАРИ
СПРАВОЧНИКИ
НАУЧНЫЙ СОВЕТ ИЗДАТЕЛЬСТВА
А. П. АЛЕКСАНДРОВ, А. А. АРЗУМАНЯН, А. В. АРЦИХОВСКИП,
И. В. ВАРАНОВ, А. А. БЛАГОНРАВОВ, И. И. БОГОЛЮБОВ,
Б. А. ВВЕДЕНСКИЙ (председатель Научного Совета), Б. М. ВУЛ,
Г. Н. ГОЛИКОВ, И. Л. КНУНЯНЦ, Ф. В. КОНСТАНТИНОВ,
Б. В. КУКАРКИН, Ф. Н. ПЕТРОВ, В. М. ПОЛЕВОЙ, А. И. РЕВИН
(заместитель председателя Научного Совета), Н. М. СИСАКЯН,
А. А. СУРКОВ, Л. С. ШАУМЯН (заместитель председателя Научного
Совета)
МОСКВА • 19 6 1
54 (03)
К78
РЕДАКТОРЫ-КОНСУЛЬТАНТЫ
А. К. БАБКО, В. Н. БЕЛОВ, И. II. БУДНИКОВ, В. Н. БУКИН, А. И. ГОЛЬ-
БИНДЕР, Л. В. ГОРДОН, Б. А. ДОГАДКИН, М. А. ЕЛЬЯШЕВИЧ, А. В. КИ-
СЕЛЕВ, А. И. КОРОЛЕВ, Л. И. ЛИНЕВИЧ, В. В. ЛОСЕВ, О. Ю. МАГИДСОН,
Г. И. МЕНЬШИКОВ, И. И. НОВИКОВ, А. Б. ПАКШВЕР, А. Н. ПРАВЕДНИ-
КОВ, Н. Г. ПУЧКОВ. А. С. ХОХЛОВ, А. И.ШЕРЕШЕВСКИЙ, Н. М. ЭМАНУЭЛЬ
РЕДАКЦИЯ КРАТКОЙ ХИМИЧЕСКОЙ ЭНЦИКЛОПЕДИИ
Заведующий редакцией — Г. Я. БАХАРОВСКИЙ; научные редакторы —
Д. Н. ВАСКЕВИЧ, р. Р. ГАЛЛЕ, Р. В. ГАРКОВЕНКО, 3. И. ГОДИН,
Н. П. МОСТОВЕНКО; литературные редакторы — Л. Д. КИРИЛЛОВА,
В. Л, КИСЛОВ; младшие редакторы — В. А. ЛЕБЕДЕВА, М. Е. ТРУХАНОВА,
К. В. ФИЛИППОВА
Редактор-библиограф — Е. И. ЖАРОВА
Художественный редактор — И. Н. САХАРОВА
Технический редактор — И. Д. КУЛИДЖАНОВА
Корректор — А. В. МАСЛОВА
Указатель составил — А. Б. ДМИТРИЕВ
ОТ РЕДАКЦИИ
Краткая Химическая Энциклопедия представляет собой научно-справочное издание по всем отраслям
химии. Энциклопедия намечена к выпуску в 4 томах по 100 авторских листов в томе. В Энциклопедии отра-
жены все разделы современной химической науки и техники: физическая химия, неорганическая химия,
органическая химия, аналитическая химия, коллоидная химия, радиохимия (в том числе химия изото-
пов), радиационная химия, геохимия, биохимия, химия природных и синтетических полимеров, химия и
технология нефти, коксохимия, химия красителей, химия лекарственных веществ и т. п.
Важное место в Энциклопедии занимают вопросы теории: строение атома и молекулы, связь реакцион-
ной способности со строением, механизм химических процессов (в том числе каталитических, цепных и т. д.),
конформационный анализ, проблемы современной органической химии (в частности, ароматичность) и мно-
гие другие.
В Энциклопедии широко освещаются новые методы физики и химии: хроматография, изотопные инди-
каторы, спектрометрия, радиоспектроскопия, протонный и ядерный магнитный резонанс, комбинационное
рассеяние света и т. д. Особое внимание уделено тем направлениям, которые развиваются быстро и приобре-
тают все большее научное и практическое значение.
Физическая химия представлена в Энциклопедии обзорами, освещающими все ее разделы (статьи «Кине-
тика», «Катализ», «Адсорбция», «Растворы», «Электрохимия» и другие), а также большим числом статей
по более узким вопросам.
В статьях по неорганической химии даны подробные сведения о химических элементах и их соединениях
(физические и химические свойства, геохимия, способы получения и применения, анализ). Большое внима-
ние уделено химии редких металлов, нашедших за последнее время широкое практическое применение, а
также различным сплавам.
Органическая химия с ее многочисленными ответвлениями представлена наибольшим числом статей.
Наряду с теоретическими вопросами Энциклопедия знакомит читатели с современными методами синтеза;
во многих статьях освещаются именвые реакции и молекулярные перегруппировки, дано современное истол-
кование их механизмов. Описаны все классы и большое число индивидуальных соединений, их структуры,
способы получения и практическое применение.
Особое место в Энциклопедии отведено природным и синтетическим полимерам, физико-механическим
свойствам, способам их получения и применения. Описаны каучуки, резина, смолы, пластмассы, пленко-
образующие вещества, лаки, целлюлоза, химические волокна и другие.
Химические методы все больше проникают в исследование разнообразных биологических процессов.
В Энциклопедии описаны важнейшие биохимические процессы (статьи «Обмен веществ», «Фотосинтез»,
«Брожение» и др.), а также многие соединения, получаемые биохимическими методами, и их значение для
жизнедеятельности и т. п.
Во всех статьях о химических элементах и различных соединениях приводятся современные способы
их промышленного получения. Общие технологические способы описываются в таких статьях, как «Абсорб-
ция», «Выпаривание», «Дистилляция», «Ректификация», «Экстракция» и многих других.
В статьях,посвященных наиболее технически важным вопросам, приведены подробные физико-химиче-
ские константы и другие справочные сведения, которые могут оказаться полезными при проектировании
и в заводской практике.
Некоторые понятия, реакции, методы, индивидуальные соединения описываются не в самостоятельных,
а в более общих статьях. Предметный указатель, помещаемый в конце каждого тома, дает возможность легко
находить о них сведения. Система ссылок позволяет связывать данный термин с другими и тем самым расши-
рить круг сведений по тому или иному вопросу (ссылки даны курсивом). Многие статьи снабжены библиогра-
фией.
Названия статей, состоящих из двух и более слов, как правило, даны на слова, в которых содержится
определяющий признак, например «Серная кислота», а не «Кислота серная», «Вюрца реакция», а не «Реакция
Вюрца» и т. д.
Всего в Краткой Химической Энциклопедии будет помещено более 5 000 статей.
Краткая Химическая Энциклопедия рассчитана на широкий круг химиков: научных работников, инже-
неров, преподавателей вузов, техникумов и средней школы, студентов и аспирантов. В Энциклопедии найдут
полезные сведения читатели и других специальностей — физики, геологи, биологи и другие.
Химические энциклопедии и словари заняли в ряде стран прочное место среди справочной литературы.
В Советском Союзе такое издание предпринято впервые. Учитывая трудности в подготовке Энциклопедии,
редакция обращается к читателям с просьбой сообщить свое мнение о достоинствах и недостатках тома в целом
и отдельных статей. Пожелания читателей редакция постарается учесть в последующих томах.
СОКРАЩЕНИЯ, УСЛОВНЫЕ ОБОЗНАЧЕНИЯ, ЕДИНИЦЫ ИЗМЕРЕНИЯ
I. ОСНОВНЫЕ СОКРАЩЕНИЯ
абс.— абсолютный
анти — приставка, напр. анти-форма,
анти-изомер, анти-С 4Н 4О 4
асимм — асимметрический (только в со-
четании с формулой), напр. асимм-
(СН3)СвНб
ат. в.'с— атомный вес
ат. % — атомный процент (после числа)
ат. теплоемкость — атомная теплоем-
кость
б. ч. — большей частью
вт. ч. — в том числе
в-во — вещество
вес. % — весовой процент (при числе)
вкл. — включительно
гл. обр. — главным образом
др. — другие
кам.-уг. — каменноугольный
к.-л. — какой-либо
к.-н. — какой-нибудь
кн. — книга
конц. — концентрированный
коэфф. — коэффициент
кпд — коэффициент полезного действия
крит. — критический (в сочетании со
словами, напр. крит. объем)
к-рый — который
к-та, к-ты — кислота, кислоты
м. б. — может быть
м. ч. — массовое число (при числе)
А — работа
а — активность
а, Ь, с; а, р, 7 — параметры кристалли-
ческой решетки
[а]£) — удельное вращение плоскости
поляризации
С — концентрация
°C — градусы стоградусной шкалы
Ср — теплоемкость при постоянном дав-
лении
Cv — теплоемкость при постоянном объе-
ме
D — соединения, по конфигурации при-
надлежащие к D-ряду глицеринового
альдегида
d — плотность (в е/слс3)
d — правовращающийся изомер; дейтрон
d|° — плотность, измеренная при 20°
и отнесенная к плотности воды при 4°
^крит. — плотность критическая
di — рацемат
АН — изменение энтальпии
е — электрон — элементарный заряд
е — электрон — частица
ехр — экспонент
F—изохронно-изотермический потенциал
/ — фугитивность
макс. — максимальный
миним. — минимальный
млн. — миллион (при числе)
млрд. — миллиард (при числе)
мп. — многие
мн. др. — многое другое
мол. в., мол. вес — молекулярный вес
мол. % — молекулярный процент (при
числе)
наз. — называется, называемый
нек-рый — некоторый
неск. — несколько
об. % — объемный процент (при числе)
ок. — около
п. п. — порядковый номер элемента
(при числе)
плотн. — плотность (обычно в з/слс3;
при числах, как правило, не ставится,
а подразумевается)
преим. — преимущественно
произ-во — производство
пром-сть — промышленность
разб. — разбавленный
разл. — разложение
рис. — рисунок
р-р, Р-ры — раствор, растворы
с. х-во — сельское хозяйство
св. — свыше
син — приставка, напр. син-форма
след. — следующий
II. УСЛОВНЫЕ ОБОЗНАЧЕНИЯ.
v — коэффициент активности
Н — энтальпия
h — Планка постоянная
Н$ —твердость по Бринеллю
ИК — инфракрасный
k — постоянная Больцмана
L — соединения, по конфигурации при-
надлежащие к L-ряду глицеринового
альдегида
1 — левовращающий изомер
X — теплопроводность
ЛДП — летальная (смертельная) доза
(п — число, указывающее на процент
смертности)
МКС (MKS) — система единиц (метр,
килограмм, секунда)
мм вод. ст. — миллиметры водяного
столба
мм рт. ст. — миллиметры ртутного
столба
МТС (MTS) — система единиц (метр,
тонна, секунда)
ц — химический потенциал
н. — нормальность раствора
н — нормальное строение органического
соединения
п — нейтрон
No — Авогадро число
с.-х. — сельскохозяйственный
табл. — таблица
т. абс. — температура абсолютная
т. возг. — температура возгонки
т. воспл. — температура воспламенения
т. всп. — температура вспышки
т. заст. — температура застывания
т. затв. — температура затвердевания
т. исп. — температура испарения
т. к. — так как
т. каплепад. — температура каплепадс-
ния
т. кип. — температура кипения
т. конд. — температура конденсации
т. н., т. наз. — так называемый
т. о., т. обр. — таким образом
т. пл. — температура плавления
т. разл. — температура разложения
т. размягч. — температура размягчения
т. стекл. — температура стеклования
темп-ра — температура (в тексте)
транс — приставка, напр. транс-форма,
транс-изомер, транс-С4Н4О4
уд. — удельный
ур-пие — уравнение
ф-ла — формула
химич. — химический
х. ч. — химически чистый
цис — приставка, напр. ipzc-форма
эдс — электродвижущая сила
25
пр — показатель преломления для D -ли-
нии Na при 25°
о — орто
об/мин — оборотов в минуту
п — пара
р — протон
р — давление
Ркрит — давление критическое
pH — водородный показатель
pf—pH при изоэлектрической точке-
рК — показатель константы ионизации
Рг — Прандтля число
Q — теплота
R — универсальная газовая постоянная
Re — Рейнольдса число
S — энтропия
СГС (CGS) — система единиц (сантиметр,
грамм, секунда)
Т — температура по абсолютной шкале,
°К
/крит. — температура критическая
U — внутренняя энергия
УФ — ультрафиолетовый
Z — изобарно-изотермический потенциал
уэ— подобно, изменяется как...
— порядка величины
=5s — приближенно равно
III. ЕДИНИЦЫ ИЗМЕРЕНИЯ. РАЗМЕРНОСТИ
а — ампер
А — ангстрем
ат — атмосфера техническая
атм — атмосфера физическая
а-ч — ампер-час
барн — единица эффективного попереч-
ного сечения ядерных процессов
бэр — биологический эквивалент рент-
гена
в — вольт
ва — вольт-ампер
вт —• ватт
вт-ч — ватт-час
Г — грамм-сила
г — грамм-масса
7—гамма (10~3.иг)
га — гектар
г-атом — грамм-атом
г-ион — грамм-ион
г-молъ — грамм-молекула
град — градусы в размерностях
гц — герц
г-экв — грамм-эквивалент
дж — джоуль
дин — дина!
кал —• калория
кв — киловольт
кет — киловатт
кГ — килограмм-сила
кг — килограмм-масса
кГм — килограммометр
кгс — то же, что и кГ
ккал — килокалория
ком — килоом
feX — килоикс
кюри — кюри
л — литр
М — моль
м — метр
мг — миллиграмм
мин — минута
мк — микрон
мкг (7) — микрограмм (10~е г, 10-3 мг)
ц — то же, что мк
мл — миллилитр
мм — миллиметр
Мом — мегом
мпуаз — микропуаз
Мэв — миллион электрон-вольт
нм3 — нормальный метр кубический газа
(отнесенный к °C и 760 мм рт. ст.)
ом — ом
пуаз — пуаз
р — рентген
рад — единица поглощенной энергии
ионизирующего излучения
сек — секунда
cAt —’сантиметр
спуаз — сантипуаз
сст — сантистокс
ст — стокс
т — тонна метрическая
фэр — физический эквивалент рентгена
ц — центнер
э — эрстед
эв — электрон-вольт
эрг — эрг
Приставки, встречающиеся при назва-
ниях основных единиц измерения
дк — дека (10)
г — гекто (102)
к — кило (103)
М — мега (10е)
Г — гига (109)
Т — тера (1012)
д — деци (10"1)
с — санти (10~2)
м — милли (10~3)
мк — микро (10-е)
ммк — миллимикро (10-9)
мкмк — микромикро (10-12)
НЕКОТОРЫЕ ПЕРИОДИЧЕСКИЕ И СПРАВОЧНЫЕ ИЗДАНИЯ
СОВЕТСКИЕ ПЕРИОДИЧЕСКИЕ ИЗДАНИЯ
Антибиотики
Атомная энергия
Биохимия
Вопросы мед. химии — Вопросы медипинской химии
Высокомолек. соединения — Высокомолекулярные соединения
Газ. пром-сть — Газовая промышленность
Геохимия
Гидролизная и лесохим. пром-сть — Гидролизная и лесохи-
мическая промышленность
ДАН СССР — Доклады Академии наук СССР
Ж. аналит. химии — Журнал аналитической химии
Ж. Всесоюзн. хим. об-ва — Журнал Всесоюзного химического
общества им. д. И. Менделеева
Ж. научн. и прикл. фотографии и кинематографии — Журнал
научной и прикладной фотографии и кинематографии
Ж. неорг. химии — Журнал неорганической химии
Ж. общ. химии — Журнал общей Химии
Ж. прикл. химии — Журнал прикладной Химии
ЖРФХО — Журнал русского физико-химического общества
Ж. структур, химии — Журнал структурной Химии
Ж. физ. химии —Журнал физической Химии
Завод, лабор. — Заводская лаборатория
Защита растений от вредителей и болезней
Изв. АН СССР. Отд. хим. наук — Известия Академии наук
СССР. Отделение Химических наук
Каучук и резина
Кинетика и катализ
Кокс И ХИМИЯ
Коллоидн. ж. — Коллоидный журнал
Лакокрас. материалы и их применение — Лакокрасочные
материалы и их применение
Нефт. х-во — Нефтяное хозяйство
Пласт, массы — Пластические массы
Радиохимия
Р. Ж. хим. — Реферативный журнал «Химия»
Сахарная пром-сть — Сахарная промышленность
Синтетич. высокополимерные материалы — Синтетические вы-
сокополимерные материалы
Спиртовая пром-сть — Спиртовая промышленность
Сталь
Стекло и керамика
Узб. хим. ж. — Узбекский химический журнал
Укр. хим. ж. —Украинский химический журнал
Усп. химии — Успехи химии
Фармакология и токсикология
Хим. волокна — Химические волокна
Хим. пром. — Химическая промышленность
Химия и технология неорг. веществ — Химия и технология
неорганических веществ
Химия и технология топлив и масел.
Цвет, металлы — Цветные металлы
Целлюлозно-бум. пром-сть — Целлюлозно-бумажная промыш-
ленность
Цемент
ИНОСТРАННЫЕ ПЕРИОДИЧЕСКИЕ ИЗДАНИЯ
Acta chem. scand. — Acta chemica scandiuavica (Дания)
Advances Carbohydr. Chem. — Advances in Carbohydrate
Chemistry (США)
Advances Catal. and Related Subjects — Advances in Cataly-
sis and Related Subjects (США)
Advances Chem. — Advances in Chemistry (США)
Agric. Chemicals—Agricultural Chemicals (США)
Amer. Dyestuff Reporter — American Dyestuff Reporter (США)
Amer. J. Pharmacy — American Journal of Pharmacy and the
Sciences Supporting Public Health (США)
Amer. Perfumer and Essent. Oil Rev. — The American Perfu-
mer and Essential Oil Review (США)
Analyst (Англия)
Analyt. chem. — Analytical chemistry (США)
Analytica chimica acta
Ann. chimica — Annali di chimica (Италия)
Ann. chimie — Annales de chimie (Франция)
Annual Rev. Biochem. — Annual Review of Biochemistry
(США)
Annual Rev. Biochem. and Allied Res. India — Annual Review
of Biochemical and Allied Research in India (Индия)
Annual Rev. Phys. Chem. — Annual Review of Physical Che-
mistry (США)
Arch, pharmaci og chemi — Archiv for pharmaci og chemi
(Дания)
Arkiv kemi — Arkiv fBr keini (Швеция)
Austral. J. Chem. — Australian Journal of Chemistry (Австра-
лия)
Beth. Angew. Chem. und Chem.-lngr.-Techn. — Beihefte zur
Angewandten Chemie und Cheniie-Ingenieur-Technik (ФРГ)
Ber. — Berichte der Deutscheu chemischen Gesellschaft
Biochem. Soc. Sympos. — Biochemical Society Symposia
(Англия)
Biochem. Z. — Biochemische Zeitschrift (ФРГ)
Biochim. et biophys. acta — Biochimica et biophysica acta
(Нидерланды)
Bitum., Teere, Asph., Peche und verw. Stoffe — Bitumen,
Teere, Asphalte, Peche und verwandte Stoffe (ФРГ)
Boll. chim. farmac. — Bollettino chimico farmaceutico (Италия)
Brennstoff-Chemie — Brennstoff-Chemie (ФРГ)
Bull. Centr. Electrochem. Res. Inst. — Bulletin of the Central
Electrochemical Research Institute (Индия)
Canad.Chem. Process. — Canadian Chemical Processing (Канада)
Canad. J. Biochem. and Physiol. —Canadian Journal of Bio-
chemistry and Physiology (Канада)
Canad. Oil and Gas Inds — Canadian Oil and Gas Industries
(Канада)
Catalyst (США)
Chem. Ber. — Chemische Berichte (ФРГ)
Chem. Bull. — The Chemical Bulletin (США)
Chem. courant — Chemische courant (Нидерланды)
Chem. Engng — Chemical Engineering (США)
Chem. and Engng News — Chemical and Engineering News
(США)
Chem. Engng Progr. — Chemical Engineering Progress (США)
Chem. Engng Progr. Sympos. Series — Chemical Engineering
Progress Symposium Series (США)
Chem. Engng Sci. — Chemical Engineering Science (Англия)
Chem. Erde — Chemie der Erde (ГДР)
Chem. Ind. — Chemische Industrie (ФРГ)
Chem.-Inform. — Chemie-Informationen. Sonderdienst der Zeit-
schrift «Praktische Chemie» (Австрия)
Chem.-lngr.-Techn. — Chemic-Ingenieur-Technik (ФРГ)
Chem. listy — Chemickd listy (Чехословакия)
Chem. and Process Engng — Chemical and Process Engineering
(Англия)
Chem. Prod. — Chemical Products and Chemical News (Англия)
Chem. prhmysl — Chemicky prhmysl (Чехословакия)
Chem. Revs — Chemical Reviews (США)
Chem. Rundschau — Chemische Rundschau (Швейцария)
Chem. Technik — Chemische Technik (ГДР)
Chem. Trade J. and Chem. Engr — Chemical Trade Journal’
and Chemical Engineer (Англия)
Chem. weekbl. — Chemisch weekblad (Нидерланды)
Chem. Zbl. — Chemisettes Zentralblatt (ГДР)
Chem. zvesti — Chemicke zvesti (Чехословакия)
Chemie (Австрия)
Chemie-Markt — Der Chemie-Markt. Sonderdienst der Zeit-
schrift «Chemische Industrie» (ФРГ)
Chernik (Польша)
Chemiker-Ztg — Chemiker-Zeitung (ФРГ)
Chemist — The Chemist (США)
Chemist-Analyst — Chemist-Analyst (США)
15
НЕКОТОРЫЕ ПЕРИОДИЧЕСКИЕ И СПРАВОЧНЫЕ ИЗДАНИЯ
16
Chemist and Druggist (Англия)
Chemistry (США)
Chemistry and Industry (Англия)
Chim. analyt. — Chimie analytique (Франция)
Chim. peintures — La chimie des peintures (Бельгия)
Chimia (Швейцария)
Chimica — Chimica. Rivista mensile per la diffusione della
cultura chimica (Италия)
Chimica e industria — La Chimica e 1’industria (Италия)
Chimie et Industrie — Chimie et Industrie (Франция)
Corros. Technol. — Corrosion Technology (Англия)
Corrosion (США)
Dtsch. Farben-Z. — Deutsche Farben-Zeitschrift (ФРГ)
Dyestuffs (США)
Erddl und Kohle (ФРГ)
ErdOl-Ztg — Erdol-Zeitung (Австрия)
Farbe und Lack (ФРГ)
Farm Chemicals (США)
Fette, Seifen, Anstrichmittel (ФРГ)
Fort Dodge Bio-Chem. Rev. — Fort Dodge Bio-Chemic Review
(США)
Fortschr. chem. Forsch. — Fortschritte der chemischen For-
schung (ФРГ)
Gas (USA)
Gas Abstrs — Gas Abstracts (США)
Gazz. chim. ital. — Gazzetta chimica italiana (Италия)
Genie chim. — Genie chimique (Франция)
Gold und Silber (ФРГ)
Helv. chim. acta — Helvetica chimica acta (Швейцария)
Ind. chim. beige — Industrie chimique beige (Бельгия)
India-Rubber J. —The India-Rubber Journal (Англия)
Industr. Chemist — The Industrial Chemist and Chemical
Manufacturer (Англия)
Industr. and Engng Chem. — Industrial and Engineering
Chemistry (США)
Industria у quimica — Industria у quimica (Аргентина)
Ingegneria chim. — Ingegneria chimica (Италия)
Isotopics (Oak Ridge) (США)
J. Amer. Chem. Soc. — Journal of the American Chemical
Society (США)
J. Amer. Oil Chemists’ Soc. — The Journal of the American
Oil Chemists’ Society (CHIA)
J. Antibiotics A — Journal of Antibiotics. Series А (Япония)
J. Antibiotics В — Journal of Antibiotics. Series В (Япония)
J. Appl. Chem. — The Journal of Applied Chemistry (Англия)
J. Biochem. — The Journal of Biochemistry (Япония)
J. Biol. Chem. — The Journal of Biological Chemistry (США)
J. Chem. Educ. — Journal of Chemical Education (США)
J. Chem. Phys. — The Journal of Chemical Physics (США)
J. Chem. Sb'c. — Journal of the Chemical Society (Англия)
J. chim. phys. et phys-chim. biol. — Journal de chimie phy-
sique et de physico-chimie biologique (Франция)
J. Electrochem. Soc. — Journal of the Electrochemical Society
(США)
J. four dlectr. et inds elcctrocbim. — Journal du four 61ectrique
et des industries dlectrochimiques (Франция)
J. Indian Chem. Soc. — Journal of the Indian Chemical Society
(Индия)
J. Indian Chem. Soc., Industr. and News Ed. — Journal of
the Indian Chemical Society, Industrial and News Edition
(Индия)
J. Inorg. and Nucl. Chem. — Journal of Inorganic and Nuclear
Chemistry (Англия)
J. Organ. Chem. —The Journal of Organic Chemistry (США)
J. Phys. Chem. — The Journal of Physical Chemistry (США)
J. Polymer Sci. — Journal of Polymer Science (США)
J. and Proc. Instn Chemists (India) — The Journal and Pro-
ceedings of the Institution of Chemists (Индия)
J. Roy. Inst. Chem. — Journal of the Royal Institute of Che-
mistry (Англия)
J. Soc. Dyers and Colourists — The Journal of the Sosiety
of Dyers and Colourists (Англия)
Kali und Steinsalz (ФРГ)
Kautschuk und Gummi (ФРГ)
Koks, smola, gaz (Польша)
Kolloid-Z. — Kolloid-Zeitschrift (ФРГ)
Kunststoffe (ФРГ)
Liebigs Ann. Chem. — Justus Liebigs Annalen der Chemie (ФРГ)
Makromolek. Chem. — Die makromolekulare Chemie (Швей-
цария)
Mikrochemie (Австрия)
Mikrochim. acta — Mikrochimica acta (Австрия)
Mitt. Chem. Forschungsinst. Wirtsch. Osterr. — Mitteilungen
des Chemischen Forschungsinstitutes der Wirtschaft Oster-
reichs (Австрия)
Mod. Plast. — Modern Plastics (США)
Monatsh. Chem. — Monatshefte filr Chemie (Австрия)
Organ. Chem. Bull. — Organic Chemical Bulletin (США)
Osterr. Chem.-Ztg — Osterreichische Chemiker-Zeitung (Австрия)
Plastics (Англия)
Plastics Ind. — The Plastics Industry (США)
Plastics World (США)
Prakt. Chem. — Praktische Chemie (Австрия)
Proc. Chem. Soc. — Proceedings of the Chemical Society (Анг-
лия)
Rev. Phys. Chem. Japan — The Review of Physical Chemistry
of Japan (Япония)
Rev. synthtsse — Revue de synthese (Франция)
Rubber Chem. and Technol. — Rubber Chemistry and Techno-
logy (США)
Rubber and Plast. Age — The Rubber and Plastics Age (Англия)
Soap and Sanit. Chemicals — Soap and Sanitary Chemicals
(США)
Soc. Analyt. Chem. Bull. — The Society for Analytical Che-
mistry Bulletin (Англия)
Spectrochim. acta — Spectrochimica acta (Англия)
Talanta
Tetrahedron
Trans, and J. Plast. Inst. — Transactions and Journal of the
Plastics Institute (Англия)
Z. analyt. Chem. — Fresenius Zeitschrift fiir analytische Che-
mie (ФРГ)
Z. anorgan. und allgem. Chem. — Zeitschrift filr anorganische
und allgemeine Chemie (ГДР)
Z. Elektrochem. — Zeitschrift filr Elektrochemie. Berichte
der Bunsengesellschaft fiir physikalische Chemie (ФРГ)
Z. Naturforsch. b — Zeitschrift fiir Naturforscliung. Ser. b —
Chemie, Biochemie, Biophysik, Biologie und verw. Gebiete
(ФРГ)
Z. phys. Chem. — Zeitschrift fiir physikalische Chemie (ГДР)
Z. phys. Chem. — Zeitschrift fiir physikalische Chemie (ФРГ)
Z. Vitamin-, Hormon- und Fermentforsch. — Zeitschrift fiir
Vitamin-, Hormon- und Fermentforschung (Австрия)
Gmelin — Gmelins Handbuch der anorganische Chemie
Houben — Weyl, Metoden der Organischen Chemie
Mellor — A Comprehensive treatise on Inorganic and theoreti-
cal chemistry by J. W. Mellor
Pascal — Noveau traite de cliimie minerale. Publ. sous la dir.
de Paul Pascal
Ullmann — Ullmanns Encykloplidie der technischen Chemie
Kirk — Encyclopedia of chemical technology. Ed. Raymond
E. Kirk and Donald F. Othmer
Thorp — Thorpe’s dictionary of applied chemistry
АБИЕТИНОВАЯ КИСЛОТА С20Н30О2, молекуляр-
ный вес 302.44 — основная составная часть канифоли,
из к-рой и получается;
Н«С\ ,СООН т. пл. 174—175°; т. кип.
248—25079,5 мм; [аЩ =
[з Тг I —104,2°(С2Н5ОН,0=10);
легко растворима в эфи-
снз[5 tLch/ch ) ре’ спиРте- бензоле, ме-
''-пз'2 таноле, ацетоне, плохо —
в петролеином эфире и
нерастворима в воде. А. к. легко окисляется; тер-
мически мало устойчива, под действием S и Se, а
также в присутствии Pd на угле дегидрируется в
ретен", вступает в диеновый синтез с малеиновым
ангидридом; сульфируется, нитруется и легко соче-
тается С нитрофенилдиазонием. г- в- Кондратьева.
АБСОЛЮТНАЯ ТЕМПЕРАТУРА — одна из тер-
модинамич. характеристик равновесного состояния,
являющаяся мерой интенсивности теплового движе-
ния. Строгое термодинамич. определение А. т., впер-
вые предложенное У. Томсоном в 1848, основывается
на втором законе термодинамики. Согласно послед-
нему, машина, совершающая обратимый цикл Карво,
может быть использована в качестве термометрия,
устройства, измеряющего отношение А. т. нагревате-
ля 7\ и холодильника Т2, причем Тх/Тг = Qi/Qz, где
Q, — количество теплоты, отнятой у нагревателя, а
Q? — количество теплоты, отданной холодильнику.
Отношение QJQt не зависит от особенностей ра-
бочего тела и температурная шкала, основанная на
приведенном ур-нии, может рассматриваться как
абсолютная шкала. Для определения этой шкалы
достаточно принять одну реперную точку (точку
отсчета) и присвоить ей определенное численное
значение темп-ры, т. е. указать конкретно, чему равна
по этой шкале темп-pa нек-рой достаточно хорошо
воспроизводимой системы. Постановлением десятой
Генеральной конференции по мерам и весам (1954)
в качестве такой точки принята темп-pa тройной
точки воды, т. е. точки фазового равновесия льда,
воды и насыщенного пара над ними (обладающего
при этой темп-ре давлением 4,579 мм рт. ст.). Этой
темп-рной точке присвоено значение 273,16°, а соот-
ветствующая шкала названа абсолютной термодина-
мич. темп-рной шкалой (ГОСТ 8550—57, «Тепловые
единицы»), В честь Томсона (лорда Кельвина) градус
этой шкалы называют градусом Кельвина и обозна-
чают °К. Величины градуса этой шкалы и стоградус-
ной международной темп-рной шкалы практически
одинаковы. При сопоставлении стоградусной междуна-
родной темп-рной шкалы с абсолютной термодинамич.
шкалой необходимо учесть, однако, что в качестве
нуля стоградусной шкалы принята темп-pa равно-
весия льда с водой, насыщенной воздухом, при дав-
лении, равном 1 атм. Эта темп-pa ниже темп-ры трой-
ной точки воды на 0,01°, т. к. растворение воздуха
в воде и повышение давления с 4,579 мм рт. ст.
до 760 мм вызывают понижение темп-ры равновесия
между водой и льдом. Таким образом, темп-pa 0°С
равна 273,15°К, или иначе: 0°К = —273,15°С. По-
этому связь между темп-рой по стоградусной шкале
(г) и А. т. (Т) дается формулой Т = 273,15° + г. Среди
используемых на практике термометрии, устройств
газовые термометры имеют шкалу, наиболее близко
отвечающую абсолютной. Вполне точное равенство
газовой и А. т. имеет место в тех частях шкалы, где
газ можно считать идеальным. Наилучшее ее при-
ближение в этом смысле дают азотный и водородный
термометры. Однако при низких температурах и
эти газы заметно отличаются от идеальных, так что
в показания термометров необходимо вводить по-
правки.
Статистич. смысл А. т. и ее связь с тепловым дви-
жением наиболее просто раскрываются на примере
идеальных газов, у к-рых, согласно Л. Больцману,
энергия поступательного движения молекул в равно-
весном состоянии равна:
£=42^=4^
i
где m — масса, a — скорость 4-й молекулы, R —
газовая постоянная. Более точное истолкование А. т.
получила в статистич. работах Гиббса.
Лит.: Гиббс Дж. В., Основные принципы статистиче-
ской механики..., пер. с англ., М.—Л., 1946; Самойло-
вич А. Г., Термодинамика и статистическая физика,
М., 1953; Путилов К. А., Курс физики, т. 1, 9 изд.,
М., 1959.
АБСОЛЮТНЫЙ ГЕОЛОГИЧЕСКИЙ ВОЗРАСТ —
см. Возраст геологический абсолютный.
АБСОРБЦИОННАЯ СПЕКТРОСКОПИЯ — мето-
ды анализа, основанные на измерении поглощения
излучения определенной длины волны (или длин волн).
Законы поглощения излучения однородных прозрач-
ных (не рассеивающих) жидкостей (растворов), газов
и твердых веществ установлены экспериментально
(см. Поглощение света); они показывают соотношение
между величиной поглощения и количеством или
концентрацией поглощающего вещества. В А. с. из-
мерения всегда производят относительно нек-рого
стандарта. А. с. в видимой и УФ, а также в ИК
областях спектра применяется для качественного
и колич. определения химич. соединений в различных
природных и промышленных объектах, установления
степени чистоты в-ва и решения других вопросов.
Возможности абсорбционного спектрального анализа
чрезвычайно велики, и этот метод получил значительно
более широкое распространение, чем эмиссионный
спектральный анализ. В аналитич. практике приме-
няется А. с. в видимой, УФ и ИК областях спектра.
В разных случаях применяемая аппаратура может
сильно изменяться. Чувствительность различных ме-
19
АБСОРБЦИЯ
20
тодов А. с. зависит от многих причин и может изме-
няться в широких пределах. См. также Спектрофото-
метрия, Колориметрия.
Лит.: Абсорбционная спектроскопия. Сб. статей, пер.
с англ., под ред. Э. В. Шпольского, М., 1953; Дункан А.
[и др.]> Применение спектроскопии в химии, пер. с англ.,
М., 1959. А. И. Бусев.
АБСОРБЦИЯ — поглощение веществ из газа
(обычно из газовой смеси) или жидкости жидкостями
или—реже — твердыми телами. Жидкость или твердое
тело, поглощающие газ или растворенное вещество,
наз. абсорбентами. А. — один из случаев
сорбции. В отличие от адсорбции, при А. поглощение
веществ происходит во всем объеме поглотителя.
Поглощение вещества может быть осложнено химич.
взаимодействием поглощаемого вещества с поглоти-
телем (см. Хемосорбция).
Технически наиболее важным видом А. является
поглощение газов жидкостями. К А. относятся так-
же извлечение растворенных в данной жидкости ве-
ществ другой жидкостью, не смешивающейся с раство-
рителем, напр. в случае воды — бензолом или эфиром
(экстракция, см. Экстрагирование). А. газов метал-
лами (напр., водорода палладием), особенно при по-
вышенных темп-pax, с образованием твердых р-ров
или отчасти химич. соединений наз. окклюзией,
А. применяют в пром-сти с целью: разделения
газовых смесей с помощью селективных поглотителей
(выделение компонентов из р-ра и получение его в
чистом виде путем десорбции, после чего поглотитель
повторно используют, напр. для А. бутадиена в произ-ве
синтетич. каучука, бензола в коксохимии, произ-ве
и др.); очистки газов от вредных примесей (H2S,
SO2, СО2, СО и др.); получения готового продукта
(напр., серной к-ты посредством A. SO3, соляной
к-ты — А. газообразного НС1). Большое значение
имеет извлечение углеводородвых газов (природных
и искусственных) из их смесей (напр., т. н. газового
бензина, ггзоз крекинга и пиролиза), а также выде-
ление индивидуальных углеводородов (от пропана
до изопентана). В этом случае в качестве абсорбентов
применяют стабильные вещества с малым мол. весом,
низкой вязкостью и малой летучестью (керосин,
газойль, вазелиновое, соляровое и веретенное масла);
см. также Газы нефтяные попутные.
При рассмотрении физич. сущности А. надо различать
равновесие А., определяемое растворимостью поглощаемого
вещества в абсорбенте (напр., в жидкости) при данных усло-
виях, и кинетику А., т. е. протекание А. во времени. Равно-
мерное распределение абсорбируемого вещества по всему
объему абсорбента обеспечивается диффузией, скорость к-рой
определяет скорость А. Поэтому А. твердыми абсорбентами
весьма замедлена. При соприкосновении жидкой и газовой
фаз у поверхности раздела образуются жидкостная и газовая
пленки. Растворимый в жидкости компонент газообразной
смеси проникает во внутренние слои абсорбента благодаря
диффузии сначала сквозь газовую пленку, а затем сквозь
жидкостную. Для осуществления диффузии необходимо, чтобы
концентрация растворенного компонента в газообразной смеси
превосходила его концентрацию над жидкостью.
Количество поглощенного вещества в единицу времени G
(кг-мол/час) определяется: для передачи вещества от газовой
фазы к поверхности: G = kiS(p — р0) (1), а для передачи ве-
щества от поверхности в жидкую фазу: G = k2S(C0 — С) (2),
где S — площадь в м2, р — парциальное давление погло-
щаемого компонента в газовой смеси в ат, р0 — равновесное
давление поглощенного газа над жидкостью у поверхности
соприкосновения в ат, Со — равновесная концентрация рас-
творенного газа в жидкости у поверхности соприкосновения
в кг-мол/м3, С—концентрация растворенного газа в жидкости
в кг-мол/м3, ki и k2 — частные коэфф. А. (коэфф, массопере-
дачи), имеющие размерность кг-мол/м2-час-ат (для kt) и
м/час (для k2).
Равновесные значения р0 и Со по закону Генри (см. Рас-
творимость) связаны соотношением: Ро == тС0 (3), где т —
коэфф. Генри в м3-ат/кг-мол. Из выражений (1), (2) и (3):
G = Kr-S(p — р*) (4), где р* — равновесное давление
поглощенного газа над жидкостью концентрацией С в ат,
КГ — коэфф. А. (массопередачи), определяемый соотношением; |
1 1m
(5), имеющий размерность кг-мол/м2-час-ат.
Частные коэфф. А. зависят от условий соприкосновения газа и
жидкости, от размеров аппарата и его основных деталей, от
гидродинамич. условий течения газа и жидкости, а также от
физико-химич. свойств взаимодействующих в-в. Эти факторы
обычно учитывают при помощи критериев подобия (см. Подобия
теория), причем в об-
щем виде зависимость
выр аж ается ур-нием:
•W = /(Re, Fr, Pr')
(6), где Nu' и Pr' —
диффузионные кри-
терии Нуссельта и
Прандтля, Re — кри-
терий Рейнольдса,
Fr — критерий Фру-
да. Конкретный вид
зависимости (6) уста-
навливается опытным
путем для каждого
типа аппарата.
Материальный ба-
ланс абсорбции выра-
жается уравнением:
где V — количество
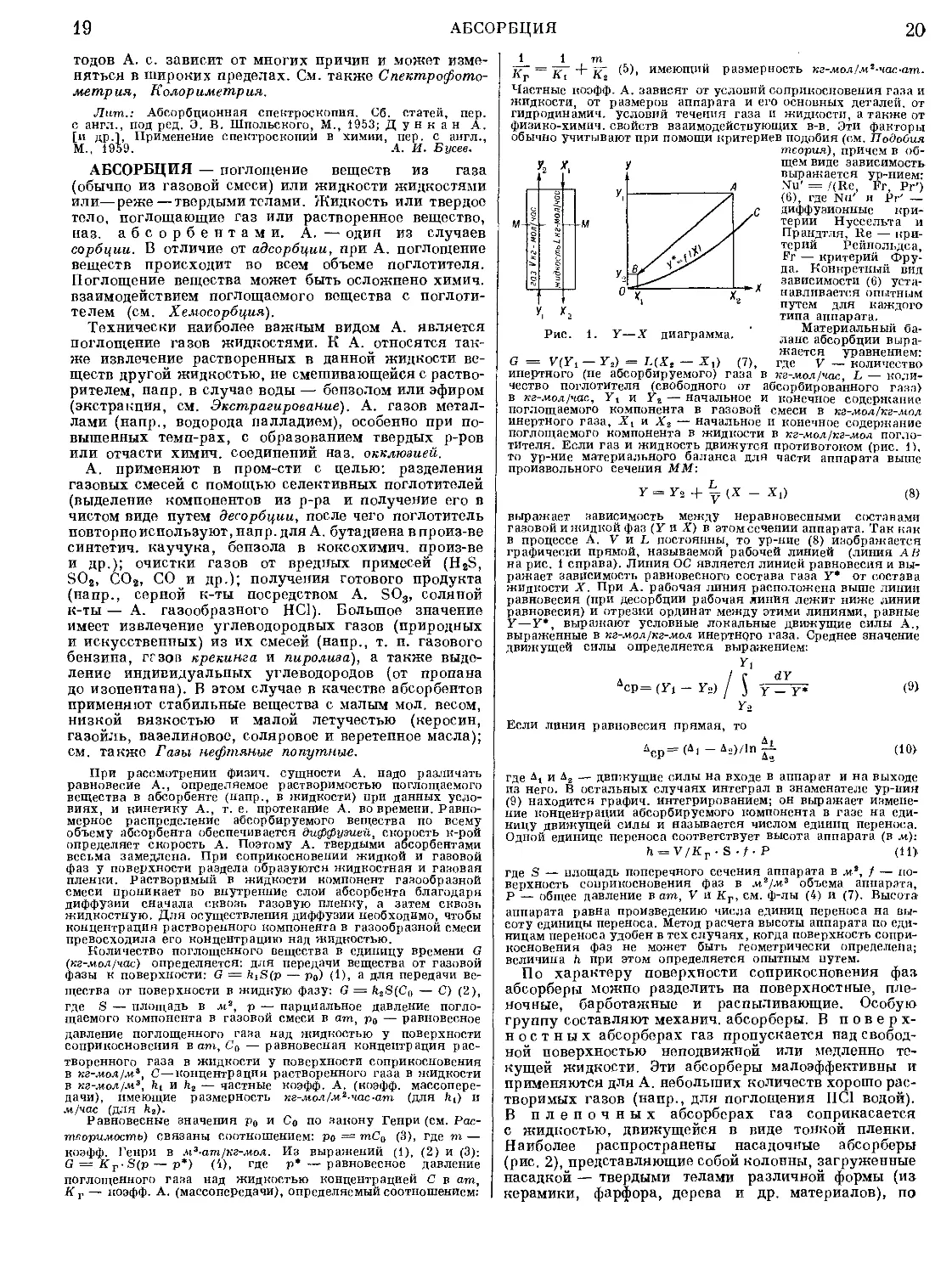
1. Y—X диаграмма.
G = V(Yt — Y2) = L(X2 — Хх) (7),
инертного (не абсорбируемого) газа в . . _
чество поглотителя (свободного от абсорбированного газа)
в кг-мол/час, Yt и У2 — начальное и конечное содержание
поглощаемого компонента в газовой смеси в кг-мол/кг-мол
инертного газа, и Х2 — начальное и конечное содержание
поглощаемого компонента в жидкости в кг-мол/кг-мол погло-
тителя. Если газ и жидкость движутся противотоком (рис. 1),
то ур-ние материального баланса для части аппарата выше
произвольного сечения ММ:
Y = ~ (X - Xi)
(8)
выражает зависимость между неравновесными составами
газовой и жидкой фаз (У и X) в этом сечении аппарата. Так как
в процессе А. V и L постоянны, то ур-ние (8) изображается
графически прямой, называемой рабочей линией (линия АВ
на рис. 1 справа). Линия ОС является линией равновесия и вы-
ражает зависимость равновесного состава газа У* от состава
жидкости X. При А. рабочая линия расположена выше линии
равновесия (при десорбции рабочая линия лежит ниже линии
равновесия) и отрезки ординат между этими линиями, равные
У—У*, выражают условные локальные движущие силы А.,
выраженные в кг-мол/кг-мол инертного газа. Среднее значение
движущей силы определяется выражением:
п
л / с dY
Дср= (У, - У.) / ) у _ у. (9)
Уз
Если линия равновесия прямая, то
д ।
АСР= (Д1 — Д2>/1П if <10>
где и Д2 — движущие силы на входе в аппарат и на выходе
из него. В остальных случаях интеграл в знаменателе ур-пия
(9) находится графич. интегрированием; он выражает измене-
ние концентрации абсорбируемого компонента в газе на еди-
ницу движущей силы и называется числом единиц переноса.
Одной единице переноса соответствует высота аппарата (в л*):
n = V/Kr-S-/-P (11>
где S — площадь поперечного сечения аппарата в л**, / — по-
верхность соприкосновения фаз в м2/м3 объема аппарата,
Р — общее давление в am, V и КГ, см. ф-лы (4) и (7). Высота
аппарата равна произведению числа единиц переноса на вы-
соту единицы переноса. Метод расчета высоты аппарата по еди-
ницам переноса удобен в тех случаях, когда поверхность сопри-
косновения фаз не может быть геометрически определена;
величина h при этом определяется опытным путем.
По характеру поверхности соприкосновения фаз
абсорберы можно разделить на поверхностные, пле-
ночные, барботажные и распиливающие. Особую
группу составляют механич. абсорберы. В поверх-
ностных абсорберах газ пропускается над свобод-
ной поверхностью неподвижной или медленно те-
кущей жидкости. Эти абсорберы малоэффективны и
применяются для А. небольших количеств хорошо рас-
творимых газов (напр., для поглощения НС1 водой).
В пленочных абсорберах газ соприкасается
с жидкостью, движущейся в виде тонкой пленки.
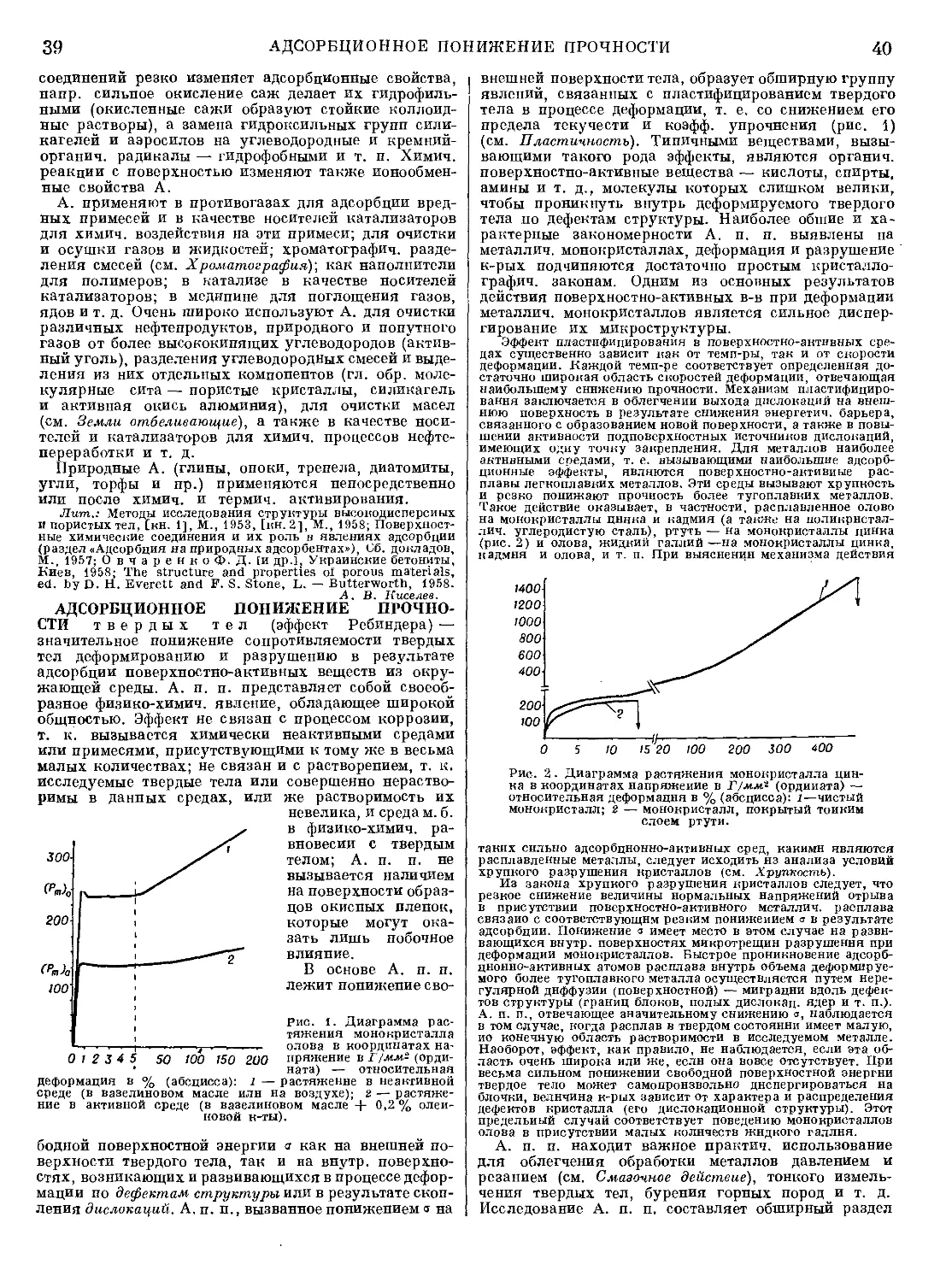
Наиболее распространены насадочные абсорберы
(рис. 2), представляющие собой колонны, загруженные
насадкой — твердыми телами различной формы (из
керамики, фарфора, дерева и др. материалов), по
21
АБСОРБЦИЯ — АБСОРБЦИЯ МАСЛЯНАЯ
22
Рис. 2. Насадочный абсор-
бер: 1 — насадка; 2 — ре-
шетка; 3 — распылитель.
поверхности к-рых стекает жидкость (см. Насадки).
Для равномерного распределения орошающей жидко-
сти по насадке применяют различные оросительные
устройства. Насадочные ко-
лонны отличаются просто-
той устройства, что особен-
но ценно при работе с агрес-
сивными средами, и сравни-
тельно небольшим гидрав-
лич. сопротивлением. Дру-
гой тип пленочного абсор-
бера — трубчатый аппарат,
состоит из вертикальных
труб, по к-рым проходит газ
и по внутр, поверхности
трубок стекает жидкость;
отвод тепла осуществляется
охлаждением труб снару-
жи. В барботажных
абсорберах газ распреде-
ляется в жидкости в виде
пузырьков и струй, на по-
верхности к-рых происходит
А. Наиболее распростране-
ны абсорберы в виде колонн
с колпачковыми (рис. 3) и ситчатыми (рис. 4) тарел-
ками. Колпачковые тарелки снабжены патрубками,
закрытыми сверху колпачками. Газ проходит через
слой жидкости, уровень к-рой на тарёлке поддержи-
вается переливными трубами. Ситчатые тарелки
имеют отверстия или щели, через к-рые проходит
газ. Жидкость на тарелке поддерживается снизу
давлением газа и перетекает по переливным трубам
или (в провальных тарелках) жидкость проходит
через те же отверстия, что и газ. В барботажных
аппаратах может быть осуществлен отвод тепла
путем установки на тарелках охлаждающих змее-
виков или выполнением тарелок в виде системы
труб, по к-рым протекает охлаждающий агент (труб-
чато-решетчатые тарелки). Барботажные абсорберы
работают интенсивно, но сложны по конструкции
и имеют довольно высокое гидравлич. сопротивление.
Рис. 3. Колонна с кол-
пачковыми тарелками:
1 —тарелки; 2— патруб-
ки; 3 — колпачки; 4 — пе-
реливные трубы.
ситчатыми тарелка-
ми: 1 — Отверстия;
2 — переливные тру-
бы.
К барботажным абсорберам относятся также наса-
дочные колонны с затопленной насадкой (наз. иногда
эмульгационными колоннами), в которых жидкость
выводится через подпорную трубу (как показано
пунктиром на рис. 2); благодаря этому жидкость
заполняет всю насадку и через нее барботирует газ.
В распыливающих абсорберах жидкость рас-
пределяется в массе газа в виде мелких капель.
Обычно эти абсорберы выполняются в виде полых
колонн, в к-рых распыление жидкости производится
сверху, а газ движется снизу вверх Распыление
жидкости производится различного типа форсунка-
ми, иногда посредством центробежных распылителей
(быстро вращающиеся
турбинки или диски).
Распыливающие аб-
сорберы применяются
гл. обр. для погло-
щения хорошо раство-
римых газов; такие
абсорберы отличают-
ся простотой и низ
ким гидравлическим
сопротивлением; они
могут работать на
сильно загрязненных
газах. Для создания
большей поверхности
соприкосновения не-
обходимо подавать
большие количества
жидкости и распылять
ее на мелкие капли.
Рис. 5. Абсорбер с трубой Вен-
тури: 1 — горло трубы; 2 — диф-
фузор; 3 — сепаратор.
Скорость газа при этом ограничена, т. к. с повыше-
нием ее возрастает унос жидкости в виде брызг.
Распыливающим абсорбером, работающим при вы-
соких скоростях газа (30—60 м)сек), является аппарат
с трубой Вентури (рис. 5). В этом аппарате жидкость
распыляется газовым потоком, проходящим с боль-
шой скоростью через горло трубы. В расширяющейся
части (диффузоре) происходит укрупнение капель,
к-рые отделяются затем от газа в сепараторе. Аппараты
Вентури работают весьма интенсивно, но обладают
довольно большим гидравлич. сопротивлением; в них
осуществим только прямоток. В механических
абсорберах развитие поверхности соприкосновения
достигается действием вращающихся деталей. Ме-
ханич. абсорберы обычно выполняются либо в виде
сосуда с мешалкой, в котором осуществляется барбо-
таж газа через слой перемешиваемой жидкости,
либо с распылением жидкости в газ вращающимися
деталями.
Лит.: Р а м м В. М., Абсорбционные процессы в химиче-
ской промышленности, М.—Л., 1951; Sherwood Т. К.,
Pigford R. L., Absorption and extraction, 2 ed., N. Y. —
L.—Toronto, 1952; Касаткин А. Г., Основные про-
цессы и аппараты химической технологии, 6 изд., М., 1955;
Циборовский Я., Процессы химической технологии,
иер. с польск., Л., 1958; Р а м м В. М., Закгейм А. Ю.,
Хим. наука и пром-сть, 1958, Мб; Поз и н М. Е., Пути ин-
тенсификации взаимодействий жидкостей с газами, в кн.:
Методы и процессы химической технологии. Сб. 1, М.—Л.,
1955; Chemical Engineers' Handbook, ed. John H. Perry, 3 ed.,
N. Y. — L. — Toronto, 1953; Morris G. A., Jack-
son J Absorption towers, L., 1953; Kirk, v. 1, N. Y.,
1947, p. 14. В. M. Рамм.
АБСОРБЦИЯ МАСЛЯНАЯ — процесс растворения
одного или нескольких компонентов газовой или
парогазовой смеси поглотительным маслом в различ-
ного типа аппаратах (см. Абсорбция). Для успешного
проведения процесса парциальное давление паров
поглощаемого компонента должно превышать давле-
ние его паров над поглотительным маслом. А. м.
способствуют: 1) повышенная концентрация погло-
щаемого компонента в газовой смеси, а также повы-
шение давления смеси; 2) пониженная концентра-
ция поглощаемого компонента и пониженное
давление его паров (за счет понижения температуры
процесса) в поглотительном масле; 3) достаточно
развитая поверхность соприкосновения между га-
зовой смесью и поглотительным маслом, движение
газовой смеси и поглотительного масла по принципу
противотока и турбулентность движения их потоков;
4) невысокий мол. вес поглотительного масла.
А. м. осуществляют: в аппаратах насадочного типа
(скрубберы), абсорберах различного типа (барботаж-
23
АВОГАДРО ЗАКОН — АВТОКЛАВЫ ЛАБОРАТОРНЫЕ
24
пых, центробежных, ступенчатых, полых, с вращаю-
щейся насадкой или вращающимися тарелками,
в виде трубы Вентури и др.), абсорбционных аппа-
ратах, в к-рых достигается высокая турбулентность
потоков жидкости и газа (колонны с «провальными»
тарелками, «пенными» аппаратами и др.). В качестве
поглотительных масел применяют, как правило,
кам.-уг. масло (продукт переработки каменноуголь-
ной смолы) или соляровое масло, получаемое при
переработке нефти.
А. м. широко применяется в коксохимия, пром-сти
для извлечения сырого бензола из газа коксового,
в нефтеперерабатывающей пром-сти, искусственного
жидкого топлива и др.
Лит.: Коляндр Л. Я., Улавливание и переработка
химических продуктов коксования, Харьков — М., 1953.
А. И. Бродович.
АВОГАДРО ЗАКОН — один из основных газовых
законов, состоящий в том, что при одинаковых темп-
ре и давлении равные объемы всех газов содержат
одно и то же число молекул. А. з. высказан в виде
гипотезы в 1811 итал. физиком А. Авогадро (и неза-
висимо от него, но в менее ясной форме, в 1814 франц,
ученым А. М. Ампером). Однако вследствие господ-
ствовавшего в науке 1-й половины 19 в. смешения
понятий атома, эквивалента и молекулы А. з. был
предан забвению и только с 1860 стал широко
применяться в физике и химии. Из А. з. вытекают
следствия: 1) молекулярный вес М газа или пара
равен произведению его плотности D по отношению
к водороду на мол. вес водорода, т. е. М = 2,016 D-,
2) грамм-молекула любого газа при нормальных
условиях (0° и 760 мм рт. ст.) занимает объем
22,416 л. А. з. строго приложим только к идеальным
газам; все реальные газы отклоняются от А. з. в той
же мере, как они отклоняются от законов Бойля —
Мариотта и Гей-Люссака. с. а. Погодин
АВОГАДРО ЧИСЛО — число молекул в грамм-
молекуле любого индивидуального в-ва или число
атомов в грамм-атоме любого элемента. А. ч. обозна-
чается через N и равно (6,02486 ± 0,'00016) • 1023
молекул на 1 г-мол (физич. шкала) или (6,02322 ±
± 0,00016) • 1023 молекул на 1 г-мол (химия, шкала),
если отношение единиц физич. и химия, шкал атом-
ных весов считать равным 1,000272. А. я. нвляется
одной из основных физич. постоянных.
АВТОКЛАВЫ ЛАБОРАТОРНЫЕ — аппараты
для проведения физико-химич. исследований в лабо-
раториях под давлением в замкну-
той системе (см. Высокие давления
и Высоких давлений техника). А. л.
представляют собой цилиндрич. тол-
стостенные металлич. сосуды, вну-
тренняя полость к-рых выполнена в
форме бутылки, стакана или пробир-
ки. Горло А. л. закрывается крыш-
кой, а герметизация дости-
гается затвором с помощью
конусного уплотнения или
уплотняющих колец (обтю-
раторов), изготовленных из
цветных металлов (медь, алюминий
и др.), пластмасс, резины, асбеста
и др. А. л. изготовляют большей
Рис. 1. Автоклав с ножевым уплотнением:
1 — манометр; 2 — головка; 3 — вентиль;
4 — крышка; 5 — ножевое уплотнение;
6 — сифонная трубка; 7 — карман для
термопары.
частью из специальных сталей, гл. обр. нержа-
веющих. Для предохранения стенок от коррозии
и контакта с реагирующими в-вами А. л. иногда
внутри покрывают эмалью, различными металлами,
органич. пластиками или др. коррозионноустойчивыми
покрытиями. Иногда в А. л. вставляют сосуд (лай-
нер) из стекла или пластмассы.
Различают след. А. л.: без перемешивания (рис. 1),
вращающиеся (рис. 2), с мешалкой, герметизирован-
ной сальником (рис. 3), с экранированным двигате-
лем (рис. 4), качающиеся (рис. 5) и др. А. л. снаб-
Рис. 2. Вращающийся автоклав с электрообогревом:
1 — манометр; 2 — головка; з — карман для термопары;
4 — электропечь; 5 — корпус; 6 — редуктор; 7 — мотор.
жают манометром, карманом для термопары или тер-
мометра, вентилями для введения и отбора веществ.
А. л. нагревают предпочтительно с помощью съем-
ных электропечей и часто
присоединяют к приборам,
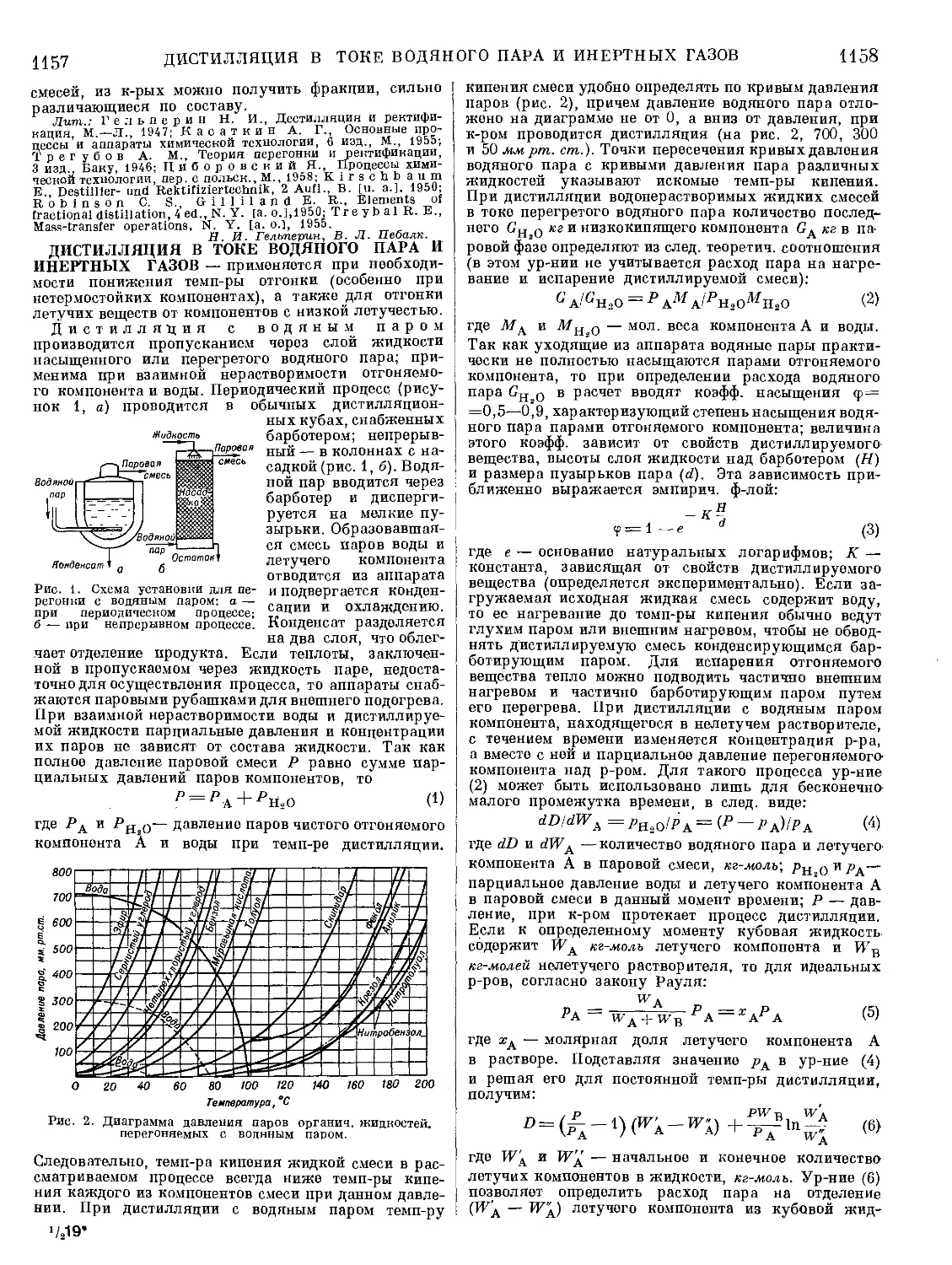
показывающим, записываю-
щим и регулирующим тем-
пературу, давление, состав
и др. параметры.
Рис. 4. Крышка авто-
клава с экранированным
двигателем: 1 — статор;
2 — ротор; 3 — гермети-
зирующая гильза; 4 —
кожух масляной бани;
5 — вал мешалки; в —
крышка автоклава.
Рис. 3. Автоклав с ме-
шалкой, герметизированной
сальником: 1 — шкив; 2 —
манометр; з— сальник; 4 —
масленка; 5 — головка; 6 —
вентили; 7 — крышка-, 8 —
карман для термопары; 9 —
мешалка.
А. л. широко применяют для каталитич. гидриро-
вания непредельных и ароматич. соединений, а также
для проведения реакций, ускоряемых давлением
(полимеризация, конденсация, окисление, гидрата-
ция и пр.) при самых разнообразных темп-pax и
давлениях (б. ч. до 300° и до 1 000 ат). Для безо-
пасности обслуживания А. л. устанавливают в спе-
циальные сейфы (кабины), изготовленные из сталь-
ных плит, бетона или кирпича (при работе с неболь-
25
АВТОЛЫ — АДАМАНТАН
26
шими А. л. и малыми давлениями можно ограни-
читься стальными щитами). Сейфы сверху должны
иметь лабиринтное перекрытие с целью задержать
Рис. 5. Установка с качающимся автоклавом: 1 — вен-
тиль; 2 — автоклав (пробирка); 3 — электропечь; 4 —
качающееся приспособление; 5 — манометр; в — карман
для термопары; i •— термопара.
в случае аварии осколки аппаратуры (при свободном
выходе газов). К работе с А. л. допускается только
обученный персонал, знакомый с аппаратурой и пра-
вилами техники безопасности.
Лит.: Ц и к л и с Д. С., Техника физико-химических ис-
следований при высоких давлениях, 2 изд., М., 1958; К о р н-
д о р ф Б. А., Техника высоких давлений в химии, Л. —М.,
1952; Болотов Б. А., Комаров В. А., Низов-
кина Т. В., Практические работы по органическому
катализу, Л., 1959; Чиркин С. И., Техника безопасности
при эксплуатации промышленных паровых котлов, сосудов,
работающих под давлением, компрессоров и трубопроводов,
М., 1955; Technique of organic chemistry, Ed. A. Weissberger,
2 ed., V. 2, N. Y„ 1956, p. 65—78. M. И. Роаенгарт.
АВТОЛЫ — моторные масла, применяемые для
смазки автомобильных, тракторных и мотоциклет-
ных двигателей с искровым зажиганием (карбюра-
торных). В СССР А. вырабатываются гл. обр. из
бакинских нефтей и сернистых нефтей восточных
районов. Будучи различными по своей химич. при-
роде и методам произ-ва, эти масла различаются
и по своим эксплуатационным качествам. А. из
восточных нефтей имеют высокий индекс вязкости
(см. в статье Вязкость), хорошие противоизносные
и антикоррозионные свойства. Для А. нормирует-
ся также отношение вязкости при 50 и 100°, а
для некоторых марок и коэффициент вязкости.
В зависимости от условий применения А. должны
иметь различную т. заст. (от —5 до —40°) и вяз-
кость (от 5 до 15 с ст при 100°). Для существующих,
а тем более перспективных форсированных двигате-
лей моторные масла без присадок (см. Присадки
к маслам) неприменимы.
Обычно А. получают кислотно-контактной и селек-
тивной очисткой масляных дистиллятов и концен-
тратов (остатков) нефтей. Для зимних масел исполь-
зуют дистилляты, для летних, более вязких, — ди-
стилляты или смесь дистиллятов с остаточными
маслами. Масла, получаемые из парафинистых неф-
тей, подвергают депарафинизации.
А. классифицируют по вязкости и способу очистки.
Выпускаются 5 марок А. сернокислотной очистки:
АК-6 (автол-6), АК-10 (автол-10), АК-15 (автол-15),
АКЗп-6 и АКЗп-10 (цифры 6, 10 и 15 показы-
вают предельную вязкость масла в сантистоксах при
100°; индекс «п» указывает на наличие присадки).
Автол-6 применяется для смазки автомобильных
двигателей в зимнее время; автол-10 — для смазки
автомобильных двигателей летом и тракторных — зи-
мой; автол-18 — для тракторных двигателей летом.
А. АКЗп-10 и АКЗп-6 представляют собой
маловязкие масла, загущенные полиизобутиленом
и содержащие присадку. Первый предназначен для
смазки автомобильных двигателей в условиях север-
ных р-нов зимой, второй является универсальным
автомобильным маслом на весь год. А. селективной
очистки выпускаются двух марок: АС-5 и АС-9,5,
применяются для смазки автомобильных двигателей
в зимний и летний периоды соответственно. Кроме
того, выпускаются масла селективной очистки с при-
садками. К А. относятся также специальные авто-
мобильные масла с присадками — «летнее» и «зимнее»,
предназначающиеся для смазки форсированных дви-
гателей типа ЗИЛ-НО, ЗИЛ-111, «Чайка» и др.
Лит.: Лосиков Б. В., Пучков Н. Г., Эн-
глин В. А., Основы применения нефтепродуктов, М., 1950;
Технические нормы на нефтепродукты (Справочная книга),
под ред. Н. Г. Пучкова, 16 изд., М., 1957.
Л. С. Поваров.
АГАР (агар-агар) — продукт, получаемый из не-
которых морских водорослей (агарофитов), харак-
терным свойством к-рого является способность давать
плотные гели. А. неоднороден; содержит 70—80%
полисахаридов, 10—20% воды, 1,5—4% минеральных
в-в. В составе полисахаридов обнаружены: D- и
Ъ-галактозы, 3,6-ангидрогалактоза, пентозы, D-глю-
куроновая кислота и пировиноградная кислота’, химич.
строение полисахаридов изучено мало. Получены две
фракции полисахаридов: агароза и амило-
пектин. Агароза — линейный полисахарид, обра-
зованный из чередующихся остатков P-D-галакто-
пиранозы и 3,6-ангидро-а-Б-галактопиранозы; об-
ладает ярко выраженной способностью к образованию
гелей. Амилопектин является, по-видимому, смесью
полисахаридов очень сложного строения, в состав
к-рых входит глюкуроновая к-та и эфирно-связанная
серная к-та, А. применяется в пищевой, особенно
в кондитерской пром-сти, в микробиологии при при-
готовлении плотных сред для выращивания микро-
организмов.
Лит.: А г a k 1 С., Proceedings 4th International congress
of biochemistry, v. 1, L., 1959, p. 15; W h 1 s 11 e r R. L.
and S m a r t C. L., Polysaccharide chemistry, N. Y., 1953.
Л. И. Линевич.
АГЛЮКОН — устаревшее назв. агликона, см. Гли-
козиды.
АДАЛИН (карбромал, бромдиэтилацетилмочевина)
(C2H5)2CBrCONHCONH2, мол. в. 237,11 — бесцвет-
ные мелкие кристаллы с слабым запахом; т. пл.
116—118°; растворяется в ацетоне, бензоле и спир-
те, мало — в воде и петролейном эфире. При на-
гревании А. со щелочью отщепляется НВг и HCN
с одновременным образованием этилкротонилмоче-
вины и а-диэтилгидантоина. Получают А. конденса-
цией галогенангидрида бромдиэтилуксусной к-ты
с мочевиной. Качеств, определение А. основано на
разложении его кипячением со щелочью, окислении
продукта реакции хлорной водой или хлорамином и
подкислении; хлороформенная вытяжка окрашивается
в желто-бурый цвет. Для колич. определения А. на-
гревают с КОН, прибавляют разбавленные HNO3
и AgNO3 и титруют избыток NO2 0,1 н. р-ром NH4SCN
с железо-аммониевыми квасцами. А. применяют в ме-
дицине как успокаивающее и снотворное средство.
Лит.: Государственная фармакопея СССР, 8 изд., М.,
1952; Машковский М. Д., Лекарственные средства,
4 изд., М„ 1960. - Е. С. Головчинская.
АДАМАНТАН (диамантан, трициклодекан) С10Н1в,
мол. в. 136,24 — бесцветные октаэдрич. кристаллы
с легким камфарным запахом; содержит систему
атомов углерода, подобную алмазу; т. пл. 269°,
возгоняется; d 1,07; nDl,568; хорошо растворяется
в ароматич. углеводородах; теплота сгорания
1451,7 ккал/моль. Частоты в ИК-спектре (в КВг):
27
АДАМКЕВИЧА РЕАКЦИЯ — АДГЕЗИЯ
28
440, 476, 799, 966, 1101, 1155, 1357, 1453, 2857, 2907
(2933) слГ1. Горячая HNO3, хромовая к-та и щелоч-
ной раствор КМпО4
Н2С—СН—СН2 не действуют на А.;
конц. II2SO4 вызыва-
ет обугливание. При
действии брома при
100° образует бром-
адамантан, с иодом
А. был обнаружен в мо-
синтетич. метод его полу-
НС—|
нгс — СН—снг
при 150° —иодадамантан.
равских нефтях. Известен
чения.
Лит.: Mecke R. und Spiesecke Н., Chem.
Ber., 1955, 88, № 12, S. 1997. Л. С. Поваров.
АДАМКЕВИЧА РЕАКЦИЯ (реакция Адамкевича —
Гопкинса) — цветная реакция на белок. При добав-
лении к р-ру белка в глиоксиловой к-те конц. серной
к-ты на границе соприкосновения жидкостей появ-
ляется темное фиолетовое кольцо. А. р. обусловлена
ннличием в белках триптофана, образующего с глиок-
силовой к-той окрашенные комплексы. Эту реакцию,
кроме триптофана, дают многие производные индола.
Чувствительность А. р. 1 : 200 000, в присутствии
солей меди чувствительность реакции возрастает
до 1:1000 000. Перекись водорода подавляет реак-
цию. А. р. открыта в 1874 Адамкевичем, химизм
реакции изучен Гопкинсом.
Лит.: Блок Р. и Б о л л и н г Д., Аминокислотный
состав белков и пищевых продуктов, пер. с англ., М., 1949.
। О. В. Пунина.
АДАМСА КАТАЛИЗАТОР — один из наиболее
активных катализаторов для реакций гидрогенизации
и восстановления различных органич. соединений
при невысоких темп-pax и давлениях, гл. обр.
в жидкой фазе, а также в реакциях изотопного обмена
дейтерия с водородом в органич. соединениях. А. к.
состоит из мелкораздробленной окиси платины,
приготовляемой нагреванием платинохлористоводо-
родной к-ты или хлороплатината аммония с нитратом
натрия с последующей экстракцией неразложившихся
солей водой и тщательным промыванием образовав-
шейся коричневой PtO2. Активность катализатора
существенно зависит от темп-ры сплавления (опти-
мальная 500—525°). В ходе реакции катализатор
восстанавливается до платиновой черни; он может
быть восстановлен водородом или другим реагентом
до начала реакции, однако в нек-рых случаях полное
восстановление приводит к уменьшению активности
катализатора и его регенерируют воздухом.
Лит.: Эллис К., Гидрогенизация органических соеди-
нений, пер. с англ., вып. 1, Л., 1934; Technique of organic che-
mistry, v. 2, 2 ed., N. Y. — L., 1956; Каталитические фотохи-
мические и электролитические реакции, пер. с англ., М., 1960.
С. Л. Питерман.
АДАМСИТ (дигидрофенарсазинхлорид) Cl2H9NAsCl,
молекулярный вес 277,58 — кристаллы желтого цве-
та (технический продукт — темно-зе-
леный), т. пл. 195°, т. кип. 410°;
нерастворим в воде, плохо раство-
рим в большинстве органич. раство-
рителей. Получают А. из дифенил-
амина и AsCl3 или из хлоргидрата
дифениламина и As2O3:
(C6H5)3NH + AsC13=HN(CeH4)3AsCl + 2НС1;
2(CeH5)-.NH • HC1 + As3O3=2HN(C(iH4) ..AsCl + 3H2O.
А. относится к группе в-в, раздражающих верхние
дыхательные пути; был предложен в конце первой
мировой войны в качестве ОБ — ядовитого дымо-
образователя; практич. применения не нашел.
Р. Н. Стерлин.
АДГЕЗИЯ (прилипание) — молекулярная связь
между поверхностями двух соприкасающихся разно-
родных твердых или жидких тел (фаз). А. измеряется
работой разрыва или сопротивлением разрыву на
единицу площади контакта при данном виде дефор-
мации (отрыв, скалывание). Если соприкасающиеся
тела одинаковы, А. при полном их соприкосновении
переходит в когезию (сцепление), характеризующую
прочность этих тел — силу сцепления молекул, ионов,
атомов вещества в данном теле. Сравнение величин
когезии и А. дает наглядное представление о соотно-
шении сил притяжения между молекулами одного
вещества (жидкого или твердого тела) и молекулами
двух разнородных жидких или твердых тел. А. твер-
дых тел обычно мала вследствие того, что истинная
площадь контакта из-за неровностей поверхности
составляет весьма малую долю от кажущейся пло-
щади соприкосновения. Только на границах раздела
двух жидкостей или твердого тела и жидкости вслед-
ствие полного контакта по всей площади соприкосно-
вения А. достигает предельно высокого значения.
Предельная А. наблюдается при горячей сварке,
паянии, лужении, склеивании. Слой жидкости (при-
поя, клея), хорошо смачивающей твердые поверх-
ности, после отвердевания образует прочный адге-
зионный шов. Таковы же случаи образования лако-
красочных покрытий с высокой А. Предельная А.
достигается также при соприкосновении твердых
тел в пластич. или эластич. состоянии при достаточно
высоких давлениях, надр. склеивание резиновым
клеем, холодная сварка металлов. Близкая к пре-
дельной А. возникает также при образовании новой
твердой фазы на самой поверхности твердого тела,
напр. в случае гальванич. покрытий (в этом случае
для прочной А. требуется высокая чистота металлич.
поверхности), а также при образовании в результате
хемосорбции и поверхностных химич. реакций окис-
ных, сульфидных и др. пленок на металлич. поверх-
ностях. Во всех этих случаях сильная (прочная) А. —
необходимое условие образования высококачествен-
ного покрытия, связующего, сварного или клеевого
шва. Если А. достаточно высока, то при возрастании
внешних сил разрыв происходит не по адгезионному
шву, а когезионно — внутри наиболее слабого из
сопряженных тел или внутри связующей прослойки.
Часто наблюдается т. н. мозаичный отрыв: по
отдельным участкам площади — чисто адгезионный,
а по другим — когезионный. Таким образом, А.
может быть измерена только в случаях заведомо
слабой А.
Термодинамич. характеристикой А. является убыль
свободной энергии на 1 см2 поверхности адгезионного
шва в изотермич. обратимом процессе. Работа А.
Wa (т. е. работа обратимого адгезионного отрыва)
^а = °1о+32О-312 (D
где а12 — поверхностное натяжение на границе фаз
1 и 2, между к-рыми происходит А., а а10 и а20 — на гра-
нице каждой из этих фаз с окружающей средой
(воздухом). Работа когезии в фазе Г. P7Ci — 2?i0,
а в фазе 2:WC = 2^20. И3 (1) видно, что А. растет
с уменьшением а12 — различия в полярностях между
фазами 1,2, и потому может быть усилена адсорбцией
поверхностно-активных добавок на границе раздела
фаз 1 и 2.
В случае соприкосновения двух слоев несмешиваю-
щихся жидкостей (в равновесии) работа А. может
быть вычислена по ур-нию (1) из прямых измерений
ai2. ’и, аго- Н° отношению к воде А. растет с увели-
чением полярности жидкости, напр., в ряду: н-гептан
(41,9), бензол (66,6), н-октиловый спирт (91,8), ни-
трометан (99), анилин (109,6) (числа в скобках —
работа А. в эрг/см2). А. жидкости к поверхности твер-
дого тела ввиду того, что его поверхностное натяжение
не может быть непосредственно измерено, опреде-
ляется по ур-нию Wa = о20 (14- COS0) путем измерений
поверхностного натяжения жидкости а20 и равновес-
29
АДГЕЗИЯ — АДЕНИН
30
пого краевого угла смачивания 0. Вследствие гисте-
резиса смачивания, т. е. неравновесности краево-
го угла, это уравнение часто неприменимо. Кроме
того, им нельзя пользоваться, когда краевой угол
не образуется в условиях полного смачивания при
aio — а12>а2<и т. е. при Wa>-2a2a, Определение А. у твер-
дых тел затруднено в связи с крайней неопределен-
ностью истинной площади соприкосновения между
ними и невозможностью непосредственного измерения
поверхностного натяжения. Ур-ние (1), применимое
к границам жидких фаз, не может быть использовано
для А. твердого тела к другому твердому телу или
к пленке вследствие сопровождающих адгезионный
отрыв различного рода необратимых явлений, зави-
сящих от скорости отрыва, от наличия неупругих
деформапий или течения в системе перед отрывом,
быстрой разгрузки при отрыве и от образования
двойного электрического слоя. Работа разделения
противоположно заряженных обкладок этого слоя
может составлять значительную часть работы А.
и резко возрастает с увеличением скорости адгезион-
ного отрыва.
А. полимерных материалов к различным твердым
телам (особенно металлам) определяется интенсив-
ностью молекулярного и химического взаимодей-
ствия на поверхности раздела. Хорошую А. прояв-
ляют поэтому полярные полимеры с большим числом
химически активных функциональных групп в макро-
молекулах.
Значительно слабее А. в случае неполярных (угле-
водородных) полимеров (полиэтилен, полиизобути-
лен). Для повышения А. в состав клея или пленко-
образующего полимера вводят активные добавки,
усиливающие адгезионную связь на поверхности
вследствие образования ориентированного адсорб-
ционного слоя, хемосорбционно связанного поляр-
ными группами с подложкой, а углеводородными
цепями проникающего в пленку. А. двух тел, в част-
ности полимеров друг к другу, определяется бли-
зостью их полярностей, т. е. интенсивностей молеку-
лярных взаимодействий в этих телах, и их совмести-
мостью, т. е. взаимной растворимостью, а также
способностью к взаимному диффузионному проник-
новению соприкасающихся гибких макромолекул или
их звеньев. Поэтому А. растет с уменьшением молеку-
лярного веса и разветвленности цепей при сохранении
гибкости и подвижности звеньев. Прочная А. твердых
тел (особенно, напр., у полимеров) при их значи-
тельной и в пределе неограниченной взаимной раство-
римости приводит со временем в результате диффузии
к непрерывному усилению связи между двумя сопри-
касающимися телами, к исчезновению поверхности
раздела между ними и превращению двух соприка-
сающихся фаз в одну, как при когезии. Примером
этого является т. н. а у т о г е з и я, или автогезия
(еамослипание), полимеров, когда происходит обра-
зование прочной связи между двумя приведенными
в контакт объемами одного и того же полимера,
напр. натурального каучука. Аутогезия связана
с диффузией цепочечных макромолекул или их
участков из одного объема в другой, прочность соеди-
нения увеличивается со временем и с повышением
темп-ры, стремясь к определенному пределу (коге-
зионной прочности). То же имеет место и в случае
термодиффузионных металлич. покрытий (вследствие
резкого возрастания скорости диффузии при повы-
шении темп-ры).
А. имеет решающее значение (вместе с непроницае-
мостью) для обеспечения высоких защитных свойств
лакокрасочных полимерных покрытий, предохраняю-
щих металлич. детали от коррозии. При образовании
таких пленочных покрытий вследствие усадки в
пленке и в подложке возникают касательные напря-
жения, возрастающие с повышением толщины покры-
тия. При этом максимальная величина таких напря-
жений является мерой А. Причиной нарушения А.
часто являются не только эти внутренние напря-
жения, но и термин, напряжения вследствие разности
коэфф, теплового расширения пленки и подложки или
сопряженных деталей, напр. стекла и металла. Если
клей или пленкообразующий материал в текучем
состоянии проникает в глубокие неровности поверх-
ности или поры твердого тела (подложки), то после
отверждения возникает связь, иногда называемая
механич. адгезией (в отличие от описанной «специ-
фической» — молекулярной А.). В таком случае,
независимо от интенсивности молекулярных связей
на поверхности раздела, для адгезионного разрыва,
напр. при скалывании, необходимо преодолеть коге-
зию в затвердевшей клеящей прослойке или пленке.
Резкое повышение трения и износа соприкасающихся
поверхностей, вызываемое А., устраняется слоем
смазки, препятствующим контакту поверхностей.
Лит.: Адам Н. К Физика и химия поверхностей,
пер. с англ., М.—Л., 1947; Кротова Н. А., О склеивании
и прилипании, М., 1956; Адгезия, клеи, цементы, припои,
|Сб. ст.]. Под ред. Н. Дебройна и Р. Гувинка, пер. с англ.,
М., 1954; В о ю ц к и й С. С., Аутогезия и адгезия высоко-
полимеров, М., 1960; его же, Высокомолекулярные соедине-
ния, 1959, 1, Jw2, с. 230; В о у д е н Ф. П. и ТейборД.,
Трение исмазка, пер. с англ., М., 1960, гл. 3; Воюцкий С. С.,
Вакула В. Л., Усп. хим., 1959, 28, вып. 6, с. 701; Ш р е й-
нер С. А. и Зубов П. И., ДАН СССР, 1959, 124,
№ 5, с. 1102; и х ж е, Коллоидн. ж., 1960, 22, вып. 4;
Klebtechnik. Die AdhSsion in Theorie und Praxis, Stuttg., 1957.
II. А. Ребиндер.
АДДЕНДЫ — см. Комплексные соединения.
АДДУКТ — молекулярное соединение, продукт
присоединения молекул друг к другу. Термин «А.»
часто не содержит понятия об определенной структуре
и характере химич. связи соединения. Обычно А.
называют молекулярные соединения, образованные
из веществ, насыщенных в валентном отношении;
например, галогенорганич. вещества типа трифенил-
хлорметана образуют А. с неорганическими солями:
(С6Н5)3 СС1 • А1С13, (CeH5)3CCl-SnCh. К А. относят со-
единения нитропроизводных с ароматич. или не-
предельными углеводородами и их производными,
напр. соединение пикриновой к-ты с пафтиламином
CeH3(NO2)3 • C1oH7NH2. Мочевина образует ряд А.
нестехиометрич. состава с алифатич. соединениями,
особенно имеющими длинную нормальную цепь,
напр. соединение декана С[2Н2в с мочевиной, в ко-
тором на 1 молекулу декана приходится 8,3 молекул
мочевины. Эти А. получают простым смешением ком-
понентов и используют в пром-сти для извлечения
определенных типов углеводородов из их смесей
при депарафинизации нефтепродуктов. Подобные А.
в большинстве случаев малоустойчивы и легко раз-
лагаются с регенерацией исходных веществ.
Af. Я. Алейникова.
АДЕНИН (6-аминопурин) C5H5N5, мол. в. 135,14 —
бесцветные кристаллы; т. пл. 360—365° (с разл.),
плохо растворим в холодной воде и в
спирте, хорошо — в горячей воде, 1N = С—NH3
нерастворим в эфире, хлороформе, I I ’ 9
растворим в кислотах и щелочах. Я| у
Из водных р-ров кристаллизуется с 8n — с—NH
тремя молекулами Н2О. В УФ-об- .4 *
ласти А. обладает характерным максимумом погло-
щения при 260 ммк (pH 7) с коэфф, молярной экстинк-
ции 13 500. А. обладает основными свойствами,
pAza 4,15, рАЯ2 9,80; образует соединения с кисло-
тами, основаниями и солями. Под действием HNO2
А. дезаминируется, превращаясь в гипоксантин
(6-оксипурин). А. является составной частью нуклеи-
новых к-т, а также адениновых нуклеотидов и в таком
виде широко распространен в живых организмах.
Получают А. при кислотном гидролизе нуклеиновых
31
АДЕНОЗИН — АДЕНОЗИНФОСФОРНЫЕ КИСЛОТЫ
32
кислот или аденин-нуклеотидов, а также синтетически,
напр. исходя из 4,6-диамино-5-фенил-азопиримидина.
Лит.: Ciba foundation symposium on the chemistry and
biology of purines. Ed. G. E. Wolstenholme and С. M. O’Con-
nor, L., 1957; В e n d ic h А., в кн.: The Nucleic acids. Che-
mistry and biology, v. 1, N. Y., 1955, p. 81.
А. В. Котельникова.
АДЕНОЗИН (9-р-Н-рибофуранозидоаденин), мол. в.
267,1, C10HJ3N5O4 — N-глюкозид, состоящий из аде-
нина и D-рибозы; бесцветные кристаллы; т. пл.
229°; [а]д = —60° (Н2О); легко растворим в воде,
N = C-NH2
CH C-N4
II II zCH
----°---
но он
СН-С-С-СН-СНаОН
I I
особенно при нагревании, нерастворим в большинстве
органич. растворителей. В УФ-обласТи А. обладает
характерным для соединений аденина максимумом
поглощения при 259,5 ммк с коэфф, молярной экстинк-
ции 14 900 (pH 6,4). А. является слабым основанием
(рКа 3,5, рА'а 12,5). При кислотном гидролизе А.
распадается на аденин и D-рибозу, под действием
HNO2 дезаминируется, превращаясь в инозин (амино-
группа замещается гидроксильной). А. образуется
при щелочном гидролизе нуклеиновых кислот, при
ферментативном отщеплении фосфатного остатка из
аденозинмонофосфорной к-ты (см. ниже), а также
синтетич. способами, напр. из 2,6-днхлораденина и
ацетохлор-О-рибофуранозы. А.— составная часть ну-
клеиновых к-т, а также адениновых нуклеотидов;
в виде этих соединений А. широко распространен
в живой природе.
Лит.: The Nucleic acids. Chemistry and biology, ed. by
E. Chargaff and J. N. Davidson, v. 1, N. Y., 1955.
А. В. Котельникова.
АДЕНОЗИНТРИФОСФАТАЗЫ (АТФ-азы) — фер-
менты , гидролитически отщепляющие один (аденозин-
трифосфатаза) или оба (апираза) лабильных фосфата
от аденозинтрифосфорной кислоты с образованием:
при действии аденозинтрифосфатазы аденозиндифос-
форной кислоты, а при действии апиразы — адено-
зинмонофосфорной кислоты (см. Аденозинфосфорные
кислоты).
Фермент Оптимальные усло- вия действия Актива- торы Ингиби- торы
pH темп-ра, °C
Мышечная АТФ-аза * 6,5 и 9,2 25 Са3+ Ou, Hg, п-ХМБ
АТФ-аза • 6,8—7,0 38 Mg2+, Мп3+ Са2+, п-ХМБ ***
АТФ-аза мито- хондрий * 8,5 28 Mg2 + Са2+ АДФ
Апираза карто- феля * * 6,5 30 Са2+ —
АТФ-аза (апира- за) насекомых *• 7,8-8,0 42 Mg3+ глютатион Са2^ п-ХМБ ***
• Катализирует реакции: АТФ + Н»О -► АДФ + Н3РО4.
** Катализирует реакции: АТФ-|-2Н2О-»АМФ+2Н3РО4.
*** п-ХМБ — п-хлормеркурбензоат.
Лит.: Methods in enzymology, ed. by S. P. Colowick and
N. 0. Kaplan, v. 2, 1955, p. 582. T. T. Болотина.
АДЕНОЗИНФОСФОРНЫЕ КИСЛОТЫ — нуклео-
тиды, представляющие собой моно-, ди- или трифос-
форные эфиры аденозина. А. к. содержат пуриновое
основание, аденин, углевод пентозу — рибозу и фос-
форные остатки; в зависимости от количества послед-
них и места их прикрепления к рибозе А. к. и разли-
чаются между собой.
Аденозинмонофосфорные кислоты (АМФ, аденило-
вые кислоты) Cj0H14N6O7P, мол. в. 347,23; известны
в виде трех изомеров: аденозин-5’-фосфат (мышечная
АМФ, А-5'-Р), аденозин-З'-фосфат (АМФ 6, А-З'-Р,
Г а'| з'| 4'
Н Н
Аденозин-5'-фосфат
прежнее название — дрожжевая адениловая кислота),
аденозин-2'-фосфат (АМФ а, А-2'-Р). Физич. свойства
АМФ приведены в таблице.
Кислота Т. пл., °C 1“1D V
А-5'-Р 192 —46,4° (Н3О) —58,7° (формамид) 0,69
А-З'-Р 197 (с разл.) 0,62
А-2'-Р 187 (с разл.) —84,3° (формамид) 0,69
• Отношение расстояния пройденного р-ром вещества
к расстоянию, пройденному одним растворителем при разде-
лении методом хроматографии на бумаге. В данном случае
разделение проводилось в системе 5% Na2HPO4 — изоамило-
вый спирт.
АМФ растворимы в воде, нерастворимы в органиче-
ских растворителях; щелочные и щелочноземельные
соли АМФ растворимы в воде, соли тяжелых металлов
нерастворимы. АМФ обладают характерным для
производных аденина максимумом поглощения при
260 ммк с коэфф, молярной экстинкции 14 200 (pH 2).
При кислотном гидролизе от АМФ легко отщепляется
аденин-, фосфат отщепляется легче от А-З'-Р и А-2'-Р,
чем от А-5'-Р; последняя, в отличие от А-З'-Р и А-2'-Р,
содержит два смежных гидроксила и дает характерные
реакции — окисляется иодной кислотой, образует
медный комплекс и т. д. А-5'-Р дезаминируется спе-
цифическим ферментом — дезаминазой А-5'-Р, пре-
вращаясь в инозиновую к-ту.
А-5'-Р получают путем гидролиза аденозинтрифос-
форной к-ты (см. ниже) или фосфорилированием адено-
зина, а А-З'-Р и А-2'-Р — при гидролизе рибонуклеи-
новой к-ты; последние получены также синтетически
из аденозина.
АМФ играют весьма важную роль в жизнедеятель-
ности организмов, входят в состав сложных биологи-
чески важных соединений (адениннуклеотидов, рибо-
нуклеиновых к-т, кофермента А, пиридиннуклеоти-
дов, ацил аденилатов). В свободном виде А-5'-Р участ-
вует в переносе фосфатных групп в так называе-
мом «адениловом цикле» (см. Обмен веществ), посред-
ством которого происходит ресинтез аденозинтрифос-
форной кислоты из аденозинмонофосфорной кисло-
ты при участии фермента аденилатканазы и про-
цессов фосфорилирования (аэробных или ана-
эробных).
Аденозиндифосфорная кислота (АДФ)С10 Н15 N6O10P,,
мол. в. 427,22. АДФ и ее щелочные соли растворимы
в воде, нерастворимы в органич. растворителях; щелоч-
ноземельные соли плохо растворимы в воде, соли тя-
желых металлов почти нерастворимы. В УФ-области
33
АДЕНОЗИНФОСФОРНЫЕ КИСЛОТЫ — АДИПИНОВАЯ КИСЛОТА
34
АДФ обладает максимумом поглощения при 259 ммк
(pH 7) с коэфф, молярной экстинкции 15 400. Один
N=C-NH2 „
X 1
НС С~ N% ОН ОН 0 0
II II СН II . а II Д II
N-C-N^---СН-С —С—СН —СН2—О-P—О—Р-ОН
II II
,Н Н ОН ОН
фосфатный остаток (Р) АДФ при кислотном гидро-
лизе отщепляется легко, второй (а) — значительно
труднее. При кислотном гидролизе АДФ легко раз-
рывается N-гликозидная связь с отщеплением аде-
нина. Под действием HNO2 АДФ дезаминируется,
прекращаясь в инозиндифосфорную кислоту; в мыш-
цах процесс дезаминирования АДФ происходит фер-
ментативным путем. Связь между остатками фосфа-
тов в молекуле АДФ относится к макроэргическим.
при ее гидролизе освобождается 7,5 ккал)моль. АДФ
содержится в живых организмах во всех тканях; ее
получают путем дефосфорилирования аденозинтри-
фосфорной к-ты с помощью фермента аденозинтри-
фосфатазы. АДФ получена также синтетически фос-
форилированием аденозин-5'-фосфата. АДФ играет
важную роль в процессах обмена веществ у живых
организмов, являясь первичным акцептором макро-
эргических фосфатов в процессах аэробного и ана-
эробного окислительного фосфорилирования.
Аденозинтрифосфорная кислота (АТФ, аденил-
пирофосфорная кислота) C10H16N5O13P3, мол. в. 507,21.
№С—NHa
---0----
он он
НС C-N4
о о
О
N-C—N-
СН II « II ? II г II
lz—сн-с—с—сн—сн2—о—р—о—Р—О—Р
1-Р-ОН
Н н
он он он
Свободная АТФ и ее щелочные соли хорошо раство-
римы в воде, плохо в органич. растворителях;
соли щелочноземельных и тяжелых металлов нера-
створимы в воде. В УФ-области АТФ обладает ха-
рактерным для соединений аденина максимумом по-
глощения при 259 ммк (pH 7) с коэфф, молярной
экстинкции 15 400. Из трех фосфатных остатков
в АТФ два остатка (р и у) легко отщепляются
при кислотном гидролизе, а a-остаток гидролизуется
трудно.
При кислотном гидролизе N-гликозидная связь
АТФ разрывается так же легко, как и р и у-фос-
фатные остатки. При мягком щелочном гидролизе
АТФ расщепляется с образованием аденозин-5'-фос-
фата и пирофосфата. АТФ окисляется иодной к-той
и образует комплексы, характерные для а-гликоле-
вой группировки соседних групп D-рибозы. Под
действием HNOa АТФ дезаминируется с образованием
инозинтрифосфорной к-ты. При гидролизе каждого
из двух легкогидролизуемых фосфатных остатков
АТФ, связанных ангидридной связью, выделяется
значительно больше энергии (от 8 до 11 ккал)моль,
в зависимости от условий), чем при отщеплении
трудногидролизуемого фосфата (2—3 ккал!моль)-, по-
этому АТФ относится кт. н. макроэргич. соедине-
ниям. Роль АТФ в обмене в-в живых организмов
чрезвычайно велика: АТФ, по-видимому, является
универсальным аккумулятором и распределителем
химич. энергии питательных веществ, в процессах
аэробного и анаэробного окислительного фосфори-
лирования. Вместе с тем АТФ является главным
донатором энергии во всех основных реакциях био-
синтеза в организме, а также и в такцх важнейших
физиология, процессах, как мышечное сокращение,
передвижение подвижных клеток, высшая нервная
деятельность. Механизм участия АТФ в сокращении
мышц был вскрыт работами В. А. Энгельгардта и
М. Н. Любимовой, показавшими, что сократитель-
ный белок мышц — миозин обладает свойствами
фермента — аденозинтрифосфатазы, отщепляющего
фосфат от АТФ. В этом процессе, по-видимому, и про-
исходит передача энергии фосфатных связей АТФ
сократительному белку мышц.
АТФ получают преим. из скелетных мышц, дрож-
жей, а также синтетически — путем фосфорилиро-
вания аденозин-5'-фосфата ортофосфорной к-той в при-
сутствии дегидратирующих агентов, как, напр., про-
изводных карбодиимида.
Лит.: Фер дм ан Д. Л., Обмен фосфорных соединений,
М.—Л., 1940- Владимиров Г. Е., Гинодман Л. М.,
Биохимия, 1953, 18, вып. 4, с. 490; Котельникова А. В.,
Усп. биол. хим., 1958, 3, с. 206 (обзор); Дыхательные ферменты,
пер. с англ., М., 1952; Габерманн В., Усп. совр. биол.,
1959, 47, вып. 1,с. 19; Phosphorus metabolism ed. by W. D. McEl
Iroy and B. Glass, v. 1—2, Balt., 1951—52; Bock R. M.
[a. O.l, Arch. Biochem. and biophys., 1956, 62, №2, p. 253;
The Nucleic acids. Chemistry and biology, ed. by E. Chargatt
and J. N. Davidson, v. 1, N. Y., 1955.
А. В. Котельникова.
АДИАБАТНЫЕ ПРОЦЕССЫ — процессы, про-
исходящие в к.-н. системе в условиях, когда она
лишена возможности как получения теплоты от окру-
жающей среды, так и передачи ее в окружающую
среду. Строго эти условия соблюдаются только в си-
стемах, полностью изолированных от окружающей
среды в отношении теплового обмена. Практически
же многие процессы, протекающие достаточно быстро,
можно рассматривать как адиабатные. При адиабат-
ном расширении или сжатии идеального газа соот-
ношение между объемом и давлением его выражается
ур-нием Пуассона pvt = const, где 7 = Cp/Cv —
отношение теплоемкостей этого газа соответственно
при постоянном давлении Ср и при постоянном объеме
Cv. Адиабатное расширение газа сопровождается
охлаждением, а адиабатное сжатие — нагреванием.
Этим А. п. отличаются от изотермич. процессов.
Если А. п. обратим, то энтропия системы сохраняет
постоянное значение (изоэнтропийный процесс); в не-
обратимом А. п. энтропия возрастает. Теорией А. п.
пользуются при термодинамич. рассмотрении горе-
ния, нек-рых процессов в химических производст-
вах и т. д. Различные закономерности, свойственные
А. п., широко используются в метеорологии. Это
связано с тем, что перемещения больших масс воздуха
обычно совершаются в условиях, близких к адиа-
батным. в. А. Киреев.
АДИПИНОВАЯ КИСЛОТА (бутандикарбоновая-1,4
кислота) СООН(СН2)4СООН, мол. вес 146,15 —
бесцветные кристаллы, т. пл. 149—150°; т. кип.
265°/100 .м.м, 216°/15 мм; растворима в воде (1,5%
при 15°), в этиловом спирте и ограниченно в эфире;
константы диссоциации: Кг = 3,90 • 10-s (25°) и
-Ка = 5,29 • 10 6 (25°); возгоняется; теплота сгорания
668,6 ккал/моль. Большинство солей А. к. растворимо
в воде. А. к. — важнейший полупродукт в произ-ве
нейлона. Эфиры А. к. широко применяются в качестве
пластификаторов и смазочных масел специально-
го назначения. Мировое производство А. к. дости-
гает нескольких сот тысяч тонн в год. Основны-
ми методами получения адипиновой кислоты являет-
ся окисление циклогексанона азотной кислотой или
кислородом воздуха в присутствии солей марганца
(катализатор):
/CHg-CHa HN0
НгС ^СНОН-------- Н00С(СНг)4С00Н
^CHj-CHg
35
АДКИНСА КАТАЛИЗАТОР —АДРЕНОКОРТИКОТРОПНЫЙ ГОРМОН
36
Ангидрид адипиновой кислоты образуется при кар-
бонилировании тетрагидрофурана:
Н2С-СН2 снй-сн„
I I +2СО------I I 2
н2с сн2 сн2 сн2
о со со
4 о 7
а из него в присутствии воды — А. к. Динитрил А. к.
получают с хорошим выходом электрохимия, гидро-
димеризацией акрилонитрила: 2СН2=СН2—CN —>
—»CN(CH2)4CN. При дегидратации А. к. уксусным ан-
гидридом образуется полимерный ее ангидрид (т. пл.
65—70°), перегонкой к-рого м. б. получен мономерный
семичленный циклич. ангидрид с т. пл. 22°. При сухой
перегонке кальциевой соли А. к. образуется цикло-
пентанои. Ниже приведены свойства нек-рых произ-
водных А. к.:
Производное А. к. Т. пл., °C Т. нип., °С/мм
Диметиловый эфир СаНцО, . . Диэтиловый эфир С8Н18О4 . . . Дифениловый эфир С16Н48О4 . Диамид CeH12O«N5 Дихлорангидрид CeHgOgCla . . Дианилид Ci8H2o02N2 Динитрил Ce_H8N2 8 28-29 105—106 220 240—241 0-1 112/10 170/17 130—132/18 295/760; 181/20
И. Л. Кнунянц.
АДКИНСА КАТАЛИЗАТОР — смесь тонкодис-
персных окисей меди и хрома; активный катализатор
для синтеза метанола из окиси углерода и водорода,
а также гидрогенизации органйч. соединений, осо-
бенно содержащих карбонильные и эфирные группы.
А. к. отличается стабильностью, высокой активностью
на единицу веса (близкой к активности платины,
палладия и никеля), возможностью применения для
гидрогенизации при невысоких темп-pax (порядка
125—150°), а также избирательностью действия
(напр., для гидрогенизации боковых цепей в аро-
матич. соединениях). Входящая в состав А. к. окись
никеля оказывает наряду с каталитич. и стабилизи-
рующее действие (препятствует восстановлению СпО
до Сп2О и Си). А. к. получают осторожным разложе-
нием смеси хромата меди и аммония при накалива-
нии и одновременном растирании, с обработкой после
разложения 10%-ным р-ром уксусной к-ты, фильтро-
ванием, промыванием, сушкой и повторным расти-
ранием.
Лит.: Adkins Н., Reactions ot hydrogen with organic
compounds over copper-chromium oxide and nickel catalysts,
Madison, 1946; M э к с т e д Э., Катализ и его промышлен-
ное применение, пер. с англ., М., 1936; Каталитические, фото-
химические и электролитические реакции, пер. с англ., М.,
1960. С. Л. Киперман.
АДРЕНАЛИН (эпинефрин) C9Ht3O3N, молекуляр-
ный вес 183,20 —гормон мозгового слоя надпочеч-
ников (L-A.); синтетически
HOf*^CH(OH)CH2NHCH, получены также D.L-A.
и D-А, По биологической
активности D-А. сходен
с природным L-A., однако его действие в 12—15 раз
слабее. L-A. — белые кристаллы, т. пл. 212—215°
(с разл.), хорошо растворим в горячей воде, плохо —
в холодной; не растворяется в эфире, хлороформе,
бензоле и абс. спирте, растворим в р-рах кислот и
щелочей, но нерастворим в аммиаке и углекислых
щелочах; [и]д—53,5°. А. дает зеленое окраши-
вание с FeCl3 и ряд. др. реакций, характерных для
фенолов, легко окисляется, восстанавливает фелинга
реактив. А. играет большую роль в процессе передачи
нервного возбуждения, влияет .на сердечно-сосуди-
стую систему и обмен веществ (в частности углеводов).
А. получают из надпочечных желез крупных живот-
ных, а также синтетически, напр. конденсацией
пирокатехина с хлоруксусной к-той. А. (в виде
солянокислых солей) используют при шоке, остановке
сердца, падении кровяного давления и др.
Лит.: Утевский А. М. Усп. биол. хим., 1950, 1,
с. 423; Асатиани В. С., Биохимическая фотометрия,
М., 1957; Напй О., Hormone, 2 Aufl., .Тепа, 1959.
, Л. И. Липевич.
АДРЕНОКОРТИКОТРОПНЫЙ ГОРМОН (АКТГ;
адренокортикотропин, кортикотропин, кортикости-
мулин) — гормон, образующийся в передней доле
гипофиза млекопитающих и стимулирующий функ-
цию коркового слоя надпочечных желез. А. г. — вы-
сокомолекулярный линейный полипептид, мол. в. ок.
4500. В состав молекулы А. г. входит 39 остатков
15 различных аминокислот. Последовательность рас-
положения остатков аминокислот в полипептидпой
цепи полностью установлена для р-кортикотропина
(А. г. свиньи), а-кортикотропина (А. г. овцы), а также
А. г. быка, к-рые выделены в чистом виде (условные
обозначения аминокислот см. ст. Аминокислоты):
/-Кортикотропин C2i0H3I4N5liO57S, мол. в. 4567
Н-сер.-тир.-сер.-мет.-глют.-гис.-фал.-сер.-трип.-глют.-лиз.-
1 2 3 4 5 6 7 8 9 10 11
-прол.-вал.-глют.-лиз.-лиз.-арг.-арг.-прол.-вал.-лиз.-вал.-
12 13 14 15 16 17 18 19 20 21 22
-тир.-прол.-асп.-глиц.-ал.-глют.-асп.-глют.-КН2-лейц.-ал.-
23 24 25 26 2 7 28 29 30 31 32
-глют.-ал.-фал.-прол.-лейц.-глют.-фал.-ОН
33 34 35 36 37 38 39
а-Кортикотропин
Н-сер.-тир.-сер.-мет.-глют.-гис.-фал.-сер .-трип.-глют.-лиз.-
12345678 9 10 11
-прол.-вал. -глют.-лиз.-лиз. -ар г.-арг.-прол. -вал.-лиз.-вал. -
12 13 14 15 16 17 18 19 20 21 22
-тир .-прол.-ал. -глиц. -глют.-асп.-асп.-глют.-ал.-сер .-глют. -
23 24 25 26 27 28 29 30 31 32 33
-ал.-фал.-прол.-лейц.-глют.-фал.-ОН
34 35 36 37 38 39
Кортикотропин быка
Н-сер.-тир .-сер.-мет.-глют.-гис.-фал.-арг.-трип.-глиц,-
1234 56789 10
-лиз.-прол.-вал.-г лиц.-лиз.-лиз.-арг.-арг.-прол.-вал.-
И 12 13 14 15 16 17 18 19 20
-лиз.-вал.-тир.-прол.-асп.-глиц.-глют.-ал.-глют.-асп.-сер.-
21 22 23 24 25 26 27 28 29 30 31
-ал.-глют.-Ш13-ал.-фал.-пр ол.-лейц.-г лют.-фал.-ОН
32 33 34 35 36 37 38 39
Препараты А. г., выделенные из гипофизов различ-
ных животных, отличаются друг от друга гл. обр.
аминокислотами в положениях 25—33.
Препараты А. г. получены в виде белого порошка,
легко растворимого в воде и 60—70%-ном спирте.
Изоэлектрич. точка для а-кортикотропина при pH
6,6; для p-кортикотропина при pH 4,6. Из водных
р-ров А. г. осаждается трихлоруксусной и сульфо-
салициловой к-тами. В водных р-рах при слабокислой
реакции А. г. устойчив к нагреванию, в щелочной
среде быстро разлагается. При действии пепсина
от карбоксильного конца полипептидпой цепи р-кор-
тикотропина вначале отщепляется И аминокислот-
ных остатков; образующийся полипептид сохраняет
биологич. активность; более глубокое расщепление
пепсином и трипсином приводит к получению неак-
тивных пептидов. Из др. препаратов А. г. также
получены полипептиды, обладающие адренокортико-
тропной активностью. Определение активности А. г.
производят различными биологич. методами: напр.,
определяют влияние препаратов А. г. на уровень
аскорбиновой к-ты в надпочечниках крыс, у к-рых
был удален гипофиз, или влияние на секрецию корти-
костероидов в кровь надпочечниками собаки, у к-рой
также был удален гипофиз. За международную
37
АДСОРБЕНТЫ
38
единипу адренокортикотропной активности принята
активность 1 мг стандартного препарата А. г. При-
меняют А, г. при лечении артрита, заболеваний, свя-
занных с корковым слоем надпочечников, и др.
Лит.: Панков Ю. А., Усп. совр. биол., 1959, 47,
вып. 3, с. 347 (библиогр. 93 назв.); С а х а ц к а я Т. С.,
Пробл. эндокрлнол. и гормонотерапии, 1957, 3, № 4, с. 95
(библиогр. 143 назв.); Behrens О. К., В г о m е г W. W.,
Ann. Rev. Biochem., 1958, 27, р. 57; В е г s i n Th., Biochemie
der Hormone, Lpz., 1959; Hanc 0., Hormone, 2 Aufl., Jena,
1959. Л. И. Линевич.
АДСОРБЕНТЫ — высокодисперсные искусствен-
ные и природные тела с большой наружной (непо-
ристые) или внутренней (пористые) поверхностью,
на к-рой происходит адсорбция веществ из соприка-
сающихся с ней газов или растворов. Адсорбционные
свойства А. определяются природой твердого тела,
химич. составом и состоянием поверхности, степенью
ее химич. и геометрия, неоднородности, величиной
уд. поверхности s, размерами и характером пор.
Ниже приведены примерные порядки величин s для
типических дисперсных тел.
Непористые А.:
Молотые кристаллы......... около 1 м“/г
Кристаллические осадки.... 1 — 10 м- 1г
Частицы дымов, сажи, аэросил . . 10 —500 м2/г
Пористые А.:
Активные окиси и гидроокиси (си-
ликагели, алюмогели, алюмоси-
ликатные катализаторы и т. п.) 10—1000 м^/г
Активные угли............. до 1000 м*/г
Наиболее важный способ получения непористых
высокодисперсиых А., применяемых в качестве напол-
нителей различных сред, — термич. разложение или
неполное сгорание углеводородов (получение саж),
сгорание элементоорганич. или галогенных соеди-
нений [получение высокодисперсного кремнезема —
аэросила — гидролизом перегретым паром галоген-
ангидридов (SiCl4, SiF4) или сжиганием кремний-
органич. соединений]. Пористые А. получают в основ-
ном след, методами: 1) Активированием грубоди-
сперсных твердых материалов путем воздействия
на них химически активных сред, напр. получение
активных углей. 2) Коллоидо-химич. путем — выра-
щиванием частиц золей, затем образованием из них
гелей с рыхлой упаковкой частиц; при высушивании
таких гелей получают А. с большим числом пор —
зазоров между частицами. К числу А., получаемых
таким способом, относятся силикагели, алюмосилика-
гели и т. п. На структуру пор получаемых сухих
гелей (ксерогелей или аэрогелей) можно влиять,
изменяя заряд частиц (pH) и поверхностное натяже-
ние среды. Особо крупнопористые аэрогели полу-
чаются при замене жидкой среды на газовую при
темп-ре выше критич. или при сублимации заморо-
женной среды. Размеры и форма коллоидных частиц
определяют в основном величину s. Рыхлая упаков-
ка крупных частиц приводит к крупнопористым Д.
с малой механич. прочностью (размер пор 100—200 А
и более), более плотная упаковка мелких частиц —
к тонкопористым адсорбентам, механически более
прочным (размер пор 20—50 А). В случае силикагелей
образующие его скелет частицы имеют шаровидную
форму (частицы диаметром 50—500 А), в ряде других
случаев — форму микрокристаллов (напр., гидроокись
и окись магния). Поры таких А. сообщаются друг
с другом. Коллоидо-химич. метод приводит к полу-
чению А. и катализаторов в виде крупинок непра-
вильной формы, а также в виде прочных шариков
размером до неск. миллиметров, скелет к-рых состоит
из множества частиц. 3) Синтезом пористых кристал-
лов типа цеолитов, обладающих свойствами молеку-
лярных сит. Эти кристаллы пронизаны каналами
точно определенных размеров с поперечником отвер-
стии приблизительно от 4 до 10 А и более, что обес-
печивает адсорбцию лишь тех молекул, к-рые обла-
дают меньшими размерами, поэтому пористые кри-
сталлы с успехом примениются для разделения
газов и паров. 4) Термич. разложением карбонатов,
оксалатов, гидроокисей, нек-рых полимеров и т. п.
при умеренных, во избежание спекания, темп-рах
(получение активных окисей, нек-рых тонкопористых
активных углей, губчатых металлов). А. получаются
также молекулярной возгонкой твердых тел в вакууме
(образование активных окисей и металлов).
А. подразделяются на 4 основных структурных
типа: 1) Непористые А., на поверхности к-рых с ро-
стом давления пара свободно происходит моно- и
полимолекулярная адсорбция. 2) Широкопористые
А., размеры пор к-рых близки друг к другу и со-
ставляют более 50А; первичный адсорбционный
процесс в этих порах близок к таковому на непори-
стых А. той же природы, но при достаточно высоких
давлениях пара он сопровождается капиллярной
конденсацией, приводящей к заполнению пор жид-
костью. 3) Тонкопористые А., с размерами пор меньше
50 А; сильно адсорбирующиеся в-ва заполняют эти
поры уже при малых давлениях. Для молекул круп-
ных размеров характерен эффект ультрапористости —
по отношению к ним тонкопористые А. полностью
или частично ведут себя как молекулярные сита.
4) Неоднороднопористые А. с размытым распределе-
нием объема пор по размерам; при адсорбции про-
являются черты, характерные как для второго, так
и для третьего типов.
Геометрич. структуру А. исследуют: 1) Адсорб-
ционным методом, позволяющим определить величину
уд. поверхности (дм. Адсорбция), объем и размеры
пор до 200— 500 А (см. Капиллярная конденсация).
2) Методом определения кажущейся и истинной
плотности (плотности зерен А. в целом и плотности
твердого остова). Разность обратных величин этих
плотностей, т, е. разность объема зерен и объема
твердого остова в зернах, дает общий объем пор (для
активных А. он равен 0,2—2 мл/г). 3) Методом вдав-
ливания ртути в поры. Этим методом находят распре-
деление крупных пор по их размерам, т. к. давление,
заставляющее ртуть входить в несмачиваемые ею
капилляры, обратно пропорционально радиусу ка-
пилляров. 4) Электронно-микроскопическим методом,
к-рый дает гл. обр. представление о размерах и
форме частиц, образующих твердый остов А. 5) Ме-
тодом рассеяния рентгеновых лучей под малыми
углами, к-рый дает представление о размерах обла-
стей неоднородности (частицах) и иногда об их форме.
6) Методом просасывания газа через спрессованные
порошки, к-рый дает представление о каналах между
крупными частицами.
Исследование химич. строения поверхности А. —
определение различных химически активных групп
на поверхности производится микрохимия, и функ-
циональным химич. анализом, методами изотопного
обмена и инфракрасной спектроскопии. Большое
значение имеет дейтеро-протонный обмен, затраги-
вающий в основном лишь активные группы поверх-
ности (напр., гидроксильные группы).
В целях повышения адсорбционных свойств, а
также для придания других спепифич. особенностей
производят модифицирование А. Термич. обработка
приводит к спеканию, в особенности тонкопористых
А.; поверхности А. становятся более однородными
(напр., графитирование саж). Обработка горячим
паром силикагелей, алюмосиликатных катализаторов
и т. п. приводит к увеличению размеров образующих
скелет А. частиц, сокращению поверхности и расши-
рению пор. Обработка углей газами-окислителями
приводит к расширению пор и увеличению их объема.
Образование и разрушение поверхностных химич.
39
АДСОРБЦИОННОЕ ПОНИЖЕНИЕ ПРОЧНОСТИ
40
соединений резко изменяет адсорбционные свойства,
напр. сильное окисление саж делает их гидрофиль-
ными (окисленные сажи образуют стойкие коллоид-
ные растворы), а замена гидроксильных групп сили-
кагелей и ааросилов на углеводородные и кремний-
органич. радикалы — гидрофобными и т. п. Химич,
реакции с поверхностью изменяют также ионообмен-
ные свойства А.
А. применяют в противогазах для адсорбции вред-
ных примесей и в качестве носителей катализаторов
для химич. воздействия на эти примеси; для очистки
и осушки газов и жидкостей; хроматографич. разде-
ления смесей (см. Хроматография)', как наполнители
для полимеров; в катализе в качестве носителей
катализаторов; в медицине для поглощения газов,
ядов и т. д. Очень широко используют А. для очистки
различных нефтепродуктов, природного и попутного
газов от более высококипящих углеводородов (актив-
ный уголь), разделения углеводородных смесей и выде-
ления из них отдельных компонентов (гл. обр. моле-
кулярные сита — пористые кристаллы, силикагель
и активная окись алюминия), для очистки масел
(см. Земли отбеливающие), а также в качестве носи-
телей и катализаторов для химич. процессов нефте-
переработки и т. д.
Природные А. (глины, опоки, трепела, диатомиты,
угли, торфы и пр.) применяются непосредственно
или после химич. и термич. активирования.
Лит.: Методы исследования структуры высокодисперсиых
и пористых тел, [кн. 1], М., 1953, [кн. 2], М., 1958; Поверхност-
ные химические соединения и их роль в явлениях адсорбции
(раздел «Адсорбция на природных адсорбентах»), Сб. докладов,
М., 1957; Овчаренко Ф. Д. 1и др.5, Украинские бетониты,
Киев, 1958; The structure and properties oi porous materials,
ed. by D. H. Everett and F. 8. Stone, L. — Butterworth, 1958.
А. В. Лиселев.
АДСОРБЦИОННОЕ ПОНИЖЕНИЕ ПРОЧНО-
СТИ твердых тел (эффект Ребиндера) —
значительное понижение сопротивляемости твердых
тел деформированию и разрушению в результате
адсорбции поверхностно-активных веществ из окру-
жающей среды. А. п. п. представляет собой своеоб-
разное физико-химич. явление, обладающее широкой
общностью. Эффект не связан с процессом коррозии,
т. к. вызывается химически неактивными средами
или примесями, присутствующими к тому же в весьма
малых количествах; не связан и с растворением, т. к.
исследуемые твердые тела или совершенно нераство-
же растворимость их
невелика, и среда м. б.
римы в данных средах, или
в физико-химич. ра-
вновесии с твердым
телом; А. п. п. не
вызывается наличием
на поверхности образ-
цов окисных пленок,
которые могут ока-
зать лишь побочное
влияние.
В основе А. п. п.
лежит понижение сво-
Рис. I. Диаграмма рас-
тяжения монокристалла
олова в координатах на-
пряжение в Г/мм* (орди-
ната) — относительная
деформация в % (абсцисса): 1 — растяжение в неактивной
среде (в вазелиновом масле или на воздухе); 2 — растяже-
ние в активной среде (в вазелиновом масле 4- 0,2% олеи-
новой к-ты).
бодной поверхностной энергии а как на внешней по-
верхности твердого тела, так и на внутр, поверхно-
стях, возникающих и развивающихся в процессе дефор-
мации по дефектам структуры или в результате скоп-
ления дислокаций. А. п. п., вызванное понижением а на
внешней поверхности тела, образует обширную группу
явлений, связанных с пластифицированием твердого
тела в процессе деформации, т. е. со снижением его
предела текучести и коэфф, упрочнения (рис. 1)
(см. Пластичность). Типичными веществами, вызы-
вающими такого рода эффекты, являются органич.
поверхностно-активные вещества — кислоты, спирты,
амины и т. д., молекулы которых слишком велики,
чтобы проникнуть внутрь деформируемого твердого
тела по дефектам структуры. Наиболее обшие и ха-
рактерные закономерности А. п. п. выявлены на
металлич. монокристаллах, деформация и разрушение
к-рых подчиняются достаточно простым кристалло-
графия. законам. Одним из основных результатов
действия поверхностно-активных в-в при деформации
металлич. монокристаллов является сильное диспер-
гирование их микроструктуры.
Эффект пластифицирования в поверхностно-активных сре-
дах существенно зависит как от темп-ры, так и от скорости
деформации. Каждой темп-ре соответствует определенная до-
статочно широкая область скоростей деформации, отвечающая
наибольшему снижению прочности. Механизм пластифициро-
вания заключается в облегчении выхода дислокаций на внеш-
нюю поверхность в результате снижения энергетич. барьера,
связанного с образованием новой поверхности, а также в повы-
шении активности подповерхностных источников дислокаций,
имеющих одну точку закрепления. Для металлов наиболее
активными средами, т. е. вызывающими наибольшие адсорб-
ционные эффекты, являются поверхностно-активиые рас-
плавы легкоплавких металлов. Эти среды вызывают хрупкость
и резко понижают прочность более тугоплавких металлов.
Такое действие оказывает, в частности, расплавленное олово
на монокристаллы цинка и кадмия (а также на поликрнстал-
лич. углеродистую сталь), ртуть — на монокристаллы цинка
(рис. 2) и олова, жидкий галлий—на монокристаллы цинка,
кадмия и олова, и т. п. При выяснении механизма действия
Рис. 2. Диаграмма растяжения монокристалла цин-
ка в координатах напряжение в Г/мм* (ордината) —
относительная деформация в % (абсцисса): 1—чистый
монокристалл; 2 — монокристалл, покрытый тонким
слоем ртути.
таких сильно адсорбционно-активных сред, какими являются
расплавленные металлы, следует исходить нз анализа условий
хрупкого разрушения кристаллов (см. Хрупкость).
Из закона хрупкого разрушения кристаллов следует, что
резкое снижение величины нормальных напряжений отрыва
в присутствии поверхностно-активного металлич. расплава
связано с соответствующим резким понижением а в результате
адсорбции. Понижение <з имеет место в этом случае на разви-
вающихся внутр, поверхностях микротрещин разрушения при
деформации монокристаллов. Быстрое проникновение адсорб-
ционно-активных атомов расплава внутрь объема деформируе-
мого более тугоплавкого металла осуществляется путем нере-
гулярной диффузии (поверхностной) — миграции вдоль дефек-
тов структуры (границ блоков, полых дислокац. ядер и т. п.).
А. п. п., отвечающее значительному снижению <з, наблюдается
в том случае, когда расплав в твердом состоянии имеет малую,
ио конечную область растворимости в исследуемом металле.
Наоборот, эффект, как правило, не наблюдается, если эта об-
ласть очень широка или же, если она вовсе отсутствует. При
весьма сильном понижении свободной поверхностной энергии
твердое тело может самопроизвольно диспергироваться на
блочки, величина к-рых зависит от характера и распределения
дефектов кристалла (его дислокационной структуры). Этот
предельный случай соответствует поведению монокристаллов
олова в присутствии малых количеств жидкого галлия.
А. п. п. находит важное практич. использование
для облегчения обработки металлов давлением и
резанием (см. Смазочное действие), тонкого измель-
чения твердых тел, бурения горных пород и т. д.
Исследование А. п. п. составляет обширный раздел
41
АДСОРБЦИЯ
42
новой, пограничной области науки — физико-хими-
ческой механики. Открыто П. А. Ребиндером в 1928.
Лит.: Р е б и н д е р II. А., в кн.: VI Съезд русских физи-
ков, М., 1928; его ж е, в кн.: Юбилейный сб., посвящ. 30-ле-
тию Великой Октябрьской социалистической революции, ч. 1,
М. — Л., 1947, с. 533—61; его же, Изв. АН СССР. Отд. хим.
н., 1957, № 11, 1284; Лихтман В. И., Усп. физ. наук,
1949, 39, вып. 3, 371; его же, там же, 1954, 54, вып. 4, с. 587;
Лихтман В. И. и Щ у к ин Е. Д., там же, 1958,
66, вып. 2,213; Л и х т м а н В. И., Ребиндер П. А.,
Карпенко Г. В., Влияние поверхностно-активной среды
на процессы деформации металлов, М., 1954; Щ у к и н Е. Д.,
Ребиндер П. А., Коллоидн. ж., 1958, 20, № 5, 645.
В. И. Лихтман.
АДСОРБЦИЯ — концентрирование вещества
(адсорбата, адсорбтива) из объема фаз на поверхности
раздела между ними, напр. из газа или р-ра на по-
верхности твердого тела "(адсорбента) или жидкости.
А. является частным случаем сорбции. А. происходит
под влиянием молекулярных сил поверхности адсор-
бента и ведет к умевыпению свободной поверхностной
энергии. Молекулы адсорбата, приближаясь из объема
газа или р-ра к поверхности раздела фаз, испытывают
притяжение со стороны этой поверхности. При со-
прикосновении с нею притяжение уравновешивается
отталкиванием. Таким образом поверхность адсор-
бента покрывается тонким (адсорбционным) слоем
молекул адсорбата. При физич. А. молекулы адсор-
бата сохраняют свою индивидуальность, при химич.
А. (см. Хемосорбция) они образуют поверхностное
химич. соединение с адсорбентом. При постоянной
темп-ре Т физич. А. увеличивается с ростом давления
газа р или концентрации р-ра. С ростом темп-ры
физич. А. уменьшается, т. к. вследствие возрастания
энергии теплового движения все большая часть моле-
кул адсорбата становится способной преодолеть
притяжение к поверхности, — происходи/ десорбция.
Физич. А. обратима, она уменьшается с уменьшением
концентрации адсорбата, хемосорбция обычно необ-
ратима. Физич. А. паров часто сопровождается ка-
пиллярной конденсацией в-ва в порах адсорбента.
В области капиллярной конденсации А. необратима
(см. Гистерезис сорбционный). Измеряемая на опыте
величина А. а, относится обычно к единице веса адсор-
бента (моль/г) и зависит не только от природы адсор-
бента и адсорбата, концентрации адсорбата в объеме
и тем-ры, во и от величины поверхности адсорбента
Так как величины s для разных адсорбентов (напр.,
для непористых и пористых) различаются на веек,
порядков, то для сопоставления пользуются абс.
величинами А. а, отнесенными к единице поверх-
ности: а = a/s микромоль/м2, или молекул/iOO А2.
Абс. величина А. (при данных Тир) зависит уже
только от природы адсорбата и от природы и струк-
туры поверхности адсорбента. Адсорбционные
силы при физич. А. имеют ту же природу, что и
межмолекулярное взаимодействие в газах, жидкостях
и твердых телах.
Адсорбционные силы притяжения подразделяются на дис-
персионные силы, вызываемые согласованием движения элек-
тронов в сближающихся частицах, и на электростатич. силы,
связанные с наличием в молекулах адсорбата постоянных элек-
трич. диполей (полярные молекулы), квадруполей или вообще
мультиполей, вызываемых неравномерным распределением
электронной плотности в молекулах, к-рые в целом неполярны
(напр., в молекулах N2, СО, СО2, С2Н4, С,Н,). В случае непо-
лярного адсорбента проявляются в основном дисперсионные
силы притяжения. У полярных адсорбентов на поверхности
существует электростатич. поле, взаимодействующее с диполями
и мультиполями молекул адсорбата, что усиливает А., в осо-
бенности, когда на поверхности расположены преим. ионы од-
ного знака или сходным образом ориентированные диполи.
Последнее наблюдается, напр., при А. полярных молекул, а
также непредельных и ароматич. углеводородов на силикагелях
с гидратированной поверхностью. В этих случаях часто обра-
зуются водородные связи между гидроксильными группами
поверхности и молекулами адсорбата. На коротких расстоя-
ниях от поверхности адсорбента силы притяжения уравнове-
шиваются силами отталкивания.
Обычно А. сопровождается выделением тепла.
Теплоты физич. А. мало отличаются от теплот кон-
денсации и составляют от 1—5 (простые молекулы)
до 10—20 (большие молекулы) ккал/моль. Теплоты
хемосорбции близки к теплотам химич. реакций
(10—100 ккал/моль).
Энергия А. зависит от природы и строения
молекул адсорбата и их ориентации у поверхности,
а также от природы и структуры адсорбирующей
поверхности. Молекулы нормальных углеводородов
располагаются вдоль поверхности, энергия их А.
поэтому линейно увеличивается с ростом числа ато-
мов углерода в них. Разветвленные молекулы адсор-
бируются хуже нормальных, т. к. часть звеньев ока-
зывается удаленной от поверхности адсорбента. Энер-
гия А. ароматич. и непредельных углеводородов на
неполярных адсорбентах (напр., ва графите) меньше,
чем соответствующих н-алканов, и, наоборот, на гид-
роокисях она больше благодаря сильному взаимо-
действию облаков электронов, образующих кратные
и ароматич. связи (it-электронов), с гидроксильными
группами поверхности адсорбента. Еще более резко
при А. на гидроокисях увеличивается энергия А.
полярных молекул, напр. спиртов и аминов.
Для приближенной оценки энергии взаимодействия <р
между силовым центром (атомом, группой атомов) неполярной
молекулы адсорбата и силовым центром неполярного адсор-
бента можно пользоваться формулой Леннард-Джонса:
—С-т-«+В-г-12, (1)
где г — расстояние между взаимодействующими силовыми
центрами, С — константа притяжения, а В — константа от-
талкивания. Поэтому энергию А., представляющую энергию
взаимодействия всех атомов молекулы адсорбата со всеми ато-
мами или ионами адсорбента, находят суммированием <р по всем
центрам молекулы адсорбата i и решетки адсорбента з: Ф =
= У<р. Константу С можно вычислить из электрич. и магнит-
i 3
ных свойств адсорбата и адсорбента, а константу В — из усло-
вий равновесия сил притяжения и отталкивания (из минимума
Ф) на расстоянии от поверхности, равном сумме ван-дер-вааль-
совых радиусов ближайших (соприкасающихся) центров адсор-
бата и адсорбента.
При А. неполярных молекул на ионных решетках учиты-
вают электростатич. индукционный потенциал притяжения,
связанный с поляризацией молекулы адсорбата в электрич.
поле у поверхности, пропорциональной поляризуемости адсор-
бата. При А. полярных молекул на ионных решетках имеет
значение электростатич. взаимодействие диполей адсорбата
с электрич. полем ионов у поверхности, зависящее от величины
диполя, его ориентации, расстояния от поверхности и величины
и распределения электрич. поля адсорбента. При А. полярных
молекул определенный вклад вносит также и поляризация ими
адсорбента, что особенно важно при А. на металлах.
Изотермы А. графически выражают зависи-
мость А. от равновесного давления газа (или равно-
весной концентрации р-ра) при постоянной темп-ре.
В зависимости от интенсивности силового поля на
поверхности адсорбента и при различных внешних
условиях могут образовываться адсорбционные слои
толщиной в одну (мономолекулярная А.), две или
неск. молекул (полимолекулярная А.) При А. га-
зов и слабо адсорбирующихся паров на поверхности
твердого тела образуется один слой (монослой) моле-
кул адсорбата.
Простейшим ур-нием изотермы мономолекулярной
А. является ур-ние Ленгмюра для локализованной
(молекулы адсорбата не могут свободно двигаться
вдоль поверхности) А. на однородной поверхности,
не учитывающее взаимное притяжение между молеку-
лами адсорбата:
0 = Кр/1 +Кр; p = tf/K(l — 6) (2)
где 0 = а/ат—степень заполнения поверхности (ат—
предельная величина А. в плотном монослое), р —
давление адсорбата, а К — константа равновесия
взаимодействия адсорбат-адсорбент. В пределе при
р —> 0, 6—>Кр (т. н. область Генри), а при р—>со,
6 —> 1 (т. е. достигается насыщение поверхности),
т. о. изотерма Ленгмюра выражается выпуклой кри-
вой с пределом 6 = 1 (а — ат).
43
АДСОРБЦИЯ
44
Простейшее ур-ние изотермы А. из газовой смеси без учета
взаимодействия адсорбат-адсорбат имеет вид:
6i = Kipi/1 4- Kipi 4- К2р2 4-..(3)
где — степень заполнения поверхности молекулами данного
компонента 1, Pi, р2, ... — парциальные давления компонен-
тов смеси, a Klf К2... — соответствующие константы равнове-
сия для взаимодействий адсорбат-адсорбент. Сильно адсорби-
рующиеся примеси (большие К2...) подавляют А. данного ком-
понента 1 уже при малых парциальных давлениях р2, ...
Учет взаимодействия адсорбат — адсорбат приводит к более
сложным ур-ниям изотермы А.; простейшим из них является
ур-ние Киселева:
р = Ki (I - 6) (1 + к„е>' (4)
Рис. 1. Изотермы мономолекуляр-
ной А. на однородной поверхности
графитированной сажи.
где Kt и Кп— константы, приближенно учитывающие притя-
жение адсорбат-адсорбент и адсорбат-адсорбат. При слабых
взаимодействиях адсорбат — адсорбат (малых Кп) и сильных
•взаимодействиях адсорбат-адсорбент (большие Ki) ур-ние (4)
переходит в (2), и изо-
терма выражается выпу-
клой кривой с пределом
6= 1 (например, А. боль-
ших молекул углеводо-
родов на графите, рис. 1,
кривая /). При слабых
взаимодействиях адсор-
бат-адсорбент (малые Ki,
большие Кп) изотерма
выражается вогнутой
кривой (А. воды на гра-
фите, кривая 3, рис. 1);
при относительно близ-
ких Ki и Кп изотерма
начинается вогнутой ча-
стью, затем переходит в
выпуклую (A. SO2 на гра-
фите, кривая 2, рис. 1);
Во всех этих случаях
взаимное притяжение мо-
лекул адсорбата по мере
роста 0 увеличивает энер-
гию А. (кривая 2, рис. 1).
При А. паров на смачиваемых, поверхностях с ро-
стом относит, давления пара p/ps (где ps — давле-
Рис. 2. Изотермы полимолеку-
лярной адсорбции паров на
графитированной саже.
иие насыщенного пара
при данной температуре)
происходит образование
полимолекулярных слоев
(рис. 2)- При переходе
от преимуществ, запол-
нения первого адсорб-
ционного слоя на одно-
родной поверхности к
преимущественному за-
полнению второго (силь-
ная А., напр. гексан на
графите, рис. 2, кривая
/) происходит резкое па-
дение теплоты А., кото-
рая для второго и после-
дующих слоев близка
к теплоте конденсации
(рис.З, кривая 2).
Простейшее приближенное ур-ние полимолекулярной А.
пара на однородной поверхности (ур-ние Брунауера, Эммета
и Теллера — т. н. ур-ние БЭТ) имеет вид:
am*-:p/Ps
’(1 — P/Ps) И 4- (С — 1) p/ps]
P/Pg_________1___С — 1 р
a (1 — p/ps) ~ атС + атС р8
Здесь c=gexp (Qi — L)/RT, где g — предэкспоненциаль-
ный (энтропийный) коэфф., Qi — теплота А. в первом слое,
a L — теплота объемной конденсации. Величина Qi—L
наз. «чистой» теплотой А. в первом слое; если она велика —
изотерма имеет форму кривой 1 (рис. 2), если она мала или даже
отрицательна — форму кривой 2; в промежуточном случае —
форму кривой з. Ур-ние БЭТ не учитывает взаимодействий
адсорбат-адсорбат вдоль поверхности. Оно применимо лишь
к изотермам, имеющим форму кривой 1. В этом случае кон-
станта ат (моль/г) повволяет определять уд. поверхность
адсорбента 8 = ат N(om, где N — число Авогадро, a —
площадь, занимаемая молекулой адсорбата в плотном монослое.
Распространенным ур-нием изотермы А. является эмпирич.
ур-ние Фрейндлиха: а — Крп, где Кип — константы. Ур-ние
Фрейндлиха пригодно для средних давлений.
Поверхности твердых тел обычно геометрически
и химически неоднородны благодаря выходу на
поверхность различных кристаллография. граней,
шероховатости, пори-
стости, наличию раз-
личных поверхност-
ных химич. соедине-
ний. Поэтому обычно
теплота А. по мере
заполнения поверх-
ности уменьшается
(рис. 3, кривая 1), а
изотермы А. имеют
выпуклую форму, так
как вначале запол-
няются места с наи-
большей энергией А.
Резко изменить энер-
гию А. и вызвать из-
менение формы изо-
термы А. может хи-
мич. модифицирова-
ние поверхности. Так,
замена слоя гидро-
гексана от заполнения поверхно-
сти сажи: 1 — с неоднородной по-
верхностью; 2 — графитированной
при 3000°; L—теплота конденсации.
ксильных групп на поверхности кремнезема на
группы —О—Si(CH3)3 столь резко снижает как
электростатич., так и дисперсионный потенциал по-
верхности, что теплота А. спиртов и углеводородов
становится меньше теплоты конденсации, и величины
А. уменьшаются в десятки раз. Примером перевода
неоднородной поверхности в однородную является
термич. обработка сажи, приводящая к ее графити-
рованию с выходом на поверхность преим. базисных
граней кристаллитов графита (рис. 3).
Геометрич. или химич. неоднородность поверхности
влияет на величину а = a s. В случае особо тонко-
пористых адсорбентов величину s надежно опреде-
лить нельзя и переход от опытпых величин а к абс.
невозможен. В этих случаях сопоставляют т. н.
приведенные изо-
термы А., совмещен-
ные при к.-л. удобном
значении plps. Различие
формы приведенных изо-
терм позволяет выявить
различие структурных и
химич. свойств сравни-
ваемых адсорбентов.
В тонких порах бла-
годаря аддитивности ди-
сперсионных сил адсорб-
ционный потенциал по-
вышен; о его распределе-
нии в объеме пор можно
судить по форме изотер-
мы А. паров. В потенци-
альной теории А. (Поля-
ки) принимается, что при
заполнении объема w =
= avm (где vm—мольный
Рис. 4. Потенциальные кривые,
полученные из изотерм адсорб-
ции паров для тонкопористых
адсорбентов: 1—сильно адсор-
бирующийся пар; 2 — слабее
адсорбирующийся пар.
объем жидкого адсорбата),
адсорбц. потенциал £ = RT In ps/p, где ps — давление
пара нормальной жидкости, а р — давление пара
над адсорбентом при величине а. Зная из изотермы
А. величины а и можно вычислить «потенциальную
кривую» z(w) (рис. 4), к-рая во многих случаях прак-
тически не зависит от темп-ры. От такой кривой для
одного пара можно перейти к кривой для другого
путем умножения на т. н. коэфф, аффиности р, что
45
АДСОРБЦИЯ
46
позволяет вычислить изотермы одного пара по изо-
терме другого при разных томп-рах.
Ур-ние потенциальной кривой Дубинина и Радушкевича:
w = w„e~h-- <0
(где С0о — объем тонких пор, a k — константа) приводит
к ур-нию изотерм А. в тонких порах.
а= К’“ гхр [— В ~ (lgp/ps)2 ] (7)
°т <3'
где В — константа, зависящая от природы адсорбента и раз-
меров пор. При расширении пор адсорбционный потенциал
падает, и ур-ние принимает вид ур-ния изотермы Фрейндлиха:
а=— ехр [ А 5- 1g р/рg] или а = Крп (8)
vm Р J
где wq, А, Кип — константы.
При А. паров в более широких порах образуются
пленки жидкости с вогнутой поверхностью, на к-рой
происходит дополнит, процесс капиллярной конден-
сации. Теплоту А. можно вычислить изизостеры
А., к-рая выражает рост давления газа, необходимый
для сохранения на поверхности постоянного коли-
чества адсорбата а при увеличении темп-ры:
= lnp~-44 + C0nSt (9)
где Q — дифференциальная теплота А. Для опреде-
ления Q из (9) необходимо исследовать изотермы А. по
крайней мере при двух темп-pax. Дифференциальную
работу А. находят из изотермы А.: А = RT In p/lp,
где — давление газа в стандартном состоянии до
А., а р — равновесное давление газа над адсор-
бентом при величине а. Энтропию А. можно опре-
делить, зная Q и А. или статистико-термодинамич.
путем на основании модели движения молекулы
адсорбата, а также из зависимости теплоемкости
адсорбц. системы от темп-ры. Зная энергию и энтро-
пию А., можно вычислить адсорбц. равновесие.
При А. из р-ров на твердых адсорбентах энергия А.
А определяется разностью энергии взаимодействия
молекулы данного компонента с адсорбентом (и с со-
седними молекулами в адсорбционном слое) и с жид-
ким р-ром, т. к. молекулярное поле жидкости велико.
Часто имеют дело с конц. р-рами, так что следует
различать полное содержание а адсорбированного
в-ва в объеме адсорбционного слоя и избыток адсор-
бированного в-ва в этом объеме по сравнению с его
содержанием в равном объеме р-ра. В расчете на
единицу поверхности этот избыток
Г = а — тс (10)
где т — толщина адсорбционного слоя, ас — кон-
центрация. При А. из разбавленных р-ров или газов
обычно с мала и Г==а. но при больших с величины
Г и а сильно различаются. Для А. из р-ров характерно
взаимное вытеснение компонентов из объема поверх-
ностного слоя. Так как сжимаемость жидких р-ров
мала, то положительная А. Г2 одного компонента
сопровождается вытеснением другого, т. е. отри-
цательной его А. Г[, а именно Г2гта + Г1от2 = 0.
Зависимость а2 компонента 2 бинарного р-ра от его кон-
центрации с2 в объеме р-ра дается приближенным ур-нием
Семенченко
,,,,
“3 1 + (К - 1) vma_cs ( °
Здесь — мольный объем, а К — константа равновесия,
равная , где Д— адсорбционный потенциал, зависящий
от с2. В случае сильно адсорбирующегося вещества 2 а2 стре-
мится к пределу а2т — плотному заполнению адсорбционного
слоя молекулами компонента 2, поэтому при увеличении с2
величина Г2 сначала возрастает (малые с2, Г2 === а2), затем про-
ходит через максимум, после чего падает до нуля при с2 —
1
=----- (рис. 5, кривые 1 и 2).
vm2
В случае слабо адсорбирующихся в-в сильно сказываются
отклонения от идеальных законов в объеме р-ра; величина Д
изменяется с изменением концентрации р-ра сложным образом,
так что А. данного компонента может измениться от положи-
тельной к отрицательной (кривая з, рис. 5). Форма изотерм А.
из р-ров определяет возможность и характер их хроматогра-
фич. адсорбционного разде-
ления (см. Хроматография).
Напр., избирательность А.
из бинарных смесей углево-
дородов на силикагеле умень-
шается в последовательно-
сти: многоядерные аромати-
ческие >• алкилароматиче-
ские > олефины > нафтены
==» парафины.
Рис. 5. Изотермы А. из
жидких растворов: 1 — пол-
ного содержания компонента
2а2; 2 — его избытка Г2 при
сильной адсорбции (толуол
3 — пример изотермы
из гептана на силикагеле);
избытка Г2 при слабой А. (толуол из
гептана на графитированной саже).
Кинетика А. данного вещества зависит от его
концентрации в объемной фазе, темп-ры, химич. при-
роды и геометрич. структуры адсорбента, природы
и концентрации др. веществ в объеме и на поверх-
ности. Скорость А. зависит от диффузии молекул
адсорбата из объема к поверхности раздела фаз
(внешняя диффузия), от миграции вдоль поверх-
ности (поверхностная диффузия) и от диффузии в по-
рах (внутренняя диффузия). Диффузия в порах про-
текает значительно медленнее, чем при А. на откры-
той поверхности. Наличие посторонних веществ сильно
тормозит все эти виды диффузии. Физич. А. газов
протекает почти мгновенно, если она не осложняется
побочными процессами — медленным проникновением
газа в капилляры и др. А. из жидких р-ров характе-
ризуется более медленным течением процесса, что
связано с меньшей скоростью диффузии в поверх-
ностный слой и в глубь пор адсорбента. Скорость
хемосорбции обычно при низкой темп-ре мала и
растет с повышением темп-ры как скорость химич.
реакции (т. н. активированная адсорб-
ция, см. Хемосорбция).
Скорость самой физической А. и' (не осложненной диффу-
зией молекул адсороата к поверхности) определяется числом
столкновений с поверхностью z, пропорциональным давлению
газа р (z — p/VZnmkT, где т — масса молекулы газа, k —
константа Больцмана) и незанятой поверхностью 1—6:
w = У '--—. (1 - е)
V2nmhT
(12)
где £ — вероятность того, что столкновение с поверхностью
приведет к А. (иеупругий удар; при физич. А. при невысоких
темп-pax обычно | = 1).
Скорость десорбции и" с однородной поверхности пропор-
циональна занятой поверхности 9 и уменьшается с ростом по-
тенциального барьера, вызванного притяжением к адсорбенту,
к-рый молекулам необходимо преодолеть, чтобы покинуть по-
верхность (т. е. с ростом теплоты A. Q):
и" = Q/RT, (13)
где k0 — константа скорости.
Таким образом, скорость А. с учетом десорбции
и = и' — и" =-- Е . р (1 _ 0) — h09e— Q/R? (14)
V2*mhT
при равновесии и' равно и" и (14) приводит к ур-нию Ленг-
мюра (2), в к-ром
к le-Q/RT
kQ V2nmkT
Неоднородность поверхности и взаимодействие между мо-
лекулами адсорбата приводят к сложной зависимости и Q
от 9. В случае хемосорбции вероятность S связана также с не-
обходимостью благоприятной ориентаций молекулы по отно-
шению к реагирующему центру поверхности адсорбента.
Кроме того, и' зависит от энергии активации А. Е', а и" — от
энергии активации десорбции Е, причем Е" == Е' Ц- Q. Хемо-
сорбция часто сопровождается диссоциацией молекул адсор-
бата, что усложняет зависимость и от 0.
В процессах А. из потока газа или жидкости (д и-
н а м и к а А.) проникновение молекул внутрь зерен
адсорбента зависит от скорости потока и размеров
47
АДСОРБЦИЯ —АДСОРБЦИЯ РАДИОАКТИВНЫХ ЭЛЕМЕНТОВ
48
пор; сужение пор затрудняет внутреннюю диффузию.
В динамич. условиях А. примеси из носителя (газа
или жидкости) происходит при прохождении смеси
в промежутках между зернами адсорбента; сам но-
ситель практически не адсорбируется. При проте-
кании газовой или жидкой смеси с начальной кон-
центрацией примеси с0 через слой адсорбента, за
слоем в течение нек-рого промежутка времени кон-
центрация примеси остается близкой к нулю, затем
она начинает расти и постепенно достигает величины е0.
Момент «проскока» примеси отвечает появлению ее
за слоем адсорбента при нек-рой минимальной кон-
центрации с, к-рую удается заметить. Время до
проскока наз. временем защитного действия слоя
(г), зависит от длины слоя адсорбента. Зависимость
концентрации за слоем (на выходе) от времени наз.
выходной кривой. Скорость А. нескольких адсорби-
рующихся в-в из потока носителя (газа или жидкости)
зависит также и от скорости перераспределения
этих веществ на адсорбенте. В первые моменты ком-
поненты смеси адсорбируются независимо, затем
менее адсорбирующийся компонент вытесняется лучше
адсорбирующимся. Первым за слоем адсорбента
выходит наиболее слабо адсорбирующийся компонент
(см. Хроматография).
Для измерения А. на твердых адсорбентах из газов
пользуются (если 8 больше 10 лг2/г) непосредственным взвеши-
ванием адсорбента, находя величину А. по увеличению веса.
Применяются пружинные весы Мак-Бэна — Бакра, коромыс-
ловые регистрирующие весы, удобные для измерения кине-
тики А. При точных измерениях А. газов применяют газовый
объемный метод (в газовой бюретке измеряют объем газа до
и после А.), а также метод сравнения натекания газа через
капилляр в пустую ампулу и в ампулу, содержащую адсорбент
(напр., пленку металла на ее стенках). Для определения А.
паров измеряют объем жидкости, испарившейся из микро-
бюретки при ее сообщении с ампулой с адсорбентом. При
использовании вакуумных статич. методов адсорбент перед из-
мерением откачивают в вакууме. Динамич. метод измерения А.
основан на пропускании газа или пара в токе газа-носителя
через трубку, содержащую адсорбент, до появления компо-
нента смеси за слоем адсорбента. А. находят по привесу трубки
(если газ-носитель практически не адсорбируется) анализом
после десорбции или исследованием хроматограмм.
А. на поверхности жидкости из газа
или из р-ра непосредственно не измеряется, а вычисляется из
измерений поверхностного натяжения с помощью Гиббса урав-
нения абсорбции.
А. из растворов на твердых адсорбен-
тах измеряют непосредственно (если s достаточно велика),
по разности введенного количества данного компонента и остав-
шегося в равновесном р-ре после А. Применяется также дина-
мич. метод.
Непосредственное измерение теплоты
А. производят в чувствительных калориметрах: теплоты А.
обычно невелики и медленно выделяются, поэтому стремятся
по возможности снизить и сделать постоянным теплообмен
с оболочкой калориметра. Удобен изотермич. способ, когда
теплота А. компенсируется ослаблением электрич. тока в нагре-
вателе калориметра, а теплота десорбции усилением этого
тока, так что темп-pa калориметра сохраняется постоянной.
При А. газов на металлич. нитях калориметром служит сама
нить, измеряется изменение ее сопротивления.
А. применяется для разделения газовых и жидких
смесей (см. Хроматография), осушки и очистки газов
(напр., очистки воздуха в противогазах) и жидкостей.
А. является первой стадией взаимодействия молекул
реагирующей смеси с поверхностью при гетероген-
ном катализе, играет существенную роль при введе-
нии наполнителей в полимеры, загустителей в смазки,
при введении смачивателей, при флотации, крашении,
модифицировании многих материалов, в полиграфич.
и строит, пром-сти. А. пленок активных металлов
и окисей влияет на работу выхода в электронных
приборах. А. играет большую роль во многих биоло-
гия. и почвенных процессах.
Лит.: Дубинин М. М., Физико-химические основы сорб-
ционной техники, 2 изд., М.—Л., 1935; Б р у и а у е р С., Ад-
сорбция газов и паров, пер. с англ., т. 1, М., 1948; Ильин Б. В.,
Природа адсорбционных сил, М.—Л., 1952; Boer J. Н.
d е, The dynamical character of adsorption, Oxf., 1953;
Поверхностные химические соединения и их роль в явле-
ниях адсорбции, Сб. трудов, под ред. А. В. Киселева, М.,
1957; Семенченко В. К., Поверхностные явления в ме-
таллах и сплавах, М., 1957; Т ре пн ел Б. М. В., Хемосорб-
ция, пер. с англ., М., 1958; Методы исследования структуры
высокодисперсных и пористых тел. Труды 2-го совещания,
13—18 июня 1956, [ред. М. М. Дубинин], вып. 1—2, М., 1958;
The structure and properties of porous materials, ed. by
D. H. Everett, F. S. Stone, L. — Butterworth, 1958; Получение,
структура и свойства сорбентов, Л., 1959; Де - В у р Я. X.,
Явления адсорбции, в |сб.]: Катализ. Некоторые вопросы
теории и технологии органических реакций, пер. с аигл., М.,
1959; К и с е л е в А. В., Усп. хим., 1946, 15, вып. 4,
456—84; Дубинин М. М., Усп. хим., 1955, 24, вып. 1,
с. 3; Родин Т., Вакуумные микровесы, в [сб.]: Катализ.
Исследование гетерогенных процессов, пер. с англ., М., 1956;
А в гу ль Н. Н. [и др.], Изв. АН СССР. Отд. хим. н., 1957,
№ 11, с. 1314; Муттик Г. Г., Ж. физ. хим., 1957, 31, вып.
1, с. 263; Киселев А. В. и Эльтеков Ю. А., там же,
с. 250; Киселев А. В., Вести. АН СССР, 1957, № 10, 43; К и-
селев А. В., Коллоидн. ж., 1958, 20, № 3, с. 338; Кисе-
лев А. В. [и др.], там же, № 4, с. 444. А. В. Киселев.
АДСОРБЦИЯ РАДИОАКТИВНЫХ ЭЛЕМЕН-
ТОВ — удержание радиоактивных элементов поверх-
ностью твердого тела (адсорбента), находящегося
в жидкости. А. р. э. является одним из важнейших
явлений, определяющих поведение радиоэлементов
в растворе. Присутствуя обычно в микроконцентра-
циях, радиоактивный элемент легко адсорбируется
стенками сосуда, а также др. поверхностями, сопри-
касающимися с раствором, что ведет к ряду экспе-
риментальных затруднений. Радиоэлемент может за-
мещать молекулы или ионы поверхностного слоя
ионообменников, стекла или кристаллич. адсорбентов
(первичная адсорбция), участвовать в построении
внешней обкладки двойного электрич. слоя на границе
раздела фаз (вторичная адсорбция) или удерживаться
на поверхности силами Ван-дер-Ваальса (специфич.
адсорбция).
Первичная А. р. э. на ионообменниках подчиняется
общим закономерностям ионного обмена. Первичная
адсорбция на кристаллич. осадках возможна в слу-
чаях, когда кристаллич. структура соединения,
образуемого радиоэлементом с противоположно за-
ряженным ионом раствора, двумерно подобна струк-
туре адсорбента. В этом случае при равновесии
твердой фазы с раствором адсорбция описывается
х D • S
соотношением {—где х— доля радио-
элемента, адсорбированного на осадке, А — число
молекул адсорбента, составляющее обменивающуюся
поверхность, v — объем жидкой фазы (пара), т —
растворимость адсорбента, Ь — коэфф, адсорбцион-
ного распределения, величина постоянная при данной
темп-ре и не зависящая от заряда поверхности.
Вторичная адсорбция происходит вблизи поверх-
ностей, имеющих заряд, противоположный заряду
ионов радиоэлемента. Вторичная адсорбция радио-
элемента подчиняется след, закономерности: =
= а , где х — доля адсорбированного радио-
элемента, к0 — константа распределения радиоэле-
мента между раствором и двойным электрич. слоем,
возрастающая с увеличением радиуса ионов радио-
элемента, а — величина, зависящая от заряда поверх-
ности и присутствия посторонних ионов, z — заряд
иона радиоэлемента, о — объем раствора.
А. р. э. на стекле включает явления, присущие
первичной и вторичной адсорбции. Знание адсорб-
ционных свойств радиоэлементов является необхо-
димым условием правильной работы с радиоактив-
ными в-вами. В ряде случаев явление адсорбции
бывает нежелательным. Его можно предотвратить
введением в раствор ионов стабильных изотопов,
одноименных с радиоактивным, посторонних много-
валентных ионов или поверхностно-активных в-в.
Во многих же случаях, наоборот, явление адсорбции
используется для разделения радиоактивных эле-
ментов, для определения состояния радиоэлементов
в растворе, очистки растворов от радиоактивных
загрязнений и т. д.
49
АДУРОЛ — АЗЕОТРОПНЫЕ СМЕСИ
50
Лит.: Бреслер С. Е., Радиоактивные элементы, 3
изд., М., 1957, с. 176—232; Никольский Б. П., Ж. неорг.
хим., 1958, 3, вып. 1, 59. Н. Э. Хандамирова.
АДУРОЛ — см. Проявляющие вещества.
АЗАСЕРИН (О-диазоацетил-Ь-серин), мол. в. 173,0,
N2CHCOOCH2CH(NH2)COOH — антибиотик, подав-
ляющий рост злокачественных образований. Вы-
делен из продуктов жизнедеятельности одного из ви-
дов Streptoinyces хроматография, адсорбцией на ко-
лонке из окиси алюминия и угля. А. — светло-желто-
зеленые иглы, т. пл. от 146° до 162° (с разложением);
уд. экстинкция Е (1%, 1 см) 1136 при % 250,5 ммк;
[аГВ’а=—0,5° (8,46%, вода, pH 5,18); [а]^ =+ 9,7°
(2н...НС1);' рКа 8,55. А. хорошо растворяется в воде,
слабо — в абс. метаноле, абс. этаноле и ацетоне;
дает положительную нингидринную реакцию и вос-
станавливает аммиачный раствор AgNO3. А. является
весьма лабильным соединением. При гидролизе в 2 М
растворе НСООН получают L-серин и гликолевую к-ту.
При действии минеральных к-т А. разлагается с выде-
лением азота; в водных р-рах в присутствии Ва(ОН)2
изомеризуется в N-диазоацетил-Ь-серин. При ката-
литич. гидрогенизации А. (палладиевая чернь) обра-
зуется О-ацетил-Ь-серин. А. получен синтетически
в 1954 по схеме:
HOCH2CH(NHCOOCH2CeH5)COOH + сн2—СО\^НС1
NH—-СО / '
— НС1 • H2NCH2COOCH2CHCOOH —
NHCOOCH2CeH5
— НС1 • H2NCH2COOCH2CHCOOH
nh2
N2CHCOOCH2CH(NH2)COOII
Известны др. методы синтеза. А. в эксперименте обна-
руживает тормозящее действие на рост мышиной
саркомы в дозах, оказывающих общетоксич. действие.
При клинич. испытаниях с различными злокачествен-
ными опухолями наряду с кратковременными улуч-
шениями у больных наблюдалась общая интоксика-
ция с нарушением функций печени и поджелудочной
ция с нарушением функций печени и
600
400
300
200 -
100 -
Р мм.
рт. ст.
600
о> 80
45
в
О 20 40 60 80 100
б Мал. % яизконипящего
S 60
|
5 40
0 20 40 60 80 100
а Мол. % низкокипящего
Рис. 1. Диаграммы фазового равновесия бинарной смеси сероуглерод — ацетон: а — зависимость давлений паров ком-
понентов и их смесей от состава последних при 35,2°; б — зависимость темп-ры кипения от составов жидкой и паровой
фаз при давлении 1 ат (абс.); в — кривая фазового равновесия при давлении 1 ат (абс.).
________________________________X
О 20 40 60 80 100
Мол. % низкокипящего
в жидкое фазе
6
железы. Антибактериальная активность А. в спектре
незначительна.
Лит.: Трахтенберг Д. М., Мед. пром. СССР,
1955, № 4, с. 37; F и s а г i S. А. [а. о.], J. Amer. Chem.
Soc., 1954, 76, р. 2878; Moore J. A. la. o.J, там же,
p. 2884. E. С. Северин.
АЗЕЛАИНОВАЯ КИСЛОТА (н-гептандикарбоно-
вая-1,7-кислота) НООС(СН2)7СООН, мол. в. 188,21 —
двухосновная предельная к-та, диморфна. Обе формы
моноклинны и имеют одинаковую т. пл. 106,5° (раз-
личаются рентгенографически); т. кип. 287°/100 мм,
237°/15 мм; при обычном давлении перегоняет-
ся выше 360°, частично превращаясь в ангидрид,
не перегоняется с водяным паром; 1,225; кон-
станты диссоциации (25°): Кг = 2,88 • 10 ь, К2 =
— 2,8 • 10 е. Растворимость А. к.: в 100 мл воды при
0° 0,1 г, при 65° 2,2 г; в 100 г диэтилового эфира при
15° 2,68 г; лучше растворима в спирте. Образуется
вместе с другими кислотами при окислении касто-
рового масла, а также олеиновой к-ты или ее мети-
лового эфира. При пиролизе кальциевой соли А. к.
наряду с другими продуктами образуется циклоок-
танон; при нагревании с глицерином получается пла-
стичная смола, с касторовым маслом — эластомер,
напоминающий Каучук. М. И. Роаенгарт.
АЗЕОТРОПНЫЕ СМЕСИ — растворы, перегоняю-
щиеся без изменения состава и темп-ры кипения,
т. е. без разделения; их часто называют нераздельно-
или постояннокипящими. Дж. Дальтон (1810) впер-
вые обнаружил, что таким свойством обладает вод-
ный р-р азотной к-ты, содержащий 68 вес. % HNO3. Г.
Роско (1859) установил, что состав А. с. зависит от
внешнего давления, опровергнув тем самым первона-
чальное объяснение явления азеотропизма образова-
нием химич. соединения между компонентами смеси.
Д. П. Коновалов (1884) показал, что А. с. существуют
в таких системах, у к-рых зависимость давления пара
р-ра от его состава при постоянной темп-ре или
зависимость темп-ры кипения от состава при постоян-
ном давлении выражаются кривыми, имеющими
экстремальные точки (точки минимума или макси-
мума). В соответствии с этим различают А. с.: 1) с ми-
нимумом темп-ры кипения (следовательно, с макси-
мумом давления пара) и 2) с максимумом темп-ры
кипения (с минимумом давления пара).
Примером смеси первого вида может служить система серо-
углерод — ацетон, для к-рой на рис. 1 представлены графики
зависимости:
р=/(х), t = <р (х, у), у = ф(х)
где х — концентрация низкокипящего компонента (CS2) в жид-
кой фазе, выраженная в мол. %; у — концентрация того же
компонента в равновесной паровой фазе в
мол. %; р — давление пара смеси ком-
понентов в мм рт.ст., t — темп-pa кипе-
ния жидкой смеси в °C.
Зависимость p=j(x) для бинарных смесей, под-
чиняющихся закону Рауля, является, как известно,
линейной. Если бы CS2 и CgHeO представляли такую
смесь, то изменение парциальных давлений паров
компонентов и полного давления наров смеси в за-
висимости от состава жидкой фазы происходило бы
по прямым, показанным на рис. 1, а. Однако откло-
нения рассматриваемой системы от закона Рауля
51
АЗЕОТРОПНЫЕ СМЕСИ
52
положительны, поэтому зависимости давлений паров
от состава жидкой фазы характеризуются кривыми,
расположенными выше прямых Рауля. Более того,
в данном случае на кривой полного давления паров р
смеси имеется экстремальная точка (относительный
максимум давления), соответствующая как раз со-
ставу А. с. (67 мол. % сероуглерода и 33 мол. % аце-
тона).
Темп-pa кипения бинарных смесей, не обладающих
азеотропной точкой, как известно, непрерывно па-
дает с возрастанием концентрации низкокипящего
компонента в жидкой фазе. Для таких смесей зависи-
мость t = <р(х, у) изображается двумя кривыми
(жидкость — пар), показанными пунктиром на
рис. 1, б. Характерной особенностью таких бинарных
смесей является, наконец, более высокая концентра-
ция низкокипящего компонента в паровой фазе,
нежели в равновесной жидкой фазе. На том же рис.
1, б показаны сплошными линиями кривые зависи-
мости t = <р(х, у) для системы CS2 — С3НвО. Обе
кривые сходятся в точке, соответствующей составу
А. с., что совершенно закономерно, т. к. у такой
смеси равновесный пар одинаков по составу с жид-
костью. Максимуму давления на кривой р = /(х)
(рис. 1, а) соответствует минимум темп-ры кипения
па кривых t — <р(х, у) (рис. 1,6). И действительно,
А. с. CS2— С3НвО под давлением 1 ат (абс.) имеет
более низкую темп-ру кипения (39,25°), чем ее ком-
поненты (т. кип. CS2 46,25°, а т. кип. С3НвО 56,25°).
Характерной особенностью рассматриваемой бинар-
ной смеси благодаря наличию азеотропной точки
является то, что при х С хаз пар содержит больше
низкокипящего компонента, чем равновесная жид-
кая фаза (у > х). За азеотропной точкой (х > хаз.)
имеет место обратная картина: у с х.
На рис. 1,е приведена диаграмма фазового
равновесия для рассматриваемой бинарной смеси
у = ф(х) приведены на рис. 2. Из рис. 2, а видно,
что отклонения данной системы от закона Рауля
отрицательны (кривые давлений расположились ниже
прямых Рауля, показанных на диаграмме), причем
на кривой полного давления имеется экстремальная
точка (минимум давления), соответствующая составу
А. с. (38,3 мол.% HNO3 и 61,7 мол.% Н2О). Мини-
муму давления на кривой р = /(х) закономерно
соответствует максимум темп-ры кипения на кривых
t = <р(х, у). Под давлением 1 ат (абс.) А. с. кипит
при более высокой темп-ре (120,5°), чем ее компо-
ненты (т. кип. HNOS 86°, т. кип. Н2О 100°). Для дан-
ной смеси характерно у <z х при х хаз и у х при
х > хаз_. Это еще нагляднее показано на диаграмме
у — х (рис. 2, в).
М. С. Вревский (1911), исследуя влияние внешних
условий на состав А. с., установил, что у А. с. с мини-
мумом темп-ры кипения повышение давления вызывает
рост концентрации компонента с большей теплотой
парообразования, а у смесей с максимумом темп-ры
кипения — рост концентрации компонента с мень-
шей теплотой парообразования. Это правило согла-
суется с тем фактом, что перемещение азеотропной
точки в диаграмме фазового равновесия с изменением
внешнего давления может происходить для одних
смесей в направлении больших, а для других сме-
сей — в направлении меньших концентраций низко-
кипящего компонента. Сказанное справедливо как
для смесей с максимумом, так и для смесей с мини-
мумом темп-ры кипения. Так, напр., А.с. С.2Н5ОН—Н2О
и С3Н,ОН — Н2О имеют минимумы темп-p кипения,
но с уменьшением внешнего давления от 760 мм до
50 мм рт. ст. у первой из них концентрация низко-
кипящего компонента возрастает от 95,57% до 97%,
а у второй — падает от 71,7% до 68,2%. В то же
время А. с. Н2О—СН2О2 и НХО3 — Н2О имеют
максимумы темп-p кипения, но с уменьшением внеш-
ние. 2. Диаграммы фазового равновесия бинарной смеси азотная кислота — вода: а — зависимость давлений
паров компонентов и их смесей от состава последних при 75°; б — зависимость темп-ры кипения от состава
жидкой и паровой фаз при давлении 1 ат (абс.); в — кривая фазового равновесия при давлении 1 ат (абс.).
CS2 — С3НвО в системе координат у — х, где по
составу жидкой фазы проще всего отсчитывается
состав равновесной паровой фазы. Кривые равнове-
сия смесей, не имеющих азеотропной точки, распо-
лагаются над диагональю квадратного поля диа-
граммы, как это показано пунктиром на рис. 1,в.
Кривая равновесия рассматриваемой бинарной смеси
закономерно пересекает диагональ квадрата в точке,
соответствующей составу А. с. (точкам по диагонали
соответствует у = х). Здесь отчетливо видно, что
прих<Схаз пар богаче, а при х > хаз пар беднее низко-
кипящим компонентом, чем равновесные с ним жид-
кие фазы.
Примером смеси второго вида может служить си-
стема азотная кислота — вода, для к-рой все три
диаграммы фазового равновесия р = /(х); t = <р(х, у)‘,
него давления от 760 мм до 50 мм рт. ст. весовая
концентрация низкокипящего компонента в первой
из них возрастает от 22,5% до 34%, а во второй —
падает от 88,1% до 87,4%. Таким образом, внешнее
давление влияет на состав А. с. Изменение коэфф,
разделения смеси и перемещение азеотропной точки
с давлепием создают благоприятные условия для
разделения А, с. последовательно при двух разных
давлениях.
Многокомпонентные смеси, подобно бинарным,
также имеют часто азеотропные точки, характери-
зующиеся преим. максимумом давления и минимумом
темп-ры кипения. Тройные смеси с минимумом давле-
ния и максимумом темп-ры кипения встречаются
очень редко. Минимум темп-ры кипения у тройных
смесей наблюдается в тех случаях, когда все три или,
53
АЗЕОТРОПНЫЕ СМЕСИ
54
по крайней мере, две бинарные смеси, а иногда даже
одна из них, содержащие те же компоненты, имеют
минимумы темп-ры кипения. При этом у тройных
смесей наблюдается более глубокий минимум темп-ры
кипения, чем у бинарных смесей, составленных из
тех же компонентов. Так, папр., из компонентов
тройной смеси С2Н5ОН — СвНв — Н2О могут быть
образованы три бинарные смеси: 1) С2Н5ОН — Н2О,
2) СвНв — Н2О и 3) С2Н5ОН — СвНв. Все эти три
бинарные смеси имеют минимумы темп-p кипения,
равные соответственно 78,2°, 69° и 68,3°, а тройная
смесь кипит при еще более низкой темп-ре (64,9°).
Бинарные смеси, имеющие азеотропные точки,
могут быть разделены методом перегонки на А. с.
и тот компонент, к-рый содержался в исходном р-ре
в избытке по сравнению с составом А. с. Перегонка
тройных смесей с азеотропными точками допускает
большее число вариантов, как в этом можно убедиться
на следующем примере. На рис. 3 в поле равносто-
ронпего треугольника построены кривые, характе-
Рис. 3. Кривые перегонки тройных смесей с азеотропными точками:
а—С3НвО — Н2О — СвНбОН; б—С2Н6ОН—С6Н6—Н2О.
С2Н5ОН
ризующие процесс перегонки тройной смеси ацетон —
вода — фенол. Из числа компонентов этой смеси вода
и фенол образуют А. с., соответствующую точке М
на рис. 3, а. Кривая, соединяющая вершину треуголь-
ника (ацетон) с точкой М, делит поле диаграммы на
две области: А и В. Как видно из диаграммы, при
перегонке жидких смесей области А невозможно
отделить воду, а при перегонке смесей области В
невозможно отделить фенол. Если из компонентов
тройной смеси могут быть образованы три бинарные
смеси с азеотропными точками, то пучок кривых
перегонки образует в поле равностороннего тре-
угольника три области. В качестве примера на рис. 3, б
приведена диаграмма для тропной смеси С2Н5ОН —
СвНв — Н2О, из компонентов к-рой могут быть обра-
зованы три бинарные смеси с • указанными выше
минимумами темп-p кипения. Состав тройной А. с.
характеризуется точкой М, а составы бинарных
А. с. — точками Mlt М2, М3 на сторонах треуголь-
ника. Линии MMlt ММ,, и ММ3 делят поле диаграммы
на три области: Л, В и С. Исходные жидкие смеси,
расположенные в каждой из трех областей, могут
быть при перегонке разделены на такие смеси, составы
к-рых расположены в пределах той же области, и на
А. с., характеризуемую точкой М. Так, смеси области
А могут быть разделены на этанол и А. с. состава М,
если их исходные составы располагаются на пря-
мой т, соединяющей точку М с вершиной треуголь-
ника, соответствующей чистому этанолу. Аналогично,
смеси областей В и С могут быть при перегонке
разделены соответственно на воду и А. с. состава Л/,
на бензол и ту же тройную А. с. С другой стороны,
из диаграммы следует, что свойством тройной А. с.
можно иногда воспользоваться для разделения би-
нарных смесей, имеющих азеотропные точки. Так,
для отгонки чистого этанола из бинарной смеси
этанол — вода (точка F на диаграмме тройной смеси)
достаточно прибавить к этой смеси такое количество
бензола, к-рое требуется для образования тройной
смеси, располагающейся на прямой т. Состав этой
тройной смеси соответствует точке Е, образованной
пересечением прямой т с прямой, соединяющей
точку F с вершиной чистого бензола. Отгонка прак-
тически чистого этанола стала возможной по той
причине, что после прибавления бензола образова-
лась тройная смесь, которая имеет более глубокий
минимум темп-ры кипения и содержит в азеотропной
точке больше воды, чем А. с. этанол — вода. При-
бавление третьего компонента для разделения бинар-
ных А. с., метод выбора к-рого в общем виде до сих
пор не разработан, часто используется на практике
(см. Ректификация азеотропная).
При наличии бинарной смеси с азеотропной точкой
и ограниченно растворимого третьего компонента не
всегда образуется тройная смесь с азеотропной точкой.
Так, изоамиловый спирт ограниченно растворим
в воде, а
смесь этанол — вода имеет азеотропную
точку. Однако тройная смесь изоами-
ловый спирт — этанол — вода не имеет
азеотропной точки; это объясняется тем,
что А. с. этанол — вода имеет бо-
лее низкую температуру кипения, чем
изоамиловый спирт — вода в присут-
ствии этанола. Это обстоятельство по-
зволяет отделить изоамиловый спирт
от смеси этанол — вода и практически
используется для удаления сивушного
масла в процессе ректификации этило-
вого спирта брожения.
Известно, что ок. 50% жидких смесей
промышленного значения имеют азео-
тропные точки. В связи с необходимостью
разделения этих смесей на практи-
чески чистые компоненты изучение свойств А. с. имеет
очень большое значение. Однако, несмотря на ряд
серьезных исследований в этой области (Д. П. Коно-
валов, М. С. Вревский, А. В. Сторонкин, В. А. Ки-
реев, В. А. Свентославский, М. Лека и др.), до сих пор
невозможно ни точно предсказать образование А. с.,
ни рассчитать заранее составы и темп-ры кипения
А. с., а также количественную зависимость состава
А. с. от величины внешнего давления. Предложенные
многочисленные эмпирич. и полуэмпирич. формулы
не отличаются высокой точностью и, в лучшем случае,
справедливы только в очень ограниченных областях.
На основании накопленных экспериментальных дан-
ных сформулированы нек-рые качественные правила,
к-рые, однако, не свободны от исключений. Так,
смеси с минимумом давления и максимумом темп-ры
кипения образуются чаще всего сочетанием следующих
компонентов: 1) кислот с водой, аммиаком, органич.
основаниями или кетонами; 2) фенолов с аммиаком,
органич. основаниями, спиртами, альдегидами, кето-
нами и сложными эфирами; 3) кетонов с галогено-
производными углеводородов; 4) полигалогенопроиз-
водных с кетонами, сложными эфирами, углеводами;
5) альдегидов или кетонов с цианистой к-той, серни-
стой к-той, ненасыщенными углеводородами. А. с.
с максимумом давления и минимумом темп-ры кипе-
ния образуются гл. обр. компонентами, содержащими
гидроксильную группу: водой, спиртами, кислотами.
С качественной точки зрения явление образования
А. с. может быть объяснено различным характером
молекулярного взаимодействия между разнородными
молекулами. Если молекулы веществ А и В оказывают
друг на друга такое же молекулярное притяжение,
как однородные молекулы А и А или В и В, то все
эти молекулы могут взаимно замещаться в любых
количественных соотношениях, не вызывая при
смешении изменения темп-ры и объема. Такие смеси
55
АЗИД СВИНЦА —АЗИДЫ КАРБОНОВЫХ КИСЛОТ
56
подчиняются закону Рауля и, разумеется, А. с. не
образуют. Для таких смесей справедливо след, усло-
вие: аАВ —]^аАав, где аА— притяжение между одно-
родными молекулами компонента А, ав— притя-
жение между однородными молекулами компонента
В, аАВ — притяжение между разнородными молеку-
лами А и В. Если молекулярное притяжение между
А и В больше, чем между А ъ А или В и В, то при
смешении освобождается потенциальная энергия,
обусловливающая повышение темп-ры и уменьшение
объема. В этом случае (а АВ д°в) молекулы в
смеси удерживаются сильнее и для их разделения тре-
буется более высокая темп-ра, способная ослабить
действие повышенного молекулярного притяжения.
Естественно, что смесь обнаружит при этом отри-
цательное отклонение от закона Рауля, а при значи-
тельном размере этого отклонения появится азеотроп-
ная точка с максимумом темп-ры кипения. Если же
молекулярное притяжение между А и В меньше, чем
между А я А или В и В, т. е. <?АВ С то
при смешении компонентов увеличится потенциальная
энергия, так как сильно притягивающие друг друга
однородные молекулы отчасти разделяются, требуя
сообщения нек-рого количества теплоты. Отрица-
тельный тепловой эффект обусловливает падение
темп-ры при смешении компонентов, а слабое притя-
жение между разнородными молекулами вызывает
нек-рое увеличение объема смеси и возрастание
упругости пара. В данном случае смесь обнаружит
положительное отклонение от закона Рауля, а при
большой степени этого отклонения появится А. с. с
максимумом давления и минимумом темп-ры кипения.
Лит.: Коновалов Д. [П.], Об упругости пара раство-
ров, 3 изд., Л., 192 8; Вревский М. С., Работы по теории
растворов, М.—Л., 1953; Swietoeuwski W., Azeotropia
1 poll azeotropia, t. 1, Warez.. 1957; Сторонки нА., Об усло-
виях термодинамического равновесия многокомпонентных си-
стем, М., 1948; Аносов В. Я. и Погодин С. А.,
Основные начала физико-химического анализа, М.—Л., 1947;
Киреев В. А., Курс физической химии, 2 изд., М., 1956;
Хорсли Л., Таблицы азеотропных смесей, пер. с англ.,
М. 1951; Lee at М., Tables azeotropiques t. 1,2 ed., Brux.
1949. H. И. Гелъперин.
АЗИД СВИНЦА Pb(N3)2 — свинцовая соль азо-
тистоводородной к-ты; инициирующее взрывчатое
вещество, бесцветные кристаллы; известны две кри-
сталлич. формы: а-орторомбическая (плоти, d 4,71),
р-моноклинная (d 4,93). А. с. плохо растворим в воде,
лучше — в р-рах нитрата и ацетата натрия и ацетата
аммония, хорошо растворим в моноэтаноламине.
Теплота взрыва А. с. 397 кал/а; при нагревании не
плавится и не горит, а детонирует; т. всп. ок. 300°;
скорость детонации 5100 м/сек при d = 4. Чувстви-
тельность А. с. к теплу и удару меньше, а инициирую-
щее действие и химич. стойкость больше, чем гремучей
ртути. При действии к-т, оснований, а также при
кипячении в воде А; с. разлагается; в присутствии
металлич. меди образуется весьма чувствительный
азид меди. При получении или перекристаллизации
А. с. (особенно jl-формы) возможен самопроизвольный
взрыв. А. с. получают при взаимодействии растворов
азида натрия и нитрата свинца; применяют в кап-
сюлях-детонаторах, как правило, совместно с др.
инициирующими взрывчатыми веществами, напр.
тринитрореаорцинатом свинца. Для определения
А. с. его обрабатывают р-ром церийаммонийнитрата
и измеряют выделяющийся при этом азот.
Лит. см. при Ст. Азиды. Б. Н. Кондриков.
АЗИДЫ — соли азотистоводородной к-ты HN3,
а также соединения, содержащие одну или неск.
групп —N3, напр. хлоразид CIN3 или тринитротри-
азидобензол Ce(NO2)3(N3)3. Большинство солен HN3
взрывчаты. Исключение составляют только А. щелоч-
ных металлов: они при нагревании разлагаются
быстро, но без взрыва. А. натрия получают действием
закиси азота на амид натрия при 200°: NH2Na -f-
+ N2O = NaN3 4- H2O. А. свинца и др. тяжелых
металлов трудно растворимы в воде, их получают
из А. натрия. Многие А. весьма чувствительны,
взрывают при легком ударе или трении даже ио
влажном состоянии. А. свинца широко используют
в качестве инициирующего взрывчатого вещества.
Значительной взрывчатостью отличаются и др. не-
органич. производные HNS : А. иода JN3, хлора
C1N3, азидотиоугольная к-та N3—С—S—Н и др.
Все органич. А., алкильные и арильные (RN3) или
ацильные (RC<^ ), в той или иной мере также обла-
XN3
дают взрывчатыми свойствами. Примерами органич. А.
могут служить: нитрогуанилазид HN=C(NHNO,)N~,
5-азидотетразол
HN-N=N-N=CN3
и циануртриазид C3N3(N3)3. Для радикала — N3 при-
нята линейная структура: —N=N= N.
Лит.: Бубнов П. Ф., Инициирующие взрывчатые ве-
щества и средства инициирования, ч. 1, М., 1940, гл. 4;О г а уР.
and Waddington Т. С., Proc. Roy. Soc., Ser. А., 1956,
235, № 1200, р. 106—119, 481—95; 1957, 241, р. 110—21; Kirk,
v. 7, N. Y., 1951, р. 591—95. А. А. Шидловский.
АЗИДЫ КАРБОНОВЫХ КИСЛОТ (азоимиды) —
//°
органич. соединения общей формулы RC^ , обычно
XN3
кристаллич. (реже жидкие) продукты, довольно легко
разлагаются, многие из них ядовиты и взрывчаты.
А. к. к. способны перегруппировываться с образова-
нием эфиров изоциановой к-ты:
RCON3 -► RN=CO + N3
Перегруппировка А. к. к. проходит в среде от ней-
тральной до сильно кислой при температуре от 20°
до 150°; заметно ускоряют ее ультразвуковые вол-
ны. А. к. к. обычно получают действием HNQ2 на
гидразиды карбоновых к-т: RCONHNH, + HNO2—>
—> RCON3 4- 2Н2О, или азида натрия на хлорангид-
риды к-т: RCOC1 + NaN3 RCON3 + NaCI; их
можно также получить при взаимодействии кетенов
или эфиров изоциановой к-ты с азотистоводородпой
к-той: RNCO -f- HN3 —> RCON3 -f- HCN; реакция про-
ходит быстро и экзотермично. А. к. к. применяют
для получения эфиров изоциановой к-ты, аминов
(образующихся при гидролизе эфиров изоциановой
к-ты), уретанов, симметрии, или песимметрич, про-
изводных мочевины, напр.: RCON3 —> RNCO + N2;
RNCO + Н2О —> RNH2 + СО2; RNH2 + RNCO ->
—► R—NHCONHR. Качественно и количественно
А. к. к. определяют по образующимся при гидролизе
N2 или амину, а также осаждением белого, плохо
растворимого азида серебра. Ниже приведены темп-ры
плавления нек-рых азидов:
Т. пл. азидов, °C
Азиды кислот
Уксусной
Масляной
Бензойной
Салициловой
о-Оксиметилбензойной
2 ,4-Диметил-З-этил-
пиррол-5-карбоновой
Из вфирных р-ров не
выделяют из-за легкой
взрываемости
31—32 (со взрывом)
27
74—75 (с разл.)
126 (с разл.)
А. к. к. и их перегруппировка были открыты Т. Кур-
циусом в 1890. См. также Курциуса реакция.
Лит.: СмитП. А., в кн..- Органические реакции, ред.
Р. Адамс [и др.1, пер. с англ., сб. 3, М., 1951, с. 322 .
Т.Н. Соркика.
57
АЗИНОВЫЕ КРАСИТЕЛИ —АЗОАМИНЫ
58
АЗИНОВЫЕ КРАСИТЕЛИ — производные фена-
айна; образуются при введении в цикл в положении
3 и 6 ауксохромов (амино- или за-
мещенных аминогрупп). Как все хи-
нониминовые красители, А. к. при
восстановлении переходят в бесцвет-
ные лейкосоединения, к-рые уже при
действии кислорода воздуха вновь
превращаются в красители. А. к. делятся на три
группы: 1) производные феназина, не имеющие
арильных остатков (I) — э у р о д и н ы; 2) неза-
мещенные аминопроизводные фенилфеназония (с а ф-
р а н и н ы, II) или нафтофеназония (р о з и н д у-
л ины, и з о р о з и н д у л и н ы, HI); 3) произ-
свн5
а
водные фенилфеназо-
ния и нафтофеназо-
ния, имеющие одну
СР или более ариламино-
вых групп. К А. к.
относятся также ин-
дулины, нигрозины и
группа анилинового черного. Технич. значение имеют
только представители сафранинов, розиндулинов и
изорозиндулинов. Среди А. к. имеются как основные,
так и кислотные красители. В пром-сти нашли
применение А. к. различных цветов, от красного
до синего.
Сафранин (Сафранин Т) (IV), основной краситель,
получают при совместном окислении бихроматом калия экви-
молекулярной смеси п-толуилеядиамина, о-толуидина и соля-
нокислого анилина:
Из кислотных А. к. наибольшее значение имеет: кислотный
темно-голубой (V). шерстопрочно-голубой Б. К классу кислот-
ных азиновых красителей относятся также: кислотный синий
(индоцианин Б), поляр синий Г, ряд нафтиловых красителей,
новазолевых кислотных синих (новазоль кислотный голу-
бой гл).
Лит.: Коган И. М Химия красителей, 3 изд., М.,
1956; В е н к ат а р а м а н К., Химия синтетических красите-
лей, пер. с англ., т. 2, Л., 1957; Ullmann, 3 Aufl., Bd 4, MUnch.—
В., 1953; Фир ц-Д а в и д Г. 9. и Бланже Л., Основ-
ные процессы синтеза красителей, пер. с нем., М., 1957.
И. Г. Лаптев.
АЗИНЫ — шестичленные гетероциклич. соедине-
ния. содержащие не менее двух гетероатомов, из
к-рых по крайней мере один представляет собой атом
азота. К А., содержащим только атомы азота в каче-
стве гетероатомов, относятся диазины (2 атома N в
кольце), триазины (три атома N в ядре) и т. д. Пред-
ставителями диазинов могут служить пиридазин (I),
пиримидин (II) и пиразин (III), отличающиеся по
взаимному положению атомов азота. К числу А.,
содержащих наряду с атомами азота и другие гете-
роатомы, принадлежат, напр., оксазин (IV) и тиа-
зин (V). Известны многочисленные соединения, у к-рых
азиновое ядро конденсировано с бензольными или
другими циклич. системами, например хиноксалин
(бензопиразин) (VI), фентиазин (VII), 9-хлордигидро-
фенарсазин ^адамсит) (VIII), пурин (конденсирован-
ные пиримидиновое и имидазольное кольца) (IX);
в частности, существуют и соединения, представляю-
щие собой два конденсированных азиновых ядра,
напр. птеридин (X) (пиримидин пиразин). Наконец,
IX X XI
к А. принадлежат и соединения типа пиперазина
(XI), не содержащие кратных связей и, в отличие от
указанных А., лишенные ароматич. свойсти (см.
Гетероциклические соединения). Как азотистые гете-
роциклич. соединения А. обладают более или менее
резко выраженным основным характером, дают соли
с к-тами, двойные соединения с пикриновой или
пикролоновой к-тами, образуют нерастворимые осадки
с реактивами на алкалоиды.
Нек-рые А., в особенности пиримидинового ряда,
играют очень большую роль в процессах жизнедея-
тельности. Так, пиримидиновые основания, напр.
2,6-диоксипиримидин (урацил), 2,6-диокси-5-метилпи-
римидин (тимин), 2-окси-6-аминопиримидин (цитозин)
чрезвычайно распространены в природе, т. к. вхо-
дят в состав нуклеиновых кислот. В частности, в со-
став ДНК (см. Нуклеиновые кислоты) входят тимин
и цитозин, а также аденин и гуанин, принадлежа-
щие к числу пуриновых оснований. Пиримидиновая
система содержится в ряде важных витами-
нов [Bj, В2, Вс (фолиевой кислоте)]. Пурино-
вые алкалоиды (кофеин, теофиллин, теобро-
ф мин) применяются в качестве тонизирующих,
антигипертонич. и антиастматич. средств.
К природным А. относятся и птеридины (X),
производными которых являются красящие
вещества крыльев нек-рых бабочек.
Многие синтетич. А. находят широкое применение
в медицине. Так, аминазин и др. производные фентиа-
зина в последние годы успешно вошли в психиатрия,
и хирургия, практику; производные барбитуровой
кислоты (люминал, веронал), содержащие пиримиди-
новую систему, принадлежат к известным снотворным
средствам. Сульфадимезин, содержащий диметилпи-
римидильную группировку, принадлежит к числу
наиболее эффективных сульфаниламидных препаратов.
Нек-рые А. имеют большое значение в качестве цен-
ных синтетич. красителей. Так, индантреновые кра-
сители содержат пиразиновую систему; сафранино-
вые и индулиновые красители являются производ-
ными феназина; метиленовый голубой представляет
собой производное тиазина и т. д.
Я. Ф. Комиссаров.
АЗОАМИНЫ — принятое в СССР назв. первичных
ароматич. аминов, применяемых в виде диазосоеди-
нений для т. н. холодного крашения, при к-ром на
волокна наносят сначала азотол, а затем диазосоеди-
нение. Образовавшиеся в результате азо-
сочетания нерастворимые в воде азокра-
сители (азопигменты) дают прочные окра-
ски, преим. на целлюлозных волокнах. А.
чаще всего — замещенные анилина, реже—
бензидина, нафтил аминов и др. аминов, обычно содер-
жащие атомы и группы Cl, NO2, СН3, ОСН3, реже
59
АЗОБЕНЗОЛ — АЗОКРАСИТЕЛИ
60
CF3, CN и некоторые другие. Ниже приведены наи-
более употребительные азоамины:
Название А. Заместители Положение
Азо амин ярко-алый Ж CF,; NH2; Cl NH3; NO2 1; 2; 5
» красный Ж 1; 4
красный О OCH3; NH2; SO2N(C.,H5)3 1; 2; 4
» красный С CH3; NH2; Cl 1; 2; 4
» красный ЗС CH3; NH,; N02 1; 2; 5
розовый О OCH,; NH,; NO2 NH,; OCH3; CN; OCH3 NH,; Cl 1; 2; 5
» бордо К 1; 2; 4; 5
» желтый О 1; 2
» оранжевый О NH,; NO2 1; 2
» ярко-оранже- вый К Cl; NH2; CF3 1; 2; 4
» оранжевый Ж NHs; Cl i; з
» оранжевый К NH,; NO2 CH3; NH,; NO, NH,; Cl; Cl i; з
» алый Ж 1; 2; 4
» алый 2Ж 1; 2; 5
алый К OCH3; NH2; NO, 1; 2; 4
А. — большей частью твердые тела с определенной
точкой плавления, горючи; плохо растворимы в воде;
лучше растворимы солянокислые и сернокислые
соли А.; в этиловом спирте, пиридине и других орга-
нич. растворителях А., как правило, хорошо раство-
римы. Цвета окрасок, получаемых с помощью А.,
зависят от строения как А., так и азотолов. Обычно
синие, коричневые, черные цвета получают при на-
личии в А. групп—NHCOAr, NlIAr,— N=NAr,
менее углубляют цвет группы СН3 и ОСН3. А. ряда
дифенила и нафталина дают более глубокие оттенки,
чем производные бензола. В названиях А. указы-
ваются цвета, получаемые с одним из азотолов, обычно
чаще применяемым. Максимальные прочности окрасок
достигаются только с определенными комбинациями
А. и азотолов; общих закономерностей не уста-
новлено. Однако известно, что группы CF3, CN,
SO2, С2Н5 и др. повышают прочность окрасок А. Спо-
собы получения А., физич. и химич. свойства, ка-
честв. реакции, методы количеств, определения — см.
Амины; см. также Азотолы, Крашение текстильных
материалов.
Лит.: Ворожцов Н. Н., Основы синтеза промежу-
точных продуктов и красителей, 4 изд., М., 1955; Чека-
лин М. А., Химия и технология органических красителей,
М., 1956; Коган И. М., Химия красителей, 3 изд., М.,
1956; Вен катар аи а н К., Химия синтетических краси-
телей, пер. с англ., т. 1, Л., 1956. М. А. Чекалин.
АЗОБЕНЗОЛ CeH5N=NC,H5, мол. в. 182,22 —
оранжево-красные ромбич. кристаллы; т. пл. 68°;
rf20 2,203 (тв.); с/68 1,0498 (распл.). Нерастворим в воде;
растворимость в 100 г растворителя: в этаноле
8,5 г (16°); в метаноле 3,95 г (16°); в лигроине 8,57 г (20°);
растворим в эфире, ледяной уксусной к-те, конц.
H2SO4; теплота сгорания 1555 ккал/моль. Транс-А.
при интенсивном освещении переходит в неустой-
чивую г^ис-форму, более глубоко окрашенную, пла-
вящуюся при 71° и самопроизвольно вновь превра-
щающуюся в транс-изомер. А. получается восста-
новлением питробензола или азоксибензола в щелоч-
ной среде (цинковой пылью, амальгамами металлов
и т. п.), электрохимич. восстановлением нитробен-
зола, окислением раствора гидразобензола в спирте
или эфире воздухом и другими окислителями, при
взаимодействии нитробензола с анилином в среде
смеси спирта и уксусной кислоты и т. д. А. образует
нестойкие продукты присоединения галогеноводоро-
дов и минеральных к-т. При восстановлении цинком
в щелочной среде превращается в гидразобензол,
в уксуснокислой среде — в анилин, оловом и соляной
кислотой — в бензидин. Окислителями А. окисляется
до азоксибензола. А. открыт Э. Митчерлихом в 1834.
2,2-АЗО - БИС - ИЗОБУТИРОНЙТРИЛ (дини-
трил-2-азо-бис-изомасляной кислоты, порофор)
(CH3)2C(CN)—N = N—(CN)C(CH3)2, мол. в. 1бЛ,22-
белые кристаллы; т. пл. 105—106° (с разл.), лег-
ко растворим в спирте, эфире, плохо — в воде. Для
А. характерны реакции с элиминироианием азо-
группы и образованием свободных радикалов (при
нагреиании):
(СН3)2С—N=N—С(СН3)3 — 2(СН3).С
I I I +N2.
CN CN CN
Эти радикалы могут соединяться между собой или
диспропорционироваться, образуя соответственно ди-
нитрил тетраметилянтарной к-ты и нитрил изомасля-
ной к-ты. Лишь немногие реакции А. протекают
с сохранением азогруппы, напр. с гидроксиламином
А. образует диамидооксил азоизомасляной к-ты:
(С Н 3)С—N= N—С(С Н3) о
I !
CN CN
+ 2NHa0H
(СН3)2С-N=N-----С(СН3)2
H«N/C^N0H H2N/C^N0H
А. получают обработкой аминонитрила H2NC(CH3)2
CN
гипохлоритом натрия. Используется А. как инициатор
свободнорадикальных реакций; его применяют как
катализатор при полимеризации аллилацетата, хло-
ристого винила, виниловых эфиров, хлорированных
терпенов, акрилонитрила, в реакциях теломериза-
ции. В отличие от перекиси бензоила, А. при ини-
циировании полимеризации непредельных соедине-
ний не обладает окисляющим действием. А. находит
также применение как порообразователь (напр.,
в производстве пористых резин). н. В. Гнучев.
АЗОКРАСИТЕЛИ — соединения, характеризую-
щиеся наличием одной или нескольких азогрупп
—N = N—.связывающих ароматич. радикалы; иногда
один из них заменяют гетероциклич. соединением
или ариламидом Р-кетокислоты. А., как правило,
содержат также замещенные или незамещенные амино-
или оксигруппы и SO3H, реже NOa, СООН, С1 и др.
заместители, влияющие на цвет и прочность А. Группы
SO3H, реже СООН и нек-рые другие придают А. рас-
творимость в воде. По числу азогрупп А. разделяют
на моноазо-, дисазо-, трисазокрасители и т. д. А.
с 3 и более азогруппами называют полиазокраси-
телями. Цвет А. зависит от строения; к углублению
цвета приводит: увеличение числа азогрупп (обычно
только до 4), замена фенильных радикалов нафтиль-
ными и увеличение числа групп ОН и NH2. Группы
SO3H, N02 и атомы С1 в зависимости от их положения
в одних случаях углубляют цвет А., в других —
повышают. Другие свойства А., в частности раство-
римость и характер взаимодействия с текстильными
волокнами, также определяются их строением.
При нагревании А., содержащие сульфогруппы,
обычно не плавятся. При темп-ре выше 140—150°
претерпевают термич. разложение; при наличии
нитрогрупп, чаще нескольких, могут взрывать, осо-
бенно в виде пылевоздушных смесей. Группы SO3H
и примеси минеральных солей (NaCl и др.) снижают
взрывчатость и пожароопасность А. Сильные восста-
новители и окислители расщепляют А., напр.:
CeH5N=NCeH4NH2 4- 2SnCl3 4- 2НС1 —►
H..NCeH4NH2 4- CeHsNHs 4- SnCl4
HO3SCeH4N=NCeH4N(CH3)2 4- HN03 (разб.) —►
+ Л--
—► HO3SCeH4№N 4- O«N\ >
X=/N(CH3)3
Расщепление А. происходит также при длительном
нагревании с водными р-рами к-т и щелочей. Для А.
61
АЗОКРАСИТЕЛИ — АЗОКСИБЕНЗОЛ
62
характерны: необратимое обесцвечивание при вос-
становлении; кривые поглощения света; частое изме-
нение окраски при изменении рН-среды.
А. чаще всего получают реакцией азосочетания
ароматич. диазосоединений с т. и. азосоставляющи-
ми — ароматич. аминами или фенолами, реже соеди-
нениями, содержащими группу СН2, активированную
соседними группами, напр. С=О и др. Азосочетание
обычно проводят при охлаждении до 0—15° в водной
среде; полученные красители осаждают из р-ров доба-
влением NaCl, высушивают и размалывают в порошок.
Для А. приняты торговые назв., рациональные назв.
применяют только для простейших А., напр. 4-ами-
поазобензол.
В зависимости от свойств, гл. обр. характера взаи-
модействия с различными окрашиваемыми материа-
лами, А. разделяют на следующие группы: 1) Кис-
лотные, чаще всего моно- и дисазокрасители, со-
держащие одну или песк. сульфогрупп; напр. кислот-
ный оранжевый (I). 2) Основные, содержат
основные, обычно аминогруппы, не содержат кислот-
творителях также растворимы комплексы протрав-
ных моноазокрасителей с Сг или Со. 8) А. д л я аце-
татного шелка, обычно моноазокрасители
ОН
/С2Н4ОН
ных групп, напр. хризоидин (II). 3) Протравные,
для к-рых характерно образование в процессе краше-
ния комплексов с ионами металлов, обычно Ст,
что обусловлено чаще всего наличием в молекулах
А. группы СООН в орто-положении к ОН-группе
или двух ОН-групп в о,о'-положениях (в соседних
ядрах) к азогруппе;
например «кислотный
Хром синий К» (III).
Реже протравные А.
содержат в орто-по-
ложении к азогруппе
одну ОН-группу и
одну СООН или NH2-
ОН
Cl
НО HNCOCHg
n=n
NaO3S m
S03Na
группу. Протравные А. значительно прочнее ки-
слотных, но менее ярки. 4) Прямые, обычно
дис- или полиазокрасители; характеризуются вытя-
нутой формой молекулы, наличием обычно более
8 конъюгированных двойных связей, расположе-
нием атомов, близким к плоскостному, напр. «конго
красный» (IV). 5) Пигменты, моно- и дисазо-
SO3Na SO3Na
красители нерастворимы в воде, т. к. не содер-
жат групп SO3H, СООН, NH2; содержат обычно
С1 или NO2, также СН3, ОСН3 и’другие группы, по-
вышающие прочности пигментов и углубляющие
их цвет; напр., «пигмент
алый» (V). 6) Лаки, нерас-
творимые в воде соли не-
которых кислотных краси-
телей; чаще всего соли На
или Са. 7) Ж и р о- и
спирторастворимые
А. близки по строению к
пигментам, но обычно не содержат С1 и NO2, нераство-
римы в воде, но растворимы в органич. растворителях,
напр. «жировой оранжевый» (VI). В органич. рас-
с небольшим молекулярным весом, производные
бензола; известны две группы последних — краси-
тели, образующие коллоидный р-р в воде, содержащие
обычно группу С2Н4ОН при азоте, напр. «алый для
ацетатного шелка Ж» (VII), и растворимые в воде
кислые сернокислые эфиры последних, напр. «со-
лацет прочный алый» (VIII). 9) А., получаемые не-
C2H4OSO3Na
C2HS
посредственно на волокне сочетанием диазосоедине-
ний с азосоставляющпми методом холодного
крашения (см. Азоамины)-, нерастворимы в воде,
обладают повышенной прочностью. 10) А., обра-
зующие в процессе крашения ковалентную связь с
волокном благодаря наличию «активных» атомов и
групп («активные» красители); напр., «красный кра-
ситель» (IX). Производство активных красителей
для шерсти начато в 1952 (р е м а л а н ы — Герма-
ния), для хлопка — в
1956 (п р о ц и о н ы— _SO3Na он nh-|< "V-Ci
Англия). Эти краси- АД N_..
тели образуют окра- \ / । J ] т
ски весьма прочные к Сг
мокрым обработкам и а°з5 SO3Na
трению. . ,х
Колич. анализ А. основан на их восстановлении
титрованными р-рами VSO4, TiCl3, CrSO4 или SnCl2
до соответствующих аминов; широко также исполь-
зуются оптич. методы: колориметрич. или спектро-
фотометрич. и др.
А. — самый многочисленный класс синтетич. кра-
сителей; среди них имеются представители почти
всех групп, на к-рые разделяют красители по их
технич. применению. Преимуществами А. являются
простота их применения и относительно низкая стои-
мость. Применяются А. для крашения текстильных
волокон, кожи, бумаги, резины, нек-рых пласти-
ческих масс и др. См. также Азосоединения, Краси-
тели синтетические, Моноазокрасители, Дисазокра-
сители, Полиазокрасители, Крашение текстильных
материалов.
Лит.: Чекалин М. А., Химия и технология органиче-
ских красителей, М., 1956; Коган И. М., Химия краси-
телей, 3 изд., М., 19э6; Венкатар аман К., Химия син-
тетических красителей, пер. с англ., т. 1, Л., 1956; Фир ц-
Д а в и д Г. Э. и Б л а н ж е Л., Основные процессы син-
теза красителей, пер. с нем. М., 1957; Л ас т о вс к и й Р. П.,
Вайнштейн Ю. И., Технический анализ в производ-
стве промежуточных продуктов и красителей, 3 изд., М., 1958.
М. А. Чекалин.
АЗОКСИБЕНЗОЛ С„Н5—N = NCeH5, мол. вес
198,23 — ярко-желтые кристаллы, иглы; т. пл. 3S0;
63
АЗОКСИСОЕДИНЕНИЯ — АЗОСОЕДИНЕНИЯ
64
СвН5 Н5С3 С3Н5
N=N N=N
I 1 1
Н5св о о
I II
d20 1,246; 1,6644. Нерастворим в воде, растворим
в спирте (17,5 г в 100 г при 16°), эфире, лигроине
(43 г в 100 г при 15°). Теплота сгорания 1529,5 кал/мол.
Известны цис- (I) и транс- формы (II) А.; первая мало
устойчива и при кратко-
временном нагревании пе-
реходит во вторую. А,—
простейшее азоксисоедине-
ние. Получается при восста-
новлении нитробензола в
разных условиях: амальгамой натрия, смесью мети-
лового спирта и едкого натра, метилатом натрия, ар-
сенитом, цинковой пылью в щелочной среде,
электрохимия, методом и др.; при этом обыч-
но наряду с А. образуются азобензол и гидра-
зобензол', получается также при взаимодей-
ствии нитрозобензола и фенилгидроксил-
амина в щелочном спиртовом р-ре. При вос-
становлении А. железом или цинком в ще-
лочной среде он превращается в азобензол и затем в
гидразобензол, при восстановлении цинком и уксус-
ной к-той — в анилин; при нагревании с конц. серной
к-той происходит изомеризация в 4-оксиазобензол.
А. является промежуточным продуктомв синтезе полу-
продуктов—азобензола И бензидина. А. А. Черкасский.
АЗОКСИСОЕДИНЕНИЯ — соединения, содер-
жащие группировку —N = N—>0 (азоксигруппу).
Хорошо изучены лишь ароматич. А. — желтые кри-
сталлы нейтрального характера, плохо растворимы
в воде и умеренно — в спирте; цвет А. обусловлен
азоксигруппой. В ряду А. имеет место явление изо-
мерии положения (для асимметричных соединений)
и геометрия, изомерии, напр. для монобромазокси-
бензола известны а- и {З-изомеры:
BrC0H4N = NCeH5 и CeH5N = NCeH4Br
I i
О О
fi-изомер вступает в реакцию замещения с бромом,
тогда как а-изомер по отношению к брому индифе-
рентен; о,о-азокситолуол существует в виде цис- и
транс- изомеров:
o-CH3CeH4N->O o-CH3CeH4N->-O
II II
o-CH3C3H4N NCjjH^CHg-o
цис-формы менее устойчивы и при нагревании пере-
ходят в транс-изомеры, к-рые обладают более низ-
кими темп-рами плавления и меньшими дипольными
моментами. При электролитич. восстановлении А.
переходят в азосоединения', с помощью (NH4)2S вос-
станавливаются в гидразосоединения и далее — в про-
изводные анилина; в присутствии концентрированной
H2SO4 перегруппировываются в га-оксиазобензолы:
CeH5N = NCeH5 —> n-HOC6H4N = NCeH3 (Валлаха пере-
I
О
группировка)', под действием света дают о-оксиазо-
бензолы. В пром-сти А. получаются в виде проме-
жуточных продуктов при взаимодействии нитрозо-
бензолов с арилгидроксиламинами, образующимися
при восстановлении нитробензолов в щелочной среде:
CeH3NO + CeH5NHOH -> C,H5N=NCeH5 + Н2О;
I
О
известны и др. способы. Большинство А. практич.
значения не имеют; азоксибензол используется для
получения азобензола и бензидина', азоксигруппа
входит в структуру нек-рых стильбеновых красителей
(солнечный желтый); к алифатич. А. относится анти-
биотик элайомицин, обладающий противотуберкулез-
ным действием. р. б. Журин.
АЗОМЕТИНОВЫЕ КРАСИТЕЛИ — красители, ха-
рактеризующиеся наличием хромофорной системы
—N=CH—, структурно аналогичной азогруппе
—N=N—, причем замена атома — N — на =СН-груп-
пу сопровождается повышением цвета. А. к. быва-
ют нейтральными (I), анионоидными (дающими комп-
лексные соединения с металлами) и катионоидны-
ми (II).
В последнее время интерес к А. к. возрос, т. к.
среди них были найдены практически ценные оптич.
десенсибилизаторы фотография, эмульсий и краси-
тели для искусственных волокон. Кроме того, А. к.
нашли применение в современной цветной фотографии.
Получают А. к. взаимодействием: 1) ароматич. или
C6H5COC(CN)=N
гетероциклич. альдегидов и аминов, с отщеплением
элементов воды; 2) ароматич. нитрозосоединений с
соединениями, содержащими активную метиленовую
группу, также сопровождающимся отщеплением эле-
ментов воды; 3) ароматич. диаминов или аминофенолов
с соединениями, содержащими активную метиленовую
группу, в присутствии окислителя. Частным случаем
этой реакции, когда окислителем служит экспони-
рованное галогенное серебро, восстанавливающееся
до металлич., является т. н. реакция цветного прояв-
ления, к-рая лежит в основе большинства методов,
применяемых в современной цветной фотографии и
кинематогр афии:
R\ Г~\
)CH2 + H2N-( ¥-N(C2H5)2 + 4AgBr----
R\ / \
----- )C=N-f ¥-N(C2H5)2+ 4Ag f 4HBr
Rj \=J
Для получения желтого изображения применяют в
качестве соединений, содержащих активную метиле-
новую группу, арилиды арилуксусных к-т, а для
получения пурпурного — производные З-алкил-(арил)-
-1-арилпиразолона-5 или ы-цианацетофенона. Эти
изображения образованы красителями типа I. А. к.,
являющиеся оптич. десенсибилизаторами, относятся
к типу II.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венкатараман К., Химия синтетических краси-
телей, пер. с англ., т. 2, Л., 1957. Н. С. Вульфсон.
АЗОСОЕДИНЕНИЯ — органич. соединения, со-
держащие азогруппу —N=N—, связанную с двумя
радикалами. В большинстве известных А. оба ради-
кала принадлежат к ароматич. ряду; известны также
А., содержащие два алифатич., один алифатич. и дру-
гой ароматич. или гетероциклич. радикалы. В ради-
калах, связанных с азогруппами, могут также на-
ходиться любые заместители. Простейшим А. алифа-
тич. ряда является азометан СН3—N=N—СН3, не-
устойчивое соединение взрывающееся при нагрева-
нии. Наибольшее значение имеют ароматич. А. Про-
стейший ароматич. А. — азобензол. Наиболее рас-
пространенный метод получения А. — азосочетание
ароматич. диазосоединений с ароматич. амино- или
оксисоединениями и веществами, содержащими ак-
тивные метильные группы, напр. АгХ2С1 + НСвН4ОН—>
—> Аг—N=N— CeH4OH + HCI. А. получаются так-
же: восстановлением металлами (Zn, Fe) нитро-,
нитрозо- и азоксисоединений в щелочной среде,
напр. 2RNO2 + 8Н -> R—N=N—R + 4Н2О; эле-
ктролитич. восстановлением нитросоединений; взаи-
модействием нитрозосоединений с первичными ами-
65
АЗОСОЧЕТАНИЕ - АЗОТ
66
нами: RNO + R'NH2 —> R—N = N—R' + Н2О; окис-
лением первичных аминов гипохлоритом или перман-
ганатом: 2RNH2 + О2 —> R—N = N—R + 2Н2О; окис-
лением нек-рых диазосоединений хлорной известью.
Для А. возможна геометрия, изомерия:
R-N R-N
II и II
R—N N—R
Все ароматич. А. — окрашенные твердые в-ва;
окраска обусловлена хромофорными свойствами азо-
группы: цвет А. зависит также от природы радикалов,
связанных с азогруппами, и от характера, числа и
расположения заместителей. А., содержащие карбок-
сильные и сульфогруппы, растворимы в воде; осталь-
ные — в воде нерастворимы, но растворяются в
спирте, эфире, бензоле. А., не содержащие кислотных
и основных заместителей, нейтральны.
А. легко восстанавливаются R—N=N—R' +
4- 4Н —> RNH2 + R'NH2. Восстановление может быть
произведено различными восстановителями: Zn или
Fe в присутствии соляной к-ты, солями 2-валентных
Sn, V, Сг или 3-валентного Ti в кислой среде, гидро-
сульфита натрия в щелочной среде и др. Восстанов-
ление А. используется для их количеств, анализа,
а также для получения нек-рых аминов. При действии
окислителей в кислой среде А. превращаются в
азоксисоединения'.
R-N=N-R 2 R-N-=N-R
1
О
Сильные окислители, напр. HNO3, расщепляют не-
которые А. с образованием диазопроизводного од-
ного радикала и нитропроизводного другого ради-
кала: R—N = N—R' + О-> RNt + R'NO2. А., со-
держащие амино- и оксигруппы, способны окрашивать
растительные и животные волокна; образуют много-
численную группу технически важных синтетич.
красителей — азокрасителей.
Лит.: Филиппычев С. Ф., Химия и технология азо-
красителей, ч. 1, М., 1938; Ворожцов Н. Н., Основы
синтеза промежуточных продуктов и красителей, 4 изд., М.,
1955; К о г а и И. М. Химия красителей, 3 изд., М., 1956;
В е нк ат ар ам ан К., Химия синтетических красителей,
пер. с англ., т. 1, Л., 1956. А. А. Черкасский.
АЗОСОЧЕТАНИЕ (сочетание) — взаимодействие
ароматич. диазосоединений с в-вами, содержащими
способный к замещению атом водорода, связанный с
атомом углерода. К этим соединениям, наз. азосостав-
ляющими, принадлежат ароматические аминосоеди-
нения, фенолы, некоторые вещества, содержащие ак-
тивную метиленовую группу (производные ацето-
уксусной к-ты, пиразолона и т. п.), и углеводороды
с сопряженными двойными связями. Наибольшее
практич. значение имеет А. с ароматич. аминами и
фенолами. Реакция состоит в том, что остаток диазо-
соединения замещает атом водорода в ароматич. ядре
в пара- или орто-положении к амино- или оксигруппе,
причем непосредственно взаимодействуют катион ди-
азония и молекула основания амина или анион фено-
лята (активные формы диазосоединения и азосостав-
ляющей). Вероятно, сначала обратимо образуется
соответств. продукт присоединения, превращающийся
затем к продукт замещения, напр.:
Возможны и другие механизмы А.
В результате А. получают азосоединения, содержа-
щие амино- и оксигруппы, обладающие окраской и
красящей способностью и составляющие один из
классов синтетич. красителей — класс азокрасите-
лей.
А. обычно проводят в водной среде в присутствии
к-т, оснований или их буферных смесей с соответств.
солями, при темп-pax от 0 до 25—30°. Большое влия-
ние на А. оказывает величина pH реакционной среды;
от нее зависят скорости А. и побочных реакций.
Скорость А. зависит прежде всего от строения ре-
агентов. Отрицательные заместители (NO2, SO3H,
Cl и т. п.) в ядре диазосоединения повышают, а поло-
жительные (ОН, ОСН3, СН3 и т. п.) понижают его ак-
тивность. Азосоставляющие, содержащие положи-
тельные заместители, более активны, чем азосостав-
ляющие с отрицательными заместителями. А. может
сопровождаться побочными реакциями: разложени-
ем диазосоединения, скорость к-рого увеличивается
при повышении pH; образованием диазоаминосоеди-
нений при А. с аминами в условиях относительно вы-
соких значений pH — в слабокислой и щелочной сре-
дах; превращением диазосоединений в неактивную
форму — антидиазотаты — в сильнощелочной среде.
А. с аминами обычно проводят в кислой среде в при-
сутствии неорганич. к-ты или смеси уксусной к-ты
и ацетата (pH от 2 до 6), с оксисоединениями — в сла-
бокислой или щелочной средах в присутствии смеси
уксусной к-ты и ацетата, бикарбоната или карбоната
натрия или их смеси (pH от 5 до 11). А. открыто
в 1864 П. Гриссом, с середины 70-х гг. 19 в. А. на-
чали применять для получения азокрасителей, и в
наст, время эта реакция лежит в основе технологии
произ-ва всех красителей этого класса.
Лит. см. к ст. Аэосоединения. А. А- Черкасский.
АЗОТ (Nitrogenium) N — химич. элемент V гр.
периодич. системы Менделеева, п. н. 7, ат. в. 14,008.
При обычных условиях А. — газ без цвета и запаха..
А. был открыт в 1772 Д. Резерфордом. Название «А.»,
обозначающее «нежизненный», «не поддерживающий
жизни», сохранилось только в русском и франц,
языках. Лат. назв. — Nitrogenium (селитрообразо-
ватель) происходит от двух слов: лат. nitrum — се-
литра и греч. fewao) — рождаю, произвожу.
Нахождение в природе. Общее содержание А. в
земной коре 1-10-2 вес. %. Наибольшая часть А. (ок.
4-1015 т) содержится в свободном состоянии в атмо-
сфере; А.—главная составная часть воздуха (75,6%
по весу или 78,09% по объему). В связанном состоя-
нии А. встречается в воздухе, водах рек, морей и оке-
анов, в атмосферных осадках. В воздухе связанный
А. содержится, в частности, в виде следов аммиака,
образующегося при разложении азотсодержащих
органич. соединений, а также в виде следов кисло-
родных соединений, возникающих за счет фотохимия,
окисления аммиака и при непосредственном синтезе
окиси азота NO в электрич. разрядах.
В земной коре А. образует три основных типа ми-
нералов, содержащих ионы CN“, NO? й NHJ. С тече-
нием времени минералы А. разрушаются, и поэтому
они встречаются только в совр. отложениях. Про-
мышленный интерес представ-
ляют натриевая (чилийская) Се-
O’ + Н* литра NaNO3 и калиевая (ин-
дийская) селитра KNO3 (см. Се-
литры). Крупные залежи селит-
ры находятся в Чили. А. вхо-
дит в состав всех живых орга-
низмов и в небольших количе-
NH2 + Н+ ствах содержится в каменном
угле (1—2,5%) и нефти (0,02—
1,5%). Очень большое значе-
2 К. X, Э. т. 1
67
АЗОТ
68
ние имеет А. в жизнедеятельности растений и жи-
вотных. В белковых телах содержится до 17% А.,
в организме человека — до 3%. В виде нейтраль-
ных и ионизированных атомов А. обнаружен в
составе газового облака комет, в туманностях
и в атмосфере Солнца. В термоядерных реакциях,
происходящих на Солнце, А. отводится весьма
важная роль: С12 + р —> N13; N13—>С13 + е+; С13 -)-
4-р->М14; №4 + р—>О15; 015->№5 + е+; N15 + р->
—>С12 -Т Не4. А. участвует в сложном процессе кру-
говорота веществ в природе. Значительное количество
А. поступает в почву в результате жизнедеятельности
азотфиксирующих бактерий, способных переводить
свободный А. воздуха в соединения, а также в ре-
зультате нек-рых других природных процессов.
Однако, благодаря извлечению из почвы растениями
огромного количества связанного А. (особенно при
интенсивном ведении с. х-ва), общий круговорот А.
нарушается. Поэтому решающее значение приобре-
тает введение А. в почву в виде азотных удобрений.
Проблема получения связанного А. была решена в
начале 20 в., с освоением промышленного синтеза
аммиака из А. воздуха и водорода.
Изотопы, атом, молекула. Природный А. состоит
из двух стабильных изотопов: N14 (99,635%) и N16
(0,365%). В верхних слоях атмосферы под действием
нейтронов космич. излучения N14 превращается в
радиоактивный изотоп углерода С14 по реакции
N14(n, р)С14. Этот процесс используют для получения
С14 (см. Углерод) в ядерных реакторах. N15 применяется
в химич. и биохимич. исследованиях в качестве изо-
топного индикатора. В пром-сти концентрирование
№5 ведут обменной реакцией в жидкой фазе: N15H3
(газ) + N14H4NO3(p-p)T=N14H3(ra3)+ N16H4NO3(p-p).
Из искусственных радиоактивных изотопов А. наи-
больший период полураспада имеет N13 (Т^ = 10,08
мин), остальные — весьма короткоживущие. Элек-
тронная конфигурация атома A.: Is2 2s2 2р3. Атомный
радиус 0,71 А, ионные радиусы N6+ 0,15 А; N3
1,48 А. Энергии ионизации (в эв): № —>• N+—> №+—>
—>N3' —>N4+—>N5+ соответственно равны 14,54; 29,60;
47,43; 77,45; 97,86. В соединениях А. формально мо-
жет проявлять переменную валентность от —3 до
+ 5. Однако в основном состоянии атом А. имеет
2s
miiiiEi
неспаренных электрона
, к-рые и опреде-
ляют его истинную валентность. Энергетически вполне
выгодно и ионное состояние с 4-ковалентным одно-
зарядно положительным A.N+|) | J | i 111, напр. в ионе
-bz/O
аммония (NH4)+ или нитрогруппе —N< . Для
Со-
того чтобы атом А. мог стать 5-валентным, необхо-
димо возбудить 2б'-электрон для осуществления его
перехода на 3«-орбиту: 2s22p3 —> 2s2p33s, что тре-
бует затраты большой энергии, к-рая не может быть
скомпенсирована образованием двух дополнитель-
ных связей. Поэтому в химических соединениях А.
практически никогда не образует 5 ковалентных свя-
зей, в отличие от его ближайшего аналога — фос-
фора.
Молекула А. двухатомна, атомы связаны тройной
связью N=N. Диаметр молекулы А., определенный
разными способами, лежит в пределах от 3,15А до
3,53 А. Энергия диссоциации молекулы N2 довольно
долго принималась равной 170,22 ккал/моль. Однако
большинство исследователей считает, что она значи-
тельно выше (225,1 ккал/моль).В соответствии с этим
термич. диссоциация N2 становится заметной лишь
при очень высоких темп-pax. Так, при 3000°К и нор-
мальном давлении диссоциированы лишь 0,1% мо-
лекул А., тогда как у Н2 и даже О2 при этих условиях
диссоциированы уже 10% молекул. Имеются ука-
зания, что в нек-рых условиях А. может образовать
молекулу состава N3 (триазон или нитрозон), а также
N6. Молекулярный А. диамагнитен.
Физические и химические свойства. Молекулярный
А. характеризуется след, константами: т. пл.—209,86°,
т. кип. —195,8°; плоти, газообразного 1,2506 г/л,
жидкого 0,808 г/см3 (—195,8°). В твердом состоя-
нии А. существует в двух модификациях: кубической
a-форме с плоти. 1,0265 г 1см3 (—252,2°), устойчивой
ниже —237,5°, и гексагональной |3-форме с плоти.
0,8792 (—210°), устойчивой выше —237,5°. Тройная
точка: Т = 63,136°К(—210,024°С); р = 94,01 мм
рт. ст.\ «крит. —147,0°; ркрит 33,5 атм, dKpaT 0,3110
г/см3. Теплота плавления 6,09 кал/г (—210°), тепло-
та испарения 47,6 кал/г (—195,55°), теплоемкость
(---кал- \ сп = 6,524 + 1,250• 10 37'+0,001 10 67'2;
Кмоль град} Р ’ 1 ’ 1 ’ ’
Ср/Ся = 1,41. Поверхностное натяжение жидкого А.
в контакте с воздухом 8,5 дин/см (—196°); давление па-
ра при различных темп-pax (в мм рт. ст.):\(—226,1°),
40(—214°), 400(—200,9°) и 760(—195,8°); диэлектрич.
проницаемость 1,000538 (25°; 760 мм рт. ст.). Рас-
творимость А. в воде (в см3 на 100 мл Н2О): 2,33(0°),
1,42(40°) и 1,32(60°). В некоторых углеводородах
(и-гексан, и-гептан и т. д.). А. растворяется лучше,
чем в воде. Растворимость в С2Н5ОН иСН3ОН при 0°
примерно такай же, как и в воде.
Большая прочность молекулярного А. является
причиной сравнительно малой его активности, т. к.
энергии активации реакций с его участием довольно
велики. Лишь с некоторыми активными металлами
(напр., литием, кальцием) А. реагирует при невысо-
ких темп-pax (если поверхность этих металлов доста-
точно чиста и активна). С большинством других прос-
тых тел А. если и реагирует, то лишь при высокой
темп-ре, а иногда также и при участии катализато-
ров. С кислородом А. заметно взаимодействует только
ок. 4000° с образованием окиси NO, к-рая при ох-
лаждении легко окисляется далее до двуокиси NO2.
Известны также: закись N2O, азотистый ангидрид
N2O3 и азотный ангидрид N2O5 (см. Азота окислы).
При действии ионизирующих излучений на смеси
А. с кислородом образуются окислщ А., а в присут-
ствии воды — азотная к-та. О кислородных соеди-
нениях А. см,- также Азотная кислота и Азотистая
кислота (и их соли — нитраты и нитриты).
С водородом А. реагирует лишь при высокой темпе-
ратуре и давлении в присутствии катализатора с об-
разованием аммиака NH3. Водородные соединения А.
образуют четыре ряда, напоминающие гомология,
ряды углеводородов. В этих соединениях скелетом
является цепь из атомов А. К I ряду (общая фор-
мула NnHn_f_2) относятся аммиак NH3, гидразин
H2N—NH2, триазан H2N—NH—NH2, тетразан
H2N—NH—NH-NH,. Ко II ряду (NnH„) принадле-
жат диимид HN=NH, триазен HN = N—NH2 и
др., к III ряду (NnHn_2)—азотистоводородная к-та
H—N=N=N и др., к IV ряду (NnHn_4) — октазон
Н— N = N— NH — N=N—NH—N = N —H. Аммиак и
гидразин получены в свободном виде; большинство
др. водородных соединений встречается лишь в виде
органич. производных. Азотистоводородная к-та HN3
образует соли — азиды.
С галогенами А. непосредственно не взаимодей-
ствует. Однако косвенным путем можно получить
соединения состава NT3, где Г = F, Cl, J (так, NFS
образуется при действии фтора на аммиад). Малая
энергия связи N—Г обусловливает термодинамич.
неустойчивость соединений NF3, что объясняет их
взрывчатость (сравнительно устойчив лишь NF3).
69
АЗОТ—АЗОТА ОКИСЛИ
70
Фтористый азот NF3 — бесцветный газ (т. пл. —216,6°,
т. кип. —120°), обладает токсич. действием. Хлори-
стый азот NC13 — желтая маслянистая жидкость с
резким запахом (т. кип. 71°), легко взрывает. Под
названием «иодистый А.» известны два соединения:
NJ3 и NJ3 • NH3 — твердые, темно-серого цвета про-
дукты, разлагающиеся в сухом виде со взрывом и
сравнительно безопасные во влажном состоянии.
Имеются сведения, что синтезировано весьма неустой-
чивое соединение А. с бромом состава NBr3 • 6NH3.
К числу соединений галогенов с А. принадлежат,
в частности, очень взрывчатые галогеназиды. I'N3:
фторазид FN3— зеленовато-желтый газ (т. пл. —154°,
т. кип. —82°), хлоразид C1N3 — бесцветный газ
(т. пл. —100°, т. кип. —15°), бромазид BrN3— крас-
ная жидкость (т. пл. —45°), иодазид JN3 — жел-
тые кристаллы. Взаимодействие NO и С12 на древесном
угле приводит к образованию хлористого нитрозила
NOC1— желтого газа (т. пл. .—64,5°, т. кип. —5,5°).
Известны также соответствующие производные фтора
NOF (т. пл. —134°, т. кип. —56°) и брома NOBr
(т. пл. —55,5°, т. кип. 2,0). С серой А. непосредствен-
но не реагирует. При взаимодействии S с жидким NH3
образуется т. н. азотистая сера N4S4 — оранжево-
желтые кристаллы, т. пл. 179°, при нагревании выше
темп-ры плавления или при ударе распадается со
взрывом. При нагревании растворов N4S4 в инерт-
ных растворителях образуется весьма неустойчивое
соединение, т. н. сернистый азот N2S3 — красная
жидкость.
При действии А. на накаленный угольный кокс
образуется циан (CN)2. С металлами А. дает нитриды;
напр., при сгорании Mg и А1 на воздухе нитриды
образуются наряду с окислами этих металлов. При
высокой темп-ре А. взаимодействует также с Mg,
Са, Sr, Ba, Ti, V, Cr, Мп, Zr, Та, Mo, W, редкоземель-
ными и щелочными металлами (соединения с послед-
ними, исключая литий, неустойчивы), а также с крем-
нием. С карбидом кальция СаС2 образует при высокой
темп-ре кальция цианамид CaCN2(Ca=N—C=N).
Нагревая смесь соды с углем на воздухе, получают
цианистый натрий: Na2CO3 + 4С + N2 = 2NaCN +
-f-ЗСО (см. Цианиды). При 1500° А. взаимодействует с
ацетиленом, образуя цианистый водород HCN. А.
входит в состав многочисленных органич. соедине-
ний (амины, аминокислоты, нитросоединения и др.).
При действии электрич. разряда на молекулярный А.
под уменьшенным давлением (1—2 мм рт. ст.) может
образоваться активный азот, представляю-
щий собой смесь неустойчивых (обладающих повы-
шенным запасом энергии) молекул и атомов А. Ак-
тивный А. получается также при разложении нитри-
дов В, Ti, Mg и Са, при взрыве смеси кислорода и СО
в присутствии А., при электрич. разряде в воздухе.
В отличие от обычного, молекулярного, активный А.
весьма энергично взаимодействует с атомарными кис-
лородом и водородом, парами серы, белым фосфором,
нек-рыми металлами. Об аналитич. определении А.
см. Азота определение.
Получение. А. в лаборатории может быть получен
несколькими способами: 1) разложением NH4NO2
при нагревании: NH4NO2=sN2.+2Н2О, практиче-
ски пользуются смесью NH4C1 + NaNO2; 2) разло-
жением оихромата аммония: (NH4)2Cr2O7 = N2 +
+ Cr2O3 + 4Н2О. Наиболее чистый А. получают осто-
рожным разложением NaN3. В пром-сти А. извлекают
из воздуха (см. Воздуха разделение).
Применение. Основная область применения А. —
синтез аммиака. Свободный А. используют во многих
отраслях пром-сти: как инертную среду нек-рых
химич. реакций, для заполнения свободного прост-
ранства в ртутных термометрах, для измерения вы-
соких темп-p, при перекачке горючих жидкостей.
2*
Жидкий А. применяют для различного рода работ,
связанных с низкой темп-рой (криостаты, вакуумные
установки и др.). Широко применяются многие со-
единения А.
Лит.: Э п ш т е й в Д. А. [и др.], Химия и техвология
связанного азота, [М.—Л.], 1934; Г е й д о в А., Эвергия дис-
социации и спектры двухатомных молекул, пер. с англ., М.,
1949; Gmelin, 8 Aufl., System-Nummer 4, Stickstoff,
В., 1934—36; Mellor, v. 8, L.— N. Y. — Toronto, 1947;
P a s с a 1, t. 10, P., 1956; К i r k, v. 9, N. Y., [1952].
АЗОТ ОСТАТОЧНЫЙ — азот веществ, остающихся
в крови или в экстрактах тканей после осаждения
белков. У взрослых людей количество А. о. в сыво-
ротке или крови колеблется от 20 до 40 мг°/0 (°/00).
Повышение А. о. в крови наблюдается при заболе-
вании почек, поражении печени, злокачеств. опухолях,
туберкулезе и т. д. Наиболее важное диагностич.
значение имеет определение А. о. при заболеваниях
почек, когда величина А. о. повышается в 2—10 раз
и больше. Общим для всех методов определения А. о.
является сжигание испытуемого образца с серной
к-той в присутствии катализаторов, после чего оп-
ределение связанного аммиака производят различ-
ными методами.
Лит.: ПредтеченскийВ. Е., Боровская В. М.
иМарголина Л. Т., Руководство по лабораторным ме-
тодам исследования, 4 изд., М, 1950; Балаховский С. Д.
и Балаховский И. С., Методы химического анализа
крови, 3 изд., М., 1953. В. И. Мазуров.
АЗОТА ОКИСЛЫ — соединения азота с кислородом.
Известны след. А. о.: закись N2O, окись NO, азоти-
стый ангидрид N2O3, двуокись NO2, ее димер (четы-
рехокись) N2O4 и азотный ангидрид N2O6.
Все А. о. в газообразном состоянии эндотермичны, т. е.
при их образовании из N2 и О2 поглощается теплота. Даже
при низких темп-pax А. о. термодинамически неустойчивы.
Однако, благодаря большой энергии связи между атомами
азота и кислорода в А. о., энергии активации реакций с их уча-
стием, как правило, высоки и А. о. (за исключением NO) срав-
нительно мало реакционноспособны при обычных условиях.
В частности, и реакции их распада «размораживаются» (начи-
нают протекать с измеримой скоростью) лишь при сравнительно
высоких темп-рах.
Закись азота N2O — бесцветный газ со
слабым приятным запахом и сладковатым вкусом;
т. кип. —89,5°, т. пл. —102,4°. Теплота образования
ДЯ°98 = 19,49 ккал/моль. Растворимость в воде:
1,048(5°); 0,629(20°) объемов газа в одном объеме воды.
С водой, с растворами к-т и щелочей N2О химически
не взаимодействует; однако формально ей соответ-
ствует малоустойчивая азотноватистая к-та H2N2O2,
легко разлагающаяся на воду и N2O. Кислородом
N2O не окисляется. При 500° заметно диссоциирует,
при 900° полностью распадается на N2 и О2. При
высокой темп-ре проявляет сильные окислительные
свойства (напр., в закиси азота, как и в кислороде,
вспыхивает тлеющая лучина). Смеси N2O с Н2, NH3 или
СО, при нек-рых соотношениях компонентов, взры-
ваются при нагревании. Получают NaO термич. раз-
ложением нитрата аммония: NH4NO3 —> N2O + 2Н2О;
процесс сопровождается выделением теплоты и про-
текает со все возрастающей скоростью, что требует
осторожности при его проведении. При вдыхании
смеси воздуха с N2O наступает состояние, близкое
к опьянению (отсюда название N2O — «веселящий
газ»). В медицине закись азота применяют в качестве
наркотич. средства.
Окись азота NO — бесцветный газ, т. кип.
—151,8°, т. пл. —163,6°. Теплота образования =
=21,6 ккал/моль. Хотя NO самый эндотермичный из
А. о., но именно этот окисел и образуется при взаи-
модействии N2 и О2; это,'в частности, связано с тем,
что реакция N2 + О2 —> 2NO не сопровождается
уменьшением энтропии (что имеет место при синтезе
всех других А. о. из простых тел), поскольку в ре-
зультате этой реакции не уменьшается число молекул
71
АЗОТА ОКИСЛИ — АЗОТА ОПРЕДЕЛЕНИЕ
72
газа. При различных темп-pax равновесные концен-
трации NO в указанной реакции составляют (в %):
2000° 3200“ 4200°
0,61 4,48 10,0
Полученная при высокой темп-ре NO при медленном
охлаждении разлагается, поэтому ее быстро охлаждают
до темп-ры ниже 1000°, при к-рой ее распад практи-
чески «заморожен». Окись азота крайне незначительно
растворима в воде: 0,0738 (0°), 0,0471 (20°) объемов
газа на один объем воды. Химически с водой и раз-
бавленными растворами кислот и щелочей не взаимо-
действует.
Молекула NO имеет нечетное число внешних элек-
тронов, в связи с чем NO парамагнитна. Для описа-
ния ее электронной структуры предложены различные
формулы, в частности: N •• О : и N :: О Окись азота
склонна к реакциям окисления — восстановления и
присоединения. При обычной темп-ре быстро окис-
ляется кислородом до NO2. Т. к. реакция 2NO + O2 =
= 2NO2 экзотермична, то при повышении темп-ры
равновесие смещается в сторону диссоциации NO2.
С хлором NO соединяется с образованием хлористого
нитрозила NOC1. Окислительные свойства NO про-
являются, напр., при ее взаимодействии с водородом:
2NO + 2На = N2 + 2Н2О (смесь равных объемов
NO с Н2 при нагревании взрывается), с сероводоро-
дом: 2NO -j- 2H2S = N2 + 2S 2Н2О и т. д. Окись
азота — основной полупродукт в производстве азот-
ной кислоты, где она получается при каталитич. окис-
лении аммиака. NO получают также непосредствен-
ным взаимодействием азота с кислородом при очень
высокой темп-ре (в пламени вольтовой дуги) и в элек-
троразрядах; в лаборатории — разложением нитритов
кислотами и др.
Двуокись азота NO2, четырехокись
азота N2O4 — бурый газ с удушливым запахом,
сжижающийся при 21,15° в светло-желтую жидкость,
замерзающую при —11,2° (бесцветные кристаллы);
при обычных условиях представляет смесь NO2 и
N2O4; в такой смеси содержится при 40° и 1 атм 31%
N О2, а при 100° и 1 атм—88% NO2, и лишь ок. 140° N2O4
целиком переходит в NO2. При дальнейшем повыше-
нии темп-ры начинает распадаться и _\О2 и ок. 600°
полностью превращается в NO и О2. Плотность газо-
образных окислов 3,3 г/л (21°), жидких—1,49г/с.м3 (0°);
г°крит. 158°, ркрит 99 атм-, давление паров: 0,58 атм
(10°), 2,38 атм (40°), 7,35 атм (70°). Теплоты образова-
ния: NO, Д77° = 8,091 ккал!молъ и N2O, МГ =
= 2,309 ккал/молъ. Молекула NO2 имеет нечетное чис-
ло внешних электронов, парамагнитна. Вопрос об элек-
тронной структуре молекул NO2 и N2O4 окончательно
не решен. С водой и р-рами щелочей NO2 взаимодей-
ствует химически: 2NO2 + Н2О = HNO3 + HNO2;
2NO2 + 2NaOH = NaNO3 + NaNO2 + H2O. Двуокись
и четырехокись азота — сильные окислители. Многие
в-ва (уголь, сера, фосфор, органич. соединения)
энергично сгорают в токе NO2. Четырехокись N2O4
с жидким аммиаком реагирует со взрывом. С соляной
к-той NO2 образует хлористый нитрозил NOC1, с
серной к-той — нитрозилсерную к-ту NOHSO4. В
пром-сти двуокись азота получают как промежуточ-
ный продукт в произ-ве азотной к-ты дальнейшим
окислением окиси азота кислородом или воздухом;
в лаборатории — разложением Pb(NO3)2 или взаимо-
действием меди с конц. HNO3. Применяют как нит-
рующий агент (см. Нитрование органических со-
единений).
Азотистый ангидрид NaO3 — красно-
бурый газ, сгущающийся при охлаждении в синюю
жидкость. Обычно приводимую в справочниках
т. кип. 3,5° нельзя считать точной из-за разложения
N2O3 на NO и NO2 при охлаждении; при сильном ох-
лаждении жидкая N2O3 застывает в белоснежные кри-
сталлы ст. пл. —102°. Теплота образования Д/Г ==
г 298
= 20,0 ккал/моль. С водой NaO3 образует азотистую
кислоту. N2O3 + Н2О = 2HNO2, с щелочами — нит-
риты. В лаборатории N2O3 получают взаимодействием
HNO3 с мышьяковистым ангидридом As2O3. Практич.
применения не находит.
Азотный ангидрид (двупятиокись) NaO5 —
единственный твердый при обычной темп-ре А. о.,
бесцветные кристаллы (ромбич. пластинки); при ком-
натной темп-ре разлагается на NO2 и О2, быстрое на-
гревание приводит к взрыву. Теплота образования
Д//°98(газ) = 3,6 ккал/молъ-, (крист.) = —10,0
ккал!молъ. В лаборатории получают взаимодействием
HNO3 с Р2О5 или жидкой NO2 с озонированным
кислородом и др. способами. Практич. применения
не находит.
Все встречающиеся на практике А. о. физиологи-
чески активны. Так, N2O в смеси с кислородом — сла-
бый наркотик; в высоких концентрациях вызывает
удушье. Окись азота NO действует на центральную
нервную систему, в больших концентрациях перево-
дит оксигемоглобин в метгемоглобин. Двуокись —
четырехокись азота раздражающе действует на лег-
кие, в тяжелых случаях вызывает отек, понижает
кровяное давление. Помимо непосредственного дей-
ствия, А. о. вызывает также и различные хронич.
заболевания при длительной работе в атмосфере,
содержащей эти окислы.
Лит. см. при ст. Азот и Азотная кислота.
АЗОТА ОПРЕДЕЛЕНИЕ — в иеорганич'еских'^'со-
единениях качественное определение проводят на-
греванием азотсодержащего в-ва с металлич. калием
или натрием с образованием цианида, обнаружива-
емого по образованию берлинской лазури (проба
Л а с с е н я). Азот в форме аммиака открывают по
желтому окрашиванию с Несслера реактивом или по
восстановлению солей двухвалентной ртути и др.
реакциям. Азот в форме нитрата открывают по синему
окрашиванию с дифениламином или по красному ок-
рашиванию с бруцином и др. реакциями; в форме
нитрита — по красному окрашиванию с Грисса ре-
активом. Для открытия азота в большинстве органич.
соединений используют метод Дюма (см. ниже).
Количественно элементарный азот в газовом анализе
почти всегда определяют как остаток вместе с ред-
кими газами после поглощения сопутствующих га-
зов различными реагентами и сожжения горючих
газов. При определении редких газов азот связывают
металлич. Li, Mg, Са и др. при высокой темп-ре (с
образованием нитридов) или сжиганием азота в элек-
трич. искре или дуге. В форме нитрата азот опреде-
ляют гравиметрия, методом с использованием гл. обр.
нитрона в качестве осадителя. Свободная HNO3 после
разбавления может быть оттитрована р-ром щелочи.
При определении небольших количеств нитратов
(напр., в питьевой воде) используют фотометрия, ме-
тод с применением фенолдисульфонового реактива
(2,4-фенолдисульфоксикислота в конц. HaSO4). Нит-
раты могут быть также определены восстановлением
цинком или Деварда сплавом до аммиака с последую-
щей его дистилляцией в определенный объем стан-
дартной к-ты. В форме нитрита азот определяют пер-
манганатометрически или гравиметрически по коли-
честву бромата серебра, восстановленного до бромида
серебра. Небольшие количества нитрита определяют
фотометрически с реактивом Грисса; в форме нитри-
дов азота (напр., в металлах) — по количеству амми-
ака, образовавшегося после растворения исследуемого
образца, а в форме окислов — либо по изменению
объема газовой смеси после восстановления, либо
73
АЗОТИСТАЯ КИСЛОТА -АЗОТНАЯ КИСЛОТА
74
титриметрически после поглощения соответствующим
растворителем.
Колич. определение азота в органич. соединениях
большей частью выполняется методами Дюма или
Къельдаля. Метод Дюма основан на измерении объема
элементарного азота, выделившегося в результате
сожжения анализируемого в-ва в атмосфере СОг
в присутствии СиО и металлич. меди. Разработаны
макро-, полумикро-, микро- и ультрамикроварианты
этого метода. Метод Къельдаля сводится к разло-
жению анализируемой пробы и связыванию всего
азота в виде сульфата аммония кипячением с серной
кислотой (часто в присутствии катализатора). По-
лученный раствор кипятят с едким натром и погло-
щают выделившийся аммиак титрованным раствором
H2SO4, избыток к-рой оттитровывают обратно едкой
щелочью. Несмотря на многочисленные вариан-
ты, метод не универсален. Существуют
другие различные методы определения
азота в зависимости от вида соедине-
ния, в котором находится азот. Спосо-
бы открытия и определения азота в
азотсодержащих группах органич. со-
единений см. в статьях Функциональный
анализ, Ван-Слайка метод.
Лиг,..: В а й б е л ь С., Идентификация
органических соединений, пер. с англ., М.,
1957; Соколов В. А., Методы анализа газов, М., 1958;
Handbuch der analytischen Chemie, hrsg. von W. Fresenius
und G. Jander, TI 3, Bd 5a, B. — Gottingen — Hdlb 1957;
Pregl F., Roth H., Quantitative organische Mikroana-
lyse, 7 Aufl., W., 1958. И. П. Ефимов.
АЗОТИСТАЯ КИСЛОТА HNOa— одноосновная сла-
бая кислота (К = 6,0 • 10 4 при 30°), известна только в
разб. водных р-рах. Изучение кристаллич. солей А. к.
показало, что анион NO2 (нитрит-ион) имеет фор-
му треугольника с углом между связями О—N—О,
равным 132°. А. к. наряду с азотной кислотой об-
разуется в р-ре при поглощении NOa водой: 2NOa +
-f-Н2О = HNOa+ HNO3. В зависимости от характера
реагирующих с ней веществ А. к. может быть либо
окислителем, либо восстановителем. Так, перекисью
водорода, перманганатом и др. HNO2 окисляется до
HNO3, а иодистым водородом восстанавливается до
NO. При действии сильных кислот А. к. разлагается
(3HNO2 = HNO3 + 2NO + Н2О). При обработке
конц. серной к-той р-ров, содержащих А. к., обра-
зуется нитрозилсерная к-та NOHSO4. С щелочами
А. к. образует солм-нитриты. Для А. к. вероятно
наличие двух способных переходить друг в друга
структур: Н—О—N=O и Н—NOa. Нитриты актив-
ных металлов (напр., NaNO2) построены, по-види-
мому, в соответствии с первой из них, нитриты мало-
активных (напр., AgNO2) — со второй. Комплекс-
ные и органические производные известны для обеих
форм А. к. Аналитически А. к. и ее соли в водных
растворах могут быть определены, например, по ро-
зовому окрашиванию с реактивом Грисса — Илосвая
(сульфаниловая кислота и р-нафтиламин), а также
по синему окрашиванию раствора с K.J и крахмалом.
Практическое применение имеют нитриты, из ко-
торых наиболее важен натрия нитрит. Все соли А. к.
ядовиты.
Лита. СМ. при ст. Азотная кислота. М. А. Миниович.
АЗОТИСТОВОДОРОДНАЯ КИСЛОТА (азоимид)
HN3 — соединение азота с водородом, имеет строе-
ние Н—N = Ji=N; расстояние N-—-N 1,14 А,
N-—>NH 1,25 А. Безводная А. к. — бесцветная жид-
кость с резким запахом, плотн. 1,13 г/см3, т. кип. 37°,
т. пл. —80°, ядовита; весьма чувствительное взрыв-
чатое в-во, легко детонирует при нагревании или очень
слабом ударе либо трении. При взрыве А. к. разла-
гается на водород и азот; теплота взрыва 1592 ккал/кг;
объем газообразных продуктов Vo = 1042 л/кг. В
парообразном состоянии А. к. также чувствительна
к внешним воздействиям и легко взрывает; в разбавл.
водных р-рах устойчива; диссоциирует на ионы Н+
и N3~ (К — 3 • 10~5). Помимо кислотной функции, для
А. к. характерна окислительная. Смесь А. к. с креп-
кой HCI при нагревании растворяет золото и платину.
А. к. получают действием на ее соли разбавл. серной
к-ты. Практич. применение имеют только соли А. к.—
азиды. А. А. Шидловскии.
АЗОТИСТЫЕ ИПРИТЫ — условное назв. группы
соединений типа fi-галогеналкил аминов общей фор-
мулы RN(GH2CH2GI)a, где R — алкильный, галоген-
алкильный или арильный радикалы. Свежепере-
гнанные А. и. — бесцветные жидкости с слабым за-
пахом, технич. продукты слабо окрашены. А. и.
плохо растворимы в воде, хорошо — в органич. рас-
творителях.
Формула Т. пл., °C Т. кип., °С/мм рта. ста. n~D d%5 Константы * А | В
CH3N (СН2СН2С1)2 C,H5N (СН2СН2С1)» N (СН.СН» = С1)3 — 60 -4-4,5 71/9 85,5/12 94/1 1,4653 1,4925 1,130“ 1.086 1,209 9,01892 9,41621 2868,9 3393,4
* По ф-ле 1g = A — В,T (А и В указаны в таблице). •• dj0.
Химич, свойства А. и. определяются наличием в
их молекулах третичного атома азота и достаточно
подвижного атома галогена. С минеральными к-тами
(HCI, H2SO4 и др.) А. и. образуют хорошо кристал-
лизующиеся, растворимые в воде соли, с пикриновой
к-той труднорастворпмые, с четкой темп-рой плавле-
ния. Растворы А. и. в полярных растворителях не-
устойчивы. При взаимодействии А. и. с хлорирую-
щими агентами происходит их дезалкилирование,
образуются нетоксичные продукты. Взаимодействие
А. и. с окислителями приводит к образованию N-ок-
сидов; при реакции с водой происходит последова-
тельное замещение атомов хлора на {гидроксил;
выделяющийся при этом HG1 образует с исходным ами-
ном солянокислую соль, медленно гидролизующую-
ся даже при нагревании. В присутствии разбавлен-
ных щелочей гидролиз протекает быстро с образо-
ванием свободных этаноламинов: RN(CH2CH2C1)2 -ф-
+ 2NaOH = RN(CHaGH2OH)2 + 2NaCl. Получение
А. и. основано на замене гидроксильных групп
в третичных ₽-этаноламинах на атомы хлора; наи-
более удобным реагентом для этой цели являет-
ся тионилхлорид: HG1 • RN(GH2GHaOH)2 + 2SOC12 =
= HCI • RN(CH2CH!C1)2+ 2SO2 + 2HC1. А. и. облада-
ют общеядовитым и сильным кожно-нарывным дей-
ствием, аналогичным действию иприта (см. Дихлор-
дизтилсулъфид), поражают дыхательные пути и желу-
дочно-кишечный тракт; скрытый период кожного дей-
ствия А. и. выражается неск. часами. Характерной
особенностью А. и. является их подавляющее дей-
ствие на клеточное деление, в связи с чем нек-рые из
них нашли применение в медицинской практике.
В частности, известный под названием эмбихина,
Х-метил-3,|3'-дихлордиэтиламин, применяется при
лечении пек-рых форм злокачеств. белокровия.
Лит.: Сартори М., Усп. хим.. 1954, 23, вып. 1,
с. 62—88. Р. II. Стерлин.
АЗОТИСТЫЙ ОБМЕН — см. Обмен веществ.
АЗОТНАЯ КИСЛОТА HNO3 — сильная однооснов-
ная к-та; в отсутствии воды малоустойчива. Строение
молекулы А. к. может быть представлено формулами:
I н—О—N\_
ХО
II H:O:N : :6
”:15:
Установлено, что все атомы А. к. расположены в од-
ной плоскости (рис. 1).
75
АЗОТНАЯ КИСЛОТА
76
Свойства А. к. При нагревании и под действием
света А. к. разлагается. На основании расчетных дан-
ных безводная А. к. плавится при
—41°, кипит при 86°. Теплота об-
разования жидкой HNO3 ЛИ°208 =
= —41,404 ккал/моль. Известны
кристаллогидраты А. к.: HNO3-3H2O,
т. пл. —18°; HNO3 • Н2О, т. пл.
—38° и эвтектики: с содержанием
32,7% HNO3 при —43°; 77,5% HNO3
при —42° и 89,95% HNO3 при —66,3°.
Теплота нейтрализации А. к. NaOH
равна 13,84 ккал/моль. С водой А. к. смешивается в
любых отношениях; т. кип. водных р-ров повышается с
увеличением концентрации, достигая максимума 121,9°
при содержании 68,4вес.% HNO3 (при 1 атм давления),
и падает при дальнейшем повышении концентрации.
При перегонке водных р-ров А. к. вначале в паровой
фазе содержатся преим. пары воды, к-рые постепенно
обогащаются HNO3, а при достижении т. кип. 121,9°
образуется азеотропная смесь — содержание HNO3
как в жидкости, так и в парах составляет 68,4%.
Таблица 1. Теплоемкость водных растворов А. к.
при различных температурах (ккал/т град)
HNO3, вес. % 2,53° 21,07° 39,49° 61,11°
15 0,838 0,842 0,851 0,862
60 0,629 0,641 0,654 0,668
95 0,456 0,460 0,461 0,460
Теплопроводность водных р-ров с увеличением со-
держания HNO3 падает; при 100° — с 0,587 ккал/м-
•час-град для чистой воды до 0,418 для 50%-ной HNO3
и 0,209 ккал/м-час-град для 98%-ной HNO3.
Таблица 2. Плотность водных растворов А. к. (при 20°)
Плотность, 2/СМ3 Содержание HNO3, г/100 г р-ра Плотность, г/см3 Содержание HNO3> 8/100 е р-ра
1.026 5 1,469 85
1,147 25 1,493 95
1,278 45 1,513 100
1,391 65
Конц. А. к. при нагревании и действии восстанови-
телей разлагается; образующиеся при этом окислы
азота растворяются, окрашивая ее в бурый цвет.
А. к. обладает сильными окислительными свойствами;
большая часть металлов превращается конц. А. к.
5 соответствующие окислы или нитраты. Нек-рые ме-
таллы, напр. Al, Fe, Сг, при взаимодействии с конц.
А. к. образуют на поверхности слои окислов, стойких
к ее действию. Устойчивы к действию А. к. Pt, Rh,
1г, Та, Ап. Конц. А. к. энергично действует на не-
металлы: сера окисляется в серную к-ту; уголь в
определенных условиях загорается, и т. п. Многие
органич. вещества при воздействии конц. А. к. раз-
рушаются и воспламеняются (солома, бумага, масла
и т. п.). Конц. А. к. действует на циклич. органич.
соединения, замещая в них один или неск. атомов во-
дорода нитрогруппой; действует на органич. вещества,
содержащие спиртовые ОН-группы с образованием
азотнокислых эфиров, и т. д. Смесь, состоящая из
1 вес. части конц. А. к. и 3,6 вес. частей конц. соля-
ной к-ты, называется царской водкой; в ней растворя-
ются благородные металлы. Разб. А. к. образует хо-
рошо растворимые, устойчивые в обычных условиях
соли — нитраты.
Получение. В лабораторных условиях А. к. полу-
чают из ее солей, действуя на них конц. H2SO4 при
нагревании; выделяющиеся пары конденсируют. Этим
способом раньше получали А. к. из чилийской се-
литры и в пром-сти. Применявшийся ранее способ
получения А. к. из атм. азота окислением его в воль-
товой дуге с последующим поглощением окислов азота
водой оставлен вследствие его нерентабельности. Сов-
ременные промышленные способы получения А. к.
основаны на окислении аммиака кислородом воздуха.
При этом сначала получают разб. А. к., к-рую при
необходимости перерабатывают в конц. А. к. пере-
гонкой с H2SO4 или действием жидких окислов азота
и чистого кислорода при давлении 50 ат. Произ-во
разб. А. к. состоит из 2 основных стадий: 1) окисления
NH3 в NO кислородом воздуха в присутствии ката-
лизаторов: 4NH3 + 5О2 = 4NO + 6Н2О; реакция про-
текает с выделением большого количества теплоты
(217 ккал/моль)', 2) окисления NO в NO2 избытком
кислорода и взаимодействия окислов азота с водой:
2NO + О2 = 2NO2, 3NO2 + Н2О 2HNO3 + NO. В
процессе абсорбции окислов азота водой образуется
также азотистая к-та, к-рая распадается: 3HNO2 =
= HNO3 -j- 2NO + Н2О. Реакция окисления NO в
NO2 и взаимодействие NO2 с водой также протекают
с выделением теплоты (29,5 и 17,5 ккал/моль соответ-
ственно).
Для ускорения окисления окиси азота применяют
повышенное давление. В зависимости от давления
произ-во разб. А. к. делят на три группы: при атм.
давлении, при повышенном давлении (до 9 ат) и
комбинированные — первая стадия проводится при
атм. давлении, а вторая (поглощение окислов азота
водой) — под повышенным. При повышенном давлении
резко сокращаются уд. объемы аппаратуры и расход
конструкционных материалов, но значительно воз-
растает расход электроэнергии. Этот недостаток ча-
стично компенсируется путем использования тепла
реакций окисления NH3 в NO и окисления NO в NO2,
а также высокой степенью рекуперации энергии сжа-
тых газов в современных турбинах. При больших мас-
штабах произ-ва А. к. целесообразно строить установ-
ки, работающие по комбинированной системе или под
повышенным давлением.
Для получения А. к. воздух и NH3 тщательно очищают от
газообразных и механич. примесей. Содержание механич.
примесей не должно превышать 0,007 мг/м‘. Воздух промывают
водой, фильтруют через сукно, а аммиак — через слой ваты
с полотном и затем их смешивают: аммиачно-воздушная смесь
содержит 10,5—12 % NH3. В такой смеси содержится значи-
тельный избыток кислорода, по сравнению с необходимым сте-
хиометрия. его количеством, что необходимо для получения
высоких выходов А. к. Аммиачно-воздушная смесь дополни-
тельно очищается от примесей на поролитовых фильтрах (по-
ристая керамика) или на фильтрах из картонных дисков и по-
ступает в верхнюю часть контактных аппаратов, где аммиак
окисляется на катализаторе при 750° (установки при атм.
давлении, далее обозначаемые I) или при 875—900° (установки,
работающие под повышенным давлением, далее обозначае-
мые II). Нитрозные газы, содержащие NO, О2, N2, Н2О (пары),
охлаждают до 160—250° в паровых котлах-утилизаторах и
далее в водяных холодильниках до 40° и направляют в си-
стему, состоящую из 6—8 башен, насаженных керамич. коль-
цами (I), или в колонны с барботажными колпачковыми или
ситчатыми тарелками (II).
До башен или колонн, а также в последних происходит
окисление NO в NO2 за счет кислорода, содержащегося в нит-
розных газах и дополнительно вводимого с воздухом. В башнях
или колоннах окислы азота поглощаются водой с образованием
разб. 45—49%-ной [I] или 55—58%-ной А. к. (II). Выделяю-
щаяся при этом теплота реакций отводится водяными холодиль-
никами. Поглощение окислов азота водой достигает 92 % (I),
98—98,5% (II). В случае (I) остающиеся окислы азота погло-
щают р-рами щелочей. Содержание окислов азота в газах,
выбрасываемых в атмосферу, не должно превышать 0,05%
(считая на NO). Вся основная аппаратура производства разб.
А. к. выполняется из кислотоупорных сталей.
Катализаторами для окисления NH3 в NO служат сетки,
изготовляемые из тройного сплава (93% Pt, 3% Rh и 4% Pd)
или двойных сплавов (90—95% Pt и 5—10% Rh). По механич.
прочности сетки из тройного сплава, разработанного совет-
скими исследователями, не уступают сеткам из сплава с 10%
Rh, а по активности превосходят на 1—2 %. Наиболее часто
сетки готовятся из нитей толщиной 0,09 мм и числом плете-
ний 1024 cjm2. В контактный аппарат загружают слой из 3 сеток
диаметром от 1,1 до 2,8 .и (I) или из 18—2 0 сеток диаметром
77
АЗОТНАЯ КИСЛОТА
78
0,5—1,15 м (II). В процессе окисления аммиака безвозвратно
теряется 0,048—0,056 г (I) и 0,160—0,180 г (II) платиноидов
на 1 га 100%-ной А. к. В СССР разработан двухступенчатый
катализатор (1-я ступень — одна платиноидная сетка; 2-ая
ступень — неплатиновый катализатор). Двухступенчатый ка-
тализатор позволяет экономить примерно 60% платиноидов
на вложениях и снизить их потери на20—30% против потерь
при применении одних платиноидных катализаторов. Сетки
постепенно загрязняются и теряют
поэтому их периодически промывают
каталитич. активность,
15%-вой соляной к-той,
Вода
Пар
Пар
'Местный забор
воздуха
Выхлопные
газы
Дальний
забор
воздуха
2
HI
77777 77
Вода (конденсат)
Рис. 2. Схема получения разбавленной азотной кислоты под давлением: 1 — башня для промывки воздуха; г — фильтр;
3 — турбокомпрессор; 4 — фильтр для воздуха; 5 — подогреватель воздуха; 6 — котел-утплизатор; 7 — испаритель
аммиака; 8 — фильтр для аммиака; 9— смеситель; 10— контактный аппарат; 11— холодильник-конденсатор; 12 — сепа-
ратор; 1з — фильтр; 14 — абсорбционная колонна; 15 — подогреватель выхлопных газов; 16 — рекуперационная турбина.
дистиллированной водой и прокаливают водородным пламе-
нем. Выход NO достигает 98% (I) или 96% (II). Лимитирующей
стадией процесса является диффузия NH3 к поверхности ка-
тализатора. В новых производствах разб. А. к. предусматри-
вается укрупненное оборудование, применяется агрегатный
принцип связи аппаратуры, используется теплота реакций
окисления NH3 в NO и NO в NOs, применяются колонны с сит-
чатыми тарелками. На рис. 2 представлена система произ-ва
разб. А. к. под давлением 8—9 ат, а на рис. 3 — комбиниро-
ванная система с давлением в абсорбционной части 3,5 ат.
Для получения конц. А. к. применяют в качестве водоот-
нимающего средства конц. H2SO4. Конц. А. к. получают в ко-
лоннах, выполненных из ферросилиция. В колонну одновре-
менно подают разб. А. к. в виде подогретой жидкости и паров,
купоросное масло и перегретый водяной пар. Выходящие из
колонны пары А. к. и воды конденсируют в холодильнике
в виде конц. HNO3, к-рая после освобождения от растворив-
шихся окислов азота выпускается в виде готового продукта.
Отработанную разб. H2SO4 концентрируют непосредственным
соприкосновением с горячими топочными газами. Другой спо-
денсируют при —10° и возвращают в произ-во. Применяют и
другие схемы произ-ва, позволяющие совмещать в одной и той
же аппаратуре получение как разбавленной, так и конц. А. к.
Большая часть получаемой А. к. обычно потреб-
ляется на месте ее произ-ва. Конц. А. к. перевозят в
алюминиевых цистернах, а разб. — в цистернах из
кислотоупорной стали. В СССР разб. А. к. выпус-
кается с содержанием HNO3: 55%, 47% и 45%, а конц.
А. к. с содержанием 98% и 97%.
Техника безопасности. Аммиачно-воздушная смесь
становится взрывоопасной при содержании NH3 от
16 до 32%; однако на практике считают опасной гра-
ницей 13% NH3b газах. На а мми а копр овод ах устанав-
ливаются отсекатели, автоматич. прекращающие пода-
чу аммиака в систему, в случае, если его концентрация
с воздухом достигнет опасной границы. При ненор-
мальной работе контактных аппаратов (при разрывах
плетений в сетках и т. п.) происходят проскоки аммиака
через катализатор. Реагируя с окислами азота, аммиак
частично разлагается, частично же образует стойкие
аэрозоли нитрита и ни-
трата аммония, к-рые
могут осаждаться на
стенках нитрозных газо-
дувок или трубокомпрес-
соров. Эти соли могут
разлагаться со взрывом.
Рис. 3. Схема получения разбавленной азотной кислоты комбинированным способом: 1 — аммиачно-воздушный венти-
лятор; 2 — фильтр; з — контактный аппарат; 4 —котел-утилизатор; 5 — подогреватель аммиачно-воздушиой смеси;
в — газозый холодильник-промыватель; 7 — компрессор нитрозных газов; 8 — окислительная башня; 9 — подогрева-
тель хвостовых газов; ю — абсорбционная башня.
соб получения конц. А. к. — прямой синтез, основан, на взаи-
модействии жидкой N2O4 с водой и кислородом по реакции:
2N2O4 + 2Н4О + О, = 4HNO3. Для этого приготовляется
смесь разб. А. к. (58% HNO3), к-рую с избытком N2O4 насосом
передают в автоклав, туда же подают под давлением 50 ат
газообразный кислород высокой чистоты. Темп-pa в автоклаве
поднимается до 70—85°, чем ускоряется процесс. Непрореаги-
ровавшие окислы азота удаляют нагреванием, затем их кон-
Поэтому нитрозные газы перед абсорбционной аппа-
ратурой промывают разб. HNO3. Жидкие окислы
азота и конц. А. к. при загрязнении органич. вещест-
вами способны разлагаться со взрывом. Поэтому тща-
тельно контролируют содержание органич. примесей
в р-рах к-ты или ее смесей с окислами азота. Макси-
79
АЗОТНЫЕ УДОБРЕНИЯ — АЗОТОЛЫ
80
мально допустимые концентрации вредных газов в
воздухе рабочих помещений: 0,02 мг/л NH3, 0,005 мг/л
окислов азота, в пересчете на N2O5, 0,02—0,03 ле/л
СО, что вполне достижимо при вентиляции.
Применение. А. к. является одним из важнейших
промышленных продуктов. Применяется в произ-ве
азотных и сложных удобрений (аммиачной, натрие-
вой и кальциевой селитры, нитрофоски и др.), в
цветной металлургии для травления и разделения
металлов, в произ-ве серной к-ты нитрозным методом
и различных органич. нитропродуктов, в ракетной
технике в качестве окислителя, как реактив в химич.
лабораториях и т. д.
Лит.: Эпштейн Д. А. [и др.], Химия и технология
связанного азота, ч. 2, Л., 1934; Атрощенко В. И. и
Каргин С. И.. Технология азотной кислоты, М.—Л.,
1949; Каргин С. И., Петунов С. П., Митро-
польский И. С., Производство азотной кислоты, М.,
1949; Иванов А. Ф., Хим. пром., 1954, .№ 7, с. 58;
Миниович М. А., Ж. прикл. хим., 1958,31,вып. 8,с. 1129.
Jf. А. Миниович.
АЗОТНЫЕ УДОБРЕНИЯ — химические соеди-
нения, содержащие в своем составе азот, способный
служить питательным элементом для растений. По-
мимо индивидуальных веществ, содержащих азот,
в качестве А. у. могут быть применены сложные, или
смешанные удобрения, включающие, кроме азота,
также калий и фосфор. Разнообразие почвенных и
клпматич. условий, биологич. особенности различных
с.-х. культур, а также экономил, соображения обус-
ловили необходимость произ-ва нескольких видов
А. у. Азот в А. у. может содержаться в нескольких
формах: в аммиачной, в нитратной, в смешанной —
аммиачно-нитратной, амидной. По этому признаку и
ирияято классифицировать А. у.
Аммиачные удобрения: жидкий аммиак,
82% N; аммиачная вода, 20—22% N; соли аммония, гл.
обр. сульфат аммония (см. Аммония сульфат), до 21%
N. Наиболее богатым по содержанию азота является
жидкий аммиак, к-рый нашел практич. применение
(так же, как аммиачная вода) в связи с разработкой
методов внесения в почву жидких удобрений. Сульфат
аммония — одно из старейших удобрений, обладает
малой гигроскопичностью, не слеживается, повышает
урожайность таких культур, как рожь, овес, карто-
фель, конопля и особенно чай и рис. На почву оказы-
вает подкисляющее действие. По мере расширения
произ-ва и применения других А. у. доля сульфата
аммония снижается. Кроме перечисленных А. у.,
К этой же группе относятся хорошо растворимый в
воде хлорид аммония, 26,1 % N, к-рый весьма удобен
длц применения. Хлорид аммония повышает урожай-
гранулированием и применением специальных мине-
ральных добавок.
К таким удобрениям относятся также известково-
аммиачная селитра, сульфат-нитрат аммония и др.
Весьма богаты азотом аммиакаты —продукты
обработки твердых солей, например NH4NO3, или
их водных растворов газообразным или жидким ам-
миаком.
Амидные удобрения — кальция циан-
амид, до 34,9% N (технический продукт 19—22% N),
и мочевина, 46,6% N. Цианамидкальция в последнее
время в качестве удобрения почти не применяется
вследствие его неэкономичности. Мочевина — высо-
коконцентрированное, безбалластное удобрение, по
эффективности не уступает нитрату аммония, приме-
няется также и для поверхностной (внекорневой)
подкормки; широко используется для подкормки
скота, а также в химическом производстве (смолы,
клеи, пластмассы и т. д.).
Наряду с перечисленными А. у. находят применение
и различные смеси, приготовляемые с целью улучше-
ния физич. свойств самих удобрений (снижение гиг-
роскопичности, устранение слеживаемости, подкис-
ляющего действия и т. д.) или для получения много-
стороннего удобрения (см. Смешанные удобрения).
Лит.: Клевке В. А., Поляков Н. Н., А р-
с е н ь е в а Л. 3., Технология азотных удобрений, М.,
1956; Справочник агронома по удобрениям, 2 изд., М., 1955;
Клевке В. А. иКонторовичЛ. М., Труды Гос.
н.-и. и проекта, ин-та азота, пром-сти, 1956, вып. 5, с. 71;
1957, вып. 7, с. 33; П р я н н ш н и к о в Д. Н., Агрохимия,
3 изд., М., 1940; Клевке В. А., Ж. прикл. хим., 1957,
30, № 12; К л е в к е В. А. и Г а м б у р г Д. Ю., Хим.
пром., 1950, № 7, с. 22 (214); Клевке В. А., М е д н и-
к о в В. Е., Ж. прикл. хим., 1960, 33, № 10; Иванов А. Ф.,
Шефм ан Ф. М., Бюл. Техн.-экон, информации, 1960 № 2;
Справочник по минеральным удобрениям, М., 1960. fertilize
technology and resources in the United States ,ed. by K. D. Ja-
cob, N. Y., 1953. В. А. Клевке.
АЗОТОЛЫ — принятое в СССР назв. азосоставляю-
щих, применяемых для получения нерастворимых в
воде азокрасителей непосредственно на текст, волок-
нах посредством азосочетания с ароматич. диазосо-
единениями. А. обладают сродством к целлюлозным
волокнам, что облегчает их применение, а также повы-
шает прочность окрасок.Применяемые в наст, время А.
разделяются на: 1) ариламиды о-оксикарбоновых к-т,
к-рые могут также содержать гетероциклы; чаще всего
используют ариламиды 2-оксинафтойной к-ты. Ниже
приведено строение нек-рых А. этой группы:
ность примерно тех же культур, что -------------------г
и сульфат аммония; почву подкисляет. Марка азотола!
На такие культуры, как табак, сахар-------------------1
ная свекла и т. п., действует отрица- Заместители
тельно; таков же эффект и при много-
А | ДМА | МНА | О | ОА । ОТ | ИА I ХА
Нет
кратном внесении. Положение
Нитратные удобрения:
натрия нитрат, до 16% N; калия ———
нитрат, до 13,8% N и 46,5% К2О; кальция нитрат,
до 15,5% N. Все нитратные удобрения оказывают
нейтрализующее действие на кислые почвы; приме-
няются гл. обр. для подкормки развившихся всхо-
дов при поверхностном внесении. Нитрат каль-
ция (собственно кальций) благотворно влияет на
улучшение структуры почвы.
Из аммиачно-нитратных удобрений наибольшее
распространение имеет аммиачная селитра (см. Ам-
мония нитрат), весьма богатое азотом соединение
(ок. 34% N), оказывающее на почву подкисляющее
действие. Используется под те же культуры, что и
аммиачные удобрения. Трудность применения из-за
слеживаемости нитрата аммония устраняется его
ОСН, N03
2; 5 3
Cl;
ОСН,
ОСН,
3; 4; 5
СН,
ОСН,
С1;
ОСН,
2 4 5; 2
2) Ариламиды (J-кетокислот, обычно ацетоуксусной
к-ты. К этой группе относится, напр., «Азотол 2Ж>>:
ОСН3
ch3coch2cohn-<21
ОСН3
А. — твердые продукты, б. ч. бесцветные, обычно
обладают характерными темп-рами плавления. В воде
практически нерастворимы, но при нагревании с
водными р-рами едких щелочей растворяются, обра-
зуя соли. Обычно А. хорошо растворимы в этиловом
81
АЗОТОМЕТР — АКАРИЦИДЫ
(5
спирте и пиридине, плохо — н эфире. А. — произ-
водные 2-окси-З-нафтойной к-ты — получают нагре-
ванием последней с соответств. аминами в органич.
растворителях, напр. толуоле или хлорбензоле, в при-
сутствии РС13: напр. С10Нв(ОН)СООН + C6H5NH2—>
—>С10Н6(ОН)СО—NHG6H5 + Н2О. А. — ариламиды
ацетоуксусной к-ты — получают нагреванием аце-
тоуксусного эфира с аминами в органич. растворите-
лях, напр. ксилоле, или взаимодействием аминов
с дикетеном. Окраска А. зависит как от их строения,
так и от диазосоединений. Ариламиды 2-окси-З-нафтой-
ной к-ты чаще всего образуют оранжевые, красные,
бордо, фиолетовые и синие окраски. Для получения
коричневого и черного цветов применяют арилами-
ды окс.икарбазолкарбоновых кислот, для темно-зеле-
ного — ариламиды 2-антрол-З-карбоновой кислоты.
Желтые оттенки образуются с А. — производными
fj-кетокислот. См. также Азокрасители, Диазосое-
динения, Азоамины, Крашение текстильных мате-
риалов.
Лит. см. при ст. Азоами-Hoi. М. А. Чекалин.
АЗОТОМЕТР — см. Волюмометры.
АЗУЛЕНЫ — небензоидные ароматические соеди-
нения, содержащие конденсированную систему 5-
и 7-членного циклов;
простейший предста-
V I V “Я ( 4- ') 5 витель — азулен (I).
/ Многие А. встреча-
X—/ юте я в природе в эфир-
ных маслах; таковы,
1 и например, гвайазулен
(1,4-димстил-7-изонронилазулен), хамазулен (1,4-ди-
метил-7-этилазулен), ветивазулен (4,8-диметил-2-изо-
пропилазулен). Большинство А. интенсивно окрашено
в синий или сине-фиолетовый цвет (максимум погло-
щения в областях ок. 700, 640 и 580 ммк). Они легко
образуют пикраты и комплексы с тринитробензолом.
Из многочисленных методов получения А. наиболее
удобен синтез из пиридина и циклопентадиена или
их гомологов (Циглер, Хафнер, 1955).
В молекуле А. 10 л-электронов, к-рые образуют
замкнутую электронную оболочку, что и обусловлива-
ет ароматичность А. (см. Ароматические системы).
Вследствие этого в молекуле А. так же, как в бензоле
и нафталине, нет чередующихся простых и двойных
связей и порядок связей является промежуточным
между простыми и двойными. Система А. обладает
значительной энергией сопряжения. Поэтому А. легко
образуются при дегидрогенизации над Pt или Pd
бпцикло-(0,3,5)-декана, бицикло-(0,3,5)-децена, цик-
лодекана, циклодеценов, а также при высоко-
темп-рной дегидрогенизации декалина, тетрагидроцик-
лопентадиена и пергидроаценафтена над окисными
катализаторами.
Облако л-электронов в молекуле А. не симметрично,
и 5-членное кольцо приобретает частичный характер
циклопентадиенильного аниона, а 7-членное — тропи-
лиевого катиона (см. II). Все атомы углерода 5-член-
ного кольца А. несут небольшой отрицательный заряд,
а 7-членного (кроме атомов, общих для обоих колец) —
положительный; в результате А.обладают дипольным
моментом (для незамещенного А. равным 1,00 D).
Биполярное строение А. проявляется в большой
легкости реакций электрофильного замещения, при-
чем первый заместитель всегда вступает в 5-член-
ное кольцо в положении 1, а второй — в положе-
нии 3 (если они не были заняты др. заместителями).
Эти реакции — нитрование, сульфирование, бромиро-
вание, ацетилирование, формилирование, азосоче-
тание, меркурирование, а также дейтерообмен —
протекают для А. значительно легче, чем для бензола,
и по своей реакционной способности А. напоминают
«суперароматические» 5-членные гетероциклы. Нуклео-
фильные реагенты вступают в положительно заря-
женное 7-членное кольцо А., но такого рода реакции
для А. менее изучены.
С биполярным строением А. связана и его способ-
ность чрезвычайно легко присоединять протон прг
растворении в минеральных к-тах с образованием
азулениевого катиона:
Простейший азулен (I) С10Н8 — синие пластин-
ки; т. пл. 98,5—99,0°; т. кип. 163°/14 мм; дипольный
момент 1,0+0,051; хорошо растворим в концентри-
рованных минеральных кислотах, из полученных р-ров
осаждается водой; легко изомеризуется в нафталин;
' гидрируется; окисляется на воздухе и КМпО4 с обра-
зованием СО2, (СООН)2 и др. Известен ряд производ-
ных А.: пикрат, т. пл. 117° (из спирта), тринитробен-
зоат, т. ил. 164°.
Лит.: Гордон М., Усп. хим., 1953, 22, вып. 8, с. 948;
Hafner К., Angew. Chem., 1958. Beilage zu Heft. 14 July
S. 419; Heilbronner E., Azulenes, в кн.: Non-benzenoid
aromatic compounds, ed. D. Ginsburg, N. Y. — L., 1959,
p. 171. M. E. Вольпин.
АЙМАЛИН (раувольфин, неоаймалин) C21H29N2O2,
мол. в. 341,48 — алкалоид (I), содержащийся в кор-
нях растения раувольфии
змеиной (Rauwolfia serpen-
tina) сем. кутровых (Аро-
супасеае). А. кристалли-
зуется с тремя молекулами
воды; безводный А. имеет т.
пл. 158—160°; [а]у=+128°
(хлороформ); образует хлоргидрат, т. пл. 253—255°,
пикрат, т. пл. 126—127° и др.; третичное однокислот-
ное индолиновое основание средней силы. Строение
А. установлено на ос-
новании изучения про-
дуктов распада, а так-
же спектральных дан-
ных. Стереохимия А.
выяснена в 1958. При
действии С2Н6ОК или
нагревании (200°) А.
изомеризуется в и з о-
аймалин (II), т. пл. П
264—265°, [a]D = + 72° (этанол) —• алкалоид, выделен-
ный из того же растения, что и А. Физиологич.
действие А. проявляется преим. в понижении кро-
вяного давления и подавлении деятельности кишеч-
ника.
Лит.: Орехов А. П., Химия алкалоидов, 2 изд.»
М., 1955; Усп. хим., 1957, 26, вып. 2, с. 239—63.
А. Е. Васильев.
АКАРИЦИДЫ — химич. средства борьбы с кле-
щами. Для борьбы с растительноядными клещами
применяют: серу и полисульфиды кальция и бария,
спирты ароматич. ряда, сложные эфиры карбоновых
и сульфокислот, эфиры сернистой к-ты, нитрофенолы,
производные циклогексиламина, ароматич. ацетали,,
фосфорорганич. соединения, азобензол и др. Серу
применяют в молотом виде, в виде коллоидных препа-
ратов или полисульфида кальция, получаемого рас-
творением серы в извести при нагревании; такой
раствор известен под названием ИСО (известково-
серный отвар). Препараты серы вытесняются раз-
личными органическими соединениями, которые зна-
чительно более эффективны и более удобны при
применении. Важнейшие органические А. приведены
в таблице.
83
АКАРОИД — АКВАМЕТРИЯ
84
Название Химическая 1 формула Внешний вид Т. пл., °C Токсичность ЛД50 (для крыс), мг(кг В каком виде применяется
химическое ' торговое
4,4"-Дихлордифе- иилметилкарбинол Ди мит (С1СеН4)2 СНОНСНз Бесцветные кристаллы 69,5—70 - -
4,4' '-Ди хлор дифе- иилтрихлорметил- карбинол Кельтан (С1СеН4)2 СНОНСС13 То же 78,5-79 730 См а чи в а ющийс я порошок или эмульсия
Этиловый эфир 4,4"-дихлорбензи- ловой к-ты Хлорбензилат (С1С0Н4)2С(ОН) СООС2Н5 Бесцветная жидкость (т. кип. 140—142° 0,06 мм) 3 000 Эмульсия
4,4"-Дихлор дифен- оксиметан Неотран (С1С0Н4О)2 сн2 Бесцветный порошок 67-68 5 800 Смачивающийся порошок
n- X лорфени л-п- хлорбензолсульфо- нат 1 Эфирсуль- фонат, ово- тран ClC0H4OSO2CeH4Cl То же 86,5 2 050 Смачив ающийся порошок с содер- жанием действую- щего начала 30—80%
4-Хлорфенил- 4-хлорбеНзилсульфид Хлорпарацид ClC6H4SCH»CeH4Cl » » 72 10 000 (ЛДюо) Смачивающийся порошок
4-ФторфеНил- 4-хлорбензилсульфид Фторпарацид FCeH4SCH2CcH4Cl » » 36 3 000, С хлористым метилом дает аэрозоли (для обработки теплиц)
Фенил-4-хлор- фенилсульфон Сульфенон CeH5SO2CeH5Cl » » 98 3 000 Смачивающиеся порошки
4-Хлорфенил- 2,4, 5-трихлор- фенилсульфон 2 Тедион ClCeH4SO2C6H2Cl3 Бесцветный порошок 145—146 — Смачив ающиес я порошки
2-(4-Третичнобутил- фенокси)-изопро- пил 2 -хлорэтил- сульфит 3 Арамит 1 (СНз). CSOOC«H4C1 1 (CH3)2 CCH2OC6H5 Бесцветная жидкость (т. кип. 175° при 0,01 мм), . Dio 1,145 3 900 Эмульсии, суспен- ЗИИ и ДуСТЫ
1 В технич. продукте содержится лишь ок. 60% n-хлорфенил-п-хлорбензолсульфоната; содержащиеся в технич. продукте
n-хлорфениловый и 2,4-дихлорфениловый эфиры бензосульфокислоты обладают более слабым действием. 3 Получают конден-
сацией трихлорбензосульфохлорида с хлорбензолом. 3 Один из наиболее активных современных А.; безопасен для насеко-
мых, токсичен для клещей во всех стадиях их развития; может быть получен из нетоксичных изомеров гексахлорцикло-
гексана.
Активными А. для борьбы с растительноядными
клещами являются также практически все фосфор-
органич. инсектициды', тетраэтилпирофосфат, тиофос,
метафос, меркаптофос, метилмеркаптофос, октаметил,
карбофос и др., а также такие соединения, как азо-
бензол, динитро-о-крезол, вторичнобутилдинитрофе-
нол и их гомологи, динитрокаприлфенилкротонат
(каротан) и др. В качестве средств борьбы с иксодо-
выми клещами с успехом могут быть использованы
такие инсектициды, как ДДТ, гексахлорцикло гексан,
хлортен, хлорированные терпены и т. п.
Лит.: Павловский Е. Н., Руководство по пара-
зитологии человека, с учением о переносчиках трансмиссивных
болезней, т. 1—2, 5 изд., М.—Л., 1946—48; Меткаф Г.,
Акарициды, в кн.: Химические средства защиты растений... .
Сб. переводов и обзоров, № 2, М., 1957, с. 120; Попов П. В.,
Справочник по ядохимикатам, М., 1956; Мельников Н. Н.,
Ж. Всес. хим. о-ва, 1960, 5, № 3, с. 2 42—49.
Н. Н. Мельников.
АКАРОИД — природная смола, образующаяся на
стволах и ветвях произрастающих в Австралии де-
ревьев (Xanthorrhoea). А. — аморфный продукт, ок-
рашенный в желтоватый или темно-красный цвет;
растворяется в этиловом спирте, кетонах,серном эфире,
хлороформе и скипидаре. На основании числа омыле-
ния (160—220) и кислотного числа (60—140) можно
судить о значительном содержании в составе А. слож-
ных эфиров и свободных к-т. Характерная особен-
ность А. — термореактивность (при нагревании пе-
реходит в неплавкое и нерастворимое состояние). А.
применяют для приготовления окрашенных в буро-
красный цвет спиртовых лаков и лаков на основе
ацетилцеллюлозы. В связи с развитием производства
синтетич. смол использование А. имеет ограниченное
значение.
Лит.: Киселев В. И., Олифа и лаки, 3 изд., М.—Л.,
1940. А. А. Благонравова.
АКВАМЕТРИЯ — методы колич. определения воды
в неорганич. и органич, веществах.
Для определения воды применяют: 1) высушивание
в сушильных шкафах до постоянного веса; 2) гете-
рогенную перегонку жидких материалов с углеводо-
родами или галогенопроизводными и измерение объема
отслаивающейся воды; 3) поглощение воды перхлора-
том магния, CaSO4, СаС12, Р2О5 и т. п. и определение
содержания ее по привесу поглотителя; 4) обработку
исследуемого в-ва карбидом кальция и измерение
объема выделившегося ацетилена. Очень часто при-
меняют т.н. реактив Фишера — иод-пиридин-метаноль-
ный р-р, в состав к-рого входит SO2. Под действием
воды происходит разрушение иод-пиридинового ком-
плекса и выделение молекулярного иода. Определе-
ние воды выполняют титриметрически. Точку эк-
вивалентности устанавливают по появлению отчетли-
вой желто-оранжевой окраски свободного иода; титр
реактива — по стандартному р-ру иода в метаноле.
При помощи реактива Фишера определяют воду и
нефтяных фракциях, красках, лаках и политурах,
пищевых продуктах и т. д. Титриметрич. метод при-
меняется также для изучения процессов, связанных с
выделением или поглощением воды. Известно много
вариантов метода. В большинстве случаев воду эк-
страгируют из растворимых соединений или взвесей
в неполярных растворителях и затем определяют тит-
рованием реактивом. При анализе окрашенных в-в,
а также нек-рых суспензий и эмульсий точку эквива-
лентности устанавливают электрометрически. Опре-
деление воды затруднительно, а иногда невозможно
в соединениях, вступающих в реакцию с Одним из
компонентов реактива (окислы и гидроокиси метал-
лов, соли 2-валентной меди и 3-валентного железа,
борная к-та и окислы бора и др.); в подобных случаях
либо пассивируют эти в-ва по отношению к реактиву,
напр. введением избытка уксусной к-ты устраняют
влияние аминов и гидразинов, либо определяют ме-
шающие в-ва в отдельных пробах и вводят соответств.
ft
85
АКВАПОЛИСОЕДИНЕНИЯ — АКОНИТОВЫЕ АЛКАЛОИДЫ
85
поправки. Затруднения возникают также при анализе
органич. соединений, взаимодействующих с реакти-
вом с образованием воды или нерастворимых про-
дуктов.
Другие химич. методы, как правило, дают возмож-
ность лишь качеств, открытия воды; они непримени-
мы в присутствии многих в-в, а также для опреде-
ления малых количеств воды в исследуемых мате-
риалах.
Лит.: Кольтгоф И. М. и Сендэл Е. Б., Коли-
чественный анализ, пер. с англ., 3 изд., М., 1948; Митчел Д.,
Смит Д., Акваметрия, пер. [с англ.], М., 1952; Кольт-
гоф И. М. [и др.], Объемный анализ, пер. с англ., т. 3, М.,
1961. И. А. Канаев.
АКВАПОЛИСОЕДИНЕНИЯ —.комплексные соеди-
нения, получаемые только из водных р-ров, со-
держащие большое количество воды, удаление к-рой
вызывает их разложение. Важнейшие А. — пара-
и метавольфраматы, пара- и метамолибдаты; в их
состав входят окисел металла и вольфрамовый (мо-
либденовый) ангидрид с отношением RaO : WO3,
равным 1:2,4 (В — одновалентный металл), и со-
ставом 5R2O-12WO3 • пН2О. Иногда этим соединени-
ям приписывают формулу: 3R2O • 7WO3 • пН2О, где
R2O : WO3 = 1 : 2,33. Для метавольфраматов (соответ-
ственно метамолибдатов) отношение R2O : WOa = 1:4;
их состав В2О • 4WO3 • пН2О.
Строение А. не установлено; часто их состав выражают
координац. формулами типа: NasHs[H2(WO4)e] • 10,5Н2О—
паравольфрамат натрия; Na,H4[H2(W2O7)e] • 2 7Н2О — мета-
вольфрамат натрия. Согласно последним исследованиям
(В. И. Спицын), в образовании А. вольфрама принимают уча-
стие H,WO(, WO®~, НаО+ и гидроксониевые формы вольфра-
мовой кислоты H(H,O)WO4, (H3O)2WO4, причем связывание
этих частиц происходит за счет водородных связей. Предло-
жена след, схема образования паравольфрамата:
Н3О+ + 3WO=" [ОН3 . . . (WO()3]s-
С6 — С13), ветхина (III) и гариина (IV, связь С7 —
С18), зонгорина (V) и нек-рых других алкалоидов.
Ко второй подгруппе А. а. относятся высокоток-
сичные в-ва, для к-рых характерно наличие гекса-
циклич. ядра, включающего 19 атомов углерода и
содержащего большое число заместителей: метоксиль-
ных, гидроксильных и ацилоксигрупп. Строение всех
этих алкалоидов оставалось неизвестным до того,
как методом рентгеноструктурного анализа было оп-
ределено строение ядра
нина. Характер и поло-
жение функциональных
групп в ядре были опре-
делены химическим пу-
тем. Таким образом были
установлены формула
ликоктонина (VI) и фор-
мулы его сложных эфи-
ров — метилликаконити-
на (VII), ликаконитина,
одного из них — ликокто-
дельсемина, авадхаридина, аяцина. Превращение в
продукты расщепления ликоктонина алкалоидов
дельфелина, элатина (VIII), эльделина, делькозина,
дельсолина привело к выяснению строения и этих
алкалоидов. Далее были установлены формулы ако-
нитина (IX) и дельфинина (X):
[H3O(wO4)3p~ + :i(H3o).wo4~
“ [ОН3 ... (WO4)3 ... (HWO4 • H3O)3(H2O)3]S-
Болыпинство А. хорошо кристал-
лизуется. Паравольфраматы мало раст-
воримы в воде. Наибольшую раствори-
мость имеет 5Li2O • 12WO3 • ЗЗНгО. Ме-
тавольфраматы хорошо растворимы
в воде (исключая соли Ag, Pb, Hg, Rb,
Т1). При нагревании до 300° А. разла-
гаются. При подщелачивании р-ров
А. образуются нормальные соли. Взаимо-
действие метавольфрамата с серной
к-той и эфиром приводит к выделению
VI—VIII
V R=R'=R"=H
tX R=C2Hs, R'=OH
X R=CH3,. R =H
VII и VIII R=
VII R'=R"=H
метавольфрамовой к-ты H2W4043 • 8Н2О.
Паравольфраматы и парамолибдаты по-
лучаются при подкислении растворов
Na2WO4 или Ха2МоО4до слабощелоч-
ной или нейтральной реакции. Мета-
вольфраматы образуются при добавле-
нии H2W04 в кипящий р-р Na2WO4
(pH ~ 3,5—4,5). Паравольфраматы являются про-
межуточными продуктами в технологии вольфрама
и его соединений.
Лит. см. при статье Гетерополисоединения.
Г. И. Пирогова.
АККУМУЛЯТОРЫ — см. Химические источники
тока.
АКОНИТОВЫЕ АЛКАЛОИДЫ — алкалоиды из
растений сем. Ranunculaceae родов Acomtum и Del-
phinium. А. а. могут быть разделены на две под-
группы. К первой относятся нетоксичные в-ва, име-
ющие пента- или гексациклич ядро, образованное
20 атомами углерода. При выяснении строения про-
дуктов дегидрирования этих А. а. селеном было опре-
делено строение основного ядра; на основании этого
и других данных предложены формулы строения ати-
зина (I) и изоатизина (II) (формулы I и II, связь
CO—CH—СНэ
со-сн
отличаются от ал-
А. а.
калоидов других групп не
только структурой, но так-
же происхождением в рас-
тениях: если большинство
алкалоидов других групп
продуктами распада и пре-
I VIII R’=R"=CHS
генетически связано
вращения белков и углеводов, то А. а., по-видимому,
связаны с дитерпенами.
Аконитин чрезвычайно ядовит. Токсич. дозы вы-
зывают паралич дыхания и оказывают прямое дей-
ствие на сердце. При внутривенном введении белым
мышам 0,2 мг/кг выживают 40%, при введении 0,3—
0,35 мг/кг гибнут 100% (10 мин.). При попадании на
слизистые оболочки даже ничтожных количеств ако-
нитин вызывает ощущение жгучей боли. Фармако-
логии. свойства дельфинина, лаппаконитина сходны
с аконитином. Продукты омыления менее токсичны.
Элатин, метилликаконитин, кондельфин, дельсемин,
аяцин, авадхаридин обладают курареподобным дей-
ствием, первые четыре применяются в медицине.
Лит.: Орехов А. П., Химия алкалоидов, 2 изд., М.,
1955; Генри I. А., Химия растительных алкалоидов,
с
87
АКРИДИН - АКРИЛАТЫ
88
т. кип. 345—346°;
пер. с англ., М., 1956; Кузовков А. Д., Статьи в Ж.
общ. хим., 1953—60; Schneider W., Arzneimittel For-
schung, 1957, Jg. 7, № 8, S. 485; M ar ion L., в ин.: XVI
Congres international de chimie..., Basel—Stuttgart, 1957 (Expe-
rimentia, suppl. 7), p. 328—42; Wiesner K., Simmons
D. L., Fowler L. R., Tetrahedron letters, 1959, № 18,
p. 1; Zechmeister L., (Hrsg.), Fortschritte der Chemie
organischer Naturstoffe, Bd 16, W., 1958. А. Д. Кузовков.
АКРИДИН C13H9N, мол. в. 179,22 — бледно-жел-
тые кристаллы; т. пл. 110—111°;
легко растворяется в большин-
стве органических растворителей;
его растворы флуоресцируют фио-
летовым светом; растворимость
в воде 1 : 20 000 (20°). А. возго-
няется; пары оказывают раздра-
жающее действие. Теплота сгора-
ния А. (в парах) 1596,5 кал/молъ.
А. — слабое основание рАкнсл
5,7 (в воде при 20°). Нумерацию
положения заместителей в молекуле А. (I) производят
аналогично антрацену; недавно предложена другая
нумерация (II). А. очень устойчив химически; он не
изменяется при нагревании до 280° с конц. НС1
или с КОН; при действии водорода в присутствии
скелетного Ni (или Zn НС1) восстанавливает-
ся в акридан (IV). К действию окислителей А.
устойчив: нагревание с хромовой кислотой дает
только 8% акридона (III). А. легко присоединяет
различные реагенты (галогены, HCN, бисульфит)
в положениях 9 и 10 (1) или 5 и 10 (II); в реакции
замещения вступает с трудом; большинство заме-
щенных А. было получено из соответств. замещен-
ных бензола или акридона. С кислотами А. образует
соли, водные р-ры к-рых обладают зеленой флуорес-
ценцией.
А. содержится в кам.-уг. смоле, в технике его по-
лучают, напр., конденсацией дифениламина с муравь-
иной или щавелевой к-тами или восстановлением ак-
ридона. А. является ингибитором коррозии металлов.
Среди производных А. имеются технически ценные
красители (см. Акридиновые красители) и важные
лекарственные в-ва (см. Акрихин и Риванол).
АКРИДИНОВЫЕ КРАСИТЕЛИ — производные
акридина или 9-фенилакридина, содержащие в поло-
жении 3 и 6 различные ауксохромные группы (ОН,
NH2, SH и др.). А. к. относятся к группе основных
и применяются для крашения кожи, бумаги, дерева
и протравленного хлопка в желтые и оранжевые тона.
Получают А. к. конденсацией ароматич. ж-диами-
нов с жирными или ароматич. альдегидами с после-
дующей циклизацией (образованием акридинового
цикла) и окислением.
При конденсации ж-толуи лендиамин а и ацеталь-
дегида образуется краситель основной желтый Н
(эухризин GGNX), а при конденсации ж-толуилен-
диамина с бензальдегидом —• краситель бензофлавин.
А. к. способны аккумулировать энергию солнечного
света, освобождая ее во время последующего пребы-
вания в темноте. Из производных акридина как кра-
сителя наибольшее значение имеют основной
желтый К (акридиновый желтый Г), основной
желтый Н (эухризин 2 ГНИКС), акридино-
вый оранжевый НО. Для получения красите-
ля основного желтого К конденсируют ж-толуилен-
диамин с формальдегидом, дифенилметановое про-
изводное подвергают
окислению:
циклизации и последующему
К А. к. относятся также линейный хинакридон
и его производные, выпуск к-рых начат в 1959 (США).
Линейный хинакридон выпускают под названием
«синквазия» илн «монастраль красный» н «мона-
страль фиолетовый». Прочности этих пигментов к
свету и различным реагентам близки к прочностям
пигментов фталоцианинового ряда. Один из методов
получения А. к. состоит в сложно-эфирной конденса-
ции двух молекул эфира янтарной к-ты через стадию
получения эфира циклогексан дион дикар боновой к-ты.
Последующее взаимодействие с анилином и окисле-
ние приводит к линейному хинакридону.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венкатараман К., Химия синтетических краси-
телей, пер. с англ., т. 2, Л., 1957; Dyestuffs manufacture. Re-
port as an index..., v. 1 — 4, L., 1948 (FIAT, № 764).
H. Г. Лаптев.
АКРИЛАМИД CH2=CHCONH2, мол. в. 71,07—
бесцветные кристаллы; т. пл. 84,5°. А. хорошо раст-
ворим в роде, метаноле, этаноле, ацетоне (соответст-
венно 215,5; 155; 86,2; 63,1 г в 100 г растворителя),
плохо растворяется в гептане, четыреххлористом уг-
лероде, бензоле. А. получают из ангидрида акриловой
к-ты и аммиака, из различных р-замещенных (окси-,
метокси-, хлор- или амино-) пропионамидов. Для
получения А. наиболее широко применяется гидролиз
акрилонитрила в присутствии конц. серной к-ты
при 90—100°:
/.О
CH.=CH-CN->CH.=CH- С"
А. взаимодействует с аммиаком, спиртами, меркапта-
нами, аминами, напр.: R2NH + GH2=CHCONH2 —>
—> R2NCH2—CH2CONH2. Большое значение приоб-
рели полимеры и сополимеры А. (см. Полиакриламид).
В. А. Сергеев.
АКРИЛАТЫ — эфиры акриловой к-ты общей фор-
мулы CH2=CHCOOR, где R — алкильный радикал.
Радикал Т. кип., °C/AtAt 20 П£)
Метил 80/760 0,9564 1,4040
Этил 43/103 0,9245 1,4068
Пропил 44/40 — 1,4130
Изопропил 108-112/760 — —
Бутил 35/8 0,9110 1,4190
Изобутил 130-134/760 — —
Изоамил 157—159/756 0,9020 1,4287 (12°)
Гексил . . . . 40/1,1 — 1,4285
Октил 57/0,05 — 1,4360
Нонил 76/0,2 1,4380
Децил 120/5 — 1,4400
Додецил 120/0,8 —— 1,4440
Тетрадецил 138/0,4 —— 1,4468
Гексадецил 148/0,04 — 1,4470 (30”)
Бензил 110-118/8 1,0630 1,5232 il8”)
Фенил 105/20 — — ,
Аллил 72/27 0,9886 1.4390
89
АКРИЛОВАЯ КИСЛОТА —АКРОЛЕИН
90
А. легко полимеризуются от действия света, тепла,
кислорода, перекисей.
В пром-сти А. получают из этилснциапгидрина и
соответствующего спирта: CH2OHCH2CN + ROH +
+ H2SO4->CH2 = CHCOOR + NH4HSO4 или взаимо-
действием смеси ацетилена, карбонила никеля и
соотв. спирта в присутствии НС1 при 40°:
4СП СП + Ni(CO), 4- 4R0H + 2НС1 ->
-> 4CH3=CHCOOR + NiCl3 + На
Обычно по этим схемам получают метил- и этил-
акрилаты, а высшие А. —путем их переэтерификации.
А. применяются для произ-ва полимеров (см. Поли-
акрилаты). И. В. Матвеева.
АКРИЛОВАЯ КИСЛОТА (пропеновая, этилен-
карбоновая кислота) СН2=СНСООН, мол. в. 72,36 —
первый член ряда алифатич. непредельных кислот с
одной двойной связью; бесцветная жидкость с острым
запахом; т. пл. 13°; т. кип. 141°/760 мм, 100°/249 мм,
40е/22мм, 20°/7,76 мм; d-" 1,0511; nff 1,4224; теплота
испарения при 136° 8,9 ккал/моль; теплота сгорания
329 ккал/моль (о = const); константа диссоциации
К = 5,6 • J05 (25°); растворима в воде, спирте, эфире.
А. к. получают омылением ее нитрила или этиленциан-
гидрина и дегидратацией гидракриловой к-ты:
HOCH3CHCN - НОСН3СН3СООН — сн3=снсоон
или оксосинтезом:
С2Н2+ СО 4- Н3О — сн2 = снсоон
В пром-сти чаще получают эфиры А. к. (см. Акри-
латы). А. к. обладает сильным корродирующим дей-
ствием. По месту двойной связи А. к. присоединяет
водород, галогены, галогеноводороды; в последнем
случае образуются [1-галогенопроизводные пропионо-
QPI вой к-ты; с водой дает гидракрило-
нс^| ХГНСООН вую к-ту: встУпает в реакции
II СН2| Дильса — Альдера, например с
НС | сн циклопентадиеном образует би-
х'СнГ 2 циклическую карбоновую к-ту
у (I). А. к. легко полимеризуется,
образуя твердый бесцветный по-
лимер — полиакриловую к-ту. В пром-сти большую
роль играют полимеры эфиров А. к. и нек-рых ее
солей (см. Полиакрилаты). Для предотвращения по-
лимеризации А. к. при хранении в нее вводят ста-
билизаторы, напр. гидрохинон и др. А. к. и ее соли
применяются как добавки к печатным краскам и
пастам, как компоненты лаков и др.
Лит.: Синтезы органических препаратов, пер. с англ., 4,
М., 1953; 10, М., 1961; Chemistry of carbon compounds, ed.
E. H. Rodd, v. 1, pt A, Amst., 1951. M. И. Роеенгарт.
АКРИЛОНИТРИЛ (нитрил акриловой кислоты,
цианистый винил) СН2=СН—CN, мол. в. 53,03—
бесцветная жидкость с характерным запахом; т. пл.
—83°; т. кип. 77,3°; d'2" 0,8060; nJ," 1,3911; изме-
нение темп-ры кипения 0,043° С/мм; т. воспл.
0:<z2,5°; теплота полимеризации 17,3±0,5 ккал/моль,
теплота сгорания 420,5 ккал/моль; теплоемкость
0,50±0,03 кал/г-град; скрытая теплота испаре-
ния 7800 кал/моль при 0—80°; смеси с воздухом от
3,05 до 17,0 об. % взрывчаты; токсичен, предельно
допустимая концентрация в воздухе 200 частей на
миллион (об.).
А. умеренно растворяется в воде, образует с ней
азеотропную смесь, т. кип. 70,5—70,7°, содержащую
87,5% А.; смешивается с большинством органич.
растворителей. При хранении и обращении с А. надо
учитывать его неустойчивость в присутствии пере-
кисей, воспламеняемость и токсичность. Для предот-
вращения полимеризации А. хранят при низкой тем-
пературе и применяют ингибиторы (гидрохинон, хи-
нон; нек-рые ароматич. нитросоединения).
А. в присутствии щелочных агентов (гидроокись
триметилбензиламмония, метилат Na или К, спир-
товой раствор КОН или 40%-ные водные растворы
NaOH и КОН) легко конденсируется с различными
органическими соединениями, обладающими по-
движным атомом водорода или активной метиленовой
группой: RH + СН2=СН— CN -> RCH2CH2—CN
(реакция цианэтилирования). А. присоединяет гало-
геноводороды, HCN, спирты и фенол, H2S и меркап-
таны; оксимы, NH3 и амины, бисульфиты щелочных
металлов не по правилу Марковникова, напр.:
НХ + СН2=СН—CN -> ХСН2—CH2CN. При электро-
химии. восстановлении А. гидродимеризуется, образуя
динитрил адипиновой к-ты. А. легко полимеризуется
в полиакрилонитрил. Получают также сополимеры
А. с бутадиеном (и его производными), винилхлори-
дом, метилметакрилатом, стиролом и др.
В пром-сти А. получают из синильнои к-ты и аце-
тилена в жидкой или паровой фазе в присутствии
катализатора: НС. .СН + HCN -> CH2=CHCN. Ре-
акция в жидкой фазе катализируется водным р-ром
полухлористой меди, содержащим NH4C1 и подкис-
ленным НС1. Основным производственным способом
получения А. является совместное окисление пропи-
лена и аммиака:
СН3=СН—CH34-NHS+O—»СН3=СН—CN4-H3O
А. находит широкое применение, особенно в произ-ве
высокомолекулярных соединений (напр., см. Бутади-
ен-нитрилъный каучук). А. применяется как промежу-
точный продукт при синтезе эфиров акриловой к-ты.
Лит.: Мономеры, Сб. ст., пер. с англ., под ред. В. В. Кор-
шака, Геб.] 1, М., 1951. Н. В. Гнучее.
АКРИХИН (атебрин) C23H30ON3C1 • 2НС1 • 2Н2О,
мол. в. 508,9 — ярко-желтые мелкие кристаллы горь-
СН3
NHCH(CH2)3N(C2H6)a
ОСН3
•2HCI - 2Н2О
кого вкуса; т. пл. 248—250° (с разл.). А. растворим
в воде (при 20° до 3%, при 100° до 60%); хорошо
растворим в спирте, почти нерастворим в эфире и
хлороформе. Р-ры А. флуоресцируют и окрашены.
При прибавлении к водным р-рам А. щелочей выде-
ляется его основание (т. пл. 86—88°). При кипячении
с водой А. гидролизуется, образуя 2-метокси-6-хлор-
акридон. Из водных р-ров А. после прибавления KJ
выделяется осадок иодгидрата основания А. Количе-
ственно А. определяют титрованием водно-эфирного
р-ра навески А. р-ром NaOH (с фенолфталеином). А.
получают конденсацией 2,4-дихлорбензойной к-ты
с п-анизидином в присутствии порошка Си и К2СО3,
циклизацией образующейся п-метоксифенилантрани-
ловой к-ты с РОС13 и реакцией полученного 2-мет-
окси-6,9-дихлоракридина с 1-диэтиламино-4-амино-
пентаном в среде фенола. А. является лечебным и
профилактич. противомалярийным препаратом и сред-
ством против лямблиоза. Его применяют также при
лечении красной волчанки и кожного лейшманиоза.
Лит.: Преображенский Н. А., Г е н к и н Э. И.,
Химия органических лекарственных веществ, М.—Л., 1953;
см. также при ст. Адалин. А. С. Елина.
АКРОЛЕИН (пропеналь, акрилальдегид), мол. в.
56,07, СН2=СНСНО — бесцветная, легко воспламе-
няющаяся слезоточивая жидкость с резким запахом;
в концентрации 70 мг/м3 воздуха опасен для жизни;
т. пл. —87,7°; т. кип. 52,5°/760 мм; d/° 0,8410;
1,4022; теплота испарения (при 52,5°, 760 мм)
6,77 ккал/моль; теплота сгорания (о = const) 389,8
ккал/моль; хорошо растворим в органич. жидкостях.
При 20° в 100 мл воды растворяется 40 г А. Максимумы
поглощения в ПК-области: 2750, 1710, 1410, 1160,
955 смг1. В присутствии оснований, хлоридов As, Sn,
91
АКТИВАЦИИ ЭНЕРГИЯ —АКТИВНОСТЬ
92
Sb, Bi и при хранении А. полимеризуется в твердую
стеклообразную массу — дисакрил; добавка гидро-
хинона стабилизирует его; легко окисляется на воз-
духе в акриловую кислоту СН2=СНСООН; с гало-
генами в воде образует а-галогенакролеин, с гало-
геноводородами — [J-галогенопропионовый альдегид;
с NaHSO3 — NaO3SCH2CH2CH(OH)SO3Na; в диеновый
синтез вступает как диенофил и как диен:
СНО
А. дает зеленое окрашивание с яичным белком в при-
сутствии KNO2 и НС1. Идентифицируется в виде
семикарбазона (т. пл. 171°) или 2,4-динитрофенил-
гпдразона (т. пл. 165°).
А. может быть получен нагреванием глицерина
с водоотнимающими средствами — KHSO4, Н3РО4,
Н3ВО3 по схеме: СН2ОНСНОНСН2ОН->СН2=СНО +
+ Н2О; промышленный способ получения — окисле-
ние пропилена над СиО или парофазная конденсация
СН3СНО с НСНО в присутствии Li3PO4:
сн3сно + сн2о — сн3 = снсно + н3о
Значительная часть продукции А. идет на произ-во
метионина, глутарового альдегида (I), 8-валеролак-
тона (II), метионина (III):
^СН8
сн
4- II
СНОС2Н5
—~СН2ОН(СН2)3СН2ОН
н*.
ОС2Н5 Н2°
СН0(СН2)3СН0
СН3=СНСНО 4- CHaSH _ CH3SCH2CH2CHO ——
CHsSCH3CH2CHOHCN ^5». CH3SCH2CH3CH(NH2)CN
CH3SCH2CH2CH(NH3)COOH
III
А. является важным промежуточным продуктом син-
теза глицерина. Продукты щелочной конденсации А. с
формальдегидом имеют эластифицирующие свойства.
Лит.: U Птапо, 3 Aufl., Bd 3, Munchen — B.f 1953,
S. 72. Ф. К. Величко.
АКТИВАЦИИ ЭНЕРГИЯ — см. Энергия активации.
АКТИВАЦИОННЫЙ АНАЛИЗ — см. Радио акти-
вационный анализ.
АКТИВИРОВАННЫЙ КОМПЛЕКС — см. Пере-
ходное состояние.
АКТИВНОСТИ КОЭФФИЦИЕНТ — отношение ак-
тивности данного компонента р-ра к его концентра-
ции; обозначается у. А. к. характеризует степень
отклонения свойств реальных р-ров от свойств иде-
альных р-ров, для к-рых он равен единице. Числен-
ное значение у зависит от способа выражения концент-
рации и от выбора стандартного состояния. В каче-
стве последнего обычно принимают состояние в бес-
конечно разбавленном растворе, иногда (в частности,
при изучении концентрированных растворов неэлект-
ролитов) состояние чистого вещества; для электроли-
тов, в связи с невозможностью дать оценку А. к. от-
дельных ионов, вводят средний А. к.
“±
7±~ vF)1/v
здесь м+ и — число катионов и анионов, образую-
щихся при диссоциации электролита (м = м+ + '< ),
a — средняя активность ионов, т — моляльность
раствора. Иногда термин «А. к.» применяют для харак-
Электро- лит т
0,001 0,005 0,01 0,05 0,1 0,5 1,0 2,0 3,0
НС1 0,966 0,929 0,905 0,830 0,796 0,757 0,809 1,01 1,32
H.,SO4 0,830 0,639 0,544 0,340 0,265 0,156 0,132 0,128 0,142
КВг 0,965 0,927 0,903 0,822 0,771 0,657 0,617 0,596 0,600
NaOH — — 0,90 0,818 0,776 0,693 0,679 0,698 0,774
ZnSO4 0,700 0,477 0,387 0,202 0,150 0,063 0,043 0,035 0,040
теристики степени отклонения свойств сжатого газа
от свойств идеального газа; в этом случае под А. к.
подразумевают отношение фугитивности (летуче-
сти) газа к его давлению; последнюю величину назы-
вают также коэффициентом фугитивности (коэффи-
циент летучести). С помощью А. к. можно устано-
вить отличие реального р-ра от идеального и оп-
ределить термодинамич. свойства первого.Для неиде-
альных р-ров А. к. может быть как меньше, так и
больше единицы. Методы вычисления А. к. те же,
что и активности. А. к. находит широкое применение:
при расчете равновесия в растворах, оценке влияния
посторонних солей на растворимость, при определе-
нии нормальных потенциалов полуэлементов, опре-
делении pH растворов и т. д. В настоящее время
имеется большое число экспериментальных данных
о А. к. различных веществ как в водных, так и в невод-
ных р-рах, ^частности для солей, кислот и ос-
нований. Значения А. к. нек-рых силь-
Не _ ных электролитов приведены в таблице (т —
концентрация в молях на 1 000 г при 25° С).
Лит. см. при ст. Активность.
М. X. Карапетьянц.
АКТИВНОСТЬ — величина, характеризующая
свойства вещества в р-ре. А. является функцией, под-
становка к-рой вместо концентрации в уравнения,
определяющие фазовые или химич. равновесия для
идеальных р-ров, делает эти уравнения применимыми
для данного р-ра. В идеальном р-ре связь между хи-
мич. потенциалом р компонента н его концентрацией
с выражается уравнением
p = Po(P, Т)-\-RT In с (1)
в к-ром ро, в отличие от р, зависит от единиц выраже-
ния концентрации. В реальных р-рах связь р =
— f(c) более сложна, и уравнение (1) неприменимо.
Для них Льюис (1907) предложил сохранить вид
зависимости (1), введя а вместо с, т. е. придав (1)
форму
H = lxo(-P. T)-\-RT\na (2)
Таким образом, введение А. является расчетным прие-
мом, основанным не на изменении функциональной
зависимости между параметрами, определяющими
состояние системы, а на изменении аргумента, при
сохранении вида зависимости, характерной для иде-
альной системы.
Несовпадение А. с концентрацией определяется
отличием данного р-ра от идеального: чем ближе по
свойствам первый ко второму, тем ближе А. к кон-
центрации; в идеальном р-ре обе величины становятся
тождественными. Степень несовпадения А. с концен-
трацией в количественной форме выражается величи-
ной коэффициента активности. Отклонения А. от
концентрации обусловлены взаимодействием между
частицами р-ра (молекулами, ионами), приводящим
в общем случае как к энергетич. изменениям системы,
так и к изменениям концентрации в результате об-
разования продуктов взаимодействия (напр., соль-
ватов). Эти факторы учитываются в А. суммарно.
Изменение А. в растворе определяется как энергией
93
АКТИВНОСТЬ ОПТИЧЕСКАЯ —АКТИВНЫЕ ЦЕНТРЫ
94
отдельных частиц, так и их числом; поэтому А. за-
висит не только от природы р-ра и его состава, но и
от темп-ры и давления, причем влияние давления
значительно лишь для газовых смесей. А. данного
компонента р-ра зависит от химич. природы и от кон-
центрации каждого из других компонентов.
А. компонента в рассматриваемом р-ре можно
вычислить из результатов экспериментальных иссле-
дований различных свойств р-рон — на основании
измерения давления пара р-ра, понижения темп-ры
замерзания, повышения темп-ры кипения, по распре-
делению вещества между двумя фазами, по величине
электродвижущей силы соответствующих элементов
и другими способами, в частности на основании дан-
ных о влиянии добавок посторонних электролитов
на растворимость труднорастворимых солей. Кроме
того, можно вычислить А. одного из компонентов р-ра
по А. другого компонента р-ра с помощью Гиббса —
Дюгема уравнения. Теоретич. расчет А., в общем слу-
чае затруднителен; лишь для разбавленных раство-
ров — в частности электролитов — расчет можно до-
вести до определения численного значения А. (см.
Ионная сила растворов).
Метод А заключается в том, что в термодинамич.
расчетах пользуются не аналитич. концентрациями
компонентов, а их А.; это дает возможность произво-
дить вычисления термодинамич. свойств компонентов
в реальных системах на основе зависимостей, относя-
щихся к идеальным системам, не исследуя физич.
причины отклонения от идеальности, т. е, не раскры-
вая природы сложных и разнообразных процессов
взаимодействия в р-рах. Метод фугитивности (лету-
чести) в применении к газам аналогичен методу А.
в применении к р-рам.
Лит.: Льюис и Рендалл, Химическая термоди-
намика, пер. с англ., Л., 1936; Шахпаронов М. И.,
Введение в молекулярную теорию растворов, М., 1956; И з-
м ай лов Н. А., Электрохимия растворов, Харьков, 1959;
Карапетьянц М. X., Химическая термодинамика,
2 изд., М.—Л., 1953. М. X. Карапетьянц.
АКТИВНОСТЬ ОПТИЧЕСКАЯ — отклонение (вра-
щение) плоскости поляризованного света при про-
хождении его через в-во, находящееся в жидком,
газообразном или твердом состоянии (кристалл)
или в растворе. А. о. является следствием асимметрия,
строения в-ва — иона, молекулы или кристалла и
связана с существованием антиподов (см. Антиподы
оптические, Асимметрическая молекула). Способно-
стью вращать плоскость поляризованного света об-
ладают многие природные и синтетич. соединения. К
их числу относятся аминокислоты, алкалоиды, са-
хара, антибиотики, витамины и др. соединения.
Для получения оптически активных в-в используют
расщепление на антиподы оптически недеятельных
соединений, а также оптически активные природные
или синтетич. в-ва, к-рые при помощи стереоспеци-
фич. реакций превращаются в новые оптически ак-
тивные в-ва. Определение вращения плоскости поля-
ризованного света производится при помощи поля-
риметра. Метод, с помощью к-рого исследуют влияние
физич. факторов (длины волны поляризованного света,
строения и агрегатного состояния в-ва или его кон-
центрации, темп-ры и др.) на величину вращения
плоскости поляризации, наз. поляриметрией.
С. В. Витт.
АКТИВНЫЕ ОСАДКИ — образования из радио-
активных атомов отдачи, осевших из газовой фазы на
поверхности твердых тел. Так, короткоживущий А. о,
радона состоит из членов ряда распада радона от
Ро218 до РЬ210; сам РЬ210 и его дочерние изотопы Bi210
и Ро210 образуют долгоживущий осадок радона. Если
материнское в-во находится в твердой фазе, то дочер-
ние атомы переходят в газовую фазу вследствие от-
дачи при радиоактивном распаде или ядерной ре-
акции. Так как атомы отдачи обычно многократно
ионизованы, т. е. имеют положительный заряд, то
лучше всего собирать их на отрицательно заряженных
(300—3000 в) электродах. Состав А. о., а следовательно,
его излучение и вид кривой распада зависят от изо-
топного состава материнского в-ва и от времени на-
копления А. о.; при прочих равных условиях коли-
чество, а значит и активность А. о. пропорциональны
количеству материнского в-ва. На этом основан ме-
тод количеств, определения, идентификации и изу-
чения радиоактивных в-в путем исследования их
А. о. (метод активных осадков). Метод А. о. исполь-
зуется при изучении зараженности атмосферы радио-
активностью, при исследовании эманац. способно-
сти различных в-в, изучении свойств короткоживущих
изотопов, продукты распада к-рых радиоактивны.
А. о. используются и для получения радиоактивных
изотопов; элементы, входящие в состав А. о., могут
быть разделены обычными радиохимия, методами.
Метод А. о. использован при выделении и исследова-
нии свойств изотопов элементов 101 и 102,- Напр.,
идентификация изотопа элемента № 102 проводилась
непрерывным улавливанием изотопа фермия, воз-
никающего при а-распаде 102-го элемента. Вначале,
при бомбардировке кюриевой мишени ионами угле-
рода, образующиеся атомы элемента № 102 вылетали
из мишени за счет отдачи, тормозились в атмосфере
гелия и притягивались отрицательно заряженной
(400 в) металлич. конвейерной лентой. При переме-
щении на конвейерной ленте атомы этого изотопа
претерпевали а-распад. Возникающие при этом до-
черние атомы Fm получали энергию отдачи, и часть
их притягивалась расположенным над лентой при-
емником — фольгой, заряженным отрицательно (600 в)
относительно ленты. Сравнивая активности разных
участков фольги и учитывая время перемещения ленты
на расстояние от начала до конца фольги, удалось
установить период полураспада этого изотопа 102-го
элемента.
Лит.: Кюри М., Радиоактивность, пер. с франц.,
М.—Л., 1947; Мурин А. Н., Введение в радиоактивность,
Л., 1955; Radiochemical studies; the fission products, ed. by
Ch, D. Coryell and N. Sugarman, book 2, N. Y. [a. o.], 1951;
Гольданский В. И., Усп. физ. наук, 1959, 67, 185.
В. И. Барановский.
АКТИВНЫЕ ЦЕНТРЫ — 1) В химической
кинетике — промежуточные продукты реакций,
обладающие особенно высокой реакционной способ-
ностью. А ц. цепных реакций являются химически
ненасыщенные осколки молекул — свободные атомы
и радикалы. Напр., в цепной неразветвленной ре-
акции водорода с хлором Н2 + Cl2 -> 2НС1 участвуют
два А. ц. — атомы водорода Н и атомы хлора С1:
1а) Н4-С12-> НС1 + СВ 16) С14-Н2->НС1-4-Н.
В цепной разветвленной реакции окисления водорода
молекулярным кислородом принимают участие три
А. ц.: атомы Н и О и радикал ОН, к-рые ведут
след, цепь превращений: 2а) ОН + Н2—>- Н2О + Н;
26) Н + О2-> ОН + О; 2в) О + Н2-> OH +Н.
Высокая активность атомов и радикалов связана а
наличием у них неспаренного электрона, благодаря
чему их взаимодействие с насыщ. молекулами требует
малой энергии активации, обычно не превышающей
10 ккал/моль. В ходе реакции могут возникнуть и
стабильные промежуточные частицы, к-рые также
образуются и расходуются в процессе; обычно это
насыщенные молекулы — А. ц. они не являются.
Появление в реакционной системе А. ц. (зарожде-
ние цепи) может происходить в результате термин,
распада молекулы, диссоциации молекулы при столк-
новении с квантом света, с частицами проникающих
излучений, а также в результате соударений двух на-
сыщенных молекул и др. При соударении атома ила
95
АКТИВНЫЙ ВОДОРОД - АКТИВНЫЙ УГОЛЬ
96
радикала с насыщ. молекулой образуется новая мо-
лекула и новый атом или радикал, т. е. один А. ц.
рождает другой (реакции продолжения цепи, напр.
1а, 16, 2а). Если в результате взаимодействия одного
А. ц. с молекулой появляется несколько новых А. ц.,
то происходит разветвление цепи (напр., 26). Исчез-
новение А. ц., т. е. обрыв цепи, происходит при столк-
новении двух атомов или радикалов, при столкнове-
нии А. ц. с молекулой ингибитора или стенкой ре-
акционного сосуда и прилипании к ней.
2) В гетерогенном катализе — участки
поверхности катализатора, на к-рых протекает ре-
акция, составляющие небольшую долю поверхности.
Ускоряющее действие катализатора связано с тем,
что реагирующие в-ва легко образуют на А. ц. по-
верхностные промежуточные соединения, что при-
водит к снижению энергии активации реакции. В
отдельных случаях на А. ц. твердой поверхности
происходит распад насыщенной молекулы на атомы
или радикалы, к-рые ведут цепь превращений в объ-
еме (гетерогенное зарождение цепей). По вопросу
о природе А. ц. катализатора нет единой точки зре-
ния (см. Катализ и Катализ гетерогенный}.
Лит.: Семенов Н. Н., О некоторых проблемах хими-
ческой кинетики и реакционной способности, 2 изд., М., 1958;
Кондратьев .В. Н., Кинетика химических газовых
реакций, М., 1958; Р Огинский С. 3., Адсорбция и ка-
тализ на неоднородных поверхностях, М.—Л., 1948.
А. Б. Гагарина.
АКТИВНЫЙ ВОДОРОД (определение) — способ
открытия и колич. определения содержания легко-
подвижного («активного») водорода в спиртах, фено-
лах, тиолах, кислотах, аминах и т. п. (с целью иден-
тификации и при исследовании их строения). Прин-
цип определения, впервые предложенный в 1902
Л. А. Чугаевым, состоит в измерении объема метана,
выделяющегося при взаимодействии исследуемого
в-ва с магнийиодметилом в среде абсолютного диизо-
амилового эфира или анизола: RHn + ziCH3MgJ —►
—> R(MgJ)n+ ziCH4; практич. метод анализа был раз-
работан Ф. В. Церевитиновым. Кроме реактива Гринь-
яра, применяется более энергично и иногда более
полно реагирующий литийалюминийгидрид: 4RH +
+ ЫА1Н4—► (LiAl)R4 + 4Н2. Точность определения
обоих методов ±3—5%. В 1947—49 А. П. Терентьевым
с сотр. был разработан новый вариант метода Чугаева—
Церевитинова, особенностью к-рого является приме-
нение в качестве реакционной среды абсолютирован-
ного диэтилового эфира; парами эфира затем произ-
водится вытеснение метана из реакционного сосуда 1,
причем эфир сполна поглощается спиртоводной смесью
л — реакционный сосуд; г — азотометр; з— эвдио-
метр: 4 — уравнительный сосуд.
труднений, связанных со спец, защитой от влаги и
кислорода, и отказаться от «холостых» опытов. Ана-
лиз можно проводить с 40—80 мг вещества, а при
использовании микроэвдиометра и с 2—5 мг.
Экспериментальные определения А. в. не всегда
дают правильные результаты. Пониженные значения
получаются для веществ, склонных к таутомерии (ено-
лов, амидов и др.). Повышенные, напр., для третичных
спиртов, к-рые реактивом Гриньяра частично дегид-
ратируются. А. в. обнаруживается и в нек-рых сое-
динениях, способных к таутомерному превращению,
напр. в ациформу, а именно у первичных и вторичных
нитро- и нитрозосоединений и др. Менее общими яв-
ляются след, методы: ацилирование, титрование окра-
шенным эфирным р-ром трифенилметилнатрия, к-рый
обесцвечивается ионами Н; для спиртов и аминов
используют реакцию с ди оксансульфотриоксидом
[d(CH2 — ch2)6-so3] с последующим алкалиметрии,
определением алкилсерной или замещенных сульф-
аминовой к-ты. Способ дает положительные резуль-
таты также и с третичными спиртами.
Лит.: Гаттерман Л. и Виланд Г., Практические
работы по органич. химии, М., 1948, с. 94—98; В а й б е л ь С.,
Идентификация органических соединений, пер. с англ., М.,
1957, с. 22—24, 317—28. Р. Р. Галле.
АКТИВНЫЙ УГОЛЬ (активированный уголь) —
пористый адсорбент, скелет к-рого построен из рых-
лых и неправильно упакованных пачек, состоящих
из сеток 6-членных углеродных колец, менее упоря-
доченных, чем в графите, и ковалентно связанных с
углеродными радикалами, с водородом, а часто и с
кислородом.
А. у. сильно адсорбируют (физически) органич. ве-
щества — углеводороды и многие их производные,
слабее — низшие спирты, аммиак и особенно плохо —
воду. Обычно А. у. обладают неоднородной поверх-
ностью и пористостью. Различают микропоры раз-
мерами 10—20 А с сильно развитой поверхностью
(до 1 000 м2/г}, поры переходных размеров (ок. 50—
500 А; с поверхностью до 100 -и2/г и макропоры с
размерами более 1 000 А и малой поверхностью (ок.
1 ж2/г). Сильная адсорбция происходит в микропорах.
В порах переходных размеров происходит полимоле-
кулярная адсорбция и капиллярная конденсация па-
ров. Макропоры служат в основном транспортными
каналами, подводящими молекулы газа и растворен-
ных веществ к внутр, частям зерен А. у. В крупных
порах (макропорах и порах переходных размеров)
в основном отлагают-
ся и катализаторы,
вносимые в А. у. для
придания ему ката-
литич. свойств, напр.
в реакциях низкотем-
пературного окисле-
ния. Иногда эти виды
пор постепенно пере-
ходят друг в друга,
в большинстве же
случаев кривая рас-
пределения объема
пор по размерам для
А. у. имеет 2—3 чет-
ких максимума.
Различают два ти-
па А. у. (по форме изо-
терм адсорбции орга-
нич. паров): тонкопо-
ристые с сильно повы-
шенным адсорбпион- Изотермы адсорбции паров бензола
шеииым адьирициип на активных углях: 1 — ТОНКОПО-
ным потенциалом В ристом и 2 — широкопористом,
порах — изотермы ад-
сорбции круто поднимаются, адсорбция достигает
предела (заполнения микропор) при малых давлениях
пара (рис., кривая 7). и широкопористые (с по-
рами, размеры которых приближаются к переход-
ным), для к-рых изотерма адсорбции вначале близка
к таковой для непористых или мало пористых видов
97
АКТИН - АКТИНИДЫ
98
угля — саж, а затем имеем гистерезис сорбционный
благодаря наложению на адсорбцию вторичного яв-
ления — капиллярной конденсации в широких порах
(рис., кривая 2). Эти изотермы в начальной адсорб-
ционной части хорошо описываются ур-ниями потен-
циальной теории адсорбции М. М. Дубинина и Л. В.
Радушкевича (подробнее см. в ст. Адсорбция). Ад-
сорбция воды, аммиака, низших спиртов и др. по-
лярных низкомолекулярных в-в при небольших дав-
лениях паров невелика; она сильно увеличивается
при окислении поверхности угля.
А. у. получают путем удаления из угля-сырца смо-
листых веществ и развития разветвленной сети пор.
Это достигается: 1) Активированием карбонизирован-
ных гранул, полученных на основе ископаемых или
древесных углей, действием газов-окислителей (пе-
регретые пары Н20, С02) при высоких темп-рах;
при этом возникают тем более крупные поры, чем
больше обгар угля. 2) Пропиткой различных органич.
материалов, напр. древесной массы, нек-рыми акти-
вирующими солями (ZnCl2, K2S), с последующим
термич. разложением (обугливанием без доступа
воздуха) и промывкой угля водой. Весьма тонкопо-
ристые А. у. могут быть получены также термич.
разложением нек-рых полимеров, напр. поливини-
лиденхлорида (сарановые угли).
А. у. применяют в сорбционной технике для улав-
ливания и возвращения в произ-во паров ценных ор-
ганич. растворителей, для разделения газовых сме-
сей, в противогазовой технике, как адсорбенты и как
основа для каталитич. и хемосорбционно-активных
добавок, в газовой хроматографии для разделения га-
зов и низших углеводородов, для очистки р-ров в воде
и других сильно полярных жидкостях от примесей
органич. веществ (широкопористые обесцвечивающие
угли), в медицине для поглощения газов и различных
вредных веществ при желудочно-кишечных заболе-
ваниях.
Лит.: Дубинин М. М., Физико-химические основы
сорбционной техники, 2 изд., М.—Л., 1935; Методы исследова-
ния структуры высокодисперсных и пористых тел, [кн. 1], М.,
1953, [кн. 2], М., 1958; The structure and properties of porous
materials, L., 1958; Дубинин M. M., Усп. хим., 1955, 24,
вып. 1 3; Николаев К. М. иДубииин М. М„ Изв.
АН СССР. Отд. хим. н., 1958, № 10, 1165—74.
А. В. Киселев.
АКТИН — белок, входящий в состав сократитель-
ных элементов мышечного волокна; извлекается во-
дой из обезжиренной и обезвоженной ацетоном мышеч-
ной ткани. Молекулы А. существуют в двух формах:
деполимеризованной, или глобулярной (приближаю-
щейся к шарообразной), и полимеризованной, или
фибриллярной (нитевидной). Мол. в. глобулярного А.
35 • 103 — 10 • 103. Взаимный переход этих форм свя-
зан с воздействием определенных концентрации р-ров
солей (до 0,1 М в случае одновалентных ионов, до
0,005 М — в случае двухвалентных) или изменением
pH; при этом обязательно также присутствие катали-
тич. количеств Mg2+. Образование фибриллярного А.
сопровождается резким повышением вязкости р-ров А.
От связанной глобулярным А. аденозинтрифосфор-
ной к-ты при полимеризации отщепляется 1 молекула
фосфата и поэтому фибриллярный А. оказывается
связанным уже с аденозиндифосфорной к-той. А. сте-
хиометрически соединяется с другим белком мышеч-
ной ткани миозином, образуя актомиозин — глав-
ный сократительный белок мышц.
Лит.: В ага п у М. [а. о.], Acta physiol. Acad, scient.
hung., 1956, 10, fasc. 2—4, p. 145; Str oh man R. C.,
Biochim. et biophys. acta, 1959, 32, №2, p. 436; см. также лит.
к ст. Актомиозин. Е. Д. Грищенко.
АКТИНИДЫ (актиноиды) — семейство из 14 эле-
ментов VII периода периодич. системы Д. И. Менде-
леева, следующих за актинием, с порядковыми номе-
рами 90—103. А. характеризуются тем, что в их
атомах прерывается заполнение наружных — шестой
(группа 6rf) и седьмой (после появления группы 7s2)
электронных оболочек, и при переходе от каждого
предыдущего А. к последующему происходит (в ос-
новном, а начиная с элемента № 96 — исключительно)
наслоение /-электронов в пятой электронной обо-
лочке (подробнее см. ниже) Название «А.» дано по
аналогии с семейством лантанидов, у к-рых проис-
ходит заполнение /-слоя в четвертой электронной
оболочке. Число возможных /-электронов (14) и опре-
деляет число переходных элементов как в 6, так
и в 7 периодах системы Менделеева. Все А. радиоак-
тивны. Среди них широкое применение нашли пока
только уран и плутоний, используемые в виде изо-
топов U335, U233 и Ри239 как горючее в ядерных реак-
торах и взрывчатое в-во в атомных бомбах.
Уран, торий и протактиний встречаются в природе и были
открыты ранее других А. Остальные А. в природе не встре-
чаются (за исключением ничтожных количеств нептуния и
плутония) и были получены в 1940—58 искусств, путем с по-
мощью ядерных реакций. О получении элемента с п. н. 103
было объявлено в 1961 и для него предложено название Лоу-
ренсий (Lawrencium, Lw, в честь Э. Лоуренса). До синтеза
трансурановых элементов (Z > 92) торий, протактиний и
уран относили соответственно к IV, V и VI группам пе-
риодич. системы в качестве аналогов гафния, тантала и
вольфрама. Само представление об А. укоренилось лишь
благодаря исследованию свойств искусственных трансура-
новых элементов. Уже задолго до начала таких исследова-
ний большинство ученых склонялось к выводу, что в седьмом
периоде системы Менделеева, как и в шестом, должно содер-
жаться 32 элемента, т. е., что этот период должен включать
семейство из 14 элементов, в к-ром происходит наслоение 5
/-электронов. Однако существовали разногласия относительно
положения первого элемента семейства, хотя большинством
признавалось неоспоримым, что этот элемент должен быть уже
заурановым. Н. Бор (1923) высказал предположение, что на-
слоение 5 /-электронов должно начаться при Z = 94, а В. Гольд-
шмидт (1924) — после элемента с Z = 96, к-рый он считал го-
мологом платины. Были выполнены расчеты энергий связи
5/- и 6 d-электронов, причем И. Сугиура и Г. Юри (1926) при-
шли к выводу о начале заполнения 5 /-электронной подгруппы
при Z = 95, а Т. By и С. Гаудсмит (1933) получили значение
Z = 93. Наряду с этим, с 1926 высказывались соображения
о возможности конкурентного наслоения 6 d- или 5 /-электро-
нов уже у тория и протактиния. В 1937 Гольдшмидт указал,
что на основе кристаллографии, данных наиболее вероятным
началом 5 /-электронной группы элементов следует считать
протактиний, и предложил для этой группы название «ториды»,
указав, однако, что не исключена и возможность начала этой
группы с тория и, впервые применив, в связи с этим, назва-
ние «актиниды».
Первое экспериментальное доказательство заполнения
5 /-электронной подгруппы в области близких к урану тяжелых
элементов было получено Э. Макмилланом и Ф. Эйблсоном
(1940), показавшими, что синтезированный ими нептуний
совсем не схож с рением, но очень напоминает уран. Обосно-
ванное предположение о наличии семейства А. было впервые
выдвинуто Г. Сиборгом и А. Валем (1942). Это предположение
было основано на опытных данных о свойствах полученных
к тому времени двух заурановых элементов — нептуния и
плутония.
В зарубежной лит-ре для элементов 90—103 и 58—71 при-
няты соответственно наименования «актиниды» и «лантаниды»
(суффикс «ид» придает значение «следующий за»). В советской
лит-ре были распространены предложенные С. А. Шукаревым
назв. «актиноиды» и «лантаноиды», отражающие аналогию
рассматриваемых элементов с актинием и лантаном (суффикс
«оид» имеет смысл «подобный»); в настоящее время термины
«актиниды» и «лантаниды» более употребительны. Основной
вклад в развитие и проверку гипотезы об актинидном харак-
тере переходных элементов VII периода был внесен Сибор-
гом и его сотрудниками в результате синтеза и исследования
химич. свойств ряда новых заурановых элементов. Прежде
чем перейти к рассмотрению свойств каждого из этих элемен-
тов в отдельности, остановимся на основных доводах в пользу
существования семейства А.
Общие свойства актинидов. Химические
свойства. Если бы U и заурановые элементы
были аналогами не редкоземельных элементов, a W,
Re, Os и т. д., то по мере перехода к более тяжелым
элементам устойчивость высших валентностей воз-
растала бы. На самом же деле, наоборот, от U к Ат
возрастают трудности перехода от низших к высшим
валентностям. Колич. характеристикой таких пере-
ходов могут служить измеренные в стандартных усло-
виях окислительно-восстановительные потенциалы (в
99
АКТИНИДЫ
100
вольтах) пар 3- и 4-валентных, 4- и 6-валентных со-
единений элементов 92—97, приведенные ниже.
Вк
-1,6
и
+ 0,63
- 0,32
Np
— 0,155
— 0,94
Ри
— 0,98
— 1,04
Am Cm
- 2,44 < - 2
— 1,34
Ш-IV
IV-VI
Положительный потенциал отвечает переходу в более
устойчивое состояние, отрицательный — в менее
устойчивое. Устойчивость 3-валентного состояния бы-
стро возрастает от U к Ат. Для Ст вообще неизвестно
никаких других состояний, кроме 3-валентного. Этот
факт особенно важен для определения структуры
VII периода системы Менделеева. Изучение ланта-
нидов и расчеты показывают, что особой прочностью
при заполнении /-мест в 4-й оболочке обладают струк-
туры с 14 /-электронами (целиком заполненная
/-группа) и с 7 /-электронами (наполовину запол-
ненная /-группа). Поэтому лантаниды—лютеций
(4/145с?6у2) и гадолиний (4/75c?6s2), обладают особо
устойчивой 3-валентностью. Высокая устойчивость
3-валентного состояния кюрия свидетельствует о том,
что в пятой электронной оболочке этого элемента
содержатся семь 5 /-электронов (5/’6<Z7sa), т. е. Ст яв-
ляется седьмым элементом в ряду аналогов лантани-
дов. Этим определяется и первый элемент среди таких
аналогов — Th, стоящий за Ас. Так, свойства кюрия
будучи сопоставлены со свойствами гадолиния, по-
могают установить, что аналогом лантанидов явля-
ются именно А., а не «ториды», «ураниды» и т. п.
3-валентный Th, 3- и 4-валентный Ра неустойчивы
в водных р-рах. Это как будто бы резко отличает
их от лантанидов. Однако существуют твердые со-
единения 3-валентного тория ThJ3, Th2S3, приготовля-
емые в восстановительных средах, а также твердые
смешанные окислы Ра, где этот элемент — второй
из А. — выступает как 3- и 4-валентный, подобно
второму лантаниду — церию. Ниже перечислены важ-
нейшие твердые соединения U, Np, Pu и Am, выде-
ленные до настоящего времени.
Окислы: UO, U2O3, UO2, U3Og, UOg; NpO, Np2O3,
NpO2 Np3O8; PuO, Pu2O3, PuO2; AmO, Am203, AmO».
Фториды: UF3, UF4, UF5, UFe; NpF3, NpF4, NpF5,
NpFe; PuF3, PuF4, PuFe; AmF3.
Хлориды: UCI3, UCI4, UCI5. UC13; NpCl3, NpCl4; PuCls;
AmCI3.
Бромиды: UBr3, UBr4; NpBrg, NpBr4; PuBr3; AmBr3.
Схема основных окислительно-восстановительных
реакций для 4 перечисленных элементов может быть
представлена след, образом:
ттш. воздух.
Zn + HCi
менее
jjIV Ja. Fe3+, MnO4 yV CeIV, Сг2О? pj VI
Np111
На
воздух
Zn + HCI
Hg-катоде
риШ
воздух,
на ртутном катоде
IV HJJOH-NaNOj N V V1
Fes+, SO2 P SnCIa P
BrOJ. CeIv. AgO
Br2, MnOp
Am11
Адсорбционные свойства и рас-
пределение между растворителями.
Химич, свойствами А. определяется их поведение при
ионообменной адсорбции. Картины хроматография,
разделения лантанидов и А. совершенно аналогичны,
что иллюстрируется рис. 1, где приведены данные о
: 102 ’7"
7О,ДМ<)1 Es
Е
О
б
CI Bk
A A
Ст Ат
10 20 30 40 50 60 70 80 90
Число капель вымывающего раствора
Рис. 1.
А. и лантанидов на смоле Дауэкс-50
__________________________p„IV HN°3 (горячая) _ V MnOp, Вг» (горячий) D VI
гидразин. SO», Zn + HCI___ru BrO7 Cr»O|“ rU
вымывании
при 87° р-ром лимоннокислого аммония (при pH 3,5
для А. и 3,28 для лантанидов). Пунктиром обозна-
чены ожидаемые положения элементов 102 и 103
при вымывании в подобных условиях. Близкая ана-
логия между поведением А. и лантанидов наблю-
дается и в картине распределения соединений этих эле-
ментов между парами разных органич. и неорганич.
растворителей.
Спектры поглощения растворов.
Характерным свойством как А., так и лантанидов,
обусловленным наличием /-электронов в их атомах,
является присутствие резких полос в спектрах по-
глощения видимого света. Эти
полосы появляются из-за того,
что под действием света опре-
деленных длин волн
происходят переходы
электронов внутри
4/-слоя с одного места
на другое; при этом
меняется энергия вза-
имодействия /-электронов. Энергии таких переходов
невелики, и они могут происходить под действием ви-
димого света. По мере заполнения /-слоя электронами
энергия перехода /-электронов с одного места на дру-
гое возрастает, и полосы смещаются в сторону ультра-
фиолетовой части спектра (т. е. в сторону меньших
длин волн). На опыте было показано сходство спектров
поглощения водных р-ров соединений 3-валентных А.
и лантанидов. Характерные спектры поглощения
C1-, Br-
—- дтШ °* плавлении^ ™ 0СГ, на аноде
Na-амальгама любые восстав. ’ *лт —--------------— Ат
I____________ ОС1-, s2oj~_________
ПГо2 ’
Ниже перечислены валентности аналогов семейств ланта-
нидов и А. Наиболее устойчивые валентности выделены жир-
ным шрифтом, наименее устойчивые помещены в скобки.
Л а н т аниды: La Се Рг Nd Pm
3 3,4 «E4J 3 3
А к т и н и д ы: Ас Th Ра 0 i Np
3 3,4 [3]4,5 з, 4, 5, 6 3, 4, 5,
Л а н т аниды: Sm Ей Gd Tb Dy
2, 3 2.3 з : 3, 4 3
Анти виды: Ри Am Cm Bk Cf
3, 5, 6 (2), 3, 4, 5 , 6 3 3, 4 3
6
101
АКТИНИДЫ
102
с резкими полосами в видимой области отмечены также
для водных р-ров 4- и 5-валентного Np и высших
валентностей Ри. Лишь для 6-валентного Np полосы в
спектре поглощения отсутствуют, что опять подтверж-
дает положение Np как четвертого А. В самом деле,
структура наружных оболочек Np (если заполнение
5/-мест начинается с Th) должна иметь вид 5/46й7№,
а в соединениях 6-валентного Np из семи перечислен-
ных электронов наружных оболочек шесть связано, и
остается лишь один /-электрон. Поэтому, естественно,
пропадают полосы, связанные с взаимодействием по
меньшей мере двух /-электронов.
Кристаллографические данные.
В пользу родства А. — элементов, следующих за ак-
тинием, — говорит изоморфизм кристаллич. структур
окислов ThO2, РаО2, UO2, NpO2, PuO2 и АшО2 и
регулярное уменьшение радиуса металлич. иона в
кристаллич. решетке этих окислов. Изоморфными
оказались также соединения типа XF4 (где X — U,
Th, Np, Pu), XF3, XC13 и XO (где X — U, Np, Pu,
Am). Вычисленные из кристаллографии, данных ион-
ные радиусы, как видно из табл. 1, меняются весьма
схожим образом в рядах лантанидов и А.
таблица 1. Ионные радиусы лантанидов и актинидов
Ланта- ниды Ионные ра- диусы, А (3-валентное состояние) Акти- ниды Ионные радиусы, А
3-валентное состояние 4-валентное состояние
La 1,06 Ас 1.11
Се 1,03 Th 1,08 0,99
Рг 1,01 Ра 1,05 0,96
Nd 0,99 С 1.03 0,93
Pm 0,98 Np 1,01 0,92
Sm 0,96 Pu 1,00 0,90
Eu 0,95 Am 0,99 0,89
Парамагнитная восприимчивость.
При образовании 3-валентных положительных ионов
лантанидов отрываются 3 внешних электрона, во
всех же внутр, электронных оболочках, кроме 4-й,
имеется четное число электронов, суммарный спин
к-рых равен нулю. Поэтому магнитные свойства всего
иона в целом определяются только строением /-слоя
четвертой электронной оболочки — числом /-электро-
нов и величиной их суммарного спина. На рис. 2
приведены данные о парамагнитной восприимчивости
для 3-валентных ионов лантанидов от церия (один
/-электрон) до гадолиния (семь /-электронов) и для
ряда ионов А. Соответствие данных указывает на то,
что число /-электронов в рассмотренных ионах А.
также меняется от одного до шести и это подтверждает
гипотезу о существовании семейства А. Подробное ис-
следование парамагнитных свойств ионов разной ва-
лентности элементов от Th до Ат показало, что в Th
и Ра энергетически более выгодно заполнение уровня
6<Z; начиная же с U, преобладает заполнение 5/-
уровня. Таким образом, нашло свое объяснение за-
метное отличие свойств первых А. (преобладание выс-
ших валентностей) от свойств первых лантанидов.
Таблица 2. Электронные структуры наружных электронных
оболочек атомов лантанидов и актинидов
Элемент Структура оболочки Элемент Структура оболочки
57 La 5d6s2 89 Ac 6d7s3
58 Се 4/26s2 90 Th 5/6d7s3 или 6d27s2
59 Рг 4/36s2 91 Pa от 5/26d7s2 до 6d37s2
60 Nd 4/46s2 92 U от 5/36d7s2 до 6d<7s2
61 Pm 4/56S3 93 Np от 5/‘6d7s2 до Rd-’7s2
62 Sm 4/66s2 94 Pu 5/67g2 или 5f56d7s2
63 Ku 4/76s2 95 Am 5/?7ss или 5/66d7s2
64 Gd 4/75d6s2 96 Cm 5/76d7s2
65 Tb 4/85d6s2 97 Bk 5/67sS (5/86d7s2)
66 Dy 4/l«6s3 98 Cl 5/107s2
67 Ho 4/n6s2 99 Es 5/>l7s2
68 Er 4/i26s2 100 Fm 5/127s2
69 Tu 4/136s2 101 Md 5/137s2
70 Vb 4/U6s3 102 5/И78 2
71 Lu 4/145d6s2 103 5/i46d7s2
На основании всего сказанного, а также спектро-
скопии U, Th, Am можно сделать вывод о строении
электронных оболочек атомов А., сопоставив это
строение с данными
для лантанидов. Со-
вокупность данных
об электронных кон-
фигурациях находя-
щихся в газе атомов
А. и лантанидов при-
водится в табл. 2.
Очевидно, что наряду
с общим сходством в ха-
рактере построения VI
и VII периодов, имеется
и весьма существенное
различие между А. и лан-
танидами — в то время, Порядковый номер
как уровни 4/-электро-
нов для всех лантанидов Рис. 3.
лежат заметно ниже, чем
уровни 5d-электронов, для А. соответств. уровни 5/ и 6d за-
метно сближаются, а для первых А. уровни Gd оказываются
даже расположены ниже уровней 5/ (рис. 3). Вследствие этого
у первых А. преобладает переменная валентность и лишь,
Порядковый номер элемента
Рис. 4.
103
АКТИНИДЫ
104
начиная с Ат, наиболее устойчивым состоянием оказывается
3-валентное. Несомненное различие химич. свойств первых А.
и первых лантанидов, признаваемое, конечно, и сторонниками
актинидной природы переходного семейства VII периода,
лежит в основе предложений др. вариантов описания положе-
ния тяжелых элементов в периодич. системе Менделеева. Так,
франц, химик М. Гайсинский считает, что переходное семейство
начинается с урана и состоит из двух подклассов — уранидов
(Г, Np, Pu, Ат) и кюридов, начинающихся с кюрия. Американ-
ский кристаллохимии В. Захарайсен склоняется к выводу, что
наслоение 5/-электронов начинается с Ра, т. е. поддерживает
«торидную» гипотезу. В качестве доводов противниками пред-
ставлений о группе А. выдвигается, в частности, заметное раз-
личие зависимости атомных плотностей от порядкового номера
редкоземельных и тяжелых элементов (рис. 4). Поскольку из
самых общих соображений ясно, что число элементов, в к-рых
происходит наслоение 5 /-электронов, ограничено 14, решаю-
щий вывод о структуре VII периода системы Менделеева может
быть сделан после синтеза и изучения химич. свойств элемента
104. Если семейство переходных элементов действительно
начинается с тория, то оно завершается элементом № 103,
а элемент № 104 должен быть уже аналогом титана, циркония,
гафния. Тогда удобным носителем для выделения этого элемента
должен быть цирконий, и в водных р-рах элемент 104 будет
только 4-валентным. Сопоставление химич. свойств этого эле-
мента и предшествующего — 103 — и послужит «решающим
опытом», окончательно доказывающим или опровергающим
существование семейства А.
Свойства отдельных актинидов. Сведения о при-
родных А. — тории, протактинии, уране, а также об
имеющем важное практич. значение плутонии см. в
отдельных статьях под соответств. названиями.
Нептуний (Neptunium) Np, п. н. 93, первый
из искусственных заурановых элементов. Предполо-
жение об образовании элемента 93 при воздействии
на уран медленных нейтронов было высказано еще
в 1935 Э. Ферми. Но лишь в 1940 Э. Макмиллан и
Ф. Эйблсон доказали существование нептуния, уста-
новив, что р -радиоактивный изотоп с периодом полу-
распада 2,3 дня, образующийся при облучении урана
нейтронами [U238 (п, у) U239£ZNp239], не принадлежит
ни к числу осколков деления, ни к ближайшим пред-
шественникам урана и, т. обр., есть изотоп нового
элемента, названного по имени след, за ураном пла-
неты, — Нептуна. В 1944 Np был впервые выделен
в чистом виде в микрограммовых количествах. Из-
вестны изотопы Np с м. ч. 231—241; наиболее долго
живущим является a-активный изотоп Np23’ (Т\/2 =
= 2,2-10е лет), рассматриваемый как родоначальник
четвертого, искусственного радиоактивного ряда изо-
топов см. ч. А = 4n + 1. Этот изотоп в ничтожных
количествах обнаружен в природе, где он образуется
по реакции U238 (п, 2п) U237 Np23’ при действии
на уран нейтронов деления или нейтронов, испуска-
емых легкими ядрами урановых руд под действием
а-частиц. Другим изотопом Np, доступным в весовых
количествах, является a-активный Np236 (Tt/ >>
5000 лет), получаемый на циклотронах в реакции
U238 (d, 4n) Np236. Элементарный Np — серебристый
металл, т. пл. 640°, существует в виде трех модифика-
ций а, р и устойчивых соответственно: ниже 278°
(а; плотн. d 20,45); при 278—570° d 19,36) и выше
570° (y; d 18,00). Металлич. нептуний получают вос-
становлением трифторида парами бария при 1300°.
Подобно урану, он энергично поглощает водород уже
при 50°, причем образуется черный рыхлый гидрид
смешанного состава — между NpH3 и NpH4, ближе
к последнему. Np обладает переменной валентностью
от +3 до +6, причем наиболее устойчивы 4- и 6-ва-
лентное его состояние; 4-валентное состояние Np
устойчивее, чем у U, а 5-валентное — устойчивее,
чем у Ри. Этим пользуются для отделения образую-
щегося в ядерных реакторах Np от U и Pu. Отличитель-
ной особенностью Np по сравнению с соседями яв-
ляется также устойчивость его 5-валентного состояния
в водных р-рах. Водные р-ры соединений Np окрашены:
Np111 — в розовый цвет; NpIV—желтовато-зеленый;
Npv — голубой; NpVI — чаще в зеленый цвет.
Америций (Americium) Am, п. н. 95, синте-
зирован в конце 1944 и начале 1945 Г. Сиборгом,
Р. Джеймсом, Л. Морганом и А. Гиорсо в виде изотопа
Ат241 путем облучения нейтронами плутония: Ри239
(n, y) Ри240 (n, y) Ри 241-Ат241. Назван по аналогии
с соответств. членом семейства лантанидов — ев-
ропием. Известны изотопы Ат с м. ч. 237—246 и изо-
мер изотопа Ат242. Наиболее продолжителен период
полураспада a-активного изотопа Am243 (Tlt = 7600
лет). Другие доступные в весовых количествах изо-
топы америция: Ат241 (а-распад; Тг/ — 458 лет),
Ат242 (Р“-распад; = 100 лет). Путь получения
изотопов Ат241—243 — длительное облучение урана
или плутония тепловыми нейтронами в ядерных реак-
торах, сопровождающееся цепочками актов радиац.
захвата нейтронов и Р -расиада. Изотоп Ат242 харак-
теризуется наивысшим среди всех А. сечением деле-
ния под действием тепловых нейтронов (6400 барн).
Элементарный Ат — серебристый мягкий металл,
d 11,9, темп-pa плавления его неопределенна, по-
скольку размягчение металла происходит уже при 850°,
а само плавление неполно даже при 1200°. Америций
обладает переменной валентностью от +3 до -|-6,
причем наиболее устойчиво 3-валентное состояние.
Подобно другим А., Ат энергично реагирует с газо-
образным водородом с образованием гидрида АтН3.
Основные данные о химич. соединениях Ат см. выше,
среди сводных данных.
Кюрий (Curium) Ст, п. н. 96, впервые получен
в 1944 Г. Сиборгом, Р. Джеймсом и А. Гиорсо в виде
изотопа Ст242 при бомбардировке плутония а-ча-
стицами: Pu239 (a, n) Ст242. Назван в честь П. и М.
Кюри. Известны изотопы Ст с м. ч. 238—250. Наибо-
лее устойчив a-активный изотоп Cm24’ (Ti, > 4 • 107
лет). Доступны в весовых количествах и мн. др. изо-
топы Ст — Cm242—246, 24s—25o Все изотопы могут быть
получены длительным облучением урана или плуто-
ния тепловыми нейтронами в ядерных реакторах.
Элементарный Ст — серебристый металл, d > 7. По-
лучается в металлич. виде, как и другие трансурано-
вые элементы, восстановлением трифторида парами
бария. Характерным химич. свойством Ст (подоб-
ным Gd) является особая устойчивость его 3-валент-
ного состояния; 4-валентное состояние Ст вообще
не было обнаружено в р-рах, хотя и доказано сущест-
вование двуокиси СтО2 и тетрафторида CmF4. Осо-
бая устойчивость 3-валентного состояния Ст и отсут-
ствие высших степеней окисления (с валентностями
5 и 6) используется для отделения Ст от Ри и Ат.
Беркелий (Berkelium) Bk, п. н. 97, синтезирован
в 1949 С. Томпсоном, А. Гиорсо и Г. Сиборгом при
облучении америция а-частицами: Am241 (a, 2n)Bk243.
Назван по месту открытия — городу Беркли (США).
Известны изотопы Bk с м. ч. 243—250 и второй изомер
изотопа Bk250. Наиболее устойчивый потому доступны
в весовых количествах Bk247 (а-распад; — 7000
лет) и Bk249 (Р^-распад; = 290 суток). Первый
получается при облучении а-частицами кюрия: Ст244
(а, р) Bk247; второй — под действием тепловых ней-
тронов реактора на U или Ри. На основании данных
о ионообменном вымывании соединений Bk можно
заключить, что этот элемент, как и соответств. ланта-
нид — тербий (№ 65), — может находиться в 3-
и 4-валентном состояниях.
Калифорний (Californium) Cf, п. н. 98,
получен впервые в 1950 С. Томпсоном, А. Гиорсо,
К. Стритом и Г. Сиборгом в ядерной рекции: Ст 242
Sa, 2n)Cf244. Назван по месту открытия (штат Кали-
юрния). Известны изотопы Cf с м. ч. 244—254. Четыре
изотопа относительно устойчивы и доступны в весовых
количествах: Cf249 (a-распад; = 400 лет), Cf260
105
АКТИНИДЫ
106
(а; Т'! = 9,3 лет) Cf23i (а; ТЧг = 660 лет) и СР52
(а-распад, деление; Tt = 2,2 года). Все эти изотопы
могут быть получены при длительном Облучении U
или Рп в ядерных реакторах. Известно лишь 3-ва-
лентное состояние Cf, проявляющееся при его соосаж-
дении на фториде и гидроокиси лантана.
Эйнштейний (Einsteinium) Es, п. н. 99,
впервые выделен в декабре 1952 в виде соединений
при ионообменном разделении радиоактивных про-
дуктов термоядерного взрыва. Первым установлен-
ным таким образом изотопом явился р -радиоактивный
Es253 {Tit = 20 дней), возникавший, видимо, вслед-
ствие захвата ядрами U238 пятнадцати нейтронов в
момент взрыва и последующих семи актов ^"-распада.
Элемент <№ 99 назван в 1955 в честь А. Эйнштейна.
В обнаружении эйнштейния и следующего элемента —
фермия — участвовала группа ученых трех америк.
лабораторий: Радиационной лаборатории Калифор-
нийского ун-та, Аргоннской и Лос-Аламосской лабо-
ратории. В дальнейшем различные изотопы Es были
получены как на реакторе (цепочки актов радиац.
захвата нейтронов и ^“-распада) — при длительном
облучении урана и плутония, так и на циклотроне —
при бомбардировке урана ионами азота по реакции:
U238 (N14, 6n) Es246. Известны изотопы Es с м. ч. 246—
256 (а также изомер Es254), из к-рых наиболее доступен
получаемый на реакторе a-активный Es254 (Tt/ =
— 280 дней). Положение Es в периодич. системе Мен-
делеева подтверждается местом эйнштейниевой фрак-
ции (подобным гольмию — см. рис. 1) при ионообмен-
ном разделении А. Как и другие 3-валентные A., Es со-
осаждается с LaF3 и La(OH)3 и экстрагируется трибу-
тилфосфатом из азотнокислых р-ров.
Фермий (Fermium) Fm, п. н. 100; соединения
Fm, а именно его a-активного изотопа Fm255 (Т1;^ =
22 часа) впервые выделены в марте 1953 при обработке
продуктов термоядерного взрыва. Назван в 1955 в
честь Э. Ферми. Среди известных изотопов Fm более
легкие (Fm250-252) получены бомбардировкой U иона-
ми О16 или бомбардировкой Cf а-частицами, а более
тяжелые (Fm253-255)— облучением U и Pu в ядерных
реакторах или (Fm256) — при бомбардировке а-ча-
стицами Es. Наиболее устойчив изотоп Fm253 ( 90%
электронный захват, 10% а-распад, Тг/ = 4,5 дня).
Для изотопов Fm с четными массовыми числами за-
метную роль в распаде уже играет спонтанное деле-
ние, по отношению к к-рому период полураспада со-
ставляет ок. 8 лет для Fm252, 200 дней для Fm254 и
всего 3 часа для Fm256, причем для последнего изо-
топа спонтанное деление оказывается преобладающим
способом распада. (Подробнее о роли разных видов
распада и в связи с этим о пределе возможного числа
элементов см. Трансурановые элементы). Данные о
ионообменном выделении Fm (рис, 1) подтверждают
его положение в периодич. системе Менделеева.
Менделевий (Mendeleevium) Md, п. н. 101,
получен в 1955 в количестве 17 атомов (!) А. Гиорсо,
Б. Харви, Дж. Чоппином, С. Томпсоном и Г. Сиборгом
в ядерной реакции Es253 (a, n)Md256. Атомы Md соби-
рались на золотой фольге, куда они вылетали под
действием бомбардирующих а-частиц, и после ряда
процедур отделялись на ионообменной колонне. Изо-
топ Md256 испытывает К-захват, с Tt. = 30 мин.
В дальнейшем в реакции Es253 (a, 2n)Md253 был полу-
чен второй изотоп менделевия. Свое название элемент
получил в честь творца периодич. системы элементов
Д. И. Менделеева. Положение Md в периодич.системе под-
тверждается данными о его ионообменном выделении.
<№10 2. В июле 1957 было объявлено о синтезе эле-
мента № 102 в виде изотопа 102 251 или 102253 рабо-
тавшей на циклотроне Нобелевского ин-та в Сток-
гольме шведско-англо-американской группой ученых,
к-рые облучали Ст244 ионами С13. По данным этой
группы, они получили a-активный изотоп с энергией
а-частиц 8,5 Мэв и Г, ок. 10 мин. Элементу было
присвоено наименование нобелий (Nobelium, No).
Тщательные попытки воспроизвести этот результат
в опытах на линейном ускорителе тяжелых ионов в
Беркли, несмотря на наличие более благоприятных,
чем в Стокгольме, экспериментальных условий, не
увенчались успехом. Между тем, осенью 1957 элемент
№ 102 (по-видимому, в виде 102 253, с энергией а-ча-
стиц 8,8±0,5 Мэв, но 7% 30 сек.) был, видимо,
получен при бомбардировке Pu239 и Pu241 ионами кис-
лорода с энергией 100 Мэв на циклотрона Ин-та атом-
ной энергии АН СССР в Москве (Г. Н. Флеров и его
сотр. С. М. Поликанов, А. С. Карамян, А. С. Пасюк,
Д. М. Парфанович и др.). В начале 1958 элемент 102
был, видимо, получен и в Беркли при бомбардировке
Ст нонами углерода: Ст246 (С12, 4n) 102 254; период
полураспада a-активного изотопа 102 254 близок к
3 сек. (работа А. Гиорсо, Т. Сиккеланда, Дж. Волтона
и Г. Сиборга). Вопрос о приоритете открытия и на-
звании элемента <№ 102 пока остается спорным.
№ 10 3. Разработка и все более широкое примене-
ние методов ускорения тяжелых ионов позволили осу-
ществить синтез элемента № 103. Если верна гипотеза
о существовании семейства А., элемент 103 должен
быть замыкающим членом этого семейства и обладать
особенно устойчивой 3-валентностью.
Для аналитич. целей используются соединения
всех А. с валентностью +3, а также соединения
NpIV’VllVI, соединения AmVllVI и соединения
Bklv А. переводят в раствор действием HNO3 или
ее смеси с HF, а также др. способами. Все А. в 3-ва-
лентном состоянии колич. соосаждаются с LaF3, вме-
сте с Pu111, IV и NpIV, а затем после окисления по-
следних броматом калия до 6-валентного состояния
отделяются от них в виде фторидов. Отделение транс-
кюриевых элементов друг от друга основано на уси-
лении их комплексообразующих свойств с ростом
порядкового номера и осуществляется хроматография,
путем на спец, ионообменных смолах-катионитах.
Разделение Np, Pu, Am и Bk и их отделение от др.
элементов и А. основано на их способности окисляться
до более высоких ступеней валентности.
Качественно А. открывают радиохимия, н спектраль-
ными методами; количественно определяют гл. обр.
по интенсивности излучения; нек-рые А., как-то:
Np111, Iv’ v и VI, Am111, v 11 VI — в больших коли-
чествах могут оцениваться калориметрически.
Лит.: Актиниды, пер. с англ., под ред. А. В. Николаева,
М., 1955; Гольданский В. И., Новые элементы в пе-
риодической системе Д. И. Менделеева, 2 изд., М., 1955;
Лаврухина А. К., Золотов Ю. А., Трансурановые
элементы, М 1958; Seaborg G. Т., The transuranium ele-
ments, New Haven, 1958; Хайд И. иСиборг Г., Транс-
урановые элементы, пер. с англ., М., 1959; С и б о р г Г. Т.,
Кац Д. Д., Химия актинидных элементов, пер. с англ., М.,
i960. В. И. Гольданский.
АКТИНИЙ (Actinium) Ас — радиоактивный хи-
мич. элемент III гр. периодич. системы Менделеева,
п. н. 89, м. ч. наиболее долгоживущего изотопа
227. Конфигурация внешних электронов атома 6<Z7s2.
Открыт в 1899 А. Дебиерном в отходах от переработки
урановых руд. Содержание А. в земной коре 6 • КН ° %
по весу. Изотопы Ас227 (Ас) и Ас 228 (MsTh II) являют-
ся членами радиоактивных рядов А. и тория (равно-
весные количества 2,06-КН г Ас и 5,05-10-8 г MsTh II
на 1 т урановой смоляной руды или тория соответ-
ственно). Ас227 = 21,8 года) испускает ^-ча-
стицы (—99%) и а-частицы (1,2%); Ac228 (Tt/ ==
=6,13 часа)—^-излучатель; Ас225 (Г,/2 = 10,0 дней)—
107
АКТИНОИДЫ — АКТОМИОЗИН
108
а-излучатель, входит в искусственно полученный ряд
нептуния.
А. — металл серебристо-белого цвета, имеет гране-
центрированную кубич. решетку с а = 5,311 А;
атомный радиус 1,88 А; т. пл. 1040°±50°. Очень
реакционноспособен; легко окисляется на воздухе
и растворяется в соляной и азотной к-тах. В химич.
соединениях А. 3-валентен; является высшим гомо-
логом лантана, сходство с к-рым весьма велико; А.
имеет те же нерастворимые соединения, что и лантан
(гидроокись, фторид, оксалат, фосфат, карбонат,
фторсиликат); летучести соответствующих галоге-
нидов Ас и La — величины одного порядка; все чи-
стые соединения А., к-рые были получены и оконча-
тельно идентифицированы, изоморфны аналогичным
солям La. Основные свойства у А. выражены сильнее,
чем у лантана; цонный радиус Ас3 1,11А; ионный
радиус La3+ 1,06 А. Вследствие их большого химич.
сходства, La обычно используется в качестве носителя
для микроколичеств А., к-рые при всех методах от-
деления почти нацело захватываются лантаном.
Окись Ас2О3 получается при прокаливании его
гидроокиси или оксалата при 1100°. При действии на
окись, сухую гидроокись или оксалат фтористым
водородом (ок. 700°), СС14 или А1Вг3 (ок. 750°) об-
разуются соответствующие галогениды А., прокали-
вание к-рых в присутствии NH3 и паров воды ведет
к образованию оксигалогенидов. Сульфид Ac2S3 можно
получить, пропуская H2S над оксалатом или окисью
А. при 1400°. Почти все соли А. белого цвета, в рас-
творах бесцветны; Ac (NO3)3 хорошо растворяется в
абсолютном спирте.
Весомые количества А. получают путем облучения
?адия нейтронами: Ra226 (п, 7) Ra227. Изотоп Ra227
T,lt = 41 мин.) переходит в Ас227. Для отделения А.
от радия и других элементов применяются экстракция
бензольным р-ром тиофенкарбонилтрифторацетона при
контролируемом pH, ионообменная хроматография
и другие методы. Обнаружение и колич. определение
А. производится путем измерения активности пре-
парата или выделенных из него дочерних веществ А.,
напр. АсК (изотоп франция) или актинона (изотоп
эманации), а также по характеристич. линиям его спек-
тра испускания, если имеется достаточное количество
анализируемого вещества. А. является опасным радио-
активным ядом с высокой удельной а-активностью.
Лит.: Актиниды, пер. с англ., под ред. А. В. Николаева,
М„ 1955; Сиборг Г., Кац Дж., Химия актинидных эле-
ментов, пер. с англ., М., 1960; Strominger D., Hol-
lander J. М. and Seaborg G. T„ Rev. mod. phys.,
1958, April, 30, № 2, pt. 2, p. 585—904. В. И. БарановскиИ.
АКТИНОИДЫ — см. Актиниды.
АКТИНОМИЦИНЫ — группа антибиотиков (св.
30), продуцируемых Actinomyces auranticus, А. пла-
вятся с разложением ок. 350°; величины удельного
вращения их лежат в пределах от —309° до —367°
(С = 0,25 в этаноле). Молекула А. состоит из двух
частей — гетероциклической и полипептидной. Гете-
роциклическая часть молекулы А. — 2-аминофенокса-
зин-3-он (I) является хромофором, благодаря чему
все А. окрашены в оранжево-
красный цвет. Все исследован-
I I ные А. имеют одинаковые
спектры поглощения, что ука-
j зывает на одинаковость строе-
ния хромофора. Многообразие
А. связано с различиями в аминокислотном соста-
ве полипептидной цепи, последовательности их со-
четания и конфигурации аминокислот. В состав по-
липептидной части молекулы А. входят след, амино-
кислоты: L-треонин, саркозин, N-метилвалин, L-npo-
лин, D-валин, D-алло-изолейцин, L-оксипролин и
К-метил-В-изолейцин. Наиболее изученными явля-
ются актиномицины А, В, С и D. Актиномицины А, В
и D содержат треонин, саркозин, пролин, валин и
N-метилвалин. Актиномицин С содержит треонин,
саркозин, пролин, валин, алло-изолейцин и N-метилва-
лин. По аминокислотному составу указанные анти-
биотики можно разделить на две группы: 1) А, В
и D не содержат алло-изолейцина, 2) С содержит эту
аминокислоту.
А. за исключением актиномицина D не являются
индивидуальными соединениями, а представляют сме-
си близких между собой соединений. Так, актиноми-
цин G — смесь 7 веществ, а актиномицины А и В со-
стоят из пяти компонентов каждый и т. д.
Антибиотик аурантин по физико-химич. свой-
ствам, аминокислотному составу и биологич. активно-
сти относится к А. Он представляет собой оранжево-
красные кристаллы, т. пл. 255°—257°, [a]g = —300°
(С = 0,02 в метаноле). Спектр поглощения ауран-
тина в УФ и видимой части характерен для А. и имеет
максимум поглощения при 443 ммк. Вещество хорошо
растворяется в хлороформе, бензоле, ацетоне, мета-
ноле и этаноле, хуже — в эфире, плохо — в этила-
цетате и амилацетате, почти нерастворим в воде. Ме-
тодом хроматографии на бумаге и окиси алюминия
установлено, что кристаллич. препарат аурантина,
в свою очередь, состоит по крайней мере из четырех
веществ А1( А2, А3, А4. Содержание Ах в различных
препаратах колеблется от 77 до 79%, А2 — от 21 до
23%, содержание А3 и А4 — невелико; т. о. аурантин
выгодно отличается от актиномицина С соотношением
компонентов; он менее токсичен и более активен.
Выделенные препараты не отличаются друг от друга,
исходного аурантина и актиномицина С по спектрам
поглощения, но отличаются по темп-ре плавления,
по коэфф, распределения на бумаге и окиси алюминия.
Антибактериальная активность аурантина определя-
лась на многих тесторганизмах: грамположительные
бактерии более чувствительны, чем грамотрицатель-
ные; специфичность действия полученных фракций
аурантина на бактерии не установлена. Аурантин, как
и некоторые другие А., активен против некоторых
опухолей.
Лит.: см. при ст. Антибиотики-полипептиды.
А. Б. Силаев.
АКТИНОН Ап — один из изотопов эманации.
АКТОМИОЗИН (S-миозин, а-миозин) — продукт
соединения актина и миозина, входящих в его со-
став в приблизительно стехиометрия, соотношении
(~1 : 5,5—1 : 3 по весу); сочетает в себе физико-химич.
и ферментативные свойства обоих белков и, кроме
того, обладает уникальной способностью реагировать
на добавление очень небольших количеств аденозин-
трифосфорной к-ты (АТФ) резким, но вполне обрати-
мым изменением коллоидного состояния (понижением
вязкости, если А. растворен, и дегидратацией, если
он находится в состоянии геля). Большинство иссле-
дователей полагает, что реакция взаимодействия А.
с аденозинтрифосфорной к-той лежит в основе со-
кратительного механизма мышц. Солевыми р-рами
(цапр., 0,6 М КС1) из поперечно-полосатой мускула-
туры извлекается т. н. натуральный А., или миозин В,
представляющий собой смесь А. и миозина. А. или
ему подобные белки (слабее, однако, реагирующие с
аденозинтрифосфорной к-той) присутствуют в мыш-
цах любого типа всех позвоночных и беспозвоночных
животных и в нек-рых других биологич. объектах,
связанных с механич. движением (хромосомные фи-
гуры делящихся клеток, плазмодии миксомицетов).
Лит.: Любимова М. Н., в кн.: Успехи современной
биохимии. Об. трудов объединенных научных конференций...,
т. 1, М., 1947, с. 141; СентДжиордьи А., О мышечной
деятельности, пер. М. Н. Любимовой, М., 1947; То но м ур а
Ю. [и др.], Усп. совр. биол., 1956, 44, вып. 3 (6), с. 362; Гри-
щенко Е. Д., там же, 1958, 46, вып. 3 (6), с. 259.
Е. Д. Грищенко.
109
АКЦЕПТОР - АЛИЗАРИНОВЫЙ КРАСНЫЙ С
110
АКЦЕПТОР (от лат. acceptor — получатель). А к-
цептором электронов в химии вообще
наз. частицу, принимающую электроны, т. е. выпол-
няющую функцию окислителя; в частности, это — так
наз. апротонные (льюисовские) кислоты — BF3,
А1С13, SnCl4 и др. В протолитической теории кислот
и оснований акцептором протонов наз.
частицу присоединяющую протон и, по Бренстеду,
играющую роль основания. В химии комплек-
сных соединений А. наз. компонент, прини-
мающий электронную пару при образовании коорди-
национной связи в процессе комплексообразования.
В химической кинетике при рассмотре-
нии сопряженных реакций A+B->NnA+C->N
вещество А наз. актором, В — акцептором и С —
индуктором. В радиационной химии А.
наз. частицу, реагирующую со свободными радика-
лами, возникающими в радиолизуемой системе; кон-
центрация вводимого А. обычно на несколько порядков
меньше концентрации веществ, подвергаемых радио-
лизу; в англ, лит-ре подобную роль играет термин
«scavenger». И. В. Верегцинский.
АЛАНИН C3H7O2N, мол. в. 89,09 — аминопропио-
новая к-та; различают а-А. и fi-A. а-А ланин
CH3CH(NH2)COOH — широко распространенная в
природе аминокислота, встречающаяся в виде L-A.
в свободном виде и в составе большинства белков;
мелкие кристаллы. Т. пл. D, L-A. 295—296° (с разл.);
хорошо растворяется в воде, плохо — в 80%-ном спир-
те, нерастворим в эфире. Т. пл. L-A. 297° (с разл.);
хорошо растворяется в воде, практически нераство-
рим в спирте; [a]g = -ф 2,7 (в воде). Т. пл. D-A. 297°
(с разл.), растворим в горячей воде; [а]3^ =—14,6°
(в 6 н. НС1). При 25° pZfai (СООН) 2,34, рАОз (NH+)
9,69; pZ 6,00. По химич. свойствам а-А. типичная
алифатич. аминокислота, дает только типичные для
аминокислот реакции. Для определения А. смеси
аминокислот (напр., гидролизата белка) обычно раз-
деляют хроматографически с последующим определе-
нием А. по окраске с нингидрином. А. можно опреде-
лить и химич. методами. А. является заменимой ами-
нокислотой (в организме образуется из др. аминокис-
лот и из пировиноградной к-ты). а-А. получают
действием аммиака на a-хлор- (или бром-)-пропионо-
вую к-ту или синтезом из ацетальдегида, синильной
к-ты и аммиака.
рА ланин NH2CH2CH2COOH — мелкие кристал-
лы; т. пл. 200°; растворим в воде, трудно — в спирте,
нерастворим в эфире и ацетоне. При 25° pKOi (СООН)
3,60; p.ffO2(NH+) 10,19; pZ 6,90. fi-A. получают дей-
ствием гипобромита на щелочной р-р сукцинимида
и др. методами; он является структурным компонентом
природных в-в (пантотеновой к-ты и дипептидов анев-
рина и карнозина), в белках не обнаружен. См. также
Аминокислоты, Белки и Обмен веществ.
Лит. см. при ст. Аминокислоты. О. С. Хохлова.
АЛАЯ КИСЛОТА С21Н13О9, мол. в. 412,36 — бес-
цветные кристаллы (технич. продукт обычно серого
цвета); растворимы в воде и разбавленных минераль-
ных к-тах, хорошо растворимы в щелочах с образо-
ванием солей; при нагревании сводными р-рами щело-
чей гидролизуется, образуя 2-амино-5-нафтол-7-суль-
фокислоту. А. к. легко вступает в реакцию азосочета-
ния с диазосоединениями, к-рую обычно проводят
в содовом р-ре; при этом получают, гл. обр., дисаао-
красители. Диазосоединения вступают преим. в о-по-
ложения к оксигруппам А. к.; побочно реакция
может протекать в «-положении к оксигруппам. Полу-
чают А. к. действием фосгена на водный р-р натриевой
(или калиевой) соли 2-амино-5-нафтол-7-сульфокисло-
ты в присутствии карбонатов натрия или калия, свя-
зывающих к-ту. Продукт осаждают из р-ра в виде
динатриевой соли. Применяется А. к. в произ-ве
преим. прямых (субстантивных) дисазокрасителей;
в содовом р-ре образует с бензолдиазонием оранжевое,
с о-метоксибензолидиазонием — красное окрашива-
ние. Количественно А. к. определяют азосочетанием
в содовом р-ре с титрованным р-ром бензолдиазония.
М. А. Чекалин.
АЛИЗАРИН (1,2-диоксиантрахинон) С14Н8О4, мол.
в. 240,20 — оранжево-красные кристаллы (иглы) три-
клинич. или ромбич. системы;
т. пл. 290°; т. кип. 430°,раствори- 9 9*"1
мость в воде 0,034 г/100 г (100°);
умеренно растворим в этиловом | |Г lj Т
спирте, хорошо — в горячем ме-
типовом спирте, бензоле, уксус- Д '
ной к-те, сероуглероде, пиридине, °
щелочах. А. находится в виде глюкозида в корнях
марены; применялся для крашения тканей; в 1826
выделен франц, химиками П. Робике и Ж. Коленом;
в 1869 синтезирован К. Гребе и Ж. Либерманом; полу-
чают сплавлением ^-сульфокислоты антрахинона со
щелочами в присутствии окислителя. А. является
важным полупродуктом синтеза более сложных кра-
сителей. С многовалентными металлами (Са, А1 и др.)
А. дает окрашенные соли, т. н. лаки, применяемые
при крашении и печатании тканей. Алюминиевый
лак А. (крапплак) применяется для приготовления
художественных красок и в полиграфии. Исполь-
зуется А. в аналитич. химии для открытия А1, Си,
Nb, F, Ga, In, Hg, Th, Ti и Zr и фотометрия, определе-
ния Al, F и др.
Лит.: Ворожцов Н. н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955.
АЛИЗАРИНОВОЕ МАСЛО — касторовое масло,
обработанное серной к-той и нейтрализованное затем
щелочью; желтая или коричневая жидкость, хорошо
растворимая в воде, спирте и эфире. А. м. состоит
гл. обр. из сложных эфиров рицинолевой к-ты
СН3(СН2)3СНСН2СН = СН(СН2)7СООН. Группа SO3H
ОН
этерифицирует гидроксильную группу или присое-
диняется по двойной связи.
Под действием H2SO4 происходит также частичное
или полное омыление триглицеридов. Образовавшаяся
свободная рицинолевая к-та дает продукты уплотне-
ния разного состава. Самым распространенным сы-
рьем для приготовления А. м. является касторовое
масло. Сульфирование проводят серной к-той плот-
ностью 1,84 при темп-ре не выше 30—35°. Продукты
сульфирования нейтрализуют р-ром NaOH или ам-
миака. Применяется А. м. в текстильной пром-сти
при крашении, печати и др. видах обработки тканей
и пряжи для улучшения прокрашивания и закрепле-
НИЯ окраски на волокне. н. В. Млодзеевская.
АЛИЗАРИНОВЫЙ КРАСНЫЙ С (натриевая соль
1,2-диоксиантрахинон-З-сульфокислоты), мол. вес
360,27 — краситель, оран- q
жево-желтые кристаллы, II |
иглы или порошок того же
цвета; хорошо растворим L I I I
в воде (р-р буровато-желто-
го цвета), водном р-ре ам- Д
миака (р-р фиолетового цве-
та) и в спирте. Краситель получают сульфированием
ализарина. А. к. образует окрашенные лаки со многими
Ill АЛИФАТИЧЕСКИЕ СОЕДИНЕНИЯ —АЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ 112
катионами; применяется для крашения шерсти и шелка
под названием «кислотный красный ализариновый» и
для открытия и фотометрия, определения Zr, Th, Al,
Ti, Ga, Be; в качестве кислотно-основного индикатора
(переход окраски от желтой при pH 3,7 к буро-розо-
вой — pH 5,2); В микроскопии. Д. Н. Васкевич.
АЛИФАТИЧЕСКИЕ СОЕДИНЕНИЯ — см. Аци-
клические' соединения.
АЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — орга-
нические соединения, молекулы которых содержат
одно или несколько колец неароматического харак-
тера. А. с. совместно с ароматическими углеводородами
и их производными составляют раздел изоцикли-
чески х, или карбоциклических, сое-
динений. А. с. подразделяются на классы и
гомологические ряды по числу и строению колец,
по наличию кратных связей и функциональных
групп в молекуле. В основе систематизации А. с.
лежит классификация цикланов (циклоалканов, цик-
лопарафинов), углеродные атомы к-рых соединены
простыми связями. А. с. подразделяются: 1) по числу
колец в молекуле — на моноциклич. (одноядерные),
бициклич., трициклич., полициклич. (многоядерные);
2) по числу атомов углерода в кольце — на цикло-
пропановые (с 3-членным циклом), циклобутановые
(с 4-членным циклом), циклопентановые (с 5-членным
циклом) и т. д.; 3) по взаимному расположению
колец — на спирановые
углеводороды, или спи-
раны (I), А. с. с конден-
сированными циклами
(II), А. с. с мостико-
выми циклами (III); 4) по наличию кратных свя-
зей в цикле или функциональных групп при нем —
на циклены, напр. IV (а также спироциклены, бици-
клены, трициклены и т. д.), циклодиены, напр. V
(спироциклодиены и т. д.), циклотриены, напр. VI,
циклотетраены, напр. VII и т. д., цикланолы, напр.
VIII, цикланоны,
У, и т. п.
напр. IX, циклановые к-ты, напр.
зей в циклах. Наличие циклов в молекуле создает
особые виды стереоизомерии, свойственные А. с.
Так, кроме обычных видов изомерии алканов, для
циклоалканов существует цис-тра нс-изомерия на-
правления заместителей по отношению к кольцу:
В конформационном анализе некоторых цикланов и
их производных дополнительно рассматривается на-
правление заместителей по отношению к оси сим-
метрии молекулы. Некоторые спираны, не имеющие
асимметрии, атома углерода, существуют в виде
двух оптических антиподов благодаря асимметрии
молекулы.
Присутствие кольца в молекуле влияет на физич.
и химич. свойства вещества, к-рые для А. с. иногда
сильно отличаются от свойств аналогичных ацикличе-
ских соединений (алифатических) с тем же числом
атомов углерода. А. с. характеризуются большим
уд. весом, показателем преломления света, темп-рой
кипения. Хотя цикланы разных классов имеют об-
щие формулы СпН2п, СПН2), _2, СпН2п_4 и т. д., они
являются насыщенными соединениями, в к-рых все угле-
родные атомы связаны простой связью; длины связей
С—С и С—Н в цикланах такие же, как и в алканах
(1,54 А и 1,09 А). Однако по химич. поведению цик-
ланы и алканы часто сильно различаются. Так, наи-
менее прочные 3-членные циклы присоединяют гало-
гены подобно олефинам с разрывом цикла:
СН2
/\ + Вг2—— ВгСН2СН2СН2Вг
Н2С—СН2
СН=СН
Нгсх zCH
сн2
циклопентен
IV
нс—сн
'сн
НС —
НС ZCH
чсн2
цик лопентадиен
нС
X ^СН
НС — Cti
ХСН2
н2с хсн—он
Н2С\ /СН2
сн2
циклоок татетоаен циклогексанол
VII
НС—сн.
X
нс
'г
'сн
НС
СН
цик логептатриеп
У!
сн2
Н2С = О
Н2С---СН2
циклопентанон
VIII IX
Циклы различаются по
своей химич. устойчивости, Н2С СН2
к-рая возрастает от 3-член- Н2С—СН-СООН
ных циклов к 6-членным;
циклы С большим ЧИСЛОМ цивлобутаниарбоновая кислоте
атомов углерода (до 39ато- х
мов) имеют примерно такую же прочность, как и
5-членные. Устойчивость циклов зависит от напряже-
ния в цикле, вызываемого: а) отклонением величины
валентного угла в цикле от угла, свойственного на-
правленным валентностям атомов углерода при оди-
нарной, двойной или тройной связях (байеровское, или
угловое, напряжение); б) притяжением или отталкива-
нием атомов, непосредственно не связанных друг
с другом (торсионное, поворачивающее напряже-
ние); в) взаимодействием л-электронов кратных свя-
циклопентаны иод действием водорода в присутствии
металлич. катализаторов легко претерпевают гидроге-
нолиз о разрывом кольца:
СНо СНо р.
I СН2^Н2-----* СНз (СН2 )3СН3
сн2-сн2
циклогексановые соединения гладко дегидрируются
па катализаторах, образуя ароматич. соединения:
/СНг-СНа ^СН-СН
^^2 ** СН JDH + ЗН2
сн2- сн2 сн=сн
а производные бицикло-(0,3,5)-декана дают при этом
азулены'.
СНо-СНоХ /<Н2 СН=СН\^Н
/ СН 'ч / С. \
Н2С | СН2—-Н2С I СН + 5Н2
СН2 сна \сн2 СН-СЬК Хсн
Циклены, содержащие в цикле двойные связи или
семициклич. двойные связи (двойная связь между
циклом и боковой цепью), изомеризуются легче, чем
олефивы: сн
НСХ ХСН2 Ц2С
II 1 — 1
нсхс/с,|2 Н2С-------сн2
Ч-сн3
из
АЛКАДИЕНЫ — АЛКАЛИМЕТРИЯ И АЦИДИМЕТРИЯ
114
Циклы в свою очередь влияют на поведение функцио-
нальных групп. Усложнение циклич. группировки
усиливает ее воздействие на кратные связи и функцио-
нальные группы.
Характерными для А. с. являются реакции взаимно-
го перехода циклов. Напр., при действии азотистой
к-ты на циклогептиламип или на аминометилгексан
получается смесь циклогептанола с циклогексилкар-
биполом, т. е. при
р-ром NaOH скачок титрования достаточно велик —
от 4 до 10 pH (рис. 1). В качестве индикатора можно
применять любой кислотно-основной индикатор с ин-
тервалом перехода, находящимся внутри скачка
титрования (метиловый оранжевый, фенолфталеин).
При титровании слабой к-ты сильной щелочью раствор
Сн2-Сн2
Н2С
Н2С
Сн2- СНа
\ HNOa
CHNH2----~
/СН2~СН2
НаС
Н2С
СН2-Сн2
СНОН +
СН—СН2ОН
О2С Сн2
Н2С\ /Сн2
•Сн2
первой реакции частично происходит сужение 7-член-
ного цикла в 6-членный, во второй — расширение
6-членного цикла в 7-членный.
Химия А. с. была развита блестящими работами
В. В. Марковникова, Н. Д. Зелинского, Н. М. Киж-
нера, Н. Я. Демьянова, С. С. Наметкина, Б. А. Ка-
занского, Л. Ружички, К. Циглера, В. Прелога.
А. с. встречаются в самых разнообразных природ-
ных органич. соединениях. Так, 3-членные циклы
встречаются в пиретринах, естественных инсектици-
дах, 4-членные — в анемонине, выделяемом из анемо-
нов, 5-членные — в хаульмугровой к-те, применяемой
в терапии проказы. Циклопентаны и циклогексаны
(нафтены, по Марковникову и Оглоблину) широко
представлены в нефтях, к-рые являются самым боль-
шим источником цикланов. Циклогексановые произ-
водные особенно широко распространены в природе;
они содержатся во многих эфирных маслах, скипидаре,
антибиотиках (стрептомицин), витаминах А и D. Из
алкалоидов были выделены производные циклогеп-
татетраена и циклооктатетраена. В состав пахучих
в-в — мускуса и цибета — входят циклич. кетоны,
содержащие 15 и 17 атомов С в цикле.
А. с. находит все возрастающее промышленное при-
менение. Циклич. углеводороды благодаря своему
большому удельному весу входят в состав ракетного
топлива. А. с., являющиеся инсектицидами, как гек-
сахлоран, альдрин, хлордан, синтетич. пиретрин,
вырабатываются пром-стью. А. с. применяются как
полупродукты в произ-ве искусств, волокна (капрон).
См. Номенклатура органических соединений, Терпены,
Смолы природные и бальзамы, Стероиды, Конформа-
ционный анализ, Изомерия, Стереоизомерия, Демьяно-
ва перегруппировка.
Лит.: Перспективы развития органической химии. Под
ред. А. Тодда, пер. с англ, и нем., М., 1959; Ч и ч и б а-
бин А. Ё., Основные начала органической химии, т. 2,
6 изд., М., 1958; Ф и з е р Л., Ф и з е р М., Органическая
химия, пер. с англ., М., 1949. М. И. Розенгарт.
АЛКАДИЕНЫ (диеновые углеводороды, диоле-
фины) — см. Непредельные углеводороды.
АЛКАЛИМЕТРИЯ И АЦИДИМЕТРИЯ — метод
колич. титриметрич. анализа, основанный на исполь-
зовании реакции нейтрализации, т. е. соединения
ионов Н+ ОН- Н2О (титрование р-ров кислот
р-рами щелочей и обратно). При титровании сильной
кислоты раствором сильной щелочи точка эквивалент-
ности практически совпадает с точкой нейтральности.
Для определения точки эквивалентности и величины
скачка титрования, т. е. интервала изменения pH
вблизи точки эквивалентности, результаты изменения
pH в процессе титрования наносятся на график в ко-
ординатах «pH — % оттитрованной кислоты (или
щелочи)». Скачок титрования, т. е. резкое изменение
концентрации иона у точки эквивалентности, зависит
от концентраций и констант диссоциации реагирую-
щих веществ. При титровании 0,1 н. р-ра НС) 0,1н.
в точке эквивалентности вследствие гидролиза соли
слабой к-ты будет не нейтральным, а слабощелочным
(рис. 2); поэтому последняя
CH2NH2
J
HNO2 Н2С ТН2
н8с. .сн2
^сн2
капля раствора щело-
чи вблизи точки экви-
валентности изменит
реакцию раствора из
слабощелочной в бо-
лее щелочную. В ка-
честве индикатора в
этом случае могут
быть применены лишь
перехода в щелочной
вещества, имеющие интервал перехода в щелочной
области; напр., при титровании уксусной к-ты р-ром
NaOH следует пользоваться фенолфталеином. При
титровании слабого основания сильной к-той раствор
pH точно эхе»
2
3
4
5
6
7
8
9
10
11
12.............;
О 20 40 60 80 100 120%
точна энв.
pH 0.1 н
О
2
4
6
8
10
12
14 С
0 20 40 60 80 100 120%
Рис. 1.
Рис. 2.
вследствие гидролиза соли
слабокислым (рис. 3). По-
pH
Оу
1
2
3
4
5
6
7
8
9
10
II
12
О 20 40 60 80 100 120%
Рис. 3.
точно энв.
в точке эквивалентности
слабого основания будет
этому можно применять только индикаторы с интер-
валом перехода в кислой области, напр. при титрова-
нии гидроокиси аммония р-ром НС1 следует пользо-
ваться метиловым оранжевым или метиловым красным.
При титровании многоос-
новных к-т (или смесей к-т
разной силы) сильной ще-
лочью имеется неск. точек
эквивалентности (для каждо-
го значения [Н+] к-ты) и со-
ответственно неск. скачков,
если последовательные кон-
станты диссоциации доста-
точно отличаются друг от
друга. В случае, когда пер-
вая константа диссоциации
Кг по крайней мере в 104
раз больше, чем К2, титро-
вание возможно с ошибкой 0,5%. Напр., можно
титровать Н3РО4 метиловым оранжевым до NaH2PO4
и с фенолфталеином до Na2HPO4. Если же титруется
смесь сильной к-ты и слабой (напр., соляной и уксус-
ной), то сначала будет титроваться сильная кислота,
а затем слабая. Чем слабее вторая кислота и чем мень-
ше ее концентрация по сравнению с первой, тем резче
скачок титрования около первой точки эквивалент-
ности. То же самое имеет место при титровании много-
кислотных оснований. Титрование Na2CO3 посредством
НС1, аналогичное титрованию двухкислотного осно-
вания, можно провести до первой точки эквивалент-
ности (pH 8,4) с фенолфталеином, однако определе-
ние конца титрования не четко; применяя метиловый
оранжевый, можно титровать до второй точки экви-
валентности (pH ок. 5), изменение окраски доста-
точно резкое.
115
АЛКАЛОИДЫ
116
Точку эквивалентности во всех реакциях нейтрали-
зации можно установить потенциометрия, методом по
скачку потенциала хингидронного, металл-окисного
или стеклянного электрода. В качестве рабочего тит-
рованного раствора к-ты наиболее часто применяют
0,1 н. р-ры НС1; р-ры HNO3, H2SO4, НСЮ4 приме-
няют только в специальных случаях. Титр раствора
к-ты можно установить весовым методом (осаждением
в виде AgCi) или титриметрич. методом, применяя
различные исходные в-ва: Na2CO3, Na4B4O7 • ЮН2О,
HgO, KJO3 или Na2C2O4 после его прокаливания до
Na2CO3. Титрованным рабочим р-ром щелочи обычно
служит 0,1 н. р-р NaOH, свободный от СО®-; послед-
нее особенно важно, если применять фенолфталеин.
Титр р-ра щелочи устанавливают по щавелевой к-те
Н2С..О4 • 2Н2О, бензойной к-те С6Н5СООН, бифтала-
ту калия С6Н4(СООН)СООК, сульфату гидразина
Na2H4 • H2SO4. Пользуясь йодатом калия, можно
связать методы нейтрализации с методом иодомет-
рии: KJO3 4- KJ + 6НС1 -> 3J2 -|- 3H2O-f- 6КС1,
т. о. количество выделившегося иода прямо пропор-
ционально количеству имевшейся в растворе кис-
лоты.
А. и а. применяются для определения кислот, ще-
лочей, смесей Na2CO3 и NaHCO3, Na2CO3 и NaOH,
жесткости воды, азота в виде гидроокиси аммония и
мн. др. случаях.
Лит.: Кольтхоф И. М. и Сендэл Е. Б., Коли-
чественный анализ, пер. с англ., 3 изд., М.—Л., 1948; Кольт-
г о ф И. М. и С т е н г е р В. А., Объемный анализ, пер.
с англ., М., т. 1—2, М., 1950—52. В. Г. Типцова.
АЛКАЛОИДЫ — азотсодержащие органич. соеди-
нения природного, чаще растительного происхожде-
ния, основного характера. Впервые А. были вы-
делены из растительного материала в начале 19 в.
(в 1806 Сертюрнер выделил морфин из опия). К наст,
времени известно более 1000 веществ этого класса.
Выделение из растений и выяснение строения А. со-
ставляет один из разделов органич. химии. Эта работа
оказалась весьма плодотворной: большое число А.
получило практич. применение, знание строения А.
позволило для многих из них разработать методы
синтеза и осуществить синтетич. получение в промыш-
ленном масштабе (кофеин, ареколин, эфедрин, тро-
пин и др.). В ряде случаев строение того или иного
А., являющегося лекарственным в-вом, служило
отправной точкой для поисков и получения новых
синтетич. лекарств, препаратов; синтез аналогов ко-
каина привел к получению новокаина, аналогичная
работа привела к получению акрихина — аналога
хинина, ряда препаратов, обладающих действием
морфина, А. кураре, папаверина и т. д. Значительную
часть современных лекарств составляют А. и вещества,
полученные в результате синтетич. поисков среди
аналогов этих А.
Простейшие А*, имеют менее 10, наиболее сложные—
более 50 атомов углерода. В состав А., кроме угле-
рода, водорода и азота, обычно входит кислород,
присутствующий в гидроксильных, метоксильных,
сложноэфирных, эфирных, лактонных и карбониль-
ных группах. В молекулах сложных А. могут содер-
жаться полициклич. системы — насыщенные, нена-
сыщенные или ароматизированные частично или
полностью. Установление строения А. часто является
сложной задачей. Работа здесь обычно сводится
к определению характера групп, в состав к-рых входят
атомы кислорода и азота, и расщеплению молекулы А.
до осколков уже известного строения. Окончательно
строение устанавливается синтезом. По спектрам по-
глощения в-ва устанавливают наличие функциональ-
ных групп и их положение. Определение величины
р/С дает возможность сделать нек-рые выводы о строе-
нии азотсодержащей части молекулы. Для выясне-
ния строения наиболее сложных А. применяют метод
рентгеноструктурного анализа. В определении строе-
ния А. достигнуты большие успехи: установлено
строение таких сложных веществ, как морфин, стрих-
нин, А. спорыньи, резерпина, предложены возможные
формулы строения аконитина и родственных ему А.
В ряде случаев установлено пространств, положение
атомов в молекуле, определена конформация (напр.,
эфедрина, псевдоэфедрина).
Большое разнообразие строения А. затрудняет их
классификацию. В наст, время общепринято считать
основным признаком при классификации А. строение
азотсодержащей части их молекул; вещества неуста-
новленного строения классифицируют по филогене-
тич. признаку, объединяя в одну группу все А.,
выделенные из близких растений. Ниже перечис-
лены основные группы А. и важнейшие, получив-
шие практич. применение представители каждой
из них.
Группа пиридина. Простейшие из А. этой группы
содержат одно пиридиновое или гидрированное пиридиновое
ядро. Ареколин — один из А. ареновой пальмы, в ветеринарии
применяется в качестве глистогонною средства, в медицине —
ограниченно. Лобелии (I) — основной А. лобелии одутлой
(Lobelia inflata) — употребляется как лекарств, препарат.
JCH2
Сн2 Сн2
СвН5~СНОН-СН2-СН .СН-СН2СОСвН5
N
I
сн3
стимулирующий дыхание. К простейшим А. этой группы отно-
сится кониин — весьма ядовитый А. болиголова (Conium
maculatum), рицинин и А. из семян клещевины (Ricinus com-
munis). Никотин, содержащийся в табаке, и анабазин, полу-
чаемый из среднеазиатского растения Anabasis aphylla, пред-
ставляют собой А., молекулы к-рых построены из двух некон-
денсированных ядер, одно из к-рых пиридиновое. Никотин
используется в качестве инсектицида и как полупродукт для
синтеза никотиновой к-ты. Анабазин — инсектицид, выпу-
скается в виде концентрированного р-ра сульфата, содержащего
побочные А. (афиллин). К производным пиридина может быть
отнесен также псевдопельтерин.
Производные тропана. Данные А. характери-
зуются наличием в молекуле бициклич. ядра (II). В медицин-
ской практике применяются: атропин и скополамин — холи-
нолитич. средства; кокаин — поверхностный анестетик.
7 1 2
сн2—<jiH—сн2
NH зСН2
I I
CHp-CH-сн2
6 d 6 4
II
СНа /СНа
Н2С« вСН 7 вСН2
СН2 СН3
HI
Группа л у пин ан а. Эти А. могут рассматривать-
ся как производные хинолизидина (III), называемого так-
же октагидропиридоколином. В медицинской практике при-
,Сн2 ^сн—сн2 сн2
Н2С< 8 >сн 7 ^сн
H2C<2>N О^<
сн2 сн2
2
2
:н2
меняются пахикарпин (IV)! как ганглиоблокирующее сред-
ство, спартеин и цитизин как препарат, возбуждающий ды-
хание.
Производные изохинолина. А. этой группы
могут быть разделены на неск. подгрупп. К производным
1-метилтетрагидроизохинолина относятся сальсолин и сальсо-
лидин, к-рые выпускаются только в СССР и применяются
для лечения нек-рых форм гипертония, болезни. Папаверин,
выделяемый из опия и получаемый синтетически, широко
используется в качестве спазмолитика; является производным
117
АЛКАЛОИДЫ
118
1-бензилизохинолина. Более сложное строение имеют А. произ-
водные
10
9
апорфина
2
1-СНз
te
е
15
12
13
5
(V) — см. Апорфиновые алкалоиды, днизохи-
нолина (VI), напр. берберин, нафтофенан-
тридина, напр. хелидонин (VII) и сан-
гвинарин. К этой
щего) средства и для лечения гипертонии; эзерин — антихолин-
эстеразное, препарат, заменяемый часто синтетич. препаратом
прозерином.
1
же
группе
относятся А.
2
и
10
Vi
опия — морфин (VIII) и близкие к нему по строению кодеин
и тебаин. Морфин и кодеин находят широкое применение в ме-
дицинской практике. А., родственные эметину, и нек-рые А.
кураре, например тубокурарин хлорид (IX), также являют-
• ся производными изохинолина.
Тубокурарин хлорид приме-
няется в хирургии. В Совет-
ском Союзе выпускаются синте-
тические препараты, заменяю-
щие этот препарат (диплацин и
парамион).
Производные хинолина. Важнейшим среди
иих является хинин (X), к-рый применяется как противомаля-
рийный препарат и в акушерской практике. Отдельную под-
группу составляют фурохинолиновые А.
Производные индола. К этой группе А. отно-
сятся в-ва нескольких типов, представителями к-рых являются:
еармин (XI); стрихнин (XII); алкалоиды спорыньи — производ-
XIII
ные лизергиновой к-ты (XIII); А., родственные иохимбину
(XIV), к к-рым принадлежит резерпин; А. группы эзерина;
алкалоиды тыквенного кураре, напр. флуорокурин (XV). При-
меняются в медицине: стрихнин как стимулятор центральной
нервной системы, А. спорыньи для стимулирования мускула-
туры матки и их гидрированные производные нак адренолитич.
препараты; резерпин — в качестве седативного (успокаиваю-
XV
НгСв--СН—;сн-сн
h2C\,^N^3x?CH2
сн2 сн2
3
XVI
Группа пиррол изид и на. В большинстве слу-
чаев А. этой группы представляют сложные эфиры оксикислот
и аминоспиртов, производных 1-метилпирролизидина (XVI).
В медицинской практике примеряется как холинолитич. и спаз-
молитич. средство выпускаемый в СССР платифиллин битар-
трат (см. Пирролизидиновые алкалоиды).
Группа имидазола. Пилокарпин — холиномиме-
тич. средство.
Группа стероидных и родственных им
алкалоидов. Практич. значение имеют гликоалкалоиды
томатов (томатин) п паслена птичьего — соласонин в качестве
сырья для производства стероидных гормонов (кортизон).
Группа пуринов. Кофеин — стимулятор централь-
ной нервной системы (XVII).
Теобромин и теофиллин —
спазмолитики, производятся
синтетически.
1 6
Н3С—N---СО
I I 7/СН
2СО 5С—N<8
I I ^сн
Н3с-N---с—
3 4 9
OCHg
XVII
XVHl
Ациклические алкалоидные амины. Эфед-
рин CeH5CH(OH)CH(NHCHa)CH8 возбуждает центральную
нервную систему. Сферофмзин
(СН3)2СНСН = CHNH(CH8)4 . NH . С (=NH)NH2 —
ганглиоблокирующее средство, выпускается только в Совет-
ском Союзе. К этой же группе относятся колхицин и близкий
к нему колхамин( XVIII), применяемый для лечения рака кожи.
Группа аконитовых алкалоидов. Кон-
дельфин, элантин, метилликаконитин иодгидрат (мелликтин)
применяются в практике для лечения нервных болезней, деле-
семин — для хирургии. Выпускаются только в СССР.
Роль и функции А. в растениях не могут считаться
выясненными; можно предполагать, что для различ-
ных А. они различны, возможно, что нек-рые А. вы-
полняют защитные функции, оберегая растения от
повреждения насекомыми или поедания травоядными,
другие являются промежуточными продуктами или
участниками биологич. синтеза (N-окиси алкалоидов),
третьи — отбросами жизнедеятельности раститель-
ного организма. В течение периода вегетации может
меняться количеств, и качеств, состав А. как в расте-
нии в целом, так и в его отдельных частях. Напр.,
цитизин в Thermopsis lanceolate содержится в семе-
нах и максимум его накопления соответствует созре-
ванию семян; известны А., к-рые накапливаются
в максимальных количествах в листьях в период наи-
более интенсивного развития надземных частей расте-
ния (элатин), другие А. содержатся в коре корней
(резерпин) и т. д.
Относительно происхождения А. до сего времени
нет общепринятой точки зрения. Было высказано
предположение, что нек-рые А. образуются в резуль-
тате превращения аминокислот; несомненно участие
в образовании, по крайней мере, части А. углеводов и
продуктов их превращения. Распространённость А.
в различных растительных семействах неоднородна:
в одних семействах большая часть видов содержит А.
(маковые, пасленовые, магнолиевые и т. д.), в других
семействах алкалоидоносы пока не обнаружены.
В алкалоидоносных растениях чаще всего находится
по нескольку А. Для близких растений среди выраба-
тываемых ими А. оказывается возможным найти А.,
119
АЛКАЛОИДЫ СПОРЫНЬИ
120
принадлежащие к одной общей группе или даже не-
скольким общим группам. Этим пользуются часто при
поисках растительного сырья для получения того или
иного А. Имеет место и обратное явление: одни и те
же А. были выделены из разных растительных се-
мейств.
В растениях, за редкими исключениями, А. содер-
жатся в виде солей органич. кислот в количестве
от 1—2% до тысячных долей % от веса высушенного
растения. При получении А. из растений приходится
решать неск. задач: экстрагировать А. из раститель-
ного материала, выделять их из экстрактов, чаще в ви-
де смесей А., выделять и очи-
щать отдельные А. Для извле-
чения А. высушенный и измель-
ченный растительный материал
подвергают экстракции (с подо-
гревом или без него) водой, под-
кисленной водой, спиртами, со-
держащими аммиак или кислоту
(чаще всего уксусную), или рас-
тительный материал подщелачи-
вают водным аммиаком, содой
или известью (что превращает
соли А. в свободные основания) и
экстрагируют А. растворителями,
не смешивающимися с водой
(дихлорэтаном, ксилолом, бензо-
лом и др.). Отдельные легколету-
чие А. получают, отгоняя их из
растительного материала вместе
с паром. Для разделения сложных смесей А., особен-
но в научно-исследовательской работе, применяют
хроматографию — адсорбционную (на А12О3) или рас-
пределительную (на целлюлозном порошке), метод
противоточного распределения и электрофорез. Для
качеств, обнаружения А. используют большое число
реактивов, способных давать с водными р-рами
солей А. осадки: фосформолибденовую, фосфорволь-
фрамовую, кремневольфрамовую к-ты, р-р иода в
иодистом калии, реактив Драгендорфа, реактив
Майера. Многие А. дают окрашивание с конц. кисло-
тами (НС1, HN03, H2SO4), реактивом Фреде, хлор-
ным железом. Для качественного обнаружения А.,
качественного анализа смесей А. с успехом приме-
няется метод хроматографии на бумаге.
Лит.: Орехов А. П.. Химия алкалоидов, 2 изд., М.,
1955; Генри Т. А., Химия растительных алкалоидов,
пер. с англ., М., 1956; Преображенский Н. А. и
Генкин Э. И., Химия органических лекарственных ве-
ществ, М.—Л., 1953; Соколов В. С., Алкалоидоносные
растения СССР, М.—Л., 1952; The alkaloids. Chemistry and
physiology, ed. by R. H. F. Manske and H. E. Holmes, v. 1—5,
N. Y., 1 950—55. А. Д. Кузовков.
АЛКАЛОИДЫ СПОРЫНЬИ (эргоалкалоиды) —
12 алкалоидов, выделенных из спорыньи Secale сог-
nutum — одной из форм паразитирующего на зла-
ках, особенно на ржи, грибка Claviceps purpurea
Tulasne. Все А. с. являются замещенными амидами
одной из двух стереоизомерных к-т — лизергиновой
кислоты или изолизергиновой, которые отличаются
конфигурацией у атома углерода, соединенного с
карбоксильной группой. Обычно А. с. рассматри-
вают как 6 пар стереоизомеров, причем левовращаю-
щие А. с., являющиеся производными лизергиновой
к-ты, имеют названия, оканчивающиеся на «ин»,
а соответствующие правовращающие А. с. — те же
наименования, но с окончанием «инин».
По строению второй части молекулы А. с. делятся
на две группы: 1) низкомолекулярная, содержит
только одну пару А. с. — эргометрин и эргометри-
нин, у к-рых один атом водорода в амиде лизергино-
вой (или изолизергиновой) к-ты замещен группой
СН(СН3)СН2ОН; 2) более высокомолекулярная, со-
держит остальные 5 пар А. с. и имеет полипептид-
ную часть, как это показано на рис.; различия заклю-
чаются в замещающих радикалах в полипептидной
Название Эмпирии, формула Ri R» Т. пл., °C (с разл.) [al (X 5461 А, хлороформ)
Эргокристин Эргокристинин CssHngOsNs -СН(СН,)г —СН3СеН5 165-170 239 - 217" + 460"
Эрготамин Эрготаминин СззНайОэКа -сн, -СН3СвН5 213-214 252 - 192" + 462"
Эргокриптин Эргонриптинин Cai>H4iO5N5 -CH(CH3)2 -СН2СН(СН,)2 212—214 240—242 — 226" + 508"
Эргонориин Эргокорнинин CS1H39O5N5 -СН(СН,)2 -СН(СНа)3 190—200 228 — 226* -f-512*
Эргозин Эргозинин CsoH3705N5 -сн, —СН3СН(СН3)2 228 228 — 193* + 522*
Эргометрин Эргометр инин Ci9H38O2N3 - — 159-163 195-197 - 160° + 520°
Содержание алкалоидов в спорынье колеблется от
0,001 до 0,1—0,2%; наряду с алкалоидами из спорыньи
выделено много других веществ, в том числе белки,
углеводы и липоиды. В связи с практическим приме-
нением ряда А. с. спорынью специально культиви-
руют и т. о. добиваются повышения содержания А. с.
ДО 0,7%.
Свойства всех А. с. в общем сходны. Левовращаю-
щие алкалоиды при кипячении со спиртовым раство-
ром щелочи переходят в правовращающие; обратное
превращение — при действии спиртового р-ра фосфор-
ной к-ты или разб. щелочи. При щелочном гидролизе
А. с. размыкаются амидные связи и образуются лизер-
гиновая (или изолизергиновая) к-та и продукты рас-
пада полипептидной части молекулы. Среди послед-
них, помимо аммиака, найдены: L-пролин, L-фенил-
аланин, L-лейцин, L-валин, пировиноградная и диме-
тилпировиноградная к-ты. При гидролизе эргометрина
образуются лизергиновая к-та и 2-аминоиропанол.
Строение лизергиновой к-ты подтверждено синтезом
(Вудворд с сотр., 1954).
Фармакология, активность левовращающих А. с.
значительно выше, чем у правовращающих, хотя
характер один и тот же. Действие на организм млеко-
питающих проявляется в основном в двух направ-
лениях: сокращающее действие на гладкую мускула-
туру (матка, кровеносные сосуды, зрачок и пр.)
и угнетение симпатич. функций вегетативной нервной
системы (противоположно адреналину). А. с. являются
антагонистами серотонина; вызывают цветные галлю-
цинации и временные психич. расстройства. В медици-
не применяются эрготамин, эргокорнин (эрготоксин) и
эргометрин (в виде солей), последний в 2 раза актив-
нее и в 3—4 раза менее токсичен, чем два первых
(по способности вызывать сокращение матки). В от-
ношении симпатолитич. действия картина обрат-
ная — эргометрин почти не активен, а эргокорнин и
эрготамин — наиболее сильно действующие. Все А. с.,
кроме эргометрина, искажают, действие адреналина.
В медицине находит применение также сумма А. с.
Лит.: Орехов А. П., Химия алкалоидов,2 изд., М., 1955,
с. 627—640; Генри Т. А., Химия растительных алкалоидов,
121
АЛКАЛОИДЫ ТЫКВЕННОГО КУРАРЕ
122
пер. с англ., М., 1956, с. 547—565; К о г n I е 1 d Е. С. [а. о.],
J. Am. Chem. Soc., 1954, 76, №20, 5256. А. Е. Васильев.
АЛКАЛОИДЫ ТЫКВЕННОГО КУРАРЕ — ал-
калоиды, входящие в состав стрельного яда кураре,
растительные экстракты, изготовляемые индейцами
тропич. областей Южной Америки; подразделяются
на три вида, отличающиеся по упаковке и составу:
трубочный (готовится в бамбуковых трубках), гор-
шечный (в глиняных горшках) и тыквенный, или
калабаш-кураре (в пустых тыквах или калебассах).
Растения, применяемые для изготовления яда, точно
не известны и различны в отдельных случаях. Дей-
ствующим началом всех трех видов яда являются
алкалоиды, найденные также в нек-рых растениях,
произрастающих в тропич. странах, что позволяет
судить о том, какие именно растения применяются
при изготовлении кураре. Трубочное и горшечное
кураре содержат гл. обр. изохинолиновые алкалоиды
как третичные (1-курин, d-хондрокурин, d-изохондро-
дендрин и др.), так и четвертичные (напр., rf-трубокура-
ринхлорид). Растительные источники тех же алкалои-
дов — различные виды семейства Menispermaceae,
особенно Hondrodendren То-
mentosum. Тыквенное кура-
ре имеет в общем тот же
характер физиологического
действия, что и первые два
вида. Большинство А. т. к.
содержится в коре растений
видов Strychnos, гл. обр
Strychnos toxifera.
Известно св. 60 А. т. к.,
к-рые представляют собой
третичные и четвертичные
основания; все они относятся
к индольным производным,
к-рые, согласно спектраль-
ным данным, могут быть
разделены на 8 групп в за-
висимости от характера за-
мещения в индольной систе-
ме: 1) С — калебассин; С-ал-
калоиды A, F, J, X, Y; С-ку-
рарип Н, каракурин VII (см. I); 2) С-алкалоиды Y,
L, Q; каракурины VIII и IX; мелинонины А, В, J; С-ма-
вакурин, гарманхлорметилат, токсиферин Ш, крип-
токурин (см. II); 3) С-курарины; С-алкалоиды G и Е;
С-гуйанин, каракурин V (см. III); 4) С-токсиферин I,
С-дигидротоксиферин, С-алкалоиды Н и S, каракурин
VI (см. IV); 5) С-алкалоиды В, С, D, R, каракурины I,
II и III (см. V); 6) С-флуорокурин, С-флуорокуринин,
псевдофлуорокурин (см. VI); 7) С-ксантокурин, мели-
нонины Е, G, М, федамацин (см. VII); 8) С-калебас-
синин, С-алкалоиды М и UB (см. VIII):
По размерам молекулы А. т. к. распадаются на две
группы: «мопомолекулярные», в молекулах к-рых
имеется ок. 20 атомов С и 2 атома N, и «бимолекуляр-
атомов С и 4 атома N.
иые», имеющие примерно 40
В первую группу входят флуорокурин, флуорокура-
рин, мавакурин и др., во вторую — С-дигидротоксифе-
рин, каракурин
(V) и др.
С- флуармурин
С-алкалоид Т
мелиноник А
Рис. 2. Генетическая связь алкалоидов тыквенного кураре.
Все А. т. к. чрезвычайно чувствительны к к-там.
В зависимости от силы действующей кислоты происхо-
дит более или менее глубокая изомеризация. Напр.,
из С-дигидротоксиферина в различных условиях
образуются: С-алкалоид D, С-курарин, С-калебассип,
а из последнего при повышении силы действующей
кислоты — изокалебассин. Важное взаимное превра-
щение могут претерпевать мавакурин и флуорокурин.
По строению А. т. к. генетически близки к алкалои-
дам ряда стрихнина и могут быть синтезированы из
продукта распада последнего—альдегида Виланда —
Гумпиха (напр., С-токоферин I) (рис. 1). Генетич.
связь разных А. т. к. между собой может быть пред-
ставлена через гипотетич. алкалоид «А» (рис. 2).
Разделение А. т. к. производят с помощью хромато-
графии на бумаге. Алкалоиды с большим значением
Rj мало токсичны и мало физиологически активны.
Найдено также, что сильно действующие алкалоиды
имеют 2 четвертичных N-атома, то есть они «бимоле-
кулярны».
По своим фармакология, свойствам А. т. к. принад-
лежат к группе мускульных релаксантов. Они пара-
лизуют окончания двигательных нервов, но не препят-
ствуют передаче возбуждения, то есть вызывают по-
терю движения без потери чувствительности. Исполь-
зование больших доз приводит к смерти в результате
остановки дыхания (паралич дыхательных мышц).
Из природных кураре-активных препаратов в ме-
дицине применяется только тубокурарин. Многие
А. т. к. значительно активнее и токсичнее тубокура-
123
АЛКАНЫ — АЛКИЛИРОВАНИЕ
124
рина, напр. С-алкалоид G в 120 раз, а С-алкалоид Е
в 250 раз эффективнее тубокурарина. 1 г С-алкалоида
Е парализует 100 000 мышей. Токсичность А. т. к.
весьма различна и колеблется от 0,3 мкг до 20 мг на 1 кг
веса мыши. Крупными достоинствами А. т. к. явля-
ются большая терапевтия, широта и практически
полное отсутствие побочного действия. Наиболее при-
годны для кураризации С-курарин, С-токсиферин 1,
С-алкалоиды Е, G, К. Отмечена связь между ток-
сичностью А. т. к. и их коэфф, распределения между
органич. фазой и водой.
Лит.: Karrer Р., Nature, 1955, 176, № 4476, 277;
К а г г е г Р. und Schmid Н., Angew. Chem., 1955, 67,
№ 14/15, 361; Karrer P., [u. а.], там же, 1958, 70, №21,
644; Karrer P., Bull. Soc. Chim. France, 1958, fasc. 1, 99.
A. E. Васильев.
АЛКАНЫ — CM. Предельные углеводороды.
АЛКЕНЫ (этиленовые углеводороды) — см. Не-
предельные углеводороды.
АЛКИЛ — обобщенное название одновалентных ра-
дикалов ряда насыщенных жирных углеводородов.
Часто А. обозначают символом R, иногда —символом
Aik. Различают первичные, вторичные и третичные
алкильные радикалы. Примером первичных А. может
служить метил СН3, вторичных—изопропил (СН3)2СН,
третичных—mpem-бутил (СН3)2С. Это обозначение со-
храняется и в случае соединений, в к-рых А. связан
С функциональными группами. я. Ф. Комиссаров.
АЛКИЛАТ (алкилбензин) — смесь изопарафиновых
высокооктановых углеводородов; по составу близок
к технич. игооктану, 0,698—0,715, начало кипения
54—89°, 50% выкипает при 105—113°, 90% — при
114—140°, конец кипения 171 —188°; давление паров
115—208 мм рт. ст. при 38°; высшая теплота сгора-
ния 11 200 ккал/кг; октановое число: по моторному
методу 87,7—95, по методу 1-С с 4 мл/кг этиловой
жидкости 105—107; сортность по методу 3-С 143—153.
А. обладает высокой приемистостью к этиловой жид-
кости. По физико-химич. и антидетонационным свой-
ствам — ценный компонент высокооктановых мотор-
ных топлив и широко применяется при их произ-ве.
Добавление 40% А. к бензину (содержащему 4 мл/кг
этиловой жидкости) с октановым числом 88,3 увели-
чивает его октановое число на 7 единиц. А. получают
алкилированием природных и промышленных углево-
дородных газов, из парафиновых углеводородов при-
родных и попутных газов обычно используют изобутан
и изомеры пентана и октана. Необходимые для алки-
лирования непредельные углеводороды — бутилены,
пропилен и амилен — получают из тех же газов путем
их пиролиза и дегидрогенизации; для произ-ва А. ши-
роко используют бутано-бутиленовые фракции газов
крекинга. При алкилировании в качестве катализа-
торов применяют H2SO4 и HF. А. не содержат непре-
дельных углеводородов и являются стабильным мо-
торным топливом.
Алкилированием бензола пропан-пропиленовой фра-
кцией газов крекинга получают другой ценный ком-
понент моторного топлива — алкилбензо л, к-рый
представляет собой смесь различных алкилированных
бензолов (этилбензол, изопропилбензол и др.). Алкил-
бензол характеризуется след, величинами: темп-ра
начала кипения 105°, конец кипения 180°, октановое
число с 4 мл/кг этиловой жидкости 99. Алкилбензолы
улучшают антидетонационные свойства базовых бен-
зинов как на бедных, так и на богатых смесях. Наряду
с этим алкилбензол утяжеляет фракционный состав
топлива и ухудшает нек-рые его эксплуатационные
свойства: понижается теплота сгорания, увеличи-
ваются гигроскопичность и токсичность топлива, по-
вышается склонность топлива к нагарообразованию
и самовоспламенению.
Лит.: Моторные топлива, масла и жидкости под ред.
К. К. Папок и Е. Г. Семенидо, 3 изд., т. 1, М., 1957.
В. В. Панов.
АЛКИЛИРОВАНИЕ — реакция введения алкиль-
ной или аралкильной группы в органич. соединение.
В зависимости от того, с каким атомом образуется
новая связь, различают С-, О-, N- и S-алкилирование,
напр. соответственно:
СвНв + СН2=СН2 — CeH5CHsCH3
С2П5ОН + SO2(OC2H5)3 — С3Н5ОС2Н5 + C2H5O(OH)SO2
C6H5NH2 + СН3С1 — CeH5NCH3H2 + ci
СН3—S—СН3 + CH3J — (CH3)3S + J
К реакциям А. может быть также отнесено образова-
ние связи С—Me, напр. тетраэтилсвинца (С2Н5)4РЬ,
этилмеркурхлорида C2H5HgCl и др. На выход и
скорость образования продуктов А. влияют многие
факторы: 1) концентрация (в газовой фазе — повыше-
ние давления) и соотношение компонентов; избыток
одного из них сдвигает равновесие и увеличивает
выход конечного продукта. В нек-рых случаях полез-
на низкая концентрация одного из компонентов,
напр. олефинов при синтезе нефтяного алкилата из
изопарафинов и олефинов, т. к. последние могут
полимеризоваться; 2) для повышения скорости А.
обычно применяют катализаторы, напр.: H2SO4, Н3РО4,
НС1, HF, А1С13, FeCl3, BF3 и др. А1С13 широко при-
меняется для создания связи С—С (см. Фриделя —
Крафтса реакция). 3) Введение алкильной группы
происходит при разных температурах. В зависимости
от нее может происходить также и изомеризация.
Напр., при алкилировании анилина метанолом в при-
сутствии H2SO4 при темп-pax до 250° образуется
N-алкилпроизводное; при дальнейшем повышении
темп-ры происходит миграция метильной группы
в ядро: CeH5NH2 + СН3ОН ?^CeH5N(CH3)22^-^
—>CH3HN—CeH4—CH3^°(CH3)2CeH3NH2. Для мно-
гих случаев известен температурный коэффициент
реакции, равный «г 2,2, например для реакции
CaHjJ -j- В качестве ре-
акционной среды иногда применяют нитробензол
СС14, СНС13.
Реакции А. могут протекать либо по механизму замещения,
либо по механизму присоединения. Механизм реакции А.,
а также ее скорость определяются: 1) строением алкилирующего
агента, 2) строением и основностью алкилируемого в-ва,
3) полярностью и сольватирующей способностью растворителя,
4) наличием катализаторов (см. Реакционная способность). Меха-
низм реакций первого типа заключается в передаче алкильной
группы от алкилирующего агента нуклеофильному в-ву или иону
в виде иона карбония (асинхронно, Sj^l, или S^2 механизм),
или в активированном комплексе (синхронно, S^2 механизм):
В-^Alkr'x—- B:A|k-HX
Алкильная группа, как правило, вступает по месту наибольшей
электронной плотности алкилируемого в-ва. Однако при А.
может иметь место перенесение нуклеофильного реакционного
центра (А. фенолов и енолят-ионов):
СН3—С=С—COOR + CH3J—*-СНа—С—СН—COOR +J
гр I II I
“ОН о СНз
В свою очередь передача алкильной группы может сопро-
вождаться перенесением реакционного (электрофильного)
центра в алкилирующей частице, вследствие чего строение
алкильной группы изменяется (А. изобутил- и фурфурилхло-
ридами):
/СН3
аС-СН3
СНз
+ HCI
125
АЛКИЛИРОВАНИЕ — АЛКИЛНАФТАЛИНЫ
126
М., 1959; Streitwieser A. Jr., Chem. Rev., 1956, 56,
№ 4, р. 571 — 752; Kirk, v. 1, N. Y., 1947, p. 532—50;
n in er J., Huisgen R., Angew. Chem., 1960. 72. № 9,
294. С. В. Bumm.
АЛКИЛНАФТАЛИНЫ — производные нафталина,
молекуле к-рых содержатся алкильные радикалы;
известны моно-, ди- и полиалкил-
Иа) нафталины. У А. заместитель мо-
2(р) жет находиться в положении 1 (а)
или 2 (Р). Физико-химич. свойства
нек-рых А. приведены в табл.
А. (кроме метилнафталина) могут быть получены
конденсацией нафталина с олефинами, спиртами и
галогеналкилами в присутствии катализаторов (H2SO4,
А1С13, ZnCl2, BF3). -Алкилирование нафталина спир-
тами в присутствии А1С13 и BF3 проводят в инертном
растворителе (например, в лигроине) при
- \ , - 40—100°; при этом образуется смесь пример-
R сн2-сн2 -RCH2-CH2: RCHa— CH2 + RH —RCH2CH3+R н0 равных количеств моно- п диалкилнафта-
линов.
В реакциях замещения алкилирующими агентами могут слу-
жить галогеналкилы, алнилониевые соединения, алкильные
эфиры неорганич., а в нек-рых случаях и органич. к-т, алифа-
тич. диазосоединения.
Ко второму типу реакций А. относятся все реакции при-
соединения олефинов, а также диазоалканов. В зависимости от
типа присоединения к олефину различают А. по нуклеофиль-
ному и по электрофильному механизму. А. ароматич. (реакция
Фриделя — Крафтса) и парафиновых углеводородов, спиртов,
воды (гидратация) в присутствии сильных к-т— А1С13, BF3,
H2SO4), а также галогеноводородных к-т (напр., гидрохлори-
рование) осуществляется по нуклеофильному механизму. Ка-
талитич. действие сильных к-т заключается в увеличении элек-
трофильности алкилируемого в-ва либо в превращении оле-
фина в ион карбония. При этом введение в молекулу олефина
заместителей (напр., Aik или OR), повышающих электронную
плотность в переходном состоянии, ускоряет реакцию, проте-
кающую по нуклеофильному механизму. А. по электрофильному
механизму происходит путем присоединения нуклеофильного
агента (напр., карбаниона) к олефину:
Скорость алкилирования по электрофильному механизму воз-
растает, если в молекуле олефина присутствуют электронно-
акцепторные заместители (—C=N, — COOR, — NO.), повы-
шающие (в результате сопряжения) электрофильность двойной
связи:
rS^CHo^CH^C-OR—ro-ch2— ch-c-or
IQ II
сг о
RO—CH2-CH-C-OR+HOR—*- RO—CHa— CH2—C—OR
II II
О о
Реакция А. по электрофильному механизму приобретает боль-
шое значение в химии металлоорганич. и фторорганич. соеди-
нений. Несмотря на большое количество исследований, меха-
низм А. олефинами изучен лишь в общих чертах.
А. широко применяется в пром-сти: для синтеза
высокооктанового топлива и его отдельных компонен-
тов, напр. изоктана и других полимерных материалов
(полиэтилен, полипропилен), для получения гомо-
логов бензола, яапр. кумола, этилбензола и др.,
жирно-ароматич. кетонов, простых и сложных эфи-
ров, аминов, а также соединений аммония и суль-
фония. См. также Метилирование.
Лит.: Реутов О. А Теоретические проблемы орга-
нической химии, М., 1956; И н г о л ь д К. К., Механизм
реакций и строение органических соединений, пер. с англ.,
S.
в
Место алкильного радикала в молекуле нафталина
при алкилировании зависит от температуры реакции
и катализатора; так, выше 200° происходит образо-
вание только ^-замещенных, при низких температу-
рах образуется смесь а- и а-А. На направление заме-
щения влияет катализатор, напр. нафталин с С6НПОН
в присутствии А1С13образует I, в присутствии BF3—П,г
I ц С-б^н
А. — ценные промежуточные продукты для синтеза
красителей и вспомогательных веществ в текстиль-
ной пром-сти. Сульфокислоты А. находят применение
в качестве смачивателей, моющих средств и эмульга-
торов, напр. некали А и БХ (смесь натриевых солей
моно-, ди- и три-вторично-бутилнафталинсульфокис-
лот). Окислением диалкилнафталинов (2,6-, 2,7- и
1.4-) получают соответств. дикарбоновые к-ты нафта-
лина, применяющиеся в синтезе кубовых красителей.
В. А. Пучков.
Название • Положе- ние заме- стителя Характер алкиль- ного ра- дикала Т. кип., °C/jhjh рт. ст. .20 “4 „20 Т. пл. пикр эта. »С
Этилнафталины* С10Н7СоН5 1* - 256,5/756 112-116/8 1,0111 1.6089 (14,2") 98,5
2** 252/760 117—118/10 0,9958 (15°) 1.6028 (15") 76-77
Пропилнафталины 1 к- 274—275/760 — — 140—141
2 н- 277-279/760 — — 89-90
Бутилнафталины С1оН7С4Нй 1 н- 281—283/760 151-152/14 - 104—105
2 н- 283-285/760 125—130/13 — — 71-73
1 трет 287—289/760 0,9629 1,5726 96
2 трет 274—276/760 0,9687 1,5768 —
Диэтилнафталин С10Нб (CgHs)? 1,4 - 138—139/8 0,993 1,601 82-83
Дипропилнафталин СюНв (СзН7)з 2,7 изо- 278—280/760 0,9683 1,5701 86
• Т. пл. 15". ** Т. пл. 7,5°,.
127
АЛКИЛСЕРНЫЕ КИСЛОТЫ - АЛКИЛФЕНОЛЫ
128
АЛКИЛСЕРНЫЕ КИСЛОТЫ—см. Алкилсулъфаты.
АЛКИЛСУЛЬФАТЫ — алкиловые эфиры серной
к-ты. Известно два ряда А.: кислые эфиры (I), наз.
алкил се р н ы м и кислотами, и средние
(нейтральные) эфиры (II), наз. диалкилсуль-
фатами:
I. CnII2n4.,0 SO2OH; II. (CnII2n 4O)2SO 2
Алкилсерные к-ты — сиропообразные гигроскопич.
жидкости, легко растворимые в воде и обладающие
свойствами сильных к-т; их соли хорошо кристал-
лизуются иногда с одной или несколькими молекула-
ми воды, напр.:
CH3OSO3ONa • Н2О; (CH3OSO3O)2Sr • 2Н2О
При перегонке алкилсерные к-ты разлагаются (100°)
с образованием диалкилсульфатов и H2SO4:
2CH3OSO3H — (CH3O)2SO2 -f- HUSO,
К- и Na-соли метилсерной к-ты разлагаются выше
200° на пиросульфат K2S2O7 и диметиловый эфир
СН3ОСН3; соли Са и Ва разлагаются с образованием
диметилсульфата и нейтральных сульфатов (CaSO4,
BaSO4). Алкилсерные к-ты идентифицируют по
темп-рам плавления аммонийных солей (табл. 1):
Таблица 1
Кислоты Мол. вес. Т. пл., °C Т. пл. ЬГН4-соли, °C
Метилсерная CH3OSO3H 112,10 —30 135
Этилсерная C2H5OSO3H 126,13 88 (разл.) 97
к-Пропилсерная C,H7OSO3H 140,16 — 132
Алкилсерные к-ты могут быть получены действием
серной к-ты на соответствующий спирт:
AlkOH 4 H»SO4 — Aik—О—so3OH 4- Н2О
От избытка H2SO4 алкилсерные к-ты отделяются
в виде бариевых или кальциевых солей, к-рые, в от-
личие от BaSO4 и CaSO4, хорошо растворимы в воде;
другой способ получения — присоединение H2SO4
к олефинам, напр.:
СН2=СН3 4 H3SO4 —- СН3СН2О—SO3OH
Алкилсерные к-ты легко гидролизуются водой с обра-
зованием спирта и H2SO4. При действии спирта на
(I) образуются простые эфиры: С2Н5О—SO2OH +
-I- С4Н9ОН -> С2Н5ОС4Н9 + H2SO4
Этилсерная к-та — промежуточный продукт при
синтезе этилового спирта из этилена (промышлен-
ный способ). Натриевые соли алкилсерных к-т, содер-
жащих не менее 8 атомов С, применяются как моющие
средства. Низшие алкилсерные к-ты служат для про-
изводства диметил- и диэтилсульфатов; являются
также катализаторами этерификации. Алкилсерные
к-ты и их соли являются алкилирующими агентами,
применяются для алкилирования в мягких условиях
аминов и фенолов.
Диалкилсульфаты (II) — маслообразные бесцветные
жидкости, не смешивающиеся с водой; очень ядовиты,
при попадании на кожу вызывают поражения. Свой-
ства нек-рых диалкилсульфатов приведены в табл. 2.
Один из главных способов получения диалкилсуль-
фатов — действие SO3 на простой эфир:
С2Н5ОС3Н5 + SO3 — (C2H5O).,SO3
Другой способ — взаимодействие SO3 со спиртом*
2СН3ОН + 2SO3 — (CH3O)3SO3 4 H2SO4
Основное применение диалкилсульфатов — алкили-
рование органич. соединений. Процесс идет в 2 ста-
дии; первая протекает легко н гладко и заканчивается
уже ок. 30°. Напр., для алкилирования фенолов:
ArONa + (CH3O)3SO3 — АгОСЫз 4 CH3OSO3Na
Вторая стадия с участием кислого эфира серной кисло-
ты происходит в более жестких условиях (110—120°):
ArONa 4 CH3OSO3Na ArOCH3 4 NaSO,
Для диэтилсульфата темп-ры алкилирования соответ-
ственно 20—55° и ок. 145°. Аналогично происходит
алкилирование аминов:
ArNH3 4 (CH3O)3SO3 _ ArNHCH3 4 CH3OSO3OH
При нагревании первичного амина с диметилсульфатом
до более высокой темп-ры происходит алкилирование
образовавшегося вторичного амина:
ArNHCH3 4 (CH3O)2SO2 — ArN(CH3).> 4 CH3OSO3OH
Регулируя темп-ру, процесс направляют в сторону об-
разования преимущественно моно- или диалкильных
производных. С третичными аминами и гетероциклич.
азотсодержащими соединениями диалкилсульфаты
образуют соли четвертичных оснований, напр.:
Ацетеленид калия с диалкилсульфатами образует
соответствующий замещенный ацетиленовый углево-
дород, напр.:
(CH3CH3CH3O)3SO3 4 NaClz CH —
- СН3СН3СН3СН4СН 4 CH3CH3CH2OSO2ONa
Диалкилсульфаты легко алкилируют щелочные соли
неорганич. к-т; так, напр., диметилсульфат с KJ
с хорошим выходом дает иодистый метил:
(CH3O)3SO3 4 KJ - CH3J 4 CH3OSO»OK
С увеличением длины и усложнением скелета алкиль-
ного радикала алкилирующая способность А. умень-
шается (падает). См. также Диметилсульфат, Диэтил-
силъфат.
Таблица 2 Дит.: Вор ож цо в Н. Н., Осно-
Диалкнлсульфаты Формула T. КИП., CC/MJW t nD
Диметилсульфат (СН3О)3 so3 188,3—188,67760 1.33.32 (15°) 1,3874 (207
Метилэтилсульфат CH3OSO,OC»H5 198—2007'42 1,228 (16,5°) 1,399 (16,57
85715
Диэтилсульфат (С2Н5О)2 so2 210/760 1,1842 (15’) 1,4025 (157
Этилпропилсульфат Этилизопропилсульфат J С2Н5О SO3OC3H7 107-108718 105718 1,140 (16,5“) 1,143 (217 1,411 (16,57
Дипропилсульфат J (C3H7O)2SO2 121720 1,1064 (207 1,4135 (207
Диизопропилсульфат 106718 (с разл.) — —
Этилбутилсульфат j C.,H5O SO2OC4He 117—118720 1,112 (187 1,415 (18")
Этилизобутилсульфат 108713 1,098 (287 —
Этилизоамилсульфат CH5O SO2OC5Hn 127-128715 1,079 (207 —
Диизобутилсульфат (C7HeO)2 so2 133-134719 1,042 (237 1,415 (207
Диизоамилсульфат (C5HnO)2SO3 145-155720 — —
вы синтеза промежуточных продуктов
и красителей, 4 изд., М., 1955; Kirk,
v. 13, N. Y., 1954, р. 506; U 1 1 mann,
3 Aufl., Bd 3, Munchen—В., 1953, S.
311. В. А. Пучков.
АЛКИЛФЕНОЛЫ — фенолы,
содержащие в молекуле алкиль-
ные радикалы. Простейшими А.
являются метилфенолы (о-, м- и
п-крезолы) и диметилфенолы (кси-
ленолы). В последнее время зна-
чение приобрели А. с длинной,
прямой или разветвленной ал-
кильной цепью. Физич. свойства
нек-рых сложных А. приведены
в таблице.
На основе замещенных фено-
лов производятся: 1) присадки,
129
АЛКИНЫ — АЛЛЕНЫ
130
Название Т. пл., °C Т. кип., °C 20 пр
760 мм 20 мм
4-н-Бутилфенол 92 248 138
4-втор-Бутилфенол 62 — — 0,969 (25”) 1.5150
4-трет-Бутилфенол 100 237 130,5 0,908 (114°) 1,5165 (22°)
4-трет-Амилфенол 93 262,5 138,5 0,962 (20°) —
2,4-Диметил-6-трет-бутилфенол 22,3 249 131 1,5183
2,6-Диметил-4-трет-бутилфенол 82,4 248 135 0,959 (26,7°) —
4-Диизобутилфенол 84 279 163 — —
2-Метил-4,6-ди~трет-бутилфенол 51 269 149,5 0,940 (26,7°) —
4-Метил-2 ,6-ди-трет-бутилфенол . . • .... 70 265 147 1,048 (20°) —
2,4, 6-три-трет-Бутилфенол . . • • 131 278 158 0,864 (26,7°) —
улучшающие качество моторных топлив, напр. (I), I действие спиртов на ангидриды двухосновных к-т или
трансформаторных и смазочных масел, папр. (II); же на эпоксисоединения с раскрытием цикла:
Он О(СН2СН20)пН
(CHJ.C
сн3
С(СН3)3
III
СН„СО. СИ.СООН
| >0 + ROH — |
ClbCO/ ch2coor
СНо—сн2
\ / +ROH~HOCH2CH3OR
превращение циануксусной кислоты в малоновый
эфир:
CNCH3COOH + 2С2Н5ОН СН2(СООС2Н5)2
Я. Ф. Комиссаров.
ОСН2СООН
HgCO NOa
У
и соединения других
двух кумулированных
связей, общей ф-лы
представитель — ал-
2) поверхностно-активные в-ва (полиоксиэтилирован-
ные А., Ш), применяющиеся в качестве смачиваю-
щих, эмульгирующих и моющих в-в; 3) стимуляторы
роста и гербициды (алкил- и арилфеноксиуксусные
кислоты, например (IV); 4) синтетические души-
стые вещества, обладающие запахом мускуса, напри-
мер (V); 5) алкил- и арилфеноло-формальдегидные
смолы.
Получают А. алкилированием фенолов спиртами
или олефинами в присутствии серной кислоты при на-
гревании, размешивают при этой температуре в те-
чение часа, дают охладиться и разбавляют водой.
После отделения водно-кислотного слоя алкилат
промывают раствором поваренной соли и оставля-
ют для кристаллизации конечного продукта. Превра-
щение фенола и изобутилового спирта достигает
90-95%.
Лит.: Huntress Е. Н., Mulliken S. Р., Iden-
tification of pure organic compounds, N. Y., 1941; Par-
dee W. A., Weinrich W., Ind. Eng. Chem., 1944,
36, № 7, p. 595—603; Исагулянц В. И., Хим. пром.
1958, №2, с. 20—27; Ш в а р ц А. и П е р р и Д ;к., Поверх-
ностно-активные вещества, их химия и технические приме-
нения, пер. с англ., М., 1953. В. А. Пучков.
АЛКИНЫ (ацетиленовые углеводороды) — см. Не-
предельные углеводороды.
АЛКОГОЛИЗ — реакция обменного разложения со
спиртами, напр. взаимодействие хлористого ацетила
с этиловым спиртом с образованием этилацетата:
СН3СОС1+ CjHsOH ->СН3СООС2Н5+ НС1. А., как
и гидролиз и аммонолиз, протекает по схеме нуклео-
фильного замещения и представляет собой типичную
сольволитич. реакцию (см. Сольволиз)', при этом спир-
ты играют такую же роль, как вода в гидролитич. про-
цессах. Примером А. может также служить переэте-
рификация сложных эфиров при действии избытка
спирта: СН3СООСН3 CjHjOH -> СН3СООС3Н, %-
-ф- СН3ОН. Известны и многие другие реакции, к-рые
могут рассматриваться как процессы А., напр.
3 К. X. Э. т, 1
АЛЛЕЛОТРОПИЯ — см. Таутомерия.
АЛЛЕНЫ — углеводороды
классов, содержащие систему
углерод-углеродных двойных
Ri\ /Н3
4С=С\ . Простейший
R2z
лен СН2—С=СН2 (т. пл. —146°, т. кип. —32°).
Наиболее общим методом получения А. является дп-
галогенирование а,Р-дигалогеналлильных соедине-
ний цинком или магнием:
(X — галоген)
X X
Аллен может быть получен пиролизом дикетена в мед-
ной трубке:
сн2=с-сн2
—I..I -----► сн.,=с=сн2 + со»
0—0=0 550”
а его гомологи — действием Na или Mg на 1,1-дибром-
циклопропаны, к-рые, в свою очередь, образуются
при взаимодействии олефинов с бромоформом в при-
сутствии оснований (см. Карбены)'.
СВг2
\с=с<(+снвгз =5^ >С=С=С<^
двойные связи в олефинах.
Плоскости, проходящие через оси л-электронных об-
лаков, образующих каждую из двойных связей в
А., взаимно перпен-
дикулярны (см. рису-
нок). Вследствие этого
реакции по двойным
связям в А. происхо-
дят независимо друг
от друга и с теми
же реагентами, что и
Направление присоединения определяется правилом
Марковникова. В том случае, если при одном из кон-
цевых атомов углерода нет заместителей, он подвер-
гается атаке в первую очередь:
СН2=С=СН2 + Н3О — [СН3=С(ОН)СН3] — СН3СОСН3
(СН3)»С=С=СН2 + Н2О — [(СН3)2С=С(ОН)СН3) —
- (СН3)2СНСОСН3
СН2=С=СН2 CH2=CBrCH3 SS- С113СВгаСН3
131
АЛЛЕНЫ — АЛЛИЛЕН
132
и т. д. Простые А. легко полимеризуются с образова-
нием, в частности, производных циклобутана:
2(СН3) 2С=С=СН2
(СН3)»С=С-СН2
I I
(С Н 3)26=0—он»
При нагревании или при действии кислых катализа-
торов алкилзамещенные А. переходят в сопряженные
диены. Кислые и основные катализаторы вызывают
перегруппировку в ацетилены, при этом сам А. пре-
вращается в метилацетилен:
n Н* > о —Н
СН3—С=С=СН2^=СН3-С-СН=СН2^=±1СНг=С-СН=СН2
U сн3 сн3
—н
СН3-С=С=СН^=±:СН3-СН-СгСН—-сн3-сн-с=с
сн/-~н снэ сн3
6
СН2=С=СНг—»~СН3-СНСН
С другой стороны, ацетиленовые производные, напр.
хлориды или спирты, способны превращаться в со-
единения алленового типа:
СНз\ On Г1 СНз\
>-с^сн ТО С=-С=СНС1
CIlZpi он,/
с„н5
С-С=С-СсИ5
С=С=С—С0Н5
сии5/ J...
с=сп-с—сен5
С(Н5/ II
Арилзамещенные А. в кислой среде образуют индены:
СН=С=СН2
Исходя из тетраэдрич. конфигурации углерода, Вант-
Гофф предсказал, что А. с различными заместителями
у каждого из концевых атомов углерода алленовой
системы должны существовать в оптически изомерных
формах:
Это предположение было экспериментально подтвер-
ждено, во-первых, получением правовращающего и
левовращающего изомеров соединения I путем дегид-
ратации спирта II в присутствии соответственно d-
или Z-камфор-Ю-сульфоновой к-ты и, во-вторых,
разделением на антиподы соединения III дробной кри-
сталлизацией его бруциновой соли:
(«) С[0Н; ^ /CioHyfa) (a) ^СюН? (а)
С-СН=С — С=С=С
С«н^он Чсе115 CoIlZ чс0н5
II 1
(а) С)(Н7 СООСН3СООН
С=С=<^
C(jll5X ^Collg
ш
Лит.: Organic chemistry, ed. Н.Gilman, v. 1,2 ed.,N. Y.—
1л., 1943; Royals E. E., Advanced organic chemistry, N. Y.,
1954; Doering W. E., La E 1 a m m e P. M., Tetrahedron,
1958, 2, № 1/2, p. 75—9; Темникова T. И., Курс тео-
ретических основ органической химии, Л., 1959, с. 252.
АЛЛИЛ ХЛОРИСТЫЙ (З-хлорпропен-1, аллилхло-
рид) СН2=СНСН2С1, мол. в. 76,53 — бесцветная
подвижная жидкость с резким запахом; т. пл.
—136,4°; т. кип. 45,1°; d|° 0,9379; Пр 1,4154; теплота
сгорания 440,9 ккал/моль’, теплота парообразования
6 940 ккал/моль’, давление пара (мм рт. ст.): 151,8
(5°); 362,5 (25°); 761,5 (45°); 1060,0 (55°); уд. теплоем-
кость (жидк.) 0,3 кал/г (30°); пределы воспламеняе-
мости смеси паров с воздухом: 3,28—11,19 об.%.
В воде А. х. растворяется 0,36 е/100 г (20°); смеши-
вается во всех отношениях с обычными органич. рас-
творителями; с водой, спиртом, муравьиной к-той обра-
зует азеотропные смеси.
А. х. легко вступает в реакции присоединения по
месту двойной связи: атом хлора очень реакционно-
способен и легко обменивается па различные группы
(ОН, NH2, CN и др.); при 500° хлор замещает водород
с образованием смеси дихлорпропенов С3Н4С1г; при
20° присоединяется хлор, образуется 1, 2, 3-трихлор-
пропан; аналогично присоединяется бром; с НС1
образуется 1,2-дихлорпропан, с HJ — пропилиодид, с
Mg в среде эфира — гексадиен-1,5. Технически важным
является присоединение к А. х. хлорноватистой к-ты
с образованием дихлоргидринов: СН2С1СНОНСН2С1
и СН2ОНСНС1СН2С1. А. х. получают хлорированием
пропилена при 500—550°:
СНа=СНСН3 + С13 - СН2=СНСНзС1 + НС1
в качестве побочных продуктов образуются другие
хлорпроизводные пропилена. А. х. применяют для
получения глицерина (через дихлоргидрины), служит
сырьем для произ-ва лекарств, препаратов и инсекти-
цидов, нек-рых видов пластич. материалов, клеевых
составов и мягчителей. е. е. Барони.
АЛЛИЛБЕНЗОЛ С6Н5СН2СН = СН2, мол. в.
118,17 — жидкость с сильным запахом; т. пл. —40°,.
т. кип. 156,3°; <7;'’ 0,8930; 1,5126; %макс 250ж.и«;
нерастворим в воде, растворим в спирте, эфире,угле-
водородах. При нагревании со спиртовой щелочью
А, перегруппировывается в а-пропенилбензол
С6Н5СН = СН2СН3; присоединяет Вг2, образуя р, у-ди-
бромиропилбензол С6Н5СН2СНВгСН2Вг; окисляет-
ся надуксусной кислотой СН3СОООН в днацетат беп-
зилэтиленгликоля СеН5СН2СН(ОСОСН3)СН2ОСОСН3;
с бромтринитрометаном CBr(NO2)3 в метиловом спирте
образует С6Н5СН2СН(ОСН3)СН2Вг (у-бром-р-мет-
оксипропилбен.зол). Получают аллилбензол из аллил-
бромида и фенилмагнийбромида: СН2=СНСН2Вг +
+ C6H5MgBr-> С6Н5СН2СН — CH2+MgBr2; конденса-
цией бензола с аллиловым спиртом СН2 = СНСН2ОН
в присутствии А1С13; восстановлением фенилалли-
лового спирта С6Н5СН(ОН)СН = СН2 натрием в
спиртовой среде. м. Ф. Турчинским.
АЛЛИЛЕН (пропин, метилацетилен) СН3С-2аСН,мол.
в. 40,06 — газ с неприятным запахом; т. пл. —101,5°,
т. кип. —23,3°; растворим в воде, хорошо растворим
в спирте, в 1 объеме эфира растворяется (объемов):
30 (16°), 100 (1°). При действии А. на аммиачные рас-
творы Ag2O и Си2О ацетиленовый водород замещается
на металл. Так, напр., с Na образует аллиленнатрий
СН3С CNa; при пропускании над флоридином
(180—360°) изомеризуется в аллен СН2=С=СН2; по-
лимеризуется под действием УФ-света; гидратируется
над Н3РО4 па кизельгуре (при 204°), превращаясь
в ацетон; присоединяет HCN (425°, катализатор ZnO
I на кизельгуре) с образованием нитрила метакриловой
I к-ты CH2=C(CH3)CN. Получают А. действием иоди-
I стого метила CH3J или диметилсульфата (CH3O)2SO2
133
АЛЛИЛМЕРКАПТОМЕТИЛПЕНИЦИЛЛИН — АЛЛОТРОПИЯ
134
на ацетиленид натрия в жидком аммиаке: CH3J -[-
CH=CNa —> NaJ -|- CH3C=CH; из карбида маг-
ния C3Mg2 и H2O (технич. метод): C3Mg2 4Н2О —>
->2 Mg(OH)2 -f- СН3С=СН. А. обладает анестези-
рующим действием; применяется в органич. синтезах;
количественно определяется броматометрич. методом.
М. Ф. Турчинский.
АЛЛИЛМЕРКАПТОМЕТИЛПЕНИЦИЛЛИН (пе-
нициллин О) — антибиотик, выделяемый из культу-
CH2=CHCH2SCH2CONHCH-CH С(СН3)2
CO-N - СНСООН
ральпой жидкости различных видов пенициллиума
(Penicillium chrysogenum, Р. notatum) при добавлении
перед выращиванием т. н. предшественника аллилмер-
каптоуксусной к-ты. Выделение А. и его очистка
производятся экстракцией. А. — сравнительно силь-
ная одноосновная к-та. Описаны его калиевая (т. пл.
204—206°), новокаиновая и 2-хлорпрокаиповая соли.
А. — высокоэффективный антибиотик, действующий
гл. обр. на грамположительные бактерии. По сравне-
нию с бензилпенициллином он менее токсичен и вызы-
вает меньше аллергич. реакций. Содержание А. опре-
деляют биологич. и химич. методами и выражают
в единицах действия. См. Пенициллины.
Лит.: Шемякин М. М. и Хохлов А. С., Химия
антибиотических веществ, 2 изд., М.—Л., 1953; S о р е г О. F.
[а. о.], J. Amer. Chem. Soc., 1948, 70 р. 2849; Ford J. H.,
Churchill В. W. and Collngs-Worth D. R.,
Antibiot, and chemotherapy, 1953, 3, p. 1149.
И. И. Иноземцева.
АЛЛИЛОВЫЙ СПИРТ CH2=CHCH2OH, мол. в.
48,08 — простейший устойчивый представитель нена-
сыщенных алкоголей; бесцветная жидкость с острым
запахом; т. кип. 96,9°; 0,8520; 1,4133; т. Крит.
271,9°; теплота парообразования 9550 кал) моль
(760 мм); границы воспламенения в воздухе 2,5—
18,0 об. % (при 100°); т. всп. 22,2°. А. с. в любых про-
порциях растворяется в воде, спирте, эфире; азео-
тропная смесь с водой содержит 72,3% А. с., кипит
при 88,89°. Давление пара 1мм рт. ст.): 4,2 (0°);
17,3 (20°); 98,8 (50°); 394,3 (80°); 850,0 (100°).
А. с. вступает во все реакции, свойственные первич-
ной спиртовой группе и этиленовой двойной связи.
Среди этих реакций практич. значение имеет присо-
единение хлорноватистой к-ты с образованием хлор-
гидринов глицерина, например: СН2(ОН)СН=СН2 -j-
+ НОС1 — СН2(ОН)СНС1СН2ОН, а также окисление в
щелочной среде с образованием глицерина:
ЗСН=(ОН)-СН=СНо + 2КМпО, 4- 4Н,0 ->
—► ЗСН»(ОН)СН(ОН)-СН»(ОН) + 2МпО3 + 2КОН
При темп-pax, близких к комнатной, А. с. стоек и не
полимеризуется. Выше 100° в присутствии кислорода
он образует вязкие полимеры, сравнительно низкого
мол. веса.
Безводный А. с. может быть получен азеотропной
перегонкой с бензолом, трихлорэтиленом и др. спо-
собами. А. с. образуется при нагревании глицерина
с безводной щавелевой к-той до 150°:
СН2(ОН)СН(ОН)СН»(ОН) + (COOH)s ->
-> СН3=СНСН2ОН + 2Н..0 + 2COs
on может быть получен селективным гидрированием
пропаргилового спирта или акролеина. В лаборатории
удобно получать нагреванием при 150° хлористого
аллила с водным р-ром Са(ОН)2. Этот же метод принят
в пром-сти: гидролиз производится 5—10%-ным р-ром
NaOH при 13—14 ат и 150°. Применяется в произ-ве
глицерина (см. выше); из него производят также проз-
рачные синтетич. смолы и пластич. материалы, гл. обр.
па основе диаллиловых эфиров двухосновных к-т,
малеиновой, фталевой и др. б. П. Гусев.
3*
АЛЛОЗА СвН12Ов, мол. в. 180,16 — одна из восьми
изомерных альдогексоз, эпимер алътрозы; бесцвет-
ные кристаллы; т. пл.: p-D-A. 128,5°, [a]D =—0,2°
(в воде) и 4-14,4° (равновесная система
в воде); p-L-A. 129°, [а]д = -(-1,9° (в во-
де) и —13,9° (равновесная система в
воде); D,L-A. 186—188° (с разл.). А. хо-
рошо растворима в воде, плохо—в спир-
те, кристаллизуется из воды и спирта.
А. — восстанавливающий сахар, обра-
зует ряд производных за счет альде-
гидной группы (фенилозазон, п-бром-
фенилгидразоп). Озазон аллозы иден-
тичен озазону альтрозы. При окислении
ОНО
н_<!;_он
I
H-с—ОН
н—с—он
I
н—с—он
сн»он
D-А. аль-
дегидная группа переходит в кислотную с образова-
нием D-аллоновой кислоты; при более глубоком
окцслении D-А. и L-А. дают одну оптически неак-
тивную алломуциновую кислоту (окисляются обе
концевые группы). А. получена синтетически циан-
гидринным методом Фишера: D-А. из D-рибозы,
L-А. из L-рибозы.
Лит.: Richtmyer N. К., в кн.: Advances in carbo-
hydrate chemistry, v. 1, N. Y., 1945, p. 37; Micheel F.,
Chemie der Zucker und Polysaccharide, 2 Aufl Lpz., 1956.
Л. И. Линевич.
АЛЛОКСАН (мезоксалилмочевина) C4H2O4N2, мо-
лекулярный вес 160,09 — белые кристаллы, розо-
веющие на воздухе, хорошо раство- q
римые в воде и спирте; водный р-р ||
бесцветен и имеет кислую реакцию.
А. кристаллизуется с 1 или 4 мо-
HN
лекулами воды; моногидрат разла- I I
гается около 170°, безводный А. — О=С С=О
при 256°. А. реагирует с аммиаком с
образованием кристаллов оксалура- и
мида H2NCONHCONH2 (катализато- °
ры HCN и цианиды). С Fe3+, Mg, Cd, Zn, Co, Ni и Mn
A. образует оранжево-желтые или красные р-ры; с Fе2+
в присутствии разбавленного р-ра NH4OH цвет р-ра
темно-синий; с ацетатами РЬ и Hg2“, а также с нитра-
тами Ag и Hg|+ дает осадки. Получают А. окислением
мочевой к-ты азотной к-той:
HN С---NH HNOq HN NH HN-----СО
I II I --X? I I + । ।
O=C C C=O 0=Cx C=O OC\ /°
XNlf XNH C NH
О
А. окисляет KJ до J2 (подобно хинонам). Применяет-
ся А. как реактив на белки, цианиды, Fe, Со, Ni, Мп,
Mg, Zn, Cd, Hg, Pb и Ag; для синтеза рибофлавина.
Нз возникновении желто-зеленой флуоресценции ос-
новано колич. определение А.
Лит.: Гетероциклические соединения, под ред. Р. Эльдер-
филда, пер. с англ., т. 6, М., 1960; W е 1 с h е г F. J., Organic
analytical reagents, v. i, 1947, p. 191. И. П. Ефимов.
АЛЛОТРОПИЯ — способность химич. элемента су-
ществовать в виде двух или большего числа простых
веществ. Явление А. обусловлено: 1) образова-
нием молекул с различным числом атомов (кислород
О2 и озон О3; модификации жидкой серы: Sx — с мо-
лекулами в виде 8-членных колец и — с молеку-
лами в виде цепочек из шести атомов; фосфор 2-атом-
ный Р2 и фосфор 4-атомный Р4 — с молекулой в виде
правильного тетраэдра и т. д.); 2) образованием кри-
сталлов различных модификаций — частный случай
полиморфизма [углерод в виде графита и алмаза; моди-
фикации твердой серы: ромбическая (SJ и моноклин-
ная (Sg); олово серое и белое; железо а, у, б и t. д. ].
Число известных химич. элементов в наст, время
превысило 100, число же известных аллотропии, моди-
135
АЛМАЗ — АЛЬДЕГИДАММИАКИ
136
фикаций достигает 400. Аллотропии, модификации
принято различать по геометрии, расположению ато-
мов. Вместе с тем возможно существование различ-
ных физич. состояний в-ва при наличии одинаковой
кристаллич. структуры. Так, железо при 769° (точка
Кюри) переходит из ферромагнитного в неферромаг-
нитное состояние, а кристаллич. решетка при этом
остается та же (кубич. грапецентрированная).
Ю. В. Мних.
АЛМАЗ — см. Углерод.
АЛЬБИХТОЛ — смесь тиофена и его гомологов
(метилтиофена, диметилтиофена и др.); прозрачная,
легкоподвижная желтоватая или зеленоватая жид-
кость, обладающая характерным запахом; на воздухе
постепенно краснеет, горит сильно коптящим пламе-
нем. А. нерастворим в воде, растворим в хлороформе,
скипидаре, бензоле, эфире; с этиловым спиртом и гли-
церином дает нестойкие эмульсии; растворяет иод,
йодоформ, камфору, смешивается с вазелином и лано-
лином. В техиич. продукте содержится не менее 9%
серы, 3,5% хлора, dp 0,890—0,925. А. готовят из лег-
ких погонов сернистой смолы кашпирских сланцев,
отличающихся высоким содержанием сернистых со-
единений; легкие погоны смолы подвергают последо-
вательной обработке слабой H2SO4 (для очистки от
пиридинов и пирролов), затем — р-ром NaOH (для ней-
трализации и очистки от фенолов и меркаптанов).
Очищенные масла обрабатывают хлорной известью
или газообразным хлором и перегоняют. А. обладает
сильными инсектицидными свойствами, может быть
использован в смесях с различными эмульгаторами и
мылами в виде альбихтоловой пасты для борьбы с вре-
дителями хлопковых полей. б. С. Ициксон.
АЛЬБОМИЦИЙ — антибиотик, образуемый лучи-
стым грибком Actrhomyces subtropicus. А. действует
на ряд грамположительных (пневмококки, стафило-
кокки) и грамотрицательных (кишечная палочка,
палочка дизентерии, пневмобактерии Фридлендера)
микробов. Чистый А. был получен пу-
тем хроматографии на пермутите и по-
следующем образовании рейнеката в
виде кирпично-красного сульфата с
активностью 700 000 единиц разведения
в 1 мг при титровании на золотистых
стафилококках. А. содержит 4,16% же-
леза, которое может быть отделено от
органич. части молекулы А. действием НС1 в аце-
тоновой среде; при этом исчезает окраска и анти-
бактериальная активность снижается в 14—15 раз.
При добавлении к инактивированному А. р-ра FeCl3
окраска и активность восстанавливаются. А. может
присоединить еще один атом железа, при этом обра-
зуется продукт с более основными свойствами.
Эквивалент А., определенный по железу, равен 1300.
Хроматографией на полистироловой сульфосмоле
А. был разделен на компоненты: а, р, у, S и е; а и
р — кислые, активные компоненты; у и S — нейтраль-
ные; у — биологически неактивен; е — основной ком-
понент, биологически неактивен; у- и е-компонен-
ты — продукты инактивации А. Все компоненты со-
держат железо и обладают поглощением в ультрафио-
летовой области спектра при 280 ммк (компонент е —
при 265 ммк). Из суммарного препарата А. после ки-
слотного и щелочного гидролиза были выделены 3-ме-
тилурацил и гетероциклич. основание, сходное с 5-ме-
тилцитозином (содержит 33% азота). В кислотном
гидролизате компонента 6 находятся серин и орнитин
в эквимолекулярных количествах. В компонентах а
и р обнаружена также и глютаминовая к-та. Впервые
А. выделен в 1951 Г. Ф. Гаузе и М. Г. Бражниковой.
Лит.: Бражникова М. Г., Ломакина Н. Н.,
Муравьева Л. И., ДАН СССР, 1954, 99, № 5, с. 827;
Бражникова М. Г., Ломакина Н. Н., Куди-
нова М. К., там же, 1956, 108, № 4, с. 677; Б р а ж н и-
нова М. Г., М и к е 1и О., Ломакина Н. Н., Био-
химия, 1957, 22, вып. 1—2, с. 111; Бражникова М. Г.
[и др J, Антибиотики, 1958, 3, № б, с. 2 9. М. Г. Бражникова.
АЛЬБУМИНЫ — простейшие представители при-
родных белков, присутствующие во всех растит, и
животных тканях; в отличие от глобулинов, с к-рыми
они составляют группу растворимых белков, раство-
ряются в полунасыщенном (50% насыщения) р-ре
сернокислого аммония и в дистиллированной воде.
Изоэлектрич. точка А. в пределах pH 4,6—4,8; мол. в.
не превышает 75 000. Все А. — глобулярные белки.
А. способны к образованию хорошо оформленных
кристаллов; в электрофоретич. поле А., как правило,
могут быть разделены на 2 и более компонентов. А.
растворимы в к-тах, щелочах, при нагревании свер-
тываются; при гидролизе образуют различные ами-
нокислоты, для состава к-рых характерно отсутствие
или относительно низкое содержание глицина (не
более 2%). А. богаты серусодержащими и дикарбоно-
выми аминокислотами. В живых тканях А. обычно
находятся в виде соединений с липидами, углеводами
и др. белками; содержатся в белке яиц, сыворотке
крови, молоке, семенах растений. А. получают из
плазмы крови фракционир. осаждением при низких
темп-pax; этот препарат широко применяют в медицин-
ской практике, особенно для питания, путем вве-
дения в кровь. Кроме того, А. получают также из
крови животных (сывороточный А.), отделением
белка яиц от желтка (яичный А.), а также из молочной
сыворотки при нагревании до 75° (молочный А.).
А. применяются в фармацевтич., кондитерской, тек-
стильной и др. отраслях промышленности и для
осветления вин.
Лит. см. при ст. Белки и Глобулины. Е. Д. Грищенко.
АЛЬГИНОВЫЕ КИСЛОТЫ — полисахариды водо-
рослей, состоящие из связанных р-1,4-связями остат-
ков D-маннуроновой к-ты (см. Гексуроновые кислоты),
образующих длинные цепи. Мол. в. порядка 50 000—
200 000. А. к. в холодной воде плохо растворимы,
н
СООН
СООН
СООН
о
о
Н J"
гели, являются
Н НН
в горячей — растворимы; образуют
сильными к-тами. Альгинаты натрия (альгин)
растворимы в воде; р-ры характеризуются большой
вязкостью; альгинаты аммония, щелочноземельных
и тяжелых металлов в воде нерастворимы. А. к.,
благодаря наличию уранозидных связей, стойки
к кислотному гидролизу, при нагревании с к-тами
образуется маннуролактон; освобождения маннуро-
новой к-ты достичь при этом не удается. В жестких
условиях гидролиза А. к. идет декарбоксилирование.
При действии горячих р-ров щелочей на А. к. послед-
ние разлагаются. Ферменты, гидролизующие А. к.,
обнаружены в нек-рых бактериях.'Для установления
строения А. к. наряду с химич. методами широко
использовались методы рентгеноструктурного анализа,
подтвердившего наличие в А. к. пиранозного кольца.
А. к. получают из бурых водорослей, где они со-
держатся, по-видимому, в виде солей Mg и Са (13—
40% от сухого веса). А. к. широко применяются в пи-
щевой пром-сти в качестве стабилизаторов нек-рых
пищевых продуктов и эмульгаторов; в виде альгина-
тов используются для изготовления искусств, шелка.
Лит.: Whistler R. L. and Smart С. L., Poly-
saccharide chemistry, N. Y., 1953. Л. И. Линевич.
АЛЬДЕГИДАММИАКИ (а-оксиамины) RCH(OH)NH2—
продукты присоединения аммиака к карбонильной
группе альдегидов.
137
АЛЬДЕГИДОКИСЛОТЫ И КЕТОКИСЛОТЫ
138
А. — мало устойчивые соединения; легко превра-
щаются в алъдимины. В отдельных случаях полу-
чаются устойчивые А., напр. трихлорацетальдегид-
аммиак CC13CH(OH)NH2. А. растворимы в воде и
спирте, нерастворимы в эфире и бензоле. А. легко
образуются при взаимодействии альдегидов с аммиа-
ком в эфирной или спиртовой среде. Реакция фор-
мальдегида с NH3 протекает сложнее; при этом обра-
зуется гексаметилентетрамин (уротропин): 6СН2О +
-ф- 4NH3 —> C6H12N4 + 6Н2О. Во влажном воздухе, а
также при действии разб. кислот А. распадаются
на исходный альдегид и NH3; поэтому его применяют
для очистки альдегидов.
Ацетальдегидаммиак реагирует с HCN с образова-
нием а-аминопропионитрила CH3CH(NH2)CN, что
позволяет осуществить переход от альдегидов через
А. к а-аминокислотам. Энергичное нагревание А.
представляет собой один из наиболее старых способов
получения гомологов пиридина, напр. 2-метил-5-
этилпиридина И др. ф. К. Величко.
АЛЬДЕГИДОКИСЛОТЫ И КЕТОКИСЛОТЫ (ок-
сокарбоновые кислоты) — органич. соединения, со-
держащие карбоксильные и карбонильные группы и
обладающие специфик, особенностями в зависимости
от взаимного расположения заместителей. По этому
признаку различают а-, р-, у- и т. д. оксокарбоновые
к-ты. Из этих соединений наибольшее значение имеют
кетокислоты жирного ряда. Единственный возможный
представитель а-альдегидокислот, а именно гли-
оксиловая кислота СНОСООН, представ-
ляет собой бесцветный густой сироп: часто содер-
жится в недозрелых фруктах. Под влиянием соседней
электрофильной карбоксильной группы альдегидная
группа в глиоксиловой к-те (аналогия с хлоралем)
легко присоединяет воду с образованием моногидрата
возможного строения НС(ОН)2СООН с т. пл. 98°.
Глиоксиловая к-та способна превращаться в смесь
щавелевой и гликолевой к-т по реакции Канниццаро,
обычно характерной для ароматич. альдегидов:
2СНОСООН -> СООНСООН -ф- СН20НС00Н. Удоб-
ный метод получения глиоксиловой к-ты заключается
в восстановлении щавелевой к-ты на Hg- или РЬ-ка-
тоде. Она находит применение в своеобразном син-
тезе ванилина.
Важнейшей типичной а-кетокислотой является п и-
рови и оградная кислота СН3СОСООН,
т. кип. 165°, т. пл. 13,6°, плотн. 1,27, значительно
более сильная к-та (К ~ 0,56), чем пропионовая к-та
(К =0,0013). Может быть получена нагреванием
винной или виноградной к-ты, а также по общим ме-
тодам синтеза кетокислот из дихлорпропионовой
к-ты или хлористого ацетила: СН3СОС1 -ф- KCN —>
->СН3СОС№5Е2.^СН3СОСООН155522!^СН3СС12СООН.
Соседство карбоксильной и карбонильной групп в
пировиноградной к-те ведет к легкому отщеплению СО2
или СО: СН3СНО4-СО2 биохимически СНзс ОС ООН ->
СН3СООН -ф- СО. Приокислении пировиноград-
ной к-ты образуется уксусная к-та: СН3СОСООН -ф-
-ф- О —> СН3СООН -ф- СО2. Пировиноградная к-та иг-
рает важную роль в процессе обмена веществ, при
распаде и биосинтезе аминокислот белка; образуется
в тканях животного организма при окислительном
распаде углеводов и жиров, в процессе дезаминирова-
ния аминокислот. Пировиноградная к-та под влиянием
ферментов присоединяет СО2 с образованием щаве-
левоуксусной к-ты, участвующей в цикле трикарбо-
новых к-т, играющем важную роль при окислении
углеводов.
P-А. и к. очень непрочны. Так, формилуксусная
к-та СНОСН2СООН не выделена, т. к. она легко
декарбоксилируется с образованием СН3СНО. Аце-
тоуксусная кислота СН3СОСН2СООН —
сироп, смешивающийся с водой во всех отношениях;
очень неустойчива, при слабом нагревании водных
р-ров, так же как и формилуксусная к-та, легко рас-
падается на ацетон и СО2; содержится в моче больных
диабетом. Из производных этой к-ты большое практич.
и теоретич. значение имеет ее этиловый эфир, т. наз.
ацетоуксусный эфир СН3СОСН2СООС2Н5, применяе-
мый в произ-ве антипирина, пирамидона, у-ацетопро-
пилового спирта (при получении акрихина, витамина
Bj и др.), для многообразных лабораторных синтезов.
Для ацетоуксусного эфира характерна кегоенольная
таутомерия:
СН3СОСН2СООС3Н3 СН3С(ОН)=СНСООСЛ13
кетоформа енольная форма
В случае у- и S-А. и к. возможна, помимо кетоеноль-
ной, дополнительная таутомерия между карбониль-
ным соединением и изомерным оксилактоном (лакто-
лом), напр.:
СН-СН2 СН2-СН2 СН2—СН3
II I I I I I
СН со сно со ♦♦ сн со
Ан Ан А„ "°
енол альдегид лактол
у-Кетокислота — пеницилловая кислота
СН2=С(СН3)СОС(ОСН3)=СНСООН, являющаяся анти-
биотиком; как в кристаллич. состоянии, так и в вод-
ном р-ре находится в лактольной форме. Простейшим
представителем у-кетокислот является левули-
новая кислота СН3СОСП2СН2СООН (т. кип.
ок. 250°, т. пл. 37°), образуется при нагревании гексоз
с конц. НС1; может быть синтезирована взаимодей-
ствием натрийацетоуксусного эфира с эфиром хлор-
уксусной к-ты с последующим котонным расщепле-
нием продукта реакции: CH3C(ON a)=CHCOOC2H5 4-
-ф-С1СН2СООС2Н5=СН3СОСН(СООС2Н5)СН2СООС2Н5-
—> СН3СОСН2СНаСООН; в отличие от Р-кетокислот,
не отщепляет СО2. Известны многие двухосновные
кетокислоты. К их числу принадлежит м е з о к с а-
левая кислота НООССОСООН, существую-
щая в виде гидрата вероятного строения
НООСС(ОН)2СООН, т. пл. 121°; может быть получена
действием воды на диброммалоновую к-ту или окисле-
нием малоновой к-ты селенистым ангидридом. При
нагревании с водой декарбоксилируется в глиоксило-
вую к-ту. Мезоксалевая к-та образуется при окисле-
нии мочевой к-ты. У рейд мезоксал евой к-ты, т. н.
аллоксан (образуется в организме при сахарном диа-
бете), гидролитически расщепляется с образованием
мочевины и мезоксалевой к-ты.
Второй член ряда двухосновных кетокислот — ща-
веле в о у к с ус н а я кислота НООССОСН2СООН,
играет важную роль в обмене углеводов в организме;
нестойка, т. к. представляет собой одновременно
а- и Р-кетокислоту. Находится почти исключительно
в енольной форме вследствие сопряжения енольной
двойной связи с обеими карбоксильными группами;
может быть выделена в цис- и транс-форме. Поэтому
правильнее говорить не о щавелевоуксусной, а об
оксифумаровой (I) и об оксималеиновой (II) к-тах
нс-сооН нс—соон
ноос—с—он но—с—соон
I и
Действительно, гидролизом ангидрида оксималеиновой
к-ты в определенных условиях можно получить окси-
малеиновую к-ту (II), т. пл. 184°, и оксифумаровую
к-ту, т. пл. 152°. Эфир щавелевоуксусной'к-ты по
своим свойствам напоминает ацетоуксусный эфир,
содержит ОК. 80% енольной формы. ,я. ф. Комиссаров.
139
АЛЬДЕГИДЫ - АЛЬДИМИНЫ
140
АЛЬДЕГИДЫ RCHO — класс органических соеди-
нений, характеризующихся содержанием карбониль-
ной группы (С=О), связанной с атомом водорода.
Кетоны RCOR' также содержат карбонильную группу,
и поэтому химич. свойства А. и кетонов во многом
сходны между собой. Обычно названия А. производят
от названий соответствующих кислот. Так, муравьи-
ной к-те соответствует простейший член ряда А. жир-
ного ряда — муравьиный А., или формальдегид
НСНО; уксусной к-те отвечает уксусный альдегид,
или ацетальдегид СН3СНО, и т. д. По женевской номен-
клатуре название А. производится от углеводородов
с прибавлением окончания «аль», напр. НСНО —
метаналь, СН3СНО — этаналь и т. д. Нек-рые А.
носят тривиальные назв., напр. фурфурол.
Из сопоставления формул аналогично построенных
спиртов RCH2OH, альдегидов RCHO и к-т RCOOH
следует, что А. по степени окисленности представ-
ляют собой тип соединений, находящихся между спир-
тами и кислотами. С этим связаны нек-рые способы
получения А. и их химич. свойства. Так, широкое
значение имеют методы синтеза А. окислением (дегид-
рированием) первичных спиртов или же восстановле-
нием производных кислот, напр. хлорангидридов.
В этой связи должен быть отмечен и метод получения
А. (в особенности ароматич. и гетероциклич. рядов)
омылением дигалогенометильных производных, напри-
мер: С6Н5СНС1Д22 С6Н5СН(ОН)2 -> С6Н6СНО + Н2О.
Промежуточному положению А. в известной мере
отвечает и способность их к реакциям окисления-
восстановления с образованием сложных эфиров:
2RCHO —> RCOOCH2R (см. Тищенко реакция), или
спирта и кислоты; 2С6Н5СНО-|- Н2О—>СеН5СН2ОН-|-
-|~ С8Н5СООН (см. Канниццаро реакция). Альдегид-
ная группа обладает слабо выраженным электрофиль-
ным характером, что, в частности, проявляется в
ориентации при замещении в ароматич. ядре; груп-
па —СНО промежуточна между алкоксигруппой
и электрофильным карбоксилом.
Помимо упомянутых, известен и ряд др. методов
получения А., напр. перегонка смешанных кальцие-
вых солей муравьиной и другой карбоновой к-ты:
RCOOCaOOCH —> RCHO -|- СаСО3; гидролиз вини-
ловых эфиров: CH2=CHOR+Н2О->СН3СНО +
-ф- ROH; действие реактива Гриньяра на эфир мура-
вьиной кислоты:
НСООС2Н6 + RMgX -*
—► RCHfOCsHsJOMgX11 =2 RCHO + С2Н5ОН + MgXOH
Известны также методы получения А. только ароматич.
и гетероциклич. рядов, напр. осторожное окисление
ароматич. углеводородов, содержащих метильные
группы СвН5СН3—> С6Н5СНО; технич. значение имеет
аналогичный способ получения простейшего ненасы-
щенного А. — акролеина — окислением пропилена:
СН3СН=СН2 —> СН2=СНСНО. При действии СО и
НС! в присутствии А1С13 на ароматические углево-
дороды образуются А.: С6Н5СН3СО-|- НС! —>
—> п-СН3СвН4СНО; по-видимому, промежуточно об-
разуется нестойкий НСОС1 и далее протекает реак-
ция типа Фриделя — Крафтса (см. Гаттермана — Коха
реакция).
Метод синтеза ацетальдегида, имеющий промыш-
ленное значение, заключается в присоединении во-
ды к ацетилену в присутствии солей окиси ртути:
HCfeCH -ф- Н2О —> СН3СН0 (см. Кучерова реакция).
В последнее время приобрел значение т. н. оксосин-
тез А’., состоящий в действии окиси углерода и водо-
рода на олефины при 100—150° и давлении более
100 am: RCH = СН2 + СО 4-Н2-> RCH2CH2CHO.
К числу доступных А. гетероциклич. ряда относится
фурфурол, который применяется для селективной
очистки нефтепродуктов и в произ-ве феиоло-альде-
гидных смол.
А. легко вступают во многие реакции присоедине-
ния, замещения и конденсации. Вследствие поляриза-
ции л-связи в карбонильной группе атом С в ней
приобретает частичный положительный заряд, а атом
кислорода в известной мере — отрицательный заряд.
Этим объясняется способность А. к присоединению
нуклеофильных реагентов. Так, А. присоединяют
HCN с образованием соответствующих оксинитрилов,
обычно называемых циангидринами: RCHO 4~HCN —>
—> RCH(OH)CN. Аналогичным образом присоеди-
няются к А. бисульфит натрия, амины и т. д. Для
А. характерны реакции замещения, при которых
кислород СО-группы обменивается на двухвалентные
остатки, содержащие азот. Так, из А. и гидроксил-
амина образуются альдоксимы: RCHO 4- NH2OH —>
—>RCN=NOH; при действии гидразина образуются
гидразоны: RCHO 4~ NH2NH2 —> RCH=NNH2. Не-
редко вместо гидразина применяют фенилгидразин
или 2,4-динитрофенилгидразин, что обычно ведет
к хорошо кристаллизующимся гидразонам. К реак-
циям замещения А. относится также их взаимодей-
ствие с РС15 с образованием RCHC12 и др.
А. склонны к реакциям полимеризации и конденса-
ции. Так, формальдегид легко превращается в пара-
формальдегид, ацетальдегид — в тример, так назы-
ваемый паральдегид. При конденсации 2 молей А.,
напр. ацетальдегида, образуется альдоль: СН3СНО 4~
4-СН3СНО —> СН3СН(ОН)СН2СНО. По этому типу
т. наз. альдольной конденсации происходит уплотнение
многих др. А. Эта реакция в определенных условиях
сопровождается отщеплением воды с образованием
кротонового альдегида (кротоновая конденсация):
СН3СН(ОН)СН2СНО—Н2О->СН3СН = СНСНО.
А. как класс очень реакционноспособных органич.
соединений находят широкое применение в произ-ве
феноло-альдегидных смол, в качестве душистых в-в
(напр., пеларгоновый А., ароматич. А. — ванилин),
как исходные продукты для синтеза других продуктов
(получение уксусной к-ты через уксусный А., уротро-
пина из формальдегида) и т. д.
Известны очень чувствительные реакции для откры-
тия А.; так, при прибавлении «фуксиносернистой к-ты»
(р-р фуксипа, обесцвеченный сернистой к-той) даже
к очень малым количествам А. появляется красное или
фиолетовое окрашивание (Шиффа реакция). По Анд-
жели—Римини реакции, к А. прибавляют нитрогидро-
ксиламин (или другое в-во, к-рое может служить источ-
ником HN=O, не существующей в свободном виде)
и к образовавшейся гидроксамовой к-те RC(O)NHOH
добавляют FeCl3, что ведет к интенсивному красно-
му окрашиванию. Я. Ф. Комиссаров.
АЛЬДИМИНЫ RCH=NH — производные альде-
гидов, представляющие собой продукты замещения
карбонильного кислорода в них иминогруппой. Назв.
«А.» обычно производится от исходного альдегида или
же от радикала, с к-рым связана имипогруппа; так,
А. строения CH3CH=NH паз. ацетальдимином, или
этилиденимином. А. легко образуются в результате
дегидратации алъдегидаммиаков; они могут быть также
получены осторожным восстановлением нитрилов дей-
ствием металлич. Na и спирта или же SnCU в НС1:
RCN —> RCH=NH. А. легко полимеризуются и
обычно находятся в тримерной циклич. форме, напр.:
3 CH3CH=NH
/NH
Н3С-СН '"сн—сн,
I I
HNv. /NH
CH
I
сн3
141
АЛЬДОБИОНОВЫЕ КИСЛОТЫ - АЛЬДОЛИ
142
Горячей водой А. гидролизуются до альдегидов,
при недостатке воды образовавшийся альдегид ре-
агирует с А.:
СНзСНО + 2CH3CH=NH -> CH3CH(N=CHCH3)3 + Н3О
При хранении и нагревании А. претерпевают кон-
денсацию типа кротоновой: 2RCH2CH=NR'—>
-> RCH2CH=C(R)CH=NR'+ R'NH2. С AgNO3 А.
образуют труднорастворимые в воде и эфире продук-
ты присоединения, напр. RCH= NH-AgNO3. Извест-
ны различные А., замещенные при азоте. См. также
Шиффовы основания. К. Величко.
АЛЬДОБИОНОВЫЕ КИСЛОТЫ -
карбоксилсодержащие дисахариды, обра-
зующиеся при окислении альдегидной
группы восстанавливающих дисахари-
дов (альдобиоз). Так, из мальтозы обра-
зуется мальтобионовая кислота, из лак-
тозы — лактобиоповая кислота, и т. д.
продуктов кислотного гидролиза ряда полисахари-
дов. Выделение компонентов А. к. для их идентифика-
ции производят обычно окислением А. к. бромом,
в условиях, в к-рых идет гидролиз; при этом из альдоз-
ного компонента образуется соответств. альдоновая
к-та, а из уроновой к-ты — сахарная.
Лита. см. при ст. Дисахариды. Л. И. Линевич.
АЛЬДОЗЫ — см. Моносахариды.
АЛЬДОЛАЗА — фермент, катализирующий распад
дифосфата фруктозы (ДФФ) на фосфодиоксиацетоп
(ФДА) и фосфоглицериновый альдегид (ФГА). Эта
(НО)2ОРОСН, ,
2 I 2 ,СН2ОРО(ОН)2
н-с-о-с.
н I 1ЧОН -
чс----с-он
НО/ I
н
ДФФ
СН2ОРО(ОН)2
со
I
СНгОН
+
СН2ОРО(ОН)2
снон
ФДА
Cf
хн
ФГА
Н-С
СООН
Н-С-ОН
НО-С-Н
Н-С-ОН
Н-С
СН2ОН
н-с-он
О
но-с-н
1Г (
н-с-он
СН2ОН
Альдобиоповая
ван) кислота.
(мальтобиопо-
А. к. — невосстана-
вливающие дисахариды.
По химич. свойствам они
близки альдоновым к-там
(легкость образования
лактонов, эпимеризация
у углерода, соседнего
с карбоксилом). Окисле-
нием восстанавливающих
дисахаридов до А.к. с
последующим гидроли-
зом последних до соот-
ветств. моносахаридов
(восстанавливающей мо-
позы и альдоповой к-ты) пользуются для идентифи-
кации дисахаридов и установления их строения.
Лит. см. при ст. Дисахариды. Л. И. Линевич.
АЛЬДОБИУРОНОВЫЕ КИСЛОТЫ — восстана-
вливающие дисахариды, компонентом к-рых яв-
ляется к.-л. уроновая к-та, полуацетальный гидроксил
к-рой связан с альдозой (I). Изомерные дисахариды,
в образовании гликозидной связи к-рых принимают
участие спиртовая группа уроновой к-ты и полуаце-
тальный гидроксил альдозы, называют псевдоальдо-
биуроновыми к-тами (II).
реакция является важным этапом анаэробного пре-
вращения углеводов в животных и растительных орга-
низмах (гликолиз, брожение). А. найдена в микро-
организмах, грибах, высших растениях, различных
тканях млекопитающих и, по-видимому, содержится
во всех клетках организмов. А. получена из мышц
в виде кристаллов. Оптимум ее действия при pH 7,5—
8,5. А. высокоспецифична по отношению к ДФФ и
ФДА. Образование фосфотриоз происходит исклю-
чительно из фруктозо-1,6-дифосфата; для реакции
синтеза обязателен только ФДА, ФГА может быть
заменен альдегидами алифатич. и ароматич. рядов
(глицеральдегид, ацетальдегид, гликолевый альдегид
и др.). В результате реакции образуются многочислен-
ные соединения, в том числе фруктозо-1-фосфат и
фосфорилированные пентозы.
Лит.: Нейл андс Дж. и Штумпф П., Очерки по
химии ферментов, пер. с англ., М., 1958; Браунштейн
А. Е., Биохимия аминокислотного обмена, М., 1949; Advances
in enzymology..., ed. by F. F. Nord, v. 6, N. Y. — L., 1946,
p. 170; v. 13, 1952, p. 7; v. 18, 1957, p. 292—93; v. 19, p. 7,
9, 24; см. также при ст. Ферменты. Н. К. Рудакова.
АЛЬДОЛИ (1,3 - о к с и а л ь д е г и д ы) — произ-
водные альдегидов, содержащие одновременно окси-
и карбонильную группу общей формулы
r3chchohcr»cho
1 1 1 1 сн
"7 неон 1 —। неон 1 неон 1 нсо— 1 —1 неон 1
носн 1 С ) 1 с носн ( ) 1 ) носн о
неон неон неон неон I
НС — ) нс нс
1 СООН df2OH 1 СООН 1 СН2ОН
/ //
Уроповые к-ты представлены обычно глюкуроновой
или галактуроновой к-той, а альдозы — различными
бесцветные жидкости, растворимые в воде и орга-
нич. веществах.
При нагревании, в особенности в присутствии ще-
лочей, А. дегидратируются, образуя а,р-невасыщеп-
ные альдегиды:
СН3СН(ОН)СН»СНО — сн3сн=снсно + П-0
Если А. содержит вторичную карбонильную груп-
пу, дегидратация не происходит и реакция под
влиянием щелочи протекает по типу Канниццаро ре-
акции. А. обладают свойствами альдегидов и спир-
тов: образуют альдоль аммиаки и анилы, присоединяют
HCN; их спиртовые группы ацилируются. Гидриро-
ванием А. легко превращаются в 1,3-гликоли.
Один из первых методов синтеза бутадиена заклю-
чался в восстановлении А. в 1,3-бутиленгликоль
Альдоль Формула Т. кип., ° С/мм "О
Ацетальдоль Пропиональдоль Бутиральдоль Изобутиральдоль СН3СН(ОН)СН,СНО С-ЩСЩОЩСЩСВДСНО С3Н7СН(ОН)СН(С3Н6)СНО (СН3)..СНСН(ОН)С(СН3).>СНО 83/20 84-86/11 100—102/12 118—120/14 1.103(20°) 0,986 0,9510 0,9482 1,4610 (20°) 1,45017 (25°) 1.4368 (13°) 1,4501 (20°)
альдопентозами, альдогексозами
и аминосахарами. А. к. являют-
ся компонентами разнообразных
веществ природного происхож-
дения: гемицеллюлоз, иммупоак-
тивпых полисахаридов бактерий,
мукополисахаридов и др. биологи-
чески важных в-в. Многие А. к.
получены синтетически. Псевдо-А. к. в качестве ком-
понентов природных в-в не обнаружены. Характерной
особенностью А. к. является прочность уронозидной
связи в кислой среде; они гидролизуются только в очень
жестких условиях, поэтому А. к. обнаружены среди
СН3СНОНСН2СН2ОН и дегидратации последнего в бу-
тадиен над фосфатным катализатором; ацетальдоль
образует при хранении паральдоль (С3Н8О2)2, т. пл.
89—91°. Ацетальдоль был одновременно открыт
А. П. Бородиным и А. Вюрцом в 1872. Идентифици-
143
АЛЬДОЛЬНАЯ КОНДЕНСАЦИЯ — АЛЬДОСТЕРОН
144
руют А. по n-бромфенилгидразонам. Получают аль-
дольной конденсацией алифатич. альдегидов, напр.;
СНдСНО + СН3СНО -> СН3СН(ОН)СН2СНО.
Лит.: Chemistry of carbon compounds, ed. E. H. Rodd,
V. 1, pt. A, Amst. [a. o.], 1951. Ф. H. Величко.
АЛЬДОЛЬНАЯ КОНДЕНСАЦИЯ — присоедине-
ние 2 молекул альдегида друг к другу с образованием
веществ, содержащих как альдегидную, так и алко-
гольную группу, и поэтому называемых альдолями.
Напр., из уксусного альдегида в присутствии щелоч-
ных агентов легко образуется р-оксимасляный альде-
гид (альдоль):
СНзСНО + СНзСНО — СН3СН(ОН)СН2СНО
А. к. является общей реакцией всех альдегидов, со-
держащих по соседству с карбонильной группой атом
углерода, связанный с атомами водорода. Склонность
к образованию альдоля падает в ряду:
снсно > кощено > СНзСНО
Такой вывод можно сделать на основании строения
альдоля, образующегося при конденсации двух раз-
личных альдегидов. Так, при конденсации уксусно-
го и пропионового альдегидов образуется;
СН3СН2СН(ОН)СН2СНО
а не:
СН3СН(ОН)СН(СН3)СНО
В А. к. легко вступают и кетоны, напр. при кон-
денсации 2 молей ацетона образуется диацетоновый
спирт:
(СНз)зСО + (СН3)зСО — (СН3)2С(ОН)СН;СОСН
Известны также продукты конденсации альдегидов и
кетонов по схеме А. к. Конденсация 2 молей карбо-
нильных соединений может не останавливаться на
стадии образования альдолей и сопровождаться от-
щеплением воды с образованием ненасыщ. альдегида;
так, напр., из уксусного альдегида можно получить
вместо альдоля кротоновый альдегид СН3СН=СНСНО;
такое направление реакции носит назв. кротоновой
конденсации.
А. к. принадлежит к процессам каталитич. нуклео-
фильного присоединения к карбонильной группе.
Роль щелочного катализатора состоит в отрыве а-во-
дородного атома в виде протона:
пещено + но rchcho <i>
Полученный анион присоединяется к СО-группе вто-
рой молекулы альдегида с образованием повой С—С
связи:
КСН.СНО + RCHCHO RCH3CH(O)CHRCHO (2)
В дальнейшем образовавшийся анион реагирует с во-
дой, что ведет к возникновению альдоля и гидро-
ксильного иона;
RCH3CH(O)CHRCHO + ЩО — RCH2CH(OH)CHRCHO + но
Первая стадия (1) является медленной, что известно
из кинетич. исследований процесса; при этом было по-
казано, что А. к. представляет собой реакцию первого
порядка относительно альдегида. Однако в нек-рых
случаях скорости медленной стадии (1) и быстрой ста-
дии (2) соизмеримы. Об этом свидетельствует А. к.
ацетона в окиси дейтерия, приводящая к диацетоно-
вому спирту, содержащему дейтерий. Можно считать
установленным, что первая стадия многих хорошо
известных процессов конденсации карбонильных со-
едииенийсвеществами, содержащими подвижные атомы
водорода (реакции с малоновым или циануксусным
эфиром, нитросоединениями и др.), представляет со-
бой А. К. Я. Ф. Комиссаров.
АЛЬДОНОВЫЕ КИСЛОТЫ (гликоновые кислоты,
оновые кислоты) — одноосновные полиоксикарбоно-
вые к-ты, карбоксильные производные углеводов —
альдоз и полиолов. Названия отдельным представите-
лям А. к. дают по альдозам, при окислении к-рых
образуются А. к. (глюконовая к-та, галактоновая
и т. д.). По количеству углеродных атомов в молекуле
А. к. делятся на тетроновые (С4), пентоновые (С5),
гексоновые (Св) и т. д. Большую группу А. к. состав-
ляют к-ты общей формулы НООС(СНОН)ПСН2ОП.
Известны также А. к., являющиеся производными дез-
оксисахаров (напр., метиипентоновые к-ты — фуко-
новая, рампоновая), А. к., содержащие кетогруппу,
и др. Большинство А. к. выделено в кристаллич. виде,
нек-рые получены в виде сиропов. А. к. хорошо рас-
творимы в воде, плохо — в спирте; в неполярных рас-
творителях растворимы лучше соответствующих аль-
доз. Водные р-ры А. к. имеют pH 2—3. А. к. раство-
римы в р-рах едких и углекислых щелочей, дают соли
с металлами и органич. основаниями. Соли А. к. (каль-
ция, бария, бруцина, стрихнина) используют для
характеристики и для разделения смесей А. к. Слож-
ные эфиры А. к. получают по Фишеру и диазометано-
вым методом. С фенилгидразином А. к. дают фенил-
гидразиды, к-рые также используются для их иден-
тификации. Получены хлорангидриды и амиды А. к.
За счет гидроксильных групп А. к. ацетилируются,
метилируются, ацетонируются. Характерная особен-
ность А. к. — способность легко давать у-и 6-лактоны,
к-рые образуются в водных р-рах А. к., где медленно
(иногда в течение месяцев) устанавливается равнове-
сие между А. к. и ее лактонами (см. Лактонов пра-
вило): напр., в р-ре D-глюконовой к-ты образуются
у- и S-D-глюколактоиы;
Z с=о СООН f=° 1
ИСОН пеон НСОН о
носн носн ~ 1 1 носн
НСОН неон нс *
1 с неон пеон
1 СН3ОН СЩОН СНоОН
При нагревании А. к. происходит изменение конфигу-
рации при 2-м углеродном атоме, ио эта эпимери-
зация идет с трудом. При окислении первичной спир-
товой группы из А. к. образуются двухосновные сахар-
ные к-ты. При восстановлении HJ из А. к. образуются
жирные к-ты. Получают А. к. окислением альдоз,
окислительным расщеплением кетоз, циангидринным
методом из альдоз с меньшим числом углеродных
атомов и др. Синтезы из веществ неуглеводного харак-
тера (напр., из 3-хлоркротоновой к-ты) используются
для получения тетроновых к-т. В промышленном масш-
табе получают только глюконовую к-ту (см. Гексоно-
вые кислоты), к-рую применяют в нек-рых медицинских
препаратах. А. к. используют для характеристики
альдоз и для синтеза различных веществ углевод-
ного характера, например альдоз, содержащих мень-
шее, чем у всходной А. к., количество углеродных
атомов.
Лит.: Vollmert В., в кн.: Handbuch der Pflanzenphysio-
logie, Hrsg. von W. Ruhland, Bd 6, B. — Hottingen — Hdlb.,
1958, S. 390; Shafizaden F.,b kh.: Advances in carbohyd-
rate chemistry, ed. by M. L. Wolfrom, v. 13, N. Y., 1958, p. 9;
Carbohydrates. Chemistry, biochemistry, physiology, ed. by
W. Pigman, N. Y., 1957. Л. И. Липевич.
АЛЬДОСТЕРОН (А4-прегнеи-11 р-21-диол-18-аль-
-3,20-дион, электрокортин, альдокортиц) С21Н28О5,
мол. в. 360,43— кортикоидный гормон (см. Кортикосте-
роиды), регулирующий минеральный обмен; образуется
в коре надпочечников; получен и синтетически. А. хо-
рошо растворяется в спирте, этилацетате, хлороформе
и воде; плохо — в углеводородах. В р-рах находится
145
АЛЬДРИН — АЛЬФА-ЧАСТИЦА
146
в таутомерных формах с значительным преобладанием
11-полуацеталя:
Лит.: Metcalf R. L., Organic insecticides, N. Y.—L.,
1955, p. 237; Попов П. B-, Справочник по ядохимикатам,
М., 1956, с. 172. Л. С. Поваров.
АЛЬТАКС (дитио-бис-бензтиазол) CI4H8N2S4, мол. в.
268,34 — бесцветные кристаллы, чешуйки (из бен-
зола); т. пл. 186°, плоти.
1,50; %макс 271 ммк; рас-
творим в CS2, СНС13, бен-
золе. При кипячении со
щелочами альтакс обра-
зует 2-меркавтобензтиазол; азотной к-той окисляется
до бензтиазола. В пром-сти А. получают окислением
2-меркаптобепзтиазола:
+ 2NaNU2 + 2HaSO4
+ 2NO+2H.2O + 2NaSO4
В водных р-рах А. гидратируется, превращаясь в мо-
ногидрат, т. пл. 108—112°; после плавления затверде-
вает и вновь плавится при 155—160°; [apg =-(-145°±2°
(в ацетоне). А. обладает максимумом поглощения при
240 ± 0,25 ммк (1g Е = 4,20) и при 308 ± 2 ммк
(1g £ = 1,92). Методы определения А. основаны на
наличии в его кольце Д4-3-кетогруппы (поглощение
ультрафиолетового света и др.), присутствии а-кетоль-
ной боковой цепи у 17-углеродного атома (восстанов-
ление голубого тетразолия в окрашенный фармазан)
и флюоресценции А. после обработки щелочью или
к-той. Биологич. определение проводится по вызывае-
мому А. снижению выделения Na из организма с мо-
чой (минералокортикоидная активность). А. эффекти-
вен при лечении заболеваний, связанных с гипофунк-
цией надпочечников.
Лит.: Юдаев Н. А., Пробл. эндонрлнол. и гормоно-
терапии, 1955, 1, № 1с. 118; Bersin Th., Biochemie der
Hormone, Lpz., 1959; Hand O., Hormone, 2 Aufl., Jena, I960;
Recent progress in hormone research, v. 11, N. Y., 1955, p. 193.
АЛЬДРИН C12H8C16, мол. в. 364,9 — бесцветные
кристаллы, иглы, почти без запаха; т. пл. 104—104,5°;
технический продукт—коричневое
воскообразное в-во, содержащее
I] I I }|С1 78% А. В воде А. нерастворим,
I СН2 CCiall хорошо растворяется в болыпин-
стве органических растворителей;
Cl li lOO.v.i при 25° растворяется: в
ацетоне 66 г, в амилацетате 30 г,
в бензоле 83 г, в н-бутиловом спирте 9 г, в четы-
реххлористом углероде 105 г, в этиловом спирте
5 г, в этилендихлориде 104 г, в метилэтилкетоне 24 г,
в пентане 3 г, в ксилоле 92 г, в керосине 24 г. В ИК-
спектре имеется характерный максимум поглощения
при 8,48 мк, что используется для колич. микроопре-
деления. А. устойчив к действию кислот и щелочей.
Промышленный метод получения А. заключается
в нагревании при 100° в течение 25 час бициклогепта-
диена с гексахлорциклопентадиеном:
Применяется А. как очень эффективный инсектицид
в виде эмульсий, концентратов, р-ров в масле и порош-
ка для борьбы с проволочниками, личинками луковой,
капустной и морковной мух, вредителями кукурузы,
сахарной свеклы и др. насекомыми; очень токсичен
для теплокровных животных — цыплят, мышей, крыс,
кроликов, собак и для человека.
Применяют А. гл. обр. в качестве ускорителя вулка-
низации каучуков. Качественной реакцией на А.
служит зеленовато-голубое окрашивание при сме-
шении бензольного р-ра А. с 1%-ным бензольным
р-ром олеата кобальта; колич. определение основано
на кипячении раствора с Na2S и титровании образовав-
шегося 2-меркаптобензтиазола щелочью.
Р. Б. Журин.
АЛЬТРОЗА C6HJ2Oc (I), мол. в. 180,16 — одна из
восьми альдогексоз, изомеров глюкозы, эпимер ал-
лозы; бесцветные кристаллы. 0-D-A.: т. пл. 103—
105°, [a]D=—69°(в воде) и —|—33,1 ° (равновесная система
ОНО
по—с—и
I
и-с-он
и—с—он
I
Н-С-ОН
I
сн»он
I
----СН-----
но—с—н
Н—с—он
О I
н—с—он о
I
------сн3
II
в воде); р-L-A.: т. пл. 107—109,5°, [а]о =—28,75°
(в воде) и —32,3° (равновесная система в воде). А.
хорошо растворима в воде, плохо — в спиртах. Кри-
сталлизуется из метилового и этилового спиртов.
При нагревании А. в водных р-рах в присутствии
НС1 образуется невосстанавливающий ангидрид А.—
D-a л ь т р о з а н (II). А. образует ряд производных
за счет альдегидной группы (фенилгидразон, бензил-
фенилгидразон). Озазон А. идентичен озазону аллозы.
При окислении А. (альдегидной группы) образуется
альтроновая к-та, при дальнейшем окислении (спир-
товой группы) — таломуциновая к-та. А. получена
синтетически циангидринным методом Фишера
(D-А. из D-рибозы и L-А. из L-рибозы), гидролизом
гликозидов неолактозы и др. способами.
Лит. см. при ст. Аллоза. Л. И. Линевич.
АЛЬФА-РАСПАД (а-распад) — тип радиоактивного
превращения, при к-ром испускаются альфа-частицы.
Поскольку а-частица характеризуется массовым чис-
лом 4 и атомным номером 2, то возникающий при
А.-р. новый химич. элемент имеет по сравнению с
исходным элементом на четыре единицы меньший
атомный вес и на две единицы меньший атомный но-
мер (см. Радиоактивность, Смещения правило).
АЛЬФА-ЧАСТИЦА (а-частица) — частица, иден-
тичная атомному ядру изотопа гелия Не4; состоит
из двух протонов и двух нейтронов. А.-ч. имеет поло-
жительный заряд, по абс. величине превышающий
в 2 раза заряд электрона, и массовое число, равное 4.
Образование А.-ч. из нуклонов сопровождается выде-
лением большой энергии (-^-28,2 Мэв), что находится
147
АЛЮМИНАТЫ — АЛЮМИНИЙ
148
в соответствии с ее высокой устойчивостью. А.-ч.
испускаются при а-распаде радиоактивных изотопов
различных элементов (Ra22e, Rn222, Ро210, U238 и др.,
см. Радиоактивность). При прохождении через ве-
щество А.-ч. сильно ионизируют атомы среды и
быстро теряют свою энергию. Длина свободного про-
бега зависит от энергии А.-ч. и природы поглощаю-
щего вещества. Так, напр., в случае металлов, воды,
спирта и т. д. для полного их поглощения практиче-
ски достаточно, чтобы толщина поглощающего слоя
составляла всего лишь несколько десятков микрон.
Длина свободного пробега А.-ч. в воздухе и в боль-
шинстве др. газов (при нормальных условиях) равна
нескольким сантиметрам.
АЛЮМИНАТЫ — соли алюминиевых кислот. Род-
ные р-ры А. содержат, вероятно, ионы [А1(0Н)4]~,
[А1(0Н)5]2-, [А1(ОН)6]3*. Поэтому же типу построены,
по-видимому, и А., получаемые из р-ров (т. н. гидро-
ксоалюмияаты), тогда как при сплавлении окислов
образуются безводные соли, производные метаалюми-
ниевой к-ты НА102. Примерами гидроксоалюминатов
могут служить К[А1(ОН)4],Ва[А1(ОН)5],Са3[А1(ОН)6]2;
их состав стехиометрически может быть представлен
соответственно: А1(0Н)3 КОН, А1(0Н)3 Ва(ОН)2,
2А1(ОН)3 • ЗСа(ОН)2. В природе наиболее распростра-
нены А. общей формулы RA12O4, где R — Mg, Са, Be,
Zn и др. Среди них различают: 1) октаэдрич. разно-
видности А.—т. наз. шпинели: MgAl2O4 (благородная
шпинель), ZnAl2O4 (ганит, или цинковая шпинель)
и др., 2) ромбич. видоизменения А.: ВеА12О4 (хризобе-
рилл) и др.
А. щелочных металлов хорошо растворимы в воде,
их водные р-ры из-за сильного гидролиза [до А1(0Н)3]
устойчивы лишь при избытке щелочи. А. менее актив-
ных металлов, по большей части, труднорастворимы.
Растворимые А., напр. NaAlO2, можно получить
действием р-ра NaOH на А1(0Н)3; нерастворимые
А. — сплавлением А12О3 с окислами соответствующих
металлов. А. находят разнообразное применение. Так,
NaA102 — промежуточный продукт в процессе полу-
чения А12О3, используется в текстильном производстве
как протрава, в бумажной пром-сти, для ионообмен-
ной очистки воды; СаА12О4 — главная составная часть
быстро твердеющего глиноземистого цемента; ВаА12О4
(растворимость в воде ок. 10%) применяют для удале-
ния из воды SO2-, СО|_, Са2+.
Лит. см. прп ст. Алюминий.
АЛЮМИНИЕВЫЕ КВАСЦЫ — см. Алюминия
сульфат и Квасцы.
АЛЮМИНИЙ (Aluminium) Al — химич. элемент
III гр. периодич. системы Менделеева; п. н. 13, ат. в.
26,98. Состоит из одного стабильного изотопа А127
(100%). Известно иеск. искусств, радиоактивных изо-
топов, из к-рых большинство короткоживущие. Един-
ственно пригодным для индикаторных исследований
является изомер А12в лет). Поперечное
сечение захвата тепловых нейтронов А. 0,215 барн.
Внешняя электронная оболочка атома А. имеет строе-
ние: 3s2 З/j. Потенциалы ионизации (в эв): А1° —
— А1+ — А12+ — А13+, равны соответственно 5,984;
18,82; 28,44. Название дано от латинского alumen —
алюминиевые квасцы (издавна применялись как про-
трава при крашении тканей). Металлич. А. впервые
получен в 1825 X. К. Эрстедом.
Содержание А. в земной коре 8,80 вес. %. По
распространенности в природе А. занимает 3-е место,
после О и Si. В свободном виде не встречается. Все
основные формы нахождения определяются большим
сродством А. к кислороду и способностью замещать
в силикатах атомы Si. Известно неск. сотен минера-
лов А., преим. алюмосиликатов. Промышленное зна-
чение для произ-ва А. имеют: боксит, состоящий
в основном из гидроокисей А. .’диаспора и бе-
мита АЮОН или гиббсита (гидраргиллита)
А1(ОН)3 (см. Алюминия окись), а также окислов и
гидроокисей Fe, Ti, Si и др.; из попутно извлекаемых
микроэлементов присутствуют Ga и V. Цвет боксита
обычно красный разных оттенков; плоти, d 2,9—3,5;
твердость по минералогии, шкале до 6; образовался
при химич. выветривании вулканич. пород в жарком
и влажном климате, а также при осаждении гидро-
окисей алюминия, железа и кремния в древних водое-
мах. Крупные месторождения находятся в СССР,
Еенгрии, Франции, Италии, США и др. местах. Алу-
нит (квасцовый камень) (Na, K)2SO4 • A12(SO4)3 •
• 4А1(ОН)3, содержит 37,0% А12О3; К2О может до
половины замещаться Na2O; сингония тригональная;
цвет белый с оттенками других цветов; d 2,6—2,8;
твердость 3,5—4; образуется при действии на магма-
тич. породы сернокислых водпых р-ров; месторожде-
ния встречаются на Кавказе, Урале, в Чехословакии,
Италии и др. местах. Нефелин (Na, К)2О А12О3 •
2SiO2; еингония гексагональная: бесцветный или
серый с различными оттенками; d 2,6; твердость 5—6,
образует крупные массы в щелочных вулканич. поро-
дах, иногда с апатитом; известные месторождения нахо-
дятся в Хибинах (Кольский полуостров), в Краснояр-
ском крае и в др. местах. При отсутствии другого сырья
для получения А12ОЭ могут служить каолин и глины.
Физические и химические свойства. Свободный А. —
серебристо-белый металл; в обычных условиях покрыт
тонкой окисной пленкой; хороший проводник тепла
и электричества А. весьма пластичен, легко поддается
обработке давлением, прокатке, ковке, штамповке и
волочению. Кристаллич. решетка А. — гранецентри-
рованная кубич. с периодом а = 4,0494А. Атомный
радиус 1,43 А, ионный радиус А13^ 0,57 А. Никаких
аллотропия, превращений у А., нет. Плоти, технич.
металла в твердом и жидком состоянии уменьшается
с увеличением его чистоты; для А. 99,996% чистоты
о!20 2,6989 и d1000 2,289; т. пл. 660°; т. кип. ок. 2500°.
Давление пара (мм рт. ст.): 0,00002 при т. пл.; 0,1
при 1123° и 1 при 1279°. Теплота плавления 2520
кал[г-атом,теплота парообразования 69600кал]г-атом
при т. кип. Ат. теплоемкость (в кал]г-атом • град)
твердого А. увеличивается от 5,99 при 0° до 7,36 при
660°, расплавленного — от 6,75 при т. пл. до 7,19
при 1 000°. Теплопроводность 0,520 кал/см сек град
при 20°. Температурный коэфф, линейного расширения
24,58 • 10~6 в интервале 20—200°. Уд. электрич. со-
противление металла чистотой 99,996% составляет
2,6548 мком • см при 20°. А. слабо парамагнитен. А.
высокой чистоты имеет пониженную механич. проч-
ность, но значительно мягче и пластичнее технич.
металла. Модуль упругости А. чистоты 99,996%
составляет 7 000 kcImm2', твердость по Бринеллю
(в кг/мм1), отожженного 17, холодцокатанвого 27; пре-
дел прочности при растяжении (в кг /мм2) соответствен-
но 5 и 11,5; относительное удлинение (в %) 49 и 5,5.
Во всех своих устойчивых соединениях А. трехвален-
тен, но при высоких темп-pax он может проявлять
валентность 1 и значительно реже 2. А. обладает очень
высоким сродством к кислороду, на воздухе покры-
вается сплошной тонкой пленкой окиси, защищающей
металл от дальнейшего окисления. Особенно быстро
А. окисляется в расплавленном состоянии. На чи-
стом А. сила сцепления окисной пленки очень велика,
и такой металл весьма стоек в отношении атм. корро-
зии, морской воды, конц. азотной к-ты и др. реагентов.
Прочность окисной пленки и защитное действие ее
сильно убывает в присутствии примесей Hg, Na, Mg,
Си и др. При этом понижается сцепление пленки
с поверхностью, нарушается непрерывность слоя
и возникают гальванич. пары в местах контакта А.
с др. фазами. При накаливании мелко измельченный
или порошкообразный А. энергично сгорает на воз-
149
АЛЮМИНИЙ
150
духе. Сжиганием А. в токе кислорода достигается
темп-pa выше 3000°.
При комнатной темп-ре в атмосфере фтора А. по-
крывается пленкой A1F3, препятствующей дальней-
шей реакции; при темно-красном калении взаимодейст-
вие происходит очень энергично. Хлор и жидкий бром
реагируют с А. как при обычной, так и при повышен-
ной темп-pax, энергичное соединение с иодом идет
лишь при нагревании. Продуктами реакции являются
галогениды А. общей формулы А1Х3 — белые кристал-
лин. в-ва. Фторид не плавится, нерастворим в воде,
мало летуч. Хлорид, бромид и иодид резко отличаются
по свойствам от фторида: они легко летучи; в расплаве,
парах и нек-рых органич. растворителях димеризо-
ваны с образованием молекул А12Х6, имеющих в газо-
образном состоянии конфигурацию сдвоенного тетра-
эдра с общим ребром; прочность димерных молекул
падает от хлорида к иодиду. Хлорид, бромид и иодпд
исключительно гигроскопичны, вследствие гидролиза
дымят на воздухе, хорошо растворимы в воде и многих
органич. растворителях, легко вступают в реакции
присоединения с водой, аммиаком, галогенидами ще-
лочных металлов. Являются катализаторами 'многих
органич. реакций. Бромид и иодид характеризуются
соответственно след, константами;
А1Вг3 <7-5 3,01; т. пл. 97°; т. кип. 255°
AIJ3 : 3,98; т. пл. 191°; т. кип. 385°
Свойства алюминия фторида и алюминия хлорида
см. в соответств. статьях.
При темп-ре красного каления А. реагирует непо-
средственно с серой и парами серы в токе водорода,
образуя сульфид A12S3 — белые игольчатые кристал-
лы, т. пл. 1100°, полностью гидролизующиеся даже
следами влаги на воздухе. С водородом А. не взаимо-
действует, но косвенным образом может быть получен
гидрид (А1Н3)Ж—белое некристаллич. в-во, разлагаю-
щееся выше 105° с выделением водорода. Известны
соединения А1Н3 с органич. молекулами, напр.
А1Н3 2N(CH3)3. Значительный интерес для химии А.
представляют двойные гидриды А. и элементов I и II
групп периодич. системы состава МеНп пА1Н3 —
алюмогидриды, существование которых стало извест-
но сравнительно недавно (1946). Алюмогидриды —
белые твердые вещества, водой разлагаются с вы-
делением водорода. Находят применение в органич.
синтезах в качестве гидрирующих агентов. Нек-рые
из них солеобразцы (напр., LiAlH4 — лития алюмо-
гидрид), другие — металлоподобны (MgH2 • 2А1Н3,
ВеН2 2А1Н3). Выше 800°А. образует с азотом нит-
рид A1N — в чистом виде белые кристаллы, т. пл.
2200° (под давлением азота 4 ат). Взаимодействие
с углеродом начинается при 650°, но протекает энер-
гично ок. 1400°, при этом получается карбид А14Сд —
в чистом виде кристаллы ярко-желтого цвета, при
обычной темп-ре медленно разлагающиеся водой с
выделением метана. При темп-ре красного каления А.
реагирует с СО и СО2; продуктами реакций являются
А14Сз, А^Од и свободный углерод. В ряду напряжений
А. располагается между Mg и Zn, его нормальный
электродный потенциал равен —1,66 е в кислом р-ре
и — 2,35 в в щелочном. Вследствие образования за-
щитной пленки А. устойчив в сильно разб. и очень
конц. HNO3, незначительно растворяется в разб.
и конц. H2SO4; в этих к-тах средних концентраций
растворяется медленно. Более сильно действует на
А. НС1; Н3РО4 совсем не реагируете А. Р-ры солей А.
вследствие гидролиза показывают кислую реакцию.
Ион А13+ в р-ре сильно гидратирован, а при выделе-
нии из р-ра солей А. многие из них образуют кристал-
логидраты с тем или иным числом молекул воды (см.,
напр., Алюминия сульфат). Большинство солей А.
хорошо растворимо в воде. А. легко растворяется
в щелочах, выделяя водород и образуя алюминаты.
Из соединений низшей валентности наиболее изу-
чены субгалогениды А1Х, особенно A1F и А1С1. Оба
соединения получаются при взаимодействии расплав-
ленного металлич. А. с соответствующим галогени-
дом А1Х3; устойчивы выше 1000°, при понижении
темп-ры разлагаются по реакции диспропорциони-
рования: ЗА1Х = 2А1 А1Х3. Субфторид играет весь-
ма важную роль при получении особо чистого А.
А. относится к аналитич. группе элементов, обра-
зующих под действием сульфида аммония гидроокиси
или основные соли. Породы, содержащие А., разла-
гают действием смеси HF, HNO3, H2SO4. Сплавы А.
можно растворять в кислотах или щелочах. От щелоч-
ных и щелочноземельных элементов, Mg, небольших
количеств Ni и Мп А. можно отделить осаждением его
р-ром NH40H в присутствии NH^Cl. Элементы серо-
водородной группы отделяют от А1 осаждением их
сероводородом из кислого р-ра. Fe, Си, Ni, Со, Сг,
Zn, Ga, Sn и др. успешно выделяют из сернокислого
р-ра, содержащего А., электролизом с ртутным ка-
тодом. Качественно А. определяют по образованию
лаков, окрашенных в красный цвет, с ализарином,
ализарином S или алюминоном. В большинстве слу-
чаев при анализе горных пород, руд, минералов ко-
лич. содержание А. устанавливают по разности, после
определения всех остальных компонентов, во взвешен-
ном прокаленном остатке (весовой формой служит
А12О3). Количественно А. осаждается также 8-окси-
хишпТином в форме A1(C9H6NO)3. Имеются фотомет-
рия. и комплексометрия, методы определения А.
Получение. В пром-сти А. получают электролизом
р-ра глинозема А12О3 в расплавленном криолите
Na3AlF6 (6—8% А12О3 и 94—92% Na3AlF6). В послед-
ние годы в электролит обычно добавляют нек-рое ко-
личество A1F3 и до 8—10% смеси CaF2 и MgF2, что
улучшает показатели процесса. Темп-pa электролиза
ок. 950°, т. е. значительно выше т. пл. А., к-рый полу-
чается поэтому жидким.
Электролитная ванна представляет собой железный кожух,
футерованный огнеупорным кирпичом и угольными плитами.
Подина ванны служит катодом; она собрана из угольн-ых бло-
ков, снабженных залитыми чугуном стальными стержнями,
свободные концы к-рых присоединяются к токоподводящим
шинам. Погруженные в электролит аноды представляют собой
алюминиевые кожухи, набитые угольной массой. Получение
А. электролизом криолито-глиноземного расплава впервые бы-
ло предложено П. Эру (Франция) и Ч. Холлом (США) в 1886.
Теория процесса разработана П. П. Федотьевым.
Из всех составляющих электролит, А12ОЯ имеет наименьший
потенциал разложения. Поэтому при прохождении тока гли-
нозем разлагается на А., к-рый разряжается и накапливается
на подине, и кислород, образующий с угольным анодом СО
и СО2. К глинозему предъявляются высокие требования по
чистоте и размерам частиц. Примеси более электроположитель-
ных элементов, чем А., разлагаются и загрязняют металл,
а более электроотрицательных — накапливаются в электро-
лите и нарушают нормальный ход процесса. При достаточном
содержании глинозема ванна работает нормально при напря-
жении ок. 4,5 в. Обеднение электролита глиноземом до нек-рого
предела (1—1,6%), зависящего от состава электролита и плот-
ности тока, сопровождается резким повышением напряжения
(анодный эффект). Для его предупреждения заранее добавляют
очередную порцию глинозема. В процессе электролиза ме-
няется состав электролита, т. е. мол. отношение NaF и A1F3;
чтобы поддерживать его постоянным, в электролит периоди-
чески добавляют A1F3.
Ванны присоединяют к источнику постоянного тока последо-
вательно, образуя серии из 150—160 ванн. Соврем, электроли-
зеры работают при силе тока до 120—130 ка. Суточная произ-
водительность такой ванны ок. 1000 кг металла. А. извлекается
обычно с помощью вакуум-ковша один раз в 3—4 дня, а из
мощных ванн — один раз в сутки. В черновом металле содер-
жатся металлич., неметаллич. и газообразные примеси. Про-
дувкой его хлором освобождаются от неметаллич. и газооб-
разных примесей, а также от Na, Са и Mg — их хлорируют,
и хлориды всплывают на поверхность. Очищенный металл раз-
ливают в чушни. В лучшем сорте первичного металла содер-
жится только 99,7% А. Более чистый металл (99,996%), отли-
чающийся особыми свойствами получают электролитич. рафи-
нированием; наиболее чистый А. (99,99999%) получен в Совет-
ском Союзе путем многократной перегонки А. через субфторид.
На получение 1 т чернового А. расходуется 1,920 т глино-
151
АЛЮМИНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
152
зема, 0,065 т криолита, 0,035 т фторида алюминия, 0,600 т
анодной массы и 17 000 квт-ч электроэнергии постоянного тока.
Техника безопасности. При электролитич. произ-ве
А. возможны поражения электрич. током, высокой
темп-рой и вредными газами (HF, СО). Мерами пре-
дупреждения против поражения электрич. током
являются: хорошая изоляция алюминиевых ванн (за-
стилка пола асфальтом, диабазом), а также пользо-
вание сухими валенками; для защиты от высокой
темп-ры применяют соответств. спецодежду (фартуки,
брюки с наколенниками, валенки). Хорошей вентиля-
цией поддерживается здоровая атмосфера.
Применение. По практич. применению А. занимает
одно из первых мест среди металлов. Важнейшая об-
ласть — производство легких сплавов на его осно-
ве (см. Алюминия сплавы). А. — одна из самых
распространенных легирующих добавок в сплавах
на основе меди, магния, титана, никеля, цинка и
железа. А. широко применяется для раскисления ста-
ли перед заливкой ее в форму, а также в процессах
получения нек-рых металлов (кальция, бария, лития)
методом алюминотермии. В виде чистого металла А.
используется для изготовления химич. аппаратуры,
электрич. проводов и конденсаторов, в строительстве,
а также для защиты др. металлов от атм. коррозии
и т. д. В значительных количествах используются
и нек-рые соединения А.
Лит.: Беляев А. И., Раппопорт М. Б., Ф и р-
с а н о в а Л. А., Электрометаллургия алюминия, М., 1953;
Беляев А. И. и Фирсанова Л. А., Одновалентный
алюминии в металлургических процессах, М., 1959; G m е-
1 1 п, 8 Aufl., System-Nummer 35, TI А—В, В.—Winheim,
1934—50; Mellor, v. 5, L., 1929; то же, 5, 1952 ; U 11 m a n n,
3 Aufl., Bd 3, Munch. —B., 1953, S. 331—428, Kirk, v. 1, N. Y.,
1947, p. 591—656; Гилл eOp ан д В. Ф. [и др.], Прак-
тическое руководство по неорганическому анализу, пер. с англ.,
М., 1957. А. И. Лайнер.
АЛЮМИНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ —
известны двух типов: полные, в к-рых все валент-
ности А1 связаны с органич. радикалами, и непол-
ные, в к-рых одна или две валентности заняты
галогеном, алкоксигруппой или водородом. А л ю-
миний-триалкилы — бесцветные жидкости,
крайне чувствительные к кислороду и влаге; низ-
шие члены ряда самовоспламеняются на воздухе;
алюминий-арилы — твердые вещества. Все
работы с А. с. выполняются в атмосфере инертных
газов (N2, Аг).
Физич. свойства важнейших А. с. приведены в табл.
Формула Т. пл., °C Т. кип., °С/Л1Л1 Плотность, г/см3
(СН3),А1 15 126/760 0,752
(С.,Н5)3А1 -52,5 207/760 0,837
(С»Н5)3А1 (С,Н5)3О — 216—218/760 —
(С.,Н5).,А1С1 — 125—126/50 —
С»Н5А1С13 32 114,5-115,5/760 —
(изо-С4Нд)зА1 4,3 47/1 0,7859
(1дзо-С4Н9)оА1Н — 116—118/1 —
(СвН5)3А1 237 — —
(UI0±±7)gAl 255—256,5 — —
Все А. с. дают прочные комплексы с эфиром, амина-
ми и т. п.; с NaR, LiR образуют комплексы типа
Me+[A1R4]“. А.с. энергично реагируют с соединениями,
содержащими подвижный водород. А. с. легко всту-
пают в реакции типа: 2R3A1 -f- AlHal3 —>3R2AlHal;
R2AlHal -|- AlHal3 —> 2RAlHal2; обратные реакции
практически идут лишь для R = СН3. Алкилалюми-
нийгалогениды могут быть симметризованы до R3A1
действием Na или NaF. При нагревании R3A1 отщеп-
ляют олефины; особенно легко отщепляется изобути-
лен: (ц30-С4Н9)3А1->^=.^ (нзо-С4Н9)2 А1Н-|-С4Н8.
Алюминийтриалкилы, не принадлежащие к типу
(пао-С4Н9)3А1, легко присоединяют а-олефины,
образуя высшие гомологи: A1R3 3R1CH=CH2 —►
—> A^CHjCHRRJj. Изобутилен легко вытесняется др.
а-олефинами. Высшие R3A1 под действием Ni дают три-
этилалюминий и соответств. а-олефин: >. A1(CH2)4R —>
—> А1(С^Н6)3RCH— СН2. При нагревании R3A1,
содержащих вторичные R, последние перегруппиро-
130°
вываются в первичные, напр.: (нзо-С3Н7)3А1 ——>
Окисление R3A1 приводит к соответствующим ал-
коголятам (RO)3A1 и может быть использовано для
получения первичных спиртов из а-олефинов. Кар-
боксилирование R3A1 (180—280°, 70—400 ат) дает
после разложения водой карбоновые к-ты с выходами
50—70%. Обменные реакции R3A1 с галогенидами
В, Si, Р, Sb, Bi, Sn, Pb, Hg, Zn, Cd приводят к полным
алкильным производным этих элементов и галогени-
дам алюминия; в случае В, Р, Т1 реакция может быть
остановлена на соединениях типа R9X2 и R29X. Гид-
риды типа R2A1H, особенно (нзо-С4Н9)2А1Н, могут
быть использованы как восстановители, близкие по
свойствам LiAlH4; (пзо-С4Н9)2А1Н восстанавливает
нитрилы и замещенные амиды к-т в альдегиды с вы-
ходами 50—90%; альдегиды и кетоны восстанавли-
ваются до спиртов. Действие (С2Н5)3А1 и(пзо-С4Н9)3А1Н
на хлориды металлов VI группы под давлением оки-
си углерода приводит к гексакарбонилам этих ме-
таллов.
А. с. получают: 1) действием галогеналкилов на
сплав Al/Mg, например 3RHal -4- 2А1 —> RAlHal2 -4-
+ R2AlHal, или 6RHal %-2А1 %-3Mg —> 2R3A1 4-
Ц- 3MgHal2; 2) реакцией ртутьорганич. соедине-
ний с А1 (100—140°), напр. 3R2Hg-- 2А1 —>2R3A1 -4-
-4- 3Hg, или с А1С13, напр. R3MgHal -- А1С13 —> R3A1—|—
MgHalCl; 3) действием замещенных этиленовых
углеводородов на дизамещенное А. с., напр. =А1Н-|-
+ RR1C=CH2 -► =А1—CHjCHjRRj, или на А1 в
присутствии водорода [применяется в пром-сти, осо-
бенно для получения (пзо-С4Н9)3А1], напр. А1
+ 172Н2 4- 3RR1C=CH2 -> (RR1CHCH2)3A1; Al ак-
тивируется измельчением и добавками иода, А1С13
и др. Каждый из этих способов является специфичным
для получения определенных А. с.
Элементарный анализ А. с. выполняется сожжением
навески, взятой в инертной атмосфере, в токе кисло-
рода с последующим определением С и Н весовым
методом, как обычно. Одновременно с С и Н может
быть определен А1 в виде А12О3; определение А1
выполняется также методами неорганич. анализа
после гидролиза или алкоголиза вещества. Содержа-
ние триалкилалюминия и диалкилалюминийгидрида
(«активного алюминия») в А. с. может быть установлено
в присутствии продуктов окисления: 1) измерением
объема выделившихся газообразных углеводородов и
водорода (или аммиака) при разложении А. с. спир-
том (или аммиаком); 2) по образованию окрашен-
ных комплексов с изохинолином, количества к-рых
измеряются титриметрически или колориметрически;
3) титрованием разб. р-ров А. с. оргапич. основа-
ниями.
А. с. применяют в качестве катализаторов полиме-
ризации олефинов при низком давлении («катализа-
торы Циглера—Натта» )и стереоспецифич. полимериза-
ции диенов. А. с. используются также для получения
ультрачистого алюминия и ряда элементоорганич.
соединений.
Лит.: Синтетические методы в области металлоорганиче-
ских соединений, под ред. А. Н. Несмеянова и К. А. Кочеш-
кова, вып. 4, М.—Л., 1945; Циглер К., Усп. хим., 1957,
26, № 10, 1187—1202; Koster В.., Annalen der Chemie,
1958, 618, Н. 1, 31—43; Захаркин Л. И. и Гаври-
ленко В. В., ДАН СССР, 1958, 118, № 4, 713; Захар-
кин Л. И. иОхлобыстин О. Ю., Изв. АН СССР.
Отд. хим. н. 1959, № 11, 1942; Z i eg 1 е г К. [u. a.], Justus
Liebigs Ann. Chem., 1960, 629, H. 1—3, 1.
О. Ю. Охлобыстин.
153
АЛЮМИНИЯ НИТРАТ — АЛЮМИНИЯ ОКИСЬ
154
АЛЮМИНИЯ НИТРАТ (алюминий азотнокислый)
A1(NO3)3 — при обычной темп-ре (от —16° до 70°) ус-
тойчив кристаллогидрат A1(NO3)3 • 9Н2О. При 70—105°
из водных растворов выделяется A1(NO3)3 8Н2О,
выше 105° — A1(NO3)3 6Н2О. Имеются указания на
возможность существования и других гидратов.
A1(NO3)3 • 9Н2О — бесцветные ромбич. кристаллы,
расплывающиеся на воздухе; теплота образования
&Н°т — —897,34 ккал/моль-, т. пл. 73,5°; выше этой
темп-ры А. п. теряет воду, а затем и окислы азота
и к 200° превращается в А12О3. Хорошо растворим
в воде (75,5 г в 100 г Н2О в расчете на безводный
А. н.), растворим в спирте. Получают А. н. растворе-
нием АГ(ОН)3 в HNO3, р-р упаривают и А. н. кристал-
лизуют. Исходный А1(ОН)3 можно получить раство-
рением А1 в водном р-ре NaOH с последующей частич-
ной нейтрализацией р-ра алюмината натрия 30%-пой
HNO3,b результате чего выпадает А1(ОН)3, к-рый отмы-
вают. Наибольшее применение А. н. находит как про-
трава при крашении в текстильной пром-сти, а также
для произ-ва катализаторов, в нефтяной пром-сти
и как лабораторный реактив.
АЛЮМИНИЯ ОКИСЬ (глипозем) А12О3 — соеди-
нение алюминия с кислородом, составная часть глин,
исходный продукт для получения алюминия.
Кристаллические формы и свой-
ства. Устойчивыми кристаллич. формами А. о.
являются а-А12О3 и у-А12О3. Первая из этих
модификаций представляет собой белые кристаллы
с ромбоэдрич. решеткой а = 5,13 А, угол а = 55,16°,
плотн. 3,96; т. пл. 2 050°; т. кип. выше3000°. Теплота
образования ДН%8 — —400 ккал]молъ. Зависимость
мол. теплоемкости от темп-ры = 23,86 0,00673 t
кал/молъ-град (для 100—1400°); а-А12О3 встре-
чается в природе в виде минерала корупда, к-рый
часто содержит в растворенном виде окислы др. ме-
таллов, придающих ему различную окраску. Про-
зрачные окрашенные кристаллы являются драгоцен-
ными камнями (сапфиры, рубины). а-А12О3 обра-
зуется при нагревании до высоких темп-р (900—
1200° и выше) гидроокиси алюминия и его солей,
а также при алюминотермия, восстановлении окислов.
Вторая кристаллич. форма, у-А12О3, — кубиче-
ская, может быть получена нагреванием алюминиевых
солей и нек-рых форм гидроокиси алюминия до 600—
900°; выше этой темп-ры необратимо переходит
в а-А12О3. Превращение у-формы в a-форму за-
капчивается при 1200° с сокращением объема на
14,3%. В литературе описано также 7 неустойчивых
форм А. о.
Известны соединения, соответствующие различной
степени гидратации А12О3. К гидроокисям относятся:
гидраргиллит (гиббсит) А1(ОН)3, моноклин-
ные кристаллы, плотп. 2,42, переходящие при 165—
200° в бемит, гидраргиллит входит в состав многих
бокситов; б а й е р и т, неустойчивая форма А1(ОН)3
(начинает разлагаться уже при 120°), моноклинные
кристаллы, получаемые искусственно. Кроме полных
гидроокисей, известны и такие, как А1ООН, в виде
двух ромбич. модификаций: диаспор а-А1ООН,
плотн. 3,3—3,5, устойчив до 350°, в интервале тем-
ператур 350—420° переходит в а-А12О3; бемит,
у-АЮОН, плотн. 3,01, выше 600° переходит в
а-А12О3.
А. о. и ее гидратированные формы нерастворимы
в воде, обладают амфотерными свойствами — взаимо-
действуют с кислотами и едкими щелочами; с NH4OH
не реагируют. Химич, активность природных соеди-
нений А. о. и гидроокисей убывает в ряду: гидраргил-
лит—> бемит—> диаспор—> глинозем; так, темп-ры
реакции трех первых из них с NaOH в р-ре состав-
ляют соответственно 100°, 180° и 200°. Активность ис-
кусственных препаратов сильно уменьшается с по-
вышением темп-ры их получения. Природный корунд
на воздухе при обычных условиях химически инертен
и не гигроскопичен. Ок. 1000° он интенсивно реа-
гирует с едкими и углекислыми щелочами, образуя
растворимые в воде алюминаты наиболее активных
одновалентных металлов. Медленнее реагирует с крем-
неземом и кислыми шлаками с образованием алюмо-
силикатов, разлагается сплавлением с KHSO4. По-
добным же образом ведет себя искусственный корунд,
образовавшийся при температуре >. 1200°. а-А12О3,
полученный из диаспора при 500—600°, разлагается
не только при темп-pax сплавления или спекания ще-
лочами, но и р-рами как щелочей, так и к-т. у-А12О3,
образовавшийся при обжиге гидратов ок. 550°, весьма
гигроскопичен и химич. активен. Гигроскопичность
и активность у-А12О3 резко снижаются с повыше-
нием темп-ры обжига. Химич, соединения А. о., об-
разующиеся при взаимодействии как с кислотами,
так и с щелочами, легко гидролизуются водой.
Получение. Исходным сырьем для получении А. о.
служат бокситы, нефелины, каолины идр. алюминий-
содержащие продукты (см. Алюминий).
Бокситы перерабатываются двумя способами. Когда
в бокситах весовое содержание глинозема в 8 раз
больше, чем содержание SiO2, сырье перерабаты-
вают по т. и. мокрому способу. При отношении
А12О3 : SiO2 <Z 7 переработку производят спеканием.
Часто комбинируют эти 2 способа.
При мокром щелочном способе (разрабо-
тан К. И. Байером на Тентелевском заводе в Петербурге в
конце 19 в.) боксит дробят, затем измельчают в шаровых
мельницах. Измельченный продукт загружают в автоклавы,
в которых производят выщелачивание оборотным р-ром (после
выделения пз него части А12ОЯ) при 225—230°. При атом
образуется алюминат натрия NaAlO2, переходящий в р-р.
В случае бокситов, содержащих гидраргиллит, выщелачи-
вание можно производить при 105° и атмосферном давле-
нии в аппаратах с мешалкой, а не в автоклавах. Алюминатные
р-ры разбавляют водой, затем производят отделение р-ра
от шлама. Р-р, содержащий алюминат натрия, подвергают
разложению, называемому в промышленности выкручиванием
(производится в аппаратах с мешалкой или с аэролифтом).
Эта операция продолжается 60—70 час., причем выделяется
ок. 1 /2 А12О3. Маточный р-р упаривается и поступает на выще-
лачивание новой партии боксита. Для затравки в р-р вносят
кристаллы А1(ОН)3. Образовавшийся осадок гидроокиси алюми-
ния отфильтровывают и прокаливают во вращающихся печах
при 1200°; в результате получается чистый глинозем.
Сухой щелочной способ основан на спекании
измельченного боксита, смешанного с содовым р-ром и извест-
няком во вращающихся печах при темп-ре ок. 1200°. Получен-
ную т. о. спекшуюся массу подвергают выщелачиванию водой.
Р-р алюмината натрия отделяют от шлама, затем освобождают
от кремнезема, осаждая последний известью в автоклаве при
6 ат. Отделенный от осадка p-j) алюмината разлагают газо-
образным СО2. Образовавшуюся гидроокись отделяют от
р-ра и прокаливают при 1200°.
Нефелин представляет большой интерес потому, что, наряду
с глиноземом, из него получаются также и др. ценные про-
дукты. Так, при переработке 4,3 т нефелинового концентрата
получается: 1 т глинозема, 1 т кальцинированной соды (и
поташа) и 7,5 т портланд-цемента. Нефелиновый концентрат
и известняк измельчают в шаровой мельнице, туда же доба-
вляют воду или содовый р-р, образовавшуюся пульпу под-
вергают спеканию в трубчатых печах при 1270—1290°. Далее
спекшуюся шихту обрабатывают примерно так же, как и в слу-
чае произ-ва А12О3 сухим способом из бокситов. Маточный
р-р используется для получения соды и поташа, а шлам — для
получения цемента. При получении глинозема из алунитов
можно одновременно получать также серную к-ту и сульфат
калия. Руду, содержащую алунит, обжигают при 500—580°
и обрабатывают р-рами едкого натра по схеме мокрого щелоч-
ного способа.
Главное применение А12О3 — в произ-ве алюминия
электролизом криолито-глиноземных расплавов. Ко-
рунд широко используется как абразивный материал
(корундовые круги, наждак — мелкозернистый ко-
рунд с примесями кварца, окислов железа и др.),
а также для изготовления керамич. резцов спеканием
технич. глинозема с небольшим количеством связую-
щего материала. Получаемые путем плавки порошка
А12О3 с добавками окислов Сг, Fe, Ti, V монокристал-
лы корунда используют для изготовления опорных
камней в точных механизмах и в ювелирных изделиях.
155
АЛЮМИНИЯ ОКИСЬ ДЛЯ ХРОМАТОГРАФИИ — АЛЮМИНИЯ СПЛАВЫ
156
Изделия из А. о. и высокоглиноземистых материалов
применяются в качестве огнеупоров, диэлектриков,
химически стойких тиглей и др. Гидроокиси исполь-
зуются для произ-ва всевозможных солей: A1F3,
Na3AlF6, A12(SO4)3 и др.; активные формы глинозема—
как адсорбенты и катализаторы.
Лит.: Лайнер А. И Производство глинозема, М.,
1961; Кузнецов С. И., Производство глинозема, Сверд-
ловск, 1956; Федотьев К. М., Гидраты глинозема,
Тр. Ин-та геол. наук. АН СССР, 1949, вып. 120, петрогра-
фическая серия, Кг 35, с. 86. См. также при ст. Алюминий.
А. И. Лайнер.
АЛЮМИНИЯ ОКИСЬ ДЛЯ ХРОМАТОГРАФИИ —
мелкокристаллический порошок, плотн. 2,55, размер
зерен 10—30 мк; величина уд. поверхности для
различных сортов колеблется в пределах от 170 до
300 м'2/г. Адсорбц. свойства А12О3 в сильной сте-
пени зависят от способа приготовления, обработки
и качества исходных материалов. Для молекулярной
хроматографии применяется А12О3, получаемая нагре-
ванием чистого гидрата окиси до ярко-красного кале-
ния или путем активирования алюмогеля из боксито-
вых руд; продукт может быть частично дезактивиро-
ван путем увлажнения на воздухе или добавлением
соответств. количества воды (1 —10%). Наилучшими
адсорбц. свойствами по отношению к катионам обла-
дает А12О3, имеющая высокое значение pH (более 9,4)
и богатая натрием. Активность окиси алюминия для
молекулярной хроматографии характеризуется с по-
мощью набора красителей, по величине теплового
эффекта смачивания растворителем или определением
влажности. Обменная емкость колеблется в пределах
от 0,07 до 0,1 мг-экв!г; ионообменной группой являет-
ся группировка NaAlO2. Схематически эквивалент
А12О3 может быть представлен так: (А12О3)Х-NaAlO2.
В таком виде препарат пригоден для хроматогра-
фического разделения катионов. А12О3 представляет
собой адсорбент с амфотерными свойствами; для хро-
матография. разделения анионов она обрабатывается
кислотой (напр., 1н. HNO3), после чего промывается
водой. Впервые применена М. С. Цветом в качестве
адсорбента для хроматография, анализа органич. ве-
ществ; в хроматографии неоргапич. в-в начала при-
меняться с 1938, однако большого распространения
не получила вследствие малой обменной емкости и
нестойкости в кислых и щелочных растворах; успешно
используется в молекулярной хроматографии, напр.
при разделении, очистке и анализе ароматич. углево-
дородов и их полинитропроизводпых, при исследова-
нии неомыляемых веществ различных тканей и орга-
нов животных, при анализе аминокислот, первичных
продуктов каталитич. окисления каротина, получе-
нии нек-рых жирорастворимых витаминов, при ана-
лизе нефтепродуктов, выделении и очистке фермен-
тов и т. п.
В хроматографии неорганич. соединений А12О3
может быть использована для маркировки сплавов,
разделения молибдена и железа, рения и молибдена,
ванадия и молибдена, вольфрама и ниобия и др.;
применяется также в качестве носителя в осадочной
хроматографии и в электрофоретич. хроматографии.
Коллоидным раствором А12О3 пользуются для получе-
ния специальной хроматографии, бумаги, обладающей
повышенной разделительной способностью.
Лит.: Шемякин Ф. М. [и др.]. Хроматографический
анализ. Введение в теорию и практику, М., 1955; Исследова-
ния в области хроматографии. Труды Всесоюзного совещания
по хроматографии, 21—24 ноября 1950 г. [ред. акад. М. М. Ду-
бинин], М„ 1952. И. П. Харламов.
АЛЮМИНИЯ СПЛАВЫ — сплавы на основе А1
с добавками Си, Si, Mg, Zn, Мп, Ni, Fe, Ti и др.
(в сумме до 20%). Промышленные А. с. обычно содер-
жат не менее 2—3 легирующих элементов (см. Сплавы).
Кремний и железо, если и не добавляются специально,
то присутствуют во всех А. с. как неизбежные примеси.
Легирующие добавки вводятся в А1, гл. обр. для повы-
шения механич. прочности. При кристаллизации
А. с. образуются повышающие механич. прочность А1
твердые растворы и интерметаллические соединения.
В кристаллич. гранецентрированной кубич. решетке
твердого р-ра на основе А1 часть атомов А1 замещена
атомами др. элементов. Интерметаллиды в А. с.
бывают двойные (CuA12, Mg2Si, MgZn2), тройные
(Al2CuMg) и более сложные, с точно неустановленной
формулой (Cux MgvSizAl,(). Вследствие большой хруп-
кости интерметаллидов их содержание в сплавах
ограничивается. Добавки и примеси снижают пластич-
ность, электро- и теплопроводность А1. Растворы
кислот, щелочей и солей действуют на А. с. примерно
так же, как и на алюминий. В большинстве сред А. с.
менее стойки против коррозии, чем чистый А]. В ат-
мосфере сухого и влажного воздуха и воздуха, за-
грязненного SO2, H2S и NH3, А. с. обладают удовлет-
ворительной стойкостью против коррозии. Устой-
чивы в пресной воде и в атмосфере водяного пара,
а также во многих расплавленных солях. Наиболь-
шая прочность А. с. достигается применением закалки
с последующим старением. При нагреве А. с под
закалку до 470—530° (темп-pa закалки должна нахо-
диться ниже точки начала плавления сплава во из-
бежание пережога, заключающегося в оплавлении
границ зерен и снижении из-за этого прочности
и пластичйости) интерметаллиды переходят в алюми-
ниевый твердый р-р. При последующем быстром охлаж-
дении в воде они не успевают выделиться из р-ра и,
следовательно, закалка фиксирует пересыщ. твердый
р-р легирующих элементов в А1. Такой раствор тер-
модинамически неустойчив и должен самопроизвольно
распадаться с выделением избытка растворенных эле-
ментов в виде интерметаллидов. В начальные стадии
распада выделяются высокодисперсные частицы ин-
терметаллидов, и прочность А. с. сильно повышается.
С повышением темп-ры распад р-ра ускоряется, и при
достаточно высокой темп-ре частицы выделяющейся
фазы в конце концов коагулируют, в результате чего
упрочнение А. с. сменяется разупрочнением. Изме-
нение структуры и свойств закаленного сплава бла-
годаря распаду пересыщенного р-ра при комнатной
темп-ре наз. естественным, а при повышенной
темп-ре — искусственным старением. Темп-pa и время
старения подбираются так, чтобы обеспечить наилуч-
шее сочетание прочности, пластичности и стойкости
против коррозии.
А. с. делятся на деформируемые и литейные (см.
табл.). Деформируемые А. с. подвергают
горячей и холодной обработке давлением — про-
катке, прессованию, волочению, ковке или штам-
повке и из них изготовляют листы, прутки, профили,
проволоку, штампованные изделия разной формы
и пр. Деформируемые А. с. должны быть весьма
пластичными, поэтому структура их состоит из твер-
дого алюминиевого р-ра и небольшого колич. Интер-
нета ллич. фаз-упрочнителей. Из деформируемых А. с.
наиболее широко известны дуралюмины, фазами —
упрочнителями в к-рых являются СпА12 и A!2CuMg.
На этих сплавах в начале 20 в. было открыто явление
старения. При отжиге (нагреве до 350—430° и мед-
ленном охлаждении) не образуется пересыщ. алюми-
ниевый р-р и дуралюмин мягок, его легко обрабаты-
вать давлением в холодном состоянии. Закалка
несколько упрочняет сплав; после нее в течение 1—3
часов дуралюмин еще достаточно пластичен. Затем,
благодаря естественному старению, в первые сутки
после закалки происходит сильное упрочнение, к-рое
через 4—6 суток практически прекращается. Предел
прочности дуралюмина после отжига, закалки и ста-
рения равен соответственно 23, 33 и 47 кг]мм2. Для
повышения стойкости против коррозии дуралюмин
плакируют (покрывают) алюминием. Под давлением
157
АЛЮМИНИЯ СПЛАВЫ — АЛЮМИНИЯ СУЛЬФАТ
158
валков при горячей прокатке алюминиевые листы
свариваются с дуралюмином; образуется лист, состоя-
щий из прочной дуралюминовой сердцевины и тонких
наружных алюминиевых слоев. По отношению к дур-
алюмину алюминий является анодом, поэтому даже
на срезах листа и около царапин, где обнажена дур-
алюминовая сердцевина, плакирующий слой оказывает
защитное действие (см. Защита металлов).
Сплавы типа авиаль (фаза-упрочнитель Mg2Si)
уступают по прочности дуралюминам, но превосходят
их по пластичности в горячем состоянии. Из авиалей
штамповкой изготовляют детали сложной формы.
Самые прочные при комнатной темп-ре А. с. типа
В95 относятся к системе А!—Zn—Mg—Си и содержат
фазы-упрочнители MgZn2 и Al2CuMg. Эти А. с. по
сравнению с дуралюминами менее пластичны и быстро
разупрочняются при нагреве выше 150°. Для повы-
шения стойкости против коррозии сплавы типа В95
плакируют сплавом А1 с 1% Zn. Плакировать их
алюминием нельзя, т. к. по сравнению с последним
они обладают более электроотрицат. потенциалом.
Для изготовления деталей, работающих при повышен-
ных темп-pax (напр., поршней), применяют ковочные
А. с. типа АК4. Жаропрочность их обусловлена при-
сутствием сложных интерметаллидов типа Al2CuMg,
(Fe, Ni)Al9 и др., замедляющих разупрочнение при
повышении темп-ры. Сплавы Al с Mg и Ми термообра-
боткой не упрочняются. Они применяются там, где
требуется сочетание высокой стойкости против кор-
розии, пластичности и хорошей свариваемости (напр.,
при изготовлении сварных резервуаров для жид-
костей и газов).
Литейные А. с. заливают в металлич. (ко-
кильные) или песчаные формы для изготовления
фасонных отливок разнообразной конфигурации. Для
того чтобы при заливке форма хорошо заполнялась
и отливка получалась без пор и трещин, литейный
Состав, термическая обработка и механические свойства некоторых сплавов алюминия
Название сплава и марка (ио ГОСТ) Содержание легирующих элементов Содержание примесей, % (не более) Термическая обработка Типичные механич. свойства
Qg, кг/мм2 «, % Н & кг/мм2
Fe Si
Алюминий 99,99%-ной чисто- 0,003 0,0025 Отжиг 420° 5 50 15
ты, AB000 Деформируемые сплавы
Дуралюмин нормальный, Д1 3,8-4,8% Си; 0,4-0,8% Mg; 0,4-0,8 % Мп 0,7 0,7 Занална с 505°; естест- венное старение 4 сутон 42 18 100
Дуралюмин повышенной прочности, Д 16 3,8-4,9% Си; 1,2-1,8% Mg; 0,3-0,9% Мп 0,5 0,5 Занална с 500°; естест- венное старение 4 сутон 47 17 105
Авиаль, АВ 0,45-0,9% Mg; 0,5-1.2% Si; 0,2—0,6% Си; 0,15—0,35% Мп 0,5 — Занална с 500°; старе- ние 160°, 15 час. 33 16 95
Высокопрочный А. с., В95 5,0-7,0% Zn; 1,8-2,8% Mg; 1,4—2,0% Си; 0,2—0,6% Мп; 0,1-0,25% Сг 0,5 0,5 Занална с 470°; старение 140°, 16 час. 60 12 150
Жаропрочный ковочный сплав, АК4 1,9—2,5% Си; 1.4—1,8% Mg; 0,5—1,2% Si; 1,1 —1,6% Fe; 1,0—1,5% Ni — — Занална с 535°; ' старение 170°, 16 час. 44 10 110
Магналий, АМг-5 4,0-5,5% Mg; 0,3-0,6% Mn; 0,5 0,5 Отжиг 400°, до 1,5 час. 27 23 70
Сплав АМц 1,0-1,6% Mn 0,7 0,6 Отжиг 400°, до 1,5 час. 13 20 30
Литейные сплавы
Силумин, АЛ2 10,0-13,0% SI 1,0 — Термообработке не под- вергается 18 6 55
Силумин, АЛ4 8,0-10,5% Si; 0,17-0,30% Mg; 0,25—0,5 % Mn 0,9 — Занална с 535°; старение 175°, 15 час. 26 4 70
Магналий, АЛ8 9,5-11,5% Mg 03 0,3 Закалка с 435° 30 12 70
ов — предел прочности при растяжении; 8 — относительное удлинение; Hg — твердость по Бринеллю.
сплав должен содержать много эвтектики. Поскольку
эвтектика значительно менее пластична, чем твердый
р-р, литейвый А. с. нельзя обрабатывать давлением
(гнуть, ковать и пр.), но его можно обрабатывать
резанием. Из литейных А. с. наиболее распростра-
нены силумины, в к-рых основная добавка—крем-
ний — образует с А1 эвтектику с т. пл. 577° (при
11,7% Si). В эвтектике Si находится в виде пластин.
Перед заливкой силумина в форму производят моди-
фицирование — обработку расплава солями натрия
(2/з NaF и !/3 NaCl в вес. долях). После модифици-
рования Si в эвтектике приобретает более желатель-
ную зернистую форму, чем обеспечивается большая
прочность и пластичность силумина. С кремнием
алюминий не образует фаз-упрочнителей. Чтобы
силумин можно было упрочнять старением, в него
дополнительно вводят элементы, образующие фазы —
упрочнители — Mg2Si, СнА]2 и др. Магналий (АЛ8) об-
ладает более высокими, чем силумин, механич. свой-
ствами и стойкостью против коррозии, но из-за мало-
го колич. эвтектики и широкого темп-рного интерва-
ла кристаллизации имеет худшие литейные свойства.
Достоинствами всех А. с. являются малая плот-
ность (2,65—2,8), высокая удельная прочность (от-
ношение предела прочности и плотности), удов-
летворительная стойкость против атмосферной корро-
зии, недефицитность, дешевизна и простота техноло-
гии их получения и обработки по сравнению с дру-
гими цветными сплавами.
Вначале производство А. с. стимулировалось гл.
обр. развитием авиации. В наст, время А. с. приме-
няются в ракетной технике, в авиа-, авто-, судо- и при-
боростроении, в ж.-д. транспорте, в химич. аппара-
тостроении, в строительстве зданий, в произ-ве
бытовых приборов и во многих др. областях. По
широте применения в нар. х-ве А. с. прочно заняли
второе место после стали и чугуна.
Лит.: Б о ч в .а р А. А., Металловедение, 5 изд., М.,
1956; Машиностроение. Энциклопедический справочник, т. 4,
М., 1947. И. И. Новиков.
АЛЮМИНИЯ СУЛЬФАТ (алюминий сернокислый)
A12(SO4)3 — при обычной температуре устойчивый
кристаллогидрат A12(SO4)3 • 18Н2О. Ниже 9,5° —
159
АЛЮМИНИЯ ФТОРИД — АЛЮМИНОТЕРМИЯ
160
A12(SO4)3 • 27Н2О. В различных условиях обезвожи-
вания отмечено образование менее богатых водой
гидратов. Обычно используется частично дегидрати-
рованный вследствие выветривания А. с., содержащий
14—14,5 молекул воды. С сульфатами щелочных ме-
таллов А. с. образует двойные соли, т. н. квасцы,
общей ф-лы MeTAl(SO4)2 12Н2О, где Me1— К, Na,
NH4 и т. д. A12(SO4)3 • 18Н2О — бесцветные кристал-
лы; d111,69; легко растворим в воде (36,15 г безводной
соли в .100 г Н2О при 20°); нерастворим в спирте;
при нагревании теряет воду не плавясь; при прока-
ливании распадается на А12О3 и SO3. Теплота обра-
зования безв. А. с. А Щ98 = —820,98 ккал]молъ.
Для получения А. с. в пром-сти в качестве сырья приме-
няют глину, богатую каолинитом А12О3 • 2 SiO2 - 2 Н2О, А. с.
может быть получен двумя способами. По первому из них глину
сначала обжигают при 700—800°, а затем обрабатывают 75%-ной
серной н-той при 100—117°. Образовавшийся раствор А. с. от-
фильтровывают от нерастворившихся примесей и упари-
вают- после охлаждения отделяют ' выпавшие кристаллы
A12(s'O4)2 18Н2О. По этому способу получают очищенный А. с.
По второму способу глину не обжигают, а обрабатывают конц.
серной к-той при 100—105°. Этим способом получают неочи-
щенный продукт,
А. с. в больших количествах применяют для очистки
воды, в произ-ве бумаги, в текстильной пром-сти,
при дублении кож, консервировании древесины и т. д.
Лит.: Позин М. Е., Технология минеральных солей,
Л.—М., 1949, см. также при статье Алюминий.
АЛЮМИНИЯ ФТОРИД (алюминий фтористый)
A1F3 — бесцветные кристаллы, ромбоэдрич. решетка,
а= 5,029 А, а = 58°31, плоты. 3,1; возгоняется без
плавления.
Теплота образования А/7298 — —311 ккал/молъ.
Давление паров достигает 760 мм рт. ст. при 1291°.
А. ф. мало растворим в воде (0,559 г в 100 г Н2О при
25°), лучше растворим в р-рах HF. Устойчив к действию
кипящего р-ра NaOH и К.ОН, холодных и горячих
к-т, кроме кипящей H2SO4, разлагается при сплавле-
нии с Na2CO3 и NaOH. С фторидами щелочных метал-
лов А. ф. образует комплексные соединения, напр.
криолит Na3AlF6. Получают А. ф. растворением
А1(ОН)3 в 15%-ном р-ре HF с последующей кристалли-
зацией и прокаливанием в токе HF или действием HF
на А12О3 при 400—700°. Обычно А. ф. содержит при-
месь оксифторида. Применяют в качестве компонента
электролита при электролитич. получении и рафи-
нировании А1.
Лит.: Рысс И. Г., Химия фтора и его неорганических
соединений, М., 1956, см. также при статье Алюминий.
АЛЮМИНИЯ ХЛОРИД (алюминий хлористый)
А1С13—бесцветные кристаллы, моноклинная базоцен-
трированная решетка, а = 5,91 А, b = 10,24А, с =
= 6,1бА, р = 108°39, плотн. d25 2,44, теплота обра-
зования Д//298 = —166,2 ккал) моль. При обычном
давлении А. х. возгоняется не плавясь; тройная
точка лежит при 192,6° и давлении пара 1715 мм рт.ст.
Вследствие гидролиза дымит на воздухе. В 100 г
воды растворяется 44,38 г А1С13 (25°); из водных р-ров
выделяется А1С13 • 6Н2О — желтовато-белые расплы-
вающиеся па воздухе кристаллы с запахом НС1.
А. х. растворим в ацетоне, пиридине, нитробензоле
и др., нерастворим в бензоле, толуоле. Твердый
безводный А. х. значительно лучше проводит электрич.
ток, чем расплавленный, что, по-видимому, связано
с переходом от ионной структуры к молекулярной,
сопровождающимся резким увеличением объема (почти
вдвое). Известны многочисленные соединения А1С13
с NH3, РС15, РОС13 и др. неорганич. и органич. соеди-
нениями.
Важнейший способ получения А. х. — действие
смеси хлора и СО на обезвоженный каолин или боксит
в шахтных печах по принципу противотока. В нек-рых
случаях для произ-ва А. х. используют металлич.
алюминий, глинозем и др. соединения А1. При полу-
чении А. х. из природного сырья в качестве примесей
образуются хлориды кремния, титана и железа.
SiCl4 и TiCl4 легко отделяют от А1С13, пользуясь
их большей летучестью. FeCl3 отделяют восстановле-
нием до металлич. железа перегонкой А1С13 над
металлич. А1. Безводный А. х. широко применяют
как катализатор в органич. синтезе, гл. обр. при
алкилировании (см., напр., Фриделя—Крафтса реак-
ция), а также при крекинге нефти и в др. процессах.
А. х. используют также при крашении шерсти для
гидролитич. расщепления примесей растит, проис-
хождения (под действием НС1, образующейся при
гидролизе А1С13 в водном р-ре).
Лит.: Томас Ч. А., Безводный хлористый алюминий
в органической химии, пер. с англ., М., 1949; См. также при
статье Алюминий.
АЛЮМИНОН (аммониевая соль ауринтрикарбоно-
вой кислоты) C22H23O9N3,
нево-красный порошок;
легко растворим в воде,
мало — в спирте даже
при нагревании, почти
не растворяется в ацето-
не; водный р-р окрашен
в красвый пвет и имеет
нейтральную реакцию.
Получают А., исходя из
сульфосалициловой к-ты
и формалина. В ацетатном растворе А. с солями А1
дает красное окрашивание или осадок (т. н. лак,
неисчезающий при добавлении NH4OH, что позво-
ляет отличать его от хромового лака); аналогично,
но при других значениях pH, А. реагируете
солями Ba, Са, Sr, Ba, Be, Sc, Се, La, Nd, Ac, Zr,
Hf, Th, Cr, In, Ga, Fe, Er. Применяют для фотометрия,
определения и обнаружения AI и фторидов (по ослаб-
лению окраски алюминиевого лака), для цветных
реакций на Th, Zr, Ga, Sc, Се и нек-рые другие эле-
менты.
Лит.: Wei cher F. J., Organic analytical reagents,
V. 2, N. Y., 1947, p. 94. В. Г. Типцова.
АЛЮМИНОТЕРМИЯ (алюмотермия) — способ по-
лучения металлов и неметаллов (а также сплавов)
восстановлением их кислородных соединений метал-
лич. алюминием. Явление открыто и впервые исполь-
зовано Н. Н. Бекетовым (1859). Алюминий для А.
обычно применяют в форме порошка или мелкой
стружки. А. основана на том, что соединение алюми-
ния с кислородом сопровождается значительно боль-
шим выделением теплоты, чем окисление многих
других металлов (см. табл.).
Теплота образования (— Д77О98) некоторых окислов
(В кшм/г-атом кислорода)
Окисел -<98 Онисел 298 Окисел —AH° 298 Окисел —AH° 298
СаО 151,9 в2о3 100,6 V»o3 74,6 MnO, 62,2
ВеО 146,0 96,6 SnO2 69,4 MoO^ 60,1
MgO 143,8 Nb.,O5 92,6 SnO 68,4 NIO 58,4
133,0 МпО 92,0 Fe3O. 66,8 CoO 57,2
ZrO2 129,1 89,9 Fe2O3 65,5 CrO, 46,1
Т1О2 109,0 Мп304 82,8 MoO2 65,0 Cu2O 39,8
SiO2 102,7 Мп2О3 77,3 FeO 63,7 Cub 37,1
Поэтому взаимодействие А1 и окисла металла
(MemOn), теплота образования которого меньше
133,0 ккал/г-атом кислорода, протекающее с выделе-
нием свободного металла (тМе) и образованием
А12О3, сопровождается выделением теплоты (Q) тем
большей, чем меньше теплота образования восста-
навливаемого окисла: MemOn 4- “ n Al = m Me ф
пА12О3 -ф- Q. Величина Q существенно влияет на ход
процесса. Для получения высокого выхода металла
161
АЛЮМИНОТЕРМИЯ — АМАДОРИ ПЕРЕГРУППИРОВКА
162
и хорошего отделения его от шлака необходимо,
чтобы количество теплоты, выделяющейся при реак-
ции, было достаточно для расплавления выделяюще-
гося металла и шлака, а именно ок. 550 ккал на 1 кг
шихты — для средненосстанавлинаемых, несколько
больше — для трудно- и меньше — для легковосста-
навливаемых окислон. При недостатке тепла металл
не успевает отделиться от шлака до застывания
последнего.
Теплопроизводительность шихты q — теплота, вы-
делившаяся при взаимодействии стехиометрия, колич.
реагирующих в-в, отнесенная к 1 кг шихты, — может
быть рассчитана при допущении независимости теп-
ловых эффектов от темп-ры по ур-нию: q = —
где Ум — сумма молекулярных (атомных) весов
исходных в-н, взятых в стехиометрич. отношении.
Если величина q ниже требуемой нормы, процесс не
получает развития. В этом случае необходим подвод
теплоты, напр., путем введения в шихту «подогре-
вающей добавки», в частности окислон с меньшей теп-
лотой образования, чем окисел восстанавливаемого
металла [МпО2, СгО3), или др. веществ, легко от-
дающих кислород (NaNO3, КС1О3, A12(SO4)3, СаО2,
ВаО2 и др.]. При выборе добавки необходимо учиты-
вать и свойства образующихся продуктов. Так, напр.,
Мн и Сг, восстанавливаемые из первых двух добавок,
переходят в сплан; Na2O, КО и A12S3, получаемые,
из след, трех добавок, разлагаются при сравнительно
низких темп-pax и испаряются, увеличивая тепловые
потери и упоен часть непрореагировавшей шихты
и продуктов реакции; СаО и ВаО, образующиеся
из соответствующих перекисей и переходящие в шлак,
уменьшают вязкость А13О3 и снижают его темп-ру
плавления, что способствует лучшему отделению ме-
талла от шлака. Подогревающие добавки могут быть
заменены проведением процесса н обогреваемой извне
печи.
Чрезмерно высокая теплопроизводительность шихты
приводит к выделению большого избытка теплоты,
и реакция протекает очень бурно, приобретая взрыв-
ной характер В этом случае интенсивность процесса
снижают введением в шихту флюсов — т. н. нейтраль-
ных добавок (СаО, CaF2, MgO и др.), не реагирующих
с алюминием, а также избытка окисла или Al. С уве-
личением теплопроизводительности шихты и степени
дисперсности ее компонентов увеличивается и темп-ра
процесса, к-рая должна быть выше темп-p плавления
продуктов реакции, но ниже темп-p их кипения. При
слишком высокой темп-ре заменяют алюминиевый
порошок на крупку или стружку с размерами частиц
до 2 им».
Алюмотермия, планка ведется в шахте, футерован-
ной магнезитом. Окислы металла и алюминий в
рассчитанных по уравнению реакции количествах
предварительно тщательно перемешивают. Реакцию
вызывают при помощи запальной смеси с низкой
темп-рой воспламенения — ок. 000°; смесь засыпается
в небольшое углубление, сделанное на поверхности
шихты. Запальные смеси состоят из тщательно пере-
мешанных тонких порошков КС1О3 + ЗА1; ЗВаО2 +
+ 2А1; ЗСаО2 + 2А1; Fe2O3 -ф- 2А1 и др. Для поджи-
гания запальной смеси используют магниевую ленту,
бикфордов шнур, высушенные полоски фильтроваль-
ной бумаги, пропитанные насыщенным р-ром селитры,
или электрич. ток (электрозапал), пропущенный
через тугоплавкую проволоку с большим омич, со-
противлением. Начавшаяся реакция распространяется
по всей шихте и не требует дополнительного нагрева.
По охлаждении из тигля извлекают продукты
плавки — металл и шлак, к-рые обычно легко отде-
ляются друг от друга. А. получила в последние годы
широкое распространение в произ-ве марганца, хрома,
ванадия, вольфрама, ферросплавов и разных лигатур
цветных и редких металлов. А. используется также
при термитной сварке рельсов, труб и др. стальных
изделий.
Лит.: Бекетов Н. Н., Исследования над явлениями
вытеснения одних элементов другими, Харьков, 1865; М у-
р а ч Н. Н., В е Р я т и и У. Д., Внеиечная металлотермия,
М., 1956; М у р а ч II. Н., Мусиенко В. Т., Алюми-
нотермия титана, М., 1958; Боголюбов В. А., Сталь,
1952, М 1; Р о с то в ц е в С. Т., Теория металлургических
процессов, М., 1956; Kubasche-wski О. and
Evans Е. LI., Metallurgical thermochemistry. [4] ed., L.,
[a. o.], 1958. А. И. Лайнер.
АЛЮМОСИЛИКАТЫ — группа силикатов, в к-рой
А13+, подобно Si4+, находится в четнерном окружении
ионов кислорода и, т. обр., входит в состав анионного
комплекса. А. являются наиболее распространенными
в земной коре соединениями. Кним относятся, в первую
очередь, полевые ш п а т ы, на долю к-рых приходит-
ся более половины веса земной коры. Главные предста-
вители этой группы — минералы ортоклаз K[AlSi3Os],
альбит Na[AlSi3O8] и анортит Ca[Al2Si2O8]. К весьма
распространенным А. относятся минералы группы
слюд, напр. мусковит KAl2[Si3AlO10][OH], Боль-
шое технич. значение имеет А. нефелин Na2[Al2Si2O8|.
Интересную группу А. образуют цеолиты, состав
к-рых может быть выражен формулой •
• пН2О, где М — Са, Na, реже Ba, Sr, К, а Э — Si,
Al в переменных отношениях. Цеолиты обладают рых-
лой кристаллич. структурой и способны обменивать
содержащуюся в них воду на другие жидкости (спирт,
аммиак и т. п.), а катионы М — на различные др.
катионы. Цеолитная вода ведет себя как сорбирован-
ная. При осторожном нагревании цеолитов она уда-
ляется постепенно, причем даже полное обезвожива-
ние не недет к разрушению основной структуры мине-
рала.
Основой структуры А. является кварц SiO2, или
Si4O8. Частичное замещение Si на А1, вследствие их
различной валентности, создает избыточный отрица-
тельный заряд, компенсируемый к.-н. катионом.
В альбите Na[AlSi3Os] происходит замещение одного
из четырех атомов Si атомом А1 с компенсац. вхожде-
нием атома Na. В нефелине Na2 [Al2Si2O8] еще один
атом Si замещается атомом А1 и в состав катиона входит
новый атом Na. В сложных А., напр. амфиболах, имеет
место дробное отношение Si: Al от 3 и выше и соот-
ветств. дробное отношение катионов. Под действием
различных природных факторов, гл. обр. СО2 и Н2О,
А. на земной поверхности постепенно разрушаются
(выветриваются), причем растворимые продукты
уносятся водой в океан, а нерастворимые частично
отлагаются на месте, частично оседают в руслах рек
или выносятся в море. Основными нерастворимыми
продуктами распада А. являются кварц и каолин —
основа- обычных глин.
В технике широко применяются алюмосили-
каты искусственные. Так, напр., нек-рые
водные А., типа природных минералов цеолитов,
обладающие способностью к ионному обмену, приме-
няются для смягчения жестких вод (пермутиты)
или же адсорбентов. Искусственные А. широко при-
меняются в качестве катализаторов, а также в качестве
носителей для др. катализаторов, во многих отраслях
химич. пром-сти (подробнее о свойствах и применении
искусственных А. см. Силикаты).
Лит.: Вернадский В. И., Очерки геохимии, 2 изд.,
М. [и др.], 1934, с. 94—98, 107—119; Бетехтин А. Г.,
Минералогия, М., 1950.
АМАДОРИ ПЕРЕГРУППИРОВКА — превраще-
ние N -гликозидов альдоз с ароматич. амином в ка-
честве агликона в кетозы с замещенной аминогруппой
у первого углеродного атома. Реакция катализи-
руется Н-ионами. Предполагают след, механизм
163
АМАЛЬГАМЫ — АМБРА
164
не perp у ппировки:
RNH» RNH RNH., RNH»
I I! I I
НС-----г —. СН — СН — сн.2
1 I I II I
неон О неон сон с=о
! ! ! !
где R — арильный радикал. Для соответствующих
N-гликозидов алифатич. аминов перегруппировки
такого рода наблюдать не удавалось. А. п. находит
применение в препаративных синтезах аминосахаров;
открыта М. Амадори в 1925.
Лит.: Hodge J. Е., в ни.: Advances in carbohydrate
chemistry, v. 10, N. Y., 1955, p. 169; The carbohydrates, ed.
W. Pigman, N. Y., 1957; Annual Rev. Biochem. 1959, 28,
P. 18. Л. И. Линевич.
АМАЛЬГАМЫ — металлич. системы, одним из
компонентов к-рых является ртуть. В зависимости
от характера взаимодействия и соотношения компо-
нентов, а также темп-ры, А. представляют: жидкие
р-ры, твердые р-ры, кристаллич. химич. соединения
(меркуриды), гетерог. смеси химич. соединения и р-ра
н ртути. При амальгамации ртуть смачивает поверх-
ность амальгамируемого металла, после чего проис-
ходит диффузия в металл и в ряде случаев — образо-
вание химич. соединения или твердого р-ра. Очень I
часто соединения металлов со ртутью при нагревании
разлагаются. Для ряда А. характерны полиморфные
превращения.
Щелочные и щелочноземельные металлы образуют
устойчивые соединения со ртутью, напр. NaHg2
(т. пл. 346°), KHg2 (т. пл. 269,7°), со значительным
выделением теплоты. Меркуриды меди, серебра, зо-
лота и платины характеризуются весьма малыми
термич. эффектами и легко разлагаются ниже темп-ры
плавления с выделением ртути. С кадмием и свин-
пом ртуть дает твердые р-ры. Эвтектики со значи-
тельным содержанием ртути образуют цинк (2,5%)
и таллий (9,3%). Низкая растворимость в ртути ха-
рактерна для металлов всех групп периодич. системы,
кроме той, в к-рую входит сама ртуть; раст-
воримости при 17° : 0,2%Ан, 0,04% Ag, 0,02%Pt.
Вследствие этого эвтектич. точка весьма близ-
ка к температуре плавления ртути. Свойства А.
золота, серебра, платины (и отчасти олова) близ-
ки между собой. Ртуть образует твердые р-ры с
золотом (16,7%), платиной (23%) и серебром (45%),
а также след, меркуриды: AuHg2, Au2Hg, Au3Hg,
PtHg, Pt2Hg, Pt3Hg, Ag3Hg4 (фазы у и f3); нек-рые
из них co своими компонентами дают твердые р-ры
огранич. концентрации. Пленки, образующиеся
на металле вследствие взаимодействия с кислоро-
дом (хемосорбция, окисление), затрудняют или пре-
кращают смачивание ртутью. А. металлов плати-
новой группы, железа, редких металлов и многие др.
по этой причине могут быть получены лишь актива-
цией водородом, электрохимия, восстановлением и т. д.
Амальгамация как метод извлечения металлов из
измельченных руд и концентратов состоит в избира-
тельном смачивании металлич. частиц ртутью. При
этом получают полужидкие А., из к-рых избыток
ртути, содержащей весьма малое количество раство-
ренного металла, удаляется фильтрацией через замшу
под давлением. Твердая А. разлагается на составные
части при нагревании; следы ртути удаляются из
металла при последующем расплавлении.
А. начали получать для практич. целей более
2 тысяч лет назад. Амальгамация применяется: для
извлечения золота и платины из руд, чаще — из
концентратов; во вторичной металлургии для перера-
ботки отходов легких металлов: амальгамный метод
применяют при электролитич. получении редких
элементов. А. используются: при золочении металлич.
изделий, в произ-ве зеркал, в зубоврачебном деле.
А. щелочных металлов, цинка й др. применяются
в лабораторной практике как восстановители.
Лит.: К урн а ко в Н. С., Собр. избр. работ, т. 2,
Л.—М„ 1939; Плаксин И. Н., Металлургия благородных
металлов, М., 1958; его же, Взаимодействие сплавов и само-
родного золота с ртутью и цианистыми растворами, М.—Л.,
1937; Haughton J. L., The constitutional diagrams of
alloys..., 2 ed., L., 1956. И. H. Плаксин.
АМБРА — воскообразное в-во, находящееся в ки-
шечнике кашалота, откуда и добывается; ее также
собирают с поверхности морей и океанов, где водятся
кашалоты. А. бывает серая и белая; более ценной
является серая А. — жирная крошащаяся масса,
серого цвета; плотн. 0,90—0,92; т. пл. ~ 60°; нераст-
ворима в воде, хорошо растворяется в спирте, эфире,
жирных и эфирных маслах. Свежие куски А. об-
ладают фекальным запахом, который со временем
исчезает и остается тонкий
характерный запах. Соеди-
нения, входящие в состав се-
рой А., являются производны-
ми политерпенов: трициклич.
тритерпеновый спирт, амб-
реин (I) (25—45%), эпикоп-
ростерин (II) и его эфиры
(30—40%), копростерин (III)
(1—5%), холестерин (0,1%),
кетоны (6—8%), половину из
них
СН,
Н
///
IV
R = HO-
R = HO-
R = O=
составляет копро-
станон-З' (IV)' кроме того,” входят жирные к-ты (5%),
этерифицированные к-ты (5—8%). Пахучим началом
А. являются продукты оки-
сления амбреина (дигидро-
-у-ионон и др.). А. приме-
няется в парфюмерии в виде
спиртового настоя для при-
дания запаха, а также в ка-
честве фиксирующего сред-
ства для удержания запаха R
других веществ; использует-
ся также в гомеопатии.
Амбре ин С33Н31ОН —
трициклический тритерценовый
спирт;
легко дегид-
ретируется при нагревании с уксусным ангидридом
или муравьиной к-той; образующаяся двойная связь
не гидрируется. При окислении амбреина КМпО4 и
СгО3 его молекула расщепляется по двойной связи
с образованием насыщенного бициклич. лактона
С17Н28О2 — амбреинолида (вещество без запаха) и мо-
ноциклич. кетона С13Н22О — дигидро-7-ионона (V).
Последний обладает тонким запахом амбры и выделен
из летучих частей серой
амбры.
При гидрировании ам- 11 ] \ | ]
бреина образуется тетра-
гидроамбреин СзоН56О, а Щ । ]г у
при озонолизе — фор- ц I Lq
мальдегид. Установлена v>
связь между амбреином и растит, дитерпенами:
маноолом (выделен из смолы Dacrydium biforme) и
склареолом (выделен из мускатного шалфея); пока-
зана идентичность структуры и конфигурации бици-
клич. систем всех растит, дитерпенов, тритерпенов
и амбреина. с. И. Вирезуб.
Амбреинолид С17Н28О2, мол. в. 265,02 — кри-
сталлич. продукт, известен в виде четырех изо-
меров: 1) т. пл. 140—141° (из эфира), [а]о = +30,8°
(СНС13); 2) изоамбреинолид, т. пл. 143°, [a]D = + 20°
(СНС13); 3) т. пл. 98°, [a]D == —13°; 4) жидкий, т. кип.
130—140°/3 мм, n'jj 1,505, [a]D =С±1°. Получают
амбреинолид окислением амбреина и др. способами,
а также из склареола. При омылении 5%-ным спирто-
вым р-ром КОН образуются оксикислоты состава
165
АМБРЕТТОЛИД — АМИДЫ КАРБОНОВЫХ КИСЛОТ
166
С16Н25(ОН)СООН, т. пл. 163—166° и 85—88°. При
нагревании с 80%-ной H2SO4 образуется ненасыщ.
кетон, обладающий запахом амбры, состава С16Н26СО,
т. кип. 125—128°/0,2 мм, d™ 1,030, njj 1,531, [а]$ =
= +68° (СНС13, с = 0,836); может быть идентифициро-
ван, напр. по семикарбазону, т. пл. 123—125°. При
восстановлении амбреинолида и его изомеров литий-
алюминийгидридом получают изомерные гликоли
состава С17Н30(ОН)2: 1) т. пл. 133°, [a]D = —26,6°;
2) т. пл. 93°, [a]D = + '17°; 3) т. пл. 89°, [a]D = + 13,7°
и 4) жидкий, [a]D =±1°.
Лит.: Lederer Е., Industr. Parfum., 1953, 8, № 6,
р. 189. С. Д. Кустова.
АМБРЕТТОЛИД [гексадецен-(7)-олид] С1вН28О2,
мол. в. 252, 4 — микроциклич. лактон (I), бесцветная
жидкость:
СИ(СН.,)6С=О
II \
СН(СН..)8—о
СГЦСГЬЦСО
|| /О, где х + у = 12
СН(СП3)уСП2
II
т. кип. 185—190716 154—156°/1 мм; d'20 0,958;
п'р 1,4815; растворим в этиловом и бензиловом
спиртах, диэтил- и диметилфталатах, бензилбензоате.
Впервые выделен из масла семян Hibiscus abelmoschus
(амбреттовое или мускусное масло). Структура А.
была подтверждена его синтезом пз серебряной соли
ю-бромамбреттоловой к-ты. В 1945 осуществлен пол-
ный синтез А. Производственный метод получения
основан на циклизации моноглицеридов смеси нена-
сыщенных к-т, к-рые образуются при дегидратации
натриевой соли 6,16-диоксипальмитиновой к-ты.
Циклизация приводит к смеси изомеров (II).
Пз смеси изомерных лактонов выделены: а-А.,
т. пл. 52—55°; fJ-A., т. пл. 26—27°; А6-нзо-А.,
т. кип. 153/2,5 льи; Д5-нзо-А., т. кип. 143/2 мм.
Все изомеры имеют сильный цветочно-мускусный
запах; у (З-изомера и природного А. запах идентичен.
Лактоны обладают способностью усиливать запах
композиции, для создания к-рых А. употребляется.
А. неустойчив к действию щелочей, образует* амбрет-
толовуго к-ту. Степень чистоты А. определяют по
эфирному числу.
Лит.: Химия и хим. технология, 1955, выв. 4 (63), с. 165 —
67; В е d о u k i a n Р. Z. Perfumery synthetics and isolates,
Toronto — N. Y. — L., 1951. H. П. Соловьева.
АМЕРИЦИЙ (Americium) Am — химич. элемент
с п. н. 95 из семейства актинидов.
АМИГДАЛИН С^Н^ОцЛ', мол. в. 457,42 — глюко-
зид, содержащийся в семенах горького миндаля,
в косточках персика,
в I
о—— сн
+Св~<+н неон
N=C I О
п носн
I
----сн
I
неон
I о
носн
неон
I
нс----
I
неон
абрикосов, слив, ви-
шен и др. растений.
А. — бесцветные кри-
сталлы; т. пл. 214—
216°; хорошо раство-
рим в воде, из кото-
рой кристаллизуется
с молекулами воды;
н2С----- сн2он растворим в спирте,
нерастворим в эфире;
[а]д =—40,57° (в воде). А. при нагревании с Ва(ОН)2
гидролизуется по CN-группе и рацемизуется до амиг-
далиновой к-ты. При полном гидролизе (кислотном
или ферментативном) из А. образуется глюкоза, бен-
зальдегид и HCN. А. получают из жмыхов горького
миндаля (2,5—3,5°/о), косточек персиков, абрикосов,
а также синтетически. Применяют А. как болеуто-
ляющее при заболеваниях желудка и кишечника.
Лит.: М i с h е е 1 F., Chemie der Zucker und Polysac-
charide, 2 Aufl., Lpz., 1956. Л. И. Линевич.
АМИДАЗЫ — ферменты, гидролитически расщеп-
ляющие амиды аминокислот. Иногда к группе А.
относят уреазу, гидролизующую мочевину, а также
креатиназу, аргиназу, гистидазу и т. д. К собственно
А. относятся аспарагиназа, глутаминаза и ю-амидаза
а-кетокислот. Аспарагиназа катализирует
реакцию расщепления L-аспарагина. Оптимум дейст-
вия фермента, полученного из различных источников,
при pH 8. Глутаминаза катализирует реакцию
расщепления L-глутамина. Оптимум действия глу-
таминазы, выделенной из бактерий Escherichia coli,
при pH 5; глутаминаза животных тканей наиболее
активна при pH 7,5—8. Аспарагиназа и глутаминаза
широко распространены в тканях животных, в расте-
ниях и микроорганизмах, о» -Амидаза a-к е то-
ки с л о т, недавно обнаруженная в тканях живот-
ных, гидролизует а-кетоаналоги аспарагина и глу-
тамина в соответствующие а-кетокислоты и аммиак.
Оптимум действия лежит между pH 8,5—9,5.
А. в чистом виде не получены, поэтому физико-
химич. характеристики препаратов этих ферментов
отсутствуют. В тканях Животных обнаружены фер-
менты, осуществляющие гидролиз амидов монокар-
боновых а-аминокислот, напр. тирозинамида, аланин-
амида, амида а-аминомасляной к-ты и т. д. В животном
организме дезамидированию подвергаются также
амиды нек-рых алифатич. и циклич. к-т, вводимые
извне и не обнаруживаемые в обычных условиях
в организме (пропионамид, бензамид, ацетамид).
Амидазная активность отмечена у ряда протеолитич.
ферментов (трипсин, химотрипсин, лейцинаминопеп-
тлдаза, папаин, фицин, бромелин и катепсины).
Лит.см. при ст. Альдолаза. Г. А. Левдикова.
АМИДИНЫ — производные органич. кислот об-
щей ф-лы RC(=NH)NR2 (R — водород, алкил,
арил); кристаллич. в-ва, растворимые в спирте и
эфире. А. наз. ио соответствующей к-те, например
СН3С(—NH)NH2 — ацетамидин. А. — сильные одно-
кислотные основания; в свободном виде неустойчивы,
используются обычно в виде хлоргидратов; N-за-
мещенные более устойчивы. Моно- и дизамещенные
А., по-видимому, способны к таутомерным превраще-
ниям: RC(=NR')NHR"=? RC (=NR")NHR'. А. полу-
чают из нитрилов (1), иминоэфиров (2), тиоамидов (3):
RON + NaNHa — RC (=NNa) NHS A12. RC (=NH) NH. (1)
RC(=NH - HCI) OC..H;,-lt5TlI4
R 'NTL
—► RC(—NH-HC1) NHR'----+ RC(=NR')NHR' (2)
RCSNH., --2+U. RC (= NR') NHa (3)
А. гидролизуются уже горячей водой, а еще быстрее
разбавленными кислотами или щелочами до амидов.
С Na и К в жидком NH3 А. дают монометал-
лические производные, например RC(=NK) NH2.
При восстановлении амальгамой натрия в спирте
образуют амины: RC(=NRZ)NHR" —>- RCH2NHR" +
+ R'NH2. Конденсацией с ацетоуксусным эфиром
из А. получаются пиримидины, с циангуанидином —
триазолы, с о-фенилендиамином — бензиминазолы,
с о-аминофеиолами —бензоксазолы. А. применяют
гл. обр. в медицине; хлоргидрат фенакаина
CH3C( = NCeH4OC2H5-n)NHCeH4OC2H5-»- НCl-
как местноанестезирующее средство; стильбамидин
HCl-H3N(NH=)OCeH4CH=CHCeH4C(=NH)NH!!-HCl-
при лечении сонной болезни. Предложено пользовать-
ся А. в качестве порообразующих агентов в произ-ве
резиновых изделий.
Лит.: S h г i n е г R. L., Neumann F. W., Chem.
Rev., 1944, 35, № 3, р. 351—425; J ohnsonF. W., D a 1 g-
1 i e s h С. E., W a 1 k e r J., в кн.: Chemistry of carbon
compounds, ed. E. H. Rodd, v. 1, prt A, Amst. — Houston —
L. — N. Y., 1951, p. 609—611. Ф. Ii. Величко.
АМИДОЛ— см. Проявляющие вещества.
АМИДЫ КАРБОНОВЫХ КИСЛОТ — производ-
ные кислот, в к-рых гидроксил карбоксильной группы
167
АМИДЫ КАРБОНОВЫХ КИСЛОТ — АМИЛЕНЫ
168
замещен NH2-rpynnoH. А. к. к. называют по соот-
ветствующим кислотам, напр. HCONH2 — формамид',
CH3CONH2 — ацетамид', СвН5СО1\Н2 — бензамид и
т. д. физич. свойства нек-рых А. к. к. приведены
в табл.:
Название Т. пл.. °C Т.кип., °C Теплота cropания, ккал/моль
Формамид HCONH2 2,5 210,7 1.133 134,9
Ацетамид CH3CONH2 .... 82—83 221,2 (20°) 1.159 288,0
Пропионамид CH3CH2CONH2 81,3 213 (20°) 439,8
Акриламид CH2=CHCONH2 84,5 — — —
Бензамид CGHf>CONH3 . . . 130 288—290 1,341 851,9
Никотинамид й J 122 — (4°) —
А. к. к. получают: дегидратацией аммониевых солей
соответствующих кислот, действием аммиака или
амина на галогенангидриды, ангидриды или сложные
эфиры, неполным гидролизом нитрилов.
Кроме жидкого формамида, А. к. к. являются крис-
таллин. в-вами; все они представляют собой нейтраль-
ные соединения; с сильными минеральными к-тами
образуют лишь очень непрочные, легко гидролизую-
щиеся соли. Нейтральный характер А'Щ-группы
в А. к. к. определяется тем, что свободная электронная
пара атома азота сопряжена с двойной связью кар-
бонильной группы:
R—С=О
'2
R-ck)
л +
С?Н2
Основные свойства аминогруппы усиливаются при
ее метилировании. Диметилацетамид дает соль с хло-
ристым водородом, устойчивую в конц. водных раст-
ворах, а также прочные, хорошо кристаллизующиеся
соли с НС1О4 и H2PtCl6. А. к. к. образуют с некоторы-
ми катионами соли, разлагающиеся водой; ртутные
соли (RCONH)2Hg более устойчивы. При кипяче-
нии с разбавленными минеральными к-тами А. к. к.
получаются карбоновые к-ты: RCONH2 -Is Н2О —>
—> RCOOH-f-NH3; аналогично, но медленнее действуют
щелочи. При действии азотистой к-ты на амиды
образуются карбоновые к-ты и выделяется азот:
RCONH2 ф- HNO2 -> RCOOH -ф- N2 4- Н2О. Алкил-
галогениды с металлическими производными амидов
дают N-замещенные амиды: RCONHNa 4- CH3J —>
-> GHSCONHCH3 4- NaJ.
Направление последней реакции зависит от природы ме-
талла. Так, натриевая соль изатина с иодистым метилом или
с диметилсульфатом в водном р-ре дает N-метилизатин:
+ NaJ
Из серебряной соли получается изомерный б-метилизатин:
Дегидратация А. к. к., напр., с помощью Р2О5
(или PjSs) приводит к соответств. нитрилам. Энергич-
ным восстановлением А. к. к. могут быть превращены
в первичные амины с тем же числом атомов углерода.
Большое препаративное значение для синтеза аминов
имеет Гофмана реакция, заключающаяся в действии
на А. к. к. гипобромитов или гипохлоритов в щелочном
р-ре: RCONH2 Д- Вг2 + 2NaOH RNH24-2NaBr +
4- СО2 -ф- Н2О. А. к. к. с РС15 образует хлорамиды,
легко распадающиеся с выделением НС1 и образова-
нием имидохлоридов:
CHsCONHs + РС15 — CH3CC1NH2 + РОС13
CH3CC1»NH2 — HCI + CH3CC1=NH
Амидная группировка NHCO играет основную
роль в построении молекул полиамидов (найлон, ка-
прон и т. п.), полипептидах, к которым относятся
многие антибиотики (напр., грамицидин), в белках.
Амиды нек-рых двухосновных карбоновых к-т легко
переходят в имиды к-т; так, диамид фталевой к-ты
при нагревании отщепляет аммиак и образует фтали-
мид. Расплавленные А. к. к. являются хорошими
растворителями для многих неорганич. и органич.
соединений; так, ацетамид CH3CONH2 применяется
в качестве растворителя для альбумоз и пептонов.
Ацетанилид CeH5NHCOCH3 — исходный продукт син-
теза важной группы лекарств — сульфаниламидных
препаратов. Ж. А. Красная.
АМИЛАЗЫ — ферменты, гидролитически расщеп-
ляющие 1,4-связи в полисахаридах типа гликогена
и крахмала. В зависимости от продуктов, образую-
щихся при гидролизе, различают a-, fl- и у-А. Первая
из них (декстриногенная) расщепляет полисахариды
до декстринов различного мол. веса; fl-A. (сахаро-
генная или мальтогенная) гидролизует полисахарид,
отщепляя дисахарид мальтозу от неальдегидных
концов молекул полисахарида, а у-А. (глюкоамилаза)
отщепляет от молекул полисахарида глюкозные ос-
татки.
а-А., в отличие от (3-А., неустойчива в кислой
среде; fl-A., в отличие от а-А., термолабильна. Раз-
личия в свойствах А. используются для их препара-
тивного разделения, а- и fl-A. различного происхож-
дения выделены в кристаллич. виде. А. широко
распространены в природе (в тканях животных,
растений, а также в бактериях). В нек-рых животных
тканях, напр. в мышцах, А. существует в связанном
состоянии.
Лит. см. при ст. Альдолаза. Е. Л. Розенфельд.
АМИЛАЦЕТАТ И ИЗОАМИЛАЦЕТАТ (ами-
левый и изоамиловый эфиры уксусной кислоты)
СН3(СН2)4ОСОСН3; (СН3)2СН—СН2СН2ОСОСН3; мол.
вес 130,18 — бесцветные жидкости. Физич. свойства
приведены в таблице.
Свойства Амилацетат Изоамилацетат
Т. пл., °C — 75,0 —78,5
Т. кип., °C 149,2 142,0
d20 и4 0,8753 0,8719
20 nD 1.40228 1,40535
Растворимость в воде, % . . 0,2 9
Азеотроп с Н2О: т. кип., °C 95,2 93,6
вес. % 59 63,7
Т. воспл., °C 25 26,7
Теплоемкость, ккал/моль . . 66,0 (30,1°) 59,73 (20°)
Ацетаты обладают характерными для сложных
эфиров химич. свойствами (гидролиз, аммонолиз,
замещение в углеводородной цепи и т. д.). А. и и.
получаются нагреванием уксусной к-ты с соответств.
спиртами в присутствии конц. H2SO4. Пзоамилацетат
широко применяется как растворитель нитроцеллю-
лозы (в произ-ве кинопленки, целлулоида и т. д.)
и в пищевой пром-сти под названием грушевой эссен-
ции. Ацетаты используют как растворители многих
органич. соединений. А. Ф. Бочков.
АМИЛЕНЫ (пентены) С5Н10— шесть изомерных оле-
финов— пентен-1 СзН,СН=СН2; цис- п транс-пентен-2
С2Н5СН = СНСН3, 2-мстилбутен-1 С2Н5С(СН3)=СН2,
169
а-и-АМИЛКОРИЧНЫЙ АЛЬДЕГИД —АМИЛОВЫЕ СПИРТЫ
170
2-мстилбутсн-2 СН3СН = С(СН3)2 и 2-мстилбутен-З
СН2=СНСН(СН3)2; присутствуют в продуктах кре-
кинга газолина. Физич. свойства А. приведены в табл.
Свойства Пентен-1 Пентен-2 3-метил- бутеп-1 2-метил- бутен-2 2-метил- бутен-3
цис транс
Т. пл., °C — 165,22 —151.37 —140,24 —137,50 —133,769 — 168.528
Т. кип., °С/760л<л< . . 29,97 37,1 36,36 31,16 38,57 20,061
% 0,6410 0,656 0,6482 0,6503 0,6623 0,6272
1,37148 1,3828 1,3793 1,3778 1.3878 1.3643
Теплота сгорания, 25°, р = const 806.85 805,34 804,26 803,17 801,68 804,93
Анилиновая точка, °C 19 смесь 18.3 — 12,8 —
Характермстич. час- тоты в ИК-области, 10,05 14.0 11,9—12,7 10,05
мк 10,98 14,6 10,36 11,24 3,3—3,5 10,98
Взрывоопасные смеси с воздухом: 1,42—8,70% А.
'Технич. продукт (т. кип. 39—40,5°, </;" 0,6509, п’д
1,3783) представляет смесь всех изомеров с примесью
изопентана. А. образуют азеотропные смеси с СН3ОН,
С2Н5ОН, (С2Н5)2О, СН3СОСН3. Для синтеза А. при-
менимы все способы получения алкенов, наиболее
важны дегидратация амиловых: спиртов над А12О3
при 380—450° или 46—65%-ной H2SO4 при 90—110°,
пиролиз амилацетатов при 500—700°. Пентен-1
получают также синтезом Фишера—Тропша. из СО
и Н2 над Со-катализатором. Смесь пентена-1 и пен-
тена-2 образуется при дегидрировании пентана над
Сг2О3/А12О3 (560°, 2—3 ат); чистый цис-пентен-2
получают гидрированием пентина-2 над коллоидным
Pd; транс-петен-2 — действием спиртового КОН
на 2-бромпентан. А. полимеризуются под действием
А1С13; с SO2 в присутствии Н2О2 дают резиноподобные
полисульфоны; образуют сополимеры с хлористым
винилом, изобутиленом, бутадиеном, изопреном. А13О3
при нагревании изомеризует каждый из А. в смесь
всех остальных с преобладанием разветвленных изо-
меров, 84—85%-ная H2SO4 превращает А. в соответств.
амиловые спирты с частичной полимеризацией. При
нагревании с СО и Н2 в присутствии Со2(СО)8 А. об-
разуют смесь изомерных гексиловых спиртов. Из
пентена-2 и 2-метилбутена-2 над V2O5 при 460—510°
получают малеиновую к-ту. Пентен-2 нагреванием
с водяным паром до 760—833° превращается в бута-
диен; из 2-метилбутена-1 над Сг2О3 при 600° получают
изопрен. Полимеры А. используются в качестве сма-
зочных масел, компонент типографских красок и др.
А. идентифицируются с трудом, гл. обр. по ИК-спект-
рам (см. табл.), пентен-1 и пентен-2 образуют с
3,5-(МО2)2СбН3СООА§ и J2 кристаллич. динитробен-
зоаты, т. пл. соответственно 76,2—77° и 124,5—125,3°,
2-метилбутен-2 дает с NOC1 нитрозохлорид, т. пл.
74—75°.
Лит.: Физико-химические свойства индивидуальных угле-
водородов, [Справочник], под ред. М.Д. Тиличеева, вып. 5—6,
М., 1954—57. Ф. К. Величко.
а-н-АМИЛКОРИЧНЫЙ АЛЬДЕГИД (жасминаль-
дегид) СбН5СН=С(С5Н11)СНО, мол. в. 202,30—блед-
но-желтая жидкость с сильным запахом, напоминаю-
щим запах жасмина; т. кип. 153—154°/10 мм', t/45
0,9715; «р 1,5552. Получают А. а. конденсацией бен-
зальдегида с и-гептиловым альдегидом (энантолом)
в присутствии щелочи:
CeH5CHO + ClJ3(CH.) jCH»CJJO ——CeHsCH=CCHO4-II«O
С5нп
В качестве побочного продукта образуется амилно-
ненальдегидСН3(СН2)5СН =С(С6Н11)СН О и более слож-
ные высококипящие продукты конденсации энантола,
к-рые легко отделяются от жасминальдегида пере-
гонкой. А. а. может быть получен в двух стереоизо-
мерных формах; продажный продукт является в ос-
новном транс-изомером. При
стоянии на воздухе А. а. лег-
ко окисляется с образовани-
ем бензойной, капроновой и ко-
ричной кислот; рекомендуется
сохранять его в темном месте
в хорошо укупоренных склян-
ках. Для предохранения от
порчи иногда добавляют 0,5%
(по весу) дифениламина. Иден-
тифицируют А. а. по произ-
водным; семикарбазон, т. пл.
117,5—118° (из бензола);
оксим, т. пл. 72,5—73°, приз-
мы (из петролейного эфира
2,4-динитрофенилгидразон, т. пл. 164°. Трудно реа-
гирует с бисульфитом; количественно определяется
оксимированием. А. а. широко применяется в пар-
фюмерной, косметич. и мыловаренной пром-сти.
Лит.: Наметкин С. С. и Шагалова Р. Ю.
в сб.: Синтезы душистых веществ. 1927—1939 гг., М.—Л.’
1939, с. 271. Т. А. Рудольфи.
АМИЛНИТРИТЫ CsHjjONO — сложные эфиры
амиловых спиртов и азотистой к-ты. Прозрачные
светло-желтые летучие жидкости с запахом фруктов,
растворимые в спирте, эфире, хлороформе, почти
нерастворимые в воде.
Амилнитриты Т. ниц., °С/760 мм ' d1 4 t 71D
Амилнитрит h-C3HuONO 104 0,8528 (20°) 1,38506 (20°)
Изоамилнитрит (CH3)2CHCH2ONO 99 0,8717 (20,7°) 1,38708 (20,7°)
трет'А милнитрит (CH3)2C(ONO)C2H5 93 0,8958 (19,5°) 1.3904 (16,8°)
А. готовятся взаимодействием соответствующих спир-
тов с HNO2 или со смесью NaNO2 + A12(SO4)3 при 0°;
легко омыляются едкими щелочами; разлагаются
соляной к-той с выделением NO, NOC1 и соответст-
вующего спирта. А. используются как агенты диазо-
тирования и в синтезе оксимов кетонов. С меркапта-
нами А. дают красное окрашивание. Вдыхание паров
А. вызывает быстрое, но непродолжительное расши-
рение кровеносных сосудов. Изоамилнитрит исполь-
зуется как спазмолитич. средство, а также как про-
тивоядие при отравлении синильной к-той.
Ф. В. Величко.
АМИЛОВЫЕ СПИРТЫ СйНцОН, мол. в. 88,15 —
алифатические насыщенные спирты; бесцветные жид-
кости с характерным «сивушным» запахом; раство-
римы в органич. жидкостях, смешиваются с расти-
тельными и минеральными маслами, растворяют
смолы и камеди; образуют азеотропные смеси с водой.
А. с. входят в состав сивушного масла. Физич. свой-
ства А. с. приведены в табл.
Пентанол-2, 2-метилбутанол-1 и З-метилбутанол-2
имеют оптически активные изомеры. Основной ис-
точник их получения — сивушные масла брожения.
Большие количества амиловых спиртов синтези-
руют хлорированием пентанов (200°, 100 ат) с по-
следующим омылением полученных хлорпарафинов
30%-ным NaOH. Перспективен метод их получе-
ния из бутиленов оксосинтезом С4Н10 + СО + Н2 —>
—>С5НИОН. При нагревании с H2SO4 А. с. дают ами-
лены и простые амиловые эфиры. При окислении пен-
танола-1 в парах над Cu-порошком при 300° обра-
зуется валеральдегид. Пентанол-2 окисляется хро-
мовой смесью до метилпропилкетона. Окислением
171
АМИЛОЗА - а-АМИНОАДИПИНОВАЯ КИСЛОТА
172
2-метилбутанола-З готовят метилизопропилкетон. Про-
пусканием паров З-метилбутанола-1 над ZnS при 430°
получают изовалеральдегид. Основное количество
А. с. идет для синтеза сложных эфиров, используемых
в парфюмерии, произ-ва порохов и пищевых эссенций.
А. с. находят также применение как растворители.
Спирты Т. пл., °C Т.кип., °С/76О мм d20 u4 20 Пр Раство- римость, г/100 мл воды
Пентанол-1 С4ЩСН3ОН —78,8 138,0 0,8144 1,4099 2,7 (22°)
Пентанол-2 С3Н7СНОНСН3 — 119,9 0,8303 1,4178 5,3 (30°)
Пентанол-3 (С2Н5)2СНОН -75 115,6 0,8218 1,404 —
2 -Метилбутанол-1 С2Н5СН(СН3)СНаОН 70 128,0 0,8193 1.4107 —
З-Метилбутанол-1 (СН3)2СНСН2СН2ОН —117,2 132,0 0,812 1,4053 2,67 (22°)
2 -Метилбутанол-2 С2Н5СОН(СН3)3 —9,1 102,3 0,809 1.4058 14 (30°)
З-Метилбутанол-2 (СН9)2СНСНОНСН3 111,5 0,819 1,4095 —
2,2-Диметил проп а- нол-1 (СН3)3ССН2ОН 53 113-114 0,812 — —
Идентифицируют А. с. по темп. пл. производных: дини-
тробензоатов, замещенных уретанов и др. А. с. пора-
жают нервную систему.
Лит.: Крендель Б. А., Основы синтеза алифатиче-
ских спиртов из нефтяных углеводородов, М., 1954; A s I п-
ger F., Chemie und Technologie der Monoolefine, В., 1957.
Ф. К. Величко.
АМИЛОЗА — см. Крахмал.
АМИЛОПЕКТИН — см. Крахмал.
АМИНАЗИН [хлорпромазин, ларгактил, хлоргид-
рат 2-хлор-10-(3-диметиламинопропил) фентиазина]
Cl7H2(1N2CI2S, молекулярный вес
... 355,34 — белые мелкие кристал-
[ || II ] лы; т- пл- 194—197°; чувстви-
телев к свету. Растворим в во-
N де, 95%-ном этиловом
CH2-CH2-CH2N(CH3L HC1 спирте И хлороформе;
г нерастворим в бензоле
и эфире. Качественное
определение А. основано на окрашивании его р-ра
в вишнево-красный цвет при прибавлении H2SO4;
при стоянии медленно темнеет. Для колич. опреде-
ления А. разлагают щелочью, выделенное основание
подкисляют точным количеством к-ты и определяют
ее избыток титрованием NaOH с метиловым красным
или метиленовым синим. Получают А. конденсацией
1-хлор-З-диметиламинопропана с 2-хлорфентиазином;
последний образуется из 3-хлордифениламина нагре-
ванием его с серой. А.— сильно действующий препарат
из группы т. н. «нейроплергических» лекарственных
в-в, оказывающих сложное и разнообразное влияние
на функции центральной и периферия, нервной
системы. Применяют в психиатрии, неврология, прак-
тике и хирургии для усиления действия анестезии
и наркоза, в качестве противорвотного средства и для
искусств, охлаждения организма; иногда вызывает
раздражение кожи и слизистой оболочки.
Лит.: Щукина М. Н., Савицкая Н. В., Ц и-
з п н Ю. С., Мед. пром. СССР, 1957, № 3, 20; см. также при
ст. Адалин. А. С. Елина.
АМИНАРСОН (карбарсон, п-карбамидофенилмы-
шьяковая кислота) n-H2NCOHN—С6Н4—AsOsH2, мол.
в 260,07 — бесцветные кристаллы; т. пл. 172—174°;
растворяется в 170 ч. холодной и 30 ч. теплой воды;
почти нерастворим в эфире и хлороформе, легко раст-
воряется в р-рах едких щелочей и карбонатов. Водные
р-ры имеют кислую реакцию. При нагревании со
щелочами А. выделяет аммиак; при подкислении ще-
лочного гидролизата выпадает осадок арсаниловой
к-ты (качеств, проба). Колич. определение основано
на разложении А. КМпО4, окислении образовавшейся
мышьяковой к-той йодистого водорода (из KJ) до
элементарного иода и определении последнего титро-
ванием тиосульфатом. Получают А. взаимодействием
натриевой соли арсаниловой к-ты с цианатом калия.
Применяют при лечении амебной дизентерии, балан-
тидиаза, трихомонадных вагинитов и иногда при
размножении трихомонад в кишечнике.
Лит. см. при ст. Адалин. А. И. Травин.
АМИНИРОВАНИЕ — введение аминогруппы NH2
в различные органич. соединения. К типичным про-
цессам А. относится действие амидов щелочных ме-
таллов на гетероциклич. основания. Напр., при реак-
ции пиридина с NaNH2 ок. 200° образуется а-амиио-
пиридин. Механизм этой реакции нуклеофильного
замещения, открытой в 1914 А. Е. Чичибабиным
и О. А. Зейде, сходен с хорошо изученным механиз-
мом взаимодействия пиридина с бутиллитием с обра-
зованием а-бутилпиридина. В последнем случае
сначала образуется довольно стойкий аддукт (продукт
присоединения) по полярной связи —N=C^ в пи-
ридине, к-рый при повышенной темп-ре отщепляет
LiH и дает а-бутилпиридин. Из пиридина (I) и NaNH2,
видимо, сначала образуется аддукт (II), находящийся
в равновесии как с исходными продуктами, так и
с полученными из него а-аминопиридином (III) и
NaH. Равновесие смещено в сторону исходных про-
дуктов, т. к. (II) легче теряет амид-ион, чем гидрид-
ион; однако равновесие нарушается вследствие пе-
реноса катиона от гидрид-иона к (III) с выделением
водорода, что ведет к смещению реакции в сторону
(III). Из таких представлений вытекает вывод, что
отщеплению гвдрид-иона от (II) должно способство-
вать прибавление окислителя. ’Действительно реак-
ция между хинолином и NaNH2 в жидком аммиаке
идет очень плохо, при прибавлении же KNO3 наблю-
дается хороший выход 2- и 4-аминохинолина.
C5II4NNH2 + NaH -< C5H4NNIlNa + Н2
Известны и нек-рые другие реакции А., не имеющие,
однако, никакого препаративного значения. Так,
при пропускании паров бензола и аммиака через
слабо накаленную трубку образуется анилин:
С6Н6 + NH3 —> C6H5NH2 + Н2. Неск. лучше реаги-
рует нафталин с NH2Na с выделением водорода и об-
разованием натриевого производного нафтиламива,
дающего после гидролиза нафтиламин. Далее взаимо-
действием бензола с гидроксиламином получают
с незначит. выходом анилин.
В рассмотренных случаях А. аминогруппа стано-
вится на место, ранее занятое атомом водорода. Не-
редко термину «А.» придают более широкий характер
и к таким реакциям причисляют также замену не
только атома водорода, но и др. атомов и групп.
Напр., легко протекающее превращение нафтолов
в аминонафтолы (см. Бухерера реакция), обмен сульфо-
группы в Р-антрахинопсульфокислоте-2 или хлора
в р-хлорантрахиноне на аминогруппу при действии
аммиака с образованием индантреновых красителей
рассматривают как реакции А.
Лит.: Органические реакции, Сб. 1, пер. с англ., М.,
1948, с. 115 —132. Я. Ф. Комиссаров.
а-АМИНОАДИПИНОВАЯ КИСЛОТА, мол. в. 161,16,
HOOC(CH2)3CH(NH2)COOH — дикарбоновая а-амино-
173
АМИНОАЗОБЕНЗОЛ — АМИНОАЛЬДЕГИДЫ И АМИНОКЕТОНЫ
174
кислота; бесцветные кристаллы; т. пл. 206°, [a]D=-f-25°
(НС1); хорошо растворима в воде, плохо — в этиловом
спирте и эфире; с FeCl3 дает красно-коричневое окра-
шивание, что используется при ее определении. По-
лучают а-А. к. из нитрила у-хлормасляпой к-ты:
Cl(CH2)3CN-|-NaCH(COOC2H5)3 —(COOC3H5)2CH(CH2)3CN —
— (COOC3H5)3CBr(CIIsj3CN
,СС\
c«hZ )nk
xcoz
(CH2)3CN
/ iloU
— (COOC2II5)3C _n —
\ /co\
N С0И4
'чсо/
— IIOOC(CH3)3CH(NH3)COOI1
Содержится а-А. к. в тканях живот-
ных организмов; образуется в печени при биологич.
распаде лизина.
Лит. СМ. при ст. Аминокислоты. О. В. Пунина.
АМИНОАЗОБЕНЗОЛ (азоамин коричневый О)
C12H12N3, мол. в 198,25 — известны 3 изомера; прак-
тическое значение имеет
лишь пара-изомер; "кри-
сталлы желтого цвета; т.
пл. 126—127°; т. кип. выше
360° или 225/120 мм (без
разл ); очень мало растворим в воде; кристалли-
зуется из этилового спирта и бевзола; константа
диссоциации К = 9,5 • 10 12 (25°). А. обладает ха-
рактерными свойствами ароматич. амина (образует
соли с к-тами, диазотируется при действии нитрита
в кислой среде и т. п.) и азосоединения (окрашен,
восстанавливается с обесцвечиванием при действии
сильных восстановителей в кислой среде, при этом
образуются анилин и n-фенилендиамин). Получают
А. диазотированием хлоргидрата анилина в присутст-
вии избытка анилина и последующим нагреванием
до 40° нейтрализованной суспензии диазоаминобен-
зола. А. является простейшим азокрасителем; впер-
вые получен в 1859; ранее применялся (под названием
анилиновый желтый) для окрашивания жиров и спир-
товых лаков; в наст, время — для крашения ацетат-
ного шелка и гл. обр. в качестве исходного продукта
в произ-ве полиазокрасителей, индулина и др. краси-
телей. Два других изомера о-А. (т. пл. 59°) и м-А.
(т. пл. 56—57°), сильно отличающиеся по свойствам
от n-изомера и получающиеся другими методами,
Не имеют практич. значения. А. А. Черкасский.
АМИНОАЛЬДЕГИДЫ И АМИНОКЕТОНЫ — ор-
ганич. соединения со смешанными функциями, свой-
ства к-рых в большой мере обусловлены взаимным
влиянием нуклеофильной аминогруппы и электро-
фильной карбонильной группы. Как известно, кар-
бонильные соединения реагируют с первичными ами-
нами с отщеплением воды и образованием соединений
типа т. н. шиффовых оснований, напр.: С3Н5СНО +
4- H2NC6H6->CliH5CH = NC6H5 + Н2О. Этим объяс-
ияется, почему А. и а., в особенности жирного ряда
с первичной аминогруппой, обычно неустойчивы
и легко превращаются в высокомолекулярные или
гетероциклич. соединения, к-рые можно рассматривать
как циклич. шиффовы основания. Так, аминоацеталь-
дегид, как и вообще а-аминоальдегиды, гладко кон-
денсируется в гетероциклич. основание, а именно
дигидропиразин, легко окисляющийся, наир., солями
Hg2+ в пиразин:
NH2 ОНС
/ \
СН2 + /СН2
СНО NH2
-2Н2О
^N’
сн., сн
1'1-
сн сн2
4N
Конденсация аминоацетона с роданистоводородной
к-той ведет к 2-меркапто-5-метилимидазолу:
1 + «
сн2 c=s
^nh2
СН3~С——NH
с c-s
При конденсации а-аминокетонов с соединениями,
содержащими активную метиленовую группу в а-
положении к СО-группе, образуются производные
пиррола. Напр., реакцией а-аминоацетоуксусного
эфира и самого ацетоуксусного эфира получен диэти-
____I ловый эфир 2,4-диметилпирролдикарбоновой-3,5 к-ты:
СН3-СО Н2С-СООС2Н5 СН3-С-----------------С-СООС„Н-
1 + I — II Ц 25
c2hsocochnh2 о=с-сн3 С2Н5ОСОС г-сна
XNH
Ввиду неустойчивости простейшие А. и а. обычно
применяют не в свободном виде, а в форме солей или
ацеталей. Так, простейший представитель а-амино-
альдегидов — аминоацетальдегид (глициновый аль-
дегид), не выделенный в чистом виде, стабилен в конц.
соляной к-те (в виде соли), его прочный ацеталь (т.
кип. 163°) может быть синтезирован действием N Н3 па
ацеталь хлоруксусного альдегида: С1СН2СН(ОС2Н6)2-г
4-NH3—>НС1 • NH2CH2CH(OC2H5)2. Простейший ами-
нокетон—аминоацетон NH2CH2COCH3, в свободном со-
стоянии не выделен, получен действием NH3 на мо-
нохлорацетон или восстановлением нзонитрозоацето-
на CH3COCH=NOH; образует прочные соли с к-тами,
напр. хлоргидрат с т. пл. 75°. При конденсации 2 мо-
лекул аминоацетона образуется 2,5-диметилдигидро-
пиразин, легко окисляемый в 2,5-диметилпиразин
(диэтиламинопентанон-4) (C2H5)2NCH2CH2CH2COCH3
(т. кип. 85—90/15 .ил1); синтезируют взаимодейст-
вием Na-производного ацетоуксусного эфира и
(C2H6)2NCH2CH2C1 с последующим кетонным расщеп-
лением продукта реакции; этот аминокетон служит
одним из промежуточных продуктов производства
антималярийных препаратов — акрихина и плаз-
мохина.
Р-аминоальдегиды и аминокетоны легко синте-
зируют присоединением NH3 к альдегидам типа
CR2 = СНСНО или к винилкетонам. Так, из окиси
мезитила образуется так называемый диацетонамин:
(СН3)2С = СНС0СН3 + NH3 -> NH2C(CH3)2CH2COCH3
(т. кип. 25°/0,2 мм). Такое направление реакции
объясняется тем, что вследствие сопряжения С=С
и С=О связей нуклеофильные реагенты способны при-
соединиться к С=С связи.
А. и а. с вторичной и в особенности с третичной
аминогруппой — гораздо более прочные соединения,
чем с 1УН2-группой. В частности, из окиси мезитила
и первичных аминов RCH2NH2 образуются стойкие
в-ва типа (CH3)2C(NHCH2R)CH2COCH3; действием
HNO2 на эти соединения можно получить соответ-
ствующие стойкие N-нитрозосоединения, реагирую-
щие с алкоголятами, лучше всего с циклогексаноля-
том Na в среде эфира с образованием диазоалкана
RCHN2. Этот метод получения диазоалканов имеет
препаративное значение и широкие границы приме-
нения. В случае присоединения NH3 к форону обра-
зуется т. н. триацетонамин (циклич. аминокетон с
вторичной аминогруппой):
/СН2-С(СН3)2
Oc[cH=C(CH3)a]a+NH3—* ОС NH
СН2-С(СН3)2
(т. кип. 205°; т. пл. 34,9°, т. пл. моногидрата 58°).
175
АМИНОАНТРАХИНОНОВЫЕ КРАСИТЕЛИ — АМИНОАНТРАХИНОНЫ
176
Аминоальдегиды ароматич. ряда более стойки, чем
аналогичные жирные соединения, и могут быть выде-
лены в свободном состоянии. Вполне стоек п-диметил-
аминобенаалъдегид, содержащий третичную амино-
группу. При конденсации о-аминобонзальдегида (т.
пл. 39—40°) с ацетальдегидом образуется хинолин:
Окраски А. к. ацетатного шелка очень прочны к свету
и к мокрым обработкам. Введение в молекулу А. к.
сн;о
-N'H.
н2;сн —2н2о
о;сн
Еще более прочны ароматич. аминокетоны; нек-рые
из них имеют большое практич. значение как полу-
продукты при произ-ве красителей, напр. 4,4'-тетра-
метилдиамииобензофенон, так наз. кетон Михлера
n-(CH3)2NCeH4COC6H4N(CH3)2-n (т. кип. выше 360°
с разл., т. пл. 179", растворим в органич. растворите-
лях за исключением эфира); в пром-сти производится
действием фосгена (в определенных условиях также
СО2) на диметиланилин и служит для получения
трифенилметановых красителей. Из аминоантрахино-
нов производят индантреновые красители (см. Амино-
антрахиноновые красители). Я. Ф. Комиссаров.
АМИНОАНТРАХИНОНОВЫЕ КРАСИТЕЛИ —
производные а-аминоантрахинона, преим. алкил- или
арилзамещенные 1,4- или 1,5-диаминоантрахинонов;
в нек-рых случаях в антрахиноновое ядро входят
и др. заместители, напр. оксигруппы. Благодаря
высоким колористич. свойствам и яркости окрасок
А. к-, широко применяются в пром-сти.
По применению А. к. подразделяют на: 1) кислот-
ные, в молекуле к-рых имеется сульфогруппа, при-
дающая красителю растворимость в воде и сродство
к волокнам животного происхождения. Эти красители
могут применяться также для нек-рых видов синто-
тич. волокон (капрон, найлон); 2) нерастворимые
в воде производные амино- и аминооксиантрахинонов,
применяемые в тонкодиспорснон форме для суспен-
зионного крашения ацетатного шелка и полиэфирных
волокон, напр. кислотный зеленый антрахиноновый
(I) или синий 3 для ацетатного шелка (II).
SO3H
Сложная хромофорная система этих соединений обра-
зуется включением свободных пар электронов атома
азота аминогруппы в л-электронную систему конъю-
гированной цепи антрахинонового ядра. При этом
перемещение электронной плотности усиливается на-
личием в цепи элоктронофильных заместителей, кар-
бонильных групп и приводит, благодаря снижению
разности уровней энергии возбужденного и нормаль-
ного состояний молекулы, к поглощению света в длин-
новолновой части спектра.
А. к. окрашивают волокна гл. обр. в фиолетовые,
синие и зеленые тона. Красные до желтых оттенки
получают обычно превращением 1-аминоантрахинон-
ироизводных в гетероциклич. соединении типа 1,9-ан-
трапиридинов (III), -антрапиридонов (IV), -антрапи-
римидинов (V), -антрапиримидонов (VI). Окраски А. к.
отличаются яркостью и высокой светопрочностью.
Повышение прочности окраски на шерсти к мок-
рым обработкам достигается введением гидрофобных
групп, утяжеляющих молекулу красителя, напри-
мер введением алкильных групп с длинной цепью.
нек-рых заместителей, напр. фтор замещенного алкила
или циангруппы в 2- и 3-положения антрахинонового
ядра, значительно повышает прочность окрасок к
дымовым газам.
Лит.: К о г а н И. М., Химин красителей, 3 изд., М.,
1956; В енкат ар ам ан К., Химин синтетических краси-
телей, пер. с англ., т. 2, Л., 1957, с. 919—936.
Г. Н. Курдюмова.
АМИНОАНТРАХИНОНЫ — промежуточные про-
дукты произ-ва синтетич. красителей (см. Антрахи-
ноновые красители, Аминоантрахиноновые красители).
1-Аминоантрахинон, 1,4-диаминоантрахинон, 1, 4, 5, 8-
тетрааминоантрахинон непосредственно используют
для крашения ацетатного шелка. А. являются сла-
быми основаниями, их соли гидролизуются водой;
диазотируются в конц. H2SO4 или смеси последней
с ледяной уксусной к-той, но не образуют диазоамино-
соединений с диазосоедпнепиями. Все А. окрашены,
окраска а-замещенных глубже, чем у р-замещенных.
2-Аминоантра хинон оранжевого цвета, 1,4,5,8-
тетраамипоантрахинон синего цвета. А. легко субли-
мируются, особенно в вакууме, нерастворимы в воде,
умеренно растворимы в большинстве органич. раство-
рителей. Смеси А., растворенные в неполярных или
малополярных в-вах, хорошо делятся при хромато-
графировании на окиси алюминия или силикагеле,
что может служить для аналитич. определений.
1-А миноантрахинон, т. пл. 2 56°; в пром-сти
готовят аммонолизом солей антрахинон-1-сульфонислоты вод-
ным р-ром аммиака в автоклавах в присутствии окислителей
(.м-нитробензолсульфонислота, мышьяковая к-та). Менее чи-
стый продукт получается при восстановлении продуктов нитро-
вания антрахинона, очищенных нагреванием с бисульфитом
натрия; наряду с 1,5-и 1,8-диаминоантрахинонами и небольшим
количеством ft-аминозвмещенных антрахинона образуется при
действии гидронсиламина на р-р антрахинона в конц. H2SO4
в присутствии небольших количеств солей железа или вана-
дия. А. используют в произ-ве кубовых, кислотных, прямых,
хемо-красителей, пигментов и красителей для ацетатного и
синтетич. волокон. 2-А миноантрахинон, т. пл. 302°,
получают аммонолизом водным р-ром аммиака 2-хлораитра-
хинона пли натриевой соли 2-антрахинонсульфокислоты; при-
меняют в произ-ве индантрона (кубовый синий О), флавант-
рона и нек-рых других кубовых красителей.
1,2-Д иаминоантрахинон, т. пл. 303—304°, полу-
чают аммонолизом 1-аминоантрахинон-2-сульфокислоты; ис-
ходный продукт производства кубового алого ЖХ. 1,4-Д и а-
миноантрахинон, т. пл. 2 68°, получают нагреванием
в автоклаве хинизарина с водным аммиаком и гидросульфитом
натрия с последующим окислением лейкосоединения нитробен-
золом или хлором; находит огранич. применение как красно-
фиолетовый краситель для ацетатного волокна и служит исход-
ным продуктом произ-ва кубового коричневого СК и 1-амино-
4-бензоиламиноантрахинона. 1,5-Диаминоантрахи-
нон, т. пл. 319°, получают аммонолизом динатриевой соли
177
АМИНОАЦЕТОН — АМИНОГАЛОГЕНАНТРАХИНОНЫ
178
1,5-антрахинонсульфокислоты, аналогично 1-аминоантрахи-
нону. 1,5-Диаминоаитрахинон применяется гл. обр. для полу-
чения 1-амино-5-бензоИламшюантрахинона — полупродукта
синтеза разнообразных кубовых красителей. Диамин ацили-
руют в нитробензоле хлористым бензоилом. При этом обра-
зуются 1-бензоил- и 1,5-ди-(беИзоиламино)-антрахиноны. Нит-
рованием последнего получают моно- и динитропроизводныс,
из к-рых восстановлением и гидролизом бензоильных групп
готовят соответственно 1,4,5-триамино-и 1,4,5,8-тетраамино-
антрахиноны. Эти в-ва можно выделить из смеси 1,5- и 1,8-ди-
бензоиламиноантрахинона, полученной из антрахинона нитро-
ванием, восстановлением и бензоилированием. 1,4,5,8-Тет-
раамии оа и трахинон готовят действием водного
р-ра аммиака под давлением на 1,4,5,8-тетрахлорантрахинон
и сульфита аммония на 2-сульфокислоту или 2 ,6-дпсульфо-
кислоту 4,8-ди амино-1,5-дпокси антр ахинон а. 1,4,5,8-Тетр а-
аминоантрахинои окрашивает ацетилцеллюлозное волокно
в синий цвет.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; В е н к а т а-
раман К., Химия синтетических красителей, пер. с англ.,
т. 1, Л., 1956, с. 114—18; Коган И. М., Химия красителей,
3 изд., М., 1956. Н. С. Докунихин.
АМИНОАЦЕТОН (амивопропавон, ацетониламин)
CH3COCH2NH2, молекулярвый вес 73,09; в свобод-
ном виде не выделев; образуется из апетовилфта-
лимида GH3COCH2N (СО)2С6Н4 при нагревании с
НС1, восставовлепием Sn и SnCl2 изонитрозоапетова
CH3COCH = NOH и витроацетова CH3COCH2NO2, дей-
ствием NH3 ва хлор-
Н3С—C==NH ацетон СН3СОСН2С1. В
не с—SH Щелочном Р"Ре А. лег-
\ / ко окисляется соля-
N ми Hg2+ и Си2+, Н2О2,
“ давая 2,4-диметилпир-
азин (I); с KSCN образует 2-меркапто-4-метилимид-
азол (II). Реакция с февилгидразином приводит
к дифенилгидразопу метилглиоксаля:
CH3COCII2NH2 4- 2CeHr)NHNH3 —
— NH3 + СИ3С(= N-NIICc1I5)CII = N-NHCGn5
Хлоргидрат A. CH3COCH2iXH2-HCl— гигроскопич.
бесцветные кристаллы, т. пл. 75° (из смеси спирта
и эфира); с KNO2 образует ацетол СН3СОСН2ОН;
может быть идентифицирован в виде двойной соли с
PtCl4:
(CH3COCH2NH3 • HCl)2PtCl4
— оранжевые иглы, т. пл. 188—189° (из спирта).
М. Ф. Турчинский.
АМИНОАЦИЛАЗЫ (ацилазы) — ферменты, ката-
лизирующие обратимый процесс гидролиза и синтеза
ряда (М-ацил)-Ь-а-аминокислот:
Н»о
R—СН-СООН------► R—СН—СООН -l-R'COOH
NHCOR' NH2
Известны след. A.: A.-I—активная по отношению
к большинству L-ациламинокислот, но неактивная
по отношению к L-ацетиласпарагивовой к-те; А.-II —
действующая только на L-апетиласпарагиновую к-ту.
A.-I выделена из почек свивьи; разработан метод
получения высокоочищенного электрофоретически го-
могенного высокоактивного препарата A.-I, к-рый
представляет собой белковое в-во с максимумом погло-
щения 240 ммк и мол. весом ок. 1,2 • 105. Оптимум
действия фермента при pH 7,5. Препараты А. нашли
широкое применение для разделевия а-аминокислот
ва оптич. изомеры путем асимметрии, гидролиза их
ацилпроизводиых. Метод разделения рацемич. а-ами-
нокислот с помощью ацилазы прост, удобен и дает
высокую степень оптич. чистоты получаемых изомеров
(99,9%). При помощи препарата А. разделено более
40 рацематов различных аминокислот.
Лит.: Methods in enzymology, ed. S. P. Colowick and
N. O. Kaplan, v. 2, N. Y., 1955, p. 115; Ци Ч ж э н-у и
Орехович В. Н., Биохимия, 1958, 23. вып. 5, с. 772;
О р е х о в и ч В. Н. [и др.], там же, 1959, 24, вып. 4, с. 667.
В. Б. Спиричев.
АМИНОБЕНЗАЛЬДЕГИД NH2C6H4CHO, мол. в.
121,14 —известен в виде 3 изомеров: о-А (т. пл. 39—40°),
м-А. (желтый аморфный порошок) и п-А. (т. пл.
70—72°). А. растворим в минеральных к-тах и обыч-
ных оргавич. растворителях. А. очень неустойчив;
нейтральвые р-ры А. быстро темнеют вследствие кон-
денсации с участием обеих функциональных групп.
А. дает оксимы, гидразоны и др. производные альде-
гидов, а также N-ацетильиые производные. Общий
способ получения А. заключается в восстановлении
соответствующих нитробензальдегидов. Своеобразный
метод синтеза п-А. состоит во взаимодействии «-нит-
ротолуола с серой или Na2S и едким натром. о-А при-
меняют для получения хинолиновых оснований:
м- и п-А. — полупродукты для синтеза нек-рых кра-
сителей И др. . ф. Комиссаров.
n-АМИНОБЕНЗОЙНАЯ КИСЛОТА (витамин Н1(
ПАВК) «-H2NC6H4COOH, мол. в. 137,11 — бес-
цветные кристаллы; т. пл. 186—187°, растворимость
(г/100 мл растворителя): в воде 0,34 (12,8°), в 90%-ном
спирте 11,3 (9,6°); нерастворима в петролейном эфире.
А. к. может быть получена окислением «-нитрото-
луола бихроматом калия и последующим восстановле-
нием п-нитробензойной к-ты:
Известны и др. методы ее получения. А. к. амфотерва;
ее соли щелочных металлов и минеральных к-т хороша
растворимы в воде. А. к. образует производные,
получаемые как превращением функциональных групп
(NH2 и СООН), так и замещением в бензольном ядре.
Для колич. определения А. к. в животных тканях
пользуются методом диазотирования и азосочетания
с N-a-пафтилэтилендиамином с последующим фото-
колориметрировавием. Этиловый эфир А. к. — анесте-
зин — обладает очень слабой витаминной активностью;
диэтиламиноэтиловый эфир — новокаин — местноане-
стезирующее средство.
А. к. применяется в качестве ростового фактора
микроорганизмов и витамина, необходимого для нор-
мального обмена веществ и жизнедеятельности че-
ловека и животных; ее антивитаминами являются
сульфаниламидные препараты. Нек-рые производные
А. к. применяются для синтеза тиоипдигоидных кра-
сителей.
Лит.: Березовский В. М., Химия витаминов, М.,
1959, см. также при ст. Аминокислоты и Витамины.
В. М. Березовский.
АМИНОГАЛОГЕНАНТРАХИНОНЫ — замещенные
антрахинона, повторяющие многие реакции и
свойства амино антрахинонов и галогенопроизводных
антрахинона. Такое сочетание свойств определяет
использование нек-рых Л. в качестве полупродуктов
синтеза красителей. В зависимости от положения за-
мещающих групп А. получают галогенированием
аминопроизводных антрахииова или их N-ацильных
замещенных, восстановлением витрогалогевопроиз-
водвых антрахинона, частичным аммонолизом полига-
логенопроизводных или циклизацией бензофенон-о-
карбоновых к-т, содержащих галоген и аминогруппу.
Из числа А. технич. значение имеют: 1-а мин о-2-х л ор-
антрахинон, образуется в качестве побочного про-
дукта при синтезе 2 -амино-3-хлорантрахинона из 3-амино-
4-хлорбензофенон-2'-карбоновой к-ты. При бромировании
образует 1-амино-2-хлор-4-бромантрахинон — исходный про-
дукт синтеза кислотного чистосинего антрахинонового К.
1-А мин о-2,4-д ибромантрахинон получают бро-
мированием 1-амицоантрахинона; служит исходным продук-
том синтеза кислотных красителей. 1-Б ензоиламино-
4-х лорантрахинон получают хлорированием 1-бензонл-
аминоантрахинона; полупродукт синтеза кубовых красите-
лей, в частности кубового коричневого К. 1-А мин о-5-х лор-
антрахинон получают восстановлением 1-хлор-5-
нитроантрахинона. С хлористым бензоилом дает 1-беИзоил-
амино-5-хлорантрахинон — полупродукт синтеза кубового зо-
179
4-АМИНОДИФЕНИЛ - АМИНОКИСЛОТЫ -
180
лотисто-оранжевого 2Ж. 2-А мин о-1-х лорантрахи-
н о н получают хлорированием 2-аминоантрахинона; служит
исходным продуктом синтеза флавантрона и желтых кубовых
красителей производных тиазола. 2-А мин о-3-х л о р ант-
рахинон получают, как указано выше, циклизацией 3-
амино-4-хлорбензофенон-2'-карбоновой к-ты или аммонолизом
2,3-дихлорантрахинона водным аммиаком в автоклавах. При-
меняют для получения 2-амино-1-бром-3-хлорантрахинона —
исходного продукта синтеза дихлориндантрона (кубового голу-
бого К), а также соответствующего последнему тетрасерно-
кислого эфира лейкосоединения—кубозоля голубого К.
1,3-Д и б р о м-2-а миноантрахинон получают броми-
рованием 2-аминоантрахинона; служит для синтеза дибромин-
дантрона. При сплавлении 1,3-дибром-2-аминоантрахииона
с незамещенным 2-аминоантрахиноном образуется 2-амино-З-
бромантрахинон. 1,4-Д и а м и н о-2,3-д ихлорантра-
хинон получают действием хлористого сульфурила на
лейко-1,4-диаминоантрахинон. Применяют для сйнтеза ди-
аминодихлор индантрона (индаптрен зеленый 2 Б) и фиолетового
кислотного красителя. Смеси А. четко разделяются при хро-
матографировании р-ров в неполярных или малополярных
растворителях на окиси алюминия или силикагеле, что может
служить для аналитич. характеристики А.
Лит.: Ворожцов Н.Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; В е н к а т а-
раман К., Химия синтетических красителей, пер. с англ.,
т. 1, Л., 1956; К о г а н И. М., Химия красителей, 3 изд., М.,
1956. Н. С. Докунихин.
4-АМИНОДИФЕНИЛ (4-фениланилин)п-КН3С6Н5СсН5,
мол. в. 120,00 — листочки (из. водного спирта); т.
пл. 50—52°; т. кип. 302°/760 ,«.и. 191°/15 мм-, раство-
рим в спирте, эфире, хлороформе, мало растворим
в холодной воде, перегоняется с водяным паром.
Получают А. восстановлением 4-нитродифенила А.
может служить промежуточным продуктом в произ-ве
кислотных антрахиноновых красителей, однако ввиду
высокой канцерогенности в наст, время не произво-
дится И не применяется. Н. С. Вульфсон.
щ-АМИНОКАПРОНОВАЯ КИСЛОТА (е-амино-
капроновая кислота, 6-аминокапроновая кислота,
е-лейцин) NH2(CH2)5COOH, мол. в 131,18 — бес-
цветные кристаллы, хорошо растворимые в воде
и нерастворимые в спирте, ацетоне и др. органич.
растворителях. А. к. получают омылением е-бензоил-
аминокапронитрила, а также этилового эфира б-фта-
лимидбутилмалоновой к-ты, но наиболее распростра-
ненным способом получения является гидролиз е-кап-
ролактама:
СН»—сн3—со
I /NH -—±1. NHS(CH3)6COOH
сн,—сн,—сн»
Благодаря наличию аминогруппы и карбоксильной
группы А. к. одновременно обладает основными
и кислотными свойствами; легко реагирует со спир-
тами, образуя эфиры, а при взаимодействии с к-тами
(напр., муравьиной) или хлорангидридами к-т дает
соответств. N-замещенные А. к. При окислении А. к.
перманганатом получают адипиновую к-ту, а при
действии азотистой к-ты вместо е-оксикислоты полу-
чают изомерные гексеновые к-ты. При нагревании
А. к. наряду с образованием линейного полиамида
происходит образование 7-членного лактама (е-кап-
ролактам), нашедшего широкое применение для
синтеза полиамидных смол, типа капрон. При взаимо-
действии А. к. с хлористым бензоилом получают N-бев-
зоил е-аминокапроновую к-ту, к-рая является исход-
ным продуктом для синтеза биологически важных
соединений— П,Б,-е-бонзоиллизина и П,Б-лизина.
В. А. Сергеев.
АМИНОКИСЛОТЫ — карбоновые к-ты, содержа-
щие одну или неск аминогрупп В зависимости от
положения аминогруппы относительно карбоксила
различают а-, (3-, ?-А. и т. д. А. широко распростра-
нены в природе. а-А. являются структурными эле-
ментами молекул белков. Из белковых гидролизатов
выделено св. 20 а-А., имеющих (за исключением про-
лина и оксипролина) общую формулу RCH(NH2)COOH.
Природные а-А. делят на след, группы.
Наименование
Формула
Глицин
Аланин
Серин
Цистеин
Цистин
Метионин
Треонин
Валин
Норвалин
Лейцин
Изолейцин
Норлейцин
Алифатические А.
Моноамино карбоновые к-ты
NH3CH»COOH
CHgCH (NH,) СООН
СН, (ОН) СН (NH,) СООН
сн; (SH) CH (NHs) СООН
S, |СН,СН (NH,) СООН]3
СН. (SCH3) СН.СН (NH-.) СООН
СН3СН (ОН) СН (NH,) СООН
(СНз), СНСН (NH,) СООН
CHSCH,CH„CH (NH,) СООН
(CH3U СНСН.СН (NH->) СООН
СН3СН.,СН (СНз) СН (NHs) COQH
СН3 (СН»)з СН (NH.) СООН
М о и оам и нод п карбоновые к-ты
Аспарагиновая I НООССН..СН (NH,| СООН
Глутаминовая | НООС (СН»), СН (NH3> СООН
Диаминокарбоновые к-ты
Лизин
Аргинин
Канаванин
Орнитин
Цитруллин
H»NCH3 (СН»)3 СН (NH.) СООН
HJNC (=NH) NH (СН»)з~СН (NH,) СООН
H,NC (=NH| NHO (CH,)» CH (NH3) CH COOH
HZNCH» (CH,), CH (NH..) COOH
h;nC (=0) NH (CH3)a CH (NH.) COOH
Циклические A.
Гомоцикличеспие к-ты
Фенилаланин । CnHsCHnCH (NH.) СООН
Тирозин ; п-НОС0Н4СН„СН (NH.) СООН
Гетероцикли ч. к-ты: триптофан (I), гистидин (II).
Иминокислоты: пролин (III), оксипролин (IV).
CH2CH(NHa)COOH
N----CHaCH(NHa)COOH
С- Д.Н
NH //
нгс-----сн2
сна снсоон
III
НОСН—сн2
I I
н„с снсоон
2 NIK
IV
Встречаются в природе и J4-A. (Р-Ал«нци). Нек-рые
А. получены синтетически, в том числе <о-А.
При обозначении последовательности расположе-
ния аминокислотных остатков в полипептидах и бел-
ках употребляются след, сокращенные обозначения:
Аланин .... . ал. Лизин . лиз.
Аргинин ...... . арг. Метионин . . . . . мет.
Аспарагин . . . . acn-NH2 Норлейцин . . . норл.
Аспарагиновая ОКСИпролин . . . опрол.
к-та . асп. Пролин . прол.
В алии . вал. Серин . сер.
Гистидин . . . . . гис. Тирозин . . . . . тир.
Глицин . . . . . глиц. Треонин . . . . • тре.
Глутамин . . . . глут-Ь1Н2 Триптофан . . . . трип.
Глутаминовая Фенилаланин . . . фал.
к-та . глут. Цистин . цистин
Изолейцин . . . . изол. Цистеин .... . цистеин
Лейцин . лейц.
А. — бесцветные кристаллич. продукты, плавя-
щиеся при высоких темп-рах (220—315°), часто с раз-
ложением. А. растворимы в воде; большинство их
нерастворимо в органич. растворителях. А. — амфо-
терные электролиты и в водных р-рах существуют
+ —
в виде диполярных ионов NH3RCOO. Рентгенострук-
турным анализом доказано, что и в твердом состоя-
нии А. представляют диполи, чем объясняется их
значительная плотность и высокие темп-ры плавле-
ния. Дипольные моменты А. — порядка 15 D. Кон-
станты ионизации А. при равновесии выражаются
в виде отрицательного логарифма констант кислот-
ности и обозначаются индексом рК. А. имеют по
нескольку констант кислотности, соответствующих
ионизациям аминных и карбоксильных групп. Так,
181
АМИНОКИСЛОТЫ
182
для глицина рКа 2,4 (карбоксил), рКаг 9,8 (амино-
группа), дляаспарагиновойк-тырАГа 2,0(а-карбоксил)7
рАа2 4,0 (Р-карбоксил), рК а 9,8 (аминогруппа) й т. д.
А. имеют характерные полосы поглощения в инфра-
красной области спектра, наблюдаемые ок. 1500 см~1
(характеристич. частота карбоксильных ионов).
Ароматич. А. и цистеин поглощают в УФ части, что
определяется наличием в их молекулах дополнитель-
ных хромофорных групп. Все А. (кроме глицина)
содержат асимметрия, атом углерода и существуют
в оптически активных модификациях. К D-ряду от-
носятся А., имеющие конфигурацию D-глицеринового
альдегида. В том случае, когда в молекуле А. имеется
два центра асимметрии и если конфигурация у вто-
рого асимметрия, атома отличается от конфигурации
у а-углеродного атома, то после значков D или L
вставляют приставку алло- (напр., L-алло-треонин).
Оптич. активность А. зависит от реакции среды
вследствие возможных изменений в состоянии равно-
весия между анионной, катионной и диполярной их
формами. При переходе к катионной форме увели-
чивается правое вращение. Все А., входящие в состав
белков, являются L-изомерамп; D-формы обнаружены
лишь в капсулах микроорганизмов и в нек-рых при-
родных антибиотиках.
Благодаря наличию аминной и карбоксильной
групп А. образуют производные, характерные как
для аминов, так и для карбоновых к-т. Пикриновая,
фосфорновольфрамовая и флавиановая к-ты обра-
зуют нерастворимые соли с ди-А. и используются
как специфич. осадители последних. Для идентифи-
кации N-концевых А. в пептидах и белках широко
используются динитрофенильные производные, к-рые
получаются при взаимодействии А. с 2,4-динитро-1-
фторбензолом. Для этой же цели используется мети-
лирование свободных аминогрупп йодистым метилом,
диазометаном, диметилсульфатом, восстановительное
метилирование формальдегидом в присутствии пал-
ладия, а также реакции N-концевых А. с гипоброми-
тами. При алкилировании А. образуются N-алкил-А.
и бетаины. А. могут ацилироваться; вступают
в реакции с арилизотиоцианатами, что дает возмож-
ность ступенчатого отщепления А. с аминного конца
полипептидной цепи и используется для изучения
последовательности А. в пептидах и белках. К мето-
дам этой группы относится отщепление N-концевых
аминокислот в виде тиотиазолидиндионов. _ _
При взаимодействии амидов а-А. с серо- । 2е
углеродом образуются дитиокарбаминовые СО ОС Н
К-ты, к-рые легко циклизуются в кислой 2 5
среде в 2-тиотиазолидиндионы. у- и б-А. могут быть
отщеплены в виде лактамов. Аминогруппы легко кон-
денсируются с формальдегидом, образуя оксиметил-
производные. Под действием азотистой к-ты происхо-
дит дезаминирование А. с образованием оксикислот.
Подобно другим карбоновым кислотам, А. дают
соли, амиды, галогенангидриды и эфиры. При нагре-
вании А. в инертном растворителе происходит декар-
боксилирование с образованием аминов. Под дейст-
вием окислителей А. дают альдегиды с укороченной
углеродной цепью. Для последовательного отщепле-
ния А. от карбоксильного конца полипептидной цепи
используется реакция с гцдразином, причем С-кон-
цевая А. отщепляется в неизменном виде, тогда как
все остальные А. дают гидразиды. Другим эффектив-
ным методом является отщепление С-концевых А.
в виде тиогидантоиновых производных. Нингидрино-
вая реакция, характерная для А., происходит при*
одновременном участии аминной и карбоксильной
групп и позволяет отличать А. от пептидов и белков.
Аллоксан дает с А. красное окрашивание, интенсив-
ность к-рого варьирует для разных А. Обе функцио-
нальные группы А. участвуют в образовании дикето-
пиперазинов и в реакции получения гидантоинов
при взаимодействии с мочевиной. А. образуют ком-
плексы с ионами (Ag, Hg, Pb, Си, Zn, Со); р-А. дают
ненасыщенные а-р-кислоты; у-, б- и е-А. легко от-
щепляют воду, превращаясь в лактамы. P-А. также
могут циклизоваться с отщеплением воды и образо-
ванием р-лактамов, однако в этом случае дегидратация
происходит значительно труднее, поэтому для получе-
ния Р-лактамов пользуются спец, методами: циклиза-
цией эфиров или ацилпроизводных P-А., присоедине-
нием кетонов к иминам или ароматич. нитрозосоедине-
ниям, действием щелочей на амиды Р-галогенозамещен-
ных карбоновых к-т и др. а-А. лактамов не образуют.
а-А. получают: О Из малонового эфира:
СН2(СООС2Н5)2 H0N0. HON=C(COOC2H5)2 -Si-
— NH»-CH(COOC»H5)2 (СНзСО)зО
Na
—• CHSCONHCH(COOC2H5)2 -
COOC2H5
CHsCONIld^=C-OC2H5 ЩЬВг C2H5
\)Na C(COOC2H5)2 —
ch8conh
—> C2H5CH(NH2)COOH 4- CO2 4- 2C2H5OH
Аналогично проводится синтез из циануксусного и ацетоуксус-
ного эфиров.
2) Действием аммиака на галогенозамещенные карбоновые
к-ты: RCH(C1)COOH 4- 2NH3 — RCH(NH2)COO 4- NH4C1.
3) По Штрекеру—Зелинскому:
RCH(OH)CN 4- NH3 — RCH(NH2)CN 4- Н2О
RCH(NH2)CN 4- 2Н2О -> RCH(NH2)COOH 4- NH3
4) Восстановлением оксимов или гидразонов кетоно- и
альдегидокислот:
a) R—С—СООН -Hi R—СН—СООН
II I
NOH NH2
б) R—C=N-NH-С„Н5 „ R—СН—СООН
। £П, iiCzi ।
соон nh2
5) Действием на кетонокислоты аммиака и водорода в при-
сутствии платины или палладия:
R—СО—СООН NH?2 Hi. R-C-COOH 4г R-сн-соон
Pt II I
NH NH2
6) Фталимидным методом Габриэля:
н2о
5
+ NH2CH2COOH 4- С2Н5ОН
соон
соон
Два последних метода позволяют получать колич. выход и
используются для синтеза А., меченных тяжелым азотом.
7) Конденсацией альдегидов: а) с глицинангидридом,
б) с гиппуровой кислотой, в) с гидантоином:
СО —NH
a) 2RC1IO 4- Н«С СН2 -~-3°,
\nH-CO 7
/СО-NH
— R—СН=С C=CHR —
\гн—СО7
восстановление 2RCHsCHCOOH
расщепление ।
NH2
б) RCHO 4- СН2—СООН - RCH = С - СО —2-
NH—COC.Hj | /°
N—С—СвН6
— RCH = ССООН 4- С.ЩС ООН
NH2
183
АМИНОКИСЛОТЫ - АМИНОНАФТОЛСУЛЬФОКИСЛОТЫ
184
н
I
RCH2-CH-N4 н2О
I /СО----»RCH2CH—СООН+NH3+COa
со—Nz I
I NHs
н
8) В нек-рых случаях А. могут быть получены карбокси-
лированием аминосоединений.
При синтезе получаются рацемич. смеси А., для разделе-
ния к-рых пользуются гл. обр. ферментативными методами.
Нек-рые а-А. ввиду сложности их синтеза выделяют из
гидролизатов (глутаминовую к-ту из казеина и клейковины
злаков, тирозин из фиброина шелка, аргинин из желатины,
гистидин из белков крови) в виде труднорастворимых солей
или др. производных. Для разделения А. широкое применение
нашли адсорбция на ионообменных смолах и высоковольтный
электрофорез. Р-, у-, 8- и е-А. получают действием NHa на соот-
ветствующие галогенозамещенные к-ты, кроме того, р-А.
получают также присоединением NHa к ненасыщенным а-,
р-к-там или конденсацией альдегидов с малоновой к-той в при-
сутствии спиртового р-ра аммиака (метод Родионова): RCHO 4-
+ НООССН2СООН + NHa — RCH(NH2)CH2COOH + Н2О +
4- СО2. 8- и е-А. получают из оксимов циклич. кетонов Бек-
мана перегруппировкой. О методах синтеза <о-А. см. <о-Алшно-
капроновая кислота и <о-Аминоэнантовая кислота.
Большое значение имеет синтез А., меченных по азоту и
углероду. Для получения А., меченных тяжелым азотом в a-по-
ложении, используется метод восстановительного аминиро-
вания, где исходными веществами служат NHa с Nls и соответ-
ствующая кетокислота, а также метод с фталимидом калия,
содержащим N16. Для синтеза меченных по углероду А. на
соответств. альдегид действуют (NH4)2CO3 и NaCN с С14. Полу-
ченный гидантоин превращают через аминозамещенный гидан-
тоин в А. с С14.
Определение общего количества А. производят по Ван-Слай-
ка методу, основанному на дезаминировании А. азотистой
к-той и измерении объема выделившегося азота. А. можно
определить титрованием щелочью после связывания NH2-rpynn
формальдегидом (Серенсена метод), а также колориметрически
по реакции с нингидрином или по Фолина методу. Для колич.
определения аминокислотного состава белков и пептидов про-
дукты гидролиза разделяют методом распределительной хро-
матографии, и А. идентифицируют с помощью нингидриновой
реакции.
А. занимают центральное место в азотистом обмене
живых организмов. Они служат источником образо-
вания необходимых для жизнедеятельности веществ —
белков, пептидов, ферментов, гормонов и др., а также
конечных продуктов азотистого обмена — аммиака,
мочевины и др. А., к-рые могут синтезироваться
в организме человека и животных, называют заме-
нимыми, а те, к-рые не синтезируются в животном
организме, получили название незаменимых А. (см.
Заменимые аминокислоты и Незаменимые амино-
кислоты). Многие А. находят применение в медицине.
Различные продукты гидролиза белков и смеси А.
используются для парентерального питания (минуя
желудочно-кишечный тракт); смеси незаменимых А.
применяются для внутривенных вливаний. Метионин
используется при лечении ожогов, малокровия, бо-
лезней печени, а также для подкормки с.-х. живот-
ных, т. к. он влияет на их рост. Гистидин применяется
при язвах желудка, триптофан — при пеллагре,
глутаминовая к-та — при ряде нервных заболеваний.
Лактамы нек-рых А. (напр., е-аминокапроновсщ к-ты)
используются для получения высокополимерных плас-
тин. материалов (капрон и др ). <о-А. применяют
в качестве исходных в-в при получении пластмасс
и искусств, волокон (см. Полиамидные волокна).
Лит.: Аминокислоты и белки. Сб. ст., пер. с англ., под
ред. А. Г. Пасынского, М., 1952; Браунштейн А. Е.,
Биохимия аминокислотного обмена, М., 1949; Блок Р. и
Боллинг Д., Аминокислотный состав белков и пищевых
продуктов, пер. с англ., М., 1949; Губен И., Методы орга-
нической химии, пер. с нем., т. 4, вып. 1, кн. 2 М. — Л.,
1949, с. 851, 1243; Кнунянц И. Л., Пер во в а Е. Я.,
Усп. хим., 1955, 24, вып. 6, с. 641; Возникновение жизни на
Земле. Сб докл. на международном совещании. Август 1957,
М., 1959; Greenberg D. М., Amino acids and proteins,
Springfield (Ill), 1951; Cohn E. J. and Eds all J. T.,
Proteins, amino acids and peptides as ions and dipolar ions,
N. Y., 1943; Advances in protein chemistry, ed. M. L. Anson,
K. Baily, J. T. Edsall, N. Y., 1954, v. 9, p. 82, 122—202; E d-
s a a 1 J. T., Aspects actuels de la biochimie des acides amines
et de protcines, Liege, 1958 (Actualites biochimiques, № 20);
Мейстер А., Биохимия аминокислот, пер. с англ., М.,
1961; J. Amer. Chem. Soc., 1960, 82, № 21, р^ 5545 —74.
АМИНОМАСЛЯНЫЕ КИСЛОТЫ — алифатические
аминокарбоновые к-ты, известные в виде несколь-
ких изомеров. В зависимости от положения и числа
аминогрупп относительно карбоксила различают а-,
Р-, у-А. к. (C4H9O2N, мол. в. *103,12) и а,|3-, а,у-ди-
аминомасляные к-ты (C4Hi0O2N2, мол. в. 118,14).
а-А миномасляная кислота — бес-
цветные кристаллы; СН3—СН2—CH2(NH2)—СООН;
т. пл.: D, L-изомеров 304—307°; D- и L-А. к. 292° (с
разл.); [а]р’: D-а-А. к. +8°; L-a-A. к. —7,86° (в воде).
а-А. к. растворима в воде, горячем спирте, нераство-
рима в эфире. Синтетически а-А. к. получают из а-
броммасляной к-ты и из а-аминопропан-а,а-дикарбо-
новой к-ты. а-А. к. вступает во все общие для ами-
нокислот реакции; количественно определяют хро-
матографически, используя нингидриновую реакцию.
L-a-A. к. найдена в крови и моче, в тканях расти-
тельного и животного происхождения.
р-А миномасляная кислота — бес-
цветные кристаллы; СН3—CHNH2—СН2—СООН;
т. пл. D, L-p-А. к. 193—194°; [a]g : D-^-A. к. +35,3°;
L-P-A. к. — 35,2° (в воде); |3-А. к. растворима в воде
и в спирте, нерастворима в эфире. При нагревании
до 200° от р-А. к. отщепляется аммиак и образуется
ненасыщенная а,|3-к-та. В организмах [3-А. к. не обна-
ружена.
у-А миномасляная кислота (пипери-
диновая к-та) NH2—СН2—СН2—СН2—СООН — кри-
сталлы; т. пл. 203° (с разл.), растворима в воде, не-
растворима в спирте, эфире, бензоле. у-А. к. легко
отщепляет воду и образует бутиролактам (а-пирро-
лидон). у-А. к. образуется при декарбоксилировании
L-глутаминовой к-ты декарбоксилазами и широко
распространена в животных и растительных тканях.
В мозге млекопитающих содержится ок. 50 мг°/0.
а,Р-Диа миномасляная кислота
CH3CH(NH2)CH(NH2)COOH; получают из а,|3-ди-
броммасляпой к-ты. В организмах не обнаружена.
а,у-Д и а м и н о м а с л я н а я кислота
CH2(NH2)CH2CH(NH2)COOH — легко растворима в
воде, плохо — в метаноле и этаноле; нерастворима
в эфире и лигроине. Медная соль а,у-А. к. интенсивно
окрашена. Получают а,у-А. к. из а-амино-у-фталь-
имида масляной к-ты. Встречается в нек-рых анти-
биотиках (полимиксины).
Лит. см. при ст. Аминокислоты. Л. П. Алексеенко.
АМИНОНАФТОЛСУЛЬФОКИСЛОТЫ — соедине-
ния, имеющие общую формулу CI0H5(NH2)(OH)(SO3H)
или C10H4(NH2)(OH)(SO3H)2. Существует неск. изо-
меров А. в соответствии с различным расположением
групп, входящих в молекулу нафталина. А. — бес-
цветные кристаллы; в большинстве случаев мало
растворимы в воде, практически нерастворимы
в обычных органич. растворителях; растворяются
в воде в присутствии щелочных реагентов (едкой
щелочи, соды, аммиака и т. п.), причем превращаются
в растворимые в воде соли. Химич, свойства А. опре-
деляются свойствами функциональных групп. В ре-
зультате замещения атомов водорода в сульфогруппах
на металлы образуются соответств. соли; соли щелоч-
ных металлов большинства А. растворимы в воде,
соли остальных металлов, как правило, мало раство-
римы. При действии на А. ангидридов и хлорангид-
ридов к-т образуются N-ацильные производные типа
185
АМИНОНАФТОЛСУЛЬФОКИСЛОТЫ — о> АМИНОПЕЛАРГОНОВАЯ КИСЛОТА
186
R(OH)(SO3H)(NHAc), где Ас обычно остаток уксусной
(СН3СО—), бензойной (С6Н5СО—), п-нитробензойной
(NO2—С6Н4—СО—) к-т или п-толуолсульфокислоты
(СН3—С6Н4—SO2—). Многие А. легко диазотируются
в обычных условиях (см. Диазотирование) и превра-
щаются в устойчивые диазосоединения. А., содержа-
щие группы —ОН и —NH.2 в орто-положении, напр.
1 -амино-2-нафтол-4-сульфокислота, диазотируются
только в отсутствии минеральной к-ты и в присутствии
катализатора — соли меди. А., содержащие группы
—ОН и —NH2 в разных циклах нафталинового ядра,
весьма активно вступают в реакцию азосочетания
с диазосоединениями; нек-рые реагируют с диазо-
соединениями только в щелочной среде, присоединяя
остаток диазосоединения к той части нафталинового
ядра, в к-рой находится группа —ОН, другие могут
реагировать как в щелочной, так и в кислой средах,
образуя соответственно изомерные окси- и амино-
азосоединения (см. таблицу). Реакции диазотирования
и сочетания используют для открытия и количеств,
определения А.
Получение А. видно на примере Аш-кислоты:
Fe+H2SO4
восстановление
Применяют А. в качестве исходных веществ в про-
изводстве азокрасителей и некоторых промежуточ-
ных продуктов. Строение, название и поведение при
синтезе азокрасителей технически важных А. при-
ведены в таблице; цифры обозначают положение за-
местителей.
Среда
сочетания
Рациональное
название
Тривиальное
название
1 - Амипо-2-н афтол- 4-с ульфокислот а 1-Амино-5-нафтол- 7-сульфокислота 1-Амино-8-н афтол- 4-с ульфокис лот а М-кислота С-кислота 1; 2; 4 NH2; ОН; SO3H 1; 2; 5; 7 NH2; X; ОН; SO3H* 1; 2; 4; 7; 8 NH2; X; SO3H; X He соче- тается Кислая или щелочная Кислая или щелочная
2-Амино-5-н афтол- 7-с ульфокислот а И-кислота 1; 2; 5; 6; 7 X; NH.,; OH; X; SO3H Кислая или щелочная
2-Амино-8-нафтол- Гамма- 1; 2; 6; 7; 8 Кислая
6-сульфокислота кислота X; NH,; SO3H; X; OH или щелочная
1-Амино-8-нафтол- 2,4-дисульфокислота СС-кислота 1; 2; 4; 7; 8 NH2; SO3H; SO3H; X; OH Щелочная
1 - Амино-8-н афтол- 3,6-дисульфокислота Аш-кислота 1; 2; 3; 6; 7; 8 NH,; X; SO3H; SO3H; X; OH Кислая или щелочная
1 -Амино-8-нафтол- 4,6-дис ульфокислот а К-кислота 1; 2; 4; 6; 7; 8 NH2; X;SO3H; SO3H; X; OH Кислая или щелочная
2-Амино-8-нафтол- 3,6-дис ульфокислота РР-кислота 1; 2; 3; 6; 7; 8 X;NH2;SO3H; SO3H; X; OH Кислая или щелочная
• X — место замещения остатком диазосоединения при
сочетании.
1-Амино-2-нафтол-4-сульфокислота и Аш-кислота
применяются в синтезе азокрасителей в качестве ди-
азосоставляющей, а все остальные А. — в качестве
азосоставляющих.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; Ворон-
цов И. И., Полупродукты анилинокрасочной промышлен-
ности, М., 1955; Фир ц-Д а в и д Г. Э. и Ел анже Л.,
Основные процессы синтеза красителей, пер. с нем., М., 1957.
А. А. Черкасский.
АМИНОНАФТОЛЫ Ci0H6(NH2)(OH) — производ-
ные нафталина, содержащие ОН- и ГШ2-группы,
кристаллич. продукты, практически нерастворимые
в холодной воде. При действии кислот А. превра-
щаются в растворимые в воде нафтиламмониевые соли,
при действии щелочных реагентов — в растворимые
в воде нафтоляты. А. очень легко окисляются на воз-
духе и при действии окислителей, причем р-ры А.
приобретают интенсивные окраски. А., содержащие
группы -ОН и -NH2 в одном ядре, при окислении
превращаются в нафтохиноны. Если же эти группы
содержатся в разных ядрах, то А. вступают в азосоче-
тание с диазосоединениями и образуют азокраси-
тели. Нек-рые А. диазо-
HO3S NO, тируются. При действии
ацилирующих средств
| || | ___г (ангидридов и хлоран-
НО Н гидридов кислот) А. пре-
3 3 вращаются в N-ацильные
производные, имеющие
четкие темп-ры плавле-
ния, используемые для
идентификации А.
А. получают восстано-
влением соответств. нит-
ро- или нитрозонафтолов (способ А), восстановле-
нием оксиазосоединений (способ Б), сплавлением наф-
тиламинсульфокислот с едким натром (способ В, про-
мышленный), действием амида натрия на нафтолы (спо-
соб Г), отщеплением сульфогруппы от аминонафтол-
сульфокислоты при нагревании с разбавленной к-той
(способ Д) или при действии амальгамы натрия (способ
Е), действием аммиака на диоксинафталины при высо-
кой темп-ре (способ Ж), действием бисульфита натрия
на нафтилендиамины (способ 3) и др. В пром-сти при-
меняют лишь 1,5-, 1,7- и 1,8-А. в качестве исход-
ных в-в для получения азокрасителей.
Положение групп Т. пл. аминофенолов (I), ацетильного (II) и бензильного (III) производ- ных, °C Способ полу- чения
он nh2 I 11 III
1 2 128—129 190 А, Б
1 4 — 187 228—229 А, Б
1 5 192 — — в, Г, 3
1 6 — 100 д
1 7 158 210—211 ж
1 8 95—97 181 193—194 В, г, д
О 1 — 235 248-249 А, Б
2 3 234 234 Ж
2 4 185 (с разл.) 179 __ в
2 5 190 218 152 А, Г, Е
2 6 212—213 Е
2 7 201 232 243-246 в, Е, Ж, 3
2 8 205—207 165 208-209 А, В
со АМИНОПЕЛАРГОНОВАЯ КИСЛОТА Р(9-амино-
нонановая кислота) H2N(CH2)8COOH, мол. в. 173, 26—
бесцветные кристаллы; т. пл. 189—190°; растворяется
в горячей воде, нерастворима в спирте, ацетоне и др.
обычных растворителях. При нагревании А. к. легко
отщепляет воду, образуя полиамидную смолу, из
к-рой можно получить синтетич. волокно (см. Поли-
амидные волокна). В промышленном масштабе может
187
АМИНОПИРИДИНЫ - АМИНОПЛАСТЫ
188
быть получена гидролизом а,а,а,со-тетрахлорнонана
в присутствии 96—98%-ной H2SO4 при 60—80° и по-
следующим аммонолизом полученной ш-хлорпеларгс-
новой к-ты 25%-ным р-ром NH4OH при 100° в течение
1 часа: C1(CH2)8GG13-G1(CH2)8GOOH-NH2(GH2)8GOOH.
а,а,а,со-тетрахлорнонан получают теломеризацией
этилена четыреххлористым углеродом, в. А. Сергеев.
АМИНОПИРИДИНЫ — замещенные /у)
пиридина, содержащие одну или неск.
аминогрупп; известны все 3 моноизо-
мера.
Моноизомер Т. пл., °C Т. кип., °C рка
2-Аминопиридин (а) 58,1-58,4 204 7,2
3-Аминопиридин (р) 64.5 250—252 6,6
4-Аминопиридин (}) 157—158 — 9,1
М о н о а м и н о п и р и д и н ы легко растворимы
в воде и многих органич. растворителях, трудно рас-
творимы в лигроине. 2- и 4-А. иногда рассматривают
как таутомерные системы:
NHg NH
СК = Си -6 = 6
N 4 Nil UU NH
Однако равновесие в обоих случаях практически
нацело смещено в сторону аминной формы; для а-изо-
мера отношение амино- к иминоформе превышает
103 : 1. 2-А. представляет собой циклич. амидин.
В соответствии с этим устойчивые соли, в к-рых про-
тон присоединен к атому азота, входящему в цикл,
А. образует лишь с одним эквивалентом кислоты.
4-А. химически весьма близок к 2-изомеру, но 3-А.
резко отличается от этих аминов, приближаясь по
химич. природе к ароматич. аминам. Он может реаги-
ровать как двухкислотное основание, несмотря на
то, что является более слабым основанием, чем два
другие изомера (см. табл.). 3-А. дает нормальные соли
диазония, между тем как при действии азотистой к-ты
в разб. соляной к-те на 2- и 4-изомеры выделяется
азот и получаются пиридоны:
NH2
Однако образуемые этими
устойчивы и в спиртовом
N-R
/
основаниями диазотаты
р-ре дают азокрасители.
Действие алкилгало-
генидов направляется
преим. на более основ-
пойдвоесвязанный атом
азота 2-аминопириди-
на.как в случае амиди-
нов. Благодаря этому получаются гл. обр. N-алкил-а-
пиридонимины I и в меньшем количестве а-алкил-
аминопиридины II. По-
следние являются пре-
обладающими продук-
тами реакции алкилга-
логенидов с натрий- или
литий - 2-аминопириди-
пом. С нитрующей сме-
сью 2-А. дает нитрими-
попиридин строения
Ша или Шб, который
под действием H2SO4
изомеризуется в смесь 5-нитро- или З-нитро-2-ами-
нопиридинов, соответственно IV (80%) иУ (10%). С
фенацилбромидом 2-А. образует
карбазиконденсированное соеди- || |
пение 2-фенилпиримидазол (VI). с6Н5
Галогенирование А. проходит в *5 у»
соответствии с правилами ориен- СН
тации.
2-А. получают по методу Чичибабина нагреванием
пиридина с натрийамидом, напр. в ксилоле; при ами-
дировании в среде диметил а нил ин а выход достигает
75—85%. 3-А. получают нагреванием 3-бромпири-
дина с NH4OH в присутствии сульфата меди или
по Гофману из никотинамида; 4-А. — сплавлением
4-феноксипиридина с хлористым аммонием или из
Г4-окиси-4-нитропиридина (см., напр., реакции 1—4).
CuSO4
(2)
2-А. применялся для синтеза сульфидина и антиги-
стаминного препарата — пирибензамипа.
Ди аминопиридины. Известны все шесть
изомеров. Практически интерес представляет 2,6-изо-
мер (т. пл. 121,5°), образующий с хлористым фе-
нилдиазонием 2,6-диамияо-З-фенилазопиридин (VII),
под названием «пиридиум»; он известен как антисеп-
тик мочевых путей. Продукт
сочетания 2,6-диаминопириди- UU— N=NCcHr
на с диазотированным 5-амино- NJ J—NH °
2-бутоксипиридином, т. наз. 2 2
неотропин, обладает бактери-
цидными свойствами. Изтри-
амипопири ди нов следует упомянуть 2,3,6-
изомер; основание неустойчиво на воздухе, сохра-
няется в виде солей; применяется в качестве про-
явителя в фотографии.
Лит.: М о ш е р Г., в кн.: Гетероциклические соединения,
ред. Р. Эльдерфплда. т. 1, пер. с англ., М., 1953, с. 42 4—433.
Й. Л. Гольдфарб.
АМИНОПЛАСТЫ — пластмассы, приготовляемые
на основе карбамидных смол (карбамидные пластики).
Карбамидные смолы, получаемые поликонденсацией
амидов многоосповных кислот с альдегидами, при-
надлежат к термореактивным полимерам. Наиболее
широко применяются смолы, получаемые конденса-
цией формальдегида с мочевиной и ее производными —
тиомочевиной, меламином, дициандиамидом и др.
Различают несколько видов А.: п р е с с м а т е-
риалы (пресспорошки — порошкообразные про-
дукты, приготовляемые из карбамидных смол с цел-
люлозным наполнением), изделия из к-рых изготов-
ляют горячим прессованием под давлением; с л о-
истые А. с прослойкой из бумаги, текстиля,
древесного шпона и др.; пористые А. Делались
попытки получать литые А. из ненаполненных карб-
амидных смол, однако, несмотря на такие их ценные
свойства, как прозрачность и твердость, литые А. не
189
АМИНОПЛАСТЫ
190
нашли широкого применения из-за склонности к обра-
зованию трещин.
Первоначальной стадией произ-ва любого вида А.
является получение карбамидных смол. Получение
смол мочевино-формальдегидных ввиду исключительной
чувствительности процесса поликонденсации к изме-
нению кислотности среды целесообразно проводить
при определенных значениях pH на каждой стадии
процесса. Это вызвано также и тем, что технич. фор-
мальдегид вследствие присутствия в нем нек-рого
количества муравьиной к-ты имеет кислую реакцию.
Регулирование pH достигается изменением соотноше-
ния мочевины и формальдегида на разных стадиях
процесса, введением тиомочевины, дициандиамида,
фенола и др. Для протекания реакции поликонденса-
ции с нужной скоростью необходима слабокислая
среда (pH 4,5—5,0), но для устранения в дальней-
шем чрезмерного нарастания вязкости Следует сни-
зить степень кислотности и поддерживать ее в пре-
делах pH 5,5—6,5. Для этого в реакционную массу
добавляют щелочные соли слабых кислот, напр. аце-
тат натрия, фосфат натрия и др., обладающие буфер-
ным действием и регулирующие pH среды. В стадии
сушки смолы во избежание желатинизации необхо-
дима нейтральная среда (pH 6,5—7,5), что дости-
гается введением соды. На последней стадии поли-
конденсации для отверждения смолы рекомендуется
снова увеличить кислотность до pH 3,5—4,5, что
достигается внесением твердых органич. кислот или
продуктов их конденсации с многоатомными спир-
тами, кислых солей, жирных кислот и т. д. Регуля-
тором кислотности реакционной массы может слу-
жить уротропин, к-рый регулирует также и скорость
реакции смолообразования, ускоряемой водородными
ионами. Используя регулирующее действие уротро-
пина, можно проводить реакцию при непродолжи-
тельном нагревании или на холоду.
Для ускорения отверждения смолы при горячем
прессовании необходимо в состав реакционной смеси
вводить ускорители. Последними обычно являются
твердые органич. кислоты (щавелевая и др.), к-рые
должны иметь темп-ру плавления, лежащую в пре-
делах темп-p горячего прессования пластика. Взаимо-
действие жидкой смолы с расплавленной к-той ведет
к резкому повышению кислотности и, следовательно,
к увеличению скорости отверждения и скорости прес-
сования. Слишком большое количество ускорителей
может снизить текучесть прессматериала. При изго-
товлении мочевино-формальдегидных смол тиомоче-
вина используется не только в смеси с мочевиной,
но и самостоятельно. Введение тиомочевины улуч-
шает водостойкость смолы и пресскомпозиций, уве-
личивает скорость отверждения, улучшает условия
обезвоживания смолы, но усиливает коррозию аппа-
ратуры.
Меламинная конденсация, в отличие от мочевинной,
характеризуется большей гидрофильностью перво-
начальных продуктов, что обусловлено более высокой
концентрацией полярных метилольных групп в ме-
ламине. Большая гидрофильность смолообразных
продуктов конденсации устраняет опасность прежде-
временной желатинизации и делает процесс конден-
сации и сушки менее чувствительным к изменению
темп-ры и pH среды. Конденсацию проводят обычно
при 70—100° при pH 5,5—6,5. Подробнее о меха-
низме конденсации, составе и строении промежуточ-
ных и конечных продуктов см. в ст. Смолы мочевино-
формалъдегидные, Смолы меламино-формалъдегидные.
Приготовление пресспорошков на основе мочевино-
формальдегидных смол может быть проведено по двум
методам. Первый метод заключается в предваритель-
ном получении водного конденсата смолы (т. наз.
сиропа) с последующей пропиткой этим конденсатом
целлюлозного наполнителя. Этот метод применяется
редко, но сохранил свое значение для пропитки ткани,
бумаги и др. наполнителей при получении слоистых А.
Второй метод, более совершенный и широко ис-
пользуемый в пром-сти, основан на пропитке напол-
нителя исходными смолообразующими реагентами.
Произ-во пресспорошков по этому методу начинается
с приготовления мочевино-формальдегидного р-ра,
состоящего из мочевины, формальдегида, уротропина
и щавелевой к-ты. Таким раствором, в к-ром преобла-
дают метилольные производные мочевины, пропиты-
вают волокнистый наполнитель. В качестве наполни-
теля для пресспорошков применяют измельченную
сульфитную целлюлозу, т. к. применение других
наполнителей не позволяет получать полупрозрач-
ные и светлоокрашенные изделия. В смесь добавляют
также стеарат цинка или кальция (в качестве смазки,
способствующей повышению водостойкости изделий),
органич. красители и минеральные пигменты (литопон
и др.). Смесь готовят при обычной темп-ре. После
смешения мочевино-формальдегидного р-ра с напол-
нителем масса в течение определенного времени
«созревает» (обычно при комнатной темп-ре), при этом
протекают процессы связывания уротропина и сво-
бодного формальдегида, увеличение кислотности и же-
латинизации массы (образование смолообразных про-
дуктов) на волокне. По окончании «созревания» массу
сушат, размалывают и просеивают. Описанный способ
имеет и второй вариант. Смешивание мочевино-фор-
мальдегидного р-ра с целлюлозным наполнителем
проводят при 50—60°, при этом происходит не только
пропитка и механич. перемешивание, по и дальней-
шая конденсация и смолообразование на волокне,
т. е. частично те же процессы, к-рые по первому ва-
рианту протекают при «созревании» массы. Приго-
товленная т. обр. масса может непосредственно посту-
пать на сушку. Это дает возможность иводить непре-
рывно-поточные методы. Прессование А. производят
при 140—150° и уд. давлении 250—350 кг/см2:
т. к. А. чувствительны к колебаниям темп-ры и дав-
ления, неравномерности обогрева, то прессование
проводят на прессах с автоматич. регулировкой дав-
ления, времени и темп-ры.
Мочевино-формальдегидные порошки делятся на
непрозрачные и прозрачные. Непрозрачные порошки
содержат минеральные пигменты (сернокислый ба-
рий, литопон и др.). По комплексу физико-механич.
свойств, структуре промежуточных и конечных про-
дуктов конденсации, технологии переработки и об-
ластям применения А. имеют много общего с фено-
пластами, но отличаются от них бесцветностью, свето-
стойкостью, отсутствием запаха.
А. стойки к действию спирта, бензина, ацетона,
хлороформа и др., по водостойкости они уступают
фенопластам. При кипячении в воде А. поглощают
ок. 4% воды за 30 мин; при этом выделяется фор-
мальдегид. При длительном нагревании А. стойки до
80°, при кратковременном — до 110—120°. Более
высокие темп-ры вызывают уменьшение прочности.
Технология, процесс произ-ва слоистых мочевино-
формальдегидных А. складывается из получения свя-
зующего раствора и пропитки слоистого наполнителя
(бумаги, ткани, древесного шпона, стеклоткани)
в пропиточно-сушильных машинах. В качестве свя-
зующего применяют водный конденсат смол, приго-
товляемый нагреванием реагирующих компонентов
в присутствии буферных солей до образования про-
зрачного и немутнеющего раствора. Для произ-ва
бумажно-слоистых пластиков применяют специаль-
ные сорта наполненных окрашенных бумаг, изготов-
ленных из сульфитной целлюлозы. Темп-pa сушки
110—120°. Прессование слоистых А. производят на
многоэтажных прессах при 140—145° и при уд. дав-
191
АМИНОПЛАСТЫ — АМИНОПУРИНЫ
192 I
лении 80—150 кг/см2 в случае бумажных и тканевых
материалов или 50—100 кг/см2 материалов на основе
древесного шпона.
Слоистые А., имея ту же механич. прочность, что
и соответствующие фенопласты, значительно усту-
пают им по водостойкости, химич. стойкости и тепло-
стойкости, их основные преимущества — светостой-
кость, окрашиваемость в яркие тона, отсутствие за-
паха, даже при повышенных темп-рах.
На основе мочевино-формальдегидных смол полу-
чается также пористый материал «мипора», отличаю-
щийся высокими тепло- и звукоизоляционными свой-
ствами и малым объемным весом (10—20 кг/л3). Для
получения мипоры применяют водную эмульсию
мочевино-формальдегидной смолы, к-рая смешивается
с пенообразователем (раствором специально очищен-
ного сульфонафтецового контакта); образовавшаяся
пена отверждается затем в формах. Сушка пены
в формах производится в сушильных камерах в те-
чение 4—5 суток при 20—40°. Отверждение мипоры
производят непрерывно на конвейере. Мипора имеет
преимущество по термостойкости перед поропластом
из стирола и широко применяется в качестве тепло-
изоляционного материала в различных конструк-
циях.
Наряду с мочевино-формальдегидными прессмате-
риалами производятся также модифицированные мо-
чевино-формальдегидные смолы для изготовления
клеев, лаков и пропитки тканей (с целью придания
им несминаемости). Напр., смола МФ-17 представляет
собой продукт конденсации мочевины с формальде-
гидом, содержащий пластификатор (диэтиленгликоль)
и наполнитель (древесная мука). Для получения клея
марки К-17 к смоле МФ-17 добавляют отвердитель
(кислые соли или кислоты) непосредственно перед
употреблением клея. Клей К-17 используется для
склеивания древесины и как связующее вещество
в различных композициях. Мочевино-формальдегид-
ные смолы, получаемые в водных р-рах, не раство-
ряются в спиртах. Для получения смол, растворимых
в спиртах, производят этерификацию начальных
продуктов конденсации (смеси моно- и диметилмоче-
вины) бутиловым, гексиловым, октиловым спиртами,
глицерином или конденсацию мочевины с формальде-
гидом ведут в присутствии спирта.
Изготовление меламино-формальдегидных пресспо-
рошков складывается из след, операций: 1) приго-
товления водного конденсата смолы; 2) смешения
конденсата с наполнителем — целлюлозой или дре-
весной мукой (используются также и минеральные
наполнители), ускорителем и красителем; 3) сушка
массы; 4) вальцевание с целью улучшения однород-
ности массы; 5) размол, просев. Наряду с чистыми
меламино-формальдегидными прессматериалами про-
изводят ирессматериалы на основе совместной моче-
вино-меламиновой конденсации с формальдегидом.
Конденсация производится в нейтральной среде при
75—95°.
Меламиновые пресскомпозиции прессуются при
150—160°. Их свойства во многом определяются ха-
рактером минерального наполнителя. Напр., приме-
нение слюды, дисперсного кремнезема, асбеста позво-
ляет значительно повысить теплостойкость, стойкость
по отношению к действию электрич. зарядов и др.
Меламино-формальдегидные пресспорошки по ряду
свойств значительно превосходят мочевино-формаль-
дегидные прессматериалы, а также феноло-формаль-
дегидные. Они обладают большей стойкостью к дейст-
вию кипящей воды, большей механич. прочностью,
лучшими диэлектрич. и химич. свойствами. Мелами-
новые пресскомпозиции бесцветны, светостойки, не-
токсичны и хорошо окрашиваются. В таблице сопо-
ставлены свойства мочевино-формальдегидных и мела-
мино-формальдегидных прессматериалов на основе
древесной целлюлозы.
Свойства Смолы
мочевино-фор- мальдегидные меламино-фор- мальдегидные
Возможность окрашивания . . Темп-ра прессования, °C ... . Время прессования, сек/мм . . Водопоглощение при 20° за 24 часа, % Теплостойкость по Мартенсу, °C Предел прочности на разрыв, кг/см- . Предел прочности на изгиб, кг/см2 . . Уд. ударная вязкость, кг-см/см'2 Диэлектрич. проницаемость при 106 Уд. поверхностное электрич. сопротивление, ом Уд. объемное электрич. сопро- тивление, ОМ'СМ ........ Пробивное электрич. напряже- ние, кв/мм ........... Неограни- ченная 145—150 60-90 0,75-2,0 120 400—600 600—700 6-10 6,5—7,5 109-Ю10 5-109—5-10Ю 10—15 Неограни- ченная 150—170 45—60 0,3-0,6 135-160 500—600 800—900 6—10 6,5—7,5 Ю10—юн 1011—5-1012 12—18
Сложные меламино-формальдегидные пластики по-
лучают путем пропитки бумаги, ткани (в т. ч.
и стеклоткани), шпона водными конденсатами смол
на специальных пропиточно-сушильных агрегатах.
Путем этерификации бутанолом получают модифи-
цированные (лаковые) меламиновые смолы, раствори-
мые в спирте. Меламино-формальдегидные компози-
ции применяют для получения пропитывающих и клея-
щих составов, а также поропластов. Преимуществом
последних является повышенная механич. прочность,
стабильность размеров и стойкость к горячей воде.
Применение А. Пресспорошки на основе
мочевино-формальдегидных смол применяются гл.
обр. для приготовления изделий широкого потребле-
ния и технич. деталей, к к-рым не предъявляют тре-
бований высокой водостойкости и хороших диэлектрич.
свойств (корпуса и ручки телефонов, штепсели, выклю-
чатели). Из них изготовляют также дверные ручки,
облицовочные плитки, плафоны, абажуры и т. д.
Слоистые А. применяют в качестве строительно-обли-
цовочного материала; из них изготовляется мебель
и осветительная арматура, изоляция. Пористые А.
используют в качестве теплоизоляционного промежу-
точного слоя в различных конструкциях. Мелами-
новые смолы часто используются для получения водо-
стойких бумаг и для повышения их прочности во
влажном состоянии. А. на основе меламино-формаль-
дегидных смол применяются для изготовления посуды,
стойкой к действию горячих жидкостей.
Лит.: Барг Э. И., Технология синтетических пластиче-
ских масс, Л., 1954; Петров Г. С., Л е в и н А. Н., Термо-
реактивные смолы и пластические массы, М., 1959; BungrF.,
Einfuhrungin die Chemie und Technologic der Kunststoffe, B.,
1959; Ullman n,3Autl., Bd 3, Milnch. —B., 1953, S. 475—49S.
О. В. Смирнова.
АМИНОПУРИНЫ — группа веществ основного ха-
рактера, представляющих собой производные пу-
рина — бициклич. основания, лежащего в основе
большой группы физиологически важных веществ.
А. образуются наряду с углеводами (D-рибозой или
дезоксирибозой) и фосфорной к-той при гидролизе
нуклеопротеидов — дезоксирибонуклеиновых кислот и
рибонуклеиновых, кислот. Из А. наибольшее значение
n=c-nh2
НС C~N^
II II /СН
N-C-NH
N=C-ОН HN-C=O
HaN9 S~N>CH = H*N<r ?~n>ch
N-C-NH N-C-NH
енольная форма нетонная форма
И
193
n-АМИНОСАЛИЦИЛОВАЯ КИСЛОТА - АМИНОФЕНОЛЫ
194
имеют 6-аминопурин, или аденин (I), и 2-амино-6-
оксипурин, или гуанин (II). А. входят в состав ну-
клеотидов, нуклеозидов, нуклеиновых кислот.
n-АМИНОСАЛИЦИЛОВАЯ КИСЛОТА см.
ПАСК.
5-АМИНОСАЛИЦИЛОВАЯ КИСЛОТА (5-амино-
2-оксибензойная кислота) (NH3)(OH)C6H3COOH, мол.
в. 153,14 — игольчатые кристаллы (из воды), т. пл.
283° (с разл.), нерастворима в спирте и холодной воде.
Получают А. к. сочетанием диазотированного анилина
с салициловой к-той с последующим восстановлением
образующегося - красителя (5-бензолазосалициловой
к-ты) р-ром дисульфида или гидросульфида натрия.
Кроме того, А. к. может быть получена также из 3-нит-
робензойной к-ты восстановлением цинком в конц.
H2SO4 или электрохимия, восстановлением-в 30%-ной
серной к-те. А. к. широко применяется как промежу-
точный продукт в произ-ве протравных азокрасителей
(напр., кислотного хром черного).
Н. С. Вульфсон.
АМИНОСАХАРА — см. Гликозамины,
АМИНОСПИРТЫ (аминоалкоголи, оксиамины) —
соединения, содержащие окси- и аминогруппу в раз-
ных положениях друг относительно друга. К А. от-
носят соединения, у к-рых окси- и аминогруппы свя-
заны с разными атомами углерода; различают пер-
вичные, вторичные и третичные аминоспирты. А. типа
RCH(OH)NH3, первоначально образующиеся при при-
соединении аммиака к альдегидам, нестойки, легко
претерпевают дальнейшие превращения. Хотя А. есте-
ственно сочетают свойства аминов и спиртов, в химич.
поведении их, в результате взаимного влияния функ-
циональных групп, наблюдаются нек-рые особенности:
так, А. труднее дегидрируются и этерифицируются,
чем обычные спирты.
А. можно получить: 1) Присоединением аммиака
i ।
или аминов к окисям олефинов, напр. СН2СН2О -|-
-|- NH3 —> H2NCH2CH2OH; образующийся этанол-
амин может присоединить еще одну или две молекулы
окиси этилена, что ведет к ди- и триэтаноламину
(HOGH3CH2)3NH и (HOGH3CH3)3N. 0-Окситриэтиламин
(С2 H5)2NCH2CH2OH, обр азующийся пр и присоединении
окиси этилена к диэтаноламину, является полупро-
дуктом произ-ва новокаина. 2) Замещением галогена
в галогенгидринах действием аммиака С1СН2СН2ОН-1-
-|- NH3 —>- NH2CH2CH2HO-|- НС1. Возможно, что при
этом сначала происходит образование окиси. 3) Вос-
становлением нитропроизводных спиртов, например
Р-нитроэтанола, образующегося при взаимодействии
нитрометана с формальдегидом в щелочной среде:
NO3CH3CH3OH-pH3->-NH3CH3CH2OH-|-2H3O. 4) Вос-
становлением эфиров аминокислот натрием и спир-
том или же алюмогидридом лития: NH2CH2GOOC2H54-
+ 2Н2 -> NH3GH3GH3OH -j- С2ЩОН.
Из А. большое практич. значение имеют этанол-
амины. Третичные А., как и третичные амины, спо-
собны присоединять алкилгалогениды с образованием
соответств. четвертичных аммониевых солей. Из со-
+
единений этого типа холин [HOGH2GH2N(GH3)3]OH_
играет крупную роль в процессе ассимиляции жиров
в организме. В тканях мозга холин превращается
в ацетилхолин, обладающий очень сильным физио-
логии. действием. Многие алкалоиды, напр. эфедрин,
кокаин, являются производными А. К числу А.
принадлежит и адреналин, один из важнейших гор-
монов. Я. Ф. Комиссаров.
АМИНОУКСУСНЫЙ АЛЬДЕГИД H3NGH3GHO —
известен лишь в виде хлоргидрата, расплывающегося
на воздухе; легко растворяется в абс. спирте; хлор-
гидрат А. а. в присутствии окислителей превращается
4 к. х, э, т± 1
в пиперазин:
СНО HC1-H2N
I + \
СН2 СН2 о _
\ I
nh2-hci осн
л
сн ^сн
II I
сн ^сн
'''Ж
Получается А. а. при гидролизе аминоацеталя соля-
ной к-той или озонированием хлоргидрата аллиламина.
H2NCH,CH(OC2H5)2 —1 НС1 • H2NCH2CHO
НС1 • HaNCH3CH=CHs HC1 • H2NCH2CHO
E. M. Рохлин,
И-АМИНОУНДЕКАНОВАЯ КИСЛОТА, мол. в.
201,31, H2N(GH2)10COOH — бесцветные кристал-
лы; т. пл. 185—186°; растворима в горячей воде и
в горячем спирте, нерастворима в др. обычно при-
меняемых органич. растворителях. При нагревании
легко отщепляет воду, образуя полиамидную смолу,
к-рая используется для получения синтетич. волокна
типа «Рилсан» (см. Полиамидные волокна). В про-
мышленном масштабе А. к. может быть получена из
а, а, а, со-тетрахлорундекана; в этом случае проводят
гидролиз тетрахлорундекана в присутствии 94—96%-
ной H2SO4 при 60—80° и последующий аммонолиз
полученной свободной 11-хлорундекановой к-ты 40%-
ным р-ром NH4OH в течение 48 час. при 25°. Процесс
протекает по ур-нию: С1(СН2)10СС13—►С1(СН2)10СООН—>
H3N(GH3)loCOOH.
а, а, а, со-Тетрахлорундекан получают по реакции
теломеризации этилена с четыреххлористым угле-
родом. В пром-сти А. к. получают также из касто-
рового масла. Касторовое масло подвергают пиро-
лизу. На образовавшуюся при этом ундециленовую
кислоту СН3=СН(СН2)8СООН действуют НВт. Полу-
ченную в результате этого бромундекановую кислоту
Вг(СН2)10СООН подвергают аммонолизу. Конечным
продуктом является 11-А. к.
о-АМИНОФЕНИЛАРСОНОВАЯ КИСЛОТА, мол. в.
217,04, H3N—С6Н5—А=О(ОН)3 — бесцветные мелкие
иглы; т. пл. 153—154°; растворима в воде, метило-
вом и этиловом спиртах и ледяной уксусной к-те,
в разбавленных к-тах и щелочах; мало растворима
в эфире; водный р-р имеет слабокислую реакцию на
конго. Получают А. к. диазотированием о-нитроани-
лина, обработкой щелочным р-ром ацсепита натрия
и последующим восстановлением нитрогруппы с по-
мощью FeSO4 в щелочном р-ре:
O2NC6H4NH„ O2NCeH4N.,Cl
Нп* ин
— O2NC6H4NH2-AsO (ОН)2 — H2N-C6H4-AsO(OH)2
Соль диазония А. к. образует с щелочным р-ром
Р-нафтола азосоединение красного цвета; в отличие от
м- и n-аминофениларсоновых к-т, орто-изомер обра-
зует желтое окрашивание (или осадок) в присутствии
салицилового альдегида с р-рами солей Ti и Th, не
содержащими избытка к-ты. Применяют для фото-
метрия. определения Ge, Sc; для цветных реакций
па Th, Ti, V; для синтетических целей.
Д. И. Кузнецов.
АМИНОФЕНОЛЫ (оксианилины) NE^Cg^OH —
кристаллич. продукты. В соответствии с относитель-
ным положением заместителей известны три изомера:
о-А., т. пл. 174°, м-А.., т. пл. 123°, n-А., т. пл. 186°.
А. — амфотерные соединения; с кислотами и щело-
чами образуют соли. Взаимное влияние обоих нук-
леофильных заместителей сказывается в том, что А.,
подобно многоатомным фенолам, особенно в щелочной
среде, легко окисляются. От о-А. можно легко перейти
195
«-АМИНОЭН АНТОВАЯ КИСЛОТА — АМИНЫ
196
к гетероциклическим соединениям — производным I низмах огромную роль играют А., содержащие кар-
1,3-оксазола, напр. к 2-метил-4,5-бензоксазолу-1,3: | боксильную группу (аминокислоты). По характеру
радикалов, связанных с атомом N, А. могут
------------------------------------N быть разделены на А. жирного, алициклич.,
|Гг.|_1г+(СНзСО)2О--------|1 О +СН3СООН +Н2О ароматич. и гетероциклич. рядов.
-^“ОН СН3 Как органич. производные NH3 все А. об-
ладают более или менее сильно выражен-
Общий способ получения А. заключается в восста-
новлении соответств. нитро- или нитрозофенолов,
лучше всего кипячением с цинковой пылью в водной
среде; n-А. в пром-сти получают восстановлением
нитробензола в условиях, при к-рых процесс оста-
навливается на стадии образования фенилгидроксил-
амина, легко перегруппировывающегося в п-А.:
C6H5NO2 -> CeH5NHOH -> n-HOCeH4NH2. Этот про-
цесс осуществляется действием цинковой пыли па
нитробензол в конц. H2SO4 или же электрохимия,
восстановлением нитробензола в конц. H2SO4. А.,
в особенности м- и n-А., применяются в произ-ве
сернистых и др. красителей. Соли n-А. и его N-ме-
тильного производного (метола) принадлежат к
числу широко распространенных в фотографии про-
явителей.
Лит.: Kiri, V. 1, N. Y., 1947; U 1 Inann, 3 Aufl.,
Bd 3, Milnchen — В., 1953. Я. Ф. Комиссаров.
(о-АМИНОЭНАНТОВАЯ КИСЛОТА (7-аминогепта-
новая кислота) NH2(GH2)6GOOH, молекулярный вес
14.5,20—бесцветные кристаллы; т. пл. 194—195°;
легко растворимы в воде, нерастворимы в спирте,
ацетоне и др. органич. растворителях. Хлоргидрат
А. к. плавится при 99—100° (перекристаллизация из
смеси спирта и эфира). При нагревании А. к. легко
отщепляет воду, и образуется полиамидная смола,
из к-рой можно получить синтетич. волокно «Энант»
(см. Полиамидные волокна). В пром-сти А. к. может
быть получена из а,а,а,со-тетрахлоргептана; в этом
случае проводят гидролиз тетрахлоргептана в при-
сутствии 94—96»/0-ной H2SO4 при 60—80° и после-
дующий аммонолиз полученной ш-хлорэнантовой к-ты
25»/0-ным р-ром NH4OH при 100° в течение 2 час.:
С1(СН2)вСС13 -> Cl(CH2)eCOOH -> NH2(CH2)eCOOH.
а,а,а,со-Тетрахлоргептан получают теломериза-
цией этилена с четыреххлористым углеродом: СС14 +
+ ЗСН2=СН2 —> СС13(СН2)6СН2С1. По другому пром,
способу А. к. полунают из фурфурола:
Н2С—СН2
I I —
н2с сн -сн2он
II
хсн2
нвс сн
I II
Н2С СН
чо/
III
—> С1СН2 (СН3)з СН2С1 —> CN (СН2)5 CN —>
V VI
—> NHo (CHaijCN —> NH2 (СН2)в СООН
VII VIII
фурфурол (I) —>тетрагидрофуриловый спирт (II) —>
дигидропиран (III) —> тетрагидропиран (IV) —>1,5
дихлорпентан (V) —> динитрил пимелиновой к-ты
(VI) —>нитрил ш-аминоэнантовой к-ты (VII) —> А. к.
(VIII).
АМИНЫ — продукты замещения одного, двух или
трех атомов водорода в NH3 органич. радикалами.
Соединения типа RNH2 наз. первичными А.; заме-
щению 2 атомов Н в NH3 соответствуют вторичные,
а соединениям типа R3N — третичные А. По числу
аминогрупп в молекуле различают моно-, ди-, три-
амины и т. д. Простейшие А. найдены среди продуктов
нормальной жизнедеятельности нек-рых растений.
Широко распространены в природе сложно построен-
ные органич. основания (см. Алкалоиды), нередко
с одним или несколькими атомами N в цикле; в орга-
ными основными свойствами и, подобно NH3, спо-
собны к образованию солей с кислотами. Третич-
ные амины могут присоединить одну молекулу
алкилгалогенида с образованием четвертичных ам-
н---------------------
мониевых солей R4NX, в которых азот связан 4
ковалентными связями с радикалами и ионной
связью с галогеном X. При действии CeH5CH2Na
на . (CH3)4NC1 образуется соединение состава
(GH3)4NGH2C6H5, несомненно обладающее солеобраз-
ным характером; его растворы проводят электриче-
ский ток, причем бензил ведет себя как анион. В
этом случае, видимо, образуется четвертичная соль
(GH3)4N[GH2C6H5]_, a (CH3)4N играет такую же роль,
как Na в C6H5GH2Na. А. жирного и ароматич. рядов
неск. различаются между собой по свойствам и спо-
собам получения. Так, один из основных методов
синтеза А, жирного ряда заключается во взаимодей-
ствии алкилгалогенидов (или других алкилирующих
агентов, напр. эфиров серной или арилсульфокислоты)
с NH3 : RX -|- NH3 —> RNH2 НХ. Получившаяся
соль А. частично разлагается аммиаком с образова-
нием свободного первичного А., к-рый может далее
реагировать с другой молекулой RX и т. д. Поэтому
при алкилировании аммиака обычно образуется смесь
А. и конечного продукта алкилирования — четвер-
тичной аммониевой соли. Ввиду малой подвижности
атомов галогенов в арилгалогенидах прямое арилиро-
вание аммиака протекает с трудом. А. ароматич. ряда
получают почти исключительно по методу, впервые
примененному Н. Н. Зининым, — восстановлением
соответственных нитросоединений, напр. C6H5NO2-|-
-|- 6Н —> CeH5NH2 -|- 2Н2О. Иногда первичные А.
разного типа получают также восстановлением нитро-
зосоединений, нитрилов, оксимов, азидов, альдими-
нов, гидразонов.
Современный промышленный метод получения пер-
вичных, вторичных и третичных А. жирного ряда
заключается во взаимодействии
^СНг спиртов с NH3 над окисью Th
Н С сн или А1. Особенно легко протекает
| | 2 образование первичных ароматич.
Н2С СН2 А. при реакции нек-рых фенолов,
Nq/ в особенности нафтолов с NH3 (см.
IV Бухерера реакция). Один из удоб-
ных методов получения чистых
первичных А. состоит в алкилировании фталимида
калия с последующим омылением продукта реак-
ции (см. Габриеля реакция): CeH4(CO)2NK-|-RX —>
-> CeH4(CO)2NR -> CeH4(COOH)2 4- RNH2. Первич-
ные А. могут быть также получены из производных
к-т с перегруппировкой промежуточных продуктов
реакции. Так, А. образуются при обработке амидов
к-т бромом и щелочью (см. Гофмана реакция) и дей-
ствием HNO2 на гидразоны к-т (см. Курциуса реакция).
Хороший способ получения чистых вторичных А. со-
стоит в нитрозировании диалкиланилинов с после-
дующим омылением продукта реакции: CsH6NR2 4-
4- HNO2 -> n-ONC6H4NR2 n-ONCeH4OH -|- R2NH.
Константы диссоциации нек-рых аминов см. в табл.
Основность А. объясняется способностью атома
азота с неподеленной электронной парой к присоеди-
нению протона с образованием иона замещенного
аммония.
Как видно из таблицы, замена атомов Н в NH3,
в особенности первых двух атомов Н, на алкильные
197
АМИНЫ — АММИАК
198
радикалы ведет к заметному усилению основности.
Первичные ароматич. А. (см. анилин в табл.) являют-
ся значительно более слабыми основаниями, чем
А. жирного ряда, что находит объяснение в умень-
шении электронной плотности у атома азота вслед-
ствие включения его свободной элек-
тронной пары в систему сопряжения _У~f~y
с л-электронами бензольного ядра. (Z У~НН2
При переходе от анилина к N-алкиль-
ным производным константа диссо-
циации несколько возрастает, у дифениламина и
трифениламина она еще более ослаблена, чем у ани-
лина; трифениламин вовсе лишен способности к
солеобразованию. Аминогруппа типично нуклеофиль-
на, и поэтому она должна была бы ориентировать
дальнейшее электрофильное замещение в аромати-
ческом ядре только в орто-, пара-положения; однако
в сильно кислых средах может наблюдаться изменение
ориентации (мета-замещение) вследствие солеобра-
зования (ониевая группа электрофильна).
Соединение К рХ
Аммиак NH3 1,8-10-5 4,75
Метиламин CH3NH2 4,4-10-1 3,36
Диметиламин (CH3)2NH 5,1 • 10 3,29
Триметиламин (CH3)3N 5,5-10_ 4 4,26
Анилин СвН5МЙ2 3,8-IO'1» 9,42
Диметиланилин C6H5N(CH3)2 . . . 1,2-10-а 8,92
Дифениламин (CeH5)0NU 7,6-10-14 13,12
Пирролидин C4H8N 1,340-3 2,89
Пиррол C4H5N l-10-ie 16
Пиперидин C5H,N 1,6-Ю-з 2,80
Пиридин C5H5N 1,7-10-» 8,77
Для А. различного типа характерно их поведение
при действии HNO3. При этом в случае первичных А.
жирного ряда выделяется азот и образуется соответств.
спирт; из вторичных алифатич. А. получаются нитроз-
амины R2NNO; жирные третичные А. при обычной
темп-ре не изменяются при действии HNO2, а при
повышенной темп-ре происходит дезалкилирование.
Эта реакция применяется для распознавания и раз-
деления жирных А. Большое значение имеет реакция
первичных ароматич. А. с HNO2; при этом образуются
диааониевые структуры ArN2X (X—анион к-ты), чрез-
вычайно реакционноспособные и поэтому применяю-
щиеся в пром-сти для введения многих заместителей
вместо NH2 в ароматич. ядро, для получения азокра-
сителей и др. Вторичные жирно-ароматич. А. дают
с HNO2 N-нитрозамины, третичные жирно-ароматич.
А. превращаются при этом в пара-нитрозопроизводные.
Реакция первичных А. алициклич. ряда с HNO2 ведет
к соответств. спиртам; однако реакция часто сопро-
вождается сужением или расширением цикла (см.
Демьянова перегруппировка). Так из циклогептил-
амина образуется циклогептанол и циклогексилкар-
бинол.
Первичные и вторичные А. различного типа легко
реагируют с хлорангидридами или ангидридами
к-т с образованием ацильных производных, напр.:
C8H5NH2 + (СН3СО)2О -► C8H5NHCOCH3 + СЙзСООН
(C.,H5)3NH + С8Н5СОС1 -► (C2H5)2NCOC8H5 + НС1
При действии хлороформа в присутствии едкой щелочи
на первичные А. образуются изонитрилы: RNH2
-|- СНС13 —>- RCN ЗНС1, к-рые обладают очень силь-
ным неприятным запахом; поэтому указанная реак-
ция пригодна для обнаружения первичных А. Для А.
известны и реакции с расщеплением С—N-связи.
Напр., третичные А. при действии бромциана дают
нестойкий продукт присоединения, при нагревании
к-рого отщепляется RBr:
R3N + BrCN —► [R3NCN]Br—> R2NCN RBr
Гетероциклич. соединения с атомом азота в цикле
можно разделить на 2 группы: насыщенные соединения
характера вторичных А., к-рые по свойствам напоми-
нают вторичные А. жирного ряда, и ненасыщенные
системы, где возможно сопряжение л-связей с непо-
деленными электронными парами атома N. Так,
пирролидин и пиперидин (см. табл.) представляют
собой значительно более сильные основания, чем
вторичные А. жирного ряда. Пиррол же является
чрезвычайно слабым основанием и обладает не резко
выраженным ароматич. характером. У пиридина
наблюдаются значительно более сильные ароматич.
свойства и основность примерно такая, какудиметил-
анилина.
А. представляют собой практически очень важный
класс соединений; они являются полупродуктами в
производстве азокрасителей и др. красителей, многих
лекарств, веществ (напр., сульфаниламидных препара-
тов, ПАСКа), высокомолекулярных соединений (амино-
пластов, винилкарбазола) и др.
Лит.: Kirk, v. 1, N. Y., 1947, р. 702 — 717.
Я. Ф. Комиссаров.
АММИАК NH3 — простейшее соединение азота
с водородом; бесцветный газ с удушливым резким за-
пахом и едким вкусом. В природе А. образуется при
разложении азотсодержащих органич. веществ.
В образовании химич. связей в молекуле А. при-
нимают участие три неспаренных электрона атома
азота и электроны трех атомов водорода. Два элек-
трона атома азота остаются при этом неразделенными:
Н
:N. + зн • = :n;h
н
В образовании связей участвуют как 2р-электроны,
так и 2я-электроны атома азота, т. е. имеет место
гибридизация атомных орбит, близкая к тетраэдрич.
гибридизации в 4-валентном углероде. Атомы водорода
располагаются в трех вершинах тетраэдра, центр
к-рого занят атомом азота. Поэтому молекула NH3
имеет форму правильной пирамиды с атомом азота
в вершине (рис. 1). Угол между связями Н—N —Н
равен 108° т. е. весь-
ма близок к тетраэдри-
ческому. Расстояния
между, ядрами N — Н
1„015 А и Н—Н 1,64
А. В„ысота пирамиды
0,38 А.
В молекуле А. атомы
Н могут иметь два
равновесных положе-
ния в параллельных плоскостях (по обе стороны
от атома N). Соответствующий переход, т. н. инвер-
сия молекулы А., делается возможным благодаря
квантово-механическому туннельному эффекту, при-
чем время перехода порядка 10-11 сек. Поэтому ста-
ционарное состояние молекулы А. представляет
собой суперпозицию двух указанных состояний с рав-
ными весами. Инверсия всех трех атомов водорода
не препятствует, однако, ориентации молекулы в
электрич. поле при измерении дипольного момента.
Связи N — Н поляриы; пары электронов, связывающие
между собой эти атомы, несколько сдвинуты к атому
азота. Дипольный момент молекулы А., равный
1,43.0, создается в основном все же не полярностью
связей, а тем, что гибридная орбита, на к-рой нахо-
дится неразделенная пара электронов атома N, вытя-
нута в сторону от ядра к вершине тетраэдра, не заня-
той атомами водорода. Поляризуемость молекулы А.
равна 22,6 • 10-25 см3. Благодаря отсутствию неспа-
ренных электронов А. диамагнитен.
Физические и химические свойства. Неразделенная
пара электронов на гибридной орбите создает у моле-
4’
199
АММИАК
200
кулы А. способность к образованию водородной связи.
Это обстоятельство, а также значительная полярность
молекул А. вызывает весьма сильное взаимодействие
между ними, вследствие чего физич. свойства А. имеют
ряд аномалий по сравнению с однотипными соедине-
ниями (РН3, SbH3, AsH3): темп-ры кипения и плавле-
ния относительно высоки, теплота испарения велика.
Теплота образования А. из элементов ЛН»98 =
= —11,04 ккал /моль, с повышением темп-ры ее значе-
ние несколько увеличивается. Энтропия А. в стан-
дартном состоянии = 46,01 кал/град-моль. Тепло-
емкость А. при постоянном давлении в состоянии
идеального газа Ср = 8,523 кал/град-молъ (25°);
т. пл. —77,75° при 1 атм; т. кип. —33,35°; в трой-
ной точке при давлении пара А. 45,2 мм рт.ст.;
т. пл. —77,70°; ф.рит. 132,4°; ^крИТ 111,5 атм;
<*крит. 0,235 г[см3; теплота испарения 5,581 ккал/молъ
(—33,43°); теплота плавления 1,351 ккал/молъ
(— 77,76°); А. очень хорошо растворим в воде (при 20°
700 объемов А. в 1 объеме воды), несколько хуже —
в спирте, ацетоне, бензоле, хлороформе.
Жидкий А. — бесцветная жидкость с плотн. 0,6814
при т. кип., сильно преломляет свет. Твердый А. —
бесцветные кубич. формы кристаллы. В жидком А.
молекулы ассоциированы, ассоциация сохраняется
вплоть до критич. темп-ры.
Собственная электролитич. диссоциация жидкого
A.: 2NH3 « NHJ4- NH? совершенно ничтожна, про-
изведение [NHJ] [NHj] = 2 • Ю-зз(—50°). Жидкий А.
практически не проводит электрич. тока. Уд. электро-
проводность 3,0 • 10-6 ом 1 • см-1 (—37°); диэлектрич.
проницаемость 25,4 (—77°). В жидком А. растворяются
щелочные и щелочноземельные металлы, нек-рые
неметаллы (Р, S, J), многие соединения как неор-
ганические (напр., нитраты, нитриты, галогениды),
так и органические. Благодаря значительной вели-
чине диэлектрич. проницаемости жидкого А. у рас-
творенных в нем соединений с полярным или ионным
характером связи проявляется способность к электро-
литич. диссоциации. Образующиеся при этом ионы
сольватируются молекулами А. Сродство к протону
у А. больше, чем у воды, благодаря чему многие
растворенные в жидком А. соединения, имеющие
в своем составе протон, становятся способными отщеп’
лять его. В результате этого электролиты, к-рые
в водных р-рах резко отличаются по степени электро-
литич. диссоциации, в жидком А. диссоциируют
часто почти одинаково (напр., НС1 и HCN). В жидком
А. проявляют кислотные свойства даже углеводы,
амиды к-т, нек-рые углеводороды. Ряд обменных р-ций
в жидком А. протекает в другом, чем в воде, направле-
нии в связи с иным характером растворимости реа-
гирующих и образующихся в-в. Конц, р-ры щелочных
металлов в жидком А. имеют металлич. электропро-
водность, они окрашены в интенсивный синий цвет.
Водный р-р А., «нашатырный спирт», — бесцвет-
ная жидкость с запахом А. Зависимость раствори-
мости NH3 в воде оттсмп-ры показана в табл. 1, а
плотности водпых р-ров от концентрации — в табл. 2.
Таблица 1 Таблица 2
Темп-па, °C Растворимость NH3, е/100 г Н2О Содержание NH3, 2/100 г h2o Плотность, г/см-' при 15°
10 66,7 5 0,978
20 52,0 10 0,959
30 39,8 15 0,942
40 31.1 20 0,925
50 22,7 25 0,910
При суммарном давлении 30 0,897
паров NH3 и Н2О над водным 35 0,880
р-ром. равном 760 мм рт. ст. 40 0,865
Растворение А. в воде сопровождается химич. взаимо-
действием, в результате чего парциальное давление
А. над его водными р-рами весьма мало длн столь
больших концентраций растворенного газа. Взаимо-
действуя с водой, А. образует гидрат NH3 • Н2О.
Наряду с этим А. в водном р-ре частично ионизиро-
ван: NH3 -ф- Н2О = NHJ' -ф- ОН-, что обусловливает
щелочной характер р-ра. Константа равновесия этой
реакции К = —= 1,75 • 10"5 (при 18°).
В 1 М р-ре А. лишь 0,4»/о молекул А. взаимодействуют
с водой, образуя ионы NHp и ОН-, и такой р-р имеет
при 18° pH 11,77. В системе NH3 — Н2О установлено
образование соединений: NH3 • Н2О, т. пл. —79,0°;
NH3 • х/2 Н2О, т. пл. —78,2°; и эвтектик: лед-ф-
-ф- NH3 . Н2О (33,23 вес. % NH3), т. пл. —100,3°;
NH3 • Н2О -ф- NH3 • */г Н2О (55,11 вес. % NH3),
т. пл. -83,3°; NH3 • Н2О -ф- NH3 (80,05 вес. <•/„ NH3),
т. пл. —92,5°. Строение NH3 • Н2О и NH3 • */2Н2О,
по-видимому, соответствует гидратам А., образован-
ным водородными связями между молекулами А. и
воды, а не соединениям NH4OH (гидроокись аммония)
и (NH4)2O (окись аммония).
При обычной темп-ре А. устойчив. Гомогенная
реакция диссоциации А. на водород и азот в газовой
фазе начинается выше 1200—1300°, в присутствии
катализаторов эта темп-pa понижается до 300—400°.
А. — весьма реакционноспособное соединение. Так
как в молекуле А. есть неразделенная пара электро-
нов атома азота, для А. особенно типичны и легко
осуществимы реакции присоединения. Наиболее важ-
ной из них является присоединение протона к моле-
куле А. при взаимодействии с водой, к-тами, газо-
образными галогенводородами, ведущее к образова-
Н
нию соединений иона аммония Н : N : Н. Ион аммо-
Н
ния имеет конфигурацию правильного тетраэдра
с 4 пространственно и энергетически равноценными
атомами водорода в вершинах. Соединяясь с ани-
онами к-т, он ведет себя подобно ионам щелочных
металлов. Эффективный ионный радиус КН, (1,43 А)
имеет промежуточное значение между радиусами
К+ и Rb+, и NHj способен изоморфно замещать
их в кристаллич. решетке. Ион аммония бесцветен.
Соли аммония — кристаллич. в-ва, в подавляющем
большинстве хорошо растворимые в воде, легко
разлагаются при нагревании (см., напр., Аммония
галогениды). Отщепление атома водорода от иона
аммония под действием аниона к-ты происходит тем
легче, чем прочнее эта к-та удерживает протон, т. е.
чем слабее выражены ее кислотные свойства. Поэтому
при одинаковости структурного типа солей аммония
их термич. устойчивость возрастает с ростом силы
к-т, напр. по ряду NH4OH, NH4CN, NH4F, NH4C1,
NH4Br, NH4J. Если анион обладает окислительными
свойствами, то разложение сопровождается выделе-
нием свободного азота или его окислов: NH4NO3 =
= N2O -ф- 2Н2О.
Известны многочисленные производные гидроокиси
аммония, образующиеся при замещении атомов Н на
какие-либо органич. радикалы (четвертичные аммо-
ниевые основания и их соли). Неразделенная пара
электронов молекулы А. создает возможность при-
соединения не только атома Н+, но и других акцеп-
торов электронных пар, напр. BF3:
н :f; н :f;
h;N:+ в :f: = h:n; в :f:
и :f: н ;f ;
так что А. является типичным основанием в смысле
определений Бренстеда и Льюиса. Распространенным
201
АММИАК
202
типом реакций присоединения А. является образова-
ние аммиакатов при действии газообразного или жид-
кого А. на соли. Кроме реакций присоединения, для
А. характерны также реакции замещения и окисле-
ния. Щелочные и щелочноземельные металлы реаги-
руют с жидким и газообразным А., образуя в зависи-
мости от условий нитриды (продукты замещения
в молекуле А. трех атомов водорода) или амиды (при
замещении металлом одного атома водорода в А.).
При нагревании в атмосфере А. многие менее активные
металлы и неметаллы (Zn, Cd, Cr, Fe, В, Si и др.)
также образуют нитриды. Жидкий А. реагирует с се-
рой по ур-нию: 10S4NH3 = 6H2SN4S4. Ок.
1000° А. реагирует с углем, образуя HCN и частично
разлагаясь на азот и водород. Большое практич.
значение имеет реакция А. с СО2, ведущая к образо-
ванию карбаминовокислого аммония: CO2 -|-2NH3 =
= NH2COONH4, к-рый ок. 140° и давлении 150 ат
распадается на воду и мочевину CO(NH2)2. А. при
обычных условиях довольно устойчив к действию
окислителей, однако, будучи подожжен, он горит
в атмосфере кислорода, образуя воду и свободный
азот, а также окислы азота. Каталитич. окислением А.
получают окись азота, далее превращаемую в азотную
кислоту.
Одновременное каталитич. окисление А. и метана
дает синильную кислоту. Последняя может быть
также получена в результате эндотермич. реакции
NH3 -ф- CH4 = HCN -|- ЗН2 при высоких темп-рах.
При пропускании над нагретой СпО или окислами
др. малоактивных металлов А. восстанавливает
их до свободных металлов.
А. обнаруживается по характерному запаху. Бу-
мажка, смоченная раствором Hg3(NO3)2, при дейст-
вии А. чернеет. Малые количества А. в воде откры-
ваются с помощью т. н. реактива Несслера — щелоч-
ного р-ра K3[HgJ4] — в присутствии А. образуется
бурый осадок NHg2 J • Н2О (следы А. дают желтое
окрашивание). Для колич. определения А. погло-
щают измеренным количеством титрованного р-ра
кислоты и оттитровывают избыток последней р-ром
щелочи с индикатором метиловым красным или мети-
ловым оранжевым.
Получение. Основным способом промышленного
получения А. является его синтез из элементов —
азота и водорода. Дополнительным источником слу-
жит улавливание А., образующегося при коксовании
углей. Исходным сырьем для получения А. служит
азот воздуха и получаемый различными способами во-
дород. В зависимости от способа получения водорода,
к нему либо добавляют азот, выделенный из воздуха
(см. Воздуха разделение), либо непосредственно полу-
чают азотно-водородную смесь. Основы промышлен-
ного синтеза А. были заложены в исследованиях
Ф. Габера в 1904—07. Синтез А. из Н3 и N2 возможен
только при высокой темп-ре на катализаторе. Реакция
ЗН3 Na 2NH3 обратима. Равновесие смещается
в сторону образования А. при повышении давления
и понижении темп-ры. В табл. 3 приведены расчетные
величины содержания А. в объемных процентах в рав-
новесной газовой смеси в зависимости от темп-ры и
давления для стехиометрия, смеси Н3 и N2.
Таблица 3
Темп-ра, °C Содержание А. в об. % при разных давлениях
1 ат 100 ат 300 ат 800 ат
400 0,405 25,37 48,18 79,34
450 0,214 16,10 35,87 62,70
500 0,1216 14,87 25,80 51,05
550 0,0736 6,82 18,23 40,19
600 0,0470 4,53 12,84 30,92
Ниже 400° скорость реакции мала. Из данных
табл. 3 видно, что даже при больших давлениях реак-
ция не может дойти до конца. В производств, усло-
виях равновесное содержание А. в газовой смеси
не достигается. В пром-сти в качестве катализатора
применяют губчатое железо (активированное окисями
калия, алюминия и др. веществами), к-рое получают
восстановлением плавленного магнетита Fe3O4, со-
держащего соответств. добавки.
Уд. .поверхность катализатора 10—15 м2/г, радиус пор
ок.100 А. Выбор размера зерен катализатора определяется
тем, что при крупных зернах их внутренняя поверхность не
используется из-за диффузионного торможения реакции,
мелкие же зерна дают большое сопротивление потоку газа.
Обычный размер зерен 6 мм. На таком катализаторе процесс
синтеза А. можно вести при 450—550°. При более высокой
темп-ре активность катализатора падает. Железные катали-
заторы необратимо отравляются сернистыми соединениями
(H2S и др.) и обратимо — кислородом и его соединениями (водя-
ным паром, СО и др.). Поэтому азото-водородную смесь тща-
тельно очищают.
Скорость синтеза А. описывается уравнением Темкина
и Пыжева.
—5----/ “Kg -
PNH3 / \ РН2 /
<о =
Здесь со — наблюдаемая скорость реакции, равная раз-
ности скоростей образования и разложения A.; Ki и К2— кон-
станты скорости образования и разложения А. , Рн и
^NH3 — парциальные давления газов, а — постоянная. Для
обычного промышленного катализатора а = 0,5. При равно-
весии (о = 0, следовательно Kt/K2 — К, где К — константа
равновесия синтеза аммиака. Kt и К2 при высоких давлениях
несколько изменяются с давлением, вследствие отклонений
от законов идеальных газов и других причин. Теоретический
вывод уравнения основан
реакции, определяющей
скорость образования А.,
является активирован-
ная адсорбция азота. За-
висимость выхода А. от
объемной скорости, вы-
ражаемой в нм3 газовой
смеси, проходящей через
1 м3 катализаторной мас-
сы в час, при различных
давлениях, показана на
диаграмме (рис. 2). При
повышении объемной ско-
Объемная скорость, тыс. нм3/м3 нас
рости растет производи-
тельность катализатора, Рис. 2.
выражаемая в кг NH3 на
1 .и3 катализаторной массы в час. Возможность повыше-
ния производительности катализатора путем увеличения
объемной скорости ограничивается падением содержания А.
в газовой смеси на выходе из реактора, что затрудняет его
выделение, а также возрастанием гидравлич. сопротивления
системы. Обычно применяемые объемные скорости 2 5 000—
70 000 нм3/м3 час.
Скорость образования А. с повышением темп-ры
растет, но выход А., достигнув максимума, понижается
вследствие возрастания скорости обратной реакции.
Чем выше содержание А. в газовой смеси, тем ниже
темп-pa, при к-рой скорость синтеза А. максимальна.
Поэтому стремятся осуществить в реакторе падение
темп-ры в направлении потока газа путем отвода
тепла реакции. Однако поскольку процесс прово-
дится автотермически, т. е. исходная газовая смесь
подогревается за счет тепла реакции, в первых по
ходу газа слоях катализатора по необходимости про-
исходит подъем темп-ры.
Промышленный синтез А. проводят при различных
давлениях: высоких (750—1000 ат), средних (200—
350 ат) и низких (150 ат). Все системы синтеза А.
работают с циркуляцией азото-водородной смеси.
Образовавшийся в колонне синтеза А. отделяют от
непрореагировавших N2 и Н2; непрореагировавшую
часть азото-водородной смеси с добавлением к ней
свежей порции снова возвращают в цикл.
Наибольшее распространение получили системы,
работающие под средними давлениями, как наиболее
экономически целесообразные.
Схема синтеза А. при среднем давлении приведена на
рис. 3. Прореагировавшая в колонне 1 газовая смесь, пройдя
203
АММИАК - АММИАКАТЫ
204
последовательно первичные конденсатор 2 и сепаратор <3,
циркуляционный насос 4 и фильтр 5, поступает в межтрубное
пространство теплообменника, расположенного вверху кон-
денсационной колонны 6, где предварительно охлаждается
Рис. 3.
до 5—10° за счет теплообмена с газом, идущим по трубкам
теплообменника в колонну синтеза. Из колонны 6 газовая
смесь поступает на дополнительное охлаждение в испари-
тель 7, где за счет испарения жидкого А. в межтрубном про-
странстве происходит дальнейшее охлаждение газовой смеси
до минус 10—15°. При этом происходит дополнительная кон-
денсация А. из газовой смеси. Смесь газа и сконденсировав-
шегося А. поступает в сепарационную часть колонны 6 для
отделения жидкого А. от газов. Газовая смесь, пройдя трубки
теплообменника колонны 6, поступает в колонну 1 синтеза А.
Жидкий А. из конденсационной колонны и первичного сепара-
тора выводится на склад. Газообразный А. из испарителя 7
через ловушку 8 направляется для получения из него HNO3,
азотнокислых солей (NH4NO3 и др.) или идет в холодильную
установку для сжижения. Колонны синтеза конструируются
так, что стенки, несущие механич. нагрузку, не подвергаются
действию высокой темп-ры. Для этого они отделяются пото-
ком холодной газовой смеси от внутр, устройств, находящихся
при высокой темп-ре, но не испытывающих действия большой
разности давлений.
Схема одной из колонн синтеза А., применяемой на уста-
новках среднего давления, изображена на рис. 4. Толстостен-
ный корпус 1 колонны высо-
той 14 .и, внутренним диамет-
ром 0,85 .и и толщиной стенки
160 мм изготовлен из слитка
ff-u холодный поток
выход газа
Рис. 4.
легированной стали, который
просверливается по оси. Свер-
ху и снизу колонна закры-
вается стальными крышками 2
и <3. В верхней части колонны
размещена катализаторная ко-
робка 4, в нижней части —
теплообменник 5. Газ посту-
пает в колонну тремя потока-
ми: основным и двумя допол-
нительными. Основной поток
(около 60% газовой смеси) на-
правляется по трубкам теп-
лообменника 5, затем смеши-
вается с первым холодным
потоком и проходит по труб-
кам теплообменника в ката-
лизаторной коробки. Перед
входом газовой смеси в слой
катализатора присоединяется
второй холодный поток, пред-
варительно прошедший тепло-
обменник 7 из двойных тру-
бок, расположенный (так же,
как и первый теплообменник,
из простых трубок) в катали-
заторной коробке 4.
Изменение соотношения
между потоками позволяет ре-
гулировать темп-ру слоя ка-
тализатора при уменьшении
его активности в ходе эксплу-
атации. Объединенный газовый поток проходит слой катали-
затора, межтрубное пространство предварительного тепло-
обменника 5 и выходит из колонны. Реакция в верхней ча-
сти катализатора протекает быстро, т. к. здесь мала кон-
центрация А.; это приводит к выделению большого колич. те-
плоты и повышению темп-ры до 550°. В дальнейшем темп-ра
понижается. В циркуляционной азото-водородной смеси со-
держится ок. 5% А., после колонны синтеза его содержа-
ние увеличивается до 18—19%. При объемной скорости
73 000 нм3/м*-час производительность описанной колонны
более 140 т А. в сутки.
А. выпускается либо в виде жидкого, либо в виде
аммиачной воды, чаще всего с содержанием 25% А.
Жидкий А. перевозят в стальных баллонах (рабочее
давление 30 ат) и в специальных ж.-д. цистернах
низкого давления (2,5 ат), содержащих охлажден-
ный А., или высокого давления (не более 12,8 ат);
жидкий А. хранят в стальных резервуарах (танках).
Техника безопасности. А. с воздухом и кислородом
образует взрывоопасные смеси; так, при комнатной
темп-ре и атмосферном давлении взрываются смеси,
содержащие: 14,5—26,8 об. % NH3, остальное — воз-
дух, или 13,5—82 об. % NH3, остальное — кисло-
род. При повышении темп-ры эти пределы расши-
ряются. Темп-ра вспышки воздушно-аммиачных сме-
сей, содержащих от 9 до 57об. % NH3,—ок. 1000°.
Жидкий А. при соприкосновении с кожей вызывает
сильные ожоги. Особенно опасно попадание А. в глаза.
При высокой концентрации его в воздухе А. оказы-
вает удушающее действие (максимально допустимая
концентрация в воздухе рабочего помещения 0,02 мг/л).
Угольный противогаз защищает от А. лишь короткое
время, поэтому в пром-сти применяют спец, противо-
газы, содержащие химич. поглотитель. Первая по-
мощь при отравлении А. — свежий воздух, кислород
и пары уксусной к-ты (вдыхание); при сильном отрав-
лении — искусств, дыхание.
Применение. А. используется для произ-ва HNO3,
азотсодержащих солей [(NH4)2SO4, NH4NO3, NaNO3,
Ca(NO3)2], мочевины, синильной к-ты. А. исполь-
зуется также при получении соды по аммиачному
способу, в органич. синтезе, для приготовления вод-
ных р-ров (нашатырный спирт), находящих разно-
образное применение в химич. пром-сти и в медицине;
в холодильных машинах. Жидкий А., а также его
водные р-ры применяют в качестве жидких удобрений,
А. представляет собой хороший растворитель для зна-
чительного класса соединений, содержащих азот,
подобно воде для кислородсодержащих в-в. Большие
количества А. идут на аммонизацию суперфосфата
и туковых смесей.
Лит..- Эпштейн Д. А. [и др.], Химия и технология
связанного азота, [М. — Л.], 1934; Общая химическая техно-
логия, под ред. С. И. Вольфковича, т. 1, М., 1952; А з-
бель И. Я., Кильштедт К. К. и Макеев Г. А.,
Производство аммиака на основе полуводяного газа, М.,
1954; Gmelin, 8, Aufl., Syst.-Nummer 4, Lfg. 2, В.,
1935; Syst.-Nummer 23, Lfg. 1—2, В., 1936; Mellor,
v. 8, L. — N. Y. — Toronto, 1947; Pascal, v. 10, P., 1956;
Ulmann, 3, Aufl., Bd. 3, Mtlnch. — B., 1953, S. 523—602;
Kirk, v. 1, N. Y., 1947, p. 771—810; Nielsen A.; An
investigation on promoted iron catalysts for the synthesis of
ammonia, 2 ed., Copenhagen, 1956. А. П. Егоров.
АММИАКАТЫ — комплексные соединения, обра-
зуемые солями и аммиаком, NH3 в А. координационно
присоединен к иону металла и входит во внутр,
сферу комплекса, напр. [Pt(NH3)6]Cl4. К образова-
нию А. способны соли большинства металлов. Мно-
гие А. весьма устойчивы. Напр., А. Сг, Со, платино-
вых металлов не отщепляют NH3 даже при нагрева-
нии до 200° и не разлагаются при действии NaOH
или НС1. Менее устойчивы А. Си, Ag, Zn и нек-рых
др. металлов. Они разлагаются разб. кислотами
и щелочами. А. щелочных н щелочноземельных ме-
таллов полностью разлагаются водой; нек-рые из них
существуют лишь при низких темп-pax, фториды же
их вообще не получены. А. получают либо взаимодей-
ствием солей с NH3 в водном растворе, либо действием
газообразного или жидкого NH3 на твердые соли.
Так как в A. NH3 испытывает действие силового поля
иона-комплексообразователя, то он способен про-
являть кислотные свойства. Напр., A. Pt,4+ мо-
гут диссоциировать по типу: [Pt(NH3)6]4+ =
xk [Pt(NH3)5NH2l34"4" Н+. Вводных растворах устой-
чивость комплексных ионов А. весьма различна и
определяется их константами нестойкости. В случае А.
205
АММИАЧНАЯ ВОДА —АММОНИЯ ГАЛОГЕНИДЫ
206
диссоциация комплексного иона, подобно диссоциа-
ции многоосновных к-т, протекает по стадиям и обра-
тимо. Напр., при взаимодействии солей Ni2+ с NH3
в водном растворе образуется ряд ионов состава от
[NiNH3]2+ до [Ni(NH3)e]2+. В таблице приводятся
значения рА (обратные логарифмы общих констант
нестойкости) для наиболее важных А. Чем больше рА,
тем устойчивее А. (при данном координационном
числе).
Комплексный ион Комплексный ион pK
(Ag(NH3)2l+ 7,03 (30°) [Mn(NH8)2p+ 1.3 (20—30°)
fCd(NH8)ep+ 5,14 (30°) tHg(NH,)4P+ 19,28 (22°)
(Co(NH3)8p+ 5,11 (30°) [Mg(NH8)8P+ —3,29 (22°)
(Co(NH8)ep+ 32,51 (30°) [Ni(NH8),P+ 8,73 (30°)
(Cu(NH,)4]’+ 12,67 (30°) [Zn(NH3)4P+ 9,46 (30°)
Особую группу составляют многоядерные А., к-рые
характеризуются наличием нескольких координа-
ционных сфер (напр., декааммин-н-амино-кобальти-
соли [(H3N)5Go— NH2Go(NH3)5]X. Замена NH3 и А.
на другие адденды приводит к образованию неодно-
родных комплексов, напр. в случае [Pt(NH3)e]4+
можно получить [Pt(NH3)6Gl]3+, [Pt(NH3)4Gl2]2+ и
т. д. Способность NH3 образовывать с нек-рыми
солями окрашенные А. используется в аналитич. хи-
мии для открытия ряда ионов (напр., Сн2+). Благо-
даря различной устойчивости в водных р-рах А.
применяются также для разделения нек-рых ионои.
См. также Комплексные соединения.
Лит.: Гринберг А.А., Введение в химию комплексных
соединений, 2 изд., Л. — М., 1951; Яцимирский К. Б.,
Васильев В. П., Константы нестойкости комплексных
соединений, М., 1959; The chemistry of the coordination com-
pounds, ed. J. C. Bailar, N. Y.— L., 1956; В j e r r u m J., Metal
ammine formation in aqueous solution. Theory of the reversible
step reactions, Copenhagen, 1957. А. К. Пикаев.
АММИАЧНАЯ ВОДА — водный р-р аммиака, об-
разующегося при коксовании каменных углей в кок-
совых печах. Кроме аммиака, в А. в. содержатся СО2,
H2S и в незначительных количествах цианистые со-
единения, фенолы, пиридиновые основания и др.
Выход NH3 составляет 0,25—0,40»/о от веса коксуе-
мых углей (в пересчете на сухие), причем 20—30°/о
NH3 растворяется в надсмольной воде, а остальное
количество извлекается из газа промыванием водой
в скрубберах или поглощается р-ром H2SO4 в сату-
раторах. А. в. получают отгонкой NH3 из надсмольной
и скрубберной воды в аммиачных колоннах. Смесь
водяных паров и NH3 из колонн поступает в дефлег-
маторы (для увеличения концентрации NH3), а потом
в конденсаторы, из к-рых стекает готовый продукт.
Для уменьшения содержания H2S и SO2 в А. в. над-
смольную и скрубберную воду предварительно подо-
гревают и диссоциаторах, после чего обрабатывают
известковым молоком для разложения солей аммония.
Содержание NH3 в А. в. составляет от 18 до 19,85»/0,
СО2 — от 70 до 100 г/л, H2S — от 30 до 50 г/л.
В А. в., получаемой из синтетич. NH3, его содер-
жание достигает 25%. Основное количество NH3,
получаемого при коксовании, используют для произ-
водства сульфата аммония. Кроме того, А. в. при-
меняют как удобрение, в производстве соды, краси-
телей и др.
Лит.; Белов К. А., Улавливание химических продук-
тов коксования, Харьков — М., 1948; Коляндр Л. Я.,
Улавливание и переработка химических продуктов коксова-
ния, Харьков — М., 1953. А. И. Бродович.
АММИАЧНАЯ СЕЛИТРА — см. Аммония нитрат.
АММОНИЙ NHJ- — неорганич. радикал, в соеди-
нениях играет роль одновалентного металла. Обра-
зует гидроокись аммония, многочисленные соли аммо-
ния и др. аммония соединения. Качественной реак-
цией для открытия иона А. служит выделение аммиака
при действии щелочей на соединения А. О строении
иона А. см. Аммиак. Примеры наиболее практически
важных солей А. — см. Аммония карбонаты, Аммо-
ния нитрат и т. д.
АММОНИТЫ — взрывчатые смеси азотнокислого
аммония с твердыми горючими и взрывчатыми веще-
ствами (ВБ). По составу А., называемые также ам-
миачно-селитренными ВВ, делят на след, классы:
1) собственно аммониты — смеси, . содержащие
в качестве одного из компонентов взрывчатые нитро-
соединения (напр., амматолы — бинарные смеси
азотнокислого аммония и тринитротолуола)', 2) а м-
м о н а л ы — смеси, содержащие металлич. А1 в виде
порошка или пудры; 3) динаммоны — смеси
аммониевой селитры с невзрывчатыми горючими в-вами
(напр., торф, древесная мука, металлы и др.); 4) по-
рошкообразные нитроглицерино-
вые В В — смеси азотнокислого аммония с горю-
чими и ВВ, содержащие до 8% жидкого или слабо-
желатинированного нитроглицерина либо других жид-
ких эфиров азотной к-ты. Все А. являются вторичными
(бризантными) взрывчатыми веществами. По сравне-
нию с другими ВВ А. малочувствительны к механич.
воздействиям (удар, трение), обладают высокой химич.
стойкостью и поэтому относительно безопасны в
произ-ве, хранении и обращении. Общими недостат-
ками А. являются; гигроскопичность, низкая водо-
устойчивость, способность к слеживанию. В резуль-
тате увлажнения и слеживания А. утрачивают свои
взрывчатые свойстиа. Введением в состав А. спец,
добавок может быть увеличена их водоустойчивость
и уменьшена слеживаемость. Взрывчатые свойства А.
зависят от состава (см. табл.), а также степени из-
мельчения компонентов и тщательности смешения.
Процесс произ-ва А. сводится к подготовке исход-
ных компонентов (сушка, измельчение, просеивание)
и их смешению. А. со значит. (> 20°/о) содержанием
тринитротолуола готовят смешением нагретых кри-
сталлов селитры с расплавл. тринитротолуолом.
Название A. Компоненты состава (кроме NH4NO3), их содержание в % Плотность, г/мл . ' Обжатие свинцового цилиндра, мм Расширение свинцовой бомбы, мл Скорость детон ации, , км/сек
Динаммон T Торф, 12,1 1,0 13 340 2,9-3,5
Аммонит № 9 Тринитротолуол, 5 Древесная мука, 8 0,9 И 310 2,5
Аммонит № 6 Тринитротолуол, 21 1,0—1,35 14-18 370 3,0-4,6
Амматол 40/60 Тринитротолуол, 60 1,45 20 310 5,0
Динафт алит № 1 Динитронафталин, 1,15 15 340 3,5-3,8
Аммонал водо- устойчивый Тринитротолуол, 15 Металлич. алюми- ний, 4,5 1,0 17 400 4,0-4,5
Аммонит ПЖВ-20 Тринитротолуол, 16 Поваренная соль, 20 1,1 13 265 2,8—3,4
А. широко применяют при взрывных работах на по-
верхности и под землей. В угольных и др. шахтах,
где выделяются горючие газы или образуется легко
воспламеняющаяся пыль, пользуются т. н. предохра-
нительными взрывчатыми веществами. А. приме-
няют также для снаряжения мин, бомб, снарядов
и др. боеприпасов и для военно-инженерных подрыв-
ных работ.
Лит.. Яременко Н. Е. и Светлов Б. Я., Тео-
рия и технология промышленных взрывчатых веществ, М.,
1957; Горст А. Г., Пороха и взрывчатые вещества, 2 изд.,
М., 1957. А. И. Гольбиндер.
АММОНИЯ ГАЛОГЕНИДЫ — соли аммония и
галогеноводородных к-т общей формулы NH4X, где
АММОНИЯ ГИДРООКИСЬ — АММОНИЯ РОДАНИД
207
X — атом галогена; бесцветные кристаллы, хорошо
растворимые в воде и жидком аммиаке (в жидком
NH3 плохо растворим NH4F), растворимость при 20°
в 100 г воды: 82,7 г NH4F, 37,2 г NH4C1, 75,5 г NH4Br,
172,3 г NH4J. В р-рах А. г. сильно диссоциированы
и показывают кислую реакцию; при нагревании дис-
социируют: NH4X = NH3-|- НХ. При действии на
А. г. щелочей также выделяется аммиак. Аммония
фторид NH4F, гексагональная решетка, а = 4,39 А;
с = 7,02 А; плоти, а!25 1,01; теплота образования
АЯ;!я- —111 ккал/моль', гигроскопичен, расплывается
на воздухе; легко разлагается, особенно во влажном
состоянии и при нагревании. Может быть получен
в кристаллич. виде при обработке р-ра HF избытком
NH3 и выпариванием р-ра в присутствии извести.
При выпаривании р-ра NH4F могут образоваться
кислые соли: NH4F • HF, NH4F • 2HF и т. д. NH4F
применяют в качестве фторирующего агента, а кис-
лые соли — для получения HF. Аммония хло-
рид (нашатырь) NH4C1, ниже 184,3° устойчива
простая кубич. решетка, а=3,8758 А (25°), d20 1,52 6;
теплота образования ДН298 = —75,38 ккал/моль.
Р-р, содержащий 77,3 г NH4G1 на 100 г Н2О, кипит
при 116°; NH4C1 мало гигроскопичен, на воздухе
устойчив. Давление паров NH4C1 при 250° состав-
ляет 49,5 мм рт. ст., а при 338° достигает 760 мм
рт. ст.', при этой темп-ре пары NH4C1 полностью
распадаются на NHS и НС1. Как пары, так и р-ры
NH4C1 сильно корродируют стальную аппаратуру.
NH4C1 получают как побочный продукт содового
произ-ва или же взаимодействием NH3 и НС1, про-
пускаемых в водный р-р NH4C1, и др. способами.
Применяют при изготовлении сухих батарей, галь-
ванич. элементов, при лужении и пайке. А м м о-
ния бромид NH4Br, о!20 2,40; теплота образова-
ния ДН298 = —64,61 ккал/моль-, т. возг. 394,6°; не
гигроскопичен, устойчив на воздухе и на свету толь-
ко в чистом состоянии. Аммония иодид NH4J,
d2a 2,51; теплота образования Д/Л98= — 48,30
ккал/моль', т. возг. 404,9°. Очень гигроскопичен.
Лит. см. при ст. Аммиак.
АММОНИЯ ГИДРООКИСЬ NH4OH — см. Ам-
миак.
АММОНИЯ КАРБОНАТЫ — аммониевые соли
угольной кислоты. В системе NH3—СО2—Н2О обра-
зуются следующие соединения аммония: бикар-
бонат NH4HCO3, карбонат (NH4)2CO3, полукарбонат
(NH4)2CO3 • 2NH4HCO3 Н2Ои карбамат NH4CO2NH2;
практич. значение приобрели первые два соединения.
Бикарбонат аммония — бесцветные, кри-
сталлы; простая ромбич. решетка, а=8,76 А, Ъ=
= 10,79 А, с=7,29 А, плоти. 1,586. Растворимость
в воде (г/100 г Н2О): 11,9 (0°), 21,7 (20°), 36,6 (40°).
Уже при комнатной темп-ре NH4HCO3 начинает выде-
лять СО2, а ок. 60° распадается нацело; давление
паров в зависимости от темп-ры (мм рт. ст.): 59 (25,4°),
278 (45°), 815 (59,3°). Получают NH4HCO3 абсорбцией
газообразных NH3 и СО2 раствором (NH4)2CO3.
Карбонат аммония — бесцветные кристал-
лы; очень неустойчив — как на воздухе, так и в р-ре
уже при комнатной темп-ре выделяет NH3, превра-
щаясь в бикарбонат. А. к. получают взаимодействием
газообразных NH3, СО2 и паров воды, взятых в стехио-
метрия. отношениях при быстром охлаждении и кон-
денсации продукта реакции. Карбонат и бикарбонат
находят применение при крашении тканей, в меди-
цине, в хлебопекарном деле.
Лит. см. при ст. Азотные удобрения. В. А. Клевке.
АММОНИЯ НИТРАТ (аммоний азотнокислый,
аммиачная селитра) NH4NO3 — бесцветные кри-
сталлы, в зависимости от темп-ры существующие
в нескольких модификациях. При темп-ре от —18°
до +32° устойчива модификация с простой ромбич.
208
решеткой, а = 5,45 А, Ъ = 5,75 А, с = 4,96 А;
плоти, d20 1,725, т. пл. 169,6 — 170°, выше — разла-
гается; при осторожном нагревании разложение
протекает по схеме: NH4NO3 = N2O + 2Н2О (на
этом основано получение N2O). При нагревании
выше 300° А. н. взрывает. Теплота образования моди-
фикации, устойчивой при комнатной темп-ре,
= — 87,27 ккал/моль. А. н. хорошо растворим в воде
(г/100 г Н2О): 150 (10°), 363 (32,3°); в жидком аммиаке:
391 г в 100 г NH3 (25°); растворяется в метаноле,
этаноле, пиридине; весьма гигроскопичен. В водных
р-рах имеет кислую реакцию. Растворение в воде
протекает с поглощением теплоты. Водные р-ры в за-
висимости от концентрации кипят при след, темп-рах:
Концентрация, вес. %. . 60 80 90 96 99
Т. кип., °C........ 113,5 128,5 147 182 222
А. н. получают нейтрализацией разб. HNO3 аммиа-
ком. Выделяющаяся при нейтрализации теплота
используется для упаривания р-ра образовавшегося
NH4NO3. Произ-во А. н. складывается из след, опе-
раций: нейтрализации HNO3 газообразным NH3; упа-
ривания р-ров NH4NO3 до состояния плава (98—
99%); кристаллизации соли из плава; сушки или
охлаждения соли. А. н. выпускают в виде мелкокри-
сталлич., чешуйчатого и гранулированного продукта.
Содержание NH4NO3 в продукте составляет 99,2—
99,5%; влаги 0,1—1,0%. При хранении и перевозке
поглощает влагу и слеживается; для предотвращения
слеживаемости А. н. гранулируют. Насыпной вес
гранулированного А. н. при плотной упаковке со-
ставляет 1,16 т/м3, при неплотной 0,83 т/м3.
Применяют А. н. гл. обр. в качестве Азотного
удобрения, а также в произ-ве взрывчатых веществ
(см. Аммониты).
Лит. см. при ст. Азотные удобрения. В. А. Клевке.
АММОНИЯ ПЕРСУЛЬФАТ (аммоний надсер-
нокислый) NH4OSO2—О—О—SO2ONH4, молеку-
лярный вес 228,21 — бесцветные кристаллы в виде
призм или белый зернистый порошок; раствори-
мость в 100 г воды: 58,2 г (0°), 74,8 г (15,5°). В чистом
сухом состоянии А. п. устойчив в течение многих
месяцев; в присутствии влаги разлагается с постепен-
ным выделением кислорода, содержащего озон; А. п.
отличается сильным окислительным действием. При
120° сухой продукт распадается с выделением кисло-
рода и образованием (NH4)2S2O7; водный р-р разла-
гается уже при комнатной темп-ре, а при более высо-
кой — быстро (NH4)2S2O8 + 2Н2О 2NH4HSO4 +
+ Н2О2. Разложение водных р-ров А. п. замедляется
при разбавлении, а также при добавлении сульфатов
щелочных металлов или алюмокалиевых квасцов.
Получают А. п. анодным окислением насыщенного
р-ра (NH4)2SO4; применяют А. п. для инициирова-
ния полимеризации виниловых соединений, как
средство для отбеливания и дезинфицирования, для
фотометрия, и титриметрич. определений Мп и Сг,
для окисления органич. в-в, в фотографии как осла-
битель. И. И. Ефимов.
АММОНИЯ РОДАНИД (аммоний роданистый)
NH4SCN, мол. в. 76,12—бесцветные или белые рас-
плывающиеся моноклииич. таблички; т. пл. 149,6°;
т. разл. > 170°; d 1,31; при нагревании до 70° частично
переходит в тиомочевину; в 100 г воды при 19° раство-
ряется 165 г; растворим в спирте, аммиаке и ацетоне.
Конц, р-ры на свету постепенно окрашиваются в крас-
ный цвет. Получают А. р. кипячением NH4CN с S
(или полисульфидами) или CS2 с аммиаком под давле-
нием (в присутствии гашеной извести); очищают
кристаллизацией. С Fe3+ А. р. дает красное окраши-
вание. Применяют А. р. в аналитич. практике для
фотометрич. определения Fe3+, Со, Мо, для титри-
метрич. определения Ag, Hg, Cl", как индикатор при
209
АММОНИЯ СОЕДИНЕНИЯ
210
титровании Fe3+F“ и др.; в фотографии, в качестве
компонента охлаждающих р-ров и для др. целей.
Д. И. Кузнецов.
АММОНИЯ СОЕДИНЕНИЯ — вещества ионного
характера, содержащие положительно заряженный
атом азота, связанный 4 ковалентными связями с орга-
нич. радикалами или с атомами и одной ионной связью
с анионом. Простейшими примерами неоргапич. А. с.
могут служить гидроокись аммония [NITiJ On , а так-
же соли аммония, напр. [NП4] С1. Неорганич. соли
аммония обычно получают действием NH3 или вод-
ного р-ра NH3 на соответствующие кислоты, напр.
NH3 + HG1—>-NH4Gl. В нек-рых случаях вместо
нейтрализации кислот прибегают к особым методам
получения солей аммония. Так, роданид аммония
образуется при нагревании водного NH3 с CS2 в авто-
клаве при 110°: 2NH3 + CS2->NH4CNS + H2S.
К органич. А. с. относятся продукты замещения ато-
мов Н у атома азота в NH4OH и солях аммония орга-
нич. радикалами, напр. |(CH3),1N] OH , [CH3NH3] ЬС1~.
В зависимости от числа органич. радикалов в А. с.
различают первичные [RNH3]+CF, вторичные
[R2NH2]+C1~, третичные [R3NH]+C1~ и четвертичные
[R4N]bCl“ А. с. Соединений, содержащих пяти-
ковалентный атом азота, не существует. А. с. пред-
ставляют собой самый типичный и распространенный
пример т. наз. ониевых соединений. Органич. А. с.
встречаются в природе. Так, во многих растениях
содержатся алкалоиды в виде солей; простейшая
четвертичная аммониевая соль [(CH3)4N]X найдена
в морских анемонах и нек-рых растениях; холин
[(GH3)3NGH2GH2OFf] OH и его ацетильное произ-
водное являются чрезвычайно важными физиологи-
чески активными соединениями.
Четвертичные А. с., содержащие 4 различных ра-
ГСвН5СН2ч + /СН3 1
дикала, например X
L с6н5/ xch2ch=ch2J
удается разделить на оптич. изомеры, что в подобных
случаях обусловлено асимметрией катиона замещен-
ного аммония. Первичные, вторичные и третичные
амины, подобно NH3, дают с кислотами соли; при
действии щелочей на эти соли выделяются исходные
свободные амипы. Прием этот широко применяется
для очистки различных аминов. Основной способ полу-
чения четвертичных аммониевых солей заключается
в действии алкилгалогенидов на третичные амины:
R3N + R'X —> [R3.\'R'| X . Вместо алкилгалогени-
дов можно применять другие алкилирующие агенты,
напр. диалкилсульфаты, эфиры арилсульфоновых
кислот. Реакция легче всего осуществляется в случае
иодидов, хуже с бромидами и еще медленнее с хлори-
дами. Скорость реакции очень сильно зависит от рас-
творителя. Так, если скорость присоединения C2H5J
к триэтиламину принять за 1, то в спирте она проте-
кает в 200 раз, в бензиловом спирте в 750 раз, в аце-
тонитриле в 5000 раз скорее. Известны также внутри-
молекулярные четвертичные А. с., напр. бетаин
(CH3)3NCH2COO_ (см. Биполярные соединения).
Особый случай получения солей аммония заклю-
чается в действии алкилгалогенидов на шифовы ос-
нования: RCH = NR' + R"X -> [RCH=NR'R"]+X-.
Образующиеся при этом соли часто называют третич-
ными аммониевыми солями. При действии р-ров едкой
щелочи на четвертичные соли образуются четвертич-
ные, очень сильные основания, к-рые вполне можно
сравнить с КОН и NaOH. Поэтому указанная реак-
ция является равновесной: (CH3)4NJ + КОН
= (CH3)4NOH + KJ. Чистые четвертичные основа-
ния обычно получают действием влажной окиси се-
ребра на четвертичные соли: (CH3)4NJ + AgOH —►
—>(СН3)4МОН + AgJ. Упаривание фильтрата после
отделения AgJ ведет к чистым основаниям, представ-
ляющим собой кристаллич. вещества, к-рые, подобно
NaOH и КОН, расплываются на воздухе. В частности,
(CH3)4NOH-5H2O плавится при 62—63°.
Гидроокиси из первичных, вторичных и третичных
аминов [CH3NH3OH, (CH3)2NH2OH, (CH3)3NHOH1
существуют только в водных р-рах и при упаривании
теряют воду и превращаются в исходные амины. Для
соединений типа R4N0H из-за отсутствия атомов
водорода при атоме азота такой процесс невозможен;
этим объясняется сравнительная стойкость четвертич-
ных оснований. Однако при нагревании оснований
они в зависимости от природы распадаются по-раз-
ному. Так, из (CH3)4NOH или (С6Н5СН2)4 NQH обра-
зуются третичные амины и спирты: (CH3)4NOH—>
—> (CH3)3N + СН3ОН. Если же при атоме азота
имеются, помимо СН3, другие группы (С2Н5, С3Н7
и т. д.), распад четвертичных оснований (гофман-
ский распад) идет с выделением олефина и воды:
[(CH3)3NCR2CR2H]+OH- (CH3)3N + GR2=GR2 +
+ Н2О. Такое направление процесса объясняется
тем, что благодаря положительному заряду на атоме
азота облегчается связывание протона в ^-положении
оксигруппой:
[(CH3)3N -^СН2-^СН3]+ОН“--------
---*-(CH3)3N + СН2=СН2 + Н2О
Предварительное метилирование исходного амина,
превращение четвертичной соли в основание и его
распад (метод исчерпывающего метилирования) ши-
роко применяется при изучении природных алкалои-
дов и других азотистых соединений.
При действии фениллития на (CH3)4NC1 отщепляется
атом водорода в a-положении и образуется триметилам-
монийметилид (CH3)3N=CH2, к-рый следует рас-
сматривать как внутримолекулярную аммониевую
соль биполярного строения (CH3)3NCH2. В случае
бензиламмониевых четвертичных солей наблюдается
легкое перемещение бензильного радикала от аммоние-
вого азота (см. Стивенса перегруппировка). Так,
гидроокись фенацилбензилдиметиламмония (I) в иод-
ном р-ре гладко переходит в диметиламинобензил-
ацетофенон (II). Такое направление процесса объяс-
няется тем, что промежуточно образующаяся неустой-
чивая внутренняя соль (А) стабилизируется пере-
группировкой в третичный амин (II):
[(CH3)2N(CH2C6H5)CH2COCeH5]+OH^-.
, CHgCgHg п
— (CH3)3N<_ (CH3)3NCH(CH3C6H5)COC6H5
ХСНСОС6Н5
L А -1 II
При восстановлении четвертичных солей, содержа-
щих двойную связь в P-положении, расщепляется
С—N-связь:
[C(iH5CH=CHCH2NR3]+ х +2Н-
— СеН5СН=СНСН3 + nr8 + нх
Неорганич. соли аммония находят широкое приме-
нение. Так, аммония сульфат и аммония нитрат
производятся в огромных количествах в качестве азот-
ных удобрений; хлористый аммоний (нашатырь)
представляет собой побочный продукт произ-иа соды
по аммиачному способу и применяется в текстильном
произ-ве, в медицине, при паянии и лужении, как
дымообразователь для маскировки в военном деле.
Роданид аммония применяется как реагент в объем-
ном анализе. Четвертичные А. с., содержащие хотя бы
одну длинную алкильную цепь, обладают поверх-
ностно-активными и антисептич. свойствами. Фенил-
триметил аммоний применяется как метилирующий
211
АММОНИЯ СУЛЬФАТ-АМОРФНОЕ СОСТОЯНИЕ
212
агент, в частности для селективного метилирования
морфина в кодеин. Четвертичные соли (иодметилаты,
иодэтилаты, иодбензилаты) находят применение для
характеристики третичных аминов. Четвертичные
основания, напр. (CH3)4NOH («тритон В»),
(CHS)2N(CH2C6H5)2OH («тритон F»), применяются
в качестве растворителей для целлюлозы, для омыле-
ния жиров и др.
Лит.: Kirk, v. 1, N. Y., 1947, р. 810—826.
Я. Ф. Комиссаров.
АММОНИЯ СУЛЬФАТ (аммоний сернокислый)
(NH4)2SO4 — бесцветные кристаллы, простая ромбич.
решетка, а — 7,78 А, b = 10,62 А, с = 5,98А,
плотн. а’20 1,77. Теплота образования ДН298 =
= —281,86 ккал/моль. При нагревании до 513° А. с.
полностью разлагается на NH3 и H2SO4, частичное
разложение с образованием кислых солей начинается
при 218°. При действии щелочей на А. с. также выде-
ляется NH3. А. с. хорошо растворим в воде (г/100 г
Н2О): 68,1 (—10°), 70,4 (0°), 76,9 (25°), 102 (100°);
в водных р-рах имеет кислую реакцию. Практич.
интерес представляет растворимость А. с. в водном
р-ре NH3 с целью применения в качестве жидкого
удобрения; с повышением концентрации NH3 раство-
римость А. с. падает: в 100 г 15%-ного р-ра NH3
растворяется 36 г (NH4)2SO4 (10°), а в 24,5%-ном
р-ре NH3 — только 18 г. Основные промышленные
способы получения А. с.: поглощение серной к-той
аммиака, содержащегося в газе коксовых печей;
нейтрализация серной к-ты газообразным синтетич.
аммиаком. А. с. в больших количествах применяют
в качестве азотного удобрения.
Лит. см. при ст. Азотные удобрения. В. А. Клевке.
АММОНИЯ ТЕТРАРОДАНОМЕРКУРИАТ. молеку-
лярный вес 469,63, (NH4)2[Hg(SCN)4] — бесцветные
кристаллы, в массе с желтым оттенком; растворим
в воде. При действии А. т. на соли Со2+ выпадает
синий осадок Co[Hg(SCN)4]. В случае разб. р-ра
соли Со2+ осадок может не выпадать часами из-за
образования пересыщенного р-ра; прибавление не-
больших количеств соли Zn2+ ускоряет выпадение
осадка; при этом образуются смешанные кристаллы
Co[Hg(SCN)4] и Zn[Hg(SCN)4], окрашенные в бледно-
голубой или темно-синий цвет, в зависимости от ко-
личества цинка. Ионы Ni2+ и Fe2b дают осадки
зеленоватого цвета, Сн2+ — желто-зеленого цвета
(в присутствии Zn2+ — фиолетового). Получают рас-
творением HgCl2 в избытке р-ра NH4SCN. При-
меняют А. т. для колич. осаждения Zn2+ в отсут-
ствие Си, Со, Ni, Fe; определение можно заканчивать
либо взвешиванием осадка Zn[Hg(SCN)4], либо раство-
рением осадка в соляной к-те и титрованием стандарт-
ным р-ром йодата калия. Реактив находит приме-
нение в микрокристаллоскопии Для обнаружения
Со И Zn. В. Г. Типцова.
АММОНИЯ ФОСФАТЫ — аммониевые соли фос-
форной (ортофосфорной) к-ты Н3РО4, образующей
моно-, Ди- и триаммонийфосфат. Моноаммоний-
фосф а т NH4H2PO4 — устойчивая соль (давление
паров при 125° не превышает 0,05 мм рт. ст.); бес-
цветные кристаллы, объемноцентриррванная тетраго-
нальная решетка, а = 7,51 А, с = 7,53 А; плотн. а!201,80,
т. пл. 190°; не гигроскопичен; растворимость в
100 г воды: 25 г (0°) и 173 г (100°). Ди аммоний-
фосф а т (NH4)2HPO4 — менее устойчив, чем моно-
аммонийфосфат; уже ок. 70° заметно теряет аммиак,
переходит в моноаммонийфосфат; растворимость в воде
при 0° почти в 2 раза выше, чем у NH4H2PO4; в р-ре
ок. 80° целиком переходит в моноаммонийфосфат.
Содержит 21,20% N и 53,76% Р2О5. Триаммоний-
фосф а т (NH4)3PO4 неустойчив, уже при 20°
выделяет NH3; давление паров аммиака при 27,5°
достигает 129 мм рт. ст.; водные р-ры имеют сильно
щелочную реакцию.
Получают А. ф. нейтрализацией фосфорной к-ты
аммиаком. Основное применение А. ф., гл. обр.
NH4H2PO4, в качестве минерального удобрения,
т. н. аммофоса; в небольших количествах исполь-
зуется и диаммонийфосфат — для приготовления пи-
щевых дрожжей, для фосфатирования металлов;
диаммонийфосфат — один из лучших антипиренов.
Лит. см. при ст. Азотные удобрения. В. А. Нлевке.
АММОНИЯ ФОСФОМОЛИБДАТ (аммоний фосфор-
номолибденовокислый) (N Н4)3Н4[Р(Мо2О7)6]жН2О,
мол. в. 1912,53 (безводного) — желтые кристаллы,
порошок, при нагревании разрушается; растворим
в щелочах, слабо растворим в холодной и горячей
воде; нерастворим в спирте и кислотах. Получают
А. ф. взаимодействием молибдата аммония с фосфор-
ной к-той в азотнокислой среде, очищают кристалли-
зацией. Применяют для обнаружения Sn; в качестве
стандарта при фотометрия, определении 1’0%; для
открытия и выделения алкалоидов осаждением.
Д. И. Кузнецов.
АММОНОЛИЗ — реакция обменного разложения
между органич. соединениями, содержащими поляр-
ную связь, и аммиаком. Типичным примером А. мо-
жет служить получение амидов карбоновых к-т дей-
ствием аммиака на хлорангидриды или ангидриды
к-т: RCOC1 + 2NH3—> RCONH2 + NH4C1; RCOOR+
+ NH3 —> rconh2 + с2н5он.
А., подобно гидролизу и алкоголизу, представляет
собой сольволитич. реакцию (см. Сольволиз), проте-
кающую по механизму нуклеофильного замещения.
Получение аминов жирного ряда взаимодействием ал-
килгалогенидов с водным NH3 (см. Гофмана реакция)
или же спиртов с NH3 в паровой фазе над дегидрати-
рующим катализатором (окись алюминия, фосфат
алюминия и др.) также может рассматриваться как
реакция А. Ароматич. галогенопроизводные реагируют
с NH3 лишь в жестких условиях. Так, хлорбензол
превращается в анилин нагреванием с избытком
водного NHS при 160—210° в присутствии Сп2О в ка-
честве катализатора. Сравнительно легко протекает А.
ароматич. нитрогалогенопроизводных, напр. превра-
щение н-нитрохлорбензола в н-нитроанилин. К легко
протекающим процессам А. с применением водного
NH3 относится превращение нафтолов в нафтиламины
(см. Вухерера реакция), превращение окиси этилена
в этаноламины и др.
Лит.: Kirk, v. 1, N. Y., 1947, p. 826 — 844.
Я. Ф. Комиссаров.
АМОРФНОЕ СОСТОЯНИЕ — состояние твердого
тела, характеризующееся след, признаками: 1) изо-
тропией формы, а также оптич., механич., электрич.
и других физич. свойств (т. е. их независимостью
от направления); 2) отсутствием четко выраженной
температурной точки плавления. По совокупности этих
признаков аморфные тела противоположны кристал-
лическим. Второй признак обычно позволяет легко
отличить А. с. от поликристаллического (см. Кристал-
лическое состояние, Анизотропия).
Указанные свойства аморфных тел объясняются
особенностями их строения. Для кристаллов харак-
терно строгое повторение одного и того же элемента
структуры (атома, атомной группы, молекулы) в трех
измерениях на протяжении сотен и тысяч периодов,
называемое дальним порядком. Для аморф-
ных же тел характерно наличие лишь ближнего
порядка в расположении частиц, возникающего
вследствие того, что, обладая конечными размерами,
атомы и молекулы тесно располагаются друг отно-
сительно друга (см. Порядок ближний и дальний).
Если бы вектор г, соединяющий центры двух частиц,
принимал любые значения, то вещество не обладало бы
никаким порядком в расположении частиц. Однако
атомы обладают конечными размерами, и, следова-
тельно, даже в простейшем случае шаровидных атомов
213
АМОРФНОЕ СОСТОЯНИЕ АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
214
должен иметь место простейший ближний порядок,
выражающийся в том, что нек-рые значения г встре-
чаются чаще, чем другие. Максимальное число центров
соседних атомов, окружающих данный атом, будет
располагаться на расстоянии, приблизительно рав-
ном 2г. Очевидно, что ближний порядок скажется
не только на ближайших соседях, но и наследующих.
По мере удаления от рассматриваемого атома ближ-
ний порядок размывается, переходя в беспорядок,
т. е. в равновероятность всех межатомных расстоя-
ний. Картина ближнего порядка усложняется для
частиц более сложной формы, в частности молекул
органич. веществ.
Ближний порядок в расположении частиц харак-
терен также и для жидкостей, но в жидкостях проис-
ходит обмен местами между соседними частицами,
затрудняющийся по мере возрастания вязкости.
Хотя и предполагается, что в аморфном твердом теле
такого обмена нет, однако четкой границы между
жидким состоянием и аморфным провести невозможно.
Аморфное тело принято рассматривать в наст, время
как переохлажденную жидкость с очень высоким
коэфф, вязкости. Иными словами, А. с. не является
термодинамически устойчивым состоянием.
В природе А. с. не столь распространено, как кри-
сталлич. В аморфном виде всегда находятся естест-
венные и искусственные смолы. Нек-рые вещества
при одних условиях могут быть получены в А. с.,
при других — в кристаллич. Возможно, напр., за-
кристаллизовать глицерин, хотя и с большим трудом;
путем расплавления кристаллов кварца и последую-
щего охлаждения легко получить аморфное кварцевое
стекло. Однако подавляющее большинство веществ
получить в А. с. трудно или вообще невозможно.
Одним из типичных представителей аморфных тел
является обычное силикатное стекло, поэтому А. с.
часто называют стеклообразным. При подо-
гревании стекло размягчается и постепенно (непре-
рывно) переходит в жидкое состояние. В обычных
условиях стекло в А. с. может существовать неопре-
деленно долгий срок, но при нек-рых условиях про-
цессы кристаллизации ускоряются, в результате чего
стекло мутнеет.
Разделение твердых тел только на две группы —
аморфные и кристаллич. — является недостаточным.
Это следует хотя бы из факта сущестиования таких
состояний в-ва, как жидкие кристаллы и т. н. газо-
кристаллы. Последние (к ним относятся высокотемпе-
ратурные фазовые модификации камфоры, этана,
гексахлорэтана, циклогексана, нек-рых парафинов
и т. п.) характеризуются наличием дальнего порядка
в расположении центров или осей частиц и беспоряд-
ком в их поворотах. Своеобразные состояния могут
возникнуть в многокомпонентных системах. Так,
в сплавах железо — кобальт дальний порядок в рас-
положении центров всех частиц может сочетаться
с, «сортовым» беспорядком в расположении соседних
атомов.
В литературе о полимерах термин «А. с.» употреб-
ляется, как правило, по отношению к телам, не яв-
ляющимся кристаллич. Однако своеобразие струк-
туры полимеров приводит к необходимости разра-
ботки более детальной классификации. Степень упо-
рядоченности в структурах полимеров может быть
самой различной. Для линейных макромолекул эта
упорядоченность возникает прежде всего вследствие
тенденции длинных молекулярных цепей уклады-
ваться в параллельные образования (пачки). Внутри
самой пачки можно встретиться с различными типами
расположения цепей, отличающимися упорядочен-
ностью взаимного расположения осей молекул, упо-
рядоченностью относительных продольных сдвигов
молекул, а также их азимутальных поворотов. При-
мером кристаллич. порядка в полимерах может слу-
жить полиэтилен, медленно закристаллизованный из
раствора. Полиакрилонитрил обладает структурой
газокристаллич. типа. Малую упорядоченность моле-
кул внутри пачек имеет полистирол. Очень своеоб-
разна структура целлюлозы. Она представляет собой
непрерывную систему параллельных молекул, уло-
женных в нестрогом трехмерном порядке. По мере
удаления цепей друг от друга корреляция ухуд-
шается. В пределах области 50—100 А имеется срав-
нительно правильная трехмерная кристаллич. ре-
шетка, но смещения отдельных цепей от «правильных»
положений достигают 1А.
Лит.: Т а м м а н Г., Стеклообразное состояние, пер.
с нем., Л. — М., 1935; К о б е к о П. П., Аморфные вещества,
М. —Л., 1952; Китайгородский А. И., Рентгено-
структурный анализ мелкокристаллических и аморфных тел,
М. — Л., 1952; его же. Структура полимеров, М., 1958;
Френкель Я. И., Собрание избранных трудов, т. 3,
М. — Л., 1959. Ю. В. Мнюх.
АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ (галь-
ванометрическое, полярометрическое, вольтамперное
титрование) — метод колич. анализа, в к-ром конеч-
ную точку титрования находят по изменению в про-
цессе титрования силы предельного диффузионного
тока, проходящего через раствор при постоянном
напряжении между индикаторным электродом и
неполяризующимся электродом сравнения. А. т.
является видоизменением полярографического метода
анализа (см. Полярография). При А. т. строят график
зависимости силы тока от объема добавленного рабо-
чего р-ра (с учетом поправки на разбавление титруе-
мого р-ра реактивом) и находят точку пересечения
двух ветвей кривой (прямых линий), соответствующую
конечной точке (точке эквивалентности, ТЭ). Выбор
материала индикаторного электрода определяется
коррозийной устойчивостью его в условиях титрова-
ния и напряжением электролиза.
Если титруемое вещество восстанавливается или
окисляется на индикаторном, электроде, то по мере
его удаления из раствора при реакциях осаждения,
окисления — восстановления, комплексообразования
или нейтрализации
ток уменьшается. Пос-
ле достижения точки
эквивалентности ток
принимает постоян-
ное значение, зави-
сящее от электро-
проводности среды
(кривая 1). В том слу-
чае, когда в электрод-
ной реакции участву-
ют оба компонента,
ток меняется как до,
так и после достиже-
ния ТЭ. Кривая ти-
трования состоит из
ТЭ{ ТЭг ГЭ3 мл прибавленного
р-ра
в ТЭ прямых линий, напр.,
двух пересекающихся
если оба вещества окисляются или восстанавли-
ваются при данном потенциале, то ТЭ отвечает
минимуму тока (кривая 2). За ходом А. т. можно
следить также по изменению силы тока восстановления
(окисления) продукта реакции или к.-л. третьего ве-
щества, взаимодействующего с одним из компонентов
реакции, т. н. «индикаторный» метод (кривая 3).
Разработаны также методы А. т. с применением двух
поляризуемых электродов. Обычно в качестве ивди-
каторного электрода применяют ртутный капельный
электрод (восстановление ионов электроотрицатель-
ных металлов, анионов, органич. соединений) или
вращающийся платиновый электрод (анодные реак-
ции окисления, восстановление сильных окислите-
лей, ионов благородных металлов и т. д.). А. т. дает
215
АМФИ — AHA
216
возможность количественно определять малые кон-
центрации вещества 10“2—10~6 жоль/л в присутствии
сравнительно больших количеств индиферентных
электролитов. Последние даже специально добавляют-
ся для устранения миграционных токов. Измерения
тока вдали от точки ТЭ позволяют исключить ошибку
за счет растворимости осадка. Анализ можно прово-
дить в мутных и окрашенных р-рах. В отличие от
полярографического анализа, точность А. т. не зави-
сит от характеристики электрода и среды (диаметр
ртутного капилляра, вязкость, темп-ра и т. д.), если
они не меняются в процессе титрования. Метод А. т.
разработан для определения большинства химич.
элементов. Метод предложен в 1927 Я. Гейровским.
Лит.: Сонгина О. А., Амперометрическое (поляро-
метрическое) титрование в анализе минерального сырья, М.,
1957; Кольтгоф И. М., Лингейм Дж. Д ж., Поля-
рография, пер. с англ., М. — Л., 1948; Milner G. W. С.,
The principles and applications of polarography and other
electro analytic al processes, L., [a. o.J, 1957; Крюкова T. A.,
Синякова С. И., Арефьева T. В., Полярографиче-
ский анализ, M., 1959, с. 505—572. А. Н. Чемоданов.
АМФИ — приставка, указывающая на наличие
двух заместителей в молекуле нафталина в положениях
2,6, напр. ам§5и-диметилнафталин (I), ам§5и-нафто-
хинон (II) и т. д. Эта же приставка применяется
для обозначения одного из трех возможных стерео-
изомеров а-дикетодиоксимов. Так, диоксимбензил
конфигурации (III) называют амд5и-диоксимом, в
отличие ОТ син- И анти-изомеров. Я. Ф. Комиссаров.
АМФОТЕРИЦИНЫ — антибиотики группы анти-
биотиков-полисное. Известны амфотерицины А и В;
в первом содержатся 4 сопряженные двойные связи,
во втором — 7. Брутто-формула амфотерицина
В C46H73NO20.
Амфотерицин А Амфотерицин В
Т. разл., °C 153 170
Эквивалент нейтрализации . . 915 929
[а]д’5 в кислом р-ре диметил-
формамида +32° +33,3°
в 0,1 н. СН3ОН-НС1 ...... —9,9° —33,6°
А. нерастворимы в безводных этаноле, н- и изо-
пропаноле, ацетоне, простых и сложных эфирах,
ароматич. и хлорированных углеводородах; А. рас-
творимы в нейтральном и подкисленном диметил-
формамиде, безводном СН3ОН, ледяной СН3СООН,
пропиленгликоле и др. растворителях. Кристаллич.
амфотерицин А образует водорастворимую Na-соль.
С СаС12 А. образуют комплексы: комплекс с амфо-
терицином А растворим в СН3ОН, с амфотерицином
В — нерастворим. А. амфотерны; амфотерицин А
стабилен при pH 6,0—7,0; амфотерицин В разлагается
в области значений pH 4,0—10,0. ИК-спектры погло-
щения обоих А. почти аналогичны. УФ-спектр погло-
щения амфотерицина В характерен для конъюги-
рованного гептаена, а амфотерици-
ОН ОН на А — для тетраенов. При смеше-
}--1 нии с N а-дезоксихолатом амфотери-
/ \—nh Чин В образует водорастворимый
Ч. Г 2 препарат, нейтральный р-р к-рого
\--/ пригоден для внутривенного вве-
Н С ОН дения при лечении микоз. Гидри-
3 рованием амфотерицина В (Pt или
Pd на угле) получено тетрадекагидропроизводное
C46H87NO2o. При ацетилировании амфотерицина В
выделены три- и тетраацетилпроизводные аминоса-
хара микозамина (I), к-рый содержится также в
молекуле нистатина.
А. образуются актиноминомицетом streptomyces
М-4575; они мало растворимы в воде и выделяются из
цельной культуральной жидкости или из отфильтро-
ванного мицелия экстракцией органич. растворите-
лями, смешивающимися с водой; обычно пользуются
низшими алифатич. спиртами, напр. изопропанолом.
А. могут быть разделены и получены в кристаллич.
виде при обработке технич. препаратов, являющихся
смесью амфотерицинов А и В, подкисленным диметил-
формамидом и СН3ОН и последующим фракционным
осаждением водой.
А. активны по отношению к нек-рым патогенным
грибам, в том числе к Candida albicans; не действуют
на бактерии. Амфотерицин В более активен, чем ам-
фотерицин А, в отношении дрожжей и дрожжеподоб-
ных грибов. Применяют А. при лечении нек-рых
грибковых заболеваний — микозов, в частности
бластомикозов.
Лит.: Sternberg Т. Н., Wright Е. Т. and О и-
r а М., в кн.: Antibiotics annual. 1955—1956, N. Y., 1956,
p. 566; Steinberg B. A., J a m b о r W. P. and S u-
y d m L. О., там же, p. 574; Gold W. la. о.], там же, p. 579;
V andeputt e J., W a c h t e 1 J. L., S t i 1 1 e r E. T.,
там же, p. 587; Dutcher J. D. (а. о.]; там же, 1956—1957,
N. Y., 1957, p. 866; Verona O., Ann. microbiol., 1957,
V. 7, p. 214; Harrell E. R. and Curtis A. C., Arch.
Dermatol., 1957, 76, № 5, p. 561; Walters D. R., Dut-
cher J. D., Wintersteiner O., J. Amer. Chem. Soc.,
1957, 79, № 18, p. 5076; В a r t n e r E., [a. o.l, в кн.: Antibio-
tics annual. 1957—1958, N. Y., 1958, p. 53.
Д. M. Трахтенберг.
АМФОТЕРНОСТЬ — способность нек-рых соеди-
нений проявлять, в зависимости от условий, как
кислотные, так и основные свойства. Кислотность и
основность любого химич. соединения зависят от
природы второго компонента, участвующего в кислот-
но-основном взаимодействии. Одно и то же вещество
при взаимодействии с сильными кислотами имеет
тенденцию проявлять свои основные свойства, тогда
как в реакциях с сильными основаниями оно может
вести себя как кислота. Так, напр., слабая борная
к-та Н3ВО3 при взаимодействии с более сильной орто-
фосфорной к-той Н3РО4 дает соль ВРО4, проявляя
тем самым основные свойства. Даже H2SO4 — силь-
ная кислота, в р-ре HF присоединяет протон с обра-
зованием иона H3SO4+, выполняя функцию основа-
ния. С другой стороны, Fe(OH)3 — основание, но при
его сплавлении с содой или поташом образуются
ферриты (напр., NaFeO2). Типичным примером амфо-
терного соединения может служить вода, образую-
щая при диссоциации ионы Н+ и ОН . Амфотерны
гидроокиси Al, Zn, Сг и нек-рых др. элементов. При
переходе в р-р они диссоциируют, в зависимости от
условий, с образованием ионов Н4" или ОН , напри-
мер Сг3+ + ЗОН Сг(ОН)3 _ Н- + СгО2~ + Н2О.
Примером гидроокиси с ярко выраженными амфотер-
ными свойствами («почти идеальная» А.) служит
Ga(OH)3, для к-рого константы диссоциации в вод-
ном р-ре по кислотному и основному типу почти равны.
Преобладание того или иного типа свойств в ряду
гидроокисей зависит от положения соответствующего
элемента в периодич. системе (см. Гидроокиси).
Изложенное выше понятие «А.» относится к прото-
литич. теории кислот и оснований Бренстеда. В теории
Льюиса понятие «А.» сохраняет свое значение,
однако А. в этом случае обусловлена способностью
данного соединения присоединять или отдавать элек-
трон с образованием гомеополярной связи (см. Кислот
и оснований теории).
Лит.: Шатенштейн А. И., Теория кислот и основа-
ний, М. — Л., 1949.
АНА — приставка, указывающая на наличие двух
заместителей в нафталине в положениях 1,5, напри-
217
АНАБАЗИН — АНАЛИТИЧЕСКАЯ ХИМИЯ
218
мер ана-диметилнафталин (I). Эта же приставка
применяется для обозначения положения 5 в хино-
I II
лине (II), т. е. при положении 5 по отношению к
атому азота. Я. Ф. Комиссаров.
АНАБАЗИН (1-а-пиперидил-Р-пиридин) C10H14N2,
мол. в. 162,23 — главный алкалоид среднеазиатского
дикорастущего растения ежовника
| | безлистного (Anabasis aphylla), сем.
s' маревых (Chenopodiaceae), в неболь-
|| | ших количествах встречающийся
Н также в табаке, впервые выделен
14 А. П. Ореховым в 1929. А. — мас-
лянистая бесцветная жидкость, темнеющая на воз-
духе; т. пл. 9°, т. кип. 276°/760 мм, 104—105°/2 мм;
Jsj; 1,0455; ngl,5430; [а]55 = —82° (без растворителя);
хорошо растворим в воде и органич. растворителях;
с водяным паром перегоняется с трудом. По химич.
свойствам А. — вторично-третичное основание, его
водные р-ры имеют щелочную реакцию. Наиболее
характерные производные А. — дипикрат, т. пл. 202—
205°, дипикролонат, т. пл. 235—237°; фторосиликат,
т. пл. 239°; хлораурат, т. пл. 180—182°. Синтезиро-
ван А. из Р-цианпиридина и 4-метокси-1-магний-
иодбутана:
(d-CsH^N-CN + JMg(CH»)4OCII3
— ₽-C5H6N-CO(CH3)4OCHs NHsOH>
— p-C5H5N-C(=NOH) (CHS)4OCH3 S.
— p-C5H5N-CH(NH2) (CH3)4OCH3 —
— [P-C5H5N-CH(NH3)(CH.)4Br • HBr] —5.-5- A
Разделение рацемата синтетич. А. произведено
с помощью 1-6,6-динитро-2,2-дифеновой к-ты. Выде-
ление А. из растит, сырья (в виде сульфата) произ-
водится экстракцией водой и керосином. По физио-
логия. активности А. представляет собой «ганглио-
нарный яд». Вследствие сильной токсичности в меди-
цине не применяется, но в ветеринарии используется
для лечения стригущего лишая и против вшивости
у животных. Сульфат суммы алкалоидов анабазиса
(в основном — А.) используется как инсектицид для
опрыскивания плодовых и овощных культур. А. мо-
жет служить сырьем для получения никотиновой к-ты.
Лит.: Садыков А. С., Химия алкалоидов. Anabasis
aphylla, Ташкент, 1956; см. также при ст. Алкалоиды.
А. Е. Васильев.
АНАЛИЗ (в химии) — о конкретных методах анализа
см. Бесстружковый метод анализа, Весовой анализ,
Газовый анализ, Качественный анализ, Количествен-
ный анализ, Объемный анализ, Почвенный анализ,
Радиоактивационный анализ, Р ентгеноспектралъный
анализ, Термический анализ, Фазовый анализ, Физико-
химический анализ и др.
АНАЛИТИЧЕСКАЯ ХИМИЯ — наука о методах
определения состава вещества. В соответствии с этой
задачей различают качественный анализ и количест-
венный анализ. В зависимости от объекта исследова-
ния А. х. принято разделять на неорганиче-
ский анализ и органический анализ.
Разделение А. х. на качественный и колич. анализ
вызвано гл. обр. различием задач, а также методов
их решения. В качественном неорганич. анализе для
открытия ионов в р-рах обычно используют различ-
ные характерные р-ции, к-рые сопровождаются к.-л.
хорошо заметными для наблюдателя эффектами, напр.
появлением окрашивания при добавлении реагента
к бесцветному испытуемому р-ру или резким измене-
нием цвета окрашенного р-ра; выделением осадка
или газа. Используются и иные характерные изме-
нения, наступающие при реакциях, а также спектры
испускания, потенциалы восстановления на ртутном
капельном электроде и т. д. В качеств, органич. ана-
лизе применяют спец, приемы исследования (см.
Элементарный анализ, Функциональный анализ). Ко-
личество элемента, находящегося в форме простого
в-ва или к.-л. соединения, ойределяют путем измере-
ния массы (веса) или к.-л. другой физич. величины
(см. таблицу).
Классификация методов количественного анализа
Измеряемая физич. величина (свойство) Методы Количество веще- ства, доступное определению*
Масса. Вес Весовые (гравиме- трические) Масс-спектр альные Макро-, микро-и ультрамикроко- лнчества Микроколичест- ва
Объем Титриметрические Газоволюметриче- ские Объемно-седимен- тометрические (по объему осадков) . 1 Макро-, микро- ? и ультрамикро- J количества Макро-, микро- и субмикроколи- чества
Плотность Денсиметрические Макро- и микро- количества
Поглощение и ис- пускание ИК-лучей. Колебания молекул Инфр акрасная спектроскопия Комбинационное рассеяние То же То же
Поглощение и ис- пускание видимых, УФ-и рентгеновских лучей. Колебания атомов. Рассеяние света Спектральные, фо- тометрия пламени Рентгеноспектраль- ные Фотометрические (колометрия и спек- трофотометрия, тур- бидиметрия, нефело- метрия) Люминесцентные и флуоресцентные Полумикро- и микроколичества Микро- и полу- микроколичества Полумикро- и микроколичества Микроколичест- ва
Показатель пре- ломления Рефр актометриче- ские интерферомет- рические Макроколичест- ва
Вращение плоско- сти поляризации Поляриметриче- ские То же
Сила диффузионно- го тока при восста- новлении или оки- слении на электроде Полярографиче- ские (вольтампер- ные) Полу микро- и микроколичества
Количество элек- тричества для элек- тродной реакции Кулонометриче- ские Микро- и суб- микроколичества
Электродный по- тенциал Потенциометриче- ские Макро- и мик- роколичества
Электропровод- ность Кондуктометриче- ские, включая вы- сокочастотное тит- рование То же
Диэлектрическая проницаемость — Микроколичест- ва
Магнитная воспри- имчивость — То же
Р адиоактивность Метод радиоактив- ных индикаторов Радиоактивацион- ный Макро-, микро- и субмикроколи- чества Микро- и суб- микроколичества
219
АНАЛИТИЧЕСКАЯ ХИМИЯ — АНАЛЬГИН
220
Продолжение
Измеряемая физич. величина (свойство) Методы * Количество веще- ства, доступное определению **
Скорость реакции Кинетические ме- тоды Макро- и мик- роколичества
Тепловой эффект реакции Тер мометрические Макроколичест- ва
Вязкость и теку- честь Вискозиметриче- ские Макроколичест- ва
Поверхностное на- тяжение Тензиметрические То же
Взаимная смеши- ваемость жидкостей — То же
Понижение точки замерзания, повыше- ние точки кипения, осмотическое давле- ние, упругость пара Криоскопич еские Э б ул и ос ко пич е- ские То же То же
* Черточки означают отсутствие специального или обще-
принятого названия. ** Макроколичества 0,05—0,5 г; полу-
микроколичества 0,01—0,05 а; микроколичества 0,1—10 мг\
ультрамикроколичества 10—100 мкг, субмикроколичества —
менее 10 мкг (градации условные).
Очевидно, между количеством элемента или в-ва
(или их концентрацией) и измеряемой физич. вели-
чиной должна существовать определенная функцио-
нальная зависимость, выражаемая аналитически или
графически. При весовых и титриметрич. методах
прямо пропорциональная зависимость соблюдается
между колич. вещества (массой) и, соответственно,
весом или объемом при любых его количествах (см.
Весовой анализ, Объемный анализ, Титриметрический
анализ). В спектральных методах (см. Спектральный
анализ) пропорциональная зависимость в известных
пределах соблюдается между колич. элемента и интен-
сивностью аналитич, спектральных линий, а во мно-
гих фотометрич. методах анализа в известных гра-
ницах — между концентрацией в-ва и светопогло-
щением (см. Поглощение света).
Измерению количества элемента (в-ва) обычно
предшествует химич. подготовка, к-рая состоит в обо-
гащении и отделении определяемого элемента в виде
подходящего соединения, удобного для соответств.
измерения. В связи с недостаточной селективностью
и чувствительностью многих методов измерения колич.
в-ва большое значение имеют методы групповых раз-
делений и методы выделения отдельных элементов
путем осаждения, перегонки, экстракции и др. (см.
также Чувствительность аналитических методов,
Чувствительность реакции). Осаждение и экстракцию
многих сопутств. ионов можно предупредить превра-
щением их в устойчивые комплексные ионы (см. Мас-
кировка). Выделить из р-ра определяемый элемент
в свободном виде или в форме окиси можно также
электролизом. Подбирая соответств. условия, удается
одни элементы осадить на электроде, а другие оста-
вить в р-ре. Для выделения небольших количеств
многих металлов применяется метод внутреннего
электролиза. Одним из наиболее эффективных мето-
дов разделения является хроматография (см. Хрома-
тография).
В органич. анализе колич. определению элементов
обычно предшествует разложение в-ва (минерализа-
ция) до СО2, Н2О, N2, NH3 и др. (аналитич. форма).
Дальнейшее определение выполняется методами не-
органич. анализа. Колич. элементарный анализ дает
брутто-формулу соединения. При установлении строе-
ния органич. соединений пользуются функциональ-
ным анализом, осуществляемым с помощью характер-
ных реакций на определенные функциональные груп-
пы: аминогруппу, алкоксигруппу, активный водород,
кратные связи и т. д. (см., напр., Ван-Слайка метод,
Активный водород, Иодное число).
Весьма актуальна дальнейшая разработка вещест-
венного анализа (химич. фазового анализа). Вещест-
венный неорганич. анализ принадлежит к числу
наименее разработанных отделов аналитич. химии.
Веществ, анализ имеет большое значение для минера-
логии, металлургии, металловедения, силикатной
пром-сти, технологии отдельных произ-в, для к-рых
уже недостаточны данные обычного общего химич.
анализа, а нужны качеств, и колич. данные о реаль-
ных формах соединений в природных и технич. ма-
териалах.
В связи с необходимостью определения следов эле-
ментов в природных и промышленных объектах про-
водятся исследования по повышению чувствительно-
сти аналитич. методов. Приобретают все большее зна-
чение микрохимии., ультрамикрохимич., а также но-
лумикрохимич. методы (см. Микрохимический анализ,
Полумикроанализ, Улътрамикрохимический анализ).
Дальнейшее развитие А. х. во многом зависит от
разработки теоретич. основ этой науки, от разработки
быстрых и точных методов открытия и определения
любых количеств элементов и соединений независимо
от присутствия др. элементов. Применение спектраль-
ного анализа, спектрофотометрии, использование
радиоактивных индикаторов и других физич. методов
открывает путь к автоматизации методов анализа и
управлению технология, процессами.
Лит.: Ко марь Н. П., Основы качественного химиче-
ского анализа, ч. 1, Харьков, 1955; Алексеев В. Н.,
Курс качественного химического полумикроанализа, 3 изд.,
М., 1958; Тредвелл Ф. П., Г о л л В. Т. Курс аналити-
ческой химии, пер. с англ., т. 1 — Качественный анализ, 9 изд.,
М. — Л., 1946; Петр ашень В. И., Качественный хими-
ческий анализ, 6 изд., М. — Л., 1948; Блок Н. И., Качест-
венный химический анализ, М.— Л., 1952 ; К о л ьтг оф И.М.»
Сендэл Е. Б. Количественный анализ, пер. с англ,,
3 изд., М. — Л.,1948; Б а б к о А. К. иПятницкийИ.В.,
Количественный анализ, М., 1956; К л я ч к о Ю. А.,
Шапиро С. А., Курс химического качественного анализа,
М., 1960; К о л ь т г о ф И. М. [и др.], Объемный анализ,
пер. с англ., т. 1—3, М.—Л. 1950—61; Али мар ин И. II.,
Петр ико ва М. Н., Неорганический ультрамикроана-
лиз, М., I960; Ляликов Ю. С., Физико-химические ме-
тоды анализа, 2 изд., М., 1951; Юинг Г. В., Инструмен-
тальные методы химического анализа, пер. с англ., М., I960;
Гиллебр а н д В. Ф. [и др.], Практическое руководство
по неорганическому анализу, пер. с англ., 2 изд., М., 1957;
Анализ минерального сырья, под ред. Ю. Н. Книповича
и Ю. В. Морачевского, 3 изд., Л., 1959; Chariot G.,
Les methodes de la chimie analytique, Analyse quantitative
minerale, P., 1961; НидерльД. и Ни дер ль В., Микро-
методы количественного органического анализа, пер. с англ.,
М.—Л., 1949; Коршун М. О. и Гельман Н. Э.,
Новые методы элементарного микроанализа, М.—Л., 1949;
Houben — Weyl, Methoden der organischen Chemie, Bd 4,
TI 2, Stuttgart, 1955; В а й б e л ь С., Идентификация орга-
нических соединений, пер. с англ., М., 1957; Бауэр К. Г.,
Анализ органических соединений, пер. с нем 2 изд., М.,
1953; Б ус е в А. И., Аналитическая химия, литература на
русском языке (1941—1952), М., 1956; Handbuch der analy-
tischen Chemie, hrsg. von W. Fresenius und G. Jander, Tl 2—3,
B. —Gottingen— Hdlb., 1940—58; Chariot G., L’ana-
lyse qualitative et les reactions en solution, 4 ed., P.. 1957
(англ. nep. — Qualitative inorganic analysis. N. Y. — L.,
1954); Журналы; Ж. аналит. химии, с 1946; Завод лаб., с 1932;
Z. analyt. Chem., с 1862; Analyst, с 1877; Analyt. Chem.,
с 1929- Analyt. Chimica Acta, c 1947; Taianta, c 1958; Analy-
tical Abst., c 1954. См. также лит. при отдельных методах
анализа. А. И. Бусев.
АНАЛЬГИН, мол. в. 351,36 — мелкие бесцветные
кристаллы; растворим в воде (1 : 1,5), мало растворим
в спирте, нерастворим
в эфире. Водный р-р н с с C_N/CH3
А. (1 : 10) прозрачен 3 | | \cH20SO2Na-H2O
и нейтрален на лак- Н3С—N СО
Мус и фенолфталеин, ^N— С6Н5
при стоянии желтеет,
не утрачивая фармакология, активности. При нагре-
вании р-ра А. в воде, подкисленной разб. соляной
221
АНГЕЛИКОВАЯ КИСЛОТА — АНГИДРОСАХАРА
222
к-той, выделяется SO2, а затем формальдегид. При
прибавлении к р-ру А. 5%-ного р-ра хлорной изве-
сти или хлорамина появляется синее окрашивание,
переходящее при нагревании в желтое. Для колич.
определения А. его водный р-р нагревают с 0,1 н.
р-ром иода, подкисляют соляной к-той и осаждают
ВаС12. Получают А. метилированием бензилиден-
аминоантипирина и взаимодействием образовавше-
гося 4-монометиламиноантипирина с формалином и
NaHSO3. Применяют А. как противовоспалитель-
ное и жаропонижающее средство, близкое по харак-
теру действия к пирамидону.
‘Лит.: Преображенский Н.А. иГенкинЭ.И.,
Химия органических лекарственных веществ, М. — Л., 1953;
см. Также при ст. Адалин. Е. С. Головчинекая.
АНГЕЛИКОВАЯ КИСЛОТА (1-метилкротоновая,
1,2-диметилакриловая, 1-этилиденпропионовая кис-
лота), мол. в. 100,12 — бесцветные кристаллы; ла-
бильная транс-форма одного из двух геометрия,
изомеров; второй изомер — стабильная цис-форма,
наз. тиглиновой к-той.
н—С—СН3 Н.С-С-П
II II
Н3С—С--СООН и3с с-соон
ангеликовая к-та тиглиновая к-та
Т. пл., °C Т. кип., °C t nD К -10-5 (25°)
Ангеликовая кислота 45 185 0,9539 1,4434 (47°) 5,0
Тиглиновая кислота. . 65 198,5 0,9641 1,4329 (47”) 0,96
А. к. перегоняется с паром и кристаллизуется
в моноклинных призмах, тиглиновая к-та кристалли-
зуется в триклиния, призмах. А. к. встречается
в виде бутилового и амилового эфиров вместе с вале-
риановой к-той в корне дягиля, в масле римской
ромашки; тиглиновая к-та — в кротоновом масле;
образуется при расщеплении многих природных сое-
динений. А. к. отделяется от валериановой к-ты пере-
кристаллизацией, от тиглиновой — растворением в хо-
лодной воде ее кальциевой соли (растворяется лучше
Са-соли тиглиновой к-ты). Обе к-ты получаются
синтетически дегидратацией а-оксиметилмасляной
к-ты. Тиглиновую к-ту можно получить с хорошим
выходом конденсацией метилмалоновой к-ты с параль-
дегидом. При нагревании, а также под действием
конц. H2SO4 и разб. щелочей при 100° А. к. изомери-
зуется в тиглиновую к-ту.
Лит.: Buckles R. Е., Mock G. V., Loc at el I L.,
Chem. Revs, 1955, 55, № 4, p. 659. M. И. Розенга-рт.
АНГИДРИДЫ (ангидриды кислот) — кислородные
соединения, к-рые могут быть получены отнятием
воды от кислородных к-т, превращаются в к-ты при
взаимодействии с водой. К А. относятся окислы эле-
ментов, обладающих неметаллич. свойствами, а также
высшие окислы нек-рых тяжелых металлов (см.
Гидроокиси). А. неорганич. к-т получают: 1) взаимо-
действием простых веществ с кислородом (напр.,
образование угольного А. СО2, фосфорного Р2О5,
сернистого SO2 при горении угля, фосфора или серы);
2) отнятием воды от к-т нагреванием (напр., 2Н3ВО3 =
= ЗН2О + В2О3; 2HNO3 + Р2О5 = 2НРО3 + N2O5);
3) разложением неустойчивых в свободном состоянии
к-т (напр., СаСО3 + 2НС1 = СаС12 + Н2СО3; Н2СО3=
= Н2О + СО2). Многие неорганич. А., напр. Р2О5,
используются как осушители. О свойствах важней-
ших А. см. Азота окислы, Мышьяка окислы, Серы
окислы, Фосфора окислы, Хрома окислы и др.
АНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ — пр оду к-
ты отщепления воды от карбоновых к-т: 2RCOOH—>
—с (RCO)2O + Н2О. Из двухосновных кислот могут
образоваться циклич. «внутренние» ангидриды, на-
пример янтарный ангидрид
сщсоон сн5со
I _> о+н2о
СИзСООИ 1НзС0/
Название «А. к. к.», например уксусный ангидрид
(СН3СО)2О, бензойный ангидрид (С6Н5СО)2О, обыч-
но производится от названия исходной к-ты. Простей-
ший А. к. к., отвечающий муравьиной к-те, чрезвы-
чайно нестоек; смешанный муравьино-уксусный ан-
гидрид СНдСОООСН сравнительно устойчив. А. к. к.
гидролизуются с образованием к-т, с галогеноводоро-
дами они дают галогенангидриды к-т, с аммиаком —
амиды к-т, со спиртами — сложные эфиры карбо-
новых к-т.
Наиболее общий способ получения А. к. к. состоит
во взаимодействии солей карбоновых кислот с их
галогенангидридами, а также с галогенангидрида-
ми минеральных кислот (РОС13, SO2’C12, HOSO2C1
и др.); RCOONa + RGOGl^RGOOGOR; 2RCOONa +
+ SOC12 —► (RCO)2O + 2NaCl + SO2. Ангидриды мно-
гих к-т, напр. фталевой и янтарной, могут быть полу-
чены непосредственно дегидратацией к-т при нагре-
вании. Один из технически важных способов получе-
ния уксусного ангидрида состоит в термич. расщеп-
лении перегретого пара уксусной к-ты. Известны
и особые методы получения нек-рых А. к. к. Так,
получил промышленное значение метод синтеза уксус-
ного ангидрида присоединением кетена к уксусной
к-те: СНдСООН + СН2=СО — (СН3СО)2О. При дей-
ствии кетена на другие карбоновые к-ты образуются
смешанные ангидриды CH3COOCOR.
А. к. к., наряду с хлорангидридами к-т, широко
применяют для ацилирования, напр. уксусный ангид-
рид в произ-ве ацетилцеллюлозы, ацетанилида, аспи-
рина и др. Фталевый ангидрид служит исходным про-
дуктом для получения некоторых синтетич. смол,
красителей и пластификаторов. В последние годы Для
облегчения процессов ацилирования рекомендовано
добавлять в реакц. смесь ангидрид трифторуксусной
к-ты; эффективность этого приема объясняется спо-
собностью (CF3CO)2O образовывать с другими А. к. к.
смешанные ангидриды CF3COOCOR, обладающие по-
вышенной ацилирующей способностью.
Я. Ф. Комиссаров.
АНГИДРОН (перхлорат магния) Mg(C104)2 —
белая пористая масса, в безводном состоянии очень
энергично поглощает влагу (до 60% от своего веса)
с образованием шестиводного кристаллогидрата
[Mg(C104)2 • 6Н2О — игольчатые кристаллы, т. пл.
145—147°]; выше 250° разлагается; растворяется
в воде с выделением теплоты, при поглощении воды
А. не расплывается — преимущество перед Р2О5.
Хранят А. в запаянных ампулах или в очень плотно
закрытых банках с притертыми пробками; получают
прибавлением MgO к 30%-ной НС1О4 до ее насыще-
ния; выкристаллизовывающийся Mg(C104)2 • 6Н2О
обезвоживают сначала при 145—147°, а затем при
220—240° в вакууме. Применяется А. для осушки
газов (Н2, О2, Cl2, N2, HCI, СО2, NH3, H2S и др.); при
микроэлементарном определении водорода; для по-
глощения воды. Регенерацию А. (обезвоживание)
производят в вакууме (0,1 мм рт. ст.) при 240°.
И. П. Ефимов.
АНГИДРОСАХАРА — ангидридные производные
сахаров, в к-рых ангидридный мостик образуется за
счет отщепления элементов воды
от двух спиртовых гидроксилов
молекулы сахара (но не полуаце-
тального гидроксила). Ангидроса-
хара являются внутренними про-
стыми эфирами сахаров; примером
является 3,6-ангидро-а-П-глюко-
пираноза:
в
н он
223
АНДЖЕЛИ - РИМИНИ РЕАКЦИЯ — АНДРОГЕННЫЕ ГОРМОНЫ
224
Аналогичные А. — ангидридные производные в ряду
многоатомных (сахарных) спиртов — наз. а н г и д р о-
альдитолами. Известны А. и ангидроальди-
толы, в к-рых ангидридный мостик замыкает цикл,
содержащий 2, 3, 4 или 5 углеродных атомов. Самыми
многочисленными и лучше всего изученными являются
А. с 3-членным гетероциклом (этиленоксидные или
эпокси-А.) и с 5-членным (тетраметиленоксидные или
гидрофурановые); 3-членные кольца А. легко гидро-
лизуются, и поэтому 2,3-, 3,4- и 5,6-анцидрогексозы
удается получить лишь в виде их производных (гли-
козидов и др.); А. с 5-членными оксидными кольцами
стойки к гидролизу. А. получают синтетически из
галогендезоксисахаров, аминодезоксисахаров й др.
производных моносахаридов. Ряд А. и ангидроаль-
дитолов являются продуктами природного происхож-
дения (стиратитол, полигалитол). А.
широко применяются в разнообразных синтезах
гликозидов, дисахаридов, гуанидинпроизводных са-
харов и др.
Лит. см. при ст. Гликомпы. Л. И. Линевич.
АНДЖЕЛИ — РИМИНИ РЕАКЦИЯ — цветная
реакция на альдегидную группу; заключается в полу-
чении гидроксамовых к-т действием на альдегиды
натриевой соли нитрогидроксамовой или бензосульфо-
гидроксамовой к-ты:
,ОН
RCHO + NaO—NHNOs — RC< + NaNO3
^NOH
Образовавшаяся гидроксамовая к-та c FeCl3 дает
интенсивно красное окрашивание. Салициловый и
аминовалериановый альдегиды, нитробензальдегид и
н-диметиламинобензальдегид, а также алифатич. у-ок-
сиальдегиды не могут быть открыты с помощью этой
реакции. Фенилацетон, дезоксибензоин, бензоин и
нек-рые др. кетоны дают красное окрашивание с
C6H5SO(OH) = NOH и FeCl3. А. — Р. р. предло-
жена для колориметрич. определения малых коли-
честв альдегидов. Открыта в 1896 А. Анджели,
усовершенствована в 1901 Е. Римини и в 1905
А. Анджели.
Лит.: Бауер К. Г., Анализ органических соединений,
пер. с нем., М., 1953. М. А. Марков.
АНДРОГЕННЫЕ ГОРМОНЫ (тестоидные гор-
моны) — группа стероидных гормонов, обладающих
биологич. активностью мужского полового гормона.
Известно неск. десятков природных стероидных в-в,
являющихся андрогенами. Наиболее активные А. г.—
тестостерон (I), андростерон (II), эпиандростерон
(III), дегидроэпиандростерон (IV) и Д4-андростен-3,17-
дион (V).
А. г. регулируют развитие мужских половых
органов, их функцию, влияют на развитие вторичных
половых признаков. Основное действие А. г. соответ-
ствует деятельности семенников (testis), в связи с чем
эти гормоны наз. тестоидными. Андрогенная актив-
ность А. г. проявляется почти на всех животных и
мало зависит от вида. Образование А. г. в половых
железах (гонадах) происходит под стимулирующим
действием гормонов— белков передней доли гипофиза,
т. н. гонадотропных гормонов. А. г. влияют не только
на половую сферу, они являются также регуляторами
роста и необходимы для жизнедеятельности организ-
мов; в женском организме А. г. содержатся в меньших
количествах. . Различают первичные и вторичные
А. г. К первичным относят гормоны, образующиеся
в железах внутренней секреции: у особи мужского
пола — в семенниках и в корковом слое надпочечников,
у особи женского пола — в яичниках и в корковом
слое надпочечников; к первичным А. г. относится
тестостерон. Вторичными называют А. г., к-рые
являются продуктами превращепия в организме
первичных гормонов; содержатся в крови и моче.
В моче обнаруживают большое число стероидных в-в
(более 40 выделено в кристаллич. виде), многие из
к-рых являются продуктами обмена А. г. Большую
группу стероидов мочи составляют кетостероиды
(андростерон, эпиандростерон).
Для выделения А. г. из биологич. жидкостей и тка-
ней обычно используют их растворимость в органич.
растворителях, способность большинства А. г. давать
нерастворимые производные за счет кето-группы (ок-
симы, 2,4-динитрофенилгидразоны и др.), легкость
образования растворимых в воде производных А. г.
с реактивом Жирара (солянокислый триметиламино-
ацетогидразид). Выходы А. г. из животного сырья
очень низки (из 100 кг семенников быка выделяют
лишь 10 мг тестостерона), поэтому большое значение
приобрели методы синтетич. получения А. г. В ка-
честве исходного в-ва в промышленном синтезе А. г.
обычно используются стерины (напр., холестерин,
нек-рые фитостерины). Разработаны синтезы А. г., ме-
ченных радиоактивными изотопами углерода и дей-
терием. Для разделения смеси А. г. и их очистки
широко используются методы хроматографии на
бумаге и на колонках с адсорбентами (А12О3) Для
определения А. г. как качественно, так и количест-
венно используются их цветные реакции с д«-динитро-
бензолом (реакция Циммермана — Бито) и с SbCl3
(реакция Пинкуса). А. г. определяются также поляро-
графия. методом. Наряду с химич. определениями
производят и биологич. испытания на кастрирован-
ных животных (см. Андрогены), используя в качестве
стандарта андростерон. Самым активным А. г. яв-
ляется тестостерон, андрогенная активность к-рого
в 7 раз превышает активность андростерона. Тесто-
стерон и нек-рые его производные применяют в меди-
цине при лечении половой недостаточности у мужчин,
климактерич. расстройств у женщин, а также при ле-
чении нек-рых сердечно-сосудистых и раковых забо-
леваний. А. г., меченные изотопами, используют
для изучения обмена стероидных веществ в орга-
низме.
Лит.: Ф и з е р Л., Ф и з е р М., Химия природных со-
единений фенантренового ряда, пер. с англ.. М. — Л., 1953;
Назаров И. Н., Бергельсон Л. Д., Химия стероид-
ных гормонов, М., 1955; Dorfman R. 1. and Ship-
225
АНДРОГЕНЫ —АНИЗИДИНЫ
226
1 е у R. A., Androgens, biochemistry, physiology and clinical
significance, N. Y. — L., 1956; H a n с O., Hormone, Praha,
1960; см. также при ст. Гормоны. Л. И. Линевич.
АНДРОГЕНЫ (андрогенные вещества) — вещества,
обладающие основной биологич. активностью муж-
ского полового гормона. Большую группу природных
А. составляют андрогенные гормоны’, тестостерон,
андростерон и др.; наиболее активным А. является
тестостерон. Нек-рые синтетич. производные природ-
ных стероидных гормонов также являются А. и приме-
няются в медицине как заменители мужского полового
гормона (метилтестостерон). Известно неск. десят-
ков синтетически полученных стероидных веществ,
обладающих андрогенной активностью, однако по
силе действия они в большинстве случаев значительно
уступаютприроднымандрогенным гормонам и практич.
применения не нашли. А. содержатся в пантах сибир-
ского пятнистого оленя, в нек-рых растениях, в за-
родышах пшеницы и др.; строение последних А. еще
не изучено, не установлено также, относятся ли они
к стероидным в-вам.
Испытание андрогенной активности
производят обычно на кастрированных животных; при
введении А. таким животным у них восстанавли-
ваются вторичные половые признаки (напр., у кап-
лунов возобновляется рост гребня). В качестве стан-
дарта при биологич. испытаниях используют гормон
андростерон, 100 мг к-рого принимается за между-
народную единицу андрогенной активности (М. е.).
Часто андрогенную активность вещества характери-
зуют «петушиной единицей» (П. е.) — количеством А.,
достаточным, чтобы вызвать увеличение гребня у кап-
луна на 20% (после двухсуточного введения в опре-
деленных условиях). А. применяют при лечении пер-
вичной половой недостаточности у мужчин, нек-рых
сердечно-сосудистых заболеваний, при терапии рака
молочных желез и др.
Лит. см. при ст. Андрогенные гормоны.
Л. И. Линевич.
АНДРОСТЕРОН CieH30O2, мол. в. 290,43 — андро-
генный гормон, вторичный мужской половой гормон
(образуется в организме из
н С О первичного полового гор-
мона тестостерона). Андро-
Н С I I стеРон содержится в моче
' и крови в мужском и в жен-
Г | ском организме, но в послед-
НО'' нем в меньшем количестве.
А. — кристаллы; т. пл. 178°
(184—185°); плохо растворим
в воде, практич. растворяется во всех органич. раство-
рителях, [а)д 4-103,5° (в метаноле), 4- 94,6° (в этано-
ле) А. дает реакции на спирты и кетоны, характерные
для кетостероидов; с реактивом Жирара (соляно-
кислый триметиламиноацетогидразид) образует рас-
творимое в воде производное; с м-динитробензолом
дает цветную реакцию (Циммермана — Бито), к-рая
используется для колич. колориметрич. и хромато-
графия. определения А. Получают А. из мочи и синте-
тически (напр., из холестерина). Как терапевтич.
средство А. потерял значение; применяется в качестве
стандарта при биологич. испытаниях: 100 мг А. при-
нимается за международную единицу андрогенной
активности (М. е.).
Лит.: Engel L. L., в кв.: Methods of biochemical
analysis, у. 1, N. Y. — L., 1954, p. 479; см. также при статье
Андрогенные гормоны. М. А. Грехова.
АНЕСТЕЗИН (бензокаин, этиловый эфир л-амино-
бензойной кислоты) n-H2N—С6Н4—СООС2НВ, мол.
в. 165,19 — белые мелкие кристаллы; т. пл. 89—
91,5°; т. кип. 305°; трудно растворим в холодной,
легче — в горячей воде, легко — в жирных маслах,
эфире, спирте (1 г в 5 мл). А. диазотируется NaNO2
в кислой среде; полученное диазопроизводное вступает
в реакции сочетания. Для качественного открытия
А. пользуются реакциями сочетания (темно-оранже-
вое окрашивание при прибавлении нескольких капель
щелочного р-ра р-нафтола к приготовленному из
А. р-ру соли диазония). Колич. определение А. осно-
вано на омылении эфирной группы спиртовым р-ром
КОН и титровании избытка щелочи 0,5 н. р-ром НС1.
Получают А. этерификацией л-нитробензойной к-ты
обычным путем с последующим восстановлением
нитрогруппы этилового эфира л-нитробензойной к-ты
различными способами, чаще всего железом в уксус-
ной или разбавленной соляной к-тах, иногда водо-
родом в присутствии катализаторов. Применяют
А. в качестве местно-анестезирующего средства, гл.
обр. для поверхностной анестезии.
Лит. см. при ст. Анальгин. А. С. Елина.
АНЕТОЛ (л-пропениланизол), мол. вес 148,20,
л-СН3О—С6Н4СН=СНСН3 — жидкость, обладающая
сладковатым вкусом и своеобразным запахом; т. кип.
235,37760 мм; 113,5714 мм; т. пл. 22,5° dlt 0,9875;
Лр 1,5591; хорошо растворим в спирте, эфире, весьма
трудно растворяется в воде. В природе А. встречается
в анисовом и фенхелевом маслах, из к-рых выделяется
фракционированной перегонкой и последующим вы-
мораживанием и отжимом на прессах; синтетически
получается изомеризацией метилхавикола со спирто-
вым р-ром КОН : л-СН3О—С6Н4СН2СН2=СН2 —►
—► л-СН3О—С6Н4СН=СНСН3. Количественно А.
может быть определен роданированием по двойной
связи. А. является основным сырьем для получения
анисового альдегида. А. А. Зеленецкая.
АНИД — см. Полиамидные волокна.
АНИЗИДИНЫ (метоксианилины, аминоанизолы)
Н3СО—СеН4—NH2, мол. в. 125,17 —метиловые эфиры
аминофенолов, известны 3 изомера:
ОСН3
мета
о-А. ! jh-A. п-А.
Т. пл., °C 5 <-12 57
Т. кип., °C 225 251 245
-20 1,0923 1.096 —
dl31 0,996 0,997 1,003
1,5754 1,5811 —
«7 nD — — 1,5559
Константа основности (25°) 1-10-9’51 — 1,5-10-9
о-А. — бесцветная жидкость, т. кип. 90—91°/4 мм;
плохо растворима в воде, очень хорошо — в спирте и
эфире. о-А. получается восстановлением о-нитроани-
зола железом в присутствии соляной к-ты;
Fe
o-CHsO-CeH4-NO3 o-CHsO-CeH4-NH3
о-А. образует соли с к-тами, вступает в реакции аци-
лирования и диазотирования; применяется для полу-
чения фармацевтич. препаратов — производных гвая-
кола, и для синтеза азокрасителей. .и-А. — масля-
нистая жидкость, т. кип. 142—143732 мм, 122713 мм;
плохо растворим в воде, хорошо — в спирте и эфире.
Получают восстановлением д«-нитроанизола. л-А. —
кристаллы в виде призм, растворим в горячей воде,
спирте, эфире. Получают л-А. восстановлением л-нит-
роанизола железом в присутствии соляной кислоты
или сульфидом натрия в водном растворе, а так-
227
АНИЗОЛ — АНИЛИН
223
же перегруппировкой фенилгидроксиламина и мета-
нольном растворе в присутствии серной кислоты:
CeH6NHOH n-CH3O—CeH4—NH2 п-А. дает
очень устойчивое диазосоединение. Применяется для
синтеза азокрасителей и некоторых лекарственных
веществ.
Лит.: Chemistry of carbon compounds, ed. by E. H. Rodd,
v. 3, pt. A, Amst. [a. o.], 1954, p. 452. E. M. Рохлин.
АНИЗОЛ (метоксибензол, фенилметилоиый эфир)
СН3ОС6Н5, мол. в. 108,13 — бесцветная жидкость
с приятным ароматич. запахом; т. пл. —37,4°; т. кип.
155,0°; й3» 0,9940; п3^ 1,5170; Хмакс2650 А (в гексане),
нерастворим в иоде, с бензолом, спиртом и эфиром
смешивается во всех отношениях. При электрохимия,
окислении А. образует бензохинон, гидрируется (ка-
талитически) до циклогексанола, хлорируется, бро-
мируется, нитруется в 2,4- и 6-положения, образуя
моно-, ди- и тризамещенные производные. Хлори-
стый ацетил СН3СОС1 в присутствии А1С13 ацили-
рует анизол с образованием п-метоксиацетофенона
п-СН3ОС6Н4СОСН3, с HCN в присутствии НС1
дает анисовый альдегид, меркурирование ацетатом
ртути приводит к n-метоксифенилмеркур ацетату
n-CH3OCeH4HgOCOCH3. А. получают из фенола
и диметилсульфата в присутствии щелочи, алкилиро-
ванием фенолята натрия иодистым метилом CH3J
и др. способами. А. применяется как растворитель
в парфюмерной пром-сти. При нагревании с SeO3
в присутствии H3SO4 А. дает темно-зеленое окраши-
вание. м. ф. Турчинский.
АНИЗОТРОПИЯ — явление, состоящее в том, что
физич. свойства тел (механич., оптич., электрич.,
магнитные и др.) в зависимости от направления ха-
рактеризуются различными величинами. Часто в-во,
изотропное в отношении одних свойств, проявляет
А. в отношении других. В случае однородной
А. зависимость физич. свойств от направления одина-
кова в различных точках среды; она обусловлена
строением тела — наличием кристаллич. структуры
или резко выраженной асимметрии молекул. Неод-
нородная (местная) А. возникает в результате
односторонних деформаций тела (напр., при механич.
обработке металлов). Поверхностный слой всякого
тела также вызывает местную А. на границе раздела
фаз (см. Поверхностные явления). Практически наи-
более важны явления А. кристаллов, жидких кристал-
лов и полимеров.
АНИЛИДОУКСУСНАЯ КИСЛОТА (фенилглико-
коль, фенилглицин) CeH5NHCH2COOH, молекулярный
вес 151,16 — кристаллы, т. пл. 127°;
легко растворима в воде, трудно —
О—С СН2 в эфире- в фенольном р-ре ассоци-
Н2С% х-С=О ирует; может растворять некоторые
N—С6Н5 неорганические соединения (окис-
лы РЬ и Zn); константа диссоциации
К = 3,9 • 10*6(25°). При медленном нагревании
А. к. до 140—150° отщепляется иода и образуется
2,5-диоксо-1,4-дифенилпиперазин, при быстрой пере-
гонке отщепляется СО2 и образуется метиланилин
CeH5NHCH3. Кипячение А. к. в 95%-ном спирте
в присутствии цианамида приводит также к 2,5-диоксо-
1,4-дифенилпиперазину. При перегонке А. к. с солями
Са (лучше с формиатом Са) образуется индол, а при
сплавлении А. к. с КОН — индоксил. В пром-сти
А. к. получают в виде железной соли обработкой ани-
лина хлоруксусной к-той в присутствии гидрата
закиси железа; побочные продукты: анилидодиуксус-
ная кислота и ее анилид. Другой способ основан
на взаимодействии формальдегида и KCN с анили-
ном: C6H6NH3 + СН3О + KCN —► CeH6NHCH2CN
—2 CeH6NHCH2COOH; промежуточным продуктом
реакции является нитрил фенилглицина. При полу-
чении по этому способу нежелательные побочные
продукты не образуются. А. к. широко применяют
при синтезе красителей; промежуточный продукт
при получении индиго. н. в Гнучев
АНИЛИДЫ — производные карбоновых кислот,
у к-рых гидроксильная группа заменена остатком
ароматич. амина (анилина или его производного)
RCONHAr. А. можно рассматривать как ароматич.
амины, у к-рых один атом водорода аминогруппы за-
мещен кислотным остатком (ацильной группой). При
нагревании с разбавл. растворами к-т и щелочей
А. гидролизуются до исходных аминов и к-т; этим
свойством А. широко пользуются для временной за-
щиты аминогруппы от нежелательного воздействия
реагентов, применяемых и последующих реакциях
(напр., перед окислением толуидинов или перед
нитрованием). Для защиты аминогрупп чаще всего
А. ацетилируют или бензоилируют одним из описан-
ных ниже способов, а по окончании реакции подвер-
гают гидролизу. А. могут быть получены прямым
взаимодействием амина и кислоты, однако эта реак-
ция обратима: ArNH2 + RCOOH --ArNIICOR -ф- Н3О.
Значительно легче А. получают при действии на
амины ангидридами или галогенангидридами кис-
лот: ArNH3 + (RCO)2O —► ArNHCOR + RCOOH или
ArNH3 + RCOX —► ArNHCOR + HX. Для получе-
ния сложных А. часто применяют взаимодействие
аминов со сложными эфирами и среде инертного
растворителя (ксилол, хлорбензол) при повышенной
температуре: ArNH3 + RCOOA1K —► ArNHCOR +
+ А1КОН.
Простейший А. — ацетанилид. Многие А., напр.
производные 2,3-оксинафтойной к-ты, т. н. азотолы,
применяют в качестве компонентов для ледяного кра-
шения. Большое значение имеют также А. 0-кето-
кислот. Напр., анилид, о-анизидид и о-хлорани.тид
ацетоуксусной к-ты применяют в качестве азосостав-
ляющих в произ-ве ряда азокрасителей, а толидид
ацетоуксусной к-ты — в качестве азотола. А. — произ-
водные ароилуксусных к-т, напр. п-нитробензоилук-
сусной, n-метоксибензоилуксуспоц и др., применяют
в качестве цветообразующих компонентов в произ-ве
цветных кинофотоматериалов, т. к. в условиях цветно-
го проявления п-диэтиламицоанилипом они образуют
желтые азометиновые красители.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955.
Н. С. Вульфсон.
АНИЛИН (аминобензол, фениламин) CeH6NH2;
мол. в. 93,12 — простейший ароматич. амин; бесцвет-
ная жидкость со своеобразным запахом; т. пл.
—6,15°; т. кип. 184,4°/760 мм, 102°,50 мм, 92°/33 мм,
71°/9 мм; давление паров: 600 лмг/175°, 50 лл/102°;
1,0268; «д' 1,5863. Теплоемкость 0,518 ккал/моль
(20°); т. всп. 76°; теплота сгорания при постоянном
давлении Ср = 815 ккал/моль; теплота парообра-
зования 9,71 ккал/моль. Константа диссоциации:
К = 4,6 • 10 10(25°), 7,6 • 10”10(40°), 1,7 • 10-9(60°).
А. смешивается во всех отношениях со спиртом, эфи-
ром, бензолом; растворим в большинстве органич.
растворителей. В 100 г воды растворяется А.: 3,4 г
(20°), 6,4 г (90°). А. смешивается с 50%-ным иодным
р-ром его хлоргидрата. В насыщенном водой при
25° А. содержится 5% воды. А. растворяет ряд ме-
таллов (К, Na, Mg, Са и др.) с образованием металлич.
производных — металламидов.
А. впервые был получен в 1826 О. Унфердорбеном при
перегонке ивдиго с известью и назван им «кристаллином».
В 1834 Ф. Рунге установил его присутствие в кам.-уг. смоле
и назвал «кианолом». В 1841 Ю. Ф. Фрицше получил А. нагре-
ванием индиго с р-ром едкого кали и назвал его А.; в 1842 году
А. был получен Н. Н. Зининым восстановлением нитробен-
зола сернистым аммонием и назван им «бензидамом». В 1843
А. В. Гофмав установил идентичность всех перечисленных
соединений.
- 229
АНИЛИН — АНИЛИНОВАЯ ТОЧКА
230
А. обладает основными свойствами (АГ=4,5 • 10 10),
более слабо выраженными по сравнению с аминами
жирного ряда, например метилами-
г,—ном (К = 5 • 10~4), что объясняет-
GZ Чу7- ся уменьшением электронной плот-
? / nh2 ности у азота аминогруппы вслед-
\=4А ствие сопряжения его пеподелен-
ной пары электронов с л-электрона-
ми бензольного ядра. При действии электроно-
фильных реагентов на А. в нем легко замещают-
ся атомы водорода бензольного ядра, находящие-
ся в о- и n-положениях; аминогруппа относится к
ориентантам 1-го рода (см. Ориентации правило). Од-
нако в сильнокислых средах получается значитель-
ное количество .и-изомера вследствие солеобразова-
ния, которое ведет к превращению аминогруппы в
аммониевую, являющуюся уже акцептором электро-
нов. Поэтому перед нитрованием А. превращают в
анилид (форм- или ацетанилид), к-рый почти цели-
ком нитруется в «-положение. С минеральными к-тами
А. образует хорошо кристаллизующиеся соли:
CeH6NHgCl, т. пл. 198°, т. кип. 245°; C6H6NH3Br,
т. пл. 286°; CeH5NH2 H3AsO4, т. пл. 140°, и др. Из
них наиболее важной является CeH6NH3Cl, применяе-
мая в ситцепечатании. Соли со слабыми неорганич.
и органич. к-тами в водных р-рах легко гидролизуются.
При действии на А. конц. серной к-ты вначале обра-
зуется сульфат, который при сильном нагревании
(200°) переходит в сульфаниловую к-ту:
CeH5NH2 • H2SO4—- h2n-^~^-so3h
При нагревании А. с органич. к-тами образуются
анилиды к-т, например: C6H6NH2 + СН3—СООН —>
-> C6H5NHCOCH3 + Н2О.
Ацетанилид производится в промышленном мас-
штабе и применяется для приготовления паранитро-
анилина, одного из важных полупродуктов в произ-ве
красителей. Очень важны продукты метилирования
А. — моно- и диметиланилин. Они с успехом полу-
чаются не только при действии на А. хлористого ме-
тила, но и при нагревании хлоргидрата А. с метило-
вым спиртом под давлением или с диметиловым эфиром
(каталитически над А12О3): CeH5NH2 + (СН3)2О—>
—> CeH5N(CH3)2 + Н2О. Моно- и диметиланилин ши-
роко применяются в произ-ве красителей, взрывча-
тых в-в и др. Продукт алкилирования А. хлоруксус-
ной к-той—фияилглицин C6H6NHCH2COOH — исполь-
зуется в производстве индиго. При нагревании А.
и хлоргидрата А. при высокой температуре и повы-
шенном давлении образуется дифениламин — важ-
ный промежуточный продукт в синтезе красите-
лей: CeH6NHr + CeH6NH3Cl -> С6Н6—NH—С6Н6 +
+ iNH^Cl. А. применяется также для получения
проявителей для фотографии (парааминофенол и др.),
ускорителей вулканизации каучука, фармацевтич.
препаратов и мн. др. Особо технически важной реак-
цией А. является диазотирование', получающийся при
этом хлористый фенилдиазоний является одним из
основных полупродуктов в синтезе азокрасителей.
При окислении А. образуется краситель, так назы-
ваемый анилиновый черный; при каталитическом
гидрировании образуется с отличными выходами
циклогексиламин, из которого в настоящее время
в промышленности получают капролактам
(см. Полиамиды).
Основным методом получения А. является восстановление
нитробензола чугунными стружками в присутствии неболь-
шого количества соляной к-ты:
4CeHsNO2 4- 9Fe 4- 4Н2О = 4CeH6NH2 4- 3Fe3O4
Промежуточными стадиями восстановления нитробензола
в этих условиях являются нитрозобензол и фенилгидрокси-
ламин CeH6NO2 С,Н6—NO С.Н.—NH—ОН ->
—> —C«HS — NH2. Соляную к-ту, применяемую в на-
•tigO
чале этого процесса в очень небольшом количестве, с успехом
можно заменить хлоридами натрия, аммония, железа, каль-
ция и др.: Fe 4-2NaCl -J-2Н2О —* [FeCl2 -J-2NaOH] -J-2Н;
[FeCl2 4- 2NaOH] —* Fe(OH)2 4- ?NaCl. Наиболее активными
являются серые сорта чугуна с
|_ большим содержанием графита;
процесс зкзотермичен. Восстановле-
г ние нитробензола ведут в т. н.
редукторах, футерованных внутри
Ю
В
Схема получения анилина восстановлением нитробен-
зола чугунными стружками: 1 — чугунные стружки;
2 — шнек для подачи чугунных стружек; 3 — резервуар
с нитробензолом; 4 — резервуар с соляной кислотой;
в — редуктор (реактор); 6 — холодильник; 7 — отстой-
ник; 8 — нейтрализатор; 9 — шламоотделитель; 10 —
осветлитель; 11 — сборник для анилина, идущего на дис-
тилляцию; А — вывод шлама; Б — непрореагировавший
нитробензол иа улавливание; В — водный анилин на пе-
реработку.
кислотоупорными материалами, снабженных мощными ме-
шалками грабельного типа. Острый пар для отгонки А. вво-
дится через полую ось мешалки. Окончательная очистка А.
производится вакуум-дистилляцией.
Наряду с этим способом применяется и каталитич. вос-
становление нитробензола над основным карбонатом меди,
нанесенным на пемзу. Каталитич. парофазное восстановление
нитробензола ведут при атм. давлении электролитич. водоро-
дом или полученным из водяного газа при 350—400°. Для
отвода значительной теплоты реакции берут большой избыток
водорода (до 50 молей на 1 моль нитробензола). Выход А. со-
ставляет 98% от теоретически возможного; побочно образуется
0,3—0,4% аминофенола, выделяемого из кубового остатка
после отгонки А. Расход меди 0,7 кг/т А. Каталитич. восстанов-
ление нитробензола может быть осуществлено и в жидкой фазе
с применением никелевых катализаторов и выходом до 90%.
Известно также получение А. из хлорбензола: СвН6С1 4-
4- NH3 —► СвН6 — NH2 4- НС1. Аммонолиз проводят в жид-
кой фазе при 200—210° и давлении до 70 ат или в паровой
фазе при 400° над катализатором, содержащим соли однова-
лентной меди. а. образуется также при пропускании паров
бензола с аммиаком через раскаленные трубки, однако этот
способ пока имеет лишь теоретич. значение.
Чувствительная качеств, реакция на А. — действие
на его водные р-ры хлорной известью; появляется
интенсинно-фиолетовое окрашивание. Колич. опре-
деление А. основано на реакции диазотирования (по
израсходованному нитриту). Эта реакция может слу-
жить и для качеств, обнаружения А. по образо-
ванию азокрасителя в результате азосочетания с р-на-
фтолом или др. диазосоставляющей (общая реакция
на первичные ароматич. амины). Содержание неболь-
ших количеств нитробензола в А. — от 0,00015
до 1%, может быть определено полярографически.
А. применяется также в аналитич. практике и, в част-
ности, при определении ароматич. углеводородов
в продуктах нефтяного происхождения по т. н. ани-
линовой точке. А. ядовит; действует на кровь, вызы-
вая превращение оксигемоглобина в метгемоглобин;
резорбируется кожными покровами; предельно до-
пустимая концентрация его в воздухе 0,005 мг/л.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М. 1955; В о л ь ф к о-
ви ч С. И., Общая химическая технология, т. 2, М., 1959;
Чичибабин А. Е., Основные начала органической хи-
мии, 5 изд., т. 2, М., 1957 И Л. Кнунянц.
АНИЛИНОВАЯ ТОЧКА у г л е в о д.о родов
(максимальная анилиновая точка) — критич. темп-ра
растворения углеводородов или смеси углеводородов
в анилине, выше к-рой указанные компоненты обра-
зуют гомогенный р-р. Углеводороды данного класса,
231
АНИОНИТЫ —АНИСОВЫЙ СПИРТ
232
близкие по мол. весу (т. е. кипящие в узких преде-
лах темп-p), обладают близкими А. т., тогда как
А. т. углеводородов разных классов сильно отли-
чаются друг от друга. Поэтому определение А. т. при-
меняется для структурно-группового анализа угле-
водородных смесей (гл. обр. нефтепродуктов). Наи-
большей растворимостью в анилине, т. е. наиболее
низкими А. т., обладают ароматич. углеводороды
(CeHe < —30°); наиболее высокие А. т. у парафи-
нов (н-С6Н14 69,1°). Нафтены и олефины занимают
промежуточное положение. Напр., для углеводородов
С8 А. т. имеет след, значение (округленно): парафины
70, нафтены 45, ароматич. 20. В пределах данного
гомологии, ряда А. т. возрастают по мере увеличения
мол. веса. На практике А. т. используются гл. обр.
для определения ароматич. углеводородов, причем
часто находят не критич. темп-ру растворения, а ми-
нимальную темп-ру, при к-рой равные объемы ани-
лина и смеси углеводородов образуют гомогенный р-р
(т. н. равнообъемную А. т.). Измерение А. т.
проводится для узких фракций, на к-рые предвари-
тельно разгоняется исследуемый нефтепродукт.
Содержание углеводородов различных классов опре-
деляется по спец, формулам, в к-рые входит в качестве
постоянного члена т. н. анилиновый коэфф., т. е.
содержание углеводородов данного класса, отвечаю-
щее разности А. т. в 1°.
При анализе летучих фракций вместо анилина удоб-
нее применять нитробензол, в к-ром критич. темп-ра
растворения углеводородов ниже, чем в анилине.
Более совершенный метод структурно-группового
анализа, использующий А. т., требует одновременного
определения показателя преломления и уд. веса
исследуемой фракции (n-d-A-метод).
Лит.: В а н-Н е с К., В а н-В е с т е н X., Состав масля-
ных фракций нефти и их анализ, пер. с англ., М., 1954; О Do-
ji е н ц е в Р. Д., Физические константы углеводородов жид-
ких топлив и масел, 2 изд., М. — Л. 1953; Химия углеводо-
родов нефти, под ред. Б. Т. Брукса [и др.], пер. с англ., т. 1,
М., 1958, с. 376. М. А. Ландау.
АНИОНИТЫ — см. Иониты.
АНИОНОТРОПИЯ (анионотропные превращения) —
явление молекулярных перегруппировок, сопровож-
дающееся миграцией отрицательно заряженных ато-
мов или групп атомов:
-С-С=С<Д^г )С-С-С< +Х' >С=С-С-
4 L 1 1 £
X — функциональная группа, галогены. Аниотроп-
ные превращения облегчаются, если X способен
образовывать устойчивый анион. Так, напр., спирты
(X—ОН) не превращаются друг в друга, эфиры же
уксусной к-ты (X— СН3С00), а еще легче галогениды
(X — С1, Вг) способны к А. Превращение облег-
чается также и в том случае, если промежуточно
образующийся катион достаточно устойчив. А. про-
текает легче в сильно ионизирующих средах, спо-
собствующих процессу диссоциации в-ва на катион
и анион. В неполярных растворителях или без раство-
рителя анионотропные превращения протекаютвнутри-
молекулярно, без предварительной диссоциации на
ионы, напр.:
,/СНЧ /СН\,
R-CH CHR—R-CH CHR
С-Х' х/
Если исходить при такого рода превращениях из
оптически • активного соединения, то оптич. актив-
ность сохраняется. При замещении группы X на др.
группу Y вновь входящая группа может соединиться
с тем углеродным атомом, с к-рым была связана
ушедшая группа X, или с другим атомом углерода
(с перенесением реакц. центра). Замещение может
быть либо одностадийным (схема II), либо двухста-
дийным (схема I), когда вслед за удалением X про-
исходит присоединение Y к катиону.
r-chx-ch=ch2 — х- + [R-CH^CH—сн2]+
R—CHY—СН=СН2 .
Y~ + [R—СН--СН—СН2]+—I
J—R—СН=СН—СН»У
|-Х- + R—СНУ—СН=СНч .
Y-+R—СНХ—СН=СН»~ I II
Х-+ R-CH=CH-CH2Y /
Если замещение протекает по схеме I, то в равных
условиях изомерные исходные вещества В—СНХ—
—СН=СН2 и R—СН=СН—СН2Х должны дать при
реакции с Y одинаковые смеси конечных продуктов.
При реакции по схеме II одинаковой смеси конечных
продуктов не должно образовываться. Этим путем
удалось экспериментально подтвердить существова-
ние двух различных механизмов замещения в анионо-
тропных системах.
Лит.: Реутов О. А., Теоретические проблемы органи-
ческой химии, м., 1956; Ингольд К. К., Механизм реак-
ций и строение органических соединений, пер. с англ., М., 1959.
О. А. Реутов.
АНИОНЫ — см. Ионы.
АНИСОВЫЙ АЛЬДЕГИД (обепин, кратежин)
п- СН30—С6Н4—СНО, молекулярный вес 136,14—мас-
лянистая жидкость, т. пл. —0,02°; т. кип. 248°;
1,1230; Ид 1,5720; растворим в 50%-ном спирте (па
7 ч. спирта 1 ч. А. а.), петролейном эфире; обладает
пряным запахом, содержится в эфирных маслах.
В пром-сти А. а. получают окислением анетола
хромовой смесью: п-СН30—СвН4—СН=СНСН3 —►
—► п-СН30—С6Н4—СНО, или же изомеризацией
эстрагола, выполняемой в щелочной среде (с после-
дующим подкислением) в анетол, который окисляют,
как описано выше: п-СН3О—С6Н4—СН2СН=СН2—►
—»-СН30—С6Н4 СН=СНСН3. Из эфирных масел выде-
ляют бисульфитным методом. На воздухе А. а. окисляет-
ся в анисовую к-ту; под действием щелочей образует
анисовый спирт и анисовую к-ту; при гидрировании
с Ni-,Pt- и Pd-катализаторами под давлением дает
анисовый спирт. Присоединяет галогеноводород, легко
хлорируется, нитруется до моно-, ди- и тринитропроиз-
водных; конденсируется с кетонами. А. а. дает желто-
оранжевую окраску с р-ром резорцина в 80%-ной
H2SO4 (чувствительность — 1-10-6). А. а. обладает
бактерицидным действием. Широко применяется в пар-
фюмерных КОМПОЗИЦИЯХ. г. М. Николаев.
АНИСОВЫЙ СПИРТ (и-метоксибензиловый спирт)
п-СН30—С6Н4—СН2ОН, мол. вес 138,16 — кри-
сталлы, т. пл. 24,5—25°; т. кип.: 258,8°/760 мм;
127—130°/8 мм; d\l 1,1129; не растворяется в воде,
растворяется в спирте, эфире и других органич. ра-
створителях. А. с. обладает своеобразным запахом,
напоминающим запах анисового альдегида; в природе
встречается в стручках ванили; может быть синтези-
рован из анисового альдегида реакцией Канниццаро
(см. Канниццаро реакция):
2СН3О-С6Н4-СНО ко— НОСН2-СеН4-ОСН3 +
+ ROOC-CeH4 — ОСН3
с луйпим выходом (85—9О°/о) получается «перекрест-
ной» реакцией Канниццаро с применением в каче-
стве второго компонента формальдегида:
СН3О—СсН4 —СНО + нсно + Н3О —
-» НОСН2-С6Н4-ОСН3 + нсоон
с хорошим выходом образуется при каталитич. вос-
становлении анисового альдегида Pt-чернью, Pd, FeCl3
или Ni (никель Ренея). А. с. дает реакции, характер-
ные для первичных спиртов; бензоильное производное,
т. пл. 38°; фенилуретан, т. пл. 96°; количественно
233
АНОДИРОВАНИЕ — АНСЕРИН
234
А. с. определяется ацетилированием в пиридине.
В парфюмерных композициях А. с. применяется для
создания запаха гелиотропа, жимолости, сирени.
Лит.: West Т. F., Stransi Н. J. and Bar-
ton D. Н. R., Synthetic perfumes, L., [1949].
А. А. Зеленецкая.
АНОДИРОВАНИЕ — электролитич. оксидирова-
ние алюминия; процесс образования окисной пленки
на алюминии в результате взаимодействия его с вы-
деляющимся на нем кислородом при прохождении
постоянного или переменного тока через р-ры серной,
хромовой или щавелевой к-т. При взаимодействии
алюминия с кислородом воздуха в обычной атмо-
сфере образующаяся при этом окисная пленка
чрезвычайно тонка, порядка 100—200 А. Химич,
оксидирование алюминия путем воздействия окисли-
телей также имеет ограниченное промышленное при-
менение, т. к. образующаяся при этом окисная пленка
по своим механич., антикоррозионным и адсорбцион-
ным свойствам значительно уступает электролитич.
пленкам. Механизм анодного окисления алюминия
сложен и точно не установлен; то же можно сказать
о составе и строении окисной пленки. Бесспорным
является факт роста пленки не по направлению от
металла к электролиту, а в глубь металла, так что
при определенных условиях тонкий алюминиевый
образец может быть насквозь электролитически окис-
лен. Можно также считать установленным, что непо-
средственно к алюминию примыкает сравнительно
тонкий и плотный, т. н. барьерный, слой пленки
(0,01—0,1 жк), а по направлению к электролиту обра-
зуется относительно толстая (до 400 мк и больше),
пористая, проницаемая для электролиза пленка.
Безводная А12О3 гигроскопична даже при нагреве
до красного каления; считают, что вода в плен-
ке химически не связана, за исключением наруж-
ных слоев пленки, которой приписывают формулу
А12О3 • Н2О. В пленках, полученных из р-ров хромо-
вой к-ты, обнаружено от 0,1 до 0,7% Сг, а в пленках
из р-ров серной и щавелевой к-т было определено
примерно 15 вес. % воды и соответственно 14% SO3
и 3% Н2С2О4.
Область применения А. алюминия широка и разно-
образна; по степени распространения анодные пленки
имеют след, назначения: защита от коррозии; защитно-
декоративная отделка, сохранение полированным алю-
минием высокого коэфф, отражения света; грунт под
лакокрасочные покрытия; специальные цели: высокие
электро- и теплоизоляционные свойства, защита от
механич. износа и др.
В зависимости от назначения выбирают тип электролита
и его режим. Наибольшее распространение имеет анодиро-
вание в 10—20%-ной H2SO4 при плотности тока 1—2 а/дм2,
напряжении 10—20 в и темп-ре 20—30°. За 10—30 мин. обра-
зуется бесцветная, прозрачная пленка толщиной 5—2 0 мк.
Для получения твердых пленок с большой износостойкостью
и адсорбционной способностью используется 7%-ная H2SO4
при искусственном охлаждении обрабатываемых изделий до
1—3°. При напряжении 120 в и плотности тока около 5 а/дм2
за 4 часа получается сероватая пленка толщиной до 200 мк.
Сернокислотное А. неприемлемо для клепаных и сварных узлов,
т. к. остатки неотмытой серной к-ты вызывают коррозию алю-
миния. Хромовая н-та не только не вызывает коррозию, но,
напротив, защищает от нее. В 5—10%-ном р-ре СгО3 при плот-
ности тока 0,15—0,3 а/дм2, напряжении 40 в и темп-ре 35°
за 30 мин. образуется серая пленка толщиной в 2—3 мк.
В 3—5%-ной Н2С2О4 при плотности тока 1—2 а/дм2, напря-
жении 40—60 в, темп-ре 18—2 0° за 1 час получается желтая
пленка толщиной до 60 мк, а при темп-ре 3—5° толщина пленки
может достигнуть 600 мк и больше. Щавелевая к-та значи-
тельно дороже серной и хромовой, в связи с чем ее применение
ограничивается нанесением пластичной окисной пленки на
проволоку, ленту или готовые изделия для электроизоляцион-
ных целей. Из разобранных трех электролитов сернокислот-
ный самый дешевый и применим для многих марок алюмини-
евых сплавов. Путем погружения анодированного в серной
кислоте алюминия в р-ры органич. или неорганич. красителей
можно окрашивать изделия в различные цвета, в том числе
имитировать их под золото.
Лит.: Ускоренные методы защиты изделий от коррозии.
Сб. ст., под ред. Г. В. Акимова, М. — Л., 1946; Аки-
мов Г. В., Томашов Н. Д., Тюкина М Н.,
Ж. общ. хим., 1942, 12, вып. 9—10, с. 433; Томашов Н.Д
и Тюкина М. Н., Тр. Ин-та физ. химии АН СССР, 1951'
вып. 2. В. И. Лайнер.
АНОМЕРЫ (анамеры) — изомеры циклических форм
сахаров, отличающиеся расположением полуацеталь-
ного гидроксила по от-
ношению к плоскости
кольца; их часто назы-
вают а- и 0-формами
сахара; a-форма сахара
D-ряда, 0-форма сахара
D-ряда. а-Формой на-
зывают изомер, у к-рого
расположение полуаце-
тального гидроксила
такое же, как у гидро-
ксила (свободного или
принимающего участие
СН2ОН
НО—
СНОН
<%н
I
СНОН
I
'НС---
. I
сн8он
Ct—форма сахара
О—ряда
fl—форма солара
D- ряда
в образовании цикла) асимметрия, углеродного атома,
определяющего принадлежность сахара к D- или
L-ряду. Согласно этой номенклатуре, a-D-форма оказы-
вается зеркальным отображениемa-L-формы. В D-ряду
a-форма обладает обычно большим правым вращением,
а 0-форма — меньшим. В L-ряду более сильным ле-
вым вращением обладает a-форма. Так как определе-
ние пространственного расположения полуацеталь-
ного гидроксила связано с большими трудностями,
часто для обозначения а- и 0-форм пользуются дан-
ными об их вращении. Формы сахара а- и 0 в водном
р-ре легко взаимопревращаются, причем в р-ре саха-
ра устанавливается равновесие. Так как а- и 0-формы
отличаются по уд. вращению, равновесие сопровож-
дается изменением [а]с р-ра. Это явление названо
мутаротацией. Изменение конфигурации у угле-
рода, связанного с полуацетальным гидроксилом
(т. н. аномерного углерода), наз. аномеризацией.
Лит.: М i с h е е 1 F., Chemie der Zucker und Polysac-
charide, 2 Aufl., Lpz., 1956. Л. И. Линевич.
АНСА (от франц, слова anse, что означает ушко,
ручка) — приставка впереди названий циклич. со-
единений, дополнительно содержащих замкнутый мо-
стик в различных положениях. Примерами би- и
трициклич. анса-соединений могут служить эфиры
двухатомных фенолов (I — III).
//
Известны и карбоциклич. акса-соединения без гете-
роатомов в циклах (IV).
сн2 СН2
(СНа)2 (СН2)а
СН2СНОН — СН2
/// IV
Я. Ф. Комиссаров.
АНСЕРИН (0-аланил-метилгистидин) Ci0H16N4O3,
мол. в. 240,27 — дипептид, образованный [.-ами-
нокислотами; содержится в
мышцах большинства позво- НС = С-СН2— СНСООН
ночных; мелкие кристаллы; I ) I
т. пл. 238—239°; раствори- ZN снз NH
мые в воде и нерастворимые сн ОС(СН ) NH
в спирте. При 25° рКа 22 2
2,64 (—СООН), рЛа2 7,04 (имидоазольная), р#аз 9,49
(NHJ), pl 8,27. А. получают из мышечной ткани
235
АНТИ — АНТИБИОТИКИ
236
в виде ртутной соли, при разложении к-рой H2S
выделяют свободный А., а также синтетически из
карбобензоокси-0-аланина и метилового эфира ме-
тил-L-гистидина. Качественное и колич. определения
А. производятся методом распределительной хромато-
графии на бумаге, а также по нарастанию количества
аминогрупп после гидролиза ртутной соли А. В отли-
чие от карнозина, А. не осаждается нитратом серебра
AgNO3 в присутствии едкого барита Ва(ОН)3; сер-
нокислая окись ртути HgSO4 в сернокислом р-ре
осаждает А. лишь при 60—70%-ной концентрации
спирта.
Лит..’ СеверинС. Е., Усп. биол. хим., 1954, №2, с. 355;
Юдаев Н. А., Усп. соврем, биол., 1950, 30, вып. 2, 176.
См. также лит. при ст. Аминокислоты. М. И. Левянт.
АНТИ — приставка, применяемая для обозначе-
ния пространственной конфигурации соединений,
содержащих атом азота, связанный двойной связью
с остальной частью молекулы и способных к существо-
ванию в двух стереоизомерных формах. Примерами
анти-соединений могут служить альдоксим (I), несим-
метричные кетоксим (II) и гидразон (III), диазотат (IV).
сн3—с-н свн6—с—СвН4СН3-п
НО—N N—ОН
I 11
СП«—С—Н СсН5—N
II II
СсН5—NH-N N—ONa
III IV
В анти-соединениях рассматриваемые замести-
тели пространственно удалены друг от друга, в
альдоксимах и гидразонах от атома Н при атоме С
с двойной связью, в кетоксимах от менее сложного
радикала и т. д. Приставка анти- аналогична при-
ставке транс- для соединений, содержащих двой-
ную связь между атомами углерода.
Я. Ф. Комиссаров.
АНТИБИОТИКИ — вещества, образуемые микро-
организмами, высшими растениями и животными тка-
нями в процессе их жизнедеятельности и обладающие
способностью убивать окружающих микробов или
препятствовать их росту, т. е. проявлять бактери-
цидное или бактериостатич. действие. Для А. весьма
характерно избирательное действие на определенные
виды микроорганизмов; каждый А. характеризуется
специфич. антимикробным спектром дейст-
вия. Нек-рые А. подавляют болезнетворные микро-
организмы, не повреждая при этом животных и расти-
тельных клеток, и могут быть использованы для ле-
чения различных заболеваний.
А. представляют собой средства химич. защиты против
окружающих микробов, выработанные организмами в эволю-
ционном процессе (явление антагонизма, отмеченное еще Л. Па-
стером и И. И. Мечниковым). Химич, основой антагонизма
является выделение одними организмами в окружающую
среду разнообразных в-в, способных подавлять другие микро-
организмы п обеспечивающих первым преимущество в борьбе
за существование. Явление антагонизма было известно давно,
однако интерес к изучению А. резко усилился в 40-х гг. 20 в.,
когда А. впервые были получены в чистом виде.
Выделение и открытие лечебных свойств тиротрицииа,
полученного Р. Дюбо (1939) из культуры почвенной бактерии
В. brevis, явились толчком к выделению и испытанию других
аптибиотич. веществ. X. Флори и Дж. Чейн обратили внима-
ние на пенициллин, образуемый плесенью Флеминга.
В 1940 они разработали метод выделения пенициллина из куль-
туральной жидкости и установили его лечебное действие при
заражении мышей гноеродными кокками. Пенициллин ока-
зался практически не токсичным в-вом. В 1942 Г. Ф. Гаузе и
М. Г. Бражникова выделили из почвенной бактерии грамици-
дин С, нашедший лечебное применение. В 1944 А. Шатц,
Е. Буги и 3. А. Ваксман открыли стрептомицин, образуемый
лучистым грибком; этот А. широко используется для лечения
туберкулеза, туляремии и др. инфекций. В 1947 Дж. Эрлих
и К. Барц выделили хлоромицетин, вскоре была выяс-
нена его химич. структура и осуществлен синтез, явившийся
первым практически важным для медицины синтезом. В по-
следующие годы была открыта новая группа близких А.,
имеющая важное значение для медицинской практики. Так,
в 1948 был открыт ауреомицин (Р. Дуггар и др.) и
в 1950 террамицин (М. Финленд). Впервые в 1953 тет-
рациклин был получен гидрированием ауреомицина в при-
сутствии платинового катализатора, при этом хлор в моле-
куле ауреомицина замещался водородом; т. о., хлортетрацик-
лин превращался в тетрациклин. Позже было установлено,
что тетрациклин можно получать брожением в культурах
грибка при добавлении к питательной среде солей брома,
к-рый тормозит включение хлора в молекулу тетрациклина
при биосинтезе. Аналогичное действие оказывает добавление
тиоцианатов.
Важная группа А. сходной химич. структуры, в основе'
к-рой лежит макроциклич. лактоновое кольцо, т. наз. мак-
рол и д о в, куда входят эритромицин, Спира-
ми ц и н магнамицин, метимицин, олеандомицин, была опи-
сана в 1952 и в последующие годы.
Первый этап поисков новых А. связан с получением
микробов-антагонистов из природных источников:
почвы, воды, навоза и др. Выделение антагонистов
может производиться различными методами: 1) обо-
гащением почвы, 2) бактериальных агаровых пласти-
нок, 3) прямого засева почвы, 4) скученной популяции
на агаровой пластинке. Содержание А. определяют
титрованием на тест-микробах, при этом находят
наибольшее разведение культуральной жидкости,
к-рое еще задерживает рост тест-микроба. Наимень-
шее количество А., задерживающее рост микробов,
принимается за единицу А. Выделение А. из культу-
ральной жидкости производится; адсорбцией на моле-
кулярных адсорбентах (угли, глины, пермутиты);
экстракцией разнообразными органич. растворите-
лями при различных значениях pH; ионным обменом
на различных катионо- и анионообменниках; осажде-
нием. Дальнейшая очистка сырцов-антибиотиков про-
изводится при помощи хроматографии (хромато-
графия на окиси алюминия, целлюлозе, ионитах),
противоточной экстракции в специальных аппара-
тах и т. п.
После получения А. в чистом кристаллич. виде
производят определенве его химич. однородности,
определяют физико-хи-мич. константы (элементарный
состав, темп-ру плавления, мол. вес, поглощение
в УФ- и ИК-областях спектра и др.), а также его анти-
бактериальную активность в отношении определен-
ного тест-микроба и его антибактериальный спектр.
Одновременно на всех этапах выделения и очистки
определяется токсичность и лечебное действие на
животных. В том случае, когда А. входит в широкую
медицинскую практику, разрабатывают промышлен-
ные методы его получения и изучают химическую
структуру.
Известны А., действующие на грамположительпые и
грамотрицательные микроорганизмы, грибки и опу-
холевые клетки животных. Грамположительные и
грамотрицательные микробы — группы микробов, раз-
личающихся по отношению их протоплазмы к краси-
телям генциан-фиолетовому или метиловому фиоле-
товому, и иоду. Грамположительные микробы об-
разуют окрашенный комплекс, не обесцвечивающийся
под действием спирта (окраска по Граму); грамот-
рицательные микробы не окрашиваются.
А. по спектру своего действия делятся па неск.
групп: 1) А., действующие на различные грам-
положительные микробы (стафилококки, стрептококки
и пневмококки и др.), т. е. обладающие сравнительно
узким спектром действия. К таким А. относятся пени-
циллин, эритромицин, альбомицин, грамицидин, ба-
цитрацин и мн. др. 2) А. с широким спектром дей-
ствия, действующие как на грамположительные,
так и на грамотрицательные организмы (кишечная
палочка, палочка дифтерии или брюшного тифа и др ).
К этой группе относятся стрептомицин, хлоромицетин,
тетрациклины, неомицин, каномицин и др. 3) А.,
действующие на грибки, — группа полиеновых А.
(нистатин, кандицидин, трихомицин и др.), анти-
мицин и др. 4) Группа А., действующих как на микро-
организмы, так и на опухолевые (раковые) клетки
237
АНТИБИОТИКИ
238
животных — актиномицины, митомицин, саркомицин,
азасерин, пуромицин и др.
По мере использования А. в качестве лечебных
средств происходит повышение устойчивости перво-
начально чувствительных микроорганизмов к их дей-
ствию. Такая приобретенная устойчивость микробов
кА. представляет собой частный случай общебиологич.
закономерности привыкания микроорганизмов к вред-
ным для них внешним факторам. Устойчивые варианты
микробов отличаются от чувствительных тем, что
у первых под влиянием А. изменяется обмен в-в,
т. е. изменяется экзиматич. система, к-рую специ-
фически блокирует данный А.
Если два А. различаются по механизму действия
на обмен веществ чувствительных бактерий, то повы-
шение устойчивости к одному из А., достигнутое
в процессе приучения бактерий, не изменяет чувстви-
тельности тех же бактерий к другому А. Таким обра-
зом, наличие или отсутствие перекрестной устойчи-
вости может быть использовано для доказательства
сходства или принципиального различия между срав-
ниваемыми препаратами. Так, перекрестная устой-
чивость микробов к различным вариантам пеницил-
лина указывает на общность биологич. механизма
действия этих препаратов. А. с широким спектром
действия (хлоромицетин, ауреомицин, террамицин)
способны одновременно поражать не одну, а неск.
ферментативных систем чувствительных микробов
и, т. о., могут отличаться механизмом действия на
различные бактерии. Для предупреждения привыка-
ния микробов к А. применяют комбинацию неск.
А., обладающих различным механизмом действия
на микробную клетку.
Выделено и описано св. 400 А., к-рые принадлежат
к разнообразным классам химич. соединений. Извест-
но, что многие А. образуются в процессе биосинтеза
в виде определенных семейств, члены к-рых в основ-
ном обладают близкой химич. структурой, но отли-
чаются нек-рыми деталями. Таково, напр., семейство
пенициллинов, члены к-рого имеют общую бицикли-
ческую систему, построенную
из сконденсированных
между собой тиазоли-
динового и 0-лактам-
ного колец, но разли-
чающиеся строением
бокового радикала R.
Строение боковой це-
6 5 2
KCONH-CH-НС'
•СНСООН
з
4
лелициллш, пи, ее положение и
длина оказывают тем
не менее существенное влияние на физико-химич.
и биологич. свойства пенициллинов (см. Пенициллины).
NH - NH
II СНОН ||
H2N-C-HNHC xhnh-c-nh2
| | (стрептиОин)
НС /СНОН
I ।/СНОН
нс—o^
I I
о HC-O--------—------CH
I I
OHC-COH HgCHNCH
1 О 1
------CH HCOH
<!:н3 носн
(стрептобиозомин)
CH
I
CH2OH
стрептомицин
Семейство стрептомицинов имеет 3 представителя:
стрептомицин, маннозидострептомицин и оксистреп-
томицин. Водными р-рами кислот стрептомицины
гидролизуются на два вещества, одно из к-рых, на-
CI Н3С Ci
он
° N Н 2
Т Т । онГ
он о но о
ауреомицин
зываемое стрептидином (1.3 дигуапидино-2,4,5, 6-
тетраоксициклогексан), является общим для всех
стрептомицинов, а другие, напр. стрептобиозамин,
являются различными дисахаридами (см., напр.,
структурную формулу стрептомицина).
След, практически важным семейством являются
тетрациклины, куда входит тетрациклин, хлортетра-
циклин, или ауреомицин, окситетрациклин, или терра-
мицин. Эти А. имеют общее нафтаценовое ядро и от-
личаются только заместителями (см., напр., струк-
турную формулу ауреомицина). Среди А. обнару-
жено много совершен-
но новых соединений и
группировок, не встре-
чавшихся ранее в при-
родных в-вах, например
хлорамфеникол, имею-
щий в своей молекуле
нитрофенильный ра-
дикал и дихлорацетиль-
ный остаток, ааасерин, представляющий собой диазо-
ацетил-А-серин. Многие из изученных А. могут быть по-
мещены в уже известные группы органич. соединений.
Напр., А. ароматического строения;
к ним можно отнести микофеноловую, гладиоловую,
анакардовую, 6-окиси-2-метилбензойную к-ты, ген-
тизиновый спирт, мицелианамид и др. Из соединений
ациклич. строения антибиотич. действием
обладают нек-рые предельные и непредельные моно-
карбоновые к-ты; аллицин, выделенный из чеснока,
представляющий собой аллиловый эфир аллитиосуль-
финовой к-ты и мп. др. А. кислородсодержа-
щие гетероциклические соединения
представлены пеницилловой, койевой, усниновой
к-тами, цитриником, гризеофульвином, патулином
и др. К группе А. азотсодержащих ге-
тероциклических соединений относятся: иоди-
нин, продигиозин, глиотоксин, пиосоединения, виола-
цеин и др. Большую и разнообразную группу пред-
ставляют А. полипептиды. Наиболее интересными
являются А. из семейства грамицидинов, бацитра-
цинов, полимиксинов, актиномицинов, альбомицинов,
эхиномицинов. Представители двух первых групп
являются полипептидами циклич. строения. Такие
А., как актиномицины, альбомицин, эхиномицин,
наряду с аминокислотами содержат ароматич. или
гетероциклич. радикалы и дисульфидные или эфирные
связи. Напр., альбомицин содержит 3-метилурацил
и 3-валентное железо, актиномицины — феноксази-
новый хромофор, этамицин — оксипиколиновую к-ту,
эхиномицин-хиноксалинкарбоновую к-ту и дисуль-
фидные связи.
Установлена структура грамицидина С, представ-
ляющего собой декапептид, состоящий из двух сим-
метричных пентапептидных половин, а также строение
таких А., как бацитрацин, этамицин, эхиномицин,
актиномицин D. Из А. б е л к о в следует упомянуть
лизоцим и нотатин. Последний представляет собой
фермент глюкозооксидазу, сам он не обладает апти-
биотич. действием, но катализирует окисление глю-
козы в глюконовую к-ту. При этом выделяется пере-
кись водорода, к-рая и подавляет бактерии. Интерес-
ной группой являются антигрибковые А. — полиены,
характерная особенность к-рых — наличие трех пиков
в ультрафиолетовой области спектра. По количеству
сопряженных двойных связей они разделяются на
тетраены, пентаены и гексаены.
Исследования в области А. проводятся в след,
направлениях: 1) поиски новых продуцентов анти-
биотич. веществ; 2) селекция продуцентов А. с целью
повышения их продуктивности; 3) изучение законо-
мерностей образования А. в культурах микроорга-
низмов; 4) выделение А. в чистом виде и их харакге-
239
АНТИБИОТИКИ-МАКРОЛИДЫ
240
ристика; 5) изучение химич. структуры А. и их син-
тез; 6) изучение химиотерапевтич. свойств антибиотич.
в-в и механизма их лечебного действия и организме;
7) изучение биохимич. основ подавляющего действия
А. на обмен веществ подавляемых ими микробов;
8) изучение технологии произ-ва А.
Лит.: Шемякин М. М. и Хохлов А. С., Химия
антибиотических веществ, 2 изд., М. — Л., 1953; Г а у з е Г. Ф.,
Лекции по антибиотикам, 3 изд., М., 1958; Красильни-
ков Н, А., Актиномицеты-антагонисты Ги антибиотические
вещества, М. — Л., 1950; Ермольева 3. В., Пеницил-
лин, М., 1946; Антибиотики, 1956—59; Florey Н. W.
[а. о.], Antibiotics. A survey of penicillin..., v. 1—2, L. —
N. Y.—Toronto, 1949; Antibiotics. Their chemistry and non-
medical uses, ed. H. S. Goldberg, Toronto — L.—N. Y., 1959;
Antibiot, and Chemotherapy, 1951—59: Antibiot, annual...,
1953—59. 1И. Г. Бражникова.
АНТИБИОТИКИ-МАКРОЛИДЫ — группа апти-
биотич. веществ, продуцируемых различными видами
Streptomyces. Общей отличительной особенностью
А.-м. является наличие макроциклич. лактонного
кольца, связанного с остатком аминосахара. Уста-
новлено строение 6 А.-м. (см. табл.) и и значительной
мере выяснено строение трех макролидов — пикро-
мицина, эритромицина-С и антибиотика А-246.
Кроме того, описаны еще недостаточно изученные
вещества, близкие к макролидам по происхождению,
микробиологич. и физико-химич. свойствам (олеаядо-
мицин, спирамиципы, лейкомицины, терциомиципы,
гризеомицин, нарбомицин, апголамицин, альбомице-
тин, нитроспорин, миамицин, антибиотик А-446). Пима-
рицин и антибиотик А-246 отличаются от других макро-
лидов тем, что в состав их молекул входит система
сопряженных двойных связей; не исключено, что
макролидной структурой обладают также и нек-рые
другие антибиотики — полиены.
Различные макролиды обладают весьма сходным
антибиотич. действием. Все они отличаются значи-
тельной бактериостатич. активностью по отношению
к большинству грамположительных бактерий. Нек-рые
из них способны подавлять также грамотрицатель-
ные бактерии видов Neisseria, Hemophilus, Brucella;
они активны и и отношении риккетсий, крупных ви-
русов и простейших. Полиеновые макролиды — пима-
рицин и антибиотик А-246 — тормозят рост многих
патогенных грибов, по почти не действуют на бакте-
рии. За исключением нарбомицина, все макролиды
активны не только in vitro, но и in vivo. У многих
микроорганизмов наблюдается перекрестная устой-
чивость к различным макролидам, что, по-иидимому,
связано с одинаковым механизмом их действия.
С другой стороны, различные бактерии (в особен-
ности стафилококки и стрептококки), ставшие устой-
чивыми к пенициллину, стрептомицину, тетрацикли-
нам, хлорамфениколу, как правило, остаются чув-
ствительными к макролидам. Это обстоятельство
приобретает большое значение, так как по мере внед-
рения антибиотиков в медицину появляется все
больше штаммов, нечувствительных к антибиотикам
массового применения.
Формула и название Т. пл., °C I [®1D
СН3 СН3 СНз СН3 с«н5сн—<!:—сн=снсоснсн2снснснсо 1 1 1 1 | ОН OR о 1 Метимицин 195—197 и 203—205 (модификации) +61° (в СН3ОН)
СНз СН3 СН3 СНз 1 III СН3СН—СНСНСН=СНСОСНСН2СНСНСНСО ОН | OR | О 1 Неометимицин 156—158 +93“ (в СНС13)
СНз СН3 СН3 СНз СН3 СН3 III III С»Н6СН —С—CHCHCOCHCHj—С—CHCHCHCHCO 111 1 1 1/ 1 1 X ОН ОН OR OR О 1 Эритромицин (X—ОН) Эритромицин В (X—Н) 135—140 и 190-193 (двойная т. пл.) 198 -73,5“ (в СН3ОН) —78“ (в С2Н5ОН)
СНз сно сн3снсн2сн—снсн=снсо<^нсн2снсн2сн—сн — СНСН2СО 1 \ / 1 1 1 1 | О OR"ОСНз ОАС О Магнамицин 212—214 —56,Г (в СНС13)
СНз сно СН3СНСН»СН=СНСН=СНСОСНСНзСНСН2СН-СН-СНСН.,СО ' 1111 I OR" ОСНз ОАС О 1 Магнамицин В 141-147 -35“ (в СНС13)
СН»ОН 1 СОаН СНОН 1 1 СНзСНСН2(СН=СН)4СНСН»СНСНСОСН-СН-С(-СН,)зСН=СНСО 1 1 1 \ / 1 1 OR"’ ОН О ОН о ! Пимарицин разлагается при 200 —
241
АНТИБИОТИКИ-ПОЛИЕНЫ — АНТИБИОТИКИ-ПОЛИПЕПТИД Ы
242
Сахарные остатки
N(CH3)3 СНз
I I
R=— СНСНСНСНз-СНСН3; R'=-CHCH3-C-CH-CHCH3
I I I 1 I I
ОН СН3О он
I-----о — I I-----о-----1
СН3 ОСОСН3СН(СН3)3
N(CH3)3 О------СНСН—СН—С(-СН3)СН3
I i II
R"=— снснсн----сн—снсн3 он
| ОН | -----0--------1
I-----О--------1
NH3
R'"=—СНСН-СН—СНСНСНз
I 1 I
ОН ОН
I--О-----1
Наиболее широко распространенным А.-м. являет-
ся эритромицин, низкая токсичность и большая
антимикробная активность к-рого позволяют ставить
его в один ряд с пенициллином и стрептомицином.
Известное практич. применение находят также маг-
памицин, олеандомицин, лейкомицины и спирами-
цины, несмотря на то, что они по токсичности примерно
в 3 раза превосходят эритромицин.
Лит.: Brink N. G. and Harman R. Е., Quarterly
Rev., 1958, 12, №2, p. 93; Фурер И. M., К а в e p И И a H. В.,
Антибиотики. Сб. переводов..., 1955, вып. 2, с. 53.
Л. Д. Бергельсон.
АНТИБИОТИКИ-ПОЛИЕНЫ — группа антибио-
тич. веществ, молекулы к-рых характеризуются нали-
чием четырех или более сопряженных двойных свя-
зей. Все они продуцируются стрептомицетами — лучи-
стыми грибами, широко распространенными в почве.
Поскольку большинство А.-п. еще мало изучено в
химическом отношении, их обычно классифицируют
по спектральным признакам. Согласно этой класси-
фикации, А.-п. разделяются в зависимости от положе-
ния УФ-максимумов на 4 группы, соответствующие
числу сопряженных двойных связей в молекуле анти-
биотика:
Тетраены
Амфотерицин А
Антибиотик РА-166
Антимикоин
Нистатин
Пимарицин
Протоцидин
Римоцидин
Эндомицин А
Хромин
Пентаены
Антибиотик РА-153
Антибиотик А-246
Капацидин
Филипин
Фунгихроматин
Эйроцидин
Фунгихромин
Гексаены
Медиоцидин
Флавацид
Эндомицин В
Гептаены
Амфотерицин В
Антибиотик РА-150
Антибиотик 1968
Аскозин
Кандидин
Кандицидин
Кандимицин
Трихомицин
Очистка и химич. исследование А.-п. представляют
значительные трудности ввиду малой стойкости этих
в-в. Элементарный состав А.-п. во многих случаях
еще точно не установлен; часто они не имеют опре-
деленной точки плавления, а разлагаются при нагре-
вании не плавясь. Поэтому не исключено, что нек-рые
из перечисленных полиенов впоследствии окажутся
идентичными. Наиболее характерной особенностью
биологич. действия А.-п. является их высокая актив-
ность в отношении грибов и дрожжей и малая актив-
ность в отношении бактерий. А.-п. приобрели боль-
шое значение для лечения вторичных микозов, вызван-
ных нарушением биологич. равновесия в организме
под влиянием пенициллина и др. антибиотиков. При
введении последних одновременно с гибелью микробов
начинают развиваться различные патогенные грибки,
чаще всего Candina. При длительном применении
антибиотиков, а также кортизона, адренокортико-
тропного гормона и антагонистов фолевой к-ты
бурное развитие этих грибков может привести к пора-
жениям кожи, слизистых и внутренних органов.
Ввиду возрастающего распространения вторичных
микозов (смертность от них в США увеличилась
с 1935 по 1950 в 10 раз) поиски А.-п. достигли в послед-
ние годы большого размаха. Наиболее важным в прак-
тич. отношении А.-п. является нистатин. Кроме
него, в медицинской практике нашли применение
также трихомицин, кандицидин, кандидин и др.
полиены. Высокая токсичность большинства А.-п.
препятствует их парентеральному введению и позво-
ляет применять их только местно.
Лит.: Соловьев С. Н., Беленький Б. Г., Ма-
лы ш к и н а М. А., Антибиотики. Сб. переводов..., 1958, №5,
с. 28; Гольдберг Л. Е., там же, 1957, № 2, с. 53.
Л. Д. Бергельсон.
АНТИБИОТИКИ-ПОЛИПЕПТИДЫ — группа по-
липептидов и нек-рых белков, обладающих антибак-
териальной активностью; состоят из аминокислот
и их производных; некоторые содержат элементы
структуры, относящиеся к гетероциклам, карбоновым
кислотам, углеводам, аминоспиртам и др. В отли-
чие от природных полипептидов и белков, в со-
став А.-п. входят L- и D-аминокислоты: орнитин
H2N(CH2)3CH(NH2)COOH, саркозин CH3NHCH2COOH,
диаминомасляная H2NCH2CH2CH(NH2)COOH и др.,
не встречающиеся в белках. Для большинства А.-п.
характерна циклопептидная структура молекулы.
Этот тип строения в одних случаях обеспечивает
поразительную устойчивость в-ва к ферментативному
и кислотному гидролизу (грамицидин С), в других,
как у полимиксинов, содержащих в своем составе
оксиаминокислоты, расщепление циклопептида про-
текает легко. А.-п. являются в-вами с ярко выражен-
ной биологич. специфичностью антиметаболитного
характера. Вопрос о влиянии D-аминокислот на
биологич. активность А.-п. до сих пор еще не полу-
чил своего полного разрешения. D-аминокислоты
широко распространены в обмене веществ микроор-
ганизмов и не являются исключительной особенностью
состава А.-п. В то же время они стабилизуют моле-
кулы антибиотика в такой мере, что последние ста-
новятся устойчивыми против действия протеолитич.
ферментов.
Низшие пептиды, вплоть до пентапептидов, обла-
дают низкой антибактериальной активностью. Уве-
личение полипептидной цепочки до декапептида ведет
к значительному увеличению активности. Переход
от линейной к циклопептидной структуре молекулы
А.-п. сопровождается увеличением антибактериальной
активности. Для проявления активности у А.-п. основ-
ного характера необходимо присутствие концевых ами-
ногрупп; карбоксильная группа является противопо-
казанной функциональной группой. Удаление кон-
цевой аминной группы от циклопептидной части моле-
кулы, напр., на 5—9 атомов углерода сопровождается
постепенным убыванием активности.
Классификация А.-п. основана на биологич. при-
знаке — наименовании продуцента, а не на химич.
природе антибиотика Продуцентами А.-п. являются
гл. обр. бактерии и в меньшей степени актиномицеты.
За малым исключением, тот или другой продуцент
образует неск. близких между собой антибиотиков,
что создает большие трудности при их идентификации.
В зависимости от условий культивирования и состава
питательной среды один и тот же продуцент может
существенно изменять характер биосинтетич. про-
цессов. Так, Вас. brevis способен синтезировать
ряд грамицидинов, схожих по спектру антимикроб-
ного действия, но различных по аминокислотному
составу. А.-п. могут быть классифицированы на:
1) антибиотики, производные аминокислоты, напр.
аза- или циклосерина', 2) антибиотики, собственно
243
АНТИБИОТИКИ ТРОПОЛОНЫ
244
полипептиды, молекула к-рых состоит только из
аминокислот — семейства грамицидинов, тироциди-
нов, бацитрацинов, лихониформинов, субтилинов
и др. мало изученные в-ва; 3) А.-n. смешанного со-
става, в молекуле к-рых наряду с пептидной частью
содержатся простетич. группировки — полимиксины,
актиномицины, алъбомицины, пуромицин и др.;
4) антибиотики-белки — пока что еще малочисленная
и слабо изученная группа в-в; практич. применение
нашли лизоцим и экмолин, антибиотич. активность
обнаружена у папаина; к этой же группе относят ряд
полипептидов и белков бактериального и живот-
ного происхождения: актиномицетин, парамеции,
стрептостазин, ингибины, лейкины, нек-рые гистоны,
сангвинины, инсектицин и ряд других в-в, еще не
получивших названия; наиболее изученным антибио-
тиком этой группы является лизоцим', 5) антибиотики-
депсипептиды — эннантины А, В, С; наиболее изу-
чен эннантин В; к ним также относятся самбуцинин,
аненацинин, фруктегинин, латеретин II и нек-рые др.;
состоят из N-метил-а-аминокислот и а-оксикислот,
соединенных между собой амидными и сложноэфир-
ными связями. Все антибиотики этой группы содержат
остаток только одной D-a-оксиизовалериановой к-ты
(СН3)2СНСН(ОН)СООН, остатки же аминокислот —
различны.
Лит.: Шемякин М. М., ХохловА. С. Химия анти-
биотических веществ, 2 изд., М.—Л., 1953, с. 445; Г а у з е Г. Ф.,
Лекции но антибиотикам, 3 изд. М., 1958; Work Т. S.,
Biochem. Soc. Sympos. Univer. Press. 1948, № 1, 61; Har-
ris J. I., Work T. S., Nature, 1948, 161, № 4099, 804;
Work T. S., Proc. Roy. Soc. of London, ser. В — Biolog.
sei., 1949, 136, № B883, p. 163; Erlanger B. F., G 0-
ode L. Nature, 1954, 174, № 4435, p. 840; Fox S. W.,
Fling M. В ol lent ack G. N. J. Biol. Chem., 1944,
155, №2, p. 465; S a 1 t о n M. R. I., Nature, 1957, 180, № 4581,
p. 338; Вышепан E. Д., Биохимия, 1954, 19, вып. 4, с. 490;
Знаменская М. П Агатов П. А., Белозер-
ский, ДАН СССР, 1948, 59, № 1, с. 95; С и л а е в А. Б.,
Степанов В. М., там же 1957, 112, № 2, с. 297; С т е п а-
н о в В. М., Силаев А. Б., П о л и и А. Н., Антибиотики,
1958, 3, 5, с. 49; О t a n i S., S a i t о J., Proc. Japan Acad.,
1954, 30, № 10, 991; Hotchkiss R. D„ Dubos R. J.,
J. Btol. Chern., 1940, 136, p. 791; Li Chon Hao, Ly-
ons W. R., Evans H. M., там же, 1940, .136, № 3, 709;
Крейг Л., Грегори Д., Барри Г., в кн.; Амино-
кислоты и белки. Сборник ст., пер. с англ., М., 1952, с. 32;
В a t ter si у A. R., Craig L. С J. Am. Chem. Soc.
1952, 74, № 16, p. 4019, 4023; P a 1 a d i n i A., C r a 1 g L. C„
там же, 1954, 74, № 3, p. 688; King T. P., C r a i g L. C.,
там же, 1955, 77, № 24 p. 6627; Хохлов А. С. [и др.1,
Антибиотики, 1960, 5, М 1, с. 3; С и л а е в А. Б. [и др.],
там же, № 3, с. 18. А. Б. Силаев.
АНТИБИОТИКИ-ТРОПОЛОНЫ — группа про-
изводных цикло-2,4,6-гектатетраен-1-он-2-ола (1)
(li1=R2=R3=R4=R5=H), обладающих антибакте-
риальным и противогрибковым действием. Известные
А.-т. (I) приведены в таблице:
Название R, К2 Rs r4 Rs Т. пл., °C УФ-спектр
хмакс.,ммк
а-Туяплицин1 . . (з-Туяплицин 1 . . СН(СН3)2 Н н н н 343 266: 320; 357
(хинокитиол) . н СН(СН3)2 н н н 52 236 ; 322; 353
7~Туяплицин 1 . . н н СН(СН3)2 н н 82 225; 323; 358; 375
Нуткатин1 . . . Стипитатовая кис- н СН(СН3)2 4 н н 94-95 238 ; 324; 357
лота а Пуберуловая кис- н СООН Н он н 302—304 262; 332: 360
лота1 Пуберулоновая н СООН н он он 317 270; 350
кислота 2. . . . строение — см. формулу (II) 298 (С разл.) 274,5; 317
R3=—СН2СН=С(СН3)2.
1 Бесцветные кристаллы. 2 Желтые кристаллы. «Имеется указание о существовании
еще одной модификации — желтых кристаллов с т. пл. 26”. ' " ”” •
Химич, свойства А.-т. определяются в основном
характерными особенностями трополонового цикла.
Они способны взаимодействовать с различными элект-
роноакцепторными реагентами. При бромировании
Аз
/зЧ
R&I 2.
но
но
СО
>О
СО
Ч /
•Н/
заместителями
в положениях
в положении 7.
а- и у-туяплицинов получены дибромпроизводные,
а при нитровании — моно- и дизамещенные продукты;
сульфирование у-туя-
плицина дает 3-суль-
фокислоту, с а-туя-
плицином реакция не
идет. 0-Туяплицин в
зависимости от усло-
вий реакции и харак-
тера реагента дает
моно-, ди- и тризаме-
щенные галогено- и
нитропроизводные с
3, 5 и 7; сульфирование идет только
Нитрозирование а- и 0-туяплицинов приводит к 5-ни-
трозоизопропилтрополонам, при восстановлении к-рых
получены амины, превращаемые после диазотирования
в галогено-, циан- и оксисоединения. Сочетанием а-
и 0-туяплицинов с солями бензол- и нафталиндиазо-
ниев получены 5-азопроизводные.
Стипитатовая и пуберуловая к-.ты из-за наличия
трополоновой группировки титруются как двухоснов-
ные, а после восстановления — как одноосновные
к-ты. Стипитатовая к-та бромируется до монобром-
производного. Пуберулоновая к-та (II) при кипячении
в водном р-ре или в разбавленной H^SC^ отщепляет
СО2 и превращается в пуберуловую кислоту. Туяпли-
цины и нуткатин при нагревании с CH3ONa в сре-
де метанола, а стипитатовая кислота при сплав-
лении со щелочью изомеризуется в соединения ряда
бензола.
А.-т. дают характерное окрашивание с FeCl3:
туяплицины — зеленое, стипитатовая к-та — красное,
пуберуловая к-та — красно-коричневое, нуткатин —
вначале красное, а с избытком FeCl3 — зеленое.
Туяплицины и нуткатин образуют растворимые в
СНС13 комплексы с медью. Туяплицины и нуткатин
найдены в различных видах кипарисовых (Cupres-
saceae); стипитатовая к-та образуется плесенью Peni-
cillium stipitatum Thom. Пуберуловая и пуберуло-
новая к-ты выделены из нескольких видов Penicil-
lium — Р. puberulum, Р. aurantio-virens и др. А.-т.
оказывают антифунгальное действие. 0-Туяплицин
подавляет, напр., дрожжи рода Candida при концент-
рации 4 мкг/мл, патогенные для человека грибы из
родов Trichophyton, Epidermophyton, Achorion при
концентрации 8—16 мкг/мл', пуберуловая и стипита-
товая к-ты активны против гриба Polyporus annosus
(первая при концентрации 10 мкг/мл, вторая —
100 мкг /мл). К туяплицинам, особенно к 0-изоме-
ру, очень чувствительны грибки, вызывающие гние-
ние древесины. Это объясняет высокую устойчи-
вость деревьев, образующих
туяплицины, к гниению. Не-
которые А.-т. обладают также
антибактериальной активно-
стью, пуберуловая к-та пода-
вляет грамположительные ба-
ктерии (в концентрациях 15—
30 мкг /мл), 0-туяплицин акти-
вен против Shigella dysenteriae
и Mycobacterium tuberculosis;
он действует также и на не-
которые спирохеты. Примене-
ния в медицинской практике
А.-т. не находят. Описаны по-
пытки использования Р-туяпли-
цина при лечении туберку-
лезных заболеваний человека.
Отмечено, что чувствительность бактерий к стреп-
томицину в присутствии 0-туяплицина повышается.
Лит.: Шемякин М. М., Хохлов А. С., Химия ан-
тибиотических веществ, 2 изд., М. — Л., 1953; Пос он П.,
245
АНТИВИТАМИНЫ - АНТИГЕНЫ
246
Химин тропонов и трополонов, пер. с англ., М., 1956, N о-
z о е Т„ в кн.: Fortschritte der Chemie organischer NaturstoHe,
Bd. 13, W., 1956, S. 232. Г. С. Розенфельд.
АНТИВИТАМИНЫ — группа органич. соедине-
ний, обладающих свойством подавлять биологич.
активность витаминов. А. имеют большое зна-
чение при изучении специфич. действия витаминов
на животных и ростовых факторов на микроорганиз-
мах. По своей структуре А. в большинстве случаев
сходны с витаминами. Иногда структурное сход-
ство является достаточно отдаленным, как это имеет
место, напр., среди антивитаминов К.
Как и антиметаболиты, А. в биокаталитич. реак-
циях подменяют истинные катализаторы — вита-
мины, проявляющие свои каталитич. функции в со-
ставе ферментных систем. По-видимому, А. наряду
с витаминами способны также входить в состав фер-
ментных систем, образуя псевдоферментные, не обла-
дающие биокаталитич. функциями, но способные
подавлять биокаталитич. активность истинных фер-
HOCH2C(CH3)2CH(OH)CONH(CH2)2COOH
III
HOCH2CtCHa)2CH(OH)CONH(CH2)2SO3H
IV
CONHCH-СООН
СН2СН2СООН
XIII
ментов, в состав к-рых входят витамины, или способ-
ные вытеснять витамины из коферментов или фермен-
тов. Одним из характерных представителей А. яв-
ляются сульфаниламидные препараты, бактериоста-
тич. активность к-рых основана на том, что они в био-
химик. реакциях, протекающих в микробной клетке,
конкурируют с витамином Ht — п-аминобензойной
к-той — и подавляют его активность. Антивитамин-
ная активность обычно является следствием геомет-
рия. и функционального сходства между витамином
и его антагонистом.
Важнейшие антивитамины и подавляемые ими витамины
Витамин Антивитамин
L-Аскорбиновая к-та — вита- мин С (I) Пантотеновая к-та — вита- мин В3 (III) n-Аминобензойная к-та— ви- тамин Ht Витамины К (производные нафтохинона) Никотинамид — витамин РР (VI) Пиридоксин — витамины Вй (VIII) Тиамин — витамин Bt (X) Фолиевая к-та — витамин Вс (XII) Рибофлавин — витамин Во (XIV) D-Аскорбиновая к-та (II) Пантоилтаурин, сульфопанто- теновая к-та (IV) Сульфаниламидные препараты Дикумарин (V) Пиридин-З-сульфо кислота (VII) 2-Этил-3-амино-4-этоксиамино- метилпиридин (IX) Пиритиамин (XI) Аминоптерин (XIII) 5,6-Диметил-9 (Г-П-РибитПн)- -изоаллоксин (XV)
Лит.: Березовский В. М., Химия витаминов М.,
1959: Вулли Д., Учение об антиметаболитах, М., 1954.
В, М. Березовский.
АНТИВУАЛИРУЮЩИЕ ВЕЩЕСТВА — см. Про-
тивовуалируюгцие вещества.
АНТИГЕНЫ — вещества, вызывающие при введе-
нии в организм образование особых глобулинов —
антигйел. А., как правило,— чужеродные для данного
организма белки, их комплексы с полисахаридами,
липоидами, нуклеиновыми к-тами, а также нек-рые
полисахариды преим. бактериального происхождения.
У разных белков антигенная активность проявляется
различно: у нек-рых (гемоглобин, протамины и т. д.)
она выражена довольно слабо, а другие (сывороточ-
ные белки, экзотоксины и др.) обладают сильно
выраженными антигенными свойствами. Одно из важ-
нейших свойств А. проявляется в их способности реа-
гировать только с теми антителами, к-рые возникают
247
АНТИГРИЗУТНЫЕ ВЗРЫВЧАТЫЕ ВЕЩЕСТВА — АНТИОКИСЛИТЕЛИ
248
при введении данного А. О взаимодействии А. с соот-
ветств. антителами судят по образованию видимых
продуктов реакции (преципитат, агглютинат) или
по биологич. эффекту (анафилактич. реакция, нейтра-
лизация вируса, токсина и т. д.). Антигенные свойства
белков довольно легко изменяются или даже утра-
чиваются под влиянием разнообразных физич. или
химич. воздействий. Измененные тем или иным спо-
собом белки (напр., соединения белков с различными
диазосоединениями, азидами и т. д. — т. н. комплекс-
ные или конъюгированные А.) широко используются
для изучения природы антигенной специфичности,
для к-рой решающее значение имеет наличие в моле-
куле А. определенных участков — детерминантов
(образованных полярными группами различных ами-
нокислотных остатков — тирозина, лизина, арги-
нина и др.). Большое значение имеет также взаимное
расположение детерминантов, зависящее от простран-
ственного расположения полипептидных цепей в бел-
ковой молекуле.
Лит.: Зильбер Л. А., Основы иммунологии, 3 изд.,
М., 1958; Гауровитц Ф., Химия и биология белков, пер.
с англ., М., 1953; Boyd W. С.. Fundamentals of immunology,
3 ed., N. Y. — L., 1956. В. И. Мазуров.
АНТИГРИЗУТНЫЕ ВЗРЫВЧАТЫЕ ВЕЩЕСТ-
ВА •— см. Предохранительные взрывчатые вещества.
АНТИДЕТОНАТОРЫ МОТОРНЫХ ТОПЛИВ —
химич. соединения, добавляемые в незначительных
количествах (менее 1%) к моторным бензинам для
повышения их детонационной стойкости. Физико-
химич. свойства топлив при добавлении к ним анти-
детонаторов практически остаются без изменения.
Ниже приведена относительная эффективность неко-
торых антидетонационных добавок по критич. степе-
ни сжатия. Действие бензола принято за единицу.
Название Формула Относительная эффективность
Бензол с«нв 1,0
Этиловый спирт С2Н6ОН 2,0
Анилин c6h5nh2 13,5
Ксилидин Сд±1з(С±13)21^ ±±2 15,0
Дифениламин (CsH6)aNH 16,0
Тетраэтилолово Sn(C2H5)4 25,0
Диэтилселен Se(C2H5)2 60,0
Диэтилтеллур Te(CoH5)o 200,0
Тетракарбонил никеля . . . Ni(CO)4 300,0
Хлористый триэтилсвинец РЬС1(С»Н5)3 450,0
Пентакарбонил железа . . . Fe(CO)5 500,0
Тетраэтилсвинец (ТЭС) . . . Pb(CaH5)4 600,0
Из всех известных А. м. т. применяется только
ТЭС в виде этиловой жидкости, в состав
к-рой входят галогенуглеводороды, способствующие
удалению из камеры сгорания твердых продуктов
сгорания ТЭС, содержащих свинец. Опыт примене-
ния пентакарбонила железа показал, что в двигателе
образуется большое количество твердых окислов
железа, к-рые приводят к быстрому износу мотора
и нарушению его нормальной работы. Эффективных
в-в для удаления твердых продуктов его сгорания
найти не удалось.
В последние годы были найдены новые А. м. т.,
важнейшим из к-рых является метилциклопента-
диенилтрикарбонилмарганец СН3С5Н4Мп(СО)з.
Лит. см. при ст. Детонационная стойкость моторного
топлива. В. В. Панов.
АНТИДОТЫ (противоядия) — химич. соединения,
способные обезвреживать попавшие в организм яды
или отравляющие вещества. К таким соединениям
относится, напр., амилнитрит, являющийся А. про-
тив HCN, токсич. действие к-рой состоит в подавлении
железосодержащих дыхательных ферментов клеток
за счет связывания 3-валентного атома железа этих
ферментов; амилнитрит превращает часть гемоглобина
крови в метгемоглобин, способный «снимать» HCN
с блокированных ею железосодержащих ферментов
с образованием цианметгемоглобина, из к-рого си-
нильная к-та постепенно выводится в виде безвред-
ных роданидов. Тиоловые соединения
(«бал» — CHaSH—CHSH—СНаОН; «унитиол» —
CH2SH—CHSH—CHjSCKjONa-НаО) применяются при
поражениях галогенарсинами, люизитом и т. п.;
последние блокируют сульфгидрильные группы не-
которых ферментативных систем, а тиоловые соеди-
нения регенерируют сульфгидрильные группы фер-
ментативных систем, восстанавливая тем самым их
нормальную биохимич. деятельность. Атропин и
нек-рые производные гидроксиламина являются А.
против ядовитых фосфорорганич. соединений. Послед-
ние подавляют фермент холиноэстеразу, регулирую-
щий содержание в организме ацетилхолина, к-рый
накапливается в организме, что и приводит к отрав-
лению. Действие производных гидроксиламина, напр.
иодметилата-пиридин-2-альдоксима и ему подобных,
выражается в основном в восстановлении активности
самой холинэстеразы. Использование А. описанных
типов позволяет нейтрализовать действие много-
кратных смертельных доз нек-рых ядов и ОВ.
Лит.: Степанский Г. А., Фармакология и токсико-
логия, 1958, 21, № 2, с. 83—90; Wilson J. В., Neurology,
1958, 8, suppl. № 1, р. 41. Р. И. Стерлип.
АНТИКАТАЛИЗАТОРЫ (отрицательные катали-
заторы) — вещества, уменьшающие скорость химич.
реакций. По характеру действия А. могут быть
разделены на след, группы: вещества, выводящие
положительные катализаторы из сферы реакции
за счет их адсорбции, образования неактивных ком-
плексных соединений или соединений, выпадающих
в осадок, а также замедляющие диффузию реагирую-
щих соединений к поверхности положительного
катализатора или продуктов реакции; вещества, отрав-
ляющие поверхность положительного катализатора
(см. Яды каталитические)', в-ва, способствующие
обрыву возникающих реакционных цепей (см. Инги-
биторы). Действие А. может быть полезным (если
они тормозят нежелательные реакции) или вредным
(если они препятствуют нужным реакциям). К полез-
ным А. относятся, напр., антидетонаторы, инги-
биторы окисления каучука (антиоксиданты) и др.
веществ, замедлители схватывания гипса. Так, напр.,
1 часть гидрохинона на 200 000 частей акролеина
предотвращает его нежелательную полимеризацию
и т. д. В отличие от положительных катализаторов,
действие А. в большинстве случаев нециклично,
а однократно, т. е. в ходе реакции они претерпевают
необратимые изменения. Вопрос о применении того
или иного полезного А. зависит от эффективности
действия (т. е. минимальной эффективной концент-
рации) и влияния на другие свойства продуктов ре-
акции, которое также может быть полезным или
вредным.
Лит.: Бейли К Торможение химических реакций,
пер. с англ., Л. — М., 1940; Abel Е., в кн.: Advances in
catalysis, v. 9, N. Y1957, p. 330. С. Л. Кипермап.
АНТИМЕТАБОЛИТЫ — вещества, к-рые вслед-
ствие их структурной близости природным продуктам
обмена веществ (напр., витаминам, аминокислотам,
гормонам) могут конкурировать с ними в биологич.
системах, что ведет к нарушению нек-рых звеньев
обмена веществ. См. также Антивитамины и Обмен
веществ.
Лит.: В ул л и Д., Учение об антиметаболитах, пер.
с англ., М., 1954.
АНТИОКИСЛИТЕЛИ (антиоксиданты, ингиби-
торы окисления) — вещества, предотвращающие или
замедляющие окисление молекулярным кислородом.
В качестве А. широко применяются ароматич. сое-
динения, содержащие фенольные ОН- или аминогруп-
249
АНТИОКИСЛИТЕЛИ-АНТИОКИСЛИТЕЛИ ПИЩЕВЫХ ПРОДУКТОВ
250
пы. Ничтожные количества этих соединений (0,01 —
0,001%) могут надолго приостановить окисление
углеводородов, альдегидов, жиров. Действие А. тесно
связано с цепным механизмом окислительных реак-
ций. Окисление органич. веществ молекулярным
кислородом протекает по цепному механизму (см.
Цепные реакции). Главную роль в этих реакциях
играет перекисный радикал RO2. Молекулы А. реа-
гируют с перекисными радикалами. В результате
каждой такой реакции активный RO2 заменяется
малоактивным радикалом из молекулы А., не способ-
ным энергично продолжать цепь. Поэтому реакция
в присутствии А. замедляется или приостанавливается.
В случае торможения фенолами, напр. гидрохиноном,
обрыв цепи происходит по реакции:
НО
ROOH + НО
О
Образовавшийся малоактивный семихинонный ради-
кал реагирует со след, перекисным радикалом:
ROOH + О
Из фенолов хорошими А. являются 2- и З-атомные
фенолы, триалкилфенолы, напр. 2,6-диметил-4-тре-
тичный бутилфенол, а также а- и р-нафтолы. Элемен-
тарные константы к скорости реакций радикалов
RO2 с нек-рыми фенолами ХН по схеме RO2 ХН =
= ROOH-|-X приведены в таблице (X, напр.,
в случае гидрохинона соответствует НО—С6Н4О—):
Окисляемое вещество ХН °C \к(л‘Моль 1-сек *)
Этиллинолеат Гидрохинон 45 2,5-106
Декановый альдегид . . Гидрохинон 5 1,6-105
Бензальдегид Гидрохинон 5 3,9-105
Тетралин 0-Н афтол 25 2,1-101
Этилбензол 0-Нафтол 70 5-Ю3
Этилбензол а-Н афтол 70 5-103
Механизм торможения ароматич. аминами заклю-
чается, по-видимому, в присоединении радикала
RO2 к молекуле А. с образованием малоактивного
радикала. Сильное тормозящее действие на окисление
углеводородов оказывает Н,Г41-дифенил-п-фенилен-
диамин, фенил-р-нафтиламин, ди-р-нафтиламин, ами-
нофенолы. Аптиокислительными свойствами обла-
дают и такие в-ва, как диалкилсульфиды. Их действие
состоит в разрушении гидроперекисей ROOH -|-
4-RjSRj —>.ROH-|- RjSOR, которые накапливаются
в окисляющемся углеводороде и, распадаясь на сво-
бодные радикалы, ускоряют дальнейшее окисление.
Диалкилсульфиды добавляются в количестве 1—2%,
они тормозят окисление при высоких темп-рах.
Реагируя со свободными радикалами, А. .неизбежно
расходуется. Поэтому торможение наблюдается до тех
пор, пока не израсходуется весь А. Продолжитель-
ность торможения процесса зависит как от концент-
рации введенного А., так и от скорости инициирова-
ния цепей, к-рое, в свою очередь, зависит от темп-ры
и глубины окисления. Одна и та же добавка А. может
тормозить окисление в течение долгих месяцев при
невысокой темп-ре и исчезать за неск. минут при
высокой из-за мощной скорости инициирования
цепей и, следовательно, высокой концентрации RO2.
Тормозящее действие зависит и от момента введения
А. в реакцию. Если ввести А. в исходное в-во, когда
процесс окисления еще не начался, то наблюдается
сильное торможение реакции. При введении его
Изменение в кинетике накопления
гидроперекиси при окислении н-де-
кана (130°) в результате введения
а-нафтола (2‘10~а м/л) в процессе
реакции.
в развившийся процесс — торможение более слабое
и непродолжительное. На рисунке показано изме-
нение в кинетике окисления н-декана при введе-
нии а-нафтола по хо-
дуреакции. Отчетливо
видно, что чем позже
введен ингибитор,тем
короче период тор-
можения. На практи-
ке эффективность дей-
ствия А. определяет-
ся по продолжитель-
ности периода тормо-
жения, вызванного
одним и тем же коли-
чеством различных в-в
при стандартных ус-
ловиях окисления.
А. имеют большое
прикладное значение.
Для стабилизации
крекинг-бензинов к
ним добавляются р-нафтол и бензил-п-аминофенол. А.,
добавленные к моторным, турбинным, трансформатор-
ным маслам, снижают образование в них к-т, смол,
шлама и повышают срок службы в 2—4 раза (см.
Ингибиторы). Для защиты каучука от старения в
него вводятся такие А., как гидрохинон, пирокате-
хин и а-нафтиламин (см. Противостарители). Пи-
щевые жиры предохраняются от порчи путем вве-
дения нетоксичных А. — бутилоксианизола, бутил-
окситолуола, эфиров галловой к-ты. См. также Анти-
окислители пищевых продуктов.
Лит,.: Крейн С. Э., Стабилизация турбинных и транс-
форматорных масел, М. — Л., 1948; Денисов Е. Т.,
Эмануэль Н. М., Усп. химии, 1958, 27, вып. 4, 385;
Pedersen С. J., Industr. and Eng. Chem., 1956 48, 1881.
E. T. Денисов.
АНТИОКИСЛИТЕЛИ ПИЩЕВЫХ ПРОДУК-
ТОВ — природные и синтетич. вещества, задержи-
вающие процесс окисления жиров и жиросодержащих
продуктов. По природе действия А. п. п. разделяются
на первичные А. п. п. и синергисты. Первичные
А. п. п., являясь ингибиторами цепного процесса
автоокисления глицеридов, прямым путем задержи-
вают окислительную порчу жиров. Под синергистами
понимают вещества, усиливающие эффективность пер-
вичных А. п. п. К синергистам относятся, напр.,
лимонная и аскорбиновая к-ты. Натуральные А. п. п.
содержатся в природных жирах, кукурузной и соевой
муке, в жмыхах, в шроте льна и сои. Среди встре-
чающихся в природе А. п. п. наибольшее значение
имеют токоферолы. Наиболее богаты токоферолами
рыбий жир, сахарная свекла, масло пшеничных заро-
дышей, коровье масло. Токоферолы проявляют анти-
окислительное действие в количествах 0,003—0,02%
от веса жира. Добавление к свиному топленому жиру
0,02% токоферолов и 0,1% аскорбиновой к-ты,
играющей роль синергиста, увеличивает стойкость
свиного топленого жира к окислению в 160 раз.
Очень эффективными натуральными А. п. п. для
жиров являются лецитины и каротин, входящие
в состав многих растительных жиров и нек-рых
овощей и ягод. Для увеличения стойкости жиров,
потерявших при рафинации щелочью большую часть
природных А. п. п., рекомендуется вводить в жиры
1—5% сырого хлопкового масла, сырого соевого
масла, пальмового масла, масла какао, богатых нату-
ральными А. п. п.
Из синтетич. А. п. п. следует в первую очередь
упомянуть о различных сложных эфирах галловой
к-ты (галлатах), среди к-рых широкое применение
нашли этил-, пропил- и додецилгаллаты. Установлено,
что при введении 0,01% от веса жира этил- или
251
АНТИОКИСЛИТЕЛЬ ДРЕВЕСНО-СМОЛЬНЫЙ — АНТИПОДЫ ОПТИЧЕСКИЕ
252
пропилгаллата в свиной жир стойкость его к окисле-
нию увеличивается в 10 раз. Пропилгаллат приме-
няется для увеличения стойкости жира в кондитер-
ских изделиях и рыбных продуктах. При введении
0,07% этилгаллата в молочный порошок стойкость
его повышается в 2,5 раза. Очень эффективным А. п. п.
является бутилоксианизол, поступающий в продажу
под названием ВНА в виде смеси З-трет-бутил-4-
оксианизола (I) с 2-трет-бутил-4-оксианизолом (II):
Преимущество ВНА заключается в том, что он очень
хорошо растворим в жирах и не разрушается при
выпечке изделий и обработке конфетных масс и эффек-
тивен в концентрациях 0,005% от веса жира. Добав-
ление 0,01% от веса жира ВНА к лярду повышает
стойкость последнего в 5—13 раз. В США ок. 50%
поступающего в продажу лярда содержит ВНА.
Широко применяется ВНА для стабилизации жира,
идущего на приготовление печенья.
В качестве А. п. п. для жиров применяется также
3,5-ди-т/>ет-бутил-4-окситолуол, выпускаемый под
названием ВНТ, «ионол» (III). Широкое применение
в качестве А. п. п. получила нордигидрогеаяретоеая
кислота — 2,3-бис(3,4-диоксибензил)-бутан (IV), вы-
деляемая из кустарника Larrea divaricate, произ-
растающего в юго-западных штатах США, или полу-
чаемая синтетически (СССР). Природная к-та, выде-
НО-тИ^гСНо-СН-СН-СНд-й^Ч-ОН
II
но-Ч^ СН3 СН3 ч^-он
IV
ляемая из кустарника, выпускается в США под
названием NDGA. Применяется также обработка
пищевых продуктов смесью А. п. п., напр., в Швеции
для стабилизации свежего молока в него вводят смесь,
содержащую 0,00005% додецилгаллата, 0,0002% про-
пилгаллата и 0,00005% NDGA.
Лит,.: Гол ант Б. Я., Петров Н. А., Повышение
стойкости жиров и жиросодержащих продуктов, М., 1958;
Эмануэль Н. М. [и др.], Мясная индустрия, 1955, № 5,
с. 44; № 6, с. 47; Г е р ч у к М. П., Иванова В. М.,
Маслобойно-жировая пром-сть, 1958, №6,с. 44; Manneck И.,
Antioxydantiem Garmisch—Partenkirchen, 1956; Smi thF. H.,
Brady D. E., Comstock R. E., Ind. and Eng. Chem.,
1945, 37, № 12, p. 1206. M. П. Герчук.
АНТИОКИСЛИТЕЛЬ ДРЕВЕСНО-СМОЛЬНЫЙ —
фракция масел, кипящая в пределах 240 — 310°,
выделяемая из древесных смол; активной частью
А. д.-с. являются диметиловые эфиры метил- и пропил-
пирогаллола, а также пирокатехин. А. д.-с. приме-
няют в качестве добавки в весьма малых количествах
(ок. 0,1% по весу) к крекингбензину с целью тормо-
жения процессов образования «фактических смол» —
источников нагарообразования в цилиндрах двигате-
лей. Различают А. д.-c.: марки А — для авиац. бен-
зинов и марки Б — для автомобильных; отношение
первоначального стабилизирующего эффекта 50 мг
А. д.-с. в 100 мл бензина к стабилизирующему эффекту
10 мг а-нафтола в 100 мл того же бензина должно
быть соответственно не менее 0,95 и 0,90.
Л. В. Гордон.
АНТИПИРЕНЫ — вещества, предохраняющие про-
питанную ими древесину, ткани и некоторые др.
органич. материалы от воспламенения и самостоятель-
ного горения. Предохраняющее действие А. опреде-
ляется к.-л. из след, факторов (или их суммой):
1) низкая темп-ра плавления с образованием тонкого
слоя глазури, затрудняющего доступ кислорода к го-
рящему материалу; 2) разложение при нагревании
с выделением инертных газов или паров, что затруд-
няет воспламенение газообразных продуктов разло-
жения древесины; 3) поглощение большого количества
теплоты на плавление, испарение и диссоциацию А.,
что предохраняет пропитанные материалы от нагре-
вания до темп-ры разложения; 4) повышенное (почти
удвоенное) углеобразование пропитанных материа-
лов вследствие образования к-т при термич. разло-
жении А., а также за счет уменьшения количества
улетучивающегося и сгорающего углерода и его сое-
динений.
Огнезащитными свойствами обладают соли, содер-
жащие Al, NH4, Са, Со, Li и др., а также кислотные
радикалы (арсенат или арсенит, борат, бромид, вана-
дат, вольфрамат, молибдат и т. п.). Наиболее употре-
бительны в наст, время А., содержащие соли аммо-
ния, щелочные и щелочноземельные соли фосфорной,
борной, серной и соляной к-т, особенно фосфорнокис-
лый и сернокислый аммоний и их смеси. Хорошими,
но малодоступными А. являются бура и борная к-та.
А. вводятся в древесину обычно путем ее пропитки
водными р-рами (50—100 или более кг безводной соли
на 1-и3) с последующей сушкой. Многие А. при введе-
нии их в древесину затрудняют ее последующую
склейку казеиновым, иногда животными и растит,
клеями, но обычно не препятствуют склейке синте-
тич. клеями.
Лит.: Копытковский Б. Ф., Способы придания
огнестойкости дереву, М. — Л., 1931; Лекторский Д. Н.,
Защитная обработка древесины, ч. 1, М. — Л.. 1951; его
ж е, Пропитка древесины, М. 1940; Способы и средства огне-
защиты древесины, [Сб. ЦНИИПО, 5], М., 1952; Т а у б-
к и н С. И., Огнезащита деревянных конструкций и сооруже-
ний, М., 1943. Д. Н. Лекторский, Е. Е. Алексеева.
АНТИПИРИН (1 -фенил-2,3-диметилпир азолон-5),
мол. в. 188,22 — бесцветные кристаллы (листочки)
слабогорького вкуса; т. пл. 110—130°, т. кип.
319°/174 мм; хорошо растворяется
в воде (1 : 1), спирте (1 : 1,5) и
хлороформе (1:1,5), хуже — в эфире
(1:75), толуоле и лигроине. А.—силь-
ное однокислотное основание, даю-
щее все характерные реакции на алкалоиды. С ионами
редкоземельных элементов (Се, La, Nd, Ргидр.)обра-
сн—со
СН3 СНз
зует комплексные катионы, осаждающиеся комплекс-
ными анионами с образованием различных по своей ра-
створимости соединений, что может быть использовано
для их разделения. Водные р-ры А. окрашиваются FeCJ3
в красный цвет, нитритами — в изумрудно-зеленый
цвет. Количественно А. определяется иодометрически
в присутствии ацетата натрия. А. получают при
действии диметилсульфата, метилового эфира бензол-
сульфокислоты или др. метилирующих средств на
1-фенил-3-метил~5-пиразолон. Применяется А. как
жаропонижающее и болеутоляющее средство; служит
исходным продуктом при синтезе пирамидона и аналь-
гина; кроме того, А. используют как реактив при
микрокристаллоскопич. определении ряда элементов,
дающих комплексные анионы, осаждающиеся анти-
пирином (Sb, Bi, Sn, Zn, Hg, Re), для фотометрия,
определения нитритов, нефелометрия, определения
HgJ2L, весового определения W, Cd, Fe(CN)|~.
Лит. см. при ст. Анальгии.
В. 1'. Типцова, А. И. Травин.
АНТИПОДЫ ОПТИЧЕСКИЕ (зеркальные изо-
меры, энантиомеры) — вещества, молекулы к-рых
относятся друг к другу, как предмет к своему зеркаль-
ному изображению. Любой асимметрия, молекуле
всегда соответствует А. о., характеризующийся оди-
наковыми физич. и химич. свойствами, по отли-
чающийся направлением (знаком) вращения плоско-
253
АНТИПОДЫ ОПТИЧЕСКИЕ
254
сти поляризованного света и обладающий хотя и
слабо разнящейся, но все же различной реакционной
способностью в реакциях с другими асимметрия, моле-
кулами или ионами (см. Асимметрический синтез).
По аналогии с зеркальноподобными правой и левой
рукой человека один из этих антиподов наз. правым
(D, dexter), другой — левым (L, laevus). Знаки D и L
обозначают конфигурации и не связаны со способом
обозначения (проекцией). Они не имеют прямого
отношения к наблюдаемому знаку вращения плос-
кости поляризованного света. Установление конфи-
гурации, т. е. выбор приставки (D или L) или отнесе-
ние к D- или, соответственно, к L-ряду, производится
путем сравнения с конфигурацией ключевого (стан-
дартного) в-ва. Для молекул, асимметрия которых
вызвана присутствием асимметрия, атома углерода,
таким в-вом служит правовращающий глицериновый
альдегид; правовращающий (-+-) глицериновый аль-
дегид принадлежит к D-ряду. Такое отнесение может
быть осуществлено как при помощи физич. (рентгено-
структурных), так и химич. методов. Химич, методы
установления конфигурации заключаются в последо-
вательном превращении оптически активного в-ва
в D- или в L-глицериновый альдегид или в оптиче-
ски активное в-во, конфигурация к-рого относительно
глицеринового альдегида была установлена ранее.
При этом пользуются такими методами, при к-рых
асимметрия, центр либо не затрагивается, либо затра-
гивается лишь строго определенным образом (стерео-
специфично). Напр.: D-конфигурация (—) винной
к-ты следует из того, что она может быть получена
из D-глицеринового альдегида:
СООН
СНО I
I НО... I ..-Н
I HCN, Н2О ’’••С--''’
Н--” 1 -•••он J...
СН2ОН н’’ | он
СН2ОН
D(+)
НО-.
онисл.
В случае невозможности полного установления кон-
фигурации производят отнесение к ряду родственных
соединений или же ограничиваются указанием знака
вращения.
Необходимо отметить, что в литературе укоренилось непра-
вильное использование приставок D и L не только для обозна-
чения конфигурации, но и для обозначения направления вра-
щения. Напр., левовращающую винную к-ту часто называют
L-кислотой, хотя она имеет Г)-ко1Тфигурацию.
Молекулы А. о. противоположной конфигурации,
соединяясь друг с другом, образуют в нек-рых слу-
чаях прочные рацемические соедине-
ния, или рацематы. Такие соединения опти-
чески неактивны. Их физич. свойства (т. пл., т. кип.,
nff, dl°t растворимость и т. п.) отличаются, иногда
весьма значительно, от свойств чистых оптически-ак-
тивных форм. Изомеры противоположной конфигура-
ции, взятые в равном отношении, образуют также
оптически неактивные рацемические смеси
(конгломераты), плавящиеся при темп-ре более низ-
кой, чем исходные оптически-активные формы.
Существование А. о. впервые было установлено
Л. Пастером в 1848. Им же было осуществлено первое
разделение в-ва (натрийаммонийтартрата) на анти-
поды и высказано предположение, что причиной энан-
тиоморфизма винной к-ты является асимметрия,
строение ее молекулы.
Для изображения пространств, строения молекулы поль-
зуются несколькими способами. Способ I. Молекула пред-
ставляется в виде комбинации тетраэдров, в центре к-рых рас-
положены атомы углерода (рис., а), тогда как заместители
лежат по вершинам. Соединение тетраэдров означает С — С
связь. Способ II. Молекула изображается, исходя из реаль-
ного пространств, строения (рис., б). При таком способе,
используемом также в конфигурационном анализе, утолщаю-
щимися линиями («клиньями») изображаются связи, напра-
вленные к наблюдателю, т. е. лежащие перед плоскостью чер-
тежа. Связи, лежащие в плоскости бумаги (связь С — С на
рис., б), изображается простой линией, а связи, лежащие за
плоскостью чертежа, — пунктирной. Способ III. Изобра-
жение при помощи проекционных формул Фишера (рис., в).
При пользовании проекционными формулами следует помнить,
что группы Ви Е лежат перед плоскостью бумаги; оперируя
с формулами Фишера, их можно поворачивать только в пло-
скости бумаги.
Разделение рацемич. (DL) соединений на А. о. про-
изводят: 1) механич. способом — напр., пинцетом раз-
деляют смесь кристаллов правой и левой формы. Этот
способ применим в том случае, когда асимметрия
молекулы вызывает появление зеркальных форм
в кристаллах. 2) Биологич. способом, при к-ром для
СЛОН жизнедеятельности бактерий использует-
I * ся лишь один из присутствующих в сме-
I ся антиподов. Другой антипод при этом
•.. I не уничтожается и может быть выделен.
3) Самопроизвольным разделением (кри-
сталлизацией). В сравнительно редких
случаях из перенасыщенного р-ра двух
•*' / X Ф°рм постепенно выкристаллизовывается
/ лишь одна из них. 4) Химическим спо-
СООН собом, основывающимся на промежуточ-
п._. ном образовании диастереоизомерных
1 ' соединений. Способ известен в двух ва-
риантах: 1) Образующиеся при реакции DLI + DII—►
DI DH , ы • DII . „
—► •— ---.—- диастереоизомеры А и В раз-
А В
деляются кристаллизацией или ректификацией и
превращаются затем в чистые DI и LI, причем
в-во, служащее для разделения (DII), может быть
регенерировано. Эта схема в различных вариантах
и усложнениях широко применяется для разделе-
ния асимметрических аминов (при помощи оптически
активных кислот), кислот (при помощи оптически
активных аминов и аммониевых оснований), спиртов
и меркаптанов (через соли их кислых эфиров двух-
основных к-т и оптически активных оснований).
2) Второй вариант этого способа основан на различии
в скоростях образования и распада (или только
распада) диастереоизомерных соединений:
ь, ь,
D1 + ВП DI • DII LI + DI1 Л LI . DII
ha kt
ki ф ks и kg ф kt
По этой схеме используют реакции этерификации,
гидролиза и каталитич. дегидратации. Ко второму
варианту близко примыкает способ, при к-ром разде-
ление базируется на различии в скоростях адсорбции
оптически активным адсорбентом D- и L-форм. Сле-
дует отметить, что не каждое асимметрически построен-
ное соединение (будь то молекула или ион) может
быть разделено на А. о. Разделение невозможно,
если А. о. превращаются друг в друга (рацемизуются).
255
АНТИПОЛИМЕРИЗАТОР ДРЕВЕСНО СМОЛЬНЫЙ АНТИТЕЛА
256
Рацемизацию можно вызвать химич. процессами,
а также обратимым изменением электронной конфи-
гурации асимметрия. частицы. Напр., амины (асим-
метрич. атом N) и карбанионы не могут быть разде-
лены на А. о. из-за того, что зеркальные изомеры этих
соединений способны превращаться друг в друга:
/В. R^ ^R
R—N; ^2 : N— R' R'— C: ^2 :C—R'
Rr/Z \rm R"Z ^R"
D L D L
Амины и карбанионы существуют только в виде опти-
чески недеятельных форм. См. также: Асимметриче-
ская молекула, Асимметрический атом углерода,
Активность оптическая, Диастереоизомеры, Глицери-
новый альдегид.
Лит.: В и т т и г Г., Стереохимия, пер. [с нем.], М. — Л.,
1934; Хот и нс ний Е. С., Стереохимия. Курс лекций,
Харьков, 1950; Organic Chemistry. An advanced treatise, ed.
H. Gilman [a. o.], 2 ed., v. 1, N. Y., 1943, p. 220—440.
С. В. Bumm.
АНТИПОЛИМЕРИЗАТОР ДРЕВЕСНО - СМОЛЬ-
НЫИ — фракция масел, получаемая при разгонке
древесных смол и содержащая компоненты с темп-рами
кипения: не более 20% до 240°, 50% до 260° и не менее
90% до 310°; влаги — не более 6%; кислотное число
не более 30, количество поглощаемых щелочью ве-
ществ (в основном фенолов) не менее 65%. По составу
А. д.-с. близок антиокислителю древесно-смольному.
Используется в произ-ве синтетич. каучука как пре-
рыватель процессов полимеризации («стоппер») для
регулирования мол. веса. л. в. Гордон.
АНТИСЕПТИКИ — вещества, способные преду-
предить или приостановить развитие микроорганиз-
мов (бактерии, грибы, актиномицеты). А. применяются
для предохранения от разрушения микроорганизмами
различных неметаллич. материалов (древесины, тек-
стильных материалов, кожи, пластич. масс, пищевых
продуктов), а также в медицине. Наиболее широко
А. используются для консервирования древесины;
такие А. должны удовлетворять след, требованиям:
иметь по отношению к грибам высокие токсич. свой-
ства и длительно сохранять их после введения в дре-
весину; не ухудшать физико-механич. свойства дре-
весины, а также соприкасающихся с ней материалов;
не содержать веществ, ядовитых для людей; быть
доступными и дешевыми. Часто требуется, чтобы А.
не имел запаха, не затруднял последующую обра-
ботку или окраску древесины. Токсичность А. харак-
теризуют так называемой предельной дозой, т. е.
концентрацией А. (в искусственной питательной сре-
де), вызывающей полную остановку роста гриба или
прекращение разрушения древесины.
А. подразделяются на три группы: минерального
происхождения, органич. происхождения (масла,
получаемые гл. обр. при сухой перегонке каменного
угля, и синтетич. А.) и комбинированные — чаще
всего смесь органич. веществ с фтористым натрием
и нек-рыми другими солями. Предельные дозы (в %%) .
наиболее распространенных А. приведены ниже
(в скобках). Наиболее известные минеральные А.;
соли фтористоводородной к-ты — фтористые натрий
и аммоний (0,6 —1,0, в зависимости от содержания
чистой соли); кремнефтористоводородной к-ты — крем-
нефтористые натрий, цинк, магний (0,65); соедине-
ния мышьяка, хрома, цинка и меди и их смеси, напр.
хлористый цинк (1,3); медный купорос (3,6); желез-
ный купорос (5,4), арсенат ципка и др. К минераль-
ным А., используемым за рубежом для консервирова-
ния древесины, относятся также нек-рые патентован-
ные составы, напр. Болидена соли и Вольмана соли.
Болидена соли состоят из мышьяковой к-ты,
двузамещенного мышьяковистокислого натрия, дву-
хромокислого натрия и сернокислого цинка. В СССР
Болидена соли, как содержащие мышьяк, не приме-
няются. Вольмана соли («Триолит», «Мино-
лит» и «Таналит») состоят из 60—73% NaF, 18—35%
нитро- или динитрофенола или их натриевых солей,
двухромокислого натрия (до 9%), растворимых соеди-
нений мышьяка (до 5%) и др. компонентов. Основные
А. органич. происхождения: кам.-уг. пропиточное
масло, или креозот (0,1—1,3); кам.-уг. антраценовое
масло (до 1,3); зеленое масло — продукт пиролиза
нефти (5,0), сланцевое генераторное масло (1,6),
карболинеумы — хлорированные кам.-уг. масла (до
1,0); оксидифенил (0,45); динитрофенол (0,25), пен-
тахлорфенол (0,45), пентахлорфенолят натрия (0,35).
Из комбинированных А. известны, напр., уралит —
85% фтористого натрия и 15% динитрофенола (0,5);
триолит — 73% фтористого натрия, 18% дитрифенола
и 9% хромокислого натрия (0,5) и др. Для антисепти-
рования пиломатериалов применяют водные р-ры
соды. За рубежом (США и др.) для пропитки древе-
сины довольно широко распространены А., содержа-
щие соединения сулемы, а также арсениты и арсенаты
меди и цинка.
Для предохранения текст, материалов от разруше-
ния микроорганизмами применяют дихлор-, тетра-
хлор- и гексахлордиоксидифенилметаны, получае-
мые конденсацией формальдегида с хлор-, 2,4-ди-
хлор- и 2,4,5-трихлорфенолами. Хорошие результаты
дает салициланилид (особенно цинксалициланилид),
а также 8-оксихинолин и 8-оксихинолят меди. Высо-
коактивными А. являются различные органич. сое-
динения ртути (ограниченно применяемые вследствие
их токсичности); используются для введения в состав
лаков. В местностях с тропич. климатом, где особенно
быстро происходит разрушение микроорганизмами
неметаллич. материалов (в т. ч. пластич. масс, орга-
нич. стекла, резины и др.), наряду с органич. соеди-
нениями ртути применяют салициланилид, цинкса-
лициланилид, 8-оксихинолят меди, 2,4,5-трихлор-
фенолят меди, бромфтординитробензол и др. В бумаж-
ной пром-сти для борьбы с слизеобразованием упот-
ребляют органич. соединения ртути, соли триалкил-
олова и нек-рые хлорированные фенолы. В пищевой
пром-сти А. применяют для консервирования пищевых
продуктов и предохранения плодов и овощей от гние-
ния, для чего используют вещества, безвредные для
организма человека; их содержание в пищевых про-
дуктах строго нормируется. Наиболее часто для этих
целей используют соли бензойной к-ты, уксусную
и салициловую к-ты и нек-рые др.
В медицинской практике А. применяют для унич-
тожения болезнетворны^: бактерий во внешней среде
(дезинфекция) и при лечении ран и гнойных процес-
сов в организме. Для этих целей пользуются иодом,
хлораминами, фенолом, алкилфенолами, нек-рыми
гетероциклич. соединениями азота (риванол), соеди-
нениями Ag, Hg и др.
Лит.: Лекторский Д. Н., Защитная обработка
древесины, ч. 1, М. — Л., 1951- Труды ЦНИИ механической
обработки древесины (ЦНИИМОД), вып. 2/8, М. — Л., 1951;
Мельников Н. Н., Владимирова И. Л., Ива-
нова С. Н., Хим. пром-сть, 1980, № 1, с. 81 — 85.
Д. Н. Лекторский, И. Н. Мельников.
АНТИТЕЛА — белки, образующиеся в организме
при попадании в кровь нек-рых чужеродных в-в —-
антигенов — и обладающие способностью избира-
тельно соединяться с теми же антигенами или (в мень-
шей степени) со сходными с ними по строению веще-
ствами. Действие А. вне организма (in vitro) может
выражаться в осаждении растворенных белков и поли-
сахаридов (преципитация), склеивании (агглютина-
ция) или растворении (лизис) бактерий или эритро-
цитов, нейтрализации токсинов, инактивации виру-
сов и блокировании действия др. антител. Действие
А. в организме (in vivo) выражается в невосприим-
257
АНТИФЕБРИН — АНТИЧАСТИЦЫ
258
чивости организма к повторному заражению возбуди-
телями ряда заболеваний (иммунитет) или в развитии
повышенной чувствительности организма к опреде-
ленным антигенам (аллергия). Содержание А. опре-
деляют различными биологич. методами или путем
осаждения соответствующим антигеном и определе-
ния количества белка в осадке. Наибольшее количе-
ство А. содержится в сыворотке крови; при фракцио-
нировании белков сыворотки значительная их часть
попадает во фракцию у-глобулинов. Выделение А.
производится обычно путем разложения нераство-
римого комплекса антиген — антитело. При фермента-
тивном гидролизе А. можно получить обломки их
молекул, сохранившие биологич. активность. Обра-
зование А. связано с биосинтезом новых молекул
белка и происходит гл. обр. в лимфатич. железах,
селезенке, костном мозге, коже и легких. Обнаруже-
ние в крови больных людей и животных тех или иных
А. позволяет судить о возбудителе заболевания (серо-
логия. диагностика). Сыворотки, содержащие А.,
и выделенные из этих сывороток у-глобулины исполь-
зуются для лечения ряда заболеваний. Массовое рас-
пространение получила искусств, стимуляция обра-
зования А. против возбудителей различных заболе-
ваний путем введения этих возбудителей в организм
в убитом или ослабленном состоянии (прививки).
Лит. см. при ст. Антигены. А. Е. Гурвич.
АНТИФЕБРИН — см. Ацетанилид.
АНТИФЕРРОМАГНЕТИЗМ — см. Магнитные свой-
ства.
АНТИФРИЗЫ — низкозамерзающие жидкости,
применяемые в установках, работающих при низких
темп-pax, а также для охлаждения двигателей. А.
должны обладать низкой темп-рой замерзания, не-
большой вязкостью, обеспечивающей свободное про-
качивание жидкости при низких темп-pax, не должны
вызывать коррозию металлов, из к-рых изготовлены
детали системы охлаждения, и не разъедать матери-
алы соединительных шлангов. А., применяемые в дви-
гателях, должны также обладать высокой теплоем-
костью и теплопроводностью, высокой темп-рой
кипения и низкой испаряемостью. В качестве А. можно
использовать водные р-ры этиленгликоля, глицерина,
спиртов и нек-рых других органич. соединений, вод-
ные р-ры солей, а в нек-рых случаях легкие нефте-
продукты.
Этиленгдиколевый А. широко применяется для
охлаждения автомобильных, тракторных и авиацион-
ных поршневых двигателей, для заполнения противо-
пожарных трубопроводов в неотапливаемых поме-
щениях, заводских' холодильных линий и проч. Уд.
теплоемкость этиленгликоля 0,56 ккал/кг при 20°.
Смешивая технич. этиленгликоль с водой в разных
соотношениях, можно получить А. с темп-рой за-
мерзания от 0° до —75°. Смесь, замерзающая при
— 75°, состоит из 66,7% этиленгликоля и 33,3% воды
(см. таблицу).
Концентра- ция этилен- гликоля, % Т.замер- зания, °C Концентр а- ция этилен- гликоля, % d;° Т.замер- зания, °C
26,4 1,0340 -10 63,1 1,0833 -60
36,4 1,0506 —20 66,7 __ -75
45,6 1,0627 -30 72,1 1,0923 -60
52,6 1,0713 -40 78,4 1,0983 -50
58,0 1,0780 -50
В технике применяются смеси, содержащие 52,6%
и 66% этиленгликоля; замерзают соответственно при
—40° и —60° (марка 40 и марка 60). К этим жидко-
стям добавляют антикоррозионные присадки — динат-
рийфосфат и декстрин. Этиленгликолевые А. имеют
значйтельные преим. перед другими: этиленгликоль
5 к. х. Э. т. 1
обладает незначительной летучестью и мало испа-
ряется из водных смесей в условиях эксплуатации.
Водные р-ры этиленгликоля имеют более низкую
вязкость, чем соответствующие водно-глицериновые
смеси, и более высокую теплоемкость и теплопровод-
ность по сравнению с водно-спиртовыми и водно-гли-
цериновыми смесями. Известны также А. из водно-
глицериновых р-ров; смесь 70% (по весу) глицерина
и 30% воды замерзает при —40°. Из одноатомных
спиртов для приготовления А. чаще всего исполь-
зуются водные р-ры метилового, этилового и изопро-
пилового спиртов. Более низкие темп-ры замерзания
имеют водные р-ры метилового спирта: смесь, содержа-
щая 50% (по весу), замерзает при —43°. Положитель-
ным качеством водно-спиртовых А. является их низ-
кая вязкость, приближающаяся к вязкости воды.
Сильная испаряемость этих смесей вызывает большие
потери спирта и приводит к повышению темп-ры замер-
зания. Известно, что растворением в воде неорганич.
и органич. солей можно значительно снизить темп-ру
ее замерзания. Наиболее низкие темп-ры замерзания
при растворении в воде хлористых солей приведены
в таблице:
Соль Содержание соли, вес. % Т. замерза- ния, °C
NII4C1 18,7 -15,8
NaCl 22,4 -21,2
MgCl2-6H2O 20,6 -33,6
CaCl2-6H2O 29,9 -55,0
К2СО3-1,5Н2О 39,9 -36,5
Солевой А. состава: 32% хлористого кальция, 7%
хлористого натрия и 61 % воды замерзает при —45°.
В практике относительно широкое распространение
получили р-ры СаС12. Отрицательными свойствами
всех солевых А. являются значительная коррозия
металлов системы охлаждения и кристаллизация
солей. В нек-рых случаях в качестве А. могут приме-
няться хорошо очищенные керосиновые фракции
низкозастывающих нефтей; однако их недостаток —
малая теплоемкость и теплопроводность; коэфф,
теплопроводности этих продуктов в 5 раз меньше,
чем у воды. Вследствие этого применение нефтяных
А. для охлаждения двигателей внутр, сгорания в усло-
виях высокой нагрузки приводит к их перегреванию.
Лит.: Моторные топлива, масла и жидкости, под ред.
К. К. Папок, т. 2, 3 изд., М., 1957, с. 441 — 477.
В. В. Панов.
АНТИЧАСТИЦЫ. Согласно законам квантовой
механики, каждой частице соответствует другая час-
тица, являющаяся как бы ее антиподом, т. е. зеркаль-
ным изображением. Впервые это положение было
высказано П. Дираком в 1929. А. во всем подобна пер-
вичной частице, отличаясь от нее только знаком элект-
рич. заряда. Ясно, что понятие «А.» взаимно, т. е.
всегда можно считать А. первичной частицей, и тогда
частица, рассматривавшаяся раньше как первичная,
будет по отношению к ней А. В нек-рых случаях пер-
вичные частицы совпадают с А. Такие частицы, оче-
видно, лишены всяких электрич. зарядов. Их иногда
называют истинно нейтральными. К таким частицам
относятся, в частности, фотон и л:°-мезон. Однако
не всякая незаряженная частица является истинно
нейтральной. Напр., нейтрон не есть истинно нейт-
ральная частица. Он, в частности, имеет магнитный
момент, тогда как у антинейтрона этот магнитный
момент имеет противоположный знак.
Частицы и А. всегда способны к аннигиляции, т. е.
могут взаимно уничтожаться, превращаясь в фотоны
или другие частицы. Возможно и обратное явление:
рождение фотонами пар, т. е. одновременно частицы
и А. Возможность аннигиляции приводит к тому,
259
АНТОЦИАНИДИНЫ
260
что в условиях нашего мира частицы и А. не могут
быть одновременно стабильными. Поэтому окружаю-
щие нас атомы содержат электроны и не содержат
позитронов; атомные ядра состоят из протонов и нейт-
ронов, антипротоны же и антинейтроны могут быть
получены только искусственно, в условиях, исключаю-
щих их контакт с соответствующими им обычными
частицами.
Благодаря этому обстоятельству предсказание Дира-
ком А. было вначале встреченос большим недоверием,
и даже сам автор считал этот результат слабой сторо-
ной своей теории. Однако уже в 1932 в космич. лучах
были открыты позитроны, к-рые затем были полу-
чены искусственно, и т. обр. правильность теории
подтверждена экспериментально. Получение анти-
нуклонов — антипротона и антинейтрона — оказа-
лось значительно более трудной задачей, поскольку
для образования пар нужна энергия, по меньшей
мере равная энергии покоя частиц, т. е. 2 тс!; в слу-
чае нуклонных пар эта энергия почти в 2 000 раз
больше, чем энергия образования электрона и пози-
трона. Антипротон и антинейтрон были искусственно
получены в 1955—56 группой амер, ученых (Э. Сегре
и др.) на ускорителе большой энергии. Что же ка-
сается нестабильных частиц, то у т. н. мезонов (частиц
с массой, промежуточной между массами электрона
и протона) частицы и А. были открыты почти одно-
временно. У гиперонов (частиц более тяжелых, чем
протоны) получение А. требует еще больших энергий,
чем у нуклонов. Пока удалось получить только .'.амый
легкий антигиперон (Л-частицу, 1959). До последнего
времени считалось, что А. отличаются от частиц
только знаком заряда. Оказалось, однако, что все
частицы обладают нек-рой степенью внутренней асим-
метрии, и для того, чтобы получить А. каких-либо
частиц, надо произвести не только изменение знака
всех зарядов, но и зеркальное отражение. См. также
Элементарные частицы. л. Д. Ландау.
АНТОЦИАНИДИНЫ — окрашенные (от оранже-
во-розового до фиолетово-розового) кислородсодер-
жащие гетероциклические соединения, встречающиеся
в растениях в виде агликонов пигментов антоциани-
нов. А. — производные полиоксифлавенола-2. В ос-
нове А. лежит система флавана (2-фенилхромана) (I).
Алкоголи дегидрофлавана — флавенолы (II) с кис-
лотами дают соли бензопирилия, называемые солями
флавилия (форма, в виде к-рой преимущ. существуют
А. и их гликозиды), к-рым приписывают строение
карбониевых (III) или оксониевых (IV) соединений
(более вероятной является карбониевая структура).
Все А. содержат по неск. оксигрупп в хромановой
части молекулы (обычно в положениях 3,5 и 7) и в фе-
нильном радикале. А. принято делить на 3 основ-
ные группы: пеларгонидина (V), ц и а -
н ид ина (VI) и дельфинидина (VII).
Каждая группа объединяет метиловые эфиры основ-
ного А., отличающиеся количеством и расположением
метильных групп. А., не подходящие ни к одной из
этих групп, встречаются редко. Особенно широкое
распространение имеют А. группы дельфинидина
(VIII): петунидин (R=CH3, R' и R"=H); мальвидин,
или сирингидин (R и R'=CH3, R"=H); гирсутидин
(R, R1 и R"=CH»).
В растениях А. содержатся в виде гликозидов.
Гликозидированы обычно ОН-группы в положении 3
(моно-, ди- или трисахаридом), реже ОН-группы в поло-
жениях 3 и 5; недавно обнаружены А., гликозидиро-
ванные в 3,7- и 3,4 -положениях. Оксигруппы А.
в нек-рых антоцианинах ацилированы кофейной,
п-кумаровой и др. к-тами. Все А. окрашены интенсив-
нее соответств. антоцианинов. С увеличением в моле-
куле А. числа ОН-групп цвет в кислой среде изме-
няется от оранжево-красного (пеларгонидин) до сине-
фиолетового (дельфинидин). Спектры метиловых эфи-
ров сходны со спектрами основных соединений.
Флавилиевые соли А. плохо растворимы в воде,
хорошо — в подкисленных спиртах и в изоамиловом
спирте. Гликозидирование увеличивает растворимость
А. в воде и уменьшает в органич. растворителях. При
действии щелочей и большого колич. воды соли А.
гидролизуются. Соли А., содержащие свободные
ОН-группы в положениях 2', 4', 5 или 7, при мягком
щелочном гидролизе образуют окрашенные в синий
и фиолетовый цвета хиноидные соединения, наз. анги-
дрооснованиями или основаниями красителей (напр.,
IX и X). При дальнейшем действии щелочей или при
сильном разбавлении р-ра основание красителя гид-
ратируется, превращаясь в бесцветный флавенол
(карбонильное основание, исевдооснование) (II). При
действии к-т на аягидрооснования и флавенолы снова
образуются флавилиевые соли. Разнообразие окрасок
цветов и плодов обусловлено гликозидированными
А. и зависит от pH, присутствия нек-рых веществ
(копигментов) и солей металлов. Получают А. из
антоцианинов, гидролизуя их 20%-ной НС1, и син-
тетически, используя общие методы синтеза хромено-
лов и солей бензопирилия из фенолов, о-оксибензаль-
дегидов, и о-оксифенилкетонов, восстановлением фла-
вонов и др. методами. Лучшие выходы дают синтезы
из о-оксибензальдегидов:
СН3СОС,,Н-
01Г
Полиоксибензальдегиды конденсируются с трудом и
их предварительно метилируют или ацилируют. Из
ацилпроизводных получают наиболее чистые препа-
раты А. Проводя конденсацию с гликозидированными
производными, получают антоцианины.
Лит.: Гетероциклические соединения, под ред. Р. Эльдер-
филда, пер. с англ., т. 2, М., 1954; Handbuch der Pflauzeaphy-
261
АНТОЦИАНИНЫ — АНТРАМИНЫ
262
siologie, hrsg. von W. Ruhland, Bd 4, B., 1958; Moderne Metho-
den der Pflanzenanalyze, hrsg. von K. Paech, M. V., Fracey,
B. [u. a.], 1955. M. H. Н&тюк. Л. И. Линевич.
АНТОЦИАНИНЫ — водорастворимые раститель-
ные пигменты цветов и фруктов, являющиеся глико-
зидами антоцианидинов и участвующие в образова-
нив розовой, красной, фиолетовой и синей окраски
различных органов растений. По современным пред-
ставлениям вызываемая А. окраска зависит не только
от строения А. и величины pH клеточного сока, но и от
характера металла, образующего комплекс с А. в рас-
АНТРАЗИН C28HI6N2, мол. в. 380,45 — желтовато-
коричневые кристаллы, иглы (из нитробензола), т. пл.
ок. 390°, сублимируется ок. 340°, растворяется в горя-
чем нитробензоле, анизоле; мало растворим в СНС13
и СС14, разб. р-ры имеют желтовато-зеленую флуорес-
ценцию; растворяется в конц. H2SO4 и HNO3. А. (I)
может быть получен сплавлением 2-аминоантрацена
(II) со смесью КОН и NaOH при 220—230°, а так-
же при энергичном восстановлении, например фосфо-
ром и HJ, индантрона (III); последний образуется
R - рамноглюкоза
nh2 кон
Сплав пение
P+HJ
K2CrO4 + H2SO4
О
in
водное. Технич.
при окислении А. хромовой
смесью с последующим
кипячением получившего-
ся продукта в хинолине; с
бромом при нагревании А.
образует октабромпроиз-
применение имеет производное А.—
r2— глюкоза
тении; красная окраска обусловлена комплексом А.
с Fe, синяя и фиолетовая — с Мо, белая — с Ni или
Си. Сахарные компоненты в А. представлены преим.
глюкозой, рамнозой, галактозой, реже—пентозами,
гентиобиозой и др. сахарами. Обычно А. гликози-
лированы в положении С3, реже в положении С3 и
С5, напр. керациапин — (])—(3-рамноглюкозидциани-
на) и дельфин — (II)—(3,5-ди-р-глюкозид дельфиниди-
на). Из растений А. экстрагируют кислыми водными
или спиртовыми р-рами, лучше всего метанолом,
содержащим 1% НС1. Обнаружение, выделение и
очистка А. производятся хроматография, методами
(на бумаге и на колонках из целлюлозного по-
рошка), что позволяет разделить сложные смеси А.
на индивидуальные компоненты.
Лит.: Reznik Н., Sitzungsber. Heidelberg. Akad. Wiss.
Math.-naturwiss. KI., Jg. 1956, Abh. 2; К и к к а в а, О гит а,
Ф у д з и т о, Кагаку, 1955, 25, №3, с. 139; Harborne J.B.,
J. Chromatogr., 1958, 1, № 6, р. 473. М. Н. Нестюк.
АНТРАГАЛЛОЛ (1,2,3-триоксиантрахивон), мол.
вес 256,22, С14Н8О5 — оранжевые кристаллы, иглы;
т. пл. 312—13° (314°); сублимируется около 290°;
мало растворим в воде и СНС13; растворяется в спирте,
эфире и СН3СООН с коричневато-желтой окраской.
Раствор в конц. H2SO4 окрашен в красно-коричне-
вый цвет, щелочной р-р зеленоватого цвета. Полу-
чают А. (I) нагреванием смеси бензойной (II) и галло-
вой к-т (III) в слабом олеуме (1—3% SO3) при 120°.
краситель индантрон, под названием «кубовый си-
ний О». в. А. Пучков.
АНТРАЗО [антрахинон-(1-азо-4)-диметиланилин со-
лянокислый] C22H17O2N3 • НС1, мол. в. 391,85 — почти
N=N \ y“N(CH3)2-HCi
Известно значительное число алкиловых эфиров А.;
2-метиловый и 1,2-диметиловый эфиры встречаются
в природе. С уксусным ангидридом и бензоилхлори-
дом А. дает О-ацетильные и О-бензоильные произ-
водные. А. галогенируется и нитруется в положе-
ние 4; при действии окислителей в р-ре H2SO4 дает
1,2,3,4-тетраоксиантрахинон; с солями тяжелых метал-
лов образует глубоко окрашенные лаки. Применяется
А. как протравной краситель под технич. названием
«ализариновый коричневый». в. а. Пучков.
б*
О
черный кристаллич. порошок с зеленым блеском;
нерастворим в воде, мало растворим (с красной окрас-
кой) в спирте, подкисленном соляной к-той; в пири-
дине растворим с бурой окраской. Получают А. соче-
танием диазотированного а-аминоантрахияона с соля-
нокислым диметиланилином; свободное основание
А. — красно-кирпичный мелкокристаллич. порошок,
т. пл. 243°. Придобавлениир-ра, содержащего [SnCl6]2~,
к нагретому розовому р-ру А. (в подкисленном соля-
ной к-той конц. р-ре NH4C1) выпадает сине-фиолето-
вый осадок. Аналогично реагирует А. с другими
элементами, образующими комплексные анионы типа
[MeIV С1„]......................
применение его для открытия и фотометрия, опреде-
ления нек-рых из названных элементов. Для откры-
тия удобно применять бумагу, пропитанную р-ром
А. и высушенную; используется для определения
Sn и Те в минералах (последнего в присутствии Se).
Лит.: Кузнецов В. И., Ж. прикл. хим.. 1940, 13,
№5; его ж е, там же, 1943, 16, № 7 — 8; его же, там
же, 1946, 1, № 4. В. Г. Типцова.
АНТРАМИНЫ (аминоантрацены) CI4HnN, мол. в.
193,25 — кристаллы желтого цвета; растворимы в
большинстве органич. растворителей; спиртовые р-ры
имеют зеленую флуоресценцию. 1-и2-А. (I и II) нерас-
творимы в воде и конц. НС1 (на холоду); 9-А. (III) хо-
рошо растворяется в соляной к-те. Свойства А. при-
ведены в таблице (см. стр. 263).
9-А. получают восстановлением 9-нитроантрацена
оловом и соляной (уксусной) к-той. 1- и 2-А. получают
либо из антролов, замещением оксигруппы на ами-
ногруппу, либо из аминоантрахинонов одним из мето-
дов восстановления. Напр., 1-А. образуется принагре-
(Те, РЬ, Re, Pt, Pd, 1г), на чем основано
263
АНТРАНИЛОВАЯ КИСЛОТА — АНТРАХИНОН
264
Название Т. пл., °C Т. пл. характерных производных, °C
1-Антрамин 2-Антрамин S-Антрамин —-130 238 145—150 N-ацетил, 198 N-ацетил, 240 N-ацетил, 273—4 N, N-диацетил, 159
вании до 220° 1-антрола с водным аммиаком в присут-
ствии СаС12. Для получения 1- и 2-А. из аминоантра-
хинонов восстановление обычно ведут Zn-пылью в ам-
миаке. 1- и 2-А. образуются также при восстановле-
нии амииоантрахинонов водородом (325—475°) над
Си — Zn-катализатором. А. способны ацилироваться,
взаимодействовать с альдегидами, диазотироваться.
А. (кроме 9-А.) дают устойчивые диазосоединения,
к-рые можно сульфировать и нитровать. 9-А. при
действии HNOa образует антрахинон и ряд более
сложных продуктов. Для 9-А. возможен таутомерный
переход в антронимин (IV). В 1- и 2-А. аминогруппа
при нагревании с р-ром NaHSO3 обменивается на
оксигруппу. При сплавлении 2-А. со щелочью обра-
зуется антрааин. А. — промежуточные продукты
в синтезе кубовых красителей и азокрасителей.
В. А. Пучков.
АНТРАНИЛОВАЯ КИСЛОТА (о-аминобензойная
кислота) п-ЩК—С6Н4—СООН, мол. в. 137,13 —
бесцветный кристаллич. порошок; т. пл. 144—145°;
возгоняется; растворимость (г/100 г растворителя):
в воде 0,35 (13,8°), в 90%-ном спирте 10,7 (9,6°),
в эфире 16,0 (6,8°), в бензоле 1,8 (11,4°), в ледяной
уксусной к-те 8,96 (13,6°), в уксусноэтиловом эфире
11,9 (10°), в сероуглероде 0,20 (10,9°); легко раство-
рима в щелочах и минеральных к-тах; константа дис-
социации КкИСЛ =1,07 • 10~s (25°). Получают А. к.
действием на щелочной р-р фталимида гипобромитом
или гипохлоритом натрия:
NaOCI
NaOH
aCOONa
nh2
С солями Cu2+ в уксуснокислом р-ре А. й. образует
ярко-зеленый осадок, в отличие от л-аминобензой-
ной к-ты, с к-рой получается только голубая окраска.
При сплавлении А. к. с небольшим избытком SnCl4
и обработке охлажденного плава водным р-ром спирта
появляется фуксино-красная окраска — отличие от
м- и n-аминобензойных к-т. В уксуснокислых р-рах
(pH 2—5) А. к. образует с Cd, Со, Си, Ni, Zn, Mo,
Pb и Hg внутрикомплексные малорастворимые соеди-
нения. Это свойство используется для весового опре-
деления перечисленных элементов; на диазотировании
А. к. основано ее применение для фотометрия, опре-
деления нитритов, используется также для цветной
реакции на CeIv.
Производные А. к. применяются в произ-ве краси-
телей и синтетич. душистых в-в (см. таблицу), ме-
тилантранилат и N-метилметилантранилат входят в
состав многих эфирных масел. Ниже приведены не-
которые производные А. к., применяющиеся в про-
мышленности.
Название Т. пл., °C Основное при- менение
N-Ацетилантраниловая к-та CH3COONH—СеН4—СООН 185 Азокрасители
N-Метилантраниловая к-та1 182 То же
CH3NH—С0Н4—СООН
N-Фенилантраниловая к-та 2 184
CbHsNH—С8Н4—СООН
5-Хлорантраниловая к-та 2 H2N—СсН3(С1)—СООН 211-212 Азокрасители, антрахинон-ак- ридоны
4-Хлорантраниловая к-та 2 235-236 Азокрасители
H2N—СвН3(С1)—соон
5-Нитроантраниловая к-та H2N—CeH3(NO2)—СООН 280 278 (с разл.) То же
Фенилглицин-о-карбоновая к-та 218-220 Индигоиды
HOOCNH—С8Н4СООН
4-Сульфоантраниловая к-та NH2CeH3(SO3H)COOIl разл. не плавясь Диазоаминолы
Метилантранилат 3 H2NC6H4COOCH3 24—25 Азокрасители Душистые веще- ства
Этилантранилат 3 а HsNCeHjCOOCsHg 13 Душистые веще- ства
1 Возгоняется. 2 Разлагается выше т. пл. 3 Запах цветов.
Лит.: Фир ц-Д авид Г. Э.иБланже Л., Основные
процессы синтеза красителей, пер. с нем., М., 1957; Bedou-
k i a n Р. Z., Perfumery Synthetics and Isolates, Toronto —
N. Y. — L., 1951, p. 54; W e 1 c h e г F. J.. Organic analy-
tical reagents, v. 2, N. Y., 1947.
АНТРАХАС (антрахинон-а-арсоновая кислота)
Cj4HeO5As, мол. в. 332,12 — почти бесцветные кри-
сталлы, иглы или светло-жел-
тый порошок; мало растворим q AsO(OH)2
в горячей воде; почти нераство- ]
рим в метиловом и этиловом
спиртах; растворим в раство- L II II I
pax NaOH, NaaCOg и в горячем
р-ре CHgCOONa со светло-бу- О
рой окраской, в конц. H2SO4—
с желтой окраской; т. разл. > 200°; очень ядовит. По-
лучают А. диазотированием а-аминоантрахинона и
последующей обработкой щелочным р-ром AsaO3;
применяют А. для осаждения малых количеств SnIV,
ZrIV, Tiiv.
Лит.: Кузнецов В. И., Заводск. лаборатория,
1945, 11, № 4. Д. И. Кузнецов.
АНТРАХИНОН С14Н8О2, мол. в. 208,22 — светло-
желтые ромбич. кристаллы; т. пл. 286°; т. кип. 379—
381°; dl (тверд.) 1,419—1,438, ра-
створяется в нитробензоле, анилине
и горячем толуоле (6% при 100°);
мало растворим в спирте; в конц.
H2SO4 растворяется с желтой окра-
ской. А. получается окислением ан-
трацена или синтетически из фталевого ангидрида и
бензола. Последний метод применяется также для
получения производных А. (2-метил-, 2-хлор- и 1,4-
диоксиантрахинонов). В химич. поведении А.имеются
нек-рые особенности, обусловленные наличием двух
СО-групп в 9,10-положениях (opmo-стоящие замести-
тели для каждого бензольного ядра): 1) А. с трудом
вступает в реакции замещения; 2) вступление заме-
стителя (SO3H, NO2 галоген) в одно из ядер не приво-
дит к заметному уменьшению реакционности второго
ядра, что затрудняет получение монозамещенных А.;
3) многие заместители в замещенных А. обладают
265 АНТРАХИНОНОВЫЕ КРАСИТЕЛИ — АНТРАХИНОНОВЫЕ КУБОВЫЕ КРАСИТЕЛИ 266
повышенной реакционной способностью. Галогениро-
вание А. проводят в олеуме; при действии хлора
при 130° образуется 1, 4, 5, 8-тетрахлорантрахинон;
бромирование при 60° дает тетрабромпроизводное.
При нитровании А. рассчитанным количеством HNO3
(d 1,38) в среде H2SO4 при 50° образуется 1-нитроант-
рахинон. Нагревание А. в конц. H2SO4 при 70—80°
с избытком HNO3 приводит к образованию смеси
динитроантрахинонов (1,5- и 1,8- и незначительное
количество 1,6-, 1,7-, 2,6- и 2,7-). При сульфировании
А. получаются антрахинонсулъфокислоты. При дей-
ствии на А. водных щелочных р-ров КСЮ3, KNO3
или Na2CrO4 образуется ализарин. Карбонильные
группы А. обладают свойствами, средними между
свойствами кетонной и хинонной групп. Как кетон А.
образует монооксим при нагревании с NH2OH • НС1
при 180°, но он не реагирует с фенилгидразином.
В отличие от бензохинона, А. не восстанавливается
при помощи H2SO3. В зависимости от условий при
восстановлении А. образуются различные соедине-
ния: Na2S2O4 -|- NaOH — антрагидрохинон; Sn-|-
-j- НС1 —> 9-антрон; Zn-|- аммиак —> антрацен. Раствор
антрагидрохинона (т. пл. 180°) в щелочи («куб»)
окрашен в красный цвет, из него при продувании
воздухом выделяется А. На этой реакции основано
применение производных А. в качестве кубовых кра-
сителей, а также качеств, определение А. Кроме
указанного А., известны 1,2-А. (т. пл. 185—190°
с разл.) и 1,4-А (т. пл. 218°, с разл.), не имеющие
технич. Применения. В. д. Пучков.
АНТРАХИНОНОВЫЕ КРАСИТЕЛИ — обширная
группа красящих вешеств, в основе строения к-рых
лежит молекула антрахинона. В более широком
смысле к А. к. относят также полициклич. краси-
тели, не являющиеся в прямом смысле производными
антрахинона, но для к-рых антрахинон или его заме-
щенные служат исходными продуктами синтеза, напр.
производные бензантрона. Основная часть применяе-
мых в наст, время прочных (особенно светопрочных)
и ярких красящих в-в относится к А. к. Широкое при-
менение в качестве красящих веществ производных
антрахинона определяется особенностями его моле-
кулы: малой реакционной способностью, чем в значи-
тельной степени объясняется высокая светопрочность
А. к.; глубокой окраской уже простейших заме-
щенных антрахинона: 1-аминоантрахинон (I) — крас-
ного, 1,4-диаминоантрахинон (II) — фиолетового, а
1,4,5,8-тетрааминоантрахинон (III)—голубого цвета;
способностью при восстановлении легко давать антра-
гидрохинон (IV), растворимый в водном р-ре щело-
чи. Последний на воздухе быстро и
Н ОН количественно окисляется в антра-
хинон, что определяет возможность
f ( II | использования многих производных
L антрахинона в качестве кубовых
красителей.
Н ОН Многие оксипроизводные антра-
п хинона, особенно содержащие не
менее двух оксигрупп в положении
1 и 2, с металлами дают интенсивно окрашенные устой-
чивые лаки. Особенное значение здесь имеет ярко-
красный алюминиевый лак 1,2-диокси антрахинона
(V) — ализарина, известный под названием крапп-
лака: этот лак, образующийся из составных частей
непосредственно на волокне методом т. н. протравного
крашения, дает яркую и прочную окраску целлюлоз-
ного волокна (кумач, турецкий красный). Алюминие-
вые лаки 1,2,6- и 1,2,7-триок- ✓Ai(OH)
сиантрахинона отличаются более I 2
желтым оттенком красного цве- О
та; нек-рые другие полиоксиан-
трахиноны имеют более глубо- [ | | |
кие оттенки. Соединения железа
и хрома с оксиантрахинонами Д
позволяют получать более глу-
бокие, но менее яркие и проч- ►
ные сравнительно с алюминием окраски. Ввиду слож-
ности и малой производительности протравное кра-
шение оксипроизводными антрахинона почти не при-
меняется.
Аминозамещенные антрахинона, их N-алкилпроиз-
водные, обычно содержащие также —ОН, —C0NH2,
—CN и др. заместители в молекуле антрахинона или
боковой цепи, все больше применяются для суспен-
зионного крашения ацетатного, полиэфирных и поли-
амидных синтетич. волокон. Алкил- и ариламино-
производные антрахинона, содержащие в боковой
цепи четвертичные аммониевые остатки, используются
как светопрочные основные красители для полиакрил-
нитрильного волокна. Сульфокислоты аминоокси- и
особенно ариламиноантрахинона являются яркими
и прочными кислотными красителями фиолетового,
синего и зеленого цвета для шерсти. Ациламиноза-
мещенные антрахинона, так же как соединения, где
остатки молекулы антрахинона сконденсированы с
другими, обычно гетероциклическими ядрами, состав-
ляют большую группу антрахиноновых кубовых кра-
сителей.
Металлич. лаки полиоксипроизводных антрахи-
нона, ациламинозамещенные, ациламинооксизамещен-
ные и более сложно построенные производные антра-
хинона применяются в качестве светопрочных пиг-
ментов в полиграфии и лакокрасочной пром-сти.
Устойчивость к воздействию темп-ры амино-, арил-
амино- и др. производных антрахинона позволяет
использовать их для сигнальных дымов.
См. также Аминоантрахиноновые красители.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венкатараман К., Химия синтетических краси-
телей, пер. с англ., т. 2, Л., 1957. Н. С. Докунихин.
АНТРАХИНОНОВЫЕ КУБОВЫЕ КРАСИ-
ТЕЛИ — кубовые красители, производные антра-
хинона. Наиболее простыми А. к. к. являются ацил-
аминозамещенные антрахинона. Сродство к целлю-
лозному волокну моноаминопроизводных антрахи-
нона, ацилированных остатком одноосновной к-ты,
недостаточно для практич. целей; поэтому большее
значение имеют производные многоосновных к-т,
напр. изофталевой или циануровой. Диаминозамещен-
ные антрахинона пригодны в качестве кубовых краси-
телей уже при ацилировании остатком одноосновной
к-ты. 1,4- и 1,5-Ди-(бензоиламино)-антрахиноны изве-
стны под названиями индантренов желтого ГК (I)
и красного 5ГК (II). При наличии в молекуле антра-
хинона одновременно оксигрупп цвет красителей
значительно углубляется, ацилированный 1,5-диа-
мино-4,8-диоксиантрахинон (III) известен как ипдан-
трен ярко-фиолетовый РК. Согласно патентным дан-
ным, при бензоилировании 1,4,5-триамино-8-окси-
антрахинона получают синий кубовый краситель.
Иногда для ацилирования полиаминозамешенных
антрахинона применяют одновременно различные,
в том числе довольно сложные, кислотные остатки.
В качестве А. к. к. ограниченное применение находят
антримиды. Большое значение имеют А. к. к., в к-рых
молекула антрахинона связана или конденсирована
с гетероциклич. ядрами, напр. кубовый бордо С (IV).
Среди последних в первую очередь следует назвать
267
АНТРАХИНОНОВЫЕ КУБОВЫЕ КРАСИТЕЛИ — АНТРАХИНОНСУЛЬФОКИСЛОТЫ 268
индатрон (V) и его замещенные. В зависимости от
степени очистки он выпускается в СССР под назва-
нием кубового синего Н, кубового синего О, кубово-
го чисто-синего. Дихлориндантрон называется кубо-
вым голубым К (VI),
а частично сульфиро-
ванный индантрон —
кубовым голубым 3.
Индантрон получают
щелочным плавлением 2-аминоатрахинона, его заме-
щенные, напр. кубовый голубой К, — хлорированием
индантрона в конц. серной к-те или конденсацией
двух молекул 2-амино-1-бром-3-хлорантрахинона.
Другим примером А. к. к. является антрахинон
с азиновым циклом — кубовый ярко-алый ЖХ (VII).
Большую группу А. к. к., от желтого до оливкового
цвета, составляют дифталоилкарбазолы, получаемые
замыканием цикла из соответств. антримидов. Высо-
кой светопрочностью обладают замещенные фталоил-
акридона, к-рые пред-
। ставлены красителя-
ми различного цве-
О N । NH | |Гснз та, например кубовый
бирюзовый ЗХ (VIII).
FIJI Практическое значе-
ние получили А. к. к.,
О производные оксазо-
|/// ла, например кубовый
красный С (IX), тиазола, имидазола и др. гетеро-
циклов. Почти все замещенные антрахинона спо-
собны при восстановлении образовать растворимые
в водном р-ре щелочи, производные антрагидрохи-
нона, из к-рых на воздухе в результате окисления
легко регенерируются исходные соединения. Необхо-
димой для красителя интенсивностью окраски и
сродством к текстильвым волокнам обладают не все
замещенные антрахиноны.
Большинство А. к. к. обладает высокой прочностью
окрасок, особенно к свету. Сложные по строению’
А. к. к. при крашении чрезвычайно быстро выбираются
из куба целлюлозным волокном, что вызывает нерав-
номерность окрасок. Поэтому их часто производят
в виде паст или порошков, содержащих мелкоизмель
ченный краситель и добав-
ки веществ, способствующих
крашению. Гидрохиноны,
образующиеся при восста-
новлении А. к. к., как пра-
вило, слабые кислоты, вслед-
ствие чего ими красят из
куба, содержащего едкий
натр; по этой причине боль-
шая часть А. к. к. непри-
менима для окраски живот-
ных волокон. В отличие от слабоокрашенного куба
индигоидных кубовых красителей, куб А. к. к. обыч-
но более глубоко окрашен, чем сами красители.
В СССР торговые назв. А. к. к. всегда начинаются
словом «кубовый». За границей наиболее прочные
к свету марки А. к. к. выпускаются под названиями
индантренов (ФРГ), каледонов, парадонов (Англия),
цибанонов, тинонов, сандотренов (Швейцария), пон-
солей, карбантренов, калколоидов (США), останов
(ЧСР) и др.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венк ат ар аман К., Химия синтетических кра-
сителей, пер. с англ., т. 2, Л., 1957; Хим. наука и пром.,
1958, 3 [№] 2, с. 174. Н. С. Докунихин.
АНТРАХИНОНСУЛЬФОКИСЛОТЫ — важней-
шие промежуточные продукты синтеза антрахиноно-
вых красителей. Все моно- и дисуль-
фокислоты антрахинона представля- О
ют собой окрашенные кристаллине- JL
окне продукты, плавящиеся во мно- [г || || 1 21
гих случаях с разложением. А. рас- IS 5 JI
творимы в воде, спирте; кристалли-
зуются с разным содержанием воды. О
Брутто формула моно-А. C14H8O5S,
мол. в. 288,28; ди-А. — C14H8O8S2, мол. в. 368,35.
Положение заме- стителей (SO3) 1 2 1,5 1,8 2,6 1,6 1,7
Т. пл., °C .... 218 — 310—311 (с разл.) 293—294 (с разл.) — 215—217
Число молекул кристаллизаци- онной воды . . 3 3 4 - 6 5 4
Вступление заместителя в один из бензольных цик-
лов молекулы антрахинона не дезактивирует другой
цикл, вследствие чего получение монозамещенного,
как основного продукта, возможно только при малой
степени превращения взятого в реакцию антрахинона.
Растворимость в воде сульфопроизводпых позволяет
легко отделять их от невступившего в реакцию исход-
ного вещества. Различная растворимость в воде нат-
риевых и калиевых солей изомерных дисульфокислот,
образующихся при сульфировании антрахинона,
открывает возможность получения также индивидуаль-
ных дизамещенных антрахинона. Сульфокислоты ан-
трахинона при хлорировании водных р-ров количест-
венно дают соответствующие хлорпроизводные, а при
нагревании с аммиаком в автоклавах — аминопроиз-
водные антрахинона, что является главным технич.
способом получения названных соединений.
269
АНТРАЦЕН — АНТРАЦЕНА СУЛЬФОКИСЛОТЫ
270
Антрахинон сульфируется только олеумом при
высокой темп-ре с образованием 2-сульфокислот,
2,6- и 2,7-дисульфокислот; в присутствии ртути основ-
ными продуктами сульфирования являются 1-моно-
сульфо-, 1,5- и 1,8-дисульфокислоты антрахинона.
1 - Сульфокислоту антрахинона получают
сульфированием антрахинона олеумом в присутствии
соединений ртути в условиях, обеспечивающих пре-
вращение примерно половины исходного в-ва. После
разбавления водой отделяют от «обратного антрахи-
нона» и добавкой КС1 или NaCl выделяют соответ-
ственно калиевую или натриевую соль. Применяют
для получения 1-амино-, 1-хлор-, в меньшей степени,
1-метиламиноантрахинона и 1-нитроантрахинон-5-
сульфокислоты. 2-С ульфо кислоту антрахино-
на получают аналогично 1-сульфокислоте, но в от-
сутствие соединений ртути; выделяют в форме нат-
риевой соли, известной под назв. «серебристой соли».
1,5- и 1,8-Д исульфокислоты антрахи-
нона получают исчерпывающим сульфированием ант-
рахинона в присутствии ртути. После разбавления
сульфомассы водой 1,5-изомер выделяют добавлением
NaCl в форме натриевой соли, а затем осаждают КС1
калиевую соль 1,8-дисульфокислоты. 2,6- и 2,7-Д и-
сульфокислоты образуются при исчерпываю-
щем сульфировании антрахинона в отсутствие ртути
и разделяются по различной растворимости натриевых
солей — менее растворима соль 2,6-изомера. 1,6- и
1,7-Д исульфо кислоты антрахинона, обра-
зующиеся в качестве побочных продуктов при полу-
чении 1,5- и 1,8-дисульфокислот, технич. значения
не имеют. С применением конц. олеума и большого
количества ртути сульфированием антрахинона не-
давно получена 1,3,5,7-т етрасульфокис-
л о т а антрахинона.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; В е н к а т а-
раман К., Химия синтетических красителей, пер. с англ.,
т. 1, Л., 1956. Н. С. Докунихин.
АНТРАЦЕН С14Н10, мол. в. 178 — бесцветные
пластинки (из спирта); т. пл. 216,6°; т. кип. 351°;
мало растворим в большинстве органич. раствори-
телей; А. (тв. и жидк.) и его растворы обладают фио-
летовой флуоресценцией. А. является конденсирован-
ной ароматич. системой, однако его химич. свойства
значительно отличаются от свойств бёнзола и нафта-
лина. Для А. характерна большая реакционная спо-
собность 9,10-, или мезо-атомов углерода; в этих
положениях легко идут реакции присоединения, что
не свойственно ароматич. соединениям. При гидроге-
низации образуется 9,10-дигидроантрацен; при хло-
рировании и сульфировании образуются смеси соот-
ветств. галоген- или сульфозамещенпых (см. Антра-
цена сульфокислоты, Антрацена галогенопроизводные).
При нитровании А. азотной к-той в смеси СН3СООН
(СН3СО)2О получаются 9-нитро- и 9,10-динитроантра-
цены. А. при окислении легко образует антрахинон,
а при озонировании дает 9,10-озонид. А. легко взаи-
модействует со свободными радикалами, напр. с бен-
зильным радикалом. Для изображения тонкого элект-
ронного строения А. предложено значительное число
формул, однако каждая из них дает лишь приближен-
ное описание свойств А. По данным рентгенострук-
турного анализа в молекуле А., в отличие от бензола,
длины С—С-связей неравноценны (см. формулы, рас-
стояния в А).
Основным источником А. является кам.-уг. смола,
в к-рой он был найден в 1832. Впервые в чистом виде
А. получен и исследован Ю. Ф. Фрицше в 1867.
В пром-сти А. выделяют из кипящих между 270’
и 360° тяжелых фракций кам.-уг. смолы (т. н. антра-
ценовое масло) с последующей очисткой от примесей
(гл. обр. карбазола и фенантрена) промыванием раз-
личными растворителями и кристаллизацией из пири-
диновых оснований. Т. обр. получают А. с содержани-
ем до 97%; возгонкой можно его получить 99,9%-ной
чистоты. Количественно А. определяется в виде антра-
хинона. А. в основном используется для получения
антрахинона.
Лит.: Ильинский М. А., Полупродукты и красители
антраценового ряда, М. — Л., 1932; Cl ar Е., Aromatlsche
Kohlenwasserstoffe, 2 Aufl., В. — [u. а.], 1952.
В. А. Пучков.
АНТРАЦЕНА ГАЛОГЕНОПРОИЗВОДНЫЕ — кри-
сталлич. продукты с характерной темп-рой плавле-
ния; монозамещенные на холоду растворимы в бен-
золе, эфире, уксусной к-те; дизамещенные растворимы
обычно в горячих бензоле и спирте; тетразамещенные
хорошо растворяются в бензоле.
Химич, поведения А. г. определяются характером
галогена и его положением в молекуле антрацена.
Наиболее легко вступает в различные реакции гало-
ген, стоящий в леао-положениях (9, 10) антрацена:
обмен на окси- и сульфогруппы, образование магний-
органич. соединений; мезогалогенозамещенные легко
окисляются с образованием антрахинона. Хлор и
бром легко взаимодействуют с антраценом с образо-
ванием мезопроизводных. 1- и 2-Галогенозамещенные
антрацена не могут быть получены непосредственным
галогенированием антрацена. Их получают из со-
ответствующих галогенантрахинонов восстановлением
Zn-пылью в аммиаке. Из фтор- и иодопроизводных
антрацена известны только мезозамещенные. 9-Иод-
антрацен получают из 9-антрилмагнийбромида и
иода в среде эфира. 9-Фторантрацен синтезируют из
антрацена действием на него хлороформенного р-ра
га-толилиододифторида.
Лит.: Ворожцов Н. Н., Основы синтеза промежу-
точных продуктов и красителей, 4 изд., М., 1955. В. А. Пучков.
АНТРАЦЕНА СУЛЬФОКИСЛОТЫ — твердые про-
дукты, не имеющие характерной темп-ры плавле-
ния; растворимы в воде и мало растворимы в органич.
растворителях. При сульфировании антрацена, в от-
личие от галогенирования и нитрования, образуются
1-(или а-) и 2-(или 0-) сульфозамещенные. Так, при
обработке антрацена олеумом в уксусной к-те полу-
чают равные количества 1- и 2-сульфокислот. При
сульфировании 75%-ной H2SO4 при 100° образуется
2-сульфокислота; добавка ртути направляет сульфо-
группу в 1-положение. В смеси H2SO4 с Na2SO4 при
105° образуется смесь дисульфокислот антрацена
(1,5-, 1,6-, 2,6- и 2,7-). Антрацен-9- (или мезо)-суль-
фокислота может быть получена из 9-нитроантрацена
кипячением с р-ром сульфита натрия. Аналогичным
путем из 9,10-дихлорантрацена получают антрацен-
9,10-дисульфокислоту. А. с. образуют функциональ-
ные производные (хлорангидриды, эфиры, амиды),
к-рые используются для их идентификации по темп-ре
плавления:
Положение
заместителя
Т.'пл., °C
хлорангидрида
амида
1
2
,5
,8
205
261
330
333
При сплавлении А. с. (а- и 0-) со щелочами сульфо-
группа заменяется оксигруппой с образованием
антролов. В отличие от а- и 0-сульфокислот, мезо-
бульфокислота антрацена неустойчива и легко гидро-
271
АНТРАЦЕНОВОЕ МАСЛО — АПАТИТ
272
лизуется с выделением антрацена. Применяются А. с.
для получения 1- и 2-ант ролов.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955. В. А. Пучков.
АНТРАЦЕНОВОЕ МАСЛО — фракция каменно-
угольной смолы, выкипающая в пре-
делах 270—360° (до 400°). См. Камен-
ноугольная смола.
АНТРИМИДЫ — не вполне строгое,
но принятое в химич. литературе назв.
соединений, содержащих, по крайней
ОН
мере, два антрахиноновых остатка, свя- /
занных иминогруппой. А. — высокоплав-
кие в-ва. Основным методом их получения являет-
ся взаимодействие соответствующих галогено-(обычно
хлор-) замещенных антрахинона с аминами в присутст-
вии металлич. меди или ее солей и щелочных агентов:
карбонатов или ацетатов щелочных металлов, окиси
магния. Процесс осуществляют нагреванием в высо-
кокипящих растворителях: нитробензоле, нафталине,
трихлорбензоле или сплавлением исходных в-в, напр.:
Большое значение приобрели А., в к-рых имино-
группа связана только с a-положением антрахинона;
они служат промежуточными продуктами произ-ва
важной группы кубовых красителей — дифталоил-
карбазолов. Последние образуются замыканием цикла
соответств. аминоантрахинонов под действием А1С13
или щелочи; ациламинозамещенные А. превращаются
в производные дифталоилкарбазола действием конц.
H2SO4. Из 1,1'-диантрахинониламиноантрахинона и
1,5-ди-(1'-антрахинониламино)-антрахинона получают
желтые, из 1,4-ди-(1'-антрахинониламино)-антрахи-
нона — коричневый, а из 1,4,5,8-тетра-(1'-аптра-
хинониламино)-антрахинона — цвета хаки кубовые
красители. Изомерные ди-(бензоиламиноантрахино-
нил)-амины служат исходными продуктами получе-
ния золотисто-оранжевого, коричневого и оливкового
кубовых красителей. Применение в технике находят
также более сложные А.
Лит.: Коган И. М., Химия красителей, М., 1956;
Венкатараман К., Химия синтетических красителей,
пер. с англ., т. 2, Л., 1957; Ворожцов Н. Н., Основы
синтеза промежуточных продуктов и красителей^ 4 изд., М.,
1955. И. С. Докунихин.
АНТРОЛОВАЯ КИСЛОТА (2-оксиантрацеп-З-кар-
боновая кислота), С15Н10О3, мол. в. 238,25 — почти
бесцветные кристаллы, т. пл. ок. 300° (с разл.), раст-
воримые в водных р-рах щелочей и карбонатов. А. к. (I)
получают из 2-хлорантрацен-З-карбоновой кислоты
(II) заменой хлора на гидроксил. В производств, усло-
виях это превращение гладко протекает в среде вод-
ного пиперидина при 155°/7 ат в присутствии меди.
А. к. может быть получена также из 2-антрола при
нагревании его с К2СО3 и СО2 при 220—230°/80 ат.
При действии на А. к. водного р-ра аммиака выше
200° (в автоклаве) в присутствии ZnCl2 образуется
2-аминоаитрацсн-З-карбоновая к-та. Арилиды А. К.
интересны как азосоставляющие для зеленых краси-
телей, образуемых на волокне. в. а. пучков.
АНТРОЛЫ (1-, 2- и 9-оксиантрацены) С14Н10О, мол.
в. 194,23—кристаллы светло-желтого цвета; известны
4 изомера А. А. хорошо растворяются в органич.
II III IV
растворителях с характерной синей флуоресценцией,
щелочные р-ры обладают зеленой флуоресценцией;
быстро окисляются на воздухе; легко образуют
эфиры и ацетильные производные. Свойства харак-
терных производных А. приведены в табл.
В химич. свойствах 1-А. и 2-А. наблюдается ана-
логия с соответствующими нафтолами; они вступают
в реакцию азосочетания с образованием азопроизвод-
ных; с азотистой к-той дают нитрозо-
О производные. 1-А. и его замещенные
JL образуют бисульфитные соединения,
if ll I Оксигруппа в А. замещается на
11 JI J аминогруппу действием аммиака при
высокой темп-ре и под давлением;
О особенно легко образование антрами-
нов идет в присутствии солей сер-
нистой кислоты (см. Бухерера реак-
ция). Под действием СО2 на 1- и 2-
антроляты Na или К (210—260°, 60 — 90 ат) образу-
ются соответственно 1-окси-2-карбоновая и 2-окси-З-
карбоновая к-ты антрацена (см. Кольбе—Шмидта реак-
ция). В технике 1- и 2-А. получают щелочным плав-
лением соответствующих к-т антрацена. При восстано-
влении антрахинона металлич. Zn, Sn, Al в кислойсреде
(конц. H2SO4, СН3СООН) образуется 9-антрон (IV),
к-рый при нагревании в щелочах переходит в 9-антрол
(III). 9-Антрон дает два ряда производных, отвечающих
обеим таутомерным формам; он легко вступает в реак-
ции за счет активной метиленовой группы в положе-
нии 10, напр. с формальдегидом в щелочной или кис-
лой среде образуется 10-метилен-9-антрон. Анало-
гично 9-аитрон реагирует с п-нитрозодиметнланили-
ном. А. применяются для синтеза красителей различ-
ных классов (полициклических, индигоидных, азо-
красителей).
Название Т. пл., °C Т, пл., °C, производного
1-Антрол (1) 150—153 (с разл.) Метиловый эфир 70 Этиловый эфир 69
2-Антрол (11) Разл. при 200 Метиловый эфир 175—8 Этиловый эфир 145—6 Ацетил 198
9-Антрол (111) 120 (при быстром нагревании) Ацетил 134
9-Антрои (IV) 154
В. А. Пучков.
АПАТИТ — минерал состава Са10В2(РО4)в, где
В—С1 или F; более распространен А. с преоблада-
нием фтора (фторапатиты). А. удлиненно-призматич.,
реже таблитчатые кристаллы гексагональной формы;
цвет преим. зеленоватый, спайность несовершенная,
блеск стеклянный, лучепреломление высокое (в сред-
нем 1,640), двойное лучепреломление низкое (0,004—
0,003). Твердость по Моосу 5; плотн. 3,18—3,41;
т. пл. фторапатита 1660°, хлорапатита 1530°. А. раст-
воряется в азотной, серной и соляной к-тах. Теоретич.
содержание Р2О5 во фторапатите 42,24%. Небольшая
часть Са в А. иногда бывает замещена Sr, Мп, а также
273
АПОРФИНОВЫЕ АЛКАЛОИДЫ — АРАБАНЫ
274
3-валентными редкими элементами (преим. цериевой
группы). Фтор или хлор бывают иногда частично
замещены SO4, СО3, О или ОН. А. широко распростра-
нен среди изверженных пород. Промышленные скоп-
ления А. встречаются относительно редко. Все изве-
стные месторождения А. могут быть отнесены: к маг-
матически инъекционным, образовавшимся при внед-
рении богатой фосфором магмы в толщу земной коры
(Хибины в СССР, Шведская Лапландия); жильным,
образовавшимся за счет газообразных выделений
остывающей на глубине магмы (Южная Норвегия);
месторождениям, приуроченным к пегматитовым жи-
лам (Канада). Хибинское месторождение А. является
крупнейшим в мире. В осадочных породах минералы
А. встречаются гл. обр. в форме фосфоритов. Апати-
товые руды б. ч. содержат в значительных количествах
примеси др. минералов. Так, в состав хибинских апа-
тито-нефелиновых руд входят: А., нефелин, пирок-
сены, титано-магнетит, ильменит, полевые шпаты и др.
Качество апатитовых руд определяется содержанием
в них фосфора в пересчете на Р2О5. Хибинская апа-
тито-нефелиновая руда содержит в среднем ок. 22%
Р2О5; после обогащения путем флотации содержание
Р2О5 повышается до 39,4—40%.
Подавляющее количество добываемого А. приме-
няют для произ-ва фосфорных удобрений, относительно
небольшую часть его — для произ-ва фосфора, фос-
форной к-ты, технич. фосфатов, в металлургии и пр.
Лит.: Ферсман А. Е., Апатит, его месторождения,
геохимия, запасы и экономика, веб.: Хибинские апатиты, т. 3,
Л., 1931; Лабунцов А. Н., Владимиров П. Н. и
Соловьянов Г. Н., в кн.: Неметаллические ископаемые
СССР. Гл. ред. Н.П. Горбунов, т. 1, М. — Л., 1936; Фи-
вег М. П., Шубин А. П., Апатиты, М. — Л., 1938; Ш е-
р е ш е в с к и й А. И., Требования промышленности к каче-
ству минерального сырья. Справочник для геологов, 2 изд.,
вып. 19, М., 1959. А. И. Шерешевский.
АПОРФИНОВЫЕ АЛКАЛОИДЫ — алкалоиды, в
основе строения к-рых лежит апорфин; обнаружены
преим. в растениях семейств Рара-
veraceae, Menispermaceae, Laureaceae
и Ranurculaceae. Главнейшие А. а.
перечислены в таблице (указаны: за-
местители, температура плавления и
уд. вращение); характеризуются, на-
'917NR8 пример, взаимодействием с ангидри-
is’^Ha Дами и хлорангидридами органич.
СН2 к-т; в результате получаются сое-
динения нейтрального характера,
что свидетельствует о наличии тетрагидрированного
пиридинового кольца.
r2
р I4 1
р2 1О|*л
о >,4СН
ному 8-винилфенантрену. Строение многих А. а. под-
тверждено полным синтезом. Апоморфин, апокодеин
и морфотебаин, получаемые при действии минеральных
к-т соответственно на морфин, кодеин и тебаин, по
строению примыкают к группе А. а. Известны А. а.,
выделяемые из растений в виде солей четвертичных
оснований. Таковы оксиметилат изокоридина и маг-
нофлорин—оксиметилат коритуберина. А. а. не нашли
широкого применения. Среди в-в группы апорфина
наибольшей известностью пользуется апоморфин соля-
нокислый, используемый в качестве рвотного.
Лит.: Юнусов С. и Прогрессов Н. Н., Ж.
общ. хим., 1952, 22, вып. 6, 1047; Т о m 1 t а М., К I к к а-
w a I., J. Pharniac. Soc. Japan, 1956, 76, 195; Cooke R. G.
and Haynes H. F., Austral. J. of Chem., 1954, 7, № 1,
99; Schmutz J., Helv. Chim. Acta, 1959, 42, fasc. 1, 335;
А г t h u r H. R. and C h e u n g H. T., J. Chem. Soc., 1959,
№ 6, 2306. См, также при ст. Алкалоиды. В. Киселев.
АПРЕССИН (хлоргидрат 1-гидразинфталазина),
мол. в. 196,65 — бесцветные кристаллы, т. разложе-
ния 273—276°; растворим в воде
(4,4 г в 100 г), мало растворим в NHNHa
спирте, нерастворим в эфире; 2%-ный JL
раствор А. в воде имеет pH 3,5— | Il I^.hci
4,5. С железом А. образует окрашен- L
ные комплексы. А. получают окис-
лением нафталина до фталоновой к-ты, к-рая с гидра-
зинсульфатом дает 1-фталазон-4-карбоновую к-ту;
последнюю декарбоксилируют и получаемый при этом
1-фталазон действием РОС13 превращают в 1-хлорфтал-
азин, из к-рого при реакции с гидразингидратом
получают основание А. А. обладает хорошо выражен-
ной способностью снижать артериальное кровяное
давление и применяется при различных формах гипер-
тония. болезни.
Лит. см. при ст. Адалин. А. И. Травин.
АПРОФЕН (хлоргидрат 0-диэтиламиноэтилового
эфира а,а-дифенилпропионовой к-ты)
(СвН5)йССН3-СОО(СН2)аН(С,.Н6)3 • НС1
мол. в. 361,92—бесцветные кристаллы, легко раство-
римые в воде; т. пл. 160—162°. С конц. H2SO4 А. дает
зеленовато-желтое окрашивание. Колич. определение
основано на разложении A. NaOH, подкислении точ-
ным колич. к-ты и определении ее избытка титрова-
нием NaOH с тимолфталеином. Получают А. конден-
сацией а,а-дифенилпропионовой к-ты с [J-диэтилами-
ноэтилхлоридом. А. применяют при спазмах сосудов
и органов брюшной полости, для стимулирования
родовой деятельности и др.
Лит. см. при ст. Адалин. А. И. Травин.
Название R1 R2 R, R. R. | Rs r7 Rs Т. пл., °C РШ
Апорфин Н н н н н | н н СН,
Бульбокапнин н н осн, он о—сн,—о н сн, 202 + 237° (СНС13)
Изокоридин н н осн. он ОСН, 1 осн, н сн, 186 4- 195° (CHCI3)
Коридин н н осн. осн, он осн, н сн, 149 -4- 205” (CHCI3)
Коритуберин н н осн, он он |осн, н сн, 240 4- 282е (спирт)
Антинодафнин н он осн, н О —сн2—о н н 210—211 4- 32,8° (спирт)
Болдин н он осн, н ОСН,| он н СН3 162-163 — 72,5°
Г.чауцин н осн, осн, н ОСНз 1 ОСНз н сн, 120 '4- 113° (спирт)
Дицентрин н осн, осн, н 0-СН,-0 н сн, 169 4-62” (СНС1,)
Лауротетанин ...... н он осн, н ОСНз I ОСНз н н 125 4- 98,5° (спирт)
Таликмидпн н осн, осн, н ОСНз 1 он н сн, . 192—193 — 84” (спирт)
Л аурелин н н ОСН3 н О—СН,-0 н сн, 114 — 98,5° (спирт)
Пукатеин н н н осн, О—СН2—О н сн, 200 — 220° (спирт)
Тудуранин н н он н ОСНз 1 ОСНз н н 125 — 148” (водный метанол)
днонаин н н н н О—СН2—О н н 122—123 — 52° (спирт)
Ремерин н н н н О—СН,—О н сн. 102—103 — 77,2° (спирт)
Таликмин н н ОСН, осн, О—СН,-0 OCHs сн, 137-138 4- 255° (спирт)
Кребанин осн.. ОСН3 н н о—сн,—о н сн, 126
Стефании осн, н н н О—СН.—О н сн, 155
Окисление оптически неактивных продуктов превра-
щения А. а. дает меллофановую к-ту, что доказывает
наличие фенантренового ядра. Распад А. а. по Гоф-
ману протекает в две стадии и приводит к замещен-
АРАБАНЫ — полисахариды, построенные из ос-
татков арабинозы. Широко распространены в расте-
ниях, -где обычно сопутствуют пектиновым веществам.
Все выделенные из растений А. построены из L-apa-
275
АРАБИНОЗА —АРБУЗОВА РЕАКЦИЯ
276
бофуранозных остатков, образующих цепи с а-1,5-
связями; к каждому второму звену цепи а-1,3-связью
присоединено по L-арабофурано'зному остатку;
Мол. веса А. порядка 6000. Очищенный препарат —
белый порошок. В воде А. образуют коллоидные р-ры;
[а]р в воде у различных А. колеблется от —60 до
—173°. А. нерастворимы в безводных спирте и аце-
тоне, но растворимы в их водных р-рах. Растворы А.
не восстанавливают Фелинга реактива. Кислотами А.
гидролизуются до L-арабинозы (с выходом 96%),
к действию щелочей стойки.
Лит.: Whistler R. L. and Smart С. L., Poly-
saccharide chemistry, N. Y_, 1953. Л. И. Линевич.
АРАБИНОЗА C5Hi0O5, мол. в. 150,14 — простой
углевод, моносахарид, относящийся к группе альдо-
пентоз (см. Пентозы). Наибольшее значение имеет
L-А. (природная А.): н
?„О„„
НОН2С-С—С —С —СНО |\он н>|н,он
н н он н т " Г
н он
L-A. — бесцветные кристаллы сладкого вкуса,
т пл. 158°; хорошо растворима в воде, значятельно
хуже в спирте, нерастворима в эфире; [а]|$=-|-108°
(в воде). Характерные производные: га-бромфенилгид-
разон, т. пл. 167° и дифенилгидразон, т. пл. 204—
205°. А. широко распространена в природе; остаток
L-А. участвует в построении молекул нек-рых
дисахаридов (напр., вицианозы), гликозидов (напр.,
генина, цикламина и др ) и многих сложных сахаров
растительного происхождения: гемицеллюлоз, ара-
бана, растительных камедей (гуммиарабик, вишне-
вый клей), пектиновых в-в, бактериальных полисаха-
ридов и др. В природных условиях остаток L-A.
в сложных сахарах образуется, по-видимому, при
окислении остатков D-галактозы у шестого углерод-
ного атома. А. получают кислотным гидролизом поли-
сахаридов, свекловичной стружки или вишневого
клея с последующим сбраживанием обычно сопрово-
ждающей А. галактозы и очисткой от других приме-
сей. L-А. в животном организме используется
мало; не сбраживается дрожжами. Другие микро-
организмы расщепляют А. с образованием молочной,
лимонной и масляной к-т.
D-A. — т. пл. 155,5—156,5°; [а]р =—108° (вводе);
в природе встречается сравнительно редко; получают
обычно из D-глюкозы.
Лит.: Carbohydrates, ed. by W. Pigman, N. Y., 1957.
Б. H. Степаненко.
АРАБИТ (арабитол) — пятиатомный спирт, мол. в.
152,15, генетически связан с арабинозой и ликсозой;
сн2он существует в виде двух стереоизомеров.
I D-A. —бесцветные кристаллы, т. пл. 102°;
хорошо растворим в воде и спирте, не-
нсон растворим в эфире; [a]D =4* 7,82° (в р-ре
неон буры). D-А. в свободном виде встре-
I чается в лишайниках в виде умбилици-
сн2он на (гликозида галактозы и D-А.) и в не-
которых грибах (ок. 10% сухого веса). Синтетиче-
ски А. получают восстановлением D-арабинозы н
D-ликсозы. По физич. свойствам L-А. аналогичен
D-А., за исключением уд. вращения, [а]в = —5,4°.
L-А. не обнаружен в природе, получен из L-ара-
бинозы при ее восстановлении и по Канниццаро
реакции. D, L-А., т. пл. 105°, в природе не встре-
чается, получен смешиванием эквимолекулярных
количеств D- и L-форм А., а также синтетически
ИЗ акролеина. Л. И. Линевич.
АРАХИСОВОЕ МАСЛО — растительное жирное
масло, получаемое из семян плодов арахиса, культи-
вируемого в странах с тропич. и субтропич. климатом.
Выход масла (в пересчете на ядро) в зависимости от
сорта и климатич. условий от 40,0 до 60,7%. Отличи-
тельная особенность А. м. — наличие высокомоле-
кулярных насыщенных жирных к-т; 2,3—4,9% ара-
хИновой, до 3% бегеновой и лигноцетиноной. А. м.
содержит также след, к-ты: 6—11% пальмитиновой,
3—6% стеариновой, 40—66% олеиновой, 18—33%
линолевой; до 1% неомыляемых в-в. Темп-pa заст.
ок. —3°, плоти. 0,91—0,96; Др 1,468—1,472; вязкость
по Энглеру 10—12 (20°); число омыления 188—197;
иодное число 83—105; родановое число 70,1—72,4.
А. м. применяется в кондитерской и маргариновой
ПрОМ-СТИ. Б. И. Хомут оз.
АРБИТРАЖНЫЙ АНАЛИЗ — контрольный ана-
лиз, проводимый с целью установления соответствия
качества сырья, готовой продукции и др. материалов
нормам ГОСТа и технич. условий. А. а. проводится
в случае возникновения разногласий между постав-
щиком и потребителем в оценке качества материала
не заинтересованной в споре лабораторией, избранной
обеими сторонами по взаимному соглашению. А. а.
производится по методике, установленной ГОСТом;
особое внимание уделяетсн квалификации исполни-
телей и технич. средствам анализа (реактивы, приборы,
посуда). Для проведения А. а. должна применяться
та же самая средняя проба материала, к-рая подверг-
лась первичному анализу. Если правильность отбора
средней пробы вызывает сомнение, то это само по себе
может быть причиной проведения А. а. (заново взятой
средней пробы).
Лит.: Дымов А. М., Технический анализ руд н метал-
лов, 5 изд., М., 1949. В. Г. Типцова.
АРБОРИЦИДЫ — см. Гербициды.
АРБУЗОВА РЕАКЦИЯ — перегруппировка эфи-
ров к-т 3-валентного фосфора под действием органич.
галогенидов с образованием соединений 5-валентного
фосфора. В результате А. р. образуется новая фосфор-
углеродная связь. Напр., при нагревании триметил-
фосфата с иодистым метилом образуется диметиловый
эфир метилфосфиновой к-ты:
(СН3О)8Р -> СН3Р(ОСН3)2
В этом случае возможно применение очень незначи-
тельных количеств CH3J. При необходимости введе-
ния к атому фосфора радикала, качественно отлич-
ного от имеющегося в фосфате, алкилгалогенвд
берется в стехиометрич. количестве:
(RO)3P + R’X
— R'P(OR)2 + RX
A. p. наблюдается в случае эфиров фосфористой
к-ты, а также эфиров алкил- и диалкилфосфицистых
к-т. При перегруппировке эфиров алкилфосфинистых
к-т образуются ’ эфиры диалкилфосфиновых к-т:
OR’ R’ о
R—Р\ + R"X -► >рС + R’X
XOR' R”Z XOR
Эфиры диалкилфосфинистых к-т в результате А. р.
277
АРБУТИН — АРГИНАЗА
278
превращаются в окиси третичных фосфинов:
R ,'R”
>P< + R-X
Rz 'O
R\
>P-OR' + R"X
Rz
НО^~~^-0—СН |
НСОН О
I
НОСН
I
НС----
I
неон
снгон
группу (напр., с FeCl3
лизуется кислотами
При наличии в молекуле фосфита нескольких разных
радикалов в процессе А. р. отщепляется наиболее
легкий.
15 качестве алкилгалогенидов можно применять
соединения, содержащие, помимо галогенов, другие
функциональные группы (галогенозамещенные эфиры
карбоновых к-т, галогенированные простые эфиры
и др.); таким способом можно синтезировать фосфорор-
ганич. соединения со смешанными функциями. При
реакции триалкилфосфитов с ацилгалогенидами обра-
зуются эфиры кетофосфиновых к-т, напр.:
сн3со о
(СН3О)3Р + СНзСОС! —> >Р< +СН3С1
H3COZ хосн3
Четыреххлористый углерод в А. р. реагирует анало-
гично алкилгалогенидам, причем образуются соеди-
нения с СС13-группой у фосфора, напр.:
н3со СС13
(СП3О)3Р + СС14 — >Р^ +СН3С1
II3COZ 'О
Обычно быстрее реагируют иодистые соединения,
медленнее — бромистые, еще медленнее — хлористые.
На примере оптически активных радикалов в исход-
ной молекуле эфира фосфористой к-ты установлено,
что А. р. сопровождается обращением конфигурации.
Кинетич. исследование А. р. показало, что процесс
является сложным, автокаталитическим. Во многих
случаях наблюдается аномальное течение А. р. Так,
аномально реагируют (пли вовсе не реагируют) вто-
ричные и третичные алкилгалогениды. При взаимодей-
ствии эфиров тритиофосфористой к-ты с алкилгало-
генидами сначала образуется сульфониевое произ-
водное, к-рое затем распадается:
P(SR)3 + R’X — (RShPS(RR')X — (RS)3PX -f- RSR'
При реакции эфиров фосфористой к-ты с а-галогени-
рованными кетонами наряду с продуктом А. р. обра-
зуется значительная примесь или даже преобладаю-
щее количество изомерного ненасыщенного эфира
фосфорной к-ты, напр.:
(СН3О)3Р + СН3СОСН2С1 — СН,СОСН3Р(О)(ОСН3)3 +
+ (СН3О)3Р(О)ОС(СН3)=СН3
А. р. широко используется в тех случаях, когда
возникает необходимость получить соединение с
С p-связями. А. р. применяется для получения
многих фосфорорганич. инсектицидов, лекарствен-
ных средств и др. физиологически активных фосфор-
органич. соединений.
Лит.: Арбузов А. Е., Избранные труды, М., 1952;
Арбузов Б.А.,вкн.: Реакции и методы исследования орга-
нических соединений, кн. 3, М-., 1954, с. 7; Химия и приме-
нение фосфорорганических соединений. Тр. первой конферен-
ции 8—10 декабря 1955 г., М., 1957.
АРБУТИН (гидрохинон-0-П-глюкознд) С12Н1вО7,
мол. в. 272.25 — гликозид группы фенолгликозидов,
встречающийся в толокнянке,
бадане и в других растениях
семейства камнеломковых и
вересковых. А. — бесцветные
кристаллы; т. пл. 199—200°;
растворим в воде, из которой
кристаллизуется с 1 молеку-
лой воды. Препараты арбути-
на, полученные из раститель-
ного материала, часто содер-
жат примеси метиларбутина.
А. дает реакции на фенольную
— синее окрашивание), гидро-
и ферментами — 0-глюкозида-
зами (эмульсином миндаля и др.) — до глюкозы и
гидрохинона. А. извлекают из листьев толокнянки
и бадана (22% А.); синтетически получен из ацето-
бромглюкозы и гидрохинона и др. способами. Исполь-
зуется для получения гидрохинона. Применяется
как мочегонное и антисептич. 'средство.
Лит.: The Carbohydrates. Ed. by W. Pigman, N. Y., 1957.
Л. И. Лиие&ич.
АРГЕНТОМЕТРИЯ — титриметрический метод ко-
личественного анализа, основанный на применении
титрованного р-ра AgNO3. При выполнении опре-
делений исследуемый р-р титруют р-ром AgNO3
или же его избыток, введенный в анализируемый
р-р, оттитровывают р-ром NaCl. Конечная точка
титрования в А. может быть установлена методом
равного помутнения. Этот метод применяли при оп-
ределении ат. веса Ag, Cl и ат. веса других элементов
в виде их чистых хлоридов; в нек-рых странах до
сих пор — для определения содержания Ag в сере-
бряной монете. Конечную точку устанавливают также
по просветлению р-ра или по отсутствию помутнения
при добавлении капли р-ра AgNO3. «Точка просвет-
ления» устанавливается с большой точностью, хотя
и не совпадает с точкой эквивалентности. Ошибка
титрования исключительно мала. Метод используется
для определения конечной точки при титрований Вт-,
J и др., образующих с Ag+ малорастворимые соли.
Часто в А. для установления конечной точки в каче-
стве индикатора применяют К2СгО4 — метод
Мора. В этом случае ионы < гС);' образуют мало-
растворимый красный осадок Ag2CrO4, по псявлению
к-рого и судят о конце титрования. Этот метод широко
применяется для определения хлоридов и бромидов;
точное определение иодидов и р щанидов невозможно.
Титрование по Мору проводится в нейтральных или
слабощелочных р-рах 7 < pH < 10,5. Чувствитель-
ность индикатора очень сильно понижается с увели-
чением концентрации водородных ионов. При опреде-
лении хлоридов, бромидов и иодидов в кислой среде
применяют метод Фольгарда. В этом слу-
чае прибавляют к анализируемому р-ру избыток
титрованного р-ра AgNO3 и затем непрореагировав-
ший нитрат серебра оттитровывают р-ром роданида.
Для установления конечной точки в А. пользуются
также адсорбционными индикаторами (см. Индика-
торы). Конечную точку титрования устанавливают
также различными электрохимическими и фотометри-
ческими методами. А. позволяет определять с большой
точностью хлориды, бромиды, иодиды, роданиды,
цианиды и вообще соли и кислоты, анионы которых
образуют с катионами серебра малорастворимые
осадки.
Лит.: Кольтхоф И. М. иСендэл Е. Б., Количест-
венный анализ, пер. с англ., 3 изд., М., 1948; Кольт-
гоф И. М. и Стенгер В. А., Объемный анализ, пер.
с англ., т. 2, М., 1952, с. 297—365. И. П. Ефимов.
АРГИНАЗА — гидролитический фермент, относя-
щийся к группе дезаминаз, мол. в. ок. 140000; А.
каталитически расщепляет L-аргинин на орнитин
и мочевину (D-аргинин практически А. не расщеп-
ляется):
NH
HOOCCHNH2(CH2)3NHC-NH2 + Н3О аргн"аза
,NH3
— HOOCCHNH2(CH2)3NH3 + о=с<
XNH3
А. действует лишь в присутствии свободных гуаниди-
новой и карбоксильной групп. Наибольшая' актив-
ность фермента при pH 10,2; в нейтральной среде
активность сохраняется, а при 6 >• pH ^9 — утра-
чивается. Активность А. повышается в присутствии
ионов Мп24', Со24', Ni2+, Cd24'; ионы Hg24' и Ag24", а так-
же нек-рые монокарбоновые L-аминокислоты алифатич.
279
АРГИ НИН — АРЕКОЛИН
280
ряда (L-орнитин, L-лизин, L-глицин и др.) тормозят
действие А. За единицу активности принимается такое
количество А.,к-рое за 1 мин. при 25°, pH 9,5 и кон-
центрации субстрата 0,285 моля образует 1 милли-
моль мочевины. А. широко распространена в тканях
животных, растений и в дрожжах; получают фрак-
ционированием водных экстрактов печени ацетоном
и сернокислым аммонием.
Лит. см. при ст. Альдолаза. Т. Т. Болотипа.
АРГИНИН (а-амино-б-гуанидиновалериановая кис-
лота)
NHS
ЛС—NH(CH»)2CH3CHCOOH
NH^ ' |
NH«
мол. в. 174,14 — мелкие кристаллы; т. пл.: L-A.
(природный) 238° (с рази.); D, L-A. 217—218° (рази.);
[а]%‘ =+ 11,37° (в воде); растворим в воде, трудно
растворим в спирте, нерастворим в эфире. При 25°
р/ 10,76, pKai 2,17 (СООН), pKai 9,04 (NH3+),
рКа 12,48 (гуанидин). А. образует труднораство-
римые соли с флавиановой, фосфорномолибденовой,
пикриновой и нек-рыми др. к-тами. Для А. специфич.
является цветная реакция с а-нафтолом и NaClO
или NaBrO в щелочной среде (реакция Сакагучи),
положенная в основу большинства методов колич.
определения А. При нагревании с щелочью А. распа-
дается на орнитин и мочевину. А. обнаружен во всех
белках. Особенно велико его содержание в протами-
нах и гистонах. L-А. является незаменимой аминокис-
лотой. А. получают выделением его из гидролизата
желатины в виде флавианата и превращением послед-
него в солянокислую соль. Синтетически А. может
быть получен из орнитина:
H»N(CH»)3CH(NH»)COOH — —°-
ZNH»
С -~кн
\осн»
— [H3N(CH»)8CH(NH2)COO]sCu----
H»N л
н ^C-NH(CHs)3CH(NHs)COO Си ——-
H»N,
— ' >C-NH(CH»)3CH(NH»)COOH
IINi7
Лит. см. при ст. Аминокислоты.
Л. Э. Локшина.
АРГОН (Argon) Аг — химич. элемент нулевой
группы периодич. системы Менделеева; п. н. 18, ат. в.
39,944, входит в число инертных газов. При обычных
условиях — газ без цвета и запаха. Впервые выделен
в 1894 Д. Рэлеем и У. Рамзаем из атм. азота. Наз-
ван от греч. ap^os — недеятельный. Содержание А.
в литосфере 4-10“ вес. %, в атмосфере 0,93 об. %.
Стабильные изотопы: Аг40, Аг38 и Аг36; их содер-
жание в атм. А. соответственно 99,600%, 0,063%
и 0,337%.
А. значительно более распространен на Земле, по срав-
нению с другими элементами нулевой группы (см. Кларки
элементов). Аномален и изотопный состав атм. А.: в отличие
от прочих легких элементов, среди изотопов А. преобладает
самый тяжелый — Аг40, с чем связано одно из нарушений хода
ат. весов в периодич. системе — ат. вес А. больше, чем у стоя-
щего за ним калия. Причина указанных явлений — образова-
ние в природных условиях Аг40 при распаде изотопа К40 по-
средством захвата орбитального электрона (К-захвата): К40 +
+ е—-Аг40. Большая часть Аг40 атмосферы очевидно и воз-
никла при распаде К40, содержащегося в земной коре. На изме-
рении отношений Аг4“/К40 в калийсодержащих минералах,
способных сохранять радиогенный Аг40, основан т. н. аргоно-
вый метод определения абс. возраста геологич. формаций (см.
Возраст геологический абсолютный).
Из искусственно радиоактивных изотопов наиболь-
ший период полураспада имеет Аг39 (Ti^ = 265 лет);
в качестве радиоактивного индикатора применяется
Аг37 (Г1/г = 34 дня). Строение наружной электрон-
ной оболочки атома А. : 3s2 Зр’. Энергия ионизации
(в :>в)\ Аг° —> Аг+—> Аг2+ соответственно равна 15,755;
27,5. При нормальных условиях А. — одноатом-
ный газ; плоти, d 1,7839 г/л; т. пл. —189,3°/760 мм;
т. кип. —185,9°/760 мм. Радиус атома А., опреде-
ляемый как половица атомного расстояния в кубич.
решетке, ранен 1,92 А(—233°). йжидк А. 1,401 г/см3
(- 185,9°), твердого 1,65 г/сж3 (—233°); %рит—122,43°,
Ркрит 49,59 кг/см2, dKplIT 530,8 г/л. Тройнаяточка А.:
Т 83,78° К; р 516,8 мм рт. cm., dTB 1,621 г/сз13,^шидк_
1,4100 г/см3. Теплота плавления 281 кал/молъ; теп-
лота испарения (в точке кипения) 1558 кал/молъ; уд.
теплоемкость газа Ср = 5,0071 кал/молъ- град (при
0°); теплопроводность 0,3889 • 10-4 кал/см-сек-град
(0°). Растворимость А. (см3 газа в 1 г воды при
давлении 1 кг/см2):0,0519 (0°); 0,0329 (20°); 0,0247 (40°);
растворимость понижается при добавлении элект-
ролитов. В органических растворителях А. раство-
рим значительно лучше, чем в воде. В металлах А.
практически нерастворим и поэтому не способен
диффундировать сквозь металлы.
В химич. отношении А. является инертным газом,
не дающим валентных соединений ни с каким другим
элементом. Получены немногочисленные химич. сое-
динения А., в к-рых его атом связан с другими ато-
мами ван-дер-ваальсовыми силами. Так, в 1896
Р. Вийяр получил кристаллогидрат Аг 6Н2О, сжи-
мая А. до 150 ат при 0° над частично замороженной?
водой. Темп-pa разложевия Аг-6Н2О 42,8°/1 ат.
В дальнейшем Б. А. Никитин получил Аг • 6Н2О
методом изоморфного соосаждения с SO2, HCI, НВг,
H2S. При выделении А. из минералов на стенках
аппаратуры, охлажденных до —193°, возникают сме-
шанные кристаллы шестиводных гидратов (Аг-6Н2О)
и моносилана (SiH4-6H2O). Получен ряд соединений
включения А. с Н2О, D2O, фенолом, гидрохиноном.
В соединении включения с фенолом Аг-2СвН5ОН
атом А. может быть изоморфно замещен НС1, НВг
или H2S. На основании изучения спектрон флюорес-
ценции в разрядных трубках сделано заключение
о существовании соединения Ar-Hg. На поверхно-
сти нек-рых твердых тел происходит адсорбция А. (он
адсорбируется лучше, чем гелий и неон). Качеств,
определение А. может быть осуществлено спектраль-
ным методом. При колич. определении А. активные
газы отделяются с помощью соответств. поглотителей.
Отделение А. от других инертных газов основывается
на различной адсорбируемости Не, No, Аг, Кг и Хе
активным углем.
В пром-сти технич. А. получают в процессе воздуха
разделения при глубоком охлаждении. От примесей
азота А. очищают дополнительной ректификацией,
а от примесей кислорода — химическими методами.
А. может быть получен как побочный продукт из про-
дувочных газов колонн синтеза аммиака. А. приме-
няют в металлургических и химических процессах,
требующих инертной среды (аргонно-дуговая сварка
алюминиевых и алюмо-магниевых сплавов), в свето-
технике (флюоресцентвые лампы, лампы накаливания,
разрядные трубки; цвет работающих аргоновых тру-
бок сине-голубой), в электронике (наполнение тира-
тронов и др.), в ядерпой технике (ионизац. счетчики
и камеры и т. п.).
Лит.: Фастовский В. Г., Редкие газы Л., 1940;
Никитин Б. А., Избр. труды, М. — Л., 1956; Кра-
мер Ф. , Соединения включения, М., 1958; fimelin,
8 Aufl., Edelgase. System-Nummer 1, Lpz. — В., 192 6;
Mellor, v. 7, L.— N. Y. — Toronto, [19561; Pascal, v. 1, P.,
1956; Ullmann, 3 Autl., Bd 6, Milnch.—B., 1955, S. 203—207;
К i r k, v. 7, N. Y., 1951, p. 408—419. Э. И. Герлинг.
АРЕКОЛИН (метиловый эфир N-метил-!,2,5,6-
тетрагидроникотиновой кислоты) C8H13O2N, мол. в.
281
АРИЛ — АРНДТА — ЭЙСТЕРТА РЕАКЦИЯ
282
155,20 — главный алкалоид семян ареновой пальмы
(Areca catechu), семейства пальмовых (Palmae), произ-
растающей в Индии, на Цейлоне и
QCOOCH3 Филиппинских островах. А. — бес-
цветное масло, т. кип. 209°; хоро-
•- шо растворим в воде и органич.
pL растворителях; летуч с водяным
3 паром, оптически неактивен. Тре-
тичное основание имеет сильно щелочную реакцию.
Ареколин легко образует соли, из которых важ-
нейшая — негигроскопичная бромистоводородная,
т. пл. 177—179° (из горячего этанола), применяется
в медицине как слюно- и потогонное средство, а
в ветеринарии — в качестве слабительного и анти-
гельминтного препарата. При гидроли-
зе сложноэфирной группы А. образует-
ся алкалоид арекаидин, также содер-
жащийся в арековой пальме. Другие
алкалоиды того же растения — гува-
цин и гуваколин — легко могут быть
переведены в А. Содержание ареколи-
на в растениях составляет 0,1—0,3% от
го сырья, выделение производится экстракцией семян
эфиром. В Советском Союзе получают А. синтети-
чески:
2 H2C=CHCOOCH3 + CH3NH2
О
соосн3
сн3
ОН
COOCHg SOCls
сн3
аСООСНд СН3ОН
НС. нВг
7
сн3
Лит. см. при ст. Алкалоиды. А. Е. Васильев.
АРИЛ — обобщенное название одновалентных ра-
дикалов ароматич. ряда, связанных с различными
функциональными группами. Для арильных радика-
лов (напр., фенил, а- и р-нафтил и т. д.) принято
сокращенное обозначение Аг.
АРИЛИРОВАНИЕ — реакция замещения водо-
рода или металла арильной группы; в более широком
смысле — реакция введения арильной группы. А.
широко используется для лабораторного и промыш-
ленного получения замещенных ароматич. соедине-
ний (фенолов и их эфиров, аминов, соединений гало-
генов). Исходными арилирующими агентами служат
галогенарилы (ур-ния 1—3), галогенониевые (4) и
диазопиевые соединения (5):
ArCl + Н-0 -, АгОН + НС1 (1)
ArJ + НО Аг' —> АгОАг' -J- HJ (2)
ArCl + NH3 —» ArNH3 -J- HC1 (3)
[Ar2Br]+BF- + HOR — ArOR + ArBr 4 HBF4 (4)
ArNaBr + H3O —. ArOH 4- Ns + HBr (5)
А. может протекать либо как ионный, либо как ради-
кальный процесс. А. по ионному механизму, характер-
ное для галогенарилов, представляет собой реакцию
сольволитич. замещения у ароматически связанного
атома углерода. Способность к такому замещению
в основном зависит от основности арилируемого в-ва
и от строения арилирующего агента (характера и
местоположения активирующих групп, а также при-
роды атома галогена) и выражена в меньшей степени,
чем у насыщенного углеродного атома. Так, замеще-
ние хлора в хлорбензоле на OR- или МН2-группы идет
с заметной скоростью лишь при 200—350°, причем
требуется применение катализаторов — соединений
меди (см. Ульмана реакция). Накопление в орто- или
пара-положениях электроноакцепторных групп уве-
личивает скорость сольволиза. Скорость замещения
галогена увеличивается в ряду: С6Н5С1; n-NO2CeH4Cl;
n-NO2C6H4Br; 2,4-C6H3(NO2)2Cl; 2, 4, 6-C6H2(NO2)3Cl
слева направо; 2,4-динитро- и 2, 4, 6-тринитрохлор-
бепзол, напр., взаимодействуют с основаниями при
комнатной темп-ре:
NOS
веса сухо- [ А. соединениями диазония и галогенония может про-
—- ------- ходить как по ионному, так и по радикальному меха-
низму (А. Н. Несмеянов). Арильные радикалы, обра-
зующиеся при распаде этих соединений, реагируя с
веществом (обычно используемым в качестве
CH3ONa растворителя), арилируют его.
(CH3OOCCHsCH2)2NCH3-----* Лит..- Реутов О. А., Теоретические про-
блемы органической химии, М., 1956; Sauer
J., Н u1sg en R., Angew. Chem., 1960, 72, № 9, 2 94.
АРМИН (этиловый пара-нитрофениловый эфир этил-
фосфиновой кислоты), мол. в. 259,20 — желтоватая
жидкость; 1,255—1,260; Пд
1,522—1,527; мало растворим С2Нб°Ч'Р—с»Н
в воде, легко — в спирте, эфире O2NC(iH4O/1|
и бензоле. Качественное опреде- °
ление А. основано на его омылении в щелочной среде
и появлении фиолетово-красной окраски после нейтра-
лизации и прибавления раствора FeCl3. Для коли-
чественного определения А. минерализуют смесью
конц. H2SO4 и HNO3, нейтрализуют аммиаком и об-
рабатывают раствором молибдата аммония; осадок
растворяют в точном количестве NaOH и титруют
раствором НС1 с фенолфталеином. А. обладает силь-
ным антихолинэстеразным действием и применяется
в качестве миотического и противоглаукоматозного
средства.
Лит. см. при ст. Адалин. А. И. Травин.
АРНДТА СПЛАВ — сплав 60% меди и 40% маг-
ния; применяется в аналитич. химии для восста-
новления нитратов в аммиак. К исследуемому рас-
твору прибавляют 20%-ный раствор MgCl2 и измелы-
ченный в порошок А. с. и отгоняют 2/3 жидкости
в титрованный раствор кислоты. Избыток последней
определяют титрованием, а содержание аммиака —
по разности.
АРНДТА — ЭЙСТЕРТА РЕАКЦИЯ - превращение
карбоновой кислоты в следующий высший гомолог
или его производное. Процесс реализуется через три
последовательные стадии:
1) получение галогенаигидрида к-ты: RCOOH —>
RCOX;
2) получение диазокетона: RCOA + CHON2 —>
RCOCHN2 + НХ;
3) перегруппировка диазокетона в кислоту или ее
производное — Вольфа перегруппировка: RCOCHN2%
+ HY — RCH2COY + N2. А.-Э. р. применима к али-
фатич., алициклич., ароматич. и гетероциклич, кар-
боновым к-там. Иногда во второй стадии реакции
вместо диазометана используют другие алифатич.
диазосоединения, при этом получаются замещенные
к-ты или их производные. Третью стадию проводят
в присутствии воды, спиртов, аммиака или аминов
283
АРНДТА-ЭЙСТЕРТА РЕАКЦИЯ — АРОМАТИЗАЦИЯ
284
и соответственно получают след, гомологи кислот, их
эфиры или амиды:
CHgCOCl
t.CH2Na
a.NH3+Ag2O
ch3ch2conh2
CH2CH2CH2COCl
CH2CHaCH2COCl
i.CHaN2
2.H2O+Ag2O
CH2CH2CH2CH2COOH
CH2CH2CH2CHaCOOH
Н3СХ
СН—СН—COCl
н3с^
n:
t,CH2Na
2.CH3OHH-Ag2O
НзСхсн-сн-сн2соосн3
НзС/ 1/СО
N.
ч?.о
o2n
2.CeHBNH2
i.CH3CHN2'
COCl „с н мн ®2
При перегруппировке в отсутствии нуклеофильных
реагентов в некоторых случаях удалось выделить со-
ответствующие кетены. Карбонильный атом угле-
рода в диазокетоне при перегруппировке Вольфа
превращается в атом углерода карбоксильной груп-
пы конечного продукта, что установлено с помощью
меченого углерода С13:
CeH6CCIINa — С0Н5СН.3СООН
I °
При перегруппировке оптически активио-
।го диазокетона, молекула которого содер-
жит асимметрич. атом углерода, соседний
с карбонильной группой, конфигурация,
как правило, сохраняется.
А.-Э. р имеет преимущество перед други-
ми методами удлинения цепи карбоновых
к-т: она не связана с применением восста-
новителей и т. о. позволяет использовать
кислоты, содержащие нитрогруппу, карб-
_ алкоксильную группу и др. заместители,
затрагиваемые при восстановлении. Сум-
марный выход по всем трем стадиям А.-Э. р. обыч-
но колеблется от 50 до 80% от теоретически воз-
можного. Метод находит применение для препара-
тивного получения (в не слишком больших количе-
ствах) карбоновых к-т и их производных из кислот
с меньшим числом атомов углерода. Перегруппиров-
ку диазокетонов в кислоты открыл Л. Вольф в
1902. Способ получения диазокетонов из хлорангид-
ридов и диазометана разработал ф. Арндт с сотруд-
никами (1927—28). В 1935 Ф. Арндт и Б. Эйстерт, ис-
пользовав перегруппировку Вольфа, разработали в
целом метод удлинения углеродной цепи карбоновых
к-т на одну метиленовую группу.
Лит.: Бахман В., Стр у в е В., в кн.: Органические
реакции, сб. 1, пер. с англ., М., 1948, с. 53—83; Кну-
нянц И. Л., Гамбарян Н. П., Рохлин В. М., Усп.
хим., 1958, 27, № 12. с. 1383. Е. М. Рохлин.
АРОМАТИЗАЦИЯ — реакция образования арома-
тич. соединений из соединений др. классов. Ароматич.
соединения могут быть получены из алифатич. соеди-
нений дегидроциклизацией, полимеризацией или кон-
денсацией. А. 6-членных алициклических соединений
производят дегидрогенизацией, дегалогенированием,
дегидратацией, изомеризацией; А. других алицик-
лич. соединений — изомеризацией в сочетании с выше-
приведенными реакциями. Парафиновые углеводо-
роды, содержащие более 6 атомов угле-
рода в прямой цепи, образуют арома-
тич. углеводороды реакцией каталитич.
дегидроциклизации, открытой в 1936
советскими химиками (Б. А. Казанским
и А. Ф. Платэ, Б. Л. Молдавским и др.).
Реакция протекает на платинированном
угле при 300°, на окиси хрома или оки-
c. лах хрома, молибдена, ванадия, нане-
сенных на окись алюминия при 500—
550°. При этом из молекулы парафиново-
го углеводорода получается содержа-
число атомов углерода ароматический
CH3
ch-co-nh
Ц I l.N2CH СООСНз- I LC1/COOCH;|
\oP-COBr a.cH3oH+Pt cr xcooch3
Галогенангидрид (обычно хлорангидрид) в виде
эфирного или бензольного р-ра медленно прибавляют
к охлажденному р-ру избытка диазометана. Образова-
ние диазокетона, как правило, заканчивается за неск.
часов при комнатной темп-ре. Перегруппировка диа-
зокетона обычно достигается нагреванием в течение
нескольких часов до 60—100° в смеси органич. раст-
ворителя (диоксана) и соответствующего нуклео-
фильного соединения (воды, спирта, аммиака, амина)
в присутствии катализатора, чаще всего окиси сереб-
ра; иногда применяют металлич. медь или платину.
В нек-рых случаях перегруппировку проводят при на-
гревании или облучении в отсутствии катализаторов.
Механизм получения лиазокетона заключается в
промежуточном образовании диазоний-бетаина (I)
с последующим отщеплением элементов галогеноводо-
рода (1). Полученный диазокетон может реагировать
с выделившимся хлористым водородом, образуя по-
бочный продукт реакции — хлорметилкетон, к-рый
становится основным продуктом в отсутствии избытка
диазометана — Ниренштейяа реакция (2). Для того
чтобы уменьшить количество хлорметилкетона, при-
меняют избыток диазометана, к-рый связывает НС1 (3):
R-Cp + CH2-nen
xCl
о
II - -
R-C-CH-N5N + HCI
c? - -
R— C-iCH-N = N
C1
'“Cl H
о
r-c-Ch2-nsn]ci
R-C-CH-NHN + HCI (I)
о
R-c-CH2Cl + N2 (2)
CH2-N=N + HCI —- CH3Cl + N2 (3)
Наиболее вероятный механизм перегруппировки
Вольфа включает промежуточное образование оксо-
карбена — TI (см. Карбены), который перегруппиро-
вывается в кетен, дающий с водой, спиртом и т. п.
к-ту или ее производное:
О
II - О +
R-C-CH-N=N—*
II р.
R\C£CН
II
—- О=С=СН — R
Н1
rch2coj
щий то же
углеводород:
С6Н14—-С6Нв + 1Н2: С7Н16-*С6Н5СН3+4Н2:
СН3СН2СН2СН2СН2СН2СН3<^ $ +1Н2
-------1 0 - СН ^С,Г1 з
Олефины и тем более ди- и полиолефины аромати-
зуются при более низких темп-pax на окисных ката-
лизаторах и не ароматизуются на платинированном
угле. А. наблюдается при пиролизе парафиновых
и олефиновых углеводородов, в том числе и низших,
даже таких, как этан или этилен. В последнем слу-
285
АРОМАТИЗАЦИЯ - АРОМАТИЗАЦИЯ НЕФТЕПРОДУКТОВ
286
чае стадия, приводящая к образованию 6-членного
циклич. углеводорода, заключается, по-нидимому,
в термич. конденсации промежуточного продукта —
диена с олефином:
СН~СН нс^ ^снг
СН2 ЧСН2—* II I — С6Н6 + 2На
+ нсх ^СН2
СН2=СН2 СН2
Ацетилен, как нашел н 1866 М. Бертло, при про-
пускании через нагретые до 500° трубки образует
частично бензол— реакция, впервые связавшая между
собой алифатич. и ароматич. классы соединений. На
активированном угле эта реакция протекает с выхо-
дом до 30% (Зелинский, Казанский), ацетилен в при-
сутствии комплексного соединения никеля дает 88%
бензола и 12% стирола. Производные ацетилена
особенно легко претерпевают А. н результате реакции
тримеризации: аллилен в присутствии H2SO4 образует
мезитилен, пропиловая к-та на солнечном свету дает
тримезиновую к-ту:
ЗСН^ССООН->1,3,5-С0Н3(СООН)3
Большое препаративное значение имеет циклоконден-
сация кетонов и альдегидов в ароматич. соединения,
напр. ацетон циклоконденсируется в мезитилен.
Моно-, ди- и полициклич. насыщенные и ненасыщен-
ные углеводороды с 6-членными кольцами в присутст-
вии катализаторов претерпевают А. В работах
Н. Д. Зелинского и его учеников подробно изучены
эти реакции, протекающие в присутствии дегидри-
рующих металлич (Pt, Pd, Ni и др.), окисных (Сг2О3,
МоО2, V2O3 и др.) и сульфидных (NiS, MoS, WS)
катализаторов: напр., С6Н12 —>С6Н6 + ЗН2 (см. Гид-
рогенизация и дегидрогенизация каталитические, Ка-
тализ). Эти реакции в ряде случаев проходят в при-
сутствии S, Se и Те. А. подвержены и многие другие
алициклич. соединения с разнообразными функцио-
нальными группами, содержащие 6-членные циклы.
Так, циклогексанол С6НиОН образует фенол С6Н5ОН.
Препаративное значение этих реакций возрастает
благодаря диеновому синтезу. Галогенопроизводные
алициклич. соединения ароматизуются при отщепле-
нии галогена или галогеноводорода. Так, гексахлор-
циклогексан при действии металлич. цинка дает
бензол, а со спиртовым р-ром щелочи — 1,3, 5-три-
хлорбензол. Реакции, приводящие к А., часто сопро-
вождаются (или им предшествуют) реакциями изо-
меризации углеродного скелета. Напр., 2,2-диметил-
гексан или ге^-диметилциклогексан при А. образуют
смесь ксилолов и толуола. А. хиноидных или полу-
хиноидных колец происходит часто путем смещения
двойных связей. Так, хинон при действии уксусного
ангидрида превращается в триацетат оксигидрохинона.
Реакции этого типа особенно часто встречаются при
синтезе или исследовании стероидов или близких
к ним веществ. Алициклич. соединения, содержащие
5,7 или больше атомов углерода в кольце, образуют
ароматич. соединения после изомеризации кольца
н 6-членное. Так, метилциклопентан при 480° под
давлением водорода на платино-алюмо-молибденовом
катализаторе превращается в бензол; циклогептан
в присутствии селена при 390° — в толуол, а цикло-
октан — в n-ксилол; циклононан на платинированном
угле образует смесь гидриндева и о-метилбензола
с другими ароматич. углеводородами, а циклодекан —
нафталин и нек-рые другие углеводороды.
Процессы А. протекают также в условиях биохи-
мия. синтеза н растит., животных и микроорганизмах.
Большое препаративное значение имеют многие реак-
ции А. В пром-сти широкое применение получили
процессы А. бензинов с целью улучшения их эксплу-
атац. свойств и получения индивидуальных ароматич.
соединений для нефтехимия, синтеза (см. Ароматиза-
ция нефтепродуктов). Образование ароматич. со-
единений при коксовании каменного угля также яв-
ляется результатом пирогенетич. А. моно- и полицик-
лич. соединений, содержащихся н нем.
Лит.: Плата А. Ф., Каталитическая ароматизация
парафиновых углеводородов, М.—.Л., 1948; Campbell N.,
в кн.: Chemistry of carbon compounds, ed. by E. H. Rodd v. 3,
part A, p. 65—80; Hanse hC., Chem. Rev., 1953, 53, p. 353—96;
Стейнер Г., в кн.: Катализ в нефтехимической и нефте-
перерабатывающей промышленности. Под ред. П. Эмметта, пер.
с англ., М., 1959, с. 2 70. М. И. Розвигарт.
АРОМАТИЗАЦИЯ НЕФТЕПРОДУКТОВ — про-
цесс обогащения различных видов нефтяного сырья
ароматич. углеводородами за счет других классов
углеводородов, протекает но многих процессах нефте-
переработки и в нек-рых из них, напр. при каталитич.
риформинге, является самостоятельной операцией
и служит для получения ароматизированных бензи-
нов и ароматич. углеводородов.
При термич. крекинге (650° и выше, 1 ат) обра-
зуются сильно ароматизированные продукты, однако
с малыми выходами. Первичными процессами А. н.
в этих условиях являются различные процессы раз-
ложения, включая разрыв колец у циклич. углеводо-
родов, в результате чего получаются высокие выходы
газа. Промежуточная стадия — образование больших
количеств олефинов, за счет к-рых путем реакций
циклизации и конденсации и происходит А. н.,
поэтому выход бензольных углеводородов в этих
условиях мало зависит от природы сырья. Одновре-
менно в термич. процессах протекают реакции обра-
зования полициклич. и полимерных продуктов, при-
водящие к появлению остаточных фракций с темп-рой
кипения выше темп-ры конца кипения исходного
сырья. Процесс крекинга нефти при высокой темп-ре,
т. н. пиролиз нефти, применяется для получения
ароматич. углеводородов и газообразных парафинов.
В каталитич. крекинге А. н. протекает одновременно
с процессом перераспределевия водорода, н резуль-
тате происходит насыщение олефияов и образование
ароматич. углеводородов, а также продуктов уплот-
нения на катализаторе. Ароматич. углеводороды,
образующиеся, напр., при каталитич. крекинге пара-
финов, накапливаются преим. но фракциях с высокой
темп-рой кипения, где содержание их повышается
с возрастанием темп-ры крекинга и может достигать
50% и более, чем затрудняется даль-
;ОСН3 нейшая переработка таких фракций.
Для произ-ва ароматизированных бен-
4 зинов каталитич. крекинг, вообще гово-
I ОСОСН Ря’ непРиг0Ден, так как для этой цели
3 требуется выполнять процесс в жест-
:ОСН3 ких условиях, что приводит к образо-
ванию больших количеств газа и кокса.
При гидрогенизации деструктивной А. н. является
результатом неполного гидрирования полициклич.
ароматич. углеводородов. Образующиеся полициклич.
углеводороды с частично гидрированными кольцами
термически нестабильны и в присутствии водорода
разлагаются с раскрытием гидрированных колец
и образованием производных менее циклизованных
ароматич. углеводородов; в результате пределы выки-
пания фракций, содержащих полициклич. углеводо-
роды, значительно понижаются. Степень А. н. зависит
287
АРОМАТИЧЕСКИЕ СИСТЕМЫ
288
от темп-ры, давления водорода и активности катализа-
тора. При темп-pax ниже 450°, высоком давлении
водорода и в присутствии активного катализатора
степень А. н. может быть весьма малой. При темп-рах
выше 500°, в присутствии умеренно активного катали-
затора реакции дегидрогенизации и А. н. протекают
легко, давая бензины с высоким содержанием арома-
тич. углеводородов.
Наиболее широкое промышленное применение
А. н., гл. обр. за счет реакций нафтенов и дегидро-
циклизации парафинов, получила в различных про-
цессах каталитического риформинга. Основой про-
мышленного использования указанных реакций яви-
лись работы Б. Л. Молдавского (1936), Б. А. Казан-
ского (1936), их учеников и сотрудников (см. Арома-
тизация}.
Лит.: Обрядчиков С. Н., Технология нефти, ч. 2,
3 изд., М. — Л., 1952; Раппопорт И. Б., Искусственное
жидкое топливо, 2 изд., М., 1955. В. В. Щекин.
АРОМАТИЧЕСКИЕ СИСТЕМЫ - - циклич. си-
стемы, характеризующиеся тем, что все их атомы при-
нимают участие в образовании единой сопряженной
системы, л-электроны к-рой образуют устойчивую,
т. н. замкнутую, электронную оболочку. Название
«ароматический» возникло по отношению к производ-
ным бензола (I) и было первоначально связано с тем,
что среди них были найдены соединения с приятным
(«ароматическим») запахом. Затем понятие А. с. очень
скоро было перенесено на нафталин (II), антрацен
(III), фенантрен (IV) и др. системы, состоящие из
конденсированных бензольных колец.
В бензоле, несмо-
тря на то, что его ча-
сто изображают фор-
мулой (I), предложен-
ной А. Кекуле, все
углерод - углеродные
связи равноценны (см.
формулу 1а) ио длины
их равны 1,40 А. Ана-
логичным образом нет
чередующихся про-
стых и двойных свя-
зей в нафталине, антрацене, фенантрене и др. А. с.
В связи с этим существует только по одному изомеру
каждой из этих А. с. Как бензол, так и другие
конденсированные системы энергетически весьма
устойчивы, что связано с весьма высокой энергией
сопряжения: 40 ккал/молъ для бензола, 75 ккал/молъ
для нафталина, 105 ккал/молъ для антрацена и т. д.
В дальнейшем понятие А. с. значительно расшири-
лось и стало применяться к самым разнообразным
циклич. системам, обладающим общими особенностями
строения и совокупностью характерных химич. и
физич. свойств. В первую очередь это относится
к многочисленным гетероциклическим А. с.: фурану
V V! VII Н VIII Н
w Н X XI XII
ролу (VII), пиразо-
лу (VIII), имидазолу
(IX), тиазолу (X),
оксазолу (XI), пири-
дину (XII), производ-
ным пирана и др., а
также к их бензо-
производным (индолу,
бензофурану, бензо-
тиофену и т. д.). Эти
гетероциклич. соеди-
н ы м и аналога-
пения являются электрон
ми бензола. Как в бензоле, так и в гетеро-
циклических А. с. (V—XII) имеется сопряженная
система бл-электронов («ароматический секстет»).
Так, в случае 5-членных систем фурана, тиофена
и пиррола ароматич. секстет л-электронов обра-
зуется за счет 4л-электронов, четырех атомов угле-
рода и 2д-электронов неподеленной пары гетеро-
атома О, S или N. В пиридине ароматич. секстет
образует 5л-электронов углеродных атомов и шестой
Л-электрон атома азота, а неподеленная пара р-элскт-
ронов азота не принимает участия в образовании
А. с. С ароматичностью этих гетероциклонов связана
их сравнительно высокая стабильность (энергия со-
пряжения тиофена составляет 31 ккал/молъ. пиррола
22,6 ккал/молъ, фурана 23 ккал/молъ}.
Для гетероциклич. А. с. мало характерны реакции
присоединепия по кратным связям, однако они про-
текают легче, чем для бензола. Так, фуран и его гомо-
логи, в отличие от бензола, легко присоединяют
малеиновый ангидрид, т. с. ведут себя в этом и в ряде
других отношений подобно диенам с сопряженными
двойными связями. Такие гетероциклич. А. с., как
фуран и пиррол, весьма неустойчивы к действию
сильных к-т, что связано с «выключением» неподе-
ленной пары электронов гетероатома из сопряжения
в результате солеобразования и, таким образом, нару-
шением ароматич. системы бл-электронов. С другой
стороны, сходство 5-членных гетероциклич. А. с.
с бензолом и их ароматичность проявляются в способ-
ности легко вступать в реакции электрофильного заме-
щения. Так, тиофен легче, чем бензол, сульфируется,
нитруется, ацилируется, мер курируется. Неустойчи-
вый к действию к-т фуран нитруется ацетилнитратом,
бро.мируется диоксандибромидом, сульфируется пири-
динсульфотриоксидом, ацилируется при действии
ангидридов к-т в присутствии хлорного олова. Пиррол
также легко подвергается электрофильному замеще-
нию при действии мягких агентов. Он легко дает
металлич. производные в результате замены атома
водорода в группе NH (напр., пирролкалий C4H4NK)
и по ряду своих свойств напоминает фенол.
В отличие от 5-членных гетероциклов, пиридин
сульфируется, нитруется, бромируется и вступает
в другие реакции электрофильного замещения значи-
тельно труднее, чем бензол, но легче подвергается за-
мещению при действии нуклеофильных агентов (обра-
зование аминопроизводных при действии амида нат-
рия и а -океппиридина при действии едкого кали).
Круг известных А. с. за последнее время значи-
тельно расширился, гл. обр. за счет большого числа
небензоидных ароматич. систем, весьма различных
по своим химич. и физич. свойствам. К их числу
относятся некоторые стабильные органич. ионы — ка-
тионы тропилия (XIII) и циклопропспилия (XIV),
анион циклопентадиенила (XV); макроциклические
системы — циклооктадеканонаен (XVI) и циклоок-
тадекагексаен -1,3,7,9,13,15 - триин - 5,11,17 (XVII);
289
АРОМАТИЧЕСКИЕ СИСТЕМЫ—АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
290
биполярные соединения — азулены (XVIII), сидноны
(XIX); металлоорганические соединения— ферроцен
вых» систем; неорганические соединения — боразол
(XXII), фосфонитрилгалогениды (XXIII) и (XXIV).
Н
I
нвЛ.-Хвн
I
н
XXII
Ci
Cl
Cl XXIV
Для всех А. с. как производных бензола, так и
нсбензоидных, характерен ряд общих особенностей
строения; к ним относятся: 1) характер углерод-угле-
родяых связей в цикле — промежуточный между
простыми и двойными (что выражается, в частности,
в величинах межатомных расстояний, промежуточных
между значениями для соответств. простых и двойных
связей); 2) равноценность всех углерод-углеродных
связей и эквивалентность атомов углерода в бензоле
и других подобных незамещенных мопоциклич. си-
стемах (тропилии, циклопентадиснил-анионе, ферро-
цене); 3) плоское или близкое к плоскому строение
цикла; 4) поглощение света в области сравнительно
больших длин волн; 5) легкая поляризуемость и отно-
сительно высокие показатели преломления; 6) ани-
зотропия диамагнитной восприимчивости.
(I другой стороны, большинство ароматич. соедине-
ний обладает рядом общих, т. н. ароматических химич.
свойств. К наиболее характерным среди них относятся:
1) сравнительная легкость образования; так, система
бензола необычайно легко образуется при дегидроге-
низации производных циклогексана, необратимом
катализе Н. Д. Зелинского, дегидроциклизации пара-
финов, циклизациях диенов, диенинов и т. д.; 2) ста-
бильность А. с., проявляющаяся, в первую очередь,
в том, что для них мало характерны реакции присо-
единения по кратным связям, типичные для олефинов;
3) легкость замещения водорода различными группами
в реакциях электрофильного замещения — нитро-
вания, сульфирования, галогенирования, ацетилиро-
вания, алкилирования, меркурирования, дейтеро-
обмена ит. д.; 4) характерные свойства нек-рых заме-
стителей в А. с., как-то: кислотные свойства фенолов,
ослабленная основность аминогруппы, устойчивость
диазосоединсний, способность к реакциям диазосоче-
тания и др.
При сравнении ароматичности в качестве эталона
обычно выбираются свойства бензола, причем при
переходе к другим А. с. одни ароматич. свойства
ослабляются, в то время как другие заметно усили-
ваются. Так, способность вступать в реакции электро-
фильного, так называемого ароматического, замещения
убывает в ряду: циклопентадпенил-анион > пиррол >
бензол пиридин > тропилий, причем катион
тропилия не подвергается электрофильному замеще-
нию даже в весьма жестких условиях. Однако способ-
ность к реакциям нуклеофильного замещения (при
действии нуклеофильных агентов) изменяется в обрат-
ном порядке, причем производные тропилия и пири-
дина вступают в эти реакции значительно легче, чем
бензол. Т. обр., для ароматич. соединений характерна
совокупность ряда свойств и ни одно из химич. или
физич. свойств в отдельности не может служить само
по себе критерием ароматичности.
Природа ароматичности и особенности строения А. с. могут
быть поняты на основании квантово-механич. рассмотрения
их по методу молекулярных орбит. С помош.ыо этого метода
было предсказано существование нек-рых из небензоидных
А. с., в частности тропилия (Э. Хюкке.ть, 1931). В методе моле-
кулярных орбит л-электроны в А. с. рассматриваются как
общие для всех углеродных атомов системы и находящиеся
на общих молекулярных орбитах. Молекулярные орбиты могут
быть связывающие (нахождение на них электронов снижает
энергию системы но сравнению с электронами на атомных орби-
тах), несвязывающие (энергия перехода электронов с атомных
на молекулярные орбиты равна нулю) и разрыхляющие (элект-
роны на них могут размещаться лишь с затратой энергии).
На каждой орбите может размещаться не более 2 электронов.
Характерной особенностью ароматич. систем является замкну-
тость электронной оболочки, т. е. нахождение всех л-электро-
нов только на связывающих молекулярных орбитах, причем
на связывающих орбитах нет вакантных мест (т. к. па каждой
из них имеется 2 л-электрона). Так, в бензоле имеется 3 связы-
вающих орбиты и 3 разрыхляющих; в основном состоянии
бл-электронов бензола располагаются на 3 связывающих
орбитах. За счет этого достигается значительное снижение
энергии системы. Точно также в катионе тропилия С7Н+ и
анионе циклопентадиенила СвНл имеется по 3 связывающих
орбиты, на к-рых располагается по бл-злектронов. Исходя
из такого рода общих соображений, Э. Хюккель сформулиро-
пиррол, фуран, тиофен, пиридин);
XXVI
XXV
XXVII
XXVIII
вал правило, согласно к-рому моноциклич. сопряженные
полиолефины обладают замкнутой электронной оболочкой и,
следовательно, ароматич. стабильностью при числе л-элект-
ронов, равном 4п + 2, где п — любое целое число. При п = 0
это составляет 2л-электрона [система циклопропенилия
(XIV) С3Н,4]. при п = 1 — бл-электронов (бензол, тропилий,
циклопентадиенил-аниои, ----- ‘----- ---J‘— --------'•
при п = 2 — 10л-электро-
нов(нафталин, азулен) и
т. д. В соответствии с этим
правилом системы цикло-
бутадиена (XXV) (4 л-эле-
ктрона), циклооктатетр-
аена (XXVI) и пенталина
(XXVII) (8 л-электронов)
и гепталина (XXVI11)
(12 л-электронов) не яв-
ляются А. с.
В неорганич. А. с.
типа фосфонитрилгало-
генидов действуют иные
закономерности, так как
здесь образование А. с.
происходит как за счет рл-электронов (атомов N), так и за счет
йл-электронов (атомов Р). Такого рода рл — dn системы обла-
дают ароматичностью при числе л-электронов 6, 8, 10, 12 и т. д.
Лит.: Перспективы развития органической химии. Под
ред. А. Тодда, пер. с англ, и нем., М., 1959; Вольпин М . Е.,
Усп. хим., 1960, 29, вып. 3, с. 298; Cl аг Е., Aromatische
Kohlenwasserstoffe. Polycyclische Systeme, 2 Aufl., В.—
Gottingen — Hdlb., 1952; Organic Chemistry. Ed. H. Gilman
[a. o.], v. 1, N. Y., 1943, p. 117—213; Baker W., McOmie
J. F. W., в кн.: Progress in organic chemistry. Ed. J. W. Cook,
v. 3, 1955, p. 44; В a d g e г G. M., The structures and reactions
of the aromatic compounds, Camb., 1954; В aker W. [a. o.],
Non-benzenoid aromatic compounds, ed. D. Ginsburg, N. Y.,
1959. M. Е. Вольпин.
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ — класс
углеводородов, содержащих в своем составе бен-
зольные ядра, к-рые могут быть конденсиро-
ванными и имЛь насыщенные или ненасыщенные боко-
вые цепи. К наиболее важным А. у. относятся бензол
С6Н6 и его гомологи общей формулы Сп112„_(;, напр.
толуол (I), п-ксилол (II), изопропилбензол (III), а также
углеводороды бензольного ряда с ненасыщенными
боковыми цепями, напр. стирол (IV), фенилацетилен
(V) и др. В соединениях, содержащих два или неск.
бензольных ядер, циклы могут быть связаны между
собой по-разному. Так, в дифенилметане С6Н5СН2С6Н5
и в трифенилметане и его производных бензольные
кольца непосредственно между собой не связаны;
соединения типа дифенила С6Н5 — С6Н5 содержат два
непосредственно связанных бензольных остатка.
К представителям А. у. с конденсированными бен-
зольными ядрами относятся нафталин (VI), антра-
цен (VII), фенантрен (VIII) и др. Известны также
А. у. со сравнительно большим числом конденсирован-
291
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ - АРСЕНАЗО
292
пых бензольных ядер, напр. гексацен (IX), 1,2-бенз-
пирен (X), коронен (XI) и др. Существуют многочис-
ленные соединения, у к-рых бензольные ядра конден-
сированы с неароматич. циклами, напр. тетрагидро-
нафталин — тетралин (XII), аценафтен (XIII) и др.
В последнее время стали известны нек-рые углево-
дороды, напр. азулены, не содержащие бензольных
ядер и тем не менее обладающие ароматич. характером
(см. Ароматические' системы).
Число возможных изомеров в случае бензола с боко-
выми углеводородными цепями равно: при 1 атоме С
вне кольца — 1 изомер (толуол), при 2 атомах С в бо-
ковых цепях — 4 изомера (напр., этилбензол, о,-м- и
п-ксилолы); в случае углеводородов бензольного ряда
состава С9Н16 — 8 изомеров и т. д. В случае нафталина
(VI), содержащего боковые цепи, число возможных
изомеров намного больше, чем у бензола. Так, известны
два монометилнафталина (а- и p-изомер, или 1- и
2-метилнафталин), так как в нафталине положения
1,4,5,8 равноценны положениям 2,3,6,7; при 2 ато-
мах С вне циклов в нафталине число возможных
изомеров равно 12 и далее очень быстро возрастает
(при 3 атомах С 32, при 4 атомах С 110).
Основным источником получения А. у. служат
продукты коксования каменного угля. Из 1 т камен-
ноугольной смолы, в состав к-рой входит по крайней
мере 300 в-в, можно в среднем выделить след, коли-
чества А. у. (в кг): 3,5 бензола, 1,5 толуола, 0,7 смеси
ксилолов; 2 нафталина; 0,15 антрацена. Большое
значение приобрели методы получения А. у. из угле-
водородов нефти (алканов и нафтенов) путем кре-
кинга, пиролиза, каталитич. дегидрирования или
дегидроциклизации (см. Ароматизация нефтепродук-
тов). Для получения нек-рых А. у. имеют практич.
значение и синтетич. методы. Так, действием этилена
па бензол в присутствии А1С13 образуется атилбенэол,
дегидрирование к-рого при 450—500° приводит к сти-
ролу (IV). При алкилировании бензола пропиленом
в присутствии А1С13 образуется изопропилбензол
(кумол) (III), из к-рого окислением получают фенол
и ацетон. Алкилировать А. у. можно также взаимо-
действием их с галогеналкилами по Фриделя —
Крафтса реакции. Вещества типа дифенила можно
синтезировать взаимодействием арилгалогенидов с на-
трием по Фиттига реакции или с медью по Ульмана
реакции. Кроме того, известны многие синтетич.
способы получения А. у. из их производных, напр.
восстановление ацетофенона в этилбензол, превра-
щение диазосоединений в А. у. кипячением со
спиртом и т. д.
А. у. обычно являются исходными продуктами для
получения многообразных ароматич. соединений,
содержащих различные заместители и имеющих боль-
шое практич. значение. Для ароматич. соединенвй
очень характерны реакции электрофильного замеще-
ния; в соответствии с этим А. у. можно непосредственно
превратить в их галогенозамещенные, нитросоедине-
ния, арилсульфокислоты. Далее реакцией А. у. с про-
изводными кислот по реакции Фриделя — Крафтса
можно синтезировать ароматич. кетоны, напр. аце-
тофенон. Непосредственно из А. у. можно получить
кетоны, альдегиды и кислоты ароматич. ряда. Так,
действием СО и НС1 на А. у. можно ввести в арома-
тич. соединения альдегидную группу (см. Каттер-
мана—Коха реакция), толуол можно окислить в бен-
зальдегид, антрацен (VII) в антрахинон (XIV), при
окислении нафталина образуется фталевая кис-
лота и т. д. Полученные т. о. ароматич. соединения
имеют самостоятельное значение. Часто продукты
первичной переработки А. у. играют большую роль
как исходные в-ва для дальнейших превращений.
Нек-рые полициклнч. А. у., напр. 1,3- и 1,2-бензпи-
рен и др., обладают канцерогенным действием.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; Olar Е.,
Aromatische Kohlenwasserstoffe. Polycyclische Systeme,2 Aut'l.,
В. — Gfittingen — Hdlb., 1952; В a d g e г G. M., The structu-
res and reactions of the aromatic compounds, Camb., 1954:
Бэйкер У., в кн.: Перспективы развития органической
химии, под ред. А. Тодда, пер. с англ, и нем., М., 1959,
с. 31—57. Я. Ф. Комиссаров.
АРОМАТИЧНОСТЬ — см. Ароматические системы.
АРРЕНИУСА УРАВНЕНИЕ — зависимость ско-
рости химич. реакции от темп-ры; установлено С. Ар-
рениусом в 1889. См. Кинетика химическая.
АРСАНИЛОВАЯ КИСЛОТА (n-аминофенил арсоно-
вая кислота) n-H2N—СвН4—AsO(OH)2, мол в.
217,04 — белый кристаллич. порошок; т. пл. 232°;
т. разл. > 280°; растворима в горячей воде, амило-
вом спирте, эфире, в р-рах щелочей, щелочных карбо-
натов и в конц. минеральных к-тах; мало растворима
в холодной воде, этаноле, уксусной к-те; нераство-
рима в ацетоне, бензоле, хлороформе; очень ядовита.
Соль диазония А. к. легко сочетается в содовощелоч-
ном р-ре с 3-нафтолом или R-солью, образуя оранже-
вое азосоединение. Получают А. к. арсенированием
анилина мышьяковой к-той; применяется для опре-
деления Zr (в 0,5 н. солянокислом или сернокислом
р-ре образуется белый осадок); для обнаружения Се
и аммиака; для синтезов.
Лит.: Портнов А. И., Ж. общ. хим., 1948, 18, вып. 4,
с. 594. Д. И. Кузнецов.
АРСЕНАЗО — группа органич. реактивов, полу-
чаемых сочетанием хромотроповой к-ты и различных
замещенных фенил ар соковой к-ты.
Арсеназо I CIfiHI1OI1N2S2AsNa2, мол. в.
592,29—темно-коричневый кристаллический порошок,
легко растворимый в
воде; окраска р-ра в AsO(OH)a ОН ОН
кислой среде красно- N=N_^\^4.
розовая, в щелоч- 1| | | Y |
ной — синевато-розо- NaO3S—SO3Na
вая. Получают арсе-
назо I сочетанием диазотированной о-аминофенил-
арсоновой кислоты с хромотроповой кислотой. Силь-
ные окислители (хлор, бром, гипохлорит, перманганат
и др.) и восстановители (Sn21, TiВ * * * * * * * * * * * * * * * * * * * * * * 31-, Na2S2O4) обес-
293
АРСЕНАТЫ И АРСЕНИТЫ — АРСОНИЕВЫЕ СОЕДИНЕНИЯ
294
цвечивают и разрушают реагент. Арсеназо 1 дает
цветные реакции с многими элементами, начиная
приблизительно с тех значений pH, при к-рых катионы
элементов гидролизуются. При этом возникают фио-
летовые окраски различных оттенков, UVI дает синюю
окраску. Арсеназо I применяется для фотометрии,
определений, цветных реакций, как индикатор при
объемных определениях, как ингредиент органич.
соосадитслей Th, Zr, Hg, UIV, Ti, Та (мияерально-
кислая среда), UV1, Al, редкоземельных элементов,
Be, Ga, In и др. (слабокислая или нейтральная среда).
Избирательность реакции достигается предваритель-
ными отделениями и применением маскирующих комп-
лексообразователей, напр. винной к-ты. Чувствитель-
ность реакции 0,1—1 мкг определяемого элемента.
Арсеназо II C32H20O22N4S4As2Na4, мол. в.
1182,56 — по внешнему виду близок к арсеназо I.
В молекуле А. II соединены две молекулы арсеназо I
в пара-положении к группе AsO(OH)2.
По свойствам арсеназо II близок к арсеназо I, но
комплексы с U, Th и др. прочнее с арсеназо I. Поэтому
повышенная кислотность, сульфаты, фосфаты, фто-
риды влияют слабее, чем при арсеназо I. Окраски
растворов арсеназо II и его комплексов близки к по-
лучаемым с А. 1.
Арссназо III C22H18O14N4S2As2Na2, мол. в.
822,27 — черный кристаллич. порошок, растворимый
в воде; в кислой среде растворы окрашены в темно-
AsO(OH)2
As0,0H^ ОН он
розовый цвет, в щелочной среде — в синий, р-ры ком-
плексов — зеленого цвета. Получают А. III сочета-
нием избытка диазопия о-чминофениларсоновой к-ты
с хромотроповой к-той при добавлении Са(ОН)2.
Арсеназо III образует исключительно прочные ком-
плексы с Th, Zr, Hf, UIV, редкоземельными элемен-
тами и не дает цветных реакций с Al, Ga, Ti, Be.
Определения Th, отличающиеся высокой избиратель-
ностью, возможны в среде 0,1—10 М НС1 в присутст-
вии больших количеств сульфатов, фосфатов, окса-
латов и др. Щавелевая к-та маскирует Zr и др., слабо
влияя на комплекс Th; т. о. можно определять от
0,1 мкг Th. Для определения Zr применяют среду
0,5—10 М или 0,5—6 М НС1О4.
Лит.: Кузнецов В. И., ДАН СССР, 1941, 31, № 9,
с. 895; его же, Ж. аиалит. хим., 1952, 7, вып. 4, с. 226;
его же, там же, 1955, 10, вып. 5, с. 276; его же, там же,
1959 14, вып. 1, с. 7; Кузнецов .В. И., Будано-
ва Л. М. иМатрасова Т. В., Заводок, лаборатория,
1956, 22, № 4, с. 406; Кузнецов В. И. и С а в в и н С. Б.,
Радиохимия. 1959, 1, вып. 5, с. 583; Саввин С. Б., ДАН
СССР, 1959, 127, № 6. В. 11. Кузнецов, В. Г. Типцова.
АРСЕНАТЫ И АРСЕНИТЫ — соли кислот мышь-
яка, соответственно 5- и 3-валентного. Известны арсе-
наты метамышьяковой HAsO3,ортомышьяковой H3AsO4
и пиромышьяковой H4As2O, к-т. Из этих кислот
в свободном состоянии получена только H3AsO4.
По химич. свойствам арсенаты похожи на соответст-
вующие фосфаты. Наибольшее практич значение
имеют ортоарсенаты, часто называемые просто арсе-
натами. Из них в воде растворимы все первичные
арсенаты общей формулы MexH2AsO4; растворимые
вторичные и третичные арсенаты образуют только
щелочные металлы и аммоний. Ортоарсенаты щелоч-
ных металлов могут быть получены растворением Ая2О5
в едких или углекислых щелочах. Пиро- и метаарсе-
наты — нагреванием двух- и однозамещенных орто-
арсенатов: 2Na2HAs04->Na4As207+H20; NaH2AsO4->-
—> AaAsO3 + Н2О. Арсениты — соли метамышьяко-
вистой HAsO2, ортомышьяковистой HgAsOg и пиро-’
мышьяковистой H4As2O5 кислот, неизвестных в сво-
бодном состоянии. В воде растворимы только арсе-
ниты щелочных металлов и аммония. Арсенаты полу-
чают аналогично арсенитам, например по реакции
As2O3 + Na2CO3 —> 2NaAsO2 + СО2. Все А. и а. очень
ядовиты. Находят применение в качестве инсектици-
дов. Наиболее широко используется кальция арсе-
нат, кальция арсенит и натрия арсенат. См. также
Мышьяк.
Лит.: II о п о в II. В., Справочник по ядохимикатам, М.,
1956. См. также лит. по статье Мышьяк.
АРСЕНИДЫ МЕТАЛЛОВ — соединения мышьяка
с металлами. Наиболее характерны А. м. типа AsMe3
(KgAs, Na3As, Cu3As2, Ca3As2, Mg3As2), а также типа
гидридов (ZnAs2, CaAs2) и др. He описаны арсениды бо-
ра, галлия и индия. А. м.—твердые, как правило, вы-
сокоплавкие продукты, большой плотности, нераство-
римые в органич. растворителях Арсениды щелочных
металлов легко гидролизуются водой, а щелочнозе-
мельных металлов водой разлагаются медленно, но
легко — разбавленными к-тами; арсениды остальных
металлов разлагаются только к-тами. Наиболее устой-
чивы А. м. VIII гр. периодич. системы, к-рые входят
в состав ряда минералов; леллингит (FeAs2). арсено-
пирит (FeAsS или FeAs2 • FeS2) и др. Леллингит и
арсенопирит являются исходным сырьем для полу-
чения мышьяка и его соединений. При действии оки-
слителей А. м. превращаются в соли мышьяковистой
и мышьяковой к-т. При гидрометаллургия, переработ-
ке нек-рых руд, содержащих А. м., выделяется весьма
ядовитый мышьяковистый водород. Р Н. Стерлин.
АРСИНЫ — органич. производные AsH3; в зави-
симости от количества органич. радикалов, связанных
с атомом As, различают первичные RAsH2, вторич-
ные R2AsH и третичные R3As. А. — бесцветные жид-
кости или кристаллы с неприятным запахом, хорошо
растворимые в органич. растворителях и плохо в воде.
Формула T. кип., °C Плотность
CH3AsH3 + 2
(СЩ)ч AsH 35,6 1,213/29"
CoH5AsHj 36 1,217/22°
(C3H5).,AsH 90 —
(CoHgjg As 140 1,151/16,7°
CeH5AsH3 148 —
(CcH5)» AsH 174/25 —
(CeH5)3 As (т. пл. 5 7°) — 1,306/64°
В отличие от азотистых аналогов — аминов, основ-
ные свойства А. выражены слабо; они усиливаются
при переходе от первичных к третичным; ароматич. А.
основными свойствами не обладают. А. очень легко
окисляются (многие даже загораются на воздухе),
образуя при этом соответствующие окиси или к-ты;
легко взаимодействуют с галогенами с образованием
галогенарсинов; с солями Ag, Au, Hg, Pt и др. метал-
лов А. образуют двойные соединения Первичные и
вторичные А. обычно получают восстановлением
соответств. галогенарсинов или кислородсодержащих
производных водородом в момент его выделения.
Основной метод получения первичных и вторичных
А. — алкилирование арсенидов калия или натрия
в жидком аммиаке, а третичных — взаимодействие
AsClg с металлорганич. соединениями: AsCl3 +
+ 3RMgCl—> R3As + 3MgCl2- А. жирного ряда ядо-
виты; наибольшей токсичностью обладают первич-
ные А., напоминающие по характеру действия AsH3.
АРСОНИЕВЫЕ СОЕДИНЕНИЯ — солеобразные
в-ва, содержащие положительно заряженный атом
мышьяка, связанный 4 ковалентными связями с ор-
ганич. радикалами и пятой ионной связью с анионом.
295
АСБЕСТ - АСИММЕТРИЧЕСКИЙ АТОМ УГЛЕРОДА
296
Примерами А. с. могут служить иодистый тетраметил-
арсоний [(СН3)4Ав]*Г", хлористый тетрафениларсо-
ний !(C6H5)4As] 'C1 . А. с. образуются при присоеди-
нении алкил- или арилгалогенидов к третичным арси-
нам. А. с., как и аммониевые, сульфониевые и другие
соединения этого типа, объединяются общим назва-
нием ониевых соединений. См. также Мышъякорганиче-
ские соединения. Н. ф. Комиссаров.
АСБЕСТ — группа минералов, способных расщеп-
ляться на тонкие и прочные волокна. По химич. со-
ставу и физич свойствам различают две подгруппы:
амфибол-А. и хризотил-А. Наибольшее значение
имеют хризотил-А. и крокидолит-А. (одна из разно-
видностей амфибола-А.). X р и з о т и л-А. — водный
силикат магния Mg6(Si40u). (ОН)6 • Н2О — относится
к весьма редкому в природе типу трубчатых кристал-
лов, стенки к-рых состоят обычно из 9 двухслойных
пачек, состоящих из кремний-кислородного и магний-
кислородного слоев. Цвет-хризотила-А. — зеленовато-
желтый до белого, твердость по Моосу 2—3, огне-
стоек, щелочеупорен, плохой проводник электриче-
ства, тепла и звука, сравнительно легко растворим
в к-тах. Крокидоли т-А. — метасиликат железа
состава 2Na2O • 6 (Fe, Mg)O-2Fe2O3-17 SiO2- 3H2O,
окрашен в темно-синий цвет, волокна тонкие и эла-
стичные (однако в сравнении с хризотилом-А. более
грубы), легко подвергаются скручиванию, по отноше-
нию к кислотам более стоек, чем хризотил-А.: хорошо
противостоит действию морской воды. Нек-рые срав-
нительные данные состава и свойств хризотилового
и крокидолитового А. приводятся в табл.:
Состав и свойства Хризоти- ловый А. Крокидо- литовый А.
SiO„ % ' . 42,0 51,0
А12о3, % 0,5—1,3 —
Ее20а, % 1-4 20,0
FeO, % 0,5—2 18,0
MgO, % 40-43 2,0
Na2O, % следы 6,11
Н2О, % 12-13,5 3,0
Плотность, 0/сЛ13 2,3-2,6 3,2-3,3
Средняя толщина волокна, мн .... 0,026 0,9-1.8
Предел прочности при разрыве в не- деформированпом состоянии, кг/мм'2 240-317 3.31
То же, в деформированном состоянии, кг/мм'2 - . 160-210 186
Т. пл., °C 1500 1150
Показатели преломления по различ- ным осям: АГр 1,522 1,698
N-m 1,512 1,699
Ng 1,508 1,706
Тсмп-ра начала потери воды, °C . . . 550-570 320-380
Темп-pa обезвоживания, °C 800 540—600
Маложелезистый хризотил А. хороший изолятор,
однако его электроизоляц. свойства, как правило,
не определяются, поскольку А. для изготовления
изделий смешивается со связующими веществами.
Ориентировочное сопротивление порядка 1-Ю6 —
8-107 ом см. Основные крупные месторождения А.:
в Советском Союзе — на Урале, в Канаде — Квебек,
в Австралии, Южно-Африканском Союзе, Южной
Родезии. Из волокон А. длиной более 8 мм (текстиль-
ный А.) изготовляют фильтры, брезенты, защитные
костюмы и пр.; волокно от 2 до 8 мм. применяется
для изготовления асбоцементных изделий, шифера,
труб, спец, картона, бумаги, тепло- и электроизоля-
ционных покрытий; волокно 0,2—2 мм входит в состав
теплоизоляц. смесей и асбоцементных строит, мате-
риалов.
Лит.: Соколов П. Н. и Шнейдер В. Е., Ас-
бест, 2 изд., М., 1959 (Требования промышленности в ка-
честву минерального сырья. Справочник для геологов. Вып. 5).
В. П. Петров, Г. Г. Воробьев.
АСИДОЛ — маслянистая темно-коричневая жид-
кость, нерастворимая в воде; смесь свободных нафте-
новых кислот. А. получают при разложении серной
к-той щелочных отходов (натриевых солей нефтяных
к-т) от выщелачивания и щелочной очистки нефтей и их
дистиллятов (керосиновых, соляровых, масляных).
При неполном разложении получают мазеобразный
продукт а с и д о л-м ы л о н а ф т, содержащий не
менее 30% неразложившегося мыла. А. выпускается
двух сортов: А.-1 и А.-2, отличающихся по содержа-
нию от минерального масла (57% и, соответственно,
45%) и кислотными числами (не более 175 и 210). Для
А. нормируются также содержание воды, минераль-
ных солей и хлоридов. А. используются в качестве
шпалопропиточного материала, растворителей смол,
для антисептики, в качестве сиккативов (ускорителей
высыхания лака), деэмульгаторов, пластификаторов
и т. д. А.-мылонафт выпускается трех сортов, отли-
чающихся кислотными числами и содержанием мине-
рального масла. Применяют А. гл. обр. в качестве
заменителя жиров в мыловарении, а также в текстиль-
ной, кожевенной и др. отраслях пром-сти.
Лит.: Технические нормы на нефтепродукты (Справочная
книга), под ред. Н. Г. Пучкова, 16 изд., М., 1957.
Л. С. Поваров.
АСИММЕТРИЧЕСКАЯ МОЛЕКУЛА — молекула,
не имеющая плоскости симметрии; отсутствие пло-
скости симметрии может быть обусловлено либо нали-
чием в молекуле элемента асимметрии, напр. асим-
метрического атома углерода, либо асимметрией моле-
кулы в целом. Последний случай характерен для моле-
кул, связи к-рых не имеют сво-
бодного вращения. Например, NO2
в динитродифеновой к-те рас- /—L \
стояния между орто-замести- С 5—\ У~МО2
телями таковы, что свободное '"==/ \=/
вращение вокруг связи, соеди- нООС СООН
няющей два бензольных ядра,
становится невозможным, а сами бензольные ядра
не могут разместиться в одной плоскости. В такой мо-
лекуле плоскость симметрии отсутствует. Молекула
динитродифеновой к-ты асимметрична в целом. Ана-
логичное положение наблюдается в случае нек-рых
алленовых и спирановых производных. Если соеди-
нение может быть разделено на оптич. антиподы, то
это является безусловным и надежным указанием на
то, что молекулы этого соединения являются асиммет-
рическими. Однако невозможность расщепления ве-
щества на антиподы не может служить доводом об
отсутствии асимметрия, строения (см. Антиподы
оптические, Асимметрический атом углерода).
АСИММЕТРИЧЕСКИЙ АТОМ УГЛЕРОДА — на-
сыщенный или отрицательно заряженный атом угле-
рода, через к-рый нельзя провести плоскость сим-
метрии. Этому условию удовлетворяет для насыщен-
ного атома углерода тот случай, когда все четыре
заместителя, с к-рыми связан этот атом углерода, раз-
личны. Для отрицательно заряженного атома углероде
(карбаниона) необходимо, чтобы все три заместителе
были различны. Напр., в молекуле этилового спирта (I
атом С не является асимметрпч., т. к. в нем есть пло
скость симметрии, проходящая между атомами К
через СН3- и ОН-группы. Если заместить в этиловох
спирте атом водорода, стоящий у вторичного атома С
то этот атом С станет асимметрич. В а-дейтеро
этиловом спирте или а-фенилэтиловом спирте (III
плоскость симметрии отсутствует. А. а. у. отмечаете)
звездочкой, напр. CgHjCH1 (ОН) СН3; он может обу
словливать явление оптич. активности. Атомы угле
рода в состоянии sp- и яр2-гибридизации, связанны
с заместителями кратными связями или несущие
как это имеет место в ионе карбония, положительны.
297
АСИММЕТРИЧЕСКИЙ СИНТЕЗ — АСКАРИДОЛ
298
заряд, обладают плоскостью симметрии и потому не
могут являться асимметрия. Следует отметить, что
вопрос о возможности существования неплоских, а
ОН он ОН
.-•С--.. .-С-.. ..-с...
Н-’ у ••Н н-’ ] Н-- у '•••С6Н5
СН3 СН3 СН3
I II III
следовательно, асимметрия, ионов карбония и сво-
бодных радикалов еще не решен окончательно. См.
также Асимметрическая молекула, Антиподы оптиче-
ские, Диастереоизомеры, Активность оптическая.
АСИММЕТРИЧЕСКИЙ СИНТЕЗ — процесс, при
к-ром образуется оптически активное соединение из
вещества до реакции оптически недеятельного и не
обладавшего центром дисимметрии. А. с. может про-
текать только под действием другого оптически актив-
ного соединения или к.-л дисимметрия, агента. В за-
висимости от природы этих агентов различают частич-
ный (парциальный) и абсолютный А. с. В первом слу-
чае агентами дисимметрии являются другие оптически
активные в-ва, синтетические или природного проис-
хождения (ферментативный А. с.). Во втором случае
используются физич. агенты: циркулярно поляризо-
ванный свет, элементарные частицы или дисиммет-
рия. решетка кристаллов, напр. кварца (дисиммет-
рия. катализаторы).
Впервые А. с. осуществил Марквальд (1904) на примере
образования оптически активной метилуксусной к-ты путем
разложения кислой соли метилэтилмалоновой к-ты и бру-
цина:
Н3С соон с1*3 ?
х —> X
H5C2Z COO • бруцин Н5С2 СООН
Маккензи осуществил А. с., исходя из эфиров кетокислот
с оптически активными спиртами, действуя на них магнпйор-
ганич. соединениями или восстанавливая водородом, напр.:
- Н —
I
СН3—С—СОО—борнил (А)
I
Н» Z 011
СН3СОСОО — борнил --- —>
I
СН3—С—СОО —борнил (В)
I
н
Н3С Н
/с\
нет хсоон
В результате асимметрирующего воздействия
оптически активного спирта образуется смесь не-
равных количеств диастереоизомеров А и В. По-
сле удаления спирта продукт реакции обладает
оптической активностью за счет возникшего но-
вого центра дисимметрии.
К частичному А. с. относятся: А. с. Марк-
вальда и Маккензи, гидрирование, бро-
мирование или гидрооксилирование двой-
ных связей С=С, С=0, CN и др., фермента-
тивный асимметрия, катализ, асимметрия,
катализ под действием дисимметрических
микрогетерогенных и гетерогенных ката-
лизаторов, воспроизводящих действие ферментов.
Абсолютный А. с. занимает особое место в связи
0Н FeSO4
ОН
с вопросом о происхождении первичных оптически
активных соединений при возникновении жизни на
Земле. Абсолютный А. с. был осуществлен (1933)
присоединением хлора к свободному радикалу три-
арилметилу при одновременном облучении циркуляр-
но поляризованным светом:
СвН5\ СвН5\
С„Н5—СвГЦ^о . —|— С1о —* С3Н3—CgHi-^C—CI
а-С10Н7/ «-CtoHj/
Широко распространенная в растительном мире пра-
вовращающая винная к-та была получена гидрокси-
лированием фумаровой к-ты под действием циркуляр-
но поляризованного света: НООС—СН=СН—СООН ->
—> НООС—СН (ОН)—СН(ОН)—СООН. Асимметрия,
адсорбция антиподов применяется для разделения
рацематов и для проведения асимметрия, ката-
лиза. Многие абсолютные асимметрия, синтезы были
проведены на катализаторах на основе дисимметрия,
кристаллов (оптически активного кварца), напри-
мер гидрогенизацией а-фенилкоричной кислоты на
никеле, нанесенном на оптически активный кварц,
была получена оптически активная а,₽-дифенил-
пропионовая кислота: С6Н5СН=С(С6Н5) СООН —>
—> СвН5СН2СН(С6Н5)СООН. В изученных до сих пор
случаях абсолютного А. с. выход оптически чистого
продукта редко превышает несколько процентов.
В частичном А. с. выход оптически чистого продукта
может превышать 90%, в случае ферментативного
катализа образуется почти исключительно один оптич.
изомер.
Помимо известного препаративного значения, А с.
применяется для установления с помощью конформа-
ционного анализа абс. конфигурации оптически актив-
ных соединений.
Лит.: Клабуновский Е. И., Асимметрический
синтез, М., 1960. Е. И. Клабуноескии.
АСКАРИДОЛ (Д2-п-ментен-1,4-диокись) С10Н1вО,
мол. в. 168,24 — густое масло с удушливым запахом;
т. пл. 2,5°; т. кип. 115°/15 мм, 96—97°/8 мм, .
83°/5.и.и, dfg0,9985; п% 1,4769; [а]^>=—0,5°. V
При нагревании до 130—135° А. самопро-
извольно разлагается со значительным по- I
вышением температуры; при действии соля- L 9.А
ной, серной, азотной, фосфорной к-т раз-
лагается со взрывом; бурно реагирует с
конц. муравьиной кислотой, образуя n-цимол; восста-
новление цинковой пылью в уксусной кислоте так-
же приводит к n-цимолу; при окислении перманга-
натом дает смесь кислот, в том числе муравьиную,
уксусную, изомасляную; в среде СС14 на холоду
присоединяет 2 атома брома; при нагревании в
ксилоле до 150° образует ангидрид аскаридола —
2,3-диоксидо-п-ментан (II), который при гидрата-
ции серной к-той переходит в аскаридолгликоль
(III) (последний получается также при восстановле-
нии А. с помощью FeSO4) и эритрол — 1,2,3,4-тетра-
окси-п-ментан (VI): гидрирование в присутствии
палладиевого катализатора приводит к п-ментандио-
лу-1,4 (V) с т. пл. 116—117°, при частичном гидри-
299
АСКАРИТ — АСПАРАГИН
300
ровании образуется п-ментен-диол-1,4 (IV). Полу-
чается А. выделением из хеноподиевого (цитвар-
ного) масла (промышленный метод). Количественно
А. определяется в хеноподиевом масле при встряхи-
вании с 60%-ной муравьиной к-той по разности объ-
емов масла; с помощью разгонки; восстановлением
с помощью TiCl3; колориметрически в присутствии
соляной к-ты, к-рая вызывает коричневую окраску.
А. содержится в эфирном масле Chenopodium ambro-
sioides var. anthelminticum. А. является единствен-
ным перекисного типа веществом, встречающимся в
природе. Применяется в качестве средства против
глистов.
Лит.: Chemistry of carbon compounds. Ed. by E. H. Rodd,
v. 2, Amsterdam [a. o.], 1953, p. 533. Л. С. Поваров.
АСКАРИТ — волокнистый асбест, пропитанный рас-
плавленным NaOH, серая или зеленовато-серая масса.
А. поглощает до 20% СО2 по отношению к своему весу;
его хранят в плотно закрытых банках с залитыми пара-
фином пробками. Применяют А. для поглощения СО2
в аналитич. практике и в пром-сти. л. п. Ефимов.
L-АСКОРБИНОВАЯ КИСЛОТА (I) (витамин С,
у-лактон 2,3-дегидро-Ь-гулоновой кислоты), мол. в.
176,13 — бесцветные кристаллы; т. пл. 190—192°
(с разл.). L-А. к. хорошо
О=С------- растворима в воде, хуже
С=О в спирте, мало растворима
। о в глицерине и в ацетоне;
СО | нерастворима в ароматич. и
н'с I алифатич.. углеводородах,
I в галогенуглеводородах и
НОСН в эфире; [а]р=+23° (Н2О)
СН=ОН и +48° (метанол). L-А. к.
получают синтетически из
I), при гидрировании к-рой образуется
D-сорбит (II); окисляя последний, получают L-cop-
бозу (Ш), а при ее ацетонировании—диацетон-L-cop-
бозу (IV). При окислении этого продукта обра-
зуется диацетон-2-кето-Ь-гулоновая к-та (V), еноли-
зацией и лактонизацией (проводится одновременно)
к-рой получают L-аскорбиновую к-ту (VI).
сон I
II О =
СОН |
НС----’
носн
сн.он
D-глюкозы (
С(СН.;)..
\7'ъ
oZ
|/СН.,ОН
L - сорбозо
О О IV
ХС(СН3)2
Мсн3)2
Диацетон - 2 - пето -
I - гупоновая кислота
Диацетон - L - сорбоза
Выделение L-А. к. в виде ее концентратов из рас-
тит. сырья практикуется в меньшем масштабе. Кислот-
ные свойства L-А. к. характеризуются след, показа-
телями: pKai 4,17, pKas 11,57. При действии сильных
щелочей у-лактонное кольцо L-А. к. гидролитически
размыкается с образованием соли кетокислоты. Со
слабыми щелочами L-А. к. легко образует нейтраль-
ные еноляты без размыкания лактонного кольца;
с высшими жирными к-тами (напр., с пальмитиновой)
L-А. к. дает сложные эфиры (применяемые в каче-
стве жирорастворимых антиоксидантов).
Для L-А. к. характерна способность к обратимым
окислительно-восстановительным превращениям с об-
разованием дегидроаскорбиновой к-ты (II). В этой
форме L-А. к. частично находится в организме;
в р-рах легко окисляется в присутствии воздуха,
особенно при каталитич. влиянии меди и серебра;
на холоду восстанавливает Фелинга реактив, нейтраль-
ный р-р перманганата калия и др.; при нагревании
с соляной к-той образует фурфурол. Способность
к окислительно-восстановительным превращениям,
сопровождающаяся перенесением атомов водорода
к акцепторам, является важнейшей каталитич. функ-
цией L-А. к. как витамина в живом организме.
L-А. к. способствует образованию дезоксирибонуклеи-
новой к-ты (см. Нуклеиновые кислоты}. Организм
человека и животных не способен сам синтезировать
А. к.; потребность в ней удовлетворяется витамином
С, вводимым с пищей. При авитаминозе у человека,
обезьян и морских свинок развивается цинга (скор-
бут). Для колич. определения L-А. к. применяют
иодный или йодатный метод титрования.
Лит.: Б е.р езовский В. М., Химия витаминов, М.,
1959; Шнайдман Л. О., Производство витаминов, М.,
1958.. В. М. Березовский.
АСКОРБИНОМЕТРИЯ — титриметрический метод
колич. анализа, основанный на использовании в ка-
честве восстановителя р-ра аскорбиновой к-ты,
стабилизированного добавлением небольших коли-
честв комплексона III и муравьиной к-ты для предот-
вращения бактериология, разложения. Нормальный
окислительно-восстановительный потенциал аскорби-
новой к-ты (+0,40 в) сильно зависит от pH раствора
(0,281 в при pH 2,04, 0,106 в при pH 5,75). Сильные
окислители, напр. бихромат или перманганат, окис-
ляют аскорбиновую к-ту до щавелевой и треоновой
к-т. Менее энергичные окислители (иод 2,6-дихлор-
фенолиндофенол и др.) окисляют ее обратимо до де-
гидроаскорбиновой к-ты. Аскорбинометрически мож-
V IV
но определить Fe3+, С1С%, JO^, ВгОу, J2, V , Се ,
кислород, растворенный в воде. В качестве индика-
тора применяют вариамин синий (4-амино-4-метокси-
дифениламин). Точку эквивалентности можно уста-
новить также по диффузионному току окисления ас-
корбиновой к-ты на платиновом вращающемся элект-
роде. Титр рабочего р-ра устанавливают иодометри-
чески. Преимуществом аскорбиновой к-ты является
возможность проводить титрование ее раствором
в открытом сосуде.
Лит.: Эрдеи Д. -Л., Ж. аналит. хим., 1953, 8, № 6.
В. Г, Тинцова.
АСПАРАГИН C4H8O3N2, мол. в. 132,12 — моно-
амид аспарагиновой к-ты; в зависимости от положе-
ния NH2-rpynni>i по отношению к амидной группе
различают а-А. HOOCCH2CH(NH2)CONH2 и 0-А.
HOOCCH(NH2)CH2CONH2. А. — бесцветные крис-
таллы; растворим в воде и нерастворим в этиловом
спирте и эфире; т. пл.: D,L-a-A. 213° (с разл.); D
и L-0-A. 234,5° (с разл.). а-А. получают синтетически,
P-А. широко распространен в животных и раститель-
ных тканях (в проростках вики, напр. до 28% су-
хого веса), входит в состав белков и полипептидов,
преим. в виде L-формы. Для L-P-A. [a]D=+ 5,41°
(в воде). L-p-А. получают из L-аспарагиновой кис-
лоты. А.—амфотерное соединение, дает соли с кис-
лотами и щелочами, при нагревании с которыми
его амидная группа гидролизуется с отщеплением
301
АСПАРАГИНОВАЯ КИСЛОТА — АСТАТИН
302
NH31 что используется для количественного опреде-
ления А. Из гидролизатов белков 0-А. может быть
выделен в виде нерастворимой медной соли. А. дает
биуретовую реакцию. [i-А. в животных организмах
является продуктом связывания аммиака, переносит
его к почкам.
Лит. см. при ст. Аминокислоты.- О. В. Лунина.
АСПАРАГИНОВАЯ КИСЛОТА (аминоянтарная
кислота) HOOCCH2CH(NH2)COOH, мол. в. 133,10 —
дикарбоновая моноаминокислота, известна в виде
следующих оптич. изомеров: L-А. к., т. пл. 270°
(с разл.); [а]р=-|-4,36о (в водных р-рах); D-А. к.,
[а]р=—25,5°; D, L-А, к., т. пл. 280° (с разл.). А. к.
плохо растворима в воде, нерастворима в спирте
и эфире При 25° р/2,77, рХа> 1,88 (—СООН); рКа*
3,65 (—СООН); pAOj 9,60 (NHJ). Методы выделения
и определения А. к. основаны на нерастворимости
ее солей Са и Ва; разработаны также хроматография.,
микробиология, и химия, методы; последние исполь-
зуют превращение А. к. в фумаровую, а затем в янтар-
ную к-ты. А. к. получают из аспарагина кислотным
гидролизом или выделяют из белковых гидролизатов.
Синтетически А. к. получают конденсацией ацетами-
номалонового эфира с эфиром хлоруксусной к-ты и
последующим гидролизом и декарбоксилированием
продуктов конденсации. Природная L-А. к. входит
в состав животных и растит, белков, причем в послед-
них обнаруживается гл. обр. в виде амида (аспара-
гина). А. к. играет важную роль в азотистом обмене
животных и растений (в реакциях переаминирова-
ния, биосинтеза мочевины, пиримидиновых основа-
ний и т. д.). '
Лит. см. при ст. Аминокислоты. В. И. Мазуров.
АСПИРИН (о-ацетилсалициловая кислота), мол. в.
180,16, о-СН3СОО—С6114 — СООН — бесцветные иглы
или пластинки слабокислого вкуса; т. пл. 136,5°;
плохо растворяется в воде и эфире, хорошо — в
водных р-рах щелочей и карбонатов. Водные р-ры
А. имеют кислую реакцию; устойчив в р-рах
кислот. При нагревании со щелочами А. рас-
падается на салициловую и уксусную к-ты, на чем
основано качественное открытие А. (запах уксусной
к-ты и осадок салициловой к-ты при подкислении
щелочного гидролизата серной к-той). Для колич.
определения А. растворяют в спирте и титруют 0,1 н.
р-ром NaOH с фенолфталеином. А. получают взаимо-
действием салициловой к-ты с уксусным ангидридом,
хлористым ацетилом или кетеном. Применяют как
жаропонижающее и болеутоляющее средство, при
невралгиях, мигрени, лихорадочных состояниях, ост-
ром ревматизме, эндо- в миокардитах и др. заболева-
ниях.
Лит. см. при ст. Акрихин. А. И. Травин.
АССОЦИАЦИЯ ИОНОВ — образование из проти-
воположно заряженных ионов (в р-рах электролитов)
особого рода частиц-ассоциатов, в к-рых ионы удер-
живаются преим. за счет электростатич. взаимодей-
ствия их электрич. зарядов в соответствии с законом
Кулона. Простейшими ассоциатами являются ионные
пары, состоящие из двух, и ионные тройники, состоя-
щие из трех ионов. Они представляют собой нейтраль-
ные или электрически заряженные, термодинамиче-
ски устойчивые частицы, находящиеся в равновесии
с простыми ионами, напр.:
Ag+ -f- NOs XZ [Ag+NO з]°; Ba8+ + Cl- TZ [Ва+С1~]+
Ag+ + [Ag+NOa]° ZZ [Ag+NOFAg+]+
NOjT -f- [Ag+NOa г ZZ INO^Ag+NOr]
Процессы А. и. подчиняются закону действующих
масс и др. термодинамич. соотношениям.
Наиболее изучены ионные пары. Их существование
и водных и неводных р-рах солей, щелочей и к-т дока-
зано измерениями электропроводности электродви-
жущих сил, оптич. свойств р-ров электролитов и др.
методами. Известны как процессы образования ион-
ных пар, сопровождающиеся полной или частичной
дегидратацией ионов, так и процессы, не сопровож-
дающиеся изменением гидратных оболочек ионов.
Ионные пары имеют очень высокие дипольные мо-
менты. Степень А. и. сильно зависит от природы раст-
ворителя и электролита, концентрации р-ра, темп-ры;
она возрастает с уменьшением диэлектрич. проницае-
мости растворителя е вследствие увеличения кулонов-
ского притяжения ионов. В воде и в других раствори-
телях с высокой е для многих электролитов валент-
ного типа I—I А. и. очень мала. Она возрастает также
с уменьшением размеров сольватированных ионов,
с ростом концентрации р-ра и имеет наиболее высокие
значения для многовалентных ионов. Из анализа
оптич. свойств р-ров электролитов установлено, что
у нек-рых ионных пар, преим. состоящих из ионов
с электронными оболочками инертных газов, притя-
жение ионов полностью является кулоновским. У мно-
гих ионных пар, не удовлетворяющих этому требова-
нию, анализ оптич. свойств указывает на наличие
дополнительного некулоновского взаимодействия ио-
нов. В растворителях с низкими е при высоких кон-
центрациях р-ра образуются ионные тройники, о чем
свидетельствует аномальный характер зависимости
эквивалентной электропроводности от концентрации
раствора, в частности появление минимумов на кри-
вых электропроводности. В растворителях с очень
низкими е наблюдается образование более крупных
ионных ассоциатов, иногда достигающих размеров
коллоидных частиц и состоящих из сотен и тысяч
ионов.
А. и. оказывает большое влияние на термодинамич.
и кинетич. свойства р-ров электролитов, играет боль-
шую роль в процессах образования комплексных
ионов. А. и. является причиной аномальной электро-
проводности, явлений переноса катионов к аноду при
прохождении тока через растворы нек-рых электро-
литов, стабилизирующего действия нек-рых электро-
литов на процесс разложения перекиси водорода и др.
явлений.
Лит.: X а р н е д Г. и Оуэн Б., Физическая химия
растворов электролитов, пер. с англ., М., 1952; Измай-
лов Н. А., Электрохимия растворов, Харьков, 1959; Сухо-
тин А. М., Вопросы теории растворов электролитов в сре-
дах с низкой диэлектрической проницаемостью. Л., 1959.
Н. Е. Хомутов.
АСТАТИН (Astatine) At — радиоактивный химич.
элемент VII гр. периодич. системы Менделеева, п. н.
85, массовое число наиболее долгоживущего изотопа
210. Стабильных изотопов не имеет. Радиоактивные
изотопы А. получаются гл. обр. при облучении вис-
мута а-частицами. Этим способом Т. Корзон, У. Мак-
кензи и Э. Сегре в 1940 впервые получил А. по реак-
ции Bi208 (а, 2п) At211. Изотоп At211 (7\/г = 7,2 часа),
распадающийся К-захватом (59,1%) и испускающий
а-частицы (40,9%), обычно используют для изучения
химич. поведения А. Наиболее долгоживущий изо-
топ At210 (Т\2 = 8,3 часа) распадается К-захватом
(99%) и испускает а-частицы (0,17%). Короткоживу-
щие изотопы А. являются членами радиоактивных
семейств U238, U235, Th282 и Np237.
А. — вероятно, металл. По свойствам близок к Ро
и J. В весомых количествах не выделен. Первая и
вторая энергии ионизации равны соответственно
9,5 зв и 18,2 зв. По отношению к водородному элект-
роду потенциал осаждения на катоде равен 1,22 ±
± 0,02 в, на аноде 1,45 ± 0,02 в. В вакууме и в обыч-
ных условиях легко возгоняется. При выпаривании
р-ров, содержащих А. и иод, улетучивается неск.
хуже последнего. А. хорошо сорбируется на поверх-
303
АСТРОХИМИЯ - АТАКТИЧЕСКИЕ ПОЛИМЕРЫ
304
ности металлов (золота, серебра, платины). Серебром
захватывается даже при высоких темп-рах (325°),
вероятно, с химич. взаимодействием. Из р-ров эле-
ментарный А. выделяется на осадках металлов (Те,
Ag), на сульфидах тяжелых металлов (Bi, Hg, Ag)
и электролитически. Из 0,25 н. р-ра HNO3 частично
может быть выделен на фольге меди или висмута.
AgJ элементарный А. не захватывает. Известные
валентные состояния А.: —1, +1, +5. Астатид-ион
At получается при восстановлении элементарного А.
сернистой к-той или водородом. Из р-ров At полно-
стью удаляется соосаждением с AgJ, T1J и PdJ2.
Ион AtO* получается при окислении бромом или би-
хромат-ионом и менее эффективно — азотной к-той и
3-валентным железом. Подобно астатид-иону, AtO’
соосаждается с AgJ. Персульфат-ион и гипохлориты
окисляют А. в AtO3, к-рый количественно соосаж-
дается с AgJO3. А. хорошо экстрагируется из 0,01 н.
HNO3 бензолом и четыреххлористым углеродом с ко-
эфф. распределения, соответственно, ок. 200 и ок. 90,
а также диизопропиловым эфиром.
От висмута и получающихся при облучении приме-
сей А. может быть отделен дистилляцией в вакууме
с поглощением на металлич. серебре или охлажденной
платине. При 310° на серебре концентрируется более
85% А. Химич, выделение А. можно осуществить
путем растворения висмутовой мишени в к-те с после-
дующим осаждением Bi в виде фосфата, не захваты-
вающего А. Представляет также интерес экстракция
элементарного А. диизопропиловым эфиром из соля-
нокислого р-ра. Определяется А. по а-излучению
радиометрически или методом фотопластинки.
Лит.: Гольданский В. И., Новые элементы в пе-
риодической системе Д. И. Менделеева, 2 изд., М., 1955,
с 100—106; Яковлев В. А., Усп. хим., 1946, 15, вып. 1,
с. 125; М е 1 1 6 г, Sappl. 2, pt. 1, L. — N. Y. — Toronto, 1956,
p. 1064—79. Б. П. Жагин.
АСТРОХИМИЯ — см. К осмохимия.
АСФАЛЬТ — смесь битума с тонкоизмельченными
минеральными материалами, придающими битуму
повышенную устойчивость при изменении темп-ры.
Битумная часть А. состоит из масел, смол и асфальте-
нов, содержащихся в них в различных соотношениях.
Различают природные и искусственные А. Природ-
ные А. делятся на собственно асфальты, асфальтиты,
асфальтовые породы. Собственно А., или гор-
ные смолы, встречаются в виде скоплений на
поверхности Земли, иногда образующих целые озера
(о. Тринидад, Венесуэла, о. Сахалин), как правило,
содержат до 40—50% минеральных в-в. Асфаль-
титы отличаются значительной твердостью и бле-
ском, почти не содержат минеральных примесей. Они
известны под различными названиями: гельсонит
(Юта и Колорадо), грэемит (3. Виргиния), альбертит
(Куба, Мексика) и др. На поверхности Земли не встре-
чаются, а заполняют ее трещины. Для асфальто-
вых пород характерно преобладание в них мине-
ральных частей — известняков и песчаников (70—
80%); встречаются в разных странах, в частности
в СССР (ок. Сызрани). Полагают, что природные А. об-
разовались из нефти в результате испарения легких
фракций с одновременным обогащением остатка смола-
ми нефтяными и асфальтенами.
Искусственные А. получаются смешением размель-
ченного известняка с битумом; содержание послед-
него в зависимости от назначения колеблется от 13%
до 60%. Природные А. либо вовсе не содержат пара-
фина, либо содержат его крайне незначительное коли-
чество; они содержат также асфалътогеновые кислоты
(полинафтеновые) и масла; в искусственных А. содер-
жание парафина может достигать нескольких процен-
тов; искусственные А. отличаются также отсутствием
асфальтогеновых к-т и значительно большим содер-
жанием масел. Важнейшие области применения А. —
дорожное и строит, дело, а также приготовление кро-
вельного толя, в электротехнике, как изоляционные
материалы, для приготовления специальных зама-
зок, клея и асфальтовых лаков. Почти во всех случаях
могут применяться как природные А., так и подходя-
щие по качеству искусственные А.
Лит.: Крейцер Г. Д., Асфальты, битумы и пеки,
М., 1952; Наметкин С. С., Собр. тр., т. 3, М., 1955.
IT. И. Галич.
АСФАЛЬТЕНЫ — наиболее высокомолекулярные
в-ва из всех выделенных компонентов нефти. Значение
мол. весов, определенных в бензоле, нафталине, кам-
фаре, колеблется в пределах 1600—6000. Легкие нефти
и их мазуты содержат небольшое количество А.,
тяжелые, богатые смолами, — значительные количе-
ства. В зависимости от химич. природы нефти и кон-
центрации А., последние могут находиться в виде
истинных или коллоидных р-ров. По элементарному
составу А. близки к нефтяным смолам; они содержат
80—86% С, содержание О, S, N колеблется в зависи-
мости от природы нефти: 0—9% S, 1—9% О и до 2% N.
Полагают, что А. являются продуктами конденсации
нефтяных смол. А. — темно-бурые или черные аморф-
ные порошки; при нагревании не плавятся, выше
300° разлагаются с образованием газов и трудно сго-
рающего кокса; нерастворимы в спирте, эфире, аце-
тоне, пентане, петролейном эфире; легко растворяются
в бензоле, хлороформе, сероуглероде, чем пользуются
для выделения их из нефти и нефтепродуктов. В про-
мышленных условиях А. выделяют жидким пропаном
(см. Деасфальтизация). Химическая природа А. изу-
чена мало; они являются веществами нейтрального
характера; перманганатом калия окисляются в ки-
слоты; с формалином и IJ2SO4 образуют формолиты;
не реагируют с РС15; не сочетаются с диазосоедине-
ниями; почти не подвергаются омылению и обладают
весьма малыми ацетильными числами; с сулемой, хлор-
ным железом образуют комплексные соединения. При
гидрогенизации деструктивной А. превращаются в по-
лициклич. углеводороды ароматич. и нафтенового
рядов. А. можно рассматривать как полициклич.
ароматич., сильно конденсированную систему, с ко-
роткими алифатич. цепями в качестве заместителей.
Среди циклич. структурных элементов молекулы,
наряду с карбоциклич., по-видимому, присутствуют
также 5- и 6-членные гетероциклич. соединения.
Такие взгляды на строение А. в наст, время стали
господствующими.
Лит.: II а м е т к и н С. С., Собр. тр., т. 3, М., 1955; Сер-
гиенко С. Р., Высокомолекулярные соединения нефти, М.,
1959. II. И. Галич.
АСФАЛЪТОГЕНОВЫЕ КИСЛОТЫ (полинафтено-
вые кислоты) — смолистые вещества, входящие в со-
став высокомолекулярных частей нефти, плотность.;,!;
растворимы в спирте и хлороформе, мало растворимы
в бензине. Na-соли А. к. трудно растворимы в воде;
А. к. содержит ок. 3% серы; кислотное число ок. 90;
число омыления ок. 120; иодное число ок. 20. При про-
должительном нагревании до 120° сначала переходят
в ангидриды, затем в вещества, неспособные омы-
литься. Cu-соли А. к. нерастворимы в бензине (в от-
личие. от нафтеновых к-т).
Лит.: Наметкин С. С., Химия нефти, [3 изд.у И.,
1955, с. 263.
АТАКТИЧЕСКИЕ ПОЛИМЕРЫ — высокомолеку-
лярные соединения, в цепи макромолекул к-рых тре-
тичные атомы углерода с различной пространственной
конфигурацией чередуются нерегулярно. У А. п. бо-
ковые заместители расположены случайным образом
относительно основной полимерной цепи. Вследствие
нерегулярности пространственной структуры А. п.
обычно аморфны, имеют меньшую механическую проч-
ность, большую растворимость и размягчаются при
более низких температурах, чем соответствующие
305
АТМОСФЕРА — ATOM
полимеры стереорегулярные (изотактические и синдио-
тактические), получаемые стереоспецифической поли-
меризацией.
АТМОСФЕРА — внешняя газообразная оболочка
Земли с постоянно убывающей концентрацией газов
до высоты 1100—1400 км. Состав сухого атм. воздуха
у поверхности Земли (в порядке убывающей концен-
трации газов) приведен в таблице:
Газы А. Содержание, ОО. % Газы А. Содержание, Об. %
n2 78,09 Кг 1 • 10“4
о2 20,95 н2 5 Ю-з
Аг 0,93 n2o 5 • 10_5
со2 0,03 Хе 8 • 10“®
Ne 1,8 . 10-8 Оз 1 • 10-е
Не 5,24 • 10-> Rn 6 • 10-18
Количество водяных паров колеблется от 0,1 до
2,8% в зависимости от сезона, климата и погоды.
Химич, элементы, характерные для А., наз. атмо-
фильными (см. Геохимическая классификация элемен-
тов). В области высот от 10 до 100 км под действием
ультрафиолетовых лучей происходит превращение
молекул кислорода в озон. Максимум концентрации
О3 наблюдается на высоте от 20 км (до 0,3 мм
рт. ст. на 1 км столба воздуха при нормальных
условиях). Начиная с высот ок. 40 км увеличивается
содержание атомарного кислорода, выше 120—150 км
кислород практически полностью диссоциирован.
Диссоциация N2 начинается на высоте ок. 200 км-,
полная диссоциация, вероятно, имеет место выше
300 км. Ла высоте от 50 до 150 км возможно образо-
вание молекул NO.
К особенностям, определяющим состав А., отно-
сятся: ее активное влияние на биосферу (см. Биогео-
химия), обменная связь с литосферой, односторонняя
связь с мировым пространством (постоянный уход
газов из А.) и постоянные процессы внутреннего гори-
зонтального и вертикального перемешивания. Важ-
ным источником газов (см. Газы природные) служат
вулканич. извержения, процессы «дыхания Земли»
(микрогазовые выделения), радиоактивный распад
и др. Наиболее легкие газы (напр., гелий) не накап-
ливаются в А. и уходят в мировое пространство.
Воздействие А. на литосферу заключается в процессах
выветривания и окисления горных пород. Живые
организмы, обязанные своим существованием А.,
способствуют изменению ее состава (напр., перера-
батывают СО2 в О2).
Промышленная деятельность человека также ока-
зывает влияние на состав А. В городах выделяется
большое количество СО, СО2, окислов свинца, JI2S,
SO2, различных углеводородов и т. д., 14% общего
количества СО2 воздуха имеет промышленное проис-
хождение. При испытании атомного и термоядерного
оружия образуются аэрозоли, создающие радиоактив-
ный слой на высоте 8—12 км (см. Радиоактивные
аэрозоли).
Состав верхних слоев А. изучают с помощью искус-
ственных спутников Земли, ракет и спец, спектраль-
ных установок. Развитие техники в этом направле-
нии позволяет решать многие важные вопросы атм.
химии, к-рая начинает приобретать самостоятельный
характер.
Лит.: X р г и а н А. X., Физика атмосферы, 2 изд., М.,
1958; Прокофьева И. А., Атмосферный озон, М.—Л.,
1951; Митра С. К., Верхняя атмосфера, пер. с англ., М.^
1955; Красовский В. И., Исследования верхней атмо-
сферы с помощью искусственных спутников и ракет, М., 1958;
Исследование мирового пространства. [Сб. статей], под ре-
дакцией Д. Р. Бейтса, пер. с англ., М., 1959.
АТМОФИЛЬНЫЕ ЭЛЕМЕНТЫ — см. Геохимиче-
ская классификация элементов.
306
АТОМ. Содержание:
Развитие учения об атоме...................... . 306
Общая характеристика строения атома .......... 307
Основы квантовой теории атома ................ 308
Теория атома водорода и водородоподобных ионов . . 311
Теория сложных атомов......................... 315
Объяснение периодичности свойств элементов на
основе теории строения атома ............... 316
Атом — мельчайшая частица элемента химического-,
сохраняющая все его химич. свойства; каждому эле-
менту соответствует определенный вид атомов. Атомы
могут существовать как в свободном состоянии, так
и в соединении с атомами того же элемента или других
элементов, образуя молекулы', все огромное разнооб-
разие химич. соединений обусловлено различными
сочетаниями атомов в молекулах. Физич. и химич.
свойства А., в том числе важнейшая для химии спо-
собность А. вступать в химич. соединение, опреде-
ляются его строением, как системы, состоящей
из движущихся электрич. зарядов и подчиняющейся
законам, характерным для систем микроскопии, раз-
меров.
Развитие учения об атоме. Гипотеза о том, что все веще-
ства состоят из атомов как мельчайших неделимых частиц —
атомистическая гипотеза — возникла еще в
Древней Греции и развивалась философами-материалистами
Левкиппом и особенно Демокритом и Эпикуром (5—3 вв. до
н. э.). Более конкретные представления об А. выработались
лишь значительно позже, в результате развития химии и фи-
зики, базирующегося на научном эксперименте. В 17 в.
Р. Бойль положил атомистику в основу своих химич. предста-
влений и объяснял все химич. изменения соединением и разъ-
единением атомов. Важную роль атомистика играла в работах
И. Ньютона. Дальнейшее развитие атомистика получила в 18 в.
в работах М. В. Ломоносова, сформулировавшего основные
положения атомно-молекулярного учения; Ломоносов разли-
чал два вида мельчайших частиц — «элементы», соответствую-
щие атомам, и «корпускулы», соответствующие молекулам.
В конце 18 в. и в начале 19 в. в результате быстрого развития
химии (работы А. Лавуазье, Ж. Пруста, К. Бертолле и др.)
была создана основа для количественной разработки атоми-
стич. учения. Дж. Дальтон сформулировал кратных отноше-
ний закон (1803) и ввел понятие атомного веса. Ж. Гей-Люсса-
ком был установлен объемных отношений закон (1808), для
объяснения к-рого А. Авогадро в 1811 ввел представление
о молекуле, как состоящей из атомов наименьшей частице
в-ва, способной к самостоятельному существованию; он пред-
положил, что в равных объемах любых газов при одних и тех
же условиях заключается одинаковое число молекул (см. Аео-
гадро закон). Однако взгляды Авогадро лишь постепенно,
к середине 19 в.л завоевали признание химиков. Четкие опре-
деления понятии А. и молекулы были даны С. Канниццаро
в 1858 и приняты в 1860 на съезде химиков в Карлсруэ:
молекула — наименьшая частица вещества, к-рая способна
существовать самостоятельно и не может дробиться дальше
без потери основных химич. свойств данного в-ва, А. — наи-
меньшая частица элемента в молекулах простых и сложных в-в.
Накопление знаний о химич. и физич. свойствах А. и особенно
установление периодич. закона (Д. И. Менделеев, 1869) яви-
лось базой для разработки учения о строении А.
Открытия в конце 19 в. — электрона, явления радиоактив-
ности, рентгеновских лучей и др. — разрушили представление
об А. как неизменной и неделимой частице и позволили по-
строить теорию А. как сложной системы, состоящей из положи-
тельных и отрицательных зарядов. Было установлено, что А.,
являющийся в целом нейтральной системой, содержит элемен-
тарные отрицательные заряды — электроны. В первоначаль-
ной модели А., предложенной Дж. Томсоном (1903), предпола-
галось, что А. состоит из положительного заряда, равномерно
заполняющего объем А., и отрицательных электронов, ко-
леблющихся внутри этого заряда. В 1911 Э. Резерфорд пред-
ложил модель А., состоящего из тяжелого положительно заря-
женного центрального ядра и движущихся вокруг него легких
электронов. Дальнейшее развитие атомной физики подтвер-
дило правильность основных представлений А. Резер-
форда. В 1913 Н. Бор построил первую квантовую теорию А.,
предположив, что электроны в А. движутся вокруг ядра по
законам классич. механики, но лишь по некоторым устой-
чивым орбитам, к-рые находятся по определенным правилам
(согласно т. н. правилам квантования). Тео-
рия Бора содержала ряд правильных общих положений,
однако не была последовательна, т. к. сочетала представления
классич. механики с чуждыми последней дополнительными
правилами квантования. Последовательная теория А. была
построена на основе механики микроскопии, систем — кван-
товой механики, созданной в 1924—28 гг. (работы М. Борна,
В. Гейзенберга, Э. Шредингера, П. Дирака и др.). Квантовая
механика позволяет предвидеть и объяснить с физич. точки
зрения разнообразные свойства А., а в наиболее простых слу-
чаях даже количественно рассчитать их. Для химии особенно
существенно объяснение основных химич. свойств А., к-рые
307
ATOM
308
находят свое истолкование в квантовой химии, в простейших
случаях — количественное, а в более сложных случаях —
качественное; при этом получают объяснение валентность
атомов и их способность образовывать различные типы хими-
ческой связи. Исключительно важной является основанная
на представлениях квантовой теории А. интерпретация пери-
одической системы элементов Менделеева.
Ниже излагаются современные представления о строе-
нии А.
Общая характеристика строения атома. А. состоит
из тяжелого центрального ядра (см. Ядро атомное),
обладающего положительным электрич. зарядом, и
электронов — значительно более легких частиц с от-
рицательным электрич. зарядом; сумма отрицатель-
ных зарядов электронов равна положительному за-
ряду ядра, А. в целом нейтрален. Двигаясь вокруг
ядра, электроны образуют электронные оболочки А.
Линейные размеры А., определяемые линейными
размерами его электронных оболочек, имеют порядок
величины 10 8 см и велики по сравнению с линейными
размерами ядра, порядок величины к-рых 10~12 см;
поэтому объем ядра (~10“8в см3} составляет очень
малую долю общего объема А. (~1024 с.«8). Экспери-
ментальной основой этой модели А. (модели Резер-
форда) явились опыты Резерфорда по рассеянию пото-
ков альфа-частиц. Нек-рые из этих положительно
заряженных частиц при рассеянии их атомами испы-
тывают очень сильные отклонения от первоначаль-
ного направления движения, что объясняется их от-
талкиванием от положительного заряда, сосредото-
ченного в очень малом объеме. Атомы различаются
зарядом и массой ядра.
Заряд ядра А. всегда является целым крат-
ным элементарного положительного электрич. за-
ряда е (равного по абс. величине заряду электрона)
и равен +Ze, где Z — порядковый номер соответствую-
щего элемента в периодич. системе; Z = 1 для ядра
А. водорода (протона), Z = 2 длн ядра А. гелия
(альфа-частицы), Z = 3 для ядра А. лития, и т. д.
Число электронов в нейтральном А. равно Z; ядро
с положительным зарядом +Ze удерживает Z электро-
нов с общим отрицательным зарндом —Ze. Теряя
электроны, нейтральный А. превращается в положи-
тельно ионизованный А. — в положительно заря-
женный ион. При потере к электронов получается
Zt-кратно ионизованный А., обозначаемый символом
элемента с индексом к-[-, напр. Fe^, Бе2+(или Fe*г),
Fe8+, ... для однажды, дважды, трижды, ... ионизо-
ванного А. железа. При максимально возможной
кратности ионизации, равной Z, получается «голое»
ядро А. данного элемента. Нейтральный А. может
не только терять, но и присоединять один или неск.
электронов, превращаясь в отрицательно заряжен-
ный ион, обозначаемый символом элемента с индек-
сом к—, где к — число присоединяемых электронов,
напр. О2 для дважды заряженного отрицательного
иона А. кислорода. Заряд ядра и, следовательно,
значение Z (равное заряду ядра, выраженному в еди-
ницах е) является основной характеристикой А. дан-
ного рода, определяющей принадлежность А. опре-
деленному элементу.
Значение Z можно определить из опытов по рассея-
нию альфа-частиц, на основе теории, разработанной
Резерфордом; его первоначальные опыты (1911) дали
правильный порядок величины заряда ядер различ-
ных атомов; впоследствии Дж. Чедвик (1920) анало-
гичным методом определил для ряда элементов значе-
ния Z с большой точностью. Впервые значения Z
были надежно установлены Г. Мозли в 1913—14 в ре-
зультате экспериментального изучения закономерно-
стей в характеристич. рентгеновских спектрах после-
довательных элементов в периодич. системе (см.
Мозли закон).
Масса ядра А. возрастает примерно пропор-
ционально массовому числу, т. е. общему числу про-
тонов и нейтронов в ядре. Т. к. масса электрона
(0,91 • 40 27 г) значительно (примерно в 1840 раз)
меньше массы протона или нейтрона (1,67 • 10~24 г),
то масса всех электронов в А. в неск. тысяч раз меньше
массы ядра, и масса А. как целого определяется в ос-
новном массой его ядра. Для изотопов определенного
элемента (Z задано, массовые числа различны) атомы
отличаются массой ядра; это практически не сказы-
вается на строении их электронных оболочек и на
их химич. свойствах, к-рые зависят от величины
заряда ядра, т. е. от Z.
Масса А. М пропорциональна его ат. весу А:
М = АМа, где Л/о — 1,66 • 10 24 г — единица атом-
ной массы, за к-рую в химической шкале
атомных масс (весов) принимается 4/1в средней массы
атомов природного кислорода, представляющего смесь
изотопов О16,01’ и О46, а в физической шкале —
4/16 массы А. основного изотопа кислорода О16. В со-
ответствии с увеличением ат. веса, приближенно рав-
ного массовому числу ядра А., от 1 (для А. водорода,
Z = 1) примерно до 250 (для атомов трансурановых
элементов, Z — 100) М изменяется от 1,66 • 10-24 г
до 4 • 10 22 г. Точные значения масс атомов могут
быть найдены методами масс-спектрометрии.
Благодаря малым размерам и большой массе при-
ближенно можно считать ядро А. точечным и непо-
движно расположенным в центре тяжести А. (общий
центр тяжести ядра и электронов близок к месту
нахождения ядра, а скорость движения ядра относи-
тельно центра тяжести А. мала по сравнению со ско-
ростями движения электронов). Т. о., ядро можно
рассматривать как неподвижный центр с зарядом
+ Ze, притягивающий электроны с зарядами —е;
теория простейшего А. — А. водорода сводится к ре-
шению задачи о движении одного электрона с заря-
дом —е вокруг неподвижного центра с зарядом -)-е.
Существенно, что последовательная теория А. прин-
ципиально не может быть построена на основе предста-
влений физики 19 в. — классич. физики. Согласно
классич. (ньютоновской) механике, рассматривающей
электроны как материальные точки, и классич. (макс-
велловской) электродинамике, электроны в А. под дей-
ствием кулоновских (электростатических) сил взаимо-
действия (сил притяжения электронов ядром и сил от-
талкивания электронов друг другом по закону Кулона)
должны двигаться с ускорением и непрерывно терять
энергию на электромагнитное излучение. Поэтому А.
с классической точки зрения является неустойчи-
вой системой. В действительности А. устойчив; объ-
яснение причин устойчивости А. дает квантовая тео-
рия А.
Основы квантовой теории атома. Как показывает
огромный экспериментальный материал современной
атомной физики, А. подчиняется квантовым законам,
к-рые впервые были сформулированы Бором в его
теории А. в виде двух основных положений (посту-
латов Бора).
1. Существование стационарных
(квантовых) состояний. А. является устойчи-
вым лишь в нек-рых состояниях — т. н. стационарных
состояниях, соответствующих определенным значе-
ниям внутренней энергии А. Любое изменение этой
энергии, связано со скачкообразным (квантовым)
переходом А. из одного стационарного состояния в
Другое.
При квантовых переходах, в соответствии с сохра-
нения энергии законом, А. получает или теряет энер-
гию, равную разности энергий двух стационарных
его состояний. Возможны переходы с излу-
чением (оптич. переходы), когда А. испускает
или поглощает электромагнитное излучение, и пере-
ходы без излучения (безызлуча-
тельные, или неоптические переходы), когда
309
ATOM
ЗЮ
происходит непосредственный обмен энергией между
А и другими системами (напр., при столкновениях
А. с другими атомами или с электронами). Характер-
ной для А. является прерывность (дискретность)
возможных значений внутренней энергии; соответ-
ственно и изменения энергии при переходах из одного
стационарного состояния в другое также дискретны.
Стационарное состояние А. с наимень-
шей энергией (Ег) наз. основным, или нормаль-
ным, состоянием, остальные стационарные
состояния (с большими энергиями Е, > Elt i = 2, 3,
4,...) — возбужденными состояниями;
для перевода А. из основного состояния в возбужден-
ное ему необходимо сообщить энергию Ег —Ег
(возбудить его).
2. Условие частот. При переходе с излу-
чением из одного стационарного состояния (с энер-
гией Л\) в другое стационарное состояние (с энергией
Е ) А. испускает (при Е{ > Ek) или поглощает (при
Ei < Ек) электромагнитную энергию в виде фотона,
характеризуемого определенной частотой колебаний v,
согласно соотношению:
Ei - Eh = hv (1)
где h — Планка постоянная.
Атомы в основном состоянии могут только погло-
щать фогопы, атомы в возбужденных состояниях
могут как поглощать, так и испускать фотоны. Сво-
бодный А. в основном состоянии может существовать
неограниченно долгое время; время пребывания А.
в возбужденном состоянии (время жизни) огра-
ничено, А. теряет энергию возбуждения, частично или
полностью, испуская фотоны. Время жизни возбу-
жденных атомов водорода имеет порядок величины
10 8 сек.
Графически значения энергии стационарных состоя-
ний и квантовые переходы условно изображают при
помощи схемы уровней энергии (см. на стр. 322
схему уровней энергии и переходов с излучением
для атома водорода). На схеме горизонтальные ли-
нии проведены на высотах, пропорциональных зна-
чениям энергии А., вертикальные линии указывают
возможные переходы. Каждому переходу с излу-
чением в спектре А. соответствует определенная
спектральная линия. Совокупности переходов свер-
ху вниз соответствует спектр испускания, снизу
вверх •— спектр поглощения. Спектр А. водорода
(как и спектры др. атомов) дискретен и состоит из
отдельных спектральных линий; такие линейча-
тые спектры типичны для атомов и позволяют де-
лать важные выводы о строении А. (см. Атомные
спектры).
Из условия частот непосредственно вытекает уста-
новленный В. Ритцем экспериментально в 1908 (на
основе изучения закономерностей в спектрах) комби-
национный принцип, согласно к-рому в спектре воз-
можны линии, частоты к-рых являются комбинациями
(суммами и разностями) частот других линий (если
Е^ — Е2 = frv12 и/12 — Е3 = hv23, TO/i\ — Е3 = ftv12+
+ hv23 = hvl3, где v13 = v12 + v2S). Принцип Ритца
оправдывается на опыте с тем большей точностью,
чем точнее измерения частот спектральных линий;
его справедливость является одним из подтвержде-
ний правильности постулатов Бора. Правильность
этих постулатов непосредственно была подтверждена
опытами Дж. Франка и Г. Герца (1914) по возбужде-
нию атомов электронным ударом. Электроны в уско-
ряющем электрич. поле приобретают кинетич. энер-
гию 1/2 mv2 = eV (т и е — масса и заряд электрона,
v — его скорость, V — приложенная разность потен-
циалов). Сталкиваясь с А., электрон может отдавать
ему порции энергии, равные энергиям возбуждения
Et — /?! (при условии, что eV^Ei — Ег). Определяя
потенциалы возбуждения при к-рых начи-
нается возбуждение соответствующих уровней, можно
находить (из соотношения = Е{ — Ег) значения
Ei — Ег. Возбуждение атомов обнаруживается по
испусканию ими фотонов; как показывает опыт, ча-
стоты испущенных фотонов соответствуют условию
частот. В частности, для А. ртути наименьший потен-
циал возбуждения (eV2 = Е2 — Ех) равен 4,89 в,
и происходит испускание наиболее интенсивной лцнии
ртутного спектра (длина волны X = 2537А =
— 2,537 • 10~5 см) в согласии с условием частот
(Е2 — Е} = hv = X?-, где с — скорость света).
Существование стационарных состояний и условие
частот следует рассматривать как твердо установлен-
ные опытные квантовые законы. Они оказались спра-
ведливыми не только для А., но и для любых микро-
скопия. систем (см. Ядро атомное, Молекула) и пред-
ставляют экспериментальную основу квантовой тео-
рии таких систем. Следует подчеркнуть, что эти основ-
ные положения теории Бора составляют ту ее часть,
к-рая не только полностью сохранилась в квантово-
механич. теории, но и получила свое обоснование.
В то же время модельные представления теории Бора
о движении электронов в А. по строго определенным
орбитам, часто используемые и в наст, время, лишь
односторонне отражают движение электронов в А.,
к-рое принципиально неправильно рассматривать
просто как движение точечных заряженных частиц
по законам классич. механики и классич. электроди-
намики. В квантовомеханич. теории А. электрон уже
не рассматривается как материальная точка, движу-
щаяся по законам ньютоновской механики. Наряду
с его свойствами как частицы (с корпускулярными
свойствами) учитываются его волновые свойства.
Согласно квантовой механике, длина волны X, соот-
ветствующая движению электрона со скоростью v
(длина волны де-Бройля), равна X = — и для элект-
рона в А. оказывается порядка линейных размеров А.
(10 8 <\м); поэтому учет волновых свойств электрона
в А. является необходимым (волновые свойства частиц
могут не учитываться лишь в тех случаях, когда раз-
меры системы велики по сравнению с длиной волны).
Квантовомеханич. моделью электрона в А. является
модель электрона, заряд к-рого распределен в про-
странстве с нек-рой плотностью (электронной плот-
ностью), образуя так называемое «электронное об-
лако».
С точки зрения квантовомеханич. описания элект-
рона при помощи волновой функции ф(а:, у, z), являю-
щейся функцией координат электрона х, у, z, электрон-
ная плотность определяется квадратом модуля вол-
новой функции | ф (х, у, z) | 2, дающим вероятность
нахождения электрона (при рассмотрении его как
точечного) в данной точке х, у, z, рассчитанную на
единицу объема. Интеграл
ЭД j I С (х, у, z) Is dx dy dz
взятый по всему пространству, т. е. полная вероят-
ность нахождения электрона где бы то ни было,
равен единице [равенство данного интеграла единице
является условием нормировки функции ф (х, у, z),
т. е. надлежащего выбора постоянного множителя,
с точностью до к-рого определяется эта функция].
В квантовомеханич. теории А. необходимо учиты-
вать наличие у электрона собственного механич.
момента — спина, к-рый с наглядной точки зрения
соответствует вращению электрона вокруг своей оси
(если электрон рассматривать как сферу малых раз-
меров). При изучении тонких эффектов взаимодейст-
1 вия ядра А. с его электронными оболочками необхо-
311
ATOM
312
димо также учитывать, что спином обладают и нек-рые
ядра (см. Ядро атомное).
Теория атома водорода и водородоподобных ионов.
В теории А. важнейшую роль играет теория простей-
шей системы, состоящей из ядра с зарядом 4-Ze и
одного электрона с зарядом — е, т. е. теория А. водо-
рода (при Z = 1) и водородоподобных ионов (при Z =
= 2, 3, 4, ...). Последние получаются при такой
ионизации более сложных атомов, когда происходит
отрыв всех электронов, кроме одного. А. водорода и
водородоподобные ионы образуют простейший изо-
электронный ряд (ряд, состоящий из нейтрального А.
исходного элемента и ионизованных атомов после-
дующих элементов, обладающих тем же числом элект-
ронов, в данном случае одним электроном):
Н, Не+, Li2+, Ве3+...
Для уровней энергии такой одноэлектронной си-
стемы (одноэлектронного А.) квантовая теория дает
(без учета спина электрона) формулу:
Za
£’п = —Лей— (n= 1, 2, 3, .. .) (2)
л 2те2р.е* „
где R = - — т. н. постоянная Рид-
берга, ап — целое число, определяющее возмож-
ные значения энергии одноэлектронного А. и назы-
ваемое главным квантовым числом.
Приближенное значение постоянной Ридберга, полу-
ченное при условии, если за величину р, принять
массу электрона т, равно/? Q4 109 700 см~г.
Для того чтобы точнее определить значение R, необходимо
брать в качестве и т. наз. приведенную массу электрона и
, тМ т
ядра (ц = ;--Гт?-, где т — масса электрона,
М + т 14- т/М
а М — масса ядра), чем учитывается движение ядра, к-рое
лишь приближенно можно считать покоящимся. В согласии
с этим, опыт дает для А. водорода Нц = 109677,58 си»-1, для А
дейтерия Rp = 109707,42 си»-1 и для иона А. гелия (Не+)
Rjje = 109722,27 си»-1. Разница Нн и RHe позволила
Бору в 1913 отличить спектральные линии Не+от спектральных
линий Н, а разница Rjj и Нр позволила в 1931 по наличию
слабых спутников у спектральных ливий А. водорода уста-
новить существование дейтерия.
Численное значение постоянной hcR = 2,179 х
X 10-11 эрг = 13,60 эв. Уровни энергии А. водорода
на схеме построены согласно ф-ле (2) при Z = 1.
Они сходятся к границе ионизации (я —* ос), соответ-
ствующей состоянию, в к-ром электрон отрывается
от ядра и может уйти в бесконечность; энергия
ионизации
ЕИОн (Н) — Ет — hcR = 13,60 эв
а ионизации потенциал Уион = ^НОн(^)/е = 13,60 в.
Выше границы ионизации возможны любые значения
энергии А. (электрон отрывается от ядра и может
иметь любую кинетич. энергию: Т = Е — Е^. Со-
гласно условию частот (1), возможные переходы опре-
деляются разностью энергии пар уровней с я = яг,
я = яй;
<3)
‘ h \nh ni/
Эта формула находится в согласии с экспериментально
установленными закономерностями в спектрах А. во-
дорода и водородоподобных ионов (см. Атомные
спектры).
Основная формула (2) для одноэлектронного А.
была впервые найдена Бором на основе его теории А.
путем рассмотрения движения электрона вокруг ядра
по круговой орбите радиуса а. При этом считаются
возможными орбиты не любого радиуса, а лишь такие,
к-рые удовлетворяют дополнительному правилу кван-
тования механич. момента А. В результате получаются
вполне определенные значения энергии А. и радиуса
круговых орбит, зависящие от главного квантового
числа я (принимающего последовательные целые
значения 1, 2, 3, ...). Уровням энергии (2) соответст-
вуют орбиты радиуса
hSflS 7)2 л Т12
“п = 4~Ж^ = °-529 И = °>529 Z ' 1°’’ с- (4)
возрастающего пропорционально квадрату главного
квантового числа я, к-рое т. о. непосредственно
определяет размер орбит. Для первой круговой ор-
биты А. водорода (я = 1, Z = 1), соответствующей
его основному уровню и имеющей наименьший радиус,
= 0,529„ А, что дает диаметр А. водорода 2at —
= 1,058 А 1А = 10 8 см. Для водородоподобных
ионов масштаб энергий увеличивается в Z2 раз,
а масштаб линейных размеров уменьшается в Z раз,
в частности для Не+ £ион (Не+) = 4 • 13,60 эв =
= 54,4 эв, а1 = 0,264А (я = 1, Z = 2) и для 1д++
£ион(Ь,'++) = 9 • 13-6 эв = 142.4 ai = °.176 А
(я = 1, Z = 3).
Правило квантования механич. момента (момента коли-
чества движения) А., представляющего, согласно боровской
модели, произведение радиуса круговой орбиты а на коли-
чество движения электрона mv, может быть записано в виде
Мр = amv = nh/2л (n = 1, 2, 3, ....) и, при учете равенства
центростремительной силы mt>2/a силе притяжения электрона
ядром Ze2/as, дает:
Mt = a2m2v3 — a2m . — = amZe2 =
Р а
4ч 2
откуда следует ф-ла (4). Полная энергия электрона в А.,
' ' -2 и потенциальной
равная сумме кинетич. энергии Т = l/t mv2
энергии U = — Ze*/а, будет
Е = f/2mv2 — Ze2/a = 1/зтг2 — mr2 =
,, , ., mW>* мр
= — VsWr2 = — 1/з 5sj
ma2 2ma2
что при подстановке МрИ аг дает (2) ср. = т.
Согласно модельным представлениям теории Бора
для одноэлектронного А., наряду с круговыми орби-
тами, возможны и эллиптич. орбиты различной формы,
соответствующие тем же значениям энергии [ф-ла (2)[
и получающиеся, если придать правилам квантования
более общий вид. Однако это не устраняет принци-
пиальные недостатки теории Бора, являющейся непо-
следовательной и не могущей объяснить ряд особенно-
стей строения А. и нек-рых его свойств. Последова-
тельная квантовомеханич. теория для А. водорода и
водородоподобных ионов дает не только ту же ф-лу (2),
что и теория Бора, но и правильную характеристику
состояний А. Эти результаты находятся путем реше-
ния основного уравнения квантовой механики — Шре-
дингера уравнения, причем, в отличие от теории Бора,
дискретность уровней энергии А. получается в этом
случае автоматически, без дополнительных предпо-
ложений. Решение Шредингером в 1926 задачи об
А. водорода явилось важным этапом в развитии тео-
рии А.
При квантовомеханич. решении задачи для одно-
электронного А. каждое его состояние характери-
зуется, наряду с дискретным значением энергии,
согласно ф-ле (2), зависящим от главного квантового
числа п, дискретными значениями орбитального меха-
нич. момента и его проекции Мр на произвольно
выбранное направление z. Этот момент создается
электроном при его движении вокруг ядра (его назы-
вают орбитальным моментом, в отличие от спинового
момента электрона, см. ниже). Возможные значе-
ния Мр определяются т. н. азимутальным
квантовым числом I, принимающим п зна-
чений, I = 0,1,2, ..., п—1, а возможные значения
М.„ — т. наз. магнитным квантовым
г?
числом т/, принимающим 21 + 1 значений,
mt = I, I—1, ..., —I. Состояния с I = 0,1,2,3,4...
313
ATOM
314
принято обозначать буквами s, р, d, f, g,.... и соот-
ветственно называть s-, р-, d-, f-, g~, ... состояниями.
Согласно квантовой механике, величина квадрата орби-
тального момента определяется ф-лой Мр = h2/4n2' I (I -f- 1),
а величина его проекции — ф-лой Мр^ ~ ^т1 ' При I *
ЛГ;. = I2, ЛГ-, = Д- I и с наглядной точки зрения вектор
к 4ла к 2л
h
Мр длины I (в единицах —) ориентируется 21 +1 способами,
давая проекции от + I до —I (пространственное квантование,
соответствующее в теории Бора различным ориентациям пло-
скости орбиты электрона, при движении по к-рой электрон
обладает механич. моментом Мр, перпендикулярным ее пло-
скости).
Для состояния с определенными значениями п, I и
т1 получается вполне определенное распределение
электронной плотности вокруг ядра. Для состояний
с п = 1,2 и 3 это распределение показано на рис.
(см. ниже); оно зависит от п, I и от абс. значения пг1
(но не от знака т(). Существенно, что при I = 0 (что
соответствует Л/р=0) распределение электронной
Распределение электронной плотности для различных
состояний атома водорода.
плотности обладает сферич. симметрией, а в центре, где
находится ядро, электронная плотность не равна нулю;
при I О (что соответствует Мр 0) электронная
плотность концентрируется в нек-рых направлениях,
а в центре обращается в нуль. Отметим, что имеется
соответствие между распределением электронной плот-
ности и воровскими орбитами; уменьшению размеров
боровских орбит соответствует концентрация элект-
ронной плотности ближе к ядру.
Распределение электронной плотности в А. может
быть экспериментально проверено путем зондирова-
ния А. рентгеновскими лучами или электронными пуч-
ками. Определяя зависимость интенсивности рассеян-
ных лучей от направления рассеяния (от угла между
падающим и рассеянным лучом), можно рассчитать
т. наз. атомный фактор, зависящий от рас-
пределения электронной плотности в А.
Важным свойством квантовых состояний одноэлект-
ронного А. является то, что заданному значению энер-
гии Л'Г[ (уровню энергии), зависящему только от я,
а не от I и т1; соответствует ряд состояний, отличаю-
щихся значениями I и mt, а следовательно, и распре-
делением электронной плотности. Это свойство наз.
вырождением уровней энергии. Число раз-
личных состояний с одной и той же энергией наз.
степенью (кратностью) вырождения, или
статистическим весом g уровня энергии.
В частности, при я = 2 получается 4 состояния с той
же энергией: I = 0, т1 = 0 и I = 1, т1 = 1, 0, —1;
степень вырождения уровня энергии с я = 2 равна
g = 4; при я = 1, / == 0, т1 = 0 и получается лишь
1 состояние — уровень энергии с я = 1 невырожден
= !)•
Три квантовых числа — я, и пг1 — дают полную
характеристику состояния электрона, если не учи-
тывать спин электрона. При учете спина нужно ввести
четвертое квантовое число, определяющее величину
проекции спинового (собственного) механич. момента
электрона и принимающее два значения ms = ±г/2.
Величина квадрата спинового момента электрона, согласно
квантовой механике, равна h2/4n2 • s (з-|- */2), где s= 1/а.
С наглядной точки зрения вектор длины ‘/2 (в единицах /i/2n)
ориентируется двумя способами — вдоль выделенного напра-
вления (параллельно) и противоположно ему (антипарал-
Лельно).
Вследствие наличия спина степень вырождения уров-
ней энергии увеличивается вдвое; при заданных зна-
чениях я, I и mt получаются два состояния, отличаю-
щиеся значениями ms. При заданных значениях я и I
получается = 2 (21 + 1) состояний, отличающихся
значениями т1 = I, I — 1,..., — ! и ms = 1/г, —х/2;
т. е. два «-состояния (I = 0), шесть р-состояний
(I = 1), десять (/-состояний (1 = 2), четырнадцать
/-состояний (I = 3) и т. д. При заданном значении
п—1
п всего получается У] 2 (21 + 1) = 2п2 состояний,
1=0
соответствующих для одноэлектронного А. одной и
той же энергии Еп; следовательно, полная степень
вырождения уровней энергии в данном случае равна
gn = 2п2 = 2, 8, 18, 32, ... при п = 1, 2, 3, 4, ...
Зависимость энергии одноэлектронного А. лишь от п имеет
место только в том случае, если не учитывать с п и н - о р-
битальное взаимодействие — магнитное вза-
имодействие момента, связанного со спином электрона, с момен-
том, связанным с движением электрона вокруг ядра (согласно
теории Бора по определенной орбите). Благодаря этому взаи-
модействию спиновый и орбитальный механич. моменты
электрона складываются в его полный механич. момент,
определяемый [по формуле = — j (j + 1)] внутрен-
ним квантовым числом принимающим значе-
ния j~l± 1/г. Энергия А. зависит при данном п от значе-
ния j, вследствие чего уровни энергии, для к-рых п>2,
расщепляются на очень близкие подуровни. Данное явление
получило название тонкой структуры и может
быть рассчитано по квантовомеханич. теории, разработан-
ной Дираком и учитывающей теорию относительности (см.
Квантовая механика); теория Дирака приводит к наличию
у электрона спина и позволяет исследовать эффекты, связан-
ные со спином. Нек-рые отступления от теории Дирака, к-рые
наблюдал В. Лэмб при исследовании тонкой структуры уровня
п = 2 А. водорода радиоспектроскопия, методом (т. наз. сдвиг
s-уровней — несовпадение подуровней п = 2, I = 0 j = х/8
и п — 2, I — 1, j = совпадающих по теории Дирака),
объясняются в квантовой электродинамике (квантовой теории
электромагнитного поля). Еще более тонкое расщепление
уровней энергии — сверхтонкая структура —
получается благодаря взаимодействию полного электронного
момента А. со спиновым моментом ядра; такое расщепление
315
ATOM
316
на два очень близких подуровня наблюдает-
ся (по методу магнитного резонанса в атом-
ных пучках, см. Радиоспектроскопия) для
основного уровня А. водорода (n = 1, I = О,
3 = 1 /2^ и обусловлено взаимодействием пол-
ного электронного момента А. со спиновым
моментом протона, определяемым квантовым
числом I — */3. При переходе между этими
подуровнями возникает излучение с длиной
волны X = 21 см, наблюдаемое длямежзвезд-
п 1 2 3 4
! 001 б 1 2 б i ^2 3
Обозначения элект-
ронов и оболочек
Число электронов
в слое............
1s
2
2s 2р
2+6=8
пого водорода методами радиоастрономии.
Теория сложных атомов. Теория сложных атомов,
содержащих два или большее число электронов, прин-
ципиально отличается от теории А. водорода вследст-
вие наличия в сложном А. взаимодействующих друг
с другом одинаковых частиц — электронов. Между
электронами имеются электростатич. взаимодейст-
вия — взаимное отталкивание по закопу Кулона —
и магнитные взаимодействия, связанные с магнитными
моментами (спиновыми и орбитальными) электронов.
Электростатич. взаимодействия велики по сравнению
с магнитными, и их учет весьма существенен при рас-
смотрении строения сложных атомов; отталкивание
между электронами уменьшает прочность связи элект-
ронов в А., напр. для нейтрального А. гелия в основ-
ном состоянии из-за отталкивания между двумя элект-
ронами энергия отрыва одного из них равна £ион(Не)=
= 24,58 эв вместо £]1он(Не+) = 54,40 эв, необходимой
для отрыва единственного электрона в ионе гелия.
Чрезвычайно важную роль для электронов в А. иг-
рают их свойства как одинаковых (тождественных)
частиц, подчиняющихся квантовой статистике Фер-
ми — Дирака, с к-рой связан Паули принцип. Согласно
этому принципу, в системе электронов не может быть
двух электронов в одинаковых квантовых состояниях,
что для сложного А. приводит к образованию элект-
ронных оболочек, заполняющихся строго определен-
ными числами электронов.
В строгой квантовомеханич. теории имеет смысл
говорить (учитывая неразличимость взаимодействую-
щих между собой тождественных электронов) только
о состояниях А. в целом. Однако приближенно можно
рассматривать состояния отдельных электронов'в слож-
ном А. и характеризовать каждый из них совокупно-
стью четырех квантовых чисел п, I, uif и т^, аналогич-
ных квантовым числам А. водорода. При этом энергия
электрона оказывается зависящей не только от га,
как в А. водорода, но и от I; от т: и т она по-прежнему
(для свободного атома) не зависит. Электроны с дан-
ными га и I в сложном А. энергетически эквивалентны—
они обладают одинаковой энергией и образуют опре-
деленную электронную оболочку; их
называют эквивалентными электронами. Т. к. в силу
принципа Паули любые два электрона не могут нахо-
диться в одинаковых состояниях, то они должны
отличаться хотя бы одним из четырех квантовых чисел
га, I, т,/, т ; для определенной электронной оболочки,
т. е; при заданных в и !, должны отличаться пары
значений гаг(, т,. Число таких пар значений, g, —
= 2 (21 + 1) (равное . числу различных состояний
электрона с заданными п и I, т. е. степени вырожде-
ния уровня энергии такого электрона), дает число
электронов, заполняющих данную оболочку. Напр.,
при I = 0 возможны 2 комбинации значений т1 и
т. (raij = 0, гаг, =-1/2 И гаг^ = 0, гаг, =—1/2), при
2 — 1 возможны 6 комбинаций (гаг/ = 1,0, —1 при
ms = !/2, — J/2), при I = 2 —10 комбинаций (гагг =
= 2, 1, 0, — 1, — 2 при гаг8 = х/2, — х/2); соответ-
ственно оболочки j (1 = 0), р (I = 1) и d (I = 2)
заполняются 2, 6 и 10 электронами.
Электроны с данным п образуют электронный слой,
к-рый состоит из га оболочек с J = 0, 1, ..., га — 1 и
заполняется 2га2 электронами. Получаются слои
3s Зр 3d 4s 4р 4d 4/
2 + 6 + 10= 18 2 + 6+ 10+ 14 = 321.
По распределению электронной плотности наиболее
внутренним является слой с п = 1 (A-слой), затем
идут слои с га = 2, 2, 4, 5, 6, ... (£-, М-, JV-, О-, Р-слои).
В данном слое электронные оболочки с меньшим I
характеризуются большей электронной плотностью
вблизи ядра. От распределения электронной плот-
ности зависит прочность связи электрона в А.: чем
ближе к ядру концентрируется электронная плотность,
тем прочнее связан электрон. Прочность связи возра-
а при данном п — с уменьше-
стает с уменьшением га,
нием I.
Энергию электрона в сложном А. можно предста-
вить, аналогично ф-ле (2), в виде:
Ьпт == — hcR
at
2’2
= — hcR ----------------
712
= - 13,60
—s—33
(5)
где вместо действительного заряда ядра (в единицах е)
входит эффективный заряд Z* = Z — сп1, а ап1 —
т. н. постоянная экранирования,
характеризующая уменьшение силы притяжения элек-
трона ядром из-за экранирования заряда ядра элект-
ронным облаком, образованным остальными электро-
нами (в первую очередь более внутренними).
При этом предполагается, что полное электрич. поле ядра
и остальных электронов, действующее на данный электрон,
обладает сферич. симметрией, как и кулоновское поле одного
ядра в одноэлектронном А., характеризуемое потенциальной
энергией U -----—; для сложного А. надо положить, что U
также является функцией только от расстояния г электрона
от ядра, однако эта функция U = U (г) будет изменяться уже
не обратно пропорционально г, а более сложным образом (т. е.
электрон движется в сферически симметричном, но некуло-
новском поле). Квантование орбитального механцч. момента
связано именно со сферич. симметрией электрич. поля и по-
этому квантовое число I сохраняет свой смысл; квантовое число
п нумерует, согласно (5), последовательные уровни энергии
с данным I.
Определение энергии сложных А. и усредненных полей,
действующих на данный электрон со стороны остальных
электронов, возможно на основе различных приближенных
методов расчета А. В методе Томаса — Ферми электроны в А.
рассматриваются как электронный газ, подчиняющийся кван-
товой статистике Ферми — Дирака; согласно этому методу,
получается электронная плотность, монотонно убывающая
с увеличением расстояния от ядра. В квантовомеханич. ме-
тоде Хартри (методе самосогласованного поля, 1928) решается
система уравнений для движения электрона в усредненном
поле остальных электронов, получающееся распределение
электронной плотности отражает структуру электронных
оболочек; уточнением этого метода является метод В. А. Фока
(метод самосогласованного поля с обменом, 1930), в к-ром учи-
тывается одинаковость электронов и к-рый является в настоя-
щее-время лучшим приближенным методом квантовомеханич.
расчета сложных атомов.
С увеличением п и I постоянные экранирования ап1
увеличиваются, в соответствии с ростом экранирова-
ния более внешних электронов более внутренними,
что приводит к уменьшению, при заданном Z, эффек-
тивного заряда Z* = Z — ant. Т. к. с увеличением п
в (5) не только увеличивается п2, но и уменьшается Z*,
то прочность связи электрона (равная —Еп1) как
функция п для сложного А. убывает быстрее, чем для
А. водорода. Для наиболее тяжелых А. энергия
связи самых внутренних электронов порядка 100000 ав,
а энергия связи самых внешних электронов порядка
нескольких эв.
Объяснение периодичности свойств элементов на
основе теории строения атома. Рассматривая последо-
вательное заполнение электронных слоев и оболочек,
317
ATOM
318
можно объяснить периодичность свойств химич. эле-
ментов. Наиболее устойчивым состоянием А. является
такое, в к-ром электроны связаны наиболее прочным
образом. Поэтому ядро А. сперва связывает электроны
с п — 1, присоединяющиеся наиболее прочно, затем
электроны с п = 2, 3, 4, и т. д. При заданном п
сперва связываются s-электроны (1 — 0), затем р-,
d-, /- электроны (£ = 1,2, 3). В табл, дана периодич.
система элементов Менделеева в форме, показывающей
порядок заполнения электронных оболочек А. Для
каждого элемента в табл, указана электронная конфи-
гурация (т. е. распределение электронов по оболочкам),
соответствующая наиболее прочной связи электронов
в нейтральном А. (нормальная или основная электрон-
ная конфигурация нейтрального А.). Для элементов
первого периода происходит заполнение оболочки 1s,
т. е. A-слоя, заканчивающееся у Не с электронной
конфигурацией 1s2, для элементов второго периода- —
оболочек 2s и 2/>, т. е.Х-слоя, заканчивающееся у Ne
с электронной конфигурацией ls22s22/>6. Для элемен-
тов третьего периода заполняются оболочки 3s и 3/>.
А. Аг имеет электронную конфигурацию 1 s22s22/>63s23/>6.
Однако затем последовательность заполнения слоев
нарушается вследствие конкуренции электро-
нов 4s и 3</, близких по энергии связи; в четвертом
периоде у переходных элементов от Sc до Zn происхо-
дит достройка более внутренней (с меньшим п) обо-
лочки 3</ и окончательное заполнение Af-слоя. В даль-
нейшем конкурируют электроны 5s и 4с? (в пятом пе-
риоде у переходных элементов от Y до Cd достраи-
вается оболочка 4с?), электроны 6s, 5с? и 4/ (в шестом
периоде у лантанидов достраивается оболочка 4/ и
у переходных элементов от Lu до Hg — оболочка 5с?),
электроны 7s, 6с? и 5/ (в седьмом периоде у актини-
дов достраивается оболочка 5/, а у элемента с Z = 103
должна начаться достройка оболочки 6с?).
В таблице приведены также значения энергии иони-
зации нейтрального А., т. е. энергии отрыва наиболее
слабо связанного электрона; они периодически изме-
няются в зависимости от Z. Для атомов элементов
с одним внешним электроном ns (начиная с Li) энер-
гии ионизации малы, для атомов элементов с двумя
внешними s-электронами они значительно больше.
По мере заполнения внешней оболочки пр (га = 2, 3,
4, 5, 6) энергии ионизации возрастают и особенно
велики для атомов инертных газов с целиком запол-
ненной р-оболочкой. Для данной группы элемен-
тов с внешними электронами ns и пр (га = 2, 3, 4, 5, 6)
энергии ионизации убывают, как правило, с увеличе-
нием га (от Li к Cs, от Be к Ва и т. д.). Для аналогич-
ных элементов с достраивающимися с?-оболочками
в четвертом и в пятом периоде энергии ионизации
близки друг к другу, а в шестом периоде неск. больше.
Параллельно с энергиями ионизации изменяются и
размеры А., к-рые тем больше, чем слабее связан
электрон; следует, однако, иметь в виду, что значе-
ния величин, характеризующих размеры А , — ра-
диуса А., поперечного сечения А. и
объема А. — зависят, в большей или меньшей
степени, от способа их определения. Это связано с тем,
что А. не имеет строго определенных границ, в соот-
ветствии с постепенным убыванием электронной плот-
ности. Периодичность химич., оптич., электрич. и
магнитных свойств А., в завйсимости от Z, связаны
со сходством в строении внешних электронных обо-
лочек, к-рыми определяются эти свойства.
Способность А. вступать в химич. соединения с об-
разованием химической связи непосредственно зависит
от строения его внешних электронных оболочек;
валентность А. определяется числом электронов
в этих оболочках, одинаковым для элементов, стоящих
в одном вертикальном столбце периодич. системы.
Для элементов с внешними s- и /^-электронами (левая
часть табл.) высшая валентность (положительная)
равна общему числу электронов в оболочках ns и пр,
совпадающему с номером группы и достигающему 7
для галогенов. Особой устойчивостью обладают инерт-
ные газы: Не с электронной конфигурацией 1s2 и Ne,
Аг, Кт, Хе, Rn с электронной конфигурацией ns3np3
(га = 2, 3, 4, 5, 6). Эти элементы благодаря упрочне-
нию заполненных оболочек s и р (сравнить значения
энергий ионизации) не образуют устойчивых химич.
соединений и обладают валентностью, равной нулю
(нульваленты). Для элементов с достраивающейся
«/-оболочкой при ее заполнении не более чем наполо-
вину высшая валентность определяется общим чис-
лом электронов ns и (га — 1)«/ (га = 4, 5, 6), также
совпадающим с номером группы и достигающим 7
для Мп, Те и Re; при заполнении «/-оболочки более
чем наполовину валентность уменьшается в соответ-
ствии с упрочнением «/-электронов, в частности Си,
Ag и Аи с заполненной оболочкой (га — 1)«/ могут
быть 1-валентны и 2-валентны [в последнем случае
наряду с электроном ns в образовании связи участвует
один из электронов (га — 1)</], a Zn, Cd и Hg 2-ва-
лентны в соответствии с наличием двух внешних
электронов ns. Для элементов с достраивающейся
/-оболочкой типичной является валентность, равная 3,
и лишь в начале достройки /-оболбчки высшая валент-
ность совпадает с общим числом электронов ns,
(га — 1)«/ и (га — 2)/ (га = 6,7), достигая для ланта-
нидов 4 у Се, а для актинидов 6 у U.
При образовании ионных связей одни
атомы (электроположительные) отдают нек-рое число
внешних электронов, равное положительной валент-
ности А., а другие атомы (электроотрицательные)
присоединяют некоторое число электронов, раввое
отрицательной валентности А., образуя обладающие
наибольшей устойчивостью заполненные электронные
оболочки, например в CaF2 А. кальция отдает два
электрона 4s, а каждый А. фтора получает по одному
электрону с образованием оболочек 3/>в, подобных
оболочке А. аргона. Электроотрицательность А при
этом может быть охарактеризована сродством к элект-
рону.
В образовании ковалентных связей
участвуют внешние электроны вступающих в соеди-
нение атомов, образуя т. наз валентные пары — пары
электронов с противоположно направленными (ком-
пенсированными или спаренными) спинами.
При этом существенно, чтобы А., вступающий в сое-
динение, находился в валентном состоя-
нии, в к-ром спины отдельных внешних электронов
параллельны и не компенсируют друг друга. Число
электронов с нескомпенсированными спинами опре-
деляет число ковалентных связей, к-рое может обра-
зовать А. При наличии двух или трех эквивалентных
электронов с / > 0 (напр., при электронной конфигу-
рации />2 или р3) А, будет находиться в валентном
состоянии и соответственно образовывать две или три
ковалентные связи. Для заполненных оболочек спины
всегда взаимно Компенсируются и поэтому оболочке s2
(как и оболочкам р3, </10 и /14) не соответствует валент-
ное состояние: валентным состоянием для А. с нор-
мальной электронной конфигурацией внешних элект-
ронов ras2 будет являться возбужденное со-
стояние, напр nsnp, в к-ром один из электро-
нов ras превратился в электрон пр. В случае А. угле-
рода нормальной электронной конфигурации 2s22/>2
соответствует 2-валентное состояние (спины «-электро-
нов спарены, спины />-электронов не спарены); 4-ва-
лентное состояние (четыре электрона с неспаренными
спинами) соответствует возбужденной электронной
конфигурации 2s2p3, к-рая сравнительно легко воз-
буждается, что объясняет 4-валеитность А углерода.
Направленность валентности объясняется тем, что
319
ATOM
320
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА В ФОРМЕ, ПОКАЗЫВАЮЩЕЙ
Пе-
риоды
Элек-
трон-
ные
обо-
лочки
1
1s
2
Не 1s3
s3
1
Н
s
13,60 24,58
2s
2
2/j
3
Li
5,39
4
Be
s3
9,32
5
В
ss
3
3s
В левой части табл, указаны номера периодов, совпадающие с главным
квантовым числом п соответствующих s- и р-электронов, и символы всех
связываемых электронов, причем фигурными скобками объединены элек-
троны, конкурирующие по энергии связи. Далее, для каждого элемента ука-
заны порядковый номер Z, знак элемента, электронная конфигурация наи-
более слабо связанных электронов и энергия ионизации нейтрального А.
(энергия отрыва первого электрона). Справа для каждого периода приведе-
ны оболочки, заполняющиеся в этом периоде (электронная конфигурация А.
соответствующего инертного газа). Внизу таблицы указаны номера групп
согласно общепринятой класснфика-
ции. Начиная с элемента Sc (4-й пе-
риод), нарушается последовательность
заполнения слоев: состояния 3d энер-
гетически более выгодны, чем состоя-
ния 4 р. Вследствие этого для элемен-
тов с порядковыми номерами Z = 21—
30 происходит заполнение 3 (/-оболоч-
ки, окончательно достраивающейся у
Си и Zn; переходные элементы Sc — Zn
смещены в таблице соответственно
вправо. Аналогичная картина имеет ме-
сто в 5—7 периодах системы Менделее-
ва, что также отражено в таблице.
7
N
s'-p:
8
О
s3p‘
9
F
S3/Ji
10
Ne
s^p’
6
; С
'р s2p-
8,30 11,26 14,5313‘,61 17,42 21,56
2s32p6
3p
12
Mg
11
Na
s s3 13 14 15 16 17 18
5,14 7,64 А1 Si Р S Cl Ar
s2p s-p'1 s-p'1 s^p1 s-p5 s3pe
5,98 8,15 10,48 10,36 13,01 15,76
3s3 3pe
4 1 4s ( 3d
5 1 5s ( 4rf 5/j
6 ( 6s 1 { 5d ' 4/ 6p
55
Cs
s
3,89
19
К
20
Ca
s 4,34 s2 6,11 31 Ga s2p 6,00 32 Ge s3p3 7,88 33 As s3p3 9,81 34 Se s3p4 9,75 35 Br ssp5 11,84 36 Kr s-p* 14,00 21 Sc ds'1 6,54 22 Ti cPs3 6,82 23 V cPs3 6,74 24 Cr ds's 6,76 25 Mn <Z5s3 7,43 26 Fe cPs3 7,87 27 Co d's'1 7,86 28 Ni dKs°- 7,63 29 Cu d*°s 7,72
37 38
Rb Sr
s S'2 39 40 41 42 43 44 45 46 47
4,18 5,69 ¥ Zr Nb Mo Те Ru Rh Pd Ag
49 50 51 52 53 54 ds'1 d'1s'1 d4s d'~s (Ps2 rf7s d*s d"> rf10s
In Sn Sb Те J Xe 6,38 6,84 6,88 7,10 7,28 7,36 7,46 8,33 7,57
s-p s-p'1 s3p3 s-p* s>5 s-pe (rf) (rf)
5,78 7,34 8,64 9,01 10,45 12,13
56
Ba
s3
5,21
71 Lu 72 Ht 73 Ta 74 W 75 Re 76 Os 77 Ir 78 Pt 79 Au
81 82 83 84 85 86 ds2 (Ps3 das2 cPs3 cPs3 cPs2 (77S2 d'Js dl0s
TI Pb Bi Po At Rn 7,88 7,98 7,87 8,7 9,0 9,22
s2p s-p2 s'2ps s'-p* S“p:’ s'1pe
6,11 7,41 7,29 8,43 10,75
87 88
1 7s Fr Ra
1 s S*"
7 { 5d 5,28
’ 4/
7p
Группа I 11 Ill IV
V VI VII о
103 104 105 106 107 108 109 no in
III IV V VI VII vin i
321
ATOM
322
ЗАПОЛНЕНИЕ ЭЛЕКТРОННЫХ ОБОЛОЧЕК
Электронная конфигурация дана в сокра-
щенной записи. Напр., для С, Z ~ 6, имеем
s2p3, что означает 2s2, 2р2, а в полной записи,
с учетом внутренних электронов, Is2 2s2 2р2;
для Сг, Z = 24, d5s, что означает 3rf54s, в
полной записи:
Is2 2s2 2рв 3s2 Зр° 3rf5 4s
для J, Z = 53, имеем s2p5, что означает
5s2 5р5, в полной записи:
1s2 2s2 2р6 3s3 Зр« 3d‘° 4s2 4р6 4rf10 5s2 5р5
Энергии ионизации даны в электрон-вольтах
(1 эв соответствует 23 082 кал/грамм-атом) и
относятся к наиболее слабо связанному элек-
трону: р-электрону для конфигурации типа
s2p* (k = 1, 2, 3, 4, 5, 6) и s-электрону для
всех остальных конфигураций (кроме конфи-
гураций, в к-рых слабее всего связан (/-элек-
трон, что отмечено символом d под значением
энергии ионизации). Энергия ионизации в эв
численно равна потенциалу ионизации, выра-
женному в вольтах. Для атомов многих тяже-
лых элементов энергии ионизации неизвестны.
30
Zn
rf10s3
9,39
3rf104s34p°
Схема уровней энергии и переходов с излуче-
нием для атома водорода.
К стр. 309.
48
Cd
d'°s2
8,99
4rf105s25pe
80
Hg
dl0s2
10,43
57 58 59 60 61 62 63 64 65 66 67 68 69 70
La Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er Tu Yb
ds2 5,61 /3S3 /3S3 /*s3 /6s2 /°s2 5,4 /’<- 5,67 fds2 (j»ds2) 6,16 /10s3 /13s3 /13s3 /14s5 6,25
(rf)
4/u5rfI06s26pe
89 90 91 92 93 94 95 96 97 98 99 100 101 102
Ac Th Pa U Np Pu Am Cm Bk Cf Es E’m Md
112 ds2 6,9 d2s2 f2dss f>ds2 /Ms3 /es2 /7s3 (/7rfs3)(/Ms3)(/10s3) (/ns3) (/13s3) (/13s3) (/14s2
II
111
6 к. X. Э. т. 1
323
атом —атомные Спектры
324
ковалентные связи образуются в направлениях,
в к-рых для электронов А. сильнее всего концентри-
руется электронная плотность. Для А. с внешними
^-электронами такими направлениями являются три
взаимно перпендикулярных направления. Распределе-
ние электронной плотности для трех р-состояний
(рт, ру, р2) может быть представлено следующим об-
разом. В каждом состоянии может находиться по
2 электрона с компенсированными спинами, что дает
устойчивую оболочку рв; в 2- или 3-валентном состоя-
нии А. в двух или трех разных электронных р-состоя-
ниях находится по одному электрону, что приводит
к образованию 2 или 3 ковалентных связей в соответ-
ствующих направлениях (напр., образование пира-
мидальных молекул аммиака NH3 атомами азота
в 3-валентном состоянии при электронной конфигура-
ции внешних электронов 2р3). Для электронной
конфигурации 2s2p3 А. углерода наиболее энергети-
чески выгодным, с точки зрения образования кова-
лентных связей, является распределение электрон-
ной плотности, соответствующее не состояниям 2s и
2р в отдельности, а т. наз. гибриднымсостоя-
н и я м (Л. Полинг); из одного s-состояния и трех
p-состояний при этом образуются четыре новых
(гибридные) и притом равноправных состояния. Это
объясняет образование атомами углерода четырех
равноценных ковалентных связей (в случае четырех
соседних атомов), направленных от центра к вершинам
тетраэдра (с углами между связями 109°28')- Наряду
с рассмотренной гибридизацией типа sp3 возможна
гибридизация других типов, в частности типа sp2
с образованием трех равноценных связей в плоскости
с углами 120° между ними (в случае трех соседних
атомов) и типа sp с образованием двух равноценных
связей в противоположных направлениях (в случае
двух соседних атомов). О гибридизации состояний
см. подробнее в ст. Квантовая химия и Химическая
связь.
Для атомов элементов с аналогичными внешними
электронными оболочками наблюдается сходство оп-
тич. спектров, связанных с возбуждением внешних
электронов. С увеличением числа внешних электро-
нов эти спектры усложняются (см. Атомные спектры).
В противоположность этому рентгеновские спектры
атомов, соответствующие переходам между уровнями
А., обусловленными внутренними электронами, и
получающиеся при удалении одного или нескольких
электронов из этих оболочек, монотонно изменяются
при изменении Z независимо от строения внешних
электронных оболочек (см. Мозли закон и Рентгено-
структурный анализ).
Магнитные свойства атомов также сходны в случае
элементов с аналогичными внешними электронными
оболочками. Атомы с целиком заполненными электрон-
ными оболочками, в частности атомы инертных газов
и щелочноземельных металлов, не обладают магнит-
ными моментами вследствие компенсации для любой
заполненной оболочки всех моментов (орбитальных
и спиновых) отдельных электронов. Для частично
заполненных оболочек такой компенсации, как пра-
вило, не происходит; атомы, обладающие подобными
оболочками, имеют магнитные моменты и являются
парамагнитными. Все атомы обладают диамагнетиз-
мом, к-рый обусловлен появлением у них магнитного
момента под действием магнитного поля (см. Магнит-
ные свойства).
Во внешних магнитных полях уровни энергии А.
расщепляются в силу того, что магнитный момент А.
в данном состоянии может различным образом ориен-
тироваться в магнитном поле (число ориентаций равно
степени вырождения соответствующего уровня энер-
гии); это т. наз. Зеемана явление можно изучать как
по расщеплению спектральных линий в оптич. спект-
рах, так и непосредственно, наблюдая переходы между
близкими подуровнями, получающимися при расщеп-
лении, методами радиоспектроскопии (частоты пере-
ходов между подуровнями соответствуют диапазону
радиочастот).
Электрич. свойства атомов характеризуются спо-
собностью электронов смещаться во внешнем элект-
рическом поле тем сильнее, чем слабее связаны эти
электроны, поэтому в основном смещаются внешние
электроны (см. Поляризация). Уровни энергии А. во
внешних электрич. полях расщепляются; это т. наз.
Штарка явление можно изучать по расщеплению спект-
ральных линий в оптич. спектрах, а также методами
радиоспектроскопии. Важное значение имеет иссле-
дование расщепления уровней энергии ионов в ком-
плексных соединениях, кристаллах и р-рах, обуслов-
ленного действием-внутренних электрич. полей, созда-
ваемых окружающими ионы частицами.
Необходимо в заключение отметить, что периодич-
ность свойств А. имеет место как для нейтральных
атомов, так и для их ионов данной кратности. Теряя
один электрон, атомы элементов данной группы ста-
новятся подобными по своим свойствам атомам эле-
ментов предыдущей группы (напр., атомы щелочно-
земельных металлов— атомам щелочных металлов) и
периодичность сохраняется. Она сохраняется и при
потере к электронов. Схожими свойствами обладают
нейтральный А. и ионы с тем же числом электронов,
т. е. члены изоэлектронного ряда; для них характерно
монотонное изменение свойств с изменением Z, част-
ным случаем к-рого является зависимость энергии от
Z для членов простейшего изоэлектронного ряда Н,
Не+, Li++,... [см. ф-лу (2)]; аналогичная зависимость
получается для сложных атомов согласно ф-ле (5),
в к-рой постоянная экранированная для членов изо-
электронного ряда мало зависит от Z.
Лит.: Фриш С. Э. и Тиморева А. В., Курс общей
физики, т. 3, М., 1959; ШпольскийЭ. В., Атомная физика,
т. 1, 4 изд., М.—Л., 1952, т. 2, 3 изд., М.—Л., 1951; Кон-
дратьев В. Н., Структура атомов и молекул, 2 изд., М.,
1959; Landolt-Bornstein, Zahlenwerten und Funktio-
nen aus Physik..., Bd 1, B.—Gottingen — Hdlb., 1959; Moore
Ch. E., Atomic energy levels, v. 1—3, Wash., 1949—58.
M. А. Ельяшевич.
АТОМАЛЬ — см. Проявляющие вещества.
АТОМНАЯ ЭНЕРГИЯ (ядерная энер-
гия) — энергия, выделяющаяся в процессе превра-
щения ядер атомов. А. э. выделяется в форме кинетич.
энергии движущихся с огромными скоростями про-
дуктов ядерной реакции (осколков ядер) и часто,
кроме того, в форме у-излучений. Кинетич. энергия
движущихся тел наиболее легко переходит в форму
тепловой энергии; этот переход имеет место и при атом-
ных взрывах, и в атомных электростанциях, являющих-
ся по существу тепловыми электростанциями с ядер-
ным «горючим» (см. Ядерная энергия).
АТОМНЫЕ СПЕКТРЫ — спектры, возникающие
при испускании или поглощении фотонов (квантов
света) свободными или слабо взаимодействующими
атомами. Такими спектрами (испускания и поглоще-
ния) обладают одноатомные газы или пары. А. с. со-
стоят из отдельных спектральных линий (линейча-
тые спектры), каждая из к-рых соответствует переходу
между определенными стационарными состояниями
атома (между уровнями энергии) и харак-
теризуется определенными значениями частоты »
фотона, волнового числа ч' = — и длины волны
X = . Частоту v обычно выражают в сек'1
(герцах), ч’ — в см1 и X — в ангстремах (1 A„=
= 10“8 см) или миллимикронах (1 ммк = 10 А).
Наряду со спектрами нейтральных атомов наблю-
даются спектры ионов. Спектр нейтрального атома
(т. наз. дуговые спектры) принято обозначать циф-
325
АТОМНЫЕ СПЕКТРЫ
326
рой I, спектры ионизованных атомов (т. наз. искро-
вые спектры) — цифрами II, III, IV, ... для последо-
вательных кратностей ионизации; значение римской
цифры всегда на единицу больше кратности иониза-
ции, напр. NI, Nil, NIII, ... означают спектры N,
N+, N2+, ... В реально наблюдаемых спектрах часто
одновременно присутствуют линии нейтрального и
ионизованных атомов, соответственно говорят, напр.,
о линиях Fel и Fell в спектре Fe. Ионы обладают
спектрами, схожими со спектрами нейтральных ато-
мов с тем же числом электронов; т. о., имеется сходство
спектров для членов изоэлектронного ряда, при этом
с увеличением порядкового номера Z спектры, при
сохранении их общего характера, смещаются в область
больших частот.
Наиболее простыми являются спектры атома водо-
рода и водородоподобных ионов (Не+, Li2+, Ве3+
и т. д.), волновые числа линий к-рых даются общей
формулой (см. ст. Атом — формулы 2 и 3):
где пк и — главные квантовые числа нижнего и
верхнего уровней энергии, комбинирующихся между
собой. При заданном пк и щ = пк + 1, пк + 2,...
получаются спектральные серии. Для
атома водорода (Z — 1) это серии Лаймана = 1),
Бальмера (nfe= 2), Пашена (nfe = 3), Брэкета (гей = 4),
Пфунда (nfe = 5), Хампфри (nft = 6). Серия Лаймана
лежит в далекой УФ-области спектра, серия Бальме-
ра — в видимой и близкой УФ-области; для линий
этой серии Бальмером в 1885 впервые была обнару-
жена закономерность в спектрах. Остальные серии
лежат в ПК-области спектра. Спектральные линии
атома водорода и водородоподобных ионов обладают
тонкой структурой, обусловленной нали-
чием у электрона спина: каждая линия расщеплена на
ряд компонент, расстояния между к-рыми (Д'/) для
атома водорода имеют порядок десятых долей еле1
(при v' -----порядка десятков тысяч с.м1), а для
водородоподобных ионов возрастают пропорциональ-
но Z4, т. е. для Не- в 16 раз и для Li2+ в 81 раз [при
возрастании ч' согласно (1) в Z2 раз].
Спектры атомов щелочных металлов, обладающих
одним внешним (оптическим) электроном сверх запол-
ненных оболочек, схожи со спектром атома водорода,
но смещены в область меньших частот. Число спект-
ральных серий увеличивается, а закономерности
в расположении линий описываются формулами,
более сложными, чем формула (1). Число серий уве-
личивается вдвое для атомов с двумя внешними элект-
ронами (Не, щелочноземельные металлы Zn, Cd и Hg).
Спектры еще усложняются для атомов, обладающих
тремя и более внешними электронами. Особенно сло-
жны спектры атомов с достраивающимися (/-оболоч-
ками (напр., спектр Fe) и /-оболочками, состоящие
из сотен и тысяч линий. Истолкование спектров мно-
гоэлектронных атомов представляет трудную задачу,
к-рая решается на основе квантовой теории строения
атома.
Современная теория А. с. исходит из приближенной
характеристики электронов в сложном атоме кванто-
выми числами п и I и позволяет определить, при
помощи векторной схемы сложения механич. момен-
тов, уровни энергии атома, соответствующие заданной
электронной конфигурации. Каждый электрон обла-
дает орбитальным механич. моментом Z{ (определяе-
мым квантовым числом (г=0,1, ..., —1) и спиновым
механич. моментом (определяемым квантовым чи-
слом si = 1/2); полный механич. момент атома J
6*
равен векторной сумме всех и 8;:
= 4“ С 4- • - • 4- 4- *1 4- 4- • • - 4- sr (2)
(г — число электронов). Для заполненных оболочек
J = 0, вследствие взаимной компенсации всех момен-
тов при их сложении (что вытекает из принципа Па-
ули), и любой электронной конфигурации, состоящей
из таких оболочек, соответствует лишь один уровень
энергии; поэтому при сложении моментов согласно (2)
нужно учитывать только электроны заполняющихся
оболочек. В простейшем случае одного внешнего
электрона J = I + s. Согласно общему квантовому
закону сложения механич. моментов (моментов коли-
чества движения или угловых моментов), при вектор-
ном сложении двух моментов и Х2 в полный момент
К для квантовых чисел klt к2 и к, определяющих
значение этих моментов, имеет место соотношение:
h = fci + fe2, fcr + fe» — 1. fel - fes (fei-sfea) (3>
к-рое с наглядной точки зрения соответствует различ-
ной взаимной ориентации векторов Л?! иХ2. Для векто-
ров I, s и J это дает j = I + 1/2,1 —1/2, т. е. два значе-
ния квантового числа J — j, определяющего значе-
ния полного механич. момента электрона *. Энергия
электрона зависит от значения /, и при данных п и I
(для одноэлектронной конфигура-
ции) получаются два уровня, расстояние между
к-рыми — дублетное расщепление —
обусловлено спин-орбитальным взаимодействием (ма-
гнитным взаимодействием моментов s и I). Дублетное
расщепление непосредственно определяет тонкую
структуру спектральных линий атомов щелочных
металлов, в частности желтой линци Na, являющейся
двойной (X = 5889,95 А, 5895,92 А); для атома водо-
рода тонкая структура зависит как от дублетного
расщепления уровней, так и от наличия вырождения
по I (от совпадения энергий состояний с данным п,
но различными (), что усложняет наблюдаемую кар-
тину расщепления.
Для случая двух или нескольких электронов сло-
жение моментов может происходить различными спо-
собами, в зависимости от того, какие взаимодействия
в атоме наиболее существенны для данной электронной
конфигурации. Если главную роль играют электро-
статич. взаимодействия электронов, то все орбиталь-
ные моменты U складываются в полный орбитальный
Г
момент L = У Z. ц все орбитальные моменты s;-—
i = l
г
в полный спиновый момент® = это обычный слу-
i = l ’
чай нормальной связи, или связи Расселя —Саундерса
(£®-связи). Значения моментов Хи® определяются
квантовыми числами L и А, в случае двух электронов
равными, согласно форм. (3): L = 1Х + /2, + 12 — 1,
..., —12 (для конфигурации dp, 1г = 2, l2 — 1, L =
— 3, 2, 1) и ® = S, + s2 = 1,0 («j = s2 =1/2). В силу
магнитного взаимодействия моментов X и ®
они складываются в полный момент J (аналогично
сложению I + s = j для случая одного электрона),
определяемый принимающим х = 2s + 1 значений
(при 8) квантовым числом J = L + 8, L 4- 8 —
— 1, ..., L — S, напр. при L = 2 и ®=1J=3, 2,1
(х = 3). Энергия атома, обладающего заданной элект-
ронной конфигурацией, зависит отмультиплет-
н о с т и х (при S = 0, х/2, 1, 3/2, 2,... х = 1, 2, 3,
4, 5,...), при данном х — от £ и при данных х и L —
* Величины, относящиеся к отдельному электрону, при-
нято обозначать строчными буквами (напр., I), а величины,
относящиеся к атому в целом, — прописными (напр., J).
В данном случае полный момент атома совпадает с полным
момевтом электрона: J=J.
327
АТОМНЫЕ СПЕКТРЫ —АТОМНЫЙ ВЕС
328
от J. Совокупность х уровней энергии с данными S и
L образует мультиплетный терм — с и н г у-
летный при х = 1 (б7 = 0), дублетный при
х = 2 (как в случае атома с одним внешним электро-
ном, для к-рого £ = s = 1/2), триплетный при
х = 3 (S — 1), квартетный при х = 4 (б7 =
— 3/2) и т. д.; в случае двух электронов получаются
сингулетные и триплетные термы (S — 0,1, х = 1,3,
что приводит к наличию двух типов уровней), в слу-
чае трех электронов — дублетные и квартетные
(S = 1/2, 3/2, х = 2,4); вообще при четном числе
электронов спин £ всегда целый (нечетная мульти-
плетность), а при нечетном числе — всегда полу-
целый (четная мультиплетность). Для каждой элект-
ронной конфигурации получается свой набор воз-
можных термов, причем, согласно правилу Г у н-
д а, глубже всего лежит терм наибольшей мульти-
плетности с максимальным значением L, возможным
при данной мультиплетности. Отдельные термы харак-
теризуются величиной мультиплетного расщепления
(расстояний между уровнями с различными J), к-рое
быстро возрастает с увеличением заряда ядра и по-
этому велико для тяжелых атомов. Термы принято
обозначать символом где значения L = 0,1,
2, 3, ... указывают буквами б7, Р, D, F,... (аналогично
буквам s, р, d, f,... для I = 0, 1, 2, 3,...), напр. 3D
при б7 = 1 (х = 3) и L = 2, а отдельные уровни терма
отмечают индексом J (3D3, 3D2, 3Dr для уровней с J —
= 1, 2, 3 терма 3D).
Если главную роль играют магнитные взаи-
модействия орбитального и спинового моментов для
каждого электрона, то имеет место случай т. наз.
(/, /) связи: сперва li + s; = jit затем ~
1 = 1
Этот случай осуществляется сравнительно редко.
Возможны и более сложные случаи связи промежуточ-
ных типов.
В А. с. проявляются далеко не все мыслимые пере-
ходы между различными уровнями, а лишь вполне
определенные, разрешенные правилами от-
бора. Для квантового числа J, определяющего
величину полного механич. момента атома (незави-
симо от случая связи), разрешены переходы с измене-
нием J не более чем на 1 (правило отбора ДА =
= 0, ± 1). Для легких атомов ДД = 0, т. е. разре-
шены переходы только между уровнями одной муль-
типлетности; для тяжелых атомов это правило отбора
нарушается и возможны т. наз. интеркомбинационные
переходы между уровнями различной мультиплет-
ности (в частности, подобному переходу соответствует
линия Hg с Х=2537А в УФ-области спектра). Важ-
ное правило отбора связано с делением электронных
конфигураций и состояний атома на четные и
нечетные, в зависимости от того, является ли
четной или нечетной сумма У, \ для всех электронов
г = 1
атома (что зависит от четности или нечетности числа р-
и /-электронов в заполняющихся оболочках); разре-
шены только переходы между четными и нечетными
состояниями (правило Лапорта), поэтому, в частности,
запрещены переходы между уровнями одной и той же
конфигурации, к-рые все являются либо четными,
либо нечетными. А. с. — одна из важнейших и наи-
более однозначных характеристик атомов. Исследова-
ние А. с. сыграло важную роль в развитии предста-
влений о строении атомов, привело к открытию спина
электрона и истолкованию периодической системы
элементов Менделеева.
Методы, основанные на применении А. с., чрезвы-
чайно широко распространены в различных областях
науки и техники. Из А. с. определяют ряд очень важ-
ных характеристик атомов и получают ценные све-
дения о строении электронных оболочек атома. Исклю-
чительно важным применением А. с. является эмис-
сионный спектральный анализ, к-рый благодаря высо-
кой чувствительности, быстроте и универсальности
завоевал прочное место в металлургии, горнорудной
пром-сти, машиностроении и во многих других отра-
слях народного хозяйства.
Лит.: Фриш С. Э., Атомные спектры, Л.—М., 1933;
Мандельштам С. Л., Введение в спектральный анализ,
М.—Л., 1946; Герцберг Г., Атомные спектры и строе-
ние атомов, пер. с англ., М., 1948; White Н. Е., Introduction
to atomic spectra, N. Y., 1934.
В. T. Алексанян, M. А. Елъягиевич.
АТОМНЫЙ ВЕС — среднее значение массы атома
химич. элемента, выраженной в относительных еди-
ницах. У анизотопного элемента, т. е. элемента, пред-
ставленного только одним природным изотопом, все
атомы имеют одинаковую массу, с к-рой и совпадает
А. в. У элемента, состоящего из нескольких природ-
ных изотопов, А. в. определяется изотопными массами,
взятыми в той пропорции, в к-рой эти изотопы состав-
ляют данный элемент. Вариации в природном изо-
топном составе химич. элементов крайне незначи-
тельны (см. Изотопы); поэтому каждый элемент, если
он искусственно не обогащен каким-либо из изотопов,
имеет практически постоянный А. в. В качестве еди-
ницы А. в. в современной химии принята х/1в часть
средней массы атома природного кислорода, к-рый
состоит из изотопов О16, О17 и О18, содержащихся в
отношении 2667 : 1 : 5,5 (для О2 воздуха). А. в элемен-
тов, выраженные в этих единицах, составляют хими-
ческую шкалу А. в. Кроме химической, рас-
пространена также физическая шкала, в
к-рой за единицу А. в. принята 1/1в часть массы изо-
топа О16. Как правило, во всей химич. литературе и,
в частности, в настоящем издании А. в. элементов,
если это специально не оговорено, приводятся по
химич. шкале.
А. в. является одной из самых фундаментальных
характеристик химич. элемента. Он служит основой
для вычисления молекулярных весов химич. соедине-
ний и для всевозможных стехиометрия, расчетов по
химич. формулам и уравнениям; входит в виде кон-
станты во многие уравнения, выражающие связь
между различнымифизико-химич. параметрами. Кроме
того, с А. в. тесно связаны вопросы строения атомов,
а также определяемые этим строением закономерности
в свойствах элементов, характеризуемые периодиче-
ской системой элементов Менделеева. Понятие «А^ в.»
приложимо не только к элементам, но и к отдельным
изотопам. А. в. изотопа тесно связан с его массовым
числом, т. е. количеством нуклонов (протонов и ней-
тронов) в ядре. Весьма важное значение имеет колич.
различие между изотопическим А. в. и соответствую-
щим массовым числом, поскольку такое различие
определяет дефект массы атомного ядра, характери-
зующий энергию связи в нем нуклонов.
Эволюция понятия А. в. Повятие А. в. нан величины,
характеризующей массу атома, вошло в химию вместе с ато-
мистической теорией и было введено Дж. Даль-
тоном в 1803. При этом А. в. элементов определялись, исходя
из тех весовых соотношений, в к-рых элементы образуют химич.
соединения, т. е. за А. в. элементов принимались их экви-
валентные веса. За единицу измерения Дальтоном был выбран
А. в. самого легкого элемента—водорода. В дальнейшем#. Бер-
целиус предложил определять А. в. элементов относительно
А. в-, кислорода, принимая его равным 100, исходя из того, что
кислород образует соединения с гораздо большим числом дру-
гих элементов, чем водород. Однако для установления А. в.
элементов по их весовым соотношениям в соединениях требо-
вались сведения о числе атомов элемента в молекуле соедине-
ния; эти числа выбирались в то время до какой-то степени
произвольно, в результате чего за А. в. часто принимались
значения, отличающиеся от действительных в кратное число
раз. В частности, молекулы всех простых газов, в том числе
водорода и кислорода, служивших эталонами измерения
А. в., считались одноатомными. В работе А. Авогадро (1811)
329
АТОМНЫЙ ВЕС
330
была установлена двухатомность молекул простых газов и
на основе Авогадро закона дан простой способ определения по
плотности газообразного соединения его мол. веса, необходи-
мого для установления истинных значений А. в. элементов,
образующих это соединение.
Однако работа Авогадро не была признана в свое время,
и лишь в 1858 ее результаты были широко использованы
в трудах С. Канниццаро, четко разграничившего понятия
атома и молекулы, а также атомного, молекулярного и экви-
валентного весов. На международном съезде химиков в 1860
результаты этих работ получили всеобщее признание, причем
были приняты единые значения А. в. элементов, за единицу
к-рых вновь был выбран А. в. водорода. В том же 1860 Ж. Ста-
сом была предложена единица измерения А. в., равная 1/ю
части А. в. кислорода, к-рая, с одной стороны, очень близка
к А. в. водорода, а с другой — соответствует более удобному
для измерения А. в. эталону; однако общепринятой эта еди-
ница стала лишь с 1906. В 1869 Д. И. Менделеев, положив
А. в. в основу классификации элементов, открыл периодич.
изменение их свойств по мере возрастания А. в. Исходя из
местоположения элементов в открытой им периодич. системе,
Менделееву удалось не только исправить ранее неверно опре-
деленные значения А. в. ряда элементов (Be, In, U, Се, Th,
La и др.), но й указать также А. в. еще не открытых тогда и им
же предсказанных элементов (Ga, Ge, Sc).
Дальнейшее уточнение понятия А. в. связано с открытием
изотопов сначала для радиоактивных элементов и свинца
(Ф. Содди, 1914), а затем для большого числа других элемен-
тов (Ф. Астон, 1920). После открытия изотопов у кислорода
(В. Джиок и Г. Джонстон, 1929) было объяснено различие
между давно применявшейся химич. шкалой А. в., в к-рой
эталоном служил природный кислород, и физической, введен-
ной в 1927 Ф. Астоном на основе масс-спектрометрич. измере-
ний, в к-рых стандартом оказался лишь один (наиболее рас-
пространенный) изотоп кислорода О16. С открытием изотопии
понятие А. в. как массы атома было перенесено на изотопы,
а А. в. элементов стали определять как средние значения
А. в. их природных изотопов. Однако для радиоактивных эле-
ментов (кроме U и Th — родоначальников естественных радио-
активных рядов) такое определение А. в. элемента неприменимо,
т. к. эти элементы либо вовсе не имеют природных изотопов
(искусств, элементы, получаемые начиная с 1940), либо образу-
ются в каждом из естественных радиоактивных рядов в изотоп-
ных комбинациях и пропорциях, присущих только данному ряду.
У ряда элементов (в особенности у легких Н, Не, С, N, О
и др.) один из изотопов распространен в значительно большей
степени, чем другие, и поэтому для них А. в. весьма близок
к А. в. наиболее распространенного изотопа. Многие элементы
имеют природные изотопы с массовыми числами, большими,
чем для нек-рых природных изотопов последующих элементов.
В этих случаях при условии, если наиболее распространен-
ными являются периферийные изотопы (т. е. с крайними
значениями массовых чисел), А. в. предыдущего элемента
может оказаться больше, чем у последующего. Этим объяс-
няются известные аномалии А. в. в периодич. системе элемен-
тов Менделеева, когда А. в. у Аг, Со и Те, соответственно,
больше, чем у К, Ni и J.
После того как было установлено, что атомное ядро со-
стоит из нейтронов и протонов, была объяснена давно подме-
ченная близость А. в. многих легких элементов к целым числам,
а именно к массовым числам их наиболее распространенных
изотопов. Такая близость обусловлена тем, что на долю ядра
приходится почти вся масса атома и что единица А. в. близка
к массе нуклона. Причина нек-рого отличия А. в. изотопов от
их массовых чисел заключается, во-первых, в уменьшении
массы ядра по сравнению с суммарной массой составляющих
его нуклонов за счет энергии их связи в ядре (дефект массы),
а во-вторых, в нек-ром отличии массы нейтрона от суммарной
массы протона и электрона. Таким образом, А. в. любого изо-
топа равен сумме масс составляющих атомное ядро нейтронов
и протонов, а также окружающих его электронов (число
к-рых равно числу протонов), за вычетом дефекта массы ядра;
при этом все массы берутся в единицах А. в.
Кислород, к-рый взят за эталон измерения А. в., относится
к числу элементов с примерно средней величиной дефекта
массы ядра, приходящегося на один нуклон (см. Дефект мас-
сы). Поэтому кислородная единица А. в. не приводит к боль-
шим отклонениям изотопических А. в. от массовых чисел (эти
отклонения меньше 0,1 единицы).
В случае же выбора за единицу А. в. массы атома Н1, ядро
к-рого состоит из одного нуклона (протона) и поэтому не имеет
дефекта массы, соответствующие отклонения почти для всех
элементов были бы значительно большими (максимальная их
величина составила бы ок. 1,9 единиц).
Методы определения. А. в. определяются физико-
химич. и физич. методами. Физико-химич. методами,
как правило, измеряется непосредственно не А. в.
элемента, а мол. вес в-ва, содержащего данный эле-
мент (см. Молекулярный вес). Для определения А. в.
дополнительно устанавливают при помощи химич.
анализа весовую долю элемента в данном соединении;
при этом А. в. представляет собой соответствующую
часть мол. веся, поделенную на число атомов данного
элемента в‘ молекуле соединения.
Указанное число атомов задается химич. формулой соеди-
нения, к-рая устанавливается, исходя из совокупности всех
химич. сведений о данном соединении и о валентности состав-
ляющих его элементов. В ряде случаев для этой цели исполь-
зуются также нек-рые дополнительные данные. Так, отноше-
ние теплоемкостей газа при постоянном давлении (Ср) и при
постоянном объеме (Cv) указывает на его атомность (т. е.
количество атомов в молекуле); сведения об изоморфизме
кристаллич. тел указывают на сходство химич. формулы дан-
ного соединения с кристаллически изоморфным ему в-вом;
величина теплоемкости простого твердого в-ва приближенно
определяет по Дюлонга и Пти закону А. в. соответствующего
элемента, а приблизительный А. в. дает возможность по дан-
ным химич. анализа соединения и его мол. весу установить
число атомов элемента в молекуле соединения; аналогичным
способом используются приближенные данные об А. в., изве-
стные, исходя из положения элемента в периодич. системе.
К физико-химич. методам определения А. в. относятся также
электрохимия, способы, к-рые позволяют, исходя из Фарадея
законов, по количеству электричества установить А. в. осажда-
емых на электродах элементов при учете значений их валент-
ности.
До 1920 для определения А. в. применяли только
физико-химич. методы; в настоящее время они хотя
и вытесняются более точными физич. методами, но
практически все еще широко используются для уточ-
нения принятых значений А. в. При этом точность
наиболее совершенных физико-химич. методов пре-
вышает 0,01%. Физико-химич. методами определяется
непосредственно А. в. природной изотопной смеси
элемента, используемой для соответств. измерений.
Наиболее важными физич. методами, применяемыми
для определения и уточнения А. в., являются масс-
спектрометрия и метод ядерных реакций. Масс-
спектрометрич. метод позволяет определять с большой
точностью массы отдельных ионов, исходя из измеряе-
мых (по отклонению движения ионов в магнитном и
электрич. полях) значений удельных зарядов, т. е.
отношений зарядов ионов к их массам; при этом изме-
ряемая масса атома находится, исходя из массы иона
за вычетом известных масс остальных атомов, состав-
ляющих ион. В методе ядерных реакций измерение
т. н. Q-величин (т.е. энергий, освобождающихся в про-
цессе ядерных превращений при переходе от одного
атомного ядра к другому — с таким же соотношением
чисел протонов и нейтронов) дает возможность опре-
делять дефекты масс и, следовательно, перейти от мас-
сового числа данного изотопа к его А. в. Физич. ме-
тоды дают значения А. в. отдельных изотопов; для
установления А. в. элемента необходимо дополни-
тельно точно знать его изотопный состав. Данные об
изотопном составе могут быть получены, в частности,
при помощи масс-спектрометрич. метода. Точность
определения А. в. физич. методами часто превышает
0,001%.
Значения А. в. элементов. Постоянные уточнения
А. в. периодически рассматриваются Международной
комиссией по А. в., к-рая каждые 2 года публикует
таблицу А. в. (эти таблицы печатаются в ряде журна-
лов, в СССР— в «Успехах химии»). Кроме того, Между-
народной комиссией по А. в. с 1949 учитываются искус-
ственно создаваемые элементы, для к-рых вместо
А. в. указываются значения массовых чисел наиболее
долгоживущих из полученных изотопов. Массовые
числа наиболее долгоживущих изотопов вводятся
также и для природных радиоактивных элементов
(кроме U и Th). В отличие от А. в., соответствующие
массовые числа в табл. Комиссии приведены в скоб-
ках. Однако, начиная с 1957, Комиссия решила отка-
заться от этой практики на том основании, что харак-
тер информации, сообщаемой посредством массовых
чисел, не имеет отношения к основной задаче таб-
лицы А. в., к-рая состоит в указании точных значений
этих констант для использования их в химич. расче-
тах. С 1957 наряду с таблицей А в. элементов, рас-
положенных в алфавитном порядке, приводится табл,
с расположением элементов по атомным номерам и,
кроме того, отдельная табл, для массовых чисел паи-
332
331
АТОМНЫЙ ВЕС —АТОМНЫЙ НОМЕР
Таблица 1. Атомные веса на 1960 г. (в порядке возрастания ат. номеров)
Поряд- ковый номер Название Сим- вол Атомный вес Поряд- ковый номер Название Сим- вол Атомный вес Поряд- ковый номер Название Сим- вол Атомный вес
1 Водород Н 1,0080 35 Бром Вг 79,916 69 Тулий Ти 168,94
2 Гелий Не 4,003 36 Криптон Кг 83,80 70 Иттербий Yb • 173,04
3 Литий L1 6,940 37 Рубидий Rb 85,48 71 Лютеций Lu 174,99
4 Бериллий Be 9,013 38 Стронций Sr 87,63 72 Гафний Hl 178,50
5 Бор В 10,82 39 Иттрий Y 88,92 73 Тантал Та 180,95
6 Углерод С 12,011 40 Цирконий Zr 91.22 74 Вольфрам w 183,86
7 Азот N 14,008 41 Ниобий Nb 92,91 75 Рений Re 186,22
8 Кислород О 16 42 Молибден Mo 95,95 76 Осмий Os 190,2
9 Фтор F 19,00 43 Технеций Tc 77 Иридий Ir 192,2
10 Неон Ne 20,183 44 Рутений Ku 101.1 78 Платина Pt 195,09
11 Натрий Na 22,991 45 Родий Rh 102,91 79 Золото Au 197.0
12 Магний Mg 24,32 46 Палладий Pd 106,4 80 Ртуть Hg 200,61
13 Алюминий Al 26,98 47 Серебро Ag 107.880 81 Таллий TI 204,39
14 Кремний Si 28,09 48 Кадмий Cd 112,41 82 Свинец Pb 207,21
15 Фосфор P 30,975 49 Индий In 114,82 83 Висмут Bi 209,00
16 Сера S 32,066 50 Олово Sn 118,70 84 Полоний Po
17 Хлор Cl 35,457 51 Сурьма Sb 121.76 85 Астатин At
18 Аргон Ar 39,944 52 Теллур Те 127.61 86 Радон Rn
19 Калий К 39,100 53 Иод J 126,91 87 Франций Fr
20 Кальций Ca 40,08 54 Ксенон Xe 131.30 88 Радий Ra
21 Скандий Sc 44,96 55 Цезий Cs 132.91 89 Актиний Ac
22 Титан Ti 47,90 56 Барий Ba 137.36 90 Торий Th 232,05
23 Ванадий V 50,95 57 Лантан La 138,92 91 Протактиний Pa
24 Хром Cr 52,01 58 Церий Ce 140.13 92 Уран U 238,07
25 Марганец Mn 54,94 59 Празеодим Pr 140,92 93 Нептуний Np
26 Железо Fe 55,85 60 Неодим Nd 144.27 94 Плутоний Pu
27 Кобальт Co 58.94 61 Прометий Pm 95 Америций Am
28 Никель Ni 58.71 62 Самарий Sm 150.35 96 Кюрий Cm
29 Медь Cu 63,54 63 Европий Eu 152.0 97 Беркелий Bk
30 Цинк Zn 65,38 64 Гадолиний Gd 157,26 98 Калифорний Cf
31 Галлий Ga 69,72 65 Тербий Tb 158,93 99 Эйнштейний Es
32 Германий Ge 72,60 66 Диспрозий Dy 162.51 • 100 Фермий Fm
33 Мышьяк As 74,91 67 Гольмий Ho 164.94 101 Менделеевпй Md
34 Селен Se 78,96 68 Эрбий Er 167,27 102
Таблица 2. Радиоактивные элементы, массовые
числа наиболее долгоживущих из известных
(I960 г.) изотопов (в порядке возрастания
ат. номеров)
Порядковый номер Название Символ Изотоп
43 Технеций Тс 99*
61 Прометий Рт 147*
84 Полоний Ро 210*
85 Астатин At 210
86 Радон Rn 222
87 Франций Fr 223
88 Радий Ra 226
89 Актиний Ac 227
90 Торий Th 232
91 Протактиний Pa 231
92 Уран u 238
93 Нептуний Np 237
94 Плутоний Pu 242
95 Америций Am 243
96 Кюрий Cm 247
97 Беркелий Bk 249*
98 Калифорний Cf 251*
99 Эйнштейний Es 254
100 Фермий Fm 253
101 Менделеевий Md 256
102
* Взят изотоп не с наибольшим, а с наиболее
точно известным периодом полураспада.
более долгоживущих изотопов радиоактивных элемен-
тов. Выше приводятся две из указанных таблиц.
Все значения А. в. в таблице 1 даны по химич. шкале.
Поскольку единица А. в. в физич. шкале в 1,000275 раз
меньше соответствующей единицы в химич. шкале,
то все физич. А. в. на 0,0275% выше приведенных
значений.
Единицы измерения А. в. Как уже было отмечено,
для измерения А. в. используются две единицы —
химическая (1/16 А. в. природного кислорода) и физи-
ческая (‘ /те массы изотопа О1в). Каждая из этих еди-
ниц обеспечивает достаточную близость значений
А. в. изотопов всех элементов к их массовым числам.
Химич, единица удобна для измерений А. в. физико-
химич. методами, поскольку в соответствующих изме-
рениях используется кислород в его природном изо-
топном составе. Но природный кислород не может
служить эталоном в физич. измерениях А. в., где
стандартом обязательно должна быть масса конкрет-
ного атома. В таких измерениях используется физич.
единица, удобная тем, что массовое число О16 является
целым, кратным для ряда других чисел (это удобно
при масс-спектрометрич. измерениях), и что соотноше-
ние чисел нейтронов и протонов в О16 (1 : 1) наиболее
распространено для изотопов других элементов (это
важно для использования метода ядерных реакций).
Однако существование двух шкал А. в. создает нек-рые
трудности. Разница между ними значительно превы-
шает точность определения А. в. как физич., так и
физико-химич. методами. Поэтому делаются попытки
создать единую шкалу А. в. При этом важно, чтобы
новая единица не привела к пересмотру огромного
числа табличных данных по атомным и мол. весам,
а также различным мольным величинам, опублико-
ванным в лит-ре.
Для унификации шкал в качестве единицы А. в.
были предложены 1/19 массы анизотопного фтора
(F19) и !/12 массы С12. Использование каждого из
этих эталонов привело бы к незначительным (почти
в пределах ошибок определения) изменениям А. в.
по химич. шкале. Однако F19 непригоден для физич.
измерений А. в. (массовое число некратное, отноше-
ние протонов к нейтронам равно 9/10). Шкала же, ос-
нованная на С12 (переводной коэфф, с химич. шкалы
0,999956), принята на Международном съезде физи-
ков в Оттаве (сент. 1960) и должна быть обсуждена
Международным съездом химиков (1961).
Лит.: Менделеев Д. И., Основы химии т. 1—2, 13
изд., М.—Л., 1947; его же, Избр. Ьоч., т. 2, (Л.), 1934;
Рабинович Е. и Тило Э., Периодическая система
элементов, пер. с нем., М.—Л., 1933; Селинов И. П.,
Усп. физ. наук, 1951, 44, вып. 4, с. 511; У и ч е р с Э., Усп.
хим., 1959, 28, вып. 5, с. 639; Guggenheim Е. A., J.
Chem. Educ., 1961, 38, № 2, 86. С. .9. Вайсберг.
АТОМНЫЙ НОМЕР — CM. Порядковый номер.
333
АТОМНЫЙ ОБЪЕМ — АТРОПОИЗОМЕРИЯ
334
АТОМНЫЙ ОБЪЕМ — объем 1 грамм-атома эле-
мента, равный частному от деления атомного веса на
плотность простого вещества в твердом состоянии.
Если элемент существует в нескольких аллотропия,
модификациях, то А. о. определяют для каждой из
них. По величине А. о. судят о пространстве, занимае-
мом атомом того или иного элемента, т. к. 1 грамм-
атом любого элемента содержит 6,0238 • 1023 атомов.
По атомным объемам входящих в молекулу атомов
судят об объеме молекулы химич. соединения, т. к.
молекулярный объем приблизительно равен сумме
атомных объемов.
АТОМЫ ОТДАЧИ — атомы, получившие опреде-
ленный импульс в процессе радиоактивного распада
или ядернои реакции. Явление аналогично, напр.,
отдаче при выстреле из орудия. Кинетич. энергия,
приобретаемая А. о., может быть вычислена по зако-
нам сохранения энергии и количества движения;
в большинстве случаев она значительно превосходит
энергию химич. связи (2—5 эв). Так, при а-распаде
энергии А. о. имеют величины порядка 102 кэв, при
испускании у-квантов 10—102 кэв. Поэтому А. о.
способны выходить из молекул химич. соединения
(см. Сцилларда — Чалмерса эффект), в к-рых они
первоначально находились, переходить в газовую
фазу из поверхностного слоя твердых тел, производить
в последних радиационные нарушения и т. д. А. о.
являются горячими атомами.
АТОФАН (цинхофен) CieHuNO2, мол. в. 249,27 —
желтоватые мелкие кристаллы горького вкуса, т. пл.
СООН 212—213°; плохо растворим в
I воде, спирте и хлороформе, луч-
ше — в кипящем спирте; легко
1 I 1_ растворяется в разб. р-рах щело-
4/-KN^CeHs
чей и к-т. При прибавлении р-ра
нитрата серебра к р-ру аммоний-
ной соли А. выпадает серебряная соль А. в виде
белого хлопьевидного осадка. Колич. определение
основано на титровании раствора А. в горячем спирте
0,1 н. р-ром NaOH в присутствии фенолфталеина
(1 мл 0,1 н. щелочи соответствует 0,02493 г А.).
Получают А. щелочной конденсацией ацетофенона
с изатином или нагреванием пировиноградной к-ты
с бензилиденанилином (образующимся из анилина
и бензальдегида). А. применяют при подагре; оказы-
вает также болеутоляющее действие.
Лит. см. при ст. Адалин. Е. С. Голотинская.
АТРОПИН C17H23O3N — алкалоид, получаемый из
корней «кавказской мандрагоры» (Scopolia carniolica)
экстракцией растворителями в щелочной (Na2COs или
NH4OH) среде; извлекается также из дурмана и бел-
ладонны; бесцветные кристаллы; т. пл. 115—116°;
сн2—сн—сн2 свнд
I к— сн3\знососн
I I / I
сн2—сн —сн2 сн»он
трудно растворим в воде (1 : 450 при 25°), легко —
в спирте, хлороформе и маслах, хуже — в эфире и
бензоле. Из солеи наиболее важен сульфат А., к-рый
широко применяется в медицине; содержит 1 молекулу
кристаллизационной воды, обезвоживается при на-
гревании до 100°, т. пл. 195—196°; хорошо растворим
в воде (1 : 1), спирте (1 : 3), плохо — в эфире и хло-
роформе; водные р-ры нейтральны. Другие соли А.:
бромгидрат, т. пл. 163—164°; оксалат, т. пл. 198°;
хлороплатинат, т. пл. 207—208°.
А. — эфир спирта тропина и &\-троповой кислоты
(а-фенил-р-оксипропионовой); оптически неактивен.
Левовращающий А., называемый гиосциамином, ши-
роко распространен в растениях и превращается
в А. при нагревании. Синтез А. сводится к получению
атроповой к-ты и далее — тропина. При конденсации
этилового эфира фенилуксусной к-ты Св Н6СН2СООС2Н6
с этилформиатом С2Н6ООСН образуется эфир формил-
фенилуксусной к-ты СвН6СН(СНО)СООС2Н6, к-рый
после восстановления и омыления дает атроповую
к-ту СвН6СН(СН2ОН)СООН. Синтез тропина осу-
ществлен, исходя из диальдегида янтарной к-ты,
метиламина и ацетондикарбоновой к-ты (производст-
венный метод):
СНаСНО Н СНаСООН
I 1 1
+ N—СНз + со -►
СН.СНО II СНаСООН
сн2—сн—-сн3 сн2—с------сн3
-< N-CHsio -> I сн.\знон
11 I I 1 /
СКъ-Ън.----СНа СН2-С-----СНа
тропинон тропин
А. парализует клетки, иннервируемые парасимпатич.
нервами, вызывает расширение зрачка, прекращает
потоотделение, учащает пульс, прекращает спазмы
кишечника. При отравлении А. наблюдается сильное
возбуждение, затем — угнетение. Применяется в ме-
дицине как мидриатич. и спазмолитич. средство.
Гиосциамин в 2 раза активнее А.
Лит. см. при ст. Алкалоиды. А. Е. Васильев.
АТРОПОИЗОМЕРИЯ — вид оптич. изомерии, воз-
можной при отсутствии асимметрии, атомов С и об-
условленной асимметрией молекулы из-за затруднен-
ности свободного вращения. Так, дифенил СвН6—СвН6
обладает свободным вращением вокруг общей оси,
т. е. оба плоских бензольных кольца могут зани-
мать любые положения между расположением в одной
плоскости (I) и во взаимно перпендикулярных плоско-
стях (И). С другой стороны, о, о-тризамещенные и
тетразамещенные дифенила, напр. динитродифеновую
кислоту (III), можно кристаллизацией солей с опти-
чески деятельными основаниями расщепить на оптич.
антиподы. В этом случае из-за пространственных эф-
фектов заместители расположены в двух плоскостях,
плоскость симметрии отсутствует и возможны две
пространственные структуры, относящиеся друг к
другу, как предмет к своему зеркальному изображе-
нию. Представления о причинах А. могут быть хорошо
иллюстрированы моделями, учитывающими межатом-
ные расстояния в молекуле. А. установлена в слу-
чае производных дифенила, динафтила, диантрила,
дипиррила и др.
С=С(СН3)2
N + (CH3)3J
VII
При достаточно больших размерах заместителя А.
возможна и при наличии двух и даже одного орто-
заместителя, напр. в случае соединений (IV) и (V).
335
АУКСИНЫ — АУТОЛИЗ
336
Удалось, кроме того, расщепить на оптич. антиподы
нек-рые nepu-производные нафталина, напр. соеди-
нение (VI). Известны и случаи отсутствия свободного
вращения у соединений с одним кольцом. Так, о-
(р,В'-диметил-а-изопропилвинил) фенилтриметиламмо-
ниииодид (VII) расщеплен на оптич. антиподы, к-рые
и при 100° рацемизируются с трудом.
Я. Ф. Комиссаров.
АУКСИНЫ — стимуляторы роста растений, обладаю-
щие высокой физиология, активностью; способны рас-
пределяться по растению вместе с питательными со-
ками. Известны А. «а» (I), «б» (II) и гетероауксины.
А. «а» С18Нз2О5, мол. в. 328,45 — бесцветные кри-
начальную концентрацию исходного в-ва; х$ — на-
чальная концентрация продукта, вызывающего А. р.
(«затравка»реакции), обычно <^1. В ур-нии (’^мно-
житель (х + #0) отражает ускоряющее действие про-
дукта-катализатора, а (1—х) передает воздействие
концентрации исходного в-ва на скорость реакции.
Кинетика накопления х во времени описывается фор-
„ 1 — е'т
мулои: х = xQ-----—, т = k(i + я0)г и показана на
ХО 4- е г
рис. 2 кривой 1. Изменение скорости реакции в за-
висимости от х изображено на рис. 1 кривой 7; макси-
мум скорости достигается при х х/2. Это уравнение
описывает кинетику омыления сложных эфиров (в от-
Н5С2СН-
сн3
сн3
(!:нс2н5
СНСН2^Н СНСООН
ОН ОН ОН
Н5С2СН-
сн3
СН3
5
сталлы; т. пл. 196°; хорошо растворим в метиловом
и этиловом спиртах и этилацетате, хуже — в эфире
и бензоле; в петролейном эфире и лигроине почти
нерастворим; в воде на холоду дает 1%-ный р-р;
оптически активен, р-р А. «а» в 96%-ном спирте пока-
зывает левое вращение; по отношению к к-там устой-
чив, щелочью быстро разлагается. А. «б» С18Н30О4,
мол. в. 310,44 — кристаллич. продукт, т. пл. 183°,
кристаллизуется из смеси абс. этилового спирта с лиг-
роином; р-р в 96%-ном спирте физиологически и опти-
чески активен; по отношению к к-там и щелочам
неустойчив; при обработке к-тами и щелочами теряет
физиология, активность. А. выделены из различных
растений (кукурузные початки и др.); синтетически
не получены.
В последние годы возможность выделения А. из растений
рядом исследователей ставится под сомнение.
Лит. см. при ст. Гетероауксины. Н. Н. Мельников.
АУРЕОМИЦИН — см. Хлортетрациклин.
АУТОКАТАЛИТИЧЕСКИЕ РЕАКЦИИ — химич.
реакции, катализируемые продуктами самих реак-
ций. Ускорение А. р. происходит, как правило, в на-
чале процесса. В даль-
нейшем оно прекра-
щается или из-за убы-
ли исходного веще-
ства, или вследствие
расходования аутока-
тализатора. Скорость
А. р. по мере ее раз-
вития проходит через
максимум (рис. 1), а
кинетика накопления
продуктов имеет S-об-
разный вид (рис. 2),
точка перегиба на
кинетич. кривой со-
ответствует максиму-
му скорости. Классич.
примером А. р. может
служить разложение
третичного амилаце-
тата, когда образующаяся уксусная к-та катализирует
дальнейшее протекание реакции: СН3СООС5Н,, +
-j- Н2О = СН3СООН + С5НЦОН. Кинетика А. р.,
в к-рой катализатором является один из конечных
продуктов, описывается одним из след, трех ур-ний:
1) ~ = k (х 4- х0) (1 — X) ...
(А. р. первого порядка), где х — относительная кон-
центрация продукта реакции в момент времени Z;
к — константа скорости реакции, умноженная на
сутствие сильных к-т или осно-
ваний), взаимодействие бромно-
Ватой и мышьяковистой кис-
лот, окисление формальдегида
СН СН2-СО-СН2СООН в газовой фазе и ряд других
I реакций.
С) Н д—.
2) jj- = k (х + х0) (1 — х)2
Скорость реакции как функция от х показана на
рис. 1 кривой 2, кинетика накопления продукта
реакции — на рис. 2 кривой 2. Максимум скорости и
точка перегиба на кинетич. кривой 2 (рис. 2) дости-
гаются при х ~ 1/3. Это ур-ние описывает, в частности,
кинетику окисления водорода с учетом ускоряющего
влияния паров воды.
gy = k (x-J- х0)2(1 — х)
(А. р. второго порядка см. кривую 3 на рис. 1 и кри-
вую 3 на рис. 2). Максимум скорости достигается при
73-
Далеко не все реакции, протекающие
Ускорение могут
3)
с ускоре-
вызывать
Рис. 1. Скорость А. р. как функция
относительной концентрации про-
дукта реакции х. Кривая 1 соот-
ветствует ур-нию 1, кривая 2—2,
кривая 3—3, kt.
нием, являются А. р.
и другие причины, та-
кие как саморазогрева-
ние реагирующей си-
стемы, когда выделяю-
щаяся теплота не успе-
вает рассеяться (тепло-
вой взрыв), израсходо-
вание ингибитора, при-
сутствующего в исход-
ном веществе, и т. д. По-
этому аутокатализ сле-
дует считать доказан-
ным только тогда, когда
спец, опытами устано-
влено ускоряющее дей-
ствие по крайней мере
одного из продуктов
реакции. Частным слу-
чаем аутокатализа яв-
ляется самоиндукция.
А. р. встречаются как
среди гомогенных, так
и среди гетерогенных
каталитич. реакций; одни А. р. протекают по ионно-
му механизму, другие — по цепному радикальному,
третьи — через образование промежуточных моле-
кулярных соединений между исходным в-вом и про-
дуктом-катализатором. Поэтому полученное эмпи-
рически ур-ние аутокатализа само по себе недоста-
точно для выяснения механизма изучаемой реакции,
однако вместе с другими данными оно помогает
установить этот механизм.
Лит.: Кондратьев В. Н., Кинетика химических
газовых реакций, М., 1958. Е. Т. Денисов.
АУТОЛИЗ (автолиз) — процесс самопереварива-
ния и растворения тканей, при к-ром происходит
распад сложных веществ (белков, углеводов, жиров^
Рис. 2. Кинетические кривые
накопления продукта реакции
в А. р. различных типов, х —
относительная концентрация
продукта реакции, т — nt,
„х0 — 0,01. Пунктиром показаны
точки перегиба на этих кривых.
Кривая 1 соответствует ур-нию
337
АУТООКСИДАЦИЯ — АЦЕТАЛИ И КЕТАЛИ
338
организма, катализируемый содержащимися в тка-
нях гидролитич. ферментами. Наиболее интенсивно
А. протекает в клетках железистых органов (под-
желудочная железа, слизистая желудка и кишеч-
ника, селезенка, почки, печень и др.). А. лежит
в основе изменения тканей при разложении трупов,
а также в различных очагах омертвения живых
организмов, напр. при прекраще-
нии притока крови к ткани, при
отмирании нек-рых тканей в ра-
стениях и др. Процессы А. проте-
кают и при нек-рых патология.
состояниях (всякого рода атрофии,
рассасывание опухолей и Др.). А.
наблюдаются также в ряде произ-
водственных процессов (созрева-
ние теста, ферментация табака,
чая, силосование кормов и др.
Показано, что распад тканевых
белков связан с дыханием; тор-
можение дыхания приводит к снижению гидролити-
веского распада белков. м. п. Черников.
АУТООКСИДАЦИЯ (самоокисление) — самопроиз-
вольное окисление в-в молекулярным кислородом
при невысоких темп-pax. В конце 19 в. было уста-
новлено, что нек-рые в-ва (альдегиды, непредельные
углеводороды) могут окисляться кислородом воздуха
при обычной темп-ре, в то время как большинство
в-в требует для этого или нагревания, или участия
катализаторов. Согласно перекисной теории Баха—
Энглера, А. протекает путем присоединения к окис-
ляющемуся в-ву RH целой молекулы О3 с образова-
нием перекиси в качестве первичного продукта окис-
ления. Представления А. Н. Баха получили даль-
нейшее развитие в цепной теории процессов окисле-
ния. Установлено, что А. органич. и нек-рых неор-
ганич. соединений (напр., сульфита натрия) проте-
кает по цепному механизму (см. Цепные реакции).
Иногда А. протекает как сопряженная реакция
(см. Индукция химическая)', напр., индиго само по
себе не окисляется кислородом воздуха, но окис-
ляется совместно с бензальдегидом. Окисление бен-
зальдегида т. о. индуцирует окисление индиго; бен-
зальдегид (индуктор) в такой сопряженной реакции
наз. аутооксидатором. Непредельные углеводороды
индуцируют А. парафиновых углеводородов. Следует
отметить, что деление процессов окисления на А.
и окисление при более высокой темп-ре и в присут-
ствии катализаторов, по современным представле-
ниям, весьма условно.
Во многих практич. случаях А. нежелательна,
т. к. приводит к порче пищевых продуктов, осмоле-
нию минеральных масел и крекинг-бензинов, старе-
нию каучука. Для предохранения в-в от А. доба-
вляются антиокислители. Особенно велика роль А.
в биохимических процессах (см., например, Обмен ве-
ществ).
Лит.: Иванов К. И., Промежуточные продукты
и промежуточные реакции автоокисления углеводородов,
М.—Л., 1949; Кнорре Д. Г. (и др.), Успехи химии, 1957,
26, вып. 4, 416—58. Е. Т. Денисов.
АФИЛЛИН (16-оксоспартеин) Cl8H24ON2, мол. в.
238,29 — алкалоид, выделенный из высококипящей
фракции суммы алкалоидов ежовника безлистного
о (Anabasis aphilla), семейства ма-
1вУ ревых (Chenopodiaceae) фракцио-
пированием по силе основности
I СН2 I (дробное осаждение хлоргидратов
\S щелочью). А. — трудно кристал-
лизующееся в-во, т. пл. 52—53°;
т. кип. 200°/4.«.«; [а]|у=4- 10,3°(водный СН3ОН);хоро-
шо растворим в органич. растворителях и трудно —
в воде и холодном петролейном эфире. Важнейшие
соли и производные А.: хлоргидрат, т. пл. 209—
210°; пикролонат, т. пл. 230—231° (с разл.); иодме-
тилат, т. пл. 212—213° (с разл.). А. проявляет свой-
ства однокислотного третичного основания; обладает
скелетом спартеина; от сопутствующего афиллидина
отличается отсутствием двойной связи С,5) — С,в).
Синтезируют А., исходя из этилового эфира а-пири-
дилуксусной к-ты:
ЯС2Н8О)3СН
H2/Pt
а) Н Вс
б) K2CO3.t
(СН3СО)2О
СН2ОН
афилпин
Хлоргидрат А. является местным анестетиком, по
силе и продолжительности действия не уступающим
новокаину.
Лит.: Садыков А. С., Химия алкалоидов Anabasis
aphylla, Ташкент, 1956; см. также при ст. Алкалоиды.
А. Е. Васильев. ~
АЦЕНАФТЕН С12Н10, мол. в. 154,20 — бесцветные
кристаллы, иглы (из спирта); т. пл. 96°, т. кип.
279°, d? 0,8314; п$а 1,6048; в 100 г
растворяется: в спирте — 4 г (20°), 40 г Н29 9На
(70°), в толуоле — 24 г (20°), 74 г (60°).
В зависимости от условий окисления [ | |
А. образует аценафтилен (I), ангидрид
нафталевой к-ты (II) или аценафтехи-
нон (III); бромируется и нитруется до 5-бром- и
5-нитроаценафтена соответственно; при нагревании
с калием дает аценафтенкалий. А. получают раз-
гонкой соответств. фракции кам.-уг. смолы; может
быть получен также бромированием а-этилнафталина
с последующей обработкой КОН и взаимодействием
нафталина с этиленом при высоких темп-pax. А.
используется для получения нафталевой к-ты и
аценафтохинона, применяемых в производстве кра-
сителей. Для колич. определения А. в кам.-уг. ма-
слах применяется «нафталевый» метод — окисле-
ние А. с помощью Na2Cr2O7 в ангидрид нафталевой
к-ты. А. в смеси с флуореном и дифениленоксидом
определяется «пикратным» методом, в к-ром исполь-
зуется образование устойчивого аддукта А. с пикри-
новой К-ТОЙ, T. ПЛ. 162°. р в Журин
АЦЕТАЛИ И КЕТАЛИ (1,1-диалкоксиалканы) —
простые эфиры а,а-гликолей, эфиры гидратирован-
ных альдегидов (ацетали) или кетонов (кетали) —
RR'C(OR")(OR"'). Ацетали могут быть симметрич-
ные (R" = R'") и несимметричные, смешанные (ра-
дикалы R" и R"1 разные). Циклические ацетали
альдегидов с 1,2-гликолями наз. диоксолан а-
м и. Ацетали — бесцветные жидкости с приятным
запахом, растворимы в органич. растворителях и
практически нерастворимы в воде. А. и к. в неболь-
шом количестве присутствуют в виноградных винах,
улучшая их букет.
Главные способы получения: 1) нагревание спиртов
с альдегидами в присутствии FeCl3 или минераль-
ной к-ты: СН3СНО + 2С2Н5ОН->СН3СН(ОС2Н5)2+
Д- 2Н2О (кетали этим способом получить не удается);
339
АЦЕТАЛЬДЕГИД — АЦЕТАНИЛИД
340
Название Т.кип., °C "D 4
Метилаль, формаль СН3(ОСН3)3 . . . Ацеталь, этилаль, ацетол, диэтилфор- маль СН3 (ОС3Н5)3 42,3 1,3504 (25°) 0,8608 (20°)
87,5 1,3748 (17,5°) 0,8319 (20°)
Диэтилацеталь СН3СН(ОС3Н5)3 .... Диэтиловый ацеталь акролеина 103,6 1,3893 (20°) 0,8254 (20°)
СН2 = СНСН (ОС3Н5)2 123,5 — 0,8543 (15°)
Ацеталь кетона СНа = С(ОС3Н5)3 . . 124—126 1,4101 (25°) 0,8778 (25°)
2) взаимодействие альдегида или кетона с ортому-
равьиным эфиром в присутствии минеральной к-ты:
(СНз) со 4- СН (ОСоН5)з -. (СН3)а С (ОС2Н5)2 -f- НООСС2Н5
3) присоединение спиртов к виниловым эфирам:
СН» = СНОС4Н8 + С3Н5ОН — СН3СН (ОС4Н8) (ОС3Н5)
4) реакцией Гриньяра:
C0H5MgBr + СН (ОС3Н5)а - С6Н5СН (OC3HS)3 -f- C2H5OMgBr
Ацеталь ацетальдегида в пром-сти получают из
ацетилена:
СН = СН 4- 2С3Н5ОН SEj СН3СН (ОС3Н5)3
А. и к. быстро гидролизуются, соответственно, до
альдегидов и кетонов разбавленными к-тами при
обычной темп-ре или кипящей водой. Щелочи их не
омыляют. Ацетали легко галогенируются:
СН3СН(ОС3Н5)3 “А СН3С1СН(ОС3Н5)2
- СНС13СН(ОС»Н5)3 ССЦСН (ОС3Н5)з
Симметрия, ацетали при нагревании с Р2О5 образуют
виниловые эфиры:
СНдСН (ОС2Н5)2 — С»Н5ОН 4- СН3 = СНОС2Н5
Алкиларилацетали при нагревании расщепляются
с выделением фенолов:
СН3СН (ОС2Н5) (ОС6Н5) — сн2 = снос2н5 4- с6н5он
Смешанные ацетали при нагревании диспропорцио-
нируются:
2СН3СН (ОС3Н5)(ОС4Н8) -
— СН3СН(ОС3Н5)2 4" СН3СН(ОС4Н9)2
Алкилацетали способны к реакциям алкоголиза:
СН3СН(ОС3Н5)3 4-2CH3OHji СН3СН(ОСН3)3 4-2С3Н5ОН
А. и к. широко применяют в парфюмерии. Ацеталь
салицилового альдегида — промежуточный продукт
в синтезе красителей. Ацетали полиальдегидов ис-
пользуются как пластификаторы. Из ацеталя акро-
леина получается глицериновый альдегид. Метилаль
используется как растворитель.
Лит.: Chemistry of carbon compounds, ed. by E. H. Rodd,
v. 1, pt A, Amst. [a. o.], 1951. Ф. К. Величко.
АЦЕТАЛЬДЕГИД (уксусный альдегид, этаналь)
СН3СНО, мол. в. 44,05 — бесцветная жидкость с рез-
ким удушливым запахом, т. пл. —124,0°; т. кип.
20,8°; 0,783; Пд 1,3316; уд. теплоемкость
(жидк.) 0,522 кал/г (0°); Cp/Cv= 1,145 (30°); тепло-
та испарения 6035 ккал/молъ', теплота сгорания
(жидк.) 278,4 ккал/молъ', гкрит 188°; ркрит 63,2 ат\
т. воспл. —35°. Смеси А. с воздухом, содержащие 3,97—
57 об. % А., взрывают при 380—400°; смеси с кислоро-
дом — при 140—143°; с водой,
1_1Г^-О\Г|_|Г|_. спиртом и эфиром смешивается
сн3нс снсн3 во всех отношениях Для А.
I I характерны все реакции альде-
О\ /С* гидов. В присутствии мине-
*1 ральных кислот А. легко по-
q_| димеризуется в жидкий п а-
3 р а л ь д е г и д (т. кип. 124°,
т. пл. 12,6°) и кристаллический метальдегид,
состоящий, по-видимому, из четырех молекул А.
При нагревании обоих полимеров в присутствии
серной к-ты происходит их полная деполимеризация.
Наиболее распространенные методы получения А.:
1) гидратация ацетилена в присутствии солей ртути
при 95° (см. Кучерова реакция):
сн=сн 4- Hgso4 — сн=сн 212
iig~so4
— HgSO4 4- CH2=CHOH Z2 CHaCHO
Равновесие этой реакции сильно сдвинуто вправо.
Процесс проводят непрерывным методом в аппаратах
колонного типа. 2) Окисление этилового спирта в па-
ровой фазе воздухом при 400—550° над металлич.
катализаторами, напр. серебром; 3) дегидрогениза-
ция этилового спирта при 250—400° над медно-цинко-
вым или медно-хромовым катализатором; 4) присоеди-
нение уксусной к-ты к ацетилену и разложение
обр азовавшегося этилидендиацетата:
сннсн 4- 2сн3соон — сн3сн(ососн3)3 —
— СНдСНО 4-(СН3СО)3О
А. можно получить также непрямой гидратацией
ацетилена через виниловые эфиры:
ROH 4- сн=сн к-°5 ROCH=CH3
ROCH=CH2 4- Н2О 5^121 СН3СНО 4- ROH
Известны другие способы получения А., напр. гид-
ратация ацетилена водяными парами над фосфорно-
кислыми или хлористыми солями меди при 250—300°
и др. А. применяют для получения уксусной к-ты,
бутадиена, ацетальдоля, ацеталя СН3СН(0С2Н5)2,
пентаэритрита, альдегидных синтетич. смол и т. д.
Лит.: Вайсбергер А. [и др.], Органические раст-
ворители, пер. с англ., М., 1958, с. 125; Казарновский
С. Н., Кузнецов Л. А., Синтетическая уксусная кислота,
М.—Л., 1946; Copenhaver J. W. and Bigelow
И. Н., Acetylene and carbon monoxide chemistry, N. Y., 1949.
АЦЕТАЛЬФОСФАТИДЫ — см. Фосфатиды.
АЦЕТАМИД (авдид уксусной кислоты) CH3C0NH2,
мол. в. 59,07 — бесцветные гексагональной формы
кристаллы, в чистом состоянии без запаха; т. пл.
82—83°; расплавленный А. затвердевает при 48,5°,
образуя лабильную ромбич. модификацию; т. кип.
221,2°; rff» 1,159; ЙГОСН = 3,l-10~i5 (25°); легко
растворим в воде, спирте (25,0 г в 100 г при 20°),
глицерине, почти нерастворим в эфире. А.—прак-
тически нейтральное соединение; образует соли
только с сильными минеральными к-тами, например
(CH3CONH3)+C1~, к-рые легко гидролизуются; водород
ГШ2-группы может быть замещен металлом, напр.
(CH3CONH)2Hg; при нагревании с Р2О5 и др. от-
щепляют воду с образованием ацетонитрила CH3CN.
С р-ром бензохинона А. дает красное окрашивание.
Получают А. нагреванием до кипения ацетата аммо-
ния в ледяной уксусной к-те: CH3C00NH4 —►
—> CH3C0NH2 + Н20. А. можно получить также взаи-
модействием уксусного ангидрида или хлористого
ацетила с аммиаком, напр.: (СН3СО)20 + 2NH3 —>
—> CH3C0NH2 + CH3C00NH4; А. применяют как
растворитель для многих органич. соединений, как
в-во, обладающее гигроскопичными свойствами. А.
используют в произ-ве кожи, бумаги и др.; ртутную
соль (CH3C0NH)2 Hg применяют для протравливания
зерна. М. Алейникова.
АЦЕТАНИЛИД (антифебрин) CeH5NHCOCH3,
мол. в. 135,16 — бесцветные блестящие листочки (из
воды) или ромбич. пластинки; т. кип. 303,8°, т. пл.
114,2°; 1,0261; уд. теплоемкость 0,3391 кал/г град.
В 100 г воды растворяется 0,5 г А. при 20° и 18 г
при 100°; хорошо растворяется в эфире, спирте, хло-
роформе, ацетоне, анилине, плохо — в ксилоле,
341
АЦЕТАТНОЕ ВОЛОКНО —АЦЕТАТЫ
342
бензоле, нерастворим в кислотах и водных р-рах
щелочей. При действии металлич. Na на горячий
р-р А. в ксилоле связанный с азотом атом водорода
замещается атомом металла. При действии газообраз-
ного НС1 в метиловом спирте А. образует два ряда
солей: 2 (C6H5NHCOCH3) • НС1 и C6H5NHCOCH3- НС1.
При ацетилировании А. образуется ^^диацетил-
анилин. А. галогенируется и нитруется в положение
2 и 4 бензольного ядра; алкилируется галогеналкила-
ми с образованием N-алкилацетанилидов. При нагре-
вании с к-тами и щелочами А. разлагается с образо-
ванием анилина и уксусной к-ты, на чем основано
его качеств, открытие. Для колич. определения на-
веску А. кипятят с соляной к-той и титруют в при-
сутствии KJ 0,1н. р-ром КВгО3 до слабожелтого
окрашивании. А. получают взаимодействием анилина
с ацетилирующими агентами (уксусная к-та; уксус-
ный ангидрид и др). А. применяют в качестве жаро-
понижающего и болеутоляющего средства в меди-
цине, полупродукта при синтезе красителей и ле-
карств (в частности, сульфамидных препаратов) и
стабилизатора Н2О2.
Лит. см. при ст. Акрихин. А. И. Травин.
АЦЕТАТНОЕ ВОЛОКНО — искусственное волокно,
получаемое из ацетилцеллюлозы. Существует два
типа А. в.: первый получают из частично омыленной
триацетилцеллюлозы, т. наз. вторичной ацетилцел-
люлозы (обычное А. в.), второй — из первичной
триацетилцеллюлозы (триацетатное волокно). А. в.,
получаемое из вторичной ацетилцеллюлозы, содер-
жит 17—22% гидроксильных и 78—83% ацетильных
групп от теоретически возможного числа этих групп
в целлюлозе. Поэтому такое А. в. обладает нек-рой
гидрофильностью и поглощает ок. 6,5% влаги (при
комнатной темп-ре и влажности воздуха 65%). Во-
локно, получаемое из триацетилцеллюлозы, содержит
ок. 97—98%, а в ряде случаев 100% теоретически
возможного числа ацетильных групп. Такое волокно
гидрофобно и при обычных условиях поглощает не
более 1—1,5% влаги. До недавнего времени в широком
масштабе производилось только А. в. из вторичной
ацетилцеллюлозы, применяемое для различных тек-
стильных изделий. Триацетатное волокно изготовля-
лось в небольшом количестве для специальных тех-
нич. изделий и для слаботочной пром-сти, т. к. по
своим электроизоляц. свойствам оно превосходит
все известные природные и искусственные волокна,
в том числе и натуральный шелк. Гидрофобность
триацетатного волокна, к-рая мешала его приме-
нению для изготовления одежды, устраняют спе-
циальной обработкой готового волокна, придающей
ему нек-рые гидрофильные свойства. Кроме того,
изготовляют воздухо- и влагопроницаемые трикотаж-
ные изделия и ткани из гидрофобных волокон, без-
вредные в гигиенич. отношении. Благодаря этому
расширилась область применения триацетатного во-
локна. Основные физико-механич. характеристики
А. в. (элементарные нити): плотн. 1,30—1,35 г/см3,
предел прочности 14—18 кг!мм2, разрывное удлине-
ние (в % к первоначальной длине) 18—25. Разрывная
длина обычного А. в. составляет 11 —14 км. Дли-
тельным вытягиванием сформованного волокна при
повышенной темп-ре можно повысить разрывную длину
до 22—25 км (упрочненное А. в.). Величина эластич.
удлинения у А. в. примерно в 2 раза выше, чем у гид-
ратцеллюлозных волокон. Разрывная прочность обыч-
ного А. в. в мокром состоянии снижается на 40—45%,
а триацетатного на 15%. А. в. термопластично
и выше 140—150° начинает деформироваться. А. в.
мало устойчиво к действию органич. полярных ра-
створителей (ацетон, сложные эфиры), в к-рых оно
сильно набухает и растворяется. Обычное А. в. по
своим эластич. свойствам и гидрофильности занимает
среднее положение между гидрофильными (хлопок,
вискозное волокно) и гидрофобными волокнами,
к к-рым принадлежат почти все синтетич. волокна,
в том числе капрон, хлорин, нитрон и др. Эксплуатац.
свойства изделий из А. в. во многих случаях выше,
чем у изделий из других видов искусств, волокна.
Хорошие эластич. свойства, умеренная гидрофиль-
ность, мягкость, красивый внешний вид обусловли-
вают широкое применение обычного А. в для изго-
товления тонких трикотажных изделий и шелковых
тканей. Произ-во А. в. до 1957 бурно развивалось
благодаря дешевизне исходного сырья, вытесняя
частично вискозное волокно и хлопок. В дальнейшем
развитие произ-ва А. в. замедлилось в связи с по-
явлением новых ценных типов синтетич. волокон.
Ацетатное штапельное волокно применяется для
частичной замены шерсти при изготовлении тонких
сукон и нек-рых трикотажных изделий. Применение
А. в. снижает сминаемость получаемых изделий.
А. в. (обычное и триацетатное) производится двумя
способами: сухим и мокрым. В обоих случаях аце-
тилцеллюлозу предварительно растворяют в подхо-
дящем органич. растворителе (вторичную ацетил-
целлюлозу — в ацетоне, триацетилцеллюлозу —
в смеси метиленхлорида и этилового спирта). Вязкий
прядильный р-р фильтруют, освобождают от возду-
ха и формуют через тонкие отверстия фильер. Для
обычного А. в. чаще применяют сухой способ формо-
вания, т. е. формуют струйки, выходящие из фильеры,
в среде горячего воздуха в прядильной шахте. Полу-
чаемые тонкие волоконца собирают в нить, к-рую
наматывают на бобину; после крутки нить пригодна
для текстильной переработки. Струи прядильного
раствора триацетатного волокна формуют в боль-
шинстве случаев по сухому способу, а иногда и по
мокрому, пропуская р-р в осадительную ванну
(с р-ром метилового спирта, уксусно-кислых солей
и др.).
Переработка обычного А. в. мало отличается от
текстильной переработки природных или вискозных
волокон. Триацетатное волокно перерабатывают при
повышенной влажности воздуха после предваритель-
ной обработки антистатич. препаратами, снижающими
его Электризуемость. Крашение А. в. производят спе-
циальными дисперсными (ацетатными) красителями.
(См. Крашение искусственных волокон).
Лит.: Литвин О. Б., Ацетилцеллюлоза и ацетатный
шелк, М., 1937; Роговин 3. А., Основы химии и техноло-
гии производства химических волокон, 2 изд., М., 1957.
А. Б. Пакгивер.
АЦЕТАТЫ — соли и эфиры уксусной к-ты; ниже
рассмотрены только соли. Щелочные и щелочноземель-
ные металлы образуют с уксусной к-той средние соли,
металлы большей валентности образуют обычно
основные соли. А. легко растворимы в воде, за исклю-
чением ацетата серебра и нек-рых основных солей,
напр. Fe111, Си11, Ti, Zr, Bi. Растворимость нек-рых А.
в воде приведена в таблице:
Соль Раствори- мость, г/100 г Н2О Соль Р астворимость г/100 г Н3О
СН3СООК CH3COONa CH3COOAg 228 (14°) 123,5 (20°); 170 (100°) 0,72 (0°); 2,52 (80°) (СН3СОО)2Си (СН3СОО)2Са (CH3COO)sHg (СН3СОО)2РЬ 7,2 (0°); 20 (100°) 37,4 (0°) 36,4 (19°); 100 (100°) 56 (25°)
А. получают растворением окисей, гидроокисей или
карбонатов металлов в разбавленной уксусной к-те,
обменной реакцией уксуснокислых солей с сульфатами
нек-рых металлов, а также действием СО2 на метил-
магнийгалогениды. При электролизе А. ион СН3СОО~
распадается на аноде на СО2 и алкил, нто доказано
343
АЦЕТИЛ ХЛОРИСТЫЙ - АЦЕТИЛАЦЕТОН
344
с помощью изотопного метода; из двух алкилов об-
разуется углеводород парафинового ряда (Кольбе
реакция). При сплавлении А. с едкими щелочами
получаются соответствующий карбонат и метан; при
нагревании до 300—400° ацетаты Li, Na, К, Mg,
Са, Ва, РЬ и Мп образуют ацетон. При нагревании
с водой А. тяжелых металлов сполна гидролизуются.
Чувствительной, хотя и не вполне специфичной каче-
ственной реакцией на А. служит образование окиси
какодила при нагревании смеси сухих А. с мышьяко-
вистым ангидридом; 4CH3COONa+As2O3—>2Na2CO34-
4- [(CH3)2As]2O + 2СО2. Окись какодила обнаружи-
вается по отвратительному своеобразному запаху.
Ацетаты Al, Fe, Сг применяются при протравном
крашении текстильных материалов; А. аммония, Na,
Zn, Mg, Al, Pb (средняя соль — «свинцовый сахар»,
основная соль — «свинцовый уксус») — в медицине;
А. Си — в гальванотехнике, а в смеси с мышьякови-
стокислой медью — для борьбы с нек-рыми вреди-
телями растений. Соли жирных кислот, в частности А.,
используются в Перкина реакции, являющейся одним
из главных методов синтеза кумарина, гетероциклич.
и ароматич. карбоновых кислот, напр. коричной:
С6Н5СНО + CHjCOONa -> С6Н5СН = CH — COONa-b
+ Н2О. Многие А. применяются для приготовления
катализаторов, напр.: А. кобальта — в оксосинтезе,
марганца — для окисления ацетальдегида в уксус-
ную к-ту, цинка — в синтезе метанола и др.
Т. И. Соркина.
АЦЕТИЛ ХЛОРИСТЫЙ (хлорангидрид уксусной
кислоты) СН3СОС1, мол. в. 78,54 — бесцветная, ды-
мящая на воздухе жидкость с резким запахом; т. пл.
—112°; т. кип. 51,8°; 1,1051; п% 1,3897. А. х. нера-
створим в воде и быстро ею гидролизуется; растворим
в ацетоне, эфире, хлороформе, бензоле, толуоле,
сероуглероде. С первичными и вторичными спиртами
А. х. образует сложные эфиры; с третичными —
алкилгалогениды: СН3СОС1 4* R3COH —> R3CC1 4*
+ СН3СООН; с формальдегидом в присутствии ZnCI2
образует хлорметилацетат; СН3СОС1 4* СН2О —>
—> СН3СООСН2С1. А. х. ацетилирует первичные и
вторичные амины; с третичными дает продукты при-
соединения. Получают А. х. взаимодействием уксус-
ной к-ты (или уксусного ангидрида) с фосгеном
в присутствии MgCl2 или СаС12; процесс ведут в жид-
кой или паровой фазе при повышенной темп-ре. А. х.
получают также действием НС1 на уксусный ангид-
рид; действием РС15, РС13, РОС13 на уксусную к-ту
и др. способами. Применяют А. х. гл. обр. для аце-
тилирования в произ-ве синтетич. красителей, многих
лекарств, веществ и др. т. И. Соркина.
АЦЕТИЛА ГИДРОПЕРЕКИСЬ (надуксусная кис-
лота) СН3СОООН, мол. в. 76,05 — бесцветная жид-
кость с резким запахом; т. пл. 0,1°; т. кип. 105°,
25°/21 мм; dp 1,226; легко растворима в воде и обыч-
ных органич. растворителях; крайне взрыв-
чата. Обычно А. г. не выделяют в чистом виде,
а применяют водные р-ры различной концентрации.
При комнатной темп-ре в отсутствие катализаторов
концентрированные р-ры А. г. не изменяются в те-
чение нескольких недель. Чистая А. г. может быть
приготовлена вымораживанием и центрифугирова-
нием 95%-ного водного р-ра; возможно получение
безводных р-ров А. г. в инертных растворителях.
А. г. получается гидролизом ацетилбензоилперекиси
или перекиси ацетила, обработкой ее щелочами,
реакцией между перекисью ацетила или уксусного
ангидрида с Н2О2 и др. способами. А. г. обладает
слабыми кислотными свойствами (см. Перекиси и
гидроперекиси органические). А. г. применяется как
окислитель для получения а-окисей и гликолей из
олефинов, для дезодорирования и обесцвечивания
гексахлорциклогексана и др.
Лит.: S w е г n D., Chem. Revs, 1949, 45, № 1, р. 1, см.
также при ст. Перекиси и гидроперекиси органические.
АЦЕТИЛА ПЕРЕКИСЬ (СН3СОО)2, ' мол^^вес
118,09 — бесцветные кристаллы с резким специфик,
запахом; т. пл. 30°; т. кип. 65°/23 мм; легко раство-
рима в органич. растворителях, в отличие от пере-
кисей других ацилов, довольно хорошо растворима
в воде; для очистки кристаллизуют при сильном
охлаждении из лигроина. В ИК-спектре (в СС14)
частота колебаний —С= 0-группы 1808 1 см1,
24 ± 2 см"1. При хранении (быстрее на свету) А. п.
разлагается. А. п. обладает всеми химич. свой-
ствами перекисей (см. Перекиси и гидроперекиси
органические), однако она не восстанавливает под-
кисленных р-ров солей хромовой к-ты и перманга-
ната калия; в водном р-ре гидролизуется, образуя
уксусную и надуксусную к-ты, быстро разлагается
щелочами. А. п. применяется в качестве окислителя,
катализатора полимеризации, напр. соединений, со-
держащих винильную группу. Применение А. п.
ограничено вследствие сильной взрывчатости; взры-
вается от удара, трения и даже от прикосновения
острым предметом.
Лит.: Tobolsky А. V. and Mesrobian R. В.,
Organic peroxides, N. Y.—L., 1954. Л. К. Лысанчук.
ге-АЦЕТИЛАНИЗОЛ re-CH3O—CeH4—COCH3, мол.
вес 150,2 — кристаллы, бесцветные пластинки с
запахом, напоминающим запах боярышника; т. пл.
37—39°; т. заст. 36°; т. кип. 258—263°, 136°/10 мм;
d)1,1 1,01818; геу,а 1,5470. В 4—5 объемах 50%-ного
спирта при 20° растворяется 1 объем А.; легко рас-
творим в органич. растворителях; Немного раство-
рим в воде. А. получают ацетилированием анизола
уксусным ангидридом в присутствии А1С13 или от-
беливающей глины — асканита:
СеН5ОСН3 4- (СН3СО)2О — 2U. сн3сосен4осн3 + сн3соон
А. не образует бисульфитного соединении; его иден-
тифицируют: по оксиму, т. пл. 80°; по семикарбазону,
т. пл. 200°, 2,4-динитрофенилгидразону, т. пл. 233°
и др. Количественно А. определяют методом окси-
мирования. Применяют А. в парфюмерии (вводится
в состав нек-рых духов) и для отдушки мыла. Тор-
говые назв. А.: «кратаегон», «обепинон», «метилэтал»,
«мимозал». с. И. Кора.
АЦЕТИЛАЦЕТОН (диацетилметан, пентандион-2,4),
мол. в. 100,12 — равновесная смесь кетонной (I) и
енольной (II) форм
СН3СОСН3СОСН3 СН3С(ОН) = сносн3
I п
содержащей от 19 до 92% енольной формы (в за-
висимости от растворителя). А. — бесцветная или
слегка желтоватая жидкость с запахом, напоми-
нающим одновременно ацетон и уксусную кислоту.
Свойства кетонной формы: т. пл. —23°; т. кип.
139°/746 мм, в-У" 0,9721; п'^ 1,4541; теплота сгорания
613,6 ккал/моль (b=const); константа диссоциации
К = 1,5 10“в (25°). Свойства енольной формы: т. пл.
—9°; геу 1,4609. Растворимость в 100 г воды: 15 г
(30°) и 34 г (80°); смешивается со спиртом, эфиром,
хлороформом, ацетоном, бензолом и др. органич.
растворителями. Наиболее характерной реакцией А.
является замещение атомов водорода метилено-
вой группы металлом: СН3СО—СН2—СОСН3 4-
+ NaOH —> СН3СО—CHNa—СОСН3 + Н2О; Na- про-
изводное, реагирующее в енольной форме, можно
алкилировать: СН3СО—CH=C(ONa)CH3 4* CjHjJ —>
-+ СН3СО— СН(С2Н6)—СОСН3 + NaJ. А. при дей-
ствии окислителей образует уксусную кислоту; при
нагревании с водой или КОН — ацетон и уксусную
кислоту. Основным способом получения А. является
конденсация ацетона и этилацетата: СН3СОСН3 4*
345
АЦЕТИЛГАЛОГЕНСАХАРА — АЦЕТИЛЕН
346
+ сн3соос2н5 -> сн3со—сн2—сосн3 + С2Н6ОН
(см. Клайзена конденсация). Известны и др. спо-
собы получения А.
А. применяют в органич. синтезе, а также для от-
крытия Fe, Сг, Со, Мп, Те (в присутствии CS2), Zr;
для фотометрия, определения Fe111 и F~. В енольной
форме А. реагирует с катионами, образуя ацетил-
ацетонаты железа, меди, алюминия, бе-
риллия, хрома, кобальта и др., являю-
щиеся внутрикомплексными (хелатны-
ми) соединениями (см. формулу).
Ацетилацетонаты легко растворимы
в органич. растворителях и являются
неэлектролитами (или лишь очень слабо
диссоциируют). Многие из них способны возгонять-
ся и даже перегоняться без разложения, напр.
ацетилацетонат алюминия А1(С5Н,О2)3 кипит при 314°,
ацетилацетонат бериллия Ве(С6Н7О2)2 — при 270°.
Константа диссоциации рК(НА) при ионной силе р-ра
Я = 0 равна 8,95 (30°); при ц = 0,1—8,82 (25°). Константы
устойчивости (ступенчатые) комплексов с различными катиона-
ми, определенные при 30° и ионной силе р-ра Ц = 0 равны:
_____^/СНз
Mef ^СН
Ь=С
ХСН»
Катион 1g К1 IgK2 Ig*G 1g Kt
АР+ 8,6 7,9 5,8
Ве-+ 7,8 6,69 —— —
Се3+ 5,28 3,98 3,38 ——
Fe3+ 9,8 9,0 7,2 —-
Ga3+ 9,5 8,4 5,7 —
ln3+ 8,0 7,1 — —
pu4+ • 10,5 9,2 8,4 6,0
Th<+ 8,8 6,3 4,2
и<+ * 8,6 8,4 6,4 6,1
uoi+ 7,74 6,43 . — —
уз+ 6,4 4,7 2,8 —
* При 25° и р. = 0,1.
См. также Таутомерия кето-енолъная.
К. В. Ваццро, И. П. Ефимов.
АЦЕТИЛГАЛОГЕНСАХАРА — СМ. о-Ацетилглико-
зилгалиды.
О-АЦЕТИЛГЛИКОЗИЛГАЛИДЫ (ацетогалогенса-
хара, ацетогалогенозы) — производные сахаров, спир-
товые группы к-рых эте-
рифицированы уксусной
к-той, а полуацеталь-
ный гидроксил заменен
галогеном.
А. известны в виде а-
и fj-форм; последние
очень нестойки, легко
изомеризуются в а-фор-
мы и для многих сахаров неизвестны. Большинство
А.—кристаллич. продукты, сравнительно трудно ра-
створимые в воде, легко — в органич. растворителях.
Стойкость А. убывает в ряду: фториды>хлориды>бро-
миды>иодиды (последние быстро разлагаются уже
при 0°, особенно в присутствии влаги, фториды можно
длительно кипятить без разложения). Щелочи отще-
пляют галоген и ацетильные группы у иодидов, бро-
мидов и хлоридов; у фторидов галоген связан проч-
нее, чем ацетильные группы.
А. обычно получают действием на ацетаты сахаров
р-ра галогеноводорода в ледяной уксусной к-те или
(при получении ацетилгликозилфторидов) действием
жидкого галогеноводорода. А. широко применяются
для синтезов, в к-рых галоген заменяется алкоксиль-
ными или ароксильными группами (для получения
ацетатов гликозидов, из к-рых ацетильные группы
легко удаляются щелочными реагентами), а также
для получения производных сахаров, в к-рых гли-
козил связан С—С-связью. Наиболее часто приме-
няются: тетра-0-ацетил-а-глюкопиранозилбромид (аце-
тобромглюкоза), т. пл. 87—88°; [а]д = -|-198° (вСНС13);
тетра-0-ацетилглюкопиранозилхлорид (ацетохлорглю-
коза), т. пл. 75—78°, [a]g = + 165,7° (в СНС13). Из-
вестны также А. ациклич. строения общей формулы
/На!
R - СН<
\ОАс
Лит.: Carbohydrates, ed. by W. Pigman, N. Y., 1957;
M i c h e e I F., Chemie der Zucker und Polysacharide, 2 Aufl.,
Lpz., 1956. Б. II. Степаненко.
АЦЕТИЛЕН (этин) СН=СН, мол. в. 26,04 — первый
член гомологии, ряда ацетиленовых углеводородов,
бесцветный газ; т. пл. —80,8° (при/>=1277 ммрт. ст.);
темп-ра возгонки твердого А. — 84,1° (760 мм);
т. кип.-83,8°; плоти, жидкого 0,463 (0°, 26,3 ат);
газообразного 1,1716 г/л (0°, 760 мм); теплота обра-
зования из элементов 58 ккал/моль; теплота сгора-
ния (р =const) 312 ккал/моль; теплоемкость при 0°
0,313 ккал/кг • град. Давление паров жидкого А.:
10,9 ат (—30°), 26,3 ат (0°), 54,1 ат (30°); теплотвор-
ность 13900 ккал/м3; «крит 35,2°; />,!рит 63,7am,
<2 крит 0,230. При нагревании до 500° и одновремен-
ном повышении давления св. 2,0am А. взрывает.
Смеси с воздухом, содержащие от 2,3 до 80,7% А.,
взрывают от искры. При разбавлении А. другими
газами — азотом, метаном или пропаном — взрыво-
опасность его уменьшается; смеси 1:1с этими газами
безопасны и под давлением. Максимальная темп-ра
кислородно-ацетиленового пламени 3150°. В таблице
приведена растворимость А. в 1 объеме растворителя.
Растворитель Темп-ра, °C Давление, мм рт. ст. Число объе- мов А.
Вода 15 760 1,15
Ацетон 15 760 25
» 15 12 ат 300
» .......... —80 760 2000
Спирт 15 760 6
Бензол 15 760 4
Уксусная к-та 18 760 6
Диметилформамид . . . 25 760 33,5
; атомы углерода и водорода
А. сравнительно хорошо растворим в хлороформе.
Молекула А. линейш
лежат на одной линии.
Расстояние между ато-
мами углерода, соеди-
ненными одной о-связью
и двумя л:-связями,
1,20 А. Расстояние ме-
жду атомами углерода
и водорода 1,057 А;
л-связи расположены
во взаимно-перпенди-
кулярных плоскостях.
А. был открыт в 1836 Э. Дэви; синтезирован в 1862 М. Бертло
из угля и водорода; получен из карбида кальция в 1862 Ф. Ве-
лером. Последний способ и в наст, время является одним из
промышленных методов получения А. В 1892 А. Муассан при-
готовил карбид кальция нагреванием смеси угля и извести
в электрич. дуговой печи. Этот способ произ-ва карбида каль-
ция, а из него А. был освоен пром-стью в 1895—96 (см. Каль-
ция карбид).
При разложении карбида кальция водой образуется
ацетилен:
СаС3 + 2Н2О -е СН;_СН + Са(ОН)2
ДНзэв =— 30,4 ккал/моль
Разложение карбида кальция водой в химич. пром-сти
обычно производится в аппаратах большой произ-
водительности (до 2000 м3/час А.). На рис. приве-
дена схема получения А. в т. н. «сухом» генераторе,
в к-ром применена система «вода на карбид». Карбид
кальция передвигается мешалкой с полки на полку,
а вода подается сверху через разбрызгивающее уст-
ройство. При этом получается А. и сухая известь.
347
АЦЕТИЛЕН
348
Применяются также генераторы системы «карбид
в воду».
Газ, получаемый из карбида кальция, содержит до
99,5% А. Технический А., получаемый из карбида
кальция, очищают от
примесей (NH3) РН3,
AsH3 и др.) ороше-
нием водой, содержа-
щей рассчитанное на
окисление этих при-
месей количество ги-
похлорита натрия.
Известь, получаемая
при разложении кар-
бида кальция водой,
получается в виде
сухого порошка [СаО
с примесью Са(ОН)2].
Схема получения ацети-
лена из карбида каль-
ция в «сухом» генерато-
ре: 1 — бункер для кар-
бида кальция; 2 — авто-
матический затвор; з — буферный бункер; 4 — шнековый
питатель; 5 — ацетиленовый генератор; 6 — шнек для уда-
ления сухой извести; 7 — охладительный скруббер; 8 — от-
стойник; о — холодильник.
Расход электроэнергии на получение А. карбид-
ным способом составляет 10 квт-ч/кг.
Важными современными методами промышленного
приготовления А. являются термоокислительный кре-
кинг и электрокрекинг метана. Электрокрекинг ме-
тана, производимый в электродуговых печах, при-
обрел промышленное значение с 1940. Метан быстро
пропускают через вольтову дугу между металлич.
электродами; постоянный ток напряжением 8000 в
и темп-pa реакционного пространства 1600°, скорость
газового потока 1000 м/сек', степень превращения
метана в А. достигает 50%:
2СН4 —► СН —СН ЗН.» ДНО298 = 91 ккал/молъ
Выходящие газы мгновенно охлаждают (до 150—200°)
вспрыскиванием воды. Газ, получаемый из печи, со-
держит ок. 13 об. % А., 45% водорода, ок. 1% эти-
лена, неразложившийся метан, следы сероводорода,
синильной к-ты, окиси углерода и др. примесей. В
результате электрокрекинга из 1000 м3 природного
газа (метан с примесью гомологов) получается: 300 кг
А., 26 кг этилена, 21 кг сажи и 1170 м3 водорода.
Расход электроэнергии ок. 9 квт-ч/кг неочищенного А.
Термоокислительный крекинг метана (с примеся-
ми гомологов) основан на его разложении за счет те-
плоты сгорания при недостаточной подаче кислорода
(СН4 : О2 = 1 : 0,65):
6СЩ + 40, —► СН=СН -I- 8На + ЗСО + СО, + ЗНаО
Темп-pa газов в печи достигает 1500°. Газ, выходящий
из реакционного пространства, как и в случае элек-
трокрекинга, . быстро охлаждается вспрыскиванием
воды. В результате термоокислительного крекинга
получают газовую смесь, содержащую (в об. %):
8% ацетилена, 54,5% Н2, 26% СО, 4% СО2, 4,5%
СН4, 0,2% гомологов А., 0,1% бензола и его гомоло-
гов и др. А. абсорбируют из газовой смеси диметил-
формамидом под давлением (ок. 8 ат.) или жидким
аммиаком. Остающаяся после абсорбции смесь газов
(т. н. синтезгаз) содержит 60% Н2, 29% СО, 5% СН4,
4% СО2 и небольшое количество примесей; газ может
быть использован для контактного получения угле-
водородов и др. целей. Перспективным методом полу-
чения А. является также пиролиз смеси метана с его
гомологами, осуществляемый с 1950. Процесс осно-
ван на рекуперации теплоты аппаратом с огнеупор-
ной насадкой, обогреваемой до 1200° сжиганием газо-
образного топлива, и последовательным пропуска-
нием через раскаленную насадку газов, подвергаемых
крекингу. В качестве исходного газа применяют
смесь метана с этаном и пропаном, разбавленную
обратным газом (после выделения А.) и водяным паром
в соотношении 1:2:6, темп-pa в печах до 1200°,
вакуум 0,5 ат. Получаемый газ содержит 10% (объем-
ных) А., выделяемого из газовой смеси абсорбцией
диметилформамидом. Остаточный газ используется
для обогрева печей.
Экономия, затраты на получение А. термоокисли-
тельным крекингом и пиролизом вполне сравнимы
с экономикой получения А. карбидным методом. Эти
способы выгодно отличаются от карбидного метода
отсутствием прямого расхода электроэнергии и яв-
ляются перспективными для районов с месторожде-
ниями природного газа, лишенных дешевой электро-
энергии. А. производится в огромных масштабах;
производственные мощности по А. в индустриальных
странах исчисляются в сотнях тысяч тонн; напр.,
в США превышают 1 млн. т в год. А. служит исход-
ным сырьем для синтеза большого числа технически
весьма важных органич. соединений. Наряду с таким
крупным потребителем А., каким является быстро
растущее произ-во хлоропренового каучука, А. нахо-
дит широкое применение для получения винилхло-
рида, ацетальдегида, уксусного ангидрида, акрило-
нитрила, винилацетата, трихлорэтилена и мн. др.
Около 70% производящегося А. расходуется на нужды
тяжелой органич. пром-сти и ок. 30% на сварку.
А. чрезвычайно реакционноспособное непредель-
ное соединение. Реакции присоединения к нему про-
текают в два этапа; вначале образуются производ-
ные этиленового ряда, к-рые затем переходят в пре-
дельные соединения:
СН = СН —-i- С1СН = СНС1 —- СНС1а—СНС12
СН = СН — ^»СН3 = СН2 — =- СН3—СН3
Хлорирование А. имеет важное промышленное зна-
чение в произ-ве различных хлорсодержащих соедине-
ний: тетрахлорэтилена, гексахлорэтана, трихлор-
этилена и др., широко используемых в пром-сти.
Присоединение НС1 в присутствии полухлористой
меди ведет к образованию винилхлорида: СН=СН +
+ НС1 —> СН2 — СНС1; присоединением синильной
к-ты под влиянием того же катализатора в кислой
среде получается винилцианид (акрилонитрил):
CHesCH -f- HCN —> СН2 = CHCN. Эти продукты
являются объектом крупного производства и употреб-
ляются для синтеза важнейших полимеров (поли-
винилхлорида и полиакрилонитрила, «нитрона»,
«орлона» и др.). Присоединение воды в присутствии
солей ртути ведет к ацетальдегиду (Кучеров):
СН = СН —> [СН2 = СН—ОН] -> СН3—СНО. При
пропускании смеси А. и паров воды при 300—400°
над фосфорнокислыми солями тяжелых металлов
имеет место т. н. прямая гидратация А. с образо-
ванием смеси, состоящей гл. обр. из ацетальдегида с
примесью ацетона; изменением условий реакции и ка-
тализатора соотношения образующихся продуктов мо-
гут быть изменены в Сторону образования ацетона с
выходом до 75% от теоретически возможного. Методы
имеют широкое промышленное значение и использу-
ются для произ-ва уксусной к-ты и ее производных.
Реакцией присоединения к кратной связи является
также карбонилирование А. (Реппе, 1944):
СН
III + СО
сн
СН
сн J ЫН
;--- • iROHl NHa _______
I Hso ♦ J
СНа=СН—СООН СН2—СН—COOR СН3=СН—СО—NH2
4СН=СН + 4С2Н5ОН + Ni(CO)4 + 2НС1 -
— 4СНа=СН—СООС2Н2 + N1C12 + Н2О
349
АЦЕТИЛЕН — АЦЕТИЛЕНДИКАРБОНОВАЯ КИСЛОТА
350
Эти реакции приводят к акриловой к-те и.ее произ-
водным, имеющим важное промышленное значение
в произ-ве пластмасс. К этим реакциям близко отно-
сится и образование хинона:
/СН=СН\
со — со со
\ch=chz
со
сн=сн
сн=сн
А. легко присоединяет сулему в р-ре 10—12%-ной
соляной к-ты с образованием p-хлорвинилмеркурий-
хлорида: CH=CH-f- HgCla-> С1СН=СН—HgCl. Дей-
ствие хлористого мышьяка на это соединение при-
водит к p-хлорвинилдихлорарсину, т. н. люизиту:
С1СН=СН—HgCl + AsClg —> С1СН=СН — AsCb + HgCl2
К А. легко присоединяются спирты, тиолы, амины
и др. вещества с подвижным атомом водорода; при-
соединение спиртов к А. происходит под давлением
под влиянием едкого кали, причем образуются вини-
ловые эфиры (Фаворский):
CH = CH + ROH -► СН2 = СН — О — R
Продукты полимеризации последних используются
в технике в качестве гидравлич. жидкостей, смазок
и др. Присоединением к А. уксусной к-ты получается
винилацетат:
+ NaJ. Ацетилениды меди CiijCj и серебра AgjCj
образуются при действии на А. аммиачных рас-
творов закиси меди или окиси серебра; в сухом состоя-
нии эти ацетилениды взрывчаты; ацетиленид меди
взрывает при нагревании его до 120°. Получение
ацетиленида меди мясо-красного цвета часто исполь-
зуется в анализе как самая чувствительная реакция
на А. (реакция Иллосвая).
А. легко присоединяет альдегиды, кетоны и окиси.
Продукт присоединения двух молекул формальдегида
к молекуле ацетилена — бутиндиол-1,4
сн = сн + 2СН3О —> носн2 — с = с—сн3он
широко используется для произ-ва бутандиола-1,4(1],
важного полупродукта произ-ва полиуретанов, а так-
же для произ-ва тетрагидрофурана (II), адипиновой
к-ты, бутадиена (III) и др.:
носна-с=с-снаон носна-сн2-сна-снаон
/ I
сна-сна
СО-СН2-СН2 -J-2C0 J I
О: -----СН3 Uri2
\со-сн2-сн2 \/
-н»о
и
О //Q
СН3—С + сн=сн — сн3—с
''ОН Ч'О-СН=СН3
используемый для произ-ва поливинилацетата и по-
ливинилового спирта. Присоединение осуществляется
пропусканием А. в уксусную к-ту, в к-рой растворен
сульфат ртути, или в газовой фазе при 250° над
катализатором, содержащим соли цинка. С пирро-
лидоном А. образует винилпирролидон
СН2 - СО.
| >NCH=CH3
СН2-CIIZ
полимеризацией к-рбго получают полив инилпирро-
лидон, являющийся заменителем плазмы крови.
Особенно интересны в теоретич. и технич. отно-
шении реакции регулируемой полимеризации А.;
известны 3 основных типа: полимеризация линейная,
происходит под влиянием солей закиси меди в при-
сутствии хлористого аммония и приводит к винил-
ацетилену (с примесью дивинилацетилена), являю-
щемуся основным сырьем в произ-ве хлоропренового
каучука:
ZCH..=CH-C-CH
/ винилацетилен
СН=СН -|- СН=СН<
*СН2=СН—С=СН=СН2
дивинилацетилен
Над активным углем А. гладко полимеризуется, об-
разуя бензол (Бертло, Зелинский), ЗСН = СН —>
—>СвНв. В р-ре цианида никеля в тетрагидрофуране А.
дает 8-, 10- и 12-членные аналоги бензола: циклоок-
татетраен С8Н8, циклодекапентаен СюН10 и циклодо-
декагексадиен С\аН1а (Реппе). В присутствии водорода
(над никелем) А. образует изобутилен (СН3)аС=СНа:
сн3 сн3
сн \ /
2 ||| + 2Н3 — С
СН |
СН2
Ряд реакций А. обусловливается подвижностью ато-
мов водорода («ацетиленовый атом водорода») при
атоме углерода, соединенным тройной связью. При
действии металлич. натрия или амида натрия (обычно
в жидком аммиаке) получаются натрийацетилениды
СН=С—Na и NaC- CNa. Действие алкилирую-
щих средств на эти металлич. производные приводит к
гомологам A. CHSJ -f- NaC=CH —► СН3—CzrCH -|-
— сна=сн-сн=сна
III
А. является эндотермич. соединением. Поэтому' при
сжигании его выделяется большое количество тепла.
На этом основана ацетилено-кислородная сварка
черных металлов. Смесь А. с воздухом горит ярким
пламенем, что иногда еще используется в целях
освещения (фонари на маяках, буях и др.). А. обла-
дает наркотич. действием. Хранят и перевозят А.
в заполненных пористой массой стальных баллонах,
в р-ре ацетона под давлением 15—25 ат. Важней-
шими гомологами А. являются: метилацетилен, или
алилен СН3—С = СН с т. кип. — 23,3°, т. пл. —
101,5°; кротопилены: бутин-1, газ, т. кип.
8,6°; бутин-2, газ, т. кип. 27°; валерилены: пен-
тин-1, т. кип. 39,7°; пентин-2, т. кип. 56,1°;
3-метилбу тин-1; т. кип. 28°, и др.
Лит.: Ч и ч и б а б и н А. Е., Основные начала органи-
ческой химии, т. 1, 6 изд., М., 1954; Н ь ю л э н д Ю. А. и
Фогт?. Р., Химия ацетилена, пер. с англ., М., 1947; Химия
ацетилена. Сб., пер. с англ, и нем., М., 1954; Федореи-
к о Н. П., Методы получения ацетилена, М., 1958; Общая
химическая технология. Под ред. С. И. Вольфковича, т. 2,
М., 1959; Стрижевский И. И., Гузов С. Г., К о-
вальскийВ. А., Ацетиленовые станции 3 изд М., 1959.
И. Л. Кнунянц.
АЦЕТИЛЕНДИКАРБОНОВАЯ КИСЛОТА, мол.
вес 114,06, НООС—С^С—СООН — простейшая
двухосновная кислота с тройной связью: т. пл.
безводной 178—179°, т. пл. С4НаОа • 2НаО 175°.
А. к. по силе приближается к серной к-те; константы
диссоциации при 25°: ^=1,82 • 10 2; К2 =4,20 • 10-6.
Получают А. к. действием спиртовой щелочи на ди-
бромянтарную кислоту: НООССНВг — СНВгСООН-]-
+ 2NaOH -> НООС—С—С—СООН 2NaBr-f-2НаО,
а также на хлор(бром)малеиновую или фумаро-
вую к-ты. А. к. тримеризуется в меллитовую к-ту.
При действии галогенводородных к-т или галогенов
образуются гл. обр. галогенфумаровая или соот-
ветственно дигалогенфумаровая к-та:
ноос X
ноос—с=с—соон + нх — 'esc'
R ''соон
(Х=С1, Br, RCO и др.)
со спиртом или фенолами образует главным образом
этокси- или феноксифумаровую к-ту. При гидрата-
351
АЦЕТИЛИРОВАНИЕ - АЦЕТИЛЦЕЛЛЮЛОЗА
352
ции А. к. превращается в щавелевоуксусную к-ту
НООС—СО—СН2—СООН; А. к. и ее эфиры часто
используются в качестве диенофилов в диеновом
синтезе:
^СН2
СН + rC0°R—Hf f'"C00R
СН с—COOR НС ^с—COOR
ЧСНа ЬН2
При конденсации эфиров А. к. с нек-рыми азотсо-
держащими соединениями (гидразины, алифатич. ди-
азосоединения) образуются гетероциклич. соедине-
ния, напр.:
О=С
I
О
I
^СН2
С=С—СООСН3 О=С ^С—СООСН,
+ NHg— NH2 HN------N
сн3
Лит.: Синтезы органических препаратов, пер. с англ.,
сб. 2, М., 1949, с. 71; Chemistry of carbon compounds. Ed. by
E. H. R о d d, v. 1, pt. A, Amst. [a. o.J, 1951, p. 110.
M. И. Роаенгарт.
АЦЕТИЛИРОВАНИЕ — см. Ацилирование.
АЦЕТИЛХОЛИНХЛОРИД СН3СООСНаСН»Й(СН3)3С1,
мол. в. 181,67 — бесцветные гигроскопич. кристаллы,
легко растворимые в воде, спирте и хлороформе и не-
растворимые в эфире. Йри обработке А. р-ром AgNO3
выпадает белый осадок, нерастворимый в HNO3
и растворимый в водном аммиаке (качественная
проба). Для колич. определения прибавляют разба-
вленные HNO3 и AgNO3 и избыток последнего ти-
труют 0,1 н. р-ром NH4SCN с железоаммониевыми
квасцами. А. получают взаимодействием окиси эти-
лена с триметиламином с последующей обработкой
хлористым водородом и ацетилированием образую-
щегося при этом холинхлорида уксусным ангидридом.
А. играет важную роль в процессе нервной деятель-
ности человека и животных, являясь передатчиком
(медиатором) нервного возбуждения. Применяется
при спазмах периферия, сосудов и артерий сетчатки,
иногда при глаукоме, атонии кишечника и мочевого
пузыря. См. также Холин и Холинэстераза.
Лит. см. при ст. Адалин. А. И. Травин.
АЦЕТИЛЦЕЛЛЮЛОЗА (ацетаты целлюлозы) —
уксуснокислые эфиры целлюлозы, получаемые обычно
действием на целлюлозу уксусного ангидрида в при-
сутствии различных катализаторов. А. может быть
получена также действием на целлюлозу кетена
(СН2=СО), но эта реакция осложняется полимери-
зацией кетена, что препятствует ее практич. исполь-
зованию. Реакция ацетилирования целлюлозы уксус-
ным ангидридом:
[СсН7Оа(ОН)3]п + пЗ(СН3СО)»О —
— [СвН7О2(ОСОСН3)3]п + пЗСНзСООН
Исходным сырьем для получения А. служит хлоп-
ковый пух, реже — древесная целлюлоза. Для более
равномерного протекания процесса целлюлозу перед
ацетилированием обрабатывают ледяной уксусной
к-той (50—100% от веса целлюлозы) в течение 3—4 ча-
сов. Конечным продуктом ацетилирования целлюлозы
является триацетат целлюлозы, называемый обычно
первичным ацетатом. При частичном омы-
лении триацетата целлюлозы получается т. н. вто-
ричный ацетат, содержащий 2,4—2,7 моля
связанной уксусной к-ты на элементарное звено цел-
люлозы.
В отсутствие катализаторов скорость реакции
ацетилирования очень мала. В качестве катализато-
ров применяют серную или хлорную к-ты, хлористый
цинк и пиридин. На практике чаще всего применяется
серная к-та (3—12% от веса целлюлозы), однако при
этом повышается скорость деструкции целлюлозы и
протекает побочная реакция образования сульфоэфи-
ров целлюлозы. Последние неустойчивы и легко
разлагаются с выделением свободной серной к-ты,
к-рая разрушает А. Поэтому А., полученную в при-
сутствии серной к-ты, подвергают стабилизации (об-
работке разбавленной серной к-той или водой при
повышенной темп-ре), в результате чего происходит'
омыление сульфоэфиров. Наиболее активным ката-
лизатором ацетилирования целлюлозы является хлор-
ная к-та (0,5—1% от веса целлюлозы); получаемая
при этом А. стабильна. Недостаток хлорной к-ты
как катализатора — желатинизация образующихся
при ацетилировании целлюлозы в гомогенной среде
(в среде уксусной к-ты) р-ров и их непригодность
для переработки. Хлористый цинк — мало активный
катализатор (применяется в количестве 80—100%
от веса целлюлозы), но в его присутствии ацетили-
рование целлюлозы можно проводить при повышен-
ной темп-ре (50—70°), однако скорость реакции при
этом низка. Наиболее мягко ацетилирование целлю-
лозы уксусным ангидридом протекает в среде пири-
дина. В этом случае полностью исключается реакция
деструкции целлюлозы и можно получить ацетаты
целлюлозы, по степени полимеризации не отличаю-
щиеся от исходной целлюлозы. Но скорость ацетили-
рования в этих условиях очень мала; этот метод при-
меняется только для научных исследований.
Ацетилирование целлюлозы проводят в среде рас-
творителя, в к-ром растворяется образующаяся А.
(гомогенное ацетилирование) или в среде разбавителя
(гетерогенное ацетилирование). В качестве разбави-
телей, применяемых с целью экономии растворителя,
применяются жидкости, не растворяющие А., но
хорошо смешивающиеся с раствором А. Ацетилирую-
щая смесь состоит обычно из уксусного ангидрида,
катализатора и растворителя или разбавителя. В ка-
честве растворителей при гомогенном ацетилирова-
нии применяют уксусную к-ту или метиленхлорид.
Разбавителями при гетерогенном ацетилировании
являются бензол, толуол или четыреххлористый
углерод. Для получения первичного ацетата аце-
тилирование целлюлозы иногда проводят в гете-
рог. среде. Состав ацетилирующей смеси: 60% уксус-
ного ангидрида, 40% разбавителя; катализатор —
хлорная кислота, 1% от веса целлюлозы. Модуль
ванны 25—30. Полученный ацетат отмывают разба-
вителем от уксусного ангидрида, затем разбавитель
отгоняют с водяным паром, а А. промывают водой,
отжимают на центрифуге и сушат.
Для получения вторичного ацетата ацетилирование
проводят в гомог. среде при 30—35°. В качестве ка-
тализатора используют хлорную или серную к-ту.
Модуль ванны 7—8. В результате ацетилирования
получается 20—22%-ный р-р триацетилцеллюлозы
в уксусной к-те или в смеси уксусной к-ты и метилен-
хлорида. Для частичного омыления триацетилцеллю-
лозы к полученному р-ру добавляют такое количество
воды, чтобы получился р-р А. в 90—95% уксусной
к-те, к-рый выдерживают при 30—45° в течение
12—24 часов (процесс созревания А.). Вторичный
ацетат высаживают из р-ра водой или разбавленной
уксусной к-той, промывают и сушат.
Основными свойствами А., к-рые обеспечили ей
широкое применение в пром-сти, являются: высокая
светостойкость, хорошие физико-механич. свойства,
негорючесть. Первичный ацетат (триацетат) раство-
рим в ледяной уксусной к-те, метиленхлориде,
хлороформе, анилине, пиридине и др. Триацетат
целлюлозы применяется для формования волокна и
пленок, используемых для произ-ва электроизоля-
ционных материалов, а также для произ-ва волокон
для текстильной пром-сти. Вторичный ацетат рас-
353
АЦЕТОН
354
творим в ацетоне, уксуснокислых эфирах низших
алифатич. спиртов (напр., этилацетате), в диоксане и
циклогексаноне. Вторичные ацетаты широко исполь-
зуются для формования ацетатного волокна, пленок
и в пром-сти пластин, масс.
Лит.: Литвин О. Б., Ацетилцеллюлоза и ацетатный
шелк, М., 1937; Роговин 3. А. и Шорыгина Н. Н.,
Химия целлюлозы и ее спутников, М.—Л., 1953 с. 419—445;
U I 1 m а п п, 3 Aull., Bd 5, Munch.—В., 1954, S. 182—201.
В. А. Деревицкая.
АЦЕТОН (диметилкетон) СН3СОСН3, мол. в.
58,08 — простейший кетон; бесцветная жидкость;
т. пл. — 95,35°; т. кчп. 56,24°; 0,7908; п2£ 1,3558;
давление пара 181,72 мм при 20°; смешивается с водой
и органич. растворителями; теплоемкость Ср =
= — 30,87 ккал/леоль(30°); теплота сгорания С„=
= - — 426,4 ккал/молъ (жидк.), 437,25 ккал/молъ (газ);
теплота парообразования 7,092 ккал/молъ (т. кип.);
т. воспл. —20°; пределы взрываемости смесей с воз-
духом 2,55—12,80%.
А. обладает типичными химич. свойствами кетонов.
Он с трудом окисляется; каталитически восстанавли-
вается до иао-С3Н7ОН, а амальгамами Mg или Zn
или Zn + СН3СООН — до пинакона:
2 (СН3)3СО - (СН,)2 С (ОН) С (ОН) (СНз)з
А. окисляет вторичные спирты в присутствии алкого-
лятов алюминия до соответствующих кетонов (см.
Меервейна—Понндорфа—Верлея реакция и Оппенауэра
реакция):
RR'CHOH + (CH8)S СО A1 (0—Ч RR'CO 4- (СН3)3 СНОН
При альдольной и кротоновой конденсациях А. про-
исходит гл. обр. образование диацетонового спирта
(1), окиси мезитила (2), форона (3) и мезитилена (4):
2(СН3)»СО — - (СН3)3С(ОН)СН3СОСН3 (1)
2(СН3)3СО — 2 (СН3)2 С = СНСОСН3 (2)
3(СН3)»СО — 2 (СН3)3 С = СНСОСН = С(СН3)3 (3)
н+ /—\СНз
3(СН3)аСО —— СНз—\ / (4)
v=/CH3
так-
С ацетиленом А. образует диметилкарбинол, который
легко может быть превращен в изопрен:
(сн3)3 со + сн = сн нс = с (он)(сн3)3 —
— Н2С = СН-С(ОН)(СН3)3 —Н3С = СН-С (СН3) = сн3
Нек-рые реакции присоединения к СО-группе
же имеют практическое значение, напр.:
(СН3)3 СО —(CH3)3C\CN —22
хон
- СН3 = С (CH3)CN -22Д СН3 = С (СН3) COOR
А. образует циклич. ацетали с соединениями, содержа-
щими вицинальные tyuc-гидроксильные группы. А.
легко в присутствии НС1 реагирует с меркаптанами,
образуя полные тиоацетали. При пиролизе А. обра-
зуется кетен:
(СН3)3СО 222 сн3 = с = о + сн4
Атомы водорода метильных групп А. легко заме-
щаются (галогенирование, нитрозирование и т. п.).
Действием хлора и щелочи А. легко превращается
в хлороформ. Он легко присоединяет СНС13, образуя
(СН3)2С(ОН)СС13 (хлоретон), применяющийся в ка-
честве антисептика.
В пром-сти А. получают исходя из пропилена,
гидратацией которого производят изопропиловый
спирт. Последний перерабатывают в ацетон окисле-
нием или дегидрированием. Окисление изопропило-
вого спирта
(СН3)«СНОН + 1/3О3 - (СН3)3 СО + Н3О + 43 ккал
проходит над серебряным катализатором при 450—
500°; реакция протекает в две стадии: 1) изопропило-
вый спирт разлагается на ацетон и водород с погло-
щением тепла; затем окисляется водород с выделением
тепла; процесс в целом экзотермичен. Побочными
реакциями являются образование СО2 (5), пропилена
(6) и уксусной к-ты (7):
(СН3)3 СНОН + 41/„СЬ — ЗСО3 + ПЬО (5)
(СН3)3 СНОН —* С3Н6 -|-Н3О (6)
(СН3)3 СНОН -I- 11/3О» — СН4 + СН3СООН (7)
Окисление проводят при значительном избытке изо-
пропилового спирта в аппарате с тонким слоем сере-
бряного катализатора; известны и другие катализа-
торы этого процесса, напр. Си, Ni, Pt; однако они
менее эффективны. Продукты реакции быстро охла-
ждают в холодильнике. Конденсат после нейтрали-
зации небольших примесей СН3СООН дистиллируют;
выделенный технический ацетон ректифицируют, а не-
прореагировавший изопропиловый спирт возвращают
на окислении. Дегидрирование изопропилового спирта
(СН3)3 СНОН — (СН3)2 СО + Н3 — 16,7 ккал
проводят в паровой фазе при темп-ре ок. 400° над
цинковым катализатором (ZnO и ZnS или сплав Zn
и Fe), нанесенным на пемзу или другой пористый
материал. Активность катализатора постепенно сни-
жается вследствие отложения на нем сажи; поэтому
периодически катализатор регенерируют. Контактные
газы промывают водой и поглощают ацетон; послед-
ний выделяют ректификацией. Известно также деги-
дрирование изопропилового спирта в жидкой фазе
при атмосферном давлении и темп-ре ок. 150° в среде
инертных растворителей с катализатором никелем
Ренея.
А. получают также одновременно с фенолом т. н.
кумол-гидропероксидным способом. Метод основан
на синтезе изопропилбензола (кумола) из бензола и
пропилена, окислении кумола воздухом в его гидро-
перекись и разложении последней до ацетона и фенола:
СвНв + СН3СН = СН3 - CeH5CH (СН3)а -А
С8Н5С (СН3)3 ООН 52 С0Н5ОН + (СН3)2 со
(подробнее см. Фенол).
Известны и др. способы получения А.: взаимо-
действие ацетилена с водяным паром при темп-ре
460° в присутствии катализаторов окисей цинка и
железа (8); разложение паров уксусной к-ты при
темп-ре ок. 400° на цериевом катализаторе (9); реак-
ция в паровой фазе между этиловым спиртом и водой
при 400—425° в присутствии смешанного катализа-
тора из окислов железа и хрома, окиси меди и карбо-
ната кальция (10);
2СНЕЕСН -I- ЗН3О (СН3)3 СО -I- СО3 -I- 2Н3 (8)
2СН3СООН (СН3)3 СО + СО3 -I- Н3О (9)
2С3Н5ОН + Н3О (СН3)3 СО + СО3 + 4Н3 (10)
А. получают также брожением крахмала под влия-
нием спец, бактерий, т. н. ацетоновое бро-
жение.
А. часто содержится в моче больных диабетом;
он образуется в результате декарбоксилирования
ацетоуксусной к-ты.
Для качеств, определения А. применяются группо-
вые цветные реакции, напр. с динитробензолом
(на группу СН3СО— или —СН2СО—); пользуются
также нек-рыми специфич. • цветными реакциями,
напр. реакцией с нитропруссидом натрия (реакция
Л е г а л я). Для идентификации используются кри-
сталлич. производные, напр. семикарбазон, т. пл. 187°.
Количественно А. определяют: 1) иодометрически,
2) оксимированием:
(СН3)3СО J- NH3OH • НС! (СН3)3 С = NOH + НС! + Н3О
355
АЦЕТОНИТРИЛ— АЦЕТОУКСУСНЫЙ ЭФИР
356
образующуюся НС1 титруют щелочью, или 3) оксиди-
метрически, окислением р-ром перманганата в щелоч-
ной среде до СО2 и Н2О.
А. широко применяется как растворитель многих
органич. веществ, в первую очередь нитро- и аце-
тилцеллюлозы и ацетилена; ввиду малой токсичности
применяется как растворитель в пищевой и фарма-
цевтической пром-сти. А. служит сырьем для синте-
зов большого числа соединений, важнейшие из к-рых
кетен, изопрен, окись мезитила,диацето новый спирт
и др.
Лит.: ВайсбергерЧ. А. [и др.], Органические рас-
творители, пер. с англ., М., 1958; Сергеев П. Г., К ру-
да а л о в Б. Д., Хим. пром., 1957, № 4, с. 201; и х да е, Химич,
наука и пром., 1956, 1, М3, 287 (библ. 29 назв.); Б а у е р К. Г.,
Анализ органических соединений, пер. с нем., М., 1953, с. 206.
А. Ф. Бочков.
АЦЕТОНИТРИЛ (нитрил уксусной кислоты, циа-
нистый метил) CH3CN, мол. в. 41,052 — бесцветная
жидкость с характерным эфирным запахом; т. пл.
—44,9°; т. кип. 81,6°; d\" 0,7828; «д1,3442; смешивается
с водой, метанолом, этанолом, СС14, эфиром, ацето-
ном и этилацетатом; с водой образует азеотропную
смесь с т. кип. 76°, содержащую 16% воды. А. обла-
дает всеми свойствами нитрилов. Наиболее важными
методами приготовления А. являются: реакция йо-
дистого метила с цианистым калием CH3J KCN —
—»- CHgCN KJ; взаимодействие цианистого калия
в водном р-ре с диметилсульфатом: (CH3O)2SO2 -f-
-f-KCN —>CH3OSO2OH -f- CHgCN; дегидратацией ацет-
амида или уксуснокислого аммония при нагревании
с P2OS; реакцией ацетилена с аммиаком над А12О3.
А. применяется как растворитель, способствующий
реакциям, сопровождающимся ионизацией, и раство-
ряющим органич. и многие неорганич. соединения;
как исходный материал для синтеза важных промыш-
ленных продуктов, а также для разделения смеси
жирных к-т экстракцией, для удаления смол, фенолов
и окрашивающих веществ из углеводородов нефти
и др. А. токсичен; предполагается, что предельно
допустимая концентрация А. в воздухе 0,002%.
Н. В. Гнучев.
АЦЕТОНОВЫЕ ТЕЛА (кетоновые тепа) — группа
промежуточных продуктов обмена веществ, включаю-
щая ацетон и химически родственные ему ацетоуксус-
ную и fJ-оксимасляную к-ты. А. т. объединяет общ-
ность происхождения, легкость взаимопревращений
в организме, возможность совокупного колич. опре-
деления. (в моче, крови). Образование А. т. в орга-
низме происходит в печени в основном из продуктов
превращения жиров, а также из аминокислот (фенил-
аланин, тирозин, лейцин и др.) и нек-рых метабо-
литов углеводного обмена (пировиноградная к-та).
Процесс образования А. т. сложен и, как все био-
химия. превращения, идет многоступенчато и с вовле-
чением промежуточных продуктов в циклич. реакции.
При нек-рых патологич. состояниях (напр., при
диабете) содержание А. т. в организме резко возра-
стает. Определение А. т. в крови и моче имеет диаг-
ностич. значение. Для определения общего содержания
А. т. р-оксимасляную и ацетоуксусную к-ты предва-
рительно переводят в ацетон, к-рый определяют коло-
риметрически, используя реакцию с 2,4-динитрофе-
нилгидразином или с салициловым альдегидом. Для
определения одной р-оксимасляной к-ты из пробы
предварительно удаляют ацетон и ацетоуксусную
к-ту, а затем переводят р-оксимасляную к-ту в аце-
тон, по к-рому судят о количестве р-оксимасляной
кислоты.
Лит.: Асатиани В. С., Биохимическая фотометрия
М., 1957; King Н. К., Sci. Progr., 1956, 44, №176, р. 669;
Lynen F. [u. a.], Biochem. Z-, 1958, 330, 4, S. 269; D e I
Campillo-CampbellA. [a. o.J, Bioch. et Bioph. Acta,
1959, 31, № 1, p. 290; Л ейтес С. M., Сов. медицина, 1956,
№ 9, c. 38. И. А. Горская.
у-АЦЕТОПРОПИЛОВЫЙ СПИРТ (4-кетопентан-
1-ол) СН3СОСН2СН2СН2ОН, мол. в. 102,13—жидкость
с характерным запахом; т. кип. 145°; 1,0071;
n'rf 1,4436; смешивается с водой, легко растворим
в спирте и в эфире. А. с. представитель класса окси-
кетонов и проявляет характерные для них свойства.
При восстановлении амальгамой Na образуется пен-
тадиол-1,4. Взаимодействие с реактивом Гринья-
ра с последующим гидролизом также приводит к
диолу, напр.: CH3MgJ + СП3СОСН2СН2СН2ОН —
—(СН3)2С(ОН)СН2СН2СН2ОН. Восстановление А. с.
амальгамой Zn и разбавленной HCI дает «-амило-
вый спирт, к-рый образуется также при пере-
гонке с КОН продукта реакции между А. с. и гидра-
зингидратом. С гидроксиламином получается оксим
пентанола-1. Взаимодействие с фенилгидразином дает
фенилгидразон. А. с. присоединяет синильную к-ту,
образуя оксинитрил: СН3 — СОСН2СН2СН2ОН -ф-
+ HCN ->СН3С(ОН)СН2СН2СН2ОН. Реакция А. с.
CN
с НВг приводит к Y'Br-пропилметилкетону. А. с. мо-
жет быть легко получен, исходя из патрацетоуксус-
ного эфира и окиси этилена:
сн3сосн3соос2н5 + сн2-сн2
\>z
СН3СОСИ-СО -OC2HS
I :......:
СН3СН2—о —и
СНзСОСН—со
I /О + С2Н5ОН
сн2-сн2
Лактон ацетоуксусного эфира гидролизуют кипяче-
нием с разбавленной к-той; при этом образуются
А. с. и СО2. А. с. представляет значительный интерес
как исходный продукт для разнообразных синтезов,
особенно лекарств, препаратов; на основе А. с. раз-
работан метод получения акрихина и витамина В2.
Н. В. Гнучев.
АЦЕТОУКСУСНАЯ КИСЛОТА — см Альдегидов
кислоты и кетокислоты. А \}О
АЦЕТОУКСУСНЫЙ ЭФИР, мол. в. " бес-
цветная легко подвижная жидкость с освежающим
запахом; т. пл. —45°; т. кип. 181°/760 мм (с ча-
стичным разложением), 100°/80 мм, 71°/12,5 мм;
константы диссоциации в воде: Кенопа 3,82 • 10е;
равное, смеси 1,90-10-“. А. э. является равновесной
смесью двух таутомерных форм этилового эфира
p-оксикротоновой к-ты (енольная форма, I) и этило-
вого эфира ацетоуксусной к-ты (кетонная форма, II):
он
I
СН3-С=СН-СООСаН5 СН3-СО-СН2-СООС2Н5
Содержание в обычном А. э. каждой из форм зависит
от темп-ры, растворителя и материала стенок сосуда,
в к-ром он хранится.
Растворитель % еноль- ной формы Растворитель % еноль- ной формы
Вода 0,4 Ацетон 7,3
25%-ный метанол . 0,83 Хлороформ .... 8,2
50%-ный » 1,52 Нитробензол . . . 10,1
50%-ный этанол . . 2,18 Укс. этил, эфир . 12,9
Метанол 6,87 Бензол 16,2
Этанол 10,52 Диэтилов, эфир . 27,1
Пропанол 12,45 Сероуглерод . . . 32,4
Пентанол 15,33 Гексан 46,4
Чистая кетонная форма выделена кристаллизацией
при сильном охлаждении р-ра А. э. в петролейном
357
АЦЕТОУКСУСНЫЙ ЭФИР —АЦЕТОФЕНОН
358
эфире; т. пл. —39°; 1,0368; п'£ 1,4425; чистая
енольная форма была получена фракционированием
А. э. из кварцевой посуды; т. пл. —44°; rf!,“ 1,0119;
nJ, 1,4480. Обычному А. э. присущи одновременно
химич. свойства как кетона, так и енола.
Чистая енольная форма дает с р-ром
хлорного железа интенсивное красно-
фиолетовое окрашивание и быстро при-
соединяет эквимолекулярное количество
брома. Чистая кетонная форма, в отли-
чие от енольной формы, не реагирует
с' хлорным железом и очень медленно
обесцвечивает бром. В зависимости от
реагента А. э. образует с колич. вы-
ходами производные либо кетонной, либо енольной
формы, что находится в соответствии с возможностью
быстрого перехода одной формы в другую (десмотро-
пия) но мере израсходования одной из форм; ниже
приведены характерные
Кетонные
реакции А. э.
Образование циангид-
рина
Образование бисульфит-
ного производного
Образование
Образование
разона
Расщепление
образованием кетонов
/О-Na
СН3-С=СН-СООС2Н5 +
СН2-СН2
\)Z
СНо-со-сн-со
I I
Н2С
хснг
э.:
реакции обеих форм А.
Енольные
реакции А. э.
Реакция с РС15 с образо-
ванием Р-хлоркротоно-
вого эфира
Красно-фиолетовая ок-
раска с ЕеС13
Быстрое обесцвечивание
эквимолекулярного ко-
личества брома
Образование натриевого
производного 0-окси-
кротонового эфира.
оксима
фенилгид-
к-тами
Образующиеся с колич. выходами оксим- и фенил-
гидразон А. э. легко отщепляют спирт и соответ-
ственно переходят в метилоксазолон (III) и метилфе-
нилпиразолон (IV):
сня- СО-СН2СООС2Н5
СН3-СО-СН2СООС3НЙ
NHaOH-^ СНз_с_СНгСООСгН5
N—ОН
C6H5NHNHo
сн3-с-сн2соос2н5
n-nhc6h5
алкоголятами, водными конц.
С металлич. натрием,
р-рами щелочей А. э. образует натриевое производное
р-оксикротонового эфира:
2СН3—СО—СНа—СООСаН5 + 2Na=
О—Na
I
=2СН3-С=СН—СООС2Н5 4- Но
При действии алкилгалогенидов на натрийацетоук-
сусный эфир реакция вследствие сопряжения связей
сопровождается перенесением реакционного центра,
причем образуются гомологи А. э., в к-рых замести-
тель находится при атоме углерода метиленовой
группы:
LO^-Na fi+ п- О
8 8 //
СН3-С=СН-СООС2Н5 + R-J —CHfc-C-CH-COOC2H5
R
Т. о., в А. э. ярко представлена таутомерия кето-
енолъная; именно на этом примере наиболее четко были
установлены основные представления об этом явле-
нии. Аналогично в А. э. могут быть введены различные
радикалы, несущие заместители: окси-, алкокси-,
карбэтокси-, амино-, диалкиламино-, алктио- и дру-
гие группировки атомов, напр.:
ONa
СН3-С=СН—СООСаН5 4-С1 . сна— соос.щ —
— сн3—со—сн—соосан5
I 4- NaCl.
СНа-СООС2Н6
Натриевое производное А. э. хорошо алкилируется
также окисью этилена, причем в зависимости от усло-
вий образуется Р-оксиэтилацетоуксусный эфир (VI)
или ацетобутиролактон (V):
сн3-со-сн-соос2н
снгсн,он
г/
'5
Гомологи А. э. используются в раз-
личных синтезах. В этих целях их
подвергают т. и. кетонному расщепле-
нию для получения кетонов (действием
к-т и разбавленных щелочей) или кислотному рас-
щеплению для получения к-т (действием конц. щелочи).
Кетонное расщепление А. э.
R
Кислотное расшепление А. э.
R
СН3—СО—СН—COOR
| NaOH
R
СНз—со—CH-COOR
R
I
СНз-СО—СН—соон
СНзСО—СН2—R + СО2
СН3—СО—СН—COONa
{ NaOH
CHgCOONa + R—CHaCOONa
А. э. впервые был синтезирован в 1863 А. Гейтером
действием металлич. натрия на этилацетат. Этот
тип реакции, названный впоследствии сложноэфир-
ной конденсацией, лежит в основе современных мето-
дов получения различных кетонокислот, дикетонов
и др. типов веществ, содержащих кетогруппу.
Технич. методом получения А. э. является действие метал-
лич. натрия или алкоголята на этилацетат:
СНо-С—сн2
3 II I
fK /СО
О
III
г сн3—с сн2
Nx /СО
n;
4^6
IV
2СН3—СООСаН5 4- 2Na —
О—Na
— СН3-С=СН-СООСаН5 +
4- CaH5ONa 4- Н2
Из образующегося т. о. натриевого
производного А. э. выделяют действием
разбавленных минеральных к-т, сушат
и перегоняют в вакууме. Обычно ме-
таллический натрий распыляют в на-
гретом ксилоле; если реакцию прово-
дят с помощью алкоголята, то послед-
ний готовят из спирта и едкого натра,
причем образующаяся вода отгоняется
с выкипающей смесью бензола и спирта. А. э. готовят также
действием спирта на дикетен:
сн2=с-о
I I
н2с-с=о
+ с2н5он—- сн3-со-снасоосан5
А. э. является важным исходным продуктом для при-
готовления различных фармацевтических препаратов
и некоторых красителей для цветной фотографии.
Так, из А. э. приготовляют пирамидон, акрихин,
у-апетопропиловый спирт и ацетобутиролактон
(для произ-ва витаминаВ;),пиразолоновые кра-
+ NaJ сители, разнообразные кетоны, к-ты, кетоно-
кислоты и пр. Водные, спиртоводные и спир-
товые растворы А. э. при действии водного
р-ра FeClg приобретают интенсивную красно-фиоле-
товую ОКраСКу. И. Л. Кнунянц.
АЦЕТОФЕНОН (метилфенилкетон) СвН5СОСН8,
мол. в. 120,14 — бесцветная маслянистая жидкость
или крупные легко плавящиеся кристаллы с запахом
черемухи; т. пл. 20,5°; т. кип. 202,3°; dj" 1,0281;
1,5342; нерастворим в воде; хорошо растворим
в спирте, эфире, хлороформе, бензоле. Атомы водорода
метильной группы А. легк</ вступают в различные
реакции: так, при хлорировании получается <о-хлора-
цетофенон; с бензальдегидом образуется, например,
359
АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — АЦИЛИРОВАНИЕ
360
СвН6СН = СНСОСвН5; с эфирами хлормуравьиной
к-ты CICOOR и с амидом натрия А. реагирует в еноль-
С н
ной форме СеН6С(ОН) = СН2, образуя Н^С=С^
\COOR
При гидрировании А. на медно-хромовом катализа-
торе получают а-фенилэтиловый спирт. В пром-сти А.
получают конденсацией бензола с хлористым ацетилом
или уксусным ангидридом в присутствии А1С13 или
FeCl3: Ce Нв + (СН3СО)2О -> Се Н6СОСН3+ СН3СООН,
или же окислением этилбензола в жидкой фазе кис-
лородом воздуха в присутствии катализаторов (Со-,
РЬ- или Mn-солей нафтеновых к-т): СвН6СН2СН3 -ф-
i- О2 —> СеН5СОСН3 -|- Н2О. А. применяется в пар-
юмерии; обладает снотворным действием.
АЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ (алифатические
соединения, соединения жирного ряда) — органиче-
ские соединения, в которых атомы углерода соединены
между собой в прямые или разветвленные цепи.
Ациклические углеводороды в большом количестве
содержатся в природном газе и нефти. А. с. играют
очень важную роль в биологич. процессах; к А. с.,
в частности, относятся жиры и продукты их метабо-
лизма, а также многие аминокислоты, входящие
в состав белков, углеводы (сахара, крахмал, клет-
чатка) и др. R эфирных маслах многих растений содер-
жатся сложные эфиры, альдегиды, спирты и др. соеди-
нения жирного ряда. R природе обнаружены все
основные классы А. с.
Изучение А. с., в особенности сравнительно просто
построенных, было начато ранее исследования позд-
нее открытых карбо- и гетероциклич. соединений.
При изучении А. с. были в свое время сформулиро-
ваны очень многие представления, играющие важную
роль в органич. химии (гомологические ряды гео-
метрия. и оптич. изомерия, таутомерия, сопряжение,
полимеризация, радикалы свободные и Др.). А. с. чрез-
вычайно разнообразны и многочисленны, что видно
уже на примере углеводородов. Так, к ним относятся
углеводороды парафинового, олефинового, диенового,
ацетиленового рядов. От углеводородов производятся
разнообразные классы соединений, напр. галогено-
производные, спирты, амины и др., а также соеди-
нения с несколькими одинаковыми или различными
функциональными группами.
От А. с. возможен переход к циклич. органич.
соединениям различного типа. Так, при высокой
темп-ре ацетилен и его гомологи полимеризуются
в ароматич. углеводороды:
<>СН
СН^ СН
+ III
сн^ сн
сн
Большое практич. значение имеет дегидроциклиза-
ция алканов в ароматич. углеводороды (см. Арома-
тизация нефтепродуктов). Известны также многие
способы превращения А. с. в алициклические соеди-
нения (действие натрия на 1,3-дибромпропан, диме-
ризация аллена, сухая перегонка Са-солей двух-
основных к-т). Примерами превращения А. с. в гете-
роциклич. соединения могут служить дегидратация
диэтаноламина в морфолин, синтез пиридина из
глутаконового альдегида и NH3 или из смеси ацети-
лена и HCN и т. д. А. с. могут быть получены синте-
тически из соединений других рядов. При катали-
тической гидрогенизации циклопентановых углево-
дородов разрывается углеродное кольцо и образуются
ациклический углеводород пентан (гидрогенолиз).
Действием уксусного ангидрида на тетрагидрофуран
получают диацетат 1,4-бутиленгликоля.
Химические свойства А. с. с прямой цепью (нор-
мальные А. с.) являются типическими для данных
классов соединений (спиртов, аминов и т. д.).
А. с. имеют большое промышленное значение.
Основными источниками А. с. являются нефть, затем
продукты растительного происхождения и, наконец,
соединения, образующиеся в различных промышлен-
ных синтезах из СО и Н2. Различные углеводороды
жирного ряда (алканы и алкены) получают гл. обр.
переработкой нефти (крекинг и пиролиз). Нормаль-
ные углеводороды выделяют из нефти и продуктов ее
переработки в виде комплексов с мочевиной, или
с помощью «молекулярных сит» (см. Адсорбенты).
Для промышленного получения очень многих А. с.
исходными продуктами служат олефины, ацетилен и
окись углерода. Так, этиловый спирт синтезируют
гидратацией этилена, аналогичная переработка про-
пилена ведет к изопропиловому спирту, к-рый легко
можно дегидрировать в ацетон. Прямым окислением
этилена воздухом получают ацетальдегид. Из окиси
углерода и водорода получают синтетич. метанол и
синтетич. бензин, из окиси углерода и ацетилена
с последующим действием спиртов производят эфиры
акриловой к-ты. Ацетилен служит также исходным
продуктом для произ-ва уксусной к-ты, хлоропре-
на и др. Многие ненасыщенные А. с. используются в
произ-ве синтетич. каучука, искусственного волокна,
пластич. масс и т. д. На переработке различных А. с.
основано также произ-во мыла, многих органич.
растворителей (эфира, хлороформа, дихлорэтана, аце-
тона и др.), антифризов, душистых веществ и т. д.
М. И. Розенгарт, Я. Ф. Комиссаров.
АЦИЛИРОВАНИЕ — метод замещения водорода
в органич. соединениях остатком карбоновой к-ты
RCO. Типичными примерами могут служить: А. ами-
нов, превращение спиртов в сложные эфиры и др.
К реакциям А. относятся формилирование, бензоили-
рование или введение др. остатков карбоновых к-т.
Б качестве ацилирующих агентов применяют свобод-
ные к-ты или, чаще, более реакционноспособные
эфиры, ангидриды или хлорангидриды к-т. Для фор-
милирования аминов применяют свободную НСООН,
а также формамид, реагирующий по схеме: RNH2 -|-
+ HGONH, — RNHGHO + NH3. Наряду с уксусной
к-той, ее ангидридом и хлор ангидридом для ацети-
лирования широко применяют и кетен: RNH2 -|-
+ СН2=С=О —> RNHCOCH3. Для бензоилирова-
ния часто прибегают к реакции Шоттена—Баумана,
заключающейся в замещении водорода в окси- или
аминогруппе действием хлористого бензоила в при-
сутствии водного раствора едкой щелочи или соды.
Нередко прибегают к временному А. («защите» NH2-
или НО-групп), чтобы сделать возможным такие
дальнейшие превращения ацилированного продук-
та, которые затруднены или невозможны при на-
личии свободных NH2- или НО-групп. Так, при
производстве сульфаниламида («белого стрептоци-
да») исходят из ацетанилида, который превраща-
ют сначала в n-CH3CONHCeH4SO2Gl и затем в
n-CH3GONHCeH4SO2NH2, от которого отщепляют
ацетильную группу и переходят к сульфаниламиду
n-NH2CeH4SO2NH2 со свободной аминогруппой.
При получении хлорангидридов из аминокислот или
пептидов для «защиты» аминогруппы обычно прибе-
гают к предварительному замещению атома водорода
в аминогруппе трифторацетильной, формильной или, в
особенности, карбобензилоксигруппой (СвН5СН2ОСО),
т. к. эти группы в дальнейшем легко отщепляются без
одновременного гидролиза пептидной связи.
Смешанные ангидриды кремневой и карбоновой
к-т, например (CH3COO)4Si, являются эффективными
ацилирующими средствами. Во многих случаях при
А. ангидридами карбоновых к-т процесс значительно
361
АЦИЛОИНЫ — АЭРОЗОЛИ
362
облегчается в присутствии ангидрида трифторуксус-
ной к-ты; при этом промежуточно образуются сме-
шанные ангидриды CF3COOCOR, представляющие со-
бой эффективные ацилирующие агенты. Помимо спир-
тов и аминов, легко ацилируются и др. соединения
с подвижными атомами водорода, напр. меркаптаны,
фенолы. К реакциям А. особого типа (С-ацилирова-
ние) можно отнести и получение ароматич. кетонов
взаимодействием ароматич. углеводородов с анги-
дридами или хлорапгидридами к-т в присутствии
А1С13 (см. Фриделя-Крафтса реакция).
Лит.: U 1 1 m а п п, 3 Aufl., Bd 3, MUnchen—В., 1953,
S. 87—92. Я. Ф. Комиссаров.
АЦИЛОИНЫ (кетолы) — органич. соединения, со-
держащие оксикетонную группировку— СН(ОН)СО—,
обычно связанную с одинаковыми радикалами. Про-
стейшим представителем А. является ацетоин,
СН3СН(ОН)СОСН3, содержащийся в рацемич. форме
во многих винах. Ароматич. А. (бензоины) обычно
получают конденсацией альдегидов в водном спирте
в присутствии KCN: СвН6СНО(1) + СвН6СНО —>
—>СеН6СОСН(ОН)СвН6(П). Механизм этой реакции,
носящей название бензоиновой конденсации, заклю-
чается в образовании циангидрина CeH6CH(CN)O, в его
прототропном превращении в CeH6C(OH)CN, к-рый
присоединяет молекулу (I) по схеме альдольной кон-
денсации с образованием CeH6CH(O)C(CeH6)(OH)CN;
в дальнейшем отщепляется CN и образуется (II).
Образование нек-рых А. жирного ряда в организмах
протекает под влиянием ферментов. Так, сбражива-
ние пировиноградной к-ты свежими дрожжами (фер-
мент карболигаза) ведет к левовращающему ацетоину
с промежуточным образованием ацетальдегида:
2СН3СОСООН —> 2СО-. + 2СН3СНО —► СН3СОСН(ОН)СН3
Общий способ получения А. жирного ряда состоит
в действии металлич. натрия на эфиры карбоновых
к-т с последующим гидролизом образовавшегося нат-
риевого соединения:
RCOOR RC(OR)ONa _|_2Na RCONa
+ 2Na RC(OR)ONa RCONa
RCOH
— || RCOCHOHR
RCOH
Широкое развитие получил метод синтеза циклич.
А. действием металлич. Na на эфиры дикарбоновых
кислот:
.COOR I I
(CH2)n\ --** (CHaln
COOR I----СНОН
Эта реакция, представляющая собой внутримолеку-
лярный ацилоиновый синтез, протекает гладко с хо-
рошими выходами и представляет практич. интерес,
т. к. от полученных циклич. оксикетонов можно
легко перейти к циклич. кетонам, находящим приме-
нение в парфюмерии.
Лит.: Органические реакции, сб. 4, пер. с англ., М., 1951,
с. 215—28. Я. Ф. Комиссаров.
АЭРОЗОЛИ — дисперсные системы, состоящие из
мелких твердых или жидких частиц, взвешенных
в газовой среде (обычно в воздухе). А., дисперсная
фаза к-рых состоит из капелек жидкости, наз. тума-
нами, а в случае твердой дисперсной фазы — ды-
мами; пыль относят к грубодисперсным А. Размеры
частиц в А. изменяются в очень широких пределах —
от нескольких миллиметров (хлопья снега, капли
дождя) до 10* ’ см. Дисперсность большинства А. зна-
чительно ниже, чем у коллоидных систем, и близка
к дисперсности эмульсий и суспензий, поэтому пра-
вильнее называть А. аэросуспензиями (дымы и пыли)
и аэроэмульсиями (туман). В туманах частицы яв-
ляются капельками шарообразной формы, в дымах
и пылях представляют собой кристаллики, их об-
ломки, аморфные образования разнообразной формы
или агрегаты-хлопья, состоящие из отдельных мел-
ких частиц.
Образование А. в различных природных и произ-
водств. процессах происходит двумя путями: диспер-
гированием и конденсацией. А. образуются при меха-
нич. измельчении и распылении твердых тел или
жидкостей: дроблении, истирании, взрывах, распы-
лении в форсунках и пульверизаторах и т. д. Так
возникают туманы в районе мощных водопадов,
шахтная пыль при бурении, отбивании и взрывании
руд и угля. Огромное облако А., состоящее из жид-
ких и твердых радиоактивных частиц, возникает
в результате взрыва атомной бомбы. Методы диспер-
гирования приводят к образованию полидисперсных
и сравнительно грубодисперсных А. Более высоко-
дисперсные и однородные по дисперсности А. полу-
чаются конденсационными методами, к к-рым от-
носятся: переходы пресыщенных паров в жидкое и
твердое состояние (напр., образование туманов и
облаков), а также химич. реакции, приводящие
к появлению новых жидких или твердых фаз (напр.,
хлористого аммония при взаимодействии газообраз-
ных хлора и аммиака). Обязательным условием
возникновения А. конденсацией является наличие
пересыщенного пара. При этих условиях А. может
образоваться: при охлаждении газовой смеси, содер-
жащей пар; при смешении газов и паров, имеющих
разные темп-ры; при адиабатич. расширении паров;
конденсацией пара на поверхностях; в результате
химич. реакции газообразных в-в. При химич. реак-
циях А. возникают, когда образуется новая фаза
с низким давлением насыщенного пара. Так, А. полу-
чается при испарении серного ангидрида во влажном
воздухе и при смешении хлористого водорода и ам-
миака: SO3 (газ) + Н2О (газ) = H2SO4 (новая жид-
кая фаза); НС1 (газ) + NH3 (газ) — NH4C1 (новая
твердая фаза).
Дисперсность А. зависит от соотношения скорости
образования ядер (центров) конденсации и скорости
их роста. Центрами конденсации, помимо групп моле-
кул, возникающих в результате флуктуационных
сгущений, являются ионы, а также дисперсные при-
меси в атмосфере, напр. пылинки, особенно если они
несут электрич. заряд. Многие свойства А. как
дисперсных систем связаны с наличием высокораз-
витой поверхности дисперсной фазы. А. характери-
зуются, прежде всего, дисперсностью и концентрацией
(весовая концентрация — количество дисперсной фазы
в единице объема, или частичная концентрация —
число частиц в единице объема).
Весовую концентрацию А. определяют, взвешивая фильтр
до и после пропускания через него измеренного объема А.
Для определения частичной концентрации А. подсчитывают
с помощью микроскопа число частиц, осевших из пробы газа
на пластинку. Применяют также метод поточной ультрамик-
роскопии, регистрируя по наблюдаемым «вспышкам» общее
число частиц п, прошедших через освещенную зону. Частич-
ная концентрация в этом случае равняется n/v, где v — про-
текший объем А. Размеры и форму частиц А. определяют при
помощи оптич. или электронного микроскопа после осаждения
пробы на соответств. поверхности. В поточном ультрамикро-
скопе размер частиц А. определяют по исчезновению видимости
частиц при уменьшении интенсивности освещения. Размеры
очень мелких заряженных частиц устанавливают также по их
подвижности в электрич. поле или по скорости их седимента-
ции, напр., фотографированием падающих частиц с небольшой
выдержкой.
Особенности А. и их отличия от коллоидных раство-
ров в значительной мере обусловлены отсутствием
жидкой дисперсионной среды, большой разницей
в плотностях дисперсной фазы и дисперсионной среды
363
АЭРОЗОЛИ
364
и малой вязкостью последней. В А. ярко выражены
явления диффузии частиц, их интенсивного броунов-
ского движения; последнее приводит к частым столк-
новениям частиц и к быстрой агрегации их в более
крупные с большой скоростью оседания. Вследствие
этого частичная концентрация А. всегда самопроиз-
вольно и быстро уменьшается. Дисперсность А. не-
прерывно изменяется также и вследствие испарения
частиц или конденсации паров на их поверхности
(на капельках). Давление насыщенного пара над
частицами А. увеличивается с уменьшением их ра-
диуса и выражается:
In pr/p0 = 2яМ1НТ?>— для незаряженных частиц;
In рг /р0 = М/ЯТр (2а/г — е2/8ет4) — для заряженных частиц
(с зарядом е).
Здесь рг и р„ соответственно — давление насыщен-
ного пара над частицей (имеющей радиус г) и над
плоской поверхностью, а — поверхностное натяже-
ние частицы (капельки), М — молекулярный вес,
R — газовая постоянная, Т — абсолютная темп-ра,
р — плотность частицы. В полидисперсных А. вслед-
ствие различия между давлением насыщенного пара
у частиц разных размеров происходит исчезновение
мелких частиц (их испарение) и укрупнение больших
частиц за счет конденсации на их поверхности пара
(изотермич. перегонка в А.).
А. проявляют значительно большую, чем дисперс-
ные системы в жидких средах, кинетич. и агрегатив-
ную неустойчивость. В отличие от коллоидных рас-
творов, в А. силы, препятствующие слиянию частиц
при столкновениях, очень слабы или совсем отсут-
ствуют. Повышение устойчивости А. (их стабилиза-
ция) осуществляется за счет приобретения электрич.
зарядов или действия диффузионных сил, т. е. диф-
фузии паров из объема между двумя ближайшими
капельками и засасывания вследствие этого в данный
объем (зазор) газа из окружающей среды. Такое
диффузионное препятствие слиянию (коалесценции)
капелек может иметь большое значение в туманах,
но исчезает при полном насыщении газовой среды
парами данной жидкости.
Частицы А., будучи нейтральными в момент обра-
зования, в дальнейшем могут приобретать заряд,
адсорбируя ионы из газоной среды. При этом все
условия, способствующие ионизации газа (ультра-
фиолетовые, рентгеновские, космич. лучи, радиоак-
тивные излучения), способствуют и возникновению
заряда у частиц А. Источником заряда частиц А.
является также трение их друг о друга или о сопри-
касающиеся твердые поверхности. В отличие от частиц
лиозолей, частицы А. не имеют диффузионного слоя
ионов (противоионов). Частицы одного и того же А.
могут иметь различные по знаку и величине заряды
или быть нейтральными. Наиболее стойкими яв-
ляются А. с одноименно заряженными частицами.
Биполярная зарядка частиц ускоряет коагуляцию А.
Луч света, попадающий в А., рассеивается взве-
шенными частицами и поглощается ими. Если
где А, — длина волны света, аг — размер частицы А.,
то свет отражается поверхностью частицы под опре-
деленным углом. Если же А/г>1, то происходит не
отражение, а рассеяние света. В этом случае свет
распределяется от частицы во всех направлениях.
Избирательность поглощения (абсорбции) света в-вом
частиц А. вместе с явлением светорассеяния и обус-
ловливает ту или иную окраску А. В ряде случаев
(произ-во сажи, цветная металлургия) необходимость
осаждения А. обусловлена ценностью вещества А.
Образование А. часто нежелательно, особенно
в производств, процессах, так как это способствует
уносу ценных в-в (пыль в металлургии, в цементной,
химич. пром-сти и др.), загрязнению продуктов и атмо-
сферы, вредно действует на здоровье людей и на меха-
низмы. А. образуют опасные взрывчатые смеси
с воздухом (угольная, сахарная, мучная пыль), сильно
поглощают свет. С другой стороны, А. широко ис-
пользуются для решения практич. задач. В медицине
А. применяют для введения в организм лекарств,
веществ ингаляцией, для дезинфекции, в с. х-ве —
для защиты растений от болезней и вредителей опы-
лением инсектицидами. А. применяют в случае пнев-
матич. окраски и металлизации различных поверх-
ностей путем нанесения распыленных металлов.
В военном деле А. применяются для целей маскировки
(дымовые завесы). Проблемы А. приобретают большой
интерес в связи с решением многих вопросов метео-
рологии, искусств, разрушения туманов и искусств,
дождевания, борьбы с запыленностью и загрязнением
воздуха городов и производств, помещений, улавли-
ванием ценных продуктов и т. д. Большое значение
приобрела задача осаждения радиоактивных А., обра-
зующихся в атомной пром-сти, и изучение распростра-
нения радиоактивных А. в атмосфере в связи с атом-
ными взрывами.
Методы борьбы с А. основываются на устранении
причин их возникновения и стабилизирующих факто-
ров и ускорении естественных процессов разруше-
ния А. (седиментация, коагуляция). Для разрушения
аэроколлоидных систем на практике применяют ме-
тоды, основанные на фильтрации А. через пористые
фильтры из войлока, бумаги, тканей, а также методы,
основанные на промывке встречным потоком жидкости
или дождя. Большое значение имеет метод осажде-
ния А. электрич. полем высокого напряжения (до
50 000 в), что вызывает ионизацию газа; частицы А.
получают дополнительный заряд и осаждаются на
противоположно заряженном электроде. Разбрасы-
вание тонкоизмельченного заряженного песка или
так наз. углекислого снега (твердой СО2) с самолета
может вызвать коалесценцию капелек в облаках и
туманах. Для разрушения А. применяют также зву-
ковые волны высокой частоты (2—10 кгц). Частицы А.
вибрируют под действием звуковых волн, благодаря
чему учащаются их столкновения, что ведет к коагу-
ляции и осаждению А. Наблюдаются случаи коагу-
ляции А. под действием высокой темп-ры.
Лит.: Гиббс В., Аэрозоли, пер. [С англ.], Л., 1929;
Уайт л о у-Гр ей Р., Паттерсон X., Дым, пер.
с англ М.—Л., 1934; Вейцер Ю. И. и Л у ч ин-
ея и й Г. П., Маскирующие дымы, М.—Л., 1947; Новые идеи
в области изучения аэрозолей. Сб. статей, М.—Л., 1949;
Амелин А. Г., Теоретические основы образования тумана
в химических производствах, М.—Л., 1951; Air pollution, N. Y.,
1952; Фукс H. А., Механика аэрозолей, М., 1955; М е 1-
d а и R., Handbuch der Staubtechnik, Bd 1, Dusseldorf, 1956;
ilreen H. L. and Lane W. R., Particulate clouds: dusts,
smokes, and mists, L., 1957; A v у A. P., Les aerosols, P.,
1956; Herzka A. and P 1 c k t h a I 1 J., Pressured packa-
ging (Aerosols), L., 1958; Рязанов В. А., Санитарная ох-
рана атмосферного воздуха, М., 1954; Елкин И. И., Э й-
дельштейн С. И., Аэрозоли антибиотиков, их получение
и клиническое применение, М., 1955; Применение аэрозолей
в сельском хозяйстве, сб. переводов иностранной периоди-
ческой литературы, М., 1955; Коротких Г. И.. Аэрозоли
и их применение в сельском хозяйстве, М., 1956.
А. Г. Амелин
БАКЕЛИТ — СМ. Смолы феноло-алъдегидные.
БАКТЕРИЦИДЫ — химические средства уничто-
жения бактерий — возбудителей заболеваний чело-
века, животных, растений и сапрофитов. К Б. отно-
сятся также вещества, к-рые не убивают бактерий,
а только задерживают их рост; такие вещества назы-
вают бактериостатическими. В качестве
Б. применяют различные неорганич. и органич. со-
единения. Из неорганич. веществ бактерицидным дей-
ствием обладают галогены, окислы азота, гипохло-
риты, хлориты, двуокись хлора, перекись водорода
и нек-рые другие (в том числе соли серебра и ртути).
Широкое применение в качестве Б. получил хлор,
к-рый в концентрации 1—3 мг/л применяется для
обеззараживания водопроводной воды; бактерицид-
ная активность хлора по отношению к кишечной
палочке проявляется при концентрации 0,021 мг/л.
Высокой бактерицидной активностью обладают также
двуокись хлора и гипохлориты щелочных и щелочно-
земельных металлов, к-рые в концентрации 1 мг/л
при экспозиции 60—70 мин обезвреживают кишечную
палочку. Наряду с хлором и гипохлоритами в ка-
честве Б. широкое применение находят различные
органич. хлорамиды, к-рые по активности прибли-
жаются к хлору (в пересчете на активный хлор),
а нек-рые хлорамиды обладают более сильными бак-
терицидным действием, чем хлор. Из хлорамидов,
получивших практич. применение следует назвать
хлорамин Б (CeH6SO2NClNa • Н2О), хлорамин Т
(п -CH3CeH4SO2NClNa • Н2О), дихлорметансульфа-
мид и др. В последнее время в качестве активных Б.
используют 1,3-дихлор-5,5-диметилгидантоин (I), три-
хлорциануровую к-ту (II) и натриевую соль дихлор-
циануровой к-ты (III), к-рые в концентрации 50-мг/л
нзс\
нзс/
С----NCf
СО ^СО
^NCl
NCl
ОС^ ''СО
I I
ON ^NCl
го
N
ОС ''''CON а
I I
C1N NCI
^ccf
III
при контакте в течение 10 сек убивают большинство
видов не споровых форм бактерий. Наиболее интерес-
ным из этих соединений является трихлорциануровая
к-та, содержащая 91,5% активного хлора. Из других
галогенов практич. применение нашел иод (в виде
5—10%-ного спиртового р-ра или в водном р-ре KJ).
Активными Б. являются фенол и его различные
производные. В водной среде на большинство бакте-
рий фенол действует в разведении 1 : 90 — 1 : 140.
Бактерицидное действие гомологов фенола с увели-
чением мол. веса повышается до известного предела,
к-рый различен для грамположительных и грамотри-
цательных микроорганизмов. В табл. 1 приведена
бактерицидная активность нек-рых гомологов и
производных фенола на двух видах бактерий.
Таблица 1
Фенольный коэффициент
Вещество
на золотистом на кишечной
стафилококке палочке
Фенол.......................
4-Метилфенол................
4-Этилфенол.................
4-Пропилфенол...............
4-Бутилфенол................
4-Амилфенол.................
4-Гексилфенол ..............
2-Оксидифенил...............
2,3
6,3
16,3
43,7
125,0
313
25
2,3
6,3
18,3
46,3
53,3
33,3
30
Бактерицидное действие фенола повышается и при
замещении водорода галогенами, напр. дихлорфенолы
имеют фенольный коэфф. 10—13, трихлорфенолы
18—25. Различные изомеры галогенофенолов обладают
различной бактерицидной активностью. Из трихлор-
фенолов наиболее активен 2,4,5-трихлорфенол. Высо-
кой бактерицидной активностью обладает также пента-
хлорфенол, но действие его несколько слабее, чем
2,4,5-трихлорфенола. Высокой бактерицидной актив-
ностью обладают также нафтолы и их галогенопроиз-
водные.
В качестве Б. и дезинфекционных средств приме-
няют также соли четвертичных аммониевых оснований,
содержащие углеводородные радикалы с длинной
цепью. Соединения такого типа обладают гл. обр.
не бактерицидным, а бактериостатич. действием; они
задерживают рост большинства бактерий в очень
больших разведениях. Так, бромистый триметилцетил-
аммоний задерживает рост золотистого стафилококка
в разведении 1 : 80 000, а брюшнотифозной палочки
в разведении 1 : 40 000. Практич. применение для
целей дезинфекции нашел препарат зифероль,
к-рый представляет собой 10%-ный водный р-р диме-
тилбензилалкиламмония хлористого в воде (где ал-
кил— смесь от С8Н15 до С18Н3,). Этот препарат задер-
живает рост многих бактерий в разведении 1 : 8ОО0 —
1 : 20 000. Такого же типа веществом является пре-
парат фомероль, содержащий вместо большого
углеводородного радикала остаток алкилфенилового
эфира полиэтиленгликоля. Следует отметить, что все
соли четвертичных аммониевых оснований, обладаю-
щие бактерицидным и бактериостатич. действием,
сильно токсичны для теплокровных животных; фун-
гицидное действие этой группы соединений незна-
чительно.
Высокой бактерицидной и бактериостатич. актив-
ностью обладают органич, соединения ртути. В табл. 2
приведена микробиология, активность нек-рых орга-
нич. соединений ртути. Эти соединения применяют
в качестве средств предотвращения роста различных
микроорганизмов. Следует указать, что ароматич.
соединения ртути обладают более сильным бактери-
цидным действием и более слабым фунгицидным, в то
367
БАЛЛИСТИТЫ — БАЛЬЗАМЫ
368
время как алифатич. соединения ртути обладают
более сильным фунгицидным действием и слабее
действуют как Б.
Таблица 2
Вещество Максимальное разведение, задерживающее рост
золотистого стафилококка кишечной палочки
Фенилмеркурхлорид 1 : 2000000 1 : 300000
Фенилмеркурбромид 1 : 3000000 1 : 350000
Фенилмеркурнитрат (основной) 1 : 4000000 1 : 500000
Мертиолят(С»Н5Н§8СсН4СООМа) 1 : 1000000 1 : 500000
З-Пиридилмеркурацетат .... 1 : 1700000 1 : 400000
З-Пиридилмеркурхлорид .... Оксимеркур-о-нитрофенол (мер- 1 : 2000000 1 : 550000
курофен) 1 : 9000000 1 : 150000
Сулема 1 : 2000000 1 : 250000
Высокой бактерицидной активностью обладает
также этилмеркурфосфат, к-рый рекомендован для
пропитки перевязочных материалов в концентрации
в десять раз меньшей по сравнению с сулемой.
Соли серебра и металлич. серебро обладают высокой
бактерицидной активностью и с успехом могут быть
применены для обеззараживания питьевой воды.
В последнее время в качестве Б. широко применяют
Р-пропиолактон, получаемый из кетена и формаль-
дегида.
Бактерицидная активность всех препаратов сильно
зависит от темп-ры (при ее повышении — возрастает)
и pH среды. Наличие органич. веществ, особенно
белкового происхождения, являющихся питательной
средой для микроорганизмов, резко снижает актив-
ность всех классов Б.; как правило, чем более сильно
действует Б. в воде, тем более резко снижается его
активность в присутствии белков.
В качестве Б. практич. применение получили также
хлорпикрин и формалин; последний используется
в виде водных р-ров (1—5%) и в парах (0,5—12 г/м3
для фумигации помещений, одежды и т. п.).
Лит.: В а ш к о в В. И., Руководство по дезинфекции,
дезинсекции и дератизации, М., 1952; Альтман Р. С.,
Левенсон К. С., Стерилизация воды препаратами серебра,
Баку, 1941; D i t z е 1 R., А г v a n Р., Symes W., Soap
and Chem. Specialties, 1956, 32, № 2, 125.
H. //- Мельников.
БАЛЛИСТИТЫ — бездымные пороха, получаемые
пластификацией коллоксилина 40—50 вес. % нитро-
глицерина (или дигликольдинитрата) без применения
легколетучего растворителя; при этом получат трудно
детонирующую твердую роговидную массу, обладаю-
щую типичными свойствами пороха. Для стабилиза-
ции таких порохов в них добавляют 1% анилина или
дифениламина. Этот тип порохов за их высокие
баллистич. свойства был назван баллиститами. Для
получения Б. компоненты пороховой массы смеши-
вают в большом избытке воды, после чего пороховую
массу подвергают горячей обработке под давлением
(напр., на вальцах) и формуют шнуры и полосы нуж-
ного сечения, к-рые затем разрезают на зерна. Б. при-
меняются как в ствольном огнестрельном оружии,
так и в ракетных стартовых и маршевых двигателях.
В качестве примера ниже приводятся составы нек-рых
порохов баллиститного типа. Ракетные по-
роха. 1. Германский R-61 : 59,8% нитроцеллю-
лозы (компонент); 35,3% диэтиленгликольдинитрата
(пластификатор); 1,1% этилфенилуретана, 0,8% ди-
фенилуретана, 0,3% несимметрич. дифенилмочевины
(стабилизаторы); 0,6% KNO3, 0,25% MgO (катали-
заторы горения); 0,35% воска и 1,5% гидроклет-
чатки. 2. Американский IRN : 51,3% нитроцеллюлозы;
43% нитроглицерина, 3,5% диэтилфталата (пластифи-
каторы); 1% диэтилдифенилмочевины (стабилизатор);
1,2% K2SO4 (катализатор горения); 0,2% сажи и
0,1% воска. Порох ствольной артил-
лерии. 1. Германский кубический: 61,7% нитро-
целлюлозы; 20% нитроглицерина, 14,5% тринитро-
толуола, 3,5% динитротолуола (пластификаторы);
0,3% диэтилдифенилмочевины (стабилизатор). 2. Италь-
янский «соленит»: 65,5% нитроцеллюлозы; 33% нитро-
глицерина (пластификатор) и 1,5% диэтилдифенил-
мочевины (стабилизатор).
Многокомпонентность ракетных порохов объяс-
няется необходимостью обеспечивать устойчивое горе-
ние пороха при малых давлениях газов и в условиях
возможного выброса через сопло двигателя продуктов
первичного термин, распада пороха. Теплота горения
Б. разных составов (для воды жидкой) может меняться
примерно от 650 до 1 250 ккал/кг. Так, для герман-
ского ракетного пороха Б-61,9Ж = 890 ккал/кг, а
для американского IRN QJ|; = 1 230 ккал/кг. В по-
следнем случае единичный импульс реактивной силы
= 230—240 кг-сек/кг.
Основные достоинства порохов баллиститного типа:
возможность широкого применения как для стволь-
ного, так и для ракетного оружия; сравнительное
постоянство физико-химич. и баллистич. свойств
орудийных порохов при их хранении; довольно широ-
кий диапазон энергетич. характеристик; быстрота
изготовления. Главные недостатки: большая коли-
чественная зависимость скорости горения в реак-
тивных двигателях от темп-ры пороха и от давления
газа; падение физико-химич. и механич. прочности
ракетных пороховых цилиндров с ростом их попе-
речных размеров; сравнительная опасность произ-ва
порохов баллиститного типа.
Лит.: В е и н е н Л., Бюрл о Э., Л е к о р ш е А.,
Пороха и взрывчатые вещества, пер. с франц., М., 1936;
У им пресс Р. Н., Внутренняя баллистика пороховых
ракет, пер. с англ., М., 1952; Sutton G. Р., Rocket pro-
pulsion elements, N. Y.—L., 1956; Хамфрис Дж-, Ракет-
ные двигатели и управляемые снаряды, пер. с англ., М., 1958.
А. С. Бакаев.
БАЛЬЗАМ ШОСТАКОВСКОГО (винилин) — поли-
винилбутиловый эфир
СН3—СН — [-СН.-СН-1 -СН-СНз
I I * I
ОС^Нв OC^Hg OC.iH()
мол. в. ок. 2 000, густая вязкая от светло-желтого
до желтого цвета жидкость со специфическим запа-
хом; мало растворим в этиловом спирте и нераство-
рим в воде; смешивается во всех отношениях с бути-
ловым и изоамиловым спиртом, хлороформом, эфи-
ром, ацетоном, а также с растит, и минеральными
маслами; на воздухе не изменяется. Водная вытяжка
имеет нейтральный характер, d 0,90—0,92, вязкость
1%-ного бензольного р-ра при 20° 0,841 сантипуаз,
zip 1,444—1,452. Потеря в весе при высушивании
Б. Ш. (90—95° в течение 4 час.) не должна превышать
20%. Б. Ш. получают полимеризацией винилбутило-
вого эфира в присутствии катализатора, напр. хлор-
ного железа, растворенного в бутиловом спирте.
Применяют Б. Ш. при фурункулах, трофич. язвах,
гнойных ранах, маститах, ранениях, ожогах, отмо-
рожениях, воспалительных процессах и язвах.
Лит.: Шостаковский М. Ф., Простые виниловые
эфиры, М., 1952; Авакова В. А. и Шостаков-
ский М. Ф., Сов. медицина 1949, № 6, с. 17; Ижев-
ский К. М., там же, 1949, № 10, с. 31; К урс а нов А. Л.
(и др.), Вестн. рентгенологии и радиологии, 1952, № 6,
с. 68; Машковский М. Д., Лекарственные средства,
4 изд., М., 1960. А. М. Хомутов.
БАЛЬЗАМЫ (от греч. fiihaapov — бальзамовое
дерево) — растительные или синтетич. сложные про-
дукты. Растительные Б. обычно густые сиро-
пообразные жидкости с пряным, ароматич. запахом,
369
БАЛЬЗАМЫ — БАРБИТУРАТЫ
370
горьким, острым вкусом и кислой реакцией. Б. —слож-
ные смеси безазотистых органич. соединений, гл. обр.
эфирных масел и растворенных в них смол, а также
ароматич. соединений (коричной, бензойной к-т и их
эфиров), альдегидов, кетонов, спиртов и т. д. На
воздухе большинство Б. постепенно твердеет. В воде
они почти нерастворимы, растворимы полностью
или ограниченно в спиртах, эфире, хлороформе,
бензоле, эфирных маслах и т. д. Многие Б. обра-
зуются в растениях как продукты нормального об-
мена и содержатся гл. обр. в особых межклеточных
вместилищах или ходах коры, реже — в древесине и
листьях; другие — являются продуктами патологич.
деятельности растений, появляющимися при повре-
ждении коры и в самом растении не содержатся.
Добывают Б. подсочкой или вываркой; применяют
в медицине, для приготовления нек-рых парфюмерных
и косметич. составов, в произ-ве лаков. Практич.
значение имеют след. Б.:
Копайский Б. — добывается из деревьев рода Copio-
fera (Copiofera officinalis L.), произрастающих в Юж. Америке.
Светло-желтая или желто-бурая маслянистая масса жидкой
или густой консистенции (в зависимости от содержания эфир-
ных масел); обладает зеленой флуоресценцией, пряным запа-
хом и горьким, острым вкусом; d 0,92—0,95, растворяется
в спирте и хлороформе, содержит 45—60% смол (смоляные
к-ты недостаточно изученного состава) и эфирное масло;
число омыления 90—106. Применяется для приготовления
лаков.
Перуанский Б. — добывается из деревьев рода
Myroxylon Pereirae (Сан-Сальвадор). Красно-коричневая или
темно-коричневая сиропообразная жидкость с запахом, напо-
минающим ванилин, пряного, острого вкуса, с кислой реак-
цией; растворим в спиртах. Состав перуанского Б.: 5—6%
коричной к-ты 56—70% бензиловых эфиров бензойной п корич-
ной к-т, 2 0—30% смол и немного ванилина; применяется
в медицине.
Толуанский В. — добывается из деревьев Toluifera
balsamum или Myroxylon toluiferum, произрастающих в Юж.
Америке. Желто-коричневая густая жидкость нежного запаха
и слегка жгучего, приятного вкуса; при длительном стоянии
затвердевает и может быть растерт в желтый порошок.
Растворяется толуанский Б. в кипящем спирте, хлороформе,
ацетоне, уксусной к-те, водном КОН. Толуанский Б. содержит
бензойную и коричную к-ты в свободном состоянии (12—15%)
и в виде эфиров (75—80%), а также ванилин, смолы, эфирные
мавпа,- применяется для лечебных и косметич. целей.
Многие природные Б., обладая теми или иными
целебными свойствами, долгое время применялись
в качестве лечебных средств; в настоящее время их
применение для этих целей весьма ограничено. Позд-
нее Б. начали называть также и нек-рые виды живиц,
не имеющих лечебного значения, к числу к-рых отно-
сятся, напр., канадский и пихтовый Б.
Канадский Б. добывается из разновидности ели
Abies balsamea (Сев. Америка) — прозрачная желтоватая
вязкая жидкость; 0,985—0,997; nj} 1,518—1,523; [a]jy
от -pl до 4-5°; кислотное число 80—90; эфирное число 4—8;
содержит 15—25% эфирных масел. Применяется канадский
Б. для приготовления фармацевтич. эластического коллодия,
для склейки оптич. стекол и для монтажа микроскопии, препа-
ратов, т. к. при высыхании не выделяет кристаллов, остается
прозрачным и имеет стабильный показатель преломления.
Пихтовый Б. — продукт переработки живицы, добы-
ваемой из пихты сибирской (Abiees sibirica). Живицу после
многократной фильтрации отмывают от водорастворимых
органич. к-т и отгоняют летучую часть (пихтовый скипидар).
Полученный желто-оранжевый прозрачный сплав (Б. пихто-
вый) состоит из смоляных к-т (70—75%), неомыляемых (20—
23%) и окисленных (4—5%) в-в, растворенных в жидком эфир-
ном масле; в составе последнего находится гл. обр. а-пинен.
Б. пихтовый растворим в эфире, ароматич. углеводородах и эти-
ловомспирте; кислотное число 80—85, число омыления 98—102;
темп-pa размягчения 65—75°, d 1,0—1,07, пр 1,52 — 1,54,
коэфф, линейного расширения 1,5 • 10“4— 2,0- 10“4 (0—
25е). Пихтовый Б., так же как и канадский Б., при высыхании
не образует кристаллов, показатели преломления этого баль-
зама и стекла близки; он термо-свето- и морозостоек. Пихтовый
Б. (в чистом виде или в р-ре, обычно в ксилоле, иногда с добав-
кой пластификаторов) применяется для склеивания оптич.
деталей, при монтировании шлифов горных пород и минералов
и для изготовления микроскопии, препаратов. Пихтовый Б.
производится в СССР. К типу Б. относят иногда и нек-рые
скипидары.
Иногда Б. называются также нек-рые синтетич.
полимеры, напр. бальзам Шостаковского, хотя состав
таких Б. совершенно иной по сравнению с природ-
ными. д, с. Поваров, Н. А. Гурич,
БАРБАМИЛ [амитал-натрий, 5-этил-5-(3-метил-
бутил)-натрий барбитурат], мол. вес 248,25 — бе-
лый порошок без запаха; хорошо растворим в во-
де и спирте. Из вод-
ного раствора Б. при
подкислении выпадает
5 - этил-5 - изоамилбар-
битуровая кислота, а
при прибавлении р-ра
AgNO3 (1 : 40) выде-
О Na
СН3
СН3
^СНСН2СН2 “I
;с—о
СН„СН2 хс—Nz
32 II I
о н
ляется осадок серебряной соли этой к-ты. Дает все ре-
акции, характерные для барбитуратов. Для колич.
определения водный р-р Б. титруют 0,1 н. соляной
к-той (индикатор — метиловый оранжевый). Полу-
чают Б. конденсацией диэфира 5-этил-5-изоамилмало-
новои к-ты с мочевиной в присутствии алкоголята
натрия и взаимодействием образовавшейся 5-этил-
5-изоамилбарбитуровой к-ты с вычисленным коли-
чеством NaOH. Применяют Б. в качестве снотворного
препарата, а также успокаивающего и противосудо-
рожного средства.
Лит. см. при ст. Адалин.
Е. С. Еоловчинская.
БАРБИТУРАТЫ — производные барбитуровой кис-
лоты, у к-рых связанные с N и С5 водородные атомы
замещены различными радикалами.
Ниже рассмотрены только двухзаме-
щенные в положении 5 производные
барбитуровой к-ты. Многие из них
обладают отчетливо выраженным
(S1OC2
HN—СО
R
. . R*
HN—СО
снотворным действием (к-рое не наблюдается у произ-
водных барбитуровой к-ты, связанной только с одним
радикалом у С5) и широко применяются в качестве
снотворных и наркотич. средств. Впервые снотворная
активность Б. была обнаружена Э. Фишером и ф. Ме-
рингом в начале 20 в. при изучении 5,5-диэтилбар-
битуровой к-ты; в 1904 Э. Фишером был опублико-
ван удобный способ получения этого препарата, на-
званного им вероналом. Внедрение веронала в меди-
цинскую практику положило начало широкому раз-
витию исследований, к-рые привели к получению
большого числа Б., отличающихся между собой ха-
рактером радикалов у С5. Наиболее сильным снотвор-
ным действием обладают Б., у к-рых R и R' представ-
ляют собой: 1) насыщенные алкилы с длиной цепи не
более 6 атомов С; 2) аллильные или циклогексиль-
ная группировки; 3) фенильный остаток. Условно,
на основании сходства фармакология, свойств, к
числу Б. относят и нек-рые близкие к ним по строе-
нию соединения, в частности 5,5-дизамещенные про-
изводные 2-тиобарбитуровой к-ты C4H4O2N2S и 2-ими-
нобарбитуровой к-ты C4H5O2N3. Нек-рые, нашедшие
практич. применение Б., приведены в табл. (стр. 373).
Б. хорошо кристаллизуются из воды или водного
спирта. По мере удлинения углеродной цепи алкила
у С5 их растворимость в воде уменьшается (напр.,
растворимость в холодной воде для веронала 0,6%,
а для амитала 0,065%), а в органич. растворителях
увеличивается. Б. — слабые к-ты, их натриевые
соли гидролизуются водой, в отличие от незамещен-
ной барбитуровой к-ты и ее 5-монозамещенных произ-
водных, к-рые хорошо титруются с фенолфталеином;
легко образуют N-алкилпроизводные при действии
галогеналкилов на Na-соли Б. и N-ацилпроизводные
при взаимодействии хлорангидридов к-т с их Ag-со-
лями. Б. подвергаются гидролитич. расщеплению под
влиянием р-ров щелочи, аммиака и даже при нагре-
вании с водой выше 100°; при этом Б. превращаются
ZR
в уреиды H2NCONHCOCH. Б. сравнительно легко
^R'
371
БАРБИТУРОВАЯ КИСЛОТА — БАРБЬЕ — ВИЛАНДА РАСЩЕПЛЕНИЕ
372
ооразуют продукты присоединения с различными лекарств, средствами основ- Формула R И Д' Название к-ты Принятое название Т. пл.. °C
ном, папаверином и др.); некоторые из csH13N3O3 них нашли применение в медицине. Для идентификации малых количеств -с,н5 -с.н5 5,5-Диэтилбарби- туровая Веронал (Na-соль— мединал) 189—191
Б. используется образование ими окра- c13HlsN3O3 шенных и-нитро- или и-галогенобензил- -с3н5 -с3н5 5-Этил-5-фенил- -барбитуровая Люминал 174—177
производных, а также оптич. свойств Б. Наиболее принятым методом синте- 18 ‘ * за Б. является конденсация соответств. -с3н5 -CH3CH2CH(CH,)S 5-Этил-5-изо амил- -барбитуровая Амитал (Na -со ль — барбамил) 155
С-дизамещенных малоновых или циан- уксусных эфиров с мочевиной ИЛИ С СцН1вЫ3О» дициандиамидом в присутствии алкого- —С2Н. —СН(СН3)СН3С8Н5 5-Этил-5-( 1 -метил- бутил ^барбитуро- вая (Na-соль) Этаминал- н атрий, нембутал 130
мена мочевины гуанидином или тиомо- c10H13BrN3O3 чевиной приводит к получению соответ- ствующих 2-иминО- или2-тио-Б. Попыт- -СН (СН3)3 -СН2СВг=СН3 5-Изопроиил-5- -(0-бромалил)- барбитуровая Квиэтал 179—185
ки получения Б. путем алкилирова- C!»HleN2O3 ния барбитуровой кислоты галогеналки- лами не дали хороших результатов, но введение аллильной группировки А । i \/ w 1,5-Диметил-5- -(1-циклогексе- нил)-барбитуровая Эвипан, гек- собарбитал (Na-соль — гексенал) 146
действием галогеналлилов в присут- ChH1sn2O2s ствии медных катализаторов в нек-рых случаях нашло практическое примене- ние. Б. оказывают угнетающее влияние -с3н5 -СН (СН3) СН2С3Н5 S — в положении 2 5-Этил-5-(1-метил- бутил)-2 -тиобар- битуровая (Na-соль) Тиопентал- натрий 159
на центральную нервную систему; ис- с н N о пользуются в медицине в качестве не 13 18 3 3 только снотворных, но и успокаиваю- щих, противосудорожных и наркотич. -СН-.СН^СНи —СН(СН3)СН2С2 Н5 5-Аллил-5-(1-метил- б ути л )-б ар бит у р о- вая (Na-соль) Секонал, секобарбитал 100
средств. Ci0h12nso3 Разные Б. обладают различной про- -СН.>СН=СНэ -стсн=сн2 5,5-Ди аллилбар- битуровая Диал 146
должительностью действия, чти свя- зано с особенностями их превращения C10hunsO3 в организме и выделения из него. -СНоСН=СН.) -СН(СН3)а 5-Аллил-5-изопро- пилбарбитуровая (Na-соль) Алюрат, апробарбитал 141
ные суточные 0,6—2 г. Большие дозы C]IH,5BrN3o3 Б. могут вызвать угнетение дыхания, а в случае быстрого внутривенного вве- -СН3СВг^СН„ СН(СН3) С3Щ 5-(Втор-бутил )- -5-(р-бромаллил)- -барбитуровая (Na-соль) Перяонтон 133
дыхания. При длительном применении C13hun2O3 Б. организм к ним привыкает. Лит.: Машковский М. Д., Лекар- ственные средства, 4 изд., М. I960- К 1 о s а -С2Н5 —> 5-Этил-5-(1-цикло- гексан-1-ил)- -барбитуровая Фанодорн, циклобарби- тал 174
J. Entwicklung und Chemie der Heilmitlel, г и w о ч Bd 3 В., 1952, S. 63—79; BudSSlnsky с13н15м2и2ь Z., Р’г о t i v а М„ Syntheticka lefiiva, Praha, 1954, с. 203—27; Н a u s с h i I d F., Pharma- kologie und Grundlagen der Toxikologie., Lpz., -СН.,-СН=СН3 5-Аллил-5-(1- -циклогексен-2-ил)- -2-тиобарбитуровая Кемит ал 142
1958, S. 729—41. U. C. 1 оловчинская. БАРБИТУРОВАЯ КИСЛОТА (малонилмочевина), мол. в. 164,12 — белые кристаллы, выветривающиеся карбинол (II), 2) окисление карбинола или получен- ного его дегидратацией олефина (III) до к-ты:
на воздухе и разлагающиеся при на-
СО—NH . гревании; безводная к-та плавится
Н2с( /СО с разложением при 245°; плохо рас-
СО—NH творяется в холодной воде, хорошо —
в горячей. Б. к. образует соли; более
сильная к-та, чем уксусная, константа диссоциации
К — 0,98 • 10“4 (25°); титруется щелочами как одно-
основная к-та. Б. к. синтезируют из мочевины и
малоновой к-ты или ее диэтилового эфира в присутст-
вии алкоголята натрия: CO(NH2).4-CH2(COOC.,H5)2 —*•
/СО —NH.
—>H2C<f ^СО + 2С2Н5ОН. Применяют Б. к.
ХЮ —NHZ
для синтезов рибофлавина, пиримидина, виолуровой
(изонитрозобарбитуровой) и мочевой кислот и др.
Производные Б. к. — широко распространенные сно-
творные средства (см. Барбитураты).
В. Г. Типцова.
БАРБЬЕ — ВИЛАНДА РАСЩЕПЛЕНИЕ — цикл
реакций, позволяющий перейти от карбоновой к-ты
(или ее эфира) к ее ближайшему низшему гомологу.
Цикл состоит из двух стадий: 1) превращение эфира
исходной к-ты (I) по реакции Гриньяра в третичный
, R"MgX
rch2coor-----—
/R
RCHg C
в °н
_/R'
RCH=C^
К
RCOOH
///
CrO3
или O3
Наиболее важными факторами реакции являются
правильный выбор реактива Гриньяра (бромистый
метилмагний, если полученный карбинол непосред-
ственно подвергается окислению; бромистый фенил-
магний, если карбинол предварительно дегидрати-
руют) и окисление раствором хромового ангидрида
в уксусной кислоте (или озоном) при низких темпе-
ратурах (10—15°). Б. — В. р. приложимо к кар-
боновым кислотам, содержащим в а-положении
к карбоксильной группе вторичный атом углерода.
Побочной реакцией на стадии окисления является
частичное превращение карбинола II в кетон типа
RCH2COR"; это превращение становится основным
373
БАРИЙ
374
направлением реакпии при наличии а-третичного
атома углерода. f
Метод предложен Барбье в 1913 и успешно приме-
нялся для выяснения строения и синтезов отдельных
к-т алифатич. и жирноароматич. рядов, однако
наибольшее значение имело предложенное в 1926
Виландом использование его для систематич. иссле-
дования стерипов и желчных к-т. Б.—В. р. явилось
важной составной частью синтезов нек-рых стероид-
ных гормонов (прогестерона, авдростерона, ацетата
кортикостерона). Метод был применен также для
решения вопроса о строении строфантидина, нек-рых
стероидных сапогенинов и их отношения к стеринам.
Однако Мейстера — Мишера метод, разработанный
в 1944—47, оказался более удобным в препаративном
отношении для превращения карбововых к-т в их
ближайшие низшие гомологи.
Лит.: Назаров И. Н. и Бергельсон Л. Д.,
Химия стероидных гормонов, М., 1955^ Физер Л. иФи-
з е р М., Химия природных соединений фенантренового ряда,
М.—Л., 1953. Н. Я. Григорьева.
БАРИЙ (Barium) Ва — химич. элемент II группы
периодич. системы Менделеева из подгруппы щелочно-
земельных металлов; п. н. 56, ат. в. 137,36. Природ-
ный Б. состоит из смеси семи стабильных изотопов,
среди к-рых преобладает Ва138 (71,66%). Искус-
ственно получены радиоактивные изотопы Б., в т. ч.
образующийся при делении урана и плутовия Ва110
(jFi/n = 12,8 двя), используемый в качестве радиоак-
тивного индикатора. Конфигурация внешних элек-
тронов атома Б. 6s2. Энергии ионизации Ва°—>Ваь
5,19э<?; Ва —>• Ва2 9,95 эе. Поперечное сечение погло-
щения тепловых вейтровов 1,17 барн на атом.
Б. в виде соединений был открыт в 1774 К. Шееле;
назван от греч. — тяжелый. Металлич. Б. по-
лучен в 1901 А. Гунтцем восстановлением окиси Б.
металлич. алюминием. Содержание Б. в земвой коре
5 • 10“2 вес. %. В свободном состоянии не встречается;
геохимич. свойства Б. определяют рассеянный харак-
тер его распространения в горных породах. Относи-
тельное концентрирование происходит при гидро-
термальных процессах; с ними связаво образование
немногочисленных минералов Б., из к-рых промыш-
ленное значение имеют барит (тяжелый шпат) BaSO4
и менее распространенный витерит ВаСО3. Барит
содержит ок. 65% ВаО, часто с примесями Sr и Са,
реже с РЬ и Ra; сингония ромбическая; бесцветный
(водяно-прозрачвый) или белый, желтый, бурый,
красный и других цветов, что обусловлено различ-
ными примесями; твердость 3—3,5, d 4,3—4,5. В и-
т е р и т содержит ок. 78% ВаО, иногда с примесью
Sr; сингония ромбическая; бесцветный или белый, но
обычно серый или желтоватый; твердость 3—3,5,
d 4,2—4,3. Оба минерала, особенно более доступный
барит, являются не только исходным сырьем для
получения различных соединений Б., во ваходят
самостоятельное широкое применение в различных
отраслях пром-сти (см. Бария сульфат, Бария кар-
бонат, Бария хлорид и др. соединения Б.).
Физические и химические свойства. Б. — мяг-
кий серебристо-белый металл (тверже РЬ, но мягче Zn);
твердость по Бринеллю 4,2 кг/мм2, по шкале Мооса 2;
решетка Б. объемноцентрированная кубическая,
а = 5,.019 А, ат. радиус 2,21 А, ионвый радиус Ва2+
1,38 А; с/20 3,76; т. пл. 710°; т. кип. 1637—
1640°; давление пара при 1200° ок. 50 мм рт. ст.-,
теплота плавления 2 070 ± 80 кал/г-ат; теплота
испарения при т. кип. 36 070 кал/г-ат; ат. теплоем-
кость 6,870 кал/г-атом-град (в интервале 0—100°);
уд. электрич. сопротивление 6 • 10 5 ом-см; термич.
коэфф, линейного расширения 1,9 - 10 5 (0—100°).
По химич. свойствам самого металла и многих его
соединений Б. сходен с кальцием и особенно с строн-
цием и радием, однако по химич. активности превос-
ходит кальций и стронций. Он быстро окисляется на
воздухе, образуя на поверхности пленку, содержа-
щую окись и перекись Б. (а также нитрид). При нагре-
вании на воздухе Б. легко воспламеняется и сгорает
красноватым пламенем; энергичнее кальция разла-
гает воду с выделением водорода. Хранят Б. так же,
как щелочные металлы, в керосине. Б. реагирует со
многими элементами. С кислородом Б. соединяется
непосредственно, образуя окись ВаО — бесцветные
кристаллы, d 5,98, т. пл. 1923°, легко переходящие
на воздухе в карбонат Б. и энергично взаимодей-
ствующие с водой с образованием Б. гидроокиси.
Теплота образования ВаО ДН298 =— 133,4 ккал/молъ.
При нагревании окиси Б. на воздухе при 500° обра-
зуется перекись ВаО2, Д.Н298 =— 150,5 ккал/молъ,
при обычных условиях существующая в виде бес-
цветных кристаллов ВаО2 - 8Н2О; нагреванием пере-
киси Б. под высоким давлением кислорода может
быть получена высшая перекись ВаО4 — вещество
желтого цвета, неустойчивое, разлагающееся уже
при 50—60°. С водородом Б. дает гидрид ВаН2,
d 4,2, т. пл. (с разл.) 675°, бурно разлагающийся
водой и к-тами с выделением водорода. Теплота об-
разования ВаН2 ДД198 =— 40,9 ккал/молъ. С азотом
Б. дает нитрид Ba3N2, к-рый образуется при нагре-
вании Б. в атмосфере воздуха в интервале 260—600°
и плавится ок. 1000°. С углеродом Б. образует кар-
бид ВаС2, аналогичный по свойствам другим карби-
дам щелочноземельных металлов; он получается при
нагревании окиси Б. с углем в дуговой электропечи.
С углеродом и азотом Б. дает термически устойчивый
цианид Ba(CN)2. С галогенами Б. соединяется непо-
средственно, давая галогениды; с фосфором образует
фосфид Ва3Р2 — устойчивое соединение, получаю-
щееся при восстановлении фосфата Б. Ва3(РО4)2
углеродом в дуговой электропечи. При нагревании
до 1 200° сульфата Б. с восстановителем образуется
сульфид BaS; наряду с сульфидом существует ряд
полисульфидов Б., к-рые получаются при взаимо-
действии BaS с S; все ови теряют серу и ок. 400°
вновь переходят в BaS. По отношению к сложным
газам (СО2, СО, SO2) и парам воды Б. ведет себя ана-
логично Са и Sr."
Во всех устойчивых соединениях Б. двухвалентен.
Однако при взаимодействии с безводным хлоридом
ВаС12 металлич. Б. дает при нагревании до 1050"
монохлорид ВаС1, разлагающийся водой с выделением
водорода. Растворяясь в жидком аммиаке, Б. обра-
зует комплексное соединение гексамин Ва (NH3)e;
раствор при этом приобретает интенсивную синюю
окраску.
Аналитическое определение бария.
Для целей анализа нерастворимые соединения Б.
переводят в раствор обработкой исследуемого мате-
риала соляной к-той или сплавлением его с содой
с последующим растворением в этой кислоте. От Са
и Sr отделяют осаждением в виде хромата в присут-
ствии укусной к-ты. Качественно В. обнаруживают
по желто-зеленому окрашиванию пламени горелки
(две характерные линии спектра Б. имеют длину
волвы 455 и 493 ммк), а также по образованию осадка
с сульфат- и хромат-иовами и по розовому окрашива-
нию р-ра в присутствии родизоната натрия; микро-
химически Б. определяют в виде BaSiF6. Количест-
венно Б. определяют весовым методом, осаждая его
в виде BaSO4 серной к-той. Для получения осадка
BaSO4 может быть использована и сульфаминовая
к-та (при этом устраняются загрязнения осадка ио-
нами Fe3+, Mg2‘\ РО?-, во неЭг2+). В присутствии Са24"
и Sr2+ Б. осаждают в виде ВаСгО4 в уксуснокислой
среде. Комплексометрически Б. определяют титро-
ванием комплексоном III (мешает Mg), используя
в качестве индикатора эриохром черный Т.
375
БАРИЙ — БАРИЯ НИТРАТ
376
Получение. Важнейшим сырьем для произ-ва соеди-
нений Б. является барит, к-рый восстанавливают
углем в пламенных печах: BaSO4 + 4С -- BaS 4-
4- 4СО; образующийся растворимый сульфид BaS
перерабатывается на другие соли Б. Технически чи-
стый металл (96—98% Б.) обычно получают нагрева-
нием в вакууме (10~3 мм рт. ст.) при 1100—1200°
смеси окиси Б. с порошком алюминия: 4ВаО 4* А1 =
= ВаО • А12О3 4- ЗВа. Б. улетучивается, осаждаясь
в холодных частях аппаратуры; перегонкой в вакууме
металл очищают до содержания в нем 99,5% Б.
Малые количества металлич. Б. получают термич.
разложением гидрида ВаН2; последний для этой
цели может быть получен из амальгамы Б., легко
образуемой электролитически. Металлич. Б. хорошо
растворяется в расплаве хлоридов щелочноземельных
металлов, поэтому произ-во электролитич. путем
чистого металлич. Б. затруднено. Однако сплавы Б.
(РЬ — Ва, Sn — Ва, РЬ — Ва — Са и др.) могут
быть получены непосредственно электролизом смеси
солей. Сплав РЬ — Ва — Са, напр., получают электро-
лизом смеси ВаС12 4- СаС12 при графитовом аноде и
катоде из расплавленного свинца; электролиз рас-
плава ведут до тех пор, пока не образуется сплав
98% РЬ и 2% (Ва 4- Са).
Применение. Технич. значение металлич. Б. не-
велико. Б., а также его сплавы с А1 и Mg применяют
в технике высокого вакуума в качестве газопогло-
тителей (геттеров). Иногда Б. используется как дезо-
ксидант при рафинировании меди. Нек-рые антифрик-
ционные сплавы содержат небольшое количество Б.
(напр., сплав 98% РЬ, 1—2% Ва и 0,5—0,7% Са).
Добавка Б. к свинцу заметно увеличивает его твер-
дость; сплавы РЬ — Ва находят применение вместо
сплавов РЬ — Sb в типографских сплавах; сплав
Ва — Ni используется в радиолампах.
Разнообразное применение находят соединения Б.
Окись ВаО используется для получения перекиси
ВаО2 и гидроокиси (см. Бария гидроокись)’, перекись —
для получения перекиси водорода (см. Водорода
перекись) и в пиротехнике для изготовления воспла-
менительных составов. Сульфид BaS является важ-
нейшим промежуточным продуктом при переработке
барита на соли Б. (см. выше); химич. обработкой
водного р-ра BaS получают бария хлорид, бария кар-
бонат и др. соли. Сульфид Б. используется также
как депилаторий для удаления волос со шкур. Фто-
рид BaF2 применяется для получения эмалей и иногда
как добавка в ванну при электролитич. рафинирова-
нии алюминия (взамен хлористого Б.) по т. паз.
трехслойному методу (для поддержания нужной
плотности электролита). Перхлорат Б., существую-
щий при обычных условиях в виде Ва (С1О4)2 • ЗН2О,
в безводном состоянии энергично поглощает влагу
из воздуха и является одним из лучших осушителей.
Титанат ВаТЮ3 обладает сегнето-электрич. свой-
ствами в широком интервале темп-ры, а также высо-
кой механич. прочностью и влагостойкостью; благо-
даря этим свойствам, а также простому способу произ-
водства является одним из наиболее практически
пригодных сегнетоэлектриков. Цирконат BaZrO3 (см.
Цирконий) является высококачественным огнеупор-
ным материалом, ценным для керамич. пром-сти.
Азид BaN6, будучи термически малостойким соеди-
нением, легко разлагается в вакууме, что исполь-
зуется для получения металлического Б. Ацетат
Ва (СН3СОО)2 • ЗН2О применяется как протрава при
крашении шерсти и ситцепечатании. Окрашенные
соли Б. являются хорошими пигментами; таковы
хромат ВаСгО4 (желтый пигмент) и манганат ВаМпО4
(зеленый пигмент). Многие соединения Б. сильно по-
глощают рентгеновские лучи и у-излучение, и поэтому
могут служить в качестве защитных материалов в рент-
। геновских установках и в ядерных реакторах (напр.,
барит BaSO4). Соединения Б. применяются как
I инертные носители при извлечении радия из урано-
i вых руд и др. объектов. Т. к. соли Ra менее раство-
римы, чем аналогичные соли Ва, то разделение Ra
и Ва можно производить многократной дробной кри-
сталлизацией (напр., бромидов Ra и Ва). Раствори-
мые соли Б. ядовиты.
Лит.: Беляев А. И., Металлургия легких металлов,
4 изд., М., 1954; Дымчишин Д. А., Производство барие-
вых солей, Л.—М., 1938; Gmelin, 8 Aufl., System-Nunnner,
30, В 1932; Mellor, v. 3, L.—N. Y.— Toronto, 1946, то же,
v. 3, 1952; Pascal, t. 4. P 1958; U 1 1 m a n n, Bd 4, Mtmch.—
B., 1953, S. 171—80; Kirk, v. 1 N. Y„ 1947, p. 458—63 v. 2,
1948, p. 307—23. А. А. ШидловстЯ.
БАРИЯ ГИДРООКИСЬ Ba(OH)2 — при обычных
условиях гидрат Ва(ОН)2 • 8Н2О, бесцветные моно-
клинные (псевдотетрагональные) кристаллы; плоти.
2,18; при 78° Ва(ОН)2 • 8Н2О плавится в собствен-
ной кристаллизационной воде; довольно хорошо
растворяется в горячей, хуже — в холодной воде.
Водный раствор имеет сильно щелочную реакцию;
соприкасаясь с воздухом, р-р поглощает СО2 и мут-
неет от выпадения нерастворимого карбоната бария.
Выше 100° из р-ра Б. г. кристаллизуется Ва(ОН)2 •
• ЗН2О; этот кристаллогидрат в сухом воздухе теряет
две молекулы воды и переходит в Ва(ОН)2 • Н2О.
Полностью обезвоживается до Ва(ОН)2 при темп-ре
красного каления. Безводная Б. г. — блестящая бе-
лая масса, состоящая из ромбич. кристаллов с плоти.
4,5; т. пл. 780°, выше переходит в ВаО; в воде раство-
ряется лучше гидроокисей других щелочноземельных
элементов; растворимость (г ВаО в 100 г Н2О): 1,5
(0°); 3,5 (20°), 11,6 (50°). Теплота образования без-
водной Б. г. ДД%8 =—226,2 ккал/моль. Получают
Б. г. гашением окиси бария, образующейся при про-
каливании ВаСО3. В небольших количествах Б. г.
получается при взаимодействии р-ров ВаС12 и NaOH
при темп-ре кипения; при кристаллизации отфиль-
трованного р-ра выделяются кристаллы Ва(ОН)2-8Н2О.
Насыщенный при обычной темп-ре водный р-р Б. г.
наз. баритовой водой; имеет большое зна-
чение в химич. анализе, в частности как реактив на
ионы SO); и СОз • Б. г. относится к числу инициаторов
полимеризации’, применяется также для выделения
сахара из патоки в пищевой пром-сти.
Лит. см. при ст. Барий. В. Е. Плющев.
БАРИЯ- КАРБОНАТ (барий углекислый) ВаСО3 —
бесцветные кристаллы, простая ромбич. решетка,
а = 6,39 А, Ъ = 8,83 А, с= 5,28 А; плотн. 4,3—4,4;
т. пл. 1,740. В природе встречается в виде минерала
витерита. Теплота образования ДН298 =— 291,3
ккал/моль. Разлагается с выделением СО2 лишь при
сильном белом калении. Парциальное дав-
ление СО2 над Б. к.
t, °C........... 1017 1097 1157 1177 1297
р, мм рт. ст...... 5 120 340 450 760
Трудно растворим в воде [20 мг/л при 18°; произве-
дение растворимости ПР = 8 • 10 9 (18°)]. Легко рас-
творим в НС1 и HNO3. Получают Б. к. из сульфида
бария по реакции: BaS 4- Н2О 4" СО2 = ВаСО3 -4
4- H2S. Вместо СО2 можно брать р-р соды. Б. к. яв-
ляется одним из важнейших соединений бария; при-
меняется для получения др. соединений бария (окиси,
нитрата, ацетата и др.); для удаления из водных р-ров
иона SO) ,ионов тяжелых и щелочноземельных метал-
лов и Mg2+. В большом количестве Б. к. используется
в стекольной пром-сти и при изготовлении эмалей
и глазурей.
Лит. см. при ст. Барий. А. А. Шидловский.
БАРИЯ НИТРАТ (барий азотнокислый) Ba(NO3)2—
бесцветные кристаллы с простой кубич. решеткой,
а = 8,13 А; плотн. 3,24; т. пл. 595°. Теплота образо-
вания Д7/)Э8=— 237,06 ккал/моль. При сильном на-
377
БАРИЯ ПЛАТИНОЦИАНАТ —БАРТА РЕАКЦИЯ
378
гревании в присутствии восстановителей (напр., маг-
ния) Б. н. разлагается: 2 Ba(NO3)2 = 2ВаО 4 2N2 4
4 5О2, причем наряду с ВаО образуется частично
и ВаО2. Б. н. негигроскопичен; растворимость в г
на 100 г воды: 8,7 (20°), 34,2 (100°). Получают Б. н.
обменной реакцией в водных р-рах между ВаС12
и NaNO3 (или HNO3) или растворением ВаСО3
в HNO3; применяют в пиротехнике для изготовления
осветительных, трассирующих и зажигательных со-
ставов, а также сигнальных составов зеленых огней.
Иногда Б. н. используется в стекольной пром-сти
вместо ВаСО3.
Лит. см. при ст. Барий. А. А. Шидловский.
БАРИЯ ПЛАТИНОЦИАНАТ (барий платиноси-
неродистый) Ba[Pt(CN)4] — бариевая соль платиноциа-
нистоводородной кислоты, одно из наиболее устой-
чивых комплексных соединений двухвалентной пла-
тины, при обычных условиях кристаллогидрат
Ba[Pt(CN4)J • 4Н2О — желто-зеленые кристаллы, ре-
шетка базоцентрированная моноклинная с парамет-
рами: а = 11,89 А, Ь = 14,08 А, с = 6,54 A, f) =
= 103° 42'; плотн. 2,09. Растворимость Б. п. в воде
3,4% (20°). При 100° теряет 2, а при 150° — 4 мо-
лекулы воды. Получают Б. п. из хлористой плати-
ны: PtCl2 + 2КС1 = KJPtClJ; K2[PtCl4J 4 4KCN =
= K2[Pt(CN)4] 4 4KC1; K2[Pt(CN)4] + BaCl2 =
= Ba[Pt(CN)4] 4 2KC1 или электрохимически из Pt
и Ba(CN)2. Под действием радиоактивного излучения
или рентгеновских лучей в кристаллах Б. п. возбу-
ждается яркая желто-зеленая флуоресценция. На
этом основано применение экранов, покрытых Б. п.,
при работе с рентгеновскими лучами и при исследо-
вании радиоактивного распада.
Лит.: [Брауэр Г.], Руководство по препаративной
неорганической химии, пер. с нем.. М., 1956 с. 722; см. также
при ст. Барий. А. А. Шидловский.
БАРИЯ СУЛЬФАТ (барий сернокислый) BaSO4 —
бесцветные кристаллы с простой ромбич. решет-
кой, а = 7,13 А, Ъ = 8,85 А, с = 5,44 А; плотн. 4,5;
т. пл. 1 580°. Теплота образования АН288 =
=—350,2 ккал/молъ. Встречается в природе в виде
минерала барита (тяжелый шпат), являющегося
основным сырьем для получения других соединений
бария. Практически нерастворим в воде [2 мг/л при
18°; произведение растворимости ПР = 0,9 • 10 10
(18°)] и разб. к-тах. Значительно растворим в конц.
H2SO4 вплоть до образования 10—12%-ных р-ров.
Б. с. растворим в аммиачных р-рах комплексона III.
Тонкодисперсный осажденный Б. с. (бланфикс, см.
Пигменты белые) получается при смешивании водных
р-ров BaS или ВаС12 с Na2SO4. Б. с. входит в состав
белой краски — литопона; используется как утяже-
литель в тиксотропных смесях при бурении глубоких
нефтяных скважин; как наполнитель (и утяжелитель)
в бумажной (добавка Б. с. делает бумагу белее),
резиновой и керамич. пром-сти. Б. с. сильно погло-
щает рентгеновские лучи и поэтому применяется
при рентгеновском исследовании желудочно-кишеч-
ного тракта.
Лит. см. при ст. Барий. А. А. Шидловский.
БАРИЯ ХЛОРАТ (барий хлорноватокислый)
Ва(С1О3)2 — при обычных условиях существует в виде
моногидрата Ва(С103)2 • Н2О, бесцветных моноклин-
ных кристаллов с плотн. 3,18; т. пл. (с разлож.)
414°. Практически негигроскопичен; при 120° теряет
кристаллизационную воду. Теплота образования без-
водного Б. х. AH288 =— 181,7 ккал/молъ. При бы-
стром нагревании разлагается: Ва(С1О3)2 = ВаС12 4
4 ЗО2 с выделением 34,8 ккал/молъ. В смеси с горю-
чими в-вами при трении, ударе или нагревании
взрывает. Растворимость Б. х. в расчете на безвод-
ную соль в г на 100 г Н2О: 27,4 (25°) и 111,2 (100°).
Получают Б. х. обменной реакцией водных р-ров
КаС1О3 и ВаС12 или электролизом водного насыщен-
ного р-ра ВаС12, применяют в пиротехнике для изго-
товления сигнальных составов зеленых огней; может
быть использован вместо NaC103 в с. х-ве для уничто-
жения сорняков.
Лит.: Шидловский А. А., Основы пиротехники,
2 изд., М., 1954; см. также при ст. Барий.
А. А. Шидловский.
БАРИЯ ХЛОРИД (барий хлористый) ВаС12 — при
обычных условиях существует в виде дигидрата
ВаС12- 2Н2О; бесцветные кристаллы, простая моно-
клинная решеткаиа = 7,136 А, Ь = 10,86 А, с =
= 6,738 А, р = 90,57°; плотн. 3,10. При нагревании
до 100° полностью теряет кристаллизационную воду.
Безводный Б. х. — ромбич. кристаллы, а = 7,823 А,
Ь = 9,333 А, с = 4,705 А; плотн. 3,92; т. пл. 960°:
т. кип. 1560°; с солями NaCl, КС1, СаС12 образует
низкоплавкие эвтектики: напр. эвтектика ВаС12 —
— NaCl при 61 мол. % ВаС12 имеет т. пл. 654°. Рас-
творимость Б. х. (в расчете на безводную соль)
в г на 100 г воды: 31,6 (0°), 35,7 (20°), 56,8 (100°); почти
нерастворим в абс. спирте и в эфире. Теплота образо-
вания ВаС12 ДН288 =— 205,56 ккал/молъ.
Получают Б. х. прокаливанием смеси барита с уг-
лем и хлоридом кальция в пламенных печах при
900—1000° по реакциям: BaSO4 4- 4С = BaS -4 4СО:
BaS 4 СаС12 = ВаС12 -4 CaS или обработкой р-ров
сульфида бария соляной к-той или хлором: BaS 4*
+ 2НС1 = ВаС12 4- H2S; BaS + Cl2 = ВаС12 4 S.
Б. х. применяют для получения др. солей бария
(сульфата, нитрата, хромата и др.); в с. х-ве для борьбы
с вредителями; в кожевенной пром-сти для утяжеле-
ния и осветления различных видов кожи; в качестве
добавки в электролитич. ваннах при получении
магния, для осаждения ионов SO; . Сплавы Б. х.
с хлоридами щелочных и щелочноземельных метал-
лов используются при отпуске и при закалке стали.
Лит. см. при ст. Барий. А. А: Шидловский.
БАРН — единица эффективного поперечного сече-
ния ядерных взаимодействий, равная 10 24 см2. Эф-
фективное поперечное сечение характеризует вероят-
ность ядерных реакций, для многих из них оно близко
по порядку величины к геометрич. сечению ядра,
к-рое для большинства ядер лежит в пределах (0,5—
2,0) 10~24 см2; этим и объясняется выбор Б. в каче-
стве единицы. Производные Б.: мегабарн (10"18 см2),
миллибарн (10 27 см2) и т. д. Обозначается барн.
БАРТА РЕАКЦИЯ — наиболее важный метод по-
лучения ариларсоновых к-т, напр. C6H5AsO(OH)2,
заключающийся во взаимодействии солей диазония
с солями мышьяковистой к-ты: ArN2Cl 4 K3AsO3 —*•
—► ArAsO(OK)2 4 КС1 4 N2. Этим методом можно
синтезировать также соединения, содержащие два
или три арильных радикала, связанных с атомом As:
ArN2Cl 4 ArAs(ONa)2 —► Ar2As(O)ONa 4 NaCl 4 N2;
ArN2Cl 4 Ar2AsONa —► Ar3AsO 4 NaCl 4 N2. В каче-
стве катализаторов применяют медь, никель, кобальт
и их соли; однако каталитич. эффект не всегда от-
четливо проявляется.
Реакция была предложена Г. Бартом в 1910 и
с тех пор подверглась значительным усовершенство-
ваниям. Так, Г. Шмидт предложил в 1920 проводить
процесс в нейтральной среде, без катализаторов,
напр.: C6H5N2C1 4 K2HAsO3 -► C6H5AsO (ОН) OK 4
4 КС1 4 N2. Большое значение имеет модификация
Е. Шеллера (1927), по к-рой первичные ароматич.
амины растворяют в метаноле или в ледяной уксусной
к-те и диазотируют в присутствии AsCl3 и следов
Cu2Cl2 : ArN2HSO4 4 AsCl3 -► N2 4 ArAsCl3OSO3H -►
—► ArAsO(OH)2. В ряде случаев целесообразно при-
менять вместо солей типа ArN2Cl более устойчивые
борфториды ArN2BF4.
Б. р. и ее модификации имеют широкие границы
приложимости. Выходы арсоновых к-т из анилина
с различными заместителями, из аминопроизводных
379
БАТОХРОМНЫЙ ЭФФЕКТ—БАЦИТРАЦИНЫ
380
нафталина, флуорена, хинолина, пиридина и т. д.
обычно достигают 48—85%. Нередко Б. р. сопрово-
ждается побочными процессами. Так, соли мышьяко-
вистой к-ты могут действовать на соли диазония как
восстановители с образованием азо- и гидразосоеди-
нений, аминов, углеводородов. Еще более сильными
восстановительными свойствами обладают арилар-
синокснды ArAsO, чем объясняется обычно более
низкий выход вторичных арсоновых к-т Ar2As(O)OH.
К побочным процессам относится и образование
соответствующих фенолов. От арсоновых к-т можно
легко перейти к другим соединениям с сохранением
С—As-связи; поэтому Б. р. является основным мето-
дом синтеза мышьякароматич. соединений.
Лит.: Гамильтон К., Морган Дж., в кн.:
Органические реакции, пер. с англ., Сб. 2, М., 1950, с. 449—460;
Фрейд л ина Р. X., Синтетические методы в области ме-
таллоорганических соединений мышьяка, М.—Л., 1945.
Я. Ф. Комиссаров.
БАТОХРОМНЫЙ ЭФФЕКТ — смещение окраски
органич. соединений в сторону углубления цвета,
т. е. переход от желтого через красный и синий
к зеленому цвету:
Зеленовато-желтый Желтый Оранжевый
Углубление Красный Повышение
цвета Фиолетовый Синий Сине-зеленый Зеленый цвета
пени зависит от ее поляризуемости. Так, Б. э. на-
блюдается при удлинении в соединениях сопряженной
цепи двойных связей (табл. 2), при введении в мо-
лекулу органич. соединений заместителей, сопряжен-
ных с основным хромофором, напр. (см. ф-лу):
I I
С2Н5 J С2Н5
и при выравнивании в последнем
электронной плотности (табл. 3).
распределения
Таблица 3
Доноры электронов Акцепторы электронов
R лмакс. ’ ммк R *макс«» •M3tK
Н 558 н 558
01, Вг 561-562
ососн. 562
сн3 563
ОСН3 572 SO3CH3 561
он 574 SO3NH2 563
NHCOCH, 577 CN 571
nh2 594 CONHo 571
NHCH3 608 СООН 572
N(CH,)S 612 NO, 584
Окраска (цвет) органич. соединения является
результатом избирательного поглощения последним
лучей определенных участков спектра и характери-
зуется длиной волны поглощенного света (табл. 1).
Значительный Б. э. наблюдается при образовании
внутрикомплексного соединения с металлом (I и II)
или молекулярных соединений типа хингидрона
ПИ—V).
Таблица 1
Поглощенный свет
длина волны К, ммк цвет Наблюдаемый цвет
400—430 430—490 490—510 510-530 530—560 фиолетовый синий сине-зеленый зеленый желто-зеле- ный желтый оранжевый красный зеленовато-желтый желтый до оранжевого красный пурпурный фиолетовый
560-590 590—610 610—730 синий зеленовато-синий сине-зеленый
Ме=А1: Красный
Ме=Fe: Фиолетовый
В проходящем свете цвет вещества всегда оказы-
вается дополнительным к поглощенному. Поэтому
при углублении цвета (Б. э.) максимум или вся полоса
поглощения смещаются в сторону более длинных волн.
Для бесцветных соединений, поглощающих свет
в УФ-части спектра, Б. э. характеризует также сме-
щение поглощения в длинноволновую область.
Таблица 2
Слабо-
желтый
IV V
Бесцветный Зеленый
Кислота Энергия возбуж- дения, ккал/моль Максимум погло- щения, ммк
сн3—сн=сн—с=о 1 он 135 208
СН3—(СН=СН)2-С=О 1 он 107 261
СН3— (СН=СН)4-С=0 1 он 85 332
Современная теория цветности органич. соединений
связывает Б. э. с изменениями в химич. строении
вещества, в результате к-рых уменьшается энергия
возбуждения его молекул, что в определенной сте-
В некоторых случаях Б. э. зависит от природы
растворителя и температуры раствора (см. так-
же Гипсохромный эффект, Цветности теории, Хро-
мофор).
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венкатараман К., Химия синтетических краси-
телей, пер. о англ., т. 1, Л., 1956; Гиллем А., Штерн Е.,
Электронные спектры поглощения органических соединений,
пер. с англ., М., 1957. В. А. Пучков.
БАЦИТРАЦИНЫ — группа антибиотиков поли-
пептидов, продуцируемых одним из штаммов бактерий
Вас. licheniformis. Описано до семи антибиотиков
этой группы; лучше других изучены бацитрацины А,
В и С. Наиболее изучен бацитрацин А—CeeH103OieN17.
При гидролизе его обнаружено 12 остатков 9 различ-
ных аминокислот; мол. в. ок. 1 410. Молекула Б.
представляет собой циклопептид с боковой цепью из
аминокислот, соединенной с циклом по лизину. Ниже
схематически показана структура Б. (условные обо-
381
БЕЗДЫМНЫЙ ПОРОХ —БЕЛКИ
382
значения аминокислот см. в ст. Аминокислоты):
. /ори.-изол.
/ D L
изол.-цистин, -леиц. -глют. -изол. -лиз. ।
L L L D L L\ I
хасп.-гист. -фал.
|LD L D
асп.
LD
При гидролизе бацитрацина В образуются те же ами-
нокислоты, кроме того, обнаружена еще одна амино-
кислота неустановленного строения. Бацитрацин С
образуется из бацитрацина А в процессе выделения.
Биосинтез Б. протекает на средах, содержащих
глюкозу, соли аммония и нек-рые аминокислоты.
Выделяют Б. из культуральной жидкости при pH ок. 7
экстракцией бутанолом или фенолом, насыщенными
водой. Из экстрактов Б. осаждают в виде трудно-
растворимых пикратов, к-рые разлагают НС1; при
этом получают хорошо растворимые в воде хлоргид-
раты; основания растворимы лучше. Б. — аморфные
в-ва, устойчивые в сухом виде, менее устойчивы в
р-рах; быстро инактивируются в кислых и особевно
в щелочных средах. Дальнейшая очистка Б. от пиг-
ментов и углеводов производится хроматографически
на окиси магния.
По спектру антимикробного действия Б. сходны
с пенициллинами, но менее активны. Наиболее акти-
вен бацитрацин А; несколько менее — бацитрацин С;
бацитрацин В в 4 раза менее активен, чем бацитра-
цин А. По токсичности Б. располагаются в ряд: А >
>.С>. В. Вс.е Б. активны против грамположительных
бактерий при разбавлении от нескольких сотен тысяч
до нескольких миллионов. Бацитрацин А подавляет
рост стафилококков в разведении 1:160000, токси-
чен для мышей в дозе 5 мг. Бацитрацин В угнетает
рост стафилококков в разведении 1:40000, токсичен
для мышей в дозе 10 мг. Бацитрацин С близок к ба-
цитрацину А. Из грамотрицательных бактерий чув-
ствительной к Б. оказалась Hemaphilus infuenzal
типа В, на к-рую смесь Б. действует прй разбавлении
1 : 100000. Б. усиливают действие других антибиоти-
ков — пенициллинов, стрептомицинов, полимиксинов,
неомицинов и ауреомицина. С последним антибиотиком
Б. образуют малорастворимый в воде препарат —
ауреотрацин, характеризующийся меньшей токсич-
ностью.
В медицинской практике Б. применяют для мест-
ного лечения гнойных процессов и др.
Лит.: Шемякин М. М., Хохлов А. С., Химия
антибиотических веществ, 2 изд., М. 1953, с. 468, 571; Г а у-
з е Г. Ф., Лекции по антибиотикам, 3 изд М., 1958, с. 221;
Craig L. С., Hausmann W. Wei si ger J.,
J. Biol. Chem. 1952, 199, № 2, p. 865; их ж e, J. Am.
Chem. Soc., 1955, 77, № 3, p. 721. А. Б. Силаев.
БЕЗДЫМНЫЙ ПОРОХ — см. Пороха.
БЕКМАНА ПЕРЕГРУППИРОВКА — перегруппи-
ровка оксимов в амиды к-т под действием кислот-
ных дегидратирующих агентов; сопровождается вы-
делением теплоты. Различают два вида Б. п.: перегруп-
пировку оксимов альдегидов или кетонов в амиды
к-т (Б. п. 1-го рода):
R—С—R' гно-с—R1 1
|| — I || I —R’CONHR или RCONHR'
NOH [ NR J
и перегруппировку, сопровождающуюся разрывом
связи С — С и образованием нитрила (Б. п. 2-го
рода). Последняя характерна для оксимов а-окси-
кетонов:
RCH(OH)C(=NOH)R' —► RCHO+R'CN+H3O
Если R и R' различны, строение продукта Б. п.
зависит от пространственной конфигурации оксима.
Установлено, что при Б. п. кетоксимов происходит об-
мен между ОН-группой и пространственно удаленным
от нее радикалом R (транс-обмен). Немногочислен-
ные исключения объясняют изменением конфигура-
ции оксима, предшествующим Б. п. При использо-
вании для Б. п. смеси пространственно изомерных
кетоксимов R — С — R' и R — С — R' обычно no-
li II
N —ОН НО—N
лучают смесь структурно изомерных замещенных
амидов RCONHR' и R'CONHR. Частным случаем
Б. п. 1-го рода, имеющим важное значение, яв-
ляется перегруппировка оксимов алициклич. кето-
нов (CH2)nC=NOH в лактамы («изооксимы»)
(CH2)nNHCO, сопровождающаяся расширением цикла
за счет внедрения в него NH-группы. Этот случай
Б. п. часто называют изомеризацией оксимов. При
обработке альдоксимов реагентами Б. п. обычно по-
лучают нитрилы (продукты дегидратации оксимов) и
лишь редко амиды к-т. Известны случаи аномальной
Б. п. (см. ниже превращения а-тетралона).
Из большого числа реагентов, применявшихся для
Б. п., главное значение имеют РС15, H2SO4, олеум
и смесь СН3СООН с (СН3СО)20, насыщенная НС1
(бекмановская смесь). Часто пользуются также поли-
фосфорной к-той (р-р Р2О5 в Н3РО4), что позво-
ляет произвести Б. п. ряда оксимов, к-рые не уда-
валось перегруппировать др. реагентами. Строение
продукта реакции иногда зависит от природы приме-
няемого реагента. Так, напр., при действии на оксим
а-тетралона (I) бекмановской смеси образуется а-наф-
тиламин (II), в то время как полифосфорная к-та
превращает а-тетралоноксим в соответств. лактам
(III), т. е. дает нормальный продукт Б. п. 1-го рода.
-NH-CO
-сн2-сн
СН2
2
III
Механизм Б. п. различен в случае различных реаген-
тов. Установлено, что перегруппировываются не
сами оксимы, а продукты их взаимодействия с при-
меняемым реагентом; в случае кислородсодержащих
к-т, напр. H2SO4, механизм Б. п. следующий:
R С R1 + H»SO4 быстро -С—R1-|-HSO4 быстро
N—ОН экзоте рмично NHOH ’
__ R—С—R' медленно
NOSOsOH
R*—С—OSO2OH тт о
II -в
R—N
RCONHR'+ HsSO4
Собственно перегруппировка является реакцией 1-го
порядка. Изучение Б. п. оксимов типа (R')(R")CHC
(=NOH)R"', имеющих асимметрии, атом С, показа-
ло, что продукты Б. п. сохраняют оптич. активность;
мигрирующая группа при Б. п. не бывает кинетически
свободной (в противном случае должны получаться
оптически неактивные продукты). Б. п. часто приме-
няют для определения строения кетонов, т. к. полу-
чающиеся продукты реакции легко идентифицируются
после их гидролиза. Б. п. находит промышленное
применение в произ-ве из оксима циклогексанона
е-капролактама, используемого для получения син-
тетич. полиамидного волокна капрон. Открыта в
1886 Э. О. Бекманом.
Лит.: Blatt А. Н., Chem. Rev., 1933, 12, № 1; Jo-
nes В., там же., 1944, 35, М3; Кнунянц И. Л. и Фаб-
ричный Б. П., Успехи химии, 1949, 18, вып. 6; их же,
в кн.: Реакции и методы исследования органических соедине-
ний кн. X М. 1954 с. 139. Б. П. Фабричный.
БЕЛИЛА — см. Пигменты белые.
БЕЛКИ (протеины) — высокомолекулярные при-
родные соединения, являющиеся поликонденсатами
383
БЕЛКИ
384
а-аминокислст. Б. — важнейшая составная часть
всех живых организмов.
Структура белков. Белки являются такими вы-
сокомолекулярными соединениями, у к-рых сотни
и даже тысячи остатков а-аминокислот соединены
друг с другом кетоимидными (пептид-
ными) связями (—СО—NH—), образуя
длинные цепи главных валентностей — полипептид-
ные цепи:
.. . -СО-NH—СН—СО—NH—CH-CO-NH—СН-СО— . . .
I I I
R' R" R'"
где R’, R", R'" ... — боковые группы или боковые
цепи у отдельных аминокислотных остатков. Пептид-
ные связи образуются путем отщепления воды от
карбоксильной группы одной аминокислоты и амино-
группы другой. Не исключена возможность, что в
основных, полипептидных цепях Б. могут присут-
ствовать в небольшом количестве другие, непептидные
связи. В состав Б. входят остатки более 20 различных
аминокислот. Структура Б. характеризуется не только
химич. составом и порядком чередования различных
аминокислотных остатков в полипептидной цепи, но
также и расположением этих цепей в пространстве,
что обусловливает существование практически не-
обозримого множества Б. с разнообразными свойст-
вами. В Б. различают 4 порядка морфологич. орга-
низации, или 4 структуры. Первый порядок, или пер-
вичная структура, — полипептидная цепь с опреде-
ленной последовательностью чередования аминокис-
лотных остатков (рис. 1). Второй порядок — та прост-
ранственная конфигурация, к-рую принимает сама
полипептидная цепь. Вторичной структурой может
быть, напр., а-спираль Полинга—Кори, имеющая 3,7
аминокислотных остатков в одном витке (рис. 2).
Третий порядок — конфигурация, возникающая в
результате складывания или закручивания структур,
соответствующих второму порядку. Четвертый по-
рядок — объединение нескольких частиц с третичной
структурой в одну более крупную частицу. Так, напр.,
полипептидная цепь (первичная структура) гемогло-
бина закручена в а-спираль (вторичная структура),
спираль свернута в клубок (рис. 3) (третичная струк-
тура) и, наконец, четыре таких клубка (рис. 4) объеди-
нены в одну крупную частицу (четвертичная структу-
ра). Наличие таких четырех поряд-
ков организации макромолекул не
является обязательным для всех Б.
Напр., четвертичная структура может
и отсутствовать.Спиралевидная конфи-
гурация в,пек-рых местах может нару-
шаться.
Формирование пространственной Рис. 3.
конфигурации макромолекул Б. в зна-
чительной степени определяется между- и внутри-
молекулярными взаимодействиями. Наиболее важ-
ными из них являются образование водородных и
дисульфидных связей. Вода- ,___„
родные связи в Б. возникают
за счет атома водорода
между атомами азота и ки-
слорода пептидных групп,
а также за счет водорода
оксигрупп оксиамипокис-
лот и кислорода пептид-
ной группы. Дисульфидные
связи (или дисульфидные
мостики) —S—S—образуют-
ся между остатками ци- Рис. 4.
стеина. В стабилизации
макромолекул Б. имеют значение и взаимодействия
между кислотными и основными группами, эфир-
ные и тиоэфирные связи, а также силы Ван-дер-Ва-
альса.
Природные Б. часто находятся в виде различных
комплексов с небелковыми компонентами (напр.,
с нуклеиновыми кислотами). Эти комплексы имеют
большое значение в жизнедеятельности организмов.
Разработаны разнообразные методы, позволяющие ус-
тановить химич. состав Б. Уже удалось выяснить пер-
вичную структуру — последовательность расположе-
ния аминокислотных остатков в цепях инсулина и
рибонуклеазы. Расшифрованы структура и строение
нек-рых биологически активных полипептидов, на-
пример гормонов: окситоцина, вазопрессина, глюка-
гона, а-кортикотропина и др. Методами рентгено-
структурного анализа установлена третичная струк-
тура миоглобина (рис. 3, заштрихован участок гема,
см. Гемоглобины). Свойства Б. могут быть объяснены
только тогда, когда будет найдена возможность уста-
новления всех четырех уровней организации их
частиц, т. к. эти свойства зависят не только от после-
довательности аминокислотных остатков в полипеп-
тидных цепях, но и в значительной степени от их
пространств, расположения и взаимодействия, обус-
ловленного вторичной, третичной и четвертичной
структурами. Большое значение поэтому имеет раз-
витие методов исследования конфигурации пептид-
ных цепей и их взаимного расположения.
Свойства Б. зависят прежде всего от их химич.
строения. Известны случаи, когда даже незначитель-
ные изменения аминокислотного состава приводят к
существенным изменениям свойств Б. Напр., замена
всего лишь одного аминокислотного остатка из трех-
сот в молекуле гемоглобина, а именно остатка глу-
таминовой к-ты на остаток валина, резко меняет
свойства этого Б. Получающийся при этом т. н. ге-
моглобин-S («серповидный») вызывает серьезное за-
болевание крови — серповидную анемию. На при-
мерах окситоцина и вазопрессина также видно, что
небольшие отличия в аминокислотном составе этих
гормонов связаны с совершенно разным характе-
ром их биологич. активности. Биологич. активность
и др. свойства Б. в значительной степени определяются
также способом закручивания пептидной цепи, прост-
ранственной конфигурацией макромолекул.
385
БЕЛКИ
386
Форма макромолекул Б. может быть весьма раз-
личной: от шарообразных частиц (глобулярные Б.)
до нитей (фибриллярные Б.).
Физические и химические свой-
ства белков. Р-ры Б. обладают рядом свойств,
характерных для лиофильных коллоидных р-ров.
Частицы Б. не проходят через полупроницаемые мем-
браны, что используется для их очистки от низко-
молекулярных соединений диализом. Наличие на
поверхности частиц Б. многочисленных полярных
групп обусловливает их значительную гидратацию.
Так, количество гидратационнтой воды, связанной с
альбуминами и глобулинами, составляет 0,2—0,6 г
на 1 г сухого веса Б. В определенных условиях Б.
образуют гели (студни). Во многих случаях Б. удает-
ся получить в кристаллич. виде. Б. в р-рах седимен-
тируют в ультрацентрифугах при ускорении порядка
200 000g; константы седиментации (s) Б. находятся
в пределах от 1-10“13 до 100-10 13 сек. Коэфф, диффу-
зии Б. 0,1-10 7—10-10 7 с.и2/сек; средний удельный
объем 0,75 см31г. Эти физико-химич. характеристики
используются для определения мол. веса Б., а также
степени асимметрии их молекул »/а, где в и а — про-
дольная и поперечная полуоси гидродинамически
эквивалентного эллипсоида, приближенно принима-
емого за форму молекулы Б. Мол. вес Б. — от 5000 до
нескольких миллионов, в/а — от 1 до 200. Для опре-
деления мол. весов и размеров молекул Б. широко
применяется метод светорассеяния. Мол. веса могут
быть определены также методом осмометрии, методом
исследования монослоев на поверхности жидкой
среды. Размеры молекул Б. определяются методом
двойного лучепреломления в потоке, измерением
коэфф, вращательной диффузии. Макромолекулы не-
которых Б. наблюдались в электронном микроскопе.
Для изучения структуры Б. широко применяется
метод рентгеноструктурного анализа и электроно-
графии.
Инкремент показателя преломления Б. (и — и0)/с
равен 0,18 см3! г (для D-натриевой линии спектра).
Б. оптически активны, в обычных условиях вращают
плоскость поляризации света влево, величина уд.
вращения [a]D колеблется от —30° до —60°, а в
нек-рых случаях, напр. для проколлагена, дости-
гает —400°. Величина [а] обусловливается наличием
асимметрии, атома углерода в аминокислотных ос-
татках, с одной стороны, и самой конфигурацией
полипептидных цепей—с другой. Б. поглощают лучи
УФ-области спектра, характерным является погло-
щение в области 280 ммк, обусловленное наличием
в Б. ароматич. аминокислот. В ИК-области спектра
Б. сильно поглощают за счет СО- и NH-групп (ок.
1600 см1 и ок. 3100—3300 см^1). Исследование
ИК-спектров и их дихроизма позволяет изучать водо-
родные связи и их направление в Б.
Б. содержат кислые карбоксильные группы и ами-
ногруппы со свойствами оснований и проявляют
в р-рах амфотерные свойства. При определенных зна-
чениях pH в р-рах Б. преобладает диссоциация тех
или других групп, что придает частицам Б. соот-
ветств. заряд и вызывает их движение в электрич.
поле (электрофорез). Электрофоретич. подвижность,
выражаемая в см2-вольт~1-сек~1, определяется плот-
ностью заряда поверхности молекул и при соблюде-
нии тождественных условий может служить харак-
теристикой Б. Электрофорез широко используется
для анализа смесей Б. и их разделения. Электромет-
рия. титрование растворов Б. позволяет судить о
природе диссоциирующих функциональных групп.
Дипольный момент Б. в р-рах равен 100—1000 D
(единиц Дебая), диэлектрич. инкремент (на 1 г Б.
в 1 л) равен 0,1—1,0; диэлектрич. проницаемость
растворов Б. всегда выше, чем растворителей.
7 К. X. Э. т. 1.
Важным свойством Б. является их способность к
денатурации. Этим понятием обозначают явления,
связанные с изменениями вторичной, третичной и
четвертичной структур Б. при воздействии различ-
ных агентов (нагревание, действие к-т, щелочей,
УФ-лучей, ионизирующей радиации, ультразвука
и др.). Денатурация Б. обычно сопровождается умень-
шением их растворимости, увеличением вязкости,
освобождением функциональных групп, потерей био-
логич. активности и др. В настоящее время слишком
общее понятие денатурация заменяется описанием
конкретных изменений молекул Б. при тех или иных
условиях.
Элементарный анализ различных Б. дает (в % на
сухой вес): 50—55 С; 6,5—7,3 Н; 21,5—23,5 О; 15,0—
17,6 N; 0,3—2,5 S. Кроме того, Б. могут содержать
небольшие количества фосфора, галогенов. При кис-
лотном, щелочном, ферментативном гидролизе Б.
распадаются до аминокислот. Т. к. в состав Б. входят
остатки разнообразных аминокислот, они содержат
большое число функциональных групп, к-рые спо-
собны вступать во многие реакции: окисления, вос-
становления, образования меркаптидов, алкилиро-
вания, арилирования, этерификации, ацилирования,
дезаминирования азотистой к-той, иодирования, фос-
форилирования, нитрования, диазотирования, диазо-
сочетания, реакцию с формалином и др. Б. осаж-
даются фосфорновольфрамовой, фосфорномолибдено-
вой, трихлоруксусной, салициловой, пикриновой и
др. кислотами, солями тяжелых металлов. Б. дают ряд
цветных реакций, обусловленных наличием нек-рых
аминокислотных остатков или химич. группировок.
К важнейшим из них относятся: биуретовая реакция
(пептидные связи), ксантопротеиноеая реакция (аро-
матич. ядра остатков тирозина, триптофана, фенил-
аланина), Миллона реакция (фенильная группа тиро-
зина), Адамкевича реакция (индольное кольцо трип-
тофана), Паули реакция (имидазольное кольцо ги-
стидина), сульфгидрильная реакция (серусодержащие
остатки аминокислот), Сакагучи реакция (гуанидино-
вая группа аргинина), нингидриновая реакция, пик-
риновая реакция.
Широкое применение в исследовании химич. строе-
ния Б. получили реакции для определения амино-
кислотных остатков, имеющих свободные а-аминные
и a-карбоксильные группы (N- и С-концевые амино-
кислоты), а также для последовательного отщепле-
ния аминокислотных остатков от полипептидной цепи.
Для определения N-концевых аминокислот исполь-
зуется реакция с 2,4-динитрофторбензолом (метод
Сангера), а также с фенилизотиоцианатом (метод
Эдмана). Последний метод используется для ступен-
чатого отщепления аминокислот с N-конца. Для оп-
ределения С-концевых аминокислот применяют ре-
акцию с гидразином (метод Акабори). Для изучения
химич. строения Б. используют и специфич. расщеп-
ление определенных пептидных связей в Б. фермен-
тами (трипсином, химотрипсином и др.), а также
последовательное отщепление аминокислотных ос-
татков с С-конца карбоксипептидазами А и В, ас
N-конца — аминополипептидазой.
Количественное определение бел-
ков. Для колич. определения Б. устанавливают общее
содержание азота по методу Кьельдаля (см. Азота
определение). Кроме того, используют колориметрия,
методы, основанные на различных цветных реакциях
Б., напр. биуретовой, а также реакции Лаури, пред-
ставляющей сочетание биуретовой реакции и реак-
ции Фолина на ароматич. аминокислоты. Концентра-
ции Б. в р-рах можно установить по поглощению в
УФ-области спектра, измерением плотности и пока-
зателей преломления р-ров. Количественно аминокис-
лотный состав Б. определяют гидролизом Б. и после-
387
БЕЛКИ
388
дующим разделением аминокислот, гл. обр. путем
различных методов хроматографии, особенно на колон-
ках из ионообменных смол. Количественное опреде-
ление аминокислот основано на нингидриновой ре-
акции. Современные автоматич. установки позволя-
ют установить полный количественный аминокислот-
ный состав Б. за 24 часа.
Выделение. Во многих случаях при экстрагирова-
нии теми или иными растворителями (обычно водными
р-рами солей, разбавленными к-тами и щелочами и
др.) Б. выделяются в виде смесей. Для выделения
Б. из смесей применяются спец, методы. Широко ис-
пользуется фракционирование Б. высаливанием. Под-
бирая соответств. концентрации соли (чаще всего
применяют сульфат аммония), pH и темп-ру среды,
можно осадить одни Б., оставляя в р-ре другие. Очень
эффективным является метод фракционирования Б.
этиловым или метиловым спиртом при низкой темп-ре
и при строгом учете условий среды (pH, концентрация
солей). С успехом применяются осаждение Б. в изо-
электрической. точке, избирательная денатурация со-
путствующих Б. примесей путем нагревания, осаж-
дение тяжелыми металлами. Для выделения Б. ши-
роко используют методы хроматографии (в частности,
на ионитах с целлюлозной основой), адсорбции, раз-
личные виды электрофореза. Нерастворимые Б. очи-
щают от растворимых примесей экстракцией водными
р-рами солей, кислот, щелочей, органич. раствори-
телями. Полученные препараты Б. освобождают от
солей диализом и при необходимости длительного хра-
нения сушат в вакууме в замороженном состоя-
нии (так называемая лиофилизация Б.). Однород-
ность препаратов Б. устанавливается методами
электрофореза, ультрацентрифугирования, хромато-
графии, исследованиями химического состава, рас-
творимости и др.
Синтез Б. вне организма. Синтез Б. удается на-
блюдать в экспериментальных условиях лишь при
использовании срезов или гомогенатов тканей, сус-
пензий клеток или их частей. Имеющиеся в литературе
сообщения о ферментативном синтезе Б. из продук-
тов их распада противоречивы и нуждаются в даль-
нейшей проверке. В определенных условиях под дей-
ствием ферментов получают из продуктов гидролиза
Б. белковоподобные вещества, т. н. пластеины. Об-
разование пластеинов связано с их нерастворимостью,
выпадением в осадок, что сдвигает равновесие реак-
ции в сторону синтеза. Пластеины по своим свойствам
сильно отличаются от естественных Б. и не могут быть
отнесены к последним. Имеются большие успехи в
химич. синтезе биологически активных полипепти-
дов, так, напр., уже синтезированы 8-членные по-
липептиды — вазопрессин и окситоцин. Путем поли-
конденсации получают полиаминокислоты с мол.
весами 100 000 и выше. Несмотря на принципиальную
возможность синтеза полипептидных цепей Б. ме-
тодами органич. химии, первый полный синтез мо-
лекул Б. с воспроизведением их вторичной, третич-
ной и четвертичной структур будет осуществлен,
очевидно, путем использования механизмов и усло-
вий биологич. синтеза Б. О синтезе Б. в организме
см. Нуклеиновые кислоты, Нуклеопротеиды.
Классификация белков. Число различных Б., вы-
деленных из органов и тканей животных, растений
и из микроорганизмов, необычайно велико. Б. отли-
чаются друг от друга по химич. (аминокислотному)
составу, по порядку чередования аминокислот в по-
липептидпых цепях, по величине молекул и их про-
странств. структуре, по биологич. активности и функ-
циям, к-рые они выполняют. Удовлетворительная
классификация Б. до сих пор отсутствует. Наиболее
распространенной остается классификация Б., ос-
нованная па рекомендациях, сделанных в 1908 ко-
митетами американских биохимич. и физиологич. об-
ществ. Все Б. делят на три группы: простые, сложные
и производные белков. К простым Б., или про-
теинам, относят такие, к-рые при полном гидролизе
дают только аминокислоты. Число простых Б. чрез-
вычайно велико. Их делят на след, группы: альбу-
мины, глобулины, проламины, протамины, гистоны,
склеропротеины, глютелины. К сложным Б.,
или протеидам, относят комплексы простых Б. с
небелковыми компонентами (напр., углеводами, фос-
форной к-той, гетероциклич. соединениями, нуклеи-
новыми к-тами и т. д.). Если небелковый компонент
является низкомолекулярным, то его иногда назы-
вают простетич. группой. В зависимости от характера
небелкового компонента протеиды делят на ряд
групп: нуклеопротеиды, содержащие нуклеиновые кис-
лоты', хромопротеиды, содержащие окрашенные в-ва;
металлопротеиды, содержащие металлы; фосфопро-
теиды, содержащие фосфорную к-ту; гликопротеиды,
содержащие углеводы; липопротеиды, содержащие
липиды. Подобное деление Б. крайне несовершенно
и даже противоречит в нек-рых случаях самому прин-
ципу, положенному в основу классификации. Напр.,
типичные представители простых Б. — альбумины —
содержат нек-рое количество углеводов и поэтому
должны быть отнесены к протеидам. Производ-
ные Б. — наименее охарактеризованная группа.
Под этим названием объединяли продукты, получаю-
щиеся в результате тех или иных изменений Б. Мно-
гие названия этой группы (напр., альбуминаты, аль-
бумозы, метапротеины) устарели и не употребляются
в современной научной лит-ре. Остаются в употреб-
лении лишь два термина — протеозы и пептоны. Про-
теозы и пептоны являются продуктами неполного
гидролиза Б., первые, в отличие от вторых, осажда-
ются из насыщенного р-ра сульфата аммония. В неко-
торых случаях Б. делят по степени асимметрии их
молекул (глобулярные, фибриллярные), по источникам
выделения (Б. крови, Б. молока, Б. тканей и т. п.),
по характеру биологич. активности (Б.-ферменты,
Б.-гормоны, Б.-токсины и т. п.).
Значение белков. Исключительная роль Б. в живых
организмах подчеркивается самим их названием —
«протеины» (от греч. слова лрсото? — первый). Белки
выполняют пластические, энергетические функции,
а также обусловливают определенную, свойственную
данному виду организмов, направленность обмена
веществ. Б. входят в состав всей живой материи, игра-
ют доминирующую роль в протоплазме клеток. Ко-
личество Б. в различных тканях и органах животных
и растений (в % от веса свежей ткани) составляет:
18—23 в мышцах, 7—9 в мозгу, 16—18 в сердце,
6,5—8,5 в крови, 1,2 — 3 в листьях растений, 10—
13 в семенах, 0,3—1 в фруктах. С Б. связаны важней-
шие функции и характерные специфич. черты живых
образований — рост, проявление наследственности,
движение (мышечное сокращение), деятельность ор-
ганов чувств, природа заболеваний, явления иммуни-
тета и мн. др. В живых организмах все химич. про-
цессы осуществляются с участием биологич. катализа-
торов— белков-ферментов. Б.-гормоны являются ре-
гуляторами многих процессов, а Б.-антитела играют
большую роль в защитных приспособлениях организ-
мов животных. Из приведенного выше примера роли
изменения молекул гемоглобина в возникновении сер-
повидной анемии видно большое значение Б. в норме
и патологии организма. Исследования Б. позволили
обнаружить причины заболеваний на «молекулярном
уровне». Л. Полингом введен термин «молекулярные
болезни», под этим подразумеваются изменения в
структуре определенного вида молекул, к-рые явля-
ются причиной тех или иных нарушений в орга-
низме. Современной наукой устанавливается все боль-
389
БЕЛКОВЫЙ ОБМЕН - БЕНЗАНИЛИД
390
шее число таких молекулярных болезней. Ведущая
роль Б. в осуществлении процессов жизни обуслов-
ливает бурное развитие их исследований, к-рое из
проблемы, в основном биохимической, выросло в одну
из решающих проблем современной биологии и меди-
цины. Изучение белковых в-в имеет значение для
познания процессов жизнедеятельности и их созна-
тельного регулирования.
Б. являются необходимыми составными частями
продуктов питания, отсутствие или недостаточное
количество их в пище вызывает серьезные заболевания.
Пищевая ценность Б. зависит от их аминокислотного
состава, от содержания в них г. н. незаменимых
аминокислот, не синтезирующихся в организмах. Б.
используются различными отраслями пром-сти. Боль-
шое значение имеют природные белковые волокна —
шелк, шерсть. С обработкой Б. имеют дело в коже-
венном произ-ве, произ-ве желатины, нек-рых видов
клеев. Из Б., в частности из казеина, получают пласт-
массы. СБ. и их переработкой постоянно имеет дело
пищевая пром-сть. Важное значение имеет произ-во
медицинских белковых препаратов: гормонов, ана-
токсинов, антисывороток, нек-рых кровезаменителей
и др., применяемых для лечебных и профилактич.
целей.
Лит.: Белки, под ред. Г. Нейрата и К. Бейли, пер. с англ.,
т. 1—3, М., 1956—59; Гауровитц Ф., Химия и биоло-
гия белков, пер. с англ., М., 1953; Аминокислоты и белки,
Сб., пер. с англ., м., 1952; Химия белка. Сб. ст., пер. с англ.,
вып. 1—2, М., 1949—52; Кнунянц И. Л., П е р-
вова Е. Я., Успехи химии, 1955, 24, .№ 6, 641; В о л ь-
кенштейнМ. В., Конфигурационная статистика полимер-
ных цепей, М. — Л 1959; Локшина Л. А., Т р о п ц-
к а н О. В., в кн.: Успехи биол. химии, т. 3, М., 1958; Э н-
гельгардт В. А., Некоторые проблемы современной
биохимии, М., 1959; Fox 8. W. and Foster!. F., Intro-
duction to protein chemistry, N. Y.— I... 1957; Advances in
protein chemistry, v. 1 —15, N. Y., 1945—60. b. О. Шпикитер.
БЕЛКОВЫЙ ОБМЕН — см. Обмен веществ.
БЕНЗА ЛЬ АЦЕТОФЕНОН (бензилиденацетофенон,
халкон) С6Н5СОСН=СНСвН5, мол. в. 208,25 — нена-
сыщенный кетон, палево-желтые ромбовидные кри-
сталлы; т. пл. 58°; т, кип. 348°; d22l,O71; нерастворим
В воде, мало растворим в спирте, растворим в эфире,
хлороформе, бензоле, сероуглероде, конц. серной
к-те Б. свойственна галлохромия: при действии к-т
или нек-рых солей, напр. SnCl4, образуются
глубокоокрашенные молекулярные соединения. В
’присутствии 60%-воп серной к-ты Б. реагирует с
альдегидами, обменивая альдегидные остатки. Полу-
чают Б. конденсацией бензальдегида с ацетофеноном
в присутствии ацетата натрия.
БЕНЗАЛЬДЕГИД С6Н5СНО, мол. в. 106,12 —
бесцветная жидкость с запахом горького миндаля;
т. пл. — 26°; т. кип. 179,0°; rfj9,5 1,049; nj/j 1,5450;
теплота сгорания 844 ккал/моль; мало растворим в
воде (0,3 г в 100 г при 20°); смешивается со спиртом,
эфиром, бензолом, хлороформом. При хранении Б.
желтеет, на воздухе быстро окисляется в бензойную
к-ту С6Н5СООН; подобно альдегидам алифатич. ряда
образует продукты присоединения к СО-связи, напр.
с бисульфитом натрия, синильной к-той, фенилгид-
разином и т. п.; с фенолом или ароматич. аминами
конденсируется в производные трифенилметана. При
кипячении с р-ром щелочи Б. образует бензиловый
спирт и бензойную кислоту {Канниццаро реакция);
с KCN конденсируется в бензоин: 2С6Н5СНО —>
—> С6Н5СН(ОН)СОСвН5; с аммиаком образует гидро-
бензамид (Cen5CH = N)2CHCeH5. Б. легко конденси-
руется с ацетангидридом, образуя коричную к-ту
(Перкина реакция).
Б. нитруется, хлорируется, сульфируется гл. обр.
в .иетя-положение. В пром-сти Б. получают прямым
окислением толуола МпО2 и H2SO4 в присутствии
катализатора CuSO4 или в паровой фазе с катализа
7*
тором V2O5; гидролизом бензилиденхлорида р-ром
Са(ОН)2 или H2SO4 при нагревании Все большее
значение приобретает каталитич. синтез Б. из бен-
зола и окиси углерода в присутствии А1С13 и НС1:
С6Н6 + СО —> СвН5СНО. Применяют Б. для синтеза
коричного альдегида, фенилуксусного альдегида и др.
душистых в-в, для синтеза красителей трифенилмета-
нового ряда И др. синтезов. Г. М. Николаев.
БЕНЗАЛЬДИАЦЕТАТ (бензилидендиацетат) мол. в.
208,22; С6Н5СН(ОСоСН3)2, бесцветные кристаллы;
т. пл. 45—50°; т. кип. 220°/760 льи, 154°/20 мм. Б.
может быть приготовлен нагреванием бензилхлорида
с ледяной уксусной к-той и уксусным ангидридом при
150—180°. При обработке замещенного Б. гидроли-
зующими агентами получаются с отщеплением аце-
тильных групп соответствующие' альдегиды. Приме-
няют Б. в органич. синтезах.
БЕНЗАМИД (амид бензойной кислоты) CeH5CONH2,
мол в. 121,14 — кристаллы, триклинные иглы (ла-
бильная форма) и моноклинные призмы (стабильная
форма); т. пл. 130°; <7;° 1,341; растворим в спирте,
кипящем бензоле, горячей воде, очень мало растворим
в холодной воде и эфире. Водный раствор Б. нейтра-
лен; с конц. к-тами образует легкогидролизующиеся
соли, напр. CeII5CONH2 НС1. Водород аминогруппы
Б. может быть замещен натрием с образованием Б.
натрия C6H5CONIINa. При нагревании с бромной
водой Б. дает лг-бромбензойную к-ту; с конц. HNO3
образует .«-нитробензамид. Кислотами и щелочами
Б. гидролизуется до бензойной к-ты и NH3; с РС15
легко образует бензонитрил. Б. получают взаимодей-
ствием хлористого бензоила с аммиаком: С6Н6СОС1 -)-
4-2NH3 —>C6H5CONH2 + NH4C1; реже — из бензой-
ного ангидрида И NH3. м. Я. Алейникова.
БЕНЗАМОН C]4H]9O4NS, мол. в. 297,38 — бес-
цветные кристаллы; т. пл. 132—134°; хорошо раство-
рим в воде и спирте,
нерастворим в эфире.
При нагревании бенз- £н с-СН2МСН3)3 - C6H5SO3
амона наблюдается по- ^q/
бурение кристаллов и
запах триметиламина. Для количественного опре-
деления Б. нагревают со смесью конц. НС1 и HNO3,
разбавляют водой и обрабатывают раствором ВаС12;
выпадает осадок BaSO4. Чистоту препарата кон-
тролируют количественным определением азота по
Кьельдалю. Б. получают восстановительным амини-
рованием фурфурола с последующей обработкой об-
разующегося при этом фурфурилампна метиловым
эфиром бензолсульфокислоты. Применяют' при лече-
нии глаукомы.
Лига. см. при ст. Адалин. А. И. Травин.
БЕНЗАНИЛИД (анилид бензойной кислоты)
CeH5NHCOCeH5, мол. в. 197,23 —- бесцветные кри-
сталлы; т. пл. 163°, т. кип. 117—119°/10л4л«; 1,32;
растворим в спирте, эфире, бензоле, нерастворим
в воде. Б. люминисцирует при облучении катод-
ными лучами. Б. является замещенным амидом и
реагирует подобно амидам карбоновых кислот. Под
действием гипохлорита в присутствии КНСО3 Б. пре-
вращается в С6Н5—NC1—СО—CeH5; с HNOS дает
смесь о- и n-нитроанилидов бензойной к-ты; с РС15
образует фенилимидхлорид C6H5-CC1 = N—СвН5; при
нагревании с серой превращается в 2-фенилбензтиа-
/NX
зол СвН4 С—С6Н5. Б. омыляется кипячением с во-
дой в присутствии к-т и щелочей до бензойной к-ты
и анилина; с К2СО3 вод давлением СО2 образует
о- и л-аминобензойные к-ты. Получают Б. из ани-
лина и бензойной к-ты при 180—225° : CeH5NH2 4-
4-CeH5COOH —> CeH5NHCOCeH5, а также действием
хлористого бензоила или бензойного ангидрида
391
БЕНЗАНТРЕН — БЕНЗИДИНОВАЯ ПЕРЕГРУППИРОВКА
392
на анилин. Б. окрашивает р-р хинона в красный цвет,
а сернокислый р-р К2Сг2О7— в фиолетовый цвет. При-
меняют Б. как пластификатор в лакокрасочной про-
мышленности. М. Я. Алейникова.
БЕНЗАНТРЕН (мезобензантрен) С17Н12, мол. в.
216,28 — бесцветные кристаллы, листочки, т. пл.
84°, нерастворим в воде, растворим
в органических растворителях, об-
з) разует р-ры с зеленой флуоресцен-
цией. На свету сухой Б. медлен-
но 1 *1 н0, быстрее во влажном состоянии
и в р-рах окисляется кислородом
воздуха в бензантрон', в темноте
кислородом воздуха не окисляется. Получают Б.
восстановлением бензантрона цинковой пылью или
триизопропилатом алюминии, а также восстановле-
нием 1,10-триметилен-9-оксифенантрена цинковой
ПЫЛЬЮ. Л. С. Поваров.
БЕНЗАНТРОН (мезобензантрон, 1,9-бензантрон-10)
С]7Н10О, мол. в. 230,27 —желтые иглы, т. пл. 170—
171°; нерастворим в воде, растворим в органич. раст-
ворителях и в H2SO4. Существует несколько способов
нумерации атомов углеродного скелета Б. (I, II,
III); более распространенным является (I):
При восстановлении щелочным раствором Na2S2O4
образуется дигидробензатрон, при восстановлении
Zn-пылью бенгантрен; хромовой к-той окислнетсн в
антрахинон-1-карбоновую к-ту (IV); при сплавлении
со щелочами дает дибензантрон (виолантрон). Элек-
трофильные реагенты замещают водород в положении
3, при этом легко образуют- >--------------
ся 3-хлор-, 3-бром-, 3-нитро- I г,—\
производные (напр., V). Дей- / \_N|-|—nh
ствие нуклеофильных реаген- \ \___/
тов ведет к замещению атомов
водорода в положении 2 или 4; I п
так, при прямом аминирова-----------------------
мии образуется 4-аминопроизводное (VI). В пром-сти
получают Б. обычно из антрахинона и глицерина в
присутствии H2SO4 и Ее-порошка:
Известны и другие способы синтеза Б. и его произ-
водные применяются в произ-ве т. наз. индантрено-
вых красителей (см. Полициклические кубовые краси-
тели). Л. С. Повароз.
БЕНЗГИДРОЛ (дифенилкарбинол) CeH5CHOHCeHt,
мол. в. 184,23 — кристаллы; т. пл. 68,5°; т. кип.
301°; нерастворим в воде, легко растворим в спирте,
эфире, хлороформе, уксусной к-те. При окислении Б.
образуется бензофенон; при гидрогенизации на Pt-ка-
тализаторе — дициклогексилметан. Б. получают вос-
становлением бензофенона Zn-пылью в щелочной
среде: С6Н5СОС6Н5 + Н2О — -► С6Н5СНОНС6Н6 +
+ Zn(OH)2, а также окислением дифенилметана
двуокисью свинца и двуокисью марганца в серно-
кислом р-ре: С6Н5СН2СеН5 2 С6Н5СНОНС6Н5. Б.—
простейший представитель бензгидролов, являющих-
ся полупродуктами в синтезе красителей.
Б. П. Гусев.
БЕНЗИДИН (4,4-диаминодифенил) H2NC6H4—
— C6H4NH2, мол. в. 184,23 — бесцветные или слегка
желтоватые блестящие кристаллы, темнеющие на
свету и на воздухе; ниже 60° из водных р-ров кристал-
лизуется с 1 молекулой воды, т. пл. кристаллогид-
рата 115—120°; т. пл. безводного 127,5—128°; т. кип.
400°/740 мм; растворимость г /100 г: в воде 0,04
(12°), 1,0 (100°); в спирте 1,0 (80°); в абс. эфире 2,2
(15°). Б. растворим в уксусной и разбавленной соля-
ной к-тах с образованием соответств. солей, напр.
солянокислого Б. (монохлоргидрата) C12H12N2 • НС1;
белые иглы, мало растворимые в воде, легко раство-
римые в разбавленной соляной к-те; солянокислого
Б. (дихлоргидрата) C12HI2N2 • 2НС1—белые листочки,
легко растворимые в воде, еще легче в спирте; выпа-
дает из водных р-ров при добавлении большого из-
бытка конц. НС1. Получают Б. изомеризацией гидр-
азобензола под действием минеральных к-т (см. Бен-
зидиновая перегруппировка). Б. легко диазотирует-
ся; сочетанием образующегося бис-диазосоединенин
с сульфокислотами аминов и фенолов получают мно-
гие важные азокрасители. Широко применяется в
аналитич. практике для определении сульфатов, воль-
фраматов, открытии и колориметрия, определении:
CrVI, Си11, Аиш, [Fe(CN)6]3~, PbIV, Hgn, Ce^.NOf,
S2C)| , Pt, Pd, Ir, Os, Ag, Vv, окисляющих Б. в силь-
нокислой среде до окрашенного в желтый цвет соеди-
нении, в нейтральной среде — до синего; используют
как окислительно-восстановительный индикатор при
объемном анализе (е0 = -|-0,92в) и как восстановитель
фосфорномолибденовой к-ты при определении фосфа-
тов. Б. — канцерогенное в-во; предельно допустимая
концентрации его в воздухе 0,001 мг!л.
В. Г. Типцова.
БЕНЗИДИНОВАЯ ПЕРЕГРУППИРОВКА — пере-
группировка гидразосоединений, происходящая под
влиянием к-т и приводящая к образованию произ-
водных 4,4'-диаминодифенила (бензидина). Название
«бензидин» дли4,4'-диаминодифенила (I) дано Н. Н. Зи-
ниным, получившим его в 1845 действием серной к-ты
на гидразобензол (II). При перегруппировке обра-
зуется также нек-рое количество 4,2'-диаминодифе-
нила (III).
Б. п. широко применнетсн в ряду гидразопроиз-
водных бензола для получении азокрасителей, диа-
минов рнда дифенила, напр. толидина, дианизидина
и др. Перегруппировка различных гидразосоединений
происходит с различной скоростью, в зависимости
от характера заместителей и их положении в бензоль-
ных ядрах, напр. о-гидразоанизол > гидразобен-
зол >• о-гидразофенетол >• .vs-гидразотолуол >• о-гид-
разотолуол. В нафталиновом ряду Б. п. может про-
исходить в отсутствие кислот. Так, 1,1'-гидразонаф-
талин при нагревании в спиртовой среде образует
393
БЕНЗИЛ — а-БЕНЗИЛДИОКСИМ
394
4,4'-Диамино-1, Г-динафтил и 1,Г-диамино-2,2'щи-
нафтил.
Б. п. является внутримолекулярной перегруппировкой,
что подтверждается экспериментальными данными: 1) при
перегруппировке смесей различных симметричных гидразо-
соединений не образуются смешанные (несимметричные) про-
изводные бензидина; 2) несимметричные гидразосоединения
дают лишь несимметричные дифенилы. Н. Н. Ворожцов мл.
рассматривает Б. п. как один из частных случаев перегруппи-
ровок N-замещенцых аминов, вызываемых действием кислот-
ных агентов. Изучение кинетики перегруппировки показало,
что скорость реакции прямо пропорциональна квадрату кон-
центрации кислоты. На этом основании был сделан вывод,
что вначале к молекуле гидразосоединения присоединяются
2 протона, а затем уже образовавшийся комплекс перегруп-
пировывается.
К. К. Ингольд допускает, что переходное состояние носит
гл. обр. ионный характер. Это связано с тем, что хотя два про-
тона, вовлекаемые в переходное состояние, в конце реакции
связаны ковалентно, два других атома водорода становятся
свободными ионами, благодаря перемещению электронов
(структуры IV и V). В переходном состоянии возможно рас-
щепление нек-рых структур на отдельные перегруппировы-
вающиеся фрагменты (напр., VI), к-рые удерживаются элект-
ростатич. связью. В переходном состоянии оба бензольных
кольца лежат приблизительно в параллельных плоскостях,
что значительно сближает парауглеродные атомы (до 4,3 А).
При таком расстоянии олектростатич. связь может быть
достаточно сильной. Образующаяся т. о. связь может быть сна-
чала ионной, но переходит в ковалентную связь по мере ее
укорачивания.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; Ингольд
К. К., Механизм реакций и строение органических соедине-
ний, пер. с англ., М., 1959. В. А. Пучков.
БЕНЗИЛ (дибензоил, дифенилглиоксаль, дифенил-
дикетон) СбНбСОСОСбН5, мол. в. 210,22 — желтые
кристаллы, призмы; т. пл. 95°; т. кип. 346—348°;
188°/12 мм\ dj5 1,23; %макс> 2550 А (в гексане); не-
растворим в воде. Растворяется в 100 г растворите-
ля (25°): 4,86 г в этиловом спирте, 59,0 г в бензоле.
При восстановлении Б. в кислой среде превращается
в бензоин СбН5СОСН(ОН)СбН5; амальгамой натрия
восстанавливается в дигидробензоин СбН5СН(ОН)—
—СН(ОН)СвН5; при нагревании со спиртовым рас-
твором КОН перегруппировывается в. бензиловую
кислоту (СбН5)2С(ОН)СООК; присоединяет калий с
образованием стильбендиолкалия СбН5С(ОК) —
= С(ОК)СбН5; с гидразинами и гидроксиламином
дает моно- и дигидразоны и оксимы; конденсируется
с аммиаком и альдегидами с образованием замещен-
ных имидазолов:
С6Н5-С----NH
Ce^5—Cx^^CR
Получают Б. окислением бензоина азотной к-той или
кислородом воздуха в присутствии CuSO4 как катали-
затора. Б. используется как инсектицид и репеллент
(см. Отпугивающие средства). м. Ф. Турчинский.
БЕНЗИЛ БРОМИСТЫЙ (со-бромтолуол, бензил-
бромид) С6Н5СН2Вг, мол. в. 171,05 — бесцветная
жидкость с резким запахом; т. пл. •—3,8°; т. кип.
198°; d22 1,438; нерастворим в воде, растворим в ор-
ганич. растворителях; лакриматор. По химич. свой-
ствам и реакционной способности очень похож на
бензил хлористый.
Получают Б. б. при бромировании толуола под
воздействием ультрафиолетовых или солнечных лучей
(см. Бензил хлористый). е. Е. Барони.
БЕНЗИЛ ХЛОРИСТЫЙ (а>-хлортолуол) С6Н5СН2С1,
мол. в. 126,61 — бесцветная жидкость с резким запа-
хом; т. пл. —41°; т. кип. 179,3° /760 мм; 106°/ 92 мм,
63°/8 мм; <ZJ5 1,043; Пд 1,5390; в воде нерастворим,
смешивается со спиртом и хлороформом; летуч с во-
дяным паром. Хлор в Б. х. отличается большей по-
движностью по сравнению с алкилгалогенидами; при
нагревании с водой постепенно гидролизуется с об-
разованием бензилового спирта С6Н5СН2ОН; щелочь
ускоряет гидролиз. С аммиаком и аминами Б. х. дает
соответствующие бензилпроизводные; с KCN — бен-
зилцианид CeH5CH2CN; легко и с высоким выходом
образует реактив Гриньяра: С6Н5СН2С1 + Mg —>
—> CeH5CH2MgCl.
В пром-сти Б. х. получают пропуская хлор в пред-
варительно нагретый до 90—100° толуол, к к-рому до-
бавлен 1% РС13; в дальнейшем толуол не подо-
гревают, так как выделяющейся теплоты реакции
(27 ккал/моль) достаточно для его подогрева. Процесс
ведут в стальных освинцованных реакторах.
В лаборатории Б. х. получают пропусканием хлора
в кипящий толуол при освещении солнечным или
ультрафиолетовым светом. Этим способом стали поль-
зоваться в пром-сти. Применяется Б. х. для произ-ва
бензилового спирта, сложных эфиров бензойной к-ты,
бензилцианида, бензилцеллюлозы. л. С. Поваров.
БЕНЗИЛАМЙН (а-аминотолуол) CeH5CH2NH2, мол.
в. 107,16 — бесцветная жидкость с щелочной реакцией
и слабым аммиачным запахом; т. кип. 184,5°; rf;"
0,9826; п$ 1,5441; константа диссоциации К =
= 2,4-10 5(25°), рА'(НА! ) = 9,62; смешивается с водой,
спиртом и эфиром; на воздухе поглощает СО2, обра-
зуя твердый карбонат. Получают Б. восстановлением
фенилнитрометана или действием аммиака на хло-
ристый бензил. Применяется Б. в органич. синтезе
и для микрокристаллоскопич. обнаружения молиб-
датов, вольфраматов, ванадатов, а также Th, Zr,
Се, La, Nd и Рг. Константа устойчивости комплекса
с Ag+ (25°, 0,5 М раствор KNO3) : lgK4 = 3,29; IgA'2 =
= 3,85. И. П. Ефимов.
БЕНЗИЛАЦЕТОН (Р-фенилэтилметилкетон), мол. в.
148,09, С6Н6(СН2)2СОСН3, — жидкость с приятным
запахом, напоминающим запах жасмина; т. кип.
235°/76О мм; 115°/13 мм; df 0,9849; п$ 1,5111.
В промышленности получают каталитич. восстанов-
лением бензилиденацетона с катализаторами (палла-
дий, платиновая чернь или никель, полученный из
формиата никеля). Б. образует бисульфитное соеди-
нение; идентифицируют по оксиму, т. пл. 87°, или
семикарбазону, т. пл. 142°; количественно определяет-
ся оксимированием. Применяют в парфюмерии для
приготовления композиций с запахом жасмина.
А. А. Зеленецкая.
а-БЕНЗИЛДИОКСИМ (а-дифенилглиоксим, нике-
лои) C14H12O2N2, мол. в. 240,26 — бесцветный мел-
кокристаллич. порошок; т. пл. 235—237° (с разл.);
почти нерастворим в воде, эфире, ледяной уксусной
к-те; в 100 г спирта растворяется 0,05 г (17°); раство-
ряется в р-рах щелочей. При действии на бензил
С6Н5СО — СОС6Н5 гидроксиламином NH2OH можно
получить три стереоизомерных диоксима:
II II II II
НО—N N—ОН N-ОН НО—N
анти-форма (а) син-форма (₽)
С(,Н4С-----С—CgHg
N—ОН N—ОН
алдЗи-форма (у)
из к-рых только анти-изомер при прибавлении к
спирто-аммиачному р-ру соли Ni дает малораствори-
мый осадок красного цвета. Применяется Б. для об-
наружения и весового определения Ni (ср. Диметил-
глиоксим). Н.П. Ефимов.
395
БЕНЗИЛИДЕНАЦЕТОН — БЕНЗИЛПЕНИЦИЛЛИН
396
БЕНЗИЛИДЕНАЦЕТОН (бензальацетон, метил-
стирилкетон) СеН5СН=СНСОСН3, мол. в. 146,19—
кристаллы светло-желтого цвета, т. пл. 42°, т. кип.
260—262°/760 л<.и, 151—153°/25 мм, d™ 1,0213;
1,035; растворяется в спирте, эфире, хлоро-
форме, бензоле, а также в серной к-те. Б. обладает
сильным запахом, напоминающим запах кумарина.
Б. получают конденсацией бензальдегида с ацетоном
в присутствии соды или иного щелочного конденси-
рующего агента:
СеН5СНО + СП:.СОСН3 —^—?СвП6СН=СНСОС113
Б. дает реакции, характерные для кетонов;
его идентифицируют по оксиму, т. пл. 116°, семикар-
базону, т. пл. 185° или n-нитрофенилгидразону, т. пл.
165—167°; количественно Б. определяют оксимирова-
нием. Применяются в парфюмерии в незначит. коли-
честве, гл. обр. для духов, вследствие раздражаю-
щего действия на кожу. Применяется в синтезе бен-
зилацетона и коричной к-ты.
Лит.: Белов В. Н. [и др.], Химан и технология души-
стых веществ, М., 1953; West Т. F., Strauss И. J.
and Barton D. Н. R_. Synthetic perfumes, L., 1949.
А А. Зеле-нецкая.
БЕНЗИЛИДЕНХЛОРИД (1,1-дихлор-1-метилбен-
зол, со, со-дихлортолуол, бензальхлорид) С6Н5СНС12,
мол. в. 161,03 — бесцветная жидкость с резким за-
пахом; т. замерзания —17°; т. пл. —16,1°; т. кип.
207°; d'\ 1,2699; rfj’ 1,2557; n'ft' 1,5515. Б. не-
растворим в воде, смешивается со спиртом и эфиром,
образует азеотропные смеси с октанолом-1, борнеолом,
хлоруксусноп и капроновой к-тами, обладает слезо-
точивым и раздражающим действием.
При нагревании с водой до 140—160° Б. гидро-
лизуется в бензальдегид; это превращение легче
протекает при действия сильных мине-
ральных кислот, например H2SO4, или
при кипячении с водной щелочью:
С„Н5СНС12 + Н,О-> С6Н5СНО + 2НС1; с
ацетатом яатрпя конденсируется в соль
коричной к-ты: С6Н5СНС12 + CH3COONa—>
—C6H5CH = CHC(JONa + 2НС1; при нагре-
вании с SbF3 дает бензилиденфторид, с ме-
таллич. натрием — стильбен, с Си — стиль-
беядихлорид, с аммиаком Б. образует гид-
робензамид CeH5CH (N = CHC6H5)2;'с алко-
голятом натрия — ацеталь бензальдегида;
с N aHS в спирте — дибензилдисульфид.
Б. получают хлорированием толуола при
темп-ре кипения в присутствии РС15 или
РС13; реакцию можно ускорить облучением
В пром-сти этот процесс ведут в освинцо-
ванной аппаратуре, т. к. железо способст-
вует хлорированию толуола в ядре. При-
меняются Б гл. обр. для произ-ва бен-
зальдегида. л. С. Поваров.
БЕНЗИЛОВЫЙ СПИРТ (фенилкарбинол)
СвН5СН2ОН, мол. в. 108,13 — бесцветная
жидкость, обладающая слабым приятным
запахом; т. кии. 205,8°, 93°/10 лг.и; rf]"l ,0455;
n'jj 1,5396; растворимость в воде 4 е/100 г
(17°); хороню растворяется в спирте, хло-
роформе и в других органич. растворите-
лях; очень легко растворяется в жидких
SO2 и NH3. Б. е. широко распространен в
природе; в свободном состоянии он нахо-
дится в маслах туберозы, иланг-иланга.
гвоздичном и яек-рых др.; во многих эфир-
ных маслах содержатся сложные эфиры
Б. с.: бензил ацетат, бензилбензоат и др. В
пром-сти Б. с. получают омылением хлори-
стого бензила нагреванием с водой и окисью
магния, окисью цинка пли свежеосажденной
окисью свинца; чаще всего омыление проводят в при-
сутствии щелочи; С6Н5СН2С1 — ° CeH5CH2OH + НС1.
Другим распространенным в пром-сти методом получе-
ния Б. с. является обработка смеси бензальдегида и фор-
малина крепким р-ром щелочи: С6Н5СНО + НСНО +
+ NaOH —> С6Н5СН2ОН + HCOONa. Последний ме-
тод имеет большое значение для парфюмерии, так
как позволяет получать Б. с высокого качества.
Идентифицируют Б. с. по фенилуретану, т. пл 78°;
зфиру фталевой кислоты, т. пл. 106—107°; количе-
ственно определяется ацетилированием в присут-
ствии пиридина при комнатной температуре. Б. с.
применяют в парфюмерии, а также как растворитель
лаков.
Лит.: Горяев М. И., Характеристика химических
соединений, входящих в состав афирных масел, Алма-Ата,
1953; West Т. F., Str ausz Н. J., Barton D. Н. R.,
Synthetic perfumes, L., [1949]. А. А. Зглеиецкая.
БЕНЗИЛПЕНИЦИЛЛИН (пенициллин G), молеку-
лярный вес 298,32, CI6HieO4N2—наиболее изученный
природный пеницил-
лин. Б. кристалли- /Ss
зуется с одной мо- CeH5CH2CONHCH—СН С(СН3)2
лекулой растворите- c0-n---СНСООН
ля из диизопропило-
вого эфира в виде
белых игл; т. нл. 80°; [a]JD =+241 ° (с = 1 в буфер-
ном растворе, pH 7,47); легко растворим в эфире,
ацетоне, метаноле и этаноле, слабо — в бензоле.
Б. довольно сильная кислота (рК ок. 2,7). Б. был
впервые синтезирован с ничтожным выходом (0,03—
0.08%) Дю-Виньи с сотр. (1946). Рациональный
синтез бензилпенициллина разработан Шиханом с
сотр. (1959):
(CH3),C-SH , Ох /СО
+ C-CH-N
H3COOC-CH-NH2-HCI Нх | ХСО
СООС(СН3)3
CH:iCOONa
zS\
(Сн3)2С сн-сн-
Н3СООС-СН-NH
D-a (I)
ZS4
(Сн3)2с ch-ch-nh2
H3COOC-CH-NH COOCtCH
О-a (ID-2HC1
zs\
(CH3)2(j: ch-ch-nh2
H3COOC-CH-NH COOH
D-a (lll)-2HCl
HCI 6 CHgCI2
>3
C1C(C6H5)3
NH(C2H5)2
/S4
(CH3)2C CH CH NHC(C6H5)3 N, N'—диизопропилнорбодиимид
H3COOC—CH~NH ioOH в смеси дионсан-водо
D-a (IV)
г /S4
(СНа)2С CH-CH-NHC(CeH5)3 NaOH
НзСООС-СН-N — СО
D-a (V;
/Sx
(СН3)2С Сн-CH-NH С(С6Н5)3 разб. HCI
HOOC-CH-N------С=О
D-a (VI) в виде диэтиламиновой соли
397
БЕНЗИЛЦЕЛЛЮЛОЗА — БЕНЗИЛЬНАЯ ПЕРЕГРУППИРОВКА
398
/Sx
(СН3)2С ch-ch-nh2-hci
НООС-СН—N—с=о
D-а (VII); т. пл. 207—208° (с разл.)
С|СОСН2С6Н5
в водном ацетоне,
содержащем NaHCOg
zs\
(СН3)2С СН-СН-NHCOCH2CeH5
НООС—СН—N— С=О
D-a (VIII) в виде N-этилпипвридиновой соли
В приведенной схеме означают: цифры в скобках — поряд-
ковый номер соединения: D — принадлежность к D-ряду
по оптич. активности: a — один из изомеров Б.
Идентичность синтетического В. с природным была
установлена с помощью ПК-спектров, темп-p плавле-
ния и смешанной пробы, оптич. вращения и микро-
биология. испытания. Шихан показал, что при аци-
лировании промежуточного соединения D-a (VII)
penicillanic acid (пеницилланиевой к-ты) обра- /
зуются пеницвллины, к-рые не могут быть получе- '
ны ферментацией, в природные пенициллины.
Б. обычно получается из культуральной жидко-
сти различных видов пенициллиума (Penicillium
chrysogenum, Р. notatum) при добавлении в ка-
честве предшественника фенилацетамида или фе-
нилуксусной к-ты. Выделение Б. из культуральной
жидкости и его очистка производится методом экстрак-
ции. Б. применяется в виде солей, из к-рых важ-
нейшими являются натриевая, калиевая, новокаино-
вая в дибензилэтилендиаминовая.
Б. — высокоэффективное антибактериальное сред-
ство; действует гл. обр. на грамположительные бак-
терии и применяется чаще всего при лечении кок-
ковых инфекций. Содержание Б. определяют биоло-
гич. и химич. методами и выражают в единицах дей-
ствия. См. также Пенициллины.
Лит.: Шемякин М. М., Хохлов А. С., Химия
антибиотических веществ, 2 изд., М., 1953; Пенициллин
и применение его в клинике. Под ред. 3. В. Ермольевой и
В. Я. Шланобарского, М., 1956; The chemistry of penicillin,
Princeton (New Jersey), 1949; Sheehan J. C., Henery-
Logan K. R., J. Am. Chem. Soo., 1959, 81, №21, p. 583
И. И. Иноземцева.
БЕНЗИЛЦЕЛЛЮЛОЗА — простые эфиры целлю-
лозы и бензилового спирта. Б получают действием
хлористого бензила на щелочную целлюлозу илв на
БЕНЗИЛЬНАЯ ПЕРЕГРУППИРОВКА — превра-
щение a-дикетонов под влиянием щелочей в а-окси-
кислоты. Впервые описана в 1838 Ю. Либихом на
примере бензила, или дибензоила (I), к-рый при на-
гревании со спиртовой щелочью превращается в бен-
зиловую, или дифенилгликолевую, к-ту (II):
СвН6 /ОН
с0н5со-сос0н5 с
СвНй^ \юон
I II
Кроме ароматич. дикетонов, Б. п. претерпевают али-
циклич., алифатич. и гетероциклич. дикетоны, а
также о-хиноны. Напр., фенантренхинон (III) в ус-
ловиях Б. п. образует 9-оксифлуорен-9-карбоновую
к-ту (IV), к-рая при окислении хромовой смесью пре-
вращается в кетонфлуоренон (V):
°\__кон /“Л/'Л +о /==\ /“Ч
\_Л"\_у-со2
Vi Xх
о о НОХ хсоон
III 17 If
Иногда дальнейшее превращение оксикарбоновых
к-т, образующихся в результате Б. п., в кетоны про-
исходит при действии серной к-ты, при этом выделяется
не СО2, а молекула муравьиной к-ты, напр.:
нооссн2со-сосн2соон — 52
НООССН2-С-СН2СООН
нсГ'соон
-> НООССН2-С-СП2СООН
IbSO,
-исоон
Таким образом при помощи Б. п. можно перейти от
а-дикетонов к кетонам, содержащим на один атом
С меньше.
Б. п. является внутримолекулярной перегруппи-
ровкой. Механизм этого превращения недостаточно
ясен. Хюккель предлагает для его объяснения след.
схему:
I II II И । . II I
НОС-ССЙН5—-НОС-ССвН5 нос-ссвн5 —- О-С-СС6Н5
C-eHj с6Н^ CeH5
VI VII II
110-
смесь целлюлозы и концентрированного р-ра едкого
натра: [С6Н-О2 (ОН)3]П + ЗгаС6Н5СН2С1 +
+ 3nNaOH [С6Н7Оа(ОСН2С6Н5)3]л +
-)-3nNaCl + ЗпН.2О. Прв получении Б. берут
значительный избыток хлорвстого бензила,
т. к. 50—60% его расходуется на побочные
реакции: образование дибензилового эфи-
ра, бензилового спирта и др. Бензилиро-
вание целлюлозы производится обычно при
130° и нормальном давлении. Ввиду гидрофильности
целлюлозы и гидрофобности хлористого бензила в
самой Б реакция идет медленно и обычно не дово-
дится до образования трехзамещепного эфира цел-
люлозы Технич. Б содержит 2,25—2,5 эфирных
групп на глюкозный остаток и растворяется в аце-
тоне, дихлорэтане, этилацетате, смесях спирта с
бензолом или толуолом и в др. органич. растворите-
лях. В воде и щелочи Б. не растворяется. Б. отли-
чается высокой химич. стойкостью, гидрофобно-
стью и высокими электроизоляционными свойствами,
термопластична, размягчается при 100—120°. Моро-
зостойкость Б. удовлетворительная, светостойкость
невысока. Б. широко применяют в произ-ве разлвчных
пластич масс, пленок, электроизоляционных покры-
тий и лаков.
Лит.: Ушаков С. Н., Эфиры целлюлозы и пласти-
ческие массы на их основе, Л.—М., 1941; Роговин 3. А.
и Шорыгина Н. Н., Химия целлюлозы и ее спутников
М.—Л., 1953. А. Б. Пакшвер.
В определенных условиях удалось получить про-
дукт присоединения одного моля КОН к бензилу,
соответствующий иону гидратной формы дибензо-
ила (VI), к-рый легко перегруппировывается в (III).
Продукт присоединения этилата калия изомеризуется
медленнее и с образованием побочных веществ. Не-
которые исследователв считают, что Б. п. аналогична
пинаколиновой перегруппировке. Интересный случай
перегруппировки наблюдается при аутоокислении
дикалиевой соли стильбендиола (VIII); наряду с бен-
зойной к-той возникает до 50% бензиловой к-ты (II):
к° ок С0Н5 /ОН
С„Н6С=СС(,Н5 + о -> С6Н5СООН + с
СвН^ \юон •
VIII II
Образование последней возможно через промежуточ-
ные ионы (VI и VII).
Лит.: Хюккель В., Теоретические основы органи-
ческой химии, т. 1, М., 1955 (с. 3421—47). Б. А. Пучков.
399
БЕНЗИМИДАЗОЛ - БЕНЗИНЫ
400
БЕНЗИМИДАЗОЛ (бензиминазол, 1,3-бенздиазол)
C,H6N2 (I), мол. в. 118,14 — кристаллы; т. пл. 170°;
растворим в воде,
спирте и др. по-
лярных раствори-
телях, плохо рас-
творим в неполяр-
ных растворите-
И' /
лях; производные: пикрат, т. пл. 223°; N-ацетил, т. пл.
ИЗ—114° (из бензола); N-бензоил, т. пл. 93° (из
спирта). Молекулы Б. ассоциированы благодаря об-
<Г
НООС
НООС
т н
разованию след, водородной связи —NH •••• N=.
13. амфотерен и растворяется в кислотах и щелочах.
С ионами нек-рых
металлов (Со, Cd,
Си, Zn, Ag) в ам-
миачном растворе
Б. образует соли,
легко гидролизу-
емые водой; при действии на Б. RMgX водород в по-
ложении 1 замещается группой MgX (где X — С1
или Вг). При нагревании с эквимолекулярным ко-
личеством RJ или R2SO4 Б. образует N-алкил-Б.
Нагревание Б. с двукратным количеством RJ приво-
дит к образованию солей 1,3-диалкилбензимидазоля
(II). N-бензоил-Б. и вообще N-ацильные производные
получают нагреванием Б. с соответствующим RCOC1
в С6Н6. Б.устойчив к действию окислителей, толь-
ко КМпО4 в жестких условиях окисляет бензоль-
вое кольцо, при этом образуется двухосновная к-та
(III). Нитрование конц. HNO3 и H2SO4 и сульфиро-
вание H2SO4 или C1SO3H приводят к 5- или 6-заме-
щенным Б. [положения 5 и 6 равноценны вследствие
таутомерии (I)].
Б. получают: 1) нагреванием о-фенилендиамина
с избытком НСООН: 2) нагреванием моно- или дифор-
NHCHO
NH2
a NHCHO
NHCHO
(I/ v
мил-о-фенилен диамин а (соответственно
и многие его производные у иминного
Дибазол) обладают высокой физиоло-
гия. активностью. Система Б. входит в
состав молекулы цианкобаламина (вита-
мина В12). Некоторые производные Б.
являются полупродуктами в произ-ве
красителей.
Лип'/.: Heterocyclic compounds. Ed. R. C. El-
derfield, V. 5, N. Y.—L., 1957; Hofmann K.,
Imidazole and its derivatives, pt. 1,N. Y.—L.,
1953. В. M. Демьянович.
БЕНЗИНЫ — сложные смеси легких
углеводородов, применяемые гл. обр.
как топливо в двигателях с искровым
зажиганием, а также в качестве раство-
рителей в различных отраслях пром-сти,
для экстракции и др. Исходным сырьем
для получения Б. служит нефть. Б.
принято подразделять по их использова-
нию на автомобильные, авиационные и
Б.-растворители, а по методам получе-
ния на Б. прямой перегонки, термич.
крекинга, каталитич. крекинга, рифор-
минга и т. д.
Автомобильные бензины представ-
ляют обычно смесь продуктов Прямой перегонки
и крекинга нефти, выкипающую не выше 205°. Основ-
ные параметры автомобильных Б.: 1) антидетонаци-
онная характеристика, выражаемая октановыми чис-
лами (ОЧ); 2) испаряемость, характеризуемая фрак-
ционным составом и давлением насыщенных паров.
Кроме того, регламентируются: кислотность, содер-
жание смол, серы, механич. примесей, воды и нек-рые
другие показатели. Улучшение антидетонационных
свойств автомобильных Б. достигается: повышением
содержания в них изопарафиновых и ароматич. угле-
водородов; добавлением к Б. антидетонатора — тет-
раэтилсвинца (ТЭС). Ввиду сильной ядовитости ТЭС,
содержащие его Б. окрашивают. Двигатели со сте-
пенью сжатия до 6 допускают применение Б. с ОЧ
не ниже 66 (68—70) (в скобках здесь и далее при-
ведены значения ОЧ, полученные по исследователь-
скому методу). Для двигателей с более высокими
степенями сжатия в СССР применяются Б. с ОЧ 72
(76), 74 (78), 76 (80). Для некоторых современных
автомобильных двигателей со степенью сжатия 8,0,
9,0 и у нек-рых до 10,5 (для автомобилей «Чайка»,
ЗИЛ-111 и др.) выпускаются две марки автомобиль-
ных Б. с ОЧ (93) и (98).
Фракционный состав и др. параметры автомобильных Б.,
выпускаемых в СССР, приведены в табл. 1. Вырабатывается
также «зональный» сорт Б., отличающийся более легким
фракционным составом и повышенным давлением насыщенных
паров для применения в холодном климате Сибири и Запо-
лярья.
От темп-ры конца кипения первой — «пусковой» (10%)
фракции зависит легкость пуска двигателя; промежуточная —
«рабочая» (50%) — фракция обусловливает «приемистость»
двигателя и чем ниже темп-pa, при к-рой она перегоняется,
тем устойчивее работа двигателя. Темп-pa выкипания конеч-
ной (90%) фракции влияет на полноту испарения и антидето-
национные свойства Б. Давление насыщенных паров является
константой, гарантирующей отсутствие паровых пробок в бен-
зинопроводах двигателей.
Автомобильные Б. обычно содержат непредельные
углеводороды — продукты термич. крекинга — и для
повышения стабильности при хранении нуждаются в
добавке антиокислителей, напр. п-оксидифениламина
или фракции древесной смолы. Срок хранения автомо-
бильных Б. с антиокислителем не более 6 месяцев.
Улучшенные по сравнению с Б. А-66, антидетонацион-
ные свойства Б. А-72 достигаются за счет введения
продуктов каталитич. крекинга или риформинга. Эти
продукты повышают т. н. чувствительность Б. к из-
менениям режима работы двигателя, что косвенно
характеризуется разницей в значениях ОЧ, получен-
ных по моторному и по исследовательскому методам.
IV и V). Б.
азота (напр.,
Таблица 1
Характеристика автомо- бильных Б. А-66 А-72 А-74 А-76 А-87 «Экстра»
Октановое число* 66 72 (76) 74 (78) 76 (80) 87 (93) (98)
ТЭС, не более г/кг Фракционный состав: 0,82 не coi (ержит 0,41 1,17 1,23
10% перегоняется не вы-
ше °C ... 50% перегоняется не вы- 79(65) 75 70 75 70 68
ше °C 90% перегоняется не вы- 145 (120) 135 105 135 120 115
ше °C 195(175) 180 165 180 180 180
Конец кипения не выше °C Давление насыщенных па- 205(190) 195 180 195 205 185
ров, не выше мм рт. ст . Кислотность не более мг 500(700) 500 500 500 500 400 (не менее)
КОН на 100мл 3 3 2 3 2 1
Смол, не более мг на 100 мл:
на месте производства . . 7 5 2 5 2 2
на месте потребления . . 20 10 2 10 2
Серы, не более % 0,15 0,15 0,10 0,15 0,1 ОД
* Цифры в скобках всюду для «зонального» Б.
401
БЕНЗИНЫ
402
Чем больше эта разница, при равных прочих каче-
ствах, тем лучше бензин.
Авиационные Б., как правило, представ-
ляют смесь бензиновых фракций и высокооктановых
углеводородных компонентов с добавкой антидетона-
тора и антиокислителя для предохранения ТЭС от
разложения кислородом воздуха. Наиболее ценными
по аитидетонационным свойствам являются изопара-
финовые, ароматич. и нафтеновые углеводороды. Пу-
тем компаундирования бензиновых фракций различ-
ного химич. состава высокооктановыми компонентами
достигается оптимальное соотношение необходимых
классов углеводородов в авиабензине. Нефтепро-
дукты, составляющие основную часть авиабензина,
наз. базовыми Б.; к ним относятся: Б. прямой пере-
гонки отборных нефтей и Б. каталитич. крекинга
и риформинга. Базовые Б. должны обладать высоким
ОЧ, иметь плавную кривую выкипания, не содержать
бутанов и более легких углеводородов, иметь низкую
темп-ру застывания (не выше —60°) и давать резкое
повышение ОЧ при добавлении ТЭС (т. н. высокая
приемистость к ТЭС). К высокооктановым компо-
нентам авиабензинов относятся: изооктан, изопентан,
алкилат, алкилбензолы и др. В СССР выпускаются
след, марки авиабензинов: Б-100/130, состоящий из
Б. каталитич. крекинга и высокооктановых компонен-
тов; Б-95/130 и Б-91/115 — в качестве базового ком-
понента содержат как Б. каталитич. крекинга, так
и прямой перегонки, в качестве высокооктановых ком-
понентов — изопарафиновые и ароматич. углеводо-
роды; Б-70—продукт прямой перегонки с высокоокта-
новыми компонентами.
Характеристика авиационных Б. Б-100/130 Б-95/130 Б-91/115 Б-70
Содержание ТЭС, не более г/кг 2,7 3,3 2,5
Содержание п-оксидифениламина
(антиокислитель), % Теплота сгорания, ие менее 0,005 0,005 0,005 —
ккал/кг ............. 10300 10300 10300 —
Фракционный состав:
начало перегонки, не ниже °C 40 40 40 40
10% перегоняются, не выше °C 75 82 82 88
50% » » » °C 105 105 105 105
90% » » » ”С 145 145 145 145
97,5% » » » СС 180 180 180 180
Давление насыщенных паров, 220
не менее мм рт. ст 240 220 ——
То же, не более 360 360 360 360
Кислотность, не более мг КОН
на 100 мл ............ 1,2 1 1 1
Серы, не более % 0,05 0,05 0,05 0,05
Начало кристаллизации, ие вы-
ше °C —60 -60 -60 -60
Цвет ярко- оранж. желтый зеленый бесцвет- ный
Для Б., применяемых в мощных авиадвигателях с
наддувом, ОЧ не в полной мере характеризует анти-
детонационные свойства топлива, вследствие чего
марки таких Б. обозначаются двумя цифрами: в чис-
лителе либо октановое число по т. н. темп-рному
методу (для ОЧ выше 90 и менее 100), либо т. н. услов-
ная сортность на бедных смесях (для Б. с ОЧ выше
100); в знаменателе — сортность на богатых смесях.
При определении сортности эталонным топливом яв-
ляется не смесь изооктана с и-гептаном, как при оп-
ределении ОЧ, а изооктан в смеси с добавками ТЭС
(см. Сортность топлив). Основные характеристики
наиболее распространенных в СССР авиабензинов
приводятся в табл. 2.
Бензины-растворители и экстра-
генты представляют собой узкие низкокипящие
фракции прямой перегонки нефти. Одним из важных
их свойств является скорость улетучивания. Б.-раст-
ворители для резиновой пром-сти выпускаются двух
марок: БР-1 («галоша») и БР-2; последний отличается
от БР-1 лишь содержанием серы (до 0,025%) и гото-
вится из сернистой нефти. Фракционный состав этих
Б. характеризуется началом кипения 80°, причем не
менее 98% должно перегоняться до 120°. Содержание
ароматич. углеводородов, несмотря на их отличные
качества как растворителей, ограничивается 3% ввиду
их токсичности. Б.-экстракционный применяют для
извлечения растительных масел из семян масличных,
жира из костей, никотина из махорочных листьев, а
также для получения быстросохнущих лаков и красок.
Начало кипения этого Б. 70°, 98% должно перегоняться
до 95°, содержание ароматич. углеводородов допус-
кается не выше 4%. Б.-растворитель для лаков и олиф
в лакокрасочной пром-сти (уайт-спирит) характери-
зуется: высокой темп-рой начала кипения (не выше
165°); 98% — перегоняется до 200°; ароматич. угле-
водородов не более 16%. Б.-растворитель марки Б-70
для технич. целей применяется в произ-ве кожзаме-
нителей, для химич. чистки тканей, для промывки
деталей механизмов при ремонте и др. Характери-
зуется низкой темп-рой начала кипения (45°); 97,5%
должно перегоняться не выше 110°; ароматич. угле-
водородов не должен содержать вовсе.
Получение бензинов основано на про-
цессах переработки нефти: прямой перегонке, кре-
кинге термич. и каталитич., риформинге, гидрофор-
минге, платформинге. Кроме того, т. п. газовые Б.
выделяют физич. методами из газов нефтяных попут-
ных. Наряду с Б. нефтяного происхождения, ограни-
ченное применение находят Б., получаемые гидроге-
Таблица 2 низациеп тяжелых нефтяных остатков
и углей или синтезом из окиси углерода
и водорода (см. Синтетическое жидкое
топливо). Б. прямой перегонки нефти
на 90—95% состоят из парафиновых и
нафтеновых углеводородов и до 5—
9% ароматических углеводородов. Среди
отечественных Б. исключением являются
Б. из верхнечусовской нефти, содер-
жащие от 30 до 50% ароматич. угле-
водородов, и из майкопской нефти — до
13%. Непредельные углеводороды не со-
держатся в Б. прямой перегонки; вслед-
ствие этого Б. обладают высокой хими-
ческой стабильностью и не осмоляются
при хранении. Б. прямой перегонки с
концом кипения 120—150° и содержа-
нием от 55 до 70% парафиновых угле-
водородов имеют ОЧ 50—65 по моторно-
му методу; Б., содержащие 50—70%
нафтеновых углеводородов — ОЧ 70—
78, а Б. с 60—65% изопарафиновых
углеводородов с метильными группами
в боковых цепях имеют ОЧ 75—80. Антидетонацион-
ные характеристики Б. прямой перегонки резко ухуд-
шаются при повышении темп-ры конца кипепия,
вследствие чего для авиабензинов строго ограничи-
вают верхний предел темп-ры выкипания фракции
97,5%. Природные нефтяные газы, содержа!цие фрак-
ции легких жидких углеводородов, являются источ-
ником получения газовых Б. Для освобождения от
этана и пропана, а также излишнего количества бу-
танов Б. стабилизируют ректификацией. Головную
фракцию стабилизированного Б. повторно фракцио-
нируют для извлечения максимального количества
бутанов, представляющих ценное нефтехимия, сырье
(см. табл. 3).
Антидетонационные свойства газовых Б. находят-
ся в зависимости от их химич. состава и, в первую
очередь, от процентного содержания изопарафиновых
углеводородов. Газовые Б. добавляют к Б. прямой
403
БЕНЗИНЫ —БЕНЗОИЛА ПЕРЕКИСЬ
404
перегонки или термического крекинга для облегче-
ния их фракционного состава и улучшения «пуско-
вых» свойств.
Таблица 3
Сырой Б., % Стабилизиро- ванный Б., % Дебутанизи- рованный Б., о/ /о
Этан 1,5
Нропзн l'i,7 — —
Изобутан 10,2 1.5 —
н-Бут ан 30,3 15,3 3,2
Изопентан 4,8 7.2 8,5
м-Пентан 15,0 21,0 24,5
Более высококипящие 23,5 55,0 63,8
Б. термич. крекинга содержат в среднем 15—25%
непредельных (олефины и диолефины) и 15—35%
ароматич. углеводородов; остальное приходится на
парафиновые и нафтеновые углеводороды; ОЧ этих
Б. в среднем (70—74). Б. каталитич. крекинга, химич.
состав к-рых связан с реакциями, протекающими в
присутствии катализатора (гл. обр. алюмосиликат-
ного), содержат высокий процент изопарафиновых
и ароматич. углеводородов и значительно меньше
по сравнению с Б. термич. крекинга, непредельных
углеводородов (см. табл. 4).
Таблица 4
Классы углеводородов Исходное сырье
газойль1, об. %1газойль2, об. %
Олефиновые 4 3
Ароматические 9 13
Нафтеновые . 7 25
Нормальные парафиновые . 17 12
Изопарафиновые 63 47
1 С преобладанием парафиновых углеводородов.
2 С преобладанием нафтеновых углеводородов.
Концентрирующиеся в головных фракциях этих Б.
изопарафиновые углеводороды обусловливают низ-
кую темп-ру выкипания 10%-ной фракции; хвостовые
же фракции, вследствие высокого содержания аро-
матич углеводородов, могут иметь более высокую
темп-ру выкипания без ухудшения аитидетонацион-
ных характеристик топлива. ОЧ Б. каталитич. кре-
кинга 84 и выше, химич. стабильность высокая. Они
являются высококачественным автомобильным топли-
вом и широко используются как основной компонент
авиабензинов,
Термич. или каталитич. преобразованием химич.
состава низкооктановых бензино-лигроиновых фрак-
ций продуктов прямой перегонки или термич. кре-
кинга получают Б. риформинга, характеризуемые
высокими антидетонационными свойствами. Термич.
риформингом различного сырья с ОЧ 38—54 получа-
ют Б. с ОЧ 80 и выше. Выход Б. составляет при этом
около 70% на загружаемое сырье Высокое содержа-
ние ароматич. соединений в Б. риформинга позволяет
получать на их основе высокооктановый компо-
нент для авиабензинов с высокой сортностью, марки
115/145. Гидроформингом из сырья с ОЧ ок. 34—50
получают Б., ОЧ к-рых составляют ок. 80 и выше.
Выход Б. на загружаемое сырье ок. 80%. Б. платфор-
минга, состоящие преим. из ароматич. и изопарафи-
новых углеводородов, также обладают высокими
ОЧ, ок. 86, пря ОЧ сырья 34—56; выход на загружае-
мое сырье 90%.
Высокооктановые компоненты Авиабензинов могут
быть получены: каталитич. полимеризацией узкой
бутан-бутеновой фракции и последующим гидриро-
ванием образовавшегося изооктена в изооктан; поли-
меризацией произвольной смеси олефиновых угле-
водородов из крекинг-газов получают т. н. полимер-
бензины с ОЧ ок. 86, являющиеся ценными компо-
нентами автобензинов; каталитич. алкилированием
бутан-бутеновой фракции газов, содержащей в необ-
ходимой пропорции алкилируемый бутеном изобутан,
получают алкилат, содержащий смесь изооктанов с
ОЧ ок. 96; каталитич. изомеризацией смеси легких
парафиновых углеводородов (низкооктановых бензи-
нов прямой перегонки с концом кипения ок. 70°) по-
лучают изобутан, применяемый для алкилирования,
а также изопентан, непосредственно вводимые в авиа-
бензины.
Гидрогенизацией деструктивной тяжелых нефтя-
ных остатков, а также кам.-уг. и буроугольных смол
могут быть получены различные моторные топлива,
в том числе Б. Выходы Б. (на органич. массу угля)
при переработке по различным схемам колеблются
примерно от 30 до 47%- ОЧ Б. (без ТЭС) от 60 до 70.
Сравнительно низкие ОЧ Б. и высокая их стоимость
ограничивают промышленное применение этого спо-
соба. Синтезом из окиси углерода и водорода может
быть получепа смесь жидких парафиновых и олефи-
новых углеводородов с прямой цепью, бензиновая
фракция к-рой — синтин — имеет низкую антидето-
национную характеристику (ОЧ 32—57), вследствие
высокого содержания неразветвленных парафиновых
углеводородов.
Получаемые из сернистых нефтей Б. содержат в
своем составе различные сернистые соединения, на-
личие которых снижает восприимчивость Б. к ТЭС.
Некоторые сернистые соединения, например H2S,
элементарная сера и низшие меркаптаны вызывают
коррозию металлов и присутствие их в Б. недопусти-
мо. Очистка Б. от нежелательных примесей является
одним из важных элементов их технологии. Необ-
ходимо удаление сернистых соединений, смолистых
веществ, органич. к-т и их солей и др. Очистка Б.
может производиться: серной к-той, щелочью, плюм-
битом натрия, гипохлоритом, действием водорода
под давлением (гидроочистка) и др., а также обработ-
кой адсорбентами, катализаторами, избирательными
растворителями. Б. газовые и прямой перегонки из
малосернистых нефтей очищаются от сероводорода
и меркаптанов щелочью. В случае высокосернистого
сырья применяют гидроочистку. Крекинг-Б. обессе-
ривают обработкой щелочью, после чего в них вводят
ингибиторы. Последнее время в США получили рас-
пространение процессы удаления из Б. нормальных
парафиновых углеводородов путем адсорбции на
высокоизбирательных адсорбентах — цеолитах («мо-
лекулярных ситах»). При этом поры адсорбента за-
полняются только молекулами углеводородов с пря-
мой цепью. Этот процесс позволяет значительно по-
высить ОЧ Б. прямой перегонки и термич. крекинга.
Лит.: Моторные топлива масла и жидкости, под ред.
К. К. Папок и Е. Г. Семенидо, 3 изд., т. 1, М., 1957; Технические
нормы на нефтепродукты, 16 изд., М., 1957; Технические
условия на нефтепродукты, М., 1960. В. А. Гетлинг.
БЕНЗОИЛА ПЕРЕКИСЬ (СвН,СОО)2, мол. в.
242,22 — бесцветные ромбические, бипирамидальные
кристаллы; т. пл. 106—108° (разлагается со вспыш-
кой); трудно растворима в воде. Растворяется
в 100 г органич. растворителя: 1,2 г в спирте,
18,5 г в ацетоне, 26,8 г в хлороформе, 16,7 г в
хлорбензоле, 14,4 г в этилацетате, 18,6 г в бензо-
ле, 8,6 г в этиловом эфире, 25,2 г в диоксане.
Б. п. и ее р-ры при нагревании часто взрывают;
теплота сгорания 6465,4 кал)г; Уф-спектры (в бен-
зине) Хмакс_ 231 ммк; Емакс. 39800, частота колеба-
ний перекисной группы 852 см1). Для очистки Б. п.
ее обычно осаждают при комнатной темп-ре из хлоро-
форменного р-ра метиловым спиртом. Чистая и сухая
Б. п. может сохраняться длительное время; щело-
405
БЕНЗОИЛАЦЁТОН - БЕНЗОИЛУКСУСНЫЕ ЭФИРЫ
406
чами разлагается медленно; очень медленно обесцве-
чивает КМпО4; при осторожном нагревании разла-
гается с образованием СО2, дифенила и небольших
количеств бензола и фенилбензоата. Свободные ради-
калы, образующиеся при разложении Б. п.
[(СвН5СОО)2—>СвН5СОО' + СвН5- + СО2], являются
инициаторами полимеризации многих ненасыщенных
соединений. При гидролизе Б. и. образуется бензойная
к-та и гидроперекись бензоила:
(CtH5COO)» + н,о — с„н5соон + С„Н5СОООН
Технич. способ получении Б. п. основан на взаимо-
действии хлористого бензоила с перекисью водорода
в щелочной среде: 2СвН5СОС1 + Н2О2 + 2NaOH —>
—> (CeH5COO)2 + 2NaCl + 2Н2О. Б. п. применяется
гл. обр. как катализатор (инициатор) реакций поли-
меризации в производстве многих технически важных
высокомолекулярных соединений (полистирола, по-
ливинилхлорида и др.). Под названием «луцидол»
Б. п. используется для дезодорации и обесцвечивания
сала, растительных масел, мыла. Качественной реак-
цией Б. п. нвлнетсн красное окрашивание от прибав-
лении капли формалина к р-ру Б. п. в конц. серной
к-те. Количественное определение Б. п. основано на
реакции ее с KJ и титровании выделившегося иода
тиосульфатом.
Лит.: Manly Т. D., Industr. Chemist., 1956, 32, № 377,
р. 271, № 378, р. 319; Но u ben — W е у 1, 4 Anil., Bd 8,
Stuttgart, 1952; Прилежаев Н., Органические перекиси
и применение их для окисления непредельных соединений,
Варшава, 1912, Л. К. Лыса-нчук.
БЕНЗОИЛАЦЕТОН (1-фенил-1, 3-бутандион)
С6Н5СОСН2СОСН3, мол. в. 162,19 — кристаллы с
сильным запахом; т. пл. 61°; т. кип. 261—2627760мм;
134—136/16 мм; 1,044. В твердом состоянии Б.
существует гл. обр. в виде енола, в расплавленном —
медленно переходит в дикетон, в паровой фазе цели-
ком превращается в енол; енольная форма Б. имеет
хелатную структуру:
СеН5ССН2ССн3^±:СеН5С=СнССН3 СКН5-С Н
О А ОН 6 н|
сн3
Б. растворим в спирте, эфире, конц. щелочах, жидком
SO2; растворы окрашены в желтый цвет.
Б. образует рнд производных с ионами металлов,
как Li, Na, К, Mg, к-рые также имеют внутрикомп-
лексную (хелатную) структуру, напр. с двухвалент-
ными металлами:
разует соли сульфокислоты. Получают действием
хлористого бензоила на водный раствор динатрие-
вой соли Аш-кислоты, содержащий избыток аце-
тата натрин; вступает в реакцию азосочетания с диа-
НО NH-СО-С0Н5
HO3S
SO3H
зосоединениями в щелочной или слабокислой среде,
причем образуются азокрасители. Технич. кислоту
выпускают в виде динатриевой соли, растворимой
в воде. Применяют в качестве исходного продукта
в произ-ве нек-рых азокрасителей.
БЕНЗОИЛУКСУСНЫЕ ЭФИРЫ - 0-кетоэфиры
общей формулы CeH6COCH2COOAlk, где Aik— алкиль-
ный радикал, обычно СН3 или С2Н5. Как все 0-ке-
тоэфиры, Б. э. склонны к кето-енольной таутомерии,
весьма реакционноспособные соединения; при кислот-
ном расщеплении образуют смесь кислот, при кетон-
ном расщеплении — жирноароматич. кетоны. Во-
дород метиленовой группы Б. э. очень подвижен,
легко замещается металлами, благодаря чему их можно
легко алкилировать и ацилировать. Конденсируясь
с альдегидами. Б. э. образуют либо алкилиденовые
(арилиденовые) производные, либо производные эфира
а,а'-диароилглутаровой к-ты (в приведенных ниже
формулах Аг — арильный радикал):
ArCOC(=CHR)COOAlk
/ ArCOCHCOO Aik
ArCOCH.COOAlk + RCHOC
CHR
ArCOCHCOOAlk
При нагревании с ароматич. аминами в инертном ра-
створителе (ксилол, хлорбензол) Б. э. дают арилиды,
широко применяющиесн в качестве нзосоставлнющих
в произ-ве азокрасителей и как желтые цветообразую-
щие компоненты в произ-ве цветных кипофотомате-
риалов:
ArCOCH2COOAlk + Ar'NH-. —
- AlCOCHoCONHAr' 4. AlkOH
При взаимодействии с арилгидразинами сперва об-
разуются арилгидразоны Б. э,, которые дальше цик-
лизуются в 1-арил-3-арилпиразолоны~5, имеющие
большое значение в произ-ве цветных кияофотома-
териалов в качестве пурпурных цветообразующих
компонент;
О О
С6Н5-С' Ме2^ "С-С6Н5
НС /О’ О. .сн
"9 9
сн3 сн3
Ar—С----СН2
II I
N COOAIk
NH
I
Ar'
Б. получают конденсацией ацетофенона с этила-
цетатом в присутствии C2H5ONa: С6Н5СОСН3 +
4- СН3СООС2Н5—>-СвН5СОСН2СОСН3 + С2Н5ОН или
этилбензоата и ацетона: СвН5СООС2Н5 + СН3СОСН3—>
—> СвН5СОСН2СОСН3 -г С2Н5ОН в присутствии
амида Na и другими способами. С FeCl3 Б. дает крас-
ную окраску. С. а-метилфенилгидразином в эфире
при 20° Б. превращается в его монометилфенилгидра-
зон, т. пл. 78—79°. Б. применяют в парфюмерии.
И. В. Матвеева.
БЕНЗОИЛ-Аш-КИСЛОТА C17H6O8NS2,
425,45 — белые кристаллы; в воде мало
растворима, нерастворима в обычных орга-
нических растворителях; в водных р-рих
едких щелочей, соды, аммиака и т. п. об-
мол. в.
Аналогичная реакция с гидроксиламином приводит
к образованию изооксазолонов:
Ar-CO-CH2 Аг —С-СН,
+ 1 -* II I
NH2 COOAIk n С=О
он ЧЪ/
При взаимодействии Б. э. с о-фенилендиамином или
его замещенными образуются кетоны, производные
бензимидазола:
NH2 Оч ZVn4
+ С-СН2СОАг R-L Г С~СН,СОАг
NH2 AlkOz vAm'
407
БЕНЗОИЛХЛОРИД — БЕНЗОИНОВАЯ КОНДЕНСАЦИЯ
408
С^О аГ
ХН
руемое цианид-ионом:
о о он
// сн- # /
2RC - НRC - CH-R
Б. к. характерна для ароматич. и гетероциклич. аль-
дегидов (бензальдегид, 3,4-метилендиоксибензаль-
дегид, 2- и 4-метоксибен.зальдегиды, фурфурол, пи-
ридин- и хинолин-альдегиды и пр.). При Б. к. тереф-
талевого альдегида образуется полимер, содержащий
бензоиновые группировки:
СОСНОН
СОСНОН
Многие ароматич. альдегиды, с трудом или вовсе не-
образующие симметричных бензоинов, легко конден-
сируются с другими альдегидами, образуя несиммет-
ричные бензоины, причем карбонильная группа в по-
следних всегда оказывается у ядра, несущего элект-
роно-донорные заместители:
о о
СвН5(^ Н+п-(СНз)21ЧСвН4сС н —Д
он о
/ //
—CjjHgCH — С — CeH4N (CHj)2-w»
Б.
к. обратима; несимметрич. бензоины могут быть
получены так же как и симметрич. — обменной реак-
цией бензоина с другим альдегидом или двух различ-
ных бензоинов:
Получают Б. э. взаимодействием галогенангидридов
ароматич. к-т с натрий- или этоксимагнийпроизвод-
ным ацетоуксусного эфира с последующим отщепле-
нием ацетильной группы от промежуточно получаю-
щегося ароилацетоуксусного эфира:
АгСОХ + CHjCOCHNaCOOAIk СН3СОСН (COAr) COOAIk —
— ArCOCH«COOAIk + СН3СООН.
Другой способ получения Б. э. — реакция сложно-
эфирной конденсации эфира ароматической кисло-
ты с этилацетатом
(или метилацета-
том), протекающ ая
в присутствии ще-
лочных конденси-
рующих агентов
(металлич. натрий, алкоголяты натрия, амид натрия):
АгСООС2Н5 + СН3СООС2Н5 — АгСОСН2СООС2Н5 -I- С2Н5ОН.
Благодаря большой реакционной способности Б. э.
широко применяются в препаративной органич.
химии, например для получения жирно-ароматич. ке-
тонов. И. с. Вульфсон.
БЕНЗОИЛХЛОРИД (хлористый бензоил, хлор-
ангидрид бензойной к-ты) СвН6СОС1, мол. в. 140,57—
бесцветная жидкость, дымящая на воздухе; т. пл.
—1°; т. кип. 197,2°; 1,2122; n'g 1,5537; т.воспл. 88°.
В парообразном состоянии Б. является лакримато-
ром; быстро гидролизуется кипящей водой и щело-
чами; холодной водой гидролизуется медленно; ра-
створяется в эфире, CeH6, CS2. Б. реагирует с соеди-
нениями, содержащими подвижной водород (спир-
тами, аминами), замещая его на бензоильную груп-
пу, например: CeH5NH2 + С5Н5СОС1—>
-> CeH5NHCOCeH5 + НС1 (см. Шотте- СеН5СОСНОНСеН5+2СН3ОС6Н4СОН^- 2 C6HsCHOHCOCfiH4OCH3:
на—Баумана реакция). Водородом В.вос-
станавливается в присутствии коллоид-
ного Pd до бензальдегида: СвН5СОС1 —> _ н mrunu
-> CeH5CHO + НС1. В лабораторных
условиях В. получают взаимодействием
бензойной к-ты с РС15, РС13 или SO2C12. В про-
мышленности В. производят из бензотрихлорида:
частичным гидролизом (катализаторы FeCl3, А1С13,
ZnCl2 и др.): СвН5СС13 + Н2О -> СвН5СОС1 + 2НС1;
нагреванием с бензойной к-той при 120—180°:
С6Н5СС13 + CeH5COOH -> 2СвН5СОС1 + НС1. Пред-
ложено также получать В. нагреванием смеси фос-
гена и бензойной кислоты: СОС12 + СвН5СООН —>-
—> СвН5СОС1 + СО2 -|- НС1. В. широко применяется
в пром-сти как бензоилирующее средство, напр. при
синтезе индигоидных красителей, для произ-ва бен-
зойной кислоты ангидрида, бензоила перекиси и др.
М. Ф. Турчинский.
БЕНЗОИН (фенил-а-оксибензилкетон), мол. в.
212,25, СвН5СОСН(ОН)СвН5 — белые или бледно-
желтые призмы без запаха; т. пл. 133—137°; т. кип.
343—3447768 мм, 194712 мм\ растворимость в 100 г
растворителя: в воде 0,03 г (25°); в пиридине 20 г;
плохо растворим в эфире; при нагревании В. раст-
воряется в спирте и ацетоне. При восстановлении
В. образуются два оптически недеятельных стерео-
изомерных гликоля: гидробензоин, т. пл. 139°, и изо-
гидробензоин, т. пл. 121°; при окислении получается
а-Дикетон-дибензоил (бензил). Спиртовой р-р В. при
встряхивании с КОН дает пурпурно-красное окраши-
вание; реактив Фелинга восстанавливается В. уже
при обычной тем-ре. В. получают конденсацией двух
молекул бензойного альдегида в присутствии KCN
(см. Бензоиновая конденсация). Применяют Б. в орга-
нич. синтезе, для флуоресцентного определения Zn,
как эталон в калориметрии и др. и. и. Ефимов.
БЕНЗОИНОВАЯ КОНДЕНСАЦИЯ — взаимо-
действие двух молекул альдегида с образованием
а-оксикетона (ацилоина), специфически катализи-
2 С6Н5СНОНСО-<О.
& чо>-соснон
Как симметрия., так и несимметрич. бензоины обычно
получают, нагревая водноспиртовые р-ры соответ-
ствующих реагентов с цианистым калием или нат-
рием. Из неароматич. альдегидов Б. к. успешно про-
текает только с фенил- и метилглиоксалями и триме-
тилуксусным альдегидом:
CN-
2RCOCOH -— RCOCOCHOHCOR;
CN-
СвН5СОН + (СНз)зССОН—-
— С«Н5СОСНОНС (СНз)з + СвН5СНОНСОС (СН3)3.
Скорость Б. к. (г?) подчиняется уравнению:
[СвН5СНО]3
V [CN-]
Реакция, соответствующая этому кинетич. ур-нию,
передается схемой:
о
//
AtC—H-f-CN-
он о
о он
АгС-Н^АгсГ
CN CN
он о-
CN Н
о
он
С— Ar + CN-
I
н н
продукт присоединения цианид-
АгС -|- АгС 22 АгС — С — Аг
'cN 'll
О- ОН
I
АгС -
I
CN
согласно к-рой,
иона к альдегиду после прототропного превращения
вступает в альдольную конденсацию со следующей
молекулой альдегида. Симметрич. и несимметрич.
бензоины широко используются в качестве промежу-
точных веществ для синтеза таких соединений, как
409
а-БЕНЗОИНОКСИМ — БЕНЗОЛ
410
\ \-C-CH—/\
\=/ Il I
НО—N ОН
дезоксибензоины, бензилы, гидробензоины, стильбены,
диарилэтиламины, оксиамины, изохиполины и др.
Лит.: Айд В., Б а к И. С., в кн.: Органические реак-
ции. Ред. Р. Адамс [и др.], [пер. с англ.], сб.4,М., 1951,с.229;
Roberts Т. G., Teagne Р. С., J. Amer. Chem. Soc.,
1955, 77, № 23, р. 6258. Н. П. Гамбарян.
а-БЕНЗОИНОКСИМ (купрон) C14H13O2N, мол. в.
277,26 — белый кристаллич. порошок, постепенно
темнеющий на свету; т. пл.
149—152°; мало растворим в
воде, растворяется в спирте
и р-рах аммиака. Получают Б.
действием солянокислого гид-
роксиламина на спиртовой р-р бензоина. Применяют
J5. для открытия и определения Си, Mo, W. Соли меди
дают зеленый осадок, нерастворимый в разбавленном
аммиаке, спирте и тартратных (виннокислых) р-рах,
но растворимый в минеральных к-тах и конц. аммиаке.
Молибдат- и вольфрамат-ионы осаждаются из сильно-
кислых р-ров. Реактив взаимодействует с ионами
металлов в анти-(а)-форме (см. Диметилглиоксим).
И. П. Ефимов.
БЕНЗОЙНАЯ КИСЛОТА СвН5СООН, мол. в.
122,05 — белые кристаллы (листочки или иглы);
т. пл. 122,37°; т. кип. 249,27760 мм, 133°/10 мм;
возгоняется при 100°, летуча с водяным паром;
1,2659; п// 1,53974; растворимость (в 100 г ра-
створителя): в воде 0,29 г (20°); в абс. спирте 58,4 г
(25°); в эфире 40,8 г (25°); в бензоле 12,17 г (25°);
теплота сгорания 774,1 ккал/моль (v = const),
771,7 ккал/моль (p=const.); константа диссоциации
К = 6,339 • 10~5 (25°); т. воспл. 121—131°. Б. к. при
нагревании до 370° (в стекле) разлагается с образо-
ванием бензола и СО2 (основные продукты), а также
фенола и СО (2—8%). Б. к. и ее соли обладают зна-
чительной бактерицидной и бактериостатич. актив-
ностью, резко возрастающей с уменьшением pH среды.
Б. к. получают окислением толуола азотной или хро-
мовой к-той, а также кислородом воздуха (в жидкой
фазе); декарбоксилированием фталевой к-ты и др.
способами. Бензоат натрия используется в пром-сти
для консервации пищевых продуктов. Бензоаты
лития, магния и кальция применяются как противо-
подагрич. и противоревматич. средства.
Замещенные Б. к. синтезируют: 1) введением за-
местителя в молекулу Б. к. (л«-нитро- и л«-сульфобен-
зойные к-ты); 2) окислением замещенных толуолов
(о- и n-изомеры нитро-,сульфо- и хлорбензойных к-т);
3) превращением имеющегося заместителя (п-амино-
и л«-хлорбензойные к-ты); 4) введением карбоксильной
группы в окси- и другие соединения. В табл, приве-
дены свойства нек-рых производных Б. к.:
Производное Т. пл., °C Т. кип., °C 4 20 nD
Ангидрид' (СвН5СО)2О 42 360 1,999 1,5767а
Хлор ангидрид СвН5СОС1 -1 197,2 1,2122 1,5537
Нитрил CeH5CN -13,8 191,1 1,0095а 1,53056а
Амид CeH3CONH3 132,5—133,5 290 — —
Анилид CeH5CONHCeH5 163 117°/10 мм 1,305 —
Перекись (СвН5СОО)2О 103,5 — — —
Метилбензоат® СбН5СООСН3 -12,4Г 199,5 1,093а 1,516
Этилбензоат® С0Н5СООС2Н5 — 211,7 1,051а 1,5029а
Фенилбензоат в СбН5СООСбН4С2Н5 — — 1,015а 1,5590а
а При 15°, 6 Своеобразный бальзамич. запах. в Запах
цветов. г Устойчивая форма;—13,9° С неустойчивая форма.
Производные Б. к. широко применяют в синтезе
красителей различных классов (напр., хлор- или
нитробензойные к-ты), фармацевтич. препаратов, как
инициаторы полимеризации (перекись бензоила), в пи-
щевой пром-сти (сахарин). Метиловый и этиловый
эфиры Б. к. обладают сильным запахом, они являются
составными частями эфирных масел; иланг-иланго-
вого, туберозы и гвоздики. Сложные эфиры Б. к.
от метилового до амилового применяются в компози-
циях для духов и одеколонов. Бензиловый и корич-
ный эфиры применяются в качестве фиксаторов
запаха и растворителей нек-рых душистых в-в. Полу-
чают сложные эфиры Б. к. обычными методами
этерификации; бензилбензоат получают переэтерифи-
кацией этилбензоата и др. способами.
См. также Бензойной кислоты ангидрид, Бензоила
перекись. Антраниловая кислота, Салициловая кис-
лота.
Лит.: Kirk, V. 2, N. Y., 1948, р. 459—476; U 1 Im а ни,
3 Aufl., Bd 4, Munchen—В., 1953, S. 272—292.
А. Ф. Бочков, А. А. Зеленецкая.
БЕНЗОЙНОЙ КИСЛОТЫ АНГИДРИД (бензой-
ный ангидрид) (СвН5СО)2О, мол. в. 226,22 — бес-
цветные кристаллы, ромбич. призмы; т. пл. 42°;
т. кип. 360°; 1,999; п'^ 1,5767; растворяется в
спирте и эфире, очень плохо в воде (0,009 г/л при
25^); гидролизуется при длительном кипячении с во-
дой, быстрее в присутствии щелочей. При пропу-
скании паров над СаСО3 при 550° Б. к. а. образует
бензол, бензойную к-ту, бензофенон и антрахинон;
окисляется ВаО2 с образованием перекиси бензоила.
С соединениями, содержащими активный водород
(спиртами, аминами), Б. к. а. реагирует, замещая
его на бензоильную группу: R—Н + (СвН5СО)2О —>
—> R—СОСвН5 -)- СвН5СООН. Б. к. а. получают
при нагревании в вакууме эквимолекулярных коли-
честв бензойной к-ты и бензоилхлорида (210—230°);
СеН5СООН + С,Н6СОС1 —-3 (С6Н5СО)2О и др. спо-
собами; применяется в качестве бензоилирующего
средства. Раствор Б. к. а. в CS2 дает с НС1О4 красно-
коричневое окрашивание. м. Ф. Турчинский.
БЕНЗОЛ СвНв — простейший представитель аро-
матических углеводородов. Б. впервые был выделен
в 1825 М. Фарадеем из жидкого конденсата светиль-
ного газа. В 1865 А. Кекуле предложил для Б. фор-
мулу 6-членного цикла (I) с чередующимися двой-
ными и простыми связями, соответствующую цикло-
гексатриену. Ф-ла Кекуле допускала изомерию орто-
дизамещенных (заместители у С-атомов с простой
или двойной связью), к-рая, однако, никогда не
наблюдалась. Поэтому Кекуле ввел в 1892 дополни-
тельную гипотезу, согласно к-рой в Б. происходит
постоянная осцилляция связей (ф-лы I и II). Фор-
17//
мула Б., предложенная Кекуле, находится в проти-
воречии с тем, что расстояния между всеми атомами G
в цикле одинаковы и равны 1,40А, тогда как расстоя-
ние между атомами С, связанными двойной связью,
должно быть 1,32 А и простой связью — 1,54 А.
Помимо ф-лы Кекуле, предлагались и другие: диаго-
нальная ф-ла Клауса (III), призматич. — Ладен-
бурга (IV), хиноидная — Дьюара (V), ф-ла Тиле
411
БЕНЗОЛ
412
(VI) — с сопряженными связями (см. Парциальных
валентностей теория). Все эти ф-лы обладают суще-
ственными недостатками. Так, ф-лы (V), как и (I),
допускают существование двух замещенных орто-
изомеров, модель Клауса не отвечает числу возмож-
ных ди- и тризамещенных Б., ф-ла (IV) противоречит
вполне установленному плоскому строению Б.; ф-ла
Тиле, во многом удовлетворительная, не позволяет
объяснить резкую разницу в свойствах Б. и цикло-
октатетраена. Установлено, что молекула Б. имеет
ось симметрии 6-го порядка и является совершенно
симметричной и плоской, что дипольный момент Б.
равен нулю, все С—С связи в Б. равноценны. Для Б.
характерно наличие секстета л-электронов, образую-
щих устойчивую единую замкнутую электронную
систему. Теплота сгорания газообразного Б. равна
789 ккал, тогда как вычисленная для гипотетич.
циклогексатриена равна 728,6 ккал. Разница в
39,6 ккал указывает на значительно большую проч-
ность полностью сопряженной ароматич. системы Б.
по сравнению с циклогексатриеном.
Несмотря на то, что строение Б. можно считать
установленным, до сих пор нет графич. изображения
ф-лы, полностью отражающей строение Б. Так,
английская химич. школа (Робинсон, Пнгольд и др.)
пользуется ф-лой с изогнутыми стрелками (VII), в осо-
бенности при наличии заместителей в бензольном
кольце. Полинг предлагает описывать Б. и ему
подобные системы набором ф-л. Для обозначения
систем с вполне сопряженными кратными связями,
в том числе и для Б., иногда прибегают к пунктир-
ному кругу внутри кольца (VIII). Обычно же по
мотивам историч. характера применяют ф-лу Кекуле,
что отнюдь не означает допущения локализованных
двойных связей в Б.
Б. — подвижная бесцветная жидкость своеобраз-
ного запаха; т. пл. 5,533°; теплота плавления
2,351 ккал/молъ; т. кип. 80,100°/760 мм; т. кип. в пре-
делах 10—1500 мм рт. cm.: t = [1211,033: (6,90665—
—1g/?)]— 220,790; теплота исп. 8,090 ккал/молъ (25°);
7,353 ккал/молъ (80,100°); п$ 1,50112; nty 1,49478;
п£" 1,49643; пр 1,51313; d™ 0,8895 (10°); 0,8790 (20°);
0,8685 (30°); 0,8134 (80°); плотность паров по воздуху
2,77; Z,i!)I1T 289,41°; Л(рит. 48,6 ат; <7Крит. 0,307;
°ь-рит 0,260 л/молъ; давление паров (мм рт. ст.):
26,6 (0°), 74,8 (20°), 268,6 (50°), 1335 (100°); теплоем-
кость 0,41441 кал/г (21,80°), 0,44422 ккал/г (63,73°);
теплота сгорания жидкого Б. 749,42 ккал/молъ.
Вязкость при 20°: абс. 0,6487 спуаз, кинематич.
0,7379 сст; поверхностное натяжение 28,88 дин/см
(20°); диэлектрич. проницаемость 2,284 (20°); т. воспл.
10,7°, взрывоопасные концентрации с воздухом
1,5—8 об. %.
Чистота Б. определяется по темп-ре замерзания
образца Т и рассчитывается по ур-пию:
1g р = 2,00000 — (7’* — Т) + 0,0032(7* — 7)],
где р — степень чистоты образца в %, 7'* — т. пл.
чистого образца.
Б. смешивается во всех отношениях с неполярными
растворителями: углеводородами (бензин, керосин,
смазочные масла, скипидар), эфирами; хуже раство-
рим в метиловом спирте; в 100 г воды при 25° раство-
ряется 0,180 г Б., нерастворим в этиленгликоле, гли-
церине; растворяет жиры, каучук, смолы (гудрон)
и др. неполярные органич. вещества, а также серу,
фосфор, иод; в 100 г Б. при 26° растворяется 0,054 г
воды. Б. дает с водой азеотропную смесь (91,17 вес.
% Б.), т. кип. 69,25°; образует двойные и тройные
азеотропные смеси со многими соединениями.
Б. легко реагирует с электрофильными реагентами,
напр. с галогенами, HNO3, H2SO4 с образованием
продуктов замещения, например CeH5Cl, CeH5NO2,
CeH5SO3H и др. При наличии одного заместителя
в зависимости от его природы дальнейшее электро-
фильное замещение может направляться преим.
в орто- и пара-положения или же в -мета-положение
(см. Ориентации правило). К заместителям 1-го рода,
ориентирующим дальнейшее замещение в орто- и
пара-положения, относятся Cl, Br, J, СН3, ОН, SH
и др.; к л«ета-ориентирующим заместителям относятся
NO2, СНО, NH4, CF3, COR, СООН, CN, SO3H и др.
Для Б. характерна устойчивость ядра, в частности
к действию окислителей; так, Б. не изменяется при
действии хромовой к-ты, КМпО4 и ряда др. окис-
лителей. При высокой темп-ре над V2O5 Б. может быть
окислен кислородом воздуха с образованием мале-
иновой к-ты. В определенных условиях активиро-
вания В. может присоединить различные реагенты.
Так, при возбуждении молекул Б. (прямое солнеч-
ное освещение, УФ-облучение), к нему присоединя-
ются 3 молекулы хлора с образованием гексахлорци-
клогексана. В присутствии катализаторов (никель,
сернистый молибден и др.) В. гладко присоединяет
водород и превращается в циклогексан. Последний
процесс имеет важное промышленное значение. Эти
реакции могут служить примерами перехода от Б.
к алициклич. соединениям. Образование Б. при на-
гревании ацетилена до ~900° или в более мягких
условиях (650°) над активным углем является приме-
ром перехода от соединений жирного ряда к аромати-
ческим. Большое значение имеют реакции алкили-
рования Б. (см. Фриделя — Крафтса реакция). Так,
действием этилена или пропилена на Б. в присутствии
А1С13 получают соответственно этил- и изопропилбен-
зол, представляющие собой полупродукты произ-ва
стирола и фенола.
При встряхивании растворов Б. с аммиачным
раствором Ni(CN)2 выделяется осадок комплексного
соединения Ni(CN)2(NH3)(CeHe). Эта реакция специ-
фична для Б. и позволяет обнаружить Б. в присут-
ствии. его гомологов.
Б. содержится в коксовом газе и частично в кок-
совой смоле, а также в нек-рых нефтях, напр. май-
копских и восточных районов СССР и др. Из коксо-
вого газа Б. извлекают растворителями (высококи-
пящими фракциями каменноугольной смолы или
нефти) после удаления смолы и аммиака. Перегонкой
в ректификац. колоннах растворителя, насыщенного
Б., получают сырой Б., который очищают обработкой
серной к-той и щелочью и многократной перегонкой
в ректификационных колоннах. Из коксового газа
Б. улавливают также адсорбцией на активном угле
или др. сорбентах и далее выделяют из адсорбента
перегонкой с водяным паром. Значительные коли-
чества Б. получают также каталитической циклиза-
цией алифатич. углеводородов нефти (см. Коксохими-
ческое производство. Ароматизация нефтепродуктов).
Б. является важнейшим продуктом химич. пром-сти.
Он применяется для получения многих полупродуктов
в произ-ве красителей, фармацевтич. препаратов
и т. д.; хлорированием Б. получают хлорбензол и
гексахлорбензол, сульфированием — бензолсулъфокис-
лоту, нитрованием — нитробензол, гидрированием —
циклогексан, окислением — малеиновый ангидрид, ал-
килированием — этилбензол и изопропилбензол, де-
гидрированием — дифенил. Б. применяют как ком-
понент моторного топлива для повышения октанового
числа; его используют как растворитель и экстраги-
рующее средство в производстве лаков, красок и др.
Б. физиологически весьма активен. В больших
концентрациях паров он действует на центральную
нервную систему. Малые концентрации паров при
многократном воздействии вызывают изменение со-
413
БЕНЗОЛИН — БЕНЗОЛСУЛЬФИНОВАЯ КИСЛОТА
414
— СН2
с6н5сн2-с( I 2 -но
хмч—сн2
става крови и нарушают нормальную функцию кро-
ветворных органов. Хроническое отравление нарами
Б. может вызвать смерть. Жидкий Б. сильно раздра-
жает КОЖу. Я. Ф. Комиссаров.
Лит. см. прист. Ароматические углеводороды.
БЕНЗОЛИН (прискол) C1OH13N2C1, мол. в. 196,69 —
бесцветные гигроскопич. кристаллы горького вкуса;
т. пл. 172—174°, легко
растворим в воде, труд-
но— в ацетоне, нераство-
рим в эфире. Б. получа-
ют в з а имодейст в нем хлор -
гидрата фенилацетиминоэтилового эфира с этилен-
диамином; применяется при мигрени, гипертонии,
расстройствах, ревматизме, нарушениях периферии,
кровообращения И др. А. И. Травин.
БЕНЗОЛПОЛИКАРБОНОВЫЕ КИСЛОТЫ —
кислоты ароматич. ряда, в к-рых два или более
атомов водорода бензольного ядра замещены на кар-
боксильные группы. КБ. к. относятся фталевые
кислоты СвН4(СООН)2, бензолтрикарбоновые кисло-
ты СвН3(СООН)3, бензолтетракарбоновые кисло-
ты СвН2(СООН)4, бензолпентакарбоновая кислота
СвН(СООН)5 и меллитовая к-та Св(СООН)в. Ниже рас-
сматриваются только трех- и более основные Б. к.;
бензолдикарбоновые к-ты — см. Фталевые кислоты’ в
таблицах данные о них приведены для сравнения.
Таблица 1. Физические свойства бензолполикарбоиовых
кислот
Кислоты Т. пл., °C Теплота сгорания, ккал/моль
о — const. р — const.
Фталевые: С0Н4(СООН)3, мол. в. 166,13 Фталевая (о-фталевая) 191* 779,3 771,6
Изофталевая (л«-фталевая) . . . 348,5 769Д 768,8
Терефталевая (п-фталевая) . . 300*’ 771,2 770,9
Бензолтрикарбоно- вые: СбН3 (СООН)3, мол. в. 210,4 Гемимеллитовая (1,2,3-бензол- трикарбоновая)
Тримеллитовая (1, 2, 4-бензол- трикарбоновая) 224—225
Тримезиновая (1, 3, 5-бензол- трикарбоновая) 375—380 768,5 767,6
Бензолтетракарбо- новые: СсНа(СООН)4, мол. в. 254,15 Меллофановая (1,2 ,3,4-бензол- тетракарбоновая) 240
Пренитовая (1,2,3,5-бензолтри- карбоновая) 253
Пиромеллитовая (1,2,4,5-бензол- тетракарбоновая) 275 778,9 777,4
Бензолпентакарбо- новая СбН(СООН)5, мол. в. 298,16 Бензолгексакарбо- новая (меллитовая) 238 288 700,8 788,2
Св (СООН)в, мол. в. 342,17
• Ангидризуется. ** Возгоняется.
Таблица 2. Константы диссоциации некоторых бензолполикарбоновых кислот
Кислота к, к2 Кз К. кв
Изофталевая 3,5 • 10-4 3,5 • Ю-з — — — —
Фталевая 1,05 • 10-3 5,2 • 10-е — —- — —.
Тримезиновая 7,5 • 10—* 1,28 • 10 * 2,05 • Ю-з — — —
Тримеллитовая .... 3,0 • 10-3 1,45 • IO”-* 6,3 • 10-е — — —
Гемимеллитовая .... 1,6 • 10-2 6,3 • 10-3 1,35 • Ю-з — — —
Пиромеллитовая . . . 1,2 • 10-2 1,34 • Ю-з 3,2 • 10-5 2,35 • Ю-з — —
Пренитовая 4,2 . Ю-з 3,1 • 10"» 3,6 • ю-s 1,55 • 10-е — —
Бензолпентакарбоно- вая 1,6 • 10-2 1,85 • 10-3 1,08 • 10-е 5,6 • 10-е 3,5 . 10~7
Меллитовая 4/J • 10-2. 6,4 • Ю-з 1,65 • 10—5 1,2- 10-6 1,27 • 10-е 1,1 • IO”’
С увеличением основности Б. к. растворимость их
в воде увеличивается. Б. к. сравнительно хорошо
растворяются в спирте, умеренно — в эфире.
Соли Б. к. и щелочных металлов и аммония легко
растворимы в воде; соли прочих металлов нераство-
римы или трудно растворимы. Б. к. могут быть пол-
ностью этерифицированы; метиловые эфиры их при
обработке LiAlH4 образуют соответствующие окси-
метилбензолы. Б. к. с рядовым расположением кар-
боксильных групп при нагревании легко образуют
ангидриды;
СООН
СООН
I
Пиромеллитовая, бензолпентакарбоновая могут давать
диангидриды, напр.:
НООС
НООС
СООН
СООН
СООН
СООН
Меллитовая к-та при действии СН3СОС1 дает триан-
гидрид, при обработке SOC12 — диангидрид. При
нагревании Б. к. с избытком извести выделяется
СО2 и образуется бензол, напр.
С(СООН)0 -► С0Н0 + 6СО2
Меллитовая к-та с сухими нейтральными аммоние-
выми солями образует парамид (триимид меллитовый
к-ты); амальгамой натрия восстанавливается до гек-
сагидромеллитовоп к-ты. При сухой перегонке мел-
литовой к-ты отщепляются СО2 и вода и образуется
диангидрид пиромеллитовой к-ты.
Б. к. обычно получают окислением гомологов бен-
зола, напр. при окислении мезитилепа образуется
тримезиновая к-та:
СНа
СООН
СООН
Б. к. синтезируют и др. методами, напр. конден-
сация формилуксусного эфира приводит к тримези-
новой к-те; получают также окислением сложных
конденсированных ароматич. углеводородов, окисле-
нием различных типов углей азотной к-той под дав-
лением и др. методами. В залежах бурых углей встре-
чается алюминиевая соль меллитовой к-ты в виде
минерала, т. н. медового камня [Св(СОО)в]А12-18Н2О.
Меллитовая к-та была также выделена из спиртовой
фракции при экстрагировании асфальта. Практи-
ческое применение находят фталевые кислоты.
БЕНЗОЛСУЛЬФИНОВАЯ КИСЛОТА** *(фенил-
сульфиновая кислота) CeH5SO2H, мол. в. 142,17 —
белые призматические или
игольчатые кристаллы (из во-
ды); т. пл. 64,5—65°, т.'разл.
i>100°; растворима в горячей
воде, особенно хорошо в спирте,
эфире, ацетоне, р-рах щелочей,
умеренно растворима в холод-
ной воде. Добавление H2SO4,
НС1, НСЮ4 к конц. р-рам ее
растворимых солей вызывает
выпадение свободной кислоты;
константа диссоциации К =
=3,5-10 2. Сухие щелочные и
аммонийные соли Б. к. не раз-
415
БЕНЗОЛСУЛЬФОКИСЛОТА — 1,2,3-БЕНЗОТРИАЗОЛ
416
лагаются при комнатной температуре, их р-ры устой-
чивы около 2 месяцев. Свободная кислота при на-
гревании ее р-ров, особенно солянокислых, подвер-
гается окислительно-восстановительному диспропор-
ционированию; окисляется сильными окислителями
HNO3, HNO2, р2О2, К2Сг2О7, Ce(SO4)2, КМпО4 и
восстанавливается сильными восстановителями SnCl2,
TiCl2, U2(SO4)3; с конц. H2SO4 образует синее окраши-
вание. Б. к. получают восстановлением хлорангидрида
бензолсульфокислоты сульфитом натрия или диазо-
тированием анилина с последующим удалением азота
в присутствии Си и SO2. В слабокислом растворе
с Fe111 Б. к. образует малорастворимый оранжевый
осадок; белые осадки образуются с TiIV,Th,UIV,
РЬП, Bi111, Ag1', в2 н. р-реНС1 образует белыеосадки
с Zr, Hf и SnIV; применяют для отделения Zr и Th
от редкоземельных элементов и Fe11; Fe111 от Со,
Ni, Сгш; для весового определения Zr, Th и Fe111.
Натриевая соль применяется в фотопромышленности.
Лит.; Алимарин И. П., Аликберов С. С.,
Заводск. лаборатория, 1957, 23, № 6, с. 658; их же, там
же, 1958, 24, № 7, с. 804; Алимарин И. П., Кузне-
цов Д. И., Вести. МГУ. Сер. математики..., химии, 1959,
№ 3, с. 189. Д. И. Кузнецов.
БЕНЗОЛСУЛЬФОКИСЛОТА CeH5SO3H, мол. в.
158,18 — кристаллич. продукт, иглы (из водного
р-ра), расплывающиеся на воздухе; т. пл. CeH5SO3H •
• V2H2O 43—44°; CeH5SO3H • Н2О 45—46°; безвод-
ной Б. 171—172°; хорошо растворима в воде и спирте,
плохо — в бензоле, нерастворима в эфире и сероугле-
роде; константа диссоциации К — 2 • 10-1 (25°). Б.
при перегонке ее р-ров или солей с перегретым водя-
ным паром при 175° распадается на бензол и H2SO4.
При метилировании солей Б. образуются эфиры,
напр. CeH5SO3Na + (CH3)2SO4 1^=1^ CeH5SO3CH3+
+ CH3NaSO4, при нитровании — преим. о- и .и-иит-
робензолсульфокислоты. Технич. применение находит
Na-соль В.; последняя разлагается при 490°, хорошо
растворима в воде (г/100 г): 57,2(30°), 125 (100°);
щелочное плавление Na-соли применяют для полу-
чения фенола. При сплавлении К-соли Б. с KCN
образуется бензонитрил:
медленно, быстрее в присутствии щелочей с образо-
ванием алкилсульфонатов. Получают Б. непосред-
ственным действием избытка хлорсульфоновой к-ты на
бензол: CeHe + 2HOSO2C1 -> CeH5SO2CI + H2SO4 +
+ НС1 или действием на бензолсульфокислоту РОС13
или РС15, напр. CeH5SO3H + РС15->CeH5SO2Cl+
+ РОС13 + НС1. Применяется Б. для получения бензол-
сульфамида (см.выше); алькильных эфиров бензосуль-
фокислоты: CeH5SO2Cl + СН3ОН -> CeH5SO2OCH3 +
+ НС1, используемых как мягкие алкилирующие реа-
гейты. м. А. Марков.
БЕНЗОПУРПУРИН 4Б C34H26OeNeS2Na2, мол. в.
724,74 — коричневый порошок, растворимый в воде
с образованием коричнево-красного р-ра; Хмак0.
501,6 ммк. Водный р-р получают сочетанием диазоти-
рованного о-толидина с нафтионовой к-той. Б. исполь-
зуется как краситель хлопчатобумажных тканей,
но окраска не отличается прочностью; применяется
в качестве окислительно-восстановительного инди-
катора при броматометрич. определении Sb; как
pH — индикатор (0,5%-ный р-р при pH 1,3 дает фиоле-
товую окраску, при pH 4,0 — красную) для открытия
Mg, Al, Hg, Ag и U. и. п. Ефимов.
БЕНЗОТИОФЕН (тионафтен) CsHeS, мол. в. 134,19—
кристаллы, по запаху напоминающие нафталин;
т. пл. 32°, т. кип. 221—222°; 1,1486. 4
Б. содержится в кам.-уг. смоле и в п?
технич. нафталине. Синтетически Б. |1 I
получают восстановлением 3-оксибен-
зотиофена: 7 i
Zn
СН3СООН
Методы синтеза Б., как правило, заключаются в по-
строении тиофенового кольца при уже имеющемся
бензольном ядре, напр.:
СН(ОСН3)2 (р2О5)х- (Н2О)У
S/CH2
сн=сн2
+H2S
CeH5SO3K + KCN • CeH5CN + K2SO3
В пром-сти В. получают сульфированием бензола
серной к-той или олеумом в жидкой фазе или про-
пусканием паров бензола в
нагретую H2SO4; одновремен-
но образуется нек-рое коли-
чество бензолдисульфокислот.
Обычно свободную Б. не вы-
деляют, но непосредственно превращают в натриевую
или калиевую соли, напр. выливанием реакционной
массы в воду нейтрализацией мелом или гашеной
известью и, после отделения сульфата кальция, обра-
боткой содой до щелочной реакции. Применяется
Б. гл. обр. для произ-ва фенола (см. выше); ограни-
ченно применяется в качестве катализатора реакций
конденсации и полимеризации.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955.
М. А. Марков.
БЕНЗОЛСУЛЬФОХЛОРИД (хлорангидрид бен-
зосульфокислоты) CeH5SO2Cl, мол. в. 176,63— масло-
образная жидкость с резким запахом; т. пл. 14,5°;
т. кип. 251,5° (с разл.), 120710 мм; 1,3842; хо-
лодной водой Б. медленно гидролизуется, быстро —
при кипячении и особенно в присутствии к-т или ще-
лочей. С NH3 или (NH4)2CO3 Б. образует бензол-
сульфамид; CeH5SO2Cl + NH3 —>CeH5SO2NH2 + HCI;
c Na2S — соль тиосульфокислоты CeH5SO2SNa; с
бензолом в присутствии А1С13 получается дифенил-
сульфон (CeH5)2SO2; водородом (Zn + H2SO4) восста-
навливается до тиофенола. Б. реагирует со спиртами
Б. обладает «ароматическими» свойствами; менее ре-
акционноспособен, чем тиофен. Нитрование, бромиро-
вание, ацилирование, меркурирование, хлормети-
лирование приводят в основном к трехзамещенным
Б. Наибольшее значение имеют 3-оксибензотиофен
и его производные в положении 2 и 3, являющиеся
промежуточными продуктами в синтезе тиоиндигоид-
ных красителей.
Лит.: Гетероциклические соединения. [Сб. статей], пер.
с англ., т. 2, М., 1954, с. 112. Е. М. Рохлин^
1,2,3-БЕНЗОТРИАЗОЛ CeH5N3, мол.
кристаллы, белые иглы (из хлороформа);
98,5°; в воде не растворяется, раство-
рим в спирте и бензоле. Получают Б.
действием KNO2 в кислой среде на
о-фенилендиамин. Б. применяется при
определении Си, Ag, Zn; с Си дает осадок
(СвН4Н3)2Си, выделяющийся из тар-
тратно-ацетатного р-ра при pH 7,0—8,5; с Ag дает
белый осадок, состав которого после высушивания
при 110° соответствует CeH4N3Ag и может быть
использован для титриметрич. определения Ag (бен-
зотриазолат серебра растворим в р-рах цианида);
в. 119,13 —
т. пл. 96 —
N
а
N
н
417
БЕНЗОТРИХЛОРИД — БЕНЗОХИНОН
418
используется в р-рах для фотографии как антивуали- I
руЮщее средство. и. П. Ефимов. |
БЕНЗОТРИХЛОРИД (<о, <о, <о-три-
хлортолуол) СвН5СС13, мол. в. 195,48 — q
бесцветная или желтоватая маслообраз- | ЯГ
ная жидкость с характерным раздра- —Cl
жающим запахом; т. пл. —5°; т. кип.
220,97760 мм; 110,7723 мм; d™ 1,3723;
п'$ 1,5573; нерастворим в воде, хорошо растворим
в спирте, эфире, бензоле; образует азеотропные смеси
с 1,4-дибромбепзолом, нитробензолом, 2-нитротолу-
олом, метилсалицилатом. Б. реагирует с бензойной
к-той с образованием бензилхлорида:
С6Н5СС13 4- С0Н5СООН — 2С0Н5СОС1 + HCI
Б. омыляется водой при нагревании до 150°, а также
при действии кислот или щелочей, образуя бензойную
к-ту СвН5СООН; с серной к-той, содержащей 4,6%
воды, дает ангидрид бензойной к-ты. Нагревание Б.
с этилатом натрия ведет к образованию эфира орто-
бензойной к-ты СвН5С(ОС2Н5)3. При нагревании с
Си при 100°, а также при гидрировании в спирте в при-
сутствии коллоидного Pd образуется толантетра-
хлорид, а с Zn в эфире — толандихлорид. При хло-
рировании в присутствии FeCl3 или FeJ3 образуется
смесь хлорбензотрихлоридов, главным образом мета-
изомер; в присутствии иода — пара-изомер. С за-
мещенными анилинами Б. реагирует с образова-
нием красителей, напр., с диметиланилином обра-
зует малахитовый зеленый. В промышленности Б.
получают хлорированием толуола (подробнее см.
Бензилиденхлорид). Применяют Б. главным образом
для получения бензоилхлорида или бензойной к-ты.
БЕНЗОФЕНОН (дифенилкетон) С6Н5СОСвН5, мол.
вес 182,21 — известен в стабильной и лабильной
модификациях; стабильная — ромбич. призмы,
т. пл. 48°; т. кип. 305°; dfg 1,0976; растворим в органич.
растворителях; нерастворим в воде; лабильная —
моноклинные призмы, т. пл. 26°; при 45° превращается
в стабильную; УФ-спектр: — 252 ммк, 12 = 330 ммк;
ИК-спектр: полосы поглощения 1240—1330 см~1,
1430—1460 см"1, 1650—1680 см"1. Хлорирование
в присутствии Fe или FeCl3 приводит к пента- и гек-
сахлор-Б., применяющимся как трансформаторные
масла; присоединение к СО-группе сильно затруд-
нено по сравнению с алифатич. кетонами. При вос-
становлении Б. получается спирт бензпинакон
(СвН5)2СОН—СОН(СвН5)2, который, теряя элементы
воды, перегруппировывается в Р-бензпинаколин
СвН5СН(ОН)С(СвН5)3 или бензгидрол СвН5СН(ОН)СвН5.
Присоединение Na к Б. приводит к относительно
устойчивому свободному радикалу (CeH5)2CONa. Иден-
тифицируется Б. в виде производных, напр. фенил-
гидразона, т. пл. 138°. При конденсации бензола с
СС14 в присутствии А1С13 образуется бензофенонди-
хлорид (СвН5)2СС12, к-рый гидролизуется водой до
Б. (промышленный способ); может быть получен так-
же конденсацией бензола с СвН5СОС1 в присут-
ствии А1С13; пиролизом бензоата кальция и т. д. Бен-
зофенон применяется в парфюмерной промышленности;
4,4'-дигалоген- или 4,4'-диалкоксибензофенон ис-
пользуется в производстве светопрочных, кислот-
ных, а также дифенилметановых красителей.
А. Ф. Бочков,
БЕНЗОФУРАН (кумарон) С8НвО, мол. в. 118,13—
бесцветная ароматно пахнущая жид-
—। кость; т. пл. —18°; т. кип. 174°; d}|
fe L г1| 1,0776; ng'7 1,5645; нерастворим в воде,
Чх'Сг растворим в спирте и эфире. В сравне-
1 нии с фураном Б. более «ароматичен»
(см. Ароматические системы). Б. обнаруживает мно-
гие характерные черты реакций виниловых эфи-
ров. При хлорировании Б. в зависимости от условий
образуются:
При бромировании Б. получается неустойчивый
2,3-дибромкумарон, который может отщеплять 1 или
2 молекулы НВг. Б. непосредственно не иодируется.
Прямое нитрование В. (в отличие от фурана) дает
2-нитропроизводное; сульфируют Б. пиридинсуль-
^отриоксидом; при озонировании образуется смесь
енолов, фенолоксикислот, феполальдегидов:
Концентрированные кислоты полимеризуют Б.
в смолообразный паракумарон (см. Кумароновая
смола). В. содержится во фракции 165—175° кам.-уг.
смолы; выделяют бромированием и последующим
разложением дибромида; применяют также селектив-
ную экстракцию гликолями или азеотропную пере-
гонку с тяжелыми бензольными фракциями. Б.
можно получать синтетически; синтезы простых про-
изводных В. основаны на замыкании фуранового
цикла. Б. является исходным материалом для про-
изводства кумароно-инденовых смол. Различные про-
изводные В. встречаются в веществах, выделяемых
из растений.
Лит.: Гетероциклические соединения. [Сб. статей], пер.
с англ., т. 2, М., 1954, с. 5. Н. В. Гнучев.
БЕНЗОХИНОН СвН4О2, мол. в. 114,06 — из-
вестны два изомерных Б.: орто-Б. (I) и пара-В.(П).
о-В. существует в двух моди-
фикациях; нестабильная фор- О О
ма — бесцветные кристаллы, >—!, ‘ /=\
призмы, переходящие при стоя- / \ O=f V=o
нии в стабильную форму — \ // \=/
коричнево-красные пластинки ц
или призмы; т. разл. 60—70°; '
нелетуч; может быть получен окислением о-амино-
фенола; применяется для открытия первичных vic-
диаминов:
и для определения цистеина. п-Б. — золотисто-жел-
тые кристаллы, призмы; т. пл. 115,7°; легко возго-
няется, летуч с паром; 1,318; растворим в горячей
воде, спирте, эфире, горячем лигроине. n-В. легко
восстанавливается в гидрохинон (промышленный спо-
соб получения последнего); с гидрохиноном образует
молекулярное соединение хингидрон. С рядом соеди-
нений Б. дает продукты присоединения к группе
\С = О, напр.:
C2H5SH
Легко образующийся монооксим В. таутомерен п-нит-
розофенолу:
419
БЕНЗПИРЕН — БЕРБЕРИН
420
Б. присоединяет агенты, содержащие подвижный
атом Н, в положение 1,4, напр.:
Cl Cl
в реакцию Дильса—Алдера вступает как активный
диенофил. В пром-сти n-Б. получают окислением
анилина хромовой смесью. n-Б. применяют также
как окислитель и как дубящий агент. Качественно
определяется пветной реакцией с ацетоуксусным эфи-
ром в присутствии конц. H2SO4, количественно—иодо-
метрически. А. Ф. Бочков.
БЕНЗПИРЕН С20Н12, мол. в. 252,10 — конденси-
рованный ароматич. углеводород; известны 2 изо-
мера: 1,2-Б. и 4,5-Б. Оба изомера находятся в кам.-уг.,
сланцевой и др. смолах, откуда они могут быть вы-
делены; их можно также синтезировать, соответст-
венно, из пирена или симметричного гексагидропирена
и ангидрида янтарной к-ты. 1,2-Б. обладает канце-
рогенными свойствами (см. Канцерогенные вещества).
1,2-Б ензпирен — бледно-желтые кристаллы,
иглы или пластинки; т. пл. 179° (из спиртобензоль-
ной смеси); т. кип. 310—312°/10 мм; d;'1 1,351; очень
мало растворим в воде, растворяется в органиче-.
ских растворителях; пикрат, т. пл. 197—198°. В
спектре флуоресценции Б. имеет J полосы с мак-
симумами при 4180, 4300 и 4580 А. Атомы углеро-
да в углеродном скелете 1,2-Б. обычно нумеруют,
как показано на I.
В присутствии пиридина Б. присоединяет тетра-
хлорид осмия в положение 4,5, образуя комплекс,
к-рый при гидролизе дает 4,5-дигидродиол. При
дегидратации этого диола получается смесь 4- и 5-окси-
производных. В реакциях замещения обычно уча-
ствует водород в положении 3: при нитровании обра-
зуется 3-нитропроизводное, с хлористым сульфури-
лом — 3-хлорпроизводное и т. д. При ацетилирова-
нии образуется 8-ацетопроизводное. Хромовой кис-
лотой Б. окисляется в смесь 1,2-бензпирен-3,8-хинона
и 1,2-бензпирен-3,6-хинона. 4,5-Б ензпирен —
бесцветные призмы, т. пл. 178—179° (из бензола);
возгоняется при 250°/3—4 мм, нерастворим в воде,
растворяется в органич. растворителях; пикрат,
т. пл. 229—230°. Существует неск. способов нумера-
ции атомов углеродного скелета
4,5-Б.; более распространен — II.
Лит.: Chemistry of carbon compounds,
ed. by E. H. Rodd, v. 3, pt. B., Amst. la. o.],
1956, p. 1519. Л. С. Поваров.
БЕНЗТИАЗОЛ C7H5NS (I), мол.
в. 138,19 — жидкость, т. кип. 223—
225°, растворим в спирте, CS2, очень
плохо растворим в горячей воде,
летуч с паром; производные: хлор-
гидрат, т. пл. 192°; сульфат, т.
пл. 157—157,5°; пикрат, т. пл. 176°.
Бромирование Б. в газовой фазе дает
2-бром-Б. При действии брома на
Б. в инертном растворителе в обыч-
ных условиях образуется кристаллич.
пербромид, из к-рого можно реге-
нерировать Б. При нагревании В. с РС15 до 160°
образуется 2-хлор-Б. Длительное нагревание (25—
45 час.) последнего со
спиртовым р-ром НС1
приводит к 2-окси-Б.
2-хлор-Б. при нагре-
вании до 150—160°
со спиртовым рас- у ((
твором аммиака или
при действии жидкого NH3 превращается в 2-ами-
но-Б. Металлирование, напр. бутиллитием, также
приводит к замещенным Б. в положение 2. При дей-
ствии RJ на Б. образуются соли бензтиазолпя (II).
Б. получают из е-аминотиофенола и НСООН:
QNH2
SH
нсоон
NHCHO
SH
Нек-рые производные и высшие аналоги Б. явля-
ются красителями. Система Б. входит в состав тио-
цианиновых красителей; 2-меркапто-бензтиазол (кап-
такс) применяют в качестве ускорителя вулкани-
зации каучука.
Лит.: Heterocyclic compounds. Ed. R. C., Elderfield, v. 5,
N. Y., 1957. В. M. Демьянович.
БЕРБЕРИН C20H18O4N4+OH- — один из наиболее
распространенных алкалоидов семейства лютиковых
(Ranunculaceae), барбарисовых (Berberidaceae), лу-
носемянниковых (Menispermaceae) и др. Четвертичное
основание (I), в растениях находится в виде солей
(окрашенные кристаллы); т. пл. 145° (свободного
основания); т. пл. сое- ___
динения с СНС13 (1 моль) л |
179°. Свободное основа-
ние Б. существует только [j1 Г
в растворах, при уда-
лении растворителя оно йВ * * 11 ] ]\ [
переходит в альдегид- СН3О—
ную форму (II). Однако 1 7
существует мнение, что ОСН3 ।
основанию соответствует
не альдегидная, а карби-
нольная форма (III).
Основание растворимо в
воде (1 : 4,5 при 21°) и
спирте (1 : 100). Водные
р-ры Б. имеют горький СН3О
вкус, оптически неактив-
ны и нейтральны на
лакмус. Важнейшие соли
Б. — иодгидрат — трудно растворим в холодной
воде (1 : 2130), нитрат и сульфат — плохо раство-
римы в соответствующих к-тах. Водный р-р хлор-
гидрата Б. с хлорной водой дает красноватое окра-
шивание. При окислении Б. щелочным раствором
421
БЕРИЛЛИЙ
422
КМпО4 образуется ряд продуктов, яз к-рых важ-
нейшие — бербериловая к-та (IV), бербераль (V)
и оксиберберин. Один из синтезов Б. осуществлен
Перкином с сотр. (1927) конденсацией ангядряда
диметоксигомофталевой к-ты с |3-пиперонилэтилами-
ном. При действии на продукт конденсации РОС13
происходит замыкание двух циклов и образуется
оксиберберин, который путем электрохимического
восстановления может быть превращен в тетрагидро-
берберин и далее — действием сла-
Известен ряд алкалоидов, структурно близких
к Б., — кавадян (1-тетрагидроберберин), нандинин
(дес-о-метил-d-тетрагидроберберин), офиокарпин
(13-окситетрагидроберберин) и др. Б. мало ядовит
и легко выводится из организма; соли Б. применяют
внутрь для повышения аппетита, для лечения малярии
(вместе с хинином) и при кровотечениях.
Лит. см. при ст. Алкалоиды. А. Е. Васильев.
БЕРИЛЛИЙ (Beryllium) Бе — химич. элемент II гр.
периодич. системы Менделеева; п. н. 4, ат. в. 9,013;
стабильный изотоп Бе9. Из искусственно радиоактив-
ных изотопов наиболее важен Be7 (7). = 53 дня),
применяемый как радиоактивный индикатор. Элект-
ронная конфигурация атома Б. Is2 2s2; энергия иони-
зации (в эв): Ве° —> Бе —> Ве2+соответственно равна
9,32 и 18,21. Поперечное сечение поглощения тепло-
вых нейтронов 0,0090 барн на атом.
В. открыт Л. Вокленом в 1798 в минерале берилле;
название «Б.» принято во всех странах, кроме Фран-
ции, где сохранилось его старое наименование Glu-
cinium (знак G1). Металлич. В. получен в 1828 Ф. Вё-
лером и независимо от него А. Бюсси при восстанов-
лении хлористого Б. металлич. калием. Содержание
Б. в земной коре 6 10 4 вес. %. Б. является типич-
ным литофильным элементом. Его геохимич. поведе-
ние определяется ограниченной возможностью изо-
морфного захвата Б. в структурах главных минералов
магматич. пород, а также подвижностью его ацидо-
комплексНых соединений, что способствовало его
экстрагированию из магмы. В. накапливается в гра-
нитных пегматитах (0,04—0,08% ВеО в промышлен-
ных месторождениях). В природе известно ок. 40 ми-
нералов В. Главнейшим является берилл
Be3Al2[SieO18] (11 —14,3% ВеО): сингония гексаго-
нальная; цвет зеленоватый или голубоватый; твер-
дость 7,5—8, плотн. 2,63—2,91; крупные месторо-
ждения находятся в США, Колумбии и Южной
Америке. В зависимости от цвета, прозрачности и
примесей, различают следующие разновидности бе-
рилла: собственно берилл, аквамарин, желтый бе-
рилл (гелиодор), изумруд (смарагд), ростерит и во-
робьевит. Другие минералы В.: хризоберилл
ВеО-А12О3 (19,8% ВеО), фенакит- BeJSiOJ
(45,5% ВеО).
Физические и химические свойства. Б. — металл
светло-серого цвета, имеет гексагональную плотно
упакованную решетку с параметрами а = 2,2856 А,
с = 3,5831 А; атомный радяус 1,13 А, ионный радиус
Be2V 0,34 А; плотн. <12Ъ 1,8477; т. пл. 1285°; т. кип.
2970°; зависимость давления пара Б. от темп-ры:
1.0g Ратм. = 6.186 + 1А54 • 10 4 Т — 16,700/Т; уд.
теплоемкость 0,481
плавления 2,8 ± 0,5
кал/г град (0—100°); теплота
ккал/г-атом; термич. коэфф, ли-
нейного расширения 13•106
(0 — 200°); электропровод-
ность составляет 40% от ме-
ди. Твердость Б. по Бринел-
лю 97—114 кг/мм2; модуль
упругости 30 000 кг/мм2,
предел прочности при рас-
тяжении для выдавленного
прутка до 60 кг/мм2, ото-
жженного — ок. 35 кг /мм2.
Уд. прочность Б. (отнесен-
ная к уд. весу) значительно
превосходит уд. прочность
других металлов и спла-
вов, однако применение Б. в
качестве конструкционного
металла затруднено из-за
его хрупкости на холоду,
что объясняется особенно-
стями строения кристаллич.
решетки Б. и наличием примесей. Хрупкость за-
трудняет также и обработку В. Заготовки из Б.
получают прессованием порошка с последующим
спеканием при 1180—1200° или горячим прес-
сованием порошка пря 1120—1150°, или плав-
кой в вакууме. Изделия из заготовок получают
обработкой давлением при повышенных темп-рах
(выдавливанием, прокаткой), иногда с последующей
обработкой резанием. Б. в жидком состоянии пол-
ностью смешивается со многими металлами, напр.
с Al, Си, Fe, Со, Ni, Zn и др., не смешивается с Mg
и, вероятно, со щелочными и щелочноземельными
металлами. Из-за большого различия в атомных
диаметрах Б. образует твердые р-ры лишь с немно-
гими металлами и в ограниченной области. Наиболь-
шая растворимость Б. в твердом состоянии наблю-
дается в сплавах с Си (2,75 вес. % В.), Ni (2,7%) и
Сг (1,7%), причем растворимость сильно уменьшается
с понижением темп-ры и сплавы в этом случае спо-
собны к дисперсионному твердению.
В химич. соединениях Б. 2-валентен. Соединения
с одновалентным Б. неустойчивы. Компактный металл
устойчив на воздухе благодаря покрывающей его
пленке окиси; выше 800° наступает заметное окисле-
ние; в холодной и горячей воде Б. практически не
изменяется. Б. растворяется в разб. НС1 и H2SO4;
с HNO3 как концентрированной, так и разбавленной
взаимодействует только при нагревании. Б. реаги-
рует со щелочами, напр.: 2КОН + Бе = К?ВеО2 +
+ Н2. С водородом Б. непосредственно не соеди-
няется; гидрид (ВеН2)х получен при разложении бе-
риллийорганич. соединений в виде твердого в-ва,
разлагающегося выше 200°. При высокой темп-ре
В. образует соединения с кислородом ВеО, азотом
Be3N2, углеродом Ве2С, хлором ВеСБ (см. Бериллия
галогениды) и др. Гидроокись Ве(ОН)2 амфотерна
(см. Бериллия окись). Соли В. большей, частью хо-
рошо растворимы в воде. К числу плохо растворимых
солей относятся фосфат Ве3(РО4)2 • zH2O и основной
карбонат (состав переменный). Р-ры солей В. вслед-
ствие гидролиза имеют кислую реакцию.
Сульфат В. BeSO4 при обычных условиях
существует в виде кристаллогидрата BeSO4 • 4Н2О —
бесцветных кристаллов, имеющих объемноцентриро-
423
БЕРИЛЛИЙ — БЕРИЛЛИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
424
ванную тетрагональную решетку: а — 8,02 А, с =
= 10,750 А; растворимость в воде (в расчете на BeSO4)
29,7% (25°), от прибавления H2SO4 растворимость
снижается и при 66% H2SO4 достигает 0,9%. Кри-
сталлогидрат BeSO4 • 4Н2О мало гигроскопичен и
легко может быть перекристаллизован из водного
раствора; при 400° теряет кристаллизационную воду.
При 550—600° BeSO4 начинает разлагаться на ВеО
и SO3.
Нитрат Б. известен в виде кристаллогидрата
Be(NO3)2 • 4Н2О, растворимость в воде (в расчете на
безводную соль) 51,2% (при 20°). Образуется при
упаривании р-ра Be(NO3)2 в вакууме или при при-
бавлении конц. HNO3 к насыщенному р-ру Be(NO3)2,
При нагревании, а также при высушивании в вакууме
образуется основной нитрат В. переменного состава-
при прокаливании — ВеО. Из насыщенного при 20°
р-ра нитрата Б., содержащего 51,2% свободной
HNO3, кристаллизуется Be(NO3)2 • ЗН2О. Действием
органич. к-т на гидроокись или карбонат В. получают
оксисоли типа Ве4О (RCOO)6, из них наиболее важны
оксиформиат Ве4О(НСОО)6 (возгоняется в вакууме
при 250—260°) и оксиацетат Ве4О(СН3СОО)в. Оксиа-
п е т а т (основной ацетат) известен в виде двух
модификаций. Ниже 148° устойчива модификация
с гранецентрированной кубич. решеткой, а — 15,72 А.
По своему характеру Ве4О(СН3СОО)6 является моле-
кулярным соединением; т. пл. 284°; т. кип. 331°;
нерастворим в воде, хорошо растворим в хлороформе
и др. органич. жидкостях. Ве4О(СН3СОО)6 исполь-
зуется для очистки Б. от других элементов перегон-
кой в связи с тем, что летучих оксиацетатов они не
образуют (оксиацетаты такого же типа известны
лишь у цинка и циркония). Разложением оксиацетата
получают ВеО высокой чистоты.
Основной карбонат Б. переменного
состава образуется при приливании к р-ру солей Б.
водного раствора Na2CO3 или (NH4)2GO3; раство-
ряется в избытке карбонатов щелочных металлов
или аммония с образованием комплексных солей
и вновь выпадает при нагревании р-ра.
Б. относится к аналитич. группе А1; осаждается
в виде гидроокиси при действии аммиачного р-ра
сульфида аммония. Минералы Б. переводят в р-р
сплавлением с содой и растворением в НС1. От А1,
Fe, Сг, Cd, Ni, Go, Zn, Mn, Bi, Pb его отделяют дей-
ствием аммиака на р-ры, содержащие этилендиамин-
тетрауксусную к-ту (комплексон III); при этом Б. ос-
тается в осадке, а примеси в виде комплексов— в р-ре.
Качественно Б. открывают спектральным путем или
по синему окрашиванию с хинализарином в щелоч-
ной среде (в отсутствии Mg и Fe), или по красно-
оранжевому цвету осадка Ве(ОН)2 в аммиачном р-ре
в присутствии куркумина. Количественно весовым
путем Б. определяют в виде ВеО после осаждения
аммиаком и прокаливания при 950°; в виде фосфата —
после осаждения при pH 5,2 и прокаливания при
800° (мешают Zr, Al, Fe, а в присутствии комплек-
сона III только Ti); кроме того, Be определяют иодо-
метрич. титрованием после осаждения его в виде
арсената в присутствии комплексона III, фотометри-
чески по реакциям с бериллоном, хинализарином; по
реакции с n-нитробензолазоарсином, флуориметрич.
путем по реакции с морином (2 • 10~4 — 25 • 10~4% Be)
и с хинализарином; радиоактивационным методом.
Получение. Известно несколько методов извле-
чения Б. из берилла. По одному из них берилл спе-
кают с известью и продукты реакции обрабатывают
конц. H2SO4; из р-ра, содержащего сульфат Б.,
удаляют различными методами примеси и осаждают
аммиаком гидроокись Б. По другому методу измель-
ченный берилл спекают с Na2[SiFe], Образовавшиеся
фторобериллаты натрия Na[BeFs] и NaJBeFJ выще-
лачивают водой и из р-ра осаждают едким натром
гидроокись Б. Дальнейшая обработка ведется с целью
получения хлорида или фторида Б.
Хлорированием ВеО в смеси с углем получают
ВеС12, к-рый в смеси с NaCl подвергают электролизу
при темп-ре ок. 350°. Металл (в виде чешуек) отмы-
вают от электролита, высушивают и переплавляют
в вакууме или в атмосфере инертного газа. Для полу-
чения BeF„ окись или гидроокись Б. сначала пере-
водят во фторобериллат аммония (NH4)2(BeF4], к-рый
подвергают термич. разложению. Из BeF2 металлич.
Б. получают восстановлением магнием; для пони-
жения темп-ры плавления шлака добавляют фториды
других металлов или берут избыток BeF2; избыток
Mg отгоняют в вакууме. Дальнейшую очистку Б.
производят дистилляцией в вакууме.
Применение. Важной областью применения Б.
являются различные сплавы, в к-рые Б. вводится
как легирующая добавка. Большое значение имеют
сплавы Си—Be, т. н. бериллиевые бронзы, содер-
жащие до 2,5% Be с добавками Ni и Со (0,2—0,5%),
приобретающие после закалки и отпуска (старения)
высокую прочность и твердость, а также хорошую
электропроводность, теплопроводность и коррозион-
ную стойкость (см. Меди сплавы). Практич. при-
менение нашли также сплавы Ni с 2—4% Б. Эти
сплавы по сопротивлению коррозии, прочности и
упругости сравнимы с высококачественными нержа-
веющими сталями, но превосходят последние по
твердости, способности к ковке и термич. обработке.
К улучшению свойств приводит введение Б. и в же-
лезные сплавы. Ничтожные добавки Б. к магниевым
сплавам повышают их сопротивление коррозии, сильно
уменьшают окисляемость сплавов во время плавки и
разливки. Сплавы с Б. находят применение в само-
летостроении, электротехнике и др. В конструкциях
атомных реакторов Б. благодаря малому поперечному
сечению захвата тепловых нейтронов используется
как замедлитель и отражатель нейтронов.
Из Б. изготовляют окошечки рентгеновских трубок,
используя его проницаемость для рентгеновских
лучей. Б. в смеси с препаратами радия служит источ-
ником нейтронов — Б. испускает нейтроны при дей-
ствии а-частиц, у-лучей и дейтронов по ядерным
реакциям: Ве9(а, n)C12; Ве9(у, n) Be8; Be9(d, п) В10.
Летучие соединения Б. и пыль, содержащая Б. и его
соединения, сильно токсичны.
Лит.: Рейфман М. Б., Бериллий, М., 1959; Фил янд
М. А. и Семенова Е. И., Свойства редких элементов,
М., 1953, с. 128—41; М е е р с о н Г. А. [и др.], Исследо-
вания в области геологии, химии и металлургии. (Доклады
советской делегации на Международной конференции по мир-
ному использованию атомной энергии, Женева, 1955), М.,
1955; М е е р с о н Г. А. [и др.], Атомная энергия, 5, вып. 6,
1958, с. 624; The Metal Beryllium, Cleveland—Ohio, 1955; Gme-
11 n, 8 Aufl., System-Nummer26, B., 1930; M e 11 о r, v. 4, L.—N.
Y. — Toronto, 1946; то же, v. 4, 1952; P a s с a I, t. 4, P., 1958;
Ullmann, 3 Aufl., Bd 4, Munchen — B., 1953, S. 319—34;
Kirk, v. 2, N. Y.,1948, p. 490—509; Kirk, Second supple-
ment volume, N. Y., 1960, p. 81 — 89. А. В. Новоселова.
БЕРИЛЛИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ —
известны двух типов: полные, в к-рых обе валент-
ности Be заняты органич. радикалами, и неполные,
в к-рых одна валентность занята, напр. галогеном.
Б. с. крайне чувствительны к кислороду, влаге и
СО2; низшие R2Be самовоспламеняются на воздухе,
а (СН3)2Ве также и в двуокиси углерода. Физические
свойства важнейших Б. с. приведены в табл.
Формула Т. пл., °C Т. кип., °C
(СН.)„Ве (CgHgJaBe ...» (С4Нэ)2Ве .... (СбН5)3Ве .... 12° т. разл. 160° Т. возг. 200° 110°/15 лш 170°/25 мм
425
БЕРИЛЛИЯ ГАЛОГЕНИДЫ — БЕРТЛО—ТОМСЕНА ПРИНЦИП
426
Твердый (СН3)2Ве полимерен и даже в парах пред-
ставляет равновесную смесь моно-, ди- и тримера;
жидкие R2Be сильно ассоциированы. R2Be образуют
устойчивые эфираты, из к-рых эфир может быть
удален лишь продолжительным нагреванием в ва-
кууме. Дифенилбериллий с фениллитием дает комплекс
Li Be (С6Н5)3; получен также LiBe(C6H5)2H. За исклю-
чением (СН3)2Ве, устойчивого вплоть до 200°, R2Be
относительно мало термостойки и при нагревании
отщепляют углеводороды, образуя алкилбериллий-
гидриды, например: (uao-C3H7)2Be —> иао-С3Н7ВеН +
+ С3Н6; пиролиз (mpem-C4H9)2Be (150—200°) про-
ходит вплоть до гидрида бериллия. С в-вами, со-
держащими подвижный атом родорода, СО2 и рядом
карбонильных соединений R2Be реагируют анало-
гично магнийорганическим соединениям; иодом и
ВеС12 дезалкилируются до алкилбериллийгалогенидов
RBeHal. Последние менее реакционноспособны, чем
R2Be, образуют комплексы с эфиром и при нагре-
вании диспропорционируют до R2Be и ВеНа12.
В. с. получают: 1) алкилированием галогенидов
Be магний- или литийорганич. соединениями; конеч-
ными продуктами являются бериллийдиалкилы:
BeHal2 + 2RLi—>2LiHal + R2Be; 2) взаимодействием
металлич. Be с ртутьорганич. соединениями: R2Hg -)-
+ Be — R2Be Hg; 3) термич. диспропорциониро-
ванием алкилбериллийгалогенидов или действием
свободных метильных и этильных радикалов на Be.
Лит.: R о с h о w Е. G., Hur d D. Т., Lewi s R. N.,
The chemistry of organometallic compounds, N. Y.—L., [10571.
БЕРИЛЛИЯ ГАЛОГЕНИДЫ - Дд“7р“ошо
растворимые в воде в-ва; в расплавленном состоянии
плохо проводят электрич. ток. Бериллия фто-
рид BeF2 имеет неск. полиморфных модификаций,
аналогичных по структуре модификациям SiO2. При
термич. разложении (NH4)2BeF4 образуется кристо-
балитоподобный C-BeFe2, кристаллизующийся в тет-
рагональной решетке с параметрами а = 6,60 А;
с =6,74 А. При 545° C-BeF2 начинает плавиться,
расплав прозрачен ок. 800°. Из расплава в присут-
ствии фторидов щелочных металлов кристаллизует-
ся кварцеподобная гексагональная модификация
Q-BeF2 .с периодами решетки: а = 4,72 А; с —
= 5,18 А; т. пл. 590 10°. Теплота образования
BeF2 ДН298 = — 241 ккал/моль. Расплавленный BeF2
затвердевает в виде стекла. Растворимость BeF2
в воде 18 моль/л при 25° (найдено экстраполяцией).
При выпаривании р-ра выделяется 5BeF2 • 2ВеО. При
нагревании с фторидами щелочных металлов образует
двойные соли — фторобериллаты, типов: MejBeFj,
Me2BeF4, MeIBeF3, MeIBe2F5 (возможно Me8Be2F7)'
С фторидами щелочноземельных металлов образует
фторобериллаты типа MeIIBeF4; фторобериллаты в
расплавленном состоянии хорошо проводят электрич.
ток. Фторид бериллия применяют для получения
металлич. бериллия.
Бериллия хлорид ВеС12 образует неск.
полиморфных модификаций; известна структура
только одной из них, получающейся в виде белых
гигроскопич. волокон при возгонке ВеС12; орторомбич.
решетка, параметры: а = 9,86 А; b = 5,36 А, с =
= 5,26 А; т. пл. 440°; т. кип. ок. 520°. Теплота обра-
зования ВеС12 ДН298 = — 112,6 ккал/моль. Раствори-
мость в воде при 0° ок. 65 г в 100 г воды. ВеС12 —
гигроскопичное и легко гидролизующееся соединение;
образует кристаллогидраты ВеС12 • 4Н2О и ВеС12 •
• 2Н2О. Хлорид бериллия — акцептор электронов,
склонен к присоединению неорганич. и органич.
молекул, атомы к-рых имеют свободные электронные
пары, напр. ВеС12 • 2(С2Н5)2О. Получают ВеС12 дей-
ствием хлора на смесь ВеО и угля при темп-ре ок. 800°
или паров СС14 на ВеО (700—800°). Применяют для
получения металлич. бериллия, а также как катали-
затор в органич. синтезе. Бериллия бромид
ВеВг2 и и о д и д BeJ2 плавятся ок. 500°, по свойствам
близки к ВеС12. Галогениды, как и др. соединения
бериллия, весьма токсичны.
Лит.: Новоселова А В., Усп. хим., 1959, 28,
вып. 1, с. 33. См. также лит. при ст. Бериллий.
А. В. Новоселова.
БЕРИЛЛИЯ ОКИСЬ ВеО — белый кристалличе-
ский порошок; гексагональная решетка с пе-
риодами: а = 2,69 А, с = 4,37 А, плотность 3,025;
т. пл. 2530°; т. кип. ок. 4120°. Теплота образования
ДЩ88 = —143 ккал/моль-, теплопроводность 397 • 10 4
кал/см сек град (1200°); линейный коэфф, термич.
расширения 10,8 • 10 6 (для 25—1000°). При прокали-
вании В. о. начинает испаряться ок. 1900°, быстрое ис-
парение наблюдается ок. 2400°. В воде В. о. нераство-
рима, в к-тах растворима, если предварительно не
была прокалена выше 440°; прокаленная не раство-
ряется в к-тах, кроме H2SO4 и HF. При нагревании
в токе водорода В. о. не восстанавливается. Получают
В. о. термич. разложением гидроокиси, сульфата,
нитрата или основного карбоната Be. Применяют
в качестве химически стойкого и огнеупорного мате-
риала для изготовления тиглей и спец, керамики;
в атомных реакторах — в качестве замедлителя и
отражателя нейтронов; в органич. синтезе — как
катализатор.
Бериллия гидроокись Ве(ОН)2 в за-
висимости от условий осаждения может существовать
в различных формах, отличающихся структурой и
растворимостью в воде. Ве(ОН)2 амфотерна; свеже-
осажденная хорошо растворима в к-тах; при раство-
рении в щелочах образует бериллаты, напр. К2ВеО2.
В воде Ве(ОН)2 плохо растворима, что используется
в аналитич. химии для определения Be.
Лит.: Материалы для ядерных реакторов, пер. с англ.,
М., 1056 (Ядерные реакторы. Материалы комиссии по атом-
ной энергии США, [т. 4], гл. 3); Тресвятский С. Г.
и Черепанов А. М., Высокоогнеупорные материалы
и изделия из окислив, М., 1957. См. также лит. к ст. Бериллий.
А. В. Новоселова.
БЕРИЛЛОН II C20H10O15N2S4Na4 • 4Н2О, мол. в.
810,60 — черно-коричневый порошок; растворим в
воде, ацетоне, этиловом
спирте; не растворяется
в бензоле, толуоле, хло-
роформе и четыреххло-
ристом углероде; водный
р-р в нейтральной и ки-
слой среде окрашен в
фиолетово-красный цвет,
в щелочной — в фиоле-
товый. Получают В. сочетанием диазотированной
Аш-кислоты с хромотороповой кислотой. Б. обра-
зует окрашенные соединения с Са, Mg, Be, Al, Fe,
Cu, Ni, Co, Mn, Cr, Sc, Th, Pa при различных
значениях pH, в частности, с Be дает голубое окра-
шивание в щелочной области; применяют для фото-
метрия. определения Be при pH 12—13,2 в присут-
ствии Al, Mg, Са, Со, Ni, Си и Fe.
Лит.: К а р а и о в и ч Г. Г., Ж. аналит. хим., 1956, 11,
№ 4, с. 400; Лукин А. М., Заварихина Г. Б., там же,
1956, И, № 4, с. 393. В. Г. Типцова.
БЕРКЕЛИЙ (Berkelium) Bk — химич. элемент
с п. н. 97 из семейства актинидов.
БЕРТИНИРОВАНИЕ — см. Полукоксование и
Термическая переработка топлива.
БЕРТЛО—ТОМСЕНА ПРИНЦИП — утверждение,
что всякая система, способная к химич. превра-
щению, будет преобразовываться преим. в такую,
переход в к-рую сопровождается наибольшим выде-
лением теплоты. Это положение было высказано
в неск. различных формах первоначально Ю. Том-
сеном (1854) и позднее М. Вертло (1867), назвавшим
его принципом наибольшей работы.
427
БЕРТОЛЛИДЫ — БЕТА-ЛУЧИ
428
Существование реакций, протекающих с поглощением
теплоты, противоречит этому принципу и показывает,
что он не является общеприменимым.
В действительности направление естественного те-
чения реакций определяется не тепловым эффектом,
а изменением соответствующего изотермич. потенциала
(свободной энергии) реагирующей системы, т. е. за-
висит не только от знака теплового эффекта реакции,
но также и от изменения энтропии при ее протекании.
Поэтому Б. — Т. п. применим в тех случаях, когда
изменение энтропии незначительно, т. е. когда макси-
мальная работа реакции близка к тепловому эффекту,
в частности, он приближенно оправдывается для
реакций в кристаллич. фазах. С другой стороны,
влияние энтропийного фактора уменьшается с пони-
жением темп-ры и становится равным нулю при
темп-ре абс. нуля. Поэтому при абс нуле Б. — Т. п.
оказывается справедливым для любых реакций.
Повышение темп-ры, наоборот, ограничивает его
применимость и способствует течению реакций, со-
провождающихся поглощением теплоты. В области
очень высоких давлений, подобно области очень
низких темп-p, изменение энтропии при протекании
процессов также незначительно. Поэтому при очень
высоких давлениях Б. — Т. п. оказывается тоже
справедливым.
Лит.: Карапетьянц М. X., Химическая термоди-
намика, 2 изд., М.—Л., 1953; Капустиной ий А. Ф.,
ДАН СССР, 1945, 18, № 4. В. А. Киреев.
БЕРТОЛЛИДЫ — см. Далыпониды и бертоллиды-
БЕССТРУЖКОВЫЙ МЕТОД АНАЛИЗА — ме-
тод качественного и количественного химического
анализа сплавов и стекол, позволяющий провести
определение их химического состава без поврежде-
ния образца. Р-р, необходимый для анализа, полу-
чают нанесением на тщательно очищенную поверх-
ность 0,1—0,5 мл подходящего растворителя, на-
пример HNO3 (1 : 1). По окончании реакции р-р сни-
мают капилляром. Если остаются нерастворившиеся
частицы (напр. карбиды), трением кончика палочки
их счищают, переводят в р-р сплавлением или агрес-
сивными растворителями и присоединяют к основному
р-ру. Для определения элемента в полученном р-ре
пользуются обычными колориметрическими реак-
циями или реакциями осаждения. Качественное
определение обычно проводят капельными реак-
циями на бумаге или в градуированных капельных
пробирках. При количественном определении ис-
пользуют стандартный образец, к-рый растворяют
в одинаковых с испытуемым образцом условиях.
Сравнивая интенсивности окрасок полученных р-ров
визуально или на фотоколориметре, находят про-
центное содержание определяемого элемента в образце.
Определение можно вести также и но стандартному
раствору, не проводя параллельного растворения
стандартного образца. Б м. а. можно использовать
для анализа готовых изделий, миниатюрных деталей,
для анализа покрытий, послойного анализа образ-
цов, а также для фазового (карбидного) анализа.
Б. м. а. отличается быстротой выполнения, малым
расходом реактивов и простотой аппаратуры. Дли-
тельность количественного анализа составляет в боль-
шинстве случаев 10—30 мин. Длительность карбид-
ного анализа 1—2 часа Относительная ошибка коли-
чественных определений обычно не превышает
± (5—10%). Большая заслуга в разработке метода
принадлежит Н. А. Тананаеву.
Лит.: Тав паев Н. А., Бесстружковый метод ана-
лиза черных, цветных и драгоценных сплавов, Свердловск —
М., 1948; Лошкарев А. Г., Завод, лабор., 1958, 24, № 8,
с. 939. А. Н. Тананаева.
БЕТ АЗИН CeHeO3NJ2, мол. в. 433,01 — белые
кристаллы, т. пл. 177—185° с разл (в зависимости
от скорости нагревания); трудно растворим в воде,
почти нерастворим в органич, растворителях, легко
растворим в водных р-рах едких щелочей, аммиака
и соляной кислоты. С NaOH
Б. дает хорошо растворимую 9*"*
двунатриевую соль (по карбо- ..
ксильной группе и фенольно- ] Г"
му гидроксилу). Водные р-ры
хлоргидрата Б. почти момен- Т
тально гидролизуются. Б. полу-
чают иодированием 0-(4-окси-
фенил)-0-аланина (0-тирозина). Применяют для ле-
чения тиреотоксикозов.
Лит.: Машковский М. Д., Лекарственные сред-
ства, 4 изд., М., 1969; Р о<д ионов В. М. [и д р.1, Ж. общ.
хим., 1957, 27, вып. 8, с. 2234. А. С. Елина.
БЕТАИНЫ — внутренние соли N-триалкилзаме-
щенных аминокислот. Молекула Б. содержит поло-
жительно и отрицательно заряженные атомы или
группы и является двухполюсным или биполярным
ирном типа
сн8
СН3—N+-CH—СОО-
CH3Z r
В зависимости от положения основной группы отно-
сительно кислотной различают а-, 0-, у- и т. п. Б.
Бетаиновую структуру имеют также внутренние соли
других ониевых оснований (оксониевых, сульфоние-
вых, содержащих кислотные группировки). Б. —
высокой давящиеся, хорошо растворимые в воде и
плохо растворимые в эфире вещества. При нагревании
а-Б. изомеризуются в эфиры диалкиламинокислот;
0- и у-Б. претерпевают перегруппировку типа Гоф-
мановской:
(CH3)3N+-CHo-CH2COO- - (CH3)3NH-O-CO-CH=CH»
Б. дают с рядом кислот и с хлоридами тяжелых
металлов нерастворимые соединения (золото-, пла-
тино-, свинцовохлористоводородные соли, железоси-
неродистые соединения и др.), что используется для
выделения Б. из биологич. объектов. Б. получают при
действии алкилгалогенидов и апкилсульфатов на
аминокислоты, взаимодействием галогенкислоты с
триалкиламином и др. способами. Превращение ами-
нокислот в Б. путем их полного метилирования — про-
цесс, характерный для азотистого обмена многих ви-
дов растений. В растениях, а также в животных ор-
ганизмах (гл. обр. беспозвоночных) обнаружены:
Б. гликоколла (бегай н), пролина (с т а х и д р и н),
триптофана (г и п о ф о р и н), орнитина (м и о-
к и н и н), никотиновой кислоты (т р и г о н е л и н)
и др. Наиболее распространен бетаин гликоколла
(CH3)3N+—СН2СОО“, мол. в. 117,15, бесцветные кри-
сталлы; растворим в воде, плохо — в эфире; т. пл.
безводного продукта 293°; при плавлении изомеризу-
ется в метиловый эфир диметилами ноуксусной к-ты;
получают из отходов сахарного произ-ва, а также син-
тетически — взаимодействием триметиламина и хлор-
уксусной к-ты. Хлорид бетаина (а ц и д о л) приме-
няют в медицине как заменитель соляной к-ты; хло-
ристоводородная соль бетаина дает при сухой пере-
гонке хлористый метил.
Лит.: Бр аунштейн А. Е.. Биохимия аминокислот-
ного обмена, М., 1949; X у б е н И., Методы органической
химии, пер. о нем., т. 4, вып. 1, кн. 2. М.—Л.. 1949.
М. И. Левчит.
БЕТА-ЛУЧИ (0-лучи) — излучение, состоящее из
электронов (или позитронов), испускаемое при бета-
распаде радиоактивных изотопов. Благодаря наличию
электрич. заряда Б.-п под действием электрич. и
магнитного полей отклоняются от прямолинейного
направления. Скорость частиц в Б.-л. близка к ско-
рости света. Б.-л. способны ионизировать газы,
инициировать радиационно-химич. процессы, вызы-
429
БЕТА-РАСПАД — БЕТОН
430’
вать люминесценцию, действовать на фотопластинки
и т. д.
БЕТА-РАСПАД (0-распад) — радиоактивное пре-
вращение атомного ядра, при к-ром испускаются
0-частицы, т. е. электроны (0 ) или позитроны (0+);
в Б.-р. включается также электронный захват, т. е.
захват атомным ядром одного из электронов окру-
жающей ядро электронной оболочки. Кроме пози-
трона (или электрона), в каждом акте Б.-р. испу-
скаются также нейтрино или, соответственно, анти-
нейтрино (см. Элементарные частицы)', при электрон-
ном захвате испускается нейтрино. Массовое число
ядра при Б.-р. не изменяется. Заряд ядра увеличи-
вается на единицу при испускании электрона и умень-
шается на единицу при испускании позитрона или
электронном захвате. При этом атом данного химич.
элемента превращается в атом другого (соседнего)
элемента. Энергия, выделяющаяся при Б.-р., по-
разному распределяется между электроном (позит-
роном) И антинейтрино (нейтрино). Поэтому энергия
вылетающих электронов (позитронов) может при-
нимать любые значения от 0 до нек-рой макс, вели-
чины £гран — т. н. граничной энергии Б.-р. (в боль-
шинстве случаев составляющей неск. Мэв). Значение
.брраи может служить характеристикой атомного
ядра данного изотопа. Другой характеристикой
Б.-р. является период полураспада 0-радиоактивного
изотопа (в большинстве случаев от долей секунды
до нескольких лет, редко много больше — прибл.
до 1012 лет). Количеств, теорию Б.-р. впервые дал
Э. Ферми (1934). См. Радиоактивность.
БЕТОН — искусственный каменный материал,
представляющий собой затвердевшую, тщательно
перемешанную смесь вяжущего вещества (см. Вяжущие
материалы и Цементы), воды и естественных или
искусственных заполнителей (песок, гравий, щебень,
шлак и т. п.) или смесь битума с минеральным порош-
ком и заполнителями (асфальтовый Б.). Изготов-
ляются также Б., в к-рых в качестве вяжущего исполь-
зуются полимеры (напр., поливинилацетат) или поли-
меры совместно с цементом (пластобетон). Б.
огнестойкий материал; степень огнестойкости Б.
зависит от вида цемента и рода заполнителей; тепло-
проводность 1,10—1,33 ккал/м час град, коэфф,
темп-рного расширения обычного Б. 0,00001. Обыч-
ный Б. непроницаем для воды, а также для мазута и
тяжелой нефти. Керосин, бензин и смазочные масла
проникают в Б. Различают след, виды Б.: 1) тяжелый
(обычный, объемный вес 2,1—2,5 т/м3) — с заполни-
телями из плотных каменных пород; применяется
в различных железобетонных конструкциях, промыш-
ленном, гражданском, гидротехническом и др. видах
строительства; 2) особо тяжелый (объемный вес св.
2,6 т/м3) — с заполнителями из барита, железных
руд, металлов и др.; применяется для защиты от
радиоактивных воздействий; 3) легкий (объемный
вес 1,2—1,7 т/м3) — с заполнителями из шлака,
пемзы, туфа и др.; применяется при кладке стен и
перекрытий с небольшой теплопроводностью и малым
весом; 4) особо легкий (объемный вес менее 1,2 т/м3)—
ячеистый (пенобетон, газобетон и т. п.), крупнопори-
стый (без песка); применяется как теплоизоляцион-
ный материал, а также при кладке стен небольших
зданий. Кроме того, изготовляются спец, виды Б. —
жароупорные, кислотостойкие и др., приготовляе-
мые с применением спец, заполнителей и вяжущих
веществ.
Прочность Б. при твердении в нормальных усло-
виях зависит в основном от качества цемента и соот-
ношения веса воды к весу цемента (водоцементное
отношение). С увеличением водоцементного отношения
выше нек-рого предела прочность Б. уменьшается;
избыток цемента ведет к образованию трещин (вслед-
ствие экзотермич. явлений и неравномерности распре-
деления темп-ры), а также к удорожанию. Б. хорошо
сопротивляется сжатию и плохо — растягивающим
усилиям. Прочность Б. на сжатие в возрасте 28 дней
наз. маркой Б.; она достигает 600 кг/см2, прочность
на растяжение меньше примерно в 8—15 раз. -Для
увеличения сопротивляемости Б. растяжению его
армируют стальными стержнями, воспринимающими
растягивающие усилия. Полученный т. о. материал наз.
железобетоном.
В физико-химич. отношении Б. представляет собой
трехфазную, полидисперсную, полиминеральную си-
стему, в образовании и развитии структурных свойств
к-рой большое значение на всех стадиях имеют кол-
лоидо-химич. процессы. При взаимодействии тонко-
молотого порошка цемента с водой развиваются
экзотермич. реакции гидратации цемента, сопрово-
ждающиеся возникновением новообразований, по-
степенно заполняющих пространства, ранее занятые
водой. Чем больше степень заполнения этих про-
странств и последующее уплотнение структуры це-
ментного камня, тем больше плотность Б. Основным
при изготовлении тяжелого Б. является придание
ему плотной структуры, обеспечивающей хорошую
сопротивляемость материала агрессивным воздей-
ствиям внешней среды и необходимую механич.
прочность. Высокая плотность Б. достигается тща-
тельным подбором соотношения его составных частей.
С целью снижения общей пористости (в особенности
фильтрующей, образующейся в результате испарения
химич. не связанной воды) производят механич. уплот-
нение различными вибрационными механич. устрой-
ствами, а при изготовлений бетонных изделий прокат-
ным способом — вибровакуумированием, центробеж-
ным способом, виброперемешиванием и др., а также
применением физико-химич. методов воздействия на
упруго-пластично-вязкие свойства уплотняемой мно-
гофазной системы. В строительстве наиболее распро-
странен цементный Б., а в дорожном строительстве —
асфальтовые Б., в к-рых в качестве вяжущего содер-
жится смесь нефтяного или природного битума с ми-
неральным порошком.
Мягкие воды, а также воды, содержащие раство-
ренные минеральные соли, углекислый газ и др.,
разрушающе действуют на Б. Различают след,
виды коррозии Б.; 1) растворение и унос водой со-
ставных частей цементного камня; 2) растворение и
унос водой продуктов обменных реакций цементного
камня и содержащихся в воде химич. веществ или
выделение этих продуктов в виде аморфной массы, не
обладающей вяжущими свойствами; 3) накопление
малорастворимых солей, кристаллизующихся в Б.
и разрушающих его. Коррозионная стойкость Б.
улучшается при использовании соотв. цементов и
заполнителей, а также при увеличении плотности
Б. Постоянный и переменный электрич. ток не раз-
рушает Б., не содержащий арматуры. Железобетон
сильно разрушается постоянным током (электролитич.
действие).
Для регулирования свойств Б. и экономии цемента
в их состав вводят след, добавки: 1) гидравлические,
повышающие стойкость вяжущих веществ как в прес-
ных, так и в соленых водах (трепел, опока, диатомит,
трасс, пемза, вулканич. пепел и туф, активные крем-
неземистые отходы, обожженная глина, доменные
гранулированные шлаки и др.); 2) микронаполни-
тели, дающие возможность расходовать меньше це-
мента (тонкоизмельченные пески, известняки, доло-
миты, природный пылевидный кварц, нек-рые виды
зол и топливных шлаков и т. д.); 3) регулирующие
сроки схватывания цементов (хлористый кальций,
растворимое стекло, сода, гипс и др.); 4) ускоряющие
431
ВЕШАНА ВОССТАНОВЛЕНИЕ — БИМОЛЕКУЛЯРНЫЕ РЕАКЦИИ
432
твердение цементов и повышающие прочность бетонов
и р-ров в первые сроки твердения (хлористый кальций,
соляная к-та); 5) поверхностно-активные органиче-
ские, предназначенные, в основном, для уменьшения
расхода вяжущего и повышения морозостойкости
р-ров и бетонов (пластифицирующие — сульфитно-
спиртовая барда и ее производные и т. д.; воздухово-
влекающие — абиетат натрия, омыленный древесный
пек, мылонафт и др.).
Лит.: Строительные материалы, под общ. ред. Б. Г. Скрам-
таева, 6 изд., М., 1953; Б у т т Ю. М.и Беркович Т. М.,
Вяжущие вещества с поверхностно-активными добавками,
М., 1954; Москвин В. М., Коррозия бетона, М., 1952;
Дес о в А. Б., Тяжелые и гидратные бетоны, М., 1956;
Некрасов К. Д., Огнеупорные бетоны, их свойства
и применение, М.—Л., 1949; Будников П. П., Избран-
ные труды, Киев, 1960. В. В. Стольников.
ВЕШАНА ВОССТАНОВЛЕНИЕ — метод пре-
вращения нитросоединений в аминосоединения с по-
мощью чугунных стружек в кислой среде. Условия
реакции многократно видоизменялись. С 1864 она
ведется в соляной к-те, позволяющей превращать
в амины все известные нитросоединения за немно-
гими исключениями. В нек-рых случаях для Б. в.
используют серную, муравьиную и др. к-ты. Для син-
теза полупродуктов анилинокрасочной промышлен-
ности (анилин, толуидин, анизидин
и т. п.) широко применяется восета- СН=СН2
новление чугунными стружками в ц3С т________=]
р-рах электролитов: солей железа, нО_1 J- Q
NH4C1 и др. в отсутствие свободной и
к-ты. В процессе восстановления
железо очень быстро окисляется
благодаря присутствию воды и нитросоединений. В
этих условиях восстанавливающими агентами являют-
ся как металлич. железо, так и его гидрат закиси, реа-
гирующие с нитросоединениями: RNO2 + 3Fe+4H2O—>
-► RNH2 + 3Fe(OH)2; RNO2 + 6Fe(OH)2 + 4H2O->
—> RNH2 -j- 6Fe(OH)3. Скорость восстановления чу-
гунными стружками в присутствии электролитов
определяется: 1) химич. составом и микроструктурой
чугуна. Наиболее активными являются мягкие серые
чугуны; 2) природой электролитов и их концентра-
цией. По активности они могут быть расположены
в ряд: NH4Cl>FeCl2>FeSO4; 3) pH реакционной
среды. Метод применяется в пром-сти при восстанов-
лении нитробензола в анилин, а также других арома-
тич. нитросоединений в амины. Открыто А. Бешаном
в 1854.
Лит.: Лукашевич В. О., Усп. хим., 1948, 17, вып. 6,
с. 692; Ворожцов Н. Н., Основы синтеза промежуточных
продуктов и красителей, 4 изд., м., 1955, с. 234.
Н. Я. Григорьева.
ВЕШАНА РЕАКЦИЯ — метод прямого введения
остатка мышьяковой к-ты OAs(OH)2 вместо атома
водорода в ароматич. соединения. Эта реакция пря-
мого арсенировация действием мышьяковой кислоты,
играющей роль электрофильного агента, по существу
аналогична сульфированию, но имеет гораздо меньшие
границы приложимости. Б. р. протекает лишь при
наличии в ароматич. ядре заместителей типа ОН и
NH2, облегчающих электрофильное замещение водо-
рода в пара-положении. В типичном случае реакции
с анилином образуется ге-аминофенилмышьяковая к-та:
CeH6NH3 + II3AsO4 —- n-NH,C6H4As(O)(OH)2
Б. р. протекает с многими фенолами, фениловыми
эфирами, аминами и их производными. Если пара-
положение занято, арсенирование направляется в
орто-, но не в мета-положение. Б. р. иногда сопрово-
ждается побочными реакциями. Так, аминофенолы
и многоатомные фенолы при действии на них H3AsO4
окисляются; при реакции с анилином при 220—230°
образуется гл. обр. (re-NH2CcH4)2As(O)OH. Реак-
ция открыта А. Бешаном в 1863.
Лит.: Органические реакции, пер. с англ., сб. 2, М., 1950,
с. 461. Я. Ф. Комиссаров.
NHC-NH-C-NHCH(CH3)21 • НС1
NH NH I
БИГУМАЛЬ (хлоргидрат А^-пара-хлорфепил-А’--
изопропил-бигуанида), мол. в. 290,21 — белый кри-
сталлич. порошок горького вкуса; т. пл. ок. 245°
(т. пл. основания 129—132°); слабо растворим в воде,
п-С1СбН4
лучше — в спирте, почти нерастворим в хлороформе
и эфире. С солями меди Б. дает нерастворимую мед-
ную соль; на этом основано его качественное открытие.
При действии на водные растворы Б. AgNO3 выпадает
осадок AgCl. Для колич. определения выделяют
1 н. р-ром NaOH из точной навески Б. его основание.
Б. получают действием изопропиламина на ге-хлорфе-
нилциангуанидин, к-рый, в свою очередь, синтезируют
из ге-хлорфенилдиазонийхлорида и дициандиамида.
Б. — лечебный и профилактич. противомалярийный
препарат; при лечении тропич. малярии значительно
эффективнее акрихина.
Лит. см. прист. Адалин. А. С. Елина.
БИКФОРДОВ ШНУР — см. Огнепроводный шнур.
БИЛИРУБИН С33НзвО6А4, мол. в. 584,68 — желч-
ный пигмент, образующийся из гемоглобина, гл. обр.
в печени. Б. — коричневые кристаллы, при нагрева-
CH.,CH2COOH CH2CHoCOOH сн3
НзСтЧ “ |гтснз _J=TCH=CH
-—L J---сн„----Ч. J—сн=< >-он
N N N
нии разлагается, не плавясь; нерастворим в воде,
эфире, глицерине, хорошо растворим в хлороформе,
хлорбензоле и др. органич. растворителях, а также
в слабых р-рах щелочей. Щелочные соли Б. хорошо
растворимы в воде, соли двухвалентных катионов
(Ва, Са) нерастворимы. Б. дает цветные реакции на
Желчные пигменты: Гмелина — с конц. HNO3, содер-
жащей HNO2; Ван-ден-Берга — с диазореактивом
Эрлиха (сульфаниловая к-та -|- NaNO2-|- HCI) и др.
р-ции, что используется для качеств, и колич. опре-
деления Б. При нек-рых патология, состояниях (жел-
туха, билирубиновый инфаркт и др.) Б. появляется
в моче, а количество его в крови возрастает. Б. полу-
чают из желчи и желчных камней, в к-рых содержится
Са-соль Б. Он получен также синтетически. Б. при-
меняют в качестве стандарта в клипич. лабораториях.
Лит.: Степаненко Б. Н., в кн.: Усп. современной
биохим. Сб. трудов...,т. 1, М., 1947, с. 269; А с а т и а н и В. С.,
Биохимическая фотометрия, М., 1957; см. также при ст. Желч-
ные пигменты. И. 3. Сергиенко.
БИМОЛЕКУЛЯРНЫЕ РЕАКЦИИ — простые, со-
стоящие из одной стадии реакции, в каждом элемен-
тарном акте к-рых участвуют две частицы (молекулы,
ионы, свободные радикалы). Скорость Б. р. пропор-
циональна числу соударений z между реагирующими
частицами. Однако реакция происходит лишь при
соударениях, в к-рых сталкивающиеся частицы дол-
жным образом ориентированы относительно друг
друга и обладают энергией, достаточной для преодо-
ления потенциального барьера реакции (см. Энергия
активации). Поэтому скорость Б. р. равна
w = zpe~
где р — стерический фактор, показывающий, какова
вероятность необходимой взаимной ориентации стал-
кивающихся частиц, Е — энергия активации реакции,
R — универсальная газовая постоянная, Т — абсо-
лютная темп-ра. Поскольку число столкновений между
частицами в единицу времени в единице объема про-
порционально произведению концентраций этих ча-
стиц, скорость Б. р. также пропорциональна произ-
ведению концентраций реагирующих частиц. Следо-
вательно, Б. р., по определению порядка реакции,
433
БИОГЕОХИМИЯ — БИОКАТАЛИЗАТОРЫ
434
является реакцией второго порядка, скорость к-рой
равна:
•ш = кС1Сз (1)
где к — константа скорости, сг и с2 — концентрации
реагирующих частиц.
В случае реакций в газовой фазе z, а следова-
тельно, и к можно рассчитать на основе молекулярно-
кинетич. теории газов. Соответствующая формула
для к имеет вид:
й = л(гА + гв)2«Р EIRt =
=(га+гв)ч/Е^^Нв) " EIRr л/моль.сеК (2)
J/ ntAmB КХХг
где гА, гв — радиусы, тА, тв — массы реагирующих
частиц, и — средняя скорость теплового движения реа-
гирующих частиц относительно друг друга, к — по-
стоянная Больцмана 1,38- 10-16 эрг/°К, N — число Аво-
гадро 6,02 • 1023. Произведение л (rA + rB)277JV/1000
является величиной порядка 1010—1011 л! моль • сек.
Поскольку р по физич. смыслу этой величины
меньше единицы, а энергия активации всегда положи-
тельна, то константа скорости Б. р. не может превы-
шать 1010—1011 л/'моль сек, а как правило является
значительно меньшей величиной. Формула (2) с извест-
ной степенью приближения может применяться для
констант скоростей Б. р. в растворах. Если Б. р. не
сопровождается никакими побочными процессами,
то уравнение (1) может быть записано в виде:
dx
-~=k(a — х) (b — х)
at
где x — концентрация продукта реакции в момент
времени I, а и Ъ — начальные концентрации реаги-
рующих в-в. Интегрирование этого уравнения при-
водит к выражениям:
(Ь — а) й/ = In (у • ) приа^б (3)
akt = ——— при а = b (4)
а—х
Определив экспериментально концентрацию одного
из продуктов Б. р. в определенный момент времени,
можно по этим формулам вычислить константу ско-
рости Б. р. Из ур-ния (3) следует, что в случае а е
в Б. р. должна иметь место линейная зависимость
величины
In |(6 — х) (а—х)] от t
по ходу реакции:
In——— = In — -Р (b — a) kt
а—х а
В случае а = Ъ должна иметь место линейная зави-
1 1
симость — от у:
Выполнение одной из этих зависимостей означает,
что рассматриваемая реакция является реакцией вто-
рого порядка. Отсюда, однако, не следует однозначно,
что эта реакция является простой Б. р., т. к. по кине-
тич. закону реакций второго порядка могут проте-
кать и нек-рые сложные химич. реакции, состоящие
из нескольких стадий. д. г. Кнорре.
БИОГЕОХИМИЯ — раздел геохимии, изучающий
геохимич. процессы с участием живых организмов.
Земная оболочка, где происходят эти процессы, наз.
биос ф.е рой; она включает всю гидросферу,
верхнюю часть литосферы и нижнюю часть атмо-
сферы. В этих пределах сосредоточено ок. 1011 т жи-
вого вещества. Самые большие его концентрации
имеются на отдельных участках земной поверхности
(фитопланктон морей и океанов, лесной и травяной
покров суши). По А. П. Виноградову, в состав жи-
вого вещества входят следующие химич. элементы
(в вес. %): 70% О, 18% С, 8% Н, 0,7% Р, 0,5% Са,
0,5% N, 0,2% К, 0,2% S.
Важной геохимич. функцией растений является
фотосинтез', в течение года все растения усваивают
ок. 175 млрд, т углерода, т. е. за 300—400 лет по-
требляется количество СО2, равное общему содержа-
нию ее в воздухе. Каждые 5—6 млн. лет растения
разлагают количество воды, равное объему всей гид-
росферы. Т. обр., живые организмы являются актив-
ными участниками круговорота веществ в природе.
Прямо влияя на состав атмосферы и связанный с нею
комплекс атмосферных явлений, живая природа тем
самым косвенно способствует изменению поверхности
литосферы: разрушению (выветриванию) горных по-
род, миграции входящих в их состав химич. элемен-
тов и последующему их рассеянию или концентриро-
ванию с образованием новых минеральных форм.
Активное влияние растений на литосферу заключается
в химич. разложении пород под действием выделяемых
кислот (напр., гуминовых) и механич. их разрушении
под действием фактора роста. В процессе жизнедея-
тельности многие организмы усваивают и концентри-
руют нек-рые химич. элементы: кремний (водоросли,
губки, наземные растения), кальций (водоросли, мол-
люски, корненожки и позвоночные), ванадий (оболоч-
ники, иглокожие), иод (губки, водоросли) и т. д.
После их отмирания образуются толщи осадочных
пород, обогащенных этими элементами, или состоя-
щих целиком из скелетов организмов (коралловые и
раковинные известняки, диатомиты и др.). Не менее
важная роль принадлежит бактериям, образующим
скопления многих марганцовых и серных руд. Ком-
плекс горных пород — продуктов органич. жизни —
наз. биолитами. Горючие (органические) био-
литы наз. каустобиолитами (торф, угли,
нефть, газы природные горючие).
Геохимич. среда в свою очередь активно влияет
на живую природу. Определенные соотношения между
химич. элементами в горных породах находят свое
отражение в почвах, растениях и животных. Измене-
ние этих отношений (обогащение или обеднение
нек-рыми элементами) может стимулировать рост
организмов или, напротив, способствовать их угне-
тению, заболеванию (биогеохимия, эндемия), появле-
нию уродливых форм и совершенно новых биологич.
видов. Так, напр., в областях развития карбонатных
пород процветает кальцефильная флора; у выходов
цинковых руд появляется особый вид фиалки Viola
cincifera; при обогащении почв селеном сельскохозяй-
ственные животные заболевают малокровием, сопро-
вождающимся выпадением волос, перьев и размягче-
нием копыт (рогов); повышенная концентрация фтора
в питьевых водах приводит к дистрофии эмали зубов
и повышенной их хрупкости (флюороз); недостаток
кальция препятствует росту всех позвоночных живот-
ных; с недостатком иода связывается нарушение функ-
ций щитовидной железы (зоб).
Из этих особенностей вытекает практическое зна-
чение Б.: использование влияния микроэлементов на
рост организмов (в частности, повышение урожай-
ности культурных растений) и геохимические поиски
руд по повышенным концентрациям элементов в водах,
почвах и золе растений (см. Геохимические методы
поисков).
Лит..' Вернадский В. И., Избр. соч., т. 5, М., 1960.
(Статьи по биогеохимии); Самойлов Я. В., Биолиты,
Л., 1929; Виноградов А. П., Почвоведение, 1945,
№ 7, с. 348—354. Г Г Воробьев.
БИОКАТАЛИЗАТОРЫ — см. Ферменты.
435
БИОМИЦИН — БИОХИМИЯ
436
БИОМИЦИН — см. Хлортетрациклин.
БИОТИНЫ — витамины группы В, производные
конденсированных имидазол-тиофановых кольцевых
систем. Известны 2 изомерных природных Б.: а-Б.
(I), выделенный из яичного желтка, и 0-Б.— вита-
мин Н (II), выделенный из молока и печени. По био-
логич. активности а- и f5-B. равнозначны; а-Б.
синтетически по получен; fJ-Б. получен синтети-
/СО
HN NH
нс — £н
,сн3
сн„
соон 3
нас сн—сн—сн
4S'
со
HNZ 4NH
НС — бн
H2(t <!:н(сн2)4соон
чески, исходя из L-цистеина, мезодибромянтарной к-ты
и др. а-Б. — бесцветные кристаллы; т. пл. 220°;
[а]р = + 51°. 0-Б. — бесцветные игольчатые кри-
сталлы; т. пл. 232—233° (с разл.); [а]^=-|-91о (0,1 н.
NaOH); растворим в воде, трудно растворим в спир-
тах, почти нерастворим в эфире и хлороформе. Бла-
годаря наличию 3 асимметрия, атомов углерода
0-Б. образует оптич. антиподы и рацематы различ-
ной пространственной конфигурации (эпибиотин, ал-
лобиотин, эпиаллобиотин). 0-Б. в р-рах устойчив
к действию разбавленных к-т и щелочей, а также к на-
греванию; при обработке конц. кислотами, щелочами
и р-рами перекиси водорода 0-Б. расщепляется.
0-Б. входит в состав ферментных систем, напр.
0-декарбоксилазы, и катализирует ряд реакций обмена
в-в в организме (0-декарбоксилирование многооснов-
ных к-т, биосинтез аспарагиновой к-ты и др.). При
недостаточности Б. в организме замедляется его рост,
поражаются кожные покровы, происходит выпадение
волос и др.
Лит. см. при ст. Витамины. В. М. Березовский.
БИОХИМИЯ (биологическая или физиологическая
химия) — наука о химич. составе организмов и их
составных частей и химич. процессах, протекающих
в организмах. Различают след, направления Б.:
1) Статическая Б., изучающая химич. при-
роду и свойства веществ, входящих в состав клеток
различных тканей и органов, межклеточных веществ,
секретов, экскретов и т. д. Это направление широко
использует методы органич. и аналитич. химии.
2) Динамическая Б., исследующая превра-
щения веществ в организме, начиная с момента по-
ступления в него питательных веществ вплоть до
образования выводимых из организма конечных про-
дуктов обмена. Основное содержание динамич. Б. со-
ставляет промежуточный (интермедиарный) обмен
веществ, связанный с обменом энергии. Промежуточ-
ный обмен (метаболизм) приводит, с одной стороны,
к переходу питательных веществ в вещества, являю-
щиеся составными химич. частями тела (ассимиляция,
анаболизм), а с другой — к распаду входящих в со-
став тела веществ до конечных продуктов обмена,
таких, как вода, углекислый газ, мочевина и т. д.
(диссимиляция, катаболизм). В ходе промежуточного
обмена процессы синтеза и распада веществ тесно свя-
заны друг с другом. 3) Функциональная Б.,
имеющая своей задачей вскрытие химич. основ функ-
циональной деятельности, напр. синтеза специфич.
веществ в клетках, выделения различных веществ
железами в ходе секреции, химич. механизма мышеч-
ного сокращения, нервного возбуждения и торможе-
ния, химич. механизма передачи наследственных
свойств и т. д. В этой области Б. происходит органич.
слияние задач и способов исследования морфологии
(изучение структуры), биохимии и биофизики (изуче-
ние химизма и физич. явлений) и физиологии (изуче-
ние функций).
В зависимости от специфики природы изучаемых
организмов Б. подразделяется на: 1) Б. живот-
ных, 2) Б. растений и 3) Б. микробов.
Это подразделение связано прежде всего с нахожде-
нием в растениях целых классов соединений, не встре-
чающихся или мало представленных в животных орга-
низмах (алкалоиды, гликозиды, терпены, смолы,
растительные пигменты и т. д.), а у микробов — с чрез-
вычайно широким кругом веществ (содержащих,
напр., D-аминокислоты, хлор, нитрогруппы, азо-
группы, четырехчленные гетероциклич. кольца и т. д.)
и процессов, не наблюдаемых в растительных и живот-
ных организмах. Растения и животные существенно
различаются способами ассимиляции. Растения син-
тезируют сложные органич. вещества из простых ве-
ществ неорганич. природы (вода, углекислый газ,
минеральные соли). Животный мир нуждается в по-
ступлении не только минеральных веществ, но и
сложных органич. веществ (белки, жиры, углеводы,
витамины). При синтезе веществ, входящих в состав
организма животного, используются сравнительно
низкомолекулярные продукты органич. природы,
напр. продукты, образующиеся при расщеплении
белков, жиров и углеводов (жирные к-ты, аминоки-
слоты, моносахариды), витамины и др. При этом ис-
пользуется энергия, необходимая для синтеза, обра-
зующаяся при процессах распада или окисления
органич. веществ. Синтетич. деятельность этого типа
обозначается как химиосинтез. Зеленые расте-
ния, способные строить свое тело из неорганич. ве-
ществ, обозначаются как аутотрофные орга-
низмы; животные организмы, нуждающиеся в введе-
нии с пищей органич. веществ, называют гетеро-
трофными. Среди микроорганизмов встречаются
аутотрофы и гетеротрофы.
Мир животных, растений и микроорганизмов ха-
рактеризуется чрезвычайным разнообразием веществ
и химич. реакций. Тем не менее имеется много общего
в основных химич. компонентах клеток (белки, нуклеи-
новые кислоты, жиры и липоиды, углеводы, витамины,
минеральные вещества), в ходе их превращений (бро-
жение, гликолиз, окислительные процессы), в агев-
тах, необходимых для протекания биохимических про-
цессов (ферменты, коферменты, активаторы, инги-
биторы), а также в отношении биохимических механиз-
мов роста, размножения и передачи наследственных
свойств (роль нуклеиновых кислот, стимуляторов
роста и др ). Эти вопросы составляют содержа-
ние так называемой общей Б., в которой изу-
чаются химич. закономерности для всех форм жиз-
ни и выясняются пути возникновения жизни и ее
развития.
Биохимич. реакции, лежащие в основе обмена
веществ и жизнедеятельности- организмов, как и
любые другие химич. реакции, полностью подчиняются
законам химии и физики, в частности законам сохра-
нения вещества, сохранения энергии и т. д. Однако
следует отметить и нек-рые особенности биохимич.
реакций. Большинство биохимич. реакций протекает
лишь при участии спец, катализаторов белковой
природы — ферментов (энзимов). Даже течение неко-
торых очень простых и идущих самопроизвольно со
значительной скоростью реакций (напр., Н2СО3 =
= Н2О -ф- СО2 или 2Н2О2 —> 2Н»О -ф- О2) чрезвычайно
сильно ускоряется соответствующими ферментами.
Для проявления активности ферментов требуется ряд
условий (определенная реакция среды, концентра-
ция электролитов и т. д.) и иногда наличие спец,
веществ, играющих роль коферментов или активато-
ров. Ферментативный характер большинства биохи-
мич. реакций придает им особую специфику и чувстви-
437
БИОХИМИЯ - БИТУМЫ
438
тельяость к изменениям состава среды. Биохимия,
превращения характеризуются также многоступен-
чатостью. Так, гликолиз, т. е. расщепление глюкозы
С„Н12ОВ до двух молекул молочной к-ты 2СзНсО3,
протекает не меньше чем через 12 промежуточных
этапов. То же самое относится к окислению высших
жирных к-т и к распаду аминокислот. На пути рас-
пада веществ встречаются реакции образования более
сложных молекул, и т. обр. реакции распада тесно
переплетаются с реакциями синтеза. Многоступенча-
тостью отличаются и процессы биохимия, синтеза.
Напр., стеариновая к-та СТ7Н35СООН или холестерин
С27Н45ОН синтезируются из уксусной к-ты через
десятки промежуточных этапов. Многоступенчатость
биохимия, превращений требует последовательного
включения ферментативных систем друг за другом.
Поэтому в биохимия, реакциях, лежащих в основе
функциональной деятельности клеток, непосредствен-
ное участие принимают различные клеточные микро-
скопия. и субмикроскопич. структуры. Биохимия,
процессы в живых организмах сопровождаются осво-
бождением химия, энергии. Однако, в отличие от
неорганизованных систем, в живых клетках только
часть освобождающейся химической энергии рассеи-
вается в виде тепла. Около половины ее аккумули-
руется в форме химической энергии некоторых со-
держащих фосфор или серу энергетических веществ.
Особенное значение при этом играет аденозинтри-
фосфорная к-та (см. Аденоаинфосфорные кислоты),
энергия гидролиза к-рой используется для осуще-
ствления синтезов, для выполнения мышечной рабо-
ты и т. п.
Ряд разделов Б. в силу большого их практич.
или теоретич. значения выделяется ныне в отдельные
научные дисциплины. Это относится к Б. витаминов,
гормонов, микробов, углеводов, антибиотиков, имму-
нохимии, химии фотосинтеза, технич. Б. нек-рых
растительных и животных продуктов. В особое на-
правление выделяется ныне гистохимия, или цито-
химия, сочетающая в себе химич. и гистоморфоло-
гич. методы изучения химич. состава и химич. изме-
нений в цитоплазме и органоидах клетки. Для меди-
цины приобрела большое значение клиниче-
ская Б., использующая химич. исследования крови,
мочи, тканей и т. д. для диагностич. целей. Интенсивно
развиваются Б. онтогенеза (эмбриохи-
мия) исравнителъная и эволюцион-
ная Б. Выясняется роль животных организмов на
земле в геология, изменениях земной коры (биогео-
химия). Многие вопросы Б (влияние на организм
лучистой энергии, возникновение в организме элект-
рич. явлений, изменения механич. свойств мышцы
при различных ее состояниях и т. д.) являются погра-
ничными между Б. и биофизикой. Биохимич.
методы исследования опираются на новейшие достиже-
ния физики и химии. Широко используются электро-
метрии., колориметрии, и спектрометрии, методы,
применение радиоактивных и тяжелых изотопов в ка-
честве меченых атомов, избирательная адсорбция,
хроматография, спец, микрометоды и ультрамикроме-
тоды, применение в качестве специфич. реагентов фер-
ментных препаратов и т. д. Биохимии, исследования
оказывают большое влияние на развитие биологии,
медицины, с. х-ва и т. д. Роль биохимии, методов
исследования в этих областях науки все время повы-
шается и приобретает все большее значение. Дости-
жения всех отраслей Б. широко используются и для
промышленных целей (получение гормонов, витами-
нов, антибиотиков, обработка кожи, технологиче-
ская переработка чая, табака, созревание вин, кон-
сервирование овощей и т. д.). В связи с этим воз-
никли различные области технической био-
химии.
Лит.: Энгельгардт В. А., Некоторые проблемы
современной биохимии. Доклад..., М., 1959; Владими-
ров Л. Е., Физиол. ж. СССР, 1957, 43, № 11, с. 1052; Сиса-
кян И. М., Биохимия обмена веществ, М., 1954 (обширная
библиогр.); Збарский Б. И., Иванов И. И., М ар-
да т е в С. Р., Биологическая химия, М., I960; Б о л д у-
ин Э., Основы динамической биохимии, пер. с англ., М.,
1949; Успехи биологической химии. Отв. ред. Б. И. Збарский
и В. Н. Орехович, т. 1 — 3, М., 1950—58; Гельман Н. С.
и Зенкевич Г. Д,, Биохимия растений. Библиографи-
ческий указатель отечественной литературы 1738—1952, М.,
1956: Annual review of biochemistry, v. 1- 27, Stanf. (California),
1932—58; Methods of biochemical analysis. Ed. by D. Glick,
v. 1—6. N. Y.—L.. 1954—58; Advances in enzymology and rela-
ted subjects of biochemistry, v. 1 — 20. N. Y., 1941 — 58; Advan-
ces in protein chemistry, v. 1 — 14. N. Y., 1944—59; Advances
in carbohydrate chemistry, v. 1 — 14, N. Y., 1945—59; H a u-
rowit* F., Progress in biochemistry since 1949, N. Y.—L.,
1959; Журналы: Биохимия (с 1936); Украшськпй б!ох1м. ж.
(с 1948); Вопр. мед. хим. (с 1955). Л. Е. Владимиров.
БИПОЛЯРНЫЕ СОЕДИНЕНИЯ (биполярные ио-
ны, Zwitterionen в нем. и нгл. литературе) — органич.
соединения, представляющие собой внутренние соли,
содержащие положительно и отрицательно заряжен-
ные атомы или группы. Типичным примером Б. с. мо-
гут служить' в-ва типа бетаина (CH3)3i\CH2COO~.
О солеобразном строении бетаина свидетельствуют
хорошая растворимость в воде, плохая — в обычных
органич. растворителях, высокая температура плав-
ления (293°). К Б. с. может быть отнесена сульф-
аниловая к-та и ряд других соединений.
Я. Ф. Комиссаров.
БИТУМЫ — общее название твердых и жидких
органич. веществ или продуктов пх' переработки,
растворимых в органич. растворителях (спирто-бен-
золе, хлороформе, сероуглероде и др.) и состоящих
из углеводородов, а часто из их кислородных, серни-
стых и азотистых производных. В технике под назва-
нием Б. обычно подразумеваются только те их разно-
видности, к-рые обладают твердой или вязкой конси-
стенцией. Различают природные и искусственные IS.
В генетич. отношении природные Б. представляют
составные части каустобиолитов, осадочных горных
пород и почв. Среди каустобиолитов различают Б. са-
пропелитов, гумитов (сланцевые, угольные, торфя-
ные) и нефтяные Б. Наибольшее промышленное зна-
чение имеют последние. В природных нефтяных Б.
при переходе от парафиновых Б. к асфальтам и ас-
фальтитам отношение С/Н возрастает обычно от 6—7
до И —12, а отношение С/ (О -|- S -|- N) уменьшается
прибл. с 800 до 50. Продукты дальнейшей карбони-
зации нефтяных Б., или так наз. пиробитумы, теряют
основной признак Б. — плавкость и способность рас-
тягиваться, и потому они помещаются вне класса
нефтяных Б., хотя и составляют с последними химико-
генетич. неразрывный ряд.
Месторождения нефтяных Б. подразделяются на
четыре типа: 1) образовавшиеся в результате про-
сачивания Б. на поверхность (асфальтовые озера и
др.); содержание чистого Б. в них может достигать
80—90%; 2) пластовые месторождения, образовав-
шиеся вследствие пропитывания Б пустот, песчани-
ков или известняков; содержат 3—10% Б..представ-
ляют промышленный интерес; 3) жильные месторо-
ждения, возникшие в результате проникновения Б.
в трещины горных пород, содержат Б., почти лишен-
ный минеральных примесей, имеют ограниченное рас-
пространение; 4) месторождения смешанного жильно-
пластового характера. Месторождения твердых Б.
(асфальтов и озокеритов) связаны с нефте- и газо-
носными областями (Азербайджан, Сев. Кавказ, Вто-
рое Баку, Средняя Азия, Предкарпатье, США, Куба,
Греция и др.).
Состав и свойства Б. В настоящее время принято
делить Б. на отдельные группы в-в только по раз-
личному отношению последних к растворителям.
Поэтому характеристика Б. основывается на их труп-
439
БИТУМЫ
440
повом составе. Наиболее распространенным методом
определения группового состава Б. является метод
Маркуссона. Этим методом установлено, что в состав
Б., кроме масел, смол, асфальтогеновых к-т и их
ангидридов, асфальтенов, входят карбены, карбоиды
и парафин. Карбены и карбоиды отличаются от ас-
фальтенов лишь более темной окраской и несколько^
повышенным содержанием кислорода. Они дают
аналогичные реакции с азотной и серной к-тами,
хлорным железом, сулемой и т. д. Характерно раз-
личие в их растворимости: карбены нерастворимы
в С С14, но легко растворимы в CS2. Карбоиды нерас-
творимы ни в каких обычно применяемых раствори-
телях. По мере перехода к более карбонизованным
членам ряда асфальтовых Б. в их составе изме-
няется содержание масел и смол и соответственно
возрастает содержание асфр'ьтенов, карбенов и кар-
бондов.
Искусственные Б. — вязко-жидкие пли мягкие и
пластичные либо твердые и хрупкие продукты пере-
работки нефти и каменного угля. Они, подобно при-
родным Б., состоят из масел, смол, асфальтенов, а
также карбенов и карбоидов, хотя колич. соотношение
их в Б. иное. Искусственные Б. отличаются от есте-
ственных значительно большим содержанием масел
и меньшим содержанием асфальтенов (см. табл. 1).
Таблица 1
Название породы или нефти, из к-рых получены битумы Асфаль- тены, % Смолы, о/ /о Масла, /О
Первомайский доломит 56,3 28,4 14,4
Стерлитамакский песчаник . . 69,4 23,4 7,7
Артемовская нефть 19,4 32,0 47,0
Уральская нефть 15,6 38,7 45,1
Мексиканская нефть 27,6 24,9 45,6
При одинаковых темп-pax размягчения искусствен-
ные Б., по сравнению с природными, содержат больше
летучих веществ и меньше карбенов и карбоидов
(не более 5%), а также вовсе не содержат асфальто-
геновых к-т и их ангидридов.
Б. представляют собой коллоидные системы, в к-рых
дисперсной средой являются масла и Смолы, а диспергирован-
ной фазой — асфальтены. В густых и твердых Б., где .мицеллы
нанимают большую часть всей системы, масла находятся
вследствие сольватации в связанном и неподвижном состоя-
нии, причем с повышением мол. весов смол, являющихся про-
межуточной средой, сольватация увеличивается. При нагре-
вании Б. и связанным с зтим разжижением его компонентов
отдельные части мицеллы постепенно переходят в растворен-
ное или пептизированное состояние. Смолы, растворяясь
в маслах, способствуют пептизации асфальтенов. Соотноше-
ние п состав фаз при этом меняются. Мицеллы Б. могут быть
агрегированы и пептизированы в различной степени и в зави-
симости от этого образуют коллоидные системы с разными
физич. свойствами (золи, звли— гели, гели). Для использования
Б. в технике большое значение имеют их реологич. свойства
(эластичность, вязкость, тиксотропия), которые определяются,
в первую очередь, структурой Б., а также их химич. приро-
дой. С увеличением количества асфальтенов пластичность Б.
возрастает. На пластичность Б. влияют и «мальтены», то есть
сумма содержания масел и смол и соотношение масел и смол
в мальтенах: чем больше масел, тем сильней выражена плас-
тичность. Эластичность Б. выявляется при достаточно боль-
шом содержании асфальтенов.
В технике Б. характеризуются: темп-рой размяг-
чения, пенетрацией (глубиной проникновения иглы),
дуктильностью (растяжимостью).
Таблица 2
Битум Темп-ра размягче- ния*, °C Пенетра- ция, мм** Дук- тильность см**
Шугуровский природный . . 42,2 5,7 100
Из бакинской нефти .... 56,0 5,3 42
Из мексиканской нефти . . 52,2 6,0 71
* Определена по методу «кольца и шара». '• При 25°.
В табл. 2 приведены некоторые свойства природ-
ных и искусственных Б.
Получение и применение технических Б. Природ-
ные Б. получают обработкой породы кипящей водой.
Как правило, такой операции подвергаются богатые
Б. песчаники; этот способ особенно часто приме-
няется для извлечения низкоплавкого Б. Песчаник
многократно вываривают в горячей воде, иногда под-
кисленной H2SO4, или обрабатывают сухим паром.
Вторым методом является экстракция с помощью
различных органич. растворителей. Основное про-
мышленное значение имеют искусственные Б. Сырьем
для их произ-ва служат мазуты, гудроны, крекинг-
остатки, экстракты от очистки масел селективными
растворителями, а также смолы полукоксования ка-
менного угля. Исходное сырье и способ произ-ва Б.
определяют их качество. Б. из парафинистых нефтей
сравнительно быстро теряют пластич. свойства вслед-
ствие кристаллизации парафина при пониженных тем-
пературах. Пластичность Б. может быть повышена по-
нижением их вязкости. Искусственные Б. получают:
окислением кислородом воздуха гудрона, крекинг-
остатков или экстрактов, если эти остатки по свой-
ствам не являются готовыми Б., полученными глубо-
ким отгоном масляных фракций из гудрона. Послед-
ним способом при помощи глубокого вакуума и пере-
гретого пара получают т. наз. остаточный Б. Гудрон,
или остаточный Б., окисляют продувкой воздуха при
высоких темп-рах (260—280°). В результате происхо-
дящих при этом реакций окисления и конденсации
нек-рая часть углеводородов масел переходит в смолы,
к-рые, в свою очередь, превращаются в асфальтены.
Чем глубже процессы окисления и конденсации, тем
больше образуется смол и асфальтенов. Однако слиш-
ком глубокое окисление или разложение может вы-
звать образование нежелательного количества карбе-
нов и карбоидов. При использовании для получения Б.
крекинг-остатков продувку воздухом обычно ведут
одновременно с продувкой паром. Качество Б., полу-
ченных из такого сырья, обычно несколько хуже, чем
Б., полученных из остатков после прямой перегонки
нефти. Б., получаемые окислением, более эластичны
и термостойки, чем остаточные. Крекинг-битумы полу-
чаются путем перегонки под вакуумом крекинг-остат-
ков. Эти Б. имеют более высокое содержание асфаль-
тенов, чем указанные выше; это придает им повышен-
ную твердость, темп-ру размягчения, большую растя-
жимость при 25°.
Нефтяная пром-сть вырабатывает широкий ассорти-
мент Б. В технике, особенно в строительстве, приме-
няются твердые, полутвердые и жидкие Б., гл. обр.
нефтяные. По степени твердости, пластичности и
темп-ре размягчения Б. (как строительный материал)
различаются марками. Твердые нефтяные Б. приме-
няются в произ-ве рулонных кровельных материалов
(покровный слой рубероида) и приклеивающих битум-
ных мастик. Полутвердые Б. находят наиболее широ-
кое применение: в произ-ве кровельных материалов
(руберойд, пергамин), гидроизоляционных материа-
лов (гидроизол, металлоизол, борулин, битумини-
рованные ткани), приклеивающих мастик, покрасоч-
ных составов, гидроизоляционных обмазок, для при-
готовления асфальтовых бетонов, строительных ас-
фальтовых р-ров, асфальтов и битумных эмульсий,
в производстве пластических масс (см. Пластики
битумные). В табл. 3 приведены нек-рые свойства
твердых и полутвердых битумов.
Для производства лаков применяют Б. с высокой
темп-рой размягчения (100—135°), малой пенетра-
цией (0,5 мм при 25°) и высокой температурой вспыш-
ки (260°).
Жидкие Б. применяются для дорожного строитель-
ства в холодном виде; вследствие частичного испаре-
441
БИТУМЫ ТВЕРДОГО ТОПЛИВА
442
ния, окисления, адсорбции каменными материалами
эти Б. увеличивают свою вязкость, что ведет к при-
обретению необходимой прочности (затвердеванию)
дорожных покрытий.
Таблица 3
Битумы Темп-ра размягче- ния, °C* Пенетр а- ция, мм** Дуктиль- ность, мм** Т. ВСПЫШ- КИ, °C
Твердые Полутвердые . . 60-90 25—50 0,5—4,0 4,1-20 1-3 40-60 230 180-200
• Определено по методу «кольца и шара». ** При 2 5°.
Жидкие Б. имеют, в зависимости от сорта, темп-ру
вспышки 65—120°; должно перегоняться до 225°
не более 10%, до 360° — не более 50%; остаток после
отбора фракций до 360° имеет пенетрацию 10,0—
30,0 мм (25°) и дуктильность 60 см (25°); вязкость
жидких Б. при истечении через отверстие диаметром
5 мм меняется от 5 до 200 сек (60°).
Лит.: Добрянский А. Ф., Геохимия Нефти, Л. — М.,
194b; Геохимический сб. [№] 1, [отв. ред. В. А. Успенский],
Л. — М., 1949; Крейцер Г. Д., Асфальты, битумы и пеки,
3 изд., М„ 1952; Михайлов В. В., Нефтяные дорожные
битумы, М„ 1949; Наметкин С. С., Собр. тр., т. 3, М.,
1955. П. Н. Галич.
БИТУМЫ ТВЕРДОГО ТОПЛИВА — продукты,
извлекаемые из торфа, бурых и каменных углей ор-
ганич. растворителями. Практич. значение получили
битумы бурых углей и торфа. Б. т. т. подразделяются
на битум А, извлекаемый холодными или кипящими
органич. растворителями, битум В, извлекаемый
органич. растворителями под давлением при повышен-
ной темп-ре (обычно выше 200°) и битум С,
извлекаемый оргапич. растворителями из
топлива, освобожденного от битума А и
обработанного затем соляной или серной
к-той. Главные составные части Б. т. т. —
восковая и смоляная; первая состоит из
алифатич. высокомолекулярных к-т и спир-
тов, эфиров этих к-т и спиртов и неомы-
ляемой углеводородной части; смоляная
часть состоит из углеводородов и оксисо-
единений. Углеводороды представлены аро-
матическими, непредельными циклич. сое-
динениями и частично углеводородами ме-
танового ряда.
Таблица 1. Характеристика компонентов смоляной части
торфяных битумов
Наименование Выход на смолистые в-ва, .% Ки- слот- ное число Иод- ное число С, % н,% 4.0 О'- й + о
Смолистые веще- ства битума . . 100,0 24,9 53,5 77,2 10,4 12,4
В том числе ком- поненты: кислые .... 7,2 114,6 40,4 68,2 8,6 23,2
омыляемые . . 7,4 119,0 40,7 70,2 9,3 20,5
неомыляемые 84,8 2,0 51,5 80,0 11.02 9,00
В буроугольных битумах содержится до
20% высокомолекулярных к-т, т. пл. 75—90°, ок.
60% эфиров высокомолекулярных спиртов и к-т
и 3—5% спиртов восковой части. Неомыляемая
часть, составляющая ок. 10% битумов, содержит угле-
водороды и кетоны. Смолы буроугольных битумов
составляют 10—15% и содержат абиетиновую ки-
слоту, битулин и др. В торфяных битумах со-
держатся высокомолекулярные жирные кислоты
(С25Н51СООН, С^ЩьСООН и др.), спирты (С27Н53ОН
и С30Нв1ОН), высшие парафиновые углеводороды,
напр. С33Нв8, С35Н72 и сесквитерпены С15Н24, а из
смоляных к-т — абиетиновая, составляющая боль-
шую часть канифоли. Смоляная часть торфяных биту-
мов (табл. 1), извлеченных бензином, составляет от 40
до 55% от их общего выхода.
Наиболее ценными экстрагируемыми продуктами
являются воски. Входящие в состав Б. т. т. различ-
ные вещества имеют неодинаковую растворимость
в органич. растворителях (табл. 2).
Таблица 2. Выход битумов при экстракции
верхового торфа (степень разложения 35%)
различными растворителями
Р астворитель
Выход биту-
ма на абсо-
лютно сухой
торф, %
Серный эфир ................
Бензин .....................
Бензол......................
Дихлорэтан..................
Спирт-бензол (1 ; 1 по объему)
3,5
5,0
8,0
12,0
10,5
Бензин растворяет восковые компоненты Б. т. т. и
преим. углеводородную часть смол. Бензол дополни-
тельно извлекает ароматическую, в том числе и
окисленную часть смол. Дихлорэтан и спирто-бензол
растворяют еще больше циклических, окисленных
высокомолекулярных соединений, содержащих азот
и серу. В табл. 3 приводятся данные о выходах и
качестве Б. т. т., получаемых .при экстракции бурых
углей различными растворителями.
Таблица 3. Влияние природы растворителей на выходы и качество
буроугольного битума
Растворитель Выход битума на органич. массу, % Извлечено битума (к макс, ана- литич. со- держанию), о/ /о Содержание Ки- слот- ное число Темп-р а капле- п аде- нин, °C
вос- ков, о/ /0 смол, о/ /о
Ацетон . 9,7 43,8 30,2 69,8 55,1 149
Бензин 9,8 44,2 90,3 9,7 21,1 86,4
Дихлорэтан . . . 14,5 64,9 76,4 23,6 31,1 87,8
Спирт-бензол . . 20,0 89,3 68,3 31,7 41,3 88,8
Извлечение битумов из твердого топлива произ-
водится преим. в периодически действующих экстрак-
торах, объединенных в батареи. Экстракцию бурого
угля производят бензолом или спирто-бензолом, а
экстракцию битуминозного торфа — бензином. Выход
битумов в количестве 8—10% является нижним пре-
делом, допускающим их рентабельное выделение.
Процесс получения Б. т. т. из бурого угля включает:
подготовку угля к экстрагированию; экстракцию ор-
ганич. растворителем; отгонку от экстракта главной
массы растворителя; освобождение от растворителя и
воды; сплавление и разливка в формы.
Скорость и полнота извлечения битумов зависят
от состава растворителя, влажности и размеров ку-
сков топлива, темп-ры и давления, при к-рых прово-
дят процесс. Уменьшение содержания смолистой
части Б. т. т., являющейся балластом (в сыром буро-
угольном битуме до 40%, в торфяном битуме 50% и
выше), может быть достигнуто перегонкой Б. т. т.
в мягких условиях (напр., под вакуумом с примене-
нием перегретого пара) или рафинированием обессмо-
ленных Б. т. т. путем их обработки СгО3, КМпО4
и др. окислителями. Дистилляция Б. т. т. приводит
к значительным потерям восков. При обработке Б. т. т.
сильными окислителями смолистые соединения (ки-
слородосодержащие и непредельные) легко окисляются.
443
БИУРЕТ — БИЦИЛЛИН
444
Образующиесн при этом значительные количества
высших карбоновых к-т ограничивают возможность
применении воска в нек-рых произ-вах.
На рисунке представлена технологи1!, схема экстракции
Сптума из бурого угля смесью спирт-бензол. Уголь конвей-
ером подается в бункер 1 и оттуда в экстракционный аппарат
2, куда поступает и смесь спирт-бензол. Образующийся эк-
стракт отводится в испаритель 3 для отгонки растворителя,
пары к-рого обогревают кожух экстракционного аппарата
и частично конденсируются. Несконденсированные пары
растворителя поступают в холодильник 4; конденсат стекает
в отстойник 5. Битумы из испарителя сливают в ванну 6.
Конденсат — смесь бензола и обводненного спирта — подается
в теплообменник 7, откуда подогретый обводненный спирт
поступает в ректификационную колонну 8; пары спирта
попадают в холодильник 9, а сконденсировавшийся спирт —
в сборники Ю—и. После отгонки растворителя уголь выгру-
жается в бункер 12.
Б. т. т. широко применяются в кабельной, электро-
технической, кожевенной, полиграфической, бумаж-
ной, текстильной, лакокрасочной промышленности,
в литейном производстве и др. отраслях народного
хозяйства.
Лит.: Раковский В. Е., Общая химическая техно-
логия торфа, М.—Л., 1949; Изучение и комплексная перера-
ботка смол и битумов бурых углей Днепровского бассейна,
ч. 2, Киев, 1958; VS е I a k V., Chemie and Technologie des
Montanwachses, Praha, 1959 В. E. Раковский
БИУРЕТ (амид аллофановой кислоты, карбамил-
мочевина, уреидоформамид) H2NCOi\HCONH2, мол. в.
103,13 — гигросконич. кристаллы; т. пл. 192,5—193°
(с разл.); гидрат (0,8 Н2О) при 110° теряет кристал-
лизационную воду, растворимость в воде сильно зави-
сит от темп-ры (1,54 г в 100 мл Н2О при 15° и 45,5 г
при 100°); в эфире мало растворим. Безводный Б. по-
лучают кристаллизацией из спирта; Б. амфотерен;
слабые кислые свойства выражаются в образовании
с амидом калия KNH2 в жидком аммиаке моно- и
трикалиевых солей, к-рые водой немедленно разла-
гаются. При нагревании Б. в отсутствие воды до 160°
и выше происходит выделение NH3 и получается циа-
нуровая кислота, при более высокой темп-ре из Б. по-
лучается меламин; при гидролизе Б. кислотами или
щелочами образуются: мочевина CO(NH2)a, циановая
HOCN и циануровая (HOCN)3 к-ты, гуанидин HN=
= С (NH2)2, NH3 и СО2. Спирты разлагают Б. с обра-
зованием аллофанатов и аммиака: (H2NCO)2NH +
+ ROH -> H2NCONHCOOR + NH3; альдегиды и
кетоны дают продукты конденсации, например:
(H2NCO)2NH+ 2110110-^ (HOCH2NHCO)2NH; глав-
ным продуктом реакции с гидразином нвляетсн уразол:
(HaNCO)2NH + H2NNHa — ОС----------МН
HN СО
'nh
Конденсация Б. с циановой к-той дает циануровую
к-ту; она же получается при взаимодействии Б.
с мочевиной. Основным методом получения Б. яв-
ляется нагревание мочевины до темп-ры 150—170°:
2H3NCONH2—> (HaNCO)2NH + NH3. Б. образует ряд
производных, в которых замещен атом водорода
в аминогруппе, например алкйлнитрозобиуреты
H3NCONHCONRNO, диаминобиурет (H2NNHCO)2NH
и др.; известны также тиобиуреты, напр. тиобиурет
H2NCSNHCONH2 и дитиобиурет (H2NCS)2 NH. С рас-
творами солей Сн2+ (обычно CuSO4) в присутствии
щелочи Б. дает биуретовую реакцию. С ионами К,
Na, Cd, Си, Hg и Ni Б. образует окрашенные ком-
плексы. В смеси с мочевиной Б. используется как
вспенивающий агент в произ-ве губчатой резины
(выделение NH3 при нагревании). Б. обладает сла-
бым диуретическим действием, понижает кровяное
давление.
Лит.: К u г z е г F., Chem. Rev., 1956, 56, № 1, р. 95.
БИУРЕТОВАЯ РЕАКЦИЯ — цветная реакция,
к-рую дают с солями меди в щелочной среде биурет,
амиды и имиды к-т, белки, полипептиды и некото-
рые др. соединения.
НО Н Н ОН
Б. р. обусловлена на-
личием в молекулах
перечисленных соеди-
нений — СО—NH —
группировки. Обра-
зующиеся при Б. р.
окрашенные соедине-
нии являются ком-
Н Н
I I
ZCO-NX /N-CO4
HN Си NH
4C=N' 'N=C/
I I I I
OH H O’
2Me+
нлексами, которым
приписывают строе-
ние, представленное,
например, формула-
ми (I) — медный ком-
R CO-CHR
I I I
СН—N N-
—СО
2 Me4
N-CHCO-
—N
I
RCH-COR
III
биуретовых комплексов
илекс из амида сали-
циловой к-ты, (II) —
комплекс из биурета L
и (III) — комплекс из
полипептида. Для медных
каждого соединения характерна окраска (красная, фис-
летовая, синяя) с максимумом поглощения в интервале
440—640 ммк. К медным биуретовым комплексам
близки по строению никелевые биуретовые комплексы,
окрашенные в желтый цвет. Б. р. используется как
цветная реакция на белок и лежит в основе количеетвен-
ных колориметрических определений. Обычно реакцию
на белок проводят, прибавляя к его раствору в NaOH
разб. раствор CuSO4; при этом появляется фиолетовое
окрашивание. Чувствительность реакции невысока.
Лит.: Гауровитц Ф_, Химия и биология белков,
пер. с англ., М.—Л., 1953, с. 17; К о р е н м а н И. М_, Ж.
аналит. хим., 1948, 3, вып. 1, с. 52; Kurzer F., Chem.
Rev., 1956, 56, № 1, р. 181.
БИЦИЛЛИН (N, N'-дибензилэтилендиаминовая
соль бензилпенициллина, дипепициллин) — белый
кристаллич. порошок, т. разл. 115—117°; =
445
БИШЛЕРА РЕАКЦИЯ - БИШЛЕРА— НАПИРАЛЬСКОГО РЕАКЦИЯ
446
= + 185° (в метаноле); легко растворим в форма-
миде, этиленгликоле, пиридине; растворим в мета-
ноле, этаноле, ацетоне; трудно растворим в воде, эфире,
/S\
R-CONHCH-НС С(СН3)2
СО — N---СНСООН NHCH2CeH3
zsx (СН2)2 4Н2О
R—CONHCH—НС С(СН3)2 |
СО — N---СНСООН-NHCH2CeHs
R=CeH5CHa
циклогексане. Соль разрушается при действии креп-
ких кислот, щелочей и пенициллиназы. Б. получают
взаимодействием водного р-ра 2 молей калиевой или
натриевой соли пенициллина с 1 молем кислой соли
1\,Н'-дибензилэтилендиамина. Б. применяют в каче-
стве лечебного препарата пенициллина длительного
действия для профилактики и лечения заболеваний,
вызванных возбудителями, чувствительными к пени-
циллину.
Лит.: Ермольева П. В. [и др.]. Антибиотики, 1956,
1, № 1, с. 37;EliasW.,Price А. Н., Мегг i о n Н. J.,
Antibiot, and Chemotherapy, 1951, 1, p. 491.
БИШЛЕРА РЕАКЦИЯ — получение производных
индола при действии ароматич. аминов в присутствии
их солей на а-галогено-, а-окси- или а-ариламино-
кетоны: *
0=С- R С6Н5NH2 О=с---R C6H5NHa-HX
X-CH-R' CgH3NH- СН-R'
В случае а-галогено- или а-оксикетонов первой ста-
дией является образование а-ариламинокетонов. Из
последних получаются затем, по-видимому, арил-
аминоанилы (I), отщепляющие ароматич. амин с обра-
зованием неустойчивых индоленинов (II), изомери-
зующихся в индолы (III):
р_(2=О R—C=NCeH3 ___
r-ch-nhc6h3+ CeHsNH2 НХ r-ch-nhc6h5
Б. р. обычно приводит к смеси изомерных индолов,
что объясняется прежде всего тем, что ариламино-
анилы находятся в таутомерном равновесии с енди-
аминами (IV). В силу этого возможен переход к изо-
мерным ариламиноанилам (V) и образование изомер-
ного индола (VI);
R —C=NCeH3 _____R —С —NHCeH3
R-CH-NHCeH3 " R-С-NHCgH3
I IV
R-CH-NHCgHg
R-C=NC6HS
В ряде случаев отмечена изомеризация а-ариламино-
кетонов, катализируемая солями ароматич. аминов:
R1—СН—Br R1—СН—NHC,H6 R’—С=О
I —► I -i I
R”_С=о R”—С=О R"—СН—NHC.H.
Показано также, что имеет место обмен остатками
ариламина между молекулами ариламинокетона и
соли ароматич. амина. Предложены и другие меха-
низмы Б. р., однако изложенный выше модифициро-
ванный механизм Бишлера, включающий образование
ендиамина, наиболее вероятен. Б. р. применяется
для синтеза различных индолов, замещенных в поло-
жениях 2 и 3, в особенности этот метод удобен для син-
теза 2-фенилиндолов и индолов с одинаковыми заме-
стителями в положениях 2 и 3.
В Б. р. были применены различные замещенные
в бензольном ядре: фенацилгалогениды, бензоины,
алифатич. а-галогенкетоны, бромлевулиновая к-та
и др. При использовании различных замещенных ани-
линов получают индолы, содержащие заместители
в бензольном ядре индольного цикла: галоген-, ацил-
амино-, алкил-, алкокси- и др. Пара- (соответственно
орто-)замещенные анилины образуют 5-(соответст-
венно 7-)замещенные индолы. Мета-замещенные ани-
лины дают смесь 4- и 6-производных индолов. Б. р. не
дает возможности получить индолы, не замещенные
в пиррольном кольце, в том числе сам индол. Б. р.
обычно проводят, нагревая смесь а-замещенного ке-
тона и ариламина до 180—200°. Во многих случаях
выходы достигают 80—90%.
Весьма близок к Б. р. синтез индолов из а-арилами-
нокетонов при нагревании с хлористым цинком:
Сй О=С-СН3 _________-----------п-СН3
Ч Х.НС6Н6 ZnClg J-ceH6
N N
СНд CH3
Здесь имеет место прямая циклизация и образуются
другие изомеры индолов, чем в Б. р. Образование
индолов из а-замещенных кетонов и ароматич. ами-
нов впервые наблюдалось Р. Мёлау в 1881; А. Биш-
лер в 1892 предложил механизм и широко исследовал
эту реакцию.
Лит..- Джулиен П., Мейер Э., Принти э., в
кн. : Гетероциклические соединения, под ред. Р. Эльдерфилда,
т. 3, М., 1954, с. 17, 70; Sumpter W., М i 11 е г F., Heterocy-
clic compounds with indole and carbazole systems, N. Y. — L.,
1954,p. 12; W e у g a n d F., Richter E., Chem., Ber., 1955,
88, 499. M. H. Преображенская.
БИШЛЕРА — НАПИРАЛЬСКОГО РЕАКЦИЯ —
один из основных методов синтеза 3,4-дигидроизохино-
линов циклодегидратацией ацильных производных
0-фенилэтиламина. Вероятно, 0-фенилэтиламиды (I) при
этом реагируют в таутомерной форме (1а). Б. — Н. р.
обычно проводят в инертном растворителе, напри-
мер толуоле, ксилоле, тетралине, с применением
РаО5, РС15, РОС13, полифосфорной к-ты в качестве
водоотнимающих средств. Реакция носит довольно
общий характер, дигидроизохинолины (II) обычно
образуются с вполне удовлетворительными выходами
при различных В в (I), с разными заместителями
в бензольном ядре, при замене последнего другими
ароматич. радикалами. Б. — Н. р. может рассматри-
ваться как внутримолекулярное электрофильное за-
мещение в ароматич. ядре; этим объясняется, почему
в случае (I) с сравнительно мало активным незаме-
щенным бензольным ядром наблюдается пониженный
выход (II), почему при циклизации метоксизамещен-
ных (I) выход (II) повышается.
От дигидроизохинолинов (II) можно легко перейти
к изохинолинам (Ш), поэтому Б. — Н. р. представ-
447
БЛАНА РЕАКЦИЯ — БЛОК-СОПОЛИМЕРИЗАЦИЯ
448
ляет удобный путь синтеза различных изохинолинов.
Дегидрирование (II) во вполне ароматич. изохино-
лины (III) легко протекает при действии окислителей
[иод, KMnO4, PdCl2, (CH3COO)2Hg и др.], а также
при помощи катализаторов скелетного Ni, Pt, Pd
на асбесте или в виде черни и др.
Реакция открыта в 1893 Бишлером и Напиральским
и широко используется на практике. В частности, по
этому методу был синтезирован папаверин и недавно
(1950) ряд его гомологов.
Лит.: Гейслер В., в кн.: Гетероциклические соеди-
нения, под ред. Р. Эльдерфилда, пер. с англ., т. 4, М., 1955,
с. 264; У э л и В. М. иГовиндачари Т. Р., в кн.:
Органические реакции, сб. 6, пер. с англ., М., 1953, с. 98.
Я. Ф. Комиссаров.
БЛАНА РЕАКЦИЯ (хлорметилирования реак-
ция) — замещение водорода ароматич. ядра хлорме-
тильной группой СН2С1:
С0Нв + СН2О + HCI Zn-G1^ CeH5CHsCl + Н2О
В качестве катализатора чаще всего служат ZnCl2,
H2SO4 или А1С13; в ряде случаев применение SnCl4
приводит к более высокому выходу. Для хлорметили-
рования обычно применяют смесь соляной к-ты и
формальдегида в виде 26—40%-ного формалина.
Аналогично формальдегиду реагирует и паральдегид:
C0HSOCH3 + СН3СНО + НС1 п-СН3ОС0Н4СНС1СН3 -I- Н2О
Продукты реакции легко отщепляют HCI, образуя
винильные производные. Скорость хлорметилирования
производных бензола действием хлорметилового эфира
в отсутствии катализаторов увеличивается под влия-
нием заместителей СН3, С2Н6, С3Н7, ОСН3, ОС2Н6
и уменьшается, если заместителями являются С1,
Вг, I, СН2С1, СООН, NO2.
Хлорметильная группа является сравнительно сла-
бым ориентантом первого рода (см. Ориентации пра-
вило). Хлорметилирование бензола дает гл. обр.
n-ксилилендихлорид; вступление второй хлорметиль-
ной группы в толуол, анизол, п- и л<-ксилол происхо-
дит в мета-положение по отношению к первой
СН2С1-группе. Хлорметилирование нафталина про-
текает в присутствии ледяной уксусной к-ты и Н3РО4.
Фенолы реагируют слишком энергично и реакция
часто приводит к полимеризации (образование ди-
арилметанов). Поэтому ОН-группу предварительно
защищают обработкой, напр. С1СООС2Н6. Полимер-
ные продукты, содержащие ароматич. ядро, также
вступают в Б. р., напр. при обработке полистирола
хлорэтиловым эфиром в присутствии ZnCl2 в среде
диоксана удается ввести в полимер группы СН2С1.
Возможно также хлорметилирование производных
фурана, напр.:
БЛАНФИКС — см. Пигменты белые.
БЛОК-СОПОЛИМЕРИЗАЦИЯ (и блок-сополикон-
денсация) — процесс синтеза высокомолекулярных
соединений из гомополимерных веществ с сравни-
тельно низкой степенью полимеризации (олиго-
меров) или из смесей олигомеров и мономеров.
Б.-с. — один из видов сополимеризации
и химич. превращений полимерных соеди-
| । нений, приводящих к изменению степени
полимеризации. В макромолекулах блок-
сополимеров длинные, монотонно построен-
w R ные (гомополимерные) участки из звень-
ев одного мономера (блоки) чередуются
с такими же блоками звеньев другого мономера.
Получаемые при соединении разнородных блоков
блок-сополимеры сочетают свойства участвующих в
реакции полимеров и мономеров. Блок-сополимеры
следует рассматривать как изомеры веществ, полу-
чаемых при взаимодействии разнородных мономе-
ров в процессах сополимеризации. Получены блок-
сополимеры полистирола и натурального каучука,
полистирола и полиметилметакрилата, полистирола
и полиакрилонитрила и мн. др.
При Б.-с. происходит взаимодействие концевых
групп или активных центров олигомеров или при их
реагировании с мономерами;
X - (AAA . . . AA) _ X 4-Y - (BBB . . . BB) — Y ->
— ---WW (AAA . . . AA) - (BBB . . . BB) -WV- . . . 4- nXY
• • • —AAAr-AAA 4-иМ ->. . . —VW-AAA—MM . . . MM—VW-. . .
где: A, В — звенья цепи олигомера; X, Y — концевые
группы; М — молекула мономера. Активными цент-
рами олигомерных блоков могут являться атомы, со-
держащие нечетное число неспаренных электронов
или группировки, способные образовывать радикалы,
а также группы, могущие участвовать в реакции поли-
конденсации или ступенчатой полимеризации. Б.-с.
может протекать по радикальному (1, 2), молекуляр-
ному или ионному (3) механизмам и, в частности, по
механизму ступенчатой полимеризации (4). Приме-
рами Б.-с. могут служить:
1) Сополимеризация теломеров (см. Теломеризация),
содержащих по концам цепи атомы галогена (С1, Вг
или J), при действии УФ-лучей:
Вг (СН3СНХ%СВг3-Вг (CHaCHX^CBrs-j1’^0?-
— Вг (СН2СНХ)пСВг2 - (CH2CHG)-A/W-....
2) Взаимодействие макрорадикалов, образующихся
при пластикации, дроблении, ультразвуковой деструк-
ции, замораживании и других интенсивных меха-
нич. воздействиях (см. Механохимия полимеров) на
смеси разнородных полимеров в твердой фазе или
р-рах, напр.
. . . -VW-CH.CH 4- СН2 - СН—VW— . • • +
— . . . -WW- сн3 - СН — СН2—СН -А/w- . ..
Аналогично можно осуществить реакции бромметили-
рования и иодметилирования. Способ бромметилирова-
ния является достаточно общим, но выходы обычно
ниже, чем при хлорметилировании. Впервые реакция
осуществлена Грасси и Мазелли в 1898; современная
модификация предложена Бланом в 1923.
Лит.: Фюзон Р.К., Мак-Кивер К.Г.,веб.: Орга-
нические реакции, общ. ред. Р. Адамс, пер. с англ., т. 1,М.,
1948,с.84; J о h n е s G. D., Industr. and Engng. Chem., 1952,
44, № 11, p. 2686. H. С. Кочеткова.
3) Присоединение друг к другу макромолекул двух
различных линейных полимеров, на концах цепей
к-рых находятся функциональные группы, напр.
поликонденс ация:
НО (СН3СН,О)лН + носн=сн3он +
+ СНаОСО - CeHs - соосн, -
- HO (CH2CH2O)n (COCeH5COOCH2CH2O)mH -I- сн3он
449
БОЙЛЯ-МАРИОТТА ЗАКОН — БОНА—ШМИДТА РЕАКЦИЯ
450
4) Ступенчатая (миграционная) полимеризация, на-
пример:
HS-(-CHaCH2—SS —)n— CH2CH2SH +
/°\ /°\
4- СН2—CH-Rm - СН-СНа —>
_>^/V-S-[(CH2)2SS]ri(CH;.)2SCH2CHOHRmCHOHCH:.-AAV-,
В тех случаях, когда участвующий в реакции
«'блок» содержит способные к цепной полимеризации
двойные связи (напр., полиэфиракрилаты), образуются
трехмерные блок-сополимеры.
Б.-с. можно получать и такие сополимеры, к-рые
не синтезируются из смеси мономеров, этим методом
удается сочетать блоки аморфных полимеров с бло-
ками полимеров, обладающих высокой степенью кри-
сталличности и т. п.
Лит.: Праведников А. Н., Липатов 10. С.,
Методы получения и свойства привитых и блои-полимеров,
М., 1958; Берлин А. А., в сб.: Успехи химии и технологии
полимеров, [№] 2, М., 1957, с. 38; Бильмейер Ф.,
Введение в химию и технологию полимеров, пер. с англ.,
М., 1958. . А. А. Берлин.
БОЙЛЯ—МАРИОТТА ЗАКОН — один из основ-
ных газовых законов, согласно к-рому при постоян-
ной темп-ре объем v данной массы газа обратно про-
порционален давлению
р, под которым газ нахо-
дится: pv — с = const.
Постоянная с пропор-
циональна абс. темп-ре
и числу грамм-молекул
газа. Согласно Б.— М. з.,
зависимость р от v в пря-
моугольной системе ко-
ординат графически вы-
ражается семейством рав-
носторонних гипербол,
ветви которых асимпто-
тически приближаются к
осям, на которых отло-
жены величины р и V.
Эти кривые наз. изотер-
мами, т. к. каждая из них относится к определенной по-
стоянной темп-ре Т (см. рис.). Б.—М. з. впервые уста-
новлен Р. Бойлем (1862), независимо открыт Э. Мариот-
том (1676). Б. — М. з. характе-
ризует свойства т. н. идеального
газа, но практически довольно
хорошо применим и к обычным
газам при невысоких давле-
ниях.
БОЛОТНЫЙ ГАЗ—газ, вы-
деляющийся со дна стоячих
водоемов. В болотном иле, без
доступа воздуха под влиянием
анаэробных бактерий происхо-
дит брожение растительных
остатков (гл. обр. клетчатки),
в результате к-рого образуется
метан СН4 и небольшие коли-
чества СО2 и N2. Горючесть
этого газа в 1776 впервые опи-
сал Вольта, он же доказал
отличие его от водорода.
1'. М. Цигуро.
БОЛЬЦМАНА ЗАКОН РАС-
ПРЕДЕЛЕНИЯ'— см. Максвел-
ла — Больцмана закон распре-
деления.
v—р-изотермы идеального газа
(Т t<zT 2СТ „СТ J
БОЛЬЦМАНА ПОСТОЯННАЯ — одна из основных
физич. постоянных, равная k — R/N, где Я — универ-
сальная газовая постоянная, N — Авогадро число. По
своему физич. смыслу к — универсальная газовая
постоянная, отнесенная к одной молекуле.
А = (1,38044 ± 0,00007) 10 »» эрг град' =
= (8,6167 +: 0,0004) • 10 5 эвград'-,
= (11605,4 ± 0,5) град эв "1.
Численное значение Б. п. не зависит от принимае-
мой шкалы атомных весов.
БОНА — ШМИДТА РЕАКЦИЯ — введение гидро-
ксильных групп в антрахиноновое ядро окислением
в присутствии серной к-ты. Реакция обычно приме-
няется для получения нолиоксиантрахинонов, содер-
жащих от 4 до 6 гидроксильных групп; могут полу-
чаться и менее гидроксилированные продукты. Оки-
слителями в Б.—Ш. р. могут служить серный ангид-
рид (в виде высокопроцентного олеума), двуокись
марганца, азотистая к-та (в виде нитрозилсерной
к-ты), двуокись свинца, мышьяковая к-та и т. п. Как
правило, для успешного окисления необходимы ката-
лизаторы, напр. селен или ртуть, но часто бывает
достаточно уже тех количеств катализирующих при-
месей, к-рые имеются в технич. серной к-те. Иногда
гидроксилирование производят в присутствии бор-
ной к-ты. Последняя, образуя сложные эфиры с ок-
сиантрахинонами, делает их более устойчивыми к оки-
слению, благодаря чему реакцию можно остановить
на требуемой стадии.
Обычно Б.—Ш. р. проводят при темп-ре не выше
60°, однако, когда окисляют в конц. серной к-те без
добавления других окислителей, требуется более
высокая темп-ра (200—300°). Часто в результате
Б.—Ш. р. получаются не полиоксиаптрахиноны,
а соответствующие им эфиры минеральных к-т—
серной и борной; эти эфиры омыляются нагреванием
с 70—80%-ной серной к-той. Такая же обработка
позволяет отщепить сульфогруппу, если одновремен-
но с Б.—Ш. р. произошло сульфирование антрахино-
нового ядра. При использовании двуокиси марганца
в качестве окислителя часто получаются гидроксили-
рованные антрадихиноны, к-рые затем восстанавли-
ваются в полиоксиантрахиноны сернистым ангидри-
дом. Механизм Б.—Ш. р. не выяснен. G помощью
Б.—Ш. р. получают ряд технически важных поли-
оксиантрахинонов, используемых в качестве протрав-
ных красителей и полупродуктов для синтеза кубовых
и суспензионных красителей: хинализарина (I), али-
зарин цианина Р (II), ализаринового яркого бордо
Р (III) и др. Б.—Ш. р. открыта почти одновременно
(1888—90) и независимо друг от друга Р. Боном и
Р. Э. Шмидтом.
8 к. х, э. т. 1
451
БОР
452
Лит.: Ворожцов Н. Н., Основы синтеза промежу-
точных продуктов и красителей, 4 изд., М., 1955, с. 588;
Коган И. М., Химия красителей, М., 1956, с. 501; Juli-
us Р., Kunz М. А., Вег., 1923, 56, А, № 2, S. 13.
Е. М. Рохлин.
БОР (Borum) В — химич. элемент III гр. периодич.
системы Менделеева; п. н. 5, ат. в. 10,82. Состоит из
двух стабильных изотопов В10 (ок. 19%) и В11 (ок.
81%). Искусственно получены радиоактивные изо-
топы В. с очень малыми периодами полураспада.
Конфигурация внешних электронов атома Б. 2s2 2р.
Энергия ионизации (в эв): В° —> В+ —> В2+ —> В3+ соот-
ветственно равна 8,296; 15,5; 37,75. Поперечное сече-
ние поглощения тепловых нейтронов 750 барн на атом.
Впервые Б. был получен Ж. Гей-Люссаком и Л. Те-
наром в 1808. Содержание Б. в земной коре составляет
310 4% по весу. В свободном состоянии Б. в при-
роде не найден. Большая летучесть соединений Б. при-
водит к его концентрированию из остаточных магма-
тич. расплавов в пегматитах, а также из газовой фа-
зы — в пневматолитах (главным образом боралюмоси-
ликат — турмалин). На земной поверхности Б. мигри-
рует и концентрируется в остаточных рассолах озер
и морей, образуя боратные месторождения. Наибо-
лее важные бораты: бура Na2B4O, • 10 Н2О, кернит
Ма2В4О7- 4Н2О, улексит NaCaB5O9 8Н2О, гидробора-
цит MgCaBgOjj • 6Н2О, иньоит Са2ВеОп 13Н2О, ка-
либорит KMg2BnO19 • 9Н2О. В галогенных породах
бораты встречаются обычно в виде скоплений белых
или бесцветных кристаллов моноклинной, ромбич. или
триклинной сингонии. Твердость различных боратов
колеблется в пределах 1 — 5; плотность 1,6—2,7. Глав-
ные месторождения находятся в ГДР (Стасфурт),
СССР (Казахстан), США (Калифорния). В комплексе
с другими солями бораты добываются как ценное
сырье для химич. пром-сти.
Физические и химические свойства. Чистый кри-
сталлич. Б. серовато-черного цвета; его структура
сложна и окончательно не расшифрована — имеются
данные о тетрагональной решетке Б., о ромбической
или моноклинной и др. Атомный радиус В 0,97 А,
ионный радиус В3 0,20 А; плотность d 2,34; по твер-
дости кристаллич. Б. занимает среди элементов вто-
рое место (после алмаза), микротвердость его ок.
3500 кг/мм?. При нек-рых условиях, напр. при вос-
становлении В2О3 магнием, получается весьма мелко-
зернистый т. н. «аморфный» порошок В., обычно за-
грязненный примесями (магний, окисные пленки).
Пикнометрия. измерения плотности аморфного Б.
показывают значительно более низкие значения
(ок. 1,73), чем кристаллического. Физич. константы
В. измерялись для не совсем чистых препаратов и
поэтому представляют собой приблизительные вели-
чины; т. пл. 2 300°; т. кип. 2 550°; теплота сублима-
ции 95 ккал/г-атом (при 25°); атомная теплоемкость
3,3 кал/г-атом град (0—100°); термич. коэфф, линей-
ного расширения 8,3 • 10 в. Б. плохо проводит элек-
трич. ток, с повышением темп-ры проводимость уве-
личивается; уд. электрич. сопротивление (Мом)
120,0 (5°), 28,1 (50°), 4,1 (100°). Б. диамагнитен.
В большинстве соединений Б. проявляет валент-
ность -|-3. Связи В—О, В—С1 и др. полярны.
В. склонен к комплексообразованию, координацион-
ное число атома В. равно 4. Химически Б. довольно
инертен, причем кристаллич. Б. менее активен, чем
аморфный. На воздухе Б. сгорает только при 700°,
образуя борный ангидрид В2О3 — аморф-
ную бесцветную стекловидную гигроскопич. массу,
переходящую ок. 294° в вязкую жидкость с т. кип.
1 500°, d 1,84; в воде В2О, растворяется с образованием
борной кислоты Н3ВО3. С водяным паром Б. медленно
реагирует при красном калении, превращаясь в Н3ВО3.
Борный ангидрид может быть получен также термич.
разложением Н3ВО3. Водород не взаимодействует
с Б. даже при высокой темп-ре; бороводороды могут
быть получены только косвенными методами. С гало-
генами Б. реагирует при нагревании, давая бесцвет-
ные летучие соединения типа ВХ;! (где X — F, С1,
Вг, J); реакция с фтором идет при комнатной темп-ре.
Общим методом получения галогенидов Б. является
прокаливание смеси угля с В2О3 в атмосфере галогена.
Галогениды Б. легко гидролизуются и дымят на
воздухе; ВХ3 -|- ЗН2О ЗНХ -|- Н3ВО3. Для них ха-
рактерны реакции присоединения с образованием
комплексных соединений типа Н [BF]4. Склонность
к комплексообразованию от F к J уменьшается, что
объясняется увеличением экранирования атома Б.
атомами галогена по мере роста радиуса послед-
них. Фторид BF3 при нормальных условиях —
газ с удушливым запахом; т. пл. — 128,7°; т. кип.
—100,4°. В лаборатории его легче всего получить на-
греванием NaBF4 или KBF4 с В2О3 в присутствии
H2SO4. Получают BF3 по реакции:
В2О3 + ЗСа!\. + 3H2SO4 = 3CaSO4 + ЗН2О -|- 2BF3
В отличие от других галогенидов Б., BF3 при гидро-
лизе образует борофтористоводородную
к-ту HBF4, устойчивую в водных р-рах. Кислотные
свойства у HBF4 выражены сильнее, чем у HF; ей
соответствует ряд бесцветных хорошо кристалли-
зующихся устойчивых солей. BF3, HBF4 и ее соли
находят широкое применение в органич. химии в ка-
честве катализаторов. Хлорид ВС13 — бесцвет-
ная, легко подвижная жидкость; d 1,43; т. пл. —107°;
т. кип. 17,5—18,5°. Бромид ВВг3— жидкость;
d 2,69; т. пл. —46,0°; т. кип. 90,6°. И о д и д BJ3 —
бесцветные, прозрачные гигроскопич. кристаллы;
d 3,3; т. пл. 43°; т. кип. 210°.
При темп-ре ок. 900° Б. соединяется с азотом,
образуя нитрид BN, к-рый может быть также
получен нагреванием В или В2О3 в токе аммиака.
BN — белый, похожий на тальк порошок; кристал-
лич. структура типа графита; d 2,34; т. пл. 3 000°
(под давлением); трудно растворим в воде; химически
устойчив, не разлагается кислотами и р-рами щело-
чей. Служит хорошим электрич. изолятором при высо-
ких темп-pax. Известна другая модификация BN
со структурой цинковой обманки; ее твердость при-
ближается к твердости алмаза. Интересным соеди-
нением является боразол B3N3He, получаемый
нагреванием диборана в токе аммиака; бесцветная
прозрачная жидкость; т. пл. —58,0°, т. кип. 55,0°,
структурная формула:
НН
HN^ ^NH
I II
нв?» ^вн
NH
По физич. и химич. свойствам боразол напоминает
бензол. Взаимодействие Б. с серой при 610° приводит
к образованию сульфида B2S3, к-рый можно
также получить нагреванием В или В2О3 в атмосфере
H2S или CS2. Сульфид Б. — бесцветная стекловид-
ная масса; d 1,55; т. пл. 310°, разлагается водой.
Нагреванием смеси селена и Б. можно получить селе-
нид B2Se3. При прокаливании борного ангидрида
или бора с углем в электрич. печи образуется кар-
бид В4С — черные блестящие кристаллы; d 2,52;
т. пл. 2 350°; микротвердость ок. 5 000 кг[мм'1\ устой-
чив к химич. воздействиям. С фосфором, мышьяком,
сурьмой Б. не реагирует даже при 700°. Сплавлением
или спеканием Б. с металлами или при взаимодейст-
вии смеси В2О3 с окислами металлов и углем в вакууме
при высокой темп-ре можно получить бориды — весьма
тугоплавкие и твердые и в нек-рых случаях — жаро-
стойкие соединения. Б. вытесняет при нагревании
453
БОР — БОРИДЫ
454
свободные элементы из многих окислов, галогенидов
и сульфидов, а также восстанавливает нек-рые соли
кислородных кислот.
Кислоты, не нвлнющиесн окислителями, на Б. не
действуют; конц. HN03 и царская водка окисляют
его до борной к-ты. При 250° конц. H2SO4 восстанавли-
вается бором до SO2, а Н3РО4 при 800° — до фосфора.
Водные р-ры галогеноводородных к-т с Б. не реаги-
руют. Газообразные HF и НС1 при красном калении
в присутствии Б. разлагаются, выделяя водород.
При сплавлении Б. со щелочами на воздухе или
в присутствии окислителей образуются бораты. Вод-
ные р-ры щелочей с Б. не реагируют. Кристаллич.
Б. довольно энергично взаимодействует с расплав-
ленной Na2O2 и смесью KNO3 с Na2CO3; расплавлен-
ная щелочь действует на него только выше 500°.
В аналитической химии Б. относится
к группе кислотообразующих элементов. Разложение
минералов, содержащих Б., производят HCI, HF или
сплавлением с Na2CO3 (К2СО3), с последующим раст-
ворением в НС1. Освовным методом отделении Б. яв-
ляется отгонка борнометилового эфира В(ОСН3)3 из
кислых р-ров. Б. можно также отогнать в виде фто-
рида. Качественно Б. открывают по буро-красному
окрашиванию куркумовой бумаги, а также по зеле-
ному окрашиванию пламени при сгорании В(ОСН3)3.
Гравиметрически Б. определяют по реакции борно-
метилового эфира с гашеной известью. Титриметри-
чески — в виде маннито-борной кислоты (борная
кислота не может быть оттитрована обычным методом
из-за низкой константы диссоциации; поэтому ее
титруют в присутствии многоатомных спиртов —гли-
церина или маннита, образующих комплекс с Н3ВО3,
усиливающий кислотные свойства). Фотометрически
Б. определяют по фиолетово-синему окрашиванию
с хинализарином или с диаминоантраруфином (4,8-
диамино-1,5-диоксиантрахинон), а также при помощи
куркумина. Имеются также потенциометрия, и другие
методы определении Б.
Получение. Элементарный Б. из природного сырья
добывают в несколько стадий. Разложением природ-
ных боратов (в одних случаях серной к-той, в дру-
гих — водОй) производит борную к-ту. При термич.
разложении последней образуется борный ангидрид.
Наиболее часто аморфный Б. получают восстановле-
нием В2О3 магнием; от примесей очищают обработ-
кой азотной и плавиковой к-тами. Чистый кристал-
лич. Б. может быть получен из галогенидов путем
термич. разложения на вольфрамовой или танталовой
проволоке (при 1300°) или восстановлением водоро-
дом (при 1000—1600°). Б. можно также получить
электролизом боратов.
Применение. Б. в небольших количествах (от ты-
сячных до десятых долей процента) вводится в сталь
и в нек-рые цветные сплавы (напр., бронзы). Присадка
к стали 0,003% Б. в виде ферробора увеличивает
глубину, на к-рую проникает закалка и тем самым
повышает прочность изделии. Небольшие добавки
Б. создают мелкозернистость сталей и цветных спла-
вов, в связи с чем улучшаются их механич. свойства.
Для повышения твердости стали (до 1 800 кг/мм2 по
Виккерсу), ее износостойкости и стойкости против
коррозии (напр., в смеси соляной и азотной к-т) при-
меняют борирование — поверхностное насы-
щение стального изделии бором на глубину 0,1 —
0,5 мм. Карбид бора как весьма твердое вещество
служит абразивным материалом.
В ндерной технике Б., карбид Б. или сплавы Б.
применяют для изготовлении регулирующих стержней
реакторов. Б. и его соединении используют также
в качестве материалов, защищающих от нейтронного
излучении. При захвате изотопом В10 нейтрона про-
исходит реакции В10 (n, a) Li7, к-ран используется
8*
для регистрации нейтровов с помощью борных счет-
чиков и борных камер, наполненных газообразным
BF3 или покрытых внутри В4С. В случае тепловых
нейтронов сечение реакции ок. 3 000 барн. Благодари
способности сильно поглощать нейтроны Б. является
чрезвычайно вредной примесью к ндерному топливу
и конструкционным материалам для реакторов.
Лит.: Бор. Тр. Конференции по химии бора и его соедине-
ний, М., 1958; & m е 1 i п, 8 Aufl., System.-Nummer 13,
Lpz.—В., 1926; M e 1 1 о r, v. 5, N. Y.—Toronto, 1946; то же,
v. 5, 1952; Ullmann, 3 Aufl., В;1 4. Munch. — В., 1953,
S. 580—612; Kirk, v. 2, N. Y., 1948, p. 584—622; Kirk,
Second supplement volume, N. Y., 1960, p. 89—126.
БОРА ГИДРИДЫ — см. Бороводороды.
БОРАТЫ — соли борных к-т. Многие Б. встре-
чаются в природе, напр. бура и др. (подробнее см. Бор).
Щелочные металлы, NH4 и’одновалентный Т1 обра-
зуют неск. видов Б., к-рые хорошо растворимы в воде
и могут быть перекристаллизованы: метабораты типа
LiBO2 8Н2О, тетрабораты Na2B4O7 • 10Н2О, пента-
бораты LiB5O7 5Н2О, декабораты К4В10О17 - 5Н2О.
Щелочноземельные металлы и Mg дают дибораты
MgB2O4 • ЗН2О, тетрабораты MgB4O7 • 9Н2О, гек-
сабораты Mg2BeOu • 13Н2О. Для двухвалентных тяже-
лых металлов известны основные Б. переменного
состава. Известны Б. типа двойных солей, у к-рых
катионами нвлнютсн щелочной металл или аммоний и
щелочноземельный или тяжелый металл. Структура
многочисленных полиборатов мало исследована. Из-
вестно только, что в состав их решетки входит ионы
ВО), к-рые соединяются друг с другом общими ато-
мами кислорода. В нек-рых Б. наряду с ВО) обнару-
жены тетраэдрич. группы ВО%. напр. в структуре
тетрабората:
Г ' z°\
О-В В—О-В В—О
L О'' "'О'"
Нек-рые из Б. находит практич. применение. В ке-
рамич. пром-сти используются Са(ВО2)2 6Н2О,
Са(ВО2)2 2Н2О, в текстильной — борат цинка. Ка-
чественное и количественное определение Б. проводит,
превращая их предварительно тем или иным способом
в борную к-ту или в растворимые Б. Далее опреде-
ляют обычным путем (см. Бор).
Лит.: Кешан А. Д., Синтез боратов в водном растворе
и их исследование, Рига, 1955; Химия боратов. Материалы
совещания по химии боратов, Рига, 1953; Кеш а н А. Д.,
В и м б а С. Г., Шварц Е. М., Изв. АН Латв. ССР, 1955,
№ 7, 8, 12; Николаев А. В., Физико-химическое изуче-
ние природных боратов. М.—Л., 194-7. Г. Н. Пирогова.
БОРИДЫ — соединении бора с относительно более
электроположительными элементами. Практически к
Б. относит соединении бора с металлами. Установлено
существование Б. различных составов: Ме4В, Ме3В,
Ме2В, Ме3В2, МеВ, Ме3В4, МеВ2, Ме2В5, МеВ4, МеВ6
и МеВ2. Один и тот же металл может давать ряд из
указанных соединений. Благодари наличию у бора
одного неспаренного 2^-электрона на внешней орбите,
атомы бора способны к прочной ковалентной связи
как в собственных кристаллах, так и в соединениях
с другими элементами, что обусловливает разнообраз-
ные и сложные кристаллич. структуры Б. При из-
бытке атомов металла в решетке атомы бора обычно
изолированы друг от друга, а при избытке атомов
бора последние образуют цепочки, сетки, каркасы.
По структурам решеток, взаимному расположению
атомов и расстоннинм между ними Кисслинг предло-
жил классификацию структурных типов Б., в к-рой
переход от одного типа к другому сопровождается
также нарастанием отношении числа атомов бора и
металла.
1. Сравнительно простые структуры с изолированными
атомами бора (МщВ — ромбическая, Fe2B — тетрагональная).
455
БОРИДЫ—БОРНАЯ КИСЛОТА
455
К этой группе относится большое число В., построенных из
полос тетраэдров атомов металла с атомами бора между этими
полосами. Расстояние В—В в них больше, чем в других бори-
дах (напр., 2,12А в Ге2В). 2. Структуры с цепями из атомов
бора (ТаВ, ГеВ — ромбические, МоВ — тетрагональная и др.).
Атомы бора располагаются в центрах тригональных призм,
имеют по шесть соседних атомов металлов и находятся в кон-
такте с двумя другими атомами бора, расположенными на
расстоянии 1,74—1,86 А. 3. Структуры со сдвоенными цепями
из атомов бора (Ta3Bi, Сг3В4, Мп3В4). Расстояние В—В в од-
ной цепи 1,74 А, а в соседней около 1,54 А. Каждый атом бора
окружен шестью атомами металла, расположенными по углам
тригональной призмы. Эти соединения менее устойчивы из-за
наличия в них двойной цепочки с различными расстояниями
В—В. 4. Структуры с сетками из атомов бора (типа А1В2 —
гексагональная). Среди диборидов (МеВ2) встречается значи-
тельное число Б. переходных металлов. Б. этого типа прочны
л стабильны. С Б. типа МеВ2 сходны по структуре Б. типа
Me2B8(W2B8 — гексагональная, Мо2В5 — ромбическая). Слои
из атомов металла, расположенные по гексагональной плотно
упакованной решетке, чередуются со слоями из атомов бора,
образующих гексагональную двухмерную сетку. Каждый
атом бора имеет трех соседей на расстоянии 1,73 А и окружен
шестью атомами металла. 5. Структурные типы с каркасом
из атомов бора (UB4 — тетрагональная, CaBe, UBt2— куби-
ческие, генсабориды щелочноземельных и редкоземельных
металлов;. Атомы бора образуют трехмерный каркас из окта-
эдров, в пустотах к-рых расположены атомы металла. Расстоя-
ние Me—В значительно больше суммы нормальных атомных
радиусов. Каждый атом бора окружен пятью атомами металла
на расстоянии 1,72 —1,74 А; вокруг атомов металла распо-
лагаются 2 4 атома бора.
Для Б. характерно наложение нескольких типов
химич. связи. Существование металлич. электро- и
теплопроводности у Б. показывает, что в химич. связи
между атомами металла и бора принимает участие
электронный коллектив; в наибольшей степени это
характерно для структур 4-го и 5-го типов. Этим
обстоятельством обусловливаются такие практически
важные свойства, как высокая электропроводность
гексаборидов, малая работа выхода электронов при
термоэмиссии, способность к автоэлектронной (хо-
лодной) эмиссии электронов. В Б. переходных метал-
лов происходит обобществление не только валентных,
но и внутренних электронов достраивающегося d-уро-
вня атомов этих металлов. Это проявляется в прочной
межатомной связи, высоких темп-pax плавления,
твердости, химич. устойчивости, жаропрочности и
жаростойкости этих Б. Среди них известно большое
число фаз с узкими облостями гомогенности. Эти
боридные фазы являются переходными между интер-
металлич. (электронными) фазами со строго опреде-
ленными составами и фазами внедрения (карбиды,
нитриды), имеющими широкие области гомогенности.
В табл, приведены составы, структуры и нек-рые
свойства Б. переходных металлов.
Наличие большого числа В. различных металлов
с разнообразными ценными свойствами создает воз-
можности их применения в различных Отраслях тех-
ники. Области применения Б. еще недостаточно уста-
новились и в этом направлении ведутся широкие
исследования. Путем диффузионного поверхностного
борирования резко повышаются твердость, износо-
устойчивость и коррозионная стойкость различных
изделий из стали, никеля, молибдена, вольфрама и др.
Известно применение В. никеля в качестве катализа-
тора в процессах гидрирования. Б. переходных ме-
таллов — хрома, циркония, титана, ниобия и тан-
тала или их сплавы, благодаря их тугоплавкости,
жаростойкости и жаропрочности могут применяться
для изготовления деталей реактивных двигателей,
лопаток газовых турбин и т. п. Гексабориды бария,
лантана, церия и др. благодаря высоким термоэмис-
сионным свойствам применяются в качестве материа-
лов для катодов электронных приборов. В химич.
отношении дибориды переходных металлов и гекса-
бориды редкоземельных металлов, как правило,
устойчивы против минеральных к-т, нек-рые даже
при нагревании, но разлагаются расплавленными
щелочами и перекисями. Моно- и тетрабориды пере-
ходных металлов разлагаются под действием водных
р-ров неорганич. к-т с выделением бороводородов.
Состав Б. Кристаллич. структура Пикно- метрия, плот- ность, г/см& Область темп-рной устойчи- вости, °C Т. пл., °C Уд. элек- тросо- проти- вление, МКОМ • см
TIB Кубнч. гранецен- трированная 5,09 600—1800 - 40
Ti2B5 Гексагональная — 1700—2100 __
ZrB Кубнч. гранецен- трированная 5,7 800—1250 — 30-35
ZrBo Гексагональная 5,82 20—3040 3040 9—11
Zl’Bia Кубич. гранецен- трированная 3,70 1650—2680 2680 60-100
HfB То же 11.6 ._
HfBo Гексагональная 10,5 20—3250 3250 10
VB Ромбическая 5,44 (рентг.) 20—1900 1900 35-40
V Bo Гексагональная 4,61 20—2400 2400 16
NbB Ромбическая — 2000 64,5
Nb3B4 То же —.
NbB2 Гексагональная 6,60 20-3000 3000 32,0
Ta2B Тетрагональная 20—1800 2040 —
TaB Ромбическая 14.0 20-2040 2040 100
Ta3B4 То же 13,5 20-1780 — —-
TaB2 Гексагональная 11,70 20-3100 3100 68
Mo2B Тетрагональная 9,10 20-2050 2000 40
MoB2 Гексагональная — 1600-2100 2100 45
Mo2B; Ромбоэдрическая 8,01 20-1600 2100 25
W2B Тетр агональная 16,0 20-1900 2770 —
a-WB То же 15,3 20—2400 2860 —,
WBo Гексагональная 13,0 — —.
w2B‘5 То же 11,0 20—2300 2300 21
Таблица составлена по данным, приведенным в обзоре
Т. В. Самсонова и Л. Я. Марковского (см. литературу в кон-
це статьи).
Известно несколько методов получения Б.: 1) син-
тез из элементов спеканием смесей порошков металла
и бора в атмосфере аргона при 1300—2000°; 2) осаж-
дение из газовой фазы при восстановлении летучих
галогенных соединений бора и металла водородом;
3) взаимодействие окисла металла с карбидом бора
или смеси окислов металла и бора с углеродом (этот
процесс предпочтительнее проводить в вакууме);
4) электролиз расплавленных смесей боратов щелоч-
ных и щелочноземельных металлов с окислами туго-
плавких металлов; 5) металлотермич. восстановление
(папр., магнием) смеси окислов металла и бора;
6) восстановление окислов металла бором. Наиболее
удобны для промышленного использования — тре-
тий, четвертый и пятый методы. Большинство указан-
ных методов приводят к получению порошкообраз-
ных Б. Для получения плотных изделий порошки
подвергают прессованию и спеканию. Изделия наи-
большей плотности получают при объединении про-
цессов прессования и спекания, т. е. горячим прессо-
ванием.
Лит.: Самсонов Г. В. и Марковский Л. Я.,
Усп. хим., 1956, 25, вып. 2, с. 190; Самсонов Г. В. и
Уманский Я. С., Твердые соединения тугоплавких метал-
лов, М., 1957; Бор. Тр. конференции по химии бора и его сое-
динений, М., 1958; Самсонов Г. В., Усп. хим., 1959,
28, вып. 2, с. 189; Kies sling A., Acta chem. scand.,
1949, 3, p. 595; его же, тамже, 1950, 4, р. 209; Anders-
son L. Н. and К i е s s 1 1 n g А., там же, p. 160; Paul R.,
В u i s s о n P. and Joseph N., Industr. and Engng Chem.,
1952, 44, ^s 5, p. 1006; Lafferty J. M., Phys, rev., 1950,
79, № 6, p. 1012; см. также при ст. Бор. В. А. Эпелъбаум.
БОРНАЯ КИСЛОТА (ортоборная кислота) Н3ВО,—
слабая неорганич. кислота, бесцветные кристаллы
в виде чешуек; триклинная решетка: а = 7,04 А,
Ь =7,04 А, с=6,5бА, а =101,10°, Р = 92,30°,
у = 120°; плотн. 1,48; т. пл. 169° (с разл.). Теплота
образования AH29S =—260,2 ккал/моль. При нагре-
вании до 50° Н8ВО8 не теряет воду, но уже при 70°
заметно обезвоживается, причем образуется метабор-
ная к-та НВО2. Конечным продуктом обезвоживания
457
БОРНЕОЛ — БОРНЫЕ УДОБРЕНИЯ
458
является борный ангидрид В2О3. Тетраборная к-та
Н2В4О, в качестве побочного продукта при обезво-
живании не образуется (разлагается). Растворимость
Н3ВО3 (в г на 100 г Н2О): 2,вв (0°), 4,9 (20°), 39,7
(100°); растворима также во многих органич. раство-
рителях. Степень диссоциации в 0,1 н. р-ре Б. к.
0,01%; константы диссоциации при 20°; ^=7,3 • 10'10;
Кг — 1,810 13; К3 = 1,6 • 10-14. Соли Б. к. — бо-
раты, являются производными либо метаборной
к-ты НВО2, либо тетраборной Н2В4О,. Со спиртами
(метиловым, этиловым и др.) в присутствии конц.
H2SO4 Б. к. образует эфиры. Известны и др. органич.
производные Б. к. В природе свободная Б. к. встре-
чается в виде минерала сассолина, в горячих источ-
никах и минеральных водах. Методы добычи Б. к.
зависят от характера месторождения. В СССР мине-
ралы обрабатывают серной к-той. После отделения
осадка фильтрат охлаждают, при этом из него кристал-
лизуется Б. к. В пром-сти и с. х-ве Б. к. применяют
для тех же целей, что и буру. В лабораторной прак-
тике Б. к. используют для приготовления буферных
растворов, в медицине — как дезинфицирующее сред-
ство. О качественном и колич. определении см. Бор.
Лит. см. при ст. Бура. Г. Н. Пирогова.
БОРНЕОЛ (борниловый спирт) С10Н13О, мол. в.
154,25 — вторичный спирт группы бициклических
терпенов. Б. имеет эндо-кон-
фигурацию; его изомер, обла-
дающий э«ао-конфигурацией,
носит название и з о бор-
неола (иао-Б.). Б. суще-
ствует в трех стереоизомер-
ных формах 1, dl и d.
камфара) содержится в выде-
СНз
H3C-C-CH3 I
----сн----сн2
СНОН
НзС'
Н2С
d-Б. (борнейская
лениях дерева Diyobalanops camphora, растущего
па Борнео и Суматре; найден в камфарном, лаван-
довом и розмариновом маслах. 1-Б. является со-
ставной частью пихтового масла (Abies sibirica).
1- и dl-Б. содержатся в валериановом масле. d-B.' —
гексагональные пластинки; т. пл. 208,5°, т. кип.
212°, растворим в спирте, эфире, бензоле, лигроине,
слабо растворим в воде, возгоняется; [а]^’=-(-37,44°
(в спирте). 1-Б.—гексагональные пластинки; т. пл.
204°, т. кип. 210° /779л<л< (возгоняется); [a]g = —34,74°
(в спирте). dl-Б., т. пл. 210,3°.
При окислении и при каталитич. дегидрировании
Б. превращается в камфару, при дегидратации обра-
зует камфен’, при действии НС1 и РС15 дает борнил-
хлорид и изоборнилхлорид. Б. получают гидратацией
пинена, содержащегося в скипидарах (промышленный
метод); восстановлением камфары; гидратацией кам-
фена; из борнилхлорида — действием металлич. маг-
ния с последующим окислением магний органич. про-
изводного кислородом воздуха. Б. применяется в про-
изводстве камфары.
l-uao-Б., т. пл. 212°; растворим в спиртах, эфире,
петролейном эфире, бензоле, толуоле; [a]D= —34,09°.
d-uao-Б., т. пл. 214°; [а]с = —1-34,14°. Металлич.
натрием изо-Б. изомеризуется в Б.; при дегидратации
образует камфен; при действии НС1 дает изоборнил-
хлорид; окислением превращается в камфару. Полу-
чается нзо-Б. восстановлением камфары металлич.
натрием; гидратацией камфена. Идентификация про-
изводится по темп-ре плавления п-нитробензоатов:
для Б. 137°, для изо-Б. 129°.
Лит.: Simonsen I. L., The terpenes у. 2, 2 ed., 1949,
p. 349; Chemistry of carbon compounds. Ed. E. H. Rodd, Amst.
[a. 0.], v. 2, 1956, p. 599. Л. С. Поваров.
БОРНИЛХЛОРИД (пиненгидрохлорид, «искусст-
венная камфара») С10Н17С1, мол. в. 172,70 — хлор-
производное камфана — бициклйч. терпена. Б. имеет
эндо-конфигурацию (I). Его изомер с экао-конфигура-
цией (VI) носит название изоборнилхлори-
д а (изо-Б). Б. — кристаллич. листочки; т. пл. 132°,
т. кип. 207—208°, возгоняется; растворим в эфи-
ре, спирте, нерастворим в воде. Б. существует в
трех стереоизомерных формах;
d-Б., [a ]D = 4-33,52° 1-Б.,
[а]д= —33,24° (в спирте) и
dl-Б. Борнилхлорид облада-
ет сильным запахом камфары.
При окислении концентриро-
ванной азотной кислотой Б.
СПз
н2с—и:-----ено
| Н3С-С-СН31
н2с---сн—сн2
образует кетопиновую кислоту
(II), разбавленной азотной к-той окисляется в кам-
фарную к-ту (III) и апокамфарную к-ту (IV); хро-
мовой к-той окисляется в 6-хлор-этш-камфару; при
восстановлении образует камфан (V); при действии
натрия дает диборнил, камфен и камфан. Медлен-
но отщепляет НС1 при кипячении с водой; при
дегидрохлорировании основаниями дает камфен; дей-
ствием солей жирных к-т с последующим омылени-
ем сложных эфиров образует изоборнеол; с магнием
образует борнилмагнийиодид. Получают Б. действием
НС1 на а-пинен, содержащийся в скипидарах (про-
мышленный метод); действием НС1 или РС15 на бор-
неол, действием НС1 на борнилацетат; хло-
рированием камфана. Применяют в произ-ве
камфары.
Изо-Б., т. пл. 161,5°; возгоняется, растворим
в эфире, петролейном эфире, слабо растворим в
спирте. При восстановлении дает камфан; при
кипячении с водой, отщепляя НС1, образует кам-
фен, при действии брома дает камфендибромид, с
уксуснокислым серебром дает изоборнилацетат, с известковым
молоком — камфенгидрат, с магнием образует магнийорганич.
производное и гидродикамфен; изо-Б. получается действием
НС1 па изоборнеол и гидрохлорированием камфена.
Лит. см. при ст. Борнеол. Л. С. Поваров.
БОРНОЭТИЛОВЫЙ ЭФИР (этилборат, триэтокси-
бор) (CjHsO^B, мол. в. 145,90 — бесцветная подвиж-
-- I.-H
'•''Cl
VI
ная жидкость приятного запаха, горящая зеленым
пламенем; т. пл. —84,8°; т..кип. 188,6°; сЦ° 0,8635;
ng 1,38076. Образует азеотроцные смеси с «-бутило-
вым спиртом, н-бутилиодидом, изоамилбромидом,
изобутилацетатом и др. Б. э. получают: 1) действием
этанола на галогениды бора; 2) взаимодействием
борного ангидрида с этанолом; 3) обработкой безвод-
ной буры этанолом и хлористым водородом; 4) дей-
ствием на спирт смешанных ангидридов — борносер-
ного B2O3(SO3)2 и борноуксусного В(СН3СОО)3; 5) оки-
слением триэтилбора. Б. э. легко гидролизуется во-
дой; разлагается действием РС16; образует с сали-
циловой к-той твердую бортрисалициловую к-ту
В(ОСвН4СООН)3; с В2О3 дает циклич. этилметаборат
(C^HjOBOja; с ВС13 образует (С2Н5О)2ВС1. При-
меняют Б. э. для получения борорганических сведи-
нений. о. Ю. Охлобыстин.
БОРНЫЕ УДОБРЕНИЯ — борсодержащие про-
дукты, применяемые наряду с азотными, калийными
и фосфорными удобрениями с целью увеличения
урожая и улучшения качества семян и др. свойств
многих растений (номерность волокна льна, содержа-
ние сахара и крахмала и др.), устранения заболева-
ния пек-рых растений (гниль сердечка свеклы, бак-
459
БОРОВОДОРОДЫ — БОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
460
териоз льна и др.), ускорения созревания нек-рых
культур (хлопка, кукурузы) и увеличения зимостой-
кости фруктов, томатов и др. К Б. у. относятся: 1)
Осажденные бораты магния, содер-
жащие (в пересчете на борную кислоту) ок. 9% лимон-
норастворимого бора (нерастворимого в воде, но раст-
воримого в 2%-ном р-ре лимонной к-ты) и ок. 32%
магния (в пересчете на MgO, частично связанного
с бором, а частично сернокислого); хорошо усваи-
ваются растениями (см. Минеральные удобрения).
Осажденные бораты магния получают обработкой
маточного р-ра (отхода произ-ва борной к-ты), содер-
жащего сульфат магния и борную к-ту, технич.
окисью магния.
2) Борнодатолитовое удо б р е н и е, со-
держащее до 14,5% водорастворимой борной к-ты,
ок. 0,5% свободной H2SO4, активную кремнекислоту
и гипс. Борнодатолитовое удобрение получают разло-
жением измельченной датолитовой руды серной кисло-
той. 3) Боросу перфосфат, содержащий 16—
18% усвояемого Р2О5 и 1,6—2,0% Н3ВО3; его полу-
чают смешением суперфосфата с борнодатолитовым
удобрением в необходимых соотношениях. 4) Б о р о -
двойной суперфосфат, содержащий 42—
43% усвояемого Р2О5 и 6—7% водорастворимой
Н3ВО3, и бораты, получаемые спеканием силикатов
бора с известняком и др. реагентами (т. н. тер-
мические борат ы), содержащие 4—16%
лимоннорастворимого бора (в пересчете на борную
к-ту). Бородвойной суперфосфат получают обработ-
кой измельченной датолитовой руды фосфорной к-той.
Б. у. применяют под многолетние травы, лен, са-
харную свеклу, овощные и плодово-ягодные куль-
туры, хлопчатник, кукурузу, эфиромасличные, под-
солнечник, сою и др. путем внесения В. у. в почву,
опыления или опрыскивания растений во время цвете-
ния, а также намачиванием семян в р-рах борной
к-ты. Норма при внесении в почву 3—9 кг/га (в пере-
счете на борную к-ту), а при поверхностном внесе-
нии — в несколько раз меньше. Основные количества
борных удобрений должны содержать бор в водо-
растворимой форме и лишь 10—15% в лимоннораство-
римой форме. Последняя предназначается для песча-
ных почв п районов с большим количеством осадков,
нуждающихся одновременно в магнии. Поэтому лимон-
норастворимые формы Б. у. должны в ряде случаев
содержать, кроме бора, и магний; эту форму Б. у.
нельзя применять для поверхностного внесения и для
намачивания семян. Эффект от применения Б. у. во
много раз превышает произведенные затраты.
Лит.: Нейве Я. В., Удобрение и урожай, 1956, № 8,
с. 58; Берлин Л. Е., там же, 1958, К» Н, с. 29; е г о ж е,
Хим. наука и пром-еть, 1957, 2, № 6. Л. Е. Берлин.
БОРОВОДОРОДЫ (гидриды бора, бораны) — сое-
динения бора с водородом состава: ВПНП _^4(напр.,
В2Н6) или B)[H1[ f; (напр., В4Н10). Первые более устой-
чивы, чем вторые. Строение Б. окончательно не уста-
новлено. Согласно теории В. В. Некрасова наряду
с гомеополярными связями в молекулах Б. важную
роль играют водородные связи;
ННН НнНнН
\/\/ \ .-\ \..-\/
в в в в-в в
/ V \ / V \ V \
ННН ННННН
Эта структура удовлетворительно объясняет нек-рые
химич. свойства Б., напр. у диборана В2Н6 углево-
дородными радикалами замещаются только те 4 атома
водорода, к-рые не образуют наряду с химич. связями
водородных. Строение высших Б. более сложно,
однако и для этих соединений электронография, и
спектроскопия, исследованиями установлено наличие
структурных элементов с водородной связью.
По физич. свойствам Б. похожи на соответствующие
углеводороды и силаны. Ниже приведены физич. свой-
ства нек-рых Б.
1 в.н, В4н1о В6нп В5Н„ В,0Н„
Т. пл., °C -165,5 — 120,8 — 123 —46,6 —65,1 +99,5
Т. кип., °C ... . —92,53 + 18 4-63 4-60 +94 +213
Плотность, ZJCJA^ . 0,447 (—112°) 0,56 (-35°) 0,63 (16°) 0,69 (0°) 0,94 (25°) 0,78 (100°)
Эти вещества бесцветны, имеют неприятный запах,
очень ядовиты. Вдыхание даже незначительных их
количеств вызывает головную боль, тошноту и может
привести к серьезным отравлениям. Во многих орга-
нич. растворителях (углеводородах, нек-рых эфирах
и др.) они растворяются без разложения. Водой и
водными растворами спиртов разрушаются: В2Н6
+ 6Н2О = 2Н3ВО3 + 6Н2. Б. легко окисляются,
на воздухе самовоспламеняются при невысоких тем-
пературах (ниже 100°). Со щелочами газообразные Б.
взаимодействуют с образованием гипоборатов по реак-
ции: В2Нв + 2КОН = К2[В2Н6О2] + Н2. Водород в
Б. может замещаться галогенами и углеводородными
радикалами. С сухим аммиаком образуются белые со-
леобразные продукты присоединения: В2Н6 + 2NH3=
= B2H6(NH3)2; В4Н10 + 4NH3 = B4H]0(NH3)4. С ами-
нами Б. реагируют с образованием комплексных и
боразотсодержащих соединений. С гидридами метал-
лов или их алкильными производными диборан обра-
зует боргидриды металлов: LiBH4, NaBH4, Ве(ВН4)а,
А1(ВН4)3 и др. Диборан получают разложением моно-
хлордиборана, взаимодействием BF3, с алюмогидри-
дом лития (4BF3 4- 3LiAlH4 = 2В2Н6 -|- 3JJF +
+ 3A1F3), гидрированием галогенидов бора в электро-
разряде или в присутствии металлов и их гидридов
(2ВВг3 + 6Н2=В2Н6 + бНВг, 2ВС13 + 2А1 + ЗН2 =
= В2Н6 + 2А1С13) и т. д. Тетраборан с примесью пен-
таборана получают взаимодействием боридов с к-тами,
причем образуется смесь Б. с водородом, от к-рого Б.
отделяются в виде жидкостей конденсацией при глу-
боком охлаждении.
Пентаборан и декаборан получаются в процессе
термич. разложения диборана при темп-pax от 100°
до 280° в токе водорода: 5В2Нв=2В5Н9 + 6Н2,
5В2Н6=В1оН14 + 8Н2. Теплоты сгорания Б. значи-
тельно превышают теплоты сгорания многих других
соединений, в частности углеводородов, в связи с чем
Б. представляют большой интерес с точки зрения их
использования в качестве ракетного топлива. Б.
применяются для покрытия металлов бором (для
повышения твердости, устойчивости к истиранию и
коррозии); могут быть использованы для вулканиза-
ции каучуков, а также в качестве катализаторов и
компонентов при получении полимеров.
Лит.: Херд Д., Введение в химию гидридов, пер.
с англ., М., 1955; Некрасов Б. В., Штуцер В. В.,
Ж. общ. хим., 1948, 18, вып. 5, с. 832; Некрасов Б. В.,
Нуре общей химии, 11 изд., М., 1954, с. 541—49; 11 а у in -
к и н Я. М., Химический состав и свойства реактивных топ-
лив, М., 1958; см. также при ст. Бор. Г. И. Пирогова.
БОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — извест-
ны полные, в к-рых все валентности бора замещены
органич. радикалами, и неполные, в к-рых одна или
две валентности замещены галогеном, алкокси-
группой и др.
Алифатич. триалкилборины R3B — бесцветные жид-
кости, крайне чувствительные к кислороду (триме-
тилбор — газ при комнатной темп-ре); низшие чле-
ны ряда самовоспламеняются на воздухе. Ароматич.
Б. с. и алкилборные кислоты — твердые вещества.
В таблице приведены физические свойства важней-
ших Б. с.
461
БОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — БРАУНА РЕАКЦИЯ
462
Формула Т. пл., °C Т. пип., °C’ Плотность, г/см9
(СНз)зВ -153 -21,8 0,625 (-100°)
(С2н5)3в —92,9 95 0,6961 (23°)
(UaHrJaB — 53 (18 мм) 0,7745 (20°)
(U3H7)aB -65,5 164,5 0,7204 (25°)
(изо-С4Н9)3В — 188 0,7380 (25°)
(С6н,)3в 142 203° (15 мм) —
(а-и10н7)аВ 203 — —
С2Н5ВС13 50,8 —
C3HSB(OH)S 40 (субл.) — —
CeH5B(OH)3 216 — ——
(CeH6)2BOH 264 — —
(^Из)зВз^ТзНз 31,5 129 —
* Т. кип. при 760 мм рт.ст., кроме особо оговоренных.
Триалкилборины с сильными электронодонорными
соединениями образуют комплексные соединения раз-
личной устойчивости. С органич. соединениями
Li образуются прочные комплексные ионы типа BR7.
Окисление R3B приводит к эфирам диалкил- и алкил-
борных к-т; при действии брома или хлора органич.
радикалы замещаются галогеном. Реакция, обратная
диспропорционированию: R3B ВС13 —> R2BCI +
RBC12, идет только при нагревании; напротив, для
R = СН3 чрезвычайно легко (—60°) происходит дис-
пропорционирование. RBC12 могут быть получены
также по реакции (RBO)3 2ВС13 —> 3RBC12 В2О3.
При нагревании Й3В способны обмениваться ради-
калами с алкильными производными др. элементов.
Гидрирование R3B может служить способом получе-
ния гидридов бора и, в частности, диборана: R3B -|-
-j- 6Н2 -> B2He -|- 6RH. В2Н6 с R3B; образует равно-
весные смеси алкилдиборанов RnB2H6_ п (п — 1—4),
состав к-рых определяется соотношением исходных
в-в. Алкилдибораны легко диспропорционируются.
Алкилборные к-ты слабее Н3ВО3, арилборные — не-
сколько сильнее. Получают их обычно окислением
R3B в RB(OR)2 с последующим гидролизом последних,
неполным алкилированием (RO)3B и (ROBO)3, гидро-
лизом ПВС12. Алкил- и арилборные к-ты легко дают
при нагревании соответствующие циклич. бороксины
(RBO)3; с HgCl2 или HgO образуются ртутьоргапич.
соединения. Диалкилборные к-ты нейтральны по
лакмусу. Пиролиз (200°) комплексов диборана и алкпл-
диборанов с аммиаком (или аминами) приводит к «не-
органич. бензолу» — боразолу:
ВН
hni^^Snh
hb^Jbh
NH
и его алкильным производным. В. с. могут быть полу-
чены несколькими способами: 1) Алкилированием га-
логенидов бора, эфиров борной и метаборной к-т,
магний-, цинк-, алюминийорганич, соединениями;
конечными продуктами являются триалкилборины
R3B (бортриалкилы), однако во многих случаях реак-
ция может быть остановлена на стадии образования
алкилгалогеноборинов или соответственно эфиров
алкилборных к-т. 2) Взаимодействием ртутьорганич.
соединений с ВНа13; реакция используется обычно
для получения ароматич. производных типа АгВНа1а
и Ar2BHal. R3B получают также из алюминийтри-
алкилов R3A1 и алкилбороксинов (R3BO)3. 3) При-
соединением диборана к олефинам с образованием
R3B; олефины с внутренней двойной связью дают
те же продукты, что и а-олефины. В2Н6 замещает
водород в бензоле (100°) с образованием (С6Н6)3В.
4) Взаимодействием бензола с ВС13 с образовани-
ем СвН6ВС12; последний с хлорбензолом и Na дает
(СвЙ6)3В.
Б. с. применяют гл. обр. для получения гидри-
дов бора, служащих сырьем для произ-ва высококало-
рийных топлив для реактивных двигателей; R3B и
нек-рые другие Б. с. применяются в качестве катали-
заторов полимеризации ненасыщенных соединений;
комплексные соединения типа NaB(CeH5)4 исполь-
зуются в аналитич. химии для осаждения ионов К,
Pb, Cs, NH4.
Лиш..- Синтетические методы в области металлоорганич.
соединений, под общ. ред. А. Н. Несмеянова и К. А. Кочеш-
кова, вып. 3, М.—Л., 1945; Херд Д., Введение в химию
гидридов, пер. с англ., М., 1955; Rochow Е. G., Н и г d
D. Т. Lewis R. N., The chemistry of organometallic com-
pounds, N. Y., [1957]. О. Ю. Охлобыстин.
БОРШЁ СИНТЕЗ — превращение фенилгидразона
циклогексанона (I) в 1,2,3,4-тетрагидрокарбазол (II)
при нагревании с разб. серной к-той. Б. с. — частный
случай получения гомологов и производных индола
из фенилгидразонов алифатич. альдегидов и кетонов
по реакции Фишера. Образование II происходит,
вероятно, через бензидиновую перегруппировку (по-
дробнее см. Фишера реакция)'.
Ранее эту реакцию наблюдал Дрексель (1888). Однако
Борше впервые показал общность этого метода и по-
лучил ряд замещенных II. Распространение метода
Борше для синтеза карбазола и его производных
тормозилось отсутствием доступных способов дегидро-
генизации тетрагидрокарбазолов. За последнее время
дегидрогенизацией II и его замещенных хлоранилом
или коричной к-той и палладиевой чернью удалось
получить карбазолы с высокими выходами. Б. с.
находит применение для синтеза замещенных карба-
зола и установления их строения. В случае циклиза-
ции .«-замещенных фенилгидразона циклогексанона
образуются два изомера — 5- и 7-замещенные тетра-
гидрокарбазола:
X
Лит.: Wheeler Т. S. and Gowan 1. Е., Каше index
of organic reactions, L., 119531; Campbell N., Bar-
clay В. M., Chem. Rev., 1947, 40, № 3, p. 359.
M. Г. Линькова.
БРАУНА РЕАКЦИЯ — расщепление С—N связи
в третичных аминах при взаимодействии с бромистым
цианом: R3N -|- BrCN —> R2NCN -|- RBr. Бромциан
с алифатич. третичными аминами бурно реагирует
уже на холоду, поэтому реакцию проводят в среде
инертного растворителя и при охлаждении. С арил-
и диариламинами реакция протекает в значительно
более жестких условиях. Если с азотом связаны три
463
БРЕДТА ПРАВИЛО — БРОЖЕНИЕ
464
различных радикала, то расщепление возможно по
трем направлениям:
Ri4 Ri^ :+ ^RjR3NCN + R3Br
Rs—N + BrCN —> \ R--N-CN :Br-^RjRgNCN + R»Br
R3Z ; R3Z : X*RiR3NCN + RjBr
Соотношение скоростей этих конкурирующих реак-
ций и определяет прочность связи радикалов с азо-
том. Большое внимание уделялось реакции броми-
стого циана с гетероциклич. соединениями, проводив-
шейся с целью определения относительной легкости
расщепления различных циклич. систем:
BrCN
BrCHgCH2N(CN)R
CN(40%)
Как правило, легкость расщепления циклич. систем
уменьшается в порядке последовательности от 3-
к 6-членному циклу; расщепление пиперидинового
кольца проходит с той же легкостью, что и отщепление
н-пропильной группы. Прочность связи различных
радикалов с азотом определяется также расщепле-
нием бензоильных производных вторичных аминов
пятихлористым фосфором:
R1>-сосвн5 —К
R/
Ri, лВ^=СС1СвН5 + RjCl
\N—CCb'CgHa.
R/ 4R.N^CC1C(jH5 + R[C1
Для определения направления реакции хлоримин
гидролизуют в амид к-ты или в амин, или же расщеп-
ляют на галогенид и нитрил. Циклич. амины превра-
щаются в дигалогениды. Получение галогенидов из
алифатич. аминов Б. р. имеет и препаративное значе-
ние, т. к. отщепляющийся радикал не перегруппиро-
вывается, как это имеет место при дезаминировании
азотистоводородной к-той.
Ряды прочностей связи радикалов с азотом, полу-
ченные обоими методами, хорошо совпадают друг
с другом и известны под названием рядов Брауна
(прочность связи увеличивается в ряду):
CHsCH=CHCHa—>СН2=СЫСН»->С0Н5СНа—>СН3->
с2н5—н-с3н7—изо~с3н7——
Б 1923 было показано, что в этом ряду следует пере-
менить местами аллильный и бензильный радикалы.
Б. р. часто применяется для определения строения
природных соединений. Реакция открыта Ю. Брауном
в 1900.
Лит.: Хейгеман X. А., в ни.: Органические реак-
ции. Сб. 7, пер. с англ., М., 1956, с. 260; X ю к к е л ь В., Тео-
ретические основы органической химии, пер. с нем., т. 2, М.,
1958. Н. П. Гамбарян.
БРЕДТА ПРАВИЛО — правило, согласно к-рому
введение двойной связи в мостиковое положение би-
циклич. системы невозможно (первоначальная трак-
товка). Примером, иллюстрирующим Б. п., может
служить ангидрид бромкамфарной к-ты (I) и бромкам-
фара (II), к-рые не отщепляют бромистого водорода
с образованием соответствующих непредельных сое-
динений. Камферхинон (III) и камфенилон (IV) не
бромируются и не вступают в реакцию дейтерообмена,
что также объяснялось невозможностью образования
кратной связи (енола). Позже (Бартлет, Несмеянов
и др.) было установлено, что заместители, стоящие у
мостикового атома углерода, не отщепляются и не заме-
щаются под действи-
ем нуклеофильных,
электрофильных и
радикальных аген-
тов. Теоретич. объ-
яснение некоторых
случаев, иллюстри-
рующих Б. п., зак-
лючается в том, что
мостиковый атом уг-
лерода не может на-
ходиться в состоянии
£р2-гибридизации, т. Ч
к. эффективное пере- '•<
крытие p-орбит не-
возможно вследствие напряженного, неплоского строе-
ния бицикла. Так, неплоский ион апокамфилкарбония
(V) должен быть на 22 ккал/молъ менее выгоден, чем
обычный (плоский) ион типа г-бутилкарбония. Невоз-
можность образования олефина или енола также связа-
на с условиями, препятствующими ,у?2-гибридизации.
Вместе с тем жесткая пространственная структура
бицикла, при к-рой мостиковая связь экранирует
«тыльную» сторону мостикового атома углерода, пре-
пятствует синхронному бимолекулярному процессу.
Б. п. не соблюдается, когда: 1) размеры циклов ве-
лики, 2) если замещению предшествует образование
ионных пар, 3) когда замещение происходит в трех-
членном переходном состоянии. Правило установлено
Бредтом в 1924.
| Лит.: Fawcett F. S., Chem. Rev., 1950, 47, № 2,
। p. 219; Applequist D. E. and Roberts J. D.,
там же, 1954, 54, № 6, p. 1065; SchOllkopf U., Angew.
Chem., 1960, 72, № 5, S. 147. С. B. Bum».
БРИЛЛИАНТОВЫЙ z ЗЕЛЕНЫЙ (оксалат тетра-
этил-4, 4-диаминотрифенилметана) С29Н33О4^, мол. в.
473,6 — зеленовато-золотистые комочки или золоти-
ОСОСООН-
сто-зеленый порошок; раствррим в воде, спирте и
хлороформе; образует р-ры интенсивно-зеленого цвета.
При прибавлении к 0,2%-ному р-ру Б. з. конц. соля-
ной к-ты появляется оранжевое окрашивание, а при
прибавлении к 0,2%-ному р-ру Б. з. р-ра NaOH выпа-
дает бледно-зеленый осадок основания. В. з. полу-
чают конденсацией диэтиланилина с бензальдегидом;
образующийся при этом 4,4'-бис-диэтиламинотрифе-
нилметан окисляют перекисью свинца или марганца;
при взаимодействии получаемого т. о. карбинольного
основания со щавелевой к-той образуется В. в. Препа-
рат в виде 0,1—2%-ного водного или спиртового р-ра
применяют как антисептич. средство.
Лит. см. при ст. Адалин. А. И. Травин.
БРОЖЕНИЕ (ферментация) — процесс разложе-
ния органич. веществ, преим. углеводов, на более
простые соединения под влиянием микроорганизмов
465
БРОЖЕНИЕ
466
или выделенных из них ферментов. Освобождающаяся
при Б. энергия необходима для жизнедеятельности
микроорганизмов, а получаемые в результате Б. про-
стые соединения используются ими для биосинтеза;
нек-рые продукты Б. используются микроорганизмами
в качестве защитных средств в борьбе с другими мик-
роорганизмами (конкурентами). Разложение органич.
веществ, происходящее без доступа кислорода, назы-
вают анаэробным, а разложение, совершаю-
щееся с участием молекулярного кислорода, — а э-
робн.ым. Известны различные типы Б. Они име-
нуются или по субстратам, которые разлагаются, на-
пример Б. пектиновое, Б. клетчатки и т. п., или по
продуктам, которые получаются в процессе Б., на-
пример спиртовое, молочнокислое и т. п. Ниже пе-
речислены некоторые широко используемые в прак-
тике типы Б.
Продукты брожения Вещества, из к-рых получаются про- дукты брожения Организмы, вызы- вающие брожение
Ацетоуксусная Уксусная к-та Ацетонобутиловые
к-та бактерии
Ацетон Крахмал, сахара, Ацетонобутиловые,
ацетоуксусная ацетоноэтиловые,
к-та, уксусная к-та изопропиловые и
уксуснокислые бак-
терпи
Бутиловый спирт Крахмал, сахара, ма- Ацетонобутиловые и
сляная к-та изопропиловые бак-
терии
Водород (газ) Сахара Маслянокислые бак-
терии
Галактоновая Галактоза Уксуснокислые баи-
к-та терин
Галактоза Дульцит То же
Глюконовая к-та Глюкоза » »
Диокси ацетон Глицерин » . э
Глицерин Углеводы Дрожжи
Изопропиловый Крахмал, сахар Ацетоноизопропило-
спирт вые бактерии
Масляная к-та Крахмал, сахара, Маслянокислые и
клетчатка, уксус- целлюлозные бак-
ная к-та терии
Маннит Фруктоза Молочнокислые бак-
терии
Молочная к-та Крахмал, сахара То же
Муравьиная к-та Глюкоза, целлюлоза Маслянокислые, цел-
люлозные бактерии
Пропиловый Пропионовая к-та Ацетонобутиловые
спирт бактерии
Пропионовая к-та Глюкоза Пропионовокислые
бактерии
Пропионовый Глюкоза, глицерин То же
альдегид
Пировиноградная Сахара » »
к-та
Уксусная к-та Крахмал, сахара Уксуснокислые бак-
терпи
Янтарная к-та Сахара Пропионовокислые
бактерии
Винная к-та Глюкоза Бактерии субокси-
дане
Лимонная к-та Сахара, тростпико- Плесневые грибы
вая или свеклович-
ная меласса
Фумаровая к-та Сахара То же
Этиловый спирт Углеводы Дрожжи'
Наиболее важными типами Б. являются: спиртовое,
лежащее в основе рнда пищевых произ-в, напр.
виноделие (см. Вино), пивоварение (см. Пиво), полу-
чение этилового спирта и др.; молочнокислое (произ-
водства молочнокислых продуктов—простокваши, аци-
дофилина, кефира и др., а также изготовление кваса,
квашение овощей, силосование кормов и др.); масля-
нокислое (происходит на дне болот и в заболоченных
почвах, а также вызывает иногда вспучивание сыра,
банок с консервами, сквашенных овощей) и метано-
вое, вызываемое смесью различных анаэробных бак-
терий. Метановое Б. имеет большое значение, в част-
ности для очистки сточных вод (при наличии в сточ-
ных водах 0,3% взвешенных органич. веществ из
33 л сточных вод может быть получено ок. 1 л СН4
в сутки).
Одним из основных субстратов многих типов Б.
служат углеводы, расщепляющиеся в анаэробных
условиях под действием ферментов и с участием фос-
форной к-ты до пировиноградной к-ты, причем первые
стадии расщепления углеводов в анаэробных усло-
виях при многих типах Б. одинаковы и идут таким же
путем, каким идет гликолиз у животных. В качестве
переносчиков фосфора и водорода здесь участвуют
аденозинтрифосфорная — АТФ и аденозиндифосфор-
ная — АДФ к-ты (см. Аденозинфосфорные- кислоты),
коферменты — кодегидрогеназа I (Ко!) и II (Roll)
и их восстановленные формы (Ко1Н2 и Ко11Н2).
Спиртовое Б. схематически представлено на рисунке.
-О-Р-О’ч.
НО.
о=р-о-сн,*снон-сон
но-"
3 = Фосфоглицериновыи
альдегид
соя снон-сиои енон’сиои сщон -----------
Глюкоза
U ^ОН
сонснонснонснонснонсн,-о-р=о
Глюкозо-6-фосфат ^ОН
/ОН
CfljOH-CO-CHoH-CHOH-CHOHCHrO-P^O ----------
Фруктозо-6-фосфат
НО. ^0Н
о«р-о-сн,-со-снон-сноя-снон-сн,-о-р=о
ЯО 1'6-фруитозодифосфат ОН
"" Z ХОН
СН2ОН-СО-СН2-0“Р=О
ЧОН
Фосфодиоксиацетон
Н,Р0,
5 Xх* Неорганический фосфат
но-. г zSoh\
О=Р-О-СН2СНОН’СОО-Ф=0 2--------------
НОХ Ч^ОН/
1,5-дифосфоглии,ерино«ая кислота
ЯОХ I6
О=Р— о—сн2— снон-соон
нох
3-фосфоглицериноеая кислота
,оН
— 0-Р=0-х
хон
-о-Р=О-
^он
7
.ОН
Н0-р=0
о
СН2ОН'СН-СООН
2-фосфоглицериновая кислота
/ ,ОН'\
8 ,'но-Р’О- —
сн,—с—соон
Энол-фосфапировиноградная кислота
on
он
СН3 —СО-СООЯ
Пировиноградная кислота
CHjCOH
Ацетальдегид
снгснаон
Этилоеый спирт
со.
Углекислота
1 — гексокиназа; г — фосфогексоизомерара; 3 — фосфо-
фруктокиназа; 4, — альдолаза; s — дегидраза триозофос-
фата; 6 — фосфорилглицерилкиназа; 7 — фосфорилгли-
церилмутаза; з — енолаза; 9 — трансфосфорилаза фос-
фоенол пировиноградной кислоты; ю — карбоксилаза;
11 — алкогольдегидраза.
Видоизмененным типом спиртового Б. является глице-
риновое Б. Глицерин образуется в небольших количе-
ствах и при обычном спиртовом Б., но его количество
увеличивается, если Б. совершается в присутствии
бисульфита (NaHSO3, KHS03) или в щелочной среде.
При молочнокислом Б. расщепление углеводов до
467
БРОМ
468
пировиноградной к-ты совершается также, как и при
спиртовом Б., но пировиноградная к-та восстанав-
ливается при этом в молочную: СН3СОСООН -|-
-4- Ко1(Н2)— ддетикодегидрогеназа СнзСН(ОН)СООН +
Koi. Маслянокислое Б. также идет по пути спир-
тового Б. до пировиноградной к-ты или до уксусного
альдегида, в ферментативном превращении к-рых
в масляную к-ту наряду с уже упоминавшимися ко-
ферментами участвуют коэнзим А (Ко А) и флавин-
адениннуклеотид (ФАД) и его восстановленная форма
(ФАД Н2). Пировиноградная к-та, уксусный альдегид
и уксусная к-та являются промежуточными продук-
тами и при ацетоноэтиловом, ацетонобутиловом, про-
пионовокислом, 2—3-бутиленгликолевом брожениях.
В практике из аэробных Б. часто используются:
глюконовокислое — для получения глюконовой к-ты
из глюкозы, лимоннокислое — для получения лимон-
ной к-ты и фумаровокислое Б. В этих случаях исполь-
зуются плесневые грибы. Глюконовая к-та образуется
при окислении глюкозы ферментом — глюкозооксида-
зой: CeH12O6 -|- Н2О -j- 1/2О2 —► С6Н12О7 -|- Н2О. Ли-
монная к-та образуется путем соединения щавелево-
уксусной к-ты с ацетил-КоА.
К Б. относится также процесс превращения сорби-
та в сорбозу под действием нек-рых видов уксусно-
кислых бактерий — сорбозное Б., превращение глю-
козы в маннит плесневыми грибами, гидролиз таннина
с образованием галловой к-ты плесневыми грибами
и т. п. Как видно из приведенных данных, понятием
Б. характеризуются весьма различные процессы.
Лит.: Федоров М. В., Микробиология, 5 изд., М.,
1955; П р е с к о т С. и Дэн С., Техническая микробиоло-
гия, пер. с англ., М., 1952; Кретович В. Л., Основы
биохимии растений, 2 изд., М., 1956; Нейландс Д. и
Штумпф П., Очерки по химии ферментов, пер. с англ.,
М., 1958; Болдуин Э., Основы динамической биохимии,
пер. с англ., М., 1949; The chemistry and biology of yeasts.
Ed. by A. H. Cook, N. Y., 1958; Industrial fermentations. Ed.
by L. A. Underkofler and R. J. Hickey, v. 1—2, N. Y., 1954.
11. А. Полесников.
БРОМ (Вгошиш) Br — химич. элемент VII гр. пе-
риодич. системы Менделеева; относится к галогенам;
п. н. 35, ат. в. 79,916. Природный Б. состоит из Двух
стабильных изотопов с массовыми числами 79 (50,56%)
и 81 (49,44%). Получено неск. радиоактивных изото-
пов. Из них наиболее интересен Вг80, на примере к-рого
И. В. Курчатовым с сотрудниками было открыто яв-
ление ядерной изомерии. Изотоп Вг82 = 35,87 ча-
са) используется в радиохимич. исследованиях. Кон-
фигурация внешних электронов атома 4s24/>5; энер-
гия ионизации (в эв): Вг°^-Вг1—>Вг2!—>Вг3+^-
—> Вг4+—> Вг5~ соответственно равна 11,844; 21,584;
35,890; ...; 119,70; сродство коэлектрону Вг° — Вг~
3,56 эр. Атомный радиус 1,19 А, ионный радиус Вг
1,96 А. Б. открыт в 1826 А. Баларом. Название эле-
мента связано с его неприятным запахом (от греч.
Рр<йр.ос — зловоние).
Содержание Б. в земной коре 1,6 • 10 4 вес.%. Рас-
сеянный в диффузном состоянии в магматич. горных
породах, Б. выщелачивается поверхностными водами
и концентрируется в калийных солях (где изоморфно
замещает часть С1), в нек-рых водорослях, остаточной
рапе, а также нефтяных водах, откуда он и добывается.
Физические и химические свойства. При обычных
условиях свободный Б. — тяжелая жидкость с резким
запахом, в отраженном свете темно-фиолетового,
почти черного, а в проходящем — темно-красного
цвета; легко образует желто-бурые пары; т. кип.
58,8°, т. пл. —7,2°. Твердый Б. — ярко-красная
масса с металлич. блеском, к-рая при —252° стано-
вится бесцветной; 1крит 311°, рк 102 ат; теплота
плавления 16,14 кал/г, теплота испарения 44,8 кал/г.
Плотность жидкого Б. 3,1023 (25°), твердого — око-
ло 3,4 (при —7,3°). Давление паров Б. в зависимости
от температуры (в мм рт. ст.): 30,0 (при —12°), 66
(0°), 173 (20°), 392(40°) и 760 (58,8°). Молекула Б.
в жидкости и в парах двуатомна. Степень диссоциа-
ции Вг2 — 2Вг 0,16% при 800° и 18,3% при 1284°,
теплота диссоциации 45,4 ккал/моль. Атомная теп-
лоемкость жидкого брома 8,6 (13—45°); твердого
5,6 кал/ г-атом-град (от —192° до —108°). Термич.
коэфф, линейного расширения жидкого Б. 11-10 4
(при 0°—30°). Уд. электросопротивление твердого Б.
больше 108 ом-см. Диэлектрич. проницаемость 3,2
(при комнатной темп-ре). Молекулярный бром диама-
гнитен.
Б. растворим в спирте, эфире, бензоле, хлороформе,
сероуглероде, четыреххлористом углероде, четырех-
хлористом титане. В 100 г воды растворяется 3,53 г
Б. (при 20°). Ниже 5,84° из водного р-ра Б. осажда-
ются гранатово-красные октаэдрич. кристаллы гид-
рата Вг2 • 8Н2О. Насыщенный водный р-р Б. (ок. 3%),
называемый бромной водой, имеет желто-
бурую окраску. Растворимость воды в Б. составляет
ок. 0,05%.
По реакционноспособности Б. занимает промежу-
точное положение между С1 и J (см. Галогены). Глав-
ные валентности: —1 (бромиды) и -|-5 (броматы). Со-
единенияс валентностью-)-1 (гипобромиты)менееустой-
чивы, чем соответствующие производные С1 (гипо-
хлориты). Существование бромитов (валентность -|-3)
еще окончательно не доказано, а перброматы (валент-
ность -(-7) вообще не получены. Б. является довольно
сильным окислителем, он вытесняет J из его соедине-
ний; сам же Б. вытесняется из своих соединений хло-
ром. С молекулярным кислородом Б. непосредственно
при обычных условиях не реагирует. Его окислы
получают косвенными методами. Окисел Вг3О8,
образующийся при действии озона на Б., представ-
ляет собой бесцветные кристаллы, устойчивые только
при низких темп-pax или в избытке озона. Твердая
коричневая двуокись ВгО2, устойчивая ниже
—40°, получается при действии тихого электрич.
разряда на охлажденную смесь Вг2 и О2. Медленное
нагревание ВгО2 в вакууме выше —40° приводит
к образованию бурой закиси Вг2О, т. пл. —17°.
С водородом Б. соединяется при нагревании, образуя
бромистый водород НВг.
Известно неск. неустойчивых соединений Б. с гало-
генами, получающихся при непосредственном взаи-
модействии элементов. Из них наиболее важны трех-
фтористый бром BrF3 (светло-желтая жид-
кость, т. пл. 8,8°, т. кип. 127°) и пятифтористый
бром BrF5 (бесцветная жидкость, т. пл. —61,3°,
т. кип. 40,5°), отличающиеся весьма высокой химич.
активностью. Напр., взаимодействие BrF5 с водой про-
текает со взрывом. BrF5 применяется как фторирую-
щий агент. Нагревание в запаянной трубке смеси серы
с Б. приводит к образованию бромистой с'е р ы
S2Br2 — гранатово-красной, маслянистой жидкости,
т. пл. —40° и т. кип. 54°/0,2 мм. Бромистая сера рас-
творима в СС14, CS2, бензоле; водой разлагается:
2S2Br2 -|- 2Н2О = SO2 -|- 3S -|- 4НВг. Известен так-
же бромистый тио пил SOBr2, получаемый
действием двуокиси серы па Б. в присутствии РС13
или при взаимодействии SOC12 и НВг. Реагируя с ме-
таллами, Б. образует бромиды. Нек-рые металлы
(А1, К, Sb) реагируют с сухим Б., другие (Fe, Zn, Bi,
Na) — с влажным; с РЬ и Ag реакция идет очень мед-
ленно, а Та и Pt по отношению к Б. устойчивы. При
взаимодействии Б. с фосфором образуется трех-
бромистый фосфор РВг3, бесцветная жидкость,
разлагающаяся водой; плотн. 2,852, т. пл. —40°,
т. кип. 4-175,3°. С азотом Б. не реагирует. Действием
избытка аммиака на Б. под уменьшенным давлением
и при —75° можно получить вещество красного цвета
состава NBr3 • 6NH3, разлагающееся со взрывом при
469
БРОМ — БРОМАТОМЕТРИЯ
47 J
—70°. Известен б р ом а я ид BrN3 — красная жид-
кость с т. пл. —45°.
Растворение Б. в воде сопровождается реакцией:
Вг2 Ц-Н2О .-* ПВгО Ц-НВг. Константа равновесия
этой реакции К = 5,8 • 10 9 (при 25°). При стоянии
на свету из бромной воды выделяется О2, при нагрева-
нии — Вг2. Б. гидролизуется в меньшей степени,
чем хлор. При добавлении кислоты гидролиз пре-
кращается, а в присутствии щелочи проходит почти
количественно. Бромноватистая к и с л о-
т а НВгО обладает слабыми кислотными свойствами
и является сильным окислителем; существует только
в водных растворах, под действием света разлагается
на НВг и О2. При взаимодействии Б. с р-рами щело-
чей при нагревании образуются бромиды и броматы —
соли бромноватой к-ты HBrO3 : 3Br2 -|- 6NaOH =
= NaBrO3 -|- 5NaBr -|- ЗН2О. Бром нова тая
кисло т а в свободном состоянии не получена, су-
ществует в водных р-рах, к-рые могут быть сконцен-
трированы только до 50,6%-ного содержания НВгО3.
На воздухе при стоянии и особенно при нагревании
р-ры НВгО3 разлагаются. Окислительные и кислот-
ные свойства НВгО3 выражены примерно в такой же
степени, как и у хлорноватой к-ты. Бромноватая к-та
получается пропусканием хлора через бромную воду:
Вг2 -|- 5С12 -|- 6Н2О = 2HBrO3 -j- 10НС1 или при вза-
имодействии бромата бария с серной к-той.
Соли бромноватой кислоты — броматы — устой-
чивы при обычных условиях; при высокой темп-ре
они проявляют окислительные свойства: КВгО3 -|-
-|- ЗС = 2КВг -|- ЗСО2. Щелочные броматы при на-
гревании разлагаются: 2КВгО3 = 2КВг -|- ЗО2. Раз-
ложение броматов менее активных металлов протекает
по ур-нию: 2Mg(BrO3)2 = 2MgO -ф- 2Вг2 -ф- 5О2. Из
броматов важнейшими являются соли калия, натрия
и бария. КВгО3 — бесцветные кристаллы, плотн.
3,25, при 400° разлагается; растворимость (в г на 100 г
воды): 3,1 (0°), 6,9 (20°), 49,7 (100°); получают электро-
литич. окислением КВг; применяют в качестве окис-
лителя. Бромат натрия NaBrO3 — бесцвет-
ные кристаллы, т. пл. 380°, плотн. 3,34, растворимость
(в г на 100 г воды): 27,5 (0°), 36,4 (20°), 90,8 (100°).
Получают электролитич. окислением NaBr и при вза-
имодействии Б. с едким натром или содой; использу-
ют в качестве окислителя. Бромат бария из
воды кристаллизуется в виде Ва(ВгО3)2 Н2О; бес-
цветные кристаллы; плотн. 4,0. При нагревании до
180—200° теряет кристаллизационную воду; при
270° начинает разлагаться. Растворимость (в г безвод-
ной соли на 100 г воды): 0,287 (0°), 0,657 (20°), 5,71
(100°). Вследствие незначительной растворимости
бромат бария может быть получен обменной реакцией
хлорида бария со щелочными броматами. Применяется
для получения бромноватой к-ты и других броматов.
Качественно свободный Б. определяют по измене-
нию окраски фуксина в красно-фиолетовый цвет или
по желтому окрашиванию слоя органич. раствори-
телей (С6Н6, СНС13, CS2, СС14). При действии конц.
H2SO4 на твердые бромиды выделяется газообразный
НВг, окрашенный в буроватый цвет в результате
частичного окисления до Вг2. Элементарный Б. мо-
жет быть определен реакцией с KJ; выделяющийся
иод титруют Na2S2O3. В весовом анализе Б. опреде-
ляют в виде AgBr, в объемном — титрованием р-ром
AgNO3 (см. также Аргентометрия). Небольшие ко-
личества бромидов в присутствии больших количеств
хлоридов можно определять путем окисления броми-
дов до свободного Б. хромовой к-той в холодном сер-
нокислом р-ре. Для колич. отделения Б. от хлора
используют окисление бромистоводородной к-ты тел-
луровой к-той или перманганатом калия с последую-
щей отгонкой свободного Вг2. Б. (и хлор) от иода от-
деляют отгонкой последнего при кипячении из р-ра,
содержащего NaNO2 (метод Гуча). Вг~ и С1“ могут быть
определены в смеси галогенионов с помощью ионо-
обменной хроматографии. Относительно определения
Б. в органич. соединениях см. Галогенов определение.
Получение. Для промышленного получения Б. ис-
пользуется озерная и морская рапа, щелока от пере-
работки калийных солей (сильвинит, карналлит) и
буровые воды (0,0065—О,4О°/о Б.) в виде бромидов.
Его выделяют пропусканием хлора при предваритель-
ном подкислении реакционной смеси (pH 3—3,5),
а затем отгоняют из р-ра водяным паром или воздухом.
Все операции при получении Б. проводят в специаль-
ных колоннах, выложенных внутри кислотоупорным
материалом. Предварительно нагретый до 70—75°
бромсодержащий р-р поступает в верхвюю часть
колонны, а снизу направляются хлор и водяной пар.
Темп-pa в реакционном пространстве поддерживается
при 110—115°. Пары Б. конденсируются в керамико-
вых холодильниках. Далее Б. отделяют от воды и
очищают. Применение воздуха вместо водяного пара
несколько упрощает аппаратуру и, кроме того, стано-
вится излишним предварительное нагревание рапы.
В этом случае бромсодержащий р-р также подкисляют,
хлорируют и направляют в верхнюю часть колонны.
Снизу идет мощный поток воздуха. Образовавшуюся
бромвоздушную смесь очищают от небольших приме-
сей хлора обработкой бромидами. Затем Б. погло-
щают раствором бромистого железа FeBr2, причем
получается FeBr3. Его восстанавливают железными
стружками до РеВг2, часть к-рого возвращают на
поглощение брома. По другому способу, к воздуху,
содержащему Б., добавляют SO2, к-рый, реагируя с
Б. в присутствии влаги, образует смесь бромистово-
дородной и серной кислот. Из полученных концен-
тратов выделяют Б. при действии хлора. Менее рас-
пространен электролиз бромных р-ров и экстракция
Б. органич. растворителями.
Техника безопасности. Б. — ядовит. Поэтому при
работе с ним необходимо соблюдать меры предосто-
рожности. При содержании Б. в воздухе ок. 0,001%
наблюдается раздражение слизистых оболочек, голово-
кружение, кровотечение из носа, слезотечение, ка-
шель. Содержание Б. в воздухе ок. 0,02% приводит
к удушью, спазмам и заболеванию дыхательных путей.
Допустимая концентрация паров Б. в воздухе
0,002 мг/л. Попадание жидкого Б. на кожу вызывает
зуд, а при длительном действии образуются медленно
заживающие язвы. При легком отравлении парами Б.
необходимо вдыхание аммиака. В тяжелых случаях
больного помещают в теплое помещение и дают вды-
хать кислород. Кожа, обожженная Б., промывается
многократно водой и р-ром соды, смазывается мазью,
содержащей бикарбонат натрия.
Применение. Б. находит довольно широкое приме-
нение. Он является исходным материалом для полу-
чения различных бромистых солей и органич. бромпро-
изводных. Наибольшие количества Б. используются
при произ-ве бромистого опгила и дибромэтана.
Нек-рые органич. производные (бромалин, ксероформ,
бромистая камфара) находят применение в медицине.
Б. и бромная вода в аналитич. лабораториях исполь-
зуются в качестве окислителей при определении мно-
гих элементов.
Лит.: Ксензенко В. И. и Ст аси не вич И. С-,
Технология брома и иода, М., 1960; U 1 1 m а п п, 3 Aufl., Bd 4,
Mtlnch. —В., 1953, S. 727—54; К i г k, v. 2, N. Y., 1948, p. 629 —
660; Руководство по препаративной неорганической химии, под
ред. Г. Бауера, пер. с нем., М., 1956, гл. 2, § 5; Гилле-
б р а н д В. Ф. [и др.], Практическое руководство по неорга-
ническому анализу, пер. с англ., М., 1957, гл. 47; Omelln,
8 Aufl., System-Nummer 7, Brom, В., 1931; Mellor, v. 2,
L.—N. Y. —Toronto, 1946; то же, v. 2, 1952; тоже, Suppl.,
v. 2, pt 1, L.—N. Y. — Toronto, [1956]. Г. H. Пирогова.
БРОМАТОМЕТРИЯ — титриметрич. метод количе-
ственного анализа, основанный на применении в ка-
471 БРОМАЦЕТОФЕНОН -
честве рабочего р-ра бромата калия как окислителя.
Раствор устойчив при хранении, его титр можно кон-
тролировать иодометрически. В кислом р-ре бромат
восстанавливается до бромида через стадию, выделе-
ния элементарного брома, к-рый и является фактиче-
ски окислителем в этих условиях. Бромат калия
применяют для титрования 3-валентных мышьяка
или сурьмы в сильно солянокислом р-ре (после от-
гонки их в виде летучих хлоридов), Т11, гидразина,
Си1, Sn11 (в отсутствие кислорода воздуха). Бромат
калия служит также для определения ряда органич.
веществ (фенол, анилин, 8-оксихинолин и нек-рые др.)
в присутствии избытка бромида калия в кислом р-ре.
Бромирование 8-оксихинолина является основой ко-
лич. определения многих катионов после осаждения
их в виде 8-оксихинолинатов (Mg, Al, Bi, Fe, In и др.).
Индикаторами являются азокрасители (метилкрасный,
метилоранжевый, бензопурпурин 4В и др.), индиго-
сульфоновые к-ты, индигокармин, хризоидин R, бордо,
к-рые необратимо обесцвечиваются от избытка эле-
ментарного брома.
Лит.: К ольт го ф И. М. и Сен дел Е. Б., Количе-
ственный анализ, пер. с англ., 3 изд., М,—Л., 1948; Кольт-
гоф И. М. [и др.], Объемный анализ, пер. с англ., т. 3,
М., 1961. В. Г. Типиова.
БРОМ АЦЕТОФЕНОН (бромметилфенилкетон), мол.
вес 199,06, СвН5СОСН2Вг — бесцветные кристаллы;
т. пл. 51°; т. кип. 133—135°/12 мм. Б. нераство-
рим в воде, хорошо растворяется в органических рас-
творителях, устойчив к действию воды, энергично
взаимодействует со спиртовым и водноспиртовым
раствором N a2S с образованием нетоксичного продукта.
Получают Б. бромированием ацетофенона. Б. обла-
дает более сильным лакримагенным действием, чем
хлорацетофенон. Р. Н. Стерли».
БРОМБЕНЗИЛЦИАНИД (нитрил фенил бромуксус-
ной кислоты) СвН5 — CH(Br)CN, мол. в. 196,060—бес-
цветные кристаллы (технич. продукт — красновато-ко-
ричневого цвета), т. пл. 25,4°; т. кип. 137°/15 мм; пло-
хо растворим в воде и хорошо — в органич. раствори-
телях, устойчив к действию воды и окислителей, раз-
лагается при нагревании выше 120—140°. Б. энергич-
но взаимодействует со спиртовым р-ром Na2S, к-рый
может быть использован как дегазатор; в присутствии
ряда металлов Б. разлагается, металлы при этом
подвергаются интенсивной коррозии. В основе тех-
нологии. метода получения Б. лежит след, ряд пре-
вращений: СвН5СН3^ СвН6СН2С1 --a-'G6H5GH2GN-^
CeH5[CH(Br)CN]. Б. обладает сильным лакримаген-
ным действием, к-рое проявляется уже при концен-
трации 3 • 10“*. мг/л, непереносимая концентрация
4 • 10 3 мг/л. Б. был предложен в качестве ОВ в
конце первой мировой войны. р. н. Стерли».
БРОМБЕНЗОЛ СвН5Вг, мол. в. 157,0 — бесцвет-
ная жидкость; т. пл. —30,6°; т. кип. 156,02°/760 мм,
43°/18 мм; 1,4951; ng 1,5572; теплота испарения
57,63 кал/г (155°); т. воспл. 51°; нерастворим в воде,
смешивается со спиртом, эфиром, хлороформом, бен-
золом. Замещение брома в Б. происходит лишь в
жестких условиях. При действии NaOH (300—370°)
бром замещается гидроксилом; замещение Вг нитриль-
ной группой происходит при нагревании с. KCN или
CuCN при 200°; замещение 8О3Н-группой и ацилом —
при нагревании с Na2SO3 и солями жирных к-т при
200° в присутствии солей меди. С ариламинами и ацил-
ариламинами в присутствии порошкообразной меди
или иодистой меди образует диариламины. Нагре-
вание с натрием в эфире и бензоле ведет к образова-
нию дифенила, с магнием легко и с высоким выходом
(более 90%) образует Гриньяра реактив CeH6MgBr,
с литием— фениллитий CeH5Li; при действии HNO3
или H2SO4 образуются соответственно бромнитробен-
БРОМИСТЫЙ МЕТИЛ 472
золы и бромсульфокислоты; при нагревании с А1С)3
получается смесь дибромбензолов, трибромбензолов
и бромдифенилов. Получают Б. бромированием бен-
зола на холоду в присутствии железных или алюми-
ниевых стружек. Наряду с монобромбензолом обра-
зуется небольшое количество о- и п-дибромбензо-
лов. Б. применяется в органич. синтезе (см., напр.,
Гриньяра реакция, Фиттига реакция).
Л. С. Поваров.
БРОМИДЫ — соединения брома с элементами, Б.
металлов (NaBr, КВг) в большинстве случаев — ти-
пичные соли. Б. неметаллов (CBr4, SiBr4) — легко-
плавкие, легколетучие вещества с молекулярной ре-
шеткой; в воде либо трудно растворимы, либо тотчас
ею разлагаются; нек-рые из Б. неметаллов являются
бромангидридами (РВг3, ВВг3). К ним относятся также
Б. многовалентных металлов (TiBr4, SnBr4, AsBr3).
Промежуточное положение между типичными солями
и бромангидридами занимают Б. Fe, Zr, Hf и т. д.
Б. образуются при непосредственном соединении эле-
ментов, при взаимодействии НВг с металлами, окис-
лами, гидроокисями и карбонатами, по обменной
реакции ВаВг2 с сульфатами соответствующих ме-
таллов. О Б. отдельных элементов см. Аммония
галогениды, Бериллия галогениды, Вольфрама галоге-
ниды, Серебра галогениды и др. г. Н. Пирогова.
БРОМИСТОВОДОРОДНАЯ КИСЛОТА — раствор
бромистого водорода в воде.
БРОМИСТЫЙ ВОДОРОД НВг—соединение брома
с водородом. Бесцветный удушливый газ, сильно ды-
мящий на воздухе; т. пл. —86,9°, т. кип. —66,8°,
гкрит. 91°. Ркрит. 84 ат- плотн. 2,17 (при — 68°). Дав-
ление пара (в атм) равно: 17,1 (при 12°), 23,8 (26°),
40,2 (50°) и 59,2 (70°). Б. в. термически устойчив, сте-
пень диссоциации составляет 0,5% (при 1000°) и
1,1% (1220°). Теплота образования AF72(IS = —8,6
ккал/моль. Молекула Б. в. имеет полярную связь;
дипольный момент 0,79D. Растворимость в воде
(г НВг на 100 г Н2О при 760 мм рт. ст.); 221 (0°),
193 (25°), 130 (100°).
Плотность водных растворов
г НВг/100 г раствора
j 65
10 1 20 30 | 40 50 | 60
1,0723 1.1579 1,2580 1,3772 1,5173 1,6787
1,7675
Раствор НВг в воде — бромистоводород-
ная кислота, бесцветная (иногда желтоватая)
жидкость с резким запахом. Ее азеотропная смесь
содержит 47,63% НВг и кипит при 124,3° (при 760
мм рт. ст.). Бромистоводородная к-та — одна из
самых сильных неорганич. кислот, кажущаяся сте-
пень диссоциации в 0,1 н. р-ре 93,5%. Довольно силь-
ный восстановитель, медленно окисляется даже при
стоянии на воздухе, выделяя свободный бром. Об-
разует соли, называемые бромидами. При попадании
на кожу-вызывает зуд и воспаление.
Б. в. получают взаимодействием паров брома с
водородом при высокой темп-ре. Темп-ру реакции
можно понизить катализатором — активированным
углем или платинированным асбестом. В лаборатории
НВг получают обычно взаимодействием РВг3 с Н2О,
бромидов с H2SO4 или тетралина с бромом. НВг
применяют для приготовления бромидов, органич.
бромпроизводных, в качестве катализатора в нек-рых
органич. реакциях. Разбавленные водные р-ры НВг
используются в медицине.
Лит. см. при ст. Бром. Г. И. Пирогова.
БРОМИСТЫЙ МЕТИЛ (бромметан) СН3Вг, мол. в.
94,95 — бесцветный газ с характерным запахом;
т. пл. —93,7°; т. кип. 3,6°; 1,730; d,8» 3,974;
473
БРОМИСТЫЙ МЕТИЛЕН — БРОМУРАЛ
474
1,4432. Б. м. растворим в большинстве органич. рас-
творителей; с холодной водой образует кристалло-
гидрат; в 100 г воды при 20° растноряется 1,75 г.
Б. м. негорюч в широком интервале концентраций
и воспламеняется только при содержании в воздухе
13,5—14,5%. Давление пара 1420 мм рт, ст. (20°);
теплота испарения 60,2 кал/г (3,6°); гкрит 194°; вес
1 л 3,9739 г (25°, 1 ат). Б. м. легко гидролизуется
щелочью до метанола, метилирует амины, обладаю-
щие достаточно сильной основностью с образованием
четвертичных аммониевых солей, с H2S дает меркап-
тан и тиоэфир. Металлы, кроме щелочных и А1 и Mg,
ивертны к чистому, сухому Б. м.; но на поверхности
Zn, Fe, Sn происходит образование этана (см. Вюрца
реакция), особенно в присутствии небольших коли-
честв спирта. Предельно допустимая кбнцентрация
в воздухе 0,005 мг[л. Б. м. получают взаимодействием
метанола и бромистого водорода. Наличие Б. м. в
воздухе определяется пробой Бейльштейна. Приме-
няют Б. м. для уничтожения насекомых-вредителей,
находящихся в с.-х. продуктах (зерно, фрукты, та-
бак, ХЛОПОК). Ю. А. Чебурков.
БРОМИСТЫЙ МЕТИЛЕН (дибромметан) СН2Вг2,
мол. в. 93,94 — бесцветная жидкость; т. пл. — 52,7°;
т. кип. 96,9°;d|" 2,4970; 1,542. Б. м. растворим
в органич. растворителях; в 100 г воды при 20° рас-
творяется 1,148 г. Б. м. получают обработкой хлори-
стого метилена стехиометрия, количеством А1Вг3.
Обладает наркотич. действием и примерно в 8 раз
токсичнее бромистого метила. Находит применение
как промежуточный продукт для синтезов и в качестве
тяжелой жидкости для разделения твердых в-в по
различию их плотностей (иммерсионный анализ).
К). А. Чебурков.
БРОМИСТЫЙ ЭТИЛ (бромэтан) С2Н5Вг, мол. в.
108,98 — бесцветная жидкость с запахом эфира;
т. пл. —119°; т. кип. 38,3°; 1,4586; nff 1,4211;
в 100 г воды при 20° растворяется 0,914 г; со спиртом
образует азеотроп, содержащий 3% спирта, т. кип.
37,6°. Б. э. получается обработкой спирта бромидами
и серной к-той; обладает свойствами, типичными для
галогеналкилов. В хирургии Б. э. применяется как
НарКОТИК ДЛЯ ИНГаЛЯЦИИ. Ю. Д. Чебурков.
БРОМНОЕ ЧИСЛО — количество брома в граммах,
присоединяющегося к 100 г органич. вещества. Б. ч.
характеризует содержание ненасыщенных соединений
в насыщенных или в ароматич. соединениях. К одной
двойной связи присоединяются два эквивалента брома.
При определении Б. ч. в присутствии в-в, вступаю-
щих с бромом в реакцию замещения, вводят поправку,
определяя количество образовавшихся ионов Вг-.
При анализе в-в, нерастворимых в воде, навеску рас-
творяют в СНС13 или СС14 и пользуются р-ром брома
в том же растворителе.
Лит.: Мейер Г., Анализ и определение строения орга-
нических веществ, пер. с нем., Харьков—Киев, 1935; В а fl-
бе л ь С., Идентификация органических соединений, пер.
с англ., М., 1957; Л асто вс кий Р. II., Вайнштейн
Ю. И., Технический анализ в производстве промежуточных
продуктов и красителей, 3 изд., М., 1958.
БРОМОНИЕВЫЕ СОЕДИНЕНИЯ — см. ₽ Галоген-
ониевые соединения.
БРОМОФОРМ (трибромметан) СНВг3, мол. в.
252,77 — бесцветная или слегка желтая жидкость;
т. пл. 7,7°; т. кип. 149,6°; 2,8912; 1,5980; на
воздухе легко разлагается, для стабилизации до-
бавляют 3—4% спирта. Б. легко вступает в реакции
радикального типа, присоединяется к олефинам, при-
чем, в отличие от хлороформа, под действием света
или перекисей разлагается на радикалы СНВг2 и
Вг, в то время как первый дает радикалы СС13 и Н.
Одно из характерных свойств Б. — легкость образо-
вания дибромкарбена при щелочном гидролизе; по-
следний далее может присоединяться к непредельным
соединениям с образованием дибромциклопропаноных
производных:
СНВгз + КОН -» СВг2 + КВг + Н2О
^>С=С<+ СВг2—*>СХ/С<
СВг2
Получают Б. обработкой хлороформа бромистым
алюминием или электролизом бромистого калия в
спирте (действие на спирт брома и щелочи). Приме-
няют Б. в качестве промежуточного продукта при
синтезе фармацевтич. препаратов (антиспазматиче-
ское, снотворное), а также для разделения минералов
(имерсионный анализ).
Лит.: Khar ash М. [а. о.], J. Am. Chem. Soc., 1947,
69, № 5, р. 1100. Ю. А. Чебурков.
БРОМСТИРОЛ С8Н7Вг, мол. в. 183,05 — известны
два изомера: а-Б. (I) и р-Б., существующий в двух
стереоизомерных формах: цис-(П) и транс-(Ш).
СВг=СН2
Н Вг
i I
с=сн
/
а-Б ромстирол (1-бром-1-фенилэтилен) — бес-
цветная жидкость с желтоватым оттенком; т. пл.
—43,5°; т. кип. 160°/75 мм; 71°/ 8 мм; 1,406;
1,5881. цис-р-Б ромстирол — бесцветная
жидкость; т. пл. —7,5°; т. кип. 108°/26 мм, 71°/6 .и.к;
г/;11 1,427; пр' 1,5590 (по другим данным, 1,6007);
хорошо растворим в спирте и эфире; нерастворим в
воде. Получают цмс-р-бромстирол нагреванием бром-
бензальацетофенона с порошкообразным NaOH:
CeH5CH=CBrCOCeH5 + NaOH
— CeH5CH=CHBr + CeH5COONa
транс-р-Б ромстирол — светло-желтая жид-
кость с запахом гиацинта; т. пл. +7°; т. кип. 219—
221°/760 мм (с небольшим разл.), 102,30°/15 мм,
т. всп. 98°; 1,416; «д’ 1,6000—1,6096; растворяет-
ся в спирте и других органических растворителях,
нерастворим в воде. ту/анс-р-Бромстирол устойчив
к действию водных растворов щелочей; спиртовый
раствор щелочи превращает его при 130—135° в
фенилацетилен СвН5С=СН и р-этокси-а-фенилэтилен
СвН6СН=СНОС2Н5. Наиболее просто /пране-р-бром-
стирол получают из коричной к-ты:
СвН3СН=СНСООН + Вг2 — СвН5СНВгСНВгСООН
— СвН5СН=СНВг + СО2 + NaBr + NaHCO3
Идентифицируют этот изомер в виде анилида корич-
ной к-ты CeH6CH=CHCONHCeH5, т. пл. 115°. Этот
анилид получают при взаимодействии магнийбром-
стирола и фенилизоцианата, цис- и /пранс-З-Бромсти-
ролы при действии брома образуют трибромпроиз-
водное СвН5СНВгСНВг2, т. пл. 32—38°. транс-р-Бром-
стирол применяют в парфюмерии для приготовления
отдушек мыла и композиций нек-рых духов и оде-
колонов И др. С. И. Кора-
БРОМУРАЛ (а-монобром-изовалероилмочевина)
(CHs)2CHCHBrCONHCONH2, мол. в. 223,09 — белые
кристаллы горьковатого вкуса со слабым запахом;
т. пл. 145—150°; плохо растворим в холодной воде
(1:450), легко растворим в горячей воде, спирте,
эфире и разбавленных р-рах щелочи. При нагревании
Б. с р-ром щелочи и подкислении выделяется СО2
и обнаруживается запах изовалериановой к-ты. Если
к р-ру Б. в разб. HNO3 добавить р-р AgNO3, выде-
ляется творожистый осадок AgBr, Для количественного
БРОМФЕНОЛЫ - БРУЦИН
476
определения Б. нагревают с NaOH. прибавляют раз- [
бавленные HNO3 и AgNO3 и титруют избыток NO^ ;
0,1 н. р-ром NH4SNC с железоаммониевыми квасцами. |
Получают Б. взаимодействием бромангидрида а-мо- '
нобромизовалериановой к-ты с мочевиной. Б. при-
меняют как снотворное и успокаивающее средство.
Лит. см. при ст. Адалин. Е. С. 1'оловчинскан.
БРОМФЕНОЛЫ ВгС6Н4ОН, мол. в. 173,02 — три
изомера (о-, м- и n-Б.); мало растворимы в воде,
растворяются в обычных органич. растворителях;
из полибромфенолов наиболее изучены 2,4-дибром-
фепол Вг2С6Н3ОН и 2,4, 6-трибромфенол Вг3С6Н2(ОН).
Свойства Б. приведены в таблице.
Изомер Т. пл. °C 1 Т. кип., °C* К”
орто- -10 194 1,5529 (80°) 9,78 • 10-1»
мета- 33 236 1135—140 (12 мм) —. 4,37 • 10-10
пара- 64 j 237—238 1,840 (15°) 1,55 • 10-10
2, 4- 40 238-239 —
2, 4, 6- 96 возгоняется 2,55 (20°) —
* Все значения, кроме оговоренных, при 760 лл< рт. rm.
»• Константа диссоциации при 2 5° в 30%-ном этиловом j
спирте.
Б. обладают более сильными кислотными свойства-
ми, чем фенол. При бромировании избытком брома о-
и n-Б. превращаются в 2,4,6-трибромфенол, щ-Б. —
в смесь 2,3,4- и 2,4,5-трибромфенолов; NaNO3 в
разб. H2SO4 нитрует о-Б. до 2-бром-4-нитрофенола,
.«-Б. — до З-бром-4-нитрофенола и я-Б. до 4-бром-
2-нитрофенола. При сплавлевии с КОН все Б. пре-
вращаются в резорцин.
я-Б. получают бромированием фенола в сероугле-
роде при темп-ре не выше 4-5°:
С6Н5ОН + Вт - п-В1СвН4ОН + НВт
о-Б. образуется при разложении перегретым паром
2-бром-4,6-фенолдисульфоки слоты:
ОН ОН
SO3H
а также при бромировании фенола в отсутствии рас-
творителя при повышенной темп-ре. Все три изомера
получаются из соответствующих бромфенилдиазони-
евых солей при нагревании их водных растворов
с NaOH:
BrC„H4N2Cl + NaOH — ВгС6Н-,ОН + N3 % NaCl
или разложением оксифенилдиазонийбромидов Од-
нобромистой медью Сц2Вг2:
HOC6H4N»Br HOCeH4Br % N-.
Б. обладают бактерицидным действием; находят
некоторое применение как фунгициды; из них полу-
чают 2, 4, 6-трибромфенол, основная висмутовая соль
к-рого (ксероформ) (С6Н2Вг3О)2 • Bi(OH) • Bi2O3 при-
меняется как антисептик. Б. используются в синтезе
нек-рых красителей. м. Ф. Турчинский.
БРОМЦИАН — см. Галогеноцианы.
БРОНЗА — см. Меди сплавы.
БРОУНОВСКОЕ ДВИЖЕНИЕ — беспорядочное,
непрекращающееся движение мелких частиц (до
5 мк), взвешенных в жидкости или газе, вызываемое
тепловым движением молекул окружающей среды.
Впервые описано Р. Броуном в 1827. Интенсивность
Б. д. зависит только от темп-ры, внутреннего трения
(вязкости) среды и от размеров частиц, усиливаясь
с ростом темп-ры и уменьшением размера и уменьшаясь
с возрастанием вязкости. В 1905—06 А. Эйнштейн
и М. Смолуховский дали полную колич. молекулярно-
статистич. теорию Б. д. Эта теория приводит для ша-
рообразных частиц к основному ур-нию Б. д.:
£2 = Д^= ——— • t
где Дж2 — среднее значение квадрата смещения ча-
стицы в определенном, но произвольно выбранном
направлении х, R — универсальная газовая постоян-
ная, Т — абс. темп-pa, т] — вязкость, а — радиус части-
цы, N — число Авогадро и t — время наблюдения. Из-
вестно, что ЯТ/&ях]аМ = D, где D — коэфф, диффузии
частиц в данной среде; отсюда следует: Дж2 = 2Dt.
Вследствие полной беспорядочности Б. д. необхо-
димо усреднять не сами смещения или их проекции,
а квадраты этих величин, т. к. смещения одинаковой
величины, но противоположные по знаку, равноверо-
ятны и при усреднении дадут 0. Усреднение квадра-
тов смещений производится из большого числа изме-
рений для одного и того же t, к-рое выбирается про-
извольно, по не меньшим, чем время затухания ско-
рости частицы в среде данной вязкости (для газа
~ 10-13 сек, для жидкости 10~8 сек).
Экспериментальная проверка основного ур-ния
Б. д., проведенная Ж. Перреном, Т. Сведбергом и др.,
полностью подтвердила его справедливость, утвердив
тем самым общность молекулярно-статистич. представ-
лений. Измерения броуновских смещений позволяют
судить о размерах коллоидных частиц, лежащих за
пределами разрешающей способности оптич. микро-
скопов. С. И. Вавилов и Е. М. Брумберг разработали
метод исследования Б. д. при помощи микро- или ульт-
рамикрофотосъемки. Точная теория этого метода дана
А. Н. Колмогоровым и М. А. Леонтовичем. Этот метод
удобен для определения вязкости в микрообъемах
жидкой среды.
Кроме рассмотренного поступательного Б. д.,
происходит и вращательное Б. д., заметное даже для
частиц диаметром в неск. десятков микрон, выражаю-
щееся средним квадратом углового смещения вокруг
мгновенной оси: ф2 = RT IMvc\asN. Б. д. нитей, стре-
лок и др. подвижных частей тонких измерительных
приборов определяет предел их чувствительности,
соответствующий порогу сообщаемой им энергии,
соизмеримой с энергией теплового движения. Посту-
пательное Б. д. является причиной диффузии, т. е.
стремления к равномерному распределению частиц,
а вращательное приводит к их дезориентации, тогда
как внешние силовые поля вызывают обратные эффек-
ты — концентрирование частиц и их ориентацию.
Изучение Б. д. послужило основанием для широ-
кого развития статистич. методов исследования в
физике и физич. химии и, в частности, метода флук-
туаций, весьма плодотворного не только в области
молекулярно-термодинамич. явлений, но и в электро-
и радиотехнике, электронной оптике и др. областях.
Лит.: Эйнштейн А., Смолуховский М.,
Брауновское движение. Сб. ст., пер. с [нем.], [Л.], 1936;
Перрен Ж., Атоми, пер. с франц., М., [1924]; С в е д-
б е р г, Коллоидная химия, 2 изд., пер. с англ., М., [1930];
Л е в п ч В. Г.. Введение в статистическую физику, 2 изд.,
М., 1954. П. А. 1‘ебиндер.
БРУЦИН (2,3-диметоксистрихпин) C23H26O4N2 4Н2О,
мол. вес 466,52 — алкалоид, содержащийся в чи-
либухе (рвотных ореш- --------
ках Strychnos), семейст- ___I
ва логаниевых (Logania- НзСОг^^б]-----
сеае), произрастающей ва „ L |l J____
Зондских и Филиппин- к
ских островах. Обычно _j I \
встречается вместе со /
стрихнином. Из 1000 кг
рвотных орешков получают 10—15 кг Б. и столько же
стрихнина. Южноамериканские виды Strychnos не со-
держат ни Б., ни стрихнина; выделенный из этих
видов тубокураринхлорид (см. Тубокурарин) является
477
БУВО СИНТЕЗ — БУМАГА
478
В
физиологии, антагонистом Б. и стрихнина. Б. — бес-
цветные кристаллы; т. пл. кристаллогидрата (4Н,О)
105° (из разб. СН3ОН); безводный Б. имеет т. пл.
178°; [a]D от —119° до —127° (хлороформ) и —80,1°
(этанол). Б. трудно растворим в холодной воде (1:320),
лучше — в кипящей (1:150), хорошо растворим в
спирте, хлороформе, умеренно — в эфире (1:134).
Как однокислотное основание Б. дает хорошо кристал-
лизующиеся растворимые в воде соли. Химич, свой-
ства Б. аналогичны свойствам стрихнина. В отличие
от последнего, Б. не дает цветной реакции с H2SO4 +
+ К2Сг2О7, но даже со следами HNO3 дает интенсив-
ное оранжево-красное окрашивание (апалитич. ре-
акция на NO.7). Разделение Б. и стрихнина основано
на различной растворимости их сульфатов и оксала-
тов. Так же, как и стрихнин, Б. является судорожным
ядом.
Лит. см. при ст. Алкалоиды.
БУВО СИНТЕЗ — синтез альдегидов реакцией
дизамещенных формамидов с гриньяровским реакти-
вом, которая может быть выражена след, уравне-
нием: RMgX + R'R"NCHO-> RCHO + R'R"NH +
-l-MgX2. Побочной реакцией в Б. с. является образо-
вание третичных аминов: 2RMgX + HCONR'R" —>
—>- R2CHNR'R" + MgO + MgX2. Реакция открыта
Л. Буво в 1904. Более удобным методом получения
альдегидов, как показал Смит с сотрудниками (1941),
является взаимодействие реактива Гриньяра с орто-
муравьиным эфиром (Бодру—Чичибабина синтез):
RMgX + HC(OR')3-RCH(0R')3 + MgXOR'
H+
RCH(OR'h + H2O P- RCHO + 2R'OH
Так, из о- и n-бромтолуола с метилформанилидом об-
радуются о- и л-толуолальдегиды е выходом соот-
ветственно 50% и 37%, тогда как реакция тех же
о- и га-бромтолуолов с ортомуравьиным эфиром дает
о- и га-толуолальдегиды с выходом соответственно
74% и 73%. Тем не менее синтез Буво является пре-
паративным методом получения ароматич. и алифа-
тич. альдегидов.
Лит.: Smith L. I. and Nichols J., J. Organ. Chem.,
1941, 6, № 4, p. 489. M. Г. Линъкова.
БУВО — БЛАНА ВОССТАНОВЛЕНИЕ — восста-
новление сложных эфиров в соответствующие спир-
ты с помощью металлич. натрия и этилового спирта.
Метод основан на кипячении р-ра эфира в этиловом
спирте в присутствии избытка натрия:
R —СО —OR' — R-CH.OH -|- ROH
В нек-рых случаях, чтобы повысить темп-ру ки-
пения реакционной смеси, употребляют более высо-
кокипящие спирты (напр., бутиловый). В 1947 Хан-
слей предложил улучшенный метод восстановления
эфиров металлич. натрием. Согласно предложенному
им механизму реакции восстановления, требуется
теоретич. количество металлич. натрия и спирта:
ONa
C16H3I-C-OCsll5 + 2Na — C16H3I-C-OCaH5
|| | — UgnslTd
О Na
ONa
I
— CI6H3I-CH-OCSH6 — CUH31-CH + C2H3ONa
Реакцию проводят при быстром прибавлении смеси
сложного эфира и спирта к энергично перемешива-
емому расплавленному натрию без растворителя или
в инертном растворителе (толуол, ксилол). Таким
способом с отличными выходами получены как пре-
дельные, так и непредельные высокомолекулярные
спирты.
В отсутствии восстанавливающего спирта в среде
инертного растворителя, по методу Буво — Блана,
с хорошими выходами получаются симметричные
ацилоины:
о
ONa
i r_____c_OR1
R-C-OR' + 2Na — R-C-OR' K U —
о
Na
ONa
I
R-C-OR1
I
R-C—OR1
ONa
R—СО
I + 2R'ONa
R—СО
R—CO
I + 2Na —
R-CO
R—С—ONa Нз£
R—С—ONa
RCO
R-CHOH
последние годы появились новые, более доступные
методы получения спиртов восстановлением кислот,
эфиров, кетонов и альдегидов. Вместо металлич.
натрия в Б.—Б. в. употребляют гидрид натрия или
литий-алюминий гидрид в инертном растворителе
(обычно эфире). Реакция открыта Л. Буво и Г. Бла-
ном в 1903.
Лит.: Н a n s 1 е у V. L., Industr. and Engng. Chem.,
1947, 39, № 1, p. 55; Мак-Эльвен С. M., в кн.: Органи-
ческие реакции, кер. с англ., сб. 4, М., 1951, с. 215: Б р а-
у п В. Г., там же, сб. 6, М.. 1953, с. 409.
М. Г. Ли-иькова.
БУГЕРА ЗАКОН ПОГЛОЩЕНИЯ СВЕТА — см.
Поглощение света.
БУМАГА — волокнистый материал, получаемый на
бумагоделательной машине в виде тонкого листа из
прочно переплетенных между собой волокон, весом
до 250 с/.и2 (материал весом 250 г/л<2 и выше называют
картоном, или плитой). Сформовавшееся на непре-
рывно движущейся бесконечной металлич. сетке
бумажное полотно непрерывно прессуется, обезвожи-
вается, сушится, уплотняется и наматывается в
лоны. Б. по форме бывает листовая и ролевая,
виду поверхности — матовая или лощеная, а по
держанию в ней проклеивающих в-в — клееная и
клееная.
Для изготовления Б. применяют: целлюлозу
древесины или однолетних растений, древесную массу,
асбест, стеклянные и синтетич. волокна, а также вто-
ричное сырье (макулатура, тряпье). В состав бумаж-
ной массы, кроме волокна, вводят в-ва: проклеи-
вающие (для уменьшения впитываемости Б.), напол-
няющие (минеральные белые и цвет-
ные пигменты для улучшения глад-
кости, блеска и впитываемости печат-
ных красок) и красители (для подцвет-
ки или окраски бумажной массы). Б.
подразделяют на печатную, писчую,
чертежно-рисовальную, электроизоля-
РУ-
НО
со-
не-
из
О
ONa
1 СИОН
CI6H31-CH + 2Na — С15Н31-СН ГА. C16H31CHaONa + C3HsONa
II I
О Na
ционную, папиросную, светочувстви-
тельную, переводную, промышленно-
техпич., оберточную и др. Всего выра-
батывается более 200 видов Б., разли-
чающихся по физико-механич. п фи-
зико-химич. свойствам.
C15H31CH3ONa + Н3О — С15Н31СНаОН % NaOH________________________
С16Н31-С-ОСаН5 + 4Na 2G^Hs°H> и-% cI5H31CHaOH + 3CaHeONa + NaOH
Физические свойства. Объемный вес
вырабатываемых Б. колеблется в пре-
делах 0,4—1,35 г/см?, толщина 4—
400 мк. Разрывная длина нек-рых вп-
479
БУМАГА
480
дов Б. достигает 16 000 м. Б. — анизотропна: в про-
дольном направлении ее механич. прочность выше,
чем в поперечном; соотношение между ними обычно
колеблется от 1:0,4 до 1:0,7. Б. гигроскопична,
при намокании в воде ее прочность резко снижается
(на 83—96%), при этом она деформируется, сохраняя
остаточную деформацию после высушивания. Благо-
прочность Б. повышается до 50—60% при введении
в ее состав синтетич. смол. Теплоемкость Б. колеб-
лется от 0,295 до 315 кал/г-град, а теплопроводность
0,04 кал/см-сек-град (20—40°). Электрич. сопротивле-
ние Б. уменьшается с повышением влажности и темп-ры
и при наличии в ее составе электролитов. Уд. элек-
тросопротивление 1015—101в ом-см и 10’—1010 ом-см
при влажности 70%, pH водной вытяжки для всех
видов электроизоляционных Б. 6,5—8,5. Диэлек-
трич. проницаемость разных видов Б. 2,5—5,0. Ди-
электрич. потери резко возрастают с увеличением
влажности, темп-ры и пористости Б. Пробивное на-
пряжение Б. падает с ростом влажности. Напр., про-
бивное напряжение при влажности 9% составляет 1/6
от пробивного напряжения в абсолютно сухом
состоянии; при влажности 2,5% и темп-ре 20—100°
пробивное напряжение снижается вдвое. Чем толще
Б., тем выше ее прочность на пробой. В сухой Б. пол
влиянием трения возникает электростатич. заряд,
к-рый снимают увлажнением Б., введением электро-
литов, заземлением, применением разрядников и т. п.
При тепловом воздействии на Б. ее механич. проч-
ность падает. Прочность Б., содержащей древесную
массу, заметно снижается через 40—50 лет.
Производство. Б. производят по непрерывно поточ-
ному методу, состоящему из след, процессов: 1) при-
готовления бумажной массы — размола волокнистых
полуфабрикатов, проклейки, наполнения и крашения
Б.; 2) отлива— образования бумажного полотна;
мокрого прессования, сушки, первичной отделки;
3) отделки — каландрирования на суперкаландрах,
тиснения и получения листов, рулонов, бобин; 4) упа-
ковки с предварительным сортированием и счетом
листов (при выработке листовой Б.).
Образование прочного, равномерного по толщине бумаж-
ного полотна достигается размолом волокон длиной до 0,5—
2,5 лсч, причем отношение толщины к длине волокна должно
быть 1 : 300. Во время размола волокон в воде они подвер-
гаются механич. измельчению, набуханию и поверхностной
фибрилляции клеточных стенок с разрушением первичной
оболочки волокна и отщеплением тончайших фибрилл и отдель-
ных цепей макромолекул целлюлозы. Этот коллоидо-химич.
процесс наз. гидратацией массы при размоле. Степень помола
бумажной массы измеряется градусами шкалы Шоппер-Риг-
лера (0—100° ШР). Измельчение волокон производят при
помощи вращающихся ножевых барабанов или конусов и
неподвижно закрепленных ножей. При длительном размоле
с повышением темп-ры до 60° уменьшается прочность Б.,
увеличивается коагуляция и выпадение смолы, находящейся
в целлюлозе. Для устраневия таких явлений применяют наруж-
ное охлаждение размалывающего оборудования. Добавки
к волокнистым материалам гидроцеллюлозы, глюкозы, кси-
лозы, карбоксиметилцеллюлозы, крахмала, мочевины, тиро-
зина (в водном р-ре) и желатины ускоряют процесс размола,
повышают степень гидратации и увеличивают прочность Б.
Для каждого вида Б. установлена норма степени
помола массы, от к-рой зависят ее физико-механич.
свойства: способность растягиваться, сопротивление
разрыву, излому, раздиранию. Впитывающая спо-
собность находится в обратной зависимости от сте-
пени гидратации массы при размоле. Воздухопрони-
цаемость Б. достигает нуля обычно при помоле 70—
85°ШР и зависит от химич. состава целлюлозы и при-
родных свойств волокна. Прозрачность Б. повышает-
ся с размолом массы. Сила сцепления между волок-
нами зависит гл. обр. от степени гидратации волокон
при размоле, а также от структуры и свойств поверх-
ности волокон. Характеристика размолотого волокна
в Б. приводится в таблице.
Для уменьшения впитывающей способности Б. ее
проклеивают введением в бумажную массу канифоль-
ного клея (из расчета 2—3% канифоли к весу волокна).
Для увеличения гладкости, уменьшения прозрач-
ности Б. и лучшего восприятия типографской краски
в ее состав добавляют наполнители (каолин, тальк,
мел, гипс, карбонаты кальция и магния, бланфикс,
литопон, двуокись титана и др.), содержание к-рых
определяется по зольности Б. В зависимости от золь-
ности различают малозольные (5%), среднезольяые
(до 15%) и высокозольные (св. 15%) Б. Чтобы устра-
нить потери наполнителей, их можно наносить в виде
суспензии непосредственно на поверхность Б. в про-
цессе ее изготовления на бумагоделательной машине
(т. н. мелование Б.). Мелование Б. производят также
на спец, красильных машинах. Для придания Б.
нужного тона в бумажную массу вводят минеральные
(охры, умбры и др.) и органич. красители. Для полу-
чения Б. более интенсивной окраски или с узорами
применяют поверхностное крашение Б., добавляя в
состав краски связующие в-ва, пигменты и т. п. При
этом в нужных случаях крашение совмещают с тис-
нением, гофрированием и креппированием Б.
Виды бумаги Степень помола. °ШР Средняя дли- на волокна, ММ Вес 1 м2, г
Тончайшая 96-98 0,5-0,8 7-14
Топкая 70-92 0,8—1,5 15-30
Жиронепроницаемая .... 70—90 0,7-1,2 40—60
Впитывающая 24—40 1,0—2,2 60—70
Отлитая из сульфитной цел-
люлозы 24—40 1,4-2,2 100—200
Электроизоляционная из
сульфатной целлюлозы . . 25—40 1,8—2,4 40-110
Писчая и печатная № 1 . . 35—45 1.2—1.6 65-80
То же К 2 45—50 1,2-1.4 65
То же № 3 50—55 1.0—1.3 60
Газетная 60-65 0,8-1,1 50
После размола волокнистых материалов, введения про-
клеивающих в-в, наполнения и окраски бумажная масса
поступает на бумагоделательную машину, являющуюся основ-
ным агрегатом при изготовлении Б.; она состоит из сеточной,
прессовой и сушильной частей, каландра, наката и привода
машины. На бумагоделательной машине производится отлив
и формование бумажного полотна, его прессование, высушива-
ние и предварительная, а иногда и окончательная отделка Б.
Различают два типа бумагоделательных машин: плоскосеточ-
ную и круглосеточную. На плоскосеточной машине бумаж-
ное полотно отливается на движущейся бесконечной металлич.
сетке, на круглосеточиой — на сетке, натянутой на вращаю-
щийся цилиндр. Вспомогательным оборудованием бумагодела-
тельной машины являются: метальные бассейны для аккуму-
лирования бумажной массы, регуляторы концентрации и
композиции массы, аппараты для очистки массы от узелков,
тяжелых частиц и механич. включений, насосы для подачи
массы и воды, вакуумные насосы и др. Влажность бумажного
полотна после сушильной части составляет 5—8%, темп-ра
80—95°. Для уплотнения Б. и придания ей гладкости и лоска
бумажное полотно пропускают через многовальный каландр
(3—10 металлич. валов). После каландра бумажное полотно
наматывается в рулоны. Электростатич. заряд с Б. снимается
заземлением. Скорость машины в зависимости от ее типа
и вида вырабатываемой Б. — 40—700 м/мин, а при выработке
газетной Б. достигает (на новейших машинах) 900—1000 м/мин.
Бумажное полотно можно формовать из сухой волок-
нистой массы; при сухом способе получения длинноволок-
нистой Б. волокно (напр., из хлопка длиной 1,5—2,5 см)
укладывают при помощи кардочесальных машин на движу-
щуюся сетку. При прохождении через клеевую ванну волокна
склеиваются и после сушки получается Б., применяемая для
приготовления восковки, микаленты, переплетного мате-
риала и т. д. Длинноволокнистая Б. обладает высокой проч-
ностью, растяжимостью и гибкостью.
Способ получения Б. из воздушной взвеси заключается
в укладывании на сетке волокон через сопло под давлением
3,5—9 ат. Волокна склеивают термопластич. порошками или
клеевой эмульсией, распыливаемыми в потоке волокон через
сопло; в отдельных случаях производят поверхностную про-
клейку бумажного полотна. Получаемая Б. отличается изо-
тропными свойствами.
Химич, обработкой Б. с поверхности ей можно при-
дать самые разнообразные свойства. Бодо- и жироне-
проницаемые Б. получают обработкой неклееной Б.
серной к-той с последующей промывкой водой, про-
питкой расплавленным парафином или битумом,
481
БУМАГА ФОТОГРАФИЧЕСКАЯ
482
синтетич. каучуком и органозолями, содержащими
синтетич. смолы. Воздухо- и газонепроницаемость
придаются Б. путем склейки ее с металлич. фольгой
и с пленками. Светочувствительные Б. получают на-
несением на Б. эмульсии, содержащей диазосоеди-
нения; при изготовлении фотографии. Б. светочув-
ствительные эмульсии наносятся на баритованную
Б. на поливочных машинах (см. Бумага фотографи-
ческая). Абразивные Б. (шлифовальные) получают на-
несением абразивных порошков на поверхность Б.,
покрытой клеевым слоем из синтетич. смолы или из
животного клея (для сухого шлифования). Таким же
способом получают бархатную и плюшевую Б. (на-
носят на Б. мелконарезанное сухое волокно шелка).
Копировальные Б. изготовляют нанесением на тон-
кую Б. расплавленной массы, содержащей черные
пигменты и красители, парафин, вазелин, олеиновую
к-ту и минеральное масло. При пропитке р-ром, со-
держащим сернокислый аммоний, буру или жидкое
стекло, Б. приобретает огнестойкость. Возможность
придания Б. желаемых свойств путем нанесения на
ее поверхность соответствующих химикатов открывает
широкие возможности создания на основе Б. самых
разнообразных изделий.
Лит.: Баранов Н. А. и ДобровольскийД.С.,
Технология бумажного производства, М.—Л., 1957; Справоч-
ник бумажника (технолога), т. j—3, М.—Л., 1955—59; К у-
л е в И. Г. и Рюхин Н. В., Справочник технической лите-
ратуры по целлюлозно-бумажному производству, М.—Л., 1950;
Кейси Д. П., Целлюлоза и бумага. Химия и хим. техноло-
гия пер. с англ. т. 1 кн. 2, М 1958. 3. В. Участкина.
БУМАГА ФОТОГРАФИЧЕСКАЯ — светочувстви-
тельный материал, состоящий из эмульсионного
слоя, нанесенного на бумажную основу (подложку).
Фотоподложка обычно предварительно покрывается
баритовым "слоем (тонкая суспензия сернокислого
бария в желатине и др.); на эмульсионный слой обычно
наносится защитное желатиновое покрытие. Эмульси-
онный слой Б. ф. — суспензия микрокристаллов гало-
генида серебра (с размерами от 0,5 до 0,01 мк и ме-
нее) в воздушно-сухом полимере, обычно желатине.
Большинство типов Б. ф. общего назначения от-
личается от аналогичных фотоматериалов на прозрач-
ной подложке значительно меньшей светочувстви-
тельностью, цветочувствительностью, ограниченной
сине-фиолетовой зоной спектра, практически полным
отсутствием вуали и возможностью достижения более
высокой контрастности. Значения максимальной плот-
ности почернений Б. ф. зависят от степени дисперс-
ности зерен галогенида серебра, от содержания его
на единице поверхности слоя, а также от качества
поверхности эмульсионного слоя и подложки (фак-
тические значения 1,75—1,80 для особоглянцевой
и 1,10—1,20 для матовой Б. ф.); при оптимальном ка-
честве поверхности значение максимальной плотности
почернения Б. ф. не превышает 2,00.
Б. ф. классифицируется по: 1) характеру химико-
фотографич. обработки (с проявлением, с видимым
печатанием); 2) составу галогенида серебра, напр.
бромосеребряная, хлоробромосеребряная и др. (см.
табл. 1); 3) назначению — для любительской, худо-
жественной и технич. фотографии и для спец, целей;
4) степени контрастности (мягкая — № 1, нормаль-
ная — №№ 2 и 3, контрастная — №№ 4 и 5, особо-
контрастная — № 6 и сверхконтрастная — № 7);
5) виду поверхности (гладкая и структурная—барха-
тистая, зернистая, тисненая); 6) степени глянца
(особоглянцевая, глянцевая, полуматовая и матовая);
7) плотности фотоподложки (тонкая, полукартон,
картон); 8) цвету окраски подложки (белая и окра-
шенная в цвет слоновой кости, кремовый и др.).
Б. ф. с проявлением составляют главную часть сов-
ременного ассортимента фотобумаг.
Светочувствительность отдельных типов Б. ф. для
проекц. и контактн. печати различается более чем
Таблица 1. Классификация фотобумаг по степени
контрастности, в зависимости от состава галогенида
серебра и характера поверхности
Наиме- Группа а- (№) Полез- ный ин- Коэфф. контрастности т
Б. ф. по л>£
нование Б. ф. галоге- ниду серебра S «5 о 2 & 5 о тервал экспози- ций L особо- глянце- вая глянце- вая матиро- ванная
Унибром, Бромо- 1 1,6-1,8 1,4—1,5 1,2—1.4 1.1—1,2
Унибром- серебря- 2 1.3-1,5 1.6-1,9 1,5—1,7 1,3-1,5
59. Фото- ная 3 1,1-1,2 2,0—2,4 1,8—2.0 1,6—1.8
бром и 4 0,9—1,0 2,5—2,9 2,1-2,5 1.9—2,1
Новобром 5 0,7—0,8 3,0—3,9 2,6—3,0 2,2-2,6
6 0,5—0,6 4.0—4,9 3,1-4,0 2,7-3,5
7 ДО 0,4 > 5,0 >4,1 > 3,6
Бром- Хлоро- 1 1,7—1,9 1.2-1,4 1,1-1,2
портрет и Кон- бромо- 2 1,4—1.6 — 1.5—1.8 1.3-1,5
сереб- 3 1.2-1,3 —- 1.9—2,3 1.6—1,9
табром ряная 4 1,0-1,1 - 2,4—2,8 2,0-2,4
Фото- Хлоро- 2 1.3—1,4 1.6-1.9 1.5—1,7 1,3-1.5
КОНТ сереб- 3 1,1-1,2 2,0—2,4 1.8-2,0 1,6-1.8
ряная 4 0,9—1,0 2,5—3,0 2,1—2,5 1,9—2,1
5 0,7—0,8 3,1—3,9 2,6—3,0 2,2-2,6
6 0,5—0,6 4,0-4,9 3,1—4,0 2,7-3,5
7 ДО 0,4 Ь> 5,0 >4.1 > 3,6
Иодо- Иодо- 1,6-1,8
КОНТ хлоро- . 1 — 1,3—1.5 1,1—1,2
бромосе- 2 1.4—1,5 — 1,6-1,8 1,3-1,5
ребря- 3 1,1-1,3 — 1,9—2,2 1,6—1,9
ная
в 1000 раз. Среди этих Б. ф. различают: черно-белые
(Унибром, Новобром, Фотоконт, Форте-Бромофорт,
Агфа-Бровира и др.), самотонирующиеся (Бромпорт-
рет, Контабром, Агфа-Портрига, Иодоконт, Аристо-
гипная и др.) и цветофотографические (Фотоцвет и др.).
Выпускаются Б. ф. и с регулируемым в процессе
печати (с помощью светофильтров) коэфф, контраст-
ности, напр. Веригем (Дюпон), Поликонтраст (Ко-
дак), Мультигрейд (Ильфорд), Мультиконтраст (Ге-
верт) и др.
В связи с различной дисперсностью и химич. со-
ставом галогенидосеребряных эмульсий содержание
серебра на единице поверхности фотобумаги, условный
фотометрия, эквивалент, разрешающая способность
и интервал изменения светочу ветвите ль ности в про-
цессе проявления (Alg А) колеблются для различных
сортов фотобумаг в широких пределах (см. табл. 2).
Таблица 2. Характеристика некоторых образцов фотобумаг
по содержанию серебра, условному фотометрическому
эквиваленту, разрешающей способности и Д 1g S
Наименование фотобумаги Содержание Ag г/м- 1 Условный фотометрии, эквивалент Х-10* Разрешаю- щая способ- ность леи-* A 1g S
Унибром № 3 1.95 0,61 82 0,25
Фотобром №4 1,25 0,35 96 —-
Бромпортрет 1,80 0,36 89 0,10
Контабром 1,75 — 0,21
Агфа-Бровира бриллиант белая Агфа-Портрига рапид 1.65 0,30 85 0,55
1,20 0,40 112 0,36
Илфорд-Мультигрейд 2.44 — — —
Илфорд-Бромида 2,17 0,(55 — 0,25
Илфорд-Пластика 1,33 0,36 112 0,28
Кодак-Бромида ......... 1,72 0,59 76 0,25
Кодак-Мёдалист 1,90 0,38 82 0,21
Кодак-Атена и -Велоне .... 0.98 — — —
Форте-Бромофорт 2,00 — 82 0,15
Гарантийный срок сохраняемости Б. ф. бромосереб-
ряных (Унибром, Новобром и др.) не менее 20 мес.,
остальных Б. ф. (Фотоконт, Контабром, Бромпортрет,
Иодоконт) — не менее 12 месяцев. Б. ф. изготовляют
483
БУМАГИ РЕАКТИВНЫЕ — БУРА
484
форматами и в виде рулонов. Форматная Б. ф. имеет
след, размеры (в см)-. 6X9, 9X12, 9X14, 10X15,
13X18, 18X18, 18X24, 24x24, 24X30, 30X30, 30X40,
40X50, 50X60.
Среди Б.ф. с видимым печатанием, или
дневных (хлоросеребряных), различают: ари-
стотипные (желатиновый слой), целлоидиновые (кол-
лодионовый слой) и альбуминные (альбуминовый слой).
От хлоросеребряных Б. ф. с проявлением они отли-
чаются наличием в эмульсионном слое избытка ионов
серебра и чрезвычайно малой светочувствительностью.
IIз этих Б. ф. производятся только аристотипные и то
в ограниченном количестве. Сохраняемость аристотип-
ной Б. ф. не более 6 мес.
Цветная Б.ф. «Фотоцвет» «Ф-1» и «Ф-2» пред-
назначена для проекционной и контактной печати
позитивов с цветных негативов (см. Цветная фото-
графия). По светочувствительности цветная Б. ф.
соответствует примерно Униброму. Она значительно
сложнее по строению однотонных Б. ф.; выпускается
только на белой картонной подложке глянцевой, ма-
товой и тисненой поверхности. По степени контраста
«Ф-2» подразделяется на нормальную и контрастную.
Выпускаемые размеры те же, что и для черно-белых
Б. ф. общего назначения. Гарантийный срок хранения
не более 2 лет.
Специальные Б. ф. подразделяются на
весьма разнообразные по своим свойствам след, глав-
ные группы: регистрирующие (осциллографная, сейс-
мографная, электрокардиографная, фототелеграфная
и др.), документные (фотостатная, рефлексная, ре-
версивная, фотокалька и др.), рентгеновские и др.
специальные сорта Б. ф., напр. для процессов «Тех-
нокопир», «Момент» (см. Диффузионные фотографи-
ческие процессы) и др.
Лит.: ГОСТ 5779 — 57. Бумага фотографическая общего
назначения (позитивная) для черно-белой фотографии. Клас-
сификация и размеры; Н е б л и т К. Б., Фотография, ее
материалы!! процессы, пер. с англ., М., 1958; КаНенелен-
б о г е и 9., Свойства и применение фотографических мате-
риалов М 1950; Васильев В. К., Ш о р М. И., Ш а м-
ш е в Л. П., Негативные и позитивные фотоматериалы, 2 изд.,
М., 1959; 3 а й д е н б е р г Я. 3., Ж. научн. и прикл. фотогр.
и кинематогр., 1958, 3, вын. 5, с. 390. В. JI. Зеликман.
БУМАГИ РЕАКТИВНЫЕ — служат для ориен-
тировочного определения значения pH растворов и
для быстрого открытия нек-рых веществ в растворах
и газах. Для определения pH Б. р. приготовля-
ют, пропитывая тонкую беззольную фильтровальную
бумагу раствором соответствующего индикатора, а
для открытия тех или иных веществ — пропитывая
р-рами реактивов, реагирующих с открываемым в-вом
<• образованием окрашенного продукта реакции. Про-
питанную бумагу сушат на воздухе, не содержащем
кислых, щелочных и иных газов. Полоску Б. р. по-
гружают в испытуемый р-р или наносят на нее каплю
этого раствора. При испытании газов или паров смо-
ченную водой или др. растворителем Б. р. вносят в
газовое пространство. Во всех случаях наблюдают
изменение окраски Б. р.
Для определения pH можно применять Б. р., про-
питанную р-ром любого индикатора, однако лучше
всего пользоваться набором индикаторов (см. табл.).
Для определения pH применяются также универ-
сальные Б. р., пропитанные р-ром смеси индикаторов
и окрашивающиеся в разные цвета при разных зна-
чениях pH (см. также Индикаторы универсальные).
Для открытия азотистой «-ты при контроле процес-
сов диазотирования и нитрозирования, брома — при
контроле процесса бромирования и др. пользуются
иодкрахмальной бумагой (белого цвета), пропитанной
р-рами KJ и крахмала (чернеет или синеет при дей-
ствии окислителей). Свинцовая бумага (белая), про-
питанная р-ром ацетата свинца, чернеет при действии
сероводорода. Бензидиновая бумага (белая), пропи-
танная уксуснокислым р-ром бензидина, окрашивается
в синий цвет при действии окислителей, в частности
хлора. Для открытия ряда др. веществ применяют
различные спец. Б. р.
Индикатор Концентра- ция и раство- ритель Изменение окраски
pH переход
Метиловый фиолето- 0,1 %, вода желтый — фиолетовый
вый 0,1—2,5
Метапиловый желтый 0,1 %, вода 0,3-2,4 лиловый — желтый
Тропеолин 00 ... . 0,2 %, вода 0,75—2,5 вишневый — желтый
Конго красный . . . Бромфеноловый св- 0,05%, вода 2,5-4,0 синий — кр асный
ний Бромкрезоловый пур- 0,1%, 60%-ный спирт 3,0—5,0 желтый — синий
пурный 0,1 %, 60%-ный спирт 5,0-7,0 желтый — фиолетовый
Дельта 0,2%, вода 5,6-7,8 желтый — синий
Лакмус Бриллиантовый жел- 0,1%, вода 6,0-8,0 красный — синий
тый 0,1%, вода 6,8-8,5 желтый — кр асный
Фенолфталеин .... 0,5%, 50%-ный спирт 9,0—10,2 бесцветный — красный
Тиазоловый желтый о,1 %, вода 11.6-13,6 желтый — красный
Лит.: Химические реактивы и препараты [Справочник],
под общ. ред. В. И. Кузнецова, М.—Л., 1953; Каря-
кин Ю. В. и Ангелов И. И., Чистые химические реак-
тивы, 3 изд., М., 1955; Л а с т о в с и и й Р. П., В а й и-
ште й и Ю. И., Технический анализ в производстве проме-
жуточных продуктов и красителей, 3 изд.. М." 1958.
А. А. Черкасский.
БУНЗЕНА — РОСКО ЗАКОН — см. Фотохимия.
БУРА Na2B4O7 10Н2О — соль тетраборной к-ты
Н2В4О7, не выделенной в свободном состоянии. В
природе Б. встречается в виде минералов тинкаля
(или просто Б.) и кернита (или разорита); содержится
во многих минеральных и нефтяных водах, в соляных
озерах, в выделениях грязевых вулканов. Обычная
Б. (десятиводвый гидрат) образует большие бесцвет-
ные прозрачные призматич. кристаллы; базоцентри-
рованная моноклинная решетка, а = 12,19А, Ь =
= 10,74 А, с = 11,89 А, р= 106°35'; плотн. 1,69—1,72;
в сухом воздухе кристаллы выветриваются с поверх-
ности и мутнеют. При нагревании до 80° декагидрат
теряет 8 молекул Н2О, при 100° медленно, а при 200°
быстро отщепляется еще одна молекула воды, в ин-
тервале 350—400° происходит полное обезвоживание.
Теплота образования безводной Na2B4O7 AHs98 =
= —777,7 ккал!моль. Растворимость Na2B4O7 ЮН2О
(в г безводной соли на 100 г воды): 1,6 (10°), 3,9 (30°),
10,5 (50°). Насыщенный водный р-р кипит при 105°.
В воде Б. гидролизуется, поэтому ее р-р имеет щелоч-
ную реакцию. Б. растворима в абс. спирте и глицерине.
Сильными к-тами полностью разлагается: Na2B4O7 +
-j-H2SO4 + 5Н2О = Na2SO4 + 4Н3ВО3. С окислами
нек-рых металлов Б. дает окрашенные бораты («перлы
буры»): Na2B4O7 + СоО = 2NaBO2 + Со(ВО2)2, что
используется в аналитич. химии для открытия этих
металлов. При медленном охлаждении р-ра обычной
Б. при 79° начинает выкристаллизовываться октаэд-
рич. Б. Na2B4O7 • 5Н2О (или «ювелирная бура»),
| плоти. 1,815, устойчивая в интервале 60—150°. Раст-
I воримость Na2B4O7 • 5Н2О (г безводной соли в 100 г
* воды): 22 (65°), 31,4 (80°) и 52,3 (100°). При медленном
I термич. обезвоживании обычной Б. получается пиро-
I бура Na2B4O7 с плотя. 2,371, т. пл. 741°.
। Обычную Б. получают из борной к-ты, из тиикаля,
кернита и нек-рых др. минералов (путем их перекристал-
। лизации), а также из воды соляных озер (фракциони-
485
БУТАДИЕН - БУТАДИЕН-МЕТИЛВИНИЛПИРИДИНОВЫЙ КАУЧУК
486
рованной кристаллизацией). В СССР производство Б.
основано па взаимодействии борной к-ты с содой. Б.
широко применяют при приготовлении эмалей, гла-
зурей, в произ-ве оптич. и цветных стекол, при сварке,
резке и пайке металлов («ювелирная бура»), в метал-
лургии, в гальванотехнике, красильном деле, бумаж-
ном, фармацевтич., кожевеином произ-вах, в качестве
дезинфицирующего и консервирующего средства и
удобрения. О качественном и колич. определении Б.
см. Бор.
Лит.: Берлин Л. Е., Производство борной кислоты,
буры и борных удобрений, М.—Л., 1950; Позин М. Е.,
Технология минеральных солей, Л.—М., 1949; см. также при
статье Бор. Г. Н. Пирогова.
БУТАДИЕН-1,3 (дивинил, эритрен), мол. в. 54,09,
СН2 = СН—СН=СН„ — бесцветный газ с характер-
1 2 3 4
ным интенсивным запахом; т. пл. —108,9°; т. кип.
—4,5°; (жидк.) 0,650, 0,6206 (жидк.),
1,4292; 1 м3 весит 2,43 кг (0°, 760 мм рт. ст.). Рас-
творимость при 15° в г на 100 г!мм рт. ст.: в воде
0,13/793, в этиловом спирте 15/807, в керосине
23,08/800, в скипидаре 23,8/798, в тетралине 28,6/818;
вязкость (жидк.) 0,178 спуаз (5°); 0,133 спуаз (30°);
гкрит 161,8°; дкрит 42,6 ат (абс.); давление паров:
Давление, мм
рт. ст . . .
— 102,8° 1 -79,7° 10 -61,3° 40 -46,8° 100 —33,9° 200 —19,3° 400
-4,5°
760
t.............
Давление, ат
1абс.) . . . .
47°
5
76° 114°
10 20
139,8° 160° 161,8°
30 40 42,6
Теплота испарения (кал/г) 96,2 (0°); 86,6 (40°); теп-
лота плавления 35,28 кал/г\ теплота полимеризации
17,4 ккал/моль (5—50°); теплота сополимеризации
17,1—17,7 ккал/моль (5—50°); теплота гидрирования
в бутан 56,56“0 ккал/моль.
Теплоемкость, хал/г .град Теплота сгора- ния, ккал/? моль Теплота образо- вания (20°), ккал/г-м-адъ
Я» идк. 0,525 602,788 -21,63
Пар 0,315 607,907 —26,75
Б. образует азеотропные смеси: с ацетальдегидом (т.
кип. —5°; 94,8% Б.); с NH3 (т. кип. —37°, 45% Б.);
взрывоопасные концентрации в воздухе 1,6—10,8
(об. %), т. всп. —40°; предельно допустимая кон-
центрация в рабочих помещениях 0,1 мг/л воздуха.
Б. присоединяет водород, галогены, галогеноводо-
роды и др.; процесс идет гл. обр. в положение 1,4,
напр. СН2 = СН — СН=СН2+Н2->-СН3—СН=СН— СН3.
Б. легко количественно присоединяет малеиновый
ангидрид с образованием ангидрида тетрагидрофта-
левой к-ты (см. Диеновый синтез):
СН2=СН-СН=СН2
СН—СО
II >0
сн—со
Эта реакция применяется для колич. определения Б.
Б. легко полимеризуется, при этом получаются кау-
чуки синтетические', с соединениями винилового ряда
легко образуются сополимеры.
В пром-сти Б. получают из спирта, бутана или аце-
тилена. Пром, получение Б. из этилового спирта было
первым по времени промышленным способом, впервые
реализованным в СССР по методу С. В. Лебедева.
Для этого пары спирта пропускают в одну стадию
над смешанным дегидратирующе-дегидрирующим ка-
тализатором. В США был принят двухстадийный про-
цесс, в к-ром сначала пары этилового спирта дегид-
рируют при 250—300° до ацетальдегида над медным
катализатором, промотированном окисью хрома. Во
вторую стадию смесь выделенного ацетальдегида и
спирта в виде паров пропускают при 325—350° над
окисью тантала или циркония; в результате конден-
сации с одновременным отщеплением воды образует-
ся Б.:
С2Н-,ОН у СНзСНО — СН3-СН-СН=СН3 + 2Н3О
Прогрессивным методом является получение Б. дегид-
рированием н-бутана и н-бутиленов;иногдадляэтойце-
ли используют бутановую фракцию, остающуюся после
извлечения бутиленов из бутанбутиленовой фракции
газов крекинга; частично применяют бутан, выделя
емый из попутных газов нефтедобычи. Процесс идет
в две стадии: сначала н-бутан дегидрируют при 590 —
600°, пропуская его пары через слой движущегося
катализатора — окись хрома, нанесенная на А12О3:
С4Н10 —> С4Н8 + Н2. н-Бугилен выделяют путем эк-
страктивной дистилляции с водным ацетоном. Дегид-
рирование очищенных бутиленов ведут при сильном
разбавлении водяным паром при 625—675° над ката-
лизатором — окислы металлов (магния, цинка, же-
леза, хрома и др.): С4Н8 —>С4Нв + Н2. Возможно
получение Б. и в одну стадию в реакторах адиаба-
тич. типа с короткими циклами контактирования:
С4Н10 —> С4Нв + 2Н2, но выход при этом ниже.
В пром-сти Б. получают из ацетилена несколькими
способами: 1) по альдольному методу вначале получают
из ацетилена ацетальдегид при 95° в присутствии
катализатора — р-ра сульфата ртути:
CHzCH + н-о - сн3сно
Затем в присутствии КОН при 30—40° получают
ацетальдоль:
2СН3СОП - СН3СН'ОН)СН2СНО
Гидрирун последний под давл. 300 ат при 90—130°
в присутствии медного или никелевого катализатора,
получают бутиленгликолъ-1,3:
СН3СН(ОН)СН-СНО + Н;, — СН.зСН(ОН)СН2СН2ОН
дегидратацией его при 280° над катализатором (моио-
кальцийфосфат и аминофосфат на угле) получают Б.:
СН3СН(ОН)СН2СН2ОН — СН3=СН-СН=СН2 + 2Н3О
2) Ацетилен при 100—120° и 5 ат реагирует с формаль-
дегидом; при этом образуется бутиндиол:
2СН.0 + С3Н3 — НОСНоС^ССНзОН
Гидрированием при 100—120° и 325 ат его переводят
в бутиленгликоль-1,4; после дегидрирования обра-
зуется Б. Возможно также получение Б. из ацетилена
через моновинилацстилен
2С,Н3 — сн3=сн-с- сн 5асн3=сн-сн=сн2
Лит.: Смирнов Н. И., Синтетические каучуки,
2 изд., Л., 1954; Синтетический каучук. Гл. ред. Г. С. Уитби,
пер. с англ., Л., 1957; Голдштейн Р., Химическая
переработка нефти, пер. с англ., М., 1952; Л и т в и н О. Б.,
Технология синтетических каучуков, Л.—М., 1950; его
же, Основы технологии синтеза каучуков, М., 1959; U 1 1-
m а п п, 3 Aufl., Bd 10, Munchen — В., 1958. S. 52.
Б УТ АД И ЕН-МЕТИЛ ВИ НИЛ ПИРИДИНОВЫЙ
КАУЧУК — продукт совместной полимеризации бу-
тадиена и 2-метил-5-винилпиридина (в соотношении
75:25 или 85:15), получаемый по эмульсионному
методу:
1 2 3 4
ЛСН2=СН-СН=СН2+ т СН2=СН-
12 3 4 12
—сн2-сн=сн-сн2-сн2-сн-снй-сн—••
А зСН
4СН2
сн3
487
БУТАДИЕН-НИТРИЛЬНЫЙ КАУЧУК
488
Темп-ра полимеризации 40—50°, продолжительность
10—20 час., глубина превращения мономеров 80—90%.
В промышленности Б.-м. к. получают по той же тех-
нологической схеме, что и бутадиен-стирольный
каучук.
Б.-м. к. — эластичная, светло-коричневая масса
(выпускаемая часто в виде ленты) с запахом пириди-
новых производных. Свойства вулканизатов с 15 и
25% метилвинилпиридина (соответственно): предел
прочности при разрыве 165 и 180 кг]см?, относитель-
ное удлинение 190 и 185%, эластичность 46 и 40%,
темп-ра хрупкости минус 65 и 55°. После старения
Б.-м. к., содержащего 15% метилвинилпиридина, в
течение 3 суток при 150° в диоктилсебацинате и тур-
бинном масле: набухание 20,6—21,3 (об. %), предел
прочности при разрыве 17—40 кг]см\ относительное
удлинение 25—70%. Вулканизаты Б.-м. к. стойки
к различным растворителям в интервале от —65°до
+ 200°, а также к воде, спиртам, альдегидам, кетонам,
сложным эфирам дикарбоновых к-т и фосфорной к-ты,
алифатич. и ароматич. углеводородам. Сочетание в
Б.-м. к. удовлетворительной морозостойкости с хо-
рошей бензиностойкостыо, а также стойкостью к раз-
личным органич. растворителям является весьма цен-
вым свойством; по комплексу этих свойств Б.-м. к.
значительно превосходит бутадиен-нитрильный ка-
учук. Напр., при набухании в этилфосфате каучук
типа СКН-40 в течение 48 час. при 70° увеличивает
вес на 113%, а Б.-м. к. только на 26%. Вулканиза-
цию Б.-м. к. осуществляют с применением галогено-
производных жирного и ароматич. рядов, к-рые реа-
гируют с азотом пиридинового кольца с образованием
соединений типа четвертичных аммониевых основа-
ний. По основным физико-механич. показателям он
уступает бутадиен-стирольному каучуку. При ста-
рении в воздухе при 125° и выше вулканизаты Б.-м. к.
быстро теряют эластичность.
Б.-м. к. применяется для изготовления стойких к
действию указанных выше жидкостей резиновых тех-
нич. изделий (прокладки, уплотнители, манжеты,
рукава и др.), работающих в интервале темп-p от
—65° до +200°.
Лит.: Синтетический каучук. Гл. рсд. Г. С. Уитби, пер. с
англ., Л., 1957; Rubber and Plast. Age, 1954, 35, № 7, p. 326;
Pritchard J.E. and Opheim M. H., Industr. and Engng.
Chem., 1954, 46, № 10, p. 2242; S vetl ik J. F., Rails-
back H. E. and Cooper W. T., там же, 1956, 48, № 6,
p. 1084. А. К. Никитич.
БУТАДИЕН-НИТРИЛЬНЫЙ КАУЧУК (CKH, Бу-
на-N, пербунан и др.) — продукт совместной поли-
меризации бутадиена и нитрила акриловой к-ты, по-
лучаемый в водной эмульсии. Содержание нитрила
акриловой к-ты в Б.-н. к. колеблется в пределах
от 17—20% в СКН-18 до 36—40% в СКН-40; при-
чем, чем больше его содержание, тем выше масло-
и бензостойкость Б.-н. к. и ниже его морозостой-
кость.
Наличие в молекуле Б.-н. к. полярной группы —CN
обусловливает его стойкость к действию минеральных
масел, бензина и др. алифатич. углеводородов и рас-
творимость в карбонильных (ацетон) и др. полярных
соединениях. Молекулы Б.-н. к. состоят из чередую-
щихся звеньев бутадиена и нитрила акриловой к-ты,
причем бутадиен полимеризуется гл. обр. в положение
1,4 и частично в 1,2:
пСН2=СН—СН=СН2 + mCH2=CHCN
—» ... -сн2—сн=сн-сн3—сн3—сн -
I
CN
-СН2-СН=СН-СН2-СН.,-СН- ...
I
CN
или -* ... -сн2—сн=сн-сн3-сн3-сн-
I
CN
-сн2-сн-сн..-сн- ...
I
3СН CN
»
4 СН2
Б.-н.к. светло-коричневая масса, выпускаемая часто
в виде ленты, с содержанием до 1% влаги и до
1,5% золы.
Основные свойства Б.-н. к.
Каучуки Показ атели^^^~~--^^ свойств СКН-18 СКН-26 СКН-40
Дефо-твердость*, г 1500—2300 1500—2300 1500-3000
Ненаполненные резины: пре- дел прочности при разры- ве, KS/CAt3 Относительное удлинение,% 30-45 50-60
500-600 500-650
Наполненные резины, 50 вес. частями канальной сажи: Предел прочности при раз- рыве, K8/GM2 250—270 280—300 300-330
Относительное удлинение,% 450—550 550-650 550—650
Истирание, cai3/квт-ч .... — 200-230 —
Эластичность по упругому отскоку, % 30—35 25-30 10-15
Темп-ра стеклования, °C. . -55 -42 -32
Темп-ра хрупкости (вулка- низаты), °C -48 -35 -25
Коэффициент морозостойко- сти (100% растяжения, темп-ра —35°) 0,11 0,07 - 0,02
Набухание за 24 ч. в смеси бензин-бензол (3 : 1),вес.% 70,0 38,0 20,0
•Дефо—показатель твердости каучука, выражаемый в
граммах той нагрузки, к-рая вызывает заданную деформацию
образца стандартных размеров в течение 30 сек.
Для диспергирования исходных мономеров в воде
и получения устойчивой эмульсии применяют в ка-
честве эмульгаторов (3—4%, считая на мономеры)
мыла на основе природных жиров и масел, соли ал-
киларилсульфокислот (некаль, сульфонол) и их ком-
позиции. Приготовление водной фазы и р-ров произ-
водят на очищевной и умягченной с помощью катио-
нитов или дистилляцией воде (150— 200 частей воды
на 100 частей мономеров). Полимеризацию иниции-
руют введением до 0,2% веществ, легко образующих
свободные радикалы-персульфаты, перекись водорода,
гидроперекись изопропилбензола (кумола) и др.
Для ускорения образования радикалов и снижения
темп-ры полимеризации вводят соли металлов пере-
менной валентности и др. легко окисляющиеся и
восстанавливающиеся соединения. Для устранения
явлений структурирования и получения желательного
мол. веса применяют регуляторы (меркаптаны, ди-
сульфиды). Сополимеризацию бутадиена с нитрилом
акриловой к-ты проводят периодически или непре-
рывно (в зависимости от рецептуры и марки каучука).
Эмульсия проходит батарею последовательно соеди-
ненных полимеризаторов, в первый из к-рых вводят
р-р инициатора. В отдельные аппараты батареи вводят
р-р- регулятора. Вследствие легкой гидролизуемости
нитрила акриловой к-ты полимеризацию проводят
при невысоких темп-рах (24—30°) и в нейтральной
среде (pH 7—8). Продолжительность полимеризации
30—60 час. Конечным продуктом является латекс
(см. Латекс синтетический), содержащий 25% неза-
полимеризованных мономеров (от поданных на поли-
меризацию). На выходе латекса, когда конверсия
достигает 75%, в полимеризатор вводят в-во, обрываю-
щее полимеризацию («стоппер»), напр. гидросульфит
натрия, и противостаритель (антиоксидант) — нео-
зон Д. Отгонка незаполимеризованных мономеров про-
489
БУТАДИЕН-СТИРОЛЬНЫЙ КАУЧУК
490
изводится под вакуумом в колонне с
водяным паром; нитрил акриловой к-ты,
содержащийся в продуктах отгонки,
улавливают водой, при этом часть его
гидролизуется. Коагуляцию латекса и
выделение Б.-н. к. проводят по той же
технологии, что и бутадиен-стирольного
каучука. Для коагуляции применяют
хлористый натрий. Сушат при 90—125°.
Б.-н. к. — масло- и бензостоек, из него
изготовляют резиновые детали и изде-
лия, предназначенные для работы в ма-
сле, бензине и др. алифатич. углево-
дородах.
Лит. см. при ст. Бутадиен-стирольный
каучук. А. Б. Никитин.
БУТАДИЕН-СТИРОЛЬНЫЙ КАУ-
ЧУК (СКС, Буна-S и др.) — продукт
совместной полимеризации бутадиена и
стирола, полученный по эмульсионно-
му методу. Б.-с. к. вырабатывается с
различным содержанием стирола или
а-метилстирола, например при соотноше-
нии исходной смеси бутадиена и стирола
70:30 (СКС-30), 50:50 (СКС-50), 90:10
(СКС-10). Средний молекулярный вес непластицирован-
ного СКС-30, определенный по вискозиметрич. методу,
200 000—300 000. Б.-с. к. имеет нерегулярную струк-
туру и не способен к кристаллизации. Ок. 30% сти-
рола образуют с бутадиеном молекулы, в к-рых пра-
вильно чередуются оба компонента; ок. 40% сти-
рола образуют молекулы, в к-рых последовательно
соединены по два звена стирола; примерно 75% бу-
тадиена полимеризуются в положении 1,4, а осталь-
ные 25% — в 1,2:
ссн6
пСН3=СН—СН=СН3 + mCH=CII3 —►
свщ
-сн3-сн=сн-сн3-сн-сн-
С.Н5
-СП3-СН=СН-СН3-СН-СН3— ... или
... -сн3-сн=сн-сн3-сн-сн2-
С«н5
-СНз-СН-СН-СН.,- ... или
I I
-СН СвН5
II
«сн3
... -СН3-СН=СН-СН3-СН-СН3-СН3-
С6н5
—СН-СН3-СН=СН-СН3— ...
С„н5
Б.-с. к. светло-коричневая масса (получаемая часто
в виде ленты).
Б.-с. к. получают 2 способами: «холодным» и «го-
рячим» (при 5° и 50°); полимер, получаемый при 5°,
имеет меньшую степень разветвленности и обладает
лучшими свойствами, его обозначают СКС-ЗОА. Для
диспергирования в воде исходных мономеров и созда-
ния устойчивых эмульсий применяют эмульгаторы
(3,3—4,2%, считая на углеводороды) — соли кислот,
природных жиров и масел, синтетич. жирных кислот
(С15—С20), кислот канифоли, соли алкиларилсульфо-
кислот (некаль й др.). Для приготовления р-ров ис-
пользуют очищенную и умягченную с помощью ионо-
обменных смол воду. Размер частиц эмульсии состав-
ляет 1 —10 мк. Для инициирования реакции полиме-
Основные свойства некоторых типов Б.-с. к.
Каучуки По к аз ате ли свойств —— СКС-30 СКС-ЗОА СКС-30 АРМ • СКС-10
Дефо-твердость * *, г Ненаполненные резины: предел прочности при разры- 2500—4000 2200—3700 400—700 2500—3000
ве, кг/см* 35—50 35-50 25—30
относительное удлинение, % Наполненные резины, 50 вес. частей канальной сажи: предел прочности при разры- 600-700 600-700 — 550—650
ве, кг/см- .......... 220—280 250—320 220—280 190—220
относительное удлинение, % 550—650 600-700 550—800 550—650
истирание, смЯ/квт-ч .... эластичность (по упругому 200-240 170-200 180—220 230-280
отскоку), % 38-44 36—40 30-34 38-45
Темп-ра стеклования, °C . . . Темп-ра хрупкости (вулкани- -60 — — -75-80
заты), °C Коэфф, морозостойкости (100% -54 — -53 от —74 до -77
растяжения) при —45° ... 0,15 — 0,20 (при —35°) 0,40-0,54
* 20 вес. ч. масла на 100 вес. ч. каучука. ** Дефо — показатель твердости
каучука, выра?каемый в граммах той нагрузки, к-рая вызывает заданную
деформацию образца стандартных размеров в течение 30 сек.
ризации применяют возбудители (0,08—0,45%), спо-
собные легко образовывать свободные радикалы: соли
надкислот (персульфаты, пербораты), перекись во-
дорода, органич. перекиси и гидроперекиси (изо-
пропилбензола, n-ментанаидр.). Для обеспечения поли-
меризации при низкой темп-ре вместе с возбудителями
применяют активаторы — вещества, обладающие вос-
становительными свойствами (сульфиты, сахара) в
комбинации с легко окисляющимися и восстанавли-
вающимися соединениями (гидрохинон, соли металлов
переменной валентности и др.), из к-рых создаются
т. н. окислительно-восстановительные (редокс) системы.
В целях предотвращения разветвления и получения
желательного мол. веса полимера применяют регу-
ляторы (меркаптаны, дисульфиды и др.), добавляемые
в количестве 0,08—0,2%, считая на углеводороды.
Эмульсионную сополимеризацию бутадиена со стиролом
проводят, кан правило, по непрерывному методу, для чего
бутадиен непрерывно смешивают в заданном соотношении со
стиролом. Параллельно готовится водная фаза растворением
эмульгаторов, стабилизаторов и других водорастворимых
соединений. Водная и углеводородная фазы непрерывно сме-
шиваются в соотношении 1 : 1—2 : 1, образуя эмульсию, посту-
пающую в батарею последовательно соединенных полимериза-
торов, каждый из к-рых представляет собой вертикальный
цилиндрич. сосуд из нержавеющей стали, 12—20 м3. Возбуди-
тель в виде р-ра подается в первый полимеризатор. Эмульсин
вытесняется из аппарата в аппарат и выходит из последнего
полимеризатора в виде латекса, содержащего незаполимери-
зованные бутадиен и стирол (ок. 40% от введенных мономе-
ров). Р-р регулятора с помощью дозировочных насосов на опре-
деленной стадии вводят в несколько полимеризаторов батареи.
Охлаждение производится водой, а при холодной полимериза-
ции холодильным рассолом или аммиаком. Продолжитель-
ность горячей полимеризации 25—30 час., холодной 14—18 час.
Степень превращения 58—60%. В латекс после последнего
полимеризатора непрерывно вводят в-во, обрывающее поли-
меризацию («стоппер»), и удаляют из латекса незаполимери-
зованные мономеры (дегазация) при остаточном давлении
350 мм рт. ст. Для предохранения каучука от старения
в латекс вводят 3% (от веса каучука) противостарителя (анти-
оксиданта), напр. неозона Д. в виде водной суспензии. Бутадиен
и стирол после очистки возвращают на полимеризацию. Дега-
зированный латекс направляют на коагуляцию для выделения
из него каучука. Латекс коагулируют к-той (напр., уксусной),
солями СаС12, MgCl2, NaCl или их комбинациями с получением
мелких зерен каучука. После коагуляции вся масса подается
на движущуюся мелкую сетку или на вибрационное сито.
После отделения от жидкости (серума) научук в виде ленты
или крошки сушится в воздушных сушилках.
Значительная часть Б.-с. к. вырабатывается в виде
маслонаполненного каучука (20—30 вес. частей масла
на 100 вес. частей полимера). Минеральное масло, со-
держащее св. 30% ароматич. соединений, вводится
в виде водной эмульсии в дегазированный латекс
холодной полимеризации перед его коагуляцией.
491
БУТАДИОН — БУТИЛЕНГЛИКОЛИ
492
В последнее время все в больших количествах выра-
батывают мягкий Б.-с.к., не требующий окислитель-
ной термопластикации на резиновых заводах. Вместо
стирола для сополимеризации иногда применяют
а-метилстирол.
Б.-с. к., имеющий в своем составе ок. 30% стирола—
СКС-30, является универсальным видом каучука,
из к-рого изготовляют автомобильные шины, транс-
портерные ленты, резиновую обувь, формовые резино-
вые детали, рукава и др. резиновые изделия. СКС-10
отличается высокой морозостойкостью, приближаясь
по этому показателю к натуральному каучуку, и ис-
пользуется для изготовления морозостойких рези-
новых изделий. СКС-ЗОА отличается улучшенными
технологич. свойствами при изготовлении на его ос-
нове резиновых смесей и меньшей истираемостью.
Лит.: Смирнов Н. И., Синтетические каучуки, 2 изд.,
Л., 1954; Д о г а д к и н Б. А., Химия и физика каучука,
М., 1947; Литвин О. Б., Технология синтетических кау-
чуков, Л.—М., 1950; его же, Основы технологии синтеза
каучуков, М., 1959; Бородина И. В., Никитин А. К.,
Технические свойства советских синтетических каучуков,
Л.—М., 1952; Синтетический каучун. Гл. ред. Г. С. Уитби,
пер. с англ., Л., 1957. А. К. Никитин.
БУТАДИОН (фенилбутазон, бутазолидин), мол.
вес 308,39, C19H20O2N2 — белые кристаллы горько-
го вкуса со слабым арома-
СН3(СН2)3СН —СО тическим запахом; т. пл.
I С6Н5 105°; почти нерастворим
СО N в воде, растворим в спир-
С те, легко растворим в аце-
6 ° тоне, эфире и этилацетате.
Б. обладает кислотными свойствами и титруется ще-
лочью по фенолфталеину; его щелочные соли хоро-
шо растворимы в воде и устойчивы при комнатной
темп-ре. Получают Б. конденсацией гидразобензола
с .диэфиром бутилмалоновой кислоты в присут-
ствии алкоголята натрия. Б. оказывает активное
аналгезирующее,, жаропонижающее и противовоспа-
лительное действие; применяют для лечения поли-
артритов и подагры.
Лит. см. при ст. Адалин. Е. С. Головчинская.
БУТАНЫ — газообразные насыщенные углеводо-
роды С4Н10, без цвета и запаха, мол. в. 58,52. Из-
вестны 2 изомера: /{-бутан СП3(СН2)2СН3 и пзобутан
(СН,)2СНСН3.
н-Бутан Изобутан
Т. пл., °C -138,4 -159,6
Т. кип., °C -0,50 -11,73
d$ “5(жидК|«) 0,5730 0,5510
nff" (жидк.) 1,3621 1,3514
*крит.> °С 152,01 134,98
РнрИТ.» ат 37,47 36,00
Теплота образования, ккал/моль 1044,8 1046,4
Вес 1 л газообразного Б. . . . 2,73204 г —
Смеси, содержащие 1,5—8,5% н-Б. или 1,8—
8,4% изобутана, с воздухом взрывают. Б. содержатся
в нефтяных газах, в природном газе и газах нефте-
переработки.
н-Б у т а н. Теплота испарения 5035 ккал}молъ (25°);
теплота сгорания 635,05 ккал!молъ (р = const, 25°).
При каталитич. дегидрогенизации на окиси хрома,
содержащей А12О3, н-Б. при 350—500° образует смесь
бутиленов, из к-рых последующей гидрогенизацией
получают бутадиен—исходный продукт для синтеза
каучука. При действии А1С13 и НС1 при 90—105°
и давлении 10—20 ат Б. изомеризуется в изобутан
(промышленный метод).
Пзобутан. Для него характерны реакции ал-
килирования и дегидрогенизации. В пром-сти изобу-
тан в смеси с этиленом (при 500° и 350 ат) образует
неогексан'. (CH3)2CHCH3+CH2=CH2->(CH3)3CCH2CHS;
при алкилировании другими углеводородами образуют-
ся гептан и октан', при каталитич. дегидрогенизации
над Сг2О3—А12О3 при 500° образуется изобутилен.
Пзобутан может быть получен гидрогенизацией СО
над Т11О2-катализатором или изомеризацией м-бутана
(см. выше). г. м. Цигуро.
БУТИЛАЦЕТАТ — см. Бутилового спирта эфиры.
БУТИЛЕНА ОКИСЬ С4Н8О, мол. в. 72,11 — об-
щее название окисей трех изомерных бутиленов;
жидкости с приятным запахом. В табл, приведены
нек-рые физич. свойства Б. о.
Название Т. кип., °C 20 nD
Окись 1,2-к-бутилена 6сн2снс2н5 58,5—59° - -
Окись 2,3-м-бутилена (окись псевдобутилеиа) Ьснснсн3 СН3 59,5—60,4* 53,6-54,1** 0,8272* 0,8053** 1,3826* 1,3736**
Окись 2-метил-1,2-пропена (окись изобутилена) ЬсН»С(СН3)2 51,5 0,8117 1.3745
* чкс-Изомер. ** трапс-Изомер,
Б. о. проявляют все химич. свойства окисей оле-
финов, напр.:
(СН3);С--СН2 + НС1 — (СН3)2С-СН2С1
Xq/ он
СН3СН- СНСН3 + NH3 СН3СН-СНСН3
Хо/ он nh,
Б. о. получают дегидрогалогенированием соответ-
ствующих галогенгидринов щелочными агентами,
напр.:
СН3СН-СНСН3 СН3СН-СНСН3
ОН С1 ' °z
а также окислением бутиленов кислородом при 130—
150° под давлением и в присутствии катализаторов
(напр., соли ванадия). Б. о. применяют в органич.
синтезе; смесь с тетрагидрофураном (1:1) употребляет-
ся для растворения высокомолекулярного поливи-
нилхлорида; вводят в нек-рые покрытия для умень-
шения коррозии металлов. н. в. Гнучев.
БУТИЛЕНГЛИКОЛИ (бутандиолы) С4Н8(ОН)2, мол.
в. 90,12 — двухатомные спирты алифатич. ряда.
Известны 4 изомерных Б. Некоторые физич. свойства
приведены в таблице.
Изомер Т. пл., °C Т. кип., ° С/мм d!? nD
1,2-Бутиленглиноль СН2(ОН)СН(ОН)СН2СН3, рацемат - 192—194; 75,0е/1 1.0059* -
1,3-Бутиленгликоль СН2(ОН)СН2СН(ОН)СН2, рацемат -50 207,5 1,0059 1,4401 (20")
1,4-Бутиленгликоль СН2(ОН)СН2СН2СН2(ОН) 20,9 203/759; 120/10 — —
2,3-Бутиленглиноль СН3-СН(ОН)СН(ОН)—СН3, рацемат 7,6 176,7/742 — —
2,3-Бутиленглиноль, мезо- форма 34,4 181,7/742 1,045 1,4364 (25°)
‘ 47.
493
БУТИЛЕНЫ — БУТИЛКАУЧУК
494
Все Б. смешиваются с водой и спиртами; 1,2-Б.
нерастворим, 1,3-, 1,4-Б. мало растворимы, а 2,3-Б.
растворим в эфире.
1,2 - Бутиле и глик оль. Рацемат получают омыле-
нием диацетата СН2(ОСОСН3)СН(ОСОСН3)СН2СН3, право-
вращающий изомер — энзиматич. восстановлением бутанол-
1-ОН-2 СН3СН2—СО—СН2ОН; [а]р'= +14,5° (в спир-
те); левовращающий изомер образуется при восстановлении
левовращающего этилового эфира а-оксимасляной к-ты
СН3СН2СН(ОН)СООС2Н, натрием и уксусной к-той в толуоле;
[а]р = —7,4° (в спирте); конфигурации оптически активных
форм не установлены. Идентифицировать 1,2-Б. мощно по
диацетилу, т. кип. 196—199°.
1,3-Б утиленгликоль. Правовращающий изомер
получают энзиматическим восстановлением альдоля; т. кип.
109°/14 -нл:; [а]р = +18° (в спирте), рацемат; производится
в промышленном масштабе восстановлением альдоля водоро-
дом при 100° и 300 ат над металлич. катализаторами (Cu,Zn,
Ni): СН3СН(ОН)СН2СНО + Н2 -» СН3СН(ОН)СН2СН2(ОН);
при нагревании с ZnC)2 до210—215° частично дегидратируется,
превращаясь в кротиловый спирт: СН2(ОП)СН2СН(ОН)СН3 ->
—< СН3—СН=СН—СН2ОН; над фосфатным катализато-
ром при270° в присутствии бутиламина дегидратируется, обра-
бутадиен:
зуя
СН3—СН(ОН)СН2—СН2(ОН)
— СН2=СН—СН=СН2
NaHPO. + C.H,NH2
270°
1,3-Б. применяют для промышленного получения
бутадиена, как растворитель, пластификатор для по-
лиэфирвых смол и заменитель глицерина; для иден-
тификации 1,3-Б. можно пользоваться фенилуретаном,
т. пл. 115—116°, [a]g = +20° (в спирте) и дифенил-
уретаном, т. пл. 127—128°, [ajyj = —51,5° (в спирте).
1.4-Б утиленгликоль. В пром-сти произ-
водят гидрированием бутиндиола водородом при
70—150° и 200—300 ат над медно-никелевым катали-
затором:
НОСН2С“. ССН2ОН-> НОСН2СН2СЫ2СН2ОН
дегидратацией получающегося по этому методу 35%-
ного водного р-ра 1,4-Б. фосфорной к-той (pH =2)
под давлением при 260—280° получают тетрагидро-
фуран. При нагревании 1,4-Б. с НС1 получают 1,4-
дихлорбутан, к-рый действием KCN превращают в
динитрил адипиновой к-ты, являющийся исходным
продуктом в синтезе нейлона; с гексаметилендиизо-
цианатом 1,4-Б. образует полиуретаны.
2,3-Б утиленгликоль. Право- и левовра-
щающий изомеры, а также мезоформа образуются при
энзиматич. распаде углеводов; рацемат получают гид-
ролизом 2,3-бутиленоксида СН3СН—СНСН3. Апетат
2,3- Б. при пиролизе превращается в бутадиен:
СНзСН(ОСО СНз) СН (ОСОСН31 - СНз ~2CH-CQP^S
СН2 СНСН СН.;
КМпО4 или СгО3 окисляются в диацетил:
СН3ОН(ОН)СН(ОН)СН3 —ЙсНз-СО-СОСН.
М. Ф. Турчинский.
БУТИЛЕНЫ (бутены) С4Н8, мол. в. 56,02 — газо-
образные ненасыщенные углеводороды, олефины, из-
вестны все возможные изомеры: a-бутилен (бутен-1,
этилэтилен) С2Н5СН=СН2; Р-бутилен (цис- и транс-
бутен), псевдобутилен (а, fj-диметилэтилен) СН3—
—СН = СН—СН3; изобутилен (изобутен, 2-метилпро-
пен, несимметрич. диметилэтилен) (СН3)2С = СН2. Фи-
зич. свойства Б. приведены в таблице.
Б. очень мало растворимы в воде, хорошо — во
многих органич. растворителях; обладают всеми
химич. свойствами олефинов: присоединяют по двой-
ной связи водород, галогены, галогеноводороды и т. д.
При окислении Б. щелочным р-ром КМпО4 образуются
гликоли, напр:
сн8- СН=СН-СН3 + О + Н.,О -► СНз -СНОН -СНОН-СН,
При окислении кислородом воздуха (над катализа-
Свойства Бутен-1 Бутен-2 Изобу- тен
цис транс
Т. пл., °C -185,35 —138,91 -105,55 -140,42
Т. кип., “С/760 .и.ч . . . -6,25 3,72 1,0 -6,90
d‘ 0,740 0,724 0,6044 0,6948
(—110°) (—78,5°) (20°) (—70°)
1,3792 1.3946 1.3862 1.3811
*КрИТ., °C 147,4 155 (смесь цис- и транс-) 144,73
РкрИТ.’ ат Теплота испарения при 40,03 •41 — 39,48
т. кип., ккал/моль . . Теплота сгорания, 4,87 5,580 5,695 5,28:1
ккал/моль Т. пл., 2 ,.4-динитробен- 649,45 647,81 646,81 64b,48
зоатов, °C 69-70,4 96,4—97.6 91,8 -92,8 108,4— 109,2
Взрывоопасные смеси с воздухом 1,7—9% по объему.
тором) образуют бутилена окись, при каталитиче-
ской дегидрогенизации Б. получают бутадиен', под
влиянием различных катализаторов, например Н3РО4
при нагревании (135—420°), происходит изомеризация
Б.: СН3—СН3—СН=СН2-> СН5СН=СН—СН3. Раз-
личные конденсирующие агенты полимеризуют Б.
В пром-сти Б. выделяют из бутан-бутиленовой
фракции газов нефтепереработки, а также получают
из бутиловых спиртов. Чистый a-Б. образуется при
пиролизе бутилацетата при 500° или н-С4Н9С1 — при
550° и при дегидрогенизации н-бутилового спирта
над свободной от кислот А12О3при375—425°. Чистый
цис-Р-Б. можно получить из 6,1-2,3-дибромбутана
кипячением с Zn в спирте или нагреванием 1-бром-
диэтилсульфона; чистый транс-Р-Б. — из мезо-2,3-
дибромбутана действием Zn в спирте или нагреванием
бромгидрата тиглиновой к-ты с р-ром соды до 70°.
Б. применяют в пром-сти для получения бутадиена
(из фракции нормальных бутан-бутиленов) — исход-
ного в-ва для получения синтетич. каучука; полиме-
ризацией смеси Б. в присутствии А1С13 получают
вязкие смазочные масла, а конденсацией смеси Б.
с изобутаном синтезируют высокооктановое моторное
топливо. Окислением смеси Б. получают ацетальде-
гид. Изобутилен применяют для произ-ва бутил-
каучука (оппанола), а также изооктана.
Лит. см. при ст. Газы природные горючие.
Н. В. Гнучев.
БУТИЛ ИЗОВАЛЕРИАНАТ — см. Бутилового спир-
та эфиры.
БУТИЛКАУЧУК — продукт совместной полиме-
ризации изобутилена и небольших количеств изо-
прена. Непредельность Б. составляет 1 — 3% от не-
предельности каучука натурального. Б. обладает ли-
нейной структурой с нерегулярным чередованием
изопреновых и изобутиленовых групп; изопреновые
звенья присоединяются почти полностью в положе-
нии 1,4. Подобно натуральному каучуку, Б. относится
к кристаллизующимся полимерам. Реакция образо-
вания полимерной пени Б.:
СН3 СН3
nC=CH, + тСН2=С-СН=СН3 —
I
сн,
сн3 сн3 сн3 сн3
-+ С-СН3—СН3—С=СН—СН.,—С—СН.,—С—СН.>— ...
I 'II
сн3 сн3 сн3
Полимеризацию проводят в р-ре монохлорпроиз-
водных углеводородов (чаще всего в р-ре хлористого
метила) при минус 90—100° в присутствии А1С13 по
непрерывному методу. Отнятие реакционного тепла
495
БУТИЛОВОГО СПИРТА ЭФИРЫ — БУТИЛОВЫЕ СПИРТЫ
493
осуществляют с помощью кипящего этилена, пода-
ваемого в рубашку и в змеевики полимеризатора. Сус-
пензию полимера после добавления стеарата Zn и
антиоксиданта подают в испаритель с горячей водой,
где происходит удаление незаполимеризованных мо-
номеров. Отделенная от воды крошка сушится, листует-
ся и упаковывается.
Б. — прозрачная, малогазопроницаемая, обладаю-
щая хорошими электроизоляц. свойствами эластич-
ная масса белого или серого цвета. Физико-механич.
свойства Б.: предел прочности при разрыве (непапол-
ненные резины) 180—220 кг/см2, относительное уд-
линение (у них же) 850—950%; у наполненных резин
(50 частей канальной сажи) предел прочности при
разрыве 170—230 кг/см2, относительное удлинение
650—800%, истирание 170—200 см3/кет-ч, эластич-
ность по упругому отскоку 8—12% при 20° и 34—
36% при 100°, темп-ра стеклования —67°, темп-ра
хрупкости вулканизатов —45°, газопроницаемость
вулканизатов (72 ч. сажи) по СО2 2,5 л/л2 за 24 часа.
Б. стоек к тепловому старению в воздушной и кисло-
родной среде, а также к свету (не при повышенных
темп-pax) и сильно действующим химич. реагентам
(перекись водорода, озон, крепкие к-ты, в т. ч. HNO3).
Б. применяется для изготовления
автомобильных камер, прорезинен-
ных тканей и различных резиновых
изделий, для обкладки химич. аппа-
ратуры и для изготовления электро-
изоляционных материалов.
Лит.: W age пег Ch. Е., Rubber
Age, 1957, 81, № 2, р. 291, см. также
лит. к Ст. Бутадиен-стирольный каучук.
А. К. Никитин.
БУТИЛОВОГО СПИРТА ЭФИРЫ
(сложные) — бесцветные жидкости с
характерным эфирным запахом; мало растворимы в
воде, хорошо — в органич. растворителях. Свойства
нек-рых сложных эфиров, бутилового спирта, при-
меняемых в пром-сти, приведены в таблице.
Таблица 1
Название Мол. вес Т. пл., °C Т.кип., °C 4» 20 пр
Бутилформиат CHS(CH2)3OOCH 102,13 —90,0 106,6 0,8917 1,3890
Бутилацет ат СНДСН2),ООСН3 116,16 -73,5 126,1 0,881 1.3941
И зо б ути л ацет ат (СН3)2СНСН#ООССН3 116,16 -98,9 118,0 0,875 1,3901
Бутилпропионат СН3(СН1)3ООССН8СН3 130,18 —88.55 146,8 0,8818* 1,4038*»
Бутилизовалеронат СН3(СНг)3ООС(СН2)3СН3 158,13 — 135—185 0,874 1,458**
• При 15°. ** При 25°.
Перечисленные эфиры образуют азеотропные смеси,
папр. бутилформиат с м-бутиловым спиртом (76%,
т. кип. 105,8°); с Н2О и бутиловым спиртом (69%
бутилформиата, 10% С4НвОН, 21% Н2О, т. кип. 83,6°).
Бутилацетат и изобутилацетат образуют азеотропы
с водой, бутиловым или изобутиловым спиртами
(табл. 2).
Таблица 2
2-Й компонент азеотропа Бутил аце- тат, % Т. кип., °C Изобутил- ацетат, % Т. кип., °C
Нго .... н-С.Н.ОН . . . uao-C.HeOH . . . 71,3 27,0 90,2 116,0 71,0 53 55 90,2 117,2 102,4
Б. с. э. обладают всеми свойствами сложных эфиров:
омыляются, способны к переэтерификации и т. д.;
получают нагреванием бутилового спирта и соответ-
ствующей к-ты в присутствии серной к-ты.
Б. с. э. растворяют нитроцеллюлозу, глифталевые
и полиэфирные смолы, хлоркаучук и др.: смешиваются
с растительными маслами; применяются как компо-
нент растворителя для нитроцеллюлозных лаков и
красок. Наиболее распространены бутил- и изобу-
тилацетат; они широко применяются в пром-сти как
растворители. Смеси паров Б. с. э. с воздухом взры-
воопасны. См. также Дибутилфталат.
Лит.: Вайсбергер А. [и др.], Органические раство-
рители, пер. с англ., М., 1958. М. Ф. Турчтккчй.
БУТИЛОВЫЕ СПИРТЫ С4Н,ОН — бесцветные жид-
кости с характерным спщэтовым запахом; триметил-
карбинол обладает камфарным запахом. Известны
следующие Б. с.: нормальный первичный бутиловый
спирт (бутанол-1) СН3СН2СН2СН2ОН; нормальный
вторичный бутиловый спирт (бутанол-2, метилэтил-
карбинол) СН3СН2СНОНСН3; изобутиловый спирт
(2-метилпропанол-1) (СН3)2СНСН2ОН; триметилкар-
бинол (2-метилпропанол-2) (СН3)3СОН.
Нек-рые физич. свойства Б. с. приведены в табл.
С этанолом и эфиром Б. с. смешиваются во всех от-
ношениях. Для Б. с. характерны все реакции спиртов.
Спирты Т. пл., °C Т. кип., °C 4° 20 пр Раствори- мость г/100 г воды Азеотропная смесь: % спирта 'т. кип., °C
н-Бутиловый .... Бторич. бутиловый Изобутиловый . . . Триметилкарбинол -79,9 -114,7 —108 25,0—25,5 117,4 99,5-100 107,5 82,9 0,8099 0,8060 0,8027 0,7887 1,3993 1,3949 1,3878 1,3954 9 (15°) 12,5 (20°) 10(15°) неограни- ченно 62,2/92,6 72,7/87,5 66,8/89,8 88,2/79,9
Бутанол-1 — уд. теплоемкость (30—80°) 0,582
кал [г', теплота сгорания 8625 кал/г\ т. воспл. 34°;
смеси с воздухом, содержащие 2—12% бутанола-1,
взрываются. В промышленности бутанол-1 получа-
ют: 1) ферментативным брожением углеводсодер-
жащих веществ; 2) оксо-синтезом из пропилена
в присутствии катализаторов — карбонилов кобаль-
та, никеля и др. при 130—180° и давлении 150—
200 ат: СН3СН=СН24-СО2+ Н2-^СН3СН2СН2СНОИ-’
—>СН3СН2СН2СН2ОН; 3) синтезом из ацетальдегида че-
рез альдоль: 2СН3СНО--СН3СТЮНСН2СНО--
->СН3СН=СНСНОСи.ваХза-СН3СН2СН2СН2ОН;
4) выделением из отходов произ-ва синтетич. каучука.
В США ферментативный метод в значительной сте-
пени вытеснен синтезом через альдоль и кротоновый
альдегид и др. Очевь перспективно получение н-бута-
нола оксо-синтезом. Бутанол-1 применяют в качестве
растворителя в произ-ве нитролаков, этилцеллюлозы,
смол и т. д. н-Бутанол используется в произ-ве слож-
ных эфиров (дибутилфталата, бутилксантата), при-
меняемых в качестве пластификаторов.
Б у т а н о л-2 — т. воспл. 24°, известны два сте-
реоизомера: d и 1; [а]д = + 13,9° (для d-изомера).
Для синтеза бутанола-2 используется бутап-бутиле-
новая фракция, получающаяся при парофазном кре-
кинге нефти, содержащая до 32% бутена-1 и бутена-2.
Гидратация бутена-1 и бутеиа-2 проводится при срав-
нительно низкой темп-ре. В присутствии 80%-ной
H2SO4 бутен-1 и бутен-2 образуют алкилсерный эфир,
из к-рого гидролизом получают бутанол-2:
СН3СН=СНСН3 | H2SO4 СН3СНСН2СН3
СН3СН2-СН=СН2 j OSO2OH
— СН3СНОНСН2СН3 + H2SO,
Бутанол-2 можно получить также гидролизом вто-
ричного бутилхлорида, к-рый образуется наряду с
497
БУТИЛФОРМИАТ — БУТИРОЛАКТОН
498
первичным бутилхлоридом при хлорировании м-бу-
тана. При окислении бутанола-2 образуется метил-
этилкетон. Применнетсн бутанол-2 в качестве эк-
страгента длн жиров и растворителя длн нитроцеллю-
лозы и лаков.
Изобутиловый спирт — уд. теплоемкость
6,6034 кал/г (30—80°); уд. теплота образования 8594
кал/г; т. воспл. 29°. Изобутиловый спирт содержится
в значительном количестве в сивушных маслах, от-
куда может быть выделен дробной перегонкой; может
быть получен также сбраживанием мелассы. Синте-
тически изобутиловый спирт получают из смеси СО
и Н2 в присутствии катализаторов—металлов VIII
группы при давлении 300—700 ат:
СО I- Н2 — СН.,СН2ОН — СП3СН2СН2ОН — (СНз)2 снсн2он
Из смеси м-пропанола, амилового спирта и др. про-
дуктов реакции изобутиловый спирт отделяют пере-
гонкой; для очистки от примесей — кипятят с нега-
шеной известью. Применяют изобутиловый спирт
в качестве растворителя длн лаков; длн масел, упот-
ребляемых в качестве тормозной жидкости; может
заменить м-бутанол при синтезе алкилированных
смол на основе мочевины.
Триметил карбинол (nipem-бутиловый
спирт) — выше 25,5° — жидкость; уд. теплоемкость
0,725 кал/г (26,8°); уд. теплота сгорании 8500 кал/г;
темп-ра воспл. 9°. Триметилкарбинол может быть по-
лучен действием 65%-ной серной к-ты на смесь изо-
бутилена с бутиленами при 5—10° и последующим гид-
ролизом сернокислого йфира:
(CHJ 2С-CH.J + H»SO4 — (СН3)»ССН3 —
OSO3OH
— (СН3)3СОН + H»SO4
Скорость поглощения бутена-1 и бутена-2 серной
к-той в этих условиях в 200— 300 раз ниже, чем ско-
рость поглощения изобутилена, что обеспечивает се-
лективность воздействии серной к-ты. Для получении
триметилкарбинола может быть также использовано
селективное воздействие (при —78°) газообразного
НС1 на смесь бутиленов и изобутилена. В этом слу-
чае реакции проходит с образованием практически
лишь одного продукта — mpem-хлористого бутила,
омылением которого получают триметилкарбинол:
(СН3)3СС1 + Н2О — (СН3)3СОН + НС1. Действие НС1
на изобутан приводит к образованию смеси первичного
и третичного хлористого бутила, при гидролизе к-рых
можно получить м-бутанол и триметилкарбинол.
Последний применяют длн получении искусственного
мускуса, в синтезе моющих средств, при денатурации
спирта (этанола) и для получении чистого изобутилена
из газов нефтепереработки.
Лит.: Kirk, V. 2, N. Y., 1948, р. 674.
Н. С. Кочеткова.
БУТИЛФОРМИАТ — см. Бутилового спирта эфиры,
п-т рет-БУТИЛЦИКЛОГЕКСИЛАЦЕТАТ, мол. в.
198,30 — бесцветная жидкость с сильным цветоч-
ным запахом, т. кип. 105—106°/9 мм; d;'1 0,9371;
nj/j 1,4511. В пром-сти Б. (I) получают из фенола:
С(СН3)3 С(СН3)3 С(СН3)3
Б. применяется в парфюмерии как душистое в-во.
Н. Ю. Лмбошиц.
БУТИНДИОЛЫ С4НвО2, мол. в. 86,05 — двухатом-
ные спирты, содержащие тройную связь; бесцветные
кристаллы, легко растворимы в воде, спиртах, мало
растворимы в эфире, хлороформе. Бутин-2-диол-1,4
НОСН2—С=С—СН2ОН; т. пл. 58°; т. кип. 238°/760 мм;
125—127°/2 мм; бутин-1-диол-3,4 СН=С—СН(ОН)—
—СН2ОН; т. пл. 39,5—40,5°.
Б. свойственны реакции ацетиленовых углеводоро-
дов: присоединении, окислении, полимеризации и др.,
и спиртов (напр., замещение ОН-группы на галоген).
Промышленное значение имеет бутин-2-диол-4. При
дегидратации образуется вначале тетрагидрофуран,
а далее бутадиен-1,3. С водой в присутствии солей
ртути Б.-1,4 образует кетобутандиол, восстановле-
нием к-рого получают 1, 2, 4-бутантриол, являющийся
заменителем глицерина:
НОСН2-С=С-СН2ОН —носн»со-сн»снаон —1
— НОСН»СН(ОН)СН»СН»ОН
При действии на бутиндиол-1,4 хлористого тионила
образуется 1,4-дихлорбутин-2, переходящий при от-
щеплении НС1 в диацетилен:
носн»-с-с-сн2он U с1сн.,-с=с-снаа
- НС - С -С -СН
Исходя из Б.-1,4, можно получать гексаметилендиа-
мин H2N—(СН2)6—NH2 — одно из исходных веществ
синтеза полиамидных смол:
носна-с^с-снаон Ьносн..-сн=сн-сн2он —
- NC-CH2-CH=CH—CH»-CN H»N-(CH»)e—nh»
В пром-сти бутиндиол-1,4 синтезируют из ацетилена
и формальдегида в присутствии катализатора ацети-
ленистой меди под давлением: СН=СН+СН2О —>
—> НОСН2С=ССН2ОН (см. Оксосинтез).
М. Я. Алейникова.
БУТИРОЛАКТОН (ангидрид у-оксимаслнной кис-
лоты), мол. вес 86,09 — бесцветная жидкость со
слабым запахом масляной кислоты;
т. пл. —42°; т. кип. 204°; 89°/12.u.w; н г_
1,13; Пр 1,4360. Б. легко раство- 2| । 2
рим в низших спиртах, эфире, слож- НгС
ных эфирах, ацетоне, бензоле, СС14, О
воде; трудно растворим в алканах
и цикланах; сам Б. является хорошим растворите-
лем длн полиакрилнитрилов и др. полимеров. В
водном р-ре Б. гидролизуется в оксимаслнную к-ту,
при 0° полностью, при 100° частично (80% лактона);
в щелочной среде гидролиз происходит быстро и пол-
ностью: О(СН2)3СО % Н2О та НОСН2СН2СН2СООН.
Б. окислнетсн (К2Сг2О7 -4H2SO4 или HNO3) в янтарную
к-ту. От действии HJ на холоду или НВг при 100° Б.
превращаетсн соответственно в у-иод- и у-броммас-
ляную к-ту. Б. восстанавливает соли серебра, превра-
щаясь при этом в янтарную к-ту. С галогеноводород-
ными к-тами и спиртами на кислотных катализаторах
Б. дает соответственные производные 4-оксимасляной
I-----' н+
к-ты: О(СН2)3СО 4- НС1 (или ВОН) —С1(СН2)3СООН
[или ВО(СН2)3СООН]. При нагревании Б. с карбо-
натами щелочных металлов или баритовой водой об-
разуются соли у-оксимасляной к-ты. Сплавлением
Б. с Na2S или NaOH получают соответственно тиоди-
и оксидимасляные к-ты, к-рые применяют как пла-
стификаторы поливинилхлорида. Безводный аммиак
или преим. амины при 230° превращают Б. в 2-пир-
г------------------------------------------1
ролидон и N-замещенные 2-пирролидоны О(СН2)3СО +
+ NH3 -► HOCH2CH2CH/3ONH2 HN(CH2)3CO. Б.
можно получить перегонкой у-оксимаслнной к-ты
и др. способами. В пром-сти Б. получают восстановле-
нием 1,4-бутандиола в жидкой (катализатор Cu/SiO2
200°) или в паровой фазе (Си/пемза; 230—250°); мо-
жет быть также получен окислением тетрагидро-
фурана. Применяют Б. как пластификатор, как рас-
творитель ацетил- и нитроцеллюлозы, полиакрилнит-
499
БУФЕРНЫЕ РАСТВОРЫ — БУФОГЕНИНЫ
500
рила, поливинилформиата и др., в качестве полупро-
дукта при различных синтезах.
Производные Б., содержащие в a-положении алкиль-
ные группы, от метильной до нонильной, алифатич.
терпеновые спирты или бензильную группировку,
обладают фруктовым или цветочным запахом. Прак-
тич. интерес для применения в парфюмерных компо-
зициях представляют след. соединения:
Радикал Т. кип., ° С/мм 20 nD
Изоамил СН2СН2СН(СН3)2 120/15 0,9662 (21°) 1,4455 (20°)
н-Гексил СН2(СН2)3СН3 146/16 0,9551 (21°) 1,4480 (21°)
Геранил СН2СНС(СН2)СН2СН=С(СН3)2 145/0,72 0,9715 (24°) 1,4842 (24°)
сн,
Цитронеллил СН2СН2СН(СН2)гСНСН3 144/0,57 0,9541 (22,5°) 1,4711 (22,5°)
СП, сн,
Бензил С,Н,СН2 150/1,55 1,1272 (22,5°) 1,5338 (22,5°)
А. А. Зелемецкая, И. В. Матвеева.
БУФЕРНЫЕ РАСТВОРЫ — растворы с опреде-
ленной концентрацией водородных ионов, смесь сла-
бой кислоты и ее соли или слабого основания и его
соли, незначительно меняющие значение pH при раз-
бавлении, концентрировании, а также при добавле-
нии небольших количеств свободной сильной к-ты
или щелочи. Для Б. р., состоящих из смеси слабой
к-ты и ее соли, концентрация водородных ионов мо-
жет быть вычислена по след, формуле:
(1)
для Б. р., состоящих из смеси слабого основания и
его соли, по формуле:
кНоО Ссоли
1= к ' ... ~с---- W
“ООН. LOCH.
^нисл и ^-’осн. — константа диссоциации слабой кис-
лоты или основания, ATji2O — ионное произведение
воды, с — общая концентрация к-ты, основания или
соли. Для приготовления Б. р. с низким значением pH
используют к-ту с большим значением для сред-
ней и слабокислой области применяют к-ту с низким
значением А^щ^да для щелочных р-ров употребляют
смеси оснований и их солей. pH Б. р., составленных
из слабой кислоты и ее соли, незначительно изменяет-
ся с темп-рой, тогда как для Б. р., составленных из
слабого основания и его соли, pH понижается с по-
вышением темп-ры, т. к. в ур-ние (2) входит величина
АГц о» к-рая чувствительна к изменениям темп-ры.
Поэтому для практич. целей лучше пользоваться
Б. р., составленными из смеси слабой кислоты и ее
соли. Б. р. можно также составлять из смеси солей
многоосновной к-ты, константы диссоциации к-рой
сильно отличаются друг от друга, напр. из смеси
одно- и двузамещенных фосфатов, а также из кислых
солей многоосновных слабы» к-т: напр., кислый вин-
нокислый калий, тетраборат натрия и др. При раз-
ведении Б. р. значение его pH мало изменяется: так,
концентрация Б. р. из смеси СН3СООН + CH3OONa
при изменении концентрации компонентов от 0,1 до
0,001 н. меняется след, образом:
Нормальность........ 0,1 0,01 0,001
pH.................. 4,63 4,67 4,73
При таких же разведениях значение pH раствора НС1
меняется от 1 до 3, а уксусной к-ты от 2,86 до 3,86.
pH буферных р-ров незначительно изменяется от
прибавления небольшого кол-ва сильной к-ты (или
щелочи):
Концентрация НС1 в воде или в буферном р-ре . . . 0 0,01 0,05 0,1
pH смеси Н2О4-НС1 .... pH-ацетатного Б. p.-f-HCl ~4,73 2,02 4,65 1,3 4,24 to сч 1 II К И а а <3 <3 м
Б. р. сохраняют постоянство pH лишь до прибав-
ления нек-рого определенного количества к-ты (или
щелочи); если концентрация добавленной к-ты (ще-
лочи) превышает предел «буферности» данного р-ра,
то pH его резко изменяется. Следовательно, Б. р.
обладают определенной «емкостью» (за единицу ем-
кости буферной смеси условно принимают емкость
такого раствора, для изменения pH к-рого на единицу
требуется введение сильной к-ты или щелочи в коли-
честве 1 г-экв на 1 л р-ра). Буферная емкость нахо-
дится в прямой зависимости от концентрации компо-
нентов Б. р., чем р-р концентрированнее, тем больше
буферная емкость; разведение не влияет заметно на
изменение pH, но сильно отражается на емкости Б. р.
На практике обычно употребляют Б. р. с общей кон-
центрацией кислоты и соли порядка от 0,05 до 0.1 н.
Эти буферные смеси устойчивы примерно в пределах
рН = рА +1. Для колориметрии, или потенциометрия,
определений pH можно употреблять Б. р. с низкой
концентрацией компонентов, и зона pH, охватыва-
емая Б. р., может быть достаточно большой. Б. р.
должны быть приготовлены из очень чистых реактивов
(квалификации «х. ч.») со всевозможной тщательно-
стью; для большей точности pH Б. р. проверяют по-
тенциометрически.
Б. р. широко пользуются в различных химич. ис-
следованиях; играют огромную роль в процессах,
протекающих в живом организме; многие из этих
жизненных процессов могут протекать только при
определенном значении pH (с незначительными коле-
баниями); это постоянство pH поддерживается в жи-
вом организме Б. р. (напр., в крови буферными сме-
сями, состоящими из карбонатов и фосфатов). Б. р.
широко используются в аналитич. химии; процессы
осаждения, разделения, экстракции и др. возможны
лишь в определенных пределах pH растворов,, что
можно создать, используя различные Б. р. Известно
большое число Б. р., с помощью к-рых можно сохра-
нять постоянство pH раствора в различных пределах.
Лит.: Кольтгоф И. М., Лайтинен Г. А.,
Определение концентрации водородных ионов и электротит-
рование, М., 1947; П ч е л и н В. А., Измерение активности
водородных ионов (pH), окислительно-восстановительных
потенциалов и потенциометрическое титрование, М., 1955;
Виноградова Е. Н., Методы определения концентра-
ции водородных ионов, 2 изд., М., 1956. И. П. Ефимов.
БУФОГЕНИНЫ — стероидные вещества, содержа-
щиеся в секретах кожных или околоушных желез жаб
(Bufo). По химич. строению Б. близки к агликонам
гликозидов сердечных (см. общую ф-лу I); по действию
яа сердце сходны с гликози-
дами сердечными.
В положении 17 стероид-
ной системы содержится нена-
сыщенное шестичленное лак-
тонное кольцо. Помимо гидро-
ксила в положении 1,4 Б.
могут содержать группы ОН
ив др. положениях (1, 2, 3).
В секретах жаб (где содер-
жатся также ядовитые в-ва алкалоидного харак-
тера, напр. буфотеин) Б. содержатся в свободном виде
501
БУХЕРЕРА РЕАКЦИЯ
502
(буфоталин) или в виде эфиров субериларгини-
на и уксусной к-ты, к-рые называют буфотокси-
п а м и (IT).
Из секретов жаб различных видов выделен ряд Б.
и буфотокспнов, незначительно отличающихся один
от другого гл. обр. расположением ОН-групп.
Строение большинства Б. полностью не расшифровано.
Ввиду высокой токсичности Б. как сердечные средства
находят ограниченное применение.
Лит.: Назаров И. И., Бергельсон Л. Д.,
Химия стероидных гормонов, М., 1955, с. 17; Фпзер Л.,
Фи вер М., Химия природных соединений фенантренового
ряда^ М., 19эЗ, е. 539; F i е s е г L., Fieser М., Steroids.
N. Y. — L., 1959. Л. Г. Равина.
БУХЕРЕРА РЕАКЦИЯ — обратимое превраще-
ние ароматич. оксисоединений в аминосоединения в
присутствии водного р-ра сульфита или бисульфита.
Практич. значение Б. р. имеет только для производных
нафталина и резорцина. Замена оксигруппы амино-
группой происходит при нагревании нафтола (I)
с водным раствором сульфита аммония и аммиака,
обратная реакция идет при обработке соответствую-
щего амина, напр. нафтиламина (II), р-ром бисуль-
фита натрия.
Он (NH4)2SO3.NH3^
Na HSO3
Многочисленными исследованиями было установ-
лено, что по Б. р. гладко реагируют лишь те произ-
водные, для к-рых возможно существование двух
таутомерных форм. Реакция идет через стадии присое-
динения бисульфита к кетоформе оксисоединения (III),
последующего обмена оксигруппы на аминогруппу
(IV) и разложения продукта присоединения щелочью.
Соединения, аналогичные продукту присоединения
бисульфита (III), были получены из гидрохинона, ре-
зорцина, 2,7-диоксинафталина и др. Существенное
влияние на замещение амино- и оксигруппы оказывает
сульфогруппа. Если 4-сульфокислоты а-нафтола и
а-нафтиламина легко обменивают ОН- и МН2-группы,
то 2-сульфокис.чоты к Б. р. не способны, т. к. послед-
ние не образуют бисульфитпых соединений. Неак-
тивны в этой реакции и 4-сульфокислоты 0-нафтил-
амина. Сульфогруппа, находящаяся в другом ядре,
мало нлияет на скорость реакции. Используя влияние
сульфогруппы, в 1,8-диаминонафталин-4-сульфокис-
лоте замещают только одну NH2-rpynny:
SO3H SO3H
Оксигруппа может быть обменена, кроме N Н2-группы,
на алкил- и диалкиламиногруппы; для этого требуются
более жесткие условия. Производные хинолина также
вступают в Б. р. Так, из 5-аминохинолина получают
5-оксмхиполин. При нагревапии с аммиаком и суль-
фитом аммония при 150—160° 8-оксихинолин коли-
чественно переходит в 8-аминохинолии. Реакция
описана в 1904 Г. Бухерером.
Лит.; Органические реакции. Сб. 1, пер. с англ., М., 1948,
с. 133; Ворожцов Н. Н., Основы синтеза промежу-
точных продуктов и красителей, 4 изд., М., 1955 (с. 399 — 418).
В. А. Пучков.
0Н + (NH4)2SO3^
~(NH4)HSO3
-(NH4)HSO3
+(NH4)Hso?
ВАГНЕРА РЕАКЦИЯ — способ определения строе-
ния непредельных органич. соединений окислением
их слабым (1%-ным) р-ром КМпО4. Первым примером
В. р. было окисление изобутилена; после отгонки
летучих продуктов глубокого окисления был полу-
чен изомасляный альдегид:
СЙз\с=сн2
сн3/
+ Н2О
-сн3\
>с—
СНз/ |
L он
сн>,
Ан
— СНз\снсно +
сн3/
54”), применяется для смазки подшипников, работаю-
щих при малых нагрузках и при темп-ре не выше
35°, для приготовления спец, смазок и защиты металла
от коррозии.
Лит.: Технические нормы на нефтепродукты (справочная
книга), под ред. Н. Г. Пучкова, 16 изд., М., 1957.
Л. С. Поваров.
ВАЗОПРЕССИН — гормон, выделенный из зад-
ней доли гипофиза и гипоталамуса (части головного
мозга) и представляющий собой циклич. пептид,
содержащий по 8 остатков L-аминокислот. В. — гиг-
Образование изомасляного альдегида происходит
вследствие пинаколиновой перегруппировки. На боль-
шом экспериментальном материале было показано,
что метод окисления соединений, содержащих этиле-
новые (несопряженные) связи, является общим и что
прв этом первичные продукты окисления пред-
ставляют собой гликоли. В. р. — один из важных
методов изучения разнообразных и сложных по струк-
туре соединений, имеющих этиленовые связи. В. р.
сыграла решающую роль в установлении строения
терпенов (пинена, лимонена, терпинеола и терпина).
Окисление, напр., камфена (I) щелочным р-ром пер-
манганата в камфениловый альдегид (III) через кам-
феногликоль (II) можно представить след, образом:
роскопич. кристаллы, хорошо растворимые в воде;
изоэлектрич. точка при pH 10,85—10,9. В. дает реак-
ции, характерные для тирозина и лизина. Полностью
установлено и подтверждено синтезом строение В.
быка C46H65N16O12S21,
мол. в. 1084, и В. свиньи
C4eHg6N43O42S2, мол. в.
1056, строение которо-
го отличается от строе-
ния В. быка только од-
2 3
I ,тир.— фал.
цистин 5 |д
^асп.— глют. - N Н2
1б 7 8
прол. - арг. - глиц. -NH2
СН
H2v I ~'С=СН2 .... „
| СН2| КМпО^
Н2С | „С(СН3)2
хн
^СН он
снУс^он_н_
Н2С. I ^С(СН3)2
Х2Н
Реакция открыта Е-. Е. Вагнером в 1887.
Лит.: Вагнер Е., К реакции окисления непредельных
углеродистых соединений, Варшава, 1888; И в г о л ь д К. К.,
Механизм реакций и строение органических соединений, пер.
с англ., М., 1959, с. 385. ,1/. Г. Линькова.
ВАГНЕРА—МЕЕРВЕЙНА ПЕРЕГРУППИРОВКА—
см. Камфеновые перегруппировки.
ВАЗЕЛИН :— однородная мазеподобная масса без
вкуса и запаха, представляющая собой смесь мине-
рального масла и твердых парафиновых углеводоро-
дов; темп-ра каплепадения 37—50°, плотн. 0,870—
0,885, растворяется при нагревании в спирте, эфире
и углеводородах — на холоду. Получают В. расплав-
лением парафина, церезина, петролатума и их смесей
(15—20%) в минеральном масле с последующей очист-
кой этой смеси серной к-той и отбеливающей глиной.
В зависимости от назначения различают неск. сортов
В.: медицинский, применяется для медицин-
ских, фармацевтич. и косметич. целей; желтый —
в ветеринарии, для технич. нужд (пропитка бумаги,
тканей и пр.), а также в качестве заменителя меди-
цинского В.; конденсаторный В., требую-
щий высокой степени очистки,—для нужд электро-
техник. пром-сти; технический В. — универ-
сальная низкоплавкая смазка (темп-ра каплепадения
ной аминокислотой (в положении 7 у В. быка —
аргинин, а у В. свиньи — лизин). В. других жи-
вотных, по-видимому, имеет строение очень близкое
В. быка или свиньи.
Основное биологическое действие
СИ В. — повышение, кровяного давления
Н2С"/| ''СН ~СПО и снижение мочеотделения; кроме
| СН2| того, В. вызывает сокращение матки
H2(\J /С(СН3)2 и выделение молока молочной же-
СН леэой в период лактации живот-
щ ного (подобно окситоцину, но в неск.
меньшей степени). В. получают из
гипофиза крупных животных; применяют гл. обр. в
виде экстрактов из задней доли гипофиза (и и т у-
итрина и адиуретина) при лечении не-
сахарного диабета и др. заболеваниях, связанных с
недостаточностью В. в организме.
Лит.: Дю-Виньо В. в сб. ст.: Современные проблемы
биохимии, М., 1957, с. 105; Behrens О. К. and Вгошег
W. W., в кн.; Annual revie.v of biochemistry, 1958, 27, p. 57;
см. также при ст. Гормоны. Ю. А. Панков.
ВАКУУМИРОВАНИЕ — техника получения и со-
хранения вакуума. Различают следующие виды ва-
куума: сверхвысокий — давление газа ниже
10 8 мм рт. ст., высокий — давление 10“3--
10 8 мм рт. ст. (средний свободный пробег молекул
значительно превышает размеры сосуда), сред-
ний — давление 10°—10" 8 мм рт. ст. (средний сво-
бодный пробег молекул того же порядка, что и раз-
меры сосуда) и низкий — давление от 760 до
1 мм рт. ст. (средний свободный пробег молекул
меньше размеров сосуда). Наибольшие разрежения,
достигаемые современными средствами, соответствуют
давлениям 10"11—1()13 мм рт. ст. В технике ограни-
чиваются давлениями 10 6 —10~7ммрт. ст. При обыч-
ных условиях (760 мм рт. ст. и 0°С) в 1 см3 газа содер-
жится ок. 1019 молекул, но даже и при таком сверхвы-
505
ВАКУУМИРОВАНИЕ
506
соком вакууме как 10*10 мм рт. ст. содержится при-
мерно 107 молекул. В сверхвысоком и высоком ва-
кууме молекулы почти не сталкиваются друг с дру-
гом, а лишь со стенками сосуда; поэтому при таком
вакууме газ не обладает внутренним трением (вяз-
костью), а только внешним, характеризующимся обме-
ном количества движения между молекулой и стенкой
сосуда. Электрич. ток в сверхвысоком и высоком
вакууме может возникать только в результате испус-
кания электронов с катода; т. к. соударения электро-
нов с молекулами крайне редки и ионизация га’за
мала, то электрич. ток почти всегда образуется в форме
электронного потока. При сверхвысоком и высоком
вакууме взаимная диффузия газов практически про-
исходит мгновенно. Скорость испарения в-ва в сверх-
высоком и высоком вакууме становится наибольшей,
когда испарившиеся молекулы не возвращаются
обратно на поверхность испарения.
Вакуум создают при помощи вакуумнасосов и спе-
циальных поглотителей газа. Простейшая схема
Рнс. 1. Схема простой вакуум-
ной установки: 1 — откачивае-
мый сосуд; 2 — вакуумметр; 3—
вакуумпровод; 4—вентиль; 5 —
вакуумнасос; в — патрубок.
сляного насоса создается
вакуумной установки по-
казана на рис. 1. Такая
установка обычно приме-
няется для получения
вакуума до 10 2 мм рт.
ст. Для получения бо-
лее высокого вакуума (до
10 8 мм рт. ст.) приме-
няется вакуумная уста-
яовка, показанная на
рис. 2; газ эвакуируют
в два приема. Вначале
при помощи форвакуум-
ного ротационного ма-
вакуум порядка 10 1 мм
рт. ст., после чего включаются в работу промежу-
точный (бустерный) и
паромасляный диффузи-
онный насосы, могущие
создать разрежение в
откачиваемом объеме до
10*8 .и.м рт. ст.
Рис. 2. Схема сложной ваку-
умной установки: 1 — отка-
чиваемый объем; 2 — иониза-
ционный манометр; 3 — иони-
зационный вакуумметр; 4 —
термоманометр; 5 — термо-
вакуумметр; в — задвижка вакуумная; г, 11 — вакуумпро-
вод; 8 — высоковакуумпый диффузионный насос; 9 — проме-
жуточный (бустерный) насос; 10 — вакуумные вентили; 12 —
масляный ротационный насос.
Низкий вакуум создают объемными насосами порш-
левого типа (газ выталкивается поступательным дви-
Рис. 3. Вихревой насос:'! — сопло; 2 — камера зави-
хрения; з — центральное сопло; 4 — диффузор; 5 —
улитка.
•жением поршня) и ротационными насосами (газ вытал-
кивается путем изменения объема всасывания и на-
гнетания при вращательном движенииротора). В ла-
бораторных условиях распространены водоструйные
насосы. При получении разрежения до 15 мм рт. ст.
может применяться вихревой насос (рис. 3), работаю-
щий по принципу образования вихря и использова-
ния вакуума, развивающегося вдоль оси вихря.
Рис. 4. Принцип рабо-
ты эжекторного насоса:
1 :— отверстие для впу-
ска пара; 2 — паровое
сопло; 3 — камера; 4 —
всасывающее отверстие;
5 — диффузор; 6 — вы-
пускное отверстие.
Рис. 3. Одноступенный масля-
ный ротационный насос: 1 — ка-
мера; 2 — бак, заполненный ма-
слом; 3 — впускной патрубок;
4—откачиваемое пространство;
5 — барабан; в — пластина; 7—
клапан; 8 — выпускной патру-
бок.
Для получения рабочих давлений от 30 до 0,1 мм рт.
ст. значительное распространение получили эжек-
торные насосы; схема такого насоса изображена на
рис. 4.
Для создания среднего вакуума широко исполь-
зуются гл. обр. эксцентрически-пластиночные масля-
ные ротационные насосы (рис. 5). Принцип действия
такого насоса состоит в засасывании воздуха при вра-
щении барабана и последующем его выталкивании.
Масляные ротационные насосы используются также
для откачки конденсирующихся паров (газобалласт-
ные насосы). Характеристика нек-рых масляных
ротационных насосов приведена в табл. 1.
таблица 1
Тип насоса Скорость отначки, л/сек Конечное давление, Мощность двигателя, кет
мм рт. ст.
ВН-1 20 3 10-з 2,8
ВН-2 7,5 3 . 10“3 1,7
ВН-4 59 1 . 10-S 7
ВН-6 155 1 . IO’8 20
ВН-494 0,21 1 • 10~з 0,15
ВН-461 0,9 1 . 10-а 0,37 ,
РВН-2 0 3,3 1 • Ю-з 0,37
Для получения больших скоростей откачки с глу-
биной вакуума до 10*4 мм рт. ст. применяют рота-
ционные зубчатые насосы (рис. 6). Два ротора-лопасти
синхронно вращаются внутри камеры в направлении,
указанном стрелками, захватывают поступающий
газ и выталкивают его. Между лопастями постоянно
сохраняется небольшой зазор (0,1—0,15 мм), не уплот-
няемый маслом, благодаря чему такие насосы допус-
кают большую скорость вращения (до 3000 об/мин)
и большую скорость откачки, св. 5000 л/сек. Насосы
работают вместе с масляными ротационными насо-
сами, к-рые для них являются форвакуумными.
507
ВАКУУМИРОВАНИЕ
508
Для получения высокого вакуума широко исполь-
зуются пароструйные масляные и ртутные диффузион-
ные насосы, принципиальное устройство которых по-
казано на рис. 7. Масло, находящееся в нижней ча-
сти насоса, подогревают
электронагревателем 1.
Образующиеся пары по
паропроводам через зон-
тичные сопла со скоро-
стью, превышающей ско-
рость звука, непрерыв-
но истекают в вакуум-
ную область, образуя
сплошную конусную за-
весу. Эвакуируемый газ
поступает через вход-
ное отверстие, увлекается
Рис. 6, Схема ротационно-зуб-
чатого насоса.
Рис.
ный
нагреватель;
входное отверстие; 4 — корпус; 5—
отражатель паров масла; в — вы-
ходное отверстие; 7 — сопло I; 8—
сопло II.
7. Пароструйный циффузион-
насос с отражателем: 1 —
2 — паропровод; 3—
Выход
охлаждающей
воды
струями сопел и уносит-
ся в область форвакуумного давления, откуда)
удаляется в атмосферу масляным ротационным насо-
сом. Паромасляные
диффузионные насо-
сы выполняются с
различными скоростя-
ми откачки (от 10 до
десятков тысяч л[сек)
и давлениями (10 е—
10 е мм рт. ст.). В
качестве рабочих жид-
костей, пары которых
используются в этих
насосах, применяют
органич. и кремний-
органич. масла, слож-
ные эфиры фталевой
и себациновой к-т,
жидкие силоксаны и
ртуть; упругость па-
ров при 20° не долж-
на превышать 10 е—
10“вмм рт. ст. Дан-
ные для нек-рых ти-
пов масляных диффу-
зионных насосов при-
ведены в табл. 2.
Для создания высо-
кого вакуума приме-
няют также ионные
насосы, принцип дей-
ствия к-рых показан на простейшей схеме (рис. 8).
В стеклянной трубке расположены два кольцевых
Поступление
охлаждающей
воды
Выпускное
отверстие
1+1/1
Рис. 8. Схема простейшего ионного насоса.
электрода, между к-рыми создается электрич. поле.
Трубка присоединена к форвакуумному насосу.
Между электродами вследствие ударной ионизации
электронами, ускоряемыми электрич. полем в напра-
влении к аноду (А), возникают положительные ионы,
направляющиеся к катоду (К). Вследствие непре-
рывного перемещения ионов газа в сторону като-
да газ из вакуумной системы непрерывно откачи-
вается. Усовершенствованный металлич. ионный
насос показан на рис. 9. В этом насосе с тремя
соленоидами (2 боковых и 1 средний) газ поступает
в середину насоса, откуда он распространяется в боко-
вые зоны. Под воздействием напряжения электрич.
и магнитного полей электроны концентрируются
в центральной зоне насоса, где и происходит иониза-
ция откачиваемого газа, перемещающегося под дей-
ствием электрич. поля к катодам и далее в форва-
Рис. 9. Усовершенствованный металлический ионный на-
сос: 1 — натекатель; 2 — боковые соленоиды; 3— сужение
анода; 4 — средний соленоид; 5 — разрядная плазма.
куумный насос. Ионными насосами достигается ско-
рость откачки до 700 л/сек и предельное давление
ДО 10е мм рт. ст.
Таблица 2
Тип насоса Скорость отначки, л/се к Конечное давление, мм рт. ст. Мощность нагревателя, кет
ММ-40А 40 1 • io-0 0,45
Д МН-20 20 1 • 10 ° 0,2
ЦВЛ-100 .... 125 1 • 10-6 0,45
ЦВЛ-15М .... 1500 1 • 10-е 2,0
Н-1С 100 1 • 10-е 0,47
Н-5С 500 1 • 10-е 1,5
Н-2Т 2000 1 • Ю-е 2,5
Н-5Т 4500 1 • 10-е 2,5
Н-8Т 8000 1 • 10-6 3.5
Для получения сверхвысокого вакуума используют
электроабсорбционные насосы, принцип действия
к-рых состоит в создании при помощи парового
диффузионного насоса давления порядка 1(+7—
10“9 мм рт. ст., после чего насос отключается и даль-
нейшее улучшение вакуума в сосуде достигается
поглощением оставшихся газов путем ионизации их
электрич. полем. Для создания сверхвысокого ва-
куума используется аппаратура из специального
стекла, из которой прогреванием до 400—500° удале-
ны газы. Затворы установки должны быть без смаз-
ки, все соединения — спайными (металл-стекло). На
рис. 10 представлена схема для сверхвысокого ва-
куума, применяемая при получении очень чистых
Рис. 10. Схема установки для получения сверхвысокого
вакуума: D — диффузионный насос; Р, и Р2 — мано-
метры; Р — поглощающий манометр; Kt, К2, Ка — за-
творы; V — резервуар с газом; Vb V2 — откачиваемые
сосуды; W — ловушка.
газов. При помощи диффузионного насоса создается
вакуум; одновременно удаляют газы прогреванием
установки, а также бомбардировкой электродов в мано-
метрах Рх и Р2 и особенно в поглощающем манометре
Р. После этого закрывается затвор и напряжение
подается на манометр, к-рый начинает теперь дейст-
вовать как поглотитель. Через 5—15 часов создается
вакуум порядка IO-10—10-11 мм рт. ст.
Измерение вакуума производится с помощью
манометров. Давления от 760 до 20 лл рт. ст. измеряются
509
вакуумирование — ВАЛЕНТНОСТЬ
510
механич. манометрами, а от 760 до 1 • 10-1 мм рт. ст. — гидро-
статич. U-образными манометрами (ртутными или масляными).
Точность измерения, а также самые низкие давления, под-
дающиеся измерению таким способом, зависят от точности
отсчета разности высот столбиков жидкости. Для ртутных
манометров при отсчете невооруженным глазом погрешность
достигает порядка 0,5 мм.
Давления от 10 до 1 • 10ммрт. ст. измеряют ртутным
компрессионным манометром Мак-Леода (рис. 11). Для изме-
рения давления газа при помощи гидростатич. манометра газ
Гис. 11. Компрессионный манометр: а и б — с подвиж-
ным резервуаром; в — с поршнем; Pt — давление в от-
качиваемом объеме; Р2 — давление в манометре; —
объем газа до сжатия; V2 — сжатый объем; R — резер-
вуар для ртути; z—I и и—и — разность уровней ртути,
равная атмосферному давлению.
объема и давления Pt сжимают до объема V2 и давления Pz.
Давление, к-рое было до сжатия Pi, определяется по закону
Бойля — Мариотта. Pt == Рг . У2/У\.
Отношение V2/V( называется степенью сжатия
компрессионного манометра. Наиболее важным свойством
этого манометра является его абсолютность, т. е. способность
непосредственно измерять давление, создаваемое газом, неза-
висимо от рода газа. Существенным недостатком является
невозможность измерения давления паров ртути, масла, воды
и т. п., тан как при сжатии эти пары конденси-
руются. Давления от 1 до 1 • 10-3 мм рт. ст.
измеряют манометрами сопротивления и тер-
моэлектрич. манометрами, действие к-рых осно-
вано на зависимости теплопроводности газа
от давления.
Для измерения давлений от 1 • 10~8 до
10-11 мм рт. ст. широко пользуются иони-
зационными манометрами (рис. 12), действие
которых основано на испускании накален-
ным катодом электронов, движущихся уско-
ренно по направлению к положительно заря-
женной сетке, играющей роль анода. Элек-
троны пролетают через редкие витки сетки,
но отталкиваются заряженным коллектором.
Ток в цепи коллектора пропорционален давле-
нию в системе, к к-рой црисоединен баллон
манометрич. лампы.
Кроме рассмотренных наиболее типичных
и распространенных способов измерения низ-
ких давлений, существуют и др. методы и
приборы, имеющие меньшее значение.
Для сохранения вакуума в приборе его
стенки должны быть непроницаемы и не вы-
Рис. 12. Иони-
зационный ма-
нометр: 1 —
дел ять к.-л. газов или паров а все детали катод, 2— сет-
прибора — плотно соединены. Наилучший ма- ка, 3 — кол-
териал для этих целей —стекло; газыс поверх- лектор,
ностей прибора и его деталей удаляют глубо-
ким эвакуированием с одновременным прогревом и сорб-
цией оставшихся газов поглотителями (активный уголь, си-
ликагель и др.), помещаемыми внутрь прибора. Улучше-
ния вакуума можно достигнуть путем установки между от-
качиваемым сосудом и насосом ловушки, охлаждаемой
жидким азотом, твердой СО8 и т. д. Сорбция газов погло-
тителями улучшается при создании электрич. поля для
ионизации газов. Скорость откачки можно измерять путем
определения количества газа, удаляемого при постоянном
давлении в течение определенного времени (метод постоян-
ного давления), или определением опытным путем (для ре-
зервуара известного объема) зависимости давления от вре-
мени откачки (метод постоянного объема).
Натекание, наблюдающееся гл. обр. в местах соедине-
ний вакуумной системы (фланцы, краны, вводы, спаи, сварные
швы и т. п.), не может быть устранено при самом тщательном
выполнении вакуумных систем. В зависимости от сложности
вакуумной системы устанавливаются пределы допустимого
натекания.
В зависимости от глубины вакуума обнаружение течи в ва-
куумных приборах производится различными способами.
Компрессионный метод обнаружения течи
заключается в создании внутри испытываемого сосуда давления,
превышающего атмосферное, и применение того или иного
внешнего указателя негерметичности (задувание или колыха-
ние пламени, образование пузырьков на мыльной пленке или
в воде и т. п.). Этим методом можно обнаружить течи до
0,0001 л • мм рт. ст. /сек. Метод искры (для обнаруже-
ния течей в стеклянных деталях) состоит в том, что при при-
косновении электрода высокочастотного трансформатора (типа
Тесла) к поверхности стекла возникает разряд. При наличии
течи свечение разряда будет красным или лиловым, при отсут-
ствии течи — серым. Этот метод широко применяется в ва-
куумных лабораториях, им можно пользоваться при давлении
в установке от 1 до 0,05 мм рт. ст. Наиболее распространен
масс-спектрометрический метод обнару-
жения течи (до 1 • 10~7 л • л(.н рт. ст./сек и менее). В основу это-
го метода положено использование масс-спектрометра; проб-
ным газом является гелий. Кроме рассмотренных методов
обнаружения течи, существуют и другие методы, но они ме-
нее производительны, недостаточно надежны и малочувстви-
тельны.
Лит.: Грошк о век и й Я., Технология высокого
вакуума, М., 1957; Дешман С., Научные основы вакуум-
ной техники, М., 1950; Иванов А., Электровакуумная тех-
нология, М_, 1944; Вакуумное оборудование и вакуумная
техника, под ред. А. Г утр и и Р. Уокерлинга, пер. с англ., М.,
1951, Королев Б. II., Основы вакуумной техники, М.,
1950; Тягунов Г. А., Основы расчета вакуумных систем,
М.—Л., 1918. А. А. Грязное.
ВАЛЕНТНОСТЬ — свойство атома данного эле-
мента присоединять или замещать определенное число
атомов другого элемента.
Понятие о В. возникло в 1853 (Э. Франкленд) и сразу же
привлекло внимание ученых, т. к. позволило объяснить многие
экспериментальные факты. В дальнейшем многие крупнейшие
ученые уделяли большое внимание выяснению сущности В.
и установлению ее закономерностей. Представление о В. яви-
лось одной из важнейших предпосылок для создания А. М. Бут-
леровым теории химич. строения (1861). Оно получило значи-
тельное развитие в связи с открытием Д. TI. Менделеевым
периодич. закона (1869). Менделеев установил связь между В.
и положением элемента в периодич. системе, ввел понятие,
о переменной В., показал взаимозависимость В. элементов
в соединениях с водородом и с кислородом. Вслед за открытием
электрона (1897) были сделаны попытки создания электронной
теории В. (Дж. Томсон). Однако только с развитием науки
о строении атома учение о В. получило физич. обоснование и
постепенно сформировалось в стройную теорию. Значительным
шагом в развитии электронных представлений В. явилась тео-
рия Косселя (1916), в основе к-рой лежала предпосылка о том,
что взаимодействие атомов сводится к передаче электронов от
одного атома (или группы атомов) к другому. Образовавшиеся
при этом оба иона приобретают электронную структуру бла-
городных газов. Так, ионы№а+ и F-воспроизводят структуру
атома неона, ионы К+ и CI-—атома аргона, ион Са2+ аналоги-
чен атому аргона, ион О2- —атому неона, и т. д. Отсюда непо-
средственно вытекало понятие о положительной п отрицатель-
ной В. элементов: положительная В. элемента равна
числу электронов, к-рое нужно отдать, отрицательная
В. — числу электронов, к-рое нужно приобрести атому для
образования оболочки инертного газа. Концепция Косселя
хорошо объясняла закономерности значительной группы ион-
ных реакций и строение ряда неорганич. соединений. Однако
при химич. взаимодействии атомов не всегда имеет место пере-
ход электронов. Согласно представлениям Льюиса (1916), атомы
могут соединяться за счет образования общей пары (дублета)
электронов, по одному от каждого атома (возможно обра-
зование нескольких общих пар). Обобществленные
электроны принадлежат обоим взаимодействующим атомам.
В. атомов определяется числом электронов, вносимых каждым
атомом на образование общих пар. Эта концепция объясняла
В. элементов в большой группе соединений с ковалентными
связями.
Однако теоретич. представления Косселя и Льюиса были
в значительной мере формальными и не всегда вскрывали
сущность В. и истинную природу химич. связи. Новым эта-
пом в развитии теоретич. химии вообще и учения о В. в
частности явилась разработка квантово-механич. теории ато-
ма (1926).
511
ВАЛЕНТНОСТЬ
512
Квантово-механическая природа валентности. Хи-
мич. свойства элементов и их В. зависят гл. обр. от
строения внешних электронных оболочек атомов.
Известно, что состояние электрона в атоме опреде-
ляется четырьмя квантовыми числами п, I, mt и ms.
Главное квантовое число п характеризует энергию
электрона или удаленность электрона от ядра; кван-
товые числа I и характеризуют пространственную
конфигурацию орбиты, по к-рой движется электрон,
или форму электронного облака. В зависимости от
значений I и т1 различают след, состояния электро-
нов: s(l = 0; = 0); p(l =1; m( = —1, 0, +1),
d(l = 2; mt = —2, —1, 0, +1, +2) и /(/ = 3; m.; =
= —3, —2, —1, 0, +1, +2, +3). Наглядная схема
формы орбит или формы облаков электронов, нахо-
дящихся в s~, р-, d- и /-состояниях, представлена
на рис. 1. Вид функции (или форма электронного
облака) для того или иного состояния устанавливается
путем решения волнового ур-ния Шредингера для
водородоподобного атома (см. Атом). Представление
о форме электронных облаков (или орбит) имеет суще-
ственное значение при рассмотрении вопроса о на-
правленности В. Квантовое число ms характеризует
величину проекция собственного момента электрона
на нек-рое направление — его спин. Эта проекция
может иметь только два значения (+1/а и —1 г),
соответственно двум возможным противоположным
ориентациям спинового момента электрона, к-рые
обычно указывают стрелками (| , J). Пару электронов
с одинаковыми квантовыми числами п, I и т( и с
противоположно направленными спинами (j ]) наз.
спаренными (или неподеленной парой электронов);
электроны с ненасыщенными спинами наз. неспарен-
ными(| J). Понятия о спине электрона, о спарен-
ных и неспаренных электронах может иметь весьма
Рис. 1. Модели электронных облаков.
существенное значение при рассмотрении вопроса
о В. данного элемента. Квантово-химич. расчеты
молекулы водорода (Н—Н) показали, что пребывание
двух электронов с противоположными спинамв ([ [)
в поле двух ядер энергетически более выгодно, чем
нахождение каждого электрона в поле своего ядра.
Объяснение В. с этой точки зрения состоит в том,
что каждый атом для образования химич. связи пре-
доставляет один неспаренный электрон. Следова-
тельно, В. атома в основном состоянии равна числу
неспаренных электронов, т. е. таких электронов,
спины которых ненасыщены. Вопрос о том, какие
из валентных электро-
нов атома являются спа-
ренными или неспарен-
ными, решается экспе-
риментально, методами
Не lss л = 1 [П]
Ne 1s22s22p6 л«| 1Й]
л=2 ЕШЩЩ]
s ~~р
спектроскопии. Дг |S22s22p"3s23p6 л=![й|
Атомы инертных газов, л=2 йшГГШТ]
все электроны к-рых спа- п=з1и|и|и|П]
рсны, нульвалентны. s р
Атомы элементов первой группы периодич. системы
имеют по одному неспаренному электрону, они одно-
валентны:
Кислород и сера, имеющие по два неспаренных элект-
рона в основном состоянии, двухвалентны:
Если электров имеет возможность в пределах одного
и того же главного квантового числа п занять различ-
ные состояния, то при дан- (
ном значении I электро- 2 3 .
ны располагаются так, что ls 2s 2д п=1 г1 .
суммарное спиновое число
максимально (правило Гун- s Р
да). Именно поэтому атом азота имеет строение 1
с тремя неспаренными электронами и суммарным
спином 3/2 и является
трехвалентным, а не 11
строение II с одним Л = 1 [П1
неспаренным электро- л=2 1И1И|* I I
ном и суммарным спи- s' р '
/а.
Валентность атомов в возбуж-
денных состояниях. В ряде случа-
-2 mt--3 ев валентность элемента оказывает-
ся больше числа неспаренных эле-
ктронов в атоме. Это наблюдается
тогда, когда спаренные валентные электроны в ре-
зультате возбуждения переходят в другое состояние,
причем возбуждение одного из спаренных электро-
нов увеличивает число неспаренных электронов на
513
ВАЛЕНТНОСТЬ
514
два и, следовательно, В. атома при этом увеличи-
вается на две единицы. Возбуждение электронов из
состояния с одним главным квантовым числом п
в состояние с другим главным квантовым числом,
особенно для низких значений квантовых чисел (1, 2),
требует большой затраты энергии; напр., в случае
гелия возбуждение электрона из состояния 1s в состоя-
ние 2s требует ок. 400 ккал!г-атом. Такие возбужде-
ния в условиях химич. реакций не имеют места. Воз-
буждения из s-состояния в p-состояние или из р- в
d-состояние в пределах одного и того же главного
квантового числа происходят со значительно меньшей
затратой энергии и имеют место во многих случаях.
Так, напр., атом углерода в основном состоянии
2s22p2 двухвалентен, но если один из двух 2.;-электро-
иов перевести на свободную 2р-орбиту, то возбужден-
ный атом углерода 2s2p3 окажется 4-валентным *:
С 2s2 2р2 |И|>|>| I --------- С 2s 2р3 ШШЕ)
s ₽ s р
Возбуждение такого валентного состояния требует
определенной энергии (160 ккал/г-атом), но она
с избытком компенсируется энергией взаимодействия
двух лишних электронов, т. е. энергией двух допол-
нительных связей. По этим причинам атом бериллия
не является нульвалентным (2s2), как атом гелия
(1s2), а переходит в возбужденное состояние (2s2p)
с В., равной 2; атом бора (2s22p) переходит в трехва-
лентиое (2s2p2) состояние:
Be 2s2 ГН| III ---------- Be 2s 2p llUI I I
2s 2p~ 2s'~2p
В 2 s2 2p IHHl I I ---------- В 2s 2p2 Hlllll I
2s 2₽ 2s 2p
Энергия возбуждения атомов из одного состоя-
ния в другое определяется экспериментально по спект-
роскопия. данным. В нек-рых случаях возбуждение
атомов в новое валентное состояние происходит путем
передачи одного из спаренных электронов другому
атому; при этом В. атома изменяется на 1, и, кроме
того, атом превращается в однозарядный положитель-
ный ион. Такое возбуждение имеет место в атомах
азота, углерода, кислорода и др. Переход же атома
азота из 3-валентного состояния в 5-валентное был бы
связан с переходом электрона из 2s- в Зя-состояние,
а переход кислорода в 4-валентное состояние —
с переходом 2р—>-3s, т. е. в этих случаях изменилось
бы главное квантовое число и требуемая энергия ока-
залась бы настолько велика, что затрата ее не могла
бы компенсироваться выигрышем энергии за счет
двух лишних ковалентных связей. Поэтому не
существует 5-валентных соединений азота и 4-валент-
ных соединений кислорода с ковалентными связями.
В этих случаях энергетически более выгодны такие
ионные валентные состояния
N 2s2 2р3 IttUlHH -------- N 2s 2р3 ГЛИПТ!
2s 2р 2s'~2p~
с образованием соединений типа NHf и др.
То или иное валентное состояние определяется,
в первую очередь, энергетикой образующихся соеди-
нений. Если сродство атома-партнера к электрону
велико и может в нек-рой степени компенсировать
энергию, необходимую для отрыва электрона, возмо-
* На этой и следующей схемах указаны только электроны
внешних оболочек.
9 К. X, Э, т. 1
жен переход:
С |И||||| I ---------С 1111 111 I валентность 3
2s 2р 2S 2р
' N |И|1II III ---------*• N 1111111 П валентность 1
2s 2р 2s 2р
О |H|HI 1111 ----------- О HUI Hill валентность 3
2 s 2р 2s~2p
Напротив, когда энергия отрыва электрона от атома-
партнера мала, могут возникать иные, отрицательно
заряженные, ионные валентные состояния тина:
С Illi 1111 1 С |lt| I 11 11~| валентность 3
2s 2р 2s 2р
N lull 11 Н1 -----------*" N lit|lt| П ♦ I валентность 2
2s 2р 2s 2р
О [llflll 111~1 1 О |lt|lt|lf| |~] валентность 1
2s "~2р 2s 2р
В соответствии с этим существуют соединения 3-ва-
лентного положительно заряженного атома углерода—
карбониевые ионы, и 3-валентного отрицательно
заряженного атома углерода — карбанионы. Т. обр.,
современная теория позволяет подойти с единой точки
зрения к вопросу о В. элементов в ионных и ковалент-
ных соединениях, понять сущность переменной В.
элементов и дать энергетич. оценку возможности
осуществления того или иного валентного состояния
атомов.
Направленность валентности. Основными факто-
рами, определяющими пространственную конфигура-
цию молекул, являются направленность В. и принцип
максимума перекрывания. Согласно законам кванто-
вой механики, наиболее прочные химич. связи обра-
зуются в направлении максимального перекрыва-
ния облаков связующих электронов. Как сказано
выше, валентные электроны, находящиеся в s-, р-, ti-
или /-состояниях, характеризуют-
ся определенными формами элект- >-------'х/----
ровных облаков (рис. 1): s-состоя- / А \
ние отвечает шаровой симметрии, ( • Ц • )
p-состояние подобно гантели, \ X У
причем рх-, р рг-функции со- 44----------------7
ставляют друг с другом прямые рИс. 2. s—.--связь,
углы. Отсюда следует, что ато-
мы с s-валентными электронами могут образовы-
вать одинаково прочные связи в любом направлении
в все направления в этом
смысле равнозначны (рис. 2).
В случае р-электронов наи-
большее перекрывание обла-
ков связующих электронов \ /
происходит в направлении —-А /,------->
оси гантели (рис. 3). Отсюда <( W-------------у—
следует, что угол между дву- — 'у У''-----'
мя связями, образованными / \
р-электронами одного атома ( )
с s- и р-электронами двух д'7
других, должен быть равен Рис. 3. Две s—р-связи.
90 . Согласно этим сообра-
жениям, углы между связями в молекулах Н2О, H2S,
SC12, NH3, РС13 и др. должны быть близки к 90°. Атомы
четвертого и последующих периодов системы элемен-
тов Менделеева (начиная со скандия) образуют связи
также с участием d-электронов. Вследствие иного
515
ВАЛЕНТНОСТЬ —ВАЛЕНТНЫЕ УГЛЫ
516
вида облаков d-электронов в этих случаях получаются
иные направления В. (квадратное или октаэдрич. рас-
положение связей).
В нек-рых случаях валентные электроны атома на-
ходятся в различных состояниях; один электрон в
«-состоянии, другой — в /^-состоянии (в атоме Be —
2«2/>), один электрон в «-состоянии и 3 электрона
в />-состоянии (в атоме С — 2«2/>3) и т. д. Здесь можно
было бы ожидать образования различных типов свя-
зей. Однако опыт показывает, что во всех этих случа-
ях (ВеС12, ВС13, СН4, СС14 и т. д.) образующиеся
связи эквивалентны; у атома Be две одинаково проч-
ные связи под углом в 180°, у В три эквивалентные
связи под углом в 120°, у С четыре связи, располо-
женные под тетраэдрич. углами (109°28'). В этих слу-
чаях состояние валентных электронов описывается
не чистыми «-, />-, rf- или /-функциями, а смешанны-
ми, так называемыми гибридными волновыми функ-
циями, которые представляют собой линейную ком-
бинацию основных функций, описывающих состоя-
ние электронов (см. Квантовая механика, Квантовая
химия).
Гибридизация сопряжена с изменением формы
электронного облака и, следовательно, с изменением
направленности В. Комбинация двух электронов
в s- и ^-состояниях всегда приводит к образованию
двух гибридных связей под углом 180°:
Комбинация одного s- и двух />-электронов приво-
дит к образованию трех гибридных связей под
углом 120°:
Рис. 5.
В атомах с d- и /-электронами гибридизация приво-
дит к образованию более сложных конфигураций.
Как правило, при образовании молекул из всех воз-
можных комбинаций в атомах осуществляется такая
гибридизация функций валентных электронов, к-рая
и приводит к образованию наиболее прочных связей.
В нек-рых случаях, однако, оказываются энергети-
чески выгодными условия, при к-рых электроны обра-
зуют связь, оставаясь в различных валентных состоя-
ниях. Это прежде всего относится к атому углерода
в непредельных и ароматич. соединениях (подробнее
см. Молекула, Химическая связь).
Некоторые особые случаи проявления валентности.
Т. обр., современная теория В. позволяет понять
зависимость В. от электронной структуры атомов,
дает физич. обоснование направленности В., правилу
насыщаемости В. и др. Однако в последние годы накоп-
лен большой экспериментальный материал, синтези-
рованы новые группы соединений, к-рые не уклады-
ваются в обычные рамки теории В., в обычные пред-
( гавления о химич. связях, осуществляемых парой
электронов в поле Двух ядер. Во всех этих случаях
число связей значительно больше, чем это позволяют
обычные правила В. Так, прочные химич. связи могут
образовываться между валентнонасыщенными моле-
кулами; напр. NH3 и BF3 образуют прочное соедине-
ние H3N • BF3 с выделением значительной энергии.
Анализ экспериментальных данных показывает, что
такого типа соединения образуются между молеку-
лами, из к-рых у одной есть атом с неподеленной
парой валентных электронов (донор), а у другой —
атом со свободными валентными орбитами (акцептор).
Такого типа связи играют большую роль в комплекс-
ных соединениях, в к-рых центральный атом предо-
ставляет свободные орбиты, а лиганды (обычно это
молекулы с атомами О, N, S) — неподеленные пары
электронов. В соединениях мостикового типа, или
в соединениях с дефицитом электронов, атомы свя-
заны с большим числом соседей, чем это позволяют
валентные возможности. Образование мостиковых
связей характерно для элементов, имеющих свободные
валентные орбиты (Al, Be, В, Pd, Fe и др.). Так, боран
ВН3, в к-ром все три валентности бора насыщены,
димеризуется с выделе-
нием значительной энер-
гии. Образуется проч-
ная молекула диборана
с мостиковой структу-
рой (рис. 6). Две груп-
пы ВН2 лежат в одной
плоскости; два других
атома Н расположены
в перпендикулярной пло-
скости и каждый из них
образует СВЯЗИ С двумя Рис. 6. Структура диборана.
атомами В, т. е. пара
электронов связи В—Н находится в поле трех ядер
(В...Н...В). Подобные связи способны образовывать
не только атомы водорода, но и атомы галогенов (в
PdCl2, FeCl3), метильная группа (в димерах триметил-
алюминия) и др.
Интересные валентные возможности элементов про-
являются в соединениях типа ферроцена, дибензол-
хрома и др. В этих молекулах атом металла (железо,
хром, никель и др.) находится посредине, в по обе
стороны от него в параллельных плоскостях располо-
жены органич. циклические молекулы, имеющие
непредельные связи (циклопентадиенил, бензол
и др.). По-видимому, связь в этих соединениях осу-
ществляется за счет л-электронов колец и свободных
валентных орбит атома металла. Изучение этих новых
типов химич. соединений ставит новые вопросы перед
теорией В.
Лит.: Coulson С. A., Valence, Oxf., [1953]; С ы р-
к и н Я. К., ДяткинаМ. Ь., Химическая связь и строение
молекул, М.—Л., 1946; В о л ь к е н ш т е й н М. В., Строе-
ние и физические свойства молекул, М.—Л., 1955; Сыр-
кин Я. К., Современное состояние проблемы валентности,
Усп. хим., 1959, 28, вып. 8, с. 903. Е. Н. Гурьянова.
ВАЛЕНТНЫЕ УГЛЫ — углы, образованные в мо-
лекулах химич. соединений линиями, соединяющими
центры атомов в направлении действующих между
ними химич. связей (см. схему). Величины В. у. опре-
деляются валентностью атомов, характером связи
между ними, их радиусами и др. (см. Стереоизоме-
рия, Молекула, Химическая связь). Определение В. у.
основано на применении современных физич. методов
исследования строения молекул: рентгеноструктур-
ного анализа, электронографии, спектрального ана-
лиза и др.
Для атомов с переменной валентностью возможны
различные значения В. у. в разных типах соединений.
Так, у атома углерода в насыщенных соединениях
нормальными являются тетраэдрич. В. у. (109°28').
В ненасыщенных органич. соединениях В. у. между
простой и двойной связями составляет я=Л20°, между
517
ВАЛЕРИАНОВЫЕ КИСЛОТЫ
518
двумя простыми связями, соседними с одной и той же
двойной, также =120° (напр., этилен), между двумя
двойными связями, а также между простой и тройной
связями 180° (напр., двуокись угле- н
рода, ацетилен и т. д.). Для соеди-
нений элементов IV группы перио-
дич. системы — аналогов углерода —
Кислоты Т. пл., °C Т.кип., °C Констан- та дис- социа- ции X х 10-5 (25°) Вязкость,
спуаз х 102
15° 30°
н-Валериановая СН3 (СН2) 3СООН — 34,5 186,35 1,56 2,359 1,724
Изовалериановая (СН3)2 СНСН2СООН -37,6 176,7 1,68 2,731 1,967
Метилэтилуксусная СНдСЩСН (СН3) СООН -80 177 — — —
Тр и метил уксусная (СН8)3 ССООН 4-35,3 — 35,5 163,7 — — —
ЛН3
Si, Ge, Sn и Pb, нормальными также являются тетра-
эдрич. В. у., наблюдаемые у тетрагалогенидов этих
элементов. Для элементов II группы нормальные В. у.
равны 180°, напр. для молекул ВеС12, ZnJ2, CdJ2 и
HgCl2. Для элементов III группы нормальные В. у. со-
ставляют 120° (тригалогениды В, Al и Ga). Для атомов
V и VI групп теоретич. следует ожидать нормальных
В. у., равных 90°. Однако силы взаимного отталкива-
ния атомов, присоединенных к одному центральному
атому (стерич. препятствия), могут привести к откло-
нениям от нормальных значений. Поэтому, а также
из-за влияния неподеленных электронных пар, на-
блюдаются значительные отклонения от теоретич.
нормального значения (90°), например: 108° (NH3),
101 ± 2° (РС13), 103 ± 3° (AsCIs), 105°3' (Н2О).
При увеличении радиуса центрального атома силы
стерич. отталкивания убывают; при этом уменьшаются
отклонения В. v. от нормальных значений (90°),
напр. 96° ± 4° -(SbClg), 92°16' (H2S), 90° (H2Se).
Увеличение размеров присоединенных атомов при-
водит к росту сил отталкивания. Особенно большие
отклонения от нормальных В. у. наблюдаются у цик-
лич. соединений с небольшим числом звеньев в коль-
це. Так, в циклопропане С3Нв и циклобутане С4Н8
углы между атомами углерода равны соответственно
60° и 90° вместо 109°28'. Так как искажение В. у.
сопряжено с затратой энергии, то подобным соедине-
ниям присуща пониженная термодинамич. устойчи-
вость.
Лит.: ВолькенштейнМ. В., Строение и физические
свойства молекул, М.—Л., 1955; II и н с к е р 3. Г., Дифрак-
ция электронов, М.—Л., 1949; Stuart Н. A., Die Struk-
tur des freien Molekills, B. — [u. a.], 1952. T. К. Ребане.
ВАЛЕРИАНОВЫЕ КИСЛОТЫ C6H10O2 — одно-
основные предельные органич. к-ты; известны все
четыре теоретически возможные В. к.: н-валериано-
вая к-та (н-бутанкарбоновая-1, пропилуксусная)
СН3(СН2)3СООН, изовалериановая к-та (2-метил-
пропанкарбоновая, изопропилуксусная, (3-метилмас-
ляная) (СН3)2СНСН2СООН, метилэтилуксусная к-та
(а-метилмасляная) СН3СН2СН(СН3)СООН и триме-
тилуксусная к-та (пивалиновая) (СН3)3ССООН.
В. к. — бесцветные жидкости с характерным за-
пахом.
н-В алериановая кислота имеет неприятный за-
пах, встречается в продуктах перегонки дерева, бурого угля.
Синтезируют ее из бромистого бутила по реакциям:
С4Н8Вг + NaCN - C4HaCN ±&2 С4НаСООН
или
C4H9Br + Mg - C4H9MgBr ±2?Яс4Н9ОООН
Нек-рые производные н-валериановой к-ты обладают сильным
дезинфицирующим действием.
Изовалериановую кислоту (имеет запах
валерианы) получают отгонкой с водяным паром из валериа-
нового корня, в к-ром она содержится в свободном виде; син-
тетически — окислением оптически неактивного изо амилового
спирта, или биохимически — при сбраживании глюкозы неко-
торыми видами грибков. Изовалериановая к-та широко при-
меняется для получения лекарственных препаратов.- валидола,
борнивала—борнилового эфира изовалериановой к-ты, необор-
нивала — эфира борйеола с гликолевой и изовалериановой
к-тами, эйборнила или вализана — борнилового эфира а-бром-
изовалериановой к-ты, бромурала — а-бромизовалерианил-
мочевины и др. В пищевой пром-сти применяется изоамиловый
эфир изовалериановой к-ты как «яблочная эссенция*.
Метилэтилуксусная кислота является оп-
тически активной; для а-формы [а]/} = 4-17,6°; d-кислота
встречается в эфирных маслах, напр. лавандовом, в пеницил-
лине. d-Кислоту получают окислением оптически активного
(ферментативного) изоамилового спирта; 1-кислоту — разло-
жением при 170° кислой соли метилэтилмалоновой к-ты и бру-
цина или перекристаллизацией соли оптически недеятельной
метилэтилуксусной к-ты с бруцином (соль 1-кислоты выпадает
раньше). Синтетич. к-та получается обычно в рацемич. форме,
напр. окислением изоамилового спирта или же действием СО
на тетрагидрофуран при 740—800 ат в присутствии карбо-
нила никеля.
Эфиры Мол. вес Т. кип., °С/Л1.и рт. ст. Плотность Запах
Метиловый 116,6 127,3/736 0,895 (15/4) 0,9097 Фруктовый
Этиловый 130,2 145,5/760 0,877 (20°) Фруктовый (сильный)
н-Пропило- вый 144,2 167,5/760 0,874 (15°) Фруктовый (мягкий)
н-Бутиловый 158,2 185,5/760 0,870 (15/5°) Яблочный
и-Амиловый 172 203,7/760 0,881 Фруктовый
Изоамиловый 172 — — Фруктовый
d-Цитронел- лиловый 240,2 194-196/30 — Роз
Линалиловый 238,3 — — Напоминает кориандр
Бензиловый р-Фенилзти- ловый 192,1 249—2'51/760 132-134/10 0,9832 (15°) Плодово- фруктовый Свежей зелени
Триметилуксусную кислоту получают оки-
слением пинаколина щелочным р-ром перманганата или луч-
ше-— гипобромитом натрия. При действии СО и Н2О на бути-
ловые спирты или бутилены при 150—250° и 200—300 ат в при-
сутствии фосфорной к-ты, содержащей в первом случае 2 %
фосфата меди, а также в присутствии катализатора, состоящего
из карбонила и иодида никеля, при 185° и 250—300 ат полу-
чают смесь метилэтилуксусной и триметилуксусной к-т с пре-
обладанием последней.
Различие в строении В. к. отражается на их реак-
ционной способности. Так, разветвленные цепи за-
519
w-ВАЛЕРИАНОВЫЙ АЛЬДЕГИД - ВАЛЛАХА ПЕРЕГРУППИРОВКА
520
трудняют этерефикацию и омыление их эфиров, что
вызвано в основном стерич. препятствиями — экра-
нирующим действием метильных групп на гидроксил.
Поэтому скорость этих реакций наиболее низкая
у триметилуксусной к-ты. Эфиры жирных спиртов
В. к., имеющие фруктовый запах, применяются
в пищевой пром-сти для приготовления фруктовых
эссенций, а эфиры с ароматич. и терпеновыми спир-
тами обладают цветочным запахом и находят нек-рое
применение в парфюмерии.
Свойства важнейших эфиров н-валериановой к-ты
приведены выше (см. таблицу на стр. 518).
Лит.: West Т. F., Straus! Н. J., Barton
D. Н. R., Synthetic perfumes, L., 1949.
М. И. 1'озенгарт, А. А. Зеленейшая.
н-ВАЛЕРИАНОВЫЙ АЛЬДЕГИД (пентанал)
СН3(СН2)3СНО, мол. в. 86,13 — бесцветная жидкость;
т. пл. — 91,5°; т. кип. 103,40°; dl° 0,810; пр°1,3944.
В. а. обладает обычными свойствами альдегидов;
образует кристаллич. производные: 4-нитрофенил-
гидразон, т. пл. 74—75°, и оксим, т. пл. 52°. Содер-
жится В. а. в эвкалиптовом, гвоздичном и др. эфирных
маслах. Получают В. а. перегонкой смеси валериа-
новой к-ты и формиата кальция, дегидрированием
амилового спирта над медным катализатором и др.
способами. И. В. Матвеева.
ВАЛИДОЛ — 25—30%-ный р-р ментола (I) в мен-
толовом эфире изовалериановой к-ты (II); масляни-
стая бесцветная жидкость с запахом ментола, хорошо
растворимая в спирте и нерастворимая в воде; плот-
ность 0,896—0,909. При взбалтывании 1—2 капель
В. с водой образуется эмульсия; при подкислении
В. серной к-той и нагревании появляется запах изо-
валериановой к-ты. Для колич. определения В. омы-
ляют при кипячении (5 час.) спиртовым р-ром КОН
в присутствии ксилола, после чего избыток щелочи
оттитровывают 0,5 н. р-ром HCI в присутствии фенол-
фталеина. В. получают взаимодействием ментола
с изовалериановой к-той в присутствии серной к-ты,
смешением образовавшегося т. обр. сложного эфира
с ментолом и перегонкой смеси. В. действует успокаи-
вающе на центральную нервную систему и
обладает рефлекторным сосудорасширяющим ,—х
эффектом; применяют при стенокардии, не- { '
врозах, истерии, морской и воздушной бо- \ /
лезни.
Лгип. см. при ст. Адалин. А. И. Травин.
ВАЛИН (а-аминоизовалериановая кислота)
(CH3)2CHCH(NH2)COOH, мол. вес 117,16 — ___,
одна из наиболее распространенных амино-
кислот. Существуют след, формы В.: 1) L-B.
(обычно просто В.), входит в состав почти всех
белков, некоторых антибиотиков (напр., грамици-
дина С) и встречается в свободном виде в животных
и растительных тканях. L-B. — бесцветные кристал-
лы; т. пл. 315° (в запаянных капиллярах); хо-
рошо растворим в воде и нерастворим в эфире;.
[а]|? = + 28,8° (3,4% в 6н. НС1) и 6,42° (в воде);
pA"ai 2,32, рАаз 9,69, р/ 5,96 (25°). В. дает общие
реакции на аминокислоты; для него характерна мед-
ная соль сине-фиолетового цвета. Определение L-B.
в смесях аминокислот производят, используя хрома-
тография., а также микробиология, метод (по измере-
нию роста молочнокислых бактерий и др. микроорга-
низмов). Получают L-В. из гидролизатов белков,
а также синтетически, общими методами получения
аминокислот, напр. действием NH3 на а-бромизова-
лериановую к-ту. В. применяют в биохимич. иссле-
дованиях (при хроматографии, изучении азотистого
обмена и др.). L-В. является незаменимой аминокис-
лотой.
2) D-В. входит в состав нек-рых антибиотиков (акти-
номицины и др.): [а]р= —29,4° (в 20%-ной НС1),т. пл.
293° (в запаянных капиллярах). D-В. не заменяет
L-В. Физич. и химич. свойства D-В. те же, что у
L-B. 3) D,L-B. получают синтетически; т. пл. 298°
(в запаянных капиллярах), р/ 6,00.
Лит. см. при ст. Аминокислоты. О. С. Хохлова.
ВАЛЛАХА ПЕРЕГРУППИРОВКА — изомериза-
ция азоксисоединений в n-окси- или о-оксипроизвод-
ные азобензола (1):
В. п., катализируемая к-тами, приводит только к п-ок-
сипроизводным. С конц. H2SO4 реакцию ведут при сла-
бом нагревании, с хлорсульфоновой — при охлажде-
нии льдом. Помимо незамещенных азоксисоединений,
хорошие результаты получаются с монозамещенны-
ми и Л1,.и'-дизамещенными производными; о,о'- и
п,п'-дизамещенные азоксибензолы не подвергаются пе-
регруппировке. Электроноакцепторные группы (ни-
тро-, галоген) способствуют В. п., электронодонорные—
затрудняют. В случае м,м -дигалогенозамещенных
азоксибензолов выходы продуктов перегруппировки
резко падают с увеличением порядкового номера
галогена, что говорит о большом влиянии стерич.
эффекта заместителя.
В отличие от В. п., катализируемой к-тами, В. п.,
протекающая на солнечном свету на схеме 1),
при облучении УФ-лучами или при нагревании с ук-
сусным ангидридом (230—240°), приводит к о-окси-
производным. В случае несимметричных азоксипро-
изводных атом кислорода мигрирует к кольцу, свя-
занному с неокисленным атомом азота, что указы-
вает на внутримолекулярный механизм перегруппи-
ровки (2):
Такой механизм подтверждается тем, что В. п. аз-
оксипроизводных, меченных по азоту, почти не сопро-
вождается выравниванием изотопного состава атомов
азота (3):
Между тем, В. п., катализируемая к-тами, связана
с выравниванием изотопного состава атомов азота,
что указывает на глубокое различие механизмов обоих
процессов. Предполагается, что начальной стадией
521
’ ВАЛЛАХА РЕАКЦИЯ —ВАЛЬДЕНОВСКОЕ ОБРАЩЕНИЕ
522
катализируемой к-тами В. п. является изомеризация
исходного азоксибензола в симметричный оксид (4):
14 15 14 15 1-1 +
C6H5-N=N-C6H5-----► С6Н5—N—N—С6н5-------
О О
I мич. равновесие. В дальнейшем все они восстанав-
| ливаются муравьиной к-той в соответствующие амины.
Так, напр., бензилиден-бис-пиперидин с му-
равьиной к-той дает с колич. выходом бен-
зилпиперидин:
Перегруппировку оксида в n-оксипроизводное можно I По В. р. аминируются алифатич., ароматич. и гетеро-
представить след, образом (5): I циклич. альдегиды и кетоны. Несмотря на большое
В. п. открыта в 1880 О. Валлахом.
Лит.: Лукашевич В. О., Курдюмова Т. Н.,
Ж. общ. хим., 1948, 18, № 11; Ше мякин М. М., М а й-
м и н д В. И., Вайчунайте Б. К., там же, 1958, 28, № 6.
11. П. Гамбарян.
ВАЛЛАХА РЕАКЦИЯ — восстановительное ами-
нирование альдегидов и кетонов первичными или
вторичными аминами и муравьиной к-той:
1<ч ,R" Rx /В"
>С=О + HN< + НСООН — >CHN< +
R,/ \R"' R'/ XR'"
-j- I RO -j-COg
В. p. имеет много общего с реакцией Лейкарта:
RR'CO + 2HCOONH4---- RR'CHNHCHO + NH3 + COa 4.
+ 2H3O
RR'CHNHCHO —A®. RR'CHNHs 4- HCOOH
В. p. иногда называют также реакцией Лейкарта—
Валлаха. Пользуясь В. р., можно получить первич-
ные, вторичные и третичные амины; механизм ее окон-
чательно не установлен. Бенневилл и Маккартне,
исследуя поведение алифатич. альдегидов в В. р.,
получили с хорошими выходами алкилированные
амины как прямой реакцией аминов и муравьиной
к-ты с альдегидами, так и через промежуточную
стадию образования непредельных аминов (енаминов)
с последующим восстановлением муравьиной к-той:
,NR' -hnr:
R-CHCHO + 2R 'NH---- RCHCH< 2-------*
I - 1 xnr;
HCOOH
-► RC=CHNR1--------- RCHCH3NR'
I - I
На основании этого исследования можно предпола-
гать, что енамины являются промежуточными соеди-
нениями в В. р.; однако Степл и Вагнер считают, что
реакция значительно сложнее, и в результате взаимо-
действия карбонильного соединения с амином перво-
начально могут образоваться оксиалкиламины (I),
алкилиден-бис-амины (II) и основания Шиффа (III);
между этими соединениями может существовать дина-
-R-N-C^°,H(1) — 1 lxR | |н2О + H2O r'
RNH + P'f.n -1 HCOOH „.,1.
R' |RNH —R-N-C-NR (II) — .* RNCH + COg +RNH I
I I ।
число других методов получе-
ния аминов, В. р., или Лей-
карта—Валлаха реакция, ши-
роко применяется в органич.
синтезе. Реакция открыта О.
В аллахом в 1891. Более под-
робно см. Лейкарта реакция.
Лит.: Богословский Б. М.,
вин.: Реакции и методы исследова-
ния органических соединений, кн. 3, М., 1954, с. 255.
ВАЛЬДЕНОВСКОЕ ОБРАЩЕНИЕ — изменение
абс. конфигурации асимметрия, центра при реакциях
замещения. Явление В. о. открыто П. Вальденом
в 1898 на примере хлорирования яблочной к-ты с по-
мощью РС15 до хлорянтарной к-ты и гидролиза послед-
ней при действии AgOH. При этом из (+)-яблочной
к-ты образуется (—)-яблочная к-та, и наоборот; сле-
довательно, одна из двух указанных реакций (хлори-
рование или гидролиз) сопровождается обращением
конфигурации оптически активного центра.
РСЬ,
НООССН«СН (ОН) СООН НО»ССНаСНС1СООН
(+)-яблочная кислота (—)-хлорянтарная кислота
AgOH t j AgOH
POU
HOOCCHaCHClCOOH HO»CCHaCH (OH)COOH
(-р)-хлорянтарная кислота (—)-яблочная кислота
Другими примерами реакций, могущих сопровож-
даться В. о., являются реакции хлористых алкилов
с KJ в ацетоне, реакция вторичных спиртов с НС1
или SOC12, реакция арилсульфонатов оптически актив-
ных спиртов с LiCl в ацетоне, реакция замещения
хлора азид-ионом. В. о. зависит от механизма реак-
ции замещения; поэтому одна и та же реакция может
в зависимости от характера второго реагента, раство-
рителя и условий сопровождаться обращением. Так,
действие AgOH или КОН на оптически активные
а-хлоркислоты приводит к оксикислотам с противо-
положными знаками вращения. Установлено, что
нуклеофильные реакции замещения, протекающие
по бимолекулярному механизму (5^2), всегда сопро-
вождаются В. о. Это явление вызвано тем, что пере-
ходное состояние (I), приводящее к обращению кон-
фигурации, энергети-
чески более выгодно, R R х
чем переходное со- I \
стояние (II), при ко- х-—-Д—.у \ 'т
тором возможно со- R'y \ R
хранение конфигура- * \
ции. В идеальном ; ц щ
случае в переходном
состоянии (I) входящая группа X и отщепляющаяся
группа Y расположены на прямой, перпендикулярной
плоскости, в к-рой находятся три заместителя В.
При таком расположении энергия отталкивания
электронов связей С—X и С—Y минимальна. При
нуклеофильных реакциях замещения, протекающих
по мономолекулярному механизму (Y^l), часто про-
523
ВАНАДАТОМЕТРИЯ — ВАНАДИЙ
исходит рацемизация, сопровождающаяся частичным
В. о. Реакции этого типа проходят через стадию обра-
зования карбониевого иона (III). Поскольку послед-
ний имеет плоское строение, следовало бы ожидать,
что результатом реакции явится полная рацемизация.
Частичное В. о. в этих случаях объясняется тем,
что карбониевый ион (III) неустойчив и не успевает
принять полностью плоскую конфигурацию. Иногда
реакции AN1 протекают с сохранением конфигура-
ции. Это явление наблюдается в тех случаях, когда
молекула содержит к.-л. группу, мешающую иону
карбония (III) принять плоскую конфигурацию.
Примером такой «закрепляющей» группы является
карбоксильная группа, при наличии к-рой конфигу-
рация а-углеродного атома, как правило, сохраняется.
Лит.: Ингольд К. К., Механизм реакций и строение
органических соединений, пер. с англ., М., 1959; Реу-
тов О. А., Теоретические проблемы органической химии,
М., 1956. Л. Д. Бергельсон.
ВАНАДАТОМЕТРИЯ — титриметрич. метод ко-
лич. анализа, основанный на применении в качестве
рабочего р-ра 5-валентного ванадия как окислителя
(или реже р-ра солей 3-валентного ванадия как вос-
становителя). В кислом р-ре соли ванадила восстанав-
ливаются до 4-валентного состояния, причем, если взят
энергичный восстановитель, то возможно частичное
восстановление также до 3-валентного состояния;
соединения 3-валентного ванадия реагируют с при-
бавляемым р-ром соли 5-валентного ванадия с обра-
зованием солей 4-валентного ванадия. Окислитель-
но-восстановительный потенциал системы VO3+/VO21~
сильно зависит от кислотности среды, изменяясь от
+0,97 в в 0,5 н. H2SO4 до +1,45 в в 27 н. H2SO4.
Раствор ванадата аммония применяют для определе-
ния Fe11, OsIV, Mov, а также для колич. определения
ряда органич. веществ (купферон, диметилглиок-
сим, 8-оксихинолин, (3-фурфуральдоксим и др.).
Напр. для определения купферона по реакции:
-NO
CeH5N< +V3O5---------- CeH5NO + VaO4 + Na+2H2O
xONH4
остаток пятивалентного ванадия титруют р-ром соли
двухвалентного железа с индикатором фенилантра-
ниловой к-ты. В. широко используется для непрямых
определений многих элементов (Na, К, Rb, Cs, Са,
Р). Индикатором в ванадатометрич. определениях
служит гл. обр. фенилантраниловая к-та.
Лит.: С ы р о к о м с к и й В. С., Кли иенко 10. В.,
Ванадометрия, Свердловск—М., 1950. В. Г. Типцова.
ВАНАДАТЫ — соли ванадиевых к-т, производ-
ных пятиокиси ванадия V2O5. В свободном виде вана-
диевые к-ты не выделены. Состав осажденных В. зави-
сит от pH среды, концентрации исходных р-ров и
времени, в течение к-рого эти соли находились в кон-
такте с маточным р-ром. Известно большое число В.
с различными соотношениями MeJO : V2O6 от 4:1
до 1 : 4. Так, напр., ортованадаты 3Na2O •
V2O5(Na3VO4) образуются при pH 11,8—12,2;
пированадаты 2Na2O • V2O5 (Na4V2O7) — при
pH 10 — 11,8; метаванадаты Na2O • V2O5
(NaVO3) — при pH 7,5—10; из слабокислых раство-
ров (pH 1,7—2,8) выделен ряд В. с соотношением
Na2O : V2O5< 1. Причина такого многообразия кроет-
ся в исключительной чувствительности ванадат-ионов
к характеру среды и легком переходе одного вида ваиа-
дат-ионов в другой. Присущий р-рам метаванадатов
желтый цвет обусловлен, по-видимому, наличием
конденсированных ионов 3VO“S (V3O9)3’. При повы-
шении кислотности обычно возрастает сложность
поливанадатов; в конечном счете выпадает пятиокись
ванадия: (VO4)3- — (V2O7)4~=± (V3O9)3~=t(HVeO17)3"=t
= V2O6 • пН2О. Между (HVeO17)3 и V2O6 • пН2О
располагается ряд очень нестойких к-т с большим
содержанием V2O5, дающих коллоидные р-ры с соста-
524
вом частиц H4VeO17 • nV2O5. Соответственно этому
могут образовываться и различные соли.
Наиболее практически важны и устойчивы мета-
ванадаты. Хорошо растворимы в воде метаванадаты
щелочных металлов и магния. Из р-ров метаванадатов
натрия выше 35° кристаллизуется безводный NaVO3,
а ниже 35° дигидрат NaVO3-2H2O. Соли кальция
и тяжелых металлов обычно труднорастворимы. Они
могут быть получены осаждением из растворов или
сплавлением окислов металлов с V8O5. Ванадий часто
выделяют из растворов в виде труднорастворимых В.
кальция, железа, свинца. Напр., В. кальция осаждают
из р-ра В. натрия известковым молоком или хлори-
стым кальцием. При pH 10,8—11 осаждается ортова-
надат Ca3(VO4)2, при pH 7,8—9,3 — пироваяадат
Ca2V2O7, при pH 5,1—6,1 — метаванадат Ca(VO3)2.
В произ-ве стремятся выделить Ca(VO3)2, как наибо-
лее богатый ванадием. В. кальция используют для
выплавки феррованадия. Метаванадат аммония
NH4VO3 осаждают из р-ров В. натрия хлористым ам-
монием в присутствии аммиака; он служит исходным
в-вом для получения других соединений ванадия.
В. применяют в текстильной пром-сти в качестве
протрав при крашении хлопчатобумажных тканей
и для фиксации анилина на шелке. О применении
в аналитич. химии см. Ванадагпометрия.
Лит.: Макарове. 3. и Р е п а А. Г., Пзв. АН СССР.
Сер. хим., 1938,_ № 2, 451; Д у м а н с к и й А. В., ЖРФХО,
192 4,54, вып. 8, /03;Меерсон Г. А. и Зел и к м ан А.Н.,
Металлургия редких металлов, М., 1955. См. также при ст.
Ванадий.
ВАНАДИЙ (Vanadium) V — химич. элемент V гр.
периодич. системы Менделеева; и. н. 23, ат. в. 50,95.
Природный В. состоит из двух изотопов: V51 (99,75%)
и V60 (0,25%); последний слабо радиоактивен (период
полураспада T1/t = 1014 лет). Искусственно полу-
чены радиоактивные изотопы В., из к-рых важней-
ший, применяемый как индикатор, V48 (Т^ = 16 дней).
Конфигурация внешних электронов атома В. 3d34s2.
Энергии ионизации (в эв): Vе —> V+ —V2+ —V3 —>
—>V4+ —> V5b 6,71; 14,1; 26,4; 48,4; 64,4 соответственно.
Поперечное сечение захвата тепловых нейтронов для
В. (природная смесь изотопов) равно 4,7 барн.
Впервые В. был обнаружен в 1801 А. М. Дель-Рио в мек-
сиканской бурой свинцовой руде и назван эритронием. Однако
позднее стали считать, что за новый элемент был принят хром.
Лишь в 1830 существование В. было твердо установлено
И. Г. Сефстрёмом, к-рый обнаружил новый элемент в железной
руде из 'Габерга (Швеция). Благодаря красивому цвету солей
этого элемента Сефстрём назвал его В. в честь древнесканди-
навской богини красоты Ванадис. В 1831 Ф. Велер показал
тождественность эритрония и В.
Содержание В. в литосфере относительно большое
(1,5 • 10 3 вес. %), но находится он преимущественно
в рассеянном состоянии.
В магматич. процессе 3-валентный В. замещает ионы А13+-
и Fe3+, накапливаясь в породообразующих алюмосиликатах
и титаномагнетптах. На поверхности В. переходит в 5-валент-
ную форму, концентрируясь в зоне окисления рудных место-
рождений (различные ванадаты), углях, нефтях, асфальтах,
битумах, горючих сланцах и других биолитах. Известно более
65 минералов В., из них важнейшие:
патронит — от VS2 до V2S5
сульванит CubVS4
алаит V2O2•Н2О
ванадинит Pb6[VO4]3Cl
деклуазит Pb(Zn, Cu)[VO4][OH]
моттрамит 5(Сп, РЬ)О • V2O6 • 2Н2О
турапит CuHVOjJOHL,
тюямунит Са[иО2ЫУО4]2 • 8Н2О
карнотит K2[UO2]2[VO4I2 • ЗН2О
роскоэлит KV2[AlSi30i0] [ОН]2
Важным промышленным источником В. (вчастности,
в СССР) служат титаномагнетитовые железные руды
(содержание V достигает 1%) и осадочные железные
руды (содержание V до 0,1%). При переработке этих
руд В. извлекается из шлаков при переделе чугуна
в сталь. Другими промышленными источниками В.
являются: патронитовые, роскоэлитовые и могтрами-
525
ВАНАДИИ
526
товые руды; карнотиты, из к-рых В. извлекают попутно
с ураном; окисленные медно-свинцово-цинковые руды,
в к-рых В. находится в виде минералов ванадинита,
деклуазита, моттрамита и др. В качестве побочного
продукта В. извлекают также при переработке ура-
нового сырья, фосфоритов, бокситов, различных орга-
нич. отложений. Вода морей содержит около 0,3 г
В. в 1000 м3.
Физические и химические свойства. В. — металл
серебристо-серого цвета, в чистом состоянии ковкий.
Кристаллизуется в системе, объемноцентрированного
куба с периодом а = 3,0382„А. Атомный радиус 1,34 Д,
ионные радиусы: V2+ 0,72 A, V3F 0,67 A, V4+ 0,61 А,
V6*' 0,4 А. Плотность В. rf15 6,11; т. пл. 1900 ± 25°;
т. кип. 3 400°; удельная теплоемкость (20—100°)
0,120 кал/г • град', теплота плавления 80 кал/г;
теплота испарения 2080 кал!г; уд. электросопротив-
ление (20°) 24,8 • 10 » ом . см; температурный коэфф,
электросопротивления (0—100°) 2,8 10~3; работа
выхода электрона 3,79 ,чв. В. переходит в состояние
сверхпроводимости при темп-ре ниже —268,7°. В.
обладает парамагнитными свойствами. Механич.
свойства металла высокой чистоты (полученного иодид-
ным методом) после отжига: модуль упругости
14100 кг/мм2, предел прочности 11,8 кг!мм2, относи-
тельное удлинение 17%, твердость по Бринеллю 65—
70, коэфф. Пуассона 0,35. Примеси кислорода, водо-
рода и азота резко снижают пластич. свойства металла
и повышают твердость и хрупкость.
В. отличается высокой химич. устойчивостью в воде,
водных р-рах минеральных солей, в морской воде;
довольно стоек в НС1 умеренных концентраций. На
него не действуют на холоду также разбавленные
HNO3 и H2SO4. Плавиковая кислота и конц. азотная
и серная к-ты, а также царская водка растворяют В.
Растворы щелочей на В. не действуют, в расплавлен-
ных щелочах в присутствии воздуха он окисляется
с образованием ванадатов. При нагревании на воз-
духе выше 300° В. становится хрупким. Растворимость
кислорода в ванадии изменяется с повышением темп-ры
от величины, меньшей0,25%, приблизительно до 1%.
Дальнейшее увеличение содержания кислорода при-
водит к перестройке решетки В. из объемноцентриро-
ванной кубич. (a-фаза) в тетрагональную ((3-фаза).
Последнее, по-видимому, является основной причи-
ной сильного влияния кислорода на твердость В.
При комнатной температуре (3-фаза сохраняет гомо-
генность в интервале 2—10,3% О. При 600—700°
происходит интенсивное окисление компактного ме-
талла с образованием пятиокиси V2O5. При нагрева-
нии В. выше 700° в токе азота образуется желто-брон-
зовый нитрид VN, т. пл. 2050°, устойчивый по отноше-
нию к воде и кислотам. С углеродом и углеродсодержа-
щими газами В. и его окислы взаимодействуют с обра-
зованием серого, с металлич. блеском, карбида VC,
т. пл. 2800°, обладающего высокой твердостью. В.
способен поглощать значительные количества водо-
рода с образованием твердых растворов и гидридов.
В. образует соединения, отвечающие валентностям
+ 2, 4-3, +4, 4-5. Соответственно этому известны
окислы VO, V2O3, VO2 и V2O5 (см. Ванадия окислы).
Соединения 2- и 3-валентиого В. неустойчивы и яв-
ляются сильными восстановителями. Практич. значе-
ние имеют соединения высших валентностей (5 и 4).
Характерен сравнительно легкий переход от V5+
к V4+ и обратно. Нормальный потенциал системы
У5+/У4+равен 4*1,01 в. Склонность В. к образованию
соединений различной валентности используется в
аналитич. химии (см. Ванадатометрия). Пятиокись
В. V2O5 имеет явно выраженный кислотный характер.
Она легко растворяется в щелочах с образованием
ванадатов — солей ванадиевых кислот, не выделенных
в свободном состоянии. Вводе пятиокись В. мало рас-
творима; В. присутствует в таких растворах в форме
комплексной ивополикислоты вероятного состава
H2[O(V2O6)2>6], В кислотах V2O6 растворяется. Формы
существования 5-валентного В. в кислых растворах
еще недостаточно изучены; вероятно, это преим. ионы
УОф, VO':'\ При нейтрализации кислых р-ров до
pH 1—2 (или подкислении р-ров ванадатов до этих
значений pH) выделяется, после нагревания раствора,
гидрат V2O5 zH2O, что используется в технологии
получения пятиокиси В. При действии перекиси
водорода на р-ры ванадатов образуются соли не выде-
ленных в свободном состоянии надкислот В., к-рые
могут быть произведены от пированадиевой кислоты
H4V2O5 путем замены части атомов кислорода на пере-
кисные группы. Общая формула надкислот H4V2O^
(х > 7). Растворы надванадатов окрашены в желтый
цвет. Ванадиевые кислоты образуют с кислотами фос-
фора, мышьяка, кремния, олова, вольфрама, молиб-
дена комплексные гетерополикислоты, в к-рых цент-
ральным атомом могут служить В. или атомы пере-
численных элементов. Известны соли этих кислот,
напр.: Me([P(V2Oe)6j; Me‘l Si(V2Oe) ] и другие.
L (w2o7)6J
Двуокись В. VO2 амфотерна с преобладанием кис-
лотных свойств. Она нерастворима в воде, но легко
растворяется в щелочах и кислотах. При растворении
VO2 в кислотах не происходит полного замещения
кислорода на кислотный радикал и образуется слож-
ный катион ванадила VO2+, окрашенный в светло-си-
ний цвет. Соли ванадила (VOSO4, VOC12 и др.) устой-
чивы в кислых р-рах. Они склонны к образованию
двойных и комплексных солей. Выделены устойчи-
вые соли типа: Me2SO4 2VOSO4; Me2SO4 • VOSO4;
2MeCNS VO(CNS)2; Me2SO3 2VOSO3; Me2C2O4 •
2VOC2O4 и др. При растворении VO2 в щелочах обра-
зуются ванадиты (или гипованадаты) — соли не выде-
ленной в свободном виде изополикислоты H2V4O9.
Их растворы окрашены в бурый цвет. Ванадиты 2-
и 3-валентных металлов нерастворимы в воде. С хло-
ром В. образует ряд хлоридов и оксихлоридов VOC13,
VC14, VCI3 и др. Из галогенидов 5-валентного В. изве-
стен только фторид VF5 (см. Ванадия галогениды).
Известны три сульфида В.: VS, V2S3 и V2S5. Высший
сульфид растворяется в растворах сернистых щело-
чей с образованием красно-бурых растворов. При
нагревании V2S5 до 500° образуется V2S3. Последний
при 1000° диссоциирует с образованием VS.
Аналитическое определение ванадия.
Для качественного определения В. порошок испы-
туемого вещества сплавляют с содой и нитратом нат-
рия или перекисью натрия и выщелачивают плав
водой. Для обнаружения В. в р-ре могут быть исполь-
зованы следующие реакции: осаждение из слабо
кислого р-ра сине-черного осадка таннинового комп-
лекса; окрашивание кислого р-ра в красновато-бу-
рый цвет при добавлении Н2О2; окрашивание кис-
лого р-ра в синий цвет при действии восстанови-
телей (HCI, SO2, H2S, Fe2+, спирта и др.); обра-
зование вишнево-красного соединения Fe (II) при
добавлении в солянокислый р-р FeCI3, диметил-
глиоксима и NH3; окрашивание р-ра в красно-
бурый цвет при добавлении сернистого аммония;
осаждение белого осадка ванадата аммония при
насыщении р-ра хлористым аммонием и др.
Для количественного определения В. предложено
большое число как весовых, так и объемных методов.
Для разложения материала и перевода В. в раствор
применяют сплавление навески с щелочными плав-
нями (К2СО3, Na2O2) с последующим выщелачиванием
водой или разложение кислотами. Большинство
аналитич. методов требует предварительного отделе-
ния В. от других элементов. Для этого В. выделяют
в осадок, действуя HgNO3, РЬ(СН3СОО)2, купфероном
527
ВАНАДИЙ —ВАНАДИЯ ГАЛОГЕНИДЫ
528
или выпаривая с HNO3. Малые количества В. могут
быть выделены аммиаком совместно с А1(ОН)3 и
Fe(OH)3. Используют также соосаждение с фосфоро-
молибдатом аммония. Кроме того, для отделения В.
от сопутствующих элементов применяют электролиз
с ртутным катодом, экстракцию эфиром и отгонку В.
в струе сухого НС1. Весовым методом В. определяют
обычно в виде Va05. Наиболее удовлетворительные
результаты получают с помощью объемных методов
определения. Они сводятся в основном к предвари-
тельному восстановлению В. (S02, FeSO4, НаО2,
нек-рыми металлами в редукторе Джонсона и др.)
с последующим титрованием его КМпО4. Используются
также потенциометрия., колориметрия, (с НаОа) и др.
методы определения В.
Получение. Несмотря на разнообразие видов руд-
ного сырья В., имеются нек-рые общие черты его
переработки. Для извлечения В. применяют: а) непо-
средственное выщелачивание рудного концентрата
р-рами кислот или щелочей; б) обжиг рудного концент-
рата (часто с добавкой поваренной соли), а затем
выщелачивание водой или разб. кислотами. Из рас-
творов методом гидролиза в интервале pH 1—3 выде-
ляют «красный кек» — гидратированную пятиокись.
При плавке ванадийсодержащих железных руд
в домне В. переходит в чугун, при переработке к-рого
в сталь получают шлаки, содержащие 10—16% VaO5.
Ванадиевые шлаки подвергают обжигу с NaCl при
800—850°, выщелачивают обожженный материал во-
дой и затем разб. серной к-той. Из р-ров выделяют
пятиокись VaO5. Высушенная и переплавленная тех-
нич. пятиокись В. служит основным материалом для
выплавки феррованадия (сплав Fe с 35—70% V) и
получения чистых соединений, необходимых для
производства металлич. В. Плавленая технич. пяти-
окись содержит 95—96% V2O5, 2,5—3,5% NaaO,
0,5—1% Fe и др. примеси. Чистую пятиокись В. полу-
чают гидролизом очищенной хлорокиси VOC13 или
термич. разложением метаванадата аммония NH4VO3.
Хлорокись получают хлорированием VaO5, а метава-
надат — растворением V2O5 в р-ре щелочи и последую-
щим осаждением малорастворимого NH4VO3 хлори-
стым аммонием. Ковкий металлич. В. может быть
получен следующими методами: а) кальциетермич.
восстановлением V2O5; б) вакуумным углетермич.
восстановлением V2O3 (последнюю получают восста-
новлением V2O5 водородом); в) магниетермич. восста-
новлением VC13 (исходным материалом для получения
хлорида служит феррованадий); г) термич. диссоциа-
цией иодида В. (метод обеспечивает получение металла
наиболее высокой чистоты). Для выплавки феррова-
надия используют восстановление углеродом, ферро-
силицием и алюминием.
Соединения В. ядовиты. Отравление возможно при
введении их внутрь организма, при вдыхании содер-
жащей их пыли. В производственных условиях следует
учитывать, что V2O5 выше 700° заметно испаряется
и дает устойчивый аэрозоль. Отравление вызывает
раздражение дыхательных путей, легочные кровоте-
чения, головокружения, нарушения деятельности
сердца, почек и т. д.
Применение. Основным потребителем В. является
черная металлургия, использующая до 95% добы-
ваемого металла в форме феррованадия. В. — актив-
ный раскислитель и один из наиболее эффективных
легирующих элементов. При введении 0,15—0,25%
В. резко повышается прочность, вязкость, сопротив-
ление усталости и износоустойчивость стали. Благо-
даря склонности В. к карбидообразоваиию, при его
присадке в сталь образуются равномерно распределен-
ные дисперсные включения карбидов, что способст-
вует измельчению зерен и препятствует их росту при
нагреве. Как правило, В. вводят в сталь в комбинации
с другими легирующими элементами: Ст, Ni, W, Мо.
Наиболее широкое применение В. нашел в производ-
стве инструментальных и конструкционных сталей.
В. применяется также для легирования чугуна,
при этом он задерживает графитизацию, стабилизи-
руя и измельчая структуру. В. является, помимо
этого, компонентом сплавов для постоянных магни-
тов. В виде чистого металла (или сплавов на его
основе) В. еще не получил широкого применения.
Благоприятные ядерные свойства в сочетании с высо-
кой темп-рой плавления, пластичностью и коррозион-
ной устойчивостью делают его ценным конструкцион-
ным материалом для ядерных реакторов.
Соединения В. используются в химич. пром-сти
( в качестве катализаторов при контактном производ-
стве серной кислоты, а также при нек-рых органич.
синтезах; применяются также в сельском хозяйстве,
медицине, текстильной, лакокрасочной, резиновой,
керамической, стекольной и фото-кинопром-ети.
Лит.: МеерсонГ. А., Зелин мая А. Н., Металлур-
гия редких металлов, М., 1955, с. 256—81; Полякова. Ю.,
Основы металлургии ванадия, М., 1959; Ростокер У.,
Металлургия ванадия, пер. с англ., М., 1959; Гилл еб-
р а н д В. Ф. [и др.], Практическое руководство по неоргани-
ческому анализу, пер.с англ., М., 1957; Mellor, v. 9,L.—N.Y.—
Toronto 1952; то же, 1947; P asc al, т. 12, P., 1958; Kirk,
v. 11, N. Y., 1955, p. 583—602. A. H. Зелик.чан.
ВАНАДИЯ ГАЛОГЕНИДЫ — с галогенами вана-
дий образует ряд галогенидов и оксигалогенидов,
в к-рых он проявляет валентности -4-2, 4-3, 4-4 и
4~5. Известны галогениды VX2 (X = Cl, J), VX3
(X = Cl, Вг, J), VX4 (X = F, Cl, J) и VX5 (X = F)
и оксигалогениды VOX (X = Cl, Br), VOXa (X =
= F, Cl, Br), VOX3 (X = F, Cl, Br) и нек-рые более
сложные оксихлориды.
При непосредственном взаимодействии V и Fa
при 300° образуется VF5 — бесцветные кристаллы,
темп-pa сублимации 111°, водой гидролизуется до
VOFS, желтовато-белых кристаллов. VF5 склонен к
образованию комплексных соединений, напр. T](VFe).
Получен также (нагреванием VC14 с HF) тетрафторид
VF4 — чрезвычайно гигроскопичное вещество, быстро
гидролизующееся до VOFa. Последнее с фторидами
ряда металлов образует двойные соли, гл. обр. типа
Mel[VOFt • Н2О].
Хлорированием V2O5 при 600—700° получен окси-
хлорид VOC13 — жидкость желтого цвета, т. кип.
127°. При нагревании ванадия в токе хлора образуется
VC14 — красно-бурая жидкость, т. пл. — 28 ± 2°, т. кип.
152°; в растворе гидролизуется: VC14 4- НаО =
— VOC12 4- 2НС1. При термич. разложении VC14 обра-
зуется красно-фиолетовый кристаллич. нелетучий
трихлорид VC13; легко растворим в воде с образова-
нием зеленого раствора, из к-рого выделяется кристал-
логидрат VC13 • 6Н2О. При нагревании выше 500°
VCI3 разлагается (диспропорционирует) с образова-
нием VC14 и VC12. Дихлорид VCla выше 1000° субли-
мирует. Хлориды играют большую роль в технологии
ванадия. Восстановлением VC14 и VC13 получается
чистый металл. Темп-ры кипения VC14 л VOC13 до-
вольно близки. Поэтому, если в процессе хлорирова-
ния окислов ванадия в присутствии угля образуется
VOC13, то он является серьезной загрязняющей при-
месью. В любом последующем процессе восстанов-
ления кислород, отдаваемый VOCI3, будет загрязнять
металлич. V, действуя как примесь внедрения, и тем
самым оказывать вредное влияние на пластичность
металла.
Иодид VJ3 и бромид VBr3 похожи по свойствам на
VC13, но менее устойчивы. Так, VJ3 известен только
в форме кристаллогидрата VJ3 • 6Н2О при 0°; VJ2
как и VC12 мало устойчив, при 1400° диссоциирует
на элементы, используется для получения чистого
металла.
529
ВАНАДИЯ ОКИСЛЫ — ВАН-ДЕР-ВААЛЬСА УРАВНЕНИЕ
530
Лит.: Ростокер У., Металлургия ванадия, пер.
с англ., М., 1959. См. также при ст. Ванадий.
ВАНАДИЯ ОКИСЛЫ — существуют в виде много-
численных фаз в системе V—О, состав к-рых изме-
няется в нек-рых пределах. При содержании кисло-
рода выше 11% (предельная растворимость О в тетра-
гональном V—(3-фазе) ок. 1150° появляется фаза на
основе VO. При более низких темп-pax и содержании
О 14,5—15,5% происходит перитектич. образование
окисной d-фазы, близкой по составу к V2O. При ком-
натной темп-ре фаза на основе VO сохраняет гомоген-
ность в пределах составов VO0,9—VOi,3; V2O3 сущест-
вует в двух модификациях: y-V2O3, гомогенная в пре-
делах составов УО^-УО^ и (J-V2O3 — в пределах
VObeu—VO!,30; VO2 также образует две модификации:
a-VO2, гомогенная в пределах VOj.so—VO2,0 и a'-VO2,
соответствующая составу УО2,17 (т. е. VeOj3). Линия
V2O5 появляется на рентгенограммах образцов, если
содержание О в них превышает соотношение УО2,23.
Окислы VO и V2O3 имеют основной характер, УО2
амфотерна,V2O5 имеет явно выраженный кислотный ха-
рактер. Наиболее важный из В. о.—пятиокись V2O3.
Моноокись ванадия VO — светло-серые
с металлич. блеском кристаллы, гранецентрирован-
ная кубич. решетка, а = 4,089 А, плотность 5,6,
т. пл. ок. 2000°. Теплота образования АН/нт =
= —106 ± 6 ккал/моль. В воде VO нерастворима;
с разб. кислотами образует растворы бледно-голубого
или фиолетового цвета, ее соли — энергичные восстано-
вители. Моноокись может быть получена спеканием
в вакууме при высоких темп-рах смеси металлич. V
и V2O3 или восстановлением водородом VOC13.
Трехокись ванадия V2O3 — блестящие
черные кристаллы, гексагональная решетка, а —
= 4,933 А, с = 13,940 А, плотность 4,84, т. пл. 1970°.
Теплота образования Дй288 = —2 90 ккал/моль. В кис-
лотах, за исключением кипящей HNO3, растворима
весьма слабо. Образуется при восстановлении У2О5
углеродом, окисью углерода или водородом.
Двуокись ванадия VO2 — сине-голубые
кристаллы, простая тетрагональная решетка, а =
= с — 2,88k, плотность 4,26—4,34, т. пл. 1545°.
Теплота образования Д#288=—-171 ±2 ккал/моль. Гиг-
роскопична, на воздухе медленно окисляется. В кис-
лых р-рах образует сложный катион ванадил УО2+,
имеющий светло-синюю окраску. При растворении
в щелочах образуются ванадиты типа В2О • xVO2.
Двуокись может быть получена окислением на воздухе
трехокиси или восстановлением пятиокиси ванадия.
Пятиокись ванадия V2O3 — красные или
красно-желтые кристаллы, простая „ромбич. решетка,
а ~ 4,36А, Ь — 11,48А, с = 3,55А, плоти. 3,36,
т. пл. 670°. Теплота образования Д77288 = —373 ± 2
ккал/моль. Является конечным продуктом окисле-
ния металлич. ванадия. Выше 700° заметно испа-
ряется с одновременной частичной диссоциацией:
2V2O5 = 4VO2 + О2; полностью переходит в VO2
выше 1125°. V2O5 удовлетворительно растворима в
воде (ок. 0,4 г/л) и легко образует коллоидные р-ры.
Возможно, что способностью сравнительно легко
выщелачиваться природными водами из коренных
месторождений может быть объяснена универсаль-
ность распространения V2O5 в осадочных породах.
Водный р-р окрашен в желтый цвет и имеет кислую
реакцию. В кислотах V2O5 растворима лучше, чем
в воде; при этом образуются сложные комплексные
кислоты. Легко растворяется в щелочах с образова-
нием ванадатов. Чистый окисел может быть получен
прокаливанием метаванадата аммония ниже темп-ры
плавления У2О5 с последующей длительной выдерж-
кой пятиокиси в струе кислорода при 400—500°.
Технич. плавленая пятиокись используется для
произ-ва феррованадия (одна из главных областей
применения V2O5). V2O5 служит катализатором в про-
из-ве серной к-ты контактным способом и во многих
процессах органич. синтеза. Ванадиевые катализа-
торы менее чувствительны к контактным ядам, чем
платиновые. На животные организмы V2O6 действует
как сильный яд.
В. о. используются в стекольном произ-ве; окра-
шенные ими стекла адсорбируют УФ-лучи. В. о.
входят также в состав глазурей для покрытия фарфо-
ровых и гончарных изделий.
Лит.: Полякова. 10., Самарин А. М., Усп. хим.,
1950, 19, вып. 5, с. 565; Полякова. Ю., Основы металлур-
гии ванадия, М., 1959; Ростокер У., Металлургия вана-
дия, пер. с англ., М. 1959. См. также при ст. Ванадий.
ВАН-ДЕР-ВААЙЬСА УРАВНЕНИЕ — уравнение
состояния реальных газов (и в некоторой степени
жидкостей) вида:
(p-pa/VS) (у =rt
гдер,У яТ — соответственно давление, мольный объем
и темп-ра газа, R — универсальная газовая постоян-
ная, а и Ъ — постоянные для данного газа величины.
При выводе ур-ния учитывается, что молекулы имеют
собственный объем: Ь — ~ jta37V (где о — диаметр
молекул, а IV — число Авогадро, т. е. Ь приближенно
равно учетверенному объему всех молекул) и что
между ними существуют силы притяжения, влияние
к-рого выражается членом a/Р2. Предполагается
также, что между молекулами происходят только
двойные соударения, а вероятность тройных соударе-
ний и образования ассоциаций мала, и что энергия
притяжения молекул гораздо меньше кТ (к — Больц-
мана постоянная, равна Я/А). Все эти условия выпол-
няются при малых давлениях и высоких темп-рах.
Графически В.-д.-В. у. изображается системой изотерм,
т. е. кривых, каждая из к-рых описывает зависимость р от v
при некоторой постоянной темп-ре. При низких темп-рах изо-
термы имеют S-образную форму в
области, где одному значению р отве-
чают три значения V (см. рис.), т. к.
В.-д.-В. у. является ур-нием третьей
степени относительно V. Эксперимен-
тальные изотермы имеют в этой обла-
сти прямолинейный участок, который
соответствует существованию двух
фаз, находящихся в равновесш^пунк-
тирные прямые ае и др.). Остальная
часть изотермы отвечает наличию
только одной фазы (левая ветвь —
жидкости, правая — газу).
На рис. область существования
двух равновесных фаз выделена пунк-
тирной кривой, при атом, чем выше
темп-ра, тем уже прямолинейный уча-
сток. При темп-ре, наз. критиче*
ской Ткрл корня ур-ния Ван-дер-Ваальса сливаются в один,
и прямолинейный участок изотермы вырождается в точку.
Эта точка изотермы ТКр является точкой перегиба. Давление
и объем, соответствующие этой точке, также наз. критическими
(см. Критическое состояние). Что касается изгиба, к-рый сле-
дует из ур-ния, то участок bd описывает совершенно неустой-
чивые состояния, неосуществимые в реальных условиях,
а участки de и ad — слабо устойчивые (метастабильные) состоя-
ния: ab — перегретую жидкость, de — переохлажденный газ.
Выражая постоянные а и b через критич. параметры,
можно привести В.-д.-В. у. к форме, в к-рой оно свя-
зывает между собой приведенные давление, объем
и темп-ру: л = р/ркр.;<р = v/vKp_; т = T/TKJf , т. е.
к форме л = 8т(3ц> — 1) — ЗЯ/ср2, наз. приведенным
В.-д.-В. у. Это ур-ние не содержит в явном виде
индивидуальных параметров газа и применимо к лю-
бому газу. Выражая связь между л, q> и т в одинако-
вой форме для различных газов, приведенное ур-ние
состояния послужило основой развития теории соот-
ветственных состояний.
В.-д.- В. у. является приближенным ур-нием. Как
правило, значения произведения pV, вычисленные
с его помощью, больше значений pV, наблюдаемых
на опыте, особенно при темп-рах, близких к критиче-
ской. Оно совершенно теряет силу в области фазового
531
ВАН-ДЕР-ВААЛЬСОВЫ СИЛЫ — ВАН-СЛАЙКА МЕТОД
532
перехода, т. к. при его выводе не учитываются изме-
нения молекулярного взаимодействия при фазовом
переходе. Однако оно верно отражает процесс, когда
это изменение взаимодействия
заторможено в силу к.-л. причин
(отсутствие центров кипения или
конденсации, см. выше об участках
аЬ и de). Уравнение выведено Ван-
дер-Ваальсом в 1873.
Лит.: ВукаловичМ. П. и Но-
виков И. И., Уравнение состоя-
ния реальных газов, М.—Л., 1948. В. И. Барановский.
ВАН-ДЕР-ВААЛЬСОВЫ СИЛЫ — см. в ст. Межмолв-
кулярное взаимодействие.
ВАНИЛАЛЬ («этилванилин», 4-окси-З-этоксибен-
зальдегид) CeHi0O3, мол. вес 166,17 — бесцветные
чешуйчатые кристаллы с запахом вани-
СНО ли; т.нл. 77—78°; хорошо растворим
JL в эфире, 1 ч. В. растворима в 5 ч.
р || спирта и в 250 ч. холодной воды; УФ-
L м спектР: Чакс. 310>° м'мк (te6 4’01);
Т 2М5 279,5 ммк (Ige 4,02); 232,0 ммк (Ige
ОН 4,20). С хлорным железом В. дает синюю
окраску; с нитритом натрия и азотной
к-той — белый осадок; с гидразинсульфатом и со-
ляной к-той — желтый кристаллич. осадок, с п-нитро-
фенилгидразином — оранжево-коричневый осадок.
В. получают из гуэтола (моноэтиловый эфир пирока-
техина), аналогично методам получения ванилина
из гваякола. В. применяется в пищевой и парфю-
мерной пром-сти. С. И. Поре.
ВАНИЛИН (4-окси-З-метоксвбензальдегид) С8Н8О3,
мол. в. 152,14 — бесцветные игольчатые кристаллы
с запахом ванили; т. пл. 81—83°; т.
СНО кип. 284—285° (в токе СО2); 170°/15.и.и;
146°/4 мм; сублимируется без разло-
Г || жения; легко растворим в спирте, эфи-
—ОСН3 ре, хлороформе, сероуглероде, ледяной
Т уксусной кислоте и в горячем лигрои-
ОЧ пе; "в 50%-ном спирте растворим в со-
отношении 1 : 4; в 70%-ном — 2:5; в
94%-пом — 1 : 2; в холодной воде — 1 : 125; в го-
рячей воде — 1 : 20. Водный раствор В. имеет
слабокислую реакцию; УФ-спектр: Хмакс 309,5 ммк
(Ige 4,08), 279,5 ммк (Ige 4,09), 231,0 ммк (]ge 4,25).
В. содержится в виде глюкозида в плодах ванили.
Наиболее важными промышленными методами полу-
чения В. являются: 1. Взаимодействие гваякола с му-
равьиным альдегидом и арильным производным гид-
роксиламина с последующим разложением образую-
щегося шиффова основания:
2. Конденсация гваякола с хлоральгидратом и по-
следующее окислительное расщепление продукта кон-
денсации (хлоральгидрцтный метод):
ОН
3. Конденсация гваякола с глиоксиловой к-той
с последующим разложением продукта конденсации
в присутствии окислителей (глиоксиловый метод):
СНО
ОН
получения В. является
Промышленным способом
также окисление лигнина.
Важнейшие производные В.: оксим, т. пл. 121 —122°;
семикарбазон, т. пл. 229—232°; фенилгидразон, т. пл.
105°; 2,4-динитрофенилгидразон, т. пл. 271°. Со
смесью уксусной и серной кислот В. дает зелено-синюю
окраску, с серной к-той в спиртовом р-ре — зеленую
окраску, переходящую при нагревании в винно-крас-
ную и фиолетовую, с водным р-ром азотнокислой
ртути и с резорцином дает фиолетовое окрашивание.
В. применяют в пищевой и парфюмерной пром-сти,
а также для изготовления нек-рых мед. препаратов.
Лит.: Kirk, v. 14, N. Y., 1955, p. 603—611. C. 11. hop.».
ВАНКОМИЦИН — антибиотик, образуемый акти-
номицетом Streptomyces orientalis; высокомолекуляр-
ное соединение (мол. в. ок. 3300) содержит ок. 55%С,
5,8%Н, 16%O, 8,5%N в 4% неионогенного С1; обла-
дает характерным спектром Хма с 282 ммк (НС1),
Хмакс 305 -и.пк (NaOH). В. почти не изучен в химич.
отношении; содержит 16—17% углеводов, поло-
вину к-рых составляет глюкоза. В. (основание) —
бесцветный аморфный порошок, хорошо растворимый
в воде и в водных полярных растворителях. Он устой-
чив при pH 3—6, не инактивируется в присутствии
аминокислот, неорганич. солей и восстановителей.
Наиболее очищенные препараты В. содержат до 20%
второго компонента, антибиотич. активность к-рого
не определена. Получены хлоргидрат, сульфат и др.
соли В.
Выделяют В. из культуральной жидкости адсорб-
цией слабоактивными ионообменными смолами, десорб-
цией смесями (СН3)2СО, Н2О, СН3СООН илиС2Н5ОН,
Н2О, СН3СООН с последующим превращением в пик-
рат, а затем в свободное основание. В. высокоактивен
в отношении спирохет и грамположительных бакте-
рий, в том числе устойчивых к обычным антибиотикам;
устойчивость к нему вырабатывается медленно. Он
умеренно токсичен: при внутреннем введении мышам
LD50 составляет 400—500 мг/кг.
Обычно В. вводят внутривенно по 0,5 г каждые
6 часов, при этом почти во всех жидкостях тела соз-
дается терапевтич. концентрация. В. выводится гл.
обр. с мочой. Опубликованы данные о клинич. приме-
нении В. для лечения заболеваний, вызванных грам-
положительными бактериями, особенно стафилокок-
ками, устойчивыми к другим антибиотикам. Побочные
реакции (сыпь) обнаружены приблизительно у 6%
пациентов. В. является одним из наиболее перспек-
тивных антибиотиков, его препараты выпускаются под
названием «Ванкоцин».
Лит.: Vancomycin. A new antibiotic, в кн.: Antibiotics
annual. 1955—1956, р. 606—33; 1956—1957, р. 75—89; 1957—
1958, р. 906—16. /7. Д. Решетов.
ВАН-СЛАЙКА МЕТОД — определение содержания
первичных аминогрупп в органич. соединениях, ос-
нованное на измерении объема азота, выде-
лившегося при реакции RNH2 + HONO —►
—► ROH + N2 + Н2О. Одновременно обра-
зующиеся из-за частичного разложения
HNO2 окислы азота поглощают щелочным
р-ром КМпО4. Метод широко применяется
для определения аминокислот.
533
ВАНТ-ГОФФА КОЭФФИЦИЕНТ - ВЕСОВОЙ АНАЛИЗ
534
Определение производится в спец, приборе (рис.), основной
частью которого является сосуд 1, соединенный с двумя во-
ронками, одна из к-рых, 2, служит
для введения р-ра исследуемого
в-ва, вторая, 3 — для 30%-ного р-ра
NaNO2. После внесения р-ров ве-
щества и реактива реакционный со-
суд продолжительное время встря-
хивают; накопившийся газ по окон-
чании реакции переводят в газовую
бюретку 4, откуда его периодически
переводят для поглощения окислов
азота в сосуд -5 с 5%-ным р-ром
КМпО4 в 2,5%-ном р-ре NaOH.
Объем газа, освобожденного от оки-
слов азота, измеряют в бюретке
4. По количеству азота рассчиты-
вают содержание первичных амино-
групп в исследуемом соединении.
Метод предложен Д. Д. ван Слай-
ком в 1910.
Лит..- Белозерский А. Н.
и Проскур яко вН. И., Прак-
тическое руководство по биохимии
растений, М., 1951, с. 120; Бай-
бе льС., Идентификация органических соединений, пер.
с англ., М., 1957, с. 211. Д. Н. Васкевич.
вант-гоффА коэффициент (изотонический
коэффициент) — поправочный множитель в термо-
динамич. уравнениях для разб. р-ров электролитов,
выражающих зависимость нек-рых их свойств от
концентрации. Введен Я. Вант-Гоффом как эмпирич.
величина; теоретически обоснован С. Аррениусом на
основе теории электролитической диссоциации. В соот-
ветствии с последней, ионы в р-рах, как и молекулы
растворенного в-ва, вызывают: понижение давления
насыщенного пара растворителя над р-ром, ДР;
повышение темп-ры кипения р-ра, ДГК; понижение
темп-ры замерзания р-ра, АГ.,; повышение осмотич.
давления, л; общее их влияние на эти свойства и
учитывается с помощью В.-Г. к., равного отношению
суммарного числа молей недиссоциированного элект-
ролита и ионов к стехиометрии, числу молей электро-
лита в р-ре. Величина В.-Г. к. выражается ур-нием;
i = 1 + a(S — 1), в к-ром а — степень электролитич.
диссоциации и Д — число ионов, на к-рые диссоци-
ирует одна молекула электролита. С уменьшением
концентрации а — 1 и i —5. С учетом В.-Г. к. свой-
ства р-ров электролитов вычисляются из соотно-
шений:
= ---, AT = Elm, AT., = iKm, n = icRT
” 1П1 + ns ’ Ь
где Ро — давление насыщенного пара чистого раство-
рителя, п, и «2 — стехиометрия, числа молей электро-
лита и растворителя в р-ре; Е — эбулиоскопия, по-
стоянная; К — крископич. постоянная; т — стехио-
метрия. моляльность электролита (число молей элект-
ролита в 1000 г растворителя); с — стехиометрия,
молярность электролита (яисло молей электролита
в 1 л р-ра); R — универсальная газовая постоянная.
ВАНТ-ГОФФА ПРАВИЛО — приближенное пра-
вило, согласно к-рому при повышении темп-ры на
10° скорость реакции увеличивается примерно в 2—
4 раза.
ВЕРАТРОВЫЙ АЛЬДЕГИД (диметиловый эфир
протокатехового альдегида, 3,4-диметокспбензальде-
СНО
гид), мол. вес 166,18 — кристаллы с
запахом ванилп; т. пл. 44,5—45°; т. кип.
283°/760лыг, 198—201° /53 мм; слабо рас-
творим в теплой воде, почти нераство-
рим в холодной; легко растворим в
спирте и эфире. При нитровании В. а.
конц. HNO3 получены 6-нитро- и немного
4-нитровератрола. При бромировании В. а. в ледяной
СН3СООН образуется 6-бром-3,4-диметоксибензальде-
гид. Получают В. а. из вератрола Г аттермана реак-
цией с HCN в присутствии А1С13; метилированием
ванилина диметилсульфатом в щелочной среде или
еще лучше метиловым эфиром паратолуол сульфокис-
лоты, или гидратом окиси триметилфениламмония.
И. В. Матвеева.
ВЕРАТРОЛ (1,2-диметоксибензол) СвН4(ОСН3)2,
мол. в. 138,17 — кристаллы с запахом пиона; т пл.
22,5°; т. кип. 206,5°; djf 1,0914; nft* 1,5287; нерас-
творим в воде, растворим в спирте, эфире и маслах.
Пикрат В. красного цвета, т. пл. 55—57°. В. встре-
чается как продукт распада природных соединений
(напр., алкалоидов). Синтетически получают В. пере-
гонкой вератровой к-ты в присутствии барита:
НООССвН3(ОСН3)2 СвН4(ОСН3)2; лучший ме-
тод — метилирование пирокатехина или гваякола
диметилсульфатом в щелочном р-ре, напр.:
СвН4(ОН)2 + (CH3)sSO4 -> CeH4(OCH3)s + H2SO4
И. В, Матвеева.
ВЕРОНАЛ (барбитал, малонал, 5,5-диэтилбарби-
туровая кислота) C8H12O3N2, мол. в. 184,20 — бес-
цветные кристаллы (призмы или
иглы) со слабым горьким вку- „ „ . /Со —NH\rn
сом; т. пл. 191°. Растворимость ( 2 \со—nh/
В. в воде: 1 : 170 (20°), 1 : 17
(100°); растворимость в 50%-ном спирте 1 : 27,5,
в 90%-ном спирте 1 : 9 (15°), в эфире 1 : 18,7, в этил-
ацетате 1 : 8,9, в хлороформе 1 : 75; легко раство-
ряется в р-рах аммиака и щелочей. Водные р-ры
В. имеют кислую реакцию. Константа электроли-
тической диссоциации (вода, 25°) 3,7 • 10~8. В. дает
качественные реакции, характерные для барбитура-
тов, выделяет аммиак при нагревании со щелочами,
образует нерастворимую серебряную соль при при-
бавлении р-ра нитрата серебра к раствору В. в щелочи
и др. Колпч. определение: к 20 лмшпирта добавляют
5 капель тимолфталеина и 0,1 н. спиртовой р-р едкого
кали до появления голубого окрашивания; затем
растворяют в нем 0,5 г В. и титруют обесцвеченный
р-р 0,1 н. спиртовым р-ром едкого кали до появления
прежнего окрашивания. В. получают конденсацией
диэфира диэтилмалоновой к-ты с мочевиной в присут-
ствии , алкоголята натрия. В. применяют в качестве
снотворного средства.
Лит. см. при ст. Адалин. Е. С. Головчинская.
ВЕСОВАЯ БЮРЕТКА — стеклянный, обычно гру-
бо проградуированный сосуд (цена делений 5—10 мл)
любой формы, снабженный краном и стеклянной
пробкой, применяемый при точных титриметрич. опре-
делениях. Количество (вес) израсходованного реак-
тива определяют взвешиванием В. б. до и после тит-
рования. Титр раствора реактива устанавливают по
весу: концентрация выражается в грамм-молях или
грамм-эквивалентах, содержащихся в 1000 г (а не
в 1000 мл) раствора. Взвешивание производят с точ-
ностью до 1 мг. Ошибки от натекания и от изменения
темп-ры полностью исключаются. Определения мо-
гут быть выполнены с погрешностью, не превышаю-
щей 0,002% (при обычных методах измерения объема
точность определения не превышает 0,1 %). Примене-
ние В. б. имеет смысл лишь тогда, когда вр.е другие
стадии определения производятся с такой же точ-
ностью, как и взвешивание. Н. А. Канаев.
ВЕСОВОЙ АНАЛИЗ — один из важнейших мето-
дов колич. анализа, в к-ром, в отличие от других
методов колич. анализа, взвешивание является не
только начальной, но и конечной стадией определе-
ния. Измерительным прибором В. а. служат аналитич.
весы. Определение начинается с отвешивания нек-рого
количества анализируемого вещества на аналитич.
весах и переведения его в раствор. Далее определяемый
компонент осаждают из раствора в виде к.-л. малораст-
воримого соединения или простого вещества (форма
осаждения), к-рые затем отделяют от маточного
р-ра и переводят посредством высушивания или про-
каливания в устойчивое соединение определенного
535
ВЕСОВОЙ АНАЛИЗ
536
состава — весовую форму. Иногда химич.
состав формы осаждения и весовой формы бывает
одинаков. Заключительной операцией В. а. является
взвешивание весовой формы. Результаты анализа
обычно выражают в процентах:
Х = аР •100/6
где X — искомое процентное содержание определяе-
мого элемента, а — вес весовой формы, Ъ — навеска,
Р — фактор пересчета; последний представляет отно-
шение ат. веса элемента (или его кратного значения)
к мол. весу весовой формы.
В. а. долгое время был одним из главных приемов
определения состава и сыграл большую роль при
установлении ряда химич. закономерностей, в том
числе закона постоянства состава химич. соединений,
закона кратных отношений, периодич. закона и др.
В. а. применяется при определении химич. состава
горных пород и минералов (силикатный анализ),
при химич. контроле различных отраслей произ-ва,
при установлении качества сырья и готовой продук-
ции многих предприятий и т. д. Основан В. а.на законе
сохранения веса веществ при химич. превращениях.
Наиболее ранняя разновидность В. а. — пробир-
ный анализ, представляющий совокупность приемов
определения содержания драгоценных металлов в ру-
дах и сплавах. Особенность этой отрасли В. а. состоит
в изолировании драгоценного металла путем проведе-
ния всех химпч. процессов «сухим путем». Пробирный
анализ позволяет определять крайне малые количе-
ства драгоценных металлов с высокой точностью.
Наиболее ответственной операцией В. а. является
превращение определяемого вещества в малораство-
римое соединение (или простое вещество) — осадок
(форма осаждения). Осадки чаще всего представляют
собой сульфиды, фосфаты, карбонаты или гидроокиси
металлов, или же различные внутрикомплексные соли
металлов с органич. реагентами — купфероном, окси-
хинолином и др. Полученные осадки должны удовлет-
ворять след, требованиям: 1) Быть практически нерас-
творимыми; в растворе после осаждения не должно
оставаться более 0,1 мг определяемого элемента, т. е.
количество, не превышающее чувствительности обыч-
ных аналитич. весов; для уменьшения растворимости
осадка часто вводят в раствор избыток осаждающего
реактива. 2) Быть практически чистым, т. е. не содер-
жать примесей посторонних веществ, не удаляющихся
при переведении осадка в весовую форму; нарушение
этого требования связано с явлением соосаждения;
для уменьшения соосаждения необходимо соблюдать
определенные условия осаждения. 3) Осадок должен
получаться в форме, удобной для отделения от раст-
вора фильтрованием. Для получения более крупно-
кристаллич. или компактных осадков, к-рые легко
фильтруются, не проходят через фильтр, осаждение
ведут при нагревании, выдерживая осадок в маточном
растворе нек-рое время перед его фильтрованием,
и т. д. Основное требование к весовой форме заклю-
чается в том, чтобы состав полученного соединения
в точности соответствовал определенной химич. фор-
муле. Желательно, кроме того, чтобы весовая форма
не была гигроскопична и вообще не изменяла химич.
состава в процессе взвешивания.
Главными вопросами теории В. а. являются зако-
номерности процессов образования твердой фазы.
Большое значение имеет правило произведения актив-
ности (растворимости), в соответствии с к-рым произ-
ведение концентраций ионов в насыщенном р-ре
малорастворимого соединения есть величина постоян-
ная ири постоянной темп-ре. Отсюда следует, что
более полное осаждение малорастворимой соли или
гидроокиси металла достигается при введении в р-р
нек-рого избытка осадителя. Величина произведения
растворимости позволяет сделать выводы о полноте
осаждения определяемого элемента и наметить возмож-
ность разделения нек-рых элементов посредством
осаждения. Важный раздел теории В. а. составляют
закономерности зарождения и роста первичных кри-
сталлов. Сложный процесс образования поверхности
раздела при осаждении состоит из двух основных
этапов: появления первичных центров кристаллиза-
ции и дальнейшего их роста. Большая скорость обра-
зования центров кристаллизации, характерная обычно
для малорастворимых соединений, служит причиной
одновременного появления в растворе множества пер-
вичных зародышей, к-рые слипаются затем в более
крупные агрегаты и выпадают из раствора в виде
аморфного осадка. Более растворимые вещества отли-
чаются меньшей скоростью образования центров
кристаллизации; при постепенном прибавлении оса-
дителя происходит дальнейший рост образовавшихся
уже в растворе первичных кристалликов и выделение
из раствора кристаллич. осадка. Эти представления
позволяют в ряде случаев выбрать оптимальные усло-
вия осаждения; так, при получении кристаллич.
осадков медленное прибавление осадителя к горячему
р-ру определяемого вещества создает наиболее бла-
гоприятные условия для роста первичных центров
кристаллизации и приводит к образованию более
крупнокристаллич. и чистых осадков. На основании
этих же представлений находит объяснение явление
соосаждения, состоящее в захвате осадком из раствора
посторонних веществ, которые сами по себе хорошо
растворимы. Причиной соосаждении для аморфных
осадков является адсорбция посторонних ионов на
поверхности осадка, а для кристаллических — т. наз.
внутренняя адсорбция, при которой посторонние ионы
адсорбируются в процессе роста кристаллов и ока-
зываются включенными внутрь этих кристаллов. Со-
осаждение сильно зависит от концентрации, темп-ры
и порядка сливания растворов; последний фактор
имеет существенное значение. Так, при осажде-
нии ионов бария ионами сульфата можно уменьшить
соосаждение посторонних катионов, медленно при-
ливая раствор серной к-ты к раствору бариевой соли;
наоборот, посторонние анионы соосаждаются в мень-
шей степени при обратном порядке сливания раство-
ров. В теории В. а. рассматриваются также вопросы
влияния маскирующих веществ (см. Маскировка)
на растворимость осадков, роль физич. факторов
в процессах осаждения и т. д.
Разновидностью В. а. является электровесовой
анализ (см. Электроанализ), в к-ром определяемые
элементы выделяют из раствора посредством электро-
лиза и затем их взвешивают. Электровесовой анализ
чаще всего применяют при определении меди, кадмия,
серебра, никеля, кобальта и др.; эти элементы осаж-
дают на катоде в виде металлов. Реже применяют
осаждение на аноде; практич. значение имеет лишь
выделение свинца в виде его двуокиси. Преимущест-
вами электровесового анализа являются практич.
нерастворимость осадков металлов, отсутствие во
многих случаях соосаждения и др. Для успешного
определения имеет важное значение поддержание
определенного напряжения и силы тока, кислотности
растворов, добавление различных комплексообразую-
щих веществ и т. д. Разновидностью электровесового
анализа является внутренний электролиз, к-рый
выполняют без применения внешнего источника тока.
В. а. обычно применяется при определении коли-
честв вещества порядка 0,02—0,1 г. Относительная
ошибка определения не превышает 0,1%, а при особо
тщательной работе может быть доведена до 0,02—
0,03%. Недостатки В. а. состоят в длительности ана-
лиза и в необходимости располагать сравнительно
большим количеством анализируемого вещества. Пос-
537
ВЕСТОНА НОРМАЛЬНЫЙ ЭЛЕМЕНТ — ВЕСЫ ЛАБОРАТОРНЫЕ
538
ледний недостаток устраняется при использовании
микро- и ультрамикрометодов, отличающихся от
рассмотренного выше обычного макро-В. а. гл. обр.
техникой эксперимента и применяемой аппаратурой.
Микрометоды пригодны для определения 0,01—10 мг
вещества. Взвешивание производится на микровесах
с точностью до 0,001—0,01 мг, осаждение — в малень-
ких пробирках, вместо фильтрования применяют цент-
рифугирование и т. д. Ультрамикрометод позволяет
определять микрограммовые количества вещества.
Все химич. реакции производятся на предметном сто-
лике микроскопа с использованием микроманипуля-
торов; для взвешивания применяются специальные
ультрамикровесы с изгибающейся кварцевой нитью или
кварцевым коромыслом и т. д. Главная трудность
при весовых микро- и ультрамикроопределениях со-
стоит в возможности потерь вещества, относительное
влияние к-рых увеличивается по мере уменьшения
количества анализируемого вещества. Микро- и ульт-
рамикрометоды применяются в нек-рых специальных
случаях.
Сравнительно новым отделом В. а. является термо-
гравиметрия, применяющаяся при изучении термич.
устойчивости веществ, определении устойчивости раз-
личных весовых форм данного элемента и др. Взвеши-
вание производится на специальных термовесах, поз-
воляющих наблюдать изменение веса вещества при
повышении темп-ры; на автоматически вычерчиваемой
кривой отчетливо выявляется изменение состава
вещества вследствие потери составных частей и обра-
зования промежуточных продуктов разложения. Ис-
пользование термовесов сокращает продолжитель-
ность анализа и дает возможность работать с гигро-
скопич. веществами; при этом отпадает необходимость
в выдерживании осадков в эксикаторе и в доведении
их до постоянного веса.
Лит.: Ко л ьтгоф И. М. и Сен да л Е. Б.. Количе-
ственный анализ, пер. с англ., 3 изд., М.—Л., 1948; Баб-
ко А. К., Пятниц кий И. В., Количественный анализ, М.,
1956; Ко р е н м ан И. М., Количественный микрохимический
анализ, М.—Л., 1949; А ли мар и н И. П., Петрикова
М. Н., Неорганический ультрамикроанализ, М., 1960; Ш а-
х о в а В. Ф., Семеновская Е. Н., Завод, лаб., 1956,
23, с. 1430. И. Ь. Пятницкий.
ВЕСТОНА НОРМАЛЬНЫЙ ЭЛЕМЕНТ — галь-
ванич. элемент для точного измерения напряжения
(обычно компенсационным методом); употребляется
также в качестве эталона разности электрич. потен-
циалов — международного вольта. Элемент легко
готовится, долговечен, дает хорошо воспроизводимую
(с точностью до 2 • 10 ’5 в) и почти не зависящую от
темп-ры эдс (темп-рный коэфф. 4 • 10~5 в/°С). При не-
больших отбираемых токах работает вполне обратимо
в интервале темп-р
от —8 до +43,4°.
Наиболее распростра-
ненная конструкция
. насыщенный _ х v
раствор coso4 В. н. э. показана на
рис. Отрицательный
электрод представ-
ляет собой амальгаму
-Cdso. |н.о кадмия (12,5%Cd), по-
Самещенную в р-р суль-
фата кадмия, насы-
_. щенного относитель-
но CdSO4 • 8/3Н2О.
Положительный электрод — паста из ртути и Hg2SO4,
помещенная в тот же р-р. При работе В. н. э. проте-
кает реакция:
Cd 4- Hg2SOi + s/3H2O = CdSO, • S/3 H»O + 2Hg
Эдс при 20° равна 1,01830 в', зависимость потенциала
от темп-ры: Е = 1,01830 —4,06-10“5 (t—20)—9,5-
• 10’’ (Z — 20)2+ 1 -10~8 (Z — 20)3 в; внутреннее сопро-
тивление В. н. э. колеблется в пределах 500—1000 ом.
Иногда для измерения напряжения используется не-
насыщенный В. н. э., не содержащий кристаллогид-
рата и наполненный р-ром CdSO4, насыщенным при 4°.
Темп-рный коэфф, такого электрода 1 • 10~5 в/°С, внут-
реннее сопротивление порядка 300 о.u, Е — 1,0186 в,
рабочий интервал темп-p от +4 до +43,4°. Воспро-
изводимость и устойчивость его эдс ниже, чем у на-
сыщенного В. Н. э. А. н. Чемоданов.
ВЕСЫ ЛАБОРАТОРНЫЕ — применяемые в лабо-
раториях приборы для измерения массы тела или
определения разности значений масс тел.
Определение массы М производится измерением
силы тяжести Р = М • g, где g — ускорение силы
тяжести. В большинстве В. л. используют комбини-
рованный метод измерений: свыше 99,9% нагрузки
уравновешивают гирями (нулевой метод), а оставшая-
ся малая разность между весом гирь и весом на-
грузки определяется по углу отклонения коромыс-
ла от положения равновесия (непосредственный ме-
тод). Это отклонение измеряется непосредственно с
помощью предварительно проградуированной отсчет-
ной шкалы.
В. и. характеризуются: 1) наибольшей допустимой (пре-
дельной) нагрузкой; 2) допустимой погрешностью показаний —
предельной разностью между действительным значением массы
взвешиваемого груза и показаниями В. л. Значение погреш-
ности характеризует верность (правильность) результатов
взвешивания (в стандартных условиях); 3) допустимой вариа-
цией (непостоянством) показаний — наибольшей допустимой
разностью показаний при неоднократном взвешивании одного
и того же груза, в стандартных условиях; значение вариации
характеризует воспроизводимость результата взвешивания
и в значительной степени — точность результатов относи-
тельных взвешиваний; 4) чувствительностью — пределом отно-
шения приращения отклонения указателя (напр., стрелки)
В. л. к приращению измеряемой величины. Часто термин
«чувствительность» используется неверно — для характеристики
точности В. л. или вместо термина «цена деления»; 5) ценой
деления. Во многих В. л. цена деления согласуется со значе-
нием допустимой погрешности или вариации показаний; 6) точ-
ностью отсчета; 7) быстродействием — числом взвешиваний
в единицу времени.
В. л. подразделяются на: а) весы аналитической
группы (аналитические, полумикроаналитические,
микроаналитические и пробирные, последние исполь-
зуются также в качестве образцовых весов для по-
верки аналитич. и технич. гирь 1-го класса); б) микро-
весы и в) общелабораторные весы (технические весы
1-го и 2-го классов, применяемые в технич. анализе,
и др.). Основные характеристики В. л. приведены
в табл. 1, 2 и 3.
Таблица 1. Основные характеристики весов ana литической
группы
Тип весов Предель- ная на- грузка, г Цена де- ления от- счетной шкалы, мг Вариация показаний (при пре- дельной нагрузке), мг Погреш- ность по- казаний, мг
Аналитические 500 2-10 9 5
200 0,1—1 0,1—0,5 1—2
100 0,05-0,2 0,1-0,2 1—2
Полумикроана- 50 0,01-0,02 0,02 0,25
литические 20 0,01 0,01 0,2
30 0,01 0,002 0,01
Пробирные 20 ' 0,001—0,005 0,002-0,005 0,01-0,04
1—2 0,01—0,02 0,01-0,02 0,04
Указанные значения погрешности показаний характери-
зуют предельную погрешность при определении абсолютного
значения массы груза, равного предельной нагрузке весов
методом прямого взвешивания и включает погрешности гирь
соответствующих классов (см. табл. 5). Погрешность показаний
микроаналитич. весов характеризует предельную погрешность
определения абсолютного значения массы грузов до 1 г в пред-
положении использования методов точного взвешивания (см.
ниже). Во всех случаях предполагается, что погрепшость от
влияния аэростатич. сил (см. ниже) исключена введением
соответствующих поправок.
К аналитич. относятся также В. л. для больших
предельных нагрузок, напр. 1; 2; 5; 10; 20 и 50 кг
539
ВЕСЫ ЛАБОРАТОРНЫЕ
540
Таблица 2. Основные характеристики микровесов
Тип весов Предель- ная на- грузка, мг Цена де- ления, мг Вариация показаний при пре- дельной нагрузке, мг Погреш- ность по- казаний, мг
Торзионные 1000 1 1 1
микровесы 200 0,2 0,2 0,2
20 0,05 0,05 0,05
Крутильные 5 0,01 0,01 0,01
микровесы 2,5 0,005 0,005 0,005
То же, с метал- 1 0,002 0,002 0,002
лической нитью 0,5 0,001 - 0,001 0,001
с пеной деления 1; 2; 5; 10; 20 и 50 мг соответственно
и вариацией показаний в пределах цены одного де-
ления.
Для специальных целей выпускаются микровесы
с ценой деления менее 1 мкг, напр., крутильноравно-
плечие весы с кварцевой нитью для предельной
нагрузки 40 мг и с ценой деления 0,01 мкг.
Таблица 3. Основные характеристики общелабораторных
весов
Тип весов Предель- ная на- грузка Цена де- ления, мг Вариация показаний при пре- дельной нагрузке', мг Погреш- ность показа- ний, мг
Равноплечие с ука- 200 г 100
зателем положения 1 кг 200
равновесия1 * 5 кг
Равноплечие с от- 20 г 5 2,5 15
счетной шкалой3 200 г 20 10 60
5 кг 250 125 500
20 кг 1000 500 2000
50 кг 2500 1250 5000
Равноплечие новы- 1 кг 10 5 30
шеиной точности 10 кг 50 25 150
с отсчетной 20 кг 100 50 300
шкалой3 50 кг 200 100 600
Равноплечие повы- 5 кг 10 5 25
шейной точности 10 кг 10 5 30
с проекционной 20 кг 40 20 80
шкалой4
Равноплечие с ци- 500 г 20 20 60
ферблатным от- 1 кг 50 50 150
счетным устройством
Квадр антные 1 кг 100 30 50
с проекционной 2 кг 1000 200 500
шкалой 3 кг 1000 200 500
1 Технические весы 2 класса. 3 Образцовые весы 3 раз-
ряда. 3 Технические весы 1 класса и образцовые 2 разряда.
4 Образцовые весы 1 разряда.
Погрешность показаний характеризует предельную по-
грешность игш определении абсолютного значения массы груза,
равного предельной нагрузке весов методом прямого взве-
шиваний и включает погрешности гирь соответствующих
классов.
К специальным В. л. относятся весы электронные,
весы для работы в вакууме, в атмосфере агрессивных
и инертных газов и весы для специальных видов ана-
лизов (напр., термогравиметрические, седиментацион-
ные и др.). По устройству В. л. подразделяются на:
а) простые равноплечие; б) равноплечие с успокоите-
лями и именованными шкалами; в) равноплечие со
встроенными гирями; г) одноплечие; д) крутильнорав-
ноплечие; е) крутильные; ж) торзионные; з) равнопле-
чие с циферблатным отсчетным устройством; и) квад-
рантные, и весы специального устройства.
Простые равноплечие весы имеют коромысло с одной опор-
ной п двумя грузоприемными призмами, к к-рым посредством
серег подвешены грузоприемные чашки или площадки. Коро-
мысло снабжено отсчетной стрелкой, служащей для опреде-
ления положения равновесия, или для отсчета отклонений
стрелки, по к-рым вычисляется положение равновесия. По
этой схеме изготовляют простейшие весы аналитич. группы
(с рейтерными шкалами) и большинство типов общелаборатор-
ных весов.
Недостатками простых равноплечих весов являются
низкая производительность, неудобство работы, зави-
симость цены деления от нагрузки, погрешности от-
счета, малый диапазон непосредственных измерений
по отсчетной шкале.
Большинство типов современных В. л. снабжаются
воздушными, магнитными и реже жидкостными успо-
коителями (апериодич. весы) и именованными шка-
лами, дающими в весовых единицах разность между
массой взвешиваемого тела и массой гирь. При при-
менении В. л. с именованными шкалами отпадает
необходимость в определении цены деления при взве-
шивании разных по массе грузов. Для ускорения
взвешивания (повышения быстродействия весов) уве-
личивают область непосредственных измерений по
шкале. Для этого применяют проекционные шкалы,
пользуясь к-рыми можно расширить диапазон непо-
средственных измерений при малом угле отклонения
коромысла. Применяя проекционные шкалы, можно
обычно обойтись без рейтерных шкал. У некоторых
микроаналитич. весов, имеющих сниженный примерно
в 10 раз угол отклонения коромысла, для отсчета
применяют также коллиматорное устройство, при
к-ром не изменяются нулевые показания при смеще-
ниях коромысла. Эти весы менее удобны и производи-
тельны, но более устойчивы к влиянию внешних
воздействий. Дальнейшее увеличение производитель-
ности достигается при применении весов с успокоите-
лями, проекционной шкалой и встроенными гирями.
Весы аналитич. группы оборудуются встроенными
гирями до 999 мг или до предельной нагрузки. Приме-
нение встроенных гирь не только упрощает взвешива-
ние и увеличивает быстродействие весов, но и способ-
ствует также повышению точности (уменьшается
износ гирь и исключается влияние воздушных пото-
ков, возникающих при открывании и закрывании
дверок). Наложение встроенных гирь производится
механизмом, рукоятки к-рого связаны с. цифровым
отсчетным устройством, расположенным рядом с экра-
ном, на к-ром проектируется изображение шкалы.
Это устройство с встроенными гирямв до полной
нагрузки дает обычно 4 знака, остальные отсчиты-
ваются по шкале. В. л., имеющие встроенные гири до
полной нагрузки, могут быть как равноплечие с двумя
или одной чашками, так и одноплечие. Равноплечие
двухчашечные В. л. со встроенными гирями позво-
ляют уравновешивать тару материалом одинаковой
с ней плотности, снижая этим погрешности от влияния
аэростатич. сил.
Равноплечие одночагаечные весы (рис. 1) отличаются тем,
что к одной из концевых призм коромысла 1 вместо чашки под-
вешена серьга с приспособлениями 2 для наложения встроен-
ных гирь з. Гири накладываются механизмом 7. Весы снаб-
жены воздушным успокоителем 4, проекционной шкалой 5
и трехпозициоц^ым арретиром 6. Отсутствие второй чашки
позволяет повернуть коромысло так, что чашка обращена
к оператору, и отгородить перегородкой переднюю, рабочую
часть витрины.
Аналогичная компоновка используется и в одноплечих
весах (рис. 2), в к-рых на заднем конце коромысла 1, опира-
ющегося на призму 2, закреплен противовес .5, уравновеши-
вающий набор гирь, подвешенных к грузоприемной призме з.
Гири помещены на стержни 4 подвески чашки. Для отсчета
применяется оптич. устройство, проектирующее на экран в
изображение шкалы 8. Весы снабжены успокоителем 7. После
наложения взвешиваемого груза гири снимаются до тех пор.
пока коромысло не придет в положение, близкое к исходному.
Такие В. л. работают всегда при предельной нагрузке. Недостат-
ками одноплечих весов являются большее влияние аэростатич.
сил при изменении плотности окружающего воздуха, большее
изменение цены деления при изменениях темп-ры и повышен-
ный износ призмы и подушек. Их достоинства — исключение
погрешности от неравноплечести и простота регулировки
(отсутствует влияние нагрузки на цену деления).
В. л. со встроенными гирями до полной нагрузки снабжаются
трехпозиционным арретиром: в положении «Закрыто» арретир
отделяет призмы от подушек; при установке арретира на пози-
541
ВЕСЫ ЛАБОРАТОРНЫЕ
542
цию «Полуоткрыто» призмы и подушки находятся в рабочем
положении, но колебание коромысла ограничено малым углом.
В этой позиции производится наложение встроенных гирь;
Вис. 1. Равноплечие одночашечные лабораторные весы:
а —схема; б — общий вид (пояснения в тексте).
ограниченный угол колебаний снижает износ призм и подушек.
Благодаря трехпозиционному арретиру отпадает необходи-
мость в предварительном взвешивании на менее точных весах.
Окончательное взвешивание производится при нахождении
арретира в позиции «Открыто».
Увеличение производительности и повышение точности
достигаются также применением витрин с выдвижными двер-
цами, к-рые не вызывают резких воздушных течений при от-
крывании и закрывании, и манипуляторов для наложения
и снятия навесок, не открывая дверец витрины.
Современные быстродействующие весы с успокои-
телями, проекционными шкалами и встроенными
Рис. 3. Крутильноравноплечие лабораторные весы
(пояснения в тексте).
гирями в 8—10 раз производительнее простых равно-
плечих В., обладают лучшими метрологич. характе-
ристиками, удобны для работы и менее чувствительны
к внешним воздействиям. Дальнейшее значительное
увеличение предела непосредственных измерений до-
стигается применением крутильноравноплечих В. л.
(рис. 3). Весы основаны на определении веса по углу
закручивания упругой металлич. нити, пропорцио-
нальному весу груза. Эта схема применяется, напр.,
в конструкции пробирных весов с предельной нагруз-
кой 2 а, ценой деления проекционной шкалы 0,01 мг
и пределом показаний по этой шкале 2,5 мг.
Равноплечее коромысло 1 подвешено на упругой металлич.
нити 2, натянутой между передней и задней втулками з и 4,
к-рые связаны соответственно с рукоятками 5 и 6. Перед нача-
лом взвешивания рукоятку 5 вращают, пока проекция начала
(нуль) проекционной шкалы 7 не совпадает с нулевой отметкой
на матовом экране 8. Затем коромысло устанавливается в исход-
ное положение равновесия вращением рукоятки в, причем
изображение отметки, отражаемой зеркалом 9, укрепленным
на коромысле, также совпадает с нулевой отметкой на экране.
Изменения нагрузки в пределах до 2,5 мг определяются по
углу закручивания нити, для чего рукоятку <5 вращают, пока
коромысло не вернется в исходное положение. Обе рукоятки
снабжены верньерами для точной установки. Весы имеют маг-
нитный успокоитель и арретир. Недостатком весов, у к-рых
опорой коромысла служит упругая нить, является малая
предельная нагрузка.
В конструкциях микровесов используются в основ-
ном схемы крутильных и торзионных весов (см. табл. 2).
По принципу действия крутильные весы отличаются
от описанных выше крутильноравноплечих тем, что
в них обычно отсутствует вторая чашка и, следова-
тельно, предельная нагрузка равна пределу показа-
ния по отсчетной шкале.
У торзионных весов (рис. 4) коромысло 1 закреплено на
горизонтальной оси 2, опирающейся на прецизионные под-
шипники. К коромыелу подвешивается чашечка 8 для взвешп-
Рис. 4. Торзионные весы: а — схема; б — общий вид
(пояснения в тексте).
ваемоготела. Спиральная пружина з, один конец к-рой закреп-
лен на оси 2, а второй иа рукоятке 4, служит для возвращения
коромысла в исходное положение равновесия. Угол закручива-
ния пружины пропорционален весу груза и отсчитывается
посредством отсчетной стрелки 5, связанной с рукояткой 4У
по шкале в, проградуированной в единицах массы. Нулевая
стрелка 7, жестко закрепленная па оеи 2, служит для контроля
установки коромысла в исходное положение равновесия. На
оси 2 укреплен также флажок 9 магнитного успокоителя.
Торзионные весы менее чувствительны, чем крутиль-
ные, но имеют большие предельные нагрузки. Показа-
ния крутильных и торзионных микровесов зависят
от местного значения ускорения силы тяжести и тем-
пературы. Для введения поправок к микровесам при-
лагают контрольный грузик, соответствующий пре-
дельной нагрузке. Значение поправки в любой точке
шкалы подсчитывается по закону прямой пропорцио-
нальности (ДР = ДРмаис . Р/Рмакс_, где ДР — иско-
мая поправка; АРмакс — поправка при предельной на-
грузке, найденная с помощью контрольного грузика;
Р — вес взвешиваемого груза и Рманс — предель-
ная нагрузка весов).
543
ВЕСЫ ЛАБОРАТОРНЫЕ
544
По схеме равноплечих весов с циферблатным от-
счетным устройством и квадрантных весов строятся
быстродействующие В. л., характеристика к-рых
приведена в табл. 3.
В равноплечих весах с циферблатным отсчетным устрой-
ством (рис. 5) коромысло 1 жестко связано с вертикальным
Рис. 5. Равноплечие лабораторные весы с циферблатным
отсчетным устройством: а — схема; б — общий вид (по-
яснения в тексте).
отсчитываются по шкале -5. У таких весов с предельной на-
грузкой t кг и ценой деления 50 мг предел показаний по
шкале равен 10 г, против 0,5—1 а для аналогичных равно-
плечих весов с отсчетной шкалой.
В квадрантных весах (рис. 6) используются только встроен-
ные гири 1 Уравновешивание нагрузки в пределах показаний
Рис. б. Квадрантные лабораторные весы: а — схема;
б — общий вид (пояснения в тексте).
по шкале достигается за счет отклонения квадранта 2, несущего
противовес 3, лепесток магнитного успокоителя 4 и проекцион-
ную шкалу 5, изображение к-рой проектируется на экран в.
Взвешиваемый предмет помещают на чашку 7. Такие весы
снабжены устройством для уравновешивания тары в широких
пределах (100 а у весов с предельной нагрузкой 1 кг).
Гири. При работе на весах сила или момент силы
тяжести взвешиваемого тела уравновешивается из-
вестной силой (моментом), создаваемым посредством
гирь или проградуированного силоизмерителыюго
устройства (пружины, упругой нити, противовеса).
Характеристики гирь, применяемых для работы
на В. л., приведены в табл. 4.
Образцовые гири I разряда служат для проверки
аналитич. и техвич. гирь 1-го класса. Аналитич. и тех-
нич. гири 1- и 2-го классов применяются для работы
на весах аналитич. группы и на технич. весах 1- и
2-го классов соответственно. Для крутильных микрове-
сов применяются гири с номинальным значением мас-
сы от 0,1 до 5 мг, проверенные с погрешностью от
±0,0002 мг до ±0,002 мг соответственно. Для В. л.
аналитич. группы нужны гири разных классов повы-
шенной точности, применяемые без введения попра-
вок. Примером требований к современным аналитич.
гирям могут служить данные, приведенные в табл. 5.
Таблица 4. Характеристики гирь для лабораторных весов
Допустимые погрешности, мг
Номинальные
значения массы
образцо-
вые гири
I раз-
ряда
аналити-
ческие
гири
технич.
гири
1-го клас-
са
технич.
гири
2 -го клас-
са
1 кг.................
500 г...............
200 г...............
100 г...............
50 г................
20 г...............
Юг............
5 г .........
2 и 1 а.......
500, 200, 100 и 50 мг
20 мг.........
10 мг.........
5 мг..........
2 мг .........
1 мг .........
200
100
50
25
20
15
10
6
4
2
2
1
0 Л
0,2
Таблица 5. Допустимые погрешности для аналитических гирь
повышенной ТОЧНОСТИ (мг)
Номинальные значения массы гирь Классы гирь
1 2 3
20 г 0,04 0,10 0,20
10 и 5 г 0,03 0,08 0,15
3,2 и 1 г 0,02 0,05 0,10
500, 300, 200 и 100 мг . . . 0,01 0,25 0,05
50, 30 и 20 мг 0,006 0,015 0,03
10, 5, 3, 2 и 1 мг 0,005 0,001 0,02
Гири выпускаются наборами (разновесами). В на-
бор входят гири одной весовой группы: килограммо-
вые (от 1 до 20 кг); граммовые (от 1 до 500 г) и милли-
граммовые (от 1 до 500 мг). В граммовые аналитич.
наборы входят гири от 1 до 100 (или 200 г), а в грам-
мовые микроаналитические наборы — массой от 1 до
20 г. Миллиграммовые наборы аналитич. гирь уком-
плектовываются рейтерами. Гири, кратные 1 или 2 еди-
ницам каждой декады (напр., единицам, десяткам и
сотням миллиграмм, единицам и десяткам грамм
и т. д.), входят в набор в удвоенном количестве (напр.,
1; 2; 2 и 5 г). Более удобны наборы аналитич. гирь
в составе 1; 2; 3 и 5 единиц каждой декады, в к-рых
гири одинакового номинала не повторяются, что дает
уменьшение погрешностей (за счет уменьшения числа
гирь, участвующих во взвешивании) и увеличение
производительности.
Материал, из к-рого изготовляются аналитич.
гири, должен быть немагнитным, обладать высокими
антикоррозийными качествами и износоустойчивостью.
Лучшие результаты дает нержавеющая немагнитная
сталь (напр., сталь IX 18Н9Т по ГОСТ 5632—51),
нержавеющая сталь с содержанием 25% Сг и 20%
Ni и сплав из 80% Ni и 20% Сг. Лучшим покрытием
для латунных гирь является родиевое и платиновое.
Гири, покрытые Ni и Сг по Ni, подвержены влияниям
магнитных полей. Золотое покрытие не обеспечивает
механич. и химич. стойкости. Подгонку гирь по массе
следует производить тем же материалом, из к-рого
сделаны гири. При необходимости применения мате-
риала более высокой плотности рекомендуется тантал;
свинец вызывает значительную нестабильность гирь.
Кварцевые аналитич. гири легко приобретают элект-
ростатические заряды, могущие значительно исказить
показания весов. При применении кварцевых и пла-
тиновых гирь нужно помнить, что они значительно
отличаются от стальных и латунных по плотности.
Взвешивание. При технических анализах и обычных
микроанализах пользуются прямым взвешиванием,
при к-ром масса тела принимается равной показаниям
весов, инструментальные погрешности к-рых не учи-
545
ВЕСЫ ЛАБОРАТОРНЫЕ— ВЕСЫ ЭЛЕКТРОННЫЕ
546
тываются. При лабораторных работах высокой точ-
ности (полумикроанализ, микроанализ и др.) приме-
няют методы точного взвешивания, позволяющие
исключить влияние основного источника инструмен-
тальной погрешности весов — неравноплечести коро-
мысла.
Метод двойного взвешивания (метод Гаус-
са) состоит в повторном прямом взвешивании после переста-
новки тела и гирь с чашки на чашку. Значение массы М опре-
деляется как М = YMi • или, т. к. — Мг —* О, можно
принять М — х/2 (Mi 4- М^), где и Мз результаты двух
прямых взвешиваний.
Метод замещения (метод Барда) заключается
в том, что тело после уравновешивания его тарным грузом
снимается с весов, и на чашку помещаются гири так, чтобы
привести весы к положению исходного равновесия. Значение
массы взвешиваемого тела определяется как алгебраич. сумма
массы гирь и показаний по шкале весов. Метод замещения
(метод Д. Й. Менделеева) состоит в том, что на одну из чашек
помещают гири в количестве, соответствующем предельной на-
грузке весов, и уравновешивают весы тарным грузом. Взвеши-
ваемое тело помещают на чашку с гирями и снимают такое
количество гирь, чтобы весы пришли в положение исходного
равновесия. Значение массы взвешиваемого тела определяется
как алгебраич. сумма массы снятых с чашки гирь и показа-
ний по шкале весов. Этот метод положен в основу одноплечих
весов.
Помимо неравноплечести коромысла, на точности
результатов взвешивания сказываются погрешности
гирь, влияние аэростатич. и электростатич. сил,
погрешности шкал весов, влияние температуры и
влажности воздуха, внешних магнитных полей и т. п.
Погрешности подгонки гирь в значительной степени
исключаются введением в расчет действительных
значений массы, указанных в свидетельствах; однако
удобнее и производительнее применять гири, подог-
нанные так, что их погрешность была меньше, чем до-
пустимая погрешность взвешивания (см. табл. 5).
Влияние аэростатич. сил возникает вследствие нера-
венства объемов взвешиваемого тела и гирь. Погреш-
ность по этой причине может быть выражена согласно
закону Архимеда как А — у (t/pr — х/рт), где у —
плотность воздуха, рг — принятая плотность мате-
риала гирь при поверке, а рт — плотность взвешивае-
мого тела. Для исключения этой погрешности в ре-
зультаты взвешивания вводят поправку (табл. 6).
Таблица 6. Значения поправок на влияние
аэростатических сил
Плотность взвеши- ваемого тела, г/мл Поправка в мг на 1 г навески
бронзовые гири; при- нятая ПЛОТ- НОСТЬ 8,4 г/мл стальные гири; при- нятая плот- ность 8,0 г/мл стальные гири; при- нятая плотность 7,8 г/мл алюминие- вые гири; принятая плотность 2,65 г/мл
22,0 20,0 18,0 16,0 14,0 12,0 10,0 8,4 8,0 7,8 6,0 4,0 3,0 2,65 2,5 2,0 1,5 1,0 0,5 0,2 — 0,0883 — 0,0829 — 0,0762 — 0,0879 — 0,0571 — 0,0429 -0,0229 0,0000 4-0,0007 --0,0090 4-0,0571 4-0,1570 — 0,2570 4-0,2945 40,3370 4-0,4570 40,6570 41,080 4-2,260 4-5,860 — 0,0955 — 0,0900 — 0,0833 — 0,0750 -0,0842 — 0,0500 — 0,0300 — 0,0071 0,0000 4-0,0029 --0,0500 4-0,1500 4-0,2500 4-0,3028 4-0,3300 4-0,4500 4-0,8500 --1.050 --2,250 4-5,850 — 0,0983 — 0,0929 — 0,0882 — 0,0789 — 0,0872 — 0,0529 — 0,0328 — 0,0099 — 0,0029 0,0000 + 0,0471 + 0,1470 — 0,2470 --0,3000 — 0,3270 --0,4470 — 0,6470 +1,055 + 2,260 --5,850 — 0,3983 — 0,3929 — 0,3882 — 0,3779 — 0,3872 — 0,3529 — 0,3329 — 0,3100 — 0,3029 — 0,3000 — 0,2528 -0,1529 — 0,0528 0,0000 + 0,0272 --0,1470 --0,3470 --0,7470 --1,850 + 5,550
Необходимо систематически определять цену де-
ления шкалы весов, пользуясь тщательно проверенной
гирей при различных значениях нагрузки (ненагру-
женные весы, при предельной нагрузке и при 0,1 ее
значения). Небольшие изменения цены деления мо-
гут быть устранены регулятором положения центра
тяжести коромысла; при больших изменениях тре-
буется юстировка весов. При работе на микроана-
литич. весах с рейтерными шкалами рейтер должен
всегда находиться в рабочем положении. Погреш-
ности при пользовании рейтерными шкалами возни-
кают за счет погрешностей самого рейтера, непра-
вильного нанесения или плохой обработки зарубок
шкалы и за счет неправильной посадки рейтера.
Влияние электрич. сил сказывается особенно сильно
при пользовании сосудами из стекла с высоким со-
держанием кремния и при низкой относительной
влажности воздуха и может существенно исказить
результаты микроанализа. Эффективным средством
борьбы с влиянием электростатич. сил является
ионизация воздуха в витрине В. л. с помощью источ-
ника излучения.
Все весы аналитич. группы и особенно микроана-
литич. весьма чувствительны к колебаниям темп-ры,
градиентам темп-ры, потокам воздуха, колебаниям
и вибрациям, магнитным полям и прочим внешним
воздействиям. Все эти причины могут значительно
исказить результаты взвешивания. Поэтому поме-
щения, в к-рых производятся точные взвешивания,
должны быть термостатированы и снабжены установ-
ками для кондиционирования воздуха (для весовых
комнат микрохимических лабораторий суточные ко-
лебания температуры должны лежать в пределах
±0,2°С). Нельзя рекомендовать применение погло-
тителей влаги, устанавливаемых в витрине весов.
Для освещения должны применяться люминесцент-
ные лампы или специальные светильники с теплоот-
водом. Пол и стены помещения не должны подвер-
гаться воздействию ударов и вибраций. Весы следует
устанавливать на тяжелых плитах, снабженных
направленным вниз стержнем с грузом. Хорошие
результаты дает установка ножек столов в коробки,
заполненные слоями свинца, песка и войлока.
Гири и объекты взвешивания должны иметь темп-ру
как можно более близкую к темп-ре в витрине весов,
для чего они выдерживаются там перед взвешива-
нием. Наиболее высокая точность достигается при
относительных (разностных) взвешиваниях, когда
изменение массы или разность двух сравниваемых
масс не превосходит предела показаний по отсчетной
шкале. При больших разностях используются рей-
терная шкала или миллиграммовые гири, что вносит
дополнительные погрешности. Погрешность опре-
деления разности масс при относительных взвеши-
ваниях может быть оценена в 1—2% от верхнего
предела показаний по проекционной шкале или
5—Ю% от верхнего предела показаний по простой
шкале, если разность не превосходит этих пределов.
При пользовании рейтерной шкалой погрешность
достигает 0,3—0,5% от предела показаний по рей-
терной шкале. Воспроизводимость показаний при
повторных взвешиваниях лежит в пределах 0,1 —
0,2 -иг для аналитич. весов; 0,01—0,02 мг для полу-
микроаналитич. весов и 0,001—0,005 -иг для микро-
аналитич. весов. Все эти данные относятся к случаю,
когда условия работы весов являются оптималь-
ными во всех отношениях.
Лит.: Рудо Н. М., Весы, М.—Л., 1957; Щедровиц-
кий С. С., Современные весоизмерительные приборы, М.,
1958; его же, Стандартизация, 1958, № 5, с. 54; его же.
Измерительная техника, 1957, № 8, с. 39; Balances, weights
and precise laboratory weighing, L. 1955 (National Phys. La-
boratory. Notes on applied science, № 7); East W., Chem. —
Ingr. — Techn., 1959, 31, H. 11, S. 725.
С. С. Щедровицкий.
ВЕСЫ ЭЛЕКТРОННЫЕ — весы, у к-рых отсчет
показаний или уравновешивание нагрузки произво-
дится с помощью электронной схемы. В. э. исполь-
зуются гл. обр. для автоматич. регистрации изме-
нений массы навески во времени, напр. при термо-
547
ВЕТИВЕРОВОЕ МАСШ) — ВЕЩЕСТВЕННЫЙ АНАЛИЗ
548
гравиметрия, анализах, исследованиях процессов
сорбции, сушки, диффузии, испарения и радиоактив-
ного распада; для изучения кинетики реакций между
твердыми и газообразными в-вами, а также в случаях,
когда управление весами и отсчет показаний должны
производиться дистанционно. Лабораторные В. э.
выполняются в виде приставок к обычным аналитич.
весам и в виде спец, конструкций, выпускаемых
серийно или изготовляемых индивидуально для про-
ведения определенных исследований. Многочислен-
ные разновидности лабораторных В. э. подразде-
ляются иа 3 типа: с датчиком, с уравновешивающим
устройством и с обратной связью.
В. э. сдатчиком — обычные аналитич. весы, у к-рых
угол отклонения коромысла преобразуется посред-
ством фотоэлектрич. емкостного, индуктивного или
следящего устройства (датчик) в пропорциональ-
ный этому отклонению сигнал, ко-
торый регистрируют самопишущим
прибором (например, электронным
потенциометром). Непрерывная ре-
гистрация показаний производится
только в пределах непосредственных
измерений по отсчетной шкале весов.
Примером В. э . с датчиком являются
весы, изображенные на ри-
сунке. Такие весы выпуска-
ются серийно с пределами
непрерывной записи 10 и
100 л«гпри цене деления диа-
граммной бумаги 0,1 и 1 мг
соответственно. Они могут
снабжаться электропечью
для нагрева навесок по за-
данной программе до 1000°
или 1450°.
В. э. с уравновешиваю-
щим устройством исполь-
зуются там, где примене-
ние гирь затруднено (весы
для работы в вакууме, в
атмосфере агрессивных га-
зов и др.). Примером явля-
ются вакуумные В. э., за-
ключенные в стеклянный
герметичный ко-
жух с трубчаты-
ми выступамидля
подвесок. Один
из выступов находится в
отверстии соленоида, яко-
рем к-рого является магнит-
ный стержень, прикреплен-
ный к подвеске. При про-
хождении тока возникает
сила, втягивающая якорь и
Электронные весы с датчи-
ком, с пределами непрерыв-
ной регистрации 10 или
100 мг п автоматическим на-
ложением встроенных
С(СН3)2СН2СНОНСН3 K2Cr2O.
гирь.
регулируется до возвращения ко-
используемая для уравновешивания нагрузки. Ток
в цепи соленоида
ромысла в исходное положение. Зависимость между
массой навески и током определяется путем граду-
ировки с помощью гирь.
Наиболее совершенными являются В. э. с обратной
связью. В них имеется датчик, дающий сигнал при
отклонении коромысла, и уравновешивающее устрой-
ство автоматически возвращающее его в исходное
положение.
Лит.: Щедровицкий С. С., Современные весоизме-
рительные приборы, М., 1958; Щедровицкий С. С.,
Моисеев Е. М., М ашиицев Е. В., Завод, лаб., 1959,
25, № 1. С. С. Щедровицкий.
ВЕТИВЕРОВОЕ МАСЛО — масло корней вети-
веровой травы (Vetiveria zizanoides), произрастаю-
щей в Индии, Конго, на Цейлоне и Яве. В. м. —
вязкая жидкость от желто-коричневого до темно-
коричневого цвета; плотн. 0,99—1,044; растворимо
в спирте. Отдельные компоненты В. м. (ветиверовый
спирт, ветиверилацетат, ветиверкетов) применяются
для получения душистых в-в.
Ветиверовый спирт — смесь спиртов, выделяе-
мая из ветиверового масла, — вязкая жидкость, содержащая
не менее 80% спиртов; d*- 0,980—1,002; пр 1,5400—1,5117;
кислотное число ок. 2 0; число омыления ок. 10. Из спиртовой
фракции выделены спирты сесквитерпенового ряда общей ф-лы
С16Н24О: первичный спирт, т. кип. 170—172/13 мм; d]8 1,0228;
пр? 1,5255; [а]р =-|-29036/; бициклич. спирт, т. кип. 152 —
154°/12 мм; cl1/ 0,0851; пр 1,5241; третичный бициклич. спирт
(не реагирует с фталевым ангидридом), т. кип. 150—155/12 мм,
d{° 0,9910; пр 1,5185. Кроме спиртов, выделяемых из В. м.,
в пром-сти душистых в-в ветиверовым спиртом наз. также син-
тетич. продукт — 4-циклогексил-4-метилпентанол-2, т. кип.
124—126°/6—7 мм; d}& 0,9629; np 1,5014, получаемый из бен-
зола:
п д»г»
J +СН3СН=СНСН2СОСН3 —
С(СН3)2СН2СОСН3 [н]
(Ni)
С(СН3)2СН2СНОНСН3
С16Н22О — смесь
из
стереоизомерных
нетонной франции
НзС\ /СНз
С
Ветиверплацетат CH3COOCt6H24 — продукт аце-
тилирования спиртов В. м., вязкая желтоватая жидкость;
dji5 0,971—0,994; кислотное число ок. 6; образует прозрачный
раствор в 9 частях 80%-ного спирта. Ветиверилацетат обладает
тонким стойким запахом, напоминающим запах цветов табака
и розы.
Ветиверкетов . _
кетонов (а- и р-ветивопы), выделяемых
В. м. а-В етиверкетон т. пл.
51—51,5°; т. кип. 144—144,5°/4 .м.и;
1,0035; пд 1,5370; [а]д = +238°15'
(с = 6,935, в спирте); запах сладкий,
характерный для В. м. 0-В етивер-
кетон, т. пл. 44—44,5°; т. кип.
141 —142°/2 лш; df° 1,0001: пр 1,5216;
nj-j 1,5309; [ц]р= —38°55' (с = 10,62,
в спирте); по запаху напоминает сти-
ракс (бальзамический запах). Наличие
этих кетонов и определяет оттенки за-
паха В. м. В промышленности душистых
веществ под тем же названием <ветп-
веркетон» известен синтетический про-
дукт, получаемый окислением 4-цикло-
гексил-4-метплпентапола-2 (см. выше,
сн2
с-снй
хсн2
НС-
н3с-с/
Дз
НС.
,СН2
о
ветиверовый спирт):
С(СН3)2СН2СОСН3
бесцветная жидкость, т. кип. 239—240°/760 мм; 96—
100°/4—5 0,9441; пр 1,4880; семикарбазон, т. пл.
156—157°. Н. П. Соловьева.
ВЕЩЕСТВЕННЫЙ АНАЛИЗ — раздел аналити-
ческой химии, разрабатывающий, в отличие от эле-
ментарного (общего, валового) анализа, методы ка-
чественного и количественного определения форм
нахождения элементов (простых и сложных веществ)
в анализируемых материалах. В литературе в настоя-
щее время нет единого взгляда на наименование этого
вида анализа и потому отдельные авторы употреб-
ляют названия фазовый анализ, «химико-минералоги-
ческий» и др. Для определения вещественного состава
анализируемого материала применяются химич., фи-
зико-химич. и физич. методы: напр., избирательное
растворение, минерало-петрографический, магнитный,
гравитационный и др. методы.
Лит.: До ливо-Добровольский В. В., К л и-
м е н к о IO. В., Рациональный анализ руд, Свердловск —
Москва, 1947; Б е р г Л. Г. Скоростной количественный фазо-
вый анализ, М., 1952; ФаинбергС. Ю., Анализ руд цвет-
ных металлов, М.. 1953; К л я ч к о Ю. А., А т л а с о в А. Г.,
Шапиро М. М„ Анализ газов и включений в стали, М.,
1953; Ю in к о С. А., Новые методы минералогического иссле-
дования окисленных руд, М., 1953; Христофоров Б. С.,
в сб.: Методы анализа руд и продуктов цветной металлургии,
№ 5, М., 1959.
549
ВЗАИМНЫЕ СОЛЕВЫЕ СИСТЕМЫ — ВЗРЫВ
550
ВЗАИМНЫЕ СОЛЕВЫЕ СИСТЕМЫ — физико-
химич. системы, в к-рых может идти реакция обмена
между солями без общих ионов, напр. по схеме:
АХ + BY AY + ВХ, где А и В — катионы,
X и Y — анионы, или между металлом М и солью
другого металла АХ по схеме: АХ + Мп А + MX.
Пары солей, формулы к-рых стоят в одной и той же
части ур-ния, наз. взаимными парами.
В. с. с. делятся на безводные (напр., система КС! +
+ NaNO3^ NaCl + KNO3 в расплавленном состоя-
нии) и водные (то же в водном р-ре). Согласно фаз
правилу, эта система в расплавленном состоянии
является тройной, а в водном р-ре — четверной.
Существуют и В. с. с., в к-рых может идти более одной
реакции.
Для графич. изображения состава тройных В. с. с. АХ 4-
4- ВУ AY 4- ВХ применяют след, метод. Состав системы
выражают т. обр., чтобы сумма концентраций катионов а Ъ
была бы равна сумме ” ’ ’ " '
концентраций анионов х 4- у, и при-
равнивают эту сумму постоянной ве-
личине, обычно 100: п 4- Ъ = 100;
х 4~ у — 100. При этом приходится
иметь дело с концентрациями экви-
валентных количеств ионов. Поэтому,
если в состав системы входят соли
с ионами разной валентности, напр.
2NaCl 4- MgSO4 MgCl2 4- Na2SO4,
то для ионов с меньшими валентностя-
ми необходимо брать соответствую-
щие кратные величины: Na2Cl2 4-
4- MgSO* MgCla 4- Na2SO4. Далее
берут квадрат и делят его стороны
на 100 равных частей (рис.). Отло-
жив от вершины BY по горизонтальной стороне х, а по
вертикальной а, получают точки D и Е; тогда отрезок D —
— ВХ = и, a Е — AY — Ь. Проведя через точки D и Е пря-
мые DD' и ЕЕ', параллельные сторонам квадрата, получают
точку F, изображающую состав системы (если дана точка F
на диаграмме и надо определить состав отвечающей ей смеси,
то указанные построения производят в обратном порядке).
Далее на перпендикулярах к квадрату состава откладывают
соответствующие темп-ры начала (или начала и конца) кри-
сталлизации и через полученные т. обр. точки проводят плав-
ные поверхности. Эти поверхности пересекают плоскостями,
параллельными плоскости квадрата; полученные кривые —
изотермы — вместе с линиями пересечения полей проектируют
на плоскость квадрата и получают т. обр. диаграмму состоя-
ния взаимной системы. Обычно ограничиваются изображением
поверхности ликвидуса, т. е. начала кристаллизации.
Если изучают В. с. с. с растворителем (наир., с во-
дой), то строят изотермич. диаграмму растворимости
(см. Растворимость).
Лит.: Курнаков Н. С., Собрание избранных работ,т.2,
Л.—М., 1939; А н о с о в В. Я. и П о г о д и н С. А., Основ-
ные начала физико-химического анализа, М.—Л., 1947; В и и -
торов М. М., Графические расчеты в технологии минераль-
ных веществ, 2 изд., Л., 1954; Бергман А. Г. и Л у ж -
н а я Н. П., Физико-химические основы изучения и использо-
вания соляных месторождений хлорид-сульфатного типа, М.,
1951; 3 д а н о в с к и и А. Б., Л я х о в с к а я Е. И., Шлей-
м о в и ч Р. Э., Справочник экспериментальных данных по
растворимости многокомпонентных водио-солевых систем,
т. 1—2, Л.—М., 1953—54; Бергман А. Г., Е в докимо-
в а К. А., Б о г у ш О. Ф., Список солевых систем (безводных,
исследованных методом термического анализа), Изв. Сектора
физ.-хим. анализа. ИОНХ, АН СССР, 1956, т. 27, с. 419—56.
С. А. Погодин.
ВЗАИМОЗАМЕСТИМОСТИ ЗАКОН — утверж-
дает, что результат фотохимии, реакции зависит только
от количества поглощенной световой энергии (77),
но не от скорости поглощения; сформулирован Р. Бун-
зеном и Г. Роско в 1862. В фотографии означает по-
, стоянство оптической
сек)
г плотности почернения
2 при проявлении в дан-
/ ных условиях незави-
0 • , / симо от освещенности
4------>7 } (Е) и выдержки («) при
~2’ -------- экспонировании, если
-в-5-4-3-2-1 о I 2 л произведение Н = Et
остается постоянным.
Для фотографии, галогеносеребряных слоев В. з.
практически действителен лишь в узком интерва-
ле выдержек. Вне его пределов наблюдается умень-
шение светочувствительности при выдержках больших
или меньших, чем оптимальные. Графически откло-
нения от В. з. обычно выражаются изоопакой, т. е.
кривой lg/7 = / (lg«) или lg/7 = / (IgE), иногда также
IgE = / (Igt) для постоянной оптич. плотности по-
чернения (см. рис.). На изоопаке различают: 7 —
область выполнения В. з. при ультракоротких выдерж-
ках; 2 — область отклонений от В. з. при коротких
выдержках; 3 — оптимум изоопаки; 4 — область от-
клонений от В. з. при длительных выдержках. Иссле-
дования отклонений от В. з. сыграли большую роль
в изучении механизма образования скрытого изобра-
жения.
Лит,: КартужанскийА. Л., Усп. физ. наук, 1953,
51, вып. 2, с. 161. К, В. Вендроаский.
ВЗВЕШИВАНИЕ — см. Весы лабораторные.
ВЗРЫВ — внезапное изменение физич. или химич.
состояния вещества, сопровождающееся крайне бы-
стрым превращением (выделением) энергии. Быстрое
выделение энергии, как правило, приводит к разо-
греву, движению и сжатию продуктов В. и окружаю-
щей среды, возникновению интенсивного скачка
давления, разрушению и разбрасыванию. В окру-
жающей среде образуется и распространяется осо-
бого рода возмущение — ударная волна. Пол-
ное количество выделившейся при В. энергии опре-
деляет масштаб явления, объемы и площади, охва-
ченные разрушением. Концентрация энергии (энер-
гия в единице объема) определяет интенсивность раз-
рушений в очаге В.
При В. исходная потенциальная энергия, как пра-
вило, вначале превращается в энергию нагретых сжа-
тых газов, к-рая, в свою очередь, при расширении
газов переходит в энергию движения, сжатия, ра-
зогрева среды. Часть энергии остается в виде внут-
ренней (тепловой) энергии расширившихся газов.
Основные виды исходной энергии В.: 1) Химиче-
ская энергия (см. далее). 2) Атомная
(я дерна я) энергия — уд. энергия (энергия
на единицу веса) при ядерных В. в 10’—108 раз выше
уд. химич. энергии (см. Ядерпые реакции), 3) Элек т-
рическая энергия — В. может возникнуть
при искровом разряде или быстром разряде через
тонкую проволоку; молния является примером по-
добного рода В. в природе. 4) Кинетическая
энергия движущихся тел — при соударениях
тел, движущихся с большими скоростями, может
внезапно выделиться тепловая энергия, достаточная
для превращения части вещества в нагретый сжатый
газ, что приводит к В. Подобного.типа В. возникают
при падении крупных метеоритов. 5) Энергия
сжатых газов — В. баллонов со сжатыми
газами, паровых котлов (часть энергии — энергия
перегретой жидкости). Примерами являются вулка-
нич. В. 6) Внезапный переход потенциаль-
ной энергии упругих деформаций
в энергию движения среды представляет собой свое-
образный В., протекающий без к.-л. участия сжатых
газов. Большинство землетрясений являются В.
такого типа. Наиболее изученными и имеющими
важнейшее практич. значение являются В., связан-
ные с внезапным выделением химич. энергии, возни-
кающие при весьма быстром химич. превращении
с выделением теплоты и образованием нагретых
сжатых газов. К ним относятся В.: взрывчатых ве-
ществ (ВВ), жидких и твердых топлив, взрывчатых
газовых смесей, к-рые могут возникнуть при опре-
деленной концентрации компонентов пылевоздушных
и пылекислородных смесей. В этой статье рассматри-
ваются именно В. химич. природы и прежде всего ВВ.
Возникновение взрыва. В химич. системах В. может
возникнуть цепным (см. Цепные реакции) или тепло-
вым путем, от удара и трения, от В. другого заряда
(в частности, капсюля-детонатора). Сущность тепло-
551
ВЗРЫВ
552
вого взрыва вскрыта Н. Н. Семеновым и заключается
в том, что при определенных условиях в веществе
нарушается тепловое равновесие: приход тепла реак-
ции становится больше теплоотдачи. В результате
в системе начинается лавинообразное нарастание
скорости реакции и темп-ры вплоть до появления
пламени и возникновения В. Возникновение В. при
ударе связано с появлением микроскопия, местных
разогревов, приводящих к развитию микроочагов
горения. В химич. системах В. обычно возникает
в нек-рой части системы и затем распространяется
на всю систему. При поджигании ВВ сначала возни-
кает медленное горение ВВ. На открытом воздухе
процесс может полностью пройти в виде медленного
горения, без развития местного повышения давления,
без развития В. Если горение проходит в условиях
замкнутого или полузамкнутого объема, то возник-
шее повышение давления может привести к суще-
ственному ускорению горения и к развитию В. Кроме
повышения давления, являющегося основным фак-
тором перехода горения во В., существенным также
является предварительный разогрев горящей систе-
мы. При сильном ударе по В В одновременно возни-
кают очаги разогрева и весьма высокое давление, что
способствует возникновению В. Если местное повы-
шение давления (из-за большой скорости горения)
станет большим, то образуется ударная волна, способ-
ная передавать разложе-
ние от слоя к слою, воз-
никнет детонация — яв-
ление распространения
по В В экзотермич. хи-
мич. реакции, возбуждае-
мой ударной волной.
Рис. 1. Схема детонационной
волны и профиль давления в
ней; I — фронт ударной вол-
ны (начало химич. превра-
щения); II — плоскость, со-
ответствующая окончанию химич. превращения; 1 — давле-
ние в исходном веществе; 2 — давление во фронте ударной
волны; 3 — давление в продуктах детонации сразу после за-
вершения химич. реакции.
Скорость распространения детонационной волны
(скорость детонации) составляет несколько тысяч мет-
ров в секунду (см. Взрывчатые вещества, табл. 2).
Характер распространения детонационного превра-
щения показан на схеме (рис. 1).
В. при детонации ВВ, порохов, твердых топлив
обладает наиболее разрушительным действием, зна-
чительно превосходя в этом отношении В. при бы-
стром горении. Именно детонацию В В используют
всегда, когда необходимо получить В. с наибольшим
механич. эффектом. Поскольку у большинства ВВ
безотказное развитие полноценной детонации из
быстрого горения происходит лишь в особых, зача-
стую довольно специфич. условиях, на практике В.
заряда В В получают, подрывая в нем капсюль-
детонатор, содержащий небольшое количество ини-
циирующего ВВ, детонацию к-рого возбудить легко.
На многих произ-вах образуются значительные
концентрации горючих газов, паров, пыли; смеси их
с воздухом при определенном соотношении компо-
нентов являются взрывчатыми. В. обычно возникает
как местная вспышка (под воздействием пламени,
искры и т. Д.), распространяющаяся затем на весь
объем, и может привести к тяжелым последствиям.
Борьба с возможностью возникновения В. в этих
условиях сводится к интенсивной вентиляции, умень-
шению «пыления», использованию специальной взры-
вобезопасной электроаппаратуры, устранению откры-
того пламени, накаленных тел, искр и пр. Катастро-
фич. В. возникали в угольных шахтах (В. метано-
воздушных смесей и смесей угольной пыли с возду-
хом). Эти В. могут быть, в частности, вызваны взры-
вами зарядов ВВ, используемых для рыхления
(отбойки) угля или породы; последствия и масштабы
В. по этой причине могут быть особенно тяжелыми и
значительными. Чтобы исключить их возникновение,
применяют особые предохранительные ВВ.
Работа взрыва. Различают формы работы В. об-
щего (фугасного) действия и бризантного действия.
К общему действию В. относятся: а) разрушения,
вызываемые ударными волнами и движением среды
на нек-ром расстоянии от очага В., в том числе и раз-
рушения, вызываемые сейсмич. колебаниями; б) выб-
рос грунта при взрывании на выброс; в) разрушение,
рыхление, дробление значительных объемов грунта
при взрывании заряда в грунте; г) образование
полости при В. в пластичном грунте и т. Д. На формы
работы общего действия тратится основная часть
энергии В., способной обратиться в работу. Величина
работы и объем, охваченный разрушениями, опре-
деляются полным количеством выделившейся энергии
Е = се, где с — вес заряда и е — уд. энергия ВВ
(количество энергии, выделяемой на единицу веса).
Значение е для ВВ, используемых на практике,
колеблется в пределах 400—1500 кал)г. Рассчитан-
ное на суммарный вес горючего и окислителя оно
меньше, чем е таких «элементарных» взрывчатых
смесей, как, напр., С + О2 — углерод, пропитанный
жидким кислородом (е = 2060 кал/г). Для практики
важно знать величину энергии единицы объема е.., =
— ре (где р — плотность заряда); объем снаряда,
мины, шпура, как правило, является ограниченным.
В этом отношении мощные В В большой плотности,
с ev = 2200 кал)см3 и выше, превосходят «элемен-
тарные» взрывчатые смеси.
Бризантное действие В. заключается в интенсив-
ном дроблении и деформации тел, непосредственно
примыкающих к заряду, продуктами детонации
высокого давления. Интенсивность дробления и
деформации определяется в основном концентрацией
энергии — энергией единицы объема во фронте дето-
национной волны, к-рая связана с величиной давле-
ния детонации. Как концентрация энергии, так и
давление детонации пропорциональны величине Ро-О2,
где Ро — плотность ВВ и D — скорость детонации.
Приближенно можно полагать давление детонации
/’дет.~14 Ро-О2, ЧТО для мощных ВВ большой плот-
ности дает значение 2,5—3,5 • 105 ат. При отраже-
нии и взаимодействии детонационных волн это дав-
ление несколько возрастает. Следует отметить, что
объем среды, на к-рый распространяется бризантное
действие данной интенсивности, пропорционален объе-
му заряда. Иногда предлагают бризантное действие
В. (и даже вообще разрушающее действие) оценивать
величиной мощности В., т. е. работой, производимой
в единицу времени. Поскольку время В. весьма мало,
то даже при умеренной работе мощность В. полу-
чается огромной. Полагая, в частности, что время
совершения работы равно времени распространения
детонации по заряду, подсчитывают, что мощность,
развиваемая при взрыве шашки тротила весом 400 г,
хравна мощности нескольких десятков крупнейших
ГЭС. В связи с этим нужно отметить, что качественно
мощность В. действительно велика: не существует
к.-л. другого механизма или устройства, к-рые
при том же весе и габаритах могли бы развивать
такую мощность, как В. заряда ВВ. Однако в колич.
отношении такой подход заведомо неправилен: нельзя
отождествлять время совершения работы с временем
распространения детонации.
Закон подобия. Для оценки разрушающего дей-
ствия В. (особенно в случае форм работы общего
действия) пользуются законом подобия при В.,
553
ВЗРЫВ
554
позволяющим переходить от В. одного масштаба
к В. другого масштаба и дающим широкие возмож-
ности моделирования явления В. Закон подобия
устанавливает связь разрушающего действия В.
с величиной выделившейся энергии Е (пропорцио-
нальной весу заряда с) и расстоянием R до очага В.
Наиболее просто формулируются условия подобия
в том случае, когда разрушения определяются вели-
чиной избыточного давления Др (на нек-ром расстоя-
нии от заряда) или скоростью движения средц и.
Согласно закону подобия, Др или и являются одно-
значной функцией величины R — приведенного рас-
стояния: R = (R — в метрах, с — в килограммах).
j/c
Вместо веса заряда легко ввести энергию. Для этого
принято в качестве с подставлять величину тротило-
вого эквивалента сэкв , т. е. такой вес заряда тро-
тила, энергия к-рого равна энергии данного взры-
ва. Приближенно сэкв.= се/етнт<( где е и етнт-—уд.
теплота В. данного ВВ и тротила (ТНТ — тротил,
или тринитротолуол). Обычно принимается етнт.=
= 1000 кал)г (для тротила большой плотности).
Используя тротиловый эквивалент, по существу
вводят своеобразную единицу энергии 1 кг ТНТ—>
—>• 103 ккал; 1 т ТНТ —> 10е ккал; 1 кт ТНТ —
—► 109 ккал. Определив опытным путем значение R,
на к-ром возникают разрушения данного вида и ин-
тенсивности, можно оценить абс. расстояние для
заряда с заданным тротиловым эквивалентом сэкв
(при условии, понятно, что разрушение определяется
значением Др и и), на к-ром возникают определенные
разрушения.
Величина давления ударной волны в функции
приведенного расстояния (от R = 1 до R = 20—50)
может быть оценена по формуле Садовского, согласно
к-рой избыточное давление Др ударной волны, рас-
пространяющейся в свободном воздухе: Др =0,84/7? +
+ 2,7/7?2 + 7,0/7?3. При весьма крупных, мощных В.
ударная волна с Др порядка 0,2—0,3 ат вызывает
значительное разрушение большинства городских
построек; ударная волна с Др порядка 1,0 am вызы-
вает полное разрушение всех зданий, кроме спе-
циальных железобетонных сейсмостойких конструк-
ций. Ориентировочно этот же порядок давления
(1—2 ат) вызывает смертельные поражения живых
организмов (ат — везде избыточная). Избыточное
давление 1 ат соответствует нагрузке iOm/м2. При
отражении ударной волны, взаимодействии ее с пре-
градами, препятствиями или другой ударной волной
величина Др может существенно увеличиться. Если
для сверхмощных взрывов величина безопасного
расстояния оценивается по формуле R = R Vе ,
то для В. средней мощности (отдельные заряды ВВ,
склады средней и малой величины) безопасное рас-
стояние принято рассчитывать по формуле R = к)/с •
В зависимости от условий расположения заряда
принимается к = 1,0—2,0 для разрушения малостой-
ких каменных и деревянных зданий; к = 2,0—4,0 —
для разрушения внутренних перегородок, рам, две-
рей, сараев и пр.; к = 4,0—8,0 — для частичного
повреждения рам, дверей, перегородок и пр. Расстоя-
ние 7? до населенного пункта во всяком случае долж-
но быть больше 10]Ес. Разлет осколков формулой
не учитывается.
Разрушающее действие взрыва в твердых средах.
При В. в сплошной пластичной среде образует-
ся полость, объем которой зависит от веса заряда и
свойств среды (в частности, может превышать объ-
ем заряда в десятки и сотни раз). Многообразная
картина деформаций и разрушений (рис. 2) полу-
чается при действии В. на большинство гор-
ных пород. В центре очага взрыва обычно образует-
ся полость (7?в), из которой вещество среды «вы-
жато» высоким давлением. К полости примыкает
слой «раздавливания» (7?р), в котором среда сплошь
разрушена интенсивной волной сжатия. К слою
раздавливания примыкает
слой «радиальных трещин»
(7?!). В этом слое волна
сжатия уже не может раз-
давить материал среды, но
он начинает двигаться в
направлении распростране-
ния волны; в среде возни-
кают растягивающие на-
пряжения, к-рые и вызы-
вают появление растрески-
вания (известно, что гор-
ные породы значительно
легче разрушаются растя-
жением, чем сжатием). На
значительных расстояниях Рис. 2. Схема разрушающего
от очага взрыва среда не действия взрыва в среде,
разрушается, но в ней
распространяются сейсмич. возмущения —. сложные
колебания, включающие продольные, поперечные,
а также поверхностные волны. Эти колебания могут
вызвать разрушение сооружений, находящихся ‘ в
грунте или на его поверхности. Сейсмически безопас-
ные расстояния могут быть оценены по формуле:
R = кс Vс’ где кс зависит от свойств грунта.
Разрушение среды растягивающими усилиями мо-
жет быть весьма существенно увеличено, если свобод-
ная поверхность сре-
ды расположена ближе
нек-рой границы от цен-
тра В. При отражении
от свободной поверхно-
сти волна сжатия транс-
формируется в волну
растяжения; если волна
Рис. 3. Схема разрушающего
действия взрыва вблизи от сво-
бодной поверхности.
достаточно интенсивна,
то это приводит к раз-
рушению среды в зна-
чительном объеме, имею-
щем форму перевернуто-
го конуса (рис. 3). Если
расстояние от заряда до
свободной поверхности
W еще меньше, то большая часть разрушенной среды
выбрасывается при В. наружу •— вверх и в стороны;
образуется воронка выброса (рис. 4). Как взрывание
для дробления (рыхления), так и взрывание на вы-
брос широко используется в практике горного дела.
Заряд (в кг), необходимый для выброса, обычно рас-
считывают по формуле, представляющей собой ча-
стный вывод закона подобия при В.; с = к W3f (п)'.
555
ВЗРЫВООПАСНЫЕ ВЕЩЕСТВА
556
[/ (п) — ОЛ + 0,6 п3], где к — зависит от свойств
среды и / (в) •— от вида образуемой воронки (от
соотношения ее радиуса г к W; если r/W — п— 1,
то / (п) = 1). Значение к колеблется от 1,0 для сла-
бых грунтов до 2,0—2,5 для самых прочных грани-
тов, базальтов и пр. Заряд рыхления может быть
рассчитан по формуле с = 1/3kW:>.
Действие В. на среду может быть изменено и уси-
лено разными способами. Если в среде заложено
несколько зарядов, то, взрывая их в соответствую-
щим образом выбранные моменты времени, можно
управлять разрушающим действием В. и, в частности,
получать направленный выброс, при к-ром отброс
среды происходит преимущественно в заданном на-
правлении. Весьма сильной формой направленного
взрыва является кумулятивное действие, получае-
мое от зарядов, снабженных специальной выемкой
с тонкой металлич. облицовкой. Образующаяся при
взрыве кумулятивного заряда кумулятивная струя
обладает сильнейшим пробивным действием.
Экспериментальные исследования в области В.
Взрывные процессы протекают при больших давле-
ниях и высоких темп-pax, когда существенно изме-
няются привычные свойства вещества. В связи с этим
исследования в этой области связаны с значитель-
ными трудностями. Давление при детонации мощ-
ных ВВ большой плотности достигает сотен тысяч
атмосфер; при отражении детонационных волн и их
столкновении величина давления дополнительно воз-
растает. В нек-рых специальных условиях, в част-
ности при формировании кумулятивной струи, дав-
ление может достигать нескольких миллионов атмос-
фер. К этому нужно добавить, что взрывные процессы
весьма кратковременны и параметры, характери-
зующие состояние вещества, крайне быстро меняются.
Так, в частности, время химич. превращения во
фронте детонационной волны у мощных чувствитель-
ных ВВ составляет порядка 10-7 сек и меньше. Для
исследования В. используются зеркальные фоторе-
гистры и лупы времени со значительным разреше-
нием во времени, искровые и теневые устройства,
спектроскопии, методы исследования, осциллографы
с датчиками, в том числе осциллографы с высоким
разрешением во времени, электронно-оптич. преобра-
зователи, импульсная рентгеновская съемка и т. д.
Лит.: СемеиовН. И., Усп. физ. паук, 1940, 23, вып. 3
и 24, вып. 4; Садовский М. А., Механическое действие
воздушных ударных волн, в сб.: Физика взрыва, 1952, №1;
Покровский Г. И., Ф е д о р о в И. С.. Действие уда-
ра и взрыва в деформируемых средах, И., 1957;КоулР.,
Подводные взрывы, пер. с англ., М., 1950; Седов Л. И.,
Методы подобия и размерности в механике, 4 изд., М., 1957;
Г> а у м Ф. А., Станюкович К. П Шехтер Б. И.,
Физика взрыва, М., 1959; Андреев К. К., Термическое раз-
ложение и горение взрывчатых веществ, М.—Л., 1957.
А. Ф. Беляев.
ВЗРЫВООПАСНЫЕ ВЕЩЕСТВА (В. в.) —
соединения или смеси, к-рые в условиях технология,
процесса, применения, хранения или транспорти-
ровки способны к быстрому самораспространяюще-
муся химич. превращению (горению или взрыву).
Отличительная особенность В. в. от собственно
ечрывчатых веществ состоит в том, что для химич.
превращения взрывчатых в-в и порохов не требуется
присутствия кислорода воздуха, в то время как для
большинства В. в. присутствие кислорода воздуха
обязательно (исключение составляет, напр., ацетилен
или жидкая синильная к-та, к-рые под действием
соответствующего импульса способны к взрывчатому
саморазложению). В. в. могут быть в газообразном
(горючие газы и газовые смеси), жидком (легко
воспламеняющиеся и горючие жидкости, пары кото-
рых способны образовывать взрывоопасные смеси
с воздухом) и твердом состоянии — диспергирован-
ном, мелкораздробленном (промышленные органич. и
неорганич. пыли, промышленные порошки, металлич.
пыль, волокнистые материалы и т. и.).
Горючие газы и газовые смеси. Взрывоопасность
газов характеризуется: концентрационными преде-
лами взрываемости (границами самовоспламенения)
в воздухе, выражаемыми обычно в процентах по
объему, темп-рой самовоспламенения (Тс), скоростью
распространения горения, или т. н. нормальной
скоростью горения. Концентрационные пределы взры-
ваемости в воздухе нек-рых газов и газовых смесей
приведены в табл. 1.
Таблица 1
Наименование газов
Пределы взрываемости
в воздухе, об. %
нижний j верхний
Аммиак.....................
Ацетилен...................
Водород....................
Метан......................
Окись углерода.............
Пропан.....................
Сероводород ...............
Смеси состава (в % по объему):
Н2 СО СН4
.50 50 — ............
— 50 50 ............
33,3 33,3 33,3 ............
15,50
2,50
4,00
5,00
12,50
2,37
4,30
27.00
80,00
74,00
15,00
74,20
9,50
45,50
6,05
7,70
5,70
71,8
22,8
29,9
Тс горючих газов и газовых смесей определяют пу-
тем впуска холодной смеси газов в эвакуированный
нагретый сосуд, раздельным нагреванием компонен-
тов смеси с их последующим смешением в потоке
внутри нагретого сосуда и адиабатич. сжатием зара-
нее заготовленной смеси. Значения Тс находятся
в зависимости от состава смеси (низшее значение Та
соответствует стехиометрии, смеси с кислородом
воздуха), объема сосуда, в к-ром ведется определение
(с увеличением объема значения Тс уменьшаются
вследствие сокращения удельной поверхности тепло-
отдачи в окружающую среду), давления и др. условий
(в том числе и от метода определения). Максималь-
ные скорости распространения горения (или взрыва)
газов в смеси с воздухом приведены в табл. 2.
Таблица 2
Наименование газов Максим, скорость горения, см/сек Содержание го- рючего газа в смеси с возду- хом, %
Водород 267,0 42.0
Ацетилен 131,0 10,0
Этилен 63,0 7,0
Пропилен 43,5 4,8
Метан 37,0 10,5
При распространении взрыва в трубах скорости примерно
удваиваются.
Легковоспламеняющиеся и горючие жидкости.
Взрывоопасность паров жидких горючих характе-
ризуется темп-рой вспышки паров (Тв) (см. Вспышки
температура), пределами взрываемости паров в воз-
духе, темп-рой самовоспламенения (Тс) паров жид-
костей в воздухе (см. Самовоспламенение) и скоростью
распространения горения или взрыва. Тв опреде-
ляется в закрытом или открытом тигле (ГОСТ 6356—52
и 4333—48 соответственно); определения в открытом
тигле дают менее точные результаты. К легковоспла-
меняющимся относятся жидкости с Тв 45° и ниже,
а к горючим — выше 45°. Противопожарными нор-
мами, принятыми в СССР, установлены две катего-
557
ВЗРЫВООПАСНЫЕ ВЕЩЕСТВА
558
рии технология, процессов с применением взрыво-
опасных жидкостей: категория А — процессы с при-
менением жидкостей с 7°в28° и ниже, категория Б —
процессы с применением жидкостей с Тв от 28 до
120°. Различными ведомственными технич. условиями
и правилами перевозки опасных грузов взрывоопас-
ность паров жидкостей оценивается различно: так,
по правилам Министерства путей сообщения, к взры-
воопасным жидкостям относят вещества с Тв от 28°
до 45°, а Министерства морского флота — от 28° до
65°.
По решению Женевского совещания экспертов
(1956) воспламеняющиеся жидкости делятся на две
группы: с Тв ниже 23° в закрытом или 26,6°воткрытом
сосуде и с 7’в от 23° до 60,5° в закрытом или от 26<2°
до 65,6° в открытом сосуде. Вообще же следует от-
нести к категории взрывоопасных жидкости с Тв
до 65° и технологии, операции, связанные с нагревом
жидкостей выше темп-p вспышки их паров.
Пределы взрываемости паров жидкости в воздухе
могут быть выражены темп-рой жидкости (темп-рные
пределы) либо концентрацией ее паров (концентра-
ционные пределы). Для оценки взрывоопасности
жидкостей в закрытых емкостях и аппаратах реко-
мендуется пользоваться темп-рными пределами, а в ус-
ловиях помещений и на воздухе, где могут образо-
ваться концентрации паров в ненасыщенном состоя-
нии, необходимо знать концентрационные пределы.
Нижним темп-рным пределом взрываемости наз.
наинизшая темп-pa жидкости, при к-рой насыщенные
пары ее с воздухом в замкнутом объеме образуют
смесь, способную воспламениться при поднесении
к ней источника воспламенения. Концентрация па-
ров при нижнем темп-рном пределе наз. нижним
концентрационным пределом взрываемости. Верх-
ним темп-рным пределом взрываемости наз. наивыс-
шая темп-pa жидкости, при к-рой насыщенные пары
се с воздухом в замкнутом объеме образуют смесь,
способную воспламениться при поднесении к ней
источника воспламенения. При более высокой темп-ре
жидкости образуется смесь насыщенных паров с воз-
духом, не способная гореть. Концентрация паров
при верхнем темп-рном пределе наз. верхним кон-
центрационным пределом взрываемости. Темп-рные
пределы взрываемости могут быть пересчитаны в кон-
центрационные по след, формулам: = P-JP 100%;
А2 = Р2)Р Ю0%, где Кг и К2 — нижний и верхний
концентрационные пределы, в процентах по объему;
Рг и Р2 — давление паров при темп-pax, соответ-
ствующих нижнему или верхнему темп-рным преде-
лам; Р — атмосферное давление (фактическое) при
определении темп-рных пределов.
Наиболее взрывоопасными являются жидкости,
пары к-рых имеют широкие пределы взрываемости
(напр., сероуглерод), а также с нижним пределом
взрываемости в 1% и менее по объему. Пределы взры-
ваемости паров нек-рых горючих жидкостей приведены
в табл. 3.
Тс паров жидкостей в воздухе для практич. целей
может быть определена различными методами. Наи-
более простой из них — метод капли (рис. 1), заклю-
чающийся в том, что в щелевую печь, нагретую на
100—150° выше ожидаемой Тс, быстро вводится
пипеткой через центральное отверстие в верхней
части испытуемая жидкость. Наблюдение за воспла-
менением ведется через вертикальную щель. За Тс
принимается наинизшая темп-pa реакционной зоны
печи, при к-рой еще наблюдается воспламенение,
причем при определении Тс рекомендуется фикси-
ровать индукционный период (время запаздывания).
В табл. 4 приводятся значения Тс и время запазды-
вания, к-рыми руководствуются при классификации
Рис. 1. Прибор для определения Тс. паров жидкостей в
воздухе методом «капли»: 1 — щелевая печь; 2 — тер-
мопара; 3 — кварцевый стакан; 4 — нагревательный
элемент; .5 — отверстие для ввода капли.
взрывоопасных смесей (группы А, Б, Г и Д воспла-
меиимости Правил устройства электроустановок Ми-
нистерства электростанций СССР).
Таблица з
Наименование Темп-рные пределы взрываемости на- сыщенных паров в воздухе, °C Концентр ационные пределы взрывае- мости паров в воз- духе, об. %
нижний верхний нижний верхний
Ацетон - 20 + 6 2,6 12,2
Бензол Нитрилакрилов ан - 14 + 12 1,1 6,8
к-та Ксилол (смесь изо- — 5 4- 25 1.85 15,1
меров) + 24 4- 50 0,93 4.5
Сероуглерод .... — 50* 4- 26* 1,0 50,0
Метиловый спирт . . + 39 6,0 34.7
н-Пропиловый спирт 4- 20 4- 53 2.02 13,55
Этиловый спирт . . . + 11 — 40 3,3 18.4
Толуол 0 --30 0,92 5,0 ,
Фурфурол + 60 -- 72 L84 3,4
Этилбензол Диэтиловый (серный) + 18 + 45 0,9 3,9
эфир — 45 + 13 2,74 44,3
* Исчислены из концентрационных пределов.
Таблица 4
Группа вос- п л а мен яе мости Температура самовоспламенения, °C
время запаздыва- ния до 5 сек время запаздыва- ния свыше 5 сек
А более 600 более 450
Б от 450 до 600 от 300 до 450
I’ от 300 до 450 от 175 до 300
д от 150 до 300 от 120 до 175
Максимальные скорости горения горючих паров
в смеси с воздухом и их зависимость от состава пока-
заны в табл. 5 и на рис. 2.
Таблица 5
Наименование Максим, скорость горения, см/сек Содержание горю- чего в смеси с воздухом, %
Сероуглерод 48,5 8,2
Бензол 38,5 3,0
Диэтиловый эфир .... 37,5 4,5
Циклогексан 35.0 2,5
н-Гексан 32,0 2,5
Ацетон 31,8 6,0
Промышленные пыли. Взрывоопасность промыш-
ленных пылей характеризуется нижним концентра-
559
ВЗРЫВЧАТЫЕ ВЕЩЕСТВА
560
ционным пределом взрываемости аэровзвеси (в г/л3)
и темп-рой самовоспламенении Гс аэровзвеси в воз-
Пыль
духе. По примерной
классификации про-
мышленных пылей
к числу взрывоопас-
ных в состоянии аэро-
взвеси относятся пы-
ли с нижним преде-
лом взрываемости до
65 г/л3, с подразде-
лением на два клас-
са: I класс до 15 г/л3,
II класс — от 16 до
65 г/л3 (табл. 6).
Рис. 2. Зависимость ско-
рости горения паровоз-
душных смесей от соста-
3 — диэтиловый эфир;
ва: 1 — сероуглерод; 2 — бензол;
4 — циклогексан; 5 — пеитан; 6 — гексан; 7 — ацетон.
Таблица 6
Нижний пре-
дел взрывае-
мости, г/л3
Серы (серный цвет)................
Нафталина.........................
Элеваторная пшеничная (аспирацион-
ная) ............................
Алюминиевая.......................
Элеваторная просяная (аспирацион-
ная) ............................
Сланцевая ........................
10,0
42,8
50
58,0
Лит.: Хитрив Л. Н., Физика горения и взрыва, М.,
1957; Пост В., Взрывы и горение в газах, пер. с нем., М.,
1952; Легковоспламеняющиеся и горючие жидкости. Справоч-
ник, под общ. ред. Н. А. Тарасова-Агалакова, М., 1956; Год-
же лл о М. Г., Взрывы промышленных пылей и их предупре-
ждение, М., 1952; Нормы и технические условия проектиро-
вания складских предприятий и хозяйств для хранения легко-
воспламеняющихся и горючих жидкостей (НиТУ 108—56),
М., 1956; Противопожарные нормы строительного проектиро-
вания промышленных предприятий и населенных мест(Н 102—
54), М., 1959; Правила устройства электроустановок, [3 изд.],
М.—Л., 1957. М. Г. Годжелло.
ВЗРЫВЧАТЫЕ ВЕЩЕСТВА (ВВ) — химич.
соединения или смеси веществ, способные к быстрому,
самораспространяющемуся превращению, с выделе-
нием большого количества тепла и образованием
газообразных продуктов. Быстрое образование газов,
нагретых до высокой темп-ры за счет теплоты реакции,
приводит к сильному повышению давления. При
последующем расширении сжатых газообразных про-
дуктов реакции их внутренняя энергия переходит
в механич. работу раскалывания, дробления и от-
броса элементов окружающей среды — происходит
взрыв. Взрывчатыми могут быть конденсированные
и газообразные вещества, а также взвеси твердых
или жидких частиц в газах. Практич. применение
имеют конденсированные ВВ, гл. обр. твердые,
а также смеси и коллоидные р-ры твердых и жидких
веществ. В дальнейшем рассматриваются только кон-
денсированные ВВ.
Количество энергии, выделяющейся при взрыве
ВВ, относительно невелико. Даже при взрыве самого
мощного ВВ (атомные и термоядерные взрывы здесь
не рассматриваются) выделяется в 5—6 раз меньше
тепла, чем при сгорании на воздухе равного коли-
чества нефти. Однако, в отличие от обычных топлив,
ВВ для превращения в газообразные продукты не
требуют участия кислорода воздуха. Именно поэтому
объемная концентрация энергии во взрывчатом ве-
ществе очень велика. В сочетании с очень большой
«скоростью химич. превращения это делает ВВ кон-
центрированным источником огромных мощностей
(см. Взрыв).
Формы взрывчатого превращения. Возможны 3 основных
режима реакций, в результате к-рых ВВ превращаются в газы:
1) медленное химич. превращение, 2) горение и 3) детонация.
Характерными для ВВ являются два последних режима, объе-
диняемые обпщм названием — «взрывчатое превращение».
Медленное химич. превращение взрывчатых и иевзрывчатых
веществ протекает в общем одинаково, реакция идет равно-
мерно во всей массе вещества; темп-ра и давление в разлага-
ющемся ВВ близки к имеющимся в окружающей среде. На-
чальная стадия такого превращения, как правило, представ-
ляет собой моиомолекуляриую реакцию. Когда отвод тепла
реакции затруднен, возникает саморазогрев ВВ и медленное
превращение само ускоряется и может перейти в горение и дето-
нацию. Такому переходу благоприятствует также накопление
в ВВ конечных или промежуточных продуктов его распада,
к-рые в большинстве случаев ускоряют реакции разложения.
Характерной чертой самораспростраияющегося взрывчатого
превращения является его макрогетерогенность: в каждый
момент реакция быстро идет лишь в узкой зоне, отделяющей
ее продукты от исходного вещества. За счет передачи энергии
соседним непрореагировавшим слоям ВВ в них возбуждается
интенсивное химнч. превращение. Таким образом, зона реак-
ции распространяется по массе ВВ. Расстояние, иа к-рое пере-
мещается фронт реакции в единицу времени, называют ско-
ростью взрывчатого превращения.
Два основных режима взрывчатого превращения — горе-
ние и детонация — отличаются прежде всего по механизму
передачи энергии реакции от слоя к слою. В случае детонации
существенную роль в передаче энергии реакции играет распро-
странение по ВВ резкого скачка давления — ударной волны;
скорость распространения (скорость детонации) составляет
несколько километров в секунду и практически не зависит от
внешних условий. Благодаря огромной скорости превращения
образующиеся газы не успевают существенно расшириться
за время реакции и в зоне реакции, независимо от наличия
прочной оболочки, возникают очень высокие давления (десятки
и сотни тысяч кг/сл2); по окружающей среде производится силь-
нейший удар, способный вблизи от заряда разрушить самые
прочные материалы. Скорость детонации меняется в нек-рых
пределах с изменением размеров заряда и плотности ВВ. Если
Рис. 1. Зависимость скорости дето-
нации от диаметра заряда.
наименьший размер заряда (напр., диаметр удлиненного ци-
линдра) меньше критич. значения, то ВВ, сжатое во фронте
ударной волны, разбрасывается раньше, чем завершится химич.
реакция (принцип Ю. Б.
Харитона), и устойчивое
распространение детона-
ции невозможно. Критич.
диаметр, величина кото-
рого позволяет оценить
скорость реакции при де-
тонации, составляет для
разных ВВ от долей мм
до десятков см. С уве-
личением диаметра заря-
да выше критического
скорость детонации ра-
стет до нек-рого предела
(рис. 1). Для многих ВВ
в интервале плотностей
заряда, имеющих прак-
тическое значение (1,0—
1,7 г/мл), скорость де-
тонации приблизительно
линейно растет с плот-
ностью.
При горении распространение реакции обеспечивается
теплопередачей от прореагировавшего слоя ВВ к соседнему.
Соответственно скорость перемещения зоны реакции (ско-
рость горения) мала и сильно зависит от внешних условий
(давление, темп-ра и др.). Скорость горения разных ВВ при
атмосферном давлении — от долей мм до десятков см в сек.
Существенное увеличение давления возможно лишь в случае,
когда горение идет в замкнутом или полузамкнутом объеме.
Для многих ВВ, обладающих нек-рой летучестью, прогретый
слой конденсированного ВВ обращается в пары в результате
испарения или слабоэкзотермич. частичного разложения,
а основной этап реакции, связанный с выделением большого
количества теплоты, происходит в газовой фазе. Закономер-
ности горения ВВ в таком случае близки к установленным
для газов. Механизм горения нелетучих ВВ более сложен.
Экзотермич. реакции идут здесь и в конденсированной фазе,
часть ВВ диспергируется с образованием взвеси несгоревших
частиц в продуктах неполного превращения. Зависимость
скорости горения ВВ от давления может быть описана ур-нием
вида: и — а 4- epv, где и — скорость горения, р — давление,
а,в и v — постоянные. Для летучих ВВ в изученном интервале
давлений м близко к 1. Скорость горения растет с увеличением
начальной темп-ры ВВ, причем для разных веществ подъем
темп-ры иа 100° ускоряет горение в 1,2—3,0 раза. Для многих
ВВ при атмосферном давлении зависимость скорости горения
от начальной темп-ры описывается ур-иием: 1/и = А —ВТ0,
где То — темп-ра ВВ, А и В — постоянные.
Одна форма взрывчатого превращения может переходить
в другую. Возможны в нек-рых условиях также промежуточ-
561
ВЗРЫВЧАТЫЕ ВЕЩЕСТВА
562
иые, как правило, неустойчивые режимы, распространяющиеся
с меньшей скоростью, чем детонация. Переходу горения в де-
тонацию благоприятствуют рост давления и темп-ры ВВ, уве-
личение поверхности горения за счет проникновения продуктов
горения в глубь порошкообразного в-ва или турбулизации
фронта горящей жидкости. Легче переходит в детонацию горе-
ние таких ВВ, для к-рых велико ускорение горения при
возрастании давления (du/dp).
Классификация ВВ. По условиям перехода го-
рения в детонацию ВВ делят на три класса: ини-
циирующие — первичные ВВ, бризант-
ные — вторичные ВВ и метательные —
пороха. Инициирующие В В горят в десятки и сотни
раз быстрее других, их горение неустойчиво и быстро
переходит в детонацию уже при атмосферном давле-
нии; горение метательных ВВ не переходит в дето-
нацию даже при давлениях в тысячи атмосфер; бри-
зантные ВВ занимают промежуточное положение.
Различия в условиях перехода горения в детонацию
определяют и области применения ВВ трех указан-
ных классов. Метательные ВВ применяют в режиме
горения для сообщения скорости пулям, снарядам
и ракетам; бризантные ВВ — в режиме детонации
для взрывных работ, а также в снарядах и др. бое-
припасах, а инициирующие ВВ используют для
возбуждения взрывчатого превращения других ве-
ществ. Основное отличие метательных и бризантных
ВВ определяется не химическим составом, а физи-
ческой структурой этих веществ. Устойчивость го-
рения порохов при высоких давлениях определяется
плотностью, газонепроницаемостью и прочностью их
зерен.
По составу ВВ разделяют на две большие группы:
индивидуальные соединения и смеси. Важнейшими
представителями первой группы являются: нитро-
соединения, среди к-рых наибольшее значение
имеют полинитросоединения ароматич. ряда и их
производные, напр. тринитротолуол (тротил), три-
нитрофенол, тринитроксилол, динитронафталин и
др.; нитроамины, напр. циклотриметилентри-
нитроамин (гексоген), циклотетраметилентетранит-
роамин (октоген), тринитрофенилметилнитроамин (тет-
рил), этилендинитроамин (эдна), динитрооксидиэ-
тилнитрамин (дина) и др.; эфиры азотной
кислоты многоатбмных спиртов и углеводов,
напр. пентаэритриттетранитрат (тэн), глицерин-
тринитрат, или нитроглицерин, диэтиленгликольди-
нитрат, нитроцеллюлоза, или пироксилин и др.;
соли азотной кислоты, например нитрат
аммония и др.; соли гремучей кислоты,
напр. гремучая ртуть", соли азотистоводо-
родной кислоты, напр. авид свинца. Известны
также и др. классы взрывчатых соединений, практи-
чески имеющие меньшее значение.
ВВ второй группы — взрывчатые смеси —
часто готовят из соединений, способных окисляться
(горючих), и веществ, содержащих значительные
количества кислорода (окислителей). При взрывчатом
превращении элементы горючего окисляются за счет
кислорода, содержащегося в окислителе. В качестве
компонентов смесей применяют как ВВ, так и в-ва,
к-рые сами по себе невзрывчаты. Важнейшими клас-
сами смесей являются: пороха — баллиститные,
пироксилиновые, смесевые; нитроглицери-
новые ВВ (см. Динамиты) — смеси нитроглице-
рина, б. ч. желатинированного небольшими количе-
ствами нитроцеллюлозы, с азотнокислыми солями и
горючими веществами; аммиачно-селитрен-
ные ВВ, или аммониты, —смеси NH4NO3
с горючими и ВВ; хлоратные и перхло-
ратные ВВ — смеси, напр. хлората калия, пер-
хлоратов калия или аммония с горючими веществами;
смеси и сплавы нитросоедипений и др. ВВ,
напр. сплавы тротила с гексогеном; дымный
порох — смеси селитры, б. ч. калиевой, серы и
угля; оксиликвиты — твердые пористые горючие в-ва,
пропитанные жидким кислородом; жидкие смеси,
например концентрированной азотной к-ты, четырех-
окиси азота или других жидких оки-
слителей с жидкими горючими.
Чувствительность ВВ. Для возбуж-
дения взрывчатого превращения не-
обходимо сообщить ограниченному объ-
ему В В определенное количество энер-
гии, называемое начальным им-
пульсом. Таким импульсом может
служить нагревание, удар, трение, дей-
ствие взрыва другого ВВ и др. Ми-
нимальная величина начального им-
пульса, обеспечивающая возникнове-
ние взрыва, характеризует чувствитель-
ность ВВ, которая должна быть огра-
ничена определенными пределами. ВВ
слишком большой чувствительности
нельзя применять из-за опасности ра-
бот с ними; при весьма малой чувст-
вительности трудно возбудить взрыв
в момент применения. Наибольшей
чувствительностью обладают иницииру-
ющие ВВ, к-рые легко воспламеняются
и детонируют от слабого механич. или
теплового импульса и служат поэтому
для возбуждения взрыва других ВВ.
Чувствительность к тепловому импуль-
су обычно характеризуют температу-
рой, до к-рой нужно нагреть ВВ, что-
бы произошло самовоспламенение (тем-
пература вспышки). Эта температура
зависит не только от свойств ВВ, но
Рис. 2. Конец
для определе-
ния чувстви-
тельности В В
к удару: 1 —
груз; 2 — ро-
ликовый при-
борчик.
и от условий опыта, которые для получения сравни-
мых результатов стандартизированы.
Чувствительность к механич. воздействиям обычно
оценивают, подвергая небольшую навеску ВВ (0,02—
0,05 г) удару падающим грузом на специальном
копре (рис. 2). Мерой чувствительности служит либо
энергия удара, при к-рой получается определенная
частота взрывов, либо частота взрывов при по-
стоянной энергии удара. Применяют и др. методы
испытаний (трение, скользящий удар и др.). Резуль-
таты таких механич. испытаний в сильной мере за-
висят не только от химич. природы вещества, но
и от его физико-механич. свойств, а также от внеш-
них условий, определяющих возникающие в ВВ
напряжения и деформации. В табл. 1 приведены
данные о темп-ре вспышки и чувствительности к удару
нек-рых ВВ.
Таблица 1
Взрывчатое вещество
Процент взрывов
при падении гру-
за в 10 иг с вы-
соты 25 см
Темп-ра
вспышки,
°C
Тротил .....................
Тетрил .....................
Гексоген ...................
Тэн.........................
Аммонит № 6.................
Гремучан ртуть .............
Азид свинца ................
4-8 290
50-60 200
70-80 230
100 215
20-30 280
— 180
— 300
Стойкость ВВ — способность сохранять физич.
и химич. свойства при длительном храпении; зависит
от строения молекул или состава ВВ, степени его
чистоты, состава примесей и от условий хранения
(темп-ра, влажность и др.). Разложение химич.
нестойкого ВВ при хранении в неблагоприятных
условиях может привести к самовоспламенению и
взрыву. Практически химическую стойкость оцени-
вают по времени достижения определенной (обычно
563
ВЗРЫВЧАТЫЕ ВЕЩЕСТВА — ВИЛЬГЕРОДТА РЕАКЦИЯ
564
небольшой) степени разложения при нагревании
навески ВВ в стандартных условиях.
Энергия взрывчатого превращения и действие
взрыва. Максимальная работа, к-рая может быть
произведена взрывом, определяется теплотой взрыв-
чатого превращения, объемом и составом газообраз-
ных продуктов реакции. Эти величины находят рас-
четом либо определяют экспериментально. В по-
следнем случае взрывают заряд ВВ в специальных
бомбах, измеряют количество выделившегося тепла
и давление газов и анализируют после охлаждения
их состав. В продуктах взрыва большинства ВВ
содержится: С02, СО, Н20, N2, NO, 02, Н2 и др. газы,
а во многих случаях—и нек-рое количество конден-
сированных веществ, напр. углерод. В табл. 2 при-
ведены данные, к-рыми обычно характеризуют ВВ.
Таблица 2
Название ВВ Теплота взрыва3, ккал/кг Объем продук- тов взры- ва®, л/кг Скорость детона- ции8, м/сек Обжатие свинцо- вого ци- линдра11, ММ Расшире- ние свин- цовой бомбы, см%
Тротил .... 1000 750 7000 16 285
Тетрил .... 1100 740 7400 18 340
Гексоген . . . 1300 890 8400 22 480
Аммонит № 6 Нитроглице- 1020 900 4500 14-18 370
рин Бездымный 1530 720 8000 •23 520
порох . . . Дымный по- 600—1200 750 -940 — — —
рох 660 280 — — —
а При постоянном объеме. ® При 0” и 1 ат. в При мак-
симальной плотности. г При плотности 1 г/см3.
При оценке механич. эффекта взрыва обычно раз-
личают дробящее действие (бризантность),
проявляющееся в непосредственной близости к за-
ряду, ифугасное действие, проявляющееся
на нек-ром расстоянии от заряда (образование по-
лости в грунте, выброс и т. п.). Дробящее действие
зависит от скорости детонации и плотности ВВ.
В первом приближении для зоны наиболее интенсив-
ного дробящего действия эффект его можно считать
Рис. 3. Определение
дробящего действия
(бризантности) ВВ:
а — перед опытом;
б — свинцовый ци-
линдр , обжатый взры-
вом.
пропорциональным максимально-
му давлению во фронте детона-
ции, которое для конденсирован-
ных В В приближенно определяет-
ся ур-нием: Р = ?D2, где р —
плотность В В и D — скорость де-
тонации. Для практической оценки
дробящего действия (бризантно-
сти) взрывают определенный за-
ряд ВВ (50 г) на свинцовом ци-
лйндре в стандартных условиях
(рис. 3). Уменьшение высоты ци-
линдра после взрыва является
сравнительной мерой дробящего
действия. Фугасное действие,
определяемое энергией взрыва,
обычно оценивают по увеличению
объема канала свинцовой бомбы
стандартных размеров (рис. 4) пос-
ле взрыва в нем небольшой
навески (10 г) ВВ; применя-
ют и другие методы оценки RCOCH3 + HN
дробящего и фугасного дей- ЧСН2-СН2
ствия.
Для возбуждения детонации I Применение высококипящего амина (напр., морфо-
ВВ служат капсюли-детонаторы, лина) позволяет проводить . реакцию в открытом
представляющие собой металлич. или бумажную сосуде. Механизм реакции окончательно не установ-
гильзу, в к-рую запрессованы небольшой (0,05— | лен. Однако опытами с разветвленными кетонами и
0,5 г) заряд инициирующего В В и несколько боль-
ший заряд мощного вторичного ВВ. При действии
луча огня или наколе инициирующее ВВ детони-
рует и вызывает взрыв вторич-
ного заряда, который возбуждает
детонацию массы ВВ, где был
помещен капсюль-детонатор. Дето-
нация одного заряда может вы-
звать взрыв другого заряда не
только при непосредственном
контакте между ними, но и на из-
вестном расстоянии. Это расстоя-
ние зависит от свойств обоих за-
рядов, среды, находящейся меж-
ду ними, и величины первого (т. н.
а б
Рис. 4. Определение
фугасного действия
ВВ всвинцовой бомбе:
а — заряд в бомбе
перед опытом; б —
канал бомбы, расши-
ренный взрывом.
активного) заряда.
ВВ широко применяют в народном хозяйстве
(добыча полезных ископаемых, строительство дорог,
тоннелей, каналов, плотин, водохранилищ и др.)
для снаряжения боеприпасов и метания снарядов,
ракет и т. п.
Произ-во, хранение, перевозка и применение ВВ
требуют спец, мер предосторожности.
Лит.: Андреев К. К. иБеляевА. Ф., Краткий курс
теории взрывчатых веществ, М., 1960; ГорстА. Г., Пороха
и взрывчатые вещества, 2 изд., М., 1957; Я р е м е н к о Н. Е.
и Светлов Б. Я., Теория и технология промышленных
взрывчатых веществ, М., 1957; Андреев К. К.. Термическое
разложение и горение взрывчатых веществ, М.—Л., 1957;
Боуден и Иоффе, Возбуждение и развитие взрыва
в твердых и жидких веществах пер. с англ., М., 1955; Г о п fa-
би и д е р А.И. в Андреев К. К., Антигризутные взрывча-
тые вещества, М,—Л., 1947; Дубнов Л. В., Предохранитель-
ные взрывчатые вещества в горной промышленности. М.—-Л.,
1953; Б а У м Ф. А., Станюкович К. П., Шехте р Б. И.,
Физика взрыва, М., 1959; Буди Яков М. А. [и др.],
Взрывчатые вещества и пороха, М., 1955; Б у б и о в П.Ф., Ини-
циирующие взрывчатые вещества, ч. 1, М., 1940- Андре-
ев К. К., Взрыв и взрывчатые вещества, М., 1956; Единые
правила безопасности при взрывных работах, 2 изд., М., 1958;
Cook М е 1 v i n A., The science of high exsplosives, N. Y. —
L., 1958. A. II. Гольбиндер.
ВИЛЬГЕРОДТА РЕАКЦИЯ — превращение ке-
тонов в амиды карбоновых кислот взаимодействием
с полисульфидом аммония (NH4)2 Sx : RCOCH3 —>
RCH2CONH2. Реакция, первоначально применяв-
шаяся к метиларилкетонам, распространена на дру-
гие алкиларилкетоны, диалкилкетоны, альдегиды,
производные альдегидов и кетонов (оксимы, имины
и т. п.), олефины, ацетилены, спирты, меркаптаны,
тиокетоны, алкилзамещенные ароматич. и гетеро-
циклич. углеводороды и др.:
RCO (СН2)ПСН3 —► R (CH»)n + jCONHa; RCHO -> RCONH2
RCH=CHCH3 -> RCH2CH2CONH2
RC-CCHj -> RCH»CH2CONH3
r2coh-ch3-. r»chconh2
R-CH(SH) -CH2CH3 -> RCH2CH3CONH2
Ar—CH3 —► ArCONH.
Обычно кетон или другое соединение нагревают
с водным р-ром желтого полисульфида аммония
в запаянной трубке при 210—230°. Добавление орга-
нич. растворителя (диоксан, пиридин) позволяет
снизить темп-ру реакции до 150—1бОэ.
Широко используется видоизменение Киндлера —
применение вместо полисульфида аммония смеси
сухого амина с серой. При этом образуются замещен-
ные тиоамиды:
CH2-CHt
\ с
/СН2”СН.-
rch2c-n )о+н2о
» чсн2-сн2
565
ВИЛЬШТЕТТЕРА КАТАЛИЗАТОР — ВИЛЬЯМСОН А СИНТЕЗЫ
566
с кетонами, молекулы к-рых содержат меченый угле-
род, доказано, что В. р. не сопровождается изме-
нением углеродного скелета (звездочкой показан
меченый атом углерода):
CeH5COCH2-CH(CH3)-CH3-> C0H5CH2CH2CH(CH3)CONH2
С0Н5СОСН3 -> CeH5CH2CONH2
Перенос функциональной группы с середины цепи
на ее конец, вероятно, связан с рядом последователь-
ных отщеплений и присоединений элементов серово-
дорода, воды или амина, т. е. с промежуточным обра-
зованием ненасыщенных соединений, напр.:
RCOCH3CH3 -I- RCSCHjCH,, ->• R-CH(SH)-CH2CH3 ->
—с R —СН=СН-CH3s-> RCH3—CH(SH)-CH3 -I-
-> rch2ch=ch2 -> rch2ch2ch2sh —►
—c rch2ch2ch=s —> RCH2CH2CONH2
В. p. применяется для получения арилалифати-
ческих к-т из доступных арилалкилкетонов, полу-
чаемых пр реакции Фриделя—Крафтса. Арилуксусные
к-ты получаются с лучшими выходами, чем их
гомологи:
(NHa)2Sn>
Н3С
-ch2conh2
СНрСООН
Н3С
СО(СН,)3СН3
(NH4)2s„
(CH2)4CONH
Н3С
Чисто алифатич. кетоны дают более низкие выходы,
причем выход тем ниже, чем дальше отстоит карбо-
нильная группа от конца цепи. Стирол и фенилаце-
тилен образуют фенилацетамид с выходами соответ-
ственно 64°/0 и 80%.
Использование В. р. может оказаться выгодным для
промышленного получения нек-рых арилуксусных
к-т, напр. нафтилуксусной кислоты:
СН2СООН
Реакция открыта в 1887 К. Вильгеродтом.
Лит.: Кармак М., Ш п и л ь м а н М. А., в кн.: Органи-
ческие реакции, сб.З под ред. Р. Адамса [и др.], пер.с англ., М.,
1951, с. 88—109; Kaltwasser Н., Chem. Techn., 1957,
9, № 7, 392. £. .If. Голдин.
ВИЛЬШТЕТТЕРА КАТАЛИЗАТОР — см. Гидро-
генизация и дегидрогенизация каталитические.
ВИЛЬЯМСОНА СИНТЕЗЫ — методы получения
симметрии, и несимметрич. простых эфиров.
Синтез простых эфиров из алко-
голятов или фенолятов и галоген-
алкилов: RX + R'OMe —> ROR' + МеХ, где
R — алкил, R' — алкил или арил, X—С1, Вг, J
или OSO2OR; Me=Na, Ag и т. п. Реакционная спо-
собность RX в этой реакции уменьшается в ря-
ду RJ>RBr>RCl, а также для R в ряду тре-
тичный ;> вторичный > первичный. В той же после-
довательности изменяется способность галогеналки-
лов к побочной реакции — образованию олефинов:
Дт Д v R'OMev /
—СН—С—X-------- АС=С\ . Поэтому при син-
II 4
тезах эфиров с третичным радикалом предпочти-
тельно вводить в ’ реакцию алкоголят третичного
спирта, а не соответствующее галогенопроизводное.
Однако синтез алкоголятов из вторичных, третичных,
алициклич. и высших спиртов часто затруднен в связи
с их малой растворимостью в исходном спирте. Из
галогенопроизводпых легче всего реагируют соеди-
нения с аллильным или бензильным радикалами;
введение фенильных радикалов в a-положение повы-
шает в этой реакции подвижность галогена:
(CeH5)3GX>(CeHs)2CHXi>C6H5CH2X. Труднее всего
реагируют винил- и арилгалогениды. Натриевые и
калиевые соли сильнокислых фенолов не реагируют
с галогеналкилами. В этих случаях пользуются
серебряными солями или вводят в реакцию соответ-
ствующий фенол, RJ и Ag2O. При синтезах эфиров-
фенолов побочным процессом является алкилирова-
ние в ядре, весьма облегчающееся при использовании
активных галогенидов в неполярной среде. Диал-
килсульфаты чаще всего применяют при алкилиро-
вании углеводов и фенолов. Условия синтеза сильно
меняются в зависимости от активности исходных
соединений, их растворимости и пр. Чаще всего при-
меняется кипячение в среде соответствующего спир-
та или в инертном растворителе (ROMe в виде
суспензии); для активных галогенидов —
нагревание RX и R'OH в присутствии не-
2 *" большого количества щелочи. Диалкил-
сульфаты, как правило, применяют в вод-
ных средах. Этот вариант синтеза широко
используется в лабораторной практике для синтезов,
простых эфиров, для Приготовления метиловых эфи-
ров углеводов (т. н. метод метилирования), для при-
готовления синтетич. катионита на основе целлю-
лозы (карбоксиметилцеллюлозы), в технике — для
синтеза алкилзамещенных целлюлоз и алкилирования
красителей по фенольным гидроксилам.
Синтез простых эфиров из спир-
тов: ROH + R'OH — ROR' + Н2О; катализато-
рами являются к-ты: H2SO4, Н3РО4, Н3ВО3, ArSO3H
ит. п., или катализаторы для реакций, аналогичных
реакции Фриделя — Крафтса: SnCi4, BF3, FeCl3,
ZnCl2 и т. п. Механизм реакции изучен для случая
катализа серной к-той:
ROH + H3SO4 —► ROSOaOH -4- Н2О (на холоду)
хлоп-' -f- XI0OV4
ROSOaOH -]- R'OH< R'OH
-H»O *ROSO3R* —ROR’ + HOSOaOR'
(при нагревании)
Если R и R' разные, то получаются смеси трех воз-
можных эфиров (ROR, ROR' и R'OR'). В таких слу-
чаях сначала проводят первую стадию реакции (на
холоду), затем вводят второй компонент (R'OH).
С возрастанием длины цепи, а также при переходе
от первичных спиртов к вторичным и третичным
возрастает тенденция к образованию олефинов вслед-
ствие внутримолекулярного выделения воды; для
спиртов выше пропилового такое направление реак-
ции является главным. Однако и в этих случаях
удается синтезировать эфиры, применяя минималь-
ные количества H2SO4. Реакция облегчается введе-
нием арильных радикалов в a-положение в молекулу
спирта, а также в случае соседства с карбонильной
группой. Для фенолов реакция крайне затруднена.
Для низших спиртов синтез проводят при 120—145°
(каталйзатор H2SO4). Добавление Al2 (SO4)3 или
PbSO4 снижает темп-ру реакции до 110°. Отгонка
567
ВИНА ЭФФЕКТ —ВИНИЛИРОВАНИЕ
568
воды с эфиром обусловливает постоянство концентра-
ции катализатора. Главное применение реакции —
промышленный синтез диэтилового эфира. Методы
синтеза эфиров предложены А. В. Вильямсоном
в 1850—52.
Лит.: Препаративная органическая химия. Под общ. ред.
И. С. Вульфсона, пер. с польск., М., 1959, с. 340; Губен И.,
Методы органической химии, пер. с нем., т. 3, вып. 1, М.—Л.,
1934, с. 164; К 1 г k, V. 5, N. Y., 1950, р. 860. Л. Ф. Бочков.
ВИНА ЭФФЕКТ — возрастание электропровод-
ности р-ров электролитов при высоких градиентах
потенциала (десятки и сотни тысяч волът/см). Суще-
ствование В. э. у р-ров сильных электролитов, за-
висимость его от концентрации и валентного типа
электролита, а также от величины градиента потен-
циала количественно объяснены в теории сильных
электролитов на основе представления о частичном
или полном исчезновении ионной атмосферы у ионов,
движущихся с высокими скоростями. У р-ров слабых
электролитов В. э. обусловлен, кроме того, возраста-
нием степени электролитической диссоциации с рос-
том градиента потенциала. Эффект открыт в 1927
М. Вином.
ВИНИЛАЦЕТАТ СН3СООСН=СН2, мол. в. 86,09—
бесцветная жидкость, замерзающая ниже —84°;
т. кип. 73°; rf|jj 0,9342; 1,3958; ц30 0,432 спуав',
в воде при 20° растворяется 2,5% В.; т. всп. от
—5 до —8° (в открытом сосуде). В промышленности
В. получают взаимодействием ацетилена с уксусной
кислотой в присутствии катализаторов: CHsCH -f-
+ СН3СООН -> СН3СООСН=СН2 (см. Кучерова
реакция). Известны жидкофазный и парофазный
методы получения В. По первому из них реакция
протекает при 60° в присутствии солей ртути; по
второму — реакционную смесь пропускают над аце-
татом цинка, нанесенным на активный уголь или
силикагель. Очищают В. перегонкой с водяным паром
или под вакуумом; в качестве ингибиторов применяют
серу, резни ат меди или дифениламин. Наиболее
важным свойством В. является его способность к по-
лимеризации и сополимеризации с другими виниль-
ными производными; при этом образуются высоко-
молекулярные полимеры, применяющиеся в пром-сти
(см. Поливинилацетат).
Лит.: Мономеры. Сборник ст., пер. с англ., под ред.
В. В. Коргаака, М., 1951; Б а р г Э. И., Технология синтети-
ческих пластических масс, Л., 1954. О. В. Смирнова.
ВИНИЛАЦЕТИЛЕН (бутенин) НС=С—СН=СН2,
мол. в. 52,03 — газ, легко сжижающийся в бесцветную
жидкость; т. кип. 5,5°; 0,6867, 0,7095—0,00114г
(в интервале минус 3 минус 80°). Зависимость давления
пара от темп-ры 29,8 мм!град', теплоемкость Ср при
298,16° К 17,49; 1 500°'К 37,2 кал/град-моль. Хими-
яеские свойства В. определяются главным образом
тройной и, в меньшей степени, двойной связью. В
зависимости от условий каталитич. гидрирование
В. дает бутан, бутилен или бутадиен. Гидратацией В.
получают метилвинилкетон. Водород концевой СН-
группы замещается обычными способами — метал-
лами, галогенами и MgX (X — галоген). Фаворского
реакция с В. приводит к винилэтинилкарбинолам
R'R"C(OH)C^CCH=CH2, используются для полу-
чения дивинилкетонов, карбинольного клея и др.
Присоединение различных реагентов к тройной связи
дает замещенные бутадиены: НС=С—СН=СН2 +
+ НА->Н3С=СН— СН=СНА (А=Н, OR, OCOR, SR и
т.д.). Особенно важное значение имеет присоединение
НС1, приводящее к хлоропрену СН2=СС1—СН=СН2—
мономеру маслостойких синтетич. каучуков. В. легко
окисляется с образованием взрывчатых продуктов.
Полимеризация В. при действии перекисных катали-
заторов или под давлением дает высыхающие масла.
В пром-сти В. получают конденсацией ацетилена в при-
сутствии солей меди в р-ре хлористого аммония при
50—100°. В. образуется в виде примеси к ацетилену
при электрокрекинге метансодержащих газов.
Лит.: Ньюлэнд Ю. иФогтР., Химия ацетилена,
пер. с англ., М., 1947. А. Е. Васильев.
ВИНИЛИДЕНХЛОРИД (хлористый винилиден,
1,1'-дихлорэтилен, асимметричный дихлорэтилен)
СН2 = СС12, мол. в. 96,95 — летучая жидкость с
слабым запахом, напоминающим запах хлороформа;
т. пл. —122,53°; т. кип. 31,7°/7СЭ мм', плотность
d 1,2695 (—10°), 1,2504 (0°), 1,2122 (20°); naDu 1,4271;
ц (жидк.) 0,358 спуаа (20°); давление пара 135 9 мм
(—10°), 215,9°з»з» (0°), 495,3 мм (20°), 1,28 ат (40°),
2,38 ат (60°); уд. теплоемкость (жидк.) 0,277 ±
±0,001 кал/г-град', теплота‘ испарения 66,8 кал/г',
теплота полимеризации 14 ± 0,5 ккал/молъ', диполь-
ный момент 1,30 D (в бензоле); пределы взрываемости
в смеси с воздухом 7—16 об. % (25°). Атомы хлора
в В. малоподвижны; при действии алкоголята нат-
рия замещается один атом хлора; при конденсации
с формальдегидом в присутствии серной к-ты обра-
зуется акриловая к-та СН2=СНСООН. В. легко
полимеризуется, особенно под влиянием перекисных
соединений, образовавшихся при соприкосновении
его с воздухом. Выпадающий при этом осадок поли-
мера может содержать значительные количества
перекисных соединений, разлагающихся при ударе
или нагревании со взрывом; для разложения пере-
кисей полимер обрабатывают горячей водой или р-рами
щелочей. В. может самопроизвольно полимеризо-
ваться.
В. легко получается дегидрохлорированием
1,1,2-трихлорэтана, спиртовыми или водными р-рами
щелочей. В пром-сти для этой цели применяют
известковое молоко: 2СН2С1—СНС12 + Са (ОН)2 —►
—► СН2=СС12+СаС12+ Н2О; процесс ведут при темп-ре
ок. 100°. Образующийся В. отгоняют и ректифици-
руют. При хранении В. стабилизируют добавками
антиоксидантов, напр. гидрохинона и др., а также
хранят при темп-pax ниже 0°. Применяют В. для
получения полимеров и сополимеров с винилхлори-
дом, акрилонитрилом и др. См. также Поливинили-
денхлорид.
Лит.: Гордон Г. Я., Хлористый винилиден и его сопо-
лимеры, М., 1957. Г. Я. Гордон.
ВИНИЛИРОВАНИЕ — введение винильной груп-
пы в органич. соединение, осуществляемое в одну
стадию. Наиболее важный метод В. — взаимодей-
ствие -ацетилена с соединениями, обладающими по-
движным атомом водорода: НС=СН + НХ —►
—►СН2=СНХ. Очень важное промышленное значение
имеет В. самого ацетилена с образованием винил-
ацетилена (I) — полупродукта синтеза хлоропрена и
хлоропренового каучука, а также В. синильной к-ты
для получения акрилонитрила (II):
НС=СН + СН=СН—► СН=С-СН=СН» (I)
СН=СН + HCN -► CH3=CHCN (П)
Препаративное значение имеет В. хлорангидридов
кислот, приводящее к хлорвинилкетонам, к-рые
в свою очередь являются кетовинилирующими со-
единениями:
О .0 „ ,0
RC< + СН=СН -> RC—СН=СНС1—RC^CH=CHX
Xci
В. спиртов, меркаптанов и аминов производят
обычно нагреванием с ацетиленом под давлением
в присутствии щелочных катализаторов. Реакции эти
имеют очень широкое распространение и применяются
для получения самых разнообразных соединений,
569
N-ВИНИЛКАРБАЗОЛ - ВИНИЛОВЫЕ ЭФИРЫ
570
например:
C6H5NHCH2CH2OH + СН=СН
ZCH2-СН2
—- CeH = NHCH„CH2OCH—СН2 —*-C6HBN |
6 5 2 ХСН—О
I
СН3
RSH + CHSCH—~ rsch=ch2—-
—- (RS)2CH-CH3 + rsch2ch2sk
RNH2 +CHSCH —- rnhch=ch2
(CH3)N + CH = CH H'2° - (CH3)3N-CH=CHa OH’
Иногда вместо ацетилена при В. употребляются
винилгалогениды. Удобный препаративный метод
винилирования — реакция с винилмагнийбромидом
(Нормана реактив), напр.:
Rx R, .он
H3C=CHMgBr+ ^.со->- >с<
R’z Rz хСН=СН2
Широкое применение в пром-сти для получения
синтетич. смол находит, помимо В. спиртов, также
В. кислот (получение поливинилацетата} и хлори-
стого водорода (получение поливинилхлорида).
Лит.: Н ь ю л э и д Ю., Ф о г т Р., Химия ацетилена, пер.
с англ., М., 1947; Иоффе С. Т., Хсп. хим., 1958, 27. с. 1010;
Miller S. I., S h k а р е n к о О., 3. Am. Chem. Soc., 1955,
77, p. 5038; R e p p e W., Liebigs Ann. Chem., 1956, 601,
№ 1—3, 81. H. П. Гамбарян.
N-ВИНИЛКАРБАЗОЛ C15 H14N, мол. в. 193,24 —
бесцветные кристаллы, темнеющие при хранении на
свету; т. пл. 65°; т. кип. 140—150°/1 мм; d’° 1,094;
ц’° 4,47 спуаз', нерастворим в воде, растворим в орга-
нич. растворителях. В. легко присоединяет водород,
H2S и меркаптаны. Наиболее важным свойством В.
является способность к полимеризации и сополимери-
зации с другими винильными соединениями (см.
Поливинилкарбазол). В лабораторных условиях В.
синтезируют: 1) из карбазола и 0-хлорэтилового
эфира п-толуолсульфокислоты, 2) из карбазола и оки-
си этилена и 3) из карбазола и этиленхлоргидрина.
В промышленных условиях В. получают винилиро-
ванием карбазола ацетиленом в присутствии сильных
щелочей при нагревании:
ОХ>сннсн—СсО
сн=сн2
Полученный В. перегоняют под вакуумом при
180—200°/5 мм; для предотвращения полимеризации
во время перегонки добавляют 0,5% фенил-0-нафтил-
амина или 1—3% КОН. В. стабилизируют аминами
и амидами (диметилформамид) и нек-рыми спиртами.
Лит.: Мономеры. Сборник ст., 1№ 2, пер. с англ., под ред.
В. В. Коршака, М., 1953; Лосев И. П., Петров Г. С.,
Химия искусственных смол, М.—Л., 1951.
ВИНИЛОВЫЕ ЭФИРЫ — ненасыщенные эфиры,
простые СН2=СНОВ (I) и сложные СН2=СН—
—OCOR (II); большинство — бесцветные жидкости,
мало растворимые в воде, смешиваются с органич.
растворителями. Многие простые В. э. образуют
азеотропные смеси с водой и со спиртами. Физич.
свойства нек-рых В. э. приведены в таблице.
Название эфира Т. кип., °С/лгм 20 nD
Винилметиловый .... 5.50/760 0,7725 1,3730 (0°)
Винилэтиловый 36,0—36,1/760 0,7531 1,3779
Винил-н-пропиловый . . 65,0—65,1/760 0,7678 1,3922
Винил-н-бутиловый . . . 93,7—93,8/760 0,7792 1,4029
Винил-мзо-бутиловый . . 83.0—83,1/760 0,7682 1,3990
Винилфениловый .... 155—156/760 0,9767 1,5225
Винилацетат 73/760 0,9342 1,3958
Винилбензоат 80/12 0,8994 (20°) 1,5259(21.5°)
Винилстеарат* 167/2 0,8517 (40°) 1,4423(30°)
• Т. пл. 30—32°.
В спектрах комбинационного рассеяния винильная
группа дает характерные линии 3020—3080 см~х
(С — Н-связь), 1610, 1640 и 1650 см~г (двойная связь).
В. э. химически весьма активны; легко присоеди-
няют галогены, галогеноводороды, озон и т. п. Гидро-
лиз, легко протекающий в кислотных (а для слож-
ных В. э. и в щелочных) средах, приводит к ацет-
альдегиду:
CH2=CH-OR СНдСНО + ROH
или
CH2=CH-OCOR СНзСНО -I- RCOOH
Присоединение агентов типа НХ (где X — анио-
ноидная группа) к В. э. (I) происходит в соответствии
с правилом Марковникова, по ионному механизму:
СН2=СН—OR-f- НВг—►СН3СН (OR) Вг. Меркаптаны
и H2S могут присоединяться как по ионному,
так и по радикальному механизмам. Взаимодействие
(I) со спиртами дает ацетали, с органич. к-тами —
ацилали. В присутствии А1С13 В. э. (I) способны
к конденсации с ароматич. соединениями, напр.:
^CHa-CH-OR
СН3—СН—OR + СсНв<^ СсН5
\h5R
Параллельно происходит алкилирование с разрывом
эфирной связи. Важнейшим свойством (I) является
их способность полимеризоваться; в большинстве
случаев реакция протекает по ионному механизму
и катализируется кислотными агентами, в то время
как ускорители свободно-радикальной полимериза-
ции (перекиси, нитрил азоизомасляной к-ты, свет,
кислород, нагревание и др.) слабо инициируют поли-
меризацию. В то же время (I) способны к сополимери-
зации с мономерами, для к-рых характерен свободно-
радикальный тип полимеризации. Полимеризация
(II) является типичным свободно-радикальным про-
цессом. В технике В. э. получают реакцией винилиро-
вания, т. е. присоединением к ацетилену соответ-
ствующих спиртов для В. э. (I) или карбоновых
кислот — для В. э. (II):
RQH
сн-гсн/ ,
RCOOH
CHS=CH-OR
СН3=СН-OCOR
Количественно В. э. определяют иодометрически,
бисульфитным методом (определение ацетальдегида,
образующегося при гидролизе В. э.), гидрированием
и др.
Синтезируют В. э. (I) гл. обр. в жидкой фазе
(150—165°) в присутствии щелочных катализаторов
(гидроокиси, алкоголяты, цианиды щелочных метал-
лов). В случае низших спиртов (С4 — С4) реакцию
проводят под давлением. В произ-ве важнейшего
из В. э. (II) — винилацетата — предпочтение от-
дается парофазному процессу, в котором применя-
ют большой избыток ацетилена. Процесс ведут при
571
ВИНИЛОВЫЙ СПИРТ — N-ВИНИЛПИРРОЛИДОН
572
180—220° над ацетатами цинка и кадмия на носителе
(силикагель, А12О3 и др.). Применяется также жид-
кофазный процесс, ведущийся при 60—65° с исполь-
зованием в качестве катализатора ртутных солей
в среде серной к-ты. Важнейшая область применения
В. э. — промышленность синтетич. полимеров. В. э.
(1)и винилацетат используются для произ-ва полимеров
и сополимеризации с другими мономерами. Омыле-
ние поливинилацетата приводит к поливиниловому
спирту. В. э. (I) могут служить для получения аце-
тальдегида по безртутному методу, присадок к сма-
зочным маслам.
Лит.: Шостаново кий М. Ф., Простые виниловые
эфиры, М., 1952. .1. Ф. Бочков.
ВИНИЛОВЫЙ СПИРТ СН2=СНОН, мол. в.
44,05 — простейший непредельный спирт, в свобод-
ном виде не выделен. При его получении, напр. на-
греванием бромистого винила с водой, образуется
изомерный ему ацетальдегид:
сн2 = снвг ——У бн2=снон -+ снасно
Такое превращение связано с выигрышем энергии
13 ккал/молъ. Виниловые эфиры СН2=СНОВ — устой-
чивы. Винилаты Li, Na и др. получены действием
этих металлов в жидком аммиаке на ртутные произ-
водные альдегидов и кетонов,напр.: Hg (СН2СНО)2—>
jbL HgLi + CH2=CHOLi.
Лит.: Несмеянов А. Н., Л у ц е н к о И. ф., X о цу-
гов Р. М., ДАН СССР, 1958, 120, М 5, с. 1049.
Б. II. Гусев.
ВИНИЛОГИЯ — широко распространенное в орга-
нич. химии явление передачи взаимного влияния
атомов и групп атомов по системе сопряженных
двойных связей. Винилоги, т. е. соединения ряда
X—(CH=CH)7i—Y, обладают сходными химич. свой-
ствами, часто напоминая первый член ряда X —
R-C
R-C-CH
II
О
— Y, где группы X и Y непосредственно связаны
между собой; напри-
мер, подвижность хло-
. Г1 ра в хлорангидридах
Ch—Ci кислот(1)С0Храняетсяи
в Р-хлорвинилкетонах
(II). Аналогично кро-
тоновый (III) и сорбиновый (IV) альдегиды вступают
в конденсации за счет w-атомов Н так же, как
ацетальдегид (V) за счет
а-атомов Н. С учетом В.
могут быть объяснены
многие явления ориен-
тации и взаимного
влияния заместителей
в ароматич. и гетеро-
циклич. соединениях;
н^-с^-сн==сн^-сн=о
н^с-сн=сн-сн=сн-сн=о
Н IV
Г»
н{-с-сн=о
напр., о- и я-дизаме-
щенные бензола явля-
ются винилогами и
обнаруживают сходные,
свойства; в то же время
свойства .и-дизвмещен-
ных резко отличаются.
Однако принцип В. не
универсален. Яркий
пример его нарушения
представляет собой орто- и пара-ориентации в реак-
циях электрофильного замещения коричной к-ты
и аналогичных соединений, где с точки зрения В.
должна была бы иметь место мета-ориентация.
Лит.: Реутов О. А., Теоретические проблемы органи-
ческой химии, М., 1956; Royals Е. Е., Advanced organic
chemistry, N. Y., 1954; F u s о n R. C., The principle of .viny-
logy, Chem. Rev., 1935, 16, № 1. Б. Л. Дяткип.
N-ВИНИЛПИРРОЛИДОН (N-винил-а-пирро ли-
дов) CeIIBNO, мол. в. 111,15 — бесцветная жидкость;
т. кип. 65—66°/1,5 мм, 71—72°/2,5 мм; d'-j" 1,0458;
1,5117; растворим в воде и почти во всех органич.
растворителях. Двойная связь В. очень активна.
При нагревании до 40—45° с
разб. минеральной к-той В. коли- „ „___
чественно расщепляется на пирро- 21 | 2
лидон и ацетальдегид (качествен- н2С СО
ная реакция обнаружения В.). При " СН = СН2
действии незначительных количеств
газообразного НС1 образуется кристаллич. димер. При
взаимодействии эквимолекулярных количеств В. и
газообразного НС1 образуется неустойчивое воско-
образное вещество, быстро разлагающееся на пир-
ролидон, ацетальдегид и HCI. В присутствии динит-
рила азоизомасляной к-ты В. присоединяет серово-
дород и меркаптаны; под влиянием HCI присоеди-
няет спирты; в диеновый синтез В. не вступает. Осо-
бый интерес представляют реакции гомополимериз ации
и совместной полимеризацив В. Полимеризация про-
текает под влиянием эфирата трехфтористого бора,
Н2О2, динитрила азоизомасляной к-ты, персульфата
калия (см. Полисинилпирролидон). Практически важ-
ной является полимеризация В. в водном р-ре под
влиянием Н2О2 с добавлением небольших количеств
NH3 в качестве активатора. Метод позволяет варьи-
ровать величину мол. веса поливинилпирролидоиа
(изменением режима полимеризации), что исполь-
зуется в пром-сти. В. не полимеризуется в присут-
ствии перекиси бензоила. В. вступает в реакцию
сополимеризации со сложными и простыми винило-
выми эфирами, стиролом, малеиновым ангидридом,
винилхлоридом, винилкарбазолом, акрилонитрилом
и др. Количественное определение В. производится
либо путем гидролитич. расщепления, либо на осно-
вании йодоформной реакции. Характерной реакцией
для В. является моментальное образование осад-
ка йодоформа при добавлении раствора щелочи и
иода.
В пром-сти В. получают из формальдегида и аце-
тилена. Образующийся вначале бутиндиол-1,4 (I)
гидрогенизацией переводят в бутандиол-1,4 (II),
этот промежуточный продукт синтеза может быть
получен также из фурфурола. Бутапдиол-1,4
в присутствии катализатора превращается в у-бу-
тиролактон (III). Воздействуя на последний аммиа-
ком, получают а-пирролидон (IV). Последняя стадия
синтеза — образование В. (V) из а-пирролидона и
ацетилена.
2СН2О + С№СН------» НОСН2С=ССН2ОН - На —
!
---► НО(СН2)4ОН—*нгс------СН2 NH3
н2сх 'СО
о
II т
___наС-----СН8 СН = СН т Н2с Сн2
НгСх „СО Н2С СО
nh n—сн=сн2
IV V
В. используется в основном для приготовления
поливинилпирролидоиа, а также сополимеров В.
с акриловой к-той, акрилонитрилом, стиролом и др.
Введение в полимерную цепочку звеньев В. повы-
шает гидрофильность полимерных материалов и их
сродство к красителям.
Лит.: Шостаковский М. Ф., Си де л ьк о fl-
ска я Ф. П., Вести. АН СССР, 1957, № 7, с. 45; Шоста-
ковский М. Ф.. Сидельковская Ф. П., Зелен-
с к а я М. Г., Изв. АН СССР. Отд. хим. наук, 1954, № 4, с. 689;
1956, № 5, с. 615; 1957, № 12, с. 1457; Reppe W., Polyvinyl-
pyrrolidon, Weinheim, 1954. Ф. П. Сиделькоиская.
573
ВИНИЛХЛОРИД — ВИНИЛЭТИНИЛКАРБИНОЛЫ
574
ВИНИЛХЛОРИД (хлористый винил, монохлор-
этилен) СН2—СНС1, мол. в. 62,50 — бесцветный газ
с слабым запахом, напоминающим запах хлороформа;
т. пл. —153,8°; т. кип. —13,8; d^15 0,9730. В. хо-
рошо растворим в хлороформе, дихлорэтане, эфире,
углеводородах нефти, очень мало растворим в воде.
Давление пара в зависимости от температуры (в мм
рт. ст.); 10 (—87,5°), 100 (—55,8°), 395,6 (—28,73°),
2258 (16,22°). 5434 (46,80°); теплоемкость (газообр.).
12,83 кал/град-моль; 1крит 156,5°; ркрит 55 ат. Уд.
теплоемкость (жидк.) В. 0,38 кал/г-град; теплота испа-
рения 71,26 кал/г (25°); г) 0,248 спуаа кал/г (—10°);
теплота образования 120 кал/г; теплота полимериза-
ции 23 ккал/моль', дипольный момент 1,48 0 (вереде
диоксана) и 1,442 (в парах); пределы взрываемости
В. в смеси с воздухом 4—22 об. % В. В. легко при-
соединяет галогены (хлор, бром) с образованием соот-
ветствующих тригалогенэтанов, напр.: СН2=СНС1 +
-г С12,—► СН2С1—СНС1»; галогеноводороды присоеди-
няются согласно правилу Марковникова, напр.:
СН2=СНС1 + НС1—>СН3—СНС12. Весьма важным яв-
ляется раскрытие двойной связи при полимеризации
и сополимеризации В. с винилацетатом, винилиден-
хлоридом и др. Процесс идет по цепному механизму,
инициаторами являются перекиси, например бензоила
перекись, гидроперекиси и др. Подробнее см. Поли-
винилхлорид, Полимеризация.
Атом хлора в В. малоподвижен, что связано с взаимодей-
ствием к-электронов и двойной связи со свободными электрон-
ными пйрами атома галогена: СН2=СН—С1 при этом связь
С = С поляризуется, а связь С—С1 упрочняется. В резуль-
тате В. значительно менее реакционноспособен в реакциях за-
мещения атома хлора по сравнению с его насыщенным анало-
гом — хлористым этилом. Однако в определенных условиях
хлор может быть замещен, напр. при получении сложных эфи-
ров и магнийорганич. соединений.
В пром-сти В. получают гидрохлорированием аце-
тилена в жидкой или паровой фазах: СН=СН +
+ НС1 —►СН2=СНС1 + НС1 или дегидрохлориро-
ванием дихлорэтана: СН2С1—СН2С1—>-СН2—CHCI +
+ НС1. Жидкофазный способ гидрохлорирования
ацетилена заключается в пропускании ацетилена и
НС1 при 60° через р-р катализатора (Сп2С12 + NH4C1)
в 12—15%-ной НС1; образующийся В. выделяют из
паровой смеси и ректифицируют. Наиболее распро-
странено парафазное гидрохлорирование ацетилена.
Следующий процесс
СН -СН + на -+ сн2 = СНС1
проводят при 120—180° над катализатором, представ-
ляющим собой активный уголь, пропитанный рас-
твором сулемы, при этом выделяется 44 кал/моль теп-
лоты.
Очищенный и тщательно высушенный ацетилен
после смешения с сухим хлористым водородом посту-
пает в трубчатый контактный аппарат, где происходит
образование В. Выделяющееся тепло отводится цир-
кулирующим в системе маслом. Контактные газы,
содержащие В., хлористый водород и немного ацети-
лена, промываются в скруббере водой. Затем газ
охлаждают до —10° и осушают. При дальнейшем
охлаждении, до —35°, В. конденсируется; для освобо-
ждения от примесей его ректифицируют. Конечный
продукт содержит не менее 99% В.; его хранят и
транспортируют в стальных емкостях без добавления
стабилизаторов.
Дегидрохлорирование дихлорэтана происходит под
действием спиртового р-ра щелочи в среде метанола
при темп-ре ок. 50°. Выделяющийся В. конденсируют
и ректифицируют, как описано выше. Дегидрохлори-
рование можно также проводить путем пиролиза
дихлорэтана при 400—500° над активным углем,
пемзой и др. пористыми материалами; при этом
заметно протекают побочные реакции, а получае-
мый В. недостаточно чист для произ-ва полимериза-
ционных смол. В. применяется для произ-ва син-
тетических смол: поливинилхлорида и его сополи-
меров.
Лит.: Шэлнт X., в кн.: Мономеры. Сб. статей, пер.
с англ., под ред. В. В. Коршака, М., 1951, с. 190; Воль-
ф к о в и ч С. И. [и др.1. Общая химическая технология,
т. 2, М., 1959, с. 409. Г. Я. Гордой.
ВИНИЛЭТИНИЛКАРБИНОЛЫ — соединения об-
R
щей формулы Сн2=СНС^ССОН, где R и R' —алкил,
\r'
арил или Н. Низшие представители В. — бесцве-
тные жидкости, полимеризующиеся при стоянии.
Формула Т. ь-ип., ° С/мм dl° 20 nD
СН»=СНС=ССН2ОН 59/12 0,9427 1.4940
СН2—СНС ССН(ОН)СН, . . . 64/18 0,9160 1,4865
СН2=СН(Г-СС(ОН) (СН,)3 . . . 52—53/10 0.8925 1.4786
СН2=СНСг£СС(,Н4ОН-о .... 102—103/10 0,9837 1.5180
В. весьма реакционноспособны, что объясняется
наличием сопряженной системы двойной и тройной
связи и гидроксильной группы. По реакционной
способности гидроксильной группы третичные В.
близки к трифенилкарбинолу: они легко этерифи-
цируются органич. кислотами, при действии хлори-
стого водорода обменивают гидроксил на хлор уже
на холоду, при действии спиртов (в присутствии
серной к-ты) образуют простые эфиры. При нагре-
вании со щелочами В. разлагаются на винилацети-
лен и карбонильное соединение:
/R'
R"COOH^CH2=CHC^CCC)CC)R„
/ 4r
ZR
СН2=СНС^ССОН ----—-----*СН2-СНС=ССС1
X R
КОН \
\ К"ОН .».CHg=CHC=CCOR"
CH2=CHCSCH + RR’CO XR
Третичные В. легко дегидратируются под влия-
нием минеральных к-т (лучше под влиянием 50%-ной
серной к-ты) с образованием дивинилацетиленовых
углеводородов:
ZR н+
СН,=СНС=СОН ,т _• CH»=CHC=CCR=CHR'
\сн/ “Нз°
В условиях гидратации по Кучерову (кислый водный
р-р сернокислой ртути) все В. образуют тетрагидро-
-у-пироны, а также (в нек-рых случаях) производные
фуранона:
ZR
СН,=.СНСНССОН
XR'
Н2О
[HgSO4]
При восстановлении В. с помощью омедненной цин-
ковой пыли затрагивается только тройная связь
и получаются диеновые спирты:
R ZR
СН^СНС-СОН-Ч сн2=снсн=снсон
\r> xr’
Под влиянием серпокислой ртути в ацетоне или лучше
в метаноле В. изомеризуются в диеинилкетоны, к-рые
575
ВИННЫЕ кислоты —ВИНО
576
далее присоединяют молекулу спирта и превращаются
в метоксикетоны:
r ,R
СН2=СНС=СОН [HgS2U CHS=CHCOCH=C СНз0Н»
\r' \r'
ZR
—> CHaOCHaCHsCOCH=C
\r'
Первичный и вторичные В. получаются при взаимо-
действии винилэтинилмагнийбромида с альдегидами:
CHs=CHC=CMgBr + RCHO CHS=CHC^CCH(OH)R
Если вместо альдегидов взять кетоны, то образуются
третичные В. Однако последние удобнее получать
конденсацией кетонов с винилацетиленом под влия-
нием едкого кали:
.R'
СН2=СНС= СН + RR'CO ГКОН]. CHS=CHC=CCOH
\r
Частично заполимеризовавшийся диметилвинилэти-
нилкарбинол под названием «карбинольный клей»,
«бальзамин» и «клей Назарова» применяют в опти-
ческой, инструментальной и электротехнич. пром-сти
как вещество, способное прочно склеивать металлы,
стекло, мрамор, пластмассы и пр.
.Пит.: Назаров И. Н., Избранные труды, М., 1861.
И. В. Торгов.
ВИННЫЕ КИСЛОТЫ (диоксиянтарные) С«Н6Ов,
мол. в. 150,09 — двухосновные кислоты; известны
три стереоизомерные формы:
( :оон СООН СООН
н— -он но—н н--он
но- - н н—он н—он
с :оон СООН соон
I II III
Конфигурации I и II являются оптич. антиподами;
конфигурация III (мезовинная к-та) оптически не-
деятельна, т. к. обладает плоскостью симметрии,
перпендикулярной центральной связи С — С.
D-В инная кислота (виннокаменная, обык-
новенная винная) (I) — моноклинные призмы, т. пл.
170°; 1,7598; растворимость (в г на 100 г рас-
творителя): в воде 139,44 (20°), в абс. этаноле 20,40
(18°), в эфире 0,30 (18°); растворяется в ацетоне;
[а]^=+11,98° (20%-ныйраствор вводе); [a]D=+0,46°
(в СН3ОН); константы диссоциации: = 1,3 • 10"3
(25°); К2 = 6,9 • 10~s (25°); ИК-спектр: полосы по-
глощения 1100—1300 см~\ 1550—1800 см1. D-В. к.
получают из винного камня, образующегося при бро-
жении виноградного сока. При пиролизе получаются
пировиноградная кислота СН3—СО—СООН и пиро-
винная (метилянтарная) НООС—СН(СН3)СН2—СООН
к-та, а также СО2, D-В. к. восстанавливается до ян-
тарной к-ты; восстанавливает аммиачный р-р AgNO3;
в щелочной среде растворяет Си (ОН)2 с образова-
нием Фелинга реактива. D-В. к. применяется в пи-
щевой пром-сти и медицине; в аналитич. химии — для
осаждения К+ в виде НООССН(ОН)СН(ОН)—COOK,
соли D-В. к. (тартраты) используются в медицине,
в аналитич. химии, при крашении тканей и т. д.
L-В инная кислота (II) — значение [a]D
равно, но противоположно по знаку для D-В. к.
Обе В. к. кристаллизуются в энантиморфных формах.
Прочие физич. свойства тождественны. Различие
химич. свойств сказывается только при реакциях
с оптически активными соединениями. L-В. к. полу-
чается расщеплением DL-B. к.
DL-B инная кислота (виноградная) —ра-
цемич. соединение кристаллизуется из воды или спирта;
из воды при темп-ре >. 73° или из спирта выпадает
безводная DL-B. к., т. пл. 205°. Кристаллогидрат
2С4НвОв • 2Н2О теряет воду при 110°. Растворимость
гидрата (в 100 г растворителя): в воде 20,6 г (20°),
в этаноле 2,08% (15°), в эфире 1,08%; константы дис-
социации: К2 == 10,2 • 10 4 (25°); К2 = 4,0 • 10~s (25°).
В 1830 И. Берцеллиус, показав, что В. к. имеет
одинаковый состав с винной к-той, ввел понятие
изомерии. В 1848 Л. Пастер впервые расщепил В. к.
на оптич. антиподы. Может быть разделена на D- и
L-В. к.: 1) фракционной кристаллизацией солей
с оптически деятельными основаниями; 2) избира-
тельной ассимиляцией D-В. к. нек-рымя бактериями;
DL-B. к. получают L-изомер.
При нагревании до 130° с соляной к-той В. к.
о
частично превращается в недеятельную мезовинную
кислоту. В. к. образуется в смеси с мезовинной к-той
при кипячении р-ра правой винной к-ты со щелочью.
Может быть получена смешением растворов, содер-
жащих равные количества правой и левой винных
кислот.
Мезовинная кислота (антивинная)
(III) — кристаллогидрат С4НвО6 • Н2О (листочки);
для безводной т. пл. 140°; 1,666; раствори-
мость в 100 г воды 125 г (20°); константы диссо-
циации: Кг = 6,0-10-« (25°); К2 = 1,4 • 10~* (25°).
Все В. к. при нагревании в кислых, нейтральных
и особенно быстро в ще- d-в к i- в
лочных средах претерпе-
вают след, превращения
(см. схему). Из приведенной
схемы виден взаимный пе-
DL - В. и. >- Мезо - в. и.
реход В. к. А. Ф. Бочков.
ВИНО — продукт спиртового брожения сока (сусла)
винограда, плодов и ягод. Главнейшими составными
частями виноградных и плодово-ягодных В. являются:
спирты (этиловый, глицерин и сорбит — шестиатом-
ный спирт в плодово-ягодных винах), органич. кис-
лоты (винная, яблочная, молочная, янтарная, лимон-
ная), азотистые, дубильные, красящие, пектиновые
и минеральный вещества. Кроме того, в В. содер-
жатся витамины А, С, Вь В2 и РР. В крепких и де-
сертных В. содержится инвертированный сахар (глю-
коза и фруктоза).
Виноградные В. приготовляются различных типов
и кондиций (по содержанию спирта и сахара): сто-
ловые сухие и полусладкие, крепкие, де-
сертные, игристые (шампанские), шипу-
чие (газированные) и ароматизированные
(вермут). Для приготовления В. употребляют вино-
град, достигший необходимой (технической) зрелости
(когда в ягоде образовалось нужное количество
экстракта, сахара и т. п., необходимое для приготов-
ления из него В. определенного типа).
Белые столовые В. готовят в основном из белого
винограда, а также из красного и черного винограда, сок к-рых
не окрашен, путем полного сбраживания сусла; в них содер-
жится 9—14% спирта и 5—8 г/л кислоты.
Для приготовления В. виноград подается на эграпомпу
(дробилку и гребнеотделитель) для раздавливания и отделения
гребней (плодоножек); полученная масса — мезга — передается
насосом в пресс. Сусло из пресса поступает на отстаивание
в чаны или железобетонные резервуары, где в течение 12 —
18 часов происходит оседание взвешенных частиц. Осветленное
сусло перекачивается насосом в емкость на брожение. При
перекачке в сусло вводится от 80 до 200 мг/л жидкого SO2 (в за-
висимости от темп-ры сусла) и 2% разводки чистой культуры
дрожжей, приученных к SO2. При нормальных условиях бро-
жение сусла в таре продолжается 6—10 дней при 18—25°.
При сбраживании сусла в крупных резервуарах темп-ра бро-
дящего сусла обычно поднимается до 30° и выше, что плохо
влияет на качество В.; в этих случаях применяют искусствен-
ное охлаждение бродящего сусла. Брожение считается закон-
ченным, когда в В. остается не более 0,1% сахара. Через
2—3 недели, когда В. вполне осветлится, его сливают в чи-
стую тару.
577
вино
578
Красные столовые В. приготовляют из красного
и черного винограда. Дробление винограда и отделение гребней
производят на эграпомпе, после чего загружают мезгу в бро-
дильный чан. Ввод сернистого ангидрида и разводка чистой
культуры дрожжей производятся по технологии белых столо-
вых вин. При брожении мезги необходимы благоприятные
темп-риые условия’ (25—28°) для деятельности дрожжей и
извлечения красящих веществ из кожицы винограда. Регули-
рование темп-ры производится при помощи теплообменников.
Бурное брожение мезги заканчивается обычно в 5—6 дней,
после чего наступает тихое брожение. Если к этому времени
В. получило интенсивную окраску и количество оставшегося
сахара не превышает 1%, чан разгружают. Молодое В. сли-
вают в емкость, установленную в помещениях, в к-рых выдер-
живается темп-ра 16—18° (для дображивания оставшегося
сахара).
Столовые розовыеВ. получают при переработке
красных сортов винограда с неокрашенным соком по техноло-
гии белых вин или купажем (смешением) белых и красных В.
Молодые столовые В. в зависимости от их качества переводят
в винные подвалы на выдержку для получения высококаче-
ственных марочных вин или передают на ускоренную
обработку для выпуска их в молодом возрасте в качестве
ординарных вин.
Столовые полусладкиеВ.,в отличие от сухих,
содержат небольшой процент сахара, вследствие его непол-
ного сбраживания в исходном сусле или мезге. Полусладкие
В. бывают 3 видов — белые, розовые и красные; содержание
в них спирта от 7 до 14%, сахара от 3 до 8%. Столовые полу-
сладкие В. неустойчивы. Для их стабилизации принимаются
меры по прекращению жизнедеятельности дрожжей (введение
повышенных доз SO? или горчицы 0,3—0,4 г/л, пастеризация,
обеспложивающая фильтрация, охлаждение). Столовые полу-
сладкие В. в основном производятся по след, схемам: 1) Вино-
град с сахаристостью не менее 23% перерабатывают по одному
из описанных выше способов приготовления столовых В. Сусло
и первый отжим с пресса направляют на брожение. После того,
как в сусле образуется 8—10% спирта, брожение останавли-
вают введением сернистого ангидрида или охлаждением, с та-
ким расчетом, чтобы в В. осталось 3—8% сахара. Полученное
В. сохраняют до розлива в помещениях с темп-рой —2°. Раз-
литое через фильтр в бутылки В. пастеризуют при темп-ре
65—70°. 2) Смешивают сухие столовые В. (купаж) с консерви-
рованным виноградным суслом с таким расчетом, чтобы
получить готовое В. требуемых кондиций (по спирту и сахару).
В., приготовленные купажем, нестойки и нуждаются в суль-
фатировании или пастеризации. Иногда эти В. выдерживают
при —2° не менее 20 дней, после чего разливают через фильтр
в бутылки и пастеризуют при 65—70°.
Крепкие и десертные В. готовят добавлением
спирта-ректификата, благодаря чему в В. приостанавливается
жизнедеятельность дрожжей и сохраняется необходимое
количество сахара; кроме того, В. приобретает требуемую кре-
пость. Сахаристость винограда для приготовления крепких
и десертных В. не должна быть ниже 23%. Виноград перера-
батывают как указано выше. Полученное сусло помещают
в емкость для брожения; когда количество сахара в бродя-
щем сусле достигнет 5—6% (иногда до 12%), в него вводят
спирт.
Крепкие В. различают по кондициям и вкусовым ка-
чествам, обусловленным технологией, применяемой при их
приготовлении. Крепкие В. типа «Мадера» содержат 18—
20% спирта и 4—6% сахара. Их характерный вкус получается
в результате выдержки в бочках в особых помещениях (мадер-
никах) в течение длительного срока (месяц и более) при высоких
темп-рах (45—70°) в условиях аэрации. Крепкие В. типа
«Портвейн» содержат 17—19% спирта и 6—12 % сахара.
Характерные вкусовые качества портвейна обусловливаются
его нагреванием при тех же темп-рах, но без аэрации. Сходная
технология применяется при приготовлении В. типа «Мар-
сала», своеобразный вкус к-рой объясняется добавлением
уваренного на голом огне сусла (бе к мес а). Оригинальный,
отличный от всех других вкус, имеют В. типа «Хере с», со-
держащие 19% спирта и 3% сахара. Специфич. вкусовые ка-
чества хереса обусловливаются технологией его приготовления.
В неполностью налитую тару с белым сухим столовым В. вносят
хересные дрожжи, к-рые образуют на поверхности В. пленку.
В результате деятельности этих дрожжей в В. образуются
альдегиды, ацетали и сложные зфиры, к-рые и обусловливают
специфич. вкус хереса.
Десертные В., готовят по технологии, сходной с тех-
нологией крепких В.; характеризуются повышенным содержа-
нием сахара (10—30% и выше) и умеренной крепостью (12 —
16%). Для их приготовления собирают виноград с возможно
большей сахаристостью. В большинстве случаев десертные
вина имеют свой специфич. аромат и вкус, к-рые обусловли-
ваются сортом винограда (мускат, токайские сор-
та, ркацители) или технологией (кагор, малага).
Помимо введения спирта для придания десертным В. особых
вкусовых качеств, при их приготовлении пользуются нек-рыми
специальными приемами. Так, при приготовлении мускатов,
токайских и иен-рых других В. для извлечения ароматич.
веществ сусло настаивают на кожице в течение 12 —18 часов;
при приготовлении кагора нагревают мезгу до 65°, а при полу-
чении малаги добавляют в В., уваренное на голом огне до 60-1—
70% сахаристости, часть сусла для придания В. характерного
вкуса прижженного сахара.
10 к. х, э. т. 1
Ароматизированное В. (вермут) готовится
из белых и красных виноградных В. с добавлением в них спирта,
сахара и ароматич. веществ (полыни, кардамона, корицы
гвоздики и пр.). Вермуты готовятся двух кондиций: крепкий
(18% спирта и 10% сахара) и десертный (16% спирта и 16%
сахара).
Виноградные В. выпускаются в продажу в молодом воз-
расте (в следующем году после их произ-ва) или выдерживаются
в винных подвалах в течение 2—5 лет. Первые называют орди-
нарными, вторые — марочными. В течение периода выдержки
В. подвергают технологии, обработке: доливке, переливке,
осветлению, оклейке (введению в В. коллоидных веществ —
рыбьего клея, желатина и др.) и фильтрации. При выдержке
В. созревают и развивают букет.
Шампанские, или игристые В., получают из
столовых путем вторичного брожения добавленного к ним
сахара и дрожжевой разводки в герметически закрытых со-
судах (бутылках или резервуарах большой емкости). При
брожении в закрытом сосуде («шампанизации») внутри сосуда
создается давление вследствие выделения углекислого газа.
В советском шампанском содержится 10,5—12,5% спирта; по
содержанию сахара для него утверждены след, кондиции:
сухое 3%, полусухое 5%, полусладкое 7% и сладкое 10%.
При произ-ве шампанского бутылочным способом готовят т. н.
тиражную смесь, состоящую из столового вина и ли-
кера, в к-рые добавляют дрожжевую разводку чистых культур,
10%-ный спиртовый р-р танина, и рыбий клей для осветле-
ния тиражной Смеси. Полученную смесь разливают в бутылки,
укупоривают пробкой, к-рую для предохранения от выталки-
вания закрепляют железной скобой. Бутылки укладывают
в штабели, где происходит брожение («шампанизация»), сопро-
вождающееся образованием углекислого газа, к-рый развивает
в бутылке давление до 5 ат и выше. По окончании брожения
бутылки не меиее 2 раз в год перекладывают, встряхивая их
при этом. В штабелях бутылки находятся в течение 2—3 лет.
Для удаления нз бутылок дрожжевого осадка, образовавше-
гося при брожении, их помещают на особые станки (пюпитры),
на к-рых осадок путем систематич. встряхивания бутылки
сводится на пробку, затем осадок вместе с пробкой удаляют,
добавляют в бутылки ликер для придания шампанскому сла-
дости, после чего немедленно укупоривают их новой пробкой,
на к-рую надевают проволочную уздечку для предохранения
пробки от выбрасывания. Эти операции производят па холоду
с возможной быстротой и принятием предупредительных
мер против потери давления. При приготовлении шампанского
резервуарным способом тиражную смесь, подготовленную для
шампанизации, подают на брожение в герметически закрываю-
щиеся металлич. резервуары емкостью в 600—1000 дкл. Когда
давление в резервуарах в результате брожения достигнет
4,5—5 ат и содержание сахара понизится до требуемых кон-
диций, брожение останавливают охлаждением содержимого
резервуара до минус 5—6°. После отстаивания при той же
темп-ре осветлившееся шампанское разливают в бутылки на
розливиых машинах под давлением 2—2,5 ат. В нроиз-ве
резервуарного шампанского применяется разработанный со-
ветскими специалистами непрерывный поточный метод, при
к-ром процесс шампанизации проводится в серии последова-
тельно расположенных резервуаров.
Плодово-ягодныеВ. готовят из культур-
ных и дикорастущих плодов и ягод (яблок, смородины,
груши, вишни, сливы и др.). Виноделие плодов и ягод
в основном проводится теми же технология, приемами
и на том же оборудовании, что и виноградное*. В пло-
дово-ягодном виноделии, в отличие от виноградного,
после дробления плодон и ягод широко применяются
пакпресса, в к-рых мезга прессуется не в деревян-
ных корзинах, а в пакетах, сделанных из редкой
прочной материи. В большинстве плодов и ягод на-
блюдается высокая кислотность, к-рая снижается
разбавлением соков водой. Натуральные и разведен-
ные водой плодово-ягодные соки имеют недостаточ-
ную сладость и для приготовления В. в них добавляют
сахар, после чего соки подвергают брожению, к-рое
прекращают при образовании 5,5% спирта; для
повышения крепости бродящей смеси в нее добавляют
спирт до требуемых кондиций. Сброженное и спирто-
ванное молодое В. обрабатывается (переливкой,
фильтрацией, охлаждением, оклейкой) в течение
40 дней. Плодово-ягодные В. готовятся сортовые
(яблочное, вишневое,, малиновое и т. д.), а также
купажные (из нескольких сортов), носящие назва-
ния: белое или красное крепкое, белое или красное
сладкое. Крепкие плодово-ягодные В. содержат
16—17% спирта и 7—9% сахара, сладкие — 16%
спирта и 10—16% сахара. Из сортов нек-рых куль-
турных яблок готовят газированный напиток — сидр
с содержанием 5—7% спирта и 5% сахара.
579
ВИНОГРАДНАЯ КИСЛОТА - ВИСКОЗА
580
Лит.: Герасимов М. А., Технология виноделия, М.,
1952; Фролов-Багреев А. М., Труды по химии и
технологии вина, т. 1, 2 изд., М., 1958; Могилян-
с кийЙ. К., Плодовое и ягодное виноделие, М., 1954; Тро-
ост Г., Технология вина, пер. с нем., М., 1958; Рибер о -
Гайон Ж., Виноделие. Преобразование вина и способы его
обработки, пер. с франц., М., 1956; Технология и технохими-
ческий контроль виноградных и плодово-ягодных вин, М., 1959,
М. А. Герасимов.
ВИНОГРАДНАЯ КИСЛОТА — см. Винные кис-
лоты.
ВИСКОЗА (вискозный прядильный раствор) —
концентрированный р-р ксантогената целлюлозы в
разбавленном р-ре NaOH. Этот р-р отличается вы-
сокой вязкостью и получил свое название от франц,
слова viscose — вязкий. Цвет чистой В. ярко-оран-
жевый. Иногда В. приобретает бурый или зеленова-
тый цвет (при наличии солей железа и др. примесей).
В. применяется для формования (прядения) вискоз-
ного волокна, вискозных неволокнистых изделий и для
произ-ва искусственной кожи (кирза и др.). Для
формования волокон и неволокнистых изделий при-
меняется В., содержащая обычно от 6,0 до 9,0%
целлюлозы в виде ксантогената целлюлозы, 5,5—7,5%
NaOH и ок. 2,0—2,5% связанной серы. Кроме того,
в В. присутствуют в виде побочных сернистых соеди-
нений натрия: тритиокарбонат, сульфид, полисульфи-
ды, сульфит, гипосульфит, пертиосоединения, а так-
же карбонаты, сернистое железо, соли кальция и рас-
творенная гемицеллюлоза. Плотность В. 1,12 при 20°.
Для получения однородной и стабильной В. ксан-
тогенат тщательно перемешивают с раствором щелочи,
растирают комки и непрерывно охлаждают (темп-ра
при растворении не должна превышать 10—12°).
После приготовления В. подвергают 2-4-кратной
фильтрации и выдерживанию под вакуумом для
удаления находящихся в р-ре пузырьков воздуха,
что необходимо для нормального прядения. Качество
В. определяют химич. анализом (содержание цел-
люлозы, едкого натра, связанной серы, т. н. гамма-
число, выражающее степень этерификации), а также
по вязкости, зрелости, фильтруемости, прозрачности
и реже по способности к прядению.
Вязкость В. колеблется в пределах 30—80 пуаз,
резко возрастает с увеличением содержания целлю-
лозы; кроме того, вязкость зависит от степени поли-
меризации целлюлозы и от содержания едкого натра
в р-ре. При увеличении степени полимеризации
целлюлозы от 350 до 800 вязкость В. (при содержа-
нии 8% целлюлозы) возрастает с 30—40 пуаз до
800—1000 пуаз. Т. к. вязкость В. сверх 60—80 пуаз
затрудняет ее переработку в современных производ-
ственных условиях, щелочную целлюлозу перед ксан-
тогенированием подвергают т. н. предсозреванию,
т. е. окислению в щелочной среде кислородом воздуха.
Снижение вязкости вискозного прядильного р-ра
с изменением содержания NaOH менее значительно.
В. имеет наименьшую вязкость, если соотношение
в ней NaOH и целлюлозы равно 0,9—1,0. При умень-
шении этого соотношения до 0,7 вязкость В. воз-
растает на 25—40%. Резкое возрастание вязкости
В. с увеличением концентрации или длины раство-
ренных молекул целлюлозы, а также с изменением
соотношения щелочи и целлюлозы объясняется тем,
что В. является высокоструктурированной жидко-
стью, и ее вязкость определяется гл. обр. структур-
ной вязкостью. Структурирование В. объясняется
наличием межмолекулярных (в основном — водород-
ных) связей между гидроксильными группами раство-
ренных целлюлозных молекул. Повышение степени
этерификации, уменьшение степени полимеризации
целлюлозы, увеличение концентрации гидроксильных
ионов в растворе ослабляют эти взаимодействия,
т. е. снижают структурную и общую вязкость В.
В том же направлении влияет повышение темп-ры.
Ксантогенат целлюлозы мало устойчив и в водных
р-рах постепенно омыляется (гидролизуется). Во
время ксантогенирования и последующего хранения
ксантогената целлюлозы в р-ре происходят парал-
лельно идущие процессы, к-рые могут быть описаны
след, суммарными химич. ур-ниями:
+[C(,H7O2(OH)a OH-NaOH]
п
- OC(,HsO4-
C=S -f-nHOH
.^SNa Jn
6NaOH + 3CS3=2Na3CS3 + NaaCO3 + 3H»O
В В. свободный сероуглерод постоянно связывается
NaOH и превращается в тритиокарбонат и др. сер-
нистые примеси. Из-за этого происходит т. н. созре-
вание В., т. е. гидролиз ксантогената или отщепле-
ние тиокарбонатных групп; степень этерификации
при этом уменьшается. Параллельно снижается
устойчивость В. к действию электролитов. Более
«зрелая» В. коагулирует (осаждается) под действием
меньшего количества электролитов и легче превра-
щается в нить при формовании в осадительной ванне,
содержащей заданное количество электролитов. Воз-
раст В. или ее «зрелость» может быть охарактеризо-
вана не только степенью этерификации ксантогената
целлюлозы в р-ре, но и индексом зрелости, т. е.
числом миллилитров р-ра электролита, добавленного
к В. до коагуляции (осаждения ксантогената цел-
люлозы). Снижение индекса зрелости по мере ста-
рения В. происходит тем быстрее, чем выше темп-ра,
ниже концентрация целлюлозы или меньше взято
сероуглерода при ксантогенировании. Большое влия-
ние на созревание В. оказывает также соотношение
содержания в ней NaOH и целлюлозы. В. наиболее
стабильна и зреет медленнее всего, если это соотно-
шение составляет 0,9—1,0. Устойчивость В. по от-
ношению к действию электролитов определяется
прочностью и величиной сольватных оболочек из
молекул воды и гидроксильных ионов вокруг гидро-
ксильных групп целлюлозных молекул (вернее моле-
кул ксантогената), находящихся в р-ре. С уменьше-
нием числа групп OSSNa (т. е. степени этерификации
целлюлозы) или концентрации гидроксильных ионов
устойчивость р-ра ксантогената целлюлозы снижается,
т. к. величина сольватных оболочек зависит от сте-
пени этерификации и наличия свободной щелочи
в р-ре.
Для достижения зрелости, пригодной для формо-
вания вискозного волокна, В. выдерживают 20—
60 часов при 16—17° в специальном помещении,
т. н. вискозном погребе. Для формования целло-
фана или укупорки для бутылок время созревания
В. при тех же условиях должно быть увеличено
до 90 часов и более, или же должна быть повышена
темп-ра в погребе. Созревание В. может быть прак-
тически прекращено снижением темп-ры в погребе
ниже 8°. Скорость созревания В. также может быть
снижена добавлением небольших количеств сульфита
натрия. Одной из важных характеристик пригод-
ности В. для формования волокна или неволокни-
стых изделий является ее способность к прядению,
к-рая зависит от темп-ры, снижаясь с ее возраста-
нием, от состава В. и от ее вязкости. С ростом вяз-
кости прядомость В. до известного предела возрастает,
после чего резко падает.
Для придания волокнам или неволокнистым изде-
лиям из В. специально заданных свойств к В. часто
добавляют в-ва, придающие им матовость (матирую-
щие), — пигменты или красители, карбонаты и др. со-
единения. Для матирования вискозных волокон в В.
можно вводить вещества, нерастворимые в осадитель-
ной ванне и не разлагаемые этой ванной, если эти
в-ва по показателю преломления сильно отличаются
от целлюлозы (1,5). Обычно для этой цели применяют
581
ВИСКОЗИМЕТРИЯ
582
высокодисперсную (размер частиц не более 1 мк)
двуокись титана, к-рая отличается химич. стой-
костью и имеет показатель преломления, равный 2,6.
В зависимости от нужной степени матовости в В.
добавляют 0,5—2% TiO2 от веса целлюлозы. Пиг-
менты, применяемые для крашения вискозных во-
локон в массе, тоже должпы быть стойки к действию
к-т и щелочей, и размер их частиц тоже не должен
превышать 1 мк ввиду большой тонипы волоконец
вискозного шелка. Кроме того, дисперсность TiO2
или пигментов должна быть весьма равномерна, т. к.
в противном случае вискозное волокно приобретает
разные оттенки в окрашенном виде. Для окраши-
вания можно использовать также водорастворимые
красители, если они устойчивы к действию кислот
и щелочей. Пока имеется очень мало водораствори-
мых красителей, пригодных для этой цели. В отдель-
ных случаях к В. добавляют Na2SO4 (при произ-ве
вискозной губки), мел или известняк (при произ-ве
пустотелых волокон или изделий), смолы (при произ-ве
водостойких волокон), белковые вещества и др.
соединения, придающие готовым изделиям те или
иные свойства. Добавление к В. специальных моди-
фикаторов, являющихся поверхностно-активными ве-
ществами (производные аминов, аминофенолов или
полиэтиленоксида), облегчает формование вискозного
волокна и позволяет получить структурно-однород-
ные волокна повышенной прочности. При введении
в В. акрилонитрила образуются цианэтиловые эфиры
целлюлозы, к-рые под действием щелочи омыляются
до монокарбоксильных производных целлюлозы. Этим
способом можно получить вискозное волокно, раство-
римое в воде или слабых р-рах щелочи. Такие волокна
применяются для создания узорчатых тканей.
Лит.: Роговин 3. А., Основы химии и технологии
производства химических волокон, 2 изд., М., 1957; Р я у-
з о в А. Н., Груздев В. А., А р т е м е н к о М. А., Техно-
логия искусственных волокон, 2 изд., М., 1952; Гетце К.,
Производство вискозного волокна, пер. [с нем.], М., 1958;
Справочник по аналитическому контролю в производстве ис-
кусственных и синтетич. волоков, М., 1957. А. Б. Пакгпвер.
ВИСКОЗИМЕТРИЯ — учение об измерении вяз-
кости, совокупность методов измерения вязкости
с учетом их сравнительных особенностей и областей
применения. Соответствующие приборы наз. в и-
скози метрам и. Вязкость должна измеряться
в условиях ламинарного, достаточно медленного
течения, поэтому к вискозиметрам предъявляются
определенные требования: 1) Конфигурация потока
в приборе должна в определенном интервале не слиш-
ком больших скоростей удовлетворять условиям
ламинарности, т. е. число (критерий) Рейнольдса
Re = vl/v (где v — средняя скорость, I — характе-
ристический линейный размер потока, напр. диа-
метр трубы, радиус шарика, ч = ц/р — кинематич.
вязкость, Г] — вязкость, р — плотность жидкости)
должно быть ниже некоторого критич. значения:
ReCReKf Для течения по трубам йекр/«2000—2400,
для установившегося движения шарика в вязкой
среде 7?еКр «= 1. Измеряемая вязкость в этих усло-
виях не должна зависеть от линейных размеров рабо-
чей части прибора, т. е. должна быть физико-химич.
константой вещества в данном агрегатном состоянии,
зависящей от темп-ры, давления, состава и строения
тела. Для того, чтобы удовлетворить это требование,
в приборе должно быть устранено пристенное сколь-
жение — слой исследуемой вязкой среды, прилегаю-
щий к твердой стенке, ограничивающей поток, должен
быть неподвижно связан с этой твердой поверхностью
(скорость слоя относительно этой поверхности v0 — 0).
Для не слишком вязких тел и в не очень узких за-
зорах (> 10 мк) это условие вследствие адгезии,
хотя бы очень слабой, практически удовлетворяется
даже на вполне гладких твердых поверхностях.
10*
В случае же высоковязких тел, особенно структури-
рованных, пристенное скольжение должно быть
устранено острой насечкой рабочих поверхностей
прибора (рис. 1).
Рис. 1. Схема прибора
с ограниченной деформа-
цией сдвига в узком за-
зоре между тангенциаль-
но смещаемыми пластин-
ками .1 и В. С — зазор
шириной Л, заполненный исследуемой системой (показано риф-
ление — насечка пластинок); Т — оболочка для термостати-
ровалия и герметизации; F — сдвигающая сила; Дл- — абсо-
лютный сдвиг.
Методы измерения вязкости жид-
костей разделяются на 3 группы: 1) Способы изме-
рения высоких вязкостей — от 102 — 104 и до 1013 пуаз
и выше, основанные на измерениях касательного
напряжения Р, поддерживающего (в условиях разви-
тия однородного сдвига с постоянной скоростью,
т. е. стационарного ламинарного течения) заданную
постоянную скорость сдвига (градиент скорости
G = dV/dy = ds/dt, где V — скорость сдвига, е —
относительный сдвиг, t — время). Вязкость вычис-
ляется из самого определения этой величины как
ц = P/G = Pldsfdi (1). При этом может быть задана
скорость сдвига G и затем измерено соответствующее
касательное напряжение Р, или, наоборот, может
быть задано Р и измеряться устанавливающаяся
(постоянная во времени) скорость сдвига G. 2) Методы
определения малых вязкостей (ниже 10—100 пуаз),
основанные на измерении средних скоростей устано-
вившегося течения в потоке заданной формы, или
скорости установившегося движения (падения) твер-
дых тел определенной формы в практически безгра-
нично вязкой среде; эти методы наиболее легко осу-
ществимы и широко распространены. 3) Методы опре-
деления малых и средних значений вязкости, осно-
ванные на измерении скоростей неустановившихся
движений: наблюдение за затуханием амплитуды
периодич. колебаний или уменьшением скорости
апериодич. движения вследствие перехода кинетич.
энергии в теплоту в результате внутреннего трения
исследуемой среды.
Измерения вязкости могут быть абсолютными или
относительными. Классич. абсолютными методами
являются: 1) метод капилляров, 2) метод вращающихся
цилиндров, 3) метод падающего шарика. В методах
2-й и 3-й группы распределение скоростей в потоке
нелинейно, т. е. соответствует неоднородному сдвигу,
а в методах 3-й группы, кроме того, и нестационар-
ному потоку. В более сложных случаях, когда рас-
пределение скоростей или закон движения, напр.,
твердого тела в вязкой среде, не могут быть получены
аналитически из общих законов гидродинамики вяз-
кой среды, абсолютные измерения вязкости становятся
невозможными. В этих случаях, исходя из подобия
вязких потоков, при одинаковых значениях Re,
можно получить правильные значения вязкости с до-
статочной точностью, если градуировать прибор при
помощи так наз. вискозиметрии, жидкостей с извест-
ными значениями вязкости (глицериноводные или
глицерино-сахарно-водные р-ры). Однако и в этом
случае вискозиметры должны удовлетворять основ-
ным требованиям В.
В 1-й группе методов измерения В. применяют:
а) Приборы, в к-рых величина деформации ограни-
чена их размерами, напр. приборы, в к-рых сдвиг
осуществляется в достаточно тонком слое толщиной Л
между двумя плоскопараллельными тангенциально
смещаемыми пластинками площадью s (рис. 1) или
осевое смещение коаксиальных цилиндров с узким
кольцевым зазором между ними, заполненным вязкой
583 •
ВИСКОЗИМЕТРИЯ
584
средой (рис. 2, в); б) более совершенные приборы, где
величины деформации не ограничены, что позволяет
наблюдать за длительным установлением стационар-,
ной скорости сдвига, иногда лишь после того, как
а б в
Рис. 2. Схемы приборов с неограниченной деформацией
в условиях, близких к однородному сдвигу (в узком
зазоре): а — коаксиальные цилиндры (rt—rt) -СГ]; б —
коаксиальные конусы по Гудибу; в — зазор в виде пло-
ского кольца достаточно большого радиуса, г, > Дг,
Н — упруго закручивающаяся нить — крутильный ди-
намометр; 3 — зеркало для оптич. отсчета угла пово-
рота внутреннего циливдра относительно внешнего.
достигнута весьма большая деформация сдвига е >> 1,
напр. в случае высоковязких и эластичных (струк-
турированных) жидкостей (полимеров и их концент-
рированных р-ров — студней). Таковы ротационные
приборы, основанные на относительном вращении
коаксиальных цилиндров радиусами гг и г2, узкий
зазор между к-рыми Аг = (г2 —г,)^^ заполнен
данной вязкой средой (рис. 2, а). Вместо цилиндров
более удобны для центрирования зазора Коаксиаль-
ные конич. поверхности (с малым углом конусности
во избежание дополнительных касательных напря-
жений из-за действия центробежной силы) (рис. 2, б).
Менее удобны вышеупомянутые приборы с плоским
кольцевым зазором, требующие большого объема
исследуемой вязкой среды.' Условия однородности
сдвига в таких приборах: Дг^г1, напр. /\г/г} С 0,05.
Приборы, основанные на методах 2-й группы,
характеризуются неоднородностью поля скоростей
сдвига, т. е. нелиней-
ностью распределения
скоростей (при условии
обеспечения стацио-
нарного течения) и
весьма многообразны,
к ним относятся:
а) Вискозиметры ро-
тационного типа, имею-
щие, в отличие от при-
боров 1-й группы, за-
зоры произвольной ши-
рины, удобны для изме-
рений вязкости высоко-
вязких жидкостей ти-
па смазочных масел при
низких температурах
(до —60°), битумов и
расплавленных сили-
катов при повышенных
Рис. 3. Ротационный вискози- температурах, а так-
метр Воларовпча РВ-7 (d — ось, же для дисперсных си-
к которой прикреплен цилиндр с, стем — глин красок
g - термостат,^- шарнкопод- клееВ; торфо;_ Широко ’
применение на практи-
ке получил ротационный вискозиметр Воларовича
РВ-7 (рис. 3).
Вискозиметр РВ-7 состоит из двух концентрич.
цилиндров с полусферич. дном: наружного неподвиж-
ного (Ъ) и внутреннего вращающегося (с), соединен-
Рис. 4. Капил-
лярный ви-
скозиметр Ост-
вальда.
ного с осью и приводимого в движение падающим
грузом (р). В пространстве между цилиндрами поме-
щается испытуемое вещество (а). Постоянная темп-ра
поддерживается с помощью термостата. Пределы
измерений РВ-7 — от 5 до 10’ пуаз. Вязкость в пуа-
зах вычисляют по формуле: ц = к (р — Pi)IN, где
р — груз, вращающий цилиндр, рг — груз, соответ-
ствующий трению в подшипниках (порядка 1—2 г),
N — число оборотов цилиндра в сек., к — постоянная
прибора, вычисляемая из измерений
вязкости эталонной жидкости и завися-
щая от размеров цилиндров.
б) Вискозиметры, основанные на
течении вязкой среды в достаточно длин-
ных и узких трубках постоянного круг-
лого сечения (капилляров). Капилляр-
ные вискозиметры представляют собой
чаще всего U-образную трубку, в узкое
колено к-рой впаян капилляр, пере-
ходящий в расширение (рис. 4). Изме-
рение вязкости при помощи капилляр-
ного вискозиметра основано на опреде-
лении времени истечения через капил-
ляр определенного объема жидкости
из рабочего пространства, ограничен-
ного кольцевыми метками айв. Су-
ществует несколько десятков модифи-
каций этих приборов, предназначаемых
для абсолютных и относительных измерений вязкости
жидкостей (от 0,001 до 100 пуаз) в широком интервале
темп-p. Точность образцовых капиллярных вискозиме-
тров ± 0,1 —0,3%, рабочих приборов ±0,5—1%. Наи-
более употребительными системами рабочих капил-
лярных вискозиметров являются вискозиметры Ост-
вальда (см. рис. 4), Уббелоде. Обычно применяют-
ся вискозиметры с истечением жидкости под дейст-
вием собственного веса и
вискозиметры с приложе-
нием внешнего давления.
При проведении измерений
капиллярные вискозиметры
помещаются в термостат.
Для измерения относи-
тельной вязкости часто при-
меняют вискозиметры Энг-
лера, Барбье, Редвуда, Сей-
болта и их разновидности.
В СССР принято обозна-
чать градусы вязкости по
Энглеру (°Е) через ВУ
(вязкость условная). Вели-
чина такой вязкости выра-
жает отношение времени
истечения 200 с.и3 иссле-
дуемой вязкой жидкости к
скорости истечения 200 см3
воды при 20°. Для пересчета условной вязкости в
вязкость динамическую т) н кинематическую при-
меняют эмпирические формулы: тд = (0,0731 Е ~
~ 0,0631/Е) . р (пуаз)', v = 0,0731 Е — 0,0631 /Е(ст).
Соотношение между кинематической и условной вяз-
костью показано в табл.
Рис. 5. Вискозиметр Энгле-
ра для определения услов-
ной вязкости.
Пуазы ВУ (по Энг- леру, Е®§) П Уазы ВУ (по Энг- леру, Eio)
0,50 2,79 1,65 6,89
0,65 3,45 2,00 7,47
0,85 4,14 2,25 8.10
1,00 4,84 2,75 10,89
1,40 5,63 3,00 11,50
в) Вискозиметры, основанные на измерении скорости
установившегося движения твердого шарика диа-
.585
ВИСКОЗНЫЕ ВОЛОКНА
586
. метром б в данной вязкой среде под действием по-
стоянной внешней силы, в простейшем случае —
силы тяжести. Расчет вязкости основан на законе
Стокса.
При падении шарика под действием собственной силы тяже-
сти вязкость приближенно вычисляется по ф-ле: у = с(р,—рг)1,
где с — константа прибора, определяемая для каждого ша-
рика по времени падения его с определенной высоты в жид-
костях известной вязкости, pt — его плотность, t — время
падения шарика, р2 — плотность жидкости. Вискозиметры
с падающим шариком получили применение и для измерений
вязкости при высоких давлениях. Для исследований вязкости
жидкостей в интервале темп-p от —60 до +150° употребляется
также вискозиметр Гёпплера, основанный на измерении ско-
рости падения шарика вдоль стенки наклонной трубы. Исполь-
зуют также методы, основанные на определении скорости подъе-
ма пузырька воздуха.
Вискозиметры 3-й группы имеют или значительно
более сложную теорию, или вообще не могут служить
для абсолютных измерений вязкости. Поэтому их
следует применять лишь в особых случаях, когда
почему-либо не могут быть применены вискозиметры
1-й и 2-й групп.
Особое положение занимают методы измерения
вязкости в весьма малых объемах нязкой среды.
Методы основаны на наблюдении броуновского дви-
жения, подвижности ионов, измерений коэфф, диф-
фузии частиц определенной формы и размера.
Для точных измерений вязкости расплавленных
металлов, помимо капиллярных вискозиметров, ис-
пользуется метод колебаний; расплавленным металлом
заполняется цилиндр из огнеупорного химически
стойкого материала, подвешиваемый на упругой
проволоке и помещаемый в печь. Вязкость металлов
вычисляется на основании измерений декремента зату-
хания цилиндра, колеблющегося нокруг оси подвеса.
Для измерения вязкости газов
и паров наиболее разработан метод капилляра,
к-рый сводится к измерению расхода и перепада
давления газа при его ламинарном движении через
капилляр. Наибольшая точность измерений дости-
гается при невысоких темп-рах и давлениях, близких
к атмосферному (вискозиметры Ренкина до 400—500°,
Халилова до 620°, Тимрота до 800° и др. позволяют
производить абс. измерения). Метод колебания диска
или цилиндра сводится к измерению декремента за-
тухания колебаний в исследуемом газе по сравнению
с воздухом. Вискозиметры такого типа просты и
компактны. Реже применяются методы падающего
тела и метод непрерывного вращения цилиндров.
Лит.: Г а тч е к Э., Вязкость жидкостей, пер. с англ.,
2 изд., М.—Л., 1935; Барр Г., Вискозиметрия, пер. с англ.,
Л.—М., 193.3; Ш в и д к о в с к и й Е. Г., Некоторые вопросы
вязкости расплавленных металлов, М., 1955; Френкель
Я. И., Кинетическая теория жидкостей, М.—Л., 1945; Сове-
щание по вязкости жидкостей и коллоидных растворов, [т.1
1—3, М.—Л., 1941—45; Панченков Г. М., Теория вяз-
кости жидкостей, М.—Л., 1947; Левич В. Г., Физико-
химическая гидродинамика, 2 изд., М., 1959; Бачинский
А. И., Избранные труды, М., 1960; Панченков Г. М.,
Ж. физ. химии, 1946, 20, № 8, с. 811; Щугаев В. и^ Со-
рок и и С., Ж. техн, физики, 1939, 9, „№ 10, с. 930; Р е б и н-
дер П. А. и Иванов а-Ч у м а к о в а Л. В., Успехи
химии и технологии полимеров, сб. 2, М., 1957; Толстой
Д. М., Коллоидн. ж., 1947, 9, „Ni 6; 1948, 10, № 2; 1950, 12,
1; е г о же, ДАН СССР, 1951, 77, М 5; Воларович
М. П., Известия Сектора физ.-хим. анализа. ИОНХ АН СССР,
1936 , 8, 125; Коллоидн. ж., 1954, 16, А6 3; Г о л у б е в И. Ф.,
Вязкость газов и газовых смесей, М., 1959.
П. А. Ребиндер.
ВИСКОЗНЫЕ ВОЛОКНА — искусственные волок-
на, получаемые путем формования вискозного пря-
дильного р-ра. Из вискозы можно формовать искус-
ственный шелк, т. е. нити бесконечной длины (состоя-
щие из многих элементарных непрерывных нитей);
искусственное штапельное волокно, т. е. отдельные
коротко нарезанные волоконца, применяемые для
произ-ва пряжи; технич. нити, напр. кордную нить.
Нити для канатов и строп обычно изготовляют из
искусственного шелка и реже — из штапельного
волокна.
Вискоза выдавливается дозирующим насосиком
через калиброванную пластинку или наперсток (филь-
еру), имеющую от 20 до 10 000 и более отверстий, раз-
мером от 0,05 до 0,09 мм, в осадительную ванну; В
зависимости от номера нити, скорости ее формования,
числа и тонины волоконец насосик подает от 10 до
1000 мл/мин раствора. В осадительной ванне, куда
попадают тонкие струйки прядильного р-ра, вискоза
осаждается и разлагается в кислотно-солевой среде.
Осадительная ванна содержит обычно от 80 до 150 г/л
серной к-ты, от 160 до 320 г/л сульфата натрия и от 5
до 100 г/л сульфата цинка. В нек-рых случаях приме-
няются ванны, содержание' только 40—70 г/л серной
к-ты; содержание сульфата натрия в таких ваннах
также может изменяться в широких пределах — от
100 г/л до 350 г/л, а содержание сульфата цинка мо-
жет доходить до 180 г/л. Иногда осадительная ванна
содержит еще сульфат магния и различные поверх-
ностно активные вещества. Темп-ра формования вис-
козных волокон обычно лежит в пределах 40—5.5°, но
иногда применяют ванны при 20—25° или же темп-ру
формования поднимают до 70—75°. В осадительной
ванне происходят след, реакции: 1) коагуляция вис-
козы- (т. е. осаждение ксантогената) под действием
электролитов, находящихся в ванне, с превращением
струйки в ксантогенатные нити (гель); 2) разложение
(омыление) ксантогената целлюлозы до гидратцеллю-
лозы под действием к-ты по реакции:
’/OCeHeOq
С—S I + nH2SO4 —- [CeH10O5]n -|- n CS2 -|- nNaHSO4
. ^SNa Jn
3) разложение различных сернистых примесей, на-
ходящихся в вискозе, с выделением сероводорода и
сероуглерода, в частности по реакции: Na2CSg +
+ H2SO4 -> Na2SO4 + CS2 + H2S. В результате всех
этих параллельно идущих реакций выделяются тон-
кие волоконца свежеосажденной гидратцеллюлозы,
а также сероводород и сероуглерод. Выделение этих
вредных газов является существенным недостатком
вискозного произ-ва и требует организации хорошей
вентиляции.
Скорость формования В. в., обычно , составляет
40—100 м{мин, а в нек-рых случаях может быть уве-
личена до 140 м)мин.
Гидратцеллюлозные волокна после выхода из-ванны
или в самой ванне подвергаются натяжению? При
этом макромолекулы или пачки макромолекул цел-
люлозы ориентируются вдоль оси волокон, и проч-
ность волокон возрастает. При формовании тех-
нич. волокон для придания повышенной разрывной
прочности пучок свежесформованных волокон после
выхода из ванны подвергают дополнительной вытяжке
на. 80—100% в горячей (95—100°) воде. После формо-
вания («прядения») волокна в виде пучка (жгута)
или в нарезанном виде подвергаются промывке для
удаления к-ты и солей, десульфурации для удаления
серы, выделившейся во время разложения примесей
в ванне, отбелке, промывке и сушке. Затем непрерыв-
ную нить (кордная или шелковая) подвергают крутке,
перемотке и др. текстильным операциям, а коротко
нарезанные штапельные волокна пакуют в кипы.
Волокна различаются между собой по тонине, раз-
рывной прочности, длине и числу волокон в нити.
Тонина волокон в СССР обычно измеряется, метрич.
номером №0 (длина волокна или нити, к-рая весит
1 г). Нить искусственного шелка и кордная нить, со-
стоящие из большого числа элементарных волоконец
п, имеют тонину №0 = №а/п, где №э — номер элемен-
тарного волокна. Разрывная прочность р4 опреде-
ляется длиной волокна, разрываемого под влиянием
собственного веса и выражается и километрах.. Раз-
587
ВИСКОЗНЫЕ НЕВОЛОКНИСТЫЕ ИЗДЕЛИЯ
588
рывное удлинение обычно выражается в процентах
к первоначальной длине В. в.
Для различных видов В. в. наиболее характерны след,
параметры: Вискозный шелк: №0 30—150; №э 2000—
5000; 15—18 км (в сухом виде), 8—10 км (в мокром виде);
разрывное удлинение от 18 до 25%. Вискозная корд-
ная нить: №0 3,7—8; №э 3300—6000; Р130—45 (в сухом
виде); разрывное удлинение от 10 до 15%. Вискозное
штапельное волокно для шерстепряде-
н и я: №э 2200—3500; pi 15—16 км (в сухом виде), 7—9 км
(в мокром виде); разрывное удлинение от 25 до 30%. Вис-
козное штапельное волокно для хлоп-
копрядения: №э 5000—6500; pi 18—25 км (в сухом виде),
8—И км (в мокром виде); длина резки от 35 мм до 45 мм. Эле-
ментарный номер В. в. в шелке, кордной нити или штапельном
волокне может быть в зависимости от требований переработки
снижен до 750—1000 или повышен до 7000—8000, а разрывная
длина в сухом виде может быть увеличена до 55 км. Разрывное
удлинение волокон и нитей также может изменяться в широких
пределах от 7% до 35%. Общий номер шелка и технич.
нитей в зависимости от назначения может быть изменен в ши-
роких пределах от 1,0 до 200. В. в. содержит в кондицион-
ных условиях (20° и 65% относительной влажности) 89% гид-
ратцеллюлозы (степень полимеризации целлюлозы 320—600)
и 11% воды. Набухание В. в. в воде (после отжима в центри-
фуге) при 2 0° достигает (в зависимости от условий и темп-ры
сушки) 85 — 110%, плотность В. в. равна 1,52 —1,53, разрывная
длина в мокром виде снижается до 50%, но может быть повы-
шена до 80% разрывной длины в сухом виде. В. в. мало устой-
чивы к действию минеральных к-т и горячих щелочей, устой-
чивы к действию ацетона, бензола. Прочность В. в. начинает
снижаться при 120°.
Для трикотажных изделий выпускается шелк с более высо-
ким удлинением и несколько меньшей разрывной длиной, шелк
для тканей и технич. целей должен иметь более высокую раз-
рывную длину и пониженное удлинение. Для удовлетворения
этих требований изменяются условия формования и отделки:
состав и темп-pa ванны, условия вытягивания волокон в ванне
или горячей воде после выхода из ванны, величина крутки,
форма паковки шелка на шпулях и т. д. Штапельное волокно
также формуется и отделывается различно, в зависимости от его
назначения: для переработки в смеси с хлопком, шерстью или
льном, для замены льна, джута, для технич. целей, или при
переработке его на искусственный мех и др. изделия. В. в.
выпускают блестящим или матированным (матовым), бесцвет-
ным или окрашенным в массе. Для придания матовости (матиро-
вания) обычно применяют двуокись титана в количестве от
0,5% до 2,0% от веса целлюлозы в р-ре; для крашения воло-
кон в массе — различные пигменты или водорастворимые
красители от 0,5% до 8% от веса целлюлозы. Штапельное
волокно иногда подвергают дополнительной отделке с целью
придания несминаемости или водоотталкивающей отделке;
чаще эти операции производятся с готовой тканью на отделоч-
ных фабриках (см. Отделка тканей специальная). Для при-
дания несминаемости В. в. или ткань обрабатывают продук-
тами конденсации мочевины или меламина с формальдегидом.
Для этой цели применяют р-ры первичных продуктов кон-
денсации мочевины или меламина с формальдегидом, содер-
жащие катализатор, например хлористый аммоний, органич.
к-ту или др. вещества, способствующие конденсации амина
с формальдегидом. После пропитки волокон или ткани соста-
вом, придающим несминаемость, их подвергают термич. об-
работке при 12 0—140°.
Вискозный шелк получают на бобинных (шпульных)
машинах в некрученом виде, на центрифугальных машинах —
в крученом, и на машинах непрерывного прядения и отделки —
в готовом отделанном, крученом и сушеном виде. Вискозное
штапельное волокно производится на прядилыю-отделочно-
сушильных агрегатах производительностью от 10 000 до
2 5 000 кг в сутки, а иногда и выше. Вискозный шелк, кордная
нить и штапельное волокно перед сушкой или перемоткой
с целью придания волокнам мягкости и гибкости подвергают
авпважной обработке или замасливанию. В обоих случаях
на волокно наносится тонкий слой жирового в-ва (от 0,5 до
2 % и выше), иногда смешанного с эмульгатором и минеральным
маслом.
Вискозный шелк нашел широкое применение для
произ-ва тонких тканей типа креповых, а также три-
котажных изделий. Вискозная кордная нить исполь-
зуется для изготовления кордной ткани, служащей
каркасом в покрышках авто- и авиашин. Поскольку
В. в. отличается более высокой теплостойкостью, оно
вытесняет при произ-ве шинных покрышек хлопчато-
бумажный корд. Из вискозного штапельного волокна
изготовляют различные штапельные ткани, сукна,
полушерстяные ткани, искусственный мех, ковровые
и др. изделия.
В. в. в мировом балансе потребления текстильных
волокон занимает второе (после хлопка) место и до-
стигает 20—23% от общего количества текстильных
волокон, т. е. около 1,4 млн. т. Произ-во В. в. со-
ставляет ок. 76—77% от общего произ-ва всех искус-
ственных и синтетич. волокон, но постепенно сни-
жается из-за роста произ-ва новых синтетич. волокон.
Широкое применение В. в. объясняется их экономвч.
эффективностью: трудовые затраты на произ-во 1 кг
пряжи из вискозного шелка или штапельного В. в.
при одинаковом номере пряжи в 2—3 раза ниже зат-
рат на произ-во 1 кг пряжи из хлопка и в 4,5—5 раз
ниже затрат труда на произ-во 1 кг шерстяной пряжи.
Основными материалами для произ-ва В. в. слу-
жит древесная целлюлоза и дешевые доступные хи-
микалии.
Лит. см. при ст. Вискоза. А. Б. Пакшвср.
ВИСКОЗНЫЕ НЕВОЛОКНИСТЫЕ ИЗДЕЛИЯ —
пленка, искусственные губки и др. изделия. Формо-
вание этих изделий принципиально не отличается от
формования волокон. Вискоза продавливается через
щели, или поступает в формы и направляется в оса-
дительную ванну, где происходит осаждение ксанто-
гената целлюлозы и его разложение до гидратцеллю-
лозы. Последнюю в виде изделия отмывают от солей
и к-ты, освобождают от серы, к-рая отлагается из-за
разложения в ванне побочных примесей, промывают,
отбеливают, красят и в нек-рых случаях пластифи-
цируют глицерином для придания изделиям мягкости.
Из вискозы можно формовать тонкую прозрачную
пленку — целлофан, укупорку для бутылок, искус-
ственную колбасную кишку, искусственную губку,
волос для матрасов и для др. целей, тубы для вазе-
лина и различных мазей и др. изделия.
В зависимости от формы вискозных изделий, фильера имеет
или длинную горизонтальную щель, от 1,0 до 1,5 м длины и
ок. 0,2 мм ширины (при произ-ве целлофана), или кольце-
образную щель — при произ-ве колбасной упаковки, или форму
туб и т. п. В нек-рых случаях вискозой заполняют прямоуголь-
ные или фасонные формы и отливают изделия в виде губки,
коробочек для лекарств и т. п. Ввиду того, что В. н. и. по тол-
щине значительно превосходят вискозные волокна, вискоза
направляемая на формование этих изделий, должна иметь
высокую зрелость (см. Вискоза); индекс зрелости достигает
обычно 3,0—5,0 мл по хлористому аммонию. Только такая
вискоза способна быстро затвердевать в виде сравнительно
толстых пленок или др. изделий без изменения формы, заданной
фильерой. Осадительная ванна, применяемая для формования
неволокнистых изделий отличается по составу от ванн, при-
меняемых для формования волокон; обычно содержится 130—
150 г/л серной к-ты и 2 00—22 0 г/л сульфата натрия. Сульфат
цинка не применяется. Темп-pa формования (в первой ванне)
ок. 40°. Обычно формование производится в двух-трех последо-
вательных ваннах до полной коагуляции и разложения ви-
скозы в средних слоях пленки или стенок изделия.
Целлофановая пленка имеет в готовом виде ширину 1200—
1500 мм и ок. 20—40 мк толщины, вес 1 м2 от 30 до 60 г. Более
толстая пленка на обычных целлофановых машинах не произ-
водится. Для того, чтобы пленка была совершенно прозрачна
и бесцветна, она должна формоваться на барабанах и валах
с очень гладкой поверхностью. Для придания мягкости пленку
обрабатывают перед сушкой р-ром, содержащим ок. 70 г'л
глицерина. После сушки готовая пленка содержит ок. 77%
гидратцеллюлозы, 12% воды и 11% глицерина. В отдельных
случаях при изготовлении технич. пленки, трубок и гибких
шлангов для бензохранилищ и для др. целей содержание гли-
церина в пленке значительно увеличивают. Целлофан, как и
любое изделие из гидратцеллюлозы, теряет в мокром виде
около половины прочности; одновременно при этом в воде от-
мывается глицерин, из-за чего после подсыхания пленка ста-
новится хрупкой. Поэтому целлофан часто покрывают тонкой
(2—4 мк толщины) защитной пленкой из ацетилцеллюлозы,
нитроцеллюлозы, полиэтилена, поливинилхлорида или др.
полимеров. Такой целлофан наз. лакированным, он приме-
няется для упаковки изделий, предохраняя их от высыхания.
Широкое применение целлофана для упаковки пищевых изде-
лий обусловлено и тем, что целлофановая пленка непроницаема
для большинства микробов, защищает пищу от загрязнения
и пыли, имеет красивый внешний вид и невысокую стоимость.
Укупорка для бутылок и упаковка для колбасных изделий
также иногда подвергаются пластификации глицерином. Для
повышения пластичности эти изделия перед применением за-
мачиваются в воде. После сушки пленка плотно облегает форму
колбасы или бутылки. Вискозная губка формуется из очень
зрелой вискозы, в к-рую перед формованием замешивают 25%
сульфата натрия, а также различные волокнистые материалы.
Вискоза формуется в прямоугольных формах в горячей воде
или водяном паре, после чего образовавшийся ксантогенатный
гель разлагается кислотой до гидратцеллюлозы. Для создания
пористости в вискозу часто замешивают карбонаты или др.
589
ВИСМУТ
590
вещества, выделяющие газы при обработке паром или кислотой.
Кроме того, в вискозу добавляют пигменты для придания губке
различных окрасок.
Лит.: Волкова. Н., Производство целлофана, М.— Л.,
1950; Роговин 3. А., Основы химии и технологии про-
изводства химических волокон, 2 изд., М., 1957, гл. 14; см.
также при ст. Вискоза. А. Б. Пакшвер.
ВИСМУТ (Bismuthum) Bi — химич. элемент V гр.
периодич. системы Менделеева; п. н. 83, ат. в. 209,00.
Природный В. состоит из одного стабильного изотопа
с массовым числом 209. Неск. инотопос В. входят в
радиоактивные семейства: Bi214 (RaC), Bi210 (RaE),
Bi212 (ThC), Bi211 (AcC) и Bi213. Важнейший из них
Bi210 (Г1/2 = 5 дням) получают облучением природ-
ного В. нейтронами в ядерном реакторе по реакции:
Bi209 (п, у) Bi210. Конфигурация внешних электронов
атома В.: 6s26p3 (см. Атом); энергии ионизации
(в эб): Bi°->Bi+ -> Bi2+ -> Bi3+ -> Bi4+ -> Bi5+соот-
ветственно равны 7,29; 16,7; 25,56; 45,4; 56,1.
В. был известен еще в 16 в., по считался разновидностью
сурьмы, олова или свинца. Лишь в середине 18 в. было уста-
новлено, что он представляет собой самостоятельный «полу-
металл».
Содержание В. в земной коре 2-10~5 вес. %. В при-
роде В. образует преим. сульфиды и тиосоли, а также
окисные и самородные формы. Геохимич. осаждение
происходит в горячей водной (гидротермалиты) и
газовой (пневматолиты) средах вместе с рудами Sn,
W, Pb, As, Au и др. При выветривании В. почти не
рассеивается, а образует скопления характерных вис-
мутовых охр (карбонаты, арсенаты, молибдаты). Ос-
новные промышленные минералы: В. самородный, бис-
мутинит Bi2S3, бисмит Bi2O3, бисмутит Bi2[CO3][ОН]4,
кехлинит Bi2(MoO4)O2. Месторождения находятся во
многих странах. Обычно В. добывается в комплек-
се с другими полиметаллич. сульфидными рудами
и при обогащении накапливается в свинцовом кон-
центрате.
Физические и химические свойства. В. — металл
серебристо-белого цвета с розоватым оттенком. Имеет
ромбоэдрич. структуру, а = 4,7458 А и Za=57°14'13”;
обычно, однако, структуру В. характеризуют как
гексагональную решетку, содержащую 6 атомов, а =
=4,5458 А, с = 11,8620 А; ат. радиус 1,82 А; ионные
радиусы: Bi3+ 1,20А, Bi5+ 0,74 A, Bi3 2,13 А; плотн.
9,80; т. пл. 271,3°, т. кип. 1560°. Существование
аллотропии, модификаций В. не доказано при давле-
нии 760 мм рт. ст.; аномалии свойств В., наблюда-
емые в интервале 75—112°, связаны с процессами де-
формации и рекристаллизации. При давлениях по-
рядка 200 000 кг/см2 и выше обнаружены другие
модификации висмута. Средняя удельная теплоем-
кость 0,030 кал/г-град (при 0—270°); теплота плавле-
ния 2717 (кал/г-атом; теплота парообразования 42700
кал/г-атом. Объемное расширение при затвердева-
нии 3,3%; темп-рный коэфф, линейного расширения
13,4 • 10~6 (0—100°). Теплопроводность твердого В.,
равная 0,02 кал/см-сек-град (при комнатной темп-ре),
ниже, чем у всех прочих металлов, за исключением рту-
ти и теллура. Уд. электросопротивление (в мком-см)'.
106,5 (при 0°), 156,5 (100°), 214,5 (200°), 267 (269°)
и 127 (при 272° — расплавленное состояние), 134,2
(400°); темп-рный коэфф, уд. электросопротивления
4,2-10 3 (0—100°). Электросопротивление В. в твердом
состоянии выше, чем в жидком (как у Ga и Sb). В.
является самым диамагнитным металлом: уд. магнит-
ная восприимчивость — 1,35 • 10 е. В. хрупок и легко
измельчается в порошок. Из В. можно изготовить
проволоку только посредством прессования; свеже-
изготовленная проволока легко изгибается, но вскоре
становится хрупкой; прочность ее на разрыв около
0,5 кг!мм2 (при диаметре проволоки 0,35 мм). Твер-
дость по Бринеллю 9,3 кг]мм2, модуль упругости
3250 кг/мм2 (для литого стержня) и 2 500 кг!мм2
(для проволоки).
В соединениях В. проявляет валентности: —3,
+ 1, +2, +3, +4, +5, из них чаще всего -[-3. Для
всех солей 3-валентного В. очень характерна склон-
ность к гидролизу с образованием основных солей;
соли 5-валентного В. являются сильными окислите-
лями. В сухом воздухе В. устойчив, во влажном по-
крывается тончайшим слоем окиси. При нагревании
на воздухе выше темп-ры плавления легко окис-
ляется, при более высоких темп-рах сгорает в трех-
окись Bi2O3 (см. Висмута окислы). В соляной и разб.
серной к-тах В. не растворяется; легко переходит
в р-р при действии HNO3 (с выделением окислов азота)
или царской водки, а также при нагревании с конц.
H2SO4 (с выделением SO2). В. непосредственно со-
единяется с галогенами и серой. Хлориды образуются
при пропускании тока С12 над порошкообразным В.,
причем при комнатной темп-ре получается BiCl2,
а при сильном нагревании — BiCl3. Другие тригало-
гениды В. также образуются при нагревании (см.
Висмута галогениды). При сплавлении В. с серой,
а также при действии H2S на р-ры солей В. получается
трисульфид. Bi2S3, плотн. 6,5—7,39, т. пл.
685° (с разл.); существование других сульфидов В. не
доказано. При действии НС1 на сплав Bi с Mg обра-
зуется висмутистый водород (висмутин) BiH3 —•
ядовитый газ, неустойчивый даже при комнатной
темп-ре.
При растворении металлич. В. или его соединений
в конц. H2SO4 и дальнейшем концентрировании р-ра
кристаллизуется кислый сульфат Bi2(SO4)3 -HjSOj-
•6Н2О, к-рый при нагревании до 350° переходит в сред-
ний сульфат Bi2(SO4)3, хорошо растворимый в к-тах;
плотн. 6,82, т. пл. 710°. Средний карбонат В. неиз-
вестен; основной карбонат образуется при обра-
ботке р-ра соли В. избытком карбоната щелочного
металла; его состав колеблется в зависимости от кон-
центрации р-ра и темп-ры.
Аналитическое определение висмута. Для аналитич.
химии важны соединения Bi111. В р-р В. переводят
последовательной обработкой объекта к-тами: азот-
ной, соляной и серной или сплавлением с Na2CO3
и растворением плава в HNO3. От элементов сероводо-
родной группы В. отделяют в виде Bi2S3 осаждением
из 18 н. р-ра H2SO4; сульфид В. нерастворим в кис-
лотах, растворим в конц. р-ре Na2S (но не K;S). От
Pb, Cd, Си, Zn В. отделяют в виде его оксибромида
гидролизом р-ра, содержащего КВт и КВгО3. От Ag,
Hg11, Pb, Cd, As, Sbv, Sn, Mn, Ni, Co, Al, Ст В. от-
деляют осаждением его купфероном из 1 н. р-ра
НС1 или HNO3.
Качественно В. открывают по окрашиванию р-ра:
в желтый цвет — в присутствии тиомочевины; в оран-
жевый — дитизона; в желто-оранжевый — йодистого
калия; в бурый — в присутствии станнита натрия;
по образованию оранжево-красного осадка с KJ и
8-оксихинолином и красного осадка с 2,5-димеркапто-
1, 3, 5-тиадиазолом. Первые два реагента используют
для фотометрия, определения В. Весовым путем В.
определяют в виде Bi2S3, BiP04, тионалидата, 8-окси-
хинолината; комплексонометрически — с комплексо-
ном III при pH 1—2 в присутствии 1- (2-пириди-
лазо)-2-нафтола, пирокатехинового фиолетового и др.
Метод применим для анализа сплавов В. со свинцом.
Получение. Висмутовые руды подвергаются обо-
гащению мокрым механич. и флотационным методами;
часто применяется также магнитная сепарация. Из
концентратов В. извлекают пиро- или гидрометаллур-
гия. способами. Практически большую часть В.
получают из отходов медеплавильных и свинцовора-
финировочных заводов.
При обжиге медных руд В. в основном остается в огарке,
в результате переработки к-рого он переходит в пыль, улавли-
ваемую фильтрами. Из свинцовых руд, нонцентратов и пылей
591
ВИСМУТА ГАЛОГЕНИДЫ — ВИСМУТА ОКИСЛЫ
592
В. выделяют в процессе рафинирования свинца эпектропитич.
способом Беттса или способом Беттертон-Кролля. Способ
Беттса основан на растворении анодного свинца и осаждении
его иа катоде. При этом В. и др. металлы, более электрополо-
жительные, чем свинец, остаются в аиодиом шламе. Катоды
отливаются из рафинированного РЬ. Электролитом служит
р-р H3SiF„ в смеси с PbSiF«. Способ Беттертои-Кролпя со-
стоит в том, что при вмешивании в расппавпеииый свинец,
содержащий В., кальция (в виде сплава со свинцом) или смеси
Са я Mg последние образуют с В. тугоплавкие соединения,
всплывающие иа поверхность ванны в виде дроссов (шпака).
Наиболее целесообразный способ извлечения В. из дроссов —
электролиз сплава по Беттсу.
Рафинирование чернового В., содержащего примеси РЬ,
Си,.Sb, As, Fe, S, Te,Sn, Zn, Se, Na, Co, Ni, Ag и Au, производят
огневыми, мокрыми или электропнтич. способами. К огневым
способам относятся: окислительная плавка, плавка с продувкой
воздуха и хлорирование газообразным хлором. Мокрые спо-
собы состоят в растворении чернового В. в HNO3 с последую-
щий гйдропизом. Эпектролитпч. рафинирование применяется
гл. обр. для отделения В. от благородных металлов. Указан-
ные способы позволяют получить В. с содержанием до 99,95%.
Для получения В. высокой степени чистоты, используемого
в качестве теплоносителя в энергетич. ядерных установках, В.
рафинируют гидрометаллургии, способом (цементацией серебра
металлическим В.) и кристаллофизич. методами (вытягиванием
металла из расплава и с помощью зонной плавки).
Применение. Металлич. В. применяется в приборах
для измерения напряженности магнитного поля. Бла-
годаря своим физич. свойствам — низкая темп-ра
плавления, хорошая теплопроводность по сравнению
с пеметаллич. теплоносителями (водой, солями, ор-
ганич. веществами), химич. стойкости и малому се-
чению захвата медленных нейтронов — В. нашел
широкое применение в энергетич. ядерных реакто-
рах в качестве теплоносителя. О применении В. в
сплавах см. Висмута сплавы. Многие соединения В.
используются в медицине (дерматол — основной гал-
лат В. СвН2(ОН)3 • COOBi(OH)2, ксероформ — ос-
новной трибромфенолят В. CeH2Br3O(BiO)2OH и др.
О применении др. соединений В. см. Висмута гало-
гениды, Висмута окислы, Висмута нитраты.
Лит.: Т о м п с о и Д ж. Г Висмут, пер. tc аигл.), М.—Л.,
1932; М о с т о В и ч В. Я. и А и и си м о в С. М., Металлур-
гия свинца, М.—Л., 1940; Б у с е в А. И», Аналитическая хи-
мия висмута, М., 1953; Сажин Н. П. иДулькинаР. А.,
Получение металлического висмута высокой чистоты, М., 1955;
G.tuelin, 8 Aufl., Syst.-Nummer 19, Wismut und radioaktive
isotope, B., 1927; M e 1 1 о r, 9, L.—N. Y.—Toronto, 1947, пере-
пад.'; 1952; Pascal, V.H, P„ 1958; К 1 г k, v. 2, N. Y., 1948,
p. 526—40; Сажин H. П., в кн.: Справочник металлурга
по цветным металлам, под ред. Н. Н. Мурача, т. 2.М., 1947;
Славинский М. П., Физико-химические свойства эле-
ментов, М., 1952. Д. Л. Абрамсон.
ВИСМУТА ГАЛОГЕНИДЫ — соединения висмута
с F, CI, Вг, J. Низшие В. г. BiCi, BiBr, BiJ малоус-
тойчивы; существование дигалогенидов не достоверно.
Наиболее изучены тригалогениды висму-
та. Фторид BiF3 в воде не растворим; получается
осаждением р-ра Bi(NO3)3 конц. р-ром K.F или дей-
ствием водной HF на Bi(OH)3 при нагревании. При
обработке BiF3 кипящей водой образуется BiOF.
Остальные тригалогениды висмута BiCl3, BiBr3 и
BiJ3 в р-ре гидролизуются с образованием нераство-
римых оксисолей состава BiOX, где X = Cl, Вг, J.
С галогенидами нек-рых металлов В. г. дают комплекс-
ные соединения с анионами[В1Х4]~,[В!ХБ]2~и [BiX6]3~
(образование K[BiJ4] — желтого' цвета, используется
в колориметрии для определения висмута). Хло-
рид BiCl3— белоснежные, расплывающиеся на воз-
духе кристаллы; плоти. 4,75; т. пл. 230°, т. кип. 447°,
теплота образования Д//298 = —90,61 ккал/моль.
Растворяется в НС1 (с образованием комплексных
хлорокислот HBiCl4 и H2BiCl5), а также в спирте,
эфире и ацетоне. Получается хлорированием метал-
лич. Bi или при растворении Bi2O3 в НС1 или Bi
в царской водке. Бромид BiBr3 — гидроскопич-
ные оранжево-желтые кристаллы, плотность 5,7;
т. пл. 217°, т. кип. 453°. Соответствующая бромиду
бромцкись BiO.Br используется в ветеринарии. И о-
ди д ВiJ3 — негигроскопичные серовато-черные кри-
сталлы;'плотность 5,64, т. пл. 439°, разлагается при
500°. Соответствующая иодиду кирпично-красная
иодокись BiOJ — фармацевтич. препарат.
• Лит. см. при ст. Висмут. Д. Л. Абрамсон.
ВИСМУТА НИТРАТЫ — нормальная и основные,
азотнокислые соли висмута. Нормальная соль при
обычных условиях существует в виде кристаллогид-
рата Bi(NO3)3 • 5Н2О — бесцветных кристаллов; плоти.
2,83. При, 30° начинает выделять HN03, при 75,5°
распадается на жидкую фазу и основную соль состава
Bi2O3 • N206 • Н20. Водой гидролизуется с образова-
нием основных солей, растворяется в к-тах при нагре-
вании; готовится выпариванием р-ра Bi в конц.
HNO3 до тех пор, пока темп-ра кипения р-ра не до-
стигнет 125°; при охлаждении выпадают кристаллы
Bi(NO3)3 • 5Н2О. Основные В. н. — продукты гид-
ролиза азотнокислых р-ров Bi(NO3)3 при разбавле-
нии их водой или под действием NaHCO3, NaNO3,
органич. оснований, солей слабых органич. к-т и пр.
Состав основных нитратов зависит от темп-ры, концен-
трации и кислотности р-ра. При добавлении холодной-
воды осаждается неустойчивая соль Bi2O3 • N2O5 •
• 2Н2О, к-рая под действием 6%-ной HN03 при стоянии
теряет 1 молекулу Н2О. Ок. 30° в р-ре В. н. с содержа-
нием 1—2% свободной HN03 образуется 6Bi2O3 -
•5N2O5 • 9Н2О; ок. 70° — 10Bi203 • 9N2O6 • 7Н2О. По-
следние две соли являются главными составными ча-
стями технич. основного нитрата. При повторной об-
работке к.-л. из них горячей водой до исчезновения
HN03 в промывных водах получается наиболее основ-
ная соль этой группы 2Bi2O3 • N2O3 • Н2О. Основной
В. н. применяется как медицинский препарат и для
живописи по фарфору.
Лит. см. при ст. Висмут. Д. Л. Абрамсон.
ВИСМУТА ОКИСЛЫ — соединения висмута с кис-
лородом. Известны следующие В. o.:Bi2O3, Bi2O4 и
Bi2O6. Висмута трехокись (окись) Bi2O3 —
основной окисел, встречается в природе в виде мине-
рала бисмита, или висмутовой охры. Тяжелый желтый
порошок, при нагревании становится оранжевым или
бурым (после охлаждения приобретает первоначаль-
ный цвет). Bi2O3 существует в нескольких моди-
фикациях. Обычная модификация имеет плотн. 8,9,
т. пл. 820°. Теплота образования Bi2O3 ДН298 =
= —137,9 ккал/моль. Bi2O3 нерастворим в воде; незна-
чительно растворим в конц. р-рах щелочей, легко —
в к-тах с образованием соответствующих солей. Р-ры
щелочей и NH4OH осаждают из р-ров этих солей гид-
роокись Bi(OH)3, к-рая при нагревании до 100° перехо-
дит в более устойчивую гидроокись висмутилa BiOОН
(в и с м у т и л BiO+ — одновалентный радикал). По-
лучают Bi2O3 добавлением избытка NaOH к р-ру
Bi(NO3)3. Применяют в произ-ве эмалей и керамич.
красок.
Висмута четырехокись Bi2O4 — мало-
характерный для Bi окисел; содержит, по-видимому,
один атом Bi в 3-валентном состоянии и другой — в
5-валентном: O==Bi—О—Bi< . Коричневый поро-
ге
шок, плотность 5,6; т. пл. 305°.
Висмута пятиокись Bi2O6 — ангидрид
не выделенной в свободном состоянии висмуто-
вой кислоты HBiO3; порошок коричнево-крас-
ного цвета; плотность 5,10; растворяется в НС1 (с
выделением С12) и в щелочах. Bi2O5 образуется при
окислении Bi2O3, взвешенной в нагретом почти до
кипения 37—38% р-ре КОН, хлором или электроли-
зом суспензии Bi2O3 в 41—46% р-ре КОН, в к-рый
добавлен мелкий порошок КС1. Щелочные соли
HBiO3 (висмутаты) по составу приближаются к MeBiO3
(Me — одновалентный металл) и являютсн сильными
окислителями, находят применение в аналитич. химии.
Лит. см. при ст. Висмут. Д. Л. Абрамсон..
593
ВИСМУТА СПЛАВЫ — ВИТАМИН Р
594
ВИСМУТА СПЛАВЫ — металлич. системы, одним
из компонентов к-рых является висмут; особенно
подробно изучены двойные сплавы висмута. Наиболее
характерные висмутиды, т. е. соединения вис-
мута с другими металлами, отвечают типу водороди-
стого висмута H3Bi, в полном согласии с положением
Bi в V группе периодич. системы. Соединения этого
типа характеризуются дистектич. точками на линиях
ликвидуса; таковы, напр., соединения: Li3Bi (т. пл.
1145°), Na3Bi (т. пл. 775°), K3Bi (т. пл. 671°), Ca3Bi2
(т. пл. 928°), Mg3Bi2 (т. пл. 823°). В системе Bi — Т1
Н. С. Курников с сотрудниками открыл в 1913 один
из самых типичных бертоллидов (см. Далътониды и
бертоллиды). Способность висмута к образованию
твердых р-ров очень невелика; непрерывные твердые
р-ры (1-го типа Розебома) найдены только в системе
Bi — Sb. С нек-рыми металлами (напр., Zn, Ga, Al,
Сг, Fe, Со) висмут дает два несмешивающихся жидких
слоя. С легкоплавкими тяжелыми металлами висмут
нередко образует эвтектики, напр. 60% Bi-j-40%
Cd, т. пл. 144°, 58% Bi +42% Sn, т. пл. 139°. Еще
ниже плавятся тройные, четверные и пятерные эв-
тектик. В. с., напр. тройная эвтектика: 51,65% Bi,
40,2% РЬ и 8,15% Cd, т. пл. 91,5°; четверная эвтек-
тика: 49,41% Bi, 27,67% РЬ, 12,88% Sn, 10,02% Cd
(очень близкая к т. н. сплаву Вуда) — т. пл. 70°.
СамЫм легкоплавким из В. с., не содержащим ртути,
является пятерная эвтектика состава 40,95% Bi,
22,10% РЬ, 10,65% Sn, 8,20% Cd, 18,10% In, т. пл. 47°.
Легкоплавкие В. с. применяют гл. обр. в автома-
тик. огнетушителях, действие к-рых основано на рас-
плавлении пробки из В. с. Легкоплавкие В. с. исполь-
зуются также как припои (особенно для спайки стекла
с металлом), для получения сложных отливок в гип-
совых формах, для предохранителей в паросиловых
установках и др. Образование трудноплавких соеди-
нений — Ca3Bi2 и Mg3Bi2 — используется в металлур-
гии для выделения висмута из чернового свинца.
Лит.: К у р н а к о в Н. С., Собрание избранных работ,
т. 2, Л.—М., 1939; Хансен М., Структуры бинарных спла-
вов, пер. [с нем.), т.1, М.—Л., 1941; Hansen М., Constitu-
tion of binary alloys, 2 ed., N. Y., 1958; D anlels E. J.,
Fusible alloys containing tin, [L., 19371 (Technical publications
of the International tin research and development council. Ser. B.,
№ 5). С. А. Погодин.
ВИСМУТОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — из-
вестны полные, в которых все валентности висмута
замещены органич. радикалами, и неполные, в к-рых
часть валентностей занята, напр., атомами галоге-
нов. Известны также гетероциклич. В. с. с атомом
висмута в цикле.
Алифатич. В. с. — бесцветные жидкости, крайне
чувствительные к кислороду и устойчивые к воде;
низшие члены ряда самовоспламеняются на воздухе.
При осторожном окислении R3Bi получаются оксиды
R3BiO. Ароматич. В. с.—твердые вещества. Физиче-
ские свойства важнейших В. с. приведены в табл.
Формула T. пл., °C T. кип., °С/лш Плотность, г/см3
(СНз)з В*. (CsHgJa Bi (СвН&)з Bi (CgHg)3 BiCl8 (CQH5)5 Bi 78° 141,5" разл. при 105° ИО 107°/79 2,300(18”) 1.82 1,585
R3Bi присоединяют галогены, образуя R3BiHal2
(R — ароматич. радикал); последние могут быть вос-
становлены в R3Bi действием гидросульфита, серни-
стого аммония, гидразингидрата. (C6H6)3BiCl2 с фе-
Нйллитием образует пентафенилвисмут. Диспропор-
ционирование типа 3R2BiHal 5±2R3Bi + BiHal3 лег-
ко Протекает в присутствии восстановителей; обрат-
ная реакция служит методом получения алкилвис-
мутгалогенидов. При нагревании R3Bi с R2'Hg или
R'Li происходит обмен радикалов: R3Bi + 3R'Li —►
—> R3'Bi + 3RLi. Дезалкилирование R3Bi, приводящее,
к соединениям типа R2BiX, может быть осуществлено
действием галогенов, Brl, CNCi, (CNS)2, Т1С13, SiCl4,-
CuCl2; органич. к-ты полностью дезалкилируют и
дезарилируют R3Bi. Сулема арилируется за. счёт
В. с., напр: (C6H6)3Bi+ HgCl2 + H2O->C6H6HgCl’+
+ BiOCl + 2C6H6. Li, Hg, Sn и Sb при нагревании с
(C6H6)3Bi фенилируются. '
В. с. получают: 1) Алкилированием галогенидов
висмута магний-, литий-, алюминий- и ртутнроргапич.
соединениями; при этом обычно образуются триалкил-
и триарилвисмуты; из соединений типа R2BiHal
и R'MgHal могут быть получены несимметричные
продукты типа R2BiR'; 2) взаимодействием сплава
Bi—Na с галогеналкилами (-арилами); при этом по-
лучается также R3Bi; взаимодействием В. е., содер-
жащих прямую связь Bi с щелочным металлом (напр.,,
R2BiNa) с R'Hal в жидком NH3, можно получать не*
симметрич. соединения R2BiR'; 3) разложением двой-
ных диазониевых солей BiHal3 порошками висмута
или меди; при этом образуются ароматич: В. с.;
4) (CH3)3Bi может быть получен по реакции: С3А14 +
+ 9НС1 + BiCl3 -► 4А1С13 + (CH3)3Bi.
Лит.: Синтетические методы в области металлоорганиче-
ских соединений, под ред. А. Н. Несмеянова и К. А. Кочеш-
кова, вып. 8, М.—Л., 1947; Gilman H.,'Yale Н. L-,
Chem. Revs., 1942, 30, .V» 2, p. 281—320; Krause E., Gros-
s e A., Die Chemie tier metallorganischen Verbindungen, B., 1937;
S. 296; W i 11 i g G., Clauss K., Liebigs. Ann. Chem.,
1952, 578, H.2, S. 136—46. O. IO. Охлобыстин.
ВИТАМИН A — то же, что ретинол.
ВИТАМИН Bj — то же, что тиамин.
ВИТАМИН В2 — то же, что рибофлавин.
ВИТАМИН В6 — то же,чтопирибоксиц(пиридоксол),
ВИТАМИН В12 — то же, что цианкобаламин.
ВИТАМИН В13 — то же, что оротовая кислота ',
ВИТАМИН В16 — то же, что пацгамовая кислота.
ВИТАМИН Вс — то же, что фолиевая кислота.
ВИТАМИН Вт — то же, что карнитин.
ВИТАМИН С — то же, что аскорбиновая кислота.
ВИТАМИН Н — то же, что биотин.
ВИТАМИН F — то же, что незаменимые ненасы-
щенные жирные кислоты.
ВИТАМИН Р — группа веществ, относящихся к
производным флавана или флавона, регулирующих
проницаемость капилляров и увеличивающих их
прочность. Важнейшими из них являются: производ-
ные флавана (I) — эпикатехин (R — Н) и эпикате-
он
—он
он
вона (II) — кверцетин (R—Н), кверцитрин (R==
рамноза), рутин (R = рамноглюкоза) или произ-
водные дигидрофлавона (III) — гесперидии (R =СН3,
R' = рамноглюкоза), эриодиктин (R=H, R' = рамно-
за), лейкоцианидин, эскулин и др.
хингаллат R—СО—/ Ч
, производные фла-
ОН
R'
Ан
СНз
W
Общим для этих сое-
динений является, нали-
OR чие двух фенольных ги-
дроксилов в орто-поло-
жении; кроме лейкоциа-
нидина — кристалличе-
ские продукты, раствориг
595
ВИТАМИН РР — ВИТАМИНЫ
596
мые в воде и этаноле (за исключением рутина и
особенно кверцетина).
Все эти соединения дают темное окрашивание с 1%-ным
этаноловым р-ром FeCl3, восстанавливают Си2+ в щелочной
среде и взаимодействуют с аммиачным р-ром AgNO3. Специ-
фичные реакции: на катехины — ярко-малиновая окраска
с 1%-ным р-ром ванилина в конц. НС1; на флавонолы — уве-
личение интенсивности желтой окраски при действии щелочей
и особенно NH4OH; при восстановлении флавонолов, а также
гесперидина и зскулина Mg в солянокислой среде образуется
оранжево-красная окраска; на пейкоцианидин — образова-
ние красно-фиолетовой окраски при нагревании с разбавлен-
ными минеральными к-тами. Для обнаружения флавоноидов
особенно пригодна хроматография на бумаге. Колич. опреде-
ление: катехинов — по реакции с ванилином или железотар-
тратным реактивом; флавонолов — по реакции с лимонно-
боратиым реактивом; гесперидина и эриодиктпна — по вос-
становлению в соответствующий антоцианидин при помощи
Mg + НС1.
Флавоноиды получают из плодов цитрусовых, из
зеленой массы гречихи, из листьев софоры и чайного
растения и околоплодников конских каштанов. Ви-
тамин Р используют для лечения различных гемор-
рагии. диатезов, капилляротоксикозов, гематурии,
глазных кровоизлияний, гипертонич. болезни и др.
Кроме того, витамин Р (совместно с антикоагулянтом)
применяется для предотвращения чрезмерного пони-
жения свертываемости крови и для повышения эффек-
тивности Действия аскорбиновой к-ты (комплексные
препараты, содержащие витамины Р и С). В связи
с отсутствием доказательств, что наличие витамина Р
необходимо для нормальной жизнедеятельности ор-
ганизма, нек-рые исследователи рассматривают ка-
пилляроукрепляющее действие флавоноидов как фар-
макологическое, а не витаминное.
Лит.: Запромётов М. Н., в сб.: Современные во-
просы советской витаминологии, М., 1955, с. 48; Бпофлаво-
ноиды и проницаемость капилляров. [Сб.]. Под ред. М. Н. За-
промётова, пер. с англ., М., 1957; Витамин Р — его свойства
и применение, М., 1959 (Витаминные ресурсы и их использо-
вание. Сб. 4); Запромётов М. Н., Физиология растений,
1958, 5, вып. 3, с. 296; Moderne Methoden der Pflanzenanalyse,
Bd3,B. — GBttingen—Hdlb., 1955. M. II. Запромётов.
ВИТАМИН РР — то же, что никотиновая кислота.
ВИТАМИНЫ (от лат. vita — жизнь) — группа
органич. веществ разнообразной структуры, необ-
ходимых человеку, животным и др. организмам. В
1880 г. Н. И. Лунин в опытах на животных показал,
что пищевые потребности организма не ограничива-
ются белками, жирами, углеводами и солями, а вклю-
чают также минимальные количества каких-то других
веществ, незаменимых для питания. Эти данные бы-
ли подтверждены многими учеными; в 1912 польский
ученый К. Функ назвал такие вещества витаминами,
а заболевания, вызываемые недостатком В., — а в и-
таминозами.
В. являются биологич. катализаторами химич. реак-
ций или реагентами фотохимич. процессов, протекаю-
щих в организме. В. участвуют в обмене веществ прея-
мущ. в составе ферментных систем, причем в организм
человека и животных В. поступают гл. обр. из внешней
среды; по сравнению с основными питательными ве-
ществами количество В., необходимых для питания
живых организмов, ничтожно. При недостатке В.
в организме развиваются те или иные заболевания.
Первоисточником В. являются гл. обр. растения.
Человек получает В. или непосредственно с расти-
тельной пищей, или косвенно — через животные
продукты, в к-рых В. были накоплены из раститель-
ных материалов во время жизни животных. Важная
роль в образовании В. принадлежит также микроор-
ганизмам, в частности микрофлоре пищеварительного
тракта, за счет жизнедеятельности к-рой, напр. взрос-
лые жвачные животные, обеспечиваются в достаточ-
ной мере витаминами группы В.
Метод обнаружения В. с помощью эксперименталь-
ных авитаминозов лег в основу исследований в этой
области. При сравнительных физиология, испытаниях
выяснилось, что не все виды животных нуждаются во
всех В. и поэтому, говоря о том или ином В., нужно
иметь в виду те организмы, к-рые нуждаются в его
поступлении в готовом виде извне. Обнаружилось,
что ряд микроорганизмов (особенно молочнокислые
бактерии и дрожжевые грибки) также нуждаются в
В., а развитие корней высших растений зависит от
притока нек-рых В. из зеленых частей, где происхо-
дит их биосинтез. Независимо от способности к само-
стоятельному синтезу все организмы, клетки и ткани
используют В. в своих обменных реакциях, что ставит
В. в разряд интегрирующих факторов живой природы,
связывающих взаимными нитями мир растений, жи-
вотных, микробов, и придает учению о В. общебиоло-
гич. интерес.
На протяжении последних 30 лет почти исчерпываю-
ще изучена химич. природа 20 отдельных В. При этом
оказалось, что многие из них представлены в природе
не единичным веществом, а группами из 3—5 и более
родственных соединений, отличающихся деталями
строения и степенью физиология, активности. Еще
больше было синтезировано искусственных аналогов
В. с целью выяснения функциональных группировок
молекул, что дало много ценного для понимания дей-
ствия В. Особенно интересным оказалось то, что некото-
рые производные В. с замещенными функциональными
группами оказывают на организм противоположное
действие по сравнению с настоящими В., являясь та-
ким образом антивитаминами.
Номенклатура и классификация В. До последнего
времени В. имели буйвенные обозначения, химич.
названия или названия, характеризовавшие их фи-
зиология. действие. В нек-рых случаях одной буквой
обозначалось несколько В., иногда весьма отличных
по химич. строению. Напр., к витаминам группы (ком-
плекса) В относили: Вх — тиамин, В2 — рибофла-
вин, Вв — пиридоксин, биотин, амид никотиновой
кислоты, фолиевую кислоту, ВХ2 — цианкобаламин,
пантотеновую кислоту и др. В 1956 комиссия по но-
менклатуре биохимия, секции Международного союза
по чистой и прикладной химии рекомендовала отойти
от буквенных и физиология, обозначений и принять
единые названия, к-рые в дальнейшем должны стать
общеупотребительными. Принято различать жиро-
и водорастворимые В., к-рые классифицируются след,
образом:
Классификация витаминов
Новая номенклатура Прежние обозначения
Жирорастворимые витамины
Ретинол
Дегидроретинол
Эргокальциферол
Холекальцифероп
а-, ₽- и ^-Токоферолы
Филлохинон
Фарнохинон
одорастворимые
В
Аскорбиновая к-та
Тиамин
Рибофлавин
Пиридоксин
Цианкобаламин
Никотиновая к-та и ее амид
Пантотеновая к-та
Биотин
Мезоинозит
n-Аминобензойная к-та
Фолиевая к-та
Холин
Витамин А
Витамин А3
Витамин D3
Витамин D3
Витамин Е
Витамин Кх
Витамин К2
витамины
Витамин С
Витамин В,
Витамин В2
Витамин В0
Витамин В,з
Витамин РР
Пантотеновая к-та
Биотип
Мезоинозит
п-Аминобензойная к-та
Витамин Вр
Холин
Кроме того, к числу В. относятся также незамени-
мые ненасыщенные жирные кислоты (витамин F),
ряд флавоноидов (витамин Р), карнитин (витамин Вт),
оротовая кислота (витамин Вхз) и пангамовая кисло-
та (витамин В16).
597
ВИТАМИНЫ — ВИТАМИНЫ К
598
Определение В. производится биологич. методами,
основанными на учете ростового эффекта и призна-
ков авитаминоза при содержании экспериментальных
животных (крысы, морские свинки, цыплята) на очи-
щенных диетах с добавлением различных доз испы-
туемого в-ва и стандартного препарата. На принципе
ростового эффекта основаны и микробиологии, методы
с использованием специальных штаммов микроор-
ганизмов, не способных к синтезу того или иного В.
Для многих В. разработаны химич. методы определе-
ния, основанные на получении очищенных экстрактов
В. и их фотометрировании или флуорометрии, с про-
ведением в необходимых случаях соответствующих
реакций. Для определения активности В. было введено
понятие т. н.международных или интернациональных
единиц (ME), к-рые представляют собой нек-рое услов-
ное количество стандартного препарата в миллиграм-
мах (мг) или микрограммах (мкг). Напр., для витамина
А 1МЕ равна 0,344 мкг его ацетата, а для витамина D—
0,025 мкг эргокальциферола. С получением чистых В.
необходимость в этих единицах отпала.
Поступая в организм, В. усваиваются (ассимили-
руются), образуя более сложные производные (эфир-
ные, амидные, нуклеотидные и др.), к-рые, как пра-
вило, соединяются с белком, образуя многочислен-
ные ферменты — типичные биологич. катализаторы,
ускоряющие разнообразные реакции синтеза, рас-
пада и перестройки веществ в организме. Наряду
с ассимиляцией в организме непрерывно идут про-
цессы разложения (диссимиляции) В. с выделением
продуктов распада. Если В. не поступают в достаточ-
ном количестве с пищей, нарушается деятельность
ферментных систем, в к-рых они участвуют, а следо-
вательно, и обмен веществ и развиваются множест-
венные формы расстройств, наблюдаемые при авита-
минозах. Эти явления могут развиться и на почве
нарушения усвоения и использования В. в организме.
Известно св. 100 отдельных ферментов, в состав к-рых
входят В. и еще большее число катализируемых ими
реакций. В. (гл. обр. водорастворимые) являются
участниками процессов распада пищевых веществ и
освобождения заключенной в них энергии (витамины
В1( В2, РР и др.). В неменьшей степени они участвуют
в процессах биосинтеза. Это касается синтеза амино-
кислот и белка (витамин В6, В12), синтеза жирных к-т
и обмена жиров (пантотеновая к-та), синтеза пури-
новых и пиримидиновых оснований и обмена нуклеи-
новых к-т (фолиевая кислота, В12), образования мно-
гих физиологически важных соединений — ацетил-
холина, глутатиона, стероидов и др. Менее ясен
каталитич. способ действия жирорастворимых В.,
но и здесь несомненно их участие в построении струк-
тур организма, напр. в образовании Костей (витаминD),
развитии покровных тканей и образовании тако! о
важного пигмента, как зрительный пурпур (витамин А),
нормальном развитии эмбриона (витамин Е) и др.
Как правило, В. не токсичны, по нек-рые из них при
дозировках, превышающих в неск. сот раз рекомен-
дуемые нормы, вызывают расстройства, называемые
гипервита микозами. К таким относятся
витамины А и D.
Практическое использование В. До недавнего вре-
мени считалось, что В. лишь защищают организм от
авитаминозов, для чего нужны сравнительно неболь-
шие количества их в пище; в последние годы установ-
лено, что при повышении содержания В. до физиоло-
гически оптимальных норм в организме создаются
такие концентрации В., к-рые повышают устойчивость
организма против всякого рода неблагоприятных воз-
действий, в том числе против инфекций. Так как содер-
жание В. в ряде продуктов (овощи, фрукты, молоко,
масло и др.) подвержено резким сезонным колебаниям,
а в нек-рых продуктах (белый хлеб, макароны) очень
мало, или они совсем лишены В. (маргарин, сахар),
повышение норм витаминного питания достигается
витаминизацией пищевых продуктов и применением
препаратов В. Широкое применение В. находят в
животноводстве в качестве средства борьбы за со-
хранение поголовья и повышения продуктивности
с.-х. животных и птицы, а также в клинич. практике
в качестве составной части комплекса лечебных ме-
роприятий при различных заболеваниях. В основе
произ-ва В. лежат методы их синтетич. получения,
особенно успешно развитые в послевоенные годы. В
большинстве — это многоступенчатый процесс тон-
кого органич. синтеза. Наряду с синтезом нек-рые В.
получают из природных источников: А — из рыбьих
жиров, Е — из растительных жиров путем молеку-
лярной дистилляции, В2 и В12 — микробиологии,
синтезом, С — из плодов шиповника, Р — из листьев
чайного растения и цветов софоры, D — из природ-
ных стеринов путем их облучения.
Лит.: Кудряшов Б. А., Физиологическое и биохими-
ческое значение витаминов, М., 1953; Березовский В. М.,
Химия витаминов, М., 1959; Б е & р А. А., Рубцов И. А.,
Синтез витаминов, М 1956; Шнайдман Л. О., Произ-
водство витаминов, М., 1958; Мережинский М. Ф.,
Механизм действия и биологическая роль витаминов, Минск,
1959; Одинцова Е. И., Микробиологические методы
определения витаминов, М., 1959; Леутский К. М.,
Витамин А, Черновицы, 1959; Номенклатура витаминов (Науч-
ная информация), Биохимия, 1957, 22, вып. 3, с. 615; Тру-
фанов А. В., Биохимия и физиология витаминов и анти-
витаминов, М., 1959; lhe vitamins chemistry, physiology,
pathology. Ed. by W. H. Sebrell, R. S. Harris, v. 1—3, N. Y.,
1954. В. H. Букин.
ВИТАМИНЫ D — см. Эргокальциферол и Холе-
кальциферол.
ВИТАМИНЫ Е — то же, что токоферол.
ВИТАМИНЫ К — группа производных 2-метил-
1,4-нафтохинона (I), замещенных в положении 3,
обладающая сильными антигеморрагич. свойствами
(способствует свертыванию крови). В. К. нераство-
римы в воде, хорошо растворимы в эфире, бензоле,
ацетоне, петролейном эфире и безводном этиловом
спирте. Для этих витаминов характерна способность
к окислительно-восстановительным превращениям;
при восстановительном ацетилировании образуют
диацетилдигидропроизводные. Известны след. В. К:
Витамин Kt (фитохинон, филлохинон, 2-метил-З-дифар-
незил-1,4-нафтохинон), R = —СН2СН=С(СН2СН2СН2С)3СН8
I I
СНз СНз
(см. I); вязкий, маслообразный продукт желтого цвета;
т. пл. —20°; т. кип. 115—145° (при 0,0002 мм рт. ст,). В. Kt
распространен в зеленых частях растений; синтетически его
получают из 2-метил-1,4-нафтогидрохинона, к-рый конден-
сируют с фитолом (его галогенопроизводными или изофитолом)
в присутствии щавелевой к-ты (или других конденсирующих
веществ) с последующим окислением.
Витамин К2, в зависимости от величины радикала
боковой цепи различают: витамин К2 (фарнохинон, 2-метил-
З-дифарнезил-1,4-нафтохинон),
R = —CH2CH=C(GH2CH2CH~C)5—СН3
сн3 сн8
(см. I); желтые кристаллы, т. пл. 53,5—54,5е;
витамин К2 — (изопреиолог фарнохннона, 2-метил-З-фарнезил-
дигеранил-1,4-нафтохинон),
R = — СН2СН=С(СН2СН2СН=С)9СН3
сн8 сн3
(см. I); желтые кристаллы, т. пл. 54°. Витамин К2 обра-
зуется под влиянием микроорганизмов, в частности выде-
лен из гниющей рыбы; синтетически его получают: фарно-
хинон— из 2-метил-1,4-нафтогидрохиноиа и полного транс-
В,Ь-фарнезилнеролидола, а изопреиолог — из того же аро-
матич. соединения и полного транс-D, L-ф ар незилгер ани лли-
налоола.
Помимо природных витаминов К, известны синтетич.
соединения, к-рые также обладают сильными антигеморрагич.
свойствами; группа таких наиболее активных соединений
также обозначается термином «Б.к» с соответствующими ин-
дексами. К ним относятся:
Витамин К3, к-рый известен в виде: 2-метил-1,4-наф-
тохинона, или метилона (II); лимонно-желтые кристаллы,
599
ВИТЕЛЛИН — ВИТТИГА РЕАКЦИЯ
600
т. пл. 106°, обладает слабым характерным запахом и жгучим
вкусом, мало растворим в воде, хорошо — в спиртах и маслах;
натриевой соли бисульфитного производного 2-метил-1,4-
нафтохинона, или викасола (III) — бесцветные кристаллы, т. пл.
154 .— 157°, горького вкуса, хорошо растворим в воде, труд-
но — р спирте, почти нерастворим в эфире. Анализ В. К3
основан на реакции восстановления хииона в гидрохинон
Н.Г; выделяющийся при атом иод титруется тиосульфатом
натрия.
Витамин К4 (2-метил-1,4-пафтогидрохинои, а также
его дцацетат, дибутират и дифосфат в виде тетранатриевой сопи)
(IV) —- бесцветные кристаллы.
Витамин К; (2-метил-4-амипо-1-нафтолгидрохлорид,
а также N-ацетат) (V) — розоватые кристаллы, т. пл. 280°
(с разл.), т. пл. N-ацетата 209°.
Витамин К, (2-метил-1,4-дпаминонафталин дпгидро-
хлориД, а также его диацетат) (VI) — бесцветные кристаллы,
т. пп. 360° (диацетата 310°).
Витамин К, (3-метип-4-амино-1-нафтолгидрохпорид)
(VII) — бесцветные кристаллы.
Сравнительные данные биологич. активности (ми-
нимальная активная доза) витаминов К (дневная
потребность- авитаминозных цыплят) в мкг: К3 0,3;
К4 0,5; Кх 1,0; Кв 1,0; К7 1,0; К2 1,6.
Лит.: Isler О. [u. a.], Helv. chim. acta, 1958, 41,
р. 786; см. также при ст. Витамины.
В. М. Березовский.
ВИТЕЛЛИН — фосфопротеид, являющийся одним
из компонентов липопротеидов яичного желтка. Строе-
ние В. не установлено, изучен его аминокислотный со-
став. В. растворим только в щелочных р-рах, его,
по-вйдимому, следует рассматривать как денатури-
рованный белок. При расщеплении В. получены фраг-
менты с высоким содержанием фосфора, названные
овотиринами я, р и р Овотирин-0 идентифи-
цирован с витеЛлиновой к-той, к-рая м. б. получена
обработкой В. NH4OH. Овотирин-0 был расщеплен на
овотприны-Pj и р2.
Состав овотиринов: а — 10,87% N, 13,76% Р; р4 —
11,33%N, 12,55%Р; 02 — 10,92%N, 12,09%Р,
3,31% Fe; р — 10,7% N, 7,9% Р.
В- содержится в яичном желтке (ок. 15%).
Лит.; Белки, пер. с англ., подред. Г. Нейрата и К. Бейли,
т. 3,М., 1958, с. 735; Лисовская Н. П., Ливанова
Н. Б., Фосфопротеины, М., 1960. Л. Л1. Гинодман.
ВИТТИГА РЕАКЦИЯ — способ получения соеди-
нений с двойной связью между атомами углерода взаи-
модействием трифенилфосфинметиленов с альдеги-
дом или кетоном. Напр., при реакции трифенилфос-
финметилена (I) с ацетоном образуется биполярное
соединение (II), соответствующее циклич. переход-
ному состоянию (Па); при нагревании (II) распадается
на трифенилфосфиноксид и олефин:
(C6H6)3P=CRR- + СН3СОСН3 —► (C6H6)3PCRR'C(CH3)3
I о-
1 и
r(CeHb)3P-?CRR’ 1
1 -----► (С6Н5)3РО+ CRR'-C(CH3)3
L о ’“'•’“'•'з'зJ
На
4- /В
Обычно исходят из f(CeH6)3PCH^ ]Х_ (X = гало-
ш ЯГ
ген), на к-рый действуютСвН51л или н-С4Н91л в эфире
или тетрагидрофуране и затем к образовавшемуся
I для его выделения прибавляют альдегид или ке-
тон. Для отщепления НХ можно также использовать
(C6H6)3GNa, C2H6MgBr, NaNH2 в ксилоле, раствор
алкоголята натрия.
Свойства продуктов отщепления НХ от III соот-
ветствуют строению I и, с другой стороны, биполярной
структуре (CeH6)3P=CRR'. Поэтому рассматрива-
емые соединения относят к группе и л и д о в («ил» —
окончания названий радикалов, напр. метил, «ид» —
окончания названия ионных соединений, напр. га-
логенид).
Триметилфосфинметилены с карбонильными соеди-
нениями дают также аддукты типа II, к-рые, однако,
не отщепляют (СН3)3РО при нагревании. Нормаль-
но участвуют в В. р. (CeH6)3P= CRR', если R и R'
представляют собой атомы водорода или алкильные
радикалы; если R является арильным радикалом,
реакция затрудняется, а (С6Н6)3Р=С(СвН6)2 вовсе
не реагирует, что объясняется делокализацией отри-
цательного заряда метиленовой группы:
(С6НБ)3РСНС6НБ —*- (С6НБ)3Р-СН=<^)
При действии НСООС2Н6 на (СвН6)3Р=СН2 обра-
зуется формильное производное (С0Н6)3Р=СНСНО
(IV), к-рое гладко реагирует с альдегидами по В. р.
Так, при реакции бензальдегида с IV образуется
коричный альдегид С0Н6СН—СНСНО. Т. о., можно
простым способом вводить группировку —СН=СН—
между альдегидной группой и остальной частью мо-
лекулы. Аналогично можно получить коричную к-ту
С0Н6СН=СНСООН взаимодействием с бензальде-
гидом (СвН6)3Р=СНСоОС2Н6.
Аналогично, исходя из 2-метиленциклогексанона,
получен триен V:
При В. р., в отличие от других методов синтеза
олефинов, положение двойной снязи не вызывает
никаких сомнений. В. р. является очень перспектив-
ным синтетич. методом, позволяет получить различ-
ные олефины и полиены.
Большое значение имеет В. р. для получения полие-
нов, в частности для синтеза витамина А(см. Ретинол).
Реакцию обычно проводят в мягких условиях, она
не сопровождается особыми осложнениями. Реакция
была предложена в 1954 Г. ВиттигомиУ. Щёлкопфом.
Лит.; Scholl k opt U. von, Angew. Chemie, 1959,
71 Jahrg., M 8, S. 260; Trippett S., в кн.; Advances in
organic chemistry. Methods and results, v. 1. N. Y., 1960, p. 83.
Я. Ф. Комиссаров,
601
ВИЦИНАЛЬНЫЕ СОЕДИНЕНИЯ — ВНУТРЕННЯЯ ЭНЕРГИЯ
602
ВИЦИНАЛЬНЫЕ СОЕДИНЕНИЯ — органич. со-
единения, содержащие заместители (преим. одина-
ковые) у соседних атомов углерода. В. с. обозначаются
приставкой «еицъ перед названием. Примерами В. с.
могут служить этиленгликоль НОСН2СН2ОН, ди-
хлорэтан С1СН2СН2С1; к В. с. можно отнести пирогал-
лол (1,2,3-, или рядовой триоксибензол). С вици-
нальным расположением заместителей связаны особен-
ности в химич. поведении подобных соединений. Так,
дегидратация euif-дитретичных гликолей (пинако-
нов) сопровождается изменением углеродного скелета
(см. Пинаколиновая и ретропинаколиноеая перегруппи-
ровки). Дигалогенид (СН3)2СС1—СС1(СН3)2 с вициналь-
ным расположением заместителей гидролизуется гораз-
до труднее, чем, напр., (СН3)2СС1—СН2—СС1(СН3)г
с атомами хлора, расположенными не у соседних ато-
мов углерода. Я. Ф. Комиссаров.
ВНУТРЕННЕЕ ВРАЩЕНИЕ — см. в ст. Изомерия.
ВНУТРЕННИЙ ЭЛЕКТРОЛИЗ — выделение ме-
таллов из р-ров в результате процесса, происходящего
внутри гальванич. элемента; применяется в аналитич.
химии как метод отделения металла От других химич.
элементов с целью колич. его определения. Для В. э.
применяются разнообразные приборы, в конструкции
к-рых всегда входят 2 различных металла, соединен-
ныхдруг с другом маленькой муфтой или проволокой,
охватывающей оба металла. При погружении такой
гальванич. пары в раствор возникает необходимая
разность потенциалов. На менее активном из двух
металлов (катоде) происходит процесс восстановле-
ния с выделением из раствора определяемого металла;
электрод, сделанный из более активного металла
(анод), окисляясь, переходит в раствор. Катодом чаще
всего служит платиновая сетка; иногда для этой же
цели применяют латунную сетку, железную проволоку
(при выделении сурьмы) и т. п. Анодом служит пла-
стинка или цилиндр из Zn, Al, Pb и др. Применяя
аноды из разных металлов, можно производить от-
деление определяемого элемента от различных ме-
шающих примесей.
Для полного устранения процесса цементации,
т. е. выделения определяемого металла на анодной
пластинке, во многих приборах в анализируемый
р-р погружают только катод, а анод опускают в элек-
тролит, не содержащий определяемого металла; ка-
толит и анолит отделяют друг от друга пористой пе-
регородкой (см. лит., 1). Преимуществами таких при-
боров являются большая точность и возможность
выделения на катоде относительно больших количеств
металла; недостатком — большое внутреннее сопро-
тивление, создаваемое гальванич. элементом, что
вызывает необходимость перемешивания р-ров элек-
трич. мешалками; несмотря на такое перемешивание,
все же требуется продолжительное время для полного
выделения металла.
В приборах второго типа (см. лит., 2) оба связан-
ных друг с другом электрода погружаются в анали-
зируемый р-р. Преимущества такого варианта В. э. —
простота прибора, отсутствие мешалок, быстрота
выполнения анализа; недостаток — возможность вы-
деления относительно малых количеств определяемого
металла (не более 20 мг). Имеется также и «компро-
миссный» прибор (см. лит., 3), в к-ром анод, пред-
варительно покрытый коллодиевой пленкой, погру-
жается непосредственно в анализируемый р-р. Этот
прибор имеет (в уменьшенных размерах) как преиму-
щества, так и недостатки приборов первых двух типов.
Выделенный внутренним электролизом металл или
непосредственно взвешивают, или растворяют и за-
канчивают определение другим способом (чаще всего
колориметрическим). Опубликованы методы В. э.
для определения Си, Bi, Cd в цинке, Ni, Со, Pb, Sn,
Sb, In, Mo.
Лит.: i) 8 and H., Analyst, 1930, 55, p. 309; Collin
E. M., там же, 1930, 55, p. 312, 495, 680; Fite J. tr., там же,
1936, 61, p. 681; Fite J. 6., Тог r a n e e S., там же, 1937,
62, p.29; Fl te J.6., там же, 1938, 63, p. 650; 1940, 65, p. 562;
Ч e p н и x о в Ю. А., Штуцер Е. В., Завод, лаб., 1939, 8,
с. 801; 1940, 9, с. 531, 723; 2) Лурье Ю. Ю., Троицкая
М. И., там же, 1936, 5, с. 1925; 1937, 6, с. 33; 1938, 7, с. 675;
Лурье Ю. Ю„ в кн.: Труды Всес. конфер. по аналит. хи-
мии, т. 1, М.—Л., 1939, с. 231; Лурье Ю. Ю., Гинз-
бург Л. Б., Завод, лаб., 1938, 7, с. И, 535; Л у р ь е Ю. Ю.,
Таль Э. М., Флигельман Л. Б., там же, 1939, 8, с.
1222; 3) Ч е р н и х о в Ю. А., Большакова Г. А., там
же, 1948, 14, с. 3; Ч е р р и х о в Ю. А., Рощина Р. Н.,
там же, 1948, 14, с. 383. ГО. ГО. Лурье.
ВНУТРЕННЯЯ ЭНЕРГИЯ — энергия данного те-
ла, обусловленная движением и взаимодействием
составляющих его молекул, атомов, ионов и образую-
щих их частиц. В. э. системы тел или многофазной
системы включает, кроме В. э. отдельных тел, еще и
энергию межмолекулярного взаимодействия, сосредо-
точенную у поверхностей раздела между этими телами,
т. е. их поверхностную энергию. Количество В. э.
тела зависит не только От природы и массы этого
тела, но также от внешних условий, в к-рых оно на-
ходится.
В. э. является однозначной функцией состояния
тела или системы, т. е. однозначной функцией зна-
чений независимых переменных, определяющих это
состояние, в частности темп-ры Т и объема v или дав-
ления р. В соответствии с законом сохранения и пре-
вращения энергии, для любого процесса перехода
тела из одного состояния (1) в другое (2) разность
В. э. Д£7 = U2 — U\ определяется разностью погло-
щенной телом теплоты Q и совершенной работы А:
SU—U S—U L=Q—A (1)
Хотя каждая из этих величин, Q и А, зависит от ха-
рактера процесса, прирост В. э. ДСЛ не зависит от
того, каким путем тело переходит из одного состояния
в другое, а только от начального и конечного состоя-
ния тела. Поэтому для любого замкнутого процесса,
возвращающего тело в первоначальное состояние, из-
менение В. э. равно нулю nQ = А. Экспериментально
может быть определено только изменение В. э. в том
или другом процессе, но не абсолютная величина ее.
На основании второго закона термодинамики В. э.
системы может быть условно разделена на свободную
энергию F и связанную энергию, определяемую про-
изведением абс. темп-ры Т на энтропию S'.
U=F+TS (2)
(см. Термодинамические потенциалы).
Зависимость В. э. тела от условий его существо-
вания в наиболее простой форме выражается для
газов. В газах в предельно разреженном состоянии
вещества межмолекулярные взаимодействия исчеза-
юще малы, и изменение В. э. при любом изменении
состояния газа сводится только к изменению энергии
самих молекул. Поэтому изменение В. э. сильно раз-
реженных газов, приближающихся к Идеальным га-
зам, при отсутствии химич. изменений, зависит только
от темп-ры (закон Джоуля): SU = f(T), так же, как
и теплоемкость газа ~ = Св= f'(T). Кинетич. теория
газов в полцом соответствии с опытными данными по-
казывает, что для газов с одноатомными молекулами
кинетич. энергия молекул 1 моля идеального газа
U = 3/2AW, а теплоемкость Св = 3/2Ик = 3/2Я
(где 7V— число Авогадро, к — постоянная Больц-
мана, В — Nk — универсальная газовая постоянная).
Так как внутриатомная энергия, кроме области очень
высоких темп-p, не зависит от темп-ры, то возраста-
ние В. э. идеального одноатомного газа с повыше-
нием темп-ры (Uт — Uа) прямо пропорционально аб-
солютной темп-ре.
В области низких темп-p, достаточно близких к
абс. нулю, В. э. кристаллич. тел, приближается к
603
ВНУТРИКОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
604
определенному постоянному значению UB, становясь
независимой от темп-ры. При Т = 0° для правильно
образованных кристаллов индивидуального чистого ве-
щества Cv = = 0 и J = 0, а величины внутрен-
ней и свободной энергии совпадают: Uo = Го.
Лит.: Ш т р а у ф Е. А., Молекулярная физика, Л.—М.,
1949; Леонтович М. А., Статистическая физика, М.—Л.,
1944-Л андау Л. и Лифшиц Е., Статистическая физи-
ка, 2 изд., М.—Л., 1940 (Теоретич. физ., т. 2); Г у г г е н-
г е й м [Э. А.], Современная термодинамика, изложенная
по методу У. Гиббса, пер. [с англ.], Л.—М., 1941.
В. А. Киреев.
ВНУТРИКОМПЛЕКСНЫЕ СОЕДИНЕНИЯ (внут-
ренние комплексные соли) — комплексные циклич.
(хелатные или клешневидные) соединения, со-
держащие внутрисферные поликоординационные за-
местители, присоединенные к центральному атому
металла как за счет главной, так и за счет побочной
валентности, и не содержащие, кроме указанных за-
местителей, никаких других аддендов.
Молекулы органич. соединений, вступающие во внутрен-
нюю сферу В. с., обязательно содержат два типа групп: 1) водо-
родсодержащие функциональные, образующие с центральным
атомом металла-комплексообразователя связь посредством
главной валентности, т. е. путем замещения в них водорода,
напр. в карбоксильной—СООН, сульфоновой — SO3H, или
оксимной - N0H группах, фенольном гидроксиле — ОН
,Н
аминогруппах —NH2 или —N< ; 2) функциональные спо-
собиые с центральным ионом металла образовать донорно-
акцепторную связь, напр. аминную незамещенную —NH,,
замещенные ^NH и ^N—, оксимную >NOH группы, спир-
товой гидроксил —ОН, карбонильную ^СО,
тиоэфирную
—S—и другие группы. Чаще всего В. с. образуются ионами
металлов, у к-рых координационное число (К.ч.) в два раза
больше его заряда. Многочисленны В. с. с центральными
атомами Со (II), Ni (II) (К. ч. 4), Ее (III), Al (III) (К. ч. 6),
тогда как В. с. Ее (II) или Со (II) (К. ч. 6) редки и обра-
зуются лишь с трехкоординационными аддендами с одним
отрицательным зарядом.
Являясь неэлектролитами и образуя в кристаллич.
состоянии молекулярную решетку, В. с. обладают
ценными свойствами, используемыми в аналитич.
химии. В. с. мало растворимы в воде, но хорошо рас-
творимы ворганич. растворителях;!, обр., из большого
объема водного р-ра их можно выделить в сравни-
тельно малый объем органич. растворителя. Т. к.
полнота экстрагирования зависит от pH среды, то
создаются широкие возможности для разделения ио-
нов металлов. При образовании осадка В. с. из вод-
ного р-ра практически не наблюдается соосаждение
содержащихся в нем примесей. Часто образование
В. с. сопровождается значительным изменением ок-
раски, что дает возможность определять элементы
фотометрия, методами. Почти всегда В. с. характери-
зуются сравнительно высоким давлением пара и низ-
кими темп-рами плавления, кипения и возгонки.
Т. к. роль внутрисферных заместителей в молекулах
В. с. часто играют молекулы органич. соединений
сравнительно высокого молекулярного веса, В. с.
характеризуются небольшим фактором пересчета,
что благоприятствует повышению точности весовых
определений.
Будучи по своему строению циклич. соединениями,
В. с. сохраняют многие характерные свойства по-
следних. В. с. ионов металла с К. ч. 6, как правило,
имеют октаэдрич. структуру, а В. с. ионов металлов,
проявляющих К. я. 4, построены по плоскому или
тетраэдрич. типам. Плоская конфигурация харак-
терна для В. с. Pt(II) и Pd(II); октаэдрическая — для
В. с. Fe(III) и А1(Ш); тетраэдрическая — для В. с.
• О способах обозначения связей в В. с. см. статью Ком-
плексные соединения.
Ве(П) *. Изомеры В. с. Pt(II) с гликоколом обладают
плоской структурой:
Н2С---NH2 nh2—сна о=с—о nh2—сн2
I X I I zP'x I
С-—О О Сх HqC—nh2 о—с=о
Октаэдрич. построено соединение А1(Ш) с ацетил-
ацетоном:
Тетраэдрич. построено соединение Ве(П) с ацетил-
ацетоном:
Октаэдрически или плоско построенные В. с. в боль-
шинстве случаев могут существовать в нескольких
изомерных формах. Так, соединения Pt(II) с ала-
нином или гликоколом образуют два геометрия,
изомера (см. выше), отличающихся друг от друга
химич. и физико-химич. свойствами (растворимостью
в воде, строением кристаллов, методами получения
и т. п.). Для многих В. с. характерно образование
лишь одной кристаллич. формы — транс-конфигу-
рации.
Образование В. с. происходит в соответствии с
правилом циклов, т. е. наибольшей устойчивостью
отличаются соединения, содержащие 5- и 6-членные
циклы, тогда как соединения с 3-, 4-, 7-, 8-членными
циклами и т. д. или совсем не образуются, или же
обладают очень невысокой устойчивостью. Так, а-
и р- аминокислоты и о-оксикетоны дают сравни-
тельно устойчивые соединения с ионами нек-рых ме-
таллов, тогда как у- и 6-аминокислоты или м- и
n-оксикетоны не образуют В. с. Разрыв цикла при
химич. превращениях происходит по месту наименее
прочной связи, напр.:
н2с-nh2 Р-с'
| \pi/ | + 2НС1--
C-О'7 VUN-СН2
(Z
/О
HO-C-CH2-NH2 Cl
ж
CL NH2-CH2-C.
он
605
ВОДА
606
и только при длительном кипячении с конц. соляной
к-той удается разорвать связь Pt — N:
О
HO-C-CH2-NH2 Cl о + 2НС1 . H»PtCh+
>Р1< II
С1/ xNH2-CH2— С-ОН
+ 2NH.CH..COOH
Кроме соединений только что рассмотренного типа, известны
производные функции, содержащие наряду с заместителями,
присоединенными за счет сил главной и побочной валентно-
стей, адденды, присоединенные только за счет проявления
одного из указанных типов валентных сил, напр. соединение
с диметилглиоксимом:
HON=C-CH3
где Д обозначает I
-ON=C-СН3
Также известны весьма многочисленные комплексные соеди-
нения, в к-рых органич. адденд может быть подвержен вторич-
ной диссоциации, напр.:
Строго говоря ни те ни другие соединения не относятся к
классу В. с.
Лит.: Гринберг А. А., Введение в химию комплекс-
ных соединений, 2 изд., Л.—М., 1951; Бейл ар Д. X.,
Химия координационных соединений, пер. с англ., М., 1960;
Шугам Е. А., Усп. хим., 1954, 23, вып. 5, с. 622; Шу-
гамЕ. А., Школь никова Л. М., там же, 1959, 28, вып.
7, с. 889; Вознесенский С. А., Внутрикомплексные
соединения и их значение для аналитической химии, Л.—
М., 1938; Martell А. Е., Calvin М., Chemistry ot the
metal chelate compounds, N. Y., [1953]. H. H. Желиговская.
ВОДА. Содержание:
Молекула и структура воды.....................606
Физические свойства...........................606
Химические свойства...........................609
Получение очищенной воды.....................610
Вода природная................................611
Вода питьевая.................................612
Вода техническая..............................613
Вода (окись водорода) Н2О — простейшее устой-
чивое соединение водорода с кислородом (11,19
Вес. %Н и 88,81 вес. %О); мол. в. 18,0160. Греч, на-
звание обсор — гидро, латинское aqua — аква. При
обычных условиях чистая В. — жидкость без запаха,
вкуса и цвета (лишь в слое толщиной более 2 м —
голубоватая). Вследствие наличия 3 изотопов водо-
рода — Н1, D и Т и 6 изотопов кислорода —О14,
О15, О16, О17, О13 и О19 имеется 36 изотопных разно-
видностей В. (см. Вода тяжелая), из к-рых 9 вклю-
чают только стабильные изотопы и содержатся в
природной В. в следующих концентрациях (в моль-
ных %): ЩО1в (99,73), ЩО17 (0,04), ЩО18 (0,20),
НЧ)О“ (0,03), H'DO17 (1,2-10“15), H'DO1S (5,7-10~s),
О2О13 (2,3-10-в), Г)2О17 (0,9-10 ») и П2О18 (4,4-10“9).
До конца 18 в. В. считали элементом; истинный ее
состав как сложного вещества был установлен А.
Лавуазье в 1783 синтезом из водорода и кислорода, а
также разложением ее при пропускании водяного
пара над раскаленным железом с образованием водо-
рода и окисла железа. В. — самое распространенное
на Земле соединение: она составляет в основном всю
гидросферу, входит в связанном состоянии в состав
различных минералов и горных пород (глина, гипс
и др.), находится в растениях и животных, составляя
от 50 до 99% их веса, присутствует в почве, а также
в атмосфере (0,1—2,8 об. %). В. занимает особое
место среди всех соединений в смысле первостепенной
ее важности в самых разнообразных процессах и яв-
лениях живой и неживой природы, а также в прак-
тич. использовании ее человеком. Ввиду этого она
представляет собой также и наиболее полно изучен-
ное соединение; нек-рые из ее свойств положены в
основу определения единиц измерения самых фунда-
ментальных физич. величин: массы и плотности, тем-
пературы, теплоты и уд. теплоемкости.
Молекула и структура воды. Три ядра в молекуле В
образуют равнобедренный треугольник с двумя про-
тонами в основании и кислородом в вершине.
Строение электронного облака молекулы В. иллю-
стрировано рис. 1. Из имеющихся в молекуле 10 элек-
тронов (5 пар) одна
пара электронов (вну- н + + Н +__________+
тренних) расположена V7//
вблизи ядра кислорода, \ \» //
а из остальных 4 пар \ ''/ /
электронов (внешних) / Xj \/\/
по одной паре обобще- - - _ *
ствлено между каждым °
из протонов и ядром Рис. 1.
кислорода, тогда как
2 пары остаются неподеленными и направлены к
противоположным от протонов вершинам тетраэдра
(см. рис. 1,6). Таким образом, в молекуле В. имеются
4 полюса зарядов, расположенные в вершинах тетра-
эдра: 2 отрицательных, созданных избытком электрон-
ной плотности в местах расположения неподеленных
пар электронов, и 2 положительных, созданных ее
недостатком в местах расположения протонов; ди-
польный момент молекулы В. составляет 1,86П. При
равновесном положении ядер молекула В. в парооб-
разном состоянии имеет след, характеристики: рас-
стояние О—Н 0,9584 А; Н — Н 1,515 А; угол НОН
104°27'; главные моменты инерции (10 40 г-c.w2): 1еА =
= 1,0243, 1ев = 1,9207, /р = 2,9470. Для низшего
колебательного уровня: расстояние О—Н 0,9568 А;
Н — Н 1,54 А; угол НОН 105°03’; главные моменты
инерции (10 40 г-с.«2): = 1,0073, 16в — 1,9296,
/°с = 3,0137. Нормальные частоты колебаний (слг-1) :
юх = 3825,03; ш2 = 1653,78; ш3 = 3935,32; поправки
на ангармоничность колебаний (см~1): = —43,89;
х22 — —19,50; г33 — —46,37; т12 = —20,02; г13 =
= —156,06; г23 = —19,81. В структуре льда межатом-
ные расстояния несколько больше: О—Н 0,99 А,
Н—Н 1,62 А, угол НОН 109,5°. Межмолекулярпое
расстояние в структуре льда составляет 2,76 А, в
соответствии с чем молекуле В. приписывается радиус
1,38 А. При этом в структуре льда каждая молекула
соединена водородными связями с 4 ближайшими к
ней другими молекулами, т. е. координационное число
молекулы равно 4, что обусловлено 4 полюсами за-
рядов в молекуле В. (рис. 1). Тетраэдрич. структура
льда является весьма рыхлой («ажурной»), т. е.
характеризуется малой упаковкой частиц. Такая
структура в смысле ближней упорядоченности сохра-
няется в основном и в жидкой В., однако межмолеку-
лярное расстояние увеличивается при плавлении
льда до 2,90 А; несколько увеличивается при плав-
лении и дальнейшем росте темп-ры координационное
число молекул, что приводит к уменьшению рыхлости,
т. е. к нек-рому уплотнению частиц за счет уменьше-
ния пустот в структуре. Существовавшая точка зре-
ния о том, что жидкая В. является смесью различных
ассоциатов типа (Н2О)8, (Н2О)4, (Н2О)2, присутствую-
щих наряду с молекулами Н2О, не подтверждена
экспериментально, тогда как сохранение в жидкой
В. структуры льда с водородными связями подтверж-
дается рентгенография, данными и результатами ис-
следования самодиффузии.
Физические свойства. Основные величины, характе-
ризующие физич. свойства В., приведены в табл. 1 и 2.
607
ВОДА
608.
Свойство
Темп-ра кипения, °C........................
Темп-ра плавления, °C......................
Крит, темп-ра, °C..........................
Крит, давление, атм........................
Крит, плотность, г/см3.....................
Теплота плавления при 1 атм, кал/г.........
Теплота сублимации льда при 0°, кал/г . . .
Уд. теплоемкость льда при 0°, кал/г-град . .
Уд. теплопроводность, кал/см • сек • град:
льда.......................................
жидкости при 0°.....................
» » 45° ....................
пара при 100°.......................
Уд. электропроводность, о-и-1'С.и_1:
льда при 0°...............................
жидкости при 0°.....................
» » 18°.....................
» » 50° ....................
Диэлектрич. проницаемость
жидкости при 20°..........................
пара при 145° и 1 атм...............
Показатель преломления для линии Na при
2 0° и 760 .u.u рт. ст.....................
Скорость распространения звука при 25°, м/сек
Коэфф, сжимаемости при давлении 1—lOOamAi:
при 0°.....................................
при 60°.............................
Таблица 1
Величина
100
0
374,15
218,53
0,325
79,4
677
0,487
ок. 5,6-10*3
1,43-10“’
1,54-Ю-3
0,0551-Ю-3
0,4-10-8
1,47-10-8
4,41-10-8
18,9-10-8
81,0
1,007
1,33299
1,496
51,1-10-8
45,5-10-8
По ряду физич. свойств В. обнаруживает нек-рые
аномалии. Так, летучесть соединений водорода с
щейся с ее ростом от 0° до 150°. В связи с особым ме-
стом, занимаемым В. в природе, многие аномалии ее
свойств играют весьма важную роль. Напр., благодаря
аномалии в плотности лед плавает на В., а при замер-
зании рек и озер на глубине сохраняется относительно
теплая (не ниже +4°) В.; вследствие высоких значе-
ний теплоты плавления и теплоемкости таяние сне-
гов происходит доста-
точно медленно и т. д. °5с0
Чистая В., будучи
в спокойном состоя-
нии, легко переохлаж-
дается и перегревает-
ся: достигнуты пере-
охлаждение до —33° °
и перегрев до +200°
при 1 атм. В., лед и
пар находятся в равно-
весии при 4,6 мм рт.
ст. и0,0100° (тройная '50
точка). В известных
пределах при увели-
чении давления темп-ра плавления В. понижается
(примерно на 1° на каждые 130 ат). При давлениях
выше 2000 ат картина усложняется вследствие поли-
морфизма льда, к-рый существует в 6 кристаллич.
модификациях. Области существования модификаций
льда I, II, III, V и VI показаны на рис. 2 (данные о
Таблица 2
Темп-ра, °C Плотность Давл. насыщ. пара, кг/см2 Теплота испарения, ккал/кг Уд. теплоемкость жидко- сти, кал/г‘град при Вязкость, с пуаз Поверхн. на- тяж. на гра- нице с возду- хом, эрг/см-
ЖИДКОСТИ, г/мл насыщ. пара, кг/м.з
Р—1 кг/см2 Р—50 кг/см2
0 0,99987 0,00485 0,00623 597,3 1,0060 1,004 1,7921 74,64
10 0,99973 0,00940 0,01251 591,7 1,3077 74,22
20 0,99823 0,01729 0,02383 586,0 0,9986 0,996 1,0050 72,75
30 0,99567 0,03037 0,04325 0,07520 580,4 — 0,8007 71,18
40 0,99224 0,05115 574,7 0,9977 0,994 0,6560 69,56
60 0,98324 0,1302 0,2934 0,2031 563,2 1,0008 0,995 0,4688 66,18
80 0,97183 0,4829 551,3 1,0045 0,999 0,3565 62,61
100 0,95838 0,5977 1,0332 539,0 пар 0,487 1.004 0,2838 58,85
120 0,9434 1,121 2,0245 526,1 0,480 1.011 0,232 54,89
150 0,9173 2,547 4,85 505,0 0,472 1,025 0,184
200 0,8649 7,862 15,85 463,5 0,470 1,071
250 0,799 19,98 40,53 409,7 0,473 1,153
87,58 пар . 1
300 — 46,21 335,4 0,479 0,757
350 — 113,6 168,55 213,3 0,486 0,630
элементами подгруппы кислорода возрастает по мере
перехода от тяжелых элементов к легким, тогда как
у В. летучесть не наибольшая, а наименьшая по срав-
нению с гидридами других членов ряда Те — Se — S,
что объясняется наличием в жидкой В. структуры с
водородными связями (аналогичны аномалии для
NH3 и HF). Аномалии в плотности заключаются в
ее увеличении при плавлении льда (от 0,9168 до
0,99987 г)см3 при 0°) и при росте темп-ры до 3,98°
(1,00000г/см3 при 4°). Аномалия в плотности объя-
сняется уменьшением рыхлости структуры льда и за-
полнением в ней пустот, к-рое происходит одновремен-
но с увеличением межмолекулярных расстояний, обус-
ловленным усилением движения молекул; начиная
с 3,98° (темп-ры максимальной плотности В.) влияние
последнего фактора преобладает и плотность в соот-
ветствии с нормальным ходом начинает уменьшаться
при дальнейшем росте темп-ры. . Необходимость за-
траты энергии на изменение структуры и уменьшение
ее рыхлости обусловливает аномально высокие теп-
лоту плавления и уд. теплоемкость В.; при плавлении
льда теплоемкость увеличивается более чем вдвое.
Аномальны у В. также зависимости вязкости от дав-
ления, уменьшающейся с его повышением при 0°—
4-30°, и теплопроводности от темп-ры, увеличиваю-
существовании льда IV не подтверждены). Плотно-
сти льдов II, III, V и VI выше, чем у В. и обычного
льда (I): 1,12 (II); 1,03 (III); 1,09 (V) и 1,13
(VI) г/см3. Тройные точки В. и льдов соответствуют
след, темперам и давлениям:
В.—I—III —22,0°, 2045 ат
I—II—III —34,7°, 2100 ат
В,—III—V —17,0°, 3420 ат
II—III—V —24,3°, 3400 ат
В.—V—VI +0,16°, 6175 ат
При давлении большем чем 21700 ат, существует лед
VII, плотность к-рого близка к 1,5 г/см3, а темп-ра
плавления при 32 000 ат составляет +192°; тройная
точка В. — лед VI — лед VII: +81,6° и 21700 ат.
Структуры льдов II—VII в общих чертах аналогичны
структуре обычного льда I, однако связи между моле-
кулами несколько искажены, а рыхлость меньше.
В. способна растворять очень многие вещества.
Газы хорошо растворимы в В., как правило, лишь
в тех случаях, когда они вступают с ней в химич. взаи-
модействие (напр., NH3, ,СО2, SO3 и др.); обычно же
растворимость газов в В. невелика. При повышении
темп-ры растворимость газов в В. уменьшается. Из не-
органич. веществ в В. растворимы очень многие соли,
кислоты и основания; их растворы являются электро-
609
ВОДА
610
литами. Из органич. веществ растворимы обычно лишь
те, в молекулах к-рых большой удельный вес состав-
ляют полярные группы (многие спирты, амины,ор-
ганич. кислоты, нитрилы, сахара и т. д.).
Химические свойства. При синтезе В. из элементов
по реакции: Н2 + х/2 О2 Н2О, теплота образования
A/ZUs — —57,7979 ккал/моль для водяного пара и
—68,3174 ккал/моль для жидкой В. В отсутствии ка-
тализаторов (Pt и др.) реакция начинает очень мед-
ленно протекать лишь при 300°, однако с повышением
темп-ры скорость резко возрастает и при 550° реак-
ция происходит уже со взрывом. Реакция обратима;
при уменьшении давления, а также при повышении
темп-ры равновесие сдвигается в сторону термич. дис-
социации В. Степень диссоциации (в %) при 1 атм:
0,034 (при 1015°); 0,74 (1711°); 8,6 (2215°); 11,1 (2483°).
Кроме термич. диссоциации, В. подвержена также
электролитич., фотохимич. и радиолитич. диссоциа-
ции. Электролитическая диссоциация В. в жидком
состоянии происходит самопроизвольно с образова-
нием ионов: Н2О Н+ + ОН Мерой электролитич.
диссоциации служит ионное произведение В.: А’в =
= где [Н+] и [ОНД — концентрации ионов
в г-ион/1000 г Н2О; оно равно 10~14 (при 22°) и лежит в
основе характеристики кислотности среды по шкале
pH (см. Водородный показатель). Электролитич.
диссоциация используется для разложения В. на
Н2 и О2 путем электролиза водных р-ров щелочей и
кислот; теоретич. потенциал разложения при 20°
и 1 атм равен 1,23 в. Фотохимич. диссоциация В. про-
исходит под действием ультрафиолетовых лучей с
длиной волны 165 ммк. Радиолитич. разложение В.
вызывается ионизирующим излучением (у- и рентге-
новские лучи, потоки а-, 0- и др. заряженных частиц,
нейтронов). В результате радиолиза образуются Н2,
Н2О2, а также свободные радикалы Н, ОН и НО2;
выход радиолиза — примерно 4 распавшихся моле-
кулы В. на каждые 100 эв поглощенной энергии из-
лучения.
В. является весьма реакционноспособным соеди-
нением, что объясняется наличием в ее молекуле двух
неподеленных пар электронов. Даже с инертными га-
зами она образует гидраты, устойчивые при очень
низких темп-рах (см. Инертные газы). В. окисляется
кислородом, находящимся в атомарном состоянии:
Н2О + О == Н2О2. С фтором В. реагирует при обыч-
ной темп-ре с выделением атомарного кислорода:
Н2О Ч- F2—> 2HF -|- О; образуются также О2, О3,
Н2О2 и F2O, являющиеся продуктами взаимодействия
атомов кислорода друг с другом, с F2 и Н2О. При раст-
ворении в В. хлора происходит реакция Н2О -f- С12 =
НС1 + НОС1;в обычных условиях гидролизуется
до половины всего растворенного хлора. Аналогич-
ные реакции происходят при растворении в В. Вг2 и
J2, однайо равновесие сильнее сдвинуто в сторону
обратного процесса,особенно в случае J2. Кроме того,
хлор (выше 100° или на холоду при действии света)
и бром (начиная с 550°) разлагают В. подобно фтору
с выделением кислорода:
2Н»О + 2С13 —- 4НС1 + О3; 2Н2О + 2Вг» —. 4НВг Ог
При пропускании паров В. через раскаленный уголь
образуется смесь СО и Н2 — водяной газ: Н2О + С =
•=*СО + Н2;при повышенной темп-ре и на катализа-
торе В. реагирует с такими соединениями углерода,
как СО и СН4: Н2О + СО СО2 + Н2 (450°, катали-
затор Fe); Н2О + СП4 = СО - ЗН» (1200—1400°
без катализатора или 700—800° с катализатором Ni
или Со). Реакции с С, СО и СН4 используются для
промышленного получения водорода. Фосфор при на-
гревании с В. под давлением в присутствии катали-
затора окисляется в метафосфорную к-ту: 6Н»О +
+ ЗР —» 2НРО3 + 5Н2.
Щелочные и щелочноземельные металлы (кроме
Mg) разлагают В. уже при комнатной темп-ре: 2Na +
+ 2Н2О -> 2NaOH + Н2;Са + 2Н2О->Са(ОН)а+ Н2
и т. д. Аналогично действуют на В. гидриды щелочных
и щелочноземельных металлов: ЫН + Н2О —> ЫОН +
+ Н2 и т. д. Магний, а также цинк (в виде пыли) разла-
гают В. при 100°. Менее активные металлы взаимодей-
ствуют лишь с водяным паром при темп-ре красного
каления: 2Fe + ЗН2О —» Fe2O3 + ЗН2, и т. д. Благо-
родные металлы (Ag, Au, Pt), а также ртуть В. не
разлагают. Реакцией В. и растворенных в ней кисло-
рода и электролитов с металлами обусловлено явле-
ние коррозии. При растворении в В. кислотных и ще-
лочных окислов образуются соответственно кисло-
родные кислоты и щелочи. Многие соли и др. соеди-
нения вступают с В. в реакцию обменного разложе-
ния — гидролиз. При растворении в В. многих соеди-
нений (солей, кислот, оснований и др.) происходит
их гидратация, т. е. присоединение молекул В. к
молекуле растворенного в-ва, причем наряду со сте-
хиометрически определенными гидратами часто обра-
зуются гидраты с неопределенным числом молекул
В., приходящихся на молекулу исходного в-ва; гид-
ратация сопровождается иногда значительными теп-
ловыми эффектами (напр., при растворении в В.
КОН выделяется 13 ккал/моль, а процесс Na3CO3 +
•4-10Н2О—> Na3CO3 - ЮН3О сопровождается выделением
22 ккал/моль). При выделении растворенных веществ
из раствора связанная в гидратах многих соединений
В. остается в их составе в качестве кристаллизацион-
ной В., образуя кристаллогидраты, к-рые представ-
ляют собой комплексные соединения. Гидратируются
в В. не только молекулы, по и ионы, причем тем силь-
нее, чем больше их заряд и меньше радиус (напр.,
протон всегда гидратирован в В. и образует ион гид-
роксоний Н3О+). Весьма важны каталитич. свойства
В. Очень многие реакции протекают только в присут-
ствии хотя бы следов В. (взаимодействие щелочных
металлов с хлором, цепная реакция Н2 с С12 и др.).
Иногда В. является каталитич. ядом (напр., для же-
лезного катализатора при синтезе NH3).
Получение очищенной воды. В., встречающаяся
в природе, всегда содержит растворенные в ней со-
ли и др. примеси (см. Жесткость воды) . В., исполь-
зуемая для многих промышленных целей, и питьевая
В. подвергаются технич. очистке (см. Водоподготовка).
Получение химически чистой В. для нек-рых исследо-
вательских целей (напр., для точного изотопного
анализа и др.) — весьма сложный процесс. Показа-
телем чистоты В. обычно служит ее электропровод-
ность. Наиболее чистая В. обладает при комнатной
темп-ре уд. электропроводностью порядка 5 • 10 8
ом~1-см~1. При контакте такой В. с воздухом ее уд.
электропроводность вследствие растворения СО2 по-
вышается примерно вдвое. Для обычных лаборатор-
ных целей пригодна В. с уд. электропроводностью
порядка 10-6 ом~‘-см~‘, к-рой обладает дистиллиро-
ванная и дважды дистиллированная В. Для получе-
ния более чистой В. используется чаще всего много-
кратная неполная перегонка дважды дистиллирован-
ной В. последовательно из щелочных и кислых р-ров
КМпО4. Высокая степень чистоты достигается полу-
чением В. пу^ем синтеза из тщательно очищенных
Н2 и О3. Получение В., практически полностью ли-
шенной примесей органич. веществ, осуществляется
действием у-излучения Со60 на трижды перегнанную
В.; органич. примеси окисляются до СО3, удаляемую
продуванием инертного газа,; образующаяся в резуль-
тате радиолиза Н2О3 разрушается ультрафиолетовым
облучением В.
Лит.: Менделеев Д. П„ Основы химии, т. 1—2,
13 изд., М.—Л., 1947; Фридман Э. X., Природа воды.
Тяжелая вода, Л., 1935; horsey N. К., Properties ot ordi-
1 nary watersubstance, N. Y., 1940; Самойлов О. H., Струн-
611
ВОДА
612
тура водных растворов электролитов и гидратация ионов,
М., 1957; В ун алович М. П., Термодинамические свой-
ства воды и водяного пара, 6 изд., М., 1958; В j errum N.,
Structure and properties of ice, Danske videnskabernes selskab.
Matematisk-physiske meddlelser, 1951, 27, 1; Вейн-
берг Б. П., Лед, M.—Л., 1940; Митчел Д ж. и С м и т Д.,
Акваметрия, пер. [с англ.], М., 1952; Internat. critical tables
of numerical data phys., chem. technol., 1929, 6, p. 77; В о л я к
Л. Д., Ж.физ.хпм.,1952, 26, вып. 3; 1954, 28, вып. 6; В у к а л 0-
*в и ч М. П., Теплоэнергетика, 1960, №7. С. Э. Вайсберг.
Вода природная — В. природных водоемов, пред-
ставляющая собой естественные р-ры, в к-рых состав п
количество растворенных веществ колеблются в ши-
роких пределах (в зависимости от источника). При-
родные В., содержащие до 0,1% растворенных
веществ, называют пресными, от 0,1 до 5% —
минерализованными, свыше 5% — рассо-
лами. Свойства В. природной зависят от состава
и концентрации содержащихся в ней веществ. Данные
о составе наиболее характерных В. природных СССР
приведены в табл. 3.
данным отдельных определений. 10) Содержанием ионов С1-»
SO1", HCOa,NOs, NOg, COf", Na+, Са3+, Mg2+,NHt, а также
кремниевой к-ты, к-рая при pH <8 находится в недиссоцииро-
ванномвиде, а при pH > 8 — в виде ионов HSiO^. 11) Содержа-
нием железа, концентрация к-рого в подземных водах может до-
стигать 50 мг/л; в этих условиях оно находится в виде иона
Fe2+ (закисное); при выходе воды ва поверхность ион Fe2^
окисляется в Fe3+, к-рый, гидролпзуясь, образует коллоид-
ную гидроокись железа Fe(OH)3, коагулирующуюся в виде
хлопьевидной суспензии. В присутствии гуминовых веществ
могут образоваться стойкие гуминовокислые соединения же-
леза. 12) Количеством растворенных О2, N2, СО2 и H2S, погло-
щаемых В. при ее выпадении в виде атмосферных осадков,
а также при проникновении в почву и др. Одним из источников
поступления О2 в воду является фотосинтез, а СО2 — окис-
ление органич. примесей в поверхностных водах. 13) Коли-
чеством микроорганизмов (бактерии, грибы, водоросли),
а также патогенных микробов и вирусов, к-рые могут попасть
в В. со сточными водами.
В. природные используют в качестве питьевой
(для питья и хозяйственных нужд) или технич. (для
производственных нужд предприятий, тепловых элек-
тростанций и др.) В.
Таблица 3
Водоисточник Сухой остаток Окисляемость по кислороду Са3+ Mgs+ SO4 ci- SiO|~ Жесткость общая Жест- кость кар- бонатная
мг/л мг-ь кв/л
Реки
Амур Волга (у г. Волгограда) .... Днепр Исеть Кама Кура Москва Нева Обь Ока (у г. Горького) Урал Чусовая 134,2 433,0 270,0 200,0 338,0 445,6 344,0 67,2 256,0 427,0 550 324,0 21.2 5,5 9,8 11,6 13,1 21,5 19,38 7,64 13,3 6,4 4,0 3,53 28,4 99,2 64,2 34,6 35,4 75,0 65,0 11,8 39,6 81,3 121,0 40,5 0,24 19,0 12.0 13,48 7,3 19,5 15,5 2,8 14,8 16,1 25,8 15,4 117,4 48.0 13,73 25,4 136,5 32,98 5,3 20,6 115,7 15,1 4,62 15,3 16,0 15,3 99,0 13,0 34,74 6.57 0,88 14,4 85,0 2,99 12,5 13,8 12.55 17,7 14.3 21,3 7,15 5,1 91.2 2,5 1,45 6,5 4.21 2,8 2.35 5,35 4.49 0,79 3,18 5,39 8,2 3,5 1.04 3,32 3,0 2,75 1.26 3,5 4,28 0,5 2,95 2,56 5.0 0,45
Озера
Байкал Балхаш Каспийское море 91,4 1775 — 15.2 38,7 360 4,2 106,0 730 4,9 572,0 3050,0 1.8 345,0 5350 10Д 8,0 1.11 11.4 76,7 ЗД8 3,57
Природные В. характеризуются: 1) Содержанием грубо-
дпсперсных примесей (частиц песка, глины и др.), определяе-
мых фильтрованием через бумагу с последующим взвешива-
нием, а также характеризуемых по прозрачности (просматри-
ванием стандартного шрифта) или мутности (путем сравнения
с образцами, замутненными стандартной взвесью). 2) Цвет-
ностью, обусловливаемой в основном растворением в В. гу-
миновых веществ, вымываемых из торфяных болот; измеряется
в градусах платинокобальтовой шкалы. 3) По вкусу и запаху:
вкус зависит от состава и количества растворенных солей;
запах В. может быть природного или производственного про-
исхождения; характеризуются по качеству и интенсивности
(по 5-балльной шкале). 4) Наличием в В. легко окисляющихся
примесей; выражается в мг/л О2, израсходованного на их
окисление в стандартных условиях. В зависимости от исполь-
зуемого реактива различают перманганатную и хроматную
окисляемость. Чаще всего окисляемость для подземных вод
равна в среднем 2 мг/л, речных 4—8, болотных и сточных
достигает 100 мг/л и выше. 5) Показателем pH, который у
большинства В. природных равен 6,6—8,6; у болотных pH
ниже, а у вод щелочного характера — выше. 6) Щелочно-
стью, которая определяется как сумма эквивалентных кон-
центраций анионов слабых кис-
лот (НСО3, HSiO^-, СО|“, гуматов).
7) Жесткостью (см. Жесткость
виды), характеризующей суммуэкви-
валентных концентраций катионов
Са2+ и Mg2+, присутствующих в
воде; щелочность и жесткость выра-
жаются в мг-экв/л. 8) Сухим остат-
ком — условным показателем, ха-
рактеризующим концентрацию ра-
створенных и коллоидных приме-
сей, остающихся при упаривании
воды и просушивании остатка_ при
105°; выражается в мг/л. 9) Общим
солесодержанием — суммарной кон-
центрацией растворенных в воде ми-
неральных солей, рассчитанной по
Вода питьевая — В., предназначаемая для питья
и хозяйственно-бытовых нужд населения, а также
коммунальных предприятий и предприятий пищевой
пром-сти; должна удовлетворять определенным сани-
тарно-гигиенич. требованиям, предъявляемым как
к самому источнику водоснабжения (ГОСТ 2761—57),
так и В., подаваемой потребителю (ГОСТ 2874—54).
Питьевая В. должна быть безопасна по бактериаль-
ному составу, безвредна и не должна обладать непри-
ятными органолептич. свойствами. Требования, предъ-
являемые к питьевой В., приведены в табл. 4, 5, 6
(ГОСТ 2761—-57 и 2874—54), причем в табл. 4 при-
ведены общие требования к питьевой В. (цветность,
прозрачность и др.), в табл. 5 — допустимые кон-
центрации различных примесей в мг/л, а в табл. 6 —
требования, предъявляемые к бактериальной чистоте
питьевой В.
Таблица 4
Цветность, условн. град. Прозрачность по шрифту, см Запах и при- вкус при 2 0°, баллов Общая жесткость, мг-зкв/л Мутность, мг/л Сухой оста- ток, мг/л
до 20а не менее 30 до 2° ДО 7а до 2а до 1 000в
а В исключительных случаях (по согласованию с органами саннадзора) допускается
цветность до 35°, мутность до 3 мг/л, общая жесткость до 14 мг-экв/л. 0 В источнике
водоснабжения не более 3 баллов. в При отсутствии в районе др. источников по согла-
сованию с органами саннадзора или при условии опреснения воды до установленной
нормы допускается использование воды с бблыпим сухим остатком. Практически выс-
шим пределом сухого остатка является 1 500—1 800 мг/л.
613
ВОДА —ВОДА ТЯЖЕЛАЯ
614
Таблица 5 ства (градирни и др.), после
Fe2+ и Fe3+ Сульфаты (SO|+) Хлориды (С1-) РЬ Ла Си Zn F чего вновь используется. В., прошедшая тот или иной тех- нологический процесс, напра- вляется на очистку от приме- сей, захваченных ею (см. Воды сточные). В. теплофикацион-
до о,за до 500 до 350 до 0,1 до 0,05 ДО 3 до 5 не ниже 0,5 и не выше 1,5
Концентрации других вредных веществ не должны прев ваемых Госсанинспекцией СССР. а При отсутствии обезжелези Таблица 6 ышать норм, устанавли- вания — не более 1 мг/л. возвращается в них ной сети'возвращается для по- вторного подогрева на ТЭЦ. В., питающая паровые котлы, в виде конденсата.
Среднее число кишечных пало- чек в источниках водоснабже- ния, намечаемых к использо- ванию Число бактерий в воде, по- даваемой потребителям (после 24-часового выра- щивания при 37°)
только с хло- рированием с хлорирова- нием и с пол- ной очисткой общее число в 1 мл кишечных па- лочек в 1 л (коли-индекс)а
до 1 000 до 10 000 до 100 ДО 3
а При использовании бродильной пробы 1 кишечная палочка^должна приходиться не менее чем на 300 Л1л (коли- титр).
При обеззараживании воды хлором содержание в
ней активного хлора должно быть в ближайшей к
насосной станции точке сети не менее 0,3 мг[л, но не
более 0,5 мг/л', хлорфенольные запахи должны отсут-
ствовать, pH воды при ее осветлении или умягчении —
в пределах 6,5—9,5; вода не должна содержать види-
мых невооруженным глазом водных организмов. Для
снабжения питьевой водой могут использоваться
лишь источники, к-рые или уже имеют зону са-
нитарной охраны или могут быть обеспе-
чены ею.
Если качество В. в источнике не удовлетворяет санитарно-
гигиенич. требованиям к питьевой В., то недостатки устраняют
обработкой ее на очистных сооружениях до подачи потреби-
телям: 1) Осветление и обесцвечивание забираемой из водоема
В. производится пропусканием ее через решетки и сита для
задержания листьев, водорослей и проч. Затем добавляют
коагулянт [A12(SO4)3, Fe2(SO4)3, FeCl, и др.] для осаждения
содержащихся в природной воде взвеси и коллоидных веществ.
Образовавшаяся взвесь задерживается в отстойниках или
осветлителях со взвешенным осадком. Из отстойников или
осветлителей вода для окончательного освобождения от взвеси
направляется на фильтры, где фильтруется через слой песка
Или антрацита и песка. Осветленная вода направляется в ре-
зервуар чистой воды. 2) Обеззараживание В. достигается ее
хлорированием. Кроме того, применяется озонирование и
облучение бактерицидными лучами. Для предотвращения
образования хлорфенольных привкусов (при наличии в исход-
ной В. примеси фенолов) и удлинения срока действия хлора
в В. вводится аммиак, образующий с хлором хлорамины. Из-
быток хлора в обработанной В. может быть удален сернистым
газом. 3) Обезжелезивание В. осуществляется путем аэрации
(окисление двухвалентного железа в трехвалентное), хлори-
рования (окисление или разрушение органич. коллоидов),
коагуляции (удаление коллоидов и взвеси) и известкования
(повышение pH) с последующим осветлением воды. 4) Дегаза-
ция В. (удаление H2S, СО2) достигается, как правило, аэра-
цией. 5) Дезодорация В. (удаление привкусов и запахов)
проводится введением аммиака перед хлорированием, хлори-
рованием повышенными дозами, обработкой порошком актив-
ного угля. 6) При недостатке в В. фтора его добавляют в виде
р-ра фтористых солей (напр., Na2SiF„). Избыток фтора может
быть удален пропусканием В. через взвешенный слой Mg(OH)a,
образующий с фтором малорастворимое соединение, или филь-
трацией через слой активной гидроокиси алюминия или син-
тетич. гидроаппатита. 7) Опреснения можно достигнуть мето-
дами ионного обмена, термическим, электрохимическим (см.
Водоподготовка) и др. 8) Умягчение В. см. Водоподготовка.
Вода техническая. Расходуемую промышленными
предприятиями на производственные нужды В. (в
качестве теплоносителя, технология, компонента в
различных производственных процессах, для питания
паровых котлов и др.) принято называть технич. В.
Она может быть использована в прямотоке (т. е. за-
бираться целиком из источника и после использова-
ния сбрасываться в него) или в обороте. В последнем
случае использованная для охлаждения и нагретая
при этом В. пропускается через охлаждающие усгрой-
Охлаждающая В. не должна давать отложений в трубах,
камерах и пр., по к-рым она циркулирует в охлаждаемой ею
аппаратуре и агрегатах, т. к. такие отложения затрудняют
теплопередачу и снижают интенсивность циркуляции В. При
прямотоке должны быть предусмотрены отстойники для за-
держания крупной неорганич. взвеси (песок), к-рая может
оседать в пазухах холодильников, печей и т. п. Более мелкая
взвесь (ил, глина) в трубках и камерах, как правило, не осаж-
дается и может лишь захватываться накипью. При обороте
В. такая взвесь оседает в бассейнах градирень и брызгальпых
устройств. При использовании для прямоточного охлаждения
В., богатой органич. веществами, стенки трубок конденсаторов
паровых турбин могут обрастать биологич. пленкой (зооглей-
ные, нитчатые и др. бактерии, микроскопии, грибки и пр.).
При использовании морской В. в прямоточных системах
охлаждения может наблюдаться обрастание ракушкой (мидия,
балянус и др.). Для борьбы с биологич. зарастаниями В. хло-
рируют (по 10 мин. каждые 1—6 час.); содержание остаточ-
ного хлора поддерживается 0,2—0,5 мг/л. Для борьбы с раку-
шечником, кроме хлорирования, может применяться также
р-р медного купороса. Содержащиеся в охлаждающей В. би-
карбонаты (в основном кальция) могут при нагревании В.
и потере углекислого газа в ее обороте разлагаться с выделе-
нием, малорастворимых карбонатов кальция и гидрата окиси
магния и образованием на стенках трубок охлаждающей
системы накипи. Для борьбы с этим добавочную В., подавае-
мую в систему, подкисляют или добавляют к оборотной В. фос-
фаты.
К В., используемой для технология, целей, требо-
вания могут быть различными. Наименьшие требо-
вания предъявляются к В., используемой для пер-
вичной промывки сырья; В. для приготовления пи-
щевых продуктов должна быть, как правило, питье-
вого качества, а при изготовлении искусственного
волокна, органич. стекла, кинопленки и др. В. не
должна содержать взвешенных веществ, солей жест-
кости, железа, марганца и др. и должна быть бесцвет-
ной. Умягчение и обессоливание В., используемой
в качестве технология, компонента в производствен-
ных процессах, производится методами, аналогичными
водоподготовке; осветление и обезжелезивание — ме-
тодами, аналогичными обработке питьевой В. (см.
выше).
Лит.: Современные методы химического анализа природ-
ной воды. Сб. ст., М., 1955; Воронков II. П., Формиро-
вание химического состава поверхностных вод степной и лесо-
степной зон Европейской территории СССР, Л., 1955; см.
также при ст. Водоподготовка. В. Т. Турчипозич.
ВОДА ТЯЖЕЛАЯ — изотопная разновидность во-
ды, HDO и D.O, в к-рой обыкновенный водород за-
менен его тяжелым изотопом — дейтерием D. Содер-
жится в природных водах и атмосферных осадках.
Отношение D : Н в реках и озерах мало колеблется
и в среднем близко к 1 : 6800; в морях — близко к
1 : 5600; наблюдались величины от 1:9000 в нек-рых
образцах снега до 1:5500 в нек-рых арктич. льдах.
Между 3 изотопными разновидностями воды устанав-
ливается химич. равновесие Н2О + D2O = 2HDO
с константой К = [HDO]2/[H2O] • [D2O] (при 25°).
Поэтому при малом содержании D в воде, напр. в при-
родных водах, В. т. присутствует в ней почти целиком
в виде молекул HDO, а при большом — в виде DSO.
По физич. свойствам В. т. существенно отличается от
обыкновенной (см. таблицу). Особенно характерны
повышенные плотность на 10,77% и вязкость на 23,2%
(при 25°). Растворимость большинства веществ в В. т.
значительно меньше, чем в обыкновенной воде, напр.
при 25° на 8,8% для КС1, на 27% для К2Сг2О7, на
36% для РЬС12. Смеси обыкновенной воды и В. т.
образуют идеальные системы, физич. свойства к-рых
615
ВОДА ТЯЖЕЛАЯ
616
Свойства обыкиовеииой и тяжелой воды
Свойство Состоя- ние Т, °C Н.О‘8а DSO‘«
Мол. вес (О1® —16) . . . . __ 18,01629 20,02948
Плоти., г/см3 Темп-ра, °C: жидк. 25 0,996781° 1,104211
плав лепи я 1 атм — 0 3,813
кипения ........ 1 атм — 100в 101.43
критическая Давление: — — 374,2 371,5
критическое, атм. . . — 218,5 218,6
пара, мм рт. ст. . . . — 25 23,75 20,05
» » » » . . . 100 760 г 719,6
Вязкость относительная жидк. 25 1,000 1,232
Показатель преломления Диэлектрич. проницае- D-линия 25 1,33250а% 1,32795
мость ЖИДК. 25 78,25 78,54
Дипольный момент, ХЮ18 Термодипамич. величины: • — — 1,86 1,87
теплоемкость, кал/моль теплота образования, жидк. 25 18,17 20,25
(—ДН), ккал/моль . . теплота плавления, газ. 1 сипм 25 57,798 59,564
ккал/моль ...... теплота испарения, 1 атм т. пл. 1,436 1,501
ккал/моль ...... энтропия (S), 1 атм 25 10,519 10,850
кал/моль • град .... свободная энергия газАатм 25 45,105 47,379
(—ДЕ), ккал/моль . . ионное произведение газ. 1атм — 54,636 56,067
[Н2О+] • [ОН ] • юы жидк. 25 1,01 0,20
а Также природная вода. 6 Речная вода: 0,997044;
Н2О18 : 1,110179 (экстраполировано). в Н2О18 : 100,09.
г Н2О18 : 757,0. «H»O18 ; 1,33258.
почти точно линейно зависят от содержания D в
атомных долях. По химич. свойствам В. т. очень близ*
ка к обыкновенной воде, но в ней наблюдается уско-
рение или замедление (в зависимости от соотношения
скорости стадий) ряда реакций, в частности прото-
литических с кислотно-основным катализом, на ве-
личины до 2—3 и более раз. Биологич. процессы в
живых организмах в В. т. замедлены.
В. т. открыта в 1932 Г.Юри и Э. Осборном; впервые
выделена электролизом в чистом виде и изучена в
1933 Г. Льюисом и Р. Макдональдом. Электролитич.
способ выделения В. т. из смесей с обыкновенной ос-
нован на том, что В. т. разлагается током в неск.
(обычно 4—6) раз медленнее, чем обыкновенная, и
потому концентрируется в остатке электролита. На-
ряду с этим способом получения В. т. в пром-сти при-
меняются и другие: фракционная перегонка жидкого
водорода (с последующим сжиганием в воду) или,
реже, воды в многотарелочных колонках, причем изо-
топы разделяются благодаря разной летучести; мно-
гократно повторенный в таких же колонках изотоп-
ный обмен воды с H2S, молекулярным водородом на
катализаторах и др., причем в первом случае исполь-
зуются также темп-рные различия в распределении
изотопов между Н2О и H2S.
Для определения содержания В. т. применяют масс-
спектрометрич. анализ воды (или лучше — выделен-
ного из нее водорода) или измерение физич. величин,
зависящих от концентрации D в воде. Чаще всего
пользуются простыми и точными денсиметрич. мето-
дами: нахождение плотности тщательно очищенной
от примесей воды в пикнометре или по времени паде-
ния ее капли заданного объема в несмешивающейся
с водой жидкости, или по темп-ре безразличного рав-
новесия поплавка, флотирующего в ней. Последний
метод позволяет находить изменения плотности в
0,3—0,5у (у = 1 -10 6 г! см? отвечает изменению содер-
жания 0,00093% D). Применяют также измерение
показателя преломления, темп-ры замерзания, вяз-
кости, теплопроводности пара и др. При денсиметрич.
анализе нужно учитывать, что изменение плотности
зависит от изотопного состава как водорода, так и
кислорода в воде. Если оба отличаются от принятого
стандарта (обычно — речная вода), то нужно один
из них привести к стандарту путем изотопного обмена
с большим избытком О2 на окисных катализаторах
пли с СО2 (для нормализации изотопного состава
кислорода), или с NH5 (для водорода) или же сочетать
определение плотности с одновременным измерением
другой физич. величины, напр. показателя преломле-
ния (см. таблицу).
В. т. производят в больших количествах (несколько,
сотен тонн в год, 1958—59), гл. обр. как эффективный
замедлитель нейтронов в ядерных реакторах, как
источник дейтронов D+ для ядерных и термонукле-
арных реакций и для получения ряда др. искусствен-
ных радиоактивных изотопов, а также в химич.,
биологич. и др. научных исследованиях, как меченую
воду и исходное вещество для получения соединений
с меченым водородом.
Другие изотопные разновидно-
сти воды. 1) Тритиевая вода НТО и Т2О (также
DTO), вода, в к-рой водород заменен его радиоактив-
ным изотопом тритием Т. В атмосфере образуется
8—9 атомов Т в минуту на 1 см? земной поверхности
в результате воздействия космич. лучей. Равновесное
содержание Т в природных водах изменяется в пре-
делах 10~15—10-1в%. Общее содержание Т во всех
водах Земли оценивают в 2—3 кг (13—20 кг НТО),
из к-рых 1/200 часть находится в атмосферной влаге.
Тритиевую воду получают искусственно, путем ядер-
пых реакций и концентрируют, как и воду дейтери-
евую, электролизом, фракционной перегонкой или
термодиффузией. По физич. свойствам тритиевая вода
сильнее, чем дейтериевая, отличается от обыкновен-
ной. Ее применяют в ядерной физике для ядерных
и термонуклеарных реакций, а также в химич., био-
логич. и др. исследованиях как меченую воду и как
источник получения соединений, меченных тритием.
Последний, как изотопный индикатор имеет перед
дейтерием преимущества большей чувствительности
и простоты определения, что обусловило его широкое
применение в последние годы; с другой стороны,
сильная радиоактивность требует предосторожности
при работе, т. к. она может вызывать побочные реак-
ции при химич. исследованиях и побочные действия
на организм при биологических.
2) Тяжелокислородная вода — изотопная разно-
видность воды, в к-рой обыкновенный кислород О1в
заменен его тяжелыми изотопами О17 и О18. В природ-
ных водах с небольшими колебаниями (до 1%) найде-
но в среднем: О18 : О1в = 1:502 и О17 : О16 = 1:2760.
Получают тяжелокислородную воду из природной
фракционной перегонкой (одновременно с концентри-
рованием дейтерия) или из кислорода, обогащен-
ного тяжелыми изотопами путем термодиффузии или
изотопного обмена его соединений. По физич. свой-
ствам тяжелокислородная вода значительно меньше
отличается от обыкновенной, чем дейтериевая, однако
плотность Н2О18 на 11,3% (при 25° и природном от-
ношении О17 : О18) выше, чем у природной воды. По
химич. поведению не обнаруживает заметных различий
от обыкновенной воды. Определение содержания тя-
желых изотопов кислорода в воде лучше всего произ-
водить масс-спектрометрически в виде СО2, кислород
к-рой уравновешен с испытуемой водой путем изотоп-
ного обмена с ней, или описанными выше денсимет-
рич. методами. Применяют тяжелокислородную воду
гл. обр. в виде разбавленных р-ров в обыкновенной
воде, как меченую по кислороду воду и как источник
препаратов с меченым кислородом для химич. и био-
логич. исследований. Всего по водороду и кислороду
вода может существовать в 18 изотопных разновид-
ностях.
617
ВОДООТТАЛКИВАЮЩИЕ ПОКРЫТИЯ — ВОДОПОДГОТОВКА
618
Лит.: Киршенбаум И., Тяжелая вода. Физические
свойства и методы анализа, пер. с англ., М., 1953; Брод-
ский А. И., Химия изотопов, 2 изд., М., 1957; Изотопный
анализ воды, 2 изд., М., 1957; Р а и к а м а К., Изотопы в
геологии, пер. с англ.-, М., 1957; Production of heavy Mater,
ed. by Сг. M. Murphy, H. C. Urey and I. Kirschenbaum,
N. Y. — Toronto — L., 1955; Pascal P., Nouveau traits
de cbimie minSrale, t. 1, P., 1956. А. И. Бродский.
ВОДООТТАЛКИВАЮЩИЕ ПОКРЫТИЯ — см.
Гидрофобные покрытия.
ВОДОПОДГОТОВКА — обработка воды, посту-
пающей из природного источника на питание паровых
котлов и для различных технологич. процессов.
Цель В. — освободить воду от грубодисперсных и
коллоидных примесей и содержащихся в ней солей
и тем самым предотвратить отложение накипи, унос
солей паром, коррозию металлов, а также загрязне-
ние обрабатываемых материалов при применении воды
в технологич. процессах. При В. для питания паровых
котлов принято различать докотловую и внутрикот-
ловую обработку воды. От того, для каких целей
предназначена вода, зависит и выбор комплекса ее
обработки. В. включает осветление, умягчение, обес-
кремнивание, обессоливание и др. виды обработки воды.
Осветление. Поступающая из открытых источников вода
содержит грубодисперсные и коллоидные прпмеси минераль-
ного и органич. происхождения. Осветление производят
коагуляцией указанных примесей сернокислыми солями:
A12(SO4)3, FeSO4 или Fe2(SO4)3. Образовавшиеся А1(ОН)3,
Fe(OH)3 выделяют в напорных фильтрах с зернистым мате-
риалом (дробленый антрацит, кварцевый песок), в открытых
отстойниках или в осветлителях со взвешенным осадном.
Умягчение. Жесткость воды может быть устранена мето-
дами осаждения и катионирования. Осаждение осно-
вано па переводе катионов кальция и магния в труднораствори-
мые соединения, выпадающие в осадок, что может быть осу-
ществлено термич. или химич. путем. Термич. умягчение осно-
вано на распаде бикарбонатов кальция н магния при подо-
греве воды с образованием труднорастворимых СаСО3
и Mg(OH)2. Термич. умягчение применяется для снижения
карбонатной жесткости в небольших промышленных котель-
ных, при подготовке воды для подпитки отопительных сетей
и сетей горячего водоснабжения. Химическое умягчение
основано на введении в воду реагентов, обогащающих ее анио-
нами СОз” и ОН-, в результате чего образуются труднораство-
римые СаСО3 и Mg(OH)2. К химич. методам умягчения отно-
сятся: 1) Обработка воды известью и содой; проводится в от-
стойниках с подогревом воды до 80 — 90° и последующим филь-
трованием через фильтры с зернистой загрузкой. 2) Обработка
воды известью проводится в тех же условиях для вод только
с карбонатной жесткостью (некарбонатная жесткость отсут-
ствует). 3) Обработка едким натром (применяется редко в связи
с высокой стоимостью). Все указанные способы обработки
воды применимы лишь для неэкранированных котлов низкого
давления (до 15 ат). 4) Для котлов низкого и среднего давле-
ния (до 39 ат) после обработки воды указанными методами
(известью, известью и содой или едким натром) производится
обработка воды фосфатами (три- и динатрийфосфатом), в ре-
зультате к-рой образуются труднорастворимые кальциевые
п магниевые соли фосфорной к-ты. При подогреве воды до
150° этот метод применим и для котлов более высокого дав-
ления.
Катионирование основано иа фильтровании воды
через слой катионита (см. Иониты), при к-ром происходит
замещение катионов Са2^ и Mg3+, содержащихся в обрабаты-
ваемой воде, иа катионы Na+, Н+ или NHj", содержащихся
в твердой фазе катионита. В связи с этим различают Na-,
Н- и КН4-катионирование. Способность различных катионитов
к обмену иоиов выражается их емкостью поглоще-
н и я по отношению к данному иону и измеряется количеством
грамм-эквивалентов катиона, поглощаемых 1 at3 катионита
(г-экв/м3). Различают рабочую емкость поглощения (по «про-
скока») и полную (до момента полного истощения). Истощен-
ный катионит регенерируют, пропуская через него раствор,
содержащий ион Nat, Н+ или NHj’ в нек-ром избытке.
В качестве катионитов в основном применяют: сульфо-
уголь, КУ-1 и КУ-2 (см. Иониты); полная емкость по-
глощения (соответственно) 500—550, 600—650 и 1500—
1700 г-эка/м3. Рабочая емкость поглощения, зависящая от
размеров зерен катионита, обмениваемого иона (Na+, Н+,
NHf), общего солесодержания, высоты слоя, характера
регенерации и скорости фильтрования, колеблется в значи-
тельных пределах. Напр., для сульфоугля, в пределах соле-
содержания 3,0-*—-32,0 мг-жв/л, она при Na-катионировании
составляет 55—70% от полной, а при Н-катионировании
40—60%.
Обескремнивание воды. Частичное уменьшение концен-
трации кремнекислоты достигается уже в процессе коагуля-
ции (примерно на 25%) и известкования (на 50%). Дальней-
шее удаление кремневой к-ты производится след, методами:
1) Обработкой воды окисью магния в отстойниках одновре-
менно с умягчением известкованием и коагуляцией коллоид-
ных частиц, содержащихся в воде, при подогреве до 40°.
Остаточная концентрация SiO2 после обработки 1,0—1,5 мг/л.
2) Фильтрованием через магнезиальный сорбент. Остаточная
концентрация SiO2 0,2—0,4 мг/л. 3) Анионированием воды
сильноосновным анионитом применяется для глубокого уда-
ления растворенной SiO2, ниже 0,04 мг/л, при одновременном
обессоливании воды.
Обессоливание воды. Устранение растворенных в воде
солей производят след, методами: 1) Ионированием — пропус-
канием воды последовательно через катионитовый и аниони-
товый(см. Йониты) фильтры. В практике В. в качестве подвиж-
ных анионов используются ОН~,Со|”,НСОз. Различают сла-
бо основные аниониты, способные к обмену своих анионов лишь
на анионы.сильных кислот (SOl~, Cl~, NOD, т. е. работающие
только в кислой среде и сильноосновные аниониты, способные
к обмену анионов слабых к-т (HCOg, HSiOg), т. е. к работе
и в нейтральной среде. Емкость поглощения анионитов по
отдельным анионам различна и зависит от кислотности аниопи-
руемой воды и тех же технич. факторов (крупности зерна
и т. д.), что и катионитов. Полная емкость поглощения слабо-
основных анионитов (марки АН д 2Ф) и сильноосновных
(марки ЭДЭ—10П) порядка 1500 г-экв/м3. Регенерация слабо-
основных анионитов проводится NaOH, Na2CO3, NaHCO3
сильноосновных — исключительно NaOH. В простейшем слу-
чае схема обессоливания методом ионирования включает
Н-катионитовый фильтр, декарбонизатор (для выделения
освобождающегося углекислого газа) и анионитовый фильтр.
Для предотвращения возможности проскоков при несвоевре-
менном выключении фильтров на регенерацию в схему в ответ-
ственных случаях вводят катиоиитовые и анионитовые фильтры
второй ступени, а при совмещении обессоливания с обескрем-
ниванием включаются фильтры третьей ступени. Метод позво-
ляет снизить общее солесодержание примерно до 1 мг/л.
2) Термическим методом с применением испарителей — тепло-
обменных аппаратов. Испарители могут быть многоступенча-
тыми с последовательным использованием пара предыдущей
ступени для испарения в последующей. 3) Электрохимическим
методом — пропуском воды через камеры, стенки к-рых
состоят с одной стороны из катионитовой, с другой стороны —
из аиионитовой диафрагмы (листового инертного материала),
в к-рой заключены мелкие зерна ионитов. Метод экономически
целесообразен для снижения солесодержания до 300—500 мг/л
(опреснения воды).
Обработка конденсата. Конденсат, входящий в состав
питательной воды, может быть загрязнен солями (за счет
уноса их с паром, подсоса охлаждающей воды через неплот-
ности конденсаторов и т. д.). Обессоливание проводится мето-
дом ионного обмена. Освобождение конденсата ст масла до-
стигается механич. очисткой отработанного пара и коагуля-
цией конденсата с последующим пропуском его через фильтры,
заполненные дробленным коксом и антрацитом; остаточная
концентрация масла 1,5—2,0 мг/л. При дальнейшем пропуске
конденсата через активный уголь или Na-катиоиит снижают
остаточное содержание до 0,5 мг/л.
Дегазация питательной воды. Освобождение воды от рас-
творенных в ней газов может быть произведено термич. я
химич. способами. Термическая дегазация осу-
ществляется в дегазаторах путем разделения воды на тонкие
струи, переливающиеся через ряд дырчатых лотков навстречу
подаваемому снизу греющему пару. Для усиления выделения
газов дегазация выполняется двухступенчатой с подводом
греющего пара дополнительно в аккумуляторный бак, куда
собирается вода из дегазационной камеры. Химическая
дегазация применяется дополнительно для повышения
эффекта после термич. дегазации и самостоятельно для под-
питочной воды теплофикационных сетей; производится путем
добавки к воде при одновременном ее подогреве сульфита и
бисульфита натрия, сернистого газа, сернистой кислоты,
гидразина (N2H4 • Н2О), гидрата закиси железа или путем
пропускания воды через фильтры, заполненные стальными
стружками.
Внутрикотловая обработка питательной и котловой воды
имеет целью устранить недостатки, связанные с отсутствием
или неполнотой докотловой обработки; к ней относится:
1) Обработка антинакнпинами — различными комбинациями
неорганич. и органич. веществ, замедляющими кристаллиза-
цию пакипеобразователей и способствующими их выпадению
в форме подвижного шлама; применяется как вынужденная
мера в котлах низкого давления при отсутствии докотловой
обработки. 2) Щелочение питательной воды (обычно едким
натром) применяется как средство повышения pH питательной
воды для защиты от коррозии оборудования питательного
тракта. 3) Фосфатирование котловой воды (Na3PO4, Na2HPO4
и NaH2PO4) проводится в котлах среднего давления (выше
15 ат) для предотвращения отложения накипи за счет оста-
точной жесткости питательной воды. 4) Сульфитирование
питательной воды (Na2SO3 или So2) с целью связывания сво-
бодного кислорода, оставшегося в питательной воде. 4) Аммо-
низация питательной воды водным р-ром аммиака для связы-
вания свободной СО2, оставшейся в питательной воде и выде-
ляющейся при распаде ее бикарбонатной и карбонатной ще-
лочности. 5) Нитратирование котловой воды NaNO3 приме-
няется для борьбы с щелочнохрупкпми разрушениями котель-
ного металла (в котлах до 70 ат).
619
ВОДОРОД
620
Состав и схема необходимой В. определяются в
каждом случае в зависимости от условий питания
котлов, их системы и давления, норм качества пита-
тельной воды, изложенных в соответствующих пра-
вилах эксплуатации, и уточняется путем технико-
экономич. сравнения возможных вариантов.
Лит.: Ш к р о б М. С., Водоподготовка, М.—Л., 1950;
Кульский Л. А., Химия и технология обработки воды,
М., 1954; Нормы и технические условия проектирования водо-
очистных станций хозяйственно-питьевых водопроводов. СН
5 4—59, М., 1959; К л я ч к о В. А. иКастальскийА. А.,
Очистка воды для промышленного водоснабжения, М., 1950;
К р у ше л ь Г. Б., Образование и предотвращение отложе-
ний в системах водяного охлаждения, М.—Л., 1955; А п е л ь-
цин И. Э., Обработка охлаждающей воды, М., 1959; Бе-
лан Ф. И., Водоподготовка, М.—Л., 1958; Руководящие
указания по химическому обессоливанию воды ионитами.
[Сост. Ф. Г. Прохоров|, М.—Л., 1957; Смирнов А. С.,
Методы анализа воды, накипи и шлама, М., 1957.
В. Т. Турчипович.
ВОДОРОД (Hydrogenium) Н — первый, наиболее
легкий и простейший по строению атома, элемент пе-
риодич. системы Менделеева: ат. в. 1,0080. При обыч-
ных условиях В. — газ, не имеющий цвета и запаха.
Впервые В. был исследован в 1766 Г. Кавендишем и наз-
ван «горючим воздухом». В 1783 А. .Лавуазье окончательно
установил сложность состава воды, а в 1787 признал «горючий
воздух» элементом и дал ему современное название, означаю-
щее «рождающий воду» (от греч. — вода, уе»»аш —
рождаю).
В. широко распространен в природе, он входит в
состав воды, глин, каменного и бурого угля, нефти,
природных газов, а также всех растительных и живот-
ных организмов. Содержание В. в земной коре (лито-
сфера и гидросфера) составляет 1 вес.%, или 16 ат. %,
в морской воде 10,72 вес.%. В свободном состоянии
В. встречается крайне редко, в небольших количест-
вах содержится в вулканич. и других природных
газах. Благодаря этим газам свободный В. присут-
ствует в количестве менее 10ат.% в атмосфере.
В космосе В. является самым распространенным эле-
ментом. Он составляет до половины массы Солнца и
большинства звезд, присутствует в атмосфере ряда
планет, в кометах, газовых туманностях и межзвезд-
ном газе (в виде: Н2, СН4, NH3, СН, NH, ОН, SiH,
PH, MgH, NaH и т. д., а в звездах в виде плазмы).
Изотопы, атом и молекула. В. имеет 3 изотопа: про-
тий (Н1), дейтерий (D, или Н2) и тритий(Т, или Н3),
с массовыми числами соответственно: 1, 2 и 3. Про-
тий и дейтерий — стабильные изотопы. Нормальный
изотопный состав природных соединений В. соответ-
ствует в среднем отношению D : Н = 1:6 800 (по
числу атомов). Тритий — радиоактивный изотоп с
мягким Р- излучением и периодом полураспада 12,262
лет. Он присутствует в атмосферном В. в количестве
ок. 4-1015 ат.% и в атм. осадках ок. 3-10 18 ат.%;
при этом расход трития за счет распада компенсирует-
ся его образованием, очевидно, из атм. азота под
действием нейтронов космич. лучей и др. естествен-
ными процессами.
Ядро атома В. содержит только один протон.
Ядра дейтерия и трития включают, кроме того, один
и два нейтрона соответственно. Атом В. имеет один
электрон; энергия ионизации Н° Н+, 13,595 эв.
Сродство к электрону Н° — Н’, 0,78 эв. Основное элек-
тронное состояние (см. Атом) отвечает нахождению
электрона на низшем энергетич. уровне, соответ-
ствующем значению квантовых чисел п = 1, I = 0,
т — 0. Магнитный момент атома В. в основном со-
стоянии равен одному боровскому магнетону, т. е.
9,23-10“21 CGSM. Квантовая, механика позволяет
рассчитать все возможные энергетич. уровни атома
В., а следовательно дать полную интерпретацию
атомного спектра. Рассчитано также распределение
вероятностей нахождения электрона по различным
направлениям от ядра и на различных расстояниях
от него. Атом В. используется в качестве модельного
в квантово-механич. расчетах энергетич. уровней
других, более сложных атомов.
Молекула В., Н2, состоит из двух атомов, соединен-
ных ковалентной связью (см. Химическая связь),
я является простейшей молекулой. Энергия диссо-
циации молекулы Щ (протия) Do = 4,4776 эв (ЮЗ
ккал/моль); межатомное расстояние ге = 0,7414-Ю8 см;
(для дейтерия Do = 4,5557 эв, ге = 0,7417-10 8 см).
Атомы колеблются друг относительно друга с основ-
ной частотой <ос = 4405,Зм”1, т. е. ок. 1,32-1014 ко-
лебаний в сек. (для молекулы D2 ше = 3118,8 см'1,
для Т2 = 2546,5 см'1). Колебания не вполне гар-
моничны; поправка на ангармоничность составляет
coeze = —125,32 см 1 (для D2 <aeze = —64,15 см'1;
для Т2 <оехе = —41,88 см'1). Молекулярный спектр В.
связан с изменением электронных и колебательно-
вращательных энергетич. уровней молекулы. Кван-
тово-механич. расчет молекулы В. дает результаты с
очень хорошим приближением. Кроме электронных
и колебательно-вращательных состояний молекулы
В., для нее различают также состояния с одинаковым
и противоположным по направлению вращением атом-
ных ядер (параллельные и антипараллельпые ядер-
ные спины). В связи с этим имеются две модификации
молекулярного В., несколько различные по физич.
свойствам: ортоводород (параллельные спины) и па-
раводород (антипараллельные спины), содержащиеся
при обычных и высоких темп-рах в отношении 3:1;
при понижении темп-ры равновесие сдвигается в сто-
рону параводорода, содержание к-рого при темп-рах
глубокого холода приближается к 100%(99,7% при
—253°) (см. Водорода пара-орто-превращение).
Физические и химические свойства. Молекулярный
В. характеризуется след, константами: т. пл. —259,1°;
т. кип. -252,6°; «крит. -240°, ркрит 12,8 атм; йкрит
31,2 г/л; плотность газа 0,0899 г/л (при 0° и 1 атм),
жидкости 70,8 г/л (—253°) и твердого В. 80,7 г/л
(—262°); плотность газа относительно воздуха 0,0695;
вязкость (в мпуавах)'. газа 87 (при 15° и 1 атм), 12,6
(—253°), жидкости 138 (—253°); уд. теплоемкость
(в кал/г-град)-. для газа Ср 3,44 (при 0—200°),
Cv 2,46 (0—200°);Cp/Cv 1, 40; то же для жидкости
Ср 2,33 (—251,8°); для твердого Ср 0,63 (—259,6°);
теплота плавления 13,89 кал/г (—259°); теплота ки-
пения 107 кал/г (—253°); коэфф, теплового расши-
рения 0,00365; теплопроводность (кал/сек • см • град)
Zo 0,00038 (при0°). Зависимость теплопроводности
от темп-ры (Г°К) выражается формулой Сутерленда:
, _ У 367 f Т \
КТ — Kq т + д4 ^273 )
Растворимость в воде при 20°: 0,0182 мл/г (1 атм),
1,785мл/г (100 атм) и 8,328мл/г (500 атм). Относитель-
ное изменение объема с изменением давления при
0°С и 99,85°С показано в табл.
Давление, атм 1 100 200 400 1000
0° 1,0000 0,0106 0,0057 0,0032 0,0017
99,85° 1,3656 0,0144 0,0076 0,0041 0,0021
В. хорошо растворим во многих металлах (Ni,
Pt, Pd и др.); наибольшая растворимость наблюдает-
ся в палладии: 850 объемов В. на 1 объем Pd. С
растворимостью В. в металлах связана его способ-
ность диффундировать через них; такая диффузия
иногда сопровождается газовой коррозией, обуслов-
ленной разрушением углеродистого сплава металла
(напр., стали) вследствие декарбонизации. При вы-
621
ВОДОРОД
622
соких темп-рах молекулярный В. диссоциирует на атомы; степень диссоциации К при 1 атм составляет:
К 2,56 • 10-34 1,22 • IO"» 0,9469 0,9996
Темп-ра, °К . . . 300 2000 5000 10000
Атомарный В. образуется также при получении его
различными химич. реакциями (напр., действием
Zn на НС1), однако существование В. в атомарном
состоянии длится лишь короткое время, атомы реком-
бинируют в молекулы Н2 с выделением теплоты, чис-
ленно равной теплоте диссоциации. Атомарный В.
обладает повышенной химич. активностью по сравне-
нию с молекулярным.
В химич. соединениях В. всегда одновалентен, яв-
ляясь в основном электроположительным элементом
(лишь в гидридах металлов он электроотрицателен);
В. — хороший восстановитель. При Обычных усло-
виях молекулярный В. сравнительно мало активен,
непосредственно соединяется лишь с наиболее актив-
ными из неметаллов (с фтором, а на свету и с хлором).
Однако при нагревании он вступает в реакции со
многими элементами. С кислородом В. образует воду:
Н2 4~ 1/2 О2—>Н2О, теплота реакции при 1 атм и 25°
Л//298 ~ '—68,3174 ккал/моль (реакция экзотермична).
При обычных темп-рах реакция протекает крайне
медленно, выше 550° — со взрывом. Пределы взрыво-
опасности водородо-кислородной смеси составляют
(по объему) от 4 до 94% Н2, а водородо-воздушной
смеси — от 4 до 74% Н2. В. отнимает кислород от
окислов металлов и используется для их восстанов-
ления: CuO + Н2 —► Си4*Н2О; Fe3O44-4H2 = 3Fe +
+ 4Н2О, и т. д.
С галогенами В. образует галогеноводороды со
след, теплотами реакций (ккал/моль); 1/2Н2 + 1/2F2—►
—►HF, АЯ"298 = -64,2; i/2Hs + VzC^-^HCl, ДЯ°т =
= —22,063; 1/гНа + 1/аВг, -+ НВт, Д//298 = нг-8,66;
Х/2Н2 + 1/2J2—► HJ, АЯ298 = 4-6,2. С фтором реакция
идет со взрывом (даже в темноте и при —252°), с
хлором и бромом — лишь при освещении или нагре-
вании, а с иодом только при нагревании. Синтез
хлористого водорода из В. и хлора имеет промышлен-
ное значение. Взаимодействие В. с азотом с образо-
ванием аммиака протекает лишь на катализаторе и
при повышенных темп-рах и давлениях. С серой,
селеном и теллуром В. реагирует со след, теплотами
реакций(ккал/моль); Н2 4- S—► Щй, ДЯ298 = —4,8 1 5;
Н2 4- Se —► H2Se, ЛЯ°298 = 4-20,5; Н2 4- Те -► Н2Те,
АЯ298 = 4-36,9. Реакция с серой протекает энергично
при нагревании до 600°, с селеном и теллуром —
значительно труднее. С чистым углеродом В. может
реагировать без катализатора только при высоких
темп-рах: 2Н2 4- С (аморфный) —► СН4, АЯ298 =
= —17,889 ккал/моль. Практич. значение имеют реак-
ции В. с окисью углерода, при к-рых образуются в
зависимости от темп-ры, давления и катализатора
различные соединения (см. Углерода окись). Ненасы-
щенные углеводороды реагируют с В., образуя главным
образом насыщенные продукты (см. Гидрогенизация
и дегидрогенизация каталитические). В. непосред-
ственно реагирует со щелочными и щелочноземель-
ными металлами, а также со многими редкоземель-
ными и нек-рыми др. металлами, образуя гидриды.
В. связывается также многими другими металлами,
однако образующиеся системы скорее являются рас-
творами В. в металле, чем химич. соединениями.
Роль В. и его соединений в химии исключительно
велика. В. легко отдает свой единственный электрон
и отрывается в виде протона от многих соединений,
находящихся п р-ре, обусловливая кислотные свойства
этих соединений (см. Кислот и оснований теории).
Находясь в соединениях, В. склонен образовывать
со многими легкими электроотрицательными элемен-
тами (F, О, N, Си даже В, Cl, S, Р) других соедине-
ний т. н. водородную связь.
Получение. Основными видами сырья для промыш-
ленного получения В. являются: природные и попут-
ные газы (см. Газы природные горючие), коксовый газ
(см. Коксохимическое производство) и газы каталитич.
риформинга (см. Газы нефтепереработки), а также
продукты газификации твердых и жидких топлив (гл.
обр. угля). В. получают также из воды электролизом.
Важнейшими способами произ-ва В. из природного
газа является взаимодействие углеводородов (гл.
обр. СН4) с водяным паром (конверсия) и их неполное
окисление кислородом. В основе первого способа
лежиткаталитич. реакция: СН4 4- Н2О —► СО 4- ЗН2.
Эта реакция эндотермпчна. Образующаяся окись уг-
лерода также подвергается конверсии: СО 4- Н2О —►
—► СО2 4* Н2.
Природный газ очищают от сернистых соединений (H2S
и др., см. Газов очистка), смешивают с водяным паром, на-
гревают и направляют на никелевый катализатор, где при «00°
происходит конверсия (см. Метан). Темп-ра поддерживается
посредством сжигания нек-рой части исходного газа. После
конверсии метана (и др. углеводородов) полученная газовая
смесь В. и окиси углерода вновь смешивается с водяным паром
и направляется на катализатор (Fe с добавкой Сг или Mg),
где при 500—550° происходит конверсия СО. Далее газ про-
ходит очистку от Со2 и остатков СО. Другой способ получе-
ния В. из природного газа — неполное окисление метана —
основан на реакции: СН4 4 */, О2 — СО 4 2Н2, идущей с вы-
делением теплоты. Дальнейшие стадии конверсии СО и очистки
аналогичны применяемым в первом способе. Целесообразно
сочетать вместе оба способа получения В. из природного
газа, т. к. при этом для протекания эндотермич. реакции
конверсии метана и др. углеводородов используется теплота,
выделяющаяся при их неполном окислении. Для проведения
такого процесса исходный природный газ смешивается с во-
дяным паром и кислородом. Реакции конверсии и неполного
окисления протекают одновременно на никелевом катализа-
торе при 800—900°. Если же при первоначальном смешении
вместо кислорода используют воздух, обогащенный кислоро-
дом, то получают В. в смеси с азотом, пригодный для синтеза
аммиака. В., получаемый из природного газа, является наи-
более дешевым.
Одним из самых распространенных способов является
произ-во В. иэ водяного и паровоздушного газов, получаемых
газификацией твердого топлива. Водяной газ содержит
до 50% Н2 и 40% СО; в паровоздушном газе, кроме Н, и СО,
имеется значительное количество N2, который используется
вместе с получаемым В. для синтеза NH,. В основе процес-
са лежит реакция конверсии углерода окиси: СО 4 Н2О —>•
-—► Н2 4 СО2.
Использование продуктов газификации угля и др. твердых
и жидких топлив для получения В. вытесняется экономически
более выгодным использованием природного газа. Важным
способом произ-ва В. является его выделение из коксового
газа, а также из газов каталитич. риформинга нефти, путем
удаления остальных компонентов газовой смеси, более легко
сжижаемых, чем В., при глубоком охлаждении газа. Коксо-
вый газ сначала очищают от сравнительно легко сжижаемых
примесей: сернистых соединений, окислов азота, СО2, паров
воды и др. Дальнейшее разделение коксового газа производят
фракционированным сжижением при глубоком охлаждении
(см. Газов разделение). После отделения этиленовой и метано-
вой фракций газ промывают жидким азотом и освобождают
от остатков СН4, а также от СО и О2. При этом получают
азотоводородную смесь, содержащую сотые доли процента СО
и О2; содержание N2 в смеси ок. 15%. Доводя содержание
N2 до 25%, получают смесь для синтеза аммиака.
Получение В. (в смеси с азотом) глубоким охлаждением
коксового газа является одним из наиболее выгодных методов
и используется всеми аммиачными заводами, имеющими
в своем распоряжении коксовый газ.
Электролиз воды используют для промышленного получе-
ния В. лишь в местах с крайне дешевой электроэнергией.
Разложение воды производят постоянным током. В качестве
электролита используется р-р едкого натра или едкого кали
(к-ты не используются во избежание коррозии стальной аппа-
ратуры). Концентрация щелочи поддерживается на уровне
максимальной электропроводности (25 вес. % для NaOH
и 3i вес. % для КОН). Получаемый В. содержит 0,5—1%
примесей (кислород и следы азота). На электролизных заводах,
кроме В. и кислорода, получают часто в качестве побочного
продукта тяжелую воду (см. Дейтерий). В прошлом был
распространен железо-паровой способ получения В., основан-
ный на реакции водяного пара с железом или с закисью же-
леза при 800—900°: Fe 4 Н2О — FeO 4 Н2 и 3FeO 4 Н2О —
— Fe30t4 Н2. Метод применялся на небольших установках
и сейчас потерял значение.
623
ВОДОРОДА ОПРЕДЕЛЕНИЕ — ВОДОРОДА ПЕРЕКИСЬ
624
Наиболее распространенными лабораторными способами
получения В. является электролиз воды, осуществляемый
в стеклянном лабораторном электролизере с использованием
в качестве электролита р-ра H2SO4, а также реакция между
металлич. цинком и соляной к-той, проводимая в аппарате
Киппа. Однако чаще в лабораторной практике, используется
готовый заводской В. в баллонах.
Аналитическое определение во-
дорода. Как правило, определение производят
с целью установления примесей: СО, СО2, О2, Na,
углеводородов. В конечном счете, после отделения
ряда примесей поглотителями и сжигания оставшегося
газа (Н2, СН4, Na и О2) по количеству образующихся
Н2О и СО2 и оставшихся N2 и О2 определяют состав
исходного газа.
Применение. В. широко используется в химич.
пром-сти, преим. для произ-ва аммиака; крупным
потребителем В. является также произ-во метанола,
синтезируемого из В. и СО. На основе синтеза из
В. и СО получают и другие спирты, а также альде-
гиды,, кетоны и др. продукты. В. применяют для гид-
рогенизации твердого и тяжелого жидкого топлива,
а также жиров и различных органич. соединений,
для синтеза НС1, для гидроочистки продуктов пере-
работки нефти, в сварке и резке металлов горячим
кислородо-водородным пламенем (темп-ра до 2800°),
а также в атомарно-водородной сварке (темп-ра до
4000°). В прошлом В. использовался для наполнения
аэростатов и дирижаблей. Очень важное применение
в атомной энергетике нашли изотопы В. (см. Дейте-
рий, Тритий, Термоядерные реакции).
Лит.: Кондратьев В. Н., Структура атомов и мо-
лекул, 2 изд., М.—Л., 1959; G m е 11 п, 8 Aufl., Syst.-Kummer 2,
В., 1927; Mellor, v. 1, L.— N. Y.— Toronto, 1946: то же
1952; Pascal, v. 1, P., 1956; Kirk, v. 7, N. Y., 1951,
p. 675—92; Малков M. П. иПавловК. Ф., Справоч-
ник по глубокому охлаждению в технике, М.—Л., 1947; Об-
щая химическая технология. Под ред. С. И. Вольфковпча, т. 1,
М., 1952; Лебедев В. В., Водород, его получение л ис-
пользование, М 1958. С. Э. Вайсберг.
ВОДОРОДА ОПРЕДЕЛЕНИЕ (в органических
соединениях) — см. Углерода и водорода определение.
ВОДОРОДА ПАРА-ОРТО-ПРЕВРАЩЕНИЕ (пара-
орто-конверсия) — переход молекул водорода Н2 из
одной формы в другую. Существование двух модифи-
каций водорода было предсказано В. Гейзенбергом и
Ф. Гундом на основе квантовой механики. К. Бонгоф-
фер и П. Хартек впервые экспериментально изучили
взаимное превращение одной формы молекул водо-
рода в другую. Существование двух модификации
молекулярного водорода связано с различной взаим-
ной ориентацией ядерных спинов атомов и, следова-
тельно, с различными значениями вращательных кван-
товых чисел. В молекулах т.н. параводорода (п-Н2)
ядерные спины антипараллельны (fj) и вращатель-
ные квантовые числа четные. Молекулы ортоводорода
(о-Н2) имеют параллельные спины ( || ) и нечетные
вращательные квантовые числа. Пара- и орто-модифи-
кации водорода обладают разным количеством вра-
щательной энергии и, следовательно, характеризуются
разными теплоемкостями, теплопроводностями, упру-
гостями пара, темп-рами плавления и др.
Каждой темп-ре соответствует определенное равно-
весное соотношение между числом орто- и пара-моле-
кул. При низких темп-рах равновесие смещено в сто-
рону п-На (при темп-ре жидкого водорода содержа-
ние п-Н2 практически уже равно 100%). Предельное
равновесное содержание о-Н2, соответствующее высо-
ким темп-рам, составляет 75%; это содержание при-
близительно достигается уже при комнатной темп-ре.
При обычных темп-рах самопроизвольное превраще-
ние одной модификации в другую, сопровождающееся
изменением ориентации спинов, не происходит, точ-
нее, идет чрезвычайно медленно. Вероятность орто-
пара-превращения при поглощении света крайне мала.
Термич. же превращение происходит лишь при 700—
800° в результате простых столкновений между
атомами, образующимися при термич. диссоциации,
и недиссбциированными молекулами водорода:
Н + n-H2=o-H2 + Н
Превращение, наблюдаемое в присутствии атомов водо-
рода, возникающих при электронном разряде или фо-
тохимически, протекает, вероятно, по аналогичному
механизму. -
Каталитич. В. п.-о.-п. может идти по двум различ-
ным механизмам. Один из них («низкотемпературный»)
состоит в прямом превращении п-Н2-— о-Н2 в резуль-
тате переориентации ядерных спинов в сильно
неоднородном поле парамагнитных катализаторов,
в качестве к-рых могут служить газы (О2, NO, NO2
и др.), ионы (Со2+, Fc2+, Ni2+, Zn2+ и др.), а также
твердые тела (Cr2O3, V2O3, V2O5 й др.). Превращение
по низкотемпературному механизму происходит также
в таких растворителях, как вода, бензол, метанол,
циклогексанол, анилин и др. По этому же механизму
идет низкотемпературное превращение на активной
угле, где оно, по-видимому, вызывается полями по-
верхностных атомов углерода с ненасыщенными валент-
ностями. Превращение по второму («высокотемпера-
турному») механизму осуществляется в результате
химич. реакции с участием атомарного водорода и
может протекать при каталитич. действии металлов,
солей и окислов. Этот механизм может быть предста-
влен одной из схем:
Наде + п-Н2=о-Н3 -|- Н-аЭс (1)
ИЛИ
пН2 = НаЭс + Надс=о-Н2 (2)
Способность молекулярного водорода к орто-пара-
превращенпю используют при выяснении механизма
таких реакций, как изотопный обмен водорода, гид-
рирование, каталитич. окисление водорода и др.,
путем-сопоставления кинетики соответствующих про-
цессов. По скорости такого превращения судят также
о концентрации свободных атомов водорода в смесях.
По способности нек-рых веществ активировать орто-
пара-превращение можно судить об их парамагне-
тизме.
Пара-орто-превращение характерно не только для
протия, но также и для других изотопов водорода —
дейтерия и трития. Равновесное отношение орто- и
пара-модификаций (для высоких темп-p) в случае
трития совпадает с протием (3 : 1), тогда как для дейте-
рия оно заметно отличается (2 : 1). Орто-пара-конвер-
сия осуществляется в случае трития значительно
легче, чем для других изотопов водорода (предельное
содержание орто-формы достигается для него уже
приблизительно около 175° К), что, очевидно, вы-
звано Р--излучением Т2.
Лит.: Фаркас А., Ортоводород, параводород и тяже-
лый водород, пер. с англ., М., 1936; Бродский А. И.,
Химия изотопов, 2 изд., М., 1957, с. 412—14; Cremer Е.,
в кн.: Handbuch der Katalyse. Hrsg. von G.—M. Schwab, Bd 1,
\\ ien, 1941, S. 325; его же, там же, Bd 6, 1943, S. 1.
Ф. С. Якушин.
ВОДОРОДА ПЕРЕКИСЬ Н2О2 — бесцветная вяз-
кая жидкость с «металлическим» привкусом; с?20 1,450;
т. пл. —0,43° (легко переохлаждается); т. кип. 150,2°;
теплота плавления 2,987 ккал/моль (—0,43°); теплота
парообразования 12,33 ккал/молъ (25°); теплоемкость
0,628 кал/г-град (25°), теплота образования в парооб-
разном состоянии А/7%8 = —32,52 ккал/молъ; вяз-
кость 1,245 спуаз (20°); поверхностное натяжение
80,4 дин/см (20°); ng 1,4067; диэлектрич. проницае-
мость 73,1 (20°); дипольный момент 2,1 • 10-18 ед.
CGSE; уд. магнитная восприимчивость —0,50 • 10-вед.
CGSM (25°). С водой смешивается в любых отношениях;
образует кристаллогидрат состава Н2О2 • 2П2О, т. пл.
625
ВОДОРОДА ПЕРЕКИСЬ — ВОДОРОДНАЯ СВЯЗЬ
626
R
катализ
Длина цепи до-
В цепном про-
—52,0°; истинных твердых растворов с водой не дает,
но твердые фазы прочно удерживают маточный рас-
твор. Структура молекулы В. п. изо-
/ /Н бражена на рисунке; угол 0 близок
О—к 100°, угол ср — к 95°; длины свя-
\ ^Н\ зей: О—НО, 97 А, О—О 1,49 А. Энер-
гия О—О связи 52 ккал/моль, энер-
гия Н—О связи 90 ккал/моль. В парах молекулы
В. п. не ассоциированы, в жидкой фазе наблюдается
сильная ассоциация за счет водородных связей.
Очень чистая В. п. достаточно устойчива (скорость
распада при 30° не превышает 0,5% в год), но в при-
сутствии следов ряда тяжелых металлов и их ионов
(Си, Fe, Мп, группа Pt и др.), ферментов, а также
под влиянием радиации, электрич. разряда и т. д.
наблюдается ее разложение по реакции: Н.2О2 =
= Н2О + х/2 О2 с выделением 23,44 ккал/моль (25°).
Механизмы процессов распада выяснены не до конца.
Гомогенное термич. и фотохимич. разложение В. п.
в парах происходит по радикальному механизму:
Н2.О2—>2ОН, ОН+ Н2О2->Н2О + НО2, НО2 + Нб2 +
+ М —> Н2О2 + О2 + М (в качестве третьего тела М
функционирует стенка, инертный газ или молекула
В. п.). Квантовый выход в случае фотохимич. процес-
са Н2О2->2ОН равен 1,7 ±0,4.
Механизм процессов распада
в конденсированной фазе рез-
ко отличен от выше описанно-
го. Термич., фотохимич., ра-
диационный и каталитич. про-
цессы распада В. п. протекают
по радикально-цепному механизму,
стигает иногда неск. тысяч звеньев.
цессе участвуют ОН и НО2-радикалы. Многие ката-
литич., в т. ч. ферментативные процессы распада
идут через образование промежуточных комплексов
В. п. с катализатором. Разложение В. п. — экзо-
термич. процесс и может проходить со взрывом.
В растворах, особенно разбавленных, В. п. обыч-
ной чистоты более устойчива. Продажные р-ры
В. п. (пергидроль, с 30% Н2О2, высококонцен-
трированная, 85—90% Н2О2) содержат, как прави-
ло, добавки стабилизаторов разложения, наиболее
эффективными из к-рых являются станнат натрия,
оксин и фосфаты.
Занимая по степени' окисленности промежуточное
положение между молекулярным кислородом и водой,
В. п. обладает как окислительными, так и восстанови-
тельными свойствами. Она окисляет нитрит в нитрат,
выделяет иод из иодида, окисляет титановую к-ту
в соединения пероксотитанила (качественная реакция
на В. п.), бензол в фенол, анилин в азоксибензол,
расщепляет ненасыщенные соединения по месту двой-
ной связи и т. д. С другой стороны, В. п. восстанавли-
вает соли золота и серебра, перманганат калия в ки-
слой среде (реакция, используемая для колич. опре-
деления В. п.) и т. д. При окислении ряда ионов:
Сг (II), Hg (I), Ti (III), Мп (II), Fe (II) образуются
гидроксильные радикалы, способствующие разложе-
нию В. п., напр. Fe2+ + Н2О2 = Fe3+ + ОН + ОН~.
Ряд реакций В. п. идет, по-видимому, через проме-
жуточное образование радикала пергидроксила: Се4*-]-
+ Н2О2 = Нб2 + Н+ + Се3+, Се4+ + НО2 = О2 +
+ Н+ + Се3+, или иона пергидроксила (окисление
сульфита в сульфат). Окисление органич. веществ
во многих случаях идет через стадию образования
органич. перекисей. Кислород воды, как правило,
не участвует в окислительно-восстановительных ре-
акциях В. п. в растворах. В то же время. изотопный •
обмен водорода между В. п. и водой проходит доста-
точно быстро, что объясняется наличием у В. п. сла-
бокислотных свойств. Константа диссоциации: Н2О2 +
+ Н2Ог=Н3О+ + НО; равна 1,39 -10-“ (20°). По той
же причине В. п. может вступать в реакции обмена:
Н2О2 + Ва (ОН)2—ВаО2 + 2Н2О, давая перекиси.
Наконец, В. п., подобно воде, способна присоеди-
няться к солям и другим соединениям с образованием
пергидратов.
Образование В. п. наблюдается при окислении и
самоокислении многих органич. соединений, водорода
(см. Перекисная теория окисления Баха—Энглера), при
катодном восстановлении кислорода и атмосферной
коррозии металлов, в фото- и радиохимии, процессах
в водных р-рах, при действии ультразвука на воду,
в биологич. процессах и т. д. Первые лабораторные и
промышленные методы получения В. п. основывались
на разложении перекисей металлов (ВаО2, Na2O2)
разб. к-тами. В настоящее время свыше 80% миро-
вого произ-ва В.п. получается электрохимия, путем —
анодным окислением кислых р-ров бисульфата аммо-
ния или серной к-ты: 2H2SO4 = H2S2O8 + 2Н+ + 2е,
и гидролиза пероксидисерной кислоты: H2S2O8 +
+ Н2О = H2SO4 + H2SO2; H2SO5 + II2O = H2SO4+
+ H2O2. Используются также реакции самоокисле-
ния продуктов восстановления антрахинона и его
алкилпроизводных,
ОН
например 2-этилантрахинона:
н2
R
4" НдОц
гидразобензола, изопропилового спирта и
а также
др. веществ. Концентрируют растворы В. п. перегон-
кой и ректификацией; хранят в таре из алюминия,
полиэтилена, стекла и т. д.
Применяют В. п. в основном для отбелки шелка,
шерсти, меха, древесной массы и т.д.; этому немало
способствует сочетание в ней высокого окислитель-
ного потенциала с эффективностью и специфичностью
действия, а также безвредность продуктов реакции.
Кроме того, В. п. применяется как в чистом виде,
так и в виде водных р-ров в аналитич. химии, в меди-
цине, в качестве пенообразователя при произ-ве пори-
стых материалов (губчатая резина, пенопласты и т. д.),
для получения органич. перекисей, для инициирова-
ния процессов полимеризации и других реакций
с участием свободных радикалов, для очистки и раз-
деления солей металлов, протравливания семян, при-
готовления и консервирования пищевых продуктов
и напитков. Являясь сильным окислителем, В. п.
получила применение в реактивной технике. В. п. не-
токсична, но ее концентрированные р-ры при попада-
нии на кожу, слизистые оболочки и в дыхательные
пути вызывают ожоги.
Лит.: Перекись водорода и перекисные соединения. Под
ред. М. Е. Позина, Л.—М., 1951; -Шамб У., Сеттер-
ф и л д Ч., Веитворс Р., Перекись водорода, пер.
с англ;, М., 1958. А. И. Чемоданов.
ВОДОРОДНАЯ СВЯЗЬ — соединение посредством
атома водорода двух атомов, входящих в состав раз-
ных молекул или одной и той же молекулы. Большей
частью атом водорода связан с одним из атомов (А)
значительно сильнее, чем с другим (В). В этом случае
группа А—Н деформируется (растягивается) сравни-
тельно мало. Однако в ряде соединений RAH...BR',
по мере усиления связи Н... Ви сокращения равновес-
ного межъядерного расстояния Н...В, свнзь А—Н
растягивается все сильнее и в предельном случае
атом Н оказывается связанным с обоими атомами А
и В в одинаковой степени [напр., ион (FIIF)- и др.].
В. с. возникает между атомами гл. обр. след, эле-
ментов: кислорода, азота, фтора, в меньшей степени
хлора, серы й др. С наличием В. с. связан ряд осо-
627
ВОДОРОДНАЯ СВЯЗЬ — ВОДОРОДНЫЙ ПОКАЗАТЕЛЬ
628
бенностей веществ. Сюда относятся ассоциация моле-
кул (спирты, вода, кислоты и др.) и обусловленные
ею повышенные значения темп-p плавления и кипения,
«аномалии» в растворимости, особенности в колеба-
тельных и электронных спектрах, в спектрах протон-
ного магнитного резонанса и др. Так, напр., темп-ра
плавления при переходе от H2S к Н2О повышается от
—83° до 0°, несмотря на то, что в сероводороде, обла-
дающем большим молекулярным весом, следовало
бы ожидать более сильного ван-дер-ваальсовского
молекулярного притяжения и, следовательно, более
высокой темп-ры плавления; в действительности В. с.
между молекулами Н2О сильнее, чем между молеку-
лами H3S, причем этот эффект является превалирую-
щим. Метан и другие гидриды группы углерода (SiH4,
GeII4, SnH4) обладают очень низкой темп-рой плавле-
ния и не проявляют способности к образованию В. с.
и по другим признакам. Группа С—Н может давать
В. с., когда атом С соединен с сильно электроотрица-
тельными атомами, например в цианистом водороде
(N = C—И), хлороформе (С13С—Н). По данным ядер-
ного магнитного резонанса, слабые В. с. могут образо-
вываться группами С—Н и таких молекул, как эти-
лен и ацетилен.
Благодаря В. с. молекулы соединяются в димеры
и полимеры; последние могут иметь линейное, развет-
вленное или циклич. строение. Так, карбоновые к-ты
в газовой и жидкой фазах существуют в основном в ви-
де димеров
Л.....н—
RC\
ХО— Н.....сг
Фтористый водород в газовой фазе образует циклич.
комплексы из нескольких молекул, напр. (HF)6.
Спирты в жидкой и твердой фазах содержат цепочеч-
ные или кольцевые полимеры. В белках, нуклеиновых
кислотах и других биологич. веществах В. с. обусло-
вливают поперечное «сшивание» цепочечных молекул
и образование сложных полимерных структур.
В зависимости от прочности В. с. расстояние между
атомами А и В в комплексе RA—Н... BR' может за-
метно варьировать. Так, расстояние О...О изменяется
примерно от 2,45 А («сильная» связь) до 2,8 А («сла-
бая» связь). Если расстояние А... В превышает иек-рое
характерное для данной пары атомов расстояние, то
В. с. между ними не образуется. Опытным путем уста-
новлено, что в ряду соединений RA—H...BR', в к-ром
варьируют R и R', с уменьшением равновесного рас-
стояния А...В закономерно увеличивается длина связи
А—Н; это удлинение, напр. для связи RO—H...OR',
составляет от 0,01 А до 0,25 А и приблизительно про-
порционально прочности В. с. Последняя обычно зна-
чительно (в 15—20 раз) слабее ковалентной связи
А—Н; ее энергия составляет, как правило, от 4 до
8 ккал/моль, что превосходит энергию ван-дер-вааль-
совского взаимодействия (ок. 1 ккал/моль). В коле-
бательных спектрах при образовании В. с. вместо
узкой полосы, отвечающей колебаниям изолирован-
ной группы А—Н (напр., О—Н в спиртах), появляется
сравнительно широкая полоса, максимум к-рой сдви-
нут в сторону длинных волн. Это смещение законо-
мерно связано с увеличением межъядерного расстоя-
ния А—Н, а также с энергией В. с. В молекулах
ароматич. соединений, особенно при внутримолеку-
лярной В. с. (напр., в ортонитрофеноле), это смещение
и ширина полосы в неск. раз шире, чем в молекулах,
не имеющих сопряженных связей. В УФ-спектрах
при образовании В. с. в нек-рых случаях наблюдается
красное, в других — фиолетовое смещение. Измене-
ния наблюдаются и в спектрах ядерного магнитного
резонанса (см. Радиоспектроскопия).
На основании опытных данных (особенно по коле-
бательным спектрам) В. с. не может быть приписана
чисто электростатич. природа. Наряду с электроста-
тич. притяжением остаточных зарядов атомов А, Н,
В в образовании В. с. существенную роль играет до-
норно-акцепторное взаимодействие атома В (донор),
имеющего неподеленную пару электронов, и атома Н
(акцептор). Весьма вероятно, что наряду с растяже-
нием связи А—Н происходит ее значительная поля-
ризация (сдвиг электронного облака от Н к А), что
способствует возникновению донорно-акцепторного
взаимодействия Н...В. В молекулах ароматич. со-
единений, кроме того, в образовании В. с., по-види-
мому, участвуют и л-электроны сопряженной системы.
Приведенная трактовка позволяет установить зави-
симость между образованием В. с. и процессами меж-
молекулярного перехода протона; по-видимому, воз-
никновение комплекса А—Н...В является первой
стадией протолитич. реакций типа RAH + BR' —>
—>RA+ + (HRR')+.
Лит.: Соколов И. Д., Усп. физ. наук, 1955, 57, вып. 2,
с. 205—78; П а у л и н г Л., Природа химической связи,
пер. с англ., М. — Л., 1947; Малышев В. И., Усп. физ.
наук, 1957, 63, вып. 1, с. 323—53; Coulson С. A., Research,
1957, 10, № 4, р. 149—59; PimentallG. С. and М с
С 1 е 1 1 a n A. L., Hydrogen bond, [San-Francisco], 1960;
Hydrogen bonding. Paper presented at the symposium on hyd-
rogen bonding held at Ljublijana, 29 July—3 August 1957,
L. — [a. o.], 1959. H. Д. Соколов.
ВОДОРОДНЫЙ ПОКАЗАТЕЛЬ — величина, ха-
рактеризующая активность или концентрацию ионов
водорода в р-рах; численно равна отрицательному
десятичному логарифму этой активности или концент-
рации, выраженной в грамм-ионах на литр: pH =
= —lgCH, где Сн — концентрация ионов водорода.
В. и. обозначается pH. В воде концентрация ионов
водорода определяется электролитич. диссоциацией
по ур-нию:
HaOz2H+4-OH~ (1)
В этом случае В. п. может быть рассчитан из кон-
станты равновесия процесса (1):
Kd~ ’ ^он-/^н2о (1а)
где Сн+ и Сон_ соответственно концентрации ионов
водорода и гидроксила в грамм-ионах на литр. По-
скольку степень диссоциации воды очень мала, то,
не внося значительной погрешности, можно считать
концентрацию недиссоциированных молекул воды
постоянной величиной и объединить ее с KD в одну
постоянную ЙГВ = 0, В этом случае ур-ние
1а примет вид:
^Н+ ‘ сОН-= (16)
7fB называется ионным произведением
в о д ы и является величиной, постоянной для данной
темп-ры. Реакция (1) сопровождается поглощением
теплоты (в количестве 13600 кал/моль), поэтому с по-
вышением темп-ры равновесие в ней смещается вправо,
т. е. степень диссоциации воды возрастает. Константа
диссоциации и ионное произведение воды измерены
несколькими независимыми методами; получены хо-
рошо согласующиеся результаты. Ионное произве-
дение А"в и концентрации Сн+ иС0Н_ в воде и водо-
родный показатель pH воды приведены в табл.
сн+ = сон- pH
0 1,139 • 10-15 3,38 • 10-8 7,972
18 5,702 • 10-15 7,64 • 10-8 7,117
25 1.008 • 10-1« 1.004 • 10-1 6,999
50 5,474 10-и 2,339 • 10-1 6,631
100 5,9 • 10-18 7,7 • 10-7 6,12
ВОДОРОДНЫЙ ЭЛЕКТРОД — ВОДЫ СТОЧНЫЕ
629
Из таблицы видно, что при 25° в чистой воде или
в любой другой нейтральной водной среде Сн+ =
= ^ОН- = = 1,004 • 10“’ и, следовательно, pH =
= 6,999. В р-рах кислот и щелочей Сн+ не будет равно
С01[., но в любом случае, как видно из уравнения (16),
будет сохраняться обратная пропорциональность ме-
жду этими величинами и, следовательно, любое повы-
шение концентрации водородных ионов вызовет соот-
ветствующее уменьшение концентрации гидроксиль-
ных ионов, и наоборот. Т. о., при данной темп-ре:
pH = lgC0H_—lg Кв (2)
Ион водорода в водных р-рах гидратируется, образуя
ион гидроксония Н3О+. Было рассчитано, что в одно-
молярных водных р-рах сильных одноосновных к-т
Сн 0+ = 1 г-ион/л, а Сн+= 10'130 г-ион/л. Растворы,
в к-рых при 25° pHi>7, являются щелочными, pH ок.
7 — нейтральными, а pH с 7 — кислыми. Методы
измерения концентраций водородных ионов многооб-
разны. Для этой цели могут быть использованы кис-
лотно-основные индикаторы (в растворах и нанесен-
ные на бумагу), кинетич. и каталитич. реакции, био-
логич. процессы, инструментальные методы и т. п.
Наиболее точно pH определяется потенциометрия,
методами, к-рые широко вошли в практику. Величина
pH характеризует активную кислотность р-ров, имею-
щую большое значение для биохимия, процессов, для
производственных процессов в пищевой, кожевенной,
текстильной, химич. и мн. др. отраслях пром-сти, при
изучении свойств природных вод и возможности их
применения и т. п. См. также Потенциометрическое
титрование.
Лит.: Бриттон X., Водородные ионы, пер. с англ.,
Л., 1936; Шатен штейн А. И., Теория кислот и основа-
ний, М.—Л., 1949; К и р е е в В. А., Курс физической химии,
2 изд., М., 1956; Виноградова Е. И., Методы опреде-
ления концентрации водородных ионов, [М.], 1950; Кольт-
г о ф И. И. н Стенгер В. А., Объемный анализ, пер.
с англ., т. 1, М., 1950; П ч е л и н В. А., Измерение актив-
ности водородных ионов (pH), окислительно-восстановитель-
ных потенциалов и потенциометрическое титрование, М.,
1955. Н. А. Канаев.
ВОДОРОДНЫЙ ЭЛЕКТРОД — электрод, потен-
циал к-рого определяется обратимо протекающей
реакцией Н2--2Н! + 2е~. При этом необходимо, чтобы
скорость всех других электрохимия, процессов (раст-
ворение материала электрода, окисление или восста-
новление компонентов р-ра) была бы мала по срав-
нению со скоростью указанной реакции. Вследствие
устойчивости потенциала В. э. на ряде металлов (Pt,
Pd, Ir, Au и др.) и удобства работы с ним, особенно
при измерении катодного перенапряжения, В. э. ши-
роко применяется в качестве электрода сравнения.
Обычно он представляет собой пластинку или прово-
локу из платинированной платины, насыщенную водо-
родом и наполовину опущенную в раствор, содержа-
щий ионы водорода. Применяемый водород и раствор
должны быть тщательно очищены от следов деполя-
ризаторов (кислород) и веществ, уменьшающих ско-
рость реакции, определяющей потенциал (соединения
мышьяка, серы, поверхностно-активные органич. в-ва).
Зависимость потенциала В. э. Е от давления водо-
рода р и активности ионов водорода в р-ре ан+ опре-
деляется формулой Нернста:
Е = Е° + In = Е° + 0,0001984 Т 1g
г р /а р /1
где Т — абс. температура, Е° — потенциал В. э. при
р = 1 атм и вн+ = 1 (для любой заданной, но по-
стоянной темп-ры условно принимается за нуль); по-
тенциалы всех остальных электродов отсчитываются
630
от потенциала этого стандартного В. э. (водородная
шкала потенциалов).
Лит.: Фрумкин А. Н. [ид р.], Кинетика электродных
процессов, М., 1952. А. Н. Че.моданов.
ВОДЫ МИНЕРАЛЬНЫЕ — воды источников или
буровых скважин, содержащие от 0,1 до 5% раство-
ренных электролитов; нек-рые из них обладают целеб-
ными свойствами, что связано с их составом и темп-рой.
Содержание отдельных компонентов в В. м. должно
быть выше след, значений (в г/кг): 0,25 СО2 (свобод-
ной), 0,001 H2S, 0,001 Li+, 0,005Ва2+, 0,010Sr2+,
0,01 Fe2*' + Fe3*', 0,002 F~, 0,005 Br“, 0,001 J~, 0,001
HAsO42“, 0,005 BO2 и для эманации радия 3,5 еди-
ницы по Махе. Важную роль при оценке физиология,
действия и генезиса В. м. играют концентрации и
соотношения ионов Na*', Са2+, Mg'2', Cl“, SO|~ и НСО3,
по к-рым классифицируют В. м. При классификации
принимается также во внимание содержание в воде
газов (СО2, H2S, СН4, N2, эманации радия) и наличие
биологически активных элементов (Fe, As, Br, J
и др.). Содержание микроэлементов в В. м. предста-
вляет интерес не только для оценки их лечебного дей-
ствия, но и с геохимич. точки зрения. Большое зна-
чение имеет наличие в В. м. слабых к-т, особенно
угольной и сероводородной.
Для характеристики В. м. широко применяются
физико-химич. методы исследования, в частности
измерения величины pH, парциального давления сво-
бодных газов и методы расчета соответствующих рав-
новесий. Для выяснения генезиса вод и условий миг-
рации элементов в них необходимо установление сте-
пени насыщенности вод по отношению к труднораство-
римым соединениям [СаСО3, CaSO4, Fe(OH)3 и др.],
а также характеристика состояния элементов с пере-
менной валентностью по величине окислительно-вос-
становительного потенциала вод. В генезисе В. м.
принимают участие как воды поверхностного проис-
хождения, так и захороненные морские воды, претер-
певающие сложные изменения состава в результате
взаимодействия с породами, влияния биологич. фак-
торов (сульфатредукции) и смешения. В распределе-
нии В. м. различных типов наблюдается связь с гео-
логия. структурами. Главные месторождения В. м.
приурочены к зонам крупных тектонич. нарушений
и молодого вулканизма. Таковы воды района Кавказ-
ских минеральных вод, Боржоми, Дарасун, Карлови
Вары, Наугейм, Виши и др.
Лит.: Вернадский В. И., История минералов земной
коры, т. 2, ч. 1, вып. 1—3, Д., 1933—36; Овчинников
А. М., Минеральные воды, М.—Л., 1947; Аленин О. А.,
Основы гидрохимии, Л., 1953. П. А. Крюков.
ВОДЫ СТОЧНЫЕ — воды, использованные про-
мышленными или коммунальными предприятиями и
городским населением, подлежащие удалению. В. с.,
как правило, спускИют в водоемы. Основные примеси,
к-рые обычно содержатся в различных В. с., указаны
в табл. 1, а в табл. 2 — предельно допустимые кон-
центрации нек-рых веществ у мест водопользования.
Условия спуска В. с. в общественные водоемы нор-
мируются соответствующими «Правилами» (НСП-101-
57), согласно к-рым при спуске В. с. состав и свойства
воды данной реки у ближайшего расположенного
ниже по течению места водопользования не должны
изменяться в большей степени, чем это предусмотрено
«Правилами». Места водопользования подразделяются
на 3 категории: 1) участки реки, используемые для
централизованного хозяйственно-питьевого водоснаб-
жения или граничащие с государственными рыбными
заповедниками; 2) участки реки, используемые для
неорганизованного хозяйственно-питьевого водоснаб-
жения,водоснабжения предприятий пищевой пром-сти,
а также участки с местами массового нереста промыс-
ловых рыб; 3) участки реки внутри населенных мест,
ие используемые для питьевого водоснабжения, но
631
ВОДЫ СТОЧНЫЕ
632
Таблица 1. Основные примеси в сточных водах
Происхождение В. с. Основные примеси
Анилннокр асочная Кислоты (серная, азотная); нитросо-
пром-сть единения; амнны; фенолы; красители
Бумажная пром-сть Органич. грубодисперсные примеси (бумажное волокно); лнгносульфо- новые к-ты; меркаптаны; сульфиды; алюминий; сульфаты; сульфиты н др.
Калийная пром-сть Калий; натрий; хлориды
Кожевенная пром-сть Органич. грубодисперсные примеси; белковые в-ва; кальций; хром; суль- фиды; полисульфиды
Коксохимическая Фенолы; жирные нйзшие к-ты; пири-
пром-сть диновые основания; углеводороды; смолы; аммиак
Лесохимическая Спирты; жирные низшие к-ты; альде-
пром-сть гиды; кетоны; смолы
Нефтедобывающая и Эмульгированные примеси; углеводо-
нефтеперер абаты- роды; нафтеновые к-ты; меркаптаны;
вающая пром-сть сероводород; натрий; кальций; хло- риды; сульфаты
Произ-во искусствен- Органич. грубодисперсные примеси;
ного волокна сероуглерод
Произ-во пластмасс Фенолы; альдегиды; кетоны; белковые
в-ва
Произ-во синтетич. Спирты; альдегиды; кетоны; углеводо-
каучука роды
Сахарная пром-сть Минеральные грубодисперсные при- меси; углеводы; белковые в-ва
Содовая пром-сть Кальций; хлориды
Текстильная Красители; жирные высшие к-ты; та-
пром-сть ннны; тяжелые металлы; хром
Хлорная пром-сть Натрий; хлориды; хлор
Хозяйственпо-быто- Оргапич. грубодисперспые примеси;
вые В. с. жиры; белковые в-ва; мочевина; ам- миак; натрий; хлориды
используемые для массового купания, организован-
ного рыбного хозяйства или находящиеся на пути
прохода рыб к нерестилищам. Для таких участков
установлены след, нормы (для 1-й, 2-й и 3-й категорий
соответственно): повышение концентрации взвешенных
веществ не более чем на 0,25 мг/л, 0,75 мг/л и 1,50 мг/л
против имеющейся в воде; пятисуточное биохимич.
потребление кислорода не должно превышать 2,0 мг[л
и 4 .мг/л (для участков 3-й категории не нормируется);
при смешении В. с. с дистиллированной водой в про-
порции, соответствующей разбавлению ее в воде реки,
не должно быть ярко выраженной окраски в столбике
высотой 20, 10 и 5 см. В. с., к-рые могут содержать
возбудителей заразных заболеваний (воды боён, коже-
венных заводов, шерстомоек и Др.), не допускаются
к спуску на участках 1-й категории, а на участках 2-й и
3-й категорий допускаются лишт» после предваритель-
ного механич. осветления и обеззараживания (хлори-
рованием или др. методами). К требованиям, одина-
ковым для всех участков, относятся: концентрация
растворенного в воде кислорода у мест водопользо-
вания не ниже 4 .мг/л; pH воды не ниже 6,5 и не выше
8,5; на поверхности воды не должно быть сплошных
плавающих пленок масла, жиров, нефтепродуктов
и т. п.; вода реки у мест водопользования не должна
приобретать каких-либо специфич. запахов или при-
вкусов. Возможные концентрации вредных веществ в
В. с., спускаемых в водоемы, устанавливаются, исхо-
дя из местных условий (смешения и разбавления В. с.
водой водоема) и предельно допустимых концентра-
ций вредных в-в в ноде водоема у места ближайшего
водопользования, получающихся при спуске В. с.
Если изменения свойств и состава воды при спуске
в водоем В. с. выходят за пределы допустимого, В. с.
должны быть очищены или обезврежены до сброса;
методы очистки или обезвреживания зависят от со-
става В. с., степени их загрязнения и др.
Таблица 2. Нормы предельно допустимых концентраций
некоторых веществ (в мг/л) у места водопользования
Азотная к-та .... 30—35
Аммиак................ 5
Бензол ............... 5
Крезол................ 5
Медь...............0,1—0,2
Меркаптаны......... 2—5
Мышьяк.............0,1—0,2
Нафтеновые к-ты . . 5—10
Нефть...............25—30
Ртуть...............0,005
Свинец.............0,1—0,2
Серная в-та........20—30
Сероводород . . . 1—3
Сернистые металлы 2—3
Сероуглерод ... 1
Смолы.............. 20—50
Соляная к-та . . 30—35
Тринитротолуол . 0,1
Фенолы.......... 0,001—0,002
Фтор............ 0,5—1,0
Хлор.............0,25—0,5
Хром............ 0,1—0,2
Цианистые соли . 0,1—0,2
Очистка В. с. производится след, способами: 1) Про-
цеягиванием — для механич. задержания грубых твердых
примесей на решетках или ситах. 2) Отстаиванием — для
удаления грубо дисперсных твердых или жидких примесей
(с плотностью больше или меньше 1); ускорение выпадения
взвеси достигается коагуляцией. При удалении всплывающих
или тонущих примесей прибегают к флотации путем добавки
флотореагента, гидрофобизирующего поверхность частиц (с по-
следующей продувкой В. с. воздухом). Если частицы обладают
гидрофобными свойствами, то для их всплывания достаточно
одной продувки воздухом (напр., флотация жиров). При на-
личии мелкой, трудно оседающей пыли или в случаях, требую-
щих проведения дополнительных процессов (дегазации, ней-
трализации и др.), отстаивание производится в прудах-от-
стойнпках большой емкости. 3) Фильтрованием — для очистки
В. с. целлюлозно-бумажных предприятий от волокон, коксо-
химии. заводов от смол и т. п. 4) Дегазацией — для выделе-
ния растворенных газов путем продувки воздухом или раз-
брызгивания. 5) Отгонкой растворенных примесей с водяным
паром (напр., извлечение фенолов) с последующим пропуском
пара через р-р, вымывающий примесь из пара (напр., р-р
NaOH для извлечения фенолов). 6) Экстракцией — для уда-
ления растворенных примесей, для чего В. с. обрабатывают
к.-ц. не смешивающимся с водой извлекающим примесь раство-
рителем. Напр., для очистки от фенолов применяют феноло-
сольван (бутилацетат), минеральные масла, бензол, четырех-
хлористый углерод, трикрезилфосфат и др. 7) Кристаллизацией,
напр. с целью выделения железного купороса. 8) Сорбцией —
для извлечения растворенных примесей активным углем,
с его последующей регенерацией, а также торфом, бурым
углем, золой, коксовой или угольной мелочью, шлаками
и т. п., не регенерируемыми и удаляемыми после использо-
вания вместе с сорбированной примесью в отвал. Иногда для
этих целей применяют отвалы золы где, кроме сорбции, про-
исходит нейтрализация кислот. 9) Ионитовыми фильтрами —
для извлечения ценных примесей. 10) Добавлением электро-
лита, с к-рым удаляемые ионы образуют труднорастворимое
соединение, выпадающее в осадок. И) Нейтрализацией —
путем пропускания В. с. через фильтры, заполненные доло-
митом, мелом, известняком и т. п., или путем добавки извест-
кового молока или кислоты (обычно серной). 12) Восстановле-
нием или окислением содержащихся в В. с. примесей или
переводом их в комплексные соединения с образованием без-
вредных или выпадающих в осадок веществ. Восстановление
производится железпой стружкой, SO2, H2S или углем; окис-
ление — кислородом, хлором или хлорной известью; ком-
плексные соединения получают, напр., переводом цианидов
в практически безвредные соединения — ферроцианиды.
13) Биохимич. окислением — одним из наиболее важных про-
цессов очистки В. с., содержащих органич. вещества (В. с. мя-
сокомбинатов, сахарных, винокурейных и т. п. предприятий,
хозяйственно-бытовые воды и др.).
Биохимич. очистка является завершающей очисткой В. с.
Содержащиеся в В. с. органич. примеси в результате жизне-
деятельности организмов превращаются в углекислый газ,
нитраты, воду и др. окислы. Сначала В. с. направляют на
механич. очистку (решетки, песколовку, отстойники), а затем
на сооружения для биохимич. очистки — биофильтры, запол-
ненные крупнозернистым материалом (кокс, котельный шлак,
кирпичный щебень и т. п.), на к-рые периодически подаются
В. с., просачивающиеся через загрузку. Наполняющий био-
фильтр материал после созревания покрывается пленкой,
заселенной бактериями и низшими организмами. Снизу, через
дренаж фильтра в загрузку поступает воздух. Биохимич.
очистка В. с. может проводиться также в аэротенках — бас-
сейнах, через к-рые протекают В. с.; к последним добавляют
активный ил — хлопьевидные колонии бактерий н низших
организмов. В аэротенки непрерывно подают воздух, необхо-
димый для окисления; он также способствует перемешиванию
жидкости и поддержанию активного пла во взвешенном со-
стоянии. После биофильтров и аэротенков В. с. направляют
во вторичный отстойник для выделения захваченных частиц
пленки или активного ила. Осадок первичных отстойников
и часть осадка из вторичных отстойников (избыточный актив-
ный ил) направляют в закрытые емкости (метантенки), где
при подогреве происходит его сбраживание с выделением ме-
тана и СО3. Ил из метантенков подсушивается на иловых пло-
щадках и является хорошим удобрением. Биохимич. окисле-
ние может применяться для завершающей и наиболее полной
очистки промышленных В. с., содержащих органич. примеси.
Очистка нек-рых промышленных В. с. должна производиться
633
ВОДЯНОЙ ГАЗ —ВОЗДУХА АНАЛИЗ
634
в смеси-с хознйственно-бытовымн В. с. (15% для фенольных
В. с., до 25—50% для текстильных В. с.). Нагрузка на соору-
жения должна быть, как правило, в 1,5—2,5 раза меньше, чем
длн одних хозяйственно-бытовых В. с.
При рассмотрении вопросов, связанных с удалением
В. с. промышленных предприятий, необходимо учиты-
вать, что очистка в большинстве случаев требует гро-
моздких сооружений, дорогих в строительстве и
эксплуатации. В ряде случаев методы очистки В. с.
новых произ-в еще не известны и требуют лаборатор-
ных и опытно-производственных исследований. По-
этому необходимо стремиться к тому, чтобы предотвра-
тить возможность попадания в В. с. вредных веществ
или чтобы содержащие вредные вещества В. с. не по-
падали в водоемы. Предотвратить попадание вредных
веществ в В. с. можно, напр., след, способами: изме-
нением технологич. процесса (напр., переходом с «мок-
рого» процесса на сухой); естественным или искусст-
венным испарением В. с. без сброса их в водоемы;
повторным использованием В. с. в производственном
цикле; регулированием сброса В. с. с помощью спец,
емкостей («накопителей») и спуском В. с. в реку в па-
водок или небольшими количествами, пропорцио-
нально расходу воды в реке. Накопитель играет также
роль очистного сооружения (отстаивание, химич. и
биохимич. процессы). Возможен также спуск особо
загрязненных В. с. в глубоко залегающие (1000 м и
более), геологически изолированные водопоглощаю-
щие пласты.
Лит.: Ш п г о р и н Г. Г. и Д е м и д о в Л. Г., Кана-
лизация, ч. 2. Очистка сточных вод, М., 1951; Лапшин
М. И., Разработка способов очистки сточных вод, М., 1952;
Производственные сточные воды. Сб. ст., вып. 1—4, М., 1948—
.1954; Указания по проектированию сооружений по очистке
промышленных сточпых вод, М., 1956; Лурье Ю. Ю.
и Р ы б н и к о в а А. И., Химический анализ производствен-
ных сточных вод, М., 1958. В. Т. Турчинович.
ВОДЯНОЙ ГАЗ — см. Газификация твердых
топлив.
ВОЗДУХ — см. Атмосфера, Воздуха разделение.
ВОЗДУХА АНАЛИЗ (в промышленных предприя-
тиях) — определение содержания в воздушной среде
предприятия вредных для здоровья и взрывоопасных
примесей.
В. а. производится с целью сигнализации об опас-
ности, гигиенического изучения производственного
процесса и др. В соответствии с этим различают экс-
прессные, скоростные и обычные
методы анализа. Для первых иногда допускается
уменьшение точности за счет скорости выполнения
анализа. Для гигиенической оценки производства
имёют значение как разовые замеры воздуха, так и
усредненные — среднечасовые, или характеризую-
щие определенный цикл производственного процесса.
В связи с этим количество воздуха, отбираемого для
. анализа, может сильно изменяться (от 50—100 см3
до 1—2 .и3). Концентрацию вредно действующего
вещества выражают в мг/л воздуха, иногда в мг/м3;
для целей санитарной техники вычисления ведут
в объемных процентах. За рубежом концентрации
выражают также и в объемных отношениях, напр.
1 ; 100 000 и т. д. Содержание многих веществ в воз-
духе производственных помещений регламентируется
в виде предельно-допустимых концентраций. Нормами
предусмотрены случаи и одновременного воздействия
нескольких вредных веществ. Особенностями В. а.
являются: 1) определение весьма малых количеств
(0,00001 — 1 мг/л воздуха) исследуемых компонентов,
часто встречающихся в сочетаниях, взаимно мешаю-
щих определению; 2) переменный состав воздушной
среды, иногда сильно изменяющийся даже во время
отбора пробы.
В. а. состоит из след, основных элементов: отбора
пробы воздуха, подготовки пробы к анализу и выпол-
нения анализа. Агрегатное состояние содержащихся
в воздухе примесей влияет на способ отбора пробы
воздуха. Так, пыль и аэрозоли обычно улавливают
ватными или бумажными фильтрами; для улавлива-
ния аэрозолей применяют также пористые стеклян-
ные фильтры; туманы, пары и газы поглощают гл.
обр. жидкостями.
Содержание пыли и аэрозоля в воздухе определяют
по привесу фильтров, к-рый выражают в мг/м3 или
числом частиц в 1 см3 (т. н. с ч е т н ы й метод).
В этом случае частицы, уловленные из воздуха на
стеклянную пластинку, подсчитывают под микроско-
пом; кроме общего числа частиц, определяют также
содержание их в отдельных фракциях (по величине
частиц). Пары и газы поглощают жидкостями, протя-
гивая через них исследуемый воздух. Быстрота и
полнота поглощения достигается обычно путем созда-
ния большой поверхности поглощения. Для этого
применяют поглотители с перистыми пластинами или
приборы, в к-рых поглощение идет в пленке жидкости
(рис. 1,а);для газов, быстро поглощающихся жидкой
средой (напр., NH3 и т. и.),
применяют более простые
поглотители, через которые ') 'Ч (WX
исследуемый воздух про- ( }
ходит мелкими пузырьками \ /
(рис. 1, б). Отобранный в II \ /
производственной обстанов- ( )
ке объем пробы воздуха ) (
приводят к нормальным Ч )
условиям. Подготовка про- (_J !
бы воздуха для анализа ° 6
производится обычными для Рис. 1.
аналитич. химии методами.
Твердые вещества вымываются подходящим раство-
рителем из фильтров, после чего объем доводится до
точно определенного в мерной колбе. При пользова-
нии жидкой средой для поглощения ее объем также
доводится до точно определенного. Во всех случаях
производят не менее двух определений. Неорганич.
вещества определяют обычно в виде ионов; органи-
ческие — либо минерализуют выпариванием пробы и
разложением ее, либо определяют специфич. реакция-
ми на определяемое соединение.
Способы определения содержания искомого вещества
весьма разнообразны. Микрометоды объемного ана-
лиза применяются для В. а. лишь ограниченно, напр.
при определении СО, СО2 и нек-рых других веществ.
Определение в воздухе взрывоопасных концентраций
нек-рых примесей (паров бензина, метана, водорода)
при содержании более 1 об. % производится посредст-
вом спец, переносных приборов. К наиболее распро-
страненным относятся колориметрич. и нефелометрич.
методы (см. Фотометрические методы анализа, Нефе-
лометрия и турбидиметрия); пробу обрабатывают
соответствующими реактивами и сравнивают визу-
ально возникающую окраску с окраской натуральной
или искусственной стандартной серии или же опре-
деляют концентрацию вещества фотоколориметриче-
ски, пользуясь калибровочной кривой. Напр., содер-
жание кремния определяют по интенсивности окраски
восстановленного силико-молибденового комплекса,
фтора — по обесцвечиванию циркон-ализаринового
лака, р-нафтола — по интенсивности окраски азо-
красителя, полученного азосочетанием с п-нитрофенил-
диазонием, сериую к-ту — нефелометрически по ин-
тенсивности мути взвешенного в растворе сульфата
бария и т. д. Для В. а. применяются также спектро-
фотометрия., люминесцентные, полярография, и кон-
дуктометрия. методы анализа. Для раздельного опре-
деления смеси углеводородов пользуются хроматогра-
фия. адсорбционным анализом.
Заметную роль в В. а. играют индикац. методы
экспресс-анализа. Этот вид анализа сочетает в себе
одновременно отбор пробы и количеств, определение.
635
ВОЗДУХА РАЗДЕЛЕНИЕ
633
При индикации происходит изменение длины окра-
шенного слоя столбика порошка в трубке или изме-
нение интенсивности его окраски, к-рое пропор-
ционально количеству сорбированного газа или паров
вещества. Такие методы основаны на цветных реак-
циях, протекающих с большой скоростью; осажден-
ный на носителе (силикагеле, фарфоре) слой реактива
должен иметь строго определенную хемосорбц. ем-
кость. По такому принципу устроены переносные газо-
анализаторы инспекторского типа, позволяющие в те-
чение нескольких минут на месте определить количе-
ство искомой примеси в воздухе.
Рис. 2.
На рис. 2 показан прибор, пользуясь к-рым можно опре-
делить неск. веществ, напр. пары бензина, бензола, толуола,
серного эфира, ацетилена, амми-
ака, сероводорода, окислов азо-
та, хлора или сернистого газа.
Принцип действия прибора со-
стоит в измерении длины окра-
шенного столбика индикаторной
трубки 3; окрашивание проис-
ходит при протягивании через
трубку исследуемого воздуха.
Длина столбика пропорцио-
нальна содержанию искомого ве-
щества в испытуемом воздухе
и измеряется ио шкале прибо-
ра. Воздухозаборное устройство
прибора состоит из резинового
сильфона 1 с пружиной внутри.
Сильфон сжимается с помощью
штока 2 до фиксированного по-
ложения. При отборе пробы
воздуха шток отпускается и
сильфон растягивается до нуж-
ного положения.
Индикация по изменению
интенсивности окраски втрубках
(колориметрия) осуществлена в
нек-рых анализаторах для окиси
углерода. Во многих экспресс-
методах воздух просасывают
черев миниатюрные жидкостные
поглотители, а определение ве-
дут по искусств, цветным шка-
лам.
К автоматич. приборам
для В. а. относится, напр.,
прибор для экспрессного
определения паров ртути
в воздухе, в к-ром ультра-
фиолетовое излучение по-
глощается парами ртути;
это поглощение измеряется
фотоэлектрич. устройством.
Известны также образцы
фотоэлектрич. анализатора воздуха (с- сигнальным
устройством) для автоматич. регистрации в воздухе
AsH3 и др. В связи с рядом специфич. особенностей
автоматич. приборы для В. а. имеют ограниченное
применение. Особое место в В. а. занимает измерение
степени радиоактивного излучения (см. Дозиметрия,
Дозиметрия химическая).
Лит.: Алексеева М. В. [и др.], Определение вредных
веществ в воздухе производственных помещений, 2 изд., М.,
1954; Выховская М. С. [и др.], Практическое руко-
водство по промышленно-санитарной химии, ч. 1, М., 1954;
Филянская Е. Д. [и др.], Линейно-колористический
метод анализа вредных газов и паров в воздухе промышленных
предприятий, М., 1958; Определения вредных веществ в воз-
духе. [Сб. статей]. Под ред. О. Д. Хализовой, М., 1957; Пре-
дельно допустимые концентрации ядовитых газов, паров и
пыли в воздухе рабочих помещений, М., 1959; Jacobs М. В.,
The chemical analysis of air pollutants, L. — N. Y., 1960 (Che-
mical analysis, v. 10). Б. E. Андронов.
ВОЗДУХА РАЗДЕЛЕНИЕ — разделение воздуш-
ной смеси на составляющие ее компоненты. Состав
газов воздушной смеси приведен в таблице 1.
В зависимости от близости к населенным пунктам
и промышленным предприятиям в воздухе колеблется
только содержание углекислого газа. Содержание
водяного пара в воздухе колеблется в широких пре-
делах.
Таблица 1
Компо- нент Объемн. % Весов. % Компо- нент Объемн. % Весов. %
N. о; Аг Ne Кг Хе 78.09 20,95 0,93 1,6- Ю-з 1,1 . 10-4 8- 10-е 75.50 23,10 1,29 1,3- Ю-з 2,9 . 10-4 3,6- 10-е О3 Rn Н., Нё СО3 2. 10—в 6- 10-18 5- Ю-з 4,6 . 10-4 0,03 3,3- 10-е 4,6-10-17 3,5-10-3 7,2. Ю-з 0,046
В районах добычи, переработки или потребления нефти,
природного газа и пр. в воздухе содержатся также небольшие
количества углеводородов, представляющих большую опас-
ность для воздухоразделительных аппаратов. Так, напр.,
темп-ра сублимации ацетилена при нормальном давлении
189,7°К; накопление его в ожижительных или разделитель-
ных установках приводит к взрывам большой силы.
Сжижение и разделение газовой смеси сопровож-
дается уменьшением энтропии, поэтому их осуществ-
ление невозможно без затраты внешней работы. Мини-
мальный расход работы А на разделение газовой смеси
в случае равновесного обратимого процесса, согласно
первому закону термодинамики, составляет: —А =
= А// — TOASC, где АН — изменение энтальпии,
Та — абсолютная темп-ра, A,S'C — уменьшение энтро-
пии системы. Реальный процесс разделения газовой
смеси, вследствие своей необратимости (потери холода
в окружающую среду, гидравлич. сопротивления
и др.), требует значительно большего расхода энергии.
Так, на получение 1 нм3 99%-ного кислорода расхо-
дуется не менее 0,5 квт-ч электрич. энергии, вместо
0,074 квт-ч по вышеприведенному ур-нию. С умень-
шением потерь снижается расход энергии, что дости-
гается в современной технике заменой рекуператив-
ных теплообменников более компактными регенера-
торами, в к-рых потери холода меньше. С ростом коли-
чества перерабатываемого воздуха от 1000 нм3{час
до 10 000 нм31час удельные потери холода в окружаю-
щую среду падают с 2,3 ккал/нм3 до 1,2 ккал/нм3.
Зависимость уд. расхода энергии от
ввт-час/нм3 часового количества перерабатывае-
мого воздуха и концентрации по-
лучаемого кислорода приведена на
рис. 1.
ма
О.7
о.е
0,5
0,4
99,5% О2
95,0% О2 .
6000 10000 20000 30000 40000 50000 чи'/чвс
Рис. 1. Зависимость удельного расхода энергии от
количества перерабатываемого воздуха и концентра-
ции получаемого кислорода.
В воздухоразделительпых установках применяют аппараты
двухкратной ректификации (рис. 2), состоящие из двух ко-
лонн 1 и 2 непрерывного действия, расположенных друг над
другом, причем дефлегматор з нижней колонны служит испа-
рителем верхней. В трубном пространстве дефлегматора-
испарителя конденсируется азот, являющийся флегмой для
обеих колони, а в межтрубном пространстве кипит кислород,
представляющий собой кубовый остаток верхней колонны.
Воздух перед входом в ректификационный аппарат сжимается
до более высокого давления, т. к. он служит одновременно
источником холода, веобходимого для проведения процесса
разделения. В промышленных установках глубокого охлажде-
ния для получения холода используется изоэнтальпическое
(Джоуля — Томсона эффект) и изоэнтропическое (с совер-
шением внешней работы) расширение сжатых газов. Для
увеличения эффекта Джоуля — Томсона сжатые газы часто
предварительно охлаждают испаряющимся аммиаком при—50°,
чем одновременно достигается и их осушка. Так, в установ-
ке для получения 99,95%-ного азота (рис. 3), для обеспечения
необходимого количества холода, помимо аммиачного охлажде-
ния, ок. 20% воздуха приходится сжимать до 100—200 ат.
Установка БР-1 для получения кислорода (рис. 4) благо-
даря применяемым высокоэффективным турбодетандерам,
а также регенераторам работает с давлением воздуха порядка
7 ат; лишь в малопроизводительных установках начальное
давление воздуха составляет 30—40 ат. При разделении воз-
духа одновременно с кислородом и азотом, в качестве побочных
637
ВОЗДУХА РАЗДЕЛЕНИЕ
638
продуктов, можпо получать и редкие газы. В определенных
условиях оказывается рентабельным и специальное выделение
из воздуха криптона и ксенона.
Получение газообразного азота. На рис.
3 приведена схема агрегата для получения
99,95%-ного азота, в к-ром холод полу-
чают за счет использования эффекта Джо-
уля—Томсона. Разделяемый воздух заса-
сывается через фильтр 1 поршневым ком-
прессором 2 и после II ступени, сжатый
до 7 ат, проходит через маслоотделитель
з в скрубберы 4 и очищается от СО2 8—
12%-ным раствором NaOH. Рециркуляцию
щелочи производят насосами 17, а подачу
свежей щелочи — насосом 16; 80% очищен-
ного от СО2 воздуха проходит через ловуш-
ку 5, предварительные теплообменники 7,
аммиачные холодильники 8, теплообмен-
ники теплой ветви 11, теплообменники
холодной ветви 12 и поступает в нижнюю
часть ректификационного аппарата 13.
Аппараты 7, 8 и 11, в которых одновремен-
но с охлаждением воздуха происходит
вымораживание водяных паров, —парные,
1пс. Аппарат переключающиеся по мере забивки льдом,
двухкратной рек- размораживание (оттаивание) теплообмен-
тификации. ников 7 производится теплым исходным
воздухом, холодильников 8 — парами пе-
регретого аммиака с давлением 7—9 ат, а теплообмен-
ников 11 — подогретым азотом или кислородом. Около 20%
воздуха, необходимого для получения холода, подвергается
духа на чистый азот и смесь азота и кислорода (38%), а в
верхней—под давлением 1,4 ат кубовая жидкость разде-
ляется на технически чистый азот (99,95%—99,98%) и 92%-ный
кислород. Между обеими колоннами расположен конден-
сатор, являющийся дефлегматором для нижней и одновре-
менно испарителем для верхней колонны. Параллельно с ос-
новным работает дополнительный конденсатор 14, из к-рого
отбирается кислород; холод его рекуперируется в теплообмен-
никах 15 и 9. Кубовая жидкость подается после дросселиро-
вания в верхнюю колонну, а флегма (жидкий азот) отбирается
из карманов нижней колонны. Азот отводится сверху ректи-
фикационного аппарата, а холод его используется последо-
вательно в теплообменниках 12, 15 (частично), 11 н 7. В слу-
чае необходимости одновременно с азотом высокой чистоты
может быть получен 99%-ный кислород (см. ниже «Получение
редких газов. Аргон*). На получение 1 нм3 азота расходуется
0,25 квт-ч электроэнергии.
Получение газообразного кислорода. Схема такой воздухо-
разделительной установки БР-1, работающей только на низ-
ком давлении, приведена на рис. 4; воздух сжимается до 6,0—
7,0 ат; ок. 20% его поступает в кислородные регенераторы 1,
а остальное количество — в азотные регенераторы 2. В реге-
нераторах воздух (прямой поток) охлаждается до 101,5° К,
что соответствует сухому насыщенному пару, освобождаясь
при атом от содержащихся в нем паров воды и СО2. В кисло-
родных регенераторах, ^переключаемых через каждые 3 ми-
нуты, обратный поток, к-рый охлаждает насадку и одновре-
менно выносит вымороженные из воздуха пары Н2О и СС>,
превышает прямой поток на 3,3%. Так как через азотные реге-
нераторы проходит меньше азота, нежели воздуха, то для
выноса из них снега и твердой С02 в БР-1 применена т. н.
дальнейшему сжатию в Ill, IV и V ступенях компрессора 2
до 120—2 00 ат, после чего проходит через маслоотделитель 6,
предварительные теплообменники высокого давления 9,
аммиачный холодильник высокого давления 10, якорный тепло-
обменник 15, змеевик (испари-
тель) в кубе нижней колонны и
дросселируется в нижнюю часть
ректификационного аппарата 13.
Аппараты 9 и Ю — парные, пе-
реключающиеся по мере их
забивки льдом и маслом. Раз-
мораживание теплообменника 9
Воздух из турбокомпрессора
Азот в^атмосферу
Кислород
Рис. 4. Схема установки БР-1 для получения газообразного кислорода.
производится подогретым кислородом, а холодильников 10 —
перегретыми парами аммиака (аналогично холодильникам 8).
Ректификационный аппарат 13 состоит из двух колонн: в
нижней—под давлением 7 ат происходит разделение воз-
«петля», посредством к-рой осуществляется дополнительное
охлаждение насадки потоком холодного (петлевого) воздуха
после регенераторов. Через азотные регенераторы, автомати-
чески переключаемые через каждые 3 мин., последовательно
проходит воздух, азот и «петлевой* воздух. Соответственно
числу цртоков установлены 3 азотных регенератора. Петле-
вой воздух отводится из середйны регенераторов с темп-рой
160—180°К. Регулированием количества петлевого воздуха
639
ВОЗДУХА РАЗДЕЛЕНИЕ
640
поддерживают на холодных концах азотных регенераторов
необходимую разность темп-р 5—6°. Из регенератора большая
часть воздуха отводится в нижнюю колонну 8, а другая часть —
петлевой воздух, регулируемая автоматич. клапанами 19,
пропускается снизу вверх через нижние слои насадки соответ-
ствующего азотного регенератора, после чего поступает в труб-
ное пространство детандерного теплообменника 5, где исполь-
зуется для подогрева воздуха, направляемого в детандер 17.
Далее петлевой воздух смешивается с основным потоком,
поступающим в нижнюю колонну из регенераторов. В нижней
колонне 8 происходит разделение на жидкий азот и обогащен-
ный кислородом воздух. Ок. 25% паров после промывки на
трех тарелках отбирается из нижней колонны, пропускается
через отделитель 6 и теплообменник 5, в к-ром подогревается
до 116—117° К, и направляется через фильтр 18 в один из
турбодетандеров 17, где расширяется от 5,8 ат до 1,4 ат.
После детандера охлажденный воздух проходит через адсор-
бер ацетилена 4 в верхнюю колонну 7.
/Видкость из куба нижней колонны 8 очищается от твердой
СО2 в керамиковых фильтр ах юн от ацетилена—в адсорберах 11,
после чего охлаждается до 96° К в переохл^ителе 9, дроссели-
руется в распределительный бачок, расположенный внутри
верхней колонны 7. Из бачка жидкость сливается на 21-ю
тарелку. Число тарелок в нижней колонне 24, в верхней — 36;
расстояние между тарелками 120 мм, тарелки — кольцевые
ситчатые; в качеетве адсорбента применяется силикагель.
Пары азота из нижней колонны конденсируются в межтруб-
ном пространстве конденсаторов 12, 13, 14 и 15, в трубах
к-рых кипит жидкий кислород, стекающий из верхней колонны.
Азот, "отводимый из колонны 7, после рекуперации его холода
в переохладителе 9, подогревателе азота з и регенераторах 2,
выпускается в атмосферу. В подогревателе 3 темп-ра азота
повышается до 95° К за счет конденсации частот паров воздуха,
подаваемого иэ нижней колонны. Повышенная темп-ра воздуха
способствует лучшей сублимации и уносу твердой СО2. Раз-
ность темп-p на теплом конце регенератора не превышает
3—5°. Газообразный кислород отбирается из конденсатора 15
и из верхней колонны 7. Рекуперация холода кислорода осу-
ществляется в кислородных регенераторах 1. Производитель-
ность установки БР-1 по воздуху составляет 62 000—
67 000 нм3/час; допускаемые колебания в пределах 36 000—
75 000 нм^/час; по кислороду: технологическому (96—98%) —
12 500 нм3/час, техническому при избыточном давлении
500 мм рт. ст.—425 нм^/час и 75 им*/час при избыточном дав-
лении до 165 ат. Максимальное давление воздуха на входе
в регенераторы 7,0 ат, содержание кислорода в отбросном
азоте 0,7—3,5%.
Основные режимы работы установки БР-1 и удельный
расход энергии на 1 At3 кислорода (при изотермич. кпд компрес-
сора 0,6) в зависимости от ее производительности приведены
в таблице 2.
Таблица 2
Количе- ство раз- деляемо- го воз- духа, нм3/час Давление воздуха Произво- дитель- ность по кисло- роду, нмЗ/час Концентра- ция после регенера- торов, % Уд. расход энергии (на вал турбо- компрессо- ра), кчт-ч/м5 О2
после турбо- компрес- сора, ат перед блоком разделе- ния, ат
Оа N2
36000 5,0 4.9 7000 96 98,5 0,38
50000 5,85 5.65 10000 97 99,1 0,41
62000 6,50 6,25 12000 96 99,3 0,43
67000 6,75 6,5 13000 98 97,9 0,47
75000 7,2 6,8 15000 96 99,2 0,45
Пусковой период установки БР-1 длится 70—80 часов при
среднем расходе воздуха ок. 40 000 шР/час и давлении перед
блоком разделения 6,8 ат. Рабочая кампания установки
БР-1 равна 9—12 месяцам.
Получение жидкого кислорода (рис. 5). Очищенный от
СОг и сжатый до 160—166 ат воздух после осушки силика-
гелем в адсорбере 1 (только на период пуска) охлаждается
в теплообменнике 2 до 5—6°, проходит через отделитель вндаи
и масла 3, после чего охлаждается в теплообменнике 4 до
минус 37—40°. По мере забивки теплообменники 4 переклю-
чаются, причем отогрев их производится азотом, поступаю-
щим из подогревателя 13. После теплообменников 4 воздух раз-
ветвляется на два потока. Один поток,, пройдя буфер 10,
поступает в детандер 11, где расширяется до 6—7 ат, охлаж-
даясь при этом до минус 155—165°. После очистки от масла
в фильтре 12 этот поток проходит испаритель 6, в к-ром ох-
лаждается до минус 170°, и направляется в куб колонны.
Другой поток воздуха поступает в теплообменник 5, затем
в соответствующую спираль испарителя 6 и дросселируется
до 5—6 ат в нижнюю колонну 7. Верхняя колонна 9 работает
под давлением 1,3—1,4 ат. Отводимый из нее 97%-ный азот
отдает свой холод в теплообменниках 5, 4 и 2. Установка
предназначена для получения жидкого 99,7%-ного кислорода,
к-рый отводится из конденсатора 8, но может быть исполь-
зована и для получения газообразного кислорода той же
чистоты. В последнем случае рабочее давление процесса не
превышает 35—40 ат.
Получение редких газов. Аргон. Выделение аргона
ректификацией атм. воздуха затруднено близостью температу-
ры кипения азота, кислорода и аргона. В воздухораздели-
Рис. 6. Получение сырого
аргона.
тельных установках аргоном
обогащена кубовая жидкость
нижней колонны (в отводи-
мом из нее азоте содержится
не более 25% от исходного
количества аргона), в к-рой
концентрация кислорода до-
стигает38—40%. Однакораз-
ность темп-p кипения кисло-
рода и аргона меньше раз-
ности темп-p кипения азота
и аргона, поэтому разделе-
ние кубовой жидкости ниж-
ней колонны затруднитель-
но. Разделение этой смеси
осуществляется в дополни-
тельной колонне 3 (рис. 6),
дефлегматор 4 к-рой охлаж-
дается дросселированной ку-
бовой жидкостью нижней ко-
лонны 1. Смесь в дополни-
тельную колонну 3 посту-
пает из верхней колонны 2.
Т. о. получают сырой 95%-
ный аргон.
При получении малых количеств аргона первоначально
специальной колонне! (рис. 7) выделяют иа смеси азот, для
чего дефлегматор 2 охлаждают дроссели-
рованным азотом, сжиженным в ис-
парителе этой же колонны. Кубовую
жидкость, представляющую собой смесь
в
N2
Аг+О2
Аг+Н2О
Исходная
Рис. 7. Выделение
азота из аргона.
Рис. 8. Получение чистого
аргона.
аргона и кислорода, невыгодно разделять методом ректифи-
кации. На практике кубовую жидкость после испарения сме-
шивают с необходимым количеством водорода, сжимают
компрессором 1 (рисунок 8) до 150 ат (для
предотвращения в последующем загрязнения
чистого аргоиа), пропускают через теплообмен-
ник 2 и контактный аппарат з, где при 770° К
кислород в присутствии медного катализа-
тора связывается с водородом. Для удале-
ния избытка водорода газы пропускают через
печь 4, наполненную окисью меди. Далее аргон
охлаждают и сушат в теплообменнике 2, после
чего нагнетают в баллоны. При большой произ-
водительности установки сырой аргон под не-
большим давлением первоначально очищают
от кислорода при 770° К над медным или пал-
ладиевым катализатором. Избыточный водород
и азот отделяют ректификацией, а жидкий аргон
во избежание загрязнения нагнетают насосом
через испаритель в баллоны. Количество приме-
сей в чистом аргоне менее 0,003% (помимо азо-
та). Недостатком очистки аргона под низким
давлением является большой расход водорода.
Помимо атм. воздуха, источником получения
аргона и других редких газов могут служить
газы продувок в цехах синтеза аммиака.
641
ВОЗДУШНЫЙ ГАЗ — ВОЗНИКАЮЩИЕ РЕАГЕНТЫ
642
Криптон и ксенон. Эти газы могут быть выделены
как побочные продукты при получении азота или кислорода.
Они накапливаются внизу верхней части колонны 1 (рис. 9);
разделение смеси О2 + Кг 4- Хе производится в дополни-
тельной колонне 2. В качестве теплоносителя в испарителе з
пары разделяемого воздуха, а в ка-
честве хладоагента в дефлег-
маторе 5 — этот же воздух,
ожиженный в испарителе.
В результате ректификации
сверху колонны 2 отводятся
кислород и азот, а в кубе ко-
лонны накапливается смесь
кислорода, азота, криптона
колонны 8 используются
Воздух
9
— В рент,
нол.
2
Рис. 9. Получение криптона и ксенона в качестве побоч-
ных продуктов.
и ксенона с концентрацией последних не выше 0,1 %. Более вы-
сокое содержание редких газов в кислороде недопустимо, т. к.
одновременно растет концентрация в нем взрывоопасных
углеводородов, загрязняющих атм. воздух. Кубовая жид-
кость непрерывно отводится в испаритель 6 и через газголь-
дер 7 направляется в контактный аппарат 8, где углеводороды
окисляются окисью меди. Образующийся при этом угле-
кислый газ поглощается щелочью в абсорбере 9, а редкие газы
после осушки направляются в ректификационную колонну,
где концентрация их повышается до 50—75%. Для тонкой
очистки смеси криптона и ксенона от кислорода и других
примесей кислород связывается водородом, а азот сорбируется
при 725°К стружками магния или активным углем, охлаждае-
мым кипящим азотом (63° К). Далее, для полного удаления
углеводородов, а также остаточного водорода газы пропус-
кают при 700°К над окисью меди. В результате получается
смесь, состоящая из 92—96% криптона, 4—8% ксенона,
0,03% азота, 0,01% кислорода, 0,001% водорода и 0,02 % угле-
кислого газа и углеводородов. При необходимости разделение
смеси криптона и ксенона производится адсорбцией их при
пониженной темп-ре активным углем и последующей фрак-
дильнике за и регенераторах 4 или 4а, пропускают через
адсорберы ацетилена 6 и колонну 7, в к-рой отмываются крип-
тон и ксенон (жидкость, стекающая в куб этой колонны,
содержит примерно в 10 раз больше криптона и ксенона, чем
в исходном воздухе). Обедненный воздух отводят сверху
колонны 7 в турбодетандер 9, где он расширяется до 1 ат
и направляется для рекуперации холода в регенератор 4
или 4а. Кубовая жидкость, пройдя ацетиленовые адсорберы ю
и конденсатор 11, подвергается в колонне 12 обогащению
до 0,1—0,3% (предельно допустимая концентрация, т. к.
одновременно в жидкости повышается и содержание углеводо-
родов — ацетилена, метана, — образующих с жидким кисло-
родом взрывоопасные смеси). Раствор редких газов отводится
снизу колонны 12 через конденсатор 13 и отделитель 14 в ис-
паритель 15, где полностью испаряется. Газовую смесь в печн 16
очищают от метана окисью меди, а углекислый газ и воду по-
глощают в адсорберах 17. После охлаждения в теплообмен-
нике 17а очищенные газы подают в концентрационную колон-
ну 18, в к-рой содержание криптона и ксенона повышается
до 35%. Дальнейшую разгонку кубовой жидкости производят
в ректификационной колонне периодич. действия 19, позво-
ляющей регулировать темп-ру в испарителе и дефлегматоре.
Первоначально дефлегматор * _
колонны поддерживают
темп-ру кипения крипто-
на. По мере выделения
кислорода в дефлегмато-
ре колонны устанавли-
вается темп-pa кипения
криптона, а в кубе —
ксенона. Отводимыйсвер-
ху колонны криптон, со-
держащий предельно
0,1% ксенона, сжижает-
ся в конденсаторе 20,
испаряется под давле-
нием в змеевиковом испа-
рителе 21 п подвергает-
ся очистке над окисью
меди в печи 22. После
поглощения продуктов
окисления в адсорбере 23
баллоны 24 наполняют
чистым криптоном. После
отгонки криптона произ-
водят отбор ксенона,
который последовательно
проходит конденсатор 25, печь 26, адсорберы 27 и 28 и ожи-
житель 29, после чего баллоны 30 заполняются ксеноном. В
охлаждают воздухом, а в кубе
He + Np
+ N2(+H2)
Воздух
Рис. И. Уста-
новка для по-
лучения не-
она.
Н2О
Н2О
За
24
Рис. 10. Выделение криптона и ксенона из воздуха.
29
ционной десорбцией насыщенного поглотителя. Т. обр. можно
получить 99%-ный криптон и 99%-ный нсенон.
Криптон и ксенон в случае необходимости могут быть
выделены из воздуха, минуя стадию его разделения на кисло-
род и азот, причем основное количество воздуха (ок. 90%)
сжимают до 1,8 ат, а ок. 10% — до 5,5 ат для получения
промывной жидкости (эти соотношения действительны при
переработке больших количеств воздуха — порядка несколь-
ких десятков тысяч л3). Принципиальная схема такой уста-
новки приведена на рис. 10. Воздух через фильтр 1 засасы-
вается турбокомпрессором 2 и после охлаждения в башенном
холодильнике з и регенераторах 5 или 5а поступает в фор-
колонну 8, где из него отмываются криптон и ксенон. Обога-
щенную редкими газами жидкость из колонны 8 дросселируют
в промывную колонну 7, а обедненный воздух, после сжиже-
ния его в конденсаторах 11 и 13, используют для орошения
колонн 7, 8 и 12. Воздух низкого давления охлаждают в холо-
, 11UVU1C “C1U и М* LJ A VZl nvGll v W* • -U
адсорбере 28 при 140°К адсорбируется криптон.
В чистом 99%-ном криптоне содержится менее
400слс8/лс3 ксенона, а в чистом 99%-ном ксеноне--
не более 500 см3/м3 криптона и столько же азота.
Расход электроэнергии на 1 л редких газов не
превышает 40 квт-н.
Неон. Как побочный продукт неон может
быть получен в воздухоразделительных установ-
ках (рис. И). Вместе с гелием, водородом и азо-
том неон накапливается под крышкой конденса-
тора 1 колонн двухкратной ректификации. Кон-
центрация неона (а также гелия и ’водорода) по-
вышается в дополнительной колонне 2. Из этой
смеси неон и гелий могут быть выделены раз-
личными методами. Азот, напр., может быть
сорбирован при 720°К стружками магния или
активным углем, охлаждаемым кипящим азотом
(63°К). Очистка от водорода, как обычно, может
быть осуществлена при 700°К окисью меди. Не-
обходимо отметить, что при адсорбции азота маг-
нием дополнительно образуются небольшие ко-
личества водорода, так как в смеси содержатся
следы влаги. Дальнейшее разделение неона и ге-
лия может быть произведено адсорбцией смеси с
последующей фракционной десорбцией ее компо-
нентов либо вымораживанием неона жидким водо-
родом. Одновременно при этом получается и гелий.
Лит.: Гельперин И. И., 3 е л и к с о н
Г. М. и Р а п о п о р т Л. Л., Справочник по
разделению газовых смесей методом глубокого
охлаждения, М. — Л., 1953; S 1 е d I е г Р.,
Schl atterer R. und Baldus H., в кн.: Chemische
Technologie, Bd 1, 2 Aufl., Mtlnch., 1958, S. 335.
II. И. Гельперин.
ВОЗДУШНЫЙ ГАЗ — см. Газификация твердых
топлив.
ВОЗНИКАЮЩИЕ РЕАГЕНТЫ — ионы осадителя,
постепенно образующиеся в анализируемом р-ре
в результате гидролиза реактива-осадителя. В. р.
используются при осаждении ряда элементов и обес-
печивают получение, как правило, осадков наиболее
полно удовлетворяющих требованиям аналитич. хи-
мии. В методе В. р. (иногда называемом «методом
гомогенного осаждения»), в отличие от обычно приме-
11 К. X. Э. т. 1
643
ВОЗРАСТ ГЕОЛОГИЧЕСКИЙ АБСОЛЮТНЫЙ
644
няемого метода осаждения, устраняется появление
в р-ре местного избытка реактива-осадителя; в ре-
зультате значительно уменьшается соосаждение со-
путствующих ионов; при пользовании В. р. обра-
зуются компактные осадки, к-рые можно отфиль-
тровывать и отмывать. Подбирая условия, регу-
лирующие скорость поступления ионов-осадителей
в раствор (pH, нагревание, использование «маскирую-
щих» веществ, добавление органич. растворителей и
т. д.), можно добиться не только осаждения того или
иного иона с целью его определения, но в ряде слу-
чаев практически полного разделения нек-рых ионов.
При использовании В. р. часто применяют «маски-
рующие» вещества, роль к-рых сводится не только
к предупреждению осаждения сопутствующих эле-
ментов, но и к улучшению химико-аналитич. свойств
осаждаемого малорастворимого соединения; улучшен
ние происходит вследствие медленного поступления
в раствор осаждающих ионов при постепенной диссо-
циации комплексного соединения, к-рое их содержит.
Напр., Ва2ь определяют в присутствии Са21" осажде-
нием диметилсульфатом в глицериново-водной среде.
Са2г не осаждается в этих условиях, т. к. он связы-
вается с глицерином в комплекс, более устойчивый,
чем комплекс с Ва2:. В. р. широко применяют
в аналитич. химии при осаждении и раздельном опре-
делении ряда элементов, напр. щелочноземельных
металлов: Ва, Sr, Са, с использованием в качестве
осадителя диметил-, метилэтил-, этилсульфата; метил-
оксалата или хромоксалата для фракционного осаж-
дения редкоземельных элементов; мочевины для
осаждения Sn и т. д. В. р. позволяют значительно
сократить продолжительность определения, так как
дают возможность сразу получать осадки с хороши-
ми аналитич. свойствами.
Лит.: Терентьев А. П., Рухадзе Е. Г., Лит-
ии н К. И., Ж. анал. хим., 1956, 11, вып. I, с. 55; и х ж е,
там же, 1959, 4, вып. 3, с. 288; Gordon L., Anal. Chem.,
1952, 24, № 3, р. 459. И. П. Ефимов.
вбЗРАСТ ГЕОЛОГИЧЕСКИЙ АБСОЛЮТНЫЙ —
время, прошедшее от к.-л. геологич. события до сов-
ременной эпохи, исчисляемое в тысячах и миллионах
лет. В. г. а. устанавливается на основании данных
о содержании радиоактивных элементов и продуктов
их распада в минералах и породах; при этом может
быть принципиально использована любая пара'радио-
активного долгоживущего и радиогенного устойчи-
вого изотопа, если для первого известны скорость и
тип радиоактивного распада. Для определения В. г. а.
древних пород используются в основном 4 метода:
свинцовый, гелиевый, аргоновый и стронциевый. В ос-
нове этих методов лежат след, ядерные превращения:
L2з3 рЬ2»в 8Не, U235 -> Pb207 -j- 7Не, Th232 -+
-> РЬ238 6Не, К4» 4- ё -+ Ar40, R b87 -> Sr87 4- fi~.
Свинцовый метод основан на распаде эле-
ментов радиоактивных рядов урана, актиноурана и
тория, в результате к-рого в радиоактивных минералах
накапливаются стабильные изотопы свинца. Зная
скорость распада U, Th и AcU и определив их содер-
жание в минерале, а также содержание и изотоп-
ный состав РЬ, можно установить возраст минерала
но отношениям Pb206; U238, Pb207: U235, РЬ207:РЬ206,
Pb208:Th232. Совпадение значений возраста, вычис-
ленных по различным изотопным отношениям, сви-
детельствует о их надежности.
Гелиевый метод основан на тех же ядер-
ных превращениях, что и Pb-метод, т. к. Не образуется
при а-распаде U, Th, AcU и продуктов их превраще-
ния и накапливается в минерале в течение геологич.
времени. Данные возраста, полученные гелиевым ме-
тодом, обычно являются заниженными вследствие
плохой сохранности Не в большинстве минералов.
Хорошей сохранностью гелия характеризуются только
минералы с плотной кристаллич. упаковкой, такие,
как магнетит, шпинель, гранат, циркон, монацит
и нек-рые др.
Аргоновый метод основан на радиоактив-
ном превращении изотопа калия К40 путем захвата
орбитального электрона, в результате чего образуется
изотоп аргона Аг40. Возраст вычисляется из отноше-
ния Аг40: К40 с учетом скоростей К-захвата и |3 -рас-
пада К40. Радиогенная природа аргона, выделенного
из минералов, проверяется масс-спектрометрически.
Метод имеет широкое применение ввиду распростра-
ненности калийсодержащих минералов (слюд, поле-
вых шпатов, сильвинов, глауконитов и др.). Надеж-
ность получаемых данных проверяется определением
возраста слюд стронциевым методом или возраста
акцессорных минералов свинцовым методом.
Стронциевый метод основан на радиоак-
тивности изотопа Rb87, содержание к-рого в рубидии
составляет 27,19%. В результате распада Rb87 в ми-
нералах накапливается радиогенный стронций Sr8’.
Возраст вычисляют по отношению Sr87 : Rb87, опре-
деленному масс-спектрометрич. методом изотопного
разбавления. Минералами, пригодными для опреде-
ления возраста стронциевым методом, являются слюды
и полевые шпаты, содержащие заметные количества
радиогенного Sr и малые количества обычного Sr
нерадиогенного происхождения. Метод эксперимен-
тально труден, чем объясняется ограниченность его
применения.
Радиоуглеродный метод применяется
для определения возраста молодых образований (до
70 тыс. лет). Метод основан на распаде естественного
радиоактивного изотопа углерода С14, к-рый обра-
зуется в атмосфере под действием космнч. радиации,
усваивается организмами и после гибели последних
убывает по закону радиоактивного распада. Возраст
может быть вычислен на основании отношения актив-
ности углерода живого в-ва или атмосферы и угле-
рода органич. остатков. Метод применим ко всем обра-
зованиям, содержащим углерод (карбонатные горные
породы, торф, ископаемые деревья, археология, дере-
вянный материал).
Кроме указанных выше методов определения В. г. а.,
существуют другие, имеющие значительно более огра-
ниченное применение: кальциевый метод (Са40 : К40),
ксеноновый (Хе : U238) и рениевый метод (Os187: Re187).
Принципиально возможно использование для опре-
деления абсолютного геологического времени следую-
щих отношений: Cr50 : V50, Те123 : Sb123, Xe129:J129,
Хе130 : Те130, Pm15»:Ndi3», Sn115:Ini15, Ba138:La138 и
Ge138 : La138, Y176 : Lu176 и Hf476 : Lu476, Nd143 : Sm147,
TI205 : Bi209.
Получение надежных и точных величин В. г. а. обу-
словлено: 1) сохранностью радиоактивных элементов и
продуктов их распада в минерале за время его сущест-
вования; 2) отсутствием первичных веществ, соответ-
ствующих продуктам распада и попавших в минерал
при его образовании; 3) точностью аналитич. опреде-
ления радиоактивных элементов и продуктов их рас-
пада; 4) точностью определения постоянных радиоак-
тивного распада. Дополнительные условия являются
специфическими для каждого метода определения воз-
раста в соответствии с химич. и геохимич. свойствами
исследуемых элементов. Основной причиной ошибоч-
ных данных, иногда получаемых радиоактивными ме-
тодами, является плохая сохранность исследуемых об-
разцов в результате воздействия на минерал вторич-
ных процессов, происходящих в природных услови-
ях и приводящих к миграции элементов, в частности
и тех, по к-рым производится определение возраста.
Развитие радиология, методов определения В. г. а.
осуществляется на основе непрерывного усовершен-
ствования химия., масс-спектрометрич., спектральных,
рентгено-спектральных, полярография., радиоактива-
645
ВОЛНОВАЯ ФУНКЦИЯ — ВОЛОКНА ТЕКСТИЛЬНЫЕ
646
ционпых и других методик. Особенно эффективным
оказалось совмещение масс-спектрометрии с методи-
кой изотопного разбавления, что позволило исполь-
зовать масс-спектрометр для определения не только
соотношений изотопов, но и их малых количеств.
На основании результатов определения возраста
минералов или пород, хорошо датированных геоло-
гически, составлена шкала абсолютного геология,
летосчисления (по Холмсу, в млн. лет): четвертич-
ный период 0—1, третичный 1—70, мел 70—135,
юра 135—180, триас 180—225, пермь 225—270, кар-
бон 270—350, девон 350—400, силур 400—440, ордо-
вик 440—500, кембрий 500—600.
Лит.: Старик И. Е., Радиоактивные методы определе-
ния геологического времени, Л.—М., 1938; Аренс Л. X.,
Радиоактивные методы определения геологического возраста,
в сб.: Физика и химия Земли, М., 1958; Kulp J. L., в сб.;
Crust of the earth (A symposium), Baltimore, 1955, p. 609 (Geol.
Soc. of America. Special papers № 62); Изотопы в геологии.
Сб. ст., пер. с англ, и нем., М., 1954; Бюллетень Комиссии по
определению абсолютного возраста геологических формаций,
вып. 1—3, М., 1955—58; Труды (1, 3, 4, 5, 7) сессий Комис-
сии по определению абсолютного возраста геологических фор-
маций, М. 1954—60; Holmes A., Trans, of the Edinb. Geol.
Soc., 1959, 17, p. 183—216. P. А. Хмельницкий.
ВОЛНОВАЯ ФУНКЦИЯ (в квантовой механике) —
служит для полного задания состояния динамич. си-
стемы, т. е. дает возможность вычислить вероятность
любого результата измерения, производимого над
системой, или определить среднее значение любой
физич. величины для данного момента времени; по зна-
чению В. ф. в данный момент можно определить В. ф.
для любого другого момента времени. Т. о., знание
В. ф. в принципе эквивалентно знанию всех имеющих
физич. смысл характеристик рассматриваемой си-
стемы (атома, молекулы, твердого тела как целого)
и их изменений во времени (в ходе химич. реакции).
Для большинства сложных молекул точное вычисле-
ние В. ф. невозможно, в связи с чем приходится поль-
зоваться приближенными методами квантовой химии.
См. Квантовая механика.
ВОЛОКНА НАТУРАЛЬНЫЕ — см. Волокна тек-
стильные.
ВОЛОКНА ТЕКСТИЛЬНЫЕ — протяженные, гиб-
кие и прочные тела с очень малыми поперечными раз-
мерами, используемые для изготовления текстильных
изделий. Большинство текстильных изделий выраба-
тывается из В. т. не непосредственно, а после из-
готовления из них нитей. В. т. чаще встречаются
в виде элементарных,
т. е. Одиночных, волокон,
не делящихся вдоль их оси на
более мелкие. Некоторые В. т.
являются комплексными; они
состоят из многих параллель-
но расположенных элементар-
ных волокон, склеенных между
собой. Длина В. т. в зависи-
мости от их вида обычно соста-
вляет от нескольких десятков
до нескольких сотен мм. Встре-
чаются В. т. очень большой
длины, определяемой многими
метрами; их называют элемен-
тарными нитями (по старой
терминологии — филаментами);
термин «В. т.» распространяют
иногда и на комплексные нити,
состоящие из нескольких эле-
ментарных. Если пучки элементарных нитей разре-
зают или разрывают на отрезки в несколько десят-
ков мм, получают штапельные волокна.
В нек-рых случаях производятся толстые элемен-
тарные нити, предназначаемые для непосредственной
выработки изделий (напр., щеток и др.); их назы-
вают м о н о в о л о к н а м и. Классификации В. т.
обычно основываются на делении по химич. составу
слагающих их веществ, происхождению или способу
выработки. Все В. т. делятся на два типа: нату-
ральные, развивающиеся в растениях, на коже
животных и т. д. (напр., шерсть, хлопок, лен и др.), и
искусственные, изготовляемые заводским пу-
тем на основе различных химич. и физич. процессов.
В советской научной и технич. литературе получил
распространение термин «химические ве-
ло к н а» в приложении ко всем искусственным волок-
нам. При этом за волокнами, полученными на заводах
из природных полимеров (целлюлозы, белков), сохра-
Рис. 1. Классификация натуральных В. т. из природных
высокомолекулярных соединений: 1 — хлопок; 2 —лен; з—
пенькз; 4 — джут; 5 — кенаф; 6 — сизаль; 7 — прочие;
8 — из кератина (шерсть); 9 — из фиброина (шелк); ю —
асбест.
Примечание. В. т., указанные под №№ 2, 3, 4,
5 и 6, сходные по их строению и свойствам, часто назы-
вают общим собирательным именем В. т. лубяные, причем в
классификацию включены лишь главнейшие из них; число
второстепенных, имеющих местное значение, достигает
12 0. В. т., указанные под №№ 8 и 9, относятся к волокнам
животного происхождения.
няется термин искусственные волокна
(напр., вискозные волокна, ацетатное волокно), а во-
локна, изготовленные из синтетич. полимеров, назы-
вают синтетическими (напр., полиамидные
волокна, полиэфирные волокна).
Классификация натуральных В. т. приведена на
рис. 1, искусственных — на рис. 2, 3 и 4, причем виды
В. т., имеющих наибольшее народнохозяйственное зна-
Рис. 2. Классификация искусственных В. т. из природных высокомолекулярных орга-
нических гстероцепных соединений; 1 — вискозное; 2 — медно-аммиачное; 3 — омылен-
ное ацетатное; 4 — ацетатные из ди- и триацетата; 3 — альгиновые; в — казеиновые;
1 — зеиновые; 8 — прочие.
чепие, на рисунках выделены толстой рамкой. Как
видно из рис. 1—4, подавляющее большинство видов
В. т. состоит из высокомолекулярных соединений. Мак-
ромолекулы этих соединений, слагающих В. т., имеют
преимущественно линейное (реже разветвленное) стро-
ение. Структура В. т. такова, что небольшие пучки
11»
647
ВОЛОКНА ТЕКСТИЛЬНЫЕ
648
вытянутых макромолекул (т. н. микрофибриллы), объ-
единенных межмолекулярными силами, удерживаются
друг около друга как с помощью этих же сил, так и
вследствие перехода отдельных молекул из одной
микрофибриллы в другую, и образуют более крупные
молекулярные комплексы — фибриллы, распо-
лагающиеся более или менее ориентированно вдоль
оси В. т. или под небольшими углами к ней. Фибриллы
удерживаются друг около друга аналогично микро-
фибриллам, однако скреплены между собой менее
прочно, между ними имеется много продольных тре-
щин, пор и т. п. Этим объясняется относительно легкое
продольное расщепление В. т. Большинство свойств
В. т. изменяется в широких пределах в зависимо-
сти от их видов, разновидностей, происхождения или
способа получения. Сведения о свойствах важнейших
В. т. приводятся в табл. 1.
Таблица 1. Основные физико-механические и геометрические характеристики
главнейших текстильных волокон (средние величины)
Виды волокон Плот- ность, г/см8 Средняя длина, мм Номер а> мм/мг Предел проч- ности, кг/мм2 Разрывное удлинение, % к перво- начальной длине
Хлопок:
средневолокнистый 1,52-1,55 25—35 4500—6000 33-39 6-8
тонковолокнистый Лен: 1,52-1,55 35—42 6000—8000 44—53 7-9
элементарное волокно . . . 1.50—1,52 15-20 3000—4000 90-110 2-2,5
техническое волокно .... Шерсть: 1,48-1,50 500—700 100-200 00—70 2,5—3
тонкая и полутонкая .... 1.30—1,33 50—100 1000—3000 16-20 30—40
полугрубая и грубая .... 1,28-1.30 50-250 200—1000 12—15 25—35
Шелк (коконная нить) 1,35-1,37 6.105—8-105 2500—4000 35-40 16—18
Асбест ...... Вискозные: 2,50-2,60 9—15 — 220—250 2—3
штапельное волокно .... 1,50—1,54 40-120 3000—6000 22-26 15-20
элементарная нить ..... 1,50-1,54 — 2000-4000 22—25° 18—24
Ацетатные элементарные нити . Полиамидные элементарные ни- 1,30—1,35 — 4500—7500 14—18 18—25
ти (капроновая, иайлоновая) . Полиэфирные элементарные ни- 1,14—1,15 — 2000—9000 45—55в 24-26
ти (лавсановая, териленовая) Полиэтиленовые элементарные 1,38—1,39 — 4500—9000 55—65в 20-30
нити Перхлорвиниловое штапельное 0,90—0,92 — — 10-20 40—60
волокно 1,45—1,60 — 2500-6000 20—25 20—30
Полиакрилонитриловые элемен- тарные нити 1,12-1,18 — 4500—9000 25-40 20-25
Стеклянные элементарные нити 2,5-2,6 — 4000—20000 120-200 2-3
а Толщина В. т. (обычно вследствие ее малости называемая «тониной») выражается
чаще всего их номером, т. е. отношением длины к весу. Номер обратно пропорционален
площади поперечного сечения волокна и его плотности. ^Упрочненные нити имеют
предел прочности до 60—70 (иногда до 75—80) хг/мма при разрывном удлинении 8—12 %.
в Упрочненные иити имеют предел прочности до 80—85 кг/мм- при разрывном удлине-
нии 12—15%.
Из физич. и физико-химич. свойств В. т. следует
прежде всего отметить способность волокон поглощать
воду (в особенности в виде паров из воздуха). В зави-
симости от количества поглощенной воды резко изме-
няются другие свойства В. т., напр. их вес, механич.
и диэлектрич. свойства. Выдержанные несколько ча-
сов в помещении в стандартных атм. условиях (65%
относительной влажности воздуха и 20°) В. т. приобре-
тают от 0,02% (фторсодержащие виниловые синтетич.
волокна) до 17% (тонкая шерсть) и даже до 30% (аль-
гинаткальциевые волокна) воды от своего сухого веса.
Все волокна в той или иной мере являются непровод-
никами тепла и электричества. Наиболее высокими
теплоизоляционными свойствами обладают асбест,
шерсть, шелк. Темп-ра, при к-рой В. т. разрушаются
от нагрева, для целлюлозных волокон составляет
150—160°, для белковых 170°, для фторсодержащих
виниловых синтетич. волокон до 300°, для стеклян-
ного волокна и асбеста 1200—1300°. Наименее устой-
чивы к нагреву хлориновые волокна — при 80° они
плавятся. Лучшими диэлектриками являются шелк,
ацетатное, стеклянное, полиолефиновые, а также
некоторые другие (синтетич.) В. т.
Механич. свойства В. т. чаще всего характеризуются
по результатам их однократного растяжения до раз-
рыва (прочность на разрыв). В качестве характеристик
механич. свойств волокон в сухом и мокром состоянии
обычно применяются: разрывная нагрузка — наи-
большее усилие, выдерживаемое В. т. при одно-
кратном растяжении до разрыва, показывающее
абс. прочность данного волокна; относительная проч-
ность, выражаемая временным сопротивлением (раз-
рывным напряжением); разрывное удлинение — уве-
личение длины растягиваемых В. т. к моменту их раз-
рыва, обычно выражаемое в процентах к исходной
длине. Вместо временного сопротивления иногда
пользуются разрывной длиной (в км), представляю-
щей отношение первого к плот-
ности. Важными характеристи-
ками, отражающими эксплуа-
тационные свойства В. т.,
являются сопротивление много-
кратным деформациям, устой-
чивость к истиранию, сминае-
мость и т. д. Следует иметь в
виду, что механич. характе-
ристики искусственных В. т.
чрезвычайно зависят от усло-
вий их производства, и приво-
димые в табл. 1 данные отно-
сятся лишь к наиболее распро-
страненным их типам.
Химич, свойства В. т., в ча-
стности их химич. стойкость,
определяются особенностями
слагающих веществ. Так, цел-
люлозные волокна очень не-
стойки к действию неорганич.
к-т, сравнительно устойчивы
к действию щелочей, нераство-
римы в обычных органич. рас-
творителях; белковые волок-
на очень нестойки к действию
щелочей и более устойчивы к
действию кислот. Большинство
синтетич. волокон, в особен-
ности хлорсодержащие, устой-
чивы к действию как кислот,
так и щелочей. Особенно высо-
кой химич. стойкостью отли-
чаются полиолефиновые, фтор-
содержащие и нек-рые другие
синтетич. В. т.
В. т. используются: для изготовления верхней
одежды (шерсть, разнообразные искусственные во-
локна, хлопок, в небольших количествах — лен и
шелк); для белья (в т. ч. трикотажного) — хлопок,
разнообразные искусственные волокна и др.; для хо-
зяйственных (бытовых) потребностей — хлопок, лен,
искусственные волокна, шерсть (в основном для ков-
ровых изделий); для веревочно-канатных изделий —
лубяные, полиамидные и др. волокна; для разнообраз-
ных технич. целей, особенно, напр., для тары — хло-
пок, джут, пенька и др.; для валяльно-войлочных
изделий — шерсть и в смеси с нею небольшие количе-
ства искусственных волокон (гл. обр. синтетических).
В последние годы широко применяется выработка
нитей (в особенности пряжи) и изделий из смеси раз-
личных волокон, обладающих разными свойствами,
что позволяет получать более разнообразные сочета-
ния последних.
В промышленно развитых странах умеренного кли-
мата из общего потребления В. т. на материалы для
649
ВОЛОКНА ТЕКСТИЛЬНЫЕ — ВОЛОКНА ХИМИЧЕСКИЕ 650
изготовления одежды расходуется примерно 35—40%,
для хозяйственных и бытовых предметов 20—25%,
в технике используется 25—30%, а остальные 5—10%
используются на прочие потребности (тару, культур-
ные нужды, медицинские цели и т. п.). Естественно,
что в отдельных странах в зависимости от социаль-
ных условий, развития техники и т. п. эти соотно-
шения могут колебаться.
Таблица 2. Производство главнейших видов текстильных
волокон в СССРа (тыс. топи)
Вид волокна пли первичного сырья 1913° 1928 1940 1955 1958 1959 1965 (план)
Хлопок-сырец . . 744 821 2237 3881 4373 4681 5700-6100
Лен 401 324 349 381 438 386 580
Шерсть (грязная) Шелковые ко ко- 192 — 161 261в 322 350 548
ны (сырые) . . Искусств. (хи- 9 14 20 25 28 30 40
мич.) волокна 0,1 0,2 И 111 166 179
а Для хлопка, льна и шелковых коконов приводится вало-
вой сбор, для шерсти — производство. 6 В современных гра-
ницах СССР. в 1956 г. г Рост в 4 раза по сравнению с 1958 г.
Таблица 3. Мировое производство главнейших
текстильных волокон (млн. тонн)
Вид волокна 1913 1940 1955 1956 1959
Хлопок-волокно а 4,96 6,70 8,75 8,69 0.74
Лубяные волокна; лен ... 0,95 0,78 1.33 1,43
джут 1.50 2 .',2 1.82 1.82 —
пенька — 0,99 1,10 1,16 —
Шерсть мытая6 0,78 1,07 1,24 1.30 1,42
Шелк-сырец 0,03 0,0(1 0,03 0,03 0,04
Искусственные (химические) во- локна всех видов 0,01 1,12- 2,53 2,68 3,09в
а Для ориентировочных пересчетов в хлопок-сырец умно-
жать на 3. 6 Для ориентировочных пересчетов в шерсть гряз-
ную умножать на 2. в В том числе природнополимерные —
2,52 млн. т и синтетические — 0,57 млн. т.
Рост произ-ва В. т. в СССР показан в табл. 2. Боль-
шие успехи в произ-ве В. т., в особенности хлопка,
В. т., в частности искусственных, и в других странах
народной демократии. Так, напр., в Польше произ-во
искусственных волокон в 1959 увеличилось по срав-
нению с довоенным в 13,5 раза. Мировое произ-во В. т.
представлено в табл. 3. На долю капиталистич. стран
пока еще падает примерно 2/3. Существенный рост
произ-ва в капиталистич. странах наблюдается только
в выработке искусственных волокон, увеличение же
произ-ва хлопка в основном определяется успехами
СССР и КНР. Крупными производителями хлопка,
кроме этих стран, являются также США, Индия,
Рпс. 4. Классификация искусственных В. т. из неорганиче-
ских соединений.
Арабская Объединенная Республика (Египетский
район) и др. страны. Основными странами-произво-
дителями являются: по льну — СССР, Польша, Бель-
гия, Франция и др.; по шерсти — Австралия, СССР,
Новая Зеландия, Аргентина и др.; по искусственным
волокнам—США, Япония, ФРГ, Англия, СССР,
Италия, ГДР и др. (порядок стран соответствует
объему произ-ва).
Лит.: Учение о волокнистых материалах. Текстильные во-
локна, под ред. Г. Н. Кукина, М.—Л., 1949; Р о г о в п н 3. А.,
Основы химии и технологии производства химических
волокон, 2 изд., М., 1957; Справочник по аналитическому
контролю в производстве искусственных и синтетических
волокон, под ред. А. Б. Пашпвера [и др.], М., 1957; Волокна
из синтетических полимеров, под ред. Р. Хилла, пер. с англ.,
М 1957; Harris М., [е d.l, Handbook of textile fibers,
Wash., 1954; Matthews’ textile fibers..., 6 ed., N. Y. — L., 1954;
Z у 1 i ns ki T. Nauka о wtbknie, Warsz., 1958; С о о k J. G-.,
Handbook of textile fibres, 2 ed., Watford, [I960]; Натан-
сон И. А., Физические и химические свойства новых син-
тетических волокон, М., 1960. Г. Н. Кукин.
Рис. 3. Классификация искусственных В. т. из синтетич. высокомолекулярных соединений: 1 — из полпгексаметплен-
адипамида и др.: найлон (анид и др.); 2 — из поликапролактама: капрон (перлон и др.); 3 — из проч, полиамидов и их
сополимеров; 4 — из полиэтилентерефталата: лавсан (терилен и др.); 5 — из сополимера гексаметплендиазоцианата и
тетраметиленгликолн: перлон «У»; в — пз полиэтилена: иолитен (курлен и др.); 7 — из полипропилена: моплен; 8 — пз
полистирола: полифайбр; 5 — из хлорсодергкащих: Пе-Це-У, хлорин (Пе-Це и др.), саран; 10 — из сополимеров хлорсодер-
жащих с другими соединениями: ацетохлорпновое (внньон «А»), санив (из сополимера винилиденхлорида п акрилонитри-
ла) и проч.; 11 — из фторсодержащих: тефлон, фторлон и проч.; 12 — из полиакрилонитрила: нитрон (орлон и др.);
13 — из сополимеров акрилонитрила: акрилан, ворел и др.; 14 — пз сополимера вини ли денцианида с винилацетатом:
дарван; 15 — винилон и др.
сделала КНР, валовый сбор хлопка-волокна к-рой
в мин. т составил: в 1949 — 0,44; в 1956 — 1,4; в
1958 — 3,5. Быстро растет производство различных
ВОЛОКНА ХИМИЧЕСКИЕ — см. Волокна тек-
стильные, Вискозные волокна, Ацетатное волокно, По-
лиамидные волокна и др.
651
ВОЛЬТАМПЕРОМЕТРИЯ — ВОЛЬФРАМ
652
ВОЛЬТАМПЕРОМЕТРИЯ — различные электро-
химии. методы анализа р-ров, основанные на измере-
нии и построении кривых зависимости силы проходя-
щего через р-р тока от напряжения между индикатор-
ным электродом и неполяризующимся электродом
сравнения. В американской литературе к В. часто
относят также многие методы исследования кинетики
электродных реакций (снятие поляризационных кри-
вых и т. д.). Различают В. при контролируемом н а-
пряжении или при контролируемой силе т о-
к а. В первом случае измеряют ток, поддерживая по-
Ыстоянным или непрерыв-
но изменяя напряжение,
накладываемое на элек-
тролитич. ячейку. Если
определяемое вещество
способно к реакции на
электроде, то на кривой
сила тока — напряжение
(точнее, сила тока —
«---------------м потенциал электрода) на-
------------------ _1_. блюдается диффузион-
. _------------------время ная волна окисления или
восстановления. Высота волны, при прочих равных
условиях, пропорциональна концентрации веще-
ства. Это явление широко используется в полярогра-
фии, при проведении амперометрического титрования
и т. д. Во втором случае задают ток и измеряют
изменение потенциала индикаторного электрода во
времени (см. рис.). Падение до нуля концентрации
определяемых частиц вблизи электрода вызывает
резкий сдвиг его потенциала. «Переходное время»
в простейшем случае определяется объемной концент-
рацией частиц; поэтому по величине переходного вре-
мени можно определять начальную концентрацию
в-ва. Измерения обычно проводят на ртутном капель-
ном электроде или на платиновом микроэлектроде,
вращающемся или неподвижном.
Лит.: Делахей П., Новые приборы и методы в электро-
химии, пер. с англ., М., 1957. А. Н. Чемоданов.
ВОЛЬФА—КИЖНЕРА РЕАКЦИЯ — см. Кижнера —
Вольфа реакция.
ВОЛЬФРАМ (Wolframium) W — химич. элемент
VI группы периодич. системы Менделеева, п. н. 74,
ат. в. 183,92. Природный В. состоит из смеси пяти ста-
бильных изотопов с массовыми числами; 180 (0,135%),
182 (26,41%), 183(14,4%), 184 (30,64%) и 186 (28,41%),
Известно также неск. искусственных радиоактивных
изотопов В.; в качестве радиоактивных индикаторов
используются: W181 (Ti/ =145 дн.), W186 (Т1/г =
= 74,5 дн.) и W18’ (Т1/г = 23,85 час.), получаемые
из В. по реакции (пу) в ядерном реакторе. Конфигу-
рация внешних электронов атома В.: 5с?4 6s2. Энергии
ионизации (в эв): W° —> W+ —>• PF2+ —>• W3+ —>• Ж4+
соответственно равны 7,98; ...; 24,08; 35,36.
В. был Открыт в 1781 К. Шееле, к-рый при обработке кисло-
тами минерала тунгстена, впоследствии названного шеелитом,
выделил «тупгстеновую кислоту», т. е. вольфрамовый ангид-
рид. Металлич. В. впервые получен в 1783 X. X. и Ф. д’Эль-
хуяр восстановлением WO3 углеродом. В США и Англии за
элементом сохранилось название tungsten, во Франции —
lungs tone.
В. мало распространен в природе; среднее содер-
жание в земной коре 1 • 10 4 вес. %. В свободном со-
стоянии не встречается, а образует собственные ми-
нералы — вольфраматы (редко — окислы, еще реже—
сульфиды) — или входит в виде изоморфной примеси
в другие, преимущественно Sn-, Мо- и Ti-минералы.
Природное концентрирование В. происходит в кислых
(гранитных) вулканич. породах. Минералы В. обра-
зуются из газовой фазы вместе с минералами Sn
(касситерит), Мо (молибденит) и золотом. Наиболее
важными являются вольфрамит и шеелит.
Вольфрамит (Fe, Мп) WO4; в случае резкого
преобладания Fei>Mn минерал называется фербери-
том, при Mn:>Fe — гюбнеритом, из микроэлементов
присутствуют Sc, Nb, Та; сингония моноклинная;
цвет темно-серый или коричнево-черный; твердость
4,5—5,5, плотн. 6,7—7,5. Шеелит CaWO4, часто
с Sr и Ва, иногда с Мо (СаМоО4); сингония тетраго-
нальная; цвет белый с различными оттенками; твер-
дость 4,5, плотн. 5,8—6,2; образуется обычно на кон-
такте гранитов с известняками. На поверхности оба
минерала переходят в вольфрамовые охры (вольфра-
маты и окислы). Крупные месторождения вольфрамо-
вых руд, кроме СССР, находятся в Китае, США, Бо-
ливии, Корее, Бирме и др. странах. В настоящее
время добываются руды с содержанием WO3 от 0,5%
и выше.
Физические и химические свойства. Компактный
В. — светло-серый металл; имеет объемноцентриро-
ванную кубич. решетку, а = 3,1649 А; ат. радиус
1,40 А; ионные радиусы: W4+ 0,68 A, We+ 0,65 А.
Плотность В. 19,3; т. пл. 3410 (самый тугоплавкий из
всех элементов, за исключением углерода); т. кип.
5930°; теплота плавления 44 кал/г', теплота испарения
1183 кал/г-, уд. теплоемкость 0,0343 кал^-град
(0—1000°); теплопроводность 0,40 кал/см-сек-град (при
0°); уд. электрич. сопротивление 5,5 • 10~6 ом см
(20°); темп-рный коэфф, электрич. сопротивления
(0—170°) 5,1 • 10 3. В. отличается низким давлением
пара при высоких темп-pax; давление пара (в мм рт.
ст.) в зависимости от темп-ры: 1,93 -10 15 (1530°);
6,55 • 10~6 (2730°); 0,76 (3940°); 7,6 (4440°); 76 (5080°) и
380 (5650°). В. характеризуется малым коэфф, термич.
расширения (4,4 • 10 е при 20—300°). Ценное свойство
В. — высокая электронная эмиссия при накаливании
металла, равная (ъма/ем2): 1,5 • 10~10 (830°); 2,3 • 10 1
(1630°); 1,0 (1730°); 298 (2230°) и 1690 (2427°). Другое
важное свойство В. — большая мощность энергии,
излучаемой поверхностью металла при высоких темпе-
ратурах; в зависимости от темп-ры ее величина соста-
вляет (в ет/ем2): 0,9 (800°); 18,0 (1600°); 64,0 (2200°);
153,0 (2700°); 245 (3030°). В. парамагнитен.
На механич. свойства В. сильное влияние оказы-
вает способ предшествующей обработки. Так, твердость
по Бринеллю равна 200—250 кг/мм2 (для спеченного
штабпка) и 350—400 кг/мм2 (для кованого штабика);
предел прочности при растяжении ПО кг!мм2 (для
отожженной проволоки) и 180—415 кг!мм2 (для не-
отожженной проволоки, в зависимости от диаметра);
модуль упругости 35000—38000 кг/мм2 (для прово-
локи) и 39000—41000 кг!мм2 (для однокристальной
нити); предел текучести 71,4—82,6 кг/мм2 (для ото-
жженной проволоки диаметром 0,1—0,5 мм) и
149,1 кг[мм2 (для неотожженной проволоки того же
диаметра).
По химич. свойствам В. близок к Мо. С другими
элементами подгруппы (Сг и U) сходство ограничено
соединениями высшей валентности. Химия В. очень
сложна. Он обладает большой склонностью к комп-
лексообразованию, примером чего служат многочис-
ленные высокомолекулярные гетерополи-, акваполи-
и изополисоединения. Кроме того, известны соедине-
ния (гл. обр., окислы), в к-рых В. одновременно нахо-
дится в нескольких валентных состояниях.
В соединениях В. может проявлять валентности
—f—2, 4-3, 4-4, 4-5 и 4-®- В высших валентных состоя-
ниях он обладает кислотными свойствами, в низших —
основными. Соединения низших степеней окисления
В. относительно неустойчивы, и двухвалентный В. из-
вестен лишь в виде галогенидов. Наиболее характер-
ными и устойчивыми являются соединения 6-валент-
ного В.
В обычных условиях металлич. В. химически весьма
стоек. Компактный металл не изменяется на воздухе
653
ВОЛЬФРАМ
654
при Комнатной темп-ре. С кислородом начинает взаи-
модействовать выше 400°. Однако в виде тонко дисперс-
ного порошка может проявлять пирофорные свойства.
Водород не действует на В. даже при очень высоких
темп-рах. С азотом металл не реагирует до 1500°; при
2300° образуется динитрид WN2 (коричнево-черный
порошок, разлагающийся водой). Твердый углерод
взаимодействует с В. с образованием карбидов W2C
и WG. Кремний и бор выше 1400° образуют соответ-
ственно силициды — W6Si3 (W3Si2) и VVSi2, бориды —
W2B, WB, WB2, W2B6. Известно неск. фосфидов В.
(W2P, или W4P2, и др.). Фтор реагирует с металлом
при комнатной темп-ре. Сухой хлор при 250—300°
образует гексахлорид WCle; в присутствии кислорода
или влаги получаются оксихлориды. С бромом в от-
сутствии влаги В. образует при темп-ре красного ка-
ления пентабромид WBr6; с иодом — дииодид WJ2
(см. Вольфрама галогениды) В. способен давать
сплавы и интерметаллич. соединевия со многими
металлами (см. Вольфрама сплавы). В обычных усло-
виях компактный металл противостоит действию воды,
но при темп-ре красного каления он легко окисляется
водяным паром. СО и углеводороды при высоких
темп-рах образуют с В. карбиды. Если реакция с СО
проводится при 275—300° и 200 атм, то получает-
ся гексакарбонил W(C0)e. С наибольшим выходом
W(G0)e образуется при взаимодействии WGle с СО
в присутствии порошкообразного А1 в абс. спирте
при 70—100° и давлении 145—200 атм. Выше 1200°
В. окисляется СО2, при 700—800°—SO2. Сероводород
реагирует с металлом при 700—800° с образованием
дисульфида WS2 (см. Вольфрама сульфиды).
В. на холоду практически не подвергается действию
HCI, H2SO,, HNO3 и HF любой концентрации, но
легко растворяется в смеси HNO3 и HF« В отсутствии
кислорода В. не раствориется в щелочах и аммиаке.
К расплавленным щелочам устойчив в отсутствии окис-
лителей, но при доступе воздуха или в присутствии
NaNO3, NaNO2, КС1О3, РЬО2 и т. п. происходит интен-
сивное окисление металла с образованием вольфра-
матов. Важнейшие из соединений В.: трехокись VVO-,
вольфрамовая к-та H2WO4 и ее соли — вольфраматы
(см. Вольфрама окислы и кислоты). При обработке
вольфраматов щелочных и щелочноземельных метал-
лов восстановителями получаются «вольфрамовые
бронзы» — кристаллич. вещества с металлич. свойст-
вами. Согласно рентгенографии, исследованиям, нат-
рийвольфрамовые бронзы представляют собой твер-
дые р-ры внедрения натрия в кристаллическую ре-
шетку WO3 и имеют состав, отвечающий формуле
NaxW03.
Аналитическое определение вольфрама.
Для аналитич. химии В. имеют значения соедине-
ния с валентностью -f-б и -f-5. В. относится к группе
кислотообразующих элементов (наряду с Si, Р и др ).
Качественно В. открывают по желто-зеленой окраске
р-ра в присутствии роданид-иона и восстановителя
(обычно SnCl2); по бурой окраске сернокислого р-ра
в присутствии гидрохинона; по образованию осадка
с а-бензоиноксимом в кислой среде и др. методами.
Весовым путем В. определяют в виде VVO3 после оса-
ждения с цинхонином или др. алкалоидом. В. также
определяют потенциометрия, титрованием с р-ром
2-валентного хрома и фотометрически по реакции W5+
с роданид-ионом в присутствии SnCi2 (или другого
восстановителя), а также по реакции с гидрохиноном.
Si от В. отделяют обработкой плавиковой кислотой и
H2SO4 или обезвоживанием с HGL От Mo, Sb, As
В. отделяют действием H2S на кислые тартратные р-ры,
от As и Р — действием магнезиальной смеси, от Ti,
Zr, Nb, Та — действием слабоаммиачного р-ра MgSO4
и NHjGl на водный р-р содового плава. В раствор В.
обычно переводит последовательной обработкой соля-
ной и азотпой к-тами с последующим растворением
образовавшегося осадка H2WO4 в аммиачном р-ре,
а иногда выщелачиванием содового или ппросульфат-
ного плава.
Получение. Главнейшим сырьем для произ-ва В. и
его соединений являются вольфрамит и шеелит. Воль-
фрамовые руды редко содержат свыше 2% WO3; их
предварительно обогащают с целью получения кон-
центратов, содержащих 50—60% WO3. Обогащение
вольфрамовых руд проводят флотацией, гравитацион-
ными методами, магнитной и электростатич. сепара-
цией, химич. обработкой. Переработка вольфрамовых
концентратов до компактного металла проводится
в три этапа: 1) химич. выделение чистой вольфрамо-
вой к-ты или ее солей; 2) восстановление WO3 до ме-
таллич. порошка; 3) превращение этого порошка
в компактный металл.
В зависимости от вида сырья, технич. требований к чистоте
и физич. качествам WO3 и масштабов произ-ва применяют неск.
методов разложения вольфрамовых концентратов. Вольфра-
мит разлагают либо спеканием или сплавлением с содой с по-
следующим выщелачиванием опека водой, либо длительным
кипячением с конц. р-рами едких щелочей. Из полученных
в результате такой обработки р-ров В. выделяют или в виде
H2W0„ или осаждением CaWO4, затем разлагаемого к-тами.
Иногда выделение В. производят кристаллизацией в виде
вольфрамата или паравольфрамата натрия. Шеелит в нек-рых
вариантах технологии разлагают НС1 при 90—100°. В осадке
при этом остается H2W04. С целью очистки этот осадок обра-
батывают водным р-ром NH3. Из полученного таким путем
р-ра В. или осаждают в виде HSWO4, или кристаллизуют
в виде паравольфрамата аммония. Шеелитовые концентраты
также перерабатывают: 1) спеканием или сплавлением с содой
в присутствии S1O2 (песка) с последующим выщелачиванием
опека водой и 2) разложением р-рами соды в автоклавах.
В обоих случаях получаются водные р-ры Na2W04, дальней-
шая обработка к-рых производится методами, описанными
выше для вольфрамита. Восстановление WO3 или H2W0,
до металлич. порошка производят нагреванием при 750 —
850° в токе сухого Н2. Структура окисла или к-ты, скорость
тока Н2, темп-ра и время восстановления оказывают существен-
ное влияние на физич. характеристики вольфрамового порошка.
Из-за трудностей проведения процессов плавки и лптьн,
в связи с высокой темп-рой плавления В., основой промышлен-
ного произ-ва компактного металла служит металлокерампч.
метод. Вольфрамовый порошок сначала прессуется в брикеты
в стальных прессформах под давлением 2—3,5 т/смг. Полу-
ченные брикеты подвергаются спеканию в две стадии. Первая
стадия — упрочнение штабика — проводится при темп-рах
до 1300° в атмосфере Н2; во второй стадии — сварке — под-
держивается темп-ра, близкая к темп-ре плавления металла
(ок. 3000°) пропусканием через штабик электрич. тока. После
этого плотность штабика становится равной 17—18,5 г/см'.
Первоначальная проковка спеченного штабика проводится
при 1300—1600° с предварительным подогревом в печи с водо-
родной атмосферой. Из полученных т. о. плотных заготовок
дальнейшей ковкой, волочением, прокаткой получают прутки,
проволоку, пластины, ленты и т. п. Когда в В. допускается
примесь углерода (напр., в произ-ве твердых карбидных спла-
вов), иногда применяется метод восстановления WO, угле-
родом при высоких темп-рах. Однако в последнее время при-
знано, что более высококачественные твердые сплавы полу-
чаются из В., восстановленного водородом. Для произ-ш
ферровольфрама, наряду с основным методом восстановитель-
ной выплавки в дуговых печах, иногда используют восстанов-
ление WO3 или обогащенных вольфрамовых руд металлами
(гл. обр. алюмотермия, способом).
Применение. Почти сто лет после открытия В. не
находил промышленного применения. Лишь во 2-й
половине 19 в. стали проводиться опыты но выясне-
нию влияния добавок В. на свойства стали. Для про-
изводства качественных сталей сейчас потребляется
ок. 85% от всего количества добываемого В. Ок. 10%
идет на произ-во карбидных и др. сплавов (см. Вольф-
рама сплавы и Карбиды). Чистый В. начали применять
в пром-сти с начала 20 в., после открытия способа
получения компактного ковкого металла. Первой
областью применения чистого В. явилось использо-
вание его в качестве нитей накаливания для электро-
ламп. Вследствие высокой темп-ры плавления и низ-
кой упругости пара при высоких темп-рах В. является
незаменимым материалом для этой цели. В. приме-
няется также для изготовления нагревателей в элект-
рич. печах, электродов для атомно-водородной сварки,
различных деталей высоковакуумных усилителен,
655
ВОЛЬФРАМА ГАЛОГЕНИДЫ — ВОЛЬФРАМА СПЛАВЫ
656
катодов генераторных ламп, эмиссионных и газораз-
рядных трубок, выпрямителей высокого напряжения,
антикатодов и катодов рентгеновских трубок, контак-
тов электроаппаратуры и др. В. и нек-рые виды стекла
имеют почти равные коэфф, термич. расширения. По-
этому В. часто используют как вводной проводник
в электровакуумных приборах. Благодаря высокой
плотности он применяется при конструировании рото-
ров гироскопов, нек-рых амортизационных приборов,
а также иногда в качестве защиты от радиоактивных
излучений.
Лит.: Зеликман А. Н., Металлургия вольфрама и
молибдена, М., 1949; Меерсон Г. А. и 3 е л и к м а н А. Н.,
Металлургия редких металлов, М., 1955; С о и г и я а О. А.,
Редкие металлы, 2 изд., М., 1955; L1 К. С. and Chung
Yu Wang, Tungsten..., 3 ed., N. Y. — L., 1955; С м и-
т e л л с К. Д ж., Вольфрам, пер. с англ., М., 1958; G m е-
1 1 п, 8 Aufl., System-Nummer 54. Wolfram, В., 1933; Mel-
lor, v. 11, L. — N. Y. — Toronto, 1931; то же, переизд., 11,
1954; Pascal, t. 14, P., 1959; Kirk, v. 14, N. Y„ 1955, p.
35—73- Agte C., Vacek J., Wolfram a molybden, Praha,
1954; Озеров P. II., Усп. хим., 1955, 24, вып. 8, с. 951.
ВОЛЬФРАМА ГАЛОГЕНИДЫ — с галогенами
вольфрам образует ряд галогенидов и оксигалогени-
дов, в к-рых он проявляет валентности —2, —f—3, -(-4,
4-5 и 4-6. Описаны галогениды типа WX2 (X — Cl,
Вт, J), WX4 (X = F, Cl, J), WX5 (X = Cl, Вг) и WX6
(X = F, Cl, Вг) и оксигалогепиды состава WOX2
(X = F), WO2X2 (X = Cl, Вг) и WOX4 (X = F, Cl,
Вг). Трифторид WF3, трихлорид WC13 и диоксиди-
фторид WOjFj известны в виде комплексных со-
лей: KWF4 • Н2О, NH4WF4 • Н2О, Me3[W2CI9], или
2WC13 • ЗМеС1, гдеМе=К+, Na+, NHf и др., Na2WO2F4
и т. п. Галогениды и оксигалогениды вольфрама по
отношению к воде неустойчивы (напр., WFe -|- 4Н2О =
= H2WO4 -j- 6HF), за исключением оксидифторида
WOF2, противостоящего действию щелочей, кислот и
царской водки. Наибольшее значение имеют хлориды
и оксихлориды. Дихлорид WC12 (серый аморфный по-
рошок), тетрахлорид WC14 (темно-коричневый поро-
шок, плотн. 4,624, нелетуч) и пентахлорид WC15
(темно-зеленые кристаллы, плотн. 3,875, т. пл. 253°,
т. кип. 288°) образуются при восстановлении гекса-
хлорида водородом. Гексахлорид WC1B (черно-фиолето-
вые кристаллы, плотн. 3,52, т. пл. 275°, т. кип. 348 + 2°)
получают взаимодействием порошкообразного W с су-
хим хлором при нагревании. Известны два оксихло-
рида: диоксидихлорид WO2C12 (светло-желтое веще-
ство, т. пл. 266°), получающийся при нагревании
WO2 или смеси WO3 с углем в токе хлора, и окситет-
рахлорид WOC14 (красные иглы, плотн. 11,92, т. пл.
204+10°, т. кип. 230°), образующийся по реакции:
W034-2WCle= 3W0CI4 (в вакууме при 180—250°).
Оба оксихлорида летучи при низкой темп-ре (ниже
300°). Водой гидролизуются с образованием H2WO4.
Благодаря низким темп-рам кипения галогениды и
оксигалогениды вольфрама (особенно WCle) могут
использоваться для нанесения вольфрамовых покры-
тий на металлы.
Лит.: Щукарев С. А., Новиков Г. И., Ж. неорг.
хим., 1956, 1, вып. 3, с. 357; Щукарев С. А., Нови-
ков Г. и. и Андреева Н. В., Ж. общ. хим., 1958,
28, вып. 7, с. 1998; см. также при ст. Вольфрам.
А. К. Пикаев.
ВОЛЬФРАМА КАРБИДЫ — см. Карбиды.
ВОЛЬФРАМА ОКИСЛЫ И КИСЛОТЫ — кисло-
родсодержащие соединения вольфрама. Вольфрам
образует неск. окислов. Из них важнейшие: WO2,
W4On (точнее WO2 72), WO2;9 и WO3. На основе этих
окислов существуют фазы с определенными областями
гомогенности, что создает условия нек-рых возмож-
ных отклонений составов окислов от указанных выше
формул. Наиболее устойчивы двуокись WO2 и трех-
окись WO3. Двуокись вольфрама — поро-
шок или кристаллы коричневого цвета, плотность
12,11, т. пл. ок. 1270°, т. кип. ок, 1700°, теплота
образования &H°sh = —130,5 ккал/моль. В воде и
водных р-рах щелочей и к-т, не являющихся окисли-
телями, не растворяется. Азотной к-той и при прока-
ливании на воздухе окисляется до WO3. Получается
восстановлением W03 водородом при 600—650° или
нагреванием выше 400° смеси порошкообразного ме-
талла с WO3. Трехокись вольфрама —
порошок лимонно- или оранжево-желтого цвета, плотн.
7,16, т. пл. ок. 1470°, теплота образования A7/2IS —
= —195,7 ккал/моль. Практически не растворяется
в воде и кислотах (за исключением плавиковой), при
действии щелочей образует растворимые вольфраматы.
Восстанавливается водородом (при 700—1000°) или
углеродом (выше 1000°) до металла. Получается про-
каливанием вольфрамовой к-ты или ее солей, а также
нагреванием W на воздухе выше 400°.
Трехокись образует неск. кислот: 1) вольфрамовую
к-ту (желтого цвета), или моногидрат H2WO4, т. е.
WO3-H2O; 2) вольфрамовую к-ту (белого цвета), или
дигидрат WO3-2H2O (по нек-рым данным, белая вольф-
рамовая к-та является гидратом с непрерывно изме-
няющимся содержанием Н2О при повышении темпера-
туры); 3) метавольфрамовую к-ту П2О • 4WO3-xH2O,
координационная формула H10[H2(W2O7)e] • 24Н2О;
4) надвольфрамовую кислоту WO3 • Н2О2 Н2О, или
H2WO5 -Н2О. Вольфрамовая к-та H2W04 — желтый по-
рошок, плотность 5,5; очень плохо растворима в воде,
почти нерастворима в к-тах (за исключением плави-
ковой). Легко растворяется в водных р-рах углекис-
лых и едких щелочей и аммиака. Из р-ров вольфра-
матов H2WO4 осаждается горячими минеральными
к-тами. При подкислении р-ра вольфрамата на холоду
выделяется дигидрат W03 • 2Н2О в виде объемистого
белого осадка, к-рый при кипячении в кислой среде
быстро переходит в моногидрат. Вольфрам обладает
способностью образовывать комплексные гетерополи-
кислоты (с Р, As, V, Si, В и др. элементами в качестве
центрального атома). К ним относятся: кремневоль-
фрамовая к-та Н8 [Si (W2O7)eJ • жН2О, желтые кри-
сталлы, растворимые в воде и эфире; фосфорновольф-
рамовая к-та Н, [Р (W2O7)e]x • Н2О, блестящие кри-
сталлы, растворимые в воде, идр. Из окислов W наи-
большее применение находит трехокись. Она приме-
няется как исходный материал при получении метал-
лич. W и его карбида, для окрашивания керамич. п
стеклянных изделий в желтый цвет, как катализатор
при гидрогенизации и крекинге углеводородов. H2WO4
служит адсорбентом, протравой в текстильной промы-
шленности и катализатором при произ-ве высокоокта-
нового бензина.
Лит.: Бергер И. И., Севастьянов Н. Г.,
Путилина Л. К., Ж. неорг. хим .1956, 1, вып. 8, с. 1713;
Карякин ГО. В. иА и гелов И. И., Чистые химиче-
ские реактивы..., М., 1955; см. также при ст. Вольфрам.
А. К. Пикаев.
ВОЛЬФРАМА СПЛАВЫ — сплавы на основе (с пре-
обладанием) вольфрама. Наибольшее практич. значе-
ние имеют сплавы вольфрама с железом, молибденом,
медью, серебром, никелем и кобальтом; перспектив-
ными являются сплавы вольфрама с рением. Важное
значение имеют сплавы на основе карбида вольф-
рама — твердые сплавы.
В о л ь ф р а м-ж е л е з о. В системе W — Fe, на-
ряду с твердыми р-рами, имеется эвтектика при 33%
W (1540°) и два интерметаллич. соединения: Fe2W
(62,2% W) и Fe,We (73,8% W). Практич. применение
нашли сплавы с 70—85% W — ферровольфрам, к-рый
используют в качестве легирующей добавки в произ-ве
сталей. Введение вольфрама в сталь приводит к упро-
чнению твердого р-ра на основе железа и образованию
тугоплавких карбидов вольфрама, вследствие чего
повышаются твердость и прочность стали. Ок. 90%
всего выплавляемого ферровольфрама расходуется на
произ-во быстрорежущей стали. Ферровольфрам полу-
657
ВОЛЬФРАМА СПЛАВЫ - ВОЛЬФРАМАТЫ
658
чают путем плавки в дуговых электрич. печах или
алюминотермии, способом (см. Алюминотермия).
В о л ь ф р а м-м о л и б д е и. Оба металла имеют
однотипные кубич. кристаллич. решетки с близкими
периодами (ок. 3,16 и 3,14 А соответственно), поэтому
образуют между собой непрерывный ряд твердых
р-ров. Изменяя соотношение между компонентами
сплава, можно получить материалы с различной твер-
достью, электросопротивлением и термич. коэфф,
линейного расширения. Максимум твердости и элект-
росопротивления соответствует 45% Мо; коэфф, ли-
нейного расширения изменяется в пределах значений,
характерных для технич. стекол, поэтому сплавы
можно соединять с ними путем пайки; вольфрамо-
молибденовые сплавы применяют для впайки токо-
цодводящих вводов в стеклянные электрич. приборы
и аппараты. Сплавы W с Мо используют в электро-
вакуумной пром-сти, а также для изготовления высо-
котемп-рных термопар, работающих в инертной или
восстановительной атмосфере. Вольфрамо-молибдено-
вые сплавы получают металлокерамич. способом ана-
логично металлич. вольфраму.
Вольфрам — медь и вольфрам — се-
ребро. Медь и серебро не сплавляются и не обра-
зуют соединений с вольфрамом, поэтому такие псевдо-
сплавы получают пропиткой пористых вольфрамсо-
держащих брикетов жидкими медью или серебром;
содержание второго компонента в сплавах составляет
5—30%. Сплавы обладают повышенной твердостью
и механич. прочностью при нагреве, высокой электро-
и теплопроводностью. Применяются для изготовления
прецизионных электроконтактов для аппаратов кон-
тактной сварки и других приборов и машин.
Вольфрам — медь — никель («тяжелый
сплав»). При отсутствии взаимной растворимости ме-
таллов в системе вольфрам—медь оба металла рас-
творимы в никеле, вследствие чего при определенных
соотношениях компонентов можно получить, исполь-
зуя методы металлокерамики, тройные сплавы с вы-
соким содержанием вольфрама (напр., 90% W, 6% Ni,
4% Си). Сплавы обладают большой плотностью (>16),
повышенной коррозионной стойкостью и высоким
коэфф, поглощения радиоактивных излучений. Приме-
няются в атомной и авиационной технике, приборо-
строении и др. областях.
В о л ь ф р а м—р е н и й. Сплавы двух самых туго-
плавких металлов являются перспективными для
использования в современной технике. В системе
имеются широкие области твердых растворов, эвтек-
тика и область о-фазы. Наибольший технич. интерес
представляют сплавы в области твердых растворов
рения в вольфраме; сплавы с содержанием до 20% Re
обладают более высокими прочностью, пластичностью
и уд. электросопротивлением, чем вольфрам; устой-
чивость против эррозии и коррозии у сплавов вольф-
рам — рений также выше, чем у вольфрама. Сплавы W
с 5 и 20% Re имеют высокие и стабильные термоэлект-
рич. свойства (напр., при 1800° ТЭДС^27 мв). Сплавы
вольфрам—рений перспективны для электровакуумной
пром-сти и изготовления высокотемп-рных термопар
(до 2500°).
К В. с. следует также отнести твердые сплавы па
основе карбида WC, широко используемые в технике
для изготовления различных видов режущего и буро-
вого инструмента. Наряду с собственно вольфрамо-
выми сплавами существует большое число жаропроч-
ных и коррозионно-устойчивых материалов (напр.,
сплавы на никелевой и кобальтовой основах), легиро-
ванных заметными количествами (до 20%) вольф-
рама.
Лит.: Меерсон Г. А. иЗеликман А. Н., Метал-
лургия редких металлов, М., 1955; Елютин В. П. [и др.],
Производство ферросплавов, 2 изд., М., 1957; Савиц-
кий Е. М., Тылкина М. А., Шишкина Л. Л., Изв.
АН СССР, Отд. техн. н. Металлургия и топливо, 1959, № 3.
Ю. А. Павлов.
ВОЛЬФРАМА СУЛЬФИДЫ — соединения вольф-
рама с серой: дисульфид и трисульфид. Дисульфид
WS2 — темно-серый порошок, плотность 7,5, встре-
чается в природе в виде минерала тунгстенита. Кри-
сталлизуется в гексагональной решетке слоистого
типа. Нерастворим в воде, легко подвергается дейст-
вию смеси HN03 и HF и расплавленных щелочей.
Получают нагреванием WS3 в отсутствии воздуха или
пропусканием паров S над нагретым W или W03.
Дисульфид, применяемый в качестве катализатора,
получают действием H2S на паравольфрамат аммония
при нагревании. Трисульфид WS3 — черный
порошок, плохо растворяется в воде п хорошо —
в р-рах гидроокисей, карбонатов и сульфидов щелоч-
ных металлов с образованием тиовольфраматов состава
Me2W03S, Me2WO2S2, Me2W0S3 или Me2WS4. Легко
дает коллоидные р-ры. При нагревании на воздухе
превращается в WO3. Получают действием соляной
к-ты на р-ры тиовольфраматов. а. к Пикаев.
ВОЛЬФРАМАТЫ — соли вольфрамовых к-т. Раз-
личают: моновольфраматы, или нормальные В., — соли
H2W04; изополивольфраматы — соли не выделенных
в свободном состоянии изополикислот вольфрама;
акваполивольфраматы — соли акваполикислот воль-
фрама; гетерополивольфраматы — соли гетерополи-
кислот вольфрама. Важнейший моновольфра-
мат — нормальный В. натрия Na2W04, и обычных
условиях существует в виде кристаллогидрата
Na2WO4 • 2Н2О, ниже 4-6° — в виде Na2WO4 • ЮН20;
плотность NajWC^ - 2Н2О 3,5, безводной соли 4,18;
т. пл. безводной соли 698°. Растворимость моноволь-
фрамата натрия в воде (в % Na2WO4): 36,5 (0°), 42,2
(20°) и 49,2 (100°). Безводный Na2W04 получают спла-
влением W03 с едким натром или содой. Другие моно-
вольфраматы (исключая соли щелочных металлов и
магния) слабо растворимы в воде. Практич. примене-
ние, кроме Na2WO4, находят также моновольфраматы
аммония (существуют только в водном растворе) и
кальция.
Изополивольфраматы образуются при
сплавлении моновольфраматов с различными количе-
ствами WO3. Известны дивольфраматы, напр. Na2W2O7,
тривольфраматы, напр. K2W3O10, и тетравольфраматы,
напр. Cs2W4013 (об их строении см. Изополисоедине-
ния). Акваполивольфраматы получают
нейтрализацией до определенных значений pH вод-
ных р-ров щелочных моновольфраматов (их строение
рассматривается в статье Акваполисоединения). Среди
акваполивольфраматов наиболее изучены мета- и
паравольфраматы. Метавольфраматы, по данным раз-
личных авторов, имеют разный состав; его часто выра-
жают ф-лой ЗМе2О • 12WO3 • ®H2O или с помощью
координационной ф-лы MeeH4 [Н2 (W2O7)e] • а:Н2О,
где Me — одновалентный металл. Метавольфраматы
образуются при подкислении р-ров щелочных моно-
вольфраматов до pH 3,5—4,5. Все метавольфраматы
растворимы в воде. Паравольфраматы имеют состав
5Ме2О • 12WO3 • хН2О, к-рый часто выражают ф-лой
Ме6Н6[Н2 (WO4)6J • хН2О; получают нейтрализацией
р-ров щелочных моновольфраматов до нейтральной
или слабощелочной реакции. Важнейший из них —
паравольфрамат аммония, выпадает из р-ра (NH4)2WO4
при нейтрализации до pH 7,3 в форме игольчатых
кристаллов (моноклинич. призм) 5 (NH4)2O • 12WO3 •
• 11Н2О или при упаривании р-ра в форме пластинча-
тых кристаллов 5 (NH4)2O • 12WO3 • 5Н2О. При нагре-
вании паравольфрамат аммония разлагается с обра-
зованием WO3, что служит одним из способов по-
лучения последнего; растворимость в воде в %
(NH4)5H6[H2 (WOj),]: 1,06 (25°) и 7,97 (70°). Гете-
659
ВОЛЮМОМЕТРЫ — ВОЛЯ — ЦИГЛЕРА РЕАКЦИЯ
660
рополивольфраматы — сложные комплексные соеди-
нения (см. Гетерополисоединения). Фосфорновольфра-
мат натрия Na2H5[P (W2O,)6J • 16Н2О — одно из важ-
нейших соединений этого типа, хорошо растворяется
в воде (47,39% в расчете на безводную соль при 20°).
В. находят широкое применение: В. натрия — при
изготовлении утяжеленных, а также огне- и водо-
устойчивых тканей, в произ-ве шелка, кожи и пигмен-
тов; В. кальция и бария входят в состав усиливающих
и фосфоресцирующих экранов для рентгенографии;
В. магния используется в качестве фосфора, испуска-
ющего синий свет; технич. вольфрамат натрия (в раст-
воре) и вольфрамат кальция (искусственный шеелит)
служат полуфабрикатами при получении вольфрамо-
вого ангидрида, используемого в произ-ве твердых
сплавов, а вольфрамат кальция применяют и в про-
изводстве ферровольфрама; паравольфрамат аммония
является важным промежуточным продуктом в техно-
логии произ-ва чистого вольфрама и его соединений.
Лит.: Карякин Ю. В.иАнгелов И. И., Чистые
химические реактивы, М., 1955; Руководство по препаративной
неорганической химии, под ред. Г. Бауера, пер. с нем., М.,
1956; С п и ц ы н В. И., Ж. общ. хим., 1950, 20, вып. 3, с. 550;
см. также лит. при статье Вольфрам. А. К. Пикаев.
ВОЛЮМОМЕТРЫ — приборы для определения со-
держания твердого или жидкого в-ва путем измерения
объема выделившегося при реакции газа.
Все В. состоят из реакционного сосуда
и измерительной бюретки, в к-рой соби-
рается выделившийся газ. В качестве за-
порной жидкости применяется ртуть или
другая жидкость, не растворяющая вы-
деляющийся газ и не реагирующая с
ним. На рис. приведена схема простей-
шего В. (1 — сосуд для разложения ве-
щества; 2 — измерительная бюретка; 3 —
уравнительная трубка). Наиболее рас-
пространенными В. являются: нитро-
метр — для колич. определения азот-
ной и азотистой к-т в растворах и соеди-
нениях; газоволюмометр — усо-
вершенствованный нитрометр (он позво-
ляет без обычных вычислении, связанных
с приведением объема газа к стандартным темп-ре и
давлению, с большой точностью и быстротой получать
результаты анализа); азотометр — для измерения
выделившегося при реакции азота, напр. при опре-
делении аммония в его солях при помощи гипобро-
мита натрия; карбометр — для определения СО2
в карбонатах путем измерения объема образующейся
при разложении СО2.
Лит.: Труды Казанского филиала АН СССР. Серия хим.
наук, вып. 3, Казань, 1956; Еремина Б. Г., Газовый
анализ, Л., 1955. Б. Г. Еремина.
ВОЛЯ — ЦИГЛЕРА РЕАКЦИЯ — бромирование
непредельных соединений с помощью N-бромамидов;
бромированию подвергается углеродный атом в
a-положении (аллильном) к двойной связи:
СН2=СНСН3 + Вг>1СОСН2СН2СО->-
-> CH2=CHCH2Br4-HNCOCH2CH2CO
В качестве бронирующего агента чаще всего при-
меняют N-бромсукцишшид; предложены также N-бром-
ацетамид, N-бромфталимид, 3-бром-5,5-диметилгидан-
тоин и перфторированные N-бромимиды янтарной
и глутаровой к-т. Общепринятым считают радикаль-
ный механизм реакции:
C4H4O2NBr---- CsHjO2N+Br
\с=с-сн2 4- С4Н4О»$--- \с=С-СН'+ c4h4o2nh
\ С=С—СН' + Вг . - - ^>С-С-СНВг
.11 \ I I
\с=С -СН'4- C4HiOsNBr-► рс=с— СНВг 4- C4H4O2N
Радикальный механизм подтверждается, в частности,
ускоряющим действием перекисей и света.
В В. — Ц. р. не вступают соединения с третичным
атомом водорода в a-положении к двойной связи, а
также непредельные сульфоны и соединения с ацети-
леновой связью. Алифатич. и алициклич. соединения
хорошо бромируются N-бромамидами; метиленовая
группа бромируется значительно легче метильной.
В случае соединений с сопряженными двойными свя-
зями необходимо присутствие перекисей, в противном
случае часто происходит осмоление:
ch3ch=chch=chcooch3
—► ВгСН2СН = СНСН = СНСООСН3
Реакция применима также к соединениям, в к-рых
двойная связь является частью гетероциклич. системы
(метилгомологи тиофена, фурана, пиридина, диоксана
и др.):
C4H4OgNBr
В случае ароматич. соединений (гомологи бензола,
нафталина, конденсированных систем) бромируется
боковая цепь или ядро в зависимости от условий
реакции:
C4H4O2NBr
перепись
C4H4O2NBrr
Al СIз, Z пС1 а
Важнейшими побочными процессами при В — Ц.р.
являются аллильные перегруппировки (особенно
в случае бициклич. терпенов) и бромирование по двой-
ной связи с образованием насыщенных дибромидов:
СН2—СН2 C.HiOJiBr СН2 — СНВг СН2—СН
I I || 4. । || 4.
СН2-С=СН2 СН2—С=СН2 СНа—С—СН2Вг
сн3-сн2
+ I I
СН2-СВг-СН2 Вг
Полученные нормальные продукты бромирования мо-
гут отщеплять НВг с образованием двойных связей:
Дегидробромирование продуктов реакции с N-бром-
амидами служит методом дегидрирования циклич. со-
единений в мягких условиях. Активация а-метилено-
вой группы может вызываться не только двойной свя-
зью, а также иными группами. Поэтому В. — Ц. р.
применима также к кетонам (гл. обр. циклическим)
и ацеталям:
СН3СН2СН(ОС3Н5)г
C4HtO2NBr
С4Н4ОгКВг
о
Вг
СН3СНВгСН(ОС2Н5)2
В а, р-ненасыщеппых кетонах, имеющих метиленовые
группы в a-положении как к карбонильной группе,
661
воски
662
таки к двойной связи (напр., Д4-3-кетостероиды), бро-
нируется преимущественно метиленовая группа в
a-положении к двойной связи:
Реакция открыта А. Волей в 1919, усовершенство-
вана А. Циглером с сотр. в 1942.
Лит.: Терентьев А. П. и Яновская Л. А.,
в кн.: Реакции и методы исследования органических соеди-
нений, кн. 6, М„ 1957, с. 289. Ю. А. Титов.
ВОСКИ — жироподобные вещества, преим. расти-
тельного и животного происхождения. В отличие от
жиров, являющихся сложными эфирами трехатом-
ного спирта — глицерина, В. состоят гл. обр. из слож-
ных эфиров, образованных высшими жирными к-тами
и высокомолекулярными одноатомными (редко двух-
атомными) спиртами. В состав эфиров наиболее часто
входят пальмитиновая СН3 (СН2)14СООН, церотино-
вая СН3(СН2)24СООН и карнаубовая С23Н4,СООН
к-ты и цетиловый СН3(СН2)14СН2ОН, цериловый
СН3(СН2)24СН2ОН и мирициловый СН3(СН2)28СН2ОН
спирты. Часть сложных эфиров, входящих в состав
нек-рых животных В. (ланолин), образована циклич.
спиртами группы стеринов — холестерином и изохо-
лестерином. Цветные, или окрашенные, В. предста-
вляют собой сложные эфиры, гл. обр. дипальмитаты
двухатомных спиртов каротиноидного характера —
ксантофилла и зеаксантина. Кроме сложных эфиров,
в В. встречаются свободные спирты, жирные к-ты,
высокомолекулярные углеводороды, а также нек-рые
красящие и душистые вещества.
В. — аморфные, пластичные, легко размягчаю-
щиеся при нагревании в-ва, плавящиеся в интервале
темп-р 40—90°. В. не смачиваются водой, водонепро-
ницаемы, неэлектропроводны, горючи, нерастворимы
в воде и холодном спирте, хорошо растворимы в бен-
зине, хлороформе и эфире. Химически В. родственны
жирам, и для их характеристики применяют те же
показатели, что и для жиров: омыления число, кислот-
ное число и иодное число. В. мало реакционноспособны
и очень устойчивы; известны случаи, когда пчелиный
В. сохранялся более тысячи лет. В отличие от жиров,
В. омыляются с трудом и только в щелочной среде.
В кислых средах гидролиз не только не идет, но, на-
оборот, при нагревании смеси высокомолекулярных
спиртов и жирных кислот, даже в присутствии воды,
происходит синтез В. При перегонке В. распадаются
на свободные к-ты и соответствующие спиртам непре-
дельные углеводороды. Для очистки природных В. их
перетапливают в присутствии воды и отбеливают при
помощи различных адсорбентов и окислителей.
Наименование В. Т. пл., °C Плотность Ки- слот- ное число Число омыле- ния Иодное число
Пчелиный .... 62-70 0,955—0,975 17-21 86-96 8-И
Шерстяной .... Ланолин (безвод- 31-42 0,924— 0,960 0,2-40 82-140 15-47
ный) 35—37 0,940—0,970 1-2 85-100 15-18
Спермацет .... 41—49 0,905—0,960 0,5 41-49 2.6—3,8
Карнаубский . . 83—91 0,990—0,999 1-8 73-86 8-13
Канделильский . Монтанный (пере- 65—69 0,969—0,993 15-16 46—65 14-37
гнанный).... 72-77 1,0 73—85 75—89 10-15
По происхождению В. можно разделить на живот-
ные, растительные и ископаемые. Промышленное зна-
чение имеют след. В.: пчелиный, шерстя-
ной, спермацет (животные В.), карнауб-
ский, канделильский (растительные В.),
озокерит и выделяемый из него церезин
(ископаемый В.) и монтанный В.
Свойства этих В. приведены в таблице.
Животные В. выделяются восковыми
железами насекомых (пчелиный воск), копчи-
ковой железой птиц и кожными железами
млекопитающих (шерстяной воск); в череп-
ных камерах кашалотов содержатся большие
количества спермацетового масла. Насекомые
используют В. в качестве строительного мате-
риала. Роль других животных В. в природе
ие выяснена окончательно; скорее всего она определяется
их несмачиваемостью и состоит в предохранении организма
от действия влаги.
Пчелиный В. добывают из сотов, обрезков вощины,
восковых наростов в ульях. Этот В. на 72% состоит из эфиров
жирных к-т, среди к-рых 33% приходится на мирицилпаль-
митат. Для пчелиного В. характерно высокое содержание
свободных к-т — до 13,5% и углеводородов — 12—12,5%.
Шерстяной В. получают при промывании шерсти
овец. В очищенном виде он носит название ланолина.
Ланолин отличается от других В. высоким содержанием сте-
ринов (холестерин, 1,8-дигидрохолестерол, оксихолестерол),
как свободных (10,5%), так и связанных в виде эфиров. Кис-
лоты, входящие в состав ланолина, относятся к т. н. лано-
ряду (ланомиристиновая, ланопальмитиновая); они отли-
чаются от изомерных им алифатич. к-т нормального ряда раз-
ветвленностью цепочки и оптич. активностью. Ланолин отли-
чается также способностью связывать до 300% воды.
Спермацет добывают из спермацетового масла каша-
лотов вымораживанием и отжиманием. При этом отделяется
жидкий В., состоящий гл. обр. из эфиров ненасыщенных к-т
и спиртов (олеилмиристоолеат), и твердый спермацет, к-рый
на 98% состоит из эфиров насыщенных к-т (преим. из цетил-
пальмитата). Спермацет получают также гидрогенизацией
спермацетового масла.
Некоторые В. являются продуктом жизнедеятельности ки-
слотоупорных бактерий, в частности микобактерий туберку-
леза и лепры; эти бактерии защищены плотной восковой обо-
лочкой, которая делает их весьма устойчивыми к внешним воз-
действиям. Из бактериальных В. выделены сложные эфиры,
образованные миколевой кислотой CS(HI72O4, содержащей
одну окси- и одну метоксигруппу, н спиртом — фтиоцеролом
С34Н„(ОН)2(ОСН3), а также эфиры вторичных спиртов
а-эйкозанола-2 CH3(CH2)i7CHOHCH3 и а-октадеканола-2
СН2(СН2)12 СНОНСНз.
Растительные В. покрывают в виде тонкого налета
или пленки стебли, листья и плоды растений; в засушливых
областях растительные В. предохраняют растения от излиш-
ней потери влаги и, возможно, от грибковых заболеваний.
Из них наиболее важен карнаубский В. — хрупкая
масса желтоватого или темно-серого цвета, к-рую получают
из листьев бразильской пальмы Copernicia cerifera. Раствори-
мость карнаубского В. в 100 а растворителя при 25—30°:
в этиловом спирте (95%-ном) 0,174 г. ацетоне 0,410 а, этило-
вом эфире 0,594 г, хлороформе 1,145 г; nj) 1,4672—1,4720
(40°) и 1,4457—1,4478 (100°); электропроводность: при 0°
14,5 • 10 6, при 160° 1,09 • 10“ (ш-1 см~‘; диэлектрич.
проницаемость 2,30 (40°). Карнаубский В. состоит из эфиров
мирицилкарнаубата и мирицилцеротата (ок. 80%), свободных
мелиссиловой и монтановой к-т (1—1,5%), свободных спир-
тов (10%), в числе к-рых карбоцериловый С27 и октакозанол
С23, не встречающиеся в других В. Кроме карнаубского В.,
известны след, растительные В.: канделильский,
добываемый из растения Pedilantlius Pavonis Boas; пальмо-
вый, покрывающий ствол пальмы Geroxilon ondlicoka,
состоящий из эфиров церотиновой к-ты и церилового спирта
п пальмитиновой к-ты и мирицилового спирта (т. пл. 102—
105°); льняной, получаемый из стеблей льна и содер-
жащий 35% эфиров, образованных цериловым спиртом и фито-
стеринами (т. пл. 61—70°).
К ископаемым В. относятся горный В., пли озо-
керит, состоящий главным образом из предельных углеводо-
родов. Очисткой озокерита получают церезин; экстрак-
цией органич. растворителями бурого угля— монтанный
В., в состав которого входит свободная ментановая к-та
С27Н55СООН и ее эфиры. Аналогичные В. получены экстрак-
цией торфа.
В. находят широкое практич. применение. Ланолин
используется для приготовления различных кремов
и мазей. Пчелиный В. идет на изготовление искусствен-
ной вощины и для др. целей. Карнаубский В. приме-
няется в произ-ве политур, мастик для вощения, при
выделке кожи, в произ-ве копировальной бумаги и др.
Различные В. используются для приготовления элек-
троизоляционных материалов, формовочных масс для
литья, лаков, восковых красок и карандашей, а так-
же при реставрации картин и др. Наряду с натураль-
ными применяют и искусственные В., получаемые эте-
663
ВОССТАНОВЛЕНИЕ — ВРЕВСКОГО ЗАКОНЫ
664
рификацией продуктов окисления церезина и парафина
жирными к-тами, а также заменители В., представляю-
щие собой продукты хлорирования нафталина.
Лит.: День ян ов Н. Я., П р я н и ш н и к о в Н. Д.,
Жиры и воска. Химия и анализ, 3 изд., М.—Л., 1932; 3 и-
н о в ь е в А. А., Химия жиров, М., 1952; Ивановский Л.,
Энциклопедия восков, пер. с нем., т. 1, Л., 1956; Deuel
Н. J., The lipids, v. 1—Chemistry, N. Y. — L., 1951; Warth
A. H., The chemistry and technology of waxes, 2 ed., N. Y.,
[1956]; Handbuch der Pflanzenphysiologie. Hrsg. von W. Ruh-
land, Bd 7, B. [u. a.], 1957. В. Б. Спиричев.
ВОССТАНОВЛЕНИЕ — химическая реакция, со-
стоящая в присоединении электронов атомами или
ионами, т. е. сопровождающаяся понижением их ва-
лентности (см. Окисление — восстановление). Прежде
характерным признаком В. считали отнятие кислорода
от окислов или солей, а также присоединение водорода
к химич. элементу или соединению. В органич. химии
и в настоящее время В. часто называют присоедине-
ние водорода к молекуле органич. соединения.
ВОССТАНОВЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕ-
НИЙ — присоединение водорода, сопровождаю-
щееся частичным или полным насыщением кратных
связей (гидрирование); расщепление простых связей
водородом (гидрогенолиз); элиминирование (удале-
ние кислорода из органич. молекулы). По характеру
действующего агента (восстановителя) В. о. с. может
быть произведено: действием водорода в присутствии
катализатора (см. Гидрогенизация и дегидрогениза-
ция каталитические)-, действием металлов в присут-
ствии соединений, содержащих подвижный атом
водорода (in statu nascendi); электролитически—со-
единениями, способными изменять валентность одного
из своих атомов: действием доноров гидрид-иона.
Наиболее распространенными приемами В. о. с.
водородом in statu nascendi является восстановление
нек-рыми металлами: Na в спиртах (см. Буво — Блана
восстановление)', Zn в р-рах к-т (замещение атома
галогена водородом в галогеналканах или имидхло-
ридах, восстановление пиррола в пирролин и пир-
ролидин и др.); Zn в р-рах щелочей (восстановлением
ароматич. нитросоединений до гидразосоединений
и др ); Zn в р-рах нейтральных соединений, напр.
синтез арилгидроксиламинов из нитросоединений,
в водном р-ре NH4C1; в присутствии кислот (восста-
новление нитросоединений в соответствующие, амины
и др.); Na или Li в жидком аммиаке или аминах
(частичное гидрирование системы сопряженных крат-
ных связей и др.).
Более энергичным восстанавливающим действием
по сравнению с чистыми металлами обладают металлич.
пары: Zn-—Си, Fe—Си, Na—К и др. Эффективно
восстанавливаются органич. соединения при действии
амальгам металлов: Na/Hg в водных или спиртовых
средах, обычно в присутствии щелочей или кислот
(восстановление карбонильной группы до спиртовой,
частичное гидрирование сопряженных систем и др.);
Al/Hg преимущественно в среде влажного диэтило-
вого эфира (см. Пинаконы) или Zn/Hg в кислых
средах (см. Клемменсена реакция). Характер В. о. с.
водородом in statu nascendi зависит от природы ме-
талла и характера среды, т. е. в конечном счете от
величины восстановительного потенциала металла
в данной среде. В. о. с. металлом в присутствии до-
нора водорода (АН), по-видимому, осуществляется
посредством серии элементарных актов, первым из
к-рых является переход электрона от металла к орга-
нич. соединению;
RX + Me-> R<«> + МеХ; R 1 <•>-р Me-» R~-р Ме+
R- 4- АН -» RH + а- или = О + Me — \ СО -ОМе
\сю>-0Ме + Me -» \.С—ОМе + Ме+
^С—ОМе + АН -> ^СН-ОМе + А~
Гидрирование карбонильных соединений, а также
нек-рых а,Р-непредельных соединений амальгамами
металлов часто сопровождается димеризацией про-
межуточно образующихся свободных радикалов (об-
разование пинаконов, гидродимеризация а,[J-непре-
дельных к-т и их производных и др.):
СНз
S\c-C-CH3
3 I I
онон
2
СНз\с=0 да
СН3Х Mg/Hg
сн
сн
2CH.,=CH-Cze:N^— №C-CH^CH.>CH.,CH»CSN
Na/Hg -
Электролитич. В. о. с. характеризуется первичным
актом перехода электрона от катода к органич. соеди-
нению. Наряду с препаративным значением этой
реакции (синтез n-аминофенола из нитробензола, по-
лучение азо- и гидразосоединений из ароматич. нитро-
соединений, восстановление пиридина в пиперидин
и др.) метод катодного восстановления нашел ши-
рокое применение в аналитич. практике (см. Поляро-
графия). В. о. с. при действии соединений элементов,
находящихся в состоянии низшей валентности (SnCL,
FeSO4, Na2S и др.), также связано с переходом элек-
трона к органич. молекуле. Под действием этих соеди-
нений восстанавливаются: имидхлориды в альдимины,
ароматич. нитросоединения в амины и др.
В отличие от описанных выше методов, В. о. с.
комплексными гидридами (LiAIH4, NaBH4 и др.)
является типично ионным процессом. Первичным
актом в данном случае является нуклеофильная
атака гидрид-иона или комплексного аниона на элек-
трофильный центр восстанавливаемой молекулы, напр.:
ZH Li*
>С=О
>с-о-L1
При действии комплексных гидридов карбоновые
к-ты или карбонильные соединения восстанавливают-
ся до соответствующих спиртов, а-окиси— в гликоли
и др. К преимуществам комплексных гидридов как
восстановителей относятся избирательность процесса
и, как правило, определенная стереохимии, конфигу-
рация образующихся продуктов реакции:
Li АI Н А
трет.
Движущей силой восстановления карбонильных
соединений алкоголятами алюминия (см. Меервейна—
Понндорфа—Берлея реакция), а также окислительно-
восстановительного диспропорционирования альдеги-
дов (см. Канниццаро реакция) также является переход
гидрид-иона от восстановителя к восстанавливаемому
соединению. См. также Окисление — восстановление.
Лит.: Уайлдс А. Л., в кн.; Органические реакции,
сб. 2, пер. с англ., М., 1950, с. 194; Браун В. Г., там же,
сб. 6, М., 1953, с. 409; Ворожцов Н. Н., Основы синтеза
промежуточных продуктов и красителей, 3 изд., М.—Л.,
1950, с. 259, 694; Препаративная органическая химия, пер.
с польск., М., 1959, с. 490. Л. С. Герман.
ВРАЩЕНИЕ ПЛОСКОСТИ ПОЛЯРИЗАЦИИ —
см. Поляриметрия.
ВРЕВСКОГО ЗАКОНЫ — вместе с законами Ко-
новалова лежат в основе термодинамич. теории двой-
ных систем типа раствор — пар; установлены
М. С. Вревским (1911). Вревский впервые доказал,
что нераздельнокипящие растворы (азеотропы) не
являются определенными химич. соединениями, т. к.
их состав изменяется с темп-рой (см. Азеотропные
смеси). Кроме того, он в обобщенном виде решил
665
ВСПОМОГАТЕЛЬНЫЕ ВЕЩЕСТВА — ВСПЫШКИ ТЕМПЕРАТУРА
666
вопрос о влиянии темп-ры испарения на состав пара
р-ров. Основной вывод облечен Вревским в форму
ур-ния:
< (b.+S)-^ + ®
у ' dt RT2
rjifi у — p-Jpz (Pl и />2 — количества первого и соот-
ветственно второго компонента в парах), £j и А2 —
теплоты испарения чистых компонентов; dQ'[di\\ и
dQ” /dN2 — дифференциальные теплоты разведения
р-ра первым и вторым компонентами. Заключенные
в скобки суммы в числителе являются соответственно
теплотами испарения 1 моля первого и второго ком-
понентов из р-ра. Эти последние величины недоступны
непосредственному измерению, в то время как L и
dQ/dN могут быть измерены.
Анализ этого уравнения приводит к формулиров-
кам первого и второго законов Вревского: 1. «При
повышении температуры раствора заданного состава
его пар обогащается тем компонентом, парциальная
молярная теплота испарения к-рого из р-ра больше».
2. «При повышении температуры растворов, кривая
давления пара которых имеет максимум (минимум),
в нераздельнокипящем растворе возрастает относи-
тельное содержание того компонента, испарение ко-
торого требует большей (меньшей) затраты энергии».
Наконец, более подробный анализ приводит к фор-
мулировке третьего закона: 3. «При изменении тем-
пературы (давления) раствора, кривая давления пара
которого имеет максимум, состав пара раствора и
состав нераздельнокипящего раствора изменяются
в одном и том же направлении. При наличии мини-
мума на кривой эти изменения происходят в противо-
положных направлениях». В. с. имеют большое зна-
чение для технология, практики процессов перегонки
и ректификации. См. также Растворы.
Лит.: Вревский М. С., Работы по теории растворов,
М.—Л., 1953. К. П. Мищенко.
ВСПОМОГАТЕЛЬНЫЕ ВЕЩЕСТВА (в тек-
стильной промышленности) — продук-
ты, благоприятно влияющие на обработку природных
или химич. волокон, на произ-во или облагоражива-
ние текстильных материалов. Строение текстильных
В. в. весьма специфично в соответствии с их целевым
назначением, но общие принципы структуры этих
веществ почти одинаковы. В. в. состоят из более или
менее длинной гидрофобной цепи атомов углерода,
к-рая может включать и гетероатомы, напр.
— (СИг)^—О—СН3—, или циклич. группировки ато-
мов: —-СН2— (СН2)Ж— С6Н4—. Гидрофобная цепь за-
канчивается одной или несколькими гидрофильными
группами (ОН, СООН, SO3H, NH2, полиэфирный
остаток и др.), к-рые могут быть также правильно
распределены по всей длине цепи. Гидрофильные
группы могут иметь кислотный или основной характер
и образовывать соли, к-рые в водных р-рах более или
менее полно диссоциируют на ионы. В зависимости
от характера образующихся в водном р-ре ионов
ионогенные в-ва могут быть подразделены на анионо-
активные и на катионоактивные. Примером первых
может служить обычное мыло, вторых — хлористый
цетилпиридиламмоний. Эти соединения распадаются
на ионы соответственно:
R-COONa RCOO- -f- Na+
' _________ +
\ СГ CieH33-N^~\ + СГ
В. в. могут быть также неионогенными. Гидрофиль-
ность их вызывается накоплением гидроксильных,
эфирных или карбамидных групп. Молекулы вспомо-
гательных веществ могут образовывать и амфотерные
ионы (амфолитные В. в.). Так, напр., в водном р-ре
цетилглицина устанавливается след, равновесие:
CieH33-NH-СНа-СООН Ci3H33-NH2-CH2COO-
Наиболыпее применение в пром-сти имеют первые
три группы вспомогательных веществ. Классификация
текстильных вспомогательных веществ приведена
в таблице:
Название
Формула*
1. Анионоактивные
вещества
Мыла (обычные) ..........
Мыла (с модифицированным али-
фатич. радикалом) ......
Алкилсульфаты, соли серно-
кислых эфиров жирных спир-
тов (сульфированные масла,
сульфированные спирты) . .
Алкил-, арил- и арилалнил- )
сульфонаты............ I
Соли высокомолекулярных [
сульфокислот ......... J
Алкилтиосульфаты ........
Алкилфосфаты
Алкилпирофосфаты ........
2. Катионоактивные
вещества
Соли аминов (первичных, вто-
ричных и третичных) ....
Соли аммония четырехзамещен-
ные ....................
Сульфониевые соединения . . .
Фосфониевые соединения . . .
(R—СОО)-Ме+
(R —X—R,—СОО)~Ме+
(R-O-SO,)-Jfe+
(R—SO3)-Me+
(R—S-SO3)-Me+
(R—0—P03)-Me+
(к=о>РзОд2’2Ме+
R—N(RiR3)HAc
[R—N(R!R2R3)]+Ac-
[R-S(R1R2)]+Ac-
[R-P(RiR2R3)]+Ac-
3. Неионогенные
вещества
Вещества, содержащие гидро-
ксильные группы .
Вещества, содержащие эфирные
группы ...................
Вещества, содержащие карба-
мидные группы ............
R-O-Rj-(OH)^
R-COO-Ri-(OH)x
R—CONH—R2—(OH)X и т.д.
R—(О—CH3—CH2)X—ОН
R-NH-(0-CH2—CH2)x-
—ОН и т. Д.
R—CO—NH—Rt—
—(CO NHRa)x-C00 Me
* В этих формулах R — алифатич. радикале 12—18 ато-
мами углерода, R;, R3, R3 — алкильный остаток с 2—4 ато-
мами углерода, X — (— СОО—; —CONH—, —О— и т. Д.):
Me — катион (натрий, калий, аммоний), К(ОН)Л — полиал-
коголь, Ас- — низкомолекулярный анион.
Анионоактивные В. в. находят применение в каче-
стве смачивателей, моющих средств, отделочных и
авиважных препаратов, выравнивателей окрасок и т. д.
Катионоактивные и неионогенные В. в. используются
для закрепления окраски тканей, в качестве выравни-
вателей, мягчителей, пластифицирующих средств и
для специальных видов заключительной отделки
тканей. Подробнее см. Выравнивающие вещества, За-
крепляющие вещества, Моющие вещества.
Лит.: Хвала А., Химические вспомогательные ве-
щества в текстильной промышленности, пер. с нем., М. — Л.,
1948; Ф р о т ш е р Г., Химия и физическая химия текстиль-
ных вспомогательных материалов, пер. с нем., т. 1, М.. 1958,
с. 31; Богословский Б. М. иЛаптев Н. Г., Химия
красителей, М., 1957. П. В. Морыганов, Б. Н. Мельников.
ВСПЫШКИ ТЕМПЕРАТУРА — минимальная тем-
пература жидкости, при к-рой ее пары образуют в за-
крытом сосуде смесь с воздухом, способную воспла-
мениться от внесенного постороннего источника зажи-
гания (искры, открытого пламени, нагретого тела).
Вспышка предварительно нагретой горючей смеси
представляет собой горение вещества путем само-
воспламенения, т. е. без участия постороннего ис-
точника зажигания.
667
ВСПЫШКИ ТЕМПЕРАТУРА — ВТОРОЙ ЗАКОН ТЕРМОДИНАМИКИ
668
В. т. определяется: а) в закрытом тигле (ГОСТ
в стандартном приборе ПВН по ГОСТ
см. рисунок) или
6356 — 52,
1421—53,
Прибор ПВН для определения тем-
пературы вспышки паров жидко-
стей (с электронагревом): 1 — на-
гревательная ванна; 2 внешний
кожух; з — тигель для испытуе-
мой жидкости; 4 — мешалка; <5 —
горелка.
б) в открытом тигле
(ГОСТ 4333—48, в
стандартном аппара-
те по ГОСТ 1369-42).
Темп-pa вспышки не
является постоянной
характеристикой дан-
ного продукта, т. к.
зависит от условий
ее определения, фор-
мы применяемого при
определении сосуда,
высоты уровня зали-
той горючей жидко-
сти, скорости нагре-
вания и т. п. В от-
крытых приборах про-
исходит свободная
диффузия паров ис-
следуемого продукта
в окружающую среду;
поэтому концентра-
ция паров в возду-
хе (при одинаковых
условиях нагревания)
меньше, а В. т. вы-
ше, чем в закрытых
приборах (обычно на
20—25°).
Прибор ПВН, предназначенный для определения В. т.
паров нефтепродуктов в интервале от 20 до 275°, может быть
использован для любой горючей жидкости. Аппарат по
ГОСТ 1369—42, предназначенный для определения темп-ры
вспышки и воспламенения масел и темных нефтепродуктов,
не рекомендуется использовать при определении В. т. для
высококипящих индивидуальных горючих жидкостей или
различных технич. смесей, поскольку данные В. т., получен-
ные в открытом тигле, имеют завышенные значения по срав-
нению с полученными в приборе с закрытым тиглем; разница
достигает нескольких десятков градусов.
В. т., определяемая в закрытом стандартном при-
боре, лежит в основе классификации жидкостей по
степени пожарной опасности. При этой темп-ре пары
горючего образуют взрывоопасные концентрации с
воздухом. При темп-ре Жидкости ниже В. т. пары ее
по загораются. Так называемая темп-pa воспламене-
ния при оценке пожарной опасности жидкостей
в расчет не принимается, поскольку она ле харак-
терна. В зарубежной практике зачастую В. т. име-
нуется «точкой воспламенения».
Для индивидуальных жидкостей В. т. при постоян-
ном давлении является величиной постоянной, ха-
рактеризующей взрывоопасность паров данной жид-
кости. Еще более точной характеристикой пожарной
опасности для жидкостей является нижний темпера-
турный предел взрываемости насыщенных паров
в воздухе, к-рый определяется в спец, герметичном
приборе.
В. т. у наиболее низкокипящих углеводородов и
бензинов минус 30—40° и ниже; В. т. керосиновых
фракций 28—60°, масляных 130—325°.
В таблице приведены значения темп-p вспышки и
нижних температурных пределов взрываемости для
нек-рых горючих жидкостей (стр. 668).
Приближенно В. т. для индивидуальных веществ
может быть рассчитана, исходя из следующей зависи-
мости: 7ВСП. = 7КИП. • К, где Гвсп иТкип - темп-ра
вспышки и темп-ра кипения в градусах абсолютной
шкалы; К — постоянная величина (колеблется в пре-
делах 0,720—0,782; практически можно принять
К ~ 0,736). В. т. может быть вычислена также по
ф-ле Торнтона, по к-рой сначала определяется давле-
ние паров (Рг) данной жидкости при В. т., с после-
Горючие жидкости Темп-ра вспышки по ГОСТ 1421 —53, °C Нижний темпера- турный предел взрываемости на- сыщенных паров в воздухе, ’С
Индивидуальные вещества п-Ксилол 26 24
н-Бутиловый спирт ....... 34 31
Метиловый спирт ........ 8 7
Этиловый спирт 13 И
Фурфурол 61 60
Хлорбензол 26 25
Циклогексанол 61 58
Этилен хлористый (дихлорэтан- -1,2) 9 8
Нефтепродукты Гидравлическая жидкость АМГ-10 92 80
Масло авиационное МК-22 . . . 259 228
Масло В М-4 212 177
Масло индустриальное «50» (машинное СУ) 200* 146
Масло соляровое 142* 116
Масло турбинное «57» ..... 193* 148
Смазка 59-Ц (предохранитель- ная СП-3) 146* 111
Топливо для быстроходных ди- зелей, специальное ДС .... 92 76
Топливо Т-1 (выщелоченное). . 31 27
• Определен а в открытом тигле.
дующим определением (по таблицам) соответствующей
темп-ры: = Робщ /[1 +4,76(А-1)], где Робщ -
давление смеси паров с воздухом в мм рт.ст.,
N — число грамм-атомов кислорода, участвующее
в сгорании одного моля горючей жидкости. Если
имеются точные данные по нижнему пределу взры-
ваемости паров индивидуальной жидкости, то еще
более точно В. т. (или нижний температурный предел
взрываемости паров в воздухе) может быть вычис-
лена из общего выражения концентрации (в %
по объему): Соб = Pt Ю0/Робш, откуда Pt =
= Соб Вобщ /100 (ммрт.ст.'). По найденному Pt опре-
деляется (по таблицам) соответствующая этому давле-
нию темп-ра, которая и будет В. т.
Лит.: Тидеман Б. Г., Сциборский Д. Б.,
Химия горения, М., 1940; Андреев К. К., Термическое
разложение, и горение взрывчатых веществ, М., 1957; Легко-
воспламеняющиеся и горючие жидкости. Справочник, М.,
1956; Наметкин С. С., Химия нефти, 3 изд., М., 1955;
Нефтепродукты и продукты переработки твердых топлив.
Технические условия, [на 1 ноября 1958 г.], М., 1958.
М. Г. Годжелло.
ВТОРОЙ ЗАКОН ТЕРМОДИНАМИКИ — прин-
цип, устанавливающий необратимость макроскопия,
процессов, протекающих с конечной скоростью. В от-
личие от чисто механических (без трения) или электро,-
динамических (без выделения джоулева тепла) обра-
тимых процессов, процессы, связанные с теплообме-
ном при конечной разности темп-p (т. е. текущие
с конечной скоростью), с трением, диффузией газов,
расширением газов в пустоту, выделением джоулева
тепла и т. д., — необратимы, т. е. могут самопроиз-
вольно протекать только в одном направлении.
Существует несколько эквивалентных формулиро-
вок В. з. т. Впервые его сформулировал Р. Клаузиус:
невозможен процесс, при к-ром тепло переходило бы
самопроизвольно от тел более холодных к телам более
нагретым. При этом самопроизвольный переход не
следует понимать в узком смысле непосредственного
перехода; переход невозможно осуществить с помощью
к.-л, машин или приборов без того, чтобы в природе
не произошло еще к.-л. изменений, т. е. невозможно
провести процесс, единственным следствием к-рого
был бы переход тепла от одного тела к другому, более
нагретому. Если бы (в нарушение положения Клау-
зиуса) такой процесс оказался возможным, то можно
669
второй закон термодинамики
670
было бы, разделив один тепловой резервуар на 2 части
и переводя тепло из одной в другую, получить 2 резер-
вуара различных темп-p. Это позволило бы, в свою
очередь, осуществить Карно цикл и получить механич.
работу с помощью периодически действующей (т. е.
многократно возвращающейся к исходному состоя-
нию) машины за счет внутренней энергии, в конечном
итоге за счет одного теплового резервуара. Поскольку
это невозможно, в природе невозможны процессы,
единственным следствием к-рых был бы подъем груза
(т. е. механич. работа), произведенный за счет охла-
ждения теплового резервуара (В. з. т. в формулировке
У. Томсона). Обратно, если бы можно было получить
механич. работу за счет внутренней энергии одного
теплового резервуара (в противоречии с В. з. т., по
Томсону), то можно было бы нарушить и положение
Клаузиуса. Механич. работу, полученную за счет
тепла более холодного резервуара, можно было бы
использовать для нагревания более теплого резер-
вуара (трением). Т. о., обе формулировки В. з. т.
эквивалентны.
Возможность использовать внутреннюю энергию
окружающих тел для получения механич. работы
означала бы возможность реализации практически
вечного двигателя, т. н. вечного двигателя 2-го рода,
работа к-рого не противоречила бы закону сохранения
энергии (напр., работа двигателя корабля за счет
охлаждения забортной воды океана — доступного
резервуара неисчерпаемой внутренней энергии).
В. з. т. можно, следовательно, формулировать так:
создание вечного двигателя 2-го рода невозможно.
Г. А. Зисман.
В современной термодинамике В. з. т. формули-
руется единым и самым общим образом как закон
возрастания энтропии. Согласно этому закону, в зам-
кнутой системе изменение энтропии 65 при любом
реальном процессе удовлетворяет неравенству 65>:
знак равенства имеет место для обратимых про-
цессов. В состоянии равновесия энтропия замкнутой
системы достигает максимума и никакие макроскопия,
процессы в такой системе, согласно В. з. т., невоз-
можны. Для незамкнутой системы направление воз-
можных процессов, а также условия равновесия могут
быть получены из закона возрастания энтропии,
примененного к составной замкнутой системе, получае-
мой путем присоединения всех тел, участвующих
в процессе. Это приводит в общем случае необратимых
процессов к неравенствам:
6Q==CT55 (1)
6Z7 — 765 — 6Л ==С О (I1)
где 6Q — переданное системе тепло, 6Л — совер-
шенная над ней работа, 6Z7 — изменение ее внутрен-
ней энергии, Т — абсолютная темп-ра; знак равен-
ства имеет место для обратимых процессов.
Важные следствия дает применение В. з. т. к систе-
мам, находящимся в фиксированных внешних усло-
виях. Напр., для систем с фиксированной темп-рой
и объемом неравенство (!') приобретает вид: 67 «''О,
где F = U — TS -— свободная энергия системы.
Т. о., в этих условиях направление реальных процес-
сов определяется убыванием свободной энергии,
а состояние равновесия — минимумом этой величины
(см. Термодинамические потенциалы). Легко убе-
диться, что приведенные в начале статьи формули-
ровки В. з. т. являются частным следствием общего
закона возрастания энтропии.
В. з. т., несмотря на свою общность, не имеет абсо-
лютного характера, и отклонения от него (флуктуа-
ции) являются вполне закономерными. Примером
таких флуктуационных процессов являются броунов-
ское движение тяжелых частиц, равновесное тепловое
излучение нагретых тел, возникновение зародышей
новой фазы при фазовых превращениях, самопроиз-
вольные флуктуации темп-ры и давления в равновес-
ной системе и т. д.
Статистическая физика, построенная на анализе
микроскопия, механизма явлений, происходящих
в макросистемах, и выяснившая физич. сущность
энтропии, позволила понять природу В. з. т., опреде-
лить пределы его применимости и устранить кажу-
щееся противоречие между механич. обратимостью
любого, сколь угодно сложного микроскопия, процесса
и термодинамич. необратимостью процессов в макро-
телах.
Как показывает статистическая термодинамика
(Л. Больцман, Дж. Гиббс), энтропия системы связана
со статистич. весом макроскопия, состояния Р : 5 —
— klnP (к — постоянная Больцмана); Р пропорцио-
нально числу различных микроскопия, реализаций
данного макросостояния и характеризует как бы сте-
пень его «размытости». Для замкнутой системы термо-
динамич. вероятность W макросостояния пропорцио-
нальна его статистич. весу и определяется энтропией
системы:
W «5 ехр (5//с) (2)
Т. о., закон возрастания энтропии имеет статисти-
чески-вероятностный характер и выражает постоян-
ную тенденцию системы к переходу в более вероятное
состояние. Состояние равновесия является максималь-
но вероятным, и за достаточно большой промежуток
времени любая замкнутая система достигнет этого
состояния и будет оставаться в нем подавляющую
часть всего времени. .
Энтропия является величиной аддитивной и пропор-
циональна числу частиц в системе. Поэтому для систем
с большим числом частиц даже самое ничтожное от-
носительное изменение энтропии существенно меняет
ее абсолютную величину; изменение же энтропии,
стоящей в показателе экспоненты в ур-нии (2), приво-
дит к изменению вероятности W в огромное число раз.
Именно этот факт является причиной того, что для
системы с большим числом частиц вероятностная
природа В. з. т. не проявляется и его следствия прак-
тически имеют характер достоверности. Крайне мало-
вероятные процессы, сопровождающиеся сколько-
нибудь заметным уменьшением энтропии, требуют
столь огромных времен ожидания, что их реализация
является практически невозможной. В то же время
малые части системы, содержащие небольшое число
частиц, испытывают непрерывные флуктуации, сопро-
вождающиеся лишь небольшим абсолютным измене-
нием энтропии. Средние размеры этих флуктуаций
являются таким же достоверным следствием статистич.
термодинамики, как и сам В. з. т.
Проиллюстрируем сказанное примером, позволяющим
оценить масштабы величин, определяющих точность В. з. т.
и отклонения от йего. Рассмотрим флуктуационный процесс,
в результате к-рого N частиц, первоначально занимающих
объем V, равный 1 ц.3 (т. е. 10~12 см3), сконцентрируется само-
произвольно в половине этого объема. Отношение статистич.
весов начального (1) и конечного (2) состояний;
Р., /Р, = v-W/fV/2)A’ = 2JV
поэтому изменение энтропии A (S/k) = NlnZ и отношение
вероятностей W2/Wt = 2^. Если время пролета частицы через
объем V, т. е. время, в течение к-рого сохраняется данная
флуктуация, х = 10*8 сек, то среднее время ожидания такой
флуктуации t 2n • х s» 10°>3 N . х. При N = 30 t ~ 10 сек;
при N = 100 t =» 1022 сек 1016 лет. Если же учесть, что при
атм. давлении число частиц в 1 ц3 составляет ^108, то время
ожидания i 10^’ лет.
Буквальное применение В. з. т. к Вселенной как
целому, приведшее Клаузиуса к неправильному вы-
воду о неизбежности «тепловой смерти Вселенной»,
является неправомерным, т. к. любая сколь угодно
большая часть Вселенной не является сама по себе
замкнутой и ее приближение к состоянию теплового
671
ВУЛКАНИЗАЦИИ ЗАМЕДЛИТЕЛИ — ВУЛКАНИЗАЦИИ УСКОРИТЕЛИ
672
равновесия, даже не говоря о флуктуациях, не яв-
ляется абсолютным.
Лит.: Френке л ьЯ.И., Статистическая физика, М.—Л.,
1948; Планк М., Введение в теоретическую физику, ч. 5,
2 изд., М.—Л., 1935; Зоммер фельд А., Термодинамика
и статистическая физика, пер. с нем., М., 1955; Л е в и ч В. Г.,
Введение в статистическую физику, 2 изд., М., 1954; Ше-
фер К., Теория теплоты, пер. с нем., ч. 1—2, М.—Л., 1933;
Леонтович М. А., Введение в термодинамику, 2 изд.,
М.—Л., 1952; Л а н д а у Л. и Л и ф ш и ц Е., Статистическая
физика, М.—Л., 1951 (Теоретическая физика, т. 4); С а м о й-
лович А. Г., Термодинамика и статистическая физика, М.,
1953. И. М. Лифшиц.
ВУЛКАНИЗАЦИИ ЗАМЕДЛИТЕЛИ (антискор-
чинги) — химич. соединения, вводимые в резиновую
смесь для предотвращения преждевременной вулка-
низации в процессе ее обработки (смешении, калан-
дровании, шприцевании и т. д.). В. з. должны ока-
зывать минимальное влияние на кинетику вулкани-
зации при темп-pax ок. 140°. В качестве замедлителей
вулканизации применяются: 1) органич. к-ты (бен-
зойная, фталевая, себациновая и др.), их ангидриды
и соли — бензоаты (кадмия, алюминия, свинца, ципка
и др.); 2) нитропроизводные различных соединений
(нитробензойная к-та, 2-4-динитрофенол, динитро-4-
гидроксидифенил; нитроспирты; нитрофенилсульфон-
амид); 3) нитрозосоединения (N-нитрозодифениламин
п др. нитрозамины); 4) хлорсодержащие соединения
(хлорбензол, хлорированные хиноны, о-хлорбензой-
ная к-та и ее соли, хлорированные парафины, содер-
жащие 40—70% С1); 5) альдегиды и продукты их
конденсации (бензальдегид, фурфурол, продукты кон-
денсации масляного альдегида с анилином); 6) окиси
металлов (РЬО, РЬО2, ZnO2, CaO, CdO) и ряд др.
соединений. Мягчители для каучуков — сосновая
смола, стеариновая к-та (в больших концентрациях),
канифоль, полидиены и др. также замедляют вулка-
низацию.
Впервые в качестве В. з. начали применять органич.
к-ты. Однако эти соединения плохо диспергируются
в резиновой смеси и значительно снижают скорость
вулканизации при высоких темп-pax. Для улучшения
диспергирования к-ты применяются в смеси с окисью
цинка или в связанном виде с в-вами основного ха-
рактера, напр. с дифенилгуанидином. Замедление
вулканизации при высоких темп-pax (> 130°) умень-
шается в случае применения к-т, к-рые разлагаются
при темп-ре вулканизации на нейтральные или менее
кислые продукты (напр., малоновая к-та). В. з. об-
ладают избирательным действием в зависимости от
ускорительной группы, типа каучука, сажи и др.
факторов. Напр., СаО является замедлителем вул-
канизациии для смесей с тетраметилтиураммоносуль-
фидом в качестве ускорителя, но в смесях с меркапто-
бензотиазолом, ди-о-толилгуанидином и альдегидами-
нами даже несколько увеличивает скорость подвул-
канизации смесей. Наиболее распространенными за-
медлителями вулканизации являются N-нитрозоди-
фениламин и фталевый ангидрид. о. Н. Беляцкая.
ВУЛКАНИЗАЦИИ УСКОРИТЕЛИ — химич. со-
единения, вводимые в резиновую смесь для ускорения
процесса вулканизации и улучшения физико-механич.
свойств вулканизатов. Первоначально применялись
в основном неорганич. ускорители: едкий натр,
сода, окиси магния, свипца и др.; паилучшими из
них считали глет и жженую магнезию. Неорганич.
ускорители по эффекту своего действия заметно усту-
пают органическим, к-рые не только значительно
увеличивают скорость вулканизации, но и уменьшают
необходимую дозировку серы, влияют на структуру
образующегося вулканизата, способствуют возник-
новению Желательных типов связей между молеку-
лами каучука.
Важнейшие органич. В. у. можно разделить на
след, группы: 1) Альдегида мины — продукты
конденсации алифатич. альдегидов с ароматич. или
алифатич. аминами. Большинство ускорителей этого
типа являются продуктами конденсации анилина и
его гомологов с формальдегидом, масляным, уксус-
ным, кротоновым и др. альдегидами в различных
соотношениях. Напр., ускоритель К-1 — продукт
конденсации 2 молекул анилина с 3 молекулами ацет-
альдегида. Ускорители этой группы обеспечивают
получение теплостойких резин. 2) Гуанидины
R-NH-C-NHR
NH ’ Один из самЬ1Х распространенных
ускорителей вулканизации — дифенилгуанидин,
к-рый обычно применяется в комбинации с др. уско-
рителями, а также ди-о-толилгуанидин и трифенил-
гуанидин. 3) Тиазолы и продукты их окисления
(дисульфиды) — наиболее распространенные ускори-
тели вулканизации. К ним относятся 2-меркапто-
бензотиазол (каптакс) (I) и 2,2-дибензотиазолилди-
сульфид (альтакс) (II). 4) Дитиокарбаматы
R. if Л
r/N—с sh • hn\r (диэтилдитиокарбамат , диэтил-
амина, диметилдитиокарбамат диметиламина, дибу-
тилдитиокарбамат цинка, диметилдитиокарбамат цин-
ка й др.). Дитиокарбаматы относятся к классу уль-
траускорителей; применяются для клеев, латексных
смесей, вулканизации тонкостенных резиновых изде-
R\ /R
лий. 5) Тиурамы >N—с—S—S—С—. Наи-
' " г Ш II II XR
S S
более распространенными ускорителями этого класса
являются тетраметилтиурамдисульфид и тетраэтил-
тиурамдисульфид. Тиурамы могут оказывать само-
стоятельное вулканизующее действие на каучук
(в отсутствии серы), дают резины с высоким сопро-
тивлением тепловому старению. Наибольшую вул-
канизующую активность тиурамы проявляют в при-
сутствии окиси цинка. Кроме дисульфидов, в качестве
ускорителей применяются моно- и полисульфиды
R-OC-SMe
тиурамов. 6) Кс антогенаты ц . Наи-
большее применение получил бутилксантогенат цинка.
Ксантогенаты являются ультраускорителями и ис-
пользуются гл. обр. для самовулканизующихся
клеев. 7) С у л ь ф е н а м и д ы. Широкое распро-
странение получили Х,Х-диэтил-2-бензотиазилсуль-
фенамид (сульфенамид ВТ, вулкацит AZ) (III) и
и К-цикло-гексил-2-бензотиазилсульфенамид (санто-
кюр) (IV). В отличие от других ускорителей, сульфен-
Ш
и
амиды обладают замедленным действием на началь-
ных стадиях вулканизации, что предохраняет рези-
новые смеси от преждевременной вулканизации при
обработке и сообщает смесям ряд других ценных
свойств (повышение прочности связи дублированных
резин, хорошее растекание в формах и др.).
Ускорители могут применяться самостоятельно или
в комбинации друг с другом. Многие ускорители
в процессе вулканизации взаимно активируются,
благодаря чему значительно повышается скорость
вулканизации и изменяются физико-механич. свой-
ства вулканизатов. Химич, природа ускорителя ока-
зывает сильное влияние на характер поперечных
связей, образующихся между молекулами каучука
673
ВУЛКАНИЗАЦИЯ
674
при вулканизации, что в значительной степени опре-
деляет физич. и механич. свойства вулканизатов.
Тиурамы, сульфенамиды и 2,2-дибензотиазолилди-
сульфид развивают полимеризац ионные процессы при
вулканизации и способствуют образованию наиболее
прочных С—С и С—S—С связей (наряду с другими
типами связей); в присутствии дифенилгуанидина
образуются гл. обр. полисульфидные связи (наименее
термически стойкие). Тип введенного в смесь ускори-
теля оказывает значительное влияние на старение
резин: напр., в присутствии дифенилгуанидина на-
блюдается ускорение старения, никелевые соли ди-
бутилдитиокарбаминовой к-ты вызывают резкое за-
медление процесса старения. Согласно современным
представлениям, ускорители взаимодействуют с серой
и каучуком через стадию образования свободных ра-
дикалов или по ионному механизму, благодаря чему
снижается энергия активации реакции каучука с се-
рой от 30—32 до 15 —25 ккал/молъ и увеличивается
скорость процесса вулканизации. Ускорители пре-
терпевают сложные химич. превращения и расхо-
дуются в процессе вулканизации.
Лит.: Догадкин Б. А., Химия и физика каучука,
М., 1947; Кошелев Ф. Ф., Климов Н. С., Общая
технология резины, М., 1958. О. Н. Беляцкая.
ВУЛКАНИЗАЦИЯ — технология, процесс резино-
вого произ-ва, при к-ром пластичный сырой каучук
превращается в эластичную резину — материал, обла-
дающий лучшими, чем каучук, физико-механич. и
эксплуатационными свойствами. В большинстве слу-
чаев В. каучуков общего назначения (натуральный,
бутадиеновый, бутадиен-стирольный) производится
серой или какими-либо другими химич. агентами,
к-рые образуют химич. связи между молекулами
каучука. В результате такого процесса образуется
пространственная молекулярная сетка со специфич.
свойствами вулканизата — наличием конечного зна-
чения модуля эластичности и неспособностью к само-
произвольному растворению в обычных растворителях
сырого каучука. В. может быть ускорена добавлением
небольших количеств органич. соединений ускори-
телей В. (см. Вулканизации ускорители). Многие
ускорители являются эффективными только в присут-
ствии активаторов — окислов металлов (напр., окиси
цинка), действие к-рых проявляется в присутствии
жирных к-т, образующих с окислами металлов соли,
растворимые в каучуке. Таким образом, в состав вул-
канизующей группы обычно входит сера, ускоритель,
активатор и к-та жирного ряда. Для предотвращения
преждевременной В. в резиновую смесь вводят вул-
канизации замедлители. При термич. разложении
вулканизующего агента или ускорителя, а также
в результате реакций между ускорителями и серой
образуются свободные радикалы, к-рые или присо-
единяются к двойным связям каучука, или отнимают
атом водорода от а-метиленовой группы углеводород-
ной цепи полимера. Свободный полимерный радикал
взаимодействует с двойной связью соседней полимер-
ной цепи, что приводит, т. о., к развитию полиме'риза-
ционной цепи, длина к-рой обычно мала. Свободный
полимерный радикал может также взаимодействовать
с другими радикалами и атомными группировками
с образованием поперечных химич. связей между мо-
лекулами каучука. В зависимости от типа полимера
и особенно от состава вулканизующей группы при
В. образуются поперечные связи различного характе-
ра —С—С—; —С—S—С—; —С—Sn—С—. Состав, кон-
центрация, распределение и энергия этих связей опре-
деляют многие важнейшие физико-механич. свойства
вулканизатов. Так, если возникают преимуществен-
но устойчивые поперечные связи (бессерная В.,
термовулканизация, радиационная В.), то это приво-
дит к образованию резин, обладающих высокой стой-
Время вулканизации
Изменение физико-меха-
нических свойств нату-
рального каучука в про-
цессе вулканизации: 1—
сопротивление разрыву;
2 — относительное удли-
нение; з — набухание;
4 — эластичность; з —
твердость.
резин из структури-
костью к различным видам старения, термомеханич.
воздействиям и утомлению. Наличие неустойчивых
поперечных связей приводит к получению резин
с низкой термостойкостью. Наряду с поперечными
химич. связями в определении физико-химич. и тех-
нологич. свойств вулканизата существенное значение
имеет взаимодействие за счет водородных, ориента-
ционных и др. видов межмолекулярных связей. При
В. нек-рые свойства вулканизуемой смеси изменяются
со временем не монотонно, а проходят через максимум
или минимум (см. рис.). Эти точки характеризуют
оптимум В., т. е. такую сте-
пень В., при к-рой достигается
наилучшее сочетание физико-
механич. свойств резин. Явле-
ние оптимума В. связано с тем,
что при В. одновременно на-
блюдается процесс соедине-
ния и деструкции молекуляр-
ных цепей каучука. Процесс
деструкции при В. связан с
термоокислительным и термич.
распадом молекулярных цепей
каучука и перегруппировкой
поперечных серных связей.
Критерием степени В. для
резин из деструктирующихся
каучуков (натуральный, цис-
полиизопрен) могут служить
набухаемость ц модуль эла-
стичности — величины, функ-
ционально связанные с числом
поперечных химич. связей ме-
жду молекулами каучука. Для
рующихся каучуков (бутадиен-стирольный, натрий-
бутадиеновый) могут быть использованы показате-
ли остаточной деформации и сопротивления раз-
диранию резины.
В большинстве случаев технич. В. заключается
в том, что резиновые смеси, содержащие элементар-
ную серу, а также в-ва, обеспечивающие необходимые
эксплуатационные качества вулканизата (усили-
тели типа сажи, наполнители типа мела, каолина;
противостарители, мягчители и т. д.), подвергают
нагреванию при 130—160°. Если к каучуку присоеди-
няется 0,5—5% серы, то образуется мягкий вулкани-
зат (автомобильные камеры и покрышки, мячи,
трубки и т. д.). Присоединение к каучуку 30—50%
серы приводит к образованию жесткого неэластичного
материала (эбонит). В. каучука серой носит название
горячей В.; она открыта в 1839 Ч. Гудьиром
(США) и в 1843 Т. Гэнкоком (Англия). Каучук может
быть свулканизован и без нагревания. В 1846 А. Паркс
предложил процесс холодной В. с помощью
хлористой серы (S2C12). Известно большое число со-
единений, к-рые не содержат серу и могут вызывать
вулканизацию. Эти соединения делятся на 2 группы:
окислительные агенты (молекулярный кислород, орга-
нич. и неорганич. перекиси, нитросоединения) и
соединения, распадающиеся с образованием свобод-
ных радикалов (органич. перекиси, поли- и дисуль-
фиды, диазосоединения, сульфенамиды и др.). Уста-
новлена также возможность В. без нагревания и без
вулканизующих агентов под действием ядерных излу-
чений (см. Вулканизация радиационная), а также под
действием только нагревания при темп-ре ок. 200° —
термовулканизация.
Нек-рые каучуки, напр. фторкаучуки, силиконо-
вые, полиуретановые и др., не содержат двойных
связей и поэтому не могут вулканизоваться в обыч-
ных условиях с помощью серы. Эти каучуки вулкани-
зуются органич. перекисями, диазосоединениями,
полиизоцианатами, полиаминами и др. Вулканизую-
675
ВУЛКАНИЗАЦИЯ РАДИАЦИОННАЯ
676
щее действие указанных соединений основано на их
способности реагировать с макромолекулярными це-
пями полимеров с образованием межмолекулярных
поперечных связей. Примером может служить В. фтор-
каучука перекисью дикумила, к-рая распадается на
свободные радикалы:
сн3 сн, сн,
II I
СвН3-С-О-О-С-СеН5 2С6Н5- С О-
1'1 I
СНз СНз СНз
Последние реагируют с фторполимерами с отрывом
атомов водорода и образованием свободных радикалов
фторполимеров:
н сн3
I I
—CF*>—С—CFa— -j- СеН5-С-О' —
I I
н сн„
Н СНз
I I
CFs-C-CF2 + CeH6-C-OH
СНз
При взаимодействии этих радикалов образуются
поперечные связи между полимерными цепями:
н
I
2-CF2-C-CFa---►
Н
I
-CF.-C-CFa-
I
-CF2-C-CF2-
I
Н
В технике В. производят нагреванием вулканизуемого
изделия в специальных формах под повышенным давлением
или же в виде неформовых изделий (в свободном виде); на-
гревают в котлах, прессах, автоклавах, индивидуальных вул-
канизаторах, аппаратах для непрерывной В. и др. В этих
аппаратах нагревание производится паром, воздухом, пере-
гретой водой, электричеством, токами высокой частоты. Хо-
лодная В. производится погружением изделия при комнатной
темп-ре в р-р S2C12 в сероуглероде или помещением в камеру,
наполненную парами S2C12. Избыток непрореагировавшей
S2C12 удаляется р-ром соды или обдувкой аммиаком. Холод-
ная В. применяется для тонкостенных изделий. Холодной В.
пользуются крайне редко, гл. обр. для неответственпых из-
делий.
Использование в технологии резины большого
числа новых синтетич. полимеров побуждает к раз-
работке новых приемов и способов В. и технич.
оформления процесса.
Лит.: Г а у з е р Э., Технология резины, пер. с нем.,
т. 1—2, М., 1936—37; Д о г а д н п и Б. А., Химия и физика
каучука, М., 1947; Синтетический каучук. Гл. ред. Г. С. Уитбп,
пер. с англ., Л., 1957; Кошелев Ф. Ф., Климов Н. С.,
Общая технология резины, М., 1958; В 1 1 1 m е у е г F. W.,
Textbook of polymer chemistry, N. Y. — L., 1957; Д о г а Д-
к и н Б. [и др.], Коллоидн. ж., 1955, 17, № 3 с. 215; Д 0-
гадкин Б. иБениска И., там же, 1956, 18, № 2,
с. 167. Б. А. Догадкин, 3. Н. Тарасова.
ВУЛКАНИЗАЦИЯ РАДИАЦИОННАЯ (сшивание
радиационное) — превращение линейных полимеров
в трехмерные сетчатые под действием ионизирующих
излучений. В. р. наблюдается в тех случаях, когда
из двух одновременно протекающих конкурирующих
радиационно-химич. реакций — реакции образования
поперечных химич. связей между цепными макромо-
лекулами и реакции расщепления главных цепей
макромолекул — преобладает первая. В результате
В. р. полимер становится нерастворимым и теряет
способность переходить при нагревании в вязко-
текучее состояние; одновременно изменяется весь
комплекс физико-механич. свойств полимера, подобно
тому как это имеет место и при обычной химич. вул-
канизации. В. р. могут подвергаться как карбоцеп-
ные, так и гетероцепные полимеры, в том числе и
элементоорганические. В. р. не подвергаются карбо-
цепные полимеры, у к-рых в цепи содержатся четырех-
замещенные атомы углерода; в этом случае преоб-
ладают реакции расщепления связей в главных цепях
макромолекул, в результате чего эти полимеры под-
вергаются деструкции (см. Деструкция полимеров).
Механизм В. р. нельзя считать полностью выяснен-
ным. Поперечные связи возникают в результате ре-
комбинации, образующихся под действием излучения
полимерных радикалов. Однако определенную роль
в этом процессе могут играть также ионно-молеку-
лярные реакции и. реакции взаимодействия радика-
лов с функциональными группами с двойными
связями и с возбужденными молекулами. Во всех
случаях В. р. сопровождается газовыделением, об-
разованием или уменьшением числа двойных связей,
а в случае кристаллич. полимеров — уменьшением
их кристалличности.
В. р. приобретает практич. значение для вулкани-
зации таких полимеров, к-рые другими способами
вообще не вулканизуются (напр., полиэтилен), а также
для проведения процесса при обычных темп-рах и
давлениях; во многих случаях радиационные вулкани-
заты имеют улучшенные технич. свойства. Несмотря
на нецепной характер процесса и небольшие радиа-
ционно-химич. выходы (не более нескольких попе-
речных связей на 100 эв поглощенной энергии излуче-
ния), для проведения В. р. требуются сравнительно
небольшие дозы (10—100 Мфэр в зависимости от
природы и исходного мол. веса полимера). Для В. р.
используют гл. обр. у-излучение Со60, смешанное
излучение ядерного реактора и ускоренные элек-
троны. Наиболее подробно исследована В. р. поли-
этилена, осуществленная в пром-сти. Полиэтилен,
подвергнутый В. р., при нагревании его выше темп-ры
плавления переходит не в вязко-текучее состояние,
как невулканизованный полимер, а в высокоэласти-
ческое (резиноподобное). В связи с этим значительно
повышается темп-рный предел его применения во
многих случаях эксплуатации. В. р. существенно
повышает стойкость полиэтилена к действию окисли-
телей и конц. кислот при повышенных темп-рах.
При В. р. сравнительно низкомолекулярного поли-
этилена наблюдается улучшение механич. характери-
стик: несколько повышается разрывная прочность,
увеличивается разрывное удлинение. В. р. полиэти-
лена проводят в листах и пленках, а иногда — непо-
средственно в изделиях (в частности, кабельных).
В. р. листов полиэтилена используют также для
упрощения технологии их формования при повышен-
ных темп-рах в вакууме, т. к. в этом случае не тре-
буется строгого контроля темп-ры, необходимого
при формовании невулканизованных листов. При
В. р. в присутствии воздуха скорость процесса сни-
жается, особенно в случае облучения тонких пленок.
В. р. силиконовых каучуков позволяет получать
вулканизаты, к-рые по сравнению с перекисными
вулканизатами обладают большей устойчивостью
в условиях повышенных темп-p и лучшими диэлек-
трич. свойствами. Кроме того, в результате В. р.
силиконовые каучуки теряют способность при низких
темп-рах кристаллизоваться, что повышает их морозо-
стойкость. Натуральный и синтетич. карбоцепные
каучуки (за исключением бутилкаучука) могут быть
подвергнуты В. р. при обычной темп-ре, причем ско-
рость процесса в значительной степени зависит от
природы каучуков. Скорость В. р. увеличивается
в ряду СКИ, СКС-30, НК, СКБ-40, СКН-26 (см.
Каучук синтетический). Сажевый наполнитель уве-
личивает скорость В. р., причем между частицами
сажи и полимерными молекулами каучука возникают
химич. связи. Радиационные вулканизаты значи-
тельно превосходят серные вулканизаты по сопро-
тивлению к термоокислительному старению, по вы-
носливости к многократным деформациям, по тепло-
стойкости. Шины, вулканизованные радиационным
методом, обладают повышенными эксплуатационными
характеристиками. Развитие промышленных методов
В. р. полимеров является важной частью общей про-
677
ВЫПАРИВАНИЕ
678
блемы использования энергии ядерных излучений
в химич. пром-сти.
Лит.: Действие ионизирующих излучений на неоргани-
ческие и органические системы. [Сборник], М., 1958, с. 325;
Труды I Всесоюзного совещания по радиационной химии
(25—30 марта 1957 г.), М., 1957; Никитина Т. С., Жу-
равская Е. В., Кузьминский А. С., Действие
ионизирующих излучений на полимеры, М., 1959; Б о в е й
Ф. А., Действие ионизирующих излучений на природные и
синтетические полимеры, пер. с англ., М., 1959; Шапиро А.,
Ионизирующие излучения и макромолекулярные вещества,
[пер. с англ.], Химия и технология полимеров. Сб. пер. из
пн. периодич. лит-ры, 1958, № 2, с. 3. Б. Л. Цетлин.
ВЫПАРИВАНИЕ — процесс концентрирования
р-ров твердых веществ в жидких растворителях путем
частичного перевода последних в парообразное со-
стояние при кипении. В подавляющем большинстве
случаев В. подвергают водные р-ры; удаляемый паро-
образный растворитель — вторичный пар. Особен-
ностью процесса В. является переход в парообразное
состояние только растворителя. Темп-ра кипения
р-ров всегда выше темп-ры кипения растворителей;
она зависит от химич. природы растворенных веществ
и растворителей и растет с увеличением конц. р-ров
и внешнего давления. Разность между темп-рами
кипения р-ра и чистого растворителя при одинаковом
внешнем давлении называется температурной
депре'ссией (б). Последняя не поддается теоре-
тич. расчету и определяется экспериментально, дости-
гая часто значительной величины и возрастая с уве-
личением концентрации р-ра и внешнего давления.
Напр., при нормальном давлении 60%-ный водный р-р
сахара кипит при 103° (б = 3°), 50%-ный р-р NaOH —
при 142,2° (б = 42,2°), 60%-ный р-р КОН — при
178,8° (6=78,8°), 75%-ный р-р NaOH — при 192°
(6 = 92°), а 80%-ный р-р КОН — при 290,5° (6 =
— 190,5°).
Выпаривание р-ров сопровождается, как правило,
ростом их плотности и вязкости; теплоемкость и
коэфф, теплопередачи при этом падают. В. произво-
дится за счет подводимого извне тепла, передаваемого
р-рам чаще всего через поверхность нагрева, реже —
путем непосредственного контакта с теплоносителем.
При темп-рах кипения 200° в качестве теплоноси-
теля используется конденсирующийся водяной пар,
а при более высоких темп-рах — высококипящие
жидкости и их насыщенные пары, а также топочные
газы.
При В. малых количеств р-ров в качестве выпарных
аппаратов используются вертикальные и горизон-
тальные цилиндрич. котлы, снабженные нагреватель-
ными рубашками и змеевиками для парового и жид-
костного обогрева или топками для газового обогрева.
В средне- и мпоготопнажных произ-вах преим. при-
меняются след, выпарные аппараты с паровым обо-
гревом: с внутренними нагревательными камерами
(рис. 1); с выносными нагревательными камерами
(рис. 2) и пленочные (рис. 4).
В аппаратах 1-го типа нагревательная камера представ-
ляет собой пучок вертикальных трубок, развальцованных
в двух трубных решетках. Греющий пар конденсируется
в межтрубпом пространстве, а в трубках циркулирует выпари-
ваемый р-р. Для создания замкнутого циркуляционного кон-
тура предусмотрен циркуляционный канал, н-рый находится:
в центре нагревательной камеры (рис. 1, I), на периферии
(рис. 1, П) или вне аппарата (рис. 1, III). Над нагреватель-
ной камерой расположен сепарационный объем, предназна-
ченный для отделения капель р-ра от вторичного пара, перед
выходом к-рого часто устанавливаются отражатели в для луч-
шего отделения капель. Нагревательные камеры аппаратов
рассматриваемого типа изготовляются из труб внутренним
диаметром 32—50 мм, высотой до 1,5—1,8 м с суммарной
поверхностью нагрева до 250 At2. Высота сепарационного про-
странства колеблется в пределах 1,8—2,4 м.
В аппаратах 2-го типа (рис. 2) возле сепаратора 1 распо-
лагаются одна или две вертикальные кожухотрубные нагре-
вательные камеры, соединенные с ним циркуляционными тру-
бами з. Вторичный пар отводится из верхнего штуцера 4.
При наличии двух нагревательных камер одна из них может
периодически отключаться при помощи вентилей 5 для ремонта
или очистки. При В. кристаллизующихся р-ров под сепарато-
ром устанавливается солеотделитель, к-рый периодич. может
отключаться для выгрузки. Рассматриваемые аппараты изго-
товляют из труб с внутренним диаметром 25—50 мм, суммар-
ная поверхность нагрева 600 м'2 и
более. На внутренней поверхности
нагревательных труб аппаратов
очень часто наблюдается инкруста-
ция (осаждение солей), что пони-
жает пароироизводительность ап-
паратов. Это явление практически
полностью устраняется в выпарных
аппаратах с принудительной цир-
куляцией раствора, осуществляе-
мой центробежными или вихревы-
ми насосами. На рис. 3 показана
конструкция такого аппарата. На
Рис. 1.
Pirc. 2.
Рис. 1. Выпарные аппараты с внутренними вертикальными
нагревательными камерами: 1 — вход греющего пара; 2 —
выход конденсата; з — нагревательная камера; 4 — циркуля-
ционный канал; 5 — сепарационное пространство; 6 — отра-
жатель; 7 — выход вторичного пара.
Рис. 2. Выпарной аппарат с двумя выносными нагревательными
камерами: 1 — сепаратор; 2 — нагревательная камера; з —
циркуляционные трубы; 4 — штуцер для выхода вторичного
пара; 5 — вентили; 6 — солеотделитель; 7 — задвижка.
Рис. 3.
параты
ной циркуляцией ра-
створа:/—нагреватель-
ная камера; 2 — се-
паратор; з — циркуля-
ционный насос.
рис. 4 показаны конструктивные модификации пленочного
выпарного аппарата. Во всех случаях высота нагреватель-
ных труб равна 6—7 м при внутреннем диаметре до 50 лич.
Высота столба кипящего р-ра ок.
2 м. Образующиеся пузырьки вто-
ричного пара, поднимаясь вверх,
сливаются постепенно в сплошную
струю. За счет поверхностного трения
паровая струя увлекает вверх тон-
кую пленку р-ра, выпариваемого на
этом пути при очень благоприятных
условиях. Время соприкосновения
р-ра с поверхностью нагрева соста-
вляет 60—90 сск. На рис. 4, I се-
паратор расположен непосредственно
над нагревательной камерой, а на
рис. 4, II парожидкостная эмульсия
из нагревательной камеры входит по
тангенциальному штуцеру в отдельно
установленный сепаратор; на рис. 4,
III нагревательная камера двух-
ходовая (первый ход снизу вверх, вто-
рой — вниз), а сепаратор располо-
жен внизу.
Пленочные выпарные аппа-
раты отличаются повышенным
коэфф, теплопередачи, но тре-
буют большой высоты здания и
подвержены инкрустации при В.
кристаллизирующихся раство-
ров. Для интенсификации про-
цесса В. внутренняя поверхность
нагревательных труб аппаратов
часто делается ребристой, бла-
годаря чему увеличивается по-
верхность нагрева и повышается
интенсивность работы выпарного аппарата. Одно-
корпусные выпарные аппараты могут работать пе-
риодически и непрерывно.
При непрерывном процессе часовой расход грею-
щего пара D кг!час определяется из ур-ния теплового
Выпарные ап-
с принудитель-
679
ВЫПАРИВАНИЕ
680
баланса:
Sid (tt-tH) + W(i-tI) + Qo кг!час
D t
l() Т— If)
где S, — часовое количество выпариваемого р-ра,
кг/час; сг — теплоемкость выпариваемого раствора,
ккал/кг°С; 4 — темп-ра кипения, а гн —нач. темп-ра
р-ра; i — теплосодержание вторичного пара, ккал/кг;
10 и !0 — теплосодержание (ккал/кг) и темп-ра насы-
щения (°C) греющего пара; Qo — потери тепла в
окружающую среду, ккал/час; W —
часовое колич. выпариваемой воды,
кг/час.
Рис. 4. Пленочные выпарные аппараты.
Если при В. концентрация р-ра возрастает от
кг/кг рр а2 кг/кг, то W — Sx (1 — кг/час. При
этом величина требуемой поверхности нагрева F вы-
ражается:
SjcHtj-tHl + WCi-t!)
F ~ ~-----гл------- **
•К (to — ti)
где К — коэфф, теплопередачи, ккал/м2 - час-°C.
Процесс периодич. В. осуществляется на практике
в двух вариантах. По первому из них аппарат одно-
кратно загружается и работает в дальнейшем с непре-
рывным понижением уровня р-ра.
Расход тепла Q в этом случае определяется из
ур-ния:
Q = aiS i
4з
J а
41
Qo ккал
где q и q2 — начальное, текущее и конечное тепло-
содержания выпариваемого р-ра при кипении, ккал/кг.
По второму варианту уровень р-ра в аппаратах под-
держивается постоянным на протяжении всего про-
цесса путем непрерывной подкачки. В этом случае
Q = (“sfa — йП1) — i (72 — 71) + (7з?2 —7i?i)j +
Ц- Qo ккал
где V — объем р-ра в аппарате, м3; q0 — теплосодер-
жание поступающего р-ра, ккал/кг; и у2 — началь-
ная и конечная плотность р-ра, кг/м3; q-^nq^ — тепло-
содержания кипящих р-ров при начальной и конеч-
ной их концентрациях, ккал/кг. Оба варианта перио-
дич. В. характеризуются нестационарным процессом
теплообмена, сопровождающимся непрерывным изме-
нением (во времени) темп-ры кипения t и коэфф,
теплопередачи К. Поэтому в обоих случаях требуемая
поверхность нагрева аппарата при продолжитель-
ности процесса т часов выражается:
Q
„ с dQ
= ^-час
о
Однокорпусные выпарные аппараты могут работать
под нормальным и избыточным давлением, а также под
вакуумом. Экономически выгодно работать под давле-
нием 1 ат; в этом случае можно использовать
вторичный пар для нагревательных целей. В. под
вакуумом неизбежно при концентрировании терми-
чески нестойких р-ров и целесо- ___
образно при наличии дешевого грею- JL
щего пара низкого давления. Для '
создания вакуума в выпарных аппа- —’
ратах обычно применяются конден- ____________(
саторы смешения с барометрич. тру- ___^2
бами (рис. 5). —•'
Конденсатор представляет со-
бой цилиндр, внутри которого
расположен ряд горизонтальных
полуперегородок с низкими по-
рогами для образования водо-
сливов. Охлаждающая вода сте-
кает сверху вниз по всем пере-
городкам навстречу подавае-
мому снизу потоку вторичного
пара из выпарного аппарата.
В результате конденсации пара
создается*вакуум, к-рый поддер-
живается непрерывным отсосом
остаточной паро-газовой’•смеси
вакуумнасосом. Смесь отрабо-
танной охлаждающей воды п
конденсата стекает по бароме-
трич. трубе, в которой удержи-
вается лишь столб воды, экви-
валентный разности между 1 ат
и абс. давлением в конденсаторе.
Конец^барометрич. трубы опу-
щен в коробку для образования
гидравлич. затвора, из которой
непрерывно стекает избыток
воды.
Расход греющего пара в
однокорпусных аппаратах
1,2—1,25 кг на 1 кг выпари-
ваемой воды. Значительная
Рис. 5. Схема вакуумвы-
парного аппарата: 1 — вы-
парной аппарат; 2 — конден-
сатор смешения; з баро-
метрическая труба; 4— ВХОД
охлаждающей воды; 5 — к
вакуумнасосу; 6 — бароме-
трическая коробка; 7—сброс
избытка воды.
экономия греющего пара
достигается в многокорпусных выпарных аппаратах,
представляющих собой ряд последовательно соединен-
ных однокорпусных аппаратов с понижающимися ра-
бочими давлениями по направлению от первого к по-
следнему. Благодаря этому вторичный пар каждого
предыдущего корпуса может быть использован для
обогрева следующего.
На рис. 6 изображена схема трехкорпусного аппарата.
Греющий пар подается по трубе 3 в нагревательную камеру 2
корпуса I, вторичный пар к-рого уходит из сепарационного
объема 1 по трубе 4 в нагревательную камеру корпуса II
Рис. 6. Схема прямоточного трехиорпусного вы-
парного аппарата: I, II, III — корпусы; 1 — се-
парационный объем; 2—нагревательная камера;
з — паропровод первичного греющего пара; 4— па-
ропровод вторичного пара; 5 — конденсатор сме-
шения; 6 — конденсатопроводы; 7 — трубопровод
слабого раствора; 8 — междукорпусные трубопро-
воды для раствора; 9 — подогреватель слабого ра-
створа; 10 — отвод конденсата; 11 — водопровод;
12 — труба к вакуумнасосу.
и т. д. Вторичный пар корпуса III уходит в конденсатор сме-
шения 5, куда по трубе 11 подается охлаждающая вода, а по
трубе 12 паровоздушная смесь отсасывается вакуумнасосом.
Исходный р-р подается по трубе 7 через подогреватель 9 в кор-
пус I, откуда он перетекает по трусе 8 в корпус II и т. д.,
681
ВЫПАРИВАНИЕ
682
выходя из корпуса III. Конденсат из всех корпусов отводится
по трубам в в подогреватель а для частичного использования
его теплосодержания п по трубе ю отводится из системы.
При В. кристаллизующихся р-ров междукорпусные трубопро-
воды часто закупориваются, поэтому практикуют параллель-
ный подвод слабого и отвод конц. р-ра из всех корпусов, со-
храняя последовательное движение пара. Оптимальное число
корпусов аппарата зависит от давления греющего пара, тем-
пературных депрессий раствора и коэфф, теплопередачи
и определяется техно-экоиомпч. расчетом. На практике число
корпусов редко бывает больше пяти. Расход греющего пара
на 1 кг выпариваемой воды падает с увеличением числа кор-
пусов (от 1,2—1,25 кг/кг в однокорпусном до 0,25—0,28 кг/кг
в пятикорпусном аппарате).
Точное значение расхода греющего пара (О) и тепловые
нагрузки отдельных корпусов могут быть найдены из системы
ур-ний их тепловых балансов:
для 1-го корпуса
D(X,-1 Wt(h-‘d + SiCt(tt — tn) + ijJ
для 2-го корпуса
Wj — ti 4- *а) VV2 (As — /2) SiСj (t2 — ti) -f- Q^q
для 3-го корпуса
Ws ~ — ^2 4" *з) = Wi (t2 — /3) 4- VV3 (X4 — f3) 4-
+ S1c1 (13 - (3) + QU„T
для 4-го корпуса
- /3 4- ti) = 4- ws) (Z3 - ti) 4- Wi (X5 - ti) 4-
4" S1C1 (ti — t$) 4- Q q
для n-го корпуса
Wn_ ! (Xn - 'n”’ - *n- 1 + = (Wl + + W> +
+ ... + wn_2)(zn_1-in)+ivn(Xn + I-fn) +
+ SjC! (tn - tn _ j) + qW
где Wt, W„ W„ W,,..., Wn — количества выпаренной воды
в отдельных корпусах аппарата; Xt, Х2, Х2, Х2. Хп—тепло-
содержания паров, греющих соответствующие корпуса
(или вторичных паров предыдущих корпусов); 1^, l^,
1^,...,/^—температуры греющих паров и их конденсатов;
(i, t2, t3, t,.tn — температуры кипения в отдельных корпу-
сах,- Si, С,, 1п — количество, теплоемкость и начальная тем-
пература исходного раствора; oj, Q1/, Q1”, Q1^,..., —
потери тепла отдельными корпусами в окружающую среду.
Принимая во внимание, что
VV1 + Ws + W, + W,. + ... + Wn = W = Sj (1 -
4 Cto '
можно из приведенной системы ур-ний определить значения
D, И\, IV'2, W3,..., IV п, необходимые для нахождения тепло-
вых нагрузок (Q) отдельных корпусов: Qt = D(X, — 1^);
Q3 —Wi (X2 — /Н); Оз (Хз —l1*1) и т. Д„ ккал/час.
Рабочие разности темп-p в отдельных корпусах равны:
= ...; = t W - tn
Полезная разность те.мп-р всего многокорпусного аппарата
составляет: д — У,8, где S8 — суммарная
депрессия темп-ры во всех корпусах. На практике обычно
стремятся к равенству поверхностей нагрева всех корпусов:
F1=F3=F3 = .-..=Fn
или
01 _ Оз __ Оз
h1At Ь2д2 Ьзаз ’ Ьпдп
где /ц, k2, kn — коэфф, теплопередачи в отдельных кор-
пусах. Этому условию соответствует следующее распределе-
ние полезной разности темп-p между корпусами:
д
At -------------------------------------------;
, , Оз , Оз 1 £? fei
1+Q1 ' ks TQ! ' ks Ql •
Вместо многокорпусных систем для экономии грею-
щего пара в ряде случаев выгодно применять одно-
корпусные аппараты с тепловым насосом, в к-ром вто-
ричный пар после нек-рого сжатия его в турбоком-
прессоре или инжекторе направляется на обогрев
того же аппарата, в к-ром он сам образовался.
На рис. 7 показаны две схемы теплового насоса. По первой
из них (рис. 7,1) вторичный пар из выпарного аппарата 1 сжн-
Рпс. 7. Схемы выпарных аппаратов с тепловым на-
сосом: 1 — выпарной аппарат; 2 — турбокомпрессор;
з — инжектор; 4 — первичный греющий или инжек-
тирующий пар; s — отвод конденсата; в — электро-
двигатель
мается в турбокомпрессоре 2 от давления Pi до давления р2
и направляется на обогрев того же аппарата. Расход энергии
на сжатие W кг/час вторичного пара при адиабатич. перепаде
тепла (12 — ч) составляет:
= кет
SW’lft
где — коэфф, полезного действия турбокомпрессора. Для
обеспечения постоянной производительности выпарного аппа-
рата требуется подвод извне нек-рого количества (Do) пара,
определяемого из ур-ния теплового баланса. Техно-экономич.
расчеты показывают, что рассматриваемая схема теплового
насоса выгодна до тех пор, пока p2/pi 2—2,5 и производи-
тельность турбокомпрессора не менее 5000 .«“/час. По второй
схеме теплового насоса (рис. 7, II) в инжекторе 3 сжимается
лишь часть (DB) вторичного пара, образовавшегося в аппарате 1.
Количество вторичного пара, засасываемое 1 кг инжектирую-
щего, называется коэфф, инжекции: U = DB/D0.
Т. обр., D = Do + DB = D„ (1 -)- 17), т. е. расход первичного
пара (Do) падает с ростом коэфф, инжекции U, определяемого
из ур-ния U = ф1/ —1,где <р — коэфф., учитывающий поте-
г й2
ри кинетич. энергии в инжекторе (обычно <р = 0,6—0,65); ht —
адиабатич. перепад тепла при расширении инжектирующего
пара до давления вторичного пара рв; й2—адиабатич. перепад
тепла при сжатии вторичного пара до давления греющего
пара рг. Расчеты показывают, что область выгодного при-
менения данной схемы теплового насоса обычно ограничи-
вается отношением рг/рв^2.
Применение аппаратов, в к-рых теплота пере-
дается через металлич. стенку (поверхность нагрева),
становится практически невозможным при В. р-ров,
химически агрессивных по отношению к доступным
конструкционным металлам. В этих случаях исполь-
зуются выпарные аппараты, изготовляемые из кисло-
тоупорного кирпича, бетона и различных горных по-
род, основанные на непосредственном контакте то-
почных газов и р-ра. В пром-сти нашли применение
два типа таких аппаратов. Первый из них представ-
ляет собой цилиндрич. башню, пустотелую или за-
полненную хордовой насадкой, в к-рой выпаривае-
мый р-р (в распыленном состоянии или пленками)
падает навстречу восходящему потоку горячих газов.
Второй тип аппарата основан на барботаже горячих
газов через слой выпариваемого р-ра. Он оформляется
в виде горизонтального цилиндра с несколькими бар-
ботажными перегородками либо в виде вертикальной
башни с барботажными устройствами. Аппараты
2-го типа имеют большую эффективность, а аппараты
1-го типа отличаются меньшим гидравлич. сопротив-
лением.
Лит.: Гельперин Н. И., Выпарные аппараты,
М.—Л., 1947; Кичигин М. А. и Костенко Г. Н.,
683
ВЫРАВНИВАЮЩИЕ ВЕЩЕСТВА — ВЫСОКИЕ ДАВЛЕНИЯ
684
Теплообменные аппараты и выпарные установки, М.—Л.,
1955; Касаткин А. Г., Основные процессы н аппараты
химической технологии, 7 изд., М., 1961; Ц и б о р о в-
с к и й Я., Процессы химической технологии, пер. с польск.,
М„ 1958. И. II. Гельперип.
ВЫРАВНИВАЮЩИЕ ВЕЩЕСТВА (в тек-
стильной промышленности) — вещества, при-
меняемые для получения равномерных окра-
сок волокнистых материалов. Действие В. в.
различно в зависимости от строения. Нек-рые
из них обладают сродством к волокну, дру-
гие — к красителю. К первым относятся анио-
ноактивные в-ва, например сульфопроизвод-
ные гомологов нафталина (некаль), алифатич.
углеводородов, кислые сернокислые эфиры
высокомолекулярных алифатич. спиртов и др.;
вторым — неионогенные соединения, напр. продукты
оксиэтилиронания и катионоактивные в-ва. Необхо-
димо отметить, однако, что такое разделение В. в.
несколько условно, т. к. в каждом конкретном слу-
чае надо учитывать химич. природу волокна и кра-
сителя.
Выравнивающее действие веществ первой группы
основано на их способности образовывать соединения
с волокном и т. о. конкурировать с красителем. По
этой причине замедляется выбирание красителя во-
локном из красильной ванны. При продолжительном
крашении, особенно при высокой темп-ре, краситель
вытесняет с поверхности ионы поверхностно-актив-
ного вещества. Описанный механизм действия В. в.
характерен для крашения белковых или полиамидных
волокон кислотными красителями; в рассматривае-
мом случае аналогично действуют в сильно кислых
средах нейтральные электролиты, напр. сульфат
натрия (глауберова соль). Однако выравнивающее
действие наблюдается только, если соль добавляют
в значительных количествах; при этом может про-
изойти осаждение (высаливание) красителя, особенно
при крашении в темные цвета.
В. в. второй группы образуют с красителем более
или менее устойчивые агрегаты и тем уменьшают их
подвижность и скорость диффузии внутрь волокна.
При высоких темп-рах, соприкасаясь с волокном, эти
аддитивные соединения распадаются и освобождают
краситель. Т. обр., В. в. замедляют скорость крашения
и способствуют более равномерному окрашиванию
волокна. Такой механизм наиболее вероятен, когда
используются красители анионного типа (прямые,
кислотные, кубовые и т. д.), а в качестве В. в. —
сравнительно высокомолекулярные катионоактивные
вещества.
Возможно и другое объяснение действия В. в. вто-
рой группы: при введении В. в. в р-ры красителей
происходит распад агрегатов красителей и образова-
ние вокруг их молекул или ионов сольватных оболо-
чек из молекул В. в. Вследствие этого снижается
сродство красителя к волокну и увеличивается срод-
ство . к растворителю. Поэтому скорость диффузии
красителя н волокне увеличивается и создаются луч-
шие условия для перераспределения молекул краси-
теля в текстильных материалах между местами с раз-
личной интенсивностью окраски. Так, напр., дей-
ствуют в процессе крашения неионогенные вспомога-
тельные вещества, напр. продукты оксиэтилирова-
ния, а также различные растворители: пиридин, эта-
ноламины, этиламины, тетрагидрофуриловый спирт,
поливинилпирролидон и др.
Наиболее часто неиопогенные выравниватели, обла-
дающие сродством к красителю, применяются при
крашении изделий из целлюлозных материалов кубо-
выми красителями. При этом между полигликолевыми
эфирными группами выравнивателя и лейкокислотой
кубового красителя образуются водородные связи,
или же взаимодействие красителя и В. в. приводит
к образованию соли из анионов кубовой к-ты и катио-
нов полиоксония. Примером может служить кубовый
краситель — индиго синий:
RCH2CH2-O~CH2-CH30-
ко
/?СН2СН2- О-СН2СН2О-
I н
О' он
I I
/С’Сх J
NH NH
ПОЛЬ полиоксония
Он
I
с-с I
I 4NH
соединение при помощи
водородной связи
Влияние продуктов оксиэтилирования на выбира-
ние кубовых красителей волокном может быть не-
сколько ослаблено добавлением в красильную ванну
анионоактивных веществ, к-рые могут образовывать
с продуктами оксиэтилирования соединения, подобно
анионам красителей. Однако опыт показывает, что
конкурирующее действие добавок анионоактивных
веществ невелико. Оно проявляется сильно только,
если в качестве выравнивателя при кубовом крашении
используются катионоактивные вещества. См. также
Вспомогательные вещества (в текстильной пром-сти).
Лит.: Хвала А., Химические вспомогательные веще-
ства в текстильной промышленности, пер. с нем., М.—Л.,
1948; Виккерст а ф ф Т., Физическая химия крашения,
пер. с англ., М., 1956; Ф р о т ш е р Г., Химия и физическая
химия текстильных вспомогательных материалов, пер. с нем.,
т. 1, 1958; S с h w е n G., Monatsh. Иг Seide, Kunstseide und
Zellwolle, 1938, 43, S. 369; Rath II., Lehrbuch der Textilclie-
mie..., B. — Gottingen — Hdlb., 1952; V a I k <5 B., Trans.
Faraday Soc., 1935, 31, № 164, p. 254; его же, Osterr. Chem.—
Ztg., 1937, Jg. 40, H. 21, S. 465.
II. В. Морыганов, Б. H. Мельников.
ВЫСОКИЕ ДАВЛЕНИЯ — давления, превышаю-
щие атмосферное. Часто встречающееся в литературе
подразделение давлений на низкие, высокие, очень
высокие и сверхвысокие в значительной степени
условно, т. к. устанавливается в зависимости от слу-
чайных факторов. Давление (Д.) имеет только нижний
предел — абс. вакуум; величина же достигнутого Д.
ограничивается возможностями техники. В природе
Д. достигает значительной величины; на больших вод-
ных глубинах до 1000 ат, в толще земли доходит
(у центра) до миллионов атмосфер; на нек-рых звез-
дах Д. достигает десятков и сотен миллионов атмосфер
(белые карлики до 1018 ат). Применение В. д. имеет
большое значение для науки и техники. Оно смещает
химич. равновесие и ускоряет многие реакции, вслед-
ствие чего используется в пром-сти для получения
синтетич. аммиака, метанола, мочевины, различных
полимеров, искусственного бензина и др. В. д. при-
меняют и в современных паровых котлах, при В. д.
добывают нефть и газ из глубинных скважин. В. д.
развивается при горении заряда в стволе артилле-
рийского орудия и в цилиндрах двигателей внутрен-
него сгорания, используется для приведения в дейст-
вие мощных гидранлич. прессов, действует в водяных
пушках — гидромониторах. Лабораторные исследова-
ния проводят при Д. до 200 000 ат; максимально
Д., достигнутое в лаборатории, равно 425 000 ат.
В. д. значительно изменяет свойства веществ. Д.
в тысячи атмосфер только уменьшает расстояние
между молекулами, при Д. в десятки тысяч — уже
начинают деформироваться электронные оболочки,
а Д. в сотни тысяч атмосфер приводит к переходу
электронов на соседние энергетич. уровни, вызывают
перестройку атома и изменение свойств элементов.
При Д. в миллионы и более атмосфер электроны пере-
ходят на коллективные уровни, а вещества — в метал-
лич. состояние; при Д. порядка 1026 ат и темп-рах
1013 градусов может существовать только вырожден-
ный электронный газ.
685
ВЫСОКИЕ ДАВЛЕНИЯ
686
Химические эффекты высоких давлений.Влияние
Д. на протекание химич. реакций выражается ком-
плексом явлений. Д. сдвигает равновесие реакции,
если реакция идет с изменением объема (см. Ле Ша-
телье принцип), и ускоряет реакции вследствие уве-
личения плотности и, следовательно, концентрации
реагирующих веществ. Ускорение происходит также
вследствие увеличения самой константы скорости
в случае, когда образование активированного ком-
плекса сопровождается уменьшением объема. Д. наи-
более сильно сдвигает равновесие реакций, протекаю-
щих в газовой фазе (см. Аммиак), а также влияет па
реакции, протекающие в твердой фазе, особенно
в случае значительного уменьшения объема твердой
фазы под Д. Так, Д. в 40 000 кг/см2 при одновремен-
ном приложении сдвига (тангенциальное усилие)
способствует синтезу сульфидов меди, фосфидов
цинка, магния и т. д. из элементов.
Зависимость константы скорости реакции от Д.
выражается:
/э In _ An
V др 'Т RT
где Ас — разность между суммой мольных объемов
исходных веществ и мольным объемом активирован-
ного комплекса. Кроме того, значительное воздей-
ствие Д. оказывает и через среду, т. е. ее диэлектрич.
проницаемость и вязкость, что особенно сильно ска-
зывается на скорости ионных реакций и реакций
полимеризации. Быстрый рост вязкости жидкостей
с Д. может привести к переходу реакции из кинетич.
в диффузионную область. Гомогенные жидкофазные
реакции по их отношению к Д. можно разбить на три
группы: 1) мономолекулярные реакции разложения,
замедляемые Д.; 2) «нормальные» бимолекулярные
реакции, ускоряемые Д.; 3) «медленные» бимолеку-
лярные реакции, значительно ускоряемые Д.
В ионных реакциях или реакциях, протекающих
через переходные состояния, сопровождаемые образо-
ванием заряженных частиц, или в полярных раство-
рителях, когда образование зарядов ведет к росту
сил взаимодействия и уменьшению объема активиро-
ванного комплекса, скорость также возрастает с ро-
стом Д. Предполагается, что Д. ускоряет те газовые
реакции, механизм к-рых включает в наиболее мед-
ленной стадии взаимодействие двух и более молекул
или радикалов. Ускорение в этом случае может про-
исходить также за счет увеличения константы ско-
рости. Влияние Д. на гетерогенные каталитич. реак-
ции зависит не только от характера наиболее медлен-
ной стадии реакции, но определяется и изменением
активности самого катализатора. Смещение равнове-
сия сложных реакций и влияние Д. на самый меха-
низм реакции сказывается и на составе продуктов
реакции и позволяет использовать Д. для получения
веществ с заданными свойствами. С повышением Д.
относительная скорость отдельных стадий реакции
может настолько измениться, что это приведет к пе-
ремене знака эффекта Д. Напр., термич. распад пара-
финов, ускоряемый небольшим Д., тормозится при
высоких Д.
Д. изменяет характер стерич. препятствий, дефор-
мируя молекулы (изменение валентных углов и т. д.),
а также ускоряет пространственно затрудненные реак-
ции, причем большую роль здесь играет изменение
объема растворителя в результате различной соль-
ватированвости реагирующих молекул и активиро-
ванного комплекса. Значительно (в тысячи раз)
быстрее под Д. идут процессы полимеризации. Наир.,
скорость полимеризации изопрена и стирола увели-
чивается пропорционально квадрату Д., а димстплбу-
тадиепа — пропорционально четвертой степени. При
полимеризации под Д. получают полимеры, свойства
к-рых значительно отличаются от свойств продуктов,
полученных при атмосферном Д.
В настоящее время насчитывается более 100 про-
цессов, к-рые осуществляют в пром-сти под Д. от
10 до 3000 ат (алкилирование, этерификация, галло-
генирование, карбоксилирование, процессы гидро-
форминга, гидрогенизация, гидратация, гидролиз,
изомеризация, нитрование, окисление, полимериза-
ция и др.), получая при этом такие ценные продукты,
как аммиак, метанол, этанол, высшие спирты, карбо-
нилы металлов, разные полимеры и т. д. Однако
качественно новых процессов в химии высоких Д.
можно ожидать только при таких Д., к-рые изменяют
химич. свойства атома, т. е. при Д. в десятки и сотни
тысяч атмосфер.
Физические эффекты высоких давлений. Сжимае-
мость веществ. При Д. порядка сотен атмо-
сфер сжимаемость газов велика, но значительно
уменьшается по мере роста Д., когда плотность сжа-
того газа становится сравнимой с плотностью жид-
кости (при 10 000 ат и 50° плотность азота равна
1,12 г[см3). При Д. в несколько тысяч атмосфер свой-
ства газа и жидкости становятся настолько схожими,
что сжимаемость газа можно передать ур-нием Тета
для сжимаемости жидкостей:
Дп=«о^п = С1п В+Р.
По По в + Ро
где v0 и р0 — начальные, a v и р — конечные объемы
и давления, С и В — константы.
Большинство жидкостей, к-рые еще не затвердевают
сжимаются на 25—30%. При
имеют значение детали моле-
при Д. в 10 000 ат,
этом Д., однако, еще
кулярпой структуры
жидкости. Так, чем
больше ОН-групп со-
держится в соедине-
нии, тем меньше его
сжимаемость (рис. 1).
К уменьшению сжи-
маемости ведет и на-
личие кислорода, за-
мена водорода галоге-
ном и т. д. При более
высоких Д. (25 000—
50 000 ат) почти все
жидкости ведут) себя
одинаково и различие
в сжимаемости редко
превышает 10%. Зна-
Рпс. 1. Сжимаемость жидкостей:
1 — глицерин; 2 — вода; 3 — эти-
ловый спирт; 4 — н-пентан.
чительно сжимаются при В. д. итвердыетела (рис. 2).
При 4 • 106 ат объем железа уменьшается в 1,7 раза.
Плотность вещества при Д. в десятки тысяч атмосфер
больше их плотности при абс. пуле и атмосферном Д.,
что указывает на деформацию молекул и атомов,
к-рая может отразиться на химич. свойствах сжатых
веществ. Сжимаемость элементов простых веществ
зависит от атомного номера элемента, из к-рого со-
стоит простое вещество, т. е. определяется поведением
внешних электронов. С ростом Д., однако, роль внеш-
них электронов в объемном поведении элементов
уменьшается, о чем можно судить по уменьшению
разницы в значениях их сжимаемости. В пределе при
бесконечно большом Д. все вещества должны следо-
вать одному общему ур-нию состояния.
Фазовые переходы в однокомпо-
неитяых системах. Темп-ра плавлении ве-
ществ значительно изменяется с Д. Опыт показывает,
что во всех исследованных случаях кривая темп-р
плавления в координатах Р—Т является плавной
кривой (рис. 3), причем для большинства исследован-
ных веществ темп-ра плавления растет с Д. Исклю-
чение составляют вода, впсмут и галлий (на рис. не
687
ВЫСОКИЕ ДАВЛЕНИЯ
688
показаны), у к-рых вначале темп-ра плавления
уменьшается с ростом Д., а при переходе твердой
фазы в другую модификацию начинается рост темп-ры
у плавления с Д. (на рис. не
показано). У гермапия это
0,7| — уменьшение с Д. прослеже-
. но до 180 000 ат. С увели-
кг/см2. /О3
Рис. 2. Сжимаемость твер-
дых тел.
чением Д. меняется наклон
кривых плавления, так что
более легкоплавкие в-ва
становятся менее легкоплав-
кими и наоборот.
Принципиально нет раз-
ницы между гетерогенным
равновесием двух твердых
фаз (полиморфные превра-
щения) или твердой и жид-
кой фазами (плавление). Од-
нако формы кривых поли-
морфных переходов весь-
ма разнообразны из-за боль-
шого различия в сжимае-
мости и объемах твердых
фаз.' В области до 50 000 ат
большинство веществ имеет
по крайней мере один по-
лиморфный переход;у орга-
нич. веществ их больше, чем у неорганич. Многие
вещества имеют несколько полиморфных модифика-
ций. Так, у висмута их 8 (рис. 4), у воды 7 (рис. 5),
а у камфары И. Возмож-
но, что нек-рые из фазо-
вых превращений обус-
ловлены электронными
переходами. Так, фазо-
вое превращение в цезии
при Д. около 45 000 ат
возможно связано с пе-
реходом валентного элек-
трона с уровня 6s на не-
заполненный уровень 5(7,
фазовый переход в це-
рии при 7 000 ат — пе-
реходом электрона из
состояния 4/ в 5й, когда
церий из 3-валентного
превращается в 4-валент-
ный.
Кроме обратимых по-
лиморфных переходов,
под Д. происходят и
необратимые превраще-
ния. При 12 000 ат и
200° желтый фосфор не-
обратимо превращается
Рпс. 3. Изменение температуры в черный, плотность ко-
плавления некоторых элемен- торого больше на 46%.
тов с давлением. Сероуглерод при 175° и
40 000 ат превращается
в черное твердое тело. При 35 000 ат и 750° смесь
метасиликата натрия и диаммонийфосфата образует
коэзит — новую модификацию кварца, более плотную
и устойчивую к действию HF. Переход гексагональ-
ной решетки графита в кубич. решетку алмаза может
происходить в области термодинамич. устойчивости
алмаза, к-рая, как показывают расчеты, находится
при Д. больших, чем 10 000 ат (при комнатной
темп-ре). Однако при низких темп-рах перестройка
решетки практич. не происходит даже при высоких Д.,
возможно, из-за кинетич. задержек. Скорость превра-
щения становится достаточной при темп-рах порядка
3000° К. Но при этом для осуществления перехода
необходимы Д. порядка 70 000 — 100 000 ат. При
Рис.
4. Диаграмма состояния
висмута.
таких условиях в 1953 в Швеции и в 1954 в США
были получены кристаллы алмаза, больший из к-рых
имел 1,2 мм в длину, что позволило начать промышлен-
ный выпуск технич.
алмазов. При усло-
виях, близких к усло-
виям синтеза алмаза
(62 000 ат и 1350° С),
был обнаружен еще
один полиморфный пе-
реход, при котором
гексагональный ни-
трид бора с решет-
кой, подобной решет-
ке графита, необрати-
мо превращается в
модификацию с куби-
ческой решеткой цин-
ковой обманки (ZnS).
Новое в-во, назван-
ное боразоном, обла-
дает твердостью алмаза и более устойчиво термически.
Фазовые переходы в многокомпо-
нентных системах. Растворимость газов
в жидкостях растет с Д., доходит до максимума и
далее начинает уменьшаться, что объясняется раз-
личным изменением с Д. мольного объема газа г>г
и парциального мольного объема газа в р-ре v . При
низких Д. моль газа занимает объем значительно
больший, чем объем жидкости. Величина парциаль-
ного мольного объема газа иг в р-ре того же порядка,
что и величина мольного объема жидкости. Вследствие
того, что сжимаемость газа больше сжимаемости
жидкости, при увеличении Д. разница между объемами
уменьшается и, когда они делаются равными, насту-
пает максимум растворимости. Когда г>г > г>г, раство-
римость газа в жидкости уменьшается с Д. При
очень высоких Д. когда сжимаемость жидкой и га-
зовой фаз становится
одинаковой, раствори-
мость газа в жидкости
почти не меняется сД.
Растворимость жид-
кости в газе при низ-
ких Д. уменьшается с
Д., проходит через ми-
нимум, далее растет
до максимума и вновь
начинает уменьшаться
(рисунок 6). Появление
экстремумов также вы- Рис. о. Диаграмма состояния
звано различным изме- воды. Примечание: эксперимен-
нением с Д. мольного ™льно обнаружу еще одан пе-
объема жидкости и пар-
циального мольного объема ее паров в газовом
р-ре (т. е. объема газовой фазы). Еще нет универсаль-
ного уравнения, с помощью к-рого можно было бы
передать Сложную форму этой кривой. В сжатых
газах могут растворяться значительные количества
жидкостей и твердого в-ва. Так, при 2000 ат в 1 нм3
этилена растворяется до 3 кг смазочного масла. Водя-
ной пар высокого Д. растворяет значительные коли-
чества солей, что приводило к отложению солей на
лопатках паровых турбин. Растворимостью кварца
в водяном паре, насыщенном нек-рыми солями, поль-
зуются для выращивания больших кристаллов кварца,
вес к-рых измеряется килограммами (гидротермаль-
ный синтез). Способность сжатых газов избирательно
растворять жидкости позволяет предложить новый
метод разделения сложных жидких смесей изотермич.
перегонкой в сжатом газе при сравнительно низких
689
ВЫСОКИЕ ДАВЛЕНИЯ
690
темп-pax. Близость свойств сжатого газа и жидкости
проявляется также и в
Рис. 6. Растворимость жидко-
го аммиака в сжатом азоте.
существовании ограниченной
взаимной растворимости
газов или явления расслое-
ния газовых смесей при
температурах больших,
чем критич, температуры
компонентов (открыто в
СССР в 1941) (см. Газовые
растворы).
Явлепия пере-
носа. Вязкость, тепло-
проводность, коэфф, диф-
фузии изменяются под Д.
Вязкость реальных газов
растет с Д.; так, вязкость
СО2 при 40° и давлении
120 ат примерно в 4 раза
больше, а этилена при
1000 ат и 24° в 12 раз
больше, чем при атмосфер-
ном Д. Теплопроводность
газов возрастает более
чем в 10 раз в пределах, когда приведенные па-
раметры достигают значений:
Р/Ркрит = °>6-0,7 и Жкрит. = 1,0 - 3,0
Вязкость жидкостей под Д. меняется тем заметнее,
чем сложнее молекула. Вязкость и-амилового эфира
при 30° и 8000 кг/сж3 возрастает в 100 раз, евгенола
при 3000 ат в 187 раз.
Влияние давления на пластич-
ность. Д. значительно повышает пластичность
твердых тел и их сопротивление разрушению. Мра-
мор, подвергнутый всестороннему гидростатич. давле-
нию в 10 000 ат, может быть растянут с удлинением
до 25% без разрушения. Пластич. деформация стали
под гидростатич. Д. в 25 000 ат значительно увели-
чивается без разрыва, к-рый в этих условиях проис-
ходит только от среза. Пластич. деформация латуни
существенно увеличивается до 3000 KsjcM2. Под гид-
ростатич. Д. деформация сдвига становится качест-
венно схожей с пластич. деформацией растяжения.
При этом может иметь место «самозалечивание» по-
явившегося нарушения. Через лист малоуглеродистой
стали, находящейся под гидростатич. Д., может быть
продавлен пуансон без высечки, причем получается
ровное отверстие. Значительное упрочнение сталей
и твердых сплавов под гидростатич. Д. используют
при создании аппаратов для исследований при В. д.
Влияние давления па поверхност-
ное натяжение. Изменение Д. сказывается
значительно на вели-
чине поверхностного
натяжения а в систе-
мах жидкость — газ
и на смачиваемости
жидкостями твердых
тел в присутствии
сжатого газа. При
этом изменяется со-
став самих фаз, а
также и работа, по-
требная для создания
единицы новой по-
верхности. Объем га-
зовой фазы меняется
с Д. сильнее, чем
объем жидкой. Вслед-
Рис. 7. Поверхностное натяже-
ние на границе жидкость — газ
в системе метанол — азот под
давлением.
ствие этого, так как
до
— величина отрицательная, поверхностное натяже-
ние на границе жидкость—газ с Д. уменьшается (рис.7).
«г/см2. 10*
Рис. 8. Относительное изменение
электросопротивления щелочных
металлов с давлением.
Рис. 9. Относительное измене-
ние электросопротивления ще-
лочноземельных металлов с
давлением.
Особенно значительное уменьшение происходит в ин-
тервале Д. в несколько сот атмосфер. При увеличе-
нии Д. до 800 ат а
в системе метанол —
метан падает с 22 до
2 дин/см. Смачивае-
мость твердых тел
жидкостями в при-
сутствии сжатых га-
зов также значительно
ухудшается.
Электрические эф-
фекты высокого дав-
ления. У большинства
твердых тел электро-
сопротивление, как
правило, понижается
с ростом Д. У элемен-
тов с ясно выражен-
ными металлич. свой-
ствами (Си, Ag, Аи
и др.) до 100 000 ат
электросопротивление
падает па 15—20%,
а у щелочных метал-
лов (Na, К, Rb) — вна-
чале падает, достигает минимума, а затем растет;
у лития, кальция и бария повышается с ростом Д.
(рис. 8 и 9). Электросопротивление цезия вначале
резко возрастает (до
53 000 ат), а при даль-
нейшем небольшом уве-
личении Д. уменьшает-
ся. Эти изменения, по-
видимому, связаны с пе-
реходом электронов на
другие уровни.Электро-
сопротивление жидких
металлов изменяется с
Д. в меньшей степени,
чем у твердых. У спла-
вов оно, как правило,
уменьшается с ростом
Д. значительно мень-
ше, чем у компонен-
тов сплава, а у некото-
рых сплавов (манганин)
увеличивается.
Непроводник — жел-
тый фосфор, при пере-
ходе под Д. в черный,
приобретает металлич.
проводимость. Полупроводник теллур при 30 000 ат
также приобретает металлич. проводимость. Рас-
четы показывают, что при Д. порядка 1,8 • 10е —
2,4 • 10е ат твердый водород обладает атомной решет-
кой типа объемноцентрированной решетки щелочных
металлов и соответственно этому — металлич. прово-
димостью. Влияние Д. на электропроводность водных
р-ров разнообразно и зависит от концентрации р-ра.
Так, электропроводность 0,0001 н. р-ра NaCl падает
с Д., а 5 н. р-ра возрастает при 3000 ат на 10%.
Электропроводность воды при изменении Д. до
127 000 ат возрастает до 0,83 омГ1 • см-1, что соот-
ветствует электропроводности 5 н. р-ра НС1. Опыт
показывает, что отношение (функция) Клаузиуса—
Моссотти [г>(е — 1)/(е — 2) = р0, где v — мол. объем,
е—диэлектрич. проницаемость и рй — мол. поляри-
зация] редко остается постоянным при увеличении
Д. Влияние Д. на диэлектрич. проницаемость разных
веществ сказывается по разному. В интервале Д.
до 1000 ат это отношение можно считать постоянным
для азота и водорода. Оно убывает для СО2, а длн
691
ВЫСОКИХ ДАВЛЕНИЙ ТЕХНИКА
692
аммиака растет уже в интервале 100 am; для поляр-
ных газов вообще меняется сильнее, чем для непо-
лярных, и для CF4 оно уменьшается на 35% при
80 ат.
Диэлектрин, проницаемость этилена растет с уве-
личением Д. Для ряда жидкостей е растет незначи-
тельно, а при очень высоких Д> и высоких частотах е
глицерина и евгенола падает, по-видимому, вслед-
ствие большого увеличения вязкости и подавления
способности молекул .достаточно быстро вращаться,
чтобы следовать за электрич. полем. Изменение е
полярных жидкостей хорошо передается ур-нием
Тета: =1 — где А и В — константы.
Д. уменьшает е твердых тел, измеренную при низкой
частоте (галогены щелочных металлов, окись магния).
Элемент с цинковым и водородным электродами в сер-
ной к-те при 9 000 ат и 20° меняет полярность. Д.
увеличивает эдс элемента Вестона, влияет на пока-
затель преломления и может вызвать пьезохрома-
тизм — изменение цвета вещества при сжатии.
У нек-рых элементов, напр. Bi при 7° К и Д.
20000—40000 ат, наступает состояние сверхпро-
водимости.
Биологические эффекты высокого давления. Д.
в 17 500 кг/см2 в течение 45 мин разрушает дифте-
рийный токсин и яд кобры. Токсин столбняка разру-
шается при 13 500 кг/сж2; туберкулезная палочка
погибает при 6000 кг/см--, чувствительны к Д. клетки
нек-рых злокачественных опухолей. Д. в 7000 кг 1см2
убивает бактерии молочнокислого брожения и сте-
рилизует мясные продукты, что может быть исполь-
зовано для консервирования без нагрева.
Лит.: Бриджмен П., Физика высоких давлений, пер.
с англ., М.—Л., 1935; его же, Новейшие работы в области
высоких давлений, пер. с англ., М., 1948; его же, Исследо-
вания больших пластических деформаций и разрыва, пер.
с англ., М., 1955; Г о н и к б е р г М. Г., Химическое равно-
весие и скорость реакций при высоких давлениях М., 1953;
его же, Вести. АН СССР, 1957, № 2, с. 50; Криче в-
с к и й И. Р., Фазовые равновесия в растворах при высоких
давлениях, 2 изд., М.—Л., 1952; Корндорф Б. А., Тех-
ника высоких давлений в химии, Л.—М., 1952; Ц и к-
л и с Д. С., Техника физико-химических исследований при
высоких давлениях, 2 изд., М., 1958; Comings Е. W.,
High pressure technology, N. Y. [a. o.], 1956; Hund F.,
Phys. Z., 1936, 37, № 22/23, S. 853; Hall H. T., J. Wash.
Acad. Sci., 1957, 47, K« 9, p. 300; Hamann S. D., Physico-
chemical effects of pressure, L., 1957; Gilchrist A., Ind.
Chem., 1958, 34, № 402, p. 423, № 404, p. 545; В о v e n-
kerk H. P. [a. o.J, Nature, 1959, 184, № 4693, p. 1094.
Д. С. Циклис.
ВЫСОКИХ ДАВЛЕНИИ ТЕХНИКА — методы со-
здания высоких давлений, конструкции и материалы
аппаратов для проведения химич. реакций и физич.
процессов под высоким давлением, методы и аппара-
тура для исследования свойств веществ при высоких
давлениях. Корпуса аппаратов и нек-рых деталей
к ним изготовляют из специальных сталей, к-рые
наряду с высокими механич. свойствами достаточно
пластичны, тепло-, жаро- и коррозионностойки и пр.
Аппараты малых размеров, работающие под давле-
нием 50—100 ат, и аппараты для исследовательских
работ изготовляют также из специального стекла.
Многие вещества при высоких давлениях и темп-рах
действуют на материалы аппаратов. Напр., азот
реагирует с железом, образуя нитриды железа, что
увеличивает ломкость металла; водород обезуглеро-
живает сталь, что ведет к понижению ее механич.
прочности; кроме того, водород под высоким давле-
нием с большой скоростью диффундирует сквозь
стенки сосуда; окись углерода образует карбонилы
металлов, разрушая стенки сосудов; ртуть проникает
через стенки сосуда, внедряясь в поры и микроскопия,
трещины и образуя на свежей поверхности амаль-
гаму, что ослабляет металл; вода при высоких давле-
ниях сильно корродирует металл, а в стекле раство-
ряется; при снижении давления вода выделяется так
быстро, что стекло при этом разрушается. Коррозия
металлов усиливается под давлением.
Расчет и конструирование сосудов высокого давле-
ния. Стенки цилиндрич. сосуда, внутри к-рого создано
избыточное давление, нагружены очень неравномерно;
наибольшим напряжениям подвержены внутренние
слои. Момент появления пластич. деформаций в стенке
может быть установлен только экспериментально.
Приближенно эта задача м. б. решена с помощью
теории прочности. При этом получают ур-ния, позво-
ляющие рассчитывать аппараты, работающие при
давлении, к-рое вызывает только упругие деформации:
— — — 1Л
а° г» у —1,73 иРв
где а0 — отношение радиусов сосуда, В и г0 — на-
ружный и внутренний радиус сосуда, as — предел
текучести материала, т—запас прочности и Ро — ра-
бочее давление. Тогда s — толщина стенки:
S~'R— r<> =;r<1 <,а _ 1,73 тр„
Если s = со, а т — 1, то
^°пред. 1,73 = 0,577
Последнее выражение показывает, что аппарат
с бесконечно толстой стенкой не может выдержать
давление большее, чем ок. 0,6 предела текучести мате-
риала, из к-рого он изготовлен, т. е. для лучших
современных сталей ок. 20 000 кг/сж2. Однако опыт
показывает, что толстостенные сосуды выдерживают
без разрыва гораздо большее давление, т. к. за пре-
делом текучести металл переходит в пластич. состоя-
ние. При этом образуется пластич. слой, эквивалент-
ные напряжения (напряжения растяжения, которыми
заменяют сложнонапряженное состояние в стенке
сосуда, находящегося под давлением) в к-ром одина-
ковы по величине. При дальнейшем увеличении дав-
ления будет увеличиваться толщина слоя. Цилиндр
высокого давления оказывается как бы разделенным
на два надетых друг на друга слоя — внутренний
пластический и наружный упругий, удерживающий
пластич. слой от разрыва. По мере увеличения давле-
ния толщина пластич. слоя растет, а упругого умень-
шается и, наконец, когда упругий слой не в состоя-
нии выдержать давления пластич. слоя, он разры-
вается. Опыты показывают, что разрыв цилиндров
высокого давления начинается именно снаружи. Про-
исходящее При пластич. деформации упрочнение
металла (см. Высокие давления} и более выгодное
использование материала стенки в пластич. слое
позволяют значительно увеличить давление в аппа-
рате. Существует и метод расчета аппаратов, рабо-
тающих с пластич. слоем. Если темп-ра снаружи и
внутри аппарата различна, то в стенках сосуда воз-
никают напряжения, величина к-рых сравнима с ве-
личиной напряжений от давления и к-рые необходимо
учитывать при расчете. В стенке сосуда, подвергше-
гося пластич. деформации, между деформированным
внутренним и внешним слоями возникают напряже-
ния, к-рые сохраняются и при отсутствии внутрен-
него давления. Такой сосуд может выдержать большие
давления, чем обычный таких же размеров. Метод
упрочнения сосудов приложением избыточного давле-
ния, вызывающего остаточные деформации, назы-
вается автоскреплением и применяется,
напр., для упрочнения стволов артиллерийских ору-
дий. Расчет автоскрепления сводится к определению
давления, необходимого для проявления пластич.
деформаций, и размеров заготовки, к-рая после
автоскрепления имела бы веданные размеры.
693
высоких давлений техника
694
Многослойные сосуды. При надевании
плотно пригнанных друг к другу и нагретых цилин-
дрич. оболочек полу-
чают после охлаждения
цилиндр, составленный
из отдельных слоев,
надетых с натягом. На
границах слоев возни-
кают напряжения, бла-
годаря чему такой со-
суд выдерживает боль-
шее давление, чем одно-
< слойный цилиндр таких
РИС. 1. витой сосуд высокого ’’ же размеров. Разно-
давления: 1 — основная труба; ВИДНОстью МНОГОСЛОИ-
2 — профиль намотки. НЫХ сосудов являются
сосуды, изготовленные
из труб, на к-рые наматывают сваренные друг с дру-
гом тонкие стальные листы или сплошную, гладкую
или профилированную ленту (рис. 1), к-рую предва-
рительно нагревают и натягивают. После охлаж-
Рис. 2. Схема внеш-
него сжатия сосуда:
1 — поршень; 2 —
СОСУД; 3 — блок; 4 —
сжимаемое вещество.
дения получают сосуд с напря-
женными стенками, способными
выдержать значительные давле-
ния.
Сосуды с перемен-
ной внешней механи-
ческой поддержкой.
Наиболее простым приемом по-
вышения механич. прочности
сосуда является создание на его
наружной поверхности сжима-
ющих напряжений, пропорцио-
нальных внутреннему давлению.
Этого достигают, сделав сосуд
коническим и поместив его в
коническую оправку
Рис. 3. Бандажный аппарат:
1 — пуансон; 2 — матрицы;
3 — поддерживающие кольца
(бандажи); 4 — исследуемое
вещество; 5 — гидросиликат
алюминия (пирофилит).
20—30 тыс. ат жидкость
(рис. 2). Поршень, вдвигаясь в
сосуд, создает в нем давление и одновременно вда-
вливает сосуд в блок. При этом на его поверхности
возникает внешнее давле-
ние, возрастающее про-
порционально внутренне-
му и аналогичное давле-
нию на поверхности кли-
на. Применяя несколько
ступеней такого устрой-
ства (т. е. изготовляя
оправку конической и рас-
полагая ее в другой
оправке), а также незави-
спмое управление движе-
нием поршня в сосуде и
сосуда в оправке, мож-
но достичь давлений в
50 000 ат. Погрузив все
устройство в сжатую до
достигают в таком аппа-
рате давления 100 000 ат.
Бандажные аппараты. Для достижения
давлений в 100 000 ат и более при темп-pax до 1500—
2000° применяют аппараты (рис. 3), в к-рых вещество,
находящееся в матрице, сжимают двумя коническими
пуансонами. Между конусными поверхностями мат-
рицы и пуансонов проложено пирофилитовое (гидро-
силикат алюминия) уплотнение, упрочняющее узкую
часть пуансона, удельная нагрузка на к-рую больше
предела упругости материала пуансона. Пирофилит
также препятствует вытеканию исследуемого вещества
в зазорах. Для упрочнения матрица укреплена тремя
бандажами, запрессованными один в другой.
’Сосуды с гидравлической под-
держкой. Эти сосуды состоят из нескольких
цилиндров, вставленных один в другой. Зазоры между
дне. 4. Сжатие пла-
стических тел: М —
наивысшее давле-
ние, достигаемое в
точке А.
ними заполнены жидкостью, находящейся под гидро-
статич. давлением несколько меньшим, чем давление
в сосуде; поэтому каждая оболочка работает под
небольшим перепадом давления.
Огромных давлений можно достичь при сжатии ве-
ществ на малых площадях, окруженных массивными
массами металла. Поместив пластич-
ный материал под пуансон (рис. 4)
из карбида вольфрама, вдавливае-
мый в блок из того же материала и
погруженный в сжатую до 30 000 ат
жидкость, удалось достичь давле-
ния в 425 000 ат.
Методы создания давлений.
Сжатие газов. Газы сжима-
ют компрессорами различной про-
изводительности (от 10 м3/час до
50 000 м3)час). Сжатие производят
до 2000 ат (в промышленности) и до
12 000 ат (в лабораторных усло-
виях). Для сжатия небольших ко-
личеств газа и обеспечения высокой
степени его чистоты в сосуд нагнетают при высоком да-
влении жидкость или какой-либо сжатый газ. Сжи-
маемый газ отделен от среды, передающей давление,
ртутью, мембраной или металлич. мехом (сильфоном).
Значительного давления достигают испарением в замк-
нутом пространстве жидкости или нагреванием сжатого
газа. Большого увеличения давления добиваются за-
мораживанием в закрытом сосуде воды. На принципе
нагрева сжатого газа построен т. н. термокомпрессор
(для сжатия инертных газов), состоящий из приемни-
ка высокого давления и нескольких последовательно
соединенных сосудов высокого давления, к-рые раз-
делены обратными клапанами; в сосуды введены элек-
троподогреватели. Для сжатия газа производят авто-
матич. попеременное включение (выключение) подо-
гревателей. При этом находящийся во всей системе
под начальным давлением газ нагревается последова-
тельно во всех сосудах. Соответственно растет давле-
ние в каждом последующем сосуде и сборнике. По-
сле окончания цикла система вновь заполняется газом
и все операции повторяются до получения необходи-
мого давления. Очень высоких давлений достигают,
сжимая газ с помощью летящего поршня. В закры-
том с одного конца стволе находится тщательно
пришлифованный к нему поршень, который полу-
чает толчок от сжатого
газа, выпускаемого из
специального сборни-
ка. Поршень в стволе
летит к его закрытому
концу и сжимает нахо-
дящийся перед ним газ,
к-рый при этом нагре-
вается до высокой тем-
пературы. Отдав газу
всю энергию, поршень
останавливается и под
действием сжатого газа
летит обратно. Весь
цикл продолжается со-
тые доли секунды,
Рис. 5. Мультипликатор на
40 000 am: 1 — конический сосуд;
2 — коническая оправка; 3 —
оправки; 4 — ЦИЛИНДР; 5 — ПОр-
шень; 6 — поршень, двигающий
поршень высокого давления; 7 —
ЦИЛИНДР; 8 — поршень высокого
давления; 9 — затвор; ю — стя-
гивающие болты.
вследствие чего стенки
ствола и поршень оста-
ются холодными. Этот
метод удобен для изу-
чения газовых реакций
при высоких давлениях
и темп-pax. Газ высо-
кого давления можно
получить также путем проведения в аппарате какой-
либо химической реакции или при электролизе.
695
ВЫСОКИХ ДАВЛЕНИЙ ТЕХНИКА — ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ
696
Рис. 6. Вентили высокого давле-
ния: а) регулировочный с конусной
шпилькой; б) мембранный: 1 — кор-
пус; 2 — ниппель; 3 — шпилька;
4 — гайка; .5 — грундбукса; 6 —саль-
ник; 7 — мембрайа; 8 — шпиндель.
Сжатие жидкостей и твердых тел.
Жидкости сжимают жидкостными насосами или гид-
равлич. компрессорами. В лабораторных условиях
часто применяют ручные рычажные насосы и винтовые
прессы. Для сжатия значительных количеств жидкости
до давления порядка 5 000—10 000 ат используют
гидравлич. компрессоры с шлифовым уплотнением.
Для сжатия небольших количеств жидкости или твер-
дого вещества до 10 000—25 000 ат (при комнатно!!
темп-ре и таких давлениях почти все жидкости за-
твердевают) применяют гидравлич. пресса. Если от-
ношение площадей поршней гидравлич. пресса до-
статочно велико, то нагнетанием под большой пор-
шень жидкости под давлением 300—400 ат можно
создать под малым
поршнем давление в
10 000 ат и более.
Для создания давле-
ний больших, чем
25 000 ат, применя-
ют мультипликаторы,
сконструированные по
принципу механич.
поддержки. На рисун-
ке 5 изображен муль-
типликатор для соз-
дания давлений в
40 000 ат с двукрат-
ной механич. под-
держкой.
Детали аппаратов вы-
сокого давления. Вен-
тили. Различают вен-
тили регулировочные,
запорные и специаль-
ного назначения. Первые
служат для плавного из-
менения расхода газов и
жидкостей под давле-
нием путем изменения
площади зазора между
седлом и шпилькой. При
давлении до 2000 ат
шпилька, притертая к седлу, имеет угол заточни 10—15°.
Для работы при более высоких давлениях в конец шпильки
закатывают шарик, к-рый свободно вращается в шпильке
и ие трется о седло. Для запорных вентилей применяют
шпильку, на конце у к-рой имеется свободно вращающийся
конус с углом в 90°. Чтобы облегчить движение шпильки
в сальнике при больших давлениях, в ряде случаев применяют
прессвентили, у которых шпилька имеет обычно только воз-
вратно-поступательное движение, но не вращается. При
работе с высокими темп-рами шпильку удлиняют и выносят
сальник в охлаждаемую зону. Применяют также бессальнико-
вые мембранные вентили. Конструкции нек-рых вентилей
изображены на рис. 6.
Затворы. На рпс. 7 приведены различные конструк-
ции затворов. В нек-рых нз них уплотнение достигается тем,
что крышку аппарата прижимают болтами к корпусу с силой,
достаточной, чтобы преодолеть внутреннее давление и создать
в прокладке напряжение, большее, чем давление в аппарате.
В одних затворах прокладка претерпевает пластич. деформа-
цию (рис. 7, а), в других — напряжение создается на площади
соприкосновения пробки и тела корпуса (рис. 7, б). В кон-
струкциях с некомпенсированной площадью (рис. 7, в, г)
давление в аппарате так действует на затвор, что вызывает
«течение'» прокладок из мягкого материала (меди, алюминия,
свинца, пластиков, резины и др.), заполняющих предназна-
ченные для этого полости затворов, и создает в них напряже-
ния, автоматически превышающие давление в аппарате, что
препятствует утечке сжатой среды.
Конструкции промышленных аппаратов высокого
давления. Конструкции аппаратов высокого давле-
ния весьма разнообразны и зависят от характера про-
текающего процесса, геометрия, размеров и рабочих
условий (давление, темп-ра).
Типичным примером, реактора высокого давления
для осуществления газовых реакций является ко-
лонна синтеза аммиака (см. Аммиак). Этот аппарат
представляет собой толстостенный цилиндр с затво-
ром высокого давления, имеющий высоту до 15 м
и диаметр более 100 см. Такой реактор рассчитан на
давление 200—300 ат при темп-ре стенок 150—275°.
Колонна снабжена катализаторной коробкой и тепло-
обменником. Аналогичным образом устроены ко-
лонны синтеза метанола, колонны предкатализа про-
цесса синтеза аммиака, реакторы процессов гидриро-
Рпс. 7. Затворы высокого
давления: а) уплотнение с
наружной прокладкой: 1 —
корпус; 2 — пробка; з —
прокладки; 4 — кольцо; 5 —
нажимной болт; 6 — вытяж-
ной болт; б) конусный за*
твор; в) самоуплотняющий-
ся затвор: 1 — прокладка; 2—пробка; 3— нажимное коль-
цо; г) затвор с некомпенсированной площадью: 1 — проб-
ка; 2 — прокладка; з — грундбукса; 4 — гайна.
вания и др. Реакторы, работающие периодически
при высоких давлениях и темп-рах, называют авто-
клавами (см. Автоклавы лабораторные)', в них
чаще всего проводят реакции в жидкой фазе и в при-
сутствии твердых компонентов. Автоклавы снабжают
мешалками, к-рые вводят внутрь через сальники.
Иногда для вращения мешалки внутрь автоклава
помещают электродвигатель. В ряде случаев статор
помещают снаружи головки из немагнитной стали,
а ротор внутри.
Работа с аппаратами высокого давления требует
особой тщательности и строгого соблюдения всех
правил техники безопасности.
Лит.: Корндорф Б. А., Техника высоких давлений
в химии, Л.—М., 1952; Цикл ис Д. С., Техника физико-
химических исследований при высоких давлениях, 2 изд.,
М., 1958; Лачинов С. С. и Курковский В. А.,
Хим. наука и пром-сть, 1956, 1, № 6, с. 610; Верещагин
Л. Ф. (и д рЛ, ДАН СССР, 1960, 132, № 5, с. 1059; Гоник-
берг М. Г., Цик л ис Д. С. и О п е к у н о в А. А.,
ДАН СССР, 1959,129| № 1, с. 88; Р я б и н и н Ю. Н., Л и в-
ш и ц Л. Д., ЖТФ, 1959, 29, вып. 9, с. 1167; Hall Н. Т.,
Rev. Scient. Instrum., 1960, 31, №2, р. 125; Comings Е. W.,
High pressure technology, N. Y. — Toronto — L., 1956;
Tongue H., The design and construction of high pressure
chemical plant, L., 1959; New itt D. M., Design of high pres-
sure plant..., Oxf., 1940. Л. C. Uuk.vuc.
ВЫСОКбМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ —
химич. соединения, мол. вес к-рых составляет вели-
чину от нескольких тысяч до нескольких миллионов,
а в отдельных случаях достигает десятков миллионов
(напр., у нек-рых белков). В состав молекул В. с.
(макромолекул) входят сотни и тысячи ато-
мов, связанных друг с другом силами главных ва-
лентностей. Атомы или атомные группировки в мо-
лекуле В. с. могут располагаться либо в виде длин-
ной цепи (линейные В. с., напр. целлюлоза),
либо в виде длинной цепи с разветвлениями (р а з-
ветвленные В. с., напр. амилопектин), либо,
наконец, в виде трехмерной сетки, состоящей из от-
резков цепного строения (сшитые В. с.). В по-
следнем случае понятия молекулярный вес и моле-
кула утрачивают свой смысл. Примером сшитых В. с.
могут служить феноло-альдегидные смолы. Если
697
ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ
698
молекулярные цепи макромолекул В. с. состоят из
большого числа повторяющихся группировок —
звеньев, имеющих одинаковое строение, то такие
В. с. наз. полимерами, напр. поливинилхлорид
(—СН2—СНС1—)п, полиоксиметилен (—СН2—О—)п,
каучук натуральный [—СН2—С(СН3) = СН—СН3—]п
и т. п.
В. с., макромолекулы к-рых содержат несколько
типов повторяющихся группировок, наз. сополи-
мерами (напр., бутадиенстирольный каучук).
Примером В. с., цепи макромолекул к-рых построены
из различных звеньев, являются белки. Часто поли-
мерами наз. все В. с., особенно имеющие линейное
строение.
В зависимости от химич. состава основной цепи
В. с. делятся на два больших класса: гетероцеп-
н ы е В. с., в основной цепи к-рых содержатся атомы
различных элементов, чаще всего углерода, азота,
кремния, фосфора, игомоцепные В. с., цепи
к-рых построены , из одинаковых атомов. Среди них
основное место занимают карбоцепные В. с.,
главные цепи макромолекул к-рых состоят только из
атомов углерода, напр. полиэтилен, полиметилмет-
акрилат, политетрафторэтилен (см. Фторполимеры),
полихлоропрен, гуттаперча и др. Примерами гетеро-
цепных В. с. являются полиэфиры простые и сложные
(полиоксиэтилен, полиэтилентерефталат, поликарбо-
наты и др.), полиамиды, смолы мочевино-формалъде-
гидные, белки, целлюлоза, кремнийорганические поли-
меры. В. с., макромолекулы к-рых наряду с углево-
дородными группами содержат также атомы неорга-
ногенных элементов, наз. элементооргани-
ческими В. с. (см. Высокомолекулярные соединения
элементоорганические). В элементоорганич. полимерах,
содержащих атомы поливалентных металлов (напр.,
Zn, Mg, Си и др.), обычные ковалентные и ионные
связи могут сочетаться с координационными (внутри-
комплексные или т. н. клешневидные металлоорганич.
полимеры). Обширную группу В. с. образуют неор-
ганические полимеры, напр. пластич. сера,
С1
полифосфонитрилхлорид (—P=N—)пи др. (см. Высо-
С1
комолекулярные соединения неорганические).
В зависимости от формы макромолекулы В. с.
делятся на глобулярные и фибрилляр-
ные. У фибриллярных В. с. молекулы по форме
представляют собой линейные или слаборазветвлен-
ные цепи. Фибриллярные В. с. легко образуют над-
молекулярные структуры в виде асимметричных пачек
молекул — фибрилл. Цепи молекул внутри каждой
фибриллы ориентированы в одном и том же направле-
нии. Примеры фибриллярных В. с. — миозин, кол-
лаген, фиброин, целлюлозные волокна, полиамиды
и др. Глобулярными наз. В. с., макромолекулы к-рых
имеют форму более или менее шарообразных клубков,
глобул; последней может быть сильно разветвленная
макромолекула. Разрушение такой глобулы невоз-
можно без химич. деструкции макромолекулы. Воз-
можно также образование глобул из фибриллярных
В. с., связанное с изменением формы молекул В. с.
Отдельная глобула может быть образована гибкой
линейной молекулой (см. Гибкость цепных молекул),
свернувшейся в клубок под влиянием сил внутримо-
лекулярного взаимодействия, напр. в р-рах линейных
В. с. при добавлении нерастворителей или в р-рах
полиэлектролитов при изменении pH среды. Обрат-
ные переходы глобулярных структур в фибриллярные
при изменении виешпих условий имеют важное значе-
ние в технике и в биологии (напр., с этим связано
явление денатурации белков). По своему происхожде-
нию В. с. делятся на природные, напр. белки,
нуклеиновые к-ты, целлюлоза, натуральный каучук,
природные смолы, и синтетические — поли-
этилен, полипропилен, полиакрилаты, феноло-альде-
гидные смолы и др.
Свойства и важнейшие характеристики В. с. Свой-
ства В. с. определяются химич. составом, строением,
взаимным расположением макромолекул (надмоле-
кулярной структурой) в конденсированной фазе
В. с. В зависимости от этих факторов свойства В. с.
могут меняться в очень широких пределах. Так,
напр., полибутадиен, построенный из гибких углерод-
ных цепей, при комнатной темп-ре представляет собой
легко деформируемый эластичный материал, в то
время как полиметилметакрилат, цепи к-рого содер-
жат сильно взаимодействующие полярные группы,
при комнатных темп-рах является твердым, стеклооб-
разным продуктом; он приобретает каучукоподобные
свойства лишь при темп-рах порядка 100°. Целлю-
лоза — полимер с очень жесткими линейными цепями,
вообще не может существовать в каучукоподобном
состоянии вплоть до темп-ры ее химич. разложения.
В рассмотренных примерах различия в химич. со-
ставе вызывают существенные различия в физич.
свойствах В. с. Однако даже при одном и том же химич.
составе в зависимости от строения больших молекул
свойства В. с. могут сильно меняться. Типичным
примером могут служить полимеры полиэтилена,
полученные путем полимеризации при низком и вы-
соком давлении. Т. наз. полиэтилен низкого давле-
ния, имеющий линейное строение, плавится при более
высокой темп-ре, чем разветвленный полиэтилен,
полученный полимеризацией при высоком давлении
(соответственно 135° и 115°). Плотность и степень
кристалличности также значительно выше в случае
линейного полиэтилена. Большие различия в свой-
ствах В. с. могут наблюдаться даже в том случае,
если различия в структуре макромолекул на первый
взгляд и невелики. Так, изотактический полистирол,
к-рый, как и атактический полистирол (см. Изотакти-
ческие полимеры), построен из линейных цепей и отли-
чается от последнего лишь регулярной последователь-
ностью третичных асимметричных атомов углерода
в цепи, представляет собой кристаллич. вещество
с т. пл. ок. 235°, в то время как атактич. полистирол
вообще не способен кристаллизоваться и размяг-
чается при темп-ре ок. 80°. В данном случае различия
в микроструктуре макромолекулярной цепи влекут
за собой и различия в надмолекулярной структуре.
Благодаря регулярному строению цепей изотактич.
полистирола в нем могут возникать надмолекулярные
образования со структурой, характерной для кристал-
лич. полимеров.
Линейные В. с., в частности линейные полимеры,
обладают специфич. комплексом физико-химич. и
механич. свойств. Основные из этих свойств: 1) спо-
собность образовывать высокопрочные анизотропные
высокоориентированные волокна или пленки (см.
Полимеры волокнообразующие)', 2) способность давать
большие, длительно развивающиеся обратимые де-
формации, характеризующиеся малыми значениями
модуля упругости (см. Эластичность полимеров)',
3) растворение, проходящее через стадию набухания
(см. Растворы высокомолекулярных соединений). Весь
этот комплекс свойств обусловлен высоким мол. ве-
сом, цепным строением и гибкостью линейных макро-
молекул В. с. При переходе от линейных цепей к раз-
ветвленным цепям, редким трехмерным сеткам и,
наконец, к густым сетчатым структурам этот комплекс
свойств становится все менее выраженным. Сшитые
В. с. нерастворимы, не плавятся, не обладают плас-
тичностью, бывают иногда довольно хрупки и менее
эластичны.
699
ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ
700
Основными химич. реакциями, в к-рые вступают
В. с., являются: 1) образование химических связей
между макромолекулами (образование трехмерных
структур, так наз. сшивание, напр. вулканизация
каучуков, дубление кожи); 2) распад макромолекуляр-
ных цепей на отдельные, более короткие цепи (см.
Деструкция полимеров, Деполимеризация); 3) реакции
боковых функциональных групп В. с. с низкомолеку-
лярными в-вами, не затрагивающие основную цепь
и приводящие к образованию полимераналогов;
4) внутримолекулярные реакции, протекающие между
функциональными группами одной макромолекулы,
напр. случаи внутримолекулярной циклизации. Реак-
ции сшивания часто протекают одновременно с реак-
циями деструкции. Примером полимераналогичных
превращений может служить омыление поливипил-
ацетата, приводящее к образованию поливинилового
спирта. Гетероцепные В. с., в отличие от карбоцепных,
обычно легко гидролизуются (см. Высокомолекулярных
соединений гидролиз). Скорость реакций В. с. с низко-
молекулярными веществами определяется в основном
скоростью диффузии низкомолекулярного в-ва в фазу
В. с. Наиболее выражено это в случае сшитых В. с.
Важной особенностью В. с. является их высокая чув-
ствительность к действию небольших количеств при-
месей или добавок, реагирующих с макромолекулами.
Это объясняется тем, что в весовой единице В. с. содер-
жится в сотни и тысячи раз меньше молекул, чем в весовой
единице низкомолекулярного вещества. Напр., чтобы превра-
тить линейное В. с. из растворимого в полностью нераство-
римое, достаточно образовать на каждую макромолекулу одну-
две поперечные связи (связь между макромолекулами). Если
для этого затрачивается на макромолекулу одна молекула
низкомолекулярного реагента и одна реакционноспособная
группа, входящая в состав макромолекулы, то требуемое
весовое количество реагента должно быть относительно очень
невелико, в сотни и тысячи раз меньше весового количества
В. с. То же самое относится и к реакциям разрыва макро-
молекул, к-рые могут быть вызваны различными физич.,
химич. и механич. факторами.
Важнейшими характеристиками В. с. являются
химич. состав, мол. вес и молекулярно-весовое рас-
пределение, степень разветвленности и гибкость
макромолекулярных цепей. Свойства В. с. существенно
зависят от этих характеристик, хотя даже сами эти
характеристики (напр., степень разветвленности) еще
не всегда поддаются колич. оценке.
Получение В. с. Природные В. с. образуются в про-
цессе биосинтеза в клетках живых организмов. С по-
мощью экстракции, фракционного осаждения и др.
методов они могут быть выделены из растительного и
животного сырья. Неорганич. природные В. с. об-
разуются в результате геохимия, процессов, проис-
ходящих в земной коре. Синтетич. В. с. получают
путем реакций полимеризации и поликонденсации.
Карбоцепные В. с. получают в результате полимери-
зации мономеров, содержащих одну или несколько
кратных углерод-углеродных связей, или мономеров,
содержащих неустойчивые карбоциклич. группировки
(напр., циклопропан и его производные). Гетероцеп-
пые В. с. получают в результате реакций поликонден-
сации, а также полимеризации мономеров, содержа-
щих непрочные гетероциклич. группировки (напр.,
окиси олефинов, лактамы и др.).
Применение В. с. Необычайно широкая гамма
свойств В. с. и большие возможности получения на
их основе материалов с желаемым комплексом свойств
обеспечивают В. с. применение во всех областях
народного хозяйства. Благодаря своей механич.
прочности, эластичности, электроизоляционным и др.
ценным свойствам В. с. применяются в машинострое-
нии, строительстве, текстильной пром-сти, электро-
и радиопромышленности, с. х-ве, медицине, автомо-
билестроении, лакокрасочной пром-сти, судостроении.
Очень широкое применение В. с. находят в быту
в качестве разнообразных текстильных и кожевенных
изделий, посуды, мебели, клеев и лаков, украшений
и мн. др. предметов. Основными типами материалов
на основе В. с. являются резины, волокна, пласт-
массы, пленки и лакокрасочные покрытия. Биологич.
значение В. с. определяется тем, что практически
из них состоят все ткани живых организмов.
Лит.: Коршак В. В., Химия высокомолекулярных
соединений, М., 1951; Тагер А., Растворы высокомолеку-
лярных соединений, М., 1952; Воюцкпй С. С., Растворы
высокомолекулярных соединений, 2 изд., М., 1960; Лосев
И. П., Трост янская Е. Б., Химия синтетических
полимеров, М., I960; Б и л ь м е й е р Ф., Введение в химию
и технологию полимеров, пер. с англ., М., 1958; Кар-
гин В. А., Слонимский Г. Л., Краткие очерки по
фпзппо-хпмпп полимеров, М., 1960; Гликман С. А.,
Введение в физическую химию полимеров, Саратов, 1959;
Meyer К. Н., Natural and synthetic high polymers, L. —
N. Y., 1950; Роговин 3. А., Шорыгина H. H.,
Химия целлюлозы и ее спутников, М., 1953; Б а т ц е р Г.,
Введение в химию высокомолекулярных соединений, пер.’
с нем., М., 1960. В. А. Вабаноз.
ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ НЕ-
ОРГАНИЧЕСКИЕ — высокомолекулярные веще-
ства, молекулы к-рых построены из неорганических
цепей, состоящих из атомов и групп атомов, соединен-
ных между собой ковалентными, а в нек-рых случаях
и координационными связями. К В. с. и. относят
многие широко встречающиеся в природе минералы:
окислы (кремнезем, кварц, корунд, рутил), сульфиды
(аурипигмент, антимонит), нек-рые самородные эле-
менты, например серу, а также соли кислородных
поликремневых кислот и поликислот других элемен-
тов. К В. с. и. относится большая часть силикатов
и алюмосиликатов (слюда, асбест, тальк, глины
и мн. др.). Ряд В. с. н. является синтетич. продук-
тами. Многие В. с. н. •— неорганич. полимеры, макро-
молекулы к-рых состоят из большого числа законо-
мерно чередующихся одинаковых атомов или групп
атомов. В нек-рых случаях макромолекулы В. с. п.
построены из различных групп атомов, чередующихся
более или менее хаотично. В полимерах линейного
строения неорганич. цепи обрамлены одновалентными
атомами (водород, галогены) или группами атомов.
Если боковые группировки содержат органич. ради-
калы, то такие полимеры относят к полуорганич. или
элемептоорганич. полимерам (см. Высокомолекулярные
соединения элементоорганические). В плоскостных и
пространственных (сетчатых) неорганич. полимерах
цепи соединены между собой поперечными ковалент-
ными или координационными связями. В плоскостных
(слоистых) и линейных В. с. н. полимерные слои и
цепи могут связываться между собой как силами
Ван-дер-Ваальса (нитрид бора, графит, черный фос-
фор), так и ионами металлов (тальк, слюда, метасили-
каты). В зависимости от состава цепей В. с. н. разде-
ляются на две основные группы: 1) г о м о ц е п н ы е,
построенные из одинаковых атомов, и 2) гетеро-
цепные, цепи макромолекул к-рых образованы
чередующимися группами из двух и более атомов
различных элементов.
Макроцепи В. с. н. могут быть построены из эле-
ментов, имеющих валентность два и выше и способ-
ных соединяться прочйыми ковалентными связями.
В образовании цепей макромолекул и скелета В. с. н.
в случае этих элементов наряду с ковалентными свя-
зями могут участвовать также и донорно-акцептор-
иые (координационные) мостиковые связи, как это
имеет место, напр., в случае хлористого палладия
PdCl2:
Одновалентные элементы участвуют в построении
полимерных цепей только в случае, если они способны
701
ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ НЕОРГАНИЧЕСКИЕ
702
к образованию координационных связей, как, напр.,
цианид серебра----->-Ag—C=N----->-Ag—C=N------
Щелочно-земельные элементы (Ва, Sr и др.) полимер-
ных цепей не образуют. Такие элементы, как гало-
гены, не способные существовать в полимерном со-
стоянии, могут, однако, входить в состав макроцепей.
Способность к образованию гомоцепных неорганич.
полимеров обнаружена у следующих элементов: бор,
углерод, кремний, германий, фосфор, мышьяк, сурьма,
висмут, сера, селен, теллур и олово, т. е. у сравни-
тельно небольшого числа элементов, имеющих харак-
тер неметаллов. Число элементов, способных к обра-
зованию гетероцепных полимеров, значительно больше.
Доказательство высокомолекулярного характера тех
или иных неорганич. соединений часто сильно за-
труднено, т. к. не всегда удается найти подходящий
растворитель, в к-ром эти соединения растворялись
бы без заметной деструкции и в к-ром проявляли бы
себя как высокомолекулярные вещества. Заключение
о полимерном характере неорганич. соединений в на-
стоящее время в большинстве случаев зависит от
результатов анализа их кристаллич. структуры (мно-
гие В. с. н. являются кристаллич. веществами)
и от теоретич. выводов о природе химич. связи в твер-
дых телах.
В отличие от органич. высокомолекулярных соеди-
нений, у к-рых преобладают линейные карбоцепные
(т. е. гомоцепные) структуры, для известных В. с. н.
преим. характерны гетероцепные пространственные
(сетчатые) структуры. Большинство В. с. н. отли-
чается от органич. полимеров повышенной термо-
стойкостью, высокими темп-рами плавления или
размягчения, большой прочностью и твердостью.
Многие из них являются полупроводниками. Недо-
статком большинства пространственных В. с. н.
является их большая хрупкость. Поэтому задача
создания эластичных термо- и химически стойких
В. с. н. является весьма актуальной.
К г о м о ц е п н ы м В. с. н. линейного и про-
странственного строения, помимо упомянутых выше
веществ, к-рые могут иметь различную пространствен-
ную структуру (графит и алмаз, см. Углерод, красный
и черный фосфор и т. д.), относятся замещенные и
незамещенные полисиланы (—SiH2—) (=SiH—)
полигерманы (—GeII2—)п, полиарсаны (—RAs—)п и
полисульфаны Х(—S—)ПХ и др.
Гетероцепные В. с. н. состоят из макроце-
пей, построенных чередованием простых групп типа
—М—X—, —М—X—М'—X—М—X— и т. п., состоя-
щих из двух, трех и значительно реже из четырех и
более различных атомов. В этих цепях атомы элемен-
тов М, М' обычно разделены атомами элементов 2 и 3
периодов периодич. системы (X). К таким «разделяю-
щим» элементам относятся кислород [элементоксаны
(—М—О—)п], азот [элементазапы (—М—N—)га и
элементазены (—M=N—)п], сера [элементтиапы
(—М—S—) ] и т. д. Количество известных гетеро-
цепных В. с. н. весьма значительно, а возможности
синтеза новых полимеров этого типа неограниченно
широки. Ниже рассматриваются основные гетероцеп-
ные неорганич. полимеры в порядке нахождения
в таблице Менделеева элемента их образующего —
наименее электроотрицательного элемента М.
В. с. н. элементов II группы известно сравнительно
немного. К ним, напр., относятся окислы, сульфиды,
нитриды и оксикарбоксилаты бериллия, магния,
цинка, полимагнийоксаны Х(—Mg—О—)ПХ‘ («це-
мент Сореля»), полицинкоксаны Х(—Zn—О—)ПХ'
и др.
Из элементов III группы наибольшей склонностью
к образованию гетероцепных В. с. н. обладают бор,
алюминий, галлий, входящие в состав полимерных
цепей в виде группировок —М—О—, —М—N—,
—M=N— и —М—Р—. К полимерным соединениям
этой группы относятся В2О3, ВО, производные по-
либорных к-т (полибороксаны), алюмо- и боросили-
каты, корунд А12О3, полимерные гидриды галлия,
нитриды бора и алюминия, а также «фосфиноборины»
(—Р—В—)„ и др.
Все элементы IV группы могут входить в состав
гетероцепей неорганич. полимеров. Особой склон-
ностью к этому обладает кремний, образующий скелет
кремнезема, поликремневых к-т, макромолекулярных
анионов силикатов, полисилоксанов (—lai—О—) , ни-
।
тридов и сульфидов кремния, полисилазанов
I I
(—Si—N—)п, полисилтианов (—Si—S—)п, а также
разнообразных полиэлементосилоксанов (—£>!—О—
—М—О—)п, где М=В, Al, Ti, Р, V, Al, Sb'и т. д.
Значительно меньшее количество аналогичных поли-
меров известно для германия и олова. К В. с. н. угле-
рода относятся: ряд карбидов и оксикарбидов, поли-
меры CS, CS2, C3S2, CSe, HCN, SCN, нек-рые неорга-
нич. производные HCN, HSCN, CN, CO и т. д. Эле-
менты IV побочной группы — титан и цирконий —
образуют преим. кислородсодержащие гетерополи-
меры, как, напр., полититаноксаны (—Ti—О—) .
Разнообразные гетероцепные В. с. н., преим. кислород-
и азотсодержащие, образуют фосфор и мышьяк,
принадлежащие к V группе. К ним относятся окислы,
сульфиды и нитриды обоих элементов, полифосфаты,
полиарсенаты, полифосфатарсенаты, полифосфорил-
амиды, имиды фосфорных кислот, оксинитрид фос-
О N
Y И Y Y HI „ „
фора (—n=p—)п и фосфам (—NH—Р—) Наиболее
интересными полимерными соединениями фосфора
являются полифосфонитрилы (полифосфазены) и, в
частности, «неорганический каучук» — полифосфони-
С1
трилхлорид (—P=N—) . В. с. н. сурьмы, висмута, ва-
Cl
надия, ниобия и тантала известны гораздо меньше.
К сравнительно немногочисленным гетероцепным
полимерам элементов VI группы в основном относятся
полимерные окислы серы, напр. волокнистый сер-
о
ный ангидрид
а также полисульфимид
(-O-S-) ,
и "
(—NH—SO2—)п, нитрид серы. Нек-рые аналогичные
В. с. н. известны для селена и теллура. Полимерными
соединениями хрома, молибдена и вольфрама являются
гомо- и гетерополикислоты и их производные.
В. с. н. элементов VII и VIII групп почти неиз-
вестны.
Многие В. с. н. имеют огромное и многообразное
применение на практике, напр. в качестве строитель-
ных материалов (стемю, цемент и др.). Нек-рые из
неорганич. полимеров, напр. алмазы и др. драго-
ценные камни, корунд, карбид бора, применяются
в технике в тех областях, где требуются материалы
высокой твердости (бурение, обработка сверхтвердых
сплавов, точная механика и т. п.). В. с. н. широко
употребляются в качестве катализаторов, биологи-
чески активных препаратов, полупроводников, пла-
мезащитных пропиток, электроизоляционных мате-
703
ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ ЭЛЕМЕНТООРГАНИЧЕСКИЕ 704
риалов, минеральных волокон и ваты, ингибиторов
коррозии, поверхностно-активных веществ и др.
Лит.: Коршак В. В. Мозгова К. К., Усп. хим.,
1959, 28, вып. 7, с. 783; Итоги науки. Химические науки.
Сб. 3 — Химия и технология синтетических высокомолеку-
лярных соединений, вып. 1, кн. 2 М., 1959, с. 886—88; Бер-
лив А. А., И а р и н и В. П., Хим. наука И пром-сть,
1956, 1, № 1, с. 44; Усп. хим., 1949, 18, вып. 5, с. 546; Эл-
лис К., Химия синтетических смол, пер. с англ., т. 2, М.—Л.,
1940, с. 1779—89; М е у е г К. Н., Natural and synthetic high
polymers, 2 ed., N. Y. — L., 1950. p. 93—139; Krebs H.,
Angew. Chemie, 1958, 70 Jarg. H. 20, S. 615; В г у d s о n J.A.,
Plastics, 1957, 22, № 240, p. 384; Тило E., Химия и техно-
логия полимеров, 1960, № 7—8, с. 73—79; Е me 1 4us Н. J.,
Proc. Chem. Soc., 1959, VIII, p. 202. См. также литературу
к ст. Высокомолекулярные соединения. М. Г. Воронков.
ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ ЭЛЕ-
МЕНТООРГАНИЧЕСКИЕ — высокомолекулярные ве-
щества, содержащие в своем составе наряду с т. н.
органогенами (С, Н, О, N и др. элементами, обра-
зующими основную массу природных органич. сое-
динений) различные другие, неорганогенные элемен-
ты. В зависимости от химич. состава основной цепи
В. с. э. делятся на три большие группы.
У В. с. э. первой группы главные цепи
макромолекул состоят из неорганогенных элементов,
чаще всего из кремния, титана, олова, свинца, герма-
ния, алюминия, бора, фосфора и др. Связь между
указанными элементами в главной цепи осущест-
вляется во многих случаях через кислород, а углерод
входит только в состав групп, обрамляющих главную
цепь. К этим соединениям относятся, напр., поли-
1111
силаны: —Si—Si—Si—Si—, полиорган o-
I I I I
силоксаны, у к-рых в основной цепи чере-
I л I
дуются атомы кремния и кислорода: —Si—О—Si—О—,
полиметаллоорганосилоксаны, мак-
ромолекулярные цепи которых включают, кроме
атомов кремния и кислорода, атомы металлов:
—Si—О—Me—О—Si—, полиорганофосфонитрилы:
—P=N—, полиалюмооксаны: —А1—О—А1—О—.
I R „ II
У В. с. э. второй группы в основных цепях
макромолекул содержатся наряду с атомами углерода
и атомы неорганогенных элементов. Связь в гл. цепи
между углеродом и неорганогенными элементами мо-
жет осуществляться и через кислород. К этой группе
В. с. э. относятся, например, полифениленсиланы (I),
полифениленсилоксаны (И) и др.
I
- И'
К этой же группе В. с. э. относятся внутрикомплекс-
ные (т. н. клешневидные) элементоорганич. полимеры,
в к-рых сочетаются координационные, ионные и ко-
валентные связи. Так, при взаимодействии ацетил-
ацетоната цинка, магния, меди и др. металлов с тетра-
кетонами образуются полимеры, содержащие груп-
пировки следующего строения:
СН3 СНа
^С-°\ ,О=СЧ
-С ,Ме С-
ЧС=О' ХО-(/
I . ’ I
СНз СН3
У В. с. э. третьей группы основные цепи
молекул составлены из углерода или углерода и кис-
лорода, а обрамляющие группы содержат наряду
с углеродом также кремний, титан, олово, свинец,
германий, алюминий, бор, фосфор и др., напр.
-сн.,-сн— -сн3-сн—
I
С=О S1R.3
I
OSnRs
Главные цепи макромолекул В. с. э. могут быть
либо в виде длинной нити (линейные В. с. э., напр.
полидиметилсилоксан), иногда более или менее раз-
ветвленной, либо состоять из соединенных друг с дру-
гом циклич. группировок, напр. полиалюмофенилси-
локсан (I), где R = СН3, С2Н5, С8Н5 и др.
I I
R R О О R
II II
I. — О—Si—О—Si—О— II. —Si-0-Ti-O-Si—
II I I :
ОО R О О
II I
—О—S1—О—А1—О— -Si-O—
I
Имеются также «сшитые» В. с. э. с трехмерной струк-
турой (II).
Методы получения В. с. э. первой группы (с неор-
ганическими главными цепями макромолекул) осно-
ваны на реакциях гидролитической поликонденсации
(гидролиза или совместного гидролиза и последую-
щей поликонденсации) или обменных реакциях (реак-
ции обменного разложения и последующей поликон-
денсации). Реакция гидролитич. поликонденсации
осуществляется между элементоорганич. полифунк-
циональными мономерами и водой:
п В.,ЭХ2 + П Н2О —► (R»90) п + 2п НХ
(где X —С1, Вг, J, F или OR, О—С—R, NH»
Э—SI, Al, Т1, В, Sn и т. д.)
В простейшем виде по стадиям она протекает так:
В2ЭХ2 + Н2О---- Н,»ЭХ + нх
'I
он
2R29X —- R.,9-0—9R2 + Н»О
i II’
ОН XX
Н,»Э—О—9R2 + Н»О —► R2—Э—О—9R2
’ I I I ।
XX X OH
2R»-Э—O-OR» —► R2-9-O-9R2—O9R2—ОЭН.» + H2O и т. Д.
x ci>H x x
Если в качестве мономера используют трифункцио-
нальные элементоорганич. соединения, напр. R9X3,
или смесь ди- и трифункциональных мономеров, то
в результате такой ступенчатой поликонденсации
образуются высокополимерные соединения с разветв-
ленной или сетчатой структурой. Реакция совмест-
ного гидролиза в щелочной среде находит применение
для получения элементоорганосилоксанов, в частности
алюмоорганосилоксанов:
R
R»SIC1» + п А1С1, H3° + NaQH. -O-Si-O-Al-O-
I I
R
Реакция обменного разложения широко применяется
для получения элементоорганич. полимеров с глав-
ными цепями молекул, построенными из кислорода
и др. элементов, напр.:
RSiCl3 + NaOH—-2 RSi(OH)»ONa + NaCl
4RSl(OH)2ONa + TiCl4 — [RSi(OH)»O]4Tl + 4NaCl
Полученный растворимый продукт общей ф-лы
[RSi(OH)2O]4Ti при нагревании конденсируется и
превращается в нерастворимый и неплавкий прост-
ранственный полимер.
705
ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ ЭЛЕМЕНТООРГАНИЧЕСКИЕ
706
Вследствие большой склонности элементоорганич.
мономеров при превращении их в полимеры образовы-
вать низкомолекулярные циклич. соединения, все
эти реакции синтеза полимеров с неорганич. цепями
макромолекул часто сопровождаются реакциями
циклизации. Если синтез органич. полимеров поли-
конденсацией приводит, как правило, к образованию
линейных, разветвленных или сшитых полимерных
молекул, то синтез полимеров с неорганич. цепями
макромолекул, осуществляемый путем совместного
гидролиза и поликонденсации или обменного разло.
жепия и поликонденсации, приводит к образованию
не только линейных или разветвленных, но при
использовании мономеров с функциональностью
больше двух, преимущественно к циклолинейпым и
циклоразветвленным молекулам (см., напр., выше
полиалюмофенилсилокс ан).
Образование циклов наблюдается при синтезе и
других полимеров, например полифенилфосфинов,
полиборорганофосфинов и полиорганофо'сфонитрилов.
Так, при действии фенилфосфина на фенилдихлорфос-
фин образуется циклотетрафосфин:
гСоЩРСЦ + 2СаН5РНг —► (C0H5P)d + 4IIC1
Бороводороды реагируют с метилфосфином с образо-
ванием циклических соединений:
ЗСН3РН2 +ЗВН3
.ВН
сн3р-^ ^РСН
НВ ВН
'Хр/
I
сн3
3
При действии хлористого аммония на трихлордиме-
тилфосфин образуются циклич. соединения такой
структуры:
(СН3)а РС13 + NH4C1 —*
(CH3)2P=N-P(CHg)2
N N
(CHg)2P- N =P(CH3)2
,P(CH3)2
N
(CHg)2fL J(CH3)2
A
Ввиду отмеченной
склонности элементо-
органич. мономеров
к циклизации при
синтезе В. с. э. боль-
шое значение приобретаютреакцииразмыкания и поли-
меризации циклов из неорганич. атомов, обрамлен-
ных органич. группами. Термич. полимеризация
циклич. соединений, содержащих группы —N=P— и
—В—Р— в циклах, легко протекает с образованием
полимеров. Циклические соединения, содержащие
атомы Si, Al, Ti, В, Р, связанные через кислород, из-за
высокой термостабильности этих циклов полимери-
зуются с трудом. Такие циклич. соединения с 6, 8 и
большим числом членов в цикле легче размыкаются
и полимеризуются под действием кислот и щелочей:
СНз СНз
I
СНз—Si—О—Si—СНз СНз
п О О 4- H<SO4—► КО (—S1—О—)4n SOsH
СНз-SI-O-SI-CHs СН3
t I
СНз СНз
С помощью этой реакции получают В. с. э. как с ли-
нейной, так и с разветвленной структурой макромо-
лекул.
Находит также применение способ получения ли-
нейных полисилоксанов, основанный на поликонден-
сации двух мономеров, содержащих по две функцио-
12 К. X. Э., т. 1
нальные группы разнообразного типа, напр.;
R r R R
। I II
п R'0-Sl-OR' 4- п Cl—Si-Ci —- —О—SI-0-S1- + 2n R'Cl
I 1 II
R R R R
Катализаторами таких реакций служат галогениды
металлов (FeCl3, СаС12). Описанные методы применимы
в большинстве случаев и для синтеза В. с. э., содер-
жащих другие элементы в цепи. Полимеры, не со-
держащие атомов кислорода в цепи (напр., полисила-
ны), могут быть получены, напр., из дигалогенозаме-
щенных силанов по Вюрцу:
R R R
I I I
2С1-Si-Cl+2Na —- Cl—Si—Si—Cl 4- 2NaCl
I I I
R R R
Полимеры второй группы (содержащие в основных
цепях макромолекул наряду с атомами углерода
атомы неорганогенных элементов) могут быть полу-
чены как полимеризационными, так и поликонден-
сационными методами. Так, поликарбосилоксановые
полимеры образуются при поликонденсации алко-
ксисиланов с эфирами гликолей:
R 0 0
СНз-о-Si—О-СНз + СНз-ft-O-СН3-О-ft-СН3 ;
I
R
R R
I I
—О—Si—О—СНз—О—S1—О— + СН3СООСН3
I I
R R
Фосфорсодержащие полиэфиры получают поликон^
денсацией дигалогенангидрида фосфорной к-ты с гли-
колями или двухатомными фенолами:
0 г 0 1
n Cl-ft-Cl + п НО—R'—ОН—- —ft—О—R'—О— -|-2пНС1
R I- R -*п
Фосфорсодержащие полимеры, не содержащие атомов
кислорода в цепи, получают сополимеризацией олефи-
нов с алкилгалогенфосфинами:
01 С1-,
п СН=СН3 + п РС13
ft к
— СН—СНз—1р—
- ft ft
+ CH3OH
Внутрикомплексные В. с. э. могут быть получены
действием ионов таких металлов, кай Zn, Си, Ni,
Со, Ве и т. д., на молекулы полифункциональных
электронодонорных соединений: бис- (а-аминокис-
лоты), бис- (Р-дикетоны) и др.
Полимеры третьей группы обычно получают поли-
меризацией мономеров, содержащих элементоорга-
нич. группировки в боковой цепи. Таким путем об-
разуются, напр., политриэтилстаннилметакрилат, по-
лимеры аллильных соединений германия, поливинил-
и полиакрилсиланы.
В. с. э. занимают промежуточное положение между
органич. и неорганич. полимерами. Наличие в струк-
туре молекул В. с. э. неорганогенных элементов и
элементов, характерных для органич. веществ, опре-
деляет их специфичность, проявляющуюся в сочета-
нии свойств, присущих неорганич. материалам (тер-
мич. стойкости, твердости), с эластичностью, термо-
пластичностью и др. свойствами органич. полимеров.
Это можно видеть на примере широко использую-
щихся в технике кремнийорганических соединении —
707
ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИИ ГИДРОЛИЗ
708
полиорганосил оксанов, свойства к-рых хорошо изу-
чены. Они могут длительно выдерживать действие
темп-p до 200° и выше. Теплостойкость полиоргано-
силоксанов значительно выше, чем у большинства
органич. полимеров, по все же ниже, чем у неорганич.
веществ. Термостабильность уменьшается с увеличе-
нием числа органич. групп, связанных с атомом
кремния, на нее влияет также тип и состав этих
групп. Высокой термич. и химич. устойчивостью
отличаются многие клешневидные (хелатные) метал-
лоорганич. полимеры.
Особенности структуры и состава молекул обуслов-
ливают не только повышенную термостабильность,
но и ряд других свойств полиорганосилоксанов,
напр. высокую светостойкость, а также устойчивость
к действию сильных электрич. полей, сравнимую с
таковой для слюды и кварца. Под _ NH ,сн . NH
действием сильного электрич. поля 211
силоксановая цепь не разрушает- н
ся, а происходит только отрыв и - —О(СН2)по-г
разрушение органич. групп, место т
к-рых занимает кислород. Присоединение кислорода,
как и отщепление боковых органич. радикалов, при-
водит к образованию сетчатых полимеров с частыми
поперечными связями, что вызывает повышение
жесткости полимера, а следовательно, и его хруп-
кости. Высокая сжимаемость кремнийорганич. жид-
ких полимеров и незначительное изменение вяз-
кости с темп-рой является типичным для полиоргано-
силоксанов. Напр., полидиметилсилоксановая резина
сохраняет эластич. свойства при —70°, а жидкие по-
лимеры не замерзают до —120°, и у них вязкость из-
меняется с понижением темп-ры в 50 раз меньше, чем
у органич. и нефтяных масел. Циклолинейная струк-
тура главных цепей макромолекул сказывается,
в частности, на повышении растворимости. Жидкие
полиорганосилоксаны и полиалкилоксититаноксаны
используются для гидрофобизации тканей, бумаги,
стекла, фарфора, строительных материалов, кожи
и т. д. Обработанные ими материалы не смачиваются
водой и не пропускают воду, но могут пропускать
воздух (см. Гидрофобные покрытия). Маслообразные
В. с. э. используются для смазок, работающих в ши-
роком диапазоне темп-p от —70° до 200° и кратко-
временно до 400°. Фосфорорганич. полимеры исполь-
зуются для понижения горючести тканей и др. орга-
нич. полимерных материалов. Введение нек-рых
В. с. э., напр. полиорганосилоксанов, в масла, в вод-
ные р-ры в количествах одной части на миллион
частей устраняет вспенивание, они широко исполь-
зуются в качестве антипенных присадок в произ-ве
пищевых продуктов, сахара и лекарственных ве-
ществ — пенициллина, стрептомицина и др. антибио-
тиков. Жидкие полимеры используют в качестве
амортизирующих и демпфирующих веществ, тепло-
носителей, применяют как диэлектрики и т. д. Поли-
органосилоксановые смолы и каучуки служат для
изготовления пластич. масс, стеклотекстолита, для
произ-ва электрич. изоляции, покровных лаков.
Пластич. массы и различные виды электрич. изоляции
из В. с. э. (в основном из кремнийорганич. полиме-
ров) применяют в произ-ве электрич. машин, транс-
форматоров, радиоаппаратуры, кабелей, электро-
ники. Они позволяют обеспечить работу электротех-
нич.’ изделий в течение 8—10 лет при 180°. Полиорга-
нофосфонитрилы и полифенилсилоксаны выдержи-
вают очень высокие темп-ры; они в сочетании с напол-
нителями, напр. алюминием, применяются в качестве
жаростойких лакокрасочных покрытий, работающих
при темп-pax до 600°.
Ценные свойства В. с. э. обеспечивают их широкое
использование не только в технике, но и в медицине,
пищевой, парфюмерной пром-сти, в быту.
Лит.: Андрианов К. А., Кремнийорганические со-
единения, М., 1955; его ж е, Теплостойкие кремнийоргапи-
ческпе диэлектрики, М.—Л., 1957; К р е ш и о в А. П.,
Кремнийорганические соединения в технике, 2 изд., М.,
1956; Андрианов К. А., Усп. хим., 1957, 26, вып. 8;
1958, 27, вып. И; Л о с е в И. П. и Т р о с т я н с к а я Е. Б.,
Химия синтетических полимеров, М., 1960, гл. V; А н д р и а-
н о в К. А., в сб.: Химия и технология полимеров, 1960,
№ 7—8. См. также литературу в ст. Высокомолекулярные
соединения. К. А. Андрианов.
ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ ГИД-
РОЛИЗ — деструкция гетероцепных высокомоле-
кулярных соединений под действием воды, водных
р-ров к-т, щелочей и солей. Реакция гидролиза за-
ключается в разрыве гетероцепной связи под влиянием
гидролизующего агента и может быть представлена
след, схемой (для случая гидролиза полиамидов и
полиэфиров):
СО(СН2)тСО— ...-. ... -NH(CH2)nNH24-НООС(СН2)тСО -...
он
СО(СН2)тСО - ---.... - О(СН3)ПОН 4- НООС(СН2)тСО-...
он
Способность высокомолекулярных соединений к гидро-
лизу определяется характером гетероцепной связи
в молекуле полимера, а также природой гидролизую-
щего агента и условиями-его воздействия. По чувстви-
тельности к гидролизу различные гетероцепные
связи располагаются приблизительно в след, порядке:
О О
—сн—о— II л
f >-C-NH- > -С-О- -S- -О-
Гликозид- Амидная Сложноэфир- Тиоэфир- Простая
ная связь связь ная связь пая связь эфирная
связь
Реакции гидролиза играют существенную роль в про-
цессах поликонденсации, сопровождающихся выде-
лением воды. При этом количество гидролизованных
(разорванных) гетеросвязей пропорционально коли-
честву гидролизующего агента. Гидролизу подвер-
гаются, в первую очередь, молекулы большей длины.
Эти особенности процесса гидролиза приводят к полу-
чению в результате поликонденсации полимера с
узким молекулярно-весовым распределением.
Наибольшее практич. значение имеет гидролиз
целлюлозы. При этом происходит разрыв гликозид-
ной связи;
Скорость гидролиза целлюлозы увеличивается в при-
сутствии минеральных к-т, к-рые являются катализа-
торами этой реакции. С увеличением степени гидро-
лиза и понижением мол. веса происходит закономер-
ное ухудшение механич. свойств целлюлозы и увели-
чивается растворимость в щелочах. Полный или
частичный гидролиз целлюлозы может быть осуще-
ствлен действием на нее конц. минеральных к-т,
разбавленных водных р-ров минеральных к-т и без-
водных к-т. Изменяя основные- параметры процесса
гидролиза целлюлозы (время, темп-ру, концентрацию
к-ты), можно получить продукты гидролиза, отли-
чающиеся по средней степени полимеризации и рас-
творимости в щелочах. Полный гидролиз целлюлозы
может быть проведен действием конц. к-т на холоду
или разбавленных к-т при высокой темп-ре под давле-
нием. В результате полного гидролиза целлюлозы,
т. е. разрыва всех гликозидных связей, образуется
D-глюкоза с выходом 92—96%. Глюкоза может быть
получена и при гидролизе растительных материалов,
содержащих целлюлозу. Гидролиз древесины яв-
709
ВЫСШИЕ ЖИРНЫЕ КИСЛОТЫ
710
ляется одним из методов получения этилового спирта;
при гидролизе растительных материалов возможно
также получение пищевых сахаров (глюкозы, кси-
лозы). В. с. г. — один из спосо-
бов изучения строения высоко-
молекулярных соединений.
Лит.: Коршак В. В., Хи-
мия высокомолекулярных соедине-
ний, М.—Л., 1950, с. 116—20;
Роговин 3. А., Шорыгин а
Н. Н., Химия целлюлозы и ее
спутников, М.—Л., 1953, гл. 5;
Коршак В. В. «Рафиков
С. Р., Синтез и исследование высоко-
молекулярных соединений, М.—Л.,
1949. Ю. С. Липатов.
ВЫСШИЕ ЖИРНЫЕ КИ-
СЛОТЫ — карбоновые к-ты
алифатического ряда, природ-
ные с четным числом углерод-
ных атомов, неразветвленного
строения; наиболее распростра-
ненные В. ж. к. содержат от
10 до 24 атомов С в молекуле;
известны и более сложные. В
животных и растительных жи-
рах и маслах В. ж. к. содер-
жатся преим. в форме сложных
эфиров глицерина, а в живот-
ных восках — в форме сложных
эфиров высших жирных спир-
тов. Из насыщенных В. ж. к.
(СПН2)1^4 СООН) наибольшее
значение имеют лауриновая,
миристиновая, пальмитиновая,
стеариновая и арахиновая; из
ненасыщенных, с одной или
несколькими двойными связями в молекуле
СпН2п_1СООН до СпН2п_эС00Н), важнейшими
ляются олеиновая, линолевая, арахидоновая,
леновая, 1
кислотах).
Схема
(от
яв-
лино-
клупанадоновая к-ты (см. статьи об этих
Природные В. ж. к. получают гидролизом глицери-
дов, содержащихся в жирах и маслах, индивидуаль-
ные В. ж. к. выделяют из их смеси дробной перегонкой
Парафин с температурой
кипения 320-450°
50-60 кг
упарка и Сушка
Ci)(Са)(с?) (^-5 Ctn(c7~C^fci0“C^fc|4“Ci^^i7 еь/ше.
78-80 кг 180-200 кг 75-80 кг
25-30 кг 150-170 кг 140-150 кг
Сульфат
натрия
300-400 кг
получения синтетических жирных кислот.
в вакууме, дробной кристаллизацией, при помощи
селективных растворителей, а в лаборатории — и хро-
матографии. методом.
Синтетические В. ж. к. с четным и нечетным числом
атомов С в молекуле (в том числе разветвленного строе-
Кислота Температура, °C Плотность 80 nD Число нейтрали- зации Подное число
засты- вания плавле- ния перегон- ки, °C/jw.w
Каприновая СщН.оО,. 31,2 31,6 146/10 0,866 (40°) 1,4286 (40°) 326,0 —
Ундекановая3 СцН..О8 44,0 28,5 176/15 0,864 (60°) — 301,0 —
Лауриновая С^Н^О» 43,9 44,2 176/15 0,875 (44°) 1,4191 (60°) 280,23 —
Тридекановаяа С13Н.26О» 41,8 40,5 236/100 — — 262,0 —
Миристиновая 54,1 53,9 196/15 0,862 (54,0°) 1,4273 (70°) 245,81 —
Пентадекановая® С15Н30О2 53,2 58,0 195/13 — — 232,0
Пальмитиновая 62,8 63,1 215/15 0,853 (63°) 1,4272 218,92 —
Гептадекановаяа С17Н34О2 61,0 60,0 277/100 0,853 (60°) 1,4342 (60°) 208,0 —
Стеариновая С18Н33О3 69,3 69,6 232/15 0,845 (70,5°) 1,4299 197,33 —
Нонаденановаяа —. 66,5 298/100 — — 188,0 —
Арахиновая С^Н^Оо 74,9 75,3 — —- 1,4250(100°) 179,63 —
Бегеновая С22Н44О2 79,7 79,9 — — 1,4270(100°) 152,29 —
Олеиновая® С^Нз^Оз — 14 232,5/15 0,898 (14°) 1,447 (60°) 198,63 89,87
Эруковая С22Н42О3 — 34 254,5/10 0,8602(55°) — 165,72 74,98
Линолевая® Ci8H3»O3 ' . . . . — -5-5,2 230-233/15 0,9206 (14°) 1,469 (20°) 200,06 181,03
Линоленовая® Ci8H30O2 — -11-11,3 230—232/17 0,9218 (15°) — 201,51 273,51
Клупанодоновая® С2»Н3$О3 — — — — — 169,76 384,03
Арахидоновая® С2сН32О2 — 49,5 — — — 184,28 333,5
Рицинолевая® Cj8H34O3 — 5 — — — 187,98 85,04 (теоретич.).
а Синтетическая насыщенная кислота. ® Ненасыщенная кислота. в Ненасыщенная оксикислота.
12*
711
ВЫСШИЕ ЖИРНЫЕ СПИРТЫ - ВЮРЦА РЕАКЦИЯ
712
ния) получают окислением парафиновых углеводо-
родов. Протекающая цепная реакция проходит через
промежуточное образование гидроперекисей и кетонов.
Число атомов С в молекуле образующейся кислоты
всегда ниже, чем в исходном углеводороде. Иницииро-
вание цепи с получением радикалов СН достигается
введением в реакцию продуктов окисления (кислот,
спиртов, кетонов, эфиров, лактонов). Катализаторами
окисления являются окислы марганца. Для окисле-
ния в пром-сти применяют нефтяной или синтетич.
парафин, т. пл. 52—54°, выкипающие при 320—450°,
с минимальным содержанием примесей масел и наф-
тенов.
Принципиальная схема получения синтетич. В.ж. к. приве-
дена на схеме (стр. 710). Смесь свежего (*/») и возвратного (2/3)
парафина загружают в окислительную колонну и в присутст-
вии катализатора (0,2% перманганата калия) при 101—105°
барботируют через него воздух (40—60 Л18»?п/час). Через
14—16 часов по окислении примерно третьей части парафина
его выгружают из колонны, отделяют отстаиванием отработан-
ный катализатор. После промывки водой для отделения водо-
растворимых кислот массу нейтрализуют щелочью и передают
сначала в отстойник, а затем в автоклав, в к-ром при 150—180°
и давлении 20 ат происходит отделение невошедшего в реакцию
парафина. Натровое мыло сырых жирных к-т пропускают затем
через термич. печь, в к-рой происходит облагораживание
кислот: дегидратация оксикислот, распад лактонов и омыление
эфиров В. ж. к. и спиртов, а при выходе из печи также отгонка
последних остатков непрореагировавшего парафина. Мыла
затем разлагают кислотой, а смесь сырых жирных кислот
подвергают фракционной дистилляции. Выход смеси кислот
70—80% от веса парафина.
В. ж. к. широко применяют для произ-ва хозяй-
ственных, туалетных и технических твердых и жидких
мыл, в произ-ве синтетич. каучука и резиновых изде-
лий, при изготовлении синтетич. смол для лаков,
эмалей и олиф, в качестве сырья для произ-ва синте-
тич. высших жирных спиртов и поверхностно-актив-
ных веществ, в составе синтетич. смазок и в других
областях.
В таблице (стр. 710) приведена характеристика наи-
более важных В. ж. к.
Лит.: Зиновьев А. А., Химия жиров, М., 1952;
Технология переработки жиров, под общ. ред. Б. Н. Тютюн-
никова, 2 изд М., 1956; Алаев Б. С., Маньков-.
с к а я н. к., ш и м а н А. М., Производство синтетических
жирных кислот, [М.], 1960. II. М. Товбин.
ВЫСШИЕ ЖИРНЫЕ СПИРТЫ — спирты, в мо-
лекулах к-рых содержится от 10 до 24, а иногда и
больше атомов углерода; твердые продукты. Природ-
ные В. ж. с. преим. одноатомные первичные, насыщен-
ные и ненасыщенный; встречаются в виде эфиров
жирных к-т или в свободном состоянии в восках,
напр. карнаубском и др., в кашалотовом жире, в шер-
стяном жире, извлекаемом из промывных вод шерсто-
моек, в пчелином воске. В. ж. с. производят также
и синтетически. Важнейшими из природных В. ж. с.
являются: цетиловый спирт, цериловый спирт, ми-
рициловый спирт, олеиновый спирт.
Для получения В. ж. с. из восков последние омы-
ляют водным р-ром едкого натра при кипячении;
спирты отделпют от образовавшегося мыла. Ресурсы
природных В. ж. с. ограничены, поэтому основное
количество их получают восстановлением высших
жирных к-т. Для этого может служить метод Буво и
Блана (см. Буво — Блана восстановление). При методе
Буво и Блана в спиртах сохраняются все двойные
связи исходных жирных к-т. В пром-сти В. ж. с.
производят главным образом каталитич. гидрогениза-
цией жирных к-т или их эфиров (метиловых, бутило-
вых) над хромо-модным катализатором при 250—300°
и 220—250 ат. Процесс ведут непрерывно в аппа-
ратах колонного типа; с хромэ-цинковым катализа-
тором по этому методу получают также и ненасыщен-
ные В. ж. с. Синтетич. В. ж. с. получают также окис-
лением нормальных парафинов (т. кип. 270—320°)
азото-кислородной смесью (4—5% О2) в присутствии
борной к-ты, к-рая образует со спиртами эфиры;
последние после отделения от невошедших в реакцию
углеводородов разлагают водой, борную к-ту реге-
нерируют и возвращают в процесс, а спирты перего-
няют. Этим методом получают смесь, состоящую из
10—15% первичных и 85—90% вторичных В. ж. с.;
по качеству они уступают спиртам, получаемым вос-
становлением жирных к-т, однако процесс произ-ва
проще. В. ж. с. предложено получать также оксо-
синтезом, из СО и Н2 на железо-медном катализаторе
при давлении 20—25 ат или на железном катализа-
торе при давлении 200—250 ат. Частично В. ж. с.
образуются в качестве попутных продуктов при
произ-ве синтетич. жирных кислот окислением па-
рафина. Они извлекаются из неомыляемых получе-
нием их борных эфиров или экстракцией метиловым
или этиловым спиртом.
В. ж. с. применяют в произ-ве алкилсульфатов —
поверхностно-активных веществ, входящих в состав
синтетич. моющих средств, вспомогательных веществ
в текстильной промышленности, косметич. изделий,
грима, мазей и др.
Лит.: Зиновьев А. А., Химия жиров, М., 1952;
Шварц А., Перри Дж., Берч Д ж., Поверхностно-
активные вещества и моющие средства, [пер. с англ.], М
1960. И. М. Товбин.
ВЫХОД ПО ТОКУ (ВТ) — доля электричества,
израсходованная непосредственно на выделение ка-
кого-либо вещества в ходе электролиза. ВТ определяет-
ся опытным путем из соотношения ВТ = m/ite — m/Qe
(см. Фарадея законы), где т — количество выделив-
шегося (или растворившегося) вещества за время г
(в секундах), в течение к-рого через электролитич.
ячейку проходил ток с силой i, е — электрохимический
эквивалент в-ва. Точную величину количества про-
шедшего электричества Q—it обычно определяют
с помощью кулометра. При умножении вышеприве-
денного соотношения на 100 получается ВТ в про-
центах. При электролитич. получении многих ве-
ществ ВТ часто меньше 100% из-за частичного рас-
хода электрич. тока на побочные процессы. При про-
ведении электролиза в водных р-рах такими побоч-
ными процессами являются выделение водорода на
катоде и кислорода на аноде. ВТ для многих случаев
электролиза зависит от плотности тока, концентрации
электролита, pH р-ра, природы материала электрода
и от др. факторов. ВТ снижается также из-за утечек
тока, потерь продуктов электролиза, из-за взаимо-
действия катодных и анодных продуктов электролиза.
В наиболее совершенных электролизерах при отсут-
ствии побочных электролитических процессов ВТ
достигают 100%. Н. Е. Хомутов.
ВЮРЦА РЕАКЦИЯ — образование парафинов при
действии металлич. натрия па галогеналкилы (обычно
иодистые или бромистые):
2RHal + 2Na —- R— R 4- 2NaHaI
В. p. co смесью 2 галогеналкилов приводит к трудно-
разделяемой смеси всех возможных продуктов реак-
ции:
RX + R'X --> R-R 4- R'—R' 4- R'—R
Механизм В. р. точно не установлен, однако имеются
данные как о промежуточном образовании натрийал-
килов (1), так и о свободнорадикальном характере
реакции (2):
RX 4- Na —* RNa; RNa -|- RX —• R-R (1)
или
RX 4- Na —- R-; 2R- —- R-R (2)
При низкой темп-ре наличие патрийалкилов можно
зафиксировать, пропуская в реакционную смесь угле-
кислый газ: RNa^>RCOONa. Побочное образо-
вание при В. р. алканов и алкенов с тем же числом
углеродных атомов так же, как и превращение опти-
713
ВЯЖУЩИЕ МАТЕРИАЛЫ — ВЯЖУЩИЕ МАТЕРИАЛЫ ГИПСОВЫЕ
714
чески активных галогеналкилов в неактивные пара-
фины, согласуется с обеими возможными схемами
реакции:
Уа .
н„с
2|
R—СН2
НСН—СН2—Cl
I
R ------
Na
н,с*-
I
R—СНг
Н:СН —СН2С1
R
СН3
R—Сн2
RCH2CH2Br
RCH2CH2
R£H2CH3 + RCH = CH2
В. р. хорошо идет с галогенидами большого мол.
веса и очень удобна для синтеза высших углеводоро-
дов. С низшими галогеналкилами В. р. осложняется
многими побочными процессами, и поэтому выходы
чистых углеводородов часто невысоки. В 1957 уда-
лось показать, что присутствие тетрафенилэтилена,
служащего, переносчиком натрия, устраняет эти
недостатки:
Na
(С0Н5)2С=С(С0Н5)2 (C0Hs)2C(Na)-(Na)C(C0H5)s
2RX + (C0H5)2C(Na)-(Na)C(C8H5)» —• R - R +
4- (C0H5),C= C (С0Н0)2 4- NaX
В среде тетрагидрофурана реакция протекает быстро и
с хорошими выходами уже при —80°. Действие те-
трафенплэтилена проверено на галогенидах типа хло-
ристого аллила или бензила, а также на бис-галоген-
метильных производных бензола, превращающихся
в соответствующие 8-, 10- и 12-членные циклич. угле-
водороды. га-(Дихлорметил)-бензол может быть пре-
вращен в линейный полимер.
В. р. можно получать не только углеводороды, но
и различные элементоорганич. соединения, напр.
тетраалкилсиланы или тетраалкилстаннаны. С тетра-
хлорсиланом реагирует даже не вступающий обычно
в В. р. малоактивный хлористый винил:
Ия
S1C14 4- СН2=СНС1 Si(CH=CH2)4
Na
SnCl4 + С4Н0С1 sn(C4H9)4
В. p. открыта в 1855 Ш. А. Вюрцем.
Лит.: Surrey A. R., Name reactions in organic che-
mistry, N. Y., 1954; Петров А. Д., Миронов В. Ф.,
Глуховцев В. Г., Изв. АН СССР. Отд. хим. наук,
1956, № 4; MOller Е., Angew. Chemie, 1957, 69, № 3,
S. 98. Н. П. Гамбарян.
ВЯЖУЩИЕ МАТЕРИАЛЫ — минеральные и орга-
нич. вещества, применяемые в строительстве для
скрепления отдельных элементов сооружения, изго-
товления бетонов, строительных р-ров, деталей и
конструкций.
Минеральные В. м. — порошкообразные
продукты, образующие при смешивании с водой пла-
стичную массу, затвердевающую в прочное кацневид-
ное тело. По теории А. А. Байкова, твердение почти
всех минеральных В. м. протекает благодаря процес-
сам растворения и возникновения насыщенного р-ра,
образования коллоидальной массы в виде студня и
частичной или полной его кристаллизации. В резуль-
тате этих физико-химич. процессов, протекающих не-
одинаково для различных видов В. м., происходит
обычно прогрессирующее нарастание прочности. Пе-
риод времени от смешения В. м. с водой до потери
им пластичного состояния характеризует время
схватывания. Большинство минеральных В. м.
имеет начало схватывания не менее 45 мин. и конец
схватывания не более 12 часов с момента их смешения
с водой.
Минеральные В. м. делятся на воздушные и гидрав-
лические. Воздушные В.м. после смешивания с
водой затвердевают и длительно сохраняют свою
прочность на воздухе; их применяют лишь для воз-
ведения наземных сооружений, не подвергающихся
действию воды. Нек-рые В. м. после затвердевания
на воздухе могут длительно сохранять свою прочность
при воздействии минеральных к-т; их используют
для создания кислотоупорных
покрытий. К таким В. м. отно-
сятся кислотоупорные цементы,
изготовляемые на основе раство-
римого стекла (силиката нат-
рия), кислотоупорных микро-
наполнителей и ускорителей
твердения (например, Na2SiFG).
Гидравлические В. м.
с водой и предварительного
сн=сн
R
2 (1)
(2)
смешивания
после
затвердевания на воздухе могут продолжать твер-
деть и длительно сохранять свою прочность в воде;
их можно применять как в наземных, так и в
подземных, гидротехнических и других сооружениях.
К воздушным В. м. относятся известь, вяжущие мате-
риалы гипсовые (строительный гипс, ангидритовый
цемент, высокообжиговый и др.) и магнезиальные
вяжущие (каустические магнезит и доломит). К гид-
равлич. В. м. относятся различные цементы: порт-
ландцемент (обычный, быстротвердеющий, пластифи-
цированный, гидрофобный, тампонажный, сульфато-
стойкий, белый и др.), пуццолановые цементы (пуц-
цолановый портландцемент, известково-пуццолано-
вый, известково-глииитный и известково-зольный
цементы), шлаковые цементы (шлако-портландцемент,
известково-шлаковый, сульфатно-шлаковый), цементы
с микронаполнителями (песчаный, карбонатный), гли-
ноземистый и сульфатно-глиноземистый цементы, рас-
ширяющийся цемент, романцемент, гидравлич. из-
весть.
В. м. изготовляются из широко распространенных
горных пород (гипсовых, известковых, известково-
глинисто-магнезиальпых, глиноземистых и кремнезе-
мистых) и отходов пром-сти (шлаков, зол, активных
кремнеземистых отходов и др.).
Органические В. м. (битумы, дегти, ас-
фальты) применяют в дорожном строительстве,
а также при гидроизоляц., кровельных и др. работах.
Лит.: Бутт Ю. М., Технология цемента и других вя-
жущих материалов, 3 изд., М., 1956; Юнг В. Н., Основы
технологии вяжущих веществ, М 1951; Крейцер Г. Д.,
Асфальты, битумы и пени, 3 изд., М., 1952. Ю. М. Бутт.
ВЯЖУЩИЕ МАТЕРИАЛЫ ГИПСОВЫЕ — воз-
душные вяжущие вещества, получаемые из природ-
ного двухводного гипса CaSO4 • 2Н2О, природного
ангидрита CaSO4 и нек-рых отходов иром-гти, со-
стоящих гл. обр. из CaSO4. Различают быстротвердею-
щие В. м. г., состоящие в основном из полуводного
гипса, и медленнотвердеющие, в к-рые входит в основ-
ном безводный гипс. К первым относится строитель-
ный гипс и его разновидность — высокопрочный гипс,
а также формовочный и медицинский гипсы, а ко
вторым — ангидритовый цемент и высокообжиговый
гипс (э с т р и х - г и п с).
Строительный гипс — наиболее распро-
страненный В. м. г. получают термич. обработкой
гипсового камня и измельчением его до или после
этой обработки. Термич. обработка ведется при
140—190°, при этом двухводный гипс превращается
в полуводный. В зависимости от условий термич.
обработки различают 2 модификации полуводного
гипса: а- и 0-полугидраты. 0-Полугидрат является
главной составной частью строительного гипса; об-
разуется при нагревании гипса при нормальном дав-
лении; он состоит из мелких кристаллов с нечетко
выраженными гранями и обладает повышенной водо-
потребностью; плотность ^-модификации 2,67—2,68.
а-Полугидрат получают нагреванием гипса под дав-
лением 1,3 ат', он состоит из крупных кристаллов
в виде длинных прозрачных игл или призм, плотность
2,72—2,73, отличается пониженной водопотребностью,
715
ВЯЖУЩИЕ МАТЕРИАЛЫ ГИПСОВЫЕ — ВЯЗКОСТЬ
716
повышенной прочностью и является главной состав-
ной частью высокопрочного гипса.
Термич. обработку гипсового камня производят в вароч-
ных котлах, вращающихся барабанах и установках для одно-
временного измельчения и обжига, а также в шахтных, камер-
ных и напольных печах. Высокопрочный строительный гипс
получают обработкой гипсового камня паром под давлением
в запарочных и самозапарочных аппаратах, в герметически
закрывающихся варочных котлах и др. установках. Изготов-
ляют два сорта строительного гипса: с остатком на сите
№ 02 (918 oms/cAt2) не более 15% (1-й сорт) и 25% (2-й сорт).
Начало схватывания строительного гипса должно наступать
не ранее 4лшн, а конец схватывания—не ранее 6 мин и не позд-
нее 30 мин после начала смешивания гипса с водой. Время от
начала приготовления гипсового теста до конца кристаллиза-
ции гипса должно быть не менее 12 мин. За конец кристаллиза-
ции принимается момент, когда вначале поднимающаяся
темп-ра твердеющего гипсового теста начинает снижаться.
Предел прочности при сжатии через 1,5 часа: для 1-го сорта
не менее 55 кг/см2 и для 2-го — не менее 40 кг/рм2; предел проч-
ности высушенных до постоянного веса образцов соответственно
100 и 75 кз/см2. У высокопрочного строительного гипса мини-
мальный предел прочности при сжатии через 3 часа составляет
90—240 кг/см2 (в зависимости от марки). Водопотребность
обычного строительного гипса для получения теста нормаль-
ной густоты 60—80%, а высокопрочного 35—45%. Испытание
прочности ведется на образцах из гипсового теста (без песка)
литой консистенции.
Строительный гипс применяют для изготовления
известково-гипсовых р-ров для штукатурных работ,
для произ-ва листов сухой штукатурки, перегородоч-
ных плит и панелей и др. строительных деталей. Гипс
применяют как в чистом виде, так и с заполнителями
(гипсобетон). Из строительного гипса изготовляют
также искусственный мрамор, декоративные изделия,
формы для отливки керамич. изделий и др. материалы.
Ангидритовый цемент состоит преим.
из безводного сернокислого кальция; его получают
обжигом природного двухводного гипса при 600—700°
и последующим измельчением продукта обжига со-
вместно с добавками — катализаторами твердения (из-
весть, сульфат или бисульфат натрия в смеси с же-
лезным или медным купоросом и др.), а также тонким
измельчением природного ангидрита с катализато-
рами.
Сроки схватывания ангидритового цемента: начало — не
ранее 30 мин., конец — не позднее 24 час. Водопотребность
для получения теста нормальной густоты составляет 30—
40%. Марки ангидритового цемента: «50», «100», «150» и «200»
(предел прочности при сжатии образцов из р-ра жесткой кон-
систенции 1 : 3 через 28 суток); его обжиг производится гл.
обр. в шахтных печах.
Ангидритовый цемент применяют для изготовления
кладочных и штукатурных р-ров, а также для полу-
чения бетонов и различных строительных деталей.
Высокообжиговый гипс (эстрих-гппс)
получают обжигом природного двухводного гипса или
природного ангидрита при 800—1000° с последующим
измельчением. При этом происходит не только обезво-
живание двухводного гидрата, но и частичное раз-
ложение CaSO4 с образованием свободной извести.
Эта известь в высокообжиговом гипсе играет роль
катализатора.
Водопотребность высокообжигового гипса для получения
теста нормальной густоты составляет 25—35%. По технич.
условиям начало схватывания должно наступать не ранее
2 часов после смешивания с чводой. Медленное схватывание
и твердение позволяют смешанную с водой рыхлую массу
высокообжигового гипса уплотнять трамбованием, примерно
через сутки после затворения водой, что значительно увели-
чивает прочность материала. Марки высокообжигового гипса:
«100», «150» и «200» (предел прочности при сжатии образцов
из теста пластичной консистенции через 28 суток).
Изделия из высокообжигового гипса хорошо со-
противляются истиранию; они мало тепло- и звуке-
проводны. Поэтому такой гипс применяется для изго-
товления полов. Высокообжиговый гипс можно также
использовать для приготовления строительных р-ров
и бетонов, искусственного мрамора и др. целей.
Лит.: Будников П. П., Гипс, его исследование и
применение, 3 изд., М.—Л., 1943; Б у тт Ю. М., Технология
цемента и других вяжущих материалов, 3 изд., М., 1956;
В и х т е р Я. И., Мак И. Л., Швагирев м. П.,
Производство гипса и гипсовых строительных деталей, М.,
1954. Ю. М. Бутят.
ВЯЗКОГО ТЕЧЕНИЯ ЭНЕРГИЯ АКТИВАЦИИ —
дополнительная энергия, необходимая для преодо-
ления межмолекулярных взаимодействий при пере-
мещении молекулы жидкости из одного состояния
временного равновесия в другое. В. т. э. а. 7?в*язк ,
отнесенная к 1 г-лолю, связана с вязкостью соотно-
шением:
т; = т;оелЕвяэк7нт (1)
где т|0 = hN]v (h — постоянная Планка, N — число
Авогадро, v — молярный объем). В. т. э. а., ДЕ*ВЯЗК ,
являющаяся изменением свободной энергии при
переходе молекулы в активированное состояние в мо-
мент ее перемещения (свободная энергия активации),
может быть представлена в виде:
ДЕ* = ДЯ* — TkS*
вязк. вязк. вязк.’
где ДЯ*язк — теплота, а АА*Н.1|; —энтропия актива-
ции вязкого течения. ДЯ*язк составляет для низко-
молекулярных жидкостей от ‘/з до 1/4 величины мо-
лярной теплоты испарения, что связано с тем, что
в процессе течения, в отличие от испарения, проис-
ходит не полный отрыв молекул друг от друга, а
лишь частичный.
Теплота активации вязкого течения определяется
из темп-рной зависимости вязкости: ДЯ* „ =
= В Для низкомолекулярных веществ величина
ДЯ*язк , подобно теплоте испарения, определяется
строением в-ва и его мол. весом. Она тем больше, чем
сильнее межмолекулярное взаимодействие и чем
больше размеры молекулы. Однако для полимерго-
мологов (см. Полимергомология) ДЯ*язк , возрастая,
стремится к пределу. Т. о., у достаточно высокомо-
лекулярных полимеров ДЯ*язк не зависит от мол.
веса, тогда как сама вязкость быстро растет с возра-
станием мол. веса. Это доказывает гибкость цепных
молекул и приводит к выводу о сегментарном (диффу-
зионном) механизме течения (см. Вязкость полимеров),
состоящем в том, что перемещение цепей осуще-
ствляется в результате случайных перемещений от-
дельных их участков — сегментов. В. т. э. а. опреде-
ляет средний размер сегмента, в то время как вязкость
связана с мол. весом всей цепной молекулы. Степень
полимеризации сегментов, определенная для ряда
полимеров, изменяется от нескольких единиц до
многих десятков. В. т. э. а. используется для оценки
гибкости цепных молекул. Однако ур-ние (1) не вы-
полняется в ряде случаев вязкого течения полимеров,
что обусловлено сложным характером взаимодействия
цепных молекул и его изменением при нагревании или
охлаждении. В последнем случае определение В. т. э. а.
теряет непосредственный физич. смысл.
Лит.: Френкель Я. II., Кинетическая теория жидко-
стей, М.—Л., 1945; Глесстон С., Лей'длер К.,
Э й р и н г Г Теория абсолютных скоростей реакций, пер.
с англ., М., 1948; Flory Р. J., J. Am. Chem. Soc., 1940,
62, 1057—70; Fox T. G. and Flory P. J., там же, 1948,
70, 2384—95; К a uzmann W. J. and E у r i n g H., там же,
1940, 62, 3113—25; Шишкин H. И., ЖТФ, 1956, 26,
вып. 7, 1461—82. Л. 3. Роговина.
ВЯЗКОСТЬ (внутреннее трение) — свойство жид-
ких, а также газообразных и твердых тел оказывать
сопротивление их течению — перемещению одного
слоя тела относительно другого — под действием
внешних сил. В. обратна текучести (подвиж-
ности, ползучести) и особенно типична для жидко-
стей. В. определяется тепловым движением, разме-
рами и формой молекул, их взаиморасположением
(«упаковкой») и действием молекулярных сил.
Количественная характеристика В. может быть рас-
смотрена на следующем примере. Слой жидкости тол-
717
ВЯЗКОСТЬ
718
щиной у0 находится между двумя одинаковыми плоско-
параллельными твердыми пластинками (рис. 1).
Нижняя пластинка А удерживается неподвижно,
а верхняя В под действием постоянной внешней
у касательной силы F
Рис. 1. Схема установившегося
развития однородного сдвига(ла-
минарного течения) в слое жид-
кости.
сдвигается параллель-
но нижней в направле-
нии оси х. Вследствие
вязкого сопротивления
слоя жидкости между
пластинками, этот сдвиг
А:г0 будет развиваться
во времени t с некото-
рой постоянной скоро-
стью г--0 = &хй/1. Разде-
ляя весь слой жидкости
на ряд тонких парал-
лельных слоев, нахо-
дим линейное распреде-
ление скоростей этих
слоев по оси у. верх-
ний слой жидкости, примыкающий к пластинке В,
вследствие адгезии, смещается вместе с ней, увле-
кая за собой нижележащий слой, движущийся
с меныпей скоростью. Этот слой, в свою очередь, увле-
кает следующий и, с другой стороны, тормозится им.
В итоге внешняя действующая сила F, приложенная
к верхнему слою, уравновешивается силой вязкого
сопротивления (внутреннего трения), и течение каж-
дого слоя проходит с постоянной во времени скоростью
(установившееся стационарное течение), но убываю-
щей линейно от слоя к слою от наибольшей величины
у0 у пластинки В к v = 0 у неподвижной пластинки А.
Работа внешней силы F, уравновешивающей устано-
вившееся течение, полностью переходит в теплоту.
Такое ламинарное, т. е. послойное, течение, при
к-ром слои жидкости движутся относительно друг
друга без перемешивания (в простейшем случае —
однородный сдвиг, рис. 1), характеризуется гра-
диентом скорости G=^ = ~ (разность скоростей
двух параллельных слоев, расстояние между к-рыми
равно единице). G равна скорости относительного
у, d /ДХл\ Д.т0 , .
сдвига G — -& \ гДе у “ вязкая (остаточная)
деформация относительного сдвига в слое.
В. определяется, по Ньютону (1687), как коэфф,
пропорциональности ц (наз. иногда коэфф. В.) в вы-
ражении, связывающем силу F, приложенную к еди-
нице площади сдвига (напряжение сдвига Р), с гра-
диентом скорости:
F п п dv
-_=P = 1G = 1S-
(1)
Отсюда В. "t\=PlG определяется как величина каса-
тельной силы, к-рая должна быть приложена к еди-
дице площади сдвигаемого слоя, чтобы поддержать
в этом слое стационарное ламинарное течение с по-
стоянным градиентом скорости, равным единице.
В области ламинарного течения ньютоновских сред
(как жидкостей, так и газов), т. е. при достаточно
малых скоростях, или, в более общем виде, при значе-
ниях числа (критерия) Рейнольдса, меньших крити-
ческого (см. Вискозиметрия), В. не зависит от напря-
жения сдвига, от толщины слоя вязкой среды, а также
от длины, ширины, а следовательно, и площади
пластинок, если размеры пластинки достаточно ве-
лики, чтобы краевыми эффектами можно было пре-
небречь. Т. о., в этой области Р пропорционально
градиенту скорости G.
При течении вязкой жидкости по длинной трубе
имеет место параболическое распределение скоростей
в плоскости осевого сечения (рис. 2). Это более слож-
ный случай стационарного вязкого течения, когда
градиент скорости не постоянен по сечению — наи-
больший у стенок и равен нулю по оси трубки. При
этом В. определяется по формуле Пуазейля — Гагена:
(2)
‘ HIQ 1 '
выводимой интегрированием дифференциального опре-
деления В. по Ньютону (1). В ф-ле (2) Др — разность
давлений на концах трубки,
по к-рой происходит течение,
г — радиус трубки, I — ее дли-
на, Q — объем жидкости, про-
текающей за единицу времени.
При возрастании скорости
потока и радиуса трубки, а
также в условиях его сложной
геометрии (резкой искривлен-
ности) течение из ламинарного 'коста™ трубеК°Й
все в большей степени превра-
щается в турбулентное (вихревое), между соседними
слоями происходит обмен веществом (перемешивание),
и измеряемая в этих условиях В. теряет свое зна-
чение физико-химич. константы жидкости (сопроти-
вление потока в большей мере определяется плотно-
стью среды, чем ее В.).
В системе CGS единицей измерения В. является
пуаз; 1 пуаз = 1 дн-сек-см~2 = 1 ел-1-г-сев-1; со-
тая доля пуаза — сантипуаз (спуаз); 1 пуаз
равен 106 микропуаз (мкпуаз). В. воды при
20° С близка к 1 спуаз. Выведенную величину ц при-
нято называть динамической вязкостью
или просто коэфф, динамич. В. Часто пользуются
величиной т. н. относительной В. т)/т)0 —
отношением В. данной жидкости к В. воды при той
же темп-ре (или отношением В. р-ра к В. чистого
растворителя) и удельной В. (г] — 'По)/Г1о> т- е-
относительным увеличением В. растворителя за счет
введения растворенного вещества.
Отношение В. жидкости к ее плотности р наз.
кинематической вязкостью: v = ц/р
(т. к. ее размерность — см2-сек~1— является кинема-
тической, т. е. содержит только длину и время), в от-
личие от динамической, или обычной В., в размер-
ность к-рой входит также и масса. Кинематич. В. опре-
деляет при данной скорости и геометрии потока, его
число (критерий) Рейнольдса, характеризующий гра-
ницу ламинарности, к-рая тем выше, чем выше т)
и чем меньше р вязкой среды. В системе CGS ки-
нематич. В. измеряется в стоксах; 1 ст= 100 ест—
= 1 см2 сек'1. Величина, обратная В.,<р=1/г] наз.
текучестью.
В. технич. продуктов иногда определяют и в условных еди-
ницах: градусах Энглера (°Е) и Барбье (°В),секундах Сайбол-
та("Я или Su) и Редвуда ("R или R-1). Эти единицы представ-
ляют собой либо отношение времени истечения жидкости
в соответствующем приборе при данной темп-ре ко времени
истечения стандартной жидкости при установленной темп-ре,
либо время истечения определенного объема исследуемой
жидкости в стандартных условиях. Полученные результаты
можно перевести в абсолютные единицы (пуазы), если измере-
ния ведутся в правильных уеловпях, т. е. в области ламинар-
ного течения. Так, переход от градусов Энглера к абсолютным
единицам определяется следующим соотношением:
*1 = (0,073ГЕ — р (пуаз)
L1
где °Е — градусы Энглера, ар — плотность жидкости в s/cai8,
а соотношение между °Е и ♦кинематической вязкостью:
. V = 0,0731 ’Е — ^51
В СССР условную вязкость выражают в единицах ВУ,
численно совпадающими с градусами Энглера.
Как физико-химич. характеристика жидкостей, В.
постоянна при изменении в широких пределах усло-
вий течения — скоростей и геометрии (формы и раз-
меров) потока и вместе с тем изменяется в зависимости
от темп-ры, давления и природы жидкости в огромном
диапазоне — от 10~3—10~2 абс. единиц у маловязких
719
ВЯЗКОСТЬ
720
жидкостей (вода, бензин при обычной темп-ре) до
1013 и выше у переохлажденных жидкостей: стекол,
битумов, смол (по механич. свойствам — изотропных
твердых тел) в интервале их размягчения и при более
низкой темп-ре. В. газов в сотни раз меньше, чем
у воды. Ожиженный гелий при весьма низких темп-рах
(2,186°К) переходит в т. н. сверхтекучее состояние
(гелий II), в к-ром В. практически равна нулю (см.
Гелий).
В. поверхностных слоев (монослоев), обычно структурная —
поверхностная В., определяется как отношение к градиенту
скорости двухмерного напряжения сдвига Р , имеющего раз-
мерность сила/длина, в отличие от обычного Р (сила/площадь),
и потому выражается в поверхностных пуазах (а • сек-1).
В. (внутреннее трение) является причиной затухания коле-
баний или замедления движений в вязкой среде. По аналогии
с В. в молекулярной физике рассматривается так наз. «вторая
вязкость», называемая также объемной В. Объемная В. ха-
рактеризует своей обратной величиной скорость деформации
всестороннего сжатия жидкости, а не деформацию ее сдвига,
как обычная, тангенциальная В. Вторая В. определяет избыточ-
ное (аномально высокое) поглощение ультразвуковых волн
в жидкостях.
Для веществ
ном состоянии
в конденсирован-
— жидкостей и твердых —
Рис. >
кости
Приведенная температура T/j
1 Вода (Шугаее и Сорокин)
— СОг ( Штанельбек)
3. Изменение вязкости жид-
t и пара в зависимости от
темп-ры.
тел,
В., определяемая по-
движностью частиц,
обратно пропорцио-
нальна коэфф, само-
диффузии (см. Само-
диффузия) и резко па-
дает с повышением
темп-ры — приблизи-
тельно обратно про-
порционально Т3 (ри-
сунок 3) или по обще-
му закону вида ц =
= A (T)ew/hT, как это
показывает теория В.
жидкостей, развитая в
основном Френкелем
(1926—1944) и Эйрингом (1936). Предэкспоненциаль-
ный множитель А (Г) по сравнению с ew^hT мало
изменяется с темп-рой (в простейшем случае А — const
или Л = Л071). W—энергия активации вязкого
течения или разрыхления жидкости, энергия обра-
зования молекулярной полости, в к-рую переска-
кивает данная молекула, срываясь после нек-рого
числа колебаний со своего временного положения
равновесия (1У — функция темп-ры и давления).
В. жидкостей в основном определяется плотностью
упаковки молекул и молекулярными силами сцепле-
ния. В соответствии с этими представлениями В.
жидкостей убывает с ростом темп-ры и повышается
с ростом давления.
Вещество Вязкость (7) • 103 пуаз) при температуре, °C
0° 10° 20° 30° 40° 50° 60°
Анилин CeHsNH.i 6.55 4,48 3,19 2,41 1,89 1.56
Ацетон СН3СОСЙ3 3,97 3,61 3.25 2,96 2,71 2,46 —
Бензол С6Не 9,00 7.57 6,47 5,66 4.82 4,36 3,95
Вода Н2б 17,92 13,10 10,09 8,00 6,54 5,49 4,69
Гексан СЙН14 3,97 3,55 3,20 2,90 2,64 2,48 2,21
Глицерин СЩОНСНОНСН.ОН .' . 121,1 39,5 14,8 5,87 3,30 1.80 1.02
Метиловый спирт СНЯОН 8,17 5,84 — 4,5 3,96 3,51
Нитробензол CeHgNOo 30,9 23,0 20,30 16,34 14,4 12,4 10,9
Пиридин CeH5N 13,6 4,3 9,58 8.29 7,24 6,39 5,69
Ртуть Hg 16,85 16,15 15,54 14,99 14,50 14.07 13,67
Уксусная кислота СН:)СООН . . . — — 12,2 10,4 9,0 7,4 7,0
Четыреххлористый углерод СС14 . 13,29 11.32 9,65 8.43 7,39 6,51 5,85
Этиловый спирт СоНзОН 17,85 14,51 11,94 9,91 8,23 7,01 5,91
Все эти соотношения качественно хорошо выражаются
классич. ф-лой Бачинского: т; = С/(э — bt), не содержащей
в явном виде темп-ры, но устанавливающей обратную пропор-
циональность между т; и свободным объемом (г — <>х) в жид-
костях, резко возрастающим с повышением темп-ры Т вслед-
ствие теплового расширения (и — молекулярный объем.
Ъ — несжимаемый объем в одном моле, С — константа, харак'
теризующая молекулярные силы сцепления). Константы ф-лы
Бачинского соответствуют константам Ъ и а ур-ния Ван-дер
Ваальса, объединяющего жидкости и реальные газы; С связано
с константой а: С М Va, где М — мол. вес вещества. В соот-
ветствии с этой ф-лой 7) зависит не только от Т, но и от давле-
ния через изменение величины (и—Ьх), убывающей при все-
стороннем сжатии с ростом давления. Для текучести эта ф-ла
дает линейное возрастание с увеличением молекулярного
объема, ф == */7) = v/C — Ь,/С.
Ф-ла Бачинского применима также к смесям жидкостей,
для к-рых аддитивны значения не а текучести 1 /т) = (и—bt)/ С
соответственно аддитивности молярных объемов. При очень
больших давлениях В. жидкостей резко возрастает, что свя-
зано с увеличением энергии активации и соответственным
уменьшением подвижности молекул. Экспериментальные дан-
ные по зависимости В. жидкостей от Т наиболее точно охваты-
ваются ф-лой с тремя константами (ф-ла Фогеля — Фуль-
чера — Таммана)
V = loo е&/(Т ~ То) или 1? Ч 'Чсо = ki/(T - То)
где т) т — наименьшее значение щ, достигаемое при повыше-
нии Т (?) —» ?) при Т —► оо ), а Т„ — та (самая низкая) темп-ра,
при к-рой г становится практически бесконечно большой
(при Т — Т„ т) = оо ). Вследствие резкого падения В. жидко-
стей с повышением темп-ры, выражают зависимость от темп-ры
Г не самой В., а ее логарифма или двойного логарифма
lglg4, к-рый (по Ле Шателье) линейно убывает с Т.
В. жидкостей следует сравнивать в соответствен-
ных состояниях, т. е. при приведенных темп-рах,
составляющих одинаковую долю от критич. темп-ры.
Данные о В. нек-рых жидкостей и сжиженных газов
приведены в табл. 1 и 2.
Таблица 2
Сжижен- ный газ Темп-ра, °C 7] • 102 пуаз । Сжижен- Темп-ра, 7] • 102 пуаз
ный газ с
Азот —209,3 0,292 Водород -258,3 0,0215
-169.1 0,085 —253,2 0,0138
-161,5 0,0740 Метан -183,1 0,210
Кислород -118,8 0,873 -104,8 0,0625
—119,0 0,0915 Этан —172,0 8,78
Воздух —183,1 0,167 -105,9 2,03
—156,8 0,0805 + 85,2 0,55
В. жидкостей и аморфных твердых тел определяет
величину периода релаксации касательных упругих
напряжений в этих телах.
Различие между В. обычных жидкостей и аморфных
стеклообразных твердых тел в основном только коли-
чественное у твердых тел она на 12—15 порядков
выше. (Об особенностях вязкости высокомолекуляр-
ных веществ см. Вязкость полимеров). Следует отме-
тить, что В. расплавленных металлов по порядку
равна В. обычных жидкостей (воды) (рис. 4). Для
органич. веществ В. растет с возрастанием мол. веса,
с введением в молекулу полярных групп и особенно
Таблица 1 п₽и наличии в молекуле ци-
клов, чем и объясняется высо-
кая В. смазочных масел (см. Вяз-
кость нефтепродуктов).
В разбавленных суспензиях,
эмульсиях й коллоидных р-рах
В., в соответствии с теорией
Эйнштейна, линейно возрастает
с увеличением степени объемно-
го заполнения v2/vl = <о среды
(1) (объем к-рой — г-J частица-
ми дисперсной фазы (2) (г2 — ее
объем, включая и сольватные
оболочки) и не зависит от разме-
ров частиц. При этом относи-
тельное повышение В. среды
СП—'По)/'По = гдеа = 2,5, для
сферич. частиц, а для анизометричпых частиц (ча-
стиц вытянутой формы) а 2,5. Для разбавленных
растворов высокомолекулярных соединений относи-
721
ВЯЗКОСТЬ — ВЯЗКОСТЬ НЕФТЕПРОДУКТОВ
722
Рис. 4. Вязкость нек-рых рас-
плавленных металлов.
тельное повышение В. растворителя (уд. В.) связапо
с молекулярным весом полимера; измеряя В. р-ров,
определяют мол. вес высокомолекулярных веществ.
Возникновение в жидкостях (дисперсных системах
или р-рах полимеров) пространственных структур,
образуемых сцеплением
частиц или макромолекул,
вызывает резкое увеличе-
ние В., к-рая при этом, в
отличие от обычной (нью-
тоновской) В., наз. струк-
турной вязкостью.
Последняя зависит от
условий течения, резко
уменьшаясь при возраста-
нии напряжения (или ско-
рости) сдвига выше неко-
торого предельного зна-
чения, соответствующего
разрушению структуры в
потоке и определяюще-
го прочность структуры
(см. Вязкость структур-
ная, Структурообразова-
ние в дисперсных системах,
Структурирование поли-
меров пространственное).
Аномалии В., т. е. ее
зависимость от условий
течения, могут вызываться также и ориентацией вы-
тянутых макромолекул или коллоидных частиц в
потоке.
В. кристаллов (особенно металлич. монокристаллов,
как и поликристаллич. твердых тел) проявляется
в процессах их ползучести (крипа), т. е. медленного
нарастания необратимых во времени (остаточных)
деформаций при действии постоянного напряжения,
особенно при малых напряжениях и высоких темп-рах
(диффузионная ползучесть, развивающаяся по меха-
низму вязкого течения). Медленное вязкое течение
в кристаллах часто маскируется пластич. течением,
т. е. сдвигами по плоскостям скольжения.
Вязкость (внутреннее трение) газов и па-
ров, в отличие от В. жидкостей, обусловливается
в основном тепловым движением, и только им, — в га-
зах и парах, близких к идеальным. В результате
проникновения молекул более быстро движущегося
слоя в слой, движущийся медленнее, и возникает
сопротивление перемещению одного слоя относи-
тельно другого. В газах в противоположность жид-
костям, вследствие иного механизма иязкого тече-
ния, кинематич. В. пропорциональна коэфф, самодиф-
фузии при длине свободного пробега, большой по
сравнению с диаметром молекулы.
Согласно кинетич. теории, связь т| с длиной свобод-
ного пробега молекул между соударениями Л и эффек-
тивным диаметром молекулы 6 (средним расстоя-
нием, на к-рое молекулы сближаются при соударе-
нии) выражается соотношениями:
и более точно
(приближенно)
Л ти
п =0,499
где и — средняя скорость теплового движения при
данной темп-ре Т, т — масса молекулы. В. данного
газа, в отличие от жидкости, не зависит от плотности
или давления и не убывает, а, наоборот, возрастает
с темп-рой.
При сжатии газа число молекул газа, переходя-
щих из одного слоя в другой, увеличивается, но
вместе с тем уменьшается длина свободного пробега
и каждая молекула менее глубоко проникает в сосед-
ние слои. Для очень разреженных газов, когда А.
становится соизмеримой с размерами сосуда, содер-
жащего газ (вискозиметра), понятие В. теряет физич.
смысл. В. газов растет с повышением темп-ры при-
ближенно пропорционально Т.
Более точной для этой зависимости является формула
Сезерланда _
„ Ут
11 в 14-с/т
учитывающая уменьшение 5 с ростом Т. При высоких Т
г.-Ут. В реальных газах при рассмотрении зависимости
В. от темп-ры и давления необходимо учитывать отклонение
от идеального состояния (поправки Ван-дер-Ваальса на соб-
ственный объем молекул и иа силы притяжения между ними).
В. нек-рых газов (в мпкропуазах) приведены в табл. 3.
Таблица 3
Газ Температура, °C
0 50 100 200 400 800
Азот N2 166 188 208 246 311 413
Аммиак NH3 93 111 128 165 —
Водород Н2 84 94 103 121 154 210
Гелий Не 18а 208 229 270 342 465
Кислород О2 192 218 244 290 360 493
Хлор С12 123 145 168 210 — —
Ацетилен С-Нл 96 111 126 —
Метан СН, 102 118 133 — — —
Этан Colle 86 101 115 — —
Пропан CgHg ....... 75 88 101 — — —
Вблизи критич. темп-ры кривые зависимости В. от
темп-ры для жидкости и пара, находящихся в равно-
весии, сопрягаются (рис. 3). Картина температурной
зависимости в области, близкой к критич., является
весьма сложной и теоретически не исследована.
Измерения В., вследствие ее чувствительности к из-
менениям состава и строения молекул, а также к на-
личию структурообразующих примесей, могут слу-
жить удобным физико-химич. методом анализа. Во
многих случаях по В. судят о готовности или качестве
продуктов и полупродуктов произ-ва, т. к. В. тесно
связана со структурой вещества и отражает физико-
химпч. изменения в материале. В. имеет большое зна-
чение в различных областях технологии и в природ-
ных, особенно в бирлогич., процессах, определяя
скорость течения жидкостей и газов и сопротивле-
ние, оказываемое ими движению взвешенных частиц.
В. среды определяет скорость диффузии в ней раство-
ренных веществ. Теплопередача в жидкостях и газах
в определенных условиях характеризуется их В.
Изменения В. сказываются на скорости химич. реак-
ций, протекающих в биологич. системах, в частности
процессов, связанных с жизнедеятельностью клетки.
В. является одной из важнейших характеристик
смазочных материалов и др. нефтепродуктов.
Лит. см. при ст. Вискозиметрия. П. А. Ребиндер.
ВЯЗКОСТЬ НЕФТЕПРОДУКТОВ — одна нз важ-
нейших констант, характеризующих эксплуатацион-
ные свойства нефтепродуктов. В. н. зависит от их
фракционного и химич. состава: нефтепродукты более
легкого фракционного состава имеют меньшую вяз-
кость; В. и. из нефтей парафинового основания ниже,
чем из нефтей ароматич. и нафтенового оснований.
Для характеристики В. н. в СССР используются
величины динамич. вязкости ц (пуазы), кинематиче-
ской v (стоксы) и условной (градусы ВУ).
Динамич. и кипематич. вязкость определяют в ка-
пиллярных и ротационных вискозиметрах. Условная
вязкость, определяемая в вискозиметрах Энглера,
представляет собой отношение времени истечения
200 мг масла при данной темп-ре t ко времени истече-
ния 200 мл воды при 20°. Условная вязкость не ха-
рактеризует истинной вязкости в-ва. Существуют
формулы и таблицы для перевода условной вязкости
723
ВЯЗКОСТЬ НЕФТЕПРОДУКТОВ — ВЯЗКОСТЬ СТРУКТУРНАЯ
724
в кинематич. и обратно (см. Вязкость и Вискозимет-
рия). В ряде зарубежных стран В. н. измеряется
в иных единицах и вискозиметрах: в США — секунды
Сайболта и вискозиметры Сайболт-Универсал и Сай-
болт-Фурол, в Англии — секунды Редвуда и виско-
зиметры Редвуд-торговый и Редвуд-адмиралтейский,
во Франции — градусы
Барбье и вискозиметры
Барбье.
Важное значение
имеет характер изме-
нения В. н. с темп-рой
(см. рис.). Положитель-
ным качеством нефте-
продукта является по-
логая вязкостно-темпе-
ратурная кривая. Су-
ществует ряд эмпирич.
формул, выражающих
зависимость В. и. от
от температуры.
темп-ры, например: lg lg(v 0,8) = А — B\gT, где
Т — абс. темп-ра; А а В — постоянные. Степень
пологости темп-рной кривой вязкости для масел при-
ближенно оценивается величиной отношения v5n : Vino
или v0 : v!nn, либо величиной температурного коэфф,
вязкости (ТКВ):
ТКВ0 ,!00 — v0 Vioo/Vso
Для этой же цели широко применяется- индекс
вязкости по Дину и Дэвису (ИВ), вычисляемой
по формуле: ИВ = (Su — IV) 100/(Su — II) =
= 100 (Su — A)/D, где Su — вязкость в сек. Сай-
болта-Универсал при 100° F (38,3° С) масла с наибо-
лее крутой кривой вязкости (масла серии L), Н —
то же для масла с наиболее пологой кривой вязкости
(масла серии Н), N — то же для исследуемого масла.
Все три масла должны обладать одинаковой вяз-
костью при 210° F (98,8° С). Для характеристики
масел применяется также вязкостно-весовая кон-
станта (В.-в. к.), отражающая связь между плот-
ностью и вязкостью, с одной стороны, и химич. соста-
вом и практич. ценностью смазочных масел — с другой.
В ряде случаев учитывают зависимость В. н. от давле-
ния, к-рая приближенно выражается ф-лой: цр= ЛоаР>
где т]р и ц0 — динамич. вязкость масла при давлении
р кг/см? и при атмосферном давлении; а — постоян-
ная величина, равная 1,002—1,004.
В таблице приведены значения вязкости некоторых
нефтепродуктов.
Название Вязкость, сст
Соляровое масло 5,0—9,0а
Вазелиновое масло 4,0—5,1а
Индустриальное масло 12 (веретенное) . . 10—14а
Индустриальное масло 45 (машинное) . . 38- 52 а
Масло для прокатных станов 26—30б
Авиационное масло (МС —ДО) 20б
Автол АН-10 не менее 10°
Масло дизельное И 10,5—12,5а
Турбинное масло 22 (турбинное Л) . . . . 20—23а
Масло приборное 6,3—8,5а
Масло трансформаторное Не более 30,0в
Масло для форвакуумных насосов .... 47—57а
Масло для замасливания хлопка 7,0-10,0а
а при 30°; 6 при 100°; в при 20°.
Лит.: Блох Л. С., Д о б р я н с к и й А. Ф., Вязкость
нефтяных продуктов, М.—Л., 1927; Технические нормы на
нефтепродукты (Справочная книга), под ред. Н. Г. Пучкова,
16 Изд., М., 1957; Наметкин С. С., Химия нефти, [3 изд.],
М., 1955. Л. С. Поваров.
ВЯЗКОСТЬ ПОЛИМЕРОВ — свойство полимерных
тел развивать необратимые изменения формы при дей-
ствии механич. напряжений. Возникающее при этом
сопротивление тела изменению его формы (пропор-
циональное скорости деформации у низкомолекуляр-
ных жидких тел) зависит от динамич. режима про-
цесса деформации. Общие колич. закономерности
В. п., вследствие их большой сложности, пока не
установлены. Вязкое течение полимеров всегда со-
провождается развитием эластич. деформаций (см.
Эластичность полимеров), т. к. перемещения длинных
и гибких цепных молекул неизбежно связаны с их
выпрямлением и ориентацией. Поэтому вязкость,
определяемая как отношение напряжения к скорости
необратимой деформации, не является у полимеров
константой, а возрастает в процессе течения. После
установления стационарного течения В. п. в даль-
нейшем не изменяется, но достигнутое значение вяз-
кости зависит от величины действующего на тело
напряжения. Возрастание вязкости в процессе тече-
ния придает полимерам специфическую для них спо-
собность вытягиваться в изотермич. условиях в нити
и пленки.
Интервал значений В. п. простирается на много
порядков величины, достигая 10!2 пуаз и более. С по-
вышением темп-ры В. п. снижается. При прочих
равных условиях вязкость полимергомологов тем
больше, чем выше степень полимеризации. Перемеще-
ние длинных, гибких цепных молекул полимера в про-
цессе вязкого течения происходит по т. наз. диффу-
зионному механизму, т. е. хаотич. перемещением
отдельных участков — сегментов гибкой молекулы,
а не всей молекулы одновременно. Благодаря такому
механизму перемещения молекул необходимая для
преодоления межмолекулярных взаимодействий вяз-
кого течения энергия активации относительно неве-
лика, в то время как одновременное перемещение
всей большой молекулы потребовало бы затраты
энергии, намного превосходящей энергию химич.
связи С—С. Поэтому при быстром воздействии боль-
ших механич. напряжений, когда перемещение цеп-
ных молекул по диффузионному механизму не успевает
развиться, возникает разрушение молекул по химич.
связям с образованием свободных макрорадикалов.
Перемещения этих обломков молекул, соединяющихся
затем хаотически в новые большие молекулы, при-
водит к текучести полимера, получившей название
«химическая текучесть». В то время как обычное
вязкое течение может развиваться лишь у линейных
полимеров, химическая текучесть обнаруживается
также и у пространственных полимеров.
В. п. широко используется в технике при перера-
ботке полимеров в изделия. Особое значение поэтому
приобретает темп-ра текучести полимеров, выше
к-рой проявляется В. п.; у кристаллич. полимеров,
имеющих темп-ру плавления большую, чем их темп-ра
текучести в аморфном состоянии, вязкое течение
появляется только выше темп-ры плавления, т. е.
после образования расплава.
Вязкость растворов высокомолекулярных соединений
сохраняет все особенности В. п., но по мере перехода
к разб. р-рам проявление ее становится все менее
ярко выраженным. См. также: Гибкость цепных мо-
лекул, Текучесть высокомолекулярных соединений.
Лит.: Каргин В. А., Слонимский Г. Л., Крат-
кие очерки по физпко-химии полимеров, М., 1960, с. 75;
Flory Р. J., Ашег. Cheni. Soc., 1940, 62, № 5, р. 1057;
Kauzmann W., Е у г i п g Н., Amer. Chem. Soc., 1940,
62, № И, р. 3113; Каргин В. А., С о г о л о в а Т. И.,
ЖФХ 1949, 23, вып. 5, с.-551; и х ж е, вкн.: Проблемы физи-
ческой химии, вып. 1, М. 1958, с. 18. Т. И. Соголова.
ВЯЗКОСТЬ СТРУКТУРНАЯ — доля вязкости дис-
персных систем (суспензий, эмульсий, коллоидных
р-ров) или истинных растворов высокомолекулярных
соединений, вызванная возникновением пространствен-
725
ВЯЗКОСТЬ СТРУКТУРНАЯ
726
ных структур в виде сеток, нитей и т. п. Возникнове-
ние таких структур связано с образованием связей
между коллоидными частицами или макромолекулами
как следствия сил межмолекулярного взаимодействия.
В. с. характерна для таких систем, как суспензии
бентонитовых глин в воде, гидрозоли А1(ОН)3,
Fe(OH)3, SiO2, бензопурпурина, мыла, р-ров жела-
тины, агара и др. Концентрация р-ра, при к-рой
начинается структурообразование, да-
лее развиваясь с увеличением концентрации, зависит
от природы растворенного в-ва и растворителя. В ре-
зультате возникновения в р-рах структур, их вяз-
кость становится зависимой от скорости сдвига (гра-
диента скорости течения) и более или менее резко
падает с возрастанием скорости сдвига (действующего
напряжения сдвига), соответственно степени разруше-
ния структуры в потоке, с последующим постепенным
восстановлением структуры при меньших скоростях те-
чения или при пребывании в покое (см. Тиксотропия).
Образование пространственных структур приводит
к тому, что вязкость ц в таких системах слагается из
двух величин: 1) истинная, ньютоновская вязкость
цу, и 2) В. с. т)3, обусловленная добавочным сопротив-
лением течению со стороны структурной сетки, в той
или иной степени разрушенной в стационарном потоке.
При достаточно высоких, а иногда даже и при очень
малых концентрациях дисперсной фазы или раство-
ренного высокомолекулярного вещества, В. с. может
быть очень велика по сравнению с т]v, Лв Л v
В ряде случаев для р-ров полимеров, особенно при
небольших концентрациях, зависимость вязкости от
градиента скорости сдвига может быть вызвана не
структурированием р-ра, а изменением гидродинамич.
сопротивления потоку со стороны макромолекул за
Счет изменения их формы в потоке (см. Структуро-
образование в дисперсных системах, Растворы вы-
сокомолекулярных соединений). Ю. С. Липатов.
ГАБРИЕЛЯ РЕАКЦИЯ — синтез первичных ами-
нов из галогеналкилов и фталимида калия с после-
дующим гидролизом образующегося N-замещенного
фталимида I:
Обычно фталимид калия нагревают с галогенидом без
растворителя или же в неполярном высококипящем
растворителе. В этих условиях фталимид калия нерас-
творим, и поэтому для завершения реакции требуется
длительное нагревание (100—180°). Установлено, что
Г. р. гораздо легче протекает в диметилформамиде,
при этом реакция облегчается, улучшаются выходы;
вместо фталимида калия можно пользоваться фтали-
мидом в присутствии поташа. Г. р. широко приме-
няется для синтеза алифатич. и жирноароматич. ами-
нов, диаминов, аминогалогенидов, аминоспиртов и
меркаптанов, аминопитрилов, а также аминоальдеги-
дов, аминокетонов и аминокислот:
/О
I II NK + Cl(CHa)5Cl
О
N(CH2)5N I
cz \СА
ЧО (Z
✓О
+ С6Н5СОСН2Вг
,0
HCON(CH3)2
-20°
Вг
Вг
I
+ ROCOCH(CH2)2CHCOOR
что имеет особенное значение при гидролизе фтали-
мидных производных, содержащих группировки, чув-
ствительные к кислотному или щелочному гидролизу.
Г. р. открыта в 1887 3. Габриелем.
Лит.: Губен И., Методы органической химии, пер.
с нем., т. 4, вып. 1, кн. 1, М.—Л., 1949; Billman J. Н.,
Cash R. V., Proc. Indian Acad. Sci., 1952, 82, p. 158; Sur-
rey A. R., Name reactions in organic chemistry, N. Y., 1954.
H. П. Гамбарян.
ГАДОЛИНИЙ (Gadolinium Gd) — химич. элемент
с п. н. 64, относится к лантанидам.
ГАЗ КОКСОВЫЙ — горючий газ, получаемый
наряду с жидкими продуктами и твердым остатком
(коксом) из ископаемых топлив при их нагревании
без доступа воздуха обычно до 900—1100° (см. Кок-
сование).
Состав Г. к. (в объемных процентах): 55—60% Н2,
24—28% СН4, 6—7% СО, 4—7% N2, 2—4% СО2,
2—4% непредельных углеводородов (этилен, пропи-
лен и др.), 0,4—1,5% С2Нв, 0,4—0,8% О2. Кроме
того, в Г. к. содержится: 5—20 г)м3 H2S, 0,1—1 г)м3
HCN, 1—4 г!м3 бензольных углеводородов, 0,05—
0,025 г/м3 нафталина, 25—50 г)м3 водяных паров,
а также незначительные количества CS2, COS и др.
Физические свойства Г. к.
Плотность (при 0° и 760 мм
рт. ст.) 0,45—0,50 кг/м3,
теплотворность (низшая) 4000—
4400 ккал/м3, теплоемкость
0,32—0,33 ккал/м3 • град, вяз-
кость (при 20°) 0,037 кг/ж • час,
т. воспл. 600—650°.
H2NCH2COC6H5 Выход Г. к. при обыч-
ных на большинстве коксо-
химия. заводов условиях
составляет 300—320 ж3 на 1 т
H2N(CHg)5NH2
-<с\
Ц NCH2COC6H5
С^о
HCON(CH3)2
90°
сухого угля. Г. к. использует-
ся для отопления коксовых и
мартеновских печей, печей сте-
кольных и керамич. заводов,'
различного рода сушилок, в
коммунальном хозяйстве, в хи-
мической пром-сти и др. В хи-
мич. пром-сти из Г. к. полу-
чают водород для синтеза NH3,
из этилена — спирт, дихлор-
этан, хлоргидрин, этиленгли-
коль. Г. к. для металлургия, предприятий может
содержать не более 3—4 г/м3 H2S, для коммунально-
бытового употребления содержание H2S и NH3 не
более 0,02 г/ж3, а нафталина — 0,05 г/м3 зимой и
0,1 г/м3 летом. Г. к., используемый химич.
предприятиями, должен- содержать: 57—
61% Н2; не более 4,6—8,0 см3/м3 окислов
+ рми азота, 0,3—1,4% H2S, 0,8%
+ RNH2 0>2 г/мз , - ' "
О,
coorV
I /С
<-° COOR
с\ I .
N-CH-(CH2)2-CH-n
Чэ о^с
Гидролиз фталимидных производных осущест-
вляется нагреванием N-замещенпого фталимида со
щелочами, -кислотами или лучше всего с гидразин-
гидратом:
NK
ЧО
НВг
COOR
COOR
H2N-CH-(CH2)2-CH-NH2
NHR
[ NR
N2H4-H2o
н„о
I ——
/NH HCi
,0
NH
,NH
о
. и , ,3 Qi, 5% Ng,
нафталина и 4 г/м3 бензольных
углеводородов. Г. к. ядовит и взрывоопа-
сен. По санитарным нормам максимальное
729
ГАЗИФИКАЦИЯ ДРЕВЕСИНЫ — ГАЗИФИКАЦИЯ ЖИДКИХ ТОПЛИВ
730
содержание отдельных компонентов Г. к. в воздухе
рабочих помещений не должно превышать: 0,03 л<е/л
СО, 0,01 мг/л H3S, 0,0003 мг/л HCN, 0,02 мг}л NH3,
0,005 мг{л фенолов и пиридиновых оснований,
0,05 мг/л бензольных углеводородов. При содержании
в воздухе от 6 до 30% Г. к. смесь является взрыво-
опасной.
Лит.: Кустов Б. И., Коксовый газ, 2 изд., Харьков —
М., 1953. А. И. Вродович.
ГАЗИФИКАЦИЯ ДРЕВЕСИНЫ — см. Газифи-
кация твердых топлив.
ГАЗИФИКАЦИЯ ЖИДКИХ ТОПЛИВ — превра-
щение жидких топлив в горючие газы путем рас-
щепления углеводородов, сопровождаемое конверсией
продуктов расщепления водяным паром, кислородом
или воздухом. В зависимости от состава получающие-
ся при Г. ж. т. газы применяются как топливо
или в качестве сырья для химич. промышленности.
Процессы, происходящие при Г. ж. т.: 1) Расщепле-
ние высокомолекулярных предельных углеводородов
с образованием низкомолекулярных предельных и
непредельных или дегидрирование предельных угле-
водородов с образованием непредельных и водорода.
Напр.: CsHla ->С2Нв + СзНв; С4Н10 С4Н8 + Н2;
2) Взаимодействие углеводородов с водяным паром
или кислородом, напр.: С5Н12 + 4Н3О —> СН4 +
+ 4СО + 8Н3; С6Н12 + 2О2СН4 + 4СО + 4Н2.
Последняя реакция — экзотермическая, предыду-
щие — эндотермические. Источниками теплоты для
осуществления процессов Г. ж. т. являются: упомя-
нутая экзотермич. реакция, сжигание части сырья,
подвергаемого газификации, и сжигание кокса, обра-
зующегося на насадке во многих процессах Г. ж. т.
Кроме описанных, протекают реакции изомеризации,
циклизации, полимеризации, саже- и коксообразо-
вания.
Скорость и характер термич. расщепления углево-
дородов зависят гл. обр. от темп-ры, времени пребы-
вания сырья в реакторе и свойств сырья. Так, при
750—800° образуется газ с содержанием до 35—40%
непредельных углеводородов (считая на сырье). При
900° и выше выход непредельных углеводородов резко
уменьшается, увеличивается количество метана, во-
дорода, смолы и кокса. При 1100° и выше процесс
полностью направляется в сторону образования во-
дорода, окиси углерода и кокса. При взаимодействии
углеводородов с водяным паром и кислородом уве-
личивается выход газа (по сравнению с процессом
расщепления), а его состав изменяется в сторону
образования СО, Н2 и СН4, уменьшается саже- и
коксообразование. Применение катализаторов в про-
цессах Г. ж. т. увеличивает скорости реакций, дает
возможность снизить темп-ру процесса на 100—150°,
уменьшает саже- и коксообразование. В качестве
катализаторов применяют природные в-ва, содержа-
щие SiO2 и А13О3 (боксит, кислые глины) или искус-
ственные смеси SiO2, AI3O3, MgO с металлами (Ni,
Со, Pt и др.). Все катализаторы в большей или мень-
шей степени отравляются сернистыми соединениями,
содержащимися в сырье.
С помощью Г. ж. т. производят: энергетич. газ для
отопления промышленных печей, водород и окись
углерода для синтеза аммиака, метанола и др., газ
для бытовых нужд. Газификации может подвергаться
любое жидкое топливо, но промышленное значение
имеет получение газа из тяжелых остатков нефтепере-
работки: мазутов и гудронов. Г. ж. т. осуществляется
по различным технологии, схемам: периодическим и
непрерывным, с катализаторами и без них, под высо-
ким и атмосферным давлением. Произ-во газа из жид-
ких топлив включает в себя подготовку топлива, его
газификацию, очистку газа и сточных вод. Подготовка
топлива заключается в его обезвоживании, подогреве'
и фильтрации. Собственно процесс Г. ж. т. ведется
на неподвижной насадке, в «кипящем» слое, в полом
сосуде, а также с применением твердого теплоноси-
теля.
Периодический каталитический спо-
соб (пример получения газа для бытовых нужд). Процесс
состоит из двух периодов: разогрева каталитич. насадки и
газификации. Насадка разогревается за счет теплоты, полу-
чаемой от сжигания кокса, образующегося на насадке в период
газификации. Во втором периоде в реактор подается перегре-
тый водяной пар и впрыскивается жидкое топливо (дистил-
лятные фракции нефтепереработки). В результате взаимодей-
ствия жидкого топлива с водяным паром в присутствии ката-
лизатора образуются газообразные продукты. Тепло газа
используют в воздухоподогревателе. Далее газ очищают,
—— Жидкое топливо — ----Водяной пар --------[аз
---- Жидкий катализатор --------Кислород
Рис. 1. Схема непрерывного каталитич. способа Г. ж. т.:
1 — подогреватель; 2 — камера смешения; з — реактор;
4 — пароперегреватель; .5 — котел-утилизатор; в — скруб-
бер; 7 — сажевый фильтр; 8 — холодильник.
охлаждают и направляют потребителю. Темп-ра процесса
900—1000°, выход из 1 кг сырья 1,7 нлг3 газа состава; 10% СО,;
3% СтНп; 0,5% О2; 19% СО; 49% Н2; 14% СН4; 5% Na.
Теплотворность газа 4200 ккал/нм3-, кпд процесса 70%.
Непрерывный каталитический способ
(пример получения газа для синтеза аммиака). Сырье (гудрон,
мазут) из подогревателя 1 (рис. 1) поступает в камеру смеше-
ния 2, куда подается небольшое количество раствора <Ja(NO3)2,
вводимого для уменьшения сажеобраэования. Вместе с паро-
кислородной смесью жидкое топливо и раствор Ca(NO3)2
впрыскивают в полый реактор з, где и осуществляется процесс
газификации. Теплота газа используется в пароперегрева-
теле 4 и котле-утилизаторе 5. Далее газ очищается, охлаж-
дается и направляется потребителю. Темп-ра процесса 1200°,
выход из 1 кг сырья 3 нм3 газа состава: 14% СО2; 0,5% H,S;
0,1% ог; 37% СО; 46% Н2; 0,3% СН4; 3% N5. Теплотвор-
ность газа 2506 ккал/нм3-, расход кислорода 0,82 нм3/кг сырья,
содержание сажи 1 г/нм3 газа, кпд 86,5%.
Непрерывный термический способ под
давлением (для получения газа для синтеза аммиака и газа
для бытовых нужд). Подогретые до 300° жидкое топливо и
кислород вместе с перегретым водяным паром впрыскиваются
форсункой в полый реактор 3 (рис. 2), где процесс газифина-
— — — Кислород
........ Вода
—— водяной пар Газ
Рис. 2. Схема непрерывного термич. способа Г. ж. т.:
1,2 — подогреватели; з — реактор; 4 — котел-утилизатор;
5 — скруббер для отмывки газа от сажи; 6 — скруббер,
7 — циркуляционный насос; 8 — сосуд для снижения
давления; э — отстойник для отделения сажи от воды.
731
ГАЗИФИКАЦИЯ ПОД ДАВЛЕНИЕМ — ГАЗИФИКАЦИЯ ТВЕРДЫХ ТОПЛИВ
732
ции осуществляется под давлением от 5 до 40 ат. Теплота
полученного газа используется в котле-утилизаторе 4. Далее
газ очищается от сажи в аппарате 5, охлаждается в скруббере в
и направляется потребителю. Вода после очистки газа от
сажи отстаивается и вновь используется в произ-ве. Сырьем
служат любые жидкие продукты нефтепереработки, а также
природный газ. Выход из 1 кг сырья 3 нм3 газа состава: 4% СО2;
47% СО; 49% Н2; 0,3% СН4; 0,3% N2. Расход кислорода
0,75 нм3/кг сырья, выход сажи 0,04 кг/кг сырья. Для получе-
ния газа для бытовых нужд на этой же установке часть сырья
впрыскивается в поток горячего газа. В зтом случае из 1 кг
сырья получают 2,3 нм3 газа состава: 7% СО2; 36% СО; 39 % Н2;
15% СН4; 1 % С2Н4; 1 % N2. Теплотворность газа 4100 ккал/нм3,
расход кислорода 0,265 нм3/нм3 газа, выход сажи 0,003 кг/кг
сырья. Способ Г. ж. т. под давлением имеет след, преимуще-
ства по сравнению с процессом при атмосферном давлении:
уменьшается расход электроэнергии на компрессию в про-
цессах переработки газов (синтез аммиака, метанола) и объем
аппаратов для произ-ва газа; газ для бытовых нужд может
транспортироваться на дальние расстояния под собственным
давлением.
Г. ж. т. получает все более широкое распростране-
ние благодаря технич. и экономия, преимуществам
по сравнению с газификацией твердого топлива (себе-
стоимость сырья меньше в 2—2,5 раза, значительно
упрощается топливоподготовка, исключается золо-
удаление, произ-во более компактно и легко автомати-
зируется, требует меньших капиталовложений и
эксплуатационных расходов).
Лит.: Паушкин Я. М., Нефт. х-во, 1954, № 8, 56;
Паушкин Я. М., Вишнякова Т.П. Производство оле-
финосодержащих и горючих газов..., М., 1960; Gerho 1 d М.,
Erdol und Kohle, 1956, № 1—4; его же там же, № И—12;
G a t t 1 к е г М., там же, 1957, № 9, S. 581; Pichler Н.
und Heike Th. Gas und Wasserfach, 1957, H. 21/22,
S. 508; В о г e 1 1 1 T. et Right A., L’Industrie Chimique,
1953, 40, № 428, 65; Wustrow W. und Kratz H.,
Erdol und Kohle, 1955, JV? 2. S. 82; Estman D. du, Industr.
and Engng. Chem., 1956, 48, № 7, p. 1118.
M. И. Дербаремдикер, К. Л. Серебренникова.
ГАЗИФИКАЦИЯ ПОД ДАВЛЕНИЕМ — см. Га-
зификация твердых топлив.
ГАЗИФИКАЦИЯ ТВЕРДЫХ ТОПЛИВ — процесс
превращения твердых топлив в горючие газы пу-
тем окисления воздухом, кислородом, водяным па-
ром или углекислым газом при высокой темп-ре. В от-
личие от коксования и полукоксования, процесс газифи-
кации характеризуется превращением всей органич.
части топлива, исключая потери, в газы. Г. т. т.
производят в газогенераторах, получаю-
щиеся газы называют генераторными. Гази-
фицируют все виды твердого топлива: каменные и
бурые угли, антрацит, кокс, полукокс, торф, древе-
сину, горючие сланцы и др. Генераторные газы при-
меняют в качестве топлива в металлургии., стеколь-
ном, керамич. и других произ-вах, для бытовых нужд,
в двигателях внутреннего сгорания и газовых тур-
бинах. В химич. пром-сти используют газы, содер-
жащие в основном СО и Н2, служащие сырьем в
производстве аммиака, метанола, высших спиртов,
моющих средств, жидкого топлива, парафина и др.
Названия генераторных газов определяются составом
газов дутья. Напр., при газификации воздухом полу-
чается: воздушный газ, аналогично—паровоздушный
(смешанный), парокислородный, водяной, полуводя-
ной, газ кислородного дутья и др.
Генераторные газы различают также по назначению:
энергетический (отопительный и силовой), техноло-
гический (для химич. синтезов), бытовой. Способы
Г. т. т. отличаются между собой видом применяемого
топлива, организацией процесса (в слое кускового
топлива — прямой или обращенный, в кипящем слое
др.), температурным режимом (с выпуском шлаков
в твердом или жидком виде), режимом давления (под
разрежением, при незначительном избыточном давле-
нии, под давлением 20—30 ат), конструкцией газоге-
нераторов и др. Большим разнообразием отличаются
и технология, схемы произ-ва. Одним из способов
Г. т. т. является процесс подземной газификации (см.
Газификация топлив подземная).
Сущность процесса. Основные химич. превращения
твердых топлив в горючие газы при любых методах
Г. т. т. в общих чертах остаются одинаковыми и могут
быть рассмотрены на примере газификации смесью
воздуха и водяного пара в плотном слое топлива. Га-
зогенератор (рис. 1) пред-
ставляет собой цилиндрич.
шахту, в к-рую сверху за-
гружается топливо, а снизу
подается дутье. Зола уда-
ляется с помощью вращаю-
щейся колосниковой решет-
ки. По мере расходования
топливо непрерывно дви-
жется нниз навстречу (про-
тивопотоком) газам и дутью.
Получающийся газ отво-
дится из верхней части газо-
генератора. В верхней части
слоя топлива, называемой
зоной подсушки, топливо
прогревается и подсуши-
вается поступающими снизу
горячими газами. В средней
части слоя, к-рая называет-
ся зоной сухой перегонки,
топливо под действием га-
зов с темп-рой 550—800°
подвергается разложению:
образуются газы, пары смо-
Рис. 1. Схема процесса га-
зификации в противоточном
газогенераторе.
лы, влага; топливо превращается в полукокс и кокс.
Ниже, в зоне газификации, где темп-ра превышает
1000°, кокс вступает во взаимодействие с газифицирую-
щими реагентами— газами дутья. В результате реакций
со свободным кислородом дутья, к-рыи в зоне газифи-
кации полностью расходуется, углерод топлива пре-
вращается в СО иСО2, водяной пар реагирует с углеро-
дом с образованием Н2, СО и СО2. Из газогенератора
выходит газ, состоящий из смеси газов, полученных
в процессах газификации и сухой перегонки, с при-
месью непрореагировавшего водяного пара, паров
смолы, пыли, сероводорода и др. Темп-ра парогазовой
смеси, покидающей газогенератор, при сухом топливе
(кокс, антрацит) достигает 600°. Как правило, перед
подачей потребителю газ подвергается охлаждению
и очистке (см. Газов очистка и Газов осушка).
В зоне газификации протекают след, реакции:
С 4- Од — СОо 4- 97650 ккал/моль (1)
2С 4- О2 = 2СО 4- 58860 ккал/моль (2)
Эти одновременно протекающие экзотермич. реакции
полного и неполного сгорания углерода являются пер-
вичными. Полученная окись углерода может вступать
во взаимодействие с кислородом:
2СО 4" Оз = 2СО2 4~ 136440 ккал/моль (3)
а СО2 может реагировать с углеродом:
СО о + С = 2СО — 38790 ккал/моль (4)
Равновесия реакций (1), (2) и (3) смещены в сторону
образования СО2 и СО при темп-рах, характерных
для процессов Г. т. т. (1000—1500°). Реакция (4) эндо-
термическая, ее равновесие смещается в сторону по-
лучения СО при повышении темп-ры; с повышением
давления выход СО понижается. Фактич. соотноше-
ние между окислами углерода зависит от темп-ры,
давления, длительности взаимодействия, реакционной
способности топлива и др. Водяной пар взаимодей-
ствует с углеродом топлива:
С 4- Н2О -= СО 4- Н3 — 28380 ккал/моль (5)
С 4- 2НаО — СОо 4~ 211® — 17970 ккал/моль (6)
Равновесие в реакциях (5) и (6) сдвигается вправо
при повышении темп-ры и понижении давления.
Продукты реакций (5) и (6) называются водяным газом.
733
ГАЗИФИКАЦИЯ ТВЕРДЫХ ТОПЛИВ
734
В смеси окислов углерода, водорода и водяного пара
протекает реакция:
СО + Н2О = СО3 Hs 4-10410 ккал/моль (7)
При темп-ре выше 1000° равновесие сдвинуто в сторону
исходных веществ. Увеличение давления не влияет на
состояние равновесия. Реакция (7) имеет и самостоя-
тельное значение для получения водорода.
Содержащийся в генераторных газах метан обра-
зуется по реакциям:
СО + ЗН2 = СП, 4- Н.О 4 49250 ккал/моль (8)
2СО 4 П3 = СН, 4 СО2 4 59100 ккал/моль (9)
а также вследствие термич. разложения органич.
массы топлива. С понижением темп-ры и повышением
давления равновесие в этих реакциях смещается в
сторону образования метана. При повышении темп-ры
и понижении давления равновесие смещается влево,
что используется в технике для получения водорода.
Скорость приведенных выше реакций газификации
зависит от скорости химич. взаимодействия веществ и
скорости диффузии. Скорость газификации лимити-
руется скоростью более медленного процесса и при
700—800° зависит от скорости реакции, поскольку
она невелика. С повышением темп-ры скорость химич.
процессов резко возрастает, скорость же диффузии
растет незначительно, вследствие чего выше 1000°
процесс газификации тормозится уже скоростью диф-
фузии. В первом случае процесс газификации опре-
деляется законами химич. кинетики и протекает
в т. наз. кинетич. области, во втором случае — в диф-
фузионной области. Скорость диффузии растет с уве-
личением скорости дутья, что является сильным сред-
ством интенсификации процессов Г. т. т. В практич.
условиях суммарный процесс газификации протекает
в промежуточной области, где его скорость зависит
одновременно от обоих факторов. Кроме повышения
темп-ры и скорости дутья, процессы газификации
интенсифицируются увеличением реакционной по-
верхности (применением мелкозернистых и пылевид-
ных топлив), увеличением давления, концентрации
кислорода в Дутье и др. Для бесперебойности процесса
газификации и устойчивости качественных и количе-
ственных показателей (напр., состав газа, расход
топлива, состав дутья и т. д.) необходимо: поддержи-
вать постоянный уровень топлива и золы в шахте
газогенератора, равномерно распределить дутье и
газы по поперечному сечению шахты, соблюдать
температурный режим и постоянство давления, что
обеспечивает работу без шлакования. При выборе
того или иного способа газификации решающее зна-
чение имеет вид топлива и назначение газа.
Генераторные газы и способы их производства.
Воздушный газ получают при дутье одним
воздухом. Сущность процесса характеризуется реак-
циями (1—4); тепловой эффект — положительный;
темп-ра в шахте газогенератора достигает 1400—
1600° и выше. В этих условиях углекислый газ почти
полностью восстанавливается до СО; зола топлива
расплавляется. Воздушный газ получается в газогене-
раторах с удалением шлака в жидком виде. Тепло-
творность воздушного газа ок. 1080 ккал/нм3 (самая
низкая среди генераторных газов). Выходящий из
газогенератора газ имеет темп-ру порядка 700—800°;
большое количество тепла уходит с газом и кпд про-
цесса невысокий (ок. 70%). Воздушный газ приме-
няется в нек-рых химич. произ-вах, где требуется
высокая концентрация СО, напр. для произ-ва муравь-
иной кислоты.
В газогенератор (рис. 2) загружается смесь топлива и
флюсов. Дутье (воздух) подается через фурмы; жидкий шлак
выпускается из летки. Такие газогенераторы могут вормальво
вксплуатпроваться на механически и термически прочных
топливах (кокс, камевные угли, тощие каменные угли и др.),
т. к. при применении др. топлив образующаяся в газогенера-
Рис. 2. Газогенератор
для газификации куско-
вых топлив с выпуском
шлака в жидком виде:
1 — загрузочная короб-
ка; 2— шахта; 3— газо-
отвод; 4— горн; 5—фур-
мы; 6 — летка.
торе мелочь нарушает равномерное газораспределение, при-
водит к загустеванию шлаков и прекращению их вытекания
из леток.
Парокислородный газ. При примене-
нии в качестве дутья парокислородной смеси или смеси
кислорода и двуокиси угле-
рода в зоне газификации одно-
временно протекают экзотер-
мич. (1) и (2) и эндотермич.
(4), (5) и (6) реакции. В ре-
зультате газ обогащается оки-
сью углерода и водородом, со-
держание которых составляет
95—97%. Высокое содержание
СО дает возможность исполь-
зовать этот газ для составле-
ния смеси газов с любым со-
отношением СО и Н,, которое
требуется в химич. синтезах.
По сравнению с процессом по-
лучениявоздушногогаза, вдан-
номслучае, благодаря наличию
водяного пара и СО2, снижает-
ся темп-ра в зоне газификации
и темп-ра выходящего газа,
что повышает кпд процесса.
Соотношение в дутье кислорода
и пара или СО2 подбирают
таким, чтобы снижение темп-ры
не уменьшило текучести шла-
ков, необходимой для беспере-
бойного их удаления. Благо-
даря высоким темп-рам газо-
генератор с удалением шлака
в жидком виде отличается вы-
сокой интенсивностью (в 3—4 раза выше, чем у
газогенераторов с удалением твердых шлаков, расход
пара в несколько раз меньше и др.). Недостатки про-
цесса: необходимость применения дорогих видов топ-
лива и повышенный удельный расход кислорода.
Кислородный газ получают при подаче
в газогенератор в качестве дутья одного технич.
кислорода.
При применении технич. 98%-ного О2 получают
газ с содержанием 95—97% СО. Произ-во кислородно-
го газа ограничивается специальными случаямп (по-
лучение А1С13 и др.).
Смешанный генераторный (паро-
воздушный) газ получают при Г. т. т. смесью воз-
духа с водяным паром. Сущность процесса заклю-
чается в одновременном протекании экзотермических
(1—3) и эндотермических (4—6) реакций. Зола в этом
процессе не расплавляется, а удаляется в твердом
виде. Для произ-ва смешанного газа используется
любое топливо. Теплотворность газа 1200—
1600 ккал)нм3 (в зависимости от вида топлива), кпд
процесса ок. 75%. Из искусственных горючих газов
смешанный газ имеет наибольшее распространение
в пром-сти, где он применяется исключительно в ка-
честве топлива и является самым дешевым.
На рпс. 3 показан разрез современного механизированного
газогенератора смешанного газа. Генератор состоит из шахты
с водявой рубашкой, загрузочного устройства, колосниковой
решетки, механизма золоудаления и устройства для подачи
дутья. Цилиндрич. шахта футерована огнеупорным кирпичом
и заключена в стальной кожух. Нижняя часть шахты выпол-
нена в виде водяной рубашки. Получающийся в водяной
рубашке пар используется для дутья. На крышке газогенера-
тора расположено загрузочное устройство, посредством к-рого
топливо непрерывно загружается в шахту и равномерно рас-
пределяется по поверхности слоя топлива. В нижней части
шахта переходит в фартук, к-рый погружен в чашу с водой,
создавая т. о. гидравлич. затвор. На две чаши установлена
колосниковая решетка, к-рая вращается вместе е чашей и
Предназначена для распределения дутья и удаления шлаков.
Водяной газ получают при подаче в раска-
ленный слой топлива водяного пара. Газ содержит
735
ГАЗИФИКАЦИЯ ТВЕРДЫХ ТОПЛИВ
736
накопле-
i в газо-
воздух
большой
вследст-
слой
83—86% СО и Нг и предназначен для синтеза химич.
продуктов. Широко распространенным способом про-
изводства водяного газа является циклич. процесс,
к-рый состоит в не-
прерывном чередо-
вании двух основ-
ных операций (фаз):
продувки воздухом
для разогрева слоя
(получение воздуш-
ного газа) и подачи
пара для получения
водяного газа. Для
быстрого
ния тепла
генераторе
подается с
скоростью,
вие чего слой то-
плива разогревается
до высокой темп-ры.
Затем подача воз-
душного дутья пре-
кращается и в газо-
генератор подается
водяной пар. Полу-
чающийся водяной
газ после охлажде-
ния и очистки от
пыли направляется
потребителю. Когда
темп-ра в газогене-
Рис. 3. Газогенератор
сметанного газа: 1 —
загрузочное устройст-
оси р уоичпис JfUipVUC'l
во; 2—шахта; з—водяная рубашка; 4 — колосниковая решетка;
.5— фартук; 6 — чаша; 7 — выгребной нож; 8—скребковый
транспортер; 9 — дутьевая коробка; 10 — привод колоснико-
. вой решетки; и — шуровочный люк.
раторе снизится настолько, что разложение водяного
пара уменьшается, подача водяного пара прекращает-
ся и снова подается воздушное дутье и т. д. Продол-
жительность всего цикла, в к-рый входят и вспомога-
тельные операции, в современных механизированных
н автоматизированных газогенераторах доведена до
2—4 мин. Для произ-ва водяного пара применяется
только тощее, термически прочное топливо (кокс,
антрацит).
Конструкция газогенератора для произ-ва водяного газа
отличается от газогенератора смешанного газа тем, что вместо
гидравлич. затвора в нем используется сухое уплотнение,
позволяющее значительно увеличить давление дутья. Особен-
ность управления газогенераторами водяного газа состоит
в большом количестве переключений потоков воздуха, пара,
водяного и воздушного газов. Все клапана, задвижки, а также
механизмы загрузки топлива действуют автоматически и для
безопасности имеют автоматич. блокировку и сигнализацию;
кпд газификации низкий — ок. 60%, что объясняется перио-
дичностью процесса. Водяной газ можно производить также
непрерывным способом путем применения парокислородного
дутья в тех же газогенераторах. Процесс аналогичен получе-
нию смешанного газа. Соотношение пара и кислорода подби-
рается таким, чтобы исключить шлакование. По сравнению
с циклич. процессом производительность газогенератора в этом
случае увеличивается в два раза, кпд достигает 85%. Сырьем
служит кокс или антрацит.
Полуводяной газ — смесь водяного га-
за со смешанным, в котором объемное соотношение
(CO-|-H2)/N2=3. Он служит сырьем для получения
азото-водородной смеси с соотношением Н2 : N2 = 3,
применяемой в синтезе аммиака. Для этого полуводя-
ной газ подвергают коннерсии по реакции (7); в ре-
зультате из окиси углерода получается эквивалент-
ное количество водорода. Полуводяной газ может
быть также получен в одном газогенераторе (непре-
рывным способом). Для этого используется воздушное
дутье, обогащенное кислородом (до 50—60%).
4. Генератор высокого
Рис.
давления: 1 — корпус; 2 —
водяная рубашка; 3 — загру-
зочный карман; 4 — золь-
ный карман; 5 — распреде-
лительное устройство; в —
колосниковая решетка; 7 —
привод распределительного
устройства и колосниковой
решетки.
Бытовой и технологический га -
з ы. Одним из наиболее современных и универсаль-
ных методов Г. т. т. является газификация
под давлением 20—30 ат на парокислород-
ном дутье и мелкокусковом топливе для получения
бытового итехнологического газов. В ка-
честве сырья используют бурые и каменные угли
(в том числе спекающиеся), антрацит и торф. Среди
способов Г. т. т. процесс под давлением является
единственным, с помощью к-рого производится быто-
вой газ. При высоком давлении значительно увели-
чиваются объемные концен-
трации реагирующих га-
зов и возрастает интенсив-
ность процесса. Характер-
ной особенностью процесса
является значительное раз-
витие реакций образования
метана (8) и (9), содержание
к-рого в очищенном от СО2
газе составляет 14—18%,
что определяет его высо-
кую теплотворность (3600—
4100 ккал/нм3). Экзотер-
мичность реакций метано-
образования обусловливает
снижение расхода кислоро-
да в 2—3 раза по сравнению
с процессом при нормальном
давлении. Объем газов со-
кращается, соответственно
уменьшаются их линейные
скорости в шахте газогене-
ратора, что позволяет при-
менять мелкокусковое топ-
ливо. Давление газа позво-
ляет уменьшить расходы на
сжатие при его применении.
Производительность мощного газогенератора (рис. 4)
32 000 мл3/час сырого газа, диаметр шахты 3,7 м. Шахта не
футерована, окружена водяной рубашкой. Устройство для
распределения топлива в шахте и многоступенчатая колос-
никовая решетка укреплены на охлаждаемом водой валу.
В этом газогенераторе используются каменный уголь класса
7—20 -ч.ч или 20—60 -м.м, пар с небольшим перегревом и технич.
98%-ный кислород. Производительность газогенераторов на
большинстве газовых заводов достигает 10 000 нм3/час, диа-
метр шахты 2,6 .м. Недостатком процесса,- протекающего не-
прерывно, является необходимость периодически произво-
дить загрузку топлива и выгрузку золы.
Обращенный процесс Г. т. т. Выше рассматривались
способы Г. т. т., в к-рых дутье и топливо движутся
в шахте газогенератора противотоком (т. и. прямой
процесс). В обращенном процессе топливо и дутье
движутся в одном и том же направлении (сверху вниз).
Выделяющиеся в зоне сухой перегонки летучие в-ва,
проходя через зону газификации, сгорают. Обращен-
ный процесс применяется для получения силового
газа из битуминозных топлив (дрова, торф, каменные
и бурые угли). Силовой газ предназначен для газовых
двигателей внутреннего сгорания (стационарных или
транспортных), нормальная эксплуатация к-рых воз-
можна только на бессмольном газе; кпд обращенного
процесса и теплотворность газа ниже, чем при прямом
процессе Г. т. т.
В описанных способах Г. т. т. сырьем служит куско-
вое топливо, к-рое в шахте газогенератора находится
в плотном слое. Одним из мощных средств интенси-
фикации процессов Г. т. т. является увеличение реак-
ционной поверхности топлива путем применения мел-
козернистых и пылевидных топлив. Газификация
таких топлив потребовала разработки иной, чем в плот-
ном слое, организации процесса: в кипящем, взвешен-
ном, вращающемся слоях, с применением твердого
теплоносителя и др. Наибольшее распространение
получила газификация в кипящем
737
ГАЗИФИКАЦИЯ ТВЕРДЫХ ТОПЛИВ
738
жении, напоминающем
Рис. 5. Генератор ГИАП
с кипящим слоем: 1 — шах-
та; 2 — колосниковая ре-
шетка; 3 — фурмы вто-
ричного дутья; 4 — дутье-
вая камера; 5 — зольная
камера; 6 — зольный бун-
кер; 7 — привод шурую-
щей штанги; 8 — топлив-
ный бункер; Р—шнек; 10—
привод шнека.
или псевдоожиженном слое (см. Псе~
едоожижение)’, осуществляется в слое частиц топлив
размером до 10 мм, находящихся в непрерывном дви-
кипение жидкости. Дутье
подается с необходимой для
этого скоростью. Например, в
генераторе системы ГИАП
(рис. 5) скорость дутья в пло-
скости колосниковой решетки
составляет 5 м/сек, В кипя-
щем слое происходит интен-
сивное перемешивание и цир-
куляция частиц топлива, что
способствует выравниванию
темп-p во всем слое, вслед-
ствие чего процессы сушки,
сухой перегонки, а также га-
зификации протекают во всем
объеме. Летучие вещества при
этом полностью разлагаются
и частично сгорают, поэтому
теплотворность газа ниже,
чем в противоточном процес-
се. Глубокое разложение про-
дуктов полукоксования при-
водит к тому, что сточные
воды не содержат фенолов и
смолы и сбрасываются без
очистки. «Кипящий» слой,
впервые примененный для
процессов Г. т. т., получил
впоследствии широкое рас-
пространение в других отра-
слях пром-стн (химич., не-
фтяной, металлургия.).
Газогенератор (см. рис. 5)
представляет собой футерован-
ную цилиндрич. шахту высотой ок. 20 At и диаметром 5 м.
В нижней конич. части установлена колосниковая решетка,
имеющая форму пологого обращенного конуса. В центре его
расположена золоспускная труба. Для вывода золы из гене-
ратора служит шурующая штанга, снабженная гребками. Зола
направляется в зольный бункер, к-рый периодически опорож-
няется. Топливо из бункера подается шнеками в нижнюю
часть кипящего слоя. Темп-ра в кипящем слое поддержи-
вается в пределах 930—980°, в зависимости от качества мине-
ральной части топлива. Вместе с газом из шахты газогенера-
тора уносится значительное количество пыли, для газифика-
ции к-рой через фурмы над кипящим слоем подается вторич-
ное дутье, вследствие чего темп-ра в верхней части шахты
достигает 1100°. Процесс в кипящем слое позволяет создавать
агрегаты мощностью до 80 000 ши3 газа в час и выше, что во
много раз больше, чем у крупных газогенераторов для куско-
вого топлива. При этом расширяется сырьевая база Г. т. т.
за счет применения дешевого топлива (мелочи бурого угля, фре-
зерного торфа и др.). Благодаря высокоразвитой реакционной
поверхности интенсивность процесса достигает 3000 кг/м9час.
В зависимости от состава дутья производится водяной, полу-
водяной или смешанный газы. Управление процессом пол-
ностью механизировано и в значительной степени автоматизи-
ровано. Недостатками процесса являются значительный унос
пыли, низкий съем газа с единицы объема шахты, необходи-
мость подсушки топлива, плохие показатели при газификации
углей с низкой реакционной способностью (каменные угли).
Газификация пылевидных топлив. Новым способом
Г. т. т. является газификации тонкоизмельченного
топлива (размер частиц
меньше 0,1 мм) в потоке
парокислородной смеси
привысокой температуре.
Огромная реакционная
поверхность пыли и вы-
сокая температура опре-
деляют исключительно
высокую интенсивность
процесса, во много раз
превосходящую сущест-
вующие способы Г. т. т.
Кроме того, применение
пылевидного топлива де-
лает процесс независи-
мым от вида и качества
Рис. 6. Газогенератор для пы-
левидного топлива: 1 — корпус;
2 — форсунки; з — газоотвод;
4 — шлаковая ванна.
сырья.
Газогенератор (рис. 6) представляет собой горизонтальный
футерованный цилиндр с конич. охлаждаемыми водой амбра-
зурами, в к-рых расположены две направленные друг против
друга форсунки. Подаваемая с большой скоростью смесь уголь-
ной пыли с кислородом и водяным паром воспламеняется не-
посредственно по выходе из форсунок. Газификация угля
происходит в факеле в благодаря высокой интенсивности
еаканчивается вблизи форсунки. Основная часть золы уно-
сится газом, остальная — расплавляется и стекает в прием-
ник. Тепло газа используется в котле-утилизаторе. Произ-
водительность каждой форсунки достигает 125 т угля в сутки-
кпд газификации достигает 95%. Состав газа: 12% СО2, 51%
СО 34% Н2, 0,1% СН4 и 2,9% N2 и H2S. Управление установ-
кой полностью автоматизировано. На установке можно гази-
фицировать и жидкое топливо, но для этого нужны форсунки
другой конструкции.
Кроме указанных, разрабатываются новые способы
газификации мелкозернистых и пылевидных топлив.
Нек-рые из них преследуют цель создания новых аэро-
динамич. схем для улучшения взаимодействия топ-
лива с дутьем, напр. осуществление процесса в слое,
Показатели Газы
смешанный бытовой водяной полуводяной
Назначение газа Топливо Химическое сырье
Способ газификации в плотном слое под давле- нием 20 ат циклический процесс с удалением шла- ка в жидком виде в кипящем слое
Топливо антрацит бурый уголь бурый уголь каменно- угольный кокс каменноуголь- ный полукокс бурый уголь
Влажность (И7Р), вес. % 5,0 16,0 26,5 3-4 11,2 12-15
Зольность (Ас), вес. % Состав газа, об. %: СО, + H2S 10,5 26,0 6,4 8-9 8,4 23—25
5,3 5,0 30,5 (0,2)* 4,7 1,0 15,1
Цп“П * 0,0 0,2 0,5 (0,7) 0,0 0,0 0,0
О2 0,2 0,6 03 (0,3) 0,2 0,0 0,0
со 27,0 26,8 16,4 (23,6) 41.4 66,2 33,0
н, 15.0 163 38,7 (55,6) 48,4 31,0 31.0
сн4 0,5 2,6 9,9 (14,1) 0,3 0,6 03
n2 ' 52,0 48,5 3,8 (5,5) 5,0 1,2 20,7
Теплотворность газа низшая, ккал/нм8 . . 1300 1480 2400 (3500) 2570 2350 1800
Выход газа с 1 кг топлива, >м<3/кг .... Производительность газогенератора по 3,75 3600 2,1 1,39 (0,97) 11150 (7750) 1,65 7500 1,94 17000 2,1 20000
газу, нм9/час ............... 2300
Напряжение поиеречн. сечения шахты по топливу, к г/м 2-час ............ 325 350 1600 570 1100 1900—3200
Кпд газификации, % 75 70 83 60 85 70
В скобках приведены показатели по газу, очищенному от СОа и HaS,
739
ГАЗИФИКАЦИЯ ТОПЛИВ ПОДЗЕМНАЯ
740
«взвешепиом» в потоке дутья в шахте с конусообраз-
ным сечением, аэрофонтанные схемы с интенсивным
перемешиванием топлива с дутьем за счет центробеж-
ных сил, возникающих при вращении потока. Другие
способы основаны на использовании различного рода
«теплоносителей», являющихся источником тепла для
проведения эндотермич. реакций газификации и пиро-
лиза летучих веществ. Напр., газификация топлива
в потоке раскаленного газового теплоносителя; в по-
токе пара и газа с использованием в качестве тепло-
носителя раскаленных мелкозернистых материалов;
в потоке дутья при прохождении сквозь слой раска-
ленного шлака.
В таблице (стр. 737—738) приведены нек-рые дан-
ные о генераторных газах и основных показателях
Г. т. т.
За последние годы в СССР и других странах обна-
ружены крупные месторождения природного горючего
газа. Выгодность его применения по сравнению
с произ-вом искусственных газов очевидна. Однако
разведанные запасы ископаемых твердых топлив зна-
чительно превосходят запасы нефти и газов. Поэтому
Г. т. т. остается одним из основных путей газоснабже-
ния промышленных предприятий и бытовых потреби-
телей в районах, бедных природными газами.
Лит.: Газификация твердого топлива. Труды третьей
научно-технической конференции, М., 1957; Шиша-
ков Н. В., Основы производства горючих газов, М.—Л.,
1948; Канторович Б. В., Основы теории горения и
газификации твердого топлива, М., 1958; Кнорре Г. Ф.,
Топочные процессы, М.—Л., 1951; Лавров Н. В., Физико-
химические основы горения и газификации топлива, М. 1957;
Чернышев А. Б., Избранные труды, М., 1956; Н u fa-
man п О., BrennstOtf-Chemle, 1959, 40, № 3, 65; J. Inst.
Fuel, 1957, 30, № 203, р. 673. М. И. Де'рба'ремдикв'р.
ГАЗИФИКАЦИЯ ТОПЛИВ ПОДЗЕМНАЯ —
использование ископаемых топлив (угля, сланца и
нефти) путем их превращения под землей, на месте
залегания, в горючий газ и вывода последнего на по-
верхность через буровые скважины. Впервые мысль
о подземной газификации угля высказал Д. И. Мен-
делеев в 1888. В 1913 В. И. Ленин в статье «Одна из
великих побед техники» охарактеризовал важное
значение этой проблемы. Химико-технологич. сущ-
ность процесса подземной газификации угля в основ-
ном такая же, как и при наземной газификации (см.
Газификация твердых топлив и Газификация жидких
топлив). В процессе газификации образуются три
основные реакционные зоны: окислительная, восстано-
вительная и сухой перегонки. Для Г. т. п. с поверх-
ности пробуриваются вертикальные скважины на
расстоянии 25—35 м одна от другой. Через одни сква-
жины в пласт подается воздушное дутье под давлением
3—4 ат. По трещинам и порам в угольном пласте
воздух проходит к другим скважинам и выходит на
поверхность. Розжиг пласта производится с помощью
электрич. Или иных нагревателей; образовавшийся
очаг горения перемещается от одной скважины к дру-
гой, и таким образом в толще угольного слоя прогорает
соединительный (сбоечный) канал. Если газопрони-
цаемость пласта мала, то применяются другие способы
сбойки: пропускание электрич. тока по пласту (элек-
тросбойка), нагнета-
ние под высоким да-
влением в пласт во-
ды или другой жид-
кости (гидроразрыв
пласта), бурение по
пласту соединитель-
ных горизонтальных
скважин и др. По
окончании сбойки
скважин начинается
собственно процесс
газификации при по-
даче дутья под дав-
лением 1,5—2 ат в
одни скважины и
пр и отводе газа через
другие. Периодиче-
ски направление по-
токов дутья и га-
за меняется (про-
цесс реверсируется)
с целью наиболее
Рис. 1. Схема подземной газифи-
кации угля в скважинах-газогене-
раторах: 1 — сбоечный канал (канал
розжига); 2 — дутьевые скважины;
3 — газоотводные скважины; 4 —
выгазованное пространство.
полного выгорания топлива. При этом между дутьевы-
ми и газоотводными скважинами образуется реак-
ционное пространство, в к-ром возникают упомянутые
выше зоны газификации. Для
Г. т. п. наклонныхпластов пробу-
риваются, кроме вертикальных,
наклонные дутьевые и газоотвод-
ные скважины (рис. 1). Сбойка
скважин требует значительных
затрат электроэнергии и вре-
мени, удорожает и осложняет
подготовку пластов для газифи-
кации. Этого недостатка лишен
метод газификации т. н. слепы-
ми скважинами, пробуренными
по угольному пласту, при к-ром
каждая скважина является инди-
видуальным газогенератором
(рис. 2), работающим незави-
симо от других. Дутье подает-
ся через выдвижную трубу, опу-
щенную в скважину, а получае-
мый газ выводится на поверх-
ность через ту же скважину по
странству. По мере выгорания угля в скважине труба
постепенно выдвигается из скважины. В зависимости
от режима и состава дутья и от качества топлива
получается газ различного состава (см. таблицу).
Наряду с подземной газификацией каменных и
бурых углей в СССР и зарубежных странах прово-
дятся работы по подземной газификации и переработке
Вода
Дутье^-1
Г аз
уголь
уголь
Рис. 2. Схема под-
земной газификации
угля в слепых сква-
жинах.
Обрушиваю-^
щояся
кровля
межтрубному про-
Топливо Дутье Состав полученных газов в % Теплотвор- ность полу- ченного газа, ккал/нм3
СО2 о2 со Н, СН. n2
Каменный уголь Воздушное 6-12.5 0,2—0,4 15-23 12,5-14 0,6-2 52— 58,5 950-1200
Бурый уголь (подмосковный) Каменный уголь То же Обогащенное кислоро- 17—18 0,4 6,0 16,0 1,3 58 800
То же дом (35—65%) Периодическое воздуш- но-паровое (в период 12—19 0,2 22—28 21 2—8 28-36 1500-1800
Бурый (подмо- паровой фазы) Парокпслородное (60% 21,5 — 15 51 6 6 2250
сковнцй) уголь * О2 и 230 г/нм3 пара) . 23,3 0,1 20 36 6,2 — ^1900
♦ В продуктах газификации содержание H2S составляло 1,5—2,7%.
741
ГАЗОВ ОСУШКА
742
горючих сланцев. В СССР работы по подземной пере-
работке сланцев проводятся комбинированным мето-
дом. Более богатые слои сланца добываются шахтным
способом и выдаются на поверхность для переработки
в сланцеперегонных печах, а образующиеся пустоты —
камеры, заполняются кусками сланца и герметизи-
руются. В камеру с поверхности земли пробуриваются
скважины для подачи дутья и отвода газа и смолы.
За счет сжигания части сланца, заполняющего камеры,
подвергается термич. обработке как топливо, запол-
няющее камеру, так и слагающее стенки и кровлю
камеры.
Проводятся также работы по бесшахтной подземной
газификации сланцев. Подготовка пласта производится
бурением скважин и бесшахтной сбойкой их. После
сбойки дутье подается в одни скважины, а газ и про-
дукты перегонки сланца отводятся через другие (па-
раллельно с процессом газификации идет и процесс
перегонки). Опыты показали большую перспективность
подземной перегонки сланцев.
К методам Г. т. п. относятся также термич. способы
добычи нефти и продуктов ее подземной перегонки
путем сжигания части нефти, заключенной в пласте,
или нагрева пласта горячими газами, получающимися
при сжигании газа или нефти в спец, горелках, опус-
каемых в скважину. Горячий теплоноситель филь-
труется по пласту нефти, нагревает и делает ее более
текучей и частично отгоняет в виде паров бензин и др.
продукты. Образующиеся при этом газообразные и
жидкие продукты выводятся на поверхность через
скважины.
Лит.: О состоянии и ближайших направлениях развития
науки по проблеме подземной газификации углей. [Сборник
ст.], М.. 1957; Л е щ и н е р Р. Е. и Юрченко В. П.
[сост.], Подземная газификация углей за рубежом [Сбор-
ник ст.]. М., 1956; К и р и ч е н к о И. П., Химические спо-
собы добычи полезных ископаемых, М., 1958; Подземная пе-
реработка топлив, М., 1960 (Тр. Ин-та горючих ископаемых.
АН СССР, т. 13); Научные труды, вып. 1. — Краткие итоги
научно-исследовательских и опытных работ по подземной
газификации углей, М., 1960. И. П. Кириченко.
ГАЗОВ ОСУШКА — процесс удаления паров воды
из газов. Г. о. необходима, напр., при глубоком охла-
ждении многокомпонентных газов в целях их разделе-
ния на фракции (см. Газов разделение и Воздуха, разде-
ление), при транспортировке горючих газов по трубо-
проводам и др. Г. о. производят физико-химич. (аб-
сорбционными и адсорбционными) и физич. спосо-
бами.
Абсорбционные способы основаны на
поглощении влаги из газов жидкими веществами,
водные р-ры к-рых имеют низкое давление паров воды.
При выборе абсорбента руководствуются его физико-
химич. свойствами по отношению к компонентам осу-
шаемого газа. В качестве абсорбентов применяют р-ры
СаС12 (35—40%), ZnCl2, глицерина (85%),- диэтилен-
гликоля (85—97%), триэтиленгликоля и др. Степень
осушки газов, достигаемая при помощи этих абсор-
бентов, различна; понижение точки росы газа: не более
17° при применении р-ра СаС12, 25—32° для ZnCl2,
25—28° — глицерина, 25—30° — диэтиленгликоля,
40—50°—триэтиленгликоля. Наиболее распростра-
ненным абсорбентом является диотиленгликолъ (СаС12
и ZnCl2 вызывают коррозию аппаратуры, глицерин
нестоек при нагревании во время регенерации).
Технологич. схема установки для осушки газа абсорбцион-
ным способом представлена на рис. 1. После предваритель-
ного охлаждения осушаемый газ поступает в абсорбер 1,
нижняя секция к-рого служит для отделения взвешенных
в газе капель жидкости. Осушенный газ по газопроводу выво-
дится из абсорбера. Навстречу потоку газа в абсорбер сте-
кает раствор абсорбента, к-рый по выходе из абсорбера по-
ступает в теплообменник 5, паровой подогреватель 4 и отгон-
ную колонну 6. При нагревании раствора глухим паром в ки-
пятильнике 7 поглощенная влага удаляется из раствора в виде
пара, к-рый выводится из колонны в атмосферу. Для преду-
преждения уноса паров диэтиленгликоля на верхнюю тарелку
отгонной колонны подается небольшое количество горячего
водяного конденсата. Регенерированный горячий раствор про-
ходит теплообменник 5 и насосом S черев холодильник 2 по-
дается в абсорбер 1. Пополнение потерь р-ра дивтиленгликоля
производится из бака 3. Установки этого типа обычно пол-
Рис. 1. Схема осушки газа абсорбционным спосо-
бом: 1 — абсорбер; 2 — холодильник; 3 — бак; 4 — подо-
греватель; 5 — теплообменник; 6 — отгонная колонна;
7 — кипятильник; 8 — насос.
ностью автоматизированы. Осушка газов иногда производится
также с помощью крепкой серной к-ты; при концентрации
к-ты 94—96% и темп-ре 30—40° остаточное содержание влаги
в газе не превышает 0,2 г/н.ч’.
Адсорбционные способы основаны на
поглощении влаги из газов твердыми веществами —
адсорбентами. В качестве адсорбентов применяют:
твердые СаС12, NaOH, КОН, бокситы, алюмогель
(активированная окись алюминия) и силикагель.
В лабораторной практике применяются также ВаО,
MgO, Р2О5. Широкое распространение в пром-сти
имеют бокситы, алюмогель и силикагель, к-рые легко
регенерируются, способны поглотить от 2 до 10%
влаги (от собственного веса), понижают точку росы
газа на 60° и более. Адсорбенты, как правило, дают
большую степень осушки газа, чем абсорбенты.
Схема установки по осушке газа адсорбционным способом
представлена на рис. 2. Газ поступает в один из адсорберов 1,
заполненных сорбентом, осушается и следует по назначению.
Рис. 2. Схема осушки газа адсорбционным способом:
1 — адсорбер; 2 — подогреватель;, з — холодильник;
4 — сепаратор.
Через определенное время, когда в сухом газе точка росы по-
высится сверх необходимого максимума, адсорбер отключают
и направляют газ во второй адсорбер. Первый адсорбер под-
вергают регенерации. Регенерация сорбента производится
газом, ответвленным от поступающего на осушку (до 20%);
в подогревателе 2 газ нагревается примерно до 200° и посту-
пает в адсорбер. Регенерация сорбента считается закончен-
ной, когда темп-ра регенерирующего газа на выходе из адсор-
бера почти достигает темп-ры входящего в адсорбер газа.
Горячий газ по выходе из адсорбера охлаждается и частично
обезвоживается в холодильнике з, проходит сепаратор 4 и
поступает вместе с основным потоком газа в адсорбер, рабо-
тающий на осушку газа. После прогрева подача пара в подо-
греватель 2 прекращается и в адсорбер поступает холодный
газ для охлаждения сорбента. После этого адсорбер переклю-
743
ГАЗОВ ОЧИСТКА
744
чается на осушку газа, а второй адсорбер — иа регенерацию.
Скорость газа в сечении адсорбера 0,1—0,3 .«/сек; потребный
объем сорбента определяется по конечному содержанию влаги
в сорбенте (сорбционной способности), производительности
по газу и продолжительности циклов.
Для Г. о. может быть использован также непре-
рывный процесс адсорбции с движущимся слоем сор-
бента, называемый гиперсорбцией.
Физические способы основаны на охла-
ждении осушаемого газа в поверхностном холодиль-
нике водой или хладоагентом, охлаждении после сжа-
тия газа и в результате внезапного расширения сжа-
того газа. Выпадающий при этом из газа конденсат
отводится через сепаратор в канализацию.
При необходимости очистки газа от H2S и СО2
и его осушки применяется гликольаминовый способ,
при к-ром в качестве поглотителя служит смесь эта-
ноламина (10—30%), диэтиленгликоля (45—85%)
и воды (5—25%). Для глубокой осушки газа часто
применяют двухступенчатую схему (сначала газ
осушается в установке абсорбционного, а затем в уста-
новке адсорбционного типа).
Лит.: Зарембо К. С. и Нусинов Г. И., Очистка,
осушка и одоризация природных газов, М.—Л., 1947; Справоч-
ник по транспорту газов. Под ред. К. С. Зарембо, М., 1954;
Касаткин А. Г., Основные процессы и аппараты хими-
ческой технологии, 6 изд., М., 1955; Campbell J. М.,
Chem. Engng. Progr., 1952, 48, № 9, р. 440.
А. П. Андрианов.
ГАЗОВ ОЧИСТКА — выделение из газовой смеси
при выбросе ее в атмосферу различных примесей
с целью сохранения нормальных санитарных усло-
вий в прилегающих к промышленным объектам райо-
нах, подготовки газов к использованию в качестве
химич. сырья или топлива, а самих примесей — как
ценных продуктов. Г. о. принято подразделять на
очистку от взвешенных частиц— пыли и тумана, и
от парообразных и газообразных примесей, нежела-
тельных при использований газов или при выбросе
их в атмосферу.
1. Очистка газов от взвешенных частиц
Содержащиеся в промышленных газах частицы
чрезвычайно разнообразны по своему составу, агре-
Рис. 1 Схема движения газа в
циклоне (типа НИИОГАЗ): 1 —
входной патрубок; 2 — винтообраз-
ная крышка циклона; з — выхлоп-
ная труба; 4 — корпус циклона;
5 — бункер; 5 — улитка для выво-
да газа из циклона; 7 — выводной
патрубок; 8 — пылевой затвор.
гатному состоянию,
а также дисперсности
(см.Азроаоли). Очистка
промышленных газов
достигается механич.
и электрич. средст-
вами.
Механическая очи-
стка газов. Очистку
производят: воздей-
ствием центробежной
силы, фильтрацией
сквозь пористые ма-
териалы, промывкой
водой или же другой
жидкостью; иногда
для освобождения от
крупных частиц ис-
пользуют их силу тя-
жести. Механич. очи-
стку газов обычно
производят методами
сухой газоочистки,
фильтрации и мокрой
газоочистки.
Сухая газо-
очистка. Весьма
распространенным ап-
паратом для этих це-
лей является циклон,
способный довольно эффективно выводить из газового
потока частицы размером от 5 мк и более. Действие
циклона основано на использовании центробежной
силы, возникающей вследствие входа загрязненного
газа с повышенной скоростью по касательной (рис. 1)
и последующего его движения по спирали. Улавли-
ванию пыли в циклонах способствует также коагуля-
ция частип. Пыль, отброшенная центробежной силой
к стенке циклона, выводится через нижнее отверстие
в бункер, а очищенный поток газа выходит из централь-
ной трубы циклона.
При вращении на частицу пыли или жидкости действует
центробежная сила; /п — 4/anr3pW2/gR, где г — радиус ча-.
стипы; р — плотность; W — окружная скорость движения
газа в циклоне; Я — радиус циклона; g — ускорение силы
тяжести.
Из этого выражения видно, что центробежная сила возра-
стает с уменьшением радиуса циклона. В широко распростра-
ненном в СССР циклоне типа НИИОГАЗ (см. рис. 1) обычно
максимальный диаметр не превышает 800 мм. Угол наклона
между винтообразной крышкой и входным патрубком у этих
циклонов равен 11°, 15° и 24°; соответственно циклоны этого
типа обозначаются: ЦН-11, ЦН-15 и ЦН-24. При большом ко-
личестве подлежащего очистке газа устанавливают группы
циклонов (по 4—8 шт.). О степени очистки газа судят по.
козфф. очистки уоч — отношению весового количества пыли
Gyn , уловленной в аппарате, к весовому количеству посту-
пившей в него пыли GBXi за то же время; ;оч определяется
по ф-ле;
Сул. gbx. — GBUX
Точ = f.---ЮО =------ъ-------100,
^вх. °вх.
где GBbIx. — весовое количество пыли в газе на выходе ив
аппарата.
Приближенные значения точ. для циклонов НИИОГАЗ
даны в табл. 1.
Таблица 1
Тип циклона Диаметр циклона, At At Точ B % п₽и диаметре частиц пыли Коэфф, гидравлич. сопротивле- ния С
5 AtK 10 At К 20 AtK
цн-и f 800 t 100 65 86 90 97 98 1 99,8 f 180
Г 800 50 85 97,5 )
ЦН-15 { 400 69 89 98,5 } 105
I 200 77 93 99,0 J
ЦН-24 f 1000 t 500 30 41 70 79 96,0 1 97,0 J 60
Стремление использовать рост центробежной силы (эффекта
осаждения пыли) с уменьшением радиуса вращения приве-
ло к распространению бата-
рейных циклонов, объединя-
ющих в одном агрегате от
нескольких десятков до со-
тен циклонов диаметром 50—
250 лш.
Сообщение газовому по-
току вращательного движе-
ния достигается в батарей-
ном циклоне при помощи
направляющего аппарата
(винта или розетки), поме-
щенного в кольцевом про-
странстве между стенкой ци-
линдрической части корпуса
циклонного элемента и вы-
ходной трубой под углом к
оси элемента. Общий вид ба-
тарейного циклона показан
на рис. 2. Приближенные
значения точ. для элемен-
тов батарейного циклона
даны в табл. 2.
Действие инерци-
онных уловите-
лей (пылевых мешков,
жалюзийных решеток,
зигзагообразных отдели-
телей и т. п.) основано
Рис. 2. Батарейный циклон
(общий вид).
на многократном изменении направления движения
запыленного потока газа, при к-ром частицы пыли (или
жидкости), продолжая двигаться по инерции в преж-
745
ГАЗОВ ОЧИСТКА
746
нем направлении, отделяются от газа. Кроме того,
используется и сила удара часгиц. Аппараты этого типа
мало эффективны и в основном служат Для улавлива-
ния довольно крупной пыли. Действие вращающихся
сухих пылеуловителей основано на использовании
центробежной силы, развивающейся при вращении
специального ротора внутри корпуса. Частины пыли
под действием центробежной силы отбрасываются
к стенкам корпуса и выводятся наружу, а очищенный
газ проходит по назначению. Подобные пылеулови-
тели у нас не получили широкого применения; за
рубежом их называют рот склонам и.
Таблица 2
Тип циклонного элемента Диа- метр, мм 7оч. в % при дна- метре частиц ПЕЛЛИ Коэфф, гид- равлич. со- противления С
5 мк 10 мк 20 мк .
С винтом < Г5° 250 63 78 91 85
Г 250 72 84 93 1
С розеткой < 25° { 150 78 88 95 1 90
1 100 82 91 96 J
Фильтрация. Для фильтрации запыленных
газовых потоков используются различные ткани из
натуральных и искусственных волокон (хлопок,
шерсть, асбест, стекловолокно, нитрон, лавсан и др.).
Фильтры такого типа называются тканевыми (рукав-
ными) фильтрами. Размеры осажденных частиц пыли
в порах ткани и на ее поверхности часто во много раз
меньше среднего диаметра нор фильтровальной ткани.
Это объясняется тем, что осаждение частиц в основном
происходит в результате их столкновения с элементами
ткани под действием спл инерции, электрич. зарядов
и др. факторов. Однако пока фильтр частично не забит
пылью, он мало эффективен по отношению к мелким
частицам. Тканевые фильтры служат для улавлива-
ния весьма тонких фракций пыли и имеют высокий
коэфф, очистки.
Рукавный фильтр (рис. 3) состоит из ряда подвешенных
в камере тканевых рукавов, заглушенных сверху. Запылен-
ный газ поступает в нижнюю часть аппарата и проходит через
ткань рукавов; на поверхности ткани и в ее порах осаждается
пыль. По мере увеличения толщины слоя пыли возрастает
сопротивление фильтра; поэтому осевшую на ткани пыль пе-
риодически удаляют путем механич. встряхивания рукавов
при помощи автоматич. устройства или путем одновременного
механич. встряхивания и обратной продувки рунавов очищен-
ным газом или воздухом. Обычно фильтр состоит из несколь-
Рис. 3. Схема многосекционного рукавного фильтра: 1 —
рукава; 2 — рама; 3 — механизм встряхивания рука-
вов; 4 — клапан переключения хода газа; 5 — вентиля-
тор; 6 — шнек; 7 — затвор для выгрузки пыли.
них однотипных параллельно работающих секций. Через
определенные промежутки одна из секций фильтра автомати-
чески переключается на 20—30 сек. распределительным меха-
низмом на очистку ткани от цыли, по.сле чего секция автома-
тически возвращается в рабочее положение, а механизм вклю-
чает на очистку след, секцию фильтра. Производительность
рукавных фильтров определяется удельной нагрузкой ткани,
равной объему запыленного газа в лг8, проходящему в 1 мин.
через 1-м2 фильтрующей поверхности ткани (скорость филь-
трации). Скорость фильтрации зависит от начальной запы-
ленности газа, характера пыли, допустимого сопротивления
фильтра (обычно 50—200 мм вод. ст.) и колеблется в пределах
0,20—2,5 м/мин. Для хлопчатобумажных тканей темп-ра газа
не должна превышать 60°, шерстяных 90°, для термостойких
тканей из синтетич. волокон 140—150° и для стеклоткани
300—350°.
Помимо тканевых, применяются фильтры из специ-
ального картона, пористой бумаги, пористой керамики,
металлокерамики, а также из слоев гравия, песка
и др. Конструкции их весьма разнообразны. Нек-рые
из них (спецкартон, бумага) не имеют приспособле-
ний для периодич. удаления пыли, и при достижении
определенной величины гидравлич. сопротивления
фильтрующий материал заменяется. Удаление пыли из
нек-рых фильтров (пористая керамика, пористый
металл) производится периодич. обратной продувкой
или промывкой, чаще всего водой. Для повышения
эффекта пылеулавливания фильтры с насадкой из
крупных материалов (керамич. и металлич. колец,
гофрированной стальной сетки и др.) смачивают ми-
неральным маслом (висциновые фильтры).
Мокрая газоочистка. Принцип дей-
ствия большинства аппаратов мокрой газоочистки
основан на тесном контакте потока запыленного газа
с жидкостью (обычно водой) с использованием сил
инерции движущихся частиц пыли, центробежной
силы, а также утяжеления частиц пыли, их коагуля-
ции. Кроме механич. воздействия, смачивание и утя-
желение частиц производятся иногда путем конден-
сации па частицах пыли содержащихся в газе паров
жидкости при охлаждении газа ниже его точки росы.
При этом происходит укрупнение частпц пыли, что
значительно увеличивает эффект пылеулавливания.
Для мокрой газоочистки применяются: скрубберы (с на-
садкой и полые, барботажные), мокрые циклоны, скоростные
турбулентные уловители,
вращающиеся промыва-
тели, В скрубберах с на-
садкой объем аппарата
заполняется кусковыми
материалами (кокс, кварц
и др.), специально изго-
товленными насадками в
виде керамич. или фарфо-
ровых колец (кольца Ра-
шига), деревянных реек
(хордовая насадка) и др.
Сверху скруббер ороша-
ется жидкостью. Выте-
кающая из скруббера за-
грязненная уловлепной
пылью жидкость часто
используется повторно
после освобождения от
пыли в отстойнике. В
полом скруббере контакт
между запыленным по-
током газа и жидкостью
достигается путем распы-
ления жидкости в объеме
Выход газа
Рис. 4. Пенный аппарат: 1 — кор-
пус; 2 — решетка; з — бункер; 4 —
приемная коробка; 5 — сливная
коробка.
скруббера форсунками. Часто улавливание пыли в скруббе-
рах производится попутно с охлаждением газа. Скорость газа
в сечении скруббера принимается в пределах 0,5—1,5 м/сек.
Скрубберы этого типа относятся к аппаратам средней степени
очистки газов от пыли (в пределах 80—90% для большинства
случаев промышленного применения). Бодее эффективными
скрубберами являются барботеры. В барботерах с колпачко-
выми распределителями скорость газа не превышает 0,25—
0,35 м/сек. В последнее время получили широкое распростра-
нение барботеры с дырчатыми решетками, в к-рых скорость
газа равна 1,5—2,5 м/сек. При таких скоростях происходит
интенсивное перемешивание жидкости газом с образованием
большого объема пены; подобные барботеры называют пен-
ными. Конструкция однополочного пенного аппарата пред-
ставлена на рис. 4. Отверстия в решетке пенного аппарата
имеют диаметр 4—8 мм, шаг 6—14 мм, процент свободного
сечения решетки по отношению к площади сечения аппарата
10—30%, скорость газа в отверстиях решетки 5—13 м/сек.
Слой жидкости на решетке составляет 20—30 мм, толщина
образующегося слоя пены 100—200 мм. В зависимости от
параметров решетки и скорости газа в отверстиях часть жид-
кости протекает через отверстия, остальное количество пере-
747
ГАЗОВ ОЧИСТКА
748
текает через сливной порог и отводится из аппарата по слив-
ному патрубку. Имеются также пенные аппараты без сливных
патрубков, у к-рых вся жидкость протекает через отверстия;
такие аппараты называются провальными. Пенные
аппараты улавливают 98—99% пыли диаметром более 5 мк,
примерно 75—80% пыли размером менее 5 мк. ДЛя очистки
газов, содержащих частицы размером 1—2 мк и меиее, пен-
ные аппараты вследствие малой степени улавливания не при-
меняются. Для пылеулавливания применяют пенные аппа-
раты с 1—2 полками.
Весьма распространены мокрые циклоны — мокрый скруб-
бер ЦС-ВТИ и мокрый прутковый пылеуловитель МП-ВТИ
(конструкции Всесоюзного теплотехнического института).
Входная скорость газа в скруббер ЦС-ВТИ 15—23 м/сек;
гидравлич. сопротивление 40—50 мм вод. ст. Наибольший
рекомендуемый диаметр ЦС-ВТИ 1700 мм. Коэфф, очистки
газа от пыли в ЦС-ВТИ диаметром 1 м приведены в табл. 3.
Таблица 3
Выход- ная ско- рость газа, м/сек Козфф. (процент) очистки уоч> в зависимости от диаметра частиц (мк)
1-9 10—14 15-25 25-35 35—45 45—60 <60
15 69,5 80,0 88.0 91,0 92,7 94,0 98,0
17 75,7 85,2 90,0 92,0 93,7 95,0 98,5
19 81,4 88,2 91,5 93,2 94,5 95,8 99,0
21 85,0 90,3 92,8 94,3 95,5 96,7 99,5
23,5 87,7 91,8 94,0 95,5 96,6 97,8 100,0
Мокрый пылеуловитель МП-ВТИ применяется в основном
для улавливания золы из дымовых газов. Входная скорость
в этих аппаратах {2м/сек;
во входном патрубке уста-
новлена орошаемая во-
дой горизонтальная прут-
ковая решетка (4—6 ря-
дов прутков диаметром
16—18 мм). Расход воды
ок. 0,15 л/л<3 газа, сопро-
тивление 60—70 мм вод.
Вход газов
Рис. 5. Схема скоростного пыле- ст-> КОЭФФ’ очистки не-
уловителя: 7 — конфузор; 2 — гор- выше, чем у
ловина; 3- диффузор.
2500, 2750, 3000 ИЗЗООлгм.
Скоростной турбулентный пылеуловитель (рис. 5) основан
на след, принципе. В поток газа подается жидкость, к-рая за
счет большой скорости газа в суженной части аппарата— гор-
ловине — дробится на мелкие капли. Благодаря высокой сте-
пени турбулентности газового потока происходит соударение
и коагуляция части пыли с каплями жидкости. При этом ча-
стицы смоченной пыли укрупняются настолько, что могут
достаточно полно улавливаться затем в циклонах. Конструк-
тивно скоростные турбулентные уловители состоят из сочета-
ния конфузора, горловины и диффузора. Скорость газа в гор-
ловине 70—120 м/сек, расход жидкости 0,3—1,0 л/м*. Чем
больше скорость газа и расход жидкости, тем выше коэфф.
Рис. 6. Принципиальная электрическая схема электро-
фильтра: 1 — рубильник; 2 — предохранители; з— регуля-
тор напряжения; 4 — трансформатор; 5 — высоковольт-
ный выпрямитель; в — изолятор; 7 — электрофильтр;
8 — заземление.
очистки и тем больше гидравлич. сопротивление. При гидрав-
лич. сопротивлении в 600—1000 мм вод. ст. скоростной тур-
булентный пылеуловитель может служить для весьма эффек-
тивного улавливания высокодисперсных частиц пыли или
тумана. Достоинством этого аппарата являются малые габа-
ритные размеры, недостатком — значительные затраты энер-
гии на подачу газа.
Вращающиеся промыватели представляют собой центро-
бежные вентиляторы особой конструкции. Внутрь вращаю-
щейся крыльчатки подается жидкость, с к-рой перемеши-
вается просасываемый через промыватель газ. Смоченная и
укрупненная т. обр. пыль „ ~
отбрасываетсяна стенки кор- 1*^4
пуса и затем вытекает из т 1 ’
аппарата в виде шлама. К
этому типу пылеуловителей
принадлежит дезинтегратор
системы Тейзена, применяе-
мый для очистки доменного
и генераторного газов. В по-
следнее время вращающие-
ся промыватели заменяются
более эффективными и эконо-
мичными аппаратами (элек-
трофильтрами, скоростными
турбулентными пылеулови-
телями и др.).
Электрическая очист-
ка газов. Этот вид очи-
стки широко применяет-
ся в пром-сти для улав-
ливания высокодисперс-
ных частиц пыли и тума-
нов и обеспечивает, при
известных условиях, вы-
сокий коэффициент очи-
стки. Принцип действия
электрофильтра (рис. 6)
заключается в том, что
навешенным в газе части-
цам сообщается электрич.
заряд;' заряженные ча-
стицы под действием
электрич. поля приходят
н движение и выводятся
из газового потока пу-
тем их осаждения на
Рис. 7. Трубчатый вертикаль-
ный однопольный мокрый элек-
трофильтр: 1 — газораспреде-
лительные решетки; 2 — осади-
тельные электроды; з — коро-
нирующие электроды.
электродах противопо-
ложного знака заряда.
Если взвешенные в газе
частицы являются жид-
костью, то после осаж-
дения она сама стекает
с электродов, если же они являются пылью, то ее
приходится удалять с электродов стряхиванием.
Установка электрофильтров состоит из двух частей элек-
трофильтра и высоковольтного агрегата, предназначенного
для питания электрофильтра выпрямленным током высокого
напряжения. Выпускаемый отечественной пром-стью злектро-
агрегат АФ-90-200 имеет максимальное напряжение 90 000 в,
номинальный ток вторичной обмотки трансформатора 200 ма
и оборудован повторным автоматич. включением.
Электрофильтры подразделяются на след, основные типы:
пластинчатые и трубчатые (по форме осадительных электро-
дов), вертикальные и горизонтальные (по ходу газа в аппа-
рате), однопольные и многопольные (по количеству электрич.
полей в аппарате), сухие и мокрые (по агрегатному состоянию
уловленных частиц, влажности газа и способу удаления улов-
ленных частиц с электродов). Трубчатый, вертикальный, одно-
польный мокрый электрофильтр представлен на рис. 7. В за-
висимости от свойств газа, темп-ры и свойств пыли электро-
фильтры изготовляются из стали, спецстали, железобетона,
кирпича, свинца, пластич. масс и др. материалов.
А. П. Андрианов.
2. Очистка газов от газообразных и парообразных
примесей
Многие газо- и парообразные примеси отравляют
катализаторы, ухудшают качество продукции, вызы-
вают коррозию и загрязнение аппаратуры или взры-
воопасны в случае их накопления (напр., ацетилен
при разделении воздуха, окись азота при разделении
коксового газа, кислород при ожижении водорода).
Горючие газы, как, напр., природные и попутные,
коксовый, газы нефтепереработки, переработки слан-
цев, генераторные газы и др., в большинстве случаев
должны быть очищены от H2S и др. сернистых соеди-
нений, Большое значение имеет очистка дымовых
749
ГАЗОВ ОЧИСТКА
750
Таблица 4
Способ очистки Реагенты. Условия процесса очистки Условия регенерации. Получаемые побочные продукты Остаточное содержание примесей Ориентировочные рас- ходные коэфф, на очист- ку 1000 нл<3 газа
Мышьяко во-содовая очистка от H2S Водный р-р окситиомышья- ковых солей натрия. Давле- ние атмосферное, темп-ра 20—40° Продувка воздухом при 40°. Плавленая сера, тиосульфат натрия 20—50мг/нм3 H2S 0,03—0,04 кг белого мы- шьяка, 2—6 кг соды, 18— 22 квт-ч электроэнергии, 80—100 кг пара
Карбонатная очистка от H2S Водный р-р Na»CO3 (5%) или К.СО3 (13%). Давление атмосферное или 9—И ат Вакуум 600—650 мм рт. ст., темп-ра 75—85°; H2S, при- годный для переработки в H2SO4 1—3 г/нм3 H2S 0,5—0,6 кг карбоната, 10 квт-ч электроэнергии, 250—340 кг пара
Очистка активным углем от H2S Активный уголь («С-уголь»), Давление атмосферное, тем- пература 20—30° Обработка угля аммиачным р-ром (NH4)2S с последую- щей промывкой и пропари- ванием угля. Плавленая сера чистотой 99,9% 20—50 мг/нм3 H2S 0,2 кг активного угля, 2—3 квт-ч электроэнер- гии, 70 кг пара
Сухая очистка болот- ной рудой от HJS Очистная масса из болотной руды с опилками. Давление атмосферное, темп-ра до 30° Отработанная масса не реге- нерируется; в ней содер- жится 40—45% серы 20—50 мг/м3 H2S 5—10 кг очистной мас- сы, 0,8—1 квт-ч элек- троэнергии, 15—20 кг пара
Этаноламиновая очистка от СО 9 и H2S Водный 15—30%-ный р-р моноэтаноламина. Давление от атмосферного до 30 ат, темп-ра 30—40° Нагревание р-ра до 120— 130°. Газ, содержащий H2S или СО3 0,002—0,2 СО2, 20—50 мг/нм3 H2S 0,05—0,5 кг амина, 10 квт-ч электроэнергии, 300—1000 кг пара
Аммиачная очистка от СО2 и H2S Водный 1,5—6 %-ный р-р NH.3. Давление атмосферное или 8—10 ат Нагревание р-ра. Газообраз- ный H.S, пригодный для переработки в H2SO, 0,5—3 г/нм3 H2S 100 кг пара на 1 кг H2S, удаленного из газа
Низкотемпературная абсорбция СО2, HeS и органич. соедине- ний серы Метиловый спирт. Давление 10—30 ат, темп-ра минус 30— минус 70° Ступенчатое снижение да- вления до 0,2 ат, темп-ра минус 60°. Углекислый газ 1—1,5% СО2, ме- нее 1 мг/нм3 сернистых соеди- нений 0,5—1 кг метилового спирта, 20—30 квт-ч электроэнергии, 40—60 кг пара
Водная очистка от СО2 Вода. Давление 12—30 ат, темп-ра 10—30° Снижение давления до ат- мосферного, дегазация воды. Углекислый газ 0,3—2% СО2 60—80 л«3 воды, 40—55 квт-ч_электрознергии
Поташная очистка от СО2 35—40%-ный водный р-р K2COS. Давление 15—30 ат, темп-ра 110—120° Снижение давления до 1,2— 1,3 ат, темп-ра 110—120°. Углекислый газ 0,5—2% СО2 0,001 кг поташа, 14 квт-ч электроэнергии, 400— 500 кг пара
Щелочная очистка от СО2 8—10%-ный водный р-р NaOH. Давление атмосферное или повышенное Регенерация р-ра не произ- водится или производится путем каустификации изве- стью. Побочных продуктов нет 0,001—0,002% СО2 1,3—1,6 кг каустика, 8— 9 кг извести, 3 квт-ч электроэнергии, 25—30 кг пара
Медно-аммиачная очистка от СО Аммиачный р-р карбонат- ных, формиатных или аце- татных солей одновалентной меди. Давление 100—300 ат, темп-ра 5—20° Атмосферное давление или вакуум,темп-ра 76—80°. Газ, содержащий СО 20—100 ем3/м3 СО 0,03 кг меди, 0,3—0,4 кг аммиака, 12 м3 воды, 20 квт-ч электроэнергии, 60—70 кг пара
Промывка жидким азотом от СО Жидкий азот. Давление 13— 30 ат, темп-ра —190° Азот не регенерируется. Газ, содержащий 30—40% СО 5—10 см3/м3 СО 300—350 л«3 N2, 85—90 квт-ч электроэнергии
Каталитическое гидрирование СО Железный или никелевый катализаторы. Давление ат- мосферное или до 300 ат, темп-ра 125—250° (никель) или 300—350° (железо) 5—10 см3/м3 СО —
Очистка от О2 ката- литич. гидрирова- нием Никелевый или палладие- вый катализаторы. Давление атмосферное или повышен- ное, темп-ра 80—120° — до 1 • Ю~7%02 —
751
ГАЗОВ ОЧИСТКА
752
Продолжение
Способ очистки Реагенты. Условия процесса очистки. Условия регенерации. Получаемые побочные продукты Остаточное содержание примесей Ориентировочные рас- . ходные коэфф, на очистку 1000 нл.з газа
Поглотительная очистка от О2 Восстановленные окислы ме- таллов (Си, Mn, Fe). Давление атмосферное, темп-ра 100—300° Восстановление газами, со- держащими Н2, СО, или природным газом <0,001% О2 —
Очистка от СоНо каталитич. гидриро- ванием Никелевый, палладиевый или др. катализатор. Давление атмосферное или до 30 ат; темп-ра 100—200° — Менее 10 см3/ма С2Н2 0,025 кг катализатора; потери ок, 1 % продукта (этилена)
Абсорбция ацетилена диметилформамидом Диметилформамид. Давле- ние 10—30 ат, темп-ра ми- нус 5 — минус 10° Снижение давления, нагре- вание. Ацетилен (не менее 85%) Менее 10 смя/м" С2Н3 -
Абсорбция ацетилена ацетоном Ацетон. Давление 3—6 ат, темп-ра минус 60—минус 65° Снижение давления, нагре- вание. Газ,содержащий 40% С2Н2 и 60% С2Н4 Менее 10 см3/м* С3Н2 -
газов от SO2. Часто производят очистку газов от окиси
углерода, окиси азота, кислорода, ацетилена. Отхо-
дящие газы ряда произ-в очищают от хлора, фто-
ристых соединений, паров органич. растворителей
и др. В больших масштабах в пром-сти производят
очистку газов от СО2.
Промышленные методы очистки газов можно свести
к трем группам: 1) с помощью твердых поглотите-
лей или катализаторов — «сухие» методы очистки;
2) с помощью жидких поглотителей (абсорбентов) —
жидкостная очистка; 3) очистка без применения по-
глотителей или катализаторов. К первой группе отно-
сятся методы, основанные на адсорбции, химич. вза-
имодействии с твердыми поглотителями и на катали-
тич. превращении примесей в безвредные или легко
удаляемые соединения. Сухие методы очистки обычно
проводят С неподвижным слоем сорбента, поглоти-
теля или катализатора, к-рый периодически должен
подвергаться регенерации или замене. В последнее
время такие процессы осуществляются также в «кипя-
щем» или движущемся слое, что позволяет непре-
рывно обновлять очищающие материалы. Жидкостные
способы основаны на абсорбции извлекаемого компо-
нента жидким сорбентом. Типичная схема жидкост-
ных способов очистки включает непрерывную цирку-
ляцию абсорбента между аппаратом, в к-ром проис-
ходит очистка газа, и регенератором, в к-ром проис-
ходит восстановление поглотительной способности
р-ра. Третья группа методов очистки основана на
конденсации примесей и на диффузионных процессах
(термодиффузия, разделение через пористую пере-
городку).
В зависимости от требуемой степени очистки газа
различают грубую, среднюю и тонкую очистку. Од-
нако количественные нормы, отвечающие такой клас-
сификации, меняются в зависимости от требований
технологии. Требуемая степень очистки газа нередко
достигается в несколько ступеней, отличающихся
условиями или даже способом проведения процесса.
Примерная характеристика нек-рых наиболее упот-
ребительных промышленных способов очистки газов
приводится в табл. 4.
Помимо способов, указанных в таблице, важное
значение имеют также способы очистки промышлен-
ных газов от органич. сернистых соединений илп
органич. серы, под к-рой понимается сумма всех
сернистых соединений, кроме H2S и SO2. Наиболее
типичные составные части органич. серы — сероугле-
род, сероокись углерода, тиофен и меркаптаны. Их
содержание в различных газах обычно колеблется
в пределах 0—1000 мг!нм3 (в пересчете на серу);
в отдельных случаях оно доходит до 2—3 г[нм3.
Обычно очистку от органич. серы проводят после
удаления H2S. Наиболее важные способы очистки:
1) Абсорбция поглотительными маслами (соляровое
масло, газойль); процесс очистки совмещается с улав-
ливанием бензольных углеводородов при повышенном
соотношении жидкость — газ; масло регенерируют
дистилляцией под вакуумом; степень очистки 65—
80%. Способ применяется к газам сухой перегонки
топлива. 2) Абсорбция органич. растворителями при
низкой темп-ре; очистка производится одновременно
с очисткой от СО2 и H2S (см. табл. 4). 3) Адсорбция
активным углем; способ эффективен для удаления
тиофена, меньше — для CS2 и мало эффективен, для
COS. Уголь регенерируют паром при 120—150°;
сероемкость — несколько процентов от веса угля.
4) Окислительный способ очистки активным углем;
основан на окислении органич. серы на поверхности
угля. Способ применим при очистке газов, содержа-
щих COS (напр., водяной и пслуводяной газы). Про-
цесс протекает при наличии в газе небольших коли-
честв О2 (0,1%) и NH3. Сероемкость активного угля
10—12%; отработанный уголь регенерируют перегре-
тым водяным паром при 350°. 5) Очистка железо-
содовым поглотителем (активная окись железа и 30%
соды); применяется для очистки водяного газа, к
к-рому необходимо добавлять 0,2—0,3%, О2. Темп-ра
процесса 180—200°, объемная скорость 150 м31час
на 1 л«3 поглотителя; сероемкость поглотителя 3—8%,
остаточное содержание серы 1—2 жг/ж3; отработанная
масса не регенерируется. 6) Очистка поглотителем на
основе ZnO (поглотитель ГИАП-10 или отработан-
ный цинкхромовый катализатор). Условия процесса:
темп-ра 300—400°, объемная скорость 1000 л!3/час на
1 м3 поглотителя; степень очистки: для коксового
газа 95%,, природного, водяного и полуводяного га-
зов ок. 100%. Сероемкость поглотителя 20—30%, от
его веса; регенерацию отработанного поглотителя
ГИАП-10 можно производить смесью воздуха с азотом
при 500—550°. 7) Каталитич. способ очистки (железо-
хромовый, никелевый, вольфрамовый или тиомолиб-
датный катализаторы); основан на каталитич. гидри-
ровании органич. серы в H2S или на окислении в SO2.
После каталитич. разложения необходима очистка
газов от образовавшихся H2S или SO2.
Лит.: Гордон Г. М. иПейсахов И. Л., Пылеула-
вливание и очистка газов, М., 1958; 3 а л о г и п Н. Г. и
Шухер С. М., Очистка Дымовых газов, 2 изд., М.—Л.,
1954; К у ч е р у к В. В., Очистка от пыли вентиляционных
и промышленных выбросов в атмосферу, М., 1955; По-
753
ГАЗОВ РАЗДЕЛЕНИЕ
.754
зин М. Е. [и др.], Пенный способ обработки газов и жид-
костей, Л., 1955; Шнеерсон Б. Л., Электрическая очистка
газов, М., 1950; Зайцев М. М., Те вер овский Е. Н.,
Хим. пром-сть, 1955, №2, с. 18; Всесоюзный трест электриче-
ской, механической и химической очистки промышленных
газов «Газоочистка». Каталог, М., 1958 (Гос. комитет Совета
Министров СССР по химии); Егоров Н. Н., Д ми-
тр и е в М. М., 3 ы к о в Д. Д., Бродский Ю. Н., Очистка
от серы коксовального и других горючих газов, 2 изд., М., 1960;
Литвиненко М.С., Очистка коксового газа от сероводо-
рода, Харьков, 1959; РозенкнопЗ. П., Извлечение дву-
окиси серы из газов, М.—Л., 1952-3 ельвенский Я. Д.,
Хим. наука и пром-сть, 1956, 1, № 6, с. 654; Kohl A. L.,
Riesenfeld F. С., Gas purification, N. Y., 1960.
Я. Д. Зельвенский.
ГАЗОВ РАЗДЕЛЕНИЕ — разделение газовых сме-
сей на их компоненты или фракции. Основные про-
мышленные методы разделения газов: фракциониро-
ванная конденсация в сочетании с ректификацией и
абсорбцией (метод глубокого охлаждения); сорбция
селективными жидкими поглотителями (абсорбция),
используемая гл. обр. при очистке газовых смесей
от СО2, H2S, JXH3 и др. (см. Газов очистка); сорбция
Рис. 1. Принципиальная схема процесса разделения углеводородных газовых смесей.
неподвижным, «кипящим» и движущимся слоем твер-
дого поглотителя; адсорбция (см. Гиперсорбция).
Наибольшее применение в пром-сти нашел метод
глубокого охлаждения, обеспечивающий четкое раз-
деление газовых смесей при минимальном расходе
энергии. Независимо от метода разделения все газо-
вые смеси предварительно очищают от окислов азота,
т. к. при низких темп-pax и повышенных давлениях
NO окисляется в NO^ и N2O3, образующие с непре-
дельными углеводородами взрывоопасные нитросо-
единения, а также от примесей, способствующих кор-
розии оборудования (H2S, NO и др.) или затверде-
вающих в аппаратуре при низких темн-рах (С10Н8,
СбНб, Н2О, СО2 и др.). Ниже дается описание процес-
сов разделения нек-рых важнейших газовых смесей.
Выделение легких углеводородов. На
рис. 1 приведена схема процесса разделения углеводородных
газовых смесей (коксовые газы и их фракции, газы крекинга
нефти, пиролиза керосина и др.). В качестве примера рас-
сматривается схема разделения газовой смеси, состоящей
(в объемных %) из: 10% С3 и выше, 25% С6Н0, 20% С2Н4,
20% СН4 и 16% Н2 + N2 + СО. Исходная смесь, сжатая до
25—30 ат, охлаждается в теплообменнике 1 остаточными га-
зами, а в холодильнике 2 аммиаком до 233° К; тяжелые угле-
водороды и пропилен сжижаются при этом целиком, а этан и
этилен частично. Отделение жидких углеводородов от конден-
сирующихся одновременно паров воды производится в сосу-
дах з. В ректификационной колонке 4 из полученной смеси
углеводородов выделяются этан и этилен, возвращаемые
в исходную смесь, а в колонне 5 под давлением 16 ат произ-
водится разделение тяжелых углеводородов. Флегмой в ко-
лонне 4 служат этан и этилен, конденсирующиеся в дефлег-
маторе 4а, а теплоносителем в испарителе 46 — водяной пар.
После аммиачного холодильника 2 исходная смесь проходит
адсорбер в и поступает в блок разделения, где первоначально
охлаждается продуктами разделения в теплообменнике 7,
а затем — в испарителе метановой колонны 0, частично сжи-
жаясь при этом. Далее, при охлаждении смеси в теплообмен-
нике s, происходит почти полное выделение всех углеводоро-
дов. Холод остаточных газов, состоящих из Н2, N8, СО и не-
большого количества СН4, после дросселирования до' 1,3 ат
используется в теплообменниках 8, 7 и 1. Конденсат, образую-
щийся в теплообменнике 8, представляет собой смесь С2Нв,
С2Н4 и СН4. После дросселирования до 2 ат часть этого кон-
денсата направляется в колонцу 9, а остальная его часть по-
ступает в ту же колонну, предварительно пройдя теплообмен-
ник 8. В колонне 9 выделяется СН4. Циркулирующий и сжа-
тый в компрессоре 10 до 50 ат метан дросселируется после
охлаждения в теплообменниках 11 и 12 до 2 ат и подается
в качестве флегмы в колонну 9. Выбор давления метанового
цикла определяется темп-рой кипения этан-этиленовой жид-
кости, отводимой из куба колонны 9 и используемой в тепло-
обменнике 12 для частичного сжижения метана. Количество
конденсирующегося в нем метана соответствует количеству
флегмы, необходимой для колонны 9. Отводимый сверху ко-
лонны метан с темп-рой 123°К содержит не более 2% при-
месей (Н2, N2, СО). Кубовая жидкость колонны 9 разделяет-
ся в ректификационной колонне 1.3
на этан и этилен, причем флегма
обеспечивается специальным этиле-
новым циклом с давлением 4 ат
(посредством компрессора 16). После
охлаждения в теплообменнике 14
этилен сжижается в испарителе ко-
лонны 13, после чего дросселирует-
ся и подается в качестве флегмы.
В отводимом из колонны этилене
(при 173° К) содержится около 1%
примесей, в том числе и ацетилен,
вредный для многих процессов поли-
меризации. Во избежание его накоп-
ления циркулирующий этилен про-
мывается в колонне 15 ацетоном
(при 203°К). Верхние тарелки этой
колонны орошаются жидким этиле-
ном; при этом пары отводимого
этилена отмываются от ацетона.
Насыщенный ацетиленом ацетон от-
водится из куба колонны 15, Дрос-
селируется до 1,3 ат и поступает
в теплообменник 17, в к-ром отдает
свой холод регенерированному аце-
тону. При этом из ацетона выде-
ляется растворившийся в нем эти-
лен, возвращаемый обратно в цикл.
Окончательная регенерация отра-
ботанного ацетона производится в
колонне 18, после чего ацетон про-
ходит холодильник 20 и насосом 19
снова подается в промывную ко-
лонну 15. Холод, необходимый для
проведения процесса разделения,
получают в результате дросселирования исходной газовой
смеси и циркулирующего метана.
Разделение коксового га «за. На рис. 2 при-
ведена схема блока разделения коксового газа под давлением
21 ат для получения водорода, метана и этилена. Выбор ра-
бочего давления определяется гл. обр. назначением агрегата
и принятыми холодильными циклами. В случае получения
азотоводородной смеси и использования для получения хо-
лода эффекта Джоуля—Томсона рабочее давление сжатого
азота равно 13—15 ат. При постепенном охлаждении коксо-
вого газа в теплообменниках 1, 2 и з и в змеевике куба метано-
вой колонны 6 из него выделяются углеводороды (Сь С2, С3
и выше), к-рые вместе с растворившимися в них газами обра-
зуют этиленовую и метановую фракции, собираемые в отдели-
телях 15 и 4 (соответственно). Ректификация этих фракций
производится соответственно в колоннах 6 и 7. После отдели-
теля 4 газовая смесь проходит азотный испаритель 5а и по-
ступает в колонну 5, где жидким азотом отмываются остаточ-
ные количества СН4, СО и др. ненонденсирующихся в данных
Условиях газов (Аг, О2). Кубовый остаток этой колонны со-
ставляет фракцию окиси углерода. Отводимая сверху колонны
смесь Н2 и N2 проходит конденсатор-дефлегматор 56, в к-ром
охлаждается кипящим под вакуумом азотом (64° К); при
этом получают 98%-ный Н2. Рекуперация холода водорода
осуществляется в змеевике 5в и теплообменниках 3, 2 и 1.
Метановая фракция дросселируется до 1,3 ат и разветвляется
на три потока: один поток проходит теплообменник 3, вто-
рой — теплообменник 14, а третий — соединяется с двумя
остальными потоками перед входом в отделитель 16, откуда
жидкость подается в колонну 6 в качестве флегмы, а пары идут
на разделение. Фракция окиси углерода дросселируется на
1,5 ат и частично подается в качестве флегмы в колонну 6,
а частично в теплообменники з и 9. Из куба колонны в-отво-
дится жидкий СН4, холод к-рого используется в теплообмен-
никах 12 и 2, а сверху отбирается смесь N, и СО, направляе-
мая для рекуперации холода в теплообменники з и 9. Этиле-
755 .
ГАЗОВ РАЗДЕЛЕНИЕ
756
новая фракция дросселируется до 1,5 ат и ректифицируется
в колонне 7. Жидкий этилен (из куба колонны) после испаре-
ния в теплообменнике 13 проходит теплообменник 11 и выво-
дится из блока, а легколетучие компоненты (СН4, СО, N2,
Н2) отбираются сверху колонны. Холод, необходимый для
процесса разделения, обеспечивается азотным холодильным
циклом с циркуляцией азота среднего давления. Азот, сжатый
до 200 ат, поступает в блок при 233° К, охлаждается в змее-
вике 8а, теплообменниках 8, 9, 10 и 11, змеевике 11а и в тепло-
обменниках 13 и 12, после чего дросселируется до 31 ат.
Часть азота, пройдя теплообменник 8, возвращается для сжа-
тия в компрессор, а оставшаяся часть после охлаждения в теп-
лообменнике 14 делится на два потока. Один поток дроссе-
лируется до 20 ат, охлаждается в змеевике и подается в каче-
стве абсорбента в колонну 5, а другой — дросселируется до
2,2 ат и после охлаждения в змеевике дросселируется до
1,1—1,2 ат в межтрубное пространство конденсатора-дефлег-
матора 56.
Получение водорода из конвертиро-
ванного газа. После тонкой очистки от углекислого
газа в конвертированном газе содержится примерно 88 об. % Н2,
4—5% N2, 6—7% СО и 0,5% СН4.-Очищенный конвертиро-
ванный газ поступает в блок разделения (рис. 3) при 233® К
и под давлением 13 ат. В теплообменнике 1 он охлаждается
водородом и азотоводородной смесью, а в теплообменнике 2 —
дополнительно фракцией окиси углерода. В испарителе азота з,
в межтрубном пространстве к-рого кипит жидкий азот, дрос-
селируемый вентилем 4 до 1,2 ат, конденсируются значитель-
ные количества Ns, СО и СН4. Часть азота высокого давления
дросселируется вентилем 5 до 12,5 ат, переохлаждается в змее-
вике до 78° К и подается в колонну о для отмывки СО, СН4
и др. высококипящих компонентов. Сверху колонны отво-
дится смесь Н2 и N2, содержащая не более 100—150 см3/м3
примесей. Для получения стехиометрия, азотоводородной
смеси для синтеза NH3 вентилем производится дозировка до-
полнительного количества N2. Для получения технически
чистого водорода часть азотоводородной смеси отводят в кон-
денсатор 7, в межтрубном пространстве к-рого испаряется
Рис. 3. Блок разделения конвертированного водяного газа.
жидкий азот под давлением 1,15 ат (64° К). В результате
конденсации азота из конденсатора 7 при 66° К отводится во-
дород, содержащий ок. 2% азота. Под давлением 200 ат азот
частично используется в теплообменниках 8—12 ран произ-ва
холода путем его дросселирования, а частично для отмывки СО.
Конденсат из конденсатора 7 и кубовая жидкость из промыв-
ной колонны 6 образуют фракцию окиси углерода.
Получение водорода из газов катали-
тического риформинга нефти. Выделение водо-
рода из газов риформинга, содержащих 60—95% водорода и
5—40% предельных углеводородов, осуществляется комбини-
рованием процессов конденсации высококипящих компонен-
тов с абсорбцией остаточных угле-
водородов пропаном. Получаемый
водород содержит 0,05% метана, а
также исходные количества окиси
углерода и азота, которые пропаном
не поглощаются. При принятых
параметрах процесса давление паров
пропана незначительно, что облег-
чает регенерацию пропана и исклю-
чает большие потери его с отходя-
щими газами в процессах абсорб-
ции и десорбции. Газовая смесь
поступает в адсорбер 1 (рис. 4), где
осушается алюмогелем (до точки
росы — 210° К). В теплообменнике
2 газы охлаждаются до 188° К;
при этом конденсируется основное
количество бутана и» пропана. Сжи-
женные углеводороды отделяются
в сепараторе з и после рекупера-
ции холода выводятся из агрегата.
Гззы последовательно охлаждаются
в теплообменниках 4, 6 и 8 до
163° К, 143° К и 103° К; образую-
щийся при этом конденсат отделяет-
ся соответственно в сепараторах 5,
7 и 9. Для рекуперации холода конденсат из сепаратора 5 на-
правляется в теплообменник 2, из сепаратора 7— в теплообмен-
ники 4 и 2, а из сепаратора 9— в теплообменники 8, 4п2. Из
сепаратора 9 остаточные газы, состоящие в основном из водо-
рода и метана, поступают в промывную колонну iot где метан
отмывается жидким пропаном при 106—97° К и давлении 21 ат.
Получаемый водород отводится для рекуперации
холода в теплообменник 8, а затем в детандер п,
• | откуда с темп-рой 89® К направляется в тепло-
1 । обменник 12 для охлаждения жидкого пропана.
Рис. 4. Получение водорода из газов каталитического
риформинга нефти.
Из теплообменника 12 водород поступает в детандер 13, а
из последнего при 103® К—в теплообменник 8, после к-рого
большая часть водорода (83—90%) отводится через тепло-
обменники 6,4 и 2 в качестве целевого продукта, а мень-
шая часть (10—17%) подается в колонну 14 для отдувки ме-
тана из насыщенного абсорбента. Десорбция метана ведется
при 130® К и давлении 1,4 ат. Регенерированный пропан
после пополнения его потерь из емкости 16 насосом 15 снова
подается в промывную колонну. Водород, используемый для
десорбции метана, пропускают через теплообменники 4 и 2,
после чего он смешивается с остаточными газами разделяемой
смеси. Необходимое для разделения газов количество холода
получают за счет расширения водорода в детандерах 11 и 13.
При малом содержании водорода в исходной смеси потребность
в холоде обеспечивается либо повышением давления, либо
рециркуляцией части водорода.
Выделение гелия иэ природного газа.
Содержание гелия в природном газе колеблется в пределах
от 1 до 7%. Процесс выделения гелия рассматривается приме-
нительно к смеси, содержащей (об. %): 1,4% Н2, 12,7% N2,
0,6—0,8% СО2, 78,2% СН4 и 7,7% С2Нв и тяжелые углеводо-
роды. Исходный газ, сжатый до 42 ат, предварительно под-
вергают очистке и осушке от водяных паров, после чего на-
правляют в блок глубокого охлаждения (рис. 5). В теплооб-
меннике 1 газы охлаждаются до 130° К; при этой темп-ре основ-
ная масса газа конденсируется. Остаточные газы дроссели-
руются до 14—21 ат и поступают в отделитель 2, где в резуль-
тате охлаждения парами азота сжижаются все углеводороды
и частично азот. Газовая фаза, состоящая из 60% гелия и
40% азота, дросселируется до 1 ат и после рекуперации ее
холода в теплообменнике 3 направляется в газгольдер. Кон-
денсат иэ отделителя 2 дросселируется до 5 ат в теплообмен-
ник 1 и используется для охлаждения исходной смеси. Далее
эти газы сжимаются до 16 ат и подаются в сеть дальнего газо-
снабжения. Смесь 60% гелпн и 40% азота концентрируют,
757
ГАЗОВАЯ ПОСТОЯННАЯ — ГАЗОВЫЕ РАСТВОРЫ
758
для чего ее сжимают до 190 ат, осушают твердым NaOH,
охлаждают в теплообменнике 3 и подают в отделитель 4, по-
груженный в жидкий азот. При 83° К практически сжижается
весь азот и в оставшейся газовой фазе содержание гелия
достигает 98,5%. При необходимости концентрация гелия
может быть повышена до 99,7—99,9% (путем адсорбции при-
месей активированным и древесным углем при 90—120° К).
Рис. 5. Выделение гелия из природного газа.
Для выделения гелия, растворившегося в жидком азоте, кон-
денсат из отделителя 4 дросселируют до 15—20 ат в емкость 9;
получающаяся при этом жидкая фаза содержит 95% азота,
а гелий почти весь переходит в газовую фазу. Смесь паров
гелия и азота добавляют к природному газу и снова подвер-
гают разделению, а жидкий азот используется для пополне-
ния ванны 1. Необходимый для процесса разделения холод
получают путем использования эффекта Джоуля—Томсона:
частично при дросселировании природного газа и частично
специальным азотным холодильным циклом, для к-рого исполь-
зуется конденсируемый азот. Циркулирующий азот сжимается
до 35—42 ат и после охлаждения в теплообменнике 5 делится
на два потока: одна часть сжижается в конденсаторе в И после
дросселирования подается в ванну 7, а вторая — расширяется
до 1 ат в детандере S, охлаждаясь при этом до 77° К, после
чего соединяется с парами азота, отводимыми из ванны 7.
Холод этих паров используется в конденсаторе азота в, от-
делителе 2 и теплообменнике 5.
Лит. см. при ст. Воздуха разделение.
И. И. Гельперин.
ГАЗОВАЯ ПОСТОЯННАЯ (точнее — универсаль-
ная газовая постоянная) — физич. постоянная R,
к-рая входит в ур-ние состояния 1 моля идеального
газа: pV = RT, где р — давление, V — объем, Т —
абс. темп-ра. Г. п. равна работе, производимой газом
при изобарич, нагревании на 1°. Г. п. входит во многие
физич. ур-ния. Значение Г. п. определяется из ур-ния
состояния идеального газа: R = PoVo/To, где р0,
Уо и Тп — соответствующие параметры газа при
нормальных условиях, для к-рых приняты значе-
ния р0 = 1 атм = 1013250 дин/см2, То — 273,15° К,
Уо = (2,24207±0,00006) • 104 см3/моль (физич. шкала).
На Международном конгрессе по мировым постоянным
в Турине (1956) принято значение R = (8,31696±
±0,00034) • 107 эрг/град-моль (физич. шкала атомных
весов). Если принять соотношение между единицами
химич. и физич. шкал атомных весов равным 1,000272,
то значения Г. п. для химич шкалы атомных ве-
сов равны: R = (8,31470±0,00034) • 107 эрг/град-моль',
R = 0,0820574±0,0000034 л-атм/град-моль', R =
= 1,98726±0,00004 кал/град-моль (калория термо-
химическая, равная 4,1840 абс. джоуля).
ГАЗОВЫЕ РАСТВОРЫ — растворы газов, жидко-
стей и твердых тел в газах. Величина давления пара
над жидкостью и твердым телом зависит от свойства
вещества и его темп-ры. Если вещество находится
в открытом сосуде в соприкосновении с воздухом,
а давление его пара меньше атмосферного, то над
этим веществом находится не чистый пар, а его смесь,
или, правильнее, раствор в воздухе. Исследуя эти
явления при давлениях, близких к атмосферному,
Дальтон пришел к выводу, что количество пара, на-
сыщающего данный объем, не зависит от природы
газа и его давления, а мольная доля растворенного
в-ва в газовой фазе N2 равна: N2 = рЦР, где />§ —
давление пара этого вещества при данной темп-ре,
Р — общее давление. Теперь известно, что даже при
малых давлениях растворимость веществ в газе зави-
сит от природы газа и его давления.
Образование газовых р-ров, как правило, сопро-
вождается изменением объема и тепловым эффектом,
т. е. явлениями, характерными и для жидких р-ров.
Эти явления становятся особенно заметными при
высоких давлениях, когда плотность газа уже сравни-
ма с плотностью жидкости. Процесс растворения
в сжатом газе также схож с растворением в жидкости.
Наблюдения, проведенные в сосуде высокого давления
с окнами, показали, что, напр., капля масла, подвешен-
ная в среде сжатого этилена, растекается в нем струй-
ками, подобно сахару в воде. Растворяться в газе
могут жидкости, твердые тела и газы. В соответствии
с этим можно считать, что газовые растворы — это
системы, в к-рых газ, жидкость или твердое тело
равномерно распределены в среде газа. Р-ры газов
в газах часто называют газовыми смесями.
Растворимость веществ в сжатых газах достигает
значительных величин. Так, в одном 1 нм? этилена
при 2000 ат растворяется до 3 кг смазочного масла.
Азот при 100° и 1000 ат растворяет до 10 мол. % бен-
зола, а этилен при 240 ат и 50° — до 17 мол. % наф-
талина. Водяной пар при давлениях и темп-рах,
превышающих его критич. параметры, растворяет
значительные количества солей, что является причиной
образования твердых отложений на лопатках паровых
турбин. Аналогичные процессы растворения в водя-
ном паре были причиной возникновения горных по-
род, содержащих Zn, W, Си, Мо и др. Большие ко-
личества жидких углеводородов растворяются в газе,
находящемся в соприкосновении с нефтью на больших
глубинах. Способность сжатых газов растворять
вещества используют в технике. Растворимостью
кварца в водяном паре, насыщенном нек-рыми со-
лями, пользуются для выращивания кристаллов
кварца весом в несколько килограммов. Аналогич-
ным методом синтезируют нек-рые минералы (гидро-
термальный синтез). Предложен метод разделения
жидких смесей, основанный на различной раствори-
мости фракций жидкой смеси в сжатых газах.
Механизм растворения веществ в сжа,тых газах
связан с взаимодействием молекул растворителя и
растворенного в-ва и принципиально не отличается
от механизма растворения в жидкости. Однако это
взаимодействие проявляется наиболее явно при высо-
ких давлениях, когда велика плотность газового
р-ра. Ввиду того, что плотность газового р-ра зависит
от давления сильнее, чем плотность жидкости, то
картина межмолекулярного взаимодействия в Г. р.
значительно сложнее, чем в жидком; сложна и форма
кривой растворимости
в координатах Р—N2,
где N2 — мольная до-
ля в-ва, растворенно-
го в газовом р-ре,
Р — давление) начи-
нается при давлении,
равном давлению на-
сыщенного пара этого
вещества, когда в
газе находится только
растворенное веще-
ство, N2 = 1.
При увеличении да-
вления (при нагнета-
нии в объем второго
в-ва в газе. Кривая (напр.,
Мольная доля растворенного вещества
Рис. 1. Кривая растворимости
веществ в сжатых газах.
компонента — газа)
количество газа в
объеме возрастает, а
количество растворенного в-ва (в первом прибли-
жении, при низких давлениях) остается постоян-
ным. Мольная доля этого в-ва в газовом р-ре но-
759
ГАЗОВЫЕ РАСТВОРЫ
760
Мольная доля растворенного вещества
Рис. 2. Фазовые равновесия в
системах жидкость — газ.
этому падает (рис. 1). Такое положение соответ-
ствует закону Дальтона, однако оно сохраняется до
сравнительно небольших давлений. Далее начинает
играть роль взаимодействие между молекулами ком-
понентов. Кроме того, поверхность жидкости или
твердого тела подвергается бомбардировке молеку-
лами газа. При этом, чем больше давление, тем больше
молекул конденсированной фазы приобретает энер-
гию, достаточную, чтобы вылететь в газовую фазу.
Количество второго компонента в газовом р-ре возра-
стает, пока не установится состояние равновесия.
В зависимости от величины этого эффекта (эффект
Пойнтинга) падение мольной доли вещества, раство-
ренного в газе, с давлением меньше, чем это следует
по закону Дальтона.
Кривая растворимо-
сти изгибается, ра-
створимость дости-
гает минимума и мо-
жет вновь начать ра-
сти.
У некоторых систем
мольная доля вещест-
ва в газовой фазе до-
стигает величины рав-
ной количеству этого
компонента в жидком
растворе, например
при растворении газа
в жидкости. В этом
случае состав газовой
и жидкой фаз такой
системы одинаков, кривая растворимости жидкости
в газе после прохождения минимума сливается с
кривой растворимости газа в жидкости и в си-
стеме наблюдают критич. явления, т. е. жидкость
и газ при давлениях, больших, чем давление кри-
тич. точки смеси, смешиваются во всех отношениях
(рис. 2). В других случаях кривая достигает макси-
мума, после чего мольная доля вновь начинает
уменьшаться, а при очень высоких давлениях уже
мало зависит от давления. Наличие максимума и
минимума растворимости и сложный ход кривой
объясняются различным изменением с давлением
парциального мольного объема растворенного в тазе
вещества и его мольного объема в конденсированной
фазе.
При низких давлениях парциальный мольный
объем в газовой фазе (аг) значительно больше моль-
ного объема в конденсированной фазе (г0). Однако
с увеличением давления гг уменьшается быстрее, чем
г’о, к-рый при этих давлениях можно считать посто-
янным. При нек-ром давлении (различном для разных
систем) эти объемы делаются равными. При этом рас-,
творимость достигает минимума. По мере дальнейшего’
роста давления vr становится меньше, чем г0, и рас-
творимость вещества в газе начинает расти с давле-
нием. Чем выше давление, тем больше становится
роль сил отталкивания в межмолекулярном взаимо-
действии в газовом р-ре и гг может начать расти с дав-
лением. Одновременно уже нельзя пренебрегать
уменьшением мольного объема растворенного ве-
щества в конденсированной фазе. При каком-то дав-
лении обе эти величины снова сделаются равными и
растворимость вещества в газе достигнет максимума.
При еще больших давлениях плотность газового
р-ра приближается к плотности жидкости и изменение
ооъемов мало отличается друг от друга. Растворимость
в этом случае почти не зависит от давления.
Давление минимума растворимости тем меньше,
чем больше величина мольного объема растворенного
стало возможным после
в-ва, и зависит также от природы газа-растворителя.
Происхождение минимума и максимума раствори-
мости вещества в газе становится понятным при рас-
смотрении общей картины фазовых равновесий в двой-
ной системе, построение к-рой
экспериментального обна-
ружения в Г. р. взаим-
ной ограниченной раство-
римости или расслоения
газовых смесей. Два сме-
шанных при атм. давлении
газа расслаиваются при
высоком давлении и темп-
ре, превышающей их кри-
тич. темп-ры, на два слоя,
в одном из к-рых преобла-
дает менее летучий ком-
Рис. 3. Общая картина фазо-
вых равновесий в двойных
системах (второй тип): D —
двойная гомогенная точка,
отвечающая минимуму темп-ры
на критич. кривой; К,—кри-
Мольная доля
тич. точка равновесия жидкость—газ при темп-ре Т,; Л' —кри-
тич. точки равновесия газ—газ; S2 — точка возврата; До,
Я2, Яа — точки минимумов растворимости; S3 — точки мак-
симумов растворимости.
понент, а в другом — более летучий. Два образовав-
шихся газовых р-ра разделены поверхностью разде-
ла, между ними существует поверхностное натяжение
и может наблюдаться баротропное явление, т. е. пере-
мена местами верхней и нижней фаз при изменении
давления.
Существует два типа равновесия газ—газ, различа-
емых по ходу критич. кривых. Кривая первого типа
(в координатах Р—Т), отходящая от критич. точки
менее летучего компонента, направляется в сторону
темп-p, более высоких, чем критич. темп-ра этого
компонента. Кривая второго типа сначала направ-
ляется в сторону низких темп-p, проходит через
минимум (находящийся при темп-ре, меньшей, чем
критич. темп-ра менее летучего компонента), а далее
направляется в сторону высоких темп-p. Эта точка
минимума, или т. н. двойная гомогенная точка, яв-
ляется центральной точкой диаграммы фазовых рав-
новесий для систем, имеющих второй тип критич.
кривой. На рис. 3 в координатах давление—мольпая
доля изображены кривые растворимости жидкости
в газе (левые кривые Т2 и Т3 с минимумами раствори-
мости Я3, Н3 и максимумом Уд), кривые растворимости
газа в жидкости (правые кривые У3 и Т3 с максимумом
растворимости газа в жидкости б'д), кривые равнове-
сия газ—газ (верхние кривые То и и кривые рав-
новесия жидкость — газ (нижние замкнутые петли).
В двойной гомогенной точке D происходит слияние
кривых равновесия газ—газ и жидкость—газ. Отсюда
и появляются максимумы растворимости жидкости
в газе и газа в жидкости. При снижении темп-ры ниже
темп-ры точки D происходит размыкание петель рав-
новесий жидкость—газ и газ—газ т. обр., что обра-
зуются две ветви кривых Т2 с точками возврата S'„.
Эти точки возврата, образовавшиеся из точки Ь, далее
при последующем снижении темп-ры и превращаются
в максимумы растворимости жидкости в газе и газа
в жидкости. При увеличении темп-ры выше точки D
критич. давление равновесия газ—газ растет, а равно-
весии жидкость—газ падает. Петли равновесия жид-
кость—газ занимают все меньшую область составов,
пока не стягиваются в- критич. точку менее летучего
компонента. Экспериментально обнаружено 14 си-
стем, содержащих преим. гелий, аммиак, двуокись
серы и другие газы, в к-рых наблюдается равновесие
газ—газ.
761
ГАЗОВЫЙ АНАЛИЗ
762
Долгое время изучение растворимости в-в в газах было
основано на представлениях Дальтона об энергетич. незави-
симости газов в смесях. При давлениях, близких к атмосфер-
ному, можно считать газы энергетически независимыми. Вслед-
ствие этого в свое время эксперимент подтвердил выводы Даль-
тона о том, что количество пара, насыщающего данный объем,
не зависит от природы газа и его давления. Однако при уве-
личении давления нельзя пренебрегать взаимодействием
между молекулами газов, плотность к-рых становится сравни-
мой с плотностью жидкостей. Даже если не учитывать этого
взаимодействия между молекулами растворенного в-ва и газа,
то само давление должно увеличивать насыщение газа паром,
действуя на жидкую фазу, подобно поршню, проницаемому
только для молекул паровой фазы. Это увеличение давления
в паровой фазе при наложении избыточного давления на жид-
кую известно под названием эффекта Пойнтинга. Закон Гибб-
са—Дальтона учитывает это изменение летучести вещества
в конденсированной фазе с давлением:
(д1п/£/бР)Т;-=у./НТ (1)
где — летучесть i-ro компонента, Р — давление, R — газо-
вая постоянная, щ — парциальный мольный объем г-го ком-
понента. На основе этого закона можно дать только качествен-
ное объяснение нисходящего участка кривой растворимости
жидкости в газе и минимума растворимости, но нельзя полу-
чить колич. совпадения с опытом, т. к. этот закон не позво-
ляет учесть значительного и сильно изменяющегося с давле-
нием взаимодействия между молекулами компонентов р-ра.
Термодинамич. расчет растворимости в-в в сжатых газах
сводится к определению летучести этого в-ва в газовой и кон-
денсированной фазах. Сведений о летучестях в-в в газовой
фазе чрезвычайно мало, и для точного их вычисления прихо-
дится определять сжимаемость р-ров. Это требует большой
затраты труда, и гораздо легче определить растворимость
веществ в газах. Для приближенного вычисления летучести
прибегают к различным допущениям. Одним из наиболее
простых является предположение о том, что молекулярные
силовые цоля обоих компонентов р-ра равны. Тогда образуется
идеальный р-р, летучести компонентов к-рого определяются
ур-нием Рауля (правило Льюиса—Репдалла). Ряд ограниче-
ний не позволяет широко пользоваться этим методом для вы-
числения растворимости жидкостей в сжатых газах. Раство-
римость полярных жидкостей в неполярных газах описы-
вается полуэмпирич. ур-нием:
ШК2 а2 RT +
где — летучесть чистого газа растворителя, К2 — коэфф.
Генри растворенного вещества в газовой фазе, N — число
Авогадро, а — радиус сферы точечного диполя, Р° — молеку-
лярная поляризация газа растворителя, a v? — его мольный
объем, у, — дипольный момент растворенного вещества. Про-
верка этого уравнения на ряде систем показала его пригод-
ность для передачи экспериментальных данных в широком
интервале темп-p и давлений. Статистич. термодинамика по-
зволяет получить для разбавленных р-ров уравнения в пред-
положении, что оба компонента фазы подчиняются уравне-
нию состояния со вторым и третьим вириальными коэфф, и
химич. потенциалы в конденсированной и газовой фазах
равны. Однако теоретич. количественный расчет раствори-
мости веществ в ся.атых газах весьма труден вследствие слож-
ности картины поведения газовых р-ров при высоких давле-
ниях.
Лит.: Кричевский И. Р., Фазовые равновесия
э растворах при высоких давлениях, *2 изд., М.—Л., 1952;
Ц и к л и с Д. С., в кн.: Тезисы докладов на совещании по
термодинамике и строению растворов, М., 1958, с. 102; R о-
b i n S., V о d а г В., Disc. Faraday Soc., 1953, № 15, р. 233;
Ewald А. Н., Jepson W. В., Rowlinson J. S.,
там же, 238. Д. С. Циклис.
ГАЗОВЫЙ АНАЛИЗ — анализ смесей газов
с целью установления их качественного и колич.
состава. По количеству газовой смеси, взятой для
анализа, различают макро- (ок. 100 мл газа), полу-
микро- (ск. 2—10 мл) и микрометоды (ок. 1 мл и
меньше); к микрометодам относят также случай опре-
делёния очень малых концентраций газа, порядка
1 мкг{л. Методы Г. а. могут быть основаны на химич.,
физико-химич. и физич. принципах. В случае при-
менения химич. методов Г. а. определяемый компонент
поглощают из газовой смеси различными поглотите-
лями, напр. СО2 может быть поглощен р-ром NaOH,
а различные непредельные углеводороды — серной
к-той разной концентрации. Приборы для выполнения
анализа химич. методами просты по конструкции,
выполнение анализа не сложно и не требует высокой
квалификации лаборанта; эти методы распространены
весьма широко. Все большее распространение при-
обретают физико-химич. и физич. методы анализа
газовых смесей; достоинством их является быстрота
выполнения, большая точность, чем у химич. методов,
возможность объективного установления результатов
определений; они дают возможность автоматизиронать
процессы Г. а. Физич. и физико-химич. методы Г. а.
предполагают знание зависимости между содержанием
определяемого компонента и физич. свойством; эта
зависимость часто выражается т. н. калибро-
вочной кривой.
Химич, методы включают: абсорбционный
анализ, когда в результате химич. реакции проис-
ходит избирательное поглощение компонента газовой
смеси жидким, иногда твердым реагентом, а содер-
жание компонента определяется по уменьшению
объема или давления. К этой же группе может быть
отнесен и газообъемный метод — определение коли-
чества вещества путем выделения газа из него в ре-
зультате реакции и измерения его объема. К химич.
методам анализа близко примыкают методы ка-
талитического сожжения, основанные
на определении горючйх компонентов газовой смеси
по количеству образовавшейся С0.2 и по уменьшению
объема вследствие конденсации паров воды; в зави-
симости от условий сожжения возможно определение
как общего содержания горючих компонентов, так
и содержания отдельных компонентов. В Г. а. при-
меняется также метод фракционирован-
ного растворения, при к-ром на анализи-
руемую газовую смесь действуют таким количеством
растворителя, к-рое может растворить только один
компонент, другие, более летучие компоненты, оста-
ются без изменения.
Физико-химич. методы анализа газов основаны на
поглощении компонента из смеси и установлении из-
менения электропроводности (кондуктометрия), силы
тока с изменением потенциала на непрерывно обнов-
ляющемся капельном ртутном электроде (полярогра-
фия)-, фотометрия, методы основаны на измерении
оптич. плотности раствора или суспензии, полу-
ченных при взаимодействии определяемого газа со
специальными реагентами (колориметрия, спектро-
фотометрия). При выполнении реакции на твердых
средах применяется линейно-колористич. метод ана-
лиза, при к-ром измеряется длина окрашенного стол-
бика реагента в индикаторной трубке (см. также
Воздуха анализ). Для анализа смесей углеводородов
особенно широко применяется хроматография, адсорб-
ционный анализ (см. Хроматография, X роматотермо-
графия), заключающийся в последовательной адсорб-
ции компонентов газовой смеси на адсорбенте и по-
следовательном выделении и определении отдельных
компонентов.
Для анализа смесей газов используются след,
физич. свойства: плотность (денсиметрия) —
анализ двухкомпонентной смеси по ее плотности;
вязкость — определение по скорости протекания
газа в капиллярной трубке или по сопротивлению газа
вращению полых шара, цилиндра или диска вокруг
перпендикулярной к их плоскости оси; темпера-
тура кипения (фракционированная дистилля-
ция) — разделение, основанное на различии темп-р
кипения и давлений насыщения при низких темп-рах;
диффузия — определение зависимости между ско-
ростью диффузии и давлением при диффузии через
пористые перегородки; термодиффузия —
разделение двухкомпонентной смеси газов, основан-
ное на возникновении градиента относительной кон-
центрации компонентов при наличии градиента темп-р;
тепловой эффект реакции (термохимии,
метод) — каталитич. сожжение и измерение повы-
шения темп-ры при помощи моста Уитстона или
термоэлемента; теплопроводность (термо-
763
ГАЗОВЫЙ АНАЛИЗ
764
кондуктометрия) — сравнение теплопроводностей ана-
лизируемого газа и газа эталона; скорость
звука (акустич. метод) — сравнение высоты тона,
издаваемого трубкой определенной длины при про-
дувании через нее исследуемого газа, с высотой тона,
полученного в тех же условиях при продувании
воздуха; поглощение прерывистого
ИК-излучения (оптйко-акустич. метод) — звучание
газа, поглощающего ИК-излучение, если это излуче-
ние прерывается со звуковой частотой; магнитная
восприимчивость (магнитный метод) — ис-
следование связи между напряженностью магнитного
поля и теплопроводностью парамагнитных газов
(эффект Зенфтлебена) и термомагнптной конвекции;
спектры поглощения и испускании
(спектральный метод) — исследование спектров в ви-
димой, УФ- и ПК-областях спектра; интерфе-
ренция (интерференционный метод) — определе-
ние преломляющей способности газов по сдвигу полос
спектра интерференции; изотопный эффект
(масс-спектральный метод) — исследование масс-
спектров анализируемой смеси газов; радиоак-
тивные излучения — измерение радиоактив-
ности, применение излучений радиоактивных элемен-
тов, а также ядерных реакций.
Г. а. производится при помощи приборов для
колич. определении содержания компонентов газовых
смесей—газоанализаторов (Г.), к-рые разделяются на
приборы ручного действия и автоматические.
Газоанализаторы ручного действия. Среди приборов
этого типа наиболее распространены абсорбционные
Г., содержание отдельных компонентов определяется
последовательным поглощением составных частей
газа в сосудах, заполненных различными раствора-
ми, напр. СО2 поглощают р-ром КОН, О2 — р-ром
пирогаллола, СО— аммиачным р-ром Си2С12 (газо-
анализатор ГХП-3). Часто абсорбцию газов сочетают
с определением горючих компонентов сожжением
на СпО в разных условиях или на высокоактивном
платиновом или палладиевом катализаторе. На СпО
при темп-ре 300—350° сжигают СО и Н2, при 850—
900° — метан и его гомологи. Так определяют, напр.,
Н2, СО и СН4; образовавшуюся в последних случаях
СО2 определяют абсорбционным методом (газоана-
лизаторы ВТИ-2, ВИАМ, ГИАП, ЛТИ, Норзе).
Рис. 1.
На рис. 1 показан типичный Г. такого типа. Погло-
щение компонентов производят в сосудах 1, напол-
ненных соответственно различными поглотительны-
ми жидкостями; изменение объема после каждого
поглощения определяют в бюретке 2; дожигание
производят в петле 3. В перечисленных Г. исследуется
100 и более миллилитров газа; для анализа смесей
объемом 1—2 мл удобен абсорбционный полумикро-
газоанализатор с бюреткой, имеющей цену деления
0,01—0,02 мл. Смесь СО2, N2, Аг, Кг п Хе объемом
2,5—3,0 мл удобно анализировать в полумикрогазо-
анализаторе манометрия, типа; СО2 и N3 определяют
абсорбцией, Аг, Кг и Хе — дистилляцией. Для ана-
лиза небольших объемов газа (1 мл и меньше), а также
для определения очень
малых концентраций еъ А
компонентов газовой Л/
смеси (1 Л1иг/л) приме- ^==5^ /2
няют микрогазоанали- [НПг ^^====^===:::==г^
заторы. Наряду с Г.
абсорбционного типа —
применяются ручныеГ., 1
основанные на исполь-
зовании физико-хими- Рис 9
ческих и физич. свойств
газов. Содержание СО2 определяют манометром 2,
измеряя разрежение в замкнутом сосуде 1 после по-
глощения СО2; степень разрежения: в системе пропор-
циональна концентрации СО2 (рис. 2). Взрывоопасные
концентрации метана и водорода определяют в приборе,
основанном на измерении диффузии этих газов через
пористую перегородку. Измерение электропроводности
растворов дает возможность быстро и точно опреде-
лять ряд газов и паров в статич. или динамич. усло-
виях; так, напр., определяют СО2 по изменению
электропроводности р-ров Ва(ОН)2 пли NaOH.
Известны Г. а., основанные на использовании поляро-
графии (определение О2), хроматографии (анализ
смесей углеводорода) И др. Б. Г. Еремина.
Автоматические газоанализаторы. Особенностью
автоматич. Г. является отсутствие ручного труда
при выполнении анализа и во многих случаях
непрерывность показаний, их регистрация (а при
необходимости и сигнализация). В автоматич. Г.
методы определения сводятся к непрерывному изме-
рению физич. величин, находящихся в закономерной
зависимости от состава. Все определения в большин-
стве случаев сводятся к электрич. измерению массы,
давления, объема, темп-ры, магнитной восприимчи-
вости и др. физич. свойств анализируемого газа.
Автоматич. Г. дают возможность контролировать
производственный процесс в потоке со скоростью,
соизмеримой со скоростью самого процесса. Это
имеет особое значение в связи с интенсификацией
химич. процессов, применением высоких темп-p и
давлений, проведением ряда процессов в псевдоожи-
женном кипящем слое и т. п.
По принципу действия автоматич. Г. могут быть
разделены на след, группы: 1) Приборы, основанные
на физич. методах анализа, выполняемых при помощи
вспомогательных химич. реакций; такие приборы
обычно называют химич. Г. Они основаны на измене-
нии объема (волюметрич. Г.) или давления (маномет-
рия. Г.) при поглощении анализируемого компонента
поглотительным раствором и последующей химич.
реакции, напр. СО2, поглощают р-ром едкой щелочи.
2) Приборы, основанные на физич. методах ана-
лиза, выполняемых при помощи вспомогательных
физико-химич. процессов, напр. изменение темп-ры
при каталитич. окислении Н2, СО, СН4, С6Н6 и др.,
изменение электрич. или оптич. свойств раствора при
пропускании через него анализируемой газовой смеси
и т. п. В эту группу входят след. Г.: термохимиче-
ские; хроматографические; электрохимические (ампе-
рометрические, деполяризационные, кулонометриче-
ские, потенциометрические, кондуктометрические);
фотоко лор иметр ическ ие.
Для иллюстрации приводится описание двух Г. этой
группы. Полярографический газоанализа-
тор типа А-1024 (рис. 3). В этом приборе применен иеза-
соряющийся неподвижный ртутный катод 1 с постоянной
поверхностью. На него с большой скоростью подается анали-
зируемый газ, а из сосуда 2 — абсорбирующий р-р. Для из-
765
газовый анализ - газы нефтепереработки
766
мерения тока применяют миллиамперметр 3. Газоанализатор
А-1024 используют для измерения содержания кислорода
в Ng, СО2, С2Н2, С2Н4и др. в пределах от 0 до 25%; наименьший
предел измерения 0—0,1%; применяется для непрерывного
определения содержания кисло-
рода в обычных технич. газах,
Рис. 3.
контроля окислительных и вос-
становительных процессов и
т. д. Фотоколориметр и-
ческий газоанализа-
тор с индикаторной
лентой (рис. 4). Свет от
источника 1 проходит через лин-
зы 2 и, отражаясь от зеркал з,
попадает на индикаторную лен-
ту. Анализируемый газ с посто-
янной скоростью через сопло 8
поступает на индикаторную лен-
ту 4 и вызывает изменение ее
окраски. Возникающая при этом
разность эдс фотоэлементов 5
и 6, пропорциональная содер-
жанию анализируемого компо-
нента, регистрируется прибором
7. Пропитывая индикаторную
ленту, напр. ацетатом свинца,
можно определять H2S. Извест-
ны приборы ФЭАВ для опреде-
ления AsH3 и ФКТ-1 для опре-
деления H2S.
3) Приборы, основанные на чисто физич. принци-
пах (напр., на измерении теплопроводности, плотно-
сти, магнитной проницаемости и т. д.); к приборам
этой группы относят-
ся газоанализаторы:
те рмокондуктометр и-
ческие, денситометри-
ческие, магнитные, ин-
фракрасные, ультра-
фиолетовые, акусти-
ческие, ионизацион-
ные, радиоспектроско-
пическпе, масс-спек-
троскопические.
Схемы приборов (рис,
5 и 6) иллюстрируют га-
зоанализаторы этой груп-
пы. Термомагнит-
ный газоанализа-
Рис. 4.
тор, рис. 5 (МКГ-2 и др.). Анализируемый газ поступает
в кольцевую камеру 1 и по кольцу направляется вверх. В со-
единительной трубке 2 газ нагревается спиралью 3. При на-
личии в газе кислорода холодный газ из кольца будет при-
тягиваться к магниту 4 сильнее, чем магнитный газ в трубке,
ив последней начинается
т£ыход газа движение газа слева на-
I право. В результате про-
порционально содержанию
кислорода изменяются тем-
пература и сопротивление
обмоток; последнее реги-
стрируется прибором б. Г.
этого типа применяются
для измерения от 0 до 100%
кислорода в газе: наимень-
ший предел измерений 0—
5%; точность ок. 2,5% от
верхнего предела измере-
ний; применяется для кон-
троля содержания кисло-
рода в хвостовых газах
заводов азотной, серной и
уксусной к-т и для дру-
гих целей. Б е здиспер-
газоанализатор с
й (ОА-2Ю2, ГИП-4
| Вход газа
Рис. 5.
сн ы й инфракрасный
положительной фильтр ацие
и др.) (рис. 6). Инфракрасное излучение от источников 1,
прерываемое обтюратором 2, поступает в две камеры: рабо-
чую 3 и сравнения 4. Через первую из них продувается ис-
следуемый газ; вторая заполнена газом сравнения. Далее
ИК-излучение поступает в приемник 5, разделенный мем-
бранным конденсатором 6. Вследствие разного поглощения
излучения в камерах 3 и 4 в приемнике 5 создается раз-
ность давлений, к-рую измеряют магнитоэлектрич. прибо-
ром 7. Анализируемый компонент в сложных газовых сме-
сях можно измерять в широких пределах концентраций
(часто от Г 0 до 100%); в смеси удается обнаруживать до
0,0005% анализируемого компонента. Г. этого типа приме-
няются для определения СО в воздухе производственных по-
мещений, СО и СО2—в азотоводородной смеси в произ-ве син-
тетич. аммиака, СН4и NHa—в циркуляционной азотоводород-
ной смеси при произ-ве синтетич. аммиака, этилового, мети-
лового и др. спиртов, в произ-ве дивинила и др.
Автоматич. Г. принято также делить на типы по
измеряемой физич. величине — объему, плотности,
магнитной проницаемости и т. д., причем к одному
типу могут относить-
ся газоанализаторы,
находящиеся в раз-
личных группах, на-
пример к фотометрия.
Г. относятся приборы,
основанные на изме-
рении физич. свойств
(оптич. плотности оки-
слов азота), и прибо-
ры, основанные на из-
мерении оптической
Вход 1 Выход
газо Т газо
Рис. 6.
плотности раствора при поглощении им анализи-
руемого компонента газовой смеси.
Наряду с универсальными методами (масс-спектро-
скопией, хроматографией и др.), позволяющими произ-
водить полный анализ многокомпонентной газовой
смеси, существуют методы, пользуясь к-рыми можно
определять только один компонент в сложной газовой
смеси, напр. методы ИК- и УФ-поглощения, фотоко-
лориметрич. и др. Нек-рые методы, напр. термо-
кондуктометр ич. , ультразвуковой, ионизационный
и др., ограничены определением одного компонента
в бинарной или псевдобинарной смеси. Для опре-
деления одного и того же компонента часто могут
быть использованы различные методы; выбор ме-
тода зависит от состава газовой смеси, от желае-
мой точности, чувствительности и скорости опреде-
ления.
Лит.: Еремина Б. Г., Газовый анализ, Л., 1955;
Бочкова О. П. и Шрейдер Е. Я., Спектральный
анализ газовых смесей, М., 1955; Филлипс К., Хромато-
графия газов, пер. с англ., М., 1958; Автоматизация химиче-
ских и коксохимических производств, под ред. Н. Я. Феста,
Н. Н. Елшина, Ю. Н. Геруляйтиса, М., 1958; Соко-
лов В. А., Методы анализа газов, М., 1958; Тр. Уральского
н.-и. хим. ин-та, 1958, вып. 6; Ш уми л о веки й Н. Н.,
Соколов А. А., Автоматика и телемеханика, 1953, 14,
№ 3, с. 328; Павленко В. А,, Рафальсон А. Э.,
Шерешевский А. М., Приборы и техника экспери-
мента, 1958, № 3; G и ё г i и Н., ТгаПё de manipulation et
d‘analyse des gases, P., 1952; Mullen P. W., Modern Gas
analysis, N. Y., 1955; Messen und Regeln in der chemi-
schen Technik, hrsg. J. Hengstenberg, B. Sturm, O. Winkler,
B.—Gottingen—Hdlb., 1957; Selected topics in modern instru-
mental analysis, ed. D. F. Boltz, N. Y. — L., 1952; Process
instruments and controls handbook, ed. D. M. Considine, N. Y.—
Toronto—L., 1957; F г a a d e D. J., Oil and Gas Journal, 1957,
55, №42, p. 93; A i k m a n A. R., Chemical Engineering, 1957,
64, № 6, p. 266; Willard H. H., M e r r i t L. L., D e a n
J. A., Instrumental methods of analysis, 3 ed. N. Y.—To-
ronto—L., 1958; Wherry T. C., Deford D. D., Control
Engineering, 1958, 5, № 2, p. 115; Lay J. O., Metallurgia,
1955, 51/№ 304, p. 109. А. К. Майзель.
ГАЗОГЕНЕРАТОРЫ — см. Газификация твердых
топлив.
ГАЗОЙЛЬ — см. Дизельное топливо.
ГАЗОПРОНИЦАЕМОСТЬ ПОЛИМЕРОВ — см.
Диффузия.
ГАЗЫ НЕФТЕПЕРЕРАБОТКИ — газы, выделяю-
щиеся при перегонке нефти или образующиеся при
крекинге, пиролизе, коксовании, деструктивной гидро-
генезации и др. процессах переработки нефти. При
перегонке нефти состав углеводородов ие меняется,
происходит лишь процесс термич. разделения ее иа
отдельные фракции: бензиновую, керосиновую, газой-
левую и т. д. Соотношение различных углеводородов
в газах прямой перегонки нефти сильно зависит от
природы взятого сырья. Другие, т. н. деструктивные
процессы, часто связаны е глубокими превращениями
углеводородов нефти, что влияет в значительной мере
и на состав получаемых газообразных углеводо-
родов. Газы деструктивной переработки нефти по
своему составу отличаются рт природных газов прежде
всего наличием в них непредельных углеводородов
и водорода, В процессах глубокой химич. переработки
767
ГАЗЫ НЕФТЯНЫЕ ПОПУТНЫЕ
768
нефти природа взятой нефти практически не влияет
на состав получаемых газов. Состав промышленных
газов в этом случае связан с характером процесса,
с глубиной превращений, претерпеваемых нефтяным
сырьем. Состав различных газов (об. %) при различ-
ных способах переработки показан в таблице.
Компоненты Пря- мая пере- гонка Пиро- лиз Жидко- фазный крекинг Паро- фазный крекинг Катали- тич. крекинг
Водород 1 12 3-4 7-9 5-8
Метан 11—46 55—57 40-50 28-30 10
Этан 3—17 5-7 17—18 13-14 3-5
Пропан 9—28 0.5 10-15 3-4 16-20
Бутаны 14—34 0,2 16-18 5-6 1—1.5 42—46
Этилен 2—3 20-23 3
Пропилен .... — 7-8 6—8 15-18 6-11
Бутилен С5 и выше (мета- — 4—5 4—5 6—10 5—6
новые и олефи- новые) 14-30 2-3 4 3 5-12
При перегонке нефти получают газ прямой пере-
гонки, представляющий собой тяжелую часть попут-
ного газа (см. Гааы нефтяные попутные), оставшуюся
растворенной в нефти. Состав и количество газа,
выделившегося при прямой перегонке, зависят не
только от состава нефти и газа, но и от условий добычи,
хранения и транспортировки; выход газов, как пра-
вило, невелик. В современных установках пиролиза
жидких нефтепродуктов получают ароматич. углево-
дороды и газообразные олефины (этилен, пропилен,
бутилен и др.). При пиролизе выход газов может
составлять 50% от исходного сырья.
Из приведенных данных видно, что Г. н. содержат
от 12% до 51% непредельных углеводородов. В газах
жидкофазного крекинга содержание метана снижается
до 40—50%, в газах парофазного крекинга—до 30% и
в газах каталитич. крекинга — до 10%. Газы каталитич.
крекинга и термич. жидкофазного крекинга по общему
содержанию предельных углеводородов близки между
собой, но резко отличаются по составу компонентов
(см. табл.). Количество непредельных газов этих
двух процессов практически одинаково, по составу
компонентов они близки. Г. н. — ценное сырье для
промышленного органич. синтеза.
Лит.: Наметкин С. С., Химия нефти, [3 изд.], М.,
1955; Некрасов А. С. иКренцель Б. А., Химиче-
ское использование нефтяных углеводородных газов, М.,
1952; Обрядчиков С. Н., Химическая переработка
углеводородных газов, М.—Л., 1946; его же, Технология
нефти, ч. 2, 3 изд., М,— Л., 1952.
Л. П. Колесникова,
ГАЗЫ НЕФТЯНЫЕ ПОПУТНЫЕ — природные
газы, сопровождающие нефть и выделяющиеся при ее
добыче. Характерной особенностью состава Г. н. п.
является наличие в них, кроме метана, также этана,
пропана, бутанов н паров более тяжелых углеводо-
родов; во многих Г. н. п. присутствуют сероводород
и негорючие компоненты:. азот, углекислый газ, а
также редкие газы — Не, Аг. Последние содержатся
в количествах, редко представляющих промышленный
интерес; в газах месторождений Апшеронского полу-
острова, Грозненских, Сахалинских, Небит-дага их
содержится ок. 10-3%. Значительные количества Не
находятся в Г. н. п. нек-рых месторождений США
[Харлей (штат Юта) — 7,16%; Клитсайд (штат Те-
хас)—до 2%].
Нефть и газ скапливаются в таких участках земной
коры («ловушках»), где физич. н геологич. условия
благоприятствуют длительному сохранению. В неф-
тяной залежи газ, сопровождающий нефть, может
находиться в растворенном виде (тяжелые углеводо-
роды) или располагаться над нефтью, образуя газо-
вую «шапку». Состав свободных газов, находящихся
непосредственно над нефтью или мигрировавших
в выше расположенные коллекторы, может сильно
отличаться от состава газов, растворенных’ в нефти;
последние обогащены тяжелыми газообразными угле-
водородами. Состав Г. н. п., выделяющихся из нефти
в процессе ее добычи, значительно отличается от
состава свободных газов, добываемых нз газоносных
пластов того же месторождения. Влиянием раство-
римости тяжелых углеводородов могут быть объяс-
нены часто наблюдаемые расхождения в составе
образцов газов, получаемых из одной и тон же нефтя-
ной скважины. Состав газов сильно зависиг от усло-
вий отбора пробы, от давления, под к-рым находится
газ в скважине, соотношения в пробе свободного газа
из залежи и газа, выделившегося из нефти при ее
подъеме в скважине. В связи с этим содержание и
состав тяжелых углеводородов в газах, отобранных
на одной и той же площади, показывают значительные
колебания. Это относится н к таким хорошо раство-
римым газам, как H2S и СО2.
При вскрытии пласта скважиной вначале начинает
фонтанировать газ газовой шапки, а затем, по мере
падения давления, начинает выделяться газ, раство-
ренный в нефти. В нек-рых случаях, Когда газ пол-
ностью растворен в нефти, он добывается вместе
с нефтью. Количество газа в кубич. метрах, прихо-
дящееся па 1 т добываемой нефти, называют газо-
вым- фактором, к-рый для различных место-
рождений неодинаков и зависит от природы место-
рождения, режима его эксплуатации и может изме-
няться от 1—2 jh3 до нескольких тысяч „и3 на 1 т до-
бываемой нефти. Состав Г. н. п. зависит от природы
нефти, в к-рой они заключены, а также от принятой
схемы отделения газа от нефти при выходе их из сква-
жины. Состав попутных газов (об. %) нек-рых нефте-
газовых месторождений СССР показан в таблице.
Месторождение Ме- тан Этан Про- пан Бута- ны Высшие углево- дороды Другие газы; Na, СО2, H„S
Туймазинское . . 41,9 20,0 17,3 7,9 3.3 9,6
Ромашкинское . . 37,0 20,0 18.5 8,2 4,7 11.6
Бугурусланское . 72.5 9,8 7.5 8,3 — 1,9
Грозненское . . . 30,8 7,5 21,5 20,4 19,8 —
Сураханское . . . 89,7 0,16 0,13 0,28 1,26 8,4
Большая часть Г. н. п. относится к т. н. «жирным»
газам, содержащим, кроме метана, тяжелые углево-
дороды (пропан, бутан и т. д.) в количестве 50 г/м3
и выше. Газы, состоящие преим. из метана и содер-
жащие до 50 г/м3 тяжелых углеводородов, называют
«сухими», или «тощими»; это, в основном, газы чисто
газовых месторождений; содержание метана в них мо-
жет составлять 90—98%. При переработке жирные
газы прежде всего подвергаются удалению бензина,
т. н. отбензиниванию, в результате к-рого из них
выделяются углеводороды, входящие в состав бензина.
Полученный при данном процессе бензин наз. газовым.
После отбензинивания Г. н. п. состоят преим. из ме-
тана, а также небольших количеств этана, пропана
и бутана.
Г. н. п. используют в качестве топлива и химич.
сырья. Энергетич. использование связано с высокой
теплотворной способностью Г. н. п., к-рая колеблется
от 9 300 до 14 000 ккал/м3 углеводородной части
газа. При электрокрекннге из метана образуется
ацетилен; при конверсии метана перегретым водяным
паром или СО2 в присутствии катализаторов — смесь
СО и Н2, применяющаяся во многих органич. синте-
зах. Этан и пропан Г. н. п. могут служить источником
получения атилена, бутилена, ацетальдегида, дру-
гих кислородсодержащих соединений. Бутан может
789
ГАЗЫ ПРИРОДНЫЕ—ГАЗЫ ПРИРОДНЫЕ ГОРЮЧИЕ
770
быть использован для получения дивинила, бутило-
вых спиртов, метилэтилкетона и др. соединений.
Лит.: Соколов В. А., Миграция газа и нефти, М.,
1956; см. также при ст. Газы нефтепереработки.
Л. П. Колесникова.
ГАЗЫ ПРИРОДНЫЕ — газы, содержащиеся в нед-
рах Земли, а также газы земной атмосферы. Г. п.
частично растворены в подземных и наземных водах
и нефтях, сорбированы углями и нек-рыми глини-
стыми породами. Г. п. выделяются из недр земли при
вулканич. деятельности по тектонич. трещинам, свя-
занным с газоносными пластами, выносятся мине-
ральными источниками. Г. п. можно подразделить
на газы биохимич., вулканич., метаморфич., воздуш-
ного и химич. происхождения, на газы радиоактивных
и термоядерных процессов.
Биохимические газы — продукты жизнедея-
тельности бактерий, образуются при превращениях органич.
веществ, восстановлении сульфатов или других минеральных
солей. В результате таких процессов могут образоваться СН4,
С2Н4, HsS, СО2, N2.
Вулканические газы выделяются из недр
земли при извержениях; растворены в расплавленной магме,
а также образуются при действии паров воды при высоких
темп-рах на вещества магмы и контактных с магмой пород.
Примерный состав вулканич. газов приведен в табл. 1.
метан. В газах, сопутствующих нефти, кроме метана, содер-
жатся значительные количества его гомологов. В табл. 2 дан
примерный состав газовых залежей разных месторождений.
В нек-рых случаях горючие газы содержат повышенное
количество гелия. Примерный состав газов, сопутствующих
нефтяным залежам из разных месторождений, приведен в ст.
Газы нефтяные попутные.
Г. п. горючие и газы попутные добываются как
ценное полезное ископаемое. Газы угольных пластов
извлекаются при разработке угольных месторождений
с целью предотвратить их выделение в горные выра-
ботки. В нек-рых странах (Бельгии, Германии, Китае
и др.) угольный газ используется как топливо. В отно-
сительно небольших количествах Г. п. содержатся
в пористых или трещиноватых породах, вмещающих
рудные и нерудные полезные ископаемые, и мешают
добыче последних, выделяясь в рудничную атмосферу.
Лит.: Белоусов В. В., Очерки геохимии природных
Газов, Л., 1937; Козлов А. Л., Проблемы геохимии при-
родных газов, М.—Л., 1950; Боксерман Ю. И., При-
Водные газы и их использование, М., 1957; Природные газы
ССР. Под ред. В. Д. Голубятникова и В. И. Рейнеке,
М.—Л., 1935; Козлова. Л., О закономерностях формиро-
вания и размещения нефтяных и газовых залежей, М.,
1959; Лидин Г. Д., Айруии А. Т., Дмитриев А. М.,
Способы извлечения и утилизации метана угольных месторож-
дений за рубежом, М., 1957. М. М. Элинсон.
Таблица 1
Наименование вулкана Состав в объемных %
COS СО SO, H»S н, Cl-f-HCl СН4 N,4-Ar о.
Килауэа (Гавайские остро- ва) Везувий (Италия) Катмаи (Аляска) Мон-Пеле (Мартиника) . . Фумаролы о-ва Уайт (Но- вая Зеландия) Шевелуч (Камчатка) . . . 23,8-62,3 34,2 17,5-70,4 15,38 88,3 48,8-19,75 5,6—3,5 0,0 0,5-0,6 1,6 0,0 0,56-0,03 0,0-12,8 0,0 0,0 0,0 0,0 1,1—0,9 0,0 0,0 23,6-3,2 0,0 2,0 0,25-0,21 7,2-7,5 3,8 8,12 9,8 0,25—0,6 0,0 2,4 0,0 Следы 0,8 0,0 0,0 0,5—12,1 5,46 0,0 0,0 63,3—13,8 48,0 57,1-12,8 55,65 1,9 39,5—62,01 0,0 11,5 1,1-1.0 13,67 0,0 10,05-16,5
Метаморфические газы образуются в процессе
превращения ископаемых углей и других горных пород под
действием теплоты и давления; содержат СН4, СОг, Hs, раз-
личные углеводороды, H2S, СО и др.
Состав газов атмосферы приведен в ст. Атмо-
сфера. Газы воздушные, находящиеся в недрах земли,
состоят из Nj и инертных газов; свободный кислород в них
отсутствует. Газы химического происхождения об-
разуются при химич. взаимодействии между газообразными
веществами, водными р-рамн и горными породами как при
нормальных условиях, так и при повышенных темперах и да-
влениях, существующих на разных глубинах земной коры,-
При этом могут образоваться Hs, СО, СО2, H2S, N„, а также
СН4 и др. углеводороды. В результате радиоактивных процес-
сов и термоядерных реакций образуется гелий, аргон, ксенон
и др. газы.
Таблица 2
Месторождение Состав в объемных %
СН4 С2Не С3Н3 СДо Ng-f-CO»
Елшанское (Саратов- ская обл.) 93,2 0,7 0,6 0,6 3,9
Бугурусланское (Оренбург, обл.) . 76,8 4,4 1,7 0,8 12,3
Абрамовское (Ста- линград. обл.). . . 98,32 0,0 0,0 0,0 1,68
Дашавское (Львов- ская обл.) 97,8 0,5 0,2 0,1 1,4
Ставропольское . . . 98,0 0,4 0,15 0,03 1,42
Б1ебелииское (Харь- ковская обл.) . . . 92,5 2,78 0,65 0,56 3,51
К Г. п. относятся также горючие газы, скопляющиеся
в породах-коллекторах в виде самостоятельных газовых зале-
жей или же сопутствующие нефтяным залежам (см. Газы
природные еорючие И Газы нефтяные попутные), а также го-
рючие газы, заключенные в угольных пластах. Происхожде-
ние природных горючих газов обусловлено биохимич. разло-
жением органич. вещества и дальнейшим метаморфизмом по-
следнего под воздействием геохимич. факторов. Кроме того,
горючие газы образуются при взаимодействии паров воды
с карбидами металлов, а также Со и Ht. Основным газом само-
стоятельных газовых залежей и угольных пластов является
13 к. X. Э. т. 1
ГАЗЫ ПРИРОДНЫЕ ГОРЮЧИЕ (переработка) —
естественные смеси углеводородов различного состава;
по способу добычи Г. п. г. разделяются: на собственно
природные газы, добываемые из чисто газо-
вых месторождений, практически не содержащих
нефти; попутные газы, растворенные в нефти
и добываемые вместе с нею, и газы газокон-
денсатных месторождений, находя-
щиеся в пластах под давлением и содержащие (в ре-
зультате т. н. «обратного» испарения) керосиновые,
а иногда и соляровые фракции нефти. Собственно
природные газы и газы газоконденсатных месторож-
дений выходят на поверхность земли под значитель-
ным давлением (50—100 ат); попутные газы выде-
ляются из нефти в сепараторах под небольшим избы-
точным давлением либо под разрежением. Природные
и попутные газы в основном состоят из алканов,
незначительного количества цикланов и ароматич.
углеводородов, небольших количеств азота и аргона,
а также следов гелия и водорода. Кроме того, иногда
в газах содержится H2S, меркаптаны и СО2. По со-
ставу Г. и. г. иногда разделяют на сухие и жирные.
К жирным относятся газы, содержащие 50—100
(и больше) г/м3 углеводородов от С8 и выше. Соб-
ственно природные газы обычно относятся к сухим
газам, попутные и газоконденсатные — к жирным.
Составы попутных и природных газов нек-рых
нефтяных и газовых месторождений см. Газы природ-
ные и Газы нефтяные попутные. Выделение тяжелых
углеводородов из природных и попутных нефтяных
газов производится: компрессией, абсорбцией, адсорб-
цией и охлаждением. Компрессионный ме-
тод разделения газов основан на сжатии газа, при
к-ром тяжелые углеводороды переходят из паровой
фазы в жидкую. Самостоятельного значения этот
метод не имеет и применяется в комбинации с другими.
771
ГАЗЫ ПРИРОДНЫЕ ГОРЮЧИЕ
772
Абсорбционный метод основан на извле-
чении тяжелых углеводородов из газов путем абсорб-
ции поглотительным маслом (см. Абсорбция масляная).
Адсорбционный метод разделения газов
(периодический и непрерывный) основан на избира-
тельном поглощении углеводородов твердыми сорбен-
тами (обычно активным углем). В периодич. уста-
новках сырой газ проходит через фильтры и поступает
в адсорберы, заполненные активным углем. Сухой
газ отводится в газопровод, а сорбированные углево-
дороды отгоняются из угля водяным паром, конден-
сируются и направляются на разделение. Влажный
уголь осушается, затем охлаждается воздухом или
газом и заново включается в цикл. Обычно установка
состоит не менее чем из двух периодически включаю-
щихся адсорберов.
При непрерывной адсорбции (см. рис.) активный
уголь непрерывно движется сверху вниз навстре-
чу сырому газу (см. Ги-
персорбция). Насыщение
активного угля происхо-
дит в адсорбционной ча-
сти колонны. Насыщенный
извлекаемыми углеводоро-
дами активный уголь по-
ступает через ректифици-
рующую часть колонны
(хроматографическое раз-
деление) в десорбционную,
где благодаря нагреву че-
рез стенку и подаче не-
большого количества пара
производится десорбция
адсорбированных угле-
водородов. Пары углеводо-
родов поднимаются из де-
сорбционной части вверх,
причем лучше сорбирую-
щийся бутан вытесняет из
Схема установки угольной
адсорбции: 1 — адсорбент; г —
секция охлаждения; з —• сек-
ция адсорбции; 4 — секция
ректификации; б — секция десорбции; в — реактиватор; 1 —
теплоноситель для нагрева десорбционной части; s — вода
для охлаждения; 9 — пар для десорбции остатков тяжелых
углеводородов.
угли пропан. В свою очередь, бутан вытесняется пента-
ном и более высококипящими углеводородами. Актив-
ный уголь из десорбционной части подается на верх ко-
лонны в трубчатый холодильник, где он охлаждается
и заново поступает в адсорбционную секцию. Для уда-
ления высших углеводородов часть угля направляется
в реактиватор, где регенерируется путем нагрева
топочными газами. Таким образом, в одной колонне
осуществляется непрерывный процесс сорбции, десорб-
ции и разделения газа с получением индивидуальных
углеводородов высокой чистоты. Процесс полностью
автоматизирован.
Разделение газов (см. также Газов раз-
деление) производится; 1) Низкотемпературной рек-
тификацией, которая состоит в предварительном
охлаждении и осушке сырого газа, направляемого
затем в колонну вместе с образовавшимся при охлаж-
дении конденсатом. Неконденси^ующиеся газы, в ос-
новном метан и этан, из верхней части колонны отво-
дятся в газопровод, а продукт из нижней части ко-
лонны поступает на дальнейшее разделение. Глубокая
осушка газа производится на специальной установке
силикагелем или алюмогелем. 2) Низкотемпературной
конденсацией в охладителях, где происходит частич-
ная конденсация сырого газа с выделением наиболее
тяжелокипящих углеводородов. Поступающий сырой
газ осушается (см. Газов осушка), затем охлаждается
и направляется в сепаратор, где из него выделяется
образовавшийся конденсат, а сухой газ после пред-
варительной очистки от H2S (см. Газов очистка)
отводигся в газопровод. Конденсат далее направляется
в ректификационную колонну, орошаемую охлаждае-
мой флегмой. Неконденсируемые газы после холо-
дильника отводятся в сеть, а бензин направляется
в газофракционирующие установки на дальнейшее
разделение.
Химическая переработка Г. п. г. При химич. пере-
Ёаботке получают след, основные продукты: 1) Н2,
О, смесь СО + Н2 (синтезгаз), HCN, ацетилен и
сажу; 2) олефины и диолефины; 3) кислородсодержа-
щие соединения; 4) хлор- и нитросоединения.
Водород, окись углерода, синтезгаз, HCN, ацетилен
и сажу получают, как правило, из сухих природных
газов. Синтезгаз образуется при конверсии метана
водяным паром либо при неполном горении метана
в кислороде. Состав получающейся при конверсии
метана водяным паром смеси газов (СН4, Н2О, СО,
Н2, СО2) зависит от темп-ры и количества пара, вво-
димого в процесс. Реакция идет со значительным
потреблением тепла, проводится обычно на никелевом
катализаторе при 700—800°. Реактор представляет
собой трубчатую печь. Реагирующие газы проходят
по вертикальным трубкам из жаропрочной стали,
заполненным катализатором. Снаружи трубки обо-
греваются горячими дымовыми газами. При непол-
ном горении метана в кислороде (наз. также кисло-
родной конверсией) процесс протекает при 1400—
1500° без катализаторов, в печах, выложенных огне-
упорным материалом. Состав сырого газа, получае-
мого при конверсии метана водяным паром и при
неполном горении метана в кислороде, приведен
в таблице.
Процесс Содержание, %
СО2 н, СО СН4 N„
Конверсия водяным паром. 8 15 75 1,5 0,5
Неполное горение в кисло- роде 3 62 32 0,5 2,5
При неполном горении образующийся газ содержит
также небольшое количество сажи. Для получения
синтезгаза сырой газ очищают от СО2, а для получе-
ния водорода — подвергают второй ступени конвер-
сии водяным паром: СО + НаО СО2 + Н2. Реак-
цию проводят на железном катализаторе при400—-450°.
При получении водорода для синтеза аммиака в про-
цесс обычно вводится нек-рое количество воздуха,
чтобы образовалась смесь состава ЗН2 + N2, а сам
процесс проводят под давлением 15—20 ат, что позво-
ляет использовать давление природного газа и умень-
шить расход энергии на сжатие азотоводородной
смеси для синтеза аммиака. Синильную кис-
лоту получают при неполном горении в воздухе
смеси природного газа и аммиака на сетке из платино-
вого сплава в качестве катализатора: 2СН4 + ЗО2 +
+ 2NH3 —> 2HCN + 6Н2О. Темп-ра в реакторе до-
стигает 1100—1200°. Продукты реакции охлаждают
и выделяют из них непрореагировавший аммиак
поглощением серной к-той, а синильную к-ту — водой.
Разделение воды и синильной к-ты производят рек-
тификацией. Термич. разложением или при непол-
ном горении природного газа получают сажу двух
видов: канальную и печную; наибольшее значение
из них имеет канальная. Как правило, для этой цели
применяется сухой или отбензиненный природный
или попутные газы. На произ-во канальной сажи во
всем мире затрачивалось более 13 миллиардов мЛ
природного газа в год.
773
ГАЗЫ ПРИРОДНЫЕ ГОРЮЧИЕ — ГАЛАКТАНЫ
774
Из природного газа электрокрекингом, термич.
крекингом или при неполном горении в кислороде
(окислительный крекинг) получают ацетилен: 2СН4=
= С2Н2 + ЗН2. При электрокрекинге природный газ
пропускают через электрич. дугу высокого напря-
жения, температура к-рой достигает 6000°. Выход
ацетилена —до 50% от исходного сырья; кроме того,
при этом образуется этилен и сажа. Термич. крекинг
проводят в периодических регенеративных печах.
Получение ацетилена при неполном горении метана
в кислороде проводят сжиганием в специальных
печах смеси, содержащей 60% СН4 и 40% О2; темп-ра
при этом достигает 1500°; получаемый газ содержит
до 8,5% ацетилена (выход ацетилена на метан 30%).
Ацетилен выделяют из продуктов горения абсорбцией
водой под давлением либо подходящими органич.
растворителями. После извлечения ацетилена газ
содержит 26% СО и 55% Н2 и является прекрасным
сырьем для получения водорода или для дальнейших
синтезов. Поэтому получение ацетилена обычно ком-
бинируется с другими процессами, при к-рых исполь-
зуется синтезгаз (см. Ацетилен). Этот способ полу-
чении ацетилена экономически наиболее выгодный
и быстро развивается.
Получение олефинов и диолефи-
нов. При нагревании Г. п. г. до 600° и выше содер-
жащиеся в них парафиновые углеводороды способны
к реакциям расщепления с разрывом связей и образо-
ванием непредельных углеводородов и водорода или
непредельных и предельных углеводородов с меньшим
числом атомов углерода в молекуле. Эти реакции при-
меняются для произ-ва этилена, пропилена, бутилена,
бутадиена и изопрена, являющихся основным сырьем
для получения спиртов, пластмасс и синтетич. каучу-
ков. Расщепление углеводородов в промышленных
условиях проводится под воздействием только темп-ры
(пиролиз) или темп-ры и катализаторов (см. Гидро-
генизация и дегидрогенизация каталитические). В за-
висимости от способа подвода тепла, необходимого
для протекания реакций, пиролиз и дегидрирование
проводят в трубчатых печах с внешним обогревом
или в печах регенеративного типа. Выход непредель-
ных углеводородов зависит от темп-ры, времени пре-
бывания сырья в реакционном пространстве, давле-
ния, отношения С/Н в исходном сырье, конструкции
печи и др. факторов. Основным продуктом термич.
пиролиза этана является этилен. При переходе от
этана к пропану и бутайу в продуктах пиролиза
наблюдается снижение выхода этилена и увеличение
выхода высших олефинов (пропилена и бутиленов).
Суммарный выход непредельных углеводородов при
термич. пиролизе составляет (в вес. %): из этана
75—77, из пропана 40—50 и из бутана ок. 50.
Каталитич. дегидрогенизацию углеводородов про-
водят гл. обр. для получения пропилена, бутилена,
изобутилена, бутадиена и изопрена из пропана, «-бу-
тана, изобутана и изопентана. Процесс проводится
обычно на хромоникелевых и хромоалюминиевых
катализаторах, что позволяет снизить темп-ру рас-
щепления углеводородов до 520—600° и вести про-
цесс более селективно. В связи с разработкой методов
получения изопренового каучука большое значение
приобретает получение изопрена из пентана или
изопентана, содержащихся в попутных газах и газах
конденсатных месторождений. Изопрен получают из
изопентана каталитич. дегидрированием.
Прямым окислением углеводородов природного
газа кислородом можно получать кислородсо-
держащие соединения: ацетальдегид, фор-
мальдегид, метанол, пропионовый альдегид, ацетон,
окиси пропилена и этилена, этиловый спирт, уксус-
ную к-ту и др. Окисление проводится чисто термич.
путем или в присутствии катализаторов. В промыш-
13*
ленных условиях окисление проводится в паровой
или в жидкой фазе. Сырьем для окисления обычно
является пропан и бутан. Катализаторы парофазного
окисления: фосфаты бора, окислы Mo, Со, Ni, Mg,
Si, Zn и др. металлов. При жидкофазном окислении,
к-рое проводится при более низких темп-рах и отли-
чается большей избирательностью, углеводороды рас-
творяют в растворителе (обычно уксусной к-те) и через
раствор пропускают кислород или воздух. Катали-
заторы жидкофазного окисления: ацетаты Мп, Со,
Сг, Ni и др. металлов. При окислении пропана и бу-
тана получается смесь различных кислородных соеди-
нений, разделение к-рой представляет большие труд-
ности. При прямом окислении метана и применении
в качестве катализаторов окислов азота получается
формальдегид с небольшой примесью метанола. Выход
кислородсодержащих продуктов при окислении ме-
тана значительно ниже, чем при окислении углево-
дородов С3 и С4. При нитровании нек-рых компонен-
тов Г. п. г., гл. обр. пропана, образуются применяе-
мые в пром-сти нитропарафины (нитрометан,
нитроэтан, 1-нитропропан и 2-нитропропан), преиму-
щественно мононитросоединения. Разделение продук-
тов нитрования производится ректификацией. Хлори-
рованием парафиновых углеводородов получают хлор-
производные (хлористый метил, хлористый метилен,
четыреххлористыи углерод, дихлорэтан и др.).
Лит.: Основы технологии нефтехимического синтеза.
[Сб. ст., под ред. А. И. Динцеса и Л. А. Потоловского], М.,
1960; Гольдштейн Р., Химическая переработка нефти,
пер. с англ., М., 1952; IV Международный нефтяной кон-
гресс, т. 4, М. 1956; Некрасов А. С. и Крен-
цель Б. А., Химическое использование нефтяных углево-
дородных газов, М., 1952; Авингер Ф., Химия и техноло-
гия парафиновых углеводородов, пер. с нем., М., 1959; Гуд-
ков С. Ф., Переработка углеводородов природных и попут-
Вых газов, М., 1960. П. А. Теснер.
ГАЛАКТАНЫ — группа гомополисахаридов, со-
стоящих из остатков галактозы. Структура Г. очень
разнообразна и мало изучена. Типичным представи-
телем этой группы полисахаридов является Г. семян
люпина— линейный полисахарид, состоящий из 100—
120 D-галактопиранозных остатков, соединенных при
помощи Р-1,4-связи:
В Г. морских водорослей D-галактопиранозные остат-
ки, по-видимому, в основном соединены через
Р-1,3-связь. Большинство этих Г. содержат сернокис-
лые остатки, этерифицирующие некоторые гидроксилы,
и L-галактозу. Галактокаролоза Penicillium Charle-
sii, в отличие от других Г., построена исключительно
из фуранозных остатков P-D-галактозы, соединенных
1,5-связями, причем ее молекула состоит из 9—10 та-
ких остатков, и с этой точки зрения она стоит на гра-
нице между олиго-и полисахаридами. Г. сопровождают
пектиновые вещества древесины и иек-рых семян
(Г. белого люпина^, являются продуктом метаболизма
нек-рых плесеней (галактокаролоза, руг/лоза) и
лежат в основе структуры технически важных про-
дуктов, добываемых из морских водорослей (агара,
каррагинана и др.). «Животный» Г. изолирован из
бычьих легких и яиц виноградной улитки.
Лит..- Advances In carbohydrate chemistry. Ed. W. Pig-
man, M. L. Woltrom, v. 1—2, N. Y., 1945—46: M i c h e e 1 F„
Chemie der Zucker und Polysaccharide, 2 Autl., Lpz., 1956;
The carbohydrates. Chemistry, biochemistry, physiology, Ed.
W. Pigman, N. Y., 1957. H. E. Рудакова.
775
ГАЛАКТОЗА — ГАЛЛИЙ
776
ГАЛАКТОЗА (цереброза) С3Н1аО3, мол. в.
180,16 — простой углевод, относящийся к группе
альдогексоз, зпимер талозы; от глюкозы отличается
только расположением Н и ОН у 4-го углеродного
атома. Из двух форм Г. D-галактоза имеет более
широкое распространение, чем L-галактоза.
СН2ОН
ОН/|——О Н Н ОНОНН
Кон н>.°"
н у------Y ОН. он н н он о
н он
a-D-Г. — кристаллы; т. пл. 167°; хорошо растворима
вводе, [а]д = + 150,7° (в воде)и +81,1 ° (равновесная система
в воде). В равновесном р-ре Г. по сравнению с таковым глю-
козы содержится больше высокореакционноспособиых форм:
ациклической и пятичленной циклической. P-D-Г. — кри-
сталлы; т. пл. 153—155°; [а]р =+ 54,4° (в воде) и+80,5° (рав-
новесная система в воде). a-L-Г. — кристаллы; т. пл. 163—165°;
[“]£>=—120° (в воде) и —78° (равновесная система в воде).
Остатки L-Г. содержатся в полисахариде слизи льняного
семени и наряду с D-Г.—в агаре и галактанах. D,L-r. — кри-
сталлы, т. пл. 143—144°; получена синтетически восстановле-
нием лактона D.L-галаКтановой к-ты и окислением ду'ль-цита.
Г. восстанавливает р-р Фелинга. Азотной к-той Г. окис-
ляется в двухосновную слизевую кислоту. Эта реакция служит
для обнаружения и колич. определения Г. и нек-рых ее про-
изводных (галактуроновой к-ты и др.). D-Г. сбраживается
лишь лактозными дрожжами и дрожжами, специально при-
способившимися к ней.
Г. широко распространена в природе, встречается
в свободном виде и в виде ди- и трисахаридов (лактоза,
из к-рой обычно Г. и получают путем гидролиза,
меллибиоза, рафиноза), гликозидов — галактозидов
(идеин, миртиллин, цереброзиды), высших полисаха-
ридов (галактаны, агар, гуммиарабик, многие расти-
тельные клеи и слизи, галактоген — полисахарид
белковой железы улитки и икры лягушек).
Лит.: The carbohydrates. Chemistry, biochemistry, phy-
siology. Ed. by W. Pigman, N. Y., 1957; Mi ch eel F.,
Chemie der Zucker und Polysaccharide, 2 АцП., Lpz., 1956.
Б. H. Степаненко.
ГАЛАКТУРОНОВАЯ КИСЛОТА — см. Гексуро-
новые кислоты.
ГАЛАЛИТ — см. Пластики белковые.
ГАЛЛИЙ (Gallium) Ga — химич. элемент III гр.
периодич. системы Менделеева, п. н. 31, ат. в. 69,72.
Состоит из двух изотопов с массовыми числами 69
(60,5%) и 71 (39,5%). Из искусственно радиоактивных
изотопов в качестве индикатора применяют Ga72
(Т,^ = 14,2 часа). Конфигурация внешних электро-
нов атома 4s24pl. Энергии ионизации (в зв): Ga° —>
—> Ga+ —> Gaa+ —> Ga3 + соответственно равны 6,00;
20,510; 30,66. Поперечное сечение захвата тепловых
нейтронов атомом 2,71 барн.
Существование Г. («зкаалюминия») было предсказано
в 1870 Д. И. Менделеевым. Элемент был открыт по искровому
спектру в пиренейской цинковой обманке и выделен в 1875
франц, химиком П. Э. Лекоком де-Вуабодраиом.
Содержание Г. в земной коре составляет 1,5-10—3
вес. °/0. Поведение Г. определяется его литофиль-
ностью и широким рассеянием, обусловленным эндо-
криптным замещением иона А13+.
Одновременно, вследствие большого сходства в строении
атомов Zn, Fe и Ga, последний проявляет халько- и сидеро-
фильные свойства. Единственный минерал Г. — галлит
CuGaS2 — очень редок. В изверженных горных породах Г.
тесно связан с А). В послемагматич. процессах пути их ми-
грации могут расходиться. В гидротермальных месторожде-
ниях Г. накапливается в виде изоморфной примеси (до 0,01—
0,02%) в сфалеритах (цинковйх обманках), а в зоне гиперге-
неза— совместно с А1 в бокситах (2-10”3— 1 • 10~2%).
Попутно с А1 он извлекается из бокситов, нефели-
новых сиенитов и попутно с Zn — из цинковых кон-
центратов. Г. наряду с германием часто содержится
в каменном угле. При газификации угля и при его
сжигании Г. вместе с Ge концентрируется в сажистых
уносах и летучих частях золы и может извлекаться
попутно с Ge.
Физические и химические свойства. Г. — серебри-
сто-белый металл, имеет ромбич. (псевдотетрагональ-
ную) решетку с параметрами: а=4,5197А, Ъ— 7,6601 А,
с=4,5257 А. Атомный радиус 1,39А, ионный радиус
Ga3+ 0,62 А. Плотность твердого металла 5,904 (20s),
жидкого 6,095 (29,8°), т. е. при отвердевании Г.,
подобно воде, расширяется; т. пл. 29,8°, т. кип. 2230°;
теплота плавления 1,336 ккал/г-атом, теплота испа-
рения 61,4 ккал/г-атом. Коэфф, линейного расшире-
ния твердого Г. 1,8 • 10 5, жидкого: 12 • 10~5 (100°)
и 9,7 • 10 5 (900°). Давление пара Г. (в мм рт. ст.):
1,0 (1349°) и 400 (1974°). Уд. теплоемкость твердого Г.
0,09 кал/г-град (0—24°), жидкого 0,098 кал/г-град
(29—100°). Уд. электрич. сопротивление твердого Г.
53,4 • 10-в ом-см (0°), жидкого 27,2 • 10~в ом-см
(30°). Вязкость (пуаз): 1,612 (98°), 0,578 (1100°),
поверхностное натяжение 735 дин/см (при 30° в ат-
мосфере На или СОа); коэфф, отражения для длин
волн 4360 А и 5890 А соответственно равны 75,6%
и 71,3%.
В химич. отношении Г. весьма сходен с алюминием,
хотя по нек-рым свойствам отличается от него. При
обычной темп-ре на сухом воздухе Г. не окисляется.
Взаимодействие с кислородом начинается выше 260°,
образующаяся пленка окиси предохраняет металл от
дальнейшего окисления. Галогены (за исключением
иода) взаимодействуют с Г. уже на холоду. Воду
металл не разлагает. При нагревании Г. растворяется
в HNO3, H2SO4, НС1 и НС1О4 с образованием иона
Ga3+; медленно взаимодействует с раствором NaOH
с выделением водорода и образованием галлата натрия
NaGaO2. Одним из лучших растворителей металла
является царская водка.
Известны три окисла Г. GaaO3 — белый порошок, су-
ществует в двух модификациях; устойчивая при обыч-
ной темп-ре a-форма имеет, подобно а-А1аО3, ромбо-
эдрич. решетку, а=5,28 А, а = 55°35', плотн. 6,44,
т. пл. 1740 ± 25°. Получают GaaO3 обезвоживанием
гидроокиси GaaO3 • zH2O. Сильно прокаленная GaaO3
плохо растворяется в кислотах и основаниях. GaO —
вещество серого цвета, получается восстановлением
GaaO3 водородом; хорошо растворяется в кислотах.
При нагревании в вакууме смеси Ga2O3 с металлич. Г.
происходит образование темно-коричневого порошка
Ga2O. Темп-ра плавления этого окисла лежит выше
660°. При 650—700° GaaO сублимирует. Из водных
р-ров солей Г. при понижении концентрации водо-
родных ионов выпадает белый желатинообразный
осадок гидроокиси GaaO3 • хН2О. Гидроокись Г. —
электролит, очень близкий к идеальной амфотерности:
вторые и третьи константы ее диссоциации по кислот-
ному и основному типам таковы: H3Ga03K2=4,8-10-11,
ЙГ3=2 • 10-ia; Ga(OH)3 ЙГа=1,6 • 10““, #3=4 • 10“ia.
Ga(OH)3 растворима в кислотах, р-рах сильных осно-
ваний, а также в р-рах NH3 [в отличие от А1(ОН)3];
при кипячении из аммиачного р-ра вновь выпадает
гидроокись. Кислотные свойства Ga(OH)3 несколько
сильней выражены, чем у А1(ОН)3. Соответственно
этому интервал pH выделения Ga(OH)3 9,7—3,4, а
для А1(ОН)3 10,6—4,1.
Из солей Г. наибольшее значение имеют хлорид
GaCl3 и сульфат Gaa(SO4)3. Хлорид образует бесцвет-
ные гигроскопич. кристаллы, т. пл. 78°, т. кип. 200°;
хорошо экстрагируется эфиром из солянокислых р-ров.
При взаимодействии GaCl3 с Ga образуется хлорид
двухвалентного Г. GaCla — бесцветные кристаллы,
с т, пл. 170,5° и т. кип. 535°. Сульфат кристаллизуется
из сернокислых р-ров в виде Gaa(SO4k- 18Н2О. Прп
нагревании почти вся вода удаляется; безводная соль
выше 520° разлагается. С сульфатами щелочных ме-
таллов и аммония образует двойные соли типа квас-
цов, напр. ^NH4)Ga(SO4)2 • 12НаО. Г. образуетмало-
растворимыи в воде и разб. кислотах ферроцианид
777
ГАЛЛИЙ — ГАЛОГЕНАЛЬДЕГИДЫ И ГАЛОГЕНОКЕТОНЫ
778
Ga4[Fe(CN)e]3, что может быть использовано для его
отделения от А1 и ряда других элементов.
Г. легко сплавляется со многими металлами. Бинар-
ные системы, к-рые он образует с Al, Zn, In, Sn,
представляют собой простые эвтектич. сплавы; в би-
нарных системах с РЬ, Bi, Cd, Hg в жидком состоя-
нии появляются смешанные расплавы. С элементами
подгруппы мышьяка образует соединения типа
GaAs, GaSb, к-рые обладают свойствами полупровод-
ников. Известны также интерметаллич. фазы в дру-
гих бинарных системах Г., напр. в системах Ga—Ni,
Ga—Си, Ga—Fe.
Аналитическое определение г а л-
л и я. Качественно Г. открывают искровым спектраль-
ным методом, а также по образованию розового лака с
хинализарином или по флуоресценции раствора окси-
хинолината Г. в хлороформе (эти реакции исполь-
зуются также для фотометрии, определения Г.).
Количественно Г. определяют осаждением аммиаком,
купфероном, таннином, пиридином и др., позволяю-
щими также отделять Г. в соответствующих условиях
от щелочных, щелочноземельных и большинства дру-
гих элементов. На реакции галогенидов Г. с родами-
ном Б основан важный фотометрии, метод определе-
ния Г. в минералах (А1 и Fe111 не мешают). От мно-
гих элементов Г. можно отделить повторной экст-
ракцией GaCl3 эфиром из среды 5—6 н. НС1; от
А1 — осаждением газообразным хлористым водоро-
дом А1С13 6Н2О из р-ра, содержащего конц. HCI и
эфир (1 : 1).
Получение. Основным источником для получения Г.
служат отходы от переработки алюминиевого и цинко-
вого сырья. При переработке бокситов по способу
Байера или по способу спекания с содой и известью Г.
вместе с А1 переходит в раствор. Получение обога-
щенных Г. продуктов основано на различии pH выде-
ления А1(ОН)3 и Ga(OH)3. Для получения галлиевых
концентратов необходимо отделить Г. от основной
массы А1, не внося существенных изменений в техно-
логию переработки боксита. В процессе Байера Г.
концентрируется в маточных р-рах после осаждения
(выкручивания) 50—60% А1 в виде А1(ОН)3. Из таких
р-ров Г. выделяют электролизом на ртутном катоде,
затем катод обрабатывают р-ром соды или щелочи и
из полученного р-ра выделяют Г. электролитически.
При содово-известковом методе переработки бокситов
Г. концентрируется в последних фракциях осадков,
выделяемых в процессе карбонизации. Дополнительное
обогащение достигается обработкой осадка гидрооки-
сей строго дозированным количеством известкового
молока. При этом основная часть А1 в виде алюмината
кальция остается в осадке, а Г. переходит в р-р, из
к-рого выделяется при пропускании СО2. Получен-
ный галлиевый'концентрат,содержащий8—12% Ga2O3,
растворяют в щелочи, и выделяют из р-ра Г. электро-
литически.
Среди других методов отделения Г. от А1 следует
отметить осаждение его в виде ферроцианида и оса-
ждение купфероном или диэтилдитиокарбаматом нат-
рия. Высокая степень очистки Г. от А1 и других при-
месей достигается экстракцией его хлорида из соляно-
кислого р-ра этиловым эфиром, этилацетатом или
бутилацетатом. Кроме алюминатных р-ров, источни-
ком Г. в произ-ве А1 может служить остаточный анод-
ный сплав процесса электролитич. рафинирования А]
по методу трехслойного электролиза. Источниками Г.
в цинковом произ-ве являются возгоны окислов
(вельц-окислы), получаемые при переработке хвостов
выщелачивания цинковых огарков. Повторенйем ряда
операций — растворения вельц-окислов в серной к-те,
ступенчатой нейтрализации р-ров й обработки гидрат-
ных осадков щелочью — получают таллиевые кон-
центраты с содержанием 2—5% Ga2O3, из к-рых затем
извлекают Г. Полученный электролизом щелочного
р-ра жидкий металлич. Г., промытый водой и кисло-
тами (соляной, азотной), содержит 99,9—99,95% Г.
Для получения более чистого металла применяют
плавку в вакууме и фракционную кристаллизацию по
методу зонной плавки или «вытягивания» слитка из
расплава.
Применение. Широкого промышленного примене-
ния Г. пока не имеет. Вследствие низкой темп-ры пла-
вления и высокой темп-ры кипения металл предложено
использовать для изготовления высокотемпературных
термометров. Г. может заменять ртуть в диффузион-
ных вакуумных насосах, а также в выпрямителях.
Небольшие добавки Г. увеличивают коэфф, преломле-
ния стекла. Таллиевые оптич. зеркала отличаются
высокой отражательной способностью и устойчивы
при высоких темп-рах. Низкотемпературные сплавы
(т. пл. ниже 60°), к-рые образует Г. с рядом металлов
(Bi, Cd, РЬ, Zn, In, Т1), могут быть использованы
в сигнальных устройствах. Предложено использо-
вание Г. в качестве жидкого теплоносителя в энерге-
тич. ядерных реакторах. Однако препятствием служит
взаимодействие Г. с большинством металлов при высо-
ких темп-рах. В последнее время Г. нашел применение
в полупроводниковой электронике в качестве при-
садки к германию, а также в форме интерметаллич.
соединений, обладающих полупроводниковыми свой-
ствами (GaAs, GaSb). Токсич. действие Г. подобно
действию ртути и висмута.
Лит.: Меерсон Г. А., Зеликман А. Н Метал-
лургия редких металлов, М., 1955; Gmelfn, 8 Aufl.,
Syst.-Nr. 36, В., 1936; Elnecke Е., Das Gallium, Lpz.,
[1937]; M e 11 о г, у. 5, L.—N. Y., Toronto, 1952; U 1 1 m a n n,
3 Aufl., Bd 7, Munch.—B., 1956; Вагнер Г. иГитзен
В., Усп. хим., 1953, 22, вып. 1; Ере ми н Н. И., Изв.
высших учебных заведений. Цветная металлургия, 1960,
№ 1, с. 123; № 2, с. 108;' Bielfeldt К. und
Laspeyres М., Z. Erzbergbau und Metallhtlttenwesen,
1959, 12, H. 4, S. 173—78.
ГАЛЛОВАЯ КИСЛОТА (3,4,5-триоксибензойная
кислота) С7НвО5, мол. в. 170,11 — в обычных усло-
виях кристаллогидрат с 1 мол. воды, мол. в. 188,13 —
бесцветные или светло-коричневые СООН
игольчатые кристаллы, темнеющие Y и
на свету; нерастворима в хлорофор-
ме и бензоле; при 100—120° теряет I
воду; т. пл. 240° (с разл.); хорошо НО—ОН
растворима в кипящей воде, спирте, Д,
хуже — в эфире, плохо растворима в и
холодной воде; константа диссоциации К==3,9 • 10-5
(25°); рК=4,41. Щелочные соли Г. к. в присутствии
кислорода воздуха буреют. Насыщенный водный р-р
реактива с каплей р-ра FeCl3 образует сине-черное окра-
шивание. Встречается Г. к. в свободном виде в расте-
ниях, содержащих дубящие вещества; получают ее
щелочным или ферментативным гидролизом таннина.
Применяют Г. к. для синтеза красителей (антрагал-
лола, галлоцианина, галлофлавина и др.), для откры-
тия Sb111, Bi111, РЬ, Си, Mo, TI, Ag, Th, Ti, U, V, Y, Ce,
редкоземельных, щелочноземельных элементов и
NO^; для определения и отделения Bi111 от РЬ, Cd,
Си, Zn, Al, Ni, Fe, щелочных и щелочноземельных
металлов; для фотометрич. определения Се; в микро-
скопии; в качестве деполяризатора при электроана-
лизе; при сухой перегонке образуется пирогаллол.
Д. И. Кузнецов.
ГАЛОГЕНАЛЬДЕГИДЫ И ГАЛОГЕНОКЕТОНЫ —
соединения, молекулы к-рых содержат карбониль-
ную (альдегидную или кетонную) группу и атомы
галогена. Г. и г. вступают в реакции, свойственные
как галогеналкилам, так и карбонильным соеди-
нениям, напр. p-галогенкарбонильные соединения
легко превращаются в а, р-ненасыщенные карбониль-
ные соединения (1); галогенальдегиды окисляются
779 ГАЛОГЕНАНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ — ГАЛОГЕНАРСИНЫ 780
в галогенокислоты (2) и т. д.
С1СН2СНаСОС2Н5 gegsNtCHjh, СНа=снСОС2н5 (1)
СС13СНО £—2 СС13СООН (2)
Характерные реакции а-галогенокетонов — Дарзана
реакция и Фаворского перегруппировка.
Накопление атомов галогена в a-положении к кар-
бонильной группе приводит к увеличению ее электро-
фильной реакционной способности, поэтому, напр.,
хлораль или гексафторацетон легко реагируют даже
со слабонуклеофильными реагентами, напр. образуют
с водой устойчивые гидраты (3):
СС13СНОН2О —- СС13СН(ОН)2 1
(CF3)aCOН2О —* (CF3)2C(OH)2 j (3)
Кроме того, у них ослаблена связь между карбониль-
ным углеродом и атомом углерода, несущим атомы
галогена, вследствие чего при действии щелочей они
претерпевают галоформную реакцию (4):
СС13СНО + NaOH — СНС13 + HCOONa (4)
Важнейшими представителями галогенальдегидов
является хлораль СС13СНО; получают хлорированием
этилового спирта; промежуточный продукт в произ-
водстве хлороформа и инсектицида ДДТ. Г. и г.
получают окислением галогензамещенных спиртов
(5) или галогенированных олефинов (6), взаимодей-
ствием магнийорганич. соединений с галогенокисло-
тами (7); присоединением галогенов или галогено-
водородных к-т к ненасыщенным альдегидам или
кетонам (8); заменой оксигруппы на галоген в оксиаль-
дегидах или оксикетонах (9), галогенированием насы-
щенных альдегидов и кетонов (10):
С1СН2СН2ОН 2, С1СНаСНО (5)
(CF3)2C=CC12 -2- (CF3)2CO (6)
CF3C00H 2Ll32-gJ. CF3COCH3 (7)
CH2=CHCHO BrCH2CH2CHO (8)
CH3COCH2OH S2L CH3COCH2C1 (9)
CH3COCH3 -21s. C1CH2COCH3, C12CHCOCH3,
C1CH2COCH3C1 и т. д., (10)
присоединением хлорноватистой или бромноватистой
к-ты к гомологам ацетилена (11) или к винилгалоге-
нидам (12):
СН3СН2С=ССНз —221 СН3СН2СНС1СОСН3 (И)
сна=снс1 2221 [cicch2<(oh j асн2сно (12)
омылением соответствующих галогензамещенных аце-
талей, к-рые образуются при галогенировании ацета-
лей (13):
снзсн(ос2н6)2-21s с12снсн(ос2н6)2 2^21
— С12СНСНО, (13)
а также присоединением галогенов к простым или
сложным виниловым эфирам с последующей обработ-
кой спиртом (14):
СН2=СНОСОСН3 21Х ВгСН2СНВгОСОСН3 —
2222Ц BrCH2CH(OC2H6)2 -Ha-s°4> ВгСН2СНО (14)
Используются Г. и г. для синтеза различных гетеро-
циклических соединений, производных диарилметана
И др. Е. М. Рохлин.
ГАЛОГЕНАНГИДРИДЫ КАРБОНОВЫХ КИС-
ЛОТ — производные карбоновых к-т, представляю-
щие собой продукты замены гидроксила карбоксиль-
ной группы галогеном RCOX (где X — галоген).
Названия Г. к. к. образуются от названий соответ-
ствующих ацилов путем добавления к ним прилага-
тельных, напр. хлористый ацетил, либо окончаний
(хлорид, бромид ит. Д.), напр. ацетилхлорид. Г. к. к,—
жидкости (низшие) или твердые вещества с резким
запахом; низшие из них дымят на воздухе. Физич.
свойства важнейших Г. к. к. приведены в таблице:
ОН или галоген Констан- ты О СН3С^ сан6с^° С1?Нз5С^ "О C«HSC^
он Т. пл., °C 16,6 -22 69,4 122
Т. кип., °C 118,1 141,1 383 249
d™ 1,49 0,992 0,847 (69°) 1,266 (d*g)
F Т. пл., °C -60 — —
Т. кип., °C d*« 20,5 44 — 159
0,993 0,972 (15°) — —
С1 Т. пл., °C -112 -94 23 —1
Т. кип., °C 51-52 80 215/15 мм 197
1,105 1,065 — 1,219
Вг Т. пл., °C —96,5
Т. кип., °C 76,7 103,5 — 218—219
d’° 1,663 1,521 — 1,570
J Т. кип., °C 104—106 127 135/25 мм
Атом галогена в Г. к. к. очень подвижен. Низшие
Г. к. к. реагируют более активно, чем высшие, али-
фатические — активнее, чем ароматические. Большин-
ство реакций Г. к. к. связано с обменом атома гало-
гена другими группами: RCOX + НА -> RCOA+ НХ,
где A=H,OH,OR, NH2, NHR, NHR2, SH, SR, CN,
Аг и др. При этом в молекулу вещества НА вступает
ацил RCO, поэтому такие реакции наз. реакциями
ацилирования.
Г. к. к. способны присоединяться по кратным свя-
зям:
RCOX + НаС=СН2 — RCO(CH2)2X
RCOX + НС=СН — RCOCH=CHX
реагировать c CH2N2 (Арндта — Эйстерта реакция)
и др. Гомология, ряд Г. к. к. начинается с галоген-
ангидридов уксусной к-ты, т. к. галогенангидриды
муравьиной к-ты (за исключением HCOF, т. кип. 100,7°,
df01,220) разлагаются в момент образования. Г. к. к.
получают взаимодействием галогенангидридов неор-
ганич. кислот (РХ3, РХ5, SOX2 и др.) с карбоновыми
к-тами. Наиболее удобным способом получения хлор-
ангидридов к-т является действие на к-ты тионил-
хлорида. В последнее время установлено, что этот
метод в присутствии небольшого количества хлори-
стого калия пригоден и длн получения галогенангид-
ридов довольно сильных к-т (трихлоруксусной, пер-
фтордикарбоновых и т. п.). Г. к. к. (хлорангидриды)
применяются в пром-сти как ацилирующие агенты.
а. Е. Васильев.
ГАЛОГЕНАРСИНЫ — органич. производные га-
логенидов мышьяка, у к-рых один или два галогена
замещены органич. радикалами; их следует рассмат-
ривать как галогенангидриды соответствующих алкил-
или арилмышьяковистых к-т. Известны хлор-, бром-,
иод- и фторорганич. производные мышьяка; наиболь-
шее значение имеют хлорпроизводные, к-рые делятся
на первичные RAsC12 и вторичные R2AsC1. Первич-
ные — подвижные жидкости с резким запахом; вто-
ричные: жирного ряда — маслянистые жидкости с рез-
ким раздражающим запахом, ароматич. ряда — кри-
сталлич. продукты. Все Г. плохо растворимы в воде
и хорошо — в органич. растворителях.
Формула T. кип.,°C d|° 20 nD
CHgAsCh 132,5 1,836 1,5677
155,3 1,66 1,5588
CeH5AsCb 257,0 1,63
(CH3)2AsC1 106,5 1,50
(CeH6)2AsCl • 333,0 1,39 1,6332
• Т. ПЛ. 38,8°.
781
ГАЛОГЕНАЦЕТОНЫ — ГАЛОГЕНОВ ОПРЕДЕЛЕНИЕ
782
Г. сравнительно быстро гидролизуются водой с об-
разованием соответствующих оксидов; скорость гид-
ролиза возрастает в присутствии щелочей. При взаимо-
действии с сероводородом или его солями образуются
нерастворимые в воде сульфиды. Первичные Г. легко
вступают во взаимодействие с органич. соединениями,
содержащими меркаптогруппы, что используется при
терапии поражений первичными Г. (см. Антидоты).
Г. легко восстанавливаются водородом с образованием
соответствующих арсинов. При взаимодействии пер-
вичных и вторичных Г. с хлором образуются алкил-
или ариларсентетрахлориды или трихлориды, раз-
лагающиеся водой до соответствующих мышьяковых
к-т. Непредельные мышьяксодержащие соединения
жирного ряда обладают в основном аналогичными
свойствами (см. Люизит).
Г. жирного ]эяда получают алкилированием солей
мышьяковистой к-ты (Мейера реакция): AsONa +
+ RX —> RAsO(ONa)2 + NaX с последующим восста-
новлением алкилмышьяковых к-т в солянокислой
среде до Г.:
RAsO(ONa)2 + SO2 4. 2НС1 — RAsCl2 4. H2SO4 -j- H2O
Г. ароматич. ряда могут быть получены в результате
взаимодействия AsCl3 с диарилртутью или арилмер-
кургалогенидами. Современными способами получе-
ния Г. ароматич. ряда являются Варта реакция
и Несмеянова реакция.
Первичные Г. жирного ряда обладают раздражаю-
щим, общеядовитым и кожно-нарывным действием.
Для Г. ароматич. ряда наиболее типично раздражаю-
щее действие на верхние дыхательные пути; непере-
носимые концентрации при экспозиции в 1 мин.:
0,010 мг)л CaHBAsCla, 0,015 мг/л CeH5AsCla, 0,001 мг[л
(CeHg)aAsGl.
Этилдихлорарсин, фенилдихлорарсин и дифенил-
хлорарсин применялись во время первой мировой
войны в качестве отравляющих веществ.
Лит.: Фрейдлина Р. X., Синтетические методы
в области металлорганических соединений мышьяка, М.—Л.,
1945; С о б о р овский Л. 3., Эпштейн Г. Ю., Химия
и технология боевых химических веществ, М.—Л., 1938.
Р. Н. Стерлин.
ГАЛОГЕНАЦЕТОНЫ — галогензамещенные аце-
тона; большей частью бесцветные или слегка окрашен-
ные жидкости, плохо растворимые в воде, хорошо —
в органич. растворителях. Пары С1СНаСОСН3 (т. кип.
119°, d|3 1,164), ВгСН2СОСН3 (т. пл. минус 54°,
т. кип. 138°, dj4 1,634) и 1СН2СОСН3(т.кип. 58,4/11 мм,
d(5 2,170) обладают сильным лакримогенным Дей-
ствием; FCH2COCH3 (т. кип. 72,5°, <7|0 0,967) раздра-
жающим действием не обладает. Эти соединения
вступают в реакции, характерные для замещенных
кетонов и галогеналкилов; исключение составляет
FCHaCOCH3, не вступающий в реакции, характерные
для галогеналкилов. Симметричные дигалогензаме-
щенные ацетона С1СНаСОСНаС1 (т. пл. 45°, т. кип.
173—173,5°, d^ 1,3876) и др. имеют более высокую
темп-ру кипения, большую плотность и более ток-
сичны, чем несимметричные; кроме лакримогенного
действия, эти соединения действуют также на кожу.
Дальнейшее введение галогена ведет к уменьшению
токсич. свойств этих производных. Моногалогенза-
мещенные ацетона получают непосредственным гало-
генированием ацетона. Дигалогензамещенные удобнее
получать окислением дигалогенгидрина глицерина.
Г. широко используются в оргапич. синтезе. Хлор-
и бромацетон применялись в качестве ОВ во время
первой мировой ВОЙНЫ. Р. И. Стерлин.
ГАЛОГЕНИРОВАНИЕ ОРГАНИЧЕСКИХ СО-
ЕДИНЕНИЙ — процесс прямого замещения атомов
водорода в органич. соединениях атомами галогенов
(F, 01, Вг). Присоединение галогенов к кратным свя-
зям с образованием вицинальных ди- и тетрагалогени-
дов также может рассматриваться как Г. о. с. Не-
редко термину Г. о. с. придают более широкий смысл—
создание свизи углерод—галоген любым образом, вклю-
чая такие процессы, как замещение ОН-группы хло-
ром, замена хлора фтором и др. (см. Галогенозаме-
щенные углеводороды). Я. Ф. Комиссаров.
ГАЛОГЕНОВ ОПРЕДЕЛЕНИЕ — качественное и
колич. определение F, С1, Вг и J, входящих в состав
различных соединений; проводит обычно после пере-
ведения анализируемого материала в раствор, в к-ром
определяют галогены в виде ионов. Нерастворимые
в воде вещества, содержащие галогены (С1, Вг, J),
разлагают либо кислотами (азотной, хлорной), либо
сплавлением с карбонатами калия или натрия. Для
разложения соединений фтора и отделения фтор ид-
иона от веществ, мешающих его определению, при-
меняют либо обработку серной или хлорной к-тами
в присутствии SiOa (кварц, стекло) с последующей
отгонкой фтора в виде кремнефтористоводородной
к-ты, либо разложение водяным паром при темп-ре
ок. 1000°, сопровождающееся выделением HF (пиро-
гидролиз). Последний способ пригоден для фторидов
урана, тория, алюминия, висмута, цинка и др. метал-
лов. Для нерастворимых фторидов и силикатов при-
меняется сплавление с карбонатами калия или натрия
и экстракция водой (о разрушении органич. веществ,
содержащих галогены, см. ниже).
Хлор, бром и иод качественно определяют после переведе-
ния анализируемого вещества в раствор в виде галогенида
серебра, по появлению характерной мути, а затем хлопьев
и осадка, при приливании к р-ру галогенида р-ра AgNO3.
Хлор может быть открыт без растворения галогенида в во-
де, по выделению газообразного НС1 (запах, покраснение влаж-
ной синей лакмусовой бумаги) при действии на хлорсодержа-
щее вещество конц. H2SO4. Содержание хлорид-иона в растворе
можно обнаружить по выделению газообразного хлора (запах,
посинение иодкрахмальной бумаги) при нагревании с конц.
H2SO4 и МцО2.
Свободный бром открывают по красно-фиолетовой
окраске раствора фуксина, предварительно обесцвеченного
NaHSO3, припиваемого к раствору анализируемого вещества,
или по желтому до красного окрашиванию слоя органич.
растворителя (бензола, хлороформа и др.), добавляемого
к р-ру бромида, вследствие извлечения растворителем брома
из водного р-ра. При действии конц. серной к-ты на твердые
бромиды выделяется газообразный НВг, окрашенный в буро-
ватый цвет, вследствие частичного окисления до свободного
брома.
И о д в р-рах иодидов можно открыть действием хлорной
воды или р-ра гипохлорита натрия. Выделяющийся свобод-
ный иод окрашивает в фиолетовый цвет прибавленный бензол,
располагающийся слоем над поверхностью р-ра иодида. Ни-
трит калия (натрия) в присутствии H2S04 окисляет иодид-
ионы до свободного иода, к-рый можно открыть по синему
окрашиванию р-ра крахмала.
Количественно С1~, Вг-, J- определяют весовыми метода-
ми (в виде галогенида серебра) или титриметрически с ис-
пользованием нитрата серебра (по Фольгарду, Мору, Фаян-
су — см. Аргентометрия) или азотнокислой (хлорнокислой)
окиси ртути (меркурометрия). Хлорид- и бромид-ионы можно
титровать основным цианидом ртути; определение заканчи-
вают ацидиметрически. Для определения иодид-иона приме-
няется окисление его в иодат с последующим выделением сво-
бодного иода, к-рый титруют тиосульфатом с крахмалом в ка-
честве индикатора (см. Иодометрия). Хлорид- и бромид-
ионы, находящиеся в смеси, определяют потенциометрия,
титрованием; бромид-ион в присутствии хлоридов — иодо-
метрически, после окисления до BrOjT. Известны многочис-
ленные потенциометрия., амперометрия, и др. электрометрия,
способы определения С1~, Вг- и J как общие, так и для ана-
лиза смесей.
Качественное определение фтора основано: 1) на обна-
ружении фторид-иона в растворе любой из реакций, приме-
няемых для его колич. определения. Наиболее распространено
открытие по обесцвечиванию окрашенных комплексов, обра-
зованных металлами (торием, цирконием и др.) с красителями
(ализаринсульфокислым натрием и др. веществами); 2) на
выделении HF при действии H2SO4 на фториды; при этом про-
исходит помутнение стекла вследствие травления HF; 3) на
выделении и гидролизе четырехфтористого кремния при взаи-
модействии HF с SiO2 (метод висячей капли). Колич. опреде-
ление фтора выполняется как весовыми методами (в виде
PbFCl, CaF2, LaF3 и др.), так и титриметрически. Известны
многочисленные варианты потенциометрии., кондуктометрия.,
амперометрия., высокочастотного, фотометрия., люминесцент-
ного определения фтора. Наиболее употребительно титрова-
ние фторид-иона нитратом тория или циркония с применением
ализаринсульфоната иатрия и других веществ в качестве
индикатора. На связывании фтора с помощью солей Fe, Al,
783
ГАЛОГЕНОВ ОПРЕДЕЛЕНИЕ—ГАЛОГЕНОЗАМЕЩЕННЫЕ УГЛЕВОДОРОДЫ
784
Т1, Се, Y основаны многочисленные методы его Определения.
Известно непрямое определение фтора в виде PbFCl арген-
тометрия. титрованием хлора после отделения и растворения
осадка.
Качественное и количественное
определение галогенов в органи-
ческих соединениях основано на окисли-
тельном или восстановительном разрушении органич.
вещества. Независимо от характера разложения гало-
гены выделяются в виде галогеноводородов или гало-
генидов металлов, в к-рых галоген открывают каче-
ственно или определяют количественно описанными
выше методами неорганич. химии. Во многих случаях
для быстрого разрушения применяют специфич.
экспресс-методы, не обладающие универсальностью.
Хлор, бром и иод в органич. соединениях часто откры-
вают пробой Вейльштейна: при внесении
вещества, помещенного на прокаленную медную про-
волоку, в окислительное пламя газовой горелки;
в присутствии галогена появляется зеленое окраши-
вание пламени. Подобное окрашивание дают также
нек-рые производные хинолина, пиридина, мочевины,
ие содержащие галогенов.
Легкоподвижный галоген в веществе может быть
определен или непосредственно или после омыления
вещества. Для разрушения фторорганич. соединений,
вследствие их специфич. свойств, часто применяют
специальные методы.
Основные способы разрушения органич. соединений
следующие: 1. Сожжение в токе кислорода в кварце-
вой трубке в присутствии платинового катализатора
(Прегль) с последующим весовым или титриметрич.
определением галоген-иона в поглотительном р-ре.
При этом требуется дополнительное восстановление
образующихся кислородных соединений галогенов.
Для фторорганич. соединений сожжение проводят
в платиновой трубке, иногда в присутствии влаги, или
в кварцевой трубке с двуокисью кремния или окисью
магния. При сожжении в кислороде возможно погло-
щение галогенов (Cl, Br, J) металлич. серебром и одно-
временное весовое определение галогена, углерода
и водорода (Коршун). 2. Восстановление металлич.
калием (натрием) в металлич. бомбе или запаянной
стеклянной ампуле при нагревании является Одним из
эффективнейших способов разложения, пригодным
для определения всех галогенов. Продолжительность
разложения — несколько минут. Возможно одновре-
менное определение нескольких галогенов титрова-
нием проб, взятых из одного и того же раствора точно
установленного объема.
3. Сожжение в кис лсроднс-вс дородном пламени — эффек-
тивный и быстрый способ разложения; применим гл. обр. для
анализа жидких и газообразных в-в, особенно для фторорга-
нич. соединений, в том числе сполна галогенированных.
4. Сожжение в колбе с пришлифованной пробкой, наполнен-
ной кислородом, в присутствии платины и поглотительного
раствора (вода, щелочь). Быстрый (несколько секунд) способ
разрушения, пригодный для определения всех галогенов,
гл. обр. в твердых нелетучих соединениях. Метод имеет огра-
ниченное применение; разработан в полумикро- и микро-
масштабе. 5. Окисление нагреванием в металлич. бомбе с пере-
кисью натрия и различными добавками (селитра, глюкоза
И др.); разложение длится неск. минут. В р-ре плава можно
определять все галогены. К недостаткам метода относятся
большое количество реагентов, необходимое для разложения,
и высокая концентрация солей в р-ре плава. 6. Окисление
мокрым путем, нагреванием вещества в запаянных стеклянных
трубках с конц. HNOS при 300° для определения хлора, брома
и иода с последующим весовым или титриметрич. окончанием
анализа. Разложение продолжается неск. часов, неприменимо
к полигалогенным соединениям; известны макро- (Кариус),
микро- (Прегль) и полумикрометоды. Эти методы выходят
из употребления как трудоемкие, длительные и неуниверсаль-
ные (см. Кариуса метод). 7. Восстановление металлами (Na,
Са, Li и др.) в органич. растворителях (Степанов); применимо
к нелетучим веществам для определения хлора, брома, иода.
8. Восстановление в токе водорода в присутствии катализа-
тора (Тер-Мейлен) с последующим титрованием галогеноводо-
родных кислот; применяется при определении хлора, брома,
иода. Известны также многочисленные специальные методы
определения галогенов в разнообразных объектах, содер-
жащих органически связанный галоген.
Лит.: Гиллебранд В. Ф. [и др.]. Практическое ру-
ководство по неорганическому анализу, пер. с англ., М.,
1957; Фтор и его соединения, пер. с англ., т. 2, М., 1956;
Houb е n-W eyl, Bd 8, 4 Autl., Stuttgart, 1952; Коршун
М. О. и Г е л ь м а н И. Э., Новые методы элементарного
микроанализа, М.—Л., 1949; Ни дер ль Дж. н Ни-
fl е р л ь В., Микрометоды количественного органического
анализа, пер. Го англ.], М., 1949; О g g С. L., Analyt. Chem.,
1956, 28, р. 768; Pregl-Roth, Quantitative organische
Mlkroanalyse, 7 Autl., W., 1958; Ч e p о н и с Н., Микро-
и полумикрометоды органической химии, [пер. с англ.], М.,
I960.
ГАЛОГЕНОЗАМЕЩЕННЫЕ УГЛЕВОДОРОДЫ —
большой класс органич. соединений, содержащих
атомы галогенов (F, Cl, Вг, J), связанные с углеводо-
родными радикалами различного типа. Примерами
Г. у. жирного, алициклич. и ароматич. рядов могут
служить соответственно хлористый метил СН3С1,
хлорциклогексан С6НИС1 и бромбензол С6Н5Вг. В за-
висимости от характера радикала, с к-рым связан
атом галогена, различают первичные, вторичные и
третичные Г. у. (X — атом галогена):
rch2x r2chx r3cx
Первичный Вторичный Третичный
Простейшим представителем вторичных Г. у. яв-
ляется изопропилгалогенид (СН3)аСНХ, третичных
Г. у. — znpezn-бутилгалогенид (СН3)3СХ.
Число возможных изомеров Г. у., начиная с моно-
галогенопроизводных жирного ряда, намного превы-
шает число изомерных соответствующих углеводоро-
дов. Так, от пропана можно произвести 2 моногало-
генида — С3Н7Х и иао-С3Н,Х. При наличии двух изо-
мерных углеводородов С4Н10 существуют 4 изомерных
моногалогенида — СН3СНаСНаСНаХ, (СН3)аСНСНаХ,
(СН3)3СХ и СН3СНаСНХСН3; последний содержит
асимметрии, атом С и поэтому известен в виде двух
оптич. антиподов.
Название Г. у. жирного ряда обычно производится
от соответствующих углеводородных радикалов с при-
бавлением слова «хлористый», «бромистый» и т. д.
По женевской номенклатуре, название Г. у. произво-
дится от названия углеводорода с указанием места
замещения галогеном: СН3СНС1СН8 (хлористый изо-
пропил — 2-хлорпропан); СН8СС1аСН3 (хлористый
изопропилиден — 2,2-дихлорпропан). Этот же способ
обозначения принят и для Г. у. ароматич. ряда, папр.
я-дихлорбензол, а-бромнафталин. Для многих Г. у.
приняты условные тривиальные обозначения, напр.
хлороформ СНС13, хлоропрен СН2=СС1СН=СНа.
Последовательное замещение атомов водорода в
СН3-группе атомами галогена ведет последова-
тельно к соединениям RCHaX, RCHXa и RCX3, что
соответствует постепенному возрастанию степени окис-
ленности исходного углеводорода: действительно, ги-
дролиз моногалогенопроизводных ведет к спиртам,
дигалогенопроизводных — к альдегидам или кетонам
и тригалогенопроизводных —к кислотам: СН3СНаХ —>
-> СН8СНаОН; СН3СХаСН3 -> СН3СОСН3; СН3СХ8 ->
->СН3СООН.
Энергия связей С-галоген падает с увеличением
атомного веса галогена и равна (в ккал): С—F ~ 104;
С—С1 ~ 70; С—Вг ~ 57 и С— J ~ 43.
По способам получения и свойствам фториды заметно
отличаются от остальных галогенидов. Нек-рыми
особенностями обладают и иодистые соединения — лег-
кое отщепление иода от вицинальных дииодидов,
напр. JCHaCHaJ, существование прочных соединений
типа CeH5JO, C6H5JOa, [(CeH5)aJ+JC1- (см. Галогено-
ниееые соединения), что объясняется сравнительно
меньшей электроотрицательностью атома иода.
Способы получения Г. у. В зависимости от типа
углеводорода, природы галогена или применяемого
галогенирующего агента, способы получения Г. у.
отличаются большим разнообразием.
1. Прямое галогенирование (заме-
щение атомов водорода галогеном). В качестве агентов
785
ГАЛОГЕНОЗАМЕЩЕННЫЕ УГЛЕВОДОРОДЫ
788
галогенирования применяются свободные галогены,
смешанные галогены, напр. ВгС1, генераторы свобод-
ных галогенов (SOaCla, CoF3 и др.), вещества, содер-
жащие положительно заряженный атом галогена,
напр. гипохлориты, N-бромсукцинимид и др. Реакция
СН4 с С1а протекает очень бурно и обычно ведет к смеси
моно- и полигалогенидов. Высшие парафины хлори-
руются более спокойно. Галогенирование соедине-
ний жирного ряда представляет собой типичный
радикально-цепной процесс
Ijv
Cig —* Cl* -4- Cl*
СГ -4- СН4 -* СНз* -4- НС1 1
> цепь
СН3- 4- С12 — СН3С1 -4- СГ J
Обрыв реакционной цепи, включающей до 1000 актов,
происходит в результате рекомбинации атомов хлора:
СГ + СГ —> С1а или вследствие реакции между ато-
мом С1 иметильным радикалом: СГ + СН3 —>СН3С1.
Металлоорганич. соединения, способные в условиях
реакции распадаться на свободные алкильные ради-
калы, сильно ускоряют процесс галогенирования.
Так, в присутствии тетраэтилсвинца можно хлориро-
вать пропилен при 0°. Фотохимия, бромирование
протекает значительно медленнее, чем хлорирование.
Фотохимия, иодирование не удается осуществить.
Для хлорирования можно также применять хлори-
стый сульфурил в присутствии перекисей как источ-
ников свободных радикалов:
RCOOOCOR — R’ + COS + RCOO’
R’ + SO2C12 — RC1 + so2cr
so2cr — SOs + СГ
RH + Cl’ — R’ + HCI
R’ + SO3C12 — RC1 + SO»C1-
Радикально-цепной механизм галогенирования со-
единений жирного ряда подтверждается кинетич. дан-
ными, эффективностью УФ-облучения или введе-
нием перекисей, а также тем, что хлорирование со-
единении жирного ряда с атомом Н у асимметрия,
атома углерода сопровождается рацемизацией. Так,
при хлорировании активного СаН5С*Н(СН3)СНаС1 об-
разуется СаН5СС1(СН3)СН2С1, лишенный оптич. актив-
ности.
Хлорирование (бромирование) ароматич. углеводо-
родов протекает по ионному механизму с участием
галоген-катиона: С1+ + СвНв —>С6Н5С1 + Н+. Такой
вывод подтверждается тем, что при применении в ка-
честве галогенирующего средства йодистого хлора
с дипольным моментом, указывающим на строение
J8+C15 , всегда образуются иодистые, а не хлористые
соединения. Далее хлорирование ароматич. соедине-
ний катализируется кислотными агентами типа BF3,
А1С13, FeCl3, роль, к-рых заключается в облегчении
гетеролитич. расщепления молекулы галогена с об-
разованием активного катиона: С1—С1 —>С1+ +
+ [A1C1J-. Однако при высокой темп-ре возможно
галогенирование ароматич. соединений и по радикаль-
ному механизму. Так, бромирование бромбензола
при 450—500° ведет к преимущественному образова-
нию л«-дибромбензола, тогда как обычно в соответ-
ствии с правилами ориентации образуется смесь
о- и и-дибромбензола. Разница в механизмах галогени-
рования соединений жирного и ароматич. рядов ведет
к тому, что галогенирование жирно-ароматич. соеди-
нений можно прибавлением FeCl3 направить в ядро,
тогда как УФ-облучение и повышенная темп-ра
способствуют галогенированию боковых цепей.
Фтор как самый электроотрицательный элемент
неспособен переходить в состояние F+; поэтому
фторирование всегда представляет собой радикалыю-
цепнои процесс. Фтор даже при разбавлении азотом
обычно чрезвычайно бурно реагирует с углеводоро-
дами. и часто с разрывом С—С-связей. Поэтому для
прямого фторирования широко применяются такие
генераторы фтора, как CoF3, в меньшей степени
PbF4. В последнее время для получения полифтори-
рованных соединении разработан новый метод, за-
ключающийся в электролизе раствора фторируемого
соединения в HF; при этом F~ окисляется до F+,
тогда как Н+ восстанавливается. В последние годы
в качестве бромирующего агента широкое приме-
нение приобрел N-бромсукцинимид (см. Воля —
Циглера реакция}. Обесцвечивание разб. раствора
брома может служить качественной реакцией на двой-
ную или тройную связь. Хотя для олефинов харак-
терно присоединение галогенов к двойной связи,
при высокой темп-ре удается осуществить замещение
атомов водорода с сохранением кратной связи, напр.
СН3СН=СН2 + С12 360~40Q°. С1СН2СН=СН2.
2. Присоединение галогенов к
кратным связям. Одни из основных способов
получения вицинальных галогенидов состоит в при-
соединении галогенов к олефинам и ацетиленовым
углеводородам:
СН2=СН2 + С1 — С1СН2СН2С1
НС=СН + 2С1а —. СНС12СНС1а
Хлорирование бензола при нагревании и УФ-облуче-
нии ведет к присоединению 3 молей хлора с образова-
нием смеси изомерных гексахлорциклогексанов.
К олефинам можно присоединить и хлорноватистую
к-ту (р-р С1а в воде) с образованием хлоргидринов:
сн3=сн2 + нею - НОСН2СН2С1
3. Присоединение галогеноводо-
родов к кратным связям. Легче всего
присоединяется HJ, труднее НС1 и HF. В случае
несимметричных олефинов в соответствии с Марков-
никова правилом атом водорода присоединяется к наи-
более «гидрогенизированному» атому С с образова-
нием вторичных и третичных алкилгалогенидов,
НаПР" СН3СН=СН2 + HJ — СНзСН.ТСНз
(СНа)2С=СН2 + НС1 — (СНз)зСС!
В случае НВг порядок присоединения зависит от
условий реакции. В отсутствии перекисей соблюдается
правило Марковникова. При наличии же перекисей
в результате раднкально-цепной реакции получают-
ся «аномальные» продукты, напр. СН,,СН.,СН,Вг из
пропилена и НВг.
4. Замещение ОН-г рупп на галоген.
Один нз наиболее общих методов получения Г. у.
заключается в замещении спиртовых групп галоге-
ном. В особенности легко осуществляется переход
от третичных спиртов к mjsem-галогенидам. Так, при
обработке (СН3)3СОН конц. НС1 на холоду образуется
(СН3)3СС1. В случае вторичных и в особенности пер-
вичных спиртов обычно требуется нагревание смеси
с НХ. В ряде случаев замещение облегчается при
применении в качестве катализатора ZnCla. Замеще-
ние ОН-групп на галоген удобно осуществлять в без-
водной среде действием галогенангидридов минераль-
ных кислот (PBr3, РОС13, SOCla и др.), лучше всего
в присутствии пиридина:
ROH + SOC12 — RC1 + SO3 + HCI
5. Обмен галогенов в Г. у. Метод приме-
няется преим. для получения фтористых и иодистых
соединений. Так, при взаимодействии хлоридов с NaJ
в ацетоне выпадает осадок NaCl и гладко образуется
соответствующий иодид (см. Финкельштейна реак-
ция}. Нагревание хлоридов, в особенности полихло-
ридов, с SbF3 (с добавкой Вга или SbCl5) ведет к по-
лучению фтористых соединений, напр. СвН5СС13 +
+ SbF3 —>CeH5GF3 + SbCl3.
6. Замена кисл’орода в карбониль-
ных соединениях на галогены. При
787
ГАЛОГЕНОЗАМЕЩЕННЫЕ УГЛЕВОДОРОДЫ
788
реакции альдегидов или кетонов с РС15 образуются
геминальные дигалогениды
(СН3)2СНСНО + РС15 — (СН3)2СНСНС12 + РОС18
СН3СОСН3 + РС15 — СН3СС12СНз + РОС1з
7. Замена аминогруппы в арома-
тическом ядре на галоген. Ароматич.
амины после предварительного диазотирования можно
превратить в галогениды. Так,от ArN2X можно перейти
к АгХ действием Си2С12 (Зандмейера реакция) или Си
(Гаттермана реакция). По этому же методу с при-
менением твердых борфторидов диазония ArN2BF4
можно ввести F в ароматич. ядро вместо NH2-rpynnr.i
(см. Шиманна реакция).
8. Присоединение СНС13 и СС14 к оле-
фин а м. Нек-рые полигалогенсоединения можно по-
лучить присоединением СНС13 или СС14 к хлоролефи-
нам в присутствии А1С13 (Принса реакция), напр.
СС12=СС12 + СНС13 -> СНС12СС12СС13. В присутствии
перекисей к СС14 присоединяется этилен; при этом
в результате радикально-цепной реакции образуются
вещества общей ф-лы С1(СН2СН2)ЯСС13 (см. Теломери-
аация).
Известны и другие частные способы получения
Г. у., напр. алкилирование солей галогеноводород-
ных кислот, хлорметилирование ароматич. соедине-
ний действием СН2О и соляной к-ты в присутствии
ZnCl2 и др.
Физические и химические свойства Г. у. Физич.
свойства моногалогенозамещенных углеводородов ука-
заны в таблице.
X х R СНз С2Н5 н-С3Н? Н-С4Н9 mpem- С4Н1) С8Н5СН2
F Т. кип. -78" -32° -3° 432’ 413’ 140°
Т. кип. —24е 4-13° 46° 78° 51’ 179’
С1| Т. пл. -97° —139 -123° —123° —29° —39®
d — 0,917 0,892 0,897 0,847 1,103
Т. кип. 4-4.6° 38° 71’ 100° 73° 198-190’
Вг1 Т. пл. -93° -119° -110° -112’ -20° -4°
d 1,732 1,459 1,354 1,283 1,189 1,443
Т. кип. 42° 72® 102° 131’ 98—99° 93®/11 мм
J | Т. пл. -66° -111° -101’ -103° -34° 4-24°
d 2,279 1,933 1,614 1,614 1,571 1,733
Г. у. легко вступают в различные реакции и по-
этому могут быть использованы для получения много-
образных веществ. Для Г. у. характерны реакции
нуклеофильного замещения. Так, они реагируют
с водой с образованием спиртов, с NH3 дают амины,
с алкоголятами — эфиры, с солями карбоновых
кислот— сложные эфиры, с NaCN—нитрилы и т. д.
При действии концентрированных р-ров щелочей
на Г. у. отщепляется НХ и образуются олефины,
напр.: СН3СН2С1 ->СН2=СН2 + HCI.
Большое значение имеют реакции Г. у. с металлами.
Так, при действии Na на моногалогениды образуется
связь между углеводородными радикалами (см. Вюр-
ца реакция). Аналогичным образом действует Na (см.
Фиттига реакция) на арилгалогениды с образованием
соответствующих бифенильных соединений.
При действии Zn в спиртовой среде на ди- и поли-
галогениды происходит дегалогенирование с обра-
зованием соединений с двойной или тройной связью,
напр.:
RCHBrCH2Br 4 Zn — RCH=CH2 4. ZnBr2
RCCI2CHC12 4- 2Zn — RC^CH -f- 2ZnCl2
В неполярных растворителях Zn реагирует с Г. у.
с образованием цинкорганич. соединений:
2RX 4 2ZH —*• R2Zn 4- ZnX2
При действии Mg на Г. у. в эфире или тетрагидро-
фуране образуются смешанные магнийорганич.
соединения: RX + Mg —> RMgX, к-рые находят ши-
рокое применение для различных синтезов. В качестве
промежуточных продуктов различных синтезов при-
обрели большое значение литийорганич. соединения
(напр., C4HeLi, CeH5Li), к-рые гладко образуются
при реакции Г. у. с литием в эфире: RX + 2Li —►
-> RLi + LiX.
Г. у. обладают алкилирующим действием. Так,
при взаимодействии Г. у. с соединениями с «актив-
ными» метиленовыми группами, реагирующими в виде
карбанионов, происходит замещение атома водорода
алкильным радикалом, напр.:
СН2(СООС2Н5)2 4- C2H5ONa — Na+ [СН(СООС2Н5)2]-
—► RCH(COOC2H5)2 4- NaX
Г. у. в присутствии кислотных катализаторов (А1С13,
BF3) могут алкилировать ароматич. углеводороды
(см. Фриделя — Крафтса реакция).
Разнообразная реакционная способность Г. у. на
примере СН3С1 наглядно показана на схеме:
СН3Й
CH3NO2
CH3SR
CHgSH
CHgMgCl
ch3nh2
CH3F
_ + NaR +Nal
_+NaNOa \ / +NaOH
4-NaSR \\ // +RONa _
i +NaSH „.. +CH2COONa
_ + Mg ,x\ +NaCN
+nh3 / \\ +C6H6 __
_ +AgF / \ .
CH3J
CH3OH
CH3OR
CH3COOR
CH3CN
С6н5сн3
CH3LI
Атом галогена у углерода с двойной связью (в том
числе и в арилгалогенидах) связан очень прочно
и с трудом вступает в обычные реакции галогенидов.
Так, гидролиз и другие реакции нуклеофильного'за-
мещения хлористого винила и хлорбензола и тому
подобных соединений крайне затруднен. Арилгало-
гениды нормально реагируют с Mg в эфире, винил-
галогениды в эфире не вступают во взаимодействие
с Mg; как было недавно показано, гриньяровы син-
тезы могут быть легко осуществлены с винилгалогени-
дами в среде тетрагидрофурана.
Г. у. находят широкое и разнообразное практич.
применение. Так, к числу Г. у. относятся многие
хорошо известные растворители (дихлорэтан, СС14,
СНС13, СС12=СС12, СН2С12, хлор- и п-дихлорбензол
и др.). Фторхлорпроизводные метана и этана (фрео-
ны) и, в частности, CF2C12 применяются в качестве
инертных хладоагеятов для холодильных машин.
Многие Г. у. обладают сильным физиологическим дей-
ствием. Так, хлористый этил, йодоформ, хлороформ
применяются в медицине; ДДТ, гексахлорциклогек-
сан представляют собой эффективные инсектициды.
Галогензамещенные этилена вызывают особый интерес
как исходные вещества для получения полимеров, так
широко применяются продукты полимеризации винил-
хлорида СН2=СНС1, хлоропрена СН2=СНСС1=СН2,
тетрафторэтилена CF2=CF2 и др.
Г. у. имеют большое значение и как исходные про-
дукты для дальнейших превращений. Так, С2Н5С1
применяется в произ-ве тетраэтилсвинца, хлористый
аллил перерабатывается на глицерин, из хлорбен-
зола получают фенол и т. д.
Для качественного определения Г. у. применяются
т. наз. проба Белыптейна; при внесении медной петли
с пробои Г. у. в пламя горелки оно окрашивается
в зеленый цвет. Для характеристики Г. у. их пере-
водят в соответствующие нитросоединения или в кри-
сталлич. производные, напр. в изотиурониевые соли:
RX 4- S=C(NH2)2 ->RSC(=NH)NH2HX.
Колич. анализ галогенов в органич. соединениях
связан с отщеплением галогена с последующим опре-
789
ГАЛОГЕНОКИСЛОТЫ — ГАЛОГЕНОНИЕВЫЕ СОЕДИНЕНИЯ
790
делением галоген-иона обычными методами. В нек-рых
случаях процесс осуществляется в мягких условиях,
иапр. действием Na" в спирте (по Степанову), нередко
требуется полная минерализация анализируемого
вещества (по Кариусу, в бомбе Парра и т. д.). См.
также Галогенов определение.
Лит.: Huntress Е. Н., The preparation, properties,
chemical behavior and identification of organic chlorine com-
pounds, N. Y., 1948; Губен И., Методы органической хи-
мии, пер. с нем., т. 3, вып. 3, М., 1935, с. 297—460.
Я. Ф. Комиссаров.
ГАЛОГЕНОКИСЛОТЫ — соединения, молекулы
к-рых содержат карбоксильную группу и атомы гало-
гена (F, Cl, Вг, J). Названия Г. производят от назва-
ний кислот с указанием положения галогена отно-
сительно карбоксильной группы, к-рое обозначается
греч. буквами а, Р, у...; замещение 1гри последнем
атоме углерода обозначается ю. Наиболее распро-
страненные методы получения Г. основаны на введении
галогена в соединение, молекула к-рого уже содержит
карбоксильную группу, или, наоборот, иа образовании
карбоксильной группы в соединении, уже содержа-
щем галоген; к таким методам относятся галогени-
рование насыщенных жирных к-т, присоединение
галогена или галогенводородной к-ты к ненасыщенным
к-там, замена оксигруппы на галоген в оксикислотах,
расщепление лактонов галогенводородной к-той, окис-
ление галогенспиртов или альдегидов, окисление
галогенированных олефинов, гидролиз трихлорметиль-
ной группы в а,а,а,<а-тетрахлоралканах и др.
СН3СН2СООН 55 СН2С1СН2СООН (на свету)
RCH2COOH 55 RCHBrCOOH
(Гелля—Фольгарда — Зелинского реакция)
RCH=CHCOOH 55 RCHC1CHC1COOH
RCH=CHCOOH 551 RCHBrCHaCOOH
RCH(OH)CHaCOOH 5.9b RCHCICHaCOOH
CH2CH2CH2COO CH2BrCH2CH2COOH
C1CH2CH2OH C1CH2COOH
ch3cf2cci=ch2 —ch3cf2cooh
Cl(CH»CH2)nCCl3 Cl(CH2CH2)nCOOH
Кроме того, фтор, а иногда и иодкарбоновые к-ты
предпочтительно приготовляют обменной реакцией
эфиров хлор-(бром)-карбоновых кислот с KF и КJ:
СН2С1СООС2Н5 — CH2FCOOC2H5; СН2С1СООК -5
—>CH2JCOOK. Галогенангидриды Г. образуются авто-
окислением галогенпроизводных этилена:
СН2=СВг2+О -+[ОСН2-СВг2] -»• СН.ВгСОВг
СНС1=СС12+О -в- [ОСНС1-СС12]-> СНС12СОС1
Г. являются более сильными к-тами, чем соответ-
ствующие к-ты, не содержащие галогена, причем это
влияние падает от фтора к иоду. Наибольшее влияние
оказывает галоген, находящийся в а-положении
к карбоксильной группе, что объясняется отрица-
тельным индукционным эффектом:
х—сн2—с_он
И
О
Г. обладают способностью вступать в реакции, свой-
ственные как галогеналкилам, так и карбоновым
к-там. Так, в а-Г. атом галогена легко вступает
в реакции обмена: RCHXCOOH 52Srch(otj)COOH;
RCHXCOOH RCH(NH2)COOH; RCHXCOOH 55S
-> RCH(CN)COOH.
В Р-Г. с этой реакцией замещения часто конкури-
рует отщепление галогеиводорода с образованием
а,Р-ненасыщеяных кислот: RCHX—СН2СООН--------
—> RCH=CHCOOH. у-Г. склонны к образованию
циклич. сложных эфиров — у -лактонов:
RCHBrCH.CH.COOH —S RCHCH.CH.COO
I___-_____I
О реакциях Г. по карбоксильной группе см. Карбоно-
вые кислоты. Высшие гомологи а-Г. (RCHXCOOH)
содержат один асимметрии, атом углерода и известны
в двух оптич. формах (d и 1) ив одной оптически
недеятельной форме (di). При исследовании реакций
обмена галогена на другие радикалы П. Вальден уста-
новил, что в зависимости от условий можно получать
один или другой оптич. антипод. Так, d-бромян-
тарная к-та с Ag2O дает d-яблочную к-ту, а с КОН —
1-яблочную к-ту. Наоборот, 1-бромянтарная к-та
с Ag2O дает 1-яблочную к-ту, а с КОН — d-яблоч-
ную к-ту. Это явление получило название вальденов-
ского обращения. Помимо самостоятельного значения,
Г. являются важными промежуточными соединениями
при синтезе карбоновых к-т с другими функциональ-
ными группами. М. Г. Линькова.
ГАЛОГЕНОНИЕВЫЕ СОЕДИНЕНИЯ — соедине-
ния, содержащие положительно заряженный атом га-
логена (Cl, Br, J), связанный ковалентными связями
с двумя органич. радикалами и ионной связью с анио-
ном, напр. [(С6Н5)2Вг]+С1~. Г. с. принадлежат к оние-
вым соединениям. Из них иодониевые соединения наи-
более легко доступны и лучше всего изучены. Все
Г. с. — твердые солеобразные соединения с высокой
темп-рой плавления (см. табл.), растворимы в поляр-
ных растворителях [СН3СОСН3, CH3NO2, CeH5NO2,
HCON(CH3)2 и др.], нерастворимы в эфире и угле-
водородах. Г. с. с анионами СП, BFj, NOj, СН3СОО_,
HSO^, SO? в большинстве случаев растворимы в воде;
соли с анионами Вг~, 1, HgBr3,PtCl|~ обычно плохо
растворимы в воде. Гидроокиси дифенилгалоген-
ониев, существующие только в р-рах, являются силь-
ными основаниями, подобными органическим основа-
ниям аммония и сульфония. При нагревании Г. с.
распадаются с образованием соответствующих гало-
генопроизводных, напр.: [Аг2Вг]1 -> АгВг+ArJ. Тер-
мич. стойкость Г. с. резко убывает в ряду
[Ar2J]+ > [Аг2Вг]+ > [Аг2С1]+
Иодониевые соединения известны следующих типов:
1) [Аг2 J р~Х~, где X — анион сильной кислоты, а Аг—
радикал ароматич. ряда с различными заместителями
или же гетероциклич. радикал ароматич. характера,
напр. тиенил; 2) соединения с атомом иода в цикле (I);
3) соединения из р-дикетоиов (II, III и IV); 4) ви-
нилиодониевые — производные ацетилена (V и VI).
. [(CICH=CH.J] X" и [С1СН=СШАг1 X-
V VI
Иодониевые соединения обычно получают: действием
влажной окиси серебра или едкого натра на экви-
молярную смесь иодо- и иодозосоединений;
ArJO + ArJO3 + AgOH —> LAr2J]OH -f- AgJOs
791
ГАЛОГЕНОНИЕВЫЕ СОЕДИНЕНИЯ
792
Катионы Анионы
Cl- Br- J- bf4- NO3-
(С3Н5)21+ 228— 229° 210° 182° 136— 137° 153— 154°
(jw-NO2CeH4)aJ+ 214“ 178- 179° 130,5° — 194°
292- 294° 245— 250° 210- 215° — 241- 242’
CP-jJCJ 229- 230° 215— 219° 135- 136° — —
CHCI=CCI-J-CH=CHCI 207° . 200° 97° —
(СвН5)2Вг+ 111- 112° 82- 83“ 81- 82° 120— 121° —
(CeHsJgCH- — — 56— 57,5° 109,5— 110° -
QP 205— 207° 194- 194,5° 165- 170° 199— 200° —
— — 130,5— 131° 169- 170° —
взаимодействием арилиодидхлоридов (или JC13) с
ароматич. соединениями ртути или олова:
2CeH5JCl2 + (CeH5)2Hg — [(CeHs)2JCl]3 -HgCl3
CeH5 JC12 + CeHsSnCls -> (C6H5)2JC1 + SnCI4
JClj 4-2CeH5HgCl — (CeH5)2JCl 4-2HgCI3
конденсацией ароматич. углеводородов с иодозосо-
едииениями в присутствии конц. H2SO4:
ArJO 4- Ar'H 4- H2SO4 —. [Ar—J—Ar1] H2SOf 4- H3O
Вместо иодозосоединения в этой реакции можно ис-
пользовать арилиодозоацетат или арилиодид в при-
сутствии окислителя (K2S2Os, ВаО2, СН3СОООН и др.).
Иодониевые соединения из Р-дикетонов получены
лишь в 1957, напр. конденсацией иодозобензола с ди-
медоном в хлороформе:
O’
H3C^CH3
CeH5JO
’0
OH
-H2O
С “”"Ung
‘6
C6H6-j
CH3
CH3
Cl’
Винилиодониевые соединения получают действием
хлорвинилмеркурхлорида или ди-р-хлорвинил-
’ути иа JC13, AtJC12 или на Р-хлорвинилиодидхлорид:
CICH=CHJCla 4- CICH=CHHgCI ->
— (CICH=CH)a JCI • HgCl2
JC13 4- (ClCH=CH)2Hg -> (CICH=CH)2JCI • HgCIa
Ar .7 Cl 2 4- CICH=CHHgCI — CICH=CH-JC1 4- HgCI2
Ar
Хлорониевые и бромониевые соединения известны
двух типов: а) [Ат2Вт]Х и [Ат2С1]Х и б) производные
дифенила:
Бромониевые и хлорониевые соединения получают
взаимодействием диазосоединений с арилгалогени-
дами. Диарилбромониевые и -хлорониевые соли обра-
зуются при разложении борфторидов арилдиазонпев
в соответствующих арилгалогенидах:
ArN2BF4 4- BrAr — [Аг2Вг] ВГ4 4- N2
Выходы бромониевых и хлорониевых солей состав-
ляют лишь 5—6%, так как преим. образуется ArF.
Диарилбромониевые соли образуются также при само-
произвольном разложении двойных солей арилди-
азониев с бромной ртутью: 2ArN2Br • HgBr2 ->
-> [Ar2Sr]Br.HgBr2 + 2N2 + HgBr2.
Диарилгалогенониевые соединения — превосходные
арилирующие реагенты, способные как к гомолитич.,
так и к гетеролитич. арилированию (с передачей
арила в виде катиона). Механизм реакции зависит
как от условий, так и от аниона Г. с. Галогениды
диарилгалогенониев распадаются обычно (в неводных
средах) гомолитически, соли же с комплексными ани-
онами (напр., борфториды)— гетеролитически (А. И.
Несмеянов). В водных р-рах все галогенониевые соли
распадаются гетеролитически. По свойствам соли
диарилгалогенониев напоминают соли арилдиазо-
ниев. С рндом металлов они реагируют с образованием
металлоорганич. соединении; при этом, аналогично
солям диазония, галогениды диарилгалогенониев об-
разуют металлоорганич. соединения с Hg, Sb, Те,
Sn, а борфториды — с TI, Pb, Bi, Sn:
[(СвН5)2СЦ .т 4- Hg — CeHsHgJ 4- CeH5Cl
2 [(С„н5)2 J] С1 4- Те — (CeHs)2TeCI2 4- 2CeH51
[(CeH5)2Br] BF4 К (С„Н5)4РЬ
С галогенидами тяжелых металлов соли диарилгало-
генониев образуют двойные соли, при разложении
к-рых порошками металлов образуются металлоор-
ганич. соединения, как и по диазометоду А. Н. Не-
смеянова:
[Ar2J] CI • SnCla 4- Sn — Ar2SnCl2 + 2АГ.Т 4- 2SnCl2
[Ar2 J] Cl • HgCI2 4- Hg — ArHgCl 4- Ar J HgCl2
3 [AraJ] Cl 4- 2B1 —— Ar3Bl 4- 3ArJ 4- B1C13
Гетеролитич. распад борфторидов диарилгалогенониев
позволяет перейти к другим классам ониевых соеди-
нений:
[(CeH5)2J] BF4 4- (C„H5)2S — [(CeH5)3S] BF4 4- CeH5J
[(С3н5)2 J] bF4 4- (CeH5)3Sb — [(CeH5)4Sb] BF4 4- CeH5 J
[(C9H5)2J] bf4 4- R3N — [C0h5nr3] bf4 4- c9h5j
При действии нитрующей смеси ди-
фенилгалогенониевые соли легко нитру-
ются с образованием ди-мета-нитропро-
изводных:
_ HNO3
х H2sp,4
Hal
no2
X"
NO3
Скорость нитрования резко падает в ряду
(CeH5)2J+ > (С6Н5)2Вг+ > (С9НБ)3С1+
Г. с. обладают широким спектром бактериостатич.
действия.
Лит.: S an d in R. В., Chem. Rev., 1943, 32, № 3, p. 249;
Несмеянов A. H., Макарова Л. Г., Тол-
793
ГАЛОГЕНОСПИРТЫ — ГАЛОГЕНЫ
794
стал Т. П., в кн.: Несмеянов А. Н., Избр. труды, т. 2,
М., 1959, с. 444; Ber In ger F. М., Forgione P. S.,
Yu dis M. D., Tetrahedron, i960, 8, № 1/2, 49.
T. П. Толстая.
ГАЛОГЕНОСПИРТЫ — соединения, содержащие
спиртовые группы и атомы галогенов. В зависимости
от относительного расположения ОН-группы и гало-
гена различают P-Г., напр. С1СН2СН2ОН, у-Г.,
напр. С1СН2СН2СН2ОН и т. д., а-Г., напр. С1СНаОН
(как и соединения с двумя ОН-группами у одного
атома углерода), чрезвычайно нестойки и легко рас-
падаются на СН2О и НС1; из а-Г. сравнительно стой-
ким является FCHjOH. Сочетая в себе свойства
спиртов и алкилгалогенидов, Г. обладают особен-
ностями, обусловленными взаимным влиянием заме-
стителей и, в частности, их способностью реагировать
между собой. Так, действие щелочи на этиленхлор-
гидрин ведет к окиси этилена: СН2ОН—СН2С1 -Ь
-|-NaOH->CH2—СН2О + NaCl+H2O. Аналогичным
образом из соответствующих Г. образуются циклич.
эфиры, особенно прочные в случае 5- и 6-членных
тетрагидрофурана и окиси пентаметилена.
Подобно алкилгалогенидам, Г. легко вступают
в реакции нуклеофильного замещения, что позволяет
синтезировать различные производные спиртов, напр.;
НОСН2СН2С1 4-NH3 — HOCH2CH2NH2 + HCI
HOCHSCH2CH2CI + NaSH — HOCHaCH2CH2SH 4- NaCl
При этом скорость реакции зависит от подвижности
галогена: наиболее реакционноспособны 0-Г., в то
время как у- и 6-Г. реагируют значительно труднее.
Г. обычно получают присоединением хлорнова-
тистой к-ты к олефинам: СН2=СН2 + НСЮ->
->СН2ОН—СН2С1 или частичным гидролизом дигало-
генозамещенных: СН2С1—СН2С1 + НОЩ-> СН2ОН—
—СН2С1 + НС1. Этиленхлоргидрин можно также по-
лучить присоединением НС1 к окиси этилена. Из
Г., содержащих 2 атома галогена, большое значение
имеют промежуточные продукты синтеза глицери-
на — дихлоргидрин глицерина (С1СН2СНОНСН2С1 и
НОСН2СНС1СН2С1), образующиеся при присоеди-
нении элементов хлорноватистой к-ты к хлористому
аллилу.
К числу Г. с 3 атомами галогена относятся трихлор-
этиловый спирт СС13СН2ОН и трибромэтиловый
спирт СВг3СН2ОН (авертин), применяемый как
наркотик. К Г. такого же физиология, действия при-
надлежит и хлорэтон, образующийся при присоеди-
нении хлороформа к ацетону в присутствии едкого
кали: (СН3)2СО + СНС13 -> (СН3)2С(СС13)ОН.
Я. Ф. Комиссаров.
ГАЛОГЕНОЦИАНЫ (циангалогениды) — галоген-
ангидриды циановой к-ты общей формулы XCN,
где X — атом галогена; молекулы Г. имеют линейную
структуру. Г. — бесцветны и весьма летучи; их пары
раздражают слизистые оболочки носоглотки и глаз,
они обладают также сильным общетоксич. действием.
Галогеноциан Мол. вес Т. пл., °C Т. кип., °C di°
FCN 45,01 возг. —72
C1CN 61,48 -6 13,8 1,186
BrCN 105,93 52 61,6 2,015
JCN 152,94 146* возг. 136 —
• В запаянном капилляре.
Г. умеренно растворимы в воде, легко в спирте, эфире
и др. органич. растворителях. Обычно хлор и бром-
циан получают действием соответствующих гало-
генов на комплексную соль цианистого цинка в вод-
ной среде: 4NaCN + ZnSO4 ->Na2[Zn(CN)4] +Na2SO4;
Na2[Zn(CN)4] + 4CI2 ->4C1CN + ZnCl2 + 2NaCl. Иод-
циан готовят из цианистой ртути: 2Ja + Hg(CN)a
т>2JCN + HgJ2. Фторциан может быть получен
действием свободного фтора на цианистое серебро.
Г. — чрезвычайно реакционноспособны, причем во
многих случаях они ведут себя как псевдогалогены,
напр. в реакциях присоединения по кратным свя-
зям: СН3СН=СН24- C1CN-> CH3CHC1CH2CN; при
взаимодействии с металлоорганич. соединениями за-
мещают органич. радикал:
RMgX 4- CICNtf
RC1 4- Mg(CN)X
RCN-j-MgClX
А1(С2Н5)з 4- 3JCN -► Al J3 4- 3C2H5CN
С аммиаком, первичными и вторичными аминами
Г. образуют цианамид н его производные: XCN +
+ 2NH3 ->NH2CN + NH4X; XCN + 2RNH2->
->RNHCN + RNH3X. С щелочами C1CN образует
цианаты, a JCN — цианиды: C1CN + 2NaOH —>
-> NaOCN+NaC14-H2O; 3JCN+ 6NaOH ->3NaCN +
4-2NaJ+NaJO3+3H2O, a BrCN в зависимости от
условий — цианаты и цианиды. Со спиртами Г. дают
имидоэфиры: XCN + С2Н5ОН-> HN=C(OC2H5)2 +
+ НХ. Под влиянием галогеноводородных к-т Г. поли-
меризуются в галогенангидриды циануровой кис-
лоты C3N3X3:
к-рые с NH3 образуют меламин, используемый в про-
изводстве синтетич. смол.
Г. применяют для синтеза нитрилов, цианамида,
циануровой к-ты и их производных и т. д. Реакция
между BrCN и третичными аминами служит для вы-
яснении структуры радикалов, связанных с атомом
азота, напр. в алкалоидах (см. Брауна реакция).
В первой мировой войне хлорциан и бромциан пы-
тались применять как боевые ОВ.
Лит.: Williams Н., Cyanogen compounds, L., 1948.
Н. В. Гнучев.
ГАЛОГЕНЫ (галоиды) — химич. элементы глав-
ной подгруппы VII гр. периодич. системы Менде-
леева: фтор, хлор, бром, иод и астатин.
Название «Г.» происходит от греч. — соль и —
производить, рождать. Неправильное название «галоиды»,
от греч. iiXs и eUoc — вид, наружность (солеподобный), ввел
в русскую химич. номенклатуру Г. И. Гесс в 1835; этим тер-
мином пользуются иногда и в настоящее время.
Атомы Г. имеют конфигурацию внешних электро-
нов s2/)5; присоединяя один электрон, приобретают
конфигурацию инертного газа s2pe. Г. характери-
зуются наибольшими среди всех элементов значе-
ниями сродства к электрону, к-рое возрастает от F
к С1, а затем падает к J. Ионизационные потенциалы
Г. падают с возрастанием порядкового номера эле-
мента Z. По мере возрастания Z возрастают атомные
и ионные радиусы Г. В газообразном, жидком и кри-
сталлич. состоянии все Г. состоят из двухатомных
молекул. В молекулах Г. атомы связаны одной двух-
электронной связью; энергии диссоциации молекул
сравнительно невелики (см. табл.), существенно
меньше, чем, напр., у молекул О2 и N2 (118,2 и
225 ккал/моль соответственно), в к-рых каждый из
атомов О и N затрачивает на образование связи два
или соответственно три электрона. С увеличением Z
и, следовательно, размера атомов Г. энергия диссо-
циации убывает, однако молекула F2 менее прочна,
чем С12 и Вг2.
Поляризуемость атомов и ионов Г. растет с увели-
чением Z. Уменьшение прочности связи внешних элек-
тронов с ядром в атомах Г. с увеличением Z вызывает
увеличение интенсивности межмолекулярного взаимо-
действия у Х2 (где X — галоген); в соответствии
795
ГАЛОГЕНЫ — ГАЛОХРОМИЯ
796
с этим возрастают по абс. величине теплоты плавле-
ния и испарения и темп-ры плавления и кипения.
Физические свойства F С1 Вг J
Порядковый номер . . . 9 17 35 53
Атомный вес Атомный радиус (тетра- 19,000 35,457 79,916 126,92
эприч.), А 0,64 0,99 1,11 1,28
Ионный радиус Х~5 А Межъядерное расстояние 1,33 1,81 1,96 2,20
в молекуле газа Хг> А Термич. диссоциация, % 1,418 1,988 2,284 2,662
XS — 2X: при 1000° К -м 0,035 0,23 2,8
» 2000° К Энергия термич. диссо- 98,9 37,2 72,4 89,5
циации, ккал/моль , . Потенциал ионизации, 37,72 57,194 45,44 36,057
вольт Сродство к электрону, 17,42 13,01 11,844 10,441
вольт Нормальный электрод- ный потенциал окисле- ния иона X- при 18°, 3,56 3,78 3,56 3,28
ВОЛЬТ 2,85 1.358 1.065 0,535
Т. пл., °C —219,62 -100,98 -7,2 4-58,75 4-113,5
Т. кип., °C —188,14 -34,15 4-184,5
Критич. темп-ра, °C . . -129 -4-144 4-311 4-553
Критич. давление, атм Теплота плавления, 55 76 102 —
ккал/моль Теплота иопарения, 1,12198 1,531 2,53 3,740
ккал/моль Теплота гидратации иона 1,56398 4,878 7,24 14,8807
X-, ккал/г-ион . . . . 128 96,9 92,2 85,8
Все Г. (о химич. свойствах At известно мало) —
активные неметаллы, непосредственно соединяются
с большинством элементов, образуя галогениды.
Высокая химич. активность Г. даже при сравнительно
низких темп-pax объясняется, отчасти, относительно
малыми энергиями диссоциаций молекул Х2 и боль-
шим сродством атомов X н электрону. Г. — окисли-
тели, причем их окислительная способность падает
от F к J. В соединениях с электроположительными
элементами проявляют валентность —1. С водо-
родом образуют галогеноводороды НХ — при обыч-
ных условиях газы, из к-рых по свойствам выде-
ляется HF; благодаря большей, чем у других гало-
геноводородов, прочности межмолекулярной водород-
ной связи HF образует полимерные молекулы (H2F2,
HeFe) и имеет аномально высокие темп-ры плавления
и кипения. Все галогеноводороды хорошо растворимы
в воде. Р-ры НС1, НВг, HJ — сильные к-ты, кажу-
щаяся степень диссоциации растет от НС1 к HJ.
Раствор Н F в воде (плавиковая к-та) — к-та средней
силы. Теплоты образования галогепводородов падают
от HF к HJ, соответственно в этом направлении пони-
жается их термич. устойчивость. Вышестоящий Г.
вытесняет все нижестоящие Г. из соединений с метал-
лами и водородом как в растворах, так и в отсутствии
растворителя.
Все соединения Г. с кислородом (кроме окислов J)
эндотермич. и чрезвычайно неустойчивы, часто раз-
лагающиеся со взрывом. Валентность Г. в соединениях
с кислородом переменна (1, 3, 4, 5, 7); валентность
7 проявляют G1 и J, ей отвечают С12О7, НС1О4 и Н5Юв.
(хлорный ангидрид, хлорная и ортоиодпая к-ты).
Бромная к-та НВгО4 и ее соли не получены, свою
высшую валентность Вг проявляет в броматах, напр.
КВгО3. Кислородные к-ты хлора более сильные
окислители, чем кислородные к-ты иода, поэтому
иод вытесняет хлор из аниона СЮ^, напр.: J2 -f-
+ 2СЮ^—>2JOy + С12. В отличие от других Г., иод
может существовать в виде иона J+, напр. в кристал-
лич. соединениях JNO3 и JC1O4, а также в виде ионов
иодила J0+ и иодония [JR2]+. См. Астатин, Бром,
Иод, Фтор, Хлор. Периодическая система алементов
Менделеева, Галогенов определение.
Лит.: Remy Н., Lehrbuch der anorganlschen Chemie, Bd
1—2, 9 Autl., Lpz., 1957—59; см. также при статьях о соответ-
ствующих элементах. В. Я. Росоловский.
ГАЛОГЕНЭФИРЫ — простые эфиры, в к-рых один
или несколько атомов водорода замещены атомами
галогенов. В зависимости от относительного распо-
ложения атомов кислорода и галогенов в Г. раз-
личают а-, 0-, у- Г. и т. д. К числу а-Г. принадле-
жит СН3ОСП2С1, примером 0-Г. может служить
О(СН2СН2С1)2. Известны Г., содержащие любые атомы
галогенов” (F, Cl, Br, J), а также полнгалогеноэфиры,
напр. (СС13)2О.
Сочетая свойства простых эфиров и галогензаме-
щенных углеводородов, Г., вследствие взаимного
влияния заместителей, отличаются характерными
особенностями, к-рые больше всего проявляются
в сравнительной легкости омыления галогенов и
в других реакциях нуклеофильного замещения.
Галоген в a-положении гидролизуется очень легко,
в 0-положении значительно труднее, галоген в у-поло-
жении очень прочен. Tajc, С1СН2—О—СН2С1 момен-
тально разлагается водой; (С1СН2СН2)2О («хлорекс»)
гидролитически прочен, применяется в качестве
растворителя, при кипячении с едкой щелочью пре-
вращается в дивиниловый эфир (0112=0112)20, чрез-
вычайно стоек к гидролизу. Изомерный хлорексу
С1СН2СН2ОСНС1СН3, содержащий один атом хлора
в a-положении, в воде легко разлагается на ат член-
хлоргидрин и ацетальдегид. Аналогичным образом
Г., содержащие два атома галогена у одного и того же
атома углерода в a-положении, легко омыляются
с образованием соответствующих сложных эфиров;
в особенности легко в этом направлении гидроли-
зуется полихлор- и полифторэфиры, напр.
CHFsCFsOCHs^CFsCOOCHj
На подвижности галогенов в а-Г. основана возмож-
ность перехода от них к высшим эфирам взаимодей-
ствием с реактивами Гриньяра: СН3ОСН2С1 + RMgX—►
-> CH3OCH2R 4- MgClX.
Наличие галогенов в Г. сказывается на их реак-
циях с участием эфирного кислорода. Так, перфтор-
эфиры, в отличие от незамещенных эфиров, не обра-
зуют оксониевых соединений, не присоединяют BF3
и т. д.
Г. обычно получают общим методом синтеза простых
эфиров — дегидратацией галогеноспиртов; так, один
из основных способов получения хлорекса заклю-
чается в действии H2SO4 на этиленхлоргидрин:
2С1СН2—СН2ОН —> С1СН2СН2—О—СН2СН2С1. Однако
для получения а-Г. этот способ непригоден, т. к.
вещества типа С1СН2ОН очень легко разлагаются
на СН2О и НС1. Для получения а-Г. прибегают
к действию НС1 на полиоксиметилен или его смесь
со спиртом:
СН2О+СН3ОН+НС1 — СН3-О-СН«С1
Моиохлорметиловый эфир можно получить также
осторожным хлорированием диметилового эфира.
0,0'-Дихлордиэтиловый эфир (хлорекс) применяется
в качестве селективного растворителя при депара-
финизации смазочных масел; при получении поли-
сульфидного каучука. Я. Ф. Комиссаров.
ГАЛОХРОМИЯ (от греч. яХс — соль и Хрш/ля —
цвет) — явление, заключающееся в появлении окраски
у органич. соединений при действии на них сильных,
как правило, конц. к-т (H2SO4, НС1, НС1О4 и т. п.)
и нек-рых солей типа SnCl4, А1С13, HgCl2 и др. (т. н.
«ансольво» кислот). Соединения, у к-рых при этом
возникает окраска, принято называть галохром-
н ы м и. Однако имеются случаи, когда солеобразо-
вание приводит не к появлению пли углублению цвета,
797
ГАЛОХРОМИЯ - ГАЛЬВАНОТЕХНИКА
798
а наоборот, к его уничтожению или повышению,
напр. n-нитрозодиметиланилин интенсивно-зеленого
цвета, его солянокислая соль — оранжевого; п-нитро-
анилин оранжевого цвета, его соли бесцветны (явле-
ние «дегалохромии»).
А. Байер ввел понятие «карбониевой» структуры, к-рая,
по его мнению, связывает в галохромных солях атом углерода
с остатком к-ты; по Байеру, соединения, способные к Г., не
обязательно должны содержать хиноидную группировку. Пер-
вую теорию Г. создал Пфейфер, считавший, что при действии
к-т или солей карбонильные соединения (кетоны и их аналоги),
способные к Г., превращаются в свободные радикалы. А сво-
бодные радикалы, как правило, почти все окрашены. Пфей-
фер систематически обследовал альдегиды, нитрозосоединения,
нитрилы, амиды, хиноны и др. и показал зависимость глубины
возникающего цвета от деталей их строения. Дильтей, и осо-
бенно Вицингер, углубили и расширили основные идеи Пфей-
фера. Вицингер обратил внимание на то, что при присоедине-
нии кислот к галохромным соединениям образуются не ради-
калы, а положительно заряженные ионы. Наличие в молекуле
заряженного атома («карбониевого» хромофора или его анало-
гов, по номенклатуре Вицингер а) и обусловливает появление
цвета. Обе теории (Пфейфера и Вицингера) не объясняют Г.
по существу и поэтому в настоящее время оставлены.
Явление Г. может быть объяснено тем, что при
взаимодействии галохромных соединений с кисло-
тами, солями и др. соединениями возникает настоя-
щая соль, состоящая из двух ионов, один из к-рых
является органическим. Такой органич. ион должен
иметь систему сопряженных связей, внутри к-рой
положительный или отрицательный заряд иона соли
делокализован; делокализация же, связанная с пере-
мещением электронов, приводит к возникновению
цвета. В молекуле, уже обладающей поляризованной
ионбидной сопряженной системой и потому окра-
шенной, солеобразование уничтожает сопряжение
или уменьшает длину сопряженной цепи (явление
дегало хромии).
Разнообразные структурные факторы и физико-
химич. свойства, напр. ассоциация ионов и др., дей-
ствие к-рых часто невозможно предвидеть, по-раз-
ному влияют на электронные процессы, вызывающие
явление Г. Таким образом, основным структурным
признаком галохромных соединений является нали-
чие системы сопряженных связей, могущей при
солеобразовании дать т. н. «сопряженные» ионы,
напр. «сопряженяо-карбониевый», «сопряженно-аммо-
ниевый», «сопряженно-азониевый» и т. д. (см. при-
меры):
сн3о-сйн4-сн=сн-сн=сн
^с=о + нх
сн3о-свн4-сн=сн-сн=сн
оранжево-желтый цвет
сн3о—с6н4-
О /'—'Х ОН
СН3О— с6н4-сн=снхсн^сна
глубоко-синий цвет
(CH3)aN-СвН4 ОН
+ нх
(CH3)2N-СвН4 СвН4
бесцветное соединение
(СНз)2М-СвнУ
^хС-С6Н5
(CH3)2N^C6H4
X- + Н2О
сине-зеленый цвет
(малахитовый зеленый)
Многие бесцветные соединения красителей можно
рассматривать как галохромные соединения. Гало-
хромные соединения используются в аналитич. хи-
мии. См. также Цветности теории.
Лит.: Bayer A., Dtsch. chem. Ges. Вег., 1905, 38, S. 589,
1156; D 11 they W., J. fiir praktische Chemie, 1925, 109,
S. 300; Pfeiffer P., Organische Molekulverbindungen,
2 Aufl., Stuttgart, 1927; Вицингер P. Л., Органические
красители, пер. с нем., Л., 1936; Аверьянов Н. Н.
иВейнберг 3. А., в сб.: Проблемы современной органи-
ческой химии, № 1, Л., 1934, с. 102; Гинзбург О. Ф.,
Порай-Кошиц Б. А., Крылова М. И. и Лота-
рейчик С. М„ Ж. общ. хим., 1957, 27 (83), вып. 2, 411;
Черкесов А. И., Ж. аналит. хим., 1960, 15, вып. 6, 651.
Б. А. Порай-Кошиц.
ГАЛЬВАНИЧЕСКИЕ ЭЛЕМЕНТЫ — см. Хими-
ческие источники тока.
ГАЛЬВАНОПЛАСТИКА — см. Гальванотехника.
ГАЛЬВАНОСТЕГИЯ — см. Гальванотехника.
ГАЛЬВАНОТЕХНИКА — область прикладной
электрохимии, относящаяся к процессам нанесения
металлич. покрытий на поверхность металлич. или
неметаллич. изделий при прохождении постоянного
электрич. тока через проводящие ток р-ры; делится
на гальваностегию и гальванопластику.
Гальванотехнич. система представляет собой элек-
тролитич. цепь, состоящую в простейшем случае из
двух электродов — катода и
анода — и электролита, содер-
жащего ионы металЯа (или
металлов), разряжающихся на
катоде. При прохождении по-
стоянного электрич. тока к
покрываемым изделиям подво-
дятся от источника постоянно-
го тока электроны и находя-
щиеся в электролите ионы реа-
гируют с ним, образуя элек-
тронейтральные атомы, кото-
рые кристаллизуются. Помимо
основных компонентов — солей
осаждающихся металлов, — в
электролиты вводят соедине-
ния, повышающие электропро-
Анодный процесс
Ni-2© — Ni+ +
Катодный процесс
Ni++ + 2©—
водность, регулирующие концентрацию ионов водоро-
да,— различные органич. соединения, существенным
образом влияющие на структуру электроосажденных
металлов. Структура и свойства электроосажденных
металлов зависят от условий кристаллизации, а эти
условия в основном определяются составом электроли-
та и режимом электролиза. Структура получается тем
мельче, чем больше относительная скорость образова-
ния центров кристаллизации. В гальванотехнике,
особенно в гальваностегии, находят широкое приме-
нение р-ры комплексных, в частности цианистых,
солей, к-рые дают осадки с тонкой структурой.
При электроосаждении металлов группы железа
из растворов простых сернокислых или хлористых
солей образуется достаточно тонкая структура; элек-
троосажденные медь, цинк, кадмий из р-ров простых
солей дают более грубую структуру, а свинец, олово,
серебро, золото и др. из р-ров простых солей (в от-
сутствии специальных добавок) кристаллизуются
в форме, не пригодной для гальванотехники. Убыль
ионов, разряжающихся иа катодах, компенсируется
поступлением их в электролит в результате раство-
рения анодов. В отдельных случаях — при хроми-
ровании, платинировании, родировании и др., при-
меняют нерастворимые аноды из устойчивых в дан-
ных условиях металлов или материалов, а убыль
разряжающихся на катодах ионов компенсируется
периодич. введением в электролит солей или других
соединений осаждающихся металлов. В табл. 1 при-
ведены наиболее распространенные составы электро-
литов и их режимы.
799
ГАЛЬВАНОТЕХНИКА
800
таблица 1
Процесс Компоненты и кон- центрация, 8/Л Темп-ра, °C Плотность тока,а/д.и2
Цинкование: в кислых электроли- тах в цианистых электроли- тах ZnSO4-7Н2О A12(SO4)3 • 18Н3О Na2SO4- 10Н2О декстрин Zn(CN)2 NaCN NaOH 215 30 50-100 10 60 85-120 65—70 18-22 18-22 « о 7 7 I •ч-l со
Кадмирование в цианистых электроли- тах Cd(CN)2 NaCN Na»SO4- 10H,O сульфированное касторовое масло 50 120 50 10—12 18-40 1-4
Лужение: в кислых электроли- тах в щелочных электроли- тах SnSO4 H2SO4 крезол (фенол) клей Na2SnO3 • ЗН2О NaOH СНз СО ONa 54 100 20-30 2,5 50—100 3-5 20—30 18—20 65-70 2-5% 15—20° 2-4
Свинцевание 2РЬСО3 • РЬ(ОН)2 HF (100%) H3BO3 клей 130 120 106 0,2 18-20 1-3
Меднение: в кислых электроли- тах в цианистых электроли- тах CuSO4.5H2O H2SO4 CuCN NaCN Na2CO3 2Й0—250 50-75 15 22,5 15-30 30-40 22-24 3-5 0,5-1,0
Никелирова- ние NiSO4 • 7H,0 NaCl H„BO3 (pH 5,3) 200 30 30 50-60 2-5®
Хромирова- ние CrO3 H2SO4 250 2,5 45-55 10-50
Серебрение AgCN KCN (своб.) K.CO. 32-40 30-45 30—90 0,001 25-30 0,5-1,5
Золочение Гремучее золото Na3HPO4. 12H3O 2 15 4 65-70 0,3-0,4
Платинирова- ние H2PtCl3 • 6H2O 4 (NH4)2HPO4- 12H3O 20 Na2HPO4-12H2O 100 65—70 0,1
Палладирова- ние PdCL • 2H2O 3,7 Na2HP04 • 12H3O 100 (NH4)s-HPOa-12H3O 20 CjHgOj 2,5 50 ОД-ОД
Родирование Rh3(SO4)3 H30O4 10 25-30 50-70 1-3
а Без перемешивания. 6 С перемешиванием. в С интен-
сивным воздушным перемешиванием.
Гальваностегия — нанесение тонких покрытий (от
долей мк до десятков и в нек-рых случаях сотен .мя),
прочно сцепленных с покрываемыми изделиями и
составляющих с ними как бы одно целое, с целью:
защиты металлич. изделий от коррозии; придания
изделиям красивого, нетускнеющего вида; защиты
поверхности трущихся деталей машин, механизмов
и приборов от механич. износа; сообщения поверх-
ности изделий повышенной электропроводности,
определенных оптич. свойств, облегчения процесса
паики и др. Различают анодные й катодные по-
крытия. К анодным относятся такие покрытия, как
цинк и кадмий, к-рые по сравнению с основным ме-
таллом (сталь, чугун) имеют более электроотрицатель-
ный потенциал, т. е. они являются анодами и электро-
химически защищают от коррозии, в то время как
медь, никель, хром и все благородные металлы яв-
ляются катодами и электрохимически не защищают
от коррозии основной металл. С этой точки зрения
цинк является наиболее эффективным металлом для
защиты стали от коррозии; примерно 40% мирового
произ-ва цинка потребляется для защиты от коррозии
черных металлов. Кадмий менее эффективно защищает
от коррозии сталь и чугун, чем цинк, т. к. разность
потенциалов между кадмием и железом меньше, чем
между цинком и железом. Кадмий имеет преимущество
перед цинком только в тех случаях, когда необхо-
димо защищать от коррозии в условиях непосред-
ственного воздействия агрессивной среды, напр.
морской воды. Химич, стойкость кадмия в агрессив-
ных средах выше, чем цинка. Кадмий применяется
также для защиты от коррозии напряженных, за-
каленных изделий, напр. стальных пружин, т. к.
хрупкость кадмированных изделий меньше, чем при
их цинковании. Однако в обычных условиях, а также
при загрязнении атмосферы промышленными га-
зами цинк защищает эффективнее, чем кадмий.
Кроме того, кадмий значительно дороже цинка, что
также ограничивает область его применения.
Олово в обычной атмосфере имеет менее электро-
отрицательный потенциал, чем железо, и электрохими-
чески не защищает его от коррозии, при наличии пор в
оловянном покрытии железо ржавеет. В р-рах органич.
к-т и в пищевых средах олово образует прочные ком-
плексные ионы и приобретает более электроотрица-
тельный потенциал, чем железо. Поэтому внутренняя
поверхность консервной тары, покрытая тонким слоем
олова, длительное время электрохимически защищена
от коррозии.
В случае, когда изделиям, помимо защиты от кор-
розии, необходимо придать красивый, нетускнеющий
вид, их покрывают никелем, хромом, часто с проме-
жуточным меднением; при этом пористость покрытия
должна быть минимальной, т. к. в этом случае покры-
тие защищает основной металл не электрохимически,
а путем изоляции его от окружающей среды. В машино-
строении, приборостроении, авиации и др. отраслях
пром-сти, начиная с 20 гг. 20 в., все большее распро-
странение получает защитно-декоративное хромиро-
вание по схеме медь — никель — хром. Главные
функции защиты основного металла от коррозии вы-
полняют медные и никелевые покрытия, поверх
к-рых наносится лишь очень тонкий слой иома
(порядка 1 мк), сохраняющий длительное время блеск
изделий. При износостойком хромировании на сталь-
ные закаленные детали (реже на алюминиевые) на-
носят сравнительно толстый слой хрома (до 200 мк).
В табл. 2 приведены данные о виде и толщине различ-
ных покрытий в зависимости от их назначения.
К покрытиям предъявляются след, основные тре-
бования: прочное сцепление с основным металлом;
плотная микрокристаллич. структура; заданная, по
возможности равномерная по всей поверхности тол-
щина; минимальная пористость (в нек-рых случаях
они должны быть совершенно беспористыми). Галь-
ванич. покрытию всегда предшествует подготовка
поверхности (удаление с поверхности покрываемых
изделий жировых загрязнении и окислов), к-рая
является составной частью всего процесса.
Жиры животные и растительные омыляют р-рами щелочей;
минеральные жиры удаляют керосином, бензином, дихлор-
этаном и др. растворителями. Во многих случаях обезжири-
вание протекает быстрее при электролитич. процессе в щелоч-
ных р-рах (изделия выдерживаются в течение 3—5 мин в
качестве катодов и 1 мин — в качестве анодов). Окислы со
стальных и чугунных изделий удаляют травлением их р-рами
серной или соляной к-ты (иногда в их смеси при повышенной
темп-ре). Продолжительность травления определяется тол-
щиной, составом и строением окисных пленок. Легированные
стали типа нержавеющих, а также углеродистые стали после
термич. обработки подвергаются влектролитич. травлению.
801
ГАЛЬВАНОТЕХНИКА
802
Таблица 2
Назначение покрытия Основной металл (сплав) Вид покрытия Тол- щина, мк
Защита от атм. кор- Сталь, чугун Цннк, кадмий 5-30
розии
То же Алюминий и его сплавы Электрохимия, оксидирование (анодирование) 5-10
» Магний и его сплавы Химич, или электрохимия, оксидирование 0,5-10
» » Цинковые сплавы Защитно-декора- тивное хромиро- вание 10-50
» » Медь и ее сплавы Олово, никель, хром 5-15
Защитно-декоратив- ная отделка Сталь, чугун Медь—никель— хром 10—50
То же Алюминий и его сплавы Электролитич. оксидирование (анодирование) с последующим окрашиванием 5-10
Защита от механич. износа Сталь, чугун, алюминиевые сплавы Хром 10—200
Восстановление Сталь, чугун Хром—железо 50-500
размеров
То же Медь и ее сплавы Медь до не- сколь- ких мм
Защита от цемента- Сталь Медь 20-50
ГЦИИ
Защита от действия Сталь Свинец 50—1000
серной к-ты
Облегчение паяния Сталь, чугун Медь, олово, свинцово-оловян- ный сплав 5—25
Защита от коррозии деталей ответствен- Медные сплавы Платина, палла- дий, родий 5-25
ных приборов
Зашита от коррозии консервной тары, пи- Железо, сталь Олово 1-5
щеварных котлов и т. п.
Обеспечение сцепле- Сталь Латунь 1-5
ния резины при го- рячем прессовании
Повышение анти- фрикционных свойств Сталь Серебро, сплавы: свинец—индий, медь—олово, свинец—олово £)— Ио
Защита поверхности от коррозии при вы- соких темп-рах Сталь, медные сплавы Алитирование (диффузионное алюминирование) 10-50
Повышение коэфф, отражения света Сталь, латунь Серебро, хром, роднй, т. н. белая бронза 5-25
3 ащитно-декор атив- ное покрытие пред- Медные сплавы Серебро, золото 1-30
метов домашнего оби- хода, ювелирных изделий и т. п.
Медные и латунные изделия травят в смеси конц. серной и азот-
ной к-т в присутствии небольшого количества соляной к-ты.
После кратковременного травления (неск. секунд) в такой смеси
изделия приобретают блестящий золотистый цвет. Перед на-
несейием защитно-декоративных покрытий (никелирование,
хромирование, серебрение и т. п.) изделия необходимо также
шлифовать и полировать для удаления различных роверхност-
ных дефектов. Количественно гальванотехнич. процессы, ре-
гулируемые законами Фарадея, могут быть вычислены след,
образом:
6OS7 . DhCrlt . _ _ 60S7 . 6OS7
DftCv; ’ 80т ’ k Ci)t ' 11 “ DACt
где t — время эпектроосаждения, мин; S — толщина покрытия,
мк; — плотность тока, а/вм3; ц — выход по тону, %;
7 — плотность осаждаемого металла, г/см3; С — электрохи-
мический эквивалент осаждаемого металла, а/а-ч. В табл. 3
приведены необходимые данные для производства подобных
вычислений.
Таблица 3
Осаждаемый металл Электро- химия, эквив., е/а-ч Плот- ность тока, а/дм* Пример- ный вы- ход по току, % Толщи- на по- кры- тия3, мк
Цинк 1,219 1-4 80—95 17,2
К адмий 2,097 1-2 90-93 24,4
Свинец Олово: Из кислых электроли- 3,885 1-2 98—99 34,2
тов Из щелочных электро- 2,214 1-2 95-98 30,3
литов Медь: Из цианистых электро- 1,107 1-2 70-80 15,2
ЛИТОВ® Из кислых электроли- 2,372 0,5-5 98 26,7
ТОВ 1,186 1-2 96-99 13,3
Никель • . . 1,095 1-5 95—98 12,6
Хромв 0,323 10-50 12-14 4,6
Железо 1,042 0,3—30 90—95 13,2
Серебро 4,025 0,3-1 98-99 38,2
Золото 7,357 0,5-2 69—70 38,1
Латунь (70% Си, 30% Zn) 2,026 0,3—1 65-73 23,8
а При ia-ч/дм2 поверхности и 100%-ном выходе по току.
6 Выход по току уменьшается по мере повышения плот-
ности тока и содержания свободного цианида в электролите.
в При декоративном хромировании 10 а/дма, при из-
носостойком хромировании 50 а/дм2; с повышением плот-
ности тока выход по тону растет.
Гальванопластика — получение точных, легко от-
деляемых металлич. копий сравнительно значитель-
ной толщины (несколько мм) с различных неметаллич.
или металлич. предметов, наз. матрицами. Гальвано-
пластика отличается от гальваностегии гл. обр. под-
готовкой поверхности.
Подготовку поверхности неметаллич. и металлич.
форм (матриц) производят по-разному. На неметаллич.
формы наносят механич., химич. или другим путем
проводящие ток слои. Механич. способ нанесения
проводящих слоев заключается в графитировании
или нанесении металляч. порошков, а химический —
в восстановлении металлов из водных р-ров солей,
реже — в восстановлении из газовой фазы летучих
соединений. Химич, восстановлением можно полу-
чать различные металлич. пленки, из к-рых наиболь-
шее распространение имеют серебряные, медные и
никелевые. Перед химич. серебрением и меднением
поверхность обрабатывают подкисленным р-ром SnCl2.
Серебрение производят из р-ра нитрата серебра,
реже—из р-ра комплексной соли Ag(NH3)2NO3; восста-
новителями служат формальдегид, моносахариды,
пирогаллол и др. При химич. меднении восстановите-
лями могут служить формальдегид, сегнетова соль,
дитионовокислый натрий и др. Для химич. никелиро-
вания чаще используют в качестве восстановителя
гипофосфит натрия. При подготовке металлич. форм
на них наносят разделительный слой из окислов или
других соединений, препятствующих прочному сцеп-
лению между основой и электролитич. осадком, путем
кратковременного погружения форм в соответствую-
щие растворы или путем поливания форм. Так, суль-
фидные пленки на железе, меди, серебре и др. полу-
чают обработкой поверхности 1%-ным р-ром сульфида
натрия, хроматные пленки образуются при воздей-
ствии слабого р-ра бихромата калия, иодидные пленки
иа серебре образуются в раз б. р-рах иода. Большее
распространение имеют гальванопластика меди и же-
леза, реже — никеля и др. Составы электролитов и их
режимы выбираются такими, чтобы гальванопластич.
изделия обладали должными механич. свойствами и, в
частности, минимальными внутренними напряжения-
ми наряду с достаточной твердостью. В табл. 4 приве-
дены наиболее распространенные составы электроли-
тов, применяемых в гальванопластике, и их режимы.
803
ГАММА-ЛУЧИ — ГАММЕТА УРАВНЕНИЕ
804
Таблица 4
Концен- трация, г/л Режим
Вид гальвано- пластики Компоненты темпе- ратура, плот- ность то- ка, а/дм%
Медная Ннкелеван Никель-кобальто- вая Железная, серно- кислая Железная, соля- нокислая CuSO4 • 5Н2О H2SO4 NiSOj- 7Н2О NiCl2 • 4Н2О Н3ВОз (pH 5,6—5,8) NiSOj- 7Н2О CoSO4 • 7112О Н3ВО3 NaCl (PH 5,6) FeSO*. 7Н2О MgSO4 • 7Н.0 NaHCO3 FeCl2 • 4H2O CaCl2 H2O 200—250 50-75 450 45 30 200 18-20 30 15 180-200 40 25-30 450 500 0,2-0,5 30—40 50—60 20-25 18-20 90—100 2-5 5-10 1-2 0,1-0,15 10—20
Часто, в особенности в полиграфии, сочетают гальва-
нопластику с гальваностегией, напр. медные гальвано-
пластич. стереотипы покрывают тонким слоем железа,
никеля или хрома, что увеличивает их износостой-
кость. Гальванопластич. процесс, как правило, ведут
из р-ров простых солей; из цианистых и других ком-
плексных солей не удается получать малонапряжен-
ные отложения значительной толщины. Помимо
старых областей, где гальванопластика применяется
давно (полиграфия, произ-во грампластинок), разра-
ботаны методы получения сложных изделии, напр.
медных волноводов на алюминиевых формах, изделий
сложной формы из никель-кобальтового сплава, отли-
чающегося высокой твердостью.
Лит.: Баграмян А. Т., Электроосаждение металлов,
М., 1950; И а й н е р В. И., Кудрявцев Н. Т., Основы
гальваностегии, 3 изд., ч. 1—2, М., 1953—57; Лайнер В. И.,
Гальванические покрытия легких сплавов, М., 1959; Г и н-
б е р г А. М., Гальванопластическое изготовление точных
полых деталей, Л., 1949; Казначей Б. Я., Гальва-
нопластика в промышленности, М., 1955; Якоби Б. С.,
Работы по электрохимии, под ред. А. Н. Фрумкина, М., 1957.
В. И. Лайнер.
ГАММА-ЛУЧИ (у-лучи) — электромагнитное излу-
чение с очень короткими длинами волн (от 1 А и
меньше), испускаемое атомными ядрами в результате
естественных и искусственных превращении или воз-
никающее вследствие торможения заряженных частиц,
аннигиляции пар частиц (напр., электронно-позитрон-
ной пары) и т. д. Г.-л. проявляют себя не только как
электромагнитные волны, но также и как поток ча-
стиц (т. н. у-квантов), причем волновые свойства (ди-
фракция, интерференция) проявляются лишь у самых
длинноволновых Г.-л., корпускулярные же свойстна
их выражены более отчетливо (фотоэффект, компто-
новское рассеяние). Энергия Г.-л. (у-квантов) выра-
жается как hv, где h — постоянная Планка, a v —
частота электромагнитной волны. Естественные радио-
активные источники испускают Г.-л. с энергией
до нескольких Мэв; в ядерных реакция® можно полу-
чить Г.-л. с большей энергией. Г.-л. с Еу порядка
сотен Мэв и даже ок. 1 Бэе получаются при торможе-
нии электронов на ускорителях заряженных частиц.
Г.-л. — одно из наиболее проникающих излучений.
При прохождении через вещество Г.-л. испытывают
поглощение, выражаемое ур-нием: Цх) = Ioe~aNx =
= Ioe~',LX, где х— толщина слоя в-ва, Г— интенсив-
ность пучка Г.-л. после прохождения слоя х, 10 —
интенсивность пучка до прохождения слоя х, N —
ядерная плотность слоя, т. е. число ядер в 1 с.и3,
о — полное эффективное поперечное сечение взаимо-
действия у-квантов с в-вом, ц — линейный коэфф,
поглощения у-квантов; он определяется гл. обр. тремя
процессами: 1) фотоэффектом (т. е. передачей энергии
у-кванта связанному электрону атома), существенным
при малых энергиях; 2) комптоновским рассеиванием
Г.-л. электронами, играющим главную роль в области
больших энергий (примерно между 0,5 и 5 Мае для РЬ
и 0,5 и 15 Мэв для А1); 3) образованием электроно-
позитронных пар в области еще больших энергий.
Т. к. все 3 эффекта возрастают с увеличением поряд-
кового номера Z элемента, то естественно, что для за-
щиты от Г.-л. используют наиболее тяжелые элементы
(напр., РЬ).
Радиоактивные в-ва, испускающие Г.-л., исполь-
зуются в технике для обнаружения внутренних де-
фектов изделий (гамма-дефектоскопия), в медицине —
для у-терапии злокачественных опухолей, в пищевой
пром-сти — для консервирования продуктов, и в др.
областях. В химии Г.-л. применяют для инициирова-
ния радиационно-химич. реакций. Источниками излу-
чения служат Со60, Cs13’ и др.
ГАММЕТА УРАВНЕНИЕ — уравнение, выражаю-
щее связь между константами равновесия или
скорости реакции замещенных производных бензола
и соответствующих незамещенных производных:
1g К/К0=р -а, где К—константа равновесия или
скорости реакции производного бензола с заместите-
лем впара- или щеша-положении по отношению к реаги-
рующей группе (opmo-положение ввиду стерич. эффек-
та не охватывается Г. у.), Ко — константа равновесия
или скорости той же реакции при отсутствии замести-
теля, р — постоянная реакции, зависящая только
от типа реакции и участвующего в ней производного
бензола, о — постоянная заместителя, определяемая
только его видом (функциональной группой) и поло-
жением в бензольном кольце.
Г. у. вытекает из т. й. «принципа линейности свободных
энергий», т. е. линейной зависимости между изменениями сво-
бодной энергии (а следовательно, и между логарифмами кон-
стант равновесия) для любых двух реакций в различных за-
мещенных производных бензола; 1g К = р • 1g К' + С, где
К и К' — константы равновесия этих реакций при любых,
но одинаковых заместителях в бензольном кольце, р и С —
постоянные величины, не зависящие от вида и положения за-
местителя. Применяя это уравнение к замещенным (К и К')
и незамещенным (Ко и Ко) соединениям, легко можно получить
выражение: 1g К/Ка = р • 1g К'/К,, где 1g К/Ко соответствует
а в Г. у.
То же соотношение выводится и для констант ско-
рости реакций, что основывается на аналогичной ли-
нейной зависимости между изменениями свободной
энергии активации (а след., логарифмами констант
скорости реакции). Поскольку р — относительная
величина, характеризующая один тип реакций по от-
ношению к другому, то, приняв какую-либо из реак-
ций за стандартную, приписывают ей р= 1. Стандарт-
ной реакцией принята ионизация бензойной к-ты
в водном р-ре при 25°, откуда о для любого замести-
теля выражается величиной lg A/Aj, где Ко—кон-
станта ионизации бензойной к-ты в водном J>-pe при
25°, а К — константа ионизации замещенной бензой-
ной к-ты.
Физич. основа Г. у. заключается в том, что каждый
заместитель, обладая определенной способностью вза-
имодействовать с электронной системой бензольного
ядра, обусловливает соответствующие изменения рас-
пределения электронной плотности в нем в момент
реакции и, следовательно, в реакционном центре со-
единения. Это изменение одинаково сказывается на
различных реакциях производных бензола. Исходя
из этого, константа о представляет собой меру влияния
заместителя на реакционную способность молекулы
ароматич. соединения. Константы о наиболее харак-
терных заместителей приведены в табл. Отрицатель-
ные значения о отвечают электронодонорной функ-
ции заместителя, положительные — электроноакцеп-
торной.
805
ГАНТЧА СИНТЕЗ - ГАТТЕРМАНА РЕАКЦИЯ
806
Заместит. 9 Заместит. 9 j Заместит. 9 Заместит. 9 Заместит. 9
п-О“ -1,00 n-(CH3)3Si -0,072 m-C„H5 -0,218 n-COOC2H5 -0,522 Для анилинов, фенолов
т-О’ - 0,71 m-CH3 —0,089 n-Cl -0,228 n-CF3 -0,551 и тиофенолов
n-NH2 —0,660 n-CH3S -0,047 n-Br -0,232 m-CHaSO -0,551 n-CH—CHC8HS 4-0,62
n-(CH,)sN ' —0,800 n-C,H5O -0,028 m-CH3CO -0,306 n-CH3SO -0,567 n-СГ) ОСдН 5 --0 68
n-OH -0,357 n-NHCOCH3 -0,015 m-F -0,3.37 n-CN -0,628 п-сд2н" - 0 73
n-СНдО -0,268 m-OH -0,002 m-J -0,352 m-CH3SO2 r-0,647 п-СКз 4-0,74
m-(CH8)2N —0,21i нет 0,0000 m-C02H -0,355 m-CN r0,678 п-СН3СО --0,87
n-(CH3)3C -0,197 n-C,H5 --0,009 7П-С1 -0,373 m-J02 -0,700 n-CN” --1,00
п-СН3 -0,170 n-F --0,062 m-Br -0,391 m-N02 -0,710 n-CH3SO2 --1,05
m-NH, —0,181 m-COg -0,10 m-COOC2H5 -0,398 n-CH3SO2 -0,728 --1,27
-0,151 n-J --0,276 m-CF3 -0,415 n-JOa -0,760
n-(CH3)2CH —0,15i m-CHaO --0,115 n-B(OH), -0,454 n-NOa , -0,778
m-(CH3)3Si —0,121 n-CO2 --0,i3 n-CeHsCu -0,459 -0,86
m-(CH3)3C -0,120 m-CHsS --0,144 n-CH8CO -0,516 m-(CH3)3N* -0,90
Г. у. не является строгим. Известны многочислен-
ные отклонения от него, обусловленные явлением
сопряжения, стерическими факторами и др. причи-
нами. Г. у. позволяет рассчитывать константы равно-
весия и константы скорости разнообразных реакций
многих важнейших ароматич. соединений. В послед-
ние годы сделаны многочисленные и большей частью
успешные попытки распространения принципа линей-
ности свободной энергии на органич. соединения
других классов: алифатич. к-т, аминов, фосфор-
органич. соединений и др.
Лит.: Hammett L. Р., Chem. Revs, 1935, 17, р. 125;
Trans. Faraday Soc., 1938, 34, pt 1, p. 158; JaffS H. H.,
Chem. Revs, 1953, 53, №2, p. 191; Hine J., Physical organic
chemistry, N. Y., 1958, p. 69. С. Э. Вайсберг.
ГАНТЧА СИНТЕЗ — образование производных пи-
ридина при конденсации двух молекул р-кетоэфира
с альдегидом и аммиаком:
Получающееся в качестве промежуточного соеди-
нения производное дигидропиридина (I) легко может
быть окислено азотной к-той в соответствующее произ-
водное пиридина (II). Вероятным механизмом является
первоначальное взаимодействие аммиака с Р-кето-
эфиром с образованием эфира Р-аминокротоновой
к-ты (III), к-рый может быть выделен и использован
как исходное соединение в этом синтезе. Одновре-
менно вводимый в реакцию альдегид конденсируется
в присутствии основного катализатора — аммиака —
со вторым молем p-кетоэфира (см. Кнёвенагеля реак-
ция) в алкилиден- или арилиден-р-кетоэфиры (IV).
NHg
СНз- С=СНСООС2Н5 + CHjCOCCOOR —•
chr'
Ш IV
R
— NH3-C=C-CH-COCH3
<!;н8 io or
v
Далее эфир аминокротоновой к-ты присоединяется
по двойной связи к образовавшемуся алкилиден- или
арилиден-р-кетоэфиру; образовавшийся ненасыщен-
ный кетон (V) циклизуется в производное дигидро-
пиридина (см. Михаэля реакция).
Применение в синтезе Гантча циануксусного и мало-
нового эфиров, нитрила Р-аминокротоновой к-ты и
различных дикетонов дало возможность получать
труднодоступные производные пиридина. Так, при
конденсации этоксиацетилацетона с цианоацетамидом
в присутствии пиперидина образуется З-циан-4-эгокси-
метил-2-окси-6-метилпиридин, являющийся промежу-
точным соединением в синтезе витамина Вв:
СО CH„CN
/ + I 2 •
сна со
Н3С—СО NHa
+ 2Н2О
Реакция открыта А. Гантчем в 1882.
Лит.: Гетероциклические соединения. [Сб. ст.1, пер. с
англ., т. 1, М.. 1953; S и г г е у A. R., Name reactions in orga-
nic chemistry, N. Y., 1954, p. 92—3. H. E. Голубева.
ГАРМИН C13H14ON2, мол. в. 213,26 — алкалоид,
содержащийся в растении могильнике (Peganum
Harmala) семейства парнолист-
никовых (Zygopbilli), произра- ----111
стающем на юге СССР. Г. — I I II
бесцветные ромбические кри- (-H3(J
сталлы; т. пл. 266° (из СН3ОН); Н сц3
оптически неактивен; слабое
третичное однокислотное основание. Разбавленные
растворы солей Г. обладают голубой флуоресцен-
цией; важнейшие соли: хлоргидрат, т. пл. 269,5—
270,5°, хлорплатинат, т. пл. 264—266°. Удаление мето-
ксильной группы из Г. приводит к алкалоиду гар-
ману — циклическому скелету алкалоидов гармалы.
Для синтеза Г. исходным в-вом служит 6-метокси-
триптофан, замыкание третьего цикла производят
по Бигилера—Напиралъского реакции. Образующееся
производное тетрагидрогармина дегидрируют в Г.
над металлич. Pd; Г. получен также конденсацией
6-метоксииндола с метилмагнийиодидом и хлорацетони-
трилом с последующей дегидрогенизацией продукта
реакции. Г. вызывает возбуждение центральной нерв-
ной системы, в больших дозах — судороги; понижает
кровяное давление и делает дыхание более поверх-
ностным, расслабляет мускулатуру кишечника и вы-
зывает сокращение мускулатуры матки; более токси-
чен для простейших, чем хинин. В медицине приме-
няется иногда для лечения паркинсонизма.
Лит. см. при ст. Алкалоиды. А. Е. Васильев.
ГАТТЕРМАНА РЕАКЦИЯ — метод введения аль-
дегидной группы в ароматич. ядро действием HCN
и НС1 в присутствии галогенидов металлов. В перво-
начальном виде реакция применялась для получения
ароматич. оксиальдегидов или их эфиров; напр., для
получения анисового альдегида (III) р-р анизола
(I) и безводного HCN в эфире насыщают хлористым
водородом, прибавляют А1С13 и размешивают при
807
ГАТТЕРМАНА — КОХА РЕАКЦИЯ — ГАФНИЙ
808
20—30°. Образовавшийся хлоргидрат альдимина (II)
гидролизуют нагреванием в водном р-ре. Выход соот-
ветствующего альдегида из фенола и крезолов со-
ставляет 30—50%, а в случае применения тимола и
HCN,HC>x
А1С13
/
C = NH-HCI
и
многоатомных фенолов выход приближается к коли-
чественному. Альдегидная группа вступает по отно-
шению к окси- (соответственно алкокси)-группе в пара-
положение; замещение в орпго-положение идет при за-
нятом «яря-месте. Как правило, в ядро вступает одна
альдегидная группа. Образование диальдегидов на-
блюдается в случае многоядерных соединений, напр.:
RO OR • RO OR
СНО СНО
Известны различные видоизменения Г. р. Адамс пред-
ложил применять для реакции Zn(CN)s вместо си-
нильной к-ты, что значительно упростило процесс.
В случае резорцина, а- и 0-нафтолов образование
альдегидов в присутствии Zn(CN)2 идет гладко и без
катализатора. Выходы оксиальдегидов близки к вы-
ходам, получаемым с HCN. Это видоизменение Г. р.
дает возможность ввести альдегидную группу в соеди-
нения, содержащие карбометоксильную группу, не
затрагивая последней. Кроме фенолов и их эфиров,
Г. р. применима к углеводородам (бензол, дифенил,
антрацен) и к нек-рым гетероциклич. соединениям
(пиррол, фуран, тиофен).
Как уже упоминалось, образование альдегидов
идет через стадию альдиминов, к-рые в отдельных слу-
чаях были выделены в виде хлоргидратов. Эта схема,
предложенная Гаттерманом, предполагает, что про-
межуточно образуется хлористый формилимин
CHC1=NH из синильной к-ты и хлористого водорода.
В более поздних работах предлагается иной механизм:
2HCN+HCI - NH=CH-N=CHC1
+ Н.0
CeHe-|-NH=CH-N=CHCl - NH—СН—N—CHC3Hg — —
-> NHa+HCOOH+CeHsCH=NH —— CeH5CHO + NH3
Известно, что из синильной к-ты и хлористого водо-
рода образуется соединение состава 2HCN • НС1,
к-рому придают строение хлорметиленформамидина.
При взаимодействии этого соединения, напр., с бен-
золом и последующем гидролизе получается бензаль-
дегид. Промежуточное соединение — арилметиленфор-
мамидин — было получено при взаимодействии резор-
цина с 2HCN • HCI, а также с хлорметиленформами-
дином.
Метод предложен Л. Гаттерманом в 1898.
Лит.: Реакции и методы исследования органических со-
единений, кн. 7, М., 1958, с. 307—65; G at termann L.,
Вег. d. Deutsch. Chem. Ges., 1898, 31, 1149; Berckel-
m a n n W., там же, 1765, S. 234. В. А. Пучков.
ГАТТЕРМАНА — КОХА РЕАКЦИЯ — метод по-
лучения ароматич. альдегидов непосредственным вве-
дением формильного остатка в ароматич. ядро при
действии СО и НС1 в присутствии соответствующего
катализатора. Метод предложен для получения альде-
гидов из толуола и изомерных ксилолов, напр.
«-толуоловый альдегид (I) получали с хорошим выхо-
дом пропусканием смеси СО и НС1 в суспензию Си2С12
и А1С1а в толуоле при 20—40°'
Г~\ CO.HCI Г~\
СНз~\=,/ AlCl3,CusCl2 СНз\. /~СН0
Позже реакция была распространена на др. гомологи
= бензола и многоядерные углеводороды.
Впервые бензальдегид из бензола синтезировал
А. Н. Реформатский в 1901, к-рый вместо А1С1а при-
менил А1Вга. Г.— К. р. может
нз° _ о_____________________быть проведена при повы-
ип3и ' СпО шенных давлениях. В этом
случае образование альдеги-
111 да идет в отсутствие актива-
тора (Си2С12), иногда без добавления НС1. В качестве
катализаторов, кроме А1Вга и А1С1а, применяются AlJa,
FeCla и фтористый бор. При формилировании по Г. —
К. р. гомологов бензола в ароматич. ядро вступаетодна
альдегидная группа, почти исключительно в «-поло-
жение к алкильной группе. В случае полиалкилбензо-
лов иногда наблюдается дезалкилирование, а также
перемещение метильной группы. Так, из «-ксилола
(II) образуется 2,4-диметилбенз альдегид (III):
т
Гаттерман рассматривал синтез альдегидов как
частный случаи реакции Фриделя—Крафтса, в к-рой
в качестве галогеносодержащей компоненты прини-
мает участие формилхлорид, неизвестный в свободном
состоянии; аналогичные взгляды высказывал Рефор-
матский. Многочисленные попытки получить формил-
хлорид не дали результата; однако А. Н. Несмеянов
и Э. И. Кан получили в свободном состоянии фторангид-
рид муравьиной кислоты — фтористый формил. Роль
Си2С12 сводится к образованию комплексов с СО и
НС1, что облегчает получение формилхлорида. Это
подтвердил Хопф, синтезировавший вещество со-
става НСОС1 • А1С1а • Cu2Cl2. Хлористый алюминий,
вероятно, участвует в образовании активного ком-
плекса с углеводородом. Возникновение подобных
комплексов было Показано еще Г. Г. Густавсеном. На
основании термодинамич. и кинетич. исследований
М. Дилк и Д. Эли пришли к выводу, что в реакции
принимает участие ион НСО+:
НС1+СО-|-А1Вг3 - НСО+ +А1Вг3С1-
НСО+ + С3Не - С3Н5СНО+Н+
Хлористая медь, по их мнению, облегчает обра-
зование активного иона. Этот механизм вполне
согласуется с экспериментальными данными. Метод
предложен Л. Гаттерманом и Ю. Кохом в 1897.
Лиш.: Органические реакции. [Сб. статей], т. 5, пер. с
англ., М., 1951, с. 271; Мачинская И. В., в кн.: Реак-
ции и методы исследования органических соединений, кн. 7,
М., 1958, с. 277. В. А. Пучков.
ГАФНИЙ (Hafnium) Hf — химич. элемент IV гр.
периодич. системы Менделеева; п. н. 72; ат. в. 178,6.
В состав природного Г. входят изотопы: Hf171 (0,163%),
Hf178 (5,21%), HP" (18,56%), HP78 (27,10%), HP79
(13,75%) и Hf180 (35,22%). Получены искусственно
радиоактивные изотопы Г., из к-рых важнейшие,
применяемые как изотопные индикаторы, Hf175
(Т1/г= 70 дням) и HP81 (Т1/г = 45дням). Конфигурация
внешних электронов атома 5cZ26s2. Энергия ионизации
Hf»->Hf+ 7,3 эв, Hf+->Hf2+ 14,8 эв. Поперечное
сечение захвата тепловых нейтронов атомом 120 барн.
Положение Г. в периодич. системе было предсказано
Д. И. Менделеевым задолго до его открытия. В 1911 об откры-
тии элемента 72 объявил Урбан, к-рый назвал его «кельтием»
и включил в группу редкоземельных элементов. Однако в дей-
ствительности Урбаном была получена смесь элементов 69 (Ти),
70 (Yt), 71 (Lu) и 72. Последний присутствовал в крайне малом
количестве. Основываясь на выводах Н. Бора, предсказавшего
строение атома 72-го элемента и его основную валентность
(+4), Д. Кестер и Г. Хевеши подвергли систематич. исследо-
ванию минералы циркония методом рентгеноспектральиого
анализа и в 1923 в норвежской руде обнаружили присутствие
809
ГАФНИЙ
810
элемента 72, назвав его «Г.» в честь города Копенгагена (по-
лати Копенгаген—Hafnia). Это название было утверждено
в 1949 международной комиссией.
Г. — типичный рассеянный элемент; он не имеет соб-
ственных минералов и в природе обычно сопутствует
цирконию. В земной коре содержится 3,2 • 10~4
вес. % Г. В большинстве циркониевых минералов со-
держится от 1—2 до 6—7% Г., во вторичных мине-
ралах—иногда до 35%. Наиболее ценным промышлен-
ным типом циркониевых месторождений, содержащих
Г., являются морские и аллювиальные россыпи мине-
рала циркона (см. Цирконий).
Физические и химические свойства. Г.—серебристо-
белый металл, существует в двух кристаллич. поли-
морфных модификациях. При обычной темп-ре устой-
чива гексагональная плотнейшая упаковка с перио-
дами решетки я=3,1946 А и с= 5,0511 А; выше 1950±
± 100° устойчива кубич. объемнонентрированная ре-
шетка. Атомный радиус Г. 1,59 А; ионный радиус
Ш4+0,75 А. Плотность Г. 13,09 при 20°; т. пл. 2222 ±
± 30°; т. кип. 5400°; атомная теплоемкость 6,27
кал/г-атом град (25—100°). Для Г. высшей степе-
ни чистоты электропроводность составляет 6% элек-
тропроводности меди; уд. электросопротивление
32,4 • 10"в ом • см (при 0°), его темп-рный коэфф.
4,43-10'3. Примеси резко увеличивают электросопро-
тивление и уменьшают темп-рный коэфф. При повы-
шении давления изменение электросопротивления при
30° прямо пропорционально давлению. По данным из-
мерения электросопротивления, при 35°К установле-
но, что Г. не является сверхпроводником. Однако при
исследовании металла недостаточной степени чистоты
обнаружена сверхпроводимость в интервале 0,3—
0,4° К. Особенность Г. — высокая эмиссионная спо-
собность. В интервале 980—1550° работа выхода
электрона составляет 3,60 ев, эмиссионная константа
22,9 а • см~г • град~г. Чистый Г. очень пластичен,
легко поддается холодной и горячей обработке (про-
катке, ковке, штамповке). Твердость по шкале Рок-
велла В 78. Коэфф, сжимаемости 0,901 • 10~в см2/кг
(при 30°). Модуль упругости для Г., отожженного
в Вакууме при 1040° и содержащего 0,72 вес. % цир-
кония, 14 • 1СГ5 кг/см-.
По химич. свойствам Г. очень похож на цирконий
вследствие почти полного сходства размеров ионов
этих элементов и одинакового расположения электро-
нов. В соединениях Г. проявляет валентность 4.
В особых условиях можно получить соединения 3- и
2-валентного Г. Химич, активность Г. неск. меньше,
чем Zr. Так, величина стандартного потенциала Г.
для реакции Hf + 2Н2О ~ ШО2 + 4Н+ + 4е состав-
ляет 1,57 в, аналогичная величина для Zr равна
1,43 в. Все реакции, характерные для Zr, протекают
и для Г. Металлич. Г. легко поглощает газы. На
воздухе покрывается пленкой окиси HfO2, образова-
ние к-рой происходит также при нагревании с кисло-
родом и в присутствии Н2О. При нагревании Г. реаги-
рует с галогенами с образованием соединении типа
ШХ4. Прп высокой темп-ре он взаимодействует с азо-
том и углеродом, образуя тугоплавкие соединения:
нитрид HfN (т. пл. 2982 ± 50°) и карбид HfC (т. пл.
3887 50°). Металлич. Г. растворяется в плавиковой
и конц. серной к-тах и расплавленных фторидах ще-
лочных металлов. Он обладает несколько более основ-
ными свойствами, чем Zr, и менее основными по срав-
нению с Th. Химия водных р-ров соединений Г. ха-
рактеризуется высокой степенью гидролиза, образо-
ванием полимеров и комплексных ионов. Склонность
к комплексообразованию у Г. проявляется несколько
меньше, чем у Zr.
Окись гафния НЮ2 — химически очень
прочное, тугоплавкое вещество белого цвета, полу-
чают прокаливанием гидроокиси, оксихлорида, ни-
трата и т. п. Ниже 400° образуется рентгеио-аморфнэя
форма, выше—'моноклинная модификация, а=5,10 А,
*=5,13 А, с=5,27 А и 0=8О°16 , при 1850—1900°
образуется тетрагональная форма. Плотн. Н1О2 9,68;
т. пл. 2780 ± 20°; теплота образования ДЯ298=
=—271,5 ккал)моль. Кристаллич. окись Г. нераствори-
ма в воде, конц. НС1 и HNO3, в растворимые соедине-
ния переводится нагреванием в конц. H2F2 и HaS04
или сплавлением с щелочами и содой. Аморфная
НЮ2 растворяется значительно лучше.
Гидроокись гафния HfO2 • а?Н2О полу-
чается при действии щелочей на растворы солей Г.
или в результате гидролиза р-ров. В зависимости от
условии осаждения получают продукт с различной
степенью гидратации. При 100° гидроокись частично
обезвоживается и образуется HfO(uH)2. Гидроокись
Г. амфотерна, преобладают основные свойства. Гаф-
наты щелочных металлов Me2HfO3 получают сухим
путем; при растворении в воде они полностью гидро-
лизуются.
Тетрафторид гафния HfF4 — белые кри-
сталлы моноклинной формы, а=9,45 А, Ъ = 9,82 А,
с = 7,60 А, р = 94°29', плотн. 7,13. При атм. давле-
нии выше 400° сублимирует; летучесть HfF4 несколько
ниже летучести ZrF4. Тетрафторид Г. легко растворим
в плавиковой к-те, практически нерастворим в воде
и холодных р-рах минеральных к-т. Получают HfF4
термич. разложением (NH4)2HfFe в токе азота при
500°. Г. образует также двойные фториды
аммония-гафния и калия-гафния MeHfF3, MesHfFe,
Me3HfF7 (пента-, гекса- и гептафторогафнаты аммония
или калия) — белые кристаллы. (Толи аммония выше
240° разлагаются с отщепление,м NH4F; K2HfFe пла-
вится ок. 600°. Растворимость гекса- и гептафторогаф-
натов аммония и калия в воде и плавиковой к-те
несколько выше аналогичных соединений Zr. Пента-
и гексафторогафнаты аммония и калия получают
растворением НЮ2 в конц. H2F2 при добавлении сте-
хиометрии. количества NH4F или K.F. При избытке
последних образуются гептафторогафнаты.
Тетрахлорид гафния HfCl4 — гигро-
скопичные белые кристаллы кубич. формы, изоморф-
ные ZrCl4; отличается от ZfCl4 несколько большей ле-
тучестью, легко летуч выше 200°. Темп-ра сублимации
317° (1 атм), т. пл. 432°. При нагревании в присут-
ствии кислорода и особенно во влажном воздухе HfCl4
превращается в HfO2. При растворении HfCl4 в воде
гидролизуется. При> кипячении разб. р-ров ШС14
образуются труднорастворимые основные хлориды.
Получают НгС14 взаимодействием элементов при
7^ 300° или хлорированием ШО2 в присутствии угля
выше 500°.
Хлорокись гафния при обычных усло-
виях существует в виде гидрата HfOCl4 • 8НаО —
тетрагональных белых игольчатых кристаллов, легко-
растворимых в воде и разб. р-рах НС1; при увеличе-
нии концентрации HCI растворимость уменьшается.
Выше 65° соединение теряет воду, затем хлор; выше
450° переходит в двуокись Г. Получают HfOCla • 8Н2О
растворением HfO2 • а:Н2О в разб. р-рах НС1 и кри-
сталлизацией соли упариванием кислых р-ров или
добавлением конц. НС1.
Сульфат гафния Hf(S04)2 — белые кри-
сталлы; выше 500° разлагается с образованпем ШОа;
хорошо растворяется в воде и разб. р-рах HaS04;
с увеличением концентрации последней растворимость
понижается. Безводный сульфат получают действием
конц. HaS04 на тетрахлорид или двуокись Г. при
нагревании. Из разб. водных р-ров осаждаются
труднорастворимые основные сульфаты, из сернокис-
лых р-ров — тетрагидрат Hf(SO4)2 • 4НаО. Для суль-
фата Г. характерно образование двойных солей:
(NH4)4Hf(SO4)4 • 5Н2О и K4Hf(SO4)4 • пН2О (п=2—7).
811
ГАФНИЙ — ГЕЙ-ЛЮССАКА ЗАКОНЫ
812
Фосфаты гафния — белые вещества, не-
растворимые в воде и большинстве минеральных к-т.
Известны: вторичный ортофосфат Г. Hf(HPO4)2, пер-
вичный ортофосфат гафнила HfO(H2PO4)2, метафос-
фат гафнила HfO(PO3)2, пирофосфат Г. ШР2О7. Прп
осаждении из сильнокислых р-ров обычно образуется
HfO(H2PO4)2. Свежеосажденныи фосфат растворяется
в конц. р-рах H2SO4, Н3РО4 и щавелевой к-ты с обра-
зованием комплексных к-т. В нейтральных и слабо-
кислых р-рах HfO(H2PO4)2 подвергается гидролизу с
образованием основных фосфатов. При нагревании вы-
ше 700° НЮ(РО4)2 превращается в HfO(PO3)2. Прока-
ливание Hf(HPO4)2 приводит к образованию Н1Р2О7.
Получение. Соединения Г. выделяют из соедине-
ний Zr обычно после завершения технология, цикла
получения последнего. Вскрытие циркониевых рудных
концентратов, содержащих Г., и получение соеди-
нений Zr являются предтехнологией гафния (см.
Цирконии). Собственная технология Г. характери-
зуется в основном методами, применяемыми для
разделения Zr и Hf, к-рые можно объединить в след,
группы: дробная кристаллизация; дробное осажде-
ние; селективное термич. разложение соединений;
сублимация, дистилляция и ректификация галогени-
дов и их производных; адсорбция и ионный обмен;
экстракция. Все эти методы базируются на использо-
вании лишь небольших различий в свойствах соеди-
нений Zr и Hf.
Самым низким коэфф, разделения (—0,02) характеризу-
ются процессы дробной кристаллизации, в к-рых используется
лишь нек-рое различие в растворимости соединений Zr и Hf.
Однако способ кристаллизации двойных фторидов калия ши-
роко применяется для выделения чистых препаратов Zr в
связи с тем, что технический K2ZrF, получается по относительно
короткой технология, схеме при переработке циркона. Более
эффективными являются методы дробного осаждения фос-
фатов, этилфосфатов и ферроцианидов Zr и Hf. Эти методы ис-
пользовались для выделения чистых препаратов Г. из полу-
продуктов, образующихся при разложении циркониевых руд
конц. H2SO4.
Высокая степень разделения достигается в процессе рек-
тификации комплексных соединений ШС14 • РОС13 и ZrCl4 -
РОС13, поскольку их темп-ры кипения различаются на — 30° •
Г. концентрируется в низкотемпературной фракции. Методы
адсорбции и ионного обмена также характеризуются большим
эффектом разделения и позволяют получать соединения Zr и Hf
высокой степени чистоты, но их основным недостатком является
низкая производительность (—0,05 a Zr в час/см2). При адсорб-
ции на активированном силикагеле используется раствор
тетрахлоридов в метиловом спирте; Г. концентрируется на
силикагеле. Насыщение катионитов производится из растворов
HCI, H2SO4, HNO3 и НС1О4. Лучшие результаты по разделению
получены при использовании в качестве десорбента растворов
H2SO4 и смеси лимонной и азотной кислот. Из современных
физико-химич. методов разделения широкое развитие полу-
чили методы экстракции, к-рые характеризуются высоким
эффектом разделения, высоким выходом и большой произ-
водительностью (10 г Zr час/с-м2). В качестве экстрагентов
применяют эфир и метилизобутилкетон, экстракция проводится
из сернокислых и солянокислых р-ров роданидов Zr и Hf;
последний концентрируется в органич. фазе, степень разделе-
ния на одной ступени составляет 10—20. Такое же хорошее
разделение достигается при экстракции из азотнокислых р-ров
трибутилфосфатом, в к-ром концентрируется Zr.
Металлич. Г. в виде мелкозернистого порошка
получают восстановлением двуокиси Г. (при 1300°),
гексафторогафната калия (при 800—900°) или тетра-
хлорида Г. (при 850°) натрием в закрытых стальных
цилиндрах. Г. может быть также восстановлен из
хлорида кальцием или магнием. Чистый, компактный,
ковкий Г. может быть получен термич. диссоциацией
иодида; при 300° из металлич. порошка получают
подид, к-рый испаряется при 600° и затем при 1100 —
1450° разлагается на накаленной вольфрамовой про-
волоке с образованием кристаллич. прутков металла.
Аналитическое определение гаф-
ния. Для анализа Г. применяют соединения с ва-
лентностью 4*4.Г. относится к аналитич. группе
А1 (осаждается сульфидом аммония в виде гид-
роокиси). В раствор Г. переводят: 1) сплавлением
образца с содой, выщелачиванием плава водой
и растворением остатка в кислотах; 2) сплавлением
образца с пиросульфатом калия или нагреванием
с конц. H2SO4 и растворением в разб. кислоте.
Иногда щелочной и кислый методы вскрытия соче-
таются. Качественное и колич. определение Hf в
присутствии Zr проводится гл. обр. методами
оптич. и рентгеновской спектроскопии. Кроме того,
известны косвенные химич., спектрофогометрич.,
а также другие физич. методы определения. К послед-
ним относятся определение плотности, метод рентге-
новской флюоресценции, полярографический и радио-
активационныи. В отсутствии Zr колич. определение
Hf проводится прямыми химич. методами, предло-
женными для Zr. Если предварительно отделить Zr
от Hf, напр. ионообменным методом, то после этого
Г. количественно осаждают из р-ра в виде гидроокиси,
фосфата, арсената, купфероната, миндалята, га-бром-
миндалята и других органич. производных.
Применение. Благодаря высокой темп-ре плавле-
ния и высокой эмиссионной способности, металлич.
Г. является хорошим материалом для изготовления
катодов электронных ламп, нитей накаливания и т. п.
Г. может применяться для получения спец, жаростой-
ких железных и никелевых сплавов, в к-рые он вво-
дится в количестве 0,05—10%, а также в качестве
материала с высоким сечением захвата нейтронов
в атомной технике. HfO2 употребляется при изготов-
лении оптич. стекол с высоким показателем преломле-
ния и для создания газонепроницаемых покрытий
электрич. нагревателей; находит применение в ка-
честве катализаторов в органич. синтезе.
Лит.: Гафний. Со. переводов, М., 1955; Меерсон Г. А.
иЗеликман А. Н., Металлургия редких металлов, М.,
1955; Smelin, Syst.-Num. 43, Hafnium, В., 1941; М е 1-
1 о г, v. 7, L.—N. Y. — Toronto, 1947, то же, 1952; U 1 m ап п,
3 Aufl., Bd 8, Munchen — В., 1957; Kirk, v. 15, N. Y., 1956;
Комиссарова JI. H., Плющев В. E., Усп. хим.,
1956, 25 вып. 10, 1197; их же, Ж. анал. хим., 1958, 13,
вып. 6, 709. Л. Н. Комиссарова.
ГВАЯКОЛ (1-окси-2-метоксибензол)СвН4(ОН)ОСН3,
мол. в. 124,13 — бесцветные или светло-желтые
призматич. кристаллы, т. пл. 98,4°, т. кип. 205°;
dll,i 1,1287; Пд 1,5383 (21,4°); хорошо растворим в
спирте, хлороформе, эфирных маслах, р-рах NaOH,
ледяной уксусной к-те, слабо— впетролеином эфире;
растворяет иод и серу. При метилировании Г. или при
перегонке его с РЬО получается ванилин; сплавле-
нием с калием превращается в пирокатехин. Пикрат
Г., т. пл. 80°; 1%-ный спиртовый р-р с FeCl3 дает
синее окрашивание, переходящее в зеленое. Г.
получают метилированием пирокатехина метил-
илидиметилсульфатом или CH3J в присутствии КОН
при 180°; Г. синтезируют также из о-анизидина диазо-
тированием и разложением водой образовавшегося диа-
зосоединения:
Ч^ОСНз S^'OCHg ЧухЧэСНз
NH2 N2Q ОН
Г. применяется для синтеза ванилина и лекарствен-
ных препаратов (тиокола и др.). и. В. Матвеева.
ГЕЙ-ЛЮССАКА ЗАКОНЫ — 1) Закон теплового
расширения газов, согласно к-рому изменение объема
данной массы газа при постоянном давлении прямо
пропорционально изменению темп-ры; может быть
записан в виде v( = г0(1 + «р0> где го — объем при
0°С, vt — объем при t°C, ар — коэфф, теплового
расширения газов при постоянном давлении. Для
всех газов при уменьшении давления коэфф. ар
стремится к одному и тому же предельному значению,
0,00367=1/273,15, не зависящему от темп-ры. Стро-
го подчиняются Г.-Л. з. лишь идеальные газы. Реаль-
ные газы тем лучше следуют этому закону, чем выше
их темп-ра и ниже давление. Если ввести понятие
813
ГЕКСАМЕТИЛЕНБЕНЗАМИД - ГЕКСАМЕТИЛЕНТЕТРАМИН
814
абсолютной температуры Т— l/ap+t=273,15 +t,
Г.-Л. з. может быть сформулирован след, обра-
зом: при постоянном давлении объем данной массы
газа прямо пропорционален его абс. темп-ре. Вместе
с законами Бойля — Мариотта и Авогадро Г.-Л. з.
составляет опытное основание уравнения состояния
идеального газа (см. Клапейрона уравнение). Установ-
лен Ж. Гей-Люссаком в 1802.
2) Закон объемных отношений, согласно к-рому при
постоянных темп-ре и давлении объемы газов, всту-
пающих в реакцию, относятся между собой и к объ-
емам газообразных продуктов реакции, как небольшие
целые числа. Этот закон сыграл важную роль в со-
здании молекулярно-кинетич. теории; находит объяс-
нение в Авогадро законе. Установлен в 1805—08.
В. И. Барановский.
ГЕКСАМЕТИЛЕНБЕНЗАМИД (N, N-гексаметилен-
бензамид, N-бензоилгексаметиленимин, гексамид)
C13H17NO, мол. в. 203,29 — бесцветные кристаллы без
запаха, т. пл. 36°, т. кип.
сн2—сн2—сн2. сое н, 190—190,5° (12 мм рт. ст.),
сн2—сн2—сн/ * ‘ п^'51, 5460, уд. теплоемкость
0,68 кал/г-град (в интервале
18—30°). Г. нерастворим в воде, растворим в спир-
те, эфире, ацетоне, бензоле, хлороформе, дизельном
топливе. Г. устойчив к действию солнечного света.
При кипячении Г. с разбавленными р-рами кислот
и щелочей образуется СвН5СООН и (CH2)eNH (гекса-
метиленимин), при нагревании с РС15 образуется
(СН2)вС12 и CeH5CN. Окисление Г. водным р-ром КМпО4
приводит к е-бензоиллейцину CeH5CONH(CH2)5COOH.
Г. получают взаимодействием гексаметиленимина
с СеН5СОС1 в присутствии разбавленного р-ра NaOH
сн2—сн2—сн2Ч
| )NH + C„H5COC1 + NaOH
СН2—СН2—СН2/
сн2—сн2—сн2
I
сн2—сн2—сн2
\NCOCeHj+NaCl+H2O
Практически Г. получают бензоилированием 40—
50%-ной азеотропной смеси гексаметиленимина с во-
дой (побочный продукт при произ-ве найлона).
Г. — безвреден при нанесении на кожу человека
и животных; при попадании на слизистую оболочку
глаз вызывает слабое жжение и гиперемию.
Г. обладает высокЬй репеллентной активностью
против гнуса (см. Отпугивающие средства). При
нанесении на кожу человека Г. отпугивает комаров
ок. 16 часов (при темп-ре воздуха 18—20°); опрыски-
вание 2%-ными водными эмульсиями Г. сельскохо-
зяйственных животных предохраняет их от укусов
слепней и комаров около 3 суток. Ткань, пропитан-
ная Г., отпугивает блох более 5 месяцев.
Лит.: Колесников Г. С. [и др.], Ж. общ. химии,
1957, 27, вып. 11—12, 3005; Бюл. комитета по делам изо-
бретений и открытий при Совете Министров СССР, i960,
[№] 18, сентябрь, с. 60. П. Б. Терентьев.
1,6-ГЕКСАМЕТИЛЕНДИАМИН NH„(CH2)eNH2,
мол. в. 116,20 — бесцветные блестящие кристаллы
в виде крупных пластинок или игл (при сублимации);
т. пл. 42 , т. кип. 204—205°, 100°/20 мм рт. ст.-,
давление паров (мм рт. ст.)'. 1,5 (50°), 25,8 (100°);
вязкость 1,21 спуаз (60°), 0,89 (80°); теплота сгорания
1062 ккал/моль. Во влажном воздухе Г. дымит; жадно
поглощает углекислый газ. Легко растворяется в воде
(960 г в 100 г воды при 30°), спирте, хлороформе,
эфире, бензоле и др. органич. растворителях; перего-
няется с водяным паром.
Аминогруппы в Г. легко могут быть замещены дру-
гими функциональными группами. Так, при дейст-
вии нитрозилхлорида на Г. при охлаждении в эфир-
ном р-ре образуется дихлоргексан. Взаимодействие
Г. с азотистой к-той приводит к получению гексан-
диола. С органич. и неорганич. к-тами Г. легко обра-
зует соли. Это свойство используется для колич.
определения Г. титрованием р-рами кислот. Полный
хлоргидрат Г. — игольчатые кристаллы (при кри-
сталлизации из воды или спирта), т. пл. 248°, легко
растворим в воде, трудно — в спирте, нерастворим
в эфире и хлороформе. Г. вступает в реакции конден-
сации и поликонденсации с двухосновными к-тами,
альдегидами, кетонами. Соли Г. при нагревании
с органич. к-тами превращаются в амиды соответст-
вующих к-т. Последнее свойство используется для
получения на основе Г. ценных полимерных продук-
тов — полиамидов. С солями металлов (золото, пла-
тина, ртуть, медь) Г. дает окрашенные труднорас-
творимые в воде комплексные соединения, нек-рые из
них растворяются в органич. веществах. Г. обладает
значительной физиология, активностью. Конц, р-ры
Г. вызывают ожог слизистых оболочек.
Наиболее распространенным промышленным ме-
тодом получения Г. является восстановление ди-
нитрила адипиновой кислоты:
NC—(СН3)4—CN + 4Н3 — NH3-CH»(CH3)4CH3NH2
Для получения его адипиновую к-ту смешивают с
аммиаком в паровой фазе, смесь пропускают при
300—375° через дегидрирующий катализатор (напр.,
силикагель), в результате образуется аммонийная соль
и амид адипиновой к-ты. При отщеплении воды от
диамида образуется динитрил адипиновой к-ты с вы-
ходом до 80%, считая на исходную адипиновую к-ту.
Динитрил гидрируют до Г. на катализаторах: медь —
кобальт (125°, 600—625 ат), кобальт на силикагеле,
никель Ренея (200 ат) и др. При гидрировании ди-
нитрила адипиновой к-ты происходит частичное
отщепление аммиака от Г. с образованием гексаме-
тиленимина; чтобы предотвратить это, гидрирование
производят в присутствии безводного аммиака. Г. по-
лучают также из бутадиена, к-рый хлорируют до
1,4-дихлор бутена. Последний обработкой цианистым
натрием переводят в 1,4-дицианбутен, гидрированием
к-рого получают динитрил адипиновой к-ты, превра-
щаемый в Г. описанным выше методом. Динитрил
адипиновой к-ты может быть получен также через
1,4-дихлорбутан, образующийся при гидрировании
1,4-бутиндиола (продукта конденсации ацетилена
с формальдегидом, по Реппе) до 1,4-бутандиола
с превращением последнего в 1,4-дихлорбутан обра-
боткой соляной к-той. Однако наиболее удобным
способом получения динитрила адипиновой к-ты яв-
ляется электрохимия. гидродимеризация акрило-
нитрила
2СН»=СН-CN % 2Н" — N=C~(CH3)4-C=N
Г. используется для получения полиамидов (напр.,
найлона).
Лит.: Солонина В., Ж. Русского физ.-хим. об-ва,
1896, 28, вып. 6, с. 556; Curtius Т. und С 1 е m m Н.
J. ftir praktische Chem., 1900, 62, S. 189—212; Neuberg C.’
Z. physiol. Chem., 1905, 45, H. 1—2, S. i 10; Ш о ф м а н С. М.,
Новости промышленности органического синтеза, 1958, №1—2,
с. 45; Волокна из синтетических полимеров, пер. с англ., М.,
1957, с. 119; Хопфф Г., Мюллер А., Венгер Ф.,’
Полиамиды, пер. с нем., М., 1958. С. В. Рогожин.
ГЕКСАМЕТИЛЕНДИИЗОЦИАНАТ — см. Изоциа-
наты.
ГЕКСАМЕТИЛЕНТЕТРАМИН (уротропин, гекс-
амин) CeH12N4, мол. в. 140,19—бесцветные ромбо-
эдрич. кристаллы; параметр решетки а = 7,02 А;
длины связей C=N 1,448±0,015А; С—Н 1,175±
±0,03 А; при 280° обугливается; выше 230° воз-
гоняется в вакууме. В 100 г воды растворяется 81,3 г Г.
(12°); с повышением темп-ры растворимость падает;
из воды кристаллизуется гидрат CeH12N4-H2O,
устойчивый ниже 14°; растворимость при комн,
темп-ре в 100 мл растворителя: в СНС13 13,4 г; в
СН3ОН 7,3 г; в С2Н5ОН 2,9 г; плохо растворим в че-
815
ГЕКСАМИДИН - ГЕКСАНДИОЛЫ
816
тыреххлористом углероде, бензоле и эфире; теплота
сгорания Cv 1006,7 ккал/г-моль; теплота образования
Д.На98 = —32,7 ккал/моль. Г. — однокислотное основа-
ние, с кислотами дает малоустойчивые соли, далее под
действием к-т разлагается на NH3 и СН2О; к щелочам
устойчив. С галогенами, нек-рыми солями и активны-
ми органич. галогенидами Г. образует комплексы,
напр.*. (CH2)eN4-2HgCl2; [(CH2)3N4"CH2C3H5]+C1 ; по-
следние используются в синтезах первичных аминов:
(CH2)eN4- RC1 + ЗНС1 + ^СаНдОН -> RNH2 • НС1 +
+ 3NH4C1 + бСН^ОСаНд^. Со многими ароматич.
соединениями Г. взаимодействует как формилирую-
щий агент, напр.:
-NH3
При нитровании Г. образуется гексоген (взрывчатое
вещество). При действии H2S на Г. получается тио-
формальдегид CH2S. В пром-сти Г. получают взаи-
модействием водного или газообразного NH3 с фор-
малином с последующим упариванием р-ра в ва-
кууме. Количественно Г. определяется титрованием
NH3, образующегося при кислотном гидролизе. При-
меняют Г. в произ-ве феноло-формальдегидных син-
тетич. смол, в произ-ве гексогена, в медицине как
антисептик и в аналитич. химии для нейтрализации
кислот, приготовления буферных р-ров, для микро-
кристаллоскопич. открытия ряда ионов, для осаж-
дения и отделения Fe, Al, Сг, Zr, Ti от двухвалентных
металлов группы H2S.
Лит.: и liman п, 3 Aufl., В<1 3, Mtlnch. — В., 1953, S. 164;
К 1г Ь, V. 7, N. Y., 1951, р. 466; Китайгородский А. И.,
Ор.ганическая кристаллохимия, М., 1955. А. Ф. Бочков.
ГЕКСАМИДИН (мисолин, примидон) C12H14O2N2,
мол. в. 218,26 — белые кристаллы (чешуйчатые пла-
стинки) горьковатого вкуса;
с0\ /свн» т. пл. 277—281°. Г. плохо рас-
2G\nh-co/Cx'C2Hs творим в воде (1:2000), лучше—
в спирте и ацетоне, почти не-
растворим в эфире, хлороформе, уксусноэтиловом
эфире; кристаллизуется из кипящего 80%-ного спир-
та (1 : 25). При нагревании Г. с 50%-ной серной
к-той (1 : 10) выделяется формальдегид. Количест-
венно Г. определяют по методу Кьельдаля, ам-
миак отгоняют в р-р борной к-ты и титруют 0,1 н.
р-ром H2SO4 с индикатором метиловым красным.
Получают Г. конденсацией диамида этилфенилмало-
новой к-ты с формамидом или с муравьиной к-той.
Применяют Г. для лечения эпилептической болезни.
Лит.: British Pharmacopoeia, 1958, L., 1958, р. 523, 891;
см. также при ст. Апрессин. Е. С. Головчинская.
ГЕКСАН (н-гексан) СН3(СН2)4СН3, мол. в. 86,18 —
бесцветная жидкость; т. пл. —94,0°; т. кип. 68,95°;
4 0,6603 (20°); 0,6770 (0°); Ир 1,37506; гКрит.234,8°,
Т’крит. 29,62 ат; Укрит. 4,266 мл/г; октановое число 25;
Г. найден в нефтях; в значительных количествах
содержится в бензинах прямой гонки и крекингди-
стиллятах нефтей; является одной из главных
составных частей синтетич. бензина, получаемого
из СО и Н; из этих продуктов может быть выделен
четкой ректификацией. Г. может быть синтезирован:
1) гидрогенизацией гексена, полученного дегидрата-
цией гексанола; 2) электролизом бутирата калия;
3) разложением водой гексилмагнийгалогенидов;
4) Вюрца реакцией и др. способами. Дегидроциклиза-
цией Г. превращается в бензол. Ввиду низкого окта-
нового числа является нежелательной составной
частью бензинов. Изомеры Г. 2,2-диметилбутан (неогек-
сан) и 2,3-диметилбутан (диизопропил) применяют для
улучшения моторного топлива. г. м. Циеуро.
ГЕКСАНДИОЛЫ СвН12(ОН)2, мол. в. 118,17 —
двухатомные спирты с углеродным скелетом н-гек-
сана; существует 9 изомерных Г.; 1,2-, 1,3-, 1,4-
и 1,5-Г. имеют по одному, а 2,3-, 2,5- и 3,4-Г. — по
два асимметрия, атома углерода; абс. конфигурации
оптич. изомеров Г. не выяснены. Все Г. растворимы
в воде, спирте и др. органич. растворителях. В табл,
приведены нек-рые свойства гександиолов.
Положение ОН-групп Т. пл., °C Т. кип., °С/Л4.Н jt d4 t nD
1,2а 110—113/6
1,3е — 123/13 0,958 (22°) 1,4461 (22°)
1,4е -20 134—135/18,5 0,982 (16°) 1,453.(16,3°)
1,5 е — 234—235/710
< 1,6 42 250/760 — —
2,3В 207/760 0,9669 (0°) —
2,4В — 210—211/750 105—105,5/0 0,9516 (21°) 1,4418 (21°)
2,5В тв. модиф. 43—44 жидк.модиф. 216—218/750 120—122/12 0,9610 (20°) 1,4475/20°
3,4® -20 мезоформа 88 87-88/15 91/20 — —
а Щр = 4- 15,2° (в спирте). 6 Рацемат. в Конфигурация
неизвестна.
По химич. свойствам Г. аналогичны другим гли-
колям; они образуют гликоляты, галогенгидрины
и т. д. Под действием H2SO4 Г. легко циклизуется,
напр.:
ZCH2- СН2
НОСН2(СН2)3СНОНСН3Н2С zo
сн2- сн-СНз
Получают Г. след, способами: 1,3-, 1,6- и 2,3-изо-
меры— гидролизом бромпроизводных, напр.:
Вг(СНа)аСНВг(СН2)аСН3+НаО -
- НО(СН2СНОН(СН2)аСН3
изомеры 1,4- и 1,5-восстановлением соответствую-
щих оксикетонов или оксиальдегидов, напр.:
СаН5СО(СНа)2СНОН—С2Н5С(ОН)(СН2)2СН2ОН
изомеры 2,5- и 3,4-гидрированием непредельных
диолов, напр. СН3СНОНС=ССНОНСН3
—СН3СНОН(СН2)2СНОНСН3;т2,4-гександиол— кон-
денсацией альдоля СНдСНОНСНО и этилмагний-
бромида CjHjMgBr; 1,2-гександиол (рацемат) — вос-
становлением натрием в спирте этилового эфира
а-оксикапроновой к-ты СН3(СН2)3СНОНСООС2Н5;
правовращающий изомер образуется при энзиматич.
восстановлении а-оксиметилбутилкетона. Промыш-
ленное применение нашел 1,6-Г. (гексаметиленгли-
817
ГЕКСАНИТРОДИФЕНИЛАМИН — ГЕКСАХЛОРБЕНЗОЛ
818
коль, «адиполь»), В пром-сти производят восстановле-
нием адипиновой к-ты:
НООС(СН2)4СООН —2НОСНа(СНа)4СНаОН
применяется в произ-ве полиэфирных и полиурета-
новых синтетич. смол. м. Ф. Турчинекм.
ГЕКСАНИТРОДИФЕНИЛАМИН (дипикриламин,
гексил, гексит) C12H5O12N7, мол. в. 439,22 — желтые
кристаллы; т. пл. 244—
245° (с разл.); плотн.
1,653; почти нераство-
рим в воде и эфире;
растворим в ацетоне,
теплой конц. уксусной
к-те, дымящей HNO3.
Г. является вторичным взрывчатым веществом', теп-
лота взрыва 1035 ккал/кг; скорость детонации
7100 м/сек при плотности 1,58 г/см3; работоспособ-
ность и чувствительность к механич. воздействиям
больше, чем у тротила, и меньше, чем у тетрила;
ядовит. Получают Г. нитрованием дифениламина или
взаимодействием динитрохлорбензола с анилином
с последующим нитрованием полученного динитро-
дифениламина. Г. применялся во время 1-й и 2-й
мировых войн, особенно в Германии, для снаряже-
ния авиабомб, мин, торпед и т. д., обычно в смесях
с другими взрывчатыми веществами. Г. применяют
в качестве реактива в микрокристаллоскопич. ана-
лизе в виде разбавленного р-ра натриевой или маг-
ниевой соли; образует характерные оранжево-красные
кристаллы с К.+, Rb+, Gs+, Т1+, Веа+, Zr2+, Pb2+ и
Hg2+, с КНфгголько при его повышенной концентрации;
Na, Li, Mg, Ba, Sr и Са не осаждаются. Для весовых
определений К, Rb и Cs применяют обычно магниевую
соль, как наиболее растворимую. Дипикриламинат
калий используют для титриметрич. и фотометрия,
(по оранжево-красной окраске ацетонового р-ра) опре-
делении калия. Аммонийная соль известна под назва-
нием «ауранция», краситель для шелка и бумаги.
Лит.: Орлова Е. Ю., Химия и технология бризантных
взрывчатых веществ, М., 1960.
Б. Н. Кендриков, В. Г. Типцова.
ГЕКСАОКСИБЕНЗОЛ (гексафенол) СвНвОв, мол. в.
174,05 — игольчатые кристаллы, к-рые при 200° раз^
лагаются не плавясь; трудно растворим в холодной
воде, спирте, эфире, бензоле; Г. сильный восстано-
витель; его растворы, окисляясь на воздухе, окра-
шиваются в фиолетовый цвет; при этом в присутствии
соды образуется тетраоксихинон (I). При испарении
р-ра Г., содержащего
Q 9 КОН, образуется кро-
НОх/Ч^ОН тонат калия; нагрева-
11 ' |Г J Т нием с цинковой пылью
HO^'-x/soh Г. превращается в бен-
I I зол и дифенил. HNO3
и О окисляет Г. в трихи-
1 11 ноил (И). Гексаацетил-
производное (т. пл. 203°) получается при нагревании
Г. с СНдСООН и CH3COONa. G FeCl3 Г. дает фиолетовое
окрашивание. Г. получают в виде фенолята С3(ОК)3
действием СО на нагретый калий. Г. образуется при
восстановлении тетраоксихинона или трихиноила, при
окислении инозита С3Нв(ОН)в, а также в качестве
побочного продукта при произ-ве металлич. калия
из поташа. И- В. Матвеева.
ГЕКСАХЛОРАН (гексахлорциклогексан) СвН3С1в,
'мол. в. 290,86 — смесь 8 изомеров 1,2,3,4,5,6-гекса-
хлорциклогексана, образующихся при присоедине-
нии хлора к бензолу.
По современным представлениям все 6 атомов углерода
молекулы циклогексана расположены не в одной плоскости,
а в виде т. н. «кресла» (рис. 1). Углеродные атомы 1, 3, 5 лежат
в одной плоскости, а 2, 4, 6— в другфй, параллельной. Рас-
стояние между плоскостями равно 0,5А, а С—С1 связи имеют
по отношению к плоскости углеродных атомов 2, 3, 5 н 6 два
направления: вертикальное, направленное параллельно оси
симметрии (обозначается буквой р), и горизонтальное (или
экваториальное), направленное к периферии молекулы (обо-
значается буквой е). Такое расположение связей и различное
распределение по ним атомов Н и С1 обусловливает наличие
изомеров у гексахлорциклогексана и различия их химич. и
физич. свойств. Энергетически устойчивыми являются 8 изо-
меров, к-рые имеют след, ориентацию связей С—С1 (рис. 2):
е peepee
Ч рререе
(или ререре)
ц реррее
6 ререее
а ррееее
6 ееееее
т ррреее
о реееее
Г. — кристаллич. продукт, без запаха, горьковато-
го вкуса; темп-ра плавления различных изомеров Г.:
а 157,5—158,5, 0 309°, у 111,8—112,8°, 6 138—139°,
е 218,5—219,3°, С 88—вЭ6, Т] 89,8—90,5°, » 124—125°.
При нагревании Г. возгоняется с образованием густо-
го белого дыма. Г. устойчив на свету, а также к
воздействию воды, различных окислителей и конц.
кислот. Под действием оснований Г. разлагается:
CeHeCle + 3NaOH ->СвН3С13 + 3NaCl + ЗН2О. При
нагревании до 220° в присутствии железных опилок
(2%) Г. бурно разлагается. Колич. определение Г.
основано на его разло-
жении спиртовым р-ром
щелочи.
Рис. 2.
• -атомы углерода
Рис. 1.
Г. — один из важнейших современных инсекти-
цидов; из изученных изомеров инсектицидными свой-
ствами обладает в основном у-изомер (токсичен, ЛД50
125 мг/кг); в технич. Г. его содержится 10—18%.
Обычно в сельском хозяйстве применяют препарат,
известный под названием линдан, содержащий
99—100% у-изомера. Линдан получают экстракцией
у-изомера из технич. Г. селективными растворите-
лями (напр., метанолом). Технич. Г. получают фото-
химич. хлорированием бензола или хлорированием
бензола в присутствии катализаторов [NaOH, Са(ОН)2,
перекиси и др.], катализирующих присоединение
хлора. Технич. Г. обладает стойким неприятным за-
пахом; применяется для борьбы с саранчой и нек-рыми
др. вредителями (огневка, свекловичный долгоносик
и т. п.), а также для борьбы с нек-рыми паразитами
животных, в частности с клещами. Линдан приме-
няется более широко Для протравливания семян,
борьбы с вредителями овощных, плодовых и технич.
культур, с картофельным жуком (в комбинация
с ДДТ) и обитающими в почве вредителями. Г. ис-
пользуют в виде дустов, смачивающихся порошков,
эмульсий с маслами, р-ров в маслах и др. (см. Пести-
цидные препараты); норма применения 1—3 кг/га.
Лит.: Безобразов Ю. Н., Молчанов А. В.,
Г а р К. А., Гексахлоран, его свойства, получение и приме-
нение, М., 1958. Н. Н. Мельников.
ГЕКСАХЛОРБЕНЗОЛ (перхлорбензол) СвС1вР
мол. в. 284,8 — бесцветные моноклинич. призматич.,
легко сублимирующиеся кристаллы; т. пл. 231°;
т. кип. 322°; daa,s 2,044; нерастворим в воде и холод-
ном спирте, растворим в горячем спирте, бензоле,
эфире, хлороформе, CSa. Хлор связан менее прочно,
чем в других менее замещенных хлорбензолах. При
180° метилат натрия превращает Г. в пентахлорфе-
нол НОС3С15 и пентахло ран изол СН3ОС3С16; анало-
гично действует раствор NaOH в метиловом спирте
при 130 —140°; при кипячении с дымящей HNO3
образуется хлоранил (I) (золотистые листочки,
т. пл. 290° н капилляре). Синтезируют Г. исчерпыва-
819
ГЕКСАХЛОРДИМЕТИЛКАРБОН AT — ГЕКСОГЕН
820
ющим хлорированием бензола в паровой фазе при
600°. В пром-ети применяют также т. н. окислительное
_ _ хлорирование бензола смесью НС1 и
\ т* кислорода или воздуха; процесс ведут
при 200—250° или 300—400° в зави-
О==( симости от катализатора (двухвалент-
/==\ ная медь, активированнан, напр., А1
Cl Ci и Fe). Г. предложен для протравлива-
т ния семян, для синтезов пентахлор-
фенола, применяющегося для консер-
вирования древесины, и пента хлортиофенола, являю-
щегося пластификатором каучука. е. е. Барони.
ГЕКСАХЛОРДИМЕТИЛКАРБОНАТ (трифосген)
ОС(ОСС13)2, мол. в. 296,87 — кристаллич. продукт;
т. пл. 78—79°; т. кип. 205—206°; практич. нераст-
ворим в воде, хорошо растворяется в спирте и эфире.
Водой на холоду Г. гидролизуется медленно, быстрее
при нагревании. Водные р-ры щелочей быстро раз-
лагают Г. до солей угольной и соляной к-т. С анили-
ном Г. реагирует с образованием мочевины, с алко-
голятами дает эфиры угольной к-ты; при 300—360°
разлагается с образованием фосгена. Г. может быть
получен хлорированием диметилкарбоната. Пары Г.
обладают удушающим действием, напоминающим дей-
ствие фосгена и дифосгена. р. н. Стерли».
ГЕКСАХЛОРЭТАН (перхлорэтан, гексоран)
СС13СС13, мол. в. 236,76 — бесцветные кристаллы со
слабым камфарным запахом. Г. существует в трех
кристаллич. модификациях; кубической, триклинной
и ромбической (устойчивы соответственно выше 71,1°,
в интервале 71,1—43,6°, ниже 43,6°); т. пл. 189°
(в запаянном капилляре), т. кип. 186°/760 мм,
73,5°/10 мм, 32,7°/1 мм; тройная точка 186°; в откры-
тых сосудах Г. возгоняется не плавясь; с£|“ 2,091.
Теплота сгорания при 18,7° (г> = const) 734,4 кал/г;
теплота возгонки кубич. формы 12,2±0,1 ккал!моль,
триклинной 14,1±0,2 ккал/моль. Г. нерастворим
в воде, умеренно растворим в спирте и эфире, хорошо—
в CS2. Для человека нетоксичен. При взаимодействии
Г. с Zn в водной среде образуется тетрахлорэтилен
(СС12=СС12). Г. устойчив к действию кислот и щело-
чей на холоду.
Получают Г. хлорированием тетрахлорэтилена при
100—200° под давлением; образуется как побочный
продукт в произ-ве СС14 из CS2. Г. применяют как за-
менитель камфары в нитроцеллюлозных пластмассах,
интенсификатор свечения пиротехнич. составов, как
глистогонное средство (фасциолин), а также в сме-
си с некоторыми металлами в качестве дымообра-
ЗОВателЯ. ’ Ф. Я. Величко.
ГЕКСЕНАЛ (эвипан-натрий, гексобарбитон-нат-
рий, циклонал-натрий) C12H15O3N2Na, молекулярный
вес 258,26— белые, расплываю-
Огп-м-СН, щиеся на воздухе кристал-
лы, слабогорького вкуса; раз-
ТХСО—лагается двуокисью углерода.
Н3С ''Na г легко растворим в воде и
спирте, мало растворим в эфи-
ре и хлороформе. Водные р-ры Г. неустойчивы,
при действии на них СО2 выделяется 5-циклогексенил-
1-ил-1,5-диметилбарбитуровая к-та. Г. дает все реак-
ции, характерные для барбитуратов’, выделяет ам-
миак при нагревании с щелочами, образует серебря-
ную соль с азотнокислым серебром и др. Для колич.
определения водный раствор Г. титруют 0,1 н.
р-ром соляной к-ты в присутствии метилового оран-
жевого. Получают Г. конденсацией эфира а-циан-
а-циклогексен-1-ил-пропионовой к-ты с монометил-
мочевиной с последующим гидролизом продукта
реакции и взаимодействия образовавшейся 5-цикло-
гексен-1-ил-1,5-диметилбарбитуровой к-ты с рассчи-
танным количеством р-ра щелочи. Г. применяют гл.
обр. как наркотич. средство (внутривенный наркоз),
в нек-рых случаях в качестве снотворного препарата,
как противосудорожное средство и др.
Лит. см. при ст. Адалин. Е. С. Голоечинская.
ГЕКСИЛОВЫЕ СПИРТЫ СвН13ОН, мол. вес
102,17 — бесцветные жидкости со слабым запахом;
мало растворимы в воде, хорошо растворимы в ор-
ганич. жидкостях. Известны 17 изомерных Г. с., из
них 7 оптически активны. Промышленное значение
приобрели 3 Г. с. (см. табл.).
Спирт Т. кип., “С ЛЬ d4 25 Пр
Гексанол-1 СН3 (СН2)4 СН2ОН 157,47 0,8224 (15е) 1,4158
2-Метилпентанол-2 (СН3)2СНСН2СНОНСН3 131,82 0,8025 1,4089
2-Этилбутанол-1 (C2H6)2CHCHSOH 146,27 0,8296 1,4205
Г. с. содержатся в сивушном масле. Синтезируют
гексанол-1 и 2-этилбутанол-1 альдольной конденса-
цией масляного альдегида и ацетальдегида с после-
дующим гидрированием образовавшегося непредель-
ного альдегида:
СН3СНаСН2СНО + СНзСНО —
,СН3СН»СН2=СНСНО + На СН3(СНа)4СН3
~^СН3ССНаСНО 4- На (CsH5)aCHCHsOH
(^НСНз
2-Метилпентанол-2 получают гидрогенизацией окиси
мезитила:
(СН3)аС=СНСОСНз+На - (СНз)аСНСНаСнОНСН8
Применяются Г. с. в качестве растворителей синте-
тич. смол в произ-ве лаков и красок и др.; гексанол-1—
в произ-ве нек-рых фармацевтич. препаратов и мою-
щих средств, в пром-сти душистых веществ.
М. Ф. Турчинский.
ГЕКСИЛРЕЗОРЦИН (1,3-диокси-4-н-гексилбензол)
С12Н18О2, мол. в. 194,27 —- бесцветный или слегка
желтоватый порошок или чешуйча-
тые кристаллы, розовеющие на ОН
воздухе; т. пл. 67—69°; т. кип. Js.
178—180° (6—7 мм). Г. растворим р |)
в спирте, ацетоне, эфире, хлоро-
форме и растительных маслах, мало
растворим в воде и петролейном СН2(СН2)4СН3
эфире. При прибавлении к спир-
товому р-ру Г. 1—2 капель р-ра FeCl3 появляется
зеленовато-желтое окрашивание. Для колич. опре-
деления Г. к спиртовому р-ру прибавляют КВг,
0,1 н. р-р КВгО3 и соляную к-ту и титруют до обес-
цвечивания 0,1 н. р-ром Na2S2O3 в присутствии K.J
и хлороформа. Для получения Г. бутилбромид кон-
денсируют с малоновым эфиром; образующийся бу-
тилмалоновый эфир омылением и частичным декар-
боксилированием превращают в капроновую к-ту,
при взаимодействии к-рой с резорцином получают
капронилрезорцин. При его восстановлении цинком
в соляной к-те образуется Г. Применяют Г. в качестве
противоглистного и бактерицидного средства.
Лит. см. при ст. Адалин. А. И. Травин.
ГЕКСОГЕН (циклотриметилентринитроамин), мол.
вес 222,13, C3HeNeOe — вторичное взрывчатое ве-
щество (бризантное), более мощное и более чувстви-
тельное, чем тротил, пикриновая кислота и тетрил;
бесцветные кристаллы орторомбич. системы, т. пл.
204,1°, d 1,816. Г. нерастворим в воде, плохо раство-
рим в спирте, эфире, бензоле, толуоле, хлороформе,
лучше — в ацетоне, конц. азотиой и уксусной к-тах,
циклогексаноне. Теплота сгорания 2307 кал/г. Теп-
лота взрыва 1300 кал/г; объем газообразных продуктов
821
ГЕКСОЗЫ
822
плотности 1,70. I
NOe
I
ZN\
Н2С сн2
OaN—N N-NOa
сн2
взрыва 908 л/кг. Скорость детонации 8350 м/сек при
сширение в свинцовой бомбе
500 cjh3. Г. разлагается серной
к-той, едкими щелочами, а также
при нагревании; т. вен. ок. 230°.
Получают Г. нитрованием гекса-
метилентетрамина (уротропина)
конц. HNO3; применяют для из-
готовления детонаторов, снаря-
жения боеприпасов и для взрыв-
ных работ в пром-сти, как пра-
вило, в смеси с другими в-вами (напр., тротилом,
алюминием), а также с добавкой флегматизаторов
(парафина, воска, церезина), уменьшающих опасность
взрыва Г. от случайных . причин.
Лит.: Горст А. Г., Химия и технология нитросоедине-
ний, М., 1940; Орлова Е. Ю., Химия и технология бри-
зантных взрывчатых веществ, М., i960. Б. Н. Кандриков.
ГЕКСОЗЫ — сахара, содержащие шесть углерод-
ных атомов в молекуле. Г. вместе со своими произ-
водными представляют самую большую группу моно-
сахаридов; их рассматривают как родоначальников
ряда производных сахаров. В зависимости от харак-
тера карбонильной группы Г. делятся на альдо-
гексозы и кетогексозы. Группа альдо-
гексоз с нормальной углеродной цепью включает
16 стереоизомерных сахаров: 8 пар D- и L-изомеров,
D-формы к-рых представлены D-рядом:
нс=о нс=о НС=О НС=О
неон но<!;н HtioH НОСН
HtioH HtioH 1 носн нос!:н
неон неон HtioH неон
неон неон неон неон
HjCOH насон Н2СОН HsioH
D-аллоза D-альтроза D-глюкоза D-манноза
НС=О НС=О нс=о нс=о
HtioH НО^Н неон НОСН
неон 1 неон носн НОСН
1 носн носн носн НОСН
неон неон неон неон
1 н2сон насон На СОН насон
D-гулоэа D-идоза D-галактоза D-талоза
Нек-рые альдогексозы (глюкоза, галактоза, манноза)
встречаются в природе в свободном виде и в виде
гликозидов, полисахаридов и фосфорных эфиров,
входят в состав гликопротеидов, гликолипидов и др.
сложных веществ. Самым распространенным сахаром
является глюкоза. В природе преобладают D-формы
альдогексоз, однако нек-рые альдогексозы, напр.
галактоза, встречаются и в виде L-формы. Нек-рые
альдогексозы получены только синтетически (аллоза).
Г. с разветвленной цепью встречаются редко (гама-
мелоза и др.). В кетогексозах кетогруппа может
занимать различное положение, но до настоящего
времени в природе обнаружены только 2-кетогексозы,
D-формы к-рых представлены ниже:
Н»СОН Н3С0Н На СОН 1 насон
о 1 с=о с=о 1 с=о
носн 1 неон HtioH носн
неон неон носн носи
неон неон неон н<к>н
H£(ioH । Н2СОН н»сон насон
D-фруктоза D-псикоза D-сорбоза D-тагатоза
З-Кетогексозы (глютоза, гадтоза) получены синтети-
чески.
Г. — кристаллы, хорошо растворимые в воде.
В водных р-рах каждая из Г. существует в виде не-
скольких таутомерных форм (а- и 0-пираноз, а-
и 0-фураноз, открытой альдегидной формы), между
к-рыми в растворах устанавливается равновесие
(см. Мутаротация). На равновесие влияют строение
Г., растворитель, темп-ра, концентрация ионов водоро-
да и др. Так, напр., глюкоза образует след, формы;
а - D-глюкопираноза & -D-глюнопираноза
Путем подбора условий кристаллизации можно
выделить нек-рые формы Г. из р-ров.
В нейтральных и слабокислых р-рах Г. довольно
устойчивы. Действие конц. сильных к-т приводит
к дегидратации с образованием ангидросахаров,
5-оксиметилфурфурола, левулиновой к-ты и др.
продуктов. В щелочных р-рах идет эпимеризация Г.
и изомеризация альдогексоз в кетогексозы (Лобри де
Бруина и Ван Экенштейна реакция). Напр., из глю-
козы образуются манноза и фруктоза и далее идет
эпимеризация у 3-го атома углерода фруктозы. Про-
исходит также расщепление углеродной цепи Г.,
внутримолекулярное окисление и восстановление и
образование производных с разветвленной углерод-
ной цепью, напр. сахариновых к-т.
Г. дают все реакции восстанавливающих сахаров,
альдоз и кетоз соответственно. Пара эпимерных аль-
догексоз и соответствующая им кетоза дают один
и тот же озазон. При окислении альдогексоз обра-
зуются одноосновные гексоновые кислоты и двухоснов-
ные гексаровые кислоты (сахарные кислоты с 6-ю
углеродными атомами в молекуле), что используется
для идентификации альдогексоз. Окисление первич-
ной спиртовой группы Г. (при защите альдегидной)
приводит к гексуроновым кислотам. Кетогексозы
окисляются с расщеплением цепи. При восстановле-
нии Г. (альдоз и кетоз) образуются г е к с и т ы.
Получают Р. из природных источников, гл.
образом гидролизом полисахаридов (глюкоза из
крахмалов, фруктоза из фруктанов, галактоза из
галактанов и т. д.) и гликозидов. Синтетически Г.
получают из веществ неуглеводного характера (фор-
мальдегида, дибромидакролеина) и из сахаров,
гл. обр. путем увеличения цепи у пентоз. Напр.,
альдогексозы получают из альдопентоз Фишера—
Килиани циангидринным методом или конденсацией
с нитрометаном, кетогексозы — конденсацией хлор-
ангидридов пептоновых к-т с диазометаном. Отдель-
ные представители Г. часто получают из изомеров,
используя их способность к эпимеризации, или глика-
левым синтезом. Все Г. применяются в качестве ис-
ходных веществ в синтезах различных сахаров и их
823
ГЕКСОКИНАЗА — ГЕКСУРОНОВЫЕ КИСЛОТЫ
824
производных; глюкозу широко применяют как пищевой
продукт, лекарственное средство и исходное сырье в
спиртово-бродильной промышленности (см. Брожение).
Лит.; The Carbohydrates, ed. W. Pigman, N. Y., 1957;
M i c h e e 1 F., Chemie der Zucker und Polysaccharide, 2 Aufl.,
Lpz., 1956; Advances Carbohydr. Chem., 1951 6, p. 83;
там же, 1956, И, p. 193. Л. И. Линевич.
ГЕКСОКИНАЗА — фермент группы фосфокиназ,
относится к белкам типа альбумина', катализирует
фосфорилирование D-глюкозы за счет одной под-
вижной группы НРО3 аденозинтрифосфорной к-ты
(АТФ) (см. Аденозинфосфорные кислоты) с образова-
нием D-глюкозо-б-фосфата и аденозиндифосфорной
к-ты (АДФ): D-глюкоза + АТФ ге«со«иг'^ D-глю-
козо-6-фосфат + АДФ. Реакция обратима; АТФ мо-
жет быть заменена инозинтрифосфорной кисло-
той. Кристаллич. Г. выделена из дрожжей (из
10 кг дрожжей получается 40 мг фермента), мол. в.
96 600; поданным электрофореза и ультрацентрифуги-
рования— гомогенна; изоэлектрическая точка при pH
4,5—4,8; оптимум действия фермента при pH 8—9.
Г. активируется Mg. Фермент фосфорилирует также
D-фруктозу, D-маннозу, D-глюкозамин. Г. широко
распространена в животных тканях, обнаружена
в клубнях картофеля. Свойства фермента несколько
меняются в зависимости от источника его получения.
Гексокиназная реакция является начальной стадией
превращения гексоз при анаэробном расщеплении их
(напр., при брожении и гликолизе) и при биосинтезе
полисахаридов.
Лит.: Владимиров Г. Е. [и др.], Биохимия, 1957,
22, вып. 6, с. 963; Диксон М., Уэбб И., Ферменты, пер.
с англ., М., 1961. См. также при ст. Ферменты.
Т. Т. Болотина.
ГЕКСОНИЙ (гексаметоний, ганглиостат, мето-
ний, дииодид гексаметилен-1,6-бис-триметиламмония)
(CH3)3N(CH2)5N(CH3)3 • 2J, мол. в. 456,21 — бесцвет-
ные кристаллы солоновато-горького вкуса, хорошо
растворимые в воде и трудно — в спирте и ацетоне;
т. пл. 277—278°. При взаимодействии водного р-ра
Г., подкисленного разб. H2SO4, с NaNO2 в присутствии
р-ра крахмала появляется сине-фиолетовое окраши-
вание. Для колич. определения Г. к его водному р-ру
прибавляют 0,1н. р-р AgNO3, подкисляют HNO3
и титруют 0,1н. р-ром NH4SCN в присутствии же-
лезоаммонийных квасцов (розовато-желтое окраши-
вание). Г. получают взаимодействием 1,6-гексаме-
тилендиамина с муравьиной к-той и формалином; на
образующийся гексамет;илен-1,6-бис-диметиламин дей-
ствуют CH3J. Применяют Г. при лечении заболеваний,
связанных с нарушениями нервной регуляции: спаз-
мах периферич. сосудов (эндартерииты, перемежаю-
щаяся хромота ит. д.), в разных стадиях гипертония,
болезни, при гипертонии, кризах, язвенной болезни
желудка, нек-рых формах бронхиальной астмы и т. п.
Лит.; Торф С. Ф., Хромов-Борисов А. В.,
Мед. пром-сть СССР, 1958, № 2, с. 18. А. И. Травин.
ГЕКСОНОВЫЕ КИСЛОТЫ (альдогексоновые кис-
лоты) СООН(СНОН)4СН2ОН, мол. в. 196,16 — сте-
реоизомерные алъдоновые кислоты с шестью угле-
родными атомами в молекуле. Г. к. содержат по 4
асимметрии, атома углерода в молекуле и образуют
16 стереоизомеров (8 пар D-и L-антиподов). Г. к.
D-ряда представлены формулами:
соон
н<!:он
н<!:он
I
неон
I
неон
СН2ОН
D-аллоно-
вая
СООН
I
НОСН
I
неон
I
неон
I
неон
<!:наон
D-альтро-
иовая
СООН
I
неон
I
носн
I
неон
I
неон
I
сн2он
D-ГЛЮКО-
новая
СООН
I
НОСН
НОСН
I
неон
н<!:он
<!:н2он
D-мавно-
иовая
соон соон СООН СООН
j неон носн неон НОСН
неон неон носн НОСН
1 носн но<!:н НОСН НОСН
неон неон неон неон
1 СН2ОН СН2ОН СН2ОН СНаОН
D-гулоно- D-идоновая D-галакто- D-талоно-
вая новая вая
Названия Г. к. даны по альдогексозам, продуктом
окисления к-рых они являются (аллоновая, альтро-
новая, глюконовая, манноновая и т. д.). Г. к. хорошо
растворимы в воде, плохо — в большинстве органич.
растворителей; дают все химии, реакции альдоновых
к-т. Темп-ры плавления нек-рых Г. к.: L-альтроно-
вой 110°, D-глюконовой 130—132°, D-галактоновой
147,5°, D-талоновой 138°; [a]g (в воде) нек-рых Г. к.:
D-аллоновой —10,1°, D-альтроновой +35,4°, L-альт-
роновой —8,1°, D-глюконовой —6,7°, D-манноновой
+ 15,6°, D-гулоновой —1,6°, D-галактоновой —13,3°,
D-талоновой +19°. Полуиают Г. к. общими методами
полуиения альдоновых кислот, напр. окислением
альдоз или циангидринным методом из пентоз. D-глю-
коновая к-та образуется при действии на p-D-глю-
козу ферментов оксидаз или дегидрогеназ глюкозы
(антибиотик нотатин и др.); a-D-глюкоза, D-манноза,
D-альтроза и D-галактоза окисляются при действии
этих оксидаз в 100—1000 раз медленнее P-D-глюкозы,
а др. альдогексозы не окисляются. Окисление D-глю-
козы при действии нек-рых ферментов идет до кето-
глюконовой к-ты. D-глюконовую к-ту в промышлен-
ном масштабе получают электролитич. окислением
D-глюкозы в присутствии КВг и мела, а также глю-
коновокислым брожением глюкозы. Глюконовокис-
лые соли нек-рых металлов применяют как лекарст-
венные препараты (кальция — при пониженной свер-
тываемости крови, аллергии, туберкулезе и др. за-
болеваниях; железа — при малокровии). Г. к., их
лактоны и соли используют для характеристики аль-
догексоз и для получения пентоз и др. сахаров.
Лит, см. прист. Альдоновые кислоты, Л. И. Линевич.
ГЕКСОНОВЫЕ ОСНОВАНИЯ — устаревшее на-
звание диаминокарбоновых кислот, содержащих по
шесть атомов углерода (см. Аргинин, Гистидин,
Лизин).
ГЕКСУРОНОВЫЕ КИСЛОТЫ СНО(СНОН)4СООН,
мол. в. 194,14 — стереоизомерные уроноеые кисло-
ты', называют Г. к. по соответствующим альдогек-
созам (см. Гексозы), от к-рых Г. к. отличаются тем, что
в положении 6 находится карбоксильная группа
СООН, вместо СН2ОН. В природе встречаются
D-глюкуроновая (I), D-галактуроновая (II), D-ман
нуроновая (III) и L-идуроновая (IV) кислоты
СНО СНО СНО СНО
1 неон неон НОСН неон
носн носн НОСН носн
неон носн НСОН неон
неон неон неон носн
соон соон соон соон
I П III IV
Г. к. входят в состав многих физиологически актив-
ных веществ; гликозиды уроновых к-т в большом
количестве содержатся в почве.
Г. к. — кристаллич. продукты, хорошо растворимы
в воде, щелочах и р-рах карбонатов и бикарбонатов,
плохо растворимы в углеводородах и в большинстве
органич. растворителей.
825
ГЕКСУРОНОВЫЕ КИСЛОТЫ —ГЕЛИ
826
Глюкуроновая кислота широко распростра-
нена в природе, она входит в состав гепарина, хондроитин-
серной кислоты, гиалуроновой кислоты и др. физиологически
активных веществ животного происхождения; является струк-
турным компонентом гуммиарабика, многих растительных ка-
медей, сапонинов, гликозидов, олигосахаридов растительного
происхождения; входит в состав специфич. полисахаридов
нек-рых микроорганизмов (пневмококков и др.). В животном
организме глюкуроновая к-та играет большую роль в процессе
выделения из организма ряда ядовитых веществ и метаболитов;
так, в виде «парных глюкуроновых к-т», т. е. гликозидов
этой к-ты, выделяются фенолы, спирты, продукты превра-
щения нек-рых гормонов и др. Галактуроновая
кислот а обнаружена в пектиновых веществах, в полиса-
харидах нек-рых слизей и в специфич. полисахаридах пато-
генных микроорганизмов. Маннуроновая кислота
входит в состав альгиновой к-ты и нек-рых гликопротеидов.
Идуроновая кислота обнаружена в мукополиса-
харидах.
Глюкуроновая к-та: Р-D-форма Галактуроновая к-та: a-D-форма (НоО) Т. пл., °C Мр ‘
154—156 156-159 160 120-130 165+167 + 11,7°; + 36,26° + 97,9°; + 50,9° + 27°; + 55,3° + 16°; 6,1° —47,9°; —23,9° ••
p-D-форма Маннуроновая к-та: a-D-форма (гидрат) P-D-форма (Da HSO)
* Первая цифра — в воде сразу после растворения, вто-
рая— через 2 часа. ** Через 12 часов.
В водных р-рах из Г. к. образуются лактоны —
гексуроны (напр., глюкурон, маннурон, галактурон
и т. д.). Галактуроновая к-та отличается от глюкуро-
новой тем, что с трудом образует лактон. Г. к. дают
реакции, характерные для альдогексоз и карбоновых
к-т; образуют соли с металлами и органич. основания-
ми. Для идентификации Г. к. применяют соли нек-рых
алкалоидов (напр., бруцина и цинхонина). В щелоч-
ных р-рах Г. к. легко разлагаются, аналогично гек-
созам. В кислой среде легко идет декарбоксилирова-
ние Г. к. с получением фурфурола, что используется
для их колич. определения. При декарбоксилировании
в присутствии металлов (Mg, Zn, Ni, Al) и в пири-
дине образуются пентозы, напр. из D-галактуроновой
к-ты — арабиноза. Глюкуроновая к-та декарбоксили-
руется до фурфурола в кислой среде, при декарбо-
ксилировании под влиянием нек-рых микроорганиз-
мов из D-глюкуроновой к-ты образуется D-ксилоза.
При действии нек-рых бактерий из D-галактуроновой
к-ты вследствие декарбоксилирования образуется
L-арабиноза. При окислении Г. к. получаются двух-
основные гексаровые кислоты (сахарные
кислоты с 6 атомами углерода); из глюконовой к-ты
образуется сахарная глюкаровая к-та, а из галакто-
новой к-ты — муциновая (галактаровая) к-та и т. д.
С фенилгидразоном Г. к. образуют гидразиды, гидра-
зоны и озазоны. При нагревании Г. к. с метиловым
спиртом одновременно идет этерификация карбо-
ксильной группы и полуацетального гидроксила.
Для получения гликозидов Г. к. (гексозидуроновых
к-т) проводят щелочной гидролиз сложноэфирной
группировки. Гликозиды Г. к. довольно устойчивы
в кислой среде; уронозидная связь гидролизуется
с большим трудом (длительное нагревание с конц.
к-тами); при этом идет также разложение Г. к.,
поэтому выделение Г. к. из природных веществ пред-
ставляет большие трудности.
Глюкурононую к-ту получают из природных ве-
ществ, напр. гидролизом полисахаридов гуммиара-
бика. Синтетически ее получают окислением произ-
водных глюкозы, восстановлением монолактона са-
харной к-ты и нек-рыми др. способами. Галактуроно-
вую к-ту получают гидролизом природных веществ,
напр. энзиматич. гидролизом пектиновых веществ
или кислотным гидролизом полигалактуронидов цит-
русовых. Синтетически галактуроновую к-ту полу-
чают обычно окислением производных галактозы (при
условии защиты альдегидной группы) или восста-
новлением монолактона соответствующей ей гекса-
ровой к-ты амальгамой натрия в кислой среде.
Лит.: The Carbohydrates, ed. W. Pigman, N. Y., 1957;
Methods of biochemical analysis, ed. D. Glick, v. 2, N. Y. —L.,
1955, p. 342; Advances Carbohydr. Chem., 1945, 1, p. 338;
1946, 2,p. 177; 1954, 9, p. 131; 1953, 8, p. 231; Artz N. E.,
Osman E. M., Biochemistry of glucuronic acid, N. Y., 1950;
Annual Rev. Biochem., 1959, 28, p. 25. Л. И. Линевич.
ГЕЛИ (от лат. gelo — застываю) — дисперсные си-
стемы с жидкой или газообразной дисперсионной
средой и образуемой частицами дисперсной фазы
пространственной структурой (сеткой). Такая сетка
придает Г. механич. свойства твердых тел. Типичные
Г. обладают пластичностью, нек-рой эластичностью,
а также тиксотропными свойствами, т. е. способ-
ностью обратимо во времени восстанавливать свою
пространственную структуру после ее механич. раз-
рушения (см. Тиксотропия).
Г. образуются из золей при их коагуляции в случаях
развития пространственной сетки в результате моле-
кулярного сцепления частиц дисперсной фазы (см.
Структурообразование в дисперсных системах). В пре-
дельном случае Г. образуются в результате коагуля-
ции золей без расслоения, с отверждением первона-
чально жидкой системы в целом (гелеобразование,
как развитие коагуляционной структуры). Г. возни-
кают в результате несьма рыхлой коагуляции по от-
носительно малому числу коагуляционных центрон
на поверхности частиц дисперсной фазы, напр. по
углам (концам) и ребрам частиц вытянутой формы
(анизометричных), особенно склонных к гелеобразо-
ванию. При этом остальная (подавляющая) часть
поверхности частиц стабилизована сольватными слоя-
ми среды. Именно поэтому типичные Г. образуются уже
при очень малом содержании дисперсной фазы, напр.,
при 0,05% в гидрозоле пятиокиси ванадия в резуль-
тате добавления гелеобразующих (коагулирующих)
электролитов наступает полное отверждение. При
коагуляции лиофобных золей образуются осадки аг-
регатов частиц. В случае более лиофильных дисперс-
ных фаз при этом возникают студенистые осадки
(к о а г е л и). Если золь или коллоидная суспензия
являются достаточно лиофильными, то образуется
типичный гель, наз. лиогелем; при этом не про-
исходит разделения фаз с появлением осадка, и вся
дисперсионная жидкая среда удерживается механи-
чески в ячейках коагуляционной структуры. Частицы
дисперсной фазы в Г., как и в золях, могут быть крис-
талликами ультрамикроскопич. или микроскопии,
размеров (в последнем случае Г., образуемые, напр.,
в суспензиях, иногда наз. псевдогелями).
Переход золь — Г. не является фазовым превраще-
нием, при этом не происходит разделения фаз с появ-
лением осадка. Он не сопровождается заметным теп-
ловым эффектом, т. к. сцепление частиц при образо-
вании Г. происходит по весьма малым участкам по-
верхности, и даже на этих участках уменьшение по-
верхностной энергии мало. Частицы слабо связы-
ваются через остаточные тонкие прослойки жидкой
среды, что и является причиной тиксотропии, плас-
тичности и сравнительно малой прочности структуры
Г. Количество дисперсионной среды, прочно связан-
ной поверхностью частиц дисперсной фазы, практи-
чески не изменяется при переходе золь — Г., и вся
жидкость, иммобилизованная, т. е. отвержденная
в структуре Г., является свободной и удерживается
механически в ячейках коагуляционной структуры.
При высушивании или уплотнении прочность Г. воз-
растает и тиксотропные свойства теряются. Разру-
шение структуры .становится необратимым, Г. пре-
827
ГЕЛИЙ
828
вращается в твердое роговидное, тонкопористое тело —
конденсационную или кристаллизационную струк-
туру ксерогеля. Такие пористые тела (к ним
относятся различные сорбенты, многие катализаторы
и их носители) иногда наз. аэрогелями; в них
дисперсионной средой является газ (воздух). Группы
типичных тиксотропных Г. образуют Г. мыл и мыло-
образных поверхностно-активных веществ. Они воз-
пикают из соответствующих золей при повышении
концентрации мыла или при понижении темп-ры.
По аналогии с золями, Г. по характеру дисперсион-
ной среды делятся на гидрогели, алкогели, бензоге-
ли и т. д.
Силы, связывающие частицы дисперсной фазы
в структуре Г. при достаточной рыхлости структуры,
ее малой прочности, могут вызывать постепенное
уплотнение Г., что приводит к выделению жидкости
из пор Г., уменьшающегося в объеме (см. Синерезис).
Этот процесс и выражается в т. наз. «старении» Г. —
длительном изменении их свойств со временем. Об-
ратный процесс перехода Г. в золь под действием
физико-химич. факторов наз. пептизацией.
Г. часто отождествляют со студнями высокомоле-
кулярных соединений. Однако, в отличие от Г.
(двухфазных структурированных дисперсных систем),
студни являются однофазными системами — доста-
точно концентрированными истинными р-рами поли-
меров. Они характеризуются развитием пространст-
венной сетки, образованной взаимодействием макро-
молекул (как, напр., в процессах вулканизации),
а в ряде случаев связанной с пространственной поли-
меризацией (как в студнях кремнекислоты в воде).
Студни, в к-рых макромолекулы связаны в пространст-
венную сетку слабыми силами, аналогичны Г. по
своим механич. свойствам, обладают тиксотропией
и повышенной эластичностью из-за гибкости макро-
молекул. Высокопрочные студни, структура к-рых
обусловлена химич. связями, возникшими в резуль-
тате пространственной полимеризации (т. наз. kohj
денсационная'структура, в отличие от коагуляционной
структуры), разрушаются внешними силами необра-
тимо, Т. е. лишены тиксотропии. И. А. Ребиндер.
ГЕЛИЙ (Helium) Не — химич. элемент нулевой
гр. периодич. системы Менделеева, п. н. 2, ат. вес
4,003. В обычных условиях Г. — одноатомный хи-
мически инертный газ без цвета и запаха.
Г. открыт в 1868 Ж. Жансеном и Н. Локьером,
обнаружившими в солнечном спектре желтую линию,
не отвечавшую ни одному из известных тогда эле-
ментов; назван от греч. — Солнце. В 1895
У. Рамзай выделил из минерала Клевеита газ, в спектре
к-рого нашел желтую линию Г. Одновременно
Г. Кайзер, также пользуясь спектральным методом,
показал присутствие Г. в атмосфере.
Г. относится к числу наиболее распространенных
химич. элементов космоса: на 10* атомов Si во все-
ленной приходится 3,5 • 10’ атомов Г. Спектральный
анализ показывает присутствие Г. в атмосфере Солнца
и звезд. Г. обнаружен также в метеоритах. В воз-
духе содержится 0,0005% Г. по объему, в нек-рых
природных газах — до 10%. Г. входит в состав
различных минералов.
Природный Г. состоит из стабильных изотопов
Не4 и Не3; их относительное содержание непостоянно
и зависит от источника получения Г., однако в смеси
всегда преобладает Не4. Содержание Не3 в смеси,
полученной из воздуха, равно 1,2 • 10“’%, из природ-
ного газа (0,2—120) -10 ’%, из радиоактивных ми-
нералов 0,2 • 10“’%, из метеоритов 17—31,5%. При
альфа-распаде радиоактивных элементов образуется
и накапливается в минералах, содержащих эти эле-
менты, изотоп Не4 (1 г урана в равновесии с продук-
тами распада дает 1,1 • 10“’ см3 Г. в год). По соотно-
шению количеств Г. и материнского вещества в мине-
рале возможно определение абс. возраста последнего
(так наз. гелиевыи метод), причем для определения
пригодны лишь нек-рые минералы с особо плотной
кристаллич. структурой (хлопинит, самарскит, эши-
нит, магнетит, колумбит и др.), способные удержи-
вать радиогенный Г. (см. Возраст геологический аб-
солютный). К накоплению ядер Не4 во Вселенной
приводит основная термоядерная реакция, служащая
источником солнечной и звездной энергии: 4Н1 =
= Не4 + 20“+ 2v. Изотоп Не3 также образуется
в нек-рых ядерных реакциях, напр. D + D = Не3+п.
В земной атмосфере Не3 постоянно образуется под
действием космич. радиации: N14+ п—>С12 + Т; Т—>
—>0“ + Не3. Известен один искусственно радиоактив-
ный изотоп Не6 (0“-активен, Т1/г = 0,8 сек), полу-
чаемый обычно при бомбардировке Be8 нейтронами.
Ядра Не3, Не4 и Не6 состоят из двух протонов
и 1, 2 и 4 нейтронов соответственно. Атомные массы
перечисленных изотопов равны: 3,0169807; 4,0038761;
6,020838 (при условии, что масса нейтрального атома
О16 равна 16,000000). В отличие от Не4 с четным
спином, ядро атома Не3 обладает спином 4/2 и маг-
нитным моментом 1,07 • 10“23 CGSM. Ядра Не4,
или альфа-частицы, чрезвычайно устойчивы (энергия
связи нуклонов 28,2937 Мэв); они широко исполь-
зуются в различных ядерных реакциях.
Атом Г. состоит из ядра и двух электронов. Радиус
атома Г., определенный различными методами, равен
(в А): 0,98 (по вязкости); 1,33 (по ур-нию Ван-дер-
Ваальса); 0,85 (из опытов по столкновению с электро-
нами). Энергия ионизации Г. больше, чем у любого
др. элемента и составляет 24,58 эв для отрыва первого
электрона (Не—>Не+) и 54,4 эв— для второго (Не+—>
—>Не2+). Г. дает два дуговых спектра, отвечающих двум
разным состояниям — ортогелию и парагелию, энер-
гии к-рых в основных состояниях отличаются на
19,77 эв. Спектр, представляющий собой серию оди-
ночных линий, относится к парагелию. Ортогелий
дает спектр, состоящий из дублетов. При обычных
условиях лишь небольшая доля Г. относится к орто-
состоянию. Кроме того, Г. дает искровой спектр, от-
вечающий ионам Не+.
Газообразный гелий. Физич. свойства Не4.
1 л Не4 при 0° и 760 мм рт. ст. весит 0,178467 г.
Удельная теплоемкость при постоянном давлении
(кал!г-град)'. при 18° Ср 1,260, Ср/С„ 1,660; при—180°
Ср 1,24, Cp/Cv 1,673. Теплопроводность 343, 10“в
кал]см-град-сек. Вязкость (мкпуаз) 189,4 (0°С) и
10,5 (4,2°К). В 1 объеме воды при 18,2° растворяется
0,0073 объема Не4, приведенного к 0°. Диэлектрич.
проницаемость 1,000074 (0° и 760 мм рт. ст.). Не4
слабо диамагнитен, хобъемн = —0,78 • 10~10. Пока-
затель преломления для желтой D-линии натрия
nD = 1,000034.
Физич. свойства Не3 довольно близки
к Не4. При 3,2°К вязкость Не3 12 мкпуаз, тепло-
проводность 3 10“6 кал!град-см-сек. Характерна вы-
сокая проникающая способность газообразного Г.
по отношению к стеклам и органич. пленкам.
Жидкий гелий — легкая бесцветная прозрачная
жидкость. О физич. свойствах жидкого Г. см. ниже.
Гелий I и гелий II. При темп-ре 2,172°К
(т. н. Х-точка) в жидком Не4, находящемся под дав-
лением насыщенных паров (37,80 мм рт. ст.), про-
исходит фазовый переход 2-го рода (рис. 1); выше
этой темп-ры Г. наз. Г. I, ниже ее— Г. II. Темп-ра
перехода смещается в сторону более низких темп-р
при увеличении давления. Характер фазового пере-
хода выясняется по наличию аномалии теплоемкости
в 1\, по температурной зависимости плотности с ха-
рактерным изломом в 7\ и по другим явлениям, ти-
829
ГЕЛИЙ
830
пичным для фазового перехода 2-го рода. Рост тепло-
емкости С при приближении к Т\ происходит по ло-
гарифмич. закону С «а In (Т—1\).
Г. I резко отличается по внешнему виду от Г. II:
Г. I бурно кипит во всем объеме, а Г. II — спокойная
жидкость с отчетливым
мениском. Такое отличие
объясняется необычайно
высокой теплопроводно-
стью Г. II, во много мил-
лионов раз превосходящей
теплопроводность Г. I,
к-рая приблизительно рав-
на 10~6 кал!град-см-сек.
Г. II обладает и другими
необыкновенными свойст-
вами. Так, он способен
образовывать на твердых
поверхностях тонкие плен-
ки, необычайно быстро
Рис. 1. Диаграмма состоя-
ния жидкого Не4.
двигающиеся против градиента темп-ры.
В Г. II имеет место т. и. термомеханич. эффект
(фонтан-эффект) (рис. 2): при подводе тепла к нижней
части трубки из ее верхнего конца бьет фонтан жид-
кого Г.; т. обр., создание разности темп-p приводит
, к течению Г. II. Имеет место и обрат-
J ный, т. н. механокалорич. эффект (рис.
1| 3); при быстром вытекании Г. из со-
J____ суда темп-ра внутри сосуда повышает-
ся, а при погружении сосуда в ван-
ну из жидкого Г., т. е. при втекании
| жидкости в сосуд, темп-ра внутри
§ j сосуда понижается.
Д-а°?° При наблюдении протекания Г. II
!—” И через узкий капилляр оказалось, что
£ его вязкость в десятки тысяч раз
Наждауный^ меньше, чем у Г. I. Вместе с тем непо-
аорошои средствениое измерение вязкости по
Рис. 2. Фонтан- затуханию крутильных колебаний по-
эффект. груженного в Г. II диска дает ре-
зультаты, мало отличающиеся от вяз-
кости Г. I. Это свойство — протекать через узкие
капилляры, не обнаруживая вязкости, открытое в
1938 П. Л. Капицей, — было названо сверхтекуче-
стью. Ойо явилось ключом к пониманию всех осо-
Рис. 3. Механо-кало-
рический эффект: Т—
термометр сопротив-
ления; Д — пробка
из спрессованного по-
рошка.
Рис. 4. Схема прибора Ка-
пицы для изучения гелие-
вой струи. Сосуд и система
с листочками погружены в
Не II; Н— нагреватель, Т —
термометр.
бенностей поведения Г. II. Во 2-м опыте Капицы
(рис. 4) в Г. погружается маленький сосудик, со-
общающийся с ванной через узкий капилляр. Внутри
сосудика помещается нагреватель. При выделении
тепла из капилляра бьет струи, обнаруживаемая
с помощью небольших крутильных весов, причем
уровень жидкости в сосудике не меняется. Для объяс-
нения этого явления следует предположить, что
имеется противоток Г., направленный внутрь со-
судика, к-рый не оказывает действия на листочки
крутильных весов. Согласно основным законам гидро-
динамики, это значит, что противоток представляет
собой потенциальное течение идеальной жидкости,
а поток жидкости из сосудика, действующий на лис-
точки, — течение вязкой жидкости. Этот опыт имел
большое значение для понимания сущности явления
теплопередачи в Г. II.
Поведение Г. может быть наглядно объяснено на
основе т. н. двухкомпонентной модели,
согласно к-рой жидкий Г. при темп-pax ниже 7\
можно рассматривать как «смесь» двух жидкостей —
сверхтекучей, не обладающей вязкостью и не увле-
кающейся колеблющимся телом, и нормальной. При
протекании Г. II по тонкому капилляру сверхтекучая
«часть» жидкости, не обнаруживающая трения, сво-
бодно проходит через него; нормальная же «часть»
задерживается, протекая через капилляр несрав-
ненно медленнее, со скоростью, соответствующей ее
вязкости и толщине капилляра. Диск же, колеблю-
щийся в жидкости, содержащей как сверхтекучую,
так и нормальную части, останавливается благодаря
трению о нормальную жидкость. Более строго сле-
дует говорить, что в Г. II при могут существо-
вать одновременно 2 движения, каждое из к-рь!х
связано со своей «эффективной массой», причем сумма
обеих масс равна полной истинной массе жидкости.
Гелий II — квантовая жидкость. Необык-
новенные свойства Г. II можно понять, исходя из квантовых
представлений. При темп-pax порядка 2—3° К де-бройлевская
длина волны атомов Г. становится сравнимой с межатомными
расстояниями. Поэтому свойства жидкого Г. должны подчи-
няться законам квантовой механики. Жидкий Г. — единствен-
ная квантовая жидкость; все другие жидкости затвердевают
раньше, чем становятся существенными квантовые явления.
То, что Г. остается жидким, связано с относительной слабо-
стью взаимодействия его атомов.
Как и для всякой квантовой системы, прежде всего возни-
кает проблема исследования энергетич. спектра квантовой
жидкости. При абс. нуле темп-ры жидкий Г. находится в наи-
низшем энергетич. состоянии, а при отличной от абс. нуля
темп-ре — в возбужденном состоянии. Всякое слабое возбуж-
дение макроскопич. системы, согласно квантовой механике,
может быть представлено как совокупность «элементарных
возбуждений», к-рые ведут себя, как нек-рые квазичастицы,
обладающие определенными энергиями и импульсами. При
малых импульсах (когда длина волны
Де-Бройля велика по сравнению с меж-
атомным расстоянием) эти возбуждения
соответствуют обычным звуковым волнам
в жидкости, т. е. представляют собой фо-
ноны, и их энергия г есть линейная функ-
ция их импульса р: г — ср (с — скорость
звука). По мере увеличения р кривая,
представляющая зависимость е от р, от-
клоняется от линейности. Л. Д. Ландау
для объяснения температурного хода тер-
модииамич. величин Г. II предположил,
что накривой е(р) имеется минимум (рис. 5).
Опытами по рассеянию нейтронов в Г.П,
при к-ром происходит рождение элементар-
ных возбуждений, такая форма спектра
полностью подтверждена. Вблизи миниму-
ма кривой Цр) ее можно представить в
виде: е — Д -J- (р — Ро)2/2ц. Соответству-
ющие возбуждения с энергией вблизи ми-
нимума называются ротонами. По нейтрон-
ным данным Д/h = 8,5°К (к — постоянная
Больцмана); р0 = 2,1 • Ю^’г-сМ’сек-1;
Рис. 5. Энерге-
тический спектр
возбуждений
Не II.
ц = 1,72 • 10~24г.
Знание энергетич. кривой позволяет вычислить термодинамич.
характеристики Г. XI (энтропию, теплоемкость и др.), объяс-
нить его свойства.
Перенос тепла в жидком Г. II имеет своеобразный
конвективный характер и принципиально отличен от
обычной теплопроводности, т. к. он осуществляется
только нормальной частью жидкости, сверхтекучая
же часть тепла не несет. Этим и объясняется механо-
калорич. эффект—понижение темп-ры Г., вытекаю-
щего через капилляр в ванну, т. к. при течении Г.
через капилляр возбуждения будут отражатьсй от
831
ГЕЛИЙ - ГЕЛИОТРОПИН
832
стенок и практически ие смогут пройти через него,
протекать же будет только сверхтекучая часть жид-
кости, не переносящая тепла. Во 2-м опыте Капицы
наряду с потоком нормальной компоненты, выте-
кающей из сосуда и действующей на крылышки,
имеется встречный поток сверхтекучей компоненты.
При этом полный поток жидкости равен нулю и уро-
вень ее в сосуде не изменяется. В Г. II невозможно
в стационарных условиях создать градиент темп-ры,
т. к. движение нормальной части (т. е. возбуждений)
от мест с большей темп-рой к местам с меньшей начи-
нается при сколь угодно малом градиенте темп-р
и происходит до тех пор, пока темп-ра не выравняется.
Аномальный характер переноса тепла в Г. II при-
водит к появлению скачка темп-ры на границе между
твердым телом и Г. II, возникающего при наличии
потока тепла от твердого тела к Г. II (или наоборот)
(т. н. Капицы температурный скачок). Особые свой-
ства сверхтекучего Г. раскрываются также в ряде
особенностей, проявляющихся при вращении сосуда
с Г. (вихрь в Г. II). Из ур-ний гидродинамики сверх-
текучего Г. следует, что в Г. II возможно распростра-
нение двух типов звуковых волн: волн сжатия (обыч-
ёО.О8
Е0.04
0,16
г/с*3
0,12
0 ____
1.0 1.8 2,6 3.4 4.2 5,0°К5,8
Т--------------
Рис. 6. Плотность Не3 и Не4.
ный, или 1-й звук)
и температурных волн
(второй звук).
Свойство сверхте-
кучести характерно
для систем частиц,
подчиняющихся ста-
тистике Бозе; поэтому
сверхтекучестью обла-
дает жидкий Г., со-
стоящий гл. обр. из
атомов Не4. Жидкий
смеси сверхтекучи, ниже этой темп-ры — верхняя фаза нор-
мальна, нижняя — сверхтекуча. Не3 и Не4 под давлением на-
сыщенных паров остаются
жидкими вплоть до абс.
нуля.
Твердый Г., по-
лучаемый под давле-
нием, — бесцветная про-
зрачная масса. Струк-
тура твердого Не4 со-
ответствует плотной ге-
ксагональной упаков-
ке. Твердый Не8 при
дайной температуре мо-
жет находиться в двух
модификациях, аир,
в зависимости от дав-
ления. a-фаза, соот-
ветствующая меньшим
давлениям, имеет куби-
ческую решетку, 0-фа-
за — гексагональную с
плотной упаковкой.
Тройная точка, где а-
и 0-фазы и жидкий Не3
находятся в равновесии, соответствует Т — 3,15°К,
р = 141 атм.
Получение и применение гелия. В промышленности
Г. получают из природных газов методами глубокого
охлаждения (см. Инертные газы).
Газообразный Г. находит широкое применение для
создания инертной среды при автогенной обработке
Рис. 7.
творов
Т^Р = 0,88° К — критич. темп-ра
смешения.
Фазовая диаграмма рас-
жидких Не3 и Не4.
Г., состоящий из ато-
мов Не3, также являющийся квантовой жидкостью, не
обладает свойством сверхтекучести, т. к. система из
частиц, подчиняющихся статистике Ферми, обладает
особыми свойствами, в частности имеет энёргетич.
спектр другого типа.
Физические свойства жидких Не3 и
Не4 приведены в табл, и на рис. 6. Как уже отмечено,
Не3, в отличие от Не4, не обладает сверхтекучестью,
по крайней мере вплоть до 0,1° К. Теплоемкость
Не3 не обнаруживает аномалий.
металлов, для консервации пищевых продуктов и др.
Благодаря исключительно малому сечению захвата
нейтронов (равному 0,0068 барн на атом), высокой
теплоемкости и химич. инертности Г. используется
в атомной энергетике и пром-сти. Поскольку Г.
обладает наиболее низкой из всех веществ темп-рой
кипения, жидкий Г. широко применяется в физич.
лабораториях в качестве хладоагента при работах
по физике низких температур. Не4, наряду с Не3,
служит основным термометрич. веществом в газовых
термометрах в интервале темп-p от 80 до 1°К. Не3 —
единственное вещество, служащее для измерений
темп-p ниже 1°К.
Лит-: Кеез о м В., Гелий, пер. с англ., М., 1949; Д о у н т
Дж. Г. и Смит Р. С., УФН, 1955, 56, вып. 3; и х ж е,
там же, 1955, 57, вып. 1;Х а л а т н и к о в И.М.,там же, 1956,
Физические свойства жидкого гелия
Изотоп Т'крит., ° к. Р Крит. ₽крит., г/см3 р (о°ю, г/см3 ткип. (760 мм рт. ст.), °К рпл. (0°К), атм г(0°К), кал/г а(0°К), дин/см т/ (2,5 °К), г/см • сек А (2,5 °К), кал/град • см • сек Щ (0°К), м/сек Р (0 °К), см~/дин
Не4 Не* 5,2 3,35 2,26 атм 890 ммрт. ст. 0,0693 0,0413 0,145 0,08235 4,215 3,19 24 ^40 3,56 1,68 0,35 0,15 3,2 • 10-5 1,8 • 10-5 Ь 10-5 3,8 10-5 237 183,4 1,2 • 10-8 3,6 • 10-s
Примечание: Т — темп-ра, р — давление, р — плотность, рпл. — давление плавления, г — теплота парообразо-
вания; а — поверхностное натяжение; г; — вязкость; X — теплопроводность; и, — скорость звука (обычного); р — адиабатич.
сжимаемость.
Характерно, что ниже 0,15° К теплоемкость линейно за-
висит от темп-ры, аналогично электронной теплоемкости в
металлах. При темп-рах ниже 1°К Не3 имеет специфич. эн-
тропийную диаграмму — энтропия жидкости меньше энтропии
твердой фазы.На кривой плавления Не3ок.0,3°К. имеется мини-
мум (ему соответствует давление 29,3атм). Ниже этой темп-ры
теплота плавления Не3 отрицательна. Не3 обладает парамаг-
нитной восприимчивостью, связанной с магнитным момен-
том ядер. Температурный ход восприимчивости для жидкости
следует закону Кюри вплоть до —-0,5° К. Ниже этой темп-ры
наблюдается отклонение от закона Кюри, связанное с упоря-
дочением ориентаций спинов ядер.
Не3 и Не4 неограниченно растворяются друг в друге выше
критич. точки смешения = 0,88° К (рис. 7). Ниже этой
темп-ры смеси расслаиваются на 2 фазы с различной концентра-
цией х == Не3 (Не3 4- Не4). С повышением концентрации я
темп-ра, соответствующая л-точке смеси, смещается в сторону
низких темп-p. При Т = 0,67°К и х = 82%л-линия пересе-
кает кривую расслоения. Выше 0,67°К обе фазы расслоившейся
59, вып. 4; е г о же, там же, 1956, 60, вып. 1; Пешков
В. П. и Зиновьева К. Н., там же, 1959, 67, вып. 2;
Atkins К. R., Liquid helium, Camb., 1959; Физика низ-
ких температур, пер. с англ., М., 1959, гл. X; D aunt J. G.,
Science, 1960, 131, Л"» 3400; Малков М. П., Алексеев
В. Н., Козлов A. JI., Технология гелия и других редких
газов, JI., 1940. И. М. Халатников, К. Н. Зиновьева.
ГЕЛИОТРОЯИН (пиперонал, метиленовый эфир
протекатехового альдегида, 3,4-метилендиоксибенз-
альдегид) С8НвО3, молекулярный вес 150,13—бес-
цветные кристаллы с запахом цветов
О СН2 гелиотропа; т. пл. 36,5—37,0°; т. кип.
А 263°; трудно растворим в холодной воде
j |Г~и (1 ’• 500), несколько лучше — в спирте,
эфире, бензоле и других органич. рас-
Т творителях, а также в смеси 20 ча-
СНО стеи 70%-ного спирта и 12 частей олив-
833
ГЕЛИОХИМИЯ — ГЕМИЦЕЛЛЮЛОЗЫ
834
нового масла; легко перегоняется с паром. При про-
должительном действии воды, света и воздуха Г.
разлагается. Г. содержится в стручках ванили,
в цветах гелиотропа и в нек-рых эфирных маслах
(Spirala ulmaria, Robina pseudoacacia и др.). Для Г.
характерно омыление метилендиоксигруппы, так, при
нагревании до 200° с разб. НС1 в запаянной трубке
образуется протокатеховый альдегид; омыление про-
исходит также при действии А1С13. Г. легко вступает
в характерные для альдегидов реакции, образуя
семикарбазон, т. пл. 230—233°; тиосемикарбазон,
т. пл. 185°; n-бромфенилгидразон, т. пл. 155°; 2,4-
динитрофенилгидразон, т. пл. 265° (с разл.); дает
монобромпроизводное, т. пл. 129,5° и мононитропро-
изводное, т. пл. 94,5°. При окислении Г. водным р-ром
КМпО4 при 70—80° образуется пиперониловая к-та (I),
т. пл. 227,5—228°; при взаимодействии Г. с 40%-ным
р-ром (NaOH), ацетоном и водой получается пиперо-
налацетон (II), т. пл. 107—108°; при действии РС15
на Г. на холоду — пипероналхлорид; при высокой
темп-ре образуются продукты хлорирования в бен-
зольном ядре. Амальгамой натрия Г. восстанавли-
вается до пиперонилового спирта (III).
В пром-сти Г. получают изомеризацией сафрола
(IV) в изосафрол (V) и окислением последнего.
I II 111
Од иЛ11
др. окислит.
Г елио-
тропин
Г. можно синтезировать также из пирокатехина:
СН2С1
Количественно Г. определяется гидроксиламиновым
методсм. Г. широко используется в парфюмерии,
косметике, в произ-ве туалетных мыл.
А. А. Зеленецкая.
ГЕЛИОХИМИЯ — см. Космохимия.
ГЕЛЛЯ — ФОЛЬГАРДА — ЗЕЛИНСКОГО РЕАК-
ЦИЯ— получение а-галогенозамещенных к-т или их
производных галогенированием карбоновых к-т в
присутствии фосфора или его галогенидов:
r—сн- соон -5я R-СН-СОХ
R' (РХ»)
r—ex—coor”
/ I
— R—СХ - СОХ\ R'
\—R-CX-COOH
R'
Первоначально для реакции употребляли эквивалент-
ные количества фосфора, причем получали галоген-
ангидриды а-галогенокислот, а из них, после обработ-
ки водой или спиртом, соответственно а-галогено-
кислоты или их эфиры. В дальнейшем было заме-
чено, что в большинстве случаев реакция галогени-
рования кислот сильно ускоряется небольшими коли-
чествами фосфора или его галогенида, действующими
каталитически. Галогенид фосфора превращает часть
кислоты в галогенангидрид, к-рый подвергается га-
логенированию гораздо легче, чем свободная кислота;
при взаимодействии галогенангидрида а-галогено-
кислоты с исходной кислотой образуется а-галогено-
кислота и регенерируется галогенангидрид незаме-
щенной кислоты:
R—СН—СООН —-3- R—СН—СОХ -5з.
I I
R' R'
R—СИ—СООН
I'
-> R-CX-COX —----------- R—СН—COX+R—СХ—СООН
! I
R' R' R'
Темп-ра при выполнении реакции обычно не пре-
вышает 100°, растворитель не применяется; выходы,
как правило, хорошие. Реакцией пользуются обычно
для получения а-бром-, иногда а-хлорзамещенных
кислот или их эфиров, напр.:
(СН3)»СНСН2СН..СООН (СН3)2СНСН2СНВгСООН
Puig
СНзСООН ВгСН2СООС3Н5
По Г. —Ф.—3. р. из p-бромпропионовой к-ты не удалось
приготовить а-, Р-дибромпропионовую к-ту. Гало-
генангидриды, промежуточно образующиеся при
Г.—Ф.—3. р., можно получить обработкой кислоты
хлористым тионилом; при последующем бромировании
в присутствии спирта без катализатора образуются
эфиры бромзамещенных к-т, напр.,
СвН5СНаСООН CeH6CHBrCOOC2H5
±>Го, 115UИ
а-Галогенозамещенные кислоты и их эфиры ис-
пользуются в качестве исходных веществ для полу-
14 К. X. Э. т. I
чения а-оксикислот, а-аминокислот, а-меркаптокис-
лот и др. (см. также Дарзана реакция,
Реформатского реакция).
Г.—Ф.—3. р. открыта К. М. Геллем
Гелио- (1881) и усовершенствована Я. Фоль-
— тропин гардом (1887) и Н. Д. Зелинским (1887).
Лит.: Вейганд К., Методы экспери-
мента в органической химии, пер. с нем.,
ч. 2, М., 1952, с. 89; S и г г е у A. R., Name
reactions in organic chemistry, N. Y., 1954,
p. 94. E. M. Рохлин.
ГЕМИН — см. Гемоглобины.
ГЕМИНАЛЬНЫЕ СОЕДИНЕНИЯ — органиче-
ские соединения, содержащие два или три замести-
теля у одного атома углерода. Для Г. с. принято
условное обозначение — приставка «гем» — впереди
названия в-ва. Например, аеле-соединением является
2,2-дихлорпропан СН3СС12СН3, в отличие от изомер-
ного 1,2-дихлорпропана С1СН2СН2С1СН3 с вициналь-
ным расположением заместителей. Для Г. с. харак-
терны нек-рые особенности в их химич. поведении.
Так, соединения, содержащие 2 или 3 карбоксильные
группы у одного атома углерода, легко декарбокси-
лируются. Вещества с двумя оксигруппами у одного
атома обычно очень нестойки и легко отщепляют
воду с образованием альдегида или кетона и др.
Я. Ф. Комиссаров.
ГЕМИЦЕЛЛЮЛОЗЫ — полисахариды, входящие
в состав клеточной стенки растительной ткани наряду
с целлюлозой и лигнином. Большинство Г. отличается
от целлюлозы лучшей растворимостью в щелочных
р-рах и способностью легче гидролизоваться разбав-
835
ГЕМОГЛОБИНЫ
836
ленными к-тами. В состав Г. входят гексозаны и пен-
тозаны — полимерные ангидриды гексоз и пентоз.
Их мол. вес значительно меньше, чем у целлюлозы;
степень полимеризации обычно не превышает 150.
Содержание Г. в древесине колеблется от 17 до 41%.
Г. содержатся в соломе злаков, шелухе семян, в куку-
рузных початках и в отрубях. В древесине хвойных
пород преобладают гексозаны, в лиственных — пен-
тозаны. Из гексозанов в Г. содержатся галактан,
маннан и фруктан. К пентозанам относятся ксилан
и арабан. Ксилан по сравнению с другими Г. наиболее
широко распространен в растительном мире. В лист-
венной древесине содержится до 25% ксилана, в хвой-
ной — до 12%; он состоит из остатков ангидрида
ксилозы и имеет след, строение:
Н |_ Н ОН Jn Н
Чистый ксилан — белый порошок, хорошо раство-
ряется в щелочном р-ре, нерастворим в спирте и дру-
гих органич. растворителях.
Часть Г. является устойчивой к обработке щелочью
и к-той. Так, напр., технич. целлюлозы после выделе-
ния из древесины всегда содержат в качестве приме-
сей часть ксилана и маннана. В произ-ве Г. называют
обычно ту часть технич. целлюлозы, к-рая раство-
ряется в 17,5%-ном р-ре NaOH при 20°. При этом
в р-р, кроме спутников целлюлозы, как ксилан и ман-
нан, переходят также низкомолекулярные фракции
самой целлюлозы со степенью полимеризации ниже
200. Часть Г., выделяющаяся из щелочного р-ра при
добавлении к-ты, наз. р-целлюлозой. Фракция Г.,
не выделяемая к-той, наз. у-целлюлозой.
Присутствие Г. в целлюлозной массе, предназначен-
ной для химич. переработки, является нежелатель-
ным. Образующиеся наряду с эфирами целлюлозы
соответствующие эфиры ксилана и маннана ухудшают
качество производных целлюлозы (ацетилцеллюлозы,
ксантогената целлюлозы). В вискозном произ-ве Г.
усложняют процесс регенерации отработанной щелочи
и ухудшают качество получаемого волокна. Наобо-
рот, в произ-ве бумаги Г. играют положительную
роль. Присутствующие в целлюлозной массе Г. улуч-
шают процесс размола, цементируют целлюлозные
волокна и тем самым улучшают механич. свойства
бумаги.
Лит.: Никитин Н. И., Химия'древесины, М,—Л.,
1951; Роговин 3. А. иШорыгина Н. Н., Химия
пеллюлозы а ее спутников, М. —Л., 1953; OttE., Spurlin
Н. М., G г а 1 I 1 I п М. W., Cellulose and cellulose derivatives,
2 ed., v. 5, pt 1—3, N. Y.—L. 1954—55. А. Г. Яшунская.
ГЕМОГЛОБИНЫ — красные пигменты эритроци-
тов крови человека и позвоночных животных, являю-
щиеся сложными белками группы хромопротеидов
(хромояротеинов) и осуществляющие перенос моле-
кулярного кислорода от легких или др. органов
дыхания к тканям. В крови человека в среднем содер-
жится 14,5% Г., его общее количество ок. 750 г;
в крови млекопитающих — ок. 12,7 г на 1 иг веса
животного. Концентрация Г. в крови животных раз-
ных видов различна; напр. в крови овцы 9% Г.,
лошади 16%. Г. различных видов позвоночных живот-
ных имеют близкие мол. веса (порядка 66 000—68 000);
отличаются химич. составом и строением белкового
компонента глобина. Г. содержат одну и туже про-
стетич. (небелковую) группу, на долю к-рой прихо-
дится ок. 4% от веса Г. Простетич. группа в молекуле
Г. представлена 4 одинаковыми железопорфирино-
выми соединениями (C34H32O4N4Fe), к-рые называют
гецами или протогемами.
Название «гем» распространяют на все железопорфири-
новые соединения с двухвалентным или трехвалентным желе1
зом; иногда их называют также геминовыми соединениями
(белки, в состав к-рых входят гемы, выделяют в группу гемо-
протеинов). Протогемамн наз. гемы, в к-рых порфириновое со-
единение представлено протопорфирином, т. е. порфином, со-
держащим 4 метильные группы, 2 винильные и 2 остатка про-
пионовой к-ты. В состав гема Г. входит один из 15 теорети-
чески возможных изомеров протопорфирина (их нумеруют
римскими цифрами) — протопорфирин IX (1, 3, 5, 8-тетра-
метил-2,4-дивинил-6, 7-карбоксиэтилпорфин). Протопорфирпн
IX входит также в состав других биологически важных
гемопротеинов—цитохромов Ь, каталазы и др.
В геме Г. 2 атома водорода у 2 пиррольных ядер
порфина IX замещены атомом двухвалентного железа,
2 дополнительные связи железа связывают его 2 дру-
гих пиррольных ядра, а 2 дополнительные
связи, направленные перпендикулярно к пло-
скости, в к-рой лежит протопорфирин и атом
железа, обусловливают, по-видимому, связь
Н.ОН гема с глобином. Точно характер связи между
гемом и глобином неизвестен. Предполагают,
что железо образует дополнительные связи
с азотом имидазола гистидиновых остатков
глобина. Гемы располагаются симметрично
поверхности Г.
Глобины Г. отличаются от других белков сравни-
тельно высоким содержанием гистидина (6—10%) и
отсутствием или низким содержанием изолейцина.
Глобины животных различных видов отличаются
аминокислотным составом, последовательностью ами-
Гем гемоглобина
Протопорфирин IX
нокислот, концевыми группами пептидных связей.
Напр., Г. человека не содержат изолейцина, а Г.
нек-рых животных содержат эту аминокислоту. После-
довательность аминокислот в Г. полностью не уста-
новлена, определены лишь концевые аминокислоты
нек-рых Г. и отдельные участки полипептидных цепей.
У нек-рых видов животных концевые остатки пред-
ставлены валином, у других — валином и метио-
нином. Методами рентгеноструктурного анализа уста-
новлены третичная и четвертичная структура нек-рых
Г. (см. Белки), форма и размеры их молекул. Известно,
что молекула Г. человека состоит из двух равных
половин, каждая из к-рых образована двумя одина-
ковыми полипептидными цепями. У человека обна-
ружены Г. различных типов, к-рые незначительно
отличаются по химич. строению. В крови взрослых
людей содержится Г., названный гемоглобином А;
Г. плода человека называют гемоглобином F. В нек-рых
случаях в крови людей были обнаружены необыч-
ные виды Г., к-рые по мере их открытия обозначались
различными буквами (С, D, Do, D1, Е, G, Н и др.).
У больных серповидной анемией в крови обнаружи-
вается гемоглобин S, а при специфич. заболевании
крови в эритроцитах появляется т. н. гемоглобин С;
эти Г. характеризуются незначительным изменением
аминокислотного состава Г.: в каждой из 2 половинок
молекулы Г. один из остатков аминокислоты (из общего
количества ок. 300) — глутаминовой к-ты— заменяет-
ся в первом случае валином, во втором — лизином.
837
ГЕМОГЛОБИНЫ
838
В Г. A, S и С обнаружена такая последователь-
ность аминокислот в отрезках полипептидной цепи
(сокращенные иазваиия аминокислот см. в ст. Ами-
нокислоты)-.
Гемоглобин А:
...гис.-вал.-лейц.-лейц.-треов.-прол.-елут.- глут.-лиз.-...
Гемоглобин S:
...гис.-вал.-лейц.-лейц.-треов.-прол.-вал.- глут.-лиз.-...
Гемоглобин С:
...гис.-пал -лейц.-лейц.-треон.-прол.-лиз.-глут.-лиз.-...
Определение различий в молекулах Г. произво-
дят путем комбинированного применения хромато-
графии и электрофореза ва бумаге для разделения
пептидов, получаемых при неполном ферментативном
гидролизе Г.; пептиды развых Г., положение к-рых
на двухмерной хроматограмме не совпадает, выделяют,
определяя затем последовательность амивокислот
в этих отрезках (метод «дактилограмм» или «отпечатка
пальцев») Небольшие откловения в строении белко-
вого компонента Г. заметно сказываются на физич.,
физико-химич., химич. и биологич. свойствах Г.
Г. — кристаллы, довольно хорошо растворимые
в воде и не растворимые в спирте, эфире и хлороформе;
форма кристаллов и их растворимость различны у Г.
животных разных видов. В эритроцитах Г. находится
в растворенном состоянии, несмотря на то что содер-
жание их обычно превосходит 30%. При изменениях
в аминокислотном составе глобина может произойти
изменение растворимости Г.; так, Г. S, растворяясь
хуже Г. А, выкристаллизовывается в эритроцитах,
вызывая их деформацию (образование серповидных
форм), что приводит к нарушению функции крови.
Р-ры Г. окрашены в темно-красный цвет и дают харак-
терные спектры поглощения в видимой и ультрафио-
летовой части спектра. Изоэлектрич. точка Г. близка
к 7. Г. разных животных имеют различную электро-
форетич. подвижность. В кислой и щелочной средах
Г. легко денатурируются, скорость денатурации также
различна у Г. животвых разных видов. В кислой
среде связь между гемом и глобином легко разры-
вается. Свободный гем Г. легко окисляется кислоро-
дом воздуха до гематина, в к-ром атом железа
трехвалентен; солянокислую соль гематина называют
гем ином; последний легко выделяется в кристал-
лич виде (кристаллы Тейхмана).
Наиболее характерным свойством Г. является
обратимое присоединение молекулярного кислорода,
СО и других газов. Соединение Г. с молекулярным
кислородом называется оксигемоглобином,
с окисью углерода — карбоксигемоглоби-
ном Реакция присоединения к Г. молекулярного
кислорода не является истинным окислением, т. к.
валентность железа в геме при этом не изменяется,
и эту реакцию правильнее называть оксигена-
цией Истинное окисление Г., при к-ром железо
гема переходит в трехвалентное состояние, происходит
при действии на Г. нек-рых окислителей; продукт
окисления Г. называют метгемоглобином
или гемиглобином. Молекула Г. содержит
4 гема и способна присоединять последовательно
4 молекулы кислорода. Если Г. обозначить символом
НЬ, то реакцию оксигенации можно представить
след, системой ур-вий; НЬ + О2 =- НЬО2; НЬО2 -|-
+ О2 = НЬ(О2)2; НЬ(О2)2 + О2 = Hb(O2)3; НЬ(О2)3 +
+ О2 = НЬ(О2)4. Такой механизм оксигенации объяс-
няет .У-образную форму кривой насыщения Г. кисло-
родом (рис. 1). При изменении парциального дав-
ления кислорода оксигемоглобин разлагается на Г.
и молекулярный кислород. Предполагают, что при
оксигенации одна из дополнительных связей железа
с имидазолами гистидина в глобипе разрывается;
при этом возникает связь между железом и молеку-
лярным кислородом Так, если в Г. из 32 остатков
14*
гистидина 8 принимают участие в образовании связей
с 4 атомами железа гемов, то в оксигемоглобине с же-
лезом связано только 4 остатка гистидина. За счет
освобождения кислотных групп имидазола увели-
чивается кислотность глобина, поэтому оксигемогло-
бин имеет изоэлектрическую точку более кислого
белка — 6,65.
сьпцения гемоглобина ки-
слородом от его парциаль-
ного давления.
1 — оксигемоглобин; 2 — гемо-
глобин; з — карбоксигемогло-
бин; 4 — метоксигемоглобин.
При присоединении к Г. кислорода и др. газов
изменяются нек-рые его свойства. На рис. 2 и в таб-
лице приведены спектральные и магнитные характе-
ристики нек-рых производных Г. Все молярные-
характеристики даны из расчета на один гем.
Соединение * макс. ммг. Е УОЛЯрНЫЙ коэфф, поглощения р. (в магнето- нах Бора)
Гемоглобин 558 13 • 103 5 46
430 125 • 103
Оксигемогло- 576 15,3 • 103
бин 541 14,5 • 103 0
414 125 * 103
Метгемогло- 630 4 10s
бин (рНг£7) 500 9,5 • 103 5,92
406 150 103
Изменения в оптич. свойствах Г. при оксигенации
используют при изучении кинетики взаимодействия
его с кислородом. Для количественного определе-
ния Г. разработано очень много способов. Чаще всего
определение производят колориметрически — измере-
нием количества образующегося из Г. гемина (гемо-
метрами Сали); используют также газометрич. опре-
деления (по количеству связанного кислорода) и др.
Для определения карбоксигемоглобина, в частности
в судебной химии и при определении профессиональ-
ных заболеваний окисью углерода, пользуются спект-
ральным методом анализа крови.
Основная биологич. роль Г. — перенос кислорода
от органов дыхания к тканям—обусловлена его обра-
тимой реакцией оксигенации. В связи с тем, что при
оксигенации меняется изоэлектрич. точка Г., он выпол-
няет и роль буфера крови при газовом обмене. Белки,
к-рые, подобно Г., способны присоединять молекуляр-
ный кислород и отщеплять его при низких давлениях
газа, наз. дыхательными белками. Близ-
кие Г. дыхательные белки, и состав которых входят
гемы, содержатся также в мышцей (их называют
мышечными Г., или миоглобинами) и
в плазме нек-рых беспозвоночных животных. Близкие
по ряду свойств гемопротеины беспозвоночных назы-
вают эритрокруоринами, или в в е к л е-
точвыми Г,
Лит.: ГауровИтц Ф., Химия и биология белков,
пер. с англ., М., 1953; Белки, пер. с англ., под ред. Г. Нейра-
та и К. Байлп, т. 3, ч. 1, М., 1958; БлюменфельдЛ. А.,
Гемоглобин и обратимое присоединение кислорода, М., 1957;
839
ГЕМОСПОРИДИН—ГЕОХИМИЧЕСКИЕ МЕТОДЫ ПОИСКОВ
840
Lemberg R. and L е g g e J. W., Hematin compounds
and bile pigments, N. Y. — L., 1949; Eisenstoffwechsei, Stutt-
gart, 1959; Annual Rev. Biochem., 1956, 25, p. 331; 1959, 28,
p. 128; Conference on hemoglobin. Publication № 557, Washing-
ton, 1958; Advances in protein chemistry, v. 12, N. ¥., 1957, p.
215; P e r u t z M. F. [a. o.l, Nature, 1960, 185, № 471, 416.
ГЕМОСПОРИДИН [метил сульфометилат Ы,Ы'-ди-
(4-диметиламинофенил)-мочевина] C21H34O9N4S2, мол.
вес 550,3 — бесцветные игольчатые кристаллы
(из водного спирта); т. пл. 215°; хорошо растворим
в воде, плохо — в спирте, эфире и бензоле; образует
с бромтимоловым синим окрашенные в синий цвет
соли, на чем основано колориметрич. определение
Г. Синтезируют Г. (I) конденсацией 4-аминодиме-
тиланилина (II) с мочевиной и последующей обработ-
кой образовавшейся М,М'-ди-(4-диметиламинофенил)-
мочевины (III) диметилсульфатом. Г. является спе-
цифическим химиотерапевтич. препаратом для лече-
ния пироплазмозов с.-х. животных. В лечебных
дозах Г. хорошо переносится животными и не вызы-
вает у них побочных явлений. Г. широко применяется
для массовой обработки с.-х. животных с лечебной
и профилактич. целью.
Лит.: Г е р ч у к М. П., Ж. общ. хим., 1941, И, вып. 11,
с. 731; Чеботарев Р. С., Пироплазмоз лошадей, Киев,
1951, с. 210. М. П. Герчук.
ГЕНЕРАТОРНЫЙ ГАЗ — см. Газификация твер-
дых топлив.
ГЕНРИ ЗАКОН — зависимость растворимости газа
в жидкости от давления, заключающаяся в том, что
при постоянной темп-ре весовая концентрация газа,
растворенного в данной жидкости, пропорциональна
его давлению над раствором. Г. з. установлен в 1803
У. Генри. Г. з. точно соблюдается только для идеаль-
ных растворов и применим лишь в области невысоких
давлений к газам, достаточно хорошо подчиняющимся
законам Бойля—Мариотта и Гей-Люссака. О раство-
римости газовых смесей в жидкости см. Дальтона
законы.
ГЕОХИМИЧЕСКАЯ КЛАССИФИКАЦИЯ ЭЛЕ-
МЕНТОВ — основана на типе строения внешних
электронных оболочек атомов и ионов химич. элемен-
тов и, как следствие из этого, на их специфич. сродстве
к тем или иным ведущим анионам; предложена в 1924
В. М. Гольдшмидтом и дополнена последующими
исследователями. Согласно Г. к. э., все химич. эле-
менты делятся на 4 неравноценные группы, распола-
гающиеся по их убывающему значению в след, по-
рядке: литофильные, халькофильные, сидерофильные
и атмофильные элементы. Г. к. э. может быть иллю-
стрирована кривой атомных объемов (см. рис. 2 в ст.
Геохимия).
Литофильные (от греч. М&о? — камень,
— люблю, имею склонность) — элементы гор-
ных пород, характеризующиеся тем, что внешняя
электронная оболочка их ионов построена по типу
инертных газов-58 электронов), что определяет их
трудную восстановляемость до элементарного состоя-
ния, специфич. сродство к кислороду и галогенам.
Литофильные элементы преимущественно парамаг-
нитны; располагаются на нисходящих участках кри-
вой атомных объемов. В природе литофильные эле-
менты образуют силикаты, окислы, галогениды и
соли кислородных кислот — сульфаты, фосфаты
и т. д., слагая ок. 95% всей земной коры. К ним
относятся: все щелочные и щелочноземельные эле-
менты (включая и Ra), В, Al, Sc; лантаниды и акти-
ниды (т. е. Ac, Th, Ра, U); С, Si, Ti и его аналоги,
Р, V, Nh, Та, О, Сг, W, галогены и Мп (а также, воз-
можно, Тс и At) — всего 54 элемента.
Халькофильные (от греч. уакхб? — медь),
по В. М. Гольдшмидту, или тиофильные (от
греч. Seiov — сера), по Дж. Р. Гиллебранду (1954),—
элементы сульфидных руд, характеризующиеся тем,
что на внешней оболочке их катионов располагаются
18 электронов; халькофильные анионы — сульфид-
ный, селенидный и теллуридный, имеют, как и лито-
фильные элементы, 8-электронную оболочку. Халь-
кофильные элементы преимущественно диамагнитны;
располагаются на восходящих
участках кривой атомных объ-
L Ln(ch \ ~*" емов. В природе образуют суль-
t п312 фиды, селениды и теллуриды (по-
следние характерны для золота),
а также тиосоли. Олово встречает-
(ЗОдСНд)^' ся не только в виде сульфидов,
но и в виде SnO2. Халькофиль-
ные элементы легко переходят в
самородное состояние (Си, РЬ,
Bi, As, S), а для нек-рых из них (Au, Ag) самород-
ное состояние наиболее характерно. Кроме вышеупо-
мянутых, к халькофильным элементам относятся:
Zn, Cd, Hg, Ga, In, TI, Ge, Sb, Se, Те, Po (всего 19 эле-
ментов).
Сидерофильные (от греч. aiSrjpoc — же-
лезо) — элементы переходного типа, характеризую-
щиеся 8—18 электронными ионами; большинству из
них свойственно самородное состояние. Сидерофиль-
ные элементы парамагнитны и ферромагнитны; зани-
мают минимумы кривой атомных объемов. Обладают
специфич. сродством к мышьяку (сперрилит PtAs2,
смальтин CoAs2, никелин NiAs, арсенопирит FeAsS),
а также к фосфору, азоту и углероду и в несколько
меньшей степени к сере (MoS2 и ReS2). К их числу
относятся все элементы VIII гр. периодич. системы,
а также Мо и Re — всего 11 элементов.
Атмофильные (от греч. атрбс — пар, испа-
рение) — элементы атмосферы, ионы или атомы
к-рых имеют 8-электронную оболочку инертных
газов. Для них характерно элементарное состояние
(для водорода, близкого к литофильным элементам,—
сродство к кислороду). Атмофильные элементы пре-
имущественно диамагнитны; располагаются в верхних
частях кривой атомных объемов. К этой группе отно-
сятся все инертные газы (Не, Ne, Аг, Кг, Хе, Rn),
азот и водород — всего 8 элементов.
По приведенной классификации все элементы рас-
пределяются по главнейшим генетич. и парагенетич.
природным ассоциациям. Понятия «биофильные» (эле-
менты живых организмов) и «талассофильные» (эле-
менты морской воды) лежат вне этой классификации.
О причинах специфич. сродства атомов элементов
к важнейшим анионам в связи со строением электрон-
ных оболочек атома см. Периодическая система эле-
ментов Менделеева, Поляризация.
Лит.: Гольдшмидт В. М., в кн.: Основные идеи
по геохимии, вып. 1. Работы по геохимии и кристаллохимии...,
1911—1930 гг., Л., 1933; Щербина В. В., Геохимия,
М.—Л., 1939; Goldschmidt V. М., Geochemische Vertei-
lungsgesetze der Elemente. II — Beziehungen zwischen den geo-
chemischen Verteiiungsgesetzen und dem Bau der Atome, Kris-
tiania, 1924, S. 1—37; Mason В r., Principles of geoche-
mistry, 2 ed., N. Y., 1958, p. 54—57. В. В. Щербина.
ГЕОХИМИЧЕСКИЕ МЕТОДЫ ПОИСКОВ п о-
лезных ископаемых — основаны на опре-
делении химич. состава различных образований
земной коры. Вокруг каждой залежи полезного иско-
паемого в результате миграции химических элементов
образуется поле повышенного содержания искомых
или сопутствующих им элементов в горных породах,
841 ГЕОХИМИЧЕСКИЕ МЕТОДЫ ПОИСКОВ — ГЕОХИМИЧЕСКИЕ ПРОЦЕССЫ 842
почвах, водах, растениях, атмосфере (рис.). Этот «ореол
рассеяния месторождения» занимает на поверхности
земли значительно большую площадь, чем рудный
выход (во многих случаях руды вообще не выходят
на поверхность). Размеры ореолов на поверхности
обычно составляют сотни и тысячи метров. Определяя
содержание элементов в названных объектах, можно
Аномальный
источник
К+ + + + + + + + ++ + 5
Ореол рассеяния в породах и почвах s
выветривания
Уровень грунтовых вод
Моренные породы
Почва
речные
о
^склоновые
о (делювий)
Источник с фоновым
содержанием
Геохимические методы поисков полезных ископаемых:
На участке рудного месторождения
(слева) металлометрическими методами в почве, коре
выветривания и в континентальных отложениях обнару-
живается повышенное (против фонового) количество руд-
ных и сопутствующих им элементов; гидрохимическими
методами устанавливается, что поверхностные и грунто-
вые воды обогащены рудными и сопутствующими эле-
ментами; биогеохимические методы позволяют установить,
что организмы, преимущественно растения, обогащены
рудными и сопутствующими элементами; атмохимические
методы показывают, что подземная и частично поверх-
ностная атмосфера содержит повышенное количество ин-
дикаторных газов. На безрудной территории
(справа) всеми этими методами устанавливается среднее
для данного ландшафта (фоновое) содержание всех хими-
ческих элементов или индикаторных газов.
найти ореолы рассеяния, в пределах к-рых легче
обычными геологич. методами найти и само месторо-
ждение. В зависимости от объекта анализа различают
4 основные группы Г. м. п.: 1) металлометрическпе
(анализируются горные породы, почвы), 2) гидрохими-
ческие (поверхностные и подземные воды), 3) биогео-
химические (организмы, преимущественно растения),
4) атмохимические (газы подземной . и надземной
атмосферы). Техника Г. м. п. (геохимич. съемка)
заключается в массовом опробовании различных при-
родных тел, выявлении участков, характеризующихся
повышенным («аномальным») содержанием элементов
по сравнению со средним для данного района («фоно-
вым»). Аномальные участки и будут, как правило,
представлять собой ореолы рассеяния, к-рые подлежат
проверке обычными геологич. и технич. методами.
Г. м. п. основаны на массовых высокочувствительных
анализах. Содержание определяемых элементов в оре-
олах обычно не превышает 10 2 %, нередко оно состав-
ляет Ю’ 3—10 5 % и в отдельных случаях опускается
ниже 10’6 %. Наибольшее применение получил полу-
количеетвенный спектрография, метод, позволяющий
определять одновременно несколько элементов. Этим
методом в СССР ежегодно анализируются миллионы
проб. Меньшее значение имеют пока колориметрия.,
полярография., люминесцентные, хроматография, и др.
методы. Все большее значение приобретают радио-
метрия. методы, использующиеся для поисков U и
Th, а также нейтронные — по активации химич. эле-
ментов, имеющих большое поперечное сечение за-
хвата, напр. В, Be и т. п.
Основное преимущество Г. м. п. состоит в скорости,
дешевизне, возможности поисков так наз. трудноот-
крываемых месторождений («слепых рудных тел»,
т. е. руд, не выходящих на поверхность, месторожде-
ний, не имеющих минералогия, признаков, и т. д.).
Г. м. п. успешно применяются для поисков месторож-
дений многих металлов (U, Си, Zn, Ph, Ag, Au, Hg,
Sb, As, Мо, W, Sn, Ni, Co, Cr, Se, V, Th), а также нефти
и газа. С помощью Г. м. п. в СССР открыты новые
месторождения нефти и газа в Ухтинском районе,
месторождения олова—на Дальнем Востоке, цветных
металлов—в Казахстане и т. д.
Лит.: Геохимические поиски рудных месторождений
в СССР, М., 1957; Геохимические методы поисков нефтяных
и газовых месторождений, М., 1959. А. И. Перельман.
ГЕОХИМИЧЕСКИЕ ПРОЦЕССЫ — геология,
процессы, приводящие к изменению химия, состава
расплавов или растворов и образовавшихся из них
горных пород и минералов, что связано с миграцией
химических элементов, выносом и рассеянием одних,
концентрацией и накоплением других. Многообразие
Г. п. обусловлено огромным различием свойств химич.
элементов и их соединений, большими интервалами
темп-p и давлений, в к-рых эти процессы протекают,
а также различиями щелочности или кислотности
среды и окислительно-восстановительных условий.
Кроме того, Г. п. «чистой» линии эволюции бывают
осложнены наложением посторонних Г. п. (метасо-
матоз и др.).
Наиболее ранним является процесс магмати-
ческой дифференциации, заключаю-
щейся в том, что при выделении из магмы кристаллов
менее растворимой твердой фазы (или удаления в виде
газа части летучих компонетов) магма обедняется
элементами, входящими в состав выделившейся фазы,
и соответственно обогащается оставшимися. Длитель-
ность этого процесса и очень большие массы перво-
начальной магмы могут приводить к очень сильному
изменению состава остаточного расплава, в к-ром
могут значительно накапливаться многие редкие
элементы. Из такого расплава образуются пегматито-
вые жилы. Из магмы выделяется жидкая фаза иного
состава, обладающая отличной от магмы вязкостью
и иным поверхностным натяжением. Другими сло-
вами, происходит расслоение магмы; такой процесс
наз. ликвацией. Таким по отношению к сили-
катному расплаву является сульфидный расплав
(сульфидов меди, никеля, кобальта и др.) в расплав-
ленном моносульфиде железа. Кристаллизация этих
двух расплавов протекает независимо друг от друга.
В результате взаимодействия магматич. расплава
с ксенолитами — обломками вмещающих пород или
с самими вмещающими горными породами, часто
отличного от магмы состава, происходит их частичное
переплавление, что, естественно, вызывает соответ-
ствующее изменение химич. состава магмы и образую-
щихся из нее горных пород. Такое «освоение» магмой
чужеродного материала носит название ассими-
ляции. Когда количество постороннего материала
велико (это может происходить при смешении двух
магм разных составов — процесс, обратный ликва-
ции), происходит явление гибридизма, возни-
кают гибридные горные породы, сочетающие в себе
особенности двух разных по составу горных пород.
«Засорение» магмы чужеродным, не освоенным ею
материалом называется контаминацией.
Кроме химич. взаимодействия с посторонним мате-
риалом, возможна также реакция с летучими компо-
нентами (водой, углекислым газом, фтористыми и
борными соединениями), накопившимися в избытке
в ходе кристаллизационной магматич. дифференциа-
ции; происходит автометаморфизм, к-рый
на примере биотитового гранита заключается в выносе
части магния и железа и в привносе воды, бора, фтора
и увеличении содержания кремневой кислоты. Этот
же процесс, протекающий при более низких темп-рах
и связанный с образованием оловянно-вольфрамито-
вых рудных жил, т. е. протекающий в меиыпих мас-
штабах и в большей степени сопровождающийся
выносом магния, железа, кальция и щелочей и прив-
носом фтора, носит название грейзенизации.
Фтористоводородная к-та, вызывающая вынос этих
элементов, образуется в результате высокотемператур-
ного (400—500°) гидролиза сублимированных SnF4,
843
ГЕОХИМИЧЕСКИЕ ПРОЦЕССЫ — ГЕОХИМИЯ
844
FeF3, BF3 и т. д. или т. наз. пневматолиза,
выражаемого реакциями типа 2FeF3+3H2O —
— Fe2O3 + 6HF, к-рые при высоких темп-рах проте-
кают вправо и сопровождаются выделением твердых
фаз: SnO2, Fe2O3, TiO2 и т. д. Привнос посторонних
веществ извне с выносом из горной породы или руд-
ной жилы одинакового по объему количества более
подвижных в этих условиях компонентов обусловлен
серией химич. реакций, приводящих к замещению
комплекса одних минералов другими, иначе говоря,
к метасоматозу. Различают «диффузионный»
метасоматоз, при к-ром компоненты перемещаются
вследствие диффузии, и инфильтрационный, если
компоненты переносятся течением р-ров, просачиваю-
щихся через породу.
Эндогенные Г. п., протекающие за счет
внутренней энергии земного шара, заканчиваются
гидротермальным процессом — кри-
сталлизацией из перегретых под давлением водных
р-ров, отлагающих в трещинах горных пород кварц,
карбонаты и рудные минералы: сульфиды железа,
меди, цинка, свинца, сурьмы, ртути и мышьяка,
а также самородное и теллуристое золото, серебро и
ряд более редких элементов (кадмий, индий и др.)
в виде примесей, присутствующих в сульфидах этих
металлов. Экзогенные Г. п. (процессы г и-
пергенеза), протекающие за счет внешней,
солнечной энергии, начинаются с выветрива-
ния, к-рое наряду с механич. разрушением горных
пород состоит и в их химич. разложении, заключаю-
щемся в выщелачивании более легко растворимых ве-
ществ, в окислении, гидратации, карбонизации и,
в конечном итоге, в переносе в-ва в виде р-ров или
коллоидных частиц. Характер выветривания опреде-
ляется физико-географич. факторами: темп-рой и
влажностью и обусловлен растительностью, при
отмирании к-рой образуется большее или меньшее
количество гумусовых веществ, способствующих вы-
ветриванию, растворению и удалению одних веществ
и накоплению других. Различают засушливое (арид-
ное) и увлажненное (гумидное) выветривание.
Весьма широко распространены Г. п. почво-
образования (педогенез), к-рые играют
большую роль в общем балансе гипергенных процес-
сов и в миграции элементов. Перенесенные водными
потоками в-ва отлагаются в морских или озерных
водоемах, либо в межгорных впадинах и т. д. Таким
образом происходит образование осадков. Различают
морские и континентальные осадки, причем каждый
из этих типов состоит из многих различных по составу
слоев, фаций. Характер осадка сильно зависит от
аридного или гумидного типа выветривания областей
сноса. Осадочная порода образуется не сразу — отло-
жившийся рыхлый осадок уплотняется (подвергается
диагенезу), превращаясь в осадочную породу,
к-рая и дальше продолжает изменяться, подвергаясь
последующим гипергенным Г. п. и метамор-
физму — дальнейшему изменению, иногда с обра-
зованием полезных ископаемых. Между двумя раз-
личивши по составу осадочными породами также могут
протекать химич. реакпии, в результате к-рых иногда
образуются своеобразные минералы. Осадочные по-
роды, прошедшие сложный путь геохимич. превраще-
ний, оказавшись под слоем более молодых осадков
на значительной глубине, под воздействием давления
и повышенной темп-ры (особенно под влиянием тек-
тонич. процессов) подвергаются метаморфизму, заклю-
чающемуся в дегидратации, декарбонизации, восста-
новлении высших окислов до низших и в образовании
рида минералов, характерных для изверженных пород,
но наряду с этим и минералов, типичных только для
метаморфич. пород, вроде натриевой слюды — пара-
гонита, и нек-рых гранатов.
Лит.: Ферсман А. Е..Избранные труды, т. 3 М.,
1955; Goldschmidt V. М., Geochemistry, Oxt., 1954;
Mason В., Principles of geochemistry, 2 ed., N. Y. — L.,
[19581; К о p ж и н с к и й Д. С., Очерк метасоматическпх
процессов, в кн.: Основные проблемы в учении о магмато-
генных рудных месторождениях, 2 изд., М., 1955, с. 335—456.
В. В. Щербина.
ГЕОХИМИЧЕСКИЙ ЛАНДШАФТ — участок зем-
ной поверхности, в к-ром за счет солнечной энергии
осуществляется миграция химич. элементов атмос-
феры, гидросферы и литосферы. Понятие о Г. л.,
введенное Б. Б. Полыновым (1946), является дальней-
шим развитием география, учения о ландшафтах,
созданного В. В. Докучаевым и развитого Л. С. Бер-
гом. Важнейшей особенностью Г. л. является биоло-
гич. круговорот атомов, заключающийся в образова-
нии и разрушении органич. вещества. Образование
органич. соединений происходит в ходе фотосинтеза;
поглощенная солнечная энергия аккумулируется
в сложных соединениях живых организмов (углево-
дах, жирах, белках и др.). При разложении органич.
веществ заключенная в них энергия освобождается,
производя в ландшафте огромную химич. работу.
Носителями этой энергии в большинстве случаев
являются природные воды, обогащающиеся такими
продуктами разложения, как СО2, органич. кислоты,
сообщающие водам высокую химич. активность.
Поэтому, чем больше в ландшафте образуется живого
вещества, тем интенсивнее в нем протекает миграция
атомов. Каждый Г. л. характеризуется миграпйей
определенных элементов. Так, в черноземных степях
интенсивно мигрирует и накапливается Са: почва и ко-
ра выветривания здесь богаты СаСО3, животные имеют
прочный скелет [СаСО3, Са3(РО4)2], в водах среди кати-
онов преобладает Са2+ (кальциевый ландшафт). Во
влажных тропиках воды кислые, здесь интенсивно миг-
рирует Si, к-рый накапливается в растениях, осадках
озер и болот в форме SiO2; в почвах и растениях много
А1, Са — редок (кислый кремнеземно-железо-алюми-
ниевый ландшафт).
Учение о Г. л. — геохимия ландшафта — является
особым научным направлением, пограничным между
геохимией и географией. В развитии этого направле-
ния особенно велика роль биогеохимии, учения о био-
геохимия. провинциях. Геохимия ландшафта исполь-
зуется при геохимических поисках полезных иско-
паемых.
Лит..- П о л ы н о в Б. Б., Геохимические ландшафты,,
в кн.: Вопросы минералогии, геохимии и петрографии [Отв.
ред. Б. М. Куплетский], М.—Л., 1946, с. 171; е г о ж е, Изб-
ранные труды, М., 1956; Перельман А. И., Очерки-
геохимии ландшафта, М., 1955. А. И. Перельман.
ГЕОХИМИЯ — наука о химич. составе Земли,
о законах распространения и распределения, а также-
способах сочетания и путях миграции химич. элемен-
тов на Земле. Поскольку основным предметом Г.
является химич. элемент в форме атома или иона, Г.
опирается на химические законы и методы исследо-
вания.
Слово «Г.» введено в науку X. Ф. Шёнбейном(1838). Первую-
сводку обширного геохимич. материала дал В. К. Кларк
(1908—1924). В. И. Вернадский впервые сформулировал задачу
Г. как науки об истории атомов на Земле. Дальнейшее развитие
Г. связано с работами А. Е. Ферсмана (ученика Вернадского),
В. М. Гольдшмидта, развивавшего в Г. кристаллохимич. пред-
ставления. и мн. др. ученых
Все космич. тела нашей галактики — звезды, пла-
неты, метеориты и др., состоят из одних и тех же
химич. элементов и их изотопов, но содержат их в раз-
ных отношениях, в связи с различными условиями
возникновения и развития космич. тел. Известно
содержание химич. элементов гл. обр во внешних
частях космич. тел — в коре Земли, в атмосфере Юпи-
тера, в обращающемся слое Солнпа и т. п. (см. Кос-
мохимия). Полнее изучен химич. состав метеоритов.
Распространенность химич элементов в общем слу-
чае являетсв функцией устойчивости атомных ядер.
*845
ГЕОХИМИЯ
846
в недрах звезд. Распространенность атомных ядер
резко падает с увеличением порядкового номера до
Z -= 28, а затем изменяется более плавно, различаясь
у отдельных атомных ядер на 13 порядков. Сравни-
тельно малая распространенность легких элементов
Базальтовый слой
(под океаном)
Земная
кора
{нонтикен-
тальная)
Земная
2.10* атм
.. 6.10* атм.
' В • .
(200 нм.)
'(океани-
ческая)
ДУНИТЫ
А
'(70 км.)‘
Осадочные породы
Базальтовый слой
континентальный
Граниты
С
(450 нм.)
.. 3,5.10* атм:
Рис. 1.
Li, Be, В и др. обусловлена неустойчивостью их
.атомных ядер из-за большого поперечного сечения
реакции с протонами, нейтронами и др. элемен-
тарными частицами, а малая распространенность
таких тяжелых элементов, как Th, U, трансураны —
а-распадом и спонтанным делением этих ядер. Наи-
более распространены атомные ядра с четным числом
протонов и нейтронов, напр. OJ°, Fe|g, Si?|, Mg?^ и др.,
меньше ядер с четным числом протонов и нечетным
числом нейтронов, или наоборот. Наименьшее рас-
пространение имеют ядра нечетно-нечетные, напр.
Dj, К|§, N}4 и др. (подробнее см. Ядро атомное).
Средний состав Земли, а особенно состав ее оболочек,
отличается от среднего состава космич. тел, однако
охарактеризуется теми же общими закономерностями.
Земля как планета — холодное тело, средняя
темп-ра 277°К, объем 1,083 • 1012 км3, поверхность
510,1 106 кле2, средний радиус 6 371,2 км, средняя
плотность 5,52. Возраст Земли, определенный по изо-
топному составу свинца, от момента отвердения
(4,5—5) 109 лет. Земля имеет оболочечное строение.
Модель твердой оболочки Земли см. на рис. 1.
Земля- состоит из концентрических (приблизительно
сферических) оболочек, или геосфер. За внешней газо-
вой оболочкой — атмосферой — идет жидкая обо-
лочка — гидросфера (всемирный океан) — и твердая
оболочка — литосфера (земная кора). Область
на Земле, занятая растительными и животными орга-
низмами, выделяется в отдельную оболочку — био-
сферу. За литосферой располагается еще более
плотный слой — мантия (промежуточная обо-
лочка). Наконец, в центре находится центральное,
наиболее плотное ядро Земли. На историю про-
исхождения Земли имеется два взгляда. Согласно
одному из них, Земля образовалась из горячего
звездного материала, по второму — из холодного
космич. материала с последующим саморазогреванием
его вследствие адиабатич. сжатия и накопления радио-
активного тепла. Последняя точка зрения более со-
временна.
Дифференциация первичного вещества Земли (веро-
ятно, близкого по составу к веществу каменных или
железо-каменных метеоритов) на оболочки произошла
под действием теплоты радиоактивного распада:
в результате накопления тепла, генерируемого радио-
активными изотопами U236, U238, Th232, К40, начались
процессы выплавления легкоплавкой фракции и дега-
зации легколетучих соединений в радиальном направ-
лении к поверхности Землиирасслоение, т. обр., веще-
ства планеты на оболочки.
В настоящее время широко принята гипотеза об
аналогичности состава ультраосновных пород внеш-
ней зоны мантии каменным метеоритам, пород внутрен-
ней зоны мантии (лежащей ближе к ядру)— железо-
каменным метеоритам и ядра Земли — железным
метеоритам (табл. 1).
Таблица 1. Средний состав Земли (в целом), каменных
и железных метеоритов (в вес. %)
Химические Земля Каменные Железные Все*
элементы метеориты метеориты метеориты
Железо Fe 36,9 15,5 90.85 22.30
Кислород О 29,3 41,0 36.7
Кремний Si 14,9 21,0 0,01 18.70
Магний Mg ..... 6,7 14,3 0,03 12,9
Алюминий А! . . . . 3,0 1,56 1,70
Кальций Са 2,9 1,80 0,02 1,68
Никель Ni 2,9 1,10 8,5 1.08
Натрий Na 0,9 0,80 — 0.67
Сера S 0,7 1,82 0.04 1.87
Титан Ti 0,50 0,12 — 0.09
Калий К 0,29 0,07 — 0.06
Фосфор Р 0,15 0,10 0.17 0,10
Марганец Мп .... 0,14 0,16 0,05 0,16
Хром Сг 0,13 0,40 0,01 0.36
Кобальт Со 0,18 0,08 0,60 0,08
Углерод С 0,06 0,16 0.03 0.15
Медь Си 0,01 0,01 0.02 0,01
* Предполагается, что железные метеориты по весу со-
ставляют около 10% веса всех каменных метеоритов.
Геосферы — оболочки земной коры, более или
менее однородные по своему составу и образовавшиеся
в сравнительно одинаковой физико-химической обста-
новке. Поэтому все явления, происходящие в геосфе-
рах, рассматриваются на основе учения о термодина-
мич. равновесии, правила фаз и других законов фи-
зич. химии с тем или иным приближением — в за-
висимости от сложности явлений, происходящих в
той. или иной геосфере, как, напр., в биосфере. Ос-
новными параметрами этих природных равновесий в
геосферах являются давление, темп-ра, число фаз, их
химич. состав, и др. В пределах внешних геосфер
между геосферами с разной интенсивностью непрерыв-
но идет обмен веществ, миграция химиче-
ских элементов. Распределение химич. эле-
ментов по оболочкам Земли имеет закономерный ха-
рактер и зависит от физико-химич. свойств самих эле-
ментов и образуемых ими соединений, в первую оче-
редь,— от строения внешних электронных оболочек
атомов и ионов, т. е. от положения элемента в пе-
риодической системе Менделеева. Геохимическая клас-
сификация элементов может быть иллюстрирована
кривой атомных объемов — рис. 2.
Геохимические процессы на Земле
поддерживаются различными источниками энергии —
гравитациогной (энергия положения), теплотой иду-
щих на Земле экзотермич. химич. реакций, за счет
облучения Земли Солнцем и под действием теплоты
радиоактивных процессов.
Из перечисленных энергетич. факторов наибольшее зна-
чение в эволюции Земли имеет теплота радиоактивных про-
цессов. За 4,5-10’ Лет 1 г пород мантии было освобождено (в
джоулях) засчет К40 — ' 880, Th232 — 392, U2” — 206, UMa —
386, всего 2 864 джоуля.
847
ГЕОХИМИЯ
848
О
химических элементов
химических элементов
преимущественно атмосферы (газы)
преимущественно силикатных горных пород
Группо
Группа
(литосферы)
Группа химических элементов,
аналогах -селенидах и теллуридах)
Группа химических элементов часто самородных
Рис. 2. Кривая атомных объемов и геохимические группы
преимущественно встречающихся в сульфидах
(и их
Содержание этих элементов составляет (в вес. %).
Метеориты каменные . .
Базальты.............
Граниты .............
и Th к
2•10-е 1 • Ю-з 8,5 • 10-а
1 •10-4 4 • 10-4 1,4
4 • 10-< 1,3 • 10-3 2,8
Помимо U, Th, К, в породах Земли известны десятки слабо-
радиоактивных изотопов — Rh8’, Sm147, In113, La138 и мн. др.
Наконец, источником геохимич. процессов является космич.
излучение под действием к-рого в атмосфере Земли создаются
Н3 (тритии Т), С14 и др. короткоживущие радиоактивные изо-
топы. В породах Земли наблюдается слабое нейтронное поле.
Развивающееся в настоящее время ядерное направление в гео-
логии (ядерная геология, или ядерная Г.) стремится полнее
выяснить роль радиоактивных процессов в химич. эволюции
Земли.
Геофизич. расчеты позволяют судить о том, что в
недрах Земли существуют темп-ры в 2000—4000°.
Темп-ра изливающейся магмы определяется в 1000—
1500°, что отвечает верхнему температурному пределу
начала кристаллизации магматич. горных пород. Об-
щая направленность глубинных геохимич. процессов
следует понижению темп-ры, выделению более туго-
плавких и труднорастворимых соединений (минера-
лов) и накоплению более легкоплавких, и особен-
но летучих соединений, таких, как Н2О, СО2, H2S
и т. д. Это отвечает последовательности кристаллиза-
ции пород базальтич. состава (ультраосновные горные
породы, габбро), среднего (диориты) и кислого со-
става (гранитоиды). Наиболее низкотемпературными
являются ультращелочные породы — нефелиновые си-
ениты. Породообразующие минералы представлены
главными компонентами, тогда как второстепенные
элементы — Li, Be, В, Zr, Nb, Та и редкоземельные —
накапливаются в остаточных расплавах, образуя
т. наз. пегматитовые жилы, или рудные жилы, в тре-
щинах уже остывших горных пород.
Ядро Земли имеет радиус 3 000 км, плотн. 11.
По массе оно составляет ~45% массы Земли. По рас-
пространенным представлениям, ядро, подобно желез-
ным метеоритам, состоит из сплава Fe (~ 92%) и Ni
8%). В железе метеоритов обнаруживается повы-
шенная концентрация Pt, Au, Ge, Мо и нек-рых дру-
гих сидерофильных элементов, обладающих
свойством неограниченного растворения в
расплавленном Fe. Ядро сжато до 106 атмо-
сфер и имеет температуру, оцениваемую
~ 2000—3000°, т. е. выше точки Кюри; по-
этому ядро Земли не обладает магнитны-
ми свойствами.
Мантия Земли (промежуточный
слой) имеет мощность 2 900 км.
Породы мантии испытывают давление, растущее
с глубиной. По скорости прохождения попереч-
ных и продольных волн через вещество мантии
Земли различают неск; зон:
элементов.
Зоны Глубина» км Давление, атм
А 30-40 10<
70 2-10*
В 200 6- Ю4
400 1,3 • 105
с 450 3,5 • 103
D 2700 1,2 • 106
При высоком давлении и повышенной
темп-ре, естественно, увеличивается плот-
ность пород, повышается темп-ра их плав-
ления, увеличивается растворимость в них
Н2О и т. п. При этих условиях могут про-
исходить химич. реакции между вещества-
ми в твердой фазе, возможны различ-
ные полиморфные превращения, например
шпинель-оливин или кварц-коэсит и др. Наконец,
при давлении порядка 105 атм у химич. элементов
происходит потеря внешних электронных оболочек
атомов и их обычных химич. свойств.
Мы принимаем, что химич. состав мантии отвечает
химич. составу каменных метеоритов, бедных SiO2 и
богатых MgO и FeO. Т. обр., эта ультраосновная по-
рода, богатая оливинами и меньше— пироксенами, со-
стоит гл. обр. из орто- и метасиликатов магния—
Mg2SiO4 и MgSiO3 с примесью, прежде всего, анало-
гичных соединений железа — Fe2SiO4 и FeSiO3. Из по-
добных пород мантии только дуниты (оливины) иног-
да выходят на поверхность Земли, заполняя зоны
глубоких разломов коры. Именно зона В, как при-
знается многими исследователями, состоит из дуни-
тов (оливинов). В зоне С (области происхождения
наиболее глубокофокусных землетрясений) и особенно
в зоне D вещество мантии, вероятно, испытывает пре-
вращения, напр. распад оливина на MgO и SiO2.
В результате дифференциации вещества мантии воз-
никла и земная кора.
Литосфера (земная кора) — твердая, наибо-
лее внешняя оболочка Земли, вначале условно выде-
ленная мощностью в 16 км. Эта оболочка, состоящая
гл. обр. из базальтов и гранитов, прикрытых чехлом
осадочных пород, отличается от нижележащих пород
мантии не только по химич. составу, но и отделяется
(по сейсмич. данным) от пород мантии поверхностью
раздела (слой Мохоровичича). Земная кора имеет раз-
личную мощность — на континентах ок. 35 км (кон-
тинентальная земная кора), на дне океана ок. 5—10 км
(океанич. земная кора), причем базальтовый слой
покрывает весь земной шар, граниты же на огромном
пространстве дна глубокого океана отсутствуют.
Средний химич. состав литосферы дан в табл. 2.
Образование земной коры, как мы допускаем, свя-
зано с процессом частичного выплавления и дегазации
вещества мантии, разделяющегося на базальтическую
магму и дуниты. Этот процесс может поддерживаться
теплом радиоактивного распада U, Th и К40. Около
1600° молекулы метасиликатов, как известно, пла-
вятся инконгруэнтно по схеме: 2MgSiO3 —> Mg2SiO4 +
+ SiO2. Все другие химич. элементы при этом про-
849
ГЕОХИМИЯ
850
Таблица 2. Средний химический состав литосферы (по А. П. Виноградову)
(мощность — 16 км, без океана и атмосферы)
Химические В атомных В весовых Химические В атомных В весовых Химические В атомных В весовых
элементы % о/ /О элементы о/ /О о/ /О элементы % %
Кислород О . . . . 58,0 47,2 Иттрий Y 6 10~4 2,8 • Ю-з Таллий Т1 . (3 • 10-3) (3 • 10-4)
Водород Н . . . . (3,0) (0,15) Церий Се 6 • 10-4 4,5 • Ю-з Тулий Ти . . 8 • 10-е 8 • Ю-з
Кремний Si ... . 20,0 27,6 Бор В 6 • 10-4 3 • 10-* Кадмий Cd . (7,6 • 10~°) (5 • 10“3)
Алюминий А1 . . . 6,6 8,80 Галлий Ga .... 4•10-4 1,5 • Ю-з Сурьма Sb . (5 • 10-3) (4 Ю-з)
Натрий Na ... • 2,4 2,64 Неодим Nd ... . 3,5 • 10 -4 2,5 . Ю-з Иод J . . . . 4 • Ю-з (3 • 10~3)
Железо Fe .... 2,0 5,10 Лантан La .... 2,5 10-4 1,8 Ю-з Висмут Bi . (1,7 • 10-3) (2 • 10-3)
Кальций Са . . . • 2,0 3,6 Скандий Sc ... . 3 • 10-4 6•10~4 Серебро Ag . 1,6 • Ю-з 1 • 10-5
Магний Mg .... 2.0 2,10 Германий G-e . . . 2• 10-4 7 • 10-4 Индий In . . (1,5 • 10-3) (1 -10-3)
Калий К ~ . 1,4 2,6 Ниобий Nb .... 2 • 10-4 1 . Ю-з Ртуть Hg . . 7 10-7 7 • 10-е
Титан Ti 2,5 • 10~‘ 6•10“! Свинец РЬ .... 1,6 • 10-4 1,6 • Ю-з Осмий Os . . 5 - 10-7 5 Ю-з
Углерод С . . . . (1,5- IO”1) (1 • 10') Мышьяк As ... . 1,5 • 10-4 5 • 10-4 Палладий Pd 1.6 • 10-7 1 • Ю-з
Фосфор Р 5 • 1(Г3 8 - IO"2 Гадолиний G-d . . . 1 10-4 1 • Ю-з Теллур Те . (1,3- 10-7) (1 • 10-3)
Азот N (2,5-10-2) (1 - 10-2) Цезий Cs 9,5 . Ю-з 7 • 10-4 Рутений Ru (1 . 10-7) (5 • 10-7)
Марганец Мп . . . 3,2 10~2 9♦10~2 Празеодим Рг . . . 9 • Ю-з 7• 10~4 Платина Pt. 5 10-8 5 • 10-7
Сера S 3,0 io-2 5 - 10-2 Самарий Sm .... 9. 10-5 7• IO"4 Золото Au . 5 • 10-8 5 10-7
Фтор F 2,8 10~2 2,7 • ДО”2 Молибден Мо . . . 6• 10-з 3 • 10-4 Родий Rh . . 1,7 • 10-8 1 •10~7
Хлор С1 • 2,6 • 10~2 4,5 10-2 Торий Th 7 • 10-5 8 • 10~< Рений Re . . 8,5 • 10-» 1 .Ю~7
Литий Li 1.9 IO-2 6,5 • Ю-з Гафний Hi ... . 5• 10-5 3,2 • 10-4 Иридий 1г . 8,5 Ю-з 1 10-7
Барий Ва 5,7 • IO"2 5 IO'2 Диспрозий Dy . . 5 • Ю-з 4,5 10~4 Протактиний Ра . (8 • 10-12) (1 • 10-ю)
Стронций Sr . . . 1 • 10-2 4 • 10~2 Эрбий Ег 5 . 10-5 4 • 10-4 Радий На . . 9- 10-12 1 • Ю-ю
Хром Ст 8•10-3 2.10-2 Бром Вт 4 • Ю-з 1,6 • 10-4 Актиний Ас . (5- 10-13) (6 • 10-1°)
Ванадий V . . . . 6 10-3 1,5- 10~2 Иттербий Yb . . . 3 • Ю-з 3 • 10-4 Полоний Ро . (2 10-13) (2 - 10-14)
Рубидий Rb . . . 7 • 10-3 3,1 • 10-2 Тантал Та .... 1,8 10-3 2•10~4 Плутоний Pu 7 • 10-17 1 • Ю-15
Цирконий Zr . . • 4 • 10-з 2 10-2 Уран U 2• 10-5 3• 10-4 Радон Rn . . (5-10-17) (7 • 10-13)
Никель Ni .... 3,2 • 10-з 8 Ю-з Европий Ей . . . 1,8 . Ю-з 1.2 • 10-4 Гелий Не
Медь Си 3,6 • 10-3 1 . 10-2 Гольмий Но . . . 1,5 • Ю-з 1.3 • 10-4
Цинк Zn Кобальт Со ... . Бериллий Be . . . 1,5 Ю-з 1,5 • 10~з 1,2- 10-з 5 • Ю-з 3- 10-3 6•10-4 Вольфрам W . . . Селен Se Лютеций Lu . . . Л in kA : 1 1 1 1 • 10-4 6• 10-3 1 • 10-4 Аргон Аг Неон Ne Криптон Кг Количественные опреде- ления ненадежны
Олово Sn 7•10~4 4 • Ю-з Тербий ТЬ .... 1 • 10-а 1,5 . 10-4 Ксенон Хе
цессе, очевидно, распределяются между фракцией
ортосиликатов и SiO2. Понижающие темп-ру плавле-
ния летучие элементы, щелочные и щелочно-земельные
металлы, U, редкоземельные элементы и др. образуют
легкоплавкие соединения с SiO2. Эта легкоплавкая
фракция вещества мантии в результате интрузив-
ной и вулканич. деятельности Земли выплавлялась
на ее поверхность, образуя ее кору, богатую летучими
веществами, водой, щелочными, щелочноземельными
и мн. редкими химич. элементами.
Химич, элементы, повышающие темп-ру плавления
вещества мантии, должны были остаться во фракции
тугоплавких ортосиликатов (темп-ры плавления к-рых
лежат выше 1800°), составляющих основу вещества
мантии (дунитов). Поскольку в среднем из силикатной
фазы метеоритов и, следовательно, первоначального
вещества мантии можно выплавить только ок. 7%
SiO2 (по-видимому, в этом процессе выплавляется
лишь какая-то их малая часть), то становится понят-
ной малая мощность земной коры нашей планеты.
В зоне земной коры легкоплавкая фракция, насыщен-
ная продуктами дегазации, — магма в результате
спада давления и остывания подвергается дальнейшей
дифференциации на многочисленные изверженные
горные породы и минералы земной коры (см. Геохи-
мические процессы).
Разделение основной магмы в земной коре происходит гл.
обр. фракционной кристаллизацией. В связи с разным време-
нем образования различных минералов и последовательностью
их отложения различают три стадии этого процесса. В ранней
стадии кристаллизации, нередко называемой магматич. ста-
дией, в первую очередь образуются твердые фазы соединений
Fe, Mg, Сг, Ti, Si; вместе с ними Ni, Со, Pt и элементы ее группы.
Продуктами первой стадии кристаллизации являются магне-
тит, хромит, титано-магнетит, платино-магнетит, сульфиды
Fe, Ni, Со и Си, руды платины и др. В результате выделения
из магмы указанных элементов она становится более кислой.
В процессе кристаллизации этой магмы (главная стадия кри-
сталлизации) идет дальнейшее удаление из расплава в твердые
фазы Mg и Fe, а также части Са, Al, Si и др. в форме тех или
иных минералов, образующих породы средней кислотности.
В течение главной стадии кристаллизации выделяется основ-
ная масса пироксенов, амфиболов, железных руд, а также
плагиоклазы и др. Продуктами остаточной кристаллизации
силикатного расплава являются полевые шпаты, биотит,
кварц. Из паров и газов образуются т. наз. грейзены — ме-
сторождения, богатые соединениями Sn, W, Sc и др. редкозе-
мельных элементов. Наконец, в позднюю низкотемп-рную
фазу кристаллизации образуются и пегматиты, богатые ред-
кими элементами — Li, Be, Th, Nb, Ta, F, редкоземельными
и др. В зависимости от состава, пегматиты делятся на типы.
Если атомное отношение (Na -4- К)/А1 меньше единицы,
то в этих плумазитовых пегматитах появляются многочислен-
ные минералы с избытком AI. Если (Na 4- К)/А1 больше 1,
то в этих пегматитах превалируют минералы, в к-рых щелочи
связаны с Fe111. При охлаждении паров и газов образуются
горячие растворы, к-рые дают начало разнообразным гидро-
термальным месторождениям цветных и редких металлов (Си,
Pb, Ag, Zn, Sn, W, Au, Sb, Hg и др.).
Распределение химич. элементов по фазам (мине-
ралам) в геохимич. процессах рассматривается в свете
кристаллохимии, законов. В первую очередь, в про-
цессах дифференциации образуются соединения с более
высокой анергией кристаллической решетки. Послед-
няя зависит от типа элементарной ячейки, радиуса
ионов (или атомов), их заряда (валентности), коорди-
национного числа и эффекта поляризации.
Ионы, размеры к-рых различаются не больше чем
на 15%, изоморфно замещаются в решетках (см. Изо-
морфизм, Ионный радиус). При общих равных усло-
виях ионы с меньшим ионным радиусом или ионы
с большим зарядом предпочтительнее входят в крис-
таллич. решетку. Гетеровалентный изоморфизм поз-
волил объяснить ассоциацию химич. элементов с очень
различными химич. свойствами, напр. U, Th и редко-
земельных в торианите и т. п. парагенетич. ассоциа-
циях элементов. Исключительную роль в составе
пород земной коры играют кристаллич. решетки из
тетраэдров(SiO4)*~, (A104)s-,(P04)3~h др.,аналогичные
им, особенно алюмосиликаты с различными способами
сочетания этих тетраэдров.
В процессе фракционной кристаллизации ионы и
атомы сортируются по своим размерам и свойствам.
Кристаллич. решетки главных породообразующих
минералов— полевых шпатов, слюд и т. д., принимают
одни ионы и не принимают других — в зависимости
от их ионного радиуса Rit заряда, поляризации.
В процессе кристаллизации силикатной магмы поро-
дообразующие минералы захватывают гл. обр. ионы
со средними величинами Rit ионы же с малыми Ri
такие, как Li, Be, В, F, или с большими — Cs
Rb, Т1 и др., концентрируются в конечных продуктах
процесса дифференциации магмы — в пегматитах,
851
ГЕОХИМИЯ
852:
пневматолитах и частично в гидротермальных рудах.
Исключительное значение в качестве индикаторов
геохимич. процессов представляют отношения в поро-
дах пар близких по свойствам химич. элементов:
Nb/Ta, Zr/Hf, W/Mo, S/Se, Rb/Tl, Cl/Br, Al/Ga,
Ni/Со, Ra/Cd и др. Распределение элементов в поро-
дах приближается к нормальному, когда данный эле-
мент находится в ряде фаз (минералах), и к логнор-
мальному, когда этот элемент преим. концентрируется
в к.-н. одном минерале породы.
Вслед за процессами образования литосферы и
дальнейшей ее дифференциации на отдельные породы
и минералы не менее грандиозным процессом является
разрушение горных пород (выветривание) и образова-
ние осадочных пород, происходящее под влиянием
Н2О, СО2, О2, жизнедеятельности организмов и др.
факторов, при участии природных водных р-ров.
В наиболее внешней части Земли формируется кора
выветривания. Средняя мощность осадочных пород
по всей поверхности Земли достигает ок. 700 м,
или, при расчете на континенты,—ок. 2000 м. Осадоч-
ные породы состоят гл. обр. из глин и сланцев (ок.
70%), песков и песчаников (ок. 20%) и карбонатов
(ок. 10%). При выветривании образуются генетич.
ряды выветривания минералов, напр. полевой шпат —
серицит — каолин —• боксит —» кварц и т. п. С вод-
ными р-рами в море выносятся легкорастворимые
соединения щелочных элементов — Li+, К+, Rb+ и
особенно Na+; щелочноземельных элементов — Са2+,
Sr2+, Mg2+, а также ионов Cl-, SO%, СО% и др. Гид-
раты тяжелых металлов — Fe, Мп и др., в известном
порядке в зависимости от условий образуют осадки
гидроокисей. Растворимость соединений, их сорбция
на глинах и др. коллоидах, pH растворов, окисли-
тельно-восстановительный потенциал и др. факторы
являются причиной природной сортировки атомов
химич. элементов в процессах, протекающих в при-
родных р-рах и приводящих к образованию различ-
ных осадочных пород.
Все разнообразие процессов осадкообразования может
быть сведено к следующей геохимич. классификации: 1) обра-
зование остаточных песков и песчаников, россыпей из устой-
чивых минералов, таких, как Si02, ТЮ2. ThO2, цирконы, мона-
циты; 2) гидролиз алюмосиликатов и накопление продуктов
гидролиза — глинистых минералов, водных Л12О3, Fe2Os,
SiO2; 3) окисление железа и марганца; гидроокиси этих ме-
таллов захватывают соединения Р, Ag, V (осадки Fe) и Ва,
Ra, Мо, Со и др. (осадки н руды Мп); 4) образование в резуль-
тате деятельности организмов, карбонатных пород — СаСО3,
MgCO3, SrCO3, РЬСО3, а также фосфатов с захватом ими F,
редкоземельных элементов и др.; 5) галогенез — выделение
солей из водных бассейнов при испарении, последовательное
отложение CaC03, NaCl, затем сульфатов и хлоридов Mg, К,
а также Rb; вместе с тем накопление Вг в остаточной рапе;
6) образование, при участии организмов, каустобиолитов —
углей, нефтей и др. битумов, сапропелей; образование СН4, H2S
и генезис осадочных сульфидов. Процессы метаморфизма и ме-
тасоматоза осадочных пород (и массивных изверженных пород)
широко развиты в земной коре. Наиболее грандиозным про-
цессом метаморфизма являетс’я переработка, под влиянием
тепла и эманаций магмы (приносящих Na и др. элементы в зону
изменений), древних осадков (гранитизация).
Гидросфера — совокупность океанов, морей
и континентальных водных бассейнов. Океан занимает
71% поверхности Земли, его средняя глубина 4 км,
масса воды 1,5-1018 т. Солевой состав океана форми-
ровался за счет продуктов дегазации вещества мантии
и выветривания горных пород и выноса солей реками.
R результате выветривания в океане накапливались
катионы Na+, Mg2+, К+, Са2+ и др. Главные анионы
морской воды — Cl , SO%, НСО3 и ДР-, а также
сама вода — продукты дегазации вещества мантии.
В настоящее время эти продукты еще поступают на
поверхность Земли, напр. из газов, к-рые в огромном
количестве выбрасываются действующими вулканами.
Помимо главных конституентов морской воды, в ней
находятся практически все остальные химич. эле-
менты, однако в очень малом количестве (см. табл,
в ст. Гидросфера}. Это объясняется тем, что при Нр
морской воды 8,2—8,5 многие химич. элементы —
Fe, Al, Ba, Мп, РЬ и т. д., дают труднорастворимые
соединения, выпадающие в осадок в прибрежной зоне
морей. Морская вода находится в равновесии с атмос-
ферой, илами дна (иловой водой) океана и живым
веществом, его населяющим, особенно фитопланкто-
ном в верхних слоях океана.
Так, напр., изменение pH регулируется буфером:
(атмосфера)
(вода)
(дно)
СО->
1
1
Н2О—Н,СО3 —IJCO - +Н+— С02 - + н+
3 j 3
Са2+ — СаСО,
В тропич. областях, где океан благодаря жизне-
деятельности организмов пересыщен СаСО3, обра-
зуется огромное количество твердых карбонатов (на
атоллах).
Арктич. воды океана, напротив, недосыщены CaCOs
и способны растворять карбонаты осадков дна. По-
этому в высоких широтах на дне океана есть лишь оса-
док SiO2, образующийся из скелетов диатомей, фора-
минифер и др. организмов. Организмы моря концент-
рируют нек-рые рассеянные химич. элементы морской
воды — Вг, F и др. Вместе с тем повышается их кон-
центрация и в илах, из к-рых позже формируются,
напр., нефтеносные межпластовые иодо-бромные воды.
В результате полного высыхания морских бассейнов
происходит садка (галогенез) солей Na, К, Mg и др.
в определенной закономерной последовательности.
Разнообразие состава речных, озерных и (особенно)
межпластовых подземных вод и источников объяс-
няется взаимодействием растворов с породами и раз-
личием в климатич. условиях. Изучение химич. со-
става природных води его изменений в связи с химич.,
физич. и биологич. процессами служит предметом
самостоятельной отрасли Г. — гидрохимии.
Атмосфера — внешняя газообразная оболочка
Земли с постоянно убывающей концентрацией газов,
простирается до высоты 1100—1400 км. На Земле-
сохранились лишь те газы, к-рые были связаны в проч-
ные химич. соединения с твердым холодным вещест-
вом планеты. В результате вторичного разогревания
вещества планеты газы и легколетучие вещества
дегазировались из твердого вещества и, с одной
стороны, образовали атмосферу, а с другой — при-
няли участие в образовании гидросферы. Часть легких
газов (Н2, Не) была потеряна Землей. Т. обр., ат-
мосфера состоит гл. обр. из газов Земли, а не газов
космич. происхождения. Состав атм. воздуха у поверх-
ности Земли приведен в таблице в ст. Атмосфера.
С высотой отношения между главными составляю-
щими атмосферы изменяются мало.
Азот содержится в породах гл. обр. в виде NHS
(его много выбрасывается фумаролами в вулканиче-
ских областях в виде NH4C1), к-рый, будучи дегази-
рован, окисляется до N2. Кислород атмосферы обра-
зуется гл. обр. в результате фотосинтеза (за счет раз-
ложения Н2О) и в атмосфере изменяет свой изотоп-
ный состав (утяжеляется) благодаря преимуществен-
ному участию более легкого изотопа (ОХ6) в окислении
органич. веществ. На высоте 30 км под влиянием фо-
тохимич. процессов образуется озоновый экран (с по-
вышенным содержанием О3), частично поглощающий
ультрафиолетовое излучение. СО2, возможно, образо-
валась в результате окисления СН4, дегазируемого
веществом мантии. Значительно больше СО2 обнару-
живается в тех осадках (карбонатах), к-рые обра-
зуются вслед за эпохами интенсивного вулканизма.
Зеленые растения действуют в одну сторону — пол-
ного элиминирования СО2 из атмосферы. Инертные
газы дегазируются из пород Земли. Не4 образуется
в породах в результате радиоактивного распада С и
Th, Аг10 — за счет превращения К.40 в Ai40. Тяжелые-.
853
ГЕОХИМИЯ — ГЕПАРИН
854
инертные газы Кг, Хе и др. образуются частично за
счет спонтанного деления U и.Th, частично — за счет
деления U на нейтронах.
Биосфера — области атмосферы, гидросферы и
частично литосферы, к-рые заняты живым веществом.
Геохимич. функция живого вещества огромна. Орга-
низмы непосредственно создают свободный кислород
в атмосфере, угли, нефти и др. битумы (в виде иско-
паемых каустобиолитов), карбонаты, диатомит, се-
литру, фосфориты; участвуют в образовании отложе-
ний серы, железных, марганцовых и др. осадочных
пород. Нек-рые организмы концентрируют F, Вг,
V, Эей др. Исключительно велико участие организмов,
а также образуемых ими органич. кислот, СО2 и О2
в выветривании горных пород. Многие процессы
миграции элементов приобретают при участии орга-
низмов циклич. характер. Биогеохнмич. процессы
изменяют состав рек, озер, почв и т. д. В свою оче-
редь, геохимич. среда влияет на распространение
и изменчивость организмов. В результате недостаточ-
ности или избыточности отдельных химич. элементов
в среде возникают т. наз. биогеохнмич. провинции;
по характеру растительно-
сти и повышенному содер-
жанию в ней того или
иного элемента применяют
биогеохнмич. метод пои-
сков руд. (См. также Био-
геохимия).
Геохимия отдель-
ных элементов
включает описание физико-
химич. свойств данного
элемента и его соединений, что служит основой пони-
мания законов его распределения и миграционной
способности в различных условиях; дает распрост-
раненность химич. элементов (см. Кларки элемен-
тов) в различных космич. и геологич. объектах и
•объясняет распределение элементов по фазам пород,
исходя из кристаллохимия, данных, изоморфизма
и изоструктурности соединений. Наиболее важной
стороной Г. отдельных элементов является изучение
поведения элемента в различных эндогенных и экзо-
генных процессах (см. Геохимические процессы). На
основе всех этих сведений дается представление
о путях миграции элемента и областях его концентра-
ции и рассеяния. Многие химич. элементы наз. ред-
кими из-за их малого распространения в природе.
Другие элементы рассеяны и не дают собственных
минералов. Наконец, многие из наиболее распрост-
раненных химич. элементов находятся в определен-
ных минералах в состоянии рассеяния, примесей,
напр. следы тяжелых металлов в кварце и т. п.
Прикладная- геохимия. На основе
изучения физико-химич. свойств ионов и атомов, зако-
нов геохимич. поведения отдельных элементов в при-
родных системах и их распределения по породооб-
разующим или рудообразующим минералам, зная
пути концентрирования и рассеяния химич. элементов
в различных геохимич. процессах, можно предсказы-
вать возможные области концентрации тех или иных
элементов. Изучение геохимич. поведения различных
химич. элементов в пределах к.-л. геологич. провин-
ции является задачей региональной Г. Исключитель-
ный интерес представляет обнаружение признаков
пород, руд, коррелирующих с концентрированием
того или иного химич. элемента (напр., повышенпе
содержания РЬ в калиевых полевых шпатах гранитов
в рудных районах или повышение содержания Sn
в биотитах гранитов оловоносных областей). Эти
геохимич. критерии используются в разведке по-
лезных ископаемых (см. Геохимические методы по-
мекав).
Лит.: Вернадский В. И., Избр. соч., т. 1, М., 1954;
Виноградов А. П., Геохимия редких и рассеянных
химических элементов в почвах, 2 изд., М., 1957; его же,
Химическая эволюция Земли, М., 1959; Goldschmidt
V. М., Geochemische Verteilungsgesetze der Elemente, Bd 1—9,
Kristiania, 1923—38; С а у к о в А. А., Геохимия, М. 1950;
Ферсман А. Е., Геохимия, т. 1—4, Л., 1933—39; П1 е р-
б и н а В. В., Геохимия, М,— Л., 1939; Р а и к а м а К.,
Изотопы в геологии, пер. с англ., М., 1956.
А. П. Виноградов.
ГЕПАРИН — кислый аминополисахарид, задержи-
вающий свертывание крови; содержится в небольших
количествах во многих тканях — в печени, легких,
селезенке и др., а также в крови; образуется в особых
клетках, называемых тучными. Строение Г. точно
не установлено. В его состав входят D-глюкозамин
и D-глюкуроновая к-та, связанные чередующимися
а-1,3- и а-1,4-гликозидными связями. Структурной
единицей Г. считают дисахарид, образованный глю-
козамином и глюкуроновой к-той. Аминогруппа
D-глюкозамина в Г. сульфатирована; сульфогруппа
этерифицирует и спиртовые гидроксилы, но точно
положение этих групп в дисахаридной единице не
установлено.
Выделяемые из тканей препараты Г. обычно неодно-
родны, в них содержатся фракции, отличающиеся
степенью полимеризации, количеством сульфогрупп
на дисахаридную единицу и силой антикоагулирую-
щего действия. Средний мол. вес Г. 16 000—20 000;
среднее содержание сульфогрупп 2—3 на дисахарид-
ную единицу. У фракций, содержащих большее
количество серы, антикоагулирующее действие выра-
жено сильнее; наиболее активные препараты различ-
ного происхожденив (овцы, свиньи, собаки) содержат
14,0—14,7% серы. При гидролитич. отщепления
от Г. всех сульфогрупп образуется полисахарид —
ф-г е п а р и н, лишенный антикоагулирующего дей-
ствия.
Препараты Г. получают обычно в виде аморфного
белого порошка, растворимого в воде и довольно
плохо растворимого в большинстве органических
растворителей. Г. растворяется в щелочах, но в щелоч-
ной среде, так же как и в кислой, он неустойчив, что
проявляется, в первую очередь, в снижении его био-
логич. активности. Соли Г. называют гепарин а-
т а м и. Гепаринат бария может быть выделен в кри-
сталлич. виде и служит для характеристики Г. С орга-
нич. основаниями (октиламин, бензидин, гистамин,
основные красители и др.), с алкалоидами (хинин,
бруцин) и белками Г. образует соли, а с липоидами —
комплексы. Основными красителями, содержащими
свободные аминогруппы (азур, бромкрезоловый синий
и др.), Г. окрашивается мета хроматически (см. Мета-
хроматическое окрашивание), что используется для
его колориметрия, определения. Для определения
Г. применяют хроматографирование на бумаге, а также
турбидиметрия, и нефелометрия, методы; последние
основаны на определении колияества нерастворимых
солей Г., напр. с казеином или 2-ДИМетиламинометил-
бензофураном. Широко используются биологич. ме-
тоды, основанные на установлении антикоагулирую-
щего действия Г. Единица действия Г. (1 Ед) соответ-
ствует 0,0077 мг международного стандартного пре-
парата Г.
855
ГЕПТАН — ГЕРАНИОЛ
856
Получают Г. в виде его натриевой соли из легких
и печени рогатого скота. Г. применяют для уменьше-
ния свертываемости крови, для предупреждения
инфарктов миокарда сердца, послеоперационных тром-
бов и в качестве естественного стабилизатора крови
при ее переливании.
• Помимо Г., в тканях содержатся другие антикоагу-
лирующие аминополисахариды (гепариноид ы):
|3-г е п а р и н, молекулы к-рого построены из остат-
ков D-галактозамина и L-идуроновой к-ты (см. Гек-
суроновые кислоты), и мукоитинсерная
к-та В—аминополисахарид из группы хондроитин-
серных кислот, по строению и свойствам близкий,
а возможно и тождественный ^-гепарину.
Лит.: Бычков С. М Усп. биол. хим., 1950, 1, с. 461;
Кузин А. М., там же, 1954, 2, 256; Advances Carbohydr.
Chemistry, 1955, р. 335; Methods of biochemical analy-
sis, v. 7, L., 1959, p. 253; см. также при ст. Мукополисахариды.
Л. И. Линевич.
ГЕПТАН (н-гептап) СН3(СН2)5СН3, мол. в. 100,21 —
бесцветная подвижная жидкость со слабым запахом;
т. кип. 98,427°/760 мм; т. пл. —90,610°; d20 0,68376;
Ир 1,38764, теплота испарения 7,575 ккал/моль
(т. кип.); теплота горения жидкого Г. (при 25°)
1167,11 ккал/моль (П2ОЖИДК.); теплота образования
(жидк.) Д//298 = 47,52 ккал/моль; теплоемкость Ср=
=34,20 кал/град моль (25°). По химич. свойствам Г.
близок к другим неразветвленным гомологам метана.
При действии хлора замещение происходит при пер-
вом и втором атоме углерода; окислы азота и 13%-ная
азотная к-та нитруют Г. преим. в положение 2. Под
действием CoF3 при 230—350° образуется перфторгеп-
тан с хорошим выходом; кислород окисляет Г., при
втором атоме С, как промежуточный продукт обра-
зуется гидроперекись. Большой интерес представляет
реакция дегидроциклизации Г,, приводящая к обра-
зованию толуола и играющая важную роль в процессах
каталитич. риформинга и ароматизации нефтяных
углеводородов. Чистый Г. может быть получен обыч-
ными методами синтеза парафинов, переработкой
смол нек-рых видов американских сосен Pinus jeffreyi
и Pinus sabiniana, а также выделен фракционировкой
из нефти или из синтетич. бензина, получаемого из
СО и Н2. Г. применяется в качестве первичного эта-
лона при определении детонационной стойкости кар-
бюраторных топлив; октановое число его принимается
равным 0; изомер Г. — 2,2,3-триметилпеятан (трип-
тан) — обладает хорошими свойствами как моторное
ТОПЛИВО. В. В. Щекин.
ГЕПТИЛОВЫЙ СПИРТ (гептанол-1), молекуляр-
ный вес 116,21,СН3(СН2)5СН2ОН — бесцветная жид-
кость с фруктово-цветочным запахом; т. пл. — 34,1°;
т. кип. 176,3°; df 0,82601; ng'1 1,42326. Содер-
жится в сивушном масле. Может быть получен ката-
литич. гидрированием гептилового альдегида (энан-
тола) или эфиров энантовой к-ты и др. способами.
Г. с. образуется наряду с другими спиртами при ката-
литич. восстановлении СО и СО2 водородом. При
окислении дает энантовую к-ту; образует сложные
эфиры, при нагревании с KHSO4 при 145° дает дигеп-
тиловый эфир, при 175° — гептилен-1. Г. с., его фор-
миат, ацетат, пропионат и бутират применяют в не-
больших количествах в парфюмерии. Г. с. образует
кристаллич. производные: фенилуретан, т. пл. 60°;
n-нитрофенилуретан, т. пл. 102° или 105°; нафтилуре-
тан, т. пл, 62° и др. Г. с. может быть количественно
определен ацетилированием в пиридине и др. методами
определения первичных спиртов.
Лит.: Синтезы органических препаратов. Сб. 1, пер. с
англ., М., 1949, с. 156. В. Н. Красева.
ГЕПТИНКАРБОНОВАЯ КИСЛОТА, мол. в. 140,18,
СН3(СН2)4С= ССООН — одноосновная карбоновая
к-та ацетиленового ряда; т. пл. +5°; т. кип. 149720 мм.
Синтезируют Г. к. из гептилового альдегида (энан-
тола):
сн3(сн»)5сно —is. сн3(сн2)5снс12 —S
— СН3(СН2)4С~СН -22- CH3(CH2)4C=CNa
— CH3(CH2)4CHCCOONa —2—Ь СН3(СН3)4С=ССООН.
Практич. интерес представляют эфиры Г. к. — бес-
цветные или слабоокрашенные жидкости с неприят-
ным резким запахом, в большом разведении напоми-
нающим запах листьев фиалки. В эфирных маслах
эфиры этого ряда не найдены; их получают этерифи-
кацией Г. к.
Эфир Т. кип.,°С/лии 4 20 Пр
Метиловый . . . Этиловый .... Пропиловый . . * Изопропиловый . Изобутиловый . . Изоамиловый . . 216/760; 107/20 115—115,5/16 133—134/17 126—127/22 138—139/23 148—149/20 0,9335 (12,3°) 0,9207 (11,5°) 0,9247 (20°) 0,9193 (20°) 0,916 (20°) 0,9114 (20°) 1,45092 (12,3°) 1,47152 (11,5°)
Наиболее интересным представителем является
метиловый эфир Г. к. («фолион»); применяется в цве-
точных композициях типа фиалки; сильно раздражает
кожу; в губных помадах недопустимы даже его следы.
ГЕРАНИЕВАЯ КИСЛОТА С10Н*Ь^' мол.^вес
168,12 — непредельная кислота, состоящая из сме-
си двух изомеров: 2,6-диметилгептадиен-1,5-карбоно-
вой кислоты НООССН=С(СН3)СН2СН2СН=С(СН,)2
и 2,6-диметилгептадиен-1,6-карбоновой кислоты
НООСНС=С(СН3)СН2СН2СН2С(СН3)=СН2; т. кип.
158°/14 мм; 0,9518; 1,4869. Г. к. встречается
в масле лимонной вербены, получается синтетически
окислением цитраля или рядом реакций из 2-метил-
гептен-2-она-6 и этилового эфира хлоруксусной к-ты.
Г. к. при действии 65—70%-ной H2SO4 легко цикли-
зуется в гидроароматич. непредельную циклогера-
ниевую к-ту, состоящую из неск. изомеров. При нагре-
вании Г. к. образуется гераниолен — смесь непре-
дельных углеводородов с двумя двойными связями
состава С9Н1б. М. И. Роэенгарт.
ГЕРАНИОЛ CioH18O, мол. в. 154,25 — жидкость
светло-желтого цвета с запахом, напоминающим запах
роз; т. пл. —15°; т. кип. 229—230°; 0,8894;
1,4766; в воде нерастворим; хорошо растворяется
в спирте и эфире. Г. стереоизомерен с неролом; он
является транс-изомером; нерол—i/uc-изомер. Как
и другие терпеновые спирты жирного ряда, Г. нахо-
дится в виде смеси лимоненной (I) и терпиноленной
(II) форм (а- и р-формы):
сн3.
>С-СН2(СН2)2С-СН3
си// II
НС—СН2ОН
СН3,
, )С=СН(СН2)2С—сн3
он,/ II
НС-СН2ОН
п
Г. — составная часть гераниевого, розового, паль-
мо-розового и нек-рых других эфирных масел. Из этих
масел его выделяют в виде твердого соединения с хло-
ристым кальцием. Последнее разлагают водой, и Г.
отгоняют с водяным паром. Синтетически Г. может
быть получен изомеризацией линалоола при нагре-
вании с уксусным ангидридом:
(СН,)2С=СН(СН2)2С(ОН)-СН=СН2 —
сн3
-» (СН3)2С=СН(СН2)3С=СНСН2ОН
Сн3
857
ГЕРБИЦИДЫ — ГЕРМАНИЙ
858
Реакция эта сопровождается одновременным окис-
лением. Идентифицируют Г. по дифенилуретану,
т. пл. 82°. Применяется Г. как компонент парфюмер-
НЫХ композиций. Д. н. Васкевич.
ГЕРБИЦИДЫ — химические средства борьбы с сор-
ными растениями. По характеру действия на растения
Г. делятся на: сплошного действия, убивающие
все виды растений, и избирательные (селек-
тивные), действующие на одни растения и безопас-
ные для других видов растений. Такое деление яв-
ляется условным, т. к. в большинстве случаев одно
и то же вещество в зависимости от концентрации и норм
расхода может проявлять себя как Г. сплошного или
избирательного действия. Каждую из указанных групп
Г. по характеру действия на растения и способам
применения делят на подгруппы: контактного
действия, системные и корневые; послед-
ние действуют на корневую систему растений или
на прорастающие семена.
К Г. контактного действия относят в-ва, поражаю-
щие листья и стебли растений при непосредственном
контакте их с препаратом. Контактным действием
обладают как Г. сплошного действия, так и избира-
тельные. К избирательным Г. относятся динитрофе-
нолы, органич. соединения ртути, цианат калия (нат-
рия), нефтяные масла, цианамид кальция, серная
к-та, сернокислая и азотнокислая медь и др., а так-
же контактные Г. сплошного действия: нефтяные и
кам.-уг. масла, богатые ароматич. углеводородами,
пентахлорфенол, пентахлорфенолят натрия, 3,6-энд-
оксогексагидрофталевая к-та, ксантогендисульфид, ал-
лилхлорфенилкарбонат и др.
К системным Г. относятся в-ва, способные распре-
деляться вместе с питательными соками по сосудистой
системе растений. Такие Г., попав на листья или корни
растения, быстро распространяются по всему расте-
нию, вызывая его гибель, что особенно ценно в борьбе
с сорными растениями, имеющими мощную корневую
систему, и с многолетними сорняками. Из Г. избира-
тельного действия в эту подгруппу входят арилокси-
алкилкарбоновые к-ты, напр. 2,4-дихлорфеноксиук-
сусная, 2-метил-4-хлорфеноксиуксусная, 2,4,5-три-
хлорфеноксиуксусная, 2,4-дихлорфеноксимасляная,
замещенные фенилуксусные и галогенопроизводные
бензойной к-ты, а также фосфорорганич. Г. и соли
четвертичных аммониевых оснований. К системным
Г. сплошного действия относят арсенит натрия, арре-
нал, хлораты, тиоцианаты, сульфамат аммония и др.
соединения подобного типа.
К Г., избирательно действующим на корневую си-
стему или на прорастающие семена, относят эфиры
фенил- и хлорфенилкарбаминовых к-т, дихлораль-
мочевину, трихлорацетат натрия, 2,4-дихлорфено-
ксиэтилсульфат натрия, пента хлорфенолят натрия,
фенил-, 4-хлорфенил- и 3,4-дихлорфенилдиметилмо-
чевину и др. Корневыми Г. сплошного действия яв-
ляются: хлорпикрин, сероуглерод, дихлорпропан,
дихлорпропен, бромистый метил, тетрахлорэтилен,
тирцианаты, соли борной и мышьяковистой к-ты,
арилдиметилмочевины и др.
Для борьбы с двудольными сорными растениями
в посевах злаков (однодольных) наиболее широкое
применение получили 2,4-дихлорфеноксиуксусная,
2-метил-4-хлорфеноксиуксусная и 2,4,5-трихлорфе-
ноксиуксусная к-ты, к-рые используются как в виде
растворимых в воде солей, так и в виде эфиров раз-
личных спиртов. Нормы расхода этих продуктов
0,4—2 кг)га (в пересчете на кислоту). Для борьбы
с однодольными сорными растениями в посевах про-
пашных культур находят применение трихлорацетат
натрия, дихлорпропионат натрия, 3-хлорфенилизо-
пропилкарбамат (хлор-ИФК) и нек-рые другие. Пред-
ставляет интерес 2-хлор-4,6-бис(этиламино)-силш-триа-
зин, к-рый при норме расхода 1—3 кг на 1 га действует
на большинство сорных растений и практически без-
опасен для кукурузы.
Для борьбы с древесно-кустарниковой раститель-
ностью чаще всего используют сульфамат аммония,
мышьяковисто-кислый натрий и 2,4,5-трихлорфен-
оксиуксусную к-ту; эти Г. иногда называют а р б о-
рицидами.
Лит.: Мельников И. И., Баскаков Ю. А.,
Бокарев К. С., Химия гербицидов и стимуляторов роста
растений, М., 1954; Альгрен Дж., Клингмэн Г.,
Вольф Д., Борьба с сорными растениями, пер. с англ.,
М„ 1953; Химические средства стимуляции и торможения
физиологических процессов растений, под общ. ред. 10. В. Ра-
китина, М., 1958; Г у н а р И. И., Березовский М. Я.,
Химические средства борьбы с сорняками, М 1952.
Н. Н. Мельников.
ГЕРМАНИЙ (Germanium) Ge — химич. элемент
IV гр. периодич. системы Менделеева, п. н. 32, ат. в.
72,60. Представлен пятью стабильными изотопами:
Ge’°(20,55%), Ge72 ( 27,35%), Ge73 (7,78%), Ge71
(36,50%) и Ge76 (7,86%). Искусственно получены
многочисленные радиоактивные изотопы Г. Нек-рые
из них образуются с небольшим выходом в качестве
осколков при делении U235 медленными нейтрона-
ми: Ge77 (Т1/2=12 час.) и Ge78 (Т1/2=1,47 часа). Конфи-
гурация внешней электронной оболочкиатома 4s24p2-
Энергии ионизации: Ge°—>Ge+—>Ge2+—>Ge3+ —>Ge1+
соответственно равны 8,13; 15,95; 34,22; 45,70 эв.
Поперечное сечение поглощения тепловых нейтронов
атомом Г. 2,35 барн.
Существование и свойства Г. предсказал в 1871 Д. И. Мен-
делеев на основании периодич. закона и наименовал этот,
неизвестный до тех пор, элемент «экасилицием». В 1886 К. Вин-
клер обнаружил в минерале аргиродите новый элемент, к-рый
назвал Г. в честь своей страны (лат. Germanium); по свойст-
вам Г. оказался вполне тождествен «экасилицию».
Содержание Г. в земной коре 7 • 10 1 вес.%. В гео-
химич. процессах Г. тесно связан с соседними по
периодич. системе элементами — Si, Zn, С, Си, Sn,
Ga, Ti, Zr, As, а такжеc Fe. Собственных минералов Г.
известно немного, все они относятся к группе сульфо-
солей: германит Cu3(Fe, Ge)S4 (10% Ge), аргиродит
AgsGeS6 (5—7% Ge), канфильдит Ag8(Sn, Ge)S6, изо-
морфен аргиродиту (до 1,8% Ge), рениерит (Си, Fe)3
(Ge, Fe, Zn, Sn)(S, As)4 (6—8% Ge), ультрабазит
28PbS • llAg2S • 3GeS2 • 2Sb2S3 (до 2,2% Ge). Эти
минералы Г. редки, их крупных скоплений до сих
пор не найдено, поэтому они почти не имеют практич.
значения как источники получения Г. Основная масса
Г. рассеяна в виде примеси в составе др. минералов,
гл. обр. силикатов, сульфидов и сульфосолей; наблю-
дается также накопление Г. в каустобиолитах (углях,
горючих сланцах, нефти). В небольших количествах
Г. обнаружен в рудничных, шахтных и нефтяных
водах, в водах горячих источников, в молоке и крови
человека, в водорослях и др. растениях. Содержание
Г. в углях различных типов колеблется от следов
до десятых долей процента, причем Г. концентри-
руется преимущественно в малометаморфизирован-
ных углях; угли антрацитового типа Г. почти не
содержат.
Подавляющая часть Г. получается из побочных
продуктов при переработке руд цветных металлов,
к-рые содержат от 0,001 до 0,1% Г. (гл. обр. цинковых
обманок и полиметаллич. руд). В качестве сырья для
извлечения Г. используются также золы от сжигания
угля, шламы от газификации и нек-рые отходы коксо-
химия. производств.
Физические и химические свойства. Компактный
Г.—серебристого цвета, по внешнему виду похож на
металл; имеет кубич. гранецентрированную кристал-
лич. решетку типа алмаза, а = 5,6574 А. Атомный
радиус 1,39 А. Ионные радиусы: Ge2+0,65 A, Ge4+
0,44 А. Имеются указания о наличии полиморфного
превращения у Г. между 117 и 560°, однако этот
859
ГЕРМАНИЙ
860
вопрос еще остается спорным. Плотность Г. 5,323
(при 25°); т. пл. 936 ± 1°; т. кип. 2700°; теплота плав-
ления 114,3 кал/г; теплота испарения 1650 кал/г;
давление паров Г. становится заметным в атмосфере
азота при 1250°; уд. теплоемкость 0,074 кал/г град
(0—100й); теплопроводность 0,14 кал/см • сек град
(25°); термич. коэфф, линейного расширения 6,1 • 10~6
(0—330°), 6,6 • 10‘6 (300—650°). Г. обладает диамаг-
нитными свойствами. Г. хрупок и не поддается холод-
ной и горячей обработке давлением до 550°. Темп-рный
интервал между 550° и точкой плавления Г. является
зоной его пластич. деформации. Твердость Г. по мине-
ралогии. шкале 6,0—6,5. Г. поддается распиловке
на пластины с помощью алмазного или металлич.
диска с применением абразива. Поверхностное натя-
жение жидкого Г. вблизи точки затвердевания в атмос-
фере аргона 600 дин/см, т. е. достаточно высокое для
того, чтобы к Г. мог быть применен особый метод
металлургии, рафииировки и кристаллизации путем
бестигельной зонной плавки.
Структура энергетич. зон в кристалле Г., опреде-
ляемая строением его атома и взаимодействием между
соседними атомами в кристаллич. решетке, является
характерной для типичного полупроводника. Она
отличается тем, что между основной зоной, т. е. зоной
тех наинизших энергетич. уровней, к-рые заполняются
валентными электронами при темп-ре абс. нуля, и
зоной проводимости, т. е. зоной уровней возбуждения,
на к-рую электроны могут переходить только, если
им будет сообщена дополнительная энергия, сущест-
вует запрещенная зона, т. е. энергетич. разрыв.
Ширина запрещенной зоны Г. 0,72 эв; этот разрыв
у металлов отсутствует, а у изоляторов много больше
(> 2 эв). Уд. электрич. сопротивление Г. при комнат-
ной темп-ре 60 ом см. Эта величина уд. сопротив-
ления достигается при глубокой очистке Г. от приме-
сей и соответствует его собственному сопротивлению.
Содержание примесей в таком Г. <; 10 7%. Примеси
сообщают Г. т. наз. примесную проводимость элект-
ронного (примеси элементов 5 группы периодич. си-
стемы— As, Sb, Р) или дырочного (примеси 3 группы—
Ga, Al, In) типа. С увеличением темп-ры уд. сопро-
тивление чистого Г. понижается. Подвижность носи-
телей зарядов в Г. (при 25°) для электронов 3600 ±
i 180 см3/в сек и для дырок 1700 -+- 30 см3/в сек;
собственная концентрация носителей зарядов 2,5 •
• 1013 см~3 (при 20°). Коэфф, преломления Г. 3,92 при
длине волны 8—12 мк. Отражательная способность
Г. 0,35 при длине волны 1,5 мк; начиная с длины волны
1,7 мк Г. становится прозрачным.
В химич. соединениях Г. может быть 2- и 4-валентен;
последние более устойчивы. Г. вполне стабилен при
обычной темп-ре; на него не действуют воздух, вода
и кислород. В ряду напряжений Г. располагается
между Си и Ag. Устойчив против действия НС1 и
разбавл. H2SO4; медленно растворяется в конц.
H2SO4; царская водка и HNO3 медленно взаимодей-
ствуют с Г., особенно при нагревании, образуя на
его поверхности пленку двуокиси. В растворах едких
щелочей Г. растворим слабо, но при одновременном
присутствии Н2О2 — легко. При взаимодействии Г.
с сильными щелочами образуются соли метагермание-
вой (H2GeO3) и ортогерманиевой (H4GeO4) кислот —
германаты, напр. Na2GeO3, являющиеся силь-
ными восстановителями. Германаты щелочных метал-
лов растворяются в воде, соли других металлов слабо
• растворимы, но легко разлагаются кислотами. Водные
р-ры германатов имеют щелочную реакцию, что яв-
ляется следствием гидролиза. Германаты получаются
также при сплавлении Г. со щелочами.
При 700° Г. быстро окисляется на воздухе и в токе
кислорода, -образуя с последним два соединения—
скись GeO и двуокись GeO», Окись Г. — темно-
серый порошок, слабо растворяется в воде и легко —
в кислотах с образованием солей 2-валентного Г.;
выше 700° заметно возгоняется. Двуокись Г.
обладает тетрагональной кристаллич. решеткой типа
рутила с параметрами: а — 4,390 А, с = 2,859 А;
известна также мета ста би льна я (т. наз. растворимая
в воде) форма GeO2, кристаллизующаяся в гексаго-
нальной решетке типа а-кварца с параметрами:
а = 4,88 А, с = 5,64 А. Плотность тетрагон, модифи-
кации 6,239, гексагон. формы 4,703; растворимость
последней в воде 0,4% при 20° и ок. 0,95% при 100°;
т. пл. гексагон. модификации GeO2 1116 ± 4°, выше
1250° заметно испаряется. Т. пл. тетрагон, формы
GeO2 1086 -|- 5°. При 600—700° GeO2 восстанавли-
вается водородом и углеродом до элементарного Г.
Двуокись Г. имеет амфотерный характер с преоблада-
нием кислотных свойств; хорошо растворима в щело-
чах с образованием германатов; в кислотах раство-
ряется с трудом.
Г. легко соединяется с галогенами и дает при нагре-
вании соответствующие тетрагалогениды. В аналитич.
практике и промышленной технологии наиболее часто
используется четыреххлористый Г.
GeCl4—бесцветная жидкость, плотность 1,87 (25°),
т. затверд. —50°, т. кип. 83°; дымит на воздухе вслед-
ствие гидролиза. Растворимость GeCl4 в конц. НС1
0,0085 молъ/л; смесь расслаивается на два слоя.
При кипячении солянокислого раствора GeCl4
дистиллируется; в водных р-рах легко подвергается
гидролизу. Бромид и иодид Г. получаются
при дейбтвии конц. НВт и HJ на GeO2. GeBr4 — бес-
цветная жидкость, т. пл. 26°, т. кип. 186°. GeJ4 —
оранжево-красные кристаллы, т. пл. 146°, т. кип.
375°. Безводный фторид Г. GeF4—бесцветный
газ, конденсируется в жидкость при —37°. В технике
применяются фторогерманаты — соли гер-
манофтористоводородной к-ты H2GeFe; при добавлении
в плавиковокислый раствор Г. фтористого калия
образуется K2GeFe, т. пл. 730°, т. кип. 835°. Из про-
изводных, содержащих одновременно кислород и
галоген, интересен оксихлорид Г. GeOCI2 —
бесцветная маслянистая жидкость, т. пл. —56°,
быстро разлагающаяся водой с образованием Ge(0H)2.
Редко используемый в технике сульфат Г.
Ge(SO4)2 получается нагреванием смеси GeCl4 с SO3
в запаянной трубке. Водой Ge(SO4)2 полностью гидро-
лизуется с образованием GeO2. Известны два суль-
фида Г. — красно-коричневый кристаллич. GeS
и белый порошкообразный GeS2. Из них GeS полу-
чается при непосредственном взаимодействии элемен-
тов при 600—700°. Плотность GeS 4,01, т. пл. 530°.
GeS2 осаждается сероводородом из кислых растворов.
Плотность GeS2 2,94, т. пл. 800°. Дисульфид Г. рас-
творяется в сернистом аммонии с образованием тио-
германата (NH4)eGe2S7; при 400—600°GeS2 восстанав-
ливается водородом с образованием GeS, последний
возгоняется при темп-ре >> 800°.
Г. не образует карбидов и, следовательно, можетбыть
расплавлен в графитовых тиглях без опасений загряз-
нения его углеродом. С фосфором Г. образует только
одно соединение — фосфид Г. GeP; это соединение
неустойчиво. Для Г., как аналога С и Si, характерна
способность образовывать германоводоро-
д ы. Известно несколько водородных соединений Г.:
моногерман GeH4 и его гомологи Ge2He и Ge3H,.
Чистый моногерман получается при обработке Mg2Ge
раствором бромистого аммония в жидком аммиаке;
может быть получен также как примесь к водороду
при воздействии на Mg2Ge кислотами. При обычвой
темп-ре GeH4—бесцветный газ, т. пл. —166°, т. кип.
—88°, легко диссоциирует выше 200° на Г. и водород.
Гомологи GeH4 — бесцветные жидкости, Ge2H6 имеет
т. пл. —109°, т. кип. 31°; Ge3Hs имеет т. пл. —106°,
861
ГЕРМАНИЙ
862
т. кип. 111°. Они образуются при обработке кислотами
Mg2Ge одновременно с GeH4 и отделяются от него
фракционной перегонкой. Оба соединения, так же
как и GeH4, не устойчивы. Для Г. характерны соедине-
ния с азотом. Коричневый нитрид Г. Ge3N4,
плотность 5,25, получается действием аммиака на Г.
или GeO2 при 700°. Ge3N4 устойчив, распадается на
элементы лишь около 1000°; вода, щелочи и разбавлен-
ные кислоты на него не действуют.
В аналитич. практике и технологии Г. большое зна-
чение имеют комплексные соединения Г. Получены
германомолибденовая, германовольфрамовая, германо-
молибденовованадиевая и германовольфрамовована-
диевая гетерополикислоты следующего состава:
H8|Ge(Mo2O7)6] nH2O, H4[GeMoi0V2O39] • пН2О. Дву-
окись Г. способна образовывать комплексные кис-
лоты с многоатомными алкоголями — глицерином,
маннитом и глюкозой. Известен оксалатный комплекс
Г. H2[Ge(G2O4)3]. При взаимодействии Г. с фенил-
флюороном в соляно- и сернокислых р-рах образуется
комплексное соединение состава
к-рое используетсн при определении малых коли-
честв Г.
Будучи по своей атомной структуре непосредствен-
ным аналогом С и Si, Г. дает органич. соединения
тех же типов. Описано более 200 германийорганич.
соединений. Большинство из них являются произ-
водными германов и тетрагалогенных соединений.
Так, при обработке диэтилцинком GeCl4 получается
тетраэтилгерман (С2Н5)4 Ge. Описаны физич. и химич.
свойства германийорганич. соединений, содержащих
в качестве радикала фенильную или пиридиновую
группы, напр. трифенилгерман, тетрафенилгерман,
гексафенилдигерман и др.
Г. образует большое число сплавов с различными
металлами, а также растворяется в нек-рых из них;
так, Fe растворяет до 20 ат. % Г., Ni — до 12 ат. %
Г., в Si Г. растворяется неограниченно. Г. образует
сплавы с Sb, Pb, Sn, Bi, Mg, Al, Go, Gu, Ni, Au; мно-
гие из этих сплавов находят применение в технике.
Аналитическое определение германия.
Г. относится к группе сероводорода (сульфид Г. осаждается
из кислых р-ров, растворим в сернистых щелочах). Г. пере-
водят в раствор обработкой пробы HF с добавлением HNO3
и Н3РО4 или выщелачиванием после сплавления с содой, ще-
лочью или Na2O2. Качественно открывают с помощью дуго-
вого спектрографического метода. Рекомендуются также не-
которые цветные реакции, напр. реакция образования ярко-
желтой германомолибденовой к-ты с последующим восстанов-
лением Мо бензидином или щелочным р-ром станнита натрия.
Специфич. реакциями на Г. являются реакции с хинализари-
ном (в присутствии Г. розово-фиолетовая окраска реактива
переходит в синюю) или же с оксинафталинхинонсульфоновой
к-той (при взаимодействии этого реактива с раствором, содер-
жащим Г., обнаруживается ярко-розовая окраска при наб-
людении в синем свете). Для определения Г. применяются
все три обычных метода — весовой, объемный и колориметри-
ческий.
Весовым методом Г. определяют в виде GeO2 после осаж-
дения GeS2 из конц. сернокислого р-ра и последующего про-
каливания осадка или в виде MgGeO4 после осаждения смесью
сульфатов магния п аммония в аммиачном р-ре и прокаливания.
Кроме того, Г. может быть осажден оксихинолином и взвешен
в виде оксихинолиновой соли германомолибденовой кислоты
(C9H5ON)4 • H4[Ge(Mot2O42)]. Для объемного определения Г.
используется его способность образовывать комплексные сое-
динения с маннитом — маннитогерманиевую кислоту, к-рую
титруют р-ром щелочи в присутствии паранитрофенола и фе-
нолфталеина пли крезол-красного (As3+ не мешает, Sn и Se
мешают).
Йодометрически Г. определяют в виде Ge2S|“, к-рый полу-
чают действием насыщенного сероводородом р-ра K^S в среде
СН8СООК. Колориметрия. Г. определяют с помощью цветной
реакции образования германомолибденовой к-ты и ее вос-
становления до молибденовой сини; или по оранжевому ок-
рашиванию с 2,3,7-триокси-9-фенил-6-флюороном в соляно-
кислом р-ре в присутствии защитного коллоида; по фиолето-
во-синему окрашиванию с хинализарином в конц. H2SO4.
Г. может быть определен также полярографпчески после вос-
становления Ge4+ до Ge2+ гипофосфатом Na или Са.
Получение. Схемы извлечения Г. из различных
видов сырья отличаются гл. обр. в первой стадии
процесса — при вскрытии сырья и выделении химич.
концентрата Г.; последующие операции разложения
концентрата, выделение индивидуальных соедине-
ний Г. и их очистка однотипны для всех схем: при
этом используют удобное сочетание свойств GeGl4 —
высокую его летучесть (для дистилляции из кон-
центрата) и малую растворимость в конц. HG1 и,
наоборот, высокую растворимость в органич. раство-
рителях (для очистки от сопутствующих примесей).
Содержащие Г. пыли, получающиеся при обжиге сульфидно-
цинковых или медно-сульфидных руд, подвергают выщелачи-
ванию H2SO4, выделяют из раствора цементацией Си, Cd и
затем концентрат Г.; последний сушат, обжигают и обрабаты-
вают конц. НС1 с дистилляцией GeCl4. При переработке кок-
сующихся углей Г. частично попадает в различные отходы, в
том числе и в аммиачные воды, откуда может быть выделен в
осадок в виде органич. комплекса; осадок подвергают озоле-
нию при низкой темп-ре и получают концентрат Г., из к-рого
дистиллируют GeCl4. При сжигании энергетич. углей или их
газификации Г. возгоняется с летучими частями золы и са-
жей. Летучие золы подвергают восстановительной плавке с
флюсующими реагентами и специальным коллектором Г. —
медью (в виде СиО) или железом (Fe2O3). Из полученного сплава
Си или Fe с Г. извлекают Г., дистиллируя его в виде GeCl4.
Летучая пыль может быть подвергнута также сплавлению с
NaOH; раствор после выщелачивания очищают от А1 и Si
и затем выделяют из него Г. в виде GeO2. Может быть также
применен способ возгонки Г. из летучих пылей в форме низ-
шего сульфида или окисла; возгоны выщелачивают кислотами
и извлекают Г. из раствора осаждением, цементацией, ионным
обменом или др. методами. Технич. GeCl4 обычно содержит
примеси Fe, Al, Си, Si, As и др. в переменных количествах; осо-
бенно сложно освободить GeCl4 от As. Для получения чистого
GeCl4 применяют либо ректификацию в кварцевых колонках
с насадкой, либо экстракцию примесей из GeCl4 конц. НС1,
либо последовательно туи другую операции. Очищенный GeCl4
гидролизуют в трижды дистиллированной Н2О, получают
GeO2, промывают ее спиртом и сушат. Содержание Си, Ni, Fe,
Мп в полученной GeO2 не должно превышать 10-в—10~’%;
Si, Sb, As, Р— не более 10~6%. Г. получают восстановлением
GeO2 водородом при 600—700°; порошкообразный Г. в зоне
повышенных темп-p печи (1050°) сплавляется в королек, к-рый
подвергают рафинировке методом зонной плавки, причем
получают Г. высочайшей степени чистоты.
Чистый Г. специальной формы, структуры и свойств,
необходимых для дальнейшего использования в ка-
честве полупроводниковых кристаллов, получают
выращиванием монокристаллов в глубоком вакууме
или атмосфере нейтрального газа по методу Чохраль-
ского или иными специальными способами; в про-
цессе выращивания монокристаллов к Г. особой
чистоты могут быть добавлены специальные легирую-
щие добавки (Sb, Ga, As, Au, Si и др.) для регули-
рования тех или иных свойств монокристалла.
Применение. Основным потребителем Г., как полу-
проводника, являются радио- и электротехника f
где его используют для изготовления кристаллич.
выпрямителей (диодов) и усилителей (транзисторов),
к-рые нашли широкое применение в радиоприемниках,
счетно-решающих устройствах, телевидении, радио-
локации, электролизе и т. д. Монокристаллич. Г.
может быть использован для дозиметрии радиоактив-
ных излучений и в качестве преобразователя свето-
вой энергии в электрическую. Однако для солнечного
света германиевые фотоэлементы имеют значительно
более низкий кпд, чем кремниевые. Подобно нек-рым
другим полупроводникам, Г. применяют для изго-
товления термометров сопротивления; термометры
с германиевым мостом весьма устойчивы при темп-рах,
близких к абсолютному нулю. Одной из важных
областей применения Г. является инфракрасная
техника, поскольку он способен пропускать инфра-
красное излучение. Высокий показатель преломления
Г., по сравнению с NaGl, обусловливает меньшую
кривизну оптич. поверхностей. Инфракрасная оптика
из Г., благодаря коррозионной устойчивости послед-
него, невосприимчива к атмосферным и темп-рным
863
ГЕССА ЗАКОН — ГЕТЕРОАЗЕОТРОПНЫЕ СМЕСИ
864
воздействиям. Для многих сплавов Г. (напр., на
медной, магниевой и алюминиевой основе) характерна
повышенная стойкость в кислых агрессивных средах.
Эвтектич. сплавы Г. с золотом способны расширяться
при затвердевании, что позволяет использовать их
для прецизионных отливок. Отмечается возможность
использования Г. в качестве легирующей добавки
к различным сортам стали, включая нержавеющую.
Из соединений Г. наиболее важны: GeO2, GeCl4
и GeH4; они используются при получении сверх-
чистого Г. для полупроводниковой техники. Являясь
одним из лучших стеклообразователей, GeO2 имеет
обширную область возможного применения при
изготовлении специальных стекол, обладающих вы-
соким коэфф, преломления и прозрачностью в ин-
фракрасной части спектра, а также эмалей и керамич.
изделий. В нек-рых случаях GeO2 обнаруживает
каталитич. свойства и является активатором для
окисных и галогенных люминофоров. Полезна добавка
GeO2 в композиции для оксидных катодов. Многие
соединения Г. как органич., так и неорганич. имеют
специфич. свойства, что обусловливает их применение
в отдельных узких целях. Примером могут служить
антивспениватели, теплоносители, смазочные и гид-
рофобизирующие вещества на основе германийорга-
нич. соединений, а также фосфаты Г. и фторгерма на ты,
к-рые находят применение для активации фосфоров
и электролитич. получения элементарного Г.
Лит.: Конвелл Е. М., Свойства кремния и германия,
в кн.: Германий. Сб. переводов..., М., 1955; Эльхонес
И. М., Аверьянова В. П. и Маслов В. И., Гер-
маний и его соединения. Области освоенного и возможного
применения, М., 1959; Меерсон Г. А., Зеликман
А. И., Металлургия редких металлов, М., 1955; Conwell
Е. М., Proc of the I. R. E., 1958, 46, № 6, 1281; Электрофизи-
ческие свойства германия и кремния. Сб. переводов, М., 1956;
Буш Г. иВинклер У., Определение характеристических
параметров полупроводников по электрическим, оптическим
и магнитным измерениям, пер. с нем., М., 1959; И о ф ф е А. Ф.,
Физика полупроводников, [2 изд.], М, —Л., 1957; Вопросы ме-
таллургии и физики полупроводников, М 1957; Gmel in,
Syst.-Nr. 45, Germanium, В., 1931; M e 11 о г, V. 7, L.—
N.Y. — Toronto, 1947; то же v. 7, 1952; U 1 1 m a n n , 3 Aufl.,
Bd 8, Milnchen — B., 1957, S. 7—13; К i г k, v. 7, N. Y., 1951,
p. 168—74; Rare metals handbook, ed. C. A. Hampel, N. Y.,
1954. H. M. Эльхонес.
ГЕССА ЗАКОН — закон постоянства сумм тепла,
являющийся основным законом термохимии. Г. з.
устанавливает, что тепловой эффект реакции не
зависит от промежуточных стадий, а зависит лишь
от начального и конечного состояния системы. Закон
открыт Г. И. Гессом в 1840 на основе эксперимен-
тальных исследований. Он представляет собой одну
из форм позднее открытого закона сохранения энергии
в применении его к химич. процессам: энергия си-
стемы является функцией состояния системы; тепло-
вой эффект химич. реакции, происходящей при
постоянном объеме, служит непосредственной мерой
изменения внутренней энергии системы, а тепловой
эффект реакции, происходящей при постоянном
давлении, служит мерой изменения энтальпии си-
стемы.
Из Г. з. вытекает ряд следствий: 1) теплота раз-
ложения химич. соединения равна по величине и про-
тивоположна по знаку теплоте его образования;
2) для двух реакций, приводящих из различных
исходных состояний к одинаковым конечным состоя-
ниям, разность между их тепловыми эффектами равна
тепловому эффекту процесса перехода из одного
начального состояния в другое; 3) для двух реакций,
приводящих из двух одинаковых исходных состоя-
ний к различным конечным состояниям, разность
между их тепловыми эффектами равна тепловому
эффекту процесса перехода из одного конечного
состояния в другое.
Пример применения Г. з.: непосредственно
измерить теплоту образования СО очень трудно, зато легко
измерить теплоты сгорания С и СО в СО2. Пользуясь Г. з.>
получаем (считая на 1 моль СО и СОг):
_ С тв. + О2 газ = СО2 газ + 94 052 кал
СО газ 1/2О2 газ = СО2 газ + 67 638 кпл_
С тв. */2О2 газ = СО газ + 26414 кал
Для (1) и (3) из этих реакций тепловой эффект зависит от
кристаллич. модификации углерода или от вида угля. Ука-
занные значения тепловых эффектов относятся к чистому уг-
лероду в форме графита.
Г. з. позволяет, т. о., находить неизвестные теплоты реак-
ций путем суммирования известных теплот реакций. В этом
состоит его большое практич. значение для термохимии и
техники.
Лит.: Hess Н., Bull. sci. publie par i’Acad. des sciences
de St.-Petersbourg, 1840, 7, № 18, 1841, 8, № 6; e г о же,
Thermochemische Untersuchungen, Lpz., 1921; Гесс Г. И.,
Термохимические исследования, М., 1958; Напусти н-
с к и й А. Ф., Термодинамика химических реакций и ей
применение в неорганической технологии, 2 изд., М.—Л.,
1936; К ар а петьянц М. X., Химическая термодинамика,
М,— Л., 1953; Колосовский Н. А., Химическая термо-
динамика, Л., 1932. В. А. Киреев.
ГЕТЕРОАЗЕОТРОПНЫЕ СМЕСИ — растворы, не-
раздельно кипящие в определенных пределах кон-
центраций; образуются жидкими компонентами с огра-
ниченной взаимной растворимостью. Компоненты
Г. с. растворяются друг в друге при каждой темп-ре
лишь в определенных пределах концентраций, обра-
зуя вне этих пределов два или больше несмешиваю-
щихся жидких слоя. Взаимная растворимость ком-
понентов Г. с., как показал В. Ф. Алексеев, возра-
стает в одних случаях с повышением темп-ры (напр.,
фенол — вода, фурфурол — вода), а в других слу-
чаях— с ее понижением (напр., эфир—вода, три-
этиламин—вода). Соответственно темп-ра, начиная
с к-рой смесь с любыми концентрациями компонентов
становится однородной, наз. в первом случае вер х-
н е й, а во втором — нижней критической
температурой растворения. Так, напр.,
фенол и вода смешиваются в любых соотношениях
выше 65,84° (верхняя критич. темп-ра растворения),
а триэтиламин и вода — ниже 18° (нижняя критич.
темп-ра растворения).
Области существования гетерогенных и гомоген-
ных жидких смесей могут быть наглядно изображены
графически, если по оси абсцисс отложить (в случае
бинарной смеси) содержание
одного из компонентов, а по
оси ординат — темп-ру. Для
смеси фенол — вода (рис. 1)
кривая расслоения делит
поле диаграммы на 2 обла-
сти, из которых внутренняя
принадлежит двухслойным, а
внешняя — однородным сме-
сям. Точки на кривой рас-
слоения характеризуют со-
ставы равновесных слоев.
Так, например, при 40° один
слой содержит раствор воды
в феноле (33% воды), а дру-
гой — раствор фенола в воде
(10% фенола). С ростом тем-
пературы составы обоих слоев
сближаются и, наконец, в т.
наз. критич. точке они становятся идентичными,
т. е. смесь становится однородной. Очевидно, что
для смесей с нижней критич. темп-рой растворения
кривая расслоения имеет обратное очертание, т. е.
она обращена к оси абсцисс не вогнутой, а выпуклой
стороной.
Существуют, ваконец, и такие смеси, к-рые обла-
дают обеими критич. темп-рами растворения, т. е.
они расслаиваются при нагревании и вновь становятся
однородными при дальнейшем повышении темп-ры
(напр., никотин—вода). Кривые расслоения таких
смесей являются замкнутыми. Возможно, что такое
----1----1--1-----1____।
О 20 40 60 80 100%
концентрация фенола
Рис. 1.
865
ГЕТЕРОАЗЕОТРОПНЫЕ СМЕСИ
866
поведение обнаруживали бы все смеси, состоящие
из компонентов с ограниченной взаимной раствори-
мостью, если бы твердая фаза не появлялась раньше
достижения нижней критич. темп-ры, а критич. точка
пар—жидкость—раньше достижения верхней критич.
темп-ры растворения. Д. П. Коновалов показал, что
в расслаивающихся системах оба жидких слоя при
равновесии обладают одинаковым насыщенным паром.
По составу последнего различаются, по крайней мере,
4 вида гетерогенных смесей, для к-рых на рис. 2
жидность-
* нритичесная -лар
точна
жидность-
жадность
состав О
состав
О
Рис. 2.
состав
представлены, наряду с кривыми расслоения, так-
же кривые состава пара (у—у). Смеси типа 1 ха-
рактеризуются тем, что состав равновесного пара
(С) является промежуточным между составами обоих
жидких слоев. К таким смесям принадлежат фур-
фурол—вода, анилин—вода, изоамиловый спирт—вода,
бутанол—вода и др. При наличии двух жидких слоев
таких смесей, находящихся в равновесии со своим
паром, образуется трехфазная система, обладающая,
согласно правилу фаз, одной степенью свободы. Сле-
довательно, при заданном полном давлении паров,
существует лишь одна темп-ра кипения, общая для
обоих слоев, определенные составы последних и
состав паров, равновесный обоим жидким слоям.
Так, под давлением в 1 ат гетерогенные жидкие
смеси фурфурола и воды кипят при 97,9° и содержат
соответственно 18,4 и 84,1 вес. % фурфурола, а их
общий равновесный пар — 35 вес. % фурфурола.
Гетерогенные жидкие смеси воды и бутанола кипят
под давлением 1 ат при темп-ре 92,25° и находятся
в равновесии с паром, содержащим 25 мол. %
спирта.
Смеси типа II (напр., вода— фенол) характери-
зуются составом пара, выходящим за пределы соста-
вов равновесных жидких слоев. Так, под давлением
150 мм рт. ст. жидкие слои содержат соответственно
83 и 53 вес. % воды, а в равновесной паровой фазе
содержится 91,1 вес. % воды. Смеси типа III харак-
теризуются тем, что при определенной темп-ре состав
пара совпадает с составом одной из жидких фаз гете-
рогенной смеси с верхней критич. темп-рой раство-
рения. Так, при 200° смесь этиловый эфир — вода
находится в равновесии с паром, имеющим тот же
состав, что и фаза, богатая эфиром. Наконец, смеси
типа IV (например, алифатич. спирты— этан) ха-
рактеризуются тем, что при определенной темп-ре
состав пара совпадает с составом одной из жидких
фаз гетерогенной смеси с нижней критич. темп-рой
растворения.
Согласно приведенному выше определению, смеси
типа I обладают давлением пара, наибольшим по
сравнению с другими однородными смесями тех же
компонентов, и наиболее низкой темп-рой кипения.
Под постоянным давлением такая смесь будет пере-
гоняться при неизменной темп-ре до тех пор, пока
полностью не израсходуется один из слоев, сохраняя
все время постоянный состав пара. По этому признаку
рассматриваемые смеси и получи-
ли название гетероазеотропных.
Зависимости состава Г. с. от дав-
ления при постоянной темпера-
туре, найденные опытным путем,
представлены на рис. 3. Смеси
типа I характеризуются диаграм-
мой (рис. 3, I), где АВ — кривая
давления пара жидкости, бога-
той компонентом В, а АС—кри-
О состав вая давления пара, находящегося
в равновесии с жидкостью, со-
став к-рой изменяется вдоль АВ,
ED и ЕС — аналогичные кривые для фазы, бога-
той компонентом А. Все точки на прямой BD соот-
ветствуют гетерогенным смесям, у к-рых обе жидкие
фазы имеют составы, представленные точками В и
D, а паровая фаза имеет постоянный состав точки С.
На рис. 3, II приведена диаграмма зависимости
давления от состава для Г. с. типа II. Здесь АВ и
СЕ — кривые давления пара однородных смесей,
AD и DE — кривые сосуществующих паровых фаз,
ВС — область гетерогенных жидких смесей, обла-
дающих равновесным составом пара в точке D. На
кривых, соответствующих гомогенным смесям, могут
быть, в зависимости от природы компонентов смесей,
экстремальные точки (минимум или максимум). Для
анализа и расчета процессов перегонки, протекающих
обычно под постоянным давлением, очень важны
О состав жадности
Рис. 5.
диаграммы фазового равно-
весия, характеризующие зави-
симость температуры кипения
жидких смесей и равновес-
ных паров от их состава. Как
показали экспериментальные
исследования, эти диаграммы
для смесей типов I и II имеют
вид, изображенный на рис. 4.
На обеих диаграммах АВ —
область существования гетеро-
генных смесей. На рис. 5 при-
ведена диаграмма фазового
равновесия Г. с., где по оси абсцисс отложен состав
жидкой смеси, а по оси ординат — состав равновес-
ной паровой фазы. На диаграмме построены линии
равновесия для смесей типов I, II и III.
Липп. см. при ст. Азеотропные смеси.
Н. И. Гельперин.
867
ГЕТЕРОАУКСИН — ГЕТЕРОЛИТИЧЕСКИЕ РЕАКЦИИ
868
ГЕТЕРО АУКСИН (Р-индолилуксусная кислота)
C10H9NO2, мол. в. 175,18—представитель группы
ауксинов', белые кристаллы; т. пл. 168—169° (с разл.);
хорошо растворим в спирте, эфире, плохо — в воде,
бензоле и бензине. Г. разлагается на свету, неорганич.
к-тами и др.; соли Г. со щелочными металлами
более устойчивы и лучше растворимы в воде. Г.
получают различными способами, напр. из грамина,
.. а также из индола и диазо-
Ц || a— CHgCOOH уксусного эфира и др. Г.
L II II обладает высокой физиоло-
NH гич. активностью и приме-
няется для стимулирования
роста растений (ускорения укоренения черенков
при вегетативном размножении растений, 50—100 мг
на 1 л воды) в виде калиевой или др. солей. Вы-
сокой физиология, активностью обладают также
нек-рые гомологи Г., из к-рых в первую очередь сле-
дует отметить индолил-у-масляную к-ту (мелкие кри-
сталлы, т. пл. 124°, умеренно растворима в воде,
хорошо — в спирте и др. органич. растворителях);
получают взаимодействием бутиролактона с магний-
под(бром)индолом:
Физиология, активность индолилмасляной к-ты не-
сколько превышает активность Г.; обычно исполь-
зуется для ускорения укоренения черенков, в кон-
центрации в 2—2,5 раза меньшей, чем концентрация
Г. Из аналогов Г. следует отметить 1-нафтилуксусную
к-ту, которую получают из нафталина и хлор уксус-
ной к-ты:
СН2СООН
+ С1СН2СООН
+ HCI
Эта кислота обладает повышенной физиологиче-
ской активностью и применяется в малых концен-
трациях.
Лит.: Мельников Н. Н., Баскаков Ю. А.,
Бокарев К. С., Химия гербицидов и стимуляторов роста
растений, М., 1954; Турецкая Р. X., Приёмы ускорен-
ного размножения растений путем черенкования, М,—Л.,
1949; Гетероциклические соединения. [СВ. статей], пер. с англ.,
под ред. Ю. К. Юрьева, т. 3, М., 1954, с. 34. Н. Н. Мельников.
ГЕТЕРОГЕННОЕ РАВНОВЕСИЕ — см. Равновесие
химическое.
ГЕТЕРОГЕННЫЕ СИСТЕМЫ— физико-химические
системы, состоящие из двух или нескольких фаз.
Г. с. имеют поверхности раздела, по к-рым сопри-
касаются однородные части системы (фазы), отли-
чающиеся друг от друга по составу, свойствам или
по тому и другому. Указанные отдельные части Г. с.
должны иметь такие размеры и содержать столь
большое число частиц, чтобы можно было применить
понятия темп-ры, давления, концентрации. Т. обр.,
Г. с. состоят из двух или более однородных частей,
к-рые, по крайней мере, в принципе, могут быть
механически отделены друг от друга. Примерами
Г. с. могут служить вода и находящийся над ней
водяной пар, насыщенный водный р-р соли с кристал-
лами той же соли в нем и водяным паром над ним,
многие металлич. сплавы, горные породы (гранит)
и др. Обычный р-р является гомогенной системой,
т. к. частицы, из к-рых он состоит, не настолько
велики, чтобы можно было говорить, напр., о темп-ре
отдельной такой частицы. Переходную область между
механич. смесями и истинными р-рами (гомогенными
системамп) занимают коллоидные системы, в к-рых
размеры частиц, находящихся в р-ре, являются
средними между размерами молекул и частицами
взвесей, грубодисперсных систем (см. Дисперсные
системы). Резкой границы между гомогенными и
гетерогенными системами провести нельзя. Исследо-
вание Г. с. проводится на основе химич. термодина-
мики, правила фаз и физико-химич. анализа. Зна-
ние природы Г. с. имеет большое значение в хи-
мии, химич. технологии, в геологии, металлургии
и т. д.
Лит.: Аносов В. Я., Погодин С. А., Основные
начала физико-химического анализа, М,—Л., 1947.
ГЕТЕРОЛИТИЧЕСКИЕ РЕАКЦИИ — реакции,
протекающие с таким разрывом ковалентной связи,
при к-ром, в отличие от гомолитических реакций,
электронная пара, осуществляющая эту связь, цели-
ком или частично остается у одного из атомов (или
группы атомов). В первом случае это приводит к обра-
зованию заряженных частиц, ионов А ;: В—-А+ + : В~
или А В —> А- : + В+, тогда как во втором случае,
когда электронная пара частично остается у одного
из атомов (или группы атомов), образуются поляри-
зованные комплексы. К Г. р. относятся также
'MgJ обратные реакции, приводящие к образованию
ковалентной (координационной) связи. Гетеро-
литич. расщепление связей энергетически мепее
выгодно, чем гомолитич., т. к. при гетеролитич.
расщеплении требуется дополнительная энергия
на раздвигание образующихся противоположно заря-
женных частиц. Эта энергия, однако, снижается при
проведении реакции в р-рах за счет взаимодействия
молекул растворителя с образующимися ассоциатами
или ионами, т. е. сольватации последних. Поэтому, в
отличие от газовой фазы, в растворах Г. р. протекают
сравнительно легко. В качестве примера можно
указать на реакции этерификации илп гидролиза
в присутствии кислотных и основных катализаторов,
присоединение галогенов к олефинам в полярных
растворителях и т. д.
Реакции замещения в органич. химии с гетеролитич.
разрывом связи принято делить на нуклеофильные
(А^) и электрофильные (>УЕ) в зависимости от харак-
тера разрыва связи С—X (X — атом водорода или
замещаемая группа). Принципиальные схемы реакций
нуклеофильного (1) и электрофильного (II) заме-
щения:
А В + ;С — А:С+ : В (I)
А : В + С — А:С + В (II)
Нуклеофильными реагентами, приносящими пару
электронов для образования связи, являются, напр.,
ион гидроксила, ион галогена, ион циана, аммиак
и др. Электрофильными реагентами, стремящимися
заполнить электронную оболочку одним или двумя
посторонними электронами, являются, напр., гало-
гены, ион водорода и др. Подробнее см. Нуклеофиль-
ные и электрофильные реагенты.
Примером реакции (1) может служить гидролиз
гало гена лкилов, а реакции (II) — нитрование аро-
матич. соединений.
В особо благоприятных случаях (использование
в качестве реагентов сильных оснований или к-т,
высокая диэлектрич. проницаемость среды и т. Д.)
гетеролитич. замещение в связях С—Н может сопро-
вождаться полным переходом электронной пары,
т. е. образованием ионов. При этом в случае нуклео-
фильных реакций образуются карбанионы (в резуль-
869
ГЕТЕРОПОЛИСОЕДИНЕНИЯ — ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
870
тате отрыва протона от молекул органич. соединения),
а в случае электрофильных реакций — карбонил ионы
(в результате присоединения протона к молекулам
органич. вещества). Какой из двух типов Г. р. осу-
ществляется, зависит от строения органич. соеди-
нения и, в частности, от распределения электронной
плотности в его молекулах, от природы действующего
реагента, а также от условий опыта. Допускается
возможность промежуточного образования донорно-
акцепторного (в общем случае тримолекулярного)
переходного комплекса. Г. р. имеют большое значение
в органич. химии; к ним относятся промышленно
важные процессы: нитрование ароматич. соединений,
сульфирование, галогенирование, алкилирование
И г. д.
Лит.: Реутов О. А Теоретические проблемы орга-
нической химии, М., 1956; Си дж ви к Н., Природа связей
в химических соединениях, пер. с англ., Л., 1936; Р е м и к А.,
Электронные представления в органической химии, пер. с англ.,
М., 1950; Хюккель В., Теоретические основы органиче-
ской химии, пер. с нем., т. 1, М., 1955; Ингольд К. К.,
Механизм реакций и строение органических соединений, пер.
с англ., М'., 1959. Ф. С. Якушин.
ГЕТЕРОПОЛИСОЕДИНЕНИЯ — комплексные сое-
динения, анион к-рых образован двумя различными
кислотообразующими окислами, причем на молекулу
одного из них приходится несколько молекул друго-
го окисла. Напр., Р2О5 • 24Мо03-пН20 — фосфорно-
молибденовая к-та; 4К2О • SiO2 • 12WO3 • 14Н2О —
кремневольфрамат калия. Строение Г. окончательно
не установлено; часто оно выражается координацион-
ными формулами типа Hs[Si(W2O,)6] — кремневоль-
фрамовая к-та; Н7[Р(Мо207)6] — фосфорномолибде-
новая к-та. Роль центрального гетероатома могут
играть JVI1, MnVI1, SVI, TeVI, Pv, Asv, Vv, SiIV, GeIV,
В1П, Cr111, Se111 и т. д., а аддендами внутренней сферы
могут являться WOf", W2O|_, МоСЦ , Мо207_,
VO3, V2O|~, СгО; , SeO|~ и Те(Ц~. Согласно последним
исследованиям (В. И. Спипын), в образовании Г.
вольфрама принимают участие H2WO4, WO;~, Н3О+,
RO4 (где R — гетероатом) и гидроксониевые формы
вольфрамовой к-ты H(H30)W04, (H30)2W04, причем
связывание этих частиц происходит при помощи
водородных связей. Гетерополисоли более устойчивы,
чем соответствующие к-ты. Г., несмотря на большой
мол. вес, хорошо кристаллизуются, образуя гидраты
с большим числом молекул воды. Исключая соли
NH4, Cs, Ag, Hg, тяжелых металлов и органич.
оснований, большинство Г. хорошо растворимы в воде.
Многие гетерополикислоты дают нерастворимые
осадки с азотсодержащими органическими основа-
ниями. Получают Г. в кислой среде: Na2HPO4 +
4- 12(NH4)2MoO4 + 23HNO3 = (NH4)3H4[P(Mo207)6] +
4- 21NH4NO3 + 6H2O 4- 2NaNO3. Щелочами ови раз-
лагаются. Иногда для получения солей применяют
нейтрализацию свободной гетерополикислоты гидро-
окисями или карбонатами соответств. металлов.
Свободные гетерополикислоты обычно получают из
соответствующих солей действием эфира и соляной
к-ты с последующим разложением эфирного комплекса
водой. Г. используют при выделении антибиотиков,
очистке керосина, в качестве катализаторов и т. д.;
в аналитической химии—для открытия As, Sn, Sb и
определения Rb и С?. См. также Комплексные соеди-
нения.
Лит.: Спицын В. И., Ж. неорг. хим., 1956, 1, вып. 3, с.
552—64; его же, там же, 1957, 2, вып. 3, с. 502—505; Н и-
кити в а Е. А., Усп. хим., 1939, 8, вып. 6, 889—930; Б а-
6 а д-3 ахряиин А. А., там же, 1956, 25, вып. 11, с.
1371 —1401; Handbuch der anorganischen Chemie, hrsg. von
R. Abegg, Bd 4, Abt. 1, H. 2, Lpz., 1921, S. 1072; Rosen-
heim A., JSnicke J., Z. anorgan. Chem., 1917, 100
304—54; J an der G., Z. phys. Chem., 1940, Abt. A, 187, H.
3, J 49—69. Г. H. Пирогова.
ГЕТЕРОЦЕПНЫЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ
СОЕДИНЕНИЯ — см. Высокомолекулярные соединения.
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — орга-
нические соединения циклич. строения, содержащие
в кольцах не только атомы углерода, но и атомы
других элементов, напр. О, N, S, Р. Типичными при-
мерами Г. с. могут служить фуран, пиридин, тиофен.
Можно считать установленным, что в образовании
циклов могут принимать участие все многовалентные
атомы; однако наибольшее значение имеют Г. с.,
в цикл к-рых входят атомы N, О и S. Г. с., в осо-
бенности содержащие в циклах атомы N и О, чрез-
вычайно распространены в природе в виде алкалоидов,
витаминов, пигментов и играют очень важную роль
в биологич. процессах. К Г. с. принадлежит примерно
половина известных природных веществ; к ним отно-
сятся такие вещества, как хлорофилл растений,
гемин крови, пигменты желчи, нуклеиновые кислоты,
почти все алкалоиды, пенициллин, витамины РР,
Вг, В6, В12, индиго и др., а также примерно половина
лекарственных веществ, применяемых в медицине.
Г. с. очень разнообразны; они отличаются между
собой числом атомов в цикле, природой и числом
гетероатомов, наличием или отсутствием двойных
связей между атомами в кольцах. Далее, Г. с. могут
содержать различные заместители; наконец, известно
очень много соединений, у к-рых гетероциклич. кольца
конденсированы с ароматич. ядрами. Эти обстоя-
тельства в сочетании с возрастающим значением
Г. с., разработанностью и перспективностью методов
их синтеза привели к тому, что общее число известных
Г. с., видимо, не уступает числу соединений жирного
ряда.
В большинстве случаев для гетероциклич. систем приняты
тривиальные названия (наир., пиррол, акридин, хинолин и
т. д.). Для систематич. обозначения Г. с. часто пользуются
т. наз. «а-н о мев к л ату ро й».
По этой системе название Г. с. состоит из двух частей:
1) названия соответствующего карбоциклич. соединения с
тем же числом атомов в цикле и 2) приставки, показывающей,
какие гетероатомы содержатся в цикле; при этом для гетеро-
атомов в цикл вводится их латинское название с окончанием1
на «а» («окса» для кислорода, «аза» для азота, «тиа» для серы и
т. д.). Так, пиридин называют азабензолом, тиофен—тиацикло-
пентадиеном -2,4, триметиленимин — азациклобутаном и т. д.
Как и в алициклич. ряду, из Г. с. наибольший
интерес представляют соединения с ненапряженными
5-членными и 6-членными циклами, отличающиеся
сравнительно большой прочностью. К представителям
пятичленных Г. с. с одним гетероатомом относятся
след, важнейшие соединения:
фуран тиофен пиррол
Примерами пятичленных
мами могут служить:
Г. с. с двумя гетероато-
пиразол
имидазол
оксазол тиозол
Большую роль играют также пятичленные Г. с.,
конденсированные с бензольными ядрами, напр.:
бензофуран бензпиррол бензотиофен
(ну марон) (индол) (тионафтен)
бензтиазол
Из производных фурана наибольший интерес вы-
зывает фурфурол (а-фурилальдегид), легко получае-
мый гидролизом растительных отходов, содержащих
пентозаны (шелуха подсолнечника, соломы), при-
871
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
872
меняемый в произ-ве феноло-альдегидных смол и др.
Важнейшим из Г. с. с пятичленным кольцом является
пиррол. Так, к производным пиррола относятся
такие алкалоиды, как никотин, атропин, кокаин.
Огромную роль играют природные вещества, содер-
жащие пиррольные ядра, комплексно связанные
с атомами металлов (с Fe в гемоглобине, с Mg в хло-
рофилле, с Со в витамине В12). а-Пирролидинкарбо-
новая к-та, или пролин (с гидрированным ядром),
является постоянной составной частью сложных
белков. Некоторые синтетич. производные пиразола
(антипирин, пирамидон, анальгин) широко при-
меняются в медицине. Тиазоловый цикл содержится
во многих природных продуктах (пенициллине, вита-
мине В,), производным тиазола является сульфанил-
амидный препарат норсульфазол. К производным
бензтиазола относятся тиоцианиновые красители и
каптакс (2-мерКаптобензтиазол), применяемый в ка-
честве ускорителя вулканизации каучука. Индольная
система содержится в индиго.
Шестичленные Г. с. имеют еще большее значение,
чем 5-членные. Важнейшими представителями шести-
членных Г. с. с одним и двумя гетероатомами, а также
конденсированных с бензольными ядрами являются
след, системы:
акридин феназин феноксазин
Пиридиновое или пиперидиновое ядро содержится
в очень многих алкалоидах (никотин, анабазин, ат-
ропин и др.). p-Пиридинкарбоновая кислота (нико-
тиновая кислота) и ее амид (витамин РР) являются
антипеллагрич. витамином. К производным хинолина
принадлежат важные лекарственные вещества (ато-
фан, совкаин, плазмохин). Синтетич. антималярий-
ный препарат — акрихин, или атебрин, представляет
собой производное акридина. В последнее время
большое значение в медицине приобрели производные
фентиазина (аминазин и др.)., оказывающие на орга-
низм разностороннее действие и применяемые в
психиатрия, практике и при хирургия, операциях ,в
каяестве веществ седативного и гипотермия, действия.
Представления о строении Г. с. были выдвинуты в 1866—71
(А. Байер, В. Кернер), вскоре после установления циклич.
строения бензола. Многие Г. с. нашли применение и были
выделены в чистом виде задолго до установления их строения.
Еще в 1776 К. Шееле выделил мочевую кислоту; в 1832 дей-
ствием H2SO4 на крахмал был получен фурфурол. С начала
19 в. продолжается выделение и установление строения алка-
лоидов, преим. принадлежащих к числу Г. с. с атомами азота
в цикле; многие важнейшие алкалоиды, напр. хинин, кодЗеин,
стрихнин, наркотин и др., были выделены еще в начале 19 в.
Г. с. можно схематияно разбить на два основных
типа. К первому из них принадлежат системы, содер-
жащие наряду с гетероатомами метиленовые группы
и представляющие собой гетероциклич. аналоги
алициклич. соединений, напр. тетрагидрофуран, диок-
сан, пиперидин. Такого рода соединения с атомами
О, N, S и т. д. в цикле в значительной степени близки
к аналогичным соединениям жирного ряда (простым
эфирам, аминам, сульфидам и др.). Ко второму типу
относятся Г. с., содержащие двойные связи и обла-
дающие более или менее резко выраженным «аро-
матическим» характером, объясняемым сопряжением
С=С связей с неподеленными электронными парами
гетероатома.
Ароматич. характер бензола в основном определяется на-
личием секстета л-электронов и выравненностью всех связей.
В случае 5-членных ненасыщенных Г. с. (фуран, тиофен, пиррол)
возможно образование секстета л-электронов за счет 4 л-элек-
тронов от 4 атомов углерода и 2 электронов неподеленной
пары гетероатома. Поэтому тиофен, обладающий ароматич.
характером, в отличие от тетрагидротиофена, легко нитруется,
сульфируется, несмотря па наличие сульфидной серы, не окис-
ляется перманганатом, не образует ни сульфоксида, ни суль-
фона; этой же причиной объясняется резко пониженная основ-
ность пиррола (X < 10-*') по сравнению с пирролидином (тет-
раметиленимином) (К — 1,3 • 10 3). В случае шестичленного
Г. с. — пиридина — «ароматич.» секстет электронов обра-
зуется за счет пяти л-электронов от 5 атомов С и одного элек-
трона азота; при этом у азота остается одна неподеленная пара
электронов, и этим объясняется, почему пиридин с резко выра-
женным ароматич. характером обладает хотя и ослабленными
по сравнению с пиперидином (К = 1,6 • 10—•>, новее же за-
метными основными свойствами (К — 1,7-10 ").
Источником Г. с. в ряде случаев могут служить
продукты превращения природных веществ. Так,
в каменноугольных смолах содержится пиридин и
его гомологи, хинолин, хинальдин, изохинолин,
акридин, индол и его гомологи, карбазол, дифенилен-
оксид и др.; в древесной смоле содержится сильван
(метилфуран), фурфурол и др. Однако синтез является
основным методом получения Г. с. Способы полу-
чения Г. с. отличаются большим разнообразием.
Часто можно исходить из соединений с открытой
цепью, содержащих заместители, допускающие замы-
кание цикла. Примерами могут служить дегидрата-
ция диэтаноламина в морфолин: HN(CH2CH2OH)2 —►
—HN(CH2CH2)2O, дегидробромирование д-бромбу-
ти ламина с образованием пиррола.
Во многих случаях для синтеза Г. с.
прибегают к особым специфичным син-
тетическим методам. Так, тиофен обра-
зуется при пропускании ацетилена или
ацетальдегида и H2S над нагретым
фентиозин глиноземом; пиридин можно синтези-
ровать пропусканием ацетилена и HCN
через раскаленные трубки; для синтеза хинолина и
его производных можно воспользоваться реакцией
Скраупа; каптакс (2-меркаптобензтиазол) произво-
дится взаимодействием смеси анилина, CS2 и серы.
В нек-рых случаях Г. с. представляют собой продук-
ты присоединения, напр. синтез 2,5-дигидротиофен-
диоксида-1 из бутадиена и SO2.
Значительный интерес вызывают процессы взаим-
ных превращений пятичленных Г. с. Так, при про-
пускании паров фурана и его производных с избытком
NH3 над А12О3 при 450° фурановый цикл превращается
в пиррольный. С другой стороны, от пиррола дей-
ствием H2S можно перейти к
тиофену и от него действием па-
ров воды к фурану (см. схему).
Г. с. ароматич. характера с
сопряженными связями обыч-
но очень легко гидрируются.
Так, пиррол может легко присоединить 2 или 4 атома
водорода с образованием соответственно Д3-пирролина
или пирролидина:
СН3СНаСН2СН3
пиррол пирролин пирролидин бутан
873
ГЕТИНАКС—ГИАЛУРОНИДАЗЫ
874
Гидрирование пиррола в жестких условиях, как
и вообще Г. с., может сопровождаться разрывом
цикла с образованием в результате гидрогенолиза
в данном случае бутана. В гидрированных Г. с.
невозможно сопряжение кратных связей, и потому
они, естественно, лишены ароматич. свойств. В соот-
ветствии с этим пирролин и пирролидин представляют
собой сильные основания, легко дающие характерные
соли. С другой стороны, дегидрирование Г. с. али-
циклич. характера (т. е. процесс, эквивалентный
ароматизации алициклич. соединений) обычно осу-
ществляется с трудом. Все же от пиперидина можно
перейти к пиридину нагреванием с солями Ag или
Си, а также каталитич. дегидрированием над палла-
дированным асбестом.
По чисто формальным признакам к Г. с. дол-
жны быть причислены и такие соединения, как
окись этилена, лактоны, лактамы, ангидриды, имиды
и циклич. эфиры двухосновных кислот, циклич.
эфиры гликолей, циклич. ацетали
(углеводы) и др. В такого рода сое-
динениях цикл очень легко расщеп-
ляется, генетически и химически R
они близки к классам соединений
жирного ряда (гликолям, оксикис-
лотам, аминокислотам и т. д.). По-
этому обычно типичными Г. с. считают такие, в ко-
ОН |
+ R-C-CHgCN—- R
X
торых цикл сравнительно стоек и сохраняется при
многих химич. превращениях.
Лит.: Гетероциклические соединения, под ред. В. Эль-
дерфилда, пер. с англ., т. 1—6, М., 1953—61; The chemistry
of heterocyclic compounds, ed. A. Welsberger, v. 1 — [13],
N. Y. — L., 1950—59; Габель Ю. О., Гетероциклические
соединения, Л. —M., 1941; Morton A. A., The chemistry
of heterocyclic compounds, N. Y.— L., 1946; Wiley R. H.,
в кн..- Organic chemistry, ed. H. Gilman [a. o.], v. 4, N. Y.—L.,
1953, p. 723. Я. Ф. Комиссаров.
ГЕТИНАКС (бумофенолит) — слоистый прессован-
ный материал, состоящий из целлюлозной бумаги,
пропитанной резольной смолой (см. Смолы фтоло-алъ-
дегидные). Ниже приведены показатели '
нек-рых свойств Г.: плотн. 1,3—1,4г/сл<3; RC~Ci +
предел прочности: на растяжение 800— II
1400 кг!см\ на изгиб 1000—1600 кг/см2, NH
на раскалывание 150—200 кг/см2; модуль
упругости при растяжении 70 103 — 100 • 103 кг/см2,
уд. ударная вязкость 15—25 кг см/см2', твердость
по Бринеллю 25—30 кг/см2", влагоемкость (увеличе-
ние веса после 24-часового пребывания в воде) 1—2%;
теплостойкость по Мартенсу 150—160°; уд. поверх-
ностное электрич. сопротивление 1010 — 1012 ом; уд.
объемное электрич. сопротивление 1010—1012 ом-см;
тангенс угла диэлектрич. потерь 0,1—0,05 (50 гц);
диэлектрич. проницаемость 5—8 (50 гц); пробивное
напряжение 15—25 кв]мм. Для получения Г. бумагу
пропитывают либо раствором резольной смолы (т. н.
лаковый метод), либо расплавленной смолой под
давлением. По второму способу резольную смолу
смешивают с бумажной массой в процессе изготовле-
ния самой бумаги (т. н. рольный метод). Г. — один
из первых слоистых пластич. материалов, нашед-
ших применение в электротехнике в качестве электро-
изоляционного и конструкционного материала; при-
меняется также в машиностроении (для изготовления
шестерен, роликов и трубок и прокладочных мате-
риалов), в мебельном произ-ве и др.
Лит.: Петров Г. С., Л е в и н А. Н., Термореактив-
ные смолы и пластические массы, М., 1959.
О. В. Смирнова.
ГЁША СИНТЕЗ — конденсация нитрилов с фено-
лами или эфирами фенолов в присутствии безводного
НС1, ведущая к образованию оксиарил- или алкок-
сиарилкетонов: RCN + СвН4(ОН)2 —RCOCeH3(OH)2.
Г. с., представляющий собой модификацию синтеза
альдегидов до Гаттерману, особенно успешно при-
меняется для синтеза полиоксиароматич. кетонов,
хотя нек-рые особенно активные нитрилы, напр.
трихлорацетонитрил, реагируют и с одноатомными
эфирами фенолов и с фенолами:
ОН
ОН
+ ClCH2CN
О=С — СН2С1
В Г. с. вступают разнообразные алифатич. и ароматич.
нитрилы, алифатич. динитрилы, кетонитрилы и др.
Кето-, альдегидо-, окси-, ацилокси- и Галогенонитри-
лы, так же как и непредельные эфиры, вступают в
т. наз. аномальную реакцию Гёша, заключающуюся
в образовании насыщенного эфира, гидролизующего-
ся и циклизующегося далее в дигидрокумарин:
Н
Он CSN
I ------- R
СНхСНг
О
С=О
I
,'СН„
сн
R' Г
Для осуществления синтеза Гёша обычно эквимоле-
кулярные количества нитрила и фенола растворяют
в абс. эфире и при 0° пропускают сухой НС1 до насы-
щения.
В качестве катализаторов обычно используют
безводные ZnCl2, SnCl4 или А1С13. Первоначальными
продуктами реакции являются хлоргидраты кети-
мина, к-рые далее гидролизуются в кетоны:
RC = N + НО------RC-C1
II
NH
Такой механизм Г. с. подтверждается как конден-
сацией имидо хлоридов с полиоксибензолами, так
и выделением в ряде случаев промежуточных
хлоргидратов кетиминов. Г. с. разработан в 1915
К. Гёшем.
Лит.: Сперри П. Е., Дюбуа А. С., в кн.; Органи-
ческие реакции. Сб. 5, пер. с англ., М., 1951, с. 284.
Н. П. Гамбарян.
ГИАЛУРОНИДАЗЫ — ферменты, гидролитически
расщепляющие гиалуроновые кислоты. Г. обнаружены
в семенниках (тестикулах) и селезенке млекопитаю-
щих, у пиявок, патогенных бактерий и в секретах
насекомых. Под действием Г. гиалуроновая к-та теряет
способность к образованию сгустка в кислой среде,
а при дальнейшем действии Г. вязкость субстрата
снижается. Конечные продукты реакции для Г.,
полученные из разных источников, различны. Напр.,
Г. семенников расщепляет гиалуроновую к-ту до
дисахарида N-ацетилгиалобиуроновой к-ты, а бак-
териального происхождения — до ненасыщенного ди-
са харида.
Активность Г. определяют след, методами: биоло-
гическим — по увеличению проницаемости тканей
под влиянием Г., по отношению к различным аген-
там, напр. нек-рым красителям; физико-химическим —
по понижению вязкости и мутности субстрата; хими-
ческим — по увеличению под влиянием Г. количества
восстанавливающих веществ в инкубационной среде,
содержащей гиалуроновую к-ту. Очищенные пре-
параты Г., имеющие высокую активность, при иссле-
875
ГИАЛУРОНОВЫЕ КИСЛОТЫ —ГИББСА РАСПРЕДЕЛЕНИЕ
876
(р о н fl-
it л и д а з а) для понижения вяз-
и отеков, а также для
довании с помощью ультрацентрифуги и при электро-
форезе оказались неоднородными. Г. активируется
NaCl, СаС12, MgCl2, желатиной; подавляется гепа-
рином, солями тяжелых металлов. Расщепление
гиалуроновой к-ты под действием Г. играет важную
роль в процессе оплодотворения, а также при про-
никновении микробов в организм животного. Г.
получают гл. обр. из семенников и селезенки млеко-
питающих. Применяют Г. из семенников
быков в лечебных препаратах
д а з а
кости эксудатов
определения гиалуроновой к-ты.
Лит.: Ferments, Hormone, Vitamins und die
Beziehnngen dieser Wirkslotfe zueinandsr, hrsg.
,R. Ammon und W. Dirscherl, Bd 1, 3 Autl.,
Stuttgart, 1959, S. 205, 240; Methods of biochemi-
cal analysis, v. 1, N. Y. — Toronto, 1954,p. 425;
Methods in enzymology, v. 1, N. Y., 1955.
ГИАЛУРОНОВЫЕ КИСЛОТЫ — кислые
аминополисахариды, построенные из остат-
ков А?-ацетил-П-глюкозамипа и D-глюку-
роновон кислоты; широко распространены
в тканях и жидкостях животных (стекло-
видное тело глаза, соединительные тка-
ни, кожа, пуповина, суставная жидкость) и
в микроорганизмах (капсулы гемолитич.
стрептококка, стафилококка и т. д.). Структурной
единицей Г. к. считают гиалобиуроновую
кислоту — дисахарид р-П-глюкуронозидо-3-D-
глюкозамин. Г. к. являются линейными высокопо-
лимерами гиалобиуроповой к-ты, аминогруппа к-рой
ацетилирована:
Участок цепи гиалуроновой к-ты (пунктиром ограничен остаток N - ацилированной гиалобиуроновой к-ты
Мол. веса Г. к. различного происхождения колеб-
лются от 1 300 000 до 4 300 000. Г. к. неограниченно
растворяются в воде, образуя вязкие р-ры, и не рас-
творяются в спирте, ацетоне, эфире и др. органич.
растворителях; [a]D от —47 до —78° (в воде),Водные
р-ры Г. к. обнаруживают явление двойного лучепре-
ломления (в потоке). С белками Г. к. образуют комп-
лексы, нерастворимые в кислой среде; в тканях и
жидкостях организмов Г. к. содержатся, по-видимому,
в виде белковых комплексов. При действии к-т гидро-
лиз Г. к. идет только до гиалобиуроновой к-ты.
Действие системы ферментов (гиалуронидазы, р-глю-
куронидазы -f- глюкозаминидазы) может привести к
полному гидролизу Г. к. до моносахаридов. Для опре-
деления Г. к. в тканях используется ее способность
к метахроматическому окрашиванию различными
красителями (толуидиновый синий и др.) и к рас-
щеплению под действием гиалуронидаз. Г. к. входят
в состав соединительной ткани, влияют па проницае-
мость тканей по отношению к различным агентам,
в том числе патогенным, принимают участие в водном
Л солевом обмене и т. д.
Лит..- Смирнова Л. Г., Клинич. мед., 1957, 35,
№ 6. с. 22; Advances in carbohydrate chemistry, v. 12, N. Y.,
1957 p. 299; Ciba foundation. Symposium on the chemistry
and biology of mucopolysacharides, L., 1958, p. 22. См. также
ст. Гиалуронидазы- Л. И. Линевич.
ГИББЕРЕЛЛИНЫ — стимуляторы роста расте-
ний, выделенные из продуктов жизнедеятельности
пек-рых грибов (Gibberella iujikuroi, Fusarium monit)
и высших растений. Выделены и охарактеризованы
след. Г.: А,—CiBH24O6, мол. в. 348,4, т. пл' 232—235°;
А2—С1ВН2вО6, мол. в. 350,41, т. пл. 235—237°; А3,
или гибберелловая к-та, — CiBH22O6, мол. в. 346,39,
т. пл. 233—235°; А4—Ci9H24O5, мол. в. 332,4, т. пл.
222°. Все эти соединения имеют близкое строение;
однако достаточно достоверно установлено строение
Ai (I) или (II) и А3 (III) или (IV):
Наиболее физиологически активен А3, стимулирую-
щий рост и развитие растений в концентрации
1—300 мг/л воды. Чаще всего А3 применяют в виде
водных р-ров, к-рыми опрыскивают растения или
наносят каплю р-ра в точки роста растения. Наиболее
характерная реакция растений
на обработку Г. —
удлинение стебля,
усиление вегета-
тивного роста, пре-
одоление карлико-
вых форм. Кроме
того, Г. способны
стимулировать цве-
тение, увеличивать
число завязей, на-
рушать период по-
коя семян, клуб-
ней и луковиц. Применение Г. может быть использо-
вано для повышения урожая или ускорения созрева-
ния (в частности, созревания трав, огородных культур
и др.).
Г. в основном получают микробиологическим спо-
собом из продуктов жизнедеятельности грибов рода
Fusarium.
Лит.: Стоу Б., Я м а к и Т., в кн.: Химические сред-
ства защиты растений. Сб. пер. и обзоров..., пер. с англ.,
[№ 3], М., 1958, с. 3; Баскаков ГО. А., там же, [№ 2],
М., 1959, с. 26. Н. Н. Мельников.
ГИББСА РАСПРЕДЕЛЕНИЕ — один из основ-
ных законов статистич. физики, определяющий ве-
роятность пребывания физич. системы в состоянии
с данной энергией. Иначе этот закон наз. кано-
ническим распределением, т. к. макро-
скопия. система рассматривается как единая механич.
система, состояние к-рой определяется заданием
всех обобщенных координат и импульсов, а измене-
ния в системе происходят в соответствии с канонич.
уравнениями механики. В классич. статистич. физике
канонич. распределение имеет вид:
dw = Ае~ Е (p’9>/llTdpdq
где dw — вероятность того, что импульсы (р) и коор-
динаты (q) частиц, из к-рых состоит система, имеют
значения между р и р + dp, а также q и q -f- dq
соответственно; Т — абс. темп-ра; к — константа
Больцмана; Е (р, q) — энергия тела как функция
всех р и q; А — постоянная, не зависящая от Е
и определяемая из условия Jdw = 1, показывающего,
877
ГИББСА УРАВНЕНИЕ АДСОРБЦИИ — ГИББСА — ГЕЛЬМГОЛЬЦ А УРАВНЕНИЯ
878
что сумма вероятностей всех возможных микросо-
стояний равна единице. В квантовой статистике
распределение Гиббса имеет вид:
и'п = Лйпе" En/kT
Здесь вместо вероятности различных значений р и
q определяется вероятность wn нахождения тела
в каком-либо re-ом квантовом состоянии с энергией
(gn — статистич. вес, т. е. число состояний с одной
и той же энергией). Постоянная А выражается из
условия: У wn = 1;
En'hT
п
Обратная величина, т. е.
iM=Sgn«-E«'ftT
п
наз. суммой по состояниям. Одной из важнейших
областей применения Г. р. является его использо-
вание в термодинамич. расчетах. Г. р. позволяет
выразить все термодинамич. потенциалы через сумму
ио состояниям, т. е. через энергетич. спектр системы.
В частности, свободная энергия системы имеет выра-
жение:
F=-RT in ^gne- E^kT
п
(где R — газовая постоянная).
Н. Э. Хандамирова.
ГИББСА УРАВНЕНИЕ АДСОРБЦИИ — урав-
нение, связывающее изменение поверхностного натя-
жения на границе раздёла фаз о с величинами адсорб-
ции компонентов 1,2, ... Гь Г2... и изменениями их
химич. потенциаловр.!, i.i2, ... при постоянной темп-ре:
— dz = 4- r2rfp2 4- ... (1)
где Гь Г2, ... — избытки компонентов 1, 2, ... в по-
верхностном слое по сравнению с их содержанием
А) и адсорбции (внизу. Б)
для поверхности водных
растворов масляной кисло-
ты (1) и KNO2(2) (по
П. А. Ребиндеру).
Рис. 1. Изотермы поверхностно-
го натяжения а(А) и величины
адсорбции Г2(Б) при адсорбции
пара н-нентана на поверхности
воды (по Джонсу и Оттевилю).
в объеме газа или р-ра. При адсорбции на жидком
адсорбенте 1, не растворяющем адсорбирующийся
из газовой фазы компонент 2, ефч = 0 и
- dz = Г»ф2 = БТГа (2)
где р2 — давление компонента 2 в газовой фазе.
Измерения ст при различных значениях р2 позволяют
вычислить Г2 (напр., в случае адсорбции паров угле-
водородов на поверхности воды, рис. 1).
При адсорбции на поверхности бинарных жид-
ких р-ров изменение концентрации р-ра с2 приводит
к изменению сь поэтому </pt]=-O. Однако можно так
выбрать положение поверхности, относительно к-рой
определяются величины Г2 и Г2, чтобы = 0. Тогда
Р 4L ______L ____________с- 4* /ч\
' dp..2 ~’ RT' d In a2 RT ' de,
где Г2 — адсорбция компонента 2 у поверхности,
для к-рой Г, = 0, а2 — его активность в растворе
(для малых значений с2а2==:с2). Измеряя ст при раз-
личных значениях с2, вычисляют Г2 (рис. 2). Величи-
на — dcAdc, наз. поверхностной активностью. Г. у. а. —
фундаментальное ур-ние термодинамики поверх-
ностных явлений. Оно применяется для определе-
ния адсорбции паров и адсорбции из бинарных
р-ров и расплавов на поверхности жидкости и для
определения поверхностного давления адсорбционных
слоев на твердых телах из изотерм адсорбции.
Лит.: Гиббс Дж. В., Термодинамические работы, пер.
с англ.. М—Л., 1950; Семенченко В. К., Поверхностные
явления в металлах и сплавах, М., 1957; Киселев А. В.,
Усп. хим., 1946, 15, вып. 4, с. 456; его же, Коллоида, ж.,
1958, 20, № 3, 338; J о n е s D. С., О t t е w I 1 1 R. Н., J.
Chem. Soc. 1955, Dec., р. 4076. А. В. Киселев.
ГИББСА—ГЕЛЬМГОЛЬЦА УРАВНЕНИЯ — ма-
тематич. выражения зависимости между: 1) работой
внутренних сил Av, теплотой процесса Qv и произ-
водной работы по темп-ре Т при постоянном объеме »:
2) работой внутренних сил А теплотой процесса
Qp и производной работы по темп-ре Т при постоян-
ном давлении />:
Лр = <?р + 7’(^)р <2>
где положительными считаются выделенная системой
теплота Qp или Qv и совершенная ею работа Ар или
Av. Г.—Г. у. характеризуют работу внутренних сил
для обратимых процессов (при постоянных темп-ре
и объеме или, соответственно, давлении). Г. — Г. у.
наз. также уравнения, дающие зависимость между изо-
хорным потенциалом F (свободной энергией по Гельм-
гольцу) или, соответственно, изобарным потенциалом
Z, их производными по темп-ре и внутренней энер-
гией U или, соответственно, энтальпией Н (см. Термо-
динамические потенциалы):
F=U + T(^jv (3)
2 = Я4-7’(Ц)р (4)
Из уравнений (3) и (4) могут быть получены уравне-
ния (1) и (2).
Г. — Г. у. объединяют первый и второй законы
термодинамики и имеют многочисленные и чрезвы-
чайно важные применения в термодинамике и физич.
химии. Напр., с помощью Г.— Г. у. выводят изобар»
реакции уравнение и изохоры реакции уравнение,
а также зависимость между электродвижущей силой
Е обратимого шльванич. элемента, теплотой q про-
текающей в нем реакции и темн-рным коэфф, электро-
движущей силы:
E = 4+TST (5)
Выражение (5) наз. Г.— Г. у. для обратимых элемен-
тов.
Лит.: Бродский А. И., Физическая химия, т. 1—2»
б иэд., М. -Л.» 1948; Раковский А. В., Введение в фи-
879
ГИББСА — ДЮГЕМА УРАВНЕНИЕ — ГИДНОКАРПОВАЯ КИСЛОТА
880
эическую химию, М., 1938; Эпштейн П. С., Курс термо-
динамики, пер. с англ., М.—Л., 1949; К и р е е в В. А., Курс
физической химии, М.—Л., 1955. В. А, Киреев.
ГИББСА—ДЮГЕМА УРАВНЕНИЕ — уравнение,
связывающее параметры термич., механич. и химич.
равновесия в многокомпонентной системе; имеет вид:
i=a
SdT+ Vdp + У n^p.^0 (1)
1=1
где З1 — энтропия, Т — темп-ра, V — объем, р —
давление, ni — число молей i-того компонента; н.; —
химич. потенциал г-того компонента, а — число
компонентов в р-ре. Г.—Д. у. особенно удобно для
исследования фазовых равновесий, т. к. все перемен-
ные здесь являются интенсивными величинами. При
постоянных темп-ре и давлении ур-ние (1) принимает
вид: ,=я
2 пг^г = ° (2)
?=1
' В случае применимости к газовой фазе законов
идеальных газов Г.—Д. у. для бинарного р-ра при-
водит к соотношению:
Nid In pt 4- N^d In р3 = 0 (3)
(здесь TVi и TV2 — мольные доли компонентов), к-рое
дает возможность по зависимости парциального
давления pi от состава р-ра для одного из компо-
нентов определить зависимость парциального давления
от состава для другого компонента.
Лит.: Гиббс Д. В., Термодинамические работы, пер.
с англ., М.—Л., 1950; Кричевский И. Р., Фазовые рав-
новесия в растворах при высоких давлениях, 2 изд., М.—Л.,
1952; К а р а петь янц М. X., Химическая термодина-
мика, 2 изд., М.—Л., 1953.
ГИБКОСТЬ ЦЕПНЫХ МОЛЕКУЛ — свойство
макромолекул полимеров, обусловленное их цепным
строением и высоким мол. весом. Уже при степенях
полимеризации 20—100, когда длина молекул пре-
вышает их поперечник приблизительно во столько
же раз, вследствие внутреннего вращения возникает
возможность заметного изменения формы молекул.
Т. о., цепные молекулы приобретают способность
изменять свою форму при сохранении запаса энергии.
Число доступных цепным молекулам различных
форм (их конформаций) очень резко возрастает по
мере увеличения их длины, т. е. степени полимери-
зации, достигая огромных значений для высокомоле-
кулярных полимеров, со степенью полимеризации
порядка тысяч или десятков тысяч. Г. ц. м. сущест-
венно зависит от природы химич. связей в главной
цепи атомов, а также от химич. состава и строения
боковых заместителей. В случае предельных угле-
водородов, когда образующие цепь С—С-связи сами
по себе допускают полную свободу внутреннего вра-
щения, гибкость цепных молекул полностью опреде-
ляется изменением взаимодействий соседних боковых
заместителей в цепи при их повороте вокруг
С—С-связи в главной цепи.Чем больше это изменение,
тем жестче цепная молекула. Поэтому изменение
состава или строения боковых заместителей приводит
к изменению Г. ц. м. и, следовательно, к изменению
свойств полимера.
Г. ц. м. приводит к особому характеру их теплового
движения, к-рый проявляется в том, что отдельные
части длинной и гибкой цепной молекулы начинают
участвовать в тепловом движении почти независимо
друг от друга; независимость движений, возрастаю-
щая по мере удаления участков молекулы друг от
друга, и приводит к тому, что большая цепная моле-
кула непрерывно изменяет свою форму, переходя от
одной конформации к другой. Подавляющее число
доступных макромолекуле конформаций, как пока-
зывает статистич. расчет, соответствует скрученным
формам молекулы. При наложении действия внешних
механич. сил макромолекулы несколько распрям-
ляются, а после прекращения воздействия вновь
скручиваются. Связанная с этим обратимая дефор-
мация полимерного тела, известная под названием
эластической (или высокоэластической), может до-
стигать значений в несколько сотен процентов (см.
Эластичность полимеров). Г. ц. м. полимеров при-
водит также и к тому, что темп-ра стеклования,
а также вязкого течения энергия активации полимер-
гомологов не зависят от степени полимеризации.
Вследствие гибкости цепных молекул возникает мно-
жество термодинамич. аномалий р-ров полимеров
и особенностей кристаллизации полимеров (см. Кри-
сталлизация высокомолекулярных соединений). Появ-
ление относительно независимого подвижного эле-
мента структуры — участка гибкой цепи (сегмента) —
приводит к тому, что одни свойства полимера опре-
деляются поведением всей цепной молекулы, в то
время как другие—только поведением сегментов.
Последнее обстоятельство позволяет определить сред-
нюю величину сегментов (напр., по величине теплоты
активации вязкого течения), т. е. оценить гибкость
молекул. Степени полимеризации сегментов колеб-
лются в зависимости от гибкости молекулы в пределах
от десятка до сотни.
Лит.: Волькенштейв М. В., Молекулы и их стро-
ение, М,—Л., 1955; его же, Конфигурационная статистика
полимерных цепей, М,—Л., 1959. Г. Л. Слонимский.
ГИГРИНОВАЯ КИСЛОТА (N-метилпирролидин-
2-карбоновая кислота, N-метилпролин), мол. в.129,17.
Известна в виде трех стереоизомеров (имеет асиммет-
рия. атом углерода). Неактивная Г. к. — бесцветные
кристаллы, т. пл. 169—170°, легко растворима в воде,
спирте, кипящем хлороформе; плохо — в холодном
этилацетате; нерастворима в эфире и бензоле.
1-изомер имеет [a]D =—80,1° (в воде). Соли Г. к.
(в т. ч. серебряная и медная) легко растворимы
в воде и спирте и не растворяются в эфире. При нагре-
вании выше т. пл. Г. к. отщепляет СО2, переходя
нгС---СН2 н2С---СН2
Il I I 2
Н2Сх *СН-СООН Н2С сн-сн2сосн3
N~CH3 XN-CH3
/ и
Н2С--СН2
H2ix+/CH-COO~
CH3-N-СН3
III
(CH2)rCOOH
в N-метилпирролидин; восстанав-
ливает при нагревании аммиач-
ный р-р окиси серебра. Г. к. (I)
может быть получена окислением
хромовой смесью алкалоида гигри-
на (II). Бетаин Г. к. — стахи-
дрин (III) — широко распространен в растительном
мире (найден в лимонной корке, в листьях апель-
синов И Т. д.). Н. В. Гнучев.
ГИДНОКАРПОВАЯ КИСЛОТА (ш-циклопентен-
2-илундекановая кислота, п = 10) С1вН28О2, мол. в.
252,2 — кристаллы, т. пл. 60,4°;
[a]g = -|- 69,3°; иодное число 100,7.
Г. к. в виде глицерида содер-
жится н хаульмугровом масле из
семян тропических растений видов
Hudnocarpus и ОпсоЬа семейства Flacourtiaceae.
Для получения Г. к. хаульмугровое масло подвер-
гают щелочному гидролизу, получают этиловые
эфиры образовавшихся кислот и после вакуумной
перегонки омыляют их. Близка к Г. к. по строению
и свойствам хаульмугровая кислота
(ш-циклопентен-2-илтридекановая кислота, п — 12)
С13Н32О2, мол. в. 280,2 — кристаллы, т. пл. 69,0°;
[а]^ =+ 69,5°; иодное число 90,5. Хаульмугровая
к-та, так же как и Г. к., содержится в виде глицерида
881
ГИДРАВЛИЧЕСКИЕ ЖИДКОСТИ — ГИДРАЗИН
882
в хаульмугровом масле; выделяется в свободном
виде таким же способом, как и Г. к. Обе кислоты
обладают антибиотич. действием; применяются для
лечения проказы (в виде препаратов хаульмугрового
масла или эфиров); оказывают благоприятное дей-
ствие при лечении кожного туберкулеза; сами кислоты
весьма токсичны.
Лит.: Шемякин М. М., Хохлов А. С., Химия
антибиотических веществ, 2 изд., М., 1953; Зелинский
Н. Д., Бондарь Л. С., Высшие жирные кислоты и их
отношение к туберкулезным бациллам, М., 1951.
А. Е. Васильев.
ГИДРАВЛИЧЕСКИЕ ЖИДКОСТИ — жидкости,
применяемые в гидравлич. передачах мощности для
приведения в действие различных механизмов и
агрегатов. В самолетах гидравлич. передачи служат
для уборки и выпуска шасси, торможения и аморти-
зации при пробегах и посадках, управления силовой
частью автопилота и проч. Гидроприводы исполь-
зуются также в автомобилях и во многих других
машинах и механизмах, где требуется передать усилия
через жидкую среду. Г. ж. должны быть стабильны
в эксплуатации, обладать пологой вязкостно-темпера-
турной кривой и низкой темп-рой замерзания, если
механизмы работают при низких темп-pax. Недопу-
скается, чтобы Г. ж. вызывали коррозию металлов
гидросистем и снижали механич. прочность и упругие
свойства уплотнительных деталей. Наличие в гид-
равлич. системах агрегатов с трущимися деталями
(поршни, детали насоса и др.) требует, чтобы Г. ж.
обладали смазочными свойствами и должной вяз-
костью. В эксплуатации получили распространение
спирто-глицериновые и нефтяные Г. ж. Спирто-
глицериновые смеси содержат этиловый спирт-ректи-
фикат и дистиллированный глицерин в различных
соотношениях. Иногда добавляют 10—15% воды.
В зависимости от состава спирто-глицериновые жид-
кости имеют т. заст. до минус 50° — минус 60° и вяз-
кость 2,5—7,5 сст (50°) и 900—17 000 ест (—45°); они
практически не действуют на резиновые и кожаные
уплотнители. Отрицательными свойствами спирто-
глицериновых смесей являются значительная корро-
зионная активность, повышенная испаряемость и
плохая смазочная способность. •
Нефтяные Г. ж. приготовляют из легких, хорошо
очищенных фракций низкозастываюгцих нефтей с до-
бавлением присадок, улучшающих стабильность и
вязкостные свойства. Применяемое для самолетов
гидравлич. масло АМ2-10 имеет т. кип. не ниже
200°, вязкость не менее 10,0 сст (+50°) и не более
1250 сст (—50°), т. заст. не выше —70. Нефтяные
Г. ж. имеют незначительную коррозионную актив-
ность и хорошие смазочные свойства; они вызывают
сильное набухание обычной резины и поэтому уплот-
нители в гидросистемах должны быть из синтетич.
маслостойкой резины. В качестве Г. ж. используются
также полиорганосилоксановые соединения (см. Крем-
нийорганические полимеры), обладающие очень хоро-
шими вязкостно-температурными свойствами и устой-
чивостью к окислению, полиалкиленгликоли и др.
синтетич. продукты. Большое внимание уделяется
получению невоспламеняющихся Г. ж. для самолетов;
среди них известны жидкости, состоящие из фтор-
углеродных, фторхлоруглеродных и фосфорорганич.
соединений.
Лит.: Моторные топлива, масла и жидкости, под ред.
К. К. Папок и Е. Г. Семенило, т. 2, 3 изд., М., 1957.
В. В. Панов.
ГИДРАЗИДЫ КАРБОНОВЫХ КИСЛОТ — соеди-
нения общей формулы CnH2n^1CONHNH2; хорошо
кристаллизующиеся соединения, растворимые в воде.
Г. к. к. обладают основными свойствами; они при-
соединяют сильные к-ты, при обработке этилатом
натрия или натрием обменивают водород на натрий.
При обработке Г.к.к.азотистой к-той образуются ази-
ды соответствующих кислот. Получают Г. к. к. обычно
действием сложных эфиров, хлорангидридов, ангид-
ридов и амидов карбоновых к-т на гидразингидрат;
можно также получить сплавлением органич. солей
гидразина, реакцией Гофмана из алкильных произ-
водных мочевины, взаимодействием эфиров изоциа-
новой к-ты с гидразином или восстановлением ацил-
нитрамидов. Для очистки от побочно образующихся
вторичных гидразидов в результате ацилирования
первоначально образующихся первичных гидразидов
последние иногда превращают в кристаллич. изопро-
пилиденовые производные нагреванием с ацетоном;
в дальнейшем Г. к. к. выделяют через солянокислые
соли.
Качественно и количественно Г. к. к. определяют
по восстановлению аммиачных р-ров AgNO3 или по
восстановлению Фелинговой жидкости. Ниже при-
ведены темп-ры плавления нек-рых Г. к. к.:
Гидразиды кислот
Т. пл., °C
Уксусной........•....................
Масляной.............................
Бензойной ...........................
о-Онсиметилбензойной.................
2,4-Диметил-3-этилпиррол-5-карбоновой .
67
44
112
128
Впервые Г. к. к. получены Т. Курциусом в 1890.
См. также Азиды карбоновых кислот.
Лит.: Смит П. А. С., в кн.; Органические реакции,
[СВ. статей], пер. с англ., Сб. 3, М., 1951, с. 322.
Т. И. Соркина.
ГИДРАЗИН (диамид) H2NNH2 — бесцветная, про-
зрачная, сильно гигроскопичная жидкость; т. пл. 2°;
т. кип. 113,5°; плотность жидкого Г. в интервале
0—50° можно рассчитать по уравнению:
d‘ = 1,0253 (1—0,00085г)
плотность твердого г/’5 1,146; теплота образования
ДН299 = 12,05 ккал/моль. В жидком состоянии мо-
лекулы Г. сильно ассоциированы. Безводный Г.
имеет высокую диэлектрич. проницаемость (51,7
при 25°) и является хорошим ионизирующим рас-
творителем для многих солей, смешивается с по-
лярными растворителями (вода, спирты, аммиак,
амины), нерастворим в углеводородах. С водой обра-
зует азеотропную смесь, содержащую 71,5 вес. % Г.,
т. кип. 120,1°. Система Г. — вода имеет две эвтектич.
точки с т. пл. ниже —50°. Гидрат Г. N2H4 • Н2О —
дымящая на воздухе жидкость, плотн. 1,03, т. кип.
118,5° при 739,5 мм рт. ст. Теплота образования
ДН298 = —57,95 ккал/моль. Водные растворы Г. обла-
дают сильными основными свойствами (К = 8,5 •
• 10 ’ при 25°), в частности поглощают двуокись угле-
рода из воздуха.
Г. термодинамически неустойчив, легко разлагается
под влиянием катализаторов, а также при нагревании
и при действии излучения. .Г. — сильный восстано-
витель. В водных р-рах Г. легко подвергается окисле-
нию кислородом воздуха; эта реакция катализируется
ионами меди, марганца, железа, кобальта, никеля и
хрома. Смеси Г. с кислородом или воздухом склонны
к взрывному сгоранию. При контакте Г. с окислами
нек-рых металлов (Си, Fe, Мо, Сг, Pb, Hg) или веще-
ствами с развитой поверхностью (уголь, асбест и др.)
может произойти его воспламенение. Г. — одно из
наиболее химически активных веществ. Замещением
атомов Н в молекуле Г. органич. радикалами полу-
чаются его многочисленные органич. производные.
С кислотами Г. образует два ряда солей, напр.
N2H4 • НС1 и N2H4 2НС1. Соли Г. бесцветны, почти
все хорошо растворимы в воде, большинство из них
плавится с разложением, соли кислот-окислителей
883
ГИДРАЗИН СЕРНОКИСЛЫЙ — ГИДРАЗИНА ПРОИЗВОДНЫЕ
884
взрывоопасны. К чиолу важнейших солей относятся:
гидразин сернокислый и солянокислый N2H4 • НС1,
т. пл. 92° и N2H4 • 2НС1, т. пл. 198°. Подобно воде
и аммиаку, Г. образует координационные соединения,
напр. СгС12 • 2N2H4, Со(СЮ4)2 • 3N2H4.
Разб. водные р-ры Г. получают окислением NH3
или мочевины гипохлоритом. Безводный Г. получают
либо обезвоживанием N2H4 • Н2О щелочами с после-
дующей вакуумной перегонкой, либо обработкой
N2H4 • H2SO4 жидким аммиаком, причем образуется
раствор Г. в жидком аммиаке; после испарения NH3
остается безводный Г. Применяют Г. в органич. син-
тезе, в произ-ве пластмасс, резины, инсектицидов,
пзрывчатых веществ, как аналитич. реактив и в ка-
честве компонента реактивного топлива. Г. и все его
производные сильно ядовиты.
Лит.: О др ит Л. и Огг Б., Химия гидразина, пер.
с англ., М., 1954; Федорова Н. М., Применение гидразина
в различных областях народного хозяйства, Л., 1957.
Л. М. ВиОавсхий.
ГИДРАЗИН СЕРНОКИСЛЫЙ (сульфат гидра-
зина) H2NNH2 • H2SO4, мол. в. 130,13 — бесцветные
ромбич. кристаллы или длинные призмы ромбич.
системы; т. пл. 254° (с разл.); плотн. 1,378; в 100 г
воды растворяется: 3,05 г (22°), 27,65 а (60°); очень
мало растворим в абс. спирте; сильный восстанови-
тель, ядовит. Получают Г. с. действием гипохлорита
натрия на мочевину в присутствии NaOH:
CO(NH2)4 + NaOCl + 2NaOH —
HjNNHs + H.>0 + NaCl -|- Na2CO3
и последующей обработкой 50%-ной H2SO4 :
H2NNH2 + H.SO4 — h»nnh2 • H so4
Г. с. является сильным восстановителем. На этом
основано его применение в неорганич. химии и в неор-
ганич. анализе. В газовом анализе Г. с. применяют
при определении гипохлоритов и хлоратов; в титри-
метрич. анализе — для установки титра иода; в элек-
троанализе — при разделении Мп и Fe; для устране-
ния растворения платино-иридиевых анодов при элек-
тролизе; его используют при определении Си, As и
ОН-группы. С соединениями, содержащими карбо-
нильную группу, Г. с. реагирует с выделением
воды и H2SO4, что используют для количественно-
го определения карбонильной группы.
Лит. см. при ст. Гидразин. И. П. Ефимов.
ГИДРАЗИНА ПРОИЗВОДНЫЕ (органические) —
продукты замещения одного или нескольких атомов
водорода в гидразине на алкильные или арильные
радикалы. По степени замещения и взаимному поло-
жению замещающих радикалов различают: 1) моно-
замещенные Г. п. — первичные гидразины (I); 2) не-
симметричные дизамещенные Г. п. — вторичные гид-
разины (II); 3) симметричные дизамещенные Г. п. —
гидразосоединения (III); 4) тризамещенные Г. п.
(IV); 5) тетразамещенные Г. п. (V):
R\
R—NH—NH, JN-NH, R—NH—NH—R'
R'z
1 II Ш
R\ R\ /4"
)N-NH-R" >N—Nz
R’z R'z
IV V
где R, R', R", R"' = H, Aik, Ar.
Последние две группы Г. п. встречаются редко.
Тетразамещенные Г. п., напр. тетрафенилгидразин,
легко диссоциируют на свободные радикалы, содержа-
щие двухвалентный атом азота:
/С6Нд СЯН5
N—N —2 N:
CgH5Z ХС6Н5 C6Hsz
Наибольшее значение в лабораторной и производ-
ственной практике имеют Г. п., содержащие два атома
водорода при одном из атомов азота (группы I и II).
Они могут быть получены алкилированием или арп-
лированием гидразина либо первичных Г. п.:
NH2NH2——2-*-CH3NHNH2-—(CH3)2NNH2
восстановлением диазосоединений, напр.
CcHsN.Cl C„HBNHNHj
восстановлением N -нитрозоаминов:
N-NO
CH3Z
Zn
СНзСООН
N—NH2‘
CHaz
расщеплением замещенных мочевин (Гофмана ре-
акция)-.
AlkNHCONH. —AlkNHCONHBr (AlkNHCON.-) —
— AlkNHN=C=О -2S. AlkNHNH2
взаимодействием аминов с галогенаминами:
C6H6NHi + FNHs — C,;HBNHNH3
Г. п. представляют собой твердые или жидкие
вещества; обычно сильно ядовиты. Первичные и вто-
ричные Г.п. легко образуют гидразоны при взаимодей-
ствии с карбонильными соединениями:
СвНв. ZR СвН5. Я
NNH2 +• О=С —•- N—N=C
CH3Z 4R' CH3Z 4R'
Г. п., как правило, являются сильными основаниями
и дают прочные соли с одним эквивалентом кислоты.
Атом водорода группы NH в первичных ароматич.
гидразинах может быть заменен атомом натрия с обра-
зованием производных типаС6Н5—NNa—NH2. При
окислении Г. п. связь C=N сохраняется или расщеп-
ляется в зависимости от условий:
CcHBNHNH3 С0Н„ + N3 4- Н.О
CcHBNHNH3 CeHsNsBr + HBr
Сильные восстановители расщепляют связь
N—N в Г. п.:
C„H5NHNH. CSHBNH, + NH3
При ацилировании Г. п. образуются гидразиды кислот:
C„HBNHNH2 +СНзСООН — CcHBNHNHCOCHs
C„HBNNaNH> 4-СН3СОС1 —♦ Ct "^NNHa
CH3COZ
Г. п. используются для идентификации н выделе-
ния альдегидов и кетонов, а также как аналитич. реа-
885
гидразобензол — ГИДРАЗОНЫ
886
генты для выделения солей нек-рых метал-
лов. Г. п. могут применяться в качестве анти-
окислителей. Наиболее широко применяются
в пром-сти первичные ароматич. гидразины
как исходные продукты для синтеза красите-
лей, душистых и ростовых веществ, лекар-
ственных препаратов—производных фенилме-
тилпиразолона, индола, карбазола.
См. также Фенилгидразин, Гидразоны, Гидразосо-
единения.
Вг
СН2-С-СООС2Н5
n-nhc6h5
Лит.: Шленк В. и Бергман Э., Органическая
химия, т. 1, пер. Гс нем.], Л., 1936, с. 262; Ullmann, Bd 8,
3 Aufl.. MOnchfn — В.. 1957, S. 708. E. M. Рохлин.
ГИДРАЗОБЕНЗОЛ C6H5NH—NHC6H5, мол. в.
184,23 — бесцветные пластинки или бипирамидаль-
ные ромбоэдры (из смеси спирта и эфира), приобре-
тающие на воздухе желто-красную окраску;
т. пл. 127—128°; очень мало растворим в воде, рас-
творим в спирте, бензоле, легко растворим в эфире,
нерастворим в уксусной к-те; Хмакс 2920 А. В эфире
Г. обладает слабоосновными свойствами; при действии
эфирного р-ра НС1 наряду с бензидинхлоргидратом
образуется небольшое количество хлоргидрата Г.
CfiH5NH—NHCeH6 • НС1; в присутствии минераль-
ных к-т превращается в бензидин (см. Бензидиновая
перегруппировка)'.
CoH5NH - NHCeH6->. HSNC,H5 - CcH5NH2
Под действием сильных восстановителей, напр.
SnCl2, Г. превращается в анилин; легко окисляется
в азобензол C6H5N = NC6H5; аутоокисление спиртово-
го р-ра, особенно в присутствии щелочи, приводит к
образованию Н2О2:
CeH5NH - NHCcH5 + О2 -> C„H5N = NC,H5 + Н2О2
В пром-сти Г. получают восстановлением нитробен-
зола цинковой пылыо в щелочной среде. Г. — проме-
жуточный продукт синтеза бензидина. Открыт Н. Н.
ЗИНИНЫМ В 1845. Д. Н. Васкевич.
ГИДРАЗОНЫ — продукты взаимодействия кар-
бонильных соединений с гидразином, содержащие
группировку атомов ^>C=N— NH2. Альдегиды и ке-
тоны, реагируя с гидразином обычно в мягких усло-
виях, часто в присутствии щелочей или кислот обра
вуют как Г. (I), так и азины (II):
R. 1с
\с=О + NH2NH» — >C=N-NH» +
R'/ ' R'z
I
R4 ,R
+ \C=N-N=C<
R'Z 'R'
[I
Аналогично получаются замещенные Г.:
Сахара при взаимодействии с арилгидразинами, как
правило, дают озазоны. Другой метод получения Г. —
сочетание солей диазония с соединениями, содержа-
щими при атоме углерода подвижные атомы водо-
рода; промежуточными продуктами при этом явля-
ются азосоединения:
CeH5N2Cl + СН2(СООС»Н5)2 —
— [C,H5-N=N-CH(COOC2H5)2] —
— CeH5NH-N=C(COOC2H5)2
Г., как правило, представляют собой хорошо кри-
сталлизующиеся и труднорастворимые вещества с бо-
лее или менее глубокой окраской. Нек-рые Г. суще-
ствуют в виде двух изомеров — сип (III; и анти (IV):
III
Вг
СН2-С-СООСгН5
CeH5HN-N
IV
Часто Г. находятся в таутомерном равновесии с соот-
ветствующими азосоединениями:
О ОН
N-NH-C6H5 N = N-CeH5
При гидролизе в присутствии кислот Г. обычно
диссоциируют на карбонильное соединение и соответ-
ствующий гидразин:
D р Ч р р ч
\c=N-n/ + Н2О — \с=О + H-.NN^
R'z XR”' R'z XR"'
Арилгидразоны в безводных условиях в присутствии
кислот или нек-рых солей часто превращаются в про-
изводные индола (Фишера реакция):
R
R'
Побочно прп этом иногда образуются нитрилы:
СН3ч CnCi СНЗЧ
>CH-CH=NNHCeH6 —)CH-CN + CeH5NH2
СНз СНз
Незамещенные Г. под действием щелочных агентов
подвергаются каталитич. разложению с образованием
углеводородов (Кижнера — Вольфа реакция):
R R
\c=NNH» — ^СН» + N.
R'z ' R'z
При восстановлении Г. получаются амины, что при-
меняется, напр., для синтеза аминокислот:
R-C-COOH 7n R-CH-COOH
II „7V I + С,Н5КН.,
N— NHCeH5 HGi NH»
При окислении незамещенных Г. образуются алифа-
тич. диазосоединения:
(CtH5)»C=NNH2 —8-2. (CcH5)..CN2
Г. карбонильных соединений, содержащих нек-рые
функциональные группы, часто легко замыкаются
в гетероциклич. соединения — производные пира-
зола и пиридазина:
,С-С--СН2 Н..СС СНг
J iooc-л — J со *
^NH
CeHs CeH5
Образование гидра-
зонов широко исполь-
зуется для обнаруже-
ния, идентификации
п выделения карбо-
нильных соединений.
В пром-сти Г. приме-
няют для получения
красителей, лекарст-
венных, душистых и
/снч
/С\
СН N
II I ------
сн nh2
4 СНО
N
I + н.»о
N
ростовых веществ —
производных фенилметилпиразолона и индола. Неко-
торые замещенные Г., напр. фтивазид, являются
аятитуберкулезными препаратами.
887
ГИДРАЗОСОЕДИНЕНИЯ — ГИДРАТАЦИЯ
888
Лит.: Шленк В. и Бергман Э., Органическая
химия, пер. [с нем.], т. 1, Л., 1936, с. 296; Вейганд К.,
Методы эксперимента в органической химии, пер. с нем.,
ч. 2, М., 1952, с. 275, 634; Thorpe, V. 6, L.— N. Y.— Toronto,
1948, р. 302. Е. М. Рохлин.
ГИДРАЗОСОЕДИНЕНИЯ — соединения, содержа-
щие гидразогруппу — NH—NH—, связанную с
двумя углеводородными радикалами R—NH—NH—R;
имеют значение преим. Г. ароматич. ряда. Алифа-
тич. Г. — подвижные бесцветные жидкости с харак-
терным запахом, смешивающиеся с водой, спиртом и
эфиром, обладающие свойствами двухкислотных осно-
ваний, с кислотами образуют устойчивые кристаллич.
соли. Ароматич. Г. — кристаллич. бесцветные веще-
ства с очень слабыми основными свойствами, нерас-
творимые в воде, растворимые в спирте, эфире, бен-
золе и т. п. Алифатич. Г. получают восстановлением
азоуглеводородов цинковой пылью в щелочной среде
или омылением соответствующих 1\’,1\т/-диацилдиал-
килгидразонов кипячением с соляной к-той. В пром-сти
ароматич. Г. получают восстановлением нитросоеди-
нений Zn-пылью в сильнощелочной среде:
ArNO3 —• ArNO —* ArNOH — ArN=NAr —*
I
О
—• ArN=NAr —• ArNH—NHAr
или при электролитич. восстановлении нитросоеди-
нений в щелочной среде. При действии сильных вос-
становителей ароматич. Г. образуют амины ArNH—
—NHAr + 2Н —> 2ArNH2; кислородом воздуха Г.
окисляются в азосоединения: ArNH—NHAr ArN =
= NAr. Большое значение имеет превращение Г. в диа-
минодифенилы при действии минеральных кислот,
т. н. бенаидиновая перегруппировка. Наиболее простое
ароматич. Г. — гидразобензол, открыто Н. Н. Зини-
ным в 1845. Ароматич. Г. не вырабатываются в каче-
стве товарных продуктов, но получаются в больших
количествах в виде промежуточных продуктов при
произ-ве бензидина и его производных (толидина,
дианизидина и т. п.), являющихся важнейшими исход-
ными в-вами для получения азокрасителей.
ГИДР АКРИЛОВАЯ КИСЛОТА (Р-оксипропиононая,
этиленмолочная) НОСН2СН2СООН, мол. в. 90,08 —
оксикислота жирного ряда, изомер молочной кислоты;
сиропообразная жидкость; хорошо растворима н воде,
спирте и эфире. Константа диссоциации К = 31,1 •
• 10-4. При нагревании Г. к. дегидратируется в акрило-
вую к-ту:
НОСН3СН2СООН СН2=СНСООН
Хромовая смесь окисляет Г. к. до СО2 и щавелевой
к-ты. Соли Г. к. (в т. ч. и серебряная) растворимы
в воде и при нагревании превращаются в соли акри-
ловой к-ты. Г. к. удобно получать, исходя из этилен-
хлоргидрина:
С1СН2СН2ОН —— CNCH2CH2OH NaOtLJie.
—. HOCH2CH2COONa НОСН .СН.СООН
Лактон Г. к. обладает бактерицидным действием.
ГИДРАСТИН C21H21OeN, мол. вес 383,41 — алка-
лоид, содержащийся в корневище желтокорня ка-
надского (Hydrastis canaden-
s*s), семейства лютиковых (Ra-
СН2 I Т ' nunculaceae), частично в сво-
— СН3 бодном, частично в связанном
Т виде. Г. — бесцветные ромбич.
СН О кристаллы; т. пл. 132° (эта-
। нол); [a]D ——67,8° (хлоро-
1| СО форм), —49,8° (абс. этанол); в
воде нерастворим, хорошо рас-
I творим в хлороформе (1 : 14
ОСН3 цри 25°) и бензоле, хуже —
в спирте (1 : 170) и эфире (1 : 175). Г. — третич-
ное основание, имеет горький вкус, обладает щелоч-
ной реакцией на лакмус; образует неустойчивые в
водных растворах соли — хлоргидрат, т. пл. 116°,
пикрат, т. пл. 184°. Раствор Г. в H2SO4 в присутствии
молибдата аммония окрашивается в оливково-зеленый
цвет. При действии на Г. разб. HNO3 он распадается
на опиановую к-ту (I) и гидрастинин (II).
СНО
ОСНз
///
Молекула Г. обладает двумя асимметрич. центрами;
синтетич. Г. получается в виде смеси двух рацематов.
Разделение одного из рацематов на антиподы с по-
мощью d-камфор-л-сульфоновой кислоты приводит
к 1-0-гидрастину, идентичному с природным про-
дуктом. Физиологии, действие
Г. проявляется в возбуждении
центральной нервной системы;
в больших дозах он вызывает
судороги и паралич; обладает
сосудосужающим действием.
Хлоргидрат гидрастинина (III) менее токсичный, чем
Г., применяется в медицине для прекращения крово-
течений, особенно маточных.
Лит. см. при ст. Ареколин и Алкалоиды.
А. Е. Васильев.
ГИДРАТАЦИЯ — присоединение воды к различ-
ным веществам, находящимся как в растворенном,
так и в свободном состояниях. Г. является частным
случаем сольватации — присоединения к веществам
какого-либо растворителя. В отличие от гидролиаа,
Г. не сопровождается образованием водородных и
гидроксильных ионов. Различают следующие случаи
Г.: Г. электролитов в растворах, Г. молекул в рас-
творах, Г. с образованием твердых гидратон, Г. окис-
лов, Г. органич. соединений, Г. высокомолекулярных
соединений.
Учение о Г. в растворах возникло в 60-х гг. 19 в. в резуль-
тате работ Д. И. Менделеева, к-рый на основе эксперименталь-
ных фактов выдвинул предположение о существовании в рас-
творах рада определенных химич. соединений молекул рас-
творенного вещества с водой, находящихся в равновесии друг
с другом и с водой. Эта идея составила основу гидратной
теории растворов (называемой в более общем виде сольватной).
Гидратная теория растворов после ее усовершенствования и
объединения Н. А. Каблуковым с теорией электролитич.
диссоциации стала важнейшей составной частью современного
учения о растворах.
Гидратация в растворах приводит к образованию
стойких и нестойких соединений воды с растворенным
веществом, вызывает изменение термодинамич. и др.
свойств растворенных веществ и воды. Степень и ха-
рактер Г. растворенного вещества сильно зависят от
его природы. Изучение Г. в растворах встречается
с большими трудностями, обусловленными недоста-
точностью знаний о природе жидкого состояния. Наи-
более полно изучена Г. электролитов.
Гидратация электролитовв раство-
рах является главной причиной их диссоциации на
ионы — она обусловливает устойчивость ионов в рас-
творах и затрудняет ассоциацию ионов.Наиболее резко
выражена Г. у сильных электролитов. В газовой
фазе диссоциация электролитов на ионы — процесс
сильно эндотермичный и не происходит самопроиз-
вольно. В разбавленных растворах энергия, выделяю-
щаяся при взаимодействии ионов с водой, в значи-
тельной степени компенсирует энергию диссоциации.
Для количественной характеристики Г. электролитов
используют теплоту Г. (изменение энтальпии &Н),
изменение изобарно-изотермич. потенциала AZ, из-
889
ГИДРАТАЦИЯ
890
менение энтропии ДА, гидратационное число и др.
величины. Теплотой гидратации наз.
тепловой эффект процесса введения в раствор моля
электролита, находящегося в виде свободных ионов
в идеальном газовом состоянии. Энтропию и изобар-
ный потенциал Г. также относят к этому процессу.
Непосредственные измерения теплот, изобарных потен-
циалов и энтропий Г. осуществить не удается. Их величины
получают путем расчета воображаемых процессов диссоциа-
ции электролита на ионы в газовой фазе и последующего их
введения в раствор. При этом используют опытные термо-
химии. данные. Так, теплота Г. соли определяется как разность
теплоты ее растворения и энергии кристаллич. решетки.
Энтропия Г. — как разность энтропий ионов в растворе и в
газообразном состоянии; для ее нахождения используют зави-
симости эдс и растворимости от темп-ры. Изменение изобар-
ного потенциала Г. вычисляется из термодинамич. соотноше-
ния, связывающего эти три величины, а также непосредст-
венно из эдс и данных об энергиях ионизации, сублимации и
кристаллич. решеток.
Теплота Г. иона не может быть непосредственно
определена опытным путем. Ее находят из теплоты Г.
электролита, равной сумме теплот Г. катиона и аниона.
При этом используют предположение о равенстве
теплот Г. J и Cs+ в одном и том же растворе, подтвер-
ждаемое многими физико-химич. данными. Сущест-
вуют и другие методы расчета ионных теплот Г.
Теплоты Г. ионов превосходят тепловые эффекты мно-
гих химич. реакций. Их абсолютные величины растут
с уменьшением кристаллографич. размеров и ростом
заряда иона. Порядок величин теплот Г. —АН
(в ккал/г-ион) ионов различной валентности иллюст-
рируется следующими данными:
Н+ 265, Сз+ 67, L1+ 127, Mg2+ 467, La“+ 796,
ОН“ 122, NOs 74, Cl~ 84, J- 67, CIO? 54, SO? ’ 265
Приведенные величины наз. химич. теплотами Г. Т. н. ре-
альные теплоты Т. для грамм-эквивалента отличаются от хи-
мич. на величину <pF — энергии, выделяемой при прохождении
заряда одного грамм-эквивалента ионов 7 через границу
раздела вакуум/раствор, имеющую скачок электрич. потен-
циала <р. Эти величины для катионов и анионов имеют проти-
воположные знаки и поэтому сумма реальных теплот Г. ка-
кого-либо электролита равна сумме химич. теплот Г.
Г. газообразных норов наряду с большими экзотер-
мич. тепловыми эффектами сопровождается боль-
шим понижением их изобарных потенциалов. По-
рядок величин изменения изобарных потенциалов Г.
ионов — AZ иллюстрируется следующими данными
(в ккал]г-ион)‘.
Н+ 255, L1+ 121, Cs+ 66, Са2+ 373,
Cd2 + 424 , 01“ 79, J~ 64
Переход ионов в гидратированное состояние сопро-
вождается значительным уменьшением энтропии. По-
рядок величин энтропий Г. — ДА виден из следующих
данных (в кал/г-ион град)'.
L1+ 22, Na+ 14,6, К+ 6,2, С1" 17,1,
J- 8, Zn3+ 57,6, SO?- 52,5
(энтропия водородного иона условно принята равной
нулю). Приведенные выше численные величины
AH, AZ и Д5 представляют изменения соответствую-
щих термодинамич. функций при переносе одного
моля ионов из газообразного состояния в стандарт-
ных условиях (давление 1 атм, 25°) в раствор с актив-
ностью тех же ионов, равной единице, при той же
темп-ре. С ростом темп-ры Г. уменьшается, что обус-
ловлено экзотермичностью процесса. Г. многих элек-
тролитов сопровождается также уменьшением объема,
т. е. объем раствора меньше суммы объемов электро-
лита и воды, израсходованных на его получение.
Часто Г. ионов характеризуют т. наз. гидрата-
ционным числом, т. е. числом молекул воды,
к-рое находится во взаимодействии с ионом и состав-
ляет его гидратную оболочку. Размер гидратной обо-
лочки ионов, в соответствии с законами гидродина-
мики, оказывает влияние на подвижности ионов,
числа переноса, коэфф, диффузии и др. свойства. Вхо-
ждение воды в состав гидратных оболочек уменьшает
концентрацию свободной воды в растворе, что ока-
зывает влияние на растворимость в ней веществ, на
распределение растворенного вещества между водой
и органич. растворителями, на активность воды в рас-
творе, самодиффузию,вязкость и др.ее свойства. Числа
гидратации (найденные каким-либо одним методом,
см. ниже) растут с ростом заряда и уменьшением раз-
меров ионов.
Учитывая влияние эффекта образования гидратных оболо-
чек на различные свойства ионов и воды в растворе, пред-
ложены различные методы нахождения гидратапионных чи-
сел. Наиболее важные из них основаны на измерении коэфф,
сжимаемости растворов электролитов, диффузии электроли-
тов, активности электролитов, а также на изучении распре-
деления вещества между водой и органич. растворителем,
на изучении адсорбции воды гелями электролита, на исполь-
зовании подвижностей ионов, на измерении диэлектрич. про-
ницаемости переменным током высокой частоты. Для нек-рых
электролитов числа гидратации, найденные разными мето-
дами, имеют близкие значения, но для многих электролитов
они значительно расходятся. Порядок величин чисел гидрата-
ции, найденных при помощи первых пяти вышеперечисленных
методов, иллюстрируется следующими данными:
LiCl (6: 7,1; 3; 10,5; 4), LiBr (5—6; 7,6; 5,6; 9,0; 4,0),
NaCl (7; 3,5; 3,5; 7,9; —), ВаС18 (16—17; 7,7; —; —; —)
При растворении поливалентных электролитов, а также
ряда электролитов типа 1—1 вязкость воды возрастает, а при
растворении нек-рых электролитов типа I—I (напр., KJ) —
уменьшается.Коэфф.активности и самодиффузии воды в первом
случае уменьшаются, а во втором возрастают. Нек-рые иссле-
дователи первое явление наз. положительной Г., а второе —
отрицательной Г. Температурные коэфф, подвижностей ионов,
вызывающих положительную Г., выше темп-рного коэфф,
вязкости воды, а при отрицательной Г. они меньше. Г. ионов
по-разному изменяет энтропию воды. В растворах сильно гид-
ратированных электролитов энтропия воды меньше, в рас-
творах слабо гидратированных электролитов она выше, чем
в чистой воде. Из этого следует, что одни электролиты усили-
вают структуру воды в р-ре, а другие ее разрыхляют. Анализ
теплоемкостей и объемов р-ров электролитов показал, что
вода, входящая в состав гидратных оболочек ионов, имеет
более низкие молярные величины теплоемкостей и более вы-
сокие величины плотностей, чем чистая вода. Для ряда элек-
тролитов эффект снижения теплоемкости воды наблюдается
в пяти молекулярных слоях, окружающих ион, а эффект
повышения плотности — в четырех слоях.
Природа гидратации электролитов выяс-
нена лишь в общих чертах. Главной причиной Г. электро-
литов является притяжение полярных молекул воды
электрически заряженными ионами. Дополнительное
притяжение возникает в результате взаимной поляриза-
ции ионов и окружающих их молекул воды (индук-
ционный эффект, см. Межмолекулярное взаимодей-
ствие), а также в результате возникновения опреде-
ленной согласованности движения электронов в на-
ружных оболочках ионов и молекул воды (диспер-
сионный эффект, см. там же). У нек-рых ионов (преи-
мущественно переходных металлов) Г. сопровождается
деформацией электронных оболочек и возникновением
связей молекул воды с ионами, аналогичных связям
в комплексных соединениях. С этим связано различие
цвета гидратированных и негидратированных ионов
меди, кобальта, хрома и др. Количественный расчет
взаимодействия ионов и молекул воды в процессах Г.
осуществлен только приближенно. Он позволил вы-
яснить нек-рые детали механизма Г. и вычислить
величины тепловых эффектов Г.
Потенциальная энергия Е взаимодействия иона
с молекулой воды в первом приближении определяется
соотношением (Е свободного иона принята равной
нулю):
Е = — cos ср
где р, — дипольный момент воды, е — заряд иона,
г — расстояние между центром диполя молекулы
воды и центром иона; ф — угол между осью диполя
и линией, соединяющей центры иона и диполя. Вы-
численное значение Е для г =г^г0 4* (rHj0, г, —
кристаллохимия, радиусы Н2О и иона) во много раз
превосходит энергию теплового движения 3Ц<Т для
многих ионов (к — постоянная Больцмана). Из этого
891
ГИДРАТАЦИЯ
892
следует, что многие ионы (в особенности с малыми
радиусами и полиза рядные) прочно притягивают
ближайшие соседние молекулы воды и образуют
с ними прочные молекулярные агрегаты, к-рые в теп-
ловом движении и при движении под действием внеш-
него электрич. поля (напр., во время электролиза)
ведут себя как отдельные частицы. Образование во-
круг попа прочно удерживаемого им слоя молекул
воды, характеризующегося пониженной способностью
этих молекул к трансляционному движению (скачко-
образное перемещение, характерное для молекул
жидкости), наз. первичной гидратацией.
Второй слой молекул воды, окружающих ион, притя-
гивается к нему значительно слабее, и трансляцион-
ное движение молекул воды в нем меньше отличается
от движения их в толще жидкости. Совокупность
изменений, вызываемых ионом во втором прилегаю-
щем к нему слое молекул воды и в более удаленных
слоях, наз. вторичной гидратацией.
Применение меченых атомов к изучению свойств гидрат-
ных оболочек ионов показало, что продолжительность жизни
молекул в гидратной оболочке иона сильно зависит от при-
роды иона. Так, с помощью изотопа О18 в качестве меченого
атома найдено, что в ионе [Сг(Н2О)в]3+ половина молекул Н8О
обменивается с молекулами раствора за 40 часов, в ионе
[Co(NH8)5H2O]3+ — за 24,5 часа, в то время как у других
трехвалентных ионов это происходит в течение времени,
меньшего 3 минут. Хотя продолжительность жизни молекул
Н2О в гидратных оболочках ионов в обычном смысле недоста-
точно велика, но, по сравнению с продолжительностью пребы-
вания отдельной молекулы Н2О на одном месте в жидкой
воде, она является очень большой и указывает на наличие
прочных связей ионов с гидратной водой. Об этом же свиде-
тельствует образование гидратированных ионов в вакууме,
напр. [К • Н2О]+. На существование первичной и вторичной
Г. указывает изучение адсорбции паров воды тонкими плен-
ками гелей солей; установлено, что теплота адсорбции воды,
отвечающая первичной Г., значительно выше, чем вторичной
Г., мало отличающейся от теплоты конденсации водяных
паров. Расчет теплоты Г., использующий представление об
образовании прочных гидратов за счет ион-дипольного взаимо-
действия и учитывающий ряд других эффектов этого взаимо-
действия, а также учитывающий понижение электростатич.
энергии ионов при переходе из газовой фазы в диэлектрик
(эффект Борна) дает величины для многих ионов, близкие к
опытным.
Степень гидратации зависит от кон-
центрации электролита в растворе. Ее изменение
с концентрацией оказывает большое влияние на за-
висимость электропроводностей, коэфф, диффузии,
химич. потенциалов, коэфф, активности и др. свойств
электролитов от их концентрации в растворах.
Г. многих электролитов зависит от степени их ассоциации,
н-рая, в свою очередь, изменяется с изменением концентрации.
Изучение энтропий Г. и энтропий ассоциации показало, что
образование ионных пар Fe3+ ОН~, Fe3+F , Fe3+Cl~, Pb2+Cl~,
[Co(NHs)J2+ Cl~ сопровождается дегидратацией ионов, в то вре-
мя как образование ионных пар Т1+С1~, Т1+ ОН _,Ва’+ (НСО3)~,
Ва2+(СН3СОО)" не сопровождается изменением Г. ионов.
Недостаточность знаний о зависимости Г. от концентрации
Является важнейшей причиной незавершенности современной
теории концентрированных растворов электролитов. Г. ионов
является одним из факторов, определяющих величины стан-
дартных электродных потенциалов различных систем и ока-
зывает большое влияние на кинетику электрохимия, разряда
ионов. Г. является одной из причин перенапряжения в электро-
химия. реакциях.
Гидратация молекул в растворах изучена слабее,
чем Г ..попов. Ряд сведений о Г. молекул получен путем
изучения двойных жидких систем различных веществ
с водой методами физико-химич. анализа, спектраль-
ными и др. методами.
Так, Д. П. Менделеев, анализируя зависимость плотности
системы, вода — спирт от состава, установил существование
гидрата С2Н8ОН • ЗН2О. Анализ вязкости и электроводности
растворов НС1 в водно-спиртовых смесях указывает на суще-
ствование также соединений С2Н5ОН • 4Н2О и С2НБОН • 8Н2О.
Установлено, что молекулы карбоновых к-т образуют с гидро-
нсплсодержащими растворителями, в т. ч. и с водой, продукты
присоединения состава 1:1 и 1:2. Определены константы
нестойкости таких соединений, порядок к-рых составляет
10~3 — 10-6. Известны также продукты присоединения воды
к фенолам состава 1:1с константами нестойкости порядка
10~*— 10~3. Из оптич. исследований (препм. из ИФ-спектров
и спектров комбинационного рассеяния) найдено, что продукты
присоединения воды к молекулам кислот и оснований обра-
вуются за счет водородных связей.
На основе термодинамич. анализа свойств растворов
Н. А. Измайловым показано, что образование продук-
тов присоединения воды к молекулам кислот и осно-
ваний является обязательной промежуточной стадией
ИХ электролитической диссоциации.
Гидратация, сопровождающаяся образованием твер-
дых гидратов. Г. простых веществ, а также солей,
кислот, оснований и др. химич. соединений, в т. ч.
органических, часто приводит к образованию твердых
продуктов — гидратов, в состав к-рых входят
молекулы воды в виде индивидуальных частиц. В хи-
мич. формулах гидратов воду пишут отдельно, напр.
CuSO4-5H2O, Ва(ОН)2 • 8Н2Ъ и т. д. Кристаллич.
гидраты известны двух типов
Кристаллогидраты (фазы опре-
деленного состава) известны для многих
кислот, оснований, для солей почти всех катионов
(исключение Pd2+, Ag+), для простых веществ и для
различных соединений. Встречаются кристаллогид-
раты, в к-рых на 1 молекулу вещества приходится
1, 2, 3, 4, 5, 6, 7. 8, 10, 12 молекул воды, но наиболее
распространены кристаллогидраты с 4, 6, 8, 2, 1 Н2О.
Для многих веществ известно несколько кристалло-
гидратов различного состава:- напр.,М£гС12 дает кри-
сталлогидраты с 2, 4, 6, 8 и 12 молекулами воды,
СаС12 с 1, 2, 4, 6 и 8 молекулами воды, NaOH с 1, 2,
Зх/2, 4, 5 и 7 молекулами воды, H2SO4 с 1, 2, 4, 6 и 8
молекулами воды.
Вода, входящая в состав кристаллогидратов, наз.
кристаллизационной. Кристаллогидраты
образуются при кристаллизации из насыщенных вод-
ных растворов, при взаимодействии веществ с рас-
творами воды в органич. растворителях, а также при
взаимодействии веществ с парами воды и др. путем.
При определенной темп-ре кристаллогидрат может
находиться в равновесии с безводным веществом (или
с другим кристаллогидратом этого вещества) и одно-
временно с водяными парами только при строго опре-
деленном давлении водяных паров к окружающем
пространстве. Это давление наз. давлением
диссоциации кристаллогидрата.
Оно индивидуально для каждого кристаллогидрата и
растет с ростом темп-ры. Если кристаллогидрат нахо-
дится на воздухе, где давление водяных паров меньше
давления его диссоциации, то происходит постепенная
потеря кристаллизационной воды — выветривание.
Равновесие в системах, содержащих кристаллогид-
раты, подчиняется правилу фаз.
Кристаллогидраты ведут себя как химич. индивиды.
Они имеют определенные темп-ры плавления (многие
плавятся без разложения), определенные растворимо-
сти, кристаллич. структуры и др. индивидуальные
свойства.
Темп-ры плавления кристаллогидратов зависят от их
природы; для солей и оснований они, как правило, значительно
выше, чем для кислот. Порядок темп-p плавления кислотных
кристаллогидратов иллюстрируется следующими данными;
HNO3 • Н2О — 33е, HNO3 • ЗН2О —18°, НСЮ4 • 2Н2О —18°,
НС104 • ЗН2О —49°. Превращение кристаллогидрата в безвод-
ное соединение или в кристаллогидрат с меньшим содержа-
нием воды часто сопровождается изменением кристалличе-
ской структуры. Прочность кристаллогидратов зависит от
их природы и от температуры. К числу непрочных относят-
ся n2S-12H2O, HCI • 12Н2О, NaCl-12H2O, NaCl - 2Н2О,
Cl2 • 8Н2О, 2CS2 • Н2О и др. Нек-рые из них существуют
при темп-pax ниже 0°. Известны кристаллогидраты, устой-
чивые при высоких темп-pax, напр. СаС12 • Н2О устойчив
даже выше 200°.
Важнейшим методом изучения строения кристалло-
гидратов является рентгенографический. С его по-
мощью установлено, что молекулы воды в каждом
кристаллогидрате располагаются в совершенно опре-
деленном порядке. В кристаллогидратах солей, как
правило, они располагаются вокруг катионов, созда-
вая окружение, подобное координационной сфере
комплексных ионов (см. Комплексные соединения).
Связь молекул воды с многими катионами в основном
8 93
ГИДРАТАЦИЯ
894
электростатическая, а с нек-рыми катионами-комп-
лексообразователями — донорно-акцепторная.
В нек-рых кристаллогидратах солей часть молекул воды
не входит в состав непосредственного окружения катионов.
Так, в CuS04 5Н2О */5 всех молекул составляет непосред-
ственное окружение иона Си2+ (располагаясь но тетраэдру),
а ‘,5 часть молекул Н2О расположена так, что каждая из них
связана с двумя ионами SO|~ и с двумя молекулами Н2О.
входящими в окружение иона Си2+. В NiSO4 • 7Н2О '/, часть
молекул Н2О не входит в состав непосредственного окружения
ионов Ni2+, а при помощи водородных связей связана с дру-
гими молекулами Н2О. Нек-рые кристаллогидраты определен-
ного состава (напр., Na2SO4 • ЮН2О) представляют твердые
растворы соли в воде. Они имеют структуру твердого льда,
в к-рой отдельные молекулы воды заменены ионами, входя-
щими в состав соли.
Нек-рые кристаллогидраты существуют в нескольких
модификациях, отличающихся оптич., химич. и физич. свой-
ствами, напр. для СтС13 6Н2О известны три модификации:
[Сг(Н2О)с JCL, (сине-зеленый, фиолетового цвета в растворе),
[Сг(Н2О)2С1]С12 • Н2О (светло-зеленый, зеленый в растворе)
и [Сг(Н2О)4С12]С1 2Н2О (зеленый, зеленый в растворе). При
взаимодействии с азотнокислым серебром Из раствора первой
модификации осаждается весь хлор, из второй -I, и из третьей
*/3 хлора. Также у СоС12 • 6Н2О известны две модификации,
отличающиеся темп-рами плавления.
Многие вещества могут быть получены в наиболее
чистом виде в форме кристаллогидратов. Поэтому
кристаллогидраты находят широкое применение в хи-
мич. пром-сти и особенно часто используются в каче-
стве реактивов. Г. полностью безводного или частично
обезвоженного сульфата кальция составляет физич.
сущность вяжущего действия гипсовых вяжущих ве-
ществ, широко применяемых в строительной пром-сти.
Гидраты — фазы неопределенного
состава. Состав таких гидратов описывается фор-
мулами вида А • гН2О, где х — число молекул воды,
приходящееся на 1 молекулу вещества А; х может
принимать различные значения, зависящие от дав-
ления водяных паров в пространстве, окружающем Г.,
и от темп-ры. В кристаллах, способных к образованию
такого типа гидратов, молекулы воды заполняют пу-
стоты в кристалле и межкристаллитные поры. Моле-
кулы воды удерживаются в них адсорбционными си-
лами. Г. или обезвоживание таких кристаллов не со-
провождается изменением их кристаллич. структуры.
Хорошо изученным примером таких веществ являются
цеолиты. Последние в зависимости от размеров пор
могут избирательно поглощать молекулы воды или
другие вещества из газовых смесей или из растворов.
Благодаря этому свойству цеолиты и им подобные
вещества используются в технике для указанной цели
и часто называются молекулярными си-
тами. Вода, адсорбированная кристаллами, часто
называется цеолитной. Г. описанного типа наблю-
дается у многих оксалатов лантанидов.
J здратация окпслов происходит при их взаимодей-
ствии как с жидкой водой, так и с ее парами. В зави-
симости от природы окислов Г. приводит к образова-
нию щелочей, кислот, амфотерных гидроокисей. Г.
окислов, ведущая к образованию гидроокисей, сопро-
вождается разложением молекул воды. Вода, пошед-
шая на образование гидроокисей, ваз. конститу-
ционной. Г. кислотных и щелочных окислов
сопровождается значительными экзотермич. тепловы-
ми эффектами. Напр., в реакции SO3(tb.)+ Н2О(ж.)=
= H2SO4 при 25° выделяется 28,63 ккал на моль,
а в реакции Na2O (чв.) 4- Н2О (ж.)=2 NaOH выде-
ляется 19,1 ккал на моль NaOH. Г. окислов составляет
основу многих технически важных процессов. Так,
Г. кислотных окислов используется для получения
кислот; напр. Г. SO3 получают серную кислоту, Г.
NO2 — азотвую кислоту. Г. окиси кальция состав-
ляет основу получения гашеной извести. Эта реакция,
а также аналогичная реакция с окисью магния играют
важную роль в процессах отвердевания известковых
и магнезиальных вяжущих материалов, широко приме-
няемых в строительной пром-сти. Г. нек-рых окислов
(преимущественно Р2О5), а также Г. нек-рых кислот
и щелочей в технике и лабораторной практике исполь-
зуется для сушки газов, жидкостей и твердых ве-
ществ.
Известны процессы Г. других неорганич. соедине-
ний и нек-рых простых веществ. Так, Г. хлора вызы-
вает образование хлорноватистой и соляной кислот в
соответствии с реакцией С12 4- Н20=НС10 4- НС1.
Г. аммиака, а также его алкилзамещенных дает гидро-
окиси, существующие только в растворах и имеющие
щелочную реакцию. Н. Е. Хомутов.
Гидратация органич. соединений включает: образо-
вание кристаллогидратов, присоединение элементов
воды по кратным связям, раскрытие цикла окисей
алкиленов, лактамов, лактонов и др., происходящее
под действием воды. Как правило, Г. протекает в при-
сутствии катализаторов — щелочных (Г. по нуклео-
фильному механизму) или кислотных (Г. по электро-
фильному механизму). Г. по нуклеофильному меха-
низму подвергаются соединения, в к-рых электрон-
ная плотность на атакуемом центре резко понижена,
причем первоначальным актом реакции является
атака атома, имеющего избыточный положительный
заряд, анионом ОН". По нуклеофильному механизму
производится Г. нитрилов кислот до амидов в присут-
ствии щелочей, присоединение воды по карбонильной
группе в органич. соединениях, содержащих элект-
роно-акцепторные заместители (хлораль, мезоксалевая
к-та, гексафторацетон), а также катализируемое
щелочами присоединение воды к а-,Р-ненасыщенным
карбонильным соединениям. Более распространенной
является Г. по электрофильному механизму, вклю-
чающему первоначальную атаку протоном или ионом
гидроксония. Так, Г. олефинов является промышлен-
ным способом получения спиртов. Различают прямую
и косвенную Г. Косвенная Г. — присоединение воды
к олефинам в жидкой фазе в присутствии сильных
неорганич. к-т (H2SO4, Н3РО4, HF и др.) — имеет след,
механизм:
' ' н3о+
й
и с-с+
I
. н2о
г: r+oh.,
ROH + Н3О+
Н2О
Одновременно происходит присоединение кислоты —
катализатора по кратной связи олефина с последую-
щим гидролизом образовавшегося сложного эфира до
спирта и исходной к-ты. В качестве побочных продук-
тов при Г. олефинов по электрофильному механизму
образуются простые эфиры. Г. лактонов и лактамов
может осуществляться как по нуклеофильному, так и
по электрофильному механизму, аналогично гидролизу
сложных эфиров и амидов.
Г. непредельных углеводородов и др. органич. сое-
динений играет огромную и непрерывно возрастаю-
щую роль в пром-сти органич. синтеза и широко
используется в препаративной органич. химии. Г.
непредельных углеводородов является важнейшим
звеном во многих процессах переработки нефтяного
сырья, попутных и природных газов, а также ацети-
лена для получения многих ценных продуктов химич.
пром-сти. Из олефинов прямой Г. получают спирты,
а комбинированием Г. и присоединения окиси угле-
рода получают карбоновые кислоты. Г. ацетилена
приводит, в зависимости от условий реакции, к ацет-
альдегиду (см. Кучерова реакция) или ацетону. Г.
окисей алкиленов может протекать как в кислой, так
и в щелочной среде, причем кислотная Г. окиси эти-
лена является одним из промышленных способов по-
лучения этиленгликоля. Г. нитрилов получают амиды.
Г. непредельных углеводородов, как правило, про-
895
ГИДРАТАЦИЯ И ДЕГИДРАТАЦИЯ КАТАЛИТИЧЕСКИЕ
896
текает при повышенных давлениях и темп-рах в при-
сутствии различных катализаторов (см. Гидратация
и дегидратация каталитические). Значительное при-
менение в пром-сти органич. синтеза получила Г.
кетенов и нек-рых органич. окисей. Этим путем из
кетена СН2=С=Ов пром-сти готовят уксусный ан-
гидрид и уксусную к-ту:
2СН3 = С = О + Н30- (СН3СО)3О
СН3 = 0 = 0 + Н30 — СНзСООН
Л. С. Герман.
Гидратация высокомолекулярных веществ. Из ана-
лиза измерений сжимаемости растворов при прохо-
ждении ультразвуковых волн, а также из измерений
тепловых эффектов в процессах поглощения воды
нек-рыми высокомолекулярными веществами найдено,
что Г. наиболее резко выражена у полиэлектролитов
и у веществ, макромолекулы к-рых содержат поляр-
ные группы, такие как—ОН, =С0, —СНО, —СООН
и др. (напр., крахмал).
Для отдельных высокомолекулярных веществ определены
стандартные величины теплосодержаний, изобарных потен-
циалов и энтропий Г. Они имеют отрицательные значения.
Для нек-рых полиэлектролитов, напр. для многих одно-
и двухзарядных катионов сульфонатов полистирола, установ-
лено, что степень Г. зависит от активности воды в растворе
и что процесс Г. протекает в две экспериментально различи-
мые стадии. Первая из них сильно экзотермична и отвечает
присоединению по одной молекуле воды к каждому иону
макромолекулы полиэлектролита, а вторая стадия слабо
экзотермична и отвечает процессу присоединения еще не-
скольких молекул воды к каждому иону. Общее число моле-
кул воды, составляющих гидратные оболочки ионов макро-
молекулы, уменьшается с ростом темп-ры и сильно зависит
от природы ионов. Наиболее высокие значения оно имеет у
сульфонзтов двухзарядных катионов с малыми кристалло-
химия. радиусами (ионы Вег+, Mg2+).
Г. полиэлектролитов оказывает большое влияние
на термодинамику и кинетику реакций ионного об-
мена. Для ряда высокополимеров установлено, что Г.
(в общем случае сольватация высокомолекулярных
соединений) является важнейшей причиной их на-
бухания в процессах поглощения воды (или другого
растворителя) (см. Набухание). Г. играет большую
роль в растворах высокомолекулярных соединений.
Подобно молекулам, ионам и макромолекулам рас-
творенных веществ коллоидные частицы (мицеллы)
способны сорбционно связывать на своей поверхности
молекулы воды. Связанная вода в коллоидных систе-
мах приобретает свойства, отличные от свободной
воды. Образование многослойной гидратной оболочки
имеет важное значение в стабилизации коллоидных
систем; способность связывать воду характеризует
гидрофильность и гидрофобность коллоидных систем.
Г. неорганич. и органич. веществ относится к числу
обратимых химич. процессов, подчиняясь закону
действующих масс и др. термодинамич. соотношениям,
при помощи к-рых могут быть рассчитаны выходы
продуктов Г. в различных экспериментальных усло-
виях
Лит.: Менделеев Д. И., Основы химии, М.—Л.,
1947; его же, Растворы, [Л.Г 1959; его же, Рассуждение
о соединении спирта с водой, СПБ, 1865; К аблуковИ. А.,
Современные теории растворов, М., 1891; Измайловы. А.,
Электрохимия растворов, Харьков, 1959; Некоторые проблемы
современной электрохимии, под ред. Дж. Бокрис, пер. с англ.,
М., 1958; К и р е е в В. А., Курс физической химии, 2 изд.,
М.—Л., 1956; Самойлов О. Я., Структура водных
растворов электролитов и гидратация ионов, М., 1957;
Райс О. К., Электронное строение и химическая связь в
неорганической химии, пер. с англ., М., 1949; И з г а р bi-
in е в Н. А., Горбачеве. В., Курс теоретической элек-
трохимии, М.—Л., 1951; Аносов В. Я., Погодин
С. А., Основные начала физико-химического анализа, М.—Л.,
1947; В о ю ц к и й С. С., Растворы высокомолекулярных
соединений, 2 изд., М., 1960; ДуманскийА. В., Учение
о коллоидах, 3 изд., М,-—Л., 1948; его же, Лиофильность
дисперсных систем, Киев, 1960. Н. Е. Хомутов.
ГИДРАТАЦИЯ И ДЕГИДРАТАЦИЯ КАТАЛИТИ-
ЧЕСКИЕ — реакции присоединения (гидратация) или
отщепления (дегидратация) воды от органич. соеди-
нений.
Гидратация каталитическая — один
из основных классов каталитич. реакций в органич.
химии — протекает без отщепления каких-либо
других групп (в отличие от реакций гидролиза). Реак-
ции гидратации можно проводить в гомогенной или
гетерогенной среде. Гомогенная, или т. в. ки-
сло т н а я, гидратация проводится обычно при
помощи жидких неорганич. к-т (серной, фосфорной).
Иногда при этом добавляют соли ртути (см. Кучерова
реакция). При гетерогенной, или т. н. п р я -
м о й, гидратации в паровой фазе применяют различ-
ные катализаторы, имеющие обычно кислотные свой-
ства (фосфаты меди, цинка, кадмия, различные окис-
лы, напр. окислы вольфрама, активированные окисью
цинка и нанесенные на силикагель, активированная
А120а с различными добавками, СнО + МпО ->
+ 4НаРО4, а также НаРО4 с различными добавками
на твердых носителях). Преимущества гетерогенной
гидратации: значительно меньшая затрата энергии,
большая легкость осуществления непрерывных про-
цессов, отсутствие необходимости в коррозионно-
устойчивой аппаратуре и др.
Основными видами реакций гидратации являются:
гидратация олефинов в спирты, ацетиленовых углево-
дородов в альдегиды и кетоны и нитрилов в амиды.
При гидратации этилена образуется этиловый спирт;
гидратация прочих олефинов протекает обычно по
правилу Марковникова; при этом образуются вторич-
ные или третичные спирты: R—СН=СН2 + Н20 —>
—>П—СН(ОН)СН3. Этот процесс лежит в основе про-
мышленного способа получения спиртов—этанола,
изопропанола, бутанола-2, триметилкарбинола. Сырь-
ем при этом служат одефины газов крекинга или др.
попутных или отходящих газов нефтяной или химич.
промышленности. Каталитическая гидратация олефи-
нов — обратимая реакция, константа равновесия ко-
торой уменьшается с температурой; поэтому ее
выгодно проводить при низких температурах и высо-
ких давлениях (при парофазных процессах обычно
150—300° и 10—300 ат). Процесс кислотной гидра-
тации олефинов:
R-CH CIL + H3SO4 — R-CH(SO3H)-CH3 +Jk2.
-- R-CH(OH)-CH3 + H2SO4
Скорость присоединения (как и скорость реакции при
парофазной гидратации) убывает в ряду: изобути-
лен >. н-бутилены > пройилен > этилен. Условия
присоединения указаны в таблице.
Углеводород Концентрация H2SO4, % Темп-ра, °C.
Этилен 90-98 до 80
Пропилен 75—90 ок. 30
н-Бутилены 75—85 15—30
Гидролиз образующихся алкилсерных к-т до спир-
тов происходит при разбавлении сернокислотного
р-ра водой и при нагревании; если обработку алкил-
серных к-т производить малым количеством воды при
100° или соответствующим спиртом, то вместо спиртов
можно получать простые эфиры, напр. диизопропило-
вый эфир из пропилена. В пром-сти применяют также
способы гетерогенной парофазной гидратации эти-
лена; катализаторами, напр., являются синяя окись
вольфрама W2O5 или НаРО4 на силикагеле. В первом
случае процесс проводят при 300—345° и давлении
300 ат, во втором — при 30° и 68 ат и рециркуляции
этилена. У гомологов этилена с повышением мол. веса
возрастает также склонность к полимеризации, по-
этому выходы спиртов максимальны для амиленов и
гексиленов. Применяются также процессы гидратации
производных олефиновых углеводородов, напр. из
897
ГИДРАТАЦИЯ И ДЕГИДРАТАЦИЯ КАТАЛИТИЧЕСКИЕ — ГИДРАТЦЕЛЛЮЛОЗА
898
трихлорэтилена получают монохлоруксусную к-ту.
М. Г. Кучеров в 1881 впервые получил уксусный аль-
дегид каталитич. гидратацией ацетилена в присутствии
H2SO4 с добавкой солей ртути. Реакцию Кучерова
широко применяют в заводском масштабе для полу-
чения ацетальдегида, этанола, уксусной к ты и этил-
ацетата. Если вместо H2SO4 пользоваться уксусной
к-той с солями ртути, то образуются винилацетат и
этилиденацетат. Из гомологов СН=СН по реакции
Кучерова получают кетоны. Известны также способы
парофазной гидратации CHsCH, причем в определен-
ных условиях можно получать также ацетон (напр.,
при 440—470° над катализаторами 2ZnO • W2O5 или
2CdO • W2O5). При гидратации нитрилов в присут-
ствии кислотных катализаторов (НС1) или в паровой
фазе над ThO2 при 420° образуются амиды. Известно
также применение в качестве катализаторов ионно-
обменных смол. Так, из ацетонитрила с высоким вы-
ходом получают ацетамид.
Дегидратация каталитическая.
Различают внутримолекулярную дегидратацию, при
к-рой одна или несколько молекул воды отщепляются
от одной молекулы органич. соединения с образова-
нием ненасыщенных или циклич. продуктов, и межмо-
лекулярную, когда молекулы воды отщепляются от
двух или многих одинаковых или различных молекул,
а образовавшиеся остатки молекул соединяются между
собой. Эту реакцию, так же как и гидратацию, можно
проводить как гомогенным, так и гетерогенным путем,
причем в первом случае катализаторами являются
минеральные или органич. кислоты, их соли или
производные, а во втором — окислы различных
металлов (А12О3, ThO2, W2O5), их смеси и нек-рые соли,
напр. Са3(РО4)2, KA](SO4)2, алюмосиликаты и др.
Реакции внутримолекулярной де-
гидратации, как правило, эндотермичны и
высокие степени превращения достигаются лишь при
высоких темп-рах и малых давлениях; важнейшими
их примерами являются: дегидратация спиртов до
олефинов; многоатомных спиртов до альдегидов, кето-
нов и диолефинов; амидов и оксимов до нитрилов,
циклодегидратация — дегидратация гликолей до оки-
сей олефинов, пятнатомных спиртов до фурфурола
и т. и. На окиси алюминия высшие спирты (до три-
метилпентанола) подвергаются дегидратации почти
без изменения углеродного скелета, тогда как для
пинаконов характерна изомеризация (т. н. пинаколи-
ноеая и ретропинаколиноеая перегруппировки). Легче
всего дегидратируются третичные спирты (при нагре-
вании с безводным бисульфитом калия, муравьиной
или щавелевой к-тами, небольшим количеством иода
и пр.). Дегидратация спиртов с последующим гидри-
рованием олефинов послужила основой для промыш-
ленного получения индивидуальных авиатоплив (нео-
гептан, неогексан, триптан и др.). При межмоле-
кулярной дегидратации константы равно-
весия реакций, как правило, меньше зависят от
темп-ры и давления. При этом может происходить
отщепление воды от двух или нескольких одинаковых
или различных молекул. В первом случае примерами
могут служить: дегидратация спиртов до простых
эфиров при сравнительно невысоких темп-рах (до
250°); дегидратация одноатомных первичных спиртов
под действием щелочи до спиртов с удвоенным числом
атомов углерода, т. н. диспиртов; дегидратация глико-
лей с образованием полных внутренних эфиров, напр.
диоксана из этиленгликоля; дегидратация однооснов-
ных к-т до ангидридов и т. п. Примерами отщепле-
ний воды от различных молекул могут быть: дегидра-
тация кислот и спиртов в присутствии А1С13, BF3,
ThO2, TiO2 до сложных эфиров; дегидратация спир-
тов в смеси с аммиаком до аминов в присутствии
А]2О3 4- Fe2O3.
15 К. х. э. 1. 1.
Реакция дегидратации составляет основу большин-
ства реакций поликонденсации, играющих огромную
роль при получении полимеров, алкидных или глиф-
талевых смол, полиамидных волокон (найлона), моче-
вино-формальдегидных смол и др. На многих катализа-
торах могут проходить дегидратация и дегидрогениза-
ция спиртов, что используется, в частности, при полу-
чении дивинила из этилового спирта по способу Лебе-
дева (катализатор — смесь дегидрирующего и дегидра-
тирующего окислов). В других случаях дегидрогениза-
ция, так же как и полимеризация олефинов, представ-
ляет собой одну из нежелательных побочных (парал-
лельных или консекутивных) реакций, почти всегда
сопутствующих реакциям каталитич. гидратации и
дегидратации. Многие исследователи относят реак-
ции гидратации и дегидратации к процессам т. н.
кислотно-основного катализа и считают, что они
протекают через промежуточное образование т. н.
карбониевых ионое.
Лит.: Сабатье П., Катализ в органической химии,
пер. с нем.. Л., 1932; Долгов Б. II., Катализ в органи-
ческой химии, 2 изд.. Л., 1959; Бер к ман С., Моррелл Д.
и Э г л о ф ф Г., Катализ в неорганической и органической
химии, пер. с англ.,кн. 2, М.—Л., 1949; Каталитические, фото-
химические и электролитические реакции, пер. с англ., М.,
I960; Гольдштейн Р., Химическая переработка нефти,
пер. с англ., М., 1952; Catalysis. Ed. Р. Н. Emmett, v. 2, N. Y.,
1955; Катализ. Труды Первого международного конгресса, М.,
1960, В. Э. Вассерберг.
ГИДРАТРОПОВЫЙ АЛЬДЕГИД (а-фенилпропио-
новый, метилфенилацетальдегид) С9Н16О, мол. вес
134,17 — жидкость, по запаху напоминающая фе-
нилацетальдегид; т. кип. 202—205°;
93о/11 мм; d™ 1.0920; nfl 1,5169; рас- ^х^СНСНО
творяется в 7—11 объемах 50%- I |Г I
ного, 6,5 объемах 60%-ного, 1,5 объ-
емах 70%-ного спирта. В промыш-
ленности Г. а. получают из ацетофенона и эфира
монохлоруксусной к-ты (Даргана реакция):
СеН5СОСН3 + C1CH.COOR R07i
СН3 СН3
— СсН5С--CHCOOR —С„Н5С снсоон —
ч'о/ ^0^
СНз
— СсН5СНСНО
Производное Г. а. — 4-диокси-4-метилпентанацеталь
а-фенилпропионового альдегида, т. кип. 115—
I2675 мм, обладает запахом резеды. Применяется
в тонких композициях для создания цветочного запаха
гиацинта, сирени и нек-рых других, а также в нек-рых
фруктовых эссенциях. д. д. Зелепецкая.
ГИДРАТЦЕЛЛЮЛОЗА — одна из структурных
модификаций целлюлозы. Г. образуется из другой
структурной модификации (природной целлюлозы)
в результате: 1) растворения целлюлозы и высажива-
ния ее из р-ра; 2) обработки целлюлозы конц. р-рами
щелочей с последующим разложением щелочной
целлюлозы; 3) этерификации целлюлозы и последую-
щего омыления эфиров; 4) интенсивного размола цел-
люлозы. Г. и природная целлюлоза, имея одинако-
вый химич. состав, различаются между собой по рент-
геноструктурным данным тем, что D-глюкопиранозные
звенья макромолекул у Г. лежат в одной плоскости,
а у природной целлюлозы они повернуты на 90°.
Кроме структурных изменений в расположении звень-
ев при превращении природной целлюлозы, в Г. про-
исходит ослабление межмолекулярных водородных
связей, из-за чего Г. быстрее и легче взаимодействует
с различными реактивами, чем природная целлюлоза.
Г. отличается повышенной гигроскопичностью и сорб-
ционными свойствами. При 65% относительной влаж-
ности и 20° природная целлюлоза поглощает 6—8%
899
ГИДРИДЫ
900
влаги, тогда как Г., полученная без варушевия мор-
фология. структуры, поглощает 10—12%, а после
переосаждения вз р-ра — 12—14% влаги. Плотность
Г. виже плотности природной целлюлозы (1,52 вместо
-1,56). Скорость химич. реакций (гидролиз, ацетилиро-
вание, окисление) и накрашивания, а также интег-
ральная теплота смачивания и растворимость Г. выше,
чем у природной целлюлозы. Переход структурной
модификации природной целлюлозы в Г. является
обратимым. При нагревании Г. в глицерине при 150—
250° она вновь превращается в структурную модифи-
кацию природной целлюлозы. При омылении ксаито-
гената целлюлозы при темп-pax выше 60° происходит
частичный переход Г, в природную целлюлозу. Хи-
мич. волокна, получаемые по вискозному или медно-
аммиачвому способу из природной целлюлозы, со-
стоят из Г., но при вагревавии этих волокон в жидко-
стях, вызывающих вабухавие, до 150° и выше струк-
турная модификация Г. вновь превращается в при-
родную модификацию целлюлозы, идентичную при-
родным волокнам (хлопок, лев).
Лит.: РоговииЗ. А. и ШорыгинаН. II., Химия
целлюлозы и ее спутников, М.—Л., 1953. А. Б. Пакшве-р.
ГИДРИДЫ — соединения химич. элементов с водо-
родом (в расширенном толковании этого термина);
делятся на три основных группы: солеобразные,
металлообразные (металлические) и летучие Г. Грани-
цы между группами не являются резкими, и могут
быть выделены две промежуточные группы: Г. пере-
ходные от солеобразных к металлич. и переходные от
металлич. и солеобразных к летучим (полимерные Г.).
Из-за своеобразия свойств Г. ряда элементов их от-
несение к той или ивой группе ивогда бывает формаль-
ным. Комиссия по номенклатуре химич. соединений
VIII Менделеевского химич. съезда рекомендовала
называть Г. только соединения водорода с относи-
тельно более электроположительными элементами,
т. е. с металлами.
К солеобразным относятся Г. щелочных и
щелочноземельных металлов. Характер связи в них
иоввый, водород содержится в форме отрицательного
нова Н~ (исключая Lift с несколько полярной связью).
Солеобразвые Г. — белые кристаллич. продукты, раз-
лагающиеся до плавления с потерей водорода (кроме
LiH, т. пл. 680°), растворимые в расплавах солей и
гидроокисей (напр., в эвтектике LiCl—КС1, т. пл.
352°), но нерастворимые в оргавич. растворителях
(исключая LiH, несколько растворимого в полярных
растворителях, яапр., в эфире). Ниже сопоставлены
теплоты образования Д/С>Оч (в ккал/моль) и плотности
й'25 (в г/см3) наиболее изученных солеобразных Г.
Гидрид ДН°98 <j2S Гидрид
LiH -21,34 0,776 CsH -10,1 3,410
NaH —13,60 1,364 Call, —45,1 1,902
КН -15,16 1,430 Srll2 -42,3 3,269
RbH -11,3 2,595 Balf2 -40,9 4,156
Давление водорода достигает 1 атм яад LiH при
865°, вад NaH при 430°, над СаН2 ок. 990°.
Получают солеобразвые Г. непосредственным взаи-
модействием соответствующих металлов с водородом
при повышенной темп-ре и давлении водорода, превы-
шающем давление разложения Г. Для получения LiH
и NaH можно обрабатывать водородом взвесь Li или
Na в парафине или керосине. СаН2 может быть полу-
чен действием Н2 иа смесь СаО + Mg (500—600°)
или СаС12 -ф Zn (800—900°). При электролизе расплав-
ленных солеобразвых Г. на аноде выделяется Н2.
В результате взаимодействия солеобразных Г. с ве-
ществами, содержащими положительно заряженный
водород (вода, кислоты, NH3, спирты и т. д.), происхо-
дит выделение Н2: NaH -b ROH —> RONa -ф Н2. Этим
же вызвано конденсирующее действие солеобразвых
Г. (более сильное по сравнению с амидами и алкоголя-
тами): С6Н5СНО 4> СН3СООСН3 + NaH -э-С6Н5СН =
= СНСООСН3 -ф- NaOH -ф- Н2. Количественно про-
текающая реакция СаН2 2Н2О-> 2Н2 -ф- Са(ОН)2
может быть использована для определения следов Н2О
и их удаления, для определения воды в кристаллогид-
ратах и для получения водорода. Окислы и галогениды
восстанавливаются солеобразными Г. до металлов
(или до металлич. Г., если восстанавливаемые металлы
способны их образовывать); галогениды неметаллов
восстанавливаются до летучих и двойных Г.:
2ВС13 + SCaH3 — B2He 4. ЗСаСЬ
ВС13 + 4L1H — LiBH4 4- 3L1C1
Г. кальция СаН2 применяют в порошковой метал-
лургии для получения порошков гидридов титана,
циркония, виобпя, тантала из их окислов. Эти Г.
могут быть превращены в металлы прокаливанием
или переплавкой в вакууме; 1,5—2%-ный раствор
NaH в расплаве NaOH применяют для снятия окисвой
пленки с металлов. LiH и NaH используют для полу-
чения боргидридов (см. Бороводороды) и алюмогидри-
дов, а также в оргавич. синтезе (См. также Лития
гидрид, Натрия гидрид).
Металлообра зные Г. по характеру связи
близки к металлам. В образовании металлич. связей
в металлообразных Г. участвуют также и электроны
атомов водорода. Значительное число Г. этой группы
относится к т. наз. «фазам внедрения». Их можно раз-
бить на две подгруппы.
Г. переходные от солеобразных
к металлическим (Г. металлов подгрупп
скандия, титана, ванадия)— твердые хрупкие порош-
ки серого или черного цвета; представляют собой
фазы переменного состава (кроме UH3). Плотность
переходных Г. ниже плотности соответствующих ме-
таллов (напр., 3,72 для TiH2, 5,56 для ZrH2). Теплоты
образования того же порядка, что и у солеобразных
Г., напр. для ZrH2 = —40,5 ккал/моль. Рас-
творение водорода в указанных металлах сначала со-
провождается образованием твердых р-рон с сохра-
нением структуры металла и увеличением размеров
его элементарной ячейки, дальнейшее повышение
содержания водорода вызывает изменение струк-
туры, связанное с образованием гидридных фаз, к-рых
может быть несколько. С повышением темп-ры содер-
жание Н в Г. уменьшается, что отличает данную под-
группу от собственно металлич. Г. Переходные Г.
образуются при взаимодействии элемептов, они яв-
ляются сильными восстановителями. Порошкообраз-
ные Г. ураиа, церия, тория самовоспламеняются во
влажном воздухе; Г. титава, циркония, виобия, тан-
тала в этих условиях устойчивы. Многие переходные
Г. катализируют реакции гидрирования органич.
соединений. Нек-рые Г. применяют для получения
чистых металлов. Г. титана и циркония входят в состав
нек-рых припоев для соединения керамич. деталей
с металлическими; Г. циркония применяется для по-
лучения циркониевого геттера в вакуумной технике.
Металлические Г. в узком’ смысле слова
представляют собой твердые р-ры водорода в металле,
К вим относятся Г. элементов подгрупп Ст, Мп, Fe,
Си и платиновых металлов. Большинство указанных
металлов поглощает небольшое количество водорода,
в относительно широком интервале ливейво зависящее
от корвя квадратного из его давления и увеличи-
вающееся с ростом темп-ры; это соответствует эндо-
термичвости процесса поглощения водорода (в отли-
чие от переходных Г.). Значительная часть водорода
окклюдирована в субмикроскопич. межкристаллитных
щелях, причем количество их при адсорбции водорода
901
ГИДРИДЫ — ГИДРИНДАН
902
увеличивается. Pd поглощает значительно больше
водорода, чем остальные металлы этой группы.
Растворенный нодород несколько меняет периоды
решеток металлов (у Pd а — 3,890 А, а у PdHOj8,
а = 4,03 А). У Pd возникают 2 гпдридные фазы, обра-
зующие гранецентрпроваиную решетку, как и сам
металлич. Pd, но отличающиеся периодами элемен-
тарной ячейки. С повышением темп-ры двухфазная
область сужается и выше 310° исчезает — система
Pd—PdH становится однофазной. Растворение во-
дорода в Pd приводит к уничтожению его парамаг-
нетизма, т. е. вакантные места полосы 4d в Pd запол-
няются электронами Н-атомов. Нек-рые из указанных
выше металлов образуют также Г. со структурой,
отличной от структуры исходного металла. Повышен-
ная летучесть Си, Ag, Аи в токе Н2 может служить
указанием на присутствие нестойких летучих Г. этих
металлов. С образованием Г. связана каталитич. ак-
тивность ряда металлов. Необходимо учитывать воз-
можность образования Г. при электролизе, т. к. раство-
рение водорода вызывает изменение механич.свойств
металла (папр., повышает его твердость и хрупкость).
Полимерные Г. (переходные к летучим)
образуются у элементов подгрупп Zn и Ga, а также
у Al, Be и, возможно, Mg. Представляют собой белые,
сильно полимеризованные вещества. Полимеризация
происходит за счет образования водородных связей.
При нагревании эти Г. разлагаются на Н2 и металл.
Полимерные Г. получают действием LiAlH4 па гало-
гениды и век-рые другие производные соответствую-
щих металлов (такие реакции могут проводиться
в эфирном р-ре). Образующиеся при этом Г. не поли-
меризованы, но сильно сольватированы. При выделе-
нии из р-ра происходит их полимеризация. Пз Г. этого
типа наиболее изучен (А1Н3)Х; в зависимости от сте-
пени полимеризации он разлагается па элементы при
100—160°, растворяется в (CH3)3N и тетрагидрофуране
с деполимеризацией и образованием сольватов. Эфир-
ный р-р А1П3 восстанавливает альдегиды, кислоты
и эфиры до первичных спиртов, кетоны — до вто-
ричных, нитрилы и нитросоединепия — до первичных
аминов.
Л е т у ч и е Г. — соединения с водородом элементов
IV, V, VI, VII главных групп периодич. системы Мен-
делеева, В и Ga. Связь в летучих Г. ковалентная пли
полярная; полярвос.ть связи меняется в зависимости
от сродства к электрону водорода и элемента. Г.,
в к-рых атом водорода несет нек-рый отрицательный
заряд (В2Н6, SiH4 и flp.J, гидролизуются водой с вы-
делением водорода. Если же водород песет положи-
тельный заряд, то в водном р-ре такой Г. диссоциирует
на ионы (напр., HCI, H2S), а при взаимодействии
с солеобразными Г., содержащими ион Н', происходит
выделение водорода. Летучие Г. легких элементов
стабильны, а тяжелых — неустойчивы. Помимо непо-
средственного взаимодействия элементов, летучие
Г. могут быть получены разложением водой илн кис-
лотами соединений соответствующих элементов с ме-
таллами и восстановлением галогенидов Г. (LiH,
1лА1Н4 и др.). О летучих Г. см. Аммиак, Бороводо-
роды, Бромистый водород, Вода, Йодистый водород,
Нремневодороды, Мышьяковистый водород, Сероводо-
род, Сурьмянистый водород, Фосфористые водороды,
Фтористый водород, Хлористый водород.
Таким образом, в коротких периодах периодич.
системы характер Г., образуемых элементами, ме-
няется по ряду: солеобразные — полимерные — лету-
чие, а в длинных периодах по ряду: солеобразные —
переходные к металлическим — собственно метал-
лические — полимерные — летучпе. Летучие и поли-
мерные Г. наиболее стабильны для легких элементов,
устойчивость солеобразпых Г. также уменьшается
в группах сверху вниз, а изменение устойчивости
15*
переходных Г. часто носит более сложный характер.
В длинных периодах склонность к образованию со-
единений с водородом постепенно уменьшается (с одно-
временным изменением характера связи от ионной
к металлич.), а затем при переходе к неметаллам снова
увеличивается (с изменением связи от металлич.
к полярной).
Двойные Г. В нек-рых случаях при взаимодей-
ствии Г. образуются двойные Г. При небольшой раз-
нице между характером связи в компонентах обра-
зуются Г. с водородной (ВеН2-2ВН3) илн ковалентной
связью (CH3SiH3). Последние обычно получаются
косвенным путем. Сюда же относятся и органич. про-
изводные простых Г. С увеличением различия компо-
нентов сначала образуются полярные (BH3NHS),
а затем ионные соединения (NH4C1, LiAlH4), при еще
более резкой разнице происходит отщепление водо-
рода: Nall 4 NH3 —> NaNH2 4 В2. Пз двойных Г.
находят практич. применение LiAlH4 (см. Лития
алюмогидрид), LiBH4, NaBH4. Получают ЫВН4 и
NaBH4 действием Г. Li и Nа на эфиры В (ОВ)3 пли дей-
ствием В2Н6 на алкоголяты Lin Na; LiAlH4 — взаимо-
действием LiH и А1С13 в эфире или тетрагпдрофурапе.
Эти Г. применяют для восстановления органич. со-
единений, т. к. они растворяются в органич. раствори-
телях и не вызывают реакций конденсации (отличие
от LiH). Растворимость (в г/100 г при °C) NaBH4:
в NH3 104 (25°), в этилендиамине 22,0 (75°), пиридине
3,1 (25°). Гидрирующее действие боргидридов умень-
шается от Li к К, поэтому можно подобрать восстано-
витель, к-рый будет восстанавливать одни группы,
не затрагивая других. NaBH4, почти не разлагаясь,
растворяется в холодной воде (55 г на 100 г Н2О при
25°). Добавка щелочей повышает, а кислот понижает
устойчивость водного р-ра NaBH4: (ВН4]_ 4 Н+ 4
4 ЗН2О —- В(ОН)3 4 4П2. Лития алюмогпдрид
LiAlH, быстро разлагается водой, средн простых и
двойных Г. он обладает наибольшей гидрирующей
способностью. При действии LiAlH4 на галогениды
образуются либо Г. (в случае галогенидов В, Si, Ge,
Sn, Sb, As), либо алюмогпдриды (Be, Mg, Ga, In, Ti,
Ag и др.). Ковалентные смешанные Г. (типа CH3SiH3)
по своим свойствам близки к соответствующим би-
нарным Г.
Лит.: X е р д Д., Введение в химию гидридов, пер. с англ.,
М„ 1955; Самсонов Г. В., У м а н с к и й Я. С., Твер-
дые соединении тугоплавких металлов, М., 1957; U 1 1 ш а п п,
3 Aufl., Bd 8, Miincli. — В., 1957, S. 714—39; Гейлорд Н.,
Восстановление комплексными гидридами металлов, пер.
с англ., М., 1959; МвхееваВ. И. и Кост М. Е., Усп.
хим., 1960, 29, вып. 1, с. 55. Л. М. 1\оаба.
ГИДРИНДАН (октагпдрнндец) С9Н1в, молеку-
лярный вес 124,22—бициклический углеводород; су-
ществует в виде двух
геометрических изоме- | Н д [ [ Н4 т
ров: цис-(1) и транс-(11у,
для (II) возможны d- и *
1-формы. 1 Ч
Свойства Изомеры
цис- dl-тпранс 1-транс
Т. ПИП., °С/Л1Л4 . . . 160/760 159/760; 62/21 159/760
< 0,8815 (20,7’) 0,8630 (20,2°) 0,863 (20°)
«lie 1,4713 1,4638 —
и-’° nD 1,4629 — —
Теплота сгорания, ккал/молъ ..... 1347,5 1345,7 —
Ниже приведены конформационные структуры
транс- и цис-гидрнндана. Для 1-шранс-изомера (а]р =
= —5,90°. При дегидрогенизации над Pt или Pd при
300—330° Г. образует индан. При нагревании цис-Г.
903
ГИДРОГЕНИЗАЦИЯ ДЕСТРУКТИВНАЯ — ГИДРОГЕНИЗАЦИЯ ЖИРОВ
904
Нонформаиии транс-гидриндана
Нон формации иис~ гидриндана
с А1Вг3 образуется транс-изомер с примесью угле-
водорода С,7Н,,. При кипячении смеси изомеров Г.
с HNO3 (d = 1,22)
получаются 5- и 8-ни-
трогидринданы, а так-
же продукты окисле-
ния и деструкции;
if ис-гидр индан обра-
зуется при гидриро-
вании инданона-2 в
уксусной кислоте с
платиновой чернью;
di-транс- и 1-транс-
изомеры получают вос-
становлением амаль-
гамой цинка в кипя-
щей НС1 транс-гицр-
инданона-2, а также
1-транс - гидриндано-
на-2 соответственно.
Смесь изомеров Г. по-
лучается при гидри-
ровании ипдена или
индана. Г. входит в
состав скелета стерои-
дов, где образует коль-
ца С и Д.
А. Ф. Бочков.
ГИДРОГЕНИЗАЦИЯ ДЕСТРУКТИВНАЯ — про-
цесс деструктивной переработки различных, бедных
водородом, низкосортных топлив — твердых горючих
ископаемых, мазутов, крекинг-остатков, смол и
т. п. — в высококачественные моторные топлива и
масла; процесс проводят в жидкой или паровой фазе
при высоких темп-рах (400—560°), под давлением
водорода (200—700 ат) в присутствии катализаторов.
Г. д. до нек-рой степени может быть отождествлена
с процессом термич. крекинга, проводимого под дав-
лением водорода, и подобно ему представляет совокуп-
ность ряда последовательных и параллельных реакций
с тем, однако, отличием, что характерные для термич.
крекинга процессы полимеризации и конденсации,
приводящие к образованию продуктов уплотнения,
подавляются в присутствии водорода. При Г. д. боль-
шое значение имеет присоединение водорода (гидри-
рования олефинов, ароматич. углеводородов и гетеро-
циклич. соединений) и деструктивное гидрирование,
т. е. реакции расщепления, сопровождающиеся при-
соединением водорода. Наряду с этим при Г. д. про-
текают реакции распада, деполимеризации и изомери-
зации.
Действие водорода в условиях Г. д. на различные
соединения неодинаково. Парафины расщепляются,
а образующиеся из них олефины интенсивно гидри-
руются, образуя парафины меньшего мол. веса; выс-
шие олефины гидрируются лишь частично и поэтому
тяжелые фракции жидкофазной Г. д, содержат значи-
тельное количество непредельных соединений. Аро-
матич. углеводороды при жидкофазной Г. д. гидри-
руются относительно медленно, протекание реакций
конденсации их с олефинами в присутствии водорода
сильно тормозится. Полифенилированные углево-
дороды дают простейшие ароматич. углеводороды,
а у конденсированных ароматич. углеводородов про-
исходит гидрирование крайнего кольца с последую-
щим его раскрытием и образованием менее конденси-
рованных углеводородов. Возникающие при этом
боковые группы отщепляются, давая более низкомо-
лекулярные углеводороды. Шестичленные нафтены
частично изомеризуются в пятичленные, частично
распадаются. Пятичленные нафтены раскрываются
с образованием углеводородов изостроения. Конеч-
ным результатом превращения азотистых, сернистых
и кислородных соединений является отщепление гете-
роатома с образованием, соответственно, аммиака,
сероводорода и воды.
Технологии, схема Г. д. определяется характером сырья
и обычно состоит из трех ступеней. Различие между схемами
переработки твердого и жидкого сырья заключается в первой
ступени, а именно в наличии в первом случае дополнительных
цехов подготовки угольной пасты и переработки шлама (оста-
ток, получаемый при жидкофазной Г. д. угля и состоящий
из золы, катализатора, непрореагировавших частиц угля и
высококипящих масел). Первая, ступень состоит в обогащении
сырья водородом, в результате"получается широкая фракция
с концом кипения выше 320°, состоящая из бензина, среднего
масла и тяжелого остатка. Следующей ступенью является
предварительное гидрирование этой фракции; образуется
бензин, содержащий в основном парафиновые и нафтеновые
углеводороды, обладающий низким октановым числом. 11о-
этому в третьей стадии процесса для повышения качества
полученного бензина проводится его ароматизация (см. Аро-
матизация нефтепродуктов).
При жидкофазной Г. д. применяется подвижный суспен-
дированный катализатор; парофазная Г. д. проводится обычно
со стационарными таблетированными катализаторами или
катализаторами на носителях, к-рые должны обладать не
только расщепляющим, но также и гидрирующим действием.
В качестве катализаторов применяются железная болотная
руда, окись железа, окиси или сульфиды молибдена и воль-
фрама, иногда с добавкой других компонентов, напр. серни-
стого никеля; промоторами служат окиси Са и Ва; носителя-
ми — окись алюминия, иногда активируемая HF, природ-
ные и синтетич. алюмосиликаты. При применении сернистых
катализаторов перерабатываемое сырье иногда дополнительно
осерняется для сохранения активности катализатора.
Из твердых топлив для Г. д. пригодны бурые угли, или
лигниты, с отношением 100 Н/С не ниже бис содержанием
золы ие более 10—13%. При Г. д. смол и нефтяных остатков
получается 70—80% моторного топлива, 18—20% газообраз-
ных углеводородов, 2—7% реакционной воды. Выход бензина
из угля ниже — до 55 %. В том случае, если полное превращение
в бензин нежелательно, Г. д. может применяться для полу-
чения других продуктов, в частности реактивных топлив и
смазочных масел, обладающих большой стабильностью, но
с индексом вязкости значительно худшим, чем у масел соль-
вентной очистки.
Процесс Г. д. усиленно разрабатывался в период
между первой и второй мировыми войнами в Германии
и первоначально применялся для безостаточной пере-
работки углей и тяжелых жидких продуктов в мотор-
ное топливо. В такой форме он не нашел широкого
применения в промышленной практике и, в частности,
в нефтепереработке, т. к. по сравнению с другими про-
цессами метод требует больших капитальных затрат
при низком октановом числе получасхмых бензинов.
После второй мировой войны интерес к Г. д. возрос в
связи с вовлечением в переработку тяжелых смолис-
тых нефтей с bucokhvi содержанием серы.
Лит.: Р апопортИ. Б., Искусственное жидкое топливо,
2 изд., М., 1955; Обрядчиков С. Н., Технология нефти,
ч. 2 3 изд., М., 1952. В. В. Щекин.
ГИДРОГЕНИЗАЦИЯ ЖИРОВ — каталитическое
превращение жидких растительных масел и жиров
морских животных в твердые продукты. Для гидроге-
низации обычно применяют подсолнечное, хлопковое,
соевое и арахисовое масла, в к-рых содержатся в виде
триглицеридов гл. обр. след, жирвые к-ты:
Кислоты Число двойных связей Т. пл., °C Т.заст., °C Иодное число
Линоленовая С37Нй9СООН 3 — И 273,51
Линолевая Ci7H3iCOOH . . 2 — 5 — 181,12
Олеиновая С37Н33СООН . . . 1 14 4 89,93
Пальмитиновая C35H3iCOOH нет 63,1 62,8 0
Стеариновая С17НззСООН . . нет 69,6 69,3 0
Для жиров морских животных, из к-рых наиболее
часто гидрируют китовый жир, характерно наличие
глицеридов высших жирных к-т с четырьмя и пятью
двойными связями. При гидрогенизации происходит
присоединение водорода по двойным связям в непре-
дельных жирных к-тах, а также образование изомеров
этих кислот, имеющих более высокую темп-ру плав-
ления. Отвержденные жиры в зависимостп от степени
насыщения, определяемой по темп-ре плавления гид-
905
ГИДРОГЕНИЗАЦИЯ ЖИРОВ
906
рированного продукта, употребляются в пищу (после
специальной рафинации), а также для технич. целей.
В зависимости от области применения гидрогенизиро-
ванные жиры характеризуются по темп-ре плавления
и твердости (пищевые жиры), по темп-ре застывания
жирных к-т, т. н. титр жира (жиры для мыловарения),
по титру и иодному числу (жиры для стеаринового
произ-ва) и др.
С практически заметной скоростью Г. ж. протекает
только в присутствии катализаторов; в пром-сти при-
меняют никелевые или смешанные медно-никелевые
катализаторы. Г. ж. является селективной, т. е. ско-
рость гидрогенизации различна в зависимости от
числа двойных связей и их положения в гидрируемом
соединении. Так, линолевая к-та (содержащая 2 двой-
ные связи) гидрируется в олеиновую к-ту (содержа-
щую 1 двойную связь) быстрее, нежели олеиновая к-та
в предельную стеариновую к-ту; двойная связь линоле-
новой к-ты в положении 15—16 гидрируется быстрее
двойной связи 12—13; медленнее всех гидрируется
двойная связь 9—10. При гидрогенизации ворваней и
рыбьих жиров сначала насыщаются непредельные
кислоты (С20 и С22) с четырьмя и пятью двойными
связями; при этом не происходит заметного образова-
ния насыщенных к-т. Лишь при иодном числе ок.
84—85, когда остаются непредельные к-ты с меньшим
числом двойных связей, начинают образовываться
пальмитиновая и стеаривовая к-ты. В смеси одинаково
ненасыщенных к-т процесс гидрогенизации предпоч-
тительно проходит у кислот, имеющих меньший мол.
вес. Наряду с преимущественным насыщением лино-
левой к-ты в известный момент начинается насыщение
кислот, содержащих одну двойную связь. Селектив-
ность Г. ж. зависит от природы жира и условий про-
цесса (темц-ра, давление, катализатор и др.) и может
резко меняться при изменении одного из этих фак-
торов.
На селективность влияет также характер процесса.
Так, при непрерывном процессе Г. ж., проходящем
в тонком слое масла на поверхности катализатора,
происходит сильное отклонение от селективности.
Селективность процесса проявляется более отчетливо
при повышенной темп-ре и ослаблении всех факторов,
стимулирующих Г. ж. Наряду с присоединением
водорода при Г. ж. постоянно происходит изомери-
зация непредельных жирных к-т. Изомерные изоолеи-
новые к-ты, образующиеся из линолевой к-ты, имеют
более высокую темп-ру плавления, напр.:
линолевая к-та
СН3(СН.,)4СН=СНСН3СН=СН(СН3)7СООН, т. пл. —5е
олеиновая к-та (9—10)
СН3(СНа)7СН=СН(СН2)7СООН, т. пл. 14°
изоолеиновая к-та (6—7)
СН3(СН3)10СН=СН(СН2)4СООН, т. пл. 30°
изоолеиновая к-та (11 — 12)
СН3(СН2)5СН=СН(СН3)„СООН, т. пл. 39°
Может также происходить изомеризация олеиновой
к-ты (I) (^мс-форма) в эллаидиновую (II) (транс-
форма), обладающую более высокой темп-рой плав-
ления:
СН3(СН,)7СН СН3(СН3)7СН
НООС(СНа)7(!!н Н^(СН,)7СООН
I, т. пл. 14° П, т. пл. 51°
Количество образующихся изоолеиновых к-т за-
висит от состава исходного жира, глубины превра-
щения и условий гидрогенизации.
Наибольшее количество таких кислот содержится в пище-
вых гидрогенизированных жирах с т. пл. 31—34°; по мере
дальнейшей гидрогенизации количество изоолеиновых к-т
снижается. Изоолеиновые к-ты способствуют образованию
смешанных глицеридов с относительно низкой темп-рой плав-
ления, что имеет положительное значение длн пищевых жи-
ров. Напротив, в жирах для произ-ва туалетных мыл наличие
изоолеиновых к-т нежелательно, так как эти кислоты дают
мыла, обладающие малой устойчивостью против размокания.
Таким образом, отверждение жира является ре-
зультатом гидрогенизации непредельных жирных к-т
и их изомеризации. Г. ж. сопровождается также
переэтерификацией, при к-рой происходит обмев ра-
дикалов между молекулами различных триглицери-
дов или в одной и той же молекуле. Получающиеся
продукты отличаются темп-рами плавления, напр.
а-олеодистеарат плавится при 39°, а fj-олеодистеарат—
при 43°.
При Г. ж. происходит ряд побочных процессов.
В результате термич. распада могут образовываться
суободные к-ты, акролеин и кетоны. Акролеин реа-
гирует с водой, образуя гидракриловый альдегид
СН2ОНСН2СНО; последний, при высокой темп-ре
взаимодействуя с водой, дает ацетальдегид, формаль-
дегид, муравьиную к-ту и метанол. В результате
гидролиза жиров получаются свободные жирные
к-ты и глицерин: С3Н5(СООВ)3 -ф- ЗН2О—>ЗВСООН -ф-
-=ф С3Н5(ОН)3. Имеющиеся в водороде СО и СО2
(примеси) в присутствии никелевого катализатора
восстанавливаются до метана и воды; этими реакциями
объясняется образование метана и влаги в водороде,
циркулирующем в системе.
В зависимости от условий гидрогенизации интен-
сивность и направление указанных процессов могут
быть различными, в результате чего получают про-
дукты с различным составом глицеридов и различ-
ными свойствами. Повышение темп-ры способствует
унеличению содержания твердых глицеридов в гид-
рогенизированных жирах; соответственно увеличи-
вается их твердость; имеет значение не только макси-
мальная темп-ра, но также и начальная, при к-рой
в гидрогенизационньш аппарат вводят катализатор.
Состав гидрогенизированного жира изменяется в за-
висимости от продолжительности повышения темп-ры
с момента введения катализатора, т. е. от продолжи-
тельности гидрогенизации в различных интервалах
темп-p. Содержание твердых глицеридов в гидроге-
визированных жирах возрастает с увеличением ко-
личества катализатора. При большом количестве
катализатора даже при низких темп-pax гидрогени-
зации получают жиры с высокой твердостью. Повы-
шение давления в реакционном аппарате ускоряет
процесс; образуются продукты, к-рые при одной
и той же степени насыщения имеют более высокую
темп-ру плавления. При одной и той же темп-ре плав-
ления жиры, гидрогенизированные под давлением,
содержат меньше твердых глицеридов и имеют мень-
шую твердость, чем гидрогенизировавные при атм.
давлении. Влияние отдельных параметров процесса
для различных растительных масел оказывается не-
одинаковым. При гидрогенизации хлопкового масла
влияние темп-ры и рода катализатора сказывается
значительно меньше, чем при гидрогенизации под-
солнечного и особенно арахисового масла.
Гидрогенизированный жир в СССР называют с а-
ломасом. В зависимости от области применения
и исходного сырья изменяются и требования к сало-
масу. Напр., саломас, применяемый в произ-ве мар-
гарина, должен иметь т. пл. 31—36°, иодное число
60—80, кислотное число не более 2—4; технич. са-
ломас имеет т. пл. жирных кислот 40—48°, иодное
число 55—65, кислотное число 5—7. При Г. ж. на-
ряду с увеличением веса вследствие присоединения
водорода происходит потеря в весе из-за термич.
разложения и гидролиза жира. В итоге вес жира
увеличивается на 0,17—0,22% при получении сало-
маса, т. пл. 32—34°, и на 0,22—0,31% при гидро-
генизации до т. пл. 41—48°.
Техника гидрогенизации жиров. Масла
перед гидрогенизацией тщательно очищают. Катализатор
должен быть свободен от соединений Pb, S и As, водород —
от H2S. Все эти соединения являются каталитич. н.чами. Мыла
и различные загрнзнепин обволакивают катализатор и тем
907
ГИДРОГЕНИЗАЦИЯ И ДЕГИДРОГЕНИЗАЦИЯ КАТАЛИТИЧЕСКИЕ
90S
снижают его активность. Лучше пользоваться электролитич.
водородом, содержащим 99,6—99,8% Н2. В пром-сти приме-
няют тоикодисперсные никелевые катализаторы, получаемые:
1) из карбоната никеля на носителе (или без носителя)
в соотношении 3:1, восстанавливаемых в масле при 240—250°;
можно также подавать компоненты катализатора в автоклав
в виде масляной суспензии' и восстанавливать их в процессе
гидрогенизации; 2) из формиата никеля на носителе, разло-
женного в масле под вакуумом при 240—260°; 3) пз карбоната
никеля, осажденного на носителе (кизельгур и др.), путем
его разложения и восстановления в сухом виде водородом
при 450°.
На большинстве заводов Г. ж. проводят в вертикальном
автоклаве из кислотоупорной или плакированной стали со
сферич. дном и крышкой. Автоклав снабжен змеевиками для
подогрева содержимого паром повышенного давления. Peai%
ция Г. ж. экзотермична, напр. теплота гидрогенизации олеи-
новой к-ты равна 38,4 ккал/моль; поэтому в аппарате имеются
также змеевики для охлаждения; через последние циркули-
рует холодное масло. Перемешивание реакционной массы
производят мешалкой турбинного типа. Через барботер,
расположенный ниже мешалки, в автоклав вводят водород
в количестве примерно в 2—4 раза больше теоретически
необходимого. Промышленное распространение получил ме-
тод гидрогенизации с катализатором, суспендированным
в жире, через к-рый продувают водород. Г. ж. обычно про-
изводятся под давлением, не превышающим 3 ат при темп-ре
в пределе 160—240°. Непрерывный процесс Г. ж. ведут в ба-
тарее, состоящей из 2 или 3 автоклавов, соединенных после-
довательно при помощи переливных труб, благодаря чему
гидрогенизируемый жир поступает непрерывно, или специаль-
ных газлифтов, действующих водородом.
Лит..- Е л о в и ч С. Ю., Ж а б р о в а Г. М., Теоретиче-
ские основы гидрирования жиров, М.—Л., 1948; Тютюн-
н и к о в Б. Н., Ю х н о в с к и й Г. Л., М а р к м а н А. Л.,'
Технология переработки жиров, М., 1950; Зиновьев А. А.,
Химия жиров, М., 1952: Венгерова Н. В., Производство
высококачественных гидрогенизированных жиров для пище-
вых и технических целей, в сб.: Пути улучшения качества
и расширения ассортимента продукции масложировой про-
мышленности, Л., 1959; Hflditch Т. Р., The chemical
constitution of natural fats, 3 ed., L., 1956; W i 11 k a F., Die
modernen Methoden zur Umformung der Fette, Lpz., 1958.
ГИДРОГЕНИЗАЦИЯ И ДЕГИДРОГЕНИЗАЦИЯ
КАТАЛИТИЧЕСКИЕ — присоединение водорода к
элементам или соединениям (гидрогенизация — Г.)
п отщепление водорода от соединений (дегидрогени-
зация — Д.), протекающие под влиянием катализа-
торов. Г. и д. к. особенно широко применяются в ор-
ганич. химии и относятся к классу окислительно-
восстановительных процессов, сопровождающихся пе-
реходом электронов (см. Катализ). Г. и д. к. связаны
динамич. равновесием, положение к-рого опреде-
ляется темп-рнымп условиями и давлением водорода;
па одном и том же катализаторе Г. идет при низких,
а Д. при высоких темп-рах; повышение давления бла-
гоприятствует Г. и тормозит Д.
Синонимами терминов Г. и Д. соответственно яв-
ляются термины гидрирование и дегидрирование.
Практически Г. чаще ведут на металлах, а высоко-
температурные реакции Д. — на окисных катали-
заторах, т. к. последние более термостойки и труднее
спекаются, чем металлы. Г. и д. к. можно проводить
на простых и на смешанных катализаторах. К простым
относятся металлы: Fe, Ni, Со, Pt, Pd, Os, Ru и неко-
торые др., имеющие гранецентрированную кубическую
или плотно упакованную гексагональную решетку,
и окислы металлов: ZnO, NiO, СоО, Сг2О3, МоО2,
PtO2 (см. Адамса катализатор) и др. К числу сме-
шанных катализаторов относятся, напр., Pt-— уголь,
Pt—AI2O3, Ni — кизельгур или высокодисперспый
никель, нанесенный на окись алюмивия (ката л и-
затор Н. Д. Зелинского). Применяют
также смеси окисей, иногда многокомпонентные,
напр. окиси Сг, AI, К, Си—Сг2О3, Ni—Сг2О3 или
сульфиды NiS, Ni3S2, MoS2, WS2 и их смеси,
алюмохромовые, алюмованадие.вые и др. катализа-
торы, к-рые обычно находятся в высокодисперсном
состоянии. Эти катализаторы получают методами сов-
местного осаждения и методами пропитки. Особую
группу представляют т. н. скелетные или сплавные
катализаторы, получаемые сплавлением нек-рых ме-
таллов (напр., Си, Ni, Со) с алюминием или кремвием
и растворением последних в щелочи; скелетные ка-
тализаторы обладают высокой дисперсностью, ста-
билизируемой адсорбированным водородом, образо-
вавшимся при растворении А] или Si в щелочи.
Г. и д. к. проводят в жидкой и паровой фазах,
однако в связи с тем, что Д. идет при более высоких
темп-рах, ее осуществляют преим. в паровой фазе.
Необходимой стадией Г. и д. к. является адсорбция
водорода на поверхности катализатора. В условиях
Г. молекулярный водород активируется, т. к. силы,
связывающие его с поверхностью катализатора, раз-
рыхляют связь Н—Н (иногда до образования про-
тона Н+, адсорбированного на поверхности); анало-
гичной деформации подвергается и гидрируемая мо-
лекула. Адсорбированный водород, гидрируемая мо-
лекула и поверхностные атомы катализатора обра-
зуют активированный комплекс (см. Переходное со-
стояние), превращающийся в продукты реакции, де-
сорбирующиеся с поверхности катализатора. При Д.
реагирующая молекула непосредственно образует-
активированный комплекс с катализатором, распа-
дающийся на продукт Д. и водород, затем десорби-
руемые с поверхности. Т. о. при Г. и Д. строение-
активированного комплекса при одинаковом угле-
родном скелете молекул веществ, подвергаемых этим
реакциям, одно и то же, что и обусловливает часто-
наблюдаемую их обратимость. Обозначая через К
атомы поверхности катализатора и точечным пункти-
ром связь с ними атомов реагирующих молекул, ак-
тивированный комплекс и его распад можно схема-
тически изобразить след, образом;
К К
rch2-ch2r
к к
„ к к
Rx' A
,с-с< —
Г"! н
rt-н
к к
к к
R CH=CHR
К Н2 к
где пунктир из черточек показывает связи, разрых-
ляющиеся при образовании комплекса пли образую-
щиеся при его распаде (Г. — гидрогенизация, Д. —
дегидрогенизация). В нек-рых случаях механизм Г.
может несколько отличаться от описанного; при из-
вестных условиях может активироваться, образуя
активированный комплекс, только Н2 или только
гидрируемая молекула, а второй компонент реаги-
рует с активированным комплексом из жидкой или
паровой фазы. Это имеет место- в случае гомогенно-
гетерогенвых реакций.
Металлич. катализаторы, в том числе и смешанные,
весьма чувствительны к ядам каталитическим, в осо-
бенности к ядам, содержащим серу. Предложен
метод «детоксикации» ядов и отравленных катализа-
торов переводом сульфидов (и др. соединений), со-
держащих атомы со свободной парой электронов,
обусловливающих отравление, в не содержащие этой
пары электронов «экранированные» структуры мето-
дами окисления, восстановления или комплексооб-
разования. Наилучшим методом борьбы с отравлением
катализаторов является тщательная очистка веществ,
подвергаемых Г. или Д. от примесей, могущих ока-
зать вредное влияние. Практич. ценность сульфидных
и многих окисных катализаторов определяется их
стойкостью по отношению к серусодержащим ядам,
что особенно важно в процессах переработки сер-
нистых нефтей и нефтехимия, синтеза на их основе.
909
ГИДРОГЕНИЗАЦИЯ И ДЕГИДРОГЕНИЗАЦИЯ КАТАЛИТИЧЕСКИЕ
910
Начало быстрого развития Г. было положено в 1897—98
многочисленными работами научных школ П. Сабатье и
Н. Д. Зелинсного, к-рыми были открыты катализаторы Г. и
изучена Г. различных органич. соединений в паровой фазе.
В 1902 С. А. Фокин и др. распространили метод Г. иа реакции
в жидкой фазе, что дало возможность Г. растительных масел.
В 1904 В. Н. Ипатьев осуществил Г. под высоким давлением
водорода. На основе дальнейшего развития те.х-никп Г. были
разработаны промышленные методы синтеза аммиака (Ф. Га-
бер, 1913) метилового спирта (Г. Патар, 1923), Г. угля (Ф. Бер-
гнус, 1913). В 1911 — 12 в практику Г. в жидкой фазе были
введены катализаторы — коллоидные металлы (Пааль, Скита).
Важнейшие закономерности избирательной Г. смесей насы-
щенных соединений или полиенов установил в 1924 С. В. Ле-
бедев, показавший, что в зависимости от строения ненасыщен-
ных молекул Г. происходит ступенчато, затрагивая в онре-
Sелейной последовательности в-ва, находящиеся в смеси.
[озднее А. А. Баландин предложил методы расчета последо-
вательности гидрирования полнненасыщспных соединений
и смесей ненасыщенных соединений, содержащих различные
типы связей (С=С, С=О, С-С и др.). Он же предложил одну
из теорий Г. органич. соединений и кинетич. ур-иие, охваты-
вающее все известные типы Г.
Различают 3 группы реакций Г.: Восстанов-
ление с одновременным п р в с о-
е д в не н ие м водорода, напр. СО 4- ЗН2 —>
->СН4-|- Н2О; 2NO+ 5H2->2NH3 + 2Н2О. II ри-
соед мнение водорода к элемен-
там пли ненасыщенным соедине-
ния Л1, напр. N2 + ЗН2 2NH3:
.сн-ськ _________ ,СН2-СН2Х
нс сн+зн2г^^н2с; сн2
чсн=сн/ сн2-CH2Z
Все реакции этого типа служат примером обратимых
реакций Г. и д. к.; так на Ni или Pt последняя реак-
ция протекает при 120—160° в сторону Г., а при
-300—320° равновесие сдвинуто п противоположную
сторону. Присоединение водорода с
раскрытием цикла, напр.
СН2-СН2Ч
^Н2_СН2/сн2+н2 —СН3(СН2)3СН3
Такие реакции принято называть гидрогено-
лизом, а при расщеплении больших молекул на
меньшие — деструктивной Г. Последние реакции пред-
ставляют типичный пример необратимых реакций Г.
В последнее время гидрогенолиз и деструктипная
гидрогенизация используются для получения много-
атомных спиртов из целлюлозы и для получения гли-
церина и гликолей из высших многоатомных спиртов.
В связи с обратимостью Г. для каждого соединения
•существует довольно узкая область темгг-р, в пределах
к-рой происходит присоединение водорода, поэтому
для каждого случая необходимо выбрать наиболее
выгодные условия (обычно при темгг-ре не выше 300°,
а с металлами при 120—160°); в ряде случаев гидро-
генизацию проводят под давлением водорода до
1000 ат, что увеличивает скорость реакции и смещает
равновесие в пужиую сторону.
Важнейшие области промышленного применения Г.:
1) Синтез аммиака, к-рый ведут обычно на плавленых
железных катализаторах, промотироваиных А12О3
а К2О при 400—500° и 100—1000 ат. 2) Синтез ме-
танола из СО и Н2 — осуществляется чаще всего на
окисных цинк-хромовых катализаторах при 370—400°
и 250—300 ат. 3) Г. растительных масел, гл. обр.
на никелевых катализаторах, получаемых из карбо-
ната никеля, выщелачиванием сплава Ni — Al или
др. способами. В зависимости от способа получения
катализатора процесс ведут при 150—250°. 4) Дест-
руктивная Г. угля, при к-рой уголь смешивают с дис-
пергированным в масле железом или никелем и обра-
батывают водородом при 400—450° и 200 ат; гидри-
рование ведут и присутстиии катализаторов MoS2,
WS2 или WS2—NiS—А12О3. Г. приобретает все боль-
шее значение в нефтепереработке (напр., процесс
гидроочистки) с целью получения высококачественных
моторных топлив. Г. веществ в жидкой фазе осущест-
вляется также путем встряхивания с платииопой или
палладиевой чернью, в атмосфере водорода (В и л ь-
штеттера метод гидрогенизации).
Этот метод применяется для соединений, содержащих
двойные, тройные, карбонильные связи, для арома-
тич. нитро- и гетероциклич. соединений, терпенов,
стеринов и др. Для нек-рых реакций, напр. Г. фта-
левого ангидрида, необходимо насыщение платиновой
черни кислородом.
Дегидрогенизация особенно широко при-
меняется в химии углеводородов, кислородсодер-
жащих и азотистых органич. соединений. Большое
значение в развитии Д. имели работы М. Бертло (Д.
спирта), П. Сабатье и Ж. Сандерена (Д. циклогек-
сана на никеле с образованием бензола и метана)
и особенно работы В. Д. Зелинского и его школы по
каталитич. Д. циклогексановых углеводородов без
побочных реакций, к-рые охватили также Д. разнооб-
разных гидроароматпч. соединений и были распрост-
ранены иа азотистые гетероциклич. соединения —
пиперидин, пироллин и их гомологи. Работами
Б. А. Казанского и А. Ф. Плате с платиновыми ка-
тализаторами, Б. Л. Молдавского и Г. Д. Камушера
(на окиснохромовом катализаторе) была открыта
возможность Д. нормальных насыщенных углево-
дородов с образованием ароматич. углеводородов
на окисных катализаторах (реакция дегидроцикли-
зации парафинов); процесс идет при 500—550°.
В 1931 Лебедев осуществил одновременную дегидра-
тацию и дегидрогенизацию этилового спирта на слож-
ных катализаторах и разработал способ промышлен-
ного получения бутадиена для произ-ва синтетич.
каучука. Позднее (1941) путем использования алюмо-
хромовых катализаторов была решена проблема Д.
нормальных парафинов в олефины, а последних —
в диолефины на' многокомпонентных катализаторах,
содержащих Сг2О3, что позволило получить исходные
материалы, необходимые для получении каучука —
бутадиен и изопрен — из природных газов и нефтя-
ного сырья. Эти открытия, помимо большого практич.
значения, расширили представления о реакционной
способности насыщенных углеводородов.
На металлах (до 300°) дегидрируются хорошо
только 6-членные нафтены, а на окисях (при 450—
600°) также и остальные нафтены и парафины. Шести-
члепные нафтены, в том числе и многоядерные, пре-
вращаются в ароматич. углеводороды:
На окисных катализаторах нафтены дегидрируютсн
с образованием циклоолефинон, напр.:
Т. к. эти реакции протекают при высокой темп-ре,
они часто сопровождаются пиролизом углеводородов.
Разработаны эффективные катализаторы Д. боковых
цепей ароматич. углеводородов, напр. для получения
стирола из этилбензола, дивинилбензола из диэтил-
бензола и т. в. Д. спиртов идет на металлах при до-
вольно низкой (120—130°) темп-ре, а на окисях часто
сопровождается параллельвой реакцией дегидратации
с образованием олефивои. При Д. первичных и вто-
911
ГИДРОДИМЕРИЗАЦИЯ — ГИДРОКСАМОВЫЕ КИСЛОТЫ
912
ричных спиртов получаются соответственно альде-
гиды и кетовы.
Дегидрогенизация в организ-
мах — одна из фаз процесса биологич. окисления,
совершается при участии биокатализаторов — окис-
лительных ферментов — дегидраз. Окисляемое ве-
щество (донор водорода) отдает свой водород окисли-
телю (акцептор водорода), способному присоединять
водород. Д. подвергаются промежуточные продукты
расщепления сахаров, аминокислоты и нек-рые др.
трудно окисляемые вне организма вещества.
'Лит.: Сабатье П., Катализ в органической химии,
иер. с нем., Л., 1932; Лебедеве. В., Жизнь и труды, Л.,
1938; Пл атэ А. Ф., Каталитическая ароматизация пара-
финовых углеводородов, М.—Л., 1948; Е л о в и ч С. Ю.
и Жаброва Г. М., Теоретические основы гидрирования
жиров, М.—Л., 1948; Беркман С., Моррелл Д.
и Э г л о ф ф Г., Катализ в неорганической и органической
химии, пер. с англ., т. 1—2, М.—Л., 1949; Катализ. Вопросы
теории и методы исследования, пер. с англ., М., 1955; Ката-
лиз. Катализаторы органических реакций, пер. с англ., М.,
1955; Зелинский Н. Д., Собрание трудов, т. 3, М., 1955;
В а л а н д и н А. А., Уч. зап. МГУ, 1956, вып. 175, с. 97;
Любарский Г. Д., Усп. хим. 1958, 27 с. 316; Д о л-
г о в Б. Н., Катализ в органической химии, 2 изд., Л., 1959;
Катализ. Некоторые вопросы теории и технологии органиче-
ских реакций, пер. с англ., М., 1959; Advances in catalysis and
related subjects, v. 9 N. Y., 1957. A. M. Рубинштейн.
ГИДРОДИМЕРИЗАЦИЯ — соединение двух ра-
дикалов, образующихся при восстановлении нена-
сыщенных соединений, напр.
2 С-с" + Н- — НС- С- С —СН
/ \ ' 111
Г. производят фотосенсибилированным гидрированием
в присутствии ртути либо, в случае фенилированных
этиленов, действием амальгамы алюминия во влажном
эфире, металлич. натрия, натрий-калиевого сплава
или р-ра натрия или калия в жидком аммиаке:
Н- + Hg — HgH' + н- — Hg + 2Н-
Н- + С-Н4 — С2Н5- — СН3СН_СН,СН3 (94%)
(CGH5)-C (C0H;)-CNa
II + 2Na — | —
СН- CHj
(С0Н5)-СН CH(CGH5)2
СН---- сн-
Гораздо больше распространена Г. производных
ненасыщенных углеводородов, гл. обр. карбонильных
соединений и производных кислот. Насыщенные
альдегиды и кетоны образуют при этом пинаконы:
R1 R'
Н’ I I
R-CO-R’ —2- R-C(OH)-C(OH)-R
Восстановление производят обычно амальгамами маг-
ния, алюминия, натрия, бинарными смесями, а также
электровосстановлением на ртутном, свинцовом, мед-
ном, никелевом, платиновом и других катодах. При
восстановлении в тех же условиях а,р-ненасыщенных
альдегидов и кетонов промежуточно образуется ра-
дикал с двумя ненасыщенными центрами (I), далее
димеризующийся либо в ненасыщенный пинакон (II),
либо в насыщенное 1,6-дикарбонильное соединение
(III) или производное фурана (IV):
RCH=CH-COR' Н » RCH-CH-iR'(OH)
I
R—СН=СН-C(OH)R’
R—CH = CH-C(OH)R'
//
r-(j:h-ch2cor*
R—CH - CH2COR'
III
RCH-CH RCH-CH2
R-CH=CH-C C(OH)R' R~CH=CH—C C(OH)R’
R'/4OH R,/4OZ
IV
Гидромеры образуются также при действии на
карбонильные соединения реактива Гриньнра в при-
сутствии незначительных количеств хлористого ко-
бальта, напр.:
С6Н5-СН=СН-СОС0Н5 + CH3MgBr —
С0Н5-СН-СН2-СОС6Н5
-* I
С,,Н5-СН-СН--СОСвН5
к-т и их производных про-
больше в них сопряжение,
радикалов, также как и во
Г. а,|3-ненасыщенных
исходит тем легче, чем
причем соединение двух
всех других случаях соединений с электрофильными
заместителями, происходит в |3-положении по отно-
шению к заместителю:
e + C±CJ-C-X + H
I I z0H
c-c=cx
X-CO-CH-C-C-CH-CO-X
I I
Осуществлена также Г. акрилонитрила; при этом
образуется динитрил адипиновой к-ты (с выходом
до 80% от теории). Г., особенно в приложении к произ-
водным акриловой кислоты, имеет большое практич.
значение, так как открывает новые пути для синтеза
технически важных производных адипиновой к-ты.
Лит.: Кнунянц И. Л., Гамбарян Н. П., Усп.
хим., 1954, 23, с. 781; Кнунянц И. Л., В я з а н к и н Н. С.,
ДАН СССР, 1957, ИЗ, № 1, 112. И. П. Гамбарян.
ГИДРОКОРИЧНЫЙ АЛЬДЕГИД (0-фенилпро-
пионовый альдегид) СвН5СН2СН2СНО, мол. в. 134,17—
бесцветная жидкость с запахом, напоминающим за-
пах жасмина и сирени; т. кип. 221—224°/744 мм;
104—105°/13 мм; 1,018; njj 1,525; не растворяется
в воде, в спирте растворяется в отношении 1 : 16,7.
Г. а. вступает в реакции, характерные для альдегид-
ной группы; образует оксим, т. пл. 93—94°, семикар-
базон, т. пл. 128°. Количественно определяется окси-
мированием. Получают Г. а. гидрированием корич-
ного альдегида над катализатором Ni (по Ренею)
в спиртовой среде при слабом нагревании, хорошем
перемешивании и пропускании количества водорода,
---R-------- ------- ---- ------------- двойной
а. в пар-
необходимого только для гидрирования
связи коричного альдегида. Применяют Г.
фюмерии, а также для отдушки мыла.
ГИДРОКОРИЧНЫЙ СПИРТ (Р-фенил^Гпропи-
ловый спирт, З-фенилпропанол-1, у-оксипропилбен-
зол) СвН5СН2СН2СН2ОН, мол. в. 126,20 — бесцветная
жидкость со слабым запахом гиацинта и розы; т. пл.
ниже —18°; т. кип. 237,4°/760 мм; 119°/12 мм;
1,0075; ng 1,5356; плохо растворим в воде, смеши-
вается с органич. растворителями. Г. с. в природе
встречается в виде эфира коричной к-ты в нек-рых
смолах и бальзамах. Г. с. может быть окислен в гид-
рокоричный альдегид СвН5СН2СН2СНО и гидрокорич-
ную к-ту СвН5СН2СН2СООН; легко дает сложные
эфиры; образует кристаллич. производные: н-нитро-
фенилуретан, т. пл. 104°; 3,5-динитробензоат, т. пл.
92°; n-нитробензоат, т. пл. 46—47,5°; фенилуретан,
т. пл. 47°; n-нитрофенилгидразон ацетоацетата гидро-
коричного спирта, т. пл. 92—93,5°. В пром-сти Г. с.
получают гидрированием коричного альдегида или
4-фенилдиоксана-1,3. Содержание Г. с. определяют
ацетилированием в среде пиридина на холоду и дру-
гими методами определения первичных спиртов.
Применяется Г. с. в парфюмерии. в. Н. Просева.
ГИДРОКСАМОВЫЕ КИСЛОТЫ — органич. со-
//°
единения общей формулы RC—NHOH, известны так-
913
ГИДРОКСИЛАМИН — ГИДРОКСИЛАМИН СОЛЯНОКИСЛЫЙ
914
/ОН
же в таутомерной форме RC=NOH (гидроксимовые
кислоты). Г. к. — кристаллич. продукты, многие из
них разлагаются со взрывом при нагревании. В рас-
творах Г. к. алифатич. ряда имеют нейтральную или
очень слабокислую реакцию (напр., для ацетгидро-
ксамовой к-ты А=3 • 10-8); ароматич. ряда — кислую
реакцию. В щелочной среде или под действием тио-
нилхлорида Г. к. претерпевают перегруппировку
Лоссеня:
RC-NHOH-----.R—N=C=O^-^— RNH» + СО»
-Н2О
при этом образуются эфиры изоциановой к-ты и за-
тем амины. При действии на Г. к. HNO2 получа-
ются соответствующие карбоновые к-ты; с основа-
ниями они образуют внутрикомплексные соли, из
к-рых наиболее характерны зеленые малораствори-
мые соли меди и ярко-красные соли трехвалентного
железа; окраска исчезает после добавления конц.
НС1 или H2SO4 и снова появляется при разбавле-
нии водой. Для качественного и колич. определе-
ния Г. к. применяют реакции образования их солей.
Г. к. обычно получают действием эфиров, ангид-
ридов, хлорангидридов и амидов карбоновых к-т на
гидроксиламин. Г. к. также можно получить окисле-
нием к-той Каро первичных и вторичных аминов,
действием 95%-ной H2SO4 на нитроуглеводороды.
При действии нитрогидроксиламина или N-бензол-
сульфогидроксиламина на альдегиды получаются Г. к.
(Анджели — Римини реакция}. Эта реакция исполь-
зуется для определения небольших количеств аль-
дегидов. Г. к. применяют как аналитич. реагент для
качественного и колич. определения ряда ионов ме-
таллов. Т. И. Сьрки-иа.
ГИДРОКСИЛАМИН NH2OH — продукт замеще-
ния группой ОН одного атома водорода в молекуле
NH3; бесцветные чешуйчатые или игольчатые крис-
таллы; плотн. 1,216, т. пл. 33—34°, т. кип. 58°/22 мм.
Межатомные расстояния и углы между связями в мо-
лекуле Г.: N—О 1,46 А, О—Н 0,96 A, N—Н 1,01 А,
Z.HON 103°, Z.HNO 105°,oZHNH 107°. Стандартная
теплота образования ДНа08 = — 25,5 ккал]молъ. Г.
легко разлагается, в особенности при нагревании,
на Na, NH3, Н2О и HNO2. Выше 100° разложение про-
текает со взрывом. Г. весьма гигроскопичен, с водой
смешивается во всех отношениях, легко растворим
в метиловом и этиловом спирте, нерастворим в выс-
ших спиртах, бензоле.
Благодаря наличию у атома азота в NH2OH одной
неподеленной пары электронов для Г., как и для
аммиака, характерны реакции присоединения. Так,
растворяясь в воде, Г. образует с ней гидрат
NH2OH • Н2О — весьма слабое основание (константа
диссоциации К = 1,7 • 10 8). С кислотами Г. обра-
зует соли гидроксиламмония. В качестве адденда Г.,
подобно аммиаку, входит в комплексные соединения
[Mg(NH2OH)2]Cl2 • 2НгО, [Co(NH2OH)e]Cl3 и др. В вод-
ных р-рах Г. и его соли ведут себя преим. как сильные
восстановители, окисляясь до N2 или N2O, но в
нек-рых случаях (напр., при взаимодействии с Мп11,
Ti111, HJ, H2S, Fe11)— как окислители, превращаясь
в NH3. Наиболее важным свойством Г. и его солей
является способность реагировать с альдегидами и
кетонами, давая оксимы (соответственно альдокси-
мы и кетоксимы), напр.,
R-CH=O + HjNOH — It -CH NOH + H.O
CH3. CH3.
\c=o + H.,NOH — )C=NOH + H,0
CH»/ ' CH3/
Г. образует разнообразные органич. производные
путем замещения в его молекуле атомов водорода ор-
ганич. радикалами.
Неорганические соли Г. Поскольку
Г. — значительно более слабое основание, чем
NH4OH, соли его обычно менее устойчивы, чем соот-
ветствующие соли аммония, и более гидролизованы
в водном р-ре. Относительно устойчивы соли Г.,
производные от сильных кислот, такие, как хлорид
и сульфат; темп-ры их разложения лежат ок. 150—
180°. Фосфат Г. уже значительно менее устойчив,
разлагается при 135° и 10—15 мм рпг. ст. Нитрит
Г. не получен — в водных р-рах он быстро разла-
гается: (NH3OH)C1 + NaNO2—>N2O + NaCl + 2H2O
(эта реакция используется для получения N2O).
Важнейшими солями Г., используемыми в лабора-
тории и пром-сти, являются хлорид (NH3OH)C1, суль-
фат (NH3OH)2SO4 и кислый сульфат (NH3OH)HSO4.
Нек-рые их свойства приведены в таблице.
Свойства Хлорид Сульфат Кислый сульфат
Молекулярный вес Т. пл., °C Растворимость, г/100 мл растворителя при 25°: в воде в метаноле в этаноле (95%) 69,5 152 (с разл.) 94,4 13,8 8,5 164,1 170 (с разл.) 63,7 нераство- рим нераство- рим 131,1 57 ок. 100 слабо растворим слабо растворим
В пром-сти соли Г. обычно получают восстановле-
нием нитрата натрия бисульфитом натрия и сернистым
газом в растворе:
NaNO» + NaHSO3 + SO» — HON(SO3Na)»
2HON(SO3Na)3 4- 4H.0 — (NH3OH)3SO4 + Na3SO4 + 2NaHSO3
В пром-сти приобретает все большее значение полу-
чение солей Г. непосредственным восстановлением
NO водородом с использованием в качестве катали-
затора платины, суспендированной в разб. кислоте;
этим способом получают довольно концентрированные
и чистые р-ры солей гидроксиламмония. Сам Г. по-
лучают отгонкой его при пониженном давлении (ок.
13 мм рт. ст.} из смеси солей гидроксиламмония
с СаО или NaOH.
Г. и его соли применяют в аналитич. химии для
очистки высших альдегидов и кетонов (через оксимы),
для получения оксимов — таких, как диметмлдиоксим
и циклогексаноноксим. Последний является проме-
жуточным продуктом в произ-ве капролактама, при-
меняемого для получения капрона. Г. и его произ-
водные ядовиты.
Лит.: Gmelin, 8 Aufl., Syst. Nr. 4, В., 1935, S. 857;
U 1 1 in a n n, 3 Aufl., Bd 8, Miinchen — B., 1957, S. 743—45;
Kirk, v. 7, N. Y., 1951, p. 764—70.
ГИДРОКСИЛАМИН СОЛЯНОКИСЛЫЙ, мол.
вес 69,50, (NH3OH)C1 — бесцветные игольчатые кри-
сталлы; плотность 1,67; другие свойства см. в ст.
Гидроксиламин; сильный восстановитель, окисляется
до N2O; ядовит, раздражающе действует на кожу.
Получают Г. с. действием HCI (d 1,18) на ацетопоксим,
а также электровосстановлением HNO3. Г. с. приме-
няют в качестве восстановителя в случаях неорга-
нич. анализа, когда нежелательно введение в раствор
сульфатов; для титриметрич. определения формаль-
дегида, фурфурола, камфоры, глюкозы; в электро-
анализе как деполяризатор; в органич. анализе для
выделения и идентификации альдегидов и кетонов;
для синтеза оксимов; в фотографии, в медицине; как
микрохимии, реактив на сульфокислоты. а-Дикетоны
реагируют с Г. с. с образованием моно- или диокси-
мов. Хиноны восстанавливаются Г. с. в щелочной
915
ГИДРОКСИЛАМИНА ПРОИЗВОДНЫЕ — ГИДРОКСОНИЙ
916
среде до соответствующих фенолов; в кислой среде,
однако, хиноны с Г. с. реагируют как кетоны, обра-
зуя моно- или диокспмы. При взаимодействии слож-
ных эфиров с Г. с. получаются гидроксамовые кислоты
уОН
В—C=NOH, к-рые с FeCl3 дают красное окрапш-
ванне. И. П. Ефимов.
ГИДРОКСИЛАМИНА ПРОИЗВОДНЫЕ — азоти-
стые соединения, содержащие органич. радикалы
вместо водорода в NH2OH при атоме кислорода
(ВС)—NII2) или атоме азота (RNH— ОН, R2N—ОН).
В табл, приведены физич. свойства нек-рых алкил-
п арилгидроксилампнов; для сравнения приведены
данные о гидроксиламине.
Соединение Формула T. пл., °C T. кип., °C/760 мм Теплота испарения при 760 .иле, ккал/моль Кажу- щаяся Р&а Т. пл. хлор- гидра- та, °C добавок к паяльным сплавам. Из- производных N-арилгидроксил- аминов важен купферон — ре- актив на Сн2+, Cd2+ и др. Ал- кил(арил)-гидроксиламины ядо- виты; О-произвоцвые являются ферментными ядами, дезактиви- рующими каталазу; N-производ- ные, попадая в кровь, превраща- ют гемоглобин в метгемоглобин, вызывая кислородное голодание. Лит.: Chemistry ol carbon com- pounds. Ed. by E. H. Rodd, v. 3, pt A, AnHt. [a, o.l, 1954. Ф. К. Величко.
Гидроксилампн . а-Метилгидроксиламин . . . 0-Метилгидроксиламин . . . аЩ-Диметилгидроксиламин Э ,0-Диметилгидроксмламин а,В.З-Триметилгидроксил- ачин Р-Фенилгидроксиламин . . . nh2oh H«NOCH3 CH3NHOH CH,NHOCH, (CH3)2 NOH (CH,),NOCH, C(H5NHOH 33-34 —86,4 38,5 —97 17,6 -97,2 82 58/22 48,1 115 42,3 100,6 30 7,71 11.88 7,44 9,67 6,41 6.0 4,6 6,0 4,8 5,2 3,7 152 149 88—90 115-116 102 123
Все алкил(арил)гидроксилампны являются осно-
ваниями, сила к-рых зависит от характера и поло-
жения заместителя (см. таблицу). Они образуют
с кислотами кристаллич. нелетучие соли, растворимые
в воде, используемые для их идентификации. N-Mo-
иоалкил(арил)гидроксиламины энергично восста-
навливают на холоду фелингову жидкость и аммиач-
ный р-р окиси серебра. Они ацилируются по атому
азота: RNH—ОН + СН3С0С1 -> RN(COCH3)OH +
-I- НС1. К-Диалкил(арил)гидроксиламины-энергичные
восстановители; эпилируются по атому кислорода:
RjN-OH + СН3СОС! -> R2N-OCOCH3 + НС1
О-Производные не восстанавливают на холоду фе-
лингову жидкость. Смешанные Г. п. восстановитель-
ными свойствами не обладают. Как правило, диза-
мощенпые гидроксиламипы стабильнее монозамещен-
ных, арилпроизводные устойчивее алкплпроизводных.
Все N-производные восстанавливаются в амины:
R2N—ОН S R2J\'H -F Н2О, с изоцианатами образуют
замещенные мочевины:
R'N’CO + RNH — ОН —. R'NHCONHR
N-Арилгпдроксилампны в водном р-ре превращаются
в нитрозо- и азоксисоединения:
CeH5NH—ОН C3H5NO geHsNHOH C3H5N=N(O)C(,H3
Под влиянием конц. H2SO4 N-арилгидроксиламины
со свободным пара-положением в ядре перегруппи-
ровываются в парааминофенолы. Эта реакция ис-
пользуется в произ-ве последних. С альдегидами
М-арилгидроксилампны дают нитроны:
ArNHOH + СвН5СНО — ArN(O)=CHC6H3 + Н2О
Наиболее удобный способ синтеза О-алкил(арил)-
гидроксиламинов состоит в алкилировании бензил-
гидроксамовой к-ты с последующим гидролизом:
C«H5CH»CONH-OH —~ CeH5CH.CONHOR —3-Я
“ — Н J
— NH2OR+ СвН5СН2СООН
N-Алкилпроизводные чаще всего получают алкили-
рованием хлоргидрата гидроксиламина в присутствии
соды:
NH.OH —RNHOH —'Н-. R..NOH
— xiJ — HJ — HJ
— [R3NOH] —[R3NOR]+
— X1J
N-Арилгидрокспламины синтезируют осторожным вос-
становлением нптросоедпнений в нейтральной среде
(напр., с помощью амальгамы А1 и воды):
CcH5NO-> C6H5NO S C,H;NH-OH
H.iO
или нагреванием эфиров фенолов с гидроксиламином*
в спирте:
2,4-(NO2)2C0H3OR -Р ХИ .ОН - 2,4-(NO2)2CeH3NHOH + ROH.
О-Арилгидроксиламииы используются в качестве
ГИДРОКСИЦИТРОНЕ Л ЛА ЛЬ (цитронеллальгид-
рат, 2,6-диметил-2-оксиоктаналь-8)
(СН3)2С(ОН)СН2СН2СН5СН(СН3)СН2СНО
мол. в. 172,25 — густая бесцветная жидкость с нежным
запахом свежей зелени, с оттенком запаха липы
и ландыша; т. кип. 108—109°/3 мм; 0,9220;
ns£ 1,4494; [а]|5 =+8°30'. В природе Г. не найден;
получают из цитронеллаля гидратацией в присутст-
вии серной или соляной к-т. При гидратации необхо-
дима предварительная «защита», альдегидной группы,
напр., образованием бисульфитного соединения, т. к.
иначе происходит нежелательный процесс образо-
вания циклич. соединений (изопулегола и ментагли-
коля). Выход ок. 40% от теоретически возможного,
считая на исходный цитронеллаль. Кристаллич. про-
изводные Г. не получены. Г. быстро окисляется на
воздухе, но его можно стабилизировать добавлением
антиоксидантов, напр. гидрохинона или дифенил-
амина. Чпетый Г. полностью должен растворяться
в 35%-ном р-ре бисульфита натрия. Г. входит в соста»
многих композиций для духов и одеколонов высших
кондиций.
Лит.: II е d о u k i a n Р. Z., Perfumery Synthetics and
Isolates, N. Y.—Toronto, 1951. А. Зелемецкая.
ГИДРОКСОНИЙ (гидроний) — комплексный ион
Н3О+, химич. соединение протопа с молекулой воды,
аналог иона аммония NHp Гидратированный ион-
Г. — основная форма существования водородных ио-
нов в водных п в спиртоводных р-рах кислот. Г. яв-
ляется структурным элементом кристаллич. моно-
гидратов кислот; установлено существование Г.
в таких комплексах, как, например, Н2О • HN О3 и
Н2О • НС1О4. Установлено также существование Г.
в газовой фазе.
Данные по парамагнитному резонансу и по ИК-
спектрам твердых моногидратов кислот показали,
что Г. имеет форму трехгранной пирамиды, в вершине
к-рой находится атом кислорода, а в углах основания
атомы водорода. Три связи ОН в Г. образованы за счет
р-электроиов кислородного атома и являются равно-
ценными. Валентные углы Н—О—Н составляют ок.
105.—110°, т. е. приблизительно равны углам Н—N—Н
в аналогичной по строению молекуле аммиака NHa
-917 ГИДРОЛАЗЫ
<108°). Длина связи ОН в Г. равна 0,98—1,02 А.
Методом ионного удара показано, что теплота образо-
вания Н3О+из Н+ и Н2О в газовой фазе составляет 169
ккал/г-ион. Это значение близко к рассчитанному
теоретически на основе пирамидальной модели. Близ-
кое значение (188,8 ккал/г-ион) получено такм;е как
разность между энергиями образования Н3О + и Н2О,
рассчитанными по методу объединенного атома путем
построения волновых функций молекул из ортонор-
мированных одноэлектронных собственных функций.
ИК-спектры Г, имеют полосы поглощения в см "1: 1134,
1670 (твердый Н2О • HNO3), 1205, 1750, 2900 (конц.
р-ры HCJ, НВт, HNO3, НС1О4 и др. кислот).
Теплота гидратации ок. 110 ккал/г-ион) кажу-
щаяся теплоемкость, кажущийся объем, коэфф, ак-
тивности, кажущаяся сжимаемость и нек-рые др.
свойства Н8О+в разб. водных р-рах близки к анало-
гичным свойствам однозарядного иона Na+. Наблю-
дается также аналогия в зависимостях этих свойств
ионов Н3О+, Li+, Na+, К+от концентрации электролита.
Электропроводность же и коэфф, диффузии Г. в воде
в 5—6 раз, а во льду в сотви раз выше, чем у др.
ионов.
Реакции Г. с анионами, а также реакции образо-
вания Г. при диссоциации молекул протекают чрез-
вычайно быстро. Напр., константа скорости реакции
Н3О+-р ОН~ = 2Н2О при 25° равна 1,3 • 1011 л моль'1-
•сек'1. Резкие различия в кинетич. свойствах Н3О+
« др. ионов (кроме ОН') объясняются специфич.
механизмом перемещения ионов Г. в растворах,
включающим стадию перехода протона от одной мо-
лекулы воды к другой. Концентрация гидроксониевых
ионов Сн+ (или Сп0) и гидроксильных ионов Сон-
в водных р-рах рассчитывается из соотношения
Ар, ~ ан+ ' аон- ~ ^н+' ^он- • f-’ где ионное
произведение воды, ац+ и «qjj — активности, а /— —
средний коэфф, активности электролита (см. Активно-
сти коэффициент). Концентрация гидроксониевых
ионов оказывает большое влияние на величины потен-
циалов некоторых окислительно-восстановительных
реакций, на скорости реакций кислотно-основного
катализа, биохимические процессы, гидролиз и т. д.
Протоны образуют комплексные ионы не только с
водой, но и с другими растворителями. Такие ионы —
диалоги иона П30-, называют ионами лиония.
Активность сольватированных ионов Г. определяет
кислотность водных растворов, а активность ионов
лиония — неводных растворов. Отрицательные ло-
гарифмы активности ионов Г.— величины pH (рН =
= —rgtfp), широко применяются для характеристики
«ислотности растворов (см. Водородный показатель).
Лит.: Измайлов Н. А., Электрохимия растворов,
Ларьков, 1959. Н. Е. Хомутов.
ГИДРОЛАЗЫ — ферменты, катализирующие про-
цессы гидролитич. расщепления веществ и участвую-
щие т. о. в обмене белков, жиров, углеводов и ну-
клеиновых к-т. В зависимости от тина связи, к-рая
подвергается действию фермента, Г. разделяют след,
•образом: 1) ферменты, расщепляющие CO=NH связи,
т. е. осуществляющие гидролиз белков, пептидов и
амидов; 2) ферменты, гидролизующие амины (гуа-
наза); 3) ферменты, гидролизующие эфирные связи
(эстеразы); 4) ферменты, гидролизующие гликозидные
связи в поли- и олигосахаридах и гликозидах (кар-
богидразы — гликозидазы и полиазы); 5) ферменты,
катализирующие гидролитич. отщепление фосфата
•от аденозинтрифосфатов, динуклеотидов и нек-рых
др. соединений (аденозинтрифосфатазы, нуклеотид-
пирофосфатазы, нуклеозиддифосфатазы и др.); 6) фер-
менты, катализирующие иеокислительное декарбокси-
лирование (декарбоксилазы).
Лит.: Д и к с о в М., Уэбб Е. С., Ферменты, пер. с
англ., М., 1961. Т. Т. Болотина.
^ВОН ’ ^НА
• f.. „
— ГИДРОЛИЗ 918
ГИДРОЛИЗ — реакции обменного разложения ме-
жду водой и различными соединениями; частный
случай сольволиза. Г. протекает как в водных р-рах,
так и при взаимодействии воды или водяных паров
с твердыми, жидкими и газообразными веществами.
Г. подвержены химич. соединения различных клас-
сов, в том числе соли, углеводы, белки, эфиры, жиры
и др. Суммарный результат Г. какого-либо соедине-
ния может быть описан одной из след, схем:
ВА-l- Н.О=В + + ОН- + НА (I)
ВА 4-Н2О=ВОН + Н++ А- (II)
ВА 4-ЩО—ВОН4-НА (III)
Детальный механизм Г. выяснен еще недостаточно;
для разных групп соединений он может быть сущест-
венно различным. Многочисленны случаи обрати-
мого Г., в особенности характерного для солей. Рав-
новесие при обратимом Г. как частный случай рав-
новесия химического подчиняется закону действующих
масс и др. термодинамич. соотношениям. Важней-
шими величинами, характеризующими гидролитич.
равновесие, являются константа гидролиза и
степень гидролиза а. Константа Г. как термодинамич.
константа равновесия гидролитич. реакции опре-
деляется соотношениями вида:
1ВОН] [НА) свон ' СНА
Г [BA] [H2OJ - евд . сН20
к-рые характеризуют реакцию типа III. Здесь
[ВОН], [НА], [ВА], [Н2О] — активности, свон,
сна, fBA, сН2о — концентрации, /вон, /нл, /вл,
^Hao — коэфф, активности участвующих в процессе
веществ. В разб. р-рах (при с-<0,01 моль/л) величи-
ны f для нейтральных молекул практически равны
единице, а для ионов они меньше единицы и зависят
от величины ионной силы раствора. В весьма разб.
р-рах (с <Z 10' 4 моль 1л) величины f ионов также близки
к единице. Активность воды в разб. р-рах мало из-
меняется с изменением концентрации растворенного
вещества и может рассматриваться как постоянная
величина. Поэтому произведение Ар[Н2О] в- разб.
р-рах также является постоянной величиной. Вели-
чину Аг[Н2О] = К очень часто называют константой
Г. и, как правило, именно ее приводят в справочной
литературе. Константа Г. может быть вычислена
из соотношения —ДЛ° = Я7’1пАр, где — Д/’° — из-
менение свободной энергии процесса Г. в стандартных
условиях. Ар определяется природой гидролитич. ре-
акции и темп-рой.
С р
Степень Г. а = —, где сг — концентрация гадро-
с
лизованиой части, с — полная концентрация в-ва.
Для вещества типа ВА величина равна концентра-
ции любого из продуктов Г. Поэтому степень Г. может
т - свон
быть определена из соотношении вида: а =----- =
с
(?н а
------Используя такие соотношения и выражение
с
для константы Г., можно легко получить уравнения,
связывающие степень Г. с константой Г. (см. ниже).
Наиболее хорошо изучен гидролиз солей —
реакция, обратная нейтрализации. Согласно широко
распространенной точке зрения, Г. солей представ-
ляет либо одну из реакций В+ + ОН_ = ВОН,
А- + Н + = НА, либо обе вместе. Реакции протекают
в том случае, когда ВОН — слабое основание и (или)
НА — слабая кислота. Если сила ВОН и НА не оди-
накова, то концентрации ионов Н+ и ОН' в растворе
соли тоже не одинаковы, и раствор имеет либо кислую,
919
ГИДРОЛИЗ
920
либо щелочную реакцию. Другая точка зрения, вы-
текающая из протолитической теории кислот и ос-
нований, рассматривает Г. солей как перенос про-
тонов от молекул воды к. анионам и от гидратиро-
ванных катионов к молекулам воды. Гидратирован-
ные катионы выполняют функцию кислоты, анио-
ны — основания, вода — либо основания, либо кис-
лоты: |А1(Н2О)е]3+ (кислота) -|- Н2О (основание) =
= | А1(Н2О)5ОН]2+ -|- Н3О+; РО?’ (основание) + Н2О
(кислота) = IIPO; -|- ОН’. Причиной Г. является
различие сродства к протону этих частиц, к-рое в свою
очередь определяется константами диссоциации соот-
ветствующих кислот и оснований и ионным произве-
дением воды.
Различают несколько случаев Г. солей. Гидролиз
солей слабых кислот и сильных основа-
ний (напр., цианидов, боратов и карбонатов щелочных ме-
таллов) отвечает схеме I: В А = В+ + А-; А- + Н2О — НА 4-
-I-OH-; характеризуется большим сродством аниона к протону
и малым сродством катиона к ОН-. Растворы средних солей
итого типа содержат свободные ионы ОН- и показывают
щелочную реакцию. Закон действующих масс для Г. одно-
основной соли ВА этого типа выражается ур-нием:
„ [НА] [ОН-] а2С WW Я2С
К= [А’] = 1^ • —fZ- ~ Г-о (°
В разб. р-рах = 1; Учет этих соотно-
ИА ид А
тений приводит к приближенной форме уравнения (1). Из
соотношения (1) видно, что в разб. р-рах степень Г. умень-
шается с ростом концентрации соли и мало зависит от ионной
силы раствора (последняя определяет коэфф, активности
ионов). Константа Г. для этого случая связана с ионным
произведением воды Kg и сконстантой диссоциации кислоты
КНА соотношением:
К-КВ^НЛ (2)
Ур-ние (2) и ему подобные получают путем комбинирования
выражений закона действующих масс для Г. соли, для дис-
социации кислоты и воды. Из соотношения (2) видно, что Г.
тем сильнее, чем меньше КНА, т. е. чем слабее кислота НА.
Гидролиз солей сильных кислот и сла-
бых оснований (напр., хлоридов, хлоратов, нитратов
аммония, 3-валентного железа и др.) протекает в соответствии
со схемой II: ВА = В+ + А~; В+ 4- Н2О = ВОН 4- Н+. Р-ры
солей этого типа имеют кислую реакцию. Константа Г. опре-
деляется соотношением:
„ [ВОН][Н+] а2с 'н^ВОН аЗс
-----[в+j '!—••• (3>
А=кв/Авон (4)
Из соотношения (4) следует, что Г. тем сильнее, чем меньше
h вон-
Гидролиз солей слабых кислот и сла-
бых оснований (напр., карбонатов, цианидов, суль-
фидов аммония, 3-валентного железа и др.) протекает в соот-
ветствии со схемой III: ВА — В+ -j- А-; В+ 4- А-4~ Н2О —
— Вин + НА. Характеризуется большим сродством аниона
соли к протону и катиона к ОН-. Сопровождается образо-
ванием недиссоциировэнных молекул слабой к-ты и слабого
основания. Гидролитич. равновесие описывается соотноше-
нием:
[НА][ВОН] «2 ^НА'^ВОН ?2
[B+][A-f “(1-4)2 ' (в+./д- “ (1 - 4)2 <5>
Приближенная форма здесь относится к весьма разб. р-рам,
для к-рых /g+ = /а- 1- Лля менее разб. р-ров —
= /д- = / ± < 1 и поэтому
1 а2
“ (1^2 • /в+ . /А- - (1 - «)2.4 (6)
где /±—средний козфф. активности электролита. Из соотно-
шений (5) и (6) видно, что степень Г. в весьма разб. р-рах не
зависит от концентрации соли, а в менее разб. р-рах она умень-
шается с ростом ионной силы раствора вследствие уменьшения
козфф. активности. Константа Г. связана с величинами кв
К„. и К соотношением
дА ilt.'M
Если Квон то в растворе соли ВА концентрации
ионов Н+ и ОН- одинаковы и раствор имеет нейтральную
реакцию (pH = 7). Если КНд > ^вон Раств0Р имеет слабо-
кислую, а если К„. — слабощелочную реакцию.
ИА ВиП
Гидролиз- солей сильных кислот и
сильных оснований (напр., хлоридов, хлоратов,
нитратов щелочных металлов и др.) в обычных условиях
практически отсутствует, что является следствием малых
величин сродства протона к аниону соли и сродства иона
ОН- к катиону соли.
Гидролиз солей многоосновных кис-
лот и оснований протекает через отдельные стадии,
каждая из к-рых характеризуется своей константой Г. Напр.,
если гидролизуется соль В2А двухосновной слабой кислоты
Н2а и щелочного металла, то Г. состоит из реакций: А2- +
+ Н2О == НА- + ОН-; НА" + Н2О = Н2А + ОН-. Для этих
стадий справедливы соотношения:
[НА-] [ОН-] [Н2А]-[ОН-]
к 1 =----[А^]---- -=---------[HF]— (8>
где Ki и к2 — константы первой и второй ступеней Г. В соот-
ветствии с соотношением (2) имеем:
к[ = кв/кНА и ^в^а (9>
Т. к. константа диссоциации Н2А всегда больше, чем НА~,
то Кх > А’2.
Г. подвергаются также кислые и основные соли. Если
ВНА — кислая соль слабой к-ты Н2А, то ее Г. обусловлен
реакцией НА- 4- Н2О = Н2А 4- ОН—. Ион НА’ ведет себя
как основание (напр., NaHCO3 4- Н2О = Na+ -j- Н2СО3 4-
4- ОН”). Если, гидролизуется основная соль сильной к-ты н
полизарядиого катиона, то гидрат последнего реагирует с во-
дой как кислота, напр.
[А1 5Н2О]ОНС12 4- Н2О=[А1 • 4Н3О](ОН)2С1 4- С1~ 4- Н3О+.
Если продукты Г. соли — летучие в-ва [напр., HaS, НСЬ
СО2, NH3) или труднорастворимые (напр., Ге(ОН)8], то Г.
может быть осуществлен количественно.
Добавка посторонней сильной к-ты к солям слабых к-т
и добавка сильного основания к солям слабых оснований
также усиливает Г. Введение в раствор избыточных продук-
тов Г. уменьшает степень Г. в соответствии с законом дей-
ствующих масс. Влияние темп-ры на Г. солей определяется в
соответствии с соотношениями (2), (4), (7) зависимостью от
темп-ры Kg, Кд и Квон. В области высоких темп-р наблю-
дается сильное возрастание К и ас ростом темп-ры для мно-
гих солей. Г. — эндотермич. реакция. Величины теплот Г.,
как правило, меньше 20 ккал/г-экв.
Продукты Г. — слабые кислоты и основания — образуются
как в виде одноядерных частиц, содержащих один гидроли-
зованный ион, так и в виде полиядерных частиц различного
состава, содержащих несколько ионов. Одноядерные частицы
образуются при Г. многих слабоосновных катионов (Mg8+,
Hg2r, Zn2-, Ni2+, Cu2+-, Fe31, А!3’’’, Се4+и др.). Они могут быть
как промежуточными так и конечными продуктами Г., подоб-
ны мн с л е д ующи м: (HgOH)+, Hg(OH)2 (FeOH)2-, (СеОН)3+.
Г. очень слабоосновных катионов (Ве2+, В13+, Й4+и др.) при-
водит к образованию многоядерных продуктов, напр.
[Be3(OH)s]3+, [Ве2(ОН)„]+, [Bi,(OH)I2]"+. Еще более сложные
полиядерные частицы образуются при Г. анионов слабых
кислот (молибденовой, кремневой, борной, ванадиевой воль-
фрамовой, хромовой, германиевой, теллуровой и др.). Напр.,
Г. иона МоО?’ приводит к образованию [Мо7О24]3’ и
[НМо,О24]6-. Во многих случаях частицы кислот и оснований,
образующихся в ходе Г., находятся в форме гидратов (см.
Гидратация). Г. солей часто приводит к образованию в виде
самостоятельной фазы осадков гидроокисей, не растворяю-
щихся при слабом подкислении р-ра. В этом случае Г. солей
наз. необратимым.
Важнейшим методом количественного изучения
гидролитич. равновесия является метод эдс.
Используется также измерение электропроводности,
распределения кислоты и основания между двумя
растворителями, измерение понижения давления
пара и понижения темп-р замерзания р-ров, изучение
поглощения света, магнитных и др. свойств р-ров
солей.
Скорости реакпий Г. солей сильно зависят от их
природы. Известны очень быстрые реакции (преим.
Г. солей с однозарядными анионами). Нек-рые соли
гидролизуются медленно (напр., FeCl3, FeF3 и др.)
и кинетика их Г. доступна изучению обычными ме-
тодами. Кинетич. параметры быстрых гидролитич.
реакций стали доступными в связи с разработкой но-
вых методов исследования: полярографии, протон-
ного магнитного резонанса, ультразвука и др.
921
ГИДРОЛИЗ ДРЕВЕСИНЫ — ГИДРОЛИЗ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
922
Г. солей используется как препаративный метод
получения основных солей и гидроокисей. Напр.,
ZrOCl2 • 8Н2О и UO2(NO3)2 • 6Н2О могут быть полу-
чены выпариванием р-ров соответствующих кислых
солей; BiOJ и Bi(OH)2NO3— нагреванием р-ров
BiJ3 и Bi(NO3)3; безводные основные соли типа
МеОНС! (где Me—Са2+, Cu2+, Zn2+, Sn2+, Fe2+ и др.)
получают из конц. водных р-ров средних солей после
введения в них окиси этилена, связывающей НС1 и
избыточную воду.
Необратимый гидролиз характерен для
многих классов химич. соединений.
Особенно распространен необратимый Г. среди
органич. соединений (см. Гидролиз, органических со-
единений, Гидролиз растительных материалов и др.).
Значение гидролиза. Г. широко ис-
пользуется в химич. пром-сти: для получения глю-
козы, ксилозы, фурфурола, этилового спирта, много-
атомных спиртов, пищевых органич. кислот и др.
веществ путем переработки многих растительных
материалов, в т. ч. отходов лесопильной и деревооб-
рабатывающей пром-сти и отходов с. х-ва (см. Ги-
дролиз растительных материалов). Г. персолей и
перекисей используется в пром-сти полимеров; он
приводит к образованию перкислот, инициирующих
процессы полимеризации. Г. жиров составляет основу
промышленного получения мыла и глицерина. Фер-
ментативный Г. применяется в пищевой, текстильной,
фармацевтич. пром-сти. Г. составляет основу процес-
сов в буферных растворах. В теплотехнике Г. служит
для очистки воды, идущей на питание паровых котлов
от корродирующих примесей, добавлением легко
гидролизуемых солей (напр., Na3PO4). В военной
технике Г. используется при дегазации отравляющих
веществ.
Г. минералов вызывает важные изменения в со-
ставе земной коры (см. Геохимические процессы).
Г. играет также большую роль в процессах жизне-
деятельности живых организмов.
Лит.: Киреев В. А., Курс физической химии, М.,
1955; Изгарышев Н. А. Горбачев С. В., Курс
теоретической электрохимии, М.—Л., 1951; Г л е с с т о н С.,
Введение в электрохимию, пер. с англ., М., 1951; Тава-
н а е в Н. А'., Теоретические основы аналитической химии,
ч. 1, 2 изд., Свердловск, 1956; К о м а р ь Н. П., Основы
качественного химического анализа Харьков, 1955; S i 1-
lenL. G., Quart. Rev., 1959, 13, №2;Lux H., Anorganisch-
chemische Experimentierkunst, 2 Aufl., Lpz., 1959.
H. E. Хомутов.
ГИДРОЛИЗ ДРЕВЕСИНЫ — см. Гидролиз рас-
тительных материалов.
ГИДРОЛИЗ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ —
расщепление связей в органич. молекуле при действии
воды с образованием 2 или более соединений, при-
чем элементы воды присоединяются по месту возни-
кающих свободных валентностей. Под Г. о. с. обычно
понимают также гидратацию циклич. соединений
(лактонов, лактамов и др.), сопровождающуюся
разрывом цикла. Гидролиз (Г.) связи углерод—гало-
ген— типичная реакция нуклеофильного замещения
галогена оксигруппой:
RHal + НОН — ROH + Hal + Н+ 1
или 1 Sv2
RHal + ОН — ROH 4- Hal- ]
RHal — R+ + Hal- 1
r+ + нон — roh + н+ г Sn1
или
R+ -j- OH- — ROH J
Механизм гидролиза галогеналкилов зависит как от
характера алкильного радикала, так и от условий
реакции. С уменьшением избыточного положитель-
ного заряда на атоме углерода, связанном с галоге-
ном, реакция по механизму SN2 затрудняется, а иони-
зация связи углерод — галоген, определяющая ско-
рость реакции по механизму SN1, облегчается. Не-
зависимо от механизма реакции скорость гидролиза
галогеналкилов с одинаковыми углеводородными ра-
дикалами, но с различными атомами галогена увели-
чивается в ряду F<Cl<Br< J. Скорость гидролиза
галогеналкилов по механизму SN2 возрастает с уве-
личением концентрации щелочи в реакционной среде,
а по механизму SN1 не зависит от наличия или от-
сутствия щелочи. Скорость мономолекулярного гид-
ролиза увеличивается в присутствии катионов Ag+
или Hg2+, к-рые связывают ион галогена, образую-
щегося при ионизации галогеналкила.
Гидролиз связи углерод — галоген облегчается
сопряжением ее с кратной связью, что видно на при-
мере гидролиза хлористого аллила или бензилхло-
рида, осуществляемого в пром-сти:
..ОК
н о
СН2=Сн-СН2-С1 2 »• сн2=сн-сн2он + н Cl
Галогенангидриды органич. кислот легко гидроли-
зуются по бимолекулярному механизму с образова-
нием соответствующих кислот:
,/ОН’
R-C —Hal
ей
R-C-OH + HHai
А
Скорость гидролиза арилгалогенидов зависит от на-
личия заместителей в ароматич. ядре и их положения.
Замещение атомов галогена на ОН-группу по меха-
низму Sn2 облегчается в соединениях, содержащих
в орто- или пара-положении к галогену заместители
2-го рода, что объясняется уменьшением электронной
плотности на атоме углерода, связанном с галогеном:
Напротив, наличие в орто- или пара-положении
к атому галогена заместителей 1-го рода делает воз-
можным гидролиз по мономолекулярному механизму,
т. к. в данном случае облегчается ионизация галогена:
Кинетика гидролиза хлорбензола щелочью также со-
ответствует мономолекулярной реакции. Этот метод
служит одним из промышленных способов получения
фенола:
СвН5С1 свн6он
Uxl
Гидролитич. расщепление сложных эфиров карбо-
новых к-т может катализироваться кислотами, ще-
лочами либо специфич. ферментом — эстеразой. Кис-
лотный гидролиз сложных эфиров является обрати-
мой реакцией (см. Этерификация) и чаще всего про-
текает по бимолекулярной схеме с разрывом связи
ацил — кислород:
R—С- OR' R-C-OR' R—С—6 —R'
Jt « 1 1
0 +0H 0 H
923 ГИДРОЛИЗ ОРГАНИЧ. СОЕДИНЕНИЙ — ГИДРОЛИЗ РАСТИТЕЛЬНЫХ МАТЕРИАЛОВ 924
R-C-6-R' R-C-O-H +ROH
А Н A H"R-C-OH+Ht
А
Известны также случап гидролиза сложных эфиров
с разрывом связи ацил—кислород по механизму
SN1: гидролиз эфиров 2, 4, 6-триметилбензойной к-ты,
а также мономопекулярный кислотный Г. с разрывом
связи алкил — кислород (гидролиз третичнобутило-
вых эфиров карбоновых к-т). Щелочной Г. сложных
эфиров карбоновых к-т является необратимой би-
молекулярной реакцией:
связи углерод — углерод легко достигается под дей-
ствием слабых р-ров щелочей или р-ров кислот (т. наз.
кетонное расщепление):
R-C-CH2-C-R' 5?R—С— СН3 + R'COOH
II II II
0 0 о
R—С —СН3—С—OR' —2—. R—С-СН3 + СО. 4- R'OH
А А А
ОН~+ R-C-R’
II
О
ОН
I
R-C-OR
I
О”
он
I
R-C + R'O”
II
—“-R—С—О' + ROH Г. сложных эфиров— ре-
q акция, широко применяю-
щаяся в пром-сти для полу-
чения спиртов и кислот, в частности щелочной ги-
дролиз жиров с целью получения глицерина и со-
лей высших алифатич. к-т. Гидролиз винилацетата
и поливинилацетата — промышленные способы син-
теза соответственно ацетальдегида и поливинилового
спирта:
СН3=СН-О-С-СН3 —СНз-С-Н 4- СНзСООН
А А
(-СН,-СН-)П —(—СН2—СН—)п -ь пСНзСООП
О-С-СНз он
А
Гликозидная связь в углеводах гидролизуется под
каталитич. влиянием неорганич. кислот либо энзи-
мов-карбогидраз (см. Гидролиз растительных мате-
риалов, Инверсия сахаров). Гидролиз простых эфиров,
как правило, происходит лишь в крайне жестких
условиях, однако эфиры, образованные спиртами
с повышенной кислотностью (эфиры нитрофенолов
или триарилкарбинолов), легко разлагаются водой:
O2N-C0Hi-OR ЯЯЯ. O3N—C0Hj—ОН + ROH
ин—
Гидролптич. расщепление связи углерод — сера (иа
примере нуклеофильного замещения сульфогруппы
на гидроксил) производят обработкой ароматич. кис-
лот щелочами; является промышленным методом
получения фенолов:
.ОН”
С другой стороны, гидролиз ароматич. сульфокислот
по электрофильному механизму достигается действием
водяного пара в присутствии неорганич. кислот и при-
водит к ароматич. углеводородам:
CcH;SO3H СцНз + HsSO4
Связи углерод — азот в амидах органич. кислот
гидролизуются аналогично сложным эфирам под
влиянием как щелочных, так и кислотных катали-
заторов. Гидролиз амидной связи в белках катали-
зируется также протеолитич. ферментами. При дей-
ствии растворов неорганич. кислот на нек-рые аро-
матич. амины происходит Г. по связи углерод — азот
и образуются соответствующие оксисоединения.
Как правило, углерод — углеродная связь устой-
чива к гидролизу. Однако в случае 1,3-дикетонов или
производных [J-кетокислот гидролитич. расщепление
Легкий гидролиз углерод—углеродной связи при дей-
ствии водных р-ров щелочей является специфич. реак-
цией полигалогенокарбонильныхсоединений (реак-
ция галоформного распада):
СС13 —С—н
А
_Н30
NaOH
СС13Н 4- H-C-ONa
А
Нек-рые а,р-ненасыщенные кислоты и кетоны под-
вергаются гидролизу по след, схеме:
(СН3)3С=СН-С-СН3 „Н2° 2 СН3-С-СН3
Il H2SC>4 II
о о
(СН3)3С=СН-СООН Ц;,0- СН3-С-СН3 + CH3C00Na
гм а о и и
Образовавшиеся в результате гидратации исходного
соединения [5-оксикарбоновые кислоты или [5-окси-
кетоны гидролизуются по реакции обратной альдоль-
ной конденсации. Гидролиз связей углерода с эле-
ментами I, II и III группы периодич. системы проис-
ходит в соответствии с полярностью этих связей
(Эл — на рис. — элемент);
»-
-С-Эл —2°>- -С-Н 4- ЭлОН
1 I
Гидролиз связи углерод — водород изучен на при-
мере нуклеофильного гидроксилирования ароматич.
соединений, содержащих заместителп 2-го рода,
а также гетероциклич. соединений ряда пиридина,
хинолина и др.:
Увеличение положительною заряда на атоме углерода,
связанном с замещением атомом водорода, облегчает
атаку аниона гидроксила. Гидролиз связей элемент —
галоген, элемент — кислород и др. в органич. соеди-
нениях принципиально не отличается от гидролиза
соответствующих связей в неорганич. соединениях.
Г. силоксанов и галогенсиланов см. Кремнийоргани-
ческие соединения, С иликоны.
Лит.: Реутов О. А., Теоретические проблемы органи-
ческой химии, М., 1956; И н г о л ь д К. К., Механизм реак-
ций и строение органических соединений, пер. с англ., М.,
1959; X юкке ль В., Теоретические основы органической
химии, пер. с нем., т. 1, М., 1955; Ворожцов Н. Н., Основы
синтеза промежуточных продуктов и красителей. 4 изд., М.,
1955; Шемякиа М. М., Щ у к и н а Л. А., Усп. химии,
1957, 26, вып. 5, с. 528. Л. С. Герман.
ГИДРОЛИЗ РАСТИТЕЛЬНЫХ МАТЕРИАЛОВ —
каталитич. процесс взаимодействия полисахаридов
растительных тканей с водой, проводимый с целью
превращения нерастворимых в воде полисахаридов
в растворимые монозы; этот процесс является осно-
вой гидролизных производств, назначение которых
производить пищевые, кормовые и технические про-
дукты из непищевого растительного сырья (опилки,
стружки и др. древесные отходы, солома, кукурузная
кочерыжка, подсолнечная лузга, костра, тростник
и др. растительное сырье). В этом сырье содержится
до 75% полисахаридов в виде целлюлозы и гемицел-
925
ГИДРОЛИЗ РАСТИТЕЛЬНЫХ МАТЕРИАЛОВ
926
люлоз, состоящих в основном из пентозанов («сплав,
арабан) и гексозанов (манвав, галактан). При гидро-
лизе получают р-ры моноз (гидролизаты), летучие
в-ва (уксусная и муравьиная к-ты и метиловый спирт)
п твердый остаток (до 30% от сырья) — гидролизный
лигнин.
Из гидролизатов можно получать: кристаллизацией
мовоз—глюкозу пищевую и ксилозу технич.; гид-
рированием — высшие полиолы (ксилит, сорбит) и их
гидрогенолизом — глицерин, этиленгликоль и про-
ннленглпколь; дегидратацией моноз — фурфурол и
левулиновую кислоту; окислением — органич. к-ты
(глюконовую, триоксиглутаровую и др.); брожением—
спирт этиловый и бутиловый, ацетон, белково-вита-
минные дрожжи и антибиотики. Из гидролизного лиг-
нина получают: путем термолиза — активные угли,
уксусную к-ту, феиол и др.; водной обработкой —
наполнители для пресспорошков; химической обра-
боткой — активированный лигвив, щавелевую к-ту
и др.; прессованием — строительные и изоляциовиые
лигноплиты.
Наиболее перспективным направлением переработки
моноз является их каталитич. превращение в полу-
продукты для тяжелого органич. синтеза, а также
биосинтез белковых веществ, витаминов и антибио-
тиков.
Гидролиз полисахаридов раститель-
ных тканей проводят при участии катализаторов —
конц. или разб. кислот, кислых солей и др.; процесс
протекает в несколько стадий с образованием сначала
промежуточных соединений, а затем простейших
сахаров — моноз. Каталитич. действие кислот тем
сильнее, чем выше степень их диссоциации; при этом
скорость гидролиза растет с увеличением темп-ры
и концентрации к-ты. По скорости гидролиза поли-
сахариды делятся на легко и трудногидролизуемые,
располагаясь в ряд: галактанжсилан>.маннан2>
целлюлоза. На скорость гидролиза и извлечение об-
разовавшихся моноз влияет также размер частиц
растительных материалов, поэтому растительное
сырье, как правило, предварительно измельчают. Гид-
ролиз легкогидролизуемых полисахаридов, как и по-
лучаемый гидролизат, называют пентозным, а непро-
гидролпзовавшийся остаток — целлолигпи-
п о м; гидролиз трудногидролизуемых полисахари-
дов (в частности, содержащихся в целлолигнине),
а также получаемый гидролпзат называют гексозным.
В присутствии конц. кислот процесс гидролиза оста-
навливается па стадии образования растворимых
в воде полисахаридов с меньшей степенью полимери-
зации — олигосахаридов', для получения моноз про-
водят инверсию (гидролиз) перешедших в раствор
олигосахаридов. При гидролизе в присутствии конц.
кислот (HCI, H2SO4, HF) выход моноз составляет
ок. 95% к теоретически возможному, процесс про-
водят прп атмосферном давлении и незначительном
расходе пара; при этом получают чистые гидроли-
заты с содержанием 15—25% моноз; применяемые
в процессе произ-ва НС1 и HF можно регенерировать.
При гидролизе разб. к-тами, проводимом при тем-
пературе выше 100° и соответствующем избыточном
давлении, наряду с образованием моноз происходит
и их частичный распад с образованием фурфурола,
органич. к-т, гуминовых и др. веществ. Реальный вы-
ход моноз определяется соотношением констант ско-
рости их образования и разложения. Для увеличения
выхода обычно стремятся образовавшиеся монозы
быстрее вывести пз сферы реакции. Гидролиз с разб.
H2SO4 дает в практич. условиях выход гексоз не более
70% от теоретически возможного при их концентрации
в гидролизатах 2—4% и требует значительного рас-
хода пара, применения громоздкой аппаратуры и т. д.
Этот метод пригоден для гидролиза гемицеллюлоз;
для гексозного же гидролиза лучше применять конц.
кислоты.
Широко применяется способ однофазного гидролиза ра-
стительных материалов в присутствии разб. H2SO4 в верти-
кальных цилиндрич. автоклавах периодич. действия емкостью
18—50 at3. После загрузки сырья в такой аппарат непрерывно
подается 0,5—1 %-ный р-р II2SO4 и пар. Процесс протекает
при 160—190° и давлении до 12 ат. Образовавшиеся монозы
переходят в р-р, непрерывно выводимый из нижней части
аппарата через фильтрующее устройство. Остаток после гид-
ролиза (лигнин) выдувают из аппарата при избыточном дав-
лении. Более совершенными являются способы непрерывного
гидролиаа, к-рые можно проводить в противоточных аппара-
тах непрерывного действия или в две сталии — обработкой
растительного материала к-той при соответствующей темп-ре
с последующим противоточным вымыванием образовавшихся
моноз.
Пищевую глюкозу и техничес-
кую ксилозу получают выделением этих моноз
из гидролизатов. Технология произ-ва состоит пз
инверсии гидролизатов, удаления пз них минераль-
ных к-т, адсорбционной и ионообменной очисткп от
минеральных и органич. примесей, упаривания р-ров
и выделения моноз прямой кристаллизацией пли (для
глюкозы) через двойное соединение с NaCl.
Для произ-ва глюкозы используют гидролизаты, по-
лучаемые при гидролизе целлолигнива конц. к-тами,
а для получения ксилозы — пентозные гидролизаты
из растительных отходов с. х-ва, лиственной древе-
сины, тростпика и др. Выход глюкозы составляет
до 30% к весу абсолютно сухого сырья. Маточный
р-р после выделения кристаллич. глюкозы исполь-
зуется для биохимич. переработки. Наибольший
выход ксилозы— до 30% к весу абсолютно сухого
сырья получается из кукурузной кочерыжки. Техяич.
ксилоза используется гл. обр. в виде очищенного
ксилозного сиропа для получения ее различных про-
изводных.
Многоатомные спирты получают вос-
становлением моноз. Из гексоз образуются соответст-
вующие гекситы СвН14Ов (сорбит, маннит, дульцит
и т. п.), а из пептоз — пентпты С5П12О5 (ксилит,
арабит и др.). Восстановление моноз протекает
с почти колич. выходом путем гидрирования под
давлением нейтральных или слабощелочных р-ров
в присутствии никелевых или др. гидрирующих ката-
лизаторов при 115—125°. Схема произ-ва многоатом-
ных спиртов — пентитов или гексптов — приведена
на рис. 1.' Общий выход спиртов достигает 85%
Я вакууму
Рис. I. Схема производства многоатомных спиртов — пен-
тятов или текстов: 1— сборник раствора моноз; 2— ком-
прессор; з — подогреватель; 4 — колонны для гидри-
рования; 5 — холодильник; 6 — газосепарэтор; 7 — сбор-
ник; 8 — ионообменные фильтры; 9 — сборник; ю — вы-
парной аппарат; 11 — кристаллизатор; 12 — центрифуга.
к весу моноз, что позволяет получать из 1 т исход-
ного абсолютно сухого сырья до 500 кг многоатомных
спиртов. Из кукурузной кочерыжки можно получать
до 25% ксилита, а из древесины хвойных пород —
до 40% сорбита. Пентиты и гекситы могут быть по-
лучены и непосредственно из полисахаридов путем
их каталитич. гидрирования в кислой среде при по-
927
ГИДРОЛИЗ РАСТИТЕЛЬНЫХ МАТЕРИАЛОВ
928
пьяненной темп-ре и под давлением; при этом идут
быстро следующие друг за другом процессы гидролиза
и гидрирования.
При гидрогенолизе высших полиолов в присутствии
никелевых или др. катализаторов в нейтральной или
слабощелочной среде при 210—230° и давлении во-
дорода св. 50 ат из гекситов образуется глицерин
и пропиленгликолъ, а из пентитов — глицерин, эти-
ленгликолъ и пропиленгликолъ. В зависимости от
условий гидрогенолиза может образовываться в не-
больших количествах и эритрит, а также низшие
спирты и др. вещества. Гликоли и глицерин выде-
ляются из смеси продуктов гидрогенолиза путем раз-
гонки, а остаток пентитов или гекситов возвращается
на повторный гидрогенолиз.
Фурфурол получают дегидратацией пентоз
(С6Н10О6-> С5Н4О2 + ЗН2О) обычно в присутствии
катализаторов — кислот, кислых солей и др. при
переработку
Рис. 2. Схема производства фурфурола: 1 — гидролиз-
ные колонки; 2 — парогенератор; 6 и ТО — холо-
дильники; 4 — сборник конденсата; 5— дистилляционная
колонна; 7 — отстойник фурфурола; 8 — сборник фурфу-
рола; 9 — обезвоживающая колонка; 10 — холодильник
для фурфурола; 11 — вакуумсборники; 12— дефлегматоры.
140—190°. Выход фурфурола зависит от содержания
пентозанов в сырье и условий проведения реакций,
определяющих скорости гидролиза пентозанов, де-
гидратации пентоз и распада фурфурола до муравьи-
ной к-ты и гуминовых веществ. Практич. значение
имеют след, способы получения фурфурола: 1) из
паров самоиспарения гидролизатов при однофазном
гидролизе разб. H2SO4; 2) совмещением пентозного
гидролиза с одновременной дегидратацией образовав-
шихся пентоз (прямой способ); из пентозных гидро-
лизатов при двухфазном Г. р. м. Прямой способ по-
лучения фурфурола (рис. 2) заключается в смачи-
вании и пропитке пентозансодержащего сырья р-ром
H2SO4 или НС1, нагревании материала выше 140°,
удалении образовавшегося фурфурола с парами, его
выделении и очистке ректификацией. Выходы фур-
фурола в зависимости от вида сырья колеблются
в пределах 6—15% к его абсолютно сухому весу.
Получение фурфурола по этом}' способ}' можно про-
изводить и в комбинации с пиролизом древесины
в лесохимии, произ-ве. Для произ-ва фурфурола прн
комплексной переработке с использованием всех
полисахаридов метод прямого получения фурфу-
рола допустим лишь при условии сохранения целлю-
лозной части сырья или же нентозансодержащее
сырье обычно подвергают двухфазному гидролизу;
фурфурол выделяется из пентозных гидролизатов
после дегидратации пентоз, а гексозные гидролизаты
направляются для биохимич. или др. переработки.
Выходы фурфурола по этому способу 40—45% от
пентоз и в зависимости от вида исходного сырья состав-
ляют к его абсолютно сухому весу до 15%.
Левулиновая кислота, образующаяся
при дегидратации гексоз через оксиметилфурфурол:
СвН12Ов - ЗН2О + СсНвО3 ->
— сн3 — со — сн2 — сн2 — соон + нсоон
получается при проведении гексозного гидролиза
в жестких условиях. Левулиновая к-та экстраги-
руется из р-ров серным эфиром, к-рый затем отго-
няется, а кислота очищается перегонкой в вакууме.
Теоретич. выход левулиновой к-ты составляет 64,5%
от гексоз.
Моно- и дикарбоновые оксикис-
лоты получают окислением моноз гидролизатов.
При окислении ксилозы HNO3 образуется триокси-
глутаровая к-та: С6Н10О6 -> C6Hi0Oe -+ С6Н8О7, про-
изводство к-рой состоит из непрерывного окисления
ксилозного сиропа HNO3, нейтрализации, выделении
триоксиглутаровокнслого кальция, его разложения
H2SO4, очистки р-ров триоксиглутаровой к-ты, их
упаривания и кристаллизации. Выход к-ты состав-
ляет до 60% от веса окисленной ксилозы. При окис-
лении ксилозы воздухом в щелочной среде образуется
D-треоновая к-та, при окислении к-рой получается
L-винная к-та с выходом до 40% от ксилозы. При
окислении глюкозы HNO3 образуются сахарная и ща-
велевая к-ты, а при окислении в щелочной среде —
D-арабоновая к-та, окисление к-рой азотной к-той
дает аработриоксиглутаровую к-ту.
Биохимическая переработка мо-
в о з гидролизатов приводит к получению различных
спиртов, белково-витаминных дрожжей, антибиоти-
ков, органич. кислот и др. продуктов. Схема полу-
чения этилового спирта путем сбраживания гексоз
гидролизатов сахаромицетами представлена на рис. 3.
Из 1 т абсолютно сухой древесины хвойных пород
получают 150—180 л этилового спирта и до 120 кг
жидкой двуокиси углерода. При сбраживании гексоз
в специальных условиях получают бутанол, ацетон,
гляколи, глицерин, молочную к-ту и т. д.
Рис. 3. Схема производства этилового спирта: 1 — ап-
парат для гидролиза; 2 — циклон для удаления лиг-
нина; 3 — испаритель гидролизата; 4 — нейтрализатор;
,5 — отстойник; 6 — теплообменник; 7 — бродильный чан;
8 — дрожжевой сепаратор; 9 — сборник бражки; 10 —
бражная колонна; 11 — ректификационная колонна; 12 —
метанольная колонна; 13 — подогреватель; 14 — конден-
сатор-холодильник; 1S — конденсатор; 16 — холодильник;
17 — газгольдер для СО2.
Дрожжи кормовые белково-ви-
таминвые получают ферментативным синтезом
белка и витаминов, протекающим в дрожжевой клетке.
Для получения дрожжей используются различные
микроорганизмы (Torulopsis utilis, Candida, Oidium
и др.), утилизирующие, кроме моноз, также и от-
дельные к-ты гидролизатов (уксусная и др.). Полу-
чение дрожжей из гидролизатов или отходов глюкоз-
ного и спиртового произ-в (рис. 4) состоит из подго-
товки субстратов (нейтрализация, очистка, обога-
щение азот- и фосфорсодержащими в-вами, охлажде-
ние), выращивания дрожжей, пх выделения и сушки.
Товарные дрожжи содержат до 10% влаги, 40—50%
929
ГИДРОЛИЗНЫЙ ЛИГНИН — ГИДРОМЕТАЛЛУРГИЯ
930
белка, комплекс витаминов группы В и эргостерин.
Белок дрожжей содержит жизненно необходимые ами-
нокислоты (лейцин, изолейцин, валин, аргинин, ли-
зин, треонин, метионин, фенилаланин, тирозив, трин-
Рис. 4. Схема производства кормовых дрожжей: 1 — бак
для выращивания чистой культуры дрожжей; 2 — чан
для выращивания дрожжей; 3 — сепараторы; 4 — вакуум-
фильтр; 5 — плазмолизатор; 6 — сборник; 7 — насос;
S — распылительная сушилка; 9 — циклон; 10 — воздухо-
дувка; 11—скруббер.
тофан, гистидин). Содержание витаминов в дрожжах
(тиамин, пантотеновая кислота, рибофлавин, биотин,
фолиевая кислота, пиридоксин, никотиновая кислота
и др.) можно повышать путем подбора соответствую-
щих рас дрожжей — продуцентов витаминов — и из-
менением условий культивирования дрожжей. Об-
лучение дрожжей УФ-лучами приводит к обогащению
их витамином D2. Выход сухих белково-витаминных
дрожжей 40—45% от веса сахара, что позволяет по-
лучить до 250 кг из 1 т сухого растительного мате-
риала.
Использование гидролизного лигнина
основано на его термич. или химич. обработке. При
пиролизе лигнина происходит его карбонизация
с получением смол и полукокса. Из смол выделяют
фенолы (выход до 10%) и ацетат кальция, а полукокс
подвергают термич. активации и получают гранули-
рованные газовые или обесцвечивающие активные
угли с выходом 10—15% от исходного лигнина. При
гидрировании гидролизного лигнина образуется то-
луол, бензол, фенолы п др. продукты. Хлорированием
гидролизного лигнина получают хлорлигнин.
При действии на лигнин НКО3 образуется нитро-
лигнин. При обработке гидролизного лигнина
щелочами он растворяется, а при последующем под-
кислении выделяется активированный лигнин, яв-
ляющийся активным наполнителем синтетич. кау-
чука. Гидролизный лигнин вахолит применение как
компонент феноло-формальдегидпых смол; после от-
мывки от кислоты, сушки и дополнительного измель-
чения гидролизный лигнин применяется в пресспо-
рошках, а также в произ-ве древесно волокнистых
плит. Теплотворность сухого лигнина 5500— 6500
ккал/кг-, он хорошо горит в топках паровых котлов
в смеси с углем или в пылевидном состоянии. Высу-
шенный до остаточного содержания влаги 15% лиг-
нин легко брикетируется и может быть использован
как топливо для коммунальных целей и газогенера-
торных двигателей. Подробнее см. Лигнин.
Лит.: Шарков В. И., Гидролизное производство,
ч 1—3, М., 1945—50; Чалов Н. В., Горячих Е. Ф.
и Л е щ'у к А. Е., Гидролизная и лесохимич. пром-сть, 1959,
№ 3, 4; О д и н ц о в П. Н., [и д р.], там же, 1957, № 8; Ш н а ii-
дер Е. Е., Шпунтова М. Е., ЧепигоС. В., там же,
1960, J8 7; Роговин 3. А., Погосов Ю. Л., там же,
1958, A'i 1; Химическая наука и промышленность, 1957, 2,
Л'1 4 (посвящен, гидролизной пром-сти, см. ст. Лебедева Н. В.,
Чепиго С. В., Чалова Н. В., Беляевского И. А., Крюч-
ковой А. П., Сухоновского С. И. и др.). С. В. Чепиго.
ГИДРОЛИЗНЫЙ ЛИГНИН — см. Лигнин и Гид-
ролиз растительных материалов.
ГИДРОМЕТАЛЛУРГИЯ — извлечение металлов из
руд, концентратов и отходов различных произ-
водств в виде их соединений водными растворами
химич. реагентов с последующим выделением из
растворов.
Основные операции гидрометаллургического извле-
чения металлов. 1. Механическая об-
работка руды — дробление, измельчение, клас-
сификация, сгущевие с целью полного или частич-
ного раскрытия зерен минералов, содержащих извле-
каемый компонент, приведения руды в состояние,
удобное для последующего химич. извлечения ме-
талла (выщелачивания). 2. Изменение хи-
мич. состава руды или концент-
рата для подготовки ее к выщелачиванию — от-
мывка растворимых солей; хлорирующий, окисли-
тельный или восстановительный обжиг; спекание;
разложение химич. реагентами. Цель—разложение
химич. соединений, трудно поддающихся выщела-
чиванию, или удаление примесей, осложняющих вы-
щелачивание. 3. Выщелачивание — получе-
ние извлекаемого компонента руды в виде водного
р-ра или концентрата; может производиться одновре-
менно с измельчением и классификацией (в мельни-
цах, классификаторах) и в специальной аппаратуре
(чаны для выщелачивания, автоклавы). 4. Обез-
воживание и промывка производятся
на фильтрах, в сгустителях и др. способами с целью
отделения металлосодержащего р-ра от измельченного
материала. 5. Подготовка растворов к
выделению из них очищенных соединений или сво-
бодных металлов — отделение взвешенных частиц
(осветление), иногда также включает химич. опера-
ции, состоящие в удалении из р-ра вредных примесей.
6. Осаждение металлов или их со-
единений из растворов в виде нераство-
римых соединений (W, U, Ag), гидролиз (А1) и крис-
таллизация соединений металла; сорбция соединений
металла ионообменными смолами (U, Мо и др. редкие
элементы, Au, Ag); жидкостйая экстракция соеди-
нений металла органич. растворителями с последую-
щей реэкстракцией в водный раствор и осаждением
из него очищенного соединения металла (U и редкие
металлы); электролиз водных р-ров (Си, Zn и др ),
восстановление более электроотрицательным металлом
(цементация — Au, Ag, Си, Cd), адсорбция углем
(Au, Ag). 7. Переработка осадка от пред-
шествующих операций с целью дальнейшей очистки
выделенного соединения или чернового металла, или
непосредственное получение готового товарного ме-
талла (в зависимости от требований к чистоте послед-
него). Дополнительная очистка осажденного соеди-
нения производится одним или несколькими из след,
методов: перекристаллизацией, разложением или рас-
творением кислотой, щелочью и др. с последующим
выделением осадка нового соединения возгонкой,
прокаливанием, плавкой и др. Готовый товарный
металл получают восстановительной плавкой, иногда
восстановлением порошка окиси пли соли с получе-
нием порошка металла, электролизом из водных пли
расплавленных сред, переплавкой чернового металла
с одновременным аффинажем и т. д.
При гидрометаллургии, процессах широко ис-
пользуется флотация как предварительная для обо-
гащения руды, так и для выделения трудновьпцела-
чиваемой фракции или для извлечения из пульпы
восстановленного в ней металлич. порошка. Так,
медь из сернокислой соли восстанавливают в пульпе,
вводя железо, и затем флотируют цементную медь.
Применяют также гравитационное обогащение, наир,
выделение крупной фракции металла, медленно рас-
творяющейся при выщелачивании (напр., при извле-
чении золота цианированием).
931
ГИДРОМЕТАЛЛУРГИЯ
932
Физико-химические- процессы растворения минера-
лов. При химич. взаимодействии металла с раствори-
телем нейтральный атом металла переходит в ионное
состояние, образуя растворимое соединение. Раст-
ворение происходит легко в случае выщелачивания
руд пли концентратов, в к-рых металл присутствует
в окисленной (ионной) форме. Данный тип руд и их
продуктов представляет наибольшую область приме-
нения Г., напр. медные и урановые руды, обожжен-
ные цинковые концентраты, продукты хлорирующего
обжига. В нек-рых случаях для извлечения металла
растворителем необходимо окисление кислородом или
другим окислителем (наир., при содовом выщелачи-
вании руд, содержащих 4-валентный уран, для пе-
ревода последнего в 6-валентный). При растворении
металлов (самородных или восстановленных) неиз-
бежно окисление для перехода металла в ионное
состояние. Окисление металла с одновременной иони-
зацией окислителя (напр., растворенного в воде, мо-
лекулярного кислорода) в случае более благородных
металлов термодинамически возможно лишь при
затрате энергии, к-ран, напр., может быть получена
при образовании комплексного иона (цианирование
золота и серебра, аммиачное выщелачивание метал-
лич. меди, никеля).
Растворение минералов с различными видами хи-
мич. связи в кристаллич. решетке (ковалентная, ме-
таллич., ионная) характерно для выщелачивания суль-
фидов, арсенидов, селенидов, теллуридов. Растворе-
ние этих минералов, если предварительно не прове-
ден окислительный обжиг, в большинстве случаен
также требует окислении в пульпе, папр. при ам-
миачном выщелачивании медно-нпкелевых сульфидных
руд в автоклаве под давлением кислорода или воз-
духа. Скорость выщелачивания определяется факто-
рами, влияющими на растворение в гетерогенной
системе, образованной раствором и измельченной
рудой или концентратом. Перенос растворителя и уда-
ление продуктов реакции происходит в объеме р-ра
за счет конвекции (турбулентной диффузии), а в слое
на границе с минералом—за счет молекулярной (теп-
ловой) диффузии. Обычно реакция, происходящая при
гидромсталлургич. извлечении, находится в диффу-
зионной области; определяющим фактором является
скорость диффузии вещества, к-рая в данных усло-
виях лимитирует реакцию (низкая концентрация,
малая скорость диффузии). Возрастание скорости
растворения минерала происходит при увеличении
его относительной поверхности (т. е. степени измель-
чения), при ускорении перемешивания и при повыше-
нии темп-ры. В случае необходимости применения
для растворения двух реагентов (растворитель и окис-
литель), растворение определяется условиями диф-
фузионной стехиометрии. При несоответствии соот-
ношению диффузионной стехиометрии (к-рое соблю-
дается редко) скорость растворения определяется
диффузией того из реагирующих веществ, к-рого в
растворе меньше, т. е. для к-рого выражение D1'™ с/гг
имеет наименьшее значение (£> — коэфф, диффузии,
с—концентрация, п—коэфф, в стехиометрии, ур-нии,
т — дробный показатель, зависящий от интенсивно-
сти перемешивания; в неподвижной среде т=0).
Абе. скорость реакции окисления минералов кис-
лородом пропорциональна корню квадратному из
парциального давления кислорода: Fo — к • А Р&г,
где к — константа, А — поверхность частиц, РОл —
парциальное давление кислорода. Влияние темп-ры
на скорость реакции выражается ур-нием Аррениуса:
dlnh Е , m _
dT =------дуз, где к — констапта скорости, Т — абс.
темп-ра, Е — кажущаяся энергия активации, R —
универсальная газовая постоянная.
Форма поверхности и размер частиц минерала
определяют функциональную зависимость количества
растворившегося металла от времени контакта с раст-
вором; поэтому они влияют на возможное извлечение
и на выбор необходимого объема аппарата для выще-
лачивания. Загрязнение р-ров примесями может
ухудшить результаты как выщелачивания, так и
последующего выделения металлов или их соединений
пз р-ра. Примеси могут вызывать образование пле-
нок на поверхности растворяемых частиц Адсорби-
рованные ионы образуют экранирующий слой, за-
трудняющий поступление реагентов и взаимодействие
нх с ион-атомамп; они могут также облагораживать
электродный потенциал, вследствие чего тормозится
анодный процесс растворения металла. В других
случаях примеси в р-ре вызывают поглощение кисло-
рода или приводят к частичному осаждению металла
(папр., действие сульфидиона).
Реагенты. Растворителями для выщелачивания со-
единений металлов являются преим. серная к-та
(U, Си, Zn), сода (U в карбонатных рудах, Mo, W),
едкий натр (глинозем, W), аммиак (Си, Ni), цианистые
соли (Au, Ag), сернистый натрий (Sb, Hg), растворы
хлора и хлоридов (благородные металлы, РЬ, редкие
металлы), тиосульфаты (Ап, Ag).
Для жидкостной экстракции применяется р-р три-
бутилфосфата и ди-2-этилгексилфосфата в керосине
н др. После экстракции очищенное соединение ме-
талла извлекается из органич. растворителя водным
р-ром, часто с добавкой к-ты или др. реагента. В ка-
честве осадителей применяют цинк (Ап, Ag, Pt, Cd),
железо (Си), медь (Ag), водород под давлением (U),
полуактивный уголь (в виде порошка, прессованный
и кусковой), аниониты, катиониты. После сорбции
соединение металла снимается растворителем с ионита
и последний регенерируется. Для повышения произ-
водительности гидрометаллургия, цехов большое зна-
чение имеют коагуляция и флокуляция, интенсифици-
рующие сгущение и фильтрацию. Это достигается при-
менением синтетич. флокулянтов. Одним из наплуч-
ших является препарат полиакриламида — сепаран-
2610. Также эффективны: продукт ковденсации кубо-
вых остатков гексаметилендиампна, таллового масла
и дихлорэтана (НОДТ), полиакриламиды, сополимер
винилового спирта и мстилолкротоиамида и поливи-
ниловый спирт, продукты щелочного гидролиза по-
лиакрилонитрила (ПАНГ-55 и 56) и продукт конден-
сации кубовых остатков гексаметилендиампна и ди-
хлорэтана.
Аппаратура. Для выщелачивания применяют чаны с ме-
шалками (реакторы) для перемешивания пульпы, к-рые
подразделяют на механич., пневматич. и пневмомеханич.
Обычно пх соединяют последовательно по несколько для
непрерывного выщелачивания. В случае применения повышен-
ного давления и повышенной темп-ры пользуются автоклавами
непрерывного и периодич. действия. Выщелачивание переме-
шиванием пульпы (агитация) — основной вид выщелачива-
ния, иногда дополняющий первую стадию выщелачивания
в цикле измельчения (мельница-классификатор). Для удешев-
ления процесса и упрощения аппаратуры иногда (для окислен-
ных медных руд, для классифицированного песка легко вы-
щелачиваемой золотой руды) применяется выщелачивание
просачиванием р-ра через слой хорошо фильтрующей загрузки
(перколяция). Для интенсификации перколяции р-р
иногда предварительно насыщают кислородом воздуха, вво-
дят вакуум под фильтрующее днище и производят предвари-
тельную промывку для удаления примесей. Для более равно-
мерного просачивания раствор заливают снизу — под филь-
трующее (ложное) днище. Выщелачивание просачиванием
применимо для руды, раздробленной в пределах до 2 с.м. При
большей степени измельчения перколяцию применяют для
выщелачивания песка, предварительно отделенного от шлама
(ила) классификацией. Выщелачивание кислыми р-рами про-
изводится в гуммированной (стальной), керамиковой или дру-
гой кислотоупорной аппаратуре; для щелочных р-ров при-
годна стальная, иногда деревянная аппаратура. Выбор аппа-
ратуры зависит от процессов выщелачивания. Последние
подразделяют: 1) на выщелачивание просачиванием через
кусковой материал без отделении шлама (чаще всего при-
меняют для извлечения меди из окисленных руд р-ром серной
933
ГИДРООКИСИ
934
к-ты, иногда для окисленных пористых золотых руд); 2) на
выщелачивание отделенного классификацией мелкого мате-
риала перемешиванием н более крупного — просачиванием
{раздельное выщелачивание окисленных медных руд в слу-
чаях, требующих более тонкого измельчения; цианирование
нек-рых золотых руд); 3) на выщелачивание полным иловым
процессом (только перемешиванием пульпы) — наиболее ши-
роко применяется для гидрометаллургии, переработки золо-
тых, серебряных, урановых, иногда медных руд, обожженных
концентратов — цинковых, молибденовых, кобальтовых; 4) на
автоклавно-щелочные процессы при повышенной теми-ре
(вольфрам, глинозем); 5) на автоклавно-окислительные для
сульфидных, нек-рых урановых и золотых руд. Возможно
и другое аппаратурное оформление выщелачивания, напр.
выщелачивание в фильтр-прессах, применяемое в частных
случаях. Новый метод жидкостной экстракции (извлечение
органич. экстрагентом из кислого р-ра) дополняет выщела-
чивание другими методами или применяется для непосред-
ственного извлечения соединения металла из руды. Для луч-
шего экстрагирования иногда предварительно производят
изменение валентности ионов экстрагируемого в-ва (UO!+
окисляют до U‘+). Экстракция производится по принципу
противотока в экстракционных колоннах. Экстракт и отхо-
дящий раствор непрерывно удаляют в разных направлениях.
Извлечение экстракцией зависит от скорости подачи р-ров,
числа ступеней экстракции, концентрации экстрагента в раз-
бавителе (напр., в керосине), устройства аппаратов. Обезво-
живание и промывка производятся в сгустителях и фильтрах.
В качестве сгустителей применяют: гребцовые с центральным
и периферия, приводом; многоярусные, в частности промыв-
ные (противоточные). В качестве фильтров применяют: вакуум-
фильтры непрерывного (барабанные и дисковые) и периодич.
действия (рамные, барабанные, нутч-фильтры), фильтр-прессы
периодич. и непрерывного действия.
Осаждение из р-ров производится в аппаратах, конструк-
ция к-рых зависит от осадителя. Для химич. (растворимых)
осадителей нужны реакторы и фильтры. Порошкообразные
осадители (цинковая, алюминиевая пыль) вводятся в смеси-
тели с р-ром, осаждение затем может продолжаться внутри
перекачивающего насоса, в трубопроводе и в течение фильтра-
ции через слой осадителя на фильтре; последняя особенно
эффективна для окончательного осаждения металла из р-ра.
Kai: правило, р-ры перед осаждением из них металла должны
быть осветлены, т. е. освобождены от взвешенных частиц,
для чего перед осадительными аппаратами устанавливают
специальные вакуум-фильтры или фильтр-прессы. Иногда
требуется химич. очистка от растворимых примесей или уда-
ление кислорода пропусканием через колонки, находящиеся
под вакуумом. Можно осаждать металл или его соединения
в самой пульпе- (золото, медь, уран), для чего, напр., в нее
вводят сетчатые корзинки с ионитами (для извлечения урана);
в других случаях в пульпу вводят осадитель (железо для меди,
уголь для золота), к-рый затем выделяют флотацией. Осажде-
ние кусковым осадителем (железо для меди; цинковая стружка
или уголь для золота) производят в желобах или в ящиках
с перегородками для зигзагообразного движения р-ра вверх
и вниз через слой осадителя. При осаждении гидролизом,
напр. глинозема из алюминатных р-ров в декомпозерах,
производится длительное и медленное перемешивание (после
введения в раствор затравки в виде гидроокиси алюминия)
в течение 100—120 час. В случае выделения цинка (из очи-
щенного р-ра после выщелачивания) или меди производится
осаждение на катоде электролизом с нерастворимыми анодами.
Лит.: Плаксин И. Н., Юхтанов Д. М., Гидро-
металлургия, М., 1949; М е е р с о н Г. А., 3 е л и к м а н А. Н.,
Металлургия редких металлов, М., 1955; Ч и ж и к о в Д. М.,
Металлургия тяжелых цветных металлов, М.—Л., 1948;
Беляев А. И., Металлургия легких металлов, 4 изд., М.,
1954; Плаксин И. И., Металлургия благородных метал-
лов, М., 1958; А г е е н к о в В. Г., К а к о в с к и й И. А.,
Электрометаллургия водных растворов. (Расчеты), М., 1947;
Кисляков И. П., Металлургия редких металлов, М.,
1957; Proceedings ot the Second United Nations International
Conference on the peaceful uses of atomic energy, v. 3, Gen.,
1958; Van ArsdaleG. D., Hydrometallurgy of base me-
tals, N. Y. — Toronto — L., 1953; Newton J., Extractive
metallurgy, N. Y. — П., 1959; International Mineral Proces-
sing Congress 1960, L., 1960. И. И. Плаксин.
ГИДРООКИСИ — химич. соединения окислов эле-
ментов с водой; один из главных классов неорганич.
химич. соединений. Часто Г. называют гидратами
окислов, однако это не соответствует природе таких
соединений, т. к. они не содержат молекул Н2О в ка-
честве структурных единиц, а включают лишь гидро-
ксильные группы ОН, связанные с атомом элемента R.
Известны Г. почти всех химич. элементов; нек-рые
из них встречаются в природе в виде минералов.
В растворах Г. подвергаются электролитич. диссо-
циации, причем отщепляются либо ионы ОН* (ос-
нования), либо Н+ (кислоты), либо — в зависимости
от условий — как ионы ОН*, так и ионы Н+ (амфо-
литы). Ослабление основных и усиление кислотных
свойств Г. связано с увеличением поляризующего дей-
ствия атома R химич. элемента, т. е. с убыванием его
радиуса и возрастанием валентности, а также с уве-
личением числа внешних электронов. Когда В имеет
малый заряд и сравнительно большой радиус, его по-
ляризующее действие на окружающие группы ОН
невелико, и они выступают в соединении как целое.
Поэтому типичными основаниями являются Г. эле-
ментов, находящихся в главных подгруппах I и [I
групп периодич. системы [КОН, NaOH, Са(ОН)2 и
т. д.], а также NH4OH и амины. По мере увеличения
поляризующего действия R основные свойства Г.
ослабляются и понвляются кислотные свойства. Из
элементов II группы Ве и Zn дают амфотерные Г. В III
группе амфотерны Г. Al, Ga, In. Амфотерность харак-
терна для большинства элементов IV группы перио-
дич. системы. Когда R имеет большой положительный
заряд и малый радиус (что типично для неметаллов),
усиление его поляризующего действия приводит к
тому, что Н становится лабильным, и преобладает
диссоциация по кислотному типу. Уже среди Г.
элементов III группы Г. бора типичная кислота
(Н3ВО3). В IV группе кислотами являются Г. угле-
рода и кремния, однако эти кислоты еще очень сла-
бые. Г. многих элементов V, VI и VII групп — силь-
ные кислоты. Внутри групп увеличение порядкового
номера ведет к усилению основных и ослаблению кис-
лотных свойств Г. Указанные закономерности наибо-
лее четко проявляются в главных подгруппах пери-
одич. системы, т. е. у ионов с 8-электронпой наружной
оболочкой. В побочных подгруппах, т. е. у ионов с мно-
гоэлектронной наружной оболочкой эти закономер-
ности имеют в общем тот же характер, но бывают ос-
ложнены из-за дополнительного эффекта поляризации,
обусловленного деформацией собственной электронной
оболочки иона. Если элемент образует несколько Г.,
то повышение его валентности ведет к ослаблению ос-
новных и усилению кислотных свойств Г.; так,
Fe(OH)2 — более сильное основание, чем Fe(OH)3,
a H2SO< — более сильная кислота, чем H2SO3. (См.
также Поляризация и П ериодическая система элемен-
тов М енделеева).
Термич. устойчивость Г. изменяется в очень широ-
ких пределах и весьма сложным образом зависит от
свойств атома, образующего Г. Так, Г. щелочных и
щелочноземельных металлов, кроме LIOH, кипят без
разложения выше 1000°, a AgOH неустойчива уже при
обычной темп-ре. В некоторых случаях, напр. у Г.
щелочных и щелочноземельных металлов, термич.
устойчивость увеличивается с возрастанием основных
свойств. Для Г. элементов с многоэлектронными обо-
лочками, как правило, характерна значительно мень-
шая термич. устойчивость, чем для Г. 8-электронных
ионов, что обусловлено дополнительным эффектом
поляризации. Кристаллич. структуры Г. находятся
в тесной связи с их химич. свойствами. Г. с сильно вы-
раженными основными свойствами имеют ионную ре-
шетку (высокотемп-рные кристаллич. модификации
NaOH, КОН). С повышением поляризующего дейст-
вия атома R, т. е. с уменьшением основных свойств
Г., появляются структуры слоистого типа [LIOH,
Mg(OH)a, Са(ОН)2]. Структуры нек-рых амфотерных
Г. построены из координационных групп Н(ОН)4
или R(OH)6, имеющих общие группы ОН; между от-
дельными группами ОН существуют гидроксильные
связи (разновидность водородной связи). Для кристал-
лов кислот характерны молекулярные структуры,
т. е. в узлах их кристаллич. решетки находятся моле-
кулы, связанные между собой водородными связями.
Поскольку такое изменение характера химич. связи
между составляющими кристаллич. решетку части-
цами ведет к уменьшению прочности решетки, некото-
рые кислоты при обычных условиях — жидкости,
935
ГИДРООЧИСТКА — ГИДРОФИЛЬНОСТЬ И ГИДРОФОБНОСТЬ
936
тогда как основные и амфотерные Г. являются твер-
дыми телами.
Г. с сильно выраженными основными или кислот-
ными свойствами хорошо растворимы в воде, слабые
основания и амфотерные Г. практически нераство-
римы. Растворимые Г. образуются при взаимодейст-
вии с водой соответствующих окислов, а также сво-
бодных щелочных и щелочноземельных металлов.
Для получения нерастворимых Г. обычно служат об-
менные реакции между р-рами соответствующих солей
и сильных кислот или оснований. Наряду с Г., име-
ющими лишь препаративное или теоретич. значение,
нек-рые Г. являются предметом основного неорганич.
синтеза и производятся в количестве многих сотен
тысяч тонн. Более подробно о конкретных свойствах
отдельных Г. см. статьи об окислах и Г. элементов.
На практике термин «Г.» часто применяют только по
отношению к основным и амфотерным Г.
Лит.: Некрасов Б. В., Курс общей химии, 12 изд.,
М., 1955; Remy Н., Lelirbuch der anorganischen Chemie,
Bd 1—2, 9 Anti,, Lpz., 1957—59.
ГИДРООЧИСТКА — процесс селективного гидри-
рования топлив, применяемый для удаления из них
непредельных, сернистых, азотистых и кислородных
соединений и примесей металлов. Сырьем для Г. слу-
жат различные нефтяные фракции, от бензина до
тяжелых вакуумных дистиллятов включительно, как
прямой гонки, так и крекинга. Г. может также при-
меняться для очистки сырья каталитич. риформинга
и каталитич. крекинга. В результате Г. происходит
почти полное гидрирование олефинов, сернистых, азо-
тистых и кислородных соединений, при этом разрыва-
ются связи в цепи или в кольце по месту вхождения
атомов серы, азота или кислорода, с образованием
соответственно сероводорода, аммиака или воды. Фрак-
ционный состав и содержание ароматич. углеводоро-
дов при этом не изменяется, но повышается стабиль-
ность топлива, сильно снижается содержание в них
серы, повышается приемистость к тетраэтилсвинцу
и несколько возрастают октановое и цетановое числа.
Процесс обычно проводится со стационарным катали-
затором, в качестве к-рого чаще всего применяется
окись молибдена на окиси алюминия; применяются
также окислы или сульфиды других элементов —
вольфрама, хрома, никеля, кобальта, часто в смеси.
Г. проводится под давлением водорода от 4 до 70 ат и
темп-рах 300—425°, иногда выше. Расход водорода
составляет 1,0—1,4 м3/м3 сырья на каждую единицу
понижения бромного числа и 9—18 м3/м3 сырья на
каждый процент снижения серы. Процесс экзотерми-
чен, и иногда подогрев сырья требуется только при
пуске установки. Сырье, поступающее на Г., подогре-
вают в трубчатой печи до 285—425° и далее подают
в реактор с неподвижным катализатором, куда нагне-
тается также свежий п циркуляционный водород.
При выходе из реактора продукты реакции охлажда-
ются и поступают в сепаратор, в к-ром происходит
их разделение; газ (водород) возвращается в реактор,
а товарные продукты поступают в отпарную колонну,
в к-рой удаляется оставшийся в продукте сероводо-
род. Выход продуктов при Г. близок к 100%. По-
скольку в Г. расходуется водород, то убыль его
должна восполняться из постороннего источника,
поэтому установки Г. часто проектируются в сочета-
нии с установками каталитич. риформинга, дающими
водород в качестве побочного продукта. За рубежом
известны различные модификации Г. под названием
«Гидрофайнинг», «Юнифайнинг», «Аутофайнинг» ит. п.
Лит.: Рапопорт И. Б., Искусственное жидкое топливо,
2 изд., М., 1955' Технологические схемы процессов перера-
ботки нефти в США, [Сб. пер. с англ., под ред. А. Г. Басистова
и др.], М„ 1956; то же, [Сб. пер. с англ., под ред. А. Л. Фей-
гина], М 1957. В. В. Щекин.
ГИДРОСФЕРА — прерывистая водная оболочка
Земли. По величине минерализации природные воды
подразделяются на пресные (до 1 г солей на 1 кг воды),
солоноватые (1—25 г/кг), морской солености (25—
50 г/кг) и соленые (св. 50 г/кг). Основу минерализации
составляют след, ионы: Na+, Mg21-, Са21-, К+, СН, SO%
и HCOj, встречающиеся в природных водах в самых
различных соотношениях. Из других составляющих
характерными для морских вод являются бром, бор
и фтор. Большинство других элементов содержится
в крайне малых концентрациях, обычно менее 1 мг/л.
Средний химич. состав Г. близок к составу океана,
т. к. последний резко доминирует среди природных
вод. Средний элементарный состав воды океана (ве-
совые %) при солености 35 г/кг приведен в таблице.
Эле- мент О/ /о Эле- мент % Эле- мент О/ /0
о н С1 Na Mg S Са К Вг С Sr В Si F 85,89 10,82 1,90 1,06 0,13 0.088 0.040 0.038 6,5 • 10-3 2,8 • Ю-з 1,3 • Ю-з 4.8-10-4 2-10-4 1.4- 10-4 Na Rb Li Alс P J As Ba Zn Mn Pb Fe Os U 0.3-7 • 10-s 2-10'6 1,2 • 10-s 1 • 10-s 5 • 10« 5 • 10—в 1,5 • 10-« 1 • 10 « 5•IO-? 5• IO-? 4•10-7 2•10-7 2•10-7 1.5 • 10-7 Se Th MO Ce Ag U La Y Ni CuB Sc Hg Au Ra 1 • 10-7 5• 10-8 5 • 10-8 4• 10-8 3-10-8 3-10-8 3•10-8 3 . 10-8 1 • 10-8 2• 10-8 4. 10-» 3 • 10-9 4 .10-“> 7 . 10-45
а Без учета растворенного азота. 6 по другим данным,
2—3• 10“7. в По другим данным, порядка 10'7.
Соленость большей части Мирового оке а-
н а колеблется в пределах 34—37 г/кг. В опресненных
участках, особенно во внутренних морях, соленость
падает и нарушается соотношение между главными
ионами. Наиболее подвержено колебаниям содержание
т. н. биогенных элементов (NO3‘, NO2, NHf,соединений
фосфора, кремния) и растворенных газов. Величина
pH воды океана 7,6—8,4, у поверхности 8,1—8,4,
содержание растворенных органич. веществ ок. 2 мг/л.
Причиной солености океана частично являются соли
первичного океана, частично — последующее вывет-
ривание литосферы и деятельность речного стока.
Реки могут иметь малую минерализацию (до
200 мг/л), среднюю (200—500 мг/л), повышенную
(500—1000 мг/л) и высокую (свыше 1000 мг/л). В ион-
ном составе речной воды при минерализации до 500 мг/л
обычно преобладают ионы НСО3 иСа2 + ; с ее повыше-
нием растет содержание ионов SO|~, Cl nNa+. Пример-
но такие же закономерности в составе наблюдаются
и у пресных озер. Высокую минерализацию (до
250 г/кг) имеют соляные озера, к-рые по направлению
процесса данной физико-химич. системы, ведущему
к выделению твердой фазы, могут быть подразделены
на карбонатные, сульфатные и хлоридные. Наиболее
разнообразны грунтовые воды, среди к-рых
могут быть пресные воды и рассолы различного со-
става, в зависимости от засушливости климата. Наи-
менее минерализованы атмосферные о с а д-
к и (обычно до 50 мг/л).
Лит.: Вернадский В. И., История минералов
земной коры, т. 2 — История природных вод, ч. 1, вып. 1—3,
Л., 1933—36; Виноградов А. П. Усп. химии, 1944,
13, вып. 1, А л е к и н О. А., в кн.: Тр. Гос. гидрология,
ин-та, вып. 33, Л., 1951; Бруевич С. В., в кн.: Элементы
химического баланса Каспийского моря, вып. 14, М.—Л.,
1941. О. А. Алехин.
ГИДРОФИЛЬНОСТЬ И ГИДРОФОБНОСТЬ — ха-
рактеристики интенсивности молекулярного взаимо-
действия иеществ и поверхностей тел с водой в дисперс-
ных системах, в к-рых вода является дисперсионной
средой. Г. и г. являются частным случаем лиофильно-
сти и лиофобности, как характеристик взаимодействия
937
ГИДРОФИЛЬНОСТЬ И ГИДРОФОБНОСТЬ — ГИДРОФОБНЫЕ ПОКРЫТИЯ
938
веществ с разными жидкостями — растворителями
различной полярности.
Гидрофильность, как и лиофильность
вообще, определяется прежде всего величиной сво-
бодной энергии связи данного вещества или поверх-
ности данного тела, напр. дисперсной фазы, с водой.
Т. обр. гидрофильность можно Оценить соответствую-
щими тепловыми эффектами, измерения к-рых при
различных т-рах позволяют с помощью методов химич.
термодинамики вычислить свободную энергию связи.
В этом смысле гидратацию следовало бы рассмат-
ривать как проявление гидрофильности. Обычно же
гидрофильность характеризуют адсорбционной связью
с водой, образованием с нею неопределенных соеди-
нений. Полная характеристика гидрофильности выра-
жается распределением количества воды по величинам
энергии связи. Для воды, адсорбционно связанной
с единицей поверхности данного твердого тела, прак-
тически учитывают только энергию связи первого слоя
молекул воды (мономолекулярного слоя), так как
энергия связи последующих слоев значительно меньше.
Гидрофильность выражается, т. обр., дифференциаль-
ными теплотами смачивания данного тела водой на
единицу его поверхности или теплотами адсорбции
водяного пара. Для этого могут быть измерены ин-
тегральные теплоты смачивания или адсорбции при
различных количествах адсорбционно связанной воды.
Гидрофобность должна рассматриваться
как малая степень гидрофильности, т. к. все вещества
обладают ею в большей или меньшей степени. В этом
смысле гидрофобные поверхности неточно называют
в технике водоотталкивающими. В действительности
даже наиболее гидрофобная (чисто углеводородная) по-
верхность парафинов адсорбирует воду, т. е. является
гидрофобной лишь в смысле слабой гидрофильности.
Г. и г. поверхностей тел могут быть оценены также по
смачиванию их водой. При этом гидрофильными считают по-
верхности, по к-рым вода растекается полностью без образо-
вания краевого угла смачивания. Все такие гидрофильные
поверхности различаются по энергиям адсорбционной связи
с водой. Гидрофильность поверхности тел, не полностью сма-
чиваемых водой и образующих с каплей воды конечный краевой
j угол 0, количественно
а/ г характеризуется величи-
-------------------------------------------- ной смачивания В, рав-
ной косинусу краевого
а32 f «Уз,------------------\ угла: В = COS0 —
где Р = - напря-
^zzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzzz/ же.1ие смачивания, T. е.
разность между уд. сво-
Рис. 1. Гидрофильная поверхность, бодными поверхностными
энергиями поверхности
данного тела (3) в равновесии с газовой (паровой) фазой (0) о30
и соответственно с водой (1) 0ЗЬ а 01О — легко измеримое по-
верхностное натяжение (уд. свободная поверхностная энер-
гия) на границе вода — пар. Абсолютная количественная
оценка Г. и г. поверхности данного твердого тела может быть
дана по величине избирательного смачивания В12 ее двумя
жидкостями противоположной полярности — водой (1) и уг-
леводородом (2) (напр., геп-
2______ таном; рис. 1, рис. 2).В1Я =
— COS0 = — 0з2 — 031 /012. При
X^i2 jr этом напряжение смачивания
} \ р12 = 032 — 031J определяющее
Xl ) знак и величину смачивания
Оз2\ °3i / В12, является разностью уд.
свободных поверхностных
j энергии на границе данного
твердого тела с углеводоро-
Рис. 2. Гидрофобная поверх- дом <т32 и с водой ц31. При
ность. 0°:gBl2s;+1, О°'0'<9О°,
Р,2Ь-0 поверхность твердого
тела является гидрофильной (рис. 1), и вода вытесняет с нее
жидкий углеводород в тем большей степени, чем меньше краевой
угол 0 и чем больше В12 (или ₽12). При 0 > В12 >—1,
9O°=s0s=18O° поверхность является гидрофобной (олеофиль-
ной) и углеводород вытесняет с нее воду в тем большей
степени, чем больше 0, т. е. чем меньше В,2 (р12) (рис. 2,
краевой угол всегда отсчитывается в водной фазе). Предельный
случай гидрофильности — полное вытеснение углеводорода
водой с поверхности данного тела (полное избирательное сма-
чивание водой), отвечает условию 0 = 0, ₽12 >: <т12. Соответ-
ственно предельная олеофильность (гидрофобность) опреде-
ляется условием полного вытеснения воды с поверхности
углеводородом [0 = 180°].
Гидрофильность твердых поверхностей резко пони-
жается в результате адсорбции поверхностно-активных
веществ под влиянием ориентированных адсорбцион-
ных слоев, их молекул или ионов, особенно резко
вследствие хемоадсорбционной связи полярных групп
молекул и ориентации углеводородных цепей в окру-
жающую среду. Такое снижение гидрофильности,
наз. гидрофобизацией, обнаруживается по резкому
I понижению смачивания водой и лежит в основе дейст-
вия флотореагентов, а также различных водооттал-
кивающих обработок волокон, тканей и др. (см.
Гидрофобные покрытия). Характерны явления гидро-
фобизации при действии щелочных солей жирных к-т
и др. анионактивных органич. веществ на поверхно-
стях ионных кристаллов с химич. фиксацией полярных
групп на поливалентных катионах, а также при дей-
ствии алкилксантогенатов на поверхности металлов
и сульфидов и йатвонактивных реагентов (алкилами-
нов и солей четырехзамещенных аммониевых основа-
ний или алкилпиридинов) на силикатах и двуокиси
кремния. Даже ничтожные примеси поверхностно-
активных веществ в виде загрязнений могут сильно
понизить гидрофильность, что дает способ оценки
чистоты гидрофильных поверхностей, напр. стекла,
к-рые при загрязнении перестают полностью смачи-
ваться водой.
Гидрофильными являются все тела, в к-рых интен-
сивность молекулярных (атомных, ионных) взаимо-
действий достаточно велика (корунд, карборунд,
алмаз и др.). Особенно резко выраженной гидрофиль-
ностью обладают минералы с ионными кристаллич.
решетками (окислы и их гидраты, карбонаты, силикаты,
сульфаты, фосфаты, галогениды, глины, а также стек-
ла). Металлы, полупроводники, а также сргшич.
вещества, особенно с преобладанием углеводородных
групп в молекуле, гидрофобны (парафин, нафталин,
далее жиры, воски, битумы и др.). При переходе от
щелочных и щелочноземельных катионов к легко поля-
рвзующимся катионам (тяжелых металлов и др.),
а также в лиотропных рядах ионов с уменьшением их
радиуса гидрофильность возрастает. Понятие гид-
рофильности применимо не только к телам (фазам),
у к-рых оно является свойством поверхности, но и
к отдельным молекулам, их группам, атомам и ионам.
Все полярные группы, входящие в состав поверхностно-
активных веществ, обладающие дипольным моментом
(—ОН —СООН, —NH2 и др.), являются гидрофиль-
ными. Именно они увеличивают растворимость в воде,
тогда как химически связанные с ними углеводород-
ные радикалы понижают ее. Результатом такого гид-
рофобно-гидрофильного баланса и является итоговая
растворимость вещества в воде. Гидрофильные (в об-
щем случае — лиофильные) тела самопроизвольно
образуют коллоидные р-ры в воде или в другой жид-
кости — коллоиды, являющиеся предельно высоко-
дисперсными термодинамически устойчивыми двух-
фазными системами.
Лит.: Ребиндер П. А. [и др.], Физикохимия флота-
ционных процессов, М.—Л. — Свердловск, 1933; Думай-
сиий А. В., Лиофильность дисперсных систем, [Киев],
1960. II. А. Ребиндеп.
ГИДРОФИЛЬНЫЕ КОЛЛОИДЫ — см. Лиофиль-
ность и лиофобность коллоидов, Гидрофильность и
гидрофобность.
ГИДРОФОБНЫЕ КОЛЛОИДЫ — см. Л иофилъ-
ностъ и лиофобность коллоидов, Гидрофильность и
гидрофобность.
ГИДРОФОБНЫЕ ПОКРЫТИЯ (иногда неточно
называемые водоотталкивающими) — тончайшие слои
нек-рых несмачивающихся водой веществ на поверх-
ностях гидрофильных материалов, придающие пос-
ледним гидрофобные свойства. Г. п. широко приме-
няются для защиты разнообразных материалов от
действия воды, ухудшающей их физико-химич. и ме-
939
ГИДРОФОРМИНГ — ГИДРОХИМИЯ
940
ханич. свойства. Г. п. получают, обрабатывая соот-
ветствующие материалы р-рами или эмульсиями гидро-
фобизующих препаратов. Последние (обычно в резуль-
тате адсорбции, сопровождающейся поверхностными
химич. реакциями) образуют на поверхности всех
частиц (или пор) материала, пришедших в соприкос-
новение с р-ром, гидрофобное, т. н. водоотталкиваю-
щее, покрытие. Гидрофобность этого покрытия обус-
ловливается тем, что в процессе обработки гидрофиль-
ные группы, входившие в состав гидрофобизующего
препарата, удаляются, и на частицах материала остает-
ся слой, наружная поверхность к-рого состоит из
одних гидрофобных углеводородных радикалов.
Для получения Г. п. чаще всего применяют: 1) мы-
ла поливалентных металлов (А1, Си, Zr и др.) часто
в комбинации с парафиново-стеариновой эмульсией;
2) катион активные соединения, в к-рых четвертичный
атом азота соединен через метиленовую связь с амид-
ной или сложноэфирной группой, содержащей длин-
ный алифатич. радикал; 3) мономерные и полимерные
.кре.мнийорганич. соединения, содержащие у атома
кремния, наряду с углеводородным радикалом, ре-
акционноспособную функциональную группу (Н, С1,
OCOR, NHR, ONa и т. д.). Кремнийорганич. Г. п.
широко применяются для гидрофобизации строитель-
ных, электро- и теплоизоляционных материалов,
тканей, бумаги, кожи, стекла, керамики и т. д. Эти
покрытия совершенно невидимы и не меняют ни по-
ристость, ни цвет, ни фактуру поверхности материала,
на к-рый нанесены. Образование гидрофобных крем-
нийорганич. покрытий, химически связанных с по-
верхностью-материалов, основано на взаимодействии
реакционноспособных функциональных групп, свя-
занных с атомом кремния (галоген, Н, OR, OCOR,
N Н2 и т. д.), с гидроксильными группами или окислами
металлов, входящими в состав гидрофобизуемого ма-
териала. Благодаря этому поверхность материала
химически связывается (через атом кислорода) с ато-
мами кремния,входящими в состав водоотталкивающей
пленки. Такое химич. связывание обусловливает вы-
сокую устойчивость Г. п. в эксплуатации. В случае
отсутствия химич. связи между гидрофобизующим
покрытием и материалом, на к-ром оно находится
(напр., при нанесении тончайшего слоя парафина),
это покрытие оказывается неустойчивым и легко уда-
ляется при стирке, действии растворителей и т. д.
(см. также Отделка тканей специальная).
Лит.: Воронков М. Г., Долгов Б. Н., При-
рода, 1954, № 5, с. 22; А н д р и а н о в К. А., Теплостойкие
кремнийорганические диэлектрики М.—Л., 1957; С и м и-
г п н П. А., 3 усман М. Н., Райхли н Ф. И.,
Защитные пропитки текстильных материалов М., 1957;
Воронков М. Г., Шорохов Н. В., Строительные
материалы 1959, № 7, с. 12; В о р о н к о в М. Г. и К а-
Л у г и и Н. В Ж. прикл. химии, 1959, 32, № 7, с. 1582;
Шварц А. и Перри Д ж., Поверхностно-активные ве-
щества, пер. с англ., М., 1953; Вельт ц ин В. и Хау-
ш и л ь д Г., О силиконах и их применении в отделке тек-
стильных изделий, пер. с нем., М., 1958; Фротшер Г.,
Химия и физическая химия текстильных вспомогательных
материалов, пер. с нем., т. 1, М., 1958; Rochow Е. G.,
An introduction to the chemistry ot the silicones, 2 ed.,
N. Y.—L 1951; Me-Grego г R. R., Silicones and their
uses, N. Y. — Toronto — L., [1954]; Ballot V., C h v a-
lovsky V. a Rathousky J., Technicke pouziti
silikond, Praha, 1959. M. Г. Воронков.
ГИДРОФОРМИНГ — один из видов каталитич.
переработки нефтепродуктов, применяемой для повы-
шения антидетонац. свойств бензинов, обогащения их
ароматич. углеводородами и снижения и них содержа-
ния серы. Г. проводится иод давлением водорода в при-
сутствии катализатора, содержащего ок. 10% окиси
молибдена на окиси алюминия. Подробнее см. Рифор-
минг.
ГИДРОХИМИЯ — наука о химич. составе природ-
ных вод и его изменениях в связи с химич., физич.
и биологич. процессами, протекающими в окружаю-
щей среде. Первыми объектами изучения Г. явились
минеральные воды, анализы к-рых начали выполнять-
ся с 18 в.Г. тесно соприкасается с геохимией, минерало-
гией, гидрологией, гидрогеологией, метеорологией,
почвоведением, гидробиологией и др. науками. Гид-
рохимия. данные учитывают при водоснабжении пить-
евой и промышленной водой, при орошении, расчете
спуска сточных вод в открытые водоемы (см. Воды
i сточные), в рыбном хозяйстве, при бальнеология.
характеристиках минеральных вод (см. Воды мине-
ральные), при изучении коррозии строительных ма-
териалов, геохимич. поисках полезных ископаемых,
в химич. технологии природных вод и для многих
др. целей. Современная Г. изучает природные воды
с различных сторон.
1. Вода как растворитель сложного комплекса ми-
нералов земной коры и взаимодействие воды с твер-
j дой фазой, атмосферой и организмами. В этом разделе
I рассматриваются растворимость веществ, встречаю-
| щихся в природе, форма содержания их в воде, ста-
1 бильность в р-ре, сорбционные, обменные, окисли-
1 тельно-восстановительные процессы и мн. др. К этому
же разделу, весьма близкому к геохимии, следует
отнести и общие вопросы круговорота веществ и миг-
рацию элементов в гидросфере.
2. Химич, состав и гидрохимия, режим определен-
ных видов природных вод, зависимость их изменений
от физико-географич. условий. Этот обширный раздел
близко примыкает к гидрологии, наиболее крупными
его частями являются химия поверхностных вод,
химия моря и химия подземных вод. Химия по-
верхностных вод изучает химич. состав
в реках, озерах, искусственных водоемах, его изме-
нения по территории и глубинам, сезонные и суточные
колебания, а также условия формирования состава
в зависимости от окружающей среды. Важное значе-
ние имеет прогноз химич. состава создаваемых в за-
сушливых областях страны водохранилищ, а также
изучение и борьба с загрязнениями, вносимыми в во-
доем сточными водами пром-сти. Практич. ценность
имеют исследования соляных озер, богатых минераль-
ным сырьем, необходимым химич. пром-сти. Химия
моря, тесно примыкающая к океанологии, наряду
с изучением солености, биогенных веществ и раство-
ренных газов в водах океанов и морей в зависимости
от гидродинамич., гидрометеорология, и гидробио-
логия. факторов, изучает формы содержания и мигра-
цию микроэлементов, генезис и процессы метаморфи-
зации органич. веществ, углекислотное и другие рав-
новесия в морской воде, процессы взаимодействия мор-
ской воды с речными водами и морскими донными осад-
ками и ряд других вопросов. Химия подзем-
ных вод включает изучение химич. состава грун-
товых, пластовых, артезианских, минеральных вод
и имеющих важное практич. значение вод нефтяных
месторождений. Важнейшими направлениями здесь
являются генетич. вопросы формирования состава
вод, изучение взаимодействия воды с окружающими
породами, происходящего под высокими давлениями
и часто при повышенных темп-рах, замедленном во-
дообмене и своеобразных микробиология, условиях.
Большое значение издавна имеет изучение минераль-
ных вод, весьма разнообразных по составу и происхож-
дению.
3. Методика гидрохимия, исследований. Этот раздел
является специальной ветвью аналитич. химии, при-
менительно к специфике анализа природных вод.
В современной Г., помимо общих методов аналитич.
химии, широко применяются методы спектроскопии,
полярографии, хроматографии, методы меченых ато-
мов и различные физико-химич. методы.
Ли т.: Аленин О. А., Основы гидрохимии, Л., 1953;
его же, Гидрохимия рек СССР, ч. 2—3, Л., 1948—49
941
ГИДРОХИНОН — ГИПЕРОНЫ
942
(Труды гидрология, ин-та, вып. 10 и 15); его же, Химический
анализ вод суши, Л., 1954; Бруевич С. В., Методика
химической океанографии, М., 1933; его же Гидрохимия
Среднего и Южного Каспия (по работам 1934 г.). М.—Л.,
1937; Бунеев А. Н., Основы гидрогеохимии минеральных
вод осадочных отложений, М., 1 956; Вернадский В. II.,
Избр. соч., т. 4, кн. 2, И., I960; Виноградов А. П.,
Усп. химии, 1944, 13, вып. 1, с. 3; В а л я ш к о М. Г., ДАН
СССР, 1939, 23, № 7, с. 688; Воронков II. П., Формиро-
вание химического состава поверхностных вод., Л., 1955;
[Воронков П. II., М у с и н а А. А. и С в и т а тн е в
А. И.], Руководство по химическому анализу морских вод,
Л., 1950; Дацко В. Г., Органическое вещество в водах
южных морей СССР, М., 1959; Драчев С. М. [и др.],
Методы химического и бактериологического анализа воды,
М., 1953; их же, Приемы санитарного изучения водоемов,
М., I960; Д зепс -Л йто вс кнй А. И., в [сб. ст.]:
Методика комплексного изучения минеральных озер, Л.—М.,
1936; К у р и а к о в Н. С., Собр. избранных работ, т. 2,
Л., 1939; Книпович Н. М., Гидрология морей и соло-
новатых вод, М.—Л., 1938; К о в д а В. А., Происхождение
II режим засоленных почв, т. 1, М.—Л., 1946; Максим о*
вич Г. А., Химическая география вод суши, М., 1955;
Методы анализа рассолов и солей. Инструкция по анализу
галург. сырья, под ред. М, Г. Валяшко [и др.], Л.—М 1950;
Овчинников А. М., Минеральные воды, М.—Л.,
1947; Прицлонский В. А. и Лаптев Ф. Ф.,
Руководство по изучению физических свойств и химического
состава подземных вод, М.—Л., 1949; Посохов Е. В.,
Соляные озера Казахстана, М., 1955; Скопинцев Б. А.,
Органическое вещество в природных водах (водный гумус),
Л., 1950 {Труды Гос. океаногр. ин-та, вып. 17 (29)1; Современ-
ные методы химического анализа природной воды. [Сб. ст.
Отв. ред. О. А. Аленин], М., 1955; Сулин В. А., Воды
нефтяных месторождений в системе природных вод, М.—Л.,
1946; Щукарев С. А. и Толмачева Т. А., Ж.
Русского физ.-хим. о-ва при Ленингр, ун-те. Часть хим., 1930,
62, вып. 4, с. 777. О. А. Алекин.
ГИДРОХИНОН (n-диоксибензол), мол. в. 110,11 —
бесцветные, чаще светло-серые кристаллы; т. пл.
169—171°; т. кип. 285—287°; плотность 1,358;
ОБ сублимируется без разложения несколь-
Око ниже температуры плавления, образуя
лабильную форму. В 100 г воды раство-
ряется 6 г (15°); хорошо растворим в горя-
I чей воде, спирте, эфире, плохо — в бензоле.
Водные р-ры Г. на воздухе окисляются и
буреют, в щелочной среде окисление ускоряется;
при окислении образуется хингидрон и затем п-хи-
пон. Г. является сильным восстановителем, он вос-
станавливает раствор Фелинга на холоду и амми-
ачный р-р AgNO3 при подогревании. Впервые Г.
был получен в 1844 Вёлером пз хинона; в технике
его синтезируют из я-фонолсульфокиелоты (илп
и-хлорфенола) сплавлением с едкими щелочами
(катализатор — соли меди или KJ) и др. способами.
Применяется Г. как проявитель в фотографии, как
антиоксидант, напр. прибавление 0,025% его стабили-
зирует акролеин в течение 6 мес. Г. используют в син-
тезе полупродуктов, применяющихся в производстве
различных органических красителей: его хлорируют,
получают алкильные эфиры, нитруют и т. д. Широко
применяют Г. в аналитической химии; в виде соеди-
нения с хиноном (хингидрона) его используют для
определения pH; применяют как восстановитель
гетерополикислот при фотометрии, определениях Р,
Si, As, Мо. для открытия вольфраматов, нитратов,
нитритов, Se, Те, Ti, W, Be и др. элементов, как
восстановитель длн Си11, Аи; в микроскопии.
Лит.: Ворожцов Н. Н., Основы синтеза промежу-
точных продуктов и красителей, 4 изд., М,, 1955; Welcher
F. J., Organic analytical reagents, v. 1, Toronto—N. Y. — L.,
1948. И. Н. Ефимов.
ГИДРОЦЕЛЛЮЛОЗА — см. Целлюлоза.
ГИОСЦИАМИН — см. Атропин.
ГИПЕРКОНЪЮГАЦИЯ — сопряжение простых и
кратных связей. Вейкер и Натан, изучая кинетику
присоединения пара-замещенных бензилбромидов к
пиридину с образованием бензилпиридинийбромидов,
показали (1935), что заместители, обладающие поло-
жительным индукционным эффектом (см. Индукцион-
ный и мезомерный эффекты), ускоряют реакцию.
Оказалось, что алкильные заместители по ускоряю-
щему влиянию на реакцию не располагаются в ряд,
соответствующий индукционному механизму электрон-
ных смещений; в действительности наблюдался об-
ратный порядок: СН3>СН3СН2>(СН3)2СН>(СН3)3С.
Из этого наблюдения был сделан вывод, что в пара-ал-
килбепзильных системах, кроме индукционного, су-
ществует дополнительный механизм смещения электро-
нов от алкильных групп. Этот механизм наиболее
сильно проявляется в метильной группе, но становится
меньшим при замене атомов водорода в метильной
группе на углеводородные радикалы.
На основании этого, Бейкер и Натан высказали
предположение о том, что электронная пара II—С-свя-
зей в метильной группе значительно меньше лока-
лизована, чем электроны С—С-связей, и способ-
на, подобно неподеленной электронной паре, всту-
пать в сопряжение с ненасыщенными системами. В
дальнейшем оказалось возможным сделать более об-
щий вывод о взаимодействии сопряженных Н—С и
С-С(С=С)-связей во всех системах типа Н—С—С=С<^
I _ 1
и Н - С—С—С—. Изучение Г. длительное время было
ограничено большей частью сопряжением Н—С- и
С=С (или С=О)-связей, вследствие чего термины
«эффект Натана—Бейкера», или Г., получили част-
ное применение к С—Н-связи. Согласно классифи-
кации А. Н. Несмеянова, Г. представляет один из ви-
дов сг, сг-сопряжения
относятся примеры
(I-IV).
В качестве конкрет-
ных примеров хими-
ческого проявления
Г. можно указать на
повышенную реакци-
онную способность
СН3-группы в молеку-
ле пропилена (сравни-
тельно с парафина-
ми) и особенно на подвижность а-водородных ато-
мов в молекулах альдегидов, кетонов, кислот и их
производных:
связей
из них к
Н—С—С=С
IV
Y-C-C-X
Н—С—С=С—
1 //
V
Y-C=C-X
VI
ill
Н Н
н-4с^-сн == сн о н-(с^-с=О>
N Ч \н
н н н
Результаты физич. исследований молекул с <7,^-со-
пряженными системами дают основание высказать пред-
положение о том, что Г., как и вообще сг, а-сопряжение,
эффект, по-видимому, динамический, проявляющийся
лишь в момент реакции. В нереагирующих молекулах
пока еще не отмечено к.-л. тонких особенностей струк-
туры, к-рые могли бы быть обусловлены <т,<г-сопря-
жением. Так, термодинамнч. оценка Г. [на основании
теплот образования олефинов, например бутенов:
С2Н5СН=СН2, СН3СН=СНСН3, (СН3)2С=СН2[ по-
казывает, что энергия Г. между алкильной группой
и двойной связью в лучшем случае величина того же
порядка, что и энергия водородной связи, и составляет
максимально несколько килокалорий па моль.
Лит..- Несмеянов А. Н., Уч. зап. МГУ, 1950,
вып. 132, кн. 7, с. 5; Ингольд К. К., Механизм реакций
и строение органических соединений, пер. с англ., М., 1959.
(гл. 2 3); Baker J. W., Nathan W. S., S h о p-
pe C. W., J. Chem. Soc., 1935, p. 1847. О. А. Реутов.
ГИПЕРОНЫ — неустойчивые элементарные ча-
стицы, масса к-рых больше массы нейтрона, но меньше
массы дейтрона. Обозначаются заглавными грече-
скими буквами. Известны след. Г.: A°,S°,S+,S~,E“,3+(
отличающиеся ио массе и заряду (индекс °, + или ~
943
ГИПЕРСЕНСИБИЛИЗАЦИЯ — ГИПЕРСОРБЦИЯ
944
указывает на нейтральность или заряд Г.; по величине
заряд Г. равен заряду электрона). Время жизни Г.
порядка 10 й1 сек (время жизни S° составляет примерно
10 19 сек). Превращение Г. при их распаде в нуклоны
(с испусканием л-мезонов) дает основание считать Г.
возбужденными нуклонами. О сходстве между Г.
и нуклонами говорит также то, что Д° может замещать
к.-л. из нуклонов в ядре, при этом возникает т. н.
А°-ядро (или гиперядро), время жизни к-рого 10 12 —
10 10 сек (см. Элементарные частицы).
ГИПЕРСЕНСИБИЛИЗАЦИЯ — повышение свето-
чувствительности фотографич. эмульсий путем допол-
нительной обработки перед экспонированием. Г.
производится: обработкой фотоматериала р-рами три-
этаноламина и других аминов, перекисью водорода
и т. п., его выдерживанием в парах ртути, а также
кратковременная предварительная засветка. В боль-
шинстве случаев повышение светочувствительности
путем Г. сопряжено с увеличением вуали, зернистости
и ухудшением сохраняемости фотографич. слоя.
Лит.: Вендровский К. В., Успехи научной фо-
тографии, 1960, 7, с. 57. К. В. Вендровский.
Исходи,
смесь
Вода
Остат. газ
Лонная фракция
Водяной пар
Конденсат
• Боковая
фракция
ГИПЕРСОРБЦИЯ — разделение газовых смесей
методом избирательной адсорбции слоем поглотителя,
движущимся навстречу газовому потоку. Схема уста-
новки с движущимся слоем твердого
поглотителя для разделения газовой
смеси на 3 фракции приведена на ри-
сунке. Основным аппаратом установки
является колонна 1, состоящая из
адсорбционной секции 2 и расположен-
ных под ней ректифи-
кационных секций 3.
Исходная смесь посту-
пает под распредели-
тельную тарелку 4 и
поднимается вверх на-
встречу гранулирован-
ному поглотителю, дви-
жущемуся вниз под
действием силы тяже-
сти. Остаточный газ
отводится сверху ад-
сорбционной секции,
а насыщенный поглоти-
тель опускается в ре-
ктификационные сек-
ции, где подвергается
десорбции. При повы-
шении темп-ры выде-
ляющиеся тяжелые ком-
поненты поднимаются
вверх в виде флегмы,
вытесняя из поглоти-
теля более легкие. В
результате в ректифи-
кационной секции про-
исходит разделение по-
фракции. Подогрев на-
глощенных компонентов на
сыщеиного поглотителя производится в отпарной сек-
ции 5 глухим паром.
Непрерывная рециркуляция регенерированного по-
глотителя из выгрузного приспособления 6 произво-
дится пневматически с помощью газодувки/; для транс-
портировки используется остаточный газ. Из бункера
8 поглотитель, пройдя холодильник 9, возвращается
в адсорбционную секцию 2.
Для определения необходимой высоты движущегося слоя
поглотителя можно воспользоваться кинетич. ур-нием процесса
адсорбции: da/dt = g{c—у), где da — бесконечно малое коли-
чество вещества, адсорбируемого единицей объема поглоти-
теля в бесконечно малый отрезок времени dt; g — коэфф,
массопередачи, характеризующий количество вещества ад-
сорбируемого единицей объема поглотителя в единицу времени
при разности концентрации поглощаемого вещества в газовом
потоке (с) и равновесной ему концентрации (у) над поглоти-
телем, равной единице. При установившемся процессе коли-
чество вещества адсорбируемого из газового потока Vr беско-
нечно малым объемом поглотителя dVa, выразится: vr dc =
= 0 (с—y)dVa. Интегрируя это ур-ние, находим необходимый
объем адсорбента:
где ск и сн — соответственно конечная и начальная концент-
рации поглощаемого вещества в газовом потоке. Если Va
и Vr рассматривать как секундные объемы и вместо Va под-
ставить HS (поскольку контакт между зернами не нарушается),
то из последнего ур-ния получаем:
сн сн
* г Г de _ йг С de
S3 j с - у ~ У J с — у
ск сн
где Н — высота колонны в м\ S — площадь поперечного се-
чения колонны в ms; w — линейная скорость газового потока
сн
С de
в м сек. Интеграл \---,
J с—у’
‘к
значение к-рого можно найти графич.
путем, выражает изменение рабочей концентрации поглощае-
мого вещества, приходящегося на единицу движущей силы,
и носит название общего числа единиц переноса. Количество
вещества G, поглощаемого в единицу времени из газового по-
тока, может быть выражено равенством: G==Vr(cH—ск),
откуда
си ск
После подстановки полученного выражения для G в при-
веденное выше ур-ние и интегрирования получаем:
и = G = G
, '"н — ск ₽®Дсср
f:S ------•---
сн
где
с к
средняя движущая сила адсорбции.
Если изотерма адсорбции в рассматриваемых пределах
концентраций прямая, то средняя движущая сила адсорбции
определяется как среднелогарифмич. разность концентраций:
» Дс1 "
Асср ~
In
- ЬС-2
ДС1
Дс3
где с, ис, - движущие силы на входе и выходе из колонны, т. е.
разности между концентрациями поглощаемого вещества
в газовом потоке внизу и вверху колонны и равновесными его
концентрациями лад поглотителем. В случае, когда Ас, Ас2* 2,
то средняя движущая сила адсорбции определяется как средне
арифметическая, т. е.
Дщ + Дс3
Дсср = 2
Коэфф, массопередачи g, зависящий от физич. свойств погло-
щаемого вещества и поглотителя, характера движения газо-
вого потока и т. д., в каждом отдельном случае может быть
надежно определен только опытным путем.
Из-за трудностей, связанных с аппаратурным
оформлением процесса, а также в связи с большим из-
мельчением поглотителя Г. пока не нашла широкого
применения в пром-сти, но используется иногда для
выделения из различных газовых смесей этилена, аце-
тилена, разделения попутных нефтяных газов и др.
В таблице приведены составы исходной газовой смеси
и полученных в результате разделения этиленовой и
метано-водородной фракции (в об. %).
945
ГИПОКСАНТИН — ГИПСОХРОМНЫЙ ЭФФЕКТ
946
Н2 | N2 | СО | О21 СН, j СО2 I С2Н2 С2Щ
Исходная смесь , .
Этиленовая фрак-
ция .............
Мет ано-водородная
фракция .........
0,2 5,8
3.6 92,7
Для разделения ок. 360 м3/час исходной смеси ис-
пользуется колонна диаметром 137 см и высотой 26 м.
Рабочее давление процесса 6,3 ат, адсорбент (уголь)
в зоне десорбции подогревается конденсирующимся
до 265° органич. теплоносителем, а затем подвергается
отдувке водяным паром давлением ок. 12 ат. Расход
пара ок. 100 кг!час.
Лит.: Серп ненова Е. Н., Промышленная адсорбция
газов и паров, М., 1956. И. И. Гельпе-рип.
ГИПОКСАНТИН (6-оксипурин) C5H4N4O, мол. в.
136,12 — пуриновое основание; бесцветные кристал-
лы; при 150° разлагается; рАа 1,98, рАа 8,94,
ОН Н ? Н
нс 1 I сн
рЛ"а 12,10; характерные максимумы поглощения в
ультрафиолетовом спектре при 248 ммк (pH 1) и
Z2 258 ммк (pH 11). Г. амфотерен; плохо растворим в
холодной воде, хорошо — в горячей, в кислотах и ще-
лочах; дает реакции на пуриновые основания. В при-
роде Г. встречается в свободном виде во многих тканях
растений и животных. Гликозиды Г. — инозин (рибо-
зилгипоксантин) и дезоксиинозин (дезоксирпбозилги-
поксантин) — являются компонентами нуклеотидов
(нуклеотиды, в состав к-рых входит Г.,называют ино-
аинфосфорными кислотами). Г. получают гидролизом
природных веществ (нуклеиновых к-т), а также син-
тетически.
Лит. см. юрист. Аденин. Е. В. Будницкая.
ГИПОСУЛЬФИТ, см. Натрия тиосульфат.
ГИППУРОВАЯ КИСЛОТА (бензоиламиноук-
сусная кислота, бензоилглицин, бензоилгликокол)
C6HsCONHCH2COOH, мол. в. 179,17 — бесцветные
кристаллы; т. пл. 187,5°; К = 15,7 • 10 5; плохо рас-
творяется в эфире и холодной воде, лучше — в горячей,
хорошо растворима в этаноле и уксусноэтиловом
эфвре, нерастворима в петролейном эфире. Щелочные
и щелочноземельные соли Г. к. хорошо растворимы
в воде, соли Ag, Си, РЬ плохо растворимы, соль Fe
нерастворима. При действии кислот, щелочей и некото-
рых ферментов Г. к. гидролизуется до бензойной
к-ты и глицина. Количественно Г. к. определяют
непосредственным титрованием ее щелочью, титрова-
нием ее производного с формальдегидом и определением
бензойной к-ты, образующейся при ее гидролизе.
Г. к. содержится в моче всех позвоночных животных
(за исключением птиц); ее получают в основном
из мочи лошадей и крупного рогатого скота. Синтети-
чески Г. к. получают действием бензойного ангидрида
или бензоилхлорида на глицин. Синтез Г. к. из бензой-
ной к-ты и глицина протекает в организме большинства
жввотных в печени и почках при участии коэнзима А.
При нек-рых патологии состояниях (гепатиты, от-
равления бензоатами, толуолом и др.) количество
Г. к. в моче возрастает. Г. к. применяют в качестве
исходного продукта в ряде синтезов, напр. изохиноли-
новых соединений и др. Количество Г. к. в моче после
нагрузки больных бензойнокислым натрием служит
диагностич. пробой на обезвреживающую работу пе-
чени (проба Крика).
Лит. см. при ст. Аминокислоты. Н. Е. Плотникова.
ГИПСОХРОМНЫЙ ЭФФЕКТ — смещение окрас-
ки органич. соединений в сторону повышения цвета,
т. е. переход от зеленого через синий и красный к жел-
тому цвету; явление — обратное батохромному эффек-
ту, т. е. при Г. э. максимум и полоса поглощения сме-
щаются в сторону более коротких волн. Причины, вы-
зывающие Г. э., излагаются в современной теории
цветности. Так, к Г. э. приводит укорочение цепи со-
пряжения двойных связей, а в красителях также вве-
дение новых заместителей, уменьшающих постоянное
(т. е. не зависящее от действия света) смещение элек-
тронов между электроположительными и электроот-
рицательными центрами молекулы. Появление этих но-
вых центров часто ведет к укорочению цепи сопря-
жения. Напр., в аурампне (I), благодаря включе-
нию в систему сопряжения неподеленной пары элек-
тронов атома азота аминогруппы, цепь сопряжения
между полярными диметиламиногруппами, существо-
вавшая в гидроле Михлера (II), прерывается, что
/
Желтый (Хмакс 420 мм::).
Синий Рмакс 603,5 ммк).
сокращает эту цепь сопряжения; происходит рез-
кое повышение окраски. Г. э. наблюдается при умень-
шении равномерности распределения электронной
плотности в хромофоре, в частности под влиянием из-
менения природы растворителя и при ослаблении
электронодонорных свойств заместителей, сопряжен-
ных с хромофором, напр. при ионизации или ацили-
ровании аминогруппы. В последнем случае смещение
неподеленной пары электронов аминогруппы к хро-
мофорной системе уменьшается за счет появления
противоположно направленного сопряжения этих
электронов с ацильным остатком, проявляющим элек-
троноакцепторные свойства (III и IV). Одной из
причин, приводящих к Г. э., является нарушение
947
ГИРОМАГНИТНОЕ ОТНОШЕНИЕ — ГИСТЕРЕЗИС СОРБЦИОННЫЙ
948
плоскостного строения молекулы, происходящее за
счет поворота части ее по одинарной связи, что ве-
дет к разобщению отдельных участков цепи сопря-
жения, т. е. уменьшает поляризацию молекулы. По-
следнее достигается введением в молекулу объемистых
ваместителей, препятствующих расположению ее от-
дельных частей в одной плоскости. Напр. (V) окрашен
в зеленый цвет, а (VI) имеет синюю окраску, т. к.
группы СН3 (см. стр. 946), занимающие большой объем,
выводят фенильные группы из плоскости, в к-рой рас-
положена молекула красителя (см. также Цветности
теории).
Лит. см. при ст. Батохромный зффект. „
В. Л. Пучков.
ГИРОМАГНИТНОЕ ОТНОШЕНИЕ — отношение
магнитного момента (М) системы (элементарной части-
цы атома или молекулы) к ее механич. вращательному
моменту (К)'.у = М/К. Существование прямой про-
порциональности между магнитным и механич. момен-
тами электрона, двигающегося по атомной орбите,
было показано еще в рамках электронной теории
Лоренца, причем для Г. о. атома было получено зна-
чение е/2тс, где е — заряд электрона (е <Z 0), т —
масса, ас — скорость света.
После открытия спина элементарных частиц выяс-
нилось, что они обладают собственными магнитными
моментами, не связанными с орбитальным движением.
Спиновое Г. о. электрона равно 2,0023 е/2тс, т. е.
вдвое больше, чем Г. о., соответствующее орбиталь-
ному магнетизму. Поэтому связь магнитных и меха-
лич. свойств атома в общем случае гораздо более слож-
на. чем это предполагалось по классич. теории.
Если выразить Г. о. атома через безразмерную ве-
личину g (g-фактор) по ф-ле у = g • е/2 тс, то
при суммарном спине атома А=0 наблюдается лишь
орбитальный магнетизм: g = 1, а Г. о. равно классич.
значению е/2тс (см. Атом). Если же, напротив,
атом находится в состоянии /, = (), то g = 2, а у —
— е/тс и наблюдаемый магнетизм имеет чисто спи-
новое происхождение. В остальных случаях g-фак-
тор принимает промежуточные значения. Эксперимен-
тальное определение Г. о. является, т. о., одним из
методов идентификации электронного состояния атома.
Лит.: Беккер Р., Теория электричества, т. 2, М.»
1941; Блохинцев Д. И., Основы квантовой механики,
М.—Л., 1949. А. И. Бурштейн, Ю. Д. Цветков.
ГИСТАМИН [|3-имидазолил-4(5)-этиламия], мол. в.
111,15, C6HVN3,— кристаллич. пластинки; т.пл.86°;
т. кип. 209—210°/18 мм; легко растворим в воде,
спирте и горячем хлороформе,
ZNH—СН нерастворим в эфире. Гистамин
Н%—<LCH2CH2NH2 Устойчив к действию горячей
t конц. соляной к-ты и холод-
ного 20%-ного раствора NaOH,
но разрушается щелочью при нагревании; с кисло-
тами образует хорошо кристаллизующиеся соли:
дихлоргидрат (т. пл. 244—246°), дифосфат (т. пл.
130—133°), дипикрат (т. пл. 238—240°) и дипикро-
лонат (т. пл. 262—264°). Получают Г. бактериаль-
ным декарбоксилированием гистидина или синтети-
чески — взаимодействием диоксиацетона с аммиа-
ком и формалином, превращением образовавшегося
4(5)-оксиметилимидазола в 4(5)-хлорметилимидазол,
замещением в нем хлора на циангруппу и восста-
новлением 4(5)-циапметплимидазола. Г. применяют
в виде мази при суставном и мышечном ревматизме,
при болях, связанных с поражением нервов (инъек-
ции под кожу), и для определения секреторной способ-
ности желудочных желез.
Лит. см. при ст. Адалин. Е. С. Головчинская.
ГИСТЕРЕЗИС МЕХАНИЧЕСКИЙ (н полиме-
рах) — отставание деформации по фазе (т. е. во вре-
мени) от напряжения при циклич. деформациях,
обусловленное неидеальной упругостью полимера.
Вследствие этого одинаковым напряжениям соответ-
ствуют разные величины деформации, а одинаковым
деформациям — разные напряжения. Г. м. наблю-
дается как в твердых, так и в высокоэластичных поли-
мерах, но в последних выражен наиболее ярко.
Кривая зависимости деформации от напряжения при
циклич. деформациях представляет собой замкнутую
гистерезисную петлю (см. рисунок). Площадь, огра-
ниченная петлей, является ме-
. рой работы, затраченной на
§ /у преодоление сил внутреннюю
j // трения, т. е. мерой количества
к / тепла, выделенного за один
§ ху цикл. Г. м. обусловливает па-
1 личие т. н. механич. потерь.
/ ________ Для идеально упругого тела
Удлинение обе ветви гистерезисной петли
Гистерезисная петля для сливаются в одну, гистерезис
резины. отсутствует. Наличие гисте-
резиса обусловлено термодииа-
мич. необратимым характером деформации полимер-
ных материалов: в процессе деформации, протекаю-
щей с конечной скоростью, равновесие не успевает
устанавливаться, и величина деформации отстает во
времени от изменения напряжения. У высокоэластпч-
ных полимеров это связано с релаксационным харак-
тером их деформации (см. Эластичность полимеров).
У полимеров, находящихся в твердом состоянии
(стеклообразном или кристаллич.), Г. м. обусловлен
тем, что, помимо обычной упругой деформации, они
дают еще деформацию упругого последействия, отста-
ющую по фазе от напряжения и обусловленную со-
хранением в твердом состоянии нек-рой ограничен-
ной свободы перегруппировок частей гибких поли-
мерных молекул.
Наиболее сильный Г. м. наблюдается в тех случаях,
когда время установлений механич. равновесия т
. близко к периоду циклич. деформации. Когда период
цикла очень велик или очень мал по сравнению с т, Г. м.
исчезает. Для высокоэластичных полимеров, при не-
больших частотах деформирования (до десятков или
сотен циклов в секунду), Г. м. максимален вблизи
темп-ры стеклования п уменьшается как при пониже-
нии темп-ры (при стекловании), так и при ее повыше-
нии. С повышением частоты циклов область максимума
Г. м. передвигается к более высоким темп-рам. Г. и.
резин, имеющий важное практич. значение, зависит
от вида каучука, а также от введения пластификаторов,
наполнителей, характера вулканизации и т. п. В ши-
нах гистерезис приводит к превращению части работы
в теплоту, к разогреву шин и уменьшению срока их
службы. Г. м. в полимерах используется для констру-
ирования демпфирующих устройств, поглощающих
энергию колебаний и приводящих, т. о., к их гашению.
Лит.: А л ф р е й Т., Механические свойства высокополи-
меров, пер. с англ., М., 1952; Т р е л о а р Л., Физика упру-
гости каучука, нер. с англ., М., 1953; К обе ко II. П.,
Аморфные вещества, М.—Л., 1952. Ю. С. Лазуркин.
ГИСТЕРЕЗИС СОРБЦИОННЫЙ — явление не-
полной обратимости процесса адсорбции, удержи-
Гистерезпспая петля для
случаевГ 1 — капилляр-
ной конденсации в порах
сорбента с жестким ске-
летом; 2 — капиллярной
конденсации и хемосорб-
ции; з — капиллярной
конденсации в порах
сорбента с нежестким
скелетом. P/Ps—относи-
тельное давление пара
жидкости.
ванне большего количества адсорбированного вещест-
| ва пористыми телами при десорбции, чем при адсорб-
949
ГИСТИДИН — гистохимия
950
ции. При Г. с. изотермы адсорбции и десорбции не
совпадают, т. е. одному и тому же давлению пара соот-
ветствуют различные величины поглощения при пря-
мом и обратном ходе процесса. Две несовпадающие
ветви изотерм адсорбции и десорбции образуют гис-
терезисную петлю (см. рис.). Причиной Г. с. может
быть капиллярная конденсация пара в открытых по-
рах, а также химич. реакции молекул газа с поверх-
ностью адсорбента (см. Хемосорбция) или медленное
обратное выделение после растворения их в веществе
адсорбента (набухание).
Лит. см. при ст. Адсорбция, Капиллярная конденсация.
ГИСТИДИН (а-амино-Р-5-имидазолилпропионовая
кислота)СвН909Хч, мол. в. 155,16 — кристаллы; т. пл.
(с разл.): D,L-r. 283°,
NH-CH D-Г. 287—288°, L-Г.
I /C-CH2CH(NH2)COOH 287o;[a]y:D-r. + 39,3°,
CH = N/ ' L-Г. —39,74° (в во-
де). Гистидин раство-
рим в воде, спирте, нерастворим в эфире. При 25°
р/ 7,6, рЛГО1 (СООН) 1,77, рКаг (имидазол) 6,0, рКа^
(NH3) 9,0. Г. дает биуретовую реакцию; характерной
для Г. является реакция с диазобензосульфокислотой
в присутствии Na2CO3 (Паули реакция). На этой
реакции основано большинство методов количествен-
ного определения Г. При обработке бромом при нагре-
вании Г. дает устойчивую в щелочной среде фиоле-
товую окраску, что также используется для количест-
венного определения Г. При декарбоксилировании Г.
образуется биологически активное вещество — гис-
тамин. L-Г. — незаменимая аминокислота, составная
часть большинства белков. В мышцах животных Г.
встречается в составе дипептидов карнозина и аневрина.
Г. получают из гидролизатов гемоглобина или сухой
крови путем осаждения его в виде ртутной или сереб-
ряной соли 3,4-дихлорбензосульфоната или флавиа-
ната. Синтезируют Г. конденсацией хлорметилимида-
зола и ацетаминомалонового или ацетаминоциан-
уксусного эфира с последующим гидролизом получен-
ных продуктов. Г. обычно получают в виде моно- или
дихлоргидрата.
Лит.: Блок Р. п Боллинг Д., Аминокислотный
состар белков и пищевых продуктов, пер. с англ., М., 1949;
Б р а у н ш т е й н А. Е., Биохимия аминокислотного об-
мена, М., 1949. Л. А. Локшина.
ГИСТОНЫ — белки основного характера, входящие
вместе с дезоксирибонуклеиновой к-той в состав де-
зоксирибонуклеопротеидов клеточных ядер; содержат
большое количество лизина и аргинина; средний мол.
в. 18 000; р/ 10—11. Электрофоретически и путем
хроматографии на катионите Г. разделяют на фракции
а-, и у-Г. Г. растворимы в кислых и нейтральных
р-рах, осаждаются аммиаком, но не растворяются в из-
бытке последнего; растворяются в избытке неорганич.
щелочей, в реактиве Миллона, содержащем H2SO4,
но нерастворимы в этом же реактиве, содержащем три-
хлоруксусную к-ту. Г. дают Миллона реакцию (Hg2+
и NO,, в кислой среде), осаждаются реактивами на ал-
калоиды, образуют нерастворимые комплексы с не-
которыми белками типй альбуминов и глобулинов. Г.
обычно получают экстрагированием клеточных ядер
или ядерных нуклеопротеидов разб. к-тами с после-
дующим осаждением путем высаливания или дове-
дения pH ~ до 10. При извлечении из клеточных ядер
Г. могут экстрагироваться в комплексе с кислым бел-
ком. Этот комплекс по своим сократительным свой-
ствам (обнаруживаемым в присутствии аденозинтри-
фосфорной к-ты) сходен с актомиозином.
Лит.: Збарский И. В., в кн.: Усп. биол. химии,
т. 1, М., 1950; Phil. Trans. Roy. Soc. London В, 1957, 241, № 678,
p. 93; Advances Enzymol. and Related Subjects Biochem.,
1955, 16, p. 441; The cell, ed. by J. Brachet A. E. Mirsky, v. 1,
N. Y.—L.. 1959. См. также лит. при ст. Белки.
Л. М. Гинодмаи.
ГИСТОХИМИЯ (гистологическая химия, гистото-
похимия) — наука, изучающая локализацию химич.
веществ и процессов в микроскопич. структурах тка-
ней и клеток растительного и животного организма,
на срезах различных органов и па изолированных
клетках. Разделом Г. является цитохимия,
изучающая локализацию веществ в клетке. Как само-
стоятельная наука Г. возникла только в 30 гг. 20 в.
(отдельные гистохимия, методы применялись и рань-
ше); развивается очень бурно и имеет большое обще-
биологич. значение. Устанавливаемые Г. закономерно-
сти способствуют развитию других биологич. наук: ги-
стологии (науки о развитии, строении и жизнедеятель-
ности животных тканей), патологической анатомии,
установлению связи между локализацией химич.
процессов в микроскопич. структурах и их специфич.
биологич. функциями. Для гистохимия, исследова-
I пий ткани предварительно фиксируют с помощью
। методов, не вызывающих изменения прижизненной
локализации биосубстратов, и изготовляют гисто-
логия. срезы. Для выявления определенных групп
веществ в срезах используются специфич., как пра-
вило, цветные реакции. Их специфичность контроли-
руется либо при помощи ферментов (напр., рибону-
клеазы при определении нуклеиновых к-т, гиалуро-
нидазы при выявлении гиалуроновой к-ты и т. д.),
либо путем блокирования соответствующих функцио-
нальных групп биосубстратов.
Гистохпмич. выявление белков производится в ос-
новном с помощью цветных реакций на аминокислоты
и входящие в состав белковой молекулы функциональ-
ные группы. Для этой цели применяют, например,
Миллона реакцию на тирозин, Сакагучи реакцию
на аргинин и др. Большое значение приобрел ме-
тод Даниэлян, основанный па выявлении тирозина,
триптофана и гистидина белка посредством азосоче-
тания с бисдиазотированным бензидином или о-диа-
пизидииом и последующем усилении окраски путем
дополнительного азосочетания с Аш-кислотой. Азосо-
четание широко используется при осуществлении
гистохимия, реакций на амино-, карбоксильные, тио-
ловые и др. функциональные группы белка. Разра-
ботаны различные способы выявления белка, в к-рых
использована способность белковых веществ коли-
чественно связывать нек-рые красители (бромфено-
ловый синий, прочный зеленый FSF).
Дезоксирибонуклеиновые кислоты
(ДНК), содержащиеся в клеточных ядрах, обнаруживаются
по методу Фёльгена, основанному нз высвобождении альде-
гидных групп в молекулах ДНК при кислотном гидролизе
и последующем выявлении альдегидов реакцией с фуисин-
сериистой к-той. Рибонуклеиновая кислота
окрашивается основными красителями о обязательным контро-
лем специфичности окрашивания (обработка рибонуклеазой).
Для гистохимия, обнаружения гликогена И др. уг-
леводов большое значение имеет реакция окисления пер-
йодатом, при к-рой в углеводах, содержащих 1,2-глпколевые
группировки, образуются альдегидные группы, выявляемые
реакцией с фуксинсерипстой к-той. Для выявления а м п н о-
полисахаридов используется их способность окраши-
ваться рядом основных красителей метахроматически.
В основе гистохпмич. обнаружения липидов лежит
высокая растворимость в них судаиовых красителей, что ведет
к избирательной окраске структур, содержащих липиды.
Для разграничения классов липидов применяется экстракция
различными растворителями, исследование в поляризованном
свете и нек-рые специальные окраски.
Локализация ферментов гистохимическп устанавливается
либо с помощью цветных реакции на продукты ферментатив-
ных реакций, либо путем использования т. н. хромогенных
субстратов — веществ, в результате ферментативного превра-
щения к-рых образуются окрашенные плохо растворимые
соединения. Наряду с применением обычной микроскопии
в Г. широко используются электронная, ультрафиолетовая и
флуоресцентная микроскопия, радпоавтография (при исполь-
зовании радиоактивных изотопов для изучения обмена ве-
ществ) и др. методы исследования.
Лит.: Гистохимические методы в нормальной и патологи-
ческой морфологии. [Сб. ст.], под ред. В. В. Португалова
и А. И. Струкова, М., 1958; Макаров П. В., Основы ци-
тологии, М., 1953; Пирс Э., Гистохимия теоретическая
и прикладная, пер. с англ., М., 1956; Браше Ж., Био-
951
ГЛАЗУРЬ — ГЛИКОГЕН
952
химическая цитология, пер. с англ., М., 1960; Кедров-
ский В. В., Цитология белковых синтезов в живот-
ной клетке, М., 1959; Фукс В. В., Гистохимия и мор-
фология нормального и повреждённого нерва, Новосибирск,
1959; Analytical cytology, ed. R. C. Mellors, 2 ed., N. Y.,
1959; Gomori G., Microscopic histochemistry. Prin-
ciples and practice, Chi., 1952; Handbuch der Histochemie, Bd 1,
Stuttg., 1958; Lillie R. D., Histopathologic technic and
practical histochemistry, N. Y. — Toronto, 1959; L i s о n L.,
Histochimie et cytochimie animales. Principes et methodes,
2 ed., P., 1953; Pearse A. G. E., Histochemistry. Theo-
retical and applied, 2 ed., L., 1960. M. Г. Шубич.
ГЛАЗУРЬ — тонкое стекловидное покрытие на ке-
рамич. изделиях, получаемое при нанесении на них ми-
неральных композиций с последующим обжигом изде-
лий при высоких темп-pax. Г. придает керамич. изде-
лиям водонепроницаемость, предохраняет от загряз-
нений, действия кислот и щелочей, а также использует-
ся в декоративных целях. По составу и характеру
строения Г. — стекловидный твердый раствор кремне-
зема и глиноземо-щелочных силикатов и окислов ме-
таллов. Г. делят на тугоплавкие и легкоплавкие.
Тугоплавкие Г. (плавкость 1230—1400°) при-
меняются для покрытия фарфора, санитарного фаянса
и полуфарфора. Основным сырьем для пих служит
кварц, полевой шпат, карбонаты кальция или магния,
каолин, измельченный фарфоровый бой. Измельчение
этого сырья производится в шаровых мельницах.
Легкоплавкие Г. (плавкость 900—1250°) при-
меняются для покрытия фаянса, каменных керамич.
и майоликовых изделий. Сырьем для этих Г. является
кварц, полевой шпат, мрамор, сода, поташ, селитра,
бура, углекислый барий, свинцовый сурик и др.,
причем соединения бария и свинца могут быть заме-
нены соединениями стронция, лития и цинка. Так
как многие компоненты легкоплавких Г. растворимы
в воде, то смесь этих материалов предварительно
сплавляется (фритуется), после чего расплав выливает-
ся в роду, а затем размалывается в шаровых или виб-
ромельницах с добавкой воды и глинистых материалов.
Для покрытия простых гончарных изделий применя-
ют свинцово-силикатные Г., содержащие в основном
свинцовый сурик. Для керамич. кислотоупорных из-
делий и канализационных труб применяют иногда т.
наз. соляную Г., к-рую получают введением в печь
при 1180—1280° поваренной соли при восстановитель-
ной газовой среде; под действием паров воды соль раз-
лагается на НС1 и Na2O; последняя входит в реакцию
с керамич. материалом и образует на его поверхности
стеклообразный слой. Для окрашивания Г. в их состав
вводят красящие окислы или соли металлов (кобаль-
та, меди, хрома, марганца, железа и др.), к-рые при
сплавлении растворяются в Г. с образованием цвет-
ных силикатов. Оттенок, сообщаемый окислами, за-
висит от состава Г., степени ее измельчения, темп-ры
печи и газовой среды обжига (окислительная или вос-
становительная). Для получения Г. белого цвета в со-
став легкоплавкой свинцовой Г. добавляют 5—10%
криолита, двуокиси олова или циркония. Непрозрач-
ной белой Г. покрывают печные кафели, изразцы, стен-
ные плитки, блюда, сосуды и др.
Нанесение размолотой в воде глазурной массы на
сырые или предварительно обожженные керамич.
пористые изделия производится: окунанием в водную
суспензию Г.; распылением из пульверизатора; кистью
или припудриванием порошком. Изделия с нанесен-
ной на них Г. подвергают сушке, а затем обжигу.
Состав Г. зависит от состава керамич. изделия, темп-ры
его обжига и цвета. Коэфф, расширения Г. должен быть
близок к коэфф, расширения керамич. материала, и
при обжиге и охлаждении Г. не должна давать тре-
щин («цека») и откалываться от поверхности изделия.
Лит.: Технология керамики и огнеупоров, под ред.
П. П. Будникова 2 изд., М., 1954; Барзаковский В.П.
иДуброво С. К., Физико-химические свойства глазурей
высоковольтного фарфора, М.—Л., 1953; Б люмен Л. М.,
Глазури, М., 1954. П. П. Будников.
ГЛ ИКАЛ И — продукты отщепления двух гидро-
ксилов (один из к-рых является полуацетальным) от
альдоз или элементов воды от 2-дезоксиальдоз (за
счет полуацетального гидроксила и водорода метиле-
новой группы), напр.:
/0Н
СН---
неон
I
носн о
НСОН
I
НС-----
СН2ОН
D-глюкоза
1 сн /он сн
II сн 1 -2ОН НОСН 1 С НСОН 1 НС 1 сн2он D-глюкаль (гликаль глюко I — !Ы) i сн2 нэ° НОСН 1 0 НСОН 1 1 нс 1 СН2ОН 2-дезокси-D-глю- коза
Непосредственно произвести отщепление такого типа
не удается; Г. получают синтетически из О-ацетил-
бромидов сахаров. Нек-рые Г. выделены в кристаллич.
виде, другие получены только в виде кристаллич.
производных (ацетатов и др.). Г. легко изомери-
зуются с миграцией двойной связи; при их окислении
перекисью водорода или иадбензойной к-той образует-
ся смесь эпимерных сахаров (напр., из глюкаля —
глюкоза и манноза), а при окислении озоном обра-
зуются альдогексозы, содержащие в цепи на один уг-
лерод меньше, чем исходный Г.
Названия Г. производят от соответствующего са-
хара, заменяя окончание «оза» на «аль», напр. Г.
D-арабинозы — D-арабиналь, Г. D-ксилозы — D-кси-
лаль и т. д. Г. широко применяются в синтезах са-
харов, дезоксисахаров и их производных.
Лит.: The Carbohydrates, ed. W. Pignian, N. Y., 1957;
M 1 e h e e 1 F., Chemie der Zucker und Polysaccharide, 2
Autl., Lpz., 1956; Advances in carbohydrate chemistry, v. 7,
N.Y., 1952, p. 210. Л. И. Линевич.
ГЛИКАМИНЫ (1-амипо-1-дезоксиальдитолы) —
производные сахарных спиртов общей формулы
CH2NH2(CHOH)nCH2OH, содержащие аминогруппу
вместо первичного спиртового гидроксила. Г. рас-
сматривают также, как производные сахарных спир-
тов, в к-рых группа СН2ОН замещена на СН2М1,
илиСН2\НН. Низшими представителями Г. являются
отаноламин и 2,3-диокси-н-иропиламип; производ-
ным Г., имеющим большое биологич. значение, являет-
ся витамин В2 — рибофлавин (производное 1-амино-
1-дезоксирибита). Ряд Г. выделен в кристаллич.
виде; большинство из них растворимо в воде и в рас-
творах к-т. Г. дают реакции па аминогруппу и на
спиртовые группы. Получают Г. каталитич. восста-
новлением гликозилиминов:
HC(=NH)(HCOH)„CH2OH H2C(NH;)(HCOH)nCH2OH
и др. способами.
Применяют Г. в качестве промежуточных продуктов
в синтезах дезоксисахаров и др. производных сахаров.
Лит.: The Carbohydrates, ed. W. Pigman, N. Y., 1957.
Л. И. Линевич.
ГЛИКОГЕН (животный крахмал) (С6Н1ОО5)П, сред-
ний мол. в. 105—10’ — полисахарид разветвленной
структуры, смесь молекул различной степени полиме-
ризации, состоит из остатков глюкозы в форме a-D-глю-
копиранозы. Подавляющее большинство глюкозных
остатков в Г. соединены при помощи а-1,4-глюкозид-
ных связей, 7—9% (в точках разветвления полиглюко-
зидных цепей) — за счет а-1,6-глюкозидных связей
и ок. 0,5—1% — за счет иных связей, 'вероятно
а-1,3-связей. Наружные ветви молекул Г. длиннее
внутренних. Наиболее полные данные о строении по-
лучены для Г. моллюсков, кроликов и лягушек.
Наиболее изученные Г. отличаются средней длиной
наружных и внутренних ветвей. Строение Г. подтвер-
953
ГЛИКОГЕН — ГЛИКОЗАМИНЫ
954
ждено энзиматич. синтезом. Г. — белый аморфный
порошок, легко растворимый в воде с образованием
(в зависимости от концентрации) опалесцирующих
или молочно-белого цвета коллоидных р-ров; [a]JJ =
= +196°. Из водных р-ров Г. осаждается спиртом, тан-
нином и сульфатом аммо-
ния. Г. способен образо-
вывать комплексы с бел-
ками. При обычных усло-
виях Г. не проявляет вос-
станавливающ. свойств,
однако, пользуясь особо
чувствительными реакти-
вами (например, динитро-
салициловой кислотой),
можно определять нич-
тожно малую восстанав-
ливающую способность
Г., что лежит в основе
химич. методов опреде-
ления мол. веса Г. Ки-
слотами Г. гидролизуют-
ся, причем вначале об-
разуются декстрины, а
затем мальтоза и глю-
коза', к действию конц.
щелочей довольно устойчив. Растворы Г. окраши-
ваются иодом в винно-красный, красно-бурый и
красно-фиолетовый цвета; окраска исчезает при ки-
пячении и вновь появляется при охлаждении.
Оттенок и интенсивность окрашивания Г. зависят от
его строения (степени разветвленности молекулы,
длины наружных ветвей и т. д.); может иметь значение
Схемастроения молекулы глико-
гена; а — «альдегидное» начало
цепи (глюкозный остаток со сво-
Оодной полуацетальной груп-
пой); треугольником обведен
участок молекулы (из 7 глюкоз-
ных остатков), изображенный
на приведенной формуле.
путем, называемым амилолизом: (СвН10 ®s)n +
+ геН2О = п (С6Н12Ов), либо при помощи фермента
фосфорилазы и солей фосфорной к-ты:(С6Н10 °5)п +
+ Na2HPO4 = (CeHloO5)n_1+(C6H11O5)OPO3Na2(3TOT
процесс называется фосфоролизом).
Лит.: Степаненко Б. Н., Изв. АН СССР. Сер.
биол., 1957, № 6, с. 706; Manners D. J. в кн.: Advances
in carbohydrate chemistry, v. 12, N. Y., 1967, p. 261.
В. H. Степаненко.
ГЛИКОЗАМИНЫ (аминосахара) — производные са-
харов, содержащие аминогруппу вместо одного из
спиртовых гидроксилов. В зависимости от положения
аминогруппы Г. делятся на 2-дезокси-2-аминоальдозы,
З-дезокси-З-аминоальдозы и т. д. Самую многочис-
ленную группу Г. составляют 2-дезокси - 2-а м и-
н о а л ь д о з ы. Из 16 теоретически возможных аль-
догексозаминов в природе встречаются: D-глюкозамин
(I), L-глюкозамин, D-галактозамин (II), D-гулозамин,
D-маннозамин, D-таллозамин.
сно
I
HCNH.
I
НОСН
I
НСОН
НСОН
I
сн.он
I
сно
I
HCNH.
I
носн
I
носн
I
НСОН
I
СН2ОН
п
н nh2
нон/^Чнон
нг\ Jhoh
о
Большинство Г. выделено в кристаллич. виде. Г.
хорошо растворимы в воде и р-рах кислот; с к-тами
образуют соли. Г. дают реакции восстанавливающих
сахаров (восстановление Фелинга реактива и аммиач-
пого р-ра AgNO3, окисление до соответ-
ствующих к-т) и аминов. С нингидрином
Г., подобно аминокислотам, дают цвет-
ную реакцию. Для колич. определения
ряда гексозаминов обычно используют
Элсона — Моргана реакцию
(цветная реакция, к-рую дают продукты
конденсации гексозаминов и ацетилаце-
тона с п-диметиламинобензальдегидом).
СН2ОН
<рн2он
сн2он
сн2он
сн2
он
Стооение участка молекулы гликогена с одной точкой ветвления
сн2он
СН2ОН
и присутствие примесей. Этой реакцией пользуются
для качественного обнаружения Г. Количественно
Г. обычно определяют после выделения его из ткани
(щелочным методом) с последующим кислотным гидро-
лизом и определением образовавшейся глюкозы (м е-
тод Пфлюгера).
Г. широко распространен в организмах животных
и представляет собой резервное вещество, важное для
энергетики организма и легко расщепляющееся с об-
разованием глюкозы, а также при гликолизе с обра-
зованием молочной к-ты. Наиболее богаты Г. печень
(до 20% сырого веса) и мышцы (до 4%), очень богаты
нек-рые моллюски (в устрицах до 14% сухого веса),
дрожжи и высшие грибы. К Г. близки крахмалы нек-рых
видов кукурузы. Г. получают путем обработки
ткани 5—10%-ной трихлоруксусной к-той на холоду
с последующим осаждением спиртом, либо обработкой
ткани 60%-ным КОН при 100°; белки при этом гидро-
лизуются, а Г. осаждается затем из гидролизата спир-
том. Расщепление Г. в организме животных происхо-
дит либо при помощи фермента а-амилазы гидролитич.
D-глюкозамин (хитозамин) и D-галактозамин (хон-
дрозамин) широко распространены, являясь струк-
турными компонентами мукополисахаридов, глико-
протеидов, аминополисахаридов и др. природных ве-
ществ. Многие вещества, в состав к-рых входят эти
гексозамины, обладают высокой биологич. активно-
стью (гепарин, у-глобулин крови, нек-рые гормоны и
др.). Маннозамин обнаружен в качестве компонента
нонулозаминовых кислот и сиаловых кислот. L-глю-
козамин и D-гулозамин входят в состав нек-рых анти-
биотиков группы стрептомицина. D-талозамин выде-
лен из препарата хондроитинсерной кислоты. Ряд
гексозаминов (D-аллозамин, D-альтрозамин и др.)
и пентозаминов получен синтетически.
З-дезокси-З-аминоальдозы — 3-ами-
но-З-дезокси-О-рибоза (III), З-амино-З-дезокси-О-глю-
коза, аллозамин, родозамин и др. являются ред-
кими сахарами; они входят в состав водорастворимых
метаболитов нек-рых низших грибов и бактерий, ряд
их производных обладает антибиотич. свойствами.
Диаминосахара обнаружены в нек-рых мик-
955
ГЛИКОЗАНЫ — ГЛИКОЗИДЫ
956
рооргапизмах. Широкое распространение имеют про-
изводные аминосахаров, содержащие карбоксильную
группу, — сиаловые и нейраминовые к-ты, к-рые вхо-
дят в состав ряда гликопротеидов и гликолипидов и
обнаружены во всех тканях животного организма.
Получают Г. гидролизом нек-рых природных ве-
ществ (напр., D-глюкозамин — гидролизом хитина),
а также синтетически, действием аммиака па ангидро-
с.ахара, перегруппировкой N-гликозидов и др. спо-
собами.
Лит.: Kent Р. W. and Whitehouse М. W.,
Biochemistry of the aminosugars, L., 1955; Methods of bio-
chemical analysis, v. 6, N. Y., 1958, p. 289; Advances in car-
bohydrate chemistry, v. 14, N. Y., 1959, p. 213.
Л. И. Линевич.
ГЛПКОЗАНЫ (ангидриды сахаров) — производ-
ные сахаров, в к-рых ангидридный мостик образуется
при отщеплении элементов воды за счет полуацеталь-
ного и одного из спиртовых гидроксилов. Г. — внут-
ренние гликозиды сахаров в пиранозной (I) (D-глю-
козан
СН2----О—
или 1,6-ангидро-0-В-глюкопираноза) пли
фуранозной форме
(II) (D-галактозан,
или 1,6 ангидро-
р- D-галактофура-
поза). Названия Г.
дают по сахарам,
ангидридами кото-
рых они являют-
ся. Хорошо изу-
чены 3 группы Г.:
1,6-апгидропиранозы и 1,6-ангидро-
и
н2с
1,2-апгидро-
фуранозы. Г. растворимы в воде, низших спиртах и пи-
ридине; нерастворимы в эфире, бензоле и углеводоро-
дах. По химич. свойствам Г. близки гликозидам; стой-
ки к щелочам, по гидролизуются к-тами, причем 1,2-
связь гидролизуется легче 1,5- и 1,6-связей. Альдегид-
ных реакций Г. не дают, и их относят к невосстанавли-
вающим производным сахаров. Получают Г. пиролизом
мопо-, олиго- и полисахаридов (глюкоза, крахмал,
целлюлоза), из арилгликозидов, нагревая их со ще-
лочью, и др способами. Изучение Г., образовавшихся
при пиролизе полисахаридов, дает возможность судить
о строении последних. Г. могут быть использованы
для синтеза сахаров, замещенных в определенных по-
ложениях.
Лит.: Peat S. Advances in carbohydrate chemistry,
V. 2, N. Y 1946, p. 37; Dialler R, J., там же, v. 7, N. Y.,
1952 p. 37. Л. II. Линевич.
ГЛИКОЗЕЕНЫ — непредельные производные са-
харов, двойную связь к-рых рассматривают как обра-
зованную в результате отщепления (гипотетического)
элементов воды или же двух гидроксильных групп от
двух соседних атомов углерода. Названия Г. произ-
водят от родоначального сахара, изменяя окончание,
________________________ например Г. гексоз — гексо-
зеены, Г. глюкозы—глюко-
зеен, Г. галактозы— галак-
тозеен и т. д. В зависимо-
сти от положения двойной
связи Г. делят на несколько
групп, например, 1,2-гексо-
зеены (I), 5,6-гексозеены (II)
и т. д. Г. дают реакции
восстанавливающих саха-
ров и непредельных соеди-
нений (присоединение гало-
гепов, каталитич. гидрирование и т. д.). Г. применяют в
качестве исходных веществ в синтезах уроновых к-т,
дезоксисахарон и др. производных сахаров.
Лит.: Polysaccharides, в ин.: The carbohydrates. Che-
mistry, biochemistry, physiology, ed. by W. Pigman, N. Y.,
1957; Micheel F., Chemie der Zucker und Polysaccharide,
2 Aufl. Lpz., 1956. Л. И. Линевич.
ГЛИКОЗИДАЗЫ — ферменты группы карбогидраз,
гидролитически расщепляющие и синтезирующие
СН СНОН
|| сон с^нон
СНОН с СНОН
СНОН СНОН
сн с
CH-OH <Чн.,
о
гликозиды и олигосахариды. Ферменты этой группы де-
лятся на а-Г. и Д-Г. (расщепляют соответственно
а- пли Д-связи). В зависимости от сахара, входящего
в состав субстрата, Г. делятся на глюкозидазы (в со-
став гликозида входит глюкоза), галактозидазы (в со-
став гликозида входит галактоза) и т. д. Активность Г.
определяется редуктометрически пли поляриметри-
чески. Наиболее щироко распространенной и хоро-
шо изученной а-глюкозидазой является мальтаза.
Д-Глюкозидазы обнаружены у растений, животных,
бактерий; паилучший источник их получения — мин-
даль. Препараты фермента, полученные до сих пор,
являются смесью Д-глюкозидаз; они гидролизуют
a-D-галактозиды, Д-В-ксилозиды, Д-Ь-арабинозиды,
a-D-маннозиды, но не действуют на а-глюкозиды.
Единица активности Д-глюкозидазы устанавливается
по гидролизу глюкозида салицина при 30° и pH 5,0.
Д-Глюкозидаза наиболее стабильна при pH 7,1; оп-
тимум действия при pH 5,0; активируется Na, К, Mg,
инактивируется окислителями. a-Галактозидазы рас-
щепляют a-галактозиды. Оптимум действия фермента
при pH 4,5 и темп-ре 30°; Д-галактозидазы (лактаза)
гидролизуют Д-галактозиды, напр. лактозу, на к-рую
a-галактозидазы не действуют. Ингибитором Д-га-
лактозидазы является Д-фенилтиогалактозид. Фермент
активируется Na; оптимальный pH действия его 7,25.
Д-Глюкуронидаза расщепляет глюкурониды, напр.
гиалобиуроновую к-ту, на глюкуроновую к-ту и N-аце-
тилглюкозамин; активируется альбумином и дезокси-
рибонуклеиновой к-той, максимальная активность при
pH 5,0,
Лит.: Диксон М. и Уэбб Э., Ферменты, пер.
с англ.. 1961. Т. Т. Болотина.
ГЛИКОЗИДЫ (гетерозиды) — производные сахаров
(моносахаридов и олигосахаридов), в к-рых гликозиль-
ная часть молекулы связана через атом кислорода, серы
или азота с радикалом органич. соединения, не яв-
ляющегося сахаром и называемого агли коном
(г е и и н о м). В зависимости от того, связывается
ли гликозильная часть молекулы с агликоном (А.)
посредством кислорода, серы или азота, различают
О-, S- и N-гликозиды (г л и
Н
R—CH(CHOH)n—С—О—А
L_—о——*
о-г.
(А—остато.
висимости от природы сахарной компоненты и агли-
кона, Г. делятся на группы; так, к группе пирано-
зидов относят Г. с 6-членным кольцом гликозиль-
ной части, к фуранозидам — Г. с 5-членным
кольцом сахара. Различают также а- и Д-Г. в зависи-
мости от а- или Д-конфигурации углерода, связан-
ного с радикалом агликона. В природе встречаются
в большинстве случаев Д-Г. В зависимости от природы
сахарной компоненты различают: пентоз иды
(Г. пентоз), напр. ксилозиды, арабинозиды, рибозиды;
гексозиды (Г. гексоз), напр. глюкозиды, галак-
тозиды и др.; б и о з и д ы (Г. дисахаридов), напр.
мальтозиды, лактозиды и т. д. При гидролизе каждой
молекулы биозида наряду с агликоном образуется
не одна, а две молекулы моносахаридов. Существуют
Г. трисахаридов и др. олигосахаридов. Агликон Г.
может принадлежать к жирному, ароматич. и гетеро-
циклич. ряду.
О-l' ликозиды — наиболее распространенная в
природе группа Г.
О-Г. делятся на: 1) а л к в л г л и к о з п л ы (метил-,
этил- и т. п.); 2) фениле л ч козиды (папр., а- и р-фе-
нилглювозиды, салицин — Г. салицилового спирта, арбутин —
р-гидрохппонглюкозид, гаультерин — Г. метилового эфира
салициловой к-ты и др.); 3) а и т р а х и н о н г л и к о з и д ы
(папр., рубэрптршювая н-та — Г. ализарина, эмодины в др.);
м
К О 3 и л
Н Ь1).
Н Н
-C-N-A
Н
-C-S-A
J
N-C
аглинока)
957
ГЛИКОЗИДЫ — ГЛИКОЗИДЫ СЕРДЕЧНЫЕ
958
4) флавонглюнозиды (напр., рутин); 5) анто-
цианины (Г. антоцианидинов)— пигменты плодов и цве-
тов; 6) цереброзиды (галактозиды сфингозина);
7) стероидные Г. (напр., Г. сердечной наперстянки,
строфанта, ландыша, а также сапонины и др.); 8) азотсо-
держащие О-гликознды (напр., амигдалин, индикан,
содержащий в качестве агликона индоксил и др.); 9) гли-
ко а л к а л о и д ы, в к-рых сахарная компонента сое-
динена О-гликозидной связью с остатком алкалоида (соланин,
демиссин) и т. д.
О-Гликозиды — кристаллич. продукты, как пра-
вило, растворимые в воде и спирте. Подавляющее
большинство Г. не гидролизуется щелочами; исклю-
метилглюнофуранозид
ОСН3
метилглюкопиранозид
чениями являются лишь нек-рые Г., агликонами
к-рых являются фенолы, енолы и спирты, содержащие
в (3-положении электроотрицательные группы (напр.,
СО, SO2). Г. обычно не обладают восстановляющей
способностью, за исключением чувствительных к ще-
лочам, а также тех, агликоны к-рых сами обладают
восстановительными свойствами. Г. гидролизуются
кислотами; скорость гидролиза зависит от строения Г.
Фуранозиды гидролизуются значительно быстрее пи-
ранозидов. Конфигурация всех асимметрия, углерод-
ных атомов, а также характер агликона оказывает
влияние иа скорость кислотного гидролиза. Г. гид-
ролизуется специфич. ферментами а- и (J-гликозида-
зами. О-Г. получают синтетически (химич. и биохи-
мич. методами) или выделяют из природных продук-
тов. Так, алкилгликозиды получают взаимодействием
сахара с избытком спирта в присутствии безводного
НС1 как катализатора (метод Фише р а); а- и
(3-алкилгликозиды — взаимодействием сахаров со
спиртами под влиянием ферментов а- и (3-гликозидаз
(метод Б у р к е л о). Фепилгликозиды синтези-
руют из ацетилпроизводных сахаров, действуя на них
фенолами в присутствии катализаторов (напр., га-то-
луолсульфокислоты) с последующим отщеплением
ацетильных групп от образовавшихся ацетилирован-
ных Г. (м е т о д Г е л ь ф е р и х а). Многие природ-
ные Г. сложного строения (флавонгликозиды, стероид-
ные и др.) выгоднее выделять из природных продук-
тов. .Многие О-Г. применяются в качестве витаминов
(Витамины К, витамин Р) и лекарственных средств
(сердечные Г., стрептомицин, альбомицин, арбутин
и др.); нек-рые имеют токсикология, значение (са-
понины, соланин).
S-Г ликозиды (тиогликозиды) —наименее рас-
пространенная группа Г., их можно синтезировать,
СН2ОН
С-----О,
Н
,1
|\он Н/?
НО SR
н он
шим природным S-Г. относится Г. черной горчи-
цы — синигрин,
напр., взаимодействием ацетатов
гликозилбромидов с тиофенола-
ми в присутствии NaOH с после-
дующим омылением ацетальных
групп образовавшегося ацетил-
производного S-Г. Эти Г. очень
стойки к кислотному гидролизу.
Щелочи расщепляют их с обра-
зованием тиосахаров. К важней-
расщепляющийся ферментом миро-
зином с выделением аллилового горчичного масла и
KHSO4. Нек-рые ароматич. тиогликозиды обладают
противомалярийным действием.
N-Г ликозиды, или вторичные (а также тре-
тичные) гликозил амины, рассматриваются как произ-
водные гликозимипа (первичного глякозиламина),
образующиеся в результате замещения одного или
двух атомов водорода в аминогруппе алифатич., аро-
матич. или гетероциклич. радикалами. В отличие от
О-гликозидов, N-Г., кроме циклической структуры,
в растворах могут, по-видимому, иметь частично
таутомерное ациклическое строепие (типа оснований
Шиффа).
Взаимными превращениями таутомерных форм N-Г.
иногда объясняют мутаротацию р-ров этих Г. Многие
N-Г. получают непосредственным взаимодействием
сахара и амина на холоду или при нагре-
вании в спиртовой, спиртоводной или вод-
ной среде в присутствии катализаторов
(СН3СООН, HCI, NH4C1 и т. д.) или без
них. Свойства N-Г. весьма различны и
зависят от характера агликонов. Подав-
ляющее большинство их очень легко гид-
ролизуется кислотами и щелочами, но
некоторые (напр., пурин- и пиримидин-N-
гликозиды) стойки к кислотам и щелочам.
К N-Г. относятся исключительно важные
р
для обмена веществ продукты расщепления нуклеино-
вых кислот и нуклеопротеидов (нуклеотиды и нуклео-
зиды), нек-рые коэнзимы (напр., аденозинтрифосфор-
ZR
(СНОН)„ О
СН---------1
I
а — N — гликозид
I
(^НОН)П
нс-он
I
шиффово
основание .
I
~2Г (СНОН)П
НС--------
I
р —N - гликозид
ная кислота), недавно открытые уридиновые коэнзи-
мы (уридиптрифосфат и др.), нек-рые антибиотич. веще-
ства. N-Г. нек-рых ароматич. аминов предложены в
качестве антиоксидантов каучука.
Лит.: The Carbohydrates, ed, by W. Pigmann, N. Y., 1957;
ConchieJ. [а.о.],вкн.: Advances in carbohydrate chemistry,
v. 12, N. Y., 1957, p. 158; Ballou E. С., там же, v. 9, 1954,
p. 59; E 1 1 i s G. P., H о n e у m a n J., там же, v. 10,
1955, p. 95. Б. H. Степаненко.
ГЛИКОЗИДЫ СЕРДЕЧНЫЕ — гликозиды расти-
тельного происхождения, характеризующиеся силь-
ным действием на сердечную мышцу. Агликоны (гени-
ны) Г. с. — стероиды, имеют структуру I (кардепо-
лиды) или II (буфадиенолиды), где Хх, Хг, Х3, Х4 —
II или ОН; В — СН3 или кислородсодержащая группа.
В ф-ле (II), кроме гидроксилов в положении 3 и 14,
возможны гидроксилы и в других положениях.
Структура и физич. свойства нек-рых агликонов
Г. с. приведены в табл. 1.
Во всех известных Г. с. гликозидная цепь, представ-
ляющая собой остаток моно-, ди-, три- или тетрасаха-
рида, присоединена к гидроксилу агликона в поло-
жении 3. Сахара Г. с. (кроме D-глюкозы и L-рамнозы)
специфичны для данной группы соединений и пред-
ставлены 6-дезоксигексозами, 2,6-дезоксигексозами и
их З-О-мети левыми эфирами (см. Дезоксисахара).
Важнейшие из них: D-аптиароза (III), D-дигиталоза
(IV), D- и L-теветоза (V), D-дигитоксоза (VI), D-ци-
959
ГЛИКОЗИДЫ СЕРДЕЧНЫЕ
960
мароза (VII), D-дигиноза (VIII), D-олеандроза (IX),
D-сарментоза (X).
в - ЗНО -он 5 н- ЭНО -он с но- :но -н сно <^н2
в 1- -он Н3СО- -н н- -ОСНз н - —он
НО- -н но- -н но- -н н- -он
в [- -он н- -он но- -н н- -он
с [ ,Н3 II ш3 V с V(L-( Нз орма) зн3 VI
но ЛЮ :но сно
с зн :н. зн ( -Н2
н -ОСН3 Н3СО- н н- -ОСНз н- осн
н — эн но- н но- -1 I но- -н
н - эн H- он но- -I 1 н- -он
с Н3 VII с V В1 III IX с Н3 к
Кроме того, при неполном гидролизе сахарного
остатка из Г. с. могут быть выделены биозы и триозы,
напр. строфантобиоза (XI) и строфантотриоза. По-
Н ОН ОСН3 Н
остаток глюкозы остаток цимаоозы
XI
рядок и конфигурации гликозидных связей в Г. с.
установлены далеко не во всех случаях. Обычно сахара
D-ряда связаны с агликоном 0-гликозидной связью,
L-ряда — а-связью.
Таблица 1
Агликон Заместители муле в фор- I Т. пл., °C [alD (в СН3ОН)
R X. Х2 1 Х3 X,
Дигитоксигенин . . сн3 н н н н 236-256 4-18=
Строфантидин . . . сно он н н н 171-175 230-232 + 42°
Гитоксигенин . . . сн3 н н н он 232 + 34”
Сциллигляукози- дйн * сно н н — — 245- 248 + 49,5”
* В формуле (II) двойная связь в положении 4,5.
Большинство Г. с. получено в кристаллич. виде,
нек-рые — в виде аморфных веществ; плавятся с раз-
ложением; растворимы в воде, спиртах, пиридине и
хлороформе, нерастворимы в эфире и бензоле. Физич.
свойства нек-рых Г. с. приведены в табл. 2.
Таблица 2
Гликозид Т. пл., °C [a’.D Агликон Сахара
Дигитоксин 233—236 (из водного + 16,5° (СНС13 4- Дигитоксиге- 3 остатка
спирта) + 5% спирта) НИИ D-дпгитонсозы
Цимарин 148 (из СН3ОН), 204—205 (безводн.) 4- 38° (СНС13 или спирт) Строфантидин D-цимароза
К-строфантин-З 195 + 32,6° » Стро ф анто биоз а
Конваллоток- син 238—245 (из этилацетЗ’ та), 212—213 (из влаж- ного спирта) 0° (спирт), —15° (пиридин) » L-рамноза
Олеапдрнн 250 —52° (СНС[3) 16-ацетилги- токсигенин L-оле андроза
Сциллигляуко- зид 164—166 + 106° (СН3ОН) Сциллигляу- козндин D-глюкоза
Характерная реакция агликонов Г. с. (и самих
Г. с.) — необратимая изомеризация в щелочных сре-
дах в соединения изо-ряда путем внутримолекуляр-
ного присоединения С(14)ОН-группы к бутенолидному
циклу:
В кислых средах или под влиянием катализаторов
(типа Фриделя—Крафтса) легко протекает дегидра-
тация агликона по третичным гидроксилам при С(5)
и С,14). Кислотный гидролиз Г. с. легко протекает
в мягких условиях, если агликон связан с 2-дезокси-
сахаром. Гидролиз гликозидной связи агликона
с сахарами, содержащими ОН-группу в положении 2,
требует более жестких условий, что затрудняет выде-
ление неизменного агликона (в связи с его лабиль-
ностью к кислотам). В этих случаях успешно приме-
няется гидролиз ацетоновым раствором НС1.
Полный синтез агликонов Г. с. не осуществлен.
Произведены лишь немногие частичные синтезы гли-
козидов (только монозидов) с сердечными агликонами
бутенолидпой группы по реакции Кёнигса—Кнорра.
Г. с. карденолидной группы найдены только в расте-
ниях; в животных (жабы) найдены буфогенные сте-
роиды (см. Буфоеенины), близкие Г. с. по действию
на сердце. В одном растении обычно содержится не-
сколько близких по структуре Г. с. Качественно
Г. с. определяют цветными реакциями с нитропрус-
сидом натрия в щелочном пиридиновом р-ре (реакция
Легаля), с динитробензойной к-той и др. Ряд реакций
Г. с. обусловлен наличием дезоксисахаров (реакция
Келлера—Килиани и др.). Г. с. добывают гл. обр. из
растений семейств Apocynaceae, Liliaceae, Scrophu-
lariaceae (род Digitalis), Cruciferae, Ranunculaceae
и др. Все Г. с. в малых дозах оказывают специфиче-
ское стимулирующее действие на сердечную мышцу,
а в несколько больших дозах являют, я сердечными
ядами, вызывая остановку сердца. Действие аглико-
нов Г. с. на сердце в общем аналогично действию са-
мих Г. с. Сахарная часть молекулы влияет на ско-
рость и продолжительность их кардиотонического
действия. Г. с. широко применяют при лечении
многих сердечных заболеваний. Важнейшими лекар-
ственными препаратами Г. с. являются галеновые
и новогаленовые препараты: настойки и экстракты
наперстянки, строфанта, ландыша майского, кен-
дыря коноплевого и др., а также индивидуальные
соединения: цимарин, К-строфантин-fJ и К-строфан-
ТОЗИД.
Биологическая активность препаратов Г. с. вы-
ражается в ЛЕД (лягушачья единица действия) и в
КЕД (кошачья единица дейст-
вия); единица действия — наи-
меньшая доза препарата, вызы-
вающая остановку сердца жи-
вотного.
Лит.: Ф и з е р Л. ифизер
М., Химия природных соединений
фенантренового ряда, пер. с англ.,
М.—Л., 1953; Karrer W., Коп-
stitution und Vorkommen der orga-
nlschen Pflanzenstoffe, Basel — Stut-
tgart, 1958; Advances in carbohyd-
rate chemistry, v. 1, N.Y., 1945, p. 147;
v. 8, 1953, p. 45; Fieser L. F
F ieser M., Steroids, N. Y.—L., 1959;
Proceeding of the Fourth Internatio-
nal Congress of biochemistry, Vienna,
1 — 6 sept., 1958, v 1, L., 1959.
А. Ф. Бочков.
961
гликокол — гликоли
962
ГЛИКОКОЛ — см. Глицин.
ГЛИКОЛЕВАЯ КИСЛОТА (оксиуксусная кислота)
НО—СН2—СООН, мол. в. 76,03 — простейшая окси-
кислота; бесцветные кристаллы; т. пл. 79—80°; хо-
рошо растворимые в воде и обычных органич. раство-
рителях. Константа диссоциации К=1,5-1О4. Содер-
жится в незрелом винограде, свекловичном соке. По-
лучают: действием азотистой к-ты на аминоуксус-
ную к-ту; осторожным окислением фруктозы; в
пром-сти — омылением хлоруксусной к-ты или ее
солей; электролитич. восстановлением щавелевой к-ты,
а также взаимодействием формальдегида с окисью
углерода и водой под давлением в присутствии кислот-
ных катализаторов. Г. к. дает все реакции, характер-
ные для гидроксильной и для карбоксильной групп.
При перегонке в вакууме образует гликолид — про-
стейший из лактидов:
-2НО /°“С0\
2 НОСН2СООН ----- СН2 Сн2
Yo-o7
Г. к. применяется как вспомогательное вещество
при крашении текстильных материалов, в кожевенной
пром-сти и как компонент составов для очистки по-
верхности металлов. Е. М. Рохлин.
ГЛИКОЛЕВОЕ ТИТРОВАНИЕ — титрование, ос-
нованное на применении гликолей и их смесей с раз-
личйыми углеводородами, высшими спиртами и т. п.
в качестве органич. растворителя, повышающего силу
растворенных в нем кислот и оснований. Выбор гли-
колевой смеси определяется гл. обр. ее растворяю-
щей способностью, изменением в ней окраски индика-
тора, доступностью. Наиболее часто применяют
смесь (1:1) этиленгликоля (пропиленгликоля) с изо-
пропиловым (или н-бутиловым) спиртом. Для титро-
вания пользуются р-рами HCI или НС1О4 в том же
растворителе. Точку эквивалентности устанавливают
потенциометрически или с помощью индикаторов:
тимолового голубого, метилового красного, бромфено-
лового голубого, ксиленцианола Ф. Метод Г. т.
успешно применяют для непосредственного титрова-
ния алифатич. и ароматич. аминов, аминокислот,
алкалоидов, солей слабых органич. к-т (в том числе
мыл и различных фармацевтич. препаратов), а также
для косвенного определения некоторых металлов. В
гликолевой среде возможно раздельное титрова-
ние некоторых аминов (например, диэтиламина и
пиридина).
Лит.: Палит Ш. Р., Дас М. Н., Сомаяд-
ж у л у Г. Р., Неводное титрование, пер. с англ., М., 1958.
ГЛИКОЛЕВЫЙ АЛЬДЕГИД (оксиуксусный аль-
дегид) НОСН2СНО, мол. в. 60,05 — бесцветные пла-
стинки; т. пл. 96—97°; 1,360, 1,391; ntf 1,4603;
растворим в воде, горячем спирте, слабо — в эфире.
Свежеприготовленные водные растворы Г. а. содержат
димерное соединение С4Н3О4, к-рое через 24 часа
полностью переходит в мономер; при окислении рас-
творами перекиси водорода, перманганата калия, аце-
тата меди образуется муравьиная к-та и вода; восста-
навливается амальгамой алюминия до этиленгликоля
СН2ОНСН2ОН. Г. а. уже на холоду восстанавливает
Фелинга реактив', с 1%-ным р-ром NaOH в водной
среде образует смесь гексоз СН2ОН(СНОН)4СНО;
реагирует подобно всем альдозам с фенилгидразином,
превращаясь в озазон:
СН2ОНСНО + 3C0H5NHNH2 —
— CH-CH=NNHCcH5 + NH3 + CbH5NH2
NNHCjHj
С уксусным ангидридом при 0° в среде пиридина кон-
16 к. X. Э. т. 1.
денсируется в диацетат 2,5-диокси-1,4-диоксана:
сн-сн-ососн3
2СНсОНСНО + (СН3СО)оО—- о<
сн-сн
СН3ОСО
Получают Г. а. нагреванием диоксималеиновой к-ты
при 50—60° с водой или пиридином:
но—с—соон
II 4- Н.,0 НОСН2СНО
НО—С—соон
а также гидролизом этилаля бромуксусного альдегида
ВгСН2СН(ОС2Н5)2 + ЗН2О — СН2ОНСНО 4- НВг +2С3Н5ОН.
С концентрированным р-ром NaOH Г. а. дает желтую
окраску, с FeCl3 — красную. м. Ф. Тушинский.
ГЛИКОЛИ (диолы, двухатомные спирты) — соеди-
нения жирного ряда, содержащие две спиртовые
группы. В зависимости от взаимного расположения
ОН-групп различают 1,2-Г., напр. НОСН2СН2ОН,
1,3-Г., напр. Н0(СН2)30Н, и т. д. 1,1-Г., т. е. соеди-
нения, содержащие две ОН-группы у одного атома
углерода [гидратные формы альдегидов и кетонов
RCH(OH)2 и RC(OH)2Rz], обычно очень нестойки и
легко отщепляют воду. Названия «Г.» производятся
от названия двухатомного радикала с окончанием
«гликоль». Так, НОСН2СН2ОН называют этиленгли-
колем, НО(СН2)3ОН — триметиленгликолем и т. д.
По женевской номенклатуре Г. называют диолами,
причем положение ОН-групп, как и возможных от-
ветвленных радикалов, обозначают цифрами. Напр.,
соединению строения (СН3)2С(ОН)СН2ОН соответ-
ствует название 2-метил-1,2-пропандиол (2-метил-
пропандиол-1,2). По характеру радикалов, с к-рыми
связаны ОН-группы, различают, напр., двупервичные
НОСН2СН2ОН, двувторичные СН3СНОНСНОНСН3
и двутретичные Г. (СН3)2С0НС0Н(СН3)2,- иначе на-
зываемые пинаконами. Известны и Г. с неодинаковым
характером ОН-групп, напр. первично-вторичный
бутиленгликоль СН3СН2СНОНСН2ОН, первично-тре-
тичный изобутиленгликоль (СН3)2С0НСН20Н и др.
Г. могут быть получены обычными методами син-
теза спиртов, напр. гидролизом дигалогенидов (через
стадию диацетатов). С другой стороны, известны и
особые методы получения Г., напр. гидратация оки-
сей алкиленов (эпоксисоединений): СН2СН2О + Н2О—►
—> НОСН2СН2ОН, окислением олефинов с образова-
нием вицинальных Г. Для бутандиола-1,4, к-рый
можно превратить в бутадиен, разработан промыш-
ленный метод синтеза из ацетилена и формаль-
дегида: HCsCH +'2СН2О -> НОСН2С=ССП2ОП ->
-> НО(СН2)4ОН.
Г. обладают всеми свойствами, характерными для
спиртов, и образуют, напр., алкоголяты, т. н. гли-
коляты, простые и сложные эфиры; гликоляты,
образованные ионами тяжелых металлов, напр.
Сп2+, — характерные комплексные соединения. Одна-
ко наличие двух ОН-групп, в особенности при со-
седних атомах углерода, ведет к значительным осо-
бенностям в химич. поведении Г. Так, с образова-
нием соединений со смешанными функциями может
реагировать лишь одна ОН-группа, напр. при дей-
ствии газообразного HCI на Г. образуются соответ-
ствующие хлоргидрины, тогда как реакция с РС15
ведет к дигалогенпроизводным. Своеобразие в свой-
ствах Г. в особенности проявляется при их дегидра-
тации. При отщеплении воды от Г. с ОН-группами,
расположенными не у соседних атомов углерода, об-
разуются ненасыщенные спирты и диолефины; напр.,
из СН3СН0НСН3СН20Н можно последовательно по-
лучить СН2=СНСН2СН2ОН и бутадиен. Однако
в случае дегидратации вицинальных Г. отщепляется
лишь одна молекула воды и образуется альдегид или
963
гликоли — гликолиз
964
кетон (СН3СНО из НОСН2СН2ОН, СН3СОС2Н5 из
СН3СНОНСНОНСН3), что объясняется изомеризацией
промежуточных енолов. Из нек-рых Г. в результате
дегидратации могут образоваться циклич. внутренние
эфиры, особо прочные в случае пяти- и шестичленных
соединений. Так, из 2 молекул этиленгликоля можно
получить диоксан (I), из одной молекулы пентаме-
тиленгликоля — окись
пентаметилена (II). Для
пинаконов характерна
дегидратация с обра-
зованием кетона, со-
провождающаяся изме-
(см. Пинаколиновая и
сн2-сн2
о
хсн2-сн./
,сн2-сн2
н2 ХО
СН2 СН2^
нением углеродного скелета
ретропинаколиновая перегруппировки). Аналогичным
образом наблюдается перегруппировка
гидратации двувторичных Г. ароматич.
ряда: СвН5СНОНСНОНС3Н5 — Н20 —►
—>• (С3Н5)2СНСНО. Для вицинальных Г.
характерна необычная легкость разры-
ва С—С-связей при действии окислите-
лей [HJO4 или (СНдСООДРЬ], напр.
НОСН2СН2ОН+ HJOi -+ 2CH2O+H2O+HJO3.
Эта реакция широко применяется и
при изучении сахаров.
Г. имеют большое практич. значение.
Так, этиленгликоль в смеси с водой при-
и при де-
(15)>
фталевые смолы, широко применяемые в лакокрасоч-
ной промышленности; продукты поликонденсации
Г. с другими двухосновными кислотами употреб-
ляются в качестве пластификаторов и т. д. Конденса-
ция этиленгликоля с окисью этилена приводит к поли-
гликолям (диэтиленгликолю НОСН2СН2ОСН2СН2ОН,
триэтиленгликолю Н ОСН2СН2ОСН2СН2ОСН2СН2ОН
и т. д.), эфиры к-рых являются хорошими раствори-
телями. я. Ф. Комиссаров.
ГЛИКОЛИЗ — ферментативный процесс расщепле-
ния углеводов в живой клетке, дающий в качестве
конечного продукта молочную кислоту, сопряженный
с образованием аденозинтрифосфорной кислоты (АТФ)
(см. Аденозинфосфорные кислоты). Г. слс::::."й
процесс, катализируемый б- ---
при
участии нескольких
Г. — сложный
более чем 20 ферментами
коэнзиматич.
СН2ОН
•О
систем, сла-
СХЕМА ГЛИКОЛИЗА
(С6н10о5)п
о
н
н
но
СН2ОН
н
он
Н
н
ОН
он
Н
н
н
Mg2+
он
он
АТФ
снэ
2CHOH
соон
н2о®
о
Н
о®
он
н
®ОСН2
н
он
АДФ
СНз
2СО
I
СООН
2(2Н)
2ДПН
2 АТФ
Mg2*\(13)
сна
и 2
2СОН
I
СООН
2АДФ
СНо
2СО®
СООН
\(12)
\ СН2ОН
2СНО©
соон''
2ДПН-Н+Н
Н
СН2ОН
ОН
. /АТФ
®ОСН2
Н
СН2О®
СНОН
СНО
* *-АДФ
сн2о®
он
(5)lMg2
(11)
\сн2о©
2CHOH
I
соон
2(2Н)
сн2о®*-"
со —-
I (7
СН2ОН
СН2О®
2СНОН
(10)
Mg2
2АТФ 2АДФ
сн2о© _________
2СНОН | /ОН
соо® ссн
он
меняется в качестве антифриза, как заменитель гли-
церина, как исходный продукт для получения моно-
и полных эфиров этиленгликоля и др.; гликольди-
нитрат применяется для производства незамерзаю-
щих динамитов. В результате поликонденсации Г.
и глицерина с фталевой кислотой образуются гли-
гающийся из большого числа реакций определенной
последовательности. Промежуточные продукты и ко-
ферменты Г. выделены и синтезированы, большинство
ферментов Г. получено в кристаллич. виде, последо-
вательность и ход реакций Г. изучены вплоть до уста-
новления кинетических констант. В точном значении
965
ГЛИКОЛИЗ — ГЛИНИСТЫЕ РАСТВОРЫ
966
слова Г. означает расщепление глюкозы, однако в ряде
животных тканей преим. расщепляется не глюкоза,
а гликоген (гликогенолиз). Окислительно-
восстановительные реакции Г. протекают без участия
кислорода. Биологич. значение Г. ясно из того, что
наряду с дыханием Г. служит источником энергии
для жизнедеятельности клетки и источником исходных
веществ для биохимич. синтезов.
Путем- сочетания вне организма (in vitro) очищен-
ных ферментов, коэнзимов и углевода вся фермент-
ная система Г. может быть искусственно воспроиз-
ведена и может быть прослежен ход реакций. Кроме
того, ввиду обратимости ферментных реакций ход
Г. можно наблюдать как в прямом, так и в обратном
направлении. Непрерывно текущий, циклически по-
вторяющийся ход Г. в клетке изображен на схеме.
До стадии пировиноградной к-ты Г. идет так же, как
спиртовое брожение. Отдельные реакции, из к-рых
слагается Г., выполняют различные биохимич. функ-
ции. Одни реакции осуществляют вспомогательную
функцию, обеспечивая ход последующих основных
превращений; другие реакции выполняют главную
функцию — генерирование богатых энергией фосфат-
ных связей (см. М акроэргические связи). Вспомога-
тельные реакции (1—5) способствуют превращению
более стойких циклич. форм гексоз в нестойкие аци-
клич. и тем облегчают разрыв шестиуглеродной
цепи гексозы на две молекулы фосфотриозы. Кроме
того, вспомогательная реакция (11) осуществляет
перегруппировку, которая подготовляет молекулу
фосфотриозы к перераспределению свободной энергии
(создает возможность образования фосфоенолпиро-
виноградной к-ты). С другой стороны, на двух эта-
пах Г. генерируются фосфатные макроэргич. связи.
Во-первых, в результате окислительно-восстанови-
тельной реакции между фосфоглицеральдегидом и
дифосфопиридиннуклеотидом (ДПН) (см. Пиридин-
нуклеотиды) освобождающаяся энергия сначала акку-
мулируется ангидридными связями 1,3-дифосфогли-
цериновой к-ты (реакция 9), а затем переносится
вместе с фосфатными группами на аденозиндифосфор-
ную к-ту (АДФ) (см. Аденозинфосфорные кислоты)
с образованием 3-фосфоглицериновой кислоты и АТФ
(реакция 10). Во-вторых, хотя реакция дегидратации
фосфоглицериновой к-ты в фосфоенолпировиноград-
ную к-ту (12) идет без окисления, эта реакция делает
возможным образование макроэргич. связи за счет
превращения первичной фосфатной группы, связанной
со спиртовым гидроксилом, в фосфориленольную
группу. При этой реакции свободная энергия моле-
кулы перераспределяется и сосредоточивается в фос-
фориленольной связи. Макроэргич. связь фосфоенол-
пирувата переносится затем на АДФ с образованием
пировиноградной к-ты и АТФ (13).
Коэнзимы Г. обычно являются компонентами не-
скольких реакций, в ходе к-рых они регенерируют, и
в силу этого не входят в общий баланс Г. Так, в реак-
ции с фосфоглицериновым альдегидом дифосфопири-
диннуклеотид (ДПН) служит акцептором водорода
(реакция 9), превращается в восстановленную форму
(ДПН-Н), а затем на завершающем этапе Г. (реак-
ция 15) ДПН-Н является донором водорода — вос-
станавливает пировиноградную к-ту
ДПН при этом регенерирует.
NH„
в молочную;
Подобным же обратимым превращениям подвергается
и АТФ, к-рая в реакциях (3) и (5) переходит в АДФ,
а в дальнейшем на этапах (10) и (13) этот коэнзим
снова регенерирует как АТФ. Таким образом, через
системы АТФ—АДФ и ДПН—ДПН-Н осуществляется
связь между отдельными этапами Г. Наряду с этим
существуют и другие коэнзимы Г., участвующие
только в одиночных реакциях. Благодаря структур-
ному подобию компонентам реакции коэнзим при
этом регенерирует в ходе самой реакции. Таковы
глюкозо-1,6-дифосфорная к-та, участвующая в реак-
ции (2), и 2,3-дифосфоглицериновая к-та, участвую-
щая в реакции (И). Наконец, необходимыми компо-
нентами реакций Г. служат также неорганич. ка-
тионы Mg2? и К+. Ряд важных сведений получен
относительно химизма ферментов Г., их простетич.
групп, природы «активного» каталитического центра,
механизма взаимодействия их с коэнзимами и суб-
стратами.
Известен ряд ингибиторов Г. — ферментных ядов,
избирательно блокирующих отдельные промежуточ-
ные реакции Г. К их числу относятся: фториды, по-
давляющие реакцию (12), иодацетат, иодацетамид,
га-хлормеркуробензоат, подавляющие реакцию (9),
и другие.
На приведенной схеме показано, что в ходе Г. на
1 моль расщепляемого углевода синтезируется 4 моля
АТФ, в связи с чем энергия системы увеличивается
на энергию 4 макроэргич. связей. Из этих 4 связей
в случае распада глюкозы 2 связи, а в случае распада
гликогена 1 связь затрачиваются на фосфорилирова-
ние углевода (реакции (3) и (5)]. Следовательно,
в результате расщепления 1 г-моля углевода имеет
место чистый выход в первом случае 2 молекул, а во
втором случае 3 молекул АТФ, используемых затем
в качестве носителя энергии для жизнедеятельности
клетки, различных биохимич. синтезов и др. Общее
количество свободной энергии, к-рое теоретически
освобождается в клетке при распаде 1 г-моля глико-
гена, составляет 52 ккал, а при распаде глюкозы соот-
ветственно 47 ккал. Стандартная свободная энергия
гидролиза связи АТФ равна 8,4 ккал. Отсюда может
быть вычислена энергетич. эффективность Г. Если
субстратом Г. служит гликоген, она равняется
3x8400/52000 — 48%; если же Г. идет на глюкозе,
эффективность процесса равняется 2x8400/47000 =
= 36%. Хотя эти величины эффективности доста-
точно велики, тем не менее выход свободной энергии
при гликолитич. распаде углевода составляет лишь
7,5% от выхода энергии при распаде такого же коли-
чества углевода в результате дыхания клетки.
Лит.; Эвгелъпрцч В. А., Изв. АН СССР. Сер.
биол., 1945, № 2, с. 182; Парнас Я. О., Избр. труды,
М., 1960; Диксон М., Уэбб 9., Ферменты, пер. с англ.,
М„ 1961;Нейландс Дж. Б. иШтумпф П.К., Очерки
по химии ферментов, пер. с англ., М., 1958: Chemical path-
ways of metabolism, v. 1, N. Y., 1954, p. 67; RobblnsE. A.,
Boyer P. D„ J. Biol. Chem., 1957, 224, № 1, 121.
С. А. НейдЗах.
ГЛИКОЛИПИДЫ — см. Липиды.
ГЛИКОПРОТЕИДЫ — см. Мукопротеины.
ГЛИНИСТЫЕ РАСТВОРЫ — основной вид про-
мывочных жидкостей, применяемых при бурении
скважин. Проходка глубоких скважин не может быть
осуществлена ' "
логичных им
ОН ОН
ОН
ОН
сн2-о-р-о-р-о-Сн2
н о о н
Дифосфопщшдинщилеотид СПЛЮ
H2NCOx^
без применения Г. р. или других ана-
промывочных жидкостей. Г. р. пред-
ставляют собой многокомпонентные,
полидисперсные, тиксотропно - струк-
турированные системы, состоящие из
воды, глины, различных добавок элек-
тролитов и органических веществ, а
также инертных наполнителей. В ка-
честве промывочных жидкостей вза-
мен Г. р. в ряде случаев применяют
воду, водные р-ры поверхностно-актив-
13*
967
ГЛИНИСТЫЕ РАСТВОРЫ — ГЛИНЫ
968
ных веществ и электролитов, а также р-ры на нефтя-
ной основе.
Промывочная жидкость в процессе бурения выпол-
няет следующие основные функции: очистка забоя от
выбуренной породы и вынос ее на поверхность, удер-
жание частиц выбуренной породы во взвешенном
состоянии при прекращении циркуляции, образова-
ние малопроницаемых для жидкости корок или пле-
нок на степках скважины (глинизация), создание
противодавления на стенки скважины в целях преду-
преждения проникновения газов, нефти, воды, об-
легчения механич. разрушения проходимых при бу-
рении твердых пород (см. Адсорбционное понижение
прочности), охлаждение долота, а в случае турбин-
ного бурения — передача энергии к забойному гид-
равлическому двигателю.
Водные суспензии глин, не обработанные химич.
реагентами, являются неустойчивыми дисперсными
системами, ухудшающими свои свойства во времени
при взаимодействии с обломками выбуренных пород
и под действием электролитов, содержащихся в при-
родных водах. Для улучшения коллоидно-химич.
свойств Г. р. в них вводятся специальные реагенты:
пептизаторы, стабилизаторы, а также реагенты, спо-
собствующие понижению вязкости.
В качестве пептизаторов используются: каустиче-
ская и кальцинированная сода, щелочные фосфаты
(тринатрвй-фосфат, гексаметафосфат), силикат на-
трия. В небольших концентрациях реагенты-пептиза-
торы повышают гидрофильность глин и способствуют
диспергированию их в воде, что улучшает структурно-
механич. и коллоидно-химич. свойства Г. р. (см.
Структурообразование в дисперсных системах. Тиксо-
тропия).
В качестве стабилизаторов используются высоко-
молекулярные природные и синтетич. продукты:
крахмал, карбоксиметилцеллюлоза, щелочные р-ры
гуматов, экстракты бурого угля и торфа, лигносуль-
фонаты сульфитно-спиртовой барды, сульфатцеллю-
лоза, полиакрилаты и др. Адсорбируясь на поверх-
ности глинистых частиц, они образуют структуриро-
ванные прочные (высоковязкие) оболочки, защищаю-
щие частицы глины в суспензии от коагуляции под
действием электролитов. В нек-рых случаях эффек-
тивность стабилизации возрастает при одновремен-
ном использовании нескольких стабилизаторов. За
исключением полиакрилатов, все перечисленные ста-
билизаторы неустойчивы при темп-рах выше 100°.
Полиакрилаты же обеспечивают достаточную стаби-
лизацию Г. р. при темп-рах до 200—250°, что позво-
ляет применять их для повышения устойчивости
Г. р. при бурении глубоких скважин с высокими за-
бойными темп-рами и, соответственно, давлениями.
Регулирование реологич. свойств Г. р. наиболее
эффективно достигается добавками реагентов-стаби-
лизаторов, понижающих структурную вязкость Г. р.
Таковы естественные дубители, получаемые экстрак-
цией таннидов из древесных пород, а также синтетич.
танниды (синтаны), продукты окисления гидролиз-
ного лигнина. Сорбция этих реагентов на поверхности
глинистых частиц приводит в основном к снижению
прочности гидрофильных коагуляционных структур.
Известь, гипс, хлористый кальций и др. соли с поли-
валентными катионами вводятся в хорошо стабилизо-
ванные Г. р. для снижения диспергируемости
в р-ре разбуриваемых глин и для повышения устой-
чивости стенок скважин, сложенных размокающими
глинами. Введение добавок этих реагентов предот-
вращает быстрое повышение вязкости Г. р. В нек-рых
специфических условиях бурения в стабилизованные
Г. р. добавляют силикат натрия в концентрациях
выше 10% для закрепления обваливающихся глини-
стых сланцев, а также нефть и продукты ее перера-
ботки для снижения липкости глинистых корок в це-
лях предупреждения прихвата инструмента, а также
образования наростов — «сальников». В качестве
инертных наполнителей для повышения плотности
утяжеленных Г. р. (с целью увеличения гидроста-
тич. давления столба раствора, уравновешивающего
горное давление) используются тонко измельченный
барит, гематит, пиритовые огарки, колошниковая
пыль и др. При бурении в кавернозных и трещинова-
тых породах с целью предупреждения уходов Г. р.
применяют различные набухающие и волокнистые
наполнители.
Вода как промывочная жидкость применяется при
бурении в устойчивых карбонатных породах. Для
улучшения вскрытия продуктивного пласта к ней
добавляют поверхностно-активные вещества, обеспе-
чивающие вытеснение воды нефтью в порах породы.
Промывочные жидкости на нефтяной основе приме-
няют при бурении в осложненных условиях. Они
представляют собой сложные коллоидно-химич. си-
стемы, состоящие из дизельного топлива или нефти
(дисперсионная среда), окисленного битума (дисперс-
ная фаза), различных мыл (стабилизаторы) и твердых
наполнителей (известь, сажа, утяжелители). Для
вскрытия продуктивных пластов широко приме-
няются также эмульсии второго рода (вода в масле),
к-рые получаются при введении воды (до 40%) в рас-
твор на нефтяной основе.
Лит.: Ребиндер П. А., Ш р е й н е р Л. А.,
Ж и г а ч К. Ф., Понизители твердости в бурении, М.—Л.,
1944; Баранов В. С., Глинистые растворы для бурения
скважин в осложненных условиях, М., 1955; РоджерсВ.Ф.,
Промывочные жидкости для бурения нефтяных скважин,
пер. с англ., М., 1960; Я т р о в С. Н., Промывочные жид-
кости в бурении скважин, М., 1960; Жиг ач К. Ф., Адель
И. Б., Загармистр О. С., Нефтяное хозяйство,
1956, №3; Ж и г а ч К. Ф., Адель И. Б., МухинЛ. К.
[и др.], там же, 1956, № 8; Ж и г а ч К. Ф., Финкель-
штейн М. 3., Могилевский Е. М., Хим. наука
и пром-сть, 1957, 2, № 1,с. 76.
И. Б. Адель, Л. К. Мухин.
ГЛИНЫ — группа осадочных горных пород, об-
ладающих способностью образовывать с водой пла-
стичную массу. Эта масса после высыхания сохра-
няет приданную ей форму, а после обжига приобре-
тает твердость камня. С большим количеством воды
Г. образуют суспензии с довольно высокой дисперс-
ностью.
В минералогич. составе Г. главную роль играют
т. н. глинистые минералы, в большинстве
своем обладающие слоистой кристаллич. структурой.
К ним относятся: каолинит
Al4[Sl4O10] [ОН]8 пли А12О3 • 2S10a • 2Н,0
монтмориллонит, минерал с сильно колеб-
лющимся составом, его типовая ф-ла SisAl4O20(OH)4 •
• пН2О, где Si может заменяться А1, Fe3', Fe2+,
Zn, Li, Си, Mg и т. д., могут присутствовать также
Na, Са и др., галлуазит, близкий по составу
к каолиниту, содержащий вдвое больше гидратацион-
ной воды, и др. минералы. Кроме собственно глини-
стых минералов, в Г. присутствуют в качестве при-
месей органич. йещества и многие минералы, в пер-
вую очередь кварц, опал, слюда, гидраты окислов
железа, алюминия и др. Каолинит — основная со-
ставная часть каолинов и многих огнеупорных Г.
Состав монтмориллонита в различных Г. может ши-
роко изменяться; окислы железа, частично замещая
А12О3, могут входить в его кристаллич. решетку,
образуя различные железистые монтмориллониты,
входящие в состав большинства обычных почвенных
Г. Монтмориллонит обладает ярко выраженной спо-
собностью к адсорбции и обмену катионов (обмену
оснований), а также способностью поглощать и выде-
лять слабо связанную воду в зависимости от влаж-
ности окружающей среды.
969
ГЛИНЫ ОТБЕЛИВАЮЩИЕ — ГЛИЦЕРИДЫ
970
Особенностью глинистых минералов является спо-
собность разбухать и распадаться на мельчайшие ча-
стицы (диаметром менее 0,01 мм) при смачивании их
водой или нек-рыми другими жидкостями. Это обус-
ловлено тем, что при увлажнении Г. молекулы воды
проникаю?? между слоями кристаллов глинистых ми-
нералов. Внутри слоя атомы связаны сильнее, чем
слои между собой. При этом слои силиката могут
быть разделены практически любым количеством воды.
Слои кристалла разделяются сначала одним, затем
двумя, тремя и т. д. слоями воды.
Образование Г. идет гл. обр. на поверхности суши
в результате процессов выветривания. Большинство минера-
лов горных пород, соприкасаясь с атмосферой и подвергаясь
механич. и химич. действию воды и воздуха, постепенно из-
меняются, разрушаются п переходят в минералы Г. Первичные
Г. образовались в геологич. эпохи интенсивного выветривания.
Перемыв первичных Г. и переотложение их материала в раз-
личных водоемах (озерах, морях и т. д.) дают накопление вто-
ричных Г. Подвергаясь метаморфизму и уплотнению в толще
земной коры, Г. теряют способность образовывать с водой
пластичную массу и превращаются в глинистые сланцы, фил-
литы и др. плотные глинистые породы.
Г. разного типа широко применяются в пром-сти и
являются одним из важнейших видов минерального
сырья. Красные (железисто-монтмориллонитовые) Г.
используются в качестве строительных материалов
(кирпично-черепичное, цементное произ-во, саманный
кирпич, строительство плотин, дорог, строительные
р-ры и др., в изделиях грубой керамики в гончарном
произ-ве). Эти Г. применяются и в произ-ве разного
рода вспученных Г. (напр., керамзита), служащих
заполнителем легких бетонов. Каолинитовые Г., от-
личающиеся высокой пластичностью и малым содер-
жанием железа, находят широкое применение в
произ-ве шамотных огнеупоров, фарфора, фаянса,
кислотоупорных изделий. Каолин после отмучивания
применяется в качестве наполнителя бумаги, резино-
вых смесей, пластмасс и др. Монтмориллонитовые Г.
вследствие высокой адсорбционной способности ис-
пользуются для очистки (обесцвечивания) нефтяных
продуктов и пищевых жиров (см. Земли отбеливаю-
щие), а вследствие способности к самопроизвольному
диспергированию в воде и коагуляционному струк-
турообразованию — для приготовления промывочных
р-ров, для бурения скважин (бентонитовые Г., см.
Глинистые растворы), в качестве связующего ком-
понента литейных песков и т. д.
Г. являются главной составной частью многих
почв и обусловливают их свойства. Нек-рые Г. яв-
ляются минеральными пигментами, напр. охры же-
лезистые (желтого цвета), умбры (бурые), волкон-
скоит (хромовая зеленая) и т. д.
Лит.: Неметаллические ископаемые СССР, т. 4, Глины
и каолин-глины отбеливающие, М.—Л., 1941; Земятчен-
с к и й П. А., Глины СССР, М.—Л., 1935; Грим Р. Е.,
Минералогия глин, пер. с англ., М., 1959; Каолины и глины
УССР, М., 1940; Райс Г., Глины — их залегание, свой-
ства и применение, пер. с англ., Л., 1932. В. Л. Петров.
ГЛИНЫ ОТБЕЛИВАЮЩИЕ — см. Земли отбе-
ливающие.
ГЛИОКСАЛЕВАЯ КИСЛОТА (глиоксиловая кис-
лота) СНО—СООН — альдегидокислота, известна
лишь в виде гидрата (НО)2СНСООН; т. пл. 98°,
при нагревании выше этой темп-ры разлагается;
перегоняется с водяным паром без разложения; легко
растворима в воде. Константа диссоциации К =
— 0,474 • 10'3. Г. к. часто содержится в недозрелых
фруктах; получается окислением этилового спирта,
этиленгликоля, гликолевой к-ты. Г. к. может быть
получена электрохимии, восстановлением щавелевой
к-ты (СООН)2-> (НО)2СНСООН или гидролизом ди-
галогенуксусных к-т С12СНСООН —> (НО)2СНСООН.
Г. к. реагирует с фенилгидразином, гидроксиламином,
семикарбазидом, образуя соответствующие производ-
ные Г. к., напр. (НО)2СНСООН-Ь CeH6NHNH2->
—*-CeHsNHN=CHCOOH. При окислении Г. к. полу-
чается щавелевая кислота, при восстановлении —
гликолевая к-та НОСН2СООН или винная кислота
НООССН(ОН)СН(ОН)СООН. При кипячении с КОН
Г. к. превращается в гликолевую и щавелевую кис-
лоты (Канниццаро реакция). Г. к. можно применять
для получения винной кислоты. е. м. Рохлин.
ГЛИОКСАЛЬ СНО—СНО, мол. в. 58,04 — диаль-
дегид, желтые кристаллы; т. пл. 15°; т. кип. 50,4°;
d|° 1,14; 1,3826; хорошо растворим в воде, спирте
и эфире. Легко полимеризуется при стоянии, особенно
в присутствии следов влаги, может быть регенериро-
ван перегонкой полимера с Р2О5. Получают Г. окис-
лением соединений, содержащих 2 атома углерода,
а также озонированием соединений, содержащих со-
пряженные двойные связи; в пром-сти — дегидроге-
низацией этиленгликоля над медным катализатором
омылением глиоксальсульфата, образующегося при
действии олеума на тетрахлорэтан, а также др. спо-
собами. Г. обладает обычными свойствами альдеги-
дов: образует диоксим (глиоксим), бисульфитное со-
единение, фенилозазон и т. п. При взаимодействии со
щелочами претерпевает внутримолекулярное превра-
щение (см. Канниццаро реакция), образуя гликоле-
вую к-ту. С конц. водным р-ром NH3 дает глиоксалин
(имидазол), с ароматич. 1,2-диаминами — производ-
ные пиразина. В синтезе нек-рых кубовых краси-
телей применяют Г. и глиоксальсульфат SO2O2CH—
CHO2SO2.
Лит.: Chemistry of carbon compounds, ed. by E. H. Rodd,
v. 1, Amst., 1952, p. 415. E. M. Рохлин.
ГЛИЦЕРИДЫ — сложные эфиры глицерина; по
числу кислотных остатков в молекуле делятся на
моно-, ди- и триглицериды; по числу различных кис-
лотных остатков — на одно- (все кислотные остатки
одинаковые), дву- и трехкислотные Г. (кислотные
остатки разные). Изомерия Г. связана с расположе-
нием кислотных остатков. Среди Г. неорганич. кислот
большое значение имеет триглицерид азотной к-ты,
неправильно называемый нитроглицерином. Г. выс-
ших и нек-рых низших жирных к-т широко распро-
странены в природе, т. к. являются основной состав-
ной частью липидов, к-рые служат источником полу-
чения индивидуальных Г. Физич. и химич. свойства
последних очень сходны, поэтому выделение их в чи-
стом виде весьма сложно; оно достигается ректифика-
цией, дробной пизкотемп-рной (до —75°) кристалли-
зацией из растворителей (ацетон, метанол, эфир
и т. д.), через соединения включения (комплексы)
с мочевиной, противоточным распределением, различ-
ными видами хроматографии и т. д. Таким путем
удается получить несколько фракций Г., различаю-
щихся размерами кислотных . остатков или числом
кратных связей в них. Эти фракпии вновь подвергают
разделению, получая в конце концов чистые Г.
Насыщенные Г. высших жирных к-т — твердые при
обычной темп-ре, Г. низших к-т и Г., содержащие
яеск. кратных связей, — жидкие. Увеличение числа
кратных связей понижает темп-ру плавления. Боль-
шинство Г. полиморфно, и это затрудняет их иденти-
фикацию. Если молекула Г. не содержит хромофора
в кислотной части, то Г. бесцветен; окраска природ-
ных жиров вызывается наличием в них каротиноидов
и продуктов окисления. Триацетин и трипропионин
немного растворимы в воде. Г. высших жирных к-т
нерастворимы в воде, но растворимы в органич. рас-
творителях; растворимость сильно зависит от темп-ры
в области ниже ~—10°. Триглицериды высших жир-
ных к-т нерастворимы в спиртах и благодаря этому
могут быть отделены от моно- и диглицеридов. Опти-
чески активные Г. в природе не найдены, однако
они могут быть получены разделением рацематов.
Являясь сложными эфирами, Г. проявляют химич.
свойства, характерные для этого класса соединений,
971
ГЛИЦЕРИДЫ — ГЛИЦЕРИН
972
но наличие трех этерифицированных или способных
этерифицироваться гидроксильных групп, из к-рых
две первичные (а и а') одинаково реакционноспо-
собны, а вторичная (|5) значительно пассивнее
[СН2ОНСНОНСН2ОН], имеет ряд особенностей.
ара'
Так, при гидролизе Г., напр. щелочью, сначала омы-
ляется кислотный остаток в [5-положении, и лишь
затем — в а- и а'-положениях. Это свойство (частич-
ный гидролиз) дает возможность получать а-моно- и
а, а'-диглицериды из ди- и триглицеридов. Подобная
же закономерность наблюдается при алкоголизе Г.
Этерификация глицерина идет в обратном порядке —
сначала этерифицируются ОН-группы в а- и а'-поло-
жениях. Кислотные остатки в Г. подвижны, может
происходить их внутри- и межмолекулярная мигра-
ция, особенно в кислой среде; [5-моно- и а, (5-дигли-
цериды легко перегруппировываются соответственно
в а-моно- ц а, а'-диглицериды. Это свойство исполь-
зуется при синтезе Г. Переэтерификация Г. эфирами
кислот применяется в технологии жиров для получе-
ния Г., содержащих нужные кислотные остатки,
однако этот процесс трудно контролировать, особенно
в отношении положения кислотных остатков в мо-
лекуле. Для образования сложноэфирной связи
используются хлорангидриды к-т, этерифицирую-
щие свободную ОН-группу, и значительно реже —
кетены.
Методы синтеза Г. определенного строения из гли-
церина распадаются на две основные группы: 1) с ис-
пользованием защитных группировок и 2) без их
использования. Первый, наиболее старый способ по-
лучения представляет собой ступенчатую этерифика-
цию и основан на различной реакционной способности
ОН-групп в глицерине (см. выше); этерификация про-
текает с высокой избирательностью. Для этерифика-
ции пользуются хлорангидридом карбоновой к-ты;
т. к. процесс идет ступенчато, то, применяя после-
довательно галогенангидриды разных к-т, можно
ввести в молекулу глицерина различные кислотные
остатки:
СН2ОН-СНОН-СН2ОН—С-4cH^OII--CIIOII--CIIOCOR'->-
^ -.I'0?-.1 CH2OR"CHOH-CHOCOR'
И т. д.
Методы с использованием за’щитных группировок
основаны на превращении части ОН-групп глицерина
в нереакпионноспособные в условиях данной реакции;
защита обычно сводится к превращению ОН-груп-
пы в группировку простого эфира, напр.:
сн2онснонсн2он + (С8Н5)з СС1 —
- СН2 ОНСНОНСН2ОС(СвН5)3.
После этерификации защитную группировку раз-
рушают и освобождают т. о. ОН-группы, напр.
CH»OCOR'CHOCOR'CH2OC(CSH5)3 —2
— CIROCOR'CHOCOR' СН2ОН
Эти методы дают вполне определенные продук-
ты. Из защитных групп применяются: изопропи-
лиденовая (Э. Фишер) — защищает две соседние ОН-
группы, бензилиденовая — защищает а- и а'-ОН-груп-
пы, и трифенилметильная (тритильная) — защищает
а-ОН-группы. Снятие изопропилиденовой защиты
происходит в кислотной среде (не дает возможности
получить [5-моноглицериды), а бензилиденовой и
тритильной — путем гидрирования (неприменимо для
получения Г. ненасыщенных к-т). Описано удале-
ние бензилиденовой защиты без миграции кислот-
ного остатка гидролизом борной к-той в присут-
ствии борных эфиров. Синтез Г. с постепенным по-
строением глицериновой части молекулы сложен,
например:
CHjOCOR' CHjOCOR' jjRr CHjOCOR' R"COONa
CO Cl CO CO
I I
CHN2 СЙ2Вг
CHoOCOR' CHSOCOR’ CH2OCOR'
—* co -^4 choh R2£0ci (iHOCOR,„
I i I
CH2OCOR” CHjOCOR" CH20C0r"
Критериями чистоты как синтетич., так и природ-
ных Г. являются обычно числа омыления, иодные и во-
дородные числа (для ненасыщенных Г.) и др., а так-
же хроматография на бумаге и спектроскопия, данные.
Для изучения полиморфизма Г. используются рент-
геноструктурные методы. Положение кислотных остат-
ков в молекуле определяется с помощью окисления,
а также избирательным ферментативным гидролизом.
Лит.: Hilditch Т. Р., The chemical constitution
of natural fats, 3 ed., L., 1956; Progress in the chemistry of fats
and other lipids, v. 1—4, L.—N. Y.—P., 1952—57.
A. E. Васильев.
ГЛИЦЕРИН (пропантриол-1,2,3; 1,2,3-триоксипро-
пан) СН2ОНСНОНСН2ОН, мол. в. 92,09 — трехатом-
нып спирт; сиропообразная бесцветная вязкая жид-
кость сладкого вкуса, без запаха. Г. может оставаться
жидким при очень низких темп-pax, однако дистилли-
рованный Г. высокой чистоты при очень медленном
охлаждении может быть получен в виде кристаллов
орторомбич. формы, т. пл. 17,9°; при переохлаждении
до минус 70—110° можно получить стекловидный Г.
Чистый Г. кипит при 290°; при малейшем загрязнении
перегоняется с разложением; без разложения перего-
няется в вакууме и с перегретым паром. Темп-ра
кипения водных р-ров Г. уменьшается с понижением
концентрации Г. (при содержании 5% воды т. кип.
160—161°); dfg 1,26362, п'^ 1,47399. Г. смешивается
во всех отношениях с водой, этиловым или метиловым
спиртом, анилином, ацетоном, растворяется в смеси
СНС13 и С2Н6ОН(1 : 1), в абс. С2Н6ОН и (CjHb^O
(2:1) и др.; нерастворим в жирах, бензине, СвНв,
CS2 и др.; при смешивании с водой происходит умень-
шение объема (контракция), достигающее наиболь-
шего значения для смеси, содержащей 57% Г.; одно-
временно повышается темп-ра. Г. гигроскопичен, он
поглощает до 40% воды (по весу); растворяет многие
органич. и неорганич. вещества: соли, едкие щелочи,
сахара, ароматич. спирты и др. Глицерино-водные
р-ры замерзают при низких темп-pax, напр. смесь,
содержащая 66,7% Г., замерзает при —46,5%; т. всп.
в открытой чашке 174°; т. воспл. 187°; т. самовозг.
393°. Теплота образования 157,9 ккал/моль (18°,
760 мм)\ теплота испарения 21,1 ккал/моль (55°),
теплота сгорания 396,8 ккал/моль (о = const),
397,1 ккал/моль (р = const); теплоемкость (кал/молъ):
0,5431 (4-2,4°), 0,5795 (26,4°); теплопроводность
(кал/см-сек-град): 0,00068 (до 100°), 0,00077 (160°);
вязкость (пуаз): 1340 (—20°), 42,2 (2,8°), 8,30 (20,3°).
Свойства Г. определяются наличием в нем трех
гидроксильных групп, двух первичных и одной вто-
ричной; первичные ОН-группы более реакционно-
способны; образуют моно-, ди- и трипроизводные.
Г. дает три ряда металлич. производных — глицера-
тов, напр. CH2OHCHOHCH2ONa; эти производные
могут получаться также при действии на Г. окислов
тяжелых металлов, напр. СиО; с галогеноводород-
ными к-тами или галогенными соединениями фосфора
образуются галогенгидрины (хлоргидрины, бромги-
дрины и др.), например СН2ОНСНОНСН2С1 или
СН2С1СНОНСН2С1; последний служит для получения
эпихлоргидрина. При окислении Г. в зависимости от
характера окислителя и условий окисления полу-
чаются различные соединения: глицериновый альдегид
СНаОНСНОНСНО, диоксиацетон СН2ОНСОСН2ОН,
973
ГЛИЦЕРИНОВАЯ КИСЛОТА - ГЛИЦЕРИНОВЫИ АЛЬДЕГИД
974
глицериновая к-та СН2ОНСНОН СООН, тартроновая
к-та (СООН)2СНОН, мезоксалевая к-та (СООН)2СО;
в кислом р-ре КМпО4 или К2Сг2О, окисляют Г. до
СО2 и воды. При действии на Г. водоотнимающих
средств или при нагревании его в присутствии щелочи
или сильных к-т образуются полиглицерины (ди-,
три- и т. д.); при действии органич. к-т, их ангидри-
дов, хлорангидридов получаются сложные эфиры
(глицериды) — моно-, ди- и триглицериды, напр.
3RCOOH С3Н6(ОН)3 С3Н5(ОСОй)3 -ф- ЗН2О при
действии на Г. минеральных к-т образуются сложные
эфиры, напр. с азотной к-той в присутствии серной —
тринитрат Г. CH2ONO2—CHONO2—CH2ONO2; с во-
доотнимающими средствами, например KHSO4, дает
акролеин СН2=СНСНО; при конденсации Г. с аль-
дегидами или кетонами — ацетали или кетали (напр.,
глицеринацетальдегид, глицеринацетон и др.).
Г. впервые был получен в 1779 Шееле при омыле-
нии жиров в присутствии окислов свинца. Основную
массу Г. получают омылением жиров. Большинство
синтетич. методов получения Г. основано на исполь-
зовании пропилена в качестве исходного продукта.
Хлорированием С3Нв при 450—500° получают аллил-
хлорид СН2=СНСН2С1, при присоединении к по-
следнему хлорноватистой к-ты НСЮ образуются
хлоргидрины, напр. СН2ОНСНОНСН2С1, к-рые при
омылении NaOH превращаются в Г. На превраще-
ниях аллилхлорида в Г. через дихлоргидрин или
аллиловый спирт основаны другие методы. Известен
также метод получения Г. окислением пропилена
в акролеин; при пропускании смеси паров акролеина
и изопропилового спирта через смешанный ZnO—MgO-
катализатор образуется аллиловый спирт; последний
при 60—70° в водном р-ре Н2О2 превращается в гли-
церин. Г. можно получить также из продуктов
гидролиза крахмала, древесной муки и т. п. и гид-
рированием образовавшихся моносахаридов или гли-
колевым брожением сахаров (гексоз); процесс может
происходить в среде сульфита или щелочи (pH 7,0
или выше).
Пром-сть выпускает след, виды Г.: сырой 1-го,
2-го и 3-го сортов и дистилл. Г. (динамитный, выс-
шего сорта, 1-го и 2-го сортов). Применяют Г. гл. обр.
в произ-ве взрывчатых веществ (см. Нитроглицерин),
синтетич. смол (см. Смолы алкидные), как мягчитель
в текстильной и кожевенной пром-сти, в бумажной и
пищевой пром-сти, как компонент в парфюмерных,
фармацевтич. и косметич. препаратах.
Лит.: Неволин Ф. В., Химия и технология произ-
водства глицерина, М., 1954; Зиновьев А. А., Химия
жиров, М., 1952; Лоури Дж. ₽., Глицерин и гликоли,
пер. с англ., [Л.], 1933. Л. И. Ловачев.
ГЛИЦЕРИНОВАЯ КИСЛОТА (а-,0-диоксипро-
пионовая кислота) СН2ОНСНОНСООН, мол. в.
106,08 — простейшая одноосновная трехатомная к-та;
обычно густой сироп, в очень чистом состоянии
твердое вещество с т. пл. 134—135°; смешивается
с водой и спиртом, нерастворима в эфире; константа
диссоциации £"=0,288 • 10-3 (25°); существуют пра-
во- и левовращающие оптически активные формы.
Г. к. проявляет химич. свойства оксикислот. Умерен-
ное нагревание Г. к. дает ангидрид типа лактидов:
носн2-снон косо носн2-нс со
\| + I — 1 1
осон носнсн2он ос сн-сн9он
"'О'
при более сильном нагревании с бисульфитом калия
получается пировиноградная к-та:
СН3ОНСНОНСООН —?-^2сН3 = С(ОН)СООН — CHgCOCOOH
При осторожном действии иодистоводородной кисло-
ты из Г. к. получают J3-J-пропионовую к-ту (один
гидроксил восстанавливается, другой замещается):
СП30НСН0НС00Н CH2JCH2COOH; более энер-
гичное восстановление дает пропионовую к-ту. Об-
работка Г. к. 3 молями РС15 приводит к а,р-дихлор-
пропионилхлориду. Оптически недеятельную Г. к.
обычно получают окислением глицерина (напр.,
HNO3); особым окислительным брожением глицерина
(Bacillus aethaceticus) можно получить правовра-
щающий стереоизомер. Оптически недеятельную Г. к.
разделяют на оба оптич. антипода кристаллизацией
солей бруцина. н. В. Гнучев.
ГЛИЦЕРИНОВЫЙ АЛЬДЕГИД (альдотриоза)
СН2ОНСНОНСНО, мол. в. 90,08 — диоксиальдегид,
простейшая альдоза; существует в виде двух оптич.
изомеров, из которых D-Г. а. (I) вращает плос-
кость поляризации вправо [а] д = ф 14° (в воде), а
L-Г. а. (II) — влево;
ено сно
н-с-он но-ci—н
CHoOH <!:н,он
I н
Конфигурационные формулы для D- и L- Г. а. соот-
ветствуют их абс. конфигурациям. D- и L-формы
Г. а. — соединения, на основании сравнения с кон-
фигурацией к-рых производят деление моносахаридов,
а также а-аминокислот на соединения D- и L-ряда.
К D-ряду относят те моносахариды, к-рые полу-
чаются из D- Г. а. путем последовательного примене-
ния циангидринного синтеза, т. е. те, у к-рых конфи-
гурация у последнего асимметрия, атома углерода
такая же, как у D-Г.а. — D-эритроза (III), D-трео-
за (IV) и т. д.
сно сно сно
н^он —» неон нос!:н
сн2он неон + нс!;он
ещон <!;н2он
ш IV
Аналогично к L-ряду а-аминокислот относят те амино-
кислоты, к-рые можно произвести от L-Г. а.: L-гли-
цериновая к-та (V), L-серин (VI), L-аланин (VII).
сно соон соон соон
I । II
НОСН —► НОСН ->H2NCH -> HSNCH
СН2ОН СН2ОН ^Н2ОН сн3
V VI VII
D- ц L-формы Г. а. получены в виде сиропа; D, L-Г. а.
кристаллизуется в виде димера, т. пл. 138,5°.
Г. а. хорошо растворим в воде, плохо в спирте и
эфире. В водных р-рах Г. а. не образует циклич.
форм в противоположность другим моносахаридам и
не дает реакций за счет полуацетального гидроксила
(образование гликозидов). К сахарам Г. а. относят
условно, на основании генетич. близости и сходства
в нек-рых свойствах. Г. а. дает реакции на альдегид-
ную (с фуксинсернистой к-той, орцином, а-нафтолом)
и спиртовые группы; с фенилгидразоном образует фе-
нилозазон; при действии спирта в присутствии НС1 —
диэтилацеталь. При окислении Г. а. получается гли-
цериновая кислота, при восстановлении — глицерин.
При действии щелочи на Г. а. идет конденсация с об-
разованием гексоз (акроза). С флороглюцином Г. а.
конденсируется, образуя нерастворимые в воде сое-
динения.
Г. а. получают окислением глицерина HNOS, Вг2,
Н2О2 и др. окислителями (смесь D, L-Г. а. с образую-
щимся одновременно диоксиацетоном, наз. глнце-
розой), электролитическим расщеплением D,L-
975
ГЛИЦЕРИНТРИНИТРАТ — ГЛУТАМИН
976
эритроновой к-ты, гидролизом ацеталя П,Ь-глицери-
нового альдегида, окислением бензаль-1,3-пропен-
диола (с последующим гидролизом) и конденсацией
формальдегида. D-Г. а. получают из 1,2-,5,6-диацетон-
D-маннита действием тетраацетата свинца или из
1,2,5,6-тетрабензоил-В-маннита. L-Г.а. получают
аналогично из L-маннита. D-Г. а., в виде фосфорного
эфира, является важным промежуточным продуктом
углеводного обмена (см. Гликолиз и Брожение).
Лит.: The carbohydrates, ed. W. Pigman, N. Y., 1957;
M i c h e e 1 F., Chemie der Zucker und Polysaccharide, Lpz.,
1956; Синтезы органических препаратов, пер. с англ. сб. 2,
М., 1949; Степаненко Б. Н., Усп. хим., 1959, 28,
вып. 5, с. 521. Л. И. Линевич.
ГЛИЦЕРИНТРИНИТРАТ — см. Нитроглицерин.
ГЛИЦИН (а-аминоуксусная кислота, гликокол)
HjNCHjCOOH, мол. в. 75,07 — одна из наиболее
распространенных аминокислот, входящих в состав
белков; бесцветные кристаллы; т. пл. 232—236°
(с разл.); d 1,61; Г. хорошо растворим в воде, нерас-
творим в абс. спирте и эфире; оптически неактивен.
Водный р-р Г. имеет почти нейтральную реакцию, pH
ок. 6,8; рК4 2,34; рХ2 9,60; р/ 5,97 (25°); константы
диссоциации (25°) Ккисл —1,5 • 10 '10, Косн =1,7-10”12,
Г. дает соли как со щелочами, так и с кислотами;
соли образуют комплексные соединения с катионами
многих металлов. Внутренняя соль 4-замещеяного
аммониевого основания Г. (CH3)3N—СН2СОО носит
название бетаина. Г. дает общие реакции на амино-
кислоты (с р-рами FeCl3 — интенсивную красную
окраску, с солями меди — темно-синюю); при нагре-
вании с нингидрином раствор становится сине-фиоле-
товым. Для разделения и идентификации Г. в смеси
с другими аминокислотами применяется метод бу-
мажной хроматографии. Для колич. определения
Г. используется цветная реакция с о-фта левым альде-
гидом (см. Циммермана реакция), а также метод фор-
мольного или гликолевого титрования.
Г. выделяют из гидролизатов белков (желатины,
фиброина шелка); его можно синтезировать общими
способами получения аминокислот, напр. из моно-
хлоруксусной к-ты и NH3. В организме Г. синтези-
руется из глиоксиловой к-ты путем переаминирования
и частично из 0-окси-а-аминокислот. Г. применяют
в качестве исходного вещества в ряде синтезов, для
получения буферных р-ров, как стандарт при колори-
метрия. определении аминокислот и для колич.
определения Си и Ag.
Константы устойчивости комплексов с различными
катионами, определенные при 25° и ионной силе
раствора р = 0, равны:
Катионы 1g К1 1g к* Катионы 1g Kt IgKj
Ag+ 3,51 3,38 Mg-+ 3,44
Ваг+ 0,77 — Mn2+ 3,44
С.^+ 1,38 — Ni:+a 6,18 4.95
Cd2+ 4,80 4.03 Pds- 5,47 3,39
СО2+ 5,23 4,02 Pd2+B 9,12 8,43
Си*+ 8,62 6,97 Sr-’+ 0,91 —
Ре2+а 4.3 — Zn=+ 5,52 4,44
Hg2+° 10,3 8,9
а При 20° и ц. = 0,01. С При 20° в среде 0,5 М
р-ра KNO3. В При 25° и ц = 0,01.
Лит. см. при ст. Аминокислоты.
Л. П. Алексеенко, И. IT. Ефимов.
ГЛИЦИН — n-оксифениламиноуксусная к-та, окси-
фенилглицин, см. Проявляющие вещества.
ГЛОБИН — см. Гемоглобины.
ГЛОБУЛИНЫ — белки, хорошо растворимые в раз-
веденных кислотах, щелочах, а также в р-рах ней-
тральных солей. Первоначально термином Г. обозна-
чали белки, к-рые, в отличие от альбуминов, осаждают-
ся из растворов при 50%-ном насыщении сернокислым
аммонием. Г. подразделяют на эв гл о бу л ины
и псевдоглобулины. Одни исследователи
называют эвглобулинами Г., нерастворимые в ди-
стилл. воде, и псевдоглобулинами Г., растворимые
в ней; другие называют эвглобулинами фракцию Г.,
осаждаемую при 33%-ном насыщении сернокислым
аммонием, и псевдоглобулинами фракцию Г., осаж-
даемую при 34—50%-ном насыщении (NH4)2SO4.
Определение Г. по их растворимости неточно; напр.,
фракция, осаждаемая при 33%-ном насыщении
(NH4)2SO4, содержит как Г., растворимые в дистилл.
воде, так и нерастворимые в ней, а при 50%-ном на-
сыщении сернокислым аммонием в осадок выпадают
не только глобулины, но и нек-рое количество альбу-
минов; в растворе, помимо альбуминов, остается за-
метное количество Г. В последние годы Г. стали назы-
вать белки, мигрирующие при электрофорезе в щелоч-
ных буферах (напр., при pH 8,6) медленнее, чем аль-
бумины. Наиболее изучены Г. плазмы крови, состав-
ляющие у здорового человека ок. 40% всех белков
плазмы. По электрофоретич. подвижности Г. разде-
ляются на а4-, а2-, 0- и у-Г. При фракционировании
плазмы этанолом на холоду из нее был выделен ряд
Г., многие из к-рых комплексированы с липидами или
углеводами. Нек-рые Г. плазмы имеют большое физио-
логия. значение., напр. фибриноген участвует
в свертывании крови, а4-, а2- и 0-липопротеиды уча-
ствуют в переносе нерастворимых в воде биологически
важных липидов, а у-Г. играют защитную роль.
Содержание Г. при различных заболеваниях обычно
возрастает, причем в крови могут появляться Г.,
отсутствующие в крови здорового организма.
Мало изучены Г. животных органов и тканей, со-
ставляющие по данным электрофоретич. анализа
ок. 90% всех извлекаемых из них белков. С одним
из Г. мышцы — миозином — связана их сократитель-
ная функция. Значительная часть белков протоплазмы
растений также относится к Г. Растительные Г.
подразделяются на растворимые в нейтральных солях,
растворимые в разб. этиловом спирте и растворимые
в разб. к-тах и щелочах.
Лит. см. при ст. Белки. А. Е. Гурвиц.
ГЛУТАМИН (глютамин) HaNOCCH.,CH,CH(NH:.)COOH,
мол. в. 146,35 — моноампд глутаминовой кислоты.
В Г. амидирован удаленный от а-аминогруппы кар-
боксил; изомер Г., в к-ром амидирована а-карбо-
ксильная группа, называется изоглутамином.
Г. в виде L-формы (его обычно называют прос-
то Г.) — бесцветные кристаллы; т. пл. 184°; плохо
растворим в воде, спирте, нерастворим в эфире
и большинстве органич. растворителей; [а]^=+6,1°
(3,6%-ный р-р в воде), рКа] 2,17, рКа2 9,13 (NH+),
р! 5,65. Г. дает общие реакции на аминокислоты,
осаждается нитратом ртути. Количественное определе-
ние Г. производят путем гидролиза амидной группы
H2SO4 или при помощи специального фермента глю-
таминазы и определения количества выделившегося
аммиака. L-Г. широко распространен в растениях,
животных и микроорганизмах, являясь аминокислот-
ным компонентом белков и полипептидов; встречается
в свободном состоянии в значительных количествах
в свекле (до 5,6% от сухого веса), проростках тыквы,
гороха и др. растениях и в животных тканях. Полу-
чают Г. обычно из свеклы и др. растений. Синтез Г.
можно осуществить из L-глутаминовой к-ты. D-Г.
получен амидированием D-глутаминовой к-ты' под
влиянием фермента из зеленого гороха, [а]д = —8,9°
(2%-ный р-р в воде), широкого распространения не
имеет. Изоглутамин получен только синтетич. путем.
977
ГЛУТАМИНОВАЯ КИСЛОТА — ГЛЮКОЗА
978
Г. играет большую роль в обмене веществ растений,
животных и микроорганизмов; он является промежу-
точным продуктом ассимиляции азота, принимает
участие в реакциях переаминирования и т. п.
Лит.: Браунштейн А. Е., Некоторые черты хими-
ческой интеграции процессов азотистого обмена. Доклад на
заключит, пленарном заседании IV Международ, биохимич.
конгресса, Вена, 6 сент. 1958 г., М., 1958; Biochemical pre-
parations, v. 1, ed. byE. Carter, N. Y., 1950, p. 44; v. 5 ed. by
D. Chemin, N. Y., 1957, p. 79; см. также при ст. Амино-
кислоты. Л. И. Линевич.
ГЛУТАМИНОВАЯ КИСЛОТА (глютаминовая кис-
лота, а-аминоглутаровая кислота), мол. в. 147,13,
HOOCCH2CH2CH(NH2)COOH—аминокислота из груп-
пы моноаминодикарбоновых аминокислот. L-Фор-
ма Г. к. (ее обычно называют просто Г. к.) — бес-
цветные кристаллы; т. пл. 247—249° (с разл.);
плохо растворима в воде и спирте, нерастворима
в эфире. Кристаллы Г. к. дают пьезоэлектрический
эффект. Р-ры Г. к. имеют кислую реакцию; рКа 2,19
(а-СООН), рКа2 4,25 (у-СООН), рАГаз 9,67, р/3,08,
|а]^ = -р31,2° (1%-ный р-р в 6 н. НС1). Г. к. дает об-
щие реакции на аминокислоты. При нагревании водных
р-ров Г. к. образуется пирролидон-2-карбоновая к-та,
с Си и Zn Г. к. дает нерастворимые соли. Количе-
ственно Г. к. определяют, напр., разложением Г. к.
до пирролидон-2-карбоновой к-ты и реакцией с нин-
гидрином, а также ферментативными и микробиоло-
гия. методами. При определении Г. к. в биологич.
объектах пользуются хроматография, способами отде-
ления ее от др. аминокислот. Г. к. входит в состав
многих белков и полипептидов и содержится в тканях
животных и растений в свободном виде. Роль ее
в организме очень велика, в частности она является
агентом, связывающим аммиак, образующийся в про-
цессе обмена веществ. L-Г. к. получают из гидролиза-
тов белков и разделением синтетич. рацемической
Г. к. Применяют L-Г. к. и ее соли (глютаминаты Са и
Mg) при лечении нек-рых психических (эпилепсия,
шизофрения) и детских заболеваний, связанных
с поражением нервной системы (полиомиелит, ослож-
нение после туберкулезного менингита и др.). На-
триевая соль Г. к., напоминающая вкус мяса, приме-
няется как приправа к пище.
D-Г. к. обнаружена в составе нек-рых полипепти-
дов микроорганизмов и получена разложением раце-
мия. синтетич. Г. к.; т. пл. 213° (с разл.); [a]jj = —12,9°
(в воде). Синтетически Г. к. получают из метилового
эфира акриловой к-ты, этилового эфира фталоилма-
лоновой к-ты и др. способами.
Лит, см. при ст. Аминокислоты. Н. Е. Плотникова. .
ГЛУТАРОВАЯ КИСЛОТА (пропандикарбоно-
вая-1,3 кислота) НООС—(СН2)3—СООН, мол. в. 84,12—
четвертый член гомология, ряда двухосновных пре-
дельных кислот; бесцветные моноклинные пластинки;
т. пл. 97,5°; т. кип. 303°/760 мм, 200°/20 мм\ кон-
станта диссоциации при 25°: Кг = 4,60 • 10~б, =
— 5,34 • 10“в. Г. к. легко растворима в воде (63,9 г
в 100 г Н2О), абс. спирте, диэтиловом эфире; содер-
жится в соке свеклы и водах от промывки овечьей
шерсти. Получают Г. к. окислением циклопентанона,
омылением триметиленцианида: NC—(СН2)3—CN 4"
-I- 4Н2О —> НООС—(СН2)3—СООН + 2NH3, разложе-
нием эфира тетракарбоновой к-ты:
С..Н,ООС. .COOC-Hj
)сн- он.,—сн< ' + 4Н3
CsHsOOC/ ' ^СООС3Н5
— НООС-(СНа)з-СООН + 4С2Н5ОН-I- 2COS
и др. способами. При нагревании Г. к. до 300—304®
образуется масляная к-та. Г. к. почти не окисляется
хромовой смесью. Кальциевая соль Г. к. при пиро-
лизе образует смесь продуктов, среди к-рых найдены
ацетон и фенол. м. И. Розенгарт.
ГЛЮКАГОН (гликогенолитический и гиперглике-
мический фактор) C1E3H22EO48N43S, мол. в. 3482 —
гормон поджелудочной железы, стимулирующий пре-
вращение гликогена в глюкозу (глюкогенная актив-
ность). Г. — кристаллич. продукт, плохо растворим
в воде и слабых водных р-рах кислот и щелочей
(pH 3,0—9,5); при pH < 3 образует фибриллы; изо-
электрич. точка Г. при pH 7,5—8,5; максимум погло-
щения при 278 ммк. Г. инактивируется при облу-
чении ультрафиолетовым светом; легко гидролизуется
протеолитич. ферментами; секреция Г. стимулируется
гормоном роста гипофиза. Г. — пептид, состоящий из
29'остатков аминокислот (см. Аминокислоты), распо-
ложенных в одиночной пептидной цепи:
NH3
Гис.-Сер.-Глют.-Глиц.-Тре.-Фал.-Тре.-Сер.-Асп.-
-Тир.-Сер.-Лиз.-Тир.-Лейц.-Асп.-Сер.-Арг. - Арг.-Ал.-
NH2 nh2
-Глют.-Асп.-Фал .-Вал.-Глют.-Трип.-Лейц.-Мет .-
-Асп.-Тре.
NH3
Биологич. методы определения Г. основаны на его
способности вызывать увеличение концентрации глю-
козы в крови после внутривенного введения, а также
на стимуляции расцада гликогена в срезах печени.
В качестве терапевтич. средства Г. не применяется
вследствие кратковременности действия.
Лит.: F о а Р. Р., Gal ansi no G., Pozza G.,
в кн.: Recent progress in hormone research, v. 13 N.Y., 1957,
p. 473; В e hr e ns О. K. and Bromer w. W„ в кн.;
Annual review ot biochemistry, v. 27, Palo Alto (California),
1958, p. 66; и x же» в кн.: Vitamins and Hormones, V. 16,
N. Y., 1958, p. 263; см. также при ст. Гормоны.
Ю. А. Панков.
ГЛЮКОЗА СвН12Ов, мол. в. 180,16 — моносахарид,
одна из восьми изомерных альдогексоз. Г. в виде
D-формы (декстроза, виноградный сахар) является
самым распространенным углеводом. D-Г. (обычно ее
называют просто Г.) встречается в свободном виде
и в виде олигосахаридов (тростниковый сахар, молоч-
ный сахар), полисахаридов (крахмал, гликоген, цел-
люлоза, декстран), гликозидов и др. производных.
В свободном виде D-Г. содержится в плодах, цветах
и др. органах растений, а также в животных тканях
(в крови, мозгу и др.). D-Г. является важнейшим
источником энергии в организме животных и микро-
организмов (см. Гликолиз). Как и др. моносахариды,
D-Г. образует неск. форм. Кристаллич. D-Г. получена
в двух формах: a-D-Г. (I) и P-D-Г. (II). a-D-Г., т. пл.
146°, [a]D = 4~ 112,2° (в воде), кристаллизуется из воды
в виде моногидрата с т. пл. 83°. P-D-Г. получают кри-
сталлизацией D-Г. из пиридина и нек-рых др. раство-
рителей,!, пл. 148—150°, [aD]=H-18,9° (в воде). В вод-
a-D-глюкопираноза
fi-D-глюнопираноэа
<рно
неон
носн
I
неон
Н(^ОН
СН2ОН
III
альдегида- D-глюкоза
ном р-ре устанавливается равновесие между несколь-
кими взаимопревращающимися формами D-Г.: а- и
p-пиранозными, а- и p-фуранозными, открытой аль-
дегидной (III) и гидратной формой. В равновесной
системе в воде [а]О=-ф52,7°. D-Г. дает общие реакции
на альдозы, она является восстанавливающим саха-
ром, образует ряд производных за счет альдегидной
группы (фенилгидразон, n-бромфенилгидразон и др.).
Озазон Г. идентичен озазону маннозы, к-рая является
979
ГЛЮКОЗИДЫ - ГНИЕНИЕ
980
впнмером Г., и озазону фруктозы. При восстановле-
нии Г. образуется шестиатомный спирт сорбит,-, при
окислении альдегидной группы Г. — одноосновная
D-глюконовая к-та, при дальнейшем окислении —
двухосновная D-сахарная к-та. При окислении только
вторичной спиртовой группы Г. (при условии защиты
альдегидной группы) образуется D-глюкуроновая кис-
лота. Образование D-глюконовой к-ты из D-Г. мо-
жет происходить при действии ферментов оксидаз
или дегидрогеназ Г. При пиролизе D-Г. образуются
гликоааны: а-глюкозан и левоглюкозан (р-глюкозан).
Для колич. определения Г. применяются колориме-
трия., йодометрия, и др. методы.
L-Г. полуяена синтетияески восстановлением лак-
тона L-глюконовой к-ты. a-L-Г. — кристаллы, т. пл.
142—143°, [a]D =—95,5° (в воде) и—51,4° (равновес-
ная система в воде). Химия, свойства L-Г. такие же,
как у D-Г.
Г. может быть полуяена гидролизом природных
веществ, в состав к-рых она входит. В пром-сти ее
полуяают гидролизом картофельного и кукурузного
крахмала кислотами. Полные синтезы Г., осуществлен-
ные, исходя из дибромакролеина, а также из глице-
ринового альдегида и диоксиацетона, имеют лишь
теоретия. интерес. Г. применяют гл. обр. как источник
легко усвояемого ценного питательного материала
при явлениях сердечной слабости, шоке и т. д.; она
входит в состав кровозаменяющих и противошоковых
жидкостей. Г. применяется также в кондитерской и
текстильной пром-сти (как восстановитель), в качестве
исходного продукта при произ-ве аскорбиновой и
глюконовых к-т, для синтеза ряда производных саха-
ров и т. д. Г., содержащаяся в виноградном соке,
соке различных фруктов и гидролизатах крахмала,
при брожении превращается в спирт и др. ценные
продукты. Произ-во спирта из древесины также осно-
вано на сбраживании Г., содержащейся в гидролиза-
тах древесины.
Лит. см. при ст. Гексозы. Л. И. Линевич.
ГЛЮКОЗИДЫ — гликозиды глюкозы; ранее этот
термин применялся для названия гликоаидов всех
сахаров.
ГЛЮКУРОНОВАЯ КИСЛОТА — см. Гексуроно-
вые кислоты.
ГЛЮТАТИОН (глутатион, у-глютаминилцистеинил-
глицин) HOOCCH(CH2)2CONHCHCONHCH2COOH,
NHa CHaSH
мол. в. 307,32 — трипептид, состоящий из трех
аминокислот: глютаминовой, цистеина и глицина.
Глютаминовая к-та, входящая в состав Г., образует
пептидную связь с цистеином за счет у-карбоксильной
группы (обычно в белках и пептидах пептидная связь
образуется за счет а-карбоксильной или а-аминной
групп). Г. — белые кристаллы, т. пл. 190—192°
(с разл.); [a]D=—9,4° (вводе); растворим в холодной
воде (в горячей — разлагается), нерастворим в абс.
спйрте, эфире и др. органич. растворителях. При
взаимодействии с кислотами Г. распадается на L-ами-
нокислоты, при кипячении с водой дает пирролидон-
карбоновую к-ту и цистеинилглицин; с Сн2О Г. об-
разует комплекс, нерастворимый в 0,5 н. H-SO4.
Г. очень устойчив к ферментативному гидролизу и
не расщепляется обычными протеиназами и пептида-
зами. При взаимодействии с фосфорновольфрамовой
к-той Г. окрашивается в синий цвет, с нитропрусси-
дом Na в присутствии щелочей — в красный, а с .ч-ди-
нитробензолом в присутствии NaOH — в зелено-
желтый, переходящий в темно-коричневый. Эти реак-
ции используются для колориметрии, определений
Г. Количественно Г. определяют также Йодометри-
чески. Наиболее точный метод определения Г. осно-
ван на его способности активировать реакцию пре-
вращения метилглиоксаля в молочную к-ту под влия-
нием фермента гипоксалазы.
Г. входит в состав активных центров нек-рых фер-
ментов (фосфоглицеральдегидраза), широко распро-
странен в животных и растительных тканях и микро-
организмах (особенно в железистых органах и тканях
растущих организмов). Благодаря наличию SH-
группы и возможности ее превращения в дисульфид-
ную (2RSH § RS—SR, пде R — остаток Г. без
SH-группы) Г. играет большую роль в окислительно-
восстановительных процессах организма, являясь
активатором некоторых тканевых протеиназ и дру-
гих ферментов, а также коферментом глиоксалазы.
Г. получают из дрожжей и синтетически — карбо-
бензоксиметодом получения пептидов. Применяют
Г. при биохимич. исследованиях, а также при
консервировании крови для удлинения срока ее
хранения.
Лит.: Браунттейн А. Е., Биохимия аминокислот-
ного обмена, М 1949; Белозерский А. Н. и Прос-
куряков Н. И., Практическое руководство по биохимии
растений, М., 1951; Glutathione. Ed. 8. Р. Colowick [а. о.],
N. Y., 1954. О. В. Троицкая.
ГЛЮТЕЛИНЫ — простые белки растительного
происхождения, содержащиеся гл. обр. в семенах
злаков. Характерным свойством Г. является их не-
растворимость в нейтральных солевых р-рах и этило-
вом спирте. Растворителем для Г. служат разб. ще-
лочи (0,2%). Г. слабо изучены и не получены в виде
индивидуальных белков вследствие сложности их
выделения в чистом виде. Кроме того, в процессе об-
работки щелочных экстрактов Г. претерпевают дена-
турационные изменения. Г. нек-рых злаков называют
глютенинами (от франц, gluten — клейко-
вина). Наиболее изучен глютенин пшеницы (изо-
электрич. точка при pH 6,0—8,0), к-рый имеет след,
аминокислотный состав (в %): 0,89 глицина; 4,65 ала-
нина; 5,95 лейцина; 4,23 пролина; 1,97 фенилаланина;
0,91 аспарагиновой кислоты; 23,42 глутаминовой
кислоты; 4,25 тирозина; 1,92 лизина; 1,76 гистиди-
на; 4,72 аргинина; 0,02 цистина; 2,36 триптофана;
0,24 валина; 0,74 серина; 1,8 оксиглутаминовой
кислоты.
Лит.: Козьмина Н. П. и Кретов и ч В. Л.,
Биохимия зерна и продуктов его переработки, 4 изд., М., 1951.
В. О. Шпикитер.
ГНИЕНИЕ — процесс разложения органич. азот-
содержащих веществ, гл. обр. белков, под влиянием
микроорганизмов. Г. — процесс, имеющий широкое
распространение и большое общебиологич. значение;
оно играет важную роль в круговороте веществ в при-
роде. Благодаря Г. из сложных органич. веществ
погибших растений и животных, из различного рода
отбросов органич. происхождения возникают простые
минеральные вещества (NH3, СО2, H2S, Н3РО4 и др.),
к-рые используются живыми организмами для син-
теза органич. веществ. При Г. вещества подвергаются
многочисленным глубоким и сложным изменениям,
процессы Г. идут различными путями и многостадийно.
В природных условиях Г. происходит под влиянием
различных гнилостных микроорганизмов (аэробных
и анаэробных), часто со сменой одних видов микро-
организмов другими. Для изучения химизма Г.
обычно выделяют чистую культуру микроорганизма,
выделяют и изучают ферменты, катализирующие от-
дельные реакции. Процессы Г. белков идут в след,
основных направлениях: гидролиз до пептидов и
аминокислот; дезаминирование аминокислот; декар-
боксилирование аминокислот (окислительное или
восстановительное); восстановление водородом, вы-
деляющимся при Г.; окисление. При Г. образуется
большое число различных веществ, напр. при дез-
аминировании аминокислот — летучие жирные к-ты.
981
ГОЛЬ’.ГПП - ГОМОГЕНТИЗИНОВАЯ КИСЛОТА
982
капроновая и изокапроновая к-ты, фенилпировино-
градная, n-оксифенилпировиноградная и др.; в ре-
зультате декарбоксилирования образуются различные
амины, напр. этилендиамин, кадаверин, путресцин,
скатол, индол, метиламины, гистамин и т. д. Органич.
основания, образующиеся при Г., наз. птомаинами.
Из серусодержащих к-т образуются метилмеркап-
таны, H2S и др. сернистые соединения. Нек-рые
продукты гниения очень ядовиты, другие обладают
неприятным «гнилостным» запахом. Строение многих
сложнопостроенных токсинов, образующихся при Г.,
не установлено.
В практике большое значение имеет борьба с Г.
при хранении пищевых продуктов. При консервиро-
вании продуктов либо уничтожается гнилостная
микрофлора путем стерилизации, применения анти-
септических в-в и использования микробов антаго-
нистов (брожение), либо создаются условия, неблаго-
приятные для процесса Г. (высушивание, заморажи-
вание и др.). Разработаны также методы консервиро-
вания древесины (пропитывание специальными веще-
ствами, покрытие лаками и т. д.). В медицине борьба
с гнилостными микроорганизмами на поверхности ран
производится различными методами асептики и ан-
тисептики. При нек-рых технологии, процессах микро-
организмы, разлагающие белки, играют положитель-
ную роль, напр. в кожевенном произ-ве при уда-
лении волосяного покрова кожи и т. д. Большую
роль играют гнилостные микроорганизмы в процессе
образования перегноя.
Лит.: Браунштейн А. Е., Биохимия аминокислот-
ного обмена, М., 1949; Березова Е. Ф., Микробы и жизнь
растений, М., 1960; П о ш о н Ж., Б а р ж а к Г. де,
Почвенная микробиология, пер. с франц., М., 1960.
Л. И. Линевич.
ГОЛЬМИЙ (Holmium) Но — химич. элемент с п. н.
67, относится к лантанидам.
ГОМБЕРГ А—БАХМАННА— ГЕЯ РЕАКЦИЯ-спо-
соб синтеза несимметричных диарилов путем взаи-
модействия диазосоединения или нитрозоацетиламина
с ароматич. или гетероциклич. соединением: ArN2X 4
4 Ar'H—>Ar—Ar' 4 N2 4 НХ, где X — OH, ONa,
OCOCH3, N(Alk)2. В качестве диазокомпоненты при-
меняют диазогидраты, диазотаты, диазоацетаты, нит-
розоацетиламины (таутомерные диазоацетатам) и диа-
зоаминосоединения. Реакцию выполняют обычно в
среде NaOH или CH3COONa. Применяя описывае-
мую реакцию, получают несимметрич. производные
дифенила и терфенилов, фенилнафталины, феннлант-
рахиноны, фенилтиофены, фенилпиридины и фенил-
ферроцены.
Реакцию с диазосоединениями выполняют, приба-
вляя по каплям р-р NaOH к охлажденной смеси р-ра
соли диазония и жидкого ароматич. соединения, пока
не образуется небольшой избыток щелочи. По мере
образования диазогидрат растворяется в ароматич.
соединении, в среде к-рого и протекает распад диазо-
соединения с образованием диарила. Успешное про-
ведение реакции зависит от экстракции диазогидрата
из водного слоя ароматич. соединением. В среде пири-
дина реакция протекает в отсутствии щелочи при
20—70°; выходы продуктов реакции составляют 20—
80%. Арилдиазоацетаты и ацетилнитрозамины, будучи
растворенными в органич. растворителях (ароматич.
соединениях), также легко (20—40°) вступают в ре-
акцию. Распад 1-арил-3,3-диалкилтриазенов идет в
присутствии ледяной СН3СООН или сухого НС1 при
60—140°. Реакция протекает с участием свободных
арильных радикалов, образующихся при распаде
диазосоединения. В первой стадии реакции органич.
растворитель не y4acTByeT:C6H5N = NOCOCH3—>СвН‘54
4 N2 4 CH3COO-. Образование диарила является
вторичным процессом — результатом взаимодействия
свободных арильных радикалов с ароматич. соеди-
нением (растворителем):
СцН£4 CuH5R - СцН5-СцН4В. + Н‘
Фенильные радикалы, образующиеся при распаде ди-
азоацетатов и диазоаминосоединений, обладают высо-
кой активностью и реагируют с алифатич. соединения-
ми: СвН: -4 HAlk—>С2Н2 4- Aik или С8Н: 4 CGL—►
->С6Н5С1 4 ССГ,.
Реакция разработана М. Гомбергом и В. Бахманном
(1924) и усовершенствована И. Элком, Г. Хевордом
и Д. Геем в 1940.
Лит.: Бахмана В. Е., Гофманн Р. А., в кн.:
Органические реакции, Сб. 2, пер. с англ., М., 1950. с. 244;
Уотерс У., Химия свободных радикалов, пер. с англ., М\,
1948; Реутов О. А., Теоретические проблемы органи-
ческой химии, М., 1956; Dernier О. С. and Edmison
М. Т„ Chem. Revs., 1957, 57, № 1, р. 77. В. А. Пучков.
ГОМО- — приставка, применяемая для обозначения
веществ, отличающихся от сравнительно сложно
построенных соединений включением дополнитель-
ной метиленовой группы СН2 — или заменой водо-
рода в метильной группе на СН3-группу. Гораздо
реже гомосоединениями называют также ближайшие
асн2соон
СООН
I
гомологи основного соединения. Примерами таких
веществ могут служить гомофталевая к-та (I), гомо-
ванилин (II), гомопирокатехин (III), гомоантипирин
(С2Н5-группа вместо СН3-группы в антипирине в
положении 5) и др. Приставка гомо- подчеркивает,
что соответствующие соединения отличаются по со-
ставу на СН2-группу от основных сравниваемых ве-
ществ (см. Гомологические ряды), я. Ф. Комиссаров.
ГОМОГЕННОЕ РАВНОВЕСИЕ — см. Равновесие
химическое.
ГОМОГЕННЫЕ СИСТЕМЫ — физико-химич. си-
стемы, состоящие из одной фазы. Внутри них нет
поверхностей раздела, по к-рым соприкасаются части
системы, отличающиеся друг от друга по составу и
по свойствам. Составные части Г. с. нельзя отделить
друг от друга механич. путем. В Г. с., состоящих из
двух и более химич. компонентов, каждый компонент
распределен в массе другого в виде молекул, атомов,
ионов. Примеры Г. с.: лед, жидкие или твердые р-ры,
смесь газов, водяной пар, жидкая вода и др. Иногда
различают физически однородные и физически неод-
нородные Г. с. У физически однородных Г. с. состав
и (или) свойства в различных местах совершенно оди-
наковы, у физически неоднородных — различны, но
в них между такими местами имеются непрерывные
переходы, и, следовательно, отсутствуют поверхности
раздела, на к-рых происходят резкие, с разрывом
непрерывности, изменения состава и (или) свойства.
Примером физически однородной системы может слу-
жить газ, не подверженный действию тяжести; напро-
тив, газ, подверженный действию тяжести, предста-
вляет физически неоднородную систему, т. к. в нем
плотность ннжних слоев заметно больше, чем у верх-
них, но переход носит непрерывный характер. Разли-
чают также системы химически однородные, т. е.
состоящие из одного химически индивидуального ве-
щества (напр., вода), и химически неоднородные, т. е.
состоящие из двух и более химически различных ве-
ществ (напр., водный р-р).
Лит. см. при ст. Гетерогенные системы.
ГОМОГЕНТИЗИНОВАЯ КИСЛОТА (2,5-диокси-
фенилуксусная кислота) С8Н8О4, мол. в. 168,15 —
промежуточный продукт обмена аминокислот трип-
983
ГОМОЛИТИЧЕСКИЕ РЕАКЦИИ — ГОМОЛОГИЧЕСКИЕ РЯДЫ
984
тофана и тирозина', кристаллы, т. пл. 152—154°; хо-
рошо растворима в воде, спирте и эфире, нераствори-
ма в хлороформе и бензоле; растворима в водных р-рах
щелочей и углекислых солеи ще-
। н лочных металлов; образует нераст-
гн слон воримую свинцовую соль. Макси-
Е j Спгьиип МуМ П0ГЛ01цения г. к. При 290 ммк
(в воде). Г. к. дает реакции фе-
I нолов (синее окрашивание с FeCl3
'-'Н и др.), обладает восстанавливающи-
ми свойствами: восстанавливает р-р Фелинга, амми-
ачный р-р AgNO3; в щелочной среде Г. к. окисляется
кислородом воздуха с образованием темноокрашен-
ных пигментов неустановленного строения. Для
колич. определения Г. к. используют ее восстанавли-
вающие свойства (иодометрич. титрование), реакцию
с фосфорномолибденовой к-той и др. Г. к. в виде свин-
цовой соли выделяют из мочи больных алкаптонурией
(при нарушениях аминокислотного обмена); синте-
тически получают из гидрохинона и монохлоруксус-
ной к-ты.
Лит.: Браунштейн А. Е., Биохимия аминокислот-
ного обмена, М., 1949; Biochemical preparation. Ed. W. W. Wes-
terteld, v. 4 N. Y.—L., 1955. Л. И. Линевич.
ГОМОЛЙТИЧЕСКИЕ РЕАКЦИИ — реакции, про-
текающие путем разрыва связи с разделением элект-
ронной пары, осуществляющей ковалентную связь.
Общая схема таких реакций может быть представлена
в следующем виде:
Z-+Rr:;X—*-R:Z+-X
или Yr<JZ+Rr:JX—«-R.'Z + Y-'X
При таком разрыве связи у каждой из частиц, состав-
ляющих молекулу, образуется по одному неспарен-
ному электрону. Это может приводить к появлению
свободных радикалов (см. Радикалы свободные) на
промежуточных стадиях реакции. Однако не исклю-
чена возможность, что нек-рые Г. р. осуществляются
через активированный комплекс без образования
кинетически независимых частиц (свободных ради-
калов). Гомолитич. распад молекулы в газовой фазе
происходит легче, чем гетеролитический (см. Гетеро-
литические реакции). Поэтому газовые реакции осу-
ществляются преим. по гомолитич. пути. К этому
типу относятся такие важные процессы, как фото-
галогенирование предельных углеводородов (металеп-
сия), сульфохлорирование, нитрование предельных
углеводородов, нек-рые реакции свободных атомов
и радикалов с парафинами, олефинами и т. д.
Многим реакциям в растворе приписывается также
гомолитич. характер, часто с допущением промежу-
точного образования свободных радикалов. В зависи-
мости от строения свободные радикалы отличаются
друг от друга устойчивостью и реакционной способно-
стью. Для гомолитич. разрыва С—С-связи молекулы
этана на два метильных радикала
СН3.-2-" СН3—*'2СН3«
необходимо затратить 82,6 ккал]молъ. Однако, если
атомы углерода, связь между к-рыми должна разор-
ваться, связаны с такими группами, как, напр., аро-
матич. кольца, то энергия такого разрыва может
оказаться значительно меньше (см. Сопряжение). Так,
для разрыва С—С-связи в гексафенилэтане требуется
лишь ок. И—12 ккал]молъ. Это объясняет легкость
(С3Нд)3СС(С6Н5)3 -*-2(СбНд)3С'*
его диссоциации в р-рах на трифенилметильные ра-
дикалы и сравнительную устойчивость последних. В
отличие от таких радикалов, простые алифатич. ради-
калы отличаются чрезвычайно высокой реакционной
способностью, обусловленной наличием у них локали-
зованного неспаренного электрона у атома углерода.
Свободные радикалы легко вступают во взаимодейст-
вие не только друг с другом (рекомбинация и диспро-
порционирование), но и реагируют с насыщенными
молекулами, вызывая их гомолитич. распад с обра-
зованием нового радикала, к-рый в свою очередь
реагирует с новой молекулой, и т. д. Так протекают
многие органич. реакции при участии свободных ра-
дикалов (см. Цепные реакции). Принято считать, что
в растворах такие реакции, как термич. и фотохимич.
разложение металлоорганич. соединений, взаимодей-
ствие галогеналкилов с металлами, термич. разложе-
ние ароматич. перекисей и перекисей диацилов, раз-
ложение азо- и диазосоединений, многие реакции
полимеризации, реакции, идущие под воздействием
излучения, и др., протекают при учаетии свободных
радикалов по гомолитич. типу. В качестве примера
можно указать на образование свободных радикалов
при разложении перекиси ацетила:
CH3f:’"C-O:-J-;O-C-CH3-*CH3- + C02 + *0-C-CH3
0 0 о
Для реакций, интерпретируемых как радикальные,
в каждом отдельном случае требуется определенное
доказательство независимого существования свобод-
ных радикалов в течение определенного промежутка
времени.
Лит.: Реутов О. А., Теоретические проблемы орга-
нической химии, М., 1956; Уотерс У., Химия свободных
радикалов, пер. с англ., М., 1948; Steacie Е. W. R.,
Atomic and free radical reactions, v. 1—2, 2 ed., N. Y., 1954;
X ю к к e л ь В., Теоретические основы органической химии,
пер. с нем., т. 1, М., 1955; Семенов Н. Н., О некоторых
проблемах химической кинетики и реакционной способности...,
2 изд., М., 1958; Ингольд К. К., Механизм реакций
и строение органических соединений, пер. с англ., М., 1959.
Ф. С. Якушин.
ГОМОЛОГИЧЕСКИЕ РЯДЫ — группы родствен-
ных органич. соединений с одинаковыми химич. функ-
циями и однотипной структурой, отличающиеся между
собой на одну или больше метиленовых групп —СН2—
в составе углеводородного радикала молекулы. Напр.,
метан СН4, этан С2Н6, пропан С3Н8, пентан СЕН12
и т. д. составляют Г. р. алканов общей формулы
С„Н2„+2; этен С2Н4, пропен С3Нвидр. — Г. р. алкенов,
ненасыщенных углеводородов с одной двойной связью
в молекуле, СпН2п; Г. р. одноатомных насыщенных
спиртов, C^Ho^^jOII; насыщенных одноосновных
карбоновых кислот, CnH2n_|_iCOOH и т. д.
К понятию о Г. р. близко примыкают понятия об
изологич. и генетич. рядах. Изологические
ряды — группы органич. соединений с одинаковым
числом атомов углерода в молекуле и с одинаковыми
функциональными группами, но с возрастающей не-
насыщенностью, напр. изологич. ряд этана: этав
СН3—СН3, этилен СН2 = СН2, ацетилен СН =СН.
Изологич. ряды известны также и у алициклич. и ге-
тероциклич. соединений, напр. циклогексан, циклогек-
сен, циклогексадиен, бензол. Генетические pa-
fl ы содержат одинаковое число атомов углерода в
молекуле, но отличаются функциональными группами,
напр. генетич. ряд этана составляют: этан СН3—СН3,
этилхлорид СН3—СН2С1, этанол СН3—СН2ОН, ацет-
альдегид СН3—СНО, уксусная к-та СН3—СООН
и др. Г. р. наряду с изологич. и генетич. рядами со-
ставляют основу естественной классификации орга-
нич. соединений (рис. 1).
Отдельные члены Г. р. по многим физич. свойствам
отличаются между собой закономерно изменяющимися
985
ГОМОЛОГИЧЕСКИЕ РЯДЫ
986
показателями. Так, темп-ры кипения в середине Г. р.
для соединений с неразветвленной цепью радикала
Изалогические ряды
Рис. 1. Схема классификации органических соединений.
отличаются между соседними гомологами прибл. на
20—30° (см. табл. 1 и 2),
Таблица 1. Температура кипения некоторых
одноосновных кислот
Кислота Т. кпп., °C Д, °C
Уксусная СНаСООН 118
Пропионовая СНаСНаСООН .... 141 Zo с)4
Масляная СН, (СН,)а СООН .... 162 Z1 <ЭО
Валериановая Сня (Сн2)я СООН . . 185 on
Капроновая СН3 (СНЯ)4 СООН . . . 205 Zu
Таблица 2. Температура кипения некоторых спиртов
Спирт Т. кип.. °C Д, °C
Этиловый СН.,СН2ОН 78,4 19,0 20,3 20,1 19,4
н-Пропиловый СН3СН2СН2ОН . . . 97,4
н-Бутиловый СН3 (СН2)2 СН2ОН . . 117,7
и-Амиловый CHS (СН2)3 СН2ОН . . 137,8
м-Гексиловый СН3 (СН2)4 СН2ОН . 157,2
У высших членов Г. р. эти различия постепенно
сглаживаются; это видно, напр., по кривым темп-р
кипения и плавления нормальных алканов (рис. 2).
Гомологич. разности состава —СН2— соответствует
и монотонное возрастание ' молекулярных объемов
(ок. 22) и теплот сгорания (150—160 ккал); гомологич.
разность молекулярной рефракции составляет 4,6
для. линии D натриевого пламени.
При общности многих химич. свойств, а также спо-
собов синтеза членов одного и того же Г. р. они имеете
с тем обнаруживают иногда очень резко выраженные
индивидуальные особенности. Так, малоновая к-та
легко декарбоксилируется: НООС—СН2—СООН —
—*СО24- СН3СООН, тогда как янтарная к-та в ана-
логичных условиях образует циклич. ангидрид:
НООС-СН2-СН2-СООН X Н2О + OC-CHS-CH3-CO
!----о-----!
Особенно заметно отклонение от типич. свойств у на-
чальных членов Г. р. Так, константа диссоциации
муравьиной к-ты НСООН равна 2,14 • Ю-4, примерно
в 12 раз больше, чем уксусной к-ты — 0,18 • 10 4 и
других ее гомологов. Метанол СН3ОН по токепч.
действию резко отличается от остальных спиртов
своего ряда; формальдегид СН2О под влиянием креп-
кой щелочи претерпевает реакцию Канниццарро, в то
время как остальные альдегиды жирного ряда осмо-
ляются.
Понятие о гомологич., изологич. и генетич. рядах,
как закономерности общего порядка, было впервые
отчетливо сформулировано Ш. Жераром в 1844—45.
Но еще в 1841 Г. Копп в систематич. исследовании
физич. свойств спиртов, иодидов, сульфидов,сложных
эфиров вплотную подошел к понятию о Г. р. В 1842
Шиль нашел, что темп-ры кипения спиртов, радикалы
к-рых разнятся на СН2, отличаются на 18°, а н 1843
на аналогичную закономерность для жирных кислот
указал Ж. Б. Дюма; однако он, так же как и Копп,
считал, что гомологич. разностью является группа
С2Н4. Правильную гомологич. разность —СН2—
нашел Жерар. Однако строго научная основа под
представление о Г. р. была подведена теорией химич.
строения органич. соединений (А. М. Бутлеров, 1861).
Усложнение молекулы внедрением в ее радикал
гомологич. разности допускает многочисленные струк-
турные варианты. В связи с местом внедрения группы
СН2 и возникающей при этом структуры молекулы
могут быть различные структурные типы гомологов.
Так, напр., в ряду циклоалканов СпН2п:
СН2 Н2С~СНа ,CHZ-CHS
н2с-сн3 н2с-сна НаС^сн2-сн2
I II III
ДН2 Н2С—СН2 н хСНа-СН2
Н2С-СН-СН3 НгС-СН-СНэ 2 ЧСН2-С.Н-СН,
« о
Л/ / у/
Углеводороды I, II, III и т. п. гомологи с посте-
пенно расширяющимся циклом, соединения типа I
и IV, 11 и V, 111 и VI и т. д. также гомологичны и
также близки друг к другу по свойствам. Оба вида
Г. р. можно дифференцировать как относящиеся: I,
II, III и т. п. — к типу ядерной гомологии и I—IV,
987
ГОНАДОТРОПНЫЕ ГОРМОНЫ — ГОРЕНИЕ
988
II—V, III—VI ит. п. — к типу цепной гомологии.
В ряду кислот СпН2п (СООН)2 также легко заметить
гомологию двух разных порядков:
ноос-соон НООС-СН2-СОоН
VII VIII
НООС--СН.--СН.--СООН
IX
НООС—СН—СООН НООС-СН-СН,-СООН
I I
СНз СНз
X XI
Г. р. VII, VIII, IX и т. д. — от щавелевой к малоно-
вой, янтарной и т. д. кислотам— представляют один
тип гомологии, тогда как VIII и X, IX и XI и т. д. —
другой.
Уточнение и развитие понятия О Г. р., последовав-
шее на основе структурной теории, продолжается и
теперь. Так, изучение соединений с системой сопря-
женных связей вызвало к жизни представление о ви-
нилогии и о винилогах, отличающихся между собой
на группу —СН=СН— или кратную ей величину.
Разработка обширной области химии высокомолеку-
лярных соединений привела к установлению понятия
о полимергомологии; в полимергомологич. рядах
различие между однотипными макромолекулами опре-
деляется количеством слагающих их структурных
звеньев; напр. групп = CF2—в полиперфторэтилене,
групп —СН2—С (СН3) СООСН3 — в полиметилмета-
крилате, —СНС1—СН2—в поливинилхлориде и т. д.
Лит.: Жданов Ю. А., Гомология в органической хи-
мии, IM.J, 1950; Чичибабин А. Е., Основные начала ор-
ганической химии, т. 1, 5 изд., М., 1953, с. 60. Р. Р. Галле.
ГОНАДОТРОПНЫЕ ГОРМОНЫ (гонадотропины) —
органич. вещества, вырабатываемые передней долей
гипофиза и стимулирующие развитие гонад (половых
желез) у животных и человека. Различают след. Г. г.:
фолликулостимулирующий (ФСГ) — активирует рост
и развитие половых клеток, и лютеинизирующий
(Л Г)— регулирует секрецию половыми железами гор-
монов (эстрона, прогестерона и тестостерона). Г. г.—
гликопротеиды с различными физико-химич. свойст-
вами (в зависимости от того, из какого вида живот-
ных они получены). Иногда к Г. г. относят лютеотро-
пин (пролактин) и хорионгонадотропин (см. Гормоны).
Наиболее изучен фолликулостимулирующий гормон
(ФСГ), получаемый из гипофизов свинеи в виде гомо-
генного препарата; мол. в. 29 000, изоэлектрич. точка
при pH 5,1—5,2, константа седиментации (в воде
при 20°) Х20и 2,49, константа диффузии (в воде при
20°) Л2О<о 7,43 • 10т; установлен аминокислотный со-
став препарата, но последовательность аминокислот
не установлена. Для ФСГ характерно присутствие
сравнительно большого количества цистина, обра-
зующего в молекуле дисульфидные мостики, к-рые,
по-видимому, важны для проявления физиология,
активности (при обработке препарата цистеином, раз-
рушающим дисульфидные связи, активность препа-
рата снижается). Помимо аминокислот, в ФСГ обна-
ружены: галактоза, манноза, фукоза и гексозамин,
образующие полисахаридную часть молекулы. Нали-
чие неизмененного углеводного фрагмента в молекуле
ФСГ необходимо для его нормального физиология,
действия; обработка ФСГ гидролитич, ферментами,
разрушающими полисахариды, инактивирует его.
Биологич. действие ФСГ обусловливается молекулой
гликопротеидов или частью ее. ФСГ — единственный
гормон гипофиза, растворимый в конц. солевых р-рах
(напр., 50%-ном сульфате аммония), что используется
для его отделения от др. гормонов. Кроме того, ФСГ
может быть отделен от Л Г хроматографически.
Лютеинизирующий гормон (ЛГ,
гормон, стимулирующий интерстициальные клетки)^ '
полученный из гипофизов животных разных видов,
имеет различные свойства (напр., мол. в. Л Г свиньи
100 000, а овцы 30 000—40 000); изоэлектрич. точка Л Г
овцы при pH 4,8. ЛГ — гликопротеид; бычий препарат
ЛГ содержит 5,9% маннозы и 6,1% гликозамина. ЛГ
кристаллизуется из 50%-ного ацетона при охлаждении
(процесс ускоряется в присутствии ионов металлов).
Г. г. определяют и стандартизуют в основном биоло-
гич. путем: ФСГ по действию на вес яичников у самок
крыс, а ЛГ по влиянию на размер простаты у самцов.
Лит.: К п р in е и б л а т Я. Д., Проблемы эндокринологии
и гормонотерапии, 1956, 2, № 6, с. 108—116; StelmanS. L.,
S е g а 1 о 1 1 А., в кн.: Recent progress in hormone research,
v. 15, N. Y.—L., 1959,'p. 115; Behrens О. K. and В ro-
mer W. W., Annual Rev. Biochem., 1958, 27, p. 92; cm.
также лит. к ст. Гормоны. Ю. А. Панков.
ГОПКАЛИТ — катализатор для окисления окиси
углерода кислородом воздуха в двуокись углерода.
Главной составной частью Г. является активная дву-
окись марганца МпО2. Известны 2 рецептуры Г.:
50% МпО2, 30% СцО, 15% Со203, 5% Ag3O или 60%
МпО2 и 40% СиО. Окисление СО до СО2 на Г. сопро-
вождается выделением значительного количества теп-
лоты: 2СО + О2 —>2СО2 ф- 136 ккал. Водяные пары
отравляют катализатор, поэтому Г. помещают между
двумя слоями осушителя, от емкости к-рого зависит
время защитного действия слоя Г. Для получения Г.
его хорошо отмытые компоненты смешивают в виде
водной суспензии, фильтруют, высушивают при уме-
ренной темп-ре, прессуют и дробят на зерна опре-
деленных размеров. Г. используется в специальных
дополнительных патронах к противогазам при нали-
чии в воздухе опасных концентраций СО, а также
в приборах для контроля за содержанием СО в поме-
щениях; приборы снабжены сигнальным приспособле-
нием, предупреждающим об опасной концентрации
СО; чувствительность прибора до 0,01 мг/л. Катали-
тич. действие Г. зависит от темп-ры; при отрицатель-
ных темп-рах оно практически прекращается.
Лит.: Алексеевский Е. В., Активная двуокись
марганца, Л., 1937; Еремина Б. Г.. Газовый анализ,
М., 1955; Беркман С., Моррелл Д. и ЭглоффГ.,
Катализ в неорганической и органической химии, пер. с англ
кн. 1—2, М.—Л., 1949; Дубинин М. М., ЧмутовК. В.,
Физико-химические основы противогазового дела, М., 1939;
Роде Е. Я., Кислородные соединения марганца, М., 1952.
С. Н. Васильев.
ГОРЕНИЕ — сложный физико-химич. процесс, ос-
новой к-рого является быстро протекающая химич.
реакция окисления, сопровождающаяся выделением
значительного количества тепла и обычно ярким
свечением (пламенем). Химич, реакция Г. в боль-
шинстве случаев является сложной и состоит из
большого числа элементарных химич. процессов
окислительно-восстановительного типа, приводящих
к перераспределению валентных электронов между
атомами взаимодействующих веществ (см, Окисление—
восстановление). Кроме того, химич. превращение
при Г. тесно связано с рядом физич. явлений —
переносом тепла и масс и, соответственно, с гидро-
и газодинамич. закономерностями. Согласно совре-
менной физико-химич. теорий Г., к процессам Г.
относят все химич. реакции, связанные с быстрым
превращением и тепловым или диффузионным уско-
рением, в том числе: раэложеиие взрывчатых веществ,
озона, ацетилена и др., взаимодействие нек-рых ме-
таллов с кислородом и галогенами, сгорание иодо-
рода в струе хлора и т. д.
Горючее и окислитель. Температура и теплота
горения
Окислителями в процессах Г. могут быть: кислород
(воздух), озон, перекиси, вещества, богатые кислоро-
дом (нитросоединения, азотная к-та, перхлораты),
галогены. Т. обр., класс веществ, способных при
определенных условиях взаимодействовать с дру-
989
ГОРЕНИЕ
990
гими веществами в качестве окислителей, весьма
обширный. В качестве горючего, способного взаимо-
действовать с окислителем, могут быть многие ве-
щества: большинство металлов в свободном виде,
сера элементарная и связанная (сероводород, колче-
дан), окись углерода, водород и огромное число
органич. соединений. Однако наибольшее практич.
значение в качестве окислителя приобрел кислород
(воздух), а в качестве горючего — углеводородные
вещества (природный газ, нефть, угли, сланцы, торф
и т. п.). Процессы сжигания таких горючих в атмо-
сфере кислорода являются и наиболее изученными.
Горючей массой общепринятых топлив являются:
углерод, водород, сера, кислород и азот, к-рые
в сумме составляют 100 вес. %, а их конечными про-
дуктами полного сгорания — СО2, Н2О, SO2 и N2 *.
Помимо горючей массы, топливо содержит балласт
(сумма влаги и золы). В процессе Г. образуются,
в зависимости от состава горючего, темп-ры и количе-
ства окислители, различные промежуточные про-
дукты (СО, СН4, SO и др.). Для полного сгорания
любого горючего вещества требуется определенное
количество окислителя — кислорода, воздуха и т. д.
Количество окислителя, рассчитанное на основании
стехиометрич. соотношения, наз. теоретически необ-
ходимым. Значение этой величины для ряда типич-
ных горючих содержится в таблице.
Горючее Теплотвор- ность Q® Теоретич. необходимое количество воздуха Теоретич. (максималь- ная) темп-ра горения Тт °C
Газообразное Водород мо леку лир- 2576 ккал/нм3
ный 2,38 нм*/нм* 2200
Окись углерода . . 3016 » 2,38 » 2340
Ацетилен 13400 » 11,9 » 2520
Метан 8558 » 9,52 » 2000
Я! и д к о е
Бензол 10030 ккал/кг 10,1 нл^8/кг 2200
Спирт этиловый . . 6600 » 6,9 » 2080
Мазут ........ 9870— 9600 » 10,7— 10,6 » 2080—2090
Твердое
Углерод (графит) . 7830 » 8,9 » 2145
Антрацит 6400 » 7,2 » 2160
Древесина (горючая
масса) 4510 » 4,76 » 2000
Практически продукты полного Г. получаются
только при избыточном количестве воздуха (окисли-
теля) по сравнению с теоретически необходимым.
При недостатке воздуха конечные продукты Г. всегда
содержат горючие вещества — Н2, СО, СН4 и т. д.;
Н2, СО могут содержаться в продуктах Г. и при из-
бытке воздуха в результате диссоциации' Н2О и СО2
при высоких темп-pax. При еще более высоких
темп-pax компонентами продуктов Г. оказываются
также атомарные водород и кислород и различные
радикалы — ОН, СН и др.
Основные характеристики горючей смеси — тепло-
творная способность Q и теоретич. темп-ра Г.
т. е. та темп-ра, к-рая была бы достигнута в стехио-
метрич. смеси при полном сгорании без теплопотерь,
при отсутствии диссоциации продуктов Г. (эту
темп-ру называют также калориметрич. темп-рой Г.):
Tt=T0+Qi'C
где То — начальная темп-ра горючей смеси, С —
средняя теплоемкость продуктов Г. между Тп и иско-
* Продуктом полного горения является также серный
ангидрид SOs, образующийся в относительно небольших коли-
чествах при горении серусодержащих горючих и при наличии
избытка кислорода.
мой Тт. В соответствии с принятым значением тепло-
емкости различают при постоянном объеме и
при постоянном давлении. При темп-pax Г. выше
2000° К, когда становится заметной диссоциация
продуктов Г. и величина Q соответственно меньше,
расчет темп-ры Г. производится по формулам ста-
тистич. термодинамики, методом последовательных
приближений. Значения Тт без учета диссоциации
приводятся в той же таблице для стехиометрич. сме-
сей ряда горючих; эти величины оказываются более
удобными для теплотехнич. расчетов по сравнению
с часто используемой другой характеристикой горю-
чего — его теплотворной способностью к-рая,
как видно из таблицы, очень сильно изменяется от
одного вещества к другому. Значения Q” для газо-
образных горючих относят обычно к л13 газа, а для
жидких и твердых — к кг веса горючего.
Теплотворную способность топлива Q определяют
сжиганием его в калориметрич. бомбе. Для прибли-
женных расчетов теплотворности не утратила своего
значения ф-ла Менделеева:
Q = 81С -|- ЗООН — 26 (О — S) ккал/кг
где С, Н, О и S — весовое содержание (в %) углерода,
водорода, кислорода и серы (горючей) в топливе.
Это т. наз. высшая теплотворность Q®, к-рая отли-
чается от (J®, наз. низшей теплотворностью, на вели-
чину теплоты, уносимой водяными парами вместе
с продуктами сгорания.
Вследствие большого разнообразия видов горючего
и окислителя конкретные характеристики и области
использования Г. весьма различны. Наиболее важ-
ным фактором, определяющим основные свойства Г.,
является агрегатное состояние горючего и окисли-
теля. По агрегатному состоянию горючего и окисли-
теля различают: 1) гомогенное — Г. газов и парооб-
разных горючих в среде газообразного окислителя
(б. ч. кислорода воздуха); 2) гетерогенное — Г. жид-
ких и твердых горючих в среде газообразного окис-
лителя, а также Г. в системе жидкая горючая смесь —
жидкий окислитель (напр., кислоты); 3) Г. взрывчатых
веществ и порохов, представляющих по существу кон-
денсированную гомогенную систему.
Основные динамические свойства Г.
Воспламенение. Скорость Г.
Самое общее свойство Г. — возможность нри из-
вестных условиях прогрессивного самоускорения хи-
мич. превращения и, как его результат,— воспламе-
нение, появление пламени. Предполагаетси, что при
этом заранее обеспечен контакт или перемешивание
горючего с окислителем в виде, напр., смеси газа и
воздуха, аэровзвеси мелко распыленных частиц твер-
дого или жидкого горючего, слоя частиц топлива,
продуваемого воздухом, и т. д.
Причиной усиорения химич. превращения может
быть как разогрев реагирующих веществ за счет
теплоты реакции, так и накопление химически актив-
ных продуктов реакции (последнее гл. обр. при реа-
гировании газообразных и парообразных горючих).
В первом случае говорят о тепловом,во втором — о
цепном механизме воспламенения. Надо, однако, ска-
зать, что термин «тепловое» в основном относится к
первопричине ускорения, поскольку химич. реакции
и при тепловом воспламенении могут быть цепными.
При тепловом воспламенении, для того чтобы на-
чался саморазогрев системы, всегда необходим ее
предварительный разогрев. Повышай темп-ру, можно
при известных условиях достичь такого состояния,
при к-ром тепловыделение за счет реакции превысит
991
ГОРЕНИЕ
992
теплоотвод из зоны реакции. С этого момента стацио-
нарное течение реакции станет невозможным; нач-
нется саморазгон реакции и произойдет воспламене-
ние. В случае цепного механизма воспламенения раз-
витие процесса происходит аналогичным образом,
с той лишь разницей, что определяющим является
«соотношение между скоростью накопления химически
активных веществ и скоростью их рассеивания за
счет диффузии.
Таким образом, процесс воспламенения в обоих
случаях — это переход от стационарной, обычно
медленно протекающей и искусственно поддерживае-
мой реакции, к самоускоряющейся лавинного типа
реакции. Этот переход совершается всегда очень
резко, практически внезапно, и прямым следствием
такого положения является еще одна характерная
черта Г.—наличие четко фиксирующихся критич.
характеристик (параметров) воспламенения: системы
значений состава смеси (или соотношений горючего
и окислителя), системы значений максимально воз-
можной темп-ры начального нагрева, критич. началь-
ного давления, системы критич. значений содержания
примесей, активирующих или флегматизирующих.
Воспламенение свойственно любым горючим систе-
мам — гомогенным, гетерогенным и более сложным
системам. Различают, однако, два способа (вида)
воспламенения: самовоспламенение и т. наз. вынуж-
денное воспламенение — зажигание. При само-
воспламенении описанные выше условия
самоускорения реакции создаются во всем объеме
данной воспламеняемой смеси. Напр., при тепловом
самовоспламенении газообразная смесь нагревается
или от горячей стенки сосуда (бомбы), или путем
быстрого сжатия смеси, или при быстром перемеши-
вании ранее нагретых компонентов смеси. При этом
фиксируется соответствующее значение начальной
темп-ры, при к-рой происходит воспламенение, и
эта темп-ра называется темп-рой самовоспламенения.
При другом типе воспламенения — зажига-
нии — условия воспламенения создаются только
в очень небольшой части смеси с помощью к.-л. высо-
котемпературного источника тепла: накаленного тела,
пламени, электрич. искры и др. Вблизи такого источ-
ника возникает пламя, к-рое затем распространяется
на весь объем горючей среды. Воспламенение сво-
дится в таком случае к саморазвивающемуся распро-
странению пламени. Аналогично тепловому меха-
низму распространения пламени возможен и процесс
чисто диффузионного («холодного») распространения
реакции в условиях цепного механизма воспламене-
ния. Для теплового зажигания, как и для самовос-
пламенения, экспериментально фиксируется мини-
мальная темп-ра высоко нагретого источника тепла,
вызывающая появление пламени. Такая темп-ра
называется темп-рой зажигания. Она также является
функцией состава смеси, начальной темп-ры и давле-
ния. Кроме того, зависит от размера источника зажи-
гания.
Для воспламенения характерна еще одна особен-
ность — явление т. н. индукционного периода, или
задержка воспламенения. Наиболее специфично это
явление для самовоспламенения. В этом случае
период задержки — это время, к-рое проходит между
моментом, когда смесь приобрела заданную началь-
ную темп-ру подогрева, и моментом заметного само-
ускорения реакции (практически появлением пла-
мени). Этот период сильно зависит от химич. свойств
смеси, кинетики предпламенных химич. процессов,
когда происходит разогрев системы и накопление
промежуточных активных веществ — неустойчивых,
легко реагирующих свободных радикалов (типа СН,
ОН и т. д.), или .более устойчивых молекулярных
соединений. Величина индукционного периода при
тепловом воспламенении очень сильно зависит от
начальной темп-ры нагрева. Иначе — каждой темп-ре
самовоспламенения соответствует своя величина ин-
дукционного периода.
Процессы воспламенения играют очень большую
роль в практике Г., особенно зажигание. Акт воспла-
менения составляет, однако, лишь начальную ста-
дию Г. После воспламенения устанавливается всегда
больший или меньший период устойчивого, квази-
стационарного Г., завершающегося или полным
исчерпанием соответствующей горючей компоненты
(компоненты, находящейся в недостатке) или уста-
новлением термодинамич. равновесия. Для этой вто-
рой стадии Г. определяющими являются соответ-
ствующие динамич. или кинетич. закономерности Г.
В силу комплексной природы Г. эти закономерности
практически никогда не тождественны кинетич. зако-
номерностям чисто химич.. взаимодействия реагентов
горючей системы. Причиной этому является то, что
при больших скоростях реакций окисления при Г.
возникает такое положение, что или подвод тепла,
как, напр., при тепловом распространении пламени,
или транспорт (диффузия) реагентов в зону реакции
будут лимитировать скорость реакции в силу крайней
медленности этих физич. процессов. Наиболее типич-
ным примером являются гетерогенные системы, реа-
гирование к-рых всегда связано с переносом газооб-
разных реагентов из газовой фазы к твердой стенке и
обратно. При высокой темп-ре, когда реакция ста-
новится очень быстрой, газовый реагент, напр.
кислород, находящийся у стенки, быстро расхо-
дуется, его становится мало, и реакция замедляется,
несмотря на имеющуюся высокую темп-py; последняя
как бы оказывается тормозящим фактором. В стацио-
нарных условиях устанавливается тогда режим горе-
ния, подчиненный процессу подвода кислорода
к стенке, а скорость Г. становится тождественной
скорости чисто физич. процесса — диффузии кисло-
рода. Соответственно этому о такого рода режимах Г.
говорят как о диффузионных или диффузионно-теп-
ловых; к их числу относится и распространение пла-
мени в гомогенных системах.
Гомогенное горение. Наиболее простой
случай гомогенного Г. представляет Г. заранее пере-
мешанных газовых смесей. Г. неперемешанных газо-
образных или парообразных веществ также является
гомогенным Г., но более сложным по своей структуре.
Реакции при Г. газов являются большей частью цеп-
ными (см. Цепные реакции), однако в обычных усло-
виях Г. определяющее значение имеет тепловое ини-
циирование реагирования (термич. активация). Усло-
вие квазистационарности элементарных реакций при
цепном превращении позволяет во многих случаях
при исследовании и описании Г. пользоваться одним
суммарным ур-нием для скорости реакции W, содер-
жащим только начальные концентрации горючего а
и окислителя Ъ и эффективную энергию актива-
ции Е:
W (а, Ь, Т) = Ко anbm exp (— E/RT)
где п и m — константы, Ко — предэкспоненциальный
.множитель в уравнении Аррениуса. Применительно
к условиям Г. величины а, b и Т связаны друг с дру-
гом, т. к. разогрев системы идет за счет расходования
реагирующих веществ: чем выше Т, тем меньше а и Ь.
В силу этого в начале процесса Г. (при воспламене-
нии) определяющее значение имеет темп-рный член
кинетич. уравнения, в конце — концентрационный,
когда скорость реакции прогрессивно уменьшается.
Для Г. заранее перемешанных смесей стадия вос-
пламенения в общем процессе Г. занимает домини-
рующее положение в силу двух обстоятельств: эта
стадия (распространение пламени) наиболее медлен-
993
ГОРЕНИЕ
994
ная, в пламени осуществляется сгорание преобла-
дающей части горючей смеси так, что последующее
квазистационарное горение или практически отсут-
ствует, или происходит быстро.
При тепловом распространении пламени различают
нормальное (тихое) распространение Г., или
дефлаграцию «.(последовательное воспламене-
ние горючей смеси происходит по механизму тепло-
проводности и, частично, за счет диффузии активных
центров), и детонацию (поджигание произво-
дится распространяющейся ударной волной). Нор-
мальное Г. в свою очередь подразделяется на лами-
нарное и турбулентное. Ламинарное пламя
обладает вполне определенной скоростью перемеще-
ния относительно неподвижного газа, к-рая зависит
от состава смеси, давления и темп-ры и определяется
только химич. кинетикой и молекулярной теплопро-
водностью. Такая скорость, называемая нормальной
скоростью пламени, является поэтому физико-химич.
константой смеси. Ламинарное пламя наблюдается
в неподвижных смесях или в потоках, движущихся
ламинарно. Величины скорости пламени обычно
составляют в воздушных средах порядка нескольких
десятков сантиметров в секунду и только для водо-
родо-воздушных смесей достигают 2,5 м/сек. В тех
случаях, когда наряду с молекулярной теплопровод-
ностью в большой степени участвует т. н. турбулент-
ный перенос тепла, при перемешивании возникает
турбулентное пламя. Скорость распростра-
нения турбулентного пламени в отличие от ламинар-
ного зависит от скорости газового потока, что является
главной и наиболее важной особенностью турбулент-
ного пламени. Турбулентное пламя имеет большое
значение в технич. процессах сжигания газообразных
и парообразных горючих.
В условиях Г. в потоке большое практич. значение
имеет стабилизация горения, удержание
пламени на горелке или в камере, поскольку обычно
скорость потока больше скорости пламени и пламя
не может самостоятельно сохранить свое положение
в пространстве. Г. стабилизируют либо непрерывным
зажиганием потока горючей смеси при помощи спе-
циального устройства (горелка, форкамера) или с по-
мощью установки поперек потока плохо обтекаемых
тел (стабилизаторов), способствующих обратной цир-
куляции горячих продуктов Г. Механизм стабили-
зации принципиально не отличается от механизма
теплового зажигания накаленными телами.
Гетерогенное горение. Класс процес-
сов Г. гетерогенного типа весьма широк. Основные
из них: Г. жидких горючих и Г. твердых топлив. Для
Г. жидких веществ большое значение имеет процесс
их испарения. Г. легко испаряющихся горючих прак-
тически относится к гомогенному Г., т. к. такие го-
рючие еще до воспламенения полностью или по-
чти полностью успевают испариться. Применительно
к жидким горючим различают 2 характеристики:
температуру вспышки и температуру самовоспламе-
нения (см. Вспышки температура и Самовоспламе-
нение').
Широко распространенной жидкой гетерогенной
системой является высокодисперсная капельная си-
стема, для к-рой определяющее значение имеют законы
воспламенения и Г. каждой отдельной капли. При
высокой темп-ре среды, когда образующиеся пары
быстро сгорают около поверхности капли, скорость
ее сгорания равна скорости испарения, к-рая в свою
очередь равна скорости подвода тепла к поверхности
жидкости. В двухфазной капельной среде возможно
распространение как ламинарного, так и турбулент-
ного пламени. Скорость его сильно зависит от степени
предварительного испарения и особенно от величины
капель.
Простейший случай Г. твердых веществ — когда
процесс не сопровождается разложением вещества
с выделением газов (напр., Г. металлов). Если при
Г. образуется твердый окисел, то дальнейший ход
процесса зависит от того, выше или ниже темп-ра
плавления окисла по сравнению с темп-рой поверх-
ности. В первом случае процесс лимитируется диф-
фузией кислорода через окисную пленку и быстро
прекращается, во-втором — продолжается беспрепят-
ственно. Именно поэтому железо легко горит в
кислороде, но в обычных условиях не горит в воз-
духе.
В технике большое значение имеет Г. твердого
топлива, гл. обр. углей. Основа любого твердого
топлива — углерод. Кроме того, оно содержит то
или иное количество органич. веществ, к-рые при
нагревании топлива разлагаются с образованием
паров и газов (гл. обр. горючих). Эту термически
неустойчивую часть топлива принято называть лету-
чей, а газы — летучими. При быстром нагреве частиц
топлива (что возможно для частиц малого размера)
летучие могут полностью не успеть выделиться и
сгорают вместе с углеродом. При медленном нагреве
наблюдается четкая стадийность начала Г. — сна-
чала выход летучих и их воспламенение, затем вос-
пламенение и Г. твердого, т. н. коксового остатка,
к-рый, кроме углерода, содержит минеральную часть
топлива золу. Первая стадия Г., как и ранее, —
самовоспламенение частиц топлива. Оно может быть
комплексным (сначала воспламенение летучих, затем
коксового остатка) или более простым, когда летучих
мало или они полностью отсутствуют (чистый угле-
род или коксовый остаток). Воспламенение происхо-
дит по законам теплового самовоспламенения. Во
всех случаях время воспламенения твердого топлива
составляет небольшую долю общего времени Г.;
в этом важное отличие от случая воспламенения газов.
Поэтому процесс гетерогенного Г. углерода — основ-
ной процесс для Г. твердых топлив и его закономер-
ности определяют закономерности Г. в целом. Эти
закономерности являются общими для любых гете-
рогенных процессов с твердыми веществами и отли-
чаются в каждом случае только типом химич. процес-
сов между газовым реагентом и твердым веществом.
Реакция кислорода с углеродом характеризуется
образованием обоих окислов углерода — СО и С02,
но соотношение между ними оказывается весьма
различным в разных условиях опыта и для разных
типов углерода. Хемосорбция кислорода, особенно
значительная при низких темп-рах, всегда сопут-
ствует реакции, и поэтому она рассматривается в ка-
честве одного из элементов механизма реакции С -|- О2.
Схема реакции до сих пор, однако, окончательно не
установлена, но предполагается, что она включает
в себя ряд последовательных элементарных реакций,
в к-рых важную роль играет образование в резуль-
тате хемосорбции промежуточного продукта в виде
т. н. поверхностного окисла типа СлОу. Последующее
разложение этого окисла и дает то или иное соотно-
шение СО и СО2. К числу особенностей Г. углерода
относится возможность протекания, наряду с основ-
ной реакцией кислорода с углеродом, также и дру-
гих, т. н. вторичных реакций: гетерогенной реакции
(С СО2 — 2СО) и гомогенной реакции (2СО -|- О2 =
= 2СО2). В присутствии кислорода имеет место кон-
куренция этих реакций, что сильно затрудняет опре-
деление истинного состава продуктов основной реак-
ции (отношения СО к СО2).
Суммарное кинетич. ур-ние реакции С -|- О2, выте-
кающее из теоретич. соображений и опыта, имеет вид:
W = к'с/с А
где к' и А — кинетич. константы. При малой вели-
995
ГОРМОНЫ
996
чине с этот закон переходит в простой кинетич. закон
первого порядка. Практич. характеристикой суммар-
ной скорости Г. твердого топлива обычно прини-
мается т. н. уд. скорость Г. — количество углерода,
сгорающее на единице геометрич. поверхности угле-
рода в секунду. Этой величиной учитывается как Г.
на внешней поверхности, так и внутри — в порах и
трещинах, куда кислород проникает путем диффузии.
Когда скорость реакции мала, диффузия как к по-
верхности тела, так и внутри его не тормозит Г., и уд.
скорость Г. отличается чисто кинетич. свойством.
В этом случае говорят о кинетич. режиме Г. Когда
диффузия становится тормозящим фактором, Г. опре-
деляется диффузионными факторами, и говорят о т. н.
диффузионном режиме Г. В связи с этим для явле-
ния Г. углерода, в процессе его развития, типичны
переходы от закономерностей Г. в основном чисто
химич. характера к закономерностям в основном
.чисто физическим.
Каталитическое, или, вернее, поверх-
ностное каталитическое, Г. газовых
смесей относится к классу гомогенно-гетерогенных
процессов Г.; химич. процесс может происходить как
в объеме, так и на твердой поверхности катализатора
(напр., на платине). В зависимости от конкретных
условий может проявляться гомогенный или гетеро-
генный тип Г. При высоких темп-рах, когда объемное
Г. идет быстро, роль поверхностно-каталитич. Г.,
как правило, мала и может быть заметной только
в случае, когда течение смеси происходит в узких
каналах, пористых материалах или мелкозернистых
засыпках из катализатора. Применяемый в технике
термин «беспламенное» Г. газовых смесей не всегда
эквивалентен понятию поверхностно-каталитич. Г.
Скорее оно является характеристикой Г. без светя-
щегося пламени и при максимально укороченной его
длине.
Лит.: Семенов Н. П., О некоторых проблемах хи-
мической кинетики п реакционной способности, М., 1954;
Кондратьев В. Н., Кинетика химических газовых
реакций, М., 1958; X и т р и и Л. Н., Физика горения и
взрыва, М., 1957; Зельдович Я. Б., Теория горения
и детонации газов М.—Л., 1944; Горение углерода, М.—Л.,
1949; Фран к-К аменецкий Д. А., Диффузия и
теплопередача в химической кинетике, М.—Л., 1947; Кнор-
ре Г. Ф., Топочные процессы, М.—Л., 1959; В у л и с Л. А.,
Тепловой режим горения, М.—Л., 1954; Канторович
Б. В. Основы теории горения и газификации твердого топ-
лива, М„ 1958; Льюис Б. и Э л ь б е Г., Горение, пламя
п взрывы в газах, пер. с англ., М., 1948; Пост В., Взрывы
и горение в газах, пер. с нем., М., 1952; Г е й д о и А. Г.
иВольфгард X. Г., Пламя, его структура, излучение
и температура, пер. с англ., М., 1959; Р а в и ч М. Б., По-
верхностное беспламенное горение, [3 изд.], М.—Л., 1949.
ГОРМОНЫ — органич. вещества, выделяемые же-
лезами внутр, секреции в кровь и тканевую жидкость
и являющиеся регуляторами важнейших функций
организма животных и человека (обмена, роста, поло-
вого развития и др.). Действие Г. очень мало зависит
от вида животного. Г., полученные из желез внутрен-
ней секреции любого позвоночного, обладают одним
и тем же физиологич. действием. Механизм действия
Г. очень сложен и еще мало изучен. Выяснено, что
действие Г. находится в сложной взаимозависимости
от центральной нервной системы и тесно связано с дей-
ствием витаминов и ферментов. Подобно этим веще-
ствам, Г. в очень небольших концентрациях прояв-
ляют высокую физиологич. активность.
По своей химич. природе Г. позвоночных могут быть
разделены на 3 группы: Г., родственные тирозину,
пептидные Г. и стероидные Г. (см. табл. 1).
Наряду с самими Г. выделены и многочисленные
продукты их метаболизма. Так, из надпочечников и
мочи было изолировано около тридцати продуктов
превращения стероидных Г. коры надпочечников (см.
Кортикостероиды). Многие из этих веществ также
обладают гормональным действием. Гормональной
Таблица 1. Химическая классификация гормонов
позвоночных
Название Г.
Железы,
образующие
Г.
Основной вид биологической
активности»
Гормоны, родственные тирозину
Фенилалкилампны
Адреналин ’’ 3
Норадреналин *’ 3
Мозговой
слой над-
почечни-
ков
Повышают кровяное давле-
ние, вызывают гликогено-
лиз, гипергликемию; нор-
адреналин — медиатор
нервного возбуждения
Иодированные тиронины
Тироксин ’’ 3 3,5, З-Трииод- тиронии ъ3 ) Щитовид- у иая же- 1 леза Стимулируют основной об- мен
Пептидные гормоны
Цикл ические октапептпды
Окситоцин’ Вазопрессин’ 1 Задняя У доля гипо- 1 физа Вызывает мышечное сокра- щение матки, периферия, вазоконстрикцию, анти- диуретич. действие
Полипе п т и д ы
Меланофор- ный Г. (интермедин, хроматотропин)2 Адренокортико- тропный Г.2 Межуточная доля гипо- физа Передняя доля гипо- физа Вызывает расширение мела- нофор в хроматофорах кожи Стимулирует функцию коры надпочечников
Инсулин *’ 2 Секретин Поджелудоч- ная железа Слизистые железы ки- шечника Регулирует обмен углеводов Стимулирует выделение пан- креатического сока
Глюкагон ’ 2 Островки Лангераиса поджелудоч- ной железы Повышает концентрацию са- хара в крови
Б е л к о в ы е е щ е с т в а
Лютеотропии 1 (пролактин) Паратиреокрин Соматотропин i (гормон роста) Ваготонин Цеитропнеин Передняя доля гипо- физа Околощито- видная железа Передняя доля гипофиза Поджелудоч- ная железа Поджелудоч- ная железа Поддерживает функцию жел- того тела и лактацию Поддерживает концентрацию Са п Р в крови Стимулирует рост, регули- рует анаболизм белков Стимулирует парасимпатиче- скую нервную систему Стимулирует дыхание
Гликопрот вины
Фолликулости- мулирующий (гонадотропный) г. Передняя доля гипо- физа Стимулирует рост фолликул, яичников и сперматогенез
Лютеинизирую- щий (гонадотроп- ный) Г. 1 Хорионгонадо- тропины 1 Тиреотропин 1 Передняя доля гипо- физа Плацента Передняя доля гипо- физа С; Стимулируют образова- ние эстрогенов у жен- ских особей и андроге- нов у мужских особей гимулирует деятельность щитовидной железы
Стероидные гормоны
Фенолы
Эстрон ’’ 3
Эстрадиол1' 3
Эстриол1’3
Яичники
Вызывают зструс и обра-
зование женских вто-
ричных половых при-
знаков
997
ГОРМОНЫ
998
Продолжение
Название Г. Железы, образующие Г. Основной вид биологической активности 1
К е т о н ы и оке и к е т о н ы
Тестостерон1, 3 Прогестерон1, 3 Семенники ’Желтое тело Вызывает образование муж- ских вторичных половых признаков, влияет на бел- ковый обмен Поддерживает беременность,
Кортизон1,3 Кортизол1’ 3 Кортикостерон1’ 3 11-Дегидрокор- тикостерон1, 3 17-Оксикортпко- стерон1’ 3 11-Дезоксикор- тикостерон1' 3 Альдостерон1’ 3 Кора над- • почечни- ков тормозит функцию фолли- кул Регулируют обмен угле- водов и белков Регулируют обмен элек- тролитов и воды
1 Выделен в кристаллич. форме. 2 Строение выяснено
полностью. 3 Получен н синтетически. 4 В этой графе приве-
дены лишь важнейшие виды активности данного Г. В дей-
ствительности биологич. действие большинства Г. отличается
исключительным многообразием. Так, напр., эстрон действует
не только на эструс и на женские половые органы, но также
на рост костей, волос и на активность ферментов коры над-
почечников. Он обладает также тимолитпч. и митогенетич.
активностью, противораковым и канцерогенным действием на
нек-рые органы, вызывает анемию и пр.
активностью обладают также нек-рые синтетич. со-
единения. К ним относятся продукты синтетич. превра-
щений нек-рых Г. (напр., производные стероидных Г.,
содержащих фтор, дополнительные двойные связи,
метильные группы или стероиды с шестичленным
кольцом, т. н. D-гомостероиды), а также нек-рые со-
единения, не обладающие структурным сходством
с Г., напр. синтетич. эстрогены, синэстрол.
Изучение Г. развивается обычно по след, пути:
первоначально удаляют орган, образующий Г., и
исследуют явления, наступающие вследствие прекра-
щения поступления Г. в организм. Затем из этого же
органа приготовляют различные неочищенные экст-
ракты и судят о содержании в них Г. по их способно-
сти снимать внешние признаки гормональной недоста-
точности. Для установления строения Г., помимо
химич. методов исследования, широко применяют
физико-химич. методы (изучение спектров, полиро-
графия, рентгеноструктурный анализ, ядерно-магнит-
ный резонанс). Строение многих Г. удалось подтвер-
дить их синтезом.
Для выделения Г. широко используются различные
методы (кристаллизация, получение различных про-
изводных, хроматография, противоточное распреде-
ление, электродиализ, электрофорез и др.), выбор
к-рых определяется химич. строением и свойствами Г.
Одно из основных затруднений при выделении Г. со-
стоит в том, что они содержатся в железах внутренней
секреции лишь в ничтожной концентрации (см. табл. 2).
Поэтому в практике часто выгоднее выделять нек-рые
Г. не из желез внутренней секреции, а из других
источников. Так, эстрон выделяют из мочи жеребцов
или жеребых кобыл, а хорионгонадотропин — из мочи
беременных женщин.
Количественно Г. определяют биологич., химич.
или физико-химич. методами. При биологич. методах
испытания пользуются специальными тестами (напр.,
испытание на рост гребня петухов — для определения
андрогенных гормонов, испытание на увеличение веса
яичников крыс — для определения эстрогенных гор-
монов, изучение изменения количества сахара в кро-
ви — для определения инсулина и т. д.). Минимальное
количество Г., обнаруживаемое в данном тесте, при-
нимают за единицу активности (напр., «петушиная
единица» для андрогенных Г., «крысиная единица»
для эстрогенных Г.). Для сравнения отдельных пре-
паратов между собой служат т. н. международные
единицы, соответствующие активности определенного
весового количества стандартного препарата дан-
ного Г. В последнее время биологич. методы испыта-
ния частично вытесняются физико-химич., из к-рых
чаще всего используются колориметрия, и спектраль-
ные методы анализа. Так, в медицинской практике
особенно широко применяют колориметрии, опреде-
ление 17-кетостероидов в моче и спектрофотометрии,
определение адреналина. В отличие от биологич. ме-
тодов испытания, физико-химич. методами удается
определить не только содержание самих Г., но и про-
дуктов их превращения. Иногда для определения Г.
пользуются и чисто химич. методами анализа (иодо-
метрич. определение тироксина).
Таблица 2. Содержание некоторых гормонов в железах
внутренней секреции
Название железы Название Г. Содержание Г. в 1 кг ткани желез
Передняя доля гипофиза свиньи Адренокорти- котропный Г. 10 мз
Щитовидная железа овцы . . . Тироксин 24 мз
Надпочечники коровы Адреналин 4- 4- норадрена- лин 0,1 г
То же Кортикосте- рон 0,7 мг
Яичники свиньи Эстрадиол 3 мкг
Желтое тело свиньи Прогестерон 5 мкз
Семенники быков Тестостерон 0,1 мг
Произ-вом Г. занимается особая отрасль фармацев-
тич. пром-сти — эндокринная. Обычно железы экст-
рагируют в свежем виде непосредственно на мясоком-
бинатах либо доставляют их в замороженном состоя-
нии на эндокринные заводы, где они подвергаются
дальнейшей обработке. Иногда железы обезвоживают,
измельчают и хранят или перевозят их в виде сухих
порошков. В ряде случаев процесс выделения Г. из
желез настолько труден и малопроизводителен, что
Г. предпочитают получать синтетич. методами из
других природных веществ; так, тестостерон синте-
зируют в промышленном масштабе из холестерина,
кортизон получают многостадийным синтезом из сте-
роидных сапонинов или алкалоидов. В эндокринной
пром-сти нашли применение также и микробиологии,
методы синтеза (введение гидроксильной группы в
стероидный скелет с помощью специфич. микроорга-
низмов).
Г. применяют при различных нарушениях нормаль-
ных функций организма. Так, пептидные Г. — при
диабете (инсулин), при заболеваниях щитовидной
железы (тиреотропин), при нарушениях в женской
половой сфере (Г., стимулирующий рост фолликул,
лютеинизирующий Г., хорионгонадотропины). Неко-
торые пептидные Г. используют также для усиления
лактации (пролактин) и стимуляции родовой деятель-
ности (окситоцин). Фенилалкиламины применяют
при шоке, внезапной остановке сердца, падении кро-
вяного давления и пр. Особенно большое значение
имеют стероидные Г., к-рые используют не только
при нарушении соответствующих функций организма,
но и при лечении нек-рых форм рака (эстрадиол при
999
ГОРЧИЧНОЕ МАСЛО — ГОРЯЧАЯ ЛАБОРАТОРИЯ
1000
раке предстательной железы, тестостерон при раке гру-
ди), а также для лечения воспалительных процессов,
бронхиальной астмы и ревматоидных артритов (корти-
зон, кортизол). Нек-рые синтетич. препараты нашли
практич. применение, т. к. они, обладая высокой гор-
мональной активностью, вызывают меньшие побочные
явления, чем сами Г. (напр., кортизон и кортизол
успешно заменяют 9-фторкортизолом, а прогестерон —
1-дегидропрогестероном, т. н. преднизолоном).
Лит.: Назаров И. Н. и Бергельсон Л. Д.,
Химия стероидных гормонов, М., 1955; Физер Л. и Фи-
зер М., Химия природных соединений фенантренового ряда,
пер. с англ., М.—Л., 1953; Ч о у Хао Л и Белковые гормоны,
в кн.: Белки, под ред. Г. Нейрата и К. Бэйли, пер. с англ.,
т. 3, ч. 1, М., 1958; Гауровитц Ф., Химия и биология
белков, пер. с англ., М., 1953; Юдаев Н. А., Биохимия
стероидных гормонов коры надпочечников, М., 1956; Н а п 60.,
НогшоПе, 2 Aufl., Jena, 1959; В er sin Th., Blochemie der
Hormone, Lpz., 1959; Fieser L., Fieser M., Steroids,
N. Y.—L., 1959; Recent progress in hormone research, ed.
by G. Pincus, v. 1—15, N. Y., 1947—59; Vitamins and hormo-
nes, ed. by R. S. Harris and К. V. Thimann, v. 1 —15, N. Y.,
1943—57; см. также Стероидные аорлюны и статьи об отдель-
ных Г. Л. Д. Бергельсон.
ГОРЧИЧНОЕ МАСЛО — растительное жирное ма-
сло семян сизой (Sinapsis nigra), а также белой гор-
чицы (Sinapsis alba). Выход Г. м. (в пересчете на се-
мена) в зависимости от сорта и климатич. факторов
от 23,2 до 46,0%. В Г. м. в виде глицеридов содер-
жится до 50,2% эруковой к-ты, 25—28% олеиновой,
14,5—20% линолевой, до 2% пальмитиновой, стеа-
риновой, линоленовой и др. кислот (каждой); содер-
жание неомыляемых веществ до 3,3%. Т. заст. Г. м.
ок. —15°; плотн. 0,91—0,92; 1,468—1,476; вяз-
кость, по Энглеру, 17—18° (15°); число омыления 170—
183,8; иодное число 92—123; число Генера 94,2—96,7;
родановое число 80—86. Г. м. применяется в консерв-
ной, хлебопекарной и кондитерской пром-сти, в мыло-
варении и текстильном произ-ве, а также для изго-
товления нек-рых фармацевтич. препаратов.
Б. И. Хомутов.
ГОРЧИЧНЫЕ МАСЛА R—N=C=S—эфиры изотио-
циановой к-ты: аллиловое Г. м.
сн2=сн - сн3 - N=C—s
бутиловое Г. м.
н3с.
)CH-N=C=S
НзС-НзСГ
бензиловое Г. м. С6Н5СН2—N = C=S и др. Аллиловое
или обыкновенное Г. м. — главная составная часть
эфирного масла, получаемого из семян черной гор-
чицы, в к-рых оно содержится гл. обр. в виде глико-
зида синигрина.
Аллиловое Г. м. — бесцветная жидкость с резким харак-
терным запахом; т. пл. —80°; т. кип.: 150°/760 мм; 44°/12 мм;
d‘‘ 1,021; нерастворимо в воде, хорошо растворимо в спирте,
эфире, бензоле, петролейном эфире. Аллиловое Г. м. было
впервые получено Н. Н. Зининым в 1845.
Г. м. токсичны; их пары раздражают слизистые обо-
лочки, жидкие — вызывают образование пузырей на
коже.
При действии света Г. м. постепенно разлагаются.
При длительном перемешивании с водой или сопри-
косновении с металлами от Г. м. отщепляется сера
с образованием алкилцианидов R—CN; при восста-
новлении Г. м. превращаются в первичные амины и
тритиоформальдегид:
R — N=C=sbRNH3+(H:>CS)3
При нагревании с водой или кислотами Г. м. гидро-
лизуются; продукты гидролиза — сероокись углерода
и первичные амины. Г. м. присоединяют спирты и мер-
каптаны, переходя в тиоуретаны R—NH—CS—OR'
и дитиоуретаны R—NH—CS—SR'; с уксусной или
бензойной к-той образуют соответственно N-алкил-
ацетамиды и N-алкилбензамиды. Г. м. легко реа-
гируют с аммиаком, превращаясь в тиозинамины
RNH—CS —NH2, к-рые при действии AgNO3 да-
ют Ag2S (метод колич. определения Г. м.). Г. м. до-
бываются из жмыхов горчицы после отжатия жирного
Г. м. Синтетически Г. м. легко получают присоеди-
нением первичных аминов к CS2 с последующим от-
щеплением H2S действием солей тяжелых металлов:
/NHR пун,
S=C==S + RNH, — S= с/ ilia.
^SH
_.==C/NHR -H-s c^NR
4S-NHR
или перегонкой диалкильных производных тиомоче-
вины:
ZNHR
SC< — R-N=C=S + RNHj
XNHR
Г. m. обладают бактерицидным и инсектицидным
действием. Аллиловое Г. м. применяют в медицине
(«горчичный спирт») и в качестве антиоксиданта
В СМазОЧНЫХ маслах. М. Я. Алейникова.
ГОРЯЧАЯ ЛАБОРАТОРИЯ — лаборатория, пред-
назначенная для работы с радиоактивными препара-
тами высокой активности (до сотен тысяч кюри).
В Г. л. производят выделение плутония и других
трансурановых элементов, переработку тепловыделяю-
щих элементов ядерных реакторов и продуктов деле-
ния, исследование физич. и химич. свойств материа-
лов, обладающих высокой активностью, приготовле-
ние мощных источников излучения, радиохимия, очи-
стку изотопов, радиохимия, анализ и т. д. Основная
специфич. особенность Г. л. — необходимость про-
ведения работ при условии биохимия, защиты персо-
нала, помещения и окружающей местности от прони-
кающего радиоактивного излучения и загрязнения
радиоактивными веществами — аэрозолями, пылью,
жидкостями, парами и т. д. Опасность облуяения
персонала исключается благодаря хорошо разрабо-
танным системам защиты, дозиметрия, контроля, сиг-
нализации и автоблокировки. Группа токсияности
и класс Г. л. определяются степенью возможной
опасности работы (вид и энергия излучения, физич.
состояние источников, количество радиоизотопов и их
относительная токсичность и т. д.).
Современные Г. л. состоят из комплекса помещений,
построенных по зональному принципу убывающей
активности. Отдельные помещения изолированы друг
от друга стальными и бетонными стенами, снабжены
саншлюзамн и другими подсобными помещениями.
В Г. л. предусматриваются хранилища для радиоак-
тивных образцов, дистанционное управление их пере-
мещением с использованием специального подъемно-
транспортного оборудования, а также приспособле-
ния для своевременного удаления твердых, жидких
и газообразных отходов. Для защиты применяются
материалы высокой плотности: свинец, сталь, барито-
вый, магнетитовый и железофтористый бетоны, магне-
титовая руда и др. Толщина защитного устройства
зависит от количества излучающего материала и от
энергии излучения. Так, напр., толщина бетонных
стен, рассчитанных на работу с 10000 кюри при энер-
гии у-излучения 1 Мэв, составляет 130 см (см.Защита
от излучений радиоактивных веществ). Непроницае-
мые трубопроводы для передачи активных жидкостей
готовятся из нержавеющей стали, монель-металла,
поливинилхлорида и др.
Работа с высокими уровнями активности произво-
дится в изолированных блоках, снабженных отдель-
ными горячими камерами; управление
всеми операциями в таких камерах производится
дистанционно. Конструкция горячих камер зависит
от характера выполняемых в них операций, а также
от типа источника излучения. Обычно они пред-
1001
ГОРЯЧАЯ ЛАБОРАТОРИЯ — ГОРЯЧИЕ АТОМЫ
1002
ставляют собой герметичные помещения размером
5 X 2,5 X 5 м3, с закругленными внутренними
углами и полом, наклоненным в сторону водосточного
канала. Оборудование вносится через расположен-
ную в задней стене Дверь, конструкция к-рой обеспе-
чивает полную герметичность при невозможности
прострела у-излучением. Внутренние поверхности
горячих камер покрываются некорродирующими и
легко дезактивируемыми материалами, такими, как,
напр., нержавеющая сталь или специальные непори-
стые пластики. Система вентиляции характеризуется
высокой скоростью воздушного потока (1—2 м/сек),
обеспечивающего 10—30-кратный обмен воздуха при
общем направлении его движения из помещений
с меньшим возможным загрязнением в помещения
с большим возможным загрязнением. Выброс воздуха
во внешнее пространство производится через доста-
точно высокую трубу, снабженную системой эффектив-
ных фильтров. Система дистанционного наблюдения
за операциями, выполняемыми внутри горячей ка-
меры, должна обеспечить достаточно широкий угол
зрения, получение перспективной картины, сохра-
нение окраски, достаточно высокую разрешающую
способность, а также удовлетворять требованиям
безопасности. Принцип наблюдения основан на раз-
личии в степени пропускания видимого света и у-излу-
чения. Самым простым является наблюдение через
слой воды, если возможность такого наблюдения
допускается природой радиоактивных материалов и
характером применяемой аппаратуры. Однако этот
способ редко может быть использован. Обычно наблю-
дение ведется при помощи перископов через каналы
в стенах, направленные наклонно вверх. Для отсче-
тов показаний стрелочных измерительных приборов
используются бинокли и зрительные трубы. Разра-
ботаны системы микроскоп—перископ для наблюдения
за малыми объектами. Часто камеры снабжаются
одним или несколькими смотровыми окнами, пред-
ставляющими собой проемы в защитной стене, запол-
ненные прозрачным материалом, толщина слоя к-рого
обеспечивает столь же эффективную защиту, что и
слой основного защитного материала. В качестве
такого рода прозрачного материала используется
вода, нек-рые концентрированные водные растворы
(напр., 75%-ный р-р ZnBr2, плотность к-рого равна
2,5—2,7), стекла различного состава в виде полиро-
ванных листов или литых блоков и др.; наиболее
часто употребляются свинцовые стекла, стабилизи-
рованные церием (плотн. 6,2). В современных Г. л.
все более широкое применение получают методы на-
блюдения за процессами внутри горячих камер при
помощи телевизионных установок.
Электрич., газовые, канализационные и др. комму-
никации внутри горячих камер заделываются в сте-
нах. Активные образцы поступают в камеры через
отдельные каналы, закрывающиеся специальными
пробками. Задача дистанционного управления в зоне
высокой активности решается либо путем применения
специальных механизмов, приспособленных для управ-
ления одной или небольшим числом операций, либо
использованием универсальных манипуляторов с ме-
ханич., электрич. или гидравлич. приводом. Мани-
пуляторы в зависимости от их сложности позволяют
либо захватывать и перемещать предмет в заданном
направлении, либо одновременно при передвижении
поворачивать предмет под любым углом. Для перво-
начальной дезактивации аппаратуры и внутренних
частей горячей камеры до допустимых значений загряз-
ненности используются манипуляторы специальных
конструкций. Окончательная дезактивация аппара-
туры производится вне рабочих помещений. Высоко-
активные жидкие отходы сливаются через водослив
в сменные баки из нержавеющей стали и перено-
сятся после заполнения в бетонные ямы, а затем
вывозятся.
Каждая горячая камера обычно предназначена для
одного определенного вида работ и оснащается соот-
ветствующим оборудованием. Так, напр., камеры для
металловедческих исследований имеют шлифовальные
станки, пирометры, рентгеновские установки, при-
боры для определения твердости, теплопроводности
и т. д. Камеры для химич. переработки активного
сырья и изучения поведения радиоактивных элемен-
тов снабжены стандартной химич. аппаратурой, по-
зволяющей готовить растворы заданной активности,
выполнять операции взвешивания, осаждения, фильт-
рования, экстракции и др.
Для работы в Г. л. с средним и низким уровнем
активности требования к защите и к степени обмена
воздуха соответственно уменьшаются и работа про-
водится согласно правилам, принятым в радиохимия,
лабораториях.
Лит.: Яковлев Г. Н. |и др.], Горячая аналитическая
лаборатория, М., 1955; Горячие лаборатории и их оборудо-
вание, |пер. с англ.], М., 1960; Организация работ с приме-
нением радиоактивных веществ, М., I960; Ядерные реакторы,
пер. с англ., т. 1—3, М., 1956—57 (Материалы комиссии по
атомной энергии США); Санитарные правила работы с радио-
активными веществами и источниками ионизирующих излуче-
ний, М., 1960; см. также, Материалы Международных кон-
ференций по мирному использованию атомной анергии, со-
стоявшихся в Женеве в 1955 и 1958 гг. К. Б. Заборенко.
ГОРЯЧИЕ АТОМЫ — атомы, возникшие в ре-
зультате ядерных превращений. Каждое ядерное
превращение сопровождается выделением энергии,
к-рая распределяется между ядром, претерпевающим
превращение, и испускаемой частицей, в соответст-
вии с законом сохранения импульса. Образовавшиеся
возбужденные атомы наз. Г. а., т. к. их энергия соот-
ветствует энергии атомов, «нагретых» до миллионов
градусов. Их наз. также атомами отдачи,
поскольку они воспринимают кинетич. энергию отдачи
материнского ядра. При испускании а-частицы энер-
гия атома отдачи достигает 10 000 эв, а для реакций
типа (п, у) — 100 эв. В момент образования Г. а.
может потерять большее число электронов, чем следует
ожидать по его положению в периодич. системе и обра-
зовать высокозарядный ион. Так, напр., при изомер-
ном переходе Вт80, образующийся ядерный изомер
имеет заряд, равный +10. Наряду с большой кинетич.
энергией для Г. а. характерно возбужденное элект-
ронное состояние.
Благодаря высокой кинетич. энергии, возбужден-
ному электронному состоянию и высокому положи-
тельному заряду Г. а. способны вступать в такие
химич. реакции, в к-рые обычные атомы не вступают.
Полученный Г. а. импульс в большинстве случаев
бывает достаточно велик, чтобы разорвать одну или
несколько связей атома в химич. соединении; при
этом Г. а. отрывается от содержащей его молекулы.
Энергия образовавшегося Г. а. (или горячего ради-
кала) в свою очередь достаточна, чтобы вызвать
возбуждение и диссоциацию еще нескольких моле-
кул. Через несколько последовательных столкнове-
ний кинетич. энергия Г. а. снижается и Г. а. всту-
пают в разнообразные химич. реакции с молекулами
или радикалами исходного соединения или раствори-
теля, что сопровождается микросинтезом новых со-
единений или возвратом Г. а. в молекулу исходного
соединения. Отношение количества Г. а., стабили-
зовавшихся в форме материнского вещества или
вообще других молекул, к общему количеству возник-
ших Г. а. наз. удержанием. При оценке по-
ведения Г. а. необходимо принимать во внимание
возможные процессы изотопного обмена, в результате
к-рых достигается равнораспределение Г. а. между
всеми химич. формами, содержащими данный атом.
Г. а. находят все большее применение при синтезе
1003
ГОРЯЧИЕ ЧАСТИЦЫ — ГРАМИН
1004
меченых' соединений. На свойствах Г. а. основан ме-
тод обогащения радиоизотопов (см. Сцилларда — Чал-
мерса аффект}. Перспективно использование реак-
ций Г. а. в процессах полимеризации, синтеза аммиа-
ка и др.
Лит.: Несмеянов Ан. Н., Борисов Е. А.,
Усп. хим., 1958, 27, вып. 2, с. 133; Мурин А. Н., Н е-
федов В. Д., Барановский В. И., П о по в Д. К.,
Усп. хим., 1957, 26, вып. 2, с. 164; Использование радиоактив-
ности при химических исследованиях [под ред. А. Валь и
Н. Боннера], пер. с англ., М., 1954, гл. 8; Радиохимия и
химия ядерных процессов, под ред. А. Н. Мурина и др., Л.,
1960. К. Б. Заборенко.
ГОРЯЧИЕ ЧАСТИЦЫ — атомы или свободные
радикалы, обладающие энергией, превышающей энер-
гию теплового движения. Горячие атомы, или атомы
отдачи, образуются при ядерных реакциях; горячие
свободные радикалы — при фотолизе, радиолизе и в
элцктрич. разряде. Избыточная энергия может иметь
величину от неск. эв до сотен тысяч эв. При столк-
новениях энергия Г. ч. либо рассеивается, либо расхо-
дуется на химич. превращения, причем в этих усло-
виях могут протекать и такие реакции, к-рые для
обычных частиц термодинамически невозможны.
ГОФМАНА РЕАКЦИЯ — превращение амидов ки-
слот в амины с потерей одного атома углерода при
I
RCH(OH)CONH2------ iRCH(OH)NH2 } ----- RCHO
* i
rch=chconh2-
RCH=CHNHCOOCH3
RCHjCHO
rc = cconh2
rc=cnh2 }
RCH2C=N
/NHCOR'
RCH
4CH2CONH2
обработке амида бромом и щелочью: RCONH, +
+ Br2 +OH-->RNH2 + СО2 + Вг- + Н2О. В спир-
товом р-ре образуются уретаны. Г. р. можно получать
с хорошими выходами алифатич., жирноароматич.,
/NHCOR’
RCH
XCH2-N=C=O
RCONH2 +
CH2-N(CH3)s
+ CH3J + KCN
ароматич. и гетероциклич. амины, диамины и амино-
кислоты. Амиды а-окси- и а, Р-ненасыщеяных к-т
в условиях Г. р. превращаются в альдегиды; амиды
замещенных пропиоловых к-т — в нитрилы; амиды
Р-ациламинокислот->-в ацилглиоксалидоны (см. фор-
мулы выше).
Первой стадией Г. р. является образование N-гало-
генамида, дающего под действием щелочей нестойкую
соль (I):
RCONHa + KOBr — RCONHBr К-— [RCONBr]~K+
Скорость Г. р. определяется превращением сильно-
основного аниона (I) в менее основной, стабильный
анион X и промежуточное соединение (II) с электрон-
ным секстетом у атома азота, стабилизирующееся
перегруппировкой в изоцианат:
[RCONX] - — RCON + X - — RN=C=O
I II
Такой механизм реакции подтверждается возможно-
стью перегруппировки, исходя из заведомого N-гало-
генамида, и выделением изоцианатов — первых ста-
бильных продуктов в апротонных условиях. При
переходе от карбонильного углерода к одновалент-
ному азоту карбанион не отщепляется в виде кинети-
чески независимой частицы, что доказывается сохра-
нением конфигурации в случае асимметрия, мигрирую-
щего радикала:
£-2^5 х
CHCONH2 ----СН —NH2
Н-СдНд^ Н.С4Н9
Интрамолекулярный хар актер перегруппировки под-
тверждается также сохранением оптич. активности
3,5-динитро-2-(а-нафтил)-бензамида при Г. р. Реакция
открыта в 1881 А. В. Гофманом.
Лит.: Уэллис Э. С., Лэн Дж. Ф., в кн.: Орга-
нические реакции, сб. 3, пер. с англ., М., 1951, с. 255; Р о-
дионов В. М., Зворыкина В. К., Кожев-
никова Н. Е., Изв. АН СССР. Отд. хим. наук, 1956,
№ 4, с. 491; Franzen V., Chemlker—Ztg, 1956, 80, № 1, S.8.
Н. II. Гамбарян.
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ — см. Бесовой
анализ.
ГРАМИН (3-диметиламинометилиндол) CUH14N,
мол. в. 170,24 — кристаллы; т. пл. 138—139°; иден-
тичен алкалоиду донаксину, выделенному из
одного из видов камыша Arunao donax; содержит-
ся также в ростках ячменя и в некоторых других
растениях. Грамин (I) обычно получают по Манниха
реакции из индола, формальдегида и диметиламина:
Г. реагирует по третичной алифатич. аминогруппе с
образованием солей с кислотами и солей четвер-
тичных аммониевых оснований (иодметилат; т, пл.
RCH—N—COR'
I xc=0
CH2—NH
176—177°). Г. является ключевым сое-
динением в синтезе триптамина, трип-
тофана, индолилуксусной кислоты (.ге-
тероауксина) и других физиологически
активных соединений индольного ряда.
При нагревании Г. или его четвер-
тичной соли с KCN в воде образуется
3-цианометилиндол, от которого можно
перейти к триптамину или к 3-индолил-
уксусной кислоте:
Г. и его четвертичные соли легко вступают в конден-
сацию с соединениями, содержащими активную мети-
леновую или метинную группу (малоновый эфир и его
производные, нитралканы, этиловый эфир цианук-
сусной к-ты и др.). Примером может служить синтез
триптофана из четвертичной соли Г. и ацетаминомало-
нового эфира:
CH2-N(CH3)2 • RX
СН(СООС2Н5)2 ь
NHCOCH3
а—рСН2-С(СООС2Н5)2
N> NHCOCH3
н
омыление и
декарбокси-
лиривание
сн2-сн-соон
NH3
1005
ГРАМИЦИДИНЫ — ГРАММ-ЭКВИВАЛЕНТ
1008
Лит.: Орехов А. Пи Химия алкалоидов, М., 2 изд.,
1955, с. 562; Джулиен П., Мейер Э., П р и и т и Э.,
в кн.: Гетероциклические соединения, под ред, Р. Эльдер-
филда, пер. с англ., т. 3, М., 1954, с. 38; Snyder Н. R.,
Pilgrim F. J., J. Amer. Chem. Soc., 1948, 70, № И,
p. 3770. M. И. Преображенская.
ГРАМИЦИДИНЫ — антибиотики-полипептиды,
продуцируемые различными штаммами Вас. brevis.
В зависимости от состава питательной среды и усло-
вий развития Вас. brevis образуются антибиотики,
сходные по строению, спектру антимикробного дейст-
вия и различающиеся по числу и характеру амино-
кислотных остатков в молекуле. В группу Г. вклю-
чают грамицидины Си J, тироцидины А, В и С и гра-
мицидин Дюбо. Последний правильнее относить к ан-
тибиотикам смешанного состава, т. к. в его состав
входит также 2-этаноламин. Ниже приведен амино-
кислотный состав нек-рых антибиотиков группы Г.
Число аминокислотных
остатков, входящих в:
Аминокислоты
L-Валин .................... 2
L-Орнитин................... 2
D-Орнитин .................. —
L-Лейцин.................... 2
D-Лейцин.................... —
L-Фенилаланин .............. —
D-Фенилаланин .............. 2
L-Пролин.................... 2
L-Глутаминовая кислота .... —
L-Аснарагиновая кислота ... —
L-Триптофан ................ —
L-Тирозин................... —
1
1
1
1
1
1
1
Для грамицидинов С и J установлено циклическое
строение.
Грамицидин С выделен Г. Ф. Гаузе и
М. Г. Бражниковой в 1942. Из спирта или ацетона
антибиотик кристаллизуется в виде бесцветных кри-
сталлов (пластинки или ущербленные призмы),
т. пл. 245—247° (по другим данным, 256—258°),
Н3С^ сн3 nh2 СН(СН3)2
СН (СН2)3 СН2 CH2-Cfih6
I I I I 7---1
NH-CHCONHCHCONHCHCONHCHCO--------n5
со D V
г-\ в 9°
j__/N—CONHCHNHCOCHNHCOCHNHCOCHNH
СН2 СН2 (СН2)3 C^i
С6Н5 ch(ch3)2nh2 н3с сн3
274,8° (из спирта), [а]р ~ —295° (из 70%-ного
спирта); т. пл. хлоргидрата 268—270° (по другим
данным, 280—282°), пикрата 226—227°; мол. вес хлор-
гидрата: вычислен 1212; экспериментально 1060—1340,
1140, 1019. Хлоргидрат растворим в спирте, ацетоне,
хлороформе, 75%-ном диоксане (водном) и бензине;
трудно растворим в воде (1 : 500), нерастворим в ми-
неральных к-тах и щелочах, эфире.
Развитие штамма грамицидина С протекает поверхностно
на мясном или казеиновом гидролизате при 40° в течение 6 су-
ток. После того как содержание грамицидина С достигает
2 г/л, нативный раствор подкисляют до pH 4,5—5,0, осадок
антибиотика просушивают и экстрагируют спиртом. Для
получения кристаллич. грамицидина С спиртовый экстракт
разбавляют водой, экстрагируют неактивные примеси эфиром,
а водно-спиртовую часть концентрируют; выпадает кристаллич.
солянокислая соль, к-рую перекристаллизовывают из спирта
или ацетона с животным углем. В 1956 грамицидин С был син-
тезирован (Швицер и Зибер).
Грамицидин С весьма активен против грамположительных
бактерий, значительно слабее действует на грамотрицательные
бактерии, отмечена активность против нек-рых патогенных
грибов и простейших. При концентрации 1 .икг/мл он подав-
ляет рост стафилококков, в концентрациях 1—50 мкг/мл —
рост стрептококков, пневмококков и нек-рых грамотрицатель-
ных бактерий кишечной группы. При парентеральном (не
через пищеварительный тракт) введении грамицидин С про-
являет сильное токсич. действие. Из-за плохой растворимости
в воде он не всасывается из желудочно-кишечного тракта
и применяется как местное средство. Грамицидин С широко
применяется при лечении гнойных процессов, в хирургии,
в акушерско-гинекологич. практике, для лечения язвенных
поражений слизистой оболочки при дизентерии. Грамицидин С
характеризуется высокой устойчивостью: сухой кристаллич.
препарат не теряет активности после нагревания до 160°,
активность сохраняется также при нагревании водных р-ров
в автоклаве при 130° или кипячении с 1%-ной НС1 в течение
часа; весьма устойчив к нагреванию с 25%-ной H2SO4. Произ-
водные грамицидина С с салициловой, п-аминосалициловой,
никотиновой и нек-рыми другими к-тами обладают активностью
против туберкулеза и нетоксичны.
Грамицидин J выделен в 1954 японскими исследо-
вателями (Отами) (аминокислотный состав см. в таблице).
Синтезирован грамицидин J2, представляющий собой цик-
логексапептид, т. пл. 279—280°, [а]р4=—313,1° (из спирта).
Грамицидин Дюбо выделен в 1939 Р. Дюбо из
тиротрицина, к-рый оказался смесью тироцидинов и Г. (см.
табл.). Грамицидиновая фракция была разделена на 3 поли-
пептида — грамицидины А, Ви Ср; в наибольшем количестве
содержится грамицидин А, тонкие листочки (из ацетопа),
т. пл. 227—228°; т. пл. грамицидина В 258—259°. Из продуктов
частичного гидролиза грамицидиновой фракции выделены
П-валил-Ь-валин, Ь-валил-Б-валин, L-валил-глицин, D-лей-
цил-глицин, Ь-аланил-Б-валин и L-аланил-Б-лейцин. При
полном гидролизе выделены D-лейцин, L-триптофан, D-
и L-валин, L-аланин глицин и 2-аминоэтанол, приблизительно
в соотношении 6 : 6 : 5 : 3 : 2 : 2. В состав грамицидина А
входят остатки валина, лейцина, триптофана, аланина и гли-
цина, в состав грамицидина В наряду с указанными амино-
кислотами — фенилаланин, а в молекулу грамицидина Ср —
остаток тирозина.
Лит.: Шемякин М. М., Хохлов А. С., Химия
антибиотических веществ, 2 изд., М.—Л., 1953, с. 446, 570;
Гаузе Г. ф.. Лекции по антибиотикам, 3 изд., М., 1958;
Schwyzer R., Chimia, 1958, 12, № 2, S. 53; Schwy-
z e r R. und Sieber P., Helv. chim. acta, 1957, 40, S. 624;
Angew. Chemie, 1956, 68, № 16, S. 518; Noda Y., J. Chem.
Soc. Japan. Pure Chem. Soc., 1959, 80, № 4, p. 411; Зна-
менская M. П. и Белозерский A. H., Антибиотики,
1957, 2, № 1, c. 36; Силаев А. Б. и Степанов В. M.,
ДАН СССР, 1957, 112, № 2, с. 297. А. Б. Силаев.
ГРАММ-АТОМ — такое количество данного эле-
мента, вес к-рого при выражении в граммах численно
равен атомному весу этого элемента. Напр., при ат.
весе железа, равном 55,85, один Г.-а. железа весит
55,85 г. В одном Г.-а. любого химич. элемента содер-
жится одинаковое число атомов (Авогадро число,
равное 6,0232-IO23 атомов для химич. шкалы атом-
ных весов). Величины Г.-а. имеют большое значение
при химических и физических вычислениях. В технич.
расчетах применяются также величины килограмм-
атом и тонна-атом, напр. 1 кг-атом железа равен
55,85 кг.
ГРАММ-МОЛЕКУЛА (грамм-моль, моль) — такое
количество данного вещества, вес к-рого при выра-
жении в граммах численно равен мол. весу этого ве-
щества. Напр., при мол. весе H2SO4, равном 98,082,
одна Г.-м. серной к-ты составляет 98,082 г. Г.-м. лю-
бых индивидуальных веществ содержат одинаковое
число молекул (Авогадро число, равное 6,0232-1023
молекул для химич. шкалы атомных весов). Для ве-
ществ, находящихся в газообразном состоянии в усло-
виях, когда применимы законы идеальных газов,
Г.-м. различных веществ при одинаковой темпера-
туре и одинаковом давлении занимают равные объемы
(см. Авогадро закон). При нормальных условиях (0°С
и давлении 760 мм рт. ст.) объем Г.-м. идеального газа
равен 22,415 л. Условно можно говорить и о Г.-м.
смеси; так, Г.-м. воздуха принимается равной 29 г.
В технич. расчетах применяются килограмм-молекула
и тонна-молекула.
ГРАММ-ЭКВИВАЛЕНТ — такое количество дан-
ного химич. элемента или соединения, вес к-рого
при выражении в граммах численно равен их экви-
валентному весу (см. Эквивалент химический). Г.-э.
1007
ГРАНОЗАН — ГРИНЬЯРА РЕАКЦИЯ
1008
химического элемента равен частному
от деления грамм-атома элемента на его валентность
в данном соединении. Напр., Г.-э. 2-валентного
железа (ат. в. 55,85) 27,92 г, 3-валентного железа
18,62 г. Г.-э. кислоты — количество данной к-ты,
содержащее один Г.-э. водорода, способного заме-
щаться металлом с образованием соли; равен част-
ному от деления грамм-молекулы к-ты на ее основ-
ность. Г.-э. основания — количество данного
основания, содержащее один Г.-э. ОН-групп. Г.-э.
соли — количество соли, полученное в результате
взаимодействия одного Г.-э. кислоты с одним Г.-э.
основания; находится делением грамм-молекулы соли
на сумму валентностей всех атомов металла, входящих
в ее молекулу. Понятие «Г.-э.» широко применяется
в объемном анализе. О значении Г.-э. в окислительно-
восстановительных процессах см. Окисление — восста-
новление.
ГРАНОЗАН (препарат НИУИФ-2) — протравитель
семян различных растений; механич. смесь, состоя-
щая из 2—2,5% этилмеркурхлорида (C2H5HgCl,
белые кристаллы, т. пл. 192,5°), 96—97% талька
и 0,6—1,2% минерального масла. Г. — порошок
белого, серого или желтоватого цвета. Действующим
началом препарата является этилмеркурхлорид. Г. по-
лучают смешением этилмеркурхлорида с тальком и
маслом в шаровой мельнице или размольно-смеси-
тельном агрегате иного типа. Г. используется для
протравливания семян (1—4 кг на тонну семян),
напр. для протравливания семян пшеницы против
фузариоза 1—2 кг/т, масличных культур (арахис,
перилла, клещевина, ляллеманция) 1—4 кг/т и т. д.
Г. эффективен в борьбе с большинством заболеваний
растений, возбудители к-рых находятся на поверх-
ности зерна, и неэффективен против возбудителей,
находящихся внутри семян (напр., пыльная головня
пшеницы и ячменя).
Лит.: Мельников Н. Н. [и др.], в кн.: Органиче-
ские инсектофунгициды, М., 1955, с. 188—200; Попов П. В.,
Справочник по ядохимикатам, М., 1956. Н. Н. Мельников.
ГРАФИТ — см. Углерод.
ГРАФИТОВАЯ КИСЛОТА — см. Бензолполикар-
боновые кислоты.
« ГРАФТ» -СОПОЛИМЕРЫ — см. Сополимеризация.
ГРЕВЕ — УЛЬМАНА СИНТЕЗЫ — термич. разло-
жение 1-фенил-1,2,3-бензотриазола (I) с образованием
карбазола (II) и выделением азота при нагревании
примерно до 360°. Впервые синтез был проведен
по схеме:
Аналогично могут также превращаться замещенные
о-аминодифениламина с образованием соответствую-
щих замещенных карбазола; с хорошими выходами
реакция протекает и в том случае, когда заместите-
лями являются алкил- или аминогруппа. В случае
отрицательных заместителей (NO2, СН3СО, CN) в
о-аминодифениламине выход замещенных карбазола
сильно понижается. Несмотря на то, что производные
о-аминодифениламина трудно доступны, синтез кар-
базола по Гребе — Ульману используется и в настоя-
щее время (см. также Борте синтез). Синтез впервые
проведен в 1896 К. Гребе и Ф. Ульманом.
Лит.: Гетероциклические соединения. [Сб. статей], под
ред. Р. Эльдерфилда, пер. с англ., т. 3, М., 1954, с. 235.
В. И. Гунар.
ГРЕМУЧАЯ КИСЛОТА (карбилоксим) C^iV-OH,
мол. в. 52,020 — существующая только в р-рах кислота,
изомерна циановой кислоте. Получают осторожным
подкислением водного раствора фульмината нат-
рия (NaONC) сероводородной водой при низких тем-
пературах. Г. к. легко полимеризуется с выделением
теплоты. Для Г. к. характерны реакции присоедине-
ния, например:
С = NOH % НС1 —' C1CH—NOH
— нсоон + H2NOH • НС1
С NOH + HsS — HSCH=NOH — HSCN % HSO
C NOH + Br3 — Br-C=NOH
Соли Г. к. взрывчаты и очень ядовиты. Подобно HCN,
Г. к. образует комплексные соли, напр. Na4[Fe(ONC)eJ-
18Н2О. Двойные соли Г. к. со щелочными и щелочно-
земельными галогенидами типа Hg(ONC)2 • NaCl при
нагревании сгорают, не взрываясь. Из солей имеют
значение гремучая ртуть Hg(ONC)2 и гремучее сереб-
ро AgONC. ф. к. Величко.
ГРЕМУЧАЯ РТУТЬ (фульминат ртути) Hg(ONC)2—
ртутная соль гремучей к-ты, инициирующее взрывчатое
вещество; бесцветные или серые кристаллыорторомбич.
системы; плотн. 4,307. Г. р. плохо растворима в воде,
лучше — в пиридине, этаноламине, а также в водных
р-рах KCN и NH3; теплота взрыва 350 ккал/кг; ско-
рость детонации 5400 м/сек (при d ок. 4). Горение
спрессованной под небольшим давлением Г. р. легко
переходит в детонацию. Возбудить детонацию в сильно
спрессованной Г. р. труднее. При увлажнении чув-
ствительность Г. р. к тепловому и механич. импульсу
сильно понижается. При нагревании Г. р. разла-
гается довольно быстро (при 80° — в течение не-
скольких дней). Т. всп. ок. 170°. Влажная Г. р.
бурно реагирует с Mg и А1 (иногда со взрывом),
медленнее с Zn. Кислоты и щелочи разлагают Г. р.,
конц. H2SO4 — со взрывом. Г. р. получают взаи-
модействием этилового спирта с р-ром азотноки-
слой ртути в HNO3; применяют в капсюлях-дето-
наторах, капсюлях-воспламенителях и т. д. В по-
следнее время Г. р. все более вытесняется другими
инициирующими взрывчатыми веществами, гл. обр.
азидом свинца.
Лит.: Бубнов П. Ф., Инициирующие взрывчатые
вещества, М., 1940. Б. Н. Кондриков.
ГРИНЬЯРА РЕАКЦИЯ — совокупность методов
синтеза органич. соединений с помощью алкил-
или арилмагнийгалогенидов. Начало применению Mg
в органич. синтезе было положено в 1899 П. Барбье.
Его ученик В. Гриньяр в 1900 усовершенствовал
метод, разделив реакцию на две самостоятельные
стадии: 1) образование смешанного магнийгалоген-
органич. соединения RMgX в эфирной среде: RX +
+ Mg —> RMgX; 2) взаимодействие RMgX с соеди-
нением, содержащим карбонильную группу, приво-
дящее к образованию новой углерод — углеродной
связи. RMgX получаются с лучшими выходами, чем
применявшиеся ранее для аналогичных целей цинк-
органические соединения; они оказались более удоб-
ными в препаративном отношении и нашли широкое
применение. RMgX очень реакционноспособны; они
реагируют с соединениями, содержащими активные
атомы водорода: с водой, спиртами, первичными и
вторичными аминами и др. Если применять CH3MgJ,
количественно выделяется СН4, напр. CH3MgJ +
+ ROH —>-СП4 + ROMgX, что позволило исполь-
зовать эту реакцию для определения активного водо-
рода. При реакции RMgX с галогеналкилами или
-арилами образуются углеводороды: RMgX + R'X—>
—>RR' + MgX2 (наряду c RR и R'R'). Вторичные
спирты получаются действием RMgX на альдегиды:
R'CHO + RMgX -► RR'CHOMgX RR'CIIOII.it.tii
на эфиры муравьиной кислоты: HCOOR + 2R'MgX —>
-► RR'CHOMgX + R'OMgX -22 R'CHOH + ROH. Из
формальдегида получаются первичные спирты, к-рые
1009
ГРИНЬЯРА РЕАКЦИЯГУАНИДИН
1010
образуются также действием RMgX на этиленхлор-
гидрин или окись этилена:
RMgX + СН2-СН2 — RCH.CH.OMgX И'-’ RCH.CH.OH
Для получения третичных спиртов Г. р. проводят
с кетонами: RCOR' + R"MgX —> RR'R"COMgX
—>• RR'R"COH + Mg(OH)X или со сложными
эфирами: RCOOR' + 2R"MgX RR"2COMgX -f-
+ R'OMgX RRJCOH + R'OH + Mg(OH)X. Г. р.
с ангидридами, хлорангидридами, амидами кислот
или чаще с нитрилами используется для синтеза
кетонов:
RCN + R'MgX — RC(=NMgX)R'
JN Н4Ы
— RC(=NH)R' --II^°— RCOR' 4- NH,
кислота
При действии СО2 на RMgX образуются карбоновые
к-ты: RMgX + СО2 -► RCOOMgX RCOOH.
Как показал Караш, добавка малых количеств
хлоридов металлов VIII группы, в особенности СоС12,
к реакционной смеси полностью изменяет направле-
ние реакции. Так, бензофенон с CH3MgBr в присут-
ствии 2 мол. % СоС12 вместо обычного нормального
продукта гетеролитич. реакции — метилдифенилкар-
бинола — образует бензпинакон:
(СвН5)2С(ОН)С(ОН)(СвН5)2
Это изменение происходит в итоге перевода реакции
на гомолитич. цепной механизм за счет образования
неустойчивого RCOC1’, гомолитически распадающегося
по схеме: RCOC? —> R • + COCI'. Радикал СОСР
играет роль переносчика цепи; при действии кетона
превращается в С0С12 за счет восстановления кето-
на в радикал кетил, к-рый далее димеризуется в
пинакон:
R'
I
2СоС1- + R'R'C =0 + MgClBr —► 2R'-C • —
OMgBr
BrMgO OMgBr
I I
— R1 — C—C-----R'+2COCb
I I
R' R1
Подобным образом в простых эфирах происходит
разрыв кислород-углеродной связи с образованием
углеводородов. RMgX нашли широкое применение
в синтезе элементоорганич. соединений. Этим путем
могут быть получены органич. соединения Au, Hg,
Be, Cd, В, TI, Si, Ge, Sn, Pb, P, As, Sb, Bi, S, Se, Те,
напр.:
SIC14 + CH3MgBr — (CH3)jSi
SnCl4 + C0H5MgBr — (C„H5)sSn
C3H8PC12 n-C8H7MgBr —> C0H3P(n-C3H7)2
AsCl3 -J- C.H5MgBr — (C2H5)3As
(CH3O)3B + C8H8MgBr — C.H5B(OCH3)3 — C0H5B(OH).
HgBr2 + CHsMgBr — CH3HgBr и т. д.
В 1954 Норман предложил применять в качестве
среды для получения RMgX тетрагидрофуран; это
позволило ввести в реакцию с магнием многочислен-
ные галогенопроизводные с винильными радикала-
ми (напр., СН2=СНС1), не реагирующие с Mg в
эфирном растворе. Г. р. представляет собой один из
наиболее универсальных синтетич. методов органич.
химии.
Лит.: Рунге Ф., Магнипорганическпе соединения,
пер. с нем., Л., 1937; ИоффеС. Т. и Несмеянов А. Н.,
Справочник по магнийорганическим соединениям, т. 1—3,
М.—Л., 1950; Иоффе С. Т., Усп. хим., 1951 20, вып.
&, с. 621; его же, там же, 1958, 27, вып. 8, с. 1010; Bar-
bier Р Ц., Compt. rend. Acad, sci., 1899, 128, №2, p. 110;
Grignard V., там же, 1900, 130, № 20, p. 1322; Nor-
mant H., там же, 1954, 239, № 22, p. 1510; Kharasch
M. S., Re i mu th O., Grignard reactions of nonmetallic
substances, N. Y., 1954. С. T. Иоффе.
ГРИССА РЕАКТИВ — раствор сульфаниловой к-ты
и а-нифтиламина в разб. уксусной к-те. С нитрит-
ионом Г. р. дает красное окрашивание. При этом
сульфаниловая к-та (1,4-аминобензолсульфокислота)
диазотируется азотистой к-той, а образовавшееся
диазосоединение с а-нафтиламином образует азокра-
ситель красного цвета:
H2N-C0H4-SO3H + NaNO» + 2СН3СООН —
— HSO3-CeH4-N=N- ООССНз + CH3COONa + Н.О
HSO8-C8H4-N=N-OOCCH8 + a-C10H7NH2 —
— HSO8-C8H4-N=N-C10H8NH2 + CHjCOOH
Реакцию обнаружения нитрит-иона проводят в ней-
тральном или уксуснокислом р-ре. Открываемый
минимум — 0,01 мкг NO3~; предельное разбавление
1:5- 10е. Сильные окислители разрушают реактив
п мешают этой реакции. В присутствий Fe3+ необхо-
димо предварительно прибавить NaF или винную
к-ту. Применяют Г. р. для открытия и фотометрия,
определения нитритов и HNO2. При отсутствии иона
NO7 Г. р. может быть применен для открытия иона
NO., после восстановления его цинковой пылью
в присутствии уксусной к-ты. Г. р. следует приготов-
лять точно по прописям, приведенным в литературе.
Лит.: К л я ч к о Ю. А., Шапиро С. А., Курс
химического качественного анализа, М., 1960, с. 591.
И. П. Ефимов.
ГРОТТУСА — ДРЕЙПЕРА ЗАКОН — см. Фото-
химия.
ГРУППОВОЙ РЕАКТИВ — реактив, образующий
с большим числом неорганич. ионов или определен-
ными классами органич. соединений характерные про-
дукты реакции (осадок, газ, растворимые окрашенные
продукты). Напр., сульфид аммония (NH4)2S яв-
ляется Г. р. для катионов Al, Or, Fe, Mn, Zn, V, Се,
Ni, Со, Be, Ti, Zr, Th и U; он образует с перечислен-
ными катионами осадки, нерастворимые в воде.
Г. р. широко используются для выделения определен-
ных групп ионов из смеси, концентрирования следов
в-в, отделения компонентов, мешающих дальнейшему
ходу анализа, в систематич. анализе и т. д. В послед-
нем случае отношение ионов к определенным Г. р.
лежит в основе аналитич. классификации ионов (см.
Качественный анализ). Г. р. следует отличать от
селективных реактивов, к-рые дают характер-
ные продукты реакции с небольшим числом (3—5)
ионов.
ГУАНИДИН (NH2)2C=NH, мол. в. 59,07 — бесцвет-
ная кристаллич. масса, расплывающаяся на воздухе
вследствие поглощения влаги и СО2 из воздуха; т. пл.
ок. 50°; сильное однокислотное основание, близкое
по силе к гидроокисям щелочных металлов. С кисло-
тами Г. образует устойчивые соли; ниже приведены
свойства нек-рых солей Г.:
Соль Т. пл., °C J30 Растворимость в г/100 г растворителя pH го (2 1% водно-
F -ра °) 4%
вода спирт 78° бензол 60°
20° 55°
Хлоргидрат 184 1,344 215 320 67 <0,05 6,4
Нитрат . . 214,2 1.436 15 47 13 < 0,05 5.7 —
Карбонат . 241 1,521 45 62 <0,1 < 0,05 — 11,2
Перечисленные соли легко растворимы в воде, нерас-
творимы или трудно растворимы в ацетоне и гексане.
Соли Г. и сильных к-т стабильны в кипящей воде.
Свободный Г. выделяется из солей лишь специаль-
ными приемами, напр. действием Ва(ОН)2 на карбо-
нат. При гидролизе Г. образуются последовательно
мочевина и аммиак. Г. легко алкилируется и ацили-
1011
ГУАНИЛОВАЯ КИСЛОТА — ГУДРОН
1012
руется; аналогично мочевине конденсируется с СН2О
с образованием синтетйч. смол; конденсацией с
R-C^C-R,
N N
нек-рыми бифункциональными соединениями полу-
чаются гетероциклич. производные, напр. с р-дике-
тонами образуется соединение (I), с Р-динитрилами—
(II). В пром-сти Г. получают из дициандиамида:
H2NC-NHCN 551^2 H.NC-NHC—NH3X —-iS.
3SIH Ан NH
— 2(NH2)sC=NH.HX
где X—Cl или NO3. Процесс проводится в автоклаве
при 160° в присутствии безводного NH3. Г. является
промежуточным продуктом при получении пиримиди-
нов, оксазолов и др. Соли Г. применяются в пром-сти:
динитрат — в качестве взрывчатого вещества, фос-
фат — в текстильной пром-сти для придания огне-
упорных свойств тканям, карбонат — в синтезе по-
верхностно-активных веществ. Продукт конденсации
Г. с формальдегидом используется как ионнообменная
смола (см. Иониты). Г. токсичен; его концентрирован-
ные растворы вызывают сильное местное раздраже-
ние КОЖИ. А. ф. Бочков.
ГУАНИЛОВАЯ КИСЛОТА Cl0Hl4O8N5P, мол. в.
363,23 — иглы; кристаллизуется с 2 молекулами
ОН
воды; т. пл. 208° (с разл.); [а]}? ——7,5 (в воде);
константы диссоциации: Кх = 4,45 • 10 3; К2 =
= 8,2-10 7; Ка = 2,0-Ю-10. Г. к. образует соль
с бруцином, т. пл. 233° (с разл.); =—26,0° (в
водном спирте). УФ-спектр имеет максимум поглоще-
ния 253 ммк (pH 7,0). Г. к. весьма распространена в
природе, является структурным элементом рибону-
клеиновой кислоты (РНК) (см. Нуклеиновые кислоты).
Известны 3 изомерные Г. к., содержащие фосфатную
группу в положениях 2,3 или 5, способные к энзи-
матич. взаимным превращениям (константы относятся
к изомеру в положении 3). При кислотном гидролизе
Г. к. отщепляется фосфатная группа с образованием
гуанозида (I) и последующим разрывом гликозидной
связи и образованием гуанина (II) и D-рибозы (III):
При pH 1,5—Г,9 Г. к. гидролизуется до 3-фосфори-
бозьг, к-рая при восстановлении дает оптически неак-
тивный 3-фосфорибитол
ннн
носн.,-с- i-c-CH,OH
I I I
но о он
(НО)гРО
Г. к. получают при щелочном гидролизе дрожжевой
РНК. Г. к. качественно и количественно определяют
методом бумажной хроматографии и электрофореза
на бумаге с выявлением пятен по их поглощению
в УФ’-свете, а также путем адсорбционной и ионооб-
менной хроматографии.
Лит. см. при ст. Нуклеотиды. А. Ф. Бочков.
ГУАНИН (6-окси-2-аминопурин) C5H5ON5, мол. в.
151,15 — пуриновое основание', кристаллы; т. пл. 365°
(с разл.); пКа.2,3,
рИа29,2 И рКа312,3. N=C-OH HN-CO
Г. нерастворим в во- h,nc C-nh или hn=C C-nh’
де, слабо растворим ^_^СН I LJ.CH
в спирте, эфире, ней-
тральных и аммиачных растворах; легко раство-
рим в кислотах и щелочах, с к-рыми образует соли.
Г. осаждается метафосфорной и пикриновой к-тами
даже из разб. р-ров, что используют для определения
Г. С диазосульфокислотой в р-ре Na2CO3 Г. дает
красное окрашивание. Раствор Г. в НС1 флуоресцеи-
рует; в кислых и щелочных р-рах Г. имеет по 2 макси-
мума абсорбции в УФ-спектре: при 275 и 248 ммк
(pH 2) и 246 и 273 ммк (pH И).
Г. широко распространен в природе, встречается
в составе нуклеиновых кислот и нуклеотидов', состав-
ляет главную часть экскрементов птиц (гуано) и
паукой; входит в состав рыбьей чешуи и кожи живот-
ных, содержится во многих растениях. В организме
млекопитающих Г. под влиянием фермента гуаназы
переходит в ксантин. Г. получают из гуано и гидро-
лизом природных соединений, в состав к-рых он вхо-
дит. Синтетически Г. может быть получен из мочевой
к-ты через трихлорпурин и из гуанидина действием
на него цианоуксусного эфира. Г. применяют для
синтеза пурвновых алкалоидов (кофеина, теофилина,
теобромина и др.).
Лит. см. при ст. Аденин. Р. А. Звягильская.
ГУАНОЗИН (вернин, 9-|3-В-рибофуранозилгуанин)
C10Hl3N5O5, мол. в. 283,26 — нуклеозид', бесцветные
Hn>C^c/N^ch
N Л-нс-с—с—сн-сн,он
I I i
н н
кристаллы; т. пл. 235°, [а]р =—72° (1,4%-ный рас-
твор в 0,1 н. водном р-ре NaOH); рКа 1,6, рХа2 9,16,
рКа 12,3; максимум поглощения в УФ-спектре
265 ммк (рН1) и X, 264 ммк (pH 12). Г. амфотерен,
при кислотном гидролизе дает гуанин и 0-рибозу.
Г. входит в состав нуклеиновых кислот и нуклеотидов,
образуется путем гидролиза нуклеиновых к-т вод-
ными р-рами слабых щелочей (NH3, пиридин) при
нагревании; получен также синтетически.
Лит.: Шабарова 3. А., Усп. хим., 1959, 28, вып. 4;
см. также при ст. Аденин. Р. А. Звягильская.
ГУДРОН — черная смолистая масса разной кон-
систенции, остающаяся после отгонки от нефти легких
и большинства масляных фракций. Выход Г. из
различных нефтей сильно колеблется и зависит
также от полноты отгона масляных фракций и в боль-
шинстве случаев составляет 15—30%, считая на
нефть. В случае недостаточно полного отгона масля-
1013
ГУДРОН КИСЛЫЙ — ГУМИНОВЫЕ кислоты
1014
ных фракций получающийся остаток называют п о-
лугудроном. Главными составными частями
Г. являются: масла — остатки высокомолекулярных
углеводородов, не отогнавшиеся при перегонке нефти;
нефтяные смолы — нейтральные вещества, являющие-
ся главными компонентами смолистых веществ нефти;
асфальтены, карбены и карбоиды — твердые асфальто-
образные вещества нейтрального характера; асфальто-
геновые к-ты и их ангидриды — смолистые вещества
кислотного характера.
Состав и свойства гудронов сильно зависят от ха-
рактера исходной нефти и полноты отгона масляных
фракций. Плотность Г. — от 0,95 до 1,00, вязкость в,
градусах ВУ — от 18 до 45 при 100°. В большинстве
случаев Г. служат полупродуктами, из к-рых получают
битумы. Для этого особенно подходят Г. с большим
содержанием смол, асфальтенов
и углеводородов, легко подвер-
гающихся реакциям окисления R
и уплотнения. Такие Г. непо-
средственно применяются в ка-
честве дорожных масел для по-
ливки и пропитки (битуминиза-
ции) дорог. Из парафинистых
Г. получаются битумы худшего
качества.
Нек-рые виды Г. выпускают-
ся в качестве товарных про-
дуктов, например полугудрон и масляный Г. марок
Л и Т. Полугудрон применяется для смазывания
грубых механизмов, а также для изготовления колес-
ной мази УС. Масляные Г. марок Л и Т, различаю-
щиеся между собой вязкостью и темп-рой вспышки,
применяются в качестве мягчителей в резиновой
пром-сти, а также в строительстве. Высокосмо-
листые Г. с плотн. 0,98—1,00 крекингом перерабаты-
вают в автомобильный бензин с выходом до 10—16%;
с той же целью применяют деструктивную гидрогени-
зацию и коксование. Выход бензина и дистиллята
дизельного топлива в процессе коксования достигает
30—40%, считая на гудрон. При деструктивной гидро-
генизации выход светлых продуктов может достигать
70—75%. Возможно также использование Г. путем
газификации в присутствии водяного пара.
Лит.: Наметкин С. С., Химия нефти, [3 изд.], М.,
1955; Пархоменко В. Е., Технология переработки
нефти и газа, 2 изд., М., 1959; Переработка нефти, т. 2, М.,
1958. Л. С. Поваров.
ГУДРОН КИСЛЫЙ — отходы при очистке нефтя-
ных продуктов конц. серной к-той; вязкая смолистая
масса черного цвета, содержащая наряду с органич.
веществом от 15 до 70% по весу свободной H2SO4.
В зависимости от характера очищаемых нефтепродук-
тов и условий очистки Г. к. обладают крайне разно-
образными составом и свойствами. Г. к. различают
по нефтепродуктам, при очистке к-рых они получены,
напр. Г. к. от очистки смазочных масел. В особую
группу выделяют смешанные старые Г. к., накопив-
шиеся в качестве отходов в земляных амбарах и наз.
обычно прудовыми гудронами. Органич. масса Г. к.
представляет собой смесь смолистых веществ, продук-
тов полимеризации непредельных углеводородов и
частично очищаемых продуктов. Концентрация со-
держащейся в Г. к. серной к-ты ниже исходной.
В ажной и трудной проблемой для нефтяной пром-сти,
полностью пока не решенной, является утилизация
Г. к., к-рая возможна в двух направлениях: 1. Регене-
рация H2SO4 и использование органич. массы. Кис-
лота отделяется от органич. массы при действии
на Г. к. растворителей, растворяющих органич.
массу, при обработке Г. к. водой или при нагревании
паром. Отделяемая при этом разб. H2SO4 концентри-
руется и снова используется для очистки. Органич.
масса Г. к. после специальной обработки может быть
использована в качестве заменителя битума, в ка-
честве топлива и для приготовления нек-рых видов
консистентных смазок. 2. Термич. разложение Г. к.
в специальных конверторах; органич. часть превра-
щается в кокс, а сера образует сернистый газ SO2,
к-рый далее поступает на упрощенную установку для
получения контактным методом H2SO4. Г. к., обра-
зующиеся при очистке соляровых масел, служат для
получения сульфонафтеновых к-т, т. наз. «контакта».
Лит.: Пархоменко В. Е., Кислый гудрон как
технологическое сырье, М.—Л., 1947; его же, Кислый
гудрон как топливо, М.—Л., 1946. Л. С. Поваров.
ГУЛЬДА — ДЖЕКОБСА РЕАКЦИЯ — образование
4-оксихинолинов из ароматич. аминов и этоксимети-
ленмалонового эфира;
С(СООС2Н5)2
СН -С2НбОН
U III
Образующийся вначале анилинометиленмалоновый
эфир (I) при нагревании циклизуется в эфир 3-хинолин-
карбоновой к-ты (II), к-рый после омыления и де-
карбоксилирования превращается в 4-оксихинолин
(III). Применение в реакции Гульда — Джекобса
этоксиметиленциануксусного,-ацетоуксусного, -бензо-
илуксусного эфиров дало возможность получать с вы-
сокими выходами 4-оксихинолины с различным поло-
жением заместителей в ароматич. ядре, в частности
нек-рые антималярийные препараты. Реакция от-
крыта Р. Г. Гульдом и В. А. Джекобсом в 1939.
Лит.: Эльдерфилд Р., в кн.: Гетероциклические
соединения, пер. с англ., т. 4, М., 1955, с. 29—31.
Н. Е. Голубева.
ГУМИНОВЫЕ КИСЛОТЫ — органич. вещества,
извлекаемые из природных продуктов (торф, бурый
уголь, каменный уголь и др.) водными р-рами щело-
чей, окрашивающимися при этом в темный цвет. При
нейтрализации щелочных р-ров Г. к. выпадают в виде
аморфных бурых осадков. В природе Г. к. образуются
из растительных остатков или в результате окис-
ления ископаемых углей и др. продуктов органич.
происхождения. На торфяной стадии образования
углей отмершие растительные остатки, попадая в поч-
ву или болрто, служат питательной средой, на к-рой
развиваются различные микроорганизмы; продуктом
их жизнедеятельности и являются Г. к. Другой путь
образования Г. к. в природе — окислительные про-
цессы, протекающие при воздействии атмосферного
или растворенного в воде кислорода на угли или дру-
гие вещества органич. происхождения (в случае
углей их называют регенерированными
Г. к.). Торфяные и буроугольные Г. к. отличаются
от Г. к., образовавшихся в процессе окисления углей,
отношением углерода к водороду (для первых оно
меньше 15, а для вторых больше).
Содержание и состав Г. к. в торфах и бурых углях
колеблются в широких пределах (см. таблицу) и за-
висят от вида растений, на базе к-рых образовался
торф, а также от степени его разложения. В бурых
углях содержание Г. к. зависит от характера угля
(землистые, плотные, полублестящие и т. п.); больше
Г. к. содержат землистые угли.
Обычно при извлечении Г. к. из торфов и бурых
углей в щелочи растворяется органич. вещества боль-
1015
ГУТТАПЕРЧА
1016
ше, чем осаждается при нейтрализации. Не осаждаю-
щиеся из раствора органич. вещества называют
фульвокислотами. Из осаждающихся Г. к.
спиртом извлекаются гим атом ел а н о в ы е кис-
лоты, остаток после экстракции спиртом представ-
ляет собой гумусовые кислоты.
Топливо Содержа- ние Г. к., % с, % н, % N, % СООН, о/ /о ОН. %
Торф степной (разл. 10%) 10—50 54—60 4-6 1.5—3,5 8—12,5 7—10,5
Бурые угли . 35—70 62—69 4,5—5,5 0,5-1,5 7,5—18 6,5-9,5
Лигниты . . 17-70 62—67 4,9-6,0 — 7-9 7-9
Примечание: В таблице приведены данные анали-
зов на органич. массу.
Г. к. — сложная смесь соединений разного состава,
свойств и строения. Химич, природа и строение Г. к.
не выяснены. Признается, что Г. к. состоят в основном
из конденсированных ароматич. ядер, что доказы-
вается реакциями нитрования и окисления. На осно-
вании исследования Г. к. методами органич. анализа
и изучения их свойств установлено наличие в них
фенольных гидроксилов, карбоксильных, карбониль-
ных и ацетогрупп, простых эфирных связей и др.
Мол. вес условно принимается 1300—1500.
Г. к. относятся к высокомолекулярным соединениям;
они легко образуют коллоидные р-ры, набухают и
пептизируются в воде, щелочных р-рах, в нек-рых
органич. веществах, напр. в диоксане и пиридине.
Щелочные (0,025 н.) р-ры Г. к. при прохождении
через катиониты освобождаются от катионов. Г. к.
при взаимодействии с углекислыми солями вытес-
няют углекислоту, образуя соответствующие соли
Г. к. Гуматы щелочных металлов растворимы в воде,
щелочноземельных — нерастворимы. Растворы Г. к.
обладают свойством обменивать ионы металлов.
В практике это свойство Г. к. используется для смяг-
чения жестких вод. Воду пропускают через фильтры,
заполненные бурым углем. Соли кальция и магния
образуют соответствующие нерастворимые гуматы
кальция и магния, к-рые задерживаются на угле.
Аммониевые соли Г. к. растворимы в воде и исполь-
зуются в с. х-ве в виде органо-минеральных удобре-
ний. Г. к. применяют как кислотостойкие наполни-
тели (сосуды для аккумуляторов), для проклеивания
и окрашивания технич. сортов бумаги, для обработки
древесины, как вещества, способствующие росту ра-
стений, и др.
Лит.: Стадников Г. Л., Происхождение углей
и нефти, М.—Л., 1937; Кухаренко Т. А., Исследование
в области гуминовых кислот углей различных стадий угле-
образования, в.ни.: Химия и генезис твердых горючих Иско-
паемых. Труды Первого Всесоюзного совещания, 1950 г., М.,
1953; Oden S., Die HuminsSuren, 2 Aufl., Dresden, 1922;
FlaigW. [U. a.], Landbouwkund. Tijdschr., 1954, 66, №5/6,
S. 307, 392. И. M. Караваев.
ГУТТАПЕРЧА — продукт естественного проис-
хождения, являющийся изомером натурального кау-
чука; добывается из млечного сока или клеточных
включений гуттаперченосных растений, произрастаю-
щих в основном на Малаккском полуострове, на остро-
вах Малайского архипелага, Филиппинских островах
и др. В Советском Союзе Г. получают в основном из
бересклета бородавчатого. В бересклетах Г. сосредо-
точена гл. обр. в клетках коры корней.
При комнатной темп-ре технич. Г. — твердый коже-
подобный продукт белого или желтовато-коричнево-
го цвета (в зависимости от происхождения и мето-
да получения), плотн. 0,95—0,97, теплоемкость 0,67
кал/г-град, коэфф, объемного расширения 0,00081/г/>ад,
.теплопроводность 3,1- 104 кал/см- сек-град, диэлек-
трич. проницаемость 2,6; уд. электросопротивление
1014 ом-см, сопротивление разрыву 170—300 кг/см2,
«р = 1,523. Г. растворяется в большинстве аро-
матических углеводородов (при нагревании) и в хло-
рированных углеводородах; кислоты, в том чис-
ле НС1 и HF, почти не действуют на Г.; она способ-
на, как и каучук, вулканизоваться серой (с уско-
рителем) с образованием прочных вулканизатов (со-
противление разрыву 200—250 г/см2, относительное
удлинение 500—550%), а при взаимодействии с кисло-
родом воздуха- превращается в смолистые хрупкие
вещества, растворимые в ацетоне. Основными компо-
нентами, входящими в состав Г., являются высоко-
молекулярный углеводород — гутта — (С5Н8)П, смолы,
влага и др. Так, напр., в Г. из бересклета со-
держится 85—91% гутты, 5—8% смол, 2,5—3,5%
нерастворимых веществ. Известны две модификации
гутты, аир, отличающиеся взаимным расположением
изопреновых остатков. Выше 70° гутта — аморфное
вещество; при быстром охлаждении гутты образуется
кристаллич. P-форма, а при медленном охлаждении —
a-форма, к-рая более стабильна. Средний мол. вес
гутты ок. 50 000. Элементарный состав гутты аналоги-
чен каучуку; оба они являются полимерами изопрена
С5Н8. Различие свойств гутты и каучука объясняется
стереоизомерией этих углеводородов, причем натураль-
ному каучуку приписывают ^ис-конфигурацию, а
гутте — щраие-конфигурацию:
--H2Cxc=c/CHa-CHaVc=cZCHa—
H3CZ хн H3CZ х-н
каучук
••-нас
м н3сч zch2—
с=с( )с=с(
H3CZ СР^-СН2 н
гутта
Высокие диэлектрич. свойства и малая влагоемкость
Г. обусловливают ее применение в качестве изоли-
рующего материала в электро- и радиопромышлен-
ности, при изготовлении подводных кабелей. Кроме
того, Г. применяют в химич. и обувной пром-сти в ка-
честве кислотоупорного и клеящего материала.
Лит..- Войновскпй А. Б., Гуттаперча, М., 1951;
Догадкин Б. А., Химия и физика каучука, М., 1947.
В. Ф. Черткова.
ДАВЛЕНИЕ НАСЫЩЕННОГО ПАРА — давление
пара, находящегося в равновесии с жидкостью или
твердым телом при данной температуре. Д. н. п. лю-
бого химического соединения определяется только
температурой; при данной температуре Д. н. п. и
его температурный коэффициент—характерные свой-
ства веществ, необходимые для многих практических
расчетов. Раньше Д. н. п. называли чаще упругостью
насыщенного пара.
ДАГЕРРОТИПИЯ — первый практич. способ полу-
чения фотографич. изображения с помощью галогени-
дов серебра; опубликован в 1839 Л. Дагерром. Для
изготовления дагерротипа полированная серебряная
пластинка выдерживалась в парах J до образования
на ее поверхности AgJ. Во время съемки под дей-
ствием света AgJ частично превращается в металлич.
серебро. Затем для усиления изображения пластинка
подвергалась действию паров ртути. В местах, где
подействовал свет, получалась амальгама серебра.
После фиксирования раствором NaCl на участках,
не подвергшихся действию света, обнажалось сереб-
ряное зеркало. Вследствие неодинакового отражения
света амальгамой и зеркальными участками серебра
при рассматривании дагерротипа под определенным
углом можно было видеть позитивное изображение.
Наиболее существенную часть способа составляло от-
крытие возможности усиления изображения, полу-
ченного действием света. Позже при изготовлении да-
герротипных пластинок стали применять смесь паров
иода и брома, что позволило значительно повысить
их светочувствительность, а для фиксирования —
вместо NaCl тиосульфат натрия. Д. просуществовала
до начала 50-х гг. 19 в., ее сменил коллодионный
Процесс. К. В. Вендровский.
ДАЛЬТОНА ЗАКОНЫ — 1) Давление смеси газов,
химически не взаимодействующих друг с другом,
равно сумме их парциальных давлений. Объяснение
закона дается молекулярно-кинетич. теорией идеаль-
ных газов. Приближенно применим и к неидеаль-
ным газам при невысоких давлениях; значительные
отступления наблюдаются в области высоких дав-
лений.
2) При постоянной темп-ре растворимость каждого
из компонентов газовой смеси в данной жидкости
прямо пропорциональна его парциальному давлению
над жидкостью и не зависит от общего давления смеси
и содержания других компонентов (т. е. каждый газ
растворяется так, как если бы он находился один
в данном объеме). Этот закон является важным допол-
нением к закону Генри, согласно к-рому раствори-
мость индивидуального газа пропорциональна его
давлению. Второй Д. з. является приближенным;
он применим к газам, поведение к-рых близко к пове-
дению идеального газа, и лишь при условии, что
растворимость газа невелика. Д. з. открыты в 1801
и 1803 Дж. Дальтоном. в. И. Еараноккий.
ДАЛЬТОНИДЫ И БЕРТОЛЛИДЫ — 1) Даль-
т о н и д ы (Д.) — определённые химич. соединения, ко-
торые характеризуются: а) рациональным (т. е. отве-
чающим простому стехиометрии, отношению компонен-
тов) максимумом на линиях (или поверхностях) ли-
квидуса и солидуса диаграммы состояния системы;
б) сингулярными точками на изотермах ее свойств;
в) способностью давать жидкие или твердые растворы
с компонентами системы (нек-рые авторы предлагают
называть эти твердые растворы дальтонидными фа-
зами или просто Д.). 2) Бертолл иды (Б.) — твер-
дые фазы переменного состава, к-рые характеризуются:
а) иррациональным (т. е. не отвечающим стехиометрии,
отношению компонентов) максимумом на линиях (или
поверхностях) ликвидуса и солидуса; б) отсутствием
сингулярных точек на изотермах свойств системы.
В двойных системах с образованием д а л ь т о-
нида (рис. 1) максимум М температур t начала
и конца кристаллиза-
ции отвечает опреде-
ленному соединению
АтВп, которое дает
твердые растворы у со
своими компонентами
А и В. На изотер-
мах состав — свойство
(напр., твердости Ht
и электропроводно-
сти Х() наблюдаются
сингулярные точки iSj
и S2, отвечающие со-
единению АтВп; точ-
ки С, D, F, G отве-
чают границам, отде-
ляющим однофазную
область у от смесей
фаз а с у и р с у
(а — твердый раствор
на основе компонента
А, р — то же на осно-
ве компонента В).
В двойных системах
с образованием бер-
толлида у (рис. 2) пологий максимум М на ли-
ниях ликвидуса и солидуса не отвечает какому-либо
рациональному отношению компонентов А и В. Изо-
термы свойств в области однородности фазы у имеют
вид плавных кривых, без сингулярных точек; точки
C,D,F и G по-прежнему отвечают границам, отде-
ляющим однофазную область у от двухфазных
а + у и у -j- р.
Понятия и термины «дальтониды» (в память Дж.
Дальтона) и «бертоллиды» (в память К. Л. Бертолле)
были предложены Н. С. Курнаковым (в 1912—13),
к-рый вместе со своими учениками открыл и исследо-
1019
ДАММАРА — ДАУТЕРМ
1020
вал многочисленные примеры Д. и б., гл. обр. в ме-
таллических двойных системах. Вопрос о природе
бертоллидов еще не-
достаточно выяснен.
Наиболее распростра-
нена трактовка бер-
толлидов как твердых
р-ров на основе либо
«мнимых» химич. сое-
динений, т. е. таких,
состав которых лежит
вне границ однород-
ности фазы у (соедине-
ние АтВп на рис. 2),
либо твердых раство-
ров на основе «мни-
мых» (т. е. не сущест-
вующих в действитель-
ности) полиморфных
модификаций компо-
нентов. Сделанные в
последнее время по-
пытки подхода к во-
просу о Д. и б. с точ-
ки зрения атомно-
структурных предста-
влений (Г. Б. Бокий)
в основных чертах
подтверждают изложенные выше взгляды.
Лит.: Курнаков Н. С., Собрание избранных работ,
т. 2, Л.—М., 1939; его же, Введение в физико-химиче-
ский анализ, 4 изд., М.—Л., 1940; Аносов В. Я. и Пого-
дин С. А., Основные начала физико-химического анализа,
М.—Л., 1947. С. А. Погодин.
ДАММАРА — бесцветная или слабоокрашенная
смола, добываемая из деревьев, произрастающих
в Вост. Индии и Новой Зеландии. Д. плавится при
84—86°, растворяется в бензоле, скипидаре, хлори-
рованных углеводородах и при нагревании в расти-
тельных маслах; кислотное число 20—60, число омы-
ления 30—70. В состав Д. входят даммароловая к-та
С81Н„(ОН)СООН, индифферентные вещества (резены),
отвечающие формулам С22Н34О2 и С32Н52О, эфирные
масла и воск. Д., освобожденная от воска растворе-
нием в бензоле с последующим осаждением спиртом,
носит название целлодаммара, к-рая исполь-
зуется в произ-ве бесцветных нитролаков. Д. при-
меняется для приготовления бесцветных типографских
лаков, красок для живописи и лаков для золочения.
Лит.: Киселев В. С., Олифа и лаки, 3 изд., М.—Л.,
1940. А. А. Благонравова.
ДАНИЭЛЯ — ЯКОБИ ЭЛЕМЕНТ — см. Химические
источники тока.
ДАРЗАНА РЕАКЦИЯ — конденсация карбониль-
ных соединений с алкилгалогенидами, содержа-
щими рядом с галогеном «активированный» атом
водорода, приводящая к образованию оксиранового
кольца. Д. к. проводят в инертной атмосфере в при-
сутствии амида натрия, едкого натра, металлич.
натрия, алкоголята натрия, поташа и др.:
в- Rx /°\ /R"
с=о+ с —- с—с
R'z н/ R,„ R,z R,„
В качестве карбонильных соединений используют
алифатич. или ароматич. альдегиды и кетоны (альде-
гиды, как правило, более активны), а в качестве
галогенидов — эфпры или амиды а-галогенокислот,
а-галогеиокетоны, галогено-метплсульфоны и др.:
R.
С=О+C1CH2COR"—* С—CHCOR"
R'z 1\ О
К.
С=О + Cl ch2so2r" —* с—~chso2r"
R"' R"' О
R R.
C=O + Cl CH2Ar —- C----CIIAr
R'Z R'z XOZ
Наиболее изучена конденсация карбонильных со-
единений с эфирами а-галогенокислот, ведущая
к синтезу глицидных эфиров. Последующий гидролиз
и декарбоксилирование приводят к альдегидам и ке-
тонам с большим числом атомов углерода:
Rx I R\ zR"
Д>О + Cl—СИ—COOR"'—* С——С—COOR"'—-
R R.z
R\ /R" -CO.. Rx zR" Rx
— c—c-cooh—c—C-H—- CH—COR"
R'z О R'z o' R'z
(R"=H. Aik, Ar)
Изомеризация глицидных эфиров и их гидролиз при-
водят к синтезу а- и p-кетоэфиров и р-альдегидо-
эфиров:
R\ zR" /R' R\
С—C-COOR"' — RCO-C-COOR’mu R'-C-C-COOR"'
R Oz V R"z
/CyO
R—CH—CH COOR' —-RCH H
4O/ 4COOR’
Механизм Д. к. лучше всего отображается схемой,
подтверждающейся как кинетич. данными, так и вы-
делением промежуточных соединений типа I. Другим
возможным путем образования окисного кольца яв-
ляется реакция присоединения промежуточного со-
единения с двухвалентным углеродом (карбеном)
к карбонильной группировке:
R\ ,Н R\ R'
R'/S ' R>C-X- R
R\ zR” R\ z°\ /R"
c=o + :C —- c—c
R'Z xR,„ R, \R,„
Д. к. открыта в 1904 Г. А. Дарзаном.
Лит.: Ньюмен М. С. н Мей джерлей н Б.Д ж.,
в кн.: Органические реакции. Сб. 5, пер. с англ., М., 1951;
В allester М., Chem. Revs., 1955, 55, № 2, р. 283; М u nch-
PetersenJ., Acta Chem. Scand., 1953, 7, К» 7, p. 1041.
H. П. Гамбарян.
ДАРЗАНА СИНТЕЗ НЕНАСЫЩЕННЫХ КЕТО-
НОВ — см. Кондакова реакция.
ДАУТЕРМ — смесь дифенила и дифенилового эфира,
применяемая в качестве теплоносителя. См. Тепло-
носители.
1021
ДАФФА РЕАКЦИЯ— ДВОЙНОЙ ЭЛЕКТРИЧЕСКИЙ СЛОЙ
1022
ДАФФА РЕАКЦИЯ — метод синтеза о-оксиаль-
дегидов ароматич. ряда, заключающийся в нагрева-
нии фенолов с гексаметилентетрамином и борной
к-той в среде глицерина (см. Реймера — Тиманна
реакция):
Фенол с гексаметилентетрамином дает вторичный
амин I, к-рый дегидрируется гексаметилентетрамином
в шиффово основание II; кислотным гидролизом по-
следнего получается о-оксиальдегид и первичный
амин. В нек-рых случаях образуется 3—5% о-кре-
зола (6-хлор-лгета-крезол); выделены также диаль-
дегиды:
Выход о-оксиальдегидов колеблется в пределах 10—
30%. В реакцию вступают фенол, его гомологи,
хлорфеволы, а- и Р-нафтолы, а также 4-оксидифенил.
Салициловая к-та дает смесь 4- и 5-альдегидосалици-
ловыХ к-т. Реакция неприменима к п- и .и-оксибензой-
ным к-там, а также нитрофенолам. Реакция открыта
в 1941 И. Даффом.
Лиш.: Dull J. С., J. Chem. Soc., 1941, Sept., p. 547;
Dull J. C., Furness V. I., там же, 1951, June, p. 1512.
Ю. А. Титов.
ДВОЙНАЯ СВЯЗЬ — связь между двумя атомами,
осуществляемая четырьмя электронами. Современные
представления о строении Д. с. сложились в основ-
ном благодаря спектроскопия, иссле-
*\о/20“ /н дованиям. В молекуле этилена пять
/ a-связей, в том числе одна С—С
zro-l С^С связь, расположены друг относитель-
\%йо"0\ но друга под углом 120° и находятся
н 'В в одной плоскости.
Вторая же связь между атомами углерода — каче-
ственно иная. Она образована за счет перекрывания
облаков двух электро-
бридизованных электронов,
нов, восьмерки которых
перпендикулярны пло-
скости молекулы эти-
лена (см. рисунок). Та-
кая связь наз. л-свя-
зью. Так как предпола-
гается, что три о-связи
атома углерода обра-
зуются с участием ги-
а л-связь — с участием
«чистого» /j-электрона (т. е. из четырех электронов
атома углерода гибридизованы одни s-электрон и
только два из трех р-электронов), то имеющая место
в молекуле этилена гибридизация электронов атомов
углерода обозначается как $р2-гибридизация. В моле-
куле этилена атом углерода находится во втором
валентном состоянии (ур2-гибридизация). В этом же
валентном состоянии углерод находится и в ароматич.
соединениях. Т. обр. принято считать, что Д. с. пред-
ставляет собой комбинацию двух связей (<т и л),
неодинаковых по своему электронному строению.
Однако до сих пор нет еще ни одного физич. или химич.
метода, с помощью к-рого можно было бы доказать
это различие.
Д. с. широко распространены в органич. соедине-
ниях, причем ими могут быть связаны друг с другом
не только атомы углерода, но и атомы азота, сурьмы,
мышьяка, а также атомы углерода с атомами кисло-
рода, азота, серы и др. В тех случаях, когда Д. с.
осуществляется между атомами, отличающимися по
своей электроотрицательности, она по-
ляризована, т. е. р-электроны, образую-
НО щие л-связь, смещены в сторону более
\_______ электроотрицательного атома. При этом
~|_j _/ \ поляризация л-связи всегда больше по-
2 У' / ляризации a-связи, связывающей те же
атомы. Вместе с тем при такой поляри-
зации р-электроны не переходят цели-
ком из электроннрй оболочки одного атома в обо-
лочку другого, как это имеет место в случае семи-
полярной связи. Примером поляризованной Д, с.
может служить связь между углеродом и кислоро-
дом в молекулах карбонильных соединений: 4c£q
(изогнутая стрелка указывает направление смещения
л-электронов, т. е. направление поляризации л-свя-
зи). В результате такой поляризации (а также от-
части и поляризации о-связи) атом углерода приобре-
тает нек-рый (неполный) положительный заряд,
а атом кислорода — нек-рый (также неполный) отри-
цательный заряд: ''c~q . Наличие этих зарядов во
многом определяет химич. свойства карбонильных
соединений.
Физич. характеристиками Д. с. являются их энер-
гии, межатомные расстояния, характеристич. частоты
колебаний групп атомов и др. (см. таблицу).
Связь Энергия связи, кал Межатомные’ рас- стояния, А Характери- стические частоты, см-"1
с = с 100 1,34»; 1,33г; 1,32« 1640»; 1590м
с = о 142а; 149б 1,21а; 1,20ж; 1,16й 1695н; 1710п;
1745Р
C = N 94 1,34й 1640; 2150°
C = S 103 1,54л 1530
а В формальдегиде. ° В других альдегидах. в В этилене
и моноолефииах. г В бутадиене и стильбене. д В бензохиие.
ж В ацетальдегиде. и В СО,. к В диазометане. л В серо-
углероде. м Сопряженная связь. н В кетонах. п В кислотах.
Р В сложных эфирах. с В изосоединениях.
Инкремент рефракции С=С равен 1,733 (для линии D
натрия); 1686; 1824; 1894 (для линий водорода На, Н, и
соответственно).
Лит.: Butlerow A. nnd Ossokin М., Ann.
Chem. und Pharmac., 1868, 145, S. 267, 271; Бутлеров A.,
Ж. Русс. физ.-хим. о-ва, 1869, 1, с. 247; его же, там же,
1870, 2, с. 187; Реутов О. А., Теоретические проблемы
органической химии, М., 1956, гл. 1; Р a n 1 i n g L., в кн.:
Theoretical organic chemistry, L., 1959 (Papers presented to
the Kekule symposium, organised by the Chem. Soc. L. 1958).
О. А. Реутов.
ДВОЙНОЙ ЭЛЕКТРИЧЕСКИЙ СЛОЙ — тонкий
поверхностный слой из пространственно разделенных
электрич. зарядов противоположного знака, обра-
зующийся на границе двух фаз. Д. э. с. может воз-
никать на поверхностях раздела твердое тело — жид-
кость, жидкость — жидкость, жидкость — газ, твер-
дое тело — твердое тело и твердое тело — газ; наи-
большее практич. значение имеет Д. э. с. на границах
твердое тело — жидкость и твердое тело — газ. В об-
разовании Д. э. с. могут участвовать как заряжен-
ные частицы (ионы, электроны), так и дипольные
1023
ДВОЙНОЙ ЭЛЕКТРИЧЕСКИЙ СЛОЙ
1024
молекулы. Простейший случай образования Д. э. с.
наблюдается в том случае, когда в состав обеих фаз
входят заряженные частицы, к-рые могут переходить
через границу раздела фаз, напр. ионы металла в слу-
чае поверхности раздела металл — раствор. При
соприкосновении металла с раствором ионы металла
переходят через границу раздела фаз и в каждой
из соприкасающихся фаз появляется избыток заря-
дов одного знака. Так, при погружении металлич.
Zn в разб. р-р ZnSO4 вначале в раствор переходит
нек-рое количество катионов Zn2+, причем поверх-
ность металла заряжается отрицательно за счет из-
бытка электронов. Эти заряды поверхности притяги-
вают из раствора ионы противоположного знака п
отталкивают одноименно заряженные ионы (рис. 1).
Рис. 1. Двойной электри-
ческий слой у поверхно-
сти металлич. цинка, по-
груженного в раствор
ZnS04.
В результате в тонком слое
раствора, прилегающем к ме-
таллу, появляется избыток по-
ложительно заряженных ионов.
Этот слой вместе с заряжен-
ной поверхностью металла и
образует Д. э. с. на границе
металл — раствор. На поверх-
ности раздела возникает элек-
трич. поле Д. э. с., к-рое тор-
мозит дальнейший переход ио-
нов цинка в раствор, и после
достижения определенной рав-
новесной разности потенциа-
лов между металлом и раство-
ром переход ионов в раствор прекращается (см.
Электродный потенциал). Д. э. с. может возникать
не только в результате перехода заряженных частиц
через поверхность раздела, но и за счет неодинако-
вой адсорбции ионов противоположного знака на
границе фаз, а также при адсорбции на поверхности
раздела электрически нейтральных молекул. Посколь-
ку положительные и отрицательные концы диполей
притягиваются к поверхности с неодинаковой си-
лой, адсорбированные диполи ориентируются пер-
пендикулярно к границе раздела и образуют Д. э. с.
При адсорбции атомов на поверхностях раздела ме-
талл— раствор и металл — газ возникновение Д. э. с.
наблюдается в том случае, если связь металл — атом
имеет дипольный характер (напр., адсорбция кисло-
рода на ряде металлов).
Простейшая модель строения Д. э. с. на поверх-
ности раздела фаз, предложенная Г. Гельмгольцем,
ограничивалась рассмотрением электростатнч. сил
притяжения между зарядами противоположного зна-
ка. Согласно этому представлению, в случае границы
металл — раствор заряженная поверхность металла
притягивает из раствора эквивалентное по числу
зарядов количество ионов противоположного знака,
и все эти ионы вплотную приближаются к поверх-
ности. Возникающий таким образом Д. э. с. можно
уподобить плоскому конденсатору молекулярных раз-
меров, причем расстояние между его обкладками
определяется радиусом ионов d. Первая количествен-
ная теория строения Д. э. с., развитая Гуи приме-
нительно к плоской поверхности раздела, весьма
сходна с предложенной позже теорией сильных
электролитов Дебая и Гюккеля (см. Электролиты).
В теории Гуи наряду с электростатнч. взаимодей-
ствием между ионами и заряженной поверхностью
учитывается беспорядочное молекулярное движение
ионов, под влиянием к-рого ионы стремятся равно-
мерно распределиться в растворе. В результате этого
внешняя обкладка Д. э. с., расположенная в растворе,
приобретает диффузное строение. В процессе дальней-
шего совершенствования теории строения Д. э. с.
были учтены, кроме того, конечный размер ионов
и наличие специфич. адсорбции ионов на поверхности,
т. е. адсорбции, обусловленной силами химич. связи
и накладывающейся на чисто электростатнч. взаимо«
действие ионов с поверхностью.
Согласно современным представлениям, внешняя
обкладка Д. э. с. состоит из двух частей: первая часть
образована ионами, Вплотную притянутыми. к по-
верхности металла («плотный», или «гельмгольцев-
ский», слой толщиной d), вторая часть — ионами, на-
ходящимися на расстояниях от поверхности, пре-
вышающих радиус иона, причем число этих ионов
убывает по мере удаления от границы раздела («диф-
фузный слой»). На рис. 2 представлено распрбделе-'
ние потенциала в плотной и диффузной частях Д. э. с.
раствор
|Р0
f-j-
Сумма зарядов плотной
и диффузной частей вне-
шней обкладки Д. э. с.
равна заряду внутрен-
ней обкладки Д. э. с.,
т. е. поверхности метал-
ла. При увеличении за-
ряда металлич. обкладки
Д. э. с. и концентрации
раствора уменьшается
доля ионов внешней об-
кладки Д. э. с., которые
расположены в диффуз-
ном слое, т. е. ионная
обкладка Д. э. с. стано-
вится менее диффузной.
Строение Д. э. с. и, в ча-
стности, степень диффуз-
ности внешней обкладки Д. э. с. оказывает существен-
ное влияние на кинетику электродных процессов, на
твердость и смачиваемость металлов, имеет большое
значение для электрокинетич. явлений и для устой-
О
Рис. 2. Распределение потен-
циала в плотной и диффузной
частях двойного слоя; <?а — раз-
ность потенциалов между ме-
таллом и раствором, Ф, — сред-
нее значение потенциала на рас-
стоянии одного ионного радиуса
от поверхности металла.
чивости коллоидных систем.
Методы исследования Д. э. с. основаны на изуче-
нии тех свойств поверхности раздела фаз, к-рые за-
висят от наличия Д. э. с. и его строения. При относи-
тельном движении твердого тела и жидкости ионы
диффузной обкладки Д. э. с. увлекаются движущейся
жидкостью (см. Электрокинетические явления), что
позволяет экспериментально определять локализо-
ванный в диффузной части Д. э. с. скачок потенциала
(ip! на рис. 2), являющийся одной из важнейших
характеристик Д. э. с. Поскольку существует про-
стая зависимость между величиной производной
поверхностного натяжения по потенциалу и плот-
ностью заряда поверхности металла, то в случае
жидких металлич. поверхностей подробные сведения
о строении Д. э. с. могут быть получены путем изу-
чения зависимости поверхностного натяжения гра-
ницы раздела металл — раствор от потенциала и
состава раствора (см. Электрокапиллярные явления).
При использовании твердых тел с высокоразвитой
поверхностью (платинированная платина, высокодис-
персный уголь) можно изучать строение Д. э. с. на
их поверхности по изменению состава раствора,
к-рое обусловлено адсорбцией ионов на границе раз-
дела фаз в процессе образования Д. э. с. при сопри-
косновении твердого тела с раствором. Наиболее точ-
ные данные о строении Д. э. с. на границе металл —
раствор получены методом измерения емкости Д. э. с.
переменным током, впервые примененным А. П. Соко-
ловым в 1887; емкость Д. э. с. может быть измерена
также с помощью постоянного тока — методом кривых
заряжения, выражающих зависимость потенциала
металлич. электрода от времени при пропускании
тока постоянной силы. Ведущая роль в развитии тео-
рии строения Д. э. с. и в разработке методов его иссле-
дования принадлежит гл. обр. А. Н. Фрумкину и
его школе, выяснившим строение Д. э. с. и механизм
электрохимия, реакций, связанных с прохождением
1025
ДВОЙНЫЕ СИСТЕМЫ
1026
ионов через Д. э. с. на границе раздела металл —
раствор. Существенный вклад в развитие представ-
лений о строении Д. э. с. сделан О. Штерном и
Д. Грэмом.
Изучение свойств и строения Д. э. с. имеет большое
значение для понимания и усовершенствования таких
практически важных процессов, как электролиз,
электроосаждение металлов, электрохимии, реакции
в хнмич. источниках тока, коррозия металлов, коагу-
ляция коллоидов, флотация, ионный обмен и др.
Лит.: Фрумкин А. Н., Усп. хим., 1955, 24, вып. 8,
с. 933; Фрумкин А. Н. [и др.], Кинетика электродных
процессов, М., 1952; Жуков И. И., Коллоидная химия,
ч. 1, Л., 1949; Коагуляция коллоидов. Сб. ст. Ред. А. И. Раби-
нович и П. С. Васильев, М., 1936; Наука о коллоидах, пер.
с аигл. Ред. Г. Р. Кройт, т. 1, М., 1955; Парсонс Р., в ни.:
Некоторые проблемы современной электрохимии, пер. с аигл.,
М„ 1958; Grahame D. С., Chem. Revs., 1941,41, № 3, р. 441.
В. В. Лосев.
ДВОЙНЫЕ СИСТЕМЫ (бинарные системы, двух-
компонентные системы) — физико-химич. системы, об-
разованные двумя компонентами. Исследование Д. с.
имеет целью установление характера взаимодействия
их компонентов; для этого необходимо знание состава
и границ существования д5аз, т. е. гомогенных ве-
ществ, образуемых компонентами системы. Наиболее
общим способом достижения этой цели является
экспериментальное построение диаграмм состояния
и диаграмм состав— свойство.
Для построения диаграмм- состояния
Д. с., к-рые обычно изучаются при постоянном (атмо-
сферном) давлении, пользуются двумя взаимно пер-
пендикулярными осями. На оси абсцисс откладывают
состав х системы, чаще всего выражаемый в весовых,
атомных или мольных процентах; на оси ординат —
темп-ры протекающих в системе фазовых йревращений
(начала и конца кристаллизации, полиморфных пре-
вращений и др.), найденные посредством термиче-
ского анализа, в частности записи кривых нагревания
и охлаждения ряда смесей компонентов изучаемой
системы, взятых в различных отношениях. Соедийив
нанесенные точки линиями, получают диаграмму со-
стояния Д. с. при постоянном давлении. Если же
система изучается при переменном давлении, то по-
следнее откладывают на третьей оси, перпендикуляр-
ной к осям состава и темп-ры, и полученную трехмер-
ную фигуру проектируют на плоскость.
Для построения диаграмм состав — свой-
ств о Д. с. на оси абсцисс откладывают состав, а на
оси ординат — значения свойства (плотности, элек-
тропроводности, твердости и др.), измеренвые при
постоянной темп-ре. Соединив нанесенные точки
линиями, получают изотермич. диаграмму состав —
свойство, или, короче, изотерму свойства.
Если свойство измерялось при переменной темп-ре,
то значения последней откладывают на третьей оси,
перпендикулярной к осям состава и свойства, и полу-
ченную трехмерную фигуру проектируют на плос-
кость. При построении диаграмм состояния Д. с.,
кроме термич. анализа и изучения физич. свойств,
широко пользуются методами микроструктуры и
рентгеновским анализом. Последовательно применяя
общие принципы физико-химич. анализа, по виду
диаграмм состояния и диаграмм состав — свойство
Д. с. делают заключения о характере взаимодействия
их компонентов.
Ниже рассмотрены простейшие диаграммы состоя-
ния Д. с., содержащих только кристаллич. и жидкие
фазы, а также диаграммы состав — свойство затвер-
девших двойных систем. О Д. с., состоящих из одних
жидких фаз, а также из жидкостей и газа (пара),
см. Жидкие системы", о Д. с. из твердых фаз и газа
(пара) см. Термическая диссоциация.
Взаимная растворимость компонентов Л и В в жид-
ком состоянии может быть либо неограниченной, либо
17 К. X. Э. т. 1.
ограниченной (предельным случаем последней яв-
ляется полное отсутствие взаимной растворимости).
При кристаллизации жидких Д. с. из них могут выде-
ляться: 1) химич. индивиды, т. е. чистые компоненты
или образуемые ими соединения, 2) твердые растворы,
т. е. твердые фазы, состав к-рых может меняться
(неограниченно или в известных пределах) без нару-
шения однородности.
Кристаллизация чистых компонентов из одной жид-
кой фазы. Если компоненты А и
одну жидкую фазу L, из к-рой
при охлаждении кристаллизуют-
ся чистые компоненты, то при-
бавление компонента В к ком-
поненту А понижает темп-ру
начала кристаллизации послед-
него, что изображает кривая
начала кристаллизации, или лик-
видуса ТАЕ, идущая вниз от
точки ТА (темп-ра кристаллиза-
ции чистого А). Точно так же
прибавление компонента А к
компоненту В понижает темп-ру
его кристаллизации, что изобра-
жает кривая ликвидуса ТВЕ,
В (рис. 1) образуют
идущая вниз от точки Тв (темп-ра кристаллизации чи-
стого В). Обе кривые ликвидуса пересекаются в точке
Е, наз. эвтектической точкой. Жидкость, со-
став к-рой отвечает этой точке, затвердевает при по-
стоянной темп-ре в очень тонкую смесь кристаллитов/1
и В, наз. эвтектикой. Растворы и сплавы, содер-
жащие больше компонента А, чем эвтектика, наз.
доэвтектическими, а содержащие больше
компонента В, чем эвтектика, — заэвтектиче-
с к и м и. Когда жидкость доэвтектич. состава xlt
состояние к-рой изображается точкой М (рис. 1),
охладится до темп-ры ликвидуса Mlt начинается
выпадение кристаллов А. Вследствие этого при даль-
нейшем охлаждении жидкость обогащается компонен-
том А; ее состав изменяется по отрезку ликвидуса
М±Е. Когда жидкость будет иметь состав и температу-
ру, отвечающие точке Е, происходит одновременная
кристаллизация А и В. Таким образом, кристаллиза-
ция доэвтектич. жидкостей начинается первичным
выделением кристаллов А и заканчивается кристал-
лизацией эвтектики. Кристаллизация заэвтектич.
жидкостей начинается первичным выделением кри-
сталлов В и заканчивается кристаллизацией эвтек-
тики. Итак, все жидкие растворы и сплавы рассматри-
ваемой системы, кроме эвтектики, отвердевают в
темп-рном интервале. Верхняя граница его дается
кривыми темп-p начала кристаллизации или линиями
ликвидуса Т аЕ и ТвЕ, а нижняя граница —
линией темп-p конца кристаллизации или линией
солидуса FG, к-рая в данном случае является
горизонтальной прямой, проходящей через точку Е.
Названные линии делят плоскость диаграммы со-
стояния на 4 фазовых поля. Верхнее (выше линии
ликвидуса ТаЕТв) отвечает одной жидкой фазе L.
Остальные 3 поля отвечают смесям двух фаз: жидко-
сти L с кристаллами А (поле ТAEF), жидкости L
с кристаллами В (поле TBEG) и кристаллов А с кри-
сталлами В (поле AFGB).
Если один из компонентов, напр. В, имеет две
полиморфные модификации и Вг (рис. 2), причем
темп-ра их превращения Тр (т. е. темп-ра, при к-рой
обе модификации одинаково устойчивы и выше к-рой
устойчива модификация В2, а ниже — модифика-
ция В±) лежит между темп-рой плавления Тв ком-
понента В и эвтектич. темп-рой, то при охлаждении
1027
ДВОЙНЫЕ СИСТЕМЫ
1028
жидкостей, состав которых лежит между точками В
и Р, первично выделяются кристаллы В2. При темп-ре
Тр они находятся в равновесии с кристаллами и
жидкостью состава Р; ниже ТР
из жидкости выпадают кри-
сталлы Blt и отвердевание за-
канчивается кристаллизацией
эвтектики Е. Из жидкостей, со-
став которых лежит между Р
и Е, первично выпадают кри-
сталлы В2, а из жидкостей, со-
став которых лежит между ТА
и Е, — кристаллы Л; в обоих
случаях затвердевание закан-
чивается кристаллизацией эв-
тектики.
Некоторые физич. свойства
затвердевших Д. с., образую-
щихся при кристаллизации двух твердых фаз из
одной жидкости, аддитивны, т. е. линейно зависят
от состава (при постоянных темп-ре и давлении).
В этом случае изотермы свойств изображаются пря-
‘в
Рис. 3.
мыми линиями, проведенными
через точки еА и е£> отвечаю-
щие величинам свойств чистых
компонентов А и В (рис. 3). Од-
нако аддитивность физических
свойств смесей двух кристаллич.
фаз обнаруживается только при
надлежащем выборе свойств и
способа выражения концентра-
ции. Весьма существенно также,
чтобы на физич. свойства систе-
мы не влияли никакие другие факторы, кроме со-
става. В частности, Очень быстро закристаллизо-
ванные эвтектич. сплавы имеют более высокую твер-
дость, чем сплавы, полученные из жидкого состояния
при медленном охлаждении.
Кристаллизация одного твердого раствора из одной
жидкости. Если компоненты А и В обладают
неограниченной взаимной растворимостью как в жид-
Рис. 4.
ком, так и в твердом состоянии,
то из одной жидкой фазы L мо-
жет кристаллизоваться только
одна твердая фаза переменного
состава — твердый раствор S.
Основные типы диаграмм состоя-
ния Д. с. с непрерывным рядом
твердых р-ров, установленные
в 1899 Б. Розебомом, изобра-
жены на рис. 4. Тип I отвечает
случаю, когда на линиях лик-
видуса I и солидуса s нет ни
максимума, ни минимума, тип
II — случаю, когда кривые лик-
видуса и солидуса имеют мак-
симум М, и тип III — когда
эти обе линии имеют минимум
т. В максимумах и минимумах
и солидуса касаются друг дру-
кривые ликвидуса г '—~~~ --------------
га, т. е. имеют Одну общую точку; отвечающий ей
но составу сплав кристаллизуется при постоянной
темп-ре.
Изотермы свойств непрерывных твердых растворов
обычно имеют вид непрерывных кривых, обращенных
выпуклостью либо вниз (рис. 5, 7), либо вверх
(рис. 5, II). При образовании твердых р-ров электро-
проводность и темп-рный коэфф, электросопротивле-
ния, а также относительное удлинение и относитель-
ное сужение площади поперечного сечения при раз-
рыве понижаются (по сравнению с чистыми компонен-
тами), и поэтому изотермы соответствующих свойств
(рис. 5,1) обращены выпуклостью вниз (к оси соста-
ва). Наоборот, модуль упругости, предел текучести,
предел прочности, твердость
и давление истечения при
образовании твердых раство-
ров повышаются, и изотермы
соответствующих свойств (рис.
5, II) обращены выпуклостью
вверх (от оси состава). Изо-
термы некоторых свойств твер-
дых растворов, например пара-
метра пространственной решет-
ки (при выражении состава
в ат. %) и уд. объема (при
выражении состава в вес. %),
весьма близки к прямым ли-
ниям.
Кристаллизация двух твердых растворов из одной
жидкости. Если взаимная растворимость компонентов
Л и В в жидком состоянии беспредельна, а в твердом
состоянии ограничена, то из жидкости кристалли-
зуются два твердых раствора, обозначенные в даль-
нейшем буквами а (твердый раствор, богатый А)
и р (твердый раствор, богатый В). Диаграмма состоя-
ния для простейшего случая,
когда аир образуют эвтектику
(тип V Розебома), изображена
на рис. 6. Из доэвтектич. жид-
ких сплавов, состав к-рых лежит
Между точками А и Е, первично
выделяется твердый раствор а,
предельная концентрация к-рого
при эвтектич. темп-ре отвечает
абсциссе точки F. Если состав
исходной жидкости лежит между
F и Е, то она обогащается ком-
понентом В и, достигнув эвте-
ктич. состава (точка Е), отвер-
девает, превращаясь в смесь
двух насыщенных твердых раст-
воров а р, имеющих составы,
Тв
Рис. 6.
воров a -f- р, имеющих составы, отвечающие точкам
F и G. Подобным же образом протекает и кристал-
лизация заэвтектич. сплавов, из которых первично
выделяется твердый раствор р. При охлаждении
ниже эвтектич. темп-ры состав обоих твердых р-ров
изменяется по кривым растворимости в твердом со-
стоянии FM (для а) и G7V (для Р).
Диаграмма состояния, изображенная на рис. 7
(тип IV Розебома), характеризуется тем, что при
охлаждении всех жидких сплавов, составы которых
соответствуют точкам отрезка PG, первично выде-
Ti
та
Рис. 7.
ляется твердый раствор р. При
темп-ре, отвечающей точке Р,
состав этого твердого раствора
отвечает точке G, а состав жид-
кости — точке Р. При дальней-
шем отнятии тепла из жидкости
Р выделяется твердый раствор
а, отвечающий по составу точке
F, а твердый раствор р раство-
ряется в жидкости, причем тем-
пература остается постоянной.
Процесс этот называется пери-
тектическим, а точка Р — пери-
тектической точкой (или прос-
то перитектикой). Если
составы сплавов соответствуют
точкам, лежащим на отрезке FG, то жидкость Р израс-
ходуется раньше, чем твердый раствор, и система
затвердеет в смесь аир. Если же составы сплавов
соответствуют точкам, лежащим на отрезке PF, то
твердый раствор р израсходуется ранее жидкости Р,
из к-рой при понижающейся темп-ре будет кристалли-
1029
ДВОЙНЫЕ СИСТЕМЫ
1030
зоваться далее твердый раствор а. При нагревании
описанные превращения протекают в обратной по-
следовательности.
Изотермы свойств Д. с. с
ограниченной взаимной раство-
римостью компонентов в твер-
дом состоянии (рис. 8) имеют
две восстающие или нисходящие
криволинейные ветви, отвечаю-
щие областям твердых раство-
ров а и Р, и одну прямоли-
нейную ветвь, отвечающую двух-
фазной области а ф р. Таковы,
например, изотермы твердости
^АтЛ^в (а также давления
истечения, рис. 8,I) и электро-
проводности лАт2л<2Хв (а также
Рис. 8.
темп-рного коэфф, электросопро-
тивления, рис. 8, II). Изломы на изотермах свойств
(точки mj, zij, ma, п2, рис. 8) отвечают границам твер-
дых растворов а и р.
Кристаллизация химических соединений. Обра-
зующиеся в Д. с. химич. соединения могут при плав-
лении либо превращаться в жидкость, имеющую
тот же состав, что и исходное твердое вещество (к о н-
груэнтное плавление), либо распадаться
на жидкость и твердую фазу, отличающиеся по со-
ставу от исходного твердого вещества (и н к о и г.р у-
энтное плавление). На рис. 9 изображена
диаграмма состояния Д. с., в к-рой образуется кон-
груэнтно плавящееся соединение АтВп (обозначен-
ное для краткости буквой С), не дающее твердых раст-
воров со своими компонентами. Ордината СМ делит
эту диаграмму на две диаграммы Л—С и С—В, вполне
подобные диаграмме, рассмотренной выше (рис. 1).
Соединение С образует 2 эвтектики: Ег с компонен-
том А и Ег с компонентом В. Если соединение С не-
сколько диссоциировано в жидком состоянии, то
на его кривой ликвидуса наблюдается плавный мак-
симум М, наз. дистектич. точкой (или просто дистек-
тикай). Если же соединение С совершенно не диссо-
циировано в жидком состоянии, то темп-ре его плав-
ления отвечает точка D, называемая сингулярной
точкой. Изотермы физич. свойств Д. с. рассматри-
ваемого типа состоят из двух ветвей, близких к прямо-
линейным, пересекающихся на ординате химич.
соединения.
Если конгруэнтно плавящееся соединение АтВп
образует непрерывные твердые растворы (напр.,
I типа) со своими компонентами Л и В, то диаграмма
состояния системы А—В имеет вид, изображенный
на верхней части рис. 10. На изотермах твердости
(и давления истечения), а также электропроводности
(и темп-рного козфф. электрич. сопротивления) на-
блюдаются сингулярные точки Sj и S2, лежащие на
ординате, отвечающей составу определенного соеди-
17»
неиия АтВп. Рациональный (т. е. отвечающий цело-
численному атомному или молекулярному отноше-
нию А : В) максимум М и сингулярные точки и 6’а
лежат на ординате, отвечающей составу д а л ь т о-
н и д а (см. Дальтониды, и бертоллиды) определен-
ного соединения АтВп. В случае образования бертол-
лида темп-рный максимум на кривой ликвидуса яв-
ляется иррациональным; т. е. не отвечает простому
стехиометрич. отношению компонентов, а на изотер-
мах свойств сингулярные точки отсутствуют.
На рис. 11 изображена диаграмма состояния Д. с.
с образованием инконгруэнтно плавящегося соедине-
ния АтВп (обозначенного для краткости буквой С,
твердые растворы отсутствуют). Из жидких сплавов,
лежащих между точками ТА и Е, первично выде-
ляются кристаллы А, а затем кристаллизуется эвтек-
тика Е. Из сплавов между Е ъ Р первично выделяются
кристаллы С, а затем—эвтек-
тика Е. Из сплавов, состав ко-
торых соответствует точкам, ле-
жащим на прямой PH, сначала
выпадают кристаллы В. Затем
при темп-ре, отвечающей отрез-
ку PH, когда жидкость имеет
состав, соответствующий точке
Р, происходит растворение В и
выделение С. Этот процесс, по-
добно уже рассмотренному (рис.
7), называется перитектическим,
а точка Р — перитектикой. Си-
стема отвердевает в смесь кри-
сталлов В и С, если состав ее
отвечает точкам, лежащим на
отрезке mH. Если же состав системы лежит между
точками т и Р, то В растворяется целиком, после
чего при дальнейшем охлаждении из жидкости
кристаллизуется С. При нагревании все описанные
превращения протекают в обратной последователь-
ности. Напр., соединение АтВп плавится при тем-
пературе, отвечающей точке т, причем образуется
жидкость состава Р, а соответствующее количе-
ство компонента В выделяется в твердом состоя-
нии. Если бы соединение АтВп плавилось конгру-
энтно, то его плавлению отвечала бы точка М (т. н.
скрытый максимум), и соответствующая часть ликви-
дуса имела бы вид, показанный иа рис. 11 пунктиром.
Нерастворимость и ограниченная взаимная раство-
римость компонентов в жидком состоянии. Если ком-
поненты А и В совершенно не растворяются друг
в друге в жидком состоянии и не
дают ни твердых растворов, ни --------------
химич. соединений, то диаграмма ia+ib
состояния имеет вид, изображен- у
ный на рис. 12. Поле диаграммы
разбивается двумя горизонталь- tA+B
ными прямыми, проведенными
через точки ТАаТв, на 3 поля а+в
(значение которых указано надпи-
сями). Выше Тв—область двух Л —в
несмешивающихся жидкостей ЬА рис. 12.
и LB, между ТА и Тв — смесь
жидкости А и кристаллов В, ниже ТА — кристал-
лы А и В, образующие два слоя. На рис. 13 изоб-
ражена диаграмма состояния системы с эвтектикой,
имеющая область MKN неполной взаимной раст-
воримости Л и В в жидком состоянии. Все сплавы,
составы к-рых отвечают точкам отрезка NO, после
кристаллизации компонента В претерпевают при
постоянной темп-ре, отвечающей этому отрезку, т. н.
моиотектическое превращение. Оно состоит
1031
ДВОЙНЫЕ СИСТЕМЫ — ДДТ
1032
Рис. 13.
в
в том, что при продолжающемся выделении В появ-
ляется второй жидкий слой Lt, состав к-рого отве-
чает точке М", количество же жид-
кого слоя L2, отвечающего по
составу точке N, уменьшается.
। После полного израсходования
жидкости £а продолжается кри-
сталлизация В ИЗ ЖИДКОСТИ Z.J.
Жидкие сплавы, составы, которых
отвечают точкам отрезка MN, при
охлаждении сначала распадаются
на два слоя (область MKN), а
затем, по достижении темпера-
туры, отвечающей отрезку МО,
претерпевают монотектич. превра-
щение. В Д. с. с областью рас-
слоения в жидком состоянии воз-
можно также образование твер-
химич. соединении; соответствую-
растворов и
типы диаграмм состояния описываются в спе-
ДЫХ
щие
циальной литературе.
Превращения в твердом состоянии. При охлажде-
нии ниже темп-ры солидуса Д. с. могут претерпевать
весьма разнообразные превращения в твердом состоя-
нии. В системах без твердых р-ров эти превращения
происходят либо вследствие полиморфизма компо-
нентов или их соединений, либо вследствие образова-
ния химич. соединений, устойчивых только ниже
темп-p конца отвердевания. В системах с твердыми
р-рами превращения в твердом состоянии могут быть
следствием: а) изменения предельной концентрации
твердых р-ров при понижении темп-ры (см. выше,
рис. 6 и 7), б) полиморфизма компонентов, в) образо-
вания химич. соединений из твердых р-ров. Напр.,
если полиморфные модификации компонентов А и В,
устойчивые при высоких темп-pax (между ТА и Т'А<
и Т'в на рис. 14), дают один твердый раствору
(напр., I типа), а модификации, устойчивые при низ-
ких темп-pax (ниже 7’д и Т’в), образуют два твердых
раствора а и 0 (напр., V типа), то диаграмма состоя-
ния получается, как сочетание рис. 4, I и рис. 6.
Кривые начала превращений в твердом состоянии
ТАе и Т'ве пересекаются в эвтектоидной точнее (ана-
логичной эвтектич. точке Е, рис. 6). При темп-ре,
отвечающей точке е, твердый раствор у, состав к-рого
дается ее абсциссой, находится в равновесии с двумя
насыщенными твердыми растворами а и 0, отвечаю-
щими по составу абсциссам точек f ng. Мелкодисперс-
ная смесь твердых р-ров а и Р, получающаяся в ре-
зультате распада твердого раствора у, по своему
строению похожа на эвтектику и потому называется
эвтектоидом. Типичный пример эвтектоида —
т. н. перлит в железоуглеродистых сплавах.
Если при охлаждении твердого раствора а обра-
зуется химич. соединение АтВп, дающее со своими
компонентами твердый раствор р, то диаграмма
состояния имеет вид, изображенный на рис. 15.
Кривая темп-p начала превращений amb имеет мак-
симум т, а кривая темп-p конца превращений cmd —
сингулярную точку, отвечающую составу соединения
АтВп. Изотермы свойств a-фазы имеют вид плавных
кривых, свойственных непрерывному ряду твердых
р-ров (рис. 5), а на изотермах свойств 0-фазы наблю-
даются сингулярные точки при составе АтВп. См.
также Физико-химический анализ.
Лит.: К у р н а к о в Н. С., Растворы и сплавы, в его
кн.: Введение в физико-химический анализ, 4 изд.. М.—Л.,
1940; его же, Собрание избранных работ, т. 1—2, Л.,
1938—39; А и ос о в В. Я. и Погодин С. А., Основные
начала физико-химического анализа, М.—Л., 1947 (гл. 4—16);
Аносов В. Я., Краткое введение в физико-химический
анализ, М., 1959; Млодзеевский А. Б., Теория фаз
(с применением к твердым и жидким состояниям), М.—Л.,
1937; Агеев Н. В., Химия металлических сплавов М.—Л.,
1941; Бочвар А. А., Металловедение, 5 изд., М., 1956;
Г и н з б е р г А. С., Экспериментальная петрография, Л.,
1951; Хансен М., Структуры бинарных сплавов, пер.
с нем., т. 1—2, М.—Л., 1941; Hansen М., Constitution of
binnary alloys, 2ed., N. У., 1958; ГаллФ. П. и ИнслейГ.,
Правило фаз и применение диаграмм состояния к изучению
силикатных систем, пер. с аигл., Киев, 1936; Меншуткин
Б. Н., Изв. Ин-та физ.-хим. анализа, 1921, 1, вып. 2, с. 473;
Timmermans J., Les solutions concentrfes. Th£orie
et applications aux m£langes binaires de composes organiques,
P., 1936; Seidell A., Solubilities, v. 1—2, 3 ed., N. Y„
1940—41, Suppi., 1952; 4 ed., v. 1, N. Y„ 1958.
С. А. Погодин.
Д Д Д (4,4' - дихлордифенилдихлорметилметан), мол.
вес 320,06 (С1СвН4)аСНСНС1а — активный контакт-
ный инсектицид, лишь немногим уступающий ДДТ',
кристаллический продукт; т. пл. 110—111°; практи-
чески нерастворим в воде, хорошо растворим в ор-
ганич. растворителях. При нагревании выше 150°
разлагается с выделением НС1; особенно легко раз-
лагается в присутствии хлориого железа. ДДД полу-
чают конденсацией дих.торацетальдегида с хлорбен-
золом в присутствии серной к-ты или олеума, в усло-
виях, близких к условиям получения ДДТ. Положи-
тельным свойством ДДД является его малая токсич-
ность для теплокровных животных (3400 лг/кг),
однако в последнее время в литературе появились
данные О высокой хронической токсичности ДДД.
В с. х-ве ДДД, так же как и ДДТ, применяют для
борьбы с различного рода вредителями; в быту —
против мух, насекомых и др. Способы применения
ДДД и примерные нормы его расхода такие же, как
Для ДДТ,
Лит. см. при ст. ДДТ. Н. Н. Мельников.
ДДТ (4,4' - дихлордифенилтрихлорметилметан)
(С1СвН4)аСНСС13, мол. в. 354,51 — важнейший совре-
менный инсектицид. Впервые синтезирован в 1874,
инсектицидные свойства открыты в 1937. Чистый
препарат — бесцветные кристаллы; т. пл. 108,5—109°
(в технич. продукте содержится 73—80% 4,4'-ДДТ).
Важнейшей примесью является 2,4'-дихлордифенил-
трихлорметилметан, не обладающий инсектицидными
свойствами; т. пл. 78—106°. ДДТ практически не-
растворим в воде, хорошо растворим во многих орга-
нич. растворителях, особенно в хлорпроизводных
углеводородов, в ароматич. углеводородах, сложных
эфирах, кетонах и простых эфирах. Растворимость
ДДТ в керосине и минеральных маслах 4—10%.
ДДТ термически устойчив (выдерживает нагревание
до 150—170°, а кратковременное и до 300—350°).
В присутствии солей 3-валентного железа термич.
устойчивость ДДТ резко уменьшается; Отрицатель-
ное действие солей железа на ДДТ может быть сни-
жено добавками (мел, карбонат магния и др.). На
свету ДДТ постепенно разлагается. Вода при обычной
темп-ре на ДДТ действует медленно, скорость гидро-
лиза увеличивается при повышении темп-ры. При
нагревании со спиртовыми р-рами щелочей и органич.
основаниями протекает след, реакция:
(С1СвН1)2СНСС1в + КОН = (CiCeH4)sC = СС1» + КС1 + н2о
1033
ДЕАСФАЛЬТИЗАЦИЯ — ДЁВНЕРА — МИЛЛЕРА РЕАКЦИЯ
1034
Эта реакция используется для колич. определения
суммы изомеров ДДТ в технич. продукте и различных
препаратах.
Получение ДДТ основано на взаимодействии хло-
раля с хлорбензолом в присутствии различных кон-
денсирующих средств:
СС13СНО + 2СвН5С1 = (С1СвН4)2СНСС13 + н2о
Наиболее хорошо изучена и получила широкое
практич. применение конденсация в присутствии сер-
ной к-ты, олеума или хлорсульфоновой к-ты. Йо
окончании реакции ДДТ отделяют от серной к-ты,
хлорбензол отгоняют и полученный продукт после
кристаллизации используют для приготовления ин-
сектицидных препаратов. Для получения технич.
ДДТ с повышенным содержанием 4,4'-изомера про-
дукт конденсации хлораля с хлорбензолом промы-
вают органич. растворителями или перекристалли-
зовывают.
ДДТ — универсальный контактный инсектицид,
к-рый проникает в тело насекомого и поражает его
нервную систему; для отравления личинок мух до-
статочно одной миллиардной грамма ДДТ на 1 см2
обрабатываемой поверхности. Для борьбы с большин-
ством вредителей растений расходуется 0,5—1,5 кг
на 1 га, для борьбы с личинками малярийного комара —
125 г/га. ДДТ — высокотоксичный препарат и для
полезных насекомых, уничтожающих растительнояд-
ных- клещей и тлей; поэтому при применении ДДТ
иногда наблюдается бурное развитие последних. В ре-
зультате массового применения ДДТ появились
устойчивые к этому препарату формы насекомых,
к-рые не погибают и при 10-кратвом увеличении доз
ДДТ.
ДДТ применяют в виде порошков, смачивающихся
порошков, представляющих собой тонкоразмолотую
смесь ДДТ, наполнителя, прилипателя и поверх-
ностно-активного агента, р-ров в различных органич.
растворителях и эмульсиях. 4—5%-ный р-р ДДТ в очи-
щенном керосине известен под названием дезин-
секталя. Р-ры ДДТ в маслах получили за послед-
ние годы широкое применение для т. н. аэрозольного
опрыскивания. Токсичность ДДТ для теплокровных
колеблется в пределах 250—400 мг!кг. ДДТ особенно
опасен для млекопитающих (в том числе и человека)
в период питания материнским молоком, поэтому
в молоке не должно содержаться ДДТ. Содержание
ДДТ в других пищевых продуктах не должно превы-
шать 1,5 мг/кг.
Лит.: Мельников Н. Н., Набоков В. А.,
Покровский Е. А., ДДТ. Свойства и применение, М.,
1954; В а ш к о в В. И., Погодина Л. Н., Сазоно-
ва Н. А., ДДТ и его применение, М., 1955.
Н. Н. Мельников.
ДЕАСФАЛЬТИЗАЦИЯ — удаление из нефти и
нефтепродуктов растворенных и диспергированных
в них асфальто-смолистых веществ. В пром-сти Д.
используется гл. обр. для очистки минеральных
масел, к-рая производится след, способами: перегон-
кой в вакууме, обработкой серной к-той с последую-
щей контактной очисткой отбеливающими землями,
обработкой отбеливающими землями с последующей
перколяционной очисткой. В настоящее время все
большее применение находит Д. при помощи специаль-
ных растворителей, для чего могут быть использованы
низкомолекулярные алканы, от этана до гексана.
Применение их основано на том, что при растворении
нефтей и нефтепродуктов происходит коагуляция
йсфельтенов и смол, благодаря чему становится
возможным удаление их из раствора.
В процессах Д. используется жидкий пропан,
содержащий не более 2% этана и 2% бутана. Часто
для Д. вместо пропана применяют смесь пропана и
пропена, выделяемых из крекинг-газов, хотя при
этом получаются несколько худшие результаты.
Количество выделяющихся асфальтовых и смолистых
веществ (или селективность) зависит от темп-ры Д.
В последнем варианте технология, процесса Д.
производится в колонне непрерывного действия.
Сырье движется в низ колонны и интенсивно обраба-
тывается на тарелках восходящим потоком сжижен-
ного пропана, в к-ром растворяются составные части
масла. Асфальтовые и смолистые вещества, нераство-
римые в пропане, отстаиваются в нижней части ко-
лонны и непрерывно выводятся. Темп-ра верха и низа
колонны устанавливается в зависимости от характера
сырья и получения желательного качества продукта.
Соотношение пропана и сырья составляет обычно
8 : 1 по объему. В результате Д. улучшаются вяз-
кость, цвет и коксуемость масла, а также получается
битум. Д. пользуются для фракционирования мазу-
тов, масляных нефтей, применяя различный темп-рный
режим в деасфальтизационной колонне, с выводом
отдельных фракций по высоте колонны.
Лит.: Технология нефти, ч. 3, 3 изд., М.—Л., 1952; К з-
личевский В. А., Современные методы производства
смазочных масел, пер. с англ., М.—Л., 1947.
П. Н. Лалич.
ДЕБАЯ — ФАЛЬКЕНГАГЕНА ЭФФЕКТ (диспер-
сия электропроводности) — повышение электропро-
водности р-ров электролитов в высокочастотных
электрич. полях по сравнению с их электропровод-
ностью, измеряемой при использовании переменного
тока низкой частоты или постоянного тока; предсказан
теоретически в 1928 П. Дебаем и Г. Фалькёнгагеном,
позднее обнаружен опытным путем. Д.—Ф. э. — ре-
зультат частичного или полного исчезновения в высо-
кочастотных полях релаксационного тормозящего
действия, т. е. той части тормозящего действия ионной
атмосферы на движение иона, к-рая обусловлена
несимметричным распределением зарядов в ионной
атмосфере движущегося иона (см. также Электропро-
водность электролитов, Электролиты).
Н. Е. Хомутов.
ДЁБНЕРА — МИЛЛЕРА РЕАКЦИЯ — один из
важных методов синтеза производных хинолина,
заключающийся в действии альдегидов на ароматич.
амины в присутствии кислот. Так, при реакции ани-
лина с паральдегидом и соляной (применяется также
серная) к-той образуется а-метилхинолин (хиналь-
дин). Прибавление небольших количеств ZnCl2 при-
водит к увеличению выхода. Д.—М. р. характери-
зуется широкими границами приложимости: хорошие
выходы наблюдаются, исходя из различных заме-
щенных ароматич. аминов (при условии свободного
о/ипо-положения), при применении смеси альдегидов
или смеси альдегида и кетона (модификация Бейера).
Механизм Д.—М. р. заключается в след.: в ре-
зультате конденсации 2 молекул ацетальдегида обра-
зуется кротоновый альдегид СН3СН=СНСНО, к-рый
далее реагирует с анилином с образованием шиффова
основания CH3CH = CHCH=NC6H5 (I); присоедине-
ние анилина к I ведет к соединению строения
C6H5NHCH(CH3)CH2CH=NC6H5 (II), к-рое с отщеп-
лением анилина циклизуется в дигидрохинальдин
(III). С другой стороны, возможно, что III образуется
в результате циклодегидратации I (продукта присо-
единения кротонового альдегида к анилину).
Далее шиффово основание I или CH3CH = NC6HS
(из ацетальдегида и анилина) реагируют с III как
акцепторы водорода с образованием хинальдина (IV)
и н-бутиланилина (этиланилина); реакции см. на
стр. 1035.
Нетрудно заметить аналогию между Д.—М. р. и
синтезом пиридиновых оснований из альдегидов и
аммиака. Д.—М. р., открытая в 1881, по существу
заключающаяся в промежуточном образовании ве-
ществ типа III, вполне аналогична синтезу Скраупа
1035
ДЕВАРДА СПЛАВ — ДЕГИДРОБЕНЗОЛ
1036
CH»NCeH5
CH3CH“CHCH»NC6H5 (/) ХСН2
+ c6h6nh2 ~I II <!;нсн3 '
//
CH3CH-CHCH-NC6H6 (/)
Х-СН3 + (+ CH3CH=NC6HS)
Nn
III
a”X H.-C4H9NHCeH5
N^-CH3 + (C2H5NHC6H5)
IV
ям. Скраупа реакция)-, разница заключается лишь
в том, что в последнем случае для дегидрирования III
в реакционную смесь вводится окислитель.
Лит.: Гетероциклические соединения, под ред. Р. Эльдер-
филда, пер. с англ., т. 4, М., 1955; Ардашев В. И., Усп.
химии, 1954, 23, вып. 1. Я.Ф. Комиссаров.
ДЕВАРДА СПЛАВ — сплав 50% меди, 45% алю-
миния и 5% цинка; сплав хорошо растирается в по-
рошок; из воды выделяет водород уже на холоду.
Д. с. приметают в аналитич. химии для восста-
новления нитратов и нитритов в аммиак, к-рый
улавливают титрованным р-ром кислоты, и избыток
последней титруют.
ДЕГАЗАЦИЯ — обезвреживание или удаление
отравляющих веществ (ОВ) с различных объектов.
Д. как понятие возникло в первую мировую войну,
когда ОВ получили широкое применение на поле боя,
причем в начальный период в виде газов (хлор, фос-
ген), а в последующий — в виде капельно-жидких
стойких отравляющих веществ, напр. дихлордиэтил-
сульфида (иприт). Обезвреживают обычно объекты,
зараженные гл. обр. стойкими жидкими ОВ. Ниже
кратко описываются способы обезвреживания, к-рые
подразделяют на физические, механические и химиче-
ские. К физическим способам относят естест-
венное испарение и смывание ОВ с зараженного
объекта различными растворителями. Испарение —
медленный процесс, и им можно пользоваться только
в случае заражения местности, на к-рой длительное
время не будут появляться люди. Смывание органич.
растворителями можно применять для удаления ОВ
с предметов вооружения, транспортных средств и др.
объектов путем тщательного протирания зараженной
ОВ поверхности ветошью или паклей, смоченной
растворителями. В качестве растворителей можно
применять бензин, керосин, дизельное топливо, спирт
и др. Расход растворителя в среднем составляет
0,5—1 л/м2 поверхности. Смывание ОВ эффективно
для металлич. или деревянных окрашенных поверх-
ностей и менее эффективно для неокрашенных дере-
вянных поверхностей. К механическим спо-
собам Д. относятся удаление зараженного слоя почвы
(снега) или изоляция зараженной поверхности путем
засыпки ее различными инертными материалами;
эти способы могут быть использованы для обезвре-
живания местности, дорог и нек-рых инженерных
сооружений. Для удаления зараженного слоя почвы
или снега могут быть применены дорожные машины.
Химическое обезвреживание производится
дегазирующими веществами; при этом ОВ разру-
шаются и превращаются в нетоксич. соединения.
Химич, способы значительно более эффективны вслед-
ствие практически полного разложения ОВ. В ка-
честве дегазирующих веществ применяют: хлорную
известь, основные гипохлориты кальция, различные
хлорамины, соду, двууглекислый аммоний и аммиач-
ную воду. Хлорная известь употребляется для Д.
III
местности и дорог кай в сухом виде
(расход 0,5—1,0 кг/м2), так и в виде
суспензий. Суспензии могут быть исполь-
зованы также для Д. зданий, средств
транспорта, машин и т. п. Монохлор-
амин Б (C6H5SO2NNaCl • ЗН2О) приме-
няют в водных или спиртоводных рас-
творах для санитарной обработки кожи человека
(открытых участков и через обмундирование). Ди-
хлорамин Б (C6H5SO2NC12) используется в виде
10%-ного р-ра в дихлорэтане для Д. средств транс-
порта и др. объектов при заражении их стойкими
ОВ; эти дегазирующ. в-ва являются одновременно
и антисептиками. Соду и двууглекислый аммоний при-
меняют для Д. одежды и белья, зараженных различ-
ными ОВ. При этом предметы одежды кипятят в 2%-ном
р-ре соды или обрабатывают в специальных каме-
рах паро-воздушно-аммиачной смесью. Аммиачную
воду можно применять для Д. различных объектов,
зараженных дифосгеном, люизитом и ОВ типа табун,
зарин и др. В ряде случаев Д. производят сернистым
натрием, едким натром, гашеной известью.
Лит.: Баратов Г. Ф., Местная противовоздушная
оборона населения в условиях химического, атомного и бак-
териологического нападения, Киев, 1959; Колосов Б. В.,
Противоатомная, противохимическая, противобактериологи-
ческая защита солдата в бою, М., 1957. И. Г. Пенвулаев.
ДЕГИДРАЗЫ — ферменты, катализирующие ре-
акцию отщепления воды от субстрата. К Д. отно-
сятся: енолаза, D- и L-серин-дегидраза, D- и Б-трео-
нин-дегидраза
RCH(OHJCH(NH2)COOH S; НаО + [RCH=C(NH2)COOH] -»
—► RCH2COCOOH 4-NH3.
Конечные продукты действия Д. серина — пировино-
градная к-та и аммиак, Д. треонина — а-кетомасля-
ная к-та и аммиак. Д., действующие на Б-амино-
кислоты, не действуют на D-формы, и наоборот.
Коэнзимом Д. серина и треонина является пирид-
оксальфосфат (см. Пиридоксин). За единицу активности
принимается такое количество фермента, к-рое необ-
ходимо для образования 1 • 10-7 моля кетомасляной
или пировиноградной к-т за 20 мин при pH 9,3 и
темп-ре 37°. Д. Б- и D-серина и Б- и D-треонина
обнаружены в животных тканях и бактериях.
Лит.: Диксон М., Уэбб Е., Ферменты, пер. с англ.,
М., 1961; Methods In enzymology, ed. S. P. Colowick and N. O.
Kaplan, v. 2, N. Y., 1955, 319. T. T. Болотина.
ДЕГИДРОАНДРОСТЕРОН — см. Дегидроэпианд-
ростерон.
ДЕГИДРОБЕНЗОЛ (циклогексадиенин) С6Н4 —
гипотетический продукт отщепления двух атомов
водорода от молекулы бензола. Д. в чистом виде не
выделен, его кратковременное существование подтвер-
ждено выделением продуктов его дальнейших превра-
щений. Для Д. возможно строение: с тройной связью
(I), биполярного сое-
динеиия (II) и бира- [ Ц1 | |Г* | |Г*
дикала (III); наиболее
вероятно строение II.
Представления о про- 11 111
межуточном образовании Д. возникли в результате
установленных многочисленных случаев «аномаль-
ного» нуклеофильного замещения в ароматич. ряду,
при к-ром нарушается принцип структурного со-
ответствия между исходными и конечными про-
дуктами. Несмотря на то, что группы ОСН3 и CF3
ориентируют дальнейшее замещение в противопо-
ложных направлениях, при действии NH2K на
о-СН3ОС6Н4С1 или o-CF3C6H4C1 образуются в рав-
ной мере мета-аминозамещенные. Действием NH2K
на С6Н5С1 с меченым атомом С14, связанным с хлором,
получена смесь равных количеств анилинов с меченым
атомом углерода в положениях 1 и 2. Такой результат
объясняется промежуточным образованием Д., к-рый
1037
ДЕГИДРОГЕНАЗЫ — ДЕГИДРОЭПИАНДРОСТЕРОН
1038
вследствие симметричности тройной связи присоеди-
няет НН2-группы в двух возможных направлениях
(цифра 14 в приведенных формулах показывает, что
в цикл входит атом углерода С*4). Аналогично ведет
себя и нафталин. Напр., при обработке а- н Р-хлор-
нафталинов амидом калия в жидком NH3 образуется
одинаковая смесь а- и р-нафтиламинов с соотноше-
нием а- и p-изомеров, равным 1:2:
-HCi
При реакции фениллития с а- или р-фторнафталином
с последующим действием СО2 выделена смесь 1-фенил-
нафтойной-2 и 2-фенилнафтойной-1 кислот. Видимо,
в обоих случаях сначала образуется 1,2-дегидронаф-
талин.
Существование Д. подтверждается также и тем,
что при обработке о-фторбромбензола амальгамой
лития получен дифенилен (IV). Еще более убеди-
тельны результаты опытов с введением Д. в реакцию
Дильса — Альдера. При обработке o-FC6H4Br амаль-
гамой Li в фуране получается с хорошим выходом
эндоокись (V), что объясняется промежуточным об-
разованием Д., который, как диенофил, реагирует
с фураном в качестве диена по схеме реакции Дильса —
Альдера (см. Диеновый синтез). Строение эндоокиси
(V) подтверждено ее изомеризацией (в р-ре НС1
в СН3ОН) в а-нефтол, в присоединении 1 моля водо-
рода с образованием вещества, легко дегидратируемого
в нафталин, и др. реакциями. Подобно фурану, реаги-
руют с Д. и другие диены (циклопентадиеи, цикло-
гексадиен).
Способность Д. вступать в диеновый синтез можно
использовать для препаративных целей. Так, трудно-
доступный углеводород триптицен (VI) можно, синте-
зировать в одну стадию
Г Jl взаимодействием антрацена
v''''] с о-фторбромбензолом и
I I L 1 JL J Mg в тетрагидрофуране.
Вполне вероятно, что со-
единения типа Д. являются
* промежуточными продук-
тами при ряде известных процессов, напр. при
гидролизе хлорбензола в фенол, при превращении .и-
и n-бензолдисульфокислоты при щелочном плавлении
в резорцин и т. д.
Лит.: В и т т и г Г., Усп. хим., 1958, 27, вып. 3,с. 291—305.
Я. Ф. Комиссаров.
ДЕГИДРОГЕНАЗЫ — ферменты, катализирую-
щие окислительно-восстановительные реакции; со-
держатся во всех живых клетках. Многие Д. выделены
в виде кристаллич. однородных белков. Д. обычно
проявляют свое действие в присутствии кофермен-
тов — коэнзимов I или II, представляющих собой
пиридиннуклеотиды.
Д. активируют атомы водорода окисляемого орга-
нич. вещества, делают их способными переходить
на другие соединения — акцепторы: ВН2 ф As
В 4' АН2. Д. специфичны, они могут переносить
атомы водорода лишь между определенными соеди-
нениями. В зависимости от химич. природы субстрата
Д. носят соответствующие названия: с у к ц и н д е-
гидрогеназа, альдегиддегидроге-
наза, алкогольдегидрогеназа, Д.
пировиноградной к-ты, Д. а-кетоглутаровой к-ты
и т. д. В зависимости от природы акцептора водорода
Д. делятся на две группы: 1) анаэробные, к-рые не
могут отдавать водород кислороду воздуха, а пере-
дают его другим акцепторам, напр. промежуточным
продуктам распада углеводов, другим дегидрогеназам
или хиноноподобным соединениям. Это наиболее
многочисленная группа Д., принимающая участие
в процессах брожения и дыхания; 2) аэробные, пере-
носящие водород непосредственно на кислород с обра-
зованием Н2О или Н2О2 в качестве конечных продук-
тов. Аэробные Д., для к-рых акцептором Н является
исключительно кислород воздуха, получили название
оксидаз.
Лит.: Михлин Д. М., Биологическое окисление,
М., 1956; его же. Биохимия клеточного дыхания, М.,
1960; Methods in enzymology, v. 1, N. Y., 1955, p. 323, 350.
P. А. Звягилъская.
ДЕГИДРОГЕНИЗАЦИЯ — см. Гидрогенизация и
дегидрогенизация каталитические.
ДЕГИДРОГЕНИЗАЦИЯ НЕФТЕПРОДУКТОВ —
каталитическое отщепление водорода от углеводородов
нефти. Реакции дегидрогенизации используются в ряде
технологич. процессов нефтепереработки: получение
непредельных углеводородов и ароматизации и др.
Дегидрогенизация применяется для получения бути-
ленов и изобутилена из бутана и изобутаиа. Процесс
ведут при 500—600°, преим. на алюмохромовых
катализаторах. Превращение бутиленов в бутадиен
на катализаторах, состоящих из окиси магния, желе-
за, меди и калия, или на окисном хромо-алюминиевом
катализаторе протекает в присутствии паров воды,
углекислого газа или при пониженном давлении я
температуре 600—650°.
Ароматич. углеводороды (бензол, толуол) полу-
чаются одновременно проводимой дегидрогенизацией
гексаметиленовых углеводородов и дегидроциклиза-
цией парафиаовых углеводородов бензино-лигроино-
вых фракций (70—180°) нефти на алюмо-молибденовом
катализаторе при 480—550° и 10—20 ат или плати-
новом катализаторе при 450—525° и 15—17 ат (см.
Риформинг)’, в обоих случаях Д. н. выполняют в при-
сутствии водорода. Большое промышленное значение
имеет каталитич. дегидрогенизация этилбензола и
изопропилбензола в стирол и метилстирол. См. также
Ароматизация нефтепродуктов.
Лит.: Химия углеводородов нефти, под ред. Б. Т. Брукса
[и др.], пер. с англ., т. 2, Л., 1958, с. 162—213.
Г. М. Циеуро.
ДЕГИДРОЭПИАНДРОСТЕРОН (А5-андростен-ЗР-
ол-17-он) С18Н22О2, мол. вес 288,42 — андроген-
ный гормон (стероидный),
кристаллы, существующие СН3
в нескольких модификаци- ___1=0
ях; т. пл. 138° (из водного Н3С I I I
метанола), 148° (из петро-
лейного эфира) и 153° (из | ( I
воды). Д. нерастворим в во-
де, хорошо растворим в спир-
те, эфире, бензоле, плохо — в СС14 и петролейном
эфире; [а]^ = 4-10,9° (в спирте). Д. дает реакции
на спирты, кетоны, двойную связь и реакции, харак-
терные для 17-кетостероидов. Для колич. определе-
ния Д. из мочи используют реакцию Циммермана
с метадинитробензолом (фиолетовое окрашивание);
кристаллич. Д. с H2SO4 в спиртовом р-ре дает сине-
фиолетовую окраску. Д. обладает слабой андрогенной
активностью (200 мкг = 1 ME).
Д. содержится в моче и крови людей, а также
в моче крупного рогатого скота (коров, быков). Полу-
чают Д. из мочи, а в промышленном масштабе —
синтетически из холестерина (см. стр. 1039), а
также из нек-рых фитостеринов. Ацетат Д. служит
основным промежуточным продуктом для получения
тестостерона, метилтестостерона, прогестеронапдр.
1039
ДЕГОТЬ ПЕРВИЧНЫЙ
1040
цевтических препаратов, Л., 1958; см. также при ст. Андро-
генные гормоны. С. А. Афиногенова.
ДЕГОТЬ ПЕРВИЧНЫЙ (смола первичная) —
продукт различной вязкости, получаемый при сухой
перегонке твердых топлив (при 500—550°) наряду
с газом, полукоксом и подсмольной водой: в отличие
от смол, образующихся при высокотемпературной
сухой перегонке (900—1100°), такие смолы принято
называть первичными. Сырьем для получения Д. п.
служат топлива, богатые летучими веществами, даю-
щие достаточно высокий выход Д. п. Химич, состав
Д. п. зависит от вида исходного топлива, а также от
метода и режима его переработки. Д. п. состоит из
продуктов низкотемпературного распада органич.
массы топлив. В состав Д. п. входят: углеводороды
различных классов, фенолы, органич. основания,
нейтральные кислородные соединения и пр. В табл. 1
приведен выход и состав Д. п., получаемых при пере-
работке различных видов твердого топлива.
Существенным ком-
понентом древес-
ных дегтей являет-
ся смесь фенолов (фе-
нол, крезолы, ксилено-
лы, гваякол и др.),
причем термин «первич-
ный» к древесному, дег-
тю обычно на приме-
няется, т. к. при пере-
работке древесины вы-
сокотемпературные ре-
жимы практически не
используются. В соста-
ве древесных дегтей со-
держатся насыщенные
кислоты жирного ряда,
от муравьиной до ка-
приловой; из высших
к-т этого ряда найдены
Р|_| пальмитиновая, арахи-
3 новая, бегеновая и др.;
। | i=NNHCONH2 установлено также при-
L J H2SO4 сутствие ненасыщенных
I (НС|Гсн3СООнТ кислот жирного ряда и
ароматич. кислот (бен-
р войной, абиетиновой и
др.). В состав нейт-
ральных соединений входят кетоны, альдегиды, окси-
кетоны, спирты, углеводороды различных классов,
углеводы и др. При переработке древесных дегтей
могут быть получены пековый крепитель (продукт,
применяемый при формовании в литейном деле),
флотореагенты, уксусная к-та, креолин, медицинский
креозот и др.
Наиболее ценными составными частями торфя-
ных дегтей являются воски (3—9%) и фенолы
(15—22%). Выделение восков целесообразно только
из Д. п. верховых (битуминозных) торфов. Перера-
ботка торфяных Д. п. может осуществляться по двум
схемам. По первой схеме из Д. п. выделяют предвари-
тельно воски, а затем уже подвергают перегонке;
по второй — деготь подвергается перегонке без пред-
варительного выделения восков. При перегонке
отбирают три фракции масел: фракция 175—225°,
являющаяся сырьем для выделения крезолов, фрак-
ция 225—300°, используемая для приготовления
антисептиков и ядохимикатов, и фракция 300—360°,
применяемая для консервирования древесины. Кроме
того, при перегонке торфяного Д. п. получается
кубовый остаток — пек. Из торфяного Д. п. могут
быть получены воски с т. пл. 70—80°, парафин,
карболовая к-та, трикрезол и др., продукты. На
основе продуктов перегонки торфяного Д. п. могут
быть получены креолин, лизол, карболи-
Таблица 1 неум, резольный клей, литейный крепи-
Наименование , показателей Исходное твердое топливо
древе- сина торф бурый уголь камен- ный уголь слайды
Выход дегтя на сухое топ- ливе, % . . Плотность, г/см3 ...... Фенолы, % Органич. кислоты, % . . . Органич. основания, % . . Нейтральные вещества6, % Парафин твердый, % . . . . 7—11 0,9-1,1 10-19 до 30а 40-60 5—9 0,95-1.05 15,0—22,0 1,5—2,0 1,0-3,5 40,0-60,0 3,0—8,0 6—10 0,90—1.00 10,0—20,0 0,1—0,15 0,8—1,4 до 65 7,0—19,0 7-12 0,96—1,08 20,0-35,0 ДО 2,5 40,0—00,0 4,0—6,0 до 18 0,95—1,0 } до 25 0,2—0,5 ДО 65
а В дегте древесины лиственных пород содержится до 15% смоляных н-т,
хвойных пород — до 30%.
6 Предельные, непредельные и ароматические углеводороды, альдегиды,
кетоны, спирты, нейтральные азотистые соединения и др.
тель и т. д.
Буроугольный Д. п. характе-
ризуется высоким содержанием парафи-
на (до 19%), сравнительно небольшим
количеством асфальтенов (3—6%) и почти
полным отсутствием карбоидов (веще-
ства, нерастворимые в бензоле). Одним
из методов переработки буроугольной
смолы является ее гидрогенизация для
получения бензина, а также для произ-ва
дизельного топлива, смазочных масел
и парафина. Буроугольный Д. п. можцо
подвергнуть также дистилляции с после-
дующей переработкой получаемых фрак-
ций на товарные продукты (фенолы, пара-
фин, жидкие топлива). Обесфеноливание
и Очистка легких и средних фракций
1041
ДЕГОТЬ ПЕРВИЧНЫЙ - ДЕЗАКТИВАЦИЯ
1042
производится кислотно-щелочным способом или с
помощью селективных растворителей. Высококипя-
щие фракции Д. п. битуминозных топлив (бурого угля,
торфа) содержат, как правило, значительные количе-
ства твердых парафинов, к-рые выделяют из парафи-
нового масла непрерывной кристаллизацией.
Каменноугольный Д. п. отличается вы-
соким содержанием фенолов, доходящим до 35%.
Схема переработки кам.-уг. Д. п. приближается
к схеме переработки торфяных Д. п. После предва-
рительной подготовки с целью отделения воды и
механич. примесей деготь дистиллируют с отбором
бензино-лигроиновой и керосиновой фракций, к-рые
затем подвергаются очистке и вторичной перегонке.
Из полученных при щелочной очистке фракций фено-
лятов удаляют паром нейтральные масла. Затем
гюноляты подвергают разложению углекислым газом
и доразложению серной к-той), в результате чего
получаются «сырые» фенолы (смесь фенола, крезолов,
ксиленолов и высококипящих фенолов), к-рые могут
быть направлены на ректификацию для получения
узких фенольных фракций или использованы в виде
фенолов для получения резольных клеев, антисеп-
тиков и др. продуктов. Мазут используется как
топливо или подвергается разгонке с получением
масел и пека. Из мазута путем его окисления (про-
дувка воздухом) может быть также получен битум
для дорожных покрытий. Из тяжелых масел с добав-
кой более низкокипящих фракций получают шпа-
лопропиточное масло. Возможна также переработка
кам.-уг. Д. п. другими способами, напр. методом
гидрогениза ции.
Сланцевый Д. и. получают полукоксованием
сланца в туннельных печах и генераторах. Наиболее
распространена переработка сланцев в газогенера-
торах. Физико-химич. характеристика Д. п., полу-
чаемых из прибалтийских сланцев, приведена в табл. 2.
Таблица 2
Показатели Деготь туннель- ных печей Генера- торный деготь
Плотность при 20° 0,97 ок. 1
Начало кипения, °C 80 130
Выкипает до 200°, % 20 5
» » 300°, % 45 25
» • 360° % Вязкость при 50°С, °Э 65 53
1,6 3,5
Сера, % • • 0,9 0.9
Теплотворность, ккал/кг 9 830 9 450
Влажность, % • 2,0 2,0
Углеводороды, % . . • ок. 30 ок. 20
Кислородные соединения, % . . • до 55 ДО 60
В том числе:
Кислот и фенолов до 25 ДО 25
Нейтральных кислородных соеди-
нений До 30 до 35
Асфальтенов и др. соединений,
содержащих кислород и серу . ДО 13 ДО 19
Нейтральные кислородные соединения сланцевого
Д. п. содержат гл. обр. альдегиды и кетоны. В состав
углеводородов входят алканы нормального строе-
ния, цикланы, алкены, диены и др. При переработке
сланцевого Д. п. можно получить жидкое топливо
различных марок, мазут, флотореагенты, ядохими-
каты, резольные смолы и др. Сланцевый Д. п. можно
перерабатывать также методом гидрогенизации с по-
лучением моторных топлив и химич. продуктов.
При переработке приволжских сернистых сланцев
получается Д. п., богатый органич. сернистыми
соединениями (тиофеном, бензотиофеном и др.). Пе-
реработка этого Д. п. производится методом дистил-
ляции с отбором легкокипящей и высококипящей
франций. При хлорировании легкокипящей фракции
получают алъбихтол. Высококипящую фракцию суль-
фируют и нейтрализуют аммиаком (см. Ихтиол)
или едким натром. В последнем случае получаются
натровые соли сульфосланцевых к-т, которые явля-
ются хорошими деэмульгаторами для нефтяных и др.
эмульсий.
Лит.: Раковский В. Е., Общая химическая техно-
логия торфа, М.—Л., 1949; Т а у А., Полукоксование углей,
пер. с нем., М.—Л., 1948; Гойхрах Н.М.и Пинягин
Н. Б., Химия и технология искусственного жидкого топлива,
2 изд., М., 1954; Сумароков В. П., Химия и технология
переработки древесных смол, М.—Л., 1953.
А. М. Кунин.
ДЕЗАКТИВАЦИЯ — очистка различных предме-
тов от радиоактивных веществ, присутствующих
в виде загрязнений. При работе с радиоактивными
веществами чаще всего загрязняются ими химич.
посуда, рабочие столы, вытяжные шкафы, боксы,
полы и стены помещений, поверхность тела работаю-
щих (особенно руки). Оборудование, на к-рое могут
попасть радиоактивные загрязнения, изготовляют
из материала, легко поддающегося Д.: из нержавею-
щей стали, пластич. масс (напр., фторопласта, поли-
этилена), стекла, кварца. С поверхности этих материа-
лов радиоактивные загрязнения могут быть удалены
смыванием водой, кислотами (обычно р-рами HNO3 и
НС1), органич. растворителями (бензин, спирт, бен-
зол, четыреххлористый углерод и т. п.) и различными
веществами, образующими комплексные соединения.
Выбор последних зависит от химич. природы радио-
активных загрязнений; обычно используются р-ры
солей лимонной, щавелевой и других к-т.
Весьма целесообразно применять покрытия из
легко смываемых лаков или стекла. В случае загряз-
нения их легко снять и уничтожить. Если (как это
часто бывает в случае пористых веществ — таких,
как дерево, линолеум) радиоактивные загрязнения
проникли в толщу материала, то Д. можно произвести
путем снятия поверхностного слоя при помощи ста-
чивания, обработки наждаком или пескоструйным
аппаратом, частичного растворения. Загрязненная
одежда чистится пылесосом; стирка ее производится
отдельно от стирки незагрязненной радиоактивностью
одежды. При невозможности полной Д. загрязнен-
ные предметы или изымаются из употребления до
тех пор, пока активность не снизится до нормы вслед-
ствие радиоактивного распада, или, в случае загряз-
нения долгоживущими радиоактивными вещест-
вами, уничтожаются. Загрязненные стены, потолки
и т. д. закрашиваются специальными красками,
содержащими тяжелые элементы (что особенно эффек-
тивно при загрязнении а- и Р-радиоактивными ве-
ществами); загрязненный линолеум, часто приме-
няемый для покрытия пола и лабораторных столов,
заменяется.
Рукн после работы с радиоактивными веществами
следует тщательно вымыть, лучше всего горячей
водой, обильно пользуясь м,ы л ом и применяя щетку
для ногтей. Радиоактивную чистоту рук следует при
этом проверять измерительным прибором. Если за-
грязнения прочно удерживаются, то мытье рук сле-
дует продолжать с применением специальных мою-
щих средств, напр. образующих комплексные соеди-
нения, разбавленных р-ров к-т, пасты двуокиси тита-
на или насыщенного р-ра перманганата калия, после
к-рого руки споласкиваются 5%-ным р-ром бисуль-
фита натрия.
При Д. уделяется особое внимание сбору и удале-
нию радиоактивных загрязнений. Обмывочные воды
помещают в специальные сосуды, радиоактивные
пыль и стружки, образующиеся при механич. обра-
ботке, тщательно собирают (лучше всего пылесосом)
и также изолируют (см. Радиоактивные отходы).
Выбор тех или Иных приемов Д. существенно зависит
1043
ДЕЗАЛКИЛИРОВАНИЕ — ДЕЗАМИНИРОВАНИЕ
1044
от характера радиоактивных загрязнении. Весь процесс
Д. проводят с применением спец, мер защиты и при
постоянном дозиметрия, контроле (см. Дозиметрия,
Защита от излучений радиоактивных веществ).
Лит.: Инструкция по работе с радиоактивными веществами
и источниками ионизирующих излучений в научных учрежде-
ниях Академии Наук СССР, М., 1958; Стефенсон Р.,
Введение в ядерпую технику, пер.с англ., М.,1956; Материалы
международной конференции по мирному использованию атом-
ной энергии, состоявшейся в Женеве 8—20 августа 1955 г.,
т. 7, [М., 1958]. В. И. Барановский.
ДЕЗАЛКИЛИРОВАНИЕ (деалкилирование) —про-
цесс отщепления алкильных групп, ранее связан-
ных с атомами О, N, S, С и др.; процесс Д. противо-
положен алкилированию, состоящему в присоединении
алкильных радикалов к атомам О, N, С и др. Д. —
часто наблюдающаяся реакция в синтетич. органич.
химии. Так, образование сложных эфиров из спиртов
и кислот можно рассматривать как О-алкилирование;
противоположный процесс омыления сложных эфи-
ров представляет собой пример Д.:
CHjCOOi СНз + Н2О -» СНзСООН + снаон
Количественное определение алкоксильных групп
нагреванием анализируемого вещества с HJ с отгон-
кой образующихся RJ (см. Цейзеля метод) основано
на процессе Д.:
ROCH, + Ш -» кон + CH3J
Бромистоводородная к-та и, в еще большей степени,
иодистоводородная к-та являются менее эффектив-
ными агентами Д., чем HJ. Эфиры фенолов, напр.
С8Н5ОСН3, очень легко подвергаются Д. при дейст-
вии А1С13. Переход от дианизидииа к продукту Д. —
3,3'-диоксибензидину, применяемому в качестве полу-
продукта в произ-ве красителей, легко осуществляется
нагреванием в автоклаве с раствором едкого кали;
Типичным примером Д. третичных аминов может
служить Брауна реакция, состоящая в действии BrCN
с образованием диалкилцианамида,легко гидролизую-
щихся во вторичные амины:
RR'R"N + BrCN — RR'NCN + R"Br
RR'NCN + НгО — RR'NH + HCNO
Для Д. третичных жирноароматич. аминов [в про-
стейшем случае для превращения CeHsN(CH3)3 в
CeHsNHCH3] можно применять многие реагенты, напр.
азотистую к-ту, фосген. В особенности легко отщеп-
ляется бензильная группа. Так, при гидрогенолизе
C8HsN(CH3)CH2CeHs образуется CeHsNHCH3. Пре-
вращение четвертичных аммониевых оснований в тре-
тичные амины (Гофманский распад) также относится
к процессам Д.
Реакции Д. легко осуществляются в случае многих
элементоорганич. соединений. Так, триалкилбораны
подвергаются Д. действием окислителей, галогенов,
галогеноводородных кислот, альдегидов:
R3B + Оз - RB(OR)3 R3B + Br, -» R,BBr + RBr
R3B + HBr -» R,BBr + RH
(C2H,)3B + C,H3CHO -> (C2H3)2BOCH2C«Hi + CH3=CH2
Аналогично можно отН4РЪ перейти к R3PbX (X—ани-
он) и к менее алкилированным производным. Ввиду
сравнительно большой прочности межуглеродных
связей в органич. соединениях Д. с отщеплением ал-
кильных групп от атомов углерода обычно наблю-
дается лишь в жестких условиях. Большое практич.
значение имеют процессы Д., к-рые происходят при
деструктивной переработке нефтепродуктов (см. Дез-
алкилирование нефтепродуктов). Я. Ф. Комиссаров,
ДЕЗАЛКИЛИРОВАНИЕ НЕФТЕПРОДУКТОВ —
процесс отщепления боковых цепей углеводородов
нефти. Д. н. ароматич. н нафтеновых углеводородов
является одной из наиболее важных реакций, проте-
кающих при деструктивной переработке нефти. Длин-
ные парафиновые цепи термически нестабильны,
так же как и высокомолекулярные парафины. Арома-
тич. и нафтеновые кольца значительно более стабильны,
и реакции, ведущие к изменению последних,— деги-
дрирование и разрыв кольца, наступают позднее,
когда дезалкилирование в значительной мере закончи-
лось. Скорость отщепления и распада боковой цепи
зависит гл. обр. от ее длины и мало зависит от при-
роды нафтенового или ароматич. кольца. Метильная
группа очень стабильна и поэтому дезалкилирование
толуола и ксилолов происходит только частично и при
очень высокой темп-jie. У различных изомеров процесс
протекает с разной скоростью, напр. из трех изо-
мерных ксилолов наиболее легко распадается о-ксилол
и наиболее трудно .и-ксилол. Дезалкилирование может
приводить к изменению соотношения алкильпых
групп различных углеводородов, благодаря чему,
напр., из толуола образуется смесь бензола и ксило-
лов. В нек-рых случаях, напр. при алкилировании
бутана бутиленами, в результате вторичных реакций,
дезалкилирования и повторного алкилирования, об-
разуется сложная смесь жидких углеводородов.
В нефтепереработке Д. н. используют для получе-
ния низкомолекулярных парафиновых, ароматич.,
нафтеновых и непредельных углеводородов, входящих
в состав бензиновых и керосиновых фракций. Иногда
Д. н. применяют для понижения вязкости тяжелых
нефтяных фракций, вязкость к-рых обусловлена на-
личием нафтеновых углеводородов с длинными боко-
выми цепями. Подобный процесс частичного термич.
Д. н. известен под названием «висбрекинг»
(Viskosity breaking). См. также Крекинг.
Лит.: Обрядчиков С. Н., Технология иефти, ч. 2,
3 ияд., М., 1952. В. В. Щекин.
ДЕЗАМИНИРОВАНИЕ — реакция, приводящая к
отщеплению или замещению аминогруппы в органич.
соединениях. Отщепление аминогруппы с выделением
аммиака осуществляется, напр., при Д. аминокислот
под влиянием специфич. энзимов или нек-рых бак-
терий. Эта реакция играет важную роль в биохимич.
процессах. Для синтезов и промышленной органич.
химии наибольшее значение имеет Д., сопровождаю-
щееся замещением аминогруппы водородом или дру-
гими группами, напр. ОН, OR, Hal, CN, СН3СОО
и т. п.
Д. производят различными методами, выбор к-рых
определяется особенностью строения исходного амина
и природой замещающей группы. Так, замену амино-
группы гидроксильной производят по-разному для
аминов различных классов; у ароматич. аминов (гл.
обр. нафталинового ряда) при помощи водных раство-
ров бисульфита натрия: ArNH3 NaHSQ<. ArSO3Na —>
—>-АгОН. Д. многих ароматич. моно- и полиаминов
[особенно содержащих электроноакцепторные группы
(Y)] легко протекает при действии гидролизующих
агентов — водных р-ров к-т и щелочей:
__ . . — — Н + ИЛИ О Н — * . 2 Z*\ W ~ "VT z"v 1
Y—Аг'—NH3 х * Y—Аг'—ОН (Y=NOs, NO и т. п.)
Наиболее универсальным методом, пригодным для
первичных аминов различных классов, является Д.
азотистой к-той. Сущность метода состоит в том, что
кислые, обычно водные, р-ры амина обрабатывают нит-
ритом Na или К, в результате чего образуются диазо-
ниевые структуры. Последние, в зависимости от строе-
ний исходного амина и состава реакционной среды,
претерпевают реакции расщепления или обменивают
диазогруппу на другие группы. Диазосоединения
1045
ДЕЗАМИНИРОВАНИЕ — ДЕЗОКСИРИБОНУКЛЕАЗА
1046
алифатич. и алициклич. рядов обычно неустойчивы и
тотчас разлагаются с выделением азота, причем али-
фатический остаток, ранее связанный с азотом, взаимо-
действует с нуклеофильными реагентами среды (на-
пример, водой, анионами к-т) или отщепляет про-
тон, переходя в олефин. Согласно современным
представлениям, распад таких солей диазония может
проходить одновременно как по бимолекулярному,
так и по мономолекулярному механизму, с промежу-
точным образованием ионов карбония. Реакции за-
мещения могут сопровождаться изомеризацией ал-
кильного остатка. У алициклич. аминов следствием
этого является изменение числа звеньев в цикле
(Демьянова перегруппировка). В ,
общем виде Д. алифатич. аминов
может быть представлено след,
схемой:
R
I । . , Н‘Х“ HNO,
-C-C-NH- ' 2
I
н
Г R
। ।
-C-C-Na
Н
V--
НгО
диазония ароматич. ряда
Соли
значительно более устойчивы и
обладают способностью замещать
диазогруппу другими функцио-
нальными группами (см. табли-
цу), что широко используется для
получения разнообразных произ- i
водных ароматич. ряда: 1
Аг-nh, 1Т Х ,г1^-ХГ-- [Ar-N.]+X--------- Ar-Z
Н2О замещение
-с=с-
I I
R
R
-кг,-х~
Sr1, Se’
-?-С-
Н R
-с-с-
R
• Бимолекулярное нуклеофильное замеще-
ние. •• Бимолекулярное электрофильное за-
мещение.
Действующий реагент Получающийся продукт Ar — Z Примечание
Н3РО2; ROH; СН2О; Na2SnO2 и др. . . Н2О ROH Cu2Cl2 (X = Cl) . . . Cu2(CN)2 • Си (X ~ Вг) . . . . Си HBF, SS-; RS~; S3- .... Na3AsO3 MeHalx + Me* . . . Ar — H Ar — OH Ar — OR Ar — Cl Ar — CN Ar — Br Ar — Ar Ar — F Ar — S — Ar; Ar — S — R; Ar — S2 — Ar Ar — AsO (OH), АгуМ%-у См. Зандмейера реакция См. Гаттермана реакция См. Гомберга — Бахманна—Гея реакция См. Шиманна реакция См. Барта реакция См. Несмеянова реакция
* Me — металл, напр. Hg.
Использование ориентирующего влияния амино-
группы и реакции Д. позволяет производить синтезы
таких производных ароматич. ряда, к-рые трудно
получить иным путем (напр., синтез 1, 3, 5-трибром-
бензола бромированием анилина с последующим Д.
2-нитро-З-бромдифенила из бензидина, 4,4-дифтор-
дифенила из бензидина, n-динитробензола из п-нитро-
анилина и др.).
Лит.: X у б е н И., Методы органической химии, пер.
с нем., т. 4, вып. 1, кн. 2, М. — Л., 1949; Корнблюм Н.,
в кн.: Органические реакции, пер. с англ. Сб. 2, М., 1950,
с. 285; Роэ А., там же, Сб. 5, М., 1951, с. 155; В о р о ж-
ц о в Н. Н., Основы синтеза промежуточных продуктов и кра-
сителей, 4 изд., М., 1955, с. 446; Ингольд К. К., Меха-
низм реакций и строение органических соединений, пер. с англ.,
М., 1959; Whitmore F. С., L a n g 1 о i s D. Р., J. Amer.
Chem. Soc., 1932, 54, № 8, р. 3441; Streitwleser А.,
17
21
СН2ОН
сн3 с=о
R
I I
-C-C-y
I I
H
ONO)
Сольволиз
образование
олефинов
СНО
I
сн3
неон
I
неон
I
СН2ОН
J. Org. Chem., 1957, 22, p. 861; Cram D. J., Me Carty
J. E., J. Amer. Chem., Soc., 1957, 79, № 11, p. 2866.
В. H. Сеткина.
ДЕЗОКСИКОРТИКОСТЕРОН (Д‘-прегнен-21-ол-3;
20-дион; 11-дезоксикортикостерон, кор-
тексон, ДОК)С21Н30О3, мол. в. 330,45 —
промежуточный продукт биосинтеза кор-
тикоидных гормонов в коре надпочечни-
ков, влияет на минераль-
ный обмен (Na и К). Д. —
кристаллы; т. пл. 141—
142°; [a]D = + 178°(в эти-
ловом спирте); максимум
поглощения при Х=240 ммк; хоро-
шо растворим в этилацетате, хло-
роформе и спирте, хуже — в
углеводородах и разб. спиртах,
плохо — в воде. Методы определе-
ния Д. основаны на его способно-
сти поглощатьУФ-свет и на восста-
навливающих свойствах, обусло-
вленных присутствием а-кетоль-
ной группировки у 17-го углерод-
ного атома. Д. определяют после
его хроматография, отделения от
других кортикостероидов. Биоло-
гическое определение Д. прово-
дится по вызываемому им сниже-
нию выделения Na из организма с
мочой (минералокортикоидная ак-
тивность). Д. получен синтети-
чески из А6-3-оксиэтиохоленовой
кислоты. Д. применяют при лече-
нии гипофункции надпочечников и некоторых других
заболеваниях.
Лит.: Юдаев Н. А., Биохимия стероидных гормонов
коры надпочечников, М., 1956; см. также при ст. Гормоны.
ДЕЗОКСИРИБОЗА (2-дезокси-П-рибоза, 2-дезокси-
D-з/штро-пентоза, тиминоза, риоодезоза) CsH^O^
мол. в. 134,1—кристаллы; т. пл. 78—82°;
растворима в воде; [a]D = —91° и —58°
(равновесная система в воде). Д. — дез-
оксисахар, входящий состав дезокси-
рибонуклеиновых кислот (см. Нуклеиновые
кислоты) в форме пуриновых и пирими-
диновых нуклеотидов. Д. дает ряд реак-
ций альдоз (образует фенилгидразон и
анилиновое производное) и неконцевых
девоксисахаров: с дифениламином после обработки кис-
лотой (реактив Дише), с триптофаном и хлорной к-той
(реакция Коена), с лейкофуксином (реакция Фейльгена),
с цистеином и H2SO4 и т. д. Озазона Д. не образует.
Д. легко разлагается при действии кислот. При наг-
ревании Д. с метиловым спиртом в присутствии НС1
образуются метиловые эфиры а- и Р-пиранозных форм
(1% HCI) и метилфуранозид (0,1% НС1). Получение
Д. из дезоксирибонуклеиновых к-т производят в нес-
колько стадий: из образующейся при мягком кислот-
ном гидролизе смеси нуклеотидов выделяют нуклеотид
гуанозина (дезоксирибозилгуанина), при гидролизе
к-рого получается Д. При гидролизе пиримидиновых
нуклеотидов Д. разлагается с образованием левули-
новой к-ты. Синтетически Д. может быть получена
из гликалей, ангидросахаров глюкозы (с промежу-
точным образованием 3-дезоксигексоновых к-т) н др.
способами.
Лит.: Biochem. prepar., 1957, 5, 75; см. также при ст.
Дегоксисахара. Л. И. Линевич.
ДЕЗОКСИРИБОНУКЛЕАЗА (ДНК-аза) — фер-
мент, гидролитически расщепляющий одну из нук-
леиновых кислот — дезоксирибонуклеиновую кислоту
(ДНК), но не гидролизующий рибонуклеиновую к-ту
(РНК). Д., выделенная из панкреатической железы,—
1047
ДЕЗОКСИРИБОНУКЛЕИНОВАЯ КИСЛОТА — ДЕЗОКСИХОЛЕВАЯ КИСЛОТА
1048
кристаллич. продукт; мол. в. 63 000; изоэлектрич.
точка 4,7—5,0; оптимальный pH действия 6,0—7,0;
активизируется ионами Mg, Мп, Со; подавляется
ионами Си, боратом, арсенатом. Действие фермента
на ДНК сопровождается быстрым уменьшением вяз-
кости субстрата, увеличением поглощения в УФ-
свете (при 260 ммк) и образованием кислотораствори-
мых фосфорных соединений или производных деаок-
сирибоаы. Среди продуктов гидролиза ДНК под дей-
ствием Д. найдены моно-, ди-, три- и олигонуклеоти-
ды. Часть олигонуклеотидов представляет собой
недиализуемую фракцию, т. наз. «сердцевину». Д.
получена из дрожжей, селезенки, зобной железы,
стрептококков.
Лит.: Нуклеиновые кислоты. Химия и биология. [Сб.
статей], под ред. Э. Чаргаффа и Дж. Дэвидсона, пер. с англ.,
М., 1957. Т. Т. Болотина.
ДЕЗОКСИРИБОНУКЛЕИНОВАЯ КИСЛОТА —
см. Нуклеиновые кислоты.
ДЕЗОКСИСАХАРА — производные сахаров,
в к-рых одна или несколько гидроксильных групп
замещены водородом. Известно ок. 100 различных
Д.; многие из них являются биологически важными
соединениями. Широкое распространение имеют дез-
оксиальдозы, важнейший представитель
которых 2-деаоксирибоаа (I) является компонентом
дезоксирибонуклеиновых к-т; многие дезоксиальдозы
входят в состав различных гликозидов, в том числе
сердечных, антибиотиков, гликопротеидов. Предста-
вителей дезоксикетоз известно лишь не-
сколько, но чрезвычайно широкое распространение
имеют их производные, содержащие амино- и карбок-
сильную группы — сиаловые кислоты п нейраминовая
кислота, к-рые входят в состав гликопротеинов и
гликолипидов. Большинство природных Д. пред-
ставлено D-формой. Д. в виде L-формы входят в состав
нек-рых полисахаридов и гликозидов (L-фукоза — II,
L-рамноза), в частности гликозидов, обладающих
антибиотич. активностью (стрептоза — III и др.). Мно-
гие D- и L-Д. получены синтетически.
По физич. свойствам Д. близки обычным сахарам.
Большинство Д. выделено в кристаллич. виде, но
нек-рые получены только в виде сиропов. Д. хорошо
растворимы в воде, умеренно — в спиртах и плохо
растворимы в большинстве органич. растворителей.
СНО СНО СНО СНО
сн2 носн неон 1 сн2
неон неон онс-сон неон
неон неон носн 1 неон
СН2ОН носн СНз неон
СН3 СНз
I II III IV
На химич. свойства Д. определяющее влияние имеет
положение дезоксигруппы, на основании чего Д.
делят на след, группы: 1) Концевые (терминал ь-
н ы е) Д., или метилозы — с метильной груп-
пой в концевом ^.-положении; широко распростра-
ненные представители—рамноза и фукоза. 2) Некон-
цевые (нетерминальные) Д. с метиленовой
группой в цепи — 2- и 3-дезоксипентозы, 2-дезокси-
гексозы и т. д.; наиболее широко распространены
и хорошо изучены 2-дезоксисахара, напр. 2-дезок-
сириооза (I); 3-дезоксиальдозы являются редкими
сахарами. Д., содержащие концевую метильную группу
и метиленовую группу в цепи, наз. дезоксиметилозами;
дидезбксисахара такого типа, напр. дигитоксоза (IV)
входят в состав сердечных гликозидов.
Концевые Д. очень близки по свойствам обычным
сахарам, они дают все реакции сахаров, за исклю-
чением тех, к-рые обусловлены наличием первичной
спиртовой группы; они не окисляются до дикарбоно-
вых кислот без расщепления углеродной цепи. При
окислении иодной к-той Д. этого типа образуется
ацетальдегид, что используется для их количествен-
ного определения. Для определения 6-дезоксигексоз
используют также превращение их при действии НС1
в метилфурфурол, к-рый осаждается флороглюцином
или тиооарбитуровой к-той.
Неконцевые Д., в особенности Д. с метиленовой
группой в a-положении по отношению к карбонильной
(2-положение в альдозах), дают ряд реакций обычных
сахаров, но по нек-рым свойствам значительно отли-
чаются от обычных сахаров и от концевых Д. Напр.,
у 2-дезоксиальдоз реакционная способность больше,
чем у соответствующих альдоз; их мутаротация идет
значительно быстрее, они легко разлагаются при
нагревании с разб. кислотами; гликозиды Д. гидро-
лизуются легче гликозидов альдоз. 2-Дезоксиальдозы
не образуют озазонов. Помимо ряда реакций альдоз,
2-дезоксиальдозы дают еще неск. цветных реакций,
к-рые используются для их качественного и колич.
определения, напр.: реакцию с дифениламином после
обработки кислотой (реакция Дише), с триптофаном
и хлорной к-той (реакция Коена), с лейкофуксином
(реакция Фейльгена), с цистеином и серной к-той,
с 3,5-диаминобензойной к-той, реакцию Келлера—
Килиани (появление синего окрашивания при сме-
шивании раствора 2-Д. в уксусной к-те, содержащей
соли закиси железа, с H2SO4). Ряд этих реакций,
к-рые дают не только свободные Д., но и связанные
Д., используются для определения дезоксирибону-
клеиновых к-т, гликопротеидов, содержащих сиало-
вые к-ты (сиалопротеинов).
Получают Д. гидролизом природных гликозидов
и полисахаридов, дезоксирибозу — гидролизом дез-
оксирибонуклеиновых к-т. Концевые Д. получают
обычно восстановлением производных сахаров, в к-рых
первичный гидроксил замещен на галоген. Неконце-
вые Д. получают из гликалей, 2,3-ангидросахаров,
нитроспиртов, из диазометилкетоз и др. способами.
Дидезоксисахара получают, комбинируя перечислен-
ные методы.
Лит.: Proceedings of the Fourth International Congress of
biochemistry, v. 1, L.—N. Y.—P., 1959, p. 107; Handbuch der
Pflanzenphysiologie, Bd 6, B., 1958, S. 39, 1023; The nucleic
acids, v. 1, N. Y., 1955, p. 48; См. также лит. при ст. Углеводы.
ДЕЗОКСИХОЛЕВАЯ КИСЛОТА (За, 12а-диокси-
холановая кислота) С24Н40О4, мол. вес 392,56 —
желчная кислота, содер-
жится в желчи в виде
гликодезоксихолевой и
тауринодезоксихолевой
кислот; играет важную
роль в процессе всасы-
вания жиров. Д. к. —
кристаллы, т. пл. 177°,
[а]$ = + 53° (в спирте);
трудно растворима в воде, хорошо— в спирте, ксилоле
и др. органич. растворителях; образует прочные ком-
плексы с молекулами растворителей. В отличие от сте-
ринов, подобно прочим желчным к-там, Д. к. не оса-
ждается дигитонином. Д. к. дает реакцию на желчные
к-ты с оксиметилфурфуролом. При нагревании в ваку-
уме и медленной перегонке Д. к. образует ненасыщен-
ную к-ту, к-рая при гидрировании превращается В насы-
щенную холановую к-ту. В отличие от других желчных
к-т, Д. к. образует с жирными к-тами, спиртами,
простыми и сложными эфирами, фенолами, холесте-
рином, ароматич. и парафиновыми углеводородами,
алкалоидами и др. веществами весьма устойчивые
молекулярные соединения — холеиновые к-ты; не
образует молекулярных соединений с парафинами,
имеющими разветвленную цепь. Для отделения Д. к.
1049
ДЕЙСТВУЮЩИХ МАСС ЗАКОН — ДЕЙТЕРИЙ
1050
от др. желчных к-т используют методы хроматографии
в сочетании с ИК-спектроскопией или специфич.
реакциями. Получают Д. к. из желчи крупного рога-
того скота и синтетически из холевой к-ты; приме-
няют для синтеза стероидных гормонов {кортизона).
Лит.: Физер Л. иФизер М., Химия природных
соединений фенантренового ряда, пер. с англ., М.—Л., 1953;
Bergstrom S., BorgstrOm в., Annual Rev. Bio-
chem., 1956, 25, p. 187; Shoppee Ch. W., Chemistry
ot the steroids, L., 1958; Fieser L., Fieser M., Ste-
roids, N. Y. — L., 1959. И. 3. Сергиенко.
ДЕЙСТВУЮЩИХ МАСС ЗАКОН — устанавли-
вает соотношение между активными массами реаги-
рующих веществ при равновесии в химич. реакциях.
Д. м. з. установлен в 1864—67 К. Гульдбергом и
П. Вааге; он представляет одно из важнейших обоб-
щений, имевших весьма большое значение для пре-
вращения химии в точную науку. Согласно Д. м. з.,
скорость, с к-рой вещество А реагирует с веществом В,
зависит от их концентрации {активности). Для
обратимой реакции: aA-f- ЬВ + ...;=: тМД- nN +...
скорость реакции, протекающей слева направо (пря-
мая реакция), дается выражением: = кг [А]“ •
• [В]6..., а скорость реакции, протекающей спра-
ва налево (обратная реакция), — выражением:
Оз = Л2 fM]m • [N]n..., где^иЛ, —величины, постоян-
ные для данной реакции при одинаковых темп-рах,
наз. константами скорости реакций (или их уд. ско-
ростями); [А], [В],..., [MJ, [N]... — концентрации
веществ А, В, ..., М, N, ..., выраженные в грамм-
ионах или грамм-молях на 1 л (или их активности).
Скорость прямой реакции уменьшается по мере ее
протекания, а скорость обратной реакции при этом
увеличивается. Когда скорости обеих реакций станут
одинаковыми, т. е. hj = v2, достигается состояние
равновесия. В этом состоянии, вследствие равенства
скоростей, концентрации веществ, участвующих в
реакции, связываются между собой соотношением:
hi g [М]"» [N]n ...
fes = [А]° [В]ь . . .
где К — величина, постоянная для данной реакции
и темп-ры; она наз. константой равновесия реакции.
Это и есть математич. выражение Д. м. з. Приведен-
ные рассуждения можно рассматривать как нагляд-
ное (но приближенное) обоснование выражения кон-
станты равновесия. Строгий вывод его достигается
термодинамич. путем. В числителе выражения кон-
станты равновесия помещают б. ч. концентрации (или
активности) веществ, указанных в правой части
ур-ния реакции, а в знаменателе — веществ, указан-
ных в левой части его. Применяют и обратную форму
записи, что, очевидно, изменяет численное значение К.
В выражение для константы равновесия обычно при-
нято включать концентрации (активности) . только
тех веществ, к-рые входят в состав данной смешанной
фазы, где происходит реакция (раствора или газа).
Активность веществ, находящихся в виде индиви-
дуальных фаз, является постоянной величиной и
принимается равной единице, напр.:
AgCl zz Ag+ + ci- К = [Ag+] [Cl-]
твердое раствор
Для реакций, происходящих в разб. р-рах или в га-
зах при низких давлениях, часто можно заменить
активности растворенных веществ их молярными
концентрациями и активности газов их давлениями,
не вводя слишком больших погрешностей. Для кон-
стант равновесия в р-рах при конечных концентра-
циях следует каждый раз указывать ионную силу
раствора. Если равновесие устанавливается быстро,
то предсказание ожидаемых результатов на основе
Д. м. з. весьма эффективно. В том случае, когда реак-
ция протекает медленно и она не достигла состояния
равновесия, предсказания не имеют ценности.
Д. м. з. широко используется при различных расче-
тах химич. процессов. Он позволяет решить вопрос,
в каком направлении возможно самопроизвольное
течение рассматриваемой реакции при заданном исход-
ном соотношении концентраций реагирующих веществ,
какой максимальный выход нужного продукта может
быть получен при тех или др. условиях и цр.
А. И. Буеве.
ДЕЙТЕРИЙ (тяжелый водород) — стабильный
изотоп водорода с массовым числом 2; обозначается D,
или иногда Н2. Наименование различных дейтероза-
мещенных соединений составляется путем прибавле-
ния слова «дейтеро» (или заменой слова «водород»
на слово «дейтерий»); применяется также система
обозначений с прибавлением буквы d, например:
DC1—хлористый дейтерий, илн хлористый водо-
род-d; NHD2 — дидейтероаммиак, или аммиак^2;
CH3C00D — уксусная дейтерокислота, или уксус-
ная к-та-d; CH2DCOOH — дейтероуксусная к-та, или
уксусная-d к-та, ит. д. В природных водородсодержа-
щих соединениях (воде, углеводородах и др.) Д. со-
держится в концентрации 1/7000—1/5000 к общему
содержанию водорода. Д. был открыт спектрально
в 1932 Г. Юри, Ф. Брикведде и Г. Мэрфи в пробе
подвергнутого ректификации водорода.
Физические и химические свойства. Ядро Д. состоит
из одного протона и одного нейтрона, в отличие от
ядра легкого изотопа водорода (протия), состоящего
из одного протона. Ат. в. Д. 2,0147411, а протия
1,0081460 (по физич. шкале). Благодаря столь силь-
ному относительному различию в массах изотопов
водорода все изотопные эффекты длр водорода и его
соединений выражены значительно сильнее, чем для
изотопов других элементов.
Т. крит., °К Т. кип., °К Т. пл. в тройной точке, °К Р насыщ. пара при 22®К, атм Теплота исп. при 194,5 мм рт. ст., кал/моль Теплота субли- мации, кал/моль
Н, 33,24 20,39 13,96 1,52 219,7 183,4
HD 35,9 22,13 16,60 0,893 263 ^228
Da 38,26 23,57 18,72 0,621 302,3 274,0
В таблице сопоставлены нек-рые физич. свойства
трех изотопных форм молекулярного водорода. Раз-
личие в химич. свойствах выражено гл. обр. в изме-
нении скорости протекания химич. реакций водород-
содержащих веществ при переходе от протия к Д.
(кинетич. изотопный эффект), а также в неравномер-
ном равновесном распределении Д. при реакциях
изотопного обмена водорода (термодинамич. изотоп-
ный эффект). Скорости отдельных реакций для Д.
и протия или их соединений различаются часто в не-
сколько раз. При этом в случае Д. возможны как
уменьшение, так и увеличение скорости реакции.
Напр., синтез НВт из элементов замедляется для Д.
примерно в 5 раз; электролитич. разложение воды
с применением железного катода замедляется в случае
окиси Д. {воды тяжелой) также примерно в 5 раз,
а при нек-рых материалах и поверхностях катода —
даже в 15—20 раз; с другой стороны, синтез NH3
из элементов в присутствии паров ртути для Д. уско-
ряется примерно в 10 раз.
При равновесии изотопного обмена в ряде систем
водородсодержащих веществ наблюдается резко не-
равномерное распределение Д.; так, напр., отношение
D/Н1 (при 20°) в системах Н2О—H2S, Н2О—РН8 и
Н2О—НС1 для воды в 2,2—2,5 раза больше, чем для
H2S, РН3 или НС1, в системах Н2О—Н2 и Н2О—HJ
для воды оно в 3,5—4 раза больше, чем для Н2 или
HJ, а в такой системе как бензол— гидрид цезия,
это отношение для бензола (по расчетным данным)
1051
ДЕЙТЕРИЙ
1052
в 8,6 раза больше, чем для гидрида цезия (см. Изотоп-
ный обмен).
Аналитич. определение Д. проводится чаще всего
путем химич. превращения анализируемого дейтеро-
соединения в воду с последующим ее изотопным
анализом. Концентрацию Д. в водороде, а также
в нек-рых других газах часто определяют масс-
спектрометрич. способом (см. Масс-спектрометрия).
Применяются также методы, основанные на измере-
нии теплопроводности, ИК- и УФ-спектров, ядер-
ного парамагнитного резонанса и т. д. (см. Изотопов
стабильных анализ).
Применение. Д. широко используется в атомной
энергетике как замедлитель нейтронов в атомных
реакторах и как термоядерное горючее. Известно,
что оптимальной замедляющей способностью в отно-
шении нейтронов обладают такие элементы и изотопы,
масса ядер к-рых наиболее близка к массе нейтрона
и к-рые слабо поглощают нейтроны. Из ядер всех
элементов и их изотопов наиболее близкой к нейтрону
массой обладает ядро протия (протон). Однако протий
сильно поглощает нейтроны. После протия наиболее
близкой к нейтрону массой ядра обладает Д., к-рый
к тому же весьма слабо поглощает нейтроны. Оба
эти свойства делают Д. наиболее эффективным замед-
лителем. В атомных реакторах Д. применяется в со-
единении с кислородом, т. е. в виде тяжелой воды.
По замедляющей способности тяжелая вода превос-
ходит обычную воду в 170 раз, бериллий — в 75 раз
и графит — в 70 раз.
В качестве термоядерного горючего Д. используется
в т. наз. водородных бомбах. Разрабатываются спо-
собы использования Д. в регулируемых термоядерных
реакциях. Наибольшее количество энергии выде-
ляется в таких термоядерных процессах, для к-рых
характерно максимальное уменьшение массы в про-
цессе реакции, т. е. когда ядра реагентов обладают
меньшими дефектами масс по сравнению с ядрами
конечных продуктов. С этой точки зрения наилучшим
термоядерным горючим является протий, поскольку
его ядро (протон) не имеет дефекта массы. Однако
протекание термоядерной реакции в протии сильно
затруднено, т. к. для инициирования процесса тре-
буется исключительно большая исходная энергия
протонов. Ядро Д. (дейтрон) имеет малый дефект
массы (0,00239 ат. единиц физич. шкалы), тогда как
для продукта реакции Не4 дефект массы составляет
0,0302; вследствие этого при термоядерной реакции:
D -ф- D —> Не4 4- у выделяется значительная энергия
(23,8 Мэе). В то же время исходная энергия частиц,
необходимая для начала реакции, при использовании
Д. значительно меньше, чем для протия, благодаря
чему с ним легче осуществить термоядерную реакцию.
Еще легче проводить ее с тритием или со смесью
трития н Д., а также смесью Li6 и Д.
Д. нашел широкое применение также в исследова-
тельских работах. В ядерной физике дейтроны наряду
с нейтронами, протонами, а-частицами и электро-
нами применяются как бомбардирующие частицы,
т. е. частицы, при облучении к-рыми исследуются
ядерные реакции. С помощью дейтеросоединении рас-
шифровываются колебательные спектры сложных
водородсодержащих веществ. Такая расшифровка
основана на том, что замена протия на Д. в молекуле,
не меняя ее электронной структуры, характера потен-
циального поля и силовых констант, приводит к опре-
деленному изменению колебательных частот, связан-
ному с изменением массы отдельных частей молекулы
и симметрии молекулы в целом. Благодаря этому
возникает возможность однозначной интерпретации
колебательного спектра молекул. В химич. исследо-
ваниях Д. широко используется в качестве меченого
атома водорода. При помощи Д. оказалось возмож-
ным установить механизм ряда химич. реакций и
выяснить пути перемещения атомов водорода между
молекулами различных водородсодержащих веществ,
а также оценить реакционную способность этих
веществ в отношении реакций водородного замещения.
Использование Д. в качестве меченого атома оказа-
лось особенно плодотворным для органич. химии
благодаря широкой распространенности водорода и
важности реакций замещения водорода в органич.
соединениях. Не менее важную роль играет его при-
менение как меченого атома в биохимич. и физиологич.
исследованиях.
Получение. Благодаря использованию Д. в атомной
энергетике разработаны промышленные способы его
получения. Трудность выделения Д. заключается
в малом его содержании в природной воде и других
соединениях и вытекающей отсюда необходимости
переработки очень больших количеств сырья. До
недавнего времени применялись в основном два
способа промышленного получения Д.: 1) электролиз
воды в сочетании с изотопным обменом между водой
и водородом и 2) ректификация воды.
Электролитический метод концен-
трирования Д. состоит в многократном повторении
процесса обогащения при электролизе воды. По-
скольку D2O подвергается электролитич. разложению
медленнее, чем Н2О (в 5 раз на обычных железных
катодах), то Д. концентрируется в электролите,
а выделяющийся водород обеднен Д. Из электролита
отбирают часть обогащенной воды и направляют на
следующую ступень электролиза, где в дальнейшем
процессе обогащения концентрация Д. в воде еще
больше повышается. Такой каскад обогащения обычно
состоит из нескольких (15—20) ступеней. Однако
такое использование электролитич. метода для про-
мышленного получения Д. неэкономично, поскольку
при этом имеют место большие потери Д., уносимого
с водородом высоких и средних ступеней каскада,
в к-рых электролизу подвергается вода с повышенным
содержанием Д. Поэтому электролитич. метод при-
меняют в сочетании с изотопным обменом между
водой и водородом. Избыток Д., содержащийся в во-
дороде, извлекается водой путем каталитич. изотоп-
ного обмена с водородом. Обедненный Д. водород
удаляется из обогатительного каскада и используется
для различных химич. целей (гл. обр. для синтеза
аммиака), а обогащенная вода направляется на элек-
тролиз. Каскад изотопного обмена служит не только
для извлечения избытка Д. из водорода, ио и для
дополнительного концентрирования. Концентрирова-
ние Д. в воде при ее изотопном обмене с водородом
основано на термодинамич. изотопном эффекте, т. е.
на неравномерном равновесном распределении Д.
между водой и водородом. Метод электролиза в соче-
тании с изотопным обменом применяется иа заводах,
получающих электролитич. водород; количество про-
изводимого Д. ограничено мощностью этих заводов,
рассчитанной исходя из потребности в электролитич.
водороде. Крупный недостаток метода — высокая
стоимость Д.
Получение дейтерия ректифика-
цией воды основано на различном давлении пара
легкой и тяжелой воды (их отношение для Н2О и
HDO при 100° составляет 1,026). Концентрирование
проводится в каскаде из нескольких ректификацион-
ных тарельчатых колонн. Исходная вода поступает
в верхнюю часть колонны 1-й ступени, а из куба
последней ступени отбирается готовый продукт.
Основные недостатки метода ректификации воды
состоят в очень больших энергетич. затратах на испа-
рение огромных масс воды и в громоздкости аппара-
туры, вследствие чего стоимость продукции, произво-
димой путем ректификации воды, наиболее высока.
1053
ДЕЙТЕРИЙ — ДЕКАЛИН
1054
Описанные выше методы произ-ва Д. вытесняются
двумя другими, более прогрессииными — ректифи-
кацией жидкого водорода и двухтемпературным
изотопным обменом между водой и сероводородом.
Получение дейтерия ректифика-
цией жидкого иодорода обладает тем
5
| Продукт
Рис. 1.
Обедненный
водород
Исходный
водород
а
преимуществом по от-
ношению к ректифика-
ции воды, что отноше-
ние давлений пара для
Н2 и HD намного выше,
чем в случае Н2О и Н DO
(а также к.-л. других
изотопных соединений
водорода), и составляет
при темп-ре кипения
Н2 (—252°) 1,7, вслед-
ствие чего процесс рек-
тификации протекает
наиболее эффективно.
Кроме того, молеку-
лярный водород не со-
держит балластных ато-
также обладает высокой
мов других элементов,
теплопроводностью, малой теплоемкостью и малой
теплотой испарения, что значительно снижает энер-
гетич. расходы при ректификации. На рис. 1 при-
ведена схема одной ступени ректификационной уста-
новки, на к-рой производится концентрирование Д.
Предварительно очищенный исходный водород после
охлаждения в теплообменнике (3) и редуцирования поступает
в виде жидкости в середину колонны (2), где смешивается с ос-
новным потоком циркулирующего водорода. Обедненный Д.
газообразный водород выходит сверху из колонны, частично
(в количестве, примерно равном исходному потоку) выводится
из системы через теплообменник, а в большей своей массе носле
теплообменника поступает в компрессор (г), затем в змеевик
куба колонны (5), где сжижается, и далее поступает через ре-
дуктор в виде флегмы на орошение колонны. Малая доля обо-
гащенного Д. водорода отбирается из куба в виде продукта.
Метод.получения Д. ректификацией жидкого водорода является
одним ив наиболее выгодных, несмотря на техиич. сложность
его промышленного использования, связанную с работой
крупных установок при исключительно низких темп-рах.
Недостатком метода является применение в качестве сырья
не воды, а водорода, промышленные ресурсы к-рого хотя и
велики, но все же ограничены.
Метод двухтемпературного об-
мена между водой и сероводородом основан на
различии равновесного распределения Д. между
водой и сероводородом при разных темп-рах. Процесс
проводится в колоннах типа ректификационных.
На рис. 2 представлена принципиальная схема соответ-
ствующего произ-ва. Исходная вода поступает в 1-ю ступень
каскада (А) и последовательно
—•—> Потоп сероводорода
Рис. 2.
брасывается. Обедненный на
проходит сверху вниз через
«холодную» 1 и «горячую» 1'
колонны в противотоке с серо-
водородом.
В холодной колонне вода
обогащается Д. за счет изо-
топного обмена с сероводоро-
дом при 20—30°. Однако для
горячей колонны, работающей
при температуре порядка 100°,
такое обогащение по отноше-
нию к сероводороду является
избыточным, поскольку не-
равномерность распределения
Д. между НаО и Н2в с ростом
темп-ры уменьшается. Поэтому
в горячей колонне вода обед-
няется, являясь источником
Д. для обогащения сероводо-
рода, к-рый затем поступает
снизу в холодную колонну J.
На выходе из нижней части
горячей колонны Г вода имеет
более низкую концентрацию
Д., чем исходная вода, и от-
выходе из холодной колонны
1 сероводород вновь направляется ка обогащение в горячую
колонну 1', циркулируя таким образом в системе и транспор-
тируя Д. из обедняемого потока воды в горячей колонне в обо-
гащаемый ее поток в холодной колонне. Нек-рая доля потока
обогащенной воды отбирается из точки а между колоннами на
питание 2-й ступени каскада и заменяется несколько более
бедным потоком воды, отходящей из 2-й ступени Ь.
Аналогично проводится дальнейшее обогащение воды
в последующих ступенях каскада. Вода, обогащенная Д., вы-
водится в конце каскада (В). В двухтемпературном обмене
с водой вместо сероводорода могут быть использованы
также и другие водородсодержащие вещества, для к-рых
степень неравномерности распределения дейтерия при обмене
с водой достаточно сильно зависит от темп-ры, однако по ряду
причин (большая скорость обмена, легкость отделения от
воды и т. д.) применение сероводорода является наиболее
целесообразным. Метод двухтемпературного изотопного обмена
является не менее выгодным, чем ректификация жидкого водо-
рода, и не связан ресурсами исходного сырья.
Лит..- Бродский А. И., Химия изотопов, 2 изд.,
М., 1957; Варшавский Я. М. и Вайсберг С. Э.,
Усп. химии, 1957, 26, с. 1434—67; Киршенб аум И.,
Тяжелая вода..., пер. с англ., М., 1953; Гальверсон Ф.,
веб.: Химия изотопов, пер. с аигл., •№ 2, М., 1948 с. 60; Упра-
вляемый термоядерный синтез. Сб. ст., М., 1958; Варшав-
ский Я. М., Вайсберг С. Э., Хим. на^ка и пром-сть,
1959, 4, № 4; Production of heavy water, ed. G. M. Murphy,
[a. o.L N. Y., [a. o.J, 1955. С. Э. Вайсберг.
ДЕЙТРОН (дейтон) — ядро атома одного из тяже-
лых изотопов водорода — дейтерия; обозначается DT,
или d; состоит из одного протона и одного нейтрона,
энергия связи к-рых в Д., равная 2,23 Мэв, значи-
тельно меньше энергии связи ядерных частиц в дру-
гих, более тяжелых ядрах; собственный момент
количества движения (спин) равен 1; магнитный
момент равен 0,857348 ядериого магнетона. Будучи
простейшей системой частиц, связанных ядерными
силами, Д. представляет большой интерес для изуче-
ния природы этих сил. В качестве бомбардирующих
частиц Д. широко используются в ядерных реакциях,
в частности в реакциях, служащих источником бы-
стрых нейтронов. Химич, свойства ионов легкого и
тяжелого водорода (протона и Д.) заметно разли-
чаются, что связано со значительным относительным
различием в их массах и, следовательно, в нуле-
вых энергиях (см. Водород, Дейтерий, Изотопные
эффекты).
ДЕКАЛИН (декагидронафталии, пергидронафта-
лин) СХ0Нц, мол. в. 135,25 — существует в виде двух
изомеров, отличающихся взаимным расположением
циклов и пространстве: транс-форма (циклы удалены
друг от друга) и цнс-форма (циклы сближены). Транс-Д.
имеет 3 конформации (а, Ь, с), возникающие при
трвмс-формв
транс-объединении: а — двух Z-форм гексаметиле-
нового цикла, Ъ — Z-формы с С-формой и с — двух
С-форм. Цис-Д. имеет 5 конформаций (а, Ь, с, d, f),
аналогично возникающих при цис-объединении Z-фор-
мы с С-формой гексаметиленового циклов. Более
устойчив транс-Д.; преобладающие конформации обо-
значены буквой а. Технич. Д. содержит 60% цис-
v. 40% транс-формы; физич. свойства приводятся
в табл. (см. стр. 1055).
Теплота изомеризации цис-формы в транс-форму
(жидк.) 2,12 ккал/моль. Д. нерастворим в воде; мало
растворим в холодной и горячей уксусной к-те;
хорошо, но ограниченно — в метиловом и этиловом
спиртах; хорошо растворим в эфире и хлороформе.
1055
ДЕКАН — ДЕКАРБОКСИЛИРОВАНИЕ
1056
Физические свойства Транс- форма Цис- форма
Т. кип., “С/760 лл1 Т. пл., °C d|° nD Вязкость (25°), спуаз.......... Поверхн. натяжение (20°), дин/см . . Критич. темп-ра, °C Теплота сгорания, ккал/моль 185,5 - 31,50 0,8700 1,4697 1,956 29,89 408,5 1500,3 ±0,1 194,6 -43,2 0,8967 1,4811 2,901 32,08 419 1502,4 ± 0,1
Д. и его гомологи содержатся в нефти; система би-
цикло-(0,4,4)-декаиа часто встречается у бициклич.
сесквитерпенов (кадииен, селииен и др.), саитонииа
и др. природных веществ. В пром-сти получают
в виде смеси изомеров при каталитич. гидрогениза-
ции нафталина с Ni, Pt или MoS3 при 100—400°.
При обычной температуре в присутствии безводного
А1С13 цис-Д. превращается в транс-Д.; при 100°
наступает изомеризация в метилгидриндаи и расщеп-
ление. Окисление кислородом воздуха при 20—120°
дает 9-декалилгидроперекись, легко подвергающуюся
дальнейшим изменениям. Каталитич. окисление воз-
духом иа ванадиевом или молибденовом катализаторе
при 500° приводит к фталевому ангидриду и продуктам
более глубокого окисления. При каталитич., дегидро-
генизации получается нафталин. Хлорирование иа
свету дает 2-хлордекалин. При нитровании HNO3 об-
разуетси 9-иитродекалии. Д. —хороший, нетоксичный
растворитель для многих органич. соединений и
высокополимериых веществ.
Лит.: Elsevier’s encyclopedia of organic chemistry, ser. 3,
V. 12 B, N.-Y. — Amst., 1948, p. 78—93. Г. M. Цигуро.
ДЕКАН (н-декан) CH3(CH2)8CH3, мол. в. 142,29 —
бесцветная жидкость; т. кип. 174,12°/760 мм;
1077100 мм; 63°/15льи; т. пл.—29,673°; df0 0,72985—
0,73005; nso° 1,41189—1,41205; гкрит 330,4°; ркрит
21,3 ат; ^крит 0,236 г/мл. Вязкость 0,91 cnyaa (20°); об-
разует азеотропные смеси с фенолом (163°, 65%), глико-
лем (161,0°, 77%) и др. Д. содержится в нефти; может
быть синтезирован из амилбромида по Вюрца реак-
ции; образуется при пиролизе нефтепродуктов; может
быть выделен: деструктивной перегонкой фракции
100—200° сапропелитовой смолы; из дизельного
топлива, получаемого из СО и Н2; из нейтральных
продуктов низкотемп-рной кам.-уг. смолы и др. Со-
ставная часть дизельных топлив. г. м. Цигуро.
ДЕКАРБОКСИЛАЗЫ — ферменты, катализирую-
щие отщепление СО2 от карбоксильной группы амино-
кислот. (а) или от кетокислот (б):
a) R-CH(NH2)-COOH — R-CH2-NH2 + СО2
б) R—СО—СООН — R-CHO + СО2
Определение активности Д. проводится в аппарате
Варбурга и выражается в миллилитрах выделяюще-
гося СО2. Д. аминокислот специфичны и действуют
только иа L-амииокислоты. Простетич. группой Д.
является пиридоксаль-5-фосфат. Д. широко рас-
пространены у бактерий, которые и являются ис-
точником получения Д. L-глутамииовой кислоты,
аргииииа, гистидина, лизина, тирозина, аспараги-
новой к-ты. Оптимум действия Д. аминокислот на-
ходится при pH 4,5—6,0. Бактериальные Д. широко
используются для колич. определения аминокислот,
а также для получения биологически активных ами-
нов. Д. аминокислот обнаружены также в раститель-
ных и животных тканях. Д. кетокислот декарбокси-
лируют пировиноградную, кетомаслиную, кетовале-
риановую и кетоглутаровую к-ты. Коферментом Д.
иетокислот является тиамиипирофосфат.
Лит.: Methods in Enzymology, v. 2, N. Y., 1955, p. 185;
Ammon R., M у r b a c k K., Fermente, Hormone, Vita-
mine und die Bezlehungen dieser Wirkstoffe zuelnander, 3 Aufl.,
Bd 1, Stuttgart, 1959. T. T. Болотина.
ДЕКАРБОКСИЛИРОВАНИЕ — процесс отщеп-
ления СО2 от карбоксильной группы. Так, при нагре-
вании карбоновой к-ты с натронной известью отщеп-
ляется СО2 и образуется соответствующий углево-
дород. Т. обр. был получен В. Мейером в 1883 из
бензойной к-ты синтетич. бензол без примеси тио-
фена. Д. облегчаетси, если ослабляется связь между
карбоксильной группой и соседним атомом углерода.
Так, Д. Р-кетокислот обусловлено возникиовеиием
циклич. переходного состояния:
СИ.,
RC'<^C=O
^•CHj
СО, + RC.
ЧОН
RCOCH3
Аналогичным образом легко декарбоксилируются
кислоты, содержащие в a-положении другие электро-
фильные группы, напр. малеиновая (I), циануксус-
иая (II), нитроуксусная кислоты (III).
СН3СООН
/
t
CH3NO2
/I/
Получение нитропарафинов и гомологов уксусной
к-ты по указанному методу имеет препаративное
значение.
В особенности легко декарбоксилируются кислоты,
у к-рых карбоксильные группы непосредственно свя-
заны с другими электрофильными группами (СООН,
RCO, СС13 и др.). Так, щавелевая к-та в присутствии
конц. H2SO4 легко распадаетсяjra СО, СО2 и воду в со-
ответствии со схемой:
0=С-ОН
I +н* —-
о с-он
и "
О=С-6-Н
о=с-ч>-н
СО+СО2+'Н2О+Н’
При нагревании пировиноградной к-ты с H2SO4
(~ 150°) образуется СН3СНО и СО2. Трихлоруксусиая
к-та в присутствии органич. оснований подвергается
т. иаз. галоформному распаду: СС13СООН -> СНС13 +
•ф- СО2. При нагревании солей перфторкарбоиовых
к-т в результате Д. образуются перфторолефины,
напр.
CFy-CF-^CF^-C^O^Na — CF3CF=CF2 + СО2 + NaF
*F
Д. облегчается также, если группа СООН нахо-
дится у ненасыщенного атома углерода. Так, одни
из способов получения стирола состоит в нагревании
коричной к-ты (~ 150°):
с,н5сн=снсоон — с8н5сн=сн2 + со2
Д. ароматич. кйслот происходит лишь в жестких
условиях, обычно нагреванием в хииолиие в присут-
ствии медного порошка. Этот же метод применяется
практически для получения фурана из пирослизевой
к-ты. В ароматич. ряду Д. облегчается при наличии
электрофильных заместителей вследствие ослабления
связи карбоксильной группы с атомом углерода
в ядре. Напр., основной способ получения симметрия-
1057
ДЕКСТРАН — ДЕКСТРИНЫ
1058
ного трйнитробензола заключается в нагревании
тринитробензойной к-ты (45—50°):
Один из общих способов Д. летучих к-т состоит
в пропускании их паров над нагретыми катализато-
рами (углекислые соли Са, Ва, А]аО3, ThO2 и др.):
2RCOOH —>• RCOR 4^ Н2О 4~ СО2. При применении
смеси разных кислот образуются несимметричные
кетоны; Д. смеси муравьиной и другой карбоновой
к-ты ведет к образованию альдегидов.
К реакциям Д., имеющим препаративное значение,
принадлежит также действие свободных галогенов
на серебряные соли карбоновых к-т, ведущее к алкил-
галогенидам с промежуточным образованием ацил-
гипогалогенитов, напр.:
CH3COOAg + С12 — СН3СООС1 — СН3С1-}-СО2.
Я. Ф. Комиссаров.
Ферментативное Д. играет важную роль в разнооб-
разных процессах обмена веществ. Существует два
типа подобных реакций: а) простое Д. (обратимая
реакция) и б) окислительное Д., где под действием
одной и той же ферментной системы (но при участии
различных ее коферментов) происходит сначала Д.,
а затем — дегидрирование субстрата, напр. при Д.
а-кетокислот (пировиноградной и а-кетоглутаровой).
Благодаря реакциям Д. в цикле трикарбоновых
к-т Кребса происходит сгорание до СО2 и Н2О активи-
рованного остатка уксусной к-ты (ацетил-кофермента
А) — общего промежуточного продукта окислитель-
ного распада углеводов, жиров и белков. Фермента-
тивное Д. аминокислот широко распространено у
бактерий. В организме животных Д. нек-рых амино-
кислот приводит к образованию физиологически ак-
— со2
тивных аминов (гистидин--------- гистамин; 5-окси-
, -С°з к
триптофан-------- 5-окситриптамин, т. е.—серотонин).
Д. фосфоглюконовой к-ты (продукта дегидрирова-
ния глюкозо-б-фосфата) открывает так называемый
пентозный цикл превращений углеводов.
Д. протекает со значительным уменьшением сво-
бодной энергии. В отдельных случаях показана со-
пряженность Д. с синтезом аденозинтрифосфата
(АТФ) из аденозиндифосфата (АДФ) и неорганич.
фосфата (Д. Р-метилглутаконил-КоА до 0-метилкро-
тонил-КоА):
НООС—СН,—С=СН—COSKoA + АДФ + Р ->
I
СН,
—> СН,—С=СН—СОКоА + СО, + АТФ
I
СН,
В других процессах предварительное Д. промежу-
точного продукта является обязательной предпо-
сылкой конденсации его в ходе синтеза таких сложных
соединений как протопорфирин гема (здесь проис-
ходит Д. глицина при конденсации его с сукци-
нил-КоА) и высшие жирные кислоты (Д. подвергается
малонил-КоА при конденсации его с ацил-КоА).
Таким образом Д. энергетически обеспечивает воз-
можность этой конденсации, и субстрат Д. функциони-
рует одновременно и как участник синтеза и как ис-
точник энергии для его осуществления.
Лит.: Houben-Weil, 4 Autl., Bd 8, Stuttgart, 1952,
S. 496. В. С. Шапот,
ДЕКСТРАН (СвН10О5)я — полисахарид, синтезируе-
мый из сахарозы с помощью нек-рых микроорганизмов,
а также бесклеточных энзимов, выделенных из куль-
тур этих микроорганизмов. Мол. вес природного Д.
достигает многих десятков миллионов. Д. имеет раз-
ветвленную структуру. В молекулах Д. глюкозные
остатки в цепях соединены а-1,6-глюкозидной свя-
зью, а связи ветвлений осуществляются в а-1,4, а-1,3
Участок цепи молекулы декстрана
и а-1,2 положениях. Д., синтезируемые различными
видами и штаммами бактерий, отличаются по строению
и свойствам. Д. оптически активен, [a]D различных Д.
колеблется от 4-199° до 4^235°. Р-ры Д. характеризу-
ются значительной вязкостью. В тканях Д. опреде-
ляют осаждением его из трихлоруксусного экстракта
избытком CuSO4 в присутствии NaOH и колориметрия,
определением избытка меди с помощью диэтилдитио-
карбамата. Д. не дает реакций с иодом. При частичной
деполимеризации Д. кислотным гидролизом, энзи-
матич. путем, термич. обработкой или действием ульт-
развуковых волн образуется!, наз. «клинический» Д.,
применяемый в качестве кровозаменителя. Энзим,
расщепляющий Д., — декстранглюкозида-
з а — был недавно обнаружен в животных тканях:
Д. широко применяется при острых потерях кро-
ви и при лечении травматич. и ожогового шока, а
также при лечении отеков нефротич. природы. Из
производных Д. особое значение имеет сульфат Д.,
обладающий противосвертывающим действием и при-
меняемый как заменитель гепарина.
Лит.: Р о з е н ф е л.ь д Е. Л., Декстран, его особен-
ности и значение, как заменителя плазмы крови, в кн.: Усп.
биологич. хим., 1958, 3, с. 366; Aberg В., Some experi-
ments on partly hydrolysed bacterial dextran, Stock., 1952;
Squire J. B. [a. o.j, Dextran. Its properties and use in
medicine, Oxt., 1955. E. Л. Розенфельд.
ДЕКСТРИНЫ (СвН10О5)л — продукты частичного
расщепления крахмала и гликогена; Д. легко раство-
римы в воде. Д. образуются из полисахаридов при их
термич. обработке, кислотном гидролизе, а также под
действием различных ферментов, расщепляющих по-
лисахариды типа крахмала и гликогена (а- и Р-амилазы
и фосфорилазы). При этом образуются соответственно
а-,0- и Ф-Д. Свойства Д. в значительной степени обус-
ловливаются их мол. весом, зависящим от глубины
распада исходного полисахарида. По мере убывания
величины молекул Д., образующихся при а-амило-
лизе крахмала, увеличиваются их восстанавливаю-
щая способность, растворимость в спирте, уменьшает-
ся уд. вращение; окраска Д. иодом меняется от сине-
фиолетовой до красно-бурой, оранжевой и желтой.
В зависимости от окраски иодом и растворимости
в спирте различают амилодекстрины, эри-
тродекстрины, ахродекстриныима-
льтодекстрины. Конечные а-Д. — олигоса-
хариды, состоящие из небольшого числа глюкозных
остатков. Конечные 0-иФ-Д.
характеризуются высокой
степенью полимеризации.
В результате действия на
крахмал а-амилаз, образуе-
мых культурами Bacillus ma-
cerans, получаются Д., назы-
ваемые а- и Р-Д. Шардинге-
ра. Это кристаллические про-
дукты, представляющие со-
бой замкнутые в кольца цепи
из 5 или 6 глюкозных остат-
ков, соединенных между собой 1,4-глюкозной свя-
зью (см. рис.). а-Д. Шардингера с иодом образуют
1059
ДЕЛЕПИНА РЕАКЦИЯ — ДЕНИЖЕ РЕАКЦИЯ
1050
•игольчатые кристаллы сине-зеленого цвета, а 0-Д.
Шардингера — коричневые призмообразные кристал-
лы. Декстрины Шардингера отличаются низкой ре-
дуцирующей способностью. Д. применяют в текстиль-
ной пром-сти как компонент красочной печати, в
картонажной, полиграфия. и обувной пром-сти как
клеющее средство и др.
Лит.: Химия и технология крахмала. Под ред. Р. В. Керр,
пер. с англ., 2изд., М., 1956; French D., в кн.; Advances
in carbohydrate chemistry. Ed. M. L. Wolfrom, v. 12, N. Y.,
1957, p. 189; Whistler R. L. and Smart Ch. L.,
Polysaccharide chemistry, N. Y., 1953. E. Л. Розенфельд.
ДЕЛЕПИНА РЕАКЦИЯ — кислотный гидролиз
четвертичных солей гексаметилентетрамина, полу-
чаемых из бензил- или алкилгалогенидов и гексаме-
тилентетрамина, с образованием первичных аминов:
RCH2X + C0HI2N4 -► [RCH2 CeHI2N4l+X"
-»• RCH2NHa - HX + CH2O + NH4C1
Если применять в качестве растворителя 95%-ный
этиловый спирт и добавлять в реакционную массу
эквимолярное количество NaJ, то выход амина сильно
возрастает, и это делает реакцию Делепнна общим пре-
паративным способом получения первичных аминов.
При этом одним из продуктов реакции является аце-
таль формальдегида. Реакция дает возможность полу-
чать галргеиамины из дигалогенидов. Так, из 1,3-ди-
бромпропана был получен 3-бромпропиламин. Реак-
ция открыта в 1895 М. Делепином.
Лит.: Surrey A. R., Name reactions in organic che-
mistry, N. Y., 1954. H. E. Голубева.
ДЕМЬЯНОВА ПЕРЕГРУППИРОВКА—расширение
или сужение на один атом углерода алициклич.
колец при дезаминировании первичных аминов азоти-
стой к-той (1):
(СН2)П CHCHaNH3
s'---СНОН
(СН2)П I
vLLci-b
(1)
< N HNO, / N
(CH2)n CHNHa----(СН2)П_1СНСН2ОН
Д. п . проводят в тех же условиях, в каких осуществ-
ляется также часто сопровождающееся перегруппиров-
кой дезаминирование первичных алифатич. аминов (2):
(СН3)2СН-CH,NH, _ (СН3)аСНСН2ОН 4- (СНз)зСОН
ОН (2)
(CH3)3CCH2NH2 — (СН3)2С-СН2СНз и т. д.
В обоих случаях промежуточно образуется соль диа-
зония, распадающаяся далее Ва азот и ион карбо-
ния. Последний стабилизируется, присоединяя гид-
роксил либо отщепляя протон, непосредственно или
после предварительной перегруппировки (3):
"х HNO, Z-----\ ♦
(CH,)n CHCH2NH2----(CH,)n CHCH2N=N—*-
(CH2)n CHCH2
(CH2)„ I
(CH2)„ 11 +(CH2)n I
OH
(3)
Такой механизм подтверждается изучением кинетики
реакции.
Расширение кольца изучено на карбоциклич. ами-
нах с 3—8 атомами в кольце. Обычно перегруппировы-
ваются амины, в к-рых аминогруппа отделена от кольца
незамещенной метиленовой группой, однако извест-
ны случаи Д. д. аминов с замещенной метиленовой
группой. Использование для Д. п. амина с оксигруп-
пой в 0-положении к аминогруппе приводит к обра-
зованию циклич. кетонов, что часто используется
при синтезе стероидов (4):
ОН
CH2NH2
Д. п. подвергаются не только карбоциклич. амины,
но и 5-членные гетероциклич. амины с кислородом,
азотом или серой в кольце (5):
СНОН
(X—О, S, NH)
Сужение имевшегося кольца в результате Д. п. про-
исходит при обработке азотистой к-той карбоциклич.
аминов с аминогруппой в боковой цепи. Реакция эта
изучена до сих пор только на примере 4-членных и
6-членных аминов (6):
СН2-СН2
сн2—сн—nh
СН2\
-*~l zCHCH2OH:
2 vngx
Д. п. открыта в 1903 Н. Я. Демьяновым.
Лит.: Демьянов Н. Я., Усп. хим., 1934, 3, вып. 4;
П у т о х и н Н. И. и Егорова В. С., ДАН СССР, 1954,
96, № 2; Smith Р. A. S. В а е г D. R. and Ege S, N.,
J. Amer. Chem. Soc., 1954, 76, № 18, p. 4564.
H. И. Гамбарян.
ДЕНАТУРИРОВАННЫЙ СПИРТ — этиловый
спирт-сырец, к к-рому добавлены вещества с непри-
ятным запахом или вкусом, полностью растворимые
в спирте и не выделяемые из него простейшими физико-
химич. методами (перегонка, вымораживание и др.).
В качестве денатурирующих средств применяют гл.
обр. пиридиновые основания. К Д. с. обычно добав-
ляют красители, придающие ему сине-фиолетовую
окраску. Д. с. обязательно снабжают этикеткой с над-
писью «яд». Применяют в качестве растворителя для
лаков И политур. Б. П. Гусев.
ДЕНИЖЕ РЕАКЦИЯ — общая реакция на третич-
ные спирты и их производные, а также этиленовые
углеводороды. При нагревании небольшого количества
испытуемого вещества с реактивом Дениже (р-р окиси
ртути в конц. H2SO4, разбавленный водой в отношении
1 : 4) появляется желтый, иногда красноватый осадок,
/Н*\
имеющий, вероятно, состав CnH2n(O^ p>SO4)3. Ре-
акция считается положительной в случае образования
осадка после кипячения пробы в течение не более
1—2 мин. Реакция основана на отщеплении воды от
1061
ДЕНСИМЕТРИЯ — ДЕПАРАФИНИЗАЦИЯ
1062
третичных спиртов с образованием этиленовых угле-
водородов, к-рые и реагируют с HgSO4. Д. р. не идет
с теми третичными спиртами (трифенилкарбинол),
к-рые вследстние особенностей строения не способны
дегидратироваться. Первичные и большая часть вто-
ричных спиртов, напр. этиловый, к-пропиловый,
изобутиловый, не реагируют с реактивом ^ениже.
Нек-рые вторичные спирты (изопропиловый спирт)
дегидратируются сравнительно легко, поэтому дают
положительную Д. р. Сложные эфиры третичных спир-
тов также дают эту реакцию, предварительно расщеп-
ляясь с образованием этиленовых углеводородов.
Реакция впервые изучена Г. Дениже в 1898.
Лит.: В а й б е л ь С., Идентификация органических со-
единений, пер. с англ., М., 1957, с. 59. М. Я. Алейникова.
ДЕНСИМЕТРИЯ — совокупность методов изме-
рения плотности жидких и твердых тел. В лаборатор-
методы, основанные на
законе Архимеда: с
помощью ареометра—
по глубине погруже-
ния плавающего арео-
метра, или же путем
гидростатич. взвеши-
вания — по результа-
там взвешивания сте-
нои практике распространены
Рис. 1. Схема плотно-
мера с плавающим по-
плавком: 1 — мост; 2 —
вторичный показываю-
щий и самопишущий при-
бор или автоматический
регулятор; з — термометр сопротивления; 4 — основной со-
суд; б — поплавок; в — индуктивный датчик перемещений;
7 —г отводящая труба; з — подводящая труба; 9— диафрагма;
10 — сосуд постоянного напора; 11 — входная труба.
клянного поплавка на специальных весах в воздухе,
дистиллированной воде и в испытуемой жидкости.
Наиболее точные результаты дают методы с исполь-
зованием пикнометров — плотность определяют по ре-
зультатам взвешивания последовательно пикномет-
ра, заполненного дистиллированной водой и испы-
туемой жидкостью.
В объемном анализе применяется денсиметрия, тит-
рование, основанное на измерении плотности раствора
во время титрования. Излом на кривой титрования
указывает на достижение конечной точки титрования.
В пром-сти наряду с периодич. определением плот-
ности, напр. ареометром, применяют непрерывные
или периодич. измерения плотности жидкости непо-
средственно в технологич. линии или производствен-
ном агрегате без участия человека. Приборы, поль-
зуясь которыми проводят такие измерения, назы-
ваются автоматическими плотномерами, или просто
плотномерами. Последние* бывают как показывающи-
ми, так и самопишущими. Пользуясь плотномерами,
можно также вести
автоматич. регули-
рование плотности
жидкостей.
Рис. 2. Схема пружин-
/ него весового плот-
номера: 1—регулятор
нулевого положения; 2 — смен-
ный груз для настройки на тре-
буемые пределы измерений; з —
коромысло; 4— стрелка; б— про-
тивовес; в — сосуд; 7 — винтовая
трубчатая пружина.
По принципу действия плотномеры делятся на следую-
щие основные группы, приведенные на схемах: 1) поплавко-
вые (ареометрия.) приборы (рис. 1); 2) весовые плотномеры,
основанные на непрерывном взвешивании определенного
объема жидкости (рис. 2); 3) гидростатич. плотномеры, в к-рых
измеряется давление столба жидкости постоянной высоты
(рис. 3); 4) плотномеры, в к-рых используется излучение
радиоактивных изотопов (радиоактивные плот-
номеры) (рис. 4 о и б); 5) ультразвуковые
плотномеры, в к-рых используется измене-
ние скорости распространения звука в жидко-
сти в зависимости от ее
К денсиметрии тес-
но примыкает опреде-
ление плотности по-
ристых и сыпучих тел.
Для этих тел разли-
чают истинную и ка-
жущуюся плотность,
так называемый н а-
сыпной вес, по-
нимая под послед-
ней отношение массы
тела ко всему зани-
маемому им объему.
Определение насыпно-
го веса для различных
веществ производят
по правилам, которые
точно оговариваются
плотности.
Рис. 3. Гидростатический плотно-
мер с сообщающимися сосудами:
1 — бак; 2 — сливиая труба; з —
цилиндр; 4 — поплавковый мано-
метр; .5 — вторичный прибор; в —
гибкий шланг; 7 — индуктивный
датчик.
Рис. 4. Радиоактивные плотномеры, а. Гамма-плотио-
мер: 1 — источник излучения; 2 — приемнин излуче-
ния; з—измерительный прибор. 6. Плотномер, основан-
ный на рассеянии т-лучей: 1 — источник излучения;
2 — экр ан; з — приемник излучения; 4 — измеритель-
ный прибор.
в каждом случае.
Д. широко применяется в исследовательских рабо-
тах и в производственном контроле в химич., нефтя-
ной, пищевой и др. отраслях пром-сти.
Лит.: Физические методы органической химии, под ред.
А. Вайсбергера, пер. с англ., т. 1, М., 1950; КивилисС. Ш.,
Техника измерения плотности жидкостей и твердых тел, М.,
1959. И. П. Ефимов.
ДЕПАРАФИНИЗАЦИЯ — процесс удаления па-
рафина и церезина из нефтяных продуктов для улуч-
шения их вязкостно-температурных свойств. При
Д. в основном используется понижение растворимости
парафина и церезина с понижением темп-ры. Практи-
чески непосредственное выделение парафина путем
охлаждения производится лишь из легких соляроиых
и веретенных фракций парафинистых нефтей. При Д.
более вязких фракций используется меньшая, посравне-
нию с другими углеводородами масел, растворимость
парафина и церезина в легких растворителях, напр.:
в ацетоне, метилэтилкетоне, дихлорэтане, трихлорэти-
лене, пропане и узкой фракции бензина (нафте).
В заводской практике применяют смеси раствори-
телей, напр. 25—50% ацетона (или метилэтилкетона),
12—25% толуола и 40—60% бензола; или смесь
дихлорэтана с 20—25% бензола. На полноту Д.
влияет предварительный нагрев смеси депарафинируе-
мого продукта с растворителем перед охлаждением.
Смесь нагревают до темп-ры на 10—15° выше темп-ры
полной растворимости парафина и церезина в этой
смеси; такой перегрев благоприятствует росту кри-
сталлов при последующем охлаждении смеси. Скорость
охлаждения раствора также влияет на процесс Д.
На рисунке приведена принципиальная схема про-
1063
ДЕПОЛИМЕРИЗАЦИЯ — ДЕРНЕРА — ГОСКИНСА РАСПРЕДЕЛЕНИЕ
1064
цесса. Депарафинируемое масло перемешивается в оп-
ределенном соотношении с растворителем, смесь сна-
чала нагревается в теплообменниках, затем охлаж-
дается; выделившиеся из раствора кристаллы парафина
Принципиальная схема депарафинизации масел с приме-
нением растворителей: 1 — смешение масла с раство-
рителем; 2 — термич. обработка смеси; з — охлаждение
водой; 4 — охлаждение раствором депарафинированного
масла; 5 — окончательное охлаждение испаряющимся
хладоагеитом; 6 — отделение твердой фазы от жидкой;
7— раствор депарафииироваииого масла; 8— раствор
петролатума; 9 — регенерация растворителя из смеси его
с депарафинированным маслом; 10 — регенерация раство-
рителя из смеси его с петролатумом; 11 —растворитель.
и церезина отделяют в виде твердой фазы — петрола-
тума — от жидкой фазы — депарафинированного мас-
ла. Растворитель регенерируют и вновь используют.
При Д. с применением пропана в качестве раствори-
теля последний является одновременно и охлаждаю-
щим агентом. Депарафинируемое масло смешивают
с 2—3 объемами пропана под давлением 12—14 ат
при 32—38°. При уменьшении давления смесь охлаж-
дается до минус 40° вследствие испарения части про-
пана. Охлажденную смесь фильтруют под давлением.
Применяют также Д. в растворе смеси SO2 с бензолом.
Нашел применение процесс карбамидной Д., основан-
ный на способности мочевины (карбамида) давать кри-
сталлич. продукты (комплексы) с к-парафинами.
Образовавшийся продукт отделяют фильтрованием
от основной массы жидкого нефтепродукта, разла-
гают водой и выделенные углеводороды нормального
строения используют в различных синтезах, напр.
моющих средств.
Лит.: Чериожуков Н. И., Очистка нефтепродук-
тов и производство специальных продуктов, 3 изд., М.—Л.,
1952 (Технология нефти, ч. 3); Чериожуков Н. И.,
Крейн С. Э. и Л о с и к о в Б. В., Химия минеральных
масел, 2 изд., М.—Л., 1959; Пархоменко В. Е., Техно-
логия переработки нефти и газа, 2 изд., М., 1959.
Ф. В. Корвнввская.
ДЕПОЛИМЕРИЗАЦИЯ — реакция отрыва моно-
мерных единиц от цепи макромолекулы. Д. — сво-
бодно-радикальный процесс, обратный реакции роста
цепи при полимеризации, является одним из видов
деструкции полимеров. В отличие от других' случаев
деструкции, когда конечные продукты представляют
собой обычно полидисперсную смесь молекул различ-
ной длины (разного мол. веса), при Д. образуется
большое количество мономера. Так, напр., щ>и сухой
перегонке капрона, полистирола, полиизооутилена,
полиметилметакрилата с хорошими выходами обра-
зуются соответственно: капролактам, стирол, изобу-
тилен, метилметакрилат. Д. часто служит методом
получения этих мономеров в лабораторной практике.
Д. используется также для регенерации отходов ме-
ханич. обработки нек-рых полимерных материалов
(напр., полиметилметакрилатов) с целью получения ис-
ходных мономеров. Деполимеризоваться с образова-
нием заметного количества мономера может только
ограниченное число полимеров. Возможность образо-
вания мономера с высоким выходом при распаде дан-
ного полимера проявляется при таких реакциях Д.,
при к-рых образующийся в результате процесса иници-
ирования полимерный радикал отщепляет молекулы,
мономера. Вследствие того что при свободно-радикаль-
ной полимеризации любого мономера такие радикалы
образуются во всех полимеризующихся системах, су-
ществует тенденция к протеканию обратной реакции,
т. е. Д. При определенной темп-ре скорости реакций
роста и отрыва мономера от цепи становятся равными,
и поэтому выше этой предельной темп-ры полимери-
зация протекать не может.
Лит.: Г р а с с и И., Химия процессов деструкции поли-
меров, пер. с аигл., М., 1959; Бильмейер Ф., Введение
в химию и технологию полимеров, пер. с англ., М., 1958.
ДЕПОЛЯРИЗАЦИЯ — снижение или устранение
поляризации электродов при работе химич. источников
тока и при электролизе; происходит под влиянием
деполяризаторов — веществ, вводимых в электролит
или в состав электродов. В качестве деполяризаторов
катода используют окислители, анода — восстанови-
тели. Происхождение термина «Д.» связано с имев-
шим распространение неправильным представлением
о роли катодных деполяризаторов, согласно к-рому
последние окисляют выделяющийся на катоде водо-
род и тем самым снижают поляризацию электрода,
возникшую в результате появления водорода на его
поверхности. В действительности деполяризаторы,
как правило, сами участвуют в электродном процессе;
поэтому в их присутствии обычно коренным образом
меняется природа электродного процесса, что приво-
дит к изменению потенциала электрода. Напр., при
работе гальванич. элемента Cu+|H2SO4|Zn~ происходят
процессы: Zn—► Zn2+ -Ь 2 е (1) на аноде и 2Н+ +
+ 2 е -> Н2 (2) на катоде. Из-за большой поляризации
при выделении водорода на медном катоде процесс (2)
может идти с конечной скоростью при значении по-
тенциала, меньшем потенциала обратимого водород-
ного электрода в том же растворе. Наблюдаемое повы-
шение потенциала медного катода при введении в рас-
твор CuSO4 обусловлено не снижением его поляриза-
ции за счет взаимодействия Си2+с водородом, напр.
по реакции Си2+ 4- Н2 —>Си -ф- 2Н+, а происходит
вследствие замены электродного процесса (2) на про-
цесс Си2+ -ф- 2 е—► Си, идущий при более положитель-
ном потенциале, чем процесс (2). Аналогично действуют
в химических источниках тока и другие деполяризаторы
(О2, МпО2, Ni2O3, РЬО2, HgO и т. п.). Наряду с этим
имеются случаи, когда деполяризаторы, не меняя при-
роды электродного процесса, увеличивают его ско-
рость и тем самым снижают поляризацию электрода.
В данном случае они действуют как катализаторы.
В настоящее время термином «Д.» обозначают более
широкий круг явлений, приводящих к снижению по-
ляризации электродов. Напр., уменьшение потен-
циала выделения металла при образовании сплава
или амальгамы наз. «Д. при выделении металла». В
литературе по коррозии металлов термин «Д.» при-
меняют для обозначения природы катодного процесса,
сопряженного с анодным процессом растворения ме-
талла при электрохимич. механизме коррозии. Напр.,
термины «коррозия с кислородной Д.» и «коррозия
с водородной Д.» означают, что такими сопряженными
процессами являются, соответственно, процессы иони-
зации кислорода и выделения водорода.
Лит.: Глесстои С., Введение в электрохимию,
пер. о аигл., М., 1951; Сочена нов В. Г., Гальванические
элементы, М.—Л., 1951; Фрумкии А. Н. [и др.],
Кинетика электродных процессов, М., 1952; Т о м а ш о в
Н. Д., Коррозия и защита металлов, ч. 1, М., 1952.
П. Д. Луковиев.
ДЕРНЕРА - ГОСКИНСА РАСПРЕДЕЛЕНИЕ -
распределение микрокомпонента между твердой и
жидкой фазами при условии медленной кристаллиза-
ции; описывается логарифмич. формулой, выведенной
Дернером и Госкинсом на основе кинетич. представ-
лений об ионном обмене между поверхностью кристал-
ла и раствором. Если х и у концентрации микро- и
1065
ДЕСЕНСИБИЛИЗАТОРЫ — ДЕСТРУКЦИЯ ПОЛИМЕРОВ
1066
макрокомпонентов в растворе после кристаллизации,
а а и b соответствующие концентрации до кристалли-
зации, то Д.—Г. р. описывается формулой 1н^ = Х1п^>
где X—константа распределения, зависящая от темп-ры,
давления, скорости кристаллизации и не завися-
щая от количества выделившейся твердой фазы. Фор-
мула Д.—Г. р. относится к случаю, когда между вы-
делившимися кристаллами и раствором нет истинного
равновесия. При этом кристалл может рассматриваться
как совокупность слоев, в к-рых содержание микро-
компонента падает или нарастает от центра к пери-
ферии, в зависимости от того, происходит ли обога-
щение или обеднение твердой фазы микрокомпонен-
том. Д.—Г. р. имеет место при изотермич. упаривании
насыщенных растворов, при изотермич. снятии пере-
сыщения растворов или переохлаждения расплавов,
содержащих изоморфные макро- и микрокомпо-
ненты.
Лит.: X л опин В. Г. и Меркулова М. С., в сб.:
Радиохимия, ГМ.], 1952, с. 115; Старик И. Е., Основы
радиохимии, М.—Л., 1959. Н. Э. Хандамирова.
ДЕСЕНСИБИЛИЗАТОРЫ — см. Десенсибилизация.
ДЕСЕНСИБИЛИЗАЦИЯ — понижение светочув-
ствительности экспонированных фотографии, материа-
лов для облегчения визуального контроля процесса
проявления. В практике применяют десенсибилизиру-
ющие красители, к-рые обладают окислительными
свойствами и препятствуют при освещении слоя обра-
зованию нового скрытого изображения. К десенсибили-
заторам предъявляют след, требования: 1) возможно
быстро и полно снижать светочувствительность слоя;
2) не уменьшать скорости проявления; 3) не вызывать
вуали; 4) не окрашивать слоя; 5) хорошо растворяться
и не разлагаться в проявляющих р-рах. При Д. осо-
бенно сильно падает светочувствительность, вызванная
оцтич. сенсибилизацией, т. к. адсорбированный де-
сенсибилизатор вытесняет молекулы сенсибилизи-
рующих красителей (см. Сенсибилизация) с поверх-
ности кристаллов гало-
генида серебра. Десенси-
билизирующие свойства
С| красителей связаны с их
строением. Наиболее ча-
сто применим'пинакрип-
тол зеленый (I), водным
р-ром (1 : 5000) к-рого
обрабатывают слой перед проявлением или в той
же концентрации вводят в проявитель. Пинакрип-
тол желтый (II) в виде водного р-ра (1 : 2000) при-
меняют только для обработки слоя перед проявле-
нием, т. к. этот сенсибилизатор разрушается суль-
фитом.
Снижение светочувствительности в результате Д.
дает возможность обрабатывать высокочувствительные
панхроматич. слои при желтом свете, а позитивные
слои — при свете электрич. лампы на расстоянии Зм
без защитных светофильтров. Продолжительность
проявления десенсибилизированных фотоматериалов
следует увеличивать на 20%.
Лит.: М и з К., Теория фотографического процесса,
пер. с англ., М.—Л., 1949. В. С. Чельцов.
ДЕСИКАНТЬ! — химич. препараты для предубо-
рочного высушивания растений с целью механиза-
ции работ при уборке картофеля, семенников сахарной
свеклы, хлопчатника, люцерны, люпина, клевера и др.
В качестве Д. применяют гербициды контактного дей-
ствия (динитроалкилфенолы — динитро-о-крезол, ди-
нитро-вторично-бутилфенол; пентахлорфенол и фе-
нолят), хлораты натрия и магния, смесь эндотала
(см. Дефолианты) с сульфатом аммония (1 : 4), соли
роданистоводородной к-ты, цианат калия и натрия,
цианамид кальция и др.
Лит. см. при ст. Дефолианты. Н. Н. Мельников.
ДЕСМОТРОПИЯ — см. Таутомерия.
ДЕСОРБЦИЯ — процесс удаления адсорбирован-
ного вещества с поверхности адсорбента. Д. обратна
адсорбции. Д. вызывается уменьшением концентра-
ции адсорбируемого вещества в окружающей адсор-
бент среде или повышением темп-ры. Д. применяется
в пром-сти и лабораторной практике для извлечения
из адсорбентов поглощенных ими газов, паров или
растворенных веществ, а также с целью регенерации
адсорбента, для чего адсорбенты обрабатывают про-
дуванием горячего водяного пара, воздуха или промы-
вают чистым растворителем (регенерация). Скорость
Д. зависит не только от темп-ры и скорости тока десор-
бирующего газа или растворителя (и его природы),
но и от особенностей структуры адсорбента, т. к. в уз-
ких порах перенос молекул десорбируемого вещества
затруднен. Часто при Д. проявляется гистерезис сорб-
ционный, причиной к-рого в области мономолекуляр-
ного покрытия поверхности являются химич. реакций
с поверхностью, а в области высоких давлений пара —
капиллярная конденсация и набухание.
А. В. Киселев.
ДЕСТРУКЦИЯ ПОЛИМЕРОВ — процесс разру-
шения макромолекул высокомолекулярных соедине-
ний, сопровождающийся изменением их структуры,
понижением мол. веса, обусловливающими измене-
ние физико-химич., механич., электрич. и других
свойств. Д. п. может происходить под действием физич.
факторов: тепла (термич. Д. п.), света (фотохимич.
Д. п.), излучения (радиационная Д. п.); в результате
механич. разрушения макромолекул при дроблении,
перетирании (собственно механич. Д. п.), при действии
ультразвука (ультразвуковая Д. п.) и др. интенсивных
механич. воздействиях; при действии химич. агентов:
окислителей (окислительная Д. п.), гидролитич. аген-
тов (гидролитич. Д. п.), нек-рых восстановителей и
т. п.; биологич. Д. п. вызывается действием бактерий,
грибков, плесени.
Д. п. часто приводит к ухудшению свойств полимер-
ных материалов; однако ее нельзя рассматривать
только как отрицательное явление. Многие процессы
Д. п. широко используются для совмещения ингреди-
ентов при произ-ве резин и пластикатов, получения
полимеров с заданной вязкостью, литьевых композиций
пластич. масс, мономеров из полимерных веществ,
регенерации каучука. Методы механич., радиацион-
ной и химич. деструкции приобретают большое зна-
чение для синтеза новых типов олок-привитых поли-
меров (см. Блок-сополимеризация).
Д. п. зависит от размера, строения и химич. природы
макромолекул, а также от физич. состояния полимера,
напр. скорость фотоокислительной инициированной
светом деструкции полимеров в аморфном состоянии
превышает скорость деструкции кристаллич. поли-
меров.
В результате физич., механич. или химич. воздей-
ствий полимер может переходить в нерастворимое со-
стояние, что чаще всего обусловлено вторичными реак-
циями образовавшихся «осколков», приводящими к
образованию разветвленных или сетчатых молеку-
лярных структур.
Механизм деструкции полимеров.
Д. п. при действии физич. или механич. агентов Про-
текает по радикально-цепному механизму (см; Цепные
реакции).
1067
ДЕСТРУКЦИЯ ПОЛИМЕРОВ
1068
В общем виде эти процессы можно представить
следующими схемами:
1. Инициирование:
X X
I I Е
. __AA/V- СН2-С-СН3-С -ЛАЛг-
• —ААЛ— СН2—С' 4~ 'СН2—С —адл— •
где: X —Н, галоген или органич. радикал; R—алкил, арил,
ацил или др. группа.
2. Развитие цепи:
X X
I I
,. . -АЛЛ- СН2-С— СН3—С- —
I I
R R
X X
I I
—* ... —АЛЛ— СН3—С'+СН2==С и т. д.
I I
R R
3. Передача цепи:
X XX
. . .-WSr-CHs-C- + • • .-АЛЛ- СН3-С-СН3-С-АЛ^. .
I I I
R R R
X X
I I
—. .-ДАД/-СН3-С-Х4-. . .—АДЛ^-СНо—С —СН, —С—АДЛ^.. .
I I I
R R R
X — подвижный атом
X
I
. • . —АЛА/— СНа-С—СН2—С —АЛЛ- • • • —*
I
. . . —ЛАЛ— СН2 — С’ -р СН,=С —АДДг- . . •
4. Обрыв цепи вследствие соединения радикалов (1)
или диспропорционирования (2):
____АЛЛ/- СНа-C(R)CHa-СХ -АЛЛг- . ..
I
R —
• . • -АЛЛ— сн2—C(R)CH„—СХ —АЛЛ- • .
I
... —АЛЛ— СН2—С—СН2—С —АЛЛ/- . ..
I I
I R
. . . -АЛЛ- СН2-С-СН»—СХ -ДАЛ— ...
... -АД/V- СНз-СН -4--СХСНа-СХ ^ЛАЛ- ...—>•
R R R (2)
-> . . . -АЛЛ- СН=СХ 4- НС(Х)СН-,-СХ -АЛА/- ...
I I I
R R R
В зависимости от природы полимера и условий дест-
рукции относительное значение процессов иницииро-
вания, развития передачи и обрыва цепи может сильно
изменяться. Так, в случае Д. п., содержащих в цепи
ароматич. ядра, четырехзамещенные (четвертичные)
атомы углерода, а также т. н. электрофильные атомы
или группы (полистирол, полиметилметакрилат, по-
лиизобутилен, политетрафторэтилен, полибутадиен
и др.), образующиеся радикалы обладают сравнитель-
но малой реакционной способностью, вследствие чего
основным процессом при деструкции становится реак-
ция развития цепи, приводящая к превращению зна-
чительной части полимера в мономер. Процесс Д. п.,
протекающий с образованием мономера, по смыслу
обратен реакции полимеризации', поэтому такой тип
Д. п. называют деполимеризацией.
Полимеры, не содержащие заместителей (напр.,
полиэтилен) или содержащие подвижный водород
(полиакрилаты), при термич. деструкции распадаются
на сравнительно большие осколки и почти не образуют
мономеров, т. к. в этом случае создаются благоприят-
ные условия для передачи цепи. При Д. п., содержа-
щих отщепляемые группы или атомы (поливинил-
ацетат, поливинилхлорид и др.), наблюдается выделе-
ние уксусной к-ты, хлористого водорода и др. про-
дуктов:
• •. —АЛЛ/- СН2—СН—СН3 —АЛА/- . • . —*
I
ОСОСН3
— ... -АЛЛ- СН=СН-СН=СН -АЛЛ/- ... 4- тСН3СООН
,.. -АЛЛ- СН3—СН-СН»—СН ^ЛАЛ- ... —
I I
X X
— .. . —АЛЛ/- СН=СН-СН=СН —АЛЛ/— ... 4- mHCl
В этих реакциях передача цепи не имеет большого зна-
чения, вследствие чего продукты Д. п. резко отличают-
ся от исходного полимера по химич. составу и строе-
нию, но мало отличаются средней длиной полимерной
цепи. Т. обр. Д. п. может протекать и без заметного
уменьшения мол. веса полимера, но в этом случае
происходят резкие химич. изменения макромолекулы.
Темп-ра Д. п., так же как и механизм распада поли-
мера, зависит от его химич. природы и прочности свя-
зей атомов и групп, составляющих макромолекулу
(см. табл.).
Термическая деструкция некоторых полимеров
Полимер Строение звена Темп-ра Продукты
цепи деструк- ции, °C деструкции
Политетра- —CFa—CFa— 400 Большой вы-
фторэтилен ход мономера
Политрифтор- —CF3—CFC1— 400 То же
хлорэтилен Полиметил- — СНа—СН—CHa— 300 90% мономера
метакрилат 1 ососн,
Полистирол —сна—сн— 1 „ СбЩ 300 65% мономе- ра, димеры,
тримеры
Полиизобу- —(CHs)aC—CHa— 240 20—50% мо-
тилен номера, ди- мер, тример,
тетрамер
Полибутадиеи -С1Ь-СН СН-СН3 20—30% мо-
-CHa-CH=CH-CHa - j- 220 номера, поли- меры
Полиакри- —сна—сн— 300-320 1 % мономера,
лонитрил 1 CN отщепление
HCN
Полиметил- —сн2—сн— 300 1 % мономера,
акрилат 1 COOR большие фраг-
менты цепей
Поливинил- —сна-сн— 140-180 Мономера нет, отщепление
хлорид 1
С1 95% НС1
Поливинил- —сна—сн— 250 1% мономера, отщепление
ацетат 1
OCOCHj 95%СН3СООН
Д. п., содержащих в основной цепи гетероатомы
(О, S, N и др.), в основном протекает по месту положе-
ния гетероатома; к таким полимерам относятся целлю-
лоза, крахмал, белки, полиамиды, полиэфиры, поли-
уретаны и т. п.
Механическая деструкция поли-
меров. Специфика этого процесса деструкции
1069
ДЕСТРУКЦИЯ ПОЛИМЕРОВ — ДЕСУЛЬФГИДРАЗЫ
1070
заключается в том, что такое разрушение макромо-
лекул не требует затраты теплоты и протекает
вследствие прямого перехода механич. энергии
Емех- в химич. энергию образовавшихся при этом
радикальных (3) или ионных (4) продуктов:
... -амг сн3-сх -л/w- • • • ——
I
R
. . . -\Мг- СХСН2- -J- -СХ-СН2 —VW- . .. ,
(3)
®мех.
.. . OOCRCOOMeOCO -AAV- ..—
... ^AV- OOCRCOO--|-+MeOCO ... (4)
Такой механо-химич. процесс может протекать при
эксплуатации пластич. масс, волокон, резин; в резуль-
тате снижается прочность материала. Механо-химич.
Д. п. при пластикации на вальцах, пластификаторах,
шнеках используется для лучшего совмещения каучу-
ков, поливинилхлорида и др. с ингредиентами компо-
зиций, а также для синтеза новых типов блок-привитых
сополимеров (подробнее см. Механохимия полимеров).
Деструкция под влиянием хими-
ческих факторов. Этот вид деструкции за-
висит от присутствия в макромолекулах реакционно-
способных атомов или групп. Химич. Д. п. активи-
руется теплотой, светом (особенно ультрафиолетовой
частью спектра), механич. воздействиями, катализа-
торами, активными радикалами. Для полимеров, со-
держащих гидроксильные или карбонильные группы,
атомы водорода и двойные связи (целлюлоза, полиакри-
латы, каучуки и т. п.), наиболее интенсивная деструк-
ция протекает при действии окислителей (кислород,
озон, перекиси и др.). В результате окислительной
деструкции, протекающей по радикально-цепному ме-
ханизму, макромолекулы разрушаются с образованием
веществ с меньшей молекулой.
Эти процессы являются причиной уменьшения проч-
ности резин, целлюлозных волокон и нек-рых типов
пластич. масс при действии атм. условий и переменных
механич. нагрузок.
Для гетероцепных полимеров (целлюлоза, белки,
полиэфиры, полиамиды и др.) наряду с окислительной
деструкцией большое значение имеет деструкция под
действием гидролизующих или омыляющих реаген-
тов (кислоты и основания, соли двух- и поливалент-
ных металлов и т. п.). Гидролитич. процессы обуслов-
ливают уменьшение степени полимеризации и в пре-
дельном случае позволяют получать исходные про-
дукты синтеза полимеров (см. Высокомолекулярных
соединений гидролиз). Гидролитич. или окислительная
Д. п. нежелательны в случае, когда полимер приме-
няется как материал для антикоррозионных покрытий
и др. Гидролитич. деструкция используется при полу-
чении из высокой олимерных соединении ценных низко-
молекулярных веществ, напр. при произ-ве глюкозы
из целлюлозы и крахмала, при получении аминокислот
из белков, синтезе жидких тиоколов из тиокольных
каучуков. А. А. Берлин.
Радиационная деструкция. При
облучении полимеров у-лучами, а также при
воздействии на них нейтронов и электронов де-
струкция происходит вследствие резкого возрас-
тания внутренней энергии и уменьшения прочности
связей в макромолекулах. Радиационная деструкция
особенно заметна в том случае, когда из двух одновре-
менно протекающих радиационцо-химич. реакций —
образования поперечных связей между макромолеку-
лами и расщепления главных цепей полимерных мо-
лекул, отщепления атомов (или групп)— преобладает
вторая. При облучении от полимеров отщепляются
легко подвижные атомы и группы. Образующиеся
при этом радикалы инициируют распад макромолекул
или приводят к образованию разветвленных или сет-
чатых структур. Радиационная деструкция харак-
терна для полимеров, содержащих в основной цепи
четырехзамещенные атомы углерода, а также связи,
активированные- поляризацией. Так, при облучении
политетрафторэтилена у-лучами наблюдается распад
макромолекул по С—С-связям и образование элемен-
тарного фтора. Целлюлоза, крахмал и др. полисаха-
риды неустойчивы к действию облучений, так как при
этом происходит поляризация связей вследствие нали-
чия ОН-групп. Полиметилметакрилат, полибутилакри-
лат, а-хлоракрилаты, полиизобутилен содержат в цепи
четырехзамещенные атомы углерода и сравнительно
легко деструктируются при действии Р- и у-лучей или
тепловых нейтронов. Распад полиметакрилатов со-
провождается отщеплением газообразных продуктов
(Н2, СН4), что с успехом используется при произ-ве
легких и прочных пенопластич. масс. Д. п. при облу-
чении наблюдается также для полимеров, содержащих
атомы галогена и водорода (поливинилхлорид, поли-
винилиденхлорид, сополимеры винил- и винилиденхло-
рида). В этом случае отщепляется галогеноводород,
и образующийся непредельный полимер претерпевает
интенсивную окислительную деструкцию. Иначе ве-
дут себя высокомолекулярные углеводороды и по-
лимеры, содержащие подвижные атомы водорода.
К числу таких веществ относятся полиэтилен, поли-
стирол, натуральный каучук, полихлорбутадиен, поли-
диметилсилоксан и др. При облучении этих полимеров
возможно отщепление водорода и образование макро-
радикалов (5), при облучении или нагревании к-рых
происходит рекомбинация с образованием сетчатых
структур (6):
• >. —ААЛ/— СНз—СНо —АЛЛ/— * —*
— ... -АЛЛ" СН—СН3 -ЛЛДг- . . + Н (5)
...—АЛА/—СН—СНз—АЛЛ/-.....АЛЛ/-СН—СНз—АЛЛ-/*•
’ —* I ’
.АЛЛ/-СН—СНз—АЛЛ/-• • • •»•—АЛЛ/-СН—СНз—АЛЛл“«.« (6)
Образование сетчатой структуры при облучении поли-
этилена повышает его теплостойкость, механич. проч-
ность и ХИМИЧ. СТОЙКОСТЬ. Б. Л. Цетлин.
Лит.: Коршаи В. В., Химия высокомолекулярных
соединений, М.—л., 1950, с. 91—134; Бильмейер Ф.,
Введение в химию и технологию полимеров, пер. с англ.,
М., 1958; Грасси Н., Химия процессов деструкции поли-
меров, пер. с англ., М., 1959; Берлин А. А., в сб.;
Успехи химии и технологии полимеров, т. 2, М., 1957; Старение
и утомление каучуков и резин и повышение их стойкости Л.,
1955; Кузьминский А. С., Усп. химии, 1955, № 7, с.
842; Грасси Н., Химия и технология полимеров, 1960,
Kt 7—8, с. 158—166; Берлин А. А., Усп. химии, 1960, 29,
вып. 10. с. 1189.
ДЕСУЛЬФГИДРАЗЫ (десульфуразы) — ферменты,
катализирующие отщепление сероводорода от серусо-
держащих аминокислот и пептидов; к Д. относятся
цистеиндесульфгидраза, гомоци-
стеиндесульфгидраза и экзоцист е-
индесульфгидраза. Цистеии-Д. катализиру-
ет отщепление H2S от L-цистеина и L-цистина с обра-
зованием аммиака и пировиноградной к-ты: ,
СН2—SH сн2 сн2 сн, сн,
I II II I Н2о I
CHNH2 -> H2S + CNH,; CNH, ->C=NH₽=: C=O+NH,
I Illi
COOH COOH COOH COOH COOH
Цистеин-Д. не действует на D-цистеин, D-цистин,
метионин и глутатион; коэнзимом ее является пиридо-
ксальфосфат, оптимальный pH действия 7,4—7,6. Фер-
мент активируется ионами Mg, Mn, Zn, подавляется
KCN, As2O3 и фенилгидразином. Цистеин-Д. содер-
жится в больших количествах в печени и в очень
небольших количествах в почках высших животных.
1071 ДЕТАЛЬНОГО РАВНОВЕСИЯ
Некоторые бактерии содержат высокоактивный фер-
мент, способный отщеплять H2S от цистеина; фермент
бактериального происхождения по своим свойствам
подобен цистеин-Д. животных тканей. Гомоцисте-
ин-Д. при действии на субстрат (гомоцистеин) об-
разует, кроме H2S, аммиак и кетомасляную кислоту.
Коэнзимом гомоцистеин-Д. является пиридоксаль-
фосфат. Экзоцнстеин-Д. катализирует реакцию от-
щепления сероводорода от пептидов, содержащих
конечный цистеин, при этом образуются пирови-
ноградная кислота и аммиак.
Лит.: Брауиштейи А. Е., Биохимия аминокислот-
ного обмена, М., 1949; Smythe С. V., в кн.: Methods in
enzymology, у. 2, N. Y., 1955, p. 315. T. T. Болотина.
ДЕТАЛЬНОГО РАВНбВЕСИЯ ПРИНЦИП — од-
но из основных положений статистич. физики, соглас-
но к-рому при условии равновесия в системе прямые
и обратные микропроцессы каждого типа компенси-
руют друг друга. Представление о детальном равно-
весии впервые было введено в теории идеальных газов.
Необходимым условием равновесия в газе является
сохранение максвелловского распределения по ско-
ростям. Столкновения между молекулами являются
тем механизмом, к-рый приводит к равновесию и под-
держивает его. Можно разделить все молекулы газа
иа группы, в каждую из к-рых входят молекулы, об-
ладающие очень близкими скоростями и находящие-
ся в одном элементе объема. Тогда, если в резуль-
тате соударения молекул групп i и к они переходят
в группы I и т, то всегда существуют такие соударе-
ния молекул групп I и т, к-рые переводят их в группы
i и к. Д. р. п. требует, чтобы выполнялось условие:
zi, к - I, т = zi, т — i, k, где Zi, k — I, m ~ тасл0 со-
ударений первого типа, a Z£ й—второго. Это
условие и приводит к распределению молекул по
скоростям по закону Максвелла. В дальнейшем было
установлено, что Д. р. п. имеет весьма общий харак-
тер. Применение его можно иллюстрировать следую-
щими примерами. Для того чтобы насыщенный
пар находился в равновесии с жидкой фазой, необхо-
димо, чтобы число молекул, испаряющихся за едини-
цу времени, было равно числу молекул, возвращаю-
щихся в жидкость за то же время. Однако, согласно
Д. р. п., этого недостаточно. Истинное равновесие
устанавливается лишь в том случае, если равенство
.числа молекул, переходящих в прямом и обратном
направлении, имеет место для каждой группы моле-
кул, обладающих близкими скоростями. Только в этом
случае распределение молекул по скоростям как в па-
рах, так и в жидкости'будет поддерживаться неизмен-
ным. Такое же положение имеет место на контакте двух
твердых тел: между ними устанавливается такая
разность потенциалов, чтобы обеспечить выполне-
ние Д. р. п. для каждой группы электронов с близкими
скоростями, пересекающих границу раздела. Д. р. п.
был с успехом применен А. Эйнштейном к анализу
процессов поглощения и излучения света атомами
вещества.
Лит.: Френкель Я. И., Статистическая физика,
М.—Л., 1948; Гайтлер В., Квантовая теории излучения,
пер. с англ., М., 1956. А. Й. Бургитейн.
ДЕТЕРГЕНТЫ — см. Моющие средства.
ДЕТОНАЦИОННАЯ СТОЙКОСТЬ МОТОРНОГО
ТОПЛИВА (антидетонационные свойства топлив) —
моторная характеристика топлива, определяющая ус-
ловия сгорания его в двигателе без детонации. Дето-
национным сгоранием в двигателе называют такое сго-
рание, при к-ром скорость распространения пламени
равна 1 500 — 2 500 м/сек, т. е. приблизительно в 100
раз выше нормальной. Детонационное сгорание мо-
торного топлива зависит как от химич. состава топ-
лива, так и от степени сжатия и конструкции мотора.
Среди ряда теорий, объясняющих причины возникно-
вения детонации, наиболее признанной считается те-
ПРИНЦИП — ДЕФЕКТ МАССЫ 1072
ория перекисей. Будучи нестойкими и реакционноспо-
собными, органич. перекиси, при какой-то критич.
их концентрации, вызывают мгновенное (взрывное)
воспламенение рабочей смеси, возникновение детона-
ционной волны и вибрацию стенок цилиндра двига-
теля (стуки). Антидетонационные свойства топлив за-
висят от количественного соотношения различных
классов углеводородов, входящих в их состав. Наи-
меньшей детонационной стойкостью обладают нор-
мальные парафиновые углеводороды, наибольшей —
изопарафиновые и ароматические; нафтеновые и оле-
финовые углеводороды занимают промежуточное по-
ложение. Детонационная стойкость нормальных пара-
финовых углеводородов снижается с увеличением
числа атомов углерода. Особенностью этих углево-
дородов является хорошая приемистость к тетраэтил-
свинцу (ТЭС) (см. Антидетонаторы моторных топлив).
Парафиновые углеводороды с разветвленными боковы-
ми цепями обладают значительно более высокой дето-
национной стойкостью. Детонационная -стойкость изо-
парафиновых углеводородов повышается с увеличе-
нием числа атомов углерода в боковых цепях, с
приближением метильных групп к центру молекулы,
при более компактном и симметричном расположении
метильных групп внутри молекулы. Ароматич. углево-
дороды отличаются высокой детонационной стойкостью
иа богатой смеси. В отличие от парафиновых углеводо-
родов, ароматич. имеют низкую приемистость к ТЭС;
длинные неразветвленные боковые цепи снижают,
а короткие и разветвленные — несколько улуч-
шают их детонационную стойкость. Из нафтеновых
углеводородов наивысшей детонационной стойкостью
обладает циклопентан. Двойная связь олефиновых
углеводородов благоприятно действует на повышение
антидетоиационных свойств, когда она расположена
ближе к центру молекулы углеводорода, а для ди-
олефинов — когда двойные связи сопряжены. Боль-
шинство встречающихся в нефтях сернистых соеди-
нений снижает детонационную стойкость моторных
топлив, все сернистые соединения понижают приемис-
тость топлив к этиловой жидкости.
Наиболее высокой детонационной стойкостью обла-
дают топлива для авиационных и форсированных ав-
томобильных поршневых двигателей, состоящие обыч-
но из базовых бензинов (т. е. являющихся основным
компонентом моторного топлива) и высокооктановых
компонентов. В качестве базовых применяют бензины
прямой перегонки, каталитич. крекинга и рифор-
минга. Для повышения октанового числа к бензинам
добавляют изопарафиновые углеводороды (изопентан,
неогексан, изооктан, триптан), а для сортности —
ароматические (бензол, толуол, этилбензол, изопро-
пилбензол).
Существует два основных метода оценки Д. с. м. т.:
на бедной рабочей смеси (избыток воздуха >1) —
по октановому числу и на богатой рабочей смеси (из-
быток воздуха 0,7—0,8) — по сортности топлива.
Лит.: Добрынин А. А., Детонация в двигателях,
М., 1949; Моторные топлива, масла и жидкости, т. 1, 3 изд.,
М., 1957; Забрянский Е. И., Зарубин А. II.,
Детонационная стойкость и воспламеняемость моторных
топлив, М., 1958. В. В. Панов.
ДЕТОНАЦИЯ — чрезвычайно быстрый экзотермич.
процесс химич. превращения вещества, распростра-
няющийся с постоянной скоростью, превышающей
скорость звука (см. Взрыв, Взрывчатые вещества).
ДЕФЕКТ МАССЫ — уменьшение массы М системы
по сравнению с суммарной массой всех отдельно
взятых ее элементарных составных частей, обусловлен-
ное энергией их связи в системе. Д. м. является след-
ствием универсального соотношения Е = Л/с2, вы-
текающего из теории относительности А. Эйнштейна,
где Е — полная энергия системы, с = 3 • 1010 см/сек —
скорость света в пустоте; при этом ДМ = МА,
1073
ДЕФЕКТЫ СТРУКТУРЫ
1074
где АЛ/= —М есть Д. м., а АТТ — энергия
связи, т. е. энергия, к-рую необходимо затратить для
разделения системы на ее элементарные составные
части. Следовательно, Д. м., являясь мерой энергии
связи, представляет собой так же, как и эта энергия,
меру устойчивости системы.
При переходе менее устойчивых систем в более устой-
чивые выделяется энергия и уменьшается масса (т. е.
Д. м. увеличивается); часть массы отдается вместе
с энергией в окружающую среду. При химич. превра-
щениях соответствующие изменения массы ничтожны
(менее 10 6%), поскольку эти превращения затрагива-
ют лишь электронные оболочки атомов с весьма ма-
лой массой, тогда как атомные ядра, в к-рых сосредо-
точена почти вся масса вещества, остаются неиз-
менными. Поэтому химич. реакции протекают в ус-
ловиях практически полного равенства масс исход-
ных и конечных продуктов. Ядерные же превращения,-
затрагивая основную массу вещества, обнаруживают
заметное ее изменение (иногда на несколько десятых
процента) и сопровождаются энергетич. эффектами,
на много порядков большими, чем при химич. реакци-
ях; при этом изменение массы на 1 физич. единицу
атомного веса соответствует энергии в 931 Мэв. Энер-
гетич. эффект ядерной реакции определяется в основ-
ном разницей Д. м. конечных и исходных ядер. Д. м.
атомных ядер известны с большой точностью и выра-
жаются в физич. единицах атомного веса: АЛ/ =
= 1,00812 Z + 1,00893 (А — Z)—A', где 1,00812 —
масса атома Н1,1,00893— масса нейтрона, Z — поряд-
ковый номер, А — массовое число, А' — атомный вес.
Д. м. ядра, приходящийся на один нуклон, т. е. удель-
ная энергия связи в массовых единицах (AM/А), об-
наруживает характерную закономерность в своем из-
менении с увеличением массового числа и порядкового
номера: сначала он быстро возрастает от нуля (для Н1)
до максимума (0,00941 физич. атомных единиц для
Ni64), а затем постепенно убывает (составляя для
U238 0,00801 физич. атомных единиц). Из этой зако-
номерности, а также из условия выделения энергии
лишь при образовании ядер с большим Д. м., чем ис-
ходные, следует, что наилучшими источниками ядер-
ной энергии могут служить либо процессы распада на-
иболее тяжелых ядер на значительно более легкие,
либо соединение наиболее легких ядер в менее легкие.
В ядерной энергетике получили использование про-
цессы обоих указанных типов (деление U235, U233
и Рц239, осуществляемое в атомных реакторах и атом-
ных бомбах, а также термоядерные реакции синтеза
Не4 из изотопов водорода, используемые в водород-
ной бомбе).
Следует иметь в виду, что термин «Д.м.»для атомных
ядер в литературе часто используют для обозначения
простой разности между атомным весом и массо-
вым числом (ДЛР = А'—А), а не истинного Д. м.
(АЛ/), причем АЛ/ = —АЛ/' Д- 0,00893А — 0,00081Z.
В этом случае вместо уд. энергии связи в массовых
единицах (ДЛ//Л) пользуются величиной ДМ'/А, на-
зываемой упаковочным коэффициентом.
С. Э. Вайсберг.
ДЕФЕКТЫ СТРУКТУРЫ — нарушения строгой
пространственной периодичности кристаллич. решет-
ки, свойственной идеальному кристаллу. Д. с. яв-
ляются неотъемлемой и характерной особенностью
строения реальных кристаллов, механич. и физико-
химич. свойства к-рых во многом определяются числом
и типом Д. с.
Простейшими и вместе с тем основными типами Д. с.
являются точечные дефекты, названные именами
ученых, впервые указавших на их существование в
кристаллах: «дефекты по Шоттки» и «дефекты по Френ-
келю». Дефекты по Шоттки представляют собой неза-
нятые узлы решетки, т. е. вакансии, возникшие в про-
цессе образования кристалла. Вакансии могут воз-
никать также в поверхностном слое уже образовав-
шегося кристалла и затем диффундировать внутрь
объема. Нек-рые атомы, образующие поверхностный
слой тела, набрав случайно достаточно большую ки-
нетич. энергию, отрываются от соседей и «испаряются»,
оставив незанятый узел решетки — вакансию. В даль-
нейшем эту вакансию мо^ет занять соседний глубин-
ный атом, вакансия же переместится на место этого
атома. В результате таких элементарных актов диффу-
зии вакансия постепенно проникает в глубь твердого
тела. Однако для образования вакансии не обязательно
должно произойти полное испарение атома из поверх-
ностного слоя. Если избыточная энергия недостаточна
для испарения, то возможно неполное испарение, при
к-ром поверхностный атом, вырвавшись из своего ок-
ружения,может перейти в новое положение над поверх-
ностным слоем, при к-ром этот атом будет иметь лишь
одного соседа снизу. Вероятность такого перехода
больше, чем полного испарения, т. к. требует меньшей
энергии активации. Образовавшаяся при этом вакан-
сия тем же механизмом миграции глубинных атомов
уйдет в глубь твердого тела.
Дефекты по Шоттки обнаруживаются как в струк-
турах, построенных из одинаковых атомов (напр.,
металлы), так и в катионной или анионной решетке
ионных кристаллов, в структурах с ковалентными
связями (германий или селен) и в молекулярных кри-
сталлах (органич. соединения). В стехиометрич. ион-
ных кристаллах эти дефекты по условию электроней-
тральности должны быть парными, т. е. на каждую
вакансию в катионной решетке должна приходиться
одна вакансия в анионной решетке.
Дефекты по Френкелю возникают в результате
элементарных актов испарения глубинных атомов
твердого тела. При этом «испаряющийся» атом ие по-
кидает объема тела, а переходит из данного узла, кри-
сталлич. решетки в соседнее междоузлие. Таким об-
разом, дефект по Френкелю представляет собой ва-
кансию и дислоцированный атом и в этом смысле всегда
является парным дефектом. Возможность образования
дефектов по Френкелю определяется неравномерным
распределением энергии между атомами твердого тела.
Те из атомов, к-рые в данный момент приобрели зна-
чительную избыточную энергию, могут покинуть поло-
жение равновесия (узел решетки) и переместиться в со-
седнее междоузлие.
Оба типа точечных Д. с. возникают в результате
термически активируемых процессов, и каждой данной
темп-ре отвечает определенная равновесная концентра-
ция этих дефектов:
лв = пое kT
где п0 — общее число атомов твердого тела в 1 см3,
п„ — число вакансий, Uv — энергия образовайия ва-
кансии. Однако число Д. с. при данной темп-ре
может быть искусственно увеличено бомбардировкой
твердого тела элементарными частицами или путем
закалки равновесного состояния, соответствующего
более высокой темп-ре, а также в результате пластич.
деформации.
Точечные Д. с. в реальных кристаллах играют ре-
шающую роль в процессах диффузии, в различных
химич. процессах, идущих как на поверхности, так и
в объеме твердых тел, в том числе и каталитич. про-
цессах, протекание к-рых существенно зависит от
степени и характера неоднородности и наличия дефек-
тов в структуре твердого тела. Вместе с тем точечные
Д. с. играют важную роль в формировании т. паз.
структурно-чувствительных свойств кристаллов: элек-
тропроводности, фотоэлектрических и термоэлектрич.
свойств.
1075
ДЕФОЛИАНТЫ — ДЕФОРМАЦИЯ ПОЛИМЕРОВ
1076
Кроме точечных Д. с., в реальных твердых телах
Имеются также линейные Д. с. и Д. с., образующие
поверхности раздела. Линейные Д. с., или дислокации,
образуются в кристаллах как в процессе их роста из
расплава или из раствора, так и в процессе пластич.
деформации под действием внешних напряжений.
Перемещение дислокаций в кристалле, их взаимо-
действие между собой и с вакансиями и дислоцирован-
ными атомами определяют упруго-пластич. и проч-
ностные свойства реальных кристаллов. Д. с., обра-
зующие поверхности раздела, представляют собой
дислокационные сетки, располагающиеся по границам
блоков мозаики в структуре реального кристалла.
В совершенно чистых кристаллах границы между зер-
нами также могут быть описаны с помощью подходя-
щим образом выбранного расположения дислокаций.
Детали строения таких Д. с. (дислокационных сеток)
и закономерности их формирования еще недостаточно
изучены (см. Дислокации).
К числу Д. с., играющих важную роль в процессах
взаимодействия деформируемого твердого тела с окру-
жающей средой, а также в процессах деформации и раз-
рушения твердых тел, относятся ультрамикротре-
щины, возникающие как на поверхности деформируе-
мого кристалла, так и внутри объема. В хрупких те-
лах эти дефекты возникают уже на самой ранней ста-
дии развития деформации и связаны в основном с тор-
можением движущихся дислокаций. При высоких
темп-рах ультрамикротрещины могут возникнуть в
результате слияния (коалесценции) дефектов по Шотт-
ки, т. е. одиночных вакансий. В пластичных телах
ультрамикротрещины — зародыши трещин хрупкого
разрушения, возникают после более или менее зна-
чительной пластич. деформации, когда в структуре
кристалла появляются труднопреодолимые препятст-
вия для двйжущихся дислокаций. Наличие в струк-
туре твердого тела ультрамикротрещин, имеющих
клиновидное сечение, обеспечивает активное взаимо-
действие деформируемого твердого тела с окружаю-
щей поверхностно-активной средой в соответствии с ме-
ханизмом адсорбционного понижения прочности.
Для полупроводниковых кристаллов большое зна-
чение имеют вторичные Д. с., к-рые возникают на ос-
нове дефектов по Шоттки и дефектов по Френкелю и
могут быть названы нарушениями идентичности в рас-
положении атомов кристалла. Сюда относятся раз-
личные дефекты в структуре полупроводников, к-рые
могут значительно изменить структурно-чувствитель-
ные свойства этих кристаллов (см. Полупроводники).
Лит.: Френкель Я. И., Введение в теорию металлов,
Л.—М., 1948; Риз А., Химия кристаллов с дефектами,
пер. с аигл., М., 1956; Барретт Ч. С., Структура метал-
лов, М., 1948; Кузнецов В. Д., Кристаллы и кристалли-
зация, М., 1954. В. И. Лихтман.
ДЕФОЛИАНТЫ — вещества, вызывающие опаде-
ние листьев растений, подобное естественному листо-
паду; при правильном их применении не оказывают
отрицательного влияния на созревание растений; могут
быть применены за 12—-15 дней до сбора урожая.
В большинстве случаев Д. в больших концентраци-
ях действуют как гербициды или десиканты. В каче-
стве Д. достаточно широкое распространение получи-
ли цианамид кальция (30 — 45 кг/га),
ГГ.Г... хлорат - пентаборат (40 — 60% хлор-
COONa ата ]уасюз и 40 — 60% пентабората
COON» Na2B10Oie • 10Н2О) натрия (8—15 кг/га),
хлорат магния (6—10 кг/га), смесь
(1 : 4) эндотала [динатриевая соль
3,6-эндоксогексагидрофталевой кислоты (I)] и сульфа-
та аммония (1—2 кг!га), бис-(этилксантоген)-трисуль-
фид (02115003)233 (2 — 3 кг/га), трибутилтритиофос-
фат (C4H9S)3PO (0,7—1,5 кг/га) и нек-рые другие.
Свойствами Д. обладают также соли и эфиры монохлор-
уксусной к-ты, толуамиды монохлоруксусной к-ты,
бутиндиол и др. Наибольшее практич. применение
в США и СССР получили цианамид кальция и хлора-
ты. Недостатком хлоратов является сравнительно мед-
ленное действие, а цианамид кальция действует только
при наличии росы. Значительно лучшим действием
обладает кислый цианамид натрия или стабилизи-
рованный свободный цианамид. Обычно при приме-
нении Д. на хлопчатнике опадает 70—95% листьев.
Лит.: Баскаков Ю. А., Мельников Н. Н.,
Хим. пром-сть, 1955, № 7; Химические средства стимуляции
и торможения физиологических процессов растений, под общ.
ред. Ю. В. Ракитина, М., 1958; Бродский Н. Г., Шес-
топал Я. В., Предуборочное химическое подсушивание
высадков сахарной свеклы, М., 1956. Н. Н. Мельников.
ДЕФОРМАЦИЯ ПОЛИМЕРОВ — способность по-
лимерных материалов значительно изменять свою
форму под действием внешних сил, проявляя при этом
специфические лишь для них закономерности сопро-
тивления деформации, обусловленные цепным строе-
нием макромолекул. Д. п. чрезвычайно резко зависит
от темп-ры, а также от динамич. режима воздействия
сил, что связано с возникновением при деформации
неравновесных состояний (см. Механические свойства
полимеров). Деформация аморфных полимеров сла-
гается из упругой, высокоэластич. и пластич. дефор-
маций. Соотношение этих деформаций определяется
природой вещества, темп-рой и скоростью воздействия
сил. Деформация аморфных полимеров при достаточно
низких темп-рах (в застеклованном состоянии) при
не очень больших напряжениях имеет чисто упругий
характер. При возрастании растягивающих напряже-
нии происходит либо хрупкое разрушение полимер-
ного стекла (при деформациях от 0,1 до нескольких
%), либо развитие больших деформаций порядка 100—
200%. Такая большая деформируемость, часто назы-
ваемая холодной текучестью, а также вынужденной
эластичностью полимеров, ведет к образованию ани-
зотропного полимера, сохраняющего свое деформи-
рованное состояние после разгружения неограниченно
долго. Полное восстановление исходной формы может
быть достигнуто нагреванием до температуры стекло-
вания.
При нагревании полимера выше темп-ры стеклова-
ния происходит изменение характера деформации.
Модули упругости, имевшие для стеклообразных поли-
меров значения порядка 100—1000 кг! мм?, резко убы-
вают, доходя до значений порядка 0,01—10 кг/мм?\
величины обратимых деформаций возрастают до сотен
процентов, а в отдельных случаях до 1000—2000%.
Подобного рода Д. п. получили название высокоэла-
стич. деформаций. Нагревание высокоэластич. поли-
мера выше темп-ры текучести приводит к появлению
пластич. свойств — Д. и. становится неограниченной
и не полностью обратимой. В переходных областях
темп-р, от стеклообразного состояния к высокоэлас-
тическому и от высокоэластического к вязко-теку-
чему (пластическому), особенно ярко проявляется
зависимость Д. п. от длительности действия сил.
Переход от одного типа деформации к другому проис-
ходит при тем более низких темп-рах, чем продол-
жительнее действие сил. Пластичный при медленном
воздействии сил полимер становится эластичным при
более быстром воздействии и даже твердым (упругим)—
при очень быстром.
Пространственные (сетчатые) полимеры не могут
развивать необратимые деформации, если при этом не
происходит химич. изменений их структуры. Химич,
изменения полимеров линейных приводят иногда к не-
возможности перехода в эластическое или вязко-те-
кучее состояние при нагревании. Полимеры в кристал-
лич. состоянии вследствие особого характера кристал-
лизации полимеров также обладают особенностями
деформации. Модули упругости при малых деформа-
циях (порядка нескольких процентов) имеют значения
1077
ДЕФОРМАЦИЯ ПОЛИМЕРОВ — ДЕЭМУЛЬГАТОРЫ
1078
ок. 1000—10000 кг/мм1; величины деформации растя-
жения зависят от темп-ры и могут достигать сотен
процентов. В процессе растяжения происходит обра-
зование резко очерченной «шейки», в к-рой полимер
чрезвычайно сильно анизотропен. Такой анизотроп-
ный полимер сохраняет свое состояние неограниченно
долго после снятия действия сил, но полностью вос-
станавливает исходную форму при темп-рах, близких
к области плавления. Кроме подобного рода деформа-
ции, также наз. вынужденно-эластической, допол-
нительно возникает и быстро обратимая деформация,
достигающая десятков и даже сотен процентов. Вы-
нужденная эластичность стеклообразных и кристал-
лич. полимеров обусловлена тем же механизмом рас-
кручивания свернутых цепных молекул, к-рый лежит
в основе обычных эластич. деформаций. Однако в этих
твердых состояниях полимеров цепные молекулы не
могут изменять свою форму при воздействии только
теплового движения. Поэтому развитие такого рода
Д. п. происходит только при воздействии достаточно
больших внешних напряжений, а восстановление ис-
ходной формы становится возможным лишь при появ-
лении подвижности цепных молекул вследствие нагре-
вания. Выше темп-ры плавления кристаллич.полимеры
становятся аморфными (эластичными или вязко-те-
кучими) и подчиняются соответствующим закономер-
ностям. Д. п. в ориентированном состоянии резко за-
висит от направления. При растяжении в направлении
ориентации молекул стеклообразные и кристаллич.
полимеры проявляют значительные обратимые де-
формации (до неск. °/0). Сочетание такой упругости
с высокой прочностью, обусловленной ориентацией
цепных молекул, делает ориентированные полимер-
ные нити незаменимым материалом для произ-ва
тканей и других текстильных изделий. Закономер-
ности деформаций растяжений и сжатия, сдвига, кру-
чения и т. п. в общем виде для полимеров пока изу-
чены не очень полно.
Химич, состав и строение молекул полимеров,
а также их молекулярный вес определяют темп-рные
области существования различных упомянутых состо-
яний того или иного полимера. Появление специфики
Д. п. связано с гибкостью цепных молекул и их дли-
ной. Поэтому в каждом полимергомологич. ряду воз-
никновение особенностей Д. п. происходит в области
определенных степеней полимеризации, ниже к-рых
деформация имеет обычный характер. Повышение мо-
лекулярного веса в случае аморфных полимеров при-
водит к повышению вязкости, а в случае кристаллич.
полимеров — к возрастанию величины деформации
при разрыве. Кроме того, рост длины молекул приво-
дит к повышению прочности (до определенного пре-
дела). Д. п. может также развиваться благодаря ме-
ханич. разрушению цепных или пространственных
(сетчатых) макромолекулярных структур на свободные
макрорадикалы. Такие обломки молекул способны пе-
ремещаться в соответствии с действием внешних сил
и впоследствии путем рекомбинации восстанавливать
молекулярную структуру тела, аналогичную исход-
ной. Этот процесс приводит к химич. текучести по-
лимеров, а также позволяет использовать Д. п. для
проведения химич. реакций в полимерах (см. Меха-
нохимия полимеров).
Лит.: Каргин В. А., СлонимскийГ. Л., Крат-
кие очерки по физико-химии полимеров, М., i960; К о б е к о
П. П., Аморфные вещества..., М.—Л., 1952; Т р е л о а р Л.,
Физика упругости каучука, пер. с англ., М., 1953; Каргин
В. А. и СоголоваТ. И., Ж. физ. хим., 1949, 23, вып. 5,
с. 530—39, 540—50, 551—62; 1953, 27, вып. 7, с. 1039—49,
вып. 8, с. 1208—12, 1213—16, вып. 9, с. 1325—29; 1955, 29,
вып. 3, с. 469—75; К о бе к о П. П., Кувшинский
Е. В., Гуревич Г. И., Изв. АН СССР. Сер. физ., 1937, № 3,
с. 329—44; Александров А. П. И Лазуркин Ю. С.,
Ж. техн, физ., 1939, 9, вып. 14, с. 1249—60; Слонимский
Г. Л.,' там же, 1939, 9, вып, 20, с. 1791—807; Die Physik der
Hochpolymeren, hrsg. von H. A. Stuart, Bd 4, B.—Gottingen—
Hdlb., 1956. T. И. Соголова.
ДЕЦИЛОВЫЙ АЛЬДЕГИД (деканаль, каприновый
альдегид) СН3(СН2)8СНО, мол. в. 156,27 — бесцветная
жидкость с запахом розы и апельсиновой корки; т. кип.
208—2097760 мм; 94712 мм; dp 0,828; nlfi 1,42977.
Д. а. образует кристаллич. производные: оксим, т. пл.
69°; семикарбазон, т. пл. 102°; тиосемикарбазон, т.
пл. 99—100s; 2,4-динитрофенилгидразон, т. пл. 104“;
азин, т. пл. 34°; димедоновое производное, т. пл.
91,7°. Д. а. может быть получен окислением или ката-
литич. дегидрированием децилового спирта, селек-
тивным гидрированием дециленового альдегида, со-
держащегося в масле листьев и цветов кориандра,
пропусканием смеси паров каприновой и муравьиной
к-т над окисью марганца. Д. а. количественно опре-
деляется оксимированием и др. методами определе-
ния альдегидов. Д. а. применяют в парфюмерии как
душистое вещество. в. И. Красева.
ДЕЦИЛОВЫЙ СПИРТ (деканол-1, 1-оксидекан)
СН3(СН2)8СН2ОН, мол. вес 158,28 — бесцветная жид-
кость с запахом цветов; т. пл. 7°; т. кип. 228—
2327760 мм, 120712 мм; dj» 0,8297; п°£ 1,43719. Д. с,
образует кристаллические производные: фенилуретан,
т. пл. 61°; n-нитрофенилуретан, т. пл. 117°; а-нафтилу-
ретан, т. пл. 71°; n-дифенилуретан, т. пл. 111°; 3,5-ди-
нитробензоат, т. пл. 57°; семикарбазон эфира пирови-
ноградной к-ты, т. пл. 111°. Д. с. может быть получен
гидрированием дециленового альдегида, выделяемого
из эфирного масла листьев и цветов кориандра; в
пром-сти получают каталитич. гидрированием эфира
каприновой к-ты над меднохромовым катализатором
при темп-ре ок. 250° и давлении 200 ат. Количественно
Д. с. определяют ацетилированием в среде пиридина
и другими методами определения первичных спиртов.
Применяют Д. с. в парфюмерии как душистое вещество;
для получения децилового альдегида; в качестве сырья
в производстве синтетических моющих средств (де-
тергентов). в. И. Красева.
Деэмульгаторы — вещества, добавляемые к
эмульсиям для их разрушения, деэмульгирования.
Характер Д. зависит от природы эмульгатора, прида-
ющего устойчивость эмульсии. Д. делятся на 3 группы:
1) Д., химически разрушающие оболочку, образуемую
эмульгатором на поверхности частиц эмульсии, напр.
сильные минеральные к-ты, разрушающие обычные
мыла, служащие эмульгаторами эмульсий типа М/В
(«масло в воде»). Выделяющаяся при этом жирная к-та
не может стабилизовать эмульсию. 2) Д., вызывающие
деэмульгирование обращением фаз (Инверсией фаз)
в эмульсии, т. е. превращением эмульсии из типа М/В в
тип В/М («вода в масле»). Таковы электролиты (соли)
с ионами-антагонистами, напр. Са++, добавляемые к
эмульсиям типа М/В, стабилизованным натриевым
мылом, напр. олеатом натрия; при этом образуется
кальциевое мыло, нерастворимое в воде и являющееся
стабилизатором обратной эмульсии В/М. При нек-ром
критич. соотношении Ca'H7Na+, недостаточном для
образования устойчивой эмульсии обратного типа,
эмульсия расслаивается. 3) Д. наиболее общего зна-
чения, достаточно сильные поверхностно-активные
вещества, вытесняющие эмульгатор с поверхности ка-
пелек, т. е. физико-химически разрушающие образуе-
мые им защитные оболочки капелек эмульсии. Обра-
зующийся при этом адсорбционный слой самого Д. (вы-
теснителя) не обеспечивает достаточно высокой устой-
чивости капелек эмульсии против их укрупнения путем
слияния (см. Коалесценция). Т. обр., Д. должен быть
более поверхностно-активным, чем эмульгатор данной
эмульсии, но обладать меньшей стабилизующей способ-
ностью. Д., как любые сильно поверхностно-активные
вещества, сами являются эмульгаторами, но значи-
тельно слабее стабилизующими эмульсии,- чем те,
1079 ДЕЭМУЛЬГИРОВАНИЕ — ДЖЭППА — КЛИНГЕМАНА РЕАКЦИЯ
1080
к-рые практически обеспечивают их устойчивость.
К таким Д. относятся неионогенные поверхностно-
активные вещества типа смачивателей, а также анион-
активные вещества, напр. алкилсульфоновые и ал-
киларилсульфоновые к-ты,частично нейтрализованные
с образованием кальциевых, магниевых или алюминие-
вых солей. Такие Д. применяются при обезвоживании
и обессоливании нефтей. Менее эффективны для этой
цели Д., получаемые сульфированием масляных и ке-
росиновых фракций нефтей, т. наз. НЧК — нейтра-
лизованный черный контакт, являющийся также от-
ходом при сернокислотной очистке нефтепродуктов
и содержащий, после нейтрализации щелочью, в ка-
честве активного начала соли сульфонафтеновых кис-
лот (контакт Петрова). П. А. Ребиндер.
Лит. см. при ст. Деэмульгирование.
ДЕЭМУЛЬГИРОВАНИЕ — процесс разрушения
(расслоения) эмульсий. Д. используется для освобож-
дения жидких сред от эмульгированных в них жид-
костей или длн выделения этих жидкостей, напр.
для разрушения таких эмульсий, как молоко, с целью
отделения жира от воды, как латекс, представляю-
щий собой водную систему со взвешенными частицами
каучука. Особое значение Д. имеет для обезвожи-
вания и обессоливания нефтей, содержащих часто до
50% и выше тонко и устойчиво эмульгированной
пластовой воды, сильно минерализованной солями
(хлоридами) натрия, кальция и магния. Д. — про-
цесс, обратный эмульгированию, направленный на
устранение стабилизующего действия тонких защит-
ных пленок на поверхности взвешенных в жидкости
капелек. Д. можно производить механич., термич.,
электрич. и химич. методами.
1. Механическое Д. — а) повышением
скорости седиментации капелек, обычно посредством
центрифугирования, применяемого, когда жидкости,
образующие эмульсию, имеют различные плотности;
центрифугирование выполняет ту же роль, что и
длительное отстаивание эмульсий; б) фильтрацией че-
рез рыхлые (пористые) слои или слои крупных зерен
твердого материала, избирательно смачиваемого эмуль
гированной жидкостью, т. е. гидрофобного для «пря-
мых» эмульсий (типа «масло в воде») и гидрофильного
для «обратных» эмульсий (типа «вода в масле»). При
этом капельки эмульгированной жидкости, прилипая
к твердым поверхностям, полностью или частично
растекаются по ним и легко сливаются друг с другом
под действием поверхностной энергии, образуй на по-
верхности сплошной слой или крупные капли, легко
отделяющиеся под действием силы тяжести. Это приво-
дит к быстрому расслоению эмульсии на два слоя. Вме-
сто фильтрации можно применять перемешивание (не
слишком интенсивное во избежание обратного эмуль-
гирования) специальными мешалками с сильно раз-
витой поверхностью, подобной поверхности, фильтру-
ющего слоя, на к-рой и происходит коалесценция ка-
пелек, их укрупнение путем слияния между собой.
В качестве гидрофобных поверхностейдля улавливания
капелек эмульгированного масла из водной среды при-
меняют фильтры из асбеста, талькового камня, угля,
а также фильтры из целлюлозы, кварца, стекла, плот-
ного известняка (мрамора), подвергнутых специальной
обработке гидрофобизирующими веществами. Кроме
того, можно использовать любой материал, покрытый
гидрофобной пленкой из тефлона. В качестве гидро-
фильных поверхностей для Д, прямых эмульсий, напр.
воды в нефтях, можно применять крупный кварцевый
песок, стекло, керамику (и кирпич). Методы фильтра-
ции и перемешивания, хотя они и представляют собой
механич. приемы, основаны на физико-химич. явле-
ниях избирательного смачивания.
2. Электрическое Д. по существу при-
водит к колебательным механич. воздействиям —
интенсивным соударениям капелек эмульсии, приоб-
ретающих разноименные заряды под влиянием пере-
менного электрич. поля высокого напряжения в спе-
циальных промышленных установках. Частицы эмуль-
сии взаимно притягиваются, сливаются и оседают
под действием силы тяжести.
3. Термическое Д. основано на понижении
вязкости жидкой среды, а также вязкости стабилизу-
ющих оболочек на поверхности капелек эмульсии
с повышение^ темп-ры. Это увеличивает скорость се-
диментации капелек и способствует их слиянию.
Все эти методы Д. приобретают высокую эффективность
в практич. использовании только при сочетании их
с химич. Д.
4. Химическое Д. состоит в добавлении
к эмульсии относительно малых количеств (0,01 —
0,1 %) специальных реагентов — деэмульгаторов, на-
значение к-рых — разрушать оболочку эмульгатора,
стабилизующую капельки эмульсии, или заменять ее
другой, менее прочной оболочкой. Такие капельки,
потерявшие устойчивость, коалесцируют (сливаются)
при сталкивании (в процессе перемешивания), и эмуль-
сия быстро расслаивается в результате седиментации
крупных капель. После прибавления деэмульгатора
расслоению эмульсий может способствовать любой из
трех вышеуказанных методов Д. п. а. Ребиндер.
Деэмульгирование нефти — один из важных тех-
нологич. процессов переработки нефти, предшест-
вующий ее перегонке. Перегонка добытой из недр
земли обводненной и засоленной нефти сложна и эко-
номически невыгодна. Применяются все четыре ука-
занных приема Д. К нестойким эмульсиям, способ-
ным расслаиваться на нефть и воду лишь вследствие
разности плотностей компонентов, применяется про-
должительное отстаивание в резервуарах. При этом
отделяются и механич. примеси. Отстаивание наиболее
экономично осуществляется при предварительном
нагревании эмульсии до 120—160° в теплообменниках
под давлением (8—12 ат) в специальных аппаратах—
водогрязеотстойниках. Подогрев снижает вязкость
нефти и ускоряет отстаивание воды. Во многих слу-
чаях смесь нефти и буровой воды образует устойчивую
эмульсию и тогда Д. успешно осуществляется химич.,
термич. и электрич. методами. В качестве деэмуль-
гаторов нефти используются органич. соединения,
способные разрушать (вытеснять) защитную пленку
природных эмульгаторов. Для этого применяются соли
Na, Са и А1 высокомолекулярных жирных кислот и
сульфокислот, нейтрализованный черный контакт
(НЧК) — контакт Петрова, получающийся нейтра-
лизацией продукта, остающегося при очистке дымя-
щей серной к-той маловязких масляных дистиллятов,
нейтрализованный кислый гудрон (НКГ), продукт
сульфирования растительных масел (СУМ), окислен-
ный керосин, а также неионогенные поверхностно-
активные вещества и др. Выбор деэмульгатора зависит
от типа эмульсии и экономики процесса.
Ф. В. Нореневская.
Лит.: Ребиндер П. А. и Поспелова К. А.,
(вступ. ст.), в кн.: Клейтон В., Эмульсии, пер.
с англ., М., 1950; Clayton W., The theory о£ emulsions
and their technical treatment, 5 ed., N. Y., [1954]; Ш в a p ц A.
и Перри Л ж., Поверхностно-активные вещества, пер.
с англ., М., 1953; Той кошуров Б. П., Серб-Сер-
бина Н. Н. и Смирнова А. М., Основы химического
деэмульгирования нефтей, М.—Л., 1946; Гурвнч Л. Г.,
Научные основы переработки нефти, 3 изд., М.—Л., 1940;
Чефранов К. А., Электрообезвоживание и электрообес-
соливание нефтей, М.—Л., 1948; Гуревич И. Л., Тех-
нология нефти, ч. 1, 2 изд., М.—Л., 1952; Пархомен-
ко В. Е., Технология переработки нефти и газа, 2 изд., М.,
ДЖЭППА — КЛИНГЕМАНА РЕАКЦИЯ — полу-
чение арилгидразонов эфиров а-кетокислот или а-дике-
тонов взаимодействием ароматич. диазосоединений с
натриевыми производными эфиров или солей [1-кето-
1081
ДЖЭППА — КЛИНГЕМАНА РЕАКЦИЯ - ДИАГРАММА СОСТОЯНИЯ
1082
кислот:
R'-C=C(R)-COOC.,H5 P'-c-c(R)-cooc2h5
iNa L 6 NTN~Ar
r-C-COOC2H5
II
N-NHAr
R'—C=C(R) —COONa rR--C-C(R)-COONa
ONa J 6 N=H~Ar
R'-C-C-R
I!) ilf-NHAr
Начальной стадией реакции является образование
соответствующего азосоединения (I или II) с последу-
ющим элиминированием в первом случае — ацильно-
го радикала, во втором — карбоксильной группы.
Чаще всего в Д.—К. р. используют ацетоуксусный
эфир или его производные, однако могут быть приме-
нены р-кетокислоты самого разнообразного строения:
C0H5N2X
СН3СОСНСООС2Н5-------сн3-с-соосан5
сн3 n-nhc6h5
CbH5NX
RCH=CHCOCHCOOC2H5 '
сосн3
—RCH=CHCOCCOOC2h5
II 2 5
N-NHC6H5
c6h6n2x
Go
COOC2H5
c6h5n2x
Ccooh
c=n-nhcgh5
COOC2H5
В Д.—К. p. вступают соединения и других типов, со-
держащие в углеводородном радикале подвижный атом
водорода, напр. соли пиридилуксусной к-ты:
I ,J-CH-R
N I
COONa
ArNgX
C-R
ll
N-NHAr
Это — способ получения 2-ацилпиридинов. В качестве
второй компоненты Д.—К. р. могут быть применены
производные самых разнообразных ароматич. аминов.
Образующиеся арилгидразоны могут быть исполь-
зованы для синтеза индолов по Фишера реакции, при-
чем особенно ценной является возможность получения
индолов, замещенных в бензольном ядре, а также воз-
можность удобного синтеза триптамина и его про-
изводных:
a-XCeH4NoCl
сн3соснсоос2н6-------------*
СНз
—- сн3-с-соос2н.
II
n-nhcbh4x
X-
СООС2Н5
XC6H4N2Cl
СООС2Н5
о
ch2ch2nh2
Широко применяется превращение образующихся
арилгидразонов в а-аминокислоты:
СН3СОСН(СН3)СООС2Н5 — СН3ССООС2Н5 —
A-nhcuh5
— CH3CH(NH2)COOCaH5
Реакция открыта в 1887 Ф. Джэппом и Ф. Клинге-
маном.
Лит.: Sumpter W. С., Miller F. М., Heterocyc-
lic compounds with indole and carbazole systems, N. Y., 1854
(The chemistry ot heterocyclic compounds, v. 8); Суворов
H. H., Мамаев В. П., Родионов В. М., в кн.: Реакции
и методы исследования органических соединений, кн. 9, М.,
1959; Феофил акта в В. и Иванов А., Ж. общ.
хим., 1943, 13, вып. 6, с. 457—66; Abramovltch R. А.
and Shapiro D., J. Chem. Soc., 1956, № 11, p. 4589.
la. H. Преображенская.
ДИАГРАММА РАСТВОРИМОСТИ — см. Раствори-
мость.
ДИАГРАММА СОСТАВ — СВОЙСТВО — см. Фи-
зико-химический анализ.
ДИАГРАММА СОСТОЯНИЯ (фазовая диаграмма)—
графич. изображение зависимости между параметрами
состояния равновесной физико-химич. системы или
между ними и нек-рыми другими параметрами си-
стемы, в частности концентрациями. Д. с. однокомпо-
нентной системы дает связь между темп-рой t и давле-
нием р; Д. с. двойной — связь между этими парамет-
рами и концентрацией одной из составных частей и
т. д. При числе параметров больше Двух для изобра-
жения Д. с. на чертеже ее проектируют на плоскость,
выбираемую т. о., чтобы нек-рое число параметров
(иногда их сумма) имело постоянное значение. Чаще
всего постоянным принимается р и получают т. н.
изобарную Д. с.; напротив, изотермиче-
ская Д. с. изображает зависимость между р и
концентрациями при постоянной t; т. н. из о кон-
центрационная Д. с. дает зависимость между
t, р и концентрациями всех составных частей, кроме
одной, концентрация к-рой принимается постоянной.
Нередко исследуют системы, в которых отсутствует
газообразная фаза (напр., сплавы
катов); такие системы наз.
конденсированными, а их
Д. с. — диаграммами кон-
денсированного состояния.
В качестве примера Д. с. од-
нокомпонентной системы на ри-
сунке приведена Д. с. воды. В
т. н. тройной точке О система
состоит из льда, жидкой воды и
пара, находящихся в равнове- '
сии. Линия ВО изображает со-
стояние равновесия льда с па-
ром, OG — жидкой воды с па- . „
ром, OD—льда с жидкой водой.
Т. наз. поля, лежащие между линиями ВО, OD и OG, изобра-
жают систему лишь в одном агрегатном состоянии (на рис.
несколько преувеличен излом между во и OG и значительно
усилен наклон влево OD, подробнее см. Вода).
металлов и сили-
0
^4.58
0,0076’
Температура в 'С
1083
ДИАГРАММА ХИМИЧЕСКАЯ — ДИАЗОАМИНОСОЕДИНЕНИЯ
1084
Основные тины Д. с. могут быть выведены теорети-
чески из кривых (поверхностей) термодинамич. по-
тенциала. Построение Д. с. реальных систем произ-
водится на основе эксперимента методами физико-
химического анализа. Значение Д. с. состоит в том, что
они позволяют делать точные выводы о физико-химич.
природе и границах существования фаз системы без
их выделения и химич. анализа. Знание Д. с. позволяет
подобрать состав системы, наиболее удовлетворяю-
щий требованиям практики, а также подходящие ус-
ловия для проведения того или иного процесса. Напр.,
Д. с. системы железо''—углерод является основой
термич. обработки стали, Д. с. водно-соляных систем
широко используются в галургии и технологии мине-
ральных солей, Д. с. силикатных систем — в ме-
таллургии (шлаки) и в технологии силикатов, Д. с.
систем из металлов и серы — в металлургии цветных
металлов и др. Описание главных типов Д. с. см.
в ст. Двойные системы, Тройные системы, Многоком-
понентные системы.
Лит.: Аносов В. Я. и Погодин С. А., Основные
начала физико-химического анализа, М.—Л., 1947.
В. Я. Аносов.
ДИАГРАММА ХИМИЧЕСКАЯ — см. Физико-хи-
мический анализ.
ДИАЗОАМИНОЛЫ — принятое в СССР название
препаратов для печати по тканям; другие названия —
рапидогены (Германия), нейтрогены
(Франция) и др. Д. — эквимолекулярные смеси диа-
зоамино- или диазоиминосоединений — триазенов с
азотолами. Д. растворимы в воде, легко воспламе-
няются, пыле-воздушные смеси нек-рых Д. — взрывча-
ты. Для получения Д. диазотируют азоамины, на
полученные диазосоединения действуют, обычно в р-ре
соды, т. н. «стабилизаторами» — вторичными или арома-
тич. первичными или вторичными аминами, содержа-
щими группы СООН или SO3H или те и другие сов-
местно. К наиболее важным стабилизаторам принад-
лежат саркозин CH3NHCH2COOH, метилтаурин
CH3NHCH3CH2SO3H, 2-амино-4-сульфобензойная и
2-метил-(или "этил-)амино-5-сульфобензойная к-ты.
При взаимодействии диазосоединений с аминами-
стабилизаторами образуются триазены, к-рые осаж-
дают из р-ров добавлением NaCl, NaOH или иным пу-
тем, отфильтровывают, высушивают и смешивают
в виде порошков с азотолами. Ниже, как пример,
приведены составы нек-рых Д.:
и диазосоединение, образующее с азотолом на ткани
нерастворимый в воде азокраситель (азопигмент).
Стабилизатор смывается при промывке окрашенной
ткани. Первоначально проявление Д. производилось
в присутствии паров муравьиной и уксусной кислот,
что вызывало коррозию аппаратуры и затрудняло при-
менение Д. в смеси с красителями других классов,
напр. кубовыми. Позднее в печатную краску стали
вводить вместо минеральных щелочей летучие орга-
нические основания, например диэтиламиноэтанол
(C2H5)2N—С2Н4ОН или аммонийные соли, легко
отщепляющие при нагревании аммиак. В обоих слу-
чаях проявление окраски достигается при про-
стой обработке паром. Найдены Д., легко проявляе-
мые в нейтральных парах. Применяют Д. обычно
для печати по целлюлозным волокнам и натуральному
шелку; они образуют яркие прочные окраски раз-
личных цветов. См. также Азотолы, Азоамины, Азо-
красители. М. А- Чекалин.
Лит. см. при ст. Азоамины.
ДИАЗОАМИНОСОЕДИНЕНИЯ (триазены) — ор-
ганич. соединения, содержащие характерную для Д.
группировку —N=N—NH—; общая формула Д.
R'—N=N—NH—R", где R' — ароматич. радикал, а
R" — любой радикал. Д.—твердые, обычно окрашенные
в желтый цвет вещества, растворимость к-рых в воде
зависит от характера заместителей в R: СООН, SO3H
и т. п. Д. устойчивы в щелочной, нейтральной и в очень
слабокислой средах. В более сильной кислой среде
Д. легко разлагаются на исходные диазосоединения
и амины. Подобно диазосоединениям, Д. разлагаются
с выделением азота при нагревании с кислотой,при
восстановлении цинком и кислотой превращаются
в арилгидразин и амин; многие Д. весьма светочув-
ствительны. Это объясняется тем, что в первой стадии
большинства реакций Д. разлагаются с образованием
диазосоединения. Д. всегда реагируют менее энергич-
но, чем соответствующие диазосоединения.
Практич. значение имеют два метода синтеза Д.г
а) взаимодействие ароматич. диазосоединения с али-
фатическим или не способным к азосочетанию аромати-
ческим первичным или вторичным амином; б) действие
на ароматический амин азотистой кислоты в отсут-
ствии избытка минеральной к-ты. В обоих случаях
реакция идет по схеме: R'—N=N—X+R"—NH2->-
—> R'—N=N—NH—R"+HX. Д. образуются также
в качестве побочных продуктов при азосочетании
Д. вводят в состав печатной краски, наносимой на
ткань; при последующей обработке ткани паром
триазеи расщепляется аа исходные амин-стабилизатор
с нек-рыми аминами. Д. образуют две таутомер-
ные формы: R'—N=N— NHR".-R'-NH—N=N—R";
состояние равновесия зависит только от характера
1085
ДИАЗОЛИ — диазометан
1086
радикалов и содержащихся в них заместителей. Д.,
полученные из вторичного амина, не способны к тау-
томерному превращению. Для открытия Д.прибавляют
соляную к-ту, разлагающую Д. до диазосоединения,
к-рое открывают на фильтровальной бумаге по по-
явлению красной окраски при действии раствора веще-
ства, легко вступающего в азосочетаиие (аш-кислоты,
Р-соли, (3-нафтола и т. п.). При колич. определении
Д. разлагают нагреванием с соляной к-той в присут-
ствии хлорида меди (I) и измеряют объем выделив-
шегося азота. Д. применяются в текстильной пром-сти
для печати по хлопчатобумажным тканям в виде сме-
сей с азотолами, т. н. диазоаминолов. Нек-рые Д.
являются инициаторами полимеризации.
Лит.: Филиппычев С. Ф., Химия и технология
азокрасителей, ч. 1, М., 1938; Саундерс К., Ароматиче-
ские диазосоединения и их техническое применение, пер.
с англ., М., 1938; Holzach К., Die aromatischen Diazo-
verbindungen, Stuttgart, 1947. А. А. Черкасский.
ДИАЗОЛИ — принятое в СССР название стабильных
форм солей диазония (см. Диазониевая структура). Д. —
твердые вещества, обычно хорошо растворимые в воде;
выше 40—50° неустойчивы; легко воспламеняются; от-
дельные Д. в виде пыле-воздушных смесей взрыв-
чаты. Соли диазония для применения в качестве Д.
обычно стабилизируют, т. е. превращают более ус-
тойчивые соединения, чаще всего в двойные соли ме-
таллов (обычно с ZnCl2), или в соли с ароматич. сульфо-
кислотами, напр. с 1,5-нафталиндисульфокислотой.
Реже диазосоединения переводят в соли с комплекс-
ными к-тами типа HBF4 и др. соли. Нек-рые более
устойчивые Д. применяют в виде солянокислых или
сернокислых солей. Ниже приведено строение
нек-рых типичных Д.
О химич. свойствах Д. см. Диазосоединения.
Д. получают диазотированием аминов и осаждением
из растворов с помощью указанных выше стабилиза-
торов. Для еще большего снижения взрывчатости или
полного ее устранения Д. смешивают с инертными со-
лями, Na2SO4 или A12(SO4)3 и др., содержание к-рых
в сухих Д. достигает 50—60%. Названия Д. соответ-
ствуют названиям азоаминов, из к-рых они получены,
напр. диазоль синий О получают из азоамина синего О.
Д. применяют для получения нерастворимых в воде
азокрасителей непосредственно на текстильных волок-
нах методом т. н. холодного крашения. Д. исполь-
зуют в виде водных р-ров для гладкого (однотонного)
крашения хлопчатобумажных или вискозных волокон
или вводят в состав печатной краски при узорчатой
расцветке тканей. См. также Азоамины, Азотолы.
Лит. см. при ст. Азоамины. М. А. Чекалин.
ДИАЗОЛИН (омерил) C29H28N2S2O3, мол. в. 564,69—
бесцветные мелкие кристаллы; т. пл. 273—275°;
трудно растворим в воде и органич. растворителях.
Основание Д. получают при конденсации N-метил-
у-пиперидона с бензилфенилгидразином. Диазолин
применяют как лечеб-
|^Ч||---J ^снз ное средство при раз-
L 1 J r н n mi личных аллергич. за-
ЬГ ю ei з >2 болеваниях (крапив-
Сн2СвН5 ница, сенная лихо-
радка, дерматиты, зу-
ды, аллергические реакции, связанные с применением
антибиотиков, и т. п.).
Лит.: Харкевич Д. А., Клинич. медицина, 1957,
№ 5^ с. 45; см. также при ст. Апрессин. А. И. Травин.
ДИАЗОМЕТАН CH2N2, мол. в. 42,043 — простей-
шее диазосоединение жирного ряда; газ желтого цве-
та, с неприятным запахом; т. пл. —145°; т. кип.
—23°; весьма токсичен и взрывоопасен. В чистом
состоянии обычно не выделяется, а используется в ви-
де раствора в эфире, с парами к-рого Д. перегоняется.
Такой раствор разлагается медленно и ок. 20° вполне
безопасен в обращении,
Н„С — N — № H2c"=N=N~ в то время как чистый
Д. при этой темпера-
1 , 11 туре взрывает и его не-
обходимо получать в токе азота. Строение Д. окон-
чательно не установлено, однако несомненно, что он
имеет линейную структуру (1), наиболее точное при-
ближение к-рой представлено формулой (II). Д,—
весьма реакционноспособное соединение; он реаги-
рует без выделения азота, присоединяясь к олефи-
нам с образованием пиразолинов, напр.:
СН2 = СН2 -I- ch2n2 -> ch2ch2n — nch2 ->
-> 8h2ch2nhn=ch
взаимодействует с магнийорганич. соединениями с об-
разованием замещенных гидразинов, напр.:
2CsH5MgBr + CH2N2->[CcH5CH2N(MgBr) - N (MgBt)CcH5]->
-> CeH5 CH2NHNHCbH5
реагирует с хлор ангидридами карбоновых кислот
(Арндта—Эйстерта реакция). Д. легко метилирует
соединения, содержащие активный атом водорода:
RCOOH -J- CH2N2 -> RCOOCHg + N3
ROH + CH2N2 -> ROCH3 + N2
взаимодействует с кетеном с образованием цикло-
бут анона:
СН2 = СО + CH2N2 -> СН2СН2СН2СО + n2
Д. легко реагирует с хлоридами металлов и неме-
таллов:
FeCl3 + 2CH2N2 -> (ClCH2)2FeCl -f- 2NS
ССЦ + 4CHSNS -> (C1CH2)4C -J- 4N2
и др. При пиролизе Д. дает свободный радикал,
метил. Старый метод получения Д. заключался в дей-
ствии гидразина и щелочи на хлороформ:
СНС13 + h2nnh2 + зкон -> CH2N2 + ЗКС1 + н2о
Новые, более удобные методы, позволяющие полу-
чать также гомологи Д., состоят в действии щелочи
на нитрозометилуретан
кон
CH3N(NO)COOC2H5—*CH2N2 4- С2Н3ОН 4- Na2CO3 4“ Н2О
на нитрозометилмочевину
кон
CH3N(NO)CONH2------ CH2N2 4- KCNO 4- 2Н2О
нитрозометил-п-толилсульфаниламид
кон
n-CH3C3H4SO2N(NO)CH3 4- ROH--------
-»• CHSNS 4- n-CHaCeHiSOsOR 4- H2O
и др.
А. Е. Васильев.
1087
ДИАЗОНИЕВАЯ СТРУКТУРА — ДИАЗОСОЕДИНЕНИЯ
1088
ДИАЗОНИЕВАЯ СТРУКТУРА — группировка,
включающая два связанных между собой атома азота,
из к-рых один 4-ковалентен,и заряжен положительно,
а другой 3-ковалентен и нейтрален: —N=N:. Подоб-
ная структура впервые предложена Бломстрандом
(1869) для ароматич. диазосоединений. Термин «диазо-
ний» (по аналогии с аммонием) был предложен А. Ган-
чем (1895). Строение Д. с. приписывают группиров-
кам, входящим в состав молекул ароматич. солей диа-
зония (I), алифатич. диазосоединений (II), азотисто-
водородной к-ты (III) и её солей, органич. азидов
(IV и V).
Н-N-N=N
III
В реально существующих молекулах «чистая» Д. с.,
как правило, отсутствует, т. к. электронные смеще-
ния приводят к нек-рому рассредоточению зарядов,
причём характер этих смещений зависит от характера
групп, связанных с Д. с. Так, напр., строение аро-
матич. диазоний-катиона (VI), диазометана (VII) и
фенилазида (VIII) лучше всего отображается след,
формулами:
* Л'
СН2—N=N:
VII
Лит.: Саундерс К., Ароматические диазосоедине-
нияи их техническое применение, пер. сангл.,М., 1938; Дья-
конов И. А., Алифатические диазосоединения, Л., 1958,
с. 37; - В о р о ж ц о в Н. И., Основы синтеза промежуточных
продуктов и красителей, 4 изд., М., 1955, с. 439.
Е. М. Рохлин.
ДИАЗОСОЕДИНЕНИЯ — соединения общей фор-
мулы RN2X, содержащие характерную группировку
из двух атомов азота, связанную только с одним угле-
водородным радикалом. В ароматич. Д. X—кислотный
остаток или гидроксил. В жирных Д. группировка X
отсутствует (см. ниже), но вследствие аналогии реак-
ций этих соединений с реакциями ароматич. Д. за
ними сохранилось название жирных Д.
Алифатические диазосоедине-
ния. Предложены две формулы строения:
R’\ /N R'
”/С\ Ч и „)С—№N
RZ XN Rz
или
R\ + -
r„/C=N=N
Ila 116
Более вероятной считается линейная формула
(Па или Пб). Наиболее изучены диааоуксусный эфир
N2=CH—СООС2Н5 и простейшее алифатич. Д. —
диазометан CH2=N2. Алифатич. Д. неустойчивы и
весьма реакционноспособны; при нагревании разла-
гаются со взрывом, реагируют с водой, галогенами,
галогеноводородами и т. п., выделяя азот:
R — СП = N2 + НОН-> RCHsOH + N3
Легко конденсируются с ненасыщенными соедине-
ниями, образуя производные пиразола или пиразо-
лина:
RCH = CHR"+ RCH = Na--
R'—СН —СН —R"
R —СН N
Алифатич. Д. широко используются для лаборатор-
ного синтеза органич. соединений.
Ароматические диазосоединения
содержат диазогруппу, соединенную с ароматич. ра-
дикалом и неорганич. остатком: Аг—N2—X, где X —
остаток сильной к-ты (Cl-, HSO^, NOf и т. п., ОН
или ОМе, где Me — одновалентный металл). Арома-
тич. Д. открыты П. Гриссом в 1858. Известно неск.
способов их получения, но практич. значение имеет
только один — диазотирование ароматич. аминов
(см. Диазотирование). При этом Д. образуются в форме
солей диазония, диссоциирующих на катион диазония
и анион галогена: ArN2Cl [Аг—N=N]X~. В зави-
симости от состояния (твердого или растворенного)
н от влияния среды, т. е. от природы растворителя и
гл. обр. от pH водного р-ра, Д. изменяют свои свойст-
ва, что связано с соответствующими изменениями стро-
ения их молекул. При этом различные формы Д. со-
ставляют равновесные системы, состояние к-рых за-
висит от характера заместителей в ядре Д. и от зна-
чения pH раствора. В течение последних десятиле-
тий наибольшим признанием пользовалась теория
строения диазосоединений А. Ганча, допускающая
существование стереоизомерных форм и удовлетво-
рительно" объясняющая поведение ароматич. Д. в
разных условиях. Однако признано, что она нуж-
дается в нек-рых дополнениях и изменениях. В част-
ности предполагается, что изомерия разных форм сое-
динения имеет структурный характер, а не про-
странственный.
Наибольшую роль в химии Д. играют три основные
формы ароматич. Д.: относительно стабильная и
реакционноспособная соль диазония [Аг—N==N]X_,
образующаяся в сильнокислом растворе; неустойчи-
вый более активный амфотерный диазогидрат, способ-
ный к образованию диазокатиона и диазоаниона
([Аг—NaNJHOH-^ ArN2OH =[Аг— N=NQ-]+H+)
и существующий в средах, близких к нейтральной;
очень устойчивый, но совсем неактивный изодиазотат
Аг—N=N—ОМе, получающийся в сильнощелочной
среде и легко изомеризирующийся при подкислении
с образованием малоактивного нитрозамина ArNHNO.
Большинство ароматич. Д. — неустойчивые и весь-
ма реакционноспособные вещества. В твердом со-
стоянии разлагаются при нагревании и при ударе,
иногда с сильным взрывом, в растворах разлагаются
более медленно при низких темп-рах, значительно
быстрее при нагревании. Разложение ароматич. Д.
происходит также при действии света. Реакции с аро-
матич. Д. протекают либо с сохранением атомов азота
в молекуле, либо с выделением азота; в последнем
случае диазогруппа заменяется, напр., группой
ArN2X + НОН —> АгОН + НХ + N«
Из реакций, протекающих без выделения азота, наи-
большее значение имеют: азосочетание с ароматич.
аминами, фенолами и соединениями, содержащими
активные метиленовые группы; образование диазо-
аминосоединений при взаимодействии с неспособ-
ными к азосочетанию аминами (ArN2X+Ar'NH2—>
->Аг—N=N—NH—Аг'+НХ); восстановление суль-
фитом в щелочной среде или хлористым оловом в кис-
лой среде до арилгидразинов (ArN2X—> ArNH—NH2).
Ароматич. Д. обменивают диазогруппу на многочис-
ленные и разнообразные группы. При нагревании
в водных кислых растворах получаются соответствую-
щие оксисоединения. При нагревании со спиртами реак-
ция может одновременно идти в двух направлениях —
с введением алкокси-группы (ArN2X+CH3OH —>
—>ArOCH3+HX+N2) и с заменой диазогруппы на атом
водорода (ArN2X + СН3СН2ОН -> АгН+СН3СНО+
+ HX-j-N2); в присутствии щелочей и восстановителей
1089
ДИАЗОТАТЫ — ДИАЗОТИРОВАНИЕ
1090
происходит преим. вторая реакция. Галогенные соли
диазония превращаются в соответств. ароматич. гало-
генопроизводные пли действии Си2С12 (Зандмейера
реакция) или порошкообразной меди (Гаттермана
реакция): ArN2Cl -> ArCl-}-N2. В присутствии CuCN
диазогруппа заменяется нитрильной группой (CN).
Галогенные соли диазония образуют с галогенными
солями нек-рых элементов двойные соли, превращаю-
щиеся при действии мелкораздробленной меди в аро-
матич. элементорганич. соединения (см. Несмеянова
реакция). Кроме того, диазогруппу в Д. можно за-
менить группами: —NO2, —NH,, —NCO, —SCN,
—SH, —S—, —SO2H, —AsO(OH)2"ii мн. др. Приведен-
ные выше реакции не исчерпывают всех возможных
превращений ароматич. Д.
Ароматич. Д. широко применяются для синтеза са-
мых разнообразных органич. соединений. Нек-рые
реакции используются в промышленности, напр. аво-
сочетание в произ-ве азокрасителей, реакции восста-
новления диазогруппы и обмена ее на галогены,
водород, группы ОН, CN и др. в произ-ве промежу-
точных продуктов, красителей и фармацевтич. пре-
паратов. На светочувствительности Д. основано при-
менение бумаги для копирования чертежей (см. Ди-
азотипия). Стойкие препараты Д. — нек-рые устой-
чивые соли диазония (см. Диазониееая структура),.
антидиазотаты и диазоаминосоединения применяются
для крашения и печатания тканей. См. также Диа-
зотаты.
Лит.: Арбузов Ю. А., Усп. химии, 1938, 7, вып. 4;
Якубович А. Я. п Гинсбург В. А., там же, 1951,
20. вып. 6; Н о 1 z а с h К., Die aromatischen Diazoverbin-
dungen, Stuttgart, 1947; Saunders К. H., The aroma-
tic diazo-cotnpounds and their technical applications, 2 ed.,
L., 1949. А. А. Черкасский.
ДИАЗОТАТЫ ароматических соеди-
нений — соли диазокислот, образующиеся в ох-
лажденных р-рах из солей диазония при изме-
нении кислой среды в щелочную. Реакция между ио-
нами диазония и гидроокисла принадлежит к числу
медленно протекающих ионных реакций; проходит
через промежуточную стадию диазогидрата, быстро
распадающегося с образованием Д. Медленность пер-
вой стадии реакции дала возможность принять смесь
соли диазония со щелочами и Д. за нестойкий, ре-
акционный Д., отличающийся от обычного, и создать
теорию сии-аи/пи-изомерии Д. На самом деле сущест-
вует только один Д. общей формулы ArN2O“Me+, где
Ме+ — ион металла. Сильные минеральные к-ты вы-
тесняют из Д. свободные диазокислоты, существую-
щие в виде двух таутомерных соединений: диазогид-
рата Аг—N=N—ОН и нитрозамина Аг—NH—NO.
Дальнейшее подкисление приводит к образованию
солей диазония. Слабые к-ты (сернистая, синильная)
взаимодействуют с Д., образуя ковалентные соеди-
нения типа Аг—N=N—Ас, вероятно, способные к
cuH-awnu-изомерпи (Ас — остаток кислоты).
Д. широко применяются в процессах крашения в
смеси с фенольными азосоставляющими (напр., азо-
толами). В таких смесях реакция не происходит,
т. к. они не содержат диазокатионов — единственной
формы диазосоединений, способной к реакции азосо-
четания, но достаточно обработать текстильный ма-
териал с нанесенными на него Д. и азотолом раство-
ром минеральной к-ты, как образуется сначала соль
диазония, а затем и азокраситель (см. Азосочетание).
Б. А. Порай-Кошиц.
ДИАЗОТИПИЯ — фотография, метод, широко при-
меняемый для копирования чертежей и текстов, а
также в полиграфии; основан на светочувствительно-
сти диазосоединений. Ароматич. диазосоединения до-
вольно быстро разлагаются при действии ультрафио-
летовых, а нек-рые также фиолетовых и синих лучей:
ArN2X4~HOH ArOH+N2-|-HX (квантовый выход
18 к. х. э. т. 1
<р = 0,07 — 0,52). Особенно светочувствительны и
находят практич. применение диазосоединения, содер-
жащие окси- или замещенную аминогруппу в орто-
или пара-положении к диазогруппе, напр. 2,1-диазо-
нафтол-4-сульфокислота (I), сульфат л-диазодифенил-
амина (II) и двойная соль хлорида 4-диазодиэтил-
анилина с ZnCl2 (III), к-рые окрашены в желтЬш цвет.
Наиболее важны
следующие методы
м 1 г, 7 с, Д ’-1)Соль арилди-
t'-2r|5'2iN г~i'2ul ’ ZnC|2 азония наносится
х==/ на поверхность бу-
111 маги, кальки или
ацетилцеллюлозной пленки. Этот материал освещают
под копируемым объектом; в освещенных местах ди-
азосоединение разлагается. Затем снимок слегка сма-
чивают р-ром фенола или ароматич. амина; неизме-
нившееся в затемненных местах диазосоединение обра-
зует с азокомпонентой краситель и получается по-
зитивное изображение (полусухой метод). 2) Соль
арилдиазония вместе с фенольной азокомпонентой
и нелетучей кислотой (винной, борной и др.), препят-
ствующей их взаимодействию, наносят на подложку.
После освещения снимок помёщают на неск. минут в ка-
меру с парами аммиака и воды — происходит нейтра-
лизация кислоты; неизменившееся диазосоединение
образует с азокомпонентой краситель, и получается
позитивное изображение (аммиачный, или «сухой»,
метод).
л-Диазопроизводные вторичных или третичных аро-
матич. аминов дают с резорцином, а- и ^-нафтолами
оранжевые или коричневые изображения, с флороглю-
цином — темно-коричневые или почти черные, с наф-
толсульфокислотами — красно-коричневые, с нафтол-
или аминонафтолдисульфокислотами и 2,3-диокси-
нафталином — синие. При применении 1,2-или 2,1-ди-
азонафтолсульфокислот с резорцином или флороглю-
цином получаются красные или красно-фиолетовые
изображения, окраска к-рых может значительно
углубляться при введении в светочувствительный слой
солей нек-рых тяжелых металлов (в присутствии со-
лей титана — до нейтрально-черной). Для предот-
вращения пожелтения фона Изображений из-за окис-
ления на воздухе азокомпонент и продуктов фотолиза
диазосоединений в светочувствительные слои или
₽ проявитель вводят слабые восстановители (тиомоче-
вину и глюкозу) и антиоксиданты (пирогаллол или
метиленовый голубой). Существуют методы диазоти-
пии, позволяющие получать и негативные изобра-
жения.
Применение методов Д. в полиграфии основано на
дублении желатины продуктами фотолиза диазосоеди-
нений или солями трехвалентного хрома, образую-
щимися при окислении этих продуктов бихроматами.
При экспонировании желатинового слоя, содержа-
щего соль арилдиазония, Освещенные места задубли-
ваются либо непосредственно, либо после обработки
р-ром бихромата калия (иногда активного диазосоеди-
нения). После промывания теплой водой получается
желатиновая матрица, к-рая используется для печати
или переносится на медную поверхность для вытрав-
ливания на ней рельефа.
Лит.: Диазотипия, М. — Л.,1937 (Тр. НИКФИ, вып. 4):
Mester L., Sei. et inds. photogr., 1955, 26, № 5, p. 161 :
№ 7, p. 249, 354. И. И. Левкоев.
ДИАЗОТИРОВАНИЕ — взаимодействие между аро-
матич. амином и HNOa в присутствии избытка не-
1091
ДИАЗОУКСУСНЫЙ ЭФИР — ДИАКАРБ
1092
органич, к-ты, в результате к-рого образуется арома-
тич. диазосоединение. Обычно при Д. пользуются
NaNO,, к-рый выделяет в кислой среде HNO2: ArNH2+
+ 2НС1 + NaNO2—> ArN^Cl- + NaCl + 2H2O, При
этом диазосоединение образуется в форме катиона
диазония. Ранее предполагалось, что реакция проте-
кает при активном участии катиона ариламмония и
HNO2 (Аг—NH++HO—NO-> Аг—Nt+2H2O).B Ha-
стоящее время принято, что в водном р-ре реагируют
основание амина и ангидрид или галогенангидрид
азотистой к-ты, образующиеся из HNO2 : 2HNO2 —
= N2O3 + Н2О или HNO2 + Н3О+ + Cl -> NOCI +
+ 2Н2О. В первой стадии образуется N-нитроз-
амин (CeH5NH, + N2O3 -> C6H5NH—NO+HNO2 или
CeH5NH2 + NOC1+ H2O -> CeHjNH—NO + H3O+ +
+ Cl-), превращающийся затем в катион диазония
(C6H5NH—NO -> CeH5N2OH -> CeH5Nt+OHOH- +
+ H3O+ й2Н2О). Скорость Д. зависит от строения ами-
на и природы аниона участвующей в реакции кислоты.
Амины, содержащие в ароматич.
ядре отрицательные заместители 3 NgOnCC
(NO2, SO3H, СООН, Cl и т. п.),
диазотируются быстрее аминов, содержащих поло-
жительные заместители (СН3, ОН, ОСН3 и т. п.). В
присутствии серной к-ты скорость Д. меньше, чем
в среде соляной к-ты. Особенно сильно увеличивает-
ся скорость Д. в присутствии ионов Вг-.
Д. проводят при относительно низких температу-
рах (от 0 до 25°) вследствие неустойчивости полу-
чающихся диазосоединений. При наибо-
лее низкой темп-ре диазотируют амины, СН3ОСОСН
содержащие положительные заместите-
ли, при наиболее высокой—ариламин-
сульфокислоты. Для полного превраще-
ния амина в диазосоединение необходимо присут-
ствие избытка неорганич. к-ты; теоретически для Д.
одного моля моноамина требуются 2 эквивалента
к-ты; на практике берут 2,3—3,0 эквивалента. В про-
изводстве Д. проводят следующим образом: амины
растворяют в воде, прибавляя необходимое количе-
ство кислоты; нерастворимые в разбавленных кис-
лотах ариламинсульфокислоты растворяют в воде
в присутствии щелочного реагента — едкого натра,
кальцинированной соды, аммиака и т. п.; при после-
дующем подкислении выпадает быстрореагирующий,
очень мелкий осадок. К раствору или суспензии амина
при определенной темп-ре прибавляют р-р NaNO2
до появления неисчезающего в течение нек-рого вре-
мени избытка HNO2, определяемого по образованию
темного пятна на иодкрахмальной бумаге, В про-
цессе Д. реакционная смесь все время должна иметь
сильнокислую реакцию (индикатор — конго). Д.
может сопровождаться побочными реакциями: раз-
ложением образующегося диазосоединения и превра-
щением его в оксисоединение; образованием диазо-
аминосоединения: RNt+RNH2—>R—N=NH—R+HA.
Д. широко применяется в производстве многочис-
ленных соединений ароматического ряда и в осо-
бенности азокрасителей. Реакция открыта в 1858
П. Гриссом.
Лит.: Саундерс К., Ароматические диазосоединения
и их техническое применение, пер. с англ., М., 1938. См. также
при ст. Азосоединения. А. А. Черкасский.
ДИАЗОУКСУСНЫЙ ЭФИР (этиловый эфир ди-
азоуксусной кислоты) N2CHCOOC2H5, мол. в. 114,10 —
лимонно-желтая жидкость со специфич. запахом;
т. пл. —24°; т. кип. 143°/76О мм, 85—86°/88лм«,
45°/12 мм; (Д7,6 1,0852; п'р'0 1,4588. Д. э. очень
летуч, перегоняется с парами воды, спирта, эфира;
трудно растворим в воде, смешивается со спиртом,
бензолом, эфиром. При действии конц. серной к-ты, а
также при нагревании, особенно в присутствии при-
месей, разлагается со взрывом; токсичен.
Д. э. получают взаимодействием хлоргидрата эти-
лового эфира глицина с азотистой к-той:
НС1 • H2NCH2COOC2H5 N^N-CH-COOCjHj
Д. э. взаимодействует с водой, спиртами, карбоновыми
к-тами, образуя соответственно окси-, алкокси- и
ацилоксиуксусные эфиры:
N2CHCOOC2H5 + ROH — ROCH»COOCoH5
(R=H, Aik, Ac)
с галогеноводородными к-тами получаются моно-,
а с галогенами — дигалогенуксусные эфиры:
N2CHCOOC2H5 + НС1 — С1СН2СООС2Н5
N2CHCOOC»H5 +J» — J2CHCOOC3H3
При реакции с альдегидами и кетонами образуются
глицидные эфиры, эфиры р-кетокислот и карбэтокси-
метиловые эфиры соответствующих енолов:
:2н5 —*- сс13сн-сн-соос2н5 + cci3coch2cooc2hs
С олефинами Д. э. образует производные пиразолина
и циклопропана, с ароматич. соединениями — про-
изводные циклогептатриена:
CH3OCO-CH-N^
+ N2CHCOOC2H5 ОСО—сн—СН
)СН3 2 сн3оси СП СП
СООСчНд .
соосн3
£н
сн-<н-СООС2Н5
СООСНз
При восстановлении Д. э. получаются, в зависимости
от применяемых восстановителей, зфир гидразона
глиоксалевой к-ты, гидразиноуксусный эфир, глицин
или уксусная кислота:
N2CHCOOC2H5
H2NN=CHCOOC2H5
H2NNHCH2COOC2Hs
h2nch2cooh
CH3COOH
Нагревание Д. э. с порошкообразной медью приводит
к образованию этилового эфира фумаровой к-ты:
2N2CHCOOCaH5 CSH5OCOCH
<^НСООС3Н3
Д. э. — одно из наиболее распространенных алифа-
тич. диазосоединений, применяемых в лаборатории
для различных синтезов.
Лит.: Chemistry of carbon compounds. Ed. by E. H. Rodd,
v. 1, pt B, Amst. [a. o.], 1952, p. 854. E. M. Рохлин.
ДИАКАРБ (диамокс, ацетазоламид, 2-ацетиламино-
1,3,4-тиадиазол-5-сульфонамид) C4HeN4O3S2, мол. в.
222,24 — белые мелкие кристаллы; т. пл. 258—
260°; растворим при 20°: в 1400 ч. воды, 166 ч. 95%-ного
спирта, в 100 ч. ацетона; нерастворим в СС14 и СНС13.
Д. растворяется в едких щелочах, из к-рых выделяется
1093
ДИАЛИЗ — ДИАДЛИЛФТАЛАТ
1094
при подкислении. При обработке. Д.0,1 н. р-ром NaOH,
а затем цинком и соляной к-той выделяется H2S.
Количественно Д. определяют
N N по его коэфф, поглощения
CH3COHNC CSO0NH0 света> который при 265 ммк
XSX равен 474 (1%-ный р-р, тол-
щина слоя 1 см). Д. получают
из гидразинсульфата и роданистого аммония. Приме-
няют Д. при сердечных и других заболеваниях, а
также для лечения глаукомы.
Лит.: British pharmacopoeia, 1958, L., 1958, р. И; Roh-
lin R. О., Clapp J. W., J. Amer. Chem. Soc., 1950, 72,
№ 11, p. 4890. См. также при ст. Апрессин. А. С. Елина.
ДИАЛИЗ — освобождение коллоидных р-ров и
р-ров высокомолекулярных веществ от истинно рас-
творенных в них низкомолекулярных веществ при по-
мощи полупроницаемой мембраны. Д. является ме-
тодом очистки коллоидных р-ров от примесей электро-
литов, всегда содержащихся в них. Для проведения
Д. между коллоидным р-ром и чистым растворителем,
чаще всего водой, помещают пористую перегородку —
мембрану, поры к-рой проницаемы для ионов и моле-
кул низкомолекулярных веществ, но не пропускают
коллоидные частицы и макромолекулы. Принцип мето-
да основан на законах диффузии. Через пористую мем-
брану диффундируют в растворитель ионы и моле-
кулы из коллоидного р-ра и заменяются молекулами
и ионами растворителя, также проникающими через
мембрану. Распределение (концентрация) ионов элек-
тролитов по обе стороны мембраны определяется мем-
бранным равновесием,к-рое впервые было описано и
теоретически объяснено Ф. Доннаном. Метод Д. впер-
вые был применен Т. Грэмом в середине 19 в.
Приборы, в к-рых производится Д., наз. диали-
заторами. Наиболее простой диализатор состоит
из полого стеклянного цилиндра, дно к-рого затяги-
вается полупроницаемой мембраной. В него наливают
подвергаемый очистке коллоидный р-р и опускают в бо-
лее широкий сосуд с дистиллированной водой, к-рую
периодически или непрерывно меняют. В таком эле-
ментарном виде прибор не удобен, т. к. диффузия про-
текает чрезвычайно медленно (для очистки требуются
недели, а иногда и месяцы). Ускорить диффузию можно
несколькими способами: увеличением поверхности
мембраны (напр., придав ей форму мешка);увеличением
градиента концентрации, что достигается постоянной
сменой воды в наружном сосуде и постоянным переме-
шиванием внутри диализатора; повышением темп-ры
р-ра; применением больших давлений (см. Ультра-
фильтрация). Ускорение процессов диффузии ионов
при Д. достигается также наложением электрич. поля
(электродиализ).
Электродиализ проводится в электродиали-
заторе, к-рый обычно (рис. 1) состоит из трех
камер, разделенных мембранами. В средней камере
Рис. 1. Электродиализа-
тор.
Рис. 2. Пятикамерный электро-
диализатор.
находится суспензия или коллоидный р-р вещества,
подвергающегося очистке, в крайних камерах — вода
и электроды. При наложении разности потенциалов
ионы электролитов переносятся в боковые камеры.
При малом градиенте потенциала электролиты, нако-
пившиеся в боковых камерах, могут диффундировать
обратно в среднюю камеру. Во избежание процесса
18*
обратной диффузии, а также для преодоления труд-
ностей, связанных с зарядами мембран (см. Мембран-
ное равновесие), применяют высокие градиенты потен-
циала (порядка 200—400 в/см). Для количеств, опре-
деления извлеченных при электродиализе веществ
можно применить два способа: 1) Раствор из боковых
камер пропускается через колонки с ионнообменными
смолами, где практически полностью поглощаются
выделившиеся вещества, т. к. они находятся в боковых
камерах в виде кислот и оснований. Затем поглощен-
ные вещества извлекаются из смол и исследуются.
2) К электродиализатору можно подключить еще две
небольшие камеры (рис. 2) с электродами, соединен-
ными с боковыми камерами через достаточно узкий ка-
нал и мембрану. Между электродами боковых камер
и присоединенными камерами накладывается допол-
нительная разность потенциалов, и извлеченные в бо-
ковых камерах электролиты переходят в дополнитель-
ные камеры и там концентрируются. Такой пятика-
мерный электродиализатор можно также применить
дляочисткинерастворимыхосадков при помощи химич.
реакций, осуществляемых непосредственно в электро-
диализаторе, напр., чтобы очистить кремневую к-ту
от примесей железа, ее помещают в среднюю камеру,
а в боковую катодную камеру постепенно добавляют
разб. соляную к-ту. Ионы хлора, попадая в централь-
ную среднюю камеру, образуют растворимое хлорное
железо, ионы к-рого под действием электрич. поля пе-
реносятся в боковые камеры. Кроме того, пятикамерный
электродиализатор может быть использован для син-
теза неорганич. веществ. Так, при синтезе алюмоси-
ликатов в боковую катодную камеру помещается р-р
соли кремневой к-ты, а в анодную — р-р соли алюми-
ния. При наложении электрич. поля ионы алюминия
и кремневой к-ты будут передвигаться навстречу друг
другу и взаимодействовать между собой в средней
камере. При помощи электродиализа можно разделить
смесь растворимых солей. Для этой цели составляют
электродиализатор из большого числа камер, отде-
ленных мембранами. В каждой камере устанавливается
определенное pH, возрастающее по мере приближения
к катодной камере. Затем в среднюю камеру вводят
анализируемую смесь солей. В каждой камере будут
выделяться те основания, произведение раствори-
мости к-рых соответствует pH этой камеры.
В качестве мембран для Д. применяются перепонки
из животного пузыря, пергаментной бумаги, в настоя-
щее время применяются также пленки, приготовленные
из р-ров нитро- и ацетилцеллюлозы (коллодий, цел-
лофан). Варьируя время сушки и подбирая раствори-
тель, эти мембраны можно изготовить с любой степенью
проницаемости, т. е. с любым диаметром пор. Приме-
няются также металлич. пористые перегородки. Д.
осложняется прилипанием коллоидных частиц к стен-
кам пор, а также коагуляцией коллоидных частиц при
соприкосновении с поверхностью мембраны. Поэтому
при выборе мембраны следует учитывать индивиду-
альные свойства коллоидного р-ра.
Д. и электродиализ применяются в пром-сти для
очистки различных веществ, напр. при произ-ве ис-
кусственных волокон (отделение отжимной щелочи
от гемицеллюлозы), для очистки желатины, а также
при изготовлении фармацевтич. препаратов.
Лит.: Жуков И. И., Усп. хим., 1943 12, вып. 4, с. 265;
Байбаев А. И. и Каргин В. А., Ж. физ. хим., 1936,
7, вып. 1; Д м и т р е н к о О. И. и К а р г и н В. А., Коллоидн.
ж., 1951, 13, вып. 4, с. 259; Каргин В. А. и Матвеева
Т. А., ДАН СССР, 1955, 105, № 2, с. 294.
3. Я. Берестнева.
ДИАЛЛИЛФТАЛАТ (диаллиловый эфир о-фта-
левой кислоты) СвН4(СООСН2—СН=СН2)2, мол. в.
244,3 — жидкость; т. пл.—70°; т. кип. 175°/10 мм;
d-,a 1,120; ri'fi 1,490—1,493; давление паров 0,01 мм
(20°), 2 мм (150°); г. воспл. 166°; практически нерас-
1095
ДИАМАГНЕТИЗМ — ДИАРЙЛМЕТАНОВЫЕ КРАСИТЕЛИ
1096
творим в воде при 20°, растворимость воды в Д. 0,2%;
растворим в спирте, нерастворим в бензине, минераль-
ных маслах, глицерине и гликолях. В тонком слое Д.
полимеризуется на воздухе при 105° в течение 48 час.
без катализатора; перекись бензоила ускоряет поли-
меризацию. Получают Д. этерефйкацией фталевого
ангидрида аллиловым спиртом в присутствии катали-
заторов: бензол- или толуолсульфокйслоты. Д. на
холоду растворяет нитроцеллюлозу, пластифицирует
алкидные смолы, хлорированный каучук, поливи-
нилхлорид, поливинилацетат, поливинилбутираль.
Пары Д. вызывают слезотечение.
Лит.: ButtreyD. N., Plasticisers, [2 ed.], L. — N. Y.,
1950; Th ini us K., Chemie, Physik und Technologle der
Weichmacher. Ein Handbuch tUr die Lack- und Plastindustrie,
B., 1960. О. Я. Федотова.
ДИАМАГНЕТИЗМ — см. Магнитные свойства.
п, п'-ДИАМИНОДИФЕНИЛДИМЕТИЛМЕТАН, мол.
вес 226,33 — кристаллы (иглы); т. пл. 132°; легко
растворим в органич. растворителях и кипящей
воде; хорошо растворим в минеральных к-тах; при
диазотировании образуется бисдиазосоединение, к-рое
при сочетании с аМинами, фенолами и др. дает дис-
азокрасители. Получают Д. (I) нагреванием под давле-
нием хлоргидрата анилина с ацетоном при 120—150°:
CeH5NH3 + (СН8)2СО 4- CeH5NH2 • НС1
— H2NCeH4—С(СН8)2—CeH4NH3
На основе Д. синтезированы ценные азокрасители
(пигменты), а также кислотные антрахиноновые кра-
сители. В. А. Пучков.
п.п'-ДИАМИНОДИФЕНИЛМЕТАН n-H2NCeH4—
—СН2—GeH4NH2-ra', мол. в. 198,27 — бесцветные
иглы (из воды) или призмы (из бензола); т. пл: 92,5°;
т. кип. 398—399°/768 мм, 257°/18мм; трудно растворим
в холодной воде, легко — в спирте и бензоле. Д. по-
лучают нагреванием водного р-ра хлоргидрата ани-
лина с водным р-ром формальдегида. Бисдиаз'отИро-
ванием Д. и сочетанием с различными азосоставляю-
щими могут быть получены дисазокрасители", кроме
того, Д. может быть использован для получения
триарилметановых красителей и антрахиноновых
красителей, напр. фуксина. н. С. Вульфсон.
п,п -ДИАМИНОСТИЛЬБЕН-о, </-ДИСУЛЬФОКИС-
ЛОТА C14H14OeN2S2, мол. в. 370,4 — желтые иглы,
почти нерастворимые в воде, под действием све-
NH,
та окрашивающиеся в коричневый цвет. В пром-сти
Д. получают восстановлением гг.тг'-динитросч'ильбен:-
о,о'-дисуйьфокислоты чугунными стружками в среде
электролита; напр., разб. р-ра соляной, уксусной,
муравьиной или серной к-т. Д. широко применяется
в качестве диазосоставляющей в произ-ве желтых и
желто-оранжевых бисазокрасителей. При сочетании
бисдиазотированной Д. с фенолом получается краси-
тель «ярко-желтый» (I), применяемый в качестве
индикатора в ацидиметрии (интервал перехода
pH 6,8—8,5) от желтого к красному. Этилированием
«ярко-желтого» хлористым этилом под давлением
получается краситель хризофенин (II), дающий проч-
ные к свету выкраски на хлопке и шерсти.
R-N=NCeH4OH-n I
R—N== NCeH4OC2H5-n II
R—N=NHCONHC0H5 III
. Д. широко применяется в произ-ве оптически от-
, беливающих веществ (бланкофоров, люминофоров),
. напр. бланкофор Р (III) получается взаимодействием
. Д. С 2 МОЛЯМИ фенилизоцианата. Я. с. Вульфсон.
; ДИАН — см. Дифенилолпропан.
! ДИАНИЗИДИН (3,3'-диметоксибензидин), мол. в.
244,30, C14HieO2N2 — бесцветные иглы (т. пл. 133°)
; или листочки (т. пл. 137°);
нерастворим в воде, легко Н3СО ОСН3
растворим в спирте, эфире, \—, .—I
ацетоне, бензоле и бензине; h,N—/ \
; образует соли, хорошо рас- " \ /1\ / 2
творимые в воде, за исклю-
чением сернокислой, которая мало растворима. Д.
получают изомеризацией 2,2'-диметоксигидразобензо-
ла под действием минеральных кислот (бензидиновая
перегруппировка). Д. легко диазотируется и широко
применяется в произ-ве бисазокрасителей, используе-
мых для непосредственного крашения хлопка. Д. при-
меняют для открытия и определения Cu2+, Vlv,
CNS-, Au3+, NOy, а также как окислительно-восста-
новительный индикатор. Д. является канцерогенным
‘ веществом, поэтому работа с ним требует особых мер
: предосторожности. в. г. Типцова.
ДИАНТРИМИД (1,Г-диантрахинониламин), моле-
кулярный вес 429,43, C2SH1SO4N — буро-красные мел-
кие кристаллы; раство-
ряется в хлороформе,
хлорбензоле, нитро-
бензоле, в конц. H2SO4
(с образованием жел-
то-зеленого раствора);
почти нерастворим в
и бензоле. Получают Д. конденсацией 1-хлорантра-
хинона н 1-аминоантрахинона в присутствии солей
меди; Д. применяют как полупродукт для синтеза
' нек-рых кубовых красителей (антрахинонкарбазо-
лов); как высокочувствительный реактив для от-
крытия и определения бора. При фотометрическом
: определении бора используется раствор реактива
; в конц. H2SO4; в зависимости от концентрации бора
(0,1—10 мкг) окраска раствора изменяется от зеленой
до голубой. Можно определять 0,1 мкг бора.
Лит.: Roth Н. und Beck W., Fresenius’ Z. t. ana-
! lyt. Chem., 1954, 141, H. 6, 404. 11. П. Ефимов.
воде, спирте, ацетоне, эфире
ДИАРИЛМЕТАНОВЫЕ КРАСИТЕЛИ — произ-
• водные дифенилметана СвН5—СН2—СвН5. Технич.
значение имеют два основных желтых красителя этой
; группы, особенно аурамин (аурамины О и Г). Первый
! получают двумя методами. По одному из них, диметил-
анилин конденсируют с формальдегидом и получен-
' ный 4,4',-бисгДиметиламинодифенилметан нагревают
при 200° с серой и хлористым аммонием в смеси с по-
варенной солью в токе сухого аммиака:
2 CeHsN(CH3)3 + СН2О —►
— (CH3)2NCeH4-CHa-CeH4N(CH3)2 “
— (CH3)2NC6H4-C-C6H4N(CH3)2 —-
S
—*(CH3)2NCeH4-C-Celi4N(CH3)2
NH
-^(CH3;2NC6H4-C==^2^>=N(CH3)2 Cl'
nh2
Вторым методом—из диметиланилина и фосгена полу-
чают тетраметилдиаминобензофенон (кетон Михлера),
к-рый сплавляют с хлористым аммонием в присут-
ствии безводного хлористого цинка при 160—170°.
1097
ДИАСТЕРЕОИЗОМЕРЫ — ДИАФОРАЗА
1098
Аурамин обладает ярким желтым цветом и высокой
красящей способностью, но малой прочностью к свету.
Его применяют гл. обр. в крашении бумаги и дерева.
Аурамин в кислых водных р-рах при темп-ре выше 70°
гидролизуется с образованием тетраметилдиаминобен-
зофенона, поэтому крашение проводят при темп-ре
пе выше 70°.
Желтый краситель с зеленоватым оттенком, изо-
мерный аурамину
H3CNHCeH3(CH3)-C=< y=NHCH3 Ci"
получают из N-метил-о-толуидина; он известен как
аурамин Г и находит более ограниченное применение.
Нек-рые Д. к. используются как индикаторы в объем-
ном анализе. В СССР в ряде отраслей пром-сти вместо
аурамина применяются красители акридинового ря-
да — основные желтые К и Н (см. Акридиновые кра-
сители).
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Веннатар аман К., Химия синтетических кра-
сителей, пер. с англ., т. 2, Л., 1957. И. Г. Лаптев.
ДИАСТЕРЕОИЗОМЕРЫ — изомеры, отличающие-
ся друг от друга конфигурацией двух (или не-
скольких) элементов асимметрии и не относящиеся
друг к другу, как предмет к его зеркальному изображе-
нию. Д. — молекулы, ионы, а также такие соли,
в к-рых различные элементы асимметрии присутствуют
как в катионе, так и в анионе. Примером может слу-
жить 1,2,3-триоксибутан-4-аль. В этом соединении
атомы 2 и 3 — асимметрические, и каждый из них мо-
жет иметь либо D-, либо L-конфигурацию. Число
возможных изомеров, определяемых числом сочета-
ний, V=2n (где п — число асимметрии, атомов), равно
четырем. Изомеры DC2- DC3(I) и DC2- LC3 (II) являют-
ся диастереоизомерами D-ряда (по атому С2 вы-
бранному в качестве ключевого атома). Изомеры LC2-
• LC3 (III) и LC2-DC3 (IV) — диастереоизомерами L-ря-
да. Изомер III является зеркальным изомером (анти-
но
СН2ОН
СНО
I
но-с-н
I
н-с-он
I
СН2ОН
D-треоза (I)
СН2ОН
СНО
H-i-OH
I
н-с-он
I
сн,он
D-эритроза (III)
СНО
I
н-с-он
I
но-с—н
I
СН2ОН
Lmpeooa (II)
СНО
I
но-с—н
I
но-с-н
I
сн2он
I- эритроза (IV)
подом) I, а изомер IV — антиподом II. Д. I и II имеют
разное пространственное строение и, в частности,
разные расстояния между атомами кислорода гидрок-
сильных групп. Поэтому химич. и физич. свойства
диастереоизомеров I и II резко различны. Соединение
(I) называют D-треоза, соединение (II) — D-эритроза,
а антиподы III и IV — L-треоза и L-эритроза. Таким
образом, у соединения, имеющего два асимметрия,
атома углерода, может быть два D-, два L- и два DL-
диастереоизомера. Если в Д. асимметрия, атомы рас-
положены так же, как в треозе или эритрозе, т. е.
рядом друг с другом, то для их характеристики исполь-
зуют приставки «треоъ- и вэритров-. При этом «трео»
обозначает «треозную» (трансоидную) D,D-конфигу-
рацию, а «эритро» — «эритрозную» В,Ь-конфигура-
цию. Напр., для винной (а,а-диоксиянтарной) к-ты
известны как /npeo-Д. (D-, L- и DL-винные кислоты),
так и эритро-Д. — мезовинная к-та:
сно соон соон
I I I
НО-С-Н НО-С-Н Н-С-ОН
I окисл. | |
Н-С-ОН ’ Н-С-ОН НО-С-Н
I I I
СН3ОН СООН соон
D-треоза D-виннан к-та L-винная н-та
СНО
I
Н-С-ОН
I
н-с-он
I
снаон
онисл.
СООН
I
ОН плоскость
....... * симметрии
Н-С-ОН
I
СООН
D-эритроза мезовиннан к-та
Несмотря на присутствие двух асимметрия, центров
эритро-Д., винная к-та (мезовинная кислота) обладает
плоскостью симметрии (внутренняя компенсация)
и потому существует только в одной (оптически недея-
тельной) форме. Приставка «мезо»- означает внутрен-
нюю компенсацию (плоскость симметрии или центр
симметрии). При действии оптически активного осно-
вания на асимметрически построенную DL-кислоту
(или наоборот) образуются диастереоизомерные соли;
DB + DLHA — DB- DHA 4- DB • LHA
т - *- . '
При взаимодействии рацемич. к-т и оснований, кроме
двух Д., образуются также их антиподы;
DLB + DLHA — DB • DHA + DB • LHA +
+ LB - DHA + LB LHA
В образовании Д. могут участвовать молекулы и ионы,
асимметрия, строение к-рых обусловлено как асим-
метрией молекулы (иона) в целом, так и наличием
асимметрия, атома. См. Антиподы оптические, Асим-
метрическая молекула.
Jium. см. при ст. Антиподы оптические. С. В. Витт.
ДИАФОРАЗА — фермент, катализирующий окис-
ление восстановленных форм пиридиннуклеотидов
в присутствии нек-рых акцепторов водорода (метиле-
новая синь, 2,6-дихлорфенолиндофенол). Существуют
две Д.: одна, катализирующая реакцию с восстанов-
ленным дифосфопиридиннуклеотидом (Ко-1-Д.), и дру-
гая, катализирующая реакцию с восстановленным
трифосфопиридиннуклеотидом (Ко-П-Д.). Р-ры Д. ок-
рашены в желтый цвет, флуоресцируют с зеленоватым
оттенком. При восстановлении под действием восста-
новленного дифосфопиридиннуклеотида или гидро-
сульфита р-ры Ко-1-Д. обесцвечиваются. Простетич,
группой Д. является алоксазинадениндинуклеотид.
Высокоочищенная Ко-1-Д. гомогенна при исследо-
вании в ультрацентрифуге и при электрофорезе;
ее мол. в. ок. 80 000 (минимальный мол. вес, рассчи-
танный по содержанию флавина, 67 000, что указывает
на наличие в каждой молекуле Д. одной простетич.
группы). Д. содержится в тканях животных, растений
и в микроорганизмах; она связана с нерастворимыми
элементами структуры клетки и при экстракции в обыч-
ных условиях не переходит в р-р, но при нагревании
суспензии до 43° и обработке ее разб. р-ром этилового
спирта и сернокислого аммония удалось получить
растворимую Д.
Лит.: Михлин Д. М., Биологическое окисление, М.,
1956; Диксон М., Уэбб Е Ферменты, пер. с англ.,
М.-, 1961; Savage N., Biochem. J., 1957, 67, W 1,
p. 146. Л. M. Гинодман.
1099
ДИАЦЕТИЛ — ДИБЕНЗИЛ
1100
ДИАЦЕТИЛ (2,3-бутандион, диметилглиоксаль)
СН3СОСОСН3, мол. в. 86,09 — желто-зеленая жид-
кость с сильным запахом хинона; т. пл. —2,4°;
т. кип. 87,5—88°; (Ц8,5 0,9808; п‘$’* 1,39331; растворим
в спирте, эфире; весьма летуч при нагревании. В 100 г
воды растворяется 25 г (15°). В природе Д. встре-
чается в лавровом дереве и различных маслах, из
к-рых он может быть выделен перегонкой с паром. Д.
содержится также в животном масле, сыре и молоке
в количестве 0,7—1,5% и обусловливает аромат и
приятный вкус масла. Д. вступает в типичные реак-
ции а-дикетонов. Так, с ароматич. диаминами обра-
зуются хиноксалины (1)
с альдегидами и аммиаком — имидазолы (2)
СН3-СО
I -t-2NH3 + RCHO--
сн3-со
(2)
И т. д.
Д. получают пропусканием винилацетилена в р-р
сульфата ртути в серной к-те и разложением нерас-
творимого продукта реакции соляной к-той при 95—
100°; нитрозированием метилэтилкетона до оксима
с последующим разложением NaHSO4 или слабыми
неорганич. к-тами; окислением 2,3-бутандиола кисло-
родом при 270° в присутствии медного катализатора.
Небольшие количества Д. легко могут быть опре-
делены в виде диметилглиоксима по характерной крас-
ной окраске, к-рую он дает с солями никеля, а также
по образованию кристаллич. аддукта с двумя молеку-
лами фосфорной к-ты. Д. используется в произ-ве
пищевых продуктов (маргарина, крема, масла, кофе
и др.); предложено применять Д. для повышения
твердости желатины при приготовлении фотография,
эмульсий и клеев, содержащих желатину.
Ю. Л. Чебурков.
ДИАЦЕТИЛЕН (бутадиин) НС^С—CesCH, мол. в.
50,06 — легко сжижающийся газ, т. пл. —36°; т. кип.
9,5°; di 0,736; n-fl 1,4386; умеренно растворим в воде,
хорошо — в органич. растворителях, в 1 объеме
диметилформамида растворяется 2 500 объемов Д.;
весьма взрывоопасен, особенно под давлением. По
химич. свойствам Д. очень сходен с ацетиленом.
Замещение метиновых водородов происходит либо
одновременно (примером является комплекс Иоцича
HalMgCsC—CsCMgHal и др.), либо замещается
только один атом водорода, напр. СН=С—C=CNa.
С формальдегидом и диэтиламином в отсутствии ката-
лизатора легко протекает Манника реакция:
НС1.С-СНСН -I- СН2О + (CH3)2NH • НС1 —
— HC=C-CsCCH2N(CH3)2 • HCI + Н2О
Присоединение к тройным связям протекает ступен-
чато; напр., со спиртами образуется соответственно
НС=С—CH=CHOR и СН3С=С—CH(OR)2; с ацето-
ном — Т. н. «дииндиол»
(СН3)2С—С^С —С=С —С(СН3)2
он он
применяющийся во многих органич. синтезах. При
гидрировании Д. образуется бутадиен', при присое-
динении хлора — гексахлорбутен-2. Присоединение
воды приводит к диацетилу СН3СО—СОСН3; ди-
алкиламины или меркаптаны присоединяются с
образованием замещенных винилацетилена.
Д. является побочным продуктом при синтезе аце-
тилена электрокрекингом метана. Его можно получить
также из бутиндиола-i ,4 отщеплением НС1 от
1,4-дихлорбутина при обработке щелочью:
НОСН2-С=С-СН2ОН —^*3.
—> С1СНа-С;;С-СН2С1 С1Г2С-ССН
1— HC1J
Другие способы получения Д. заключаются в окисле-
нии HC=CNa кислородом воздуха в присутствии ка-
тализатора Fe2O3 или же КМпО4 в жидком аммиаке.
ДИАЦЕТОНОВЫЙ СПИРТ (4-окси-4-метилпен-
танон-2, диметилацетонилкарбинол), мол. в. 116,16,
(СН3)2С(ОН)СН2СОСН3 — бесцветная жидкость;
т. кип. 163,5—164,5°/760 мм (с частичным разл.),
63—64°/11 мм; d25 0,9306; 1,4219; с водой, спир-
том и эфиром смешивается во всех отношениях;
выделяется из водных р-ров при добавлении щелочей;
Хмакс 231 ммк, ИК-спектр показывает отсутствие водо-
родной связи; слабо токсичен. Д. с. образуется при аль-
дольной конденсации ацетона в присутствии щелочных
агентов; обладает свойствами как спирта, так и кетона.
При действии кислотных веществ от Д. с. легко от-
щепляется вода, при этом получается окись мезитила;
спиртовая группа может быть этерифицирована; при
восстановлении образуется соответствующий гликоль,
а при окислении NaOBr—Р-оксинзовалериановая
к-та; щелочами и аминами Д. с. расщепляется с обра-
зованием ацетона; реагирует с NH4CNS и аммиаком
или аминами, причем получаются замещенные дигид-
ропирамидины. с. с. Юфит.
ДИБАЗОЛ (хлоргидрат 2-бензилбензимидазола)
C14H13N2C1, мол. в. 244,73 — бесцветный со слабо-
желтоватым или Серова-
а тым оттенком гигроскопи-
1 ^°ССН2СвН3-НО ческий порошок горько-
NHх соленого вкуса; т. пл.
182—186°; легко раство-
рим в горячей воде, трудно — в холодной и в аце-
тоне, нерастворим в эфире. Растворы Д. в воде
имеют кислую реакцию. Для колич. определения
навеску Д_, высушенного до постоянного веса, рас-
творяют в спирте и титруют 0,1 и. р-ром NaOH в при-
сутствии фенолфталеина. Д. получают нагреванием
о-фенилендиамина с фенилуксусной к-той или N.N'-ди-
фенацетил-о-фенилендиамина с конц. соляной к-той.
Д. применяют при спазмах сосудов (коронарная
недостаточность, гипертонии, кризы) и гладкой му-
скулатуры внутренних органов (язвенная болезнь
желудка, спазмы привратника и кишечника), а также
при лечении нервных заболеваний, гл. обр. остаточных
явлений полиомиелита, периферия, паралича лицевого
нерва и т. п.
Лит. см. при ст. Апрессин. А. И. Травин.
ДИБЕНЗИЛ (симм. дифенилэтан) СвН5СН2СН2СвН6,
мол. в. 182,25 — игольчатые кристаллы (из спирта);
т. пл. 52,0°; т. кип. 284,7°; d™ 0,9661; п% 1,5704;
нерастворим в воде, растворим в спирте, эфире, серо-
углероде и др. органич. растворителях. При ката-
литич. гидрировании Д. образуется дицнклогексялэтан
СвНпСН2СН2СвНп; дегидрирование окисью свинца
приводит к образованию стильбена С3Н6СН=СНСвН5;
под действием серы при 200° превращается в антра-
цен; разлагается пиролитически при 500° на толуол
и стильбен. КМпО4 и СгО3 в уксусной к-те окисляет
Д. в бензойную к-ту; с HNO3 (d 1,075) при 100° обра-
зует 1,2-дифенил-1-нитроэтан CeH5CH2CH(NO2)CeH5,
конц. HNO3 (d 1,42) при 70—80° превращается в
п, n'-динитродифенилэтан NO2CeH4CH2CH2CeH4NO2; в
присутствии иода хлорируется, давая п, п'-дихлор-
дифенилэтан С1С3Н4СН2СН2СвН4С1. С бромом при 150°
образует 1,2-дибром-1,2-дифенилэтан, при более низ-
кой температуре—н,п'-дибромдифенилэтан. Получают
1101
ДИБЕНЗОЛХРОМ— ДИБУТИЛАДИПИНАТ
1102
Д. конденсацией 1,2-дигалоидэтанов с бензолом в при-
сутствии А1.С13; действием Na на хлористый бензил;
реакцией бензилмагнийбромида CeH5MgBr с FeCl3
И CuCl2. М. Ф. Турчииский.
ДИБЕНЗОЛХРОМ — простейший представитель
соединений, в к-рых переходный металл связан с дву-
мя ароматич. ядрами; коричнево-черный порошок;
т. пл. 284—285°; умеренно растворим в органич. рас-
творителях, нерастворим в воде; диамагнитен (катион
парамагнитен), дипольный момент равен нулю. Д.
легко окисляется до катиона. Катион Д. может быть
получен т. наз. восстановительным методом Фриде-
ля — Крафтса: ЗСгС13 + 2А1 + А1С13 + 6СвНв —>
-> 3[Сг(СвН6)2]+А1С14~. При восстановлении катиона
с помощью гидросульфита натрия (или другого вос-
становителя) образуется нейтральный Д.:
2[Сг(С0Н6)2]+ + SaO®- + 4ОН- 2СГ(С6НО)3 + 2SO»~ + 211,0
Бензольные кольца в молекуле Д. не сохраняют
своей реакционной способности; с ними не идут ни-
какие реакции замещения. Со-
гласно рентгенографическим
исследованиям, дибензолхром
имеет «сэндвичевую» структу-
ру с параллельными бензоль-
ными кольцами и атомом хрома
между ними в центре симмет-
рии (см. рис.). Расстояния
С—С равны 1,38±0,05 А, рас-
стояния С—Сг равны 2,19 ±0,1
А. Следует отметить, что Ф.
Хайн еще в 1919—20, действуя
бромистым фенилмагнием на
хлористый хромил и соли хро-
ма, получил ряд хроморганич. соединений, имею-
щих структуру Д. Однако эти соединения долгое
время считались не «сэндвичевыми», а обычными
хроморганич. соединениями три-, тетра- и пентафе-
нилхромом. Получен в 1955 Е. О. Фишером.
Лит.: Fischer Е. О., Hainer W., Z. Naturtorsch.,
1955,10 В, Н. 12, S. 665; F is с Не г Е. О., S е us D.,
Chem. Вег., 1956, 89, № 8, S. 1809; Hein F г., там же, S.
1816. О. А. Реутов.
ДИБЕНЗОФУРАН (окись дифенилена) С12Н8О,
мол. в. 168,185 —бесцветные пластинки; т. пл.
86—87°; т. кип. 287°; растворим в
хНЧ. спирте, эфире, бензоле; умеренно
Р '[ А 9 растворим в горячей воде: образует
пикрат, т. пл. 94°; молекулярное
и ' соединение с 1,3,5-тринитробензо-
лом, т. пл. 86°. Электрофильное замещение (суль-
фирование, галогенирование, ацилирование, но не
нитрование и метилирование) идет в положение
2; при нитровании Д. образуется тринитродибензо-
фуран; метилирование приводит к четырехзамещен-
ным Д. При сплавлении Д. (I) с NaOH образуется
2..2'-дпоксидифенил (II):
Восстановление Д. металлич. Na в спирте или цикло-
гексаноле, а также гидрирование над платиновым
катализатором приводит к 1,2,3,4-тетрагидро-Д.,
к-рый можно дегидрировать в Д. действием КМпО4,
Se или Вг2. Наиболее употребительным способом
получения Д. является пиролиз фенола над РЬО2:
Д. может быть также получен: пиролизом дифенило-
вого эфира или о-фенилфенола над бокситом или же
замыканием цикла 2,2'-дизамещенных дифенилов.
Д. выделен из антраценового масла кам.-уг. смолы.
Система Д. входит в молекулу морфина; сам Д. обла-
дает слабым анестезирующим действием.
В. М. Демьянович.
ДИБРОМБЕНЗОЛ СвН4Вг2, мол. в. 235,92 — трп
изомера: 1,2- и 1,4-дибромбензолы — кристаллич. про-
дукты, 1,3-Д. — жидкость; все изомеры нераствори-
мы в воде, растворимы в органич. растворителях.
Положение атомов Вг Т. пл., °C Т. кип., °C d?’5 4 п])’5
1.2 (о) 1.8 224 1.9557 1.6117
1.3 (м) —7 219,5 1.9523 1,6083
1,4 (п) 86,9 218—219 • 2,261 ♦♦ —
* Возгоняется. При 17°.
1,4-Д. с примесью 1,2-изомера получают броми-
рованием бензола при нагревании в присутствии же-
лезных стружек С6Н6 -f- 2Вг2 С6Н4Вг2 -ф- 2НВг.
1,2-Д. применяют в качестве трансформаторного
масла, как растворитель жиров, восков или резин;
1,4-дибромбензол используют как промежуточный
продукт В синтезах. М. Я. Алейникова.
ДИБРОМОКСИН (бромооксин, 5,7-дибром-8-окси-
хинолин) C9H5ONBr2, мол. в. 302,98 —блестящие,
почти бесцветные или светло-жел- дг
тые кристаллы; т. пл. 196°; субли- iг
мируется; почти нерастворим в горя-
чей воде и разб. минеральных к-тах, gr_I | I
эфире, бензоле, сероуглероде; легко
растворим в спирте, конц. минераль-
ных к-тах. Получают бромированием
8-оксихинолина. При pH 1,3—1,9 образует осадки: с
Сп2+—зеленовато-желтый, с Fe3+—зеленовато-черный,
с Ti4+—коричнево-желтый, с V3+— коричневый. При-
меняют для весового определения Al, Си, Fe, Pb, Ti,
Ga, Zr, Co. В. Г. Tunuoea.
1,2-ДПБРОМЭТАН (бромистый этилен),мол. в.187,89,
СН2Вг —СН2Вг — бесцветная жидкость, т. пл. 9,97°;
т. кип. 131,7°; 2,1806; л201,5380; давление пара
8,5 мм при 20°; 295,3 мм при 100°; теплоем-
кость жидкого 0,173 кал/г (21,3°), теплота испарения
46,24 кал/г (130,8°); «Крвт 309,8°; ркрит. 70,6 ат.
Растворим в четыреххлористом углероде, бензоле,
эфире, спирте; в 100 г воды при 20° растворяется
0,404 е. Д. негорюч и достаточно устойчив при обыч-
ных темп-рах, однако под действием света происходит
слабое разложение. При нагревании до 340—370°
разлагается на бромистый винил и бромистый водо-
род. Д. при гидролизе образует этиленгликоль, при
действии цинка — этилен и бромистый цинк, с аммиа-
ком — этилендиамин и его высшие гомологи. Д. ядо-
вит и вызывает поражения кожи.
Получают Д. бромированием этилена. Д. приме-
няют в качестве добавки к моторному топливу для
удаления из цилиндров двигателя свинца, к-рый
образуется при разложении тетраэтилсвинца. Он
используется также для синтеза фарма цевтич. препа-
ратов, напр. пиперазина. , Ю. А. Чебурков.
ДИБУТИЛАДИПИНАТ (ди-и-бутиловый эфир ади-
пиновой кислоты) С4Н2ООС—(СН2)4—СООС4Н9,
мол. в. 258,4 — прозрачная почти бесцветная жид-
кость со слабым запахом; т. пл. —38°, т. кип. 168—
170°/17 мм; dig 0,9605; п8^ 1,4350; т]20 5,58 сот, т]80
1,79 сот; т. воспл. 115°; в воде растворяется 0,025%
(20°); растворим во всех обычных растворителях.
Получают Д. этерификацией адипиновой к-ты бути-
1103
ДИБУТИЛОВЫЕ ЭФИРЫ — ДИВИНИЛБЕНЗОЛ
1104
новым спиртом в присутствии кислотных катализа-
торов, напр. H2SO4, бензол- или толуолсульфокислоты.
Д. хорошо растворяет природные смолы, за исключе-
нием шеллака; хорошо пластифицирует поливинил-
хлорид и его сополимеры, сообщая пленкам на их
основе гибкость при низких темп-рах, пластифицирует
также этил- и нитроцеллюлозу, полистирол, синте-
тйч. каучук. Значительная летучесть Д. препятствует
его широкому применению.
Лит. см. при ст. Диаллилфталат. О. Я. Федотова.
ДИБУТИЛОВЫЕ ЭФИРЫ, мол. в. 130,22 —бес-
цветные жидкости. н-Д ибутиловый эфир
СН3(СН2)3—О—(СН2)3СН3, т. пл. —95,37°; т. кип.
141,97°; с/;° 0,76889; ng 1,39925; в воде при 17° рас-
творяется менее 0,01%; образует азеотропные смеси:
с водой (67%Н2О), т. кип. 92,9, с бутиловым спиртом
(ок. 88% Д. э.), т. кип. 117,25°; с бутиловым спиртом
и водой, т. кип. 91°; с изобутиловым спиртом и водой,
т. кип. 89°; т. всп. 38°, вязкость: 0,741 спуаа (15°),
0,602 спуаа (30°); поверхностное натяжение (дин/см):
23,40 (15°), 21,99 (30°). Получают н-Д. э. нагреванием
бутилового спирта с конц. серной к-той (препаратив-
ный метод); кипячением с дымящей серной к-той, со-
держащей 20% SO3; пропусканием паров бутилового
спирта над обезвоженвыми квасцами при 190° и др.
способами. Ограниченно применяют как раствори-
тель; используют также в синтезе Гриньяра. Преиму-
ществом н-Д. э. по сравнению с диэтиловым эфиром
является меньшая летучесть. При длительном хране-
нии н-Д. э. образуются перекиси, в связи с чем перед
употреблением берут пробу на содержание перекисей;
последние устраняются встряхиванием с р-рами
солей двухвалентного железа или с р-ром сульфита
натрия.
Диизобутиловый эфир (СН3)2СНСН2—
—О—СН2СН(СН3)2, т. кип. 122—122,5°; d\* 0,7616;
образует азеотропную смесь с водой (23% Н2О),
т. кип. 88,6°. Получают диизобутиловый эфир нагре-
ванием изобутилового спирта с конц. серной к-той
(препаративный метод), а также нагреванием изобу-
тилового спирта под давлением в присутствии окиси
алюминия или окиси тория при темп-ре около 300°;
применяют в качестве растворителя питроцеллюлоз-
ных лаков.
Ди-втор, - бутиловый е ф и р (6,1)
CH,CH,CH(CHS) — О — СН(СН.,)СН2СН3; т. кип. 120—122°;
dj1 0,756; получают нагреванием d,1-emop.-бутилового спирта
с конц. H2SO4 до 103—104°, а также действием диэтилсвинца
на ди-а-хлорэтиловый зфир (СН3—СНС1)2О; практического
значения не имеет. Известны также смешанные дибутиловые
ефиры, содержащие различные бутильные радикалы.
Л. С. Поваров.
ДИБУТИЛСЕБАЦИНАТ (ди-н-бутиловый эфир се-
бациновой кислоты) С4Н9ООС—(СН2)8—СООС4Н9,
мол. вес 314 — бесцветная жидкость без запаха;
т. пл. —8—11°; т. кип. 344—345°/760 мм, 180°/3 мм;
di0 0,936; n'g 1,4391; г]20 9,3 спуаа; г]25 7,9 спуаа; раство-
римость в воде 0,005% (25°); воды в Д. 1,6% (20°);
т. воспл. 178°. Получают Д. этерификацией себаци-
новой к-ты бутиловым спиртом в присутствии бензол-
нли толуолсульфокислоты. Применяется Д. ^качестве
пластификатора поливинилбутираля, поливинилхло-
рида и его сополимеров с другими винильными соеди-
нениями; пластикаты обладают гибкостью при низких
темп-рах. Используются также для пластификации
лаковых покрытий на основе полистирола, хлоркау-
чука, мочевино-формальдегидных смол. Д. пластифи-
цирует простые и сложные эфиры целлюлозы: этил-
и бензилцеллюлозу, ацетобутират- и ацетопропионат-
целлюлозы, полиакрилаты.
Лит.: Doolittle А. К., The technology of solvents and
plasticizers, N. Y.—L., [1954], p. 910, 977; Buttrey D. N.,
Plasticizers, 2 ed., L., 1957; T h 1 n 1 u s IE,, Chemie, Physik
und Technologie der Welchmacher. Bln Handhuch ftir die Lack-
und Plastlndustrle, B., I960, О. Я. Федотова.
ДИБУТИЛФТАЛАТ (ди-н-бутиловый эфир о-фта-
левой кислоты) СвН4(СООС4Н9)2> мол. в. 278,25 —
бесцветная жидкость с тонким фруктовым запахом;
т. пл. —35°; т. кип. 330—340° (с частичным разл.),
206°/10 мм (без разл.), 182°/5 мм; с/25 1,047—1,050;
ng 1,490—1,493; »]20 25 спуаа; г]25 20 спуаа; давление
паров 0,0001 мм (25°), 1,1 мм (150°); скорость испаре-
ния 2,21 • 10“* г/см2 час (100°); растворимость в воде
0,1% (20°); растворимость воды в Д. 0,3—0,5% (25°).
Получают Д. этерификацией фталевого ангидрида
в избытке бутилового спирта в присутствии кислотных
катализаторов, напр. H2SO4, бензол- или толуолсуль-
фокислоты. Д. — один из основных пластификаторов;
применяют для пластификации поливинилхлорида и
его сополимеров с винилацетатом и др. винильными
соединениями, нитроцеллюлозы, ацетобутирата и
ацетопропионата целлюлозы, полистирола, полиметил-
метакрилата (особенно в смесях для литья под давле-
нием), поливинилбутираля, поливинилацетата, сопо-
лимера винилиденхлорида с акрилонитрилом, син-
тетич. каучуков: неопрена, бутадиенстирольного, бу-
тадиеннитрильиого и др. Недостатком Д. является его
относительно высокая летучесть (0,22 мг/см2/час при
Лит. см. при ст. Дибутилсебацинат. О. Я. Федотова.
ДИВИНИЛАЦЕТИЛЕН (гексадиен-1,5-ин-З), мол. в.
78,11, Н2С=СН—С=С—СН=СН2—тример ацетиле-
на; бесцветная жидкость, желтеющая на свету; т. кип.
85,0°, т. пл. —87,8°; d|° 0,7759; ng 1,5047. Д. по-
глощает кислород из воздуха с образованием взрыв-
чатых перекисей. Чистый Д. выше 105° разлагается
со взрывом; при разбавлении выдерживает более высо-
кие темп-ры. При гидрировании Д. образуется н-гексан,
можно получить также и продукты неполного гидри-
рования, в частности гексен-3; присоединение брома
идет вплоть до 1,2,3,4,5,6-гексабромгексена-3, далее
происходит замещение. Гидратация Д. приводит
к тексадиен-1,5-ону-3. Образуется Д. при полимери-
зации ацетилена в присутствии смеси Си2С12 и NH4C1
в кислотном р-ре в виде примеси к винилацетилену,
а также при термич. полимеризации ацетилена и поли-
меризации в электрич. разрядах. Полимеры Д. могут
быть использованы как высыхающие масла, клеи,
пластификаторы и т. д.
Лит.: Н ь ю л э я д Ю., Фогт Р., Химия ацетилена,
пер. с англ., М., 1947. А. Е. Васильев.
ДИВИНИЛБЕНЗОЛ (диэтенилбензол), мол.в. 130,18,
СвН5(СН=СН2)2 — известны три изомера Д.
Изомер Т. пл., °C Т. кип., °C п£>
1,2 (о) 1,3 (.«) 1,4 (п) -52,25 31 78,5/11 мм 210,55/760 мм* ок. 18О/760лии** 0,934 (21°) 0,9294 (20°) 0,913 (40°) 1.5760 (21°) 1.5746 ( 22°) 1,5280 (40°)
• При этой темп-ре полимеризуется.
*• 85 — 86°/16 мм; 52°/3 -ад.
Д. вступает в большинство реакций, характерных
для олефинов; с азотистой к-той количественно об-
разует псевдонитрозит:
аСН=СН2
СН=СН2
+ 2N2O3
0j-CH(NO)~CH2N02
L-CH(NO)-CH2NO2
Реакция используется для колич. определения. Харак-
терной особенностью Д. является его склонность к по-
лимеризации; 1,4-Д. полностью полимеризуется в те-
чение нескольких дней при комнатной темп-ре; 1,2-Д.
полимеризуется в тех же условиях в течение несколь-
ких недель. Полимеры Д. — прозрачные стеклообраз-
ные материалы, нерастворимы в органич. раствори-
1105
ДИВИНИЛКЕТОНЫ
1106
телях. При нагревании или под действием света,
а также под влиянием инициатора полимеризация
ускоряется. Замедляется полимеризация дифенилами-
ном, дитретичным бутил- или амилгидрохиноном и др.;
гидрохинон и пирокатехин действуют на Д. гораздо
слабее. Бромирование в хлороформе приводит к обра-
зованию бесцветных кристаллических тетраброми-
дов; т. пл. тетрабромидов 1,2-Д. 71—74°; 1,3-Д. 64°;
1,4-Д. 57°.
Основным методом получения Д. (как смеси, так
и индивидуальных изомеров) является каталитич.
дегидрирование диэтилбензолов при 600—630° в при-
сутствии ZnO, СаО и др. с разбавлением водяным па-
ром. Вследствие близости темп-p кипения изомеров
и большой скорости полимеризации Д. выпускают в
производственных условиях в виде 25—45%-ного рас-
твора в этилстироле. При хлорировании диэтилбен-
зола СвН4(С2Н5)2 (80—120° в темноте в присутствии
РС15) образуется дихлордиэтилбензол СаН4(С2Н4С1)2;
последний обработкой щелочью переводят в диэтилол-
бензол СаН4(С2Н4ОН)2, к-рый при нагревании с гид-
рохиноном и KHSO4 образует Д. Применяют Д. в ка-
честве компонента, образующего поперечные связи
при сополимеризации со стиролом, акриловой и мет-
акриловой кислотами и др. винильными соединениями;
для получения полистирольных смол с пониженной
хрупкостью, повышенной твердостью и термостой-
костью; нерастворимых ионообменных смол с ограни-
ченной набухаемостью, а также смол, применяемых
в качестве диэлектриков.
Лит.: Баландин А. А. [и др.], Ж. прикл. хим.,
1959, 32, № И, с. 2566; J. Polymer Sci., 1952, 8, № 5, р. 481;
Marquardt R. Р., L u с е Е. N., Analyt. Chem., 1951,
23, № 4, р. 629; Де К а т А., в кн.: Химия и технология
полимеров. Сб. переводов, № 3, М., 1957; S a b е t а у S.,
Compt. rend. Acad, sci., 1931, 192, № 18, p. 1109.
A. К. Никишин.
ДИВИНИЛКЕТОНЫ — соединения общей фор-
Rk Ж5
мулы ^0=0-00-0=0^ где R — алкил, арил или
R3 R4
Н. Низшие представители Д. — желтые подвижные
жидкости с чрезвычайно резким запахом и лакрима-
торным действием, легко полимеризуются при стоя-
нии, причем с увеличением мол. веса радикалов ско-
рость полимеризации падает.
Формула Т. кип.. ° С/мм Т. пл., °C df° 20 пр
СН2 — СНСОСН = СН2 СН2 = СНСОСН = СНСН3 СН3СН = СНСОС (СН3) = сн2 (СН3)3 С = СНСОСН = С(СН.)2 СвН5СН = СНСОСН = снсвн5 49/100 30-31/7,5 47/11 197,2/743 28 ИЗ 03811 0,8959 0,8851 0,8850 1,4485 1,4690 1,4730 1,4998
Д. способны к разнообразным реакциям присоеди-
нения, к-рые, как правило, имеют резко выраженный
ступенчатый характер. Активность двойной связи
в реакциях присоединения сильно снижается с введе-
нием заместителей в винильную группу. Так, под влия-
нием HgSO4 вода присоединяется к Д, со свободной
винильной группой с образованием неустойчивых
непредельных 0-оксикетонов, к-рые под влиянием
кислот циклизуются в тетрагидро-у-пироны:
/ R нО
CH,=CHCQCH=C
4R’
ZR н*
---«~СН2ОНСН2СОСН = С -2-*-
4R'
Весьма легко, особенно в присутствии щелочных ка-
тализаторов, присоединяются к Д. спирты, причем
легче' всего реакция идет с первичными и труднее
всего с третичными спиртами:
CH2=CHCOCH^CR'R2—2Я ROCH2CH2COCH=CR'R8 —ОД.
R1 R2
roch2ch2coch2cor
Вторичные амины, цианистый водород и соединения
типа малонового и ацетоуксусного эфиров присо-
единяются к Д. только по незамещенной винильной
группе:
СН2—.CHCOCR1=CR2R3 —Д— R2NCH2CH2COCR1=CR2R3
1 CHS(COOC2H5)2
(C2H5OCO)2CHClbCH,COCRi^=CR2Ra
Хлористый водород и сероводород присоединяются
к Р-замещенным Д. в обратном порядке, т. е. в первую
очередь по замещенной винильной группе, образуя
неустойчивые Р-хлоркетоны и Р-меркаптокетоны:
ch3=chcoch2ccir»-S5L ch2=chcoch=cr3 —
CH2=CHGOCH2CR2(SH)
При действии аммиака и первичных аминов на Д.
получаются различные пиперидоны:
сня=ссосн=снсн3
сн3
CH,NH.
(СН3)2С=СНСОСН=СН2—::
При окислении Д. перекисью водорода в присутствии
щелочи образуются моно- или диокиси:
СН,=ССОСН=Сн СИ,И,°'. н,с—с-со-сн-снсн.
сн, Л„з V
Интересной реакцией является циклизация Д. в цик-
лопентеноны под влиянием фосфорной, муравьиной
и др. кислот:
О О
R, Ri и т.д.~Н, амил или арил
Д. с незамещенной винильной группой в эту реакцию
не вступают, но образуют упомянутые выше тетра-
гидро-у-пироны.
Д. с незамещенной винильной группой получаются:
1) Изомеризацией винилэтинилкарбинолов под влия-
нием HgS04 в среде метанола, причем вначале обра-
зуются (3-метоксикетоиы, к-рые при перегонке в ва-
кууме в присутствии л-толуолсульфокислоты дают Д.:
R1RsC(OH)Cz=CCH=CH3
н+ 3
— RiR2O=CHC0CH2CH20CH3 -i— RiR2C=CHCOCH=CH3.
2) Из замещенных винилацетилена Маннмха реакцией
получают ацетиленовые амины, к-рые гидратируют,
в (3-аминокетоны, легко отщепляющие вторичный амин,
образуя Д.:
R1CH=CR2C=CH R'CH=CR2C=:CCH.1XR3
HNK2 "
— R1CH=CR2COCH2CH2NR3 ~ —ОЧ
— RICH=CB/COCH=CH2.
1107
ДИГИДРОЖАСМОН — ДИЕНОВЫЙ СИНТЕЗ
1108
Д. с замещенными впнильнымн группами полу-
чаются: 1) Конденсацией кетонов с альдегидами или
кетонами под влиянием кислых или щелочных агентов:
СНаСОСН,
2(СН3)аСО |2С,Н,СНО
(СН3)2С=СНСОСН=С(СНа)2 С,Н5СН=СНСОСН=СНС,На
2) Гидратацией дивиннлацетиленовых углеводородов
в воднометанольных р-рах в присутствии HgSO4:
СН2=СНС^СС(СН3)=СН3 СН3СН=СНСОС(СН3)=СН3.
Лит.: Назаров И. Н., Усп. хим., 1945, 14, вып. 1, 3;
Назаров И. Н. и Торгов И. В,, Изв. АН СССР. Отд.
хим. и., 1947, № 5, 495; Назаров И. Н., Избранные тру-
ды, М., 1960. Ф. П. Сидельковская.
ДИГИДРОЖАСМОН (З-метил-2-пентилцнклопен-
тен-2-он-1) СцН]8О, мол. в. 166,25 — жидкость с за-
пахом жасмина; т. кип. 101—102°/5 мм; d}| 0,9201;
n'p 1,4811. Д. (I) — продукт неполного гидрирования
жасмона. Методы синтеза Д. сложны и не могут слу-
жить основой для производственного способа получе-
ния. Поэтому в парфюмерной пром-сти применяется
аналог Д. — 2-гексилциклопентен-2-он-1, получив-
ший условное название «Д.» (II).
СНз
(СН2)4СН3 (СН2)5СН3
1----1=0 I---Lo
1 и
Последний является жидкостью с резким и своеобраз-
ным запахом, при разбавлении отдаленно напоминает
запах жасмина; т. кип. 100—105°/4—5 мм; 0,921;
Пр 1,475; получают дегидратацией ундекалактона (IV)
иля ундециленовой к-ты (III) (внутримолекулярное
ацилирование). В последнем случае процесс ведут
в присутствии ортофосфорной к-ты или хлористого
цинка.
CH,=CH(CH2)SCOOH—* Н29 СН2(СН2)6СН3 Н2С
н2Сч Хо н2с
со
п> IV
В качестве дегидратирующего агента рекомендуется
применять также полифосфорную к-ту. Количественно
Д. может быть определен оксимированием.
А. А. Зеленецкая.
ДИГИДРОКАРВЕОЛ (1-метил-4-метоэтенил-цикло-
гексанол-2) С10Н18, мол. в. 138,25 — моноцикличе-
ский терпен ментанового ряда. Обыч-
ный правовращающий Д. — смесь диа-
НО—стереомеров; т. кип. 224—225°/760 мм,
II 112°/14 мм; df» 0,9274; 1,48168;
jK [a]D = -Ь30,56°. Природный Д. имеет
т. кип. 100—10277—8 -m.w; 0,9368;
1,48364; [a]D=—6,14°. При окислении перман-
ганатом калия Д. превращается в п-ментантриол (1);
при нагревании с НС1 Д. образует дипентен-бисгидро-
хлорид; с H2SO4 на холоду легко переходит в п-мен-
тандиол-2,8; при нагревании с H2SO4 дает терпинен
Н°-Т1 Г4)
ГСН,ОИ
он
I !! in
(II). Обычный Д. можно расщепить на два диастерео-
мера (а- и p-формы) переводом в дигидрокарвнлксан-
тогенамид C17H10OCSNH2.
Д. получают восстановлением d-карвона (III) нат-
рием в абс. спирте; его выделяют из тминного, укроп-
ного и других масел. В составе природных эфирных
масел Д. применяют в парфюмерной и мыловаренной
ПрОМ-СТИ. м. Я. Алейникова.
ДИГИДРОСТРЕПТОМИЦИН C21H41O12N„ мол. в.
583,62 — аналог антибиотика стрептомицина; обла-
дает таким же антибактериальным действием, но не-
NH /СНОН NH
h2n-c-hnhc chnh-c-nh2
НС СНОН
. /
СНОН
I /
НС-0
н он НС-О
I / I
о сн2—с—сон
I---------сн
I
СНз
I
------СН
I
CHgHN-СН
о неон
I
носн
I
--------сн
I
сн,он
сколько менее токсичен. Д. получают каталитически
(катализаторы PtO2, Pd, «скелетный» Ni) или электро-
химия. восстановлением стрептомицина, а также вос-
становлением с помощью борогидридов натрия, калия,
никеля и кобальта, SnCl2 и др. В процессе восстанов-
ления альдегидная группа стрептомицина превра-
щается в оксиметильную, что обусловливает более
высокую стабильность Д. Имеются указания, что Д.
образуется непосредственно при биосинтезе актино-
мицетом Streptomyces humidus n. sp. Д. обычно вы-
деляют в виде его солей, к-рые легко
—CI-I получаются в кристаллич. состоянии.
И Наиболее распространенной является
ХСЧСН2)3СН3 сернокислая соль Д. (белые пластинки,
О разлагается при 255—265°); хорошо
и растворима в воде, почти нерастворима
в СН3ОН, С2Н5ОН и других органич.
растворителях; [a]g = —88° (1%-ный р-р в воде).
Д., подобно стрептомицину, подавляет рост многих
грамположительных, грамотрицательных и кислото-
стойких бактерий; он является одним из эффективных
противотуберкулезных средств, Д. широко применяет-
ся при лечении различных форм туберкулеза, туляре-
мии, бруцеллеза, коклюша, чумы и других инфекций;
Д. хорошо переносится больными, чувствительны-
ми к стрептомицину. Д. часто применяется в виде
комбинаций с различными лекарственными средства-
ми (антибиотиками, сульфаниламидами, витаминами).
Лит.: Шемякин М. М. и Хохлов А. С., Химия
антибиотических веществ, 2 изд., М.—Л., 1953; Вейс Р, А.,
М а м и о ф е С. М., Антибиотики, 1950, вып. 4 (17), с. 5;
Городецкая А. В., Савицкая Н. М., там же, 1954,
вып. 5 (43), с. 3; М а м и о ф е С. М., С и и и и и н а 3. Т.,
Хохлов А. С., Мед. пром-сть СССР, 1957, № 11, с. 16;
Ashton &. С., Poster М. С. and Fatherley М.,
Analyst, 1953, № 931, р. 581; Weiss Р. J., Antibiot, and
chemotherapy, 1956, 6, № 11, p. 653; Titsuoka S. [a. oj,
Pharmac. Bull., 1957, 5, № 4, p. 343. С. M. Мамиофе.
ДИГЛИМ — см. ДиэпгиленгЛиколь.
ДИЕНОВЫЕ ИНСЕКТИЦИДЫ — см. Хлорцикло-
диены.
ДИЕНОВЫЙ СИНТЕЗ — реакция присоединения
диенов (соединений, содержащих две сопряжен-
ные двойные связи) к т. н. диенофилам — нена-
сыщенным соединениям, кратная связь к-рых акти-
вирована соседней электрофильной группой: СО,
1109
ДИЕНОВЫЙ СИНТЕЗ
1110
СООН, CN, NO2 и др. Д. с. получают соединения,
содержащие циклогексеновое или ненасыщенное ге-
тероциклич. кольцо:
Примерами Д. с. являются реакции бутадиена с малеи-
новым ангидридом или с п-бензохиноном:
^сна
сн
I
СН
хна
сн-со
II >0
сн-со
^сн,
сн
I
сн
^СНо
(I)
(2)
В качестве «диенов» применяются не только произ-
водные бутадиена, но и соединения ряда винилацети-
лена, а также циклич. и гетероциклич. диены, обра-
зующие при Д. с. соединения с мостиковыми связями:
Х = СН2 или О
С «диенами», содержащими гетероатомы, Д. с. приво-
дит к гетероциклич. системам:
^СН2
СН
СН
СН2
II
СНОС2Н5
t^Xoc2H5
В качестве диенофилов могут быть использованы как
производные олефинов, так и соединения, содержа-
щие ацетиленовую связь, а также группы N = C,
NzhC, N = N и N = O, напр. ацетилендикарбоновая
к-та или цианугольный эфир:
Q соон
соон
^СНо
сн
I
сн
^сн,
с—соон
ill
с—соон
Систематич. изучение Д. с. позволило установить
след, основные закономерности: скорость Д. с. падает
с увеличением числа и объема заместителей в диене и
диенофиле; Д. с, приводит к ifuc-аддуктам (см. 1 или 2);
при Д. с. компоненты ориентируются преимуще-
ственно в соответствии с т. н. «правилом накопления
ненасыщенности», согласно к-рому ненасыщенная
группа X диенофила располагается так, чтобы рас-
стояние между нею и л-электронами диена было
наименьшим:
но не
X
Так, например, при реакции малеинового ангидрида
с циклопентадиеном продукт реакции имеет эндо-
конфигурацию, несмотря на то, что эиао-изомер энер-
гетически более выгоден:
Вопрос о механизме Д. с. еще недостаточно изучен. Пз фак-
тических данных следует, что Д. с. протекает без участия
свободных радикалов. Вме-
сте с тем представления
о ионном механизме Д. с.,
согласно которым реакция
инициируется присоедине-
нием анионоидного конца
диена к катионоидному
углеродному атому диено-
фила с образованием про-
межуточного иона (I), не могут объяснить закономерности
Д. с. (в частности, правило цис-присоединения). Более вероят-
но, что начальной ступенью Д. с. являет-
ся циклич. переходное состояние (II), в СНо ^Х
к-ром в поле 6 атомов углерода нахо- НС^ ^-СН
дятся 6 электронов размыкающихся л-свя- i f ]j п
зей. Согласно этим представлениям, раз- j|
рыв л-связей и образование о-связей осу- СН2
ществляются в одном акте. Этот механизм СН9
удовлетворительно согласовывается с ос-
ионными закономерностями Д. с. н объ- //
ясняет, в частности, тот факт, что диены
типа нис-пиперилена (III), не способные Н
из-за стерических препятствий существо- I
вать в цис-конфигурации, не вступают н С
В Д. с. ХСН3
4
Структурная направленность дне- £
нового синтеза определяется в ос- ^Н
новном электрической природой за- Хн
местителей. Так, напр., при реакции ш
2-метоксибутадиена с акролеином образуется лишь
один из двух возможных структурных изомеров:
Сен,-'5
сн3о—с сн2+« сн3о-<'>
нс (СН -з Ч^Л-сно
ЧСН2 Ч,^сн=о
что объясняется наличием в молекулах диена и диено-
фила частичных зарядов, определяющих их взаимную
ориентацию в переходном состоянии.
Первыми примерами Д. с. могут считаться работы
В. Н. Ипатьева (1887) и затем С. В. Лебедева (1909)
по исследованию димеризации изопрена:
ХСН2
сн3-с +
СН
чсн2
II
сн-с=сн2
сн3
В препаративном и теоретич. отношении Д. с. был
подробно изучен О. Дильсом и К. Альдером, открыв-
шими в 1928 реакцию бутадиена с малеиновым ангид-
ридом. Д. с. с малеиновым ангидридом и бензохино-
ном обычно осуществляется уже при комнатной темп-ре
и дает высокий выход. С менее активными диенофи-
лами Д. с. проводят при кипячении в инертном раст-
1111
ДИЗЕЛЬНОЕ ТОПЛИВО — ДИИЗОПРОПИЛОВЫЙ ЭФИР
1112
ворителе (чаще всего в бензоле или толуоле) или при
нагревании компонентов выше их темп-ры кипения
в закрытом сосуде. Д. с. нашел применение при синтезе
полициклич. соединений, в том числе стероидов, а так-
же при исследовании витамина D и других сложных
природных соединений, содержащих диеновые и по-
лиеновые группировки. На взаимодействии малеинр-
вого ангидрида с бутадиеном основано колич, опре-
деление последнего в газах при произ-ве синтетич, ка-
учука.
Лит.: К л е т ц е л ь М. С., в кн.: Органические реакции,
сб. 4, пер. с англ., М., 1951; Холмс Г. Л., там же, 1951;
Бутц Л. В. и Ритина А. В., там же, сб. 5, М.; А 1 -
der К., в кн.: Neuere Methoden der praparativen organischen
Chemie, [Bd 1], B., 1944. Л. Д. Бергельсон.
ДИЗЕЛЬНОЕ ТОПЛИВО — средние и тяжелые
фракции нефти, используемые в качестве топлива
для двигателей с воспламенением от сжатия (дизелей).
Д. т. разделяется на три группы: 1. Топливо для
быстроходных форсированных транспортных и ста-
ционарных двигателей (от 1000 об/мин и более), при
переменных нагрузках и сравнительно частых оста-
новках. При получении таких топлив используются
керосино-газойлевые фракции прямой перегонки и
каталитич. крекинга нефти с пределами выкипания
270—400°. Основными параметрами, характеризую-
щими их пусковые свойства и качество сгорания,
являются цетановое число и фракционный состав.
Такие показатели, как вязкость, коксуемость, содер-
жание серы, темп-ры помутнения и застывания и
другие, характеризуют качество распыливания топ-
лива, .износ топливной аппаратуры, коррозионный
износ деталей двигателя, нагарообразование, а также
темп-рные условия применения, транспортировки и
хранения. По ГОСТу, эти топлцва выпускаются четы-
рех марок: ДА — предназначенное для применения
при внешних темп-pax воздуха ниже —30°; ДЗ —
для темп-p до —30°; ДЛ — для темп-p не ниже 0°
и ДС — предназначенное для быстроходных судовых
двигателей, работающих в закрытых помещениях
с повышенной темп-рой воздуха. 2. Для тяжелона-
груженных стационарных и судовых двигателей (от
600 до 1000 об/мин) применяют дистиллятное топливо
средней вязкости (соляровое масло), к-рое ввиду
ограниченности его ресурсов м. б. успешно заменено
топливами ДЛ и ДС предыдущей группы, если нет
ограничений в отношении норм пожарной безопас-
ности (темп-ра вспышки). 3. Для тихоходных двига-
телей — стационарных и судовых (до 600 об/мин),
применяют высоковязкие топлива тяжелого фрак-
ционного состава, вырабатываемые из мазутов прямой
перегонки или крекинга нефти путем разбавления их
керосино-газойлевыми фракциями. По ГОСТу, такие
топлива выпускаются трех марок: ДТ-1 — для дви-
гателей, не оборудованных подогревом (до 600 об/мин),
а также как пусковое топливо для более тихоходных
дизелей; ДТ-2 — для двигателей до 300 об/мин;
ДТ-3 — до 200 об/мин. Д. т. марок ДТ-2 и ДТ-3
нуждаются в подогреве как для улучшения прокачи-
вания, так и лучшего распыливания; при наличии
подогрева эти топлива могут заменить ДТ-1 в более
быстроходных дизелях. ДТ-2 и ДТ-3 применяются
также в калоризаторных двигателях.
Одним из важнейших свойств Д. т., от к-рого зави-
сит характер его сгорания в дизеле, является темп-ра
самовоспламенения. В двигателе самовоспламенение
топлива наступает через нек-рое время с момента
начала впрыска топлива в камеру сгорания до начала
интенсивного горения. Чем короче период задержки
воспламенения, тем лучше условия работы двигателя.
Время запаздывания, а также темп-ра, при к-рой про-
исходит самовоспламенение, зависят от химич. со-
става Д. т. Показатель, характеризующий склонность
дизельного топлива к самовоспламенению, наз. цета-
новым числом. Чем выше цетановое число Д. т., тем
меньше время запаздывания его воспламенения, легче
пуск, равномерное нарастание давления в цилиндрах
двигателя и, следовательно, мягче его работа.
Максимальными цетановыми числами обладают
парафиновые углеводороды с прямой цепью атомов
углерода. Снижают цетановое число разветвление
углеводородной цепи и двойные связи в ней. Цетано-
вое число нафтеновых углеводородов значительно
ниже, чем н-парафиновых, и зависит от числа колец,
длины боковой цепи и ее разветвленности. Самое
низкое цетановое число и самый большой период за-
держки воспламенения — у ароматич. углеводородов.
Требования к цетановому числу Д. т. меняются
в зависимости от числа оборотов двигателя: для тихо-
ходных двигателей менее чем 600 об/мин цетановые
числа топлив не нормируются; от 600 до 1000 об/мин
оптимальные цетановые числа 35—40; для двигателей
от 1000 до 1500 об/мин и выше — 45—50.
Второй важной характеристикой Д. т.,также влияю-
щей на пусковые свойства и характер сгорания, яв-
ляется фракционный состав. Требования к нему опре-
деляются условиями работы двигателя, при к-рых
Д. т. впрыскивается в воздушную среду, нагретую
до 500 — 700° и находящуюся под давлением 35—
40 кг/см2. Для таких условий воспламенения при-
годны нефтяные фракции, выкипающие в интервале
от 170—200° до 380—400° (газойль).
На улучшение самовоспламеняемости и полноту
сгорания Д. т. наряду с подбором сырья надлежащего
химич. состава влияют также глубина и способ
очистки. Существенное значение может представить
гидрогенизационная или селективная очистка, позво-
ляющая снизить содержание нежелательных арома-
тич., непредельных углеводородов и серусодержащих
соединений. Разработаны присадки, улучшающие
воспламеняемость и полноту сгорания Д. т. (нитраты,
ВНИИНП-111 и др.), однако использование таких
присадок является пока ограниченным. Д. т. могут
быть получены синтетически разными способами.
Д. т. из окиси углерода и водорода с цетановым чис-
лом ок. 80 используются в качестве высокоцетановой
добавки к нефтяному Д. т. Имеют пока весьма огра-
ниченное распространение Д. т., получаемые гидро-
генизацией твердых топлив, а также коксованием и
полукоксованием углей; первые — в силу высокой их
стоимости, вторые — из-за низкого качества.
Лит.: Л о с и к о в В. В., Пучков Н. Г., Энглив
Б. А., Основы применения нефтепродуктов, 2 изд., М., 1959;
Технические нормы на нефтепродукты, под ред. Н. Г. Пучкова,
М., 1957; Технические условия на нефтепродукты, М., 1960.
ДИИЗОАМИЛОВЫЙ ЭФИР (изоамиловый эфир)
[(СН3)аСНСНаСНа]аО, мол. в. 168,28 — бесцветная
жидкость с эфирным запахом; т. кип. 173,4°/7б0 мм,
60,0°/10 мм; 0,7777; n2£ 1,4085; теплота парообра-
зования при темп-ре кипения 8,40 ккал/моль; раство-
рим в органич. растворителях, малорастворим в воде,
образует с ней азеотроп (46,6% Д. э.), т. кип. 97,4°.
Получают Д. э. из изоамилового спирта при нагрева-
нии с HaSO4 (см. Диэтиловый эфир). Применяют
Д. э. как высококипящий растворитель; при опреде-
лении активного водорода по Церевитинову и при
Гриньяра реакции. а. Е. Васильев.
ДИИЗОПРОПИЛОВЫЙ ЭФИР (изопропиловый
эфир) [(СН3)аСН]аО, мол. в. 102,17—бесцветная летучая
жидкость с эфирным запахом; т. пл. — 85,89°; т. кип.
68,27°/760 мм; d|° 0,7281; zip 1,3689; растворим
в органич. растворителях, растворяет многие органич.
вещества. В Д. э. при 20° растворяется 0,87% воды,
растворимость его в воде 0,94%; образует азеотроп
с водой (95,53% Д. э.), т. кип. 62,2°. Теплота парооб-
разования при темп-ре кипения 6,968 ккал/моль;
теплоемкость Ср (19,9°)51,7 кал/молъ; темп-ра вое-
шз
ДИИЗОПРОПИЛФТОРФОСФАТ — ДИКМАНА РЕАКЦИЯ
1114
пламенения (в закрытой чашке) 7,8°. Получают Д. э.
из изопропилового спирта при нагревании с H2SO4
(см. Диэтиловый эфир)', образуется как побочный про-
дукт при произ-ве изопропилового спирта из пропи-
лена. Применяется Д. э. гл. обр. как растворитель.
По физиологич. действию аналогичен диэтиловому
эфиру, но более токсичен; пары его незначительно
раздражают глаза и слизистые оболочки носоглотки.
А. Е. Васильев.
ДИИЗОПРОПИЛФТОРФОСФАТ (фторангид-
рид диизопропилового эфира фосфорной кислоты)
(uao-C3H7O)2 РГ , мол. в. 184,14 — бесцветная по-
движная жидкость с очень слабым запахом; т. пл.
—82°; т. кип. 67,5°/12зии; df» 1,0682; 1,3832; дав-
ление насыщенного пара 0,9 ммрт. ст. при 20°; лету-
честь (максимальная концентрация) 9,2 мг/л при 20°;
плохо растворяется в воде (1,5%), хорошо — в орга-
нич. растворителях. Водой Д. гидролизуется очень
медленно (реакция носит автокаталитич. характер)
с образованием диизопропилфосфорной к-ты и HF;
энергично взаимодействует с водными р-рами аммиака,
и аминов с образованием соответствующих солей диизо-
пропилфосфорной и фтористоводородной к-т, Д. может
быть получен действием фтористого натрия на диизо-
пропилхлорфосфат; последний получают взаймодей-
ствием изопропилового спирта с РС13:
Г уН -]
РС13 4- ЗС3Н7ОН — (С3Н7О)аР^С1
— (С3Н7О)»РОН + изб-С3Н7С1
с последующим превращением полученного диизо-
пропилфосфита в хлорофосфат:
п /,°
(С3Н7О),РОН (С3н7О)2р^С1
Д. очень токсичен; судорожно-паралитич. яд с резко
выраженным миотич. действием (сужение зрачка).
Концентрация 0,082 мг/л вызывает сильный миоз.
Лит.: Сартори М., Усп. химии, 1964, 23, вып. 1,
с. 62—88. Р. Н. Стерлин.
ДИИОДТИРОЗИН (иодгоргон, иодглобин, 3,5-ди-
иод-4-оксифенилаланин) C9H9O3NJ2, мол. вес 433 —
. бесцветные кристаллы,
। известны в двух фор-
h \ мах: d, 1 и 1. Изомер
НО -С 'y-CH2CH(NH2)COOH d, 1 получают иодиро-
ванием d, 1-тирозина;
j т. пл. 200° (с разл.),
растворимость в воде
при 15° 1 : 2164. 1-дииодтирозин получают аналогич-
но из 1-тирозина; т. пл. 213° (с разл.), [а]р=2,27°
(в 25%-ном NH4OH); при 15° растворяется в 347
частях воды. 1-изомер образуется также в щито-
видной железе и служит исходным веществом при
образовании тироксина. Д. тормозит выработку ти-
реотропного гормона передней доли гипофиза и акти-
визирует деятельность щитовидной железы; приме-
няют при гипертиреозе, а также совместно с метил-
тиоурацилом для уменьшения «зобогенного» действия
последнего.
Лит. см. при ст. Апрессин. А. И. Травин.
ДИКАИН (пантокаин, тетракаин хлоргидрат, хлор-
гидрат диметиламиноэтилового эфира п-бутиламино-
ын/ги 1 бензойной К-ТЫ)Ci5H»5N2O3C1,
1 с г>зСпз молекулярный вес 300,84—
белые мелкие кристаллы;
I |1 т. пл. 147—150°; дикаин
растворим в воде и спир-
Г<ЭГМГН„Ъ,И(СН 1 .НГ1 те’ нерастворим в эфире и
СОО(СНг)г МСН3)г НО беН80Ле. Водные растворы
Д. нейтральны, устойчивы при хранении, при
кипячении не разлагаются. При прибавлении конц.
HN03 к водному раствору Д. (1 : 50) появляется
желтая окраска, а при прибавлении к такому же
р-ру Д. разб. HNO3 и AgNO3 выделяется осадок
AgCl. Количественно Д. определяют титрованием
точной навески 0,1 н. NaOH в смеси воды и хлоро-
форма (индикатор — фенолфталеин). Д. получают
из n-аминобензойной к-ты N-алкилированием ее
натриевой соли бромистым бутилом и этерификации
образующейся при этом n-бутиламинобензойной к-ты
Р-диметиламиноэтанолом. Д. — сильное местноанесте-
зирующее средство, превышающее по активности
новокаин и кокаин; токсичнее кокаина в 2 раза, но-
вокаина — в 10 раз.
Лит. см. при ст. Адалин. А, С. Елина.
2,5-ДИКЕТОПИПЕРАЗИНЫ — шестичленные ге-
тероциклич. соединения, производные пиперазина,
к-рые можно рассматривать как циклич. ангидриды
аминокислот:
ZCO-NHX
R-НС СН-Р’
XNH-CO/ 4
Д. легко могут быть получены циклизацией а-ами-
нокислот либо их эфиров:
R
I , ZCO-NH4
H2N СН —COOR—— R-HC СН—R + R—ОН
4NH-COZ
R — Н : алкил
Эфиры дипептидов также легко циклизуются в Д.:
/СО—NH4
H2NCH2CONHCH2COOR—- Н2С СН2 + R-OH
4NH-COZ
Д,, выделяемые из гидролизатов белков, являются
продуктами последующей циклизации аминокислот,
образовавшихся при гидролизе.
Д. — бесцветные кристаллы, нейтральные соеди-
нения, сравнительно легко растворимые в горячей
воде и плохо—в холодной. Под действием электрич.
тока они не мигрируют ни к катоду, ни к аноду, чем
пользуются для их отделения от аминокислот. Разб.
минеральные к-ты и щелочи гидролизуют Д. до ди-
пептидов. При энергичном гидролизе они расщеп-
ляются до аминокислот. Натрием в амиловом спирте
Д. восстанавливаются до пиперазинов. С ангидри-
дами и галогенангидридами кислот образуют N-ациль-
нЫе производные. Реагентом на Д. является водный
щелочной р-р пикриновой к-ты, к-рая восстанавли-
вается до пикраминовой к-ты, имеющей красно-ко-
/СО-NHx /CO-NH\
/. Н2с СН2 //. СНд-НС Сн_Сн
XNH-COZ 'NH-COZ
ричневый цвет. Простейшие Д. — глицинангидрид (I),
,т. пл. 311—312° (с разл.) и аланинангидрид (II).
Н. Е. Голубева.
ДИКМАНА РЕАКЦИЯ — внутримолекулярная
конденсация эфиров двухосновных кислот в циклич.
fj-кетоэфиры, катализируемая основаниями:
/СН,\С00(? /СО
NaOl% н2С CHCOOR
Нг /COOR нгс ~ СН2
Спо
В качестве оснований чаще всего применяют алкого-
ляты, реже — трйфеНилметилнатрий, гидрид натрия
й др. Образующиеся p-кетоэфиры далее гидроли-
1115
ДИКУМАРИН — ДИМЕДОН
1116
зуются и декарбоксилируются в циклич. кетоны. На-
личие алкильных заместителей в а- и 0-положениях
затрудняет циклизацию, причем из двух возможных
продуктов предпочтительно получается тот, образо-
ванию к-рого благоприятствуют пространственные
отношения:
сн3х /CH2CH2COOR
СН3\ /СНа—CHCOOR
он
ОН
сн3 xch2coor
со
Д. р. имеет широкое распространение и дает воз-
можность синтеза разнообразных алициклич., поли-
циклич. и гетероциклич. полиэфиров. Циклизация
диэфира (I) приводит к производным пиперидона (II)
СН2—СН2 СН2—СНах
RN\ XCH2COOR---------RN"f CHCOOR
CH2—COOR CH2—СОХ
По Д. р. могут быть получены и гетероциклич.
тоны с атомами серы и кислорода в кольце:
СН2 СН2
SO SO
I I ------ I I
MeOCO—CH, CH, MeOCO—CH CH2
/ “ \ /
MeO—C=O C=O
Изучение кинетики реакции, а также определение
изотопного эффекта при циклизации эфиров фенилен-
диацетата, меченных в различных положениях тяже-
лым углеродом, показало, что Д. р. протекает по
ке-
Ci
О
СС1
Cl
Cl
лым углеродом, показало, что Д. р. протекает
схеме:
^-ch2coor + r5
^-CH2COOR
CHCOOR
ch2coor
(»
CHCOOR
/U-CH2COOR
CHCOOR
(2)
СНг °R
личественное определение Д. основано на его вза-
имодействии с 0,1 н. NaOH в присутствии спирта и
титровании избытка ще-
лочи 0,1 н. соляной кис-
лотой в присутствии ме-
тилового красного.
Д. получают взаимо-
действием этоксимагний-
малонового эфира с хлорангидридом ацетилсалици-
ловой к-ты.
Д. задерживает свертывание крови и применяется
при лечении тромбозов, тромбофлебитов, эмболий,
инфаркта миокарда и т. п.
Лит. см. при ст. Апрессин. А. И. Травин.
ДИЛЬДРИН С12Н8С1вО, мол. в. 380,9 — белое
вещество без запаха; т. пл. 175—176°; 1,54; технич.
продукт — рыжевато-коричневые
хлопья, содержащие 76% Д.; в
воде нерастворим; в 100 мл
органич. растворителя при 25°
растворяется: в ацетоне 26 г, в
бензоле 56 г, в этиловом спирте
4 г, в дихлорэтане 70 г, в пента-
не 2 г, в ксилоле 52 г. В ИК-спектре Д. имеет
характерный максимум поглощения при 10,98 мк,
используемый для колич. микроопределения. По
устойчивости к кислотам и щелочам аналогичен аль-
дрину; при обработке уксусной и бромистоводородной
к-тами образуется 6,7-бромацетоксипроизводное, к-рое
при действии цинка превращается в альдрин. Полу-
чают Д. окислением альдрина надуксусной или над-
бензойной к-тами (промышленный метод). Применяют
как эффективный инсектицид в порошке или же
в виде растворов в ксилоле; очень токсичен, ЛД50
для крыс 25—30 мг/кг.
Лит. см. при ст. Альдрин. Л. С. Поваров.
ДИЛЬСА-АЛЬДЕРА РЕАКЦИЯ — см. Диеновый
синтез.
ДИМЕДОН (5,5-диметилциклогександион-1,3), мол.
вес 140,18, С8Н12О2 — бесцветные или светло-жел-
тые кристаллы, цвет которых зависит от условий
кристаллизации и примененного растворителя. По-
видимому, окраска кристаллов зависит также от
преобладания одной из таутомерных форм (I или
II). Т. пл. 145—148°; в 100 г воды рас-
C\COOR творяется 0,416 г (25°), 3,8 г (90°); в 100 г
С=О w спирта растворяется при нагревании 6,6 г;
растворяется также в нагретом бензоле,
эфире, этилацетате и хлороформе. Безводный
Д. вполне устойчив, водные растворы его разлагаются
и окисляются даже в темноте; титруется щелочами
как одноосновная кислота; константа диссоциации
К ~ 0,71 • 10~5 (25°). При действии солей диазо-
ния на щелочные растворы Д. образуются интен-
сивно окрашенные азосоединения. Д. синтезируют
конденсацией малонового эфира и окиси мезитила
в присутствии этилата натрия:
CHCOOR
CHCOOR
СН, OR СНа
причем определяющей скорость реакции
стадия (2) — образование С—С связи. Д. р. открыта
в 1894 В. Дикманом.
Лит.: Органические реакции. Сб. 1, пер. с анг., М., 1948,
355—57; Reed R. I., Thornley М. В., J. Chem. Soc.,
1954, р. 2148; Carrick W. L., Fry A., J. Amer. Chem.
Soc., 1955, 77, № 16, p.4381. H. II. Гамбарян.
ДИКУМАРИН (дикумарол, антитромбозин, тром-
бозан, 3,3-метилен-бис-4-оксикумарин) С19Н12Ов, мо-
лекулярный вес 336,3 — бесцветные или
слегка желтоватые кристаллы слабо-
горького вкуса; т. пл. 285—287°; раство-
рим в едких щелочах, хлороформе и пи-
ридине, нерастворим в воде, спирте, аце-
тоне и эфире. Дикумарин—токсически
действующее начало загнивающего дон-
ника (Melilotus officinalis), вызывающее
кровотечение у животных, поедающих это растение.
Качественно Д. определяют растворением сплава,
состоящего из равных весовых частей Д. и КОН,
в горячей воде и подкислением раствора соляной ки-
слотой, при этом образуется белый осадок салицило-
вой кислоты; при прибавлении 1 капли раствора
FeCl3 появляется сине-фиолетовое окрашивание. Ко-
СН2
является
н3<4 сн3
но-с^
сн2
+
СН2-СООС2Н5
СО-ОС2Н3
Н,С^ _/СН3
сн2
н3сХс/Сн3
HgC-^ "^CH-COOCaHg
О=
Н2С
О=Сх. /С=О
сн2
/С=О
сн2
Н3Схс/СН3
Н2с^ ^СНа
>?.=он
сн
II
1117
ДИМЕДРОЛ — п-ДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИД
1118
Применяют Д. для идентификации альдегидов по
темп-рам плавления образуемых производных, а также
для их определения весовыми или титриметрич. ме-
тодами. Со всеми альдегидами (кетоны реагируют,
как правило, только в особых условиях) Д. образует
хорошо кристаллизующиеся продукты конденсации.
При этом две молекулы его конденсируются с молеку-
лой альдегида. Образующийся кристаллич. альдиме-
дон (Ш) при нагревании теряет воду, образуя циклич.
ангидрид (IV):
СО F СО
НоС-^ ~"С--СН—С-^ А?.на
нс. II II Lch,
•,1<А /С-он но-с. сГ 3
Н3с/ ^Сн2 сн2 СНЭ
III
R
I
СО сн со
H’i ? V 1н”
A A А°"’
нзс СН2 О СН2 СН3
IV
Для индентификации используют продукты обеих
реакций. С формальдегидом образуется формальди-
медон, т. пл. 191—191,5°; с ацетальдегидом — про-
дукт, т. пл. 141—142°. и. п. Ефимов.
ДИМЕДРОЛ (хлоргидрат р-диметиламиноэтилового
эфира бензгидрола) (CeH5)2CHOCH2CH2N(CH3)2-HCl,
мол. в. 291,83 — бесцветные кристаллы; т. пл. 166—
168°, хорошо растворим в воде и спирте, нерастворим
в эфире. Выделенное из хлоргидрата основание Д.
перегоняется при 150—155°/5 мм. С конц. H2SO4 Д.
дает желтое окрашивание; при кипячении с разведен-
ной НС1 в течение 3 мин и охлаждении выделяются
кристаллы бензгидрола (т. пл. 62—67°). Колич.
определение Д. основано на выделении основания Д.
щелочью, извлечении основания эфиром и определе-
нии С1~ в оставшемся водном р-ре. Д. может быть син-
тезирован, напр. конденсацией р-диметиламиноэтил-
хлорида (или его хлоргидрата) с бензгидролом.
Д. — один из основных противогистаминных пре-
паратов (см. Л екарственные вещества синтетические),
его применяют при лечении различных аллергич.
заболеваний (крапивница, сенная лихорадка и др.)
или осложнений, вызванных повышенной чувстви-
тельностью организма к нек-рым лекарствам, химич.
воздействиям и т. п.
Лит. см. при ст. Апрессин. О. Ю. Магидсон.
2,3-ДИМЕРКАПТОПРОПАНОЛ, мол. в. 124,23
HOCH2CHSHCH2SH, — густая, вязкая маслянистая
жидкость с запахом меркаптана; т. кип. 89°/0,5 мм;
d25 1,2385, пр’ 1,5720; в 100 мл воды растворяется
8,7 г Д.; растворим в растительных маслах; в воде
постепенно разлагается. Получают Д. бромирова-
нием аллилового спирта с последующей обработ-
кой NaSH под давлением или гидрированием трисуль-
фида оксипропилена. Применяют при анализе как
маскирующее средство для связывания металлов,
образующих сульфиды; как противоядие при отравле-
нии солями As, Bi, Н2О, Au и др. тяжелых металлов
(но не свинца); для устранения нарывного действия
льюизита на пораженной коже и слизистой оболочке.
См. также Антидоты. Д. И. Кузнецов.
3,4-ДИМЕРКАПТОТОЛУОЛ H3CCeH3(SH)2, мол. в.
156,25 — бесцветный или слабоокпашенный порошок;
т. пл. 31—32°; т. кип. 185—187°/84 мм; растворяется
в разб. щелочах; гигроскопичен; следует хранить
хорошо закрытым в сухом месте; имеет неприятный
запах. Получают Д. восстановлением хлорангидрида
толуол-3,4-дисульфокислоты цинком в соляной к-те;
применяют для открытия и определения олова (розо-
вый или красный осадок); для открытия Pd11, SeIV,
TeIV, Au, WVI, Revn в 10 н. p-pe HCI; Osvin, Rh111
или RhIV, As111, Ir, Ru, GeIV, Cu1, Hg11 в 6—8 н.
р-ре HCI; для открытия Мо, Bi. Применяют как коа-
гулянт коллоидной се]эы и сульфидов металлов в кис-
лом р-ре. Д- И. Кузнецов.
ДИМЕТИЛАМИН (CH3)2NH — простейший пред-
ставитель вторичных аминов жирного ряда; при ком-
натной темп-ре — газ, с резким неприятным запахом,
легко сжижающийся при охлаждении или под давле-
нием в бесцветную жидкость; т. кип. 6,9°; т. пл. —
92,2°; dj 0,6804; константа диссоциации А = 5.1-101;
рА 3,29; хорошо растворим в воде и органич. рас-
творителях; образует кристаллич. соли с кислотами;
т. пл. хлоргидрата 171 , бромгидрата 133,5°, иодгид-
?ата 155°, нитрата 74°, перхлората 200—203°, пикрата
58°. Д. образуется при гниении белковых веществ,
содержится в селедочном рассоле. Д. обладает химич.
свойствами, характерными для вторичных аминов.
Так, при действии азотистой к-ты на Д. образуется
N-нитрозодиметиламин, представляющий собой жел-
тое масло, т. кип. 153°. Как и другие вторичные амины,
Д. (в виде соли) можно вводить в Манниха реакцию
для замещения активного атома водорода группи-
ровкой CH2N(CH3)2.
Наиболее простой способ получения Д. (в смеси
с метил- и триметиламином) заключается во взаимо-
действии метилового спирта с аммиаком под давлением
в газовой фазе над катализатором типа глинозема.
Д. может быть получен также нагреванием формаль-
дегида с хлористым аммонием; образующаяся перво-
начально соль метиламина реагирует далее с формаль-
дегидом с образованием соли Д.:
2НСНО + NHjCl — CH3NH2 • HCI 4- HCOOH
CH,NH2 • HCI + 2HCHO - (CH„)2NH • HCI + HCOOH
Для получения чистого Д. без примесей первичного
и третичного аминов действуют конц. р-ром щелочи
на л-нитрозодиметиланилин:
C«HsN(CH3)2 + HNO2 - n-ONC,H4N(CHa)2 -
- (CHs)2NH + n-ONCaH4OH
Д. применяется для введения группы (CH3)2N —
в различные органич. соединения. Так, в случае при-
соединения окиси этилена к Д. образуется диметил-
аминоэтанол (CH3)2NCH2CH2OH, применяющийся в
синтезе дикаина — препарата местноанестезирующего
действия. Д. используется и при получении очень эф-
фективного лекарственного вещества — аминазина,
содержащего группировку (CH3)2NCH2CH2CH2—,
и др. На организм действует раздражающе, подобно
аммиаку. Я. ф. Комиссаров.
zz-ДИМЕТИ Л АМИН О АЗОФЕНИЛАРСОНОВАЯ
КИСЛОТА (CH3)2NCeH4N = NCeH4—AsO(OH)2, мол. в.
349,20 — красный порошок, растворимый в к-тах
(с красной окраской) и в щелочах (с желтой окраской);
солянокислая соль кристаллизуется в темно-фиоле-
товых блестящих иглах; ядовита. Получают Д. диазо-
тированием арсаниловой к-ты и последующим соче-
танием соли диазония с диметиланплином в соляно-
кислой среде; применяют для фотометрия, определе-
ния Th; для открытия Zn (в присутствии Мо, W, Ti,
Се, Sn), Та И фториД-ИОНЭ. Д. И. Кузнецов.
п-ДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИД С,НиОМ,
n-(CH3)2NCeH4CHO, мол. в. 149,30 — твердое веще-
ство; т. пл. 75°; т. кип. 176—177°/17 мм; растворим
в обычных органич. растворителях. Для очистки Д.
растворяют в разб. соляной к-те и прибавляют р-р
едкого натра. Как третичный амин дает соли, напр.
хлоргидрат с т. пл. 107—109°, как альдегид — оксим,
фенилгидразон, семпкарбазон и др.
1119
n-ДИМЕТИЛАМИНОБЕНЗИЛИДЕНРОДАНИН — ДИМЕТИЛГЛИОКСИМ
1120
Д. образуется при действии щелочи на про-
дукт конденсации диметиланилина с хлоралем
n-(CH3)2NCeH4CHOHCCl3, а также при формилиро-
вании диметиланилина N-метилформанилидом. Луч-
ший способ получения Д. — конденсация диметил-
анилина, формальдегида и п-нитрозодйметиланйлина:
n-(CH3)2NC3H4NO -f-2НСНО -}- C3H5N(CH3)s —
— n-(CH3)»NC3H4N=CHCeH4N(CH3)2-n + НСООН
n-(CH3)2NC3H4N=CHC0H4N(CH3)2 + HCHO-i*
— n-(CH3)»NC3H4CHO -f-n-(CH3)2NC,H4N=CH2
Применяется в произ-ве нек-рых красителей, а также
как реактив, дающий характерные цветные реакции
со многими алкалоидамй, а также с нек-рыми гете-
роциклич. и ароматич. соединениями и в качестве
реактива на фосген.
Лит.: Синтезы органических препаратов, под ред. Гиль-
мана [и др.], М., 1949, с. 193—96. Я. Ф. Комиссаров.
л-ДИ МЕТ ИЛАМ И НОБЕ Н 3 ИЛ ИДЕ Н РОДАН И Н
C12H12ON2S2, мол. в. 264,35 —красные кристаллы,
,, _____г=п разлагающиеся выше
। । / 200°; нерастворим в
S=Ck_' ХО=СН —< У—N(CH3)2 воде, малорастворим
в ацетоне, спирте (при
нагревании), в эфире, хлороформе, бензоле; раство-
ряется в сильных к-тах с образованием желтых
р-ров. Получают конденсацией п-ди метил аминобен-
зальдегида с роданином. С солями Ag+ в кислом р-ре Д.
образует красно-фиолетовый осадок или окрашивание
(в зависимости от концентрации соли Ag+). Анало-
гично реагирует е Pd, Hg, Au, Pt, Си1, на чем основа-
ны методы их открытия и фотометрия, определения.
Лит.: Banerjl Samir К. et Dey Arun К.,
Chimie Analytlgue, 1958, 40, № 6, p. 202—203.
В. Г. Tunuoca.
ДИМЕТИЛАНИЛИН CeH5N(CH3)2, мол. в. 121 —
третичный жирноароматич, амин; жидкость, в чистом
виде бесцветна; т. заст. 4-1,96°; т. пл. 2,5°; т. кип.
192,5—193,57760 мм, 153,4°/250 мм, 100734 мм,
77°/13 мм; d\a 0,9557; п*$ 1,55819; растворяется в
большинстве органич. растворителей, в воде 1—1,4°/0
(12°); константа диссоциации в воде А — 1,0-10~9 (20°).
Характерные реакции — образование п-нитрозоди-
метиланилина и Михлера кетона. В пром-сти Д. го-
товится обработкой анилина метиловым спиртом и
серной к-той под давлением. Д., свободный от примеси
других аминов, получается при контактном алкили-
ровании анилина диметиловым эфиром. Д. приме-
няется в синтезе основных красителей (малахитовый
зеленый, метиленовый голубой и др.), проявителей
для цветной фотографии, взрывчатых веществ (те-
трила); ранее применяли для получения диметиламина
(через нитрозодиметиланилин). в. А. пучков.
2,3-ДИМЕТИЛБУТАДИЕН-1,3, мол. в. 82,14 —
диеновый углеводород (диолефин) с двумя сопряжен-
ии Ли ными двойными связями; т. пл.
I 3 । 3 -76,0°; т. кип. 68,5-68,7°; d|«
CHS=C---С=СН3 0,7262; 1,4391.
Д. свойственно большинство реакций, характерных
для олефинов, и все реакции диолефинов (бутадиена,
изопрена). Д. полимеризуется легче, чем его низшие
гомологи, образуя т. наз. метилкаучук, названный так
за наличие еще одной метильной группы по, сравне-
нию с натуральным каучуком. Д. служил основой для
получения в небольших количествах синтетич. кау-
чука в Германии в период первой мировой войны.
Этот каучук отличался низкими технич. свойствами.
Д. получали восстановлением ацетона с помощью алю-
миния до пинакона и последующей дегидратацией по-
следнего, напр. над окисью алюминия при 427—470°;
он ОН
2(СН3)2СО А- +-Na-— (СН3)2С-d(CH3)3 А-а-°3.
— СН2=С(СН3)-(СН3)С=СН2
Д. может быть получен также и др. способами,
напр. нагреванием тетраметилдихлорэтана со спирто-
вым р-ром КОН при 130°; нагреванием диметилизо-
пропенилкарбинола с разб. соляной к-той при 100°;
нагреванием 2,2,3-триметил-3,4-ди-броммасляной к-ты
в присутствии пиридина. Попытки применить Д.
вместо изопрена при получении бутилкаучука иэ
изобутилена не дали положительных результатов.
Колич. определение Д. аналогично определению
бутадиена. А. к. Никитин.
ДИМЁТИЛГИДРАЗИН C2H8N2, мол. в. 60,10 —
жидкость. Известны: 1) Несимметричный Д.
(CH3)2NNH2;t. кип. 63°; d™ 0,7914; ntf 1,40753; очень
гигроскопичен; растворим в воде, спирте, эфире. Д.
образует устойчивые соли, напр. кристаллич. хлор-
гидрат, т. пл. 81—82°. Для получения Д. действуют
азотистой к-той на соль диметиламина и образовав-
шийся нитрозодиметиламин восстанавливают, напр.,
Zn-пылью в среде СН3СООН (выход ок. 80%):
(CH3)»NH • НС1 + hno2 — (CH3)3NNO + HCI + Hso
(CH3),NNO + 2H3 - (CH3)iNNH2 + H3O
Д. приобрел практич. значение в качестве ракетного
топлива.
2) Симметричный Д. CH3NHNHCH3 значительно
менее доступный, т. кип. 81°; 0,8274; пг£ 1,4209;
т. пл. дихлоргидрата 165—167°. Обычный способ полу-
чения симметричного Д. состоит в ацилировании гид-
разина, в метилировании диацилгидразина диметил-
сульфатом и в омылении образовавшегося диацил-
диметилгидразина:
NHSNH3 + 2RCOC1 - RCONHNHCOR -
- RCON(CH3)N(CH3)COR - CH3NHNHCH3
Лит.: Синтезы органических препаратов, пер. с англ,
сб. 2, М , 1949, с. 199—205. Я. Ф. Комиссаров.
ДИМЕТИЛГИДРОХИНОН (диметиловый эфир гид-
рохинона, 1,4-диметоксибензол) п-СН3О—Св Н4—ОСН3,
мол. в. 138,16 — бесцветные кристаллы со своеобраз-
ным запахом свежего сена или меда с посторонним
пряным оттенком; т. пл. 55—56°; т. кип. 205°. Полу-
чают Д. метилированием гидрохинона диметилсульфа-
том, метилсернокислым натрием или метиловым эфи-
ром толуолсульфокислоты, напр.:
f^V-OH (CH3)2SO2 rs*'Xjr-OCH3
н3со-Ч>
Полученный Д. для очистки перегоняют с паром
и перекристаллизовывают из водного спирта. Приме-
няют Д. в парфюмерных композициях и в особен-
ности для отдушки мыла. А. а. Зеленечкая.
ДИМЕТИЛГЛИОКСИМ (диацетилдиоксим, реак-
тив Чугаева) C4H8O2N2, мол. в. 116.12 — бесцветные
кристаллы; т. пл. 238—240°; в 100 г воды растворяется:
0,05 г (25°) и 0,93 г (100°); легко растворяется в спирте,
эфире и р-рах щелочей; константа диссоциации рА'
(НА) при 25° в 50%-ном водном р-ре диоксана равна
12,84; в 75%-ном р-ре диоксана 19,5; получается
действием гидроксиламина на диацетил или на его
монооксим. Из трех известных стереоизомеров Д. —
анти-(а) I, син-(р) II и амфи-(у) III — только анти-
форма (а-Д.) представляет (см. ф-лу)
Н3С—С—С—СН3
II I
НО—N N-ОН
I
Н3С—С------С—сн3
N-OHHO-N
И
Н3С—С—С—СН3
II II
НО—N N-ОН
III
селективный реагент на Ni. При действии этой формы
Д. на соли Ni образуется нерастворимый кристаллич.
1121
ДИМЕТИЛДИХЛОРСИЛАН — ДИМЕТИЛСУЛЬФАТ
1122
осадок красного цвета, содержащий 20,32% Ni и
отвечающий формуле (C4H7O2N)2Ni. Диметилглиокси-
мат никеля является енутрикомплексным соедине-
нием. Осадок обладает хорошими химико-аналитич.
свойствами: практически нерастворим в воде, осаж-
дается достаточно чистым, легко отфильтровывается,
обладает интенсивной красной окраской. Применяют
Д. для открытия и определения (весовым, титримет-
рич. и фотометрич. методами) Ni, Pd, Pt, Fe11, Co,
Rh и др.; для открытии Bi, Си, Re, Sn,'V. Комплекс
c Feir применяют как окислительно-восстановитель-
ный индикатор для титрования феррицианидом в ам-
миачном р-ре (£0 — +0,25 в, pH 9,4); окисленная
форма — бесцветная (или желтая), восстановленная —
красная.
Константы устойчивости (ступенчатые) комплексов с раз-
личными катионами, определенные при 25° в среде 50%-ного
водного р-ра диоксана, равны:
Катион 1g к, 1g к2 Катион 1g к, ig кг
Cd 2+ 5,7 5,0 N12+ 11,16 10,54
Со2+- 9,80 9,14 Ni3+* 14,6 13,8
CU 2+ 11,94 11,36 Pd3+ 7,3 ——
La3+ 6,6 5,9 Zn2+ 7,7 6,2
* При 30° в среде 75%-ного водного р-ра диоксана.
Впервые был описан как реактив на никель
Л. А. Чугаевым в 1905. И. п. Ефимов.
ДИМЕТИЛДИХЛОРСИЛАН (CH3)2SiCl2, мол. в.
129,07 — жидкое кремнийорганическое соединение;
т. пл. —86°; т. кип. +70,1°; 1,0663—1,0637; ng
1,4002. Д. обладает свойствами хлорангидридов,
мгновенно гидролизуется водой, легко реагирует
с гидроксилсодержащими соединениями. Д. может
быть получен из хлористого метила и кремния в при-
сутствии катализатора — металлич. меди в виде
сплава с кремнием: 2СН3С1 + Si—> (CH3)2SiCl2. Кроме
того, Д. может быть получен также при помощи маг-
нийорганич. соединений: 2CH3MgCl(Br) + SiCl4 ->
(CH3)2SiCl2 + 2MgCl2(ClBr).
В обоих синтезах одновременно образуются метил-
трихлорсилан и триметилхлорсилан. Д. применяют
для синтеза высокополимерных соединений (полиор-
ганосилоксанов), вт. ч. силиконового каучука. Колич.
определение производят четкой разгонкой и определе-
нием омыляемого хлора.
Лит.: Андрианов К. А., Кремнийорганические
соединения, М., 1955; Клебанский А. Л., Фихтен-
гольц В. С., Ж. общ. хим., 1956, 26, вып. 9, с. 2502;
1957, 27, вып. 9—10, с. 2475, 2648. А. К. Никитин.
ДИМЕТИЛОВЫЙ ЭФИР СН3—О—СН3, мол. в.
46,07—бесцветный газ, т. пл. —138,5°; т. кип. —23,65°;
d20 2,091 г/л.; в 1 объеме воды при 18° растворяется
37 объемов Д. э.; т. воспл. —41,1° (в закрытом сосуде).
Д. э. сравнительно инертен и разлагается только
при красном калении, не реагирует с Na, сильными
к-тами и щелочами при умеренной темп-ре. Харак-
терное свойство Д. э., как и других простых эфиров, —
способность давать при низкой темп-ре соли оксо-
ния с галогеноводородными к-тами:
Д. э. образует комплексные соединения с НС1, НВг,
HJ и Вг2; т. пл. соответственно: —97,1°; —13°; —22°;
—80°. Д. э. может быть получен из диметилсульфата
нагреванием с окисью меди или из метанола с помощью
серной к-ты:
<Г 1ПП°
СН3ОН4- H2SO4 —— CH3OSO2OH4- н2о
15^1— СН3ОСН3 + H3SO4
CH3OSO2OH+ СН3ОН-
1---- CH3OSO2OCH3 4- H2O
CH3OSOsOCH3 4- CH3OH — CH3OCH3 4- CH3OSO,OH
Метилсерная к-та, потребляемая во 2-й стадии реак-
ции, регенерируется в 3-й стадии, но вода, к-пая удер-
живаемся в процессе, приводит к такому разбавлению,
что реакция перестает быть эффективной. Д. э. яв-
ляется слабым наркотиком; применяется для метили-
рования ароматич. аминов, для получения диметил-
сульфата, также в качестве растворителя для экстрак-
ции и др.
Лит.: Mayor Y., Industr. chlmlque, 1952, № 422,
sept., 259. Ю. А. Чебррков.
ДИМЕТИЛПИРОН (2,6-диметил-у-пирон) С7Н8О2,
мол. в. 124,14 (I) — бесцветные иглы; т. пл. 132°;
т. кип. 248—249°/713 мм; возгоняется; хорошо рас-
творим в воде, спирте, эфире, хлороформе (34,37 г
в 100 г СНС13); плохо растворим в СС14 (1,04 г в 100 г
СС14); 9-10-14, й(киол =0,8-10-14; дипольный
О-
О
момент 4,05 D. С органич. и минеральными к-тами
Д. образует сопи пи-
рилия (II), например
хлоргидрат (к 1 мо-
лекуле Д. присоеди-
нены 1 молекула НС1
и 2 молекулы воды),
т. пл. 83—85°, безвод-
ного 154°; бромгидрат,
т. пл, 194—196°. При действии CH3MgJ на Д. об-
разуется- пироксониевое основание, к-рое с НС1О4
образует прочную соль:
н3сЛ^М?СНз н3с^оЛсн3
//
Д. не образует фенилгидразона. Получают Д. дей-
ствием HCI на диацетилацетон и др. методами:
О
СО СО У
НгС^СН2 _ НС" 'СН __ /Ч
со со снгС Схсн Н3С"\Асн3
н3с сна он но 3
Лит.: Фред Д., в ки.: Гетероциклические соединения,
под ред. Р. Эльдерфвлда, пер. с англ., т. 1, М., 1953, с.
288. В. М. Демьянович.
ДИМЕТИЛСУЛЬФ AT (CH3)2SO4 — бесцветная жид-
кость; т. кип. 188,3—188,6°/7б0 мм, 74°/11 мм,
т. пл. —26,8°; dj5 1,3332; ng 1,3874; в воде мало
растворим (2,8% при 16°); смешивается во всех отно-
шениях со спиртом и эфиром; ограниченно растворим
в углеводородах; при перегонке частично разлагается.
Д. омыляется водой; присутствие кислоты ускоряет
гидролиз; с аммиаком без растворителя реагирует
почти со взрывом. Д. широко применяется как мети-
лирующее средство для окси-, карбокси-, меркапто-,
амино- и иминогрупп, обычно в щелочной среде;
при этом реагирует одна метильная группа, при про-
должительном нагревании до 100—105° иногда всту-
пает в реакцию вторая СН3-группа; при метилирова-
нии соединений, чувствительных к воздействию ще-
лочи, пользуются бикарбонатом натрия; возможно
1123
ДИМЕТИЛТЕРЕФТАЛАТ — ДИМЕТИЛФОРМАМИД
1124
метилирование в неводных средах, напр. фенолов
в среде ксилола; с азотистыми основаниями, пиронами
и нек-рыми другими типами соединений образует
метилсульфонаты, напр. с хинолином:
SO4CH3
Д. легко реагирует с алкил- и арилмагнийгалогени-
дами, натрацетоуксусным и натрмалоновым эфирами,
ацетиленидами и другими металлоорганич. соедине-
ниями; при алкилировании солей HCN, HSCN, HNO3,
галогеноводородных и др. кислот образуются соот-
ветственно ацетонитрил, метилтиоцианат, нитроме-
тан, метилгалогениды, эфиры и т. д.; можно применять
для введения СН3-группы в ароматич. ядро по Фри-
делю—Крафтсу; сульфирует нек-рые ароматич. соеди-
нения (трифениламин, эфиры фенолов). Д. полу-
чается с выходом до 95% при медленной перегонке
в вакууме смеси СН3ОН и 60%-ного олеума (1 : 4
по весу); из SO3 и диметилового эфира (СН3)2О; при
частичном хлорировании диметилсульфита и др. спо-
собами. Д. не имеет запаха, очень ядовит,
проникает в организм через органы дыхания, может
всасываться через неповрежденную кожу, работы
с ним ведут под тягой; для разрушения паров в воз-
духе рекомендуется аммиак, а для снятия с кожи и
аппаратуры — водный раствор аммиака.
Г. М. Цигуро.
ДИМЕТИЛТЕРЕФТАЛАТ (диметиловый эфир те-
рефталевой кислоты) СвН4(СООСН3)2, молекулярный
вес 194,19 — бесцветные кристаллы призматической
формы, т. пл. 141—142°; выше точки плавления
возгоняется; 1,63; в 100 г горячей воды раство-
ряется 0,33 г Д.; растворим в диэтиловом эфире,
умеренно растворим в холодном этиловом спирте. Д.
может быть получен при пропускании хлористого
водорода в суспензию терефталевой к-ты в метиловом
спирте или при нагревании терефталевой к-ты с ме-
тиловым спиртом в присутствии серной к-ты. Очистку
Д. производят сублимацией в токе инертного газа,
а также перегонкой в вакууме в присутствии этилен-
гликоля или MgO. Применяют Д. в качестве исход-
ного компонента для синтеза полиэфиров и в част-
ности полиэтиленгликольтерефталата (полиэтиленте-
рефталата). Последний перерабатывается на волокно
и пленку (см. Полиэфиры, Полиэфирные волокна).
О. Я. Федотова.
N-ДИМЕТИЛ-п-ФЕНИЛЕНДИАМИН СОЛЯНО-
КИСЛЫЙ (n-диметиланилин хлоргидрат), мол. в.
209,12 [H3NCeH4N(CH3)2]^Clj — белый или серова-
тый кристаллич. порошок; гигроскопичен, раство-
рим в воде и спирте; в кислых р-рах с окислителями
дает красное окрашивание; обладает сильным физио-
логии. действием (кровяной, сосудистый и нервпый
яд). Д. окисляется солями Fe3f в присутствии H2S
с образованием метиленовой голубой. Получают вос-
становлением n-нитрозодиметиланилина или метило-
вого оранжевого с помощью SnCl2. Применяют для
определения H2S и сульфидов V, С12, Вг2, озона в воз-
духе, в микроскопии для открытия оксидаз, а также
как внешний индикатор при титриметрич. определе-
нии бария с помощью хромата. в. г. Тип-цова.
ДИМЕТИЛФЛУОРОН (п-диметиламинофенилфлуо-
рон) C19H17O5N, мол. в. 363,39 — кирпично-красные
кристаллы с зеленым отливом, растворимые в подкис-
ленном этаноле и нерастворимые в воде; кислый
этанольный р-р — темно-красного цвета; окраска
сильнокислых водных р-ров — желтая, щелочных —
малиновая. Д. образует с Та окрашенное комплексное
соединение. Окрашенные р-ры стабилизируются же-
латином.
Д. применяется для фотометрия, определения малых
количеств Та после его отделения от сопутствую-
щих элементов экстракци-
ей в виде фторкомплекса
из сильнокислых растворов
фторидов. Для определения
используется 0,05%-ный
этанольный раствор Д. Ком-
плексное соединение Д. с
Та имеет максимум погло-
щения при Х=500 ммк, а
максимум поглощения само-
го Д. приХ = 470л4лтк. Фотометрически Та определяют
при X = 530 ммк. Пользуясь Д., определяют Та в
присутствии больших количеств Nb при добавлении
оксалата и Ti при добавлении Н2О2. Открываемый
минимум — 3 мкг Та в 10 мл р-ра (0,1 н. по НС1),
содержащего 0,4% оксалата аммония и 1 мл 1%-ного
р-ра желатины. Реагент пригоден для определения
ряда других элементов.
Лит.: Назаренко В. А., Шустова М. Б.,
Завод, лаб., 1957, 23, № 11, 1283; и х ж е, в кн: Сб. научных
трудов Гиредмета, 2, М., 1959, с. 51. И. П. Ефимов.
ДИМЕТИЛФОРМАМИД (формилдиметиламин)
(CH3)2NCOH, мол. в. 73,09 — бесцветная подвижная
жидкость со слабым специфич. запахом; т. пл. —61°;
т. кип. 153°/760 мм, 76°/39 мм', d9t 0,9683, d45 0,9445;
rift 1,4269; уд. электропроводность 1,83-10-в ом 1
(25°). Д. смешивается с водой, спиртом, ацетоном,
эфиром, сероуглеродом, галогеносодержащими и аро-
матич. соединениями; алифатич. насыщенные угле-
водороды растворяет лишь при нагревании; хорошо
растворяет ацетилен (при 25° 31,4 объема) и ацетиле-
новые соединения, винилакриловые смолы, нитроцел-
люлозу, многие красители и полупродукты для син-
теза красителей, красители для кожи; на холоду рас-
творяет лигнин. Д. перегоняется без разложения при
обычном давлении; в нормальных условиях не гидро-
лизуется; для гидролиза необходимо действовать из-
бытком горячей щелочи; не действует на обычно ис-
пользуемые конструктивные металлы.
Получают Д. взаимодействием метилформиата и
диметиламина при обычном или несколько повышен-
ном давлении: НСООСН3 -|- (CH3)2NH —>СН3ОН +
+ (CH3)2NCOH (промышленный метод), из диметил-
амина и окиси углерода под давлением (CH3)2NH +
+ СО -> (CH3)2NCOH (промышленный метод); при
действии смеси СО и NH3 на метанол или этиленгли-
коль при 300° и давлении от 300 до 1000 ат в при-
сутствии солей сильных оснований и слабых к-т
(напр., К- или Na-соли таких кислот, как муравьиная,
уксусная, угольная, борная и т. д.); в последнем слу-
чае наряду с Д. образуется монометилформамид (про-
мышленный метод). Д. очищают от примесей мономе-
тилформамида и метанола перегонкой в присутствии
малолетучих или нелетучих слабых к-т, напр. бор-
ной, мышьяковистой или солей бисульфита калия, или
тринатрийфосфата или же обработкой ионнообмен-
ными смолами с последующей перегонкой.
Д. применяют в качестве растворителя в процессе
формирования полиакрилонитрильного волокна («ни-
трона», «орлона»); в качестве селективного раствори-'
теля вместо ацетона при выделении ацетилена из
газовых смесей; для растворения красителей при кра-
шении кожи, бумаги, древесины, вискозы и др.;
для абсорбции HCI, SO2 и др. кислотных газов; в ка-
честве добавки для ускорения желатинизации при
произ-ве взрывчатых веществ; в смеси с дибутилформ-
амидом используется для растворения многих органич.
веществ.
Лит.: U 1 ш ап п, 3 Aufl., Bd7, Mllnchen—В., 1956,S. 676;
Общая химическая технология, под ред. С. И. Вольфковича,
т. 2, М., 1959; Вайсбергер А. [и др.], Органические
растворители, пер. с англ., М., 1958, с. 215, 436.
Л. С. Поваров.
1125
ДИМЕТИЛЦИКЛОГЕКСАН — ДИНИТРОРЕЗОРЦИН
1126
ДИМЕТИЛЦИКЛОГЕКСАН СвН10(СН3)2> мол. в.
112,21 — известны все 4 изомера; три из них суще-
ствуют в цис- и транс-формах. В таблице приведены
нек-рые физич. свойства Д.
Изомеры Т. пл., °C Т. кип., °C d’« 20 Пр
1,1- -33,49 119,54 0,7809 1,4290
1,2-
г<ис- -50,02 129,73 0,7963 1,4360
транс- 1,3- . . -88,19 123,42 0,7760 1,4270
цис- —75,57 120,09 0,7660 1,4249
транс- 1,4- -90,10 124,45 0,7847 1,4309
Нис- -87,43 124,32 0,7829 1,4297
транс- —36,92 119,35 0,7625 1,4209
Для всех изомеров теплоемкость Ср — в пределах
0,329—0,339 кал/молъ (25°); теплота сгорания
1245,65—1248,31 кал/г-град (жидк. 25°). Д. нераство-
рим в воде, растворим в спирте, эфире. При нагрева-
нии с Pt-катализатором Д. дегидрируется до соответ-
ствующего ксилола; при нагревании с А1С13 изоме-
ризуются. Д. можно получить каталитич. гидрирова-
нием соответствующих ксилолов; изомеризацией цик-
лоалканов состава С8 в присутствии А1С13 и др. спо-
собами. Д. присутствует в нефтях различных место-
рождений и является составной .частью моторных
ТОПЛИВ. Д. А. Кондратьев.
ДИМОРФИЗМ — см. Изоморфизм.
ДИМРОТА РЕАКЦИЯ — реакция электрофиль-
ного замещения атома водорода в ароматич. ядре
с образованием С—Hg-связи при взаимодействии аро-
матич. соединений с солями ртути: Aril HgX2 —►
—> ArHgX НХ; обычно осуществляется в воде,
спирте, уксусной к-те или сплавлением реагентов.
Чаще всего в качестве меркурирующего агента исполь-
зуют ацетат ртути; иногда вместо солей ртути приме-
няют желтую HgO. Меркурирование ароматич. соеди-
нений в зависимости от условий реакции может идти
по ионному и по радикальному механизму. Меркури-
рование ионизированными ртутными солями [ Hg(N О3)2,
Hg(C104)2] в растворах сильных к-т проходит по
ионному типу; при этом наблюдается ориентация,
обычная для электрофильного замещения:
NO2C3H5 4- HgC104 — jw-N03C6H4HgC104 4- Н+
Меркурирование слабодиссоциированным ацетатом
ртути в неполярных растворителях протекает гомо-
литически; независимо от характера имеющегося
в ядре заместителя образуются преим. о- и «-замещен-
ные изомеры:
.хОСОСНз
о-Сн3ОСО-С6Нг,: • Н + ОСОСН3 —-
—-o-CHgOCOHg-СбЩСОСНз + СИдСООН
Условия Д. р. широко варьируют в зависимости от
характера ароматич. соединения. Мета-ориентанты
в ароматич. ядре затрудняют реакцию. Кетоны мерку-
рируются без растворителя при темп-ре не ниже 150°,
углеводороды и их галогенопроизводные — выше 100°
и дают побочно полимеркурированные продукты
реакции; нафталин меркурируется легче. Фенолы реа-
гируют с Hg(OCOCH3)2 в воде при 100°. Амины мерку-
рируются уже на холоду:
CeH5NH3 4. Hg(OCOCH3)3 _ch3cqoh'
— CeHsNHHgOCOCH3 — n-CH3COOHgC6H4NH3
Фуран, тиофен, селенофен дают с солями ртути
а-меркурпроизводные при обычной темп-ре. Пиридин
меркурируется при нагревании с ацетатом ртути до
180°. Реакция открыта О. Димротом в 1900.
Ф. И. Величко.
ДИНАМИТЫ (нитроглицериновые взрывчатые ве-
щества) — бризантные взрывчатые вещества, содержа-
щие значительные количества нитроглицерина. Д. со-
стоят из смеси нитроглицерина с порошкообразным
наполнителем: инертным (кизельгур, тальк) — т. н.
гурдинамиты, или активным (древесный уголь, смесь
древесной муки с селитрой и т. п.) — т. н. смешан-
ные Д. В смешанных Д. нптроглицерин недостаточно
прочно связан с поглотителем и может быть легко
вытеснен водой. Значительно более устойчивы жела-
типдинамиты (А. Нобель, 1875), изготовляемые на
основе нитроглицерина, желатинированного неболь-
шим количеством (до 10%) коллоксилина (см. Нитро-
целлюлоза). Один из наиболее употребительных со-
ставов таких Д.— 62,5% нитроглицерина, 2,5% кол-
локсилина, 27% NaNO3 и 8% древесной муки. Рас-
твор в нитроглицерине 7—10% коллоксилина —
эластичная каучукоподобная масса — наз. грему-
чим студнеми является одним из самых мощ-
ных взрывчатых веществ (теплота взрыва 1550 ккал/кг,
скорость детонации ок. 8000 м/сек, расширение в свин-
цовой бомбе до 600 см3). Для понижения темп-ры
затвердевания Д. (замерзшие Д. менее чувствительны
к детонации и более опасны в обращении) в них
вводят динитраты гликолей (реже др. нитроэфиры или
нитросоединения). Д. применяют в виде патронов
(d — 2 — 3 см, I = 10 — 20 см), представляющих
собой бумажные гильзы, заполненные тщательно пере-
мешанной массой.
В конце 19 в. внедрение Д. произвело революцию
во взрывном деле; их применяли почти при всех видах
взрывных работ. Однако значительная опасность в об-
ращении и высокая стоимость Д. привели к их замене
более безопасными и дешевыми взрывчатыми веще-
ствами, гл. обр. аммиачно-селитренными (см. Аммо-
ниты). Б. Н. Кондриков.
ДИНЕЗИН [дипаркол, хлоргидрат М-(2-диэтилами-
ноэтил)-фентиазина] Ci8H23N2SC1, мол. в. 334,92 —
бесцветные кристаллы, т. <.
пл. 184—186°; хорошо рас-
творим в воде, спирте, хло- | О Й J HCI
реформе, нерастворим в эфи-
ре. Для колич. определения ।
спиртовой раствор Д. тит- CH2CH2N(C,H5)2
руют0,1 н. раствором NaOH
в присутствии фенолфталеина. Д. получают конден-
сацией фентиазина с Р-диэтиламиноэтилхлоридом.
Применяют Д. для лечения паркинсонизма, торзион-
ной дистонии и т. п.
Лит. см. при ст. Апрессин. А. Травин.
ДИНИТРОРЕЗОРЦИН C8H4O8N2, мол. в. 200,11 —
из четырех возможных известны два изомера: 2,4-ди-
нитрорезорцпн (I) и 4,6-динитрорезорцин (II).
Свойства 2,4-Д. 4,6-Д.
Т. пл., "С Плотность Константа диссоциации (25°) . Растворимость в воде, % . . . 147-148 8,85 • 10-i 0,63 (57,7°) 2,02 (76,5°) 213,7 1.786 (пики.) 1,781 (рентг.) 1,05 • 10 -4 6,0 • 10-S 0,008 (25°) 0,1 (96,3°)
4,6-Д. легко растворим в эфире, хлороформе и го-
рячей ледяной уксусной к-те, труднее — в бензоле,
спирте и кипящей воде, нерастворим в холодной воде;
растворимость в спирте 0,12 г в 100 мл (25°).
2,4-Д. при быстром нагревании вспыхивает. При
восстановлении сульфидом аммония или хлористым
1127
2,4-ДИНИТРОФЕНИЛГИДРАЗИН — ДИОКСАН
1128
оловом, в соляной или уксусной к-те получается
4-нитро-2-аминорезорцин; при нагревании с HNO3
(даже с очень разбавленной) образуется 2,4,6-три-
нитрорезорцин. Калие-
вая соль 2,4-Д. метили-
руется диметилсульфа-
том. 4,6-Д. при восста-
новлении металлич. или
хлористым оловом в со-
ляной к-те превращает-
ся в 4,6-диамйнорезор-
цин; смесью азотной и серной К-т нитруется до
2,4,6-тринитрорезорцина, но в конц. HNO3 ие изме-
няется даже при кипячении.
С простейшими алифатич. альдегидами 2,4;Д. и
4,6-Д. не реагируют. При действии паратолуолоуль-
фохлорида гидроксил замечается на хлор в 4,6-Д.,
но не в 2,4-Д.
Для получения 2,4-Д. резорцин нитрбзируют азоти-
стой к-той до 2,4-динитрозорезорцина, к-рый затем
окисляют до 2,4-динитрорезорцийа 50%-ной пере-
кисью водорода на холоду или 15%-ной при 40°.
Другой способ получения этого изомера состоит
в нитровании резорцина азотной к-той (d 1,5), сначала
до 2-нитрорезорцина; вторую ' нитрогруппу вводят
при 0°. Положения 4 и 6 предварительно защищают
введением сульфогруппы. 4,6-Д. получают нитрова-
нием диацетилрезорцина азотной к-той (d 1,48) при
10—15°. Свинцовые соли 2,4-Д. (нормальная и основ-
ная) и 4,6-Д. (основная) нрименяются в воспламени-
тельных зарядах капсюлей-детонаторов.
Л. И. Хмельницкий,-
2,4-ДИНИТРОФЕНИЛГИДРАЗИН CeHeO4N4, мол.
вес 198,14 — кристаллы красного цвета с фиолето-
NHNH вой флуоресценцией; т. пл. 194—198°
I 2 (с разл.); нерастворим в воде, мало рас-
1^Дг-NOa творим в спирте, эфире, сероуглероде,
L J бензоле, лучше растворяется в анилине,
этилацетате и в умеренно разб. мине-
^О ральных кислотах. При нагревании со
щелочами превращается в динитрофе-
нол. Получают Д. действием гидразина на 2,4-дини-
трохлорбензол. Применяется Д. для идентификации
альдегидов и кетонов по темп-рам плавления полу-
чающихся динитрофенилгидразонов, а также для ка-
чественного и колич. определения карбонильных
соединений. И. П. Ефимов.
ДИНИТРОФЕНОЛЫ CeH3(NOa)aOH, мол. вес
184,1 — бесцветные или желтоватые кристаллы, обла-
дающие сильными кислотными свойствами; известны
все шесть изомеров (см. табл.).
Положение нитрогрупп Т. пл., °C Константа дис- социации (25°)
2,3 144-145 1,3 • 10-S
2,4 113,1 108 8,3 • Ю-з •
2,5 0,7 • 10-5
2,6 64 1,7 • 10~4
3,4 134 4,3 • IO”8
3,5 126 2,1 • 10-’
* По другим данным 1,0 • 10~4.
Д.мало растворимы в холодной воде, лучше в горя-
чей, растворимы в органич. растворителях. Д. обра-
зуют соли со щелочными металлами и аминами. При
восстановлении нитрогруппы образуются соответст-
вующие амины. Практич. значение имеет 2,4-Д.,
продукт восстановления к-рого применяется в произ-ве
синтетич. красителей. Изомеры 2,5-, 2,3- и 3,4-Д. яв-
ляются индикаторами для колориметрии, определения
концентрации водородных ионов; 2,4-Д. — индикатор
при ацидиметрия, титровании (pH 2,6—4,2). Полу-
чают Д. нитрованием фенолов или нитрофенолов.
2,4-Д. производят в пром-сти омылением 2,4-динитро-
хлорбензола конц. р-ром NaOH; описано также по-
лучение Д. нитрованием бензола при 50° 50—55%-ной
HNO3 в присутствии солей ртути.
Д. обладают взрывчатыми свойствами, ядовиты,
сильно раздражают кожу и слизистые оболочки, резко
повышают обмен веществ в клетках и вызывают
сильное повышение темп-ры тела. Нек-рые алкилза-
мещевные 4,6-Д. применяют для борьбы с сорными
растениями (контактные гербициды), зимующими ста-
диями вредителей плодовых деревьев в период покоя
(инсектициды) и для искореняющего опрыскивания
(фунгициды). Практич. применение нашли: 2-метил-
4,6-динитрофенол (т. пл. 86,4°, ЛД60 50 мг/кг); 2-вто-
рично-бутил-4,6-динитрофенол (т. пл. 42°, ЛД50
60 мг!кг); 2-вторично-амил-4,6-динитрофенол (жид-
кость, ЛД50 60 ме/кг). Наиболее активен 2-вторично-
бутил-4,6-динитрофенол. Для этих же целей приме-
няют также 2-циклогексил-4,6-динитрофенол (т. пл.
106°, ЛД50 1 80 мг/кг). В гомология, ряду алкилдинит-
рофенолов физиология, активность возрастает при
переходе от метилдинитрофенола к бутилдинитрофе-
нолу; далее она падает.
Получают алкилзамещениые Д. нитрованием О-ал-
килфенолов нитрующей смесью или алкилированием
спиртами или непредельными углеводородами п-фенол-
сульфокислоты с последующим нитрованием алкил-
фенолсульфокислоты.
АЛкилзамещенные Д. чаще всего применяют в виде
хорошо растворимых в воде солей с щелочными метал-
лами или органич. аминами. К таким фенолятам для
улучшения смачиваемости растений обычно добавляют
небольшие количества поверхностно-активных ве-
ществ; для уменьшения взрывоопасности их часто
выпускают в виде 40—50%-ных разб. препаратов:
в качестве разбавителя применяется вода, неорганич.
соли (сульфат натрия), а иногда масла и др. органич.
растворители. В качестве инсектицида и фунгицида
для борьбы с сорняками применяется также продукт
нитрования нек-рых сланцевых фенолов под назва-
нием нитрофен. Нормы расхода алкилзамещен-
ных Д. весьма различны; так, расход 2-вторично-бу-
тил-4,6-динитрофенила 1—3 кг/га, других — почти
в 3 раза больше. Д. широко применяются в качестве
гербицидов в борьбе с павеликами.
Лит.: Фирц-Давнд Г. Э. и Б л а н ж е Л.,
Основные процессы синтеза красителей, пер. с нем.,М., 1957;
Ворожцов Н. Н., Основы синтеза промежуточных
продуктов и красителей, 4 изд., М., 1955; Мельников Н. Н.,
Баскаков Ю. А., Бокарев К. С., Химия герби-
цидов и стимуляторов роста растений, М., 1954.
Н. Н. Мельников.
ДИОКСАН (пара-диоксан, 1,4-диоксан, диэтилен-
диоксид), мол. вес 88,10 — бесцветная горючая
жидкость с легким запахом; т. пл.
11,80°; т. кип. 101,32°/760 мм; за-
висимость давления паров от темп-ры:
1g р = 7,8642—1866,Т/Т; dl» 1,03375;
пВ 1,42241; гкрит. 311,8’; дкрит. 49,4 ат;
теплота испарения 8,55 ккал/моль (при
/О.
Н2С
2l I
НЙС (ЗН2
т. кип.); т. всп. 5° (в закрытом сосуде); диэлек-
трическая проницаемость е25" 2,209; дипольный
момент р, 0,45 D; криоскопическая константа 4,63°;
вязкость 7]ао 1,31 спуаз.' Взрывоопасные смеси
с воздухом содержат 1,97—22,5% Д. (по объему).
Д. смешивается с водой, спиртом и эфиром; образует
азеотропные смеси с водой (т. кип. 87,8°, 81,6% Д.)
и со спиртом (т. кип. 78,13°, 9,3% Д.). По химич.
свойствам Д. подобен алифатич. простым эфирам;
устойчив к действию кислот, •' щелочей, металлич.
натрия, аммиака; образует оксониевые соединения,
напр. диоксандибромид С4Н8О2 • Вга и диоксансуль-
1129
ДИОКСИАЦЕТОН - ДИПИРИДИЛЫ
ИЗО
фотриоксид С4Н8О2 • SO3, используемые в качестве
Хромирующего, соответственно сульфирующего аген-
тов; при стоянии в Д. накапливаются взрывчатые
перекиси; хранят его над SnCl2 или FeSO4.
Д. получают: 1) перегонкой этиленгликоля с H2SO4:
2НОСН2СН2ОН -> О(СН2)2ОСН2СН2 4- 2НгО; 2) из ₽-
хлорэтилового эфира: (С1СН2СН2)2О -h 2NaOH —►
—>• O(CH2)2OCH2CH2-|-2NaCH-H2O;3) из окиси этилена:
।-----ч NaOH 1-----------1
2СН2СН2О------> О(СН2)2ОСН2СН2. Главные примеси в
технич. продукте: СН3СООН, СН3СНО.
Д. — отличный растворитель для ацетилцеллю-
лозы, смол, каучуков, минеральных и растительных
масел; в смеси со спиртом — для нитроклетчатки;
входит как растворитель в состав различных красок
и детергентов; используется при очистке нефтяных
масел. С тетранитрометаном в спирте Д. дает желтое
окрашивание.
Лит.: Вайсбергер А. [и др.], Органические раство-
рители, пер. с англ., М., 1958, с. 120. Ф. Л. Величко.
ДИОКСИАЦЕТОН (кетотриоза) СН2ОНСОСН2ОН,
мол. в. 90,08 — диоксикетон, простейшая кетоза.
Д. кристаллизуется в виде мономера (т. пл. 65—71°)
и димера (т. пл. 80°). Димер плохо растворим в спирте,
эфире и ацетоне; при нагревании он переходит в моно-
мер, к-рый хорошо растворяется в перечисленных
растворителях; в воде Д. растворим очень хорошо.
В растворах Д., так же как и другая триоза—глицери-
новый альдегид (в противоположность другим саха-
рам), не образует циклич. форм и не дает реакции
полуацетального гидроксила (не образует гликозида).
Д. дает реакции на кетоны и спирты, напр. восстанав-
ливает раствор Фелинга, дает цветные реакции с фено-
лами (резорцин, р*нафтол, салициловая и галловая
к-ты) и с алкалоидами (кодеин). С фенилгидразином
Д. образует фенилозазон. При восстановлении Д.
образуется глицерин. Получают Д. окислением гли-
церина Н2О2, NaOBr и др., а также при ферментатив-
ном дегидрировании глицерина нек-рыми микро-
организмами. В виде фосфорного эфира Д. является
промежуточным продуктом углеводного обмена (см.
Гликолиз и Брожение).
Лит.: The carbohydrates, ed. W. Pigman, N. Y., 1957;
M i c h e e 1 F., Chemie der Zucker und Polysaccharide,
2 Aufl., Lpz., 1956. Л. И. Линевич.
ДИОКСИВИННАЯ КИСЛОТА C4HeO8, НООС —
C(OH)2—С(ОН)2—СООН, мол. в. 182,09 — бесцветные
мелкие кристаллы, легко растворимые в воде; т. пл.
114—115° (с рази.); при нагревании водного р-ра
Д. к. разлагается с выделением СО2. Динатриевая
соль Д. к. кристаллизуется с 3 молекулами воды;
в 100 г воды растворяется 0,04 г (0°) динатриевой соли.
Д. к. можно получить окислением HNO3 конц. р-ра
винной к-ты. Применяется Д. к. для открытия и опре-
деления натрия — при прибавлении конц. р-ра Д. к.
к насыщенному р-ру соли натрия образуется кристал-
лич. осадок Na-соли.
Продукт взаимодействия Д. к. с фенилгидразином—
озазон — применяется в виде динатриевой соли
(I), мол. в. 330,28, светло-жел-
CeH5NH—N=C— COONa тые кристаллы, т. пл. 200°
C0H6NH-N=C-COONa (с Разл-)! в 100 г воды раство-
j ряется 2,0 г (20 ); в аналитич.
химии применяют для микро-
химия. определения Са и открытия Mg. В присут-
ствии Саа+ озазон при перемешивании стеклянной
палочкой образует осадок кальциевой соли. Эта
реакция применяется для отличия природной воды
от дистиллированной и для определения чистоты
дистилл. воды. Озазон применяется также для микро-
химия. определения Са и открытия Mg. Конденсацией
Д. к. с фенилгидразинсульфокислотой получают тар-
тразин— желтый краситель для шерсти из группы
пиразолоновых красителей. И. П. Ефимов.
ДИОКСИФЕНИЛАЛАНИН [ Р-(3,4-диоксифенил)-
а-аланин, а-амино-[}-(3,4-диоксифенил)-пропионовая
кислота] CeHjjO4N, мол. в. 197,17— сн qhCOOH
бесцветные кристаллы; т. пл.: L-изо- i г|
мера 285,5°, D-изомера 288°, D,L-изо-
мера 271—272° (с разл.); [а] у.' L-изо- I
мера—12,7° (в 4%-й НС1) и > 39,5° 4^°*^
(в воде), D-изомера 4-11,9° (в 4%-й он
НС1). Д. плохо растворим в воде,
бензоле, сероуглероде, растворим в разбавленных
р-рах минеральных к-т, нерастворим в этаноле, эфире,
хлороформе, уксусной к-те и петролейном эфире. Д.
окисляется на воздухе в нейтральных и слабощелоч-
ных р-рах, образуя окрашенные продукты. Р-ры Д.
дают цветные реакции с NaOH, FeCl3, фосфорноволь-
фрамовой к-той и реактивом Миллона; восстанавли-
вает AgNO3, реактив Фелинга и др. При окислении в
организмеД. образует меланин ы—бурые пигмен-
ты кожи и волос. Д. можно выделить в небольших
количествах при эцзиматич. окислений L-тирозина;
синтетически Д. получают диазотированием 3-амино-
тирозина нитритом в разбавленной H2SO4. Д. является
промежуточным звеном при биосинтезе адреналина
из тирозина. л. П. Алексеенко.
ДИОКТИЛСЕБАЦИНАТ (ди-н-октиловый эфир
себациновой кислоты) С8Н17ООС—(СН2)8—СООС8-Н17,
мол. в. 426,69 — бесцветная жидкость со слабым запа-
хом; т. пл. —55°; т. кип. 248/4 мм; d20 0,91 3; «у
1,4496; г;20 19,65 сст , т|80 4,04 ест.; т. воспл. 213°;
растворимость в воде 0,025%; растворяется в боль-
шинстве органич. растворителей. Д. получают эте-
рификацией себациновой к-ты н-октиловым спиртом
(катализатор — бенэолсульфокислота). Применяют Д.
в качестве пластификатора поливинилхлорида и сопо-
лимеров винилхлорида с винилацетатом и др. моно-
мерами; придает пленкам на основе этих полиме-
ров гибкость при низких температурах, пластифици-
рует также простые эфиры целлюлозы и природ-
ные смолы за исключением шеллака. Д. стоек к на-
греванию и действию света, однако легко экстраги-
руется из пленки маслом, поэтому не может приме-
няться в тех случаях, когда необходима маслостой-
кость.
Лит. ей. при ст. Диаллилфталат. О. Я. Федотова.
ДИОКТИЛФТАЛАТ (ди-н-октиловый эфир о-фта-
левой кислоты) С8Н4(СООС8Н17), мол. в. 390,46 —
бесцветная жидкость; т. пл. —40°; т. кип. 340°;
«у 1,482; т]20 40 спуаз; т. воспл. 219°; давление паров
0,01 мм (150°); летучесть 0,25% за 24 часа при 105°;
растворим в петролейном эфире, бензине и мине-
ральных маслах, нерастворим в воде; в Д. раство-
ряется 0,6% воды (25°); Д. — устойчивый высококипя-
щий пластификатор, высокой светостойкости. Полу-
чают Д. этерификацией о-фталевого ангидрида н-окти-
ловым спиртом в присутствии бензолсульфокисло-
ты. Применяется для пластификации полистирола,
поливинилбутираля, смешанных и простых эфиров
целлюлозы, поливинилхлорида и его сополимеров
с винилацетатом и другими винильными соедине-
ниями.
Лит. см. при ст. Диаллилфталат.
ДИПИРИДИЛЫ (C5H4N)2, мол. в. 156,18 — орга-
нич. соединения, содержащие в молекуле два пириди-
новых ядра; возможны 6 изомерных Д.; из них наи-
более доступны а, а - и Y, у'-изомеры. Д. получают:
взаимодействием соответствующих бромпиридинов с
порошком меди (Ульмана реакция); нагреванием
пиридина с FeCl3, Na или NaH; действием на пири-
дин Zn-пылью и (СН3СО)2О, окислением фенантро-
линов. Д. реагирует. подобно пиридину. Бромзаме-
щенные Д. можно превратить по Ульману в три-,
тетра- и пентапиридплы.
1131
ДИПОЛЬНЫЙ МОМЕНТ
1132
а,а'-Д ипиридил (2,2'-дипиридил), бесцвет-
ный кристаллич. порошок; т. пл. 69,5°; т. кип. 272,5°;
в 100 г воды растворяется 0,5 г
Д.; хорошо растворим в спирте,
эфире, бензоле и хлороформе.
Константа диссоциации рА'(НА+)
при 25° в 0,1М р-ре KNO3 равна
4,43; в 0,ЗЗМ р-ре KNO3 4,43.
Константы устойчивости (ступенчатые) комплексов с раз-
личными катионами, определенные при 25° в среде 0,1 М
р-ра KNO3, равны:
Катионы 1g к, 1g If.
С<Р+ 4,5 3,5
Fe2+ а 4,43
Zn2+ 5,4 4,4
а В среде 0,33М р-ра.
С солями Fe2+ образует след, комплексный ион
Ре(С10Н8Кг)з+, интенсивно окрашенный в красный
цвет; этот комплекс осаждает анионы CI()4 ,Br ,
J- и др.; применяется для цветных и микрокристалло-
скопич. реакций на элементы, способные образовы-
вать комплексные анионы (Cd, Zn, Та, Hg, Bi и др.).
Применяют Д. для качеств, и количеств, определения
железа; комплекс Д. с Fe2+ применяют как окисли-
тельно-восстановительный индикатор (Z?o = + 1,33 в).
у,у'-Д ипиридил (4,4'-дипиридил), бесцветный
кристаллич. порошок; т. пл. безводного Д. 114°,
дигидрата 73°; т. кип. 304,8°;
сублимируется; мало растворим
в холодной воде, лучше в горя-
чей; растворим в спирте, эфи-
ре, хлороформе. у,у'-Д. не дает
красного окрашивания с Fe2+, можно применять как
окислительно-восстановительный индикатор, прини-
мающий под действием восстановителей сине-фиолето-
вую окраску; Ео = —0,44 в (pH 8—12). у,у'-Д. обла-
дает инсектицидными свойствами, действуя как кон-
тактный и внутренний яд. в. Г. Типцова.
ДИПОЛЬНЫЙ МОМЕНТ — произведение заряда
на расстояние (ц=ег) в диполе, т. е. в системе, состоя-
щей из двух равных по величине и противоположных
по знаку зарядов,-Ье и —е, находящихся на нек-ром
расстоянии г друг от друга. В данной статье рассмат-
риваются только постоянные Д. м. полярных моле-
кул, характеризующие электрич. асимметрию их
строения. Д. м. —- векторная величина; в химич.
литературе ее направление принято считать от плюса
к минусу. Заряды в молекулах имеют порядок вели-
чины 1010 электростатич. единиц (заряд электрона
равен 4,8 • 10 10 электростатич. единиц), а межатом-
ные расстояния — 10 8 см, поэтому порядок вели-
чин Д. м., измеряемый в дебаях (Л), составляет
10-10 • 10-8 = 10‘18 электростатич. единиц.
П. Дебай положил начало статистич. теории пове-
дения полярных диэлектриков (газов и неассоцииро-
ванных непроводящих жидкостей) в электрич. поле.
Теория Дебая просто постулирует наличие постоян-
ных Д. м. у молекул таких диэлектриков, но не объяс-
няет их происхождения. Согласно этой теории, в от-
сутствии электрич. поля направления векторов Д. м.
молекул распределены равномерно; внешнее электрич.
поле стремится ориентировать их, но вследствие теп-
лового вращения молекул устанавливается не пол-
ная их ориентация, а лишь неравномерное статистич.
распределение с преимущественной ориентацией Д. м.
вдоль линий поля. Кроме того, вследствие поляризую-
щего действия поля на упруго-связанные заряженные
частицы в молекулах (электроны и атомы) происходит
их сдвиг, и молекулы приобретают наведенный Д. м.;
последний исчезает при снятии поля, и его следует
отличать от рассматриваемых здесь постоянных Д. м.
Характеристикой способности молекул полярных
диэлектриков поляризоваться во внешнем электрич.
поле служит молярная поляризация Р, к-рая, в соот-
ветствии с изложенным выше, состоит из трех частей:
ориентационной (Рор ), атомной (Рат ) и электронной
(Рэл ) поляризации:
^ = Л,р.+ ^ат.+ ^эл.
Происхождение постоянного дипольного момента
молекул. Природу сил, удерживающих частицы в мо-
лекуле в состоянии электрич. асимметрии и приводя-
щих к наличию у молекулы постоянного Д. м., можно
понять лишь с точки зрения квантово-химич. теории.
До привлечения квантовой теории происхождение Д. м.
двухатомных молекул, а также Д. м. отдельных химич. свя-
зей многоатомных молекул объяснялось различием в электро-
отрицательностях атомов, образующих связь. Вследствие Этого
различия пара валентных электронов смещена в сторону более
электроотрицательного атома, и центр зарядов электронов
оказывается смещенным относительно центра положительных
зарядов ядер (положение центра зарядов определяется так
же, как для центра тяжести, с заменой величин масс величи-
нами зарядов). Такие связи наз. полярными или «частично
ионными». В простых симметричных молекулах, образован-
ных из двух одинаковых атомов (Н2, О2, N2, С12 и др.), центры
зарядов обоих знаков совпадают, и Д. м. равны нулю.
В двухатомных молекулах с неодинаковой электроотрицатель-
ностью атомов (НС1, НВг, JC1 и др.) центры зарядов не сов-
падают; такие полярные молекулы электрически несиммет-
ричны и имеют Д. м. Сложные многоатомные молекулы, от-
дельные связи к-рых могут быть полярны, как правило, не
имеют Д. м., если у них есть центр симметрии ГСН4, СС14,
SiFe, CeHe, n-CeH4Cl2, l,3,5-CeH3(NO2)3 н др.’], и имеют
Д. м., если центр симметрии отсутствует (Н2О, NH3, СН8С1,
о-СвН4С12 и др.). Однако электроотрицательность атомов
является не единственной причиной существования Д. м.
С помощью квантовой теории Паулингом, Мулликеном, Кол-
соном были установлены еще три фактора, определяющие
появление Д. м. химич. связей.
1. В результате различия объемов атомов, образующих
связь (вернее, объемов, ограниченных атомными электронными
орбитами), может появиться в чисто-ковалентной связи
т. наз. гомеополярный диполь (М-гомЛ Наглядно
появление гомеополярного Д. м. можно представить т. обр.,
что при образовании связи между атомами разных размеров
центр пространства, расположенного внутри области пере-
крывания атомных орбит, образующих молекулярную свя-
зывающую орбиту, оказывается смещенным по отношению
к центру зарядов ядер. Зависимость величины гомеополяр-
ного Д. м. от соотношения (К) зарядов ядер атомов пока-
зана на рис. (именно это
соотношение главным об-
разом
шения
орбит,
речь). Когда связь обра-
зуется между атомными ор-
битами одинаковых разме-
ров (К=1), цгом ==0. Чем
больше различие в заря-
дах, т. е. в размерах ато-
мов, образующих связь,
тем больше гомеополярный
Д. м. У НС1, например, он
близок к 1 D.
2. В ряде случаев химич.
связи между атомами обра-
зуются не за счет «чистых»
8-, р- или d-электронов, а
с участием электронов,
находящихся в более слож-
ных состояниях, описы-
ваемых гибридизованными
ность, Химическая связь).
менением симметрии электронных орбит в атомах, что обусло-
вливает появление т. наз. атомного диполя. Так,
четыре симметричные (относительно ядра) электронные орбиты
(в, Рх> Ру, Рг) атома углерода образуют четыре эквивалентных
орбиты sp3, каждая из к-рых не симметрична относительно
ядра. Такое же положение имеет место у атома углерода в со-
стояниях sp2,sp, а также у нек-рых других атомов. Д. м. химич.
связи, обусловленный асимметрией атомных орбит, может
в нек-рых случаях достигать значительной величины. Раз-
личие Д. м. связей С—Н в метане, этилене и ацетилене, атомы
углерода в к-рых, соответственно, находятся в состояниях
гибридизации sp3, sp2 и зр, обусловлено в значительной мере
различием атомных Д. м.
3. Вследствие гибридизации появляется асимметрия
электронного облака не только валентных элек-
тронов, но также инеподеленных электронных
(К) зарядов ядер атомов пока-
0.6
2,5
2.2
К
определяет соотно-
размеров атомных
о к-ром идет здесь
Зависимость гомеополярного дп-
польного момента (ц) от отноше-
ния зарядов (К).
функциями (см. Атом, Валент-
Гибридизация сопровождается из-
1133
ДИПОЛЬНЫЙ МОМЕНТ
1134
п а р. В ряде случаев (в соединениях азота, кислорода, серы
и др.) неподеленные электронные пары принимают непосред-
ственное участие в образовании химич. связей, причем состоя-
ние этих неподеленных пар изменяется и описывается уже не
симметричными или фр-функциями, а гнбридизованной
(ф8 4- афр)-функцией, асимметричной по отношению к ядру.
Д. м., появляющийся в результате асимметрии орбиты непо-
деленной пары электронов, может достигать значительной
величины (1,5—2 О) и в ряде случаев оказывать существенное
влияние на суммарный Д. м. молекулы.
Дипольные моменты отдельных связей и групп и
их векторное сложение в многоатомных молекулах.
Д. м. многоатомной молекулы является векторной
суммой всех Д. м. отдельных химич. связей, имею-
щихся в структуре молекулы (правило векторной
аддитивности Д. м. связей). Большинство Д. м. свя-
зей, приводимых в литературе, вычислено не теорети-
чески (абс. расчет здесь очень труден), а путем сопо-
ставления опытных Д. м. ряда молекул. Векторные
суммы Д. м. связей хорошо совпадают с опытными
Д. м. в тех случаях, когда взаимное влияние атомов,
связей и групп проявляется достаточно слабо. В этих
случаях имеет место аддитивность Д. м. отдельных
связей и, следовательно, оправдано приписывание
каждому виду химич. связи определенного вектора
Д. м.,' сохраняющего приближенное постоянство
в разных соединениях. Здесь имеется аналогия с вычи-
слением молекулярной рефракции и теплоты обра-
зования таких молекул в виде суммы нек-рых постоян-
ных инкрементов, приписанных отдельным видам
связей. В молекулах с сильным взаимным влиянием
наблюдаются отклонения от правила векторной адди-
тивности, если для Д. м. связей берутся средние
значения, характерные для связей в молекулах адди-
тивного типа. Это используется для суждения о допол-
нительной поляризации в подобных молекулах за
счет т. наз. «мезомерных» сдвигов электронов, о стери-
ческом влиянии и других явлениях.
Д. м. молекул всех парафинов (в т. ч. изо- и цикло-
парафинов) оказались равными или близкими нулю
(Д. м. изобутана, напр., равен всего 0,13 D). Этого и
следовало ожидать из тетраэдрич. симметрии атомов
углерода и практически одинаковой полярности всех
связей С—Н в парафинах (связи С—С в них при-
знаются неполярными). Молекулы ненасыщенных и
ароматич. углеводородов с центром симметрии (С2Н2,
С2Н4, СвНв, нафталин, антрацен, дифенил), естест-
венно, не имеют Д. м. Оправдалась на опыте предска-
занная квантово-химич. расчетом слабая полярность
несимметричных ароматич. углеводородов (фульвен,
азулен).
Распределение общего Д. м. молекулы по Д. м.
отдельных связей носит отчасти условный характер;
так, обычно считают, что Д. м. сосредоточен в одной
или нескольких наиболее полярных связях; напр.,
в случае производных углеводородов условно счи-
тается, что весь Д. м. молекулы сосредоточен в поляр-
ном заместителе, а Д. м. углеиодородного остатка
равен нулю. Это, во-первых, означает, что приписы-
ваемый полярному заместителю Д. м. равен раз-
ности истинного Д. м. заместителя и Д. м. замещенной
им связи С—Н, а во-вторых, что все вызванные заме-
щением дополнительные эффекты поляризации угле-
водородного остатка уже включены в Д. м., припи-
сываемый заместителю. Отсюда получаются разные
ряды значений Д. м. заместителей в ароматич. соеди-
нениях, парафинах и т. п.
Многоатомным заместителям, содержащим несколько
полярных связей, обычно приписывается некий
общий (суммарный) вектор Д. м., имеющий не
только определенную постоянную величину, но и
определенное направление. Для т. наз. «регулярных»
групп — заместителей (—NO2, —CN, —СС13, все гало-
гены), направление Д. м. совпадает с осью группы,
т. е. с направлением ее связи с углеводородным ядром.
У «нерегулярных» заместителей (группы —СООН,
—COOR, —COR, —СНО, —ОН, —OR, —NH2,
—NHR, —NR2, —CH2Hal, —CHHal2 и др.) вектор
Д. м. составляет нек-рый угол (<р) с этим направле-
нием (от углеводородного ядра к заместителю). Угол
нерегулярности <р не следует смешивать с валентным
углом центрального атома группы. Нерегулярная
группа может совершать свободное или заторможен-
ное вращение вокруг своей оси, и в расчетах вектор-
ных сумм Д. м. связей этот факт следует учитывать.
Таблица 1. Дипольные моменты
соединений алифатического ряда
Соединение Диполь- ный мо- мент, D Соединение Диполь- ный мо- мент, D
Спирты Кетоны
СН3ОН 1,67 СН„СОСН3 2,73
С2Н5ОН 1,70 С2Н5СОСН3 2,76
H-C3H7OH 1,66 н-С3Н7СОСН3 2,71
U3O-C3H7OH 1,65 н-С4Н9СОСН3 2,70
н-С1Н9ОН 1.66 н-С|)Н1ЯСОСН3 2,70
н-СеН13ОН 1,64 H-(Jg Н j gUUCiig 2,69
н-С7Н15ОН н-С5НпСНОНСНя 1.70 1.70 Хлористые алкилы
н-С, Н, СНОНСг н5 1.70 2,03
(н-С3Н7)2СНОН 1,70 CgUgGi
h-CsHi7OH 1,67 H-U3H7CI 2,15
1.60 H-UiHsUl 1,90
1.63 н-С^НцС! 1.92
h-Ci2H25OH 1,62 Н-С7Н j 5 <J1 н-С5НпСНС1СН8 1,85 2,03
(h-C3H7)2CHC1 2,04
Таблица 2. Дипольные моменты полярных групп
для днзамещенных бензолов типа X — С6Н4 — У
Группы Дипольные моменты, D
X Y мета- пара-
опит расчет опыт расчет
С1 Cl 1,48 1,58 0 *0
Вг Вг 1.50 1,52 0 0
J J 1,27 1,30 0 0
no2 NO, 3,79 4,01 0 0
Cl Br 1,52 1,55 0 0,06
Cl J 1.14 1,46 0,5 0,28
Ci no2 3,39 3,49 2,55 2,43
Вг NO, 3,41 3,49 2,61 2,49
сн, no2 4,17 3,82 4,44 4,41
Таблица 3. Дипольные моменты полярных групп
в ряду парафиновых и ароматических углеводородов
Группа Дипольные моменты, D
С2Н5 с6н5
пары раствор пары раствор ф°
F 1,92 __ 1,57 1,43 0
С1 2,05 1,8 1,72 1,58 0
Вг 2,01 1,9 1,75 1,52 0
J 1,87 1,8 1,60 1,30 0
СН3 0 — 0,36 0,4 0
ОСН3 —— — 1,35 1,25 66
он 1,69 1,7 1,4 1,45 66
nh2 1,31 1,39 1,48 1,53 48
N (СН3)2 — — 1,61 1,58 34
СООН 1,73 1.7 — 1,7 ——
СООСН3 1,76 1.9 — 1.9 —
СНО 2,73 2,5 (3,1) 2,8 ——
СОСН3 2,78 — 3,01 2,9
CN 4,0 3,57 4,39 4,02 0
NO3 3,68 3,3 4,21 4,01 0
NO — — — 3,14
Многочисленные опытные данные подтверждают,
что изложенный выше метод вычисления Д. м. моле-
кул по правилу векторной аддитивности Д. м. связей
применим к молекулам аддитивного типа. Так, одно
из основных допущений этого метода, что Д. м. сосре-
1135
ДИПОЛЬНЫЙ МОМЕНТ
1133
доточен в полярных группах, доказывается тем, что
Д. м. алифатич. производных почти не зависят от
длины и разветвленности алифатич. радикала (см.
табл. 1). Векторные суммы Д. м. регулярных поляр-
ных групп в двузамещенных бензолах (табл. 2)
весьма близки к опытным значениям, если рассмат-
ривать только те случаи, когда отсутствует сильное
взаимное влияние (напр., орто-эффект или мезомер-
ный сдвиг электронов).
В табл. 3 дана сводка принятых значений Д. м.
полярных групп для парафинового и ароматич. ря-
дов. Опытная величина Д. м. зависит от того, измерена
ли она для газов или для растворенных веществ, а
также ет рода растворителя. Поэтому в табл. 3 даны
2 ряда значений, соответственно для паров и для ве-
ществ, растворенных в бензоле. Сравнение вычислен-
ных и опытных Д. м., строго говоря, имеет смысл
только в пределах одного из этих рядов, но не между
рндами. Значения углов нерегулярности (ср) вычис-
лены из Д. м. пара-производных типа X—СвН4—Y,
где X — данный заместитель, a Y — какой-либо
регулярный заместитель, не взаимодействующий с X.
Применение метода Д. м. при различных исследо-
ваниях строения молекул, природы химической связи
и взаимного влияния связей, атомов и групп в моле-
кулах. Метод Д. м. используется гл. обр. для выяс-
нения типа химич. связи (полярная, ковалентная),
геометрии строения молекул (изомерия, валентные
углы и др.), свободы и заторможенности вращения
атомных групп, взаимного влияния атомов и групп
(мезомерныи сдвиг электронов, орто-эффект и др.),
а также для обнаружения донорно-акцепторных свя-
зей в комплексах.
Таблица 4. Дипольные моменты Изомеров
Соединение Дипольные моменты, D
Дигалогениды этилена UUC~ транс-
1,2-дихлорэтилен 1,2-дибромэтилен 1,2-дииодэтилен 1,8 1.35 0,75 0 0 0
Комплексы платины a ₽
Ptci, t(C1H5),SlI PtCl3 [(CgHjJjSJj 9,5 9,0 2,41 2,35
Дизамещенные бензола орто- мета- пара-
С0Н.С1, CgH.Br C6H.Ja CeH4 (NO3)3 СД(ОН), .' C6H. (OCH3). C,H.(NH,)3 C6Ht (COOCH3)2 ClCeH.Br ClCeH.J C1CoH.no, BrCoH.NO. CH3CeH.NOa CH.CeH.OH CH3C„H.OCH3 NO,CeH.NH, 2,27 2,0 1,69 6,0 2,16 1,31 1,44 2,5 2,2 1,85 4,30 4,20 3,66 1,41 1,0 4,35 1,48 1,50 1,27 3,79 1,59 1,79 1,52 1.14 3,39 3,41 4,17 1,54 1,17 4,85 0 0 0 0 2,47 1,73 1,55 2,2 0 0,5 2,55 2,61 4,44 1,57 1,20 6,2
Для выяснения геометрия, структуры молекул вычисляют
Д. м. для различных моделей молекул по правилам сложения
векторов Д. м. связей и сравнивают его с экспериментальной
величиной. За правильную структуру принимается та, для
к-рой рассчитанный момент наиболее близок к эксперименталь-
ной величине. Так, наличие Д. м. у молекул НаО, H3S, H3Se,
SCla, SOj, (CH3)aO и др. подтвердило, что эти молекулы имеют
/Н
не линейную (И -> О >- Н), а угловую (Off ) конфигура-
ХН
цию; валентные связи атомов направлены под углом ~ 100°.
Данные о Д. м. позволили подтвердить пирамидальную кон-
фигурацию молекул NH3, N(CH3)3, SbCl3 и др. Хорошей ил-
люстрацией значения метода Д. м. для идентификации изо-
меров могут служить величины Д. м., приведенные в табл. 4.
Вследствие полной компенсации Д. м. связей С—Hal в транс-
изомерах опыт дает для Д. м. значение нуль. У цис-изомеров
два вектора Д. м.связи С—Hal складываются под углом 120°,
давая заметный момент. Аналогичное положение наблюдается
и для а- и р-изомеров в комплексах платины. По Д. м. можно
идентифицировать орто-, мета- и пара-изомеры, если замести-
тели регулярны (<р = 0). Для этого экспериментальная вели-
чина Д. м. сравнивается с рассчитанной по формуле:
р. = |/тЗ + т| -I- 2m1ms cos О
где т, ит, — Д. м. замещающих групп (табл. 3), а в — угол
между ними (60° — орто-, 120° — мета-, 180° — пара-).
Метод Д. м. весьма эффективен при идентификации тау-
томеров. Так, для молекулы известного ускорителя вулкани-
зации каучука — 2-меркапто-бензтнозола — возможны две тау-
томерные структуры:
aNX r^V^N\H
с-SH и [ I C“S
I n
Вычисленный Д. м. для I равен 2,0 D, для II — 4,5 D. Экспе-
риментально найденный Д. м. 4,67 D однозначно свидетель-
ствует в пользу того, что в бензольном растворе вещество имеет
структуру II. Аналогичные примеры можно привести для син-
й анти-изомеров диазосоединений, для полпзамещенных аро-
матич. производных и т. д.
В случае нерегулярных заместителей, когда возможно их
вращение вокруг осей, расчет векторных сумм требует не
только знания величин Д. м. групп и углов между их осями,
но и учета поворота вектора Д. м. каждой группы вокруг ее
оси относительно некоей нулевой плоскости в молекуле, . а
также знания потенциальных барьеров вращения для этих
групп. Проще всего вопрос решается для полностью затор*
моженного вращения, когда барьер много больше средней
тепловой энергии на одну степень свободы (|/г kT). Тогда
расчет ведется для жесткой, энергетически наивыгоднейшей
конфигурации молекулы, принятой для нее из структурных
соображений. С этим случаем встречаются, иапр., когда
сопряжение двойных связей в ядре и заместителе делает
энергетически наивыгоднейшим их компланарное (плоское) рас-
положение (стирол, бензальдегид и др.). В идеализированном
случае полностью свободного вращения (высота барьера равна
нулю) измеренный Д. м. представляет статистически усред-
ненную по всем возможным конфигурациям величину (ix).
Расчет его ведется путем усреднения квадрата моментального
значения векторной суммы Д. м. групп по всем азимутам их
вращения (ввиду того, что в формулу Дебая входит квадрат
Д. м.). В реальном случае заторможенного вращения Д. м.
зависит от темп-ры, и сравнение измеренных при ряде темп-р
Д. м. с результатами расчета для модели со свободно вращаю-
щимися группами позволяет делать выводы о характере вра-
щения этих групп, высоте барьера и т. п.
Для палочной модели молекулы с двумя нерегулярными,
свободно вращающимися группами на концах Л. Мейер (1930)
вывел формулу:
р. = V(т^ cos -j- m-j cos ?2)а -j- (mi sin ?i)2 -p (m2 sin
где тг и ma — величины Д. м., a (pi и ср8 — углы нерегуляр-
ности полярных групп. Эта модель и формула применялись
при исследовании свободного вращения в 1,2-дигалогенэтанах
в парах в широком интервале темп-p, причем предлагались
также формулы, выведенные с учетом высоты барьера враще-
ния.
Для более общей модели молекулы в виде ядра с присоеди-
ненными к нему заместителями, когда ядро и оси этих групп
образуют жесткую систему, а векторы Д. м- нерегулярных
групп свободно вращаются вокруг своих осей, Цан (1932)
обобщил эту ф-лу след, образом:
? = |/~т=+
где т0 — векторная сумма осевых составляющих Д. м. не-
регулярных групп, Д. м. ядра и Д. м. всех регулярных групп,
а второй член — квадрат алгебраич. суммы кзадратов пер-
пендикулярных составляющих Д. м. всех п нерегулярных
групп. К случаю, описываемому этой моделью и формулой,
можно отнести все полизамещенные ароматич., гетероциклич.
н циклопарафиновых соединений.
Для модели молекулы в виде длинной зигзагообразной
цепочки, нек-рые связи к-рой полярны, где возможно свобод-
ное вращение любой части молекулы вокруг каждой связи,
как вокруг оси (при сохранении постоянными всех валентных
углов), средний квадрат результирующего Д. м. равен, по
Г. Эйрингу (1932), сумме квадратов Д. м. полярных связей
цепочки плюс всевозможные удвоенные парные произведения
этих Д. м. на произведения косинусов всех углов, на к-рые
1137
ДИПОЛЬНЫЙ МОМЕНТ
1133
изменяют свое направление жесткие связи, соединяющие
данную пару Д. м. связей, при переходе от одного диполя
к другому. Смайс (1949) применил это правило к расчету д. м.
3» (г-дихлорэтилового эфира и получил 2,58 D в полном согла-
сии с опытным значением 2,57 D.
Таблица 5. Дипольные моменты молекулярных соединений
донорно-акцепторного типа
Соединение Дипольный момент
соедине- ния, ц донора, ц' ц—И'
А1С13 • (С2Н5)2О ....... 6,45 1,14 5,31
А1С13 C,H5NH2 6,82 1,39 5,43
А1С13 • CeH5NO2 8,99 4.01 4,98
А1Вг3 • (С2Н5)2О 8,39 1.14 5,25
А1Вг3 • CjjHsN 8,03 2.23 5,50
А1Вг3 • (С«Н5)2О BF3 • (С2Н5)2О 6,56 1,14 5,42
4,92 1,14 3,78
ВС1„ • (СаН5)2О ВС13 • CjjHjCN 5,96 7,64 1.14 3,57 4,82 4,07
GaCl, • C6H5CN 8,65 4,02 4.63
GaCl,-n= CHaC6H1NO2 . . 9,16 4.44 4,72
'lid* • U2H5UW • 6,05 3,57 2,48
TiCl. - CeHjCN 6,16 4,02 2,14
Метод Д. м. оказался весьма чувствительным индикатором
на образование донорно-акцепторных связей в комплексах.
В табл. 5 приведены Д. м. ряда таких комплексов; в последней
графе указан прирост Д. м. (ц—р?) в этих молекулярных соеди-
нениях за счет появления новой связи донорно-акцепторного
типа (сам акцептор — галогенид металла — построен симмет-
рично и не имеет Д. м. в свободном состоянии).
Если положенная в основу расчета векторной суммы Д. м.
связей и групп геометрич. модель молекулы хорошо обосно-
вана и не вызывает сомнений, и тем не менее наблюдается за-
метное расхождение между этой суммой н опытным Д. м.
молекулы, тогда найденная разность может быть использована
для суждения о дополнительном взаимном влиянии атомов
и групп в молекуле, не учтенном при расчете суммы д. м.
связей. Напр., Д. м. пара-нитроанилина равен 6,19 D; век-
торная сумма составляет 5,16 D. Прирост полярности молекулы
с 1,03 Й, несомненно, связан с дополнительным мезомерным
сдвигом электронов, возникающим только в этой молекуле и
отсутствовавшим в молекулах анилина и нитробензола, из
Д. м. к-рых были взяты составляющие вектора Д. м. групп
КН2 и NO2:
оч т /“Л
Примеров такого использования метода Д. м. очень много.
Накопление полярных связей или групп у одного и того
же центрального атома (напр., при переходе СН3С1 -► СН2С1, —
-► СНС13) приводит, как правило, к уменьшению полярности
втих связей, и метод Д. м. также позволяет вынести суждение
о величине этого эффекта при должном учете другого влияю-
щего фактора — изменения валентных углов. В тех случаях,
когда известны Д. м. реагирующих молекул и их суммарные
концентрации, можно использовать измерения Д. м. для су-
ждения о Д. м. комплексов и о равновесии между комплексом
и его компонентами в растворе, о степени ассоциации и поляр-
ности ассоциатов в растворах, о сольватации полярных моле-
кул молекулами растворителя, а также следить за протеканием
реакций в растворах. Растворитель в этих случаях должен
быть неполярным. В качестве примера можно указать на иссле-
дование равновесия комплексообразования в системе хлоро-
форм — эфир, в результате которого было найдено, что Д. м.
комплекса больше суммы Д. м. компонентов (за счет донорно-
акцепторной связи). Была изучена ассоциация в спиртах,
причем обнаружено, что в области низких концентраций
молекулы спирта ассоциируют с понижением Д. м., а затем
с ростом концентрации Д. м. ассоциатов растет. Обнаружен
т. наз. «.диоксановый эффект», т. е. повышение Д. м. аминов,
спиртов, фенолов и др. при их растворении в диоксане за счет
образования водородной связи вида —О—Н...О<^ или
—Н...О\. Найдено, что молекулы уксусной к-ты в раство-
рах ассоциированы в двойники пониженной полярности и
только в очень слабых растворах наступает их диссоциация;
при этом удалось определить Д. м. мономера уксусной к-ты.
Методы определения дипольных моментов. Практи-
чески все применяемые методы определения Д. м.
основаны на измерении эффекта ориентации полярных
молекул во внешнем электрич. поле. Вычисление
Д. м. на основании результатов измерений может быть
строгим только в том случае, если в условиях опыта
молекулы полярного соединения могут свободно
ориентироваться во внешнем поле, вернее, принимать
равновесное статистич. распределение углов ориен-
тации, зависящее только от темп-ры, напряженности
поля и величины Д. м. Явления ассоциации, комплек-
сообразования и другие виды взаимодействия между
полярными молекулами должны быть исключены.
Поэтому измерения Д. м. ведутся либо в газах и парах
при давлениях порядка 20— 700 мм рт. ст., либо
в разб. р-рах в неполярных растворителях (бензол,
гексан, четыреххлористый углерод, диоксан и др.)
при концентрациих порядка 0,2—2,0 мол. %.
Измерения в газах трудны методически, но дают
результаты без систематич. ошибок. Измерения в рас-
творах проще и легче и применяются в большинстве
работ, но результаты их систематически занижены
против газовых; это можно, впрочем, отчасти учесть
специальной поправкой. В чистых жидких полярных
веществах измерение Д. м. дает очень неточные резуль-
таты, особенно для больших Д. м. и ассоциированных
жидкостей. В твердых телах Дипольные молекулы
обычно уже жестко ориентированы, и измерение
Д. м. невозможно.
Первый, наиболее часто применяемый метод определения
Д. м. пригоден длн газов, паров и разбавленных р-ров и со-
стоит в измерении полной молярной поляризации (Р) и моле-
кулярной рефракции (Я) исследуемого вещества (о методах
измерения см. Поляризация и Рефракция молекулярная). Опыт
сводится н измерению плотности, нонцентрации и диэлектриче-
ской проницаемости ряда разб. р-ров исследуемого вещества
в неполярном растворителе при Одной темп-ре (в случае газов
и паров измеряют диэлектрич. проницаемость при нескольких
давлениях). Значение Р затем экстраполируют к нулевой
концентрации (или к нулевому давлению в случае газов и
паров) с целью устранить влияние остаточного взаимодействия
между полярными молекулами. Для вычисления Д. м. ис-
пользуется только та часть молярной поляризации, к-рая
зависит от ориентации Д. м. во внешнем поле, т. е. ориента-
ционная поляризация (РОр_); она входит в Р вместе с элек-
тронной (Рзл.) и атомной (Рат ) поляризацией вещества:
Р ~ Рор. + ^вл. + Рат.
Электронную поляризацию находят, экстраполируя мо-
лекулярную рефракцию вещества к нулевой частоте света;
атомную поляризацию оценивают приближенно, для чего
имеется ряд эмпирич. правил (прямых методов измерения
Рат не имеется). Для большинства полярных веществ Р
мало по сравнению с Р0Л
гают как членом Рат
рефракции к нулевой частоте; при этом обе эти небольшие
ошибки частично компенсируются, и точность расчета полу-
чается вполне удовлетворительной. Следовательно, условно
принимают Рэл- 4- Рат. равной просто молекулярной рефрак-
ции вещества при D-линии натрия. Т. обр.:
^*ор. = Р Р D
Из ориентационной поляризации Ро„ можно вычислить Д. м.
(ц), пользуясь формулой Дебая: 1
4 и.2
где N — число Авогадро, k — постоянная Больцмана и Т —
абс. темп-ра. При подстановке вначений констант получается
формула для вычисления ц:
И = 0,0128 ур^т
ат.
и РОр .; учитывая это, обычно пренебре-
, так и экстраполяцией молекулярной
Большие Д. м. (2—5 D) могут быть измерены с точностью
±0,01—0,02 D. Если Д. м. малы, то ориентационная поляри-
зация мала и тогда небольшие ошибки в определении Рэл
и Рат могут значительно повлиять на результат; поэтому ма-
лые моменты (0,5—0,8 D) измерены недостаточно надежно.
Второй метод определения Д. м. применим только к газам
и состоит в измерении температурной зависимости молярной
поляризации, к-рая в общей форме имеет вид:
Р = А + В/Т
где А = Р№. + Рат, В = 4nNp.a/9 h
(применению этого метода к растворам мешает недостаточная
изученность температурной зависимости влияния растворителя
на молярную поляризацию различных соединений). В случае
неполярных молекул В — 0 и Р не зависит от Т, т. е. прямая
Р— X- параллельна оси абсцисс; если молекула имеет Д. м.,
прямая идет наклоинок оси абсцисс. Экстраполяция прямой н
1139
ДИПРОФЕН — ДИСАЗОКРАСИТЕЛИ
1140
1/7’= 0 дает величину А, т. е. Рэл 4-Рат> в то время как тан-
генс угла наклона дает В. Отсюда
Г
Этот метод основан на предположении, что ц не зависит от Т.
С практич. точки зрения только в случае сравнительно про-
стых и легколетучих соединений можно изучать в широком
интервале темп-p зависимость ориентационной поляризации
паров вещества и, следовательно, измерять Д. м.
Существуют также методы определения Д. м., не связан-
ные с измерением диэлектрич. проницаемости и поляризации.
В методе молекулярного пучка газовый пучок направляется
через поперечное неоднородное электрич. поле, к-рое смещает
полярные молекулы в сторону тем в большей степени, чем
выше Д. м. Этим методом были измерены Д. м. галогенидов
щелочных металлов. В последние годы получил значительное
развитие новый радиоспектроскопич. метод определения Д. м.
Этот метод дает возможность определить Д. м. с точностью
до 0,002 D, но пока он применим только к сравнительно про-
стым молекулам в газовой фазе. Поскольку для исследования
строения молекул весьма существенно знание не только
суммарного Д. м. молекул, но также Д. м. отдельных связей,
то в последние годы начал развиваться метод определения
Д. м. отдельных связей по измерению интенсивности инфра-
красных спектров.
Лит.: Омайс Ч. Ф., Диэлектрическая постоянная и
структура молекул, пер. с англ., М., 1937; Smith J. W.,
Electric dipole moments, L., 1955; Физические методы органи-
ческой химии, пер. с англ., т. 3, М., 1954; Landolt-
В ornste in, Bd 1, TI 3, В. [u. a.], 1951, S. 386—508;
Волькенштейн M. В., Строение и физические свойства
молекул, М.—Л., 1955. В. Г. Васильев, Е. Н. Гурьянова.
ДИПРОФЕН (хлоргидрат 0-ди-н-пропиламивоэти-
лового эфира дифенилтиоуксусной кислоты), мол.
вес 392,01, (CeH5)aCHCOSCHaCH2N(C3H7)a • НС1 —
бесцветные кристаллы горького вкуса, т. пл. 132—
134°. Д. растворим в воде (1 : 1000 при 40° и 1 : 4
при 100°), спирте, ацетоне, бензоле, этилацетате,
дихлорэтане и хлороформе; малорастворим в эфире.
Количественное определение основано на разложении
Д. NaOH, экстракции основания эфиром, подкисле-
нии щелочного р-ра, извлечении к-ты эфиром и тит-
ровании 0,1 н. р-ром NaOH в присутствии фенолфта-
леина. Получают Д. конденсацией р-ди-н-пропилами-
ноэтилмеркаптана с хлор ангидридом дифенилуксусной
к-ты. Д. применяют при спазмах кровеносных сосу-
дов (эндартериитах, болезни Рейно и др.) и гладкой
мускулатуры внутренних органов (желудка, кишеч-
ника, мочевыводящих путей, бронхов).
Лит. см. при ст. Апрессин. А. И. Травин.
ДИСАЗОКРАСИТЕЛИ — азокрасители, содержа-
щие две азогруппы— N = N—. В зависимости от мето-
дов получения различают три основные группы Д.:
первичные, получаемые сочетанием двух молей
одинаковых или разных ароматич. диазосоединений
с одним молем азосоставляющей. Примером красителя
этой группы является кислотный сине-черный (см.
табл.); вторичные, для получения к-рых диазо-
соединение сочетают с ароматич. или гетероциклич.
амином, полученный амино-моноазокраситель диазо-
тируют и сочетают с азосоставляющей, напр. кислот-
ный синий К.; производные ароматических
n-диаминов, к-рые диазотируют и сочетают
с двумя одинаковыми или разными азосоставляющими.
В этой группе наиболее многочисленны прямые Д.,
напр. прямой коричневый КХ.
В табл, приведены некоторые Д.
По технич. свойствам Д. делятся на кислотные,
протравные, прямые, пигменты и основные красители
(последние не имеют большого значения). Цвет Д.,
как правило, глубже, чем моноазокрасителей, и они
обычно менее ярки. Среди Д. имеются красители всех
цветов, в том числе большое число глубокоокрашен-
ных черных, синих и коричневых. Прочность Д. часто
выше прочности моноазокрасителей. Так, окраски
многими кислотными Д., особенно вторичными, зна-
чительно прочнее окрасок моноазокрасителей, в част-
ности к мокрым обработкам (воде, стирке и др.).
Прямые Д. обладают обычно большим сродством
к целлюлозному волокну, чем моноазокрасители, од-
нако светопрочность их невысока. Значительно больше
светопрочность у медных комплексов прямых Д.,
содержащих в о,о'-положениях к азогруппе две
группы ОН или одну ОН-группу и одну группу
—ОСН3, —СООН, — ОСЩСООН или—NHa. Комп-
лексообразование может производиться при изготов-
лении красителя или в процессе крашения непосред-
ственно на волокне. Примером такого рода красителя
Название
Строение
Прочность в баллах а в кон-
центрации 3%
к волокну! к свету
к мыльному
раствору®
Кислотный сине-черный
Кислотный синий К
Прямой фиолетовый
светопрочный 2К
Прямой коричневый КХ
шерсть
то же
хлопок
то же
5/4/2
4/4/4
3/1
3/2
а См. Красители синтетические', пятибальная шкала. ® 1-я цифра — степень посветленпя окраски; 2-я — степень
вакрашивания шерсти (для прямых красителей степень закрашивания хлопка); 3-я — степень закрашивания хлопка.
1141
ДИСАХАРИДЫ — ДИСЛОКАЦИИ
1142
является прямой фиолетовый светопрочный 2К. Дис-
азопигменты превосходят многие моноазопигменты
в отношении термостойкости, светопрочности, нераст-
воримости в органич. растворителях, миграционной
устойчивости (отсутствие миграции пигмента из окра-
шенной в неокрашенную часть изделия из резины,
пластич. масс и т. п.). О физич. и химич. свойствах,
качественных реакциях, методах колич. определения
и литературу см. Азокрасители. См. также Красители
синтетические. М. А. Чекалин.
ДИСАХАРИДЫ (биозы) — группа олигосахаридов,
в к-рых содержится по два остатка моносахаридов,
связанных гликозидной связью. Д. рассматривают
как гликозиды, в к-рых агликон представлен моносаха-
ридом. При действии кислот и ферментов гликозидаз
Д. гидролизуются до моносахаридов. Моносахариды,
входящие в состав большинства известных Д., пред-
ставлены гексозами или гексозой и пентозой. Помимо
обычных альдоз и кетоз, в образовании Д. могут
принимать участие уроновые к-ты (см. Алъдобиуроно-
вые кислоты), аминосахара, сиаловые к-ты и др.
В зависимости от того, содержится ли в Д. свободный
полуацетальный гидроксил или же полуацетальные
гидроксилы обоих моносахаридных компонентов при-
нимают участие в образовании гликозидной связи,
Д. делят на 2 группы:
1. Восстанавливающие Д. (типа маль-
тозы, гликозидо-гликозы, гликозидо-альдозы или
гликозидо-кетозы)—Д., компоненты к-рых связаны
гликозидной связью (а или 0), образованной за счет
отщепления воды от полуацетального гидроксила
одного моносахарида и какого-либо из спиртовых
гидроксилов второго моносахарида. Представителями
Д. являются мальтоза (I) и лактоза (II).
СН2ОН СН2ОН
Н /ы" °\н н ZL °\он
I 1 г г
н он н он
Д. этого типа по свойствам близки моносахаридам —
альдозам или кетозам: образуют таутомерные формы,
мутаротируют и восстанавливают р-р Фелинга. При
их окислении образуются алъдобионовые кислоты—Д.,
в к-рых карбонильная группа окислена до карбок-
сильной. Восстанавливающие Д. образуют озазоны,
а- и 0-гликозиды и дают другие реакции, обусловлен-
ные наличием полуацетального гидроксила.
2. Невосстанавливающие Д. (типа
трегалозы, гликозидо-гликозиды) — Д., компоненты
к-рых связаны гликозидной связью (a,a-, а,0-
или Р,а-), образованной за счет отщепления воды от
полуацетальных гидроксилов обоих моносахаридных
компонентов. Типичными представителями этих Д.
являются трегалоза (III) и сахароза (IV).
Строение Д. устанавливают: идентификацией моно-
сахаридов, образующихся при гидролизе; определе-
нием формы (пиранозной или фуранозной), в виде
к-рой моносахариды входят в Д., и положением гидрок-
силов, принимающих участие в образовании гликозид-
ной связи. Конфигурацию полуацетальных гидрокси-
лов, участвующих в образовании гликозидных связей,
устанавливают ферментативным путем, с использо-
ванием а- и 0-гликозидаз. Д. получают из природ-
ных материалов, напр. сахарозу — из свеклы, лак-
тозу — из молока. Многие Д. получают при неполном
гидролизе природных полисахаридов, олигосахари-
дов и гликозидов, напр. при ферментативном гидро-
лизе крахмала и гликогена — мальтозу, при гидро-
лизе целлюлозы — целлобиозу, из трисахарида ген-
циозы — генцибиозу и т. д. Синтетически Д. полу-
чают: изомеризацией Д., напр. при щелоч-
ной изомеризации лактозы получается лактулоза,
из восстанавливающих Д. через гликали получают
их эпимеры и т. д.; конденсацией моносаха-
ридов и др.
Лит.: The Carbohydrates, ed. W. Pigman, N. Y., 1957;
M i c h e e 1 F., Chemie der Zucker und Polysaccharide, 2 Aufl.,
Lpz., 1956. Л. И. Линевич.
ДИСЛОКАЦИИ в кристаллах — линей-
ные дефекты структуры реальных кристаллов, обра-
зующиеся в процессе роста кристаллов или в процессе
пластич. деформации. В простейшем случае пластич.
деформация кристалла может быть макроскопически
представлена как результат скольжения атомных
плоскостей друг по другу, подобно сдвиганию колоды
карт, причем сдвиг в каждой данной плоскости сколь-
жения не охватывает одновременно всю эту плоскость;
напротив, сдвиг распространяется постепенно, от
одного участка данной плоскости к другому.
На рис. 1 показан кристалл с простой кубич. решет-
кой, у к-рого сдвиг произошел только по одной части
плоскости скольжения, тогда как на остальной части
сдвиг отсутствовал;
линия АВ (располо-
женная внутри кри-
сталла) является гра-
ницей той зоны ABCD
плоскости скольже-
ния, где произошел
сдвиг. Здесь же на
торцевой поверхно-
сти кристалла пока-
зано расположение
атомов в плоскости,
Рис. 1. Незавершенный
сдвиг, который создает
краевую Д. Единичный
сдвиг захватывает пло-
щадь ABCD. Проходя-
щая внутри кристалла граница зоны сдвига (линия АВ) являет-
ся краевой Д. Вектор Ь — вектор Бюргерса. На торцевой по-
верхности кристалла схематически показано расположение
атомов при наличии краевой Д.
перпендикулярной к линии АВ. Несовершенная об-
ласть строения кристалла вдоль этой линии являет-
ся одним из главных типов Д.; она называется ли-
нейной, или краевой Д. Величина сдвига b (рис. 1)
наз. вектором Бюргерса. В случае краевой Д. век-
тор Ь перпендикулярен направлению Д. .
1143
ДИСЛОКАЦИИ
1144
Пластич. сдвиг в кристалле осуществляется не одно-
временным перемещением всех атомов (ионов), лежа-
щих в данной плоскости (что потребовало бы весьма
значительных напряжений), а последовательным пере-
мещением Д. от одной группы атомов к другой, т. е.
представляет собой последовательное «пересоедине-
ние» связей между рядами атомов (ионов), лежащих
по одну сторону от плоскости скольжения, и нх бли-
жайшими соседями по другую сторону от этой плос-
кости. На рис. 2 показано последовательное движение
Рис. 2. Простое трансляционное скольжение в
кубич. решетке: перемещение краевой Д. в пло-
скости скольжения через весь кристалл отвечает
сдвигу на одно атомное расстояние. Стрелками
указано приложенное скалывающее напряжение.
Д. через кристалл с выходом ее на противоположную
грань: в результате происходит элементарный сдвиг
на одно атомное расстояние. Перемещением Д. опи-
сываются и другие формы пластич. деформации:
двойникование, образование сбросов и т. д.
Д. в кристалле весьма подвижны; сопротивление
решетки их движению по плоскостям скольжения
невелико. Его можно преодолеть, приложив неболь-
шое напряжение, — на несколько порядков величины
меньше, чем это следует из классич. теории идеальной
кристаллич. решетки. Связано это с тем, что атомы,
находящиеся в равновесном положении впереди Д.,
могут сохранить свои положения, только если они
способны оттолкнуть Д. назад; аналогично, атомы,
расположенные позади Д., толкают ее вперед. В пер-
вом приближении эти силы уравновешивают друг
друга, и решетка вообще не оказывает сопротивления
движению Д. Более точный расчет показывает, что
эти силы уравновешиваются не полностью; чтобы
привести Д. в движение, нужно приложить к образцу
нек-рое усилие.
Д. в кристаллах возникают в процессе образования
и роста кристаллов из расплава или раствора. Даже
при условии соблюдения самых тщательных пред-
осторожностей вырастающий кристалл всегда содер-
жит значительное число Д. Их движение приводит
к появлению первых пластич. сдвигов уже при весьма
малых напряжениях. С ростом напряжений внутри
кристалла появляются многочисленные новые Д.,
не связанные уже с происхождением кристалла;
их перемещение по плоскостям скольжения ведет
к дальнейшему развитию пластич. деформации. Плот-
ность Д. (число дислокационных линий, пронизываю-
щих площадку в 1 см2, взятую внутри кристалла)
в хорошо отожженных недеформированных моно-
кристаллах может составлять 104—10е линий на 1 см2;
в предельно наклепанном материале она достигает
Ю14—101а.
По мере развития пластич. деформации в кристалле
возникают и накапливаются разного рода препятствия
для дальнейшего движения Д. Такими препятствиями
являются сместившиеся из положений устойчивого
равновесия атомы, вакансии, упругие напряжения,
создаваемые другими Д., в частности связанные с мест-
ным поворотом и изгибом плоскостей скольжения,
и т. д. Они затрудняют движение и дальнейшее раз-
множение Д.; чтобы их преодолеть, необходимо уве-
личивать внешнее напряжение, — в этом состоит
механизм упрочнения кристаллов в процессе пластич.
деформации. Существенными препятствиями для дви-
жения Д. служат также инородные атомы, а в поли-
кристаллах — границы зерен. При повышенных
темп-рах в результате усиления теплового движения
атомов образовавшиеся препятствия могут быстро
ликвидироваться либо легко преодолеваться, тем
самым снимается упрочнение. При низких темп-рах
преодоление дислокационных препятствий сущест-
венно затруднено и упрочнение велико.
В силу самой своей дислокационной природы пла-
стич. деформация протекает в кристалле крайне неод-
нородно; она может локализоваться в отдельных
небольших районах кристалла, это означает, что тор-
мозимые препятствиями Д. скапливаются в данных
участках и образуют различные специфич. конфигу-
рации. В результате взаимодействия Д. внутри таких
групп, а также между различными группами в кри-
сталле возникают резкие локальные концентрации
напряжений. Уже на ранних этапах деформации эти
высокие локальные напряжения могут привести,
напр., к слиянию нескольких близко расположенных
и поджатых Друг к другу Д. — образуется полое
дислокационное ядро, служащее по существу зароды-
,шем микротрещины. С ростом деформации и напряже-
ний подобные зародыши развиваются, включая в свою
полость все новые и новые Д., превращаются в мак-
роскопии. трещины и в конце концов приводят к раз-
рушению образца. При низких темп-рах, когда даже
небольшие препятствия трудно преодолимы для дви-
жущихся Д., уже на ранних стадиях деформации
могут образовываться трещины, быстро приводящие
к разрушению кристалла. В этих условиях в кри-
сталлах проявляется хрупкость, т. е. пластич. дефор-
мация, предшествующая разрыву, оказывается мала.
С повышением темп-ры препятствия становятся все
более и более «прозрачными», т. е. все легче преодо-
лимыми для Д., в результате пластичность кристал-
лов растет. В этом случае требуется значительная
деформация (т. е. большие искажения решетки),
чтобы возникли условия, необходимые для формиро-
вания трещин разрушения.
Взаимодействуя друг с другом, Д. могут расщеп-
ляться, образовывать новые типы Д., в том числе
неподвижные, служащие препятствием для скольже-
ния других Д. При пересечении Д. возникают различ-
ные точечные дефекты структуры: уступы на линиях
Д., вакансии, дислоцированные ионы. Д. способны
адсорбировать инородные атомы и служат своеобраз-
ными каналами для их миграции, взаимодействуют
со свободной поверхностью кристалла, рассеивают
электроны и т. д. Поэтому присутствие Д. заметно
сказывается на разнообразных свойствах кристаллов:
оптических, полупроводниковых, магнитных, плот-
ности, электропроводности и др.
Если граница между претерпевшей и непретерпев-
шей сдвиг областями плоскости скольжения парал-
лельна вектору сдвига Ь, то эта граница образует
винтовую Д. (рис. 3). Присутствие винтовой Д.
обусловливает рост кристаллов при малых пересыще-
ниях (т. е. при малой вероятности появления зароды-
шей роста на гранях кристалла): выход винтовой Д.
образует специфич. зародыш — ступеньку, к-рая не
1145
ДИСЛОКАЦИИ — ДИСПЕРГИРОВАНИЕ
1146
Рис. 3. Винтовая Д. в простой
кубнч. решетке (линия АВ). На
торцевой поверхности металла
схематически показано располо-
жение атомов при наличии вин-
товой Д.
может зарасти и с каждым своим оборотом дает но-
вый атомный слой. Ряды параллельных Д. могут об-
разовывать внутренние
границы раздела в кри-
сталлах. Так, изображен-
ный на рис. 4 ряд крае-
вых Д., расположенных
одна над другой, отве-
чает повороту решетки
одной части кристалла
относительно другой его
части, т. е. образует гра-
ницу двух разориенти-
рованных блоков. Дру-
гие конфигурации Д.
отвечают линиям сколь-
жения (рис. 5), границам
двойников, зерен и фаз
в кристаллич. телах.
Перемещением Д. или
их сеток (границ бло-
ков и зерен) и появле-
нием новых дислока-
ционных границ могут
быть описаны с точки зрения теории Д. также
процессы ползучести, отдыха и рекристаллизации
(кроме высоких темп-p, когда эти процессы сущест-
венно связапы с явлением самодиффузии), полигони-
зация и ряд фазовых превращений в твердых телах
(напр., мартенситное превращение в сталях). На ос-
нове дислокационных моделей развития опасных де-
фектов формулируются условия
разрушения кристаллич. тел, опи-
сывается переход от хрупкого
разрушения к пластич. течению
Рис. 4. Вертикаль-
ный ряд краевых Д.
(простая наклонная
граница блоков).
Рис. 5. Горизонтальный ряд
краевых дислокаций, лежащих
в общей плоскости скольжения
(линия скольжения).
(темп-рный порог хладноломкости), анализируются
процессы усталости и явление понижения прочности
в присутствии поверхностноактивных сред (эффект
Ребиндера).
Основными методами изучения Д. в кристаллах
служат: электронно-микроскопич. наблюдение рас-
положения атомных плоскостей в кристаллах; деко-
рирование Д. примесями с наблюдением в видимом
или ИК-свете; металлография, травление точек выхода
Д. на поверхность кристалла; изучение фигур спи-
рального роста с применением фазовоконтрастной
микроскопии, многолучевой интерферометрии и ион-
ного проектора; поляризационно-оптич. определение
.внутренних напряжений, рентгеновские исследова-
ния, а также измерения электропроводности, плот-
ности и комбинации этих методов.
Лит.: Р и д В. Т., Дислокации в кристаллах, пер. с англ.,
М., 1957; Коттрелл А. X., Дислокации и пластическое
течение в кристаллах, пер. с англ., М., 1958; Проблемы совре-
менной физики. Сб. переводов..., №9, М., 1957; Инден-
б о м В. Л. и Метелкин И. И., Кристаллография,
1958, т. 3, вып. 1; Р и з А., Химия кристаллов с дефектами,
пер. с англ., М., 195S; Лихтман В. И., Физика в школе,
I960, № 3. В. Й. Лихтман, Е. Д. Щукин,
ДИСПЕРГИРОВАНИЕ — тонкое измельчение твер-
дых или жидких тел в окружающей среде, в ре-
зультате которого образуются порошки, суспензии,
эмульсии. Наряду с противоположным процессом —
конденсацией или кристаллизацией — Д. служит
одним из двух путей получения коллоидных и вообще
дисперсных систем. Д. требует затраты работы тем
большей, чем больше вновь образующаяся .поверх-
ность и поверхностная энергия на границе диспер-
гируемого тела с окружающей средой. Поэтому усло-
вия Д. определяются не только структурой и составом
диспергируемого тела, но г» физико-химич. особенно-
стями окружающей среды.
Д. жидкостей обычно наз. распылением, если оно
происходит в газовой форме, и эмульгированием,
когда оно проводится в другой (несмешивающейся
с первой) жидкости. Д. твердых тел происходит
в результате механич. деформирования с разрушением
тела в предельно напряженном состоянии по наиболее
слабым местам — дефектам структуры, развиваю-
щимся в напряженном состоянии. Работа Д. твердых
тел значительно выше энергии развивающейся поверх-
ности вследствие необходимости упругого или пла-
стич. деформирования частиц до разрушения, а для
жидкостей — вследствие затраты работы на преодоле-
ние вязкого сопротивления. Однако затрачиваемая
на деформирование работа также приблизительно
пропорциональна поверхностной энергии. По мере
перехода ко все более мелким частицам их проч-
ность возрастает. Этим объясняется резкое снижение
эффективности Д. для частиц диаметром 1—0,1 мк
(практич. предел механич. Д.). Для дальнейшего
Д. в таких частицах твердых тел должны возникнуть
новые дефекты структуры, что возможно в результате
ударного,действия на весьма больших скоростях или
высокочастотного вибрационного воздействия. Кроме
того, по достижении достаточно высокой степени раз-
дробления частицы начинают слипаться между собой,
и дальнейшее их Д. прекращается. Небольшие добавки
адсорбирующихси поверхностно-активных веществ
облегчают Д. тел, т. к. они понижают поверхностное
натяжение на вновь возникающих границах раздела
фаз, а также образуют в ряде случаев структурирован-
ные адсорбционные слои с повышенной вязкостью
и упругостью, препятствующие обратному слипанию
мелких частиц. При Д. твердых тел поверхностно;
активные добавки (понизители твердости) проникают
в мельчайшие поверхностные трещинки в процессе
их развития при механич. воздействии. Такие добавки
(диспергаторы, эмульгаторы, смачиватели и др.) могут
служить и стабилизаторами образующихся частиц,
препятствуя их коагуляции и удерживая их в состоя-
нии тонкой суспензии или эмульсии.
При понижении поверхностной энергии данного
тела на границе с окружающей средой до очень низ-
ких значений порядка 0,1 эрг[см~г, при обычной
темп-ре Д. становится самопроизвольным процессом
и происходит без затраты внешней работы под влия-
нием теплового движения (броуновского движения
отщепляющихся частиц) вследствие возрастания энтро-
пии системы при более равномерном распределении
в ней диспергируемой фазы. Таковы случаи коллоид-
ного растворения, напр. бентонитовых глин в воде
или мыл в соответствующих жидких средах.
, На практике Д. проводят в специальных машинах
для тонкого измельчения — мельницах различных
конструкций (шаровые мельницы для получения
частиц до 100—10 Л1к, вибрационные, виброкавита-
ционные и струйные мельницы — до 10—1 мк и т. в.
коллоидные мельницы — для получения еще более
мелких частиц). Интенсивное Д. может быть произве-
дено также с помощью звуковых и ультразвуковых
колебаний, Используют также местное нагревание
до высоких темп-p с возникновением значительных
градиентов тёмп-ры и, т. о., высоких термич. напря-
1147
ДИСПЕРСИОННАЯ СРЕДА - ДИСПЕРСИОННЫЙ АНАЛИЗ
1148
жений. Эффективное Д. может быть также произведено
при перетирании весьма концентрированных, а по-
тому сильно структурированных суспензий и эмуль-
сий. При этом даже в частицах малых размеров могут
быть возбуждены предельные скалывающие напря-
жения. При тех же расходах энергии и скоростях Д.
менее концентрированных суспензий практически
не происходит или протекает значительно менее эф-
фективно. Тонкий помол — Д. в мельницах — при-
обретает решающее значение в новой технологии полу-
чения строительных материалов (быстротвердеющие
цементы), красителей, пигментов, наполнителей, ком-
понентов твердых сплавов, огнеупоров, керамики
и металлокерамики, электродных масс и др. Широкое
применение в технике имеет Д. в виде тонкого шлифо-
вания, при чистовой обработке поверхностей твердых
тел (металлов, стекла). Большое значение имеют также
вредные виды Д. — износ, особенно при трении в ма-
шинах. Методы борьбы с износом сводятся к повыше-
нию твердости поверхностей трения и к смазке, т. е.
к разъединению этих поверхностей — к снижению
усилий, вызывающих Д. Применяется Д. и для акти-
вирования веществ в твердом состоянии, т. е. для
повышения интенсивности их взаимодействия с окру-
жающей средой или с другими веществами. В природе
Д. играет важнейшую роль при выветривании, будучи
одним из путей переноса веществ в процессах образова-
ния почв, глинистых пород и др.
Лит.: Ребиндер П. А., Ш р е~й н е р Л. А. и
Ж и г а ч К. Ф., Понизители твердости в бурении, М.—Л.,
1944; Ребиндер П. А., в кн.: Юбилейный сборник, по-
священный тридцатилетию Великой Октябрьской социалисти-
ческой революции, ч. 1, М.—Л., 1947, с. 533; Коллоидн. я;,,
1958, 20, вып. 5; Л и х т м а н В. И., Ребиндер П. А. и
Карпенко Г. В., Влияние поверхностно-активной среды
на процессы деформации металлов, М., 1954.
П. А. РебииЭер.
ДИСПЕРСИОННАЯ СРЕДА — см. Дисперсные
системы.
ДИСПЕРСИОННЫЙ АНАЛИЗ — совокупность
методов определения дисперсности — размеров ча-
стиц в дисперсных системах и распределения частиц
по размерам (фракциям) и уд. поверхности дисперс-
ных тел.
Д. а. в грубодисперсных системах (см. таблицу),
сыпучих телах, порошках, грубых нзвесях с части-
цами диаметром более 100—70 мк проводится с по-
мощью сухого или мокрого ситового анализа — после-
довательным просеиванием через набор сит с опре-
деленными, все уменьшающимися отверстиями. В бо-
лее тонкодисперсных суспензиях Д. а. проводят
путем фильтрации через специальные фильтры с
порами определенных размеров. Прямые определе-
ния размеров частиц диаметром от десятков мк и
до десятых долей мк производят при помощи оптич.
микроскопов с окулярными микроскопии, сетками
(окулярмикрометрами). Подсчет большого числа ча-
стиц в разных местах препарата позволяет найти
распределение числа частиц или их массы (объема)
по размерам (фракциям). Частицы, размеры к-рых
лежат за пределом разрешающей способности оптич.
микроскопов (десятые доли мк и менее), измеряются
с помощью электронных микроскопов. Современные
электроннооптич. методы обработки микрофотографий
или электронно-микроскопич. снимков позволяют
автоматически на основании просмотра большого числа
полей дисперсного препарата получить полную кри-
вую распределения частиц по размерам. Средний
размер частиц в области коллоидной дисперсности
(0,1—0,001 мк) может быть определен с помощью
ультрамикроскопов по определению числа мерцаний
в данном объеме в покое или в потоке. Методы опре-
деления размеров частиц приводят лишь к прибли-
женной оценке их среднего диаметра, т. к. практически
все дисперсные системы полидисперсны.
Системы
Диаметр частиц,
см
Грубодисперсные ..............
Тонкодисперсные (суспензии,
эмульсии).....................
Коллоидные растворы...........
Молекулярно-ионные растворы . .
1—ю-2
10-5-10 4
10-4-10-7
10-7—10-8
Методы анализа
Диаметр опреде-
ляемых частиц, см
Ситовой ......................
Отмучивание ..................
Микроскопический..............
Седиментационный..............
Фильтрационный................
Центрифугирование ............
Броуновское движение..........
Ультрамикроскопин.............
Электронная микроскопия ......
Ультрафильтрация..............
Измерения диффузии............
Ультрацентрифугирование.......
1-10-а
10 1—10-4
10-2-10-8
10-2—10-4
10-3-10-5
10-4-10-3
10-4 —10 7
10-5—10-7
10-8-10-7
10-5—10-7
10-5-10-8
10-6-10-’
В суспензиях и эмульсиях, а также в дымах (пы-
лях) и туманах с размерами частиц мейее 100 мк
обычно порядка 10 мк, полная картина распределения
частиц по размерам проще всего определяется с по-
мощью метода седиментометрии (см. Седиментацион-
ный анализ), основанного на измерении скорости
установившегося оседания частиц под действием силы
тяжести в соответствии с законом Стокса. Для частиц
(напр., капелек в эмульсиях) с плотностью, меньшей
плотности среды, наблюдается скорость обратного
оседания (всплывания) частиц. Для частиц с разме-
рами менее 1—0,1 мк вместо свободного оседания
наблюдают оседание под действием центробежной
силы в центрифугах. С помощью ультрацентрифуги
напряжение поля тяжести земли g = 981 дин/г может
быть увеличено в 105 и более раз, что позволяет про-
водить полный Д'. а. не только для коллоидных ча-
стиц — мицелл, но и для макромолекул, т. е. на-
ходить распределение частиц полимера в его р-ре
по мол. весу.
Измерение кипетики накопления осадка частиц
с помощью седимеитометра как при свободном осе-
дании, так и в центрифугах, позволяет проводить
полный Д. а. полидисперсных систем, т. е. находить
функции распределения числа частиц п или массы т
(объема и) дисперсной фазы по размерам, т. е. по фрак-
циям, соответствующим определенному малому интер-
валу диаметров частиц.
Для анализа высокодисперсных систем применяется
также ультрафильтрация через тонкопористые пере-
городки (мембраны) с определенной величиной пор.
Д. а. может быть проведен и с помощью оптич. мето-
дов, основанных на измерении интенсивности рассе-
янного или поглощенного света (см. Нефеломет-
рия и турбидиметрия, Фотометрические методы
анализа).
Для достаточно монодисперсных фаз или, что то
же самое, для отдельных фракций полидисперсных
фаз диаметр частиц, включая и сольватную (гидрат-
ную оболочку), может быть вычислен из измерения
скорости диффузии: S = RT/NAD3nr\,rpfiD— коэффи-
циент диффузии, НА—число Авогадро, Т]— вязкость,
или по седиментационному равновесию, т. е. устанав-
ливающемуся распределению частиц в постоянном
силовом поле (с помощью ультрацентрифуги). Для
Д. а. полидисперсных систем применяется также рент-
генографии. анализ.
Для определения уд. поверхности пористых тел
и порошков применяют методы, основанные на пзме-
1149
ДИСПЕРСНАЯ ФАЗА — ДИСПЕРСНОСТЬ
1150
рении адсорбции паров или растворенных веществ
из раствора, исходя из площади, приходящейся
в насыщенном мономолекулярном адсорбционном
слое на молекулу или ион адсорбирующегося вещества.
Используют также радиохимия, методы, связанные
с измерением кинетики изотопного обмена и адсорб-
ции веществ с мечеными атомами. Сюда же относятся
метод определения уд. поверхности пористого тела
или слоя порошка по проницаемости — по скорости
фильтрации газа или жидкости — и метод измерения
скорости растворения твердых дисперсных тел в жид-
костях, основанный на пропорциональной зависи-
мости между этой скоростью и величиной поверхности.
Полный Д. а. пористых тел (с открытой пористо-
стью) может быть произведен методами капилляр-
ной конденсации и капиллярного продавливания
жидкости, например ртути (метод ртутной поро-
метрии) через пластинку из данного дисперсного (по-
ристого) тела или пузырьков газа через пористую пла-
стинку, погруженную в жидкость. Полный Д. а.
является обязательным для характеристики иссле-
дуемых систем в современной коллоидной химии,
а также в физико-химии и технологии полимеров.
Лит.: фигуровский Н. А., Седиментометрпческий
анализ, М.—Л., 1948; Методы исследования структуры высоко-
дисперсных и пористых тел, М., 1953 (Тр. совещания 25—29
июня 1951 г.); The physics of particle size analysis, L., 1954
(Brit. J. Appl. Phys. Suppl., № 3). II. А. Ребиндер.
ДИСПЕРСНАЯ ФАЗА — см. Дисперсные системы.
ДИСПЕРСНОСТЬ — характеристика размеров
частиц в дисперсных системах. Д. обратно пропорцио-
нальна размеру (диаметру) б частиц дисперсной фазы
и определяется их уд. поверхностью з1( т. е. отноше-
нием поверхности частицы s к ее объему t^: s = aS2;
1ц = рб3; sx = s/Vi = y/6, где a, p и у = a/p —
безразмерные коэфф., определяемые формой частиц;
для частиц формы, близкой к сферической, a = л,
р = л/6, у = 6 и Д. выражается: sx = 6/6 (1).
Определение Д. выражением (1) справедливо только
в простейшем случае монодисперсных систем, частицы
к-рых одинаковы или близки по размерам. В общем
случае полидисперсных систем, в к-рых частицы
дисперсной фазы могут сильно различаться_по разме-
рам, Д. нельзя определять как sx = 6/6, где 6 — сред-
ний диаметр всех частиц, т. к. частицы малых размеров
(тонких фракций) в основном и определяют Д. (уд.
поверхность) всей дисперсной фазы. Так, если имеются
две фракции одинаковой массы, одна — грубая
6Х = 100 мк, а другая — тонкая 62 = 1 мк, то sx
тонкой фракции будет в 100 раз больше, чем у грубой
фракции, она и будет практически определять
всей дисперсной фазы, так что наличием равной
по массе грубой фракции можно пренебречь. При
вычислении же среднего по массе диаметра частиц
получим 6 = 50 мк, что даст величину Д. в 50 раз
меньше действительной.
Д. полидисперсных систем характеризуется функ-
цией распределения объема (или массы) частиц по
их размерам /(6). Функция или, соответственно,
кривая распределения (см. рис.), определяемая экспе-
риментально методами
дисперсионного анали-
за, позволяет легко
вычислить или же най-
ти графически Д., как
уд. поверхность диспер-
сной фазы в полиди-
сперсной системе. Д.
полидисперсной систе-
мы характеризуется,
еще и степенью поли-
дисперсности, т. е. размытостью кривой распре-
деления— ее основанием 6„,„v — 6min— полным ин-
UlaX HiAIl
тервалом диаметра частиц и их вероятнейшим диа-
метром 60, соответствующим максимуму дифферен-
циальной кривой распределения. Если имеется не-
сколько фракций со средними диаметрами 61F 62, ... и
соответственно массами m1( т2, ..., то:
1 M8i 1 о2 /
где пг0 = т± + пг2 + ... Д. как уд. поверхность
может быть определена и по измерениям адсорбции.
Тогда, зная slt можно из (1) определить эквива-
лентный диаметр частиц, соответствующий данной
Д.: 6 = 6/S1.
Д. — основная характеристика дисперсных систем;
ее размерность в системе CGS см2 см :t = см 1.
Для грубодисперсных систем 6>;100 мк (10~2 см)
и Д., т. е. sx С 600 см'1; для тонкодисперсных систем
100 мк > 6 > 1 мк и Sj лежит между 600 и 6 • 104 см'1,
а для коллоидных, т. е. предельно высокодисперсных
систем (6 между 1 мк и 10~3 мк), Д. находится между
6 • Ю4 и 6 • 107 см'1. Уд. поверхность часто выражается
поверхностью единицы массы т дисперсной фазы
(дисперсного тела) в см2/г: slm = sx/p, где р = m/v —
плотность тела. Наибольшая Д. коллоидных фаз,
напр. в наиболее высокодисперсных золях с разме-
рами частиц порядка 4 ммк (107 см1), по порядку
величины составляет sx = 107 <- 108 см'1, что соот-
ветствует внутренней уд. поверхности наиболее вы-
сокопористых тел, таких активных адсорбентов,
как активированные угли, силикагели, у к-рых sx
достигает 103 м2/г.
Физико-химич. смысл определения Д. как уд.
поверхности состоит в том, что Д. характеризует
в данной системе относительную роль поверхностных
явлений по сравнению с объемными, возрастающую
с ростом Д. Так, плотность свободной поверхностной
энергии при переходе к дисперсным телам не остается
постоянной при заданных Т, р, а возрастает пропор-
ционально Д. Соответственно пропорционально Д.
адсорбента растет и адсорбируемое им из окружающей
среды количество вещества.
Д. характеризует отношение числа молекул п^
находящихся в поверхностном (мономолекулярном)
слое частиц дисперсной фазы, к числу молекул nv
образующих объем частиц: sx = k1ns /nv
где v = М/р — молярный объем, М — мол. вес
вещества частицы, /УА — число Авогадро. Для во-
дяного тумана 1сг 3 • 107 см'1; для наибольшей Д.
отношение ns /nv близко к 0,3. Так как поверхность
раздела в дисперсной системе является общей для
обеих фаз, то Д. может быть определена не только для
дисперсной фазы, sj = в/и±, но и для дисперсионной
среды, s2 = s/v2; т. обр. s2 = sx . —. Если в разб.
дисперсных системах (обычных золях) отношение
объемов фаз Pj/^ssO.Ol, то s2 в 100 раз меньше,
чем У; наоборот, в предельно конц. дисперсных
системах, пенах, конц. эмульсиях или в конц. суспен-
зиях (пастах), в к-рых дисперсионная среда находится
в виде достаточно тонких прослоек между частицами,
капельками или пузырьками, t^/^2 70/30, 98/2,
99/1 или даже более, и s2 в неск. раз, а иногда в десятки
и до сотни раз превышает sx, т. е. в этом случае Д.
среды (отношение числа ее поверхностных молекул
к числу объемных молекул) выше, чем у дисперсной
фазы.
В отличие от «массивных» частиц (т. е. лишенных
пористости внутренней поверхности), у к-рых Д.
определяется их размером, у тонкопористых тел
(в виде зерен, напр., угля или силикагеля) Д. зависит
от размера пор, а не от размера зерен, если они
гораздо больше размера пор, т. к. внешняя поверх-
ность весьма мала по сравнению с внутренней. При
1151
ДИСПЕРСНЫЕ СИСТЕМЫ
1152
образовании рыхлых агрегатов частиц в процессах
коагуляции Д., определяемая как уд, поверхность,
не изменяется, но резко уменьшается, если ее опре-
делять по размеру агрегатов сравнительно, с первич-
ными частицами. У волокнистых (фибриллярных)
дисперсных тел, состоящих из отдельных волокон
(нитей), диаметром сечения б: = 4/6 и не зависит
от длины нитей. У пленочно-листовых (ламинарных)
тел, состоящих из отдельных пленок (листов) тол-
щиной б — бумага, фотопленка и др., s = 2/6 и не
зависит от протяженности (ширины, длины) пленки.
(См. Дисперсионный анализ, Дисперсные системы).
Лит. см. в ст. Дисперсионный анализ.
П. А. Ребиндер.
ДИСПЕРСНЫЕ СИСТЕМЫ — микрогетерогенные
системы с сильно развитой внутренней поверхностью
раздела между фазами; состоят из двух или большего 1
числа фаз, причем по крайней мере одна из них
(дисперсная фаза) обладает достаточно,
высокой дисперсностью и распределена в окружающей
сплошной дисперсионной среде — газе,
жидкости или твердом теле, в виде мелких частиц
(кристалликов, капелек или'пузырьков). Д. с. вслед-
ствие сильно развитой межфазной поверхности раз-
дела обладают рядом особых характерных свойств:
избытком свободной энергии, повышенной химич.
активностью и адсорбционной способностью, й яв-
ляются обычно термодинамически неустойчивыми.
Д. с. изучаются к о л л о и д н ой хим и’.е й
(физико-химией Д. с. и поверхностных слоев). Гомо-
генные системы, состоящие из отдельных молекул,
атомов или ионов, принято называть молекул ярно-
или ионно-дисперсными, относя к йим всё истинные
р-ры — однофазные системы переменного состава с лю-
бым числом компонентов. Такие системы не от-
носятся к Д. с.
Разнообразные Д. с. широко распространены в при-
роде и часто встречаются в технике. Все горные
породы, грунты и почвы, туманы и облака, выпадаю-
щие из них атм. осадки (дождь, снег), космич. пыль,
а также все ткани живых организмов и продукты их
жизнедеятельности являются Д. с. Образование,
разрушение и свойства Д'. с. играют решающую роль
в генезисе горных пород и почв, в развитии живых
организмов, в технологич. процессах в различных
отраслях пром-сти. Такие процессы, как тонкое
измельчение твердых материалов (см. Диспергирова-
ние), разрушение горных пород при бурении и взры-
вании, обогащение полезных ископаемых (прежде
всего флотация), обезвоживание (деэмульгирование)
нефтей, обработка металлов, смазочное действие,
печатание в полиграфии, отмывание загрязнений при
помощи моющих средств и т. д., целиком основаны
на использовании характерных особенностей Д. с.
Твердые материалы современной техники — металлы
и сплавы, бетоны, материалы на основе полимеров,
волокнистые материалы, резины, так же как и при-
родные материалы — дерево, кожа, растительные и
животные волокна и ткани на их основе — являются
твердыми Д. с. (дисперсными структу-
рами).
Д. с. классифицируются по ряду признаков: 1) дис-
персности, 2) агрегатному состоянию фаз, образую-
щих Д. с., 3) лиофильности, т. е. по интенсивности
молекулярного взаимодействия на границе раздела
между фазами, образующими Д. с., 4) отсутствию
или наличию пространственной дисперсной структуры
(сетки или каркаса) и 5) топографическому приз-
наку — по форме частиц и характерным геометрия,
особенностям распределения фаз в Д. с.
По основной характеристике Д. с. — их ди с-
персиости, они делятся на грубодисперсные
системы с размерами '.частиц более 1 мк (10~* см)
и тонкодисперсные (коллоидные) с размерами ча-
стиц меньше 1 мк или даже меньше 0,1 мк. Частицы
грубодисперсных систем видны в обычный оптич.
микроскоп, не проходят через бумажный фильтр,
не диффундируют. Для тонкодисперсных систем
характерны интенсивное броуновское движение частиц,
а .также необходимость применения специальных
методов исследования — ультрамикроскопии и элект-
ронной микроскопии (т. к. размеры частиц выходят
за пределы разрешающей способности оптич. микро-
скопов), а также ультрацентрифугирования, ультра-
фильтрации и диализа. Кроме того, в области коллоид-
ной дисперсности с дальнейшим уменьшением раз-
меров частиц начинает теряться представление об
агрегатном состоянии дисперсной фазы в обычном
объемном смысле.
По агрегатному состоянию обеих
фаз различают суспензии и дымы (Д. с. с твердой ди-
сперсной фазой и соответственно жидкой или газовой
дисперсионной средой), эмульсии — Д. с., образуемые’
капельками одной жидкости в другой, туманы — Д. с.
с жидкой дисперсной фазой в газовой среде (см..
Аэрозоли). Своеобразную группу Д. с. составляют,
пены — ячеисто-пленочные системы, в к-рых пузырьки
газа разделены тонкими пленками жидкой среды,
образующими непрерывный каркас, и Д. с., образуе-
мые газовыми или жидкими включениями в твердых
фазах, а также частицами твердой фазы, сросшимися
друг с другом (как, напр., в мелкозернистых поли-
кристаллич. твердых телах) или разделенных тонкими
прослойками второй твердой фазы (как, напр., в ке-
рамич. и стеклокерамик, телах разного рода). К таким
Д. с. относятся и пористые тела (сорбенты), напр.
активный уголь, силикагель, ряд ионообменных
смол.
По интенсивности молекулярного взаимодействия
между фазами на их поверхности раздела Д, с! раз-
деляются на лиофильные и лиофобные. Лиофоб-
ные Д. с., где это взаимодействие является слабым
Вследствие' большой разности полярностей образую-
щих их веществ, обладают большим избытком свобод-
ной энергии на единицу площади поверхностного
слоя на границе между фазами (высоким межфазным
поверхностным натяжением) и, следовательно, яв-
ляются термодинамически неустойчивыми. Они са-
мопроизвольно коагулируют или коалесцируют, в них
происходит рост мелких частиц (капелек) вследствие
изотермич. перегонки вещества дисперсной фазы от
мелких частиц к более крупным, т. к. более мелкие
частицы, в соответствии с законом Кельвина—Томсона,
обладают большой растворимостью (большим давле-
нием насыщенного пара). Лиофильные Д. с.
характеризуются интенсивным молекулярным взаи-
модействием между фазами, значительной (но не
безгранично^) взаимной растворимостью образующих
эти фазы веществ вследствие небольшого различия
в их молекулярной природе (полярности). В лиофиль-
ных Д. с. уд> свободная поверхностная энергия на
границе фаз очень мала, хотя и не равна нулю. Лио-
фильные Д. с. образуются самопроизвольно в виде
двухфазных, предельно высокодисперсных систем —
коллоидных р-ров. К таким лиофильным Д. с. отно-
сятся эмульсии, образующиеся вблизи критич. темп-ры
растворения, когда имеет место неограниченное сме-
шение фаз, или при любой темп-ре в присутствии
больших количеств поверхностно-активных веществ.
Таковы, напр., растворимые масла или эмульсолы —
минеральные масла с большим количеством коллоидно-
растворенных поверхностно-активных веществ — мыл
карбоновых к-т или сульфокислот с нек-рым избытком
соответствующей органич. к-ты; такие масла само-
произвольно эмульгируются в воде или в водио-
щелочной среде с образованием коллоидных р-ров
1153
ДИСПЕРСНЫЕ СИСТЕМЫ — ДИСТИЛЛЯЦИЯ
1154
или тонкодисперсных эмульсий, широко применяемых
в качестве замасливателей, смазочно-охлаждающих
жидкостей для обработки металлов, в препаратах для
борьбы с с.-х. вредителями. В отличие от лиофильных,
лиофобные Д. с. требуют стабилизации, т. е. введения
веществ, адсорбирующихся на поверхности частиц
дисперсной фазы и образующих защитные слои,
препятствующие сближению частиц друг с другом
(см. Мицелла, Стабилизация дисперсных систем).
По степени структурированности
различают: а) бесструктурные Д. с. со свободными J
частицами дисперсной фазы — золи (коллоидные р-ры),
агрегатное состояние к-рых определяется дисперсион-
ной средой; б) структурированные Д. с. — гели;
к ним относятся также пленочно-ячеистые Д. с. —
дисперсные структуры, наз. пенами, или конц. эмуль-
сии. При достаточно прочной пространственной
сетке такие дисперсные структуры приобретают ряд
свойств твердых тел, независимо от агрегатного со-
стояния дисперсионной среды и дисперсной фазы
в отдельности. Сюда же относятся аэрогели или
ксерогели — пористые тела с системой сообщаю-
щихся тонких открытых пор (капиллярно-пористые
тела типа углей, силикагелей, получающихся удале-
нием свободной воды из студней кремнекислоты, и др.
сорбенты). Структурированные Д. с. (к ним относятся,
в частности, и ткани живых организмов) характери-
зуются при неразрушенной структуре довольно вы-
сокой вязкостью структурной и упругостью формы.
При разрушении пространственной структуры струк-
турированные Д. с. переходят в Д. с. со свободными
частицами, такое превращение золь — гель является
обратимым (тиксотропия); в покое структура по-
степенно снова развивается в результате сцепления
частиц при соударениях в броуновском движении
(см. Структурообразование в дисперсных системах).
По топографическому признаку
наряду с Д. с., частицы к-рых обладают малыми
размерами по трем координатным направлениям
(т. е. частицы, к-рые можно приближенно рассмат-
ривать как эквивалентные по дисперсности шарикам
или кубикам), различают Д. с., в к-рых частицы
дисперсной фазы по форме могут быть и отдельными
листочками или чешуйками, имея по двум направле-
ниям большие размеры (длину и ширину) и лишь
очень малую толщину. Частицы дисперсной фазы
могут представлять собой и нити (волокна) произ-
вольно большой длины, но малой толщины. В обоих
случаях в пленочных (ламинарных) и волокнистых
(фибриллярных) Д. с., т. е. в системах с резко анизо-
диаметричными частицами, дисперсность дисперсной
фазы любой массы (объема) не зависит от того, состоит
ли дисперсная фаза из множества таких частиц или
из одной тонкой пленки или тонкой нити, к-рые
в этом случае вместе с окружающей средой также
следует рассматривать как своеобразную Д. с., уд.
поверхность к-рой обратно пропорциональна толщине
пленки или нити.
По этому признаку, т. е. по распределению в про- :
странстве, надо также различать обычные трехмерные
Д. с., в к-рых частицы дисперсной фазы распределены
в объеме дисперсионной среды, двухмерные и одно-
мерные Д. с., в к-рых эти частицы расположены в виде
одночастичного слоя (дисперсного покрытия) на
поверхности данного тела (фазы) или внутри нити
(волокна). Мономолекулярные слои поверхностно-
активных веществ, напр. на поверхности воды, пред-
ставляющие собой вещество в особом двухмерном
состоянии, могут в определенных условиях распа-
даться на отдельные «островки» коллоидных разме-
ров, образуя двухмерную коллоидную (двухфазную)
систему (см. также Оптические свойства коллоидных
систем, Электрические свойства дисперсных систем).
*/219 К. X. Э. т. 1
Лит.: Песков Н. П. и Александрова-
Прейс Е. М., Курс коллоидной химии, 2 изд., М.—Л.,
1948; Alexander J., Colloid chemistry, 4 ed., N. Y.,
[1945]; его же, Colloid chemistry, v. 1—7, N. Y., 1926—50;
Freundlich H., Kapillarchemie, 4 Aufl., Bd 1—2, Lpz.,
1930—32; Наука о коллоидах, под ред. Г. Кройта, пер. с англ.,
т. 1, М., 1955. 17. А. Ребиндер.
ДИСПРОЗИЙ (Dysprosium) Dy — химич. элемент
с и. н. 66, относится к лантанидам.
ДИСПРОПОРЦИОНИРОВАНИЕ — процессы са-
моокисления-самовосстановления. При Д. одновре-
менно образуются соединения, в к-рых элемент на-
ходится в более окисленном и более восстановлен-
ном состоянии по сравнению с первоначальным.
Легкость Д. связана с близостью энергетич. уровней
валентных электронов в обоих состояниях атома.
Примером Д. с изменением валентности могут служить
процессы самоокисления-самовосстановления нек-рых
соединений Cl, Sn, Hg, S, Np, Pu, Am, напр.:
(+5» (4-7)
4KC1O3 -► KC1 -f- 3KC1O4
(-2) (0) (4-2? +4) (4-1) (0) (+S>
4SnO — Sn 4- Sn3O4; Hg2O Hg + HgO
(4-4> (—2) (4-6) (+6)
3R.-SOON —*-R—S—SO—R + RSO2-OH + H2O
<+5> (-G> <4-41
2NpO2 -» NpOl+ + NpO
3Pu4+ 4- 2HgaO —> 2Pu3+ + PuO2 + 4H+
<4-5> (4-6) 9.
3PuOa + 4H+ —>• PuS+ + 2PuO2 + 2H3O
(4-5) 1 (4-6) 01. (4-4)
2AmOa —► AmOj 4- AmOa
(последний сразу же восстанавливается до Am3).
Реакции Д. могут протекать и без изменения валент-
ности:
2НСО- COS- + н2со3 6В2Н5С1 - 2В2Н, + 2ВС13
ЗСеН10 - 2СвН12 + С,Н, ЗСвН„ - СвН>2 + 2С,Н.
2СН2О + Н2О СНзОН + НСООН
C«HtNH — NHC«HS - C«HSN = NC«H5 + 2CeH5NH2
См. также Канниццаро реакция, Тищенко реакция.
Д. И. Кузнецов.
ДИСТЕКТИКА (дистектическая точка) — плавный
максимум на диаграммах состояния двойных систем,
отвечающий составу химич. соединения, несколько
диссоциированного в точке плавления. См. Двойные
системы.
ДИСТИЛЛЯЦИЯ (перегонка) — процесс разде-
ления жидких смесей на фракции различных составов
путем их частичного испарения с последующей кон-
денсацией образовавшихся паров.
В металлургии под термином Д. понимается получение
цветных металлов (цинка, магния, ртути и др.) из руд или
Рудных концентратов путем их перевода в парообразное
состояние с последующей конденсацией.
смеси, непрерывного
Рис. 1. Схема процесса про-
стой дистилляции.
Д. подразделяют на простую и равновесную.
Простая Д. производится путем частичного
испарения кипящей жидкой
отвода и конденсации обра-
зовавшихся паров. Полу-
ченный конденсат наз. ди-
стиллятом, а неиспарив-
шаяся жидкость — кубовым
остатком. На рис. 1 пред-
ставлена схема простой Д.
В дистилляционном кубе
1 кипит исходная жидкая
смесь. Образующиеся пары
непрерывно отводятся в
конденсатор 2, откуда конденсат, являющийся ди-
стиллятом, стекает в приемник 3. При теоретич.
рассмотрении процессов Д. исходят из того, что
парообразование происходит очень медленно и что
между жидкостью и паром устанавливается равно-
весие. В процессе простой периодич. Д. содержание
низкокипящих компонентов в жидкой и паровой
фазах непрерывно падает. Если количество загру-
1155
ДИСТИЛЛЯЦИЯ
1156
женной в куб жидкой смеси с концентрацией xt
молярных долей низкокипящего компонента равно wf
кг-молей, а количество кубового остатка с концентра-
цией х0 равно w0 кг-молей, то средняя концентрация
дистиллята определяется ур-нием
х _ta,i3ci~ta,oyo
Д W — W
1 о
При этом соотношение между количеством кубового
остатка и его концентрацией описывается уравнением
(1)
In — = \
*<1
dx
V — х
где у — концентрация пара, равновесного жидкости,
содержащей х молярных долей низкокипящего компо-
нента. В общем случае решение интеграла последнего
ур-ния производится графически при помощи диа-
граммы равновесия у — х. В тех случаях, когда
зависимость у от х может быть выражена простым
ур-нием, ур-ние (2) может быть проинтегрировано.
Так, напр., в случае бинарных смесей, подчиняющихся
закону Рауля:
__ ЧХ________
У 1 -+-Ж (а — 1)
где а = Р^/Рв— коэфф, разделения — отношение дав-
ления паров низкокипящего компонента А к давле-
нию паров вышекипящего компонента В при одина-
ковой темп-ре. Подставляя значение у в ур-ние (2),
получаем ур-ние (4):
а
Ш1 __ «О Zi — Ж0 _ Ж! у — 1
Х1 ' Хо 1 —
Простая Д., осуществляемая по схеме, изображен-
ной на рис. 1, является периодич. процессом. Полу-
непрерывная Д. предполагает непрерывное поступле-
ние в дистилляционный куб w[ кг-молей/час исходной
смеси с концентрацией хг и отвод такого же количества
дистиллята. В этом случае значение х определяют
из ур-ния
где wt — количество смеси, загруженной в куб,
кг-молей, аг — продолжительность процесса в часах;
связь между wQ и х0 описывается ур-нием
__ С dx
~ V ~ х
(6)
где у — концентрация пара, равновесная текущей
концентрации кубовой жидкости.
При фракционной Д. (дробной перегонке) (рис. 2),
применяемой для разделения жидкостей на фрак-
ции, кипящие в узких
интервалах темпера-
тур, дистилляты раз-
ных составов отводят
в несколько сборни-
ков (последовательно
во времени). При этом
в сборник I поступа-
ет первая по времени
Рис. 2. Схема процесса фракцион- порпия дистиллята,
ной дистилляции. наиболее богатая низ-
кокипящими компо-
нентами, в сборник II — менее богатый дистиллят, в
сборник III — еще менее богатый дистиллят и т. д.
В каждом из этих дистиллятов (фракциях) преобладает
один или несколько компонентов исходной смеси
с повышающимися темп-рами кипения. Для улучше-
ния разделения простая Д. часто комбинируется
с противоточной дефлегмацией (рис. 3). Образующиеся
при этом пары частично конденсируются, конденсат
непрерывно возвращается в куб, а остаток паров
отводится в виде ди-
стиллята. Этим дости-
гается большее обо-
гащение дистиллята
низкокипящими ком-
понентами, т. к. при
частичной конденса-
ции (дефлегмации) па-
ров преим. конденси-
руются вышекипящие
компоненты. Практич.
применение рассмот-
ренных процессов Д.
Рис. 3. Схема комбинированного
процесса простой дистилляции с
противоточной дефлегмацией.
ограничено смесями с
высокими, коэффициентами разделения. Дистилляци-
онные кубы обычно представляют собой вертикаль-
ные или горизонтальные котлы, снабженные различ-
ными поверхностями теплообмена для парового или
газового нагрева. Выбор металла (сталь, чугун, цвет-
ные металлы и т. д.) для кубов обусловливается гл.
обр. агрессивностью дистиллируемых жидкостей.
При газовом нагреве для увеличения активной
поверхности куба и выпаривания дистиллируемой
жидкой смеси в тонком слое часто применяют кубы
с вогнутыми днищами, широко используемые в уста-
новках для дистилляции смол. Ввиду ограниченной
поверхности нагрева дистилляционных кубов с паро-
выми рубашками более целесообразны кубы с внутрен-
ними нагревательными змеевиками или трубчатыми
нагревательными элементами. При высоких темп-рах
кипения дистиллируемой жидкости для обогрева
часто используют высококипящие жидкие органиче-
ские теплоносители (дифенил, дифениловый эфир
и др.), к-рые отличаются более мягким режимом
нагрева, большим коэфф, теплопередачи и гибкостью
регулировки процесса.
Равновесная Д. (однократное испарение),
часто применяющаяся в нефтяной пром-сти, харак-
терна испарением части жидкости и продолжительным
контактом образовавшихся паров с неиспарившей-
ся жидкостью до достижения фазового равнове-
сия. Разделяемая смесь
проходит по трубам, обо-
греваемым снаружи то-
почными газами (рису-
нок 4). Образовавшаяся
при этом парожидкостная
смесь, близкая к равно-
весному состоянию, по-
ступает в сепаратор для
механич. Отделения ЖИД- рис 4 схема процесса равно-
кости от пара. В нек-рых весной дистилляции.
случаях нагрев смеси
в трубах происходит под избыточным давлением;
в этом случае жидкость перед входом в сепаратор
дополнительно испаряется за счет дросселирования.
Пары из сепаратора поступают в конденсатор. По-
скольку при такой Д. образовавшийся пар и жид-
кость близки к равновесию, то из ур-ния материаль-
ного баланса можно определить соотношение между
количествами образовавшегося пара П и оставшейся
ЖИДКОСТИ Wg
U
Х1 — х2
У2 — Xi
(7)
где xL и х2 — начальная и конечная концентрация
жидкости, у2 — концентрация пара, равновесного
с жидкостью состава х2. Равновесная Д. редко приме-
няется для двухкомпонентных смесей, но часто дает
хорошие результаты в случае многокомпонентных
1157
ДИСТИЛЛЯЦИЯ В ТОКЕ ВОДЯНОГО ПАРА И ИНЕРТНЫХ ГАЗОВ
1158
Жадность
Рис. 1. Схема установки для пе-
регонки с водяным паром: а —
при периодическом процессе;
б — при Г-~:_г------
смесей, из к-рых можно получить фракции, сильно
различающиеся по составу.
Лит..: Гельперин Н. И., Дестилляция и ректифи-
кация, М,—Л., 1947; Касаткин А. Г., Основные про-
цессы и аппараты химической технологии, 6 изд., М., 1955;
Трегубов А. М„ Теория перегонки и ректификации,
3 изд., Баку, 1946; Циборовский Я., Процессы хими-
ческой технологии, пер. с польск., М., 1958; Kirschb a u m
Е Destillier- und Rektifiziertechnik, 2 Aufl., В. [и. а.]. 1950;
Robinson С. S., Gilliland Е. R., Elements of
fractional distillation, 4 ed., N.Y. [a. 0.1,1950: Tre у b al R. E„
Mass-transfer operations, N. Y. [a. o.l, 1955.
H И. Гельперин, В. Л. Пебалк.
ДИСТИЛЛЯЦИЯ В ТОКЕ ВОДЯНОГО ПАРА И
ИНЕРТНЫХ ГАЗОВ — применяется при необходи-
мости понижения темп-ры отгонки (особенно при
нетермостойких компонентах), а также для отгонки
летучих веществ от компонентов с низкой летучестью.
Дистилляция с водяным паром
производится пропусканием через слой жидкости
насыщенного или перегретого водяного пара; при-
менима при взаимной нерастворимости отгоняемо-
го компонента и воды. Периодический процесс (рису-
нок 1, а) проводится в обычных дистилляцион-
ных кубах, снабженных
барботером; непрерыв-
ный — в колоннах с на-
садкой (рис. 1, б). Водя-
ной пар вводится через
барботер и дисперги-
руется на мелкие пу-
зырьки. Образовавшая-
ся смесь паров воды и
летучего компонента
отводится из аппарата
и подвергается конден-
сации и охлаждению,
иидичсипиш ирицсььс; ~
непрерывном процессе. Конденсат разделяется
на два слоя, что облег-
чает отделение продукта. Если теплоты, заключен-
ной в пропускаемом через жидкость паре, недоста-
точно для осуществления процесса, то аппараты снаб-
жаются паровыми рубашками для внешнего подогрева.
При взаимной нерастворимости воды и дистиллируе-
мой жидкости парциальные давления и концентрации
их паров не зависят от состава жидкости. Так как
полное давление паровой смеси Р равно сумме пар-
циальных давлений паров компонентов, то
р = /’а + 7’н2о
(1)
Температура, °C
Рис. 2. Диаграмма давления паров органич. жидкостей,
перегоняемых с водяным паром.
Следовательно, темп-ра кипения жидкой смеси в рас-
сматриваемом процессе всегда ниже темп-ры кипе-
ния каждого из компонентов смеси при данном давле-
нии. При дистилляции с водяным паром темп-ру
кипения смеси удобно определять по кривым давления
паров (рис. 2), причем давление водяного пара отло-
жено на диаграмме не от 0, а вниз от давления, при
к-ром проводится дистилляция (на рис. 2, 700, 300
и 50 мм рт. ст.). Точки пересечения кривых давления
водяного пара с кривыми давления пара различных
жидкостей указывают искомые темп-ры кипения.
При дистилляции водонерастворимых жидких смесей
в токе перегретого водяного пара количество послед-
него СНг0 кг и низкокипящего компонента GA кг в па-
ровой фазе определяют из след, теоретич. соотношения
(в этом ур-нии не учитывается расход пара на нагре-
вание и испарение дистиллируемой смеси):
ga/gh2o ==-paA7a/-ph2o^h2o (2)
где А/л и А7Нго — мол- веса компонента А и воды.
Так как уходящие из аппарата водяные пары практи-
чески не полностью насыщаются парами отгоняемого
компонента, то при определении расхода водяного
пара СНг0 в расчет вводят коэфф, насыщения <р=
=0,5—0,9, характеризующий степень насыщения водя-
ного пара парами отгоняемого компонента; величина
этого коэфф, зависит от свойств дистиллируемого
вещества, высоты слоя жидкости над барботером (Н)
и размера пузырьков пара (d). Эта зависимость при-
ближенно выражается эмпирич. ф-лой:
_ кн
<Р = 1 - е d (3)
где е — основание натуральных логарифмов; К —
константа, зависящая от свойств дистиллируемого
вещества (определяется экспериментально). Если за-
гружаемая исходная жидкая смесь содержит воду,
то ее нагревание до темп-ры кипения обычно ведут
глухим паром или внешним нагревом, чтобы не обвод-
нять дистиллируемую смесь конденсирующимся бар-
ботирующим паром. Для испарения отгоняемого
вещества тепло можно подводить частично внешним
нагревом и частично барботирующим паром путем
его перегрева. При дистилляции с водяным паром
компонента, находящегося в нелетучем растворителе,
с течением времени изменяется концентрация р-ра,
а вместе с ней и парциальное давление перегоняемого-
компонента над р-ром. Для такого процесса ур-ние
(2) может быть использовано лишь для бесконечна
малого промежутка времени, в след, виде:
dDfdW h =?Н20/Ра=(Р — ?А)/рА (4)
где dD и dWA —количество водяного пара и летучего-
компонента А в паровой смеси, кг-моль; рн 0 и рА—
парциальное давление воды и летучего компонента А
в паровой смеси в данный момент времени; Р — дав-
ление, при к-ром протекает процесс дистилляции.
Если к определенному моменту кубовая жидкость
содержит WA кг-моль летучего компонента и И7В
кг-молей нелетучего растворителя, то для идеальных
р-ров, согласно закону Рауля:
WA
Ра — И’А-;- И’н РА = х АР А (5)
где хА — молярная доля летучего компонента А
в растворе. Подставляя значение рА в ур-ние (4)
и решая его для постоянной темп-ры дистилляции,
получим:
+ (б>
где W'A и WA — начальное и конечное количество
летучих компонентов в жидкости, кг-моль. Ур-ние (6)
позволяет определить расход пара на отделение
(ЖА — ЖА) летучего компонента из кубовой жид-
*/s19*
1159
ДИСТИЛЛЯЦИЯ МОЛЕКУЛЯРНАЯ
1160
кости. Если дистиллируемый р-р не является идеаль-
ным, то определение D значительно усложняется,
т. к. требуется знание давления пара компонента А
над растворами разных концентраций, и осуществ-
ляется графич. интегрированием.
Непрерывный процесс дистилляции в токе водяного
пара в колоннах с насадкой (рис. 1, б) в принципе
похож на процесс абсорбции. Дистилляцию в токе
водяного пара целесообразно вести при максимально
допустимых темп-рах, т. к. в этом случае возрастает рд,
а следовательно, сокращается расход пара (см.
ур-ние 2). При необходимости большего понижения
темп-ры дистилляции процесс ведется под вакуумом.
При дистилляции в токе водяного пара обычно ис-
пользуется перегретый водяной пар (во избежание
его конденсации в аппаратуре). К недостаткам про-
цесса дистилляции в токе водяного пара относятся:
большой расход барботирующего пара и охлаждаю-
щей воды, частичная потеря отгоняемого продукта
с конденсационной водой, необходимость в нек-рых
случаях его сушки и т. и.
Большинство перечисленных недостатков устра-
няется при дистилляции в токе инерт-
ных газов. Компоненты раствора в этом случае
будут испаряться в поток газа даже если раствор не
кипит, парообразование при испарении может про-
исходить при любых темп-рах, вне зависимости от
внешнего давления, что позволяет вести процесс
дистилляции в токе инертных газов при весьма низких
темп-рах. Расход инертного газа на отгонку задан-
ного количества летучих веществ может быть опре-
делен по ур-нию (2) при замене в нем и 0
парциальными давлениями отгоняемого компонента и
газа. К недостаткам дистилляции в токе инертных
газов, ограничивающим область ее практич. приме-
нения, относятся трудность полного извлечения отго-
няемого вещества из газового потока, а также громозд-
кость подогревателей и конденсаторов в связи с низ-
ким коэфф, теплоотдачи газов.
Лит. см. при ст. Дистилляция.
Н. И. Гельперин, В. Л. Пебалк.
ДИСТИЛЛЯЦИЯ МОЛЕКУЛЯРНАЯ — процесс
разделения жидких смесей путем свободного испа-
рения в вакууме (10~3—101 ммрт. ст.) при темп-ре
ниже точки их кипения; осуществляется при рас-
положении поверхностей испарения и конденсации
на расстоянии, меньшем длины свободного пробега
молекул перегоняемого вещества (20—30 мм). Бла-
годаря вакууму молекулы образовавшегося пара
движутся с минимальным числом столкновений от
испаряющей поверхности к конденсирующей. В целях
полной конденсации пара на конденсирующей поверх-
ности поддерживается значительно более низкая
темп-ра (на 50—100°), чем на поверхности испарения.
При Д. м., в отличие от простой, полное давление
паров над разделяемой жидкой смесью ниже равно-
весного при данной темп-ре, поэтому жидкость испа-
ряется ниже точки кипения. При Д. м. изменение
состава пара по сравнению с составом жидкости
определяется различием скоростей испарения компо-
нентов, к-рые могут быть-1вычислены по ф-ле Ленг-
мюра (1):
п = РРу' 2nMRT
где п — скорость испарения компонента жидкой
смеси, молъ/сек', Р — давление паров этого компо-
нента, дин/см2; М — мол. вес; R — газовая постоян-
ная (8,3, • 107 эрг/°К); Т — темп-ра, °К; F — поверх-
ность испарения, см2. На практике скорость Д. м.
составляет от 70 до 90% от ее теоретич. величины.
По ур-нию Ленгмюра относительное количество ком-
понентов в дистилляте пропорционально отношениям
PJ У Ми Р2/ УМ2, ...,Рп/ У Mn. W Р1( Р2.....Рп-~
давления паров первого, второго и n-го компонентов;
Mlt М2, ..., М п — мол. веса первого, второго и
n-го компонентов. Следовательно, достигаемое при
Д. м. разделение пропорционально отношению
аМ = р; V <2)
Как видно из ур-ния (2), Д. м., в отличие от обыч-
ной, позволяет разделить смеси, компоненты к-рых
обладают одинаковой упругостью паров. И наоборот,
если упругости паров компонентов смеси при данной
темп-ре таковы, что PJ УМг — Р2/ УМ2 и т. д.,
то смесь не может быть разделена методом Д. м. В
нек-рых случаях путем изменения темп-ры процесса
можно все же подобрать условия, когда и такие
смеси будут разделяться Д. м. Эффективность про-
цесса Д. м. характеризуется «теоретич. молекуляр-
ными тарелками» (ТМТ); под этим термином пони-
мают степень разделения, при к-рой относительные
молярные концентрации компонентов в дистилляте
в точности соответствуют относительным скоростям
испарения этих компонентов (ам), при условии, что
состав жидкости на поверхности испарения идентичен
составу всей дистиллируемой жидкости. Т. к. нет
теоретич. методов определения числа ТМТ, дости-
гаемого в молекулярных кубах различных конструк-
ций, то эффективность процесса разделения и сравни-
тельная характеристика различных кубов устанав-
ливается лишь на основе экспериментальных данных.
При данной темп-ре жидкости и соответствующем ей
давлении паров скорость Д. м. растет с понижением
давления в аппарате.
Процесс переноса вещества при Д. м. состоит из
след, стадий: диффузии молекул более летучего ком-
понента из глубины слоя жидкости к поверхности
испарения; испарения молекул с поверхности; пере-
хода молекул с поверхности испарения на поверх-
ность конденсации; конденсации молекул на поверх-
ности конденсатора. Лимитирующей обычно является
первая стадия (диффузия молекул), поэтому для интен-
сификации процесса необходимы условия, обеспечи-
вающие наибольшую скорость диффузии к поверх-
ности испарения. В современных молекулярных
кубах это обеспечивается тем, что процесс испарения
протекает в очень тонкой пленке
жидкости (0,01—0,05 мм), уменьша-
ющей время нахождения вещества
на поверхности испарения и опас-
ность его термического разложения.
Конденсация молекул на поверхно-
сти конденсатора при большой раз-
ности температур (обычно 50—100°)
Рис. 1. Аппарате
горизонтальной
поверхностью ис-
парения: 1 — по-
верхность испаре-
ния; 2 — поверх-
ность конденса-
ции; 3 — вакуум;
4 — нагреватель-
ный прибор; 5 —
хладо агент.
происходит почти мгновенно.
Аппараты для Д. м. разделяют на
аппараты с горизонтальной поверх-
ностью испарения, аппараты с «па-
дающей» пленкой и «центробежные»
аппараты (с вращающимся испари-
телем). Аппараты с гори-
зонтальной поверхно-
стью испарения (рис. 1),
над к-рой располагается конденсатор,
применяются при однократной пери-
одич. молекулярной дистилляции.
Жидкая смесь во время дистилляции находится
в неподвижном состоянии, а толщина ее слоя отно-
сительно велика (от 1 мм до нескольких сантимет-
ров). Конструкция аппарата проста, но имеет суще-
ственные недостатки: дистиллируемое вещество под-
вергается длительному воздействию высокой темпе-
ратуры; продолжительность диффузии компонентов к
1161 ДИСТИЛЛЯЦИЯ МОЛЕКУЛЯРНАЯ — ДИТИОКАРВАМИНОВОЙ КИСЛОТЫ СОЛИ 1162
поверхности испарения весьма велика. Аппараты
с «падающей» пленкой (рис. 2) характерны
малой толщиной пленки (0,1—0,5 мм) и сокращенным
временем пребывания дистиллируемой смеси на по-
верхности нагрева (1—3 мин). Дистиллируемая жид-
кость стекает по вертикальной
поверхности цилиндра-испари-
теля, а дистиллят собирается
на конденсационной поверх-
ности, расположенной вокруг
испарителя. Эффект разделе-
ния такого аппарата составляет
Рис. 2. Аппарат с падающей пленкой: 1 — испаритель; 2 —
конденсатор; 3 и в — вход и выход воды; 4 — распределитель
жидкости; 5 — проволочная спираль; 7 — отвод дистиллята;
з — отвод остатка.
Рис. 3. Центробежный аппарат для молекулярной дистил-
ляции: 1 — ротор-испаритель; 2 — конденсатор; 3 — посту-
пающая смесь; 4 — желоб; 5 — отвод остатка.
Примечание. С поверхности конич. конденсатора
дистиллят непрерывно отводится по спец, устройствам.
0,3—0,5 ТМТ. Центробежные аппараты
(рис. 3) отличаются наименьшей толщиной жидкой
пленки (в среднем 0,05 мм) и временем ее пребывания
на поверхности нагрева (0,03—1,2 сек). Испаритель
представляет собой быстровращающийся конус
(в нек-рых конструкциях диск), на к-рый подается
разделяемая смесь. Центробежная сила прижимает
пленку жидкости к поверхности конуса (ротора)
и перемещает ее от центра к периферии (вверх).
Пары перегоняемого вещества собираются на непо-
движном конденсаторе, расположенном параллельно
поверхности ротора. Остаток после перегонки сбра-
сывается в специальный кольцевой желоб и выводится
из куба. Толщина жидкой пленки в аппарате изме-
няется от (1,08 мм внизу до 0,04 мм у верха ротора.
При диаметре ротора в 1,5 м и поверхности нагрева
ок. 4 м2 производительность аппарата составляет
750 кг!час. Эффективность центробежных молекуляр-
ных аппаратов составляет 0,8—0,95 ТМТ. В промыш-
ленной практике наибольшее распространение полу-
чили аппараты с «падающей» пленкой и центробеж-
ные. При необходимости иметь большой эффект раз-
деления (измеряемый несколькими ТМТ) прибегают
к последовательной установке нескольких аппаратов.
В промышленной и лабораторной практике Д. м.
применяется для разделения и очистки высокомолеку-
лярных и термически нестойких органич. веществ.
Так, напр., этот процесс находит применение для
очистки эфиров фталевой, себациновой, стеариновой,
олеиновой и других к-т, для выделения витаминов из
рыбьего жира и различных растительных масел,
для произ-ва медицинских препаратов, стеролов и
стеринов, в произ-ве пластификатов, вакуумных
масел, смазок и др.
Лит.: Жаворонков Н. М., Малюсов В. А.,
Хим. пром., 1950, № 11,с.29; Ks 12,с. 20; М а т р о з о в В. И.,
Аппаратура для молекулярной дистилляции, М., 1954; его
ж е, в кн.: Исследования сублимационных и дистилляционных
аппаратов и гидродинамики мешзлок. М., 1954, с. 63—87;
Жаворонков Н. М., Майер А. И., в кн.: Методы
19 К. X. Э. т 1
и процессы химической технологии, сб. 1, М.—Л., 1955,
с. 5—44; Малюсов В.. А., Малофеев Н. А.,
Жаворонков Н. М., Хим. пром., 1958, № 1, с. 31: М а-
люсов В. А., Умник Н. Н., Жаворонков Н. М„
там же, 1958, №5, с. 36; Hickman К. С. D., TrevoyD.J.,
Ind. Engng. them., 1952, 44, № 8, p. 1882; Burrows G.,
Trans. Instn. Chem. Engrs., 1954, 32, № 1, p. 23—34.
H. И. Гельперин, В. Л. Пебалк.
ДИТИЗОН (дифенилтиокарбазон), мол. в. 264,32,
C6HSN=N—CS—NHNHC6HS — кристаллы сине-чер-
ного цвета, перекристаллизовываются из смеси хло-
роформа и этилового спирта; нерастворимы в воде,
очень мало растворимы в этиловом спирте и эфире
(раствор темно-красного цвета), несколько лучше —
в хлороформе или четыреххлористом углероде (рас-
твор зеленого цвета); растворимы в H2SO4, в рас-
творах едких щелочей и карбонатов щелочных метал-
лов. Получают Д. обрабатывая спиртовой р-р дифе-
нилтиокарбазида (из фенилгидразина и сероуглерода)
р-ром КОН и подкисляя продукт реакции. Со многими
катионами Д. образует окрашенные внутрикомплекс-
ные соли (дитизонаты), растворимые с красной или
малиновой окраской в СНС13, СС14 и CS2. В виде
0,003—0,005%-ного р-ра в хлороформе Д. широко
применяется для разделения и фотометрия, определе-
ния Ag, Bi, Cd, Со, Си, Hg, Zn, Pb, In и др.; для
экстракционного обогащения металлов при определе-
нии их следов методом спектрального анализа.
Реакции ионов металлов с Д. отличаются очень высо-
кой чувствительностью.
Лит.: Иванчев Г., Дитизон и его применение в микро-
анализе и анализе следов элементов, пер. с нем., М., [19611.
Д. И. Васкевич.
ДИТИЛИН (миорелаксин, сукцинилхолин, дииод-
метилат бис-fJ-диметиламиноэтилового эфира янтарной
К-ТЫ) (СНз)3ХСН2СН2ОСОСН2СН2СООСН2СН2Й(СН3)3 • 2J-,
мол. в. 544,23 — бесцветные кристаллы; т. пл. 247—
248°. Д. хорошо растворим в воде, трудно — в
спирте и ацетоне, нерастворим в эфире, бензоле и
толуоле. Водлые растворы Д. имеют нейтральную
реакцию. Ёсли к смеси Д. и резорцина прибавить
конц. H2SO4 и осторожно нагревать, появляется
красновато-коричневое окрашивание; по охлаждении
полученной смеси и подщелачивании ее р-ром NaOH
образуется оранжево-красный р-р с интенсивной
зеленой флуоресценцией. Для колич. определения
к водному р-ру Д. прибавляют несколько мл разб.
уксусной к-ты и 5 капель 0,5%-ного водного р-ра
эозиннатрия и титруют 0,1 н. р-ром AgNO3 до изме-
нения окраски от розовой до пурпурно-красной. Д.
получают взаимодействием (3-диметиламиноэтанола
с хлорангидридом янтарной к-ты и обработкой обра-
зующегося при этом р-диметиламиноэтилового эфира
янтарной к-ты иодистым метилом. Д. применяют гл.
обр. при кратковременных хирургия, вмешатель-
ствах, для расслабления скелетных мышц.
Лит.: Дитилин и опыт его клинического применения (Сб.
ст.). Под ред. А. Л. Мнджояна, Ереван, 1957; См. также при
ст. Апрессин. А. И. Травин.
ДИТИОКАРВАМИНОВОЙ кислоты соли —
активные фунгициды для борьбы с болезнями расте-
ний. Наибольшее практич. применение получили
соли этиленбисдитиокарбаминовой к-ты — натриевая,
цинковая и марганцовая, а также цинковая и желез-
ная соли диметилдитиокарбаминовой к-ты, обладаю-
щие наиболее сильным действием на широкий круг
возбудителей болезней растений, в том числе на
мильдью винограда, паршу яблонь, серую гниль
винограда, болезни сливы и др. Средняя норма
2—4 кг/га (в зависимости от вида растений).
Диметилдитиокарбамат цинка
(ДДКЦ) — белые кристаллы; т. пл. 244°; плохо
растворим в воде (при 25° в 1 л 0,065 г), мало ядовит
для животных; применяется в виде смачивающегося
порошка (водная суспензия), содержащего 50—70%
действующего начала. ДДКЦ получают взаимодей-
1163
ДИТИОФОС — ДИФЕНИЛГУАНИДИН
1164
ствием диметиламина, сероуглерода и едкого натра;
затем на образовавшуюся натриевую соль действуют
сернокислым цинком:
(CH3)2NH + CS3 + NaOH — (CH3)2NCSSNa + H2O
2 (CH3)aNCSSNa+ ZnSO4 —' [(CH3)2NCSS]2Zn 4- Na2SO4
Диметилдитиокарбамат железа —
черные кристаллы, плохо растворим в воде; ЛД50
4 г/кг (для крыс); его применяют аналогично ДДКЦ.
Этиленбис (дитиокарбамат) цинка
(ЦНИЭБ) — белый порошок; практически нераство-
римый в воде (0,001 г/л); ЛД30 5,2 г/кг. ЦНИЭВ
получают так же, как и ДДКЦ, только в качестве
исходного вещества берут этилендиамин:
CH..NH3 CH.,NHCSSNa
I ' + 2CS3 + 2NaOH — I + 2H2O
CH2NH2 CH.NHCSSNa
CHaNHCSSNa CH.,NHCSS4
I + ZnSO4 — | ' >Zn + Na"SO4
CH»NHCSSNa CIL.NIICSS/
ЦНИЭВ применяют в виде смачивающегося порошка
с содержанием действующего начала 50— 70%; для
борьбы с большинством болезней растений исполь-
зуется в виде водных суспензий 0,25—0,35% (по
действующему началу). Этот препарат обладает не-
большим стимулирующим действием, что приводит
к нек-рому повышению урожая; кроме того, он исполь-
зуется также в качестве протравителя семян злаков
и овощных культур против головни и др. заболеваний.
Этиленбис (дитиокарбамат) мар-
ганца (МАИЭВ) применяется и получается анало-
гично этиленбис(дитиокарбамату)цинка и обладает
тем же действием. Применяется также хорошо раство-
римая в воде натриевая соль этилен-
бис (дитиокарбаминовой) кислоты
(НАВАМ), к-рая используется как системный фунги-
цид. Довольно широкое применение в качестве сред-
ства стерилизации почвы от сорняков, нематод,
насекомых и грибов находит натриевая соль м е-
тилдитиокарбаминовой кислоты
(ВАПАМ), к-рая используется для поливки почвы
(в виде 2—3%-ного р-ра за 2—3 недели до посадки
возделываемой культуры). Получают ВАПАМ из
монометиламина, сероуглерода и едкого натра в вод-
ной среде; в кристаллич. состоянии он недостаточно
стабилен и разлагается при хранении; в водном р-ре
может храниться неограниченно долгое время.
Среди солей дитиокарбаминовой кислоты имеются
пенные аналитич. реагенты для фотометрии, определе-
ния многих тяжелых металлов, их отделения и обога-
щения. См. также Диэтилдитиокарбаминат натрия.
Лит.: Frear D. Е. Н., Chemistry ot the pesticides,
3 ed., Toronto — N. Y. — L., 1955; Zbirovsky M., M y-
S k a J., Insekticidy, tungicidy, rodenticidy, Praha, 1957.
H. H. Мельников.
ДИТИОФОС — см. Фосфорорганические инсектициды.
ДИТРАЗИН (гетразан, цитрат-1-диэтилкарбамоил-
4-метилпиперазина) CieH29O8N3. мол в. 391,43 —
,СН2~СН2\
H3C-N< )n-CO-N(C2H5)2-C6H8O7
'сн2-сн/
бесцветные кристаллы; т. пл. 136—137,5° (с разл.).
Д. хорошо растворим в воде и спирте, нерастворим
в бензоле и эфире. Исходными продуктами для полу-
чения Д. являются диэтилкарбамоилхлорид и пипе-
разин. Д. обладает противоглистными свойствами и
применяется применении аскаридозов и филяриидозов.
Лит. см, при Ст. Апрессин. А. И. Травин.
ДИФЕНИЛ (фенилбензол) СвН5—CeHs, мол. в.
154,21 — бесцветные моноклинич. кристаллы; т. пл.
71°; т. кип. 254 — 255°/760 мм; 216,6°/300 мм;
145°/22 мм; rff' 1,156 (тв.); жидкого: 0,846 (250°);
0,800 (300°); 0,749(350°); ng 1,5873; давление насы-
щенного пара (ат): 0,926 (250°); 2,51 (300°); 5,74(350°);
8,8 (380°); гкрит 526,7°; ркрит 41,3 ат; теплосо-
держание (ккал/кг): 95,1(260°); 122,5(300°); 157,5(350°);
теплота . парообразования (ккал/кг): 75,1 (260°);
67 (300°); 59,5(350°); теплосодержание насыщенного
пара (ккал/кг): 170,2(260°); 189,5(300°); 217,0(350°);
теплоемкость (ккал/кг град): 0,69(260°); 0,70(300°);
0,70(350°); теплота сгорания Ср 1610,1 кал/моль; Со
1508,7 кал/моль; т. воспл. 113°; легко растворяется в
спирте, эфире, бензоле и многих органич, раствори-
телях, нерастворим в воде.
Д. — типичный ароматич. углеводород: при суль-
фировании Д. 94%-ной H2SO4 в присутствии BF3
получается дифенил-4,4/-дисульфокислота, а при его
нитровании образуются 2- и 4-нитродифенил и 2,4-ди-
нитродифенил. Д. содержится в антраценовом масле,
выделяемом из кам.-уг. смолы. В пром-сти Д. полу-
чают дегидрированием бензола при 750—8009. Д. —
полупродукт в произ-ве красителей; его производным
является бензидин. Оксидифенилы (о- и п-) служат
сырьем для синтетич. смол. Сам Д. применяется
в смеси с дифениловым эфиром в качестве высокотем-
пературного теплоносителя — даутерма (см. Тепло-
носители),
Лит.: Каган С. 3., Чечеткин А. В., Органиче-
ские высокотемпературные теплоносители и их применение
в промышленности, М.—Л., 1951. В. А. Пучков.
ДИФЕНИЛАМИН CeH5—NH—CeHs, мол. в.169,22-
белые кристаллы со слабым характерным запа-
хом, темнеющие на свету; т. пл. 54,0°; т. кип. 302°,
179°/22 мм; нерастворим на холоду в воде; в 100 а
спирта растворяется 56 г (19,5°), растворим в эфире,
СН3ОН, СвНв, CS2, лигроине и других органич. рас-
творителях; растворим в конц. минеральных к-тах,
при разбавлении растворов выпадает в неизмененном
виде. Получают нагреванием анилина с его хлористо-
водородной солью в автоклаве; известны и другие
способы получения. Д. применяют для разнообразных
синтезов (карбазола, акридина и др.); в произ-ве
азо- и трифенилметановых красителей; пироксилино-
вых порохов для их стабилизации. В присутствии
нитритов и нитратов, а также других окислителей
(Vv, Сг2О2,~, МпСЦ и др.) Д.окисляется с образованием
окрашенного в синий цвет соединения; это явление
используется для колориметрии, определения азот-
ной к-ты и др. окислителей; как окислительно-восста-
новительный индикатор (Ео = 4-0,76 в), напр.,
при бихроматометрич. определении железа.
1, Г-ДИФЕНИЛГИДРАЗИН СОЛЯНОКИСЛЫЙ
(CeH5)2NNH2 • НС1, мол. в. 220,68 — кристаллич.
порошок белого или серого цвета; в 100 г воды раство-
ряется 20 г (15°); легко растворяется в спирте; хранят
в запаянных ампулах. Восстановлением дифенилнит-
розамина получают дифенилгидразин (C6Hs)2NNO2i.
—► (C6H6)2NNH2, к-рый с конц. НС1 образует соляно-
кислую соль. Применяется Д. для идентификации
сахаров. И. И. EjOujtoe.
ДИФЕНИЛГУАНИДИН C,H6NH—С—HNCeH5,
NH
C13H13N3, мол. в. 217,27 — бесцветные кристаллы
(из спирта); т. пл. 147° (технич. продукта 145—147°);
плотн. 1,13; немного растворим в холодной воде или
эфире, растворим в бензоле. В пром-сти Д. получают
действием сероуглерода на анилин, образующийся
тиокарбанилид окисляется РЬО в присутствии аммиака
до Д.:
2C6H5NH2 + CS2 — (C0H6NH)sCS + H.,S
(C„H6NH)2CS + NH, + PbO —
CgH5NH- C-HNCcH5-|-PbS+ H2O
1165 ДИФЕНИЛЕНОВАЯ ПЕРЕГРУППИРОВКА — Г<Л-ДИФЕНИЛ-ГТ-ПИКРИЛГИДРАЗИН 1166
Применяют Д. в качестве вулканизации ускорителя
каучука. Л- л. Васкевич.
ДИФЕНИЛЕНОВАЯ ПЕРЕГРУППИРОВКА — см.
Бензидиновая перегруппировка.
ДИФЕНИЛКАРБАЗИД (1,5-дифенилкарбогидра-
зид) CeH5NHNH—СО—NHNHCeH6, мол. в. 242,28 —
почти бесцветные или розоватые мелкие кристаллы;
т. пл. 172—175°; мало растворим даже в горячей
воде; нерастворим в эфире и хлороформе; растворим
в спирте (при нагревании), в ацетоне и ледяной '
уксусной к-те. Получают Д. из мочевины и фенилгид-
разина при нагревании смеси до 100°: 2CeH6NHNH2 -|-
4- CO(NH2)2 —► CeHsNHNH—СО—NHNHCeH5. При-
меняют Д. для открытия CrVI, Си, Pb, Cd, Fe, Hg,
Mg, Mo, V; для фотометрия, определения CrVI, Hg11,
Pb; как окислительно-восстановительный индикатор !
при титровании бихроматом; в качестве адсорбцион-
вого индикатора при меркурометрич. определениях
хлоридов, цианидов. И. П. Ефимов.
ДИФЕНИЛКАРБАЗОН CeH6NHNH-CO-N=NC0Hls,
мол. в. 240,27 — оранжево-красный кристаллич. по-
рошок; т. пл. 157° (с разд.); нерастворим в воде,
растворим с оранжевой окраской в спирте, хлоро-
форме и бензоле. Получают из дифенилкарбазида
нагреванием со спиртовым раствором КОН. При-
меняют Д. для открытия Cd, Сг, Си, Fe, Hg и Мо;
для фотометрия, определения Hg, Ag, Pb, Zn; в ка-
честве адсорбционного индикатора в меркурометрии
при определении галогенов и цианидов.
ДИФЕНИЛМЕТАН С6Н5—СН2—СД15, ' мол.^вес
168,25 — бесцветные кристаллы; т. пл. 26—27°; т. кип.
261—262°/760 мм, 14Г/27 мм; d? 1,0060 (жидк.);
с/;0 1,090 (тв.); легко растворим в спирте, эфире,
хлороформе, жидком SO2; нерастворим в воде и жид-
ком аммиаке, волновое число •* (спирт абс.) 3712;
3814; 3845; 3942; 4009; 4112 см1. В технике Д. полу-
чают конденсацией толуола на катализаторах ThO2,
СоО и др.; можно получить также восстановлением
бензофенона, конденсацией СН2С12 и СвНв в присут-
ствии А1С13 и др. способами. Д. дегидрируется в тетра-
фенилэтилен или флуорен. При гидрировании Д.
(Н J и Р) были получены бензол и толуол или дицикло-
гексилметан (CeH11)2CH2 (PtO2 в СН3СООН или Ni
и высокое давление). Д. окисляется в бензофенон;
РС15 при 170° хлорирует СН2-группу; Вг2 без катали-
затора и при высокой темп-ре образует дифенилбром-
и дифенилдибромметаны. С SbCl3 Д. образует аддукт
С13Н(2 2SbCl3. При нагревании Д. с HNO3 получен
2,4,2',4'-тетранитродифенилметан, т. пл. 172° (при-
меняется для идентификации). Д. применяется как
растворитель в лакокрасочной пром-сти и для отдушки
МЫЛЭ. И, В. Матвеева.
ДИФЕНИЛМОЧЕВИНА (карбанилид) (CeH5NH)2CO,
мол. в. 212,24 — бесцветные кристаллы, приз-
мы; т. пл. 238—239°; т. кип. 260°; df* 1,239;
пр 1,583; при 20° в воде растворяется 0,015%; малора-
створима в спирте, бензоле, хлороформе, растворима
в эфире. При нагревании с водой в присутствии
NaOH Д. распадается на анилин, СО2 и N,N',N"-TpH-
фенилгуанидин (CeH,,NH)2C=N—СвН5. Д. нитрует-
ся и сульфируется в о- и «-положении к NH-rpyn-
пе; при нагревании с СО2 под давлением и после-
дующем гидролизе образуется смесь о- и п-аминобен-
зойных кислот. Д. получают нагреванием анилина
с мочевиной при 170°: 2CeHsNH2,-|- (NH2)2CO —►
—(CeH5NH)2CO + 2NH3; действием фосгена СОС12
на анилин и др. Д. применяют как дешевое N-ациль-
ное производное анилина, напр. при получении
амида сульфаниловой к-ты и др.
ДИФЕНИЛОВЫЙ ЭФИР (фенотсибензолТдифе-
нилоксид) СвН6—-О—СвН6, мол. в. 170,2 — кристаллы
19*
с запахом герани; т. пл. 26—27°; т. кип. 259,3°/760 мм,
127°/10 мм; d'i" 1,148 (тв.); жидкого 0,884 (250°);
0,831 (300°); 0,779 (350°); п$ 1,5809; динамич. вяз-
кость (х • 10е (кг-сек/м2): 34 (280°), -28 (300°); давле
ние насыщенного пара (ат); 0,848 (250°); 2,32
(300°); 5,24 (350°); 8,00 (380°); «врвт 532^; р
35,2 ат; теплосодержание (ккал/кг): 116,5 (260°);
142,3 (300°), 175,7 (350°); теплота парообразования
(ккал/кг): 65,7 (260°), 61 (300°), 55,5 (350°); тепло-
содержание насыщенного пара (ккал/кг): 182,2 (260°),
203,3 (300°), 231,2 (350°); теплоемкость (ккал/кг-град);
0,63 (260°); 0,65 (350°); 0,68 (350°); темп-ра вспышкв
при медленном нагревании 119° (в закрытом сосуде);
легко растворим в бензоле, ледяной СН3СООН,
спирте; нерастворим в воде. В пром-сти Д. э. получают
нагреванием фенолята с хлорбензолом в присутствии
порошкообразной меди: C6H6ONa + С;Н5С1—
—>СвН6—О—CeH5-|-NaCl. Д. э. —побочный про-
дукт при произ-ве фенола гидролизом хлорбензола.
В смеси с дифенилом Д. э. образует т. н. дифениловую
смесь, применяемую как теплоноситель во многих
произ-вах (см. Теплоносители); применяют также для
отдушки мыла.
Лит.: Каган С. 3., Чечеткин А. В., Органи-
ческие высокотемпературные теплоносители и их применение
в промышленности, М. — Л., 1951. И. В. Матвеева.
ДИФЕНИЛОЛПРОПАН (4,4-диоксидифенилдиме-
тилметан, диан) (СН3)2С(С6Н4ОН)2, мол. в. 228,29 —
бесцветные кристаллы; т. пл. 156—157° (технич.
продукт 150—152°); т. кип. 250—252/13 мм; раство-
рим в метиловом, этиловом, изопропиловом и бутило-
вом спиртах, в уксусной к-те, ацетоне и в диэтиловом
эфире. Растворимость в углеводородах и воде при
нормальной темп-ре незначительная. Получен впер-
вые Дианиным в 1891. В пром-сти Д. получают кон-
денсацией фенола с ацетоном в присутствии серной
или соляной к-ты или же хлористого водорода (что
дает более чистый продукт) СН СОСН3 + 2С Н5ОН—►
СН3
-► НОС6Н4—С—СвН40Н + Н2О. При синтезе Д. ис-
i
сн3
пользуют катализаторы — низкомолекулярные мер-
каптокислоты и меркаптаны, сероводород, сульфиды,’
полисульфиды, селениды и, теллуриды щелочных
металлов, хлористый сульфурил, фосген, катализа-
торы Фриделя — Крафтса. Д. можно получить из
гидроперекиси кумола в присутствии серной к-ты,
а также на основе реакции изопропенилацетата и
фенола. Получение Д. проводят обычно при темп-рах,
не превышающих 40° (в отсутствии катализатора до
70—80°). Технич. продукт очищают перекристалли-
зацией из растворителей (хлорбензол, толуол, уксус-
ная к-та) или растворением в щелочах с последующим
осаждением кислотами (серной, соляной, муравьи-
ной, угольной). Д. широко применяется в качестве
исходного продукта для получения эпоксидных и
феносмол, поликарбонатов, антиоксидантов, герби-
цидов.
Лит.: Дианин А., Ж. Русск. физ.-хим. о-ва 1891,'
23, № 7, с. 488; Schnell Н., Angew. Chemie, ’ 1956,
№ 20, S. 633; Hanzlik V. [и др.], Chem. prilmysl, 1956,
6, S. 201; L е i b n i t z Е. und Naumann K., Chem. Tech-
nik, 1951, 3, № 1. А. А. Елаганравова.
^^ДИФЕНИЛ-^-ПИКРИЛГИДРАЗИН (а,а-ди-
фенил-Р-пикрилгидразин) (NO2)3CeH2NHN(CeH6)2, мол.
вес 395,34 — призматические кристаллы (при кристал
лизации из этилацетата); т. пл. 172—173° (с разл.);
легко растворим в хлороформе и бензоле, плохо —
в спирте, эфире, четырехлористом углероде, этилаце
тате (на холоду). Р-ры Д. в спирте имеют оранжевую
окраску. Восстановление Д. двухлористым оловом
в присутствии соляной к-ты приводит к образованию
1167
ДИФЕНИЛТИОМОЧЕВИНА — ДИФОСГЕН
1168
дифениламина и 1,2,3,5-тетрааминобензола. Для
получения Д. используют взаимодействие дифенил-
гидразина с хлористым пикрилом в хлороформе или
бензоле. При окислении двуокисью свинца Д. в хло-
роформе или бензоле в присутствии сернокислого
натрия получается а,а-дифенил-Р-пикрилгидразил,
являющийся устойчивым свободным радикалом:
no2 no2
С6Н5\ //~\ С6Н5\ \
N-N-C )-NOs----------- N-N-Z У-NOg
Свн5 [!( СвН5/ *
NO, NO,
Д ифенилпикрилгидр азил представ-
ляет собой интенсивно окрашенное в фиолетово-
черный цвет соединение, т. пл. 138°, легко раство-
римое в хлороформе, трудно — в бензоле и почти нера-
створимое в спирте; растворы имеют глубокую фиоле-
товую окраску. Д. легко соединяется со свободными
радикалами с образованием слабоокрашенных со-
единений. Получил применение как «улавливатель»
свободных радикалов, для измерения скорости ини-
циирования ценных реакций в жидкой фазе (напр.,
полимеризации) и для определения выхода свобод-
ных радикалов при радиолизе органич. жидкостей.
Дифенилпикрилгидразил применяют в качестве стан-
дарта при исследовании спектров электронного
парамагнитного резонанса.
Лит.: Goldschmidt S., Renns К., Вег., 1922, 55,
S. 628—643; Уотерс У., Химия свободных радикалов, пер.
с англ., М., 1948. X. С. Багдасарьян, И. В. Матвеева.
ДИФЕНИЛТИОМОЧЕВИНА (тиокарбанилид)
CeH5NH—CS—NHCeH6, мол. в. 228,32 — блестя-
щие бесцветные листочки; т. пл. 154°; нерастворима
в воде, разб. кислотах и щелочах, хорошо раство-
ряется в спирте и эфире. При действии кислот Д.
-образует фенилгорчичное масло: (CeHsNH)2CS—>
—> CeH.5NCS + C8H5NH2. Получают Д. действием се-
роуглерода на анилин; применяют в качестве флото-
реагента; как полупродукт при изготовлении дифе-
нилгуанидина (ускоритель вулканизации каучука),
.для определения Ru и Os. н.п. Ефимов.
ДИФЕНИЛХЛОРАРСИН (хлорангидрид дифенил-
мышьяковистой кислоты) (C6H5)2AsC1, мол. в. 264.58 —
^бесцветные кристаллы; т. пл. ок. 38°; т.
кип. 333°; d\- 1,3870; лй 1,6332. Д. перас- / \
творим в воде, хорошо растворяется в /
•большинстве, органич. растворителей, в
хлорпикрине и фосгене', легко реагирует с водой
и щелочами (в том числе и сернистыми); достаточно
легко окисляется. Получают Д. восстановлением
дифенилмышьяковой к-ты сернистым газом в р-ре
конц. соляной к-ты: (C6H5)2AsO(OH) + SO2 + HCI —*
—> (C6H5)2AsC1 + H2SO4. Д. обладает резко выражен-
ным раздражающим действием на верхние дыхатель-
ные пути (вызывает неудержимое чихание и кашель).
Непереносимая концентрация 0,001 мг/л, в больших
концентрациях Д. может вызвать тяжелые поражения
верхних дыхательных путей. Применялся в качестве
ОВ во время первой мировой войны.
Лига. см. при ст. Отравляющие вещества.
Р. Н. Стерлин.
ДИФЕНИЛЦИАНАРСИН (цианангидрид дифенил-
мышьяковистой к-ты) (CeH5)2AsCN, мол. в. 255,14 —
бесцветные кристаллы; т. пл. 31,5°; т. кип. 346°;
rfp 1,3160, rq~ 1,6153; нерастворим в воде; хорошо
растворяется в органич. растворителях, довольно
быстро гидролизуется .горячей водой до тетрафенил-
диарсиноксида; сравнительно легко окисляется до
дифенилмышьяковой к-ты. Основной способ получения
Д. — обменная реакция между дифенилхлорарсином
л водным р-ром NaCA при 60°. Д. — сильное раздра-
жающее вещество; непереносимая концентрация
0,00025 мг/л. Применялся в качестве ОВ в первой
мировой войне.
Лит. см. при ст. Отравляющие вещества.
Р. Н. Стерлин.
ДИФЕНИН (дилантия, фенитоин, 5,5-дифенилги-
дантоин-натрий) С15НцА2О2Аа, мол. вес 281,26 —
бесцветные или с желтоватым
оттенком кристаллы, без запа- ^О—NH \
ха, слегка горького вкуса. Д. | /С(С6Н5).г
легко растворим в воде, спирте N=C(ONar
и уксусной к-те; почти нераство-
рим в эфире, хлороформе, бензоле и петролейном эфи-
ре. Водные растворы Д. опалесцируют, т. к. легко раз-
лагаются углекислым газом; при этом выпадает
5,5-дифенилгидантоин (т. пл. 286—287°); при pH
11,7 растворы прозрачны. Для колич. определения
высушенный при 105° в течение 4 час Д. растворяют
в воде, подкисляют и выделившийся 5,5-дифенилги-
дантоин извлекают эфиром; остаток после упаривания
вытяжки высушивают при 105° в течение 4 час и
взвешивают. Д. получают из бензальдегида, к-рый
последовательно превращают в бензоин, а затем
в бензил; при конденсации последнего с мочевиной
образуется 5,5-дифенилгидантоин, к-рый превращают
в Д. Препарат обладает противосудорожными свой-
ствами, не вызывая снотворного эффекта. Применяют
при лечении эпилепсии.
Лит. см. при ст. Апрессин. А. И. Травин.
4,4'-ДИФЕНОХИНОН (бифенохинон) С12Н8О2, мо-
лекулярный вес 184,20—хинон дифенилового ряда; кри-
сталлы золотисто-желтого цве-
та; т. пл. 165° (с разл.); нелетуч, / \ / q
с водяным паром не перегоняет- ° \ / \ /
ся. Д. нерастворим в холодной
вОде, умеренно растворим в теплом спирте и хло-
роформе, довольно трудно — в кипящем бензоле,
легко — в теплом нитробензоле. Д. обладает свойствами
хинонов и имеет высокий окислительно-восстанови-
тельный потенциал (в спирте при 0° 0,954 в), т. к.
содержит ненасыщенную сопряженную хиноидную
систему, проходящую через оба ядра. Д. является
сильным окислителем. 4,4 -Д. получают окислением
4,4'-диоксидифенила (окисление можно довести до
п-бепзохинона):
РьО» /“Ч Рь02 /==*\ ~
/ /—ОН—- О=< W >О—-2О=< >=о
\/ -нго \____________/ \____/ 4=/
Д. образует с фенолами соответствующие хингидроны:
обладает бактериостатич. активностью.
Н. В. Гнучев.
ДИФОСГЕН (трихлорметиловый эфир хлоруголь-
уОСС13
ной кислоты) ОС< , мол. в. 197,85 — бесцвет-
но
ная, тяжелая, подвижная жидкость, слегка дымящая
на воздухе, с характерным запахом прелого сена;
т. пл. —57°; т. кип. 128°; <7!,5 1,644; п)) 1,4566; плохо
растворяется в воде, хорошо — в органич. раствори-
телях и сам является хорошим растворителем для
многих органич. соединений. Д. медленно гидроли-
зуется водой; гидролиз ускоряется при нагревании
и в присутствии щелочей; энергично взаимодействует
с аммиаком и аминами с образованием мочевины или
। ее производных. При 300—350° в присутствии гало-
генидов металлов (FeCl3, А1С1Э и др.) Д. разлагается
с образованием в основном фосгена. В присутствии
галогенидов металлов (FeCl3, А1С13 и др.) разложение
Д. протекает с образованием СО2, СС14 и др. Метод
получения Д. основан на хлорировании метилхлор-
формиата С1СООСН3. Д. обладает сильным удушаю-
щим и заметным раздражающим действием. Непере-
1169 ДИФОСФОПИРИДИННУКЛЕОТИДЫ - ДИФРАКЦИЯ РЕНТГЕНОВСКИХ ЛУЧЕЙ 1170
носимая концентрация 0,075 мг/л. Характерным при
отравлении Д. является скрытый период действия
(до 6—8 час); очень опасно длительное воздействие
малых концентраций, суммарное действие к-рых мо-
жет привести к тяжелым поражениям (куммулятив-
ный эффект). Д. применялся в годы первой мировой
войны в качестве ОВ.
Лит. см. при ст. Отравляющие вещества. @
ДИФОСФОПИРИДИННУКЛЕОТИДЫ — см. Пири-
диннуклеотиды.
ДИФРАКЦИЯ РЕНТГЕНОВСКИХ ЛУЧЕЙ —
один из видов рассеяния рентгеновских лучей; пред-
ставляет собой результат сложения амплитуд вторич-
ных волн, рассеянных электронами вещества, без
изменения частоты (когерентно). Дифрагированные
лучи составляют часть всего рассеянного веществом
рентгеновского излучения. Наряду с Д. р. л. при
падении лучей на вещество происходит рассеяние
лучей с незначительно увеличенной длиной волны
(эффект Комптона), а также при надлежащих усло-
виях могут возникнуть характеристич. лучи со спек-
тром длин волн, свойственным данному атому. Явле-
ние дифракции доказывает волновую природу рент-
геновских лучей. Д. р: л. осуществляется на кристал-
лах. Мысль о возможности использования кристалла
в качестве природной дифракционной решетки для
рентгеновских лучей была впервые высказана М. Лауэ
в 1912. В том же году был поставлен В. Фридрихом
и др. соответствующий опыт, подтвердивший пра-
вильность высказанной идеи.
Открытие Д. р. л. послужило основой для развития
двух областей физики — рентгеноструктурного ана-
лиза, т. е. изучения строения вещества при помощи
рентгеновских лучей, и рентгеноспектралъного ана-
лиза, т. е. изучения спектров рентгеновских лучей
при помощи кристалла. Задачей теории Д. р. л.
является подсчет интенсивности рентгеновского излу-
чения, дифрагированного веществом, в зависимости
от угла рассеяния 20 (т. е. угла между первичным
и вторичным лучами). При анизотропии объекта иссле-
дования интенсивность рассеяния зависит также от
ориентировки объекта по отношению к лучу.
В случае Д. р. л. одноатомным газом результат
опыта зависит исключительно от распределения
электронов в атоме. Расчетом было показано и опы-
том подтверждено, что интенсивность рассеяния мо-
нотонно убывает с углом 0. В направлении первич-
ного луча рассеяние максимально и пропорционально
числу электронов в атоме — все электроны рассеи-
вают волны в одной и той же фазе. В случае Д. р. л.
молекулярным газом результат опыта зависит от
т. н. атомных факторов (см. Атом) и расстояний
между атомами в молекулах. В этом случае распре-
деление интенсивности по углам уже не будет моно-
тонно убывать. На кривой интенсивности могут воз-
никнуть максимумы. Картина, заснятая на фотопла-
стинку, будет состоять из первичного пятна и не-
скольких концентрич. колец. Исследуя распределение
интенсивности, можно определить строение молекул.
Однако из-за необходимости больших экспозиций,
достигающих сотен часов, Д. р. л. редко применяется
для этой цели. Такого же типа дифракционные кар-
тины могут быть получены со значительно меньшими
экспозициями при помощи дифракции электронов
(см. Электронография). С большим трудом поддается
расчету Д. р. л. жидкостями и твердыми аморфными
телами. Из-за близости молекул друг к другу здесь
уже нельзя считать независимым рассеяние разными
молекулами.
Наиболее интересной является Д. р. л. кристал-
лами. Одиночные кристаллы (монокристаллы) состоят
обычно из слегка (от нескольких секунд до нескольких
минут ДЖи) дезориедуироваииых блоков. Все атомы
внутри блока рассеивают когерентно. Таким образом,
интенсивность рассеяния кристалла равна произве-
дению числа блоков на интенсивность рассеяния от-
дельным блоком, состоящим из большого числа эле-
ментарных ячеек кристалла. Суммирование амплитуд
волн, рассеянных атомами кристаллич. блока, при-
водит к следующему результату. В отличие от аморф-
ных тел, кристалл рассеивает (дифрагирует) лучи
лишь в строго определенных направлениях. Эти
направления определяются законом, установленным
в 1913 Ю. В. Вульфом и независимо от него У. Брэг-
гом. Закон Вульфа—Брэгга говорит, что Д. р. л.
кристаллом можно рассматривать как селективное «от-
ражение» от систем узловых плоскостей кристаллич.
решетки. Эти системы характеризуются кристаллогра-
фия. индексами h, k, I, имеющими следующий смысл.
Если выбрать один из узлов пространственной ре-
шетки за нулевой и построить произвольную систему
параллельных равностоящих плоскостей, то пло-
скость, ближайшая к нулевому узлу, будет отсекать
на осях кристалла, выбранных за основные, доли
длин ребер aJh, b/k, c/Z; здесь а, Ь, с — длины ребер
выбранной элементарной ячейки. Индексы h, к, I
являются целыми числами, не содержащими общего
множителя. Если обозначить кратчайшее расстоя-
ние между соседними плоскостями семейства hkl через
cZftfe(, то закон Вульфа — Брэгга можно записать так:
2rfhfei sin0 = пк, где к — длина волны рентгеновско-
го луча, п — целое число, называемое порядком отра-
жения, и 0 — половина угла отклонения рассеянного
луча от первичного. Однако в данном случае 0 имеет
и другой смысл: это угол, образованный падающим,
а следовательно, и отраженным лучом с плос-
костью hkl (см. рис. 1).
Если кристалл повора-
чивать, то, подставляя
по очереди в отража-
ющее положение те или
иные системы плоско-
стей, мы можем полу-
чить все дифракцион-
ные лучи, свойственные
данному кристаллу.
Как видно из уравне-
ния, одна и та же си-
стема плоскостей hkl
дать несколько лучей (разные h). Явления Д. р. л.
кристаллом можно наблюдать и иным способом,
а именно, используя сплошной спектр рентгеновских
лучей, т. е. такой, к-рый содержит непрерывный
набор длин волн. В этом случае неподвижный кри-
сталл создает одновременно множество дифрагиро-
ванных лучей, т. к. для многих систем плоскостей
в спектре найдется подходящая длина волны к, удо-
влетворяющая закону Вульфа—Брэгга для тех углов 0,
к-рые образуют системы плоскостей неподвижного
кристалла с падающим лучом. Согласно уравнению
Вульфа—Брэгга, дифракционные ' лучи возникают
под строго определенными углами. Но это справедливо
для бесконечного и идеального кристаллов. В реаль-
ном кристалле дифракция имеет место в небольшом
интервале углов около точного значения 0. Расшире-
ние угла зависит также от наличия напряжений
и неоднородностей в объекте и от темп-ры.
Интенсивность дифрагированного луча зависит от
ряда причин и, в первую очередь, от т. н. структурного
фактора;последний определяется атомными факторами
атомов кристалла, расположением атомов внутри
элементарной ячейки кристалла, а также характером
тепловых колебаний атомов. Естественно, структур-
ный фактор зависит от симметрии расположения
Рис. 1.
может
1171 ДИФФЕРЕНЦИАЛЬНЫЙ СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД Ц72
атомов в ячейке. Интенсивность- дифрагированного
луча зависит также от размеров и формы объекта,
от совершенства кристалла (в частности, от величины
блоков), условий опыта (расходимость луча, степень
монохроматичности луча, перемещение объекта во
время съемки и пр.) и от поглощения объекта. К свое-
образным эффектам приводят тепловые колебания
атомов. Они не только влияют на интенсивность
лучей, дифрагированных согласно закону Вульфа—
Брэгга, но и создают собственную картину рассеяния
(правда, в 1000—10000 раз более слабую), наклады-
вающуюся на дифракционную картину, к-рую создал
бы кристалл с покоящимися атомами.
Д. р. л. от поликристаллич. тел приводит к возник-
новению резко выраженных конусов вторичных
лучей. Осью конуса является первичный луч, а угол
раствора конуса равен 40 (см. рис. 2). Каждый конус
соответствует определенному семейству плоскостей hkl.
В создании конуса лу-
чей участвуют все кри-
сталлики, плоскости hkl
к-рых расположены под
углом 0 к падающему
лучу. Если кристалли-
ки очень малого раз-
Рис. 2. мера и на единицу
объема их приходится
большое число, то конус лучей будет сплошным. Если
кристаллики имеют размеры порядка 10'1—10-3 см,
то конус будет состоять из отдельных лучей. При
вполне беспорядочной ориентации кристалликов число
и интенсивность лучей, приходящихся на тот или
иной участок поверхности конуса, одинаковы. Конус
обладает осевой симметрией. Текстура, т. е. наличие
предпочтительной ориентировки в расположении кри-
сталликов, приводит к нарушению этой симметрии.
Дифракционная картина, снятая на пластинку, уста-
новленную перпендикулярно падающему лучу, будет
состоять в случае текстуры из неравномерно зачернен-
ных колец. О методах съемки дифракционных картин
(рентгенограмм) и использовании их для определения
строения вещества см. Рентгеноструктурный анализ.
На Д. р. л. существенно сказывается неупорядо-
ченность структуры. При помощи этого явления
можно изучать дефекты структуры, возникающие
благодаря примесям, напряжениям и пр. Весьма
своеобразна Д. р. л. полимерными веществами. Соот-
ветствующие дифракционные картины являются в не-
котором смысле промежуточными между картинами
аморфного и кристаллич. веществ. Промежуточные ме-
зоморфные состояния (газокристаллическое, жидко-
кристаллическое) с успехом изучаются методом Д. р. л.
Особую область применения имеет Д. р. л. под ма-
лыми углами (доли минут, минуты отклонения от
первичного пучка). Этим методом исследуется неодно-
родность структуры в масштабе сотен ангстрем, могут
быть определены размеры пор или мельчайших
частиц, из к-рых построено твердое тело.
Д. р. л. может наблюдаться и от обычных дифрак-
ционных решеток, используемых для световых лучей.
Коэфф, преломления рентгеновских лучей немного
меньше 1. Было показано, что при падении рентгенов-
ских лучей на стекло под очень малыми углами сколь-
жения (6'10" и меньше) лучи претерпевают полное
внутреннее отражение. Для наблюдения Д. р. л.
от решетки заставляют лучи падать на дифракцион-
ную отражательную решетку (200 линий на 1 мм)
под углами, меньше указанного. Несмотря на малость
длины волны, в этих условиях удается добиться
прекрасного разрешения и измерить с большой точ-
ностью длины волн рентгеновских лучей.
Лит.. Жданов Г. С., Основы рентгеновского структур-
ного анализа, М.—Л., 1940; Китайгородский А. И.,
Рентгеноструктурный анализ, М.—Л., 1950; его же, Рент-
геноструктурный анализ мелкокристаллических и аморфных
тел, М.—Л., 1952; Корсунский М. И., Физика рентге-
новых лучей, М,—Л., 1936; Б р э г г У. Л., Общий обзор, пер.
с англ., М.—Л., 1938 (Кристаллическое состояние, т. 1);
Джеймс Р., Оптические принципы дифракции рентгенов-
ских лучей, пер. с англ.. М._ 1950; Комптон А. и Али-
сон С., Рентгеновские лучи. Теория и эксперимент пер.
с англ., М,—Л., 1941; Friedrich W. [u. a.J, Ann. Physik,
1913, 41, № 10; 42, № 15; Zachariasen W. H., Theory
of X-ray diffraction in crystals, N. Y.—L., 1945; Handbuch der
Physik, hrsg. v. H. Geiger und K. Scheel, Bd 23, TI 2, 2 Aufl.,
B., 1933. А. И. Китайгородский.
ДИФФЕРЕНЦИАЛЬНЫЙ СПЕКТРОФОТОМЕТРИ-
ЧЕСКИЙ МЕТОД — оптич. метод анализа, поль-
зуясь к-рым можно определять большие концентрации
веществ, дающих окрашенные р-ры (в отличие от
обычных фотометрии, методов, при к-рых определяют
обычно менее 1—2% вещества). Д. с. м. используется
также в случае применения реактивов, имеющих
собственное поглощение. В качестве нулевого р-ра
при спектрофотометрии, измерении берут эталонный
р-р, содержащий определяемый элемент в неск. мень-
шей концентрации, чем он находится в анализируе-
мом р-ре. Точность метода повышается, если отно-
шение интенсивностей световых потоков, прошедших
через анализируемый и эталонный р-ры, близко к 1.
Концентрация вещества в р-ре, не содержащем опре-
деляемого вещества (нулевом), должна быть такой,
чтобы прибор можно было настроить на нуль. Кон-
центрацию вещества .в анализируемом р-ре вычис-
ляют по формуле: Сх = Dx F -|- С где Сх и D —
концентрация и оптич. плотность анализируемого р-ра
и Cj — концентрация нулевого р-ра; F = &C/D.
Для нахождения коэфф. F берут два раствора с раз-
личной концентрацией определяемого элемента
(С2—C!=AC), к-рые отличались бы на какую-то
величину D (0,05; 0,1). Измеряют значение D для
более конц. раствора по отношению к раствору менее
конц. и вычисляют значение F по приведенной фор-
муле. Из 6—7 измерений находят среднее значение
для величины F. Видоизменение Д. с. м. состоит в сле-
дующем: готовят два раствора, один с концентрацией
Сх и второй с такой же концентрацией определяемого
элемента и добавлением к нему небольшого извест-
ного количества стандартного р-ра определяемого
элемента (Ст 4- Са). Оптич. плотность этих двух
растворов измеряют по отношению к нулевому р-ру,
которым является раствор испытуемого объекта
(соответственно Dx и Dx + a); тогда CX=DX • Ca/Da,
где Da — разность между Dx+a и Dx
Д. с. м. может быть применен в случае работы
с реагентами, имеющими собственное поглощение
в области поглощения определяемого соединения.
Оптич. плотности растворов реагента Z>t и окрашен-
ного соединения Z>2 измеряют по отношению к нуле-
вому р-ру, содержащему растворитель. Истинная
кривая поглощения соединения вычерчивается в коор-
динатах Оист — %, где X—длина волны; значения DnCT
являются разностью D2—Dt. Если реагент берется
в большом избытке (1 : 50; 1 : 100), то количеством его,
вошедшим в соединение с определяемым элементом,
можно пренебречь при определении небольших
количеств последнего. При работе с окрашенными
реагентами можно использовать и 1-й вариант Д. с. м.,
когда в качестве нулевого р-ра взят раствор реа-
гента и прибор настроен на нуль по этому раствору.
Лит.: Добкина Б. М., М а л ют и н а Т. М.., Завод,
лаб., 1958, 24, № 11, с. 1336; Hiskey С. F Analyt.
Chem., 1949, 21, № 12, р. 1440; Banks С. V Sponer
J. L., O’L a u g h 1 1 n J. W., там же, 1958, 4, № 4, p. 458.
В. M. Пешкова, М. И. Громова.
ДИФФУЗИОННЫЕ ФОТОГРАФИЧЕСКИЕ ПРО-
ЦЕССЫ — методы образования позитивного фотогра-
фия. изображения с помощью диффузии из негатива
в контактируемый с ним приемный слой позитивного
1173
ДИФФУЗИОННЫЙ ДВОЙНОЙ СЛОЙ — ДИФФУЗИОННЫЙ ток
1174
материала веществ, не израсходованных при проявле-
нии негатива и находящихся в последнем (или посту-
О Непроявленные кристаллы AgBr
• Проявленные кристаллы Ад Вг
Зародыши проявления
пающих в него) в строго дозированном количестве.
Так, Д. ф. п. с солями серебра (см. рис.) состоит из
3 стадий. 1) Проявление негатива:
2) Растворение неэкспонированного AgBr и диффу-
зия его в приемный слой в форме тиосульфатного
комплекса: AgBr 4-2S20g_—>Ag (SgOjJV + Br-. 3) Про-
явление позитива:
+ 4 S2O32'
—2Ag н-О
Отдельные стадии Д. ф. п. могут быть проведены
раздельно или одновременно. В последнем случае
в состав проявляющего р-ра вводят растворитель
галогенного серебра, чаще всего тиосульфат натрия
(гипосульфит). Разработано два типа Д. ф. п. с солями
серебра. 1) Быстрое копирование документов и чер-
тежей непосредственно в конторских помещениях.
Для этого на негативном материале экспонируют кон-
тактным способом копируемый документ, затем экспо-
нированный негатив контактируют с позитивным ма-
териалом (светочувствительным слоем к приемному
слою последнего), предварительно смочив их в про-
являюще-фиксирующем р-ре, и плотно отжимают.
По истечении нек-рого времени оба материала раз-
нимают и получают готовый полусухой позитив,
окончательно высыхающий за 1—1,5 мин. 2) Полу-
чение фотографии. снимка с любого объекта в форме
готового позитивного отпечатка непосредственно из
фотоаппарата в течение 1 мин. Для этого применяется
специальный фотоаппарат, снабженный темной ка-
мерой, в к-рой происходит контактирование негатив-
ного и позитивного материалов и распределение между
ними проявляюще-фиксирующего состава. Выпущен-
ный в СССР аппарат подобного рода и комплект к нему
носит название «Момент».
Лит.: Вейденбах В. Н., Ж. научной и прикл.
фото- и кинем., 1958, 3, вып. 4, 306; Н е б л и т К. Б., Фо-
тография, ее материалы и процессы, пер. с англ., М., 1958;
Спасокукоцкий Н. С., Дейчмейстер М. Д.,
Хим. наука и пром-сть, 1958, 3, № 5, 607; Современное раз-
витие фотографических процессов. [Со. статей, под ред.
Н. И. Кириллова], М., 1960, гл. 8. И. С. Спасокукоцкий.
ДИФФУЗИОННЫЙ ДВОЙНОЙ СЛОЙ — см.
Двойной электрический слой.
ДИФФУЗИОННЫЙ ПОТЕНЦИАЛ (потенциал
жидкостного соединения) — разность потенциалов,
возникающая на границе двух растворов разных элек-
тролитов или двух растворов одного электролита,
но разной концентрации. Так, при соприкосновении
двух растворов данного электролита с различными
концентрациями происходит диффузия электролита
из более концентрированного раствора в менее кон-
центрированный. Если подвижности катиона и аниона
неодинаковы и один из них движется быстрее другого,
то на границе растворов возникает скачок потен-
циала — Д. п. Величина Д. п. обычно не превышает
нескольких десятков милливольт и в простейших
случаях может быть рассчитана теоретически. Для
границы растворов одно-одновалентного электролита
с активностями и а2, ED=(i— 2t+) In р , где
Ed — Д. п., г+ — число переноса катиона, R —
универсальная газовая постоянная, Т — абс. темп-ра,
F — число Фарадея. Для границы двух растворов
одно-одновалентных электролитов с одинаковыми кон-
центрациями, содержащими общий ион, ED = -^Т]п
где и — эквивалентные электропроводности
этих растворов. В общем случае величина Д. п.
зависит не только от состава соприкасающихся рас-
творов, но и от типа границы между растворами;
устойчивость этой границы определяет воспроизводи-
мость величины Д: п. Хорошо воспроизводимые зна-
чения Д. п. (с точностью до 0,02 мв) получаются при
пользовании Т-образной трубкой, причем оба рас-
твора непрерывно поступают в 2 отростка трубки и
удаляются через 3-й общий. При точном определении
эдс электрохимии, цепей необходимо либо рассчи-
тывать Д. п. по одной из приведенных формул, либо
устранять его йли, по крайней мере, сводить до мини-
мума. Последнее достигается на практике включением
между обоими растворами солевого мостика, пред-
ставляющего собой насыщенный р-р КС1 (иногда
NH4NO3, KN03 или LiCH3COO). Благодаря большой
концентрации соли в солевом мостике ее ионы при-
сутствуют в месте контакта жидкостей в большом
избытке и практически переносят через границу весь
ток. Если ионы этой соли имеют одинаковые подвиж-
ности (как, например, в случае перечисленных со-
лей), т. е. числа переноса аниона и катиона близ-
ки к 0,5, то Д. п. невелик. См. также Подвижность
ионов.
Лит.: Глесстон С., Введение в электрохимию, пер. с
англ., М., 1951. Ю. В. Плесков.
ДИФФУЗИОННЫЙ ток — ток в электрохимии,
цепи, величина к-рого определяется скоростью диф-
фузии к электроду реагирующих на нем ионов или
молекул. Диффузия протекает в слое раствора, при-
легающем к поверхности электрода (диффузионный
слой). Плотность Д. т. выражается уравнением:
i=nFD (Сд—Cs)/8 (1), где п — число электронов,
участвующих в электрохимии, реакции, F — число
Фарадея, D — коэфф, диффузии реагирующих ча-
стиц, Со и Cs — их концентрации в объеме раствора
и у поверхности электрода. Для вычисления плот-
ности Д. т. по уравнению (1) надо знать толщину
диффузионного слоя 6, к-рая может быть рассчитана
теоретически для частного случая вращающегося ди-
скового электрода: 6— 1,62-Z) /з v «о /2 (ч — кине-
матич. вязкость раствора, ш — угловая скорость
вращения электрода). При уменьшении концентрации
реагирующих частиц у поверхности электрода Д. т.
возрастает, достигая нек-рого предельного значения
(см. ур-ние 1), называемого предельным Д. т. (см.
Предельный ток). В случае нестационарного электро-
лиза на плоском электроде
nFVP
(Со-С8),
где
t — время электролиза. Выражения для Д. т. лежат
в основе полярографии, метода анализа (см. Поляро-
1175
ДИФФУЗИЯ
1176
графия, Полярографический анализ). Приведенные
формулы справедливы для электролиза в присутствии
избытка индифферентного электролита.
Лит.." Л е в и ч В. Г., Физико-химическая гидродинамика,
М., 1952; Фрумкин А. Н. [и др.], Кинетика электродных
процессов, [МЛ, 1952. Ю. В. Плесков.
ДИФФУЗИЯ — самопроизвольный процесс пере-
носа вещества, приводящий к установлению равно-
весного распределения концентраций в результате
беспорядочного теплового движения молекул, атомов,
ионов и коллоидных частиц в газах, жидкостях или
твердых телах. В многокомпонентных фазах (фазах
переменного состава — в газовых, жидких или твер-
дых растворах) Д., иногда наз. гетеродиффу-
зией, представляет собой процесс направленного
переноса вещества в сторону выравнивания разностей
концентраций в различных участках данной фазы
при постоянной темп-ре. Хаотическое тепловое дви-
жение частиц вещества вызывает упорядоченный,
направленный перенос его в области, где позникают
градиенты концентрации (строго говоря, градиенты
химич. потенциалов), или где распределение концен-
траций отличается от равновесных.
В однокомпонентной системе Д., наз. самодиффузией,
она выражается в самом тепловом движении частиц
этого вещества и может быть изучена с помощью мече-
ных атомов (молекул). Наличие градиента темп-ры
в объеме тела также приводит к Д., причем каждому
данному значению градиента отвечает определенное
равновесное распределение концентраций диффунди-
рующего вещества вдоль этого градиента (см. Термо-
диффузия). Открыто явление восходящей Д., связан-
ное с наличием в объеме твердого тела градиента
внутренних напряжений. В этом случае имеет место
диффузионный поток из областей, где отсутствуют
внутренние напряжения, в области максимально
напряженные, в соответствии с общим принципом
Ле Шателье—Брауна.
Скорость Д. зависит от плотности и вязкости среды,
от темп-ры, природы диффундирующих частиц, воз-
действия разного рода внешних сил и т. д. В случае
газов при нормальном давлении и темп-ре Д. проте-
кает достаточно быстро, так что в сосудах с линей-
ными размерами порядка нескольких дециметров она
завершается в течение минут. В случае жидкостей
(растворов) для тех же расстояний требуются уже
дни и недели. В твердых телах, напр. в металлах,
скорость Д. еще во много раз меньше. С повышением
темп-ры скорость Д. всегда возрастает, причем у твер-
дых тел в значительно большей мере, чем у жидкостей
или газов.
Закономерности Д. описываются законами Фика.
Первый закон Фика определяет величину переноса
вещества в процессе Д.:
dm — — D~Sdt
ах
(dm — масса переместившегося вещества, — пло-
, de
щадь сечения, dt — время, — градиент концен-
трации в направлении переноса). Знак минус в этом
уравнении указывает на то, что перемещение вещества
происходит в направлении убыли концентрации.
Коэфф. D, называемый коэфф. Д., определяет собой
количество вещества, продиффундировавшего через
1 см2 поверхности за единицу времени при градиенте
концентрации, равном единице. Коэфф. Д. в абсо-
лютной системе единиц измеряется в см2/сек и яв-
ляется константой, не зависящей от времени и глу-
бины проникновения диффундирующего вещества
в нек-ром диапазоне концентраций.
Второй закон Фика (выводимый из первого при допу-
щении независимости D от концентрации) связывает
изменение концентрации диффундирующего вещества
во времени с ее изменением в пространстве:
8с ____________________ 82с
dt U 8х2
Это ур-ние, являясь общим дифференциальным уравне-
нием Д., имеет определенное решение лишь при нали-
чии известных граничных условий. В нек-рых случаях
такими условиями являются: 1) с = 0 при t = О
(для всех значений г); 2) при х = 0, с = с0 и дли-
тельно не меняет своего значения. При этих гранич-
ных условиях решение второго ур-ния Фика дается
в виде:
X
2VDt
о
где интеграл называется трансцендентной функцией
Крампа и определяется по специальным таблицам.
Процесс Д. имеет место также в коллоидных раство-
рах, где он обусловлен броуновским движением. Эйн-
штейн и Смолуховский теоретически показали, что
в случае Д. коллоидных частиц коэфф. Д. выражается:
О=х2/21, где х2 — среднее значение квадрата сме-
щения частиц, t — время. Для шарообразных частиц
можно показать, что D = кТ/бтсцг, где к = константа
Больцмана, Т — абс. темп-ра, ц — вязкость дис-
персионной среды иг — радиус частицы.
Следовательно, x2/t = RT/3m]rN, где N — число
Авогадро. Это ур-ние имеет большое значение прежде
всего для прямого экспериментального определения
числа Авогадро, что впервые было сделано Перреном.
Кроме того, используя выражение О = kT/dm^rN
для изучения процесса Д. в истинных (молекулярных)
растворах, оказывается возможным определять раз-
меры молекул (г). По крайней мере, для сравнительно
больших молекул (тетрабромэтан, тетрахлорэтан,
этилбензоат и г. п.) этот метод дает вполне удовлетво-
рительные результаты.
Д. в твердых телах связана с механизмом образо-
вания вакансий и дислоцированных атомов (см. Де-
фекты структуры). В результате тепловой подвиж-
ности (колебаний около положения равновесия) ато-
мов, ионов, расположенных в узлах кристаллич.
решетки, некоторые из них, обладающие в данный
момент избыточной энергией, могут покинуть свое
положение равновесия и переместиться в соседние
междоузлия. Ранее занимаемые ими места в решетке
окажутся вакантными, кроме того, образуется соот-
ветствующее число дислоцированных атомов, г. е.
атомов, расположенных в междоузлиях. Такой про-
цесс, по Френкелю, следует рассматривать как внут-
реннее «испарение» атомов твердого тела. Число ва-
кансий, т. е. пустых мест в решетке, вообще может
и не соответствовать числу дислоцированных атомов,
т. к. вакансии могут образовываться с поверхности
твердого тела в результате полного или неполного
«испарения» поверхностных атомов. Каждой данной
темп-ре соответствует определенное равновесное число
вакансий тем большее, чем выше темп-ра. Вблизи
темп-ры плавления число вакансий может достигать
1% числа атомов твердого тела. Наличие пустых мест
в кристаллич. решетке реального кристалла обеспе-
чивает возможность процесса Д. в твердых телах.
Зависимость коэфф. Д. от темп-ры дается выражением:
D—Ae~u/Rr, где и — энергия активации процесса Д.,
составляющая несколько десятков ккал/моль. Помимо
обмена местами атомов с незанятыми узлами решетки—
вакансиями, Д. в твердых телах может осуществляться
посредством прямого обмена местами двух соседних
атомов (механизм Хевеши), а также движением атомов
и ионов через междоузлия, напр. когда в кристаллич.
1177
ДИФФУЗИЯ — ДИХЛОРДИЭТИЛСУЛЬФИД
1178
теле диффундируют атомы (ионы) с радиусом, гораздо
меньшим радиуса атомов (ионов) основного компо-
нента, находящихся в узлах решетки.
Д. в полимерах протекает в результате теплового
движения не только самих цепных молекул полимера,
но и их отдельных участков (самодиффузия), что
способствует адгезии полимера к полимеру или ауто-
гезии (слипанию) двух кусков данного полимерного
материала. Диффундировать в полимерах могут и
частицы посторонних веществ. Газопаропроницае-
мость полимеров (напр., в виде пленок) является
результатом последовательно протекающих процес-
сов растворения газа в пограничном слое пленки,
Д. растворенного вещества (газа) через полимер и
выделения газа с другой стороны пленки. Она опре-
деляется как P=D (c1—c2)/h, где Р — коэфф, газо-
проницаемости, D — коэфф. Д., с-'-^ - = — —гра-
диент концентрации газа в толще полимера. Коэфф,
проницаемости полимера Р показывает, сколько сл«э
газа (при 0°) проходит через 1 см2 полимерной пленки
толщиной в 1 с.и за 1 сек при разности давлений газа
в 1 ат. Это ур-ние применимо к полимерам при усло-
вии, что процессы Д. и растворимости протекают по
законам Фика и Генри. Практически это достигается
лишь в области малых парциальных давлений газов.
В большинстве случаев (паропроницаемость и Д.
твердых растворенных веществ) коэфф. Р и D имеют
явно выраженную зависимость от концентрации и
времени. Газопроницаемость полимера зависит от
гибкости цепных макромолекул, от физич. состояния
полимера, от природы диффундирующих частиц.
С уменьшением гибкости молекул, напр. при кристал-
лизации или поперечном сшивании макромолекул
(вулканизация), с ростом межмолекулярных сил и
плотности упаковки полимера газопроницаемость
уменьшается. То же самое имеет место при переходе
полимера из высокоэластич. состояния к стеклооб-
разному. Наиболее газопроницаемыми полимерами яв-
ляются полисилоксаны (P v = 9-10 6 см2-сек"1-ат'1),
наименее проницаемыми — гидратцеллюлоза и поли-
хлорвинилиден (Рл-=3-10"12 см2 • сек1 ат'1).
Газопроницаемость является весьма важной харак-
теристикой полимерного материала и играет суще-
ственную роль в процессах вулканизации, дубления,
окисления, при защите изделий от коррозии и в др.
областях.
Диффузионные процессы имеют огромное значение
в самых различных областях науки и техники. Метод
Д. применяется для определения мол. веса раство-
ренных веществ (и частичного веса диспергированных
веществ). В биологии диффузионные процессы яв-
ляются определяющими в явлениях проницаемости
тканей, клеточных оболочек, всасывания и поглоще-
ния питательных веществ, выделения продуктов об-
мена; особое значение имеет осмос, т. е. Д. через
перегородку. Явление Д. определяет механизм и
кинетику таких важных процессов, как окисление,
сорбция, набухание, пептизация и т. д., физико-химич.
процессы в конденсированных фазах (кристаллиза-
ция, рекристаллизация, старение сплавов и т. д.).
В технике Д. имеет многочисленные и важные при-
менения, определяя в значительной степени скорости
производственных процессов — дубления кож и кра-
шение тканей, цементацию и азотирование сталей,
образование защитных диффузионных металлич. по-
крытий и т. д.
Лат.; Дюкло Ж., Диффузия в жидкостях, пер.
[с франц.], М.—Л., 1939; Френкель Я. И., Кинетиче-
ская теория жидкостей, М.—Л., 1945; Б уг аков В. 3.,
Диффузия в металлах и сплавах, М.—Л., 1949; БэррерР.,
Диффузия в твердых телах, пер. с англ., М., 1948; W е s t С. J.',
[а. о], Permeability of organic materials to gases, v. 1—2,
Wisconsin, 1948; Горбунов H. С., Диффузионные покрьь
тия на железе и стали, М., 1958; Рейт л ингер С. А., Усп.
химии, 1951, 20, вып. 2, 213.
В. И. Лихтман, С. А. Реитлингер.
ДИХЛОРГИДРИН ГЛИЦЕРИНА СЭН6ОС12, мол. в.
129,00 — известны два изомера: 1,3-, или а,у-Д. г.
СН2С1СНОНСН2С1 — бесцветная жидкость, т. кип
174°; d17 1,3506; 1,4800 и 1,2-, или а,Р-Д. /
СН2С1СНС1СН2ОН — бесцветная жидкость, т. кип.
183°; 1,3534; n’J 1,4875. Д. г. растворим в воде
(1 г в 9 г воды), эфире, спирте, глицерине, бензоле.
Технич. продукт является смесью обоих изомеров,
преобладает 1,2- Д. г., т. кип. 174—176°; d20 1,34—
1,38; т. воспл. 74°. Д. г. под действием водного р-ра
NaOH гидролизуется с образованием глицерина:
CH2ClCHOHCH2Cl+2NaOH-> СН2ОНСНОНСН2ОН -ф-
+ 2NaCl; на этой реакции основан метод промышлен-
ного синтеза глицерина. Д. г. является исходным
продуктом для синтеза глицериновых эфиров и эпи-
хлоргидрина. Получают Д. г. присоединением хлора к
аллиловому спирту (гл. обр. 1,2-Д. г.), действием НС1
на эпихлоргидрин (гл. обр. 1,3-Д. г.); технич. способ
получения Д. г. основан на взаимодействии хлорно-
ватистой к-ты и хлористого аллила при 28°:
СН2 = СНСН3С14-2НОС1 ->
-» СН»С1СНОНСН»С1+СНзОНСНС1СН2С1
В полученном продукте содержится ок. 92% Д. г.
Применяют Д. г. в качестве растворителя для
лаков. к. П. Гусев.
ДИХЛОРДИЭТИЛСУЛЬФИД [Р, Р'-дихлордиэтил-
сульфид,ди-(2-хлорэтил)-сульфид, иприт] S(CH»CH.2C1)2,
мол. в. 159,08 — органич. соединение, относящееся
к классу тиоэфиров; в чистом виде — бесцветная
жидкость; т. пл. 14,5°; т. кип. 217°/760 мм (с частич-
ным разл.), 117°/2б мм, 93°/10 мм; dp 1,280; давле-
ние пара 0,055 мм рт. ст. (10°), летучесть (макси-
мальная концентрация) 0,6—0,7 мг/л (20°); вес 1 л
пара 7,1 г, плотность пара (по воздуху) 5,5; раство-
римость в воде 0,05% (0,48 г/л); легко растворяется
во всех обычных органич. растворителях, выше 20°
(критич. темп-ра растворения) смешивается во всех
отношениях с бензином и керосином. Технический
Д. — темно-коричневая, почти черная жидкость, об-
ладающая неприятным своеобразным запахом, имеет
кислую реакцию, интенсивно корродирует металлы.
Чистый Д. — нейтральное вещество;
обычной температуре не действует.
Д. очень медленно гидролизуется во-
дой; при значительном избытке воды
образуется тиодигликоль (Р,Р'-ди-
оксидиэтилсульфид) и соляная к-та.
Скорость гидролиза резко возра-
стает при нагревании, перемешива-
нии, а также в присутствии едких
щелочей. При взаимодействии Д. с
Na2S образуются дитиан (I) и полимерные ве-
щества типа(—S—СН2—СН2—S—СН2—CH2)re, ас NH3
и первичными аминами — тиазан (II) или его N-ал-
кильные производные. Атомы С1 в Д. при действии
соответствующих солей могут обмениваться на Вг,
J, SCN и CN группы; с хлором и др. хлорирующими
агентами Д. превращается в неустойчивые полихлор-
производные; хлорирование Д. в присутствии воды
приводит к образованию 0, Р'-дихлордиэтилсульфок-
сида О = S(CH2CH2C1)2. Энергичные окислители
(HNO3, Н2О2 и др.) переводят Д. в р, Р'-дихлордиэтил-
сульфон и p-хлорэтаносульфокислоту. Хлорирование
и окисление лежат в основе процессов дегазации Д.
С солями тяжелых металлов (HgCl2, HgJ2, AuCl
и др.) Д. образует комплексные соединения тппа
HgJ2 • S(CH2CH2C1)2; эти реакции могут быть ис-
пользованы для обнаружения (индикации) Д. Полу-
чают Д. взаимодействием этилена с хлоридами серы
на металлы при
s<ch2~ch2>s
Сп<2—Сп2
zCH,-CHoXc.
HN< - ->S
хсн.-сн/
1179
ДИХЛОРМЕТИЛОВЫЙ ЭФИР — ДИХЛОРЭТАН
1180
(SC12, S2C12) или действием на тиодигликоль конц.
соляной к-ты.
Д. — протоплазматич. яд, обладает многосторон-
ним физиология, действием; в капельно-жидком,
паро- и туманообразном состоянии вызывает пораже-
ние кожи и глаз; вдыхание паров Д. вызывает тяже-
лые поражения верхних дыхательных путей и лег-
ких, а прием внутрь — поражение пищеварительного
тракта и общее отравление; обладает «скрытым» перио-
дом действия (до 12 и более часов); абсолютная смер-
тельная кожно-резорбтивная доза ок. 80 мг/кг веса.
Д. применялся в конце первой мировой войны в
качестве ОВ общетоксич. и кожнонарывного действия.
Для защиты органов дыхания от действия Д. ис-
пользуется противогаз, а для защиты кожных по-
кровов различного типа — защитная одежда.
Лит.: Соборовский Л. 3., Эпштейн Г. Ю.,
Химия и технология боевых химических веществ, М.—Л.,
1938. Р. Н. Стерлин.
ДИХЛОРМЕТИЛОВЫЙ ЭФИР [хлор (хлорметокси)
метан] С1СН2ОСН2С1 — бесцветная жидкость; т. кип.
105°; d~/' 1,3150; 1,4446; Д. э. легко гидролизуется
водой до формальдегида и соляной к-ты; с аммиаком
образует гексаметилентетрамин (уротропин); с фе-
нолом — парахлорметилфенол, со спиртовым р-ром
КОН—диэтоксидиметиловый эфир и диэтилацеталь
формальдегида:
zch2oc2h5
\ch»oc2hs
.CII..C1
о< ' + 2СаН6ОН
\сн.,С1
+ 2НС1
С»Н5ОН °<СН2С1>а с»Н5ОСН»С1 СзН5-Н. С2Н5ОСН2ОС»Н5
Получают хлорированием метанола или действием на
параформ НС1 или А1С13
2 (СНаО)з + 6НС1 — 3 (С1СН2)2О + ЗН2О
Д. э. — сильный яд, раздражает дыхательные пути;
концентрации 0,47 мг/л для человека смертельны при
1—2 мин экспозиции. Применяется для хлорметили-
рования нек-рых фенолов при получении феноло-
формальдегидных смол. ю. а Чебурков.
2,3-ДИХЛОРНАФТОХИНОН Ci„H4O2Cl2, мол. в.
227,06—желтые кристаллы; т. пл. 193°; плохо раство-
рим в воде, лучше — в органич. растворителях; ядовит
(ЛД50 для белых крыс 1,3 г/кг). Д. получают хлори-
рованием нафтионовой к-ты в 60%-ной H2SO4 при 80°
в присутствии железного катализатора:
а также хлорированием нафтохинона в органич. ра-
створителях и а-нафтола в 50—80%-ной H2SO4
в присутствии FeSO4 при повышенной темп-ре.
В пром-сти Д. готовят в виде 50%-ного смачивающе-
гося порошка для опрыскивания зеленых растений;
концентрация 0,05—0,2%. Д. применяют для борьбы
с болезнями растений, особенно с истинно-мучни-
стыми грибами, а также с лишайниками п водорос-
лями. Н. Н. Мельников.
ДИХЛОРОКСИН (5,7-дихлор-8-оксихинолин, хлор-
оксин) C9H5ONC12, мол. в. 214,05 — блестящие
QI белые мелкие кристаллы; т. пл. 179—
I 180°; практически нерастворим в воде,
растворим с желтой окраской в конц.
JI \ минеральных к-тах, щелочах, эфире, се-
роуглероде; легко растворим в спирте,
ОН бензоле. Получают Д. хлорированием
8-оксихинолина; осаждает те же эле-
менты, что и %>-оксихинолин; в отличие от последнего,
не применим для броматометрич. определений. Реагент
используется для определения Си, Fe, Ti, с к-рыми
образует в 0,2—0,1 н. минеральнокислом p-ре соот-
ветственно зеленый, темно-зеленый и оранжево-ко-
ричневый Осадки. В. Г. Типцова.
2,4-ДИХЛОРФЕНОКСИУКСУСНАЯ КИСЛОТА
(2,4-Д) С12С6Н3ОСН2СООН, мол. в. 241,06 — важней-
ший современный гербицид для борьбы с двудольными
сорнымирастениями (широколистными). Чистая Д.к.—
белые кристаллы, без запаха (технич. препараты
часто имеют неприятный фенольный запах); т. пл.
141—142е; в 1 л воды при 20° растворяется 540 мг
Д. к.; константа диссоциации 2,3 • 10 3; устойчива
при хранении как в твердом виде, так и в водных
р-рах. Для борьбы с сорными растениями чаще всего
используют соли натрия, калия и органич. аминов,
а также эфиры с различными спиртами. Раствори-
мость натриевой соли 27,51 г/л при 0° и 50,6 г/л при
30°; растворимость аминных солей значительно выше.
Так, растворимость наиболее часто используемой для
борьбы с сорными растениями диметиламинной соли
Д. к. более 200 г в 100 г воды. По гербицидному дей-
ствию аминные соли Д. к. на 25—30% превосходят
соли щелочных металлов.
Д. к. может быть получена след, способами:
]. СвН5ОН-I-2C1., — CbCeII,,OII+ 2IICI
С12С0Н3ОН + NaOH — Cl,C0H3ONa + Н-0
Cl»CeH3ONa + ClCH2COONa — Cl2'ceH3OCH2COONa -f- NaCl
II. C6H5ONa-J- ClCHoCOONa — C6H6OCH»COONa -|-NaCl
CeH5OCH,COONa + 2С1» — C1»C0H3OCH3COOH + 2HC1
Норма расхода различных солей Д. к. 0,75—2 кг/га.
Соли Д. к. применяют в виде водных р-ров, а нерас-
творимые в воде эфиры — в виде эмульсий или масля-
ных р-ров.
Лиш. см. при ст. Гербициды. Н. Н. Мельников.
2,6-ДИХЛОРФЕНОЛИНДОФЕНОЛЯТ НАТРИЯ
C12H6O2NO2Na, мол. в 290,09 — темно-зеленый поро-
шок, растворим в воде и
спирте, водный р-р окрашен
в синий цвет (окисленная „
форма) при подкислении пе-
реходит в красный (рА 5,7).
Применяют Д. н. для ко-
лич. определения аскорбиновой к-ты, а также в ка-
честве окислительно-восстановительного индикатора,
Е = 0,217 в (30°, pH 7). При действии сильных восста-
новителей, напр. аскорбиновой к-ты, синяя окраска
раствора становится бесцветной. и. п. Ефимов.
ДИХЛОРЭТАН (1,2-дихлорэтан, хлористый этилен)
С1СН2СН2С1, мол. в. 98,97 — бесцветная жидкость
с запахом, напоминающим хлороформ; т. пл. —35,87°;
т. кип. 83,483°/760 Л4Л4,2О°/63 им«;а^1,26000;b2d5 1,44759;
теплота испарения (при т. кип.) 7,654 ккал/моль;
«крит 288,4°; 1,28164; теплота плавления
2,112 ккал/моль; теплоемкость 30,86 ккал/моль
(26,72°). Эбулиоскопия, постоянная 3,44. Вязкость
0,887сш/аа (15°). Поверхностное натяжение32,23Эин/сл«
(20°); электропроводность 3 10-10 ом-см'-'1 (25°);
диэлектрич. проницаемость 10,36 (25°); диполь-
ный момент 2,06 D; растворимость в воде 0,81 вес. %
(20°); растворимость воды в Д. 0,15 вес. % (20°);
т. воспл. 21,1°; азеотропная смесь с водой содержит
82,9 вес. % Д., т. кип. 71,5°.
Д. получают взаимодействием хлора и этилена;
СН2=СН2 4- С12 —>С1СН2СН2С1; побочно получаются
полихлорэтаны, гл. обр. трихлорэтан. При нагре-
вании с водой, раствором соды или суспензией гаше-
ной извести, Д. образует этиленгликоль; действием
щелочи или пиролизом превращается в хлористый
винил. При нагревании Д. с аммиаком под давлением
дает этилендиамин, с цианистым натрием Д. об-
разует динитрил янтарной кислоты. При нагревании
1181
ДИЦИАНДИАМИД — ДИЭЛЕКТРИКИ
1182
с растворами полисульфидов натрия Д. образует
каучукбподобное вещество — тиокол',
ClCH»CH..Cl-J-Na2S4 —
-► . . . -CH.,-CH.,-S-S-CHO-CH.-S-S- . ..
'НИ ’ II II
s s s s
Д.—сильное наркотич. средство, при приеме внутрь
или вдыхании паров вызывает отравление; допусти-
мая концентрация паров в воздухе производственных
помещений 0,01 %. Д. мало огнеопасен. Пределы взрыв-
чатости в воздухе 6,20—15,90 об. %.
Д. широко применяют в качестве растворителя
(в экстракционных процессах, при очистке текстиль-
ных материалов, в лакокрасочной пром-сти), как со-
ставную часть антидетонаторных смесей, в качестве
фумиганта, как азеотропную добавку при дистилля-
ции, а также как сырье в произ-ве тиоколов и др.
Лит.: Дихлорэтан, Сб., под ред. Ю. П. Руденко, М.—Л.,
1939; ВайсбергерА. [и др.1,Органические растворители,
пер. с англ., М., 1958. Е. М. Рохлин.
ДИЦИАНДИАМИД (циангуанидин) C2H4N3, моле-
кулярный вес 84,08 — бесцветные кристаллы; т. пл.
209°; о!20 1,386; растворимость в
NH=C<N 2 ЮО г воды: 4,13 г (25°); 11,8 г
'NH—C ,N (49,8°); растворим в спирте, жид-
ком аммиаке; плохо растворим в
эфире, нерастворим в бензоле. Д. получают из циан-
амида кальция:
н2о со2 н2о
CaCN« —- Ca(HCN2)2 iNHaC(=NH)NHCN
С водяным паром при 160—170° Д. образует амме-
лид, СО2 и N Н3 :
2NH»C( = NH)NHCN+4H.>0— N= C(OH)N = C(OH)N = CNH3 +
4-CO24-4NH3
с сильными кислотами — соли гуанилмочевины:
-NIL. ,NH3C1
HN = C< ' 4-H..O4-HC1—HN = C<
'NHCN . 'NH-CONH.
При нагревании выше темп-ры плавления разла-
гается с выделением аммиака и образованием мелами-
на и др. веществ; при нагревании с К2СО3 при 300°
— соли гуанидина. Соли ароматич. аминов дают с Д.
соли бигуанидина:
NH2C( = NH)NHCN+RNHS—» NHSC(=NH)NHC( = NH)NHR
С формальдегидом Д. образует смолы; с 0-дикетоиами,
Р-кетоэфирами, малоновыми эфирами дает пирими-
дины (используемые в синтезе барбитуровых к-т):
,соос..н5
r2c4
ХСООС2Н5
NHs\c-nhcn
,CONH4
— r2c< >c=ncn
XCONH/
Идентифицируют по соли серебра. Д. — промежу-
точный продукт синтеза меламина, .эпоксидных смол,
дубителей синтетических (синтанов) и взрывчатых
веществ (нитрогуанидин).
Лит.: Williams Herbert Е., Cyanogen compounds:
their chemistry, detection and estimation, 2 ed., L. — ra. o.]
1948. , Ф. H, Величко.
ДИЦИКЛОГЕКСИЛ (циклогексилциклогексан)
С6НЦ—C6HU, мол. в. 166,30 — маслянистая жид-
кость со слабым запахом; т. пл. 3,5—4°; т. кип.
239—240°; dj" 0,8835; «)) 1,4800; нерастворим в воде;
малорастворим в спирте; смешивается с эфиром и уг-
леводородами. Д. дегидрируется при 300° над пла-
тинированным углем, образуя дифенил. Получают
Д. из бромциклогексана и натрия в среде эфира:
2CgHuBr4- 2Na—*-СвНи—C6Hn + 2NaBr и др. спо-
собами. В пром-сти Д. производят гидрированием
дифенила при 110—120 ат и 200—250° в присутствии
никеля. Применяется в пром-сти как растворитель.
М . Ф. Турчинский.
ДИЦИКЛОПЕНТИЛ (циклопентилциклопентан)
С5Н9—С5Н9, мол. в. 130,19 — бесцветная подвижная
жидкость со слабым скипидарным запахом; т. кип. 188—
190°/760 мм, 66,5°/12 мм; dj» 0,8656; ng1 1,46429. Для
получения Д. предложены след, способы: 1) действие
натрия на бром- или иодциклопентан в эфире; вы-
ход 12—17%, главным продуктом реакции является
циклопентен; 2) восстановление по Кижнеру цикло-
пентилциклопентанона; 3) гидрирование 2,2-дицикло-
пентенила над Ni-на А12О3. Д. устойчив к действию
брома, но медленно окисляется перманганатом; при
пропускании над платинированным углем (300°) не
дегидрируется. Д. содержится в нек-рых нефтях.
О. Р. Охлобыстин.
ДИЭЛЕКТРИКИ — материальная среда, непро-
водящая электричества. Термин «Д.» введен Фара-
деем. В действительности, каждая материальная среда
обладает нек-рой, хотя иногда и весьма малой, элек-
тропроводностью (уд. сопротивление 109—1018 ом-см).
Однако в Д. процессы смещения электрич. зарядов
внутри атомов и молекул и потери энергии, связан-
ные с этими процессами, доминируют над процессами
проводимости. Поэтому в первом приближении можно
считать, что в Д. нет свободных зарядов, к-рые могут
быть сдвинуты электрич. полем на большие расстояния.
Д. построены из нейтральных молекул или атомов
(жидкости, газы, нек-рые твердые тела) или из отри-
цательных и положительных ионов (ионные кри-
сталлы), закрепленных в определенных положениях
равновесия в кристаллич. решетке. При воздействии
электрич. поля электрич. заряды, из к-рых состоят
молекулы, атомы и ионы Д., смещаются в пределах
молекулы, атома или элементарной ячейки. В отсут-
ствии электрич. поля электрич. момент любого объема,
выделенного внутри Д., и достаточно большого по
сравнению с размером молекулы или элементарной
ячейки, равен нулю. При наличии поля, когда заряды
разных знаков смещаются в противоположных напра-
влениях, электрич. момент объема отличен от нуля;
в этом случае говорят, что Д. поляризован. Величина
электрич. момента единицы объема Д. наз. поля-
ризацией диэлектрика. Само явление сме-
щения зарядов также наз.
поляризацией. Ее величи-
на JJ определяется равен-
ством;
где — заряд, находя-
щийся в данной точке г,
Ri — радиус-вектор, про-
веденный в эту точку из
произвольно выбранного
центра.
Электрические свойства
Д. обычно изучаются на
плоском конденсаторе, за-
полненном Д. Поляриза-
ция создает в таком кон-
денсаторе поле, обратное
внешнему, которое при не-
изменных зарядах на эле-
ктродах ослабляет поле
внутри Д. (рис. 1). Отно-
Н-] Связанный заряд f+j
‘'1 W Диполь
@ Свободный заряд
Рис. 1. Поле плоского кон-
денсатора: Eq — напряжен-
ность поля в вакууме; Ер —
напряженность поля поляри-
зации.
шение напряженности по-
ля в вакууме к напряженности поля в Д. при
одном и том же расположении электродов и той же
величине заряда на электродах наз. диэлектрической
проницаемостью. Диэлектрич. проницаемость — важ-
нейшая макроскопия, характеристика Д. Она не
является константой и может зависеть от частоты
приложенного переменного поля и от темп-ры. Вектор
1183
ДИЭЛЕКТРИКИ
1184
I), определяемый равенством D = eZ?, где е — диэлек-
трич. проницаемость, а Е — напряженность элек-
трич. поля, наз. электрич. смещением. Можно пока-
зать, что D = Е -р 4лР, где Р — поляризация Д.
Когда под действием электрич. поля смещаются за-
ряды внутри атома или молекулы, то атомы и моле-
кулы превращаются в диполи. Диполи обладают
дипольным моментом, к-рый равен произведению
е г, где е — заряд, г — расстояние между зарядами
(или центрами зарядов). Если заряды связаны упру-
гими силами, противодействующими смещению, то
дипольный момент пропорционален полю т = аЕ,
где а носит название поляризуемости атома или моле-
кулы. Многие Д., однако, состоят из молекул, к-рые
сами являются диполями и без наложения электрич.
поля. В этом случае поле ориентирует молекулярные
диполи, в результате чего электрич. момент объема
Д. становится отличным от нуля.
Различают три основных типа поляризации: поляри-
зация электронного смещения, ионная поляризация
и дипольная поляризация. Электронная по-
ляризация обусловлена смещением электрон-
ных оболочек относительно положительных ядер.
Поляризацией этого типа обладают все вещества.
Нек-рые вещества (напр., алмаз, инертные газы, кис-
лород и др.) имеют только электронную поляризацию.
В области радиочастот, включая сантиметровые волны,
диэлектрич. проницаемость этого класса веществ не
зависит от частоты. С увеличением темп-ры е слабо
уменьшается. Ионная поляризация вызы-
вается смещением положительных и отрицательных
ионов относительно своих положений равновесия.
Ионной поляризацией обладают ионные кристаллы,
такие, как галогениды щелочных металлов, рутил,
титанаты нек-рых металлов, напр. кальция, магния,
цинка. Диэлектрич. проницаемость ионных кристал-
лов может быть велика (е титаната кальция равна
150) и не изменяется с частотой в диапазоне радио-
частот. Дипольной поляризацией, к-рую
называют также релаксационной, обладают Д., в со-
став к-рых входят молекулярные диполи. В отсут-
ствии поля дипольные молекулы распределены хаоти-
чески; они участвуют лишь в тепловом движении и
обладают энергией кТ. При наложении поля диполи,
взаимодействуя с полем, приобретают нек-рую пре-
имущественную ориентацию вдоль направления поля
и Д. поляризуется. В этом случае поляризация,
а следовательно диэлектрич. проницаемость зависят
от темп-ры. Эффективная диэлектрич. поляризуе-
мость, рассчитанная на молекулу, равна рР/ЗкТ,
где и. — электрич. момент молекулярного диполя.
Дипольной поляризацией обладают многие органич.
жидкости (спирты, производные бензола), вода, неко-
торые полимеры и, в определенном диапазоне темп-р,
дипольные кристаллы (папр., твердая НС1). Диполь-
ной поляризацией обладают также нек-рые ионные
кристаллы. Электрич. поле, действующее на молекулу
впутри Д., отличается от среднего макроскопического.
В наиболее простом случае неполярных жидкостей и
газов и кубич. двухатомных кристаллов это локальное
поле определяется уравнением: Е = Ес^ ф-
где ЕСр. — среднее макроскопическое поле. В
этом случае связь между диэлектрич. проницае-
г — 1 4~
мостью и поляризуемостью имеет вид: с-^ 9 = па
(ур-ние Клаузиуса—Моссоти), где п — число частиц
в единице объема.
При наложении переменного электрич. поля в Д.
имеют место потери энергии, связанные с процессами
поляризации. Потери энергии в образце Д. выра-
жаются формулой:
w ^гЭфф.“С tg 8
где и = 2л/, / — частота приложенного напряжения,
С — емкость данного образца, б^фф. — эффективное
значение приложенного напряжения, tgS — отно-
шение активной составляющей тока в Д. к реактив-
ной. В технике за величину, характеризующую по-
тери, принимают не абсолютное значение этих потерь,
а значение tgS, к-рому они пропорциональны. Для
однородных Д. с электронной и ионной поляриза-
цией tgfi изменяется обратно пропорционально ча-
стоте приложенного поля и увеличивается с увели-
чением темп-ры. В случае наличия дипольной поля-
ризации зависимость tg<5 от частоты, а также от тем-
пературы, имеет максимум. При комнатной темп-ре
и звуковых и радиочастотах значение tgd для
наиболее распространенных Д. лежит в пределах
IO-2—ПУ5.
Каждый Д. имеет нек-рую, хотя иногда и весьма
малую, электропроводность (проводимость). Носите-
лями заряда в Д. могут быть как электроны, так и
ионы. Большинство Д., применяемых в технике, обла-
дает ионной электропроводностью. Величиной, харак-
теризующей электропроводность Д., является удель-
ная объемная проводимость — проводимость куба
из Д. с длиной ребра в 1 см при наложении поля па-
раллельно ребру. Электропроводность газов обусло-
влена ионизацией атомов под влиянием ионизирую-
щих облучений. В обычных условиях проводимость
газов очень мала. Однако при высоких напряжениях
начинается ударная ионизация и проводимость резко
возрастает (газовый разряд).
Носителями тока в диэлектрич. жидкостях являются
ионы, а также более крупные коллоидные частицы,
к-рые, как известно, несут на себе заряд. Ионная
электропроводность жидкостей делится на два класса:
собственная и примесная. Присутствие примесей
очень сильно увеличивает проводимость жидкостей.
Так, напр., удельная проводимость промышленного
трансформаторного масла равна 0,5 • 10-12oaj-1- см 1,
в то время как проводимость того же масла после
тщательной очистки равна 0,5 • 10“15 ом1 • см1.
И собственная и примесная электропроводности обус-
ловлены диссоциацией молекул на ионы. Неполярные
жидкости слабо диссоциированы, собственная электро-
проводность их мала и общая электропроводность
определяется гл. обр. примесями. Непосредственно
из измерений разделить собственную и примесную
электропроводность невозможно.
Ионная электропроводность жидкостей сильно зави-
сит от темп-ры. Если электропроводность обусловлейа
движением ионов одного типа, то зависимость выра-
жается формулой: у=уоехр(—U0/kT), где у — про-
водимость, U„ — энергия сцепления иона с молеку-
лами жидкости, Т — абсолютнаятемп-ра, у0 — коэфф.,
постоянный для данной жидкости, слабо зависящий от
температуры, к — постоянная Больцмана. Ионная
электропроводность жидкостей связана с ее вязкостью
экспериментально установленным законом Вальдена,
согласно к-рому произведение электропроводности
жидкости на ее вязкость не зависит от темп-ры. Теоре-
тич. вывод закона Вальдена приводит к выражению
. / Ui~
= Л ехр (----5^)
где у — проводимость, 1] — вязкость жидкости,
Ux и /7 2 — энергии связи между ионом и молекулой
жидкости и между молекулами жидкости соответ-
ственно, А — коэфф, постоянный для данной жид-
кости, зависимость к-рого от темп-ры определяется
зависимостью степени диссоциации от темп-ры. Т. обр.,
закон Вальдена должен оправдываться, если£/1=Е/2.
В действительности закон Вальдена плохо выпол-
няется для жидкостей с ярко выраженной примесной
электропроводностью и слабополярных, где связь
1185
ДИЭЛЕКТРИКИ
1186
между молекулами жидкости мала. Электропровод-
ность жидких Д„ обусловленная движением колло-
Рис. 2. Зависимость проводимости
NaCl от температуры (для двух
образцов).
ла, обусловленная одйим
ляется выражением:
идных частиц, носит
название катафорети-
ческой. Для катафо-
ретич. проводимости
закон Вальдена спра-
ведлив.
Ионная электропро-
водность твердых Д.
изучена теоретически
и экспериментально
главным образом для
ионных кристаллов.
Идеальная ионная
решетка не обладает
электропроводностью.
Проводимость появ-
ляется при наличии
в решетке дефектов:
ионов в междоузлии
и вакантных мест в
узлах решетки. Тем-
пературная зависи-
мость проводимости
для ионного кристал-
сортом дефектов, опреде-
7 = 7о ехр-(^т-^
где W — энергия образования дефекта, U — энергия
активации, необходимая иону для перехода в сосед-
нее положение равновесия, у0— коэфф., слабо зави-
сящий от темп-ры. Экспериментальная зависимость
проводимости от темп-ры имеет вид, представленный
на рис. 2. Прямолинейная зависимость ]gy = /(^,)
имеет излом. Существование этого излома обусло-
влено тем обстоятельством, что при нек-рой темп-ре
концентрация дефектов в кристалле за счет примесей
превышает равновесную концентрацию дефектов, за-
висящую от темп-ры, как ехр —(^.)- Начиная с этой
темп-ры и ниже, концентрация дефектов остается
постоянной и экспоненциальный член оказывается
равным ехр — (^). В этой области темп-p проводи-
мость наз. структурно-чувствительной. В нек-рых
кристаллах (особенно при высоких темп-pax) подвиж-
ными являются не один, а неск. сортов ионов. В этом
случае в выражении для проводимости будет стоять
не один член, а сумма. Значительное число Д. по-
строено из молекул, не диссоциированных на ионы
(напр., полимеры), или же ионов, обладающих очень
небольшой подвижностью. Эти Д. обычно содержат
в себе примеси ионного характера, к-рые и дают
весьма заметную электропроводность.
Для каждого Д. существует нек-рое значение
напряженности поля, при к-рой происходит пробой.
Эта напряженность поля носит название электрич.
прочности. Пробой газов (или газовый разряд) начи-
нается, когда свободные электроны, имеющиеся
в газе, приобретают в электрич. поле энергию, доста-
точную для ионизации молекул газа. Напряженность
поля при пробое газов зависит от природы газа, но
порядок величины один и тот же. Для воздуха при нор-
мальном давлении и однородном поле = 30 кв/см.
Механизм пробоя жидких Д. зависит от того,
насколько хорошо очищена и обезгажена жидкость.
При наличии в жидкости примесей с большой прово-
димостью между электродами перед пробоем обра-
зуется мостик из этих примесей. Ток через жидкость
усиливается, увеличивается выделение тепла, и жид-
кость вблизи мостика вскипает. При этом образуется
газовый канал, вдоль к-рого происходит разряд.
Напряженность электрич. поля при пробое неочи-
щенных и необезгаженных жидкостей может коле-
баться в пределах 10э—105 в/см. При очень высокой
степени очистки электрич. прочность жидкостей
возрастает до 10е в/см. В чистых жидкостях пробой,
очевидно, возникает в результате ударной ионизации
молекул жидкости электронами, т. е. имеет нечто
общее с процессом пробоя в газах. Электрич. проч-
ность твердых Д. больше, чем жидкостей и газов.
Для нек-рых Д. она достигает значения 107 в)см.
Класс диэлектри- ков и тип поляри- зации Наименование вещества Диэлектрическая прони- цаемость
। °C частота, гц €
Неполярные га- Азот 0 С 3 10® 1,00058
зы * (электрон- Аргон 0 < 10’ 1,000545
ная поляризация) Воздух 0 с 3 • 10е 1,000590
Полярные газы Ацетон 100 То же 1,0159
(электронная и дипольная поля- ризация) Водяные пары . 110 » 1,0126
Неполярные Бензол 20 104 2,28
жидкие диэлект- Циклогексан . . 20 104 2,052
рики и твердые Гелий —268,9 6 10® 1.048
диэлектрики, не содержащие ио- Водород .... Кремнийоргани- —253 105 1,22
нов (электронная поляризация) ческое масло . Тр ансформ атор- ное масло . . . Полистирол . . Полиэтилен . . Политетр афтор - этилен .... Парафин .... Церезин .... 20 12 11 III 2,2 2,24 2,4 —2,65 2,3 2,0 2,0 —2,5 2,25-2,50
Полярные жид- Ацетон -80 31,0
кости Нитробензол . . Хлорбензол . . о-Крезол .... Спирт этиловый Спирт метиловый Глицерин .... Вода 20 20 24 14,7 20 20 20 10« 10* 4 • 108 X = оэ 4 • 108 3 108 108 36,1 5,94 5,8 26,8 33,1 43 80
Дипольные кри- Лед - 20 300 ~ 75
сталлы (электрон- ная и дипольная поляризация) НС1 -160 60 • 103 ~ 16
Ионные кри- СаГ2 20 10’ 7,36
сталлы (электрон- КС1 20 104 5,03
ная и ионная по- AgBr 20 10в 12,2
ляризация) Т1С1 NaCl Рутил TiO2 . . . | оптич. оси . . ]) оптич. оси . . 20 20 20 20 10» 10» 108 108 46,9 6,12 86 170
Стекла (элект- ронная и ионная поляризация) — — — 3—20
Сегнетоэлект- рики (спонтанная поляризация) — — — 200—50000
* При атмосферном давлении.
В настоящее время различают два вида пробоя
твердых Д. — тепловой и электрический. Тепловой
пробой достаточно хорошо изучен как теоретически,
так и экспериментально. При увеличении напряже-
ния, приложенного к Д., ток через него растет. Вместе
с этим растет количество тепла, выделяемого током,
и, следовательно, темп-ра внутри Д. Так как прово-
димость сильно зависит от темп-ры, то сопротивление
Д. падает и ток еще увеличивается. В конце концов
1187 ДИЭЛЕКТРИЧЕСКАЯ ПОСТОЯННАЯ — ДИЭЛЕКТРИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ Ц88
в Д. устанавливается нек-рая равновесная темп-ра,
при к-рой количество тепла, отведенного в секунду
от образца, равно выделенному. При определенном
значении напряжения, однако, равновесие между
теплоотдачей и теплообразованием нарушается.
Темп-ра и ток в Д. начинают расти до тех пор, пока
не наступит тепловое разрушение вещества. Электрич.
прочность при тепловом пробое имеет порядок 104—
105 в/см. Тепловой пробой имеет место, если проводи-
мость Д. относительно высока, и наблюдается в стек-
лах и фарфоре при повышенных темп-рах и длитель-
ном приложении напряжения, в нек-рых полимерах,
слоистых изоляционных материалах — как тексто-
лит и гетинакс.
Если проводимость Д. мала или мало время при-
ложения напряжения, то тепловой пробой не может
развиться и происходит электрич. пробой. Электрич.
пробой возникает в результате процессов электрон-
ного размножения внутри Д. под влиянием сильного
электрич. поля. К таким процессам относятся: удар-
ная ионизация атомов или ионов кристаллич. решетки
электронами, автоэлектронная эмиссия из валентной
зоны, автоэлектронная эмиссия из катода. Наиболее
общепризнанные теории электрич. пробоя твердых Д.
(валентных и ионных кристаллов) основаны на пред-
положении возможности ударной ионизации.
Диэлектрич. материалы очень широко исполь-
зуются в технике, прежде всего для электрич. изо-
ляции токоведущих частей электрич. машин и аппа-
ратов. Из жидких Д. наиболее распространено транс-
форматорное масло, представляющее собой смесь
различных углеводородов. Из твердых Д. в качестве
электрич. изоляции широко распространены слюда,
искусственные смолы, к-рые получаются путем поли-
меризации органич. веществ, воскообразные веще-
ства — такие, как парафин и церезин, битумы (про-
дукты окисления нефти). На основе искусственных
смол (бакелит, глифталь) изготовляются электроизо-
ляционные лаки для пропитки бумажной и хлопчато-
бумажной изоляции. Для изготовления жестких элек-
троизоляционных конструкций применяются различ-
ные пластмассы: гетинакс, текстолит, полихлорвинил,
полистирол, плексиглас (полиметилметакрилат), фто-
ропласт и многие другие.
Для радиоконденсаторов и радиодеталей широко
используются керамич. материалы на основе рутила
TiOa и титанатов металлов (калия, магния и др.).
Конденсаторная керамика имеет сравнительно боль-
шую диэлектрич. проницаемость и малый tg6.
Для радиоаппаратуры большое значение имеют Д.,
обладающие пьезоэффектом (см. также Сегнетоэлек-
трики). Пьезоэффект заключается в том, что прило-
жение к кристаллу механич. силы вызывает появле-
ние поверхностных зарядов и, наоборот, приложение
разности потенциалов вызывает изменение размеров
кристалла. Этим свойством обладают диэлектрич.
кристаллы малосимметричной структуры, напр. кри-
сталлич. кварц. В таблице приведены диэлектрич.
проницаемости для Д. различных классов.
Лит.: Сканави Г. И., физика диэлектриков (Область
слабых попей). М.—Л., 1949; его же, Физика диэлектриков
(Область сильных попей), М.—Л., 1958; Ф р е п и х Г., Тео-
рия диэлектриков, пер. с англ., М., 1960; Smyth С. Р-,
Dielectric behavior and structure, N. Y. [a. o.J, 1955; Уайт-
хед С., Пробой твердых диэлектриков, пер. [с англ.], M.—Л.,
1957. В. А. попорот.
ДИЭЛЕКТРИЧЕСКАЯ ПОСТОЯННАЯ — см. Ди-
электрическая проницаемость.
ДИЭЛЕКТРИЧЕСКАЯ ПРОНИЦАЕМОСТЬ — от-
ношение напряженности поля в вакууме к напря-
женности среднего макроскопич. поля в однородной
изотропной среде при неизменных зарядах, создаю-
щих поле. Д. п показывает также, во сколько раз
уменьшается сила взаимодействия электрцч. зарядов
при переносе их из вакуума в однородную изотропную
сРеДУ, если расстояние между зарядами остается неиз-
менным. Д. п. связана с величиной поляризации
диэлектрика Р соотношением:
4к
где 8— Д. п., а Е— напряженность среднего мак-
роскопич. поля. Коэфф, пропорциональности между
поляризацией и полем, равный , наз. диэ-
лектрич. восприимчивостью вещества. В большом
диапазоне значений напряженности поля Д. п. не
зависит от величины напряженности — электрич.
смещение линейно зависит от поля (D—&E, см.
Диэлектрики). Однако имеется класс веществ,
для к-рых это соотношение не линейно: эти вещества
наз. сегнетоэлектриками.
Д. п., измеренная в постоянном электрич. поле, наз. ста-
тической; она определяется установившимся значением поля-
ризации диэлектрика. Процесс установления поляризации
в диэлектриках требует определенного времени. При мгно-
венном включении поля поляризация диэлектрика изменяется
по экспоненциальному закону:
Р = Ро [1 — exp (— t/x)]
где PQ — установившееся значение поляризации, t — время
с момента включения поля, г — время релаксации. В пере-
менном электрич. поле, когда период изменения поля сравним
с временем релаксации, Д. п. является комплексной функцией
частоты поля (дисперсия):
s (ш) = е' (со) — je" (ш)
Мнимая часть Д. п. определяет собой диэлектрич. потери.
Тангенс угла диэлектрич. потерь, равный отношению активной
составляющей тока, протекающего через диэлектрик, к реак-
тивной составляющей этого тока, может быть определен через
действительную и мнимую части Д. п.; tg д = г”/е' (если
не учитывать потери, связанные с электропроводностью).
Время релаксации дипольной поляризации соизмеримо
с периодом колебаний поля в диапазоне радиочастот, мнимая
и действительная части Д. п. зависят от частоты. Можно
показать, что 8' и 8" могут быть определены через выражения
(уравнения Дебая):
t Ss ' еОО •qq)’
e’ = eoo + t _|_ ,„2-2 ; e = t +
где es — статич. Д. n., sco — Д. п., соответствующая ионной
и электронной поляризации (может быть определена измере-
нием при частоте много большей частоты дисперсии), т — время
релаксации. <в = 2л/. где / — частота приложенного поля.
Во всем диапазоне радиочастот Д. п., обусловлен-
ная поляризацией, вызванной смещениями электро-
нов или ионов, не зависит от частоты и слабо зависит
от темп-ры. Если же диэлектрик построен из молекул,
представляющих собой диполи, то в нем возникает
дипольная поляризация (см. Дипольный момент).
Для таких диэлектриков Д. п. существенно зависит
от темп-ры и частоты.
Величина Д. п. связана со структурой вещества.
Она зависит как от индивидуальных свойств атомов,
молекул и ионов, из к-рых построено вещество, гак
и от его строения. Величины Д. п. для диэлектриков
разных классов приведены в таблице в ст, Диэлек-
трики. _ Е. А. Нонорова.
ДИЭЛЕКТРИЧЕСКИЕ СВОЙСТВА ПОЛИМЕ-
РОВ — совокупность свойств, величинами к-рых опре-
деляется пригодность полимеров для электротехнич.
и радиотехнич. изделий. Д. с. п. характеризуются
электропроводностью, диэлектрич. проницаемостью
и электрич. прочностью.
Ионная электропроводность о экспоненциально воз-
растает с повышением темп-ры. Источником ионов
в полимерах-диэлектриках являются низкомолеку-
лярные примеси, связанные молекулярными силами
с макромолекулами; о резко возрастает выше темп-ры
стеклования полимеров их текучести, когда время
релаксации макромолекул становится малым и соиз-
меримым с длительностью измерения о. Величина о
существенно зависит от химич, состава и строения
1189
ДИЭТИЛАМИН — ДИЭТИЛДИТИОФОСФАТ НИКЕЛЯ
1190
полимеров и в интервале темп-р ниже Tg имеет зна-
чение от IO’12 ом 1 см-1 до 10~19 ом~г см"1; выше
Т верхний предел о в нек-рых полимерах достигает
l(Veo.w 1 • см-1. Электропроводность возрастает под
действием ядерных излучений; возрастание электро-
проводности в этом случае обусловлено появлением
электронной проводимости и зависит от строения
полимеров.
Обобщенная диэлектрич. проницаемость е=е' -f- ге"
является комплексной величиной, где вещественная
часть е,' обусловлена деформационной и ориентацион-
ной поляризацией, а мнимая е" характеризует ки-
нетику процесса установления ориентационной ди-
польной поляризации (i — коэфф, при мнимой еди-
нице). С макроскопич. точки зрения в" является
мерой диэлектрич. потерь. В технике обычно вместо
е" пользуются величиной tgd=e"/e' (тангенс угла
диэлектрич. потерь). По зависимости е' и е" от ча-
стоты и темп-ры полимеры сходны с молекулярными
жидкостями и кристаллами и по этим свойствам могут
быть разделены на аморфные и кристаллические, а
также на полярные и неполярные.
Для неполярных полимеров, аморфных и кристал-
лических (полистиролы, поливинилнафталин, поли-
этилен, политетрафторэтилен) в' в оеновном опреде-
ляется деформационной электронной поляризацией
и ее значение (2,0—2,6) не зависит от частоты / в пре-
делах от 0 до 1011 гц, уменьшаясь с повышением
темп-ры. в" по величине близко к 5 • 104, но в опре-
деленном для каждого полимера температурно-частот-
ном диапазоне может проходить через максимум,
причем в зависимости от строения полимера ечакС_
имеет величину от 7 • 10 4 до 7 • 10-3. В случае аморф-
ных неполярных полимеров имеется лишь одна об-
ласть, в к-рой е" проходит через максимум, в то
время как для кристаллич. наблюдается до трех таких
областей; е' и в" полярных полимеров имеют в ос-
новном дипольную природу; в' может достигать
значения 10 и выше. В области максимального зна-
чения в” может превышать 1. Зависимость в" от
темп-ры и частоты для полярных полимеров обладает
одним, двумя или даже тремя областями максимумов.
Для всех полимерных диэлектриков температурно-
частотные зависимости в' и е," обусловлены диполь-
ной природой полимера или его примесей и релакса-
ционным характером установления поляризации. Сорб-
ция полимерами низкомолекулярных веществ при-
водит к смещению областей е^акс к более низким
темп-рам или высоким частотам. Это влияние особенно
характерно для потерь, наблюдаемых выше Tg. Уста-
вовлено влияние одноосного и двуосного растяже-
ний полимеров на величину в' и в" и положение об-
ласти максимума в" в зависимости от темп-ры и
частоты. Особенно значительно такое изменение для
полистирола, полиамидов и др. при двуосном растя-
жении.
Пробой диэлектрика характеризуется напряжен-
ностью электрич. поля Еп^ , к-рое для различных по-
лимеров имеет порядок сотен киловольт на 1 мм тол-
щины образца. Характерным является резкое сни-
жение Еп^ выше Tg и более высокие значения Еп$
для полярных полимеров по сравнению снеполярными.
Лит..* Т а р е е в Б. М., Электротехнические материалы,
6 изд., М.—Л., 1958; К о б е к о П. П., Аморфные вещества,
М. —Л. 1952;«Действие излучений на полупроводники и изоля-
торы, М., 1954; Сканави Г. И., физика диэлектриков,
М. — Л .. 1958; Михайлов Г. П., Усп. химии 1955, 24,
вып. 7; Th urn Н., WUrstlin F., Kolloid-Z., 1958,
156, H. 1, 21—7; Mti 1 1 er F. H., Krum F. Hutt К
там же, 1957, 152, H.l; X и п п e л ь А. Р., Диэлектрики
и их применение, пер. с англ., М.—Л., 1959; Михай-
лов Г. П., Сажин Б. И., Усп. хим., 1960,29, вып 7-
Mich ailowG.P., Makromolek. Chem., 1960, 35 A.
Г. П. Михайлов.
ДИЭТИЛАМИН (C2H6)2NH, мол. в. 73,14 — бес-
цветная жидкость с резким запахом; т. пл. —50°;
т. кип. 55,2°/760 мм, 6°/100 мм; 0,7056; nj/j
1,38637; константа диссоциации ^=12,6 • 10(25°);
4крИТ. 223,3°; ркрит 36,58 ат; теплота испарения
6,66 ккал/молъ (58°). Д. легко растворим в воде,
спирте и эфире. Д. обладает всеми свойствами вто-
ричных аминов. Применяется Д. в различных про-
мышленных синтезах, например в произ-ве диэтил-
амида никотиновой к-ты (кордиамина), употребляе-
мого в качестве стимулятора сердечной деятельно-
сти; при получении ускорителей вулканизации кау-
чука (тиурам); в синтезе новокаина и др. В пром-сти
Д. получают при взаимодействии этилового спирта
с аммиаком при 300—450°, 15—30 ат в присутствии
смешанного катализатора Al2O3(SiO2)Cr2O3 по реак-
ции: 2С2Н6ОН + NH3 -> (C2H6)2NH + 2Н2О. Д.
можно получить разложением диэтиланилина. Д.
идентифицируется: по монохлоргидрату, т. пл. 226°;
оксалату, т. пл. 220°; пикрату, т. ил. 155° и др.
ДИЭТИЛАНИЛИН CeH6N(C2H6)2, мол. в. 149,23 —
жидкость слабо-желтого цвета, т. заст. — 38,8°;
т. кип. 215,5°/760 мм, 147,3°/100 мм, 129°/60 мм;
0,93507; nl£’7 1,5439; растворяется в спирте, эфи-
ре, нерастворим в воде; константа диссоциации: К —
=4,5- 10-8 (20°). Д. .образует с HNO3 п-нитрозопро-
изводное; легко вступает в конденсацию с альдеги-
дами, фосгеном. Д. получают из анилина действием эти-
лового спирта и соляной кислоты с последующим
разделением образующей смеси аминов ректифика-
цией. Д. применяется гл. обр. в произ-ве трифенил-
метановых красителей, напр. основного ярко-зеле-
ного, патентованного голубого и др., и для получения
диэтиламина. в. А. Пупков.
ДИЭТИЛДИТИОКАРБАМИНАТ НАТРИЯ, мол. в.
171,25 — белые кристаллы; хорошо растворим в воде,
плохо — в спирте. Получают
Д. н. из диэтиламина и серо- xN_r^S
углерода. Впервые применен \gNa-3H»O
в 1908 для определения меди, 2
с которой образует в нейтральном, щелочном и
кислом р-рах желто-коричневый осадок или окра-
шивание; реагирует со всеми элементами, даю-
щими в водных р-рах устойчивые сульфиды, в том
числе с Se и Те, с образованием внутрикомплексных,
малорастворимых в воде соединений, отличающихся,
как правило, по цвету от соответствующих сульфидов.
Диэтилдитиокарбаминаты металлов хорошо извлекают-
ся обычными органич. растворителями с образованием
экстрактов, окрашенных в различные цвета (от жел-
того до коричневого, для Мо — красного), чем исполь-
зуются для отделения, обогащения и фотометрич. опре-
деления следов элементов (Си, Bi, Со, Ni, V, Gr и др.);
применяется для определения Pt, Pd, In, Ga, Mn, Pb,
Zn и t. д. и разделения элементов, основанного на
различной устойчивости диэтилдитиокарбаминатов прв
различных pH среды в присутствии маскирующих ве-
ществ (тартрат натрия, цианид калия, комплексон III
и др.).
Лит.: Чернихов Ю. А., Добкина Б. М., Зав.
лаб., 1949, 15, № 10, 1143—49; Bode Н., Fresenius Z. ' 1.
analyt. Chem., 1954, 142, Н. 6, 414; его же, там же,
143, Н. 3, 182; е г о ж е, тамже, 1955, 144, Н. 1,90, Н. 3, 165.
В. Г. Типиова.
ДИЭТИЛДИТИОФОСФАТ НИКЕЛЯ c8H20O4S4P2Ni,
мол. в. 429,15 — кристаллы фиолетового цвета; т. пл.
104 — 105°; хорошо рас-
творим в большинстве v=11su\ //>
(бенщмге, эК^иТло^
этане, хлороформе), умеренно — в воде, относительно
мало— в спирте, ледяной уксусной к-те на холоду, но хо-
1191
ДИЭТИЛЕНГЛИКОЛЬ — ДИЭТИЛОВЫЙ ЭФИР
1192
рошо при нагревании. Получают Д. н. при добавлении
насыщенного р-ра хлорида никеля к р-ру диэтилдитио-
фосфорной к-ты. Д. н. легко окисляется иодом в кис-
лых и нейтральных р-рах до дисульфида, а в сильно
щелочных р-рах — до сульфата; это свойство исполь-
зуется для определения концентрации его водных
р-ров и определения нек-рых металлов; образует
с многими элементами группы сероводорода мало-
растворимые соединения постоянного состава. При-
меняют Д. н. для открытия Мо, для связывания Пи
в комплексное соединение при открытии осмия тио-
мочевиной, определения Си, Bi, Pd в присутствии
Pt и др., Cd в присутствии Zn, РЬ в присутствии Ва,
Са, Zn и др. В виде титрованного р-ра Д. н. исполь-
зуют в амперометрич. и потенциометрич. титровании
Си, Pb, Pd и т. д. Диэтилдитиофосфаты многих эле-
ментов хорошо экстрагируются, образуя окрашенные
р-ры, на чем основано фотометрии, определение Си,
Bi, Pd и нек-рых др.
Лит.: Б у с е в А. И., Иванютин М. И., Ж. аналит.
хим., 1956, 11, вып. 5, 523; их же, там же, 1956, 13, вып. 1,
18; и х же там же, 1958, 13, вып. з, 312, вып. 6, 647; и х ж е,
Вести. МГУ. Серия мат., мех., астр., физ., хим. 1958, № 2,
с. 177; и х ж е, там же, 1957, № 5, с. 157. В. Г. Типцова.
ДИЭТИЛЕНГЛИКОЛЬ (дигликоль, |3,|У-диокси-
диэтиловый эфир) НОСН2СН2—О—СН2СН2ОН, мол. в.
106,12 — густая бесцветная жидкость; т. пл. —8°;
т. кип. 245°/760 мм, 133°/14 мм, 119°/7 мм; 1,1197;
п‘-$ 1,4472; смешивается с водой, спиртом, ацетоном;
мало растворим в бензоле, толуоле, четыреххлористом
углероде. Д. получают при взаимодействии этилен-
гликоля с окисью этилена:
НОСН»СН-,ОН + СН2СН26—(НОСН3СН.,)3О
он образуется также из этиленгликоля и этиленхлор-
гидрина:
НОСН..СН»ОН+С1СН3СН«ОН — (НОСН2СН»)»О+НС1
Оба способа являются промышленными. Химич, свой-
ства Д. подобны свойствам этиленгликоля. Д. при-
меняют как растворитель масел, нитроцеллюлозы,
полифенольных смол; в качестве пластификатора;
Д. входит в состав антифризов; в газовой пром-сти
используется в качестве осушителя и в комбинации
с этаноламином HOCH2CH2NH2 для обессеривания
газов. Диметиловый эфир диэтиленгликоля («диглим»)
СН3ОСН2СН2—О—СН2СН2ОСН3; т. кип. 160,5—
161°/756,6 мм; <7)1 0,9514; n'/'j 1,4097; применяется как
растворитель. М. Ф. Турчинский.
ДИЭТИЛЕНГЛИКОЛЬДИНИТРАТ (нитродигли-
коль) 02NOCH2CH2OCH2CH2ON02, мол. в. 196,12 —
вторичное (бризантное) взрывчатое вещество; бесцвет-
ная сиропообразная жидкость; при охлаждении кри-
сталлизуется в двух модификациях — стабильной (т. пл.
4-2°) и лабильной (т. пл. — 10,9°); <716 1,39; пр* 1,4517.
Теплота сгорания 2 792 кал/г. Вязкость Д. при
20° 8,1 спуаз, летучесть больше, чем нитроглицерина
и меньше, чем нитрогликоля. Д. плохо растворим
в воде (0,4% при 25°), этиловом спирте, СС14 и CS2,
смешивается во всех отношениях с нитроглицерином,
нитрогликолем, ацетоном, хлороформом, бензолом и
метиловым спиртом; хорошо желатинирует нитро-
целлюлозу. Скорость омыления в присутствии щело-
чей и кислот меньше, чем скорость омыления нитро-
глицерина. Получают Д. этерификацией диэтилен-
гликоля смесью конц. серной и азотной к-т.
Д. применяют для изготовления нек-рых типов бал-
листных порохов и промышленных взрывчатых ве-
ществ.
Лит.: Наум Ф., Нитроглицерин и нитроглицериновые
взрывчатые вещества, М.—Л., 1934; Орлова Е. Ю.,
Химия и технология взрывчатых веществ, М., 1960.
Б. Н. Кендриков.
ДИЭТИЛКАДМИЙ Cd(C2Hs)2, мол. в. 160,53 —
бесцветная жидкость; т. кип. 61 — 62°/20 мм; плотн.
1,65. Д. разлагается под действием воды, воздуха и
света. Получают из CdBr2 и C2H6MgBr. В присутствии
малых количеств кислорода или нек-рых кислород-
содержащих веществ инициирует радикальную поли-
меризацию ряда мономеров. В сочетании с TiCl4
образует катализатор, обладающий нек-рой стерео-
специфичностью; на этом катализаторе могут быть
получены 1,4-т.ранс-полибутадиен и небольшие коли-
чества изотактич. полистирола.
Лит.: Furukawa J., Tsuruta Т., Fueno Т.,
Makromolek. Chem., 1959, 30, 109.
А. А. Арест-Якубович.
ДИЭТИЛМАЛОНАТ (малоновый эфир, диэтило-
вый эфир малоновой кислоты) С2Н6ООССН2СООС2Н6,
мол. в. 160,18 — бесцветная маслянистая жидкость
с приятным фруктовым запахом, практически нерас-
творимая в воде, смешивающаяся со спиртом и
эфиром во всех отношениях; т. пл. — 49,9°; т. кип.
198,9°/760 мм, 92°/18 мм; d2a 1,0553; nft 1,41428. Д.
является очень реакционноспособным соединением;
соседство двух карбэтоксильных групп обусловли-
вает высокую подвижность водорода метиленовой
группы, благодаря чему легко образует металлич.
производные. При действии на натрий- или этоксимаг-
нийпроизводное Д. галоидными алкилами или аци-
лами получаются моно- или диалкил- и ацилпроизвод-
ные, применяемые для синтеза высших алифатич. или
жирноароматич. к-т, кетокислот, кетонов и др. При
конденсации Д. с альдегидами и кетонами образуются
а,|3-непредельные дикарбоновые эфиры или произ-
водные тетракарбоновой к-ты, а с мочевиной Д. и
его алкилпроизводные образуют барбитуровую кис-
лоту и ее 5-алкилзамещенные, многие из к-рых на-
ходят широкое применение в качестве снотворных
средств. Д. может быть получен этерификацией мало-
новой кислоты обычными способами; в пром-сти полу-
чают цианированием монохлоруксуснои к-ты с по-
следующим гидролизом нитрильной группы в обра-
зующейся циануксусной к-те и этерификацией полу-
чающейся при этом малоновой к-ты.
ДИЭТИЛОВЫЙ ЭФИР (этиловый эфир, серный
эфир) (С2Н6)2О, мол. в. 74,12 — важнейший предста-
витель простых эфиров; бесцветная, легкоподвижная,
летучая жидкость со своеобразным запахом; т. пл.
—116,3° (устойчивая форма), —123,3° (неустойчивая
форма); т. кип. 34,481°/760 мм; d2n 0,7135; пр 1,3527;
ц20 0,242 спуаз; теплота парообразования прв 20°
6,488 ккал/моль, при темп-ре кипения 6,380 ккал/моль;
теплоемкость Ср 39,88 кал/моль (15°); давление пара
1 мм (—74,3°), 10 мм (—48,1°), 40 мм (—27,7°), 200 мм
(2,2°), 400 мм (17,9°); гкрит 193,8°; ркрит 36,3 ат;
криоскопии, постоянная 1,79°; эбулиоскопии, по-
стоянная 1,84°; т. воспл. 9,4° (в закрытой чашке);
т. всп. 188°; взрывоопасные смеси с воздухом от 1.71
до 48,0 об. %. Д. э. хорошо растворим в органич.
растворителях и сам растворяет очень многие органич.
вещества; резина в Д. э. набухает; вещества, имеющие
кристаллич. ионную структуру, в Д. э. нерастворимы;
в воде при (20°) растворяется 6,590 вес. % Д. э;,
при 25° в Д. э. растворяется 1,468 вес. % воды; об-
разует азеотроп с водой (98,74% Д. э.), т. кип. 34,15°.
При хранении Д. э. под действием кислорода воз-
духа в незначительном количестве образуются взрыв-
чатые перекиси и ацетальдегид:
(С3Н5)2О + ЗО3 — 4СН3СНО + Н3О3
(С3Н5)2О + Н3О3 —- С3Н5О-ОС3Н5 + Н3О
СН3СНО+ Н3О — СН3СНОН-О-О-СНОНСН3 и т. д.
Образование перекисей замедляется при хранении
в темноте и на холоду. При взаимодействии Д. э.
с конц. минеральными к-тами получаются оксониевые
соединения типа [(С2Н6)2ОН]Х-;эфираттрехфторисгого
1193 ДИЭТИЛПАРАФЕНИЛЕНДИАМИНСУЛЬФАТ — ДИЭТИЛФТАЛАТ
1194
бора (С2Н6)2О • BF3 применяется
В пром-сти Д. э. готовят из
действуя на него H2SO4 при
140° (отсюда его старое название
«серный эфир»):
с2н5онч- H3SO4 — C2H5OSO2OH+H,O
2C3H5OSO3OH + с3н-оон —-
— (С3Н5)2О + H2SO4
Д.э. широко применяется'как рас-
творитель, но гл. обр. в лаборатор-
ной практике (напр., при Гринь-
яра реакции). Применение Д. э.
в пром-сти затруднено из-за его
большой огне- и взрывоопасности.
Широко используется Д. э. в ме-
дицине для ингаляционного нар-
коза (для этой цели выпускают
особо чистый, свободный от пе-
рекисей «наркозный» эфир). Частое
воздействие Д. э. на человека вы-
зывает привыкание. Д. э. ядовит;
максимально допустимая концен-
трация его в воздухе 0,3 -мг/л.
См. .также Вильямсона синтезы.
Лит.: Вайсбергер А. [и др.), Органические раство-
рители, пер. с англ., М., 1958; Роберт-Нику М. Ц.,
Химия и технология химико-фармацевтических препаратов,
2 изд., М., 1959. А. Е. Васильев.
ДИЭТИЛПАРАФЕНИЛЕНДИАМИНСУЛЬФАТ
(сульфат п-амино-М,1Ч-диэтиланилина), мол. в. 262,32,
n-(C2H6)2N—СвН4—NH2 • H2SO4 — бесцветные кри-
сталлы; т. пл. 182—183°; очень хорошо растворим в
воде, плохо — в спиртах, нерастворим в эфире. Свобод-
ное основание (C2H6)2N—СвН4—NH2, мол. в. 166,24,
бесцветная до светло-желтого цвета маслообразная
жидкость, темнеющая на воздухе, т. кип. 260—
262°; с неорганич. кислотами образует устойчивые
соли. В щелочных растворах Д. выделяет свободное
основание, к-рое в присутствии окислителей с веще-
ствами, содержащими активную метиленовую или
метильную группу, образует азометиновые или хи-
нониминовые красители:
CeH5COCH2CONHCeH5 + n-(C3H5)2NC,H4NH3 + 4Ag+ —
СеН5-СО
/ C=NCeH4N(C2H5) -)-4Ag
CeH5NHCO
Получают Д. нитрозированием М,М-диэтиланилина
с последующим восстановлением полученного п-нит-
розо-М,1Ч-диэтиланилина. Под маркой «ППН» Д.
используется в кинофотопром-сти в составе прояви-
теля для цветных многослойных фотоматериалов.
См. также Проявляющие вещества, Цветное про-
явление. р. б. Журки.
ДИЭТИЛСТИЛЬБЭСТРОЛ [стильбэстрол, транс-
-3,4-ди-(я-оксифенил)-гексен-3, тране-k -диокси-а,
а'-диэтилстильбен] CISH20O2, мол. в. 268,36 — бес-
цветные- кристаллы; т. пл. 168—171°; трудно рас-
творим в воде, растворим в водных растворах
едких щелочей, спирте, эфире и арахисовом масле.
При растворении Д. в 1 мл конц. H2SO4 появляется
интенсивная оранжевая окраска, к-рая при разба-
влении водой бледнеет и через неск. минут исчезает.
Колич. определение Д. основано на ацетилировании
точной навески Д. уксусным ангидридом в пири-
как катализатор. I дине и определении веса полученного диацетата Д.
этилового спирта, Обычно Д. получают из фенола:
а также из а-фенилмасляной к-ты. Д. применяют при
расстройствах эндокринной системы, а также для
лечения рака предстательной и молочной желез. Для
инъекций Д. выпускается в виде дипропионата (т. пл.
104—107°), из к-рого приготовляют растворы в ра-
стительных маслах.
Лит.: Wilds A. L., Biggerstaff W. R., J.
Amer. Chem. Soc., 1945, 67, № 5, p. 789; Dongorozi S.,
C 1 г J a M., Rev. Chim., 1955, 6, № 11, p. 599; см. также при
ст. Адалин. А. С. Елина.
ДИЭТИЛСУЛЬФАТ (C2H6)2SO4 — жидкость; т. пл.
— 26°; т. кип. 210°/760 мм, 96°/15 мм; rfj5 1,1842;
п1£ 1,4025. При перегонке, особенно под атм. давле-
нием, Д. частично разлагается, что вызывается при-
сутствием обычно самых незначительных количеств
H2SO4. В воде Д. практически нерастворим, но легко
гидролизуется теплой водой; растворим в спирте, при
нагревании раствора превращается в диэтиловый эфир
и этилсерную к-ту; с эфиром смешивается во всех
отношениях; ограниченно растворим в углеводоро-
дах. Получают Д. при насыщении H2SO4 (моногидрат)
этиленом под давлением 10 ат и обычной темп-ре;
образуется с хорошим выходом также при действии
на С2Н6ОН дымящей H2SO4, смеси H2SO4 с Р2О6 или
SO3. Д. применяется для этилирования окси-, кар-
бокси-, меркапто-, амино- и иминогрупп в присут-
ствии щелочей или карбонатов. При низких темп-рах
(50—60°) реагирует только одна этильная группа,
вторая вступает в реакцию лишь ок. 150°.
Лит.: De L о i s у Е., Damiens A., Chimie et Indu-
strie, 1923, № 5 (Numero special), 644, Сьюте p 4. [M.J,
Химия органических соединений серы, пер. с англ., ч. 1,
М., 1950. Г. М. Цигуро.
ДИЭТИЛФТАЛАТ (диэтиловый эфир о-фталевой
кислоты) СвН4 (СООС2Н6)2, мол. в. 222— бесцветная
жидкость; т. пл. — 40°; т. кип. 295°/760 мм, 158°/10 мм;
1,118; tip 1,501; т. воспл. 152°; вязкость 10,06
спуаз (25°); поверхностное натяжение 35,3 дин [см
(20,5°); растворимость Д.: в воде 0,1% (18°), 0,15%
(20°); в минеральном масле 2,8%; растворимость воды
в Д. 0,7% (20°), минер, масла в Д. 1,1%; летучесть
3,4 мг[см2[час при 100°. Д. совмещается с ацетилцел-
люлозой, ацетобутиратом целлюлозы, нитроцеллю-
лозой, этилцеллюлозой, полистиролом, полиметил-
метакрилатом, поливинилацетатом, поливинилбути-
ралем. При кипячении Д. с мочевиной или тиомочеви-
ной и этилатом натрия в спиртовом р-ре образуется
фталимид. При гидрировании Д. при высоком давле.
1195
ДОДЕЦИЛМЕРКАПТАН — ДОЗАТОРЫ
1196
нии в присутствии никеля Ренея получается смесь
цис- и транс-гексагидродиэтилфталатов. При нагре-
вании с этиленгликолем сначала при 190°, потом
в вакууме при 300° получаются полимеры гликоль-
фталата. Колич. определение Д. проводится омыле-
нием его спиртовой щелочью с последующим титро-
ванием избытка последней. Д. входит в состав поли-
меров, перерабатываемых экструзией, с целью улуч-
шения поверхности изделий; в небольших количе-
ствах иногда добавляют в феноло-формальдегидные
смолы для повышения ударной прочности. Д. яв-
ляется пластификатором для полимеров, денатури-
рующим средством для спиртов, фиксатором запаха
духов.
В пром-сти Д. получают нагреванием молекуляр-
ных количеств этанола и фталевого ангидрида в авто-
клаве при 15—20 ат, снабженном дистилляционной
колонной. Выделяющаяся при реакции вода уда-
ляется в виде азеотропной смеси с бензолом, к-рый
непрерывно вводится в реакционную смесь. Получают
Д. также нагреванием фталевого ангидрида: с 5-крат-
ным количеством абс. этанола, содержащего 3% НС1;
с этанолом в присутствии безводной CuSCh; с 85%-ным
этанолом в присутствии безводного СаС12 и неболь-
ших количеств НС1; с тетраэтилортосиликатом при
160° и др. способами.
Лит.: Buttrey D. N., Plasticizers, 2 ed., L., [1957];
В a i 1 e у W. J. and GoldenH.K., J. Amer. Chem. Soc.,
1953, 75, № 19, p, 4780; L 1 d d e 1 1 H. F., Analyst, 1957,82,
p. 375; Doolittle A. К The technology of solvents and plas-
ticizers, N. Y. —L., 1954; Th in ins K., Chemie, Physik und
Technologie der Weichmacher, B., 1960. О. Я. Федотова.
ДОДЕЦИЛМЕРКАПТАН (лаурилмеркаптан)
CH3(CH2)10CH2SH, мол. в. 202,39 — бесцветная мас-
лянистая жидкость; т. кип. 169°/39 мм, 124°/5 мм,
95—96°/1—1,5 мм; 0,8540; «р 1,4568; нерастворим
в воде, растворяется в спирте, эфире, углеводородах.
Различными окислителями (H2SO4, J2 -ф- NaOH, О» в
присутствии NH3). Д. превращается в дисульфид
(C12H26)2S2; HNO3 при нагревании окисляется в доде-
цилсульфокислоту Ci2H26SO3H; дает солис тяжелыми
металлами (Pb, Hg). Получают Д. из бромистого
н-додецила и тиомочевины нагреванием в спирте;
образующуюся бромистоводородную соль додецил-
изотиомочевины разлагают щелочью при нагреванив:
n-CjeHs5Br + S=C(NH»)3 — CI2H45S— ОС -НВг
'KH2
/.NH
2C12H»5-Sf • НВг -I- 2NaOH — 2C!,H25SH -f- 2H„O 4-
XNH2
,nh.
+ 2NaBr 4- NH=C(
'NH-CN
Под действием окислителя (K2S2O3) Д. легко превра-
щается в радикал C12H25S, к-рый инициирует по-
лимеризацию мономера (напр., дивинила). Приме-
няют Д. для регулирования процессов полимериза-
ции в произ-ве синтетич. каучука по эмульсионному
методу. М. Ф. Турчипский.
ДОЗА ИОНИЗИРУЮЩЕГО ИЗЛУЧЕНИЯ — мера
энергии излучения, поглощенной единицей среды
на облучаемом участке. Различают локальную
Д. и. и. — дозу в данной точке, и интегральную
Д. и. и. — количество энергии излучения, поглощен-
ной во всем облучаемом объеме. Д. и. и., отнесенная
к единице времени, наз. мощностью дозы. За единицу
измерения Д. и. и. принят рад (rad, radiation absorbed
dose) — Д. и. и., равная 100 эрг)г облученного веще-
ства (обозначается рад). Эта единица применима
к любым средам и любым видам излучения. Инте-
гральная Д. и. и. выражается в грамм-радах, т. е.
дозой в радах, умноженной на массу облучаемого
вещества, выраженную в граммах. Дозы рентгенов-
ских и у-лучей с энергиями до 3 Мэв (3 • 1,60 • 10 е эрг)
в воздухе могут измеряться в рентгенах (обо-
значается р). В интервале энергий фотонов от 0,3
до 3 Мэв при дозе в воздухе в 1 р одним граммом воды
или аналогичной ей среды (напр., мускульной ткани)
поглощается 93 эрг, или 5,87 • 1013 эв[см3, т. е. 1р
в этом случае соответствует 0,93 рада. При энергиях,
меньших 0,3 Мэв, это соотношение заметно изме-
няется (см. рис.). В работах по радиобиологии в ка-
честве единиц для измерения доз корпускулярных
излучений (fj-излучение, нейтроны и т. nJ приме-
няются физич. и биологич. рентген-эквиваленты
(обозначается фэр и бэр). Фэр — такая Д. и. и.,
при к-рой в 1 см3 мускульной ткани поглощается
Зависимость количества энергии, поглощенной в 1 г
мускульной ткани и воды при дозе в 1 р от энер-
гии фотонов.
93 эрга энергии излучения, а бэр— Д. и. и., вызы-
вающая такой же биологич. эффект, как и 1 р жест-
кого рентгеновского или у-излучения. В радиацион-
ной химии дозу часто выражают в эв[см3. Различные
единицы Д. и. и. связаны между собой соотношением:
1 рад = 1,07 фэр = 6,25 • 1013 эв/г = 100 эрг/г.
Лит. см. при ст. Дозиметрия. А. М. Кабакчи.
ДОЗАТОРЫ — устройства для измерения заданных
количеств твердых, жидких и газообразных веществ,
применяемые в целях учета при произ-ве, отпуске,
потреблении и хранении различных материалов.
Д. изготовляются периодич. и непрерывного дей-
ствия и применяются в виде самостоятельных агрега-
тов со своими питателями и транспортерами либо
являются частью производственного агрегата поточ-
ной линии или автомата. По принципу действия Д.
делятся на весовые, объемные, скоростные и дроссе-
лирующие. Для твердых тел (кокс, цемент, известняк,
сода и т. п.) основным методом измерения является
весовой; объемный метод применим лишь для порошко-
образных неслеживающихся и не образующих ком-
ков сыпучих материалов. Жидкости (бензин, спирт,
керосин и т. п.) вследствие их текучести, практич.
несжимаемости и постоянства объемного веса можно
измерять весовым, объемным, скоростным и дроссе-
лирующим методами. Для измерения количества газа
применяются объемный, скоростной и дросселирую-
щий методы.
Весовые Д., применяемые для измерения коли-
чества и расхода твердых кусковых и порошкообраз-
ных материалов, существуют в виде приборов пе-
риодич. и непрерывного действия, с ручным или авто-
матич. управлением. Порционные весы автоматически
отсекают определенные количества взвешиваемого
материала, поступающего из питательного бункера,
и после взвешивания выбрасывают порцию в прием-
ный бункер. Счетчик весов суммирует вес материала,
прошедшего через весы. Автоматич. порционные
весы подразделяются на весы для вагонеток, весы
1197
ДОЗАТОРЫ
1198
с опрокидывающимся ковшом и весы с открывающимся
дном ковша. Весовые Д. непрерывного действия
(рис. 1), автоматически суммирующие вес материала,
Рис. 1. Схема весового дозатора для непрерывного дози-
рования с электрорегулировкой: 1 — ленточный транспор-
тер; 2 — опора; з •— электродвигатель; 4 — весовой меха-
низм;! 5 — коромысло; 6 — электровибрационный пита-
тель; 7 — передвижная гиря; 8 — электроустройство;
9 — приемная воронка.
мая установка (рис.
проходящего через них на ленточном или ковшовом
транспортере, обычно состоят из бункера с затворами
и транспортера с весовым механизмом, регулирующим
выход дозируемого вещества. Регулирование подачи
вещества на транспортер может также осуществляться
вибратором с пневматич. или электромагнитным при-
водом, управляемым весовым механизмом.
Объемные Д. применяются гл. обр. для
периодич. или непрерывного измерения количества
жидкостей и газов. Периодич. Д. для жидкостей
основаны на использовании резервуаров с определен-
ной емкостью (мерники, сосуды с поплавком, опроки-
дывающимся ковшом, сосуды с вытеснителем и т. п.).
Используются также счетчики, по показаниям к-рых
производится отпуск жидкости вручную или авто-
матически. Примером может служить бензораздаточ-
прерывные объемные Д.
делятся на барабанные,
поршневые, кольцевые,
дисковые и шестеренча-
тые, в устройство к-рых
входят счетчики жидко-
сти и регулирующие
органы (клапаны, венти-
ли и т. п.). Барабанные
Д. снабжены барабан-
ными счетчиками, к-рые
приводятся в действие
жидкостью, притекаю-
щей открытой струей
в барабан; применяются
для измерения расходов
от 0,025 до 12 м^/час
с точностью ± 0,5%,
в трубопроводах с да-
Рис. 2. Схема бензоразда-
точного дозатора периодиче-
скоро действия: 1 — резер-
вуар; 2 — воздушный кран;
3 — фильтр; 4 — насос; 5 —
г азоотделитель; в — трубопровод; 7 — воздушник; 8 — тон-
кий фильтр; 9 — бензосчетчик; 10 — спускной кран; 11 —
смотровое стекло; 12 — указатель уровня; 13 — воздушный
клапан; 14 — шланг; 15 — кран.
влением до 0,4 кг/см2. Поршневой Д. снабжен
поршневым расходомером, поршень к-рого совер-
шает прямолинейное переменно-возвратное движение
В замкнутом цилиндре; применяются преим. для до-
зирования нефтепродуктов в закрытых системах
под давлением, измеряют расходы от 0,01 до 100 м3[час
с точностью ± 1%. Кольцевой Д. имеет кольцевой
расходомер, для приведения в действие к-рого тре-
буется значительное давление жидкости. Применяется
для учета расходов от 1,8 до 160 мР/час с точностью
± 1%. Дисковые Д. снабжены дисковыми расходо-
мерами, рабочим органом к-рых является поршень
в виде качающегося диска, совершающий круговое
колебательное движение; они учитывают расход
жидкости от 0,03 до 100 м?[час с точностью ± 1%.
Шестеренчатый Д. имеет расходомер, к-рый состоит
из двух сцепляющихся шестерен. Применяется для
учета расходов от 0,002 до 600 м3[час с точностью
± 1%. Для примера на рис. 3 показан дисковый Д.,
к-рын автоматически производит дозирование двух
Рис. 3. Схема дискового объемного дозатора: 1 — ди-
сковый расходомер; 2 — золотник; 3 — насос; 4 —
волюметр; А и Б — резервуары; В — трубопровод.
жидкостей посредством поршневого насоса и волю-
метра с пневматич. приводом, действующими от вра-
щающегося расходомера. Дисковый расходомер воз-
действует на золотник, через к-рый сжатый воздух
периодически подается в приводы насоса и волюметра,
нагнетающие дозируемые жидкости из резервуаров
А и Б в трубопровод В. Подобные Д. применяются
для огнеопасных жидкостей. Для дозирования агрес-
сивных жидкостей (кислоты) применяются мембран-
ные Д. с расходомерами и регулирующей аппарату-
рой. Для измерения количества жидкостей со взве-
шенными частицами (суспензий) применяются ков-
шевые Д., работающие в открытых системах (при
атм. давлении). Дозирование полужидких, вязких,
густых и тестообразных масс производится Д. в виде
•шприца, поршневого насоса или формовочного колеса.
Объемные Д. для периодич. измере- 4
ния расхода газов обычно состоят из г-------
резервуаров определенной емкости,
заполняемых при определенном да-
влении и темп-ре. Порции газа до-
зируются при помощи газомеров,
снабженных специальными приспо- хчР'У-И/
соблениями для прекращения пода- /У—
чигаза. Объемные Д. для непрерыв-
ного измерения расхода газов пред- Рис. 4. Мокрый
ставляют собой газомеры или рас- пус°сежидк7е^ью'
ходомеры, связанные между собой а — четырехка-
и с регулирующими органами (кла- мерный барабан;
паны, вентили, задвижки} посред- t ~ цилиндр; 4 —
’ ’ ’ г « отводящий трубо-
ством механической, гидравлической провод,
или электрической системы. Объемные
газомеры подразделяются на мокрые (рис. 4) — для
учета количества газа, от 0,5 до 10 000 м3)часу и
сухие — для учета количества газа от 0,1 до
1000 м2[час.
1199
ДОЗАТОРЫ — ДОЗИМЕТРИЯ
1200
Скоростные
рывного измерения
Д., применяющиеся для непре-
количества жидкости от 0,2
Рис. 5. Винтовой расходомер: 1 — корпус; 2 — струевы-
прямитель; з — колесо с винтовыми лопастями; 4 —
счетчик.
до 10 м3/час (точность zt0,2%), работают на принципе
измерения скорости потока
Рис. 6. Типы дроссельных при-
боров: а — острая диафрагма;
б — сопло; в — труба вентури.
и подразделяются на
крыльчатые и винтовые.
У крыльчатого Д. крыль-
чатка расходомера при-
водится во вращение
струями жидкости, вы-
ходящими из направляю-
щих отверстий и уда-
ряющими о лопасти. Вин-
товой Д. снабжен винто-
вым расходомером (рис.
5), к-рый имеет аксиаль-
ное колесо с лопатками,
приводимое во вращение
потоком жидкости, на-
правленным по его оси.
Аксиальное колесо при-
водит во вращение свя-
занный с ним червячной
передачей счетчик, ско-
рость вращения к-рого
пропорциональна расхо-
ду жидкости. Д. этого
типа применяются для
учета расходов от 1 до 4000 м3)час с точностью ±2%.
Дросс елирующие Д. применяются в химич.
пром-сти для измерения больших расходов жидкости,
4
газа или пара и загрязненных вод.'
Д. этого типа основаны на измерении
перепада давлений при протекании
вещества в трубопроводе с местным
сужением. Дросселирующие устрой-
ства исполняются трех видов (рис. 6):
острая диафрагма (наиболее распро-
страненная), сопло и труба Вентури.
При прохождении среды через су-
женное отверстие увеличивается ско-
рость потока, часть потенциальной
энергии потока переходит в кине-
тическую. Величина перепада дав-
ления (Рг и Р2) до и после сужения
зависит от количества протекающего
газа или жидкости, что дает воз-
можность вычислить их расход. Дрос-
селирующие Д. монтируются с рас-
Рис. 7. Ротаметр: 1 — вход среды; 2 —
ивмерительиая стеклянная труба со шка-
лой; 3 — поплавок; 4 — выход среды.
ходомерами (дифманометры), к-рые подразделяются
на поплавковые (применяемые при высоких давле-
ниях — до 350 кг/см?) и колокольные (для давлений
до 30 кг[см-) расходомеры и кольцевые весы (для
давлений до 50 кг^см*). Особую группу Д. постоян-
ного перепада представляют ротаметры (рис. 7), дей-
ствие к-рых основано на прохождении жидкости
или газа через кольцевое отверстие переменного
сечения. В расширяющуюся кверху трубу помещают
поплавок. Измеряемая среда, перемещаясь снизу
вверх, поднимает поплавок до тех пор, пока вес
его не уравновесится разностью давлений потока
до и после поплавка. Высота перемещения поплав-
ка будет пропорциональна количеству протекающей
среды, к-рое отсчитывается по шкале прибора, гра-
дуированной в единицах расхода. Ротаметры слу-
жат для измерения мгновенного расхода газов или
жидкостей от 0,5 л/час до 10 000 м31час. Разраба-
тываются новые типы измерительных расходомеров
и Д., основанных на индукции, ультразвуке, иони-
зации и др. эффектах.
Лит.: Щепкии С. И., Коитропьио-измеритепьиые и
регулирующие приборы в химических производствах, М.,
1945; Лоскутов В. И., Лабораторные приборы для изме-
рения расходов жидкостей и газов, М., 1950; Павлов-
ский А. Н., Измерение расхода и количества жидкостей,
газа и пара, М., 1951; Топерверх Н. И. и Шер-
ман М. Я., Теплотехнические измерительные и регулирую-
щие приборы иа металлургических заводах, М., 1951.
А. А. Гряэиов.
ДОЗИМЕТРИЯ — методы измерения и расчетов
доз в полях источников ионизирующих излучений,
а также измерений активности радиоактивных пре-
паратов. Д. основана на законах взаимодействия
с веществом заряженных частиц, коротковолнового
электромагнитного излучения и нейтронов. Области
применения: медицина, атомная пром-сть, разделы
науки и техники, встречающиеся с ионизирующими
излучениями.
Ионизирующие излучения, проходя через вещество,
поглощаются и рассеиваются. Первичными про-
цессами в веществе являются возбуждение и иони-
зация атомов и молекул. Характер вторичных про-
цессов всецело определяется природой облученного
вещества; могут наблюдаться: увеличение электро-
проводности, возникновение люминесценции, химич.
реакции, нагревание и пр. Для описания колич.
действия излучения вводится понятие — доза иони-
зирующего излучения.
В Д. различают внешние и внутренние источники
облучения. Внешними источниками могут быть:
радиоактивные препараты, нейтронные источники,
ускорительные установки, ядерные реакторы. При-
родными источниками внешнего облучения являются
космич. лучи и у-излучение горных пород. Внутрен-
нее облучение происходит в р-рах радиоактивных
веществ, с к-рыми имеют дело при переработке про-
дуктов ядерных реакций, или в растворах, специально
предназначенных для проведения химич. процессов
(см. Радиационная химия). Внутреннее облучение
живых организмов происходит за счет;содержащихся
в биосфере и входящих в ткани радиоактивных С14,
К40, Ra226 и др., а также за счет радиоактивных
веществ, введенных для лечения или исследования,
или попавших при аварии. Облучение всего тела
человека дозой 400—500 бэр приводит к смерти.
Недельная доза в 0,1 бэр принимается за предельно-
допустимую для лиц, работающих с излучением. При-
родный фон создает дозу ок. 0,1 бэр [год.
Основная задача Д. в полях внешних источников
заключается в указании значений дозы на поверх-
ности и на различных глубинах облучаемой среды;
в установлении оптимальных условий облучения дан-
ного объема среды. Расчет мощности дозы в поле
у-излучателей различной конфигурации в общем
1201
ДОЗИМЕТРИЯ — ДОЗИМЕТРИЯ ХИМИЧЕСКАЯ
1202
случае сложен. Для точечного источника мощность
дозы Р на расстоянии г см будет равна
Р = е~ |1Г р.час
где А — активность препарата (в мкюри), р.— коэфф,
поглощения излучения в среде, Г — гамма-постоян-
ная изотопа, т. е. мощность дозы в p/час на расстоя-
нии 1 см от точечного препарата активностью 1 мкюри.
Для наиболее распространенных у-излучателей, Соео
и Cs137, гамма-постоянные равны соответственно
12,9 и 3,44 р)час-мкюри-см. Для получения равномер-
ного поля доз часто применяется размещение препа-
ратов на поверхности цилиндров. Источник, содержа-
щий 10 000 кюри Со60, при диаметре и высоте цилиндра
в 20 см, дает в центре мощность дозы ок. 300000 р)час.
Мощность дозы в поле fS-излучателя пропорциональ-
на его активности и зависит от энергии ^-спектра,
формы и размеров препарата и глубины погружения
в среду. Напр., доза на одну Р-частицу, падающую на
см2 данной области среды при переходе от одного из
самых мягких среди наиболее распространенных
Р-излучателей — S35 (максимальная энергия fj-спект-
ра 0,169 Мэв) к самому жесткому — Рг144 (макси-
мальная энергия 3 Мэв), изменяется от 15 • 10“® рад
до 2,8 • 10~® рад. В пучке электронов, получаемом
с помощью ускорителя, мощность дозы может быть
очень высока. При напряжении 3000 кв и силе тока
4 ма в пучке сечением 100 см2 можно получить мощ-
ность дозы ок. 107 р/сек.
Д. внутренних источников включает: расчеты и из-
мерения дозы и установление предельнодопустимых
концентраций при попадании радиоактивных веществ
внутрь организма, исследование движения веществ,
в организме и законов их выведения. В радиационной
химии рассчитывают выход химич. реакций в среде,
содержащей радиоактивные вещества. Эти задачи
приводят к необходимости измерений концентраций
радиоактивных веществ в воздухе, воде, различных
средах.
В среде, содержащей равномерно распределенный
а- или р-излучатель, мощность дозы равна: Р —
= 2140 • сЕ рад/час, где Е — средняя энергия а- или
Р-частиц (в Мэв), с — концентрация радиоактивного
вещества (в мкюри)г). При расчете биологич. дей-
ствия вводится коэфф. 7] — относительная биологич.
эффективность излучения: Р = 2140 т\сЕ бэр/час. Для
рентгеновских и у-лучей Т)т = 1; Для а-частиц т)а =
= 10 — 20; для P-излучения Яз = 1.
Биологич. действие тепловых нейтронов обуслов-
лено происходящими в тканях ядерными реакциями
Н1(п,у)Н2 и N14 (п, р)С14, при к-рых возникают у-лучи
и протоны. Максимальная доза получается на глу-
бине 0,3 см и равна 7,6 • 10~10 бэр)н (где н — ней-
трон). Биологич. действие быстрых нейтронов обус-
ловлено ядрами отдачи (Н, С, N, О), приведенными
в движение при упругих столкновениях с нейтро-
нами.
Ионизирующие излучения могут быть обнаружены
и измерены по тем физич. и химич. процессам, к-рые
происходят при их взаимодействии с веществом.
В связи с этим используют ионизационные, фотогра-
фии., сцинтилляционные, калориметрии., химич. и
нек-рые др. методы. Наиболее распространены иони-
зационные методы. В дозиметрии, практике удобны
стеночные ионизационные камеры; ионизация в них
обубловлена частицами, выбиваемыми из стенок ка-
меры при поглощении в них излучения. Стенки камер
изготовляют из тканеэквивалентного по химич. со-
ставу материала, а камеру заполняют воздухом или
тканеэквивалентным газом. Стеночные камеры гра-
дуируют в рентгенах по эталонным камерам или с по-
мощью образцовых у-излучателей. Сила тока в каме-
рах, в зависимости от условий их эксплуатации,
лежит в пределах от 10-1в до 10~4 а)см3. Измерения
малоинтенсивных излучений затруднены наличием
токов утечки, флуктуации, ионизации, фона. Фон
космич. лучей дает в 1 см3 воздуха при атм. давле-
нии ок. 2 пар ионов в секунду; у-излучение горных
пород 1—8 пар ионов в секунду, радиоактивные за-
грязнения стенок 10~4 а-частиц в минуту на 1 см2
поверхности стенок.
Для измерения излучений малой мощности
(до 10“в р)час) используются счетные трубки. Для
измерений дозы быстрых нейтронов применяются
пропорциональные счетчики с тканеэквивалентными
стенками, наполненные этиленом. Дозы тепловых
нейтронов измеряют счетчиками, наполненными BF3.
Недостаток счетчиков — зависимость чувствитель-
ности от спектра излучения. В сцинтилляционных
методах Д. используется способность нек-рых веществ
светиться (люминесцировать) под действием излуче-
ний; возникающие при этом вспышки регистрируются
с помощью фотоэлектронных умножителей. В случае
Р- и у-лучей используются органич. тканеэквивалент-
ные Сцинтилляторы: стильбен, антрацен, пластмассы.
В Д. быстрых нейтронов возможно применять в каче-
стве сцинтиллятора смесь ZnS с парафином или пласт-
массой. Фотографии, методы Д. основаны на измере-
нии почернения фотопленок под действием излуче-
ния. Преимущества фотографии. Д.: возможность
накопления дозы, широкие пределы чувствительноств
(от 0,1 до 105 р), удобство для относительных измере-
ний. Недостатки — сложность обработки, зависи-
мость чувствительности от энергии частиц или кван-
тов и от процесса обработки пленок. Химич, методы
Д. основаны на измерении выхода химии, реакций,
происходящих под действием излучений. Их преиму-
щества: возможность измерений очень высоких доз
и при весьма высоких мощностях дозы (до 10’ р)сек)
(см. Дозиметрия химическая). Калориметрии, методы
Д. служат для определения абс. значения мощности
потока рентгеновских, |3- или у-лучей. Они основаны
на измерении количества теплоты, выделяемой при
поглощении излучения в калориметрах.
Важный, раздел Д. — дозиметрии, контроль без-
опасности работы с излучениями и радиоактивными
веществами. Работа с последними часто приводит
к образованию аэрозолей. Определение их концентра-
ции производится путем осаждения из определенного
объема воздуха и последующего измерения активности
осадка; возможно прокачивание воздуха через фильтр
или электростатич. осаждение. Расчетные методы Д.
применяются для определения необходимой защиты
от излучения реакторов, ускорителей, препаратов.
В современной дозиметрии, аппаратуре широко ис-
пользуются различные радиотехнич. устройства.
Лит.: Аглинцев К. К., Дозиметрия ионизирующих
излучений, 2 изд., М., 1957; Радиационная дозиметрия, [Под
ред. Дж. Хайна и Г. Браунелла], пер. с англ., М., 1958;
Горшков Г. В., Гамма-излучение радиоактивных тел
и элементы расчета защиты от излучения, М.—Л., 1959;
Гусев Н. Г., Справочник по радиоактивным Излучениям
и защите, М., 1956; Калугин К. С. [и др.], Практическое
руководство по дозиметрии, М 1959. Н. К. Аглинцев.
ДОЗИМЕТРИЯ ХИМИЧЁСКДЯ — один из мето-
дов измерения энергии ионизирующего излучения,
поглощенной единицей массы облучаемой среды.
Основана на определении химич. изменений, происхо-
дящих в результате действия излучения. Если из-
вестны радиационно-химич. выход G, т. е. число
молекул или ионов данного продукта, образующихся
при поглощении веществом 100 эв энергии излучения,
концентрация этого продукта с и плотность вещества,
подвергавшегося облучению, d, то величина дозы
ионизирующего излучения определяется из соотно-
шения: D = kc)Gd, где к — коэфф., величина к-рого
зависит от выбранной системы единиц. Д. х. приме-
20 к. X. Э. т. 1.
1203
ДОЗИМЕТРИЯ ХИМИЧЕСКАЯ — ДРЕВЕСИНА
1204
няется для определения доз различных видов ионизи-
рующих излучений (рентгеновские и у-лучи, быстрые
электроны, протоны и ядра отдачи и др.) и доз сме-
шанного излучения (см. Доза ионизирующего излу-
чения). Д. х. позволяет определять дозы от внешнего
и внутреннего источников в объемах любой формы.
Измерение дозы при помощи Д. х. может произво-
диться силами и средствами обычной химич. лабора-
тории. По своей чувствительности Д. х. уступает
другим методам дозиметрии, в частности ионизацион-
ным, и применяется гл. обр. для измерения доз в пре-
делах от 10 до 108 рад. К системам, применяемым
в Д. х., предъявляются след, требования: 1) незави-
симость величины радиационно-химич. выхода в за-
данном интервале доз при действии данного вида
излучения от изменений концентрации исходного
продукта, темп-ры и мощности дозы; 2) устойчивость
к действию света и кислорода воздуха; 3) легкость
определения химич. изменений, происходящих под
действием ионизирующих излучений.
В качестве дозиметрич. систем в Д. х. применяются:
водные р-ры различных веществ, гели, стекла спе-
циального состава, пластмассы, кристаллы щелоч-
ных галогенидов, галогенопроизводные углеводородов
и нек-рые др. вещества. Наибольшее распростране-
ние получила ферро сульфатная дозиметрич. система
(дозиметр Фрике), пригодная для измерения доз
от 103 до 4 • 104 рад. Она представляет собой насы-
щенный воздухом раствор 0,001—0,01 М FeSO4 или
соли Мора и 0,001 М NaCl в 0,8 н. растворе H2SO4.
Под действием излучений Fe2+ в этом растворе окис-
ляется до Fe3T. Кон-
центрацию образующе-
гося Fe3+ удобнее все-
го определять спектро-
фотометрически. Моляр-
ный коэффициент экс-
тинции (см. Поглощение
света) Fe3+ в 0,8 н.
р-ре H2SO4 при 20° и
3040 А равен 2095 ± 20.
При действии фотонов
и быстрых электронов
с энергиями от 0,03
до 20 Мэв G=15,5. Ве-
личина G практически
не зависит (в пределах 3—5%) от изменений кон-
центрации Fe2+ в пределах от 10~4 до 10-3 М, концен-
трации кислоты — от 0,1 до 5 М, мощности дозы —
от 10 до 1010 радДек и темп-ры — от 2 до 60°.
При действии [3-излучения трития величина G сни-
жается до 12,9. На рис. показана зависимость вы-
хода окисления Fe2+ от энергии протонов Е.
Для определения доз до 10е рад применяются разб.
р-ры Се (SO4)2 в 0,8 н. р-ре H2SO4. Под действием
излучений Се4+ восстанавливается до Се3+. При
действии у-лучей и быстрых электронов G = 2,32.
Величина G практически не зависит от изменений
концентрации Се4+ от 10“5 до 3,2 • 10~2М и мощности
дозы в широких пределах. Для определения доз,
начиная с 10 рад, используются насыщенные (при-
мерно 0,3%) водные р-ры трихлорэтилена в смеси
с индикатором на НС1, выделяющийся при облучении,
и водные р-ры солей бензойной к-ты. Последние под
действием излучений разлагаются с образованием
продуктов, люминесцирующих в УФ-свете. В Д. х.
нашли широкое применение метафосфатные стекла,
содержащие небольшие количества серебра. Под
действием излучений они приобретают желтую ок-
раску, интенсивность к-рой в интервале от 6 • 104
до 7 • 106 рад пропорциональна полученной дозе.
Серебро, содержащееся в метафосфатных стеклах,
под действием излучений переходит из ионной в ато-
Зависимость выхода окисления
Fe!+ от энергии протонов Е.
ОН
N(CH2CH2Cl)2
марную форму. Атомы серебра, возоужденные
УФ-светом, являются центрами люминесценции в види-
мой части спектра. Благодаря этому становится воз-
можным определять дозы, начиная с 10 рад. Кри-
сталлы щелочных галогенидов, содержащие гидриды
соответствующих металлов и галогенопроизводные
углеводородов, применяются в химич. дозиметрах,
предназначенных для определения степени радиацион-
ного поражения. Для определения пространствен-
ного распределения поглощенной энергии излучения
предлагались гели агар-агара и желатины, содержа-
щие различные красители, а для определения больших
доз (до 10s рад) — пленки из пластмассы.
Лит.: Кабакчи А. М. и Грамолин В. А.,
Усп. хим., 1958, 27, вып. 4, с. 459. А. М. Кабакчи.
ДОННАНА РАВНОВЕСИЕ — см. Мембранное рав-
новесие.
ДОНОРНО-АКЦЕПТОРНАЯ СВЯЗЬ — см. Коор-
динационная связь.
ДОПАН [4-метил-5-ди- (3-хлорэтил) -аминоурацил]
CBH13N3O2C12, мол. в. 266,14 — бесцветные кристал-
лы горького вкуса; т. пл.
180—484°; мало растворим
в спирте и ацетоне, практи-
чески нерастворим в воде,
бензоле и эфире. При упа-
ривании на водяной бане
р-ра0,01 г Д. в1 мл конц. HNO3 на дне чашки остается
желтое или желто-оранжевое пятно, к-рое при дей-
ствии газообразного NH3 окрашивается в оранжево-
красный цвет. При растворении 0,01 г Д. в 1,5 мл
спирта и прибавлении к 10 каплям раствора на часо-
вом стекле 2 капель 10%-ной H2SO4 и 1 капли реак-
тива Драггендорфа K[BiJ]4 выпадает оран-
жевый кристаллич. осадок. Для колич. определения
к спиртовому р-ру Д. прибавляют металлич. натрий,
упаривают раствор, разбавляют его водой, подкис-
ляют разб. HNO3, прибавляют 0,1 н. р-р AgNO3
и титруют 0,1 н. NH4SCN в присутствии железоам-
монийных квасцов. Д. получают взаимодействием
4-метил-5-аминоурацила с окисью этилена и после-
дующей обработкой образующегося при этом 4-ме-
тил- 5-ди- (р-оксиэтил)-аминоурацила тионилхлоридом.
Препарат применяют для лечения хронич. миелоид-
ной лейкемии и лимфогранулематоза.
Лит. см. при ст. Апрессин. А. И. Травин.
ДРЕВЕСИНА — ткань древесных и травянистых
растений, состоящая из • клеток с одревесневшими
оболочками. Химич, состав и свойства Д. в основном
определяются составом и стро-
ением тех отмерших и лишен-
ных протоплазмы и ядра кле-
ток, из к-рых Д. состоит на
90—95%. Основная масса ли-
гнина расположена в межклет-
ном веществе срединной пла-
стинки айв первичной стенке
Ъ (см. рис.).
Схема поперечного сечения древе-
сного волокна: А — общий вид
клетки; В — схематический разрез;
а — срединная пластинка; 5 — пер-
вичная стенка; с — внешняя часть
вторичной стенки; d — вторичные
спои; е — третичный спой.
Главная масса многочисленных концентрич. вто-
ричных слоев d построена гл. обр. из целлюлозы', по
микрохимич. реакции с флороглюцином (красное
окрашивание) в них также обнаруживают присутст-
вие лигнина. В клеточных стенках Д. различают еще
внешнюю часть вторичной стенки с и внутренний
незначительный по толщине и нелигнифицированный
третичный слой. В состав древесных тканей входя!
1205
ДРЕВЕСИНА — ДРЕВЕСИНЫ ЭНЕРГОХИМИЧЕСКАЯ ПЕРЕРАБОТКА
1206
также легко экстрагируемые разными растворите-
лями вещества — смолы и дубители. Смолы содер-
жатся в смоляных ходах, а дубильные вещества яв-
ляются внутриклеточными включениями.
Содержащаяся в Д. вода частью заполняет полости
клеток (свободная влага), частью пропитывает кле-
точные стенки (гигроскопическая влага). Максималь-
ное содержание последней для Д. разных пород со-
ставляет 25—30% («точка насыщения волокон»).
Содержание воды оказывает различное влияние на
объемный вес, усадку, механич. прочность, тепло-
творность и др. свойства Д. Максимум набухания Д.
достигается в точке насыщения волокон. Свеже-
срубленная Д. обычно содержит 60—100% воды
(по отношению к абсолютно сухому весу, принимае-
мому за 100%). Абсолютно сухая Д. разных пород
весьма сходна по элементарному составу и содержит:
49,4—50,2% углерода, 6,1—6,9% водорода, 43,6—
45,2% кислорода и обычно 0,9—1,3% азота. Сред-
ний зольный остаток 0,3—1%, в том числе 10—25%
растворимых в воде поташа и небольшого количества
соды, и 75—90% нерастворимых — СаСО3, угле-
кислых, кремнекислых и фосфорнокислых солей
магния и железа. Главнейшей состав'ной частью
Д. является целлюлоза (СвН10О5)п; обычное содержа-
ние ее 40—50% (Д. дуба 32—37%, Д. сосны 43—52%,
в даурской лиственнице — самой распространенной
породе Восточной Сибири — 30—45%).
Способность Д. подвергаться гидролизу (осахарива-
нию) при действии 64—80 %-ной серной и 41—42%-ной
соляной к-т (при обычной темп-ре) или при нагре-
вании с 0,5—1%-ной H2SO4 при 170—180° зависит
от содержания целлюлозы и других полисахаридов
в ее составе (см. Гидролиз растительных материалов).
Последние имеют, как и целлюлоза, цепное строение
и являются полимерными ангидридами гексоз и
пентоз. Объединяемые общим названием гемицеллю-
лозы, они классифицируются как гексозаны (маннан,
галактан) и пентозаны (ксилан, арабан). Наши обыч-
ные хвойные Д. содержат больше гексозанов (8—14%),
нежели лиственные (0,8—6,0%). Последние, наобо-
рот, содержат больше пентозанов (18—29%); чем хвой-
ные (6—12%, у можжевельника до 15%). Суммарное
приближенное содержание гемицеллюлоз в Д. раз-
личных пород (в%): сосна обыкновенная 19—20, ель
обыкновенная 20—21, пихта сибирская 22—23, бе-
реза 28, береза тяньшаньская 41, тополь 18, дуб зим-
ний до 29, осина 23.
При изучении строения гемицеллюлоз интересными
оказались открытые в главной цепи ксилана осины
ответвления, состоящие из единиц 4-метокси-П-глю-
куроновой к-ты
Ксилан бородавчатой березы состоит примерно из
22 ксилопиранозпых единиц со связями 1—4', ксилан
бука — из линейной цепи приблизительно 70 . (3-D-
ксйлопиранозных единиц, связанных в положении
1—4, из к-рых примерно каждая десятая имеет в от-
ветвлении 4-метокси-Ь-глюкуроновую к-ту. Содер-
жание уроновых к-т (глюкуроновой и галактуроно-
вой) составляет в хвойных породах ок. 3%, в лист-
венных ок. 5% от веса Д. Суммарное содержание
полисахаридов (целлюлозы % гемицеллюлоз) в Д.,
например, хвойных пород составляет около 65%
(см. таблицу), из них легко гидролизуемых до 20%.
Данные анализов некоторых образцов абсолютно сухой
древесины (в %)
Порода древесины Целлюло- за * Лигнин (72%-ная H2SO4) t 05 СО ф К К к со ф X - <и 2 X Зола Экстракт
вфир- ный вод- ный
Сосна 51,9 28,2 11.2 9.3 0.2 1.6 0,6
Ель Лиственница сибир- 58,3 29,0 10,1 9,8 0,2 1.1 1,8
ская Лиственница даур- 42,8 29,5 9,3 — 1,0 1.8 5,1
ская 35.2 27,4 7.4 8,7 0.5 1.7 17.7
Пихта сибирская . . 48,0 29,9 5.3 17.8 0,7 0.9 1.4
Кедр сибирский . . 50,0 30,1 8.6 11,8 0,1 2,4 1.5
Дуб 35.7 21.5 20,6 0,1 0,2 0,6 10,9
Осина 52,4 20.3 22,6 0,5 0,2 1.6 2.2
Бук 42,2 20,8 29.3 7.6 0.5 0,5 0,6
Клен 41,5 23,1 25,6 7,7 0,3 0,5 0,5
* Чистая по азотнокислотному методу.
Примечание. В зависимости от условий и места про-
израстания содержание может отклоняться от приведенных
цифр.
Лигнин в хвойных породах является в основном
смесью продуктов конденсации производных метил-
пирокатехина, в лиственных — производных диметил-
пирогаллола. Исследования указывают на близость
строения основного вещества лигнина хвойных к кето-
форме Р-оксикониферилового спирта:
НО
СН2СОСН2ОН
ОСН3
Содержание лигнина в Д. наиболее распространенных
хвойных пород составляет 26,5—30%; в лиственных —
17—25%. Лигнин, по-видимому, химически связан
с углеводами Д. по типу полуацетальных связей.
При действии диоксана, фенолов, ледяной уксусной
к-ты (при 100—115°) и спиртов в присутствии малых
количеств кислых катализаторов лигнин извлекает-
ся и переходит в раствор. В технике целлюлозного
произ-ва делигнификация Д. производится б. ч.
варкой щепы с сернистой к-той и бисульфитом каль-
ция или щелочной варкой.
Основные отрасли пром-сти, занимающиеся химич.
переработкой древесины: произ-во целлюлозы древес-
ной, гидролиз растительных материалов, сухая
перегонка Д., древесины энергохимическая перера-
ботка, произ-во дубящих веществ, канифоли, скипи-
дара и др. Из Д. получают также древесные пластики
и древесные плиты; Д. подвергают консервированию
и огнезащитной обработке (см. также Лесохимия).
Лит.: Никитин Н. И., Химия древесины и целлю-
лозы, М.—Л., 1951; Ванин С. И., Древесиноведение,
3 изд., М.—Л., 1949; Wood chemistry, 2 ed., ed. by L. E. Wise
and E. C. Jahn, v. 1—2, N. Y., 1952; H a g g 1 u n d E.,
Chemistry of wood, N. Y., 1951. H. И. Никитин.
ДРЕВЕСИНА ПЛАСТИФИЦИРОВАННАЯ — см.
Древесные пластики.
ДРЕВЕСИНЫ ЭНЕРГОХИМИЧЕСКАЯ ПЕРЕ-
РАБОТКА — комплексный пирогенный процесс пере-
работки древесины, дающий наряду с газом химич.
продукты (смолы, кислоты и т. д.); осуществляется
газификацией в генераторах прямого процесса (см.
Газификация твердых топлив) или в специальных
топках (скоростных топках-генераторах ЦКТИ си-
стемы В. В. Померанцева). При газификации сравни-
тельно крупной и влажной щепы (влажность 42—45%)
преим. хвойных пород (75% хвойной и 25% листвен-
1207
ДРЕВЕСНО-ПЛАСТИЧЕСКИЕ МАССЫ — ДРЕВЕСНЫЕ ПЛИТЫ
1208
ной древесины) из 1 т сухой массы получаются 1300—
1500 нм3 газа с теплотворной способностью
1550 кал/нм3, 75 кг осадочной смолы, 30 кг раствори-
мой смолы и 30 кг технич. уксуснокальциевой соли
(60%-ной). При газификации лиственной древесины
в виде мелкой и подсушенной щепы (осина влажностью
25%) из 1 т сухой массы получают: 1300 и.«3 газа
(QHH3m = 1500 кал/нм3), 90 кг осадочной смолы,
100 кг растворимой смолы и 35 кг уксуснокальци-
евой соли. При сжигании хвойной щепы в скоростных
топках-генераторах ЦКТИ из 1 т сухой массы полу-
чают: 4000 кг пара, 55 кг осадочной смолы, 45 кг
растворимой смолы и 22 кг технич. уксуснокальцие-
вой соли (60%-ной).
При газификации древесной щепы весь газ, обра-
зующийся в зоне газификации кокса, вместе с про-
дуктами пирогенного распада и влагой выходит из
генератора в виде парогазовой смеси. При сжигании
древесной щепы в скоростных топках часть газа из
активной зоны, где сгорает кокс, подается в швель-
шахту и вместе с продуктами пирогенного распада
и остатком влаги выходит из топки в виде парогазо-
вой смеси. Подсушка топлива производится в верх-
ней части шахты дымовыми газами котельного агре-
гата. Обработка парогазовых смесей в обоих случаях
одинакова: осаждение смоляного тумана в электро-
фильтрах или центробежных смолоотделителях, улав-
ливание уксусной к-ты 10—14%-ным водным р-ром
уксуснокальциевой соли в скрубберах или прямая
конденсация паровой фазы с получением пироли-
зата — р-ра продуктов термич. распада (кислоты,
спирты, растворимая смола и т. д.). В состав осадочной
смолы входят к-ты жирного ряда, фенолы и нейтраль-
ные вещества. Наибольший интерес представляет
фенольная часть смоляных масел осадочной смолы;
они состоят из 15% одноатомных фенолов, ок. 30%
двух- и трехатомных фенолов и 40% неполных мети-
ловых эфиров этих многоатомных фенолов. В состав
осадочной смолы входят: 25—30% 1,6-ангидро-1,5-
глюкопиранозы (левоглюкозана), 15% новолаков (про-
изводные пирокатехина и других фенолов), 20%
лактонов оксикислот (с преобладанием лактонов),
5—7% пирокатехина и его производных, 7% этилен-
гликоля, 5% гликолевого альдегида и др. Смолы,
получаемые указанными выше способами, являются
ценным сырьем для получения фенольных и других
продуктов: термореактивных клеев для древесностру-
жечных плит, разнообразных крепителей для литей-
ного произ-ва, гербицидов, понизителей вязкости,
ускоряющих бурение нефтяных скважин, неиногенных
поверхностно-активных веществ (моющих средств),
обычных и специализированных флотореагентов
и т. д. Энергохимич. переработка древесины — один из
важнейших путей использования разнообразных дре-
весных отходов.
Лит.: К о р о т о в С. Я. [и др.1, Энергохимическое ис-
пользование древесины, Л., 1958; Померанцев В. В.
|и др.], Опыт энергохимического использования древесных
отходов, [М., 1956] (Комитет по участию СССР в междунар.
энергетич. объединениях. Доклад на 5-й мировой энерге-
тпч. конференции 1956 г.); Лямин В. А., Тр. ин-та лесохоз.
проблем. АН Латв. ССР, 1955, вып. 8, с. 167; Т и щ е н к о Д. В.,
там же, с. 149. А. А. Лиееровский.
ДРЕВЕСНО-ПЛАСТИЧЕСКИЕ МАССЫ—см. Дре-
весные пластики.
ДРЕВЕСНЫЕ ПЛАСТИКИ — пластифицирован-
ные древесные материалы с улучшенными физико-
механич. свойствами, получаемые комбинированной
механич., термич. и химич. обработкой древесного
сырья. Д. п. подразделяются на три группы: древе-
сина прессованная, древеснослоистые пластики, дре-
весно-пластич. массы..
Древесина прессованная — материал, полу-
чаемый обработкой давлением при повышенной темп-ре (пла-
стификацией), а иногда в сочетании с пропиткой цельных или
склеиваемых в процессе пластификации заготовок древесины.
Вначале древесину с влажностью 8—15% уплотняют поперек
волокон для повышения ее механич. прочности. В целях
увеличения пластичности древесины и во избежание вредных
деформаций производится постепенное повышение давления до
110—150 кг/смг по мере прогрева до 90—100°. Затем для ста-
билизации свойств уплотненной древесины ее выдерживают
при повышенной темп-ре (до 120°)-. Прессование древесины
можно сочетать с продольным и торцовым гнутьем (гнутопрес-
сованная Д. п.). Гнутью подвергают пропаренную древесину
(при 80—90°), затем уплотняют и высушивают до равновесной
влажности. По плотности различают Д. п.: малого веса 0,90,
легкую 1,1, среднего веса ок. 1,2 и тяжелую 1,3—1,4 г/см3.
Физико-механич. свойства Д. п. в 1,5—2,5 раза выше, чему
исходной древесины. Для повышения влагостойкости и ста-
билнности формы Д. п. заготовки древесины предварительно
пропитывают синтетич. смолами и др. Получить влагостой-
кую Д. п. можно и без пропитки, путем усиления теп-
ловой обработки во второй стадии пластификации (при вы-
держивании); при этом в древесине образуются смолообразные
продукты изменения лигнина и гемицеллюлов. Д. п. широко
используется в деталях машин, несущих ударные нагрузки, и
как антифрикционный материал, в особенности гнутопрессо-
ванная в текстильном машиностроении и других произ-вах.
Древеснослоистые пластики (лигнофоль,
монолит и др.) получают из фанерного шпона в виде плит или
листов плотностью 0,9—1,43 г/см3. Листы шпона пропитывают
(иногда промазывают) р-рами термореактивных синтетич. смол,
высушивают, собирают в пакеты и подвергают горячему
прессованию в многоэтажных гидравлич. прессах в условиях,
обеспечивающих заданное уплотнение и полимеризацию смолы
со связыванием слоев в монолит (давление 100—175 кг/см2,
темп-ра 120—150°, выдержка в прессе 0,5—4 мин на 1 мм
толщины, охлаждение часто под давлением). При параллель-
ном расположении волокон шпона в пакете получают материал
для изготовления подшипников и втулок, в частности рабо-
тающих в водной и абразивной средах. При расположении
волокон в соседних листах шпона под углом получают мате-
риал для изготовления бесшумных шестерен. Добавки гра-
фита, масла, фольги улучшают антифрикционные свойства
пластиков, а увеличение количества связующего — их диэлек-
трич. свойства. Большой прочностью и эластичностью обладает
арктилит, для получения к-рого сочетают шпон с про-
резиненной тканью и медной сеткой. Для различных видов
пластиков предел прочности при сжатии вдоль волокон со-
ставляет 1000—2000, при растяжении 1100—3500, при
изгибе 1400—3500 кг/см1, при скалывании по клеевому шву
140—250, по древесине 150—350 кг/см2; уд. ударная вязкость
равна 25—150 кг-см/см2.
Древесно-пластические массы — цельио-
прессованные профильные изделия и плиточные материалы,
изготовляемые горячим прессованием в прессформах измель-
ченной древесины (опилок, стружек, волокон, обрезков шпона),
пропитанной р-рами синтетич. смол и высушенной. В нек-рых
случаях древесину предварительно подвергают частичному
гидролизу кислотой или пропаркой под давлением или же
обработке щелочью. Цельно прессов энные древесно-пластич.
изделия изготовляются в виде втулок и вкладышей (взамен
цветных металлов) деталей электрооборудования и др. Повы-
шенную прочность имеют цельнопрессованные изделия из
обрезков шпона и, особенно, полученные продольным прессо-
ванием навитого на стержень пропитанного шпона. Нек-рые
строительные изделия (паркетные плитки и др.) и детали машин
изготовляют прессованием измельченной древесины при высо-
ких темп-рах без связующих веществ. В этом случае глубокая
термообработка повышает пластичность древесины, а роль
связующих веществ играют смолообразные продукты изме-
нения нек-рых компонентов древесины (гемицеллюлоз и лиг-
нина). К числу плиточных древесных материалов относятся
древесные плиты различных типов.
Лит.: Пластические массы в машиностроении. Сб. тр.
Уральского совещания по пластмассам, М., 1955; Шей-
дин И. А., С м и р н о в А. В., Демидова Л. А.,
Технология древесных пластиков, М.—Л., 1956; X у х р я н-
с к и й П. Н., Прессование и гнутье древесины М.—Л.,
1956; Нысенко Н. Т. и Генель С. В., Пластифи-
кация цельной древесины, М.—Л., 1958. Н. Т. Нысенко.
ДРЕВЕСНЫЕ ПЛИТЫ — древесно-стружечные и
древесно-волокнистые плиты, получаемые из отходов
древесины, соломы, хлопка и др. растительных мате-
риалов.
Древесно-стружечные плиты делятся на
необлицованные с объемным весом у — 650—850 кг/см3,
облицованные у = 650—850 кг/см3. Древесноволок-
нистые плиты делятся на сверхтвердые у не менее
950 кг/м3, изоляционно-отделочные у = 250—350 кг/м3, изо-
ляционные у до 250 кг/м3.
При изготовлении древесно-стружечных плит частицы
древесины с влажностью 4—5% смешивают с 50—60%-ным
водным р-ром мочевино-формальдегидной, феноло-формальде-
гидной или иной синтетич. смолы (расход сухой смолы 6—12%
от веса сухой стружки), формуют «ковер» требуемой толщины
и подвергают периодическому или непрерывному горячему
прессованию при 120—160°. При периодич. прессовании «ко-
вер» предварительно уплотняют в холодном прессе. Древесно-
стружечные плиты изготовляют одно- и трехслойиыми, в виде
1209
ДРОБЛЕНИЕ — ДУАЛИСТИЧЕСКАЯ СИСТЕМА
1210
листового и профильного материала или в виде готовых щито-
вых деталей. Наружные слои трехслойных плит изготовляют
из более тонной стружки с применением большего количества
связующих, а середину — из грубо измельченной стружки
с меньшим количеством связующих. Плиты могут быть обли-
цованы шпоном, бумагой, фанерой, листовыми пластинами.
Толщина плит 5—100 мм, предел прочности при статич. из-
гибе 7—500 кг/см2, коэфф. теплопроводности 0,045—
0,075 ккал/м-град-час.
При изготовлении древесно-волокнистых плит древесину
размалывают на волокно и затем формуют из волокна на отли-
вочных машинах в л ажно^ полотно. Пр и произ-ве изоляционных
плит отрезки влажного полотна высушивают в роликовой
сушилке, при произ-ве полутвердых и твердых плит — прес-
суют в многоэтажном гидравлич. прессе при уд. давлении
35—50 кг/см2 и темп-ре 160—200°. Для придания плитам
водоустойчивости в массу, подаваемую на отливочную машину,
вводят парафиновую или канифольную эмульсию. Толщина
плит 3—25 мм, предел прочности на статич, изгиб от 100 до
500 кг/см2 для плит, изготовляемых в прессах, и от 2 до
20 кг/см2 для плит, изготовляемых в сушилках; коэфф, тепло-
проводности сверхпористых плит 0,04—0,06 и полутвердых
0,075—0,1 ккал/м • град-час.
Д. п. применяют для устройства перегородок, облицовки
стен, потолков, настила черного и чистого полов и др.
Лит.: Отливанчик А. И., Производство древесно-
стружечных плит на лесопильных и деревообрабатывающих
предприятиях, М.—Л., 1959; Кауфман Б. Н. [и др.],
Производство и применение древесно-стружечных плит за
рубежом,М., 1958; Солечник Н. Я., Производство древесно-
волокнистых плит, М.—Л., 1959; Новосельский Н. Л.,
Кунин В. М. и Дроздов И., Строительные плиты
из органического волокна, М., 1956; Шварцман Г. М.,
Производство древесно-стружечных плит, М., 1959; Справоч-
ник по стружечным плитам, пер. с финск., М.—Л., I960;
Sciieibert W., Spanplatten, Lpz., 1958.
В. П. Павлов.
ДРОБЛЕНИЕ — см. Измельчение.
ДРОБНАЯ КРИСТАЛЛИЗАЦИЯ — способ раз-
деления и очистки веществ, основанный на преиму-
щественном переходе одного из компонентов в твер-
дую фазу при кристаллизации из раствора или рас-
плава. Д. к. может применяться как в том случае,
когда разделяемые вещества имеют близкую раство-
римость и присутствуют в соизмеримых количествах
(при этом каждый компонент образует самостоятель-
ную твердую фазу), так и в том случае, когда один
из компонентов, присутствуя в микроколичествах,
не образует самостоятельной твердой фазы, а изоморф-
но соосаждается с макрокомпонентами. Д. к. — много-
стадийный процесс. На первой стадии исходный рас-
твор делят на две фракции: концентрат (твердая
фаза, обогащенная одним из компонентов) и хвосты
(раствор, обедненный этим компонентом). Для этого
производится частичная кристаллизация компонен-
тов раствора путем охлаждения, добавления веществ,
понижающих растворимость, или же изотермич. испа-
рением. На второй стадии Д. к. каждую из фракций,
полученных в результате первой стадии, делят вновь
на две фракции и т. д.; образуется кристаллизацион-
ный каскад.
Эффективность разделения компонентов в резуль-
тате одной стадии характеризуется относительным
обогащением твердой фазы а или же относительным
обеднением жидкой фазы р одним из компонентов.
Эффективность разделения в результате п стадий
дробной кристаллизации равна а". Число ступеней,
необходимых для полного разделения, определяется
величиной а. Если Д. к. происходит с образованием
каждым компонентом самостоятельной твердой фазы,
то величина а зависит от соотношения количеств
разделяемых компонентов в исходном растворе и раз-
личия растворимостей компонентов. Если же при
Д. к. один из компонентов соосаждается с другим,
то а зависит от состава раствора и условий кристалли-
зации. Учет факторов, влияющих на величину а,
и требование максимального изменения свободной
энергии системы на каждой ступени разделения
позволяют построить термодинамически наиболее
выгодный каскад. Однако часто оказывается необхо-
димым столь большое число фракций, что практич.
осуществление такого каскада оказывается невыгод-
ным и в таком случае построение каскада изменяется
по пути его приближения к простейшему типу. Д. к.—
трудоемкий и длительный процесс; применяется для
разделения химически близких веществ (соединений
Nb и Та, Ra и Ва, лантанидов и т. д.), а также для
получения чистых веществ (в тех случаях, когда
отсутствуют более совершенные методы).
Лит.: Несмеянов Ан. Н., Лапицкий А. В.
и Руденко Н. П., Получение радиоактивных изотопов,
М., 1954; Бреслер С. В., Радиоактивные элементы,
3 изд., М.,1957;Р яб чико в Д. И., ТерентьеваВ.А.,
Способы разделения редкоземельных элементов, Усп. хим.,
1947, 16, вып. 4. Н. Э. Хандамирова.
ДРОБНОЕ ОСАЖДЕНИЕ — способ разделения
смеси веществ, близких по химич. свойствам и раство-
римости. Д. о. состоит в последовательном переводе
компонентов смеси в осадок отдельными порциями
(фракциями). При добавлении осадителя к смеси
двух солей в растворе в первую очередь осаждается
компонент, образующий наименее растворимое соеди-
нение. Затем, когда большая часть его будет в осадке
и отношение концентраций ионов компонентов достиг-
нет значения, равного отношению произведений рас-
творимости образующихся соединений, в осадок начнет
переходить также и второй компонент, причем каждая
последующая фракция осадка будет богаче вторым
и беднее первым компонентом. Возможность полного
разделения смеси двух веществ зависит от соотно-
шений первоначальных концентраций их в растворе,
а также от значений произведений растворимости
соответствующих соединений. Недостатками Д. о.
являются большая трудоемкость, связанная с много-
кратным повторением операций охлаждения, раство-
рения, упаривания и т. д., и возможность загряз-
нения осадка за счет соосаждения. В связи с этим
Д. о. используется лишь в тех случаях, когда от-
сутствуют другие, более совершенные методы раз-
деления.
Лит.: Кольтгоф И. М. и Сендая Е. Б., Коли-
чественный анализ, пер. с англ., 3 изд. . М.—Л., 1948; Хло-
пни В. Г., Избр. труды, т. 1, М.—Л., 1957.
II. Э. Хандамирова.
ДУАЛИСТИЧЕСКАЯ СИСТЕМА — одна из ран-
них химич. теорий, в основе к-рой лежало предполо-
жение о двойственности (дуализме) состава химич.
соединений (высказанное в конце 18 в. А. А. Ла-
вуазье). Д. с., разработанная шведским химиком
Я. Берцелиусом в 1812—18, сводила причину химич.
сродства к действию электростатнч. сил между про-
стыми и сложными частицами. Согласно Д. с., ча-
стицы всех элементов принимались двояко заряжен-
ными противоположным по знаку заряда, но не рав-
ным количеством электричества. В силу этого частица
каждого элемента, соответственно преобладающему
в ней электричеству — униполярна. По убывающей
величине отрицательного заряда частиц все элементы
располагались в один ряд (электрохимия, ряд напря-
жений), причем самым электроотрицательным эле-
ментом считался кислород (фтор был тогда в свобод-
ном виде неизвестен). Степень химич. сродства счита-
лась пропорциональной интенсивности полярности;
т. е. количеству электричества на полюсах частицы.
По мнению Берцелиуса, этот фактор не постоянен
и обычно возрастает с повышением темп-ры; этим
он объясняет, что многие соединения образуются
лишь при нагревании. Связь между элементарными
частицами, за счет притяжения и нейтрализации их
противоположных полюсов, приводит к образованию
сложной частицы первого порядка, к-рая сохраняет
униполярность ввиду неравноценности зарядов ее
некомпенсированных полюсов. Поэтому частица пер-
вого порядка способна образовать дальнейшие соеди-
нения — второго порядка. Т. к. всегда погашаются
сначала наибольшие по величине заряды, электрич.
силы постепенно ослабляются, чем лимитируется
1211
ДУБЛЕНИЕ— ДУБЛЕНИЕ ФОТОГРАФИЧЕСКОЕ
1212
образование частиц высших порядков. Обмен электрич.
энергией, лежащий в основе любого химич. акта,
составляет причину также и сопровождающих его
явлений — выделения теплоты, света. Если сложные
вещества подвергнуть действию электрич. тока, то
их частицам возвращается первоначальная поляр-
ность и они выделяются в свободном виде. Этим
давалось рациональное объяснение явлениям электро-
лиза, изучение к-рых дало начальный толчок Берце-
лиусу к созданию Д. с. Неотделима от Д. с. также
номенклатура и метод обозначения химич. соедине-
ний и реакций.
Д. с. сохранилась до наших дней в таких, напр.,
терминах, как «углекислая известь», к-рая считалась
соединением электроотрицательной углекислоты СО2
с электроположительной известью СаО и изображалась
формулой СаО-СО2, или «сернокислая магнезия» —
MgO-SOa и т. д. Формулы многих минералов до сих
пор нередко пишут, как сумму соответствующих
окислов, напр. ортоклаз К2О • А12О3 • 6SiO2, совре-
менная формула K[AlSi3O8]2.
Начало крушения Д. с. связывают с открытием
реакции металепсии (Ж. Дюма, 1834), состоящей
в эквивалентном замещении электроположительного
водорода электроотрицательным хлором, что в корне
противоречило Д. с. Этот и многие другие факты,
гл. обр. из области органич. химии, сильно подорвали
доверие к Д. с. и она была оставлена. Лишь многие
десятки лет спустя, после раскрытия сложной струк-
туры атома, наука смогла вернуться к вопросу о при-
роде химич. сродства. В свете современных воззре-
ний Д. с. Берцелиуса предстает как гениальная до-
гадка, намного опередившая научный уровень своего
времени и в силу этого оказавшаяся не в состоянии
преодолеть возникшие перед ней трудности.
Лит.: Berzelius J. J., Bssai sur la thgorie des pro-
portions chlmlques et sur 1'lnfluence chlmique de I'dlectricitd,
P., 1819; Lehrbuch der Chemie, Bd 3, Abt. 1, Dresden, 1827;
ЛаденВург А., Лекции по истории развития химии,
пер. с [нем.], Одесса, 1917. Р. Р. Галле.
ДУБЛЕНИЕ — обработка кожи дубящими вещест-
вами, к-рые в процессе Д. распределяются в обраба-
тываемом материале и частично связываются с его
функциональными группами (—NH2,—СООН и др.).
Д. применяется также при обработке желатины для
фотографич. целей (см. Дубление фотографическое)
и казеина (см. Пластики белковые). При Д. между
структурными элементами белка и молекулами дуби-
теля образуются различные виды связи: водородные,
электровалентные и ковалентные. В результате Д.
волокна коллагена связываются («сшиваются») моле-
кулами дубителя в структуру с большим числом попе-
речных связей. При этом кожный покров животного
превращается в выдубленную кожу, для к-рой ха-
рактерна совокупность след, свойств: уменьшение
деформируемости обводненной дермы; сохранение
пористости кожи в процессе сушки; уменьшение
склеиваемости; повышение прочности при растяже-
нии; уменьшение влагоемкости при набухании в воде;
повышение термич. стойкости коллагена и его стойко-
сти к химич. и ферментативным воздействиям; умень-
шение общей упорядоченности структуры коллагена.
От Д. следует отличать т. н. псевдодуОление —
обработку голья в р-рах обезвоживающих солей (напр., суль-
фатов), в водоотнимающих жидкостях (напр., спирте или
ацетоне) или в слабых р-рах серной или соляной к-т и ней-
тральных солей, обычно NaCl (пикелевание). При этом
голье приобретает многие свойства, характерные для выду-
бленной кожи: уменьшение деформируемости в мокром со-
стоянии, сохранение пористости после высушивания и др.
В отличие от Д. эти свойства под действием воды исчезают.
Важнейшими видами Д. являются: красное, хромо-
вое, замшевое, формальдегидное и комбинированное.
Красное Д. производится водными р-рами раститель-
ных дубильных веществ (таннидов) и их заменителей. Д. ве-
дется в слабокислой или нейтральной средах при темп-ре
не выше 40°. Методами красного Д. вырабатывают кожаные
изделия всех видов. Хромовое Д. осуществляется вод-
ными р-рами основных солей трехвалентного хрома; этим
способом чаще всего получают кожи для верха обуви. Зам-
ше в о е Д. выполняется вминанием в набухший в воде полу-
фабрикат высоконепредельного жира (ворвани) с последующим
окислением. По окончании Д. несвязанный избытон жира
удаляется. Формальдегидное Д. ведется в слабо-
щелочной среде с помощью формалина; применяется при
произ-ве белой кожи. Комбинированное Д. — произ-
водится обработкой несколькими дубителями разного типа
(чаще всего комбинируется хромовое и красное Д.), что при-
водит к повышению носкости и термостойкости кожи крас-
ного Д.
Сущность процесса Д. заключается в следующем.
При хромовом Д. возникают связи между карбоксиль-
ными группами белка и комплексными солями трех-
валентного хрома (I); при Д. таннидами — между
ОН-группами • таннидов, пептидными группами,
остатками лизина и особенно аргинина в белке (II).
Особой прочностью отличаются пятичленные циклы
(III), возникающие при хромовом Д., и семичленные
циклы (IV) — при Д. таннидами. При Д. синтанами
получаются продукты, образованные за счет ионной
(V) и водородной связей.
О НО ОН-о
г /У I I
Б-4-С-О--Сг-ОН-Сг-О-С
NH * С1- (°Н2)з (ОН2)3
IN П Q I
Б
I
NH3 СГ
zNH3t..O--T ।
Б %.......н
I
ОН...о
Сг-о-£-
(ОН2)3
U!
н
-с=о о-с-
I II
NH-O-----с-
I I
/СООН
Б;
nh; "o3sr
V
Б '•остаток молекулы белка
Т “остаток молекулы таннида
R- многоядерный арил ’ 1
Д., как правило, проводят в цилиндрич. сосудах (бараба-
нах), вращающихся вокруг полой горизонтальной оси. При
вращении барабана происходит интенсивное перемешивание,
ускоряющее процесс Д. В нек-рых случаях Д. производят
в неподвижных чанах. В зависимости от назначения кожи
режим Д. сильно меняется. Переменными являются темп-ра
процесса, его продолжительность, реакция среды, порядок
введения растворов и др. При хромовом Д. чаще всего при-
меняют предварительное пикелевание. В зависимости от
назначения Д. кожи проводится обычно при темп-рах от
20—25° (начальная) до 40—45° (конечная). Известны одно-
ванный и двухванный процессы хромового Д. В первом случае
белок обрабатывают основными солями трехвалентного хрома;
во втором — голье сначала обрабатывают р-ром бихромата
калия (хромпика) в кислой среде, а затем р-ром тиосульфата
натрия (Na2S2O8).
Лит.: Михайлов А. Н., Химия дубящих веществ и
процессов дубления, М., 1953; Химия кожевенного и мехового
производства, под ред. Н. В. Чернова, М., 1957; Технология
кожи и меха, под ред. Н. В. Чернова, М., 1959; Справочник
кожевника, т. 1—3, М., 1952—54.
ДУБЛЕНИЕ ФОТОГРАФИЧЕСКОЕ — обработка
галогено-серебряных желатиновых слоев с целью
понижения их набухания в воде и водных р-рах,
повышения их механич. прочности и термостойкости,
а также сохраняемости и бактериальной устойчи-
вости. Д. ф. можно производить до или после нане-
сения эмульсионного слоя на подложку или в про-
цессе проявления и фиксирования. При концентра-
ции желатины выше 1—2% Д. ф. приводит, в основ-
ном, к образованию прочных межмолекулярных
мостиков между смежными цепями молекул желатины,
за счет поливалентных ионов металлов (Cr, А1) или
органич. групп (метиленовой и др.). Это приводит
к повышению вязкости желатины. В сильно разбав-
ленных р-рах образуются преим. внутримолекулярные
связи, в результате чего вязкость понижается и
Д. ф. не происходит. При Д. ф. возрастает хрупкость
слоя, что понижает прочность кинопленок; это устра-
няется введением пластификаторов.
1213
ДУБЯЩИЕ ВЕЩЕСТВА
1214
Фотографии, дубители разделяются на две основные
группы: оказывающие и практически не оказываю-
щие влияния на скорость проявления, независимо
от степени задубленности. К первой группе относятся
неорганич. дубители (напр., хромокалиевые квасцы,
уксуснокислый хром, алюмокалиевые квасцы и др.);
ко второй — различные органич. дубители (формалин,
глиоксаль, диацетил и др.). Процесс Д. ф. поддается
предварительному приближенному расчету, при кон-
кретно выбранных условиях («число дубления» жела-
тины, концентрация желатины в растворе, pH среды,
условия «последующего задубливания» — относитель-
ная влажность, темп-ра). При процессе последующего
задубливания (продолжающемся в воздушно-сухом
слое) относительная влажность определяет необра-
тимо-достигаемый предел задубливания, а темп-ра —
скорость его достижения.
Лит.: Михайлов А. Н., Химия дубящих веществ
и процессов дубления, М., 1953; М и а К., Теория фотогра-
фического процесса, пер. с англ., М.—Л., 1949; Анто-
нов С. М., Зеликман В.Л. иМархилевич К. И.,
Кинопленка и ее обработка, М., 1950; Зеликман В. Л.,
Л е в п С. М., Основы синтеза и полива фотографических
эмульсий, М., I960; Зеликман В. Л., Усп. научн. фо-
тогр., 1960, 7, с. 161. В. Л. Зеликман.
ДУБЯЩИЕ ВЕЩЕСТВА (дубители) — химические
соединения, водные р-ры к-рых применяют для дена-
турации белков, содержащихся в кожевенном сырье,
желатине или казеине (см. Дубление, Дубление фото-
графическое и Пластики белковые). Для Д. в. харак-
терна способность видоизменять коллоидное состояние
белков: вызывать затвердение, противодействовать
набуханию в воде, в нек-рых случаях осаждать их
из р-ров (напр., клей или желатину) и др. Дубящими
свойствами обладают вещества различной химич.
природы. Различают минеральные и органич. Д. в.
Важнейшими неорганич. Д. в. являются
основные соли трехвалентного хрома, чаще всего
сульфат хрома 1/OfPV I ’ пРиме‘
няются также ' алюминиево-калиевые квасцы
К2А12 (SO4)4 • 24Н2О и др. Минеральные Д. в. при-
меняют преим. при выделке кож верха обуви, а также
при выработке жестких кож и юфти — в смеси с
органич. дубителями (таннидами и их заменителями).
Органические Д. в. бывают растительного и
животного происхождения, а также синтетические.
Наиболее важную и обширную группу органич.
Д. в. составляют дубители растительного происхожде-
ния, к-рые находятся в коре, древесине и корнях раз-
личных растений. Для их выделения растительный
материал измельчают и экстрагируют горячей водой.
При этом в состав водной вытяжки входят Д. в. —
т а н н и д ы (Т) и недубящие вещества (сахар, про-
стейшие фенолы и органич. к-ты) — нетанниды
(НТ). Отношение содержания в водной вытяжке
таннидов к общему количеству содержащихся в ней
таннидов и нетаннидов, выраженное в процентах,
называется доброкачественностью. В таб-
лице приведено содержание таннидов и нетаннидов
в ряде дубильных материалов.
Дубильный материал
Древесина дуба..........
Древесина квебрахо . . . .
Кора ели................
» ивы.................
» лиственницы.........
1 » дуба ...............
» эвкалипта ..........
Корень бадана...........
» кермека............
» тарана ............
% НТ
2,5—3,5
2,4
8—14
8-12
6—10
6-8
2-18
12—17
8—16
7-24
Полученные растворы обычно упаривают для повы-
шения концентрации дубителя, а иногда для удобства
транспортировки и хранения высушивают. Различают
жидкие дубильные экстракты (45—50% воды), твердые
(17—20% воды) и порошкообразные (5—7% воды).
Танниды растворимы в воде, водный р-р имеет слабо-
кислую реакцию, сильно вяжущий вкус; с солями
трехвалентного железа дают черно-зеленое или сине-
черное окрашивание; р-ры таннида с р-рами жела-
тины образуют осадок или муть. Танниды являются
производными многоатомных фенолов. При нагрева-
нии таннидов до 180—200° они разлагаются с образо-
ванием пирогаллола или пирокатехина. Эта реакция
была использована для построения первой эмпирич.
классификации таннидов. К таннидам пирогаллового
ряда относят китайский таннин, танниды листьев
сумаха и др.; к таннидам пирокатехинового ряда —
танниды коры ивы, ели, лиственницы и др.; известны
танниды смешанного класса: танниды коры и древе-
сины дуба, коры ели, корней бадана и тарана, древе-
сины каштана и др.
Рациональная классификация растительных Д. в.,
основанная на характере связей между отдельными
частями в молекуле таннида, была опубликована
в 1911 Г. Г. Поварниным. По этому признаку все
танниды можно разбить на гидролизуемые (э с т е-
ротанниды) и конденсированные (к о т а н-
н ид ы). Нек-рые растительные Д. в., имеющие
свойства обоих типов, иногда выделяют в особую
группу — таннидов смешанного характера. Гидро-
лизуемые танниды содержат 50—53% углерода,
конденсированные — более 60%. Гидролизуемые тан-
ниды содержат сложноэфирную группировку; к их
числу относят галлилгексозы и эллаготанниды. В со-
став гидролизуемых таннидов обязательно входит
глюкоза или другая гексоза, которые образуют слож-
ный эфир с галловой к-той, депсидами (напр.,
НООС—СвН4—О—СО—С6Н4ОН) или эллаговой к-той.
Последняя образуется окислительной конденсацией
двух молекул галловой к-ты и обычно встречается
в виде лактона (I). Сложноэфирные и гликозидные
связи между компонентами молекул таннидов гидро-
лизуемого типа расщепляются не только при обра-
ботке кислотами, но и в присутствии ферментов —
эстераз и карбогидраз. Важнейшим дидепсидом,
обнаруженным в составе гидролизуемых таннидов,
является метадигалловая кислота (II).
При нагревании конденсированных таннидов с разб.
к-тами происходит не гидролиз, а их конденсация.
Образующиеся при этом вещества — ф л о б а.ф е-
н ы — выпадают из раствора в виде темно-красного
или бурого осадка. В молекулах конденсированных
таннидов особенно распространены структуры, род-
ственные дифенилпропану СвН6(СН2)3 С6Н6. В ре-
зультате окислительной конденсации подобного рода
соединений образуются последовательно оксидифе-
нилпропан (III), флаван (IV) и флавон (V):
1215
ДУБЯЩИЕ ВЕЩЕСТВА — ДУЛЬЦИТ
1216
Различные полиоксипроизводные флавана (т. н,
катехины) и флавона являются важнейшими струк-
турными элементами молекулы конденсированных
таннидов. При нагревании катехинов в воде, даже
без доступа кислорода', они теряют способность кри-
сталлизоваться и превращаются в неограниченно
растворимое аморфное коричневое соединение, яв-
ляющееся типичным таннидом. При более энергичном
воздействии на раствор, напр. при нагревании в при-
сутствии разб. минеральной к-ты, эти танниды выпа-
дают в осадок в виде флобафенов. Вещества типа
катехинов имеют мол. вес ок. 300; мол. вес таннидов
превышает 1000, а мол. вес флобафенов много выше.
Основной тип реакции таннидов с белком—образова-
ние прочных водородных связей между фенольными
оксигруппами и функциональными группами белка,
гл. обр. группами, содержащими азот.
При произ-ве белой кожи в качестве дубителя
в ограниченных количествах применяют формаль-
дегид, к-рый реагирует с аминогруппами коллагена,
связывая отдельные цепи структуры белка. Для
выделки замши применяют Д.в. животного
происхождения — ворвань (китовый жир).
Синтетические Д. в. (синтаны) в по-
следние годы широко применяются в произ-ве кожи
наряду с растительными. Синтаны являются соеди-
нениями ароматич. ряда, содержащими несколько
ядер в одной молекуле. Различают синтаны 2 типов:
вспомогательные и заменители таннидов. Вспомога-
тельные синтаны, к-рые вводятся в дубящую смесь
в количестве ок. 20% от содержания в них таннидов,
применяют для диспергирования осадков и коллоид-
ных частиц в р-рах растительных дубителей, а также
для ускорения диффузии таннидов в толщу кожевен-
ного полуфабриката. Вспомогательные синтаны из-
готовляют гл. обр. из углеводородного сырья (напр.,
из нафталина или сырого антрацена, реже для этой
цели используют фенол или крезолы). Исходный про-
дукт сульфируют, конденсируют с формалином и
иногда частично нейтрализуют:
При получении вспомогательных синтанов из продук-
тов, содержащих три конденсированных ядра, напр.
антрацена, обработка формалином не является обя-
зательной. В отличие от одноядерных ароматич. суль-
фокислот, многоядерные прочно фиксируются бел-
ками кожи и способствуют их гидролизу. Этот недо-
статок вспомогательных синтанов обычно компенси-
руется дополнительным дублением кожи основными
солями хрома. К вспомогательным синтанам относят
также лигносульфоновую к-ту (сульфит-целлюлоз-
ный экстракт), содержащую, ароматич. ядра и сульфи-
рованную боковую цепь. В нек-рых случаях лигно-
еульфоновая к-та применяется при дублении кожи
в смеси с растительными таннидами; она фиксируется
белком также через сульфогруппы.
Синтаны — заменители таннидов вырабатываются
из кристаллич. фенольного сырья (фенола, крезолов)
или из технич. фенольных смесей, выделенных из
надсмольных и подсмольных вод пиролиза углей,
сланцев и др.; их обычно вводят в дубящую смесь
(до 50% от содержания растительных дубителей).
В молекулах синтанов-заменителей сульфировано
не более 50% фенильных ядер; при фиксации белками
образуются прочные водородные связи между феноль-
ными группами и функциональными группами белка
(аналогично фиксации таннидов). Для пройз-ва син-
танов-заменителей фенольное сырье конденсируют
с образованием метиленовых или сульфоновых мости-
ков (VI); для сообщения полученному продукту
растворимости его сульфируют или подвергают олиго-
сульфированию (VII).
В нек-рых случаях сульфоны дополнительно обра-
батывают формалином
или (VI) конденсируют с формальдегидом и ароматич.
сульфокислотой (напр., VIII); при этом получают
IX или X. Конечные продукты обладают дубящими
свойствами.
В некоторых случаях фенольным полупродук-
там сообщается растворимость диспергированием
посредством вспомогательных синтанов или лигно-
сульфоновой кислоты без дополнительной конден-
сации.
Наилучшими дубящими свойствами обладают син-
таны-заменители, вообще не содержащие сульфо-
групп; их получают конденсацией резорцина и дру-
гих многоядерных фенолов с формальдегидом.
Лит,: Михайлов А. Н., Химия дубящих веществ п
процессов дубления, М., 1953; Арбузов Г. А., Ш и п-
к о в П. Ф., Товароведение растительных дубильных мате-
риалов, М., 1932; Справочник кожевника, т. 2, М., 1952; The
chemistry of vegetable tannins. A symposium, Croydon, 1956;
Кубе лк а В. и Бинко И., Синтетические дубители,
пер. с чешек., М., 1959.
ДУЛЬЦИТ (галактит) СвН14Ов, мол. в. 182,18 —
шестиатомный спирт, продукт восстановления галак-
1217
ДУРОЛ — ДУШИСТЫЕ ВЕЩЕСТВА
1218
тозы; кристаллы; т. пл, 188,5; плохо растворим
в воде и спирте, в эфире нерастворим; оптически
неактивен. В природе Д. находится в раз-
। личных растениях (жасмине, бересклете,
Н—с—он морских водорослях) и в дрожжах. Осо-
НО с н бен но много Д. в высохшем соке нек-рых
-1 тропич. растений (манна Gymnosporia dif-
но—с—н 1еха содержит 54% Д.). Синтетически Д.
I получают восстановлением D-галактозы
-С—он амальгам0й натрия. Д. входит в состав
СН2ОН ряда сред, применяемых при бактериоло-
гия. исследованиях.
Лит.: С а г г С. J. and Krantz J. С., в кн.: Advances
in carbohydrate chemistry. Ed. by W.W. Pigman and M. L. Wol-
from, v. 1, N. Y., 1945, p. 175; Lohmar R. and G о e p p
R. M., там же,’v. 4, N. Y., 1949, p. 211. E. Л. Розенфельд.
ДУРОЛ (1,2,4,5-тетраметилбензол) C10H14, молеку-
лярный вес 134,22 — листочки с запахом камфары;
т. пл. 79,7°; т. кип. 196°; <7|1,3 0,8380,
9^з 1,034 (твердого); п31,3 1,47896;
<\-СН3 ”31,3 1’49369! 'крит. 402,5°, ?крит
Н3С-*%. J ' 28,6 ат; легко растворим в спирте,
эфире и бензоле, с трудом—в горя-
СН3 чей уксусной кислоте; возгоняется,
летуч с водяным паром. Д. с 1,3,5-
тринитробензолом дает молекулярное соединение, т. пл.
92—98°. Получают Д. из толуола, ксилолов или кумола
действием СН3С1 в присутствии А1С13, восстановле-
нием 1,4-диметил-2,5-ди(хлорметил)бензола, алкили-
рованием метилбензола. Д. выделен из нефти и кам.-уг.
смолы. При окислении Д. образуется тетракарбоновая
к-та или ее ангидрид, применяющиеся для синтеза
эпоксидных смол, полиэфиров и др.; при хлорирова-
нии замещается водород в СН3-группах; продукт
реакции применяется как инсектофунгицид и др.;
аминопроизводные Д. используются в произ-ве син-
тетич. красителей; винильные производные — в
произ-ве полимеризационных смол.
Лит.: Kirk, Second supplement volume, N. Y., 1960,
p. 610—655. Л. С. Поваров.
ДУСТЫ — см. Пестицидные препараты.
ДУШИСТЫЕ ВЕЩЕСТВА — органические соеди-
нения, обладающие характерным приятным запахом,
применяемые при произ-ве различных парфюмерных
и косметич. изделий, мыла, пищевых и др, продуктов
для придания им определенного запаха. Д. в. широко
распространены в природе. Они находятся в эфирных
маслах, душистых смолах и др. сложных смесях
органич. веществ, выделяемых из природных продук-
тов как растительного, так и животного происхожде-
ния. Многие Д. в. получаются синтетически.
С древнейших времен и до 19 в. в качестве арома-
тич. продуктов применялись только природные смеси—
эфирные масла, выделяемые из растений отгонкой
с водяным паром, настои и экстракты, получаемые
обработкой растительного или животного сырья
растворителями, душистые смолы, бальзамы и нек-рые
др., без разделения их на отдельные компоненты.
Химич, изучение состава эфирных масел и др. при-
родных ароматич. продуктов и выделение из них
индивидуальных соединений уже в 19 в. привело
к установлению строения ряда душистых в-в и син-
тезу их на основе использования простейшего химич.
сырья. Первыми были синтезированы ванилин, (}-<fie-
нилэтиловый спирт, индол и др. Целесообразность
производственного получения того или иного Д. в.
синтетически или путем переработки природного
сырья решается технико-экономич. соображениями.
Нек-рые Д. в. оказывается все же целесообразнее
получать пока из природных продуктов, тогда как
для многих Д. в. уже в настоящее время исполь-
зуются гл. обр. синтетич. методы. Примером может
служить мускус, для получения 1 кг к-рого из желез
самца кабарги потребовалось бы уничтожить ок.
30 тыс. этих животных. Вместе с тем синтезировано
и освоено в пром-сти несколько соединений, обладаю-
щих мускусным запахом; к числу последних отно-
сятся: мускус амбровый, мускус-кетон, мускус-ксилол,
тибетолид (см. Мускус). Попытки классифицировать
Д. в. по запаху делались неоднократно, но успеха
не имели, т. к. такое распределение Д. в. по группам
встречает значительные практич. трудности’и лишено
серьезного научного основания. Классификация Д. в.
по их назначению также является весьма условной.
Очень часто одни и те же Д. в. имеют различное на-
значение, напр. для парфюмерии, для кондитерских
изделий и т. п.
Наиболее рационально классифицировать Д. в.
по группам в соответствии с систематикой органич.
соединений. Такая классификация Д. в. могла бы
позволить связывать их запах с наиболее важной
химич. характеристикой — строением молекулы. Од-
нако эта зависимость установлена еще не настолько
строго, чтобы можно было с полной определенностью
предвидеть запах того или иного соединения на основе
данных о его строении. Тем не менее для отдельных
групп органич. соединений можно установить нек-рые
частные закономерности влияния структурных осо-
бенностей молекулы на запах. Для большинства
Д. в. характерно наличие в их молекуле функцио-
нальных групп: карбинольной (ОН), карбониль-
.. \
ной \С=О, сложноэфирной—CY и нек-рыхдр.
/ XOR
Для сложных эфиров, напр., характерен фруктовый
или фруктово-цветочный запах. Накопление в одной
молекуле нескольких одинаковых, а у соединений
алифатич. ряда даже и разных функциональных групп
приводит обычно не к усилению запаха, а к его ослаб-
лению и даже исчезновению. Это можно наблюдать,
например, при сравнении одноатомных спиртов, об-
ладающих запахом, с лишенными запаха много-
атомными спиртами, а также при сравнении запаха
сложных эфиров монокарбоновых и дикарбоновых
кислот и т. д.
Значительное влияние на запах оказывает величина
молекулы. Обычно соседние члены гомология, ряда
обладают сходным запахом, причем сила его посте-
пенно меняется при переходе от одного члена ряда
к другому. При определенной величине молекулы
запах исчезает. Так, соединения алифатич. ряда,
имеющие более 17—18 атомов углерода, как правила,
лишены запаха. Сила аромата зависит также и от
степени разветвления цепи атомов углерода; обычно
альдегиды, имеющие разветвленную цепь атомов угле-
рода, обладают более сильным и часто более прият-
ным запахом, чем изомерные им альдегиды нормаль-
ного строения. Большое влияние на запах органич.
соединения оказывают положение заместителей в мо-
лекуле, наличие кратных связей, а также простран-
ственные факторы. Ббльшая часть соединений, на-
шедших применение в парфюмерной пром-сти, содер-
жит в составе своей молекулы циклич. группировку.
В конечном счете запах определяется строением всей
молекулы в целом.
Специфика общего строения молекулы особенно
наглядно вид^а при рассмотрении Д. в. группы мак-
роциклич. соединений, т. е. соединений, содержащих
большие циклы. Эти соединения были найдены в при-
родных продуктах: животного и растительного проис-
хождения. Так, при изучении душистых начал про-
дуктов, выделяемых из желез нек-рых животных —
мускусной кабарги (мускон, I), цибетовой кошки
(цибетон, II), — было установлено, что они являются
макроциклич. кетонами, а Д. в. с мускусным запахом,
1219
ДУШИСТЫЕ ВЕЩЕСТВА
1220
выделяемые из нек-рых эфирных масел — семян
абельмоша (амбреттолид, III), корней ангелики (тибе-
толид, IV), —- оказались многочленными лактонами:
О=С------СН2 СН-(СН,)7Х
(СН2)2-СН-СН3 ' СН-(СН2)/С-° "
СН-(СН2)7-С^О сн2
СН-(СН2)5-С=О <СН2)|з'^О IV
Запах макроциклич. соединений определяется гл.
обр. числом звеньев в цикле. Так, мускусным запахом
обладают не только приведенные выше хорошо из-
вестные макроциклич. лактоны и кетоны, но также
и другие макроциклич. соединения, относящиеся
к самым разнообразным классам. Различие в запахе
у таких аналогов проявляется лишь в его силе и от-
тенках. Строение всей молекулы в целом в этих соеди-
нениях оказывает значительно большее влияние
на запах, чем наличие в молекуле тех или иных груп-
пировок атомов.
Соединения, очень близкие по своему строению
(напр., цис- и транс-изомеры), иногда обладают раз-
личными оттенками запаха. С другой стороны, ве- -
щества, очень далекие по своей химич. природе, мо-
гут иметь сходный запах. Так, кроме упомянутых
выше макроциклич. соединений, мускусным запахом
обладают нек-рые алкилнитропроизводные ароматич.
ряда, нек-рые алкилированные бензиловые спирты,
не имеющие нитрогруппы, представители замещенных
полиалкилинданов, тетрагидронафталинов, нафтин-
данов, индаценов и ряд других соединений, сильно
различающихся по строению.
Ниже приведены лишь нек-рые из этих продуктов,
применяющиеся в качестве Д. в. с мускусным запахом:
мускус амбровый (V); 2,4,6-триизопропилбензиловый
спирт (VI); фантолид (VII)
Целесообразность применения в парфюмерии того
или иного вещества, обладающего приятным запахом,
решается не только на основании установления цен-
ности запаха этого соединения, но также с учетом
других его свойств — химич. устойчивости, летучести,
токсичности; принимается во внимание также наличие
удобных и экономически выгодных методов его полу-
чения.
В парфюмерной пром-сти широко используются
Разнообразные сложные эфиры. Нек-рые эфиры али-
атич. к-т имеют запах цветов, напр.: линалилаце-
тат—запах цветов бергамота, фенилэтилацетат — запах
фруктов и зелени, метиловый эфир гептинкарбоновой
к-ты СН3(СН2)4С= ССООСН3 — запах свежих ли-
стьев фиалки; эфиры кислот ароматич. ряда обычно
обладают слабым запахом бальзама и служат в парфю-
мерных композициях фиксаторами запаха. Практич.
применение нашли эфиры к-т: уксусной СН3СООН
(особенно для пищевых продуктов), валериановой
СН3(СН2)3СООН, лауриновой СН3 (СН2)10СООН, са-
лициловой о-НО—СвН4—СООН, антраниловой
o-H2N—СвН4—СООН, коричной С6Н6СН=СНСООН.
IX
СОН
Н3СЛСН3
К другим продуктам, широко применяемым в пар-
фюмерной пром-сти, относятся ванилин (VIII) (запах
ванилй), дифениловый
эфир СвН5ОСвН5 (запах СНО СН3
герани), терпинеол, гл.
образом а-изомер (IX)
(запах сирени), &-фе- ОСН3
нилэтиловый спирт i
С„Н5СН2СН2ОН (запах ОН
розы), диэтилфталат
С„Н5(СООС2Н5)2.
Практическое применение в качестве Д. в. на-
ходят также соединения, приведенные в таблице:
Соединение № фор- мулы Запах
Ацетил анизол X Обепина и гелиотропина
Гелиотропин XI Цветов гелиотропа
Гераниол XII Розы
Гидроксицитр онелл аль XIII Липы и ландыша, свежей зелени
Дециловый альдегид XIV В больших разведениях запах розы и апель- синовых корок
Дигидрожасмон XV В разбавлении напоми- нает запах жасмина
Жасмин альдегид * Жасмина
Изоэвгенол XVI Гвоздики
Ионон XVII Кедра, в разбавлении— цветов фиалки
Индол XVIII В сильном разбавлении запах цветов жасмина
Коричный альдегид XIX Корицы
Коричный спирт XX Гиацинта
Кумарин XXI Свежего сена
Лауриновый альдегид (додециловый альде- гид) XXII В очень больших разве- дениях запах цветов; отдаленно — фиалки
Линалоол XXIII Ландыша
«-Метил ацетофенон XXIV Миндаля и свежего сена
изо-а-Метилионон XXV Нежный запах фиалки
(«Ир алия»)
Метиловый эфир р-наф- тола (Яра-яра) XXVI Отдаленно напоминает запах черемухи
Нониловый спирт XXVII В разведении запах розы
Обепин (анисовый аль- дегид) Ундекалактон XXVIII Цветов боярышника
XXIX Персика
Фенилуксусный альде- XXX Гиацинта
гид
Цикламенальдегид XXXI Цветов цикламена
Цитр аль XXXII Лимонов
Цитронеллол XXXIII Розы
Эвгенол и другие XXXIV Гвоздики
• CtH5CH = С(С3Нц)СНО.
II
n-CHgO—С6Н4—С-СН3
X
(СН3)2С=СН(СН2)2С=СНСН2ОН
СН3
XI/ d
(сн3)г^(сн2)3снсн2сно
он сн3
XIII
н2с—сн
I и
Н2С С(СНа)5СН3
СО XV
СНО
XI
СН3(СН2)8СНО
XIV
СН=СН-СН3
С^ХоеНз
он XVI
1221
ДЭКИНА РЕАКЦИЯ — ДЮМА МЕТОД
1222
В произ-ве духов и одеколонов главным процессом
является составление т. наз. «парфюмерной компози-
ции» путем смешивания в определенных пропорциях
различных Д. в. и ароматных продуктов. Для полу-
чения «парфюмерной жидкости» эту композицию рас-
творяют в спирте определенной крепости в зависимости
фенолов из природных оксиальдегидов. Так, из о-ва-
нилина в атмосфере инертного газа получается с
80%-ным выходом монометиловый эфир пирогаллола:
сч/сн3
XVII р
аСН=СНСОСН3
сн3
СвН5СН=СНСНО С6Н5СН=СНСН2ОМ
СН3(СН2)10СНО
XXII
Реакция открыта Н. Д. Дэкиным в 1909.
Лит.: Surrey A. R., Name reactions in organic che-
mistry, N. Y., 1954, p. 45—46. H. E. Голубева.
ДЭКИНА — ВЕСТА РЕАКЦИЯ — взаимодействие
а-аминокислот с уксусным ангидридом в присутствии
пиридина с образованием а-ацетамидоалкилметилке-
тонов:
R-CH-COOH + (СН3СО)2О — R—СН—СОСНд
I I 4“ СО2 -р Н2О
NH3 NHCOCHa
1СНз)2С=СН(СН2)2С(ОН)СН=СН2
XXIII СНз
н3сЧхсн3 сн3
,-сн=ссосн3
-сн3
XXV
СН3(СН2)7СН2ОН
XXVII
СН з (СН ,)6 СН -(СН 2 )2-С=О
I---о——J
XXIX
СН3С6Н4-сО-СНа
XXIV
осн3
XXVI
Применение в реакции ангидридов бензойной, про-
пионовой и масляной к-т дало возможность получать
различные а-ациламидоарилкетоны и а-алкилкетоны.
Так, при взаимодействии р-фенил-а-аланина с пропио-
новым ангидридом был получен 1-фенил-2-пропиоами-
допентанон-3:
с«н5сн2снсоон
| + (СН3СН,СО)2О —
NH3
Л-Н3СО-С6Н4-СНО
XXVIII
Со Н 5 СНа СН СО СН2 СН3
I
NHCOCH2CH3
4- СО3 + Н3О
с6н5Сн2сно
XXX
СН2-СН-СНО (СН3)2С=СН(СН2)2(р=СНСНО
сн3 сн3
II XXXII
Вместо пиридина возможно применение безводного
ацетата натрия. Механизм Д. — В. р. включает ката-
лизируемое основаниями образование из N-ацил-
а-аминокислоты оксазолона (I) (см. Эрленмейера —
Плохла реакция) с последующим его ацилированием
и расщеплением;
сн
H3CZ чсн3
XXXI
(СН3)2С=СН(СН2)2СН сн2сн2он
XXXIII СНз
сн2-сн=сн2
•ОСН3
0Н XXXIV
R—СН—СООН (СН3СО)2О R—СН-СООН___
NH2 NHCOCH3
СОСН3
_ R-СН-СО , R-C - СО г R—СН~СОСН3
NO NO NHCOCH3
Ч / Ч /
с-сн3 с-сн3
1
от вида приготовляемых духов или одеколона. Пар-
фюмерная пром-сть потребляет Д. в. с самыми раз-
личнымр оттенками запаха (цветочные, мускусный,
амбровый, санталовый и т. п.). Насколько велик
ассортимент Д. в., находящих применение в парфю-
мерии, можно видеть уже из того, что дли приготовле-
ния каждой сложной парфюмерной композиции тре-
буется обычно несколько десятков различных Д. в.
и ароматных продуктов. Так, напр., композиция
для духов «Красная Москва» составляется из 34 раз-
личных компонентов.
Лит.: Белов В. Н., Скворцова Н. И., Природа,
1954, № И, с. 33; Б е л о в В. Н. [и др.], Химия и техноло-
гия душистых веществ, М., 1953; Белов В. Н. [и др.],
Усп. химии, 1957, 26, вып. 1, с. 96: Р у т о в с к и й Б. Н.,
Эфирные масла, т. 1, М.—Л., 1931; West Т. F., Strausz
Н. J. and Barton D. Н. R., Synthetic perfumes, L., [1949];
Bedoukian P. Z., Perfumery synthetics and isolates,
N. Y.—L. — Toronto, 1951. H. И. Скворцова.
ДЭКИНА РЕАКЦИЯ — взаимодействие щелочно-
го раствора о- или n-оксибензальдегидов или кетонов
с разб. перекисью водорода с образованием поли-
оксифенолов:
но-свн4-сно НО-СеН4-ОН + нсоон
Реакция находит применение при получении поли-
Реакция открыта в 1928 Н. Д. Дэкиным и Р. Вестом.
Лит.: Cornforth J. W. and Elliott D. F.,
Science, 1950, 112, 534; Surrey A. R., Name reactions in
organic chemistry, N. Y., 1954, p. 47. H. E. Голубева.
ДЮЛОНГА И ПТИ ЗАКОН — эмпирич. правило,
согласно к-рому ат. теплоемкость (т. е. произведение
уд. теплоемкости на ат. вес) простого тела в кристал-
лич. состоянии приблизительно одинакова для всех
элементов и составляет ок. 6,2 кал[град. Закон при-
ближенно оправдывается для большинства металлов
при комнатной темп-ре. Многие легкие элементы,
в особенности неметаллы, при комнатной темп-ре
имеют значение ат. теплоемкости много меньше
6,2 кал)град (напр., алмаз 1,36, бор 3,5, бериллий 3,82
и др.). Элементы, приблизительно подчиняющиеся
Д. и П. з. при комнатной и более высоких темп-рах,
совершенно перестают следовать ему в области низ-
ких темп-p. Закон установлен П. Дюлонгом и А. Пти
в 1819; в свое время сыграл положительную роль в
первоначальном определении атомных весов.
Лит.: Ш т р а у ф Е. А., Молекулярная физика, Л.—М.,
1949; Л е онт они ч М. А., Статистическая физика, М.—Л.,
1944.
ДЮМА МЕТОД — способ определения содержания
азота в органич. соединениях; заключается в сожжении
вещества в атмосфере СО2 нагреванием с СиО и изме-
рении выделяющегося азота. См. Азота определение.
ЕВРОПИЙ (Europium) Eu — химич. элемент из
семейства лантанидов с п. н. 63 и ат. в. 152,0.
ЕНАМИНЫ — а, ^-ненасыщенные амины, в моле-
кулах к-рых атом азота связан непосредственно
с атомом углерода, соединенным с соседним атомом
углерода двойной С=С связью. Е. (I) могут нахо-
диться в состоянии таутомерного равновесия с ими-
нами (II). Третичные Е. (III) не могут превращаться
в иминоформу. Прй действии кислот все Е., в том
числе и третичные, образуют соли иммония (IV).
-С=С< = -с-сн-
Iх II I
NHR NR
I II
-с=с<^-с-сн-
1Х II I
nr2 +NR»X-
III IV
Е. получают обычно взаимодействием аминов с кар-
бонильными соединениями. При этом альдегиды об-
разуют алкилидендиамины (V), к-рые при перегонке
отцепляют молекулу амина с образованием Е. (VI):
\CH-CHO+2NHR2 -ЛсН-CHNR., -Ac=chnr.,
/ Z | Z
NRs
V VI
Кетоны (реакция изучена гл. обр. для циклич. кето-
нов) присоединяют одну молекулу амина к карбо-
нильной группе; после отщепления воды от продукта
присоединения (VII) путем азеотропной отгонки
получают Е. (III):
XCII-СО-1-NHRi —>ХсН—С—NR-i ->\C=C-NR„
z Z ! Z j
ОН
VII III
Е. легко обнаруживаются по спектрам в УФ-обла-
сти Хмакс.=220—235 ммк, в ИК-области =1620—
1670 см-i.
Е. называют аналогично аминам, напр. [5-амино-
кротоновый эфир (VIII), диэтиламиноэтилен (IX),
1-пирролидиноциклогексен (X).
К числу Е. относят и нек-рые гетероциклич. соеди-
нения, в молекулах к-рых атом азота и по меньшей
мере один из двух атомов углерода, связанных двой-
ной связью, являются звеньями одного цикла. На-
пример, Х-метил-Д2-метилениндолин (XIII):
СН<С = СН-СООС„Н-
О I 6 0
nh2
VIII
CH2=CH-N(C2H5)2
X
XI
Е. мало устойчивы, обладают повышенной реакцион-
ной способностью. Они могут быть использованы для
различных синтезов; выделение Е. в чистом виде
не является необходимым. Широко изучены реак-
ции Е. с различными электрофильными реагентами.
В этих реакциях атака электрофильного реагента
направляется, как правило, не на атом азота, а на
^-углеродный атом:
Образовавшаяся соль иммония (XIV) дает при гид-
ролизе карбонильное соединение, содержащее в
a-положении заместитель R. На этой основе разра-
ботаны препаративные способы получения а-замещен-
ных альдегидов и кетонов по общей схеме:
.Xnh .н х I пп
с- сн/ z -» -с=с/ — -с-с-^10
И Х Iх II I
О N +N R
Z \ Z \
I /
_СО-С —1-НХ+Н -N^
R
В качестве RX используют галогеналкилы игалогенар-
алкилы, а-галогенэфиры, а-галогенокетоны, хлоран-
гидриды карбоновых к-т, хлористый циан, соли диа-
зония, трпнитробромбензол, цианурхлорид. Е. взаи-
модействуют и с а, [5-ненасыщенными соединениями —
эфирами акриловой и кротоновой к-т, винилкетонами,
акролеином, а также изоцианатами и кетеном, напр.
с
акрилонитрилом:
-С=СН- --CH..- CHCN — С—СН СН-СНСХ -
I II I
N +N
z Н..0
-> -CCCIL-CH..CN -Л - С -СН-СН . -CILCN
I I ‘ ' И I
N О
Наилучшие выходы замещенных альдегидов и кетонов
получаются при использовании в качестве вторичных
аминов, дающих Е. с карбонильными соединениями,
пирролидина, пиперидина и морфолина.
Лит.: Kloubek J., Chem. Listy, 1959, 53, 821; Сое-
ne n M., Angew. Chem., 1949, 81, II; S t о г k &., Landes-
m a n H. K., J. Amer. Chem. Soc., 1956,78, Ki 19, 5128; К u e h-
n e M. E., J. Amer. Chem. Soc., 1959, 81, № 20,5400.
Л. II. Беленький.
ЕНОЛАЗА — фермент, катализирующий обрати-
мую реакцию превращения D-(—)-2-фосфоглицери-
новой к-ты (2-ФГК) в фосфоенолпировиноградную
(ФЕПК):
соон соон
I I
НСОРО(ОН)2—г СОРО(ОН). + Н,0
I и
СН,ОН СН,
1225
ЕНОЛЫ
1226
Эта реакция является важным этапом в процессе
ав аэробного (гликолитического) расщепления угле-
водов, доставляющего энергию живой клетке (см.
Гликолиз). В результате указанной реакции бедная
энергией фосфоэфирная связь 2-фо сфо глицериновой
кислоты превращается в богатую энергией (макро-
эргическую) енолфосфатную связь. Константа равно-
весия реакции:
к = ФЕПК/2-ФГК = 2,5
Е. — белок, мол. в. 63 700; обладает фермента-
тивной активностью только в присутствии ионов
Mg3+, Mn2+, Zn2+ и Fe2+, с к-рыми Е. образует металл-
ферментные комплексы в соотношении 1 атом металла
на 1 молекулу фермента. Наиболее сильным активирую-
щим действием обладает Mg2+, к-рый, по-видимому,
и является ионом, активирующим Е. в физиология,
условиях. Константа диссоциации комплекса Mg-
фермент 0,61 •10~3М при pH 7,34. Са2+ и Sr2+ образуют
с Е. неактивные комплексы, в связи с чем они кон-
курентно тормозят ее активность. Конкурентное тор-
можение Е. вызывают также ионы F-; действие по-
следних проявляется только в присутствии ионов
Mg2+ и фосфата, так что истинным ингибитором в
этом случае, по-видимому, является комплекс
Mg2+ — — фосфат.'
Один моль Е. при 25° катализирует превращение
15 000 молей 2-фосфоглицериновой к-ты в минуту;
константа Михаэлиса для этой к-ты составляет 10'4А/,
оптимум действия Е. при pH 7,5. Активность Е. оп-
ределяют количественно по увеличению концентра-
ции фосфоенолпировиноградной к-ты; последняя мо-
жет быть измерена спектрофотометрически по по-
глощению при 240 ммк или же йодометрическим ме-
тодом.
Е. — широко распространенный фермент; присут-
ствует во всех клетках, способных осуществлять гли-
колитическое расщепление углеводов; выделена в
кристаллич. состоянии из дрожжей путем фракцио-
нирования бесклеточного сока спиртом и ацетоном
с последующей кристаллизацией в виде ртутного
комплекса из насыщенного раствора.
Лит.: Лейланде Дж., Ш т у м п ф И., Очерки по химии
ферментов, пер. с англ., М., ГЭ58; Methods In Enzymology.
Ed. by S. P. Colowlck and N. 0. Kaplan, v. 1, N. Y., 1955,
p. 427. В. Б.- Спиричев.
ЕНОЛЫ—органич. соединения, несущие гидроксиль-
ную группу при углерод — углеродной двойной связи.
Е. находятся в таутомерном равновесии с соответ-
ствующими карбонильными соединениями (см. Тауто-
мерия кето-енольная): —С=С^^.—С—CHcf'. Поло-
I 4 II 4
ОН О
жение таутомерного равновесия определяется прежде
всего строением вещества и характером растворителя.
Эта зависимость выражена ф-лой КТ = Е • L, где
КТ — константа кето-енольного равновесия, представ-
ляющая собой отношение концентраций енольной и
карбонильной форм, Е — константа енолизации, ха-
рактерная для каждого таутомерного соединения,
L — константа, определяющая енолизующую спо-
собность растворителя. Карбонильная форма энерге-
тически более выгодна, чем енольная (на 13—
17 ккал/моль), поэтому попытки получения простейших
Е. приводят к соответствующим альдегидам или кето-
нам, в к-рых содержание Е. ничтожно мало (2,5• 10-4%
ацетона, 2,5-10-2 % циклогексанона, 4,8-10 4 % ци-
клопентанона). Однако в отдельных случаях енольная
форма становится устойчивой, т. к. вышеприведен-
ная разница в энергии компенсируется энергетич.
выигрышем вследствие образования системы сопря-
женных связей и установления водородной связи за
счет возникающей гидроксильной группы. Указанная
стабилизация наблюдается в Р-дикарбонильных соеди-
нениях (Р-дикетонах, Р-кетоальдегидах, Р-кетокарбо-
новых к-тах и их производных) (1), соединениях ряда
бензилглиоксаля (2), циклич. а-дикетонах (3), Р-аце-
тиленовых альдегидах и кетонах (4) и жирноарома-
тич. кетонах, в состав к-рых входит мезитильный
радикал (5);
Н
снз-с-снг-с-сн3 СН3--С<2>х-сн3 (I)
0 о <А оЬ
тТ"
СНз С С CH2CfiHs,—
о о
r-C=C-CH2-COR’
R-9H-C-R R-C=C-R (5)
СНзО СНзОН
____________________(СН3 Значение константы енолиза-
r=CH —?___у- ции & для различных таутомер-
3 \ / ных соединений изменяется в ши-
'Сн рокихпределах, составляя, напр.,
3 0,02 для метилдибензоилметана
и 5075 для димедона (Е ацетоуксусного эфира при-
нята за 1). Что касается енолизующей способности
растворителя L, то эта величина возрастает с умень-
шением полярности последнего. Содержание Е в равно-
весной смеси зависит также от темп-ры и агрегатного
состояния вещества, а в растворе — от концентрации
вещества. Подвижность кетоенольных систем, т. е.
время установления равновесия, зависит от струк-
турных факторов, растворителя, температуры, присут-
ствия катализаторов (обычное стекло, протонные
и апротонные к-ты, основания). В нек-рых случаях,
напр. для ацетоуксусного эфира, удается выделить Е.
в чистом виде. Кетоенолы могут существовать в цис- и
трамс-формах соответственно взаимному расположе-
нию енольного гидроксила и карбонильной группы.
Обычно цис-Е. более стабильны, т. к. в транс-Е. не
может образоваться внутримолекулярная водородная
связь. В ряде случаев, напр. для этилового эфира фор-
милфенилуксусной к-ты, удается выделить обе формы.
Иногда структурные факторы допускают существова-
ние только одной из форм. Так, Е. циклич. р-кетокар-
боновых к-т существуют исключительно в цис-(§), а
Е. циклич. р-дикетопов — только в транс-формах (7):
Большинство реакций Е. сопровождается смещени-
ем таутомерного равновесия, причем многие из реак-
ций могут быть объяснены также через карбониль-
ную форму. Так, замещение гидроксильной группы
хлором действием РС16 может осуществляться по
1227
ЕНОЛЫ
1228
схемам:
То же самое относится к конденсации Е. с азотистыми
основаниями:
он
jAHsNHNH»
-С(ОН)-СН<^
HN-NHCcH,
|C0H5NHNHs
-с-сн/
II 4
N-NHC0H3
Однако имеются реакции, определенно свойственные
только енольной форме. Енольный гидроксил ацили-
руется хлорангидридами к-т в пиридине и метили-
руется диазометаном. Е. легко присоединяют галоге-
ны по кратной связи с образованием а-галогенокар-
бонильных соединений:
-С=С< Г-С(ОН)-СХ< т —-С-СХ<
он L х J (!)
Эта реакция во многих случаях может быть применена
для колич. определения Е., т. к. скорость ее значи-
тельно больше скорости таутомерного превращения.
Подвижный водород в Е. может быть определен мето-
дом Чугаева — Церевитинова (см. Активный водо-
род), однако для этого лучше использовать цинк-
органич. соединения, т. к. магнийорганические ката-
лизируют енолизацию. При этом образуются Zn-
и Mg-еноляты. Еноляты щелочных металлов могут
быть получены действием на Е. самих металлов, их
алкоголятов или гидроокисей. Окрашенные еноляты
тяжелых металлов образуются при обработке Е.
соответствующими солями в водном р-ре. При взаимо-
действии Е. с солями тяжелых металлов в спирте
происходит образование клешнеобразных соединений,
характерные окраски которых позволяют применить
эту реакцию для качественного определения Е. С дру-
гой стороны, указанные комплексы могут быть ис-
пользованы как для выделения Е., так и для вы-
деления и разделения катионов тяжелых металлов.
Могут быть также получены и тетраалкиламмоние-
вые еноляты. Во всех енолятах атом металла свя-
зан с атомом кислорода, и кетоенольная таутоме-
рия по схеме —С=С<——С—С С не имеет места,
I II I
ОМе О Me
что было показано А. Н. Несмеяновым на примере
Li-, Na- и Mg-енолятов дифенилпропиомезитилена
(СвН5)2СН—СН=С(ОМе)—СвН2(СН3)2. В то же время
ацилирование и алкилирование енолятов приводит к
образованию производных как по кислороду, так и по
углероду. Такая двойственная реакционная способ-
ность связана с явлением переноса реакционного цент-
ра. Различными способами могут быть получены еноля-
ты неустойчивых простейших Е., напр. Li-алкоголят
винилового спирта СНа=СН—OLi. Другим обширным
и важным классом производных этих гипотетич.
спиртов являются виниловые эфиры. Следует также
отметить, что нек-рые превращения альдегидов и ке-
тонов, напр. катализируемое кислотами галоидиро-
вание ацетона, осуществляются с предварительной
енолизацией карбонильной группы (см. также Элъ-
текова правило).
Лит.: Темникова Т. И., Курс теоретических основ
органической химии, Л., 1959; Реутов О. А., Теоретиче-
ские проблемы органической химии, М., 1956; Henect а Н.,
Chemie der Beta-Dicarbonylverbindungen, В., 1950.
Б. Л. Вяткин.
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
КАК ПОЛЬЗОВАТЬСЯ УКАЗАТЕЛЕМ
В алфавитно-предметный указатель включены в качестве рубрик названия статей тома и тер-
мины, упоминаемые в тексте статей. Предметные рубрики в ряде случаев дополнены подрубриками,
причем первая подрубрика отделена от рубрики запятой, а последующие — знаком тире и начи-
наются с новой строки. За рубрикой и подрубрикой следуют номера страниц тома. Номера страниц,
на которых помещены статьи, выделены полужирным шрифтом. Например:
Бензойная кислота 409
— азид 56
— гидразид 882
В рубриках, состоящих из нескольких слов (сложных рубриках), при повторениях первое слово
заменяется удлиненным тире. Например:
Весы лабораторные
— аналитические
— квадрантные
Цифры, латинские и греческие буквы при наименованиях органических соединений, а также
обозначения п, о, втор., трет, и другие (в начале и внутри сложного наименования) не принимаются
во внимание при расположении рубрик по алфавиту.
Инверсия (перестановка) прилагательного применяется для группировки материалов. На-
пример:
Красители
----азиновые
----азометиновые
Названия солей начинаются с катиона. Например:
Натрий, нитрат, а не Нитрат натрия
Кислоты, спирты, альдегиды и т. п. даны по их наименованиям. Например:
Бутиловый спирт
Валериановые кислоты
Гидратроповый альдегид
Эфиры, нитрилы и т. д. приведены под названием соответствующей кислоты, если они не имеют
общераспространенного названия. Например: Амилацетат, Акрилонитрил.
Рубрики в необходимых случаях сопровождаются отсылками.
Отсылки, напечатанные курсивом, означают, что соответствующие статьи будут помещены в сле-
дующих томах энциклопедии. В Указателе при группировке материала названия отдельных статей
даны в виде подрубрик с выделением номера страницы полужирным шрифтом. Например:
в томе — Азота окись
в Указателе — Азот, окись
Абиетиновая кислота 17
Абсолютная температура 17
Абсолютный геологический возраст —
см. Возраст геологический абсолютный
Абсорбенты 19
Абсорберы 20
Абсорбционная спектроскопия 18
Абсорбционный анализ 762
Абсорбция 19
——масляная 22
Авадхаридин 86
Авертин 793
Авиаль 157
Авиационные бензины 401
Авитаминозы 595
Авогадро закон 23
Авогадро число 23
Автогезия — см. Аутогезия
Автоклавы лабораторные 23, 696
Автолиз — см. Аутолиз
Автолы 25
Автометаморфизм (в геохимии) 842
Автомобильные бензины 400
Агар (агар-агар) 26
Агароза 26
Агликон 956, 958
Аглюкон — см. Агликон
Адалин 26
Адамантан 26
Адамкевича — Гопкинса реакция — см.
Адамкевича реакция
Адамкевича реакция 27
Адамса катализатор 27
Адамсит 27
Адгезия 27
Адденды — см. Комплексные соединения
Аддукт 30
Адениловые кислоты — см. Аденозин-
монофосфор ные кислоты
Адениловый цикл 32
Аденин 30
Аденозин 31
Аденозиндифосфорная кислота 32
Аденозинмонофосфорные кислоты 32
Аденозинтрифосфатазы 31
Аденозинтрифосфорная кислота 33
Аденозинфосфорные кислоты 32
Адиабатные процессы 34
Адипиновая кислота 34
— диамид 35
1231
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1232
— дианилид 35
— диметиловый эфир 35
— ди-н-бутиловый эфир — см. Дибутп-
ладипинат
— динитрил 35
— дифениловый эфир 35
— дихлорангидрид 35
— диэтиловый эфир 35
Адиуретин 504
Адкинса катализатор 35
Адреналин 35, 996, 998
Адренокортикотропин — см. Адренокор-
тикотропный гормон
Адренокортикотропный гормон 36, 996,
998
Адсорбат 41
Адсорбенты 37
Адсорбтив 41
Адсорбционное понижение прочности
твердых тел 39
Адсорбционные силы 41
Адсорбция 41; динамика 46; кинетика 46
--- активированная 46
--- радиоактивных элементов 48
Адурол — см. Проявляющие вещества
Азасерин 49
Азелаиновая кислота 49
Азеотропные смеси '50
Азид свинца 55, 562
Азидотиоугольная кислота 56
Азиды 55
— карбоновых кислот 56
Азимутальное квантовое число 313
Азиновые красители 57
Азины 57
Азоамин алый Ж 59
--- алый 2Ж 59
алый К 59
бордо К 59
- — желтый О 59
— коричневый О 173
- — красный Ж 59
- — красный О 59
--- красный С 59
- — красный ЗС 59
- — оранжевый Ж 59
- — оранжевый К 59
- — оранжевый О 59
- — розовый О 59
-— ярко-алый Ж 59
--- ярко-оранжевый К 59
Азоамины 58
Азобензол 59
2,2-Азо-бис-изобутиронитрил 59
2-Азо-бис-изомасляная кислота, динит-
рил — см. 2,2-Азо-бис-изобутиронит-
рил
Азоимид — см. Азотистоводородная кис-
лота
Азоимиды — см. Азиды карбоновых кис-
лот
Азокрасители 60
Азоксибензол 62
Азоксисоединения 63
Азометиновые красители 63
Азосоединения 64
Азосочетание 65, 64
Азот 66
— атом 67
— водородные соединения А. 68
— двуокись 71
— закись 70
— изотопы 67
— молекула 67
— нахождение в природе 66
— окисление 76
— окислы 70
— окись 70
— определение 72
— получение 69, 636
— применение 69
— физич. и химич. свойства 68
— четырехокись 71
Азот активный 69
--- иодистый 69
--- остаточный — см. Белки, осажде-
ние
--- сернистый 69
--- фтористый 68
--- хлористый 69
Азотистая кислота 73
Азотистая сера 69
Азотистоводородная кислота 73
Азотистые иприты 74
Азотистый ангидрид 71
Азотистый обмен — см. Обмен веществ
Азотная кислота 74
• — получение 75
— применение 79
— свойства 75
— техника безопасности 78
Азотнокислый алюминий — см. Алюми-
ний, нитрат
Азотнокислый аммоний — см. Аммоний,
нитрат
Азотные удобрения 79
Азотный ангидрид 72
Азотол 2Ж 80
Азотолы 80
Азотометр — см. Волюмометры
Азулены 81, 289
Аймалин 82
Акарициды 82
Акароид 83
Акваметрия 83
Акваполивольфраматы 658
Акваполисоединения 85
Аккумуляторы — см. Химические источ-
ники тока
Аконитин 86
Аконитовые алкалоиды 85
Акридан 87
Акридин 87, 871
Акридиновые красители 87
Акридон 87
Акрилальдегид — см. Акролеин
Акриламид 88, 167
Акрилаты 88
Акриловая кислота 89
Акрилонитрил 89
Акрихин 90
Акролеин 90
— дизтиловый ацеталь 339
Активации энергия — см. Энергия акти-
вации
Активационный анализ — см. радиоак-
тивационный анализ
Активированный комплекс — см. Пере-
ходное состояние
Активированный уголь — см. Актив-
ный уголь
Активности коэффициент 91
Активность 92
--- оптическая 93
Активные осадки 93
Активные центры 94
Активный азот 69
Активный водород (определение) 95
Активный уголь 96
Актин 97
Актиниды 97
— адсорбционные свойства и распреде-
ление между растворителями 100
— ионные радиусы 101
— кристаллография, данные 101
— парамагнитная восприимчивость 101
— спектры поглощения 100
— химич. свойства 98
— электронные структуры 102
Актиний 106
Актинодафнин 273
Актиноиды — см. Актиниды
Актиномицины 107
Актинон 108
Актомиозин 108
Акцептор 109, 516
Алаит 524
р-Аланил-метилгистидин — см. Ансерин
Аланин 109, 180
Алая кислота 109
Ализарин 110
Ализариновое масло 110
Ализариновый красный С НО
Алифатические аминокислоты 180
Алифатические диазосоединения 1087
Алифатические соединения — см. Ацик-
лические соединения
Алициклические соединения 111
Алкадиены — см. Непредельные углево-
дороды
Алкалиметрия и ацидиметрия 113
Алкалоиды 115
--- аконитовые 85
--- апорфиновые 273
--- спорыньи 119
--- тыквенного кураре 121
Алканы — см. Предельные углеводороды
Алкены — см. Непредельные углеводороды
Алкил 123
N-Алкил-аминокислоты 181
Алкилат 123
Алкилбензин — см. Алкилат
Алкилбензол 123
Алкилирование 124
Алкилнафталины 126
Алкилсерные кислоты — см. Алкилсуль-
фаты
Алкилсульфаты 127
Алкилфенолы 128
Алкины —,см. Непредельные углеводороды
Алкоголиз 129
Алкогольдегидрогеназа 1037
Аллелотропия — см. Таутомерия
Аллены 130
5-Аллил-5- (1-метил-бутил)-барбитуровая
кислота — (Na-соль) — см. Секонал
5-Аллил-5-изопроцилбарбитуровая кис-
лота (Na-соль) — см. Алюрат
5-А ллил-5-(1 - циклогексен-2-ил)-2-тиобар-
битуровая кислота — см. Кемитал
Аллил хлористый 132
Аллилакрилат 88
Аллилбензол 132
Аллилен 132, 350
Аллилмеркаптометилпенициллин 133
Аллиловый спирт 133
Аллилхлорид — см. Аллил хлористый
Аллоза 134, 821
Аллоксан 134, 181
D-Аллоновая кислота 823
Аллотропия 134
Аллофановая кислота, амид — см. Би-
урет
Алмаз — см. Углерод
Алунит 148
Альбихтол 135
Альбомицин 135
Альбумины 136
Альгиновые кислоты 136
Альдегидамины 671
Альдегидаммиаки 136
Альдегиддегидрогеназа 1037
Альдегидокислоты и кетокислоты 137
Альдегиды 139
— оксо-синтез 139
Альдимины 140
Альдобионовые кислоты 141
Альдобиуроновые кислоты 141
Альдогексозы 821
Альдогексоновые кислоты — см. Гексо-
новые кислоты
Альдозы — см. Моносахариды
Альдокортин — см. Альдостерон
Альдолаза 142
Альдоли 142
Альдольная конденсация 143
Альдоновые кислоты 143
Альдостерон 144, 997
Альдотриоза — см. Глицериновый аль-
дегид
Альдрин 145
Альтакс 146
Альтроза 146, 821
D-Альтрозан 146
D-Альтроновая кислота 823
Альфа-распад 146
Альфа-частица 146
Алюминаты 147
Алюминиевые квасцы — см. Алюминий,
сульфат, Квасцы
Алюминий 147
— аналитич. определение 150
— бромид 149
— галогениды 149
— гидрид 149
— иодид 149
— карбид 149
— нитрат 153
— нитрид 149
— окись 153
— — для хроматографии 155
— — кристаллич. формы и свойства 153
— — получение 154
— — применение 154
— оксидирование 233
— получение 150
— применение 151
— сплавы 155
— субгалогениды 150
— субфторид 150
— сульфат 158
— сульфид 149
— техника безопасности 151
— физич. и химич. свойства 148
— фторид 159, 149
— хлорид 159, 149
Алюминийарилы 151
Алюминийорганические соединения 151
Алюминийтриалкилы 151
Алюминон 160
Алюминотермия 160
1233
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1234
Алюмогидриды 149
Алюмосиликаты 162
Алюмотермия — см. Алюминотермия
Алюрат 372
Амадори перегруппировка 162
Амальгамы 163
Амбра 164
Амбренн 164
Амбреинолид 164
Амбреттолид 165
Америций 165, 104
Амигдалин 165
Амидазы .166
Амидины 166
Амидные удобрения 80
Амидол — см. Проявляющие вещества
Амиды карбоновых кислот 166
Амилазы 168
Амилацетат и изоамилацетат 168
Амилены 168
а-н-Амилкоричный альдегид 169, 1220
Амилнитриты 170, 248
Амиловые спирты 170
Амилодекстрины 1058
Амилоза — см. Крахмал
Амилолиз 954
Амилопектин 26 — см. также Крахмал
4-трет-Амилфенол 129
Аминазин 171
Аминарсон 171
Аминирование 172
а-Аминоадипиновая кислота 172
Аминоазобензол 173
Аминоалкоголи — см. Аминоспирты
Аминоальдегиды и аминокетоны 173
Аминоаниэолы — см. Анизидины
Аминоантрахиноновые красители 175
Аминоантрахиноны 176
Аминоантрацены — см. Антраммны
Аминоацетон 177
Аминоацилазы 177
Аминобензальдегид 177
n-Аминобензойная кислота 178
— этиловый эфир — см. Анестезин
о-Аминобензойная кислота — см. Ан-
траниловая кислота
Аминобензол — см. Анилин
Аминогалогенантрахиноны 178
а-Аминоглутаровая кислота — см. Глу-
таминовая кислота
а-Амино-$-гуанидиновалериановая кис-
лота — см. Аргинин
1-Амино-1-дезоксиальдитолы — см. Гли-
камины
4-Аминодифенил 179
а-Аминоивовалериановая кислота — см.
Валин
а-Амино-р-5-имидазолилпропионовая кис-
лота — см. Гистидин
(у-Аминокапроновая кислота 179
Аминокетоны — см. Аминоальдегиды и
аминокетоны
Аминокислоты 179
— лактамы 182, 183
Аминомасляные кислоты 184
9-Аминононановая кислота — см. со-Ами-
нопеларгоновая кислота
1 - Амино-8-нафто л-2,4-дисульфокислота
185
1 -Амино-8-нафтол-3,6-дисульфокислота
185
1-Амино-8-нафтоя-4,6-дисульфокислота
185
2-Амино-8-нафтол-3,6-дисульфокислота
185
1-Амино-2-нафтол-4-сульфокислота
185
1 - Амино-5-нафтол-7-сульфокислота 185
1 - Амино-8-нафтол-4-сульфокислота 185
2-Амино-5-нафтол- 7-сульфокислота 185
2-Амино-8-нафтол-6-сульфокислота 185
Аминонафтолсульфокислоты 184
Аминонафтолы 186
5-Амино-2-оксибензойная кислота — см.
5-Аминосалициловая кислота
(о-Аминопеларгоновая кислота 186
Аминопиридины 187
Аминопласты 188
Аминопропанон — см. Аминоацетон
а-Амино-р-(3,4-диоксифенил)-пропионо-
вая кислота — см. Диоксифенилаланин
6-Аминопурин — см. Аденин
Аминопурины 192
n-Аминосалициловая кислота — см.
ПАСК
5-Аминосалициловая кислота 193
Аминосахара — см. Гликозамнны
Аминоспирты 193
а-Аминотолуол — см. Бензиламин
а-Аминоуксусная кислота — см. Глицин
Аминоуксусный альдегид 193
И-Аминоундекановая кислота 194
o-Аминофениларсоновая кислота 194
п-Аминофениларсоновая кислота — см.
Арсаниловая кислота
Аминофенолы 194
(о-Аминоэнантовая кислота 195
Аминоянтарная кислота — см. Аспара-
гиновая кислота
Амины 195
Амита л 372
Амитол-патрий — см. Барбамил
Амматолы 206
Аммиак 198
— аналитич. определение 201
— молекула 198
— ркисление 76, 77
— получение 201
— применение 204
— техника безопасности 204
— физич. и химич. свойства 198
Аммиак жидкий 79, 199, 204
Аммиакаты 204
— удобрения 80
Аммиачная вода 205, 79
Аммиачная селитра — см. Аммоний, нит-
рат
Аммиачные удобрения 79
Аммонал водоустойчивый 206
Аммоналы 206
Аммониевые основания четвертичные 209
Аммоний NH't" 205
4
— бикарбонат 207
— бромид 207
— галогениды 206
— гидроокись — см. Аммиак
— иодид 207
— noil 200
— карбонаты 207
— нитрат 79, 207
— окись 200
— персульфат 208
— роданид 208
— соединения 209
— сульфат 79, 211
— тетрароданомеркурмат 211
— фосфаты 211
— фосфомолибдат 212
— фторид 207
— фторобериллат 424
— хлорид 79, 207
Аммоний карбаминовокислый 201
Аммониты 206, 562, 563
Аммонолиз 212
Аморфное состояние 212
Амперометрическое титрование 214
Амфи (приставка) 215
Амфотерицины 215
Амфотерность 216
Ана (приставка) 216
Анабазин 217
Анализ 217
--- абсорбционный 762
--- арбитражный 276
--- весовой 534
---вещественный 548
--- газовый 761; фракционированного
растворения метод 762
--- дисперсионный 11, 47
--- электровесовой 536
Аналитическая химия 217
Аналитические весы 538
Анальгин 220
Анаэробное брожение 465
Ангеликовая кислота 221
Ангидриды (кислот) 221
--- карбоновых кислот 221
Ангидритовый цемент 715
Ангидроальдитолы 223
Ангидрон 222
Ангидросахара 222 — см. также Трико-
заны
Анджели — Римини реакция 223
Андрогенная активность 225
Андрогенные гормоны 223, 997, 998
Андрогены 225
Д6-Андростен-Зр-ол-17-он — см. Дегид-
роэпиандростерон
Андростерон 225
Анестезин 225
Анетол 226
Анид — см. Полиамидные волокна
Анизидины 226
Анизол 227
Анизотропия 227
Анилидоуксусная кислота 227
Анилиды 228
Анилин 228, 247
Анилиновая точка 230
Аниониты — см. Иониты
Анионоактивные гещества 666, 683
Анионотропия 231
Анионы — см. Ионы
Аниотропные превращения—см. Анионо-
тропия
Анисовый альдегид 232, 1220
Анисовый спирт 232
Аннигиляция частиц и античастиц 258
Анодирование 233
A-номенклатура гетероциклических со-
единений 870
Аномеризация 234
Аномеры 234
Анонаин 273
Анса (приставка) 234
Ансерин 234
Ансольво кислоты 796
Анти (приставка) 235
D-Антиароза 958
Антибиотики 235
Антибиотики-макролиды 239
Антибиотики-полиены 241
Антибиотики-полипептиды 242
Антибиотики-трополоны 243
Антивитамины 245
Антивуалирующие вещества — см. Про-
тивовуалирующие вещества
Антигены 246
Антигиперон 259
Антигризутные взрывчатые вещества —
см. Предохранительные взрывчатые ве-
щества
Антидетонаторы моторных топлив 247
Антидетонационные свойства топлив —
см. Детонационная стойкость мотор-
ного топлива
Антидоты 247
Антикатализаторы 248
Антиметаболиты 248
Антинейтрон 259
Антинуклоны 259
Антиокислители 248
— пищевых продуктов 250
Антиокислитель древесно-смольный 251
Антиоксиданты — см. Антиокислители
Антипирены 251
Антипирин 252
Антиподы оптические 252, 93
Антиполимеризатор древесно-смольный
255
Антипротон 259
Антисептики 255
Антискорчинги — см. Вулканизации за-
медлители
Антитела 256
Антитромбозин — см. Дикумарин
Антифебрин — см. Ацетанилид
Антиферромагнетизм — см. Магнитные
свойства
Антифризы 257
Античастицы 258
Антоцианидины 259
Антоцианины 261
Антрагаллол 261
Антраэин 262
Антразо 262
Антрамины 262
Антраниловая кислота 263
Антрахас 264
Антрахинон 264
Антрахинон-а-арсоновая кислота — см.
Антрахас
Антрахннон-(1-а°о-4)-диметиланплин со-
лянокислый — см. Антразо
Антрахиноновые красители 265
Антрахиноновые кубовые красители 266
Антрахинонсульфокислоты 268
Антрацен 269
--- галогенпроизводные 270
--- сульфокислоты 270
Антраценовое масло 271
Антримиды 271
Антроловая кислота 271
Антролы 272
Апатит 272
Апираза 31
Апорфин 273
Апорфинозые алкалоиды 273
Апрессин 274
АпробарбитаЛ - см. Алюраг
1235
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1236
Апрофен 274
Арабаны 274
Арабиноза 275
Арабит 275
Арабитол — см. Арабит
Арамит 83
Арахидоновая кислота 709
Арахиновая кислота 709
Арахисовое масло 276
Арбитражный анализ 276
Арборициды — см. Гербициды
Арбузова реакция 276
Арбутин 277
Аргентометрия 278
Аргиназа 278
Аргинин 279, 180, 183
Аргиродит 858
Аргон 279
— выделение из воздуха 639
Ареколин 280
Арил 281
Ацилирование 281
Арктилит 1208
Армин 282
Арндта сплав 282
Арндта—Эйстерта реакция 282, 780
Ароматизация 284
--- нефтепродуктов 286
Ароматические диазосоединения 1088
Ароматические системы 287 — см. также
Азулены
Ароматические углеводороды 290
Ароматичность — см. Ароматические сис-
темы
Аррениуса уравнение 292
Арсаниловая кислота 292
Арсеназо 292
Арсенаты и арсениты 293
Арсениды металлов 294
Арсениты — см. Арсенаты и арсениты
Арсины 294
Арсониевые соединения 294
Асбест 295
•-- текстильный 295; физ.-механич. и
геометрия, характеристики 647
Асидол 296
Асидолмылонафт 296
Асимметрическая молекула 296
Асимметрический атом углерода 296
Асимметрический синтез 297
Аскаридол 298
Аскарит 299
L-Аскорбиновая кислота 299
Аскорбинометрия 300
Аспарагин 300
Аспарагиназа 166
Аспарагиновая кислота 301, 180
Аспирин 301
Ассимиляция (в геохимии) 842
Ассоциация ионов 301
Астатин 302
Астрохимия — см. Космохимия
Асфальт 303
Асфальтены 304, 439
Асфальтиты 303
Асфальтогеновые кислоты 304
Атактические полимеры 304
Атебрин — см. Акрихин
Атизин 85
Атмосфера 305, 852
Атмофильные элементы — см. Геохими-
ческая классификация элементов
Атом 306
— заряд ядра 307
— квантовая теория А. 308
— магнитные свойства 323
— масса 308
— масса ядра 307
— периодичность свойств 316, 324
— развитие учения об А. 306
— размеры 317
— состояния возбужденные 309, 310,
318
— состояния стационарные 308, 309, 310
— строение 307
— теория атома водорода и водородо-
подобных ионов 311
— теория сложных атомов 315
— уровни энергии, вырождение 314
— электрич. свойства 324
Атомаль — см. Проявляющие вещества
Атомистическая гипотеза 306
Атомная поляризация 1132, 1138
Атомная энергия 324
Атомно-молекулярное учение 306
Атомные спектры 324
Атомный вес 328
— единицы измерения 331
— значения на 1960 г. 332
— определение, методы 329
— физич. шкала 328
— химич. шкала 328
— эволюция понятия 328
Атомный диполь 1132
Атомный номер — см. Порядковый номер
Атомный объем 333
Атомы горячие 1002
--- отдачи 333, 94, 1002
Атофан 333
Атропин 333, 248
Атропоизомерия 334
Ауксины 335
Аурамин 1096, 1097
Аурантин 108
Ауранция 817
Ауреомицин — см. Хлортетрациклин
Ауринтрикарбоновая кислота, аммоние-
вая соль — см. Алюминон '
Аутогезия 29
Аутокаталитические реакции 335
Аутолиз 336
Аутооксидация 337
Аутотрофные организмы 436
Афиллин 337
Ахродекстрины 1058
Аценафтен 338
Ацетазоламид — см. Диакарб
Ацетали и кетали 338
Ацетальдегид 339
Ацетальдоль 142
Ацетальфосфатиды — см. Фосфатиды
Ацетамид 340
— физич. свойства 167
Ацетанилид 340
Ацетатное волокно 341, 647
Ацетатные красители 62, 342
Ацетаты 342, 351
Ацетил хлористый 343
Ацетила гидроперекись 343
Ацетила перекись 344
2-Ацетиламино-1,3,4 тиадиазол-5-сульфо-
намид — см. Диакарб
п-Ацетиланизол 344, 1220
N-Ацетилантраниловая кислота 264
Апетилацетон 344
Ацетилгалогенсахара — см. о-Ацетил-
гликозилгалиды
о-Ацетилгликозилгалиды 345
Ацетилен 346
Ацетилендикарбоновая кислота 350
Ацетиленовые углеводороды — см. Не-
предельные углеводороды
Ацетилирование — см. Ацилирование
О-Ацетилсалициловая кислота — ем. Ас-
пирин
Ацетилхолинхлорид 351
Ацетилцеллюлоза 351
Ацетогалогенозы — см. о-Ацетилглико-
зилгалиды
Ацетогалогенсахара — см. о-Ацетилгли-
козилгалиды
Ацетол 339
Ацетон 353
Ацетониламин — см. Аминоацетон
Ацетонитрил 355
Ацетоновое брожение 354
Ацетоновые тела 355
у-Ацетопропиловый спирт 356
Ацетоуксусная кислота — см. Альдегидо-
кислоты и кетокислоты
Ацетоуксусный эфир 356
Ацетофенон 358
Ацидиметрия — см. Алкалиметрия и
ацидиметрия
Ацидол 428
Ациклические соединения 359
Ацилазы — см. Аминоацилазы
Ацилирование 360, 780
Ацилоины 361, 407
Аш-кислота 185
Аэробное брожение 465
Аэрогели 827
Аэрозоли 361
Аяцин 86
Б
Базовые бензины 401
Байерит 153
Бакелит — см. Смолы феноло-алъдегидные
Бактериостатики 365 1
Бактерициды 365
Баллиститы 367
Бальзам канадский 369
--- копайский 369
— - перуанский 369
--- пихтовый 369
--- толуанский 369
--- Шостаковского 368, 369
Бальзамы 368
Барбамил 370
Барбитал — см. Веронал
Барбитураты 370
Барбитуровая кислота 371
Барбье — Виланда расщепление 371
Барда метод взвешивания 545
Барий 373
— азид 375
— аналитич. определение 374
— ацетат 375
— бромат 469
— гидрид 374
— гидроокись 374, 376
— карбид 374
— карбонат 376
— манганат 375
— нитрат 376
— нитрид 374
— окись 374
— перекись 374
— перхлорат 375
— платиноцианат 377
— полисульфиды 374
— получение 375
— применение 375
— сульфат 377
— сульфид 374
— титанат 375
— физич. и химич. свойства 373
— фосфид 374
— фторид 375
— хлорат 377
— хлорид 378
— хромат 375
— цианид 374
— цирконат 375
Барий азотнокислый — см. Барий, нит-
рат
--- платиносинеродистый — см. Ба-
рий, платиноцианат
--- сернокислый — см. Барий, сульфат
--- углекислый — см. Барий, карбонат
--- хлористый — см. Барий, хлорид
--- хлорноватокислый — см. Барий,
хлорат
Барит 373
Баритовая вода 376
Барн 378
Барта реакция 378
Батохромный эффект 379
Бацитрацины 380
Бачинского формула 719
Бегеновая кислота 709
Бездымный порох 563 — см. также По-
роха
Бейльштейна проба 783
Бекмана перегруппировка 381
Белила — см. Пигменты белые
Белки 382
— выделение 387
— гормоны 996
— денатурация 697
— значение 388
— классификация 387
— количественное определение 386
— осаждение 70
— синтез 387
— структура 383
— физич. и химич. свойства 385
— элементарный состав 386
Белки дыхательные 838
Белковый обмен — см. Обмен веществ
Бемит 153
Бензальацетофенон 389
Бензальдегид 389
Бензальдиацетат 390
Бензальхлорид — см. Бензилидснхлорид
Бензамид 390, 167
Еензамон 390
Бензанилид 390, 409
Бензантрен 391
Бензантрон 391
Бензгидрол 391
— 3-диметиламиноэтиловый эфир, хлор-
гидрат — см. Димедрол
1,3-Бензидиазол — см. Бензимидазол
Бензидин 392
Бензидиновая бумага 484
1237
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1238
Бензидиновая перегруппировка 392, 887
Бензил 393
--- бромистый 393
--- хлористый 393
Бензилакрилат 88
Бензиламин 394
Бензилацетон 394
2-Бензилбензимидазол, хлоргидрат — см.
Дибазол
Бензилбутиролактон 499
а-Бензилдиоксим 394
Бепзилиденацетон 395
Бензилиденхлорид 395
Бензиловый спирт 395
Бензилпенициллин 396
—N, N'-дибензилэтплсндиаминовая
соль — см. Бициллин
Бензилцеллюлоза 397
Бензильная перегруппировка 398
Бензимидазол 399
Бензиминазол — см. Бензимидазол
Бензины 399
•— авиационные 401
--- автомобильные 400
--- базовые 401
Бензины-растворители 401
Бензины-экстрагенты 401
— получение 402
Бензоила перекись 404
Бензоиламиноуксусная кислота — см.
Гиппуровая кислота
Бснзоилацетон 405
Бензоил-Аш-кислота 405
N-Бензоилгексаметиленимин — см. Гек-
саметиленбензамид
Бензоилгликокол — см. Гиппуровая кис-
лота
Бензоилглицин — см. Гиппуровая кислота
БенЗоилуксусные эфиры 406
Бензоилхлорид 407
Бензоин 407
Бензоиновая конденсация 407
а-Бензоиноксим 409
Бензойная кислота 409
— азид 56
— амид 390, 409
— ангидрид 409. 410
— анилид 309, 409
— гидразид 882
— метилбензоат 409
— нитрил 409
— перекись 409
— фенилбензоат 409
— хлорангидрид 407, 409
— этилбензоат 409
Бензокаин — см. Анестезин
Бензол 410, 247, 287
---неорганический — см. Боразол
Бензолгексакарбоновая кислота 413
Бензолин 413
Бензолпентакарбоновая кислота 413
Бензолполикарбоновыс кислоты 413
Бензолсульфиновая кислота 414
Бензолсульфокислота 415
Бензолсульфохлорид 415
Бензопиразин — см. Хиноксалин
Бензопурпурин 4Б 416
Вензотиофен 416, 870
1, 2, З-Бензотриазол 416
Бензотрихлорид 417
Бензофенон 417
Бензофуран 417, 870
Бензохинон 418
Бензпирен 419
Бензпиррол 870, 1220
Бензтиазол 419, 870
Берберин 420
Берилл 421
Бериллиевые бронзы 424
Бериллий 421
— аналитич. определение 423
— бромид 426
— галогениды 425
— гидрид 422
— гидроокись 426
— иодид 426
— карбонат основной 423
— нитрат 423
— окись 426
— оксиацетат 423
— получение 423
— применение 424
— сплавы 424
— сульфат 422
— физич. и химич. свойства 422
— фторид 425
— хлорид 425
Бериллийорганические соединения 42 4
Бериллон II 426
Беркелий 426, 104
Бертинирование — см. Полукоксование и
термическая переработка топлива
Бертло — Томсена принцип 426
Бертоллиды — см. Дальтониды и бер-
толлиды
Бесстружковый метод анализа 427
Бетазин 427
Бетаины 428
Бета-лучи 428
Бета-распад 429
Бетон 429
— коррозия 430
Бешана восстановление 431
Бешана реакция 431
Бигумаль 432
Бикфордов шнур — см. Огнепроводный
шнур
Билирубин 432
Бимолекулярные реакции 432
Бинарные системы — см. Двойные систе-
мы
Биогеохимия 433, 437
Биозиды 956
Биозы — см. Дисахариды
Биокатализаторы — см. Ферменты
Биолиты 434
Биомицин — см. Хлортетрациклин
Биосфера 433, 845, 853
Биотины 435
Биохимические газы 769
Биохимия 435
Биполярные ионы — см. Биполярные со-
единения
Биполярные соединения 438
Бисмит 589
Бисмутинит 589
Бисмутит 589
Бис-(этилксантоген)-трисульфид 1075
Битумы 438
--- буроугольные 441
--- искусственные 439
--- твердого топлива 441
--- торфяные 441
Биурет 443
Биуретовая реакция 444
Бифенохинон — см. 4,4'-Дифенохинон
Бициллин 444
Бишлера реакция 445
Бишлера — Напиральского реакция 446
Блана реакция 447
Бланфикс — см. Пигменты белые
Блок-сополиконденсация 448
Блок-сополимеризация 448
Бодру — Чичибабина синтез 477
Бойля — Мариотта закон 449
Боксит 147
Болдин 273
Болидена соли 255
Болотный газ 449
Больцмана закон распределения — см.
Максвелла-Больцмана закон распреде-
ления
Больцмана постоянная 449
Бона—Шмидта реакция 450
Бор 451
— аналитич. определение 453
— бороводороды 459
— бромид 452
— гидриды — см. Бороводороды
— иодид 452
— карбид 452
— нитрид 452
— получение 453
— применение 453
— сульфид 452
— физич. и химич. свойства 451
— фторид 452
— хлорид 452
Бора теория атома 306, 310, 312
Боразол 289, 452, 461
Бораны — см. Бороводороды
Бораты 451, 454
---термические 459
Бориды 454
Борирование стали 453
Борная кислота 456, 451
Борнеол 457
Борнивал 518
Борниловый спирт — см. Борнеол
Борнилхлорид 457
Борнодатолитовое удобрение 459
Борноэтиловый эфир 458
Борные удобрения 458
Борный ангидрид 451
Бороводороды 459
Бородвойной суперфосфат 459
Борорганические соединения 460
Боросуперфосфат 459
Борофтористоводородная кислота 452
Борше синтез 462
Брауна реакция 462
Брауна ряды 463
Бредта правило 463
Бризантность 563
Бризантные взрывчатые вещества 561
Бриллиантовый зеленый 464
Брожение 464
--- анаэробное 465
--- ацетоновое 354
--- аэробное 465
Бром 467
— аналитич. определение 469
— двуокись 468
— закись 468
— окислы 468
— определение 782
— получение 470
— применение 470
— техника безопасности 4 70
— физич. и химич. свойства 467
Бром пятифтористый 468
---’трехфтористый 468
Бромазид 69, 469
Броматометрия 470
Броматы 469
Бромацетофенон 471
Бромбензилцианид 471
Бромбензол 471
Бромдиэтилацетилмочевина — см. Ада-
лин
Бромиды 472
Бромистая сера 468
Бромистоводородная кислотч 472
Бромистый водород 472
Бромистый метил 472
Бромистый метилен 473
Бромистый тионил 468
Бромистый этил 473
Бромметан — см. Бромистый метил
Бромметилфенилкетон — см. Бромаце-
тофенон
Бромная вода 468
Бромноватая кислота 469
Бромноватистая кислота 469
Бромное число 473
Бромонйевые соединения — см. Галоге-
нониевые соединения
Бромооксин — см. Дибромокснн
Бромосеребряная бумага фотографиче-
ская 482
Бромоформ 473
Бромстирол 474
ю-Бромтолуол — см. Бензил бромистый
Бромурал 474
Бромфенолы 475
Бромциан — см. Галогеноцианы
Бромэтан — см. Бромистый этил
Бронза — см. Меди сплавы
Бронзы бериллиевые 424
--- вольфрамовые 653
Броуновское движение 475
Бруцин 476
Брэгга — Вульфа закон 1170
Буво синтез 477
Буво — Блана восстановление 477
Бугера закон поглощения света — см.
Поглощение света
Бульбокапнин 273
Бумага 478
— производство 479
— физич. свойства 478
Бумага бензидиновая 483
--- иодкрахмальная 483
--- свинцовая 483
Бумага фотографическая 481
--- бромосеребряная 482
---иодохлоробромосеребряная 482
--- с видимым печатанием 483
--- специальная 483
---хлоробромосеребряная 482
--- хлоросеребряная 482
•—- цветная 483
Бумаги реактивные 483
Бумофенолит — см. Гетинакс
Буна-Эн — см. Бутадиен-нитрильныЙ
каучук
Буна-Эс — см. Бутадиен-стирольный ка-
учук
Бунзена — Роско закон — см. Фотохимия
Бура 484, 451
Буркело метод 957
1239
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1240
Буроугольный деготь 1040
Бутадиен-1,3 485
Бутадиен-метилвинилпиридиновый кау-
чук 486
Бутадиен-нитрильный каучук 487
Бутадиен-стирольный .каучук 489
Бутадиин — см. Диацетилен
«Бутадион» 491
Бутазолидин — см. Бутадион
Бутандикарбоновая-1,4 кислота — см.
Адипиновая кислота
Бутандиолы — см. Бутиленгликоли
2,3-Бутандион — см. Диацетил
Бутанолы 496
Бутаны 491
Бутенин — см. Винилацетилен
Бутены — см. Бутилены
Бутилакрилат 88
n-Бутиламинобензойная кислота, димет ил-
аминозтилового эфира хлоргидрат —
см. Дикаин
Бутилацетат — см. Бутилового спирта
эфиры
5-(втор-Вутил)-5- (В-бромаллил)-барбиту-
ровая кислота, Na-соль — см. Пернок-
. тон
Бутилена окись 492
Бутиленгликоли 492
Бутилены 493
Бутилизовалерианат — см. Бутилового
спирта эфиры
Бутилкаучук 494
Бутилнафталины 126
Бутилового спирта эфиры 495
Бутиловые спирты 496
Бутилпропионат 495
4-втор-Бутилфенол 129
4-трет-Бутилфенол 129
Бутилфенолы 129
Бутилформиат — см. Бутилового спирта
эфиры
n-mpem-БутилцикЛогексилацетат 497
Бутийдиолы 497
Бутины 350
Бутиральдоль 142
Бутиролактон 498
Буферные растворы 499
Буфогенины 500
Буфотоксины 501
Бухерера реакция 501
Бытовой газ 736
Бэр 1196
Бюргерса вектор дислокации 1142
В
Вагнера реакция 503
Вагнера—Меервейна перегруппировка —
см. Камфеновые перегруппировки
Ваготонин 996
Вазелин 503
Вазопрессин 504, 996
Вакуум 504
— измерение 508
Вакуумирование 504
Валентность 510, 317
— квантово-механич. природа 510
— направленность 514
— особые случаи проявления 515
Валентность атомов в возбужденных со-
стояниях 512
Валентные углы 516
Валерианаты 519
Валериановые кислоты 517
— н-амиловый эфир 518
— бензиловый эфир 518
— н-бутиловый эфир 518
— изоамиловый эфир 518
— линалиловый эфир 518
— ментоловый эфир 519
— метиловый эфир 518
— н-пропиловый эфир 518
— р-фенилзтиловый эфир 518
— а-цитрбнеллиловый эфир 518
— этиловый эфир 518
н-Валериановый альдегид 519
Валерилены 350
Валидол 519
Вализан 518
Валин 519, 180
Валлаха перегруппировка 520, 63
Валлаха реакция 521
Вальденовскоэ обращение 522, 790
Ванадатометрия 523
Ванадаты 523
Ванадиевые кислоты 526
Ванадий 524
— аналитич. определение 526
— галогениды 528
— гидриды 525
— двуокись 526, 529
— карбид 525
— моноокись 529
— нитрид 525
— окислы 529
— оксигалогениды 528
— получение 527
— применение 527
— пятиокись 525, 529
— сульфид 526
— трехокись 529
— физич. и химич. свойства 525
Ванадил 526, 529
Ванадинит 524
Ванадиты 526
Ван-ден-Берга реакция 432
Ван-дер-Ваальса уравнение 530
Ван-Дер-Ваальсовы силы — см. Межмо-
лекулярное взаимодействие
Ванилаль 531
Ванилин 531
Ванкомицин 532
Ван-Слайка метод 532, 183
Ванг-Гоффа коэффициент 533
Вант-Гоффа правило 533
Вентури аппараты 22
Вератровый альдегид 533
Вератрол 534
Вернин — си. Гуанозин
Веронал 534, 372
Вес насыпной 1062
«Веселящий газ» 70
Весовая бюретка 534
Весовой анализ 534
Вестона иориальный элемент 537
Весы лабораторные 538
--- аналитические 538
--- квадрантные 543
---микровесы 539
--- полумикроаналитические 538
--- пробирные 538
--- электронные 546
Ветиверилацетат 548
Ветиверкетон 548
Ветиверовое масло 547
Ветиверовый спирт 548
Ветхин 86
Вещественный анализ 548
Взаимные солевые системы 549
Взаимозаместимости закон (в фотохи-
мии) 549
Взвешивание 544
Взрыв 550
— возникновение 550
— работа 552
— разрушающее действие в твердых
средах 553
— экспериментальные исследования в
области В. 555
Взрываемости концентрационные преде-
лы 557
Взрываемости температура 557
Взрывоопасность водородсодержащих
смесей 621
--- ныли промышленной 558
Взрывоопасные вещества 555
Взрывчатого превращения скорость 560
Взрывчатые вещества 559
— классификация 561
— стойкость 562
— формы взрывчатого превращения 560
— чувствительность 562
— энергия взрывчатого превращения
и действие взрыва 563
Взрывчатые вещества бризантные 561
--- инициирующие 561
--- метательные 561
--- нитроглицериновые 206
Викасол 599
Вильгеродта реакция 564
Вильштеттера метод гидрогенизации —
си. Гидрогенизация и дегидрогениза-
ция каталитические
Вильямсона синтезы 565
Вина эффект 567
Винилацетат 567, 570
Винилацетилен 567, 349
Винилбензоат 570
Винил-изо-бутиловый эфир 570
Винил-н-бутиловый эфир 570
Впнилиденхлорид 568
I Винилин — см. Бальзам Шостаковского
Винилирование 568
N-Винилкарбазол 569
Винилметиловый эфир 570
Винил-н-пропиловый эфир 570
Виниловые-эфиры 569
Виниловый спирт 571
Винилогия 571
N-Винил-а-пирролидон — см. N-Винил-
пирролидон
N-ВиниЛпирролидон 571
Винилстеарат 570
Винилфениловый эфир 570
Винилхлорид 573
Винилэтиловый эфир 570
Винилзтинилкарбинолы 574
Винные кислоты 575
Вино 576
--- виноградное 576
--- плодово-ягодное 578
Виноградная кислота — см. Винные кис-
лоты
Висбрекинг нефтепродуктов 1044
Вискоза 579
— гамиа-число 579
— индекс зрелости 580
Вискозиметрия 581
Вискозиметры 581, 582
Вискозные волокна 585, 647
Вискозные неволокнистые изделия 588
Вискозный прядильный раствор— си.
Вискоза
Висмут 589
— бромид 591
— галогениды 591
— иодид 591
— карбонат основной 590
— нитраты 592
— окислы 592
— определение 590
— открытие 590
— получение 590
— прииенение 591
— пятиокись 592
— сплавы 593
— сульфат 590
— трехокись 590, 592
— трисульфид 590
— физич. и химич. свойства 589
— фторид 591
— хлорид 591
— четырехокись 592
Висмутиды 593
Висмутин — си. Висмутистый водород
Висиутистый водород 590
Висмуторганические соединения 593
Витамин А — см. Ретинол
Витамин В, — см. Тиамин
Витамин В2 — см. Рибофлавин
Витамин В, — см. Пиридоксин (пиридок-
сол)
Витамин В12 — см. Цианкобаламин
Витамин В13 — см. Оротовая кислота
Витамин В15 — см. Пангамовая кислота
Витамин Вс — см. Фолиевая кислота
Витамин Вт — см. Карнитин
Витамин С — см. Аскорбиновая кислота
Витамин Н — см. Биотины
Витаиин F — см. Незаменимые ненасы-
щенные жирные кислоты
Витамин Р 594
Витамин РР — см. Никотиновая кислота
Витамины 595
Витамины D — см. Эргокальциферол и
Холекалъциферол
Витамины Е — см. Токоферолы
Витамины К 598
Вителлин 599
Витерит 373
Виттига реакция 599
Вицинальные соединения 601
Внутреннее вращение — см. Изомерия
Внутреннее трение — см. Вязкость
Внутренний электролиз 601
Внутренняя энергия 602
Внутрикомплексные соединения 603
Внутрикоиплексные элементоорганиче-
ские полимеры 703, 706
Вод морских химия 940
Вод поверхностных химия 940
Вод подземных химия 940
Вода 605
— дегазация 618
— дезодорация 613
— жесткость 611
— ионное произведение 628
1241
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1242
— катионирование 617
— молекула 606
— обезжелезивание 613
— обеззараживание 613
— обескремнивание 617
— обессоливание 618
— обесцвечивание 613
— осветление 613, 617
— растворяющая способность 608, 610
— структура 606
— тройная точка 17
— умягчение 617
— физич. свойства 606
— химич. свойства 609
— цветность 611
— щелочность 611
Вода конституционная 893
--- кристаллизационная 892
---минерализованная 611
--- очищенная, получение 610
--- питьевая 612
--- пресная 611
'-- природная 611
--- техническая 613
--- тритиевая 616
--- тяжелая 614; применение 616
--- тяжелокислородная 616
Водоотталкивающие покрытия — см. Гид-
рофобные покрытия
Водоподготовка 617
Водород 619
— аналитич. определения 623
— взрывоопасность 621
— изотопы 619
— молекула 619
— определение см. Углерода и водорода
определение
— пара-орто-превращени? 623
— перекись 624
--- получение 626
—• применение 626
— получение 622, 755
— применение 623
— теория атома 311, 619
— физич. и химич. свойства 620
Водород активный (определение) 95
--- атомарный 621
—- бромистый 4 72
Водородная связь 626
Водородный показатель 628
Водородный электрод 629
Воды минеральные 630
--- сточные 630; очистка 632
Водяной газ — см. Газификация твер-
дых топлив
Возбужденные состояния атома 309, 310,
318
Воздух — см. Атмосфера, Воздуха раз-
деление
Воздуха анализ 633
Воздуха разделение 635
Воздушные вяжущие материалы 713
Воздушный газ — см. Газификация твер-
дых топлив
Возникающие реагенты 642
Возраст геологический абсолютный 643
— определение 64 3, 644
Волновая функция 645
Волокна натуральные — см. Волокна
текстильные
Волокна текстильные 645
— классификация 646
— относительная прочность 648
>— разрывная длина 648
— разрывная нагрузка 648
— разрывное удлинение 648
Волокна химические — см. Волокна тек-
стильные, Вискозные волокна, Ацетат-
ное волокно, Полиамидные волокна
и др.
---штапельные 645
--- элементарные 645
Волокно перхлорвиниловое штапельное
647
Вольмана соли 256
Вольтамперное титрование — см. Ампе-
рометрическое титрование
Вольтамперометрия 651
Вольфа — Кижнера реакция — см. Киж-
нера — Вольфа реакция
Вольфрам 651
— аналитич. определение 653
— бориды 653
— галогениды 655
— двуокись 655
— динитрид 653
— карбиды 653
— кислоты 655
— окислы 655
— оксихлориды 655
— получение 654
— применение 654
— силициды 653
— сплавы 656
— сульфиды 658
— трехокись 656
— физич. и химич. свойства 652
— фосфиды 653
— хлориды 655
Вольфраматы 658
Вольфрам — железо 656
Вольфрамит 652
Вольфрам — медь 657
Вольфрам — медь — никель 657
Вольфрам —молибден 657
Вольфрамовая кислота 656
Вольфрамовые бронзы 653
Вольфрам — рений 657
Вольфрам — серебро 657
Волюмометры 659
Воля — Циглера реакция 659
Ворвань 1215
Воск канделильский 661, 662
--- карнаубский 661, 662
--- льняной 661, 662
--- монтанный 661, 662
--- пальмовый 661, 662
---пчелиный 661, 662
--- шерстяной 662
Воски 661
--- животные 662
--- ископаемые 662'
--- растительные 662
Воспламенение 990
Воспламенения точка — см. Вспышки
температура
Восстановление 663
--- органических соединений 663
Вращение плоскости поляризации — см.
Полярим етрия
Вревского законы 664
Вспомогательные вещества (в текстиль-
ной пром-сти) 665
Вспышки температура 666
Второй закон термодинамики 668, 17
Вулканизации замедлители 671
--- ускорители 671, 673
Вулканизация 673
--- горячая 674
--- перекисями 675
--- радиационная 675
--- холодная 674
Вулканические газы 769
Выветривание 843
Выпаривание 677
Выпарные аппараты 678
Выравнивающие вещества (в текстильной
пром-сти) 683
Высокие давления 684
— биологич. эффекты 691
— физич. эффекты 686
— химич. эффекты 685
— электрич. эффекты 690
Высоких давлений техника 691
Высокомолекулярные соединения 696
— гидролиз 708
— получение 699
— применение 699
— свойства и важнейшие характеристики
698
Высокомолекулярные соединения
--- неорганические 700
--- природные 698
---разветвленные 696
--- синтетические 698
--- сшитые 696
--- элементоорганические 703, 697
Высшие жирные кислоты 709
Высшие жирные спирты 711
Выход по току (ВТ) 712
Вюрца реакция 712
Вяжущие материалы 713
--- воздушные 713
--- гидравлические 714
--- гипсовые 714
--- минеральные 713
Вязкого течения энергия активапии 716
Вязкости коэффициент 717
Вязкость 716; температурный коэффи-
циент 723
---динамическая 718
---кинематическая 718
— нефтепродуктов 722
•— относительная 718
—- паров 721
--- полимеров 724
— структурная 724
--- удельная 718
Г
Габриеля реакция 727
Гадолиний 728
Газ бытовой 736
--- генераторный 731, 733, 734
---коксовый 728; разделение 754
--- полуводяной 735
--- технологический 736
Газификация древесины — см. Газифи-
кация твердых топлив
--- жидких топлив 729
--- под давлением — см. Газификация
твердых топлив
--- пылевидных топлив 738
--- твердых топлив 731
--- топлив подземная 739
Газоанализаторы 763, 764
Газобетон 429
Газов осушка 741
— абсорбционные способы 741
— адсорбционные способы 742
— физич. способы 743
Газов очистка 743
--- механическая 743
--- химическая 748
--- электрическая 748
Газов разделение 753, 771
Газовая постоянная 757
Газоволюметр 659
Газовые потоки, фильтрация 745
Газовые растворы 757
Газовый анализ 761
— фракционированного растворения ме-
тод 762
Газовый фактор 768
Газогенераторы — см. Газификация твер-
дых топлив
Газойль — см. Дизельное топливо
Газокристаллы 213
Газопапопроницаемость полимеров —
см. Диффузия
Газопроницаемость полимеров — см.
Диффузия
Газы, сжатие 694
— электропроводность 1184
Газы атмосферы 305
--- биохимические 769
--- вулканические 769
--- газоконденсатных месторождений
770
--- метаморфические 769
--- нефтепереработки 766
--- нефтяные попутные 767
--- природные 769
---природные горючие 770
--- химического происхождения 769
Галактаны 774, 835
Галактит — см. Дульцит
Галактоза 775, 821
D-Галактозамин 954
Галактокаролоза 774
D-Галактоновая кислота 824
Галактуроновая кислота — см. Гексу-
роновые кислоты
Галалит — см. Пластики белковые
Галлий 775
— аналитич. определение 777
— гидроокись 776
— окислы 776
— получение 777
— применение 778
— сульфат 776
— физич. и химич. свойства 776
— хлорид 776
Галлит 775
Галловая кислота 778
Галлуазит 968
Галогеназиды 69
Галогенальдегиды и галогенокетоны 778
Галогенаигидриды карбоновых кислот
779
Галогенарсины 780
Галогеиацетоны 781
Галогенводороды 621, 795
Галогенирование органических соеди-
нений 781
Галогенокетоны — см. Галогенальдегиды
и галогенокетоны
1243
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1244
Галогенов определение 782
Галогенозамещенные углеводороды 784
— физич. и химич. свойства 787
Галогенокислоты 789
Галогенониевые соединения 790
Галогеноспирты 793
Галогеноцианы 793
Галогены 794
— определение 782
— физич. свойства 795
— химич. свойства 795
Галогенэфиры 796
Галоиды — см. Галогены
Галоформная реакция 779, 924
Галохромия 796
Гальванические элементы — см. Хими-
ческие источники тока
Гальванометрическое титрование — см.
Амперометрическое титрование
Гальванопластика — см. Гальванотехника
Гальваностегия — см. Гальванотехника
Гальванотехника 798
Гамамелоза 821
Гамма-кванты 803
Гамма-кислота 185
Гамма-лучи 803
Гаммета уравнение 804
Ганглиостат — см. Гексоний
Гантча синтез 805
Гариин 86
Гармин 806
Гаттермана реакция 806
Гаттермана — Коха реакция 807
Гаусса метод взвешивания 545
Гафний 808
— аналитич. определение 811
— гидроокись 810
— окись 809
— получение 811
— применение 812
— тетрафторид 810
— тетрахлорид 810
— сульфат 810
— физич. и химич. свойства 809
— фосфаты 810
— хлорокись 810
Гваякол 812
Гей-Люссака законы 812
Гексабарбитон-натрий — см. Гексенал
Гексадецилакрилат 88
Гексадиен-1,5-ин-З — см. Дивинилацети-
лен
Гексаметиленбензамид 813
1,6-Гексаметилендиамин 813
Гексаметилендиизоцианат — см. Изоциа-
наты
Гексаметилентетрамин 814
Гексаметоний — см. Гексоний
Гексамид — см. Гексаметиленбензамид
Гексамидин 815
Гексамин — см. Гексаметилентетрамин
Гексан 815
Гександиолы 816
Гексанитродифениламин 817
Гексанол-1 820
Гексаоксибензол 817
Гексаровые кислоты 822, 825
Гексафенол — см. Гексаоксибензол
Гексахлоран 817
Гексахлорбензол 818
Гексахлордиметилкарбонат 819
Гексахлорциклогексан — см. Гексахлоран
Гексахлорэтан 819
Гексенал 819
Гексил — см. Гексанитродифениламин
Гексилакрилат 88
w-Гексилбутиролактон 499
Гексиловые спирты 820
Гексилрезорцин 820
Гексит — см. Гексанитродифениламин
Гексобарбитал — см. Эвипан
Гексоген 820, 562, 563
Гексозаны 835
Гексозиды 956
Гексозы 821
Гексокиназа 823
Гексоний 823
Гексоновые кислоты 823
Гексоновые основания 824
Гексоран — см. Гексахлорэтан
Гексуроновые кислоты 824
Гели 826
Гелий 827; получение 756, 832; приме-
нение 832
--- газообразный 828
--- жидкий 832
Гелиотропин 832, 1220
Гелио химия — см. Космохимия
Гелля — Фольгарда — Зелинского реак-
ция 833, 789
Гельфериха метод 957
Гематин 837
Гемиглобин — см. Метгемоглобин
Гемимеллитовая кислота 413
Гемин — см. Гемоглобины
Геминальные соединения 834
Гемицеллюлозы 834
Гемоглобины 835
Гемоспоридин 839
Гемы 836
Генераторный газ — см. Газификация
твердых топлив
Генетические ряды 984
Генины — см. Глюкозиды сердечные
Генри закон 839
Геологический возраст абсолютный 643,
644
Геологическое летоисчисление абсолют-
ное 645
Геосфера 846
Геохимическая классификация элементов
Геохимические методы поисков полезных
ископаемых 840
Геохимические процессы .842, 847
Геохимический ландшафт 844
Геохимия 844
--- прикладная 853
--- элементов 853
Гепарин 854
Гепаринаты 854
Гепариноиды 855
Гептадекановая кислота 709
Гепталин 290
Гептан 855
н-Гептандикарбоновая-1,7-кислота — см.
Азелаиновая кислота
Гептанол — см. Гептиловый спирт
Гептиловый спирт 855
Гептинкарбоновая кислота 855
— изоамиловый эфир 856
— изобутиловый эфир 856
— изопропиловый эфир 856
— метиловый эфир 856
— пропиловый эфир 856
— этиловый эфир 856
Гераниевая кислота 856
Геранилбутиролактон 499
Гераниол 856, 1220
Гербициды 857
Германаты 859
Германий 858
— аналитич. определение 861
— бромид 860
— гетерокислоты 861
— двуокись 860
— иодид 860
— нитрид 861
— окись 859
— оксихлорид 860
— получение 862
— применение 862
--- Г. как полупроводник 862
— сульфат 860
— сульфид 860
— физич. и химич. свойства 858
— фосфид 860
— фторид 860
Германий четыреххлористый 860
Германийорганические соединения 861
Германит 858
Германоводороды 860
Гесперидии 594
Гесса закон 863
Гетероазеотропные смеси 864
Гетероауксин 867
Гетерогенное горение 993
Гетерогенное равновесие — см. Равнове-
сие химическое
Гетерогенные системы 867
Гетеродиффузия 1175
Гетерозиды — см. Гликозиды
Гетеролитические реакции 868
Гетерополивольфраматы 658
Гетерополисоединения 869
Гетеротрофные организмы 436
Гетероцепные высокомолекулярные со-
единения — см. Высокомолекулярные
соединения
Гетероциклические аминокислоты 180
Гетероциклические соединения 870
-А-номенклатура 870
Гетинакс 873
Гетразан — см. Дитразин
Гёша аномальная реакция 874
Гёша синтез 873
Гиалобиуроновая кислота 875
Гиалуронидазы 874
Гиалуроновые кислоты 875
Гиббереллины 875
Гиббса распределение 876
Гиббса уравнение адсорбции 877
Гиббса — Гельмгольца уравнения 878
Гиббса — Дальтона закон 761
Гиббса — Дюгема уравнение 879
Гибкость цепных молекул 879
Гибридизация состояний 323, 515
Гибридизм (в геохимич. процессах) 842
Гигриновая кислота 880
Гидантоины 182
Гиднокарповая кислота 880
Гидравлические вяжущие материалы 714
Гидравлические жидкости 881
Гидразиды карбоновых кислот 881
Гидразин 882
--- сернокислый 883
--- солянокислый 883
Гидразина производные 883
1-Гидразинфталазина хлоргидрат — см.
Апрессин
Гидразобензол 885
Гидразоны 885
Гидразосоединения 887
Гидракриловая кислота 887
Гидраргиллит 153
Гидрастин 887
Гидрастинин 888
— хлоргидрат 888
Гидратационное число 889
Гидратация 888
— степень 891
— теплота 889
Гидратация, вторичная 891
--- высокомолекулярных веществ 895
--- гетерогенная 896
--- гомогенная 896
--- каталитическая 896
--- кислотная 896
--- молекул в растворах 891
--- окислов 893
--- органических соединений 894
--- первичная 891
--- твердых гидратов 892
--- электролитов 888
Гидратация и дегидратация каталитиче-
ские 895
Гидратная теория растворов 888
Гидратроповый альдегид 898
Гидратцеллюлоза 898, 586
Гидраты 892
Гидриды 899
--- бора — см. Бороводороды
--- двойные 902
--- летучие 901
--- металлообразные 900
--- переходные 900
--- полимерные 901
--- солеобразные 899
Гидриндан 902
Гидрирование органических соединений
Гидроборацит 451
Гидрогенизация деструктивная 903
---жиров 904
--- и дегидрогенизация"каталитические
907
Гидрогенолиз 909
--- органических соединений 663
Гидродимеризация 911
Гидрокоричный альдегид 912
Гидрокоричный спирт 912
Гидроксамовые кислоты 912, 915
Гидроксиламин 913
— неорганические соли 914
— производные 915
— сульфат 914
— сульфат кислый 914
— хлорид 914
Гидроксицитронеллаль 916, 1220
Гидроксоалюминаты 147
Гидроксоний 916
Гидролазы 917
Гидролиз 918
---древесины — см. Гидролиз расти-
тельных материалов
---необратимый 921
--- обратимый 918
--- органич. соединений 921
--- полисахаридов 925
--- растительных материалов 924
--- солей 912
1245
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1246
Гидролизный лигнин — см. Лигнин, Гид-
ролиз растительных материалов
Гидрометаллургия 930
Гидроний — см. Гидроксоний
Гидроокиси 933
Гидроочистка 935
Гидросфера 935, 851
Гидротермальный процесс (в геохимии)
843
Гидротермальный синтез 758
Гидрофильность и гидрофобность 936
Гидрофильные коллоиды — см. Лиофиль-
ность и лиофобность коллоидов, Гид-
рофильность и гидрофобность
Гидрофобность — см. Гидрофильность и
гидрофобность
Гидрофобные коллоиды — см. Лиофиль-
ность и лиофобность коллоидов, Гидро-
фильность и гидрофобность
Гидрофобные покрытия 938
Гидроформинг 939
Гидрохимия 939
Гидрохинон 941
— диметиловый эфир — см. Диметилгид-
рохинон
Гидрохинон-|3-Г-глюкоЗид — см. Арбу-
тин
Гидроцеллюлоза — см. Целлюлоза
Гиосциамин — см. Атропин
Гипервитаминозы 597
Гипергенез — см. Экзогенные геохимиче-
ские процессы
Гиперконъюгация 941
Гипероны 942
Гиперсенсибилизация 943
Гиперсорбция 943, 771
Гиперядро 943
Гипоксантин 945
Гипосульфит — см. Натрий, тиосульфат
Гипофория 428
Гиппуровая кислота 945
Гипс строительный 714
--- высокообжиговый (эстрих-гипс) 714,
715
Гипсовые вяжушие материалы 714
Гипсохромный эффект 946
Гири 543
Гиромагнитное отношение 947
Гистамин 947
Гистерезис механический (в полимерах)
947
-- сорбционный 948, 41
Гистидин 949, 180, 183
Гистологическая химия — см. Гистохи-
мия
Гистоны 949
Гистотопохимия 950
Гистохимия 950
Гптоксигенин 959
Глазурь 951
Глауцин 273
Гликали 952
Гликамины 952
Гликоген 952, 950
Гликогенолиз 965
Гликогенолитический и гипергликемиче-
ский фактор — см. Глюкагон
Гликозамины 954
Гликозаны 955
Гликозеены 955
Гликозидазы 955
Гликозиды 956
--- сердечные 958
Гликозиламииы 956
Гликокол — см. Глицин
Гликолевая кислота 961
Гликолевое титрование 961
Гликолевый альдегид 961
Гликоли 962
Гликолиз 964
Гликолипиды — см. Липиды
Гликоновые кислоты — см. Альдоновые
кислоты
Гликопротеиды — см. Мукопротеины
Гликопротеины 996
Глинистые минералы 968
Глинистые растворы 966
Глинозем — см. Алюминия окись
Глины 968
— отбеливающие — см. Земли отбели-
вающие
Глиоксалевая кислота 969, 137
Глиоксиловая кислота — см. Глиоксале-
вая кислота
Глиоксаль 970
Глицериды 970
Глицерин 972
Глицериновая кислота 973
Глицериновый альдегид 974
Глицеринтринитрат — см. Нитроглице-
рин
Глицероза 974
Глицин (гликокол) 975, 180
Глицин (оксифенилглйцин) — см. Прояв-
ляющие вещества
Глобин — см. Гемоглобины
Глобулины 975
Глобулы 697
Глутамин 976
Глутаминовая кислота 977, 180, 183
Глутаровая кислота 977
Глутатион — см. Глютатион
Глюкагон 978, 996
Глюкоза 978, 821
--- пищевая, получение 926
D-Глюкозамин 954
Глюкозиды 979
D-Глюконовая кислота 823
Глюкуроновая кислота — см. Гексуро-
новые кислоты
Глютамин — см. Глутамин
Глютаминаза 166
у-Глютаминил-цистеинилглицин — см.
Глютатион
Глютаминовая кислота — см. Глутами-
новая кислота
Глютатион 979
Глютелины 980
Глютенины 980
Гмелииа реакция 432
Гниение 980
Гольмий 981
Гомберга—Бахмана—Гея реакция 981
Гомеополярный диполь 1132
Гомо (приставка) 982
Гомогенного осаждения метод — см.
Возникающие реагенты
Гомогенное горение 992
Гомогенное равновесие — см. Равновесие
химическое
Гомогенные системы 982, 868
Гомогентизиновая кислота 982
Гомолитические реакции 983
Гомологические ряды 984
Гомоцепные высокомолекулярные соеди-
нения 697
--- неорганические 701
Гомоциклические аминокислоты 180
Гомоцистеиндесульфгидраза 1070
Гонадотропины — см. Гонадотропные
гормоны
Гонадотропные гормоны 987
Гопкалит 988
Горение 988
--- гетерогенное 993
--- гомогенной 992
Горения максимальная скорость
--- газов в смеси с воздухом 556
--- паров в смеси с воздухом 558
Гормон адренокортикотропный 36, 996,
998
--- лютеинизирующий 987, 996
--- меланофорный 996
---фолликулостимулирующий 987, 996
Гормоны 995
---андрогенные 223, 997, 998
--- гонадотропные 987
Горчичное масло 999
Горчичные масла R—N=C—S 999
«Горчичный спирт» 1000
Горячая лаборатория 1000
Горячие атомы 1002
Горячие камеры 1000
Горячие частицы 1003
Гофмана реакция 1003
Гравиметрический анализ — см. Весовой
анализ
Грамин 1004
Грамицидины 1005
Грамм-атом 1006
Грамм-молекула 1006, 23
Грамм-моль — см. Грамм-молекула
Грамм-эквивалент 1006
Гранозан 1007
Графит — см. Углерод
Графитовая кислота — см. Бензолполи-
карбоновые кислоты
«Графт»-сополимеры — см. Сополимери-
зация
Гребе — Ульмана синтезы 1007
Грейзенизация 842
Гремучая кислота 1007
Гремучая ртуть 1008, 562
Гремучий студень 1126
Гриньяра реакция 1008
Грисса реактив 1010
Гроттуса — Дрейпера закон — см. Фо-
то химия
Групповой реактив 1010
Гуанидин 1010, 672
Гуаниловая кислота 1011
Гуанин 1012
Гуанозин 1012
Гудрон 1012
--- кислый 1)13
D-Гулоза 821
D-Гулоновая кислота 824
Гульда — Джекобса реакция 1014
Гуминовые кислоты 1014
Гумусовые кислоты 1015
Гуттаперча 1015
д
2,4-Д — см. 2,4-Дихлор!еноксиуксусная
кислота
Давление насыщенного пара 1017
Дагерротипия 1017
«Дактилограмм» метод 837
Дальтона законы 1017
Дальтониды и бертоллиды 1017
Даммара 1019
Даниэля — Якоби элемент — см. Хими-
ческие источники тока
Дарзана реакция 1019
Дарзана синтез ненасыщенных кетонов—
см. Кондакова реакция
Даутерм 1020
Даффа реакция 1021
Двойная гомогенная точка 760
Двойная связь 1021
Двойного взвешивания метод 545
Двойной электрический слой 1022
Двойные системы 1025
Двухатомные спирты — см. Гликоли
Двухкомпонентные системы — см. Двой-
ные системы
ДДД 1032
ДДТ 1032
Деалкилирование — см. Дезалкилирова-
ние
Деасфальтизация 1033
Дебая — Фалькенгагена эффект 1034
Дёбнера — Миллера реакция 1034
Деварда сплав 1035
Дегазация 1035
Дегалохромия 797
Дегидразы 1036
Дегидратация внутримолекулярная 897
---каталитическая — см. Гидратация и
дегидратация каталитические
Дегидроандростерон — см. Дегидроэпиан-
дростерон
Дегидробензол 1036
Дегидрогеназы 1037
Дегидрогенизация каталитическая — см.
Гидрогенизация и дегидрогенизация
каталитические
Дегидрогенизация нефтепродуктов 1038
2,3-Дегидро-Ь-гулоновая кислота, у-лак-
тон — см. L-Аскорбиновая кислота
11-Дегидрокортикостерон 997
Дегидроэпиандростерон 1038
Деготь буроугольный 1040
--- древесный 1040
--- каменноугольный 1041
--- первичный 1039
--- сланцевый 1041
—— торфяной 1040
Дезактивация 1042
Дезалкилирование 1043
---нефтепродуктов 1044
Дезаминирование 1044
Дезинсекталь 1033
Дезоксиальдозы 1047
2-Дезокси-2-аминоальдозы 954
З-Дезокси-З-аминоальдозы 954
Дезоксикетозы 1047
Дезоксикортикостерон 1046, 997
Дезоксиметилозы 1047
Дезоксирибоза 1046
2-Дезокси-Б-рибоза — см. Дезоксирибо-
за
Дезоксирибонуклеаза 1046
Дезоксирибонуклеиновая кислота 950 —
см также Пуклеиновые кислоты
Дезоксисахара 1047
Дсзокспхолсвая кислота 1048
1247
алфавитно-предметный указатель
1248
2-Дезокси-Б-эритро-пентоэа — см. Дезо*
ксирибоза
Действующих масс закон 1049
Дейтерий 1050, 619
— получение 1052
— применение 1051
— физич. и химич. свойства 1050
Дейтон — см. Дейтрон
Дейтрон 1054, 1051
Декагидронафталин — см. Декалин
Декалин 1054
Декан 1055
Деканаль — см. Дециловый альдегид
Деканол-1 — см. Дециловый спирт
Декарбоксилазы 1055
Декарбоксилирование 1056
Деклауазит 524
Декстран 1057
Декстранглюкозидаза 1058
Декстрины 1058
Делепина реакция 1059
Делькозин 86
Дельсемин 86
Дельсолин 86
Дельфелин 86
Дельфинидин 259
Дельфинин 86
Демьянова перегруппировка 1059,
1045
Денатурированный спирт 1060
Дениже реакция 1060
Денсиметрия 1061
Депарафинизация 1062
Деполимеризация 1063, 1068
Деполяризаторы 1064
Деполяризация 1064
Дерматол 591
Дернера — Госкинса распределение 1064
Десенсибилизаторы — см. Десенсибили-
зация
Десенсибилизация 1065
Десиканты 1065
Десмотропия — см. Таутомерия
Десорбция 1066, 41
Десульфгидразы 1070
Десульфуразы — см. Десульфгидразы
Детального равновесия принцип 1071
Детергенты — см. Моющие средства
Детонационная стойкость моторного топ-
лива 1071
Детонация 1072, 993
— скорость 560
Дефект массы 1072
Дефекты структуры 1073
Дефлаграция 993
Дефолианты 1075
Деформация пластическая 1144
---полимеров 1076
Децилакрилат 88
Дециловый альдегид 1078, 1220
Дециловый спирт 1078
Деэмульгаторы 1078
Деэмульгирование 1079
Джоуля закон 602
Джэппа — Клингемана реакция 1080
Диагенез 843
Диаграмма растворимости — см. Раство-
римость
--- состав — свойство — см. Физико-
химический анализ
--- состояния 1082, 1025
--- химическая — см. Физико-химиче-
ский анализ
Диазаминол красный 1083
--- оранжевый 1083
--- синий 1083
Диазаминолы 1083
Диазоаминосоединения 1084
О-Диазоацетил-Ь-серин — см. Азасерин
Диазоли 1085
Диазолин 1085
Диазоль оранжевый О 1085
--- розовый О 1085
--- синий 0’1085
Диазометан 1086
Диазониевая структура 1087
Диазосоединения 1087, 64
Диазотаты 1089
Диазотипия 1089
Диазотирование 1090
Диазоуксусная кислота, этиловый
эфир — см. Диазоуксусный эфир
Диазоуксусный эфир 1091
Диакарб 1092
Диал 372
Диализ 1093 |
Диализаторы 1093 *
1,1-Диалкоксиалканы — см. Ацетали и
кетали
5,5-Диаллил-барбитуровая кислота — см.
Диал
Диаллилфталат 1094
Диамагнетизм — см. Магнитные свой-
ства
Диамантан — см. Адамантан
Диамид — см. Гидразин
Диаминоантрахиноны 176
4,4-Диаминодифенил — см. Бензидин
п, п'-Диаминодифенилдиметилметан 1095
п,п'-Диаминодифенилметан 1095
Диаминокарбоновые кислоты 180
Диаминомасляные кислоты — см. Амино-
масляные кислоты
Диаминопиридин 188
Диаминосахара 954
п,п'-Диаминостильбен-о,о'-Дисульфокис -
лота 1095
Диаммонийфосфат 211
Диамокс — см. Диакарб
Диан — см. Дифенилолпропан
Дианизидин 1096
1,1’-Диантрахинониламин — см. Диан-
тримид
Диантримид 1096
Диарилгалогенониевые соединения 792
Диарилметановые красители 1096
Диаспор 153
Диастереоизомеры 1097
Диафораза 1098
Диацетил 1099
Диацетилдиоксим — см. Диметилглиок-
сим
Диацетилен 1099
Диацетилметан — см. дцетилацетон
Диацетоновый спирт 1100
Дибазол 1100
Дибензил 1100
Дибензоил — см. Бензил
Дибензолхром 1101, 289
Дибензофуран 1101
Дибромбензол 1102
Дибромметан — см. Бромистый метилен
Дибромоксин 1102
1,2-Дибромэтан 1102
Дибутиладипинат 1102
Дибутиловые эфиры 1103
Дибутилсебацинат 1103
Дибутилфталат 1104
Дивинил — см. Бутадиен-1,3
Дивинилацетилен 1104
Дивинилбензол 1104
Дивинилкетоны 1105
Ди-втор .-бутиловый эфир 1103
D-Дигиноза 958
Дигидрожасмон 1107, 1220
Дигидрокарвеол 1107
Дигидрострептомицин 1108
Дигидрофенарсазинхлорид - см. Адам-
сит
D-Дигиталоза 958
Дигитоксигенин 959
Дигитоксин 959
Дигитоксоза 958, 1047
Дигликоль — см. Дизтпленгликоль
Диглим — см. Диэтиленгликоль
Диеновые углеводороды — см. Непре-
дельные углеводороды
Диеновый синтез 1108
Диенофилы 1108
Диены 1108
Дизельное топливо 1111
Диизоамиловый ефпр 1112
Ди-изо-амилсульфат 127
Диизобутиловый эфир 1103
Ди-изо-бутилсульфат 127
4-Диизобутилфенол 129
Диизопропиловый эфир 1112
Ди-изо-пропилсульфат 127
Диизопропилфторфосфат 1113
3,5-Дииод-4-оксифенилаланин — см. Ди-
иодтирозин
Дииодтирозин 1113
Дикаин 1113
2,5-Дикетопиперазины 1114
Дикмана реакция 1114
Дикумарин 1115
Дикумарол — см. Дикумарин
Дилантин — см. Дифенин
Дильдрин 1116
Дильса — Альдера реакция — см. Дие-
новый синтез
Димедон 1116
Димедрол 1117
2,3-Димеркаптопропанол 1117
3,4-Димеркаптотолуол 1117
1,2-Диметилакриловая кислота — см.
Ангеликовая кислота
Диметиламин 1118
n-Диметиламиноазофениларсоновая кис-
лота 1118
п-Диметиламинобензальдегид 1118
п-Диметиламинобензилиденроданин 1119
З-Диметиламинометилиндол — см. Гра-
мин
п-Дйметиламинофенилфлуорон — см. Ди-
метилфлуорон
Диметиланилин 1119
n-Диметиланилин хлоргидрат — см. Ди-
метил-п-Фенилендиамин солянокислый
Диметилацетонилкарбинол — см. Диацс-
тоновый спирт
2,3-Диметилбутадиен-1,3 1119
Диметилгидразин 1120
0, 0-Диметилгидрокси ламин 915
а, р-Диметилгидроксиламин 915
Диметилгидрохинон 1120
Диметилглиоксаль — см. Диацетил
Диметилглиоксим 1120
Диметилдихлорсилан 1121
Диметилкетон — см. Ацетон
Диметиловый эфир 1121
2,6-Диметил-2-оксиоктаналь-8 — см. Гид-
роксицитронеллаль
Диметилпироп 1122
2,6-Диметил-у-пирон — см. Диметилпи-
рон
2,2-Диметилпропанол-1 171
Диметилсульфат 1122, 127
Диметилтерефталат 1123
2,4-Диметил-б-трет-бутилфенол 129
2,6-Диметил-4-трет-бутилфенол 129
Диметил-п-Фенилендиамин солянокислый
1123
Диметилфлуорон 1123
Диметилформамид 1124
Диметилциклогексан 1125
5,5- Диметилциклогександион-1,3 — см.
Димедон
1,5- Димети л-5-( 1 -цик логексенил)-барби-
туровая кислота — см. Эвипан
2,4-Диметил-3-этил-пиррол-5-карбоно-
вая кислота, азид 56
— гидразид 882
3,3'-Диметоксибензидин — см. Дианизи-
дин
1,2-Диметоксибензол — см. Вератрол
2,3-Диметоксиитрихнин — см. Бруцин
Димит 83
Диморфизм — см. Изоморфизм
Димрота реакция 1125
Динамиты 1126
Динамическая биохимия 435
Динамическая вязкость 718
Динаммоны 206
Динафталит № 1 206
Динезин 1126
Динитрорезорцин 1126
2,4-Динитрофенилгидразин 1127
Динитрофенолы 1127
Диоксан 1128
1,2-Диоксиантрахинон — см. Ализарин
Диоксиацетон 1129
п-Диоксибензол — см. Гидрохинон
Диоксивинная кислота 1129
1,3-Диокси-4-н-гексилбензол — см. Гек-
силрезорцин
4,4-Диоксидифенилдиметилметан — см.
Дифенилолпропан
0, р-Диоксидиэтиловый эфир — см. Ди-
этиленгликоль
а-, 0-Диоксипропионовая кислота — cmj
Глицериновая кислота
Диоксифенилаланин ИЗО
2,5-Диоксифенилуксусная кислота — см.
Гомогентизиновая кислота
За, 12а-Диоксихолановзя кислота — см.
Дезоксихолевая кислота
Диоксиянтарные кислоты — см. Винные
кислоты
Диоксоланы 338
Диоктилсебацинат ИЗО
Диоктилфталат ИЗО
Диолефины 773
Диолы — см. Гликоли
Дипаркол — см. Динезин
Дипенициллин 444
Дипикриламин — см. Гексанитродифе-
ниламин
Дипиридилы ИЗО
Дипольная поляризация 1183
Дипольный момент 1131
1249
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1250
Дипропилнафталин 126
Дипропилсульфат 127
Дипрофен 1139
Дисазокрасители 1140
Дисахариды 1141
Дислокации в кристаллах 1142, 1075
Диспергирование 1145 .
Дисперсионная среда — см. Дисперсные
системы
Дисперсионный анализ 1147
Дисперсная фаза — см. дисперсные си-
стемы
Дисперсность 1149
Дисперсные системы 1151
Дисперсные структуры 1151
--- лиофильные 1152
--- лиофобные 1152
Диспрозий 1154
Диспропорционирование 1154
Дистектика 1154, 1029
Дистектическая точка — см. Дистектика
Дистилляция 1154
--- в токе водяного пара и инертных
газов 1157
--- молекулярная 1159
Дитизон 1162
Дитилин 1162
Дитио-бис-бензтиазол — см. Альтакс
Дитиокарбаминовой кислоты соли 1162,
672
Дитиокарбаминовые кислоты 181
Дитиофос — см. Фосфорорганические со-
единения
Дитразии 1163
Дифенил 1163
Дифениламин 1164, 247
1,1'-Дифенилгидразин солянокислый
1164
Дифенилглиоксаль — см. Бензил
а-Дифенилглиоксим — см. а-Бензилдио-
ксим
Дифенилгуанидин 1164
Дифенилдикетон — см. Бензил
Дифенилен, окись — см. Дибензофуран
Дифепиленовая перегруппировка — см.
Бензидиновая перегруппировка
Дпфенилкарбазид 1165
Дифенилкарбазон 1165
Дифенилкарбилол — см. Бензгидрол
Дифенилкетон — см. Бензофенон
Дифенилметан 1165
Дифенилмочевина 1165
Дифенилмышьяковистая кислота, циан-
ангидрид — см. Дифенилцианарсин
Дифениловый эфир 1165
Дифенилоксид — см. Дифениловый эфир
Дифенилолпропан 1166
Дифенилпикрилгидразил 1167
N, М-Дифенил-М'-пикрилгидразин 1166
Дифенилтиокарбазон — см. Дитизон
Дифенилтиомочевина 1167
Дифенилтиоуксусная кислота, 0-ди-н-про-
пиламиноэтилового эфира хлоргид-
рат — см. Дипрофен
Дифенилхлорарсии 1167
Дифенилцианарсин 1167
Дифенилэтан симметричный — см. Ди-
бензил
Дифенин 1168
4,4’-Дифенохинон 1168
Дифосген 1168
Дифосфопиридиннуклеотиды — см. Пи-
р идинн у кл еоти ды
Дифракция рентгеновских лучей 1169
Дифференциальный спектрофотометриче-
ский метод 1172
Диффузионные фотографические процес-
сы 1172
Диффузионный двойной слой — см. Двой-
ной электрический слой
Диффузионный потенциал 1173
Диффузионный ток 1174
Диффузия 1175
4,4"-Дихлорбензиловая кислота, этило-
вый эфир 83
Дпхлоргидрин глицерина 1178
4,4-Д их лордифенилдихлорметилметан —
см. ДДД
4,4 "-Дихлордифенплметил-карбинол 83
4,4 "-Дихлордифенилтрихлорметилкар-
бинол 83
4.4’-Дихлордифенилтрихлорметилме-
тан — см. ДДТ
4,4”-Дихлордифеноксиметан 83
Дихлордиэтилсульфид 1178, 74
1,1-Дихлор-1-метилбензол — см. Бензи-
лиденхлорид
Дихлорметиловый эфир 1179
2,3-Дихлорнафтохинон 1179
Дихлороксин 1179
5,7-Дихлор-8-оксихинолин — см. Дихлор-
оксин
со, о)-Дихлортолуол — см. Бензилиден-
хлорид
2,4-Дихлорфеноксиуксусная кислота 1180
2,6-Дихлорфенолиндофенолят натрия
1180
Дихлорэтан 1180
1,1'-Дихлорэтилен — см. Винилиденхло-
рид
Ди-(2-хлорэтил)-сульфид — см. Дихлор-
диэтилсульфид
Дицентрин 273
Дициандиамид 1181
Дициклогексил 1181
Дициклопентил 1182
Дише реакция 1048
Диэлектрики 1182
— пробой 1185, 1186
— электропроводность 1185
Диэлектрическая постоянная — см. Ди-
электрическая проницаемость
Диэлектрическая проницаемость 1187,
1182
— значения 1186
Диэлектрические материалы 1187
Диэлектрические свойства полимеров
1188
Дпэтенилбензол — см. Дивинилбензол
Диэгиламин 1190
М-(2-Диэтиламиноэтил)-фентиазин хлор-
гидрат — см. Динезин
Диэтиланилин 1190
Диэтилацеталь 339
5,5-Диэтилбарбитуровая кислота — см.
Веронал
Диэтилдитиокарбаминат натрия 1190
Диэтилдитиофосфат никеля 1190
Диэтиленгликоль 1191
Диэтилепгликольдинитрат 1191
Диэтилен-диоксид — см. Диоксан
Диэтилкадмий 1191
1-диэтилкарбамоил-4-метилпиперазин,
цитрат— см. Дитразин
Диэтилмалонат 1192
Диэтилнафталин 126
Диэтиловый эфир 1192
диэтилпарафенилендиаминсульфат 1193
Диэтплселен 247
диэтилстильбэстрол 1193
Диэтилсульфат 1194, 127
Диэтилтеллур 247
диэтилформаль 339
диэтилфталат 1194
дНК-аза — см. Дезоксирибонуклеаза
додецилакрилат 88
додецилмеркаптан 1195
додециловый альдегид — см. Лаурино-
вый альдегид
Доза ионизирующего излучения 1195
Дозаторы 1196
--- весовые 1196
--- дросселирующие 1199
--- объемные 1197
--- скоростные 1199
Дозиметр Фрике 1203
Дозиметрия 1200
---химическая 1202
ДОК — см. Дезоксикортикостерон
Доннана равновесие — см. Мембранное'
равновесие
Донор 516
Донорно-акцепторная связь — см. Коор-
динационная связь
Допан 1204
Доэвтектические растворы и сплавы 1026
Драггендорфа реактив 1204
Древесина 12 34
---пластифицированная — см. Древес-
ные пластики
Древесины энергохимическая переработ-
ка 1206
Древесно-волокнистые плиты 1209
Древесно-пластические массы — см. Дре-
весные пластики
Древесно-слоистые пластики 1 208
Древесно-стружечные плиты 1208
Древесные пластики 1207
Древесные плиты 1208
Древесный деготь 1040
Дробление — см. Измельчение
Дробная кристаллизация 1209
Дробное осаждение 1210
Дробящее действие 563
Дрожжи кормовые белково-витаминные
928
Дросселирующие дозаторы 1199
Дуалистическая система 1210
Дубители — см, Дубящие вещества
Дубление 1211
--- фотографическое 1212
Дублетное расщепление 326
Дубящие вещества 1213
--- синтетические 1215
Дуктильность 439, 441
Дульцит 1216
Дуралюмин 157
Дурол 1217
Пусты — см. Пестицидные препараты
Душистые вещества 1217
Дымный порох 563 — см. также Пороха
Дымы 1221, 361
Дыхание — см. Обмен веществ
Дэкина реакция 1222
Дэкина — Веста реакция 1222
Дюлонга и Пти закон 1222
Дюма метод 1222, 73
Е
Европий 1223
Енамины 1223
Енолаза 1224
Енолы 1225
Ж
Ж асмин альдегид — см. а-н-Амилкорич-
ный альдегид
Железо, диметилдитиокарбамат 1163
— пентакарбонил 247
Железобетон 430
Жесткость воды 611
Жидкости, сжатие 695; электропровод-
ность 1184
--- гидравлические 881
--- промывочные 967
Жирара реактив 224, 225
Жирного ряда соединения — см. Аци-
клические соединения
Жирорастворимые азокрасители 61
Жиры, гидрогенизация 904
3
Зажигание 991
Замедлитель нейтронов 1051
Заряд ядра атома 307
Заэвтектические растворы и сплавы 1026
Зелинского катализатор 907
Земли отбеливающие 39
Земля, мантия 845, 848
— ядро 845, 847
Зенфтлебена эффект 763
Зеркальные изомеры — см. Антиподы
оптические
Зифероль 366 -
Зонгорин 86
И
D-Идоза 821
D-Идоновая кислота 824
D-Идуроновая кислота 824
Изоаймалин 82
Изоамилакрилат 88
Изоамилацетат — см. Амилацетат и изо-
амилацетат
Изоамилбутиролакгон 499
Изоамилнитрит 170
Изоамиловый эфир — см. Диизоамило-
вый эфир
Изоатизин 85
Изоборнеол 457
Изобутан 491
Изобутеи 494
Изобутилакрилат 88
Изобутилацетат 495
Изобутилен 493
1251
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1252
ИзобутиловыЙ спирт 497
Изобутиральдоль 142
Изовалериановая кислота 518
Изоглутамин 976
Изокоридин 273
Изолейцин 180
Изологические ряды 984
Изоолеиновая кислота 905
Изополивольфраматы 658
Изопропилакрилат 88
5-Изопропил-5- <{$-бромалил)-барбитуро-
вая кислота — см. Квиэтал
Изопропиловый эфир — см. Диизопро-
пиловый эфир
Изорозиндулины 57
Изостера адсорбции 45
Изотермы адсорбции 42
Изотонический коэффициент — см. Вант-
Гоффа коэффициент
Изотопный обмен 1050
Изофталевая кислота 413
Изохинолин 871
Изоциклические соединения — см. Али-
циклические соединения
Изоэвгенол 1220
И-кислота 185
Илиды 600
Иллосвая реакция 350
Имидазол 287, 870
р-Имидазолил-4(5)-этиламин — см. Ги-
стамин
Иминокислоты 180
Ингибиторы окисления — см. Антиокис-
лители
Индол — см. Вензпиррол
р-Индолилуксусная кислота — см. Ге-
тероауксин
Инерционные уловители 744
Инициирующие взрывчатые вещества 561
Инконгруэнтное плавление 1029
Инсулин 996
Интермедин — см. Меланонофорный гор-
мон
Иньоит 451
Иод, определение 782
Иодазид 56, 69
Иодглобин — см. Дииодтирозин
Иодгоргон — см. Дииодтирозин
Йодистый азот 69
Иодкрахмальная бумага 483
Иодохлоробромосеребряная бумага фото-
графическая 482
Ионизирующее излучение, доза 1195, 1201
Ионная поляризация 1183
Ионное произведение воды 628
Ионные пары 301
Ионные радиусы лантанидов и актинидов
102
Ионные связи 318
Ионные тройники 301
Ионон 1220
Иприт — см. Дихлордизтилсульфид
Иприты азотистые 74
Ир алия — см. изо-а-Метилионон
К
Калиборит 451
Калибровочная кривая 762
Калий, нитрат 79
Калифорний 104
Кальций, нитрат 79
— цианамид 80, 1075
Каменноугольный деготь 1041
Камфара искусственная — см. Борнил-
хлорид
Канаванин 180
Канадский бальзам 369
Канделильский воск 661, 662
Каноническое распределение 876
Канфильдит 858
Канцерогенные вещества 419
Каолинит 968
Капиллярная конденсация 41, 45
Каприновая кислота 709
Каприновый альдегид — см. Дециловый
альдегид
Капроновая элементарная нить 647
Карбамидные пластики 188
Карбамидные смолы 188
n-Карбамидофенилмышьяковая кислота—
см. Аминарсон
Карбамилмочевина — см. Биурет
Карбанилид — см. Дифенилмочевина
Карбанион 1003
Карбарсон — см. Аминарсон
Карбены 439
Карбилоксим — см. Гремучая кислота
Карбоиды 439
Карбоксигемоглобин 837
Карболинеумы 256
Карбометр 659
Карбоновые кислоты, азиды 56
— амиды 166
— ангидриды 221
— галогенангидриды 779
— гидразиды 881
Карбоновые оксикислоты 928
Карбоцепные высокомолекулярные со-
единения 697
Карбоциклические соединения — см. Али-
циклические соединения
Карбромал — см. Адалин
Карнаубский воск 661, 662
Карнотит 524
Касторовое масло 194
Катализгетерогенный,активные центры 95
Катализатор Адамса 27
--- Адкинса 35
--- Зелинского 907
Каталитическое горение — см. Поверх-
ностное каталитическое горение
Каталитическое сожжение 762
Катионирование воды 617
Катионоактивные вещества 666
Каустобиолиты 434
Каучук маслонаполненный 490
Квадрантные лабораторные весы 543
Квантовая жидкость 830
Квантовое число азимутальное 312
--- внутреннее 314
--- главное 311
--- магнитное 312
Квасцы 159
Кверцетин 59 4
Кверцитрин 594
Квиэтзл 372
КЕД (кошачья единица действия) 960
Кельтан 83
Кемитал 372
Кернит 451, 459
Кетали — см. Ацетали и кетали
Кетогексозы 821
Кетокислоты — см. Альдегидокислоты и
кетокислоты
Кетолы — см. Ацилоины
Кетоновые тела — см. Ацетоновые тела
4-Кетопентан-1-ол — см. у-Ацетопропи-
ловый спирт
Кетотриоза — см. Диоксиацегон
Кехлинит 589
Кинематическая вязкость 718
Кинетика адсорбции 46
--- химическая, активные центры 94
Кислород газообразный, получение 637
--- жидкий, получение 638
Кислородная конверсия 772
Кислородсодержащие соединения 773
Кислотные азокрасители 61
К-кислота 185
Кислотный сине-черный 1139
Кислотный синий К 1139
Клаузиуса — Моссоти уравнение 690,
1183
Клешневидные злементоорганические по-
лимеры 703, 706
Клупанодоновая кислота 709
Коагели 826
Ковалентных связей образование 318
Когезия 28
Коена реакция 1048
Коксовый газ 728;
— разделение 754
Коллоидные системы 868
Конваллотоксин 959
Конгруэнтное плавление 1029
Кондельфин 86
Конденсация капиллярная 41, 45
Контаминация 842
Конформации макромолекул 879
Копайский бальзам 369
Коридин 273
Коритуберин 273
Коричный альдегид 1220
Коричный спирт 1220
Кортексон — см. Дезоксикортикостерон
Кортизол 997
Кортизон 997
Кортикостерон 997, 998
Кортикостимулин — см. Адренокортико-
тропный гормон
Кортикотропин — см. Адренокортико-
тропный гормон
Корунд 153, 154
Котанниды 1214
Кошачья единица действия — см. КЕД
Красители
--- азиновые 57
---азо- 60
--- азометиновые 63
--- акридиновые 87
--- аминоантрахиноновые 175
--- антрахиноновые 265
--- антрахиноновые кубовые 266
--- ацетатные 62, 342
---диарилметановые 1096
--- дисазо- 1140
---жирорастворимые 61
--- кислотные 61
--- основные 61
--- протравные 61
--- спирторастворимые 61
Кратежнн — см. Анисовый альдегид
Крахмал, радиоационная деструкция
--- животный — см. Гликоген
Кребанин 273
Крекинг нефти 286
Крика проба 945
Криптон, выделение из воздуха 641
Кристаллизация дробная 1209
Кристаллогидраты 892
Кристаллы, дислокации в К. 1142;
образование и рост 1143
--- жидкие 213
Критическая температура растворения
864
Крокидолит-асбест 295
Кротонилены 350
Крутильные микровесы 539
Ксантогенаты 672
Ксенон, выделение из воздуха 641
Ксерогель 827
Ксероформ 591
Ксилан 835
Ксилидин 247
Ксилоза техническая 926
Кумарин 1220
Кумарон — см. Бензофуран
Купрон — см. а-Бензоиноксим
Курциуса реакция 56
Къельдаля метод 73
Кюрий 104
Л
Лаборатория горячая — см. Горячая ла-
боратория
Лавсановая элементарная нить 647
Лаки 61
Лактамы аминокислот 182, 183
Ламинарное пламя 993
Ланолин 662
Лантаниды, ионные радиусы 101
Ларгактил — см. Аминазин
Лассеня проба 72
Лаурелин 273
Лаурилмеркаптан — см. Додецилмер-
каптан
Лауриновая кислота 709
Лауриновый альдегид 1220
Лауротетанин 273
Левулиновая кислота 138, 928
Легаля реакция 354
Лед 606, 608
ЛЕД (лягушачья единица действия) 960
Лейкоцианидин 594
Лейцин 180
Лен (волокно) 647
Летоисчисление геологическое абсо-
лютное 645
Летучесть — см. Фугитивность
Лигнин 1206
--- гидролизный 929
Лигнофоль 1208
Лидаза 875
Лизин 180
Ликаконитин 86
Ликвация (в геохимии) 842
Ликвидуса линии 1026
Ликоктонин 86
Линалоол 1220
Линдан 818
Линейные высокомолекулярные соеди-
нения 696
1253
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1254
Линейные дефекты структуры — см. Дис-
локации в кристаллах
Линолевая кислота 709, 904
Лиогель 826
Лиофильность 936
Лиофильные дисперсные системы 1152
Лиофобность 936
Лиофобные дисперсные системы 1152
Липиды 950
Литосфера 845, 848
— средний химич. состав 849, 850
Литофильные элементы 839
Лоуренсий 107
«Луцидол» 405
Льняной воск 662
Люминал 372
Лютеотропин 996
Лягушачья единица действия — см. ЛЕД
м
Магматическая дифференциация 847
Магналий 157
Магнамицин 239
Магний, бораты осажденные 459
— перехлорат — см. Ангидрон
— хлорат 1075
Магнитное квантовое число 312
Макромолекула 697
— конформации 879
— сегмент 880
Малонал — см. Веронал
Малонилмочевина — см. Барбитуровая
кислота
Малоновый эфир — см. Диэтилмалонат
Мальтены 439
Мальтодекстрины 1058
Маннан 835
Манниха реакции 1004
D-Манноза 821
D-Манноновая кислота 823
Маннуроновая кислота 825
Мантия Земли 845, 848
Марганец, этиленбис (дитиокарбамат)
1163
Масло ализариновое НО
--- антраценовое 271
--- арахисовое 276
--- ветиверовое 547
--- горчичное 999
--- касторовое 194
Масляная кислота, азид 56
— гидразид 882
Масса атома 308
— дефекты 1072
Масса ядра атома 307
Мединал 372
Межмолекулярная дегидратация 897
Мезобензантрен — см. Бензантрен
Мезовинная кислота 576
Мезэксалевая кислота 138
Мезоксалилмочевина — см. Аллоксан
Меламино-формальдегидные прессма-
териалы 192
Меланин ИЗО
МеланофорныЙ гормон 996
Меллитовая кислота — см. Бензолгекса-
карбоновая кислота
Меллофановая кислота 413
Менделевий 105
Менделеева метод взвешивания 545
Д2-п-Ментен-1,4-диокись — см. Аска-
ридол
Ментол 519
Мербензантрон — см. Бензантрон
Метаванадаты 523
Метавольфраматы 85
Металламиды 228
Металлические гидриды 900
Метальдегид 339
Метамолибдаты 85
Метаморфизм (в геохимии) 843
Метаморфические газы 769
Метасоматоз 843
Метательные взрывчатые вещества 561
Метгемоглобин 837
Метилакрилат 88
Метилаль 339
2-Метил-4-амино-1-нафтол гидрохлорид
599
3-Метил-4-амино-1-нафтолгидрохлорпд
599
Метилантранилат 264
N-Метилантраниловая кислота 264
Метилацетилен — см. Аллилен
п-Метилацетофенон 1220
Метилбензоат 409
Метилбутанолы 171
Метил-бутены 169
З-Метилбутин-1 350
Метилгидроксиламины 915
2-Метил-1,4-диаминонафталин дигидро-
хлорид 599
2-Метил-4,6-ди-трет-бутилфенол 129
Метилдитиокарбаминовая кислота, Na-
соль 1163
2-Метил-3-дифарнезил-1,4-нафтохинон
598
4-Метил-5-ди (р-хлорэтил)-аминоура-
цил — см. Допан
3,3-Метилен-бис-4-окси кумарин — см.
Дикумарин
3,4-Метилендиоксибензальдегид — см. Ге-
лиотропин
изо-а-Метилионон («Иралия») 1220
1-Метилкротоновая кислота — см. Ан-
геликовая кислота
Метилликаконитич 86
1 -Метил-4-метоэтенил-циклогексанол- 2—
см. Дигидрокарвеол
2-Метил-1,4-нафтогидрохинон 599
2-Метил-1.4-нафтохинон 598
Метилозы 1047
2-Метилпентанол-2 820
3-Метил-2-пентилциклопентен-2-он-1 —
см. Дигидрожасмон
№-Метилпирролидип-2-карбоновая ки-
слота — см. Гигриновая кислота
N-Метилпролин — см. Гигриновая ки-
слота
Метилсерная кислота 127
Метилстирилкетон — см. Бзнсилиден-
апетон
Метилсульфометилат ^№-ди-(4-димегил-
аминофекил)-мочевина — см. Гемо-
споридин
N-Метил-!, 2, 5, 6,-тетрагидроникоти-
новая кислота, метиловый эфир — см
Ареколин
Метилфенилацетальдегид — см. Гидра-
троповый альдегид
Метилфенилкетон — см. Ацетофенон
Метилэтилсульфат 127
Метилэтилуксусная кислота 518
Метимицин 239
Мети нон 598
Метионин 180
Метоксианилины — см. Анизидины
п-МетоксибенЗиловый спирт — см. Ани-
совый спирт
Метоксибензол — см. Анизол
Метоний — см. Гексоний
Меченый атом, дейтерий 1051
Миграция химических элементов *846
Микровесы крутильные 539
--- торзионные 539
Микрофибриллы 647
Минерализованная вода 611
Минеральные вяжущие материалы 713
МиоЬлобины 838
S-Миозин — см. Актомиозин
а-Миозин — см. Актомиозин
Миокинин 428
Миорелаксин — см. Дитилин
Миристиновая кислота 709
Мисолин — см. Гексамидии
М-кислота 185
Мозаичный отрыв 28
Молекулы цепные, гибкость 879
«Молекулярные болезни» 388, 389
Молекулярные сита 893
Молекулярный вес 23
Моль — см. Грамм-молекула
Молярная поляризация 1132, 1138
Монастраль красный — см. Синквазия
Монастраль фиолетовый — см. Синква-
зия
Моноаминодикарбоновые кислоты 180
Моноаминопиридины 187
Моноаммонийфосфат 211
а-Монобром-изовалерианил-мочевина —
см. Бромурал
Моноволокна 646
Моновольфраматы 658
Монозы, биохимич. переработка 928
Монолит 1208
Монотектическое превращение 1030
Монохлорэтилен — см. Винилхлорид
Монтанный воск 662
Монтмориллонит 968
Морфолин 871
Мостиковые связи 516
Моттрамит 524
Мочевина, удобрение 80
Мочевино-формзльдегидные прессмате-
риалы 192
Мукоитинсерная кислота В 855
Мукополисахариды 950
Мультиплетность 326
Мутарогация 234
Мышьяковистая кислота, хлорангидрид-
дифенил — см. Дифенилхлорарсии
н
Надуксусная кислота—см. Ацетила гид-
роперекись
Найлоновая элементарная нить 847
Насыпной вес 1062
«Натана — Бейкера эффект» 942
Натрий, бромат 469
— 2,6-дихлорфенолиндофенолят 1180
— диэтилдитиокарбаминат 1190
— нитрат 79
— фторобериллаты 423
— хлорат-пентаборат 1075
Нафталин 287
1-Нафтилуксусная кислота 867
р-Нафтол.. метиловый эфир (Яра-яра)
1220
Нашатырный спирт 199
Нашатырь — см. Аммоний, хлорид
Неионогенные вещества, 666, 683
Нейтрогены 1083
Нейтроны, замедлитель 1051
Нембутал 372
Неоаймалин — см. Аймалин
Необорнивал 518
Неометимицин 239
Неон, выделение из воздуха 642
Неотран 83
Неотропин 188
Нептуний 103
Нернста формула 629
Нетанниды 1213
Нефелин 148
Нефтепродукты, ароматизация 286
— вязкость 722
— дегидрогенизация 1038
— дезалкилирование 1044
Нефть, диэмульгирование 1080
— крекинг 286
Никелон — см. я-Бензилдиоксим
Никель, диэтилдитиофосфат 1190
— тетракарбонил 247
Никотинамид 167
Нингидриновая реакция 181
Нитратные удобрения 79
5-Нитроантраниловая кислота 264
Нитроглицерин 563
Нитроглицериновые В В 206
Нитрогуанилазид 56
Нитродигликоль — см. Диэтиленгли-
кольдинитрат
Нитрозил хлористый 69
Нитролигнин 929
Нитрометр 659
Нитропарафины 774
Нитрофен 1128
НИУИФ-2 — см. Гранозан
№ 102 105
№ 103 106
Нонадекановая кислота 709
Нонилакрилат 88
Нониловый спирт 1220
Норадреналин 996, 998
Норвалин 180
Нордигидрогваяретовая кислота 251
Норлейцин 180
Нормана реактив 569
Нуклеофильные и электрофильные реа-
генты 868
Нуткатин 243
О
Обепин — см. Анисовый альдегид
Овотирины 599
Озотран 83
Озокерит 662
Окислы, гидратация 893
Оксазин 57, 871
Оксазол 287, 870
1,3-Оксиальдегиды — см. Альдоли
6-Окси-2-аминопурин — см. Гуанин
1255
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1256
Оксиамины — см. Аминоспирты
а-Оксиамины — см. Альдегидаммиаки
Оксианилины — см. Аминофенолы
2-Оксиантрацен-З-карбоновая кислота —
см. Ан*гроловая кислота
1,2 и 9-Оксиантрацены— см. Антролы
Оксигемоглобин 837
Оксигенация 837
1-Оксидекан — см. Дециловый спирт
Оксикетоны 997
Оксикислоты дикарбоновые 928
---- монокарбоновые 928
17-Оксикортикостерон 997
Оксималеиновая кислота 138
у-Оксимасляная кислота, ангидрид —
см. Бутиролактон
о-Оксиметилбензойная кислота
— азид 56
— гидразид 882
4-Окси-4-метилпентанон-2 — см. Диапе-
тоновый спирт
4-Окси-З-метоксибензальдегид — см. Ва-
нилин
1-Окси-2-метоксибензол — см. Гваякол
Оксипролин 180
3-Оксипропионовая кислота — см. Гид-
ракриловая кислота
6-Оксипурин — см. Гипоксантин
Окситоцин 996
Оксиуксусная кислота — см. Гликоле-
вая кислота
Оксиуксусный альдегид — см. Гликоле-
вый альдегид
n-Оксифеииламиноуксусная кислота —
см. Проявляющие вещества
Оксифенилглицин — см. Проявляющие ве-
щества
Оксифумаровая кислота 138
4-Окси-З-этоксибенэальдегид — см. Ва-
нилаль
Оксокарбоновые кислоты — см. Альде-
гидокислоты и кетокислоты
16-Оксоспартеин — см. Афиллин
Октагидринден — см. Гидр индан
Октилакрилат 88
Олеандрин 959
П-Олеандроза 958
Олеиновая кислота 709, 904
Олефины, получение 773
Омерил — см. Диазолин
Ониевые соединения 790
Оновые кислоты — см. Альдоновые ки-
слоты
Онтогенеза биохимия 437
Опиановая. кислота 888
Оппанол 494
Оптическая активность 93
Оптические антиподы 252
Органические соединения
— восстановление 663
— галогенирование 781
— гидратация 894
— гидрирование 663
— гидрогенолиз 663
— гидролиз 921
Ориентационная поляризация 1132, 1138
Орнитин 180
Ортоборная кислота — см. Борная ки-
слота
Ортованадаты 523
Ортоводород 620, 623
Осаждение дробное 1210
Осмос VI7 7
Основные азокрасители 61
Отбора правила 327
«Отпечатка пальцев» метод — см. «Дак-
тилограмм» метод
п
ПА БК — см. n-Аминобензойная кислота
Пальмитиновая кислота, 709, 904
Пальмовый воск 661, 662
Пантокаин — см. Дикаин
Пар насыщенный, давление 1017
Параводород 620, 623
Паравольфраматы 85
Пара-диоксан — см. Диоксан
Паральдегид 339
Парамид 414
Парамолибдаты 85
Паратиреокрин 996
Паровоздушный газ 734
Парокислородный газ 734
Пары, вязкость 721
Патронит 524
Педогенез 843
Пеларгонидин 259
Пенетрация 439, 441
Пенициллин О — см. Аллилмеркапто-
метилпенициллин
--- G- — см. Бензилпенициллин
Пенный аппарат 746
Пенобетон 429
Пентадекановая кислота 709
Пенталин 290
Пентаметилен, окись 963
Пентанал — см. Валериановый альдегид
Пеитандион-2,4 — см. Ацетилацетон
Пентанолы 171
Пентены — см. Амилены
Пентины 350
Пентозаны 835
Пентозиды 956
Пептидные гормоны 996
Пептизаторы 967
Пептоны 388
Пербунан — см. Бутадиен-нитрильный
каучук
Пергидроль 625
Пергидронафталин — см. Декалин
Перегонка — см. Дистилляция
Перегруппировки
--- Амадори 162
--- Бекмана 381
—- бензидиновая 392, 887
--- бензильная 398
--- Вагнера — Меервейна — см. Кам-
феновые перегруппировки
--- Валлаха 520
--- Демьянова 1059, 1045
--- дифениленовая — см. Бензидино-
вая
Периодическая система элементов Мен-
делеева 319—322
Перитектика 1028
Перноктон 372
Перуанский бальзам 369
Перхлорбензол — см. Гексахлорбензол
Перхлорэтан — см. Гексахлорэтан
Пивалиновая кислота 517
Пигменты 61
Пигменты — Синквазия 88
Пикелевание 1211
Пимарицин 239
Пиненгидрохлорид — см. Борнилхлорид
Пиперазин 58, 871
1-а-Пиперидил-р-пиридин — см. Анаба-
зин
Пиперидиновая кислота 184
Пиперонал — см. Гелиотропин
Пиразин 57, 871
Пиразол 287, 870
Пиранозиды 956
Пиридазин 57
Пиридин 287, 871
Пиридиум 188
Пиримидин 57, 871
Пированадаты 523
Пировиноградная кислота’137
Пиромеллитовая кислота 413
Пиррол 287| 870, 872
Пирролидин 872
Пирролин 872
Питуитрин 5q4
Пихтовый бальзам 369
Плавление инконгруэнтное 1029
--- конгруэнтное 1029
Пластбетон 429
Пламя ламинарное 993
--- турбулентное 993
Пластики древесные 1207
--- карбамидные 188
Пластичность, влияние высокого давле-
ния 689
Пневматолиз 843
Поверхностное каталитическое горение
995
Поверхностное натяжение, влияние вы-
сокого давления 689
Пойнтинга эффект 759, 761
Полиакрилонитрил, термич. деструкция
1068
Полиакрилонитр иловые элементар ные
нити 647
Полиалюмооксаны 703
Полиамидные элементарные нити 647
Полибутадиен, деструкция термическая
1068
Поливинилацетат, деструкция термичес-
кая 1068
Поливипилиденхлорид, деструкция ра-
диационная 1069
Поливинилхлорид, деструкция радиа-
ционная 1069
— деструкция термическая 1068
Полидиметилсилоксан, деструкция ра-
диационная 1069
Полиизобутилен, деструкция термичес-
кая 1068
Полимеры 697
— вязкость 724
— газопаропроницаемость — см. также
Диффузия
— гистерезис механический 947
— деструкция 1066
--- механическая 1068
--- радиационная 1069
---термическая 1068
---химическая 1070
— диэлектрич. свойства 1188
— химич. текучесть 724
Полимеры атактические 304
--- кремнийорганические 707
--- фосфорорганические 706, 707
—- элементоорганические 703
--- элементоорганические внутрикомп-
лексные (клешневидные) 703, 706
Полиметаллоорганосилоксаны 703
Полиметилакрилат, деструкция радиа-
ционная 1069
— деструкция термическая 1068
Полиметилметакрилат, деструкция тер-
мическая 1068
Полинафтеновые кислоты — см. Асфаль-
тогеновые кислоты
Полиорганосилоксаны 703, 707
Полиорганофосфонитрилы 703
Полисахариды, гидролиз 925
Полисиланы 703
Полисилоксаны 705
Полистирол, деструкция радиационная
1069; деструкция термическая 1068
--- атактический 698
--- изотактический 698
Политетрафторэтилен, деструкция ра-
диационная 1069
— деструкция термическая 1068
Политрифторхлорэтилен, деструкция тер-
мическая 1068
Полифениленсилоксаны 703
Полихлор бутадиен, деструкция радиа-
ционная 1069
Полиэтилен, деструкция радиационная
1069
Полиэтиленовые элементарные нити 647
Полиэфирные элементарные нити 647
Полуводяной газ 735
Полугудрон 1013
Поляризация 1132, 1138
--- атомная 1138
--- дипольная 1183
--- диэлектрика 1182
--- ионная 1183
--- ориентационная 1132, 1138
--- молярная 1132, 1138
---релаксационная — см. Поляриза-
ция дипольная
-— электронная 1132, 1138, 1183
Поляриметрия 93
Полярометрическое титрование — см.
Амперометрическое титрование
Порофор — см. 2,2-Азо-бис-изобутиро-
нитрил
Порох бездымный 563
--- дымный, 561. 563
--- ствольной артиллерии 368
Пороха 563
-— ракетные 367
Порядок ближний и дальний 212
Потенциал ионизации 311
Почвообразование 843
Д4-Прегнен-11 р-21-диол-18-аль 3,20-ди-
он — см. Альдостерон
д4-Прегнен-21-ол-3,20-дион — см. Дез-
оксикортикостерон
Пренитовая кислота 413
Пресная вода 611
Прессматериалы, мочевнно-формальде-
гидные и меламино-формальдегидные
192
Прилипание — см. Адгезия
Примидон — см. Гексамидин
Природные высокомолекулярные соеди-
нения 698
Прискол — см. Бензолин
Приставки:
-— амфи- 215
1257
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1258
--- ана- 216
--- анса- 234
--- анти- 235
--- гомо- 982
Прогестерон 997, 998
Пролактин — см. Лютеотропин
Пролин 180
Промывочные жидкости 967
Пропандикарбоновая 1,3 кислота — см.
Глутаровая кислота
Пропантриол-1.2,3 — см. Глицерин
Пропеналь — см. Акролеин
п-Пропениланизол — см. Анетол
Пропеновая кислота — см. Акриловая
кислота
Пропилакрилат 88
Пропилнафталины 125
н-Пропилсерная кислота 127
Пропин — см. Аллилен
Пропиональдоль 142
Пропионамид 167
Протеиды 388
Протеины — см. Белки
Протеозы 388
Противоядия — см. Антидоты
Протий 619
Протогемы 836
Протокатеховый альдегид
— диметиловый эфир — см. Вератровый
альдегид
— метиленовый эфир — см. Гелиотропин
Протопорфирин 836
Поотравные азокрасители 61
Проционы 62
Прочность твердых тел, адсорбционное
понижение 39
Прямой коричневый КХ 1139
---фиолетовый светопрочный 2 К 1139
Прямые азокрасители 61
Псевдоальдобиуроновые кислоты 141
Псевдогели 826
Псевдоглобулины 976
Псевдодубление 1211
D-Псикоза 821
Птеридин 58
Пуаз 718
Пуазейля — Гагена формула 718
Пуберуловая кислота 243
Пуберулоновая кислота 243
Пукатеин 273
Пурин 57
Пфлюгера метод 953
Пчелиный воск 661, 662
Пылеуловители сухие 745, 747
Пыли промышленные, взрывоопасность
558
Пыль 361
— газоочистка мокрая 746
— очистка электрич. 748
— улавливание 744
— фильтрация 745
Пьезоэффект 1187
р
Работы наибольшей принцип 426
Равновесия детального принцип 1071
Рад 1195
Радиационная деструкция 1069
Радиоактивные вещества, защита от
излучений 1000
Радиоактивные элементы, адсорбция 48
Разветвленные высокомолекулярные сое-
динения 696
Разорит — см. Кернит
Ракетные пороха 367
Рапидогены 1083
Распространенность химических элемен-
тов — см. Литосфера, средний химич.
состав
Рассолы 611
Растворение, критическая температура
864
Растворы, гидратная теория 888; соль-
ватная теория 888
--- буферные 499
•-- газовые 757
---глинистые 966
--- доэвтектические 1026
--- заэвтектические 1026
---твердые 1026
--- эвтектические 1026
Растительные воски 662
Растительные материалы, гидролиз 924
Раувольфин — см. Аймалин
Рацемические смеси 253
Рацемические соединения 253
Реактив групповой — см. Групповой ре-
актив
--- селективный — см. Селективный ре-
актив
Реакции аутокаталитические 335
Реакция, константа равновесия 1049
Ребиндера эффект — см. Адсорбционное
понижение прочности твердых тел
Резерфорда модель атома 306
Ремаланы 62
Ремерин 273
Рениерит 858
Рентгеновские лучи, дифракция 1169
Рибодезоза — см. Дезоксирибоза
Рибонуклеиновая кислота 950
9-0-П-Рибофуранозидоаденин — см. Аде-
нозин
9-₽-Б-Рибофуранозилгуанин — см. Гуа-
нозин
Ридберга постоянная 311
Рильсан 1S4
Рицинолевая кислота 709
Родионова метод 183
Розиндулины 57
Ронидаза 875
Роскоэлит 524
Ротаметр 1199
Ротоклоны 745
РР-кислота 185
Ругулоза 774
Рутин 594
С
Сажа 772
Салициловая кислота, азид 56
Саломас 906
Самовоспламенение 991
Самовоспламенения температура 556, 557
Самодиффузия 1175, 1177
Самоокисление — см. Аутооксидация
D-Сарментоза 958
Сафранины 57
Сахара, ангидриды 222 — см. также
Гликозаны ч
Сверхтекучесть 829
Сверхтонкая структура 314
Свинец, азид — см. Азид свинца
Свинцовая бумага 483
Связи мостиковые 516
Связь двойная 1021
--- ионная 318
— ковалентная 318
Себациновая кислота, ди-н-бутиловый
эфир — см. Дибутилсебацинат
— ди-н-октиловый эфир — см. Диоктил-
себацинат
Секобарбитал — см. Секонал
Секонал 372
Секретин 996
Селективный реактив 1010
Сера бромистая 468
Серенсена метод 183
Серин 180
Сернистый азот 69
Серный эфир — см. Диэтиловый эфир
Сжатие газов 694
---жидкостей и твердых тел 695
Сжимаемость веществ 686
Сидерофильные элементы 840
Сидпоны 289
Силумин 157
Сингулярная точка 1029
Синильная кислота 201, 772
Синквазия (пигменты) 88
Синтаны 1215
Синтезгаз 347
Синтетические высокомолекулярные сое-
динения 698
Синтин 404
С-кислота 185
Сланцевый деготь 1041
Смачивание 937
Смешанный генераторный газ 734
Смола первичная — см. Деготь первич-
ный
Смолы карбамидные — см. Аминопласты
Солевые системы взаимные 549
Соли, гидролиз 918
Солидуса линии 1026
Сольватная теория растворов 888 ‘
Соматотропин 996
Сополимеры 697
D-Сорбоза 821
Сосуды высокого давления 692
Сочетание — см. Азосочетание
Спектральные серии 325
Спектроскопия абсорбционная 17
Спектрофотометрический метод диффе-
ренциальный 1172
Спермацет 662
Спирт денатурированный ЮбО
Спин 311
Спин-орбитальное взаимодействие 314
Спирторастворимые азокрасители 61
Спирты многоатомные 926
Сплавы алюминиевые 155
---бериллиевые 424
--- вольфрамовые 656
--- доэвтектические 1026
--- заэвтектические 1026
--- эвтектические 1026
СС-кислота 185
Сталь, борирование 453
Стахидрин 428
Стеариновая кислота, 709, 904
Стеклообразное состояние 213
Стеклянные элементарные нити 647
Стерический фактор 432
Стероидные гормоны 996
Стефании 273
Стильбэстрол — см. Диэтилстильбэстрол
Стинитатовая кислота 243
Строительный гипс 714
Строфантидин 959
К-Строфантин-р 959
Строфантобиоза 959
Строфантотриоза 959
Структура, дефекты 1073
— линейные дефекты 1142, 1075
Структурная вязкость 724, 721
Структурообразование 725
Сукциндегидрогеназа 1037
Сукцинилхолин — см. Дитилин
Сульванит 524
Сульфенамиды 672
Сульфенон 83
4-Сульфоантраниловая кислота 264
«Сухие» газы 768, 770
Сциллигляукозид 959
Сциллигляукозидин 959
Сцинтилляторы 1202
Сшитые высокомолекулярные соединения
696
т
D-Тагатоза 821
Таликмидин 273
Таликмин 273
D-Талоза 821
D-Талоновая кислота 824
Танниды 1213
Твердые растворы 1026
Твердые тела, сжатие 695
D-и L-Теветоза 958
Тедион 83
Текучесть 716, 718
Температура абсолютная 17
Температурная депрессия 677
Температурные шкалы 17
Теплота гидратации 889
Теплотворная способность топлива 990
Терентьева метод определения — см. Ак-
тивный водород
Терефталевая кислота 413
— диметиловый эфир — см. Диметил-
терефталат
Териленовая элементарная нить 647
Термические бораты 459
Термовулканизация 674
Термодинамики второй закон 17, 668
Термодиффузия 1175
Термоядерное горючее, дейтерий 1051
Тестоидные гормоны —. см. Андроген-
ные гормоны
Тестостерон 997, 998
1, 4, 5, 8-Тетрааминоантрахинон 177
Тетрадецилакрилат 88
Тетракаин хлоргидрат — см. Дикаин
Тетраэтил-4, 4-диаминотрифенилметана-
оксалат — см. Бриллиантовый зеленый
Тетраэтплолово 247
Тетраэтилсвинец 24?
Тетрил 562, 563
1259
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1260
Технологический газ 736
Тиазин 57, 871
Тиазолы 287, 672, 870
Тиглиновая кислота 221
Тиминоза — см. Дезоксирибоза
Тинкаль 484
Тиокарбанилид — см. Дифенилтиомоче-
вина
Тиоловые соединения 248
Тионафтен — см. Бензотиофен
Тиопенталнатрий 372
Тиофен 287, 870
Тиофильные элементы 840
Тиреотропин 996
Тирозин 180, 183
Тироксин 996, 998
Тиронины иодированные 996
Титрование амперометрическое 214
--- гликолевое 961
Тиурамы 672
Толуанский бальзам 369
Тонкая структура 314, 325
Топливо, газификация подземная 739;
теплотворная способность 990
--- дизельное 1111
--- жидкое, газификация 729
--- моторное, антидетонаторы 247; де-
тонационная стойкость 1071
--- пылевидное, газификация 733
--- твердое, газификация 731
Торзионные микровесы 539
Торфяной деготь 1040
Точечные дефекты структуры 1073
«Тощие» газы — см. «Сухие» газы
Транс-4,4’диокси-а, а-диэтилстильбен —
см. Диэтилстильбэстрол
Транс-3, 4-д- (п-оксифенил)-гексен-З —
см. Диэтилстильбэстрол
Треонин 180
2- (4-Третичнобутилфенокси)-нзопропил-
2-хлорэтил-сульфит 83
Триазены — см. Диазоаминосоединения
Триаминопиридины 188
Триаммонийфосфат 211
Триацетатное волокно 341
Трибромметан — см. Бромоформ
Трибутил тритиофосфат 1075
Тригонелин 428
Тридекановая кислота 709
3, 5, З-Трииодтиронин 996
Тримезиновая кислота 413
Тримеллитовая кислота 413
а, 0, р-Триметилгидроксиламин 915
Триметилкарбинол 497
Триметилуксусная кислота — см. Пива-
линовая кислота
Тринитротриазидобензол 55
1, 2, З-Триоксиантрахиион — см. Антра-
галлол
3, 4, 5-Триоксибензойная кислота — см
Галловая кислота
1, 2, З-Триоксипропан — см. Глицерин
Триолит 256
Триптофан 180, 183
Тритий 619
«Тритон В» 211
«Тритон F» 211
Трифосген — см. Гексахлордиметилкар-
бонат
сооко-Трихлортолуол — см. Бензотрп-
хлорид
Трициклодекан — см. Адамантан
Триэтилсвинец хлористый 247
Триэтоксибор — см. Борноэтиловый
эфир
Тройная точка воды 17
Тромбозан — см. Дикумарин
Тропилий 288
Тротил 562, 563
Тудуранин 273
Туманы 361
Туранит 524
Турбулентное пламя 993
Туяплицины 243
Тэн 562
Тюямунит 524
Тяжелый водород — см. Дейтерий
У
Углеводороды, анилиновая точка 230
--- ароматические 290
--- галогензамещенные 784
Ударная волна 550, 560
Удельная вязкость 718
Удержание горячих атомов 1002
Удобрения азотные 79
--- амидные 80
--- аммиачные 79
--- борные 458
--- жидкие 79
--- нитратные 79
Уксусная кислота
— азид 56
— амид — см. Ацетамид
— амиловый эфир — см. Амилацетат и
изоамилацетат
— гидразид 882
— изоамиловый эфир — см. Амилацетат
и изоамилацетат
— нитрил — см. Ацетонитрил
— соли — см. Ацетаты
— хлорангидрид — см. Ацетил хло-
ристый
Уксусный альдегид — см. Ацетальдегид
Улексит 451
Уловители инерционные 744
Ультрабазит 858
Умягчение воды 617
Ундекалактон 1220
Ундекановая кислота 709
Уралит 256
Уреидоформамид — см. Биурет
Уротропин — см. Гексаметилентетрамин
ф
Фазовая диаграмма — см. Диаграмма
состояния
Фазовые переходы 686
Фазовые равновесия, в системах
жидкость — газ 759
Фазы 1025
Фанодорн 372
Фасциолин 819
Фейльгена реакция 1048
Феназин 871
Фенакит 422
Фенантрен 287
Фенилакрилат 88
Фенилаланин 180
Фенилалкиламины 996, 998
Фениламин — см. Анилин
4-Фениланилин — см. 4-Аминодифенил
N-Фенилантраниловая кислота 264
Фенилбензоат 409
Фенилбромуксусная кислота, нитрил —
см. Бромбензилцианид
Фенилбензол — см. Дифенил
Фенилбутазон — см. Бутадион
1-Фенил-1-3-бутандион — см. Бензоил-
ацетон
6-Фенилгидроксиламин 915
Фенилгликоколь — см. Анилидоуксус-
ная кислота
Фенилглицин — см. Анилодоуксусная
кислота
Фенилглицин-о-карбоновая кислота 264
1-Фенил-2, З-диметилпиразолон-5 — см.
Антипирин
Фенилкарбинол — см. Бензиловый спирт
Фенплметиловый эфир — см. Анизол
Фенил-а-оксибензилкетон — см. Бензоин
З-Фенилпропанол-1 — см. Гидрокорич-
ный спирт
p-Фенил-н-пропиловый спирт — см. Гид-
рокоричный спирт
а-Фенилпропионовый альдегид — см
Гидратроповый альдегид
p-Фенилпропионовый альдегид — см. Гид-
рокоричный альдегид
Фенилсульфиновая кислота — см. Бен-
золсульфиновая кислота
Фенилуксусный альдегид 1220
Фенил-4-хлорфенил-сульфон 83
2-Фепилхроман 259
р-Фенилэтилметилкетон —см. Бензилаце-
тон
Фенитоин — см. Дифенин
Феноксазин 871
Феноксибензол — см. Дифениловый эфир
Фенолы 996
Феитиазин 57, 871
Ферментация — см. Брожение
Фермий 105
Ферроцен 289
Фибриллы 647, 697
Фибриллярные высокомолекулярные
соединения 697
Фибриноген 976
Фика законы 1175
Фильтры для газовых потоков 745
Фишера метод 95 7
Фишера реактив 84
Флавенолы 259
Фогеля — Фульчера — Таммана фор-
мула 720
Фолион — см. Гептинкарбоновая кисло-
та, метиловый эфир
Фолликулостимулирующий гормон (ФСГ)
987, 996
Фомероль 366
Формаль 339
Формальдегидное дубление 1212
Формамид 167
Формилдиметиламин — см. Диметил-
формамид
Фосфонитрилгалогениды 289
Фосфор трехбромистый 468
Фосфоролиз 954
Фотографические процессы диффузион-
ные 1172
Фотографическое дубление 1212
Фотохимические реакции, закон взаимо-
эаместимости 549
Фракционированное растворение в га-
зовом анализе 762
Фриделя — Крафтса реакция 808
Фрике дозиметр 1203
Фруктан 835
Б-Фруктоза 821
о-Фталевая кислота 413
— диаллиловый эфир — см. Диаллил-
фталат
— ди-н-бутиловый эфир — см. Дибутал-
фталат
— диэтиловый эфир — см. Диэтилфталат
Фтор, определение 782
Фторазид 69
Фторапатит 272
Фтористый азот 68
Фторобериллаты 425
Фторогерманаты 860
Фторпарацид 83
4-Фторфенил-4-хлорбензилсульфид 83
Фугасное действие 563
Фугитивность 92
Фульвокислоты 1015
Фульминат ртути — см. Гремучая ртуть
Фуран 287, 870
Фуранозиды 956
Фурфурол 927
Фэр 1196
X
Халкон — см. Бензальацетофепон
Халькофильные элементы 840
Хаульмугровая кислота 880 !
Хемосорбция 46
Химиосинтез 436
Химическая кинетика 94
Химическая текучесть полимеров 724
Химических элементов распространен-
ность — см. Литосфера, средний хи-
мич. состав
Химическое деэмульгирование 1080
Химия аналитическая 217
Хинокитиол 243
Хиноксалин 57
Хинолин 871
Хитозамин — см. Б-Глюкозамин
Хлопок (волокно) 647
Хлор, определение 782
Хлоразид 55, 56, 69
Хлоранил 818
4-Хлорантраниловая кислота 264
5-Хлорантраяиловая кислота 264
Хлорапатит 272
Хлорбензилат 83
9-Хлордигидрофенарсазин 57
Хлористый азот 69
Хлорлигнин 929
Хлор (хлорметокси) метан — см. Ди-
хлорметиловый эфир
Хлорметилирование 447
Хлоробромосеребряная бумага фото-
графическая 482
Хлороксин — см. Дихлороксип
Хлоросеребряная бумага фотографичес-
кая 482
Хлорпарацид 83
Хлорпромазин — см. Аминазин
З-Хлорпропен-1 — см. Аллил хлористый
1261
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1262
ю-Хлортолуол — см. Бензил хлористый
Хлоругольная кислота, трихлорметило-
вый эфир — см. Дифосген
N i-napa~X лорфенил-N 5-изопропил-бигуа-
нид, хлоргидрат — см. Бигумаль
4-Хлорфенил-2, .4, 5-трихлорфенилсуль-
фон 83
п-Хлорфенил-п-хлорбензосульфат 83
4-Хлорфенил-4-хлорбензилсульфид 83
2-Х лор-10-(3-диметиламинопр опил)-фен-
тиазина хлоргидрат — см. Аминазин
Хондрозамин — см. D-Галактозамин
Хорионгонадотропины 996
Хризоберилл 421
Хризотил-асбест 295
Хроматотропин — см. Меланофорный
гормон
Хромовое дубление 1212
ц
Цветная бумага фотографическая 483
Цветности теории 947
Целлодаммара 1019
Целлолигнин 925
Целлофан 588
Целлюлоза 835
— ацетат — см. Ацетилцеллюлоза
— ксантогенат 586
— радиационная деструкция 1069
Центропнеин 996
Цереброза — см. Галактоза
Церезин 662
Циангалогениды — см. Галогеноцианы
Циангуанидин — см. Дидиандиамид
цианидин 259
Цианистый винил — см. Акрилонитрил
Цианистый метил — см. Ацетонитрил
Циануртриазид 56
Цикламенальдегид 1220
Циклические аминокислоты 180
Циклобарбитал — см. Фанодорн
циклобутадиен 290
Циклогексадиенин — см. Дегидробензол
Циклогексилциклогексан — см. Дицик-
логексил
Циклоны 743
Циклонал-натрий — см. Гексенал
Циклооктадекагексаен-1, 3, 7, 9, 13,
15-триин-5, 11, 17, 288
Циклооктадеканонаен 288
Циклооктатетраен 290
Циклопентадиенил 288
со-Циклопентен-2-илтридекановая ки-
слота — см. Хаульмугровая кислота
со-Циклопентен-2-илундекановая ки-
слота — см. Гиднокарповая кислота
Циклопентилциклопентан — см. Ди-
циклопентил
Циклопропенилий 288
Циклотриметилентринитроамин — см.
Гексоген
Цимарин 959
D-Цимароза 958
Циммермана — Бито реакция 225
Цинк, диметилдитиокарбамат 1162
— этиленбис (дитиокарбамат) 1163
Цинхофен — см. Атофан
Цистеин 180
Цистеиндесульфгидраза 1070
Цистин 180
Цитохимия 950
Цитраль 1220
Цитронеллальгидрат — см. Гидрокси-
цитронеллаль
Цитронеллилбутиролактон 499
Цитронеллол 1220
Цитруллин 180
ч
Четвертичные аммониевые основания 209
Чугаева реактив — см. Диметилглиок-
сим
Чугаева — Церевитинова метод опреде-
ления — см. Активный водород
ш
Шеелит 652
Шелк (волокно) 647
Шерсть (волокно) 647
Шерстяной воск 662
Шиффовы основания 173
Шредингера уравнение 312
щ
Щавелевоуксусная кислота 138
Щелочность воды 611
Эвгенол 1220
Эвглобулины 976
Эвипан 372
Эвипан-натрий — см. Гексенал
Эвтектика 1026
Эвтектическая точка 1026
Эвтектоид 1031
Эйборнил 518
Эйнштейний 105
Экзогенные геохимические процессы 843
Экзоцистеиндесульфгидраза 1070
Экранирования постоянная 316
Экстракция 19
Элатин 86
Электрическая очистка газов 748
Электрическая прочность 1185
Электрические эффекты высокого давле-
ния 690
Электрический слой двойной 1022
Электрическое деэмульгирование 1079
Электровесовой анализ 536
Электродиализ 1093
Электрокортин — см. Альдостерон
Электролиз, выход по току 712
--- внутренний 601
Электролитическое восстановление ор-
ганических соединений 664
Электролиты, гидратация 888, 890
Электронная оболочка 315
Электронная поляризация 1132, 1138,
1183
Электронное облако 310
Электронный слой 315
Электропроводность газов 1184
---диэлектриков твердых 1185
--- жидкостей 1184
Электрофильтр 747, 748
Элементоорганические высокомолеку-
лярные соединения 697, 703
Элементы химические, миграция 846
— периодич. система 319—322
Элсона — Моргана реакция 954
Эльделин 86
Эмбриохимия — см. Биохимия
Энант 195
Энантиомеры — см. Антиподы оптиче-
ские
Энглера градусы 718
Эндогенные геохимич. процессы 843
3, 6-Эндоксогексагидрофталевая кислота,
динатриевая соль — см. Эндотал
Эндотал 1075
Энергия адсорбции 42
--- внутренняя 602
--- ионизации 311, 317
--- свободная, принцип линейности 804
Энтропия 669
Эпикатехин 594
Эпикатехингаллат 594
Эпинефрин — см. Адреналин
Эргоалкалоиды — см. Алкалоиды спо-
рыньи
Эргозин 120
Эргозинин 120
Эргокорнин 120
Эргокорнинин 120
Эргокриптин 120
Эргокриптинин 120
Эргокристин 120
Эргокристинин 120
Эргометрин 120
Эргометринин 120
Эрготамин 120
Эрготаминин 120
Эриодиктин 594
Эритокруорины 838
Эритородекстрины 1058
Эритрен — см. Бутадиен-1,3
Эритромицин 239
Эрлиха реакция 432
Эруковая кислота 709
Эскулин 594
Эстеротанниды 1214
Эстрадиол 996, 998
Эстриол 996
Эстрих-гипс 714, 715
Эстрогенные гормоны 998
Эстрон 996
Этаминалнатрий 372
Этаналь — см. Ацетальдегид
Этилакрилат 88
Этилаль 339
Этилантранилат 264
5-Этил-5-(1-метил-бутил)-барбитуровая
(Na-соль) кислота — см. Этаминалнат-
рий
5-Этил-5-(1 -циклогексан-1 -ил) -барбиту-
ровая кислота — см. Фанодорн
Зтилбензоат 409
Этилборат — см. Борноэтиловый эфир
2-Этилбутанол-1 820
Этилбутилсульфат 127
Этилванилин — см. Ванилаль
Этилен бромистый — см. 1, 2-Дибромэтан
--- хлористый — см. Дихлорэтан
Этиленбис (дитиокарбаминовая кислота,
Na-соль) 1163
Этиленкарбоновая кислота — см. Акри-
ловая кислота
Этиленмолочная кислота — см. Гидрак-
риловая кислота
Этиленовые углеводороды — см. Непре-
дельные углеводороды
1-Этилиденпропионовая кислота — см.
Ангеликовая кислота
5-Этил-5-изоамилбарбитуровая кисло-
та — см. Амитал
Этил-изо-амилсульфат 127
Этил-изо-бутилсульфат 127
Этил-изо-пропилсульфат 127
5-Этил-5-(3?метил-бутил)-натрий барби-
турат — см. Барбамил
5-Этил-5-(1-метил-бутил)-2-тиобарбитуро-
вая кислота — см. Тиопенталнатрий
Этилнафталины 126
Этиловая жидкость 247
Этиловый спирт 247
Этиловый эфир — см. Диэтиловый эфир
Этилпропилсульфат 127
Этилсерная кислота 127
5-Этил-5-фенил-барбитуровая кислота —
см. Люминал
Этилфосфиновая кислота, этиловый пара-
нитрофениловый эфир — см. Армин
Этилэтилен 493
Этин — см. Ацетилен
Эуродины 57
Эфир сульфонат 83
Я
Ядро атома, заряд и масса 307
Ядро Земли 845, 847
Янтарная кислота, бис-р-дпметилами-
ноэтилового эфира дииодметилат —
см. Дитилин
Яра-яра — см. р-Нафтол, метиловый
эфир
ОПЕЧАТКИ И ИСПРАВЛЕНИЯ в 1-м томе КРАТКОЙ ХИМИЧЕСКОЙ ЭНЦИКЛОПЕДИИ
Ко- лонка Статья Строка статьи Напечатано Должно Сыть
35 Адкинса Катализатор 12—13 сверху Входящая в состав А. к. окись никеля Входящая в состав окись хрома
130 Аллены 4 сверху Ri\ /Нз )с=с( Rjt 414 В1\ /Вз \с=с=с< Ra^ 'R,
346 Ацетилен 5 сверху т. КИП. 83,8°; т. кип. —83,8°
» То же 7 сверху 58 ккал/моль 54,94 ккал/моль
353 Ацетон 6 сверху — — 30,87 ккал/моль == 30,87 кал/моль
357 Ацетоуксусный эфир 33—34 сверху и соответственно переходит в метил- оксазолон (Ш) и соответственно переходит в метилизо- оксазолон (Ш)
411 Бензол 24 снизу теплота сгорания жидкогоВ. 749,42 ккал/моль теплота сгорания жидкого Б. 782,2 ккал/моль
> То же 5 снизу Б. дает с водой азеотропную смесь Б. дает с водой гетероаэеотропную смесь
458 Борноэтиловый эфир 4 сверху т. кип. 188,6° т. кип. 118,6°
491 Бутаны 18 снизу Теплота образования, ккал/моль 1044,8; 1046,4 Теплота образования, ккал/моль 30,15; 32,15
» То же 12 снизу Теплота испарения 5035 ккал/моль Теплота испарения 5,035 ккал/моль
573 Винилхлорид 2 сверху СН2 - СНС1 СН2 = СНС1
614 Вода тяжелая 12 снизу К = [HDO]2/[H«O] • [D2O] (при 25°) К = [HDO]2/[H2O1 • [D2O] = 3,80 (при 25°)
812 Гвоякол 3 сверху Призматич. кристаллы, т. пл. 98,4° Призматич. кристаллы, т. пл. 28,4
855 Гептан То же 5—6 сверху 7 сверху теплота горения жидкого Г. (при 25°) 1167,11 ккал/моль ДН2в8 = 47,52 ккал/моль Теплота сгорания жидкого Г. (при 25°) 1151,27 ккал/моль = ~~ ^3,63 ккал/моль
Ш1 Дизельное топливо И сверху 270 -400° 200-400°
1117 3,4-Димеркаптопропанол 12 сверху Аз, В1, Н2О As, Bl, Hg
1118 п- Диметиламиноазофе- ниларсоновая кислота 2 снизу ; для открытия Zn (—21 (0) (+2! +4) 4SnO -* Sn + SngOi* (4-4' ‘ (-2) (4-6) ; для открытия Zr (4-2> (0) (4-3» 4-4)
1154 Дпсп ропорционирование 12 сверху 4SnO -> Sn -j- Sn2O.;
» То же 13 сверху 3R—SOON -> R—S-SO-R + -f-RSOa-OH~f-HaO (4-5) (-6) (4-4) 3R-SOOH -> R-S-SO»R b 4- RSOaH-l-HaO (+5> , <+0> <+41
» То же 14 сверху 2NpOj - NpO^+ NpO (4-6) 2NpOj - NpOj+4- NpOa (4-fl)
» То же 15 сверху 3Pu4+ 4- 2Hg,0 - 2Pu3++ PuO^- 4-4H+ 3Pu’+ 4- 2HaO - 2Pu’+ 4- PuOJ+ 4- 4H+
1165 Дифенилметан 2—3 сверху т. пл. 26—27°; т. кип. 261—2627760 мм t. пл. 25,24°; т. кип. 264,277760 мм
1225 Енолы 6—5 снизу ацетона, 2,5 . 10-2 % циклогексанона, 4,8 • 10~1% циклопентанона в ацетоне, 2,5 • 10-2 % в циклогексаноне, 4,8-10-1% в циклопентаноне
Краткая химическая энциклопедия. Ред. кол.
И. Л. Кнунянц (отв. ред.) и др., т. 1 —
М., «Советская Энпиклопедия»>. 1961.
т. (Энциклопедии. Словари. Справочники).
т. 1. А — Е. 1961. 1262 стб. с илл.
Сдано в набор 22 ноября 1960 г. Том подписан к печати 17 мая 1901 г.
Государственное научное издательство «Советская Энциклопедия». Москва. Ж-28. Покровский бульвар, д. 8.
Т-04284. 17/V 1961 г. Тираж 70 тыс. экз. Заказ № 1451. Формат 82X108716. Объем 64,78 усл. п. л., 39,5 физич. и. л.
Уч.-изд. л. 116,49. Цена 1 экз. книги Зр. 30 к.
Ленинградский Совет народного хозяйства. Управление полиграфической промышленности. Типография № 1 «Печатный
Двор» имени А. М. Горького. Ленинград, Гатчинская, 26.