/














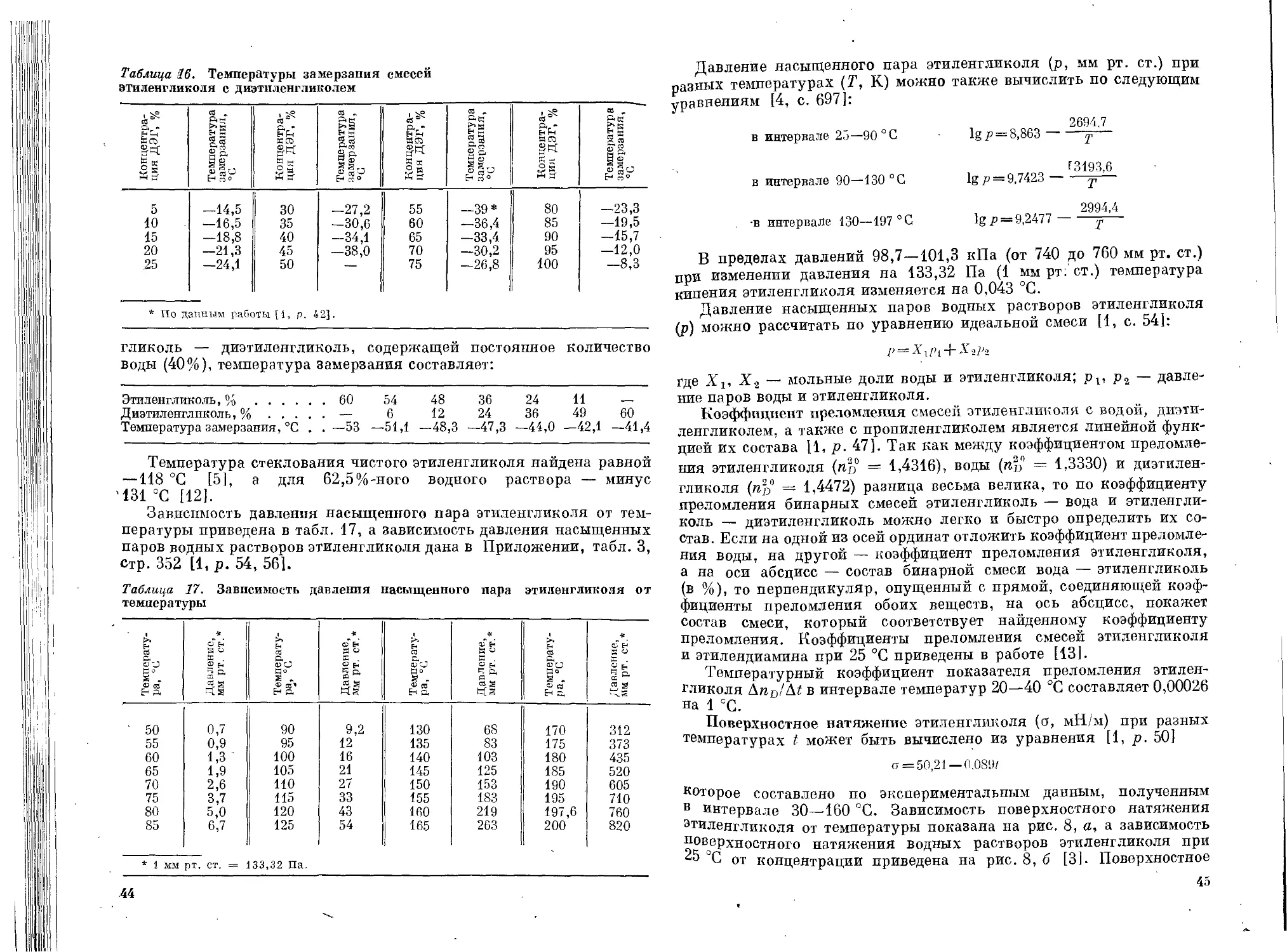
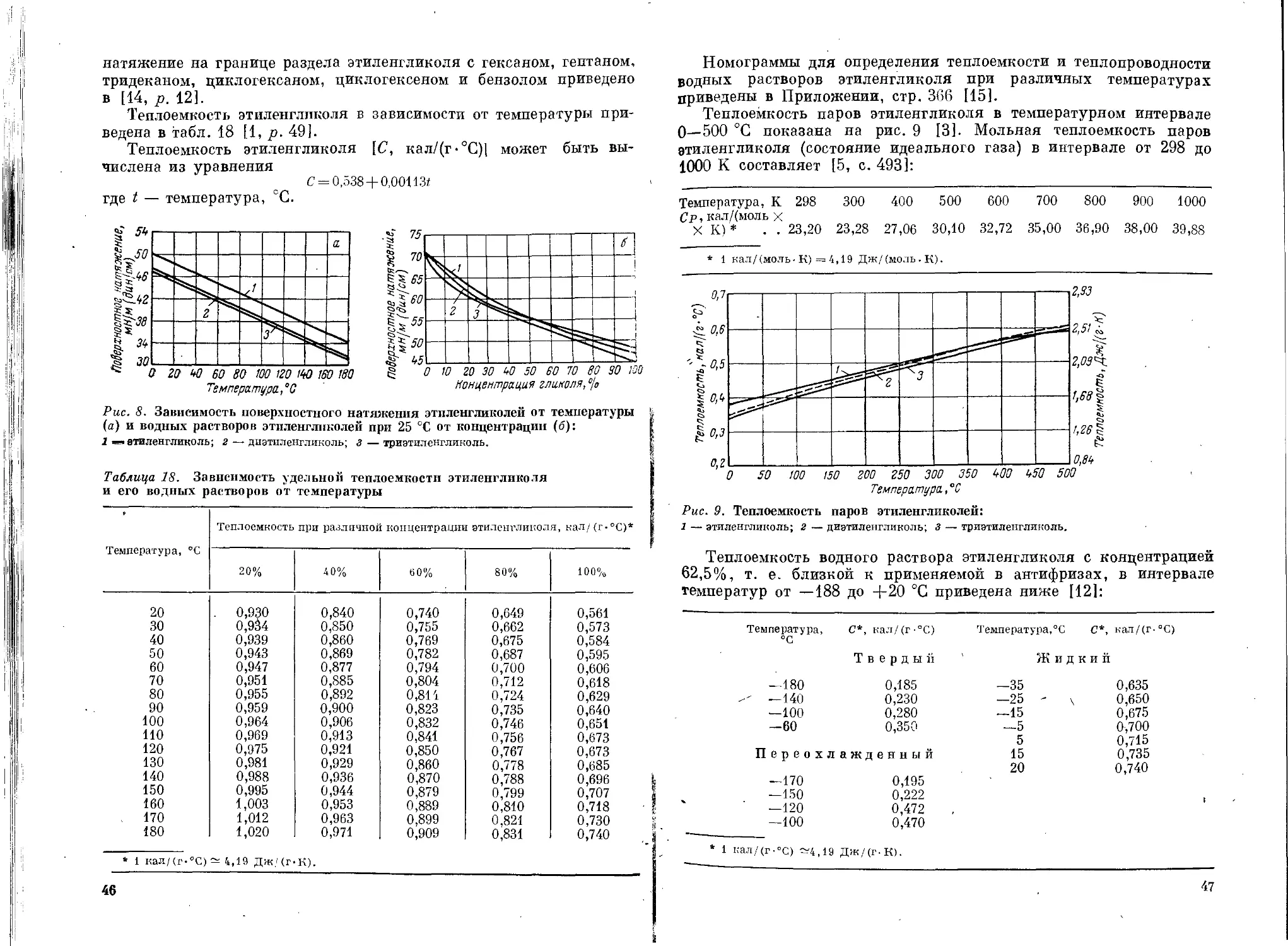
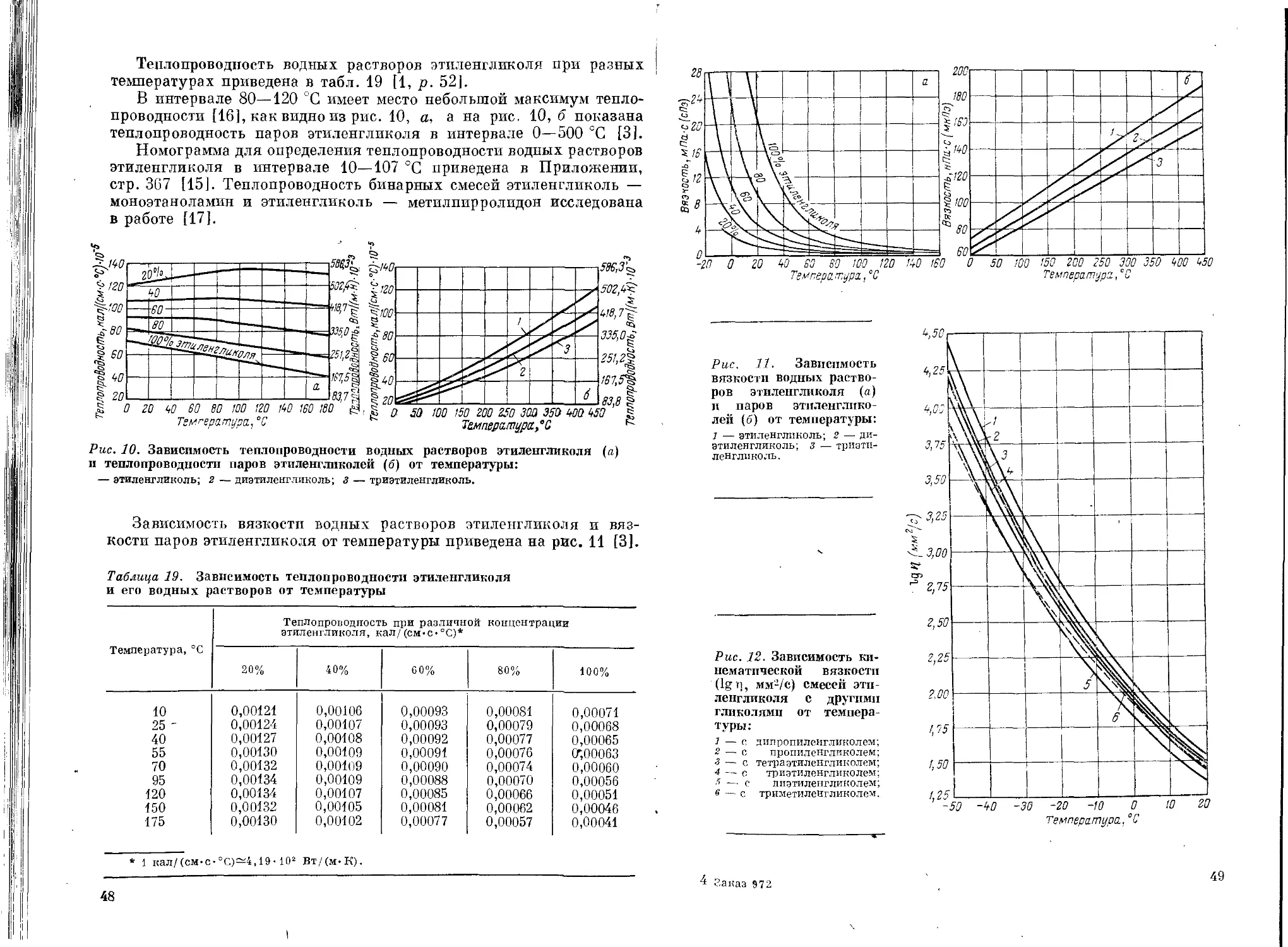
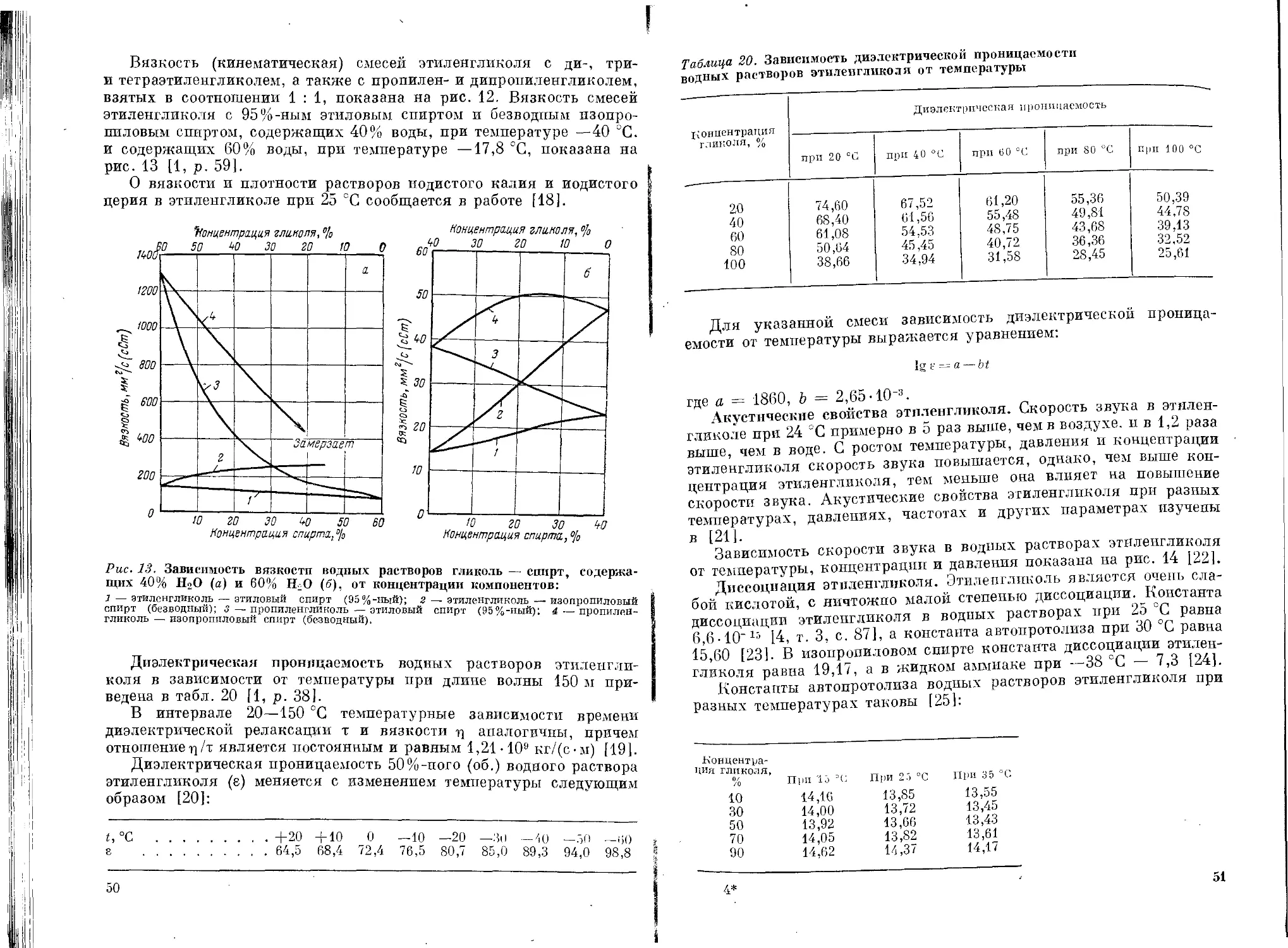
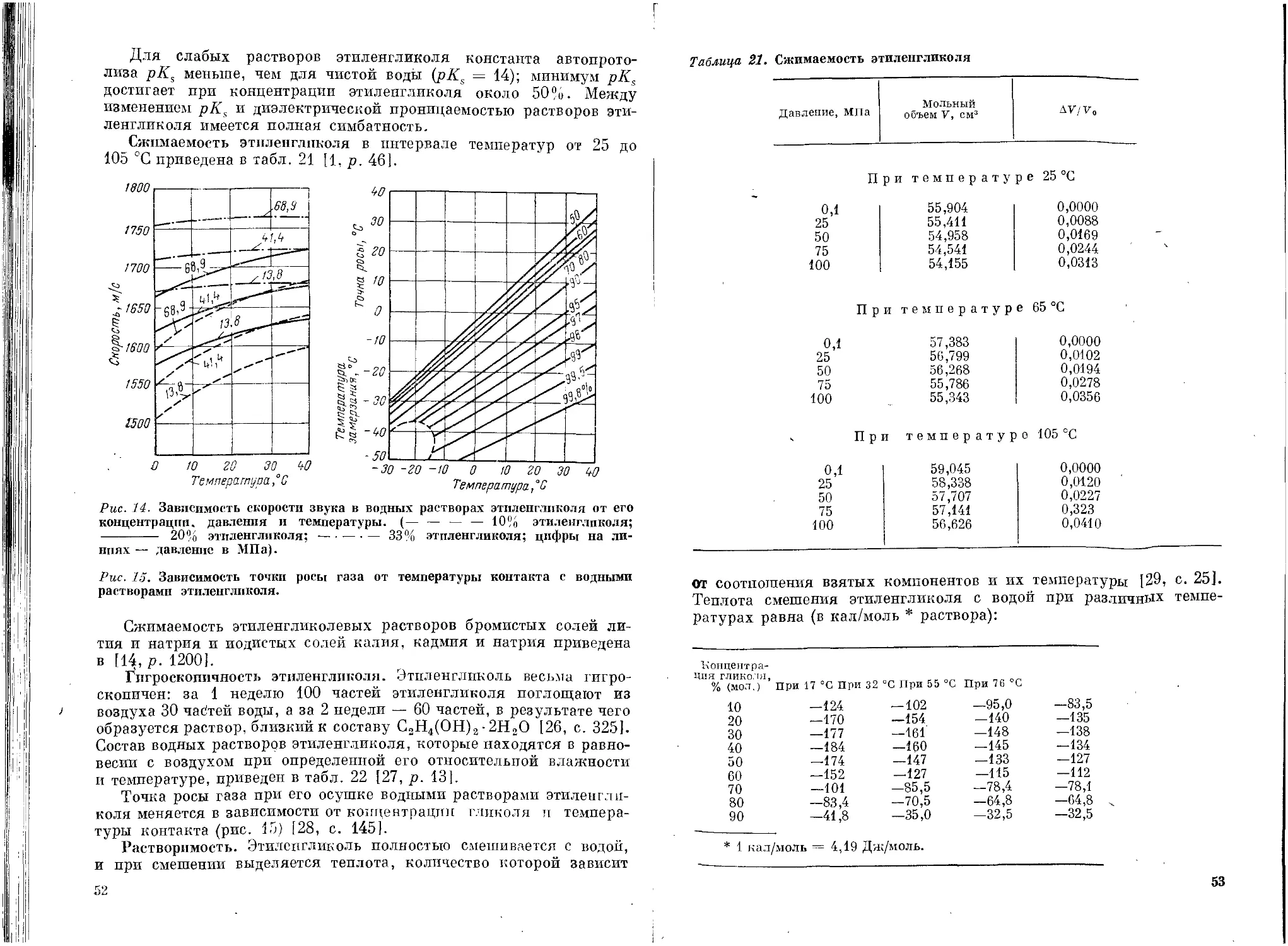

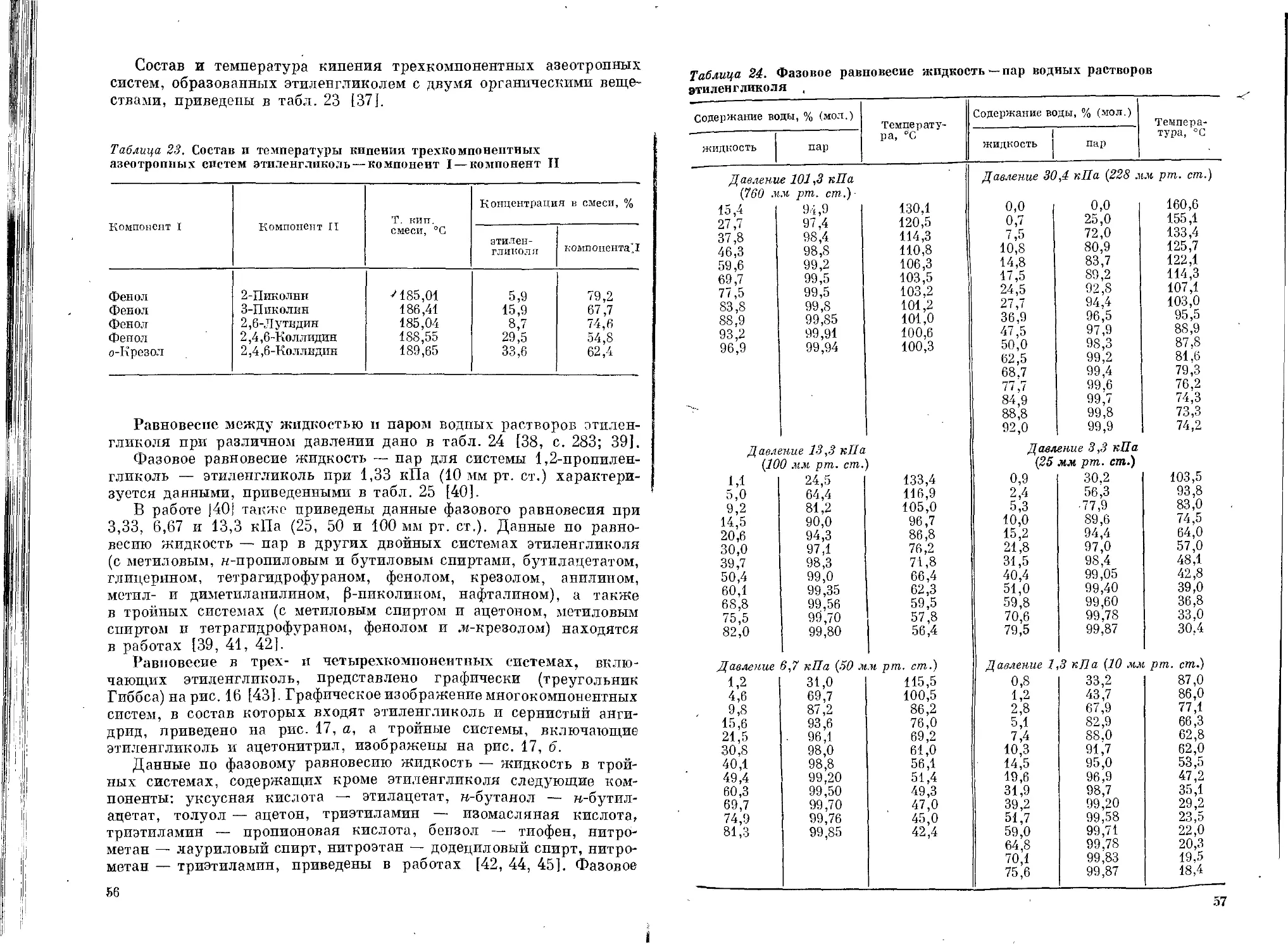
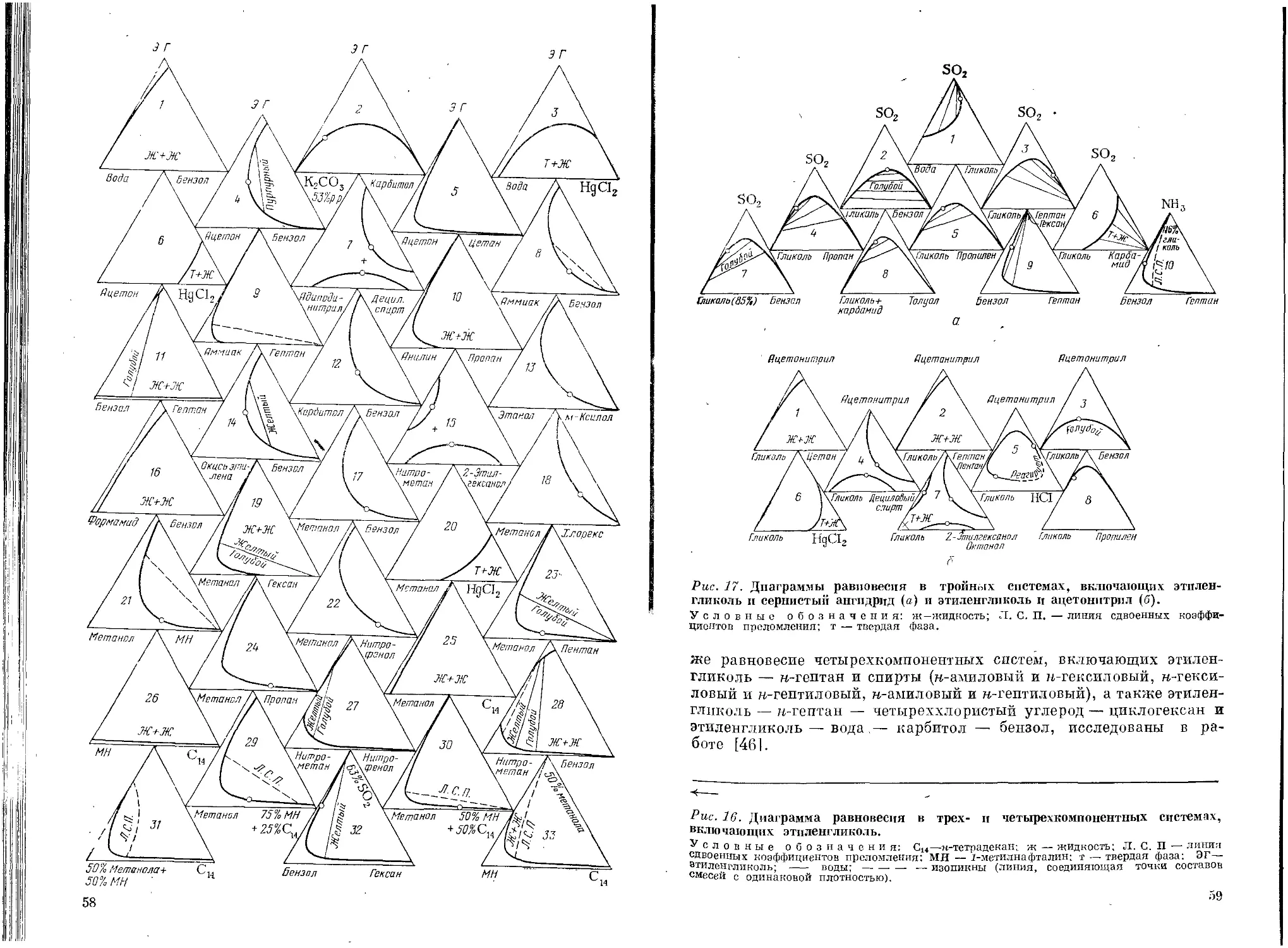





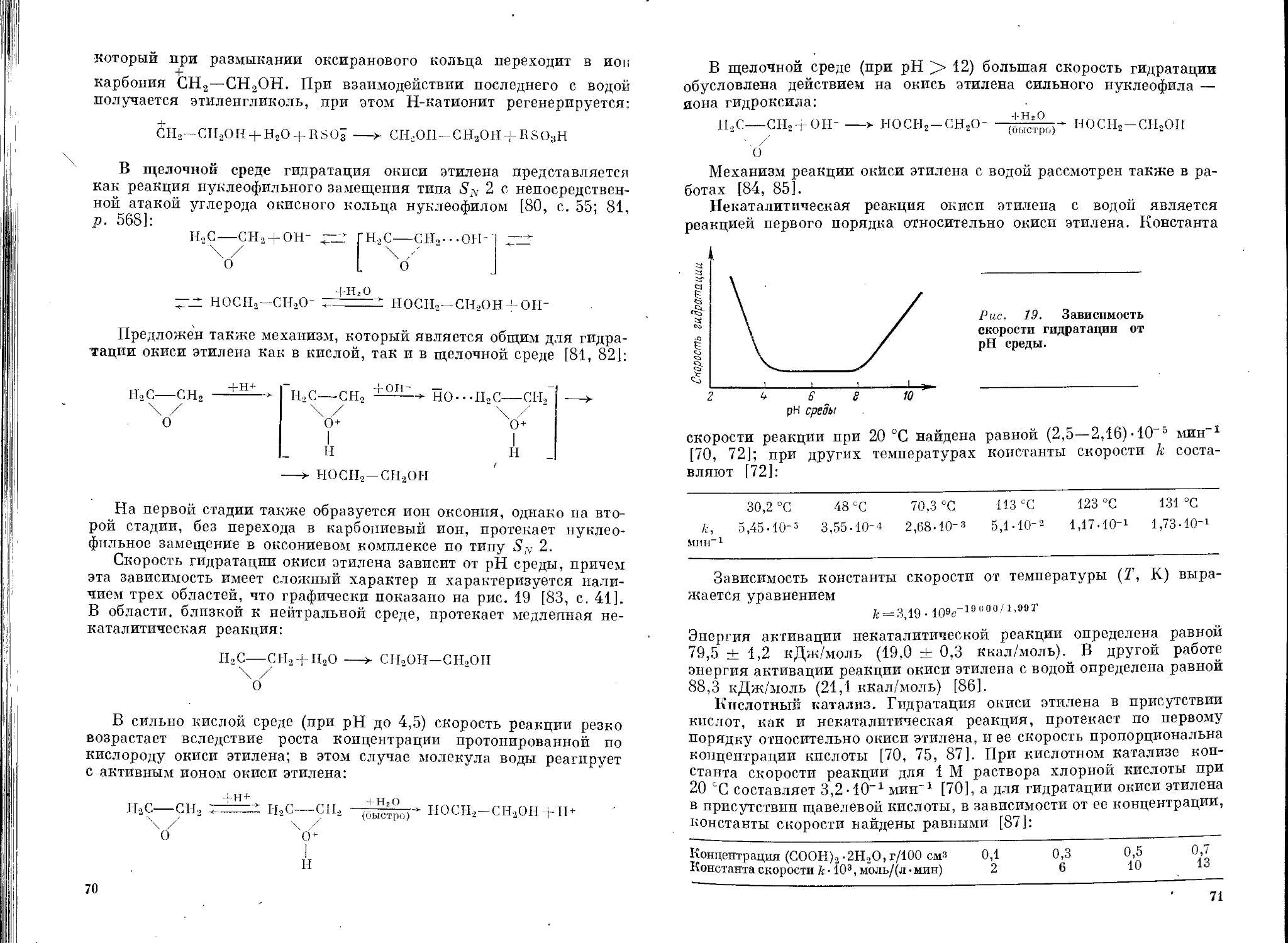






















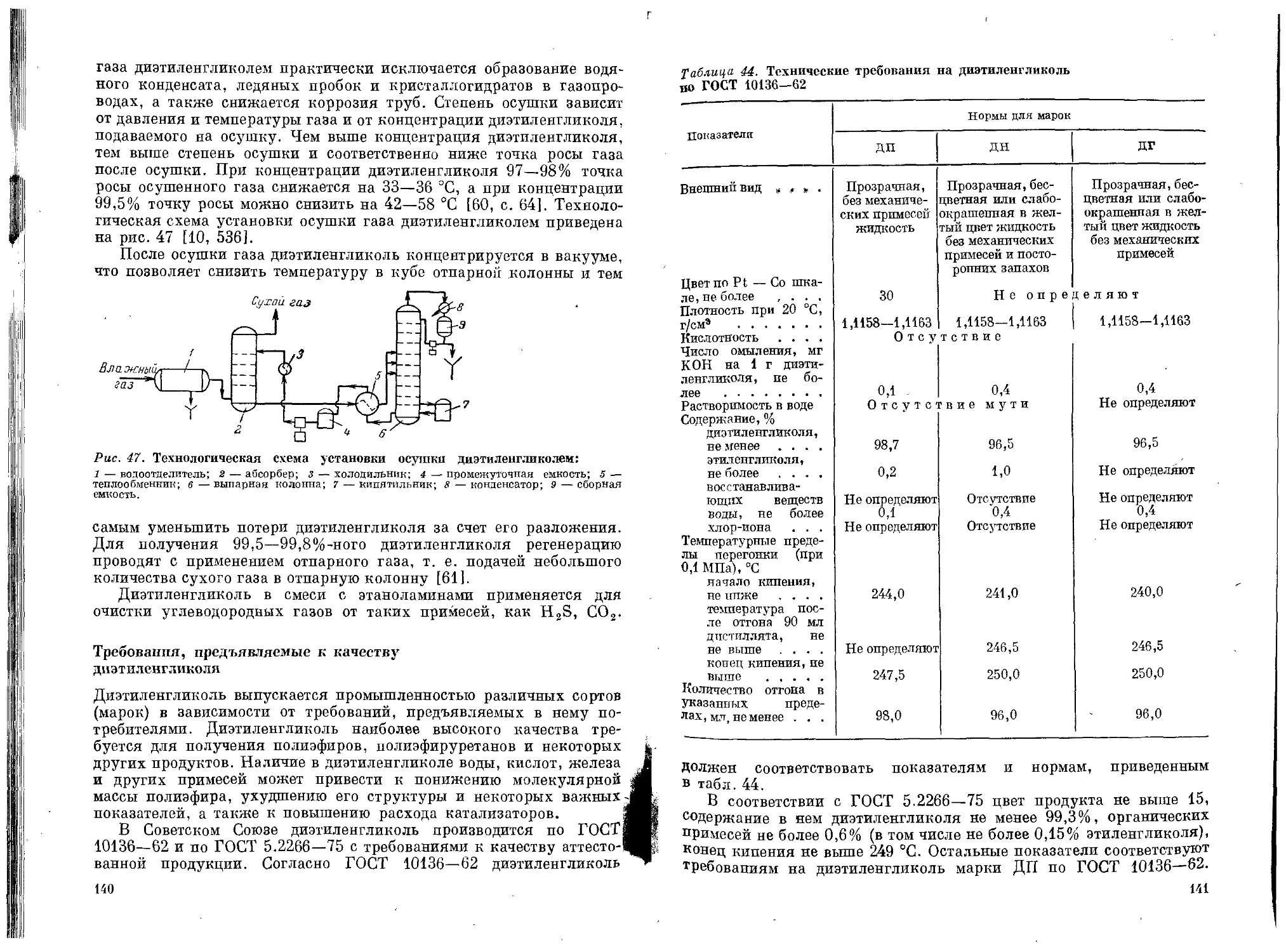



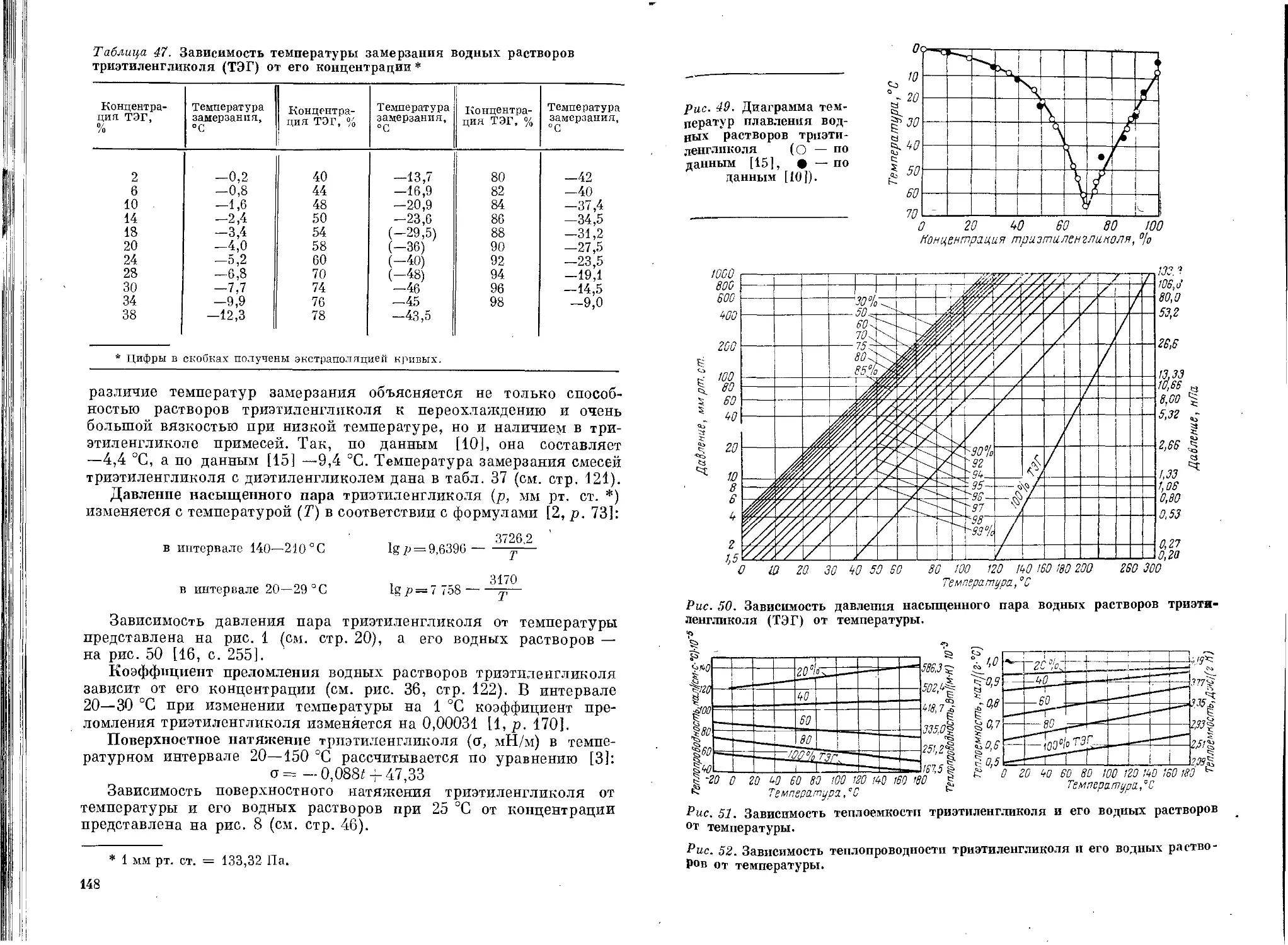

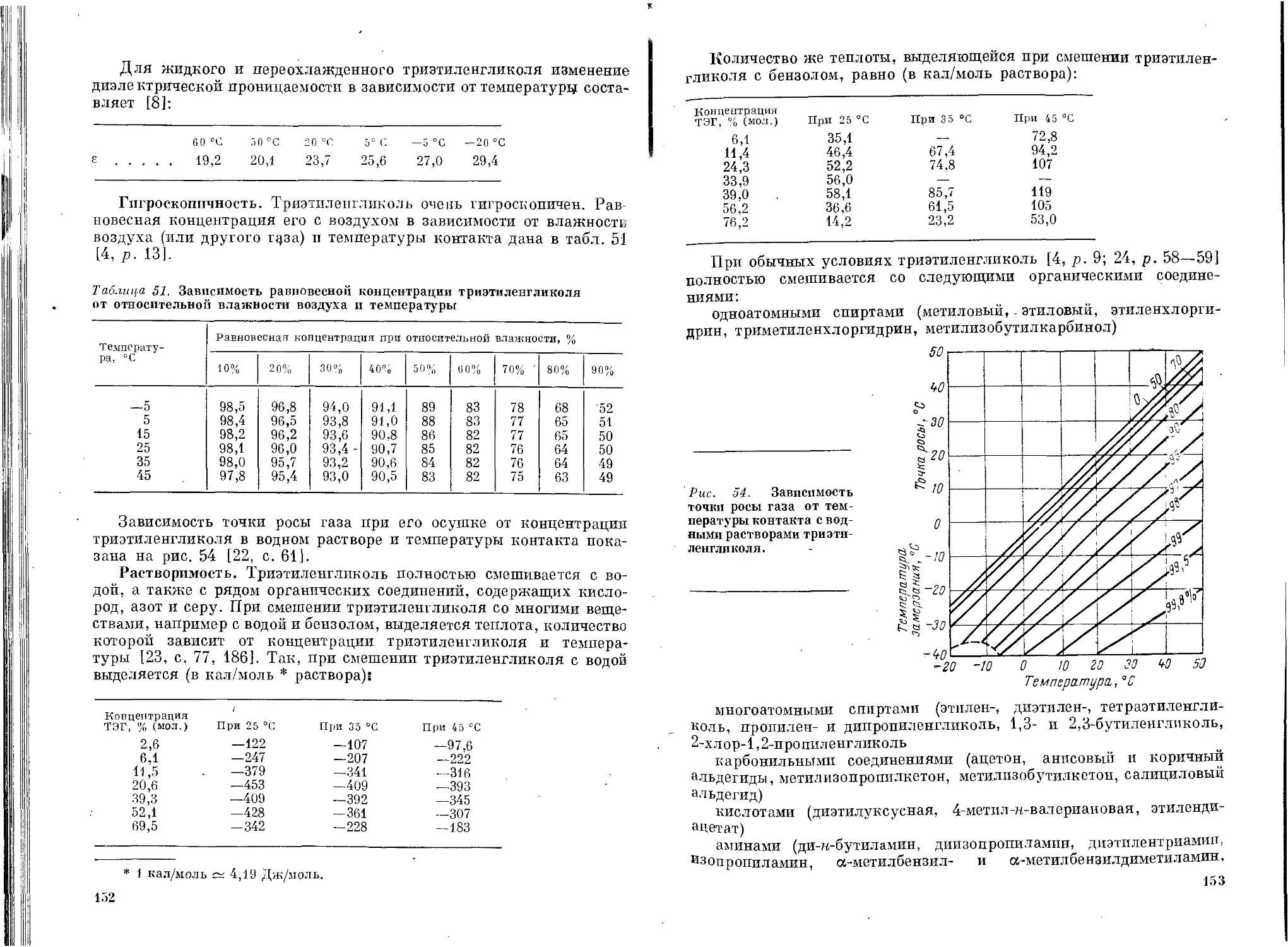
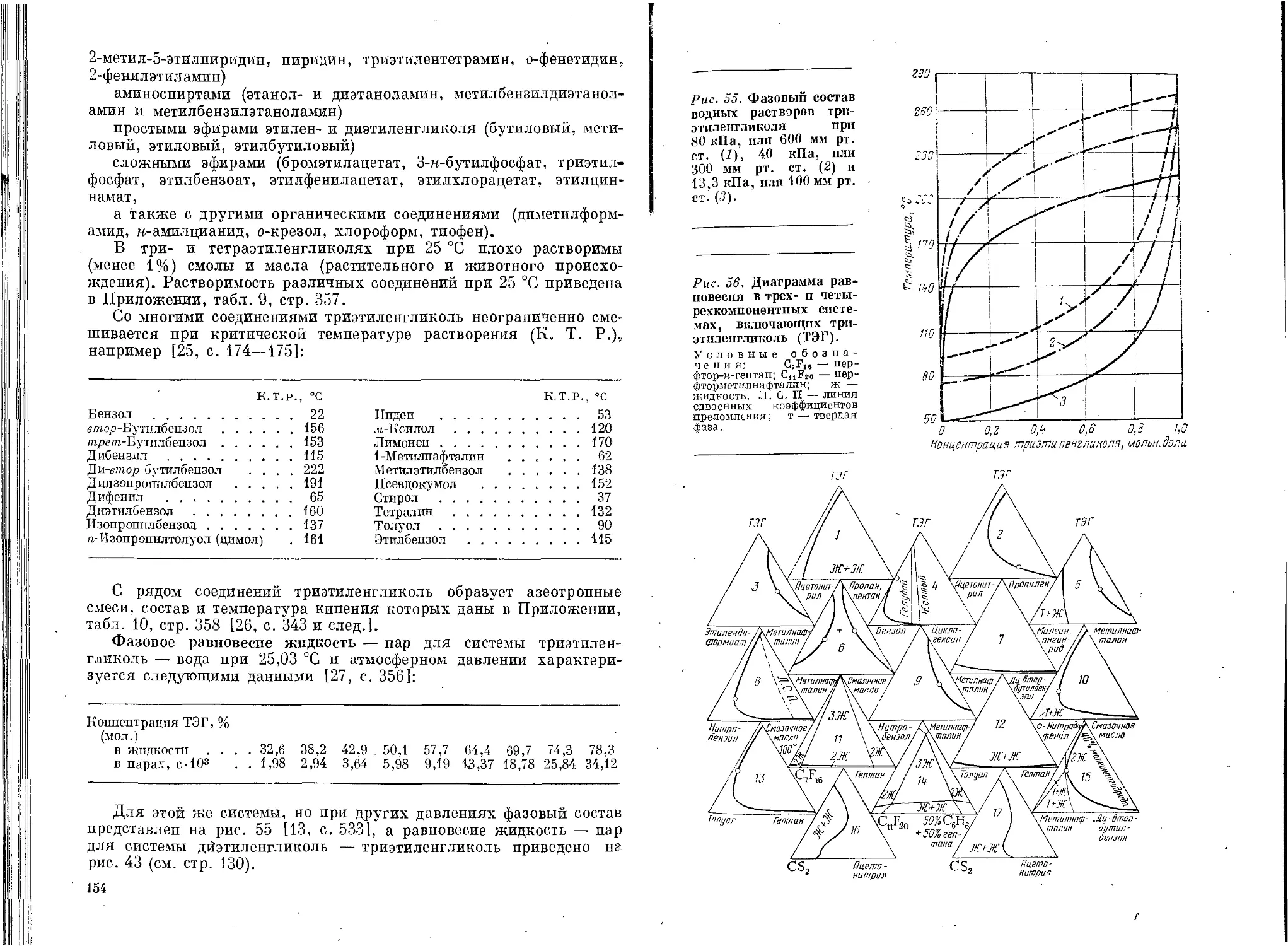








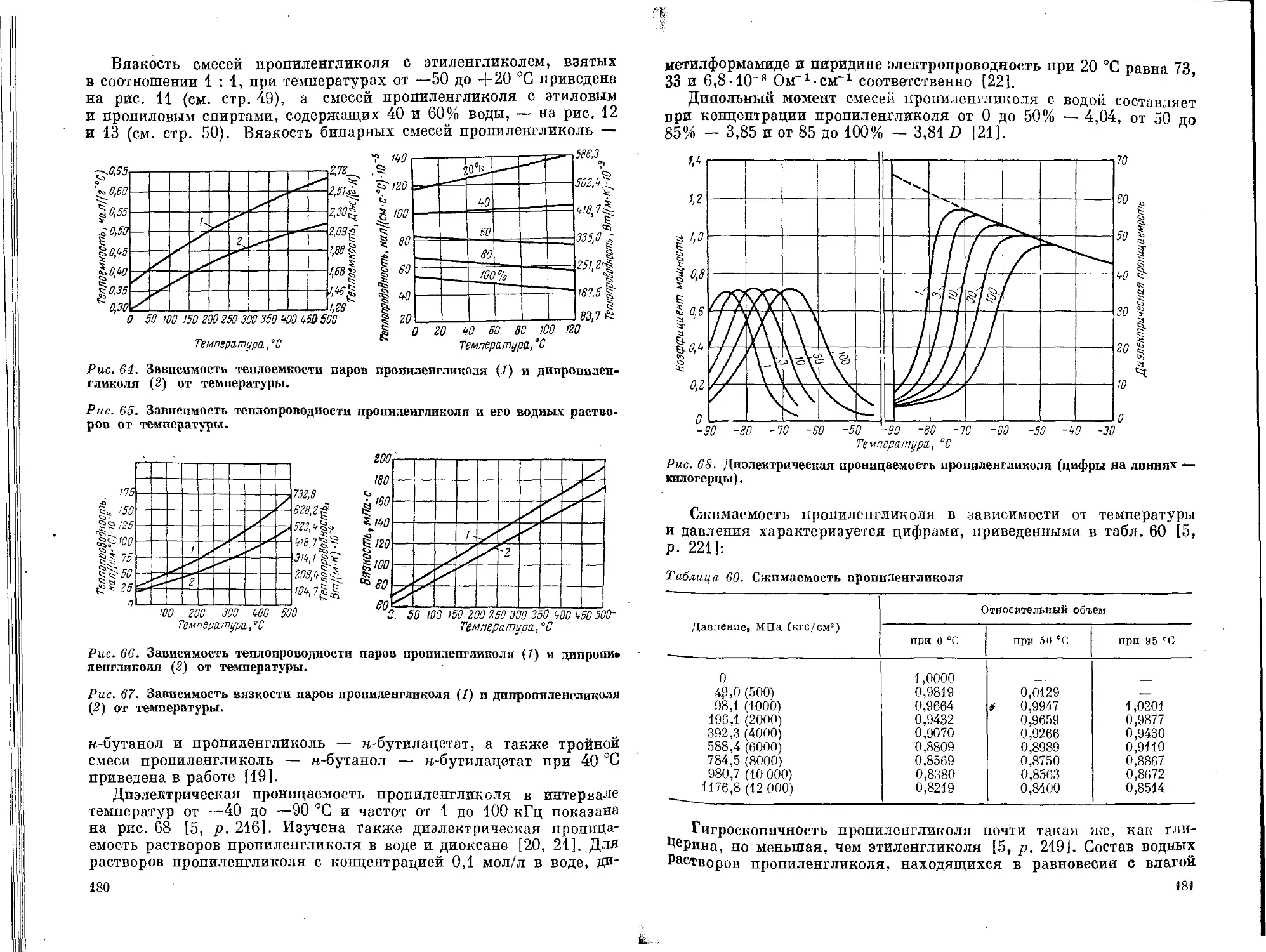
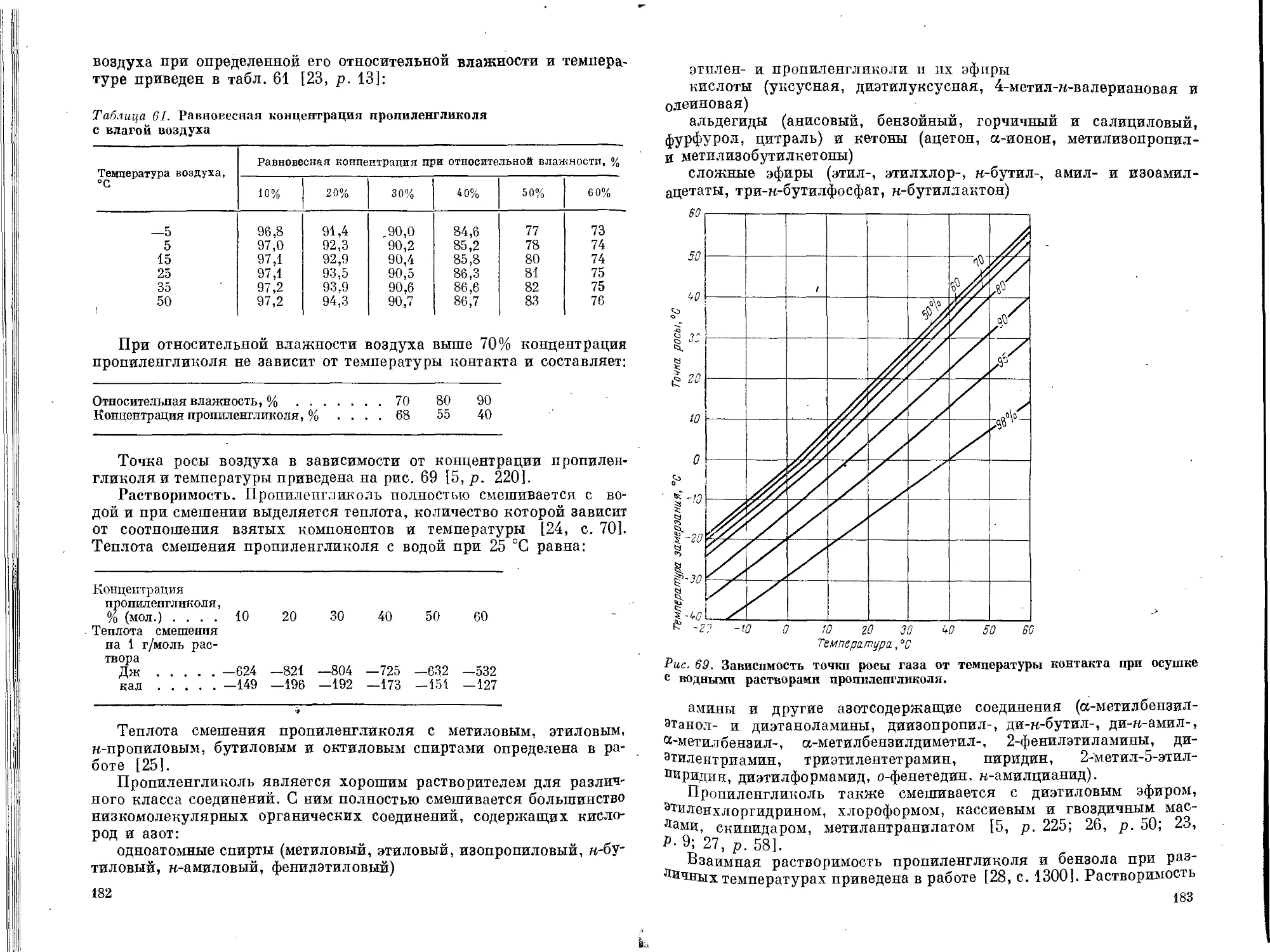
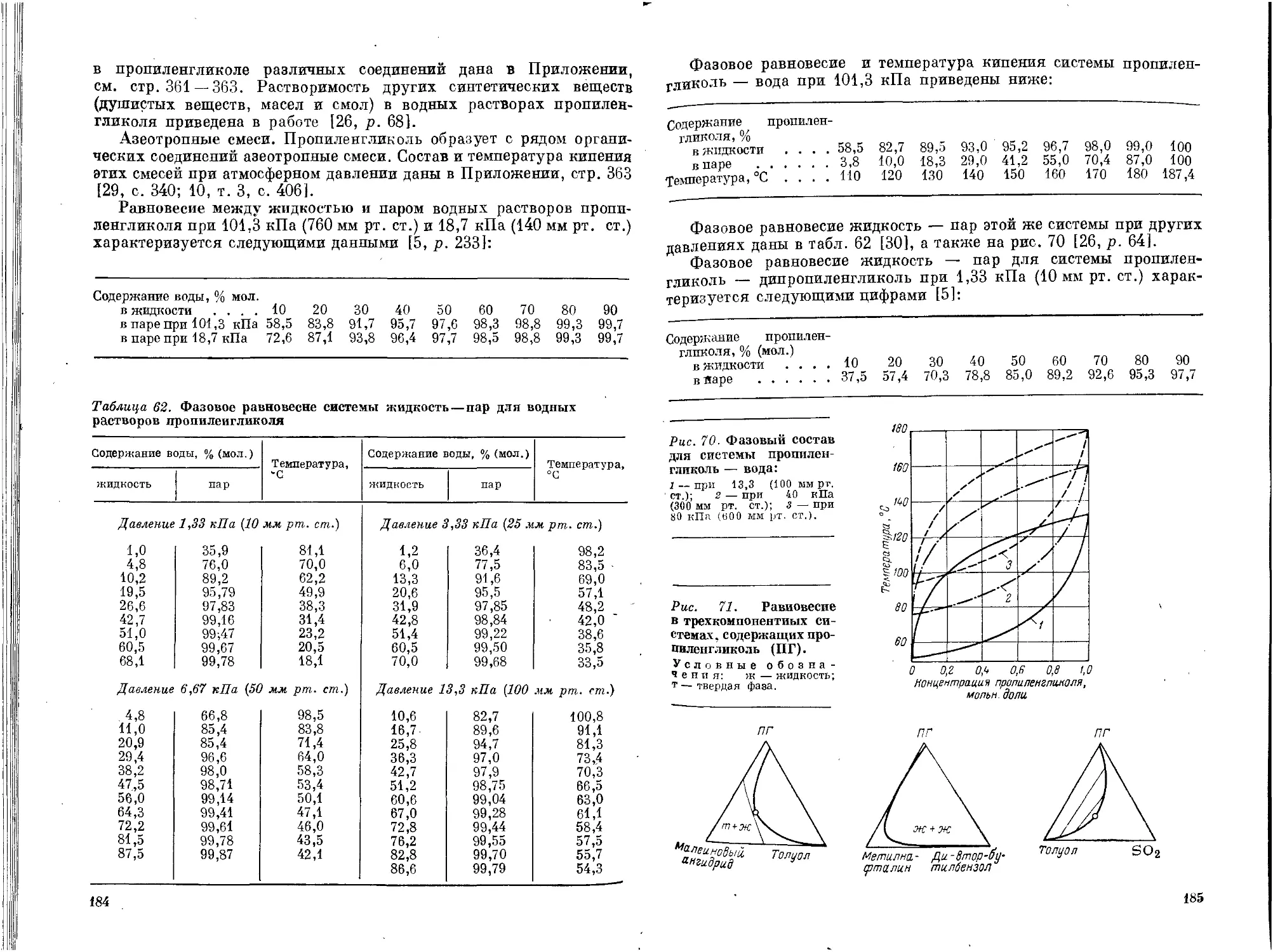

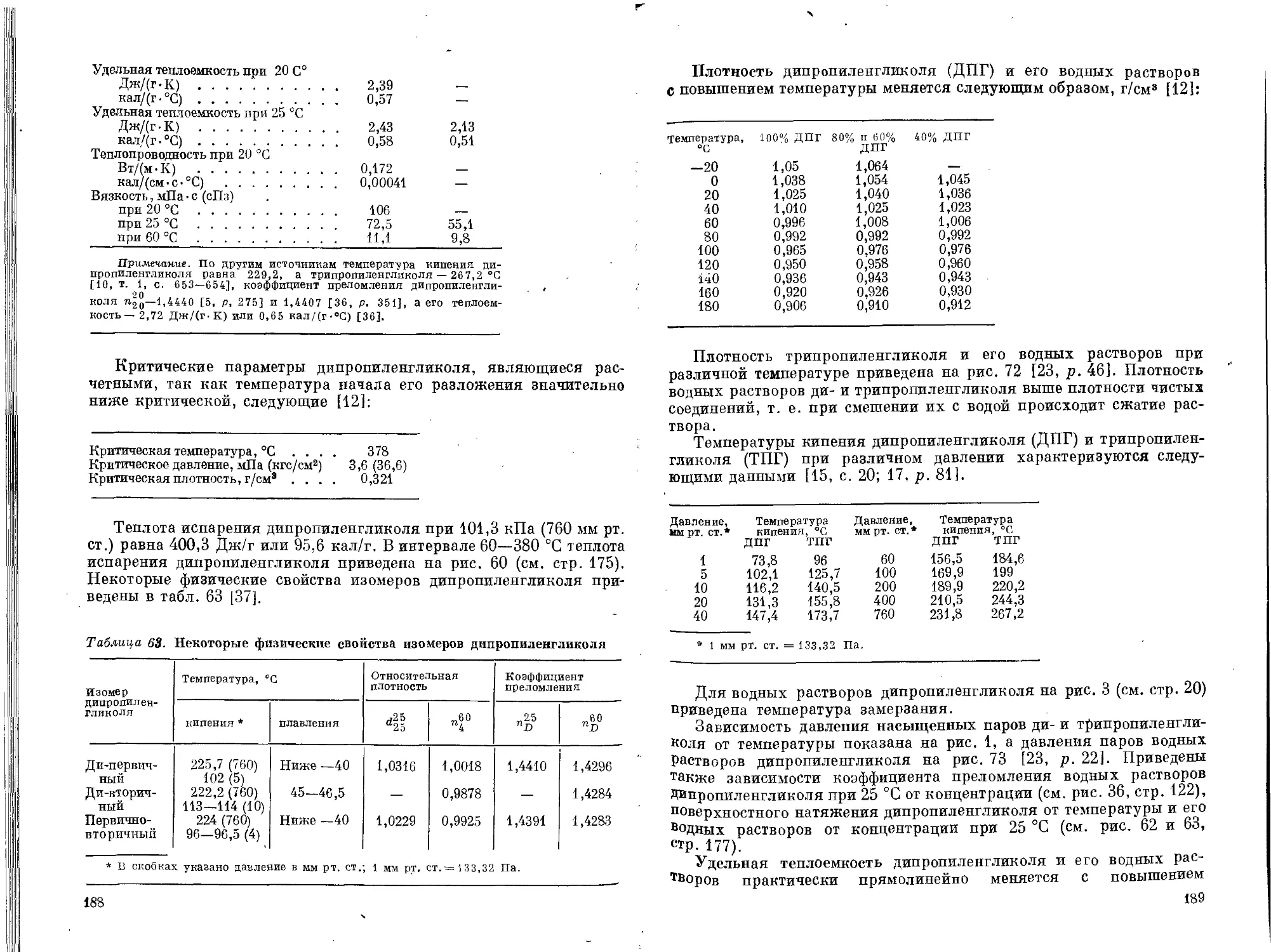



























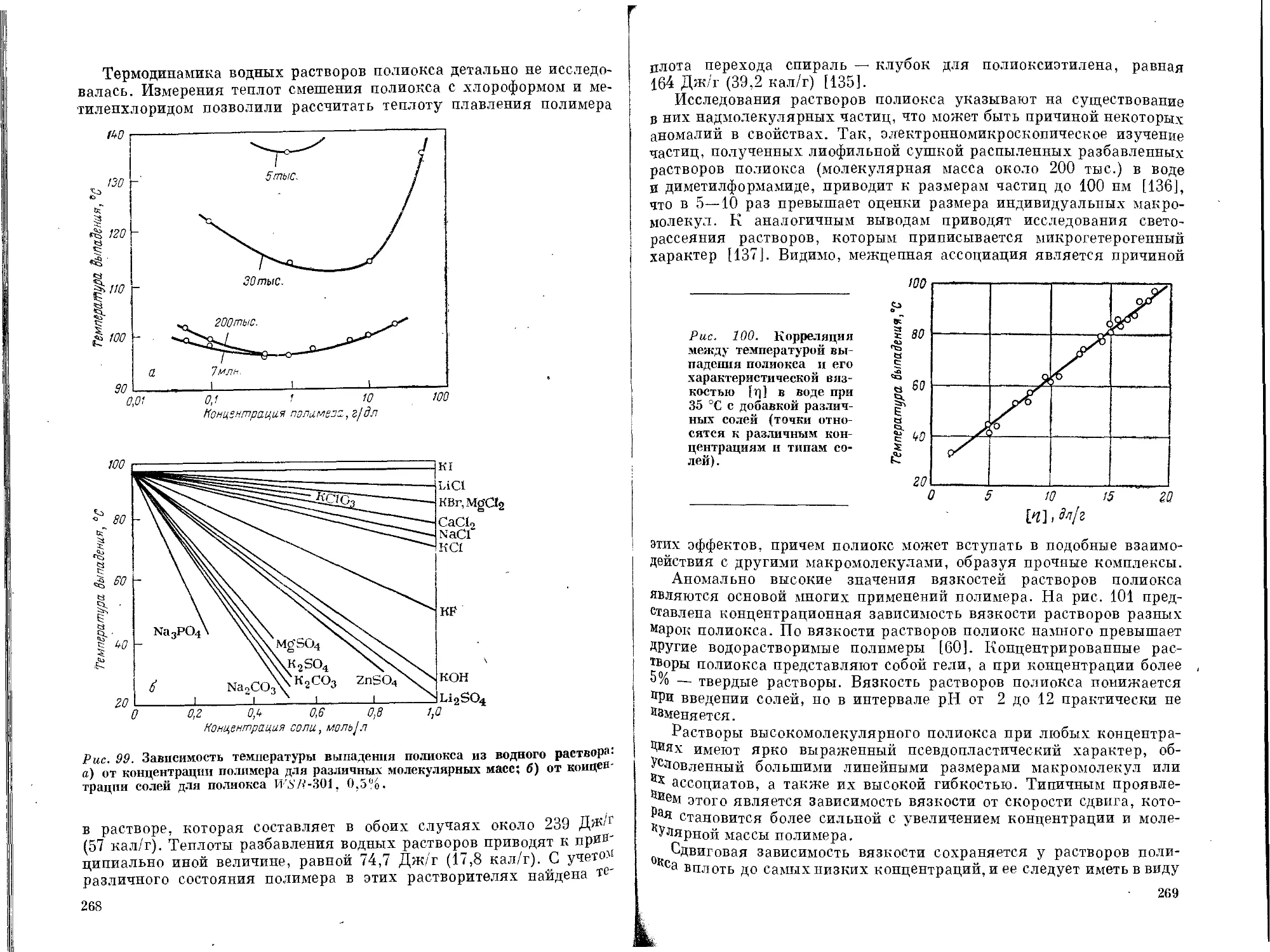





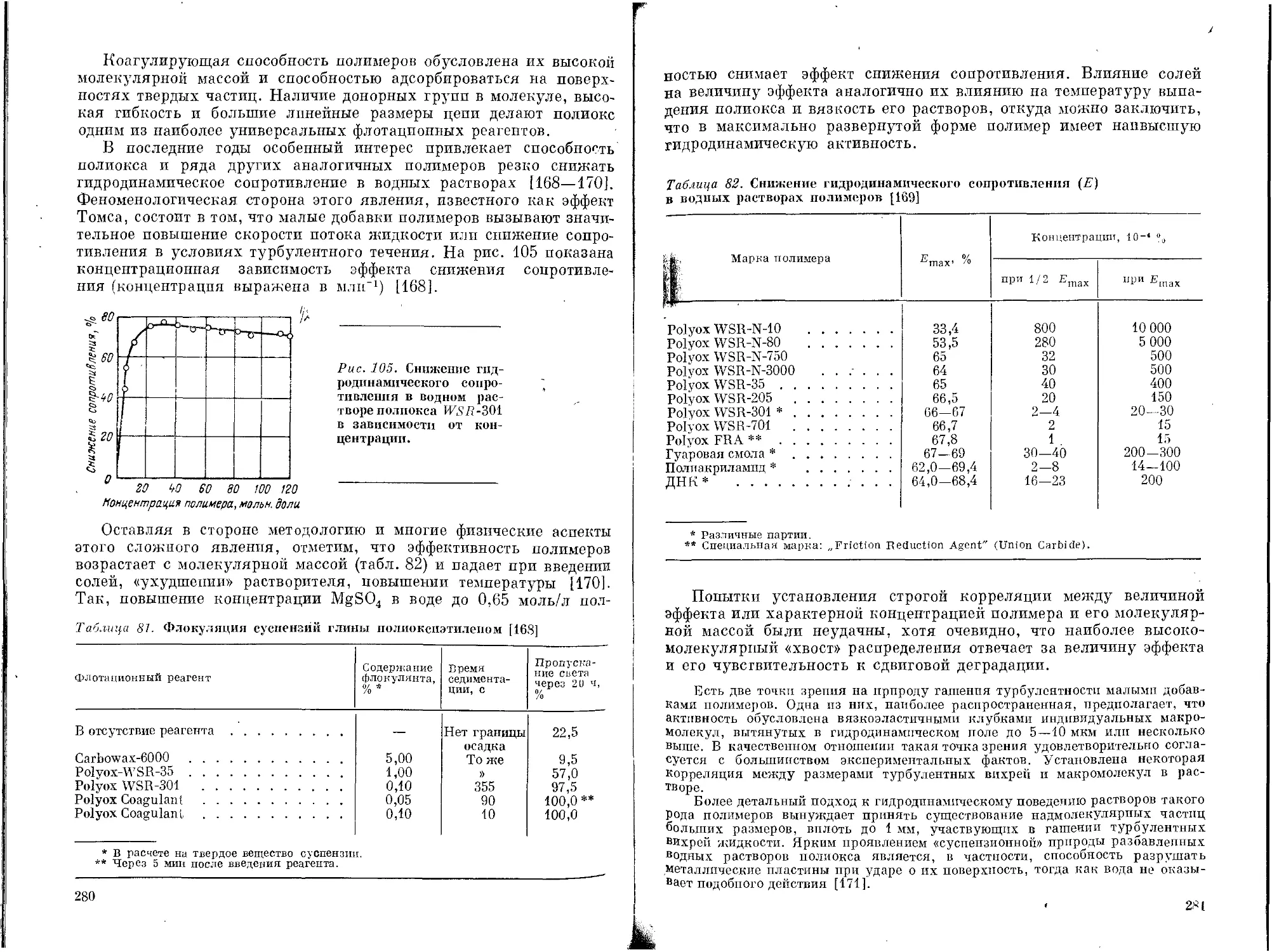





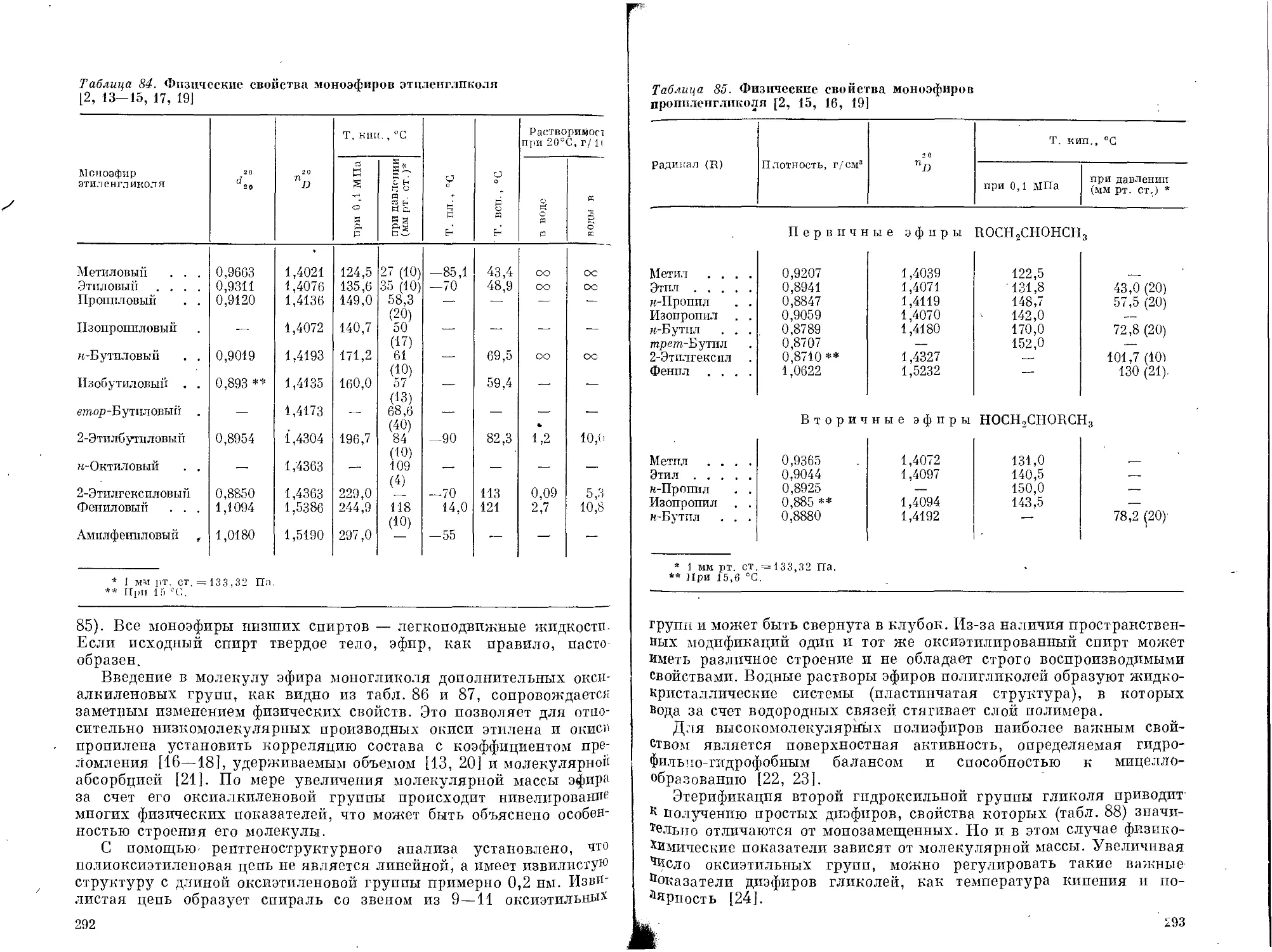
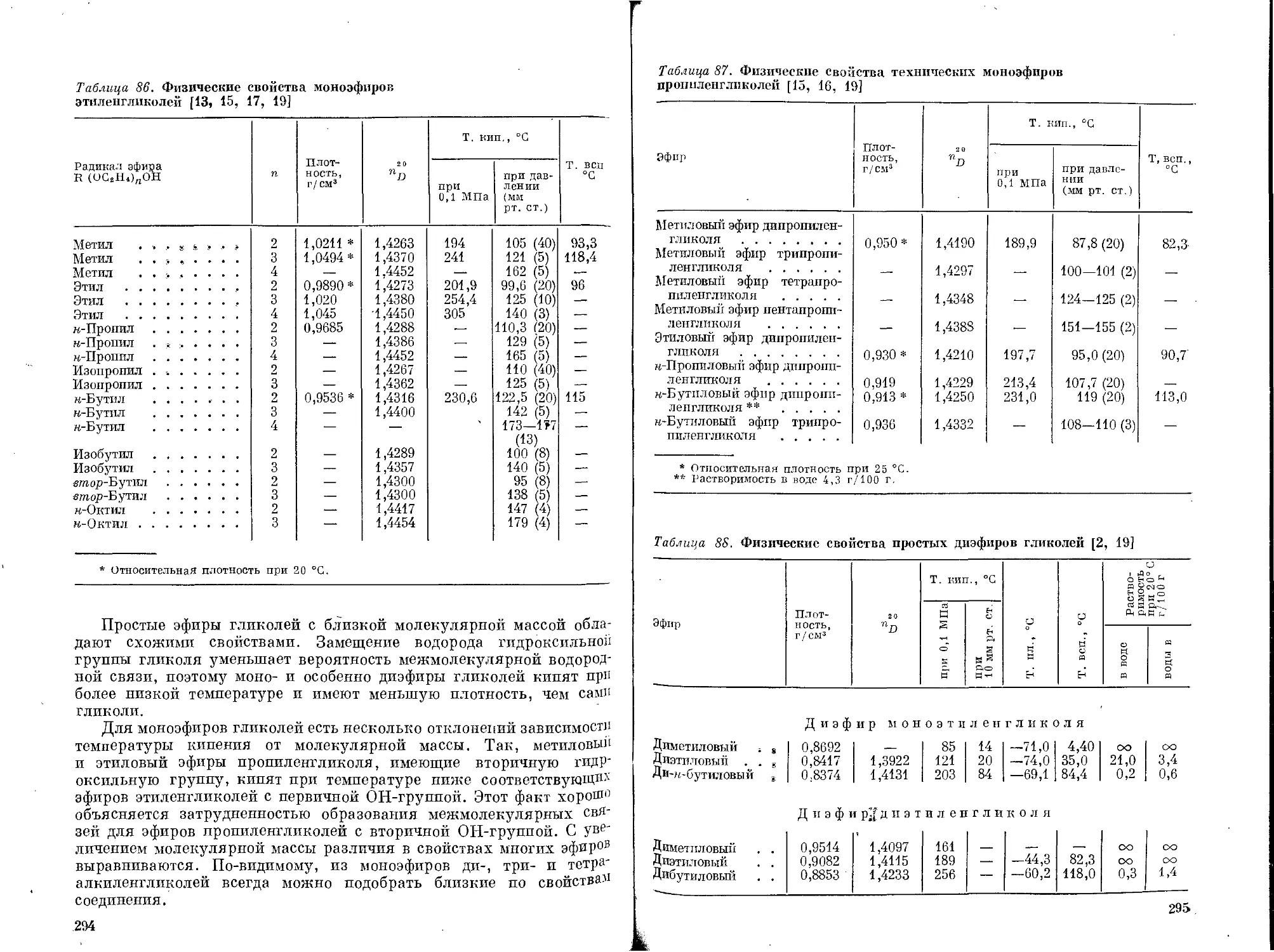

































Текст
Москва
Издательство
«Химия»
1976
О. Н. Дымент,
К. С. Казанский
А. М. Мирошников
Гликоли
и другие
производные
окисей
этилена
и пропилена
Под общей редакцией
О. Н. Дымента
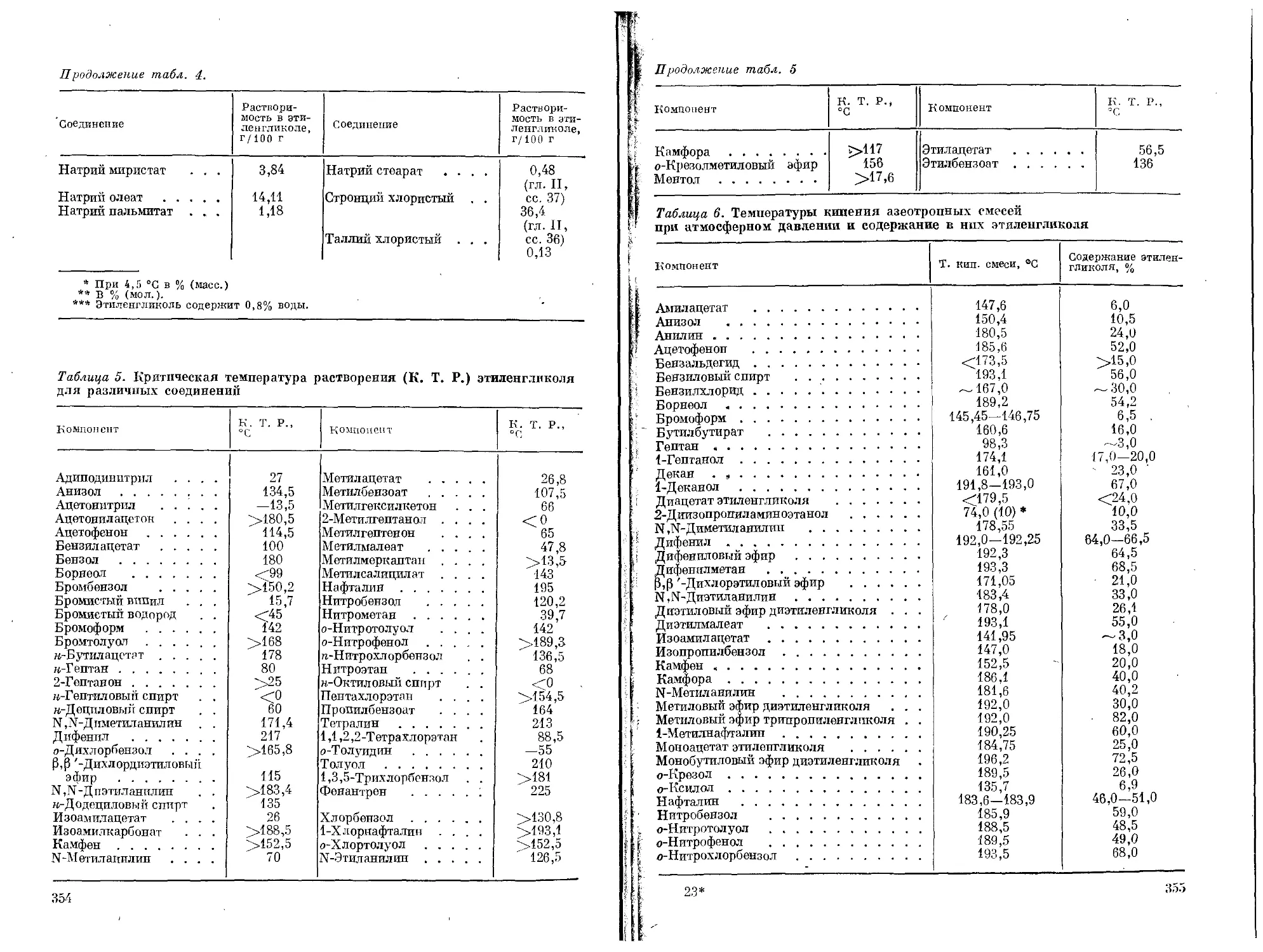
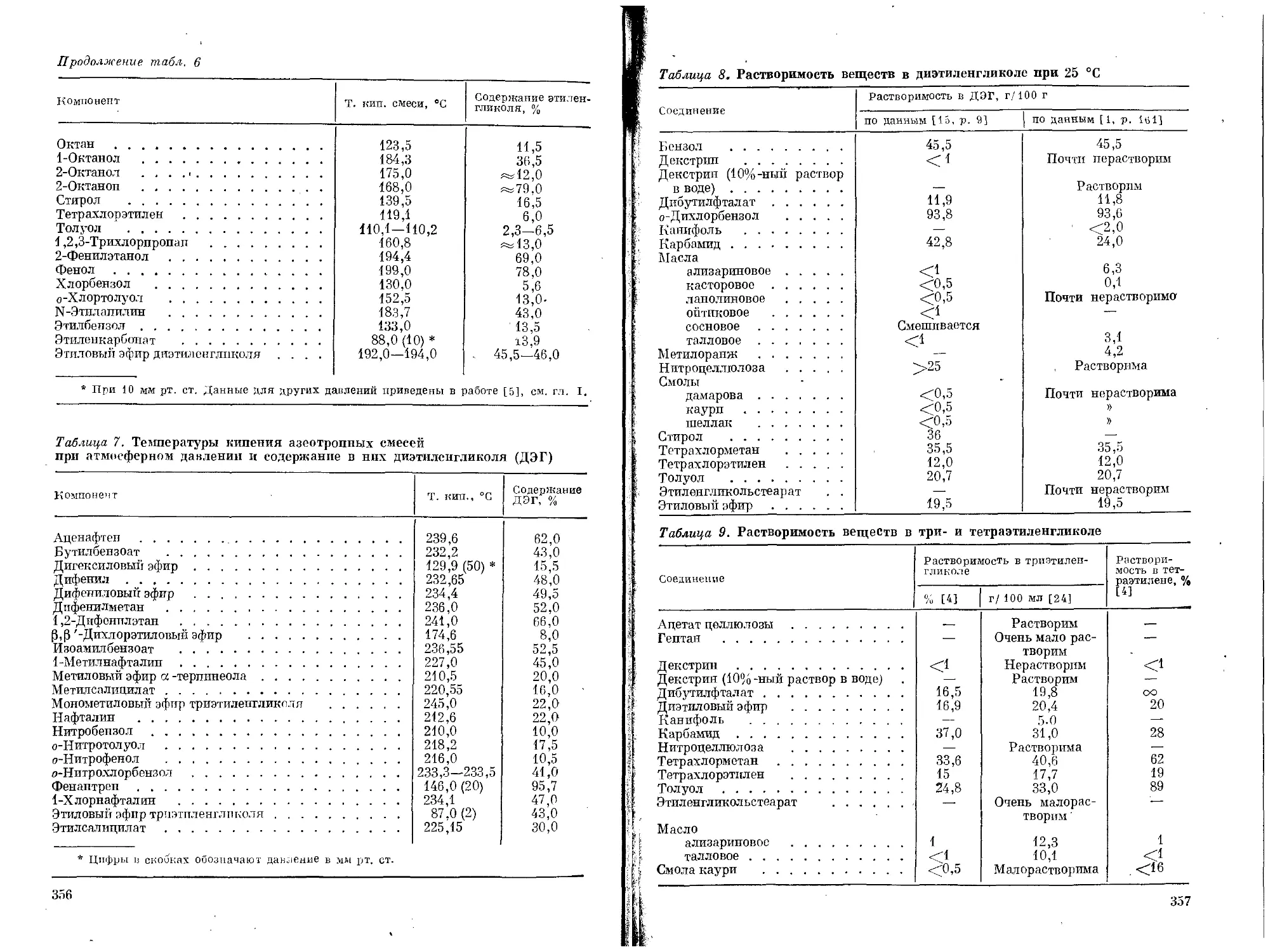
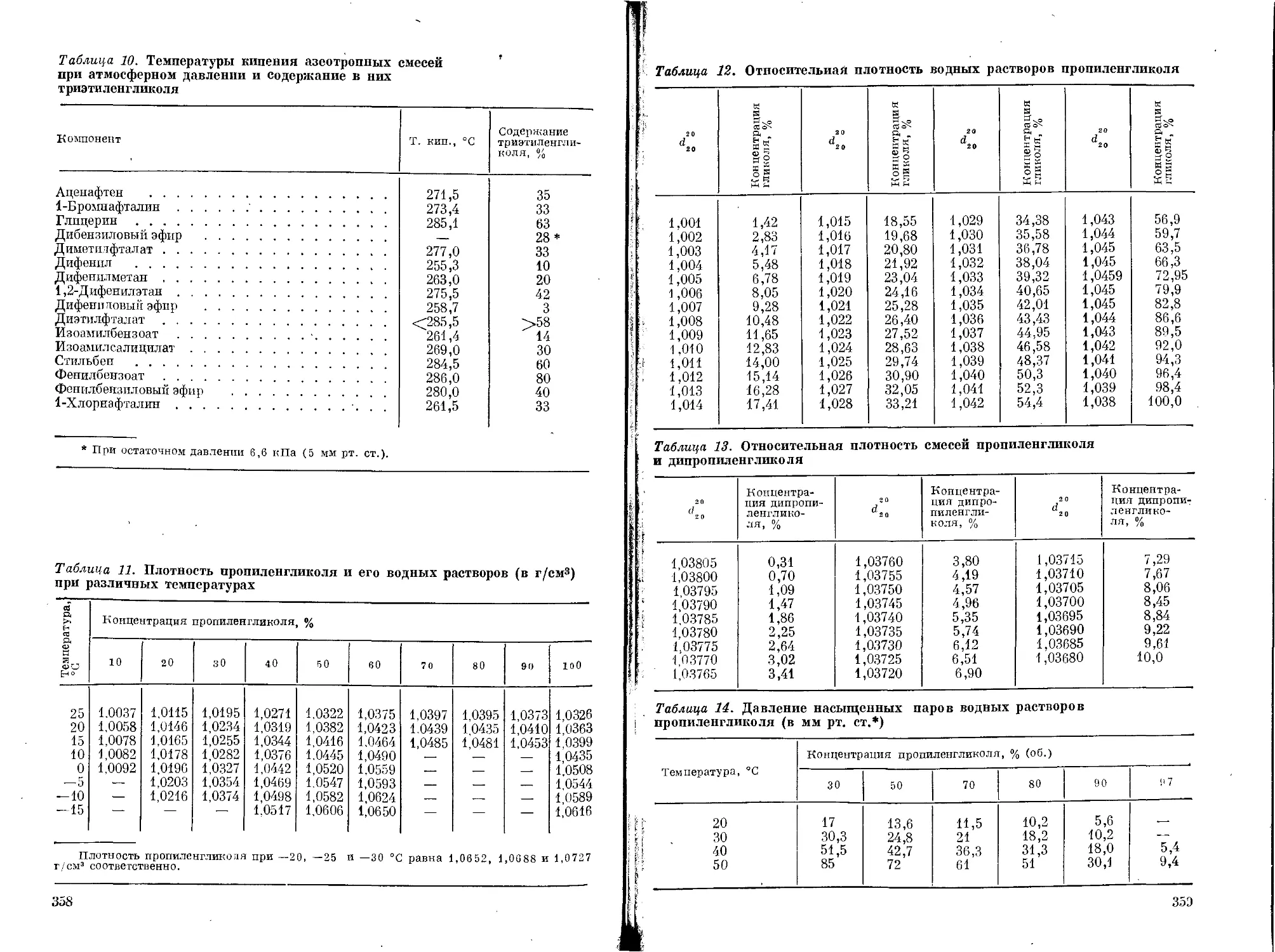
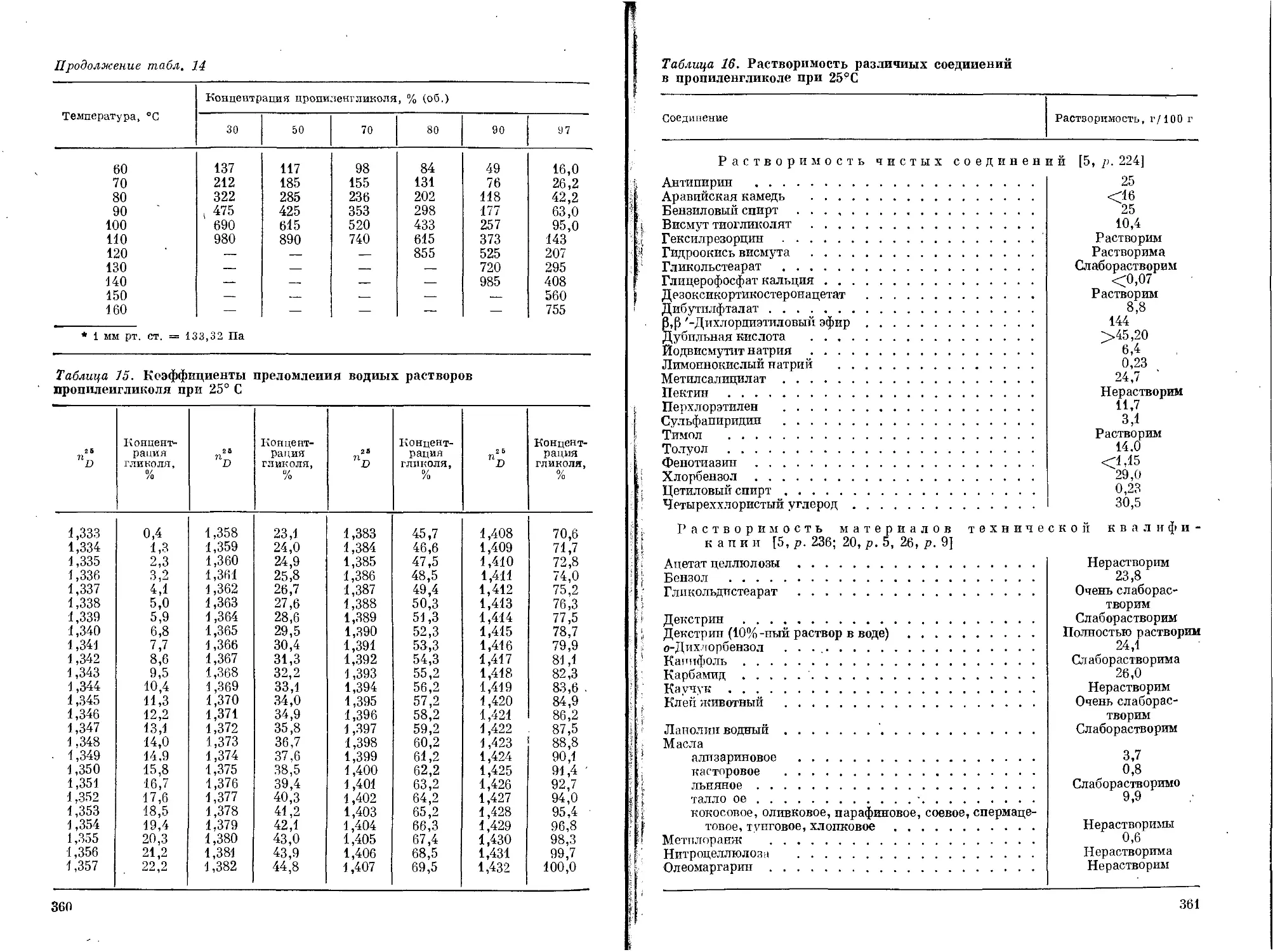
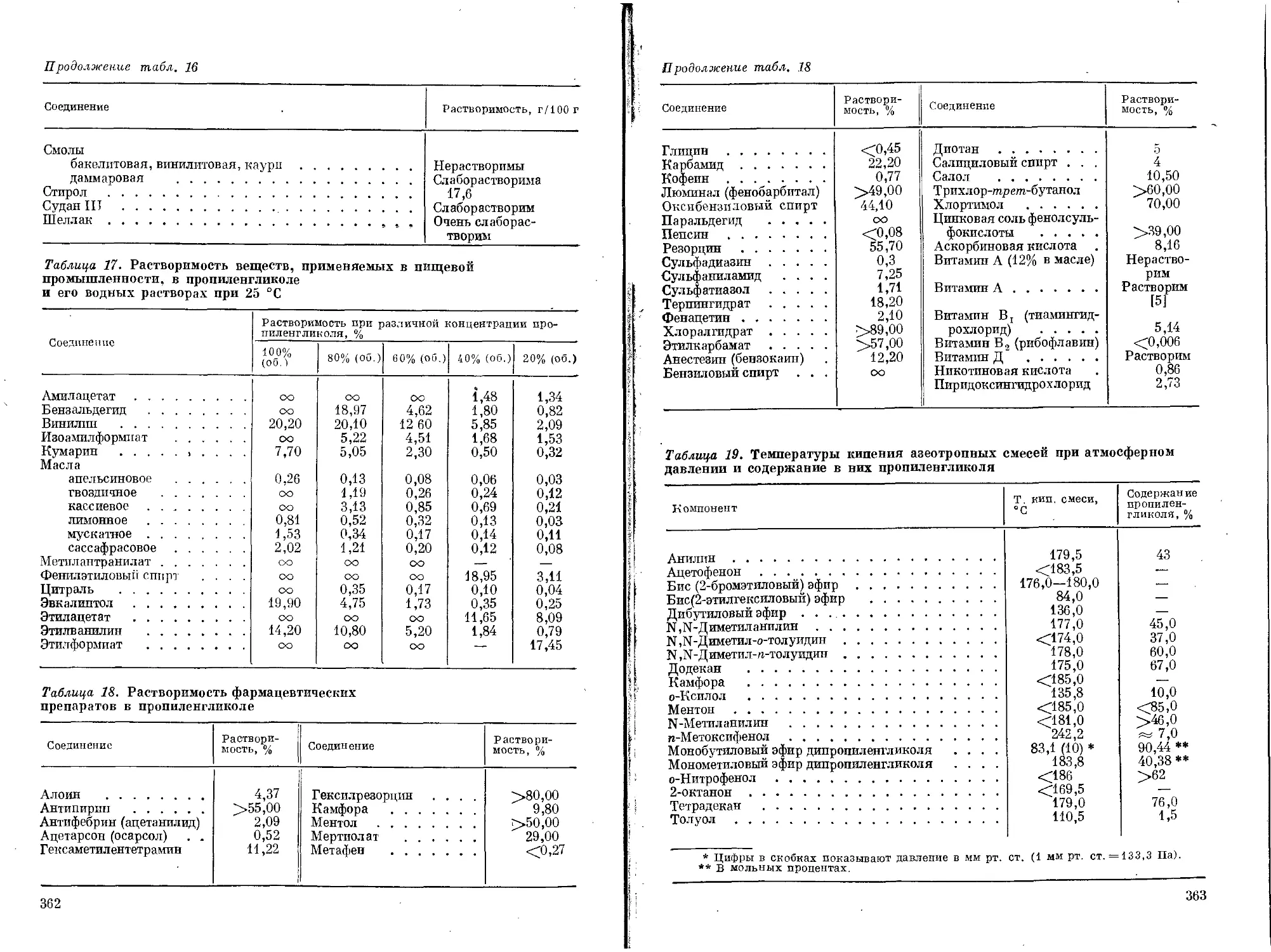
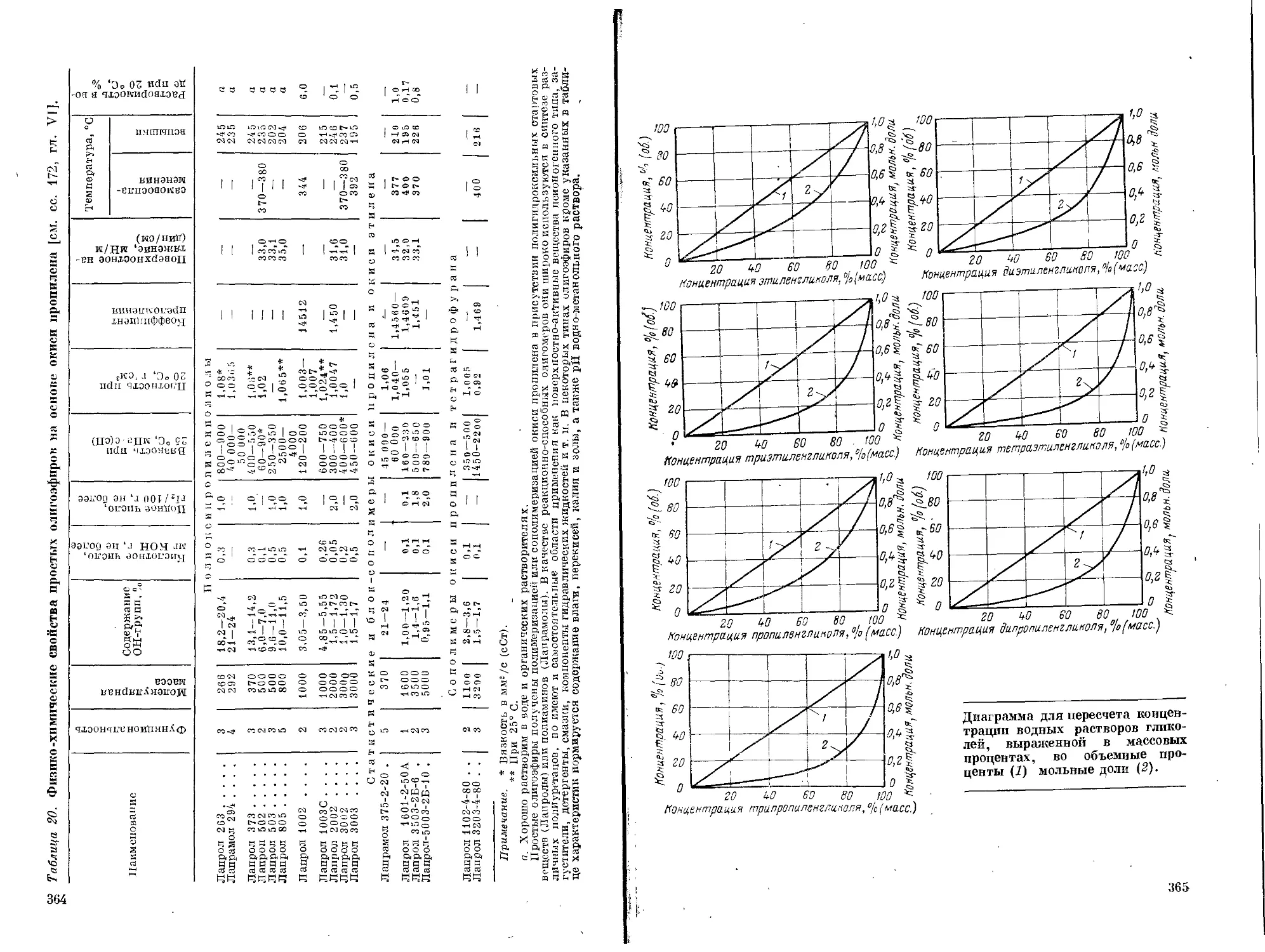
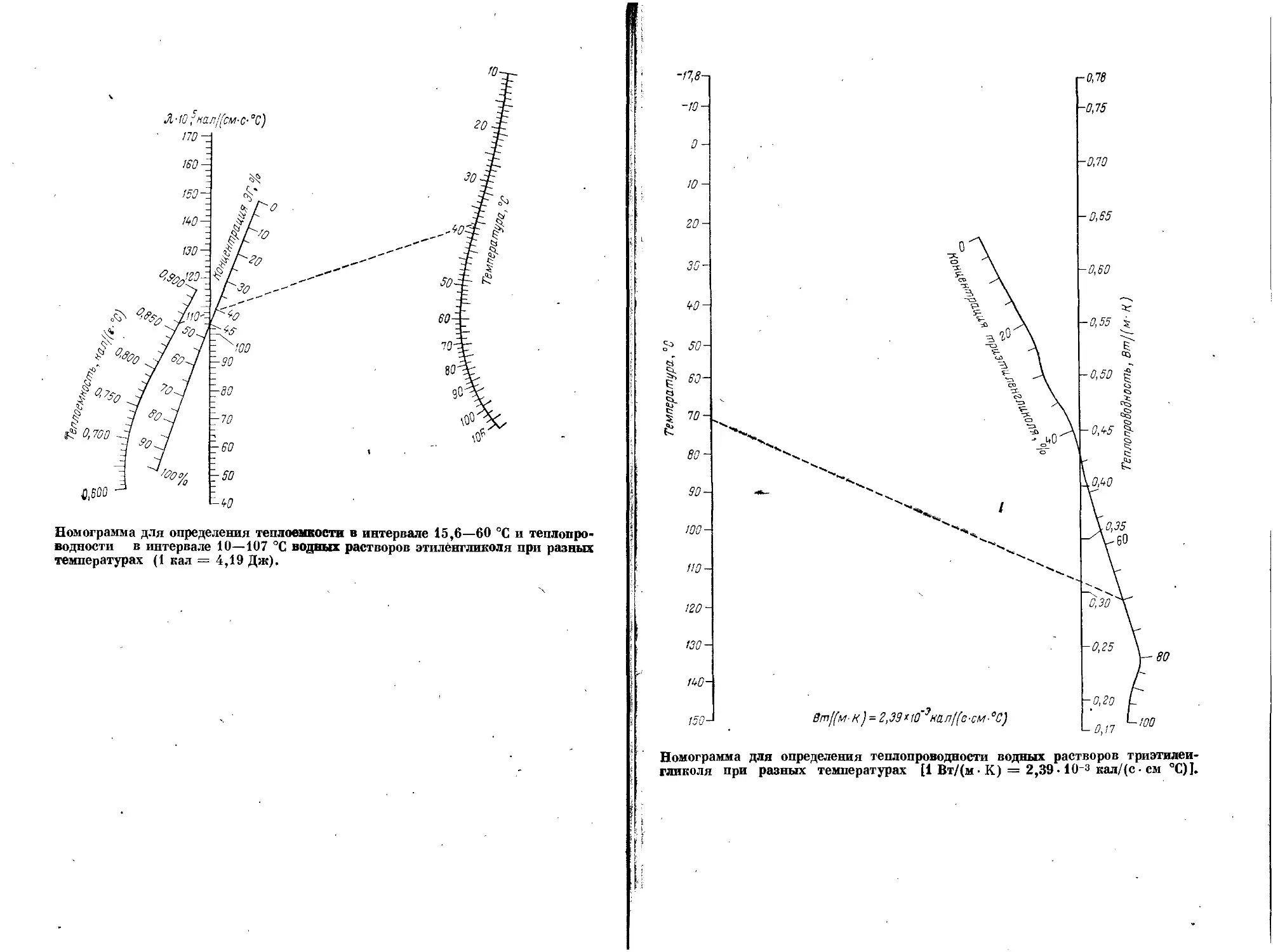
6
8
15
16
17
Ц
30
33
34
35
38
38
>0
>8
58
'4
34
12
18
)1
>5
'6
8
0
7
7
3
3
Дымент О. Н., Казанский К. С., Мирошников А. М.
Гликоли и другие производные окисей этилена и про-
пилена. Под общей ред. О. Н. Дымента. М., «Химия»,
1976.
В книге описаны физические и химические свойства,
методы получения, области применения, условия транспор-
тирования, хранения и методы анализа ряда наиболее важных
производных окисей этилена и пропилена: этиленгликоля,
ди-, три- и тетраэтиленгликолей, пропилен-, дипропилен-
и трипропиленгликолей, эфиров гликолей и полимеров окиси
этилена и окиси пропилена. Рассмотрены также токсические
свойства указанных продуктов, условия обращения с ними.
Книга предназначена для инженерно-технических ра-
ботников нефтехимической промышленности и промышлен-
ности органического синтеза, работающих в области произ-
водства и применения указанных продуктов. Она предста-
вляет интерес и для работников научно-исследовательских
и проектных организаций, преподавателей и студентов
химических вузов и техникумов.
• 376 стр., 110 рис., 102 табл., список литературы 730 ссылок.
31407-095
Д 050(01)-76 У /Ь
© Издательство «Химия», 1976 г.
Содержание
Предисловие ...................................................... 6
Введение ........................................................ 8
Литература ........................................................15
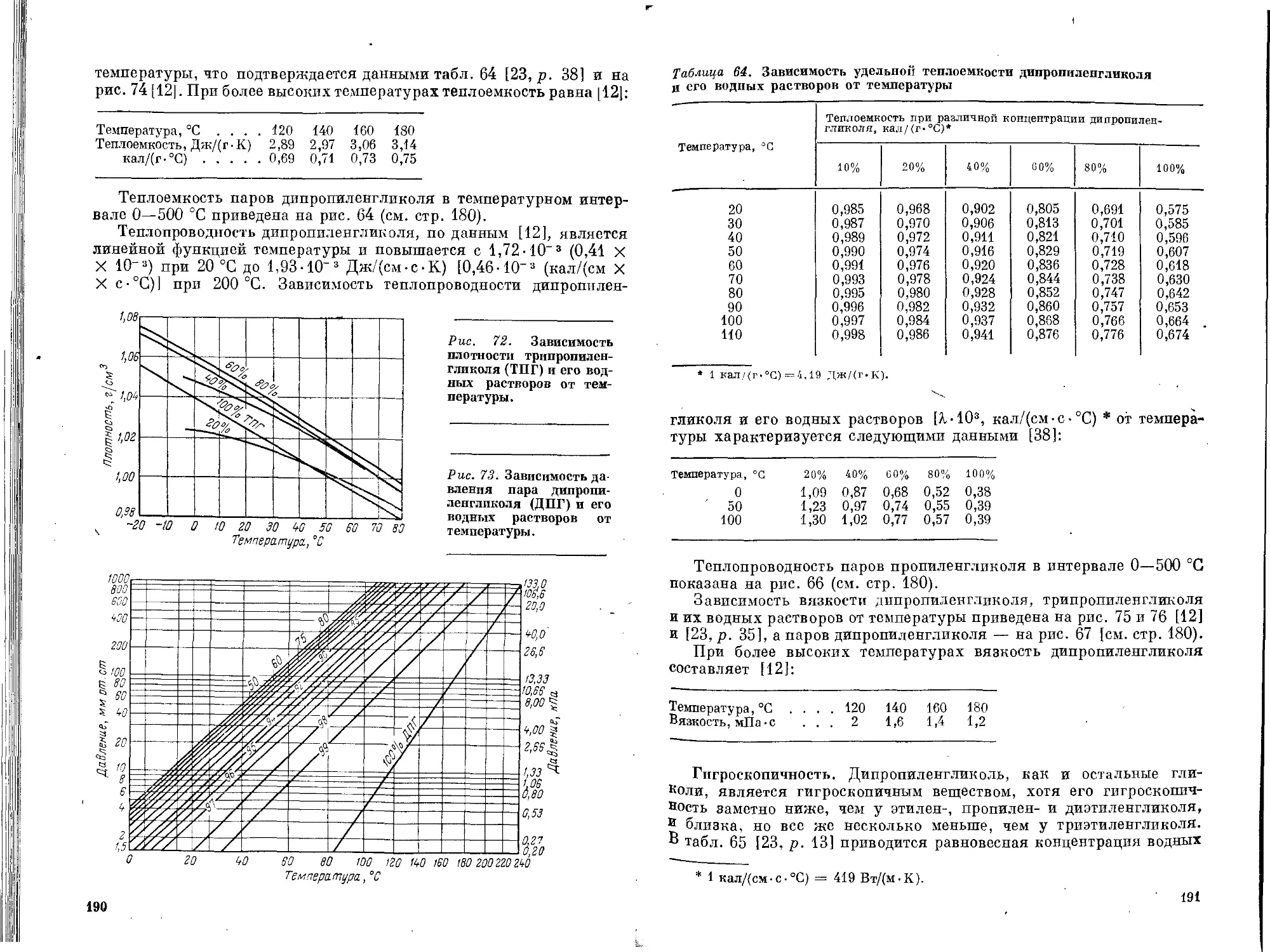
v
Глава I. Общие свойства гликолей..........................................16
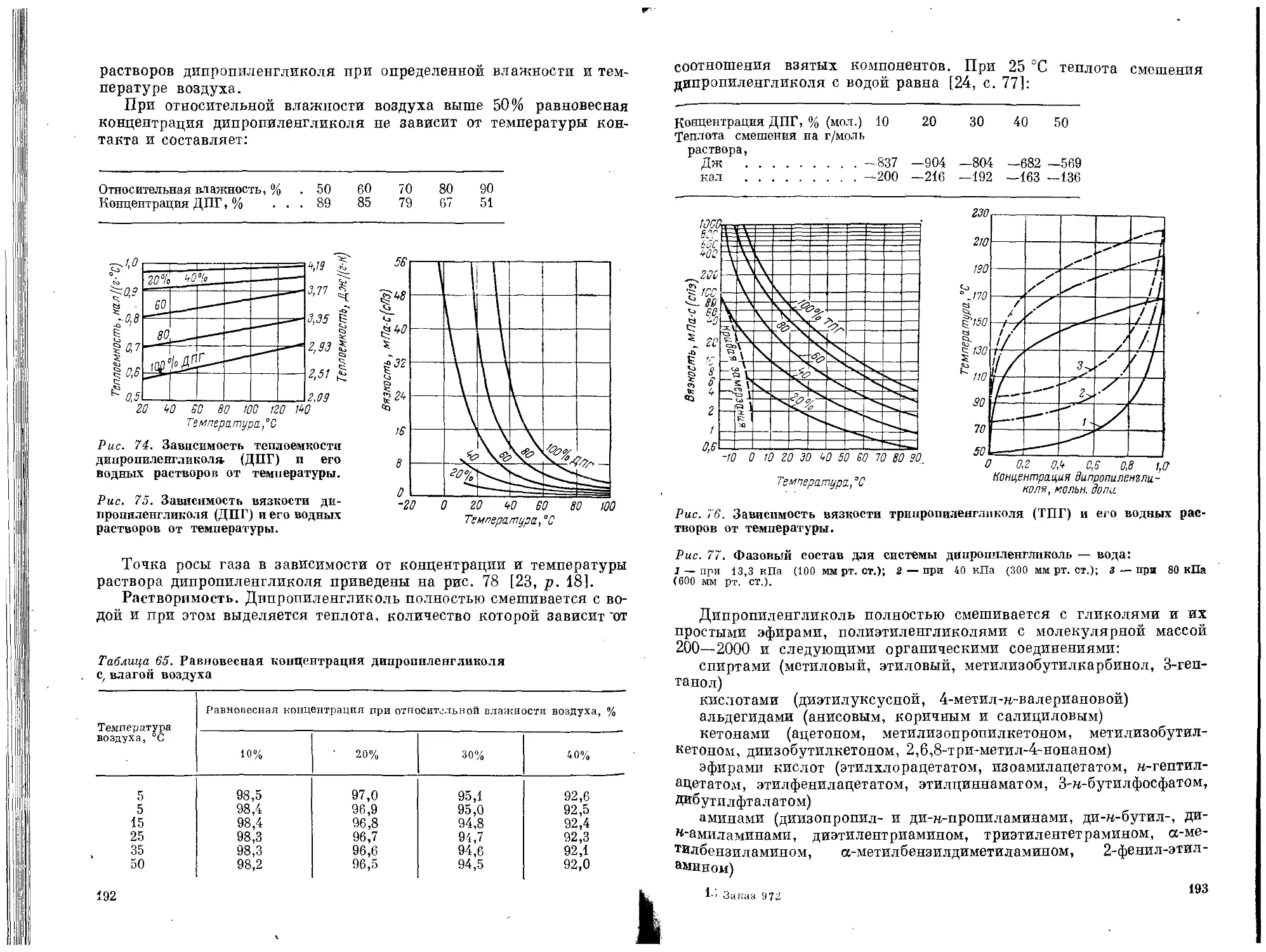
Физические свойства...............................
Химические свойства...............................
Токсические свойства .............................
Техника безопасности при работе с гликолями.......
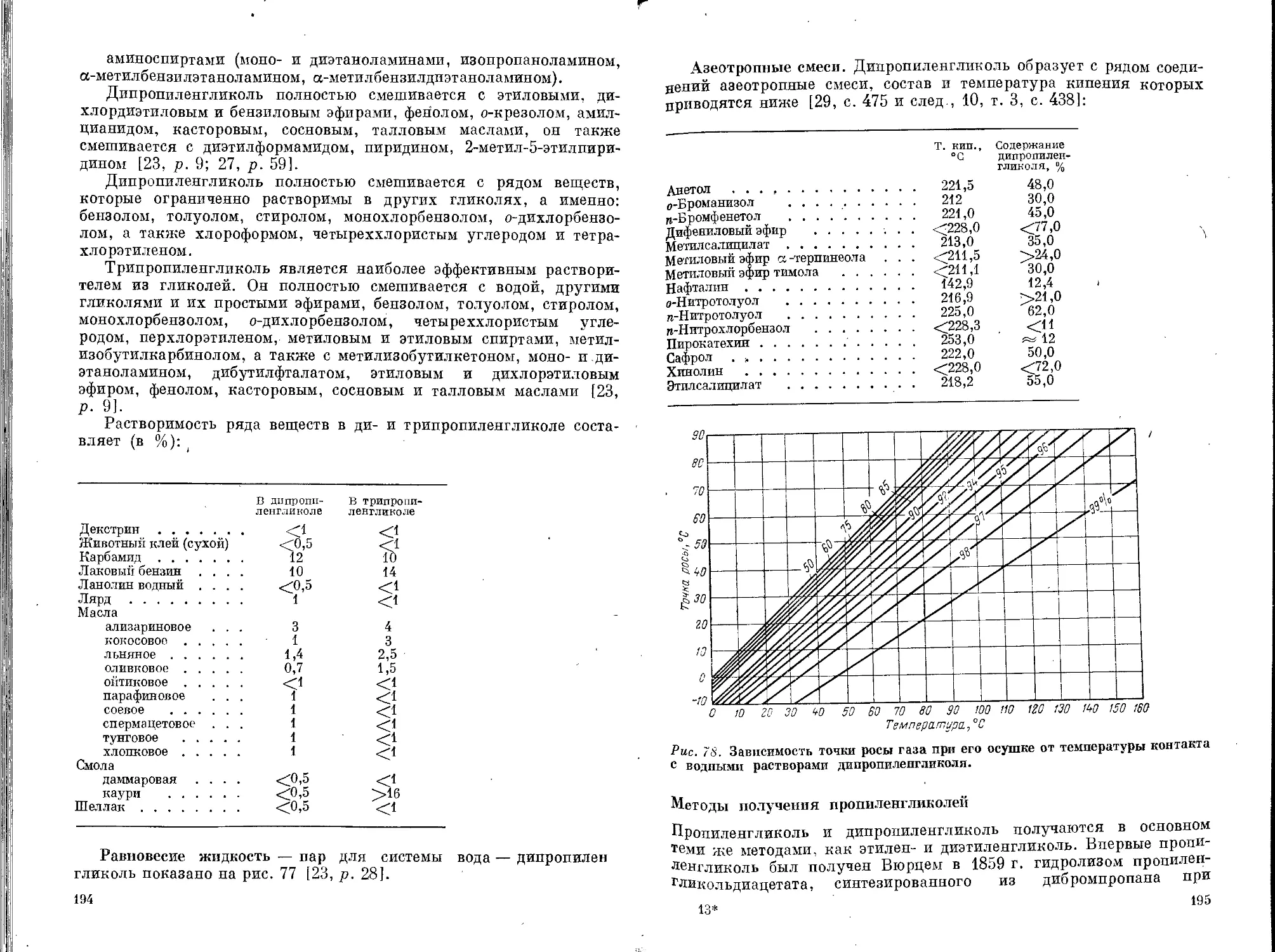
Хранение и транспортирование гликолей.............
Литература .......................................
17
24
30
33
34
35
Глава II. Этиленгликоль ....................................... 38
Физические свойства .................................38
Методы получения этиленгликоля.......................60
Получение этиленгликоля гидратацией окиси этилена ... 68
Катализ, механизм и кинетика реакции.............68
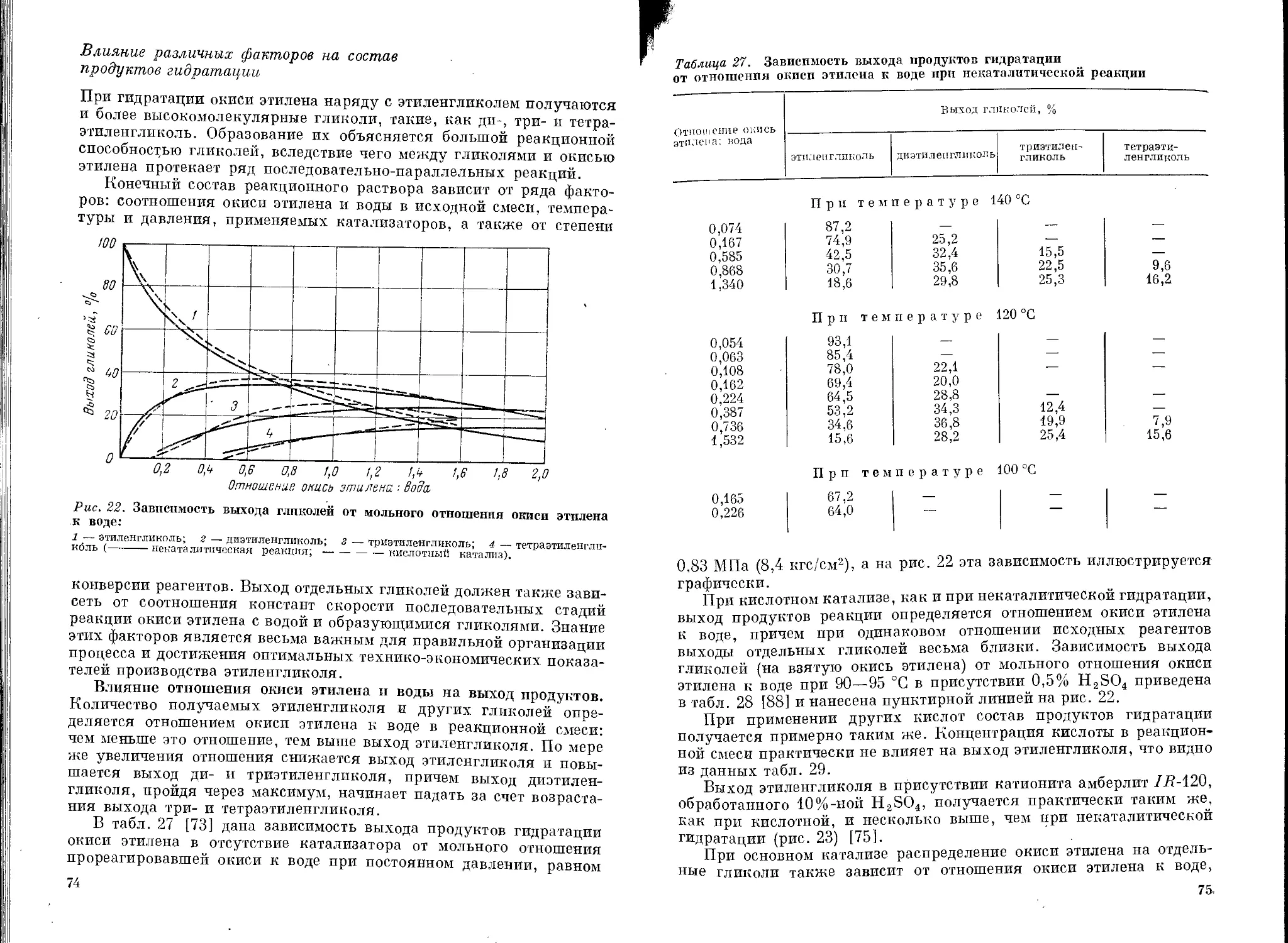
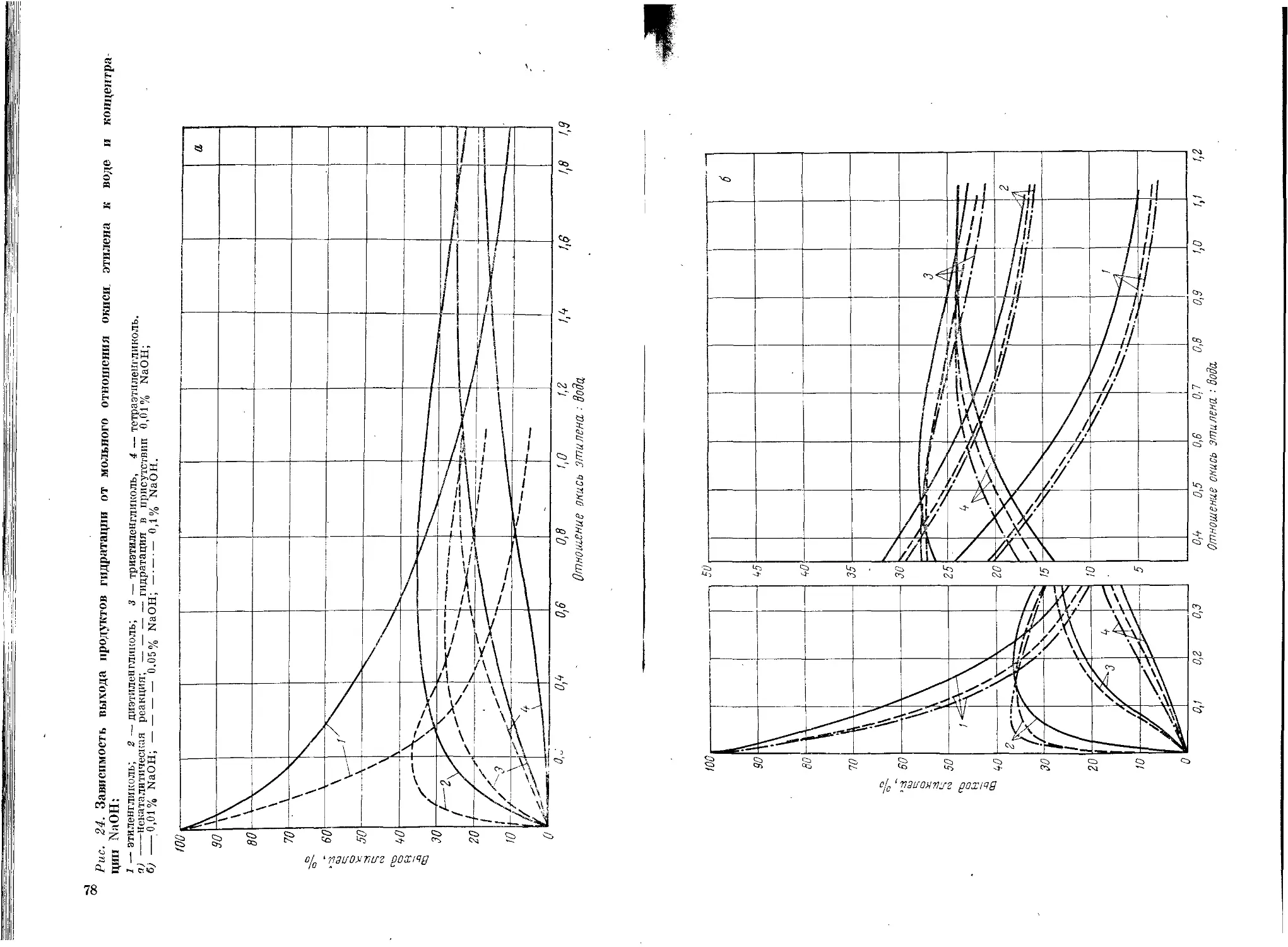
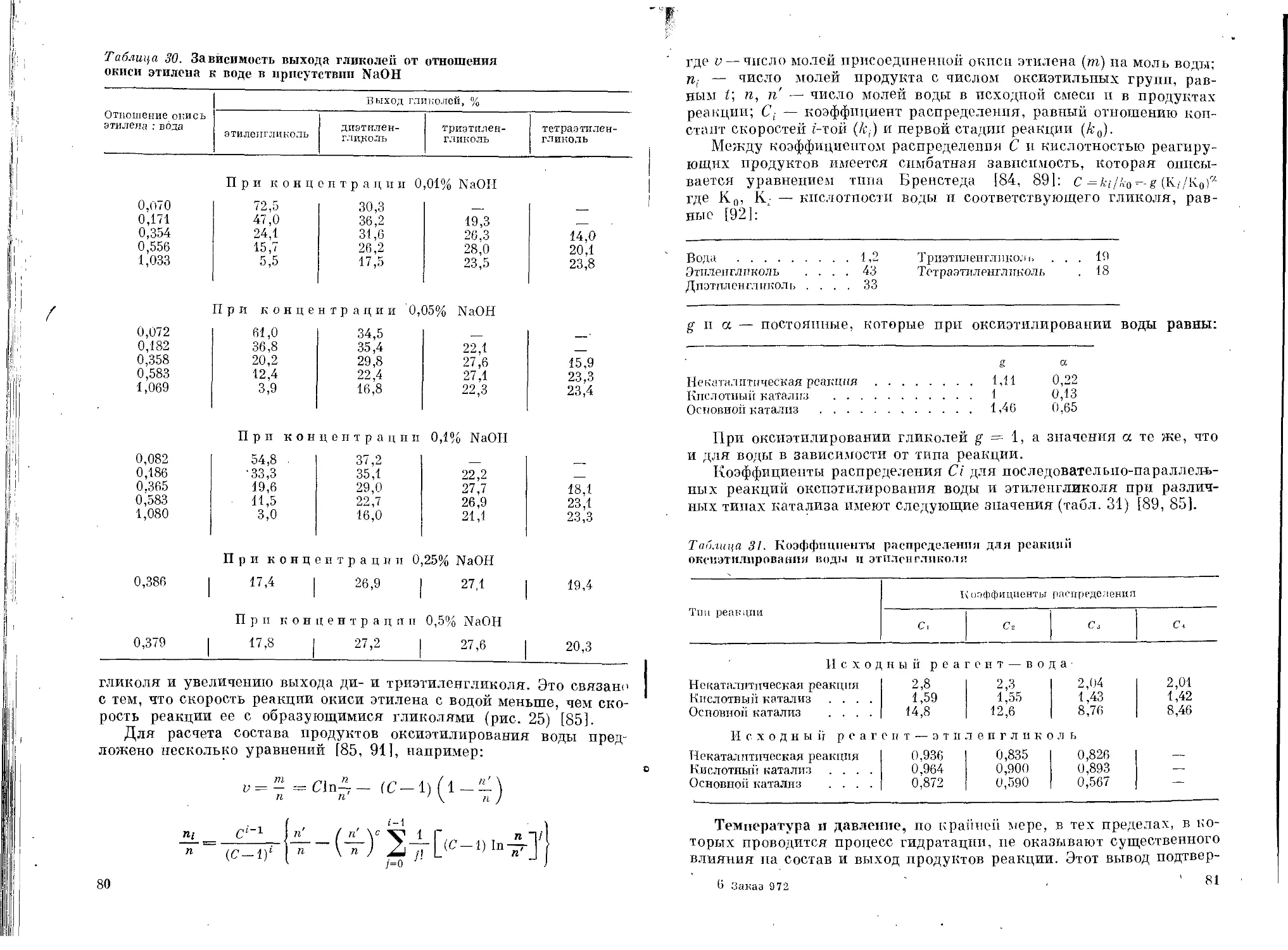
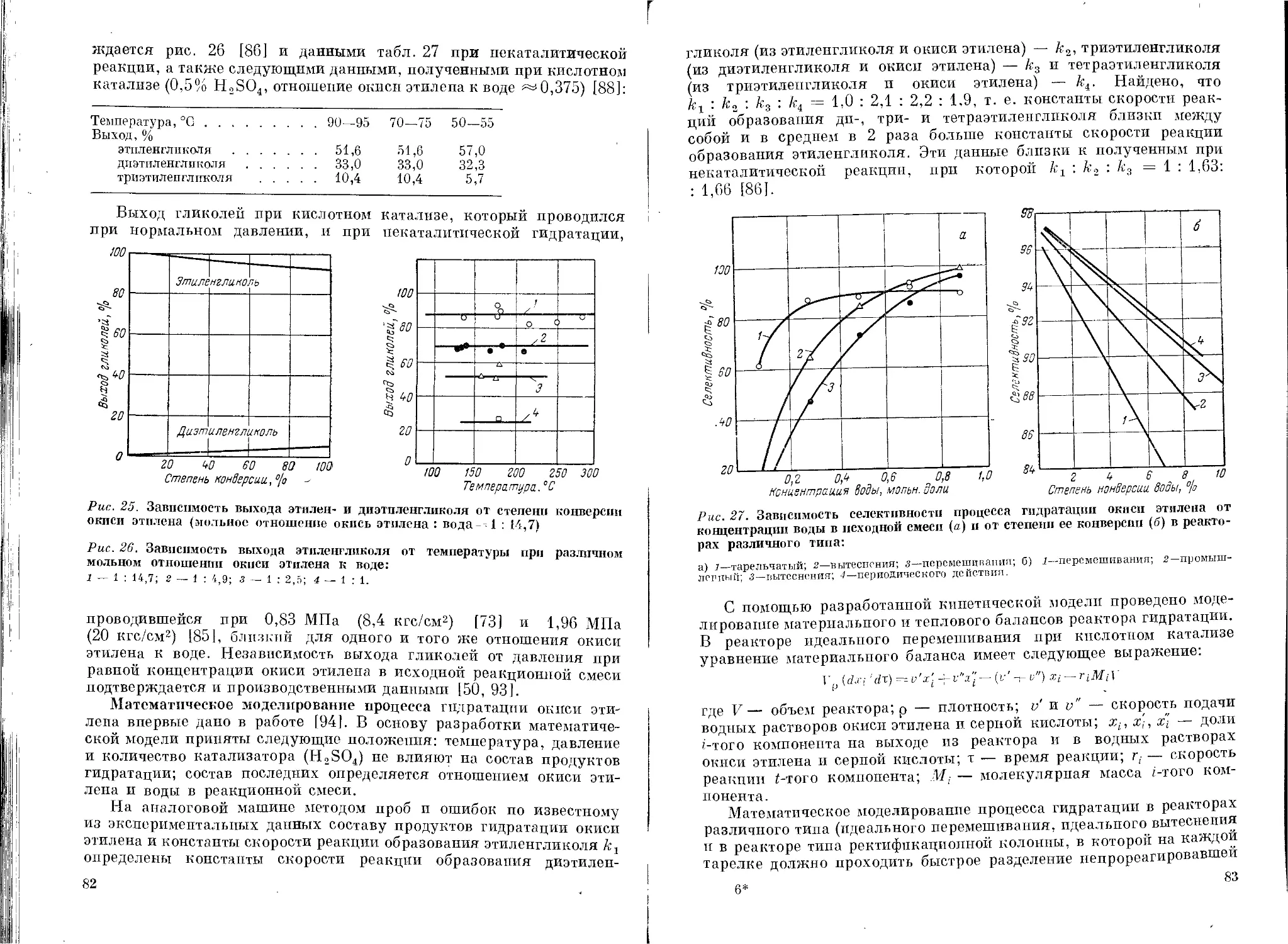
Влияние различных факторов на состав продуктов
гидратации ......................................74
Технологическая схема производства этиленгликоля нека-
талитической гидратацией окиси этилена ............. 84
Получение этиленгликоля кислотно-каталитической гидрата-
цией окиси этилена...................................92
Применение этиленгликоля ............................98
Антифризы .......................................101
Полиэфирные волокна ............................105
Гидравлические жидкости .........................106
Требования, предъявляемые к качеству этиленгликоля . . 108
Литература ..........................................110
Глава III. Диэтиленгликоль
...........................................117
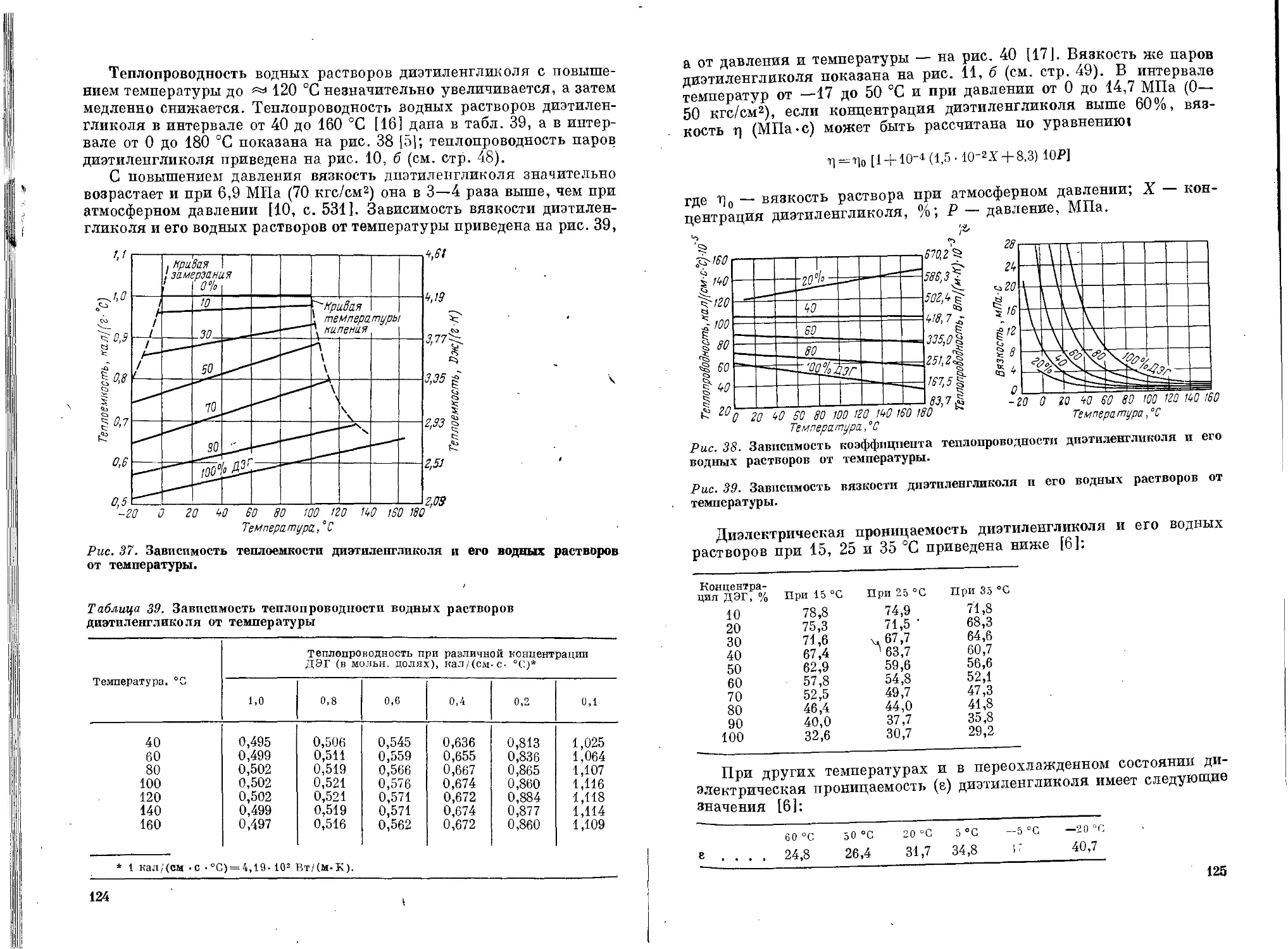
Физические свойства..................................117
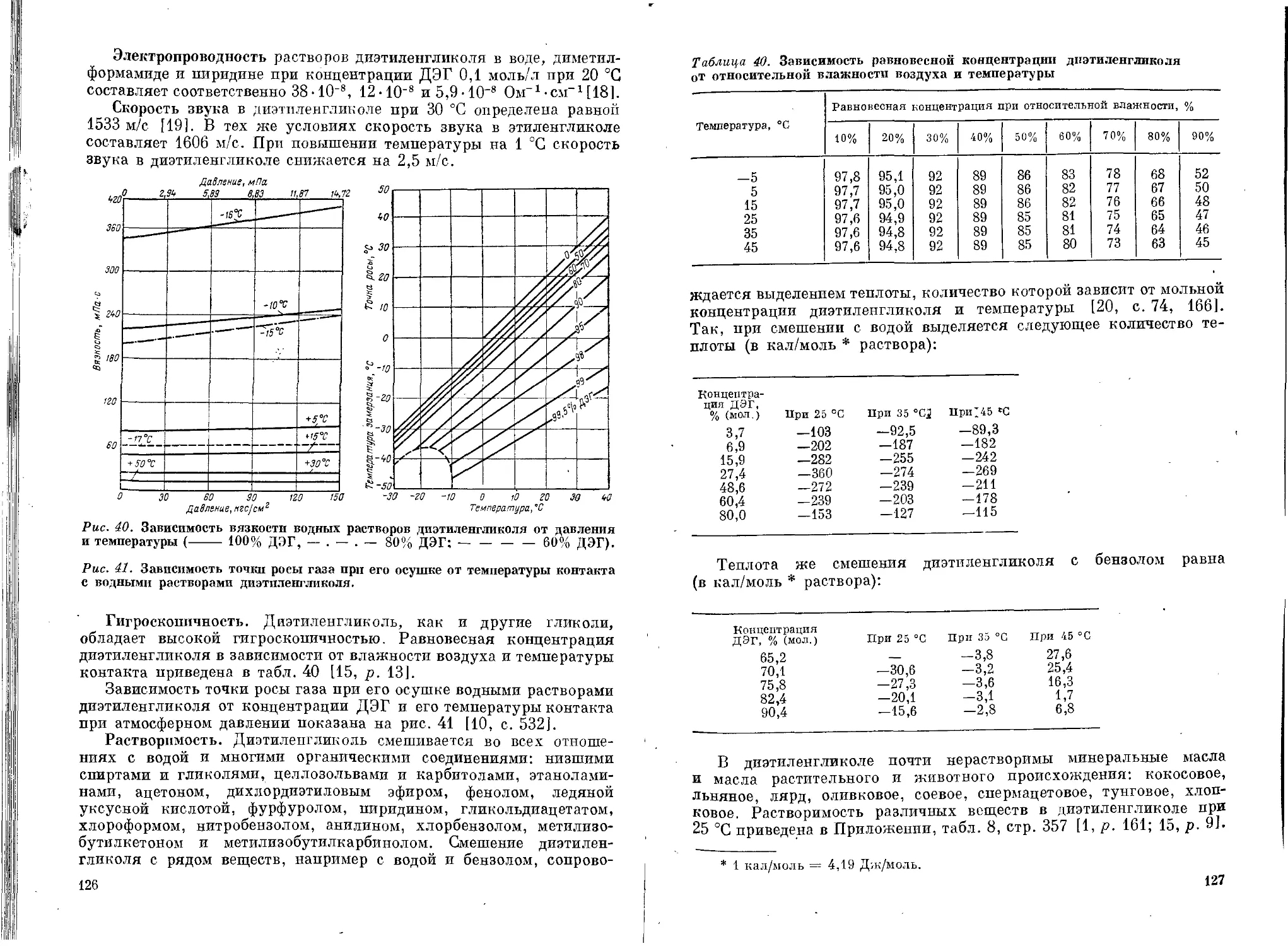
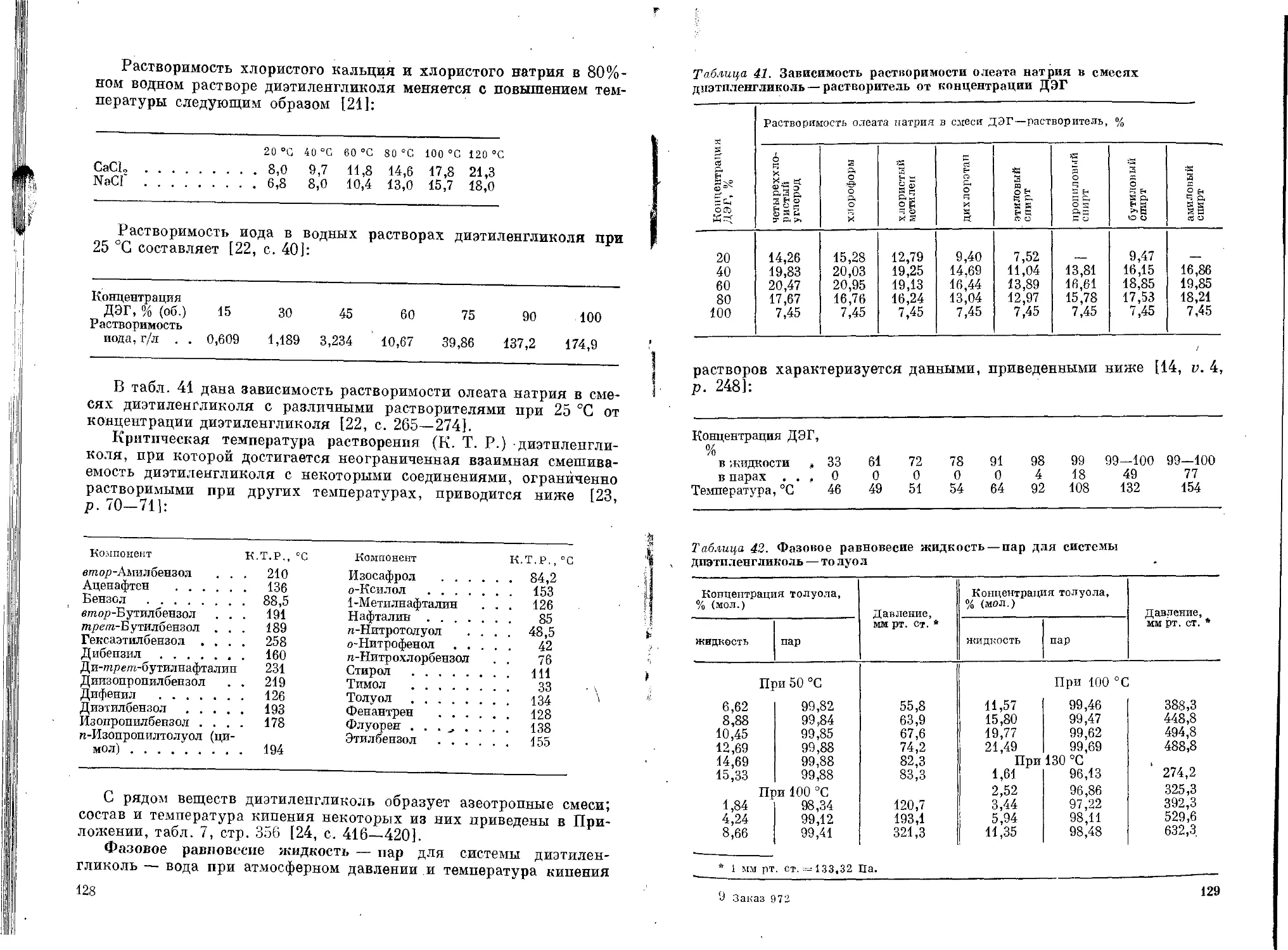
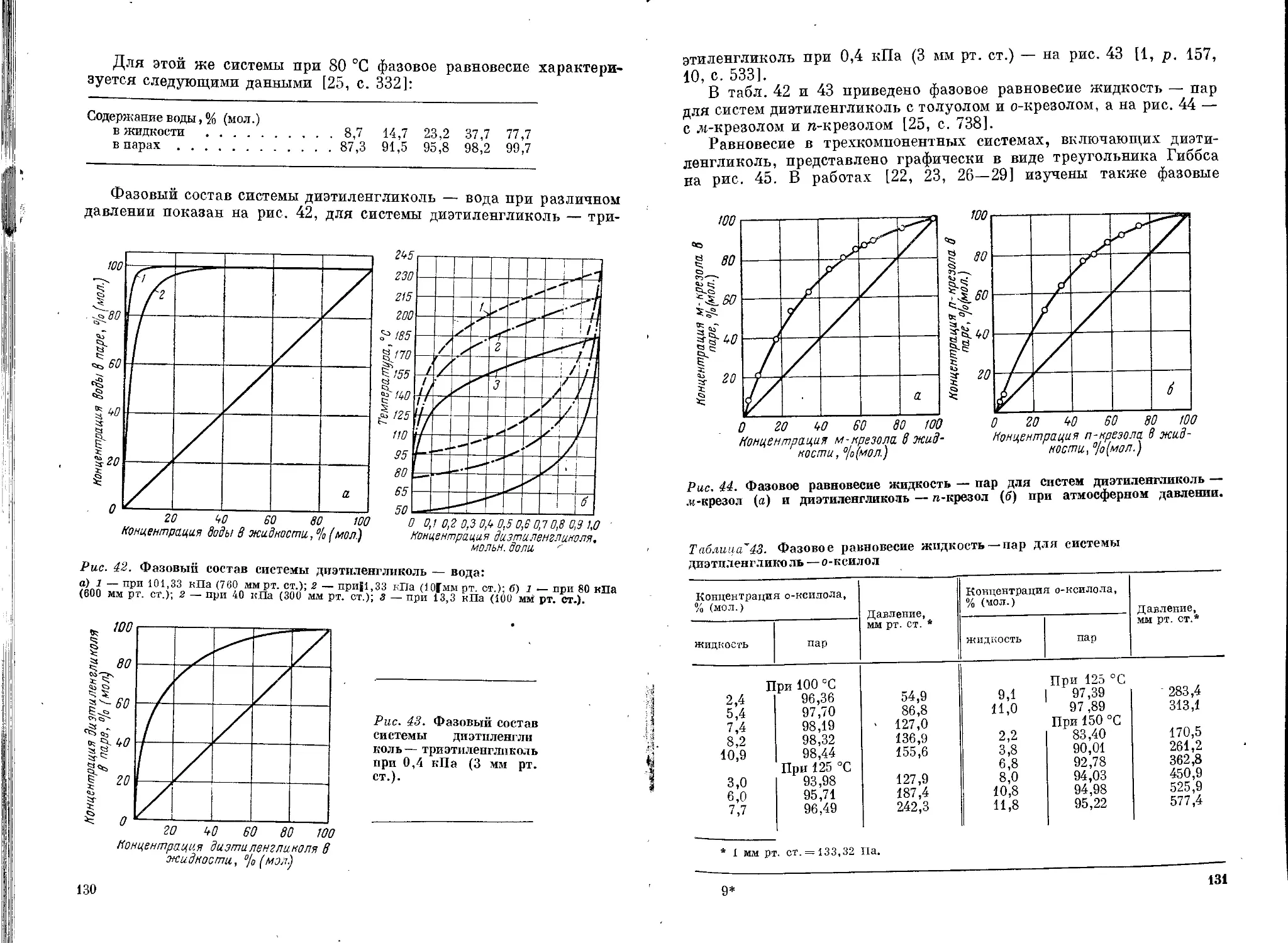
Получение диэтиленгликоля ........................ .133
1*
3
Применение диэтиленгликоля ...................136
Требования, предъявляемые к качеству диэтиленгликоля . . 140
Литература ...................................142
Глава IV. Триэтиленгликоль и тетраэтиленгликоль................145
Физические свойства триэтиленгликоля...............145
Физические свойства тетраэтиленгликоля.............156
Получение три- и тетраэтиленгликолей................158
Применение триэтиленгликоля ......................163
Применение тетраэтпленгликоля ....................167
Требования, предъявляемые к качеству три- и тетраэтилен-
гликолей ...........................................168
Литература .........................................169
Глава V. Пропиленгликоль, дипропнленглпколь и трипропиленгликоль 172
Пропиленгликоль ...................................172
Физические свойства............................173
Дипропиленгликоль и трипропиленгликоль.............186
Физические свойства ...........................187
Методы получения пропиленгликолей..................195
Получение пропиленгликоля гидратацией окиси пропи-
лена ..........................................199
Получение пропиленгликолей в промышленных условиях 203
Применение пропиленгликоля, дипропиленгликоля и три-
пропиленгликоля ...................................204
Требования, предъявляемые к качеству пропиленгликоля
и дипропиленгликоля ........................... 210
Ли тература .......................................210
Глава VI. Полимеры окиси этилена и окиси пропилена . . . ....214
Основные методы полимеризации а-окисей............215
Катионная полимеризация ...................... 215
Анионная полимеризация ....................... 217
Полимеризация координационного типа............220
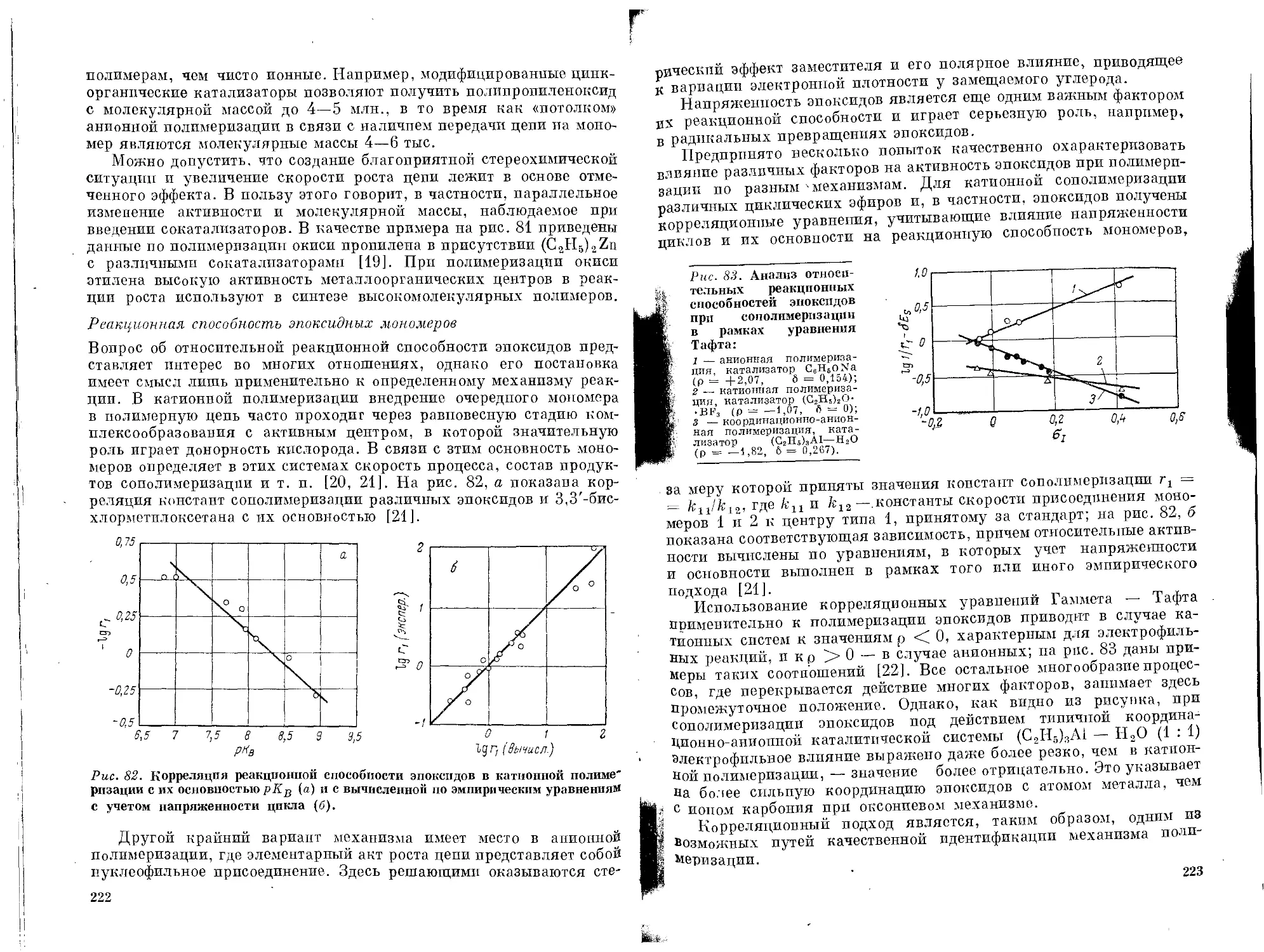
„ Реакционная способность эпоксидных мономеров . . . 222
Олигомерные продукты на основе а-окисей...........224
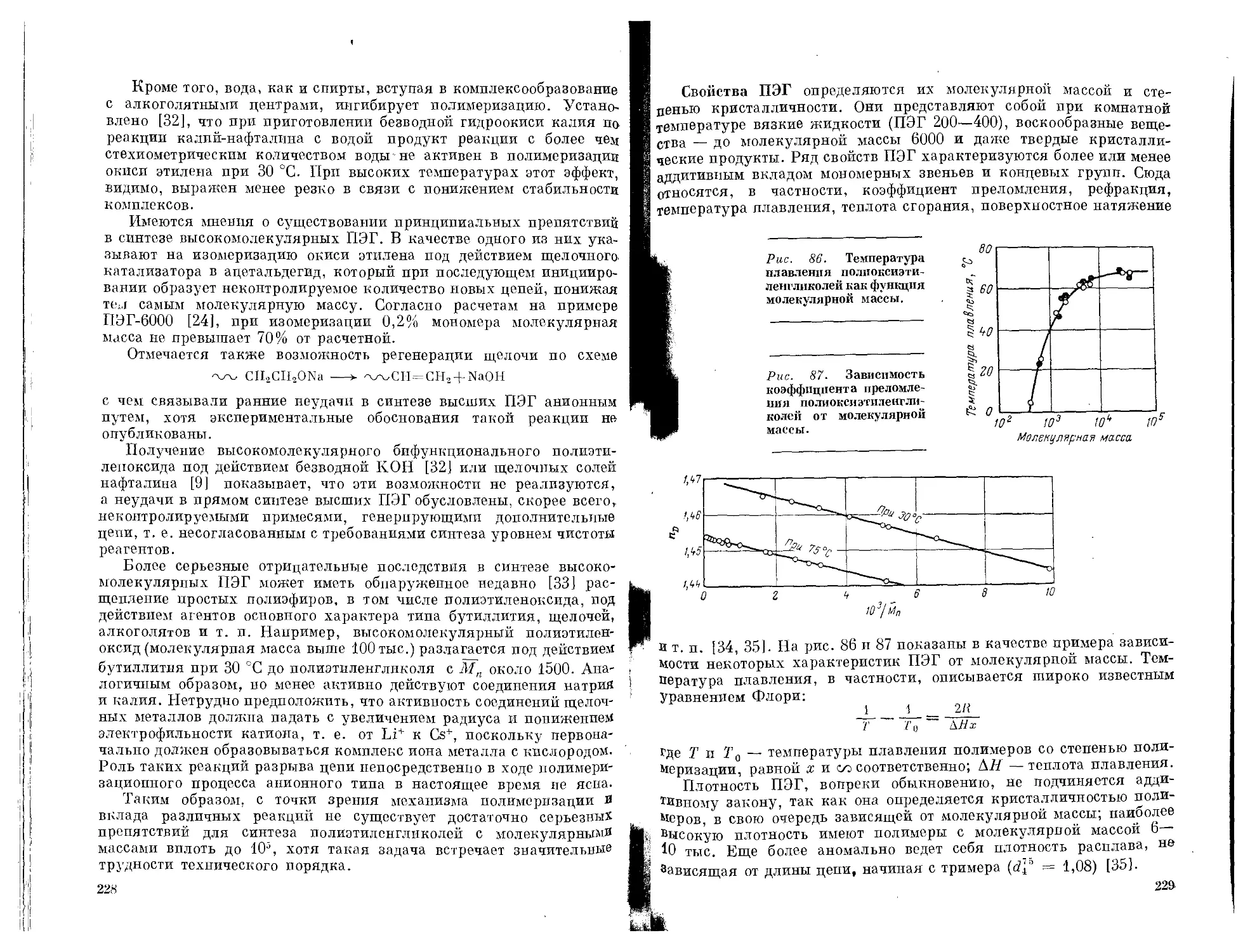
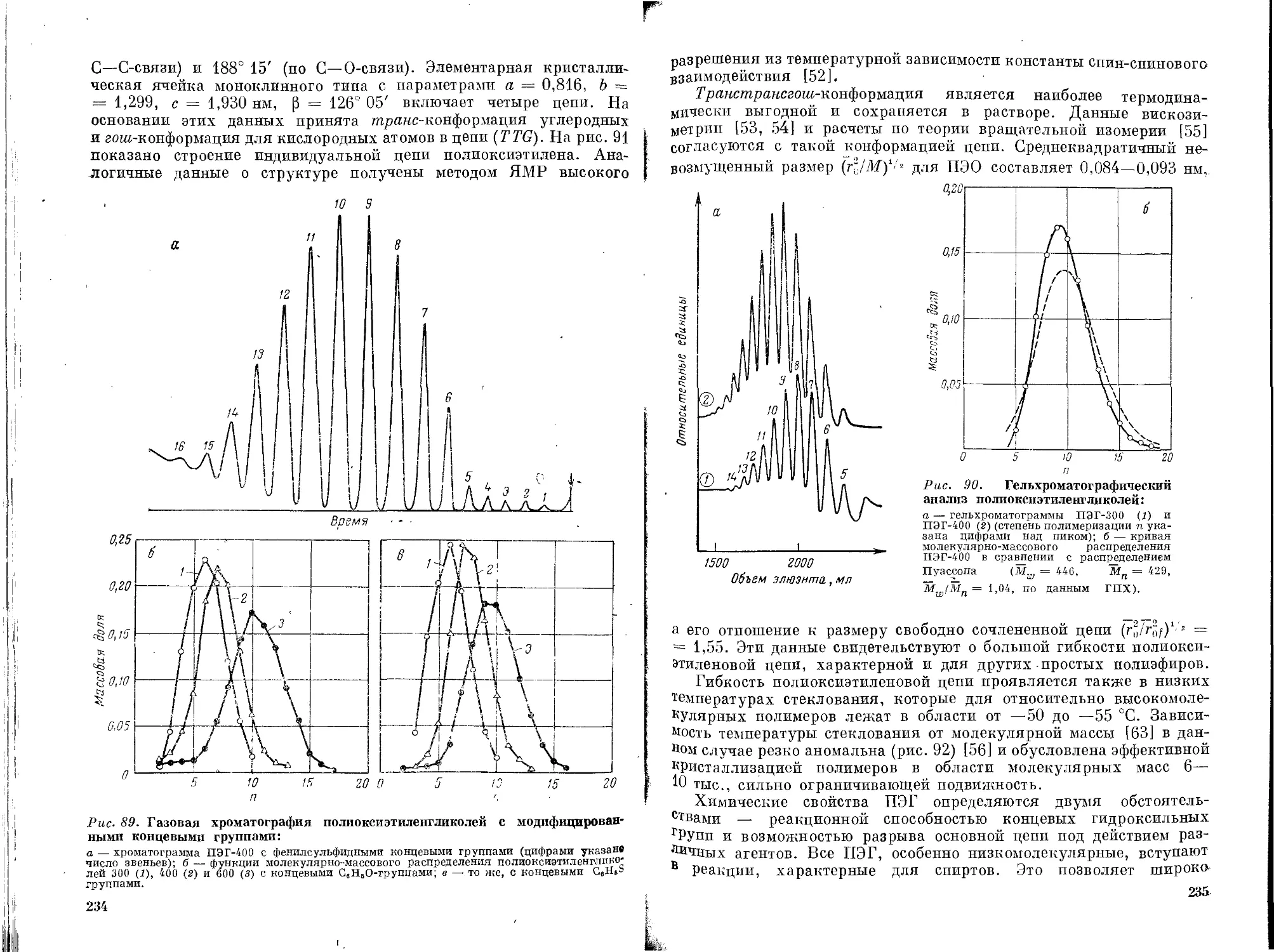
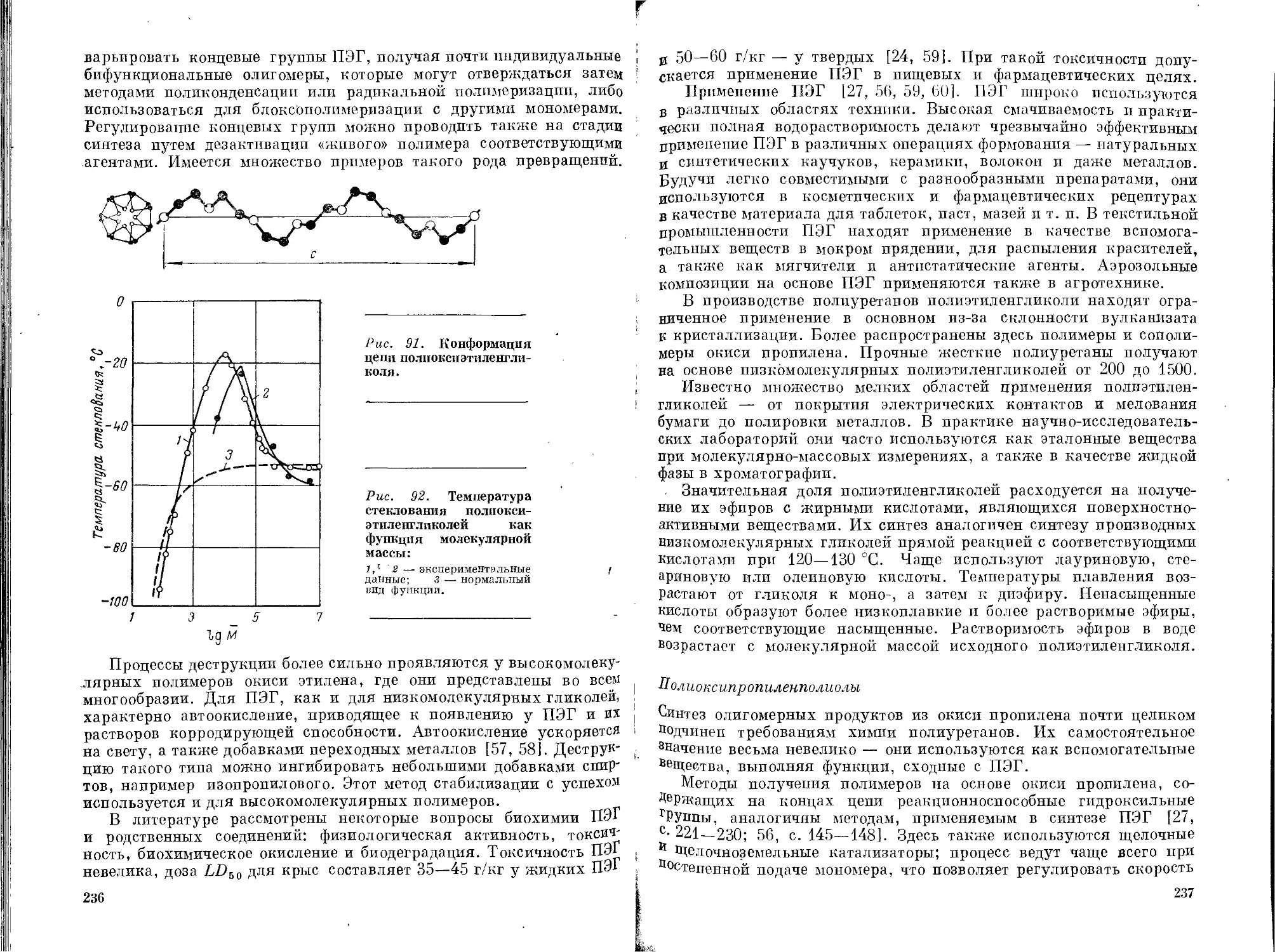
Полиэтиленгликоли .............................224
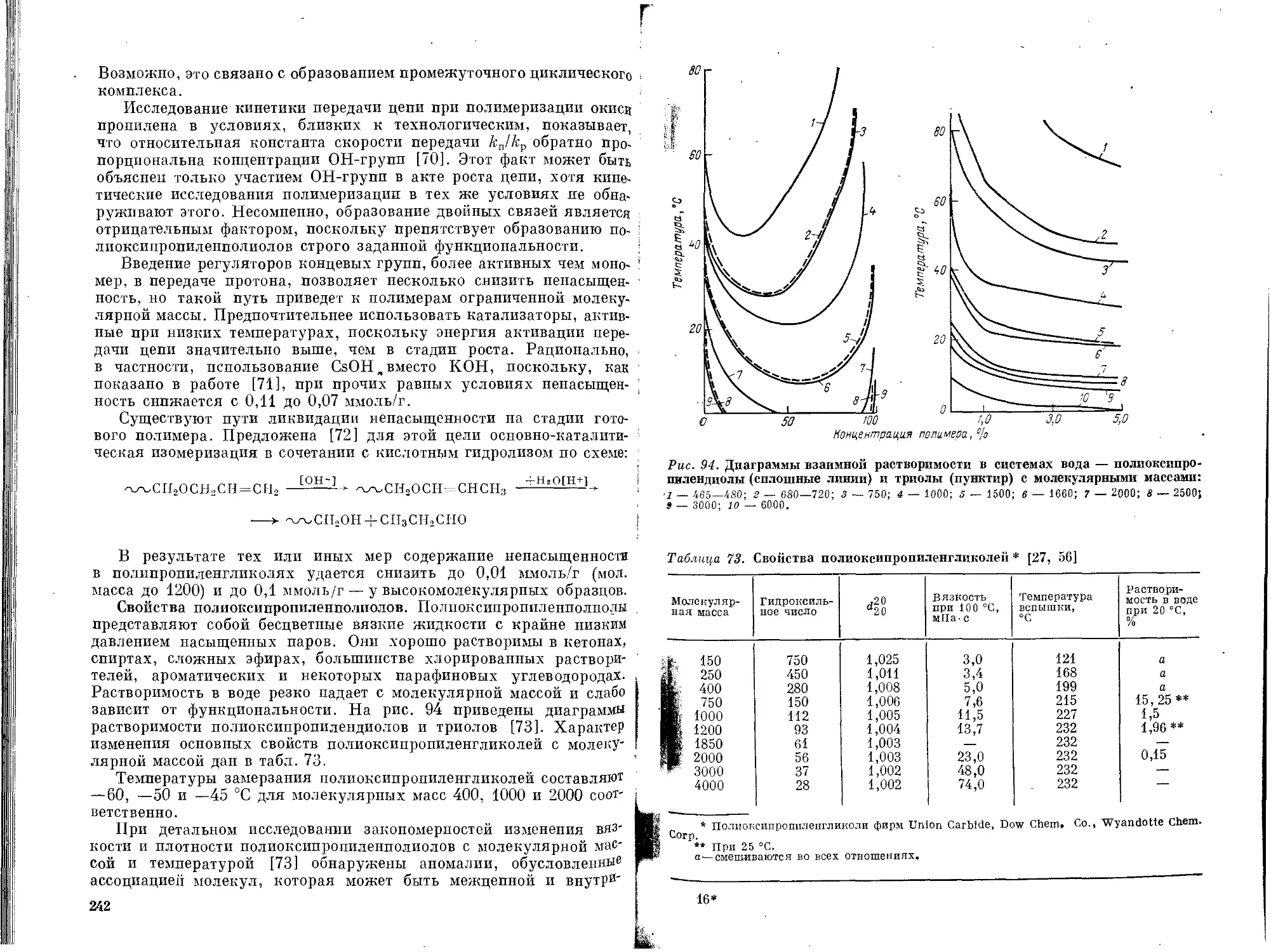
Полиоксипроппленполиолы .......................237
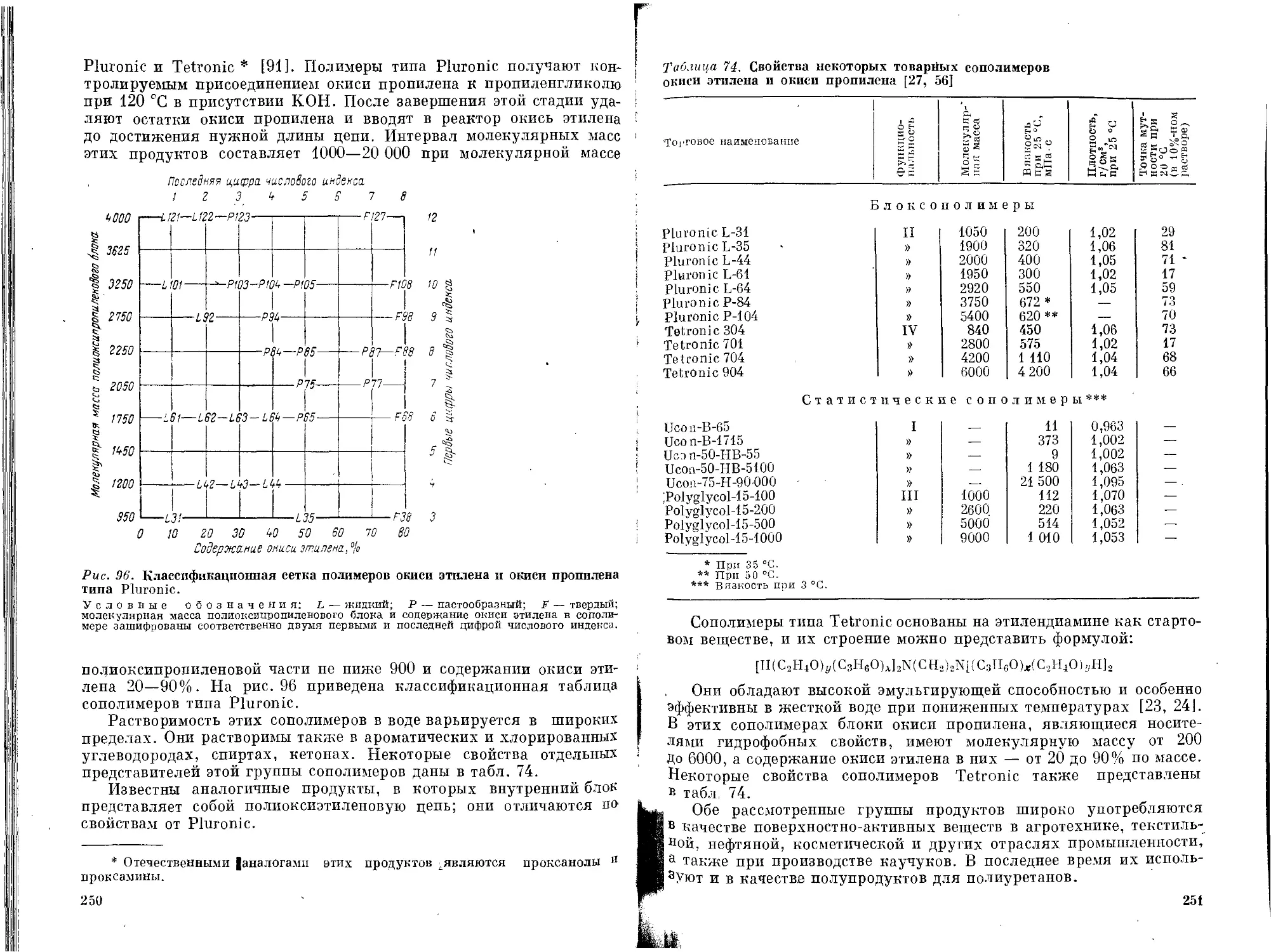
' Сополимеры окиси этилена и окиси пропилена .... 249
Высокомолекулярные полимеры а-окисей..............254
Стереорегулярный полиоксипропилен .............254
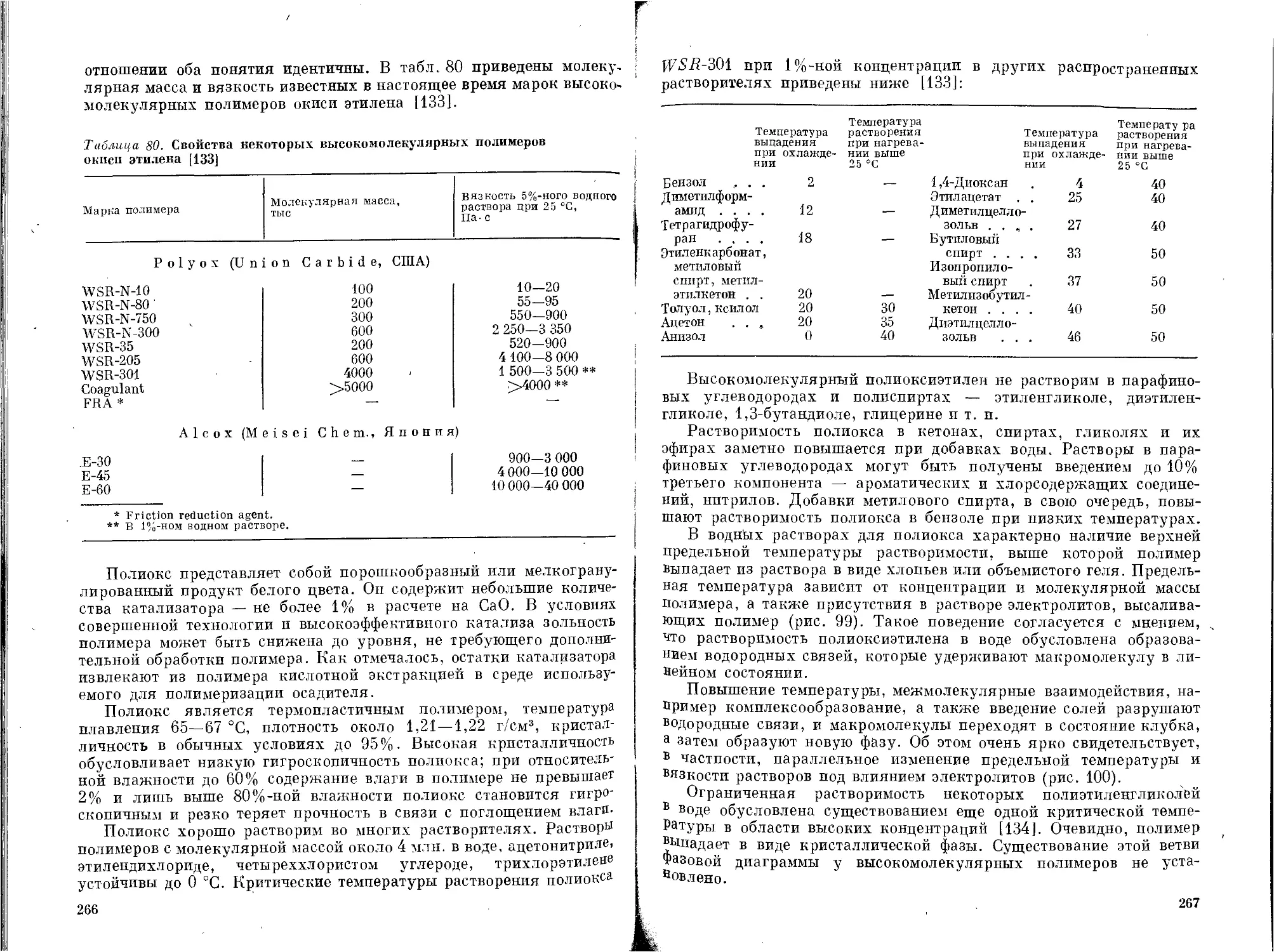
Высокомолекулярный полиоксиэтилен .............262
Литература .......................................283
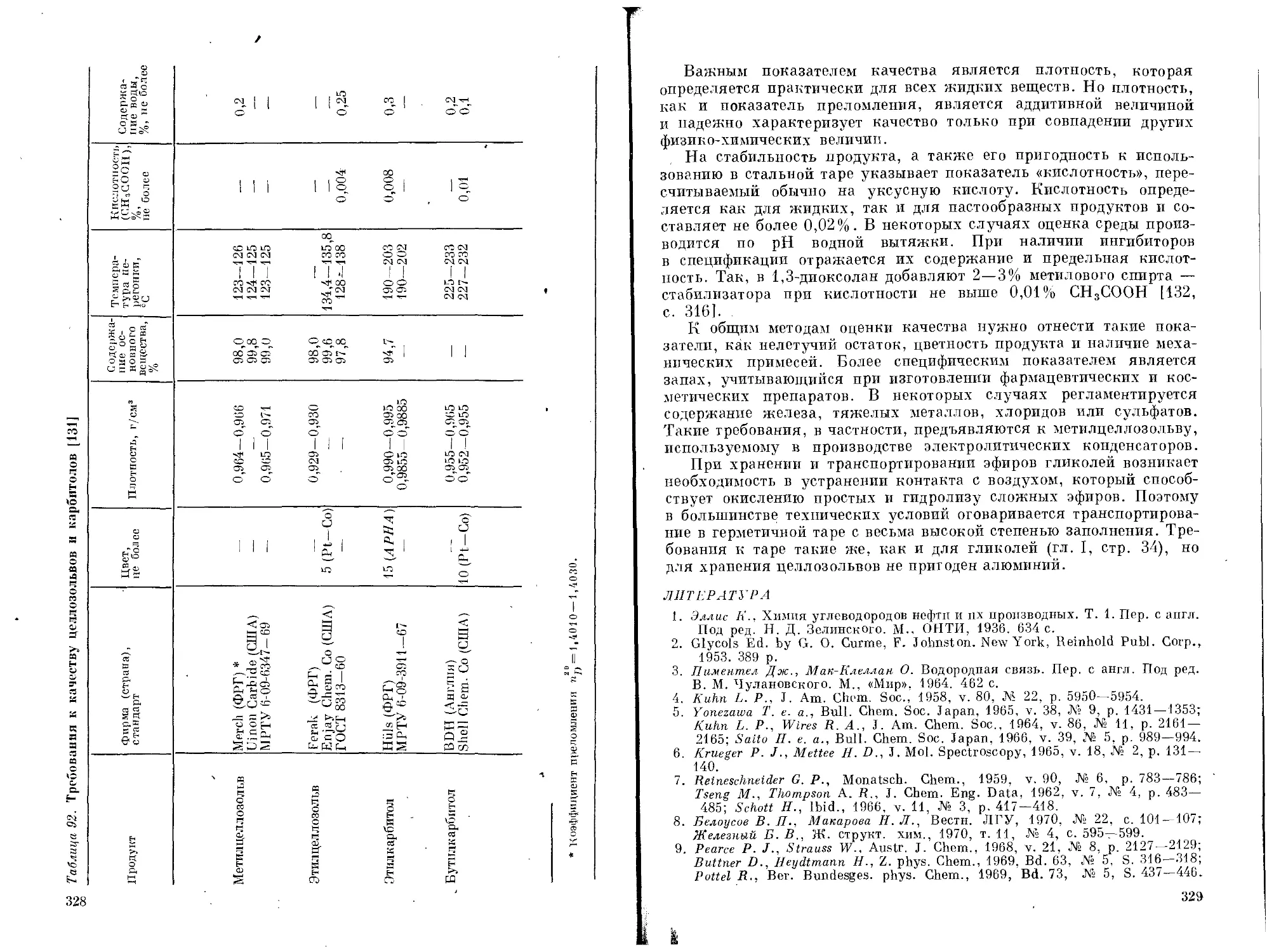
Глава VII. Эфиры гликолей ..........................................289
Физические свойства эфиров гликолей................289
Простые ациклические эфиры.....................289
Простые циклические эфиры......................297
Сложные эфиры .................................298
V Смешанные сложные и простые эфиры 300
4
Г Химические свойства эфиров гликолей...............300
Токсические свойства эфиров гликолей . (...........305
Методы получения эфиров гликолей...................306
Синтезы на основе окисей олефинов.............306
Синтезы через галогенпроизводные..............308
Синтезы на основе гликолей....................309
Синтезы на основе олефинов....................310
Получение эфиров гликолей из а-окисей и спиртов .... 312
Теоретические основы ..........................312
Технология получения простых эфиров гликолей .... 317
Производство этилцеллозольва ..................318
Производство этилкарбитола ....................320
Применение эфиров гликолей.........................322
Эфиры низкомолекулярных гликолей...............322
Эфиры гликолей средней молекулярной массы .... 325
Эфиры цолигликолей ............................325
Требования, предъявляемые к качеству эфиров гликолей 327
Литература ........................................329
Глава VIII. Методы анализа гликолей и их эфиров............. • 335
Химические методы анализа.......................335
Качественный анализ .........................335
Количественный анализ .......................336
Хроматографические методы ......................340
Другие методы анализа гликолей и их производных .... 347
Литература .....................................349
П риложения ...............................................351
Предметный указатель ......................................368
Предисловие
Гликоли и продукты, полученные на их основе, а также другие
производные окисей этилена и пропилена являются весьма важными
и крупнотоннажными продуктами тяжелого органического синтеза.
По темпам роста производства они занимают одно из ведущих мест
среди других продуктов химической промышленности, так как нашли
широкое применение во многих отраслях народного хозяйства:
химической, нефтеперерабатывающей, нефтегазовой, автомобильной,
машиностроительной, текстильной, мебельной, строительной, пище-
вой и др.
Между тем, на русском языке отсутствует систематизированная
литература по гликолям, эфирам и другим соединениям, полученным
из окисей олефинов. Книга Лоури «Глицерин и гликоли», вышедшая
в 1933 г. (перевод монографии, изданной в США в 1928 г.), не только
устарела, но и тогда содержала весьма скудные сведения о гликолях
(одна небольшая глава из 15).
Монография Glycols. Ed. by G. О. Curme, F. Johnston, New
Jork, Reinhold Publ. Corp., изданная в 1953 г., содержит много
сведений о свойствах гликолей, но очень мало материалов по тех-
нологии их производства. Некоторые данные, особенно по масштабам
производства гликолей и областям их применения, уже устарели.
Опубликованная в США в 1962 г. книга по многоатомным спиртам
(Ibert Mellan. Polyhydric Alcohols. Washington. Spartan Books),
хотя и содержит главы по этиленгликолю и пропиленгликолю,
недостаточно освещает технологию производства этих гликолей,
в ней совершенно отсутствуют материалы по ди- и триэтиленглико-
лям, ди- и трипропиленгликолям, а также другим производным
окисей этилена и пропилена.
В связи с развитием производства гликолей и других произ-
водных окисей этилена и пропилена возникла насущная потребность
в технической литературе, освещающей свойства, методы и техно-
логию получения, хранения, анализ, а также другие вопросы, каса-
ющиеся получения и применения названных продуктов. Такая
литература необходима инженерно-техническому персоналу заводов,
6
особенно вновь создаваемых производств, работникам научно-иссле-
довательских и проектных организаций.
В предлагаемой книге приведены также составы азеотропных
смесей, растворимость, равновесие систем жидкость — пар, взятые
из различных справочников, изданных на русском языке. В книге
использована литература, опубликованная по 1975 г. Введение
и главы I—V написаны к. т. н. О. Н. Дыментом, глава VI — к. х. н.
К. С. Казанским, глава VII — к. т. н. А. М. Мирошниковым и
глава VIII — к. т. н. А. Ф. Чудновым.
Авторы будут весьма благодарны всем читателям, которые выска-
жут критические замечания по книге и свои пожелания.
Введение
Окись этилена и окись пропилена, а также их основные производ-
ные — гликоли, эфиры гликолей, аминоспирты, неионогенные по-
верхностно-активные вещества, полиэфиры — являются в настоящее
время крупнейшими по масштабам производства продуктами нефте-
химического синтеза. Этиленгликоль служит основой для получения
низкозамерзающих жидкостей — антифризов — применяемых для
охлаждения автомобильных и других двигателей, как хладо- и тепло-
носители, антиобледенители. Этиленгликоль широко используется
и в качестве растворителя, пластификатора, увлажнителя, а также
для изготовления низкозамерзающих взрывчатых веществ, гидравли-
ческих жидкостей, в электролитических конденсаторах. Особенно
перспективным является его применение для производства синтети-
ческих волокон и пленок.
Пропиленгликоль, как и этиленгликоль, применяется для при-
готовления антифризов, в качестве растворителя, пластификатора
и увлажнителя. Практическая безвредность пропиленгликоля поз-
воляет использовать его в пищевой, косметической и фармацевти-
ческой отраслях промышленности. Особенно широкое применение
пропиленгликоль нашел в производстве полиэфиров.
Диэтиленгликоль используется в качестве селективного раствори-
теля при выделении ароматических углеводородов из их смеси с пара-
финами и нафтенами, для осушки газов, как пластификатор, увлаж-
нитель, для производства полиэфирных и эпоксидных смол, взрыв-
чатых веществ. Эфиры гликолей — целлозольвы и карбитолы —
являются прекрасными растворителями и широко используются
в лакокрасочной промышленности.
При взаимодействии окиси этилена и окиси пропилена с алкил-
фенолами, жирными спиртами и кислотами получается разнообраз-
ный ассортимент неионогенных поверхностно-активных веществ
(ПАВ), нашедших применение в различных отраслях промышлен-
ности (нефтедобывающей и нефтеперерабатывающей, химической и
текстильной, цветной металлургии). Одним из существенных преиму-
ществ ПАВ является то, что они легко подвергаются биологической
очистке в сточных водах.
8
Алканоламины (моно-, ди- и триэтаноламины, соответствующие-
изопропаноламины, а также производные окисей и аминов) нашли
применение в химической, нефтяной, газовой, нефтеперерабатыва-
ющей и нефтехимической и других отраслях промышленности для
очистки газов от примесей (СО2, H2S), для производства моющих
средств, в качестве стабилизаторов и ингибиторов коррозии и т. п.
В последние годы все большее применение получают полимеры
окиси этилена и окиси пропилена и их сополимеры, свойства и на-
значения которых зависят от молекулярной массы полимера, спо-
соба его получения, а также от типа катализатора. Так, полиэтилен-
гликоли с молекулярной массой от 200 до 20 000 (карбоваксы)
являются водорастворимыми смазочными материалами, раствори-
телями смол, красителей, косметических и фармацевтических пре-
паратов, полупродуктами для ПАВ и алкидных смол, пластификато-
рами для ряда веществ. Высокомолекулярные полиэтиленоксиды
с молекулярной массой до 8 000 000 (полиоксы) — кристаллические
водорастворимые полимеры окиси этилена — применяются для
получения различных пленок" для снижения коэффициента трения
растворов, как загустители разнообразных жидкостей, флокулянты.
Сополимеры окиси этилена и окиси пропилена, обладающие одновре-
менно гидрофобными (блоки полипропиленоксида) и гидрофильными
(блоки полиэтиленоксида) свойствами, являются весьма важными де-
эмульгаторами и используются для разрушения нефтяных эмульсий.
Большое значение приобрели простые полиэфиры на основе окиси
пропилена, являющиеся исходным сырьем для производства пено-
полиуретанов. Последние обладают малой плотностью, высокими
тепло- и звукоизолирующими свойствами, стойкостью при низких
и достаточно высоких температурах и поэтому нашли широкое при-
менение в строительстве, авиа-, авто- и вагоностроении, производство
мебели, одежды. Весьма перспективным оказалось использование
окиси пропилена для производства ряда важных продуктов, напри-
мер аллилового спирта, синтетического глицерина и синтетического
каучука.
Несмотря па то, что окись этилена и окись пропилена, а также
их основные производные были открыты более 100 лет тому назад
(в 1859—1860 гг.), промышленное производство их началось значи-
тельно позднее. Производство этиленгликоля в промышленном
масштабе было начато в Германии в период первой мировой войны
с целью изготовления взрывчатого вещества динитрогликоля в связи
с нехваткой глицерина для производства динамита. В США окись
этилена и этиленгликоль были выпущены на рынок в 1925 г., а окись
пропилена стала промышленным продуктом лишь в 1950 г. Промыш-
ленное же производство пропиленгликоля было начато в США
в 1931 г. К этому же периоду относится начало промышленного
производства простых эфиров этиленгликоля (1926 г.) и этанол-
аминов: в 1928 г. — триэтаноламина и в 1931 г. — моно- и диэтанол-
аминов. Неионогенные поверхностно-активные вещества — окси-
этилировапные спирты, кислоты и некоторые другие соединения,
9
появились на рынках Германии в 1933 г. и США в 1934 г. [1, р. 7;
2, v. 1., р. 509; v. 16, р. 595; 3, с. 30].
В отличие от США, где этилен для окиси этилена и этиленгликоля
производили из газов нефтепереработки, попутного и природного
газа, в Германии этилен получали гидрированием ацетилена, дегидра-
тацией этилового спирта и при низкотемпературном фракциониро-
вании коксового газа. В настоящее время в ФРГ и в других странах
Западной Европы этилен и пропилен в основном являются продук-
тами пиролиза нефтяного сырья.
В период между первой и второй мировыми войнами из капита-
листических стран только США и Германия вырабатывали значитель-
ные количества окиси этилена и ее производных — этилен- и ди-
этиленгликоля, эфиров гликолей и этаноламинов. Резкий рост
производственных мощностей и выработки этих продуктов, а также
окиси пропилена и ее производных начался после второй мировой
войны не только в США и ФРГ, но п в странах Западной Европы,
Японии, Канаде. При этом характерной тенденцией было укрупнение
мощностей заводов и установок. Например, в США мощность круп-
ных производств окиси этилена составляла 50—100 тыс. т/г в 1961 —
1962 гг. и 250—350 тыс. т/г в 1967—1968 гг. В ФРГ и Англии мощ-
ность установок по производству окиси этилена в 1967 г. составляла
от 20 до 65 тыс. т/г, а в 1969 г. достигла 40—100 тыс. т/г. В Японии
мощность установок возросла с 9—12 тыс. т/г в начале 60-х гг.
до 50—60 тыс. т/г в конце этих годов [5, с. 38, 43]. В результате
строительства крупных установок производственные мощности по
получению окиси этилена резко возросли и составили (в тыс. т/г):
1964 г. 1969 г. 1970 г.
США 1325 1630 1744
ФРГ 170 445 —
Англия .... 117 130 (1967 г.) 330
Япония — 340 (1971 г.)
К началу 1970 г. мощности по производству окиси этилена в капи-
талистических странах превысили 3 млн. т/г, в том числе в Западной
Европе — 970 тыс. т [6]. В 1975 г. они достигли 4,42 млн. т/г: в Аме-
рике 2,35, Западной Европе 1,61, Японии п других странах
Азии 0,46 ]7].
Весьма быстрыми темпами росли мощности по производству
окиси пропилена. Они составили (в тыс. т/г) [8]:
США .....................
Япония .............
ФРГ .....................
Англия ..................
Франция .................
1964 г. 1970 г.
272 654
70 102
58 141
44 63
21 179
10
Общая мощность производства окиси пропилена в капиталисти-
ческих странах возросла с 1209 тыс. т в 1970 г. до 1984 тыс. т
в 1972 г. или на 64%, в том числе (в тыс. т):
США ................
Западная Европа . . .
Япония......... . .
С 654 до 831 (прирост 27%)
С 412 до 903 (прирост 119%)
С 102 до 193 (прирост 89%)
В 1975 г. мощности по окиси пропилена в США возросли до
1193 тыс. т [7]. Данные о масштабах производства окиси этилена,
окиси пропилена и ряда продуктов их переработки в США, Японии
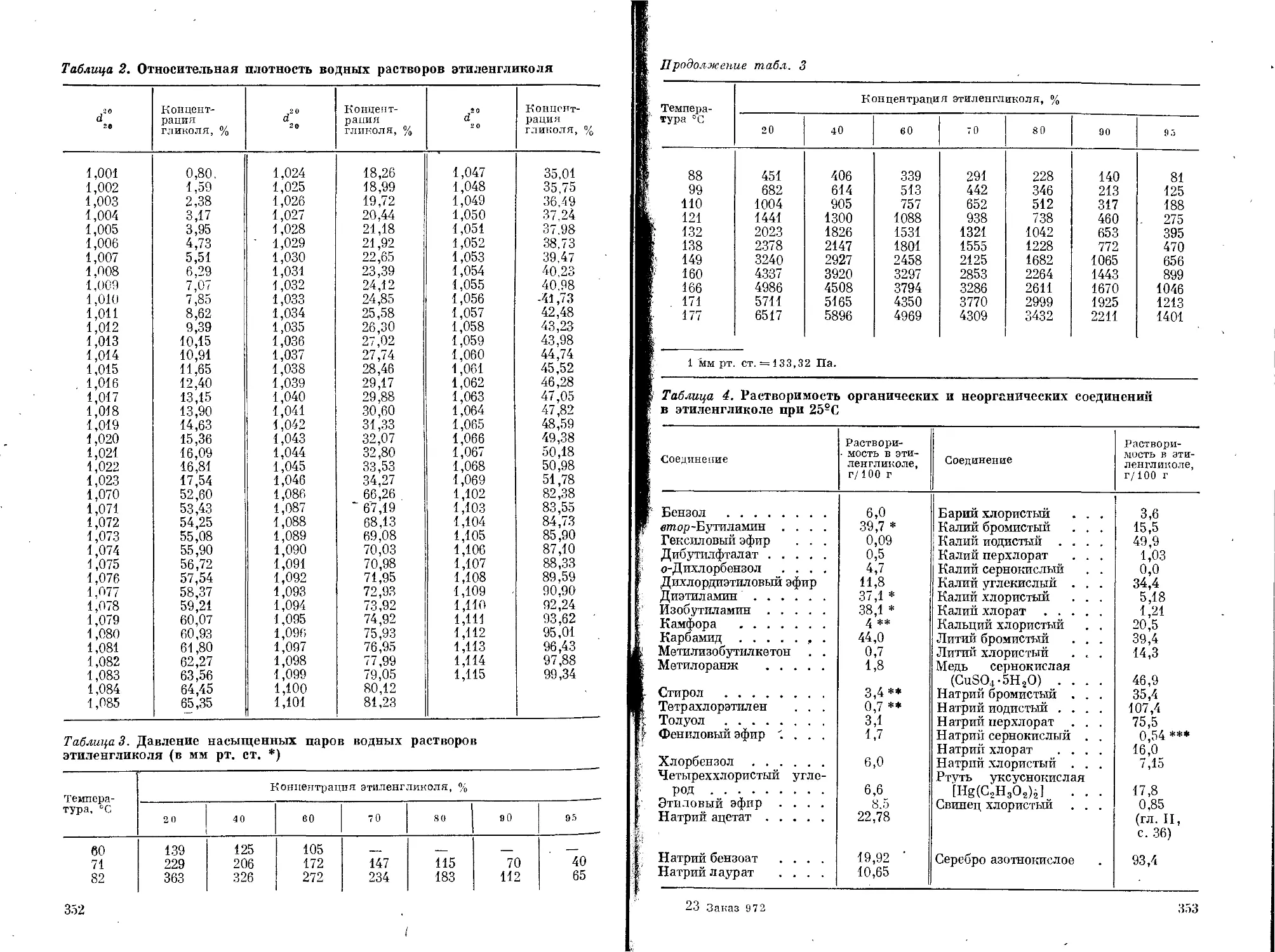
и некоторых странах Западной Европы приведены в табл. 1, 2 и 3.
Высокие темпы роста производства окиси этилена и ее производ-
ных, в частности этиленгликоля, после второй мировой войны в зна-
чительной степени связаны с бурным ростом автомобилестроения
и в некоторой степени авиастроения, ставших крупнейшими потре-
бителями антифризов на основе этиленгликоля. С развитием авто-
мобильной и авиационной промышленности увеличился спрос на
высококачественные бензины, что в свою очередь привело к развитию
в нефтеперерабатывающей промышленности процессов крекинга.
В результате крекинга наряду с бензином получали значительные
количества непредельных углеводородов, в частности этилена и про-
пилена. Таким образом, одновременно с ростом потребности в про-
дуктах на основе окисей этилена и пропилена создавалась и реальная
сырьевая база для их получения.
Бурное развитие нефтегазодобывающей и нефтегазоперерабатыва-
ющей промышленности, ставших основными поставщиками сырья
для производства олефинов, непрерывный рост и совершенствование
установок для пиролиза этана и пропана и агрегатов газоразделения
привели к снижению цен на этилен и пропилен. Так, в США с i960
по 1966 г. цена на этилен снизилась почти вдвое, в то же время цена
окиси этилена долгое время оставалась стабильной, что обеспечивало
высокую прибыль при продаже окиси этилена и ее производных
и стимулировало рост их выработки.
Представления о структуре потребления окисей этилена и про-
пилена в США и Японии можно получить из данных, при-
веденных в табл. 4—7.
В США в 1940 г. около 75%, а с учетом одновременно получаемых
ди- и триэтиленгликолей — более 83% вырабатываемой окиси эти-
лена потреблялось в производстве этиленгликоля, основная часть
которого расходовалась на получение антифриза (в 1950—1965 гг.
около 75—80%). В последующие годы доля окиси этилена, перера-
батываемой в этиленгликоль, снизилась до 55—60%, однако выра-
ботка этиленгликоля непрерывно увеличивалась в связи с быстрым
ростом его потребления на производство полиэфирных волокон
и пленок.
Заметное снижение доли окиси этилена, используемой на произ-
водство акрилонитрила (см. табл. 4), связано с широким примене-
11
Потребность
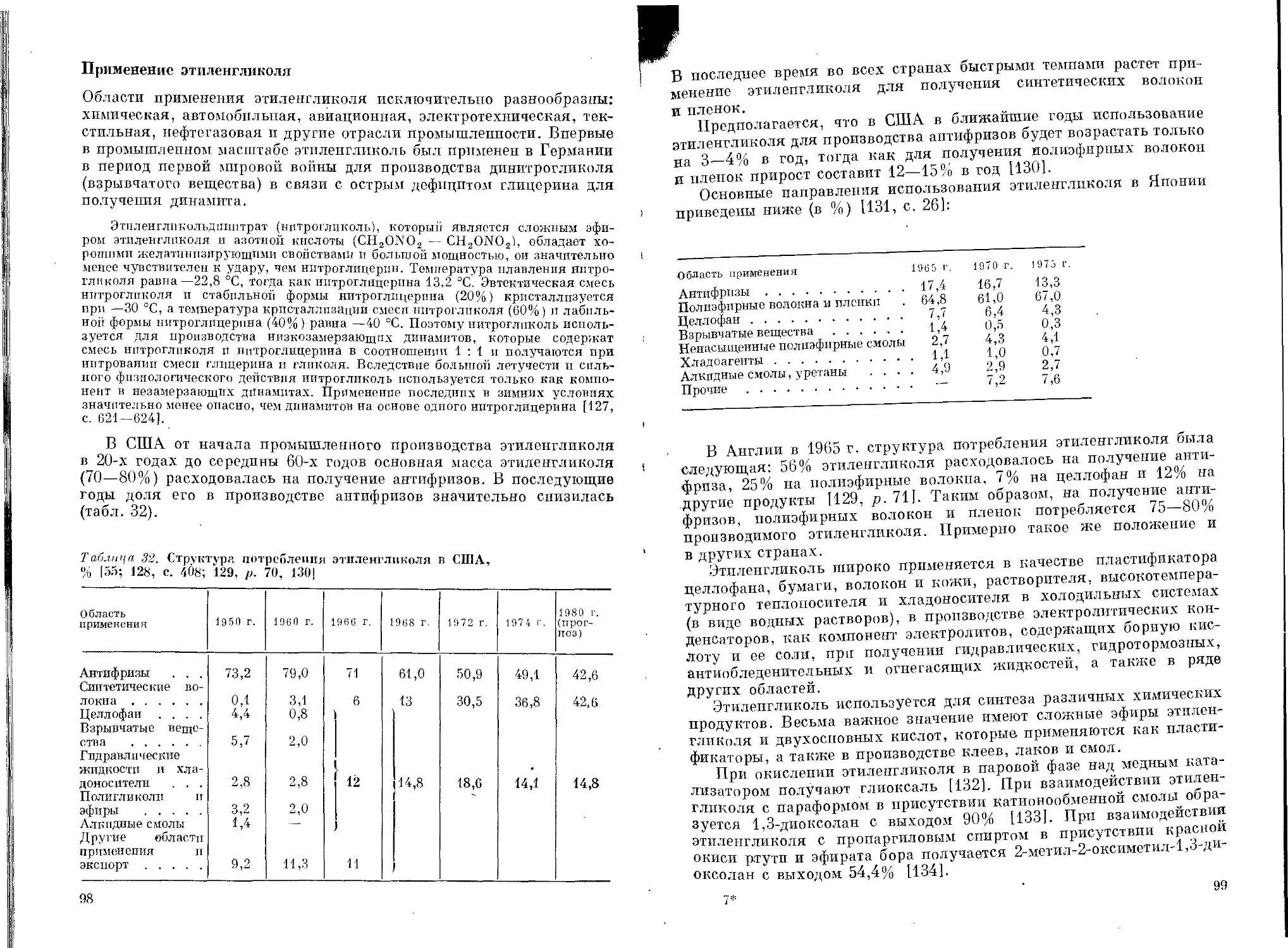
Таблица 3. Масштабы производства окиси этилена и этиленгликоля
в странах Европы (в тыс. т/г) [9, 12, 13, 17—20]
Годы ФРГ Франция Англия Италия Нидер- ланды ФРГ Франция
Оке сь эти лена Этиленгликоль
1960 г. 79,2 28,5 46,0 6,7 — 19,4
1966 г. 162,9 70,0 97,3 58,6 12,0 76,5 56,4
1970 г. 225,0 107,0 177,4 70,0 105,0 156,9 * 78,3 *
1972 г. 244,4 — 198,1 75,1 151,0 — —
1973 г. 271,4 — 210,6 72,9 — —
1974 г. 286,5 — — 76,0 — — —
* 1969 г.
Таблица 4. Структура потребления окиси этилена в США (в %)
J4, с. 12; 6—8; 21: 22, р. 519] ~—
Область применения 1945 г. 1955 г. 1964 г. 1968 г. 1974 г. 1980 г. (прогноз)
Этиленгликоль 72,0 65,6 60,8 54 57 62
Диэтиленгликолъ 7Л 6,8 5,7 • 1 Q * 1 R *
Триэтиленгликоль 1,1 1,9 2,0
Эфиры гликолей 7,4 4,8 6,7 7 7 6
Этаноламины 6,3 7,5 7,1 7 6 6
Акрилонитрил 5,8 5,6 3,2 — — —
Поверхностно-активные вещества . . — 7,9 10,6 13 и 11
Прочие — — 2,3 10 и 15
* Также и тетраэтиленгликоль.
Таблица 5. Структура потребления окиси этилена в Японии (в %)
[14: 15, с. 25]
Область применения 1 96 5 г. 1967 г. 1970 г. 1975 г..
Этиленгликоль 64,7 64,0 65,5 67,3
Полиэтиленгликоли 3,4 4,2 3,4 2,7
Эфиры гликолей .... 4,8 3,2 4,3 4,2
Этаноламины 6,3 6,8 6,7 5,0
Поверхностно-активные вещества . . . 20,2 20,4 16,9 17,0
Прочие 0,6 1,4 3,2 3,8
13
Таблица 6. Структура потребления окиси пропилена в США (в %)
[4, с. 33; 7; 8]
Область применения 1965 г. 1967 г. 1970 г. 1972 г. 1977 г. (прогноз)
Пропиленгликоль 33,7 33 29 29 26
Полппропиленглпколь и другие полиэфирполиолы 37,2 45 56 56 64
Дипропиленгликоль .... — — 4 — —
Поверхностно-активные вещест- ва 5
Прочие 29,1 22 6 15 10
Таблица 7. Структура потребления окисп пропилена в Японии (в %)
[14; 15, с. 36]
Область применения 1965 г. 1968 г. 1970 г. 1975 г.
Пропиленгликоль 28,0 30,8 28,2 25,0
Полипропиленгликоль 70,8 67,5 68,2 71,5
Прочие 1,2 1,7 3,6 3,5
нием новых, более экономичных способов получения акрилонитрила,
вначале из ацетилена и синильной кислоты, а в последние годы —
из пропилена и аммиака.
В ближайшие годы ожидается дальнейший рост производства
окиси этилена и особенно окиси пропилена. Согласно прогнозам,
среднегодовой прирост производства окиси этилена в США составит
около 7,5% и к концу 70-х годов ее выработка достигнет 2,7 млн. т
[7, 21]. На том же уровне (8—9% в год) предполагается рост произ-
водства окиси этилена в Японии, в результате чего ее выработка
достигнет 700 тыс. т в 1980 г. [16].
Предполагается, что в США наиболее высокими темпами (13—
15% в год) будет расти переработка окиси этилена в этиленгликоль
для производства полиэфирных волокон и пленок, тогда как пере-
работка окиси этилена в стандартный этиленгликоль для получения
антифриза возрастет только на 3—4% в год. В итоге в 1979—1980 гг.
расход этиленгликоля на производство полиэфирных соединений
и антифриза сравняется [21]. В связи с непрерывным ростом потре-
бления окиси пропилена на получение полиэфиров для производства
полиуретанов, ожидается, что ее выработка будет возрастать на 12%
в год в США и на 14—15% в год в Японии [7, 8, 16].
Необходимо, однако, иметь в виду, что к указанным прогнозам
следует относиться с осторожностью, так как спад производства
в капиталистических странах, особенно в автомобильной промышлен-
ности, тенденция к увеличению выпуска малолитражных автомоби-
14
лей, потребляющих не только меньше бензина, но и меньше анти-
фриза, рост цен на нефтяное сырье и как следствие повышение цен
па этилен и пропилен могут существенно повлиять на объем произ-
водства окиси этилена п окиси пропилена и продуктов их пере-
работки.
ЛИТЕРАТУРА
1. Glycols. Ed. by G. О. Curine, F. Johnston, New York, Reinhold Puhi. Corp.,.
1953. 389 p.
2. Kirk — Othmer. Encyclopedia of Chemical Technology. Ed. 2nd. New York —
London — Sydney, Interscience Publ., 1963—1972. •
3. Шенфельд H. Неионогенные моющие средства. Пер. с нем. Под ред. А. И. Гер-
шеновича. М., «Химия», 1965. 488 с.
4. Химическая промышленность США. Т. 2. Под ред. Г. Л. Коренькова. М.,.
НИИТЭхим, 1972. 613 с.
5. Производство олефинов (этилена, пропилена) и их производных в основных
капиталистических странах. М., ЦНИИТЭнефтехим, 1970. 82 с.
6. Stobaugh И. В. е. a.. Hydrocarb. Process., 1970, v. 49, № 10, р. 105—115.
7. Chem. Eng. News, 1973, v. 51, № 21, p. 8, 10; Chem. Market. Report., 1975,
v. 208, № 11, p. 9; 1976, v. 209, № 18, p. 9; Gans Al., Ozero B., Hydro-
carb. Process., 1976, v. 55, № 3, p. 73—77.
8. Stobaugh R. B. e. a., Hydrocarb. Process., 1973, v. 52, № 1, p. 99—108.
9. Химическая и резиновая промышленность капиталистических стран.
Изд. 2-е. М., НИИТЭхим, 1962. 599 с.
10. Synthetic Organic Chemicals. United States Tariff Commission. Washington,
Gov. Print. Off., 1947—1974.
11. Chem. Market. Report.. 1973, v. 203, № 9, p. 9; № 20, p. 3. 20; 1974, v. 205,
Al 7, p. 16; 1975, v. 207, № 7, p. 18; 1976, v. 209, Al 7, p. 15; Chem.
Market. Abst., 1975. v. 67, Al 8, p. 130.
12. Хим. пром, за рубежом, 1970, Al 2, с. 41—72; 1972, № 10, с. 51—57.
13. Производство химических продуктов в зарубежных странах в 1965—
1968 годах. М„ НИИТЭхим, 1970. 192 с.
14. Yamamoto К., Chem. Economy and Eng. Rev., 1974, v. 6, № 7, p. 22—28.
15. Химическая промышленность Японии. Вып. 2. М., НИИТЭХим, 1974. 92 с.
16. Chem. Economy and Eng. Rev., 1973, v. 5, № 8, p. 56—57; 1974, v. 6, № 3,
p. 67—68; 1975, v. 7, № 1—2, p. 57.
17. Jose Collado Bravo, «Jon», 1974, v. 34, № 396, p. 511 — 521; Chem. Market
Abst., 1975, v. 67, Al 6, p. Ill; 1976, v. 68, № 3, p. 119.
18. Хим. пром, за рубежом, 1967, № 11, с. 72—73; 1968, № 7, о. 31—42, 87—90;
1968, № 11, с. 39—45; 1969, № 6, с. 45—60.
19. Хим. пром, за рубежом, 1971, № 2, с. 3—35; № 7, с. 74—84; № 9, с. 77—93;
1972, № 2, с. 80; № 5, с. 3—22; № 9, с. 75—76; 1973, № 1, с. 59-63;
1974, № 8, с. 50—97; 1975, № 6, с. 48—50.
20. Europ. Chem. News Chemscope, 1974, v. 21, 88 p.; 1975, 104 p.
21. Chem. Eng. News, 1975, v. 53, № 24, p. 12—13.
22. Ethylene and its Industrial Derivatives. Ed. by S. A. Miller. London, Ernest
Benn Lim., 1969. 1321 p.
Глава I
Общие свойства гликолей
Гликоли являются двухатомными спиртами, т. е. органическими
соединениями жирного ряда, которые имеют две гидроксильные
группы при разных атомах углерода. Общая формула этих соедине-
ний С,гН2п(ОН)2. Названия двухатомных спиртов производятся
от названия двухатомного радикала с окончанием «гликоль», на-
пример этиленгликоль, пропиленгликоль, бутиленгликоль и т. д.
Рассматривая двухатомные спирты как диоксипроизводные угле-
водородов, полученные замещением в них водорода при разных ато-
мах углерода гидроксильными группами, их также называют по
исходным углеводородам. Например, этиленгликоль будет назы-
ваться 1,2-диоксиэтаном, пропиленгликоль — 1,2-диоксипроланом
или 1,3-диоксипропаном в зависимости от того, у каких атомов
углерода водород замещен па гидроксильную группу, и т. д.
По женевской и льежской номенклатурам двухатомные спирты
называют диолами, обозначая цифрами положения ОН-групп и воз-
можных ответвленных радикалов. Например, этиленгликоль будет
называться 1,2-этандиолом, пропиленгликоль CHSCHOHCH2OH —
J,2-пропандиолом, а пропиленгликоль СН3ОНСН3СН3ОН— 1,3-про-
пандиолом. Бутиленгликоль строения СН3С(СН3)ОНСН2ОН будет
называться 2-метил-1,2-пропандиолом или 2-метилпропандиолом-1,2
[1, с. 479].
Низшие полигликоли — ди-, три- и тетраэтиленгликоль, ди-
и трппропиленгликоль — имеют одну или несколько эфирных групп
и поэтому иногда их именуют эфирами. Например, диэтиленгликоль
СН3ОНСН3—О—СН2СН2ОН называют 2,2'- или р,р'-диоксидиэти-
ловым или бис-2-оксиэтиловым эфиром, а триэтиленгликоль
СН2ОНСН2—О—СН2СН2—О—СН2ОН — бис(оксиэтил)гликолевым
эфиром. Низшие полигликоли можно рассматривать и как производ-
ные соответствующих спиртов. В этом случае диэтиленгликоль
называется 2,2'-оксидиэтанолом, а один из дипропиленгликолей
(их три изомера) — 1,1~оксиди-2-пропанолом.
Этиленгликоль является начальным членом гомологического ряда
гликолей, в который входят пропиленгликолп, бутиленгликоли,
16
а также другие гликоли с большей молекулярной массой. Этилен-
гликоль имеет две первичные гидроксильные группы. Пропилен-
гликоль, например, имеет или 2 первичные группы (1,3-пропилен-
гликоль), или одну первичную и одну вторичную ОН-группы (1,2-про-
пиленгликоль). Бутиленгликоль же может иметь 2 первичные (1,4-бу-
тиленгликоль) или 2 вторичные гидроксильные группы (2,3-бутилен-
гликоль), либо одну первичную и одну вторичную ОН-группы
(1,2-бутиленгликоль и 1,3-бутиленгликоль). По мере увеличения
молекулярной массы число изомерных форм гомологического ряда
гликолей непрерывно возрастает.
В зависимости от характера радикалов, с которыми связаны
ОН-группы, гликоли могут быть двупервичными, двувторичными
и первично-вторичными. Кроме того, гликоли могут быть первично-
третичными (изобутиленгликоль) и двутретичными, которые назы-
ваются пинаконами, например:
СН3
HO-CIT--C-CHS (CH3)2C-C(CII3)2
I I I
ОН но он
Гликоли, содержащие ОН-группы у соседних атомов углерода,
например, этиленгликоль или 1,2-пропиленгликоль, являются при-
мерами вицинальных соединений. Такие гликоли обычно получаются
гидратацией а-окисей олефинов.
Физические свойства
Низшие гликоли — это прозрачные, бесцветные, довольно вязкие
гигроскопические жидкости, не имеющие запаха и обладающие
сладким вкусом. Температуры кипения и плавления, вязкость и плот-
ность гликолей значительно выше, а давление паров значительно
ниже, чем одноатомных спиртов с тем же числом атомов углерода.
В табл. 8 приводятся некоторые физические свойства низших глико-
лей по данным [1, с. 418; 2, с. 492; 3, р. 4; 4, р. 2, 287; 5, р. 2; 6,
с. 621; т. 2, с. 548; 7, и. 10, р. 638]. Зависимость давления паров
ряда гликолей от температуры приведена на рис. 1 [8, р. 73], а их
относительная гигроскопичность на рис. 2 [3, р. 5].
Из изомерных гликолей двупервичные имеют более высокую
температуру кипения, чем первично-вторичные; самая низкая темпе-
ратура кипения у дитретичного изомера. Двупервичные гликоли
обладают также наиболее высокой плотностью и коэффициентом
преломления. Гликоли с нормальной цепью кипят при более высокой
температуре, чем с разветвленной цепью атомов углерода.
У низших полиэтиленгликолей по мере увеличения числа окси-
этиленовых групп значительно повышаются температура кипения,
вязкость и температура замерзания. В меньшей степени повышаются
2 Заказ 972 17
Таблица 8, Физические свойства гликолей
ПоказаЮли Этиленгликоли Пропилен
этилен- гликоль диэти- лен- гликоль трпэти* ленгли- коль тетра- • этилен- гликоль 1,2- пропи- ленгли- коль дипро- пилен- гликоль
Брутто-формула Молекулярная С2Н6О2 свн14о4 с8н1803 CgHgOj СеН 14О3
масса Относительная 62,07 106,12 150,18 194,20 76,09 134,17
плотность Температура кипе- ния, °C при 101,3 кПа (760 мм рт. 1,116 1,118 1,126 1,125 1,034 1,024
ст) Давление насы- щенного пара 197,3 244,8 278,3 307,8 188,2 231,8
прп20°С,Па . . 8,0 <1,3 <1,3 <1,3 10,7 <1,3
мм рт. ст. . . Температура замер- 0,06 <0,01 <0,01 <0,01 0,08 <0,01
зания,°C .... -13 —8 -7,2 -5,6 -60 <—50
Коэффициент 20 1,4411
преломления по 1,4316 1,4472 1,4559 1,4598 1,4326
Вязкость при
20 °C, мПа-с 20,9 35,7 47,8 39,9 (25 °C) 56,0 106
Примечание. Физические свойства гликолей по различным источникам несколько
по [6], но наиболее часто указывается 197,6 °C [3, 7, 11], хотя приводятся значения
по [6], но в [1,5] она равна 197,2 и 197,4 °C Т. кип. низших полиэтиленгликолей
тетраэтиленгликоля—327,3 °C. Разные значения можно найти и для других показателей
плотность и коэффициент преломления, причем по мере увеличения
числа оксиэтиленовых групп разница в этих показателях умень-
шается.
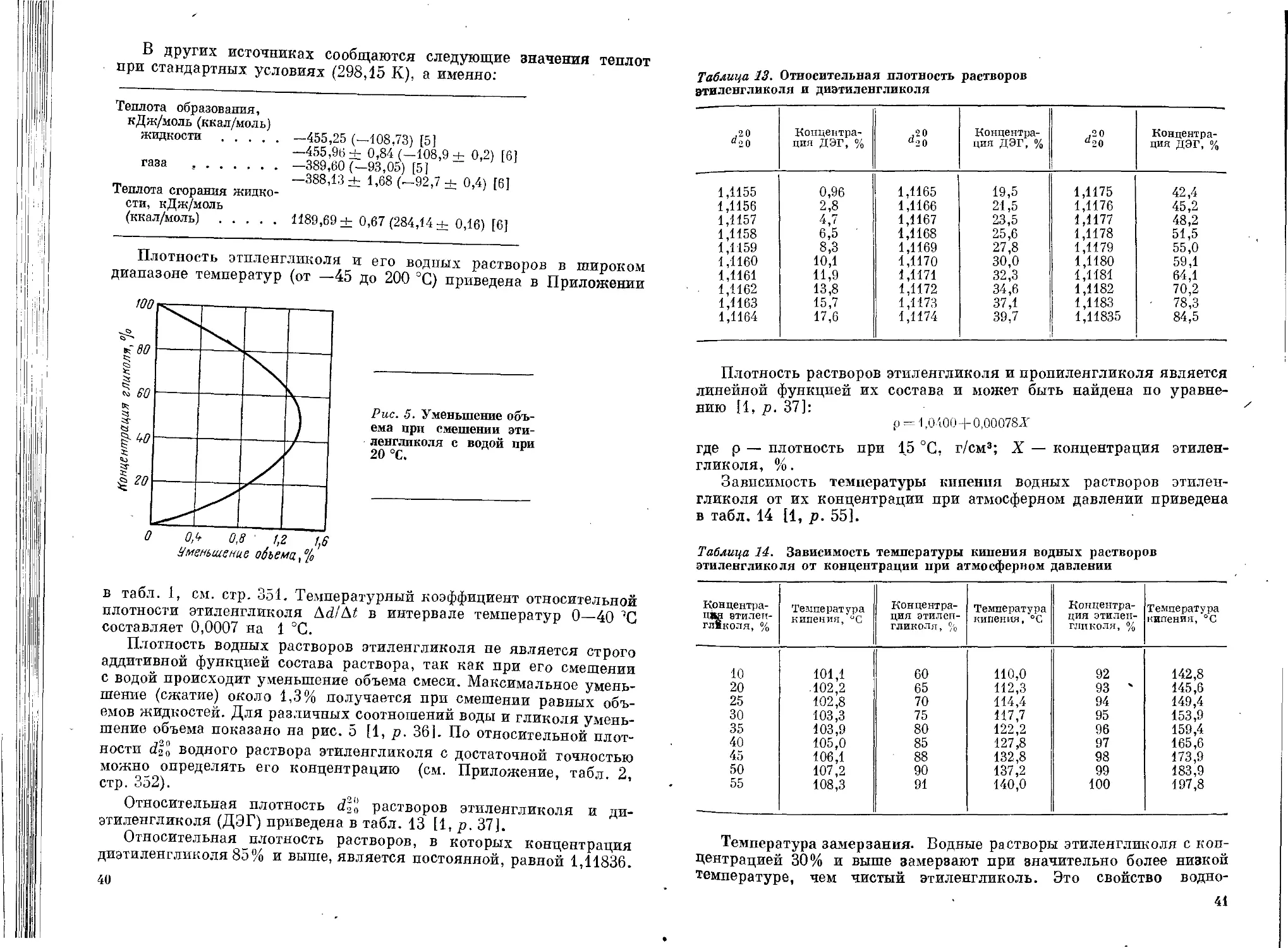
Низшие гликоли полностью смешиваются с водой, а также с орга-
ническими соединениями, растворимыми в воде: алифатическими
спиртами, альдегидами, кетонами, кислотами, аминами, с некоторыми
ароматическими гидроксилсодержащими соединениями, например
фенолом, резорцином, многоатомными спиртами (глицерином). При
числе атомов углерода в молекуле гликоля выше 7 растворимость его
в воде ухудшается.
Предельные углеводороды практически нерастворимы в гликолях;
растворимость ароматических углеводородов в гликолях увеличи-
вается по мере возрастания молекулярной массы гликоля. Напри-
мер, в этиленгликоле растворяется 5,7% бензола, в диэтиленгли-
18
гликоли В утиленгликоли
трипро- пилен- гликоль 1,3- пропи- ленгли- коль 1,2- раце- мат 1,3- раце- мат 1,4- раце- мат 2,3-(85% мезоформа, 15% раце- мат) 2,3-лево- вращающий 2,3-право- вращающий 2,3-мезо- форма , 2,3-раце- 1 мат 1
С9Н20О4 С3Н8О, о2
192,25 76,09 90,1 2
1,016 1,055 1,006 1,006 1,020 1,009 0,991 0,987 (25 °C) 1,004 .1,003
267,2 214,2 192 207,5 230 182 178 — 182 177
<1,3 <0,01 — — 8,0 0,06 — 22,6 0,17 — — — —
— — — <-50; —77 20,9; 16 19 • 18 — 34,4 7,6
1,4420 (25 °C) 1,4389 1,4391 (18 °C) 1,4401 1,4460 1,4377 1,4318 (26 °C) 1,4306 (25 °C) 1,4325 (35 °C) 1.4363 (25 °C)
55,1 (25 °C) — — 89 (35 °C) 71,5 (25 °C) 121 (25 °C) 41,1 (25 °C) 65,6 (35 °C) —
отличаются между собой. В таблице т. кип, этиленгликоля, равная 197,3 °C, приведена
197,2 и 197,4 °C [1,5]; для Прониленгликоля т. кип, равная 188,2 °C, также приведена
согласно [11, р. ЗОв] равна: диэтиленгликоля— 245,8 °C; триэтиленгликоля—288,4 °C;
(т. пл., <7, и др.).
коле — 31,3%, а три- и тетраэтиленгликоль полностью с ним сме-
шиваются. В пропиленгликоле растворяется 19,2% бензола, т. е.
в 3 раза больше, чем в этиленгликоле, а дипропиленгликоль уже
полностью смешивается с бензолом.
Одним из важных свойств гликолей является то, что в некоторых
из них, например пропиленгликоле, могут' растворяться вещества
как гидрофильного, так и гидрофобного характера, что позволяет
гомогенизировать несмешивающиеся компоненты.
Гликоли являются хорошими растворителями для многих смол,
масел, красителей, лекарственных препаратов и пищевых веществ
(в двух последних случаях применяется пропиленгликоль, практи-
чески не обладающий токсическими свойствами). Полигликоли (ди-,
три-, и тетраалкиленгликоли) являются более эффективными раство-
рителями, чем моногликоли.
2*
19
30 50 50 60 70 go
Относительная Влажность
воздуха, °1о
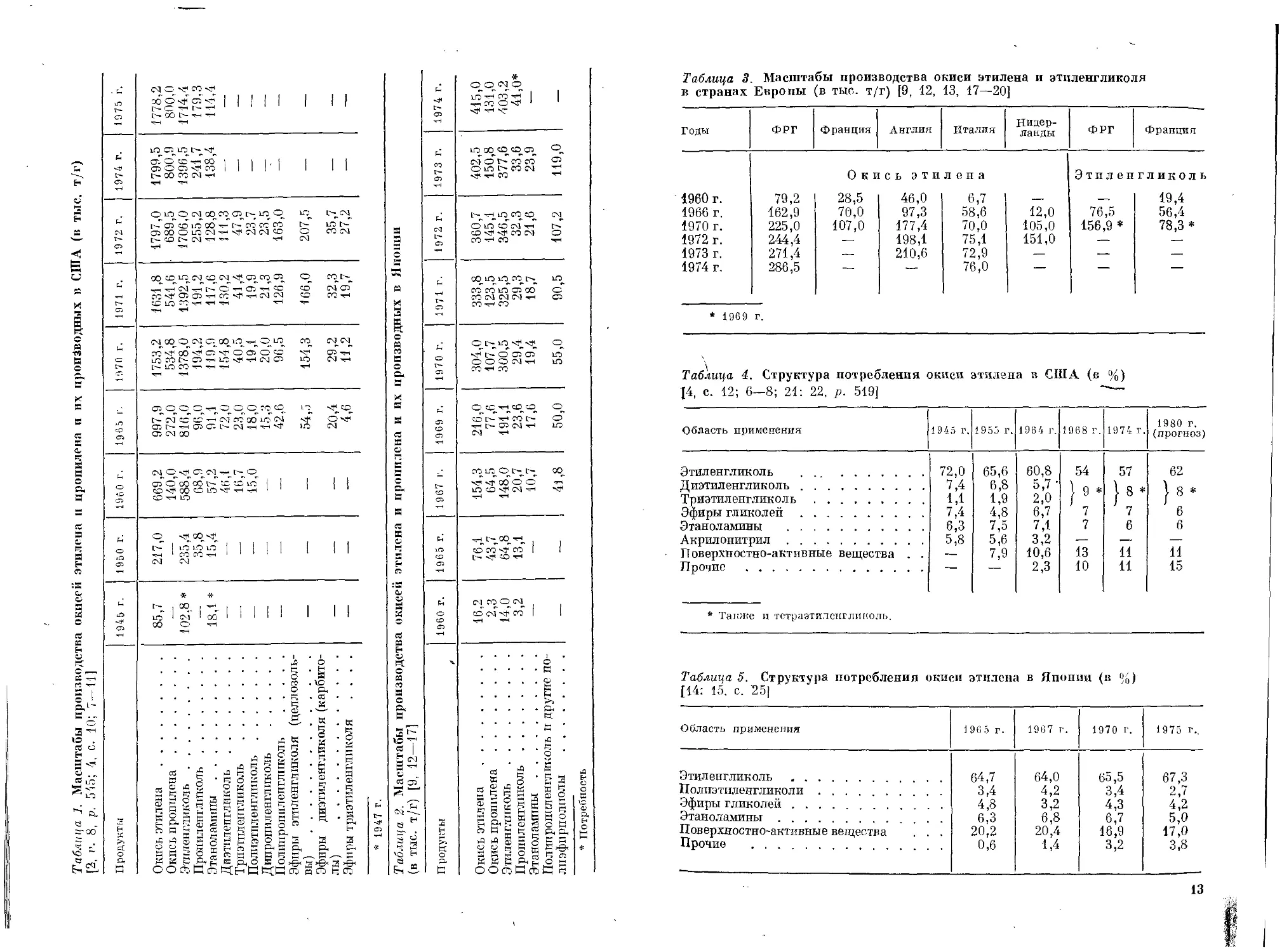
Рис. 1. Зависимость давления паров гликолей от температуры:
7 — пропиленгликоль; 2 — этиленгликоль; з — дипропиленгликоль; 4 — диэтиленгликоль;
S — трипропиленгликоль; в — триэтиленгликоль; 7 — тетраэтиленгликоль.
Рис. 2. Относительная гигроскопичность гликолей при 20—25 °C:
7 _ этиленгликоль; 2 —. диэтиленгликоль; 3 — пропиленгликоль; 4 — триэтиленгликоль
6 — дипропиленгликоль.
Рис. 3. Зависимость температуры замерзания водных растворов гликолей от
концентрации гликоля:
7 _триэтиленгликоль; 2 — дипропиленгликоль; з — диэтпленгликоль; 4 — пропилен»
гликоль; 5 — этиленгликоль.
Исключительно важным свойством гликолей является их способ-
ность понижать температуру замерзания водных растворов, в кото-
рых они ассоциируют с водой, образуя гидраты. Температура замер-
зания этих гидратов ниже той, которая теоретически рассчитана
для смеси гликоля с водой при концентрации, соответствующей
составу гидрата. Чем ниже дипольный момент гликоля, тем больше
его способность к ассоциации и понижению температуры замерзания
раствора [9, р. 10]. Характерным свойством гликолей и их раство-
ров является способность к переохлаждению. На рис. 3 приводится
зависимость температуры замерзания водных растворов гликолей
от их концентрации [10, с. 536].
Химические свойства *
Химические свойства гликолей определяются наличием в их моле-
куле двух гидроксильных групп. Гликоли могут вступать во все
реакции, характерные для спиртов, причем в зависимости от того,
реагирует ли одна ОН-группа или обе, образуются два ряда соеди-
нений. Только из этиленгликоля получаются производные с двумя
первичными атомами углерода. Другие вицинальные гликоли, на-
пример пропиленгликоль, могут давать производные как первичного,
так и вторичного атома углерода.
При действии на гликоли щелочных металлов образуются гли-
коляты:
CH>OHCII2OH-l-2Na -> CH2ONaCH.2ONa + Н2
2CH2OHCH2OH + 2Na > 2CH2OHCII3ONa+H2
Гликоляты могут быть получены и при взаимодействии гликолей
со щелочами:
CII2OHCH2OH + NaOII —► CH2OHCH2ONa-]-H2O
При взаимодействии гликолей с окисями олефинов в зависимости
от соотношения взятых реагентов образуются полигликоли различ-
ного состава:
/°\ °
СН2ОПСН2ОН СН2ОНСН2-О-СН2СН2ОН‘!^=22-1*
диэтиленгликоль
—> СН2ОНСИ2—О—СН2СН2—О—CH2CH2OH
триэтиленгликоль
о
гтт TrtH,C-ОН.
СН3ОНСН2ОН -----------> НОСН2СН2—О—(—СН2СН2О—)n—Н
полиэтиленгликоли
* в разделе использована литература [1, с. 482; 3, р. 28; 4, р. 114: 7, р. 638;
9, р. 2; 11, р. 293; 12, р. 289]. ' г
1
20
С органическими кислотами и их ангидридами этиленгликоль
реагирует с образованием одно- и двузамещенных сложных эфиров:
СН2ОНСН2ОН-
+СН3СООН
+2СН3СООН .
СН2ОНСН2ОСОСН3
сн3ососн2сн2ососн3
Сложные эфиры образуются также при взаимодействии этилен-
гликоля с двухосновными кислотами или их ангидридами и эфирами.
Например, с метиловым эфиром терефталевой кислоты образуется
этиленгликольтерефталат:
СН3ООС—СООСН3-|-2СН2ОНСН2ОН-->
СН2ОНСН2ООС-/^Л—СООСН3СН2ОН + 2СН3ОН
Гликоли при реакции с низкомолекулярными двухосновными
кислотами или их эфирами могут давать циклические соединения:
С2Н5ОС=О
СН2ОНСН2ОН+ |
С2Н5ОС=О
щс/ \с=О
I I +2С2Н5ОН
Н2С^ /С=О
о
1,4-диоксан-2,3-дион
Циклические эфиры гликолей и угольной кислоты — алкилен-
карбонаты образуются при реакции гликолей с фосгеном:
н.с7 \
СН2ОНСН2ОН+СОС12-----> “I ;c=O-j-2HCI
Н2сх Z
о
этиленкарбонат
Ациклические сложные эфиры угольной кислоты образуются
при взаимодействии этиленгликоля со сложными эфирами хлор-
угольной кислоты:
СН2ОНСН2О11 + 2НОСОС1 —> ROCOOCH2CH2OOCOR + 2HC1
Сложные эфиры получаются и при взаимодействии этиленгликоля
с минеральными кислотами. Так, с борной кислотой образуется
циклический этиленгликольборат
О
сн2онсн2он+н3во3—> Нз? ^вон + гноО
н2сх /
о
а с азотной кислотой — зтиленгликольдинитрат:
CII2OHCH2OH + 2HNO3 —► O2NOCH2CH2ONO24-2H2O
При действии на этиленгликоль галогеноводородных кислот
обычно замещается на галоген одна ОН-группа с образованием
этиленгалогенгидрина:
СН3ОНСН2ОНЧ-НС1 —> СН2ОНСН2С1 + Н,О
Однако при действии на этиленгликоль пятихлористого фосфора
можно заместить на хлор обе ОН-группы, в результате чего обра-
зуется дихлорэтан:
СН2ОНСН2ОН4-2РС15------► СН2С1СН2С1-|-2РОС13-|-2НС1
При взаимодействии этиленгликоля с диалкилсульфатами в при-
сутствии щелочи получаются простые моно- и диэфиры этиленгли-
коля — целлозольвы:
сн2онснаон косн2сн2он
—> ROCH2CH2OR
+R2SO4; 4-Каон
—NaRSO,; -Н,О *
При реакции этиленгликоля с альдегидами и кетонами в при-
сутствии хлорного железа или фосфорной кислоты образуются
циклические ацетали — диоксоланы:
СН2ОНСН2ОН + НСНО—► Нз? 'с/ +Н2О
Н2сх / \н
о
1,3 диоксолан
Н \ /СН3
СН2ОНСН2ОН4-СН3СОСН3—► 3| X +Н2О
н2сх / \СНз
о
2,2-диметил-ЦЗ-
диоксолан
С пропиленгликолем формальдегид образует 4-метил-1,3-диок-
солан
О
СН3СНОНСН2ОН + НСНО—> СНз-Н9 ^СН2 + Н2О
Н2СХ /
о
23
22
а с диэтиленгликолем — триоксоциклооктан:
,СНаСНа—О.
сн2онсн2осн2сн2он+нсно —> о; ^сн2
^СНзСНз-О/
При взаимодействии этиленгликоля с вицинальными диальдеги-
дами получаются бициклические соединения:
О О
HSC^ ^СН \на
2СН2ОНСН2ОН + СНОСНО --------> I | I +2Н2О
н.2с сн сн2
1,4,5,8-нафтодиоксан
В щелочной среде (обычно в присутствии КОН) гликоли реаги-
руют с ацетиленом; при этом образуются виниловые и дивиниловые
эфиры (реакция Фаворского — Шостаковского):
СН2ОНСН2ОН +™-Ch[KOhU CH2=CHOCH2CH2OH - +сн=сн
—> СН2=СНОСН2СН.2ОСН=СН2
Продуктами взаимодействия гликолей с акрилонитрилом являются
циановые эфиры гликоля. В зависимости от количества акрило-
нитрила и катализатора можно получить моно- или дициановый
эфир [13]:
+CHZ=CHCN .
+ 2CHs=CHCN .
СН2ОНСН2ОП—
CH2OHCH2OCH2CH2CN
C2H4(OCH2CH.,CN)2
Дегидратация гликолей в зависимости от условий реакции про-
текает различно. При нагревании этиленгликоля с хлористым пин-
ком получается ацетальдегид с малым выходом:
СН2ОНСН2ОН * СПа=СНОН —> сн3-спо
При нагревании этиленгликоля в присутствии щелочи образуется
диэтиленгликоль, а при нагревании этиленгликоля с небольшим
количеством серной или другой кислоты (в качестве катализатора)
получается циклический эфир диоксан:
FNaOHl /СН2СН2ОП
2СНаОНСН2ОН - 1ЛаОН1^ 0/ +Нз0
4 СН2СНаОН
[кислота! /СН2СН2.
2СН2ОНСНаОП - КИСЛ0Та]^ (У чО+2НаО
4 сн2сн/
Диоксан получается также при дегидратации диэтиленгликоля
и высокомолекулярных этиленгликолей. При дегидратации пропи-
24
ленгликоля, в присутствии указанных выше катализаторов, обра-
зуется смесь 2,5- и 2,6-диметил-1,4-диоксанов:
СН3 СН3
СН-СНа. /СН2—сн.
2СН3СНОНСНаОН —► с/ ХО-|-с/ Ч) + 2НаО
' \ciL-CH/ СН2-СН/
I I
СН3 СН3
Согласно работе [14], основным продуктом превращения про-
пиленгликоля при 120 °C в присутствии катионита КУ-2 и некоторых
кислот является циклический ацеталь пропионового альдегида
и пропиленгликоля:
О
/ Х СН,
2СН3СНОНСН2ОН---->С2Н3—НСХ | +Н2О
\ „СН—СН3
, 0
Над фосфорной кислотой, нанесенной на цеолит, при 250 °C
в основном получается пропионовый альдегид. Над фосфорнокислым
алюминием при этой же температуре образуются аллиловый спирт
и ацетон. Над окисью алюминия при 300 °C образуется ацетон, а при
280 °C — аллиловый спирт (основной продукт), ацетон и 2,5-ди-
метилдиоксан [15].
При действии дегидрирующих катализаторов из гликолей можно
получить ряд продуктов. Например, при каталитическом дегидри-
ровании диэтиленгликоля образуется 2-диоксанон:
zCH2-CH2
НОСН2СН2—О-СНаСНаОН —> с/ )о+2На
\сн2----с/
При дегидрировании пропиленгликоля над цинкмедьхромовым
катализатором с хорошими выходами получается ацетол:
СН3СПОНСН2ОН ---> С1Т3— СО—СН,ОН + Па
При пропускании же пропиленгликоля при 250—320 °C над
медью и окисью никеля, нанесенных на алюмосиликат, образуются
а-метиллевулиновый альдегид, метилглиоксаль, пропионовый аль-
дегид, пропионовая кислота и ряд других веществ [16].
При повышенной температуре (200—300 °C) и pH среды, равной
5—7, из высших этиленгликолей образуются низшие [17]:
СН2ОНСН2—О—СПаСНа— О—СН,СНа01[4-Н,0 —>
триэтиленгликоль
—> снаонсна—О-СНаСН2ОН+СН2ОНСП2ОН
диэтиленгликоль этиленгликоль
25
При окислении гликолей в зависимости от условий и применя-
емых окислителей окисляется одна или обе гидроксильные группы.
Так, при окислении этиленгликоля возможно образование гликоле-
вого альдегида, гликолевой кислоты, глиоксаля, глиоксиловой
кислоты, щавелевой кислоты и двуокиси углерода.
При окислении гликолей молекулярным кислородом получаются
перекисные соединения, формальдегид, муравьиная кислота и слож-
ные эфиры. При окислении пропиленгликоля возможно также обра-
зование большого числа различных соединений: ацетон, пропионовый
альдегид, пировиноградный альдегид, ацетол, молочная кислота,
формальдегид, ацетальдегид, двуокись углерода и др. При окислении
этиленгликоля иодной кислотой количественно образуется формаль-
дегид
СН2ОПСН2ОН + НЮ4 —> 2НСНО + НЮз + Н2О
а при окислении пропиленгликоля — формальдегид и ацетальдегид:
СН3СНОНСН2ОН-|-Н1О4 —► ПСНО + СН3СНО + ШО34- Н2О
При действии на этиленгликоль бихромата калия или перманга-
ната калия происходит его полное окисление:
3CH2OHCH2OH+5K2Cr2O7+20H2SO4 —>
—» 5K2SO4+5Cr2(SO4)34-6CO2 + 29H2O
Аналогичным образом бихромат калия действует и на пропилен
гликоль, а также на ди- и триэтиленгликоль.
Водородная связь и ассоциация гликолей. В гликолях, как
и в других гидроксилсодержащих соединениях, образуются водо-
родные связи. Однако вследствие наличия двух ОН-групп, в отличие
от спиртов и воды, образуются два типа водородных связей: меж-
молекулярные и внутримолекулярные. В ИК-спектре жидкого эти-
ленгликоля при комнатной температуре наблюдается только широ-
кая полоса в области частот 3353 см'1. В разбавленном растворе
этиленгликоля в четыреххлористом углероде наблюдаются две интен-
сивные полосы при частотах 3604 и 3635 см-1. Полоса 3353 см'1
относится к ОН-группе, образующей межмолекулярные водородные
связи, полоса 3604 см-1 — к ОН-группе с внутримолекуляр-
ной водородной связью и полоса 3635 см-1 — к свободной
ОН-группе [181. При низкой температуре (—15 СС) в разбавленном
растворе этиленгликоля в СС14 полоса свободной ОН-группы найдена
при 3644 см-1, а слабо выраженный дублет при 3604 и 3614 см-1
свидетельствует о существовании двух типов внутримолекулярных
водородных связей [19]:
ч
°н Н2/С-СН2 3644СН-1 Н2С-СНг
сн,—сн2 Ох ..О-^Н С)^ ''%о
3644см'1 о—н 3604 см"1 Н 3614см"1 Н
транс гош (l) гош (11}
I
™ Конформер II с двумя водородными связями является более
стабильным. При повышении температуры до комнатной дублетная
структура исчезает. По данным разных авторов, частота валентных
колебаний свободной ОН-группы в этиленгликоле v (ОН) составляет
3636—3645 см'1, а ОН-группы, участвующей в образовании водород-
ных связей, у' (ОН) равна 3603—3619 см-1 и Av(OH) = v(OH) —
— v' (ОН) равна 26—33 [20, с. 169].
Для пропиленгликоля определено v/0H) = 3634 ± 6 см-1 и
vfoHi — 3596 см'1 [21].
Соотношение между транс- и го/и-конформерами зависит от тем-
пературы и диэлектрической проницаемости растворителя. В раство-
рителях с низкой диэлектрической проницаемостью в этиленгликоле
содержится до 20% транс-конформера. В твердом этиленгликоле
обнаружен только гогн-конформер [22].
Гликоли находятся в ассоциированном состоянии за счет меж-
молекулярных связей. Определено, что максимальная степень ассо-
циации жидких этилен- и пропиленгликоля равна 2,9, а бутан-
диола-2,3 — 4,1. При повышении температуры степень ассоциации
снижается: при 10 °C она составляет 2,15 для этиленгликоля и 2,59
для пропиленгликоля, а при 100 °C — 1,87 и 2,40 соответственно.
Для димера и тримера этиленгликоля предложена следующая
структура [23]:
СН»СН2ОН
!
НОСН2СН2-О—II н '
: ; • I > '
Н-О—СН2СН2ОН НОСН2СН2—С\ ,О—СП2СН2ОН
н
При смешении гликолей с водой, аминами и другими соедине-
ниями, имеющими электроотрицательные атомы, также образуются
межмолекулярные водородные связи. «Аномальные» свойства рас-
творов гликолей объясняются образованием ассоциатов определен-
ного молекулярного состава. Смешение этиленгликоля с водой со-
провождается выделением тепла и сжатием полученной смеси, причем
максимальное выделение тепла наблюдается в растворе, отвечающем
составу С2Н4(ОН)3-2Н3О [4, р. 42; 24, с. 68]. Образование гидрата
С2Н4(ОН)2 • 2Н2О подтверждается изменением диэлектрической про-
ницаемости и вязкости водно-гликолевых растворов [25]. Эвтекти-
ческая точка системы этиленгликоль — вода также близка к составу
С2Н4(ОН)2• 2Н2О (см. гл. II, стр. 42).
Аналогичное явление происходит при смешении пропиленгли-
коля с водой: максимальное выделение тепла и наибольшая плот-
ность наблюдаются для растворов, близких к такому составу:
С3Н6(ОН)2 2Н2О [4, р. 214; 24, с. 70].
С дициклогексиламином гликоли (этилен-, диэтилен-, пропилен-
гликоль и некоторые другие) образуют аддукты такого состава:
27
26
1 молекула гликоля и 2 молекулы амина [26]. Водородная связь
и ассоциация гликолей подробно изучены в работах [2, 277].
Радиолиз и фотолиз гликолей. При поглощении энергии ионизи-
рующего излучения и под действием световой радиации гликоли
претерпевают изменения вплоть до разрыва связи углерод — углерод
в молекуле. Состав получающихся продуктов и их радиационно-
химический выход g (абсолютное количество молекул, образующихся
при поглощении 100 эВ * энергии ионизирующего излучения) зави-
сят от ряда факторов: концентрации гликоля, мощности дозы, тем-
пературы, pH среды, содержания в растворе кислорода или других
газов, ионов [28—30].
В продуктах радиолиза этиленгликоля в зависимости от условий
его проведения найдены альдегиды (муравьиный, уксусный, глико-
левый, янтарный, глиоксаль), кислоты (уксусная, щавелевая, гли-
колевая, глиоксалевая), спирты (метиловый, этиловый, эритрит)
и другие кислородсодержащие соединения (ацеталь, этилацетат,
метилдиоксолан), а также газообразные продукты (водород, кисло-
род, окись углерода, метан, этан, этилен). Кроме того, образуется
и вода.
Одним из основных факторов, определяющих состав и выход
продуктов радиолиза, является концентрация этиленгликоля. В лю-
бой среде с увеличением концентрации этиленгликоля выход карбо-
нильных соединений возрастает. Так, в насыщенном воздухом кислом
растворе (0,4 М H2SO4) и концентрации этиленгликоля 0,02 М при
дозе облучения 1018 эВ/мл и мощности дозы 1,2-1015 эВ/(мл-с)
выход продуктов радиолиза составляет: g (СН2ОНСНО) = 4;
g (СН3О) = 0,5 и g(H2O2) = 3,8. При концентрации же этилен-
гликоля 0,5 М радиационно-химический выход гликолевого альде-
гида и формальдегида повышается до 7,4 и 1,0 соответственно, а вы-
ход перекиси водорода остается практически неизменным и равен 3,5.
В деаэрированных щелочных (0,1 М КОН) растворах, в отличие
от кислых и нейтральных, гликолевый альдегид не образуется.
В них практически отсутствуют перекись водорода и янтарный
альдегид и преобладает ацетальдегид. При дозе облучения
1,1 • 1018 эВ/мл, мощности дозы 1,2-1015 эВ/(мл-с) и концентрации
этиленгликоля 0,01, 0,1 и 1,0 М выход ацетальдегида соответственно
равен 3,2, 12,5 и 36,5. В щелочной среде, при прочих равных усло-
виях, общий выход карбонильных соединений выше, чем в кислой.
Температура весьма существенно влияет на выход продуктов
радиолиза. С ее повышением от 20 до 170 °C при радиолизе нейтраль-
ного деаэрированного раствора этиленгликоля (5-10“2 М), мощности
дозы 4-1015 эВ/(мл-с) выход гликолевого альдегида повысился
с 0,5 до 12, формальдегида с 2 до 13, а выход водорода снизился
с 2,2 до 0,5.
В определенных условиях при у-радиолизе этиленгликоля полу-
чается 2-метил-1,3-диоксолан (с высоким выходом). Вначале обра-
* 1 эВ = 1,6021 • 10" I» Дж.
28
дуется ацетальдегид, который при взаимодействии с гликолем пре-
вращается в диоксолан [30]. Степень разложения пропиленгликоля
примерно такая же, как этиленгликоля, если радиолиз проводится
при одинаковых условиях [31]. -
Гликоли претерпевают изменения также под действием световой
радиации. При действии ультрафиолетового света ртутной лампы
на этиленгликоль в качестве первичных продуктов образуются вода
и ацетальдегид, а в результате вторичных реакций — кротоновый
альдегид и этилацетат. При фотолизе пропиленгликоля первичными
продуктами являются вода, пропионовый альдегид и ацетон; при
гидрировании карбонильных соединений атомом водорода, получа-
ющимся в процессе фотораспада, образуются изо- и н-пропанол.
В газовой фазе обнаружено значительное количество метана, этана,
окиси углерода и воды [32].
Аутоокисление гликолей. Гликоли способны к аутоокислению,
т. е. к самопроизвольному окислению молекулярным кислородом
при невысоких температурах. Самым стойким при хранении является
моноэтиленгликоль; ди-, три- и тетраэтиленгликоли из-за наличия
эфирных групп больше подвержены окислению кислородом
воздуха [33].
Наиболее подробно изучено аутоокисление диэтиленгликоля.
Процесс окисления имеет цепной характер, он ускоряется в присут-
ствии веществ, способных генерировать радикалы, и тормозится при
добавке антиокислителей. При окислении диэтиленгликоля наблю-
дается индукционный период, продолжительность которого (от 1
до 200 ч) зависит от чистоты исходного гликоля. Индукционный
период сокращается до нуля при введении в диэтиленгликоль не-
большого количества веществ, являющихся источниками радикалов,
например 0,02—0,5% 2,2'-азо-бис-изобутиронитрила, гидропереки-
сей (треш-бутилового спирта, кумола) и перекисей (ацетила, бен-
зоила, метилэтилкетона). Источником свободных радикалов может
быть некоторое количество аутоокисленного диэтиленгликоля. Так,
индукционный период для свежего диэтиленгликоля, равный 170 ч,
снизился до 0,5 ч при добавке к нему аутоокисленного гликоля.
Начальными продуктами аутоокисления являются гидропере-
кисные соединения, наиболее вероятно — моногидроперекись ди-
этиленгликоля, конечными и основными продуктами окисления —
муравьиная кислота (преимущественно в форме сложного эфира)
и формальдегид. Кроме того, образуются вода, этиленгликоль,
гликолевый альдегид, глиоксаль и диоксолан. Скорость аутоокисле-
ния диэтиленгликоля зависит от таких факторов, как температура,
парциальное давление кислорода, pH среды, начальное содержание
перекисных соединений.
С повышением температуры резко нарастает эквивалентная кис-
лотность продукта, т. е. сумма свободной и связанной в виде слож-
ного эфира кислоты. За 100 ч при 35 °C эквивалентная кислотность
практически не изменилась, за то же время при 95 °C она повысилась
более чем в 20 раз. В обоих случаях парциальное давление кислорода
29
и другие условия были постоянными. Скорость накопления гидро-
перекиси также растет с повышением температуры, однако при
температуре выше 75 °C концентрация гидроперекиси снижается,
так как скорость ее разложения превышает скорость образования.
Константа скорости распада гидроперекиси составляет 1,1 • 10-е с-1
при 26 °C, 1,6 • 10-5 с1 при 55 °C и 9,0-10-5 с-1 при 75 °C. Скорость
окисления диэтиленгликоля возрастает с повышением парциального
давления кислорода и интенсивности перемешивания.
Весьма своеобразно влияние pH среды. В нейтральной и щелоч-
ной средах, а также в кислой при pH *=*1,5 скорость аутоокисления
мала. Она резко возрастает (в 15—20 раз) при pH от 2 до 4,5. Эффек-
тивным ингибитором аутоокисления диэтиленгликоля является ги-
дрохинон, примерно 0,1%. Разбавление диэтиленгликоля водой,
концентрация ДЭГ ниже 90% (мол.), несколько снижает скорость
аутоокислепия.
Токсические свойства
Гликоли являются веществами с относительно низкой токсичностью.
Вследствие весьма малой летучести гликолей при комнатной темпе-
ратуре опасности острого отравления при вдыхании их паров нет.
хотя хроническое отравление, по-видимому, возможно [34, с. 302J.
Токсические свойства гликолей приведены в табл. 9 [7, р. 641].
Некоторые гликоли вызывают отравление при попадании в орга-
низм через рот, т. е. они обладают оральной токсичностью. Этилен-
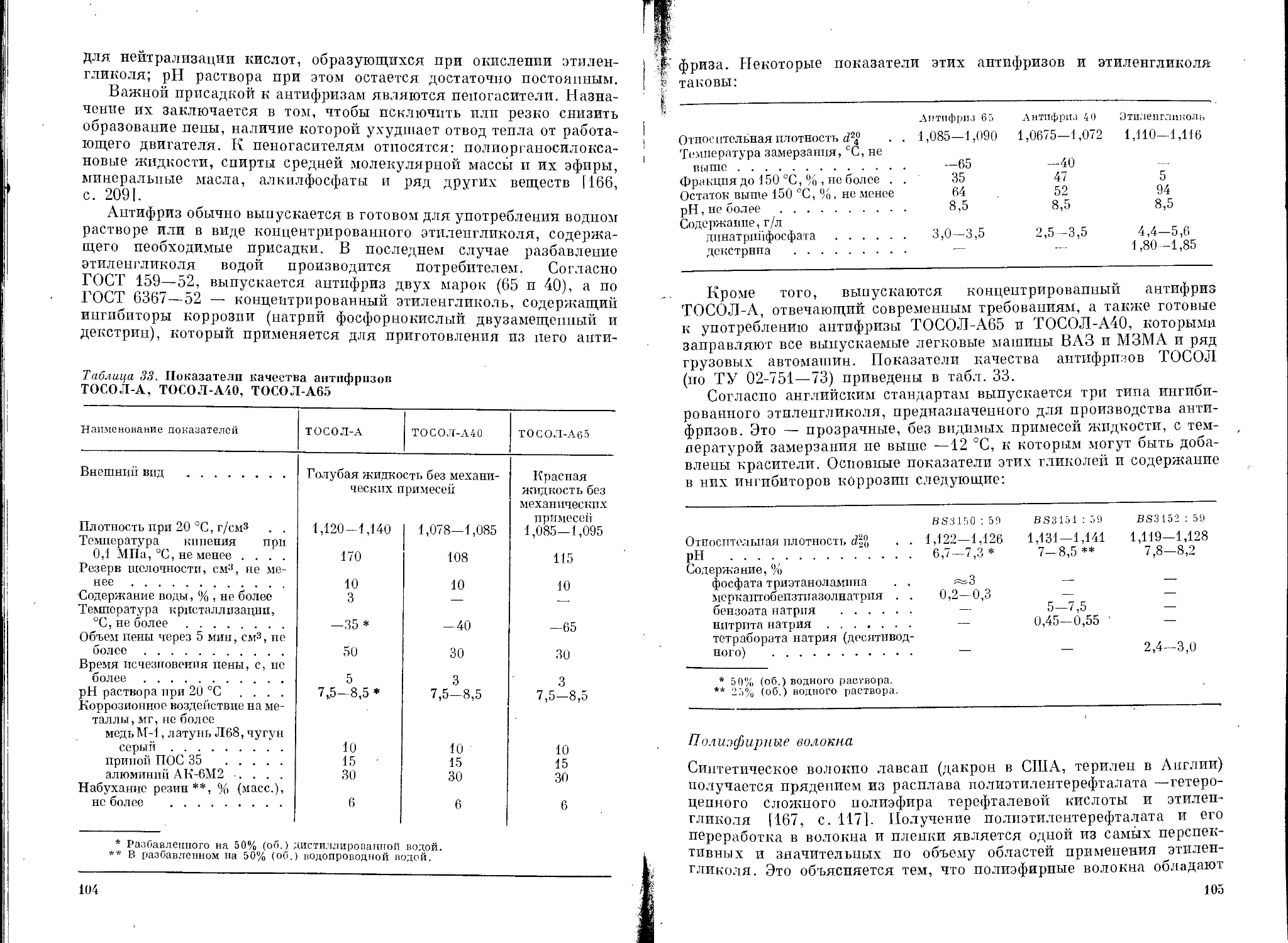
Таблица 9. Токсические свойства гликолей
Гликоль лД*о при введении через рот (крысы), мл/кг Допустимая доза ** при введении через рот (крысы), г/кг ЛДво *** при кожной аппликации (кролики), мл/кг
Этиленгликоль 7,4 (5,5) 0,18 (30 дней) >20
Диэтиленгликоль 28,3 (14,8) 0,18 (30 дней) 11,9
Триэтиленгликоль 28,2 (21,0) 0,83 (30 дней) >20
Тетраэтиленгликоль 28,9 (34,0) 1,88 (2 года) >20
Пропиленгликоль 34,6 (32,5) >20
Дипропиленгликоль 14,8 (14,8) — >20
* лд6„ количество гликоля в мл на 1 кг живой массы, принятое через рот кото-
рое приводит к гибели 50% животных в пределах до 14 дней; в скобках приведены дан-
ные [5, р. 49] в г/кг.
** Допустимое содержание гликоля в пище при указанной в скобках продолжитель-
ности испытаний.
*** Кожно-резорбтивная доза гликоля при контакте с жидкостью в течение 24 ч
30
гликоль при попадании в организм через рот проявляет себя как
сосудистый и протоплазматический яд, действуя главным образом
на центральную нервную систему и почки. У обезьян при концен-
трации этиленгликоля в воздухе 0,5 мг/л отмечено раздражение
верхних дыхательных путей и проявление наркотического действия,
которое быстро проходило. Раздражающее действие жидкого этилен-
гликоля на слизистые оболочки ничтожно.
У людей длительное воздействие больших концентраций паров
этиленгликоля вызывает легко обратимые явления: раздражение
глаз, верхних дыхательных путей, повышенную сонливость, кратко-
временный наркоз. Прием этиленгликоля внутрь является весьма
опасным. При употреблении антифриза (содержащего около 60%
этиленгликоля) отмечено много случаев тяжелых (и даже смертель-
ных) отравлений. При тяжелом отравлении поражается преимуще-
ственно нервная система (смерть через 12—48 ч у 35—40% отравлен-
ных) либо почки (смерть через 5—23 дня у 35—40% отравленных).
Признаки отравления (сильная головная боль, головокружение,
дрожь в конечностях, двоение в глазах, тошнота, повышенная сон-
ливость и др.) появляются через 2—3 ч после приема этиленгли-
коля [34, с. 302]. Считается, что смертельная доза этиленгликоля
при приеме его внутрь составляет около 1,4 г/кг, или 100 г [35,
р. 1497].
Предполагается, что в организме этиленгликоль подвергается
окислению по схеме [34, с. 302]:
Этиленгликоль — > Гликолевый альдегид-->
--->- Гликолевая —
кислота ______ Глиоксалевая ____ Щавелевая
кислота кислота '
---> Глиоксаль —
Токсическое действие диэтиленгликоля слабее, чем этиленгли-
коля [34, с. 307]. Однако при приеме внутрь он тоже представляет
большую опасность. Смертельная доза для человека составляет 15—
100 мл [36, р. 136].
Три- и тетраэтиленгликоль являются менее токсичными, чем
диэтиленгликоль, что объясняется весьма низкой летучестью этих
веществ. Они даже рекомендуются для очистки кондиционированного
воздуха и его стерилизации [4, р. 303; 34, с. 103]. Эти гликоли обла-
дают также низкой хронической оральной токсичностью. Малая
токсичность триэтиленгликоля показана при испытаниях его на
животных. На основании проведенных исследований три- и тетра-
этиленгликоль В'США признаны безопасными для применения в веще-
ствах, которые употребляются в ограниченных количествах, напри-
мер в косметических и фармацевтических препаратах. Однако эти
данные неудовлетворительно согласуются с результатами исследо-
вания токсикологического действия смеси ди- и триэтиленгликоля,
— приведенными в работе [39].
1
31
Пропиленгликоль, в отличие от этиленгликоля, практически
не токсичен, не опасен при вдыхании паров и случайном приеме
внутрь [34, с. 306]. Токсичность пропиленгликоля и глицерина
одного и того же порядка [4, р. 317]. В США с 1942 г. пропилен-
гликоль признан безопасным для применения в пищевых продуктах,
фармацевтических и косметических препаратах. Что касается ди-
и трипропиленгликоля, токсичность их мало изучена. Вдыхание
их паров из-за малой летучести продуктов, по-видимому, опасности
не представляет. Ди- и трипропиленгликоли не оказывают раздра-
жающего действия на кожу и глаза кроликов даже при длительном
и повторном контакте [34, с. 103].
Максимальные концентрации гликолей в промышленной сточной
воде, поступающей на биохимическую очистку, приводятся
в табл. 10, а предельно допустимые концентрации (ПДК) гликолей
в воде водоемов — в табл. 11 [37, с. 216—242 и 302—304].
Экспериментальными данными определены следующие предельно .
допустимые концентрации гликолей в воздухе рабочей зоны: для
этиленгликоля — 0,1 мг/м3, для диэтиленгликоля — 0,2 мг/м3 [37,
с. 48, 81]. Однако длительное изучение состояния здоровья лиц,
работающих с этиленгликолем, показало возможным увеличить
предельно допустимую концентрацию этиленгликоля в рабочей зоне
до 5 мг/м3 [38].
Таблица 10. Максимальные концентрации гликолей в сточных водах
Гликоль Потребность в кислороде, мг О г на 1 мт гликоля Максимальная концентрация гликоля, мг/л Возможность биологического разрушения
ХПК к Й с й и % ‘них : “нпя
To'%k
Этиленгликоль 1,50 0,54 1,26 84,0 1 1000 Поддается рас-
Диэтиленгликоль 1,27 0,06 0,176 13,8 200 200 । паду Практически не разрушаются
Трпэтпленгликоль Тетраэтиленгли- 1,6 0,5 — 31,2 10
КОЛЬ 1,65 0,5 — 30,3
Пропиленгликоль — — — 1000 Поддается рас- паду
БПК5 — биохимическая потребность в О2 за 5 сут. Б11КП — полная потребность в О2
(примерно за 20 сут); ХПК — химическая потребность в О2, определенная бихроматным
методом;
MKg — максимальная концентрация вещества, которая не нарушает биохимических
процессов за любое время; МКб 0 с —максимальная концентрация вещества, не влияю-
щая па работу биологических очистных сооружений.
32
Таблица 11. ПДК гликолей в воде водоемов
Гликоль
Этиленгликоль ......................
Диэтиленгликоль.....................
Триэтиленгликоль ...................
Пропиленгликоль ....................
Концентрация, мг/л
14 о в 'U'd'%inu й" й пдкв 1
450 1 1 1
2050 1 1 1
1000 10 200 По ВПК
1000 10 200 По ВПК
ППКорл — подпороговая концентрация (1 балл) вещества в водоеме, определяемая
органолептически; ППКС р в — то же, но определяемая по влиянию на санитарный
режим водоема; ППКТ — то же, но определяемая по токсикологическим характеристи-
кам; ПДКВ — предельно допустимая концентрация вещества в воде водоема.
Для три- и тетраэтиленгликолей, а также для пропиленгликолей
ПДК не определены, так как считается, что установление норм для
них не обязательно в связи с их малой летучестью и низкой токсич-
ностью [35].
Техника безопасности при работе с гликолями
При работе с этиленгликолем, ди-,' три- и тетраэтиленгликолями,
а также ди- и трипропиленгликолями должны соблюдаться опре-
деленные правила, обеспечивающие безопасность обращения с про-
дуктами. К этим правилам в первую очередь относятся: герметиза-
ция аппаратуры, емкостей для хранения, тары, недопущение про-
ливов (особенно горячих продуктов), защита органов дыхания инди-
видуальными средствами при попадании значительных количеств
паров и аэрозолей в атмосферу, защита рук и других участков кожи
при работе с гликолями (особенно с горячими). Пролитые гликоли
Должны смываться обильным количеством воды [39].
При отравлении этиленгликолем или диэтиленгликолем рекомен-
дуются следующие меры первой помощи: вызвать рвоту, промыть
желудок водой или насыщенным раствором соды.
Гликоли являются пожаро- и взрывоопасными веществами. Пока-
затели их пожарной опасности приведены в табл. 12 [40].
Пожароопасность гликолей снижается с разбавлением водой.
Растворы этиленгликоля, содержащие более 15% воды, не вспыхи-
вают, а антифриз, в котором имеется «*40% воды, не загорается
в пламени горящих дров или бензина [4, р. 38].
Этиленгликоль и другие гликоли, являющиеся горючими гидро-
фильными жидкостями, рекомендуется тушить тонкораспылённой
водой, химической или воздушно-механической пеной, газовыми
огнегасительными составами. В качестве первичных средств тушения
3 Заказ 972 33
Таблица 12. Показатели пожарной опасности гликолей
Гликоль Температура*, °C Область воспламе- нения, % (об.) Температур- ные пределы воспламене- ния, °C
вспышки воспламе- нения самовос- пламене- ния нижний верхний
Этиленгликоль Диэтиленгликоль Триэтиленгликоль Тетраэтиленгли- коль Пропиленгликоль Дипропиленгли- коль ' . Трипропиленгли- коль 120 (115,6) 135 (137,8) 154 (160) 174 (185) 107 (101,7) 118 (126,7) (140,6) (118,3) (143,3) 170 (165,6) (190,6) (104,4) (126,7) (154,4) 380 345 371 348 ** 421 3,8-6,4 0,62—6,8 0,9—9,2 2,6-12,6 112 118 124 170
* В скобках приведены данные по [5].
** По данным [41].
рекомендуются огнетушители ОЖ-7 с зарядом 4—6% водного рас-
твора пенообразователя ПО-11, порошковые и газовые огнетуши-
тели [40].
Хранение и транспортирование гликолей
Специфические физические свойства гликолей (гигроскопичность,
относительно высокая вязкость) и способность их к аутоокислению
требуют соблюдения ряда условий при хранении и транспортирова-
нии. Длительно хранить гликоли следует при возможно более низкой
температуре во избежание окисления. Этиленгликоль, ди- и три-
этиленгликоль рекомендуется хранить при температуре не ниже
—4 °C, а три- и тетраэтиленгликоль, пропиленгликоль и дипро-
пиленгликоль — не ниже 2 °C [5, р. 52]. Сроки хранения, согласно
действующим стандартам и техническим условиям, для гликолей,
не имеющих добавок антиокислителей, установлены в зависимости
от сорта или марки следующие [41]:
Этиленгликоль
Дпэтиленгликоль .
Триэтиленгликоль
Тетраэтиленгликоль
От 5 месяцев до 1 года
От 3 до 6 месяцев
От 2 до 6 месяцев
3 месяца
Емкости для хранения гликолей должны иметь теплоизоляцию
и устройство для их обогрева в связи с большой вязкостью гликолей
при низких температурах и возникающих из-за этого затруднений
34
при их перекачивании. Обогревать можно с помощью змеевиков
(наружных) или рубашки. Возможна также циркуляционная система
непосредственного обогрева подогретым гликолем.
Для хранения гликолей применяются вертикальные или гори-
зонтальные сосуды любой вместимости. Для длительного хранения
рекомендуются емкости из нержавеющей стали или алюминия.
Возможно хранение и в стальных емкостях, покрытых изнутри 5—6
слоями лака на основе фенольных или винильных смол [3, р. 19;
5, р. 51]. Гликоли, содержащие ингибиторы коррозии или не содер-
жащие их, но предназначенные для процессов, где незначительные
примеси железа и некоторое изменение цвета не имеет существенного
значения, можно хранить в сосудах из углеродистой стали.
Емкости должны иметь дыхательный клапан, чтобы исключить
образование вакуума при опорожнении емкости и повышение давле-
ния при ее заполнении. В качестве «подушки» применяется сухой
инертный газ, а при кратковременном хранении можно использовать
воздух, осушенный чешуйчатым хлористым кальцием или другим
эффективным осушителем.
Трубопроводы, арматуру и насосы для перекачивания гликолей
выполняют из легированной стали или алюминия и его сплавов;
для ингибированных продуктов или в случае, когда окрашивание
гликолей не имеет существенного значения, они могут быть изгото-
влены из обычной стали.
Открытые склады гликолей должны соответствовать противо-
пожарным нормам устройства складов горючих веществ (обваловка
вокруг склада с удалением растительности внутри ее) и обеспечены
всеми необходимыми средствами пожаротушения.
Транспортирование этиленгликоля производится в бочках и ци-
стернах, изготовленных из алюминия или легированной стали,
а также в стеклянных бутылях, окрашенных снаружи в черный цвет.
Этилецгликоль 1-го и 2-го сортов по согласованию с потребителем
можно перевозить в стальных бочках и цистернах. Этиленгликоль
концентрированный (95%-ный), в который введена противокорро-
зионная присадка^ перевозят в стальных.бочках.
ТХиэтиленгликоль транспортируется в алюминиевых бочках или
цистернах; допускается перевозка дизтиленгликоля марки ДГ
в стальных цистернах или оцинкованных бочках. Три- и тетраэти-
ленгликоль транспортируются в бочках или цистернах из алюминия
или легированной стали, либо в окрашенных в черный цвет стеклян-
ных бутылях. Триэтиленгликоль может перевозиться в стальных
цистернах и бочках или оцинкованных бочках. Любой вид тары,
применяемой для перевозки гликолей, должен быть герметич-
ным [41].
ЛИТЕРАТУРА
1. Чичибабии А. Е. Основные начала органической химии. Изд. 7-е. Т. 1.
Под ред. П. Г. Сергеева и А. Л. Либермана. М., Госхимиздат, 1963. 912 с.
2. Краткая химическая энциклопедия. Т. 1. Под ред. И. Л. Кнунянца. М.,
«Советская энциклопедия», 1961. 1262 с.
3* 35,
3. Ethylene and Propylene Glycols. Imperial Chemical Industries (ICI). Bir-
mingham, Heavy Organic Chemicals Division, 1970. 33 p;
4. Glycols. Ed. by G. 0. Curme, F. Johnston. New York, Reinhold Puhi. Corp.,
1953. 389 p.
5. Glycols. Properties and Uses. Midland, (Michigan), The Dow Chem. Co.
1961. 64 p.
6. Справочник химика. Изд. 2-е. М.—Л., «Химия», 1963—1966.
7. Kirk — Othmer. Encyclopedia of Chem. Techn. Ed. 2nd. v. 10, New York —
London — Sydney, Interscience Publ., 1966, 938 p.
8. Iordan T. E. Vapour Pressure of Organic Compounds. New York — London
Interscience Publ. Inc., 1954. 266 p.
9. Mellon I. Polyhydric Alcohols. Washington. Spartan Books, 1962. 208 p.
10. Катц Д. Л. и др. Руководство по добыче, транспорту и переработке при-
родного газа. Пер. с англ. Под ред. IO. П. Коротаева и Г. В. Пономарева.
М., «Недра», 1965. 676 с.
11. Monick I. A., Alcohols. New York, Reinhold Book Corp., 1968. 594 p.
12. Propylene and its Industrial Derivatives. Ed. by E. G. Hancock. London,
Ernest Benn Lim., 1973. 517 p.
13. Терентьев А. П., Кост A. И. В кн.: Реакция и методы исследования орга-
нических соединений. М.— Л., Госхимиздат, 1952, с. 4—208.
14. Чернышева Д. А., Полянский Н. Г., Ж. орг. хим., 1971, т. 7, вып. 1, с. 212;
Чернышева Д. А., Полянский И. Г., Труды Московского ин-та хим. машино-
строения, 1972, вып. 46, с. 137—140.
15. Котляревский И. А., Домнина Е. С., Изв. СО АН СССР, 1959, № 8, с. 43—49;
Домнина Е. С. В кн.: Материалы конференции молодых научных сотрудни-
ков. Вып. 3. Благовещенск, 1960, с. 19—22.
16. Долгов Б. Н., Ивановский В. И., Вест. ЛГУ, 1959, № 16, с. 128 —133.
17. Пат. ФРГ 898894 (1953).
18. Matsuura Н., Miyazawa Т., Bull. Chem. Soc. Japan, 1967, v. 40, № 1,
p. 85—94.
19. Krueger P. I., Mettee H. D., J. Mol. Spectroscopy, 1965, v. 18, № 2,
p. 131 — 140.
20. Успехи органической химия. T. 5. Пер. с англ. Под ред. И. Л. Кнунянца.
М.. «Мир», 1968. 316 с.
21. Morantz D. I., Waite М. S., Spectrochim. Acta, 1971, v. 27, part A, № 7,
p. 1131—1137.
22. Pachler K. G., Wessels P. L., J. Mol. Struct., 1970, v. 6, № 6, p. 471—478;
Krishnan K., Krishnan R. S., Proc. Indian Acad. Sci., 1966, v. 64, part A,
№ 2, p. 111—123.
23. Thomas L. H., Meatyard R., J. Chem. Soc., 1966, part A, № 1, p. 92—96.
24. Белоусов В. П.. Морачевский А. Г. Теплоты смешения жидкостей. Л., «Хи-
мия», 1970. 256 с.
25. М arenas М., Deheret G., С. г. Acad. Sci., 1970, v. 270, № 26, р. 2097—2100.
26. Nakagawa Masazumi e. a., Bull. Chem. Soc. Japan, 1960, v. 33, № 4,
p. 433—436.
27. Малышев В. И., Мурзин В. И. В кн.: Исследование по экспериментальной
и теоретической физике. М., изд. АН СССР, 1959, с. 134—148; Yoneza-
wa Т. е. a., Bull. Chem. Soc. Japan, 1965, v. 38, № 9, p. 1431—1435: Sai-
to H. e. a., Ibid, 1966, v. 39, № 5, p. 989—994.
28. Achmad M. e. a., J. Chem. Soc., Ser. B, 1969, v. 9, p. 945—946; Карта-
шева Л. И., Сосновский О. А., Пикаев А. К., «Химия высоких энергий»,
1971, № 4, с. 362—363; Карташева Л. И., Пикаев А. К., там же, 1973,
т. 7, № 1, с. 36—40; № 5, с. 434—438; Ветров В. С. и др., там же. № 4,
с. '380—381.
29. Hiroto Kozo е. а., «Нихон кагаку дзасси» (J. Chem. Soc. Japan, Pure Chem.
Sec.), 1960, v. 81, № 1, p. 29—33; РЖхим, 1960, 95785; Seidler F., Son-
ntag C., 1.. Naturforsch., 1969, Bd. 24, № 6, S. 780—781; Sonntag C., Tho-
mas E., Ibid, 1970, Bd. 25. № 12, S. 1405—1407.
30. Venter P. I., Linde H. I., Basson R. A., J. Cherii. Soc., Chem. Communs,
1972, № 3, p. 187-188.
36
31. Петряев Е. П. и др. Вести. Белорусского ун-та. Сер. 2, 1971, № 2, с. 6—8.
32. Pfordte К., Leuschner G., Arm., 1961, Bd. 643, № 1—3, S. 2—6; Linde H. J.,
Sonntag C., Photochem. a. Photobiol., 1971, v. 13, № 2, p. 147—155.
33. Lloyd W. G., Tailor F. G., Ind. Eng. Chem., 1954, v. 46, № 11, p. 2407—2416;
Lloyd W. G., J. Am. Chem. Soc., 1956, v. 78, № 1, p. 72—75; Lloyd W. G.,
J. Chem. Eng. Data, 1961, v. 6, № 4, p. 541—547.
34. Вредные вещества в промышленности. Под ред, Н. В. Лазарева. Изд. 6-е.
Л., «Химия», 1971, Т. 1, 832 с.; доп. том. Изд. 5-е. 1969, 536 с.
35. Industrial Hygiene and Toxicology. V. 2. Ed. by A. F. Patty. Ed. 2nd.
New York, Interscienee Publ., 1962. 2377 p.
36. Dreisbach R. H. Handbook of Poisoning. Ed. 6th. Los Altos (Calif.), Lange
Medical Publications, 1969. 477 p.
37. Предельно допустимые концентрации вредных веществ в воздухе и воде.
Изд. 2-е. Л., «Химия», 1975. 456 с.
38. Дубейковская Л. С. и др., Гигиена труда и проф. заболеваний, 1973, № 10,
с. 1—4.
39. Сонина Ю. П., Кочеткова П. А. В кн.: Токсикология новых промышлен-
ных химических веществ. Вып. 9. Л., «Медицина», 1967, с. 146—152.
40. Дарковский А. К., Зубов А. И. Противопожарная техника на предприятиях
химической промышленности. М., Госхимиздат, 1961. 244 с; Пожарная
опасность веществ и материалов, применяемых в химической промышлен-
ности. Под ред. И. В. Рябова. М., «Химия», 1970. 336 с.
41. ГОСТ 19710—74, этиленгликоль; ГОСТ 10136-62, ГОСТ 5.2266—75,
дпэтнленгликоль; ТУ 6-01-874—73, триэтиленгликоль; ТУ 6-02-371—73,
тетраэтиленгликоль.
Глава II
Этиленгликоль
Физические свойства
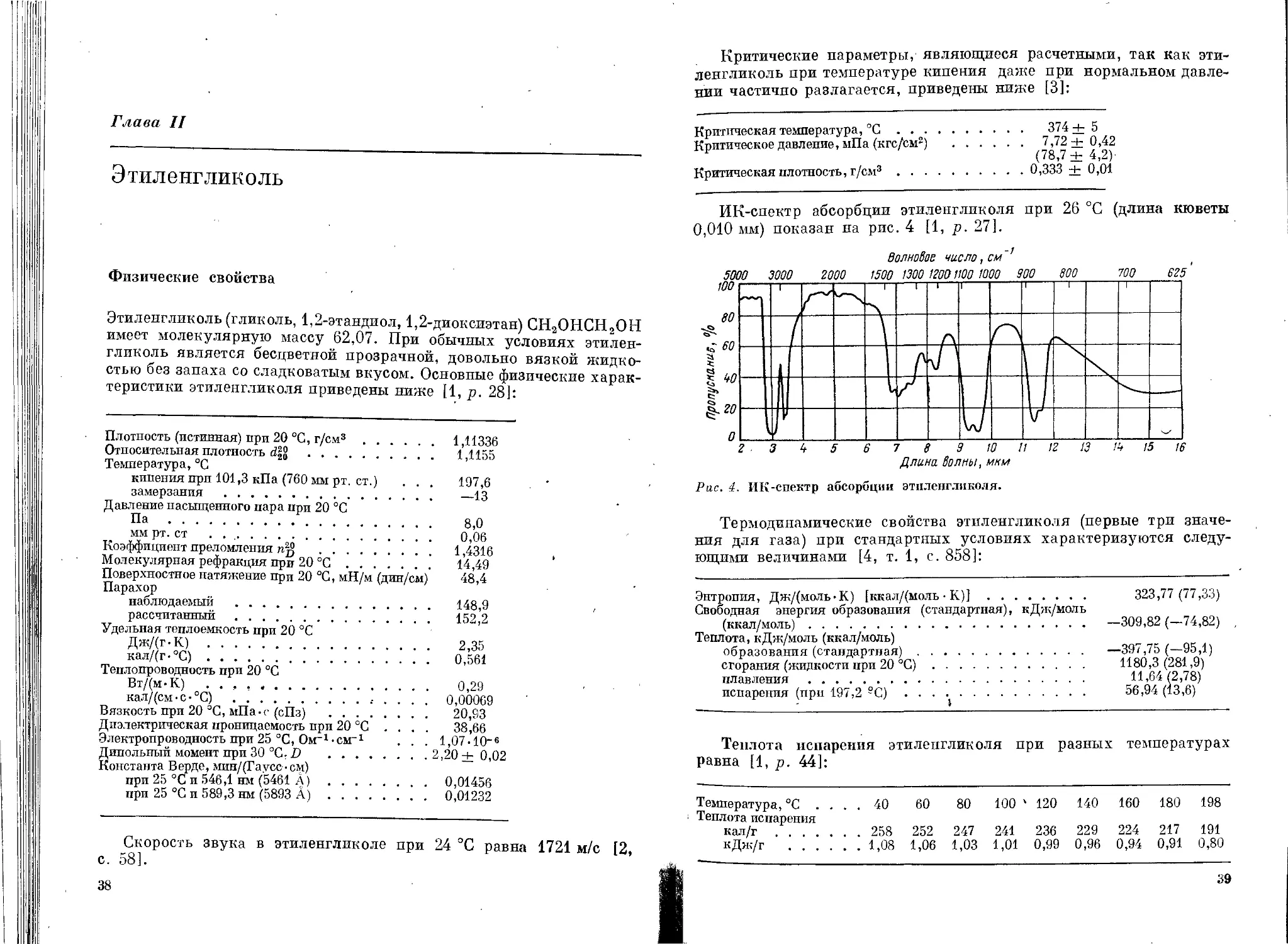
Этиленгликоль (гликоль, 1,2-этандиол, 1,2-диоксиэтан) СН2ОНСН2ОН
имеет молекулярную массу 62,07. При обычных условиях этилен-
гликоль является бесцветной прозрачной, довольно вязкой жидко-
стью без запаха со сладковатым вкусом. Основные физические харак-
теристики этиленгликоля приведены ниже [1, р. 28]:
Плотность (истинная) при 20 °C, г/см3......... 1,11336
Относительная плотность ...................... 1,1155
Температура,°C
кипения при 101,3 кПа (760 мм рт. ст.) . . . 197,6
замерзания ............................... —13
Давление насыщенного пара при 20 °C
Па ........................................... 8,0
мм рт. ст . . ............................... 0,06
Коэффициент преломления ..... 1,4316
Молекулярная рефракция при 20 °C...... 14,49
Поверхностное натяжение при 20 °C, мН/м (дин/см) 48,4
Парахор
наблюдаемый ............................... 148,9
рассчитанный ................................ 152,2
Удельная теплоемкость при 20 °C
Дж/(г-К) ................................... 2,35
кал/(г-°С)................................... 0,561
Теплопроводность при 20 °C
Вт/(м-К) ................................... 0,29
кал/(см • с • °C) ....................... 0,00069
Вязкость при 20 °C, мПа-с (сПз) ................. 20,93
Диэлектрическая проницаемость при 20 °C .... 38,66
Электропроводность при 25 °C, Ом"1-см*1 . . . 1,07-10*6
Дипольный момент при 30 °C, D ................2,20 + 0,02
Константа Верде, мин/(Гаусс• см)
при 25 °C и 546,1 нм (5461 А) ............ 0,01456
при 25 °C и 589,3 нм (5893 А)............. 0,01232
Скорость звука в этиленгликоле при 24 °C равна 1721 м/с [2,
с. 58].
38
Критические параметры, являющиеся расчетными, так как эти-
ленгликоль при температуре кипения даже при нормальном давле-
нии частично разлагается, приведены ниже [3]:
Критическая температура, °C................... 374+5
Критическое давление, мПа (кгс/см2) .......... 7,72+0,42
(78,7+4,2)'
Критическая плотность, г/см3 ................ 0,333 + 0,01
ИК-спектр абсорбции этиленгликоля при 26 °C (длина кюветы
0,010 мм) показан на рис. 4 [1, р. 27].
Рис. 4. ИК-спектр абсорбции этиленгликоля.
Термодинамические свойства этиленгликоля (первые три значе-
ния для газа) при стандартных условиях характеризуются следу-
ющими величинами [4, т. 1, с. 858]:
Энтропия, Дж/(моль-К) [ккал/(моль • К)]................
Свободная энергия образования (стандартная), кДж/моль
(ккал/моль)............................................
Теплота, кДж/моль (ккал/моль)
образования (стандартная)..............................
сгорания (жидкости при 20 °C).....................
плавления ........................................
испарения (при 197,2 9С)..........................
323,77 (77,33)
—309,82 (-74,82)
—397,75 (-95,1)
1180,3 (281,9)
11,64 (2,78)
56,94 (13,6)
Теплота испарения этиленгликоля при разных температурах
равна [1, р. 44]:
Температура, °C .... 40 60 80 100 ' 120 140 160 180 198
Теплота испарения
кал/г ................. 258 252 247 241 236 229 224 217 191
кДж/г ............. 1,08 1,06 1,03 1,01 0,99 0,96 0,94 0,91 0,80
39
В других источниках сообщаются следующие значения теплот
при стандартных условиях (298,15 К), а именно:
Теплота образования,
кДж/моль (ккал/моль)
жидкости ...............—455,25 (—108.73) [5]
—455,96 + 0,84 (—108,9 + 0,2) [6]
газа ...—389,60 (—93,05) [5]
-388,13+ 1,68 (-92,7 + 0,4) [6]
Теплота сгорания жидко-
сти, кДж/моль
(ккал/моль) .......... 1189,69+ 0,67 (284,14+ 0,16) [6]
Плотность этиленгликоля и его водных растворов в широком
диапазоне температур (от —45 до 200 °C) приведена в Приложении
Рис. 5. Уменьшение объ-
ема при смешении эти-
ленгликоля с водой при
20 °C.
в табл. 1, см. стр. 351. Температурный коэффициент относительной
плотности этиленгликоля Д<5/Д£ в интервале температур 0—40 'С
составляет 0,0007 на 1 °C.
Плотность водных растворов этиленгликоля не является строго
аддитивной функцией состава раствора, так как при его смешении
с водой происходит уменьшение объема смеси. Максимальное умень-
шение (сжатие) около 1,3% получается при смешении равных объ-
емов жидкостей. Для различных соотношений воды и гликоля умень-
шение объема показано на рис. 5 [1, р. 36]. По относительной плот-
ности водного раствора этиленгликоля с достаточной точностью
можно определять его концентрацию (см. Приложение, табл. 2,
стр. 352).
Относительная плотность djo растворов этиленгликоля и ди-
этиленгликоля (ДЭГ) приведена в табл. 13 [1, р. 37].
Относительная плотность растворов, в которых концентрация
диэтиленгликоля 85% и выше, является постоянной, равной 1,11836.
40
Таблица 13. Относительная плотность растворов
этиленгликоля и диэтиленгликоля
,20 + о Концентра- ция ДЭГ, % У20 ®2 0 Концентра- ция ДЭГ, % .20 ^20 Концентра- ция ДЭГ, %
1,1155 0,96 1,1165 19,5 1,1175 42,4
1,1156 2,8 1,1166 21,5 1,1176 45,2
1,1157 4,7 1,1167 23,5 1,1177 48,2
1,1158 6,5 ' 1,1168 25,6 1,1178 51,5
1,1159 8,3 1,1169 27,8 1,1179 55,0
1,1160 10,1 1,1170 30,0 1,1180 59,1
1,1161 11,9 1,1171 32,3 1,1181 64,1
1,1162 13,8 1,1172 34,6 1,1182 70,2
1,1163 15,7 1,1173 37,1 1,1183 • 78,3
1,1164 17,6 1,1174 39,7 1,11835 84,5
Плотность растворов этиленгликоля и пропиленгликоля является
линейной функцией их состава и может быть найдена по уравне-
нию И, р. 37]:
р = 1,0400 + 0,00078Х
где р — плотность при 15 °C, г/см3; X — концентрация этилен-
гликоля, %.
Зависимость температуры кипения водных растворов этилен-
гликоля от их концентрации при атмосферном давлении приведена
в табл. 14 И, р. 55].
Таблица 14. Зависимость температуры кипения водных растворов
этиленгликоля от концентрации при атмосферном давлении
Концентра- ция этилен- гликоля, % Температура кипения, °с Концентра- ция этилен- гликоля, % Температура кипения, °C Концентра- ция этилен- гликоля, % Температура кипения, QG
10 101,1 60 110,0 92 142,8
20 .102,2 65 112,3 93 ' 145,6
25 102,8 70 114,4 94 149,4
30 103,3 75 117,7 95 153,9
35 103,9 80 122,2 96 159,4
40 105,0 85 127,8 97 165,6
45 106,1 88 132,8 98 173,9
50 107,2 90 137,2 99 183,9
55 108,3 91 140,0 100 197,8
Температура замерзания. Водные растворы этиленгликоля с кон-
центрацией 30% и выше замерзают при значительно более низкой
температуре, чем чистый этиленгликоль. Это свойство водно-
41
этиленгликолевых растворов позволило получить на их основе низко-
замерзающие жидкости (антифризы) и определило тем самым наиболее
широкую область их применения. Следует отметить, что такими же
свойствами обладают водные растворы и других гликолей, однако
при равной концентрации растворы, содержащие этиленгликоль,
имеют температуру замерзания ниже, чем растворы других гликолей
(см. рис. 3, стр. 20).
Зависимость температуры замерзания (начала кристаллизации) от
концентрации в области от 2 до 58% и от 80 до 98% приведена
в табл. 15 [1, с. 40]. Данные других источников [1—11] отличаются
от значений, приведенных в таблице.
Таблица 15. Зависимость температуры замерзания водных
растворов этиленгликоля от его концентрации
Концентра- ция гликоля, % Температура замерзания, °C Концентра- ция гликоля, % Температура замерзания, °C Концентра- ция гликоля, % Температура замерзания, °C
2 —0,6 28 -13 54 -41
. 4 —1,3 30 -15 56 -44
6 -2,0 32 —17 58 -48
8 —2,7 34 -18 80 —47
10 -3,5 36 -20 82 —43
12 —4,4 38 —22 84. -40
14 -5,3 40 -24 86 —36
16 -6,3 42 -26 88 —33
18 -7,3 44 —28 90 —29
20 -8 46 -31 92 -26
22 —9 48 -33 94 -23
24 —И 50 -36 96 -19
26 -12 52 -38 98 -16
На рис. 6 показана диаграмма температуры замерзания водных
растворов этиленгликоля по данным [7, с. 441]. При замерзании
водных растворов этиленгликоля образуется рыхлая кашицеобраз-
ная масса, объем которой больше первоначального только на 0.25—
0,30%. Точка Б, отвечающая составу 33,3% воды и 66,7% этилен-
гликоля и температуре замерзания —75 °C, близка к составу гидрата
С2Н4(ОН)2 • 2Н2О. При охлаждении раствора с содержанием воды
больше 33,3% (кривая АВ) до температуры замерзания выпадают
кристаллы льда в виде мягкой взвешенной шуги, а этиленгликоль
остается в жидком состоянии. При охлаждении концентрированных
растворов этиленгликоля (содержание воды менее 33,3%), при тем-
пературе замерзания выпадают кристаллы этиленгликоля, а вода
остается в жидком состоянии. В точке В одновременно кристал-
лизуются этиленгликоль и вода.
Температура замерзания водных растворов этиленгликоля опре-
делялась и в ряде других работ. Так, в работе [8] найдено, что водно-
42
гликолевые растворы образуют три эвтектические смеси с темпера-
турой замерзания —51,0, —63,6 и —49,4 °C. По данным [9], наблю-
далась только одна эвтектическая точка с температурой замерзания
минус 64 — минус 70 °C и с содержанием этиленгликоля 66%. Близ-
кие показатели получены в работе [10]: температура замерзания
—65 °C при содержании этиленгликоля 70%. Результаты одной из
последних работ [11], посвященной изучению фазового равновесия
растворов этиленгликоля в воде, приведены на рис. 7. Как и в ра-
боте [8], обнаружены три эвтектические точки, соответствующие
температурам замерзания —49,03, —63,65 и —42,93 °C и содержанию
этиленгликоля соответственно 58,25, 63,46 и 80,25%.
5 кристаллы льоа + кристаллы
р? этиленгликоля
-30 \----i_____। ।_____।
о го w во so wo
Концентрация гликоля, %
Температура,
Рис. 6. Диаграмма температур замерзания водных растворов этиленгликоля.
Рис. 7. Диаграмма фазового равновесия водных растворов этиленгликоля
(— — — линия солидуса).
Сравнение данных различных авторов показывает, что наиболь-
шие расхождения наблюдаются в области высоких концентраций
этиленгликоля, примерно от 58 до 80%. Это объясняется тем, что
при низких температурах жидкость становится очень вязкой, склонна
к переохлаждению, и различить точку перехода от высоковязкого
состояния к твердому затруднительно. Недостаточная чистота исход-
ного гликоля также могла повлиять на результаты определения
температуры замерзания водно-гликолевых растворов.
Температуры замерзания смесей этиленгликоля с диэтиленгли-
колем (ДЭГ) приведены в табл. 16 [9].
Температуры замерзания тройных смесей вода — этиленгли-
коль — диэтиленгликоль и вода — этиленгликоль — триэтилен-
гликоль определены в работе [9]. Найдено, что замена этиленгликоля
Равным количеством ди- или триэтиленгликоля приводит к повыше-
нию температуры замерзания раствора. Для смеси вода — этилен-
43
Таблица 16. Температуры замерзания смесей
этиленгликоля с диэтиленгликолем
Концентра- ция ДЭГ, % Температура замерзания, °C Концентра- ция ДЭГ, % Температура замерзания, °C Концентра- ция ДЭГ, % Температура , замерзания. г Концентра- ция ДЭГ, % 1 Температура замерзания, °C
5 -14,5 30 —27,2 55 —39* 80 -23,3
10 —16,5 35 -30,6 60 —36,4 85 —19,5
15 -18,8 40 —34,1 65 —33,4 90 —15,7
20 —21,3 45 —38,0 70 —30,2 95 —12,0
25 -24,1 । 50 75 —26,8 100 -8,3
* По данным работы [1, р. 42].
гликоль — диэтиленгликоль, содержащей постоянное количество
воды (40%), температура замерзания составляет:
Этиленгликоль, % ............ 60 54 48 36 24 11 —
Диэтиленгликоль, %.............— 6 12 24 36 49 60
Температура замерзания, °C . . —53 —51,1 —48,3 —47,3 —44,0 —42,1 —41,4
Температура стеклования чистого этиленгликоля найдена равной
—118 °C [5], а для 62,5%-ного водного раствора — минус
'131 °C [12].
Зависимость давления насыщенного пара этиленгликоля от тем-
пературы приведена в табл. 17, а зависимость давления насыщенных
паров водных растворов этиленгликоля дана в Приложении, табл. 3,
Стр. 352 [1, р. 54, 56].
Таблица 17. Зависимость давления насыщенного пара этиленгликоля от
температуры
Температу- ра, °C Давление, мм рт. ст.* Температу- ра, °C Давление, мм рт. ст.* Температу- ра, °C Давление, мм рт. ст.* Температу- ра, °C Давление, мм рт. ст.*
50 0,7 90 9,2 130 68 170 312
55 0,9 95 12 135 83 175 373
60 1,3 100 16 140 103 180 435
65 1,9 105 21 145 125 185 520
70 2,6 НО 27 150 153 190 605
75 3,7 115 33 155 183 195 710
80 5,0 120 43 160 219 197,6 760
85 6,7 125 54 165 263 200 820
* 1 мм рт. ст. = 133,32 Па,
Давление насыщенного пара этиленгликоля (р, мм рт. ст.) при
разных температурах (Т, К) можно также вычислить по следующим
уравнениям [4, с. 697]:
, 2694,7
в интервале 25—90 °C lgp = 8,863 у
Г 3193,6
в интервале 90—130 °C lgp = 9,7423 j,
2994,4
•в интервале 130—197 °C lgp = 9,2477 у
В пределах давлений 98,7—101,3 кПа (от 740 до 760 мм рт. ст.)
при изменении давления на 133,32 Па (1 мм рт; ст.) температура
кипения этиленгликоля изменяется на 0,043 °C.
Давление насыщенных паров водных растворов этиленгликоля
(р) можно рассчитать по уравнению идеальной смеси [1, с. 541:
j>^XlPl + X2i>2
где Хг, Хг — мольные доли воды и этиленгликоля; рг, р2 — давле-
ние паров воды и этиленгликоля.
Коэффициент преломления смесей этиленгликоля с водой, диэти-
ленгликолем, а также с пропиленгликолем является линейной функ-
цией их состава [1, р. 47]. Так как между коэффициентом преломле-
ния этиленгликоля (пЬ° = 1,4316), воды (zip = 1,3330) и диэтилен-
гликоля (njj — 1,4472) разница весьма велика, то по коэффициенту
преломления бинарных смесей этиленгликоль — вода и этиленгли-
коль — диэтиленгликоль можно легко и быстро определить их со-
став. Если на одной из осей ординат отложить коэффициент преломле-
ния воды, на другой — коэффициент преломления этиленгликоля,
а на оси абсцисс — состав бинарной смеси вода — этиленгликоль
(в %), то перпендикуляр, опущенный с прямой, соединяющей коэф-
фициенты преломления обоих веществ, на ось абсцисс, покажет
состав смеси, который соответствует найденному коэффициенту
преломления. Коэффициенты преломления смесей этиленгликоля
и этилендиамина при 25 °C приведены в работе [13].
Температурный коэффициент показателя преломления этилен-
гликоля Дтгс/Д7 в интервале температур 20—40 °C составляет 0,00026
на 1 °C.
Поверхностное натяжение этиленгликоля (о, мН/м) при разных
температурах t может быть вычислено из уравнения [1, р. 50]
а = 50,21 — 0.089/
которое составлено по экспериментальным данным, полученным
в интервале 30—160 °C. Зависимость поверхностного натяжения
этиленгликоля от температуры показана на рис. 8, а, а зависимость
поверхностного натяжения водных растворов этиленгликоля при
45 °C от концентрации приведена на рис. 8, б [3]. Поверхностное
45
44
натяжение на границе раздела этиленгликоля с гексаном, гептаном,
тридеканом, циклогексаном, циклогексеном и бензолом приведено
в [14, р. 12].
Теплоемкость этиленгликоля в зависимости от температуры при-
ведена в табл. 18 [1, р. 49].
Теплоемкость этиленгликоля [С, кал/(г-°С)| может быть вы-
числена из уравнения
С = 0,538 + 0,00113/
где t — температура, °C.
Рис. 8. Зависимость поверхностного натяжения этиленгликолей от температуры
(а) и водных растворов этиленгликолей при 25 °C от концентрации (б):
1 «этиленгликоль; г — диэтиленгликоль; 3— триэтиленгликоль.
Таблица 18. Зависимость удельной теплоемкости этиленгликоля
и его водных растворов от температуры
» Температура, °C Теплоемкость при различной концентрации этиленгликоля, кал/(г-°С)*
20% 40% «0% 80% 100%
20 . 0,930 0,840 0,740 0,649 0,561
30 0,9-U 0,850 0,755 0,662 0,573
40 0,939 0,860 0,769 0,675 0,584
50 0,943 0,869 0,782 0,687 0,595
60 0,947 0,877 0,794 0,700 0,606
70 0,951 0,885 0,804 0,712 0,618
80 0,955 0,892 0,814 0,724 0,629
90 0,959 0,900 0,823 0,735 0,640
100 0,964 0,906 0,832 0,746 0,651
НО 0,969 0,913 0,841 0,756 0,673
120 0,975 0,921 0,850 0,767 0,673
130 0,981 0,929 0,860 0,778 0,685
140 0,988 0,936 0,870 0,788 0,696
150 0,995 0,944 0,879 0,799 0,707
160 1,003 0,953 0,889 0,810 0,718
, 170 1,012 0,963 0,899 0,821 0,730
180 1,020 0,971 0,909 0,831 0,740
* 1 кал/(г-°C)4,19 Дж/(г-К).
46
Номограммы для определения теплоемкости и теплопроводности
водных растворов этиленгликоля при различных температурах
приведены в Приложении, стр. 366 [15].
Теплоемкость паров этиленгликоля в температурном интервале
0—500 °C показана на рис. 9 [3]. Мольная теплоемкость паров
этиленгликоля (состояние идеального газа) в интервале от 298 до
1000 К составляет [5, с. 493]:
Температура, К 298 300 400 500 600 700 800 900 1000
Ср, кал/(моль х
X К) * . . 23,20 23,28 27,06 30,10 32,72 35,00 36,90 38,00 39,88
Рис. 9. Теплоемкость паров этиленгликолей:
1 — этиленгликоль; г — диэтиленгликоль; 3 — триэтиленгликоль.
Теплоемкость водного раствора этиленгликоля с концентрацией
62,5%, т. е. близкой к применяемой в антифризах, в интервале
температур от —188 до -]-20 °C приведена ниже [12]:
Температура, С*, кал/(г •°C) Температура,°C с*, кал/(г- °C)
°C
Твердый ' Жидкий
—180 0,185 —35 0,635
—140 0,230 —25 - у 0,650
—100 0,280 -15 0,675
-60 0,350 —5 0,700
5 0,715
П е р е о х л ажденный 15 0,735
20 0,740
—170 0,195
—150 0,222
—120 0,472
—100 0,470
* 1 кал/(г-°С) — 4,19 Дж/(г-К).
47
Теплопроводность водных растворов этиленгликоля при разных
температурах приведена в табл. 19 [1, р. 52].
В интервале 80—120 °C имеет место небольшой максимум тепло-
проводности [16], как видно из рис. 10, а, а на рис. 10, б показана
теплопроводность паров этиленгликоля в интервале 0—500 °C [3].
Номограмма для определения теплопроводности водных растворов
этиленгликоля в интервале 10—107 °C приведена в Приложении,
стр. 367 [15]. Теплопроводность бинарных смесей этиленгликоль —
моноэтаноламин и этиленгликоль — метилпирролидон исследована
в работе [17].
Теплопроводность, калЦом-с- °С)-10
Рис. 10. Зависимость теплопроводности водных растворов этиленгликоля (а)
и теплопроводности паров этиленгликолей (б) от температуры:
— этиленгликоль; 2 — диэтиленгликоль; з — триэтиленгликоль.
Зависимость вязкости водных растворов этиленгликоля и вяз-
кости паров этиленгликоля от температуры приведена на рис. 11 [3].
Таблица 19. Зависимость теплопроводности этиленгликоля
и его водных растворов от температуры
Температура, °C Теплопроводность при различной концентрации этиленгликоля, кал/(см*с»°C)*
20% 40% 6 0% 80% 100%
10 0,00121 0,00106 0,00093 0,00081 0,00071
25 ' 0,00124 0,00107 0,00093 0,00079 0,00068
40 0,00127 0,00108 0,00092 0,00077 0,00065
55 0,00130 0,00109 0,00091 0,00076 СТ,00063
70 0,00132 0,00109 0,00090 0,00074 0,00060
95 0,00134 0,00109 0,00088 0,00070 0,00056
120 0,00134 0,00107 0,00085 0,00066 0,00051
150 0,00132 0,00105 0,00081 0,00062 0,00046
175 0,00130 0,00102 0,00077 0,00057 0,00041
* 1 кал/(см-с-°C)—4,19 • 102 Вт/(м-К).
48
Рис. 11. Зависимость
вязкости водных раство-
ров этиленгликоля (а)
и паров этиленглико-
лей (б) от температуры:
1 — этиленгликоль; 2 — ди-
этиленгликоль; 3 — триэтп-
ленгликоль.
Рис. 12. Зависимость ки-
нематической вязкости
(1gг], мм-/с) смесей эти-
ленгликоля с другими
гликолями от темпера-
туры:
1 — с дипропиленгликолем;
2 — с пропиленгликолем;
з — с тетраэтиленгликолем;
4 — с триэтиленгликолем;
'> — с лиэтиленгликолем;
в — с триметиленгликолем.
4 Заказ 972
49
Вязкость (кинематическая) смесей этиленгликоля с ди-, три-
и тетраэтиленгликолем, а также с пропилен- и дипропиленгликолем,
взятых в соотношении 1 : 1, показана на рис. 12. Вязкость смесей
этиленгликоля с 95%-ным этиловым спиртом и безводным изопро-
пиловым спиртом, содержащих 40% воды, при температуре —40 °C.
и содержащих 60% воды, при температуре —17,8 °C, показана на
рис. 13 [1, р. 59].
О вязкости и плотности растворов подпетого калия и йодистого
церия в этиленгликоле при 25 °C сообщается в работе [18].
Рис. 13. Зависимость вязкости водных растворов гликоль — спирт, содержа-
щих 40% Н2О (а) и 60% Н_О (б), от концентрации компонентов:
1 — этиленгликоль — этиловый спирт (95%-ный); 2 — этиленгликоль — изопропиловый
спирт (безводный); з—пропиленгликоль — этиловый спирт (95%-ный); 4 — пропилен-
гликоль — изопропиловый спирт (безводный).
Диэлектрическая проницаемость водных растворов этиленгли-
коля в зависимости от температуры при длине волны 150 м при-
ведена в табл. 20 [1, р. 38].
В интервале 20—150 °C температурные зависимости времени
диэлектрической релаксации т и вязкости ц аналогичны, причем
отношениец/т является постоянным и равным 1,21 • 109 кг/(с-м) [19].
Диэлектрическая проницаемость 50%-пого (об.) водного раствора
этиленгликоля (s) меняется с изменением температуры следующим
образом [20]:
г, °C ..................+20 +10 О -10 -20 —зо -30 —30 -но
с .................... 64,5 68,4 72,4 76,5 80,7 85,0 89,3 94,0 98,8
50
Таблица 20. Зависимость диэлектрической проницаемости
водных растворов этиленгликоля от температуры
^концентрация гликоля, % Диэлектрическая проницаемость
при 20 °C при 40 °C при 6 0 СС при 80 °C При 100 °C
20 74,60 67,52 61,20 55,36 50,39
40 68,40 61,56 55,48 49,81 44,78
60 61,08 54,53 48,75 43,68 39,13
80 50,64 45,45 40,72 36,36 32.52
100 38,66 34,94 31,58 28,45 25,61
Для указанной смеси зависимость диэлектрической проница-
емости от температуры выражается уравнением:
Ig 8 ~a—bt
где а = 1860, b = 2,65-Ю’3.
Акустические свойства этиленгликоля. Скорость звука в этилен-
гликоле при 24 °C примерно в 5 раз выше, чем в воздухе, и в 1,2 раза
выше, чем в воде. С ростом температуры, давления и концентрации
этиленгликоля скорость звука повышается, однако, чем выше кон-
центрация этиленгликоля, тем меньше она влияет на повышение
скорости звука. Акустические свойства этиленгликоля при разных
температурах, давлениях, частотах и других параметрах изучены
в [21].
Зависимость скорости звука в водных растворах этиленгликоля
от температуры, концентрации и давления показана на рис. 14 [22].
Диссоциация этиленгликоля. Этиленгликоль является очень сла-
бой кислотой, с ничтожно малой степенью диссоциации. Константа
диссоциации этиленгликоля в водных растворах при 25 °C равна
6,6-10'15 [4, т. 3, с. 87], а константа автопротолиза при 30 °C равна
15,60 [23]. В изопропиловом спирте константа диссоциации этилен-
гликоля равна 19,17, а в жидком аммиаке при —38 °C — 7,3 [24].
Константы автопротолиза водных растворов этиленгликоля при
разных температурах таковы [25]:
-Концентра-
ция гликоля, О/ /0 При '15 °C При 23 °C При 3 5
10 14,16 13,85 13,55
30 14,00 13,72 13,45
50 13,92 13,66 13,43
70 14,05 13,82 13,61
90 14,62 14,37 14,17
4*
51
Для слабых растворов этиленгликоля константа автопрото-
лиза pKs меньше, чем для чистой воды (pKs = 14); минимум pKs
достигает при концентрации этиленгликоля около 50%. Между
изменением pKs и диэлектрической проницаемостью растворов эти-
ленгликоля имеется полная симбатность.
Сжимаемость этиленгликоля в интервале температур от 25 до
105 °C приведена в табл. 21 [1, р. 46].
Рис. 15. Зависимость точки росы газа от температуры контакта с водными
растворами этиленгликоля.
Рис. 14. Зависимость скорости звука в водных растворах этиленгликоля от его
концентрации, давления и температуры. (— — — — 10% этиленгликоля;
-------- 20% этиленгликоля; — • — • — 33% этиленгликоля; цифры на ли-
ниях — давление в МПа).
Сжимаемость этиленгликолевых растворов бромистых солей ли-
тия и натрия и иодистых солей калия, кадмия и натрия приведена
в [14, р. 1200].
Гигроскопичность этиленгликоля. Этиленгликоль весьма гигро-
скопичен: за 1 неделю 100 частей этиленгликоля поглощают из
воздуха 30 чабтей воды, а за 2 недели — 60 частей, в результате чего
образуется раствор, близкий к составу С2Н4(ОН)2 2Н2О [26, с. 325].
Состав водных растворов этиленгликоля, которые находятся в равно-
весии с воздухом при определенной его относительной влажности
и температуре, приведен в табл. 22 [27, р. 13].
Точка росы газа при его осушке водными растворами этиленгли-
коля меняется в зависимости от концентрации гликоля и темпера-
туры контакта (рис. 15) [28, с. 145].
Растворимость. Этиленгликоль полностью смешивается с водой,
и при смешении выделяется теплота, количество которой зависит
52
Таблица 21. Сжимаемость этиленгликоля
Мольный
Давление, МПа объем V, см3 ду/Vo
П ри темп ер ату ре 25 °C
0,1 55,904 0,0000
25 55,411 0,0088
50 54,958 0,0169
75 54,541 0,0244
100 54,155 0,0313
При температуре 65 °C
0,1 57,383 0,0000
25 56,799 0,0102
50 56,268 0,0194
75 55,786 0,0278
100 55,343 0,0356
При температуре 105 °C
0,1 59,045 0,0000
25 58,338 0,0120
50 57,707 0,0227
75 57,141 0,323
100 56,626 0,0410
от соотношения взятых компонентов и их температуры [29, с. 25].
Теплота смешения этиленгликоля с водой при различных темпе-
ратурах равна (в кал/моль * раствора):
Концентра- ция гликоля, % (мол.) При 17 °C При 32 °C При 55 °C При 76 °C
10 —124 —102 -95,0 —83,5
20 —170 —154 -140 —135
30 —177 —161 —148 —138
40 —184 —160 -145 -134
50 -174 _147 —133 —127
60 —152 —127 -115 —112
70 —101 -85,5 —78,4 —78,1
80 —83,4 —70,5 -64,8 —64,8
90 —41,8 —35,0 —32,5 —32,5
* 1 кал/моль = 4,19 Дж/моль.
53
Таблица 22. Равновесная концентрация этиленгликоля с влагой воздуха
Температура воздуха, СС Равновесная концентрация при относительной влажности, %
10% 20% 3 0% 4 0% 50% 60% 70% 80% 90%
—5 97,5 93,4 89,3 85.7 82 78 72 63 48
5 97,3 93,2 89,1 85,4 82 76 69 60 42
15 97,1 93,0 88,9 85,0 81 75 66 57 37
25 96,8 92,8 88,6 84,7 80 73 64 55 36
35 96,6 92,7 88,4 84,3 79 72 63 53 35
45 96,4 92,5 88,2 84,0 78 71 62 51 34
В работе [29] приведены также теплоты смешения этиленгликоля
с ацетоном, тетрагидрофураном, диоксаном, метиловым спиртом
и глутаронитрилом. Теплота растворения хлористого калия в смесях
этиленгликоля с водой и спиртом приведена в работе [30].
Этиленгликоль при 20—25 °C полностью смешивается с рядом
низкомолекулярных органических соединений, содержащих кисло-
род, азот и серу [1, р. 47; 27, р. 9; 31, р. 59; 32, р. 185, 225]:
одноатомными спиртами (метиловый, этиловый, изопропиловый,
н-бутиловый, изоамиловый, 3-пентиловый, диацетоновый, 3-гепти-
ловый, бензиловый, фурфуриловый)
многоатомными спиртами (ди-, три- и тетраэтиленгликоли, 1,2-
и 1,3-пропиленгликоли, дппропиленгликоль, глицерин)
алициклическими и ароматическими гидроксилсодержащими со-
единениями (циклогексанол, фенол, о-крезол)
карбонильными соединениями (ацетил ацет он, ацетон, бензальде-
гид, метилэтилкетон, фурфурол, циклогексанон)
кислотами (уксусная, диэтилуксусная, 4-метил-н-валериа-
новая)
аминами (анилин, ди-м-бутиламин, оксиэтилэтплидендиамин, ди-
изопропил амин, диметиламин, диэтилентриамин, а-метилбензил-
амин, а-метилбензилдиметиламин, триметилентетрамин, о-феиети-
дин, 2-фенилэтиламин)
амидами (формамид, диэтилформамид)
аминоспиртами (этанол- и изопропаноламин, а-метилбензилэта-
ноламин, а-метилбензилдиэтаноламин), а также с другими азот-
содержащими соединениями (пиридин, 2-метил-5-этилпиридин)
простыми эфирами этилен- и диэтиленгликоля (метиловый, эти-
ловый, изопропиловый, бутиловый, этилбутиловый)
сложными эфирами (этилформиат, триэтилфосфат, бутиллактат).
Этиленгликоль полностью смешивается со скипидаром и серни-
стым ангидридом, по практически не растворяется или же раство-
ряется в весьма небольших количествах в углеводородах, не смеши-
54
вается с хлор- и нитроуглеводородами. Растворимость этиленгликоля
в углеводородах [33, с. 1263] составляет [в % (мол.)]
при х-Л ...........................
при 67,4 °C.........................0,943
В циклогексане
при 46,1 °C .........................0,038
при 67,4 °C.........................0,943
В гептане
при 42,7 °C..........................0,034
при 67,9 °C.........................0,103
В этиленгликоле при комнатной температуре очень плохо рас-
творимы (менее 1%) масла растительного и животного происхожде-
ния, а минеральные масла нерастворимы совсем. Растворимость
талового масла, например, составляет 1,1 г/100 г, а ализаринового —
3,3 г/100 г. В этиленгликоле нерастворимы или очень мало раство-
римы (менее 0,5%) смолы (даммара, канифоль, каури, копал, шел-
лак), животный клей, ланолин водный, декстрин (10%-ный водный
раствор декстрина смешивается с этиленгликолем полностью).
Каучук, ацетил- и этилцеллюлоза, полихлорвинил в гликоле
нерастворимы, а нитроцеллюлоза в нем набухает. Растворимость
в этиленгликоле некоторых органических и неорганических соеди-
нений при 20—25 °C приведена в Приложении, табл. 4, стр. 353.
Растворимость солей в водных растворах этиленгликоля дана в ра-
ботах [30, 33, 34]. Особенно хорошо растворяются азотнокислые
соли. Так, при 25 °C четырехкомпонентные смеси с концентрацией
около 50% этиленгликоля, 10—20% воды и 0,5—1% циклогексена
содержат 30—35% (масс.) азотнокислых солей марганца, никеля,
кобальта, меди, цинка или кадмия [34].
Растворимость двуокиси углерода при 25 °C и парциальном
давлении СО2, равном 0,1 МПа (760 мм рт. ст,), имеет минимум
25,7 ммоль/л при концентрации гликоля около 30% (мол.), тогда
как в воде и чистом гликоле она составляет соответственно 33,6
И 39,4 ммоль/л [35]. Данные по смешиваемости этиленгликоля и рас-
творимости в нем ряда соединений относятся к комнатной темпера-
туре. Однако при критической температуре растворения этилен-
гликоль смешивается со значительно большим числом соединений.
Критическая температура растворения (К. Т. Р.) этиленгликоля,
т. е. температура, при которой достигается его неограниченная
взаимная смешиваемость с компонентами, ограниченно раствори-
мыми при других температурах, для различных соединений при-
водится в Приложении (табл. 5, стр. 354) [36, р. 225].
Азеотропные смеси. Этиленгликоль с большим числом соедине-
ний образует азеотропные смеси. Температуры кипения некоторых
смесей при атмосферном давлении и содержание в них этиленгликоля
приводятся в Приложении, стр. 356 [37].
55
Состав и температура кипения трехкомпонентных азеотропных
систем, образованных этиленгликолем с двумя органическими веще-
ствами, приведены в табл. 23 [37].
Таблица 23, Состав и температуры кипения трехкомпонептных
азеотропных систем этиленгликоль — компонент I — компонент II
Компонент I Компонент II Т. кип. смеси, °C Концентрация в смеси, %
этилен- гликоля компонента^!
Фенол 2-Пиколни <485,01 5,9 79,2
Фенол З-Пиколин 186,41 15,9 67,7
Фенол 2,6-Лутидин 185,04 8,7 74,6
Фенол 2,4,6-Коллидин 188,55 29,5 54,8
о-Крезол 2,4,6-Коллидин 189,65 33,6 62,4
Равновесие между жидкостью и паром водных растворов этилен-
гликоля при различном давлении дано в табл. 24 [38, с. 283; 39].
Фазовое равновесие жидкость — пар для системы 1,2-пропилен-
гликоль — этиленгликоль при 1,33 кПа (10 мм рт. ст.) характери-
зуется данными, приведенными в табл. 25 [40].
В работе |40| также приведены данные фазового равновесия при
3,33, 6,67 и 13,3 кПа (25, 50 и 100 мм рт. ст.). Данные по равно-
весию жидкость — пар в других двойных системах этиленгликоля
(с метиловым, н-пропиловым и бутиловым спиртами, бутилацетатом,
глицерином, тетрагидрофураном, фенолом, крезолом, анилином,
метил- и диметилапилином, ^-пиколином, нафталином), а также
в тройных системах (с метиловым спиртом и ацетоном, метиловым
спиртом и тетрагидрофураном, фенолом и л-крезолом) находятся
в работах [39, 41, 42].
Равновесие в трех- и четырехкомпонентных системах, вклю-
чающих этиленгликоль, представлено графически (треугольник
Гиббса) на рис. 16 [43]. Графическое изображение многокомпонентных
систем, в состав которых входят этиленгликоль и сернистый анги-
дрид, приведено на рис. 17, а, а тройные системы, включающие
этиленгликоль и ацетонитрил, изображены на рис. 17, б.
Данные по фазовому равновесию жидкость — жидкость в трой-
ных системах, содержащих кроме этиленгликоля следующие ком-
поненты: уксусная кислота — этилацетат, м-бутанол — м-бутил-
ацетат, толуол — ацетон, триэтиламин — изомасляная кислота,
триэтиламин — пропионовая кислота, бензол — тиофен, нитро-
метан — лауриловый спирт, нитроэтан — додециловый спирт, нитро-
метан — триэтиламин, приведены в работах [42, 44, 45]. Фазовое
56
Таблица 24. Фазовое равновесие жидкость—пар водных растворов
этиленгликоля ,
Содержание воды, % (мол.) Температу- ра, “С Содержание воды, % (мол.) Темпера- тура, °C
! жидкость пар 1 ЖИДКОСТЬ ] пар
Давление 101,3 кПа Давление 30,4 кПа (228 мм рт. ст.)
(760 мм рт. ст.)
15,4 94,9 130,1 0,0 0,0 160,6
27,7 97,4 120,5 0,7 25,0 155,1
37,8 98,4 114,3 7,5 72,0 133,4
46,3 98,8 110,8 10,8 80,9 125,7
59,6 99,2 106,3 14,8 83,7 122,1
69,7 99,5 103,5 17,5 89,2 114,3
77,5 99,5 103,2 24,5 92,8 107,1
83,8 99,8 101,2 27,7 94,4 103,0
88,9 99,85 101,0 36,9 96,5 95,5
93,2 99,91 100,6 47,5 97,9 88,9
96,9 99,94 100,3 50,0 98,3 87,8
62,5 99,2 81,6
68,7 99,4 79,3
77,7 99,6 76,2
84,9 99,7 74,3
88,8 99,8 73,3
92,0 99,9 74,2
Давление 13,3 кПа Давление 3,3 кПа
(100 мм рт. ст. ) (25 мм рт. ст.)
1,1 24,5 133,4 0,9 30,2 103,5
5,0 64,4 116,9 2,4 56,3 93,8
9,2 81,2 105,0 5,3 77,9 83,0
14,5 90,0 96,7 10,0 89,6 74,5
20,6 94,3 86,8 15,2 94,4 64,0
30,0 97,1 76,2 21,8 97,0 57,0
39,7 98,3 71,8 31,5 98,4 48,1
50,4 99,0 66,4 40,4 99,05 42,8
60,1 99,35 62,3 51,0 99,40 39,0
68,8 99,56 59,5 59,8 99,60 36,8
75,5 99,70 57,8 70,6 99,78 33,0
82,0 99,80 56,4 79,5 99,87 30.4
Давление 6,7 кПа (50 м .и рт. ст.) Давление 1 3 кПа (10 мм рт. ст.)
1,2 31,0 115,5 0,8 33,2 87,0
4,6 69,7 100,5 1,2 43,7 86,0
9,8 87,2 86,2 2,8 67,9 77,1
15,6 93,6 76,0 5,1 82,9 66,3
21,5 . 96,1 69,2 7,4 88,0 62,8
30,8 98,0 61,0 10,3 91,7 62,0
40,1 98,8 56,1 14,5 95,0 53,5
49,4 99,20 51,4 19,6 96,9 47,2
60,3 99,50 49,3 31,9 98,7 35,1
69,7 99,70 47,0 39,2 99,20 29,2
74,9 99,76 45,0 51,7 99,58 23,5
81,3 99,85 42,4 59,0 99,71 22,0
64,8 99,78 20,3
70,1 99,83 19,5
75,6 99,87 18,4
57
58
Рис. 17. Диаграммы равновесия в тройных системах, включающих этилен-
гликоль и сернистый ангидрид (а) и этиленгликоль и ацетонитрил (б).
Условные обозначения: ж-жидкость; Л. С. П. — линия сдвоенных коэффи-
циентов преломления; т — твердая фаза.
же равновесие четырехкомпонентных систем, включающих этилен-
гликоль — м-гептан и спирты (н-амиловый и н-гексиловый, м-гекси-
ловый и w-гептиловый, м-амиловый и м-гептиловый), а также этилен-
гликоль — н-гептан — четыреххлористый углерод — циклогексан и
этиленгликоль — вода,— карбитол — бензол, исследованы в ра-
боте [461.
Рис. 16. Диаграмма равновесия в трех- и четырехкомпонентных системах,
включающих этиленгликоль.
Условные обозначения: Сы—н-тетрадекан; ж — жидкость; Л. С. П — линия
сдвоенных коэффициентов преломления: МН — 1-метилнафталиш т — твердая фаза: ЭГ—
этиленгликоль; -- воды;---------— изопикны (линия, соединяющая точки составов
смесей с одинаковой плотностью).
59
Таблица 25. Фазовое равновесие жидкость — пар системы
1,2-пропиленгликоль — этиленгликоль при 1,33 кПа
Содержание 1,2-пропилеи- гликоля, % (МОЛ.) Темпер а- тура, °C Содержание 1,2-пропи- ленгликоля, % (мол.) Темпера- тура, °C
ЖИДКОСТЬ пар жидкость пар
4,8 6,6 91,2 47,6 56,6 86,8
10,4 13,6 90,8 . 53,0 62,1 86,0
21,5 27,6 89,5 59,5 68,3 85,6
21,9 28,1 89,3 72,0 79,2 84,9
29,7 37,2 88,4 82,3 88,0 81,1
38,0 46,5 87,3 92,0 94,5 83,6
Методы получения этиленгликоля
Методы получения этиленгликоля, рассмотренные в этом разделе,
можно разделить на лабораторные, часть из которых прошла про-
верку на опытных установках и, возможно, найдет промышленное
применение, и промышленные, которые в свое время нашли при-
менение, однако в дальнейшем потеряли значение в связи с большими
успехами, достигнутыми в процессе получения окиси этилена и ее
гидратации в этиленгликоль. Следует заметить, что такое разделение
методов получения этиленгликоля является в известной степени
условным, так как по мере развития науки и техники — открытия
новых катализаторов и экономичных способов получения исходных
продуктов, существенного усовершенствования уже известных спо-
собов — лабораторный способ может стать промышленным, а способ,
по которому производится продукт, может потерять свое промышлен-
ное значение.
В начале раздела рассмотрены методы получения этиленгликоля
из этилена или его производных, затем из метанола или формальде-
гида п дано описание процессов получения этиленгликоля по мето-
дам, которые были реализованы в промышленности. В настоящее
время в промышленности этиленгликоль производится практически
только одним методом — гидратацией окиси этилена.
1. Впервые этиленгликоль был получен Вюрцем в 1859 г. из
дибромэтана в две ступени. В результате нагревания дибромэтана
с ацетатом серебра образуется этиленгликольдиацетат, при гидролизе
которого калиевой щелочью получается этиленгликоль:
.... у, т, 2Cn3COOAg гплгп оп сыпгтт т~2К0Н
CHgHrCl^Hr ____2AgBr СН3 COO CIT2 CrJo С 00 СИд *
—> СН2ОНСН2ОН + 2СН3СООК
2. Этиленгликоль получается и непосредственно гидролизом ди-
бромэтана (или других дигалогенидов этилена, у которых атомы
60
галогена находятся при разных атомах углерода), водой и окисью
свинца [47] или водными растворами карбонатов и бикарбонатов
щелочных металлов:
СН2ВгСН3Вг+2НгО ---> СН2ОНСН2ОН + 2НВг
СН2ВгСН2ВгН-Н2О + К2СО3 > СН2ОНСН2ОН + 2КВг+ СО2
Поскольку дихлорэтан значительно доступнее и дешевле ди-
бромэтана, то большое число работ было посвящено гидролизу
дихлорэтана карбопатом или бикарбонатом натрия, а также слабо-
щелочными солями [48, 49]. Гидролиз дихлорэтана протекает с боль-
шим трудом, поэтому процесс рекомендуется проводить при повы-
шенных температуре (до 200 °C) и давлении (до 9,81 МПа или
100 кгс/см2) [49].
Этот способ нашел промышленное применение в Германии в период первой
и второй мировых войн [50, с. 90]. Гидролиз проводили водным раствором соды
по реакции:
CH2ClCH2Cl + Na2CO3 + H2O > CH2OHCH2OH-]-2NaC14- СО2
Наряду с основной реакцией протекает и побочная реакция образования
хлористого винила. Избыток дихлорэтана и низкая щелочность раствора способ-
ствуют снижению образования хлористого винила. Содержащийся в дихлор-
этане в виде примеси дихлордиэтиловый эфир гидролизуется в диэтилен, лпколь:
CH2ClCH2OCH2CH2Cl + Na2CO3+H2O —>
—> CII2OHCH2OCH2CH2OI-I +2NaCl + CO2
Реакцию гидролиза осуществляли периодически в стальном автоклаве при
140—160 °C п 2,35 МПа (24 кгс/см2). Продолжительность операции составляла
«=П5 ч. Образовавшаяся реакционная масса содержала (в среднем) 11% этилен-
гликоля, 0,5% соды, 20% NaCl, 68,5% воды. Для осветления этой массы в нее
добавляли 2,5—3%-ный раствор хлористого магния, который коагулировал
коллоидные частицы окиси железа (появляющиеся в результате коррозии обору-
дования).
После фильтрования раствор дважды выпаривали в вакууме при 60 и 80 °C
с выделением основной массы NaCl. Концентрирование раствора и выделение
из пего этиленгликоля проводили па насадочной колонне периодического дей-
ствия при остаточном давлении 2 кПа (15 мм рт. ст.), температуре в верху ко-
лонны 95 °C и флегмовом числе, равном 2. Полученный этиленгликоль («гли-
зантин») выкипал в пределах 195—245 °C и имел относительную плотность
1,111 —1.113. На 1 т товарного этиленгликоля расходовалось 2.1 т дихлорэтана
и 2,03 т соды.
3. Гидролиз этиленхлоргидрина карбонатами или бикарбонатами
Щелочных металлов при умеренной (70—80 °C) или более высокой
температуре (110—170 °C) приводит к образованию этиленгликоля
с выходом, достигающим 90% [51]:
СН2ОНСН2С1 + NaHCO3 ;=-► CH2OHCH2OH-|-NaCl+CO2
Способ непосредственного гидролиза этиленхлоргидрина в эти-
ленгликоль не нашел широкого промышленного применения из-за
сложности выделения гликоля из солевых растворов, хотя именно
по этому способу в 1925 г. в США было налажено первое промыш-
ленное производство этиленгликоля [52]. Оказалось, что и.з
61
этиленхлоргидрина выгоднее получать этиленгликоль, если вести
процесс в 2 ступени по реакциям:
2СП2ОНСН2С1+Са(ОП)2 —> 2ЩС—СН2+ СаС12 + 2Н2О
О
П2С--СН2+Н2О----> СН2ОИСН,ОН
чо"
«Хлоргидриппый» метод в последнее время почти полностью
вытеснен гидратацией окиси этилена, получаемой каталитическим
окислением этилена. Таким образом, все способы получения этилен-
гликоля из хлорпроизводных этилена утратили промышленное
значение, так как их использование сопряжено с расходом значи-
тельных количеств хлора, получением больших количеств химически
загрязненных сточных вод и рядом других недостатков.
4. Получение этиленгликоля гидролизом этиленгликольацетатов.
Основанием для этого метода послужила разработка процесса окис-
ления этилена кислородом в уксусной кислоте с образованием эти-
ленгликольацетата или этиленгликольдиацетата (в зависимости от
выбранного катализатора). Гидролиз полученных ацетатов приводит
к получению этиленгликоля:
синтез ацетатов гликоля
2С2Н4л O2J-2CH3COOH -> 2СН2ОНСН2ООССН3
2C2II4 + O2+4CH3COOII > 2СН3СООСН2СН2ООССН3 + 2Н2О
гидролиз полученных ацетатов
СН2ОНС112ООССП3 + Н2О -> СН2ОНСН2ОН+СН3СООН
СН3СООСН2СН2ООССН3 + 2И2О > СН2ОНСР£2ОН-г2СП3СООН
Поскольку уксусная кислота, расходуемая на первой стадии,
регенерируется на второй и возвращается на первую, можно считать,
что этиленгликоль получается из этилена, кислорода и воды, а уксус-
ная кислота расходуется только для возмещения ее потерь. Схему
получения гликоля через этиленгликольацетаты можно представить
следующим образом [53]:
62
Синтез этиленгликольацетата протекает при умеренной темпера-
туре (до 100 °C) в жидкой фазе (под давлением) или в паровой фазе
(при нормальном давлении) в присутствии катализатора (соединений
палладия) и сокатализаторов (солей щелочных и щелочноземельных
металлов, меди), а также кислот. Выход этиленгликольацетата,
содержащего около 5% этиленгликольдпацетата, достигает 95%
от теоретического. При применении в качестве катализатора ТеО2
и НВг в оптимальных условиях (145—160 °C, 2,8 МПа или
28,9 кгс/см2, рецикл производных муравьиной кислоты) селектив-
ность процесса превышает 98%, степень конверсии этилена соста-
вляет около 60% . При этом образуется 60% этиленгликольдиацетата,
35% этиленгликольацетата и 5% этиленгликоля [54].
Гидролиз эфиров этиленгликоля проводится горячей водой в аппа-
рате противоточного типа: в верхнюю часть аппарата подают эфир
и кислотный катализатор, а в нижнюю часть — водяной пар. Сверху
отбирают образующуюся уксусную кислоту и водяной пар, а снизу —
этиленгликоль, который дополнительно обрабатывают щелочным
раствором для полного гидролиза остатков эфира.
Гидролиз этиленгликольдиацетата рекомендуется проводить
при 80—160 °C и 0,03—0,52 МПа (0,35—5,25 кгс/см2) в присутствии
катализатора серной или фосфорной кислоты (менее 0,01 моль на
моль эфира) в колонне или в каскаде реакторов [53, 54]. Степень
конверсии эфира и селективность процесса составляют более 99%.
Таким образом, общая селективность процесса получения этилен-
гликоля из этилена через гликольацетаты составляет более 90%
(согласно [62] 97%). Это значительно выше, чем при синтезе этилен-
гликоля из этилена через окись этилена, в котором селективность,
с учетом образующихся ди- и триэтилепгликолей, составляет около
70% на исходный этилен. По такому способу строится завод мощ-
ностью 363 тыс. т/г этиленгликоля [54, 55].
5. По реакции Вагнера этиленгликоль получается окислением
этилена слабым (1%-ным) щелочным раствором перманганата калия.
Предполагалось образование этиленгликоля из этилена за счет
присоединения к нему атома кислорода из перманганата и молекулы
воды. Однако, как ‘показали исследования реакции непредельных
соединений с КМпО4, меченного 18О, более справедлива такая реак-
ция присоединения кислорода:
л-2
2ОН“\ /
---> ХС---С 4-2МпО?“
/[ |\
*ОН *011
Предложен ряд способов гидроксилирования этилена в этилен-
гликоль перекисями, гидроперекисями, хлоратом калия, органичес-
кими надкислотами в присутствии четырехокиси осмия (OsO4) или
осмиевых соединений, а также соединений других переходных
металлов (V, Mo, W и др.), как катализаторов [57, 58]. При окислении
63
этилена феррицианидом в присутствии OsO4 при 25 °C выход этилен-
гликоля составляет 94=96 от теоретического [59]. Протекающая в
этом случае реакция может быть выражена уравнением:
CH2-CH2-|-2Fe(CN)|" + 2OH- > СН2ОН—CH2OH + 2Fe(CN)’"
При пропускании смеси этилена и кислорода через слабощелочной
раствор (pH 8—10) OsO4 при давлении 6,9 МПа (70 кгс/см2) и отно-
шении С2Н4 : О2, равном 1 : 19, выход этиленгликоля составляет 95%.
Этиленгликоль с выходом 88—97% от теоретического получается
при гидроксилировании этилена перекисью водорода в растворе
mpm-бутилового спирта при температуре около 0 °C в присутствии
OsO4. Гидроксилирование этилена может проводиться также водным
33%-ным раствором Н2О2 в присутствии стабилизаторов — перекисей
алифатических и циклоалифатических гликолей, их неполных эфи-
ров, несмешивающихся с водой спиртов и углеводородов [60].
Синтез этиленгликоля прямым окислением этилена сильными
окислителями (перманганат калия, перхлорат, перекись водорода)
хотя и дает высокий выход этиленгликоля, промышленного примене-
ния не нашел, так как использование этих окислителей привело бы
к высокой себестоимости этиленгликоля. Следует отметить высокую
токсичность OsO4, которая является одним из лучших катализаторов
процесса.
6. Этиленгликоль можно получать при электрохимическом окис-
лении этилена в разбавленном растворе H2SO4; максимальный выход
продукта 24—27% [61].
7. Предложен новый способ прямого синтеза этиленгликоля
методом окисления этилена при 7,2 МПа (73 кгс/см2) и 160 °C в при-
сутствии катализатора Т1(ОН)3 в 0,6 н. растворе НС1 [54, 62]. Выход
этиленгликоля достигает 89%, одновременно получается 6% ацет-
альдегида и 5% других продуктов. Для осуществления непрерыв-
ного процесса образующийся Ti+ окисляется в Т13+ солью трех-
валентного железа (или двухвалентной меди) в присутствии кисло-
рода (аналогично тому, как это осуществляется в процессе синтеза
ацетальдегида из этилена):
Ti+ + 2Cu2+-► Ti3+-)-2Cu+
2Cu ++2H+ + 0,5O2 ► 2Cu2+-)-H2O
Расход этилена на этиленгликоль значительно ниже («=> на 20%),
чем в применяемом в промышленности способе гидратации окиси
этилена. В связи с большой коррозионностью реакционной среды
для промышленной реализации процесса, по-видимому, необходима
аппаратура из титана (как и в производстве ацетальдегида).
8. Сущность метода получения этиленгликоля из этиленкарбо-
ната [63] заключается в том, что гидратацию окиси этилена проводят
64
в присутствии двуокиси углерода и катализатора по следующей
схеме:
СН2ОИ-С112ОН
СП2-О\
I
СН2-СГ
+нго
-СО2
При выбранных условиях скорость реакции окиси этилена с дву-
окисью углерода значительно выше таковой с водой, поэтому этилен-
гликоль образуется в основном через этиленкарбонат. Диэтилен-
гликоль получается при этом в незначительных количествах. Про-
цесс проводят при 80—120 °C на стадии образования этиленкарбоната
и 160—220 °C на стадии разложения его в этиленгликоль; давление
составляет 1,5—5,9 МПа (15—60 кгс/см3).
Мольное соотношение окиси этилена и воды колеблется от 1 : 1
до 1 : 1,3 (предпочтительнее 1 : 1,1). Продолжительность процесса
около 1 ч. Катализаторами служат галогениды щелочных металлов,
четвертичного аммония или аминов, а также смеси веществ, при
взаимодействии которых получается нужный катализатор. Опти-
мальное количество катализатора 0,3 — 1,5% от общей массы окиси
этилена и воды. Выход этиленгликоля достигает 97%, а диэтилен-
гликоля — около 3% на взятую окись этилена; конверсия окиси
этилена практически полная.
Реакционная масса после удаления из нее основного количества
СО2 подвергается переработке для выделения катализатора, этилен-
и диэтиленгликоля и остатков СО2, которая после компримирования
возвращается на синтез.
Описанный способ представляет определенный интерес: получа- .
емый в результате реакции раствор этиленгликоля содержит незна-
чительное количество воды, так как исходные вещества (окись эти-
лена и вода) берутся почти в эквимольных соотношениях. Поэтому
для отгонки воды не требуется сооружения выпарных установок
И значительно снижается расход водяного пара. Однако по сравне-
нию с применяемым в промышленности методом некаталитической
гидратации окиси этилена этот способ имеет ряд недостатков: в про-
цессе, кроме воды, используется двуокись углерода, а также катали-
затор, для приготовления которого требуется дополнительная аппа-
ратура. Дополнительная аппаратура необходима и для выделения
солей из реакционного раствора, и для компримирования СО2.
91 Этиленгликоль образуется при каталитическом гидрировании
этиленкарбоната, при этом сопутствующим продуктом является
метиловый спирт [64]:
О
Н2с/ \
I / с=о + зн2 —>. СН2ОН-СН2ОН+СНзОН
Н2СХ '
о
3 Заказ 972
65
В присутствии промышленного меднохромового катализатора
при 29,4 МПа (300 кгс/см2) и 240 СС степень конверсии этиленкарбо-
ната равна 100%, а выход этиленгликоля и метилового спирта соста-
вляет 71,5 и 92—93% (мол.) от теоретического. Кроме основных
продуктов гидрирования образуется ряд побочных: диэтиленгликоль
(₽=* 16,5%), этиловый спирт (4—5%), метилцеллозольв (3%), а также
небольшие количества газообразных продуктов — СО, СО2, СН3ОН
и СН4.
10. Этиленгликоль можно получить радиационно-химическим ме-
тодом из метилового спирта [65]. Для этой цели используют у-излу-
чение отработанных тепловыделяющих элементов ядерных реакторов
(ТВЭЛ) и других источников, а также источники р-излучения.
Метиловый спирт для синтеза может содержать до 30% воды. Наряду
с этиленгликолем образуются формальдегид, вода, окись углерода,
водород, метан. Выход этиленгликоля составляет 65—70% от теоре-
тического, а формальдегида — около 20%. Присутствие кислорода
приводит к снижению выхода этиленгликоля и повышению выхода
формальдегида. Для повышения выхода этиленгликоля предложено
проводить процесс в присутствии закпси азота. Реактор мощностью
138 МВт может обеспечить 50 тыс. т в год этиленгликоля с достаточно
низкой себестоимостью.
11. Этиленгликоль с выходом 62% образуется при термическом
или фотолитическом разложении перекиси ди-пгреш-бутила в рас-
творе метилового спирта, содержащего небольшие количества эти-
лендиаминотетрауксусной кислоты.
12. Этиленгликоль совместно с глицерином можно получить из
формальдегида [66]. Сущность метода заключается в том, что при
конденсации формальдегида в присутствии окислов металлов И
и IV групп, а также триметил- и триэтиламина образуется смесь
гликолевого и глицеринового альдегидов, гидрированием которых
получают этиленгликоль и глицерин:
1ЮСН2СНО + Н2 —> НОСН2СН2ОН
1ЮСН2СНОНСНО + П2 ---> СП2ОПСНОНСН2ОН
При конденсации формальдегида образуются и другие окси-
альдегиды, вплоть до гексоз С6Н]2О6, а также муравьиная кислота,
метиловый спирт (по реакции Канницаро — Тищенко), ацетали.
Побочные высокомолекулярные продукты образуются также при
гидрировании оксиальдегидов, чему способствует присутствие форм-
альдегида.
13. Этиленгликоль получается при гидрогенолизе многоатомных
спиртов — ксилита и глюкозы; одновременно образуются пропилен-
гликоль и глицерин [67]. При давлении 19,6 МПа (200 кгс/см2),
температуре 240—250 °C, в присутствии катализатора 50% Ni +
+ 0,25% СаО на кизельгуре выход этиленгликоля на превращенный
ксилит составляет 33,8—39,7%. Ксилит и глюкозу получают гидро-
лизом природных веществ растительного происхождения, поэтому
66
гидрогенолиз многоатомных спиртов может рассматриваться как
способ получения этиленгликоля из растительного сырья (см. гл. V,
стр. 198).
14. Этиленгликоль, наряду с другими полифункциональными
кислородсодержащими соединениями, получается из окиси углерода
и водорода:
2СО + ЗН, >• СН2ОН-СН2ОП
При применении в качестве катализатора ацетонилацетонатди-
карбонила родия процесс проводится при 344,4 МПа (3500 кгс/см2)
и 190—230 °C. В кислородсодержащих продуктах содержится 64%
многоатомных спиртов, состав которых следующий (в %):
Этиленгликоль ...........76,5
Пропиленгликоль..........11,75
Глицерин ................11,75
Кроме перечисленных продуктов образуются также метиловый
спирт, метилформиат и вода [54]. Хотя процесс является весьма
сложным (высокое давление, многокомпонентная смесь продуктов
реакции), его достоинство в том, что синтез-газ — доступное и деше-
вое сырье. Можно ожидать, что при разработке катализаторов, кото-
рые позволят значительно снизить давление, процесс найдет про-
мышленное применение.
15. Получение этиленгликоля из метанола. Процесс получения
этиленгликоля по этому методу, который был осуществлен в про-
мышленных условиях в США, включает следующие стадии [68]:
получение формальдегида окислением метанола
2CH3OH-j-02 --> 2HCHO + 2II2U
получение гликолевой кислоты из формальдегида, окиси углерода
и воды
НСПО-гСО4-П2О —> ПОСН2СООН
получение метилового эфира гликолевой кислоты этерификацией
гликолевой кислоты метанолом
HOCH2COOH-f-CH3OH ----> НОСН2СООСНз + Н2О
восстановление метилового эфира гликолевой кислоты
НОСН2СООСНз + Н2 —> НОСН2СН2ОН-|-СН3ОН
На рис. 18 приведена принципиальная схема получения этиленгликоля
из метапола. Стадия окисления метанола в формальдегид не показана,
так как этот процесс является хорошо известным и широко применяемым в про-
мышленности. Кроме того, формальдегид может быть получен не только из
метанола.
Раствор гликолевой кислоты, вода п серная кислота, которая является
катализатором реакции образования гликолевой кислоты, в мольном соотноше-
нии 2:1: 0,2 поступают из смесителя 1 на орошение скруббера 2. В нижнюю
часть последнего подаются пары формальдегида и воды в отношении 1 : 1. После
скруббера 2 смесь вместе с окисью углерода, которая вводится с большим
5* 67
Рис. 18. Принципиальная
схема получения этилен-
гликоля из метанола:
I — смеситель; 2 — скруб-
бер; 3 — реактор; 4 — рек-
тификационная колонна;
5 — этерификатор; в — ре-
актор гидрирования.
J
избытком, направляется в реактор 3. Здесь при давлении 68,6 МПа (720 кгс/см2),
температуре 200 °C и времени контакта 5 мин образуется гликолевая кислота
с выходом 90—95%.
Из реактора 3 раствор гликолевой кислоты поступает в ректификационную
колонну 4, работающую при остаточном давлении 10—13 кПа (80—100 мм рт. ст.).
Снизу колонны выводятся гликолевая кислота в виде жидкости, а сверху —
гликолевая кислота выводится в виде паров вместе с парами воды, которые
возвращаются в процесс. Далее гликолевая кислота этерифицпруется метанолом
обычным способом, п полученный сложный эфир направляется в реактор гидри-
рования 6. При давлении 3,0 МПа (31 кгс/см2), температуре 200 °C и объемной
скорости 20 000 с-1 в присутствии катализатора медь — хромит бария обра-
зуется этиленгликоль с выходом 90—95%; здесь же регенерируется и метанол.
При давлении выше 9,81 МПа (100 кгс/см2) для гидрирования эфира можно
использовать также катализатор окись меди — окись магния.
Из реактора гидрирования смесь гликоля с метанолом и остатки непро-
реагировавшего метилового эфира гликолевой кислоты поступают на ректифи-
кацию. Расход основных видов сырья на 1 т этиленгликоля составляет (в т):
Формальдегид.............0,59
Окись углерода...........0,57
Водород ..................0,068
Серная кислота............0,041
В 1962—1967 гг. фирма Du Pont de Nemours выпускала около
77 тыс. т в год этиленгликоля из формальдегида, однако в 1968 г.
производство этиленгликоля по этому методу было прекращено [69].
Основными недостатками метода являются многостадийность, слож-
ность аппаратурного оформления из-за высоких давлений и большая
коррозионность среды.
Получение этиленгликоля гидратацией
окиси этилена
Катализ, механизм и кинетика реакции
Еще в 1860 г. Вюрц обнаружил, что при нагревании окиси этилена
с водой образуется этиленгликоль. При комнатной температуре
скорость реакции весьма мала. Так, за 22 сут вступает в реакцию
только половина окиси этилена, взятой в виде 0,5%-ного водного
раствора [70]. При нагревании выше 100 °G реакция протекает
значительно быстрее. Взаимодействие окиси этилена с водой уско-
ряется также кислотами (как минеральными, так и органическими),
являющимися катализаторами реакции гидратации. Ускорение этой
68
реакции водородными ионами исследовано в работах [71]; в них
отмечалось, что анионы сильных кислот не оказывают существенного
влияния на скорость реакции гидратации.
В присутствии щелочей скорость реакции окиси этилена с водой
также возрастает, однако значительно увеличивается количество
полигликолей — продуктов взаимодействия окиси этилена с этилен-
гликолем и с другими образующимися в процессе реакции глико-
лями [72, 73]. Катализаторами реакции гидратации могут являться
также ионообменные смолы (катиониты) [74—76] -
В настоящее время считается общепризнанным, что в водных
растворах окиси этилена образуется ее промежуточная оксониевая
форма, которая имеет значительно большую реакционную способ-
ность, чем исходная молекула [77, с. 26; 78, с. 257]:
Н,С—СН2 + НОН
хо/
~Н2С—СН2 ОН--
\)'+
—> носн2—СН2ОЫ
При кислотном катализе на первой стадии происходит прото-
нирование окиси этилена, которое сопровождается смещением
электронной плотности и сильной активацией ее молекулы [79,
с. 391]
п2с—сн2 + и+ > Н2С—сн2
О 0+ ' .
I
и
благодаря чему облегчается атака углеродного атома окиси этилена
нуклеофилом — молекулой воды:
н2с—сн2+н2о
Н2С—сп2-
\)б+
I
. н
..он2 ] -> НОСН2-СН2ОН2
длг носн2-сн2он+н+
Реакцию можно рассматривать как мономолекулярную, протека-
ющую по механизму нуклеофильного замещения типа Sx 1. Ускоре-
ние реакции гидратации в присутствии кислот объясняется тем, что
при увеличении концентрации водородных ионов смещается вправо
реакция образования оксониевого иона — активной формы окиси
этилена. Считается, что гидратация окиси этилена в присутствии
Н-катионитов и кислот протекает по одинаковому механизму [76,
с. 118]. Вначале образуется оксониевый ион
Н2С—CH2+RSO3H Н2С—CH2 + RSOg
н
6»
который при размыкании оксиранового кольца переходит в ион
карбония СН2—СН2ОН. При взаимодействии последнего с водой
получается этиленгликоль, при этом Н-катионит регенерируется:
СН2-СИ2ОН4-Н2О-f-RSOjj —> CH.2OI1-GH2OH+RSO3H
В щелочной среде гидратация окиси этилена представляется
как реакция нуклеофильного замещения типа 2 с непосредствен-
ной атакой углерода окисного кольца нуклеофилом [80, с. 55; 81,
р. 568]:
Н2С—СН2 + ОН- ГН2С—сн2---он~
Х'б/ L б
+Н2О
—— НОСН2-СН2О- z: HOCH2-CH2OH-LOH-
Предложен также механизм, который является общим для гидра-
тации окиси этилена как в кислой, так и в щелочной среде [81, 82]:
Н2С—сн2
—> НОСН2-СН2ОН
На первой стадии также образуется ион оксония, однако па вто-
рой стадии, без перехода в карбопиевый ион, протекает нуклео-
фильное замещение в оксониевом комплексе по типу 2.
Скорость гидратации окиси этилена зависит от pH среды, причем
эта зависимость имеет сложный характер и характеризуется нали-
чием трех областей, что графически показано на рис. 19 [83, с. 41].
В области, близкой к нейтральной среде, протекает медленная не-
каталитическая реакция:
Н2С—СН2 + Н2О > СН2ОН-СН2ОП
В сильно кислой среде (при pH до 4,5) скорость реакции резко
возрастает вследствие роста концентрации протонированной по
кислороду окиси этилена; в этом случае молекула воды реагирует
с активным ионом окиси этилена:
Н2С—СН2 н2с—си2 —НОСН2-СН2ОН + П+
н
70
В щелочной среде (при pH 12) большая скорость гидратации
обусловлена действием на окись этилена сильного нуклеофила —
иона гидроксила:
щс—сн2+он- —> иосн2-сн2о- НОСН2-СН2ОП
О
Механизм реакции окйси этилена с водой рассмотрен также в ра-
ботах [84, 85].
Некаталитическая реакция окиси этилена с водой является
реакцией первого порядка относительно окиси этилена. Константа
Рис. 19. Зависимость
скорости гидратации от
pH среды.
скорости реакции при 20 °C найдена равной (2,5—2,16)-10-5 мин-1
[70, 72]; при других температурах константы скорости к соста-
вляют [72]:
30,2 °C 48 °C
к, 5,45-10-5 3,55-IO-4
мин-1
70,3 °C
2,68-Ю-з
ИЗ °C
5,1-10-2
123 °C 131 °C
1,17.10"1 1,73-Ю’1
Зависимость константы скорости от температуры (Т, К) выра-
жается уравнением
fc = 3,19 • Ю9е-1»,’0°/1’99Г
Энергия активации некаталитической реакции определена равной
79,5 ± 1,2 кДж/моль (19,0 ± 0,3 ккал/моль). В другой работе
энергия активации реакции окиси этилена с водой определена равной
88,3 кДж/моль (21,1 ккал/моль) [86].
Кислотный катализ. Гидратация окиси этилена в присутствии
кислот, как и некаталитическая реакция, протекает по первому
порядку относительно окиси этилена, и ее скорость пропорциональна
концентрации кпслоты [70, 75, 87]. При кислотном катализе кон-
станта скорости реакции для 1 М раствора хлорной кислоты при
20 СС составляет 3,2-10"1 мин-1 [70], а для гидратации окиси этилена
в присутствии щавелевой кислоты, в зависимости от ее концентрации,
константы скорости найдены равными [87]:
Концентрация (СООН)2-2Н2О,г/100 см3 0,1 0,3 0,5 0,7
Константа скорости к-103, моль/(л-мин) 2 6 10 13
71
i
i
I
На рис. 20 дана зависимость концентрации окиси этилена от
продолжительности реакции в присутствии серной кислоты при
различном pH раствора, а на рис. 21 — зависимость константы
скорости реакции от нормальности серной кислоты и температуры.
Прямые линии рисунков подтверждают первый порядок реакции
относительно концентрации как окиси этилена, так и кислоты.
Энергия активации гидратации окиси этилена при кислотном
катализе определена равной 75,4 кДж/моль [18 ккал/(г-моль)].
Катализ катионитами. Гидратация окиси этилена в присутствии
гетерогенного катализатора (сульфополистирольного катионита
амберлит /7?-120) также протекает по реакции первого порядка
Рис. 21. Зависимость константы скорости реакции от нормальности Серной
кислоты и температуры.
Рис. 20. Зависимость изменения концентрации окиси этилена от продолжитель-
ности реакции при различном pH среды и при 30 °C.
относительно окиси этилена, причем константа скорости реакции растет
с увеличением количества взятого катионита и уменьшением размера
его частиц [75]. Для различных температур константа скорости
реакции к приведена ниже (взято 100 г сырого катионита /7?-120
на 1 л раствора):
При 30 °C
/с-10®, мин-1 29,5
При 50 °C
114
При 70 °C При 90 °C
259 627
Энергия активации реакции в присутствии катионита определена
равной 41,9 кДж/(г-моль) или 10 ккал/(г-моль). Скорость реакции
увеличивается примерно в 2 раза при повышении температуры на
20 °C, тогда как при гомогенном кислотном катализе она увеличи-
вается в 2 раза при повышении температуры на 10 °C. По-видимому,
это объясняется тем, что скорость реакции в присутствии катионита
определяется скоростью адсорбции на поверхности и диффузии
72
окиси этилена в порах смолы, а также скоростью десорбции и обрат-
ной диффузии продуктов реакции из пор смолы в раствор.
Основной катализ. В щелочной среде гидратация окиси этилена
является реакцией первого порядка относительно окиси этилена
и константа скорости ее при 20 °C определена равной 4,85X
X 10“3 л/(моль-мин) [72]. Это значение найдено для 0,135—1,092 М
растворов окиси этилена. Константы скорости реакции для растворов
окиси этилена с мольностыо 1,1 —1,3 в зависимости от мольной
концентрации NaOH (Муаон) приведены ниже:
А. 103 ю3, *OH'JD3-
мХаОН МИН-1 МИН-1 л/(моль-МИН)
0,257 1,04 1,02 3,97
0,511 2,32 2,30 4,51
1,010 4,93 4,91 4,86
1,030 5,46 5,44 5,28
1,540 8,69 8,67 5,63
2,020 11,68 11,66 5,77
k — константа скорости, найденная экспериментально;
h0 — константа скорости некаталитической реакции:
&ОН “ константа скорости основного катализа для 1 М NaOH,
равная
— : WNaOH
Константа скорости гидратации окиси этилена возрастает с увели-
чением концентрации щелочи на 0,1 М приблизительно на 2,5%.
Зависимость константы скорости гидратации окиси этилена при-
щепочном катализе от температуры приведена в табл. 26.
Таблица 26. Зависимость константы скорости реакции гидратации
окиси этилена от температуры в присутствии NaOH
Мольность Температура, °C Константа скорос- ти k0H, л/(моль«мин)
ХаОН СН1«О
0,10 1,010 20,0 3,90-10-3
0,10 1,050 48,0 6,40-10-2
0,05 0,421 70,3 3,66-1 о-1
Энергия активации определена равной 75,8 + 4,2 кДж/моль
(18,1 ± 1 кал/моль).
Константа скорости основного катализа выражается уравнением
/с = 1,23.10’1е’18,)оо/1’99Г
73
Влияние различных факторов на состав
продуктов гидратации
При гидратации окиси этилена наряду с этиленгликолем получаются
и более высокомолекулярные гликоли, такие, как ди-, три- и тетра-
этиленгликоль. Образование их объясняется большой реакционной
способностью гликолей, вследствие чего между гликолями и окисью
этилена протекает ряд последовательно-параллельных реакций.
Конечный состав реакционного раствора зависит от ряда факто-
ров: соотношения окиси этилена и воды в исходной смеси, темпера-
туры и давления, применяемых катализаторов, а также от степени
Рис. 22. Зависимость выхода гликолей от мольного отношения окиси этилена
к воде:
1 — этиленгликоль; 2 — диэтиленгликоль; 3 — триэтиленгликоль; 4 — тетраэтиленглп-
кбль (-----некаталитическая реакция;-----------кислотный катализ).
конверсии реагентов. Выход отдельных гликолей должен также зави-
сеть от соотношения констант скорости последовательных стадий
реакции окиси этилена с водой и образующимися гликолями. Знание
этих факторов является весьма важным для правильной организации
процесса и достижения оптимальных технико-экономических показа-
телей производства этиленгликоля.
Влияние отношения окиси этилена и воды на выход продуктов.
Количество получаемых этиленгликоля и других гликолей опре-
деляется отношением окиси этилена к воде в реакционной смеси:
чем меньше это отношение, тем выше выход этиленгликоля. По мере
же увеличения отношения снижается выход этиленгликоля и повы-
шается выход ди- и триэтиленгликоля, причем выход диэтилен-
гликоля, пройдя через максимум, начинает падать за счет возраста-
ния выхода три- и тетраэтиленгликоля.
В табл. 27 [73] дана зависимость выхода продуктов гидратации
окиси этилена в отсутствие катализатора от мольного отношения
прореагировавшей окиси к воде при постоянном давлении, равном
74
Таблица 27. Зависимость выхода продуктов гидратации
от отношения окиси этилена к воде при некаталитической реакции
Отяо1нение окись этилена; вода Выход гликолей, %
этиленгликоль ди эти л еп гл пи о л ь триэтилен- гликоль тетраэти- ленгликоль
При температуре 140 °C
0,074 87,2 — — —
0,167 74,9 25,2 — —
0,585 42,5 32,4 15,5 —
0,868 30,7 35,6 22,5 9,6
1,340 18,6 29,8 25,3 16,2
При тем п е р а т у р е 120 °C
0,054 93,1 — — —
0,063 85,4 — — —
0,108 78,0 22,1 — —
0,162 69,4 20,0
0,224 64,5 28,8 — —
0,387 53,2 34,3 12,4 —
0,736 34,6 36,8 19,9 7,9
1,532 15,6 28,2 25,4 15,6
При температуре 100 °C
0,165 67,2 — — —
0,226 64,0 — — —
0,83 МПа (8,4 кгс/см2), а на рис. 22 эта зависимость иллюстрируется
графически.
При кислотном катализе, как и при некаталитической гидратации,
выход продуктов реакции определяется отношением окиси этилена
к воде, причем при одинаковом отношении исходных реагентов
выходы отдельных гликолей весьма близки. Зависимость выхода
гликолей (на взятую окись этилена) от мольного отношения окиси
этилена к воде при 90—95 °C в присутствии 0,5% H2SO4 приведена
в табл. 28 [88] и нанесена пунктирной линией на рис. 22.
При применении других кислот состав продуктов гидратации
получается примерно таким же. Концентрация кислоты в реакцион-
ной смеси практически не влияет на выход этиленгликоля, что видно
из данных табл. 29.
Выход этиленгликоля в присутствии катионита амберлит /77-120,
обработанного 10%-ной H2SO4, получается практически таким же,
как при кислотной, и несколько выше, чем при некаталитической
гидратации (рис. 23) [75].
При основном катализе распределение окиси этилена на отдель-
ные гликоли также зависит от отношения окиси этилена к воде,
75,
Таблица 28. Зависимость выхода гликолей от отношения
окиси этилена к воде в присутствии H2SO4
Отношение окись этиле- на : вода Выход гликолей, %
этилен- гликоль диэтилен- гликоль триэтилен- гликоль тетраэтп- ленгликоль высшие пб- лигликоли
0,095 82,3 12,7
0,126 77,5 17,5 —
0,150 76,5 18,5
0,238 65,7 27,0 2,3
0,375 51,6 33,0 10,4
0,476 47,2 34,5 13,0 0,3
0,714 35,0 33,0 19,2 5,7 2,1
1,640 15,7 26,0 19,8 19,0 14,5
Рис. 23. Зависимость выхода этиленгликоля при гидратации окиси этилена
в присутствии различных катализаторов от мольного отношения окиси этилена
к воде:
I — без катализатора, 100—140 °C; 2 — H2SO4, 50—90 °C; 3 — амберлит ГВ-120, 80 СС'
4 — амберлит ГВ-120, 20—50 °C; 5 — NaOH, 50 °C.
Таблица 29. Зависимость выхода продуктов гидратации
от концентрации H2SOi
Концентрация HaSO4, % (масс.) Отношение окись этилена : вода Выход гликолей, %
этилен- гликоль диэтилен- гликоль триэтилен- гликоль
0,5 0,435 48,5 34,2 12,3
2,0 0,435 50,0 30,8 14,2
0,5 ' 0,408 50,5 33,4 11,1
5,0 0,409 50,0 29,2 8,8
однако выход этиленгликоля ниже, а выход полигликолей выше,
чем при кислотном катализе или при некаталитической гидратации.
В отличие от кислотного катализа состав продуктов гидратации
зависит и от количества щелочи, взятой в качестве катализатора:
с его увеличением выход этиленгликоля уменьшается, а выход поли-
гликолей возрастает. Зависимость выхода (на прореагировавшую
окись этилена) продуктов гидратации при 120 °C от мольного отно-
шения окиси этилена к воде и концентрации NaOH приведена
в табл. 30 и на рис. 24 [73].
Влияние некоторых солей, например NaCl, аналогично действию
щелочных катализаторов: в присутствии хлористого натрия выход
этиленгликоля значительно ниже, чем при гидратации окиси этилена
в нейтральной или кислой среде [75]. Это объясняется каталити-
ческим действием щелочи, которая выделяется при взаимодействии
окиси этилена с хлористым натрием:
Н2С—CH2 + NaCl—> CH2ClCH2OH-f-NaOH
О
При гидратации окиси этилена в присутствии солей слабых кис-
лот и сильных оснований наряду со щелочным и некаталитическим
гидролизом протекает реакция с участием соли. При этом образуется
этиленгликолевый эфир соответствующей кислоты [89]. При быстром
гидролизе последнего образуется этиленгликоль (с количественным
выходом), и катализатор регенерируется. Таким образом, выход
этиленгликоля при нуклеофильном катализе солями слабых кислот
выше, чем при других типах катализа и при некаталитической гидра-
тации, причем он увеличивается с повышением концентрации и нук-
леофильности аниона и уменьшением концентрации гидроксил-иона.
Например, при гидратации окиси этилена под давлением СО2 в вод-
ном растворе, содержащем 0,5—5% NaHCO3, выход этиленгликоля
достигает более 90 % при отношении окиси этилена к воде около
0,17 [90]. Однако наличие значительных количеств солей в водном
растворе этиленгликоля усложняет процесс его выделения.
Влияние степешГ конверсии окиси этилена. Повышение степени
конверсии окиси этилена приводит к уменьшению выхода этилен-
77
Рис, 24. Зависимость выхода продуктов гидратации от мольного отношения окиси этилена к воде и концентра
цип NaOH:
1 — этиленгликоль; 2 — диэтилепгликоль; з — триэтиленгликоль, 4 — тетраэтиленгликоль
------некаталитическая реакция;----------гидратация в присутствии 0,01% NaOH'
б; ---0,01% NaOH;------------0,05% NaOH;----------0,1% NaOH.
Выход гликолей,
Таблица 30. Зависимость выхода гликолей от отношения
окиси этилена к воде в присутствии NaOH
Отношение окись этилена : вода Выход гликолей, %
этиленгликоль диэтилен- гли^оль триэтилен- гликоль тетраэтилен- гликоль
При к о н ц о и т р а ц и и 0,01% NaOH
0,070 72,5 30,3 — -
0,171 47,0 36,2 19,3 —
0,354 24,1 31,6 26,3 14,0
0,556 15,7 26,2 28,0 20,1
1,033 5,5 17,5 23,5 23,8
При концентрации 0,05% NaOH
0,072 61,0 34,5 — — •
0,182 36,8 35,4 22,1 —
0,358 20,2 29,8 27,6 15,9
0,583 12,4 22,4 27,1 23,3
1,069 3,9 16,8 22,3 23,4
При кон центрацип 0,1% NaOH
0,082 54,8 37,2 — —
0,186 зз,з 35,1 22,2 —
0.365 19,6 29,0 27,7 18,1
0,583 11,5 22,7 26,9 23,1
1,080 3,0 16,0 21,1 23,3
При концентрации 0,25% NaOH
0,386 ] 17,4 | 26,9 1 2U 1 19,4
При к о н ц е н т р а ц п п 0,5% NaOH
0,379 | 17,8 | 27,2 27,6 I 20,3
гликоля и увеличению выхода ди- и триэтиленгликоля. Это связано
с тем, что скорость реакции окиси этилена с водой меньше, чем ско-
рость реакции ее с образующимися гликолями (рис. 25) [85].
Для расчета состава продуктов оксиэтилирования воды пред-
ложено несколько уравнений [85, 91], например:
v = - = с1пД- (с-1) (1 - - 'J
п п ' \ п J
П[ _ С
~~ (С-1У
80
где и — число молей присоединенной окиси этилена (т) на моль воды;
п- — число молей продукта с числом оксиэтильных групп, рав-
ным п, п' — число молей воды в исходной смеси п в продуктах
реакции; CL — коэффициент распределения, равный отношению кон-
стант скоростей г-той (4,) и первой стадии реакции (Аг0).
Между коэффициентом распределения С и кислотностью реагиру-
ющих продуктов имеется епмбатная зависимость, которая описы-
вается уравнением типа Бренстеда [84, 89]: с =ki/k0^-g (К,7К0)а
где К0, К- — кислотности воды и соответствующего гликоля, рав-
ные [92]:
Вода .................1,2
Этиленгликоль .... 43
Дпэтпленглпколь .... 33
Т риэтиленглпкоа i>
Тетраэтиленгликоль
19
18
g п а — постоянные, которые при оксиэтилировании воды равны:
Некаталптическая реакция............... 1,11 0,22
Кислотный катализ ..................... 1 0,13
Основной катализ ...................... 1,46 0,65
При оксиэтилировании гликолей g = 1, а значения а те же, что
и для воды в зависимости от типа реакции.
Коэффициенты распределения Ci для последовательно-параллель-
ных реакций окспэтилирования воды и этиленгликоля при различ-
ных типах катализа имеют следующие значения (табл. 31) [89, 85].
Таблица 31. Коэффициенты распределения для реакций
оксиэтилнрованпя воды и этиленгликоля
Тип реакции 1\ оэффициенты распределения
с, с2 Сз Сг
Исходный р е а гонт — вода1
Не каталитическая реакция 2,8 2,3 2,04 2,01
Кислотный катализ .... 1,59 1,55 1,43 1,42
Основной катализ .... 14,8 12,6 8,76 8,46
Исходный реагент — этиленгликоль
Некаталптическая реакция 0,936 0,835 0,826 —
Кислотный катализ .... 0,964 0,900 0,893 —
Основной катализ .... 0,872 0,590 0,567 —
Температура и давление, по крайней мере, в тех пределах, в ко-
торых проводится процесс гидратации, не оказывают существенного
влияния па состав и выход продуктов реакции. Этот вывод подтвер-
6 Заказ 972 • '
ждается рис. 26 [86] и данными табл. 27 при иекаталитической
реакции, а также следующими данными, полученными при кислотном
катализе (0,5% H2SO4, отношение окиси этилена к воде ^0,375) [88]:
Температура, °G Выход, % . . . 90—95 70—75 50-55
этиленгликоля .... . . . 51,6 51,6 57,0
диэтпленгликоля . . . . . . 33,0 33,0 32,3
триэтилепгликоля . . . . . 10,4 10,4 5,7
Выход гликолей при кислотном
при нормальном давлении, и при
катализе, который проводился
иекаталитической гидратации,
Рис. 26. Зависимость выхода этиленгликоля от температуры при различном
мольном отношении окиси этилена к воде:
1 — 1 : 14,7; 2 — 1 : 4,9; 3 — 1 : 2,5; 4 — 1:1.
Рис. 25. Зависимость выхода этилен- и диэтпленгликоля от степени конверсии
окиси этилена (мольное отношение окись этилена : вода- 1 : 14,7'1
проводившейся при 0,83 МПа (8,4 кгс/см2) [73] и 1,96 МПа
(20 кгс/см2) [85], близкий для одного и того же отношения окиси
этилена к воде. Независимость выхода гликолей от давления при
равной концентрации окиси этилена в исходной реакционной смеси
подтверждается и производственными данными [50, 93].
Математическое моделирование процесса гидратации окиси эти-
лена впервые дано в работе [94]. В основу разработки математиче-
ской модели приняты следующие положения: температура, давление
и количество катализатора (H2SO4) не влияют на состав продуктов
гидратации; состав последних определяется отношением окиси эти-
лена и воды в реакционной смеси.
На аналоговой машине методом проб и ошибок по известному
из экспериментальных данных составу продуктов гидратации окиси
этилена и константы скорости реакции образования этиленгликоля кг
определены константы скорости реакции образования диэтилен-
82
гликоля (из этиленгликоля и окиси этилена) — к2, триэтиленгликоля
(из диэтиленгликоля и окиси этилена) — к3 и тетраэтиленгликоля
(из триэтилепгликоля и окиси этилена) — ki. Найдено, что
кг : к2 : к3 : к4 = 1,0 : 2,1 : 2,2 : 1,9, т. е. константы скорости реак-
ций образования дп-, три- и тетраэтиленглпколя близки между
собой и в среднем в 2 раза больше константы скорости реакции
образования этиленгликоля. Эти данные близки к полученным при
некаталитической реакции, при которой : к2 : кя = 1 : 1,63:
: 1,66 [86].
Рис. 27. Зависимость селективности процесса гидратации окиси этилена от
концентрации воды в исходной смеси (а) и от степени ее конверсии (5) в реакто-
рах различного типа:
а) 1—тарельчатый; 2—вытеснения; з—перемешивания; б) I—перемешивания; 2—промыш-
ленный; 3—вытеснения; 4—периодического действия.
С помощью разработанной кинетической модели проведено моде-
лирование материального и теплового балансов реактора гидратации.
В реакторе идеального перемешивания при кислотном катализе
уравнение материального баланса имеет следующее выражение:
Гр (dxs ’dx) ~ v'x't (г' -г v") xt — г/М/Г
где V — объем реактора; р — плотность; v' и v" — скорость подачи
водных растворов окиси этилена и серной кислоты; xt, х'-, х{ — доли
i-того компонента на выходе из реактора и в водных растворах
окиси этилена и серной кислоты; т — время реакции; г(- — скорость
реакции i-того компонента; М,: — молекулярная масса г-того ком-
понента.
Математическое моделирование процесса гидратации в реакторах
различного типа (идеального перемешивания, идеального вытеснения
и в реакторе типа ректификационной колонны, в которой на каждой
тарелке должно проходить быстрое разделение непрореагировавшеи
6* S3
окиси этилена и раствора гликолей) показало, что наибольшая
селективность процесса достигается в реакторе идеального вытесне-
ния [95]. При большом избытке воды (мольное соотношение окись
этилена : вода = 1 : 9 и выше) разница в селективности в реакторах
идеального вытеснения и идеального перемешивания уменьшается
(рис. 27, а). Однако увеличение избытка воды является невыгодным,
так как приводит к повышению энергетических расходов и снижению
производительности оборудования. Селективность процесса в реак-
торах различного типа в сравнении с имеющимися промышленным
приведена на рис. 27, б. Математическая модель установки получения
этиленгликоля, включая стадии приготовления исходной шихты,
гидратации и ректификации, полученная методом динамического
программирования, приведена в работе [96].
Тепловой эффект реакции окиси этилена с водой составляет
96,3 кДж/моль (23 ккал/моль) [86]. Эта величина определена по
стандартным теплотам образования окиси этилена, воды и этилен-
гликоля. Для окиси этилена (газ) она равна 51,04 кДж/моль
(12,19 ккал/моль), а для этиленгликоля, в зависимости от концентра-
ции водного раствора, она составляет [97, р. 122]:
Число молей
воды на
1 моль
гликоля
Теплота образования
(жидкого), кДж/моль
(ккал/моль)
0 —454,6 (—108,58)
0,5 —455,4 (—108,76)
1 —455,9 (—108,88)
5 —458,4 (—109,50)
Число молей воды на 1 моль гликоля Теплота образования (жидкого), кДж/моль (ккал/моль)
10 -459,6 (-109,77)
15 —460,0 (—109,88)
25 —460,4 (—109,97)
50 -460,7 (-110,04)
Технологическая схема производства этиленгликоля
некаталптической гидратацией окиси этилена
Получение этиленгликоля в промышленных условиях включает
следующие стадии: приготовление исходного водного раствора окиси
этилена в воде, гидратация окиси этилена, упаривание водного
раствора этиленгликоля и ректификация раствора гликолей с вы-
делением товарных продуктов (моноэтиленгликоля и побочно полу-
чаемых ди- и триэтиленгликоля). Технологическая схема производ-
ства этиленгликоля мощностью около 3 т/ч (Германия, Anorgana).
показана на рис. 28 [93, р. 28].
Окись этилена, предварительно охлажденная до температуры
около —10 °C, из сборника 1 насосом 2 подается в смеситель 3. Верх-
няя его часть представляет собой насадочную колонну, заполненную
кольцами Рашига (15 X 15 мм), и служит для абсорбции паров
окиси этилена, которые могут выделиться при неполном поглощении
ее водой в нижней части смесителя.
В верхнюю часть смесителя из сборника 5 насосом подается
водяной конденсат с температурой около 20 °C, полученный после
84
1 5 9 12, 13, 19, 20, 21, 22 — сборшпш; 2, I — насосы; 3 — смесит», ль, 6 — тснлообмепн::.; 7 - гдаигор;
10, 23 — подогреватели; 11, 17 — выпарные аппараты; 74, 15, 16, 18 — колонны.
выпаривания и ректификации слабого раствора гликоля. Соот-
ношение окиси этилена и воды поддерживается равным 1 : 6 (по
объему).
Шихта из смесителя 3, содержащая 13% окиси этилена, с по-
мощью насоса 4 подается при- давлении до 2,45 МПа (25 кгс/см2)
через теплообменник 6 в гидрататор 7. В теплообменнике 6 шихта
подогревается реакционной жидкостью, выходящей из гидрататора,
до 160—180 °C, при этом сама реакционная жидкость охлаждается
до 90 °C. Температуру шихты, поступающей в гидрататор, можно
менять за счет пропускания части ее помимо теплообменника.
Гидрататор представляет собой толстостенный сосуд, рассчитанный для
работы при высоком давлении и повышенной температуре. Внутри аппарата
размещены 2 змеевика для подогрева или охлаждения реакционной массы.
Внутренний диаметр гидрататора 810 мм, высота 10 м. Гидрататор вначале был
заполнен стальными кольцамгг рапптга 35 х 35 мм, чтобы агрессивные продукты,
содержащиеся в реакционной массе, в первую очередь действовали па развитую
поверхность колец, а пе на поверхность гидрататора. В дальнейшем кольца
были удалены из-за малой эффективности их действия.
Рабочее давление в гидрататоре 1,5—2,1 МПа (15—21 кгс/см2),
температура 200—210 °C в верхней части и около 180 °C в нижней
части гидрататора (на некоторых заводах гидратация проводилась
при 160—180 °C и 1,1—1,3 МПа (11 —13 кгс/см2). Продолжительность
процесса около 40 мин.
Реакционная жидкость из гидрататора перед поступлением в рас-
ширитель 8 дросселируется и при этом из нее выделяется часть
газов — ацетальдегид, кротоновый альдегид и др. Затем жидкость,
в которую непрерывно вводится 30%-ный раствор щелочи для под-
держания pH на уровне 7—8, попадает в емкость 9.
Слабый раствор гликолей из емкости 9 с помощью насоса через
подогреватель 10 подается на трехкорпусную выпарную установку 11.
Подогреватель обогревается вторичным паром, выходящим из по-
следнего корпуса. Конденсат из подогревателя собирается в сбор-
нике 5.
В первом корпусе раствор нагревается до 214 °C паром 2,1 МПа
(21 кгс/см2). Выходящий из этого корпуса пар давлением 0,8—
0,9 МПа (8—9 кгс/см2) и температурой 170 °C поступает на обогрев
второго корпуса. Второй корпус работает при давлении 0,3—0,4 МПа
(3—4 кгс/см2). и раствор в нем нагревается до 170 °C; пары воды из
этого корпуса с температурой 129 °C поступают на обогрев третьего
корпуса. Последний работает при давлении 66,7 кПа (500 мм рт. ст.),
и выходящий из него сырой концентрированный гликоль содержит
5—15% воды; он собирается в сборнике 12.
Для интенсификации выпаривания применена принудительная
циркуляция вязкого раствора гликолей центробежным насосом.
Из сборника 12 гликоль с помощью насоса прокачивается через
напорный сборник 13 и подается в ректификационную колонну 14
для полной отгонки воды. Колонна работает при остаточном давлении
вверху 1,3 кПа (10 мм рт. ст.). Колонна диаметром 2000 мм имеет
86
т
г 31 колпачковую тарелку, расстояние между ними 250 мм. Питание
вводится на 9, 11, 13, 15 и 19 тарелки, считая снизу.
Дистиллят колонны 14 собирается в сборнике 5 л используется
для приготовления водно-оксидной шихты. Обезвоженный гликоль-
сырец из куба колонны 14 с температурой 145 °C подается насосом
в колонны 15 (в схеме предусмотрены две колонны) для получения
этиленглпколя-ректпфиката (характеристика этих колонн такая
же, как у колонны 14). Ректификация проводится при остаточном
давлении 0,7 кПа (5 мм рт. ст.) вверху колонны, что соответствует
температуре 80 °C. Товарный этиленгликоль собирается в сбор-
нике 19. Кубовая жидкость колонны 15 с температурой 195 °C насо-
сом подается в колонну 16 диаметром 1600 мм. Колонна имеет 33 та-
релки, расстояние между ними 333 мм. Питание подается на 6, 8, 10,
12 м 14 тарелки, считая снизу.
Сверху колонны при остаточном давлении 0,5 кПа (3,5 мм рт. ст.),
что соответствует температуре около 115 °C, отбирается товарный
диэтиленгликоль, который собирается в сборнике 20. Кубовый
остаток колонны 16 направляют па склад и, по мере накопления,
перерабатывают на двух последовательно работающих насадочных
колоннах (18-1 и 18-11) при остаточном давлении 0,7 кПа
(5 мм рт. ст.). Диаметр колонн 800 мм, высота 6,5 м, насадка —
кольца Рашига 50 X 50 мм высотой слоя 3,5 м. Каждая
колонна имеет нагревательный змеевик п по два выносных
выпарных аппарата 17, обогреваемых паром с давлением 2,1 МПа
(21 кгс/см2).
Кубовая жидкость колонны 16 насосом через подогреватель 23
подается в первый выпарной аппарат 17, из которого пары гликолей
поступают в колонну 18-1. Дистиллят этой колонны, содержащий
некоторое количество диэтиленгликоля, возвращается в сборник 12
концентрированного гликоля-сырца, а триэтиленгликоль в смеси
с более высокомолекулярными гликолями из нижней части колонны
насосом подается во второй выпарной аппарат 17. Пары из этого
аппарата поступают во вторую колонну 18-11, дистиллят которой
является товарным триэтиленгликолем и собирается в сборнике 21.
Кубовые остатки колонны 18-11 — технический тетраэтилепгликоль
собирается в сборнике 22. Выпарные аппараты 17 работают пери-
одически. Перед сливом полигликолей аппараты продувают водяным
паром (для отдувки остатков три- и тетраэтнленгликолей).
Практически вся аппаратура описанной установки для получения
этиленгликоля была изготовлена из углеродистой стали. Вакуум
в колоннах создавался индивидуальными паро-эжекционными уста-
новками. На такой установке с двумя параллельно работающими
гидрататорами (один — внутренним диаметром 810 мм и второй —
/60 мм) перерабатывали 2500 кг/ч окиси этилена и получали 3197 кг/ч
гликолей, в том числе: этиленгликоля — 2830 кг (88,5%), диэти-
ленгликоля— 296 кг (9,3%) и триэтиленгликоля— 71 кг (2,2%).
Суммарный выход гликолей составляет от 92.5 до 95—96% от теоре-
тического.
87
Расход сырья, материалов и энергетических средств на 1 т эти-
ленгликоля и выход побочных продуктов составляли:
Окись этилена, кг Охлаждающая вода, мэ . По данным [93] 869 По данным [98] 860
320 240
Пар высокого давления, т . . . . 6,6 4,5 *
Пар низкого давления, т 0,3 0,15
Электроэнергия, кВч 63 20
Сжатый воздух, мэ ........ Азот,м3 Диэтиленгликоль, кг 14 12 105
Триэтилеигликоль, кг 25
* Пар среднего давления.
По схеме получения этиленгликоля Dow Chemical of Canada [991
окись этилена и вода в соотношении 1 : 8 смешиваются под давле-
нием, и смесь подогревается до 100 °C. За счет теплоты реакции
температура в гидрататоре повышается до 165—185 °C. Первый
выпарной аппарат трехкорпусной выпарпой установки работает при
0,98 МПа (10 кгс/см2), а третий — при 9.8 кПа (0,1 кгс/см2).
Этиленгликоль высокой степени чистоты, предназначенный для
производства синтетического волокна, выделяется в колонне высо-
той 24,4 м. Верхняя часть колонны выполнена из высоколегирован-
ной стали, чтобы исключить попадание железа в этиленгликоль.
С целью получения продукта с малым количеством примесей и вы-
кипающего в узких пределах, этиленгликоль отбирается с одной
из верхних тарелок ректификационной колонны (а не сверху).
Описанная технологическая схема получения этиленгликоля
(Апогдаиа. Германия) в принципе сохранилась до сих пор, однако
в нее внесены существенные изменения и улучшения по аппаратур-
ному оформлению процесса. Значительно повысилась и мощность
установок: в пастоящее время единичная мощность агрегата для
получения этиленгликоля составляет 100 тыс. т (и более) в год.
На современных установках аппаратура, как правило, изготовляется
из высоколегированной стали, что не только резко увеличивает
межремонтные пробеги аппаратов, но и обеспечивает получение
продуктов более высокого качества. Окись этилена с водой смеши-
вается непосредственно в трубопроводе.
В связи с тем что в аппаратах типа идеального перемешивания
селективность процесса гидратации ниже, чем в аппаратах идеаль-
ного вытеснения, наблюдается тенденция проводить гидратацию
в трубчатых аппаратах с большим числом последовательно соеди-
ненных секций. Для уменьшения уноса этиленгликоля с вторичным
паром на выпарных аппаратах устанавливают колонны с небольшим
числом ректификационных тарелок, а для снижения расхода пара
на выпаривание на ряде установок число корпусов увеличено до
четырех.
Для повышения качества и снижения возможности разложения
88
гликолей при их выделении применяют пленочные и роторные аппа-
раты, а также принудительную циркуляцию кубовой жидкости.
Это позволяет понизить температуру кипения и время пребывания
гликолей в испарителях и кипятильниках, улучшить коэффициент
теплопередачи.
В связи с тем что к качеству этиленгликоля, который потреб-
ляется для производства полиэфирных волокон и пленок, предъ-
являются весьма высокие требования, разработан ряд мер с целью
повышения качества этиленгликоля (особенно этиленгликоля, кото-
рый получается в виде разбавленного водного раствора при выделе-
нии окиси этилена из контактных газов). Количество этого этилен-
гликоля может быть весьма значительным — до 20% от получаемой
окиси этилена.
Этиленгликоль, полученный как побочный продукт при произ-
водстве окиси этилена, может содержать такие примеси, как альде-
гиды, ацетали, сложные эфиры, перекиси, полимерные продукты
и др. Некоторые из них придают продукту неприятный запах и
окраску при нагревании этиленгликоля. Такой этиленгликоль можно
использовать для получения антифриза, в качестве теплоносителя,
для осушки газов, однако его нельзя применять в производстве
синтетических волокон и пленок, так как содержащиеся в нем при-
меси усложняют процесс получения полиэфиров и ухудшают каче-
ство товарных продуктов — волокон и пленок.
Для очистки этиленгликоля, а также других гликолей от нежела-
тельных примесей предложены различные методы: дистилляция
водных растворов, обработка щелочью, кислотой, активированным
углем, отбеливающими глинами или ионообменными слюдами, ги-
дрирование и т. п. Согласно [100], разбавленный («*20%) водный
раствор этиленгликоля, который получается как побочный продукт
в производстве окиси этилена, концентрируется до «* 85%, под-
щелачивается 45%-ным раствором NaOH до pH «* 13 (с целью
превращения некоторых веществ в нелетучие) и подается в колонну
предварительной дистилляции. Здесь при остаточном давлении 6,7—
12,0 кПа (50—90 мм рт. ст.) и температуре в кубе колонны 130—
160 °C отгоняется «*98% раствора, а полимеры и другие нелетучие
примеси выводятся с низа колонны.
Дистиллят колонны поступает в колонну обезвоживания для
отгонки воды и летучих примесей при остаточном давлении 13,3—
17,3 кПа (100—130 мм рт. ст.), а безводные гликоли из куба колонны
передаются на следующую колонну. В этой колонне выделяется
чистый этиленгликоль при остаточном давлении 2,7 кПа
(20 мм рт. ст.), а кубовые остатки направляются на выделение ди-
и триэтиленгликоля. Полученный этиленгликоль выкипает при
197,0—197,4 °C, содержит 0,03—0,04% воды, 0,0008% уксусной
кислоты и не имеет неприятного запаха. Цвет его по Pt—Со шкале
<5, причем он мало меняется при кипячении с добавкой соляной
кислоты, и по всем показателям полученный продукт отвечает тре-
бованиям производства полиэтилентерефталата.
89
По другому способу [101], этиленгликоль, полученный в каче-
стве побочного продукта при производстве окиси этилена, подщела-
чивается и перегоняется при пониженном давлении для отделения
от тяжелокипящих примесей. Вода отгоняется вместе с легкокипя-
щпми примесями, а кубовый остаток при 80 °C обрабатывается акти-
вированным углем, после чего подвергается ректификации при
температуре в кубе колонны не выше 200 °C (лучше при 180 °C).
Полученный этиленгликоль пригоден для производства синтети-
ческого волокна. Очистку этиленгликоля можно также проводить
методом противоточной дистилляции с водяным паром.
Для уменьшения в этиленгликоле содержания железа и других
металлов, а также слабых органических кислот его пропускают
через сильнокислотный Н-катионит (например, сульфированный
полистирол, модифицированный дивинилбензолом). В результате
такой очистки содержание железа снижается в 10 раз (с 8-10-5 до
7 -10-6%), золы в 5—10 раз (с 0,0005—0,001 до 0,0001%) и кислот-
ность в 1,5 раза (с 0,0035 до 0,0021%). Регенерацию смолы проводят
после отложения на ней 3—4% железа 20%-ной серной кислотой
и последующей промывкой обессоленной водой [102].
Цветность этиленглпколя может быть резко уменьшена при
перегонке его в вакууме в присутствии окислов меди, магния, ртути,
никеля, железа или свинца. Лучшие результаты получаются при
применении 0,001—0,5% (от массы гликоля) окиси свинца. Цвет
этиленгликоля можно улучшить (снизить с 80 до с' 10 по Pt — Со
шкале), пропуская его при 80—100 °C через слой сильнокислотного
катионита (леватит 5-115) [103].
Предложен способ очистки гликолей методом гидрирования [104].
Гликоль, содержащий 5 — 10% воды, гидрируется при температуре
100—200 °C и давлении выше 19,6 МПа (200 кгс/см2) в присутствии
катализатора, содержащего 12—20 ч. Ni, 3—7 ч. Си и 0,2—1 ч. Мп
или Ст, нанесенных на носитель (пемза, кизельгур), или без пего.
После гидрирования и фильтрования цветность этиленгликоля
равна нулю.
Этиленгликоль высокой степени чистоты получается при
ректификации сырого продукта, к которому добавлены неболь-
шие количества га-амипофенола или а- либо Р-нафтола [105].
Для снижения содержания альдегидов в техническом этилен-
гликоле рекомендуется разбавить его водой с небольшим коли-
чеством соляной кислоты, продукт выпарить и подвергнуть ректи-
фикации [106].
Очистка этиленглпколя. При многократном использовании этилен-
гликоля в нем накапливаются примеси, затрудняющие дальнейшее
его употребление. Например, возвратный этиленгликоль в произ-
водстве полиэтилентерефталата содержит ряд примесей: метиловый
спирт, воду, диметилтерефталат, высшие эфиры, ацетали, остатки
катализатора, а также примеси, содержащиеся в исходном гликоле
(диэтиленгликоль, ацетальдегид и др.). Для очистки от этих при-
месей предложена следующая схема [107]. Возвратный гликоль
90
испаряется при остаточном давлении — 26,7 кПа (200 мм рт. ст.)
и температуре ниже 175 °C.
Пары отделяются в сепараторе, конденсируются и поступают
на 24-ю тарелку ректификационной колонны, а в жидком этилен-
гликоле остается около 20% твердых примесей (катализатор, соли
терефталевой кислоты). В колонне при таком же остаточном давле-
нии, температуре 165—170 °C внизу и 90—100 °C наверху отго-
няются легкокппящие продукты (вода, метиловый спирт, ацеталь-
дегид), а с 10-й тарелки колонны выводятся пары этиленгликоля.
Полученный этиленгликоль (99,9%-ный) пригоден для производства
антифриза. Для получения продукта еще более высокого качества
этиленгликоль пропускается через активированный уголь, после
чего он вполне отвечает требованиям производства полиэтилен-
терефталата.
Для полноты регенерации этиленгликоля жидкость из сепара-
тора испаряется при остаточном давлении 6,7 кПа (50 мм рт. ст.)
и температуре 125—130 °C, пары конденсируются и конденсат при-
соединяется к возвратному этиленгликолю, поступающему на очи-
стку, а отходы, содержащие 10% этиленгликоля, выводятся из
системы.
Возвратный этиленгликоль в производстве полиэфиров можно
очистить методом обработки его в течение 15 мин при 60 С концен-
трированной серной или фосфорной кислотой, взятых в количестве
0,2%, последующей нейтрализации содой и ректификации. Цветность
очищенного продукта составляет 20—50 против 120—5000 исход-
ного [108].
По другому способу [109], этиленгликоль, загрязненный в произ-
водстве полиэфиров, вначале разбавляется водой (0,1—0,5 ч. воды
на 1ч. гликоля), подкисляется минеральной кислотой (H2SO4,
Н3РО4, НС1) до pH 2 для разложения солей терефталевой кислоты,
и осадок терефталевой кислоты отфильтровывается через слой инфу-
зорной земли. Фильтрат (разбавленный раствор этиленгликоля)
подается в колонну, в которой отгоняется вода при нормальном
давлении и температуре в кубе до 180 °C. При этом тяжелокипящие
ацетали гидролизуются в легкокипящие продукты, отгоняющиеся
в виде азеотропных смесей вместе с водой. После отгонки воды кубо-
вый остаток колонны нейтрализуют щелочью до pH 7,5—8 и под-
вергают ректификации при пониженном давлении. Полученный эти-
ленгликоль (кислотность 0,003% и цветность 5) вполне годен для
получения антифриза, а также для других целей.
Этиленгликоль с концентрацией 99% получается'также следу-
ющим образом: возвратный этиленгликоль (80—90%-ный) в резуль-
тате ректификации освобождается от метилового спирта; к кубовому
остатку добавляется от 2 до 25% воды. Водный раствор этилен-
гликоля подвергается ректификации при остаточном давлении
13,3 кПа (100 мм рт. ст.) для удаления вместе с водой легкокипящих
примесей. Затем при остаточном давлении 0,13 кПа (1 мм рт. ст.)
выделяется этиленгликоль [110].
91
Отработанный этиленгликоль может быть очищен при кипячении
в течение 20 мин. после добавки к нему 10% воды и 0,001—0,1%
окиси магния с последующей ректификацией [111].
Комбинированная очистка этиленглпкол*я обработкой водой и
отбеливающими глинами заключается в том, что [112] загрязненный
гликоль смешивается с водой в соотношении 1 : 10—1 : 50, воду
отгоняют при пониженном давлении, при этом с ней удаляется ряд
примесей, а к кубовому остатку добавляют 0,2—0,3% обработанной
минеральной кислотой порошкообразной отбеливающей глины. Затем
гликоль перегоняется при остаточном давлении 1,3—13,3 кПа (10—
100 мм рт. ст.). В полученном гликоле содержание альдегидов сни-
жается более чем в 10 раз и составляет 0,0001—0,0002% против
0,0038% в неочищенном продукте.
Этиленгликоль, загрязненный метилвппилкетоном и нитрилом
молочной кислоты, после очистки им акрилонитрила рекомендуется
регенерировать при нагревании (140—180 °C) в присутствии толуол-
сульфокислоты. Затем раствор нейтрализуется безводными основа-
ниями и подвергается ректификации [113].
Очистку этиленгликоля от формальдегида предложено проводить
за счет перевода формальдегида в формаль, например при пропуска-
нии загрязненного гликоля через сильнокислую ионообменную
смолу при 70 °C, и последующем выделении чистого гликоля ректи-
фикацией. Формаль при этом выделяется в смеси с водой [114].
Очистку этиленгликоля от пропиленгликоля рекомендуется про-
водить при нагревании до 140—160 °C с дегидратирующими катали-
заторами, в присутствии которых пропиленгликоль превращается
в пропионовый альдегид и 2,6-диметил-1,4-диоксан, легко отгоня-
ющиеся вместе с водой [115].
Получение этиленгликоля кислотно-каталитической
гидратацией окиси этилена
Кислотный катализ позволяет проводить процесс гидратации при
умеренных температурах (50—70 °C) и низком давлении и тем самым
упростить аппаратурное оформление процесса. С целью увеличения
скорости реакции кислотный катализ можно проводить и при повы-
шенной температуре (180—200 °C). В этом случае давление в реакторе
будет высоким, 1,5—2,0 МПа (15—20 кгс/см2), но объем реактора
во много раз уменьшится, его конструкция упростится, масса и сто-
имость снизятся. Однако применение кислот имеет и ряд отрица-
тельных сторон, главными из которых являются коррозия обору-
дования и необходимость в дополнительной аппаратуре для нейтра-
лизации кислоты, выделение образующихся при нейтрализации
кислоты солей или выделение гликолей из солевых растворов.
Для ускорения гидратации окиси этилена можно применять
различные минеральные и органические кислоты: серную, фосфор-
ную, щавелевую и др. Выбор кислоты в качестве катализатора опре-
деляется рядом факторов, из которых степень ее агрессивности
92
к материалам аппаратуры, легкость выделения кислоты или обра-
зующейся после ее нейтрализации соли из реакционного раствора
являются основными. Кислотно-каталитическая гидратация окиси
этилена нашла применение в промышленности [116—118], причем
в качестве катализатора чаще всего употребляются серная и фосфор-
ная кислоты.
По одному из первых патентов [118] процесс проводится в авто-
клаве при температуре ниже 100 °C в присутствии 0,2% H2SO4.
Для получения минимального количества полигликолей рекомен-
дуется брать 20-кратный (в молях) избыток воды. Полученный рас-
твор гликоля нейтрализуется щелочью, содой или мелом.
Этиленгликоль может быть получен гидратацией окиси этилена,
находящейся в контактных газах [118]. Процесс проводится в наса-
дочном скруббере, в нижнюю часть которого подаются контактные
газы после реактора окисления этилена в окись этилена, а в верхнюю
часть — циркулирующий водный раствор, содержащий кислотный
катализатор. Часть раствора с образовавшимся гликолем выводится
из системы и заменяется свежим. В присутствии 0,5% H2SO4 при
температуре 50—70 °C гидратация проходит за 30 мни, а в присут-
ствии 0,5% HCI и температуре 100 °C — за 20 мин. Принципиальная
схема непрерывного способа получения этиленгликоля в присутствии
серной кислоты состоит в следующем.
Окись этилена или 20%-ный (по объему) водный раствор ее охла-
ждается в холодильнике до температуры ниже 10 °C и затем посту-
пает в гидрататор колонного типа, куда подается также вода, под-
кисленная серной кислотой. Для снижения образования полигли-
колей вода подается в таком количестве, чтобы концентрация
полученного гликоля составила 10%. Содержание серной кислоты
в реакционном растворе составляет 0,1—0,5%. Смешение водных
растворов окиси этилена и серной кислоты происходит на перфориро-
ванных свинцовых тарелках, расположенных в верхней части гидра-
татора. Остальная часть аппарата заполнена насадкой. Вытекающий
с низа гидрататора раствор поступает в реакционную камеру, и здесь
завершается конверсия окиси этилена. Окись этилена, которая
может испариться вследствие повышения температуры за счет экзо-
термичности реакции, конденсируется в конденсаторе и возвра-
щается в гидрататор.
Полученный тем или иным способом кислый раствор этиленгли-
коля нейтрализуется щелочью, содой, мелом или другими веще-
ствами основного характера. Выделение гликоля из водного рас-
твора, содержащего соли, довольно затруднительно, особенно если
часть кислоты (например, серная кислота) образует с окисью эти-
лена соединения, как, например, этиленгликольсульфат. Для раз-
ложения последнего необходима обработка раствора избыточным
количеством щелочи, подогрев и выдержка нагретого раствора
и повторная его нейтрализация кислотой. Нейтрализованный таким
образом водный раствор этиленгликоля, содержащий сульфат на-
трия, подается в трехкорпусную вакуумную выпарную установку
93
для отгонки основной массы воды и затем на ректификацию. По мере
концентрирования раствора из него начинает выпадать сульфат
натрия, который, осаждаясь на теплопередающей поверхности,
снижает коэффициент теплопередачи. В связи с этим возникает
необходимость в устройстве солеотделителей и в периодических
остановках для очистки от соли аппаратуры и трубопроводов. Вместе
с солями теряется некоторое количество этиленгликоля.
Для исключения указанных выше затруднений предложена
очистка раствора гликоля от кислоты с помощью анионитов, на-
пример, амберлита /7?-4, анекса. при комнатной температуре [119].
Анионит сорбирует как свободную, так и связанную кислоту, поэтому
упаривание и ректификация очищенного раствора не представляют
трудностей. Активность отработанной анионообменной смолы вос-
станавливается в результате последовательной промывки ее водой
2—4%-ным раствором соды н снова водой.
Для упрощения технологии получения этиленгликоля при кис-
лотном катализе предложено использовать в качестве катализа-
торов такие кислоты, которые являются достаточно стойкими при
умеренной температуре гидратации, но разлагающимися на летучие
продукты при более высокой температуре упаривания и ректифика-
ции раствора гликолей, например трихлоруксусную кислоту [120].
Гидратацию проводят при 60 °C в противоточном скруббере, в ниж-
нюю часть которого подается окись этилена, а сверху — 0,5%-ный
раствор трпхлоруксусной кислоты. Вытекающий из скруббера вод-
ный раствор гликолей выдерживается при 100 °C, при этом три-
хлоруксусная кислота разлагается на хлороформ и двуокись угле-
рода (образуются также следы соляной кислоты). Далее раствор
нейтрализуется углекислым кальцием, упаривается и направляется
на ректификацию.
Предложенный процесс имеет такие недостатки, как образование
соляной кислоты и необходимость ее нейтрализации, дополнительные
операции для выделения хлороформа. Использование сернистого
ангидрида, который удаляется из реакционного раствора при от-
гонке, в известной мере исключает эти недостатки [121]. При моль-
ном отношении воды к окиси этилена, равном 10,5 ('^ 4,3 : 1 по
массе), концентрации SO2 в воде 0,1%, температуре 25—35 °C и атмо-
сферном давлении гидратация проходит за 4 ч с выходом этилен-
гликоля 87%. Данных о применении SO2 в качестве катализатора
в промышленности не имеется.
Одним из первых промышленных процессов получения этилен-
гликоля с применением серной кислоты в качестве катализатора
является следующий [117]. Абсорбция окиси этилена из газов пря-
мого каталитического окисления этилена производится водой, содер-
жащей 1% H2SO4, при давлении 0,3 МПа (3,2 кгс/см2) последова-
тельно в двух гуммированных насадочных колоннах. Одновременно
с абсорбцией протекает гидратация окиси этилена. По достижении
концентрации гликоля в растворе около 20% часть его непрерывно
выводится на дальнейшую переработку, а основная часть, в которую
94
вводится свежая вода с 1% H2SO4, возвращается на орошение
колонн.
Выведенный из системы раствор нейтрализуется каустической
содой и направляется на вакуум-выпаривание и обессоливание;
концентрация гликоля в растворе повышается до 90%. Полная
отгонка воды и выделение чистого этиленгликоля производятся
на непрерывнодействующих ректификационных колоннах, а ди-
и триэтиленгликоля — на периодической ректификационной колонне.
Ректификация гликолей протекает при остаточном давлении 8 кПа
(60—70 мм рт. ст.). Выход этиленгликоля составляет 80—85% от
суммы гликолей. На 1 т этиленгликоля в качестве побочных про-
дуктов образуется 138 кг диэтиленгликоля и 21 кг триэтиленгликоля.
По данным [68, р. 374], концентрация серной кислоты при гидра-
тации окиси этилена составляет 0,5—1%, температура процесса
50—70 °C, а продолжительность около 0,5 ч. Отношение воды к окиси
этилена равно 6 : 1 (по массе), расход окиси этилена на 1 т гликоля
составляет 816,5 кг. При этом выход этиленгликоля составляет
88—93% при практически полной конверсии окиси этилена. Серная
кислота удаляется из раствора при пропускании его через колонну
с анионитом.
Имеются сведения о применении в промышленных условиях
в качестве катализатора фосфорной кислоты, которая является
менее агрессивной, чем серная, а ее соли — менее коррозионными,
чем сернокислые соли. Кроме того, фосфорнокислые соли после их
выделения из раствора гликолей можно использовать как удобрения
или питательные вещества.
Гидратацию окиси этилена, как и при некаталитической реакции,
целесообразно проводить в аппаратах типа идеального вытеснения.
Для отделения гликоля от солей часто используются роторные
испарители, которые по сравнению с трубчатыми дают малый перепад
давления, и их поверхность меньше загрязняется отложениями
солей, что позволяет перегонять гликоли при более глубоком вакууме
и низкой температуре.
Гидратация окиси этилена в присутствии катионитов. Осуще-
ствление процесса гидратации в присутствии катионитов должно
было сохранить преимущества кислотно-каталитической гидратации
(большая скорость реакции при низкой температуре), устранив
при этом ее недостатки (коррозия, образование солей, необходимость
их отделения). Катализаторами реакции окиси этилена с водой
являются сильнокислотные катиониты: сульфополистиролыше
(77?-120 и КУ-2), сульфофенолоформальдегидный (КУ-1), активная
форма — SO3H, фосфорнокислый катионит (КФ-1), активная
форма — РО(ОН)2 [75, 76, 122]. На скорость гидратации окиси
этилена влияет не только природа, но и количество катионита, его
структура и размер частиц. Однако срок службы катионитов КУ-1
и КУ-2 оказался весьма небольшим, и уже через 5 — 6 ч их емкость
снижается па 70—80%, причем обработка катионитов соляной
кислотой практически не увеличивает емкость [122]. Отсутствие
95
в литературе данных о промышленном применении катионитов в ка-
честве катализаторов процесса гидратации окиси этилена или других
алкиленокспдов, вероятнее всего, объясняется нестабильностью их
свойств в водных растворах а-окисей олефинов.
Парофазпая гидратация окиси этилена. Процесс жидкофазной
гидратации окиси этилена, главным преимуществом которого яв-
ляется практически полная конверсия окиси этилена, имеет некото-
рые недостатки, а именно: некаталитическая гидратация осуще-
ствляется при повышенном давлении; при кислотном катализе
реакционная аппаратура должна изготавливаться из материалов,
стойких к агрессивному действию разбавленных кислот, кроме того,
требуется дополнительная аппаратура для нейтрализации кислоты
и для вывода из системы образующихся солей; большие энергети-
ческие затраты при концентрировании разбавленных растворов
этиленглпколя.
Предполагалось, что гидратация окиси этилена в паровой фазе
над твердыми катализаторами позволит устранить недостатки жидко-
фазной гидратации. В одной из первых работ [123] были испробованы
в качестве катализаторов фосфорная кислота, силикагель, окись
алюминия, окись тория и серебро, однако они оказались катализа-
торами изомеризации окиси этилена и неактивными катализаторами
ее гидратации. Более эффективным катализатором гидратации ока-
залась окись серебра, нанесенная па окись алюминия: при степени
конверсии окиси этилена, равной 22—24%, селективность превра-
щения ее в гликоль составила около 80%. При увеличении степени
конверсии до 40% селективность снижалась до 40%.
При применении в качестве катализатора цеолитов (температура
от 30 до 150 °C, мольное отношение воды к окиси этилена, равное
10 : 1) выход этиленгликоля достигает 90,5% при степени конверсии
окиси этилена лишь 11,3%. Повышение степени конверсии окиси
этилена до 17—24%) снижает выход этиленгликоля до 78—69% [124].
В качестве катализатора парофазпой гидратации окиси этилена
предложен катионит амберлит 7/7-120 [124]. Процесс рекомендуется
проводить при температуре от 115 до 200 °C, давлении от 0,14 до
0,55 МПа (1,4—5,6 кгс/см2) и мольном отношении паров воды и окиси
этилена от 5 до 20. Реакция протекает с большой скоростью (время
контакта — сотые и десятые доли секунды). При температуре 160—
165 °C и мольном отношении воды к окиси этилена, равном 20 : 1,
выход этиленгликоля достигает 89% при степени конверсии окиси
этилена около 14%. При отношении воды к окиси этилена, равном
(5—10) : 1, выход этиленгликоля составляет 80—85%, а степень
конверсии окиси этилена — максимум 17%.
Наиболее высокие показатели получены при применении в ка-
честве катализатора фосфатов кальция и меди: смеси Cag(PO4)2
и СаНРО4 с соотношением СаО к Р2О5, равным 2,7—2,8, содержащей
0,1 — 1% фосфата меди [1251. Катализатор получается совместным
осаждением фосфатов кальция и меди из растворов хлористых солей
этих металлов растворами ортофосфорной кислоты и гидроокиси
86
аммония. Осадок солей тщательно промывают, сушат, размалывают
и просеивают. Полученный порошок с добавкой графита формуется
в таблетки для проведения процесса в стационарном слое или же
определенные фракции катализатора используются для процесса
в псевдоожиженном слое.
В псевдоожиженном слое катализатора при мольном отношении
воды и окиси этилена, равном (6,4—6,8) : 1, времени контакта 7—14 с
и температуре 275—-299 °C селективность превращения окиси эти-
лена в этиленгликоль значительно выше, чем при жидкофазной
гидратации, и составляет 97—99,3% при степени конверсии 91,3—
94,3%. Уменьшение отношения воды к окиси этилена до 4 снижает
селективность на 10%, а дальнейшее снижение этого отношения до 2
приводит к резкому падению селективности до 56%. При этом сте-
пень конверсии практически не изменяется, оставаясь на уровне
90-93%.
В стационарном слое катализатора получены худшие показатели;
даже при очень большом отношении водяного пара к окиси этилена
(50,3 : 1) селективность не превышает 91%, т. е. на 7—8% ниже, чем
в псевдоожиженном слое; степень конверсии при этом достаточно
высокая (93—97%).
В качестве катализатора парофазнон гидратации окиси этилена
предложен также фосфат кальция [126]. При температуре 232 °C,
времени контакта 10,1 с, мольном отношении воды к окиси этилена,
равном 12,4 : 1, выход этиленгликоля составляет 95—97% на про-
реагировавшую и 37—40% па пропущенную окись этилена.
Процесс гидратации окиси этилена в паровой фазе имеет ряд
недостатков. При применении в качестве катализаторов цеолитов
и Н-катионптов высокая селективность достигается при большом
соотношении паров воды и окиси этилена и очень низкой степени
конверсии окиси этилена. Высокая селективность и конверсия при
малом отношении воды к окиси этилена получаются при проведении
процесса парофазной гидратации окиси этилена в псевдоожиженном
слое смешанного фосфатного катализатора (фосфатов кальция и
меди). Однако при этом весьма велико время контакта, и поэтому
в промышленных условиях потребуются большие количества катали-
затора и соответственно реакторы значительного объема. Отсутствие
данных по механической прочности и стабильности катализатора
не позволяет судить о практической возможности его использо-
вания.
Учитывая способность окиси этилена к полимеризации под вли-
янием различных веществ, можно полагать, что активность гетеро-
генных катализаторов по мере их эксплуатации будет снижаться
из-за отложения на них продуктов полимеризации или изомеризации
окиси этилена. По-видимому, этими причинами объясняется то, что
в литературе отсутствуют сведения об осуществлении процесса паро-
фазной гидратации окиси этилена в промышленных условиях.
I Заказ 972
97
Применение этиленгликоля
Области применения этиленгликоля исключительно разнообразны:
химическая, автомобильная, авиационная, электротехническая, тек-
стильная, нефтегазовая и другие отрасли промышленности. Впервые
в промышленном масштабе этиленгликоль был применен в Германии
в период первой мировой войны для производства динитрогликоля
(взрывчатого вещества) в связи с острым дефицитом глицерина для
получения динамита.
Этиленглнкольдпиптрат (нитрогликоль), который является сложным эфи-
ром этиленгликоля и азотной кислоты (CHaONO2 — CH2ONO2), обладает хо-
рошими желатинизирующими свойствами и большой мощностью, он значительно
менее чувствителен к удару, чем нитроглицерин. Температура плавления нитро-
гликоля равна—22,8 °C, тогда как нитроглицерина 13,2 °C. Эвтектическая смесь
нпгроглггколя и стабильной формы нитроглицерина (20%) кристаллизуется
при —30 °C, а температура кристаллизации смеси нитрогликоля (60%) и лабиль-
ной формы нитроглицерина (40%) равна —40 °C. Поэтому нитроглпколь исполь-
зуется для производства низкозамерзающих динамитов, которые содержат
смесь нптроглпколя и нитроглицерина в соотношении 1 : 1 и получаются при
нитровании смеси глицерина и гликоля. Вследствие большой летучести и силь-
ного физиологического действия нитрогликоль используется только как компо-
нент в незамерзающих динамитах. Применение последних в зимних условиях
значительно менее опасно, чем динамитов на основе одного нитроглицерина [127,
с. 621-624].
В США от начала промышленного производства этиленгликоля
в 20-х годах до середины 60-х годов основная масса этиленгликоля
(70—80%) расходовалась на получение антифризов. В последующие
годы доля его в производстве антифризов значительно снизилась
(табл. 32).
Таблица 32. Структура потребления этиленгликоля в США,
% |55; 128, е. 408; 129, р. 70, 130]
Область применения 1950 г. 1960 г. I960 г. 1968 г. 1972 г. 1974 г. 1980 г. (прог- ноз)
Антифризы . . . Синтетические во- локна Целлофан .... Взрывчатые веще- ства Гидравлические жидкости и хла- доносители . . . Полигликолп и эфиры Алкидные смолы Другие области применения и экспорт 73,2 0,1 4,4 5,7 2,8 3,2 1,4 9,2 79,0 3,1 0,8 2,0 2,8 2,0 11,3 71 6 12 ) 11 61,0 13 14,8 50,9 30,5 18,6 49,1 36,8 14,1 42,6 42,6 14,8
98
в последнее время во всех странах быстрыми темпами растет при-
менение этиленгликоля для получения синтетических волокон
и пленок.
Предполагается, что в США в ближайшие годы использование
этиленгликоля для производства антифризов будет возрастать только
на 3—4% в год, тогда как для получения полиэфирных волокон
и пленок прирост составит 12—15% в год [130].
Основные направления использования этиленгликоля в Японии
приведены ниже (в %) [131, с. 26]:
Область применения 1965 г. 1970 г. 197 5 г.
Антифризы 17,4 16,7 13,3
Полиэфирные волокна и пленки 64,8 61,0 67,0
Целлофан 7,7 6,4 4,3
Взрывчатые вещества 1,4 0,5 0,3
Ненасыщенные полиэфирные смолы 2,7 4,3 4,1
Хладоагенты • 1,1 1,0 0,7
Алкидные смолы, уретаны . . . . 4,9 2,9 2,7
Прочие . — 7,2 7,6
В Англии в 1965 г. структура потребления этиленгликоля была
следующая: 56% этиленгликоля расходовалось на получение анти-
фриза, 25% на полиэфирные волокна, 7% на целлофан и 12% на
другие продукты [129, р. 71]. Таким образом, на получение анти-
фризов, полиэфирных волокон и пленок потребляется 75—80%
производимого этиленгликоля. Примерно такое же положение и
в других странах.
Этиленгликоль широко применяется в качестве пластификатора
целлофана, бумаги, волокон и кожи, растворителя, высокотемпера-
турного теплоносителя и хладоносителя в холодильных системах
(в виде водных растворов), в производстве электролитических кон-
денсаторов, как компонент электролитов, содержащих бориую кис-
лоту и ее соли, при получении гидравлических, гидротормозных,
антиобледенительных и огнегасящих жидкостей, а также в ряде
других областей.
Этиленгликоль используется для синтеза различных химических
продуктов. Весьма важное значение имеют сложные эфиры этилен-
гликоля и двухосновных кислот, которые применяются как пласти-
фикаторы, а также в производстве клеев, лаков и смол.
При окислении этиленгликоля в паровой фазе над медным ката-
лизатором получают глиоксаль [132]. При взаимодействии этилен-
гликоля с параформом в присутствии катионообменной смолы обра-
зуется 1,3-диоксолан с выходом 90% [133]. При взаимодействии
этиленгликоля с пропаргиловым спиртом в присутствии красной
окиси ртути и эфирата бора получается 2-метил-2-оксиметил-1,3-ди-
оксолан с выходом 54,4% [134].
7*
99
Окислением этиленгликоля кислородом воздуха в среде жидкого
катализатора с хорошими выходами получается щавелевая кислота,
а из этиленгликоля, едкого натра (кали) и воды при повышенном
давлении и температуре получаются соли щавелевой кислоты [135].
При взаимодействии этиленгликоля с акролеином в присутствии
кислотного катализатора и ингибитора полимеризации получают
этиленгликольацеталь [136]. Из этиленгликоля и муравьинокислых
солей металлов I и II групп периодической системы получаются
гликоляты металлов [137].
Моиовиниловый эфир этиленгликоля образуется с высоким выхо-
дом из этиленгликоля и ацетилена в присутствии едкого кали, как
катализатора, по реакции Фаворского — Шостаковского. При гидра-
тации этого эфира почти с количественным выходом образуется
ацетальдегид и регенерируется этиленгликоль, возвращаемый на
синтез эфира [138].
Аминокарбинолы типа [H2N(CH2)„]2CHOH получаются в резуль-
тате реакции этиленгликоля с непредельными аминами [139]. При
нагревании этиленгликоля в жидкой фазе с аммиаком при 242—
248 С'С под давлением водорода 20,6 МПа (210 кгс/см2) образуется
пиперазин [140]. Этиленгликоль предложен в качестве исходного
сырья для получения бис(Р-оксиэтил)терефталата из нитрила тере-
фталевой кислоты [140]. Путем конденсации этиленгликоля с пиро-
мелитовым диангидридом образуется диэфир, который используется
в качестве отверждающего агента для эпоксидных смол [142].
Бис-р,р1-дикарбо-у-хлоркротилоксиэтиловый эфир этиленгли-
коля, являющийся пластификатором поливинилхлорида, получается
после обработки у-хлоркротпловым спиртом продукта взаимодей-
ствия этиленгликоля и акрилонитрила [143]. Эффективным стабили-
затором поливинилхлорида является продукт взаимодействия эти-
ленгликоля, малеинового ангидрида, окиси дибутилолова, моно-
додецил- и монобутилмалеатов [144].
N-Феноляты диоксиалкилдифеиилсулъфонов, получаемые в среде
этиленгликоля, являются стабилизаторами полиолефинов [145].
Композиция из этиленгликоля и гомополимера или сополимера
акриламида является пластификатором и растворителем при изгото-
влении из полимера пленки, волокна или клеев [146]. Органозоли
SiO2 в этиленгликоле могут быть применены как наполнители поли-
меров [147].
Этерификацией терефталевой кислоты смесью этиленгликоля
и диэтиленгликоля, а затем глицерином, получается электроизоля-
ционный термостойкий (до 155 °C) лак, обладающий хорошей адге-
зией и эластичностью и устойчивый к влаге и химическим
реагентам [148].
Этиленгликоль применяется для изготовления лекарственных
препаратов. Дипикотиновый эфир этиленгликоля, например, пони-
жает содержание холестерина и упрочняет кровеносные сосуды;
эфиры этиленгликоля и салициловой кислоты обладают противо-
лихорадочным и противоврспалительным действием, а стероидные
100
третичные эфиры этиленгликоля — антибактериальным действием.
Бактерицидной актнвностыо обладают эфиры этиленгликоля и суль-
фаметилфенилкарбаминовой кислоты [149].
Этиленгликоль используется при экстракции различных веществ
и очистке продуктов от примесей, например как селективный раство-
ритель для экстракции индена и кумарона из легких масел каменно-
угольной смолы, толуола из различных фракций, бутадиена из его
смеси с изобутиленом и другими углеводородами [150, р. 26]. .Эти-
ленгликоль может быть применен в качестве разделяющего агента
для выделения тетрагидрофурана из его водных растворов методом
экстрактивной ректификации [151]. Для разделения моно- и ди-
алкилфосфатов употребляется этиленгликоль с несмешивающимся
с ним углеводородом [152]. Этиленгликоль применяется для очистки
акрилонитрила от примесей карбонильных соединений, которые
образуют с ним высококипящие эфиры [153]. В смеси с mpem-бутило-
вым спиртом этиленгликоль предложен как растворитель при синтезе
адиподинитрила из 1,4-дихлорбутапа и цианида натрия [154].
Этиленгликоль используется в производстве гербицидов, а также
как среда при их применении [155]. С целью получения противо-
корневых препаратов этиленгликоль вместе с гербицидными веще-
ствами вводится в битумы для дорожных покрытий, уплотнения
трубных соединений, изоляции труб. Ацеталь хлораля в смеси
с этиленгликолем является гербицидным препаратом против одно-
дольных сорняков. Этиленгликоль служит эффективным стабили-
затором акарицидов. Свойства и способы получения некоторых
наиболее широко применяемых продуктов на основе этиленгликоля
даны ниже.
Антифризы
Крупнейшим потребителем этиленгликоля является производство
антифризов — низкозамерзающих жидкостей, применяемых для
охлаждения двигателей внутреннего сгорания автомобилей и других
машин, в качестве хладопосителя в рефрижераторах, теплоносителя
в теплообменной аппаратуре и т. д. Этиленгликолевый антифриз
используется также и в летний период, поскольку позволяет повы-
сить рабочую температуру жидкости в системе охлаждения и тем
самым увеличить температурный перепад между охлаждающей
жидкостью и окружающим воздухом, т. е. увеличить теплоотдачу
от двигателя. Указанные обстоятельства позволяют форсировать
работу двигателя и полнее использовать его мощность, а также
способствуют экономии топлива вследствие его более полного испа-
рения и сгорания.
К жидкости для охлаждения двигателей внутреннего сгорания предъ-
является совокупность таких требования [7, с. 673; 156, р. 540; 157]:
1) высокая теплоемкость и теплопроводность: чем выше теплоемкость, тем
больше тепла будет отведено от стенок цилиндра и головки блока данным коли-
чеством циркулирующей охлаждающей жидкости;
101
2) низкая температура замерзания, что имеет весьма важное значение для
большинства районов Советского Союза; применение антифриза с низкой темпе-
ратурой замерзания позволяет безопасно эксплуатировать автомашины практи-
чески при любых отрицательных температурах окружающего воздуха, не сливая
жидкость из радиаторов на время стоянок;
3) высокая температура кипепия, что обеспечивает нормальную работу
двигателя в летнее время, особенно в горных районах с пониженным атмосфер-
ным давлением, а также уменьшает вероятность образования паровых пробок,
приводящих к резкому ухудшению охлаждения двигателя и использованию
его мощности;
4) высокая температура воспламенения; этот показатель важен для обеспе-
чения безопасности при пользовании антифризом;
5) малая вязкость, особенно при низких температурах (высокая вязкость
антифриза затрудняет его циркуляцию в системе охлаждения и снижает тепло-
передачу от двигателя к жидкости и от нее в окружающую среду);
Конце^траци-Я, % (об.)
Рис. 29. Зависимость
температуры замерзания
водных растворов неко-
торых спиртов от их
концентрации:
1 — метиловый спирт; 2 —
этиленгликоль; з — глице-
рин.
,6 ) малая вспенпваемость (при вспенивании жидкости снижается коэффи-
циент теплопередачи, возможно образование паровых пробок, перегрев двига-
теля, а также выброс жидкости);
7) низкая коррозионная агрессивность по отношению к металлам, сплавам,
припоям и покрытиям, применяемым в системе охлаждения двигателя; этот
показатель является одним из решающих при оценке качества антифриза, так
как продукты коррозии могут ухудшить теплопередачу в системе охлаждения,
нарушить нормальную циркуляцию жидкости, не говоря уже о выводе из строя
элементов системы охлаждения и потере антифриза;
8) инертность по отношению к резиновым шлангам и уплотнительным дета-
лям из резины или пластических масс, применяемым в системе охлаждения,
а также инертность или слабое воздействие на лаковое покрытие машины;
9) отсутствие запаха или малый замах, низкая токсичность и, наконец,
доступная цена.
Для получения низкозамерзающих охлаждающих жидкостей,
кроме этиленгликоля, применялись также глицерин, метиловый,
этиловый и изопропиловый спирты. На рис. 29 приведены темпера-
туры замерзания водных растворов этиленгликоля, глицерина и ме-
тилового спирта [116, р. 62]. В настоящее время эти жидкости почти
полностью вытеснены этиленгликолем, так как по комплексу
•свойств — низкой температуре замерзания, большой теплоемкости,
102
высокой температуре кипения, относительно низкой вязкости, не-
высокой стоимости и ряду других показателей, этиленгликолевый
антифриз наиболее полно отвечает предъявляемым требованиям.
Основные компоненты антифриза (вода и этиленгликоль) спо-
собны вызывать коррозию железа и других металлов. Коррозия
может ускоряться образованием электрохимических пар из-за разно-
образия применяемых в охлаждающих системах металлов и сплавов.
Выбор для антифриза эффективных ингибиторов коррозии металлов
является весьма сложной задачей: ингибитор, эффективный для
одного металла, может оказаться неэффективным или даже вредным
для другого.
В качестве ингибиторов коррозпи применяются как органические,
так и неорганические продукты. Наиболее часто указываются амины,
амиды, меркаптобензтиазол, бензтриазол, бензоаты, салицилаты,
тетрабораты, бораты, нитриты, фосфаты, молибдаты, арсениты,
метасиликаты, карбонаты, органосилоксаны и их различные комби-
нации [156, 158]. Одной из комбинаций, нашедшей широкое при-
менение в антифризах, является смесь триэтаноламинфосфата и на-
триймеркаптобензтиазола [159].
Широко применяются бораты, например бура, особенно в ком-
бинации с другими компонентами. Предполагают двоякое действие
буры: увеличение буферности системы (поддержание pH на уровне
7—9,5) и собственно ингибирование. Вместе с бурой в антифриз
вводятся также нитрит натрия, часто меркаптобензтиазол или его
натриевая соль для предотвращения коррозии меди и ее сплавов,
и силикат натрия — для предотвращения коррозии алюминия [160].
Хроматы, которые являются хорошими ингибиторами коррозии
многих металлов при действии на них воды и ее солевых растворов,
для применения в антифризах не рекомендуются не только из-за
их токсичности, но также из-за взаимодействия хроматов с этилен-
гликолем, особенно на свету.
Варианты комбинаций ингибиторов весьма разнообразны [160,
161], а их содержание колеблется от десятых долей процента до
нескольких процентов. Например, один ингибитор содержит 0,25—
1% арсенита, 0,5—2% метабората и 0,1—0v&% меркаптобензтиазола
щелочного металла, а также 0,2—2% бората алканоламина [162].
В качестве ингибиторов предложены также соли II группы металлов,
например бораты или тетрабораты магния и кальция [163].
В ряде случаев в состав присадок включается цветной индикатор,
изменяющий цвет при потере антифризом антикоррозионных свойств
[164]. Для уменьшения просачивания антифриза через неплотности
в состав присадок предложено включить трибутоксиэтил-
фосфат [165].
Эффективность действия ингибиторов зависит от pH среды, для
поддержания которого на уровне 7,5—9,5 в ингибитор вводят веще-
ства, образующие буферные растворы. К ним относятся одно- и дву-
замещениыё фосфаты, бура или другие соединения бора, карбонаты
и т. п. Они обеспечивают в антифризе большой резерв щелочности
103
для нейтрализации кислот, образующихся при окислении этилен-
гликоля; pH раствора при этом остается достаточно постоянным.
Важной присадкой к антифризам являются пеногасители. Назна-
чение их заключается в том, чтобы исключить пли резко снизить
образование пены, наличие которой ухудшает отвод тепла от работа-
ющего двигателя. К пеногасителям относятся: полиорганосилокса-
новые жидкости, спирты средней молекулярной массы и их эфиры,
минеральные масла, алкилфосфаты и ряд других веществ [166,
с. 209].
Антифриз обычно выпускается в готовом для употребления водном
растворе или в виде концентрированного этиленгликоля, содержа-
щего необходимые присадки. В последнем случае разбавление
этиленгликоля водой производится потребителем. Согласно
ГОСТ 159—52, выпускается антифриз двух марок (65 и 40), а по
ГОСТ 6367—52 — концентрированный этиленгликоль, содержащий
ингибиторы коррозии (натрий фосфорнокислый двузамещепный и
декстрин), который применяется для приготовления из пего анти-
Таблица 33. Показатели качества антифризов
ТОСОЛ-А, ТОСОЛ-А40, ТОСОЛ-А65
Наименование показателей ТОСОЛ-А ТОСОЛ-Л40 ТОСОЛ-А65
Внешний вид Голубая жидкость без механи- Красная
Плотность при 20 °C, г/см3 . . Температура кипения при ческих примесей жидкость без механических примесей
1,120—1,140 1,078—1,085 1,085—1,095
0,1 МПа, °C, не менее .... Резерв щелочности, см3, не ме- 170 108 115
нее 10 10 10
Содержание поды, % , не более Температура кристаллизации, 3 — —
°C, не более Объем пены через 5 мин, см3, не —35 * —40 -65
более Время исчезновения пены, с, не 50 30 30
более 5 3 3
pH раствора при 20 °C .... Коррозионное воздействие на ме- таллы, мг, не более медь М-1, латунь Л68, чугун 7,5—8,5 * 7,5-8,5 7,5—8,5
серый 10 10 10
припой ПОС 35 15 • 15 15
алюминий АК-6М2 . . . . Набухание резин**, % (масс.), 30 30 30
не более 6 6 6
* Разбавленного на 50% (об.) дистиллированной водой.
** В разбавленном на 50% (об.) водопроводной водой.
104
фриза. Некоторые показатели этих антифризов и этиленгликоля
таковы:
Антифриз 65 Антифриз 4 0 Этиленгликоль
Относительная плотность d24° . . Температура замерзания, СС, не 1,085—1,090 1,0675—1,072 1,110—1,116
выше -65 -40 —
Фракция до 150 °C, % , не более . . 35 47 5
Остаток выше 150 °C, %, не менее 64 52 94
pH, не более Содержание, г/л 8,5 8,5 8,5
динатрппфосфата 3,0—3,5 2,5—3,5 4,4—5,6
декстрина — —. 1,80—1,85
Кроме того, выпускаются концентрированный антифриз
ТОСОЛ-А, отвечающий современным требованиям, а также готовые
к употреблению антифризы ТОСОЛ-А65 и ТОСОЛ-А40, которыми
заправляют все выпускаемые легковые машины ВАЗ и МЗМА и ряд
грузовых автомашин. Показатели качества антифризов ТОСОЛ
(по ТУ 02-751—73) приведены в табл. 33.
Согласно английским стандартам выпускается три типа ингиби-
рованного этиленгликоля, предназначенного для производства анти-
фризов. Это — прозрачные, без видимых примесей жидкости, с тем-
пературой замерзания не выше —12 °C, к которым могут быть доба-
влены красители. Основные показатели этих гликолей и содержание
в них ингибиторов коррозии следующие:
В 83150 : 5 9 В S3 151 : 5 9
Относительная плотность .
pH .............................
Содержание, %
фосфата триэтаноламина . .
меркаптобепзтпазолнатрия . .
бензоата натрия ............
нитрита натрия.............
тетрабората натрия (десятивод-
ного) ......................
1,122—1,126
6,7—7,3 *
«=3
0,2—0,3
1,131—1,141
7—8,5 **
BS3152 : 59
1,119—1,128
7,8—8,2
5-7,5
0,45-0,55
2,4-3,0
* 5 0% (об.) водного раствора.
* * 25% (об.) водного раствора.
Полиэфирные волокна
Синтетическое волокно лавсан (дакрон в США, терилен в Англии)
получается прядением из расплава полиэтилентерефталата —гетеро-
цепного сложного полиэфира терефталевой кислоты и этилен-
гликоля [167, с. 117]. Получение полиэтилентерефталата и его
переработка в волокна и пленки является одной из самых перспек-
тивных и значительных по объему областей применения этилен-
гликоля. Это объясняется тем, что полиэфирные волокна обладают
105
рядом достоинств: высокой прочностью, эластичностью, термо-
и светостойкостью, стойкостью к кислотам, окислителям и восстано-
вителям, несмипаемостью, способностью сохранять приданную
форму в процессе переработки волокна, хорошими электроизоля-
ционными качествами, устойчивостью к действию моли и микро-
организмов. Полиэтилентерефталатные пленки, кроме того, почти
непроницаемы для таких газов, как кислород и азот.
Синтез полиэтилентерефталата включает две основные стадии:
получение дигликолевого эфира терефталевой кислоты переэтери-
фикацией диметилтерефталата этиленгликолем и поликонденсация
дигликолевого эфира терефталевой кислоты. Для обеспечения пол-
ноты реакции переэтерификация диметилтерефталата проводится
в избытке этиленгликоля при повышенной температуре (180 °C
вначале и 200—210 °C в конце) и в присутствии небольших количеств
(0,05—0,1%) слабощелочных катализаторов — окислов свинца,
кальция, магния, солей низших жирных кислот и двухвалентных
металлов, например ацетатов свинца, кадмия, цинка и др. В про-
цессе переэтерификации отгоняются выделяющийся метиловый спирт
и избыток этиленгликоля.
После фильтрования от нерастворившегося катализатора п дру-
гих примесей диглпколевый эфир терефталевой кислоты подвергается
поликонденсации при 270—280 °C и остаточном давлении менее
133,3 Па (1 мм рт. ст.). Б таких условиях отгоняется выделившийся
этиленгликоль, а также образующиеся в качестве побочных про-
дуктов димеры, тримеры и тетрамеры этиленгликольтерефталата.
В последнее время стал широко применяться синтез дигликоле-
вого эфира терефталевой кислоты непосредственной этерификацией
терефталевой кислоты высокой степени чистоты (99,9%) этилен-
гликолем. Этерификация проводится при избытке этиленгликоля
(2,5—3 моль на 1 моль кислоты) и температуре 190—200 °C. Скорость
реакции возрастает, если процесс проводится при 0,3—1,2 МПа
(3—12 кгс/см2) и более высокой температуре (250—260 °C).
Ассортимент изделий из волокна лавсан весьма обширен: ткани,
трикотажные и вязаные изделия, тюль, бельтинг, автокорд, канаты,
фильтровальные и парусные ткани, рыболовные сети, пожарные
рукава, электроизоляция и ряд других изделий.
Гидравлические. жидкости
Значительное количество этиленгликоля расходуется на получение
гидравлических жидкостей, применяемых в гидроприводах металлур-
гического, металлообрабатывающего, транспортного и подъемного
оборудования, в землеройных и сельскохозяйственных машинах,
автомобилях, самолетах. Давление жидкости в системе гидропривода
достигает 19,6—24,5 МПа (200—250 кгс/см2), а температура от минус
50—60 до плюс 100, а иногда и 200 °C.
К жидкостям для гидравлических систем предъявляются такие
требования [168, с. 13]: хорошие смазывающие свойства, стабиль-
10G
/
ность при эксплуатации и храпении, низкая температура замерзания
и пологая кривая изменения вязкости в широком интервале темпе-
ратур, совместимость с материалами системы и низкая летучесть,
высокие температуры кипения, вспышки и самовоспламенения,
минимальная вспепиваемость и малая плотность (при применении
на самолетах, подводных лодках и др.), нетокспчпость и отсутствие
действия на кожные покровы. Для гидравлических систем, работа-
ющих вблизи открытого огня или других источников воспламенения,
жидкость должна быть негорючей.
В зависимости от условий эксплуатации некоторые из перечислен-
ных требований могут меняться. Например, температура замерзания
жидкости, которая применяется в автомобилях и самолетах, должна
быть весьма низкой и может быть более высокой при использовании
жидкости в машинах и оборудовании, находящихся в помещении.
Водпо-гликолевые негорючие гидравлические жидкости содержат
кроме воды и этиленгликоля следующие компоненты: другой гликоль
или смесь гликолей, водорастворимый загуститель, который придает
раствору необходимые вязкостные свойства, а также ингибиторы
коррозии и различные присадки, улучшающие ее противоизпосные,
антипенные, буферные и прочие необходимые показатели. Вода
является компонентом, обеспечивающим негорючесть жидкости,
ее содержание должно быть не менее 35%.
В качестве загустителей (вязкостных присадок) рекомендуются
статистические сополимеры окиси этилена и окиси пропилена, поли-
этиленгликоли, полиоксиэтилен, оксиэтилированные и оксипропи-
лированные кислоты и алкилфенолы, натриевая соль карбокси-
метилцеллюлозы, полиметакрплат калия, полиакриловая кислота,
а также ряд других материалов [169—172]. Одним из основных
требований к вязкостным присадкам является термостабильность
и стабильность к механической деструкции.
В качестве ингибиторов коррозии рекомендуются алкиламино-
нитриты, меркаптобензтиазолпатрий, фосфаты и силицилаты алка-
ноламинов, бура, морфолин и его соединения и др. [169 —171].
Одна из первых негорючих гидравлических жидкостей содержала
37% воды, 49% этиленгликоля, 17% сополимера окиси этилена
и окиси пропилена в соотношении (3—1) : 1 с молекулярной массой
17 000, а также ингибиторы коррозии — 2% диэтил- или триэтил-
аминофосфата, 1,5% диизопропил- или диизобутиламинонитрита
И 0,2% меркаптобензтиазолнатрий [169]. Состав другой гидравли-
ческой жидкости таков: 50% воды, 19% этиленгликоля, 6% ди-
пропиленгликоля, 0,50% димеризованного льняного масла и 1%
морфолина — ингибитора корррозии, 0,06% натриймеркаптобенз-
тиазола (50%-пого), по 0,5% фосфорной кислоты (75%-ной) и сили-
коновой антипенпой присадки; кроме того, введено 21 % сополимера
окиси этилена и окиси пропилена, взятых в соотношении 3,5 : 1
с молекулярной массой 19 500 [170]. Еще один вариант негорючей
жидкости, в которой содержится: от 10 до 30% сополимера в за-
висимости от требуемой вязкости окиси этилена (75%) и окиси
107
пропилена (25%), 40—60% воды, 20—30% этиленгликоля, 3% ингиби-
тора на основе буры, 5% соли оксокислот С10 и изопропиламина
и до 2,5% калиевой соли бутплфосфорной кислоты [1711-
Ниже приводятся показатели двух водно-глпколевых негорючих
гидравлических жидкостей Houghto Safe 620 II] и Ucon Hygrolube
275-cp (II) [168, c. 2891:
i ii
Плотность, г/см3 .................. 1,055 1,080 (d|g)
(15,6 °C)
Вязкость, мм2/с при температуре, °C
17,8 1100 902
21,1 88 —
37,8 43,2 60,5
54,4 25,3 —
65,6 15,6 27,3
Индекс вязкости......................... 80 155
Температура, °C
застывания ........................ —28,9 —31,6
вспышки .........................Нс вспыхивает
воспламенения ...................Не воспла- —
меняется
Удельная теплоемкость, кДж/(кг-К) 3,35 2,93
Коэффициент теплопроводности,
Вт/(м-К) .....................: . . 0,43 0,45
Коэффициент сжимаемости от 0 до 20,7
МПА (210,9 кг/см2) ................2,46 ХЮ'7 —
pH ................................ 9 • —
Требования, предъявляемые к качеству
этиленгликоля
Технические требования на этиленгликоль определяются областью
его применения. Для получения антифриза содержание воды в эти-
ленгликоле может быть больше, чем в продукте для производства
полиэфиров. В первом случае этиленгликоль разбавляется водой,
а во втором — наличие значительных количеств влаги приведет
к загрязнению полиэфира низкомолекулярными гликолями.
Этиленгликоль, используемый в производстве электролитических
конденсаторов, должен обладать высоким удельным сопротивлением,
что не имеет значения для многих других областей его применения.
Особенно высокие требования предъявляют к этиленгликолю для
производства синтетических волокон и пленок, так как повышенное
содержание примесей в нем может не только вызвать затруднения
в процессе получения полиэтилентерефталата, но и ухудшить каче-
ство готовых изделий. В связи с указанным в большинстве стран
выпускается этиленгликоль нескольких марок.
Этиленгликоль, который производится в СССР по ГОСТ 19710—74,
должен соответствовать показателям и нормам, указанным
в табл. 34.
.108
Таблица 34. Технические требования на этиленгликоль
по ГОСТ 19710-74
— Нормы
Показатели с государствен- ным знаком качества 1 сорт 2 сорт
Внешний вид Бесцветная прозрачная Бесцветная
жидкость без осадка пли слегка
желтоватая
прозрачная жидкость без
осадка
Цвет по Pt—Со шкале, не более
в обычпом СОСТОЯНИИ . . . 5 50 80
после кипячения с НС1 . . 30 Не нормируется
Плотность при 20 °C, г/см3 . . Коэффициент преломления при 1,1127—1,1140 1,1100—1,1150 1,4300—1,4320 Не менее 1,1100 1,4300—1,4320
20 °C 1,4316-1,4320
Кислотность Должен выдерживать испытание *
Содержание, % 99,7 98,5 96,0
этиленгликоля, не менее
диэтпленгликоля, не более 0,15 1,0 3,2
воды, не более 0,15 0,5 0,8
золы, не более 0,001 0,003 0,030
хлор-пона, не более . . . Не опреде- 0,0001 0,0010
ляется
железа, не более 0,00003 0,0005 0,0005
тяжелых металлов .... От сутствпе
Температурные пределы пере- гонки (при 0,1 МПа), °C 193
начало кипения, не ниже 196 194
конец кипения, не выше 199 200 200
Количество отгона в указанных пределах, % , не ниже .... 95 96 99
* 5 мл этиленгликоля в 10 мл дистиллированной воды не должны розоветь после
прибавлении 1—2 капель фенолфталеина и перемешивании.
Спецификации на этиленгликоль некоторых иностранных фирм
приводятся в табл. 35 [150, р. 6; 173].
Фирмой ICI кроме этиленгликоля марки S выпускается этилен-
гликоль марки LC с низким содержанием хлоридов и электропровод-
ностью при 20 °C, равной 2.5-10'7 Ом-1, для электролитических
конденсаторов и марки PR с низким содержанием железа и других
примесей — для полиэфиров.
Фирма Kuhlmann производит также этиленгликоль марки С
и D. Они выкипают в более узких пределах (195—200 °C), содержат
не более 0,10% воды и их кислотность не выше 0,005%. Для этил н-
гликоля марки С регламентируется электрическое сопротивление
(электропроводность); в этиленгликоле марки D должны отсутство-
вать восстанавливающие вещества, зола, хлориды, и он должен
Давать отрицательную реакцию с нитратом серебра и аммиаком.
109
Таблица 35. Спецификации на этиленгликоль иностранных фирм
Показатели Фирма, марка
ICI стан- дартный, марка 8 Shell Kuhlmann марка Т General Anilinland Film Corp.
Цвет, не более Относительная плотность Кислотность (в расчете па уксусную кислоту), % , 10 10 10 15
1,116—1,118 (15,5/15,5 °C) 1,115—1,116 (20/20 °C) 1,113—1,1145 (20 °C) 1,1151—1,1156
не более Содержание, % , не более 0,002 0,003 0,01 0,003
воды 0,3 0,3 0,2 0,25
золы 0,002 0,002 0,01 —
сульфатов .... — Отсутствие — —
хлор-иона Температурные пределы перегонки при 0,1 МПа, °C начало кипения, не Отсутствие
ниже конец кипения, не вы- 195 195 193 196
гае Количество отгона в ука занных пределах, % , 203 199 200 206
не ниже Температура кристаллиза- ции, °C, не выше (50% 95
об. водного раствора) — -30 — —
Согласно стандарту БДС 9194—72 Народной Республики Болга-
рии выпускается 3 сорта этиленгликоля. Продукт 1 сорта предназна-
чен для производства синтетического волокна. Он должен содержать
не менее 99,90% этиленгликоля и не более: 0,05% суммы ди- и три-
этиленгликоля, 0,04% воды, 0,001% золы, 0,00001% железа, 0,0007%
альдегида (в расчете на ацетальдегид); иметь кислотность не выше
0,0006% (в расчете на H2SO4) и выкипать в пределах 196,5—199,0 °C.
ЛИТЕРАТУРА
1. Glycols. Ed by G. О. Curme, F. Johnston. New York, Reinhold Publ. Corp.,
1953. 389 p.
2. Пиментал Д., Мак-Клеллан О. Водородная связь. Пер. с апгл. Под ред.
В. М. Чулановского. М., «Мпр», 1964. 462 с.
3. Gallant R. W., Hydrocarb. Process., 1967, v. 46, .Y 4, p. 183—196.
4. Справочник пипка. T. 1—3. Изд. 2-е. М.—Л., «Химия», 1963—1966.
5. Сталл Д., Вестрам Э.. Зинке Г. Химическая термодинамика органических
соединений. Пер. с англ. М., «Мир», 1971. 807 с.
6. Gardner Р. I., Hussian К. S., J. Chem. Thermodyn., 1972, v. 4, № 6-
р. 819-827.
7. Моторные п реактивные масла и жидкости. Под ред. К. К. Папок п Е. Г. Се-
меппдо. М., «Хпмпя», 1964. 704 с.
8. Ewert М., Bull. Soc. chim. Belg., 1937, v. 46, № 3, p. 90—103.
110
9. Dykyi I., Kus’ka V., Seprakova MChem. Zvesti, 1956, v. 10, X» 4
p. 193-203.
10. Миронов К. E., Дерябина Л. Д., ЖПХ, 1962, т. 35, вып. 6, с. 1338—1342.
11 Ott I. В., Goates I. R., Lamb I. D., J. Chem. Thermodyn., 1972, v. 4, X» 1
p. 123—126.
12 Milnes M. V., Zanella F. M., J. Chem. Phys., 1965, v. 42, № 8, p. 2817—
2819.
13. Gladden I. K., J. Chem. Eng. Data, 1972, v. 17, № 4, p. 468—470.
14. Timmermans I. The Physico-Chemical Constants of Binary Systems in Con-
centrated Solutions. New York — London, Interscience. Publ. Inc., 1959—
1960. V. 2-1272 p.; V. 3-1322 p.; V. 4 — 1332 p.
15. Tans A. M., Brit. Chem. Eng., 1967, v. 12, № 1, p. 93.
16. Ганиев Ю. А., ЖФХ, 1969, t. 43, вып. 1, c. 239—240; Расторгуев Ю. A.,
Газдиев M. А., Пнж.-физ. ж., 1969, т. 17, № 1, с. 72—79; Расторгуев Ю. Л.,
Газдиев М. А., ЖФХ, 1970, т. 44, вып. 12, с. 3092—3095; Ганиев Ю. А.
В кн.: Теплофизические свойства жидкостей. М., «Наука», 1970, с. 95—98.
17. Резниченко Е. П., Труды Новочеркасского политехнического ин-та, 1972,
т. 257, с. 111—114.
18. Grickard К., Skinner I. F., J. Phys. Chem., 1969, v. 73, № 6, p. 2060—2062.
19. Левин В. В., Подловчеико Т. А., Ж. структ. хим., 1970, т. 11, № 4,
с. 766—767.
20. Travers F., Douzon Р., J. Phys. Chem., 1970, v. 74, № 10, p. 2243—2244.
21. Ергопуло E. В., Белинский Б. А. В кн.: Тепловое движение молекул и меж-
молекулярное взаимодействие в жидкостях и растворах. Самарканд, 1969,
с. 244—247; в кн.: Теп.тофпзпческпе свойства жидкостей. М., «Наука»,
1970, с. 45—47; Артемченко М. И., 7КФХ, 1963, т. 37, № 1, с. 3—7.
22. Hebert I., J. Acoust. Soc. Am., 1971, v. 49, № 3, part 2, p. 930—931.
23. Крешков A. IT. Основы аналитической химии. Физико-химические (инстру-
ментальные) методы анализа. М., «Химия», 1970, 472 с.
24. Tissier Claude, С. г. Acad. Sci., 1971, v. 272, № 6, р. 527—529; Bau-
mann W. M., Simon W., Helv. chim. Acta, 1969, v. 52, № 7, p. 2060—2064.
25. Banerjee S. K., Kundu К. K., Dus M. N., J. Chem. Soc., Ser. A, 1967, № 1,
p. 166—169.
26. Лоури Д. Глицерин и гликоли. Пер. с англ. Под ред. Ю. С. Залькинда.
М. И. Серякова. Л., Госхимиздат, 1933. 396 с.
27. Glycols. Properties and Uses. Michigan, The Dow Chem. Co., 1961. 65 p.
28. Гликоли и опыт их применения в нефтяной и газовой промышленности. М.,
ВНИИОЭНГ. 1970. 152 с.
29. Белоусов В. П.. Морачевский А. Г. Теплоты смешения жидкостей. Л., «Хи-
мия», 1970. 256 с.
30. Егорова И. В. К кн.: Материалы Всесоюзного симпозиума по термохимии
растворов электролитов и неэлектролитов. Иваново, 1971, с. 171—172;
Крестов Г. АНеделька Б. Е., там же, с. 131 —139.
31. Mellanl. Compatibility and Solubility. New York — London, Noyes Develop.
Corp., 1968. 304 p.
32. Francis A. W. Liquid — Liquid Equilibriums. New York — London, Inters-
cience Publ., 1963. 288 p.
33. Справочник по растворимости. T. 1, 2. Под ред. В. В. Кафарова. М.—Л.,
изд. АН СССР. 1962—1963.
34. Banateanu Gh'. 'e. a., Bull. Inst. Petrol., gaze si geol., 1966, v. 14, p. 123—141.
35. Hayduk W., Malik V. K., J. Chem. a. Eng. Data, 1971 v. 16, № 2,
p. 143—144.
36. Francis А. И7. Critical Solution Temperatures. Washington, Am. Chem. Soc.,
1961. 264 p.
3c Огородников С. К., Лестева T. M., Коган В. Б. Азеотропные смеси. Л.,
«Химия», 1971. 848 с.
38. Коган В. Б., Фридман В. М., Кафаров В. В. Равновесие между жидкостью
и паром. М,—Л., «Наука», 1966.'1426 с.
39. Соколов Н. М., Цыганкова Л. Н.. Жаворонков II. М. Теорет. основы хим.
техпол., 1971, т. 5, № 6, с. 900—904.
111
40. Соколов Н. М. и др., Хим. пром., 1972, № 7, с. 499—501.
41. Chandramouli V. V., Laddha G. S.., J. Sci. and Ind. Res., 1962, v. 21, № 8,
p. 266—274; Shukla N. P., Indian Chem. Eng.. 1968, № 1, p. 48—49; Brus-
set II., Chim. et ind.-genie chim., 1968, v. 99, № 2, p. 207 — 219.
42. Mainkar S. V., Mene P. S., Indian J. Technol., 1970, v. 8, № 9, p. 320—322;
Соколов H. M. и др., Хпм. пром., 1973, № 5, с. 13 —16.
43. Francis A. W., J. Chem. a. Eng. Data, 1965, v. 10, № 1, p. 45—49; № 2,
p. 145 -151; № 3, p. 260—264; № 4. p. 327—330.
44. Orel Aluf, J. Chem. a. Eng. Data, 1967, v. 12. A° 1, p. 1—4; Sims L. L.,
Bolme D. W., Ibid., 1965, v. 10, № 2, p. 111 — 113.
45. Журавлев E. Ф., Ерофеева JI. Ф., Уч. зап. Пермского ун-та, 1968, № 178,
с. 10—17; Бурдыкина Р. И., Журавлев Е. Ф.. ЖФХ, 1968, т. 42, № 8,
с. 1925—1929; Зарецкий М. II. и др., ЖПХ, 1970, т. 43, № 10, с. 2269—
2274; Журавлев Е. Ф., Корнеева А. II., ЖОХ, 1971. т. 41, № 1, с. 28—34.
46. Александрова М. В. а др., ЖПХ, 1966. т. 39, вып. 5, с. 1041 —1049; Стар-
ков А. А. и др., «Труды МИТХТ пм. №. В. Ломоносова», 1958, вып. 8
с. 63-73.
47. Элътеков А. II., ЖРХО, 1878, т. 10, вып. 5, с. 21'1—222.
48. Пат. США 1206202 (1916); 1237076 (1917); 1259758 (1918); 1709605 (1929);
1928240 (1933).
49. Клебанский А. Л., Долгополъский И. М., ЖПХ. 1934, т. 7, вып. 5,
с. 790—805.
50. Коган Л. М. Производство и переработка окиси этилена в Германии.
№.— Л., Госхпмпздат. 1947. 121 с.
51. Пат. США 1737545 (1929); 1895517 (1933); 2378104 (1945).
52. Stanley II. М., Chem. a. Ind., 1965, № 23. р. 1192—1199.
53. Mitsutani A., Chem. Economy and Eng. Rev., 1973, v. 5, Al 3, p. 32—35-
пат. США 3647892 (1972); 3809724 (1974).
54. Brownstein A. M., Hydrocarb. Process., 1974, v. 53, № 6, p. 129 —132;
Chem. Eng. Progr., 1975, v. 71, № 9 p. 72 — 76; пат. США 3872164 (1975).
55. Anderson E. V., Chem. Eng. News, 1975, v. 53, A” 24, p. 12—13.
56. Wiberg К. B., Salgebarth K. A., J. Am. Chem. Soc., 1957, v. 79, № 11,
p. 2822—2824.
57. Успехи органической химии. T. 1. Пер. с англ. Под ред. И. Л. Кнунянца.
№.. Издатинлпт, 1963. 398 с.
58. Gurjar V. G., Chem. Process a. Eng. (India). 1971, v. 5, № 4, p. 25—36.
59. Mayell J. S., I and EC, Prod. Research and Develop., 1968, v. 7, Al 2,
p. 129—136.
60. Milas AT. A., Sussman S., J. Am. Chem. Soc., 1937, v. 59. Al 11, p. 2345—
2347; Франц, пат. 1411211 (1965); пат. США 33117592 (1967).
61. Калинин М. А., Стендер В. В., ЖПХ, 1946, т. 19, вып. 10—11, с. 1045—
1058; Bhattacharyya S., Muthanna М., Patankar A., J. Sci. and Ind. Rese-
arch, 1952, № 11B, p. 365—368.
62. Заявка ФРГ 1643081 (1971).
63. Англ. Пат. 1177877 (1970); пат. США 3629343 (1971); Шапиро А. Л. и др.,
((Химия и технология топлив и масел», 1971. А» 5. с. 14 —18.
64. Гринберг Д. II., Шапиро А. Л., Левин С. 3.. ЖПХ, 1970, т. 43, вып. .11,
С. 2584—2586; Левин С. 3.. Шапиро А. Л., Гуревич Г. С. Авт. свпд. № 269925
(1970); Открытия. Изобр. Пром, образцы. Товарп. знаки, 1970, А» 16, с. 21.
65. Ким Хаи Сил и др., Хпм. пром., 1963, № 7, с. 12—15; яп. пат. 25563 (1969);
Борисов А. В., Тимофеев В. Д., Журп. ВХО пм. Д. И. Менделеева, 1973.
т. 18, № 3, с. 32.3—327.
66. Имянитов II. С., Рудковский Д. М. Авт. свпд. № 145232 (1960); Бюлл.,
изобр., 1962, А1» 5, с. 25; герм. пат. 725842 (1912); С. А., 1943, v. 37, 5984;
’ Гидроформилирование. Под ред. Н. С. Имянптова п В. Ю. Ганкпна. Л..
«Химия», 1972. 253 с.
67. Швейц, пат. 343384. 344400 (1960); Васюнина Н. А. и др., Хим. пром.,
А° 2, с. 82—86; Балахонцева В. Н., Гелъперин Н. И., там же, с. 86—88;
Васюнина II. А., Баландин А. А., Маматов Ю. Л., «Кинетика и катализ»,
1963, т. 4, № 1, с. 156 — 162.
112
68. Faith W., Keyes D., Clark R. Industrial Chemicals. Ed. 3d, New York —
Sydney, John Wiley and Sons, 1965. 852 p.; Goldstein R. F., Waddams A. L.
The Petroleum Chemicals Industry. Ed. 3d. London, Spon, 1967. 523 p.
69. Stobaugh R. W„ Ray G. S., Spinek R. A., Hydrocarb. Process, 1970, v. 49,
№ 10, p. 105—116.
70. Bronsted I. N., Kilpatrick M., Kilpatrick M., J. Am. Chem. Soc., 1929,
v. 51, № 2, p. 428—461.
71. Henry L., C. r. Acad. Sci., 1907, v. 144, № 25, p. 1404—1406; Smith L.,
Woae G., Widhe T., Z. phys. Chem., 1927, Bd. 130, S. 154—166.
72. Lichtenstein H. I., Twigg I. H., Trans. Faraday Soc., 1948, v. 44, part 11,
p. 905—909.
73. Davis P. C., Waaden С. E., Kurata F., Chem. Eng. Progr., Symp. Ser.,
1952, v. 48, № 4, p. 91—97.
74. Reed L. M., Wenzel L. A., O'Hara I. B., Ind. Eng. Chem., 1956, v. 48, № 2,
p. 205—208; Hamilton G. E., Meltzner A. B., Ind. Eng. Chem., 1957, v. 49,
Ks 5, p. 838—846; Полянский H. Г., «Успехи химии», 1962, т. 31, вып. 9,
с. 1046—1075; 1970, т. 39, вып. 3, с. 50.
75. OthmerD. F., Thaker М. S., Ind. Eng. Chem., 1958, v. 50, № 9, p. 1235—1244.
76. Полянский H. Г. Катализ ионитами. M., «Химия», 1973. 214 с.
77. Окись этилена. Под ред. П. В. Зимакова и О. Н. Дымента. М., «Химия»,
1967. 320 с.; Гетероциклические соединения. Под ред. Р. Э. Эльдерфильда.
Т. 1. Пер. с англ. Под ред. Ю. К. Юрьева. М., Издатинлит, 1953. 556 с.;
Инголъд К. К. Механизм реакций и строение органических соединений.
Пер. с англ. Под ред. И. Л. Кнунянца и И. Ф. Комиссарова. М., Издат-
инлит, 1959. 674 с.
78. Реутов О. А. Теоретические основы органической химии. М., изд. МГУ,
1964. 700 с.
79. Лебедев Н. Н. Химия и технология основного органического и нефтехими-
ческого синтеза. М., «Химия», 1971. 840 с.
80. Лебедев И. Н., Манаков М. Н., Швец В. Ф. Теория технологических процес-
сов основного органического и нефтехимического синтеза. М., «Химия»,
1975. 478 с.
81. Ethylene and its Industrial Derivatives. Ed by S. A. Miller. London, E. Benn,
1969. 1321 p.
82. Parker R. E., Isaaks N. S., Chem. Rev., 1959, v. 59, № 4, p. 737—799.
83. Исаакс H. Практикум по физической органической химии. Пер. с англ.
Под ред. В. С. Петросяна. М., «Мир», 1972. 290 с.
84. Heterocyclic Compounds with Three — and Four-membered Rings. Part. 1.
Ed. by A. Weisberger. New York, Interscience Publ., 1964. 646 p.; Prit-
chard J. G., Long F. H., J. Am. Chem. Soc.,1956, v. 78, № 12, p.,2667—2670;
№ 23, p. 6008—6013.
85. Лебедев H. H., Савелъянов В. П., Баранов Ю. И., ЖПХ, 1969, т. 42, № 8,
с. 1815—1824; Лебедев Н. Н., Савелъянов В. П., Швец В. Ф., Теорет. и экспе-
рим. химия, 1970, т. 6, № 1, с. 111—116.
86. Mikl М. е. а., «Юкагаку», 1966, V. 15, № 5, р. 257—262.
87. Ferrero М. Р., Flamme L. R., Fourez А. М., Ind. chim. Belg., 1954, v. 19,
№ 2, p. 113—119.
88. Matignon C., Moureu H., Dode M., Bull. Soc. chim. France, ser. 5, 1934,
v. 1, № 10, p. 1308—1317.
89. Швец В. Ф. Докторская диссертация. М., МХТИ им. Д. И. Менделеева, 1974;
Лебедев Н. Н. и др. В кн.: Всесоюзное совещание по проблеме механизма
гетеро литических реакций. Л., 1974, с. 114—115.
90. Лебедев И. Н., Швец В. Ф.. Ромашкина Л. Л. Авт. свид. № 321519 (1973);
Открытия. Изобр. Пром, образны. Товарн. знаки, 1973, А" 14, с. 204;
Швец В. Ф., Ромашкина Л. Л., Лебедев Н. И. Авт. свид. № 418464 (1974);
Открытия. Изобр. Пром, образцы. Товарн. знаки, 1974, № 9, с. 79.
91. Weibull В., Nycander В., Acta Chem. Scand., 1954, № 8, р. 847—858.
92- Hine F., Hine M., J. Am. Chem. Soc., 1952, v. 74, № 21, p. 5266—5271;
Shigematsu H., Nlshikawa I., «Когё кагаку дзасси», 1962, v. 65, № 6;
p. 945—949. РЖХим, 1963, 8Ж7.
8 Заказ 972
ИЗ
93. Final Report № 1059. British Intelligence Objectives Subcommittee. Lon-
don, H. M. Stationery Office, 1947. 55 p.; Wetter F., «Schweizer Archiv
fur angewandte Wissenschaft und Technik», 1956, April, S. 120—126.
94. Parker W. A., Prados F. W., Chem. Eng. Progr., 1964, v. 60, № 6,
p. 74-78.
95. Miller I. H., Corrigan T. E., Hydrocarb. Process., 1967, v. 46, № 4, p. 176—
178; Corrigan T. E.-, Lessels G. A., Dean M. I., Ind. Eng. Chem., 1968, v. 60,
№ 4, p. 62—64.
96. Badea L., Ivanus Gh., «Revista de chimie», 1969, v. 20, № 12, p. 749—754.
97. Selected Values of Chemical Thermodynamic Properties. Circular of National
Bureau of Standards 500. Washington, D. S., 1952.
98. Cranfield J., Petrol. Petrochem. Intern., 1973, v. 13, № 11, p. 28—30.
99. Weaver D. G., Smart I. L., Ind. Eng. Chem., 1959, v. 51, № 8, p. 894—900.
100. Пат. США 224399 (1968); Изобр. образцы. Товарн. знаки, 1968, № 25,
с. 166; яп. пат. 9926 (1970); РЖХим, 1971, 7Н57.
101. Яп. пат. 10324 (1970); РЖХим, 1971, 7Н58; яп. пат. 19608 (1973); РЖХим,
1974, 10НЗЗ.
102. -Пат. США 2772237 (1956); РЖХим, 1959, 35889; пат. США 19608 (1973);
РЖХим, 1974, 6Н193.
103. Англ. пат. 779063 (1957); РЖХим, 1959, 39575; пат. ФРГ 1668052 (1975);
РЖХим, 1975, 21Н23.
104. Пат. ФРГ 1025832 (1961); РЖХим, 1962, 24Л42.
105. Англ. пат. 1140588 (1969J; С. А., 1969, v. 70, 87024.
106. Яп. пат. 11848 (1966); РЖХим, 1968, 8Н61.
107. Пат. США 3367847 (1968); РЖХим, 1969, 9Н41.
108. Англ. пат. 794382 (1958); РЖХим, 1959, 65270.
109. Пат. США 2788373 (1957); РЖХим, 1959, 65269.
110. Англ. пат. 801723 (1958); РЖХим, 1960, 85744; пат. США 3408268-(1966);
РЖХим, 1970, ЗН41.
111. Пат. США 3311544 (1967).
112. Пат. ФРГ 1282632 (1969); РЖХим, 1970, 15Н57.
ИЗ. Пат. ФРГ 1189071 (1965); РЖХим, 1967, 24Н39; пат. ФРГ 1250427 (1968);
РЖХим, 1969, 19Н47.
.114. Пат. ГДР 43911 (1966); РЖХим, 1967, 17Н29.
115. Пат. США 2775623 (1956); РЖХим, 1958, 61865.
116. Monick I. A. Alcohols. New York — London, Reinhold Book Corp., 1968.
594 p. .
117. Sherwood P. W., Petrol. Refiner, 1949, v. 28, № 3, p. 129—134; Sher-
wood P. W., Petrol. Process., 1954, v. 9, № 10, p. 1592—1597; Sher-
wood P. W., Ind. Chem., 1959, v. 35, № 409, p. 126—131; № 412,
p. 278—282.
118. Пат. США 1875312 (1932); С. A., 1932, v. 26, 5962; пат. США 2135271 (1938):
пат. США 2255411 (1941).
119. Пат. США 2409441 (1946).
120. Пат. США 2472417 (1949).
121. Пат. ФРГ 1043308 (1959).
122. Рабовская Н. С., ЖФХ, 1959, т. 33, вып. И, с. 2467—2470.
123. Cartmell В. К. е. a., Ind. End. Chem., 1948, v. 40, № 3, p. 389—392.
124. Пат. США 3028434 (1962); пат. США 3091647 (1963).
125. Пат. США 2770656 (1956); РЖХим, 1958, 54865.
126. Фрейдлин Л. X., Шарф В. 3. Авт. свид. № 114336 (1958); Бюлл. изобр.,
1958, № 7, с. 14.
127. Орлова Е. Ю. Химия и технология бризантных взрывчатых веществ. Л.,
«Химия», 1973. 688 с.
128. Химическая и резиновая промышленность капиталистических стран.
Изд. 2-е. М., НИИТЭхим, 1962. 559 с.
129. Waddams A. L. Chemicals from Petroleum. London, John Muray, ed. 2d,
1968, 244 p.; ed. 3d, 1973, 326 p.
130. Chem. Eng. News, 1973, v. 51, № 21, p. 8, 10; 1975, v. 53, № 24, p. 12—13.
131. Химическая промышленность Японии. Вып. 2. М., НИИТЭхим, 1974. 92 с.
114
г ж
132. Bohmjalk I. F., McNamee R. W., Barry P. In: Modern Chemical Processes.
V. 2. New York, Reinhold Publ. Corp., 1952, p. 55—63.
133. Брикенштейн А. А. и др., Хим. пром., 1966, № 7, с. 20—21.
134. Атавин А. С., Дмитриева Л. П., Васильев Н. И. Авт. свид. № 182172
(1966); Изобр. Пром, образцы. Товарн. знаки, 1966, № 11, с. 23; РЖХим,
1967, ЗН201.
135. Англ, пат. 1129544 (1968); РЖХим, 1969, 11Н69; яп. пат. 28764 (1972);
РЖХим, 1973, 19Н34; Isshiki Т„ Chem. Eng., 1973, v. 80, № 4,
р. 96—97.
136. Яп. пат. 5368 (1969); РЖХим, 1970, ЗН60.
137. Имянитов Н. С. и др. Авт. свид. № 332071 (1972); Открытая. Изобр.
Пром, образцы. Товарн. знаки, 1972, № 10, с. 88.
138. Якубов Р. Д. и др. В кн.: Химия ацетилена. Под ред. М. Ф. Шостаковского.
М., «Наука», 1968, с. 488—493.
139. Яп. пат. 6731 (1966); РЖХим, 1967, 23Н125.
140. Пат. США 3068232 (1962); РЖХим, 1964, 14Н182.
141. Яп. пат. 2656 (1965); РЖХпм, 1967, 18Н92.
142. Пат. США 3225065 (1965); РЖХим, 1967, ЗН170.
143. Коренькова О. П. и др. Авт. свид. № 213340 (1968); Изобр. Пром, образцы.
Товарн. знаки, 1968, № 10, с. 88.
144. Яп. пат. 5983 (1960); РЖХим, 1962, 9Л51.
145. Яп. пат. 21820 (1967); РЖХпм, 1968, 15Н151.
146. Пат. США 2780610 (1957); РЖХпм, 1959, 40726.
147. Пат. США 2921913 (1960); РЖХпм, 1962, 8К148.
148. Пат. ПНР 43451 (1960); РЖХим, 1962, 13П260.
149. Пат. США 2827970 (1959); РЖХим, 1959, 83406; яп. пат. 2358 (1967);
РЖХим, 1968, 18Н372; пат. США 3318918 (1967); РЖХим, 1968, 23Н153;
яп. пат. 3291 (1968); РЖХим, 1969, 10НЗЗЗ.
150. Ethylene and Propylene Glycols. Birmingham, Imperial Chemical Indu-
stries (TCI) Lim., 33 p.
151. Шнитке В. А., Коган В. Б., ЖПХ, 1968, т. 41, вып. 6, с. 1305—
1313.
152. Англ. пат. 778081 (1957); РЖХпм, 1959, 50568.
153. Пат. ФРГ 1094736 (1951); РЖХим, 1962, 9Л91.
154. Яп. пат. 2158 (1959); С. А., 1960, v. 52, 14531.
155. Пат. ФРГ 1003497 (1958); РЖХим, 1959, 45393; пат. США 2832716 (1958);
РЖХим, 1959, 759611; пат. США 3231398 (1966); РЖХим, 1967,
16Н750.
156. Kirk-Othmer Encyclopedia of Chemical Technology. V. 2. Ed. 2nd. New
York, Interscience Publ. Inc., 1963.
157. Howard F. L., Brooks D. B., Streets R. E. Automotive Antifreezes. Washing-
ton, National Bureau of Standards Circular 576, 1956. 23 p.
158. Брегман Дж. Ингибиторы коррозии. Пер. с англ. Под ред. Л. И. Антро-
пова. М.—Л., «Химия», 1966. 310 с.
159. Squires А. Т. In: The Protection of Motor Vehicles from Corrosion. London,
SCI, 1958, p. 229.
160. Англ. пат. 1064473 (1967); РЖХим, 1967, 23П220.
161. Бельг, пат. 660075 (1965); С. А., 1966, v. 64, 4848; пат. США 3231502,
3282846 (1966); РЖХпм, 1968, 21П226; С. А., 1966, v. 64, 9496е; пат. США
3362910 (1968).
162. Пат. США 3046230 (1962); РЖХим, 1966, 18П131; пат. США 3090757 (1963);
РЖХим, 1965, ЗП229.
163. Пат. США 3046229 (1962); РЖХим, 1964, ЗК108; пат. США 3228884,
3231501 (1966): С. А., 1966. V. 64, 17101е.
164. Пат. США 2937146 (1960); РЖХим, 1961, 13М421; норвеж. пат. 99593 (1962);
РЖХим, 1963, 4П286; швейц, пат. 185678 (1963); РЖХим, 1965, 1П259;
. пат. США 3079343 (1963); РЖХим, 1965, 9П227.
165. Пат. США 3042620 (1962); РЖХим, 1963, 24П239.
166. Тихомиров В. К. Пены. Теория и практика их получения и разрушения.
М., «Химия», 1975. 264 с.
8*
115
167. Роговин 3. А. Основы химии и технологии химических волокон. Т. 2. М.,
«Химия», 1974. 344 с.
168. Хаттон Р. Е. Жидкости для гидравлических систем. Пер. с англ. Под ред.
В. В. Вайнштока. М.—Л., «Химия», 1965. 364 с.
169. Пат. США 2602780 (1952).
170. Пат. ФРГ 1220069 (1967).
171. Франц, пат. 1486440 (1967).
172. Пат. США 2455117 (1948); 2841560 (1958); 2758976, 2768141 (1956); 3346501
(1967); 3379644 (1968).
173. Чернышев А. К. и др. Справочный материал по качеству химической про-
дукции. М., ГИАП, 1972. 492 с.; Shell. Monoethylene Glycol. Data she-
et 2F 001; Monoethylene Glycol. PB 01—1, Service Publicite Kuhlmann.
Глава III
Диэтиленгликоль
Физические свойства
Диэтиленгликоль (дигликоль, этиленгликоль, 2,2'- или (3,|3'-диокси-
диэтиловый эфир) СН2ОНСН2ОСН2СН2ОН является вязкой, весьма
гигроскопичной, бесцветной прозрачной жидкостью со сладким
вкусом, практически без запаха. Молекулярная масса диэтилен-
гликоля 106,12. Основные физические характеристики диэтилен-
гликоля приведены ниже [1, р. 155] *:
Плотность (истинная) при 20 °C, г/см3......... 1,1161
Относительная плотность ...................... 1,1184
Температура,°C
кипения при 101,3 кПа (760 мм рт. ст.) . . . 245,0
замерзания ............................... —8
Давление насыщенного пара при 20 °C
Па .......................................... <1,3
мм рт. ст.................................. <0,01
Коэффициент преломления га...................... 1,4472
Молекулярная рефракция
наблюдаемая ............................ 25,39 [4]
вычисленная ..............................25,36
Поверхностное натяжение при 25 °C, мН/м (дин/см) 48,5
Удельная теплоемкость при 20 °C
Дж/(г-К) ................................... 2,093
кал/(г-°С)......................... . . 0,500
Теплопроводность при 20 °C
Вт/(м-К) ...................'.............0,25 [5]
кал/(см-с-°С) ............................ 0,00059
Вязкость при 20 °C, мПа-с (сПз) .............. 35,7
Диэлектрическая проницаемость при 15 °C .... 32,6 [6]
Электропроводность при 20 °C, Ом-1-см"1 .... 3,1-10_« [7]
Дипольпый момент при 25 °C, D ................2,69
Константа Верде, мин/(Гс-см)
при 546,1 нм (5-461 А)........................ 0,01490
при 589,3 нм (5893 А)..................... 0,01264
* В других источниках приводятся показатели, отличающиеся от указанных
ниже, например т. кип. 244,8 °C и т. замерз. —10,45 и —6,5 °C [2, т. 2, с. 665],
теплопроводность 0,21 Вт/(м-К) [3].
117
Критические параметры, являющиеся расчетными, так как ди-
этиленгликоль при температуре кипения и нормальном давлении
частично разлагается (температура начала разложения его 164,5 °C),
приведены ниже [5]:
Критическая температура, °C .... 407+5
Критическое давление, МПа (кгс/см2) 4,7+0,4(47,8+4,2)
Критическая плотность, г/см3 .... 0,336 + 0,01
Термодинамические свойства диэтиленгликоля характеризуются
следующими величинами [5]:
Теплота, кДж/моль (ккал/моль)
образования при 25 °C ............
сгорания при постоянном давлении
и 20 °C .......................
плавления .....................
испарения при 101,33 кПа (760 мм
рт. ст.) ......................
испарения при 1,3 кПа (10 мм рт.
ст.)...........................
—626,8 (—149,7) [8]
2374 (567)
13,46 (3,22) [9]
37,01 (8,84)
68,87 (16,45)
Зависимость теплоты испарения диэтиленгликоля от температуры
в интервале 40—440 °C приведена на рис. 30.
Плотность диэтиленгликоля и его водных растворов в темпера-
турном интервале от —40 до 75 °C и от 0 до 180 °C приведена на
рис. 31 [5, 10, с. 534]. Температурный коэффициент относительной
плотности диэтиленгликоля в интервале 5—40 °C составляет 0,00071
на 1 °C [1, р. 155].
Относительная плотность смесей ди- и триэтиленгликоля пока-
зана на рис. 32 [1, р. 158], а смесей диэтиленгликоля и этиленгли-
коля приведена в табл. 13 (см. стр. 41).
Зависимость температуры кипения диэтиленгликоля от давления
характеризуется следующими данными [11, с. 15]:
явление, рт. ст. * Т. кип., °C Давление, мм рт. ст. * Т. КИП. °C
1 91,8 60 174,0
5 120,0 100 187,5
10 133,8 200 207,0
20 148,0 400 226,5
40 164,3 760 244,8
* 1 мм рт.ст. = 133,32 Па.
Зависимость температуры кипения водных растворов диэтилен-
гликоля при атмосферном давлении от концентрации приведена
на рис. 33 [10, с. 536].
118
Температура замерзания (начала кристаллизации) водных рас-
творов диэтиленгликоля показана на рис. 34 [1, р. 156]. Раствор,
содержащий около 66% (масс.) или 62% (об.) диэтиленгликоля,
замерзает при —52 °C. По данным [9], водные растворы диэтилен-
гликоля имеют более низкую температуру замерзания (табл. 36).
Рие. 30. Зависимость
теплоты испарения эти-
ленгликоля (7), диэтилен-
гликоля (2) и триэтилен-
глпколя (3) от темпера-
туры.
Температура., °C
По данным [12], эвтектическая смесь диэтпленгликоля и воды
содержит 70% ДЭГ и замерзает при —63,5 °C. Различия температур
замерзания объясняются, как и для смесей этиленгликоля с водой,
Рис. 31. Зависимость плотности диэтиленгликоля и его водных растворов от
температуры:
а) в интервале ог —40 до +75 °C; б) в интервале от 0 до 180 °C.
тем, что вблизи эвтектической точки вязкость смеси резко повы-
шается, что затрудняет нахождение истинной температуры замерза-
ния. Кроме того, смеси склонны к переохлаждению; не исключено
также влияние примесей в исходном диэтиленгликоле.
Температура замерзания смесей диэтиленгликоля с этиленглико-
лем и триэтиленгликолем приведена в табл. 37 [9].
119
Давление насыщенного пара диэтиленгликоля (р, мм рт. ст. *)
в интервале 80—165 °C можно вычислить по уравнению [2, т. 1,
с. 702]:
1g/> = 8,1527
2727,3
— Т
Температурный коэффициент давления насыщенного пара ди-
этиленгликоля в интервале 56,4—57,8
Концентрация три.этиленгликоля,%
Рис. 32. Относительная плотность смесей дп-
и трпэтиленгликоля.
Рис. 33. Зависимость температуры кипения водных растворов диэтиленгликоля
(7) и триэтиленгликоля (2) при атмосферном давлении от концентрации гликоля.
Концентрация дизтиленгликоля,01о
Рис. 34. Температура за-
мерзания водных раство-
ров диэтиленгликоля.
составляет 0,049 °C на 0,133 кПа (1 мм рт. ст.) [1, р. 155]. Зависи-
мость давления насыщенного пара диэтиленгликоля от температуры
приведена на рис. 1 (см. стр. 20), а давления насыщенных паров
водных растворов диэтиленгликоля— на рис. 35 [13, с. 255].
Коэффициент преломления диэтиленгликоля в интервале 20—
30 °C изменяется на 0,00028 при изменении температуры на 1 °C [1.
* 1 мм рт. ст. = 133,32 Па.
120
Таблица 36. Зависимость температуры замерзания водных растворов
диэтиленгликоля (ДЭГ) от его концентрации *
Концентра- ция ДЭГ, % Температура замерзания, °C Концентра- ция ДЭГ, % Температура замерзания, °C Концентра- ция ДЭГ, % Температура замерзания, °C
2 -0,3 60 —41,4 80 (-43)
ю -1,9 62 —44,9 82 (-41)
Е 20 —5,1 64 (-48,3) 84 -39
К 30 —9,9 66 (-51,5) 86 -37
К 40 -16,9 68 (-55,5) 88 —34
К 50 —26,6 70 (-58) 90 -30
52 —29,3 72 (-62) 92 —25,5
К 54 -32,3 74 94 -21,3
56 —35,4 76 — 96 —17,0
V 58 -38,4 78 — 98 —12,7
* Цифры в скобках получены экстраполяцией кривых.
Таблица 37. Зависимость температуры замерзания смесей диэтиленгликоля
(ДЭГ) с этиленгликолем (МЭГ) и триэтиленгликолем (ТЭГ)
от концентрации диэтиленгликоля *
Концентра- ция ДЭГ, % температура замерзания, °C Концентра- ция ДЭГ, % Температура замерзания, °C
с МЭГ с ТЭГ С МЭГ С ТЭГ
55 -39 (-28)
5 —14,5 -6,8 60 -36,4 —
10 —16,5 . -9,3 65 —33,4 —
15 -18,8 -11,6 70 -30,2 -24,2
20 —21,3 -13,9 75 -26,8 —19,8
25 -24,1 —16,1 80 -23,3 —16,4
30 —27,2 (-18,3) 85 —19,5 —13,9
35 —30,6 (—20,3) 90 -15,7 -11,8
40 —34,1 (-22) 95 —12,0 —10 0
45 -38,0 (-24) -
50 — —26
* Цифры в скобках получены экстраполяцией кривых.
Р- 155). Коэффициенты преломления водных растворов диэтилен-
гликоля при 20 °C приведены ниже [14, и. 4, р. 248]:
Концентрация
ДЭГ, % . . 33 61 72 78 91 98 99
........ 1,3685 1,3992 1,4142 1,4205 1,4357 1,4422 1,4450
121
Рис. 35. Зависимость давления насыщенных паров водных растворов диэтилен-
гликоля от температуры.
Рис. 36. Зависимость коэффициентов преломления водных|растворов диэтилен-
гликоля и других гликолей от их концентраций:
1 —• этиленгликоль; 2 — пропиленгликоль^ 3 — диэтиленгликоль i 4 —— дипропиленгликоль*
g — триэтилонгликоль.
На рис. 36 дана зависимость коэффициента преломления водных
растворов диэтиленгликоля от концентрации при 25 °C для сравне-
ния показана зависимость и для других гликолей [15, р. 48].
Коэффициент преломления смесей диэтиленгликоля с этилен-
гликолем является линейной функцией их состава (см. стр. 45).
Для смесей диэтиленгликоля с триэтиленгликолем коэффициенты
преломления при 20 °C имеют следующие значения [14, v. 2, р. 1137]:
Концентрация
ДЭГ, % . . 18 56 73 81 90 93 97
tC° .......... 1,4588 1,4547 1,4541 1,4530 1,4516 1,4497 1,4482
Зависимость поверхностного натяжения диэтиленгликоля от тем-
пературы, а его водных растворов при 25 °C от концентрации пока-
зана на рис. 8 (см. стр. 46). Поверхностное натяжение на разделе
фаз диэтиленгликоля с некоторыми углеводородами при различных
температурах характеризуется следующими данными, мН/м (дин/см)
[14, и. 2]:
При 20 °C При 40 °C При 60 °C
Гептан . . . 10,126 10,133 9,961
Гексан . . . 9,929 9,860 9,704
Циклогексан . . . . . 8,345 8,098 7,746
Бензол . . . 0,511 0,447 0,313
Зависимость теплоемкости диэтиленгликоля и его водных раство-
ров от температуры приведена на рис. 37 [10, с. 534] и в таблч38 [15,
р. 38], а теплоемкость паров диэтиленгликоля — на рис. 9 (см.
стр. 46).
Таблица 38. Зависимость теплоемкости диэтиленгликоля
и его водных растворов от температуры
Температура, °C Теплоемкость при различной концентрации ДЭГ, кал/(г.°С)*
10% 2 0% 40% 60% 80% 100%
20 0,951 0,924 0,851 0,741 0,637 0,548
30 0,956 0,928 0,857 0,753 0,649 0,558
40 0,961 0,933 0,862 0,764 0,662 0,567
50 0,965 0,938 0,869 0,775 0,673 0,576
60 0,970 0,943 0,874 0,787 0,686 0,583
70 0,974 0,948 0,879 0,799 0,699 0,592
80 0,979 0,953 0,885 0,810 0,711 0,601
90 0,984 0,958 0,891 0,822 0,724 0,610
100 0,988 0,963 0,897 0,834 0,736 0,619
НО 0,993 0,968 0,904 0,846 0,749 0,629
* 1 кал/(г.°С) = 4,19 Дж/(г-К).
123
Теплопроводность водных растворов диэтиленгликоля с повыше-
нием температуры до *=* 120 °C незначительно увеличивается, а затем
медленно снижается. Теплопроводность водных растворов диэтилен-
гликоля в интервале от 40 до 160 °C [16] дана в табл. 39, а в интер-
вале от 0 до 180 °C показана на рис. 38 [5]; теплопроводность паров
диэтиленгликоля приведена на рис. 10, б (см. стр. 48).
С повышением давления вязкость диэтиленгликоля значительно
возрастает и при 6,9 МПа (70 кгс/см2) она в 3—4 раза выше, чем при
атмосферном давлении [10, с. 531]. Зависимость вязкости диэтилен-
гликоля и его водных растворов от температуры приведена на рис. 39,
Рис. 37. Зависимость теплоемкости диэтиленгликоля и его водных растворов
от температуры.
Таблица 39. Зависимость теплопроводности водных растворов
диэтиленгликоля от температуры
Температура. °C Теплопроводность при различной концентрации ДЭГ (в мольн. долях), кал/(см-с- °C)*
1,0 0,8 0,6 0,4 0,2 0,1
40 0,495 0,506 0,545 0,636 0,813 1,025
60 0,499 0,511 0,559 0,655 0,836 1.064
80 0,502 0,519 0,566 0,667 0,865 1,107
100 0,502 0,521 0,576 0,674 0,860 1,116
120 0,502 0,521 0,571 0,672 0,884 1,118
140 0,499 0,519 0,571 0,674 0,877 1,114
160 0,497 0,516 0,562 0,672 0,860 1,109
* 1 кал/(см -с °С)=4,19-102 Вт/(м-К).
124
а от давления и температуры — на рис. 40 [17]. Вязкость же паров
диэтиленгликоля показана на рис. 11, б (см. стр. 49). В интервале
температур от —17 до 50 °C и при давлении от 0 до 14,7 МПа (0____
50 кгс/см2), если концентрация диэтиленгликоля выше 60%, вяз-
кость ц (МПа-с) может быть рассчитана по уравнению!
ц=ц0 [14- ю-4 (Ц5 .10-2x4-8,3) ЮР]
где р0 — вязкость раствора при атмосферном давлении; X — кон-
центрация диэтиленгликоля, %; Р — давление, МПа.
Рис. 39. Зависимость вязкости диэтиленгликоля и его водных растворов от
температуры.
Рис. 38. Зависимость коэффициента теплопроводности диэтиленгликоля и его
водных растворов от температуры.
Диэлектрическая проницаемость диэтиленгликоля и его водных
растворов при 15, 25 и 35 °C приведена ниже [6]:
Концентра- ция ДЭГ, % При 15 °C При 25 °C При 35 °C
10 78,8 74,9 71,8
20 75,3 71,5 ' 68,3
30 71,6 ч 67,7 64,6
40 67,4 ’63,7 60,7
50 62,9 59,6 56,6
60 57,8 54,8 52,1
70 52,5 49,7 47,3
80 46,4 44,0 41,8
90 40,0 37,7 35,8
100 32,6 30,7 29,2
При других температурах и в переохлажденном состоянии ди-
электрическая проницаемость (е) диэтиленгликоля имеет следующие
значения [6]:
60 °C 50 "С 20 °C 5 °C — 5 °C —20 °C
24,8 26,4 31,7 34,8 ! 40,7
125
Электропроводность растворов диэтиленгликоля в воде, диметил-
формамиде и пиридине при концентрации ДЭГ 0,1 моль/л при 20 °C
составляет соответственно 38-10"8, 12-Ю"8 и5,9-10'8 ОдГ1 -см-1 [18].
Скорость звука в диэтпленгликоле при 30 °C определена равной
1533 м/с [19]. В тех же условиях скорость звука в этиленгликоле
составляет 1606 м/с. При повышении температуры на 1 °C скорость
звука в диэтиленгликоле снижается на 2,5 м/с.
Рис. 40. Зависимость вязкости водных растворов диэтиленгликоля от давления
и температуры (---- 100% ДЭГ, - . - . - 80% ДЭГ;--------------60% ДЭГ).
Рис. 41. Зависимость точки росы газа при его осушке от температуры контакта
с водными растворами диэтпленгликоля.
Гигроскопичность. Диэтиленгликоль, как и другие гликоли,
обладает высокой гигроскопичностью. Равновесная концентрация
диэтиленгликоля в зависимости от влажности воздуха и температуры
контакта приведена в табл. 40 [15, р. 13].
Зависимость точки росы газа при его осушке водными растворами
диэтиленгликоля от концентрации ДЭГ и его температуры контакта
при атмосферном давлении показана на рис. 41 [10, с. 532].
Растворимость. Диэтиленгликоль смешивается во всех отноше-
ниях с водой и многими органическими соединениями: низшими
спиртами и гликолями, целлозольвами и карбитолами, этанолами-
нами, ацетоном, дихлордиэтиловым эфиром, фенолом, ледяной
уксусной кислотой, фурфуролом, пиридином, гликольдиацетатом,
хлороформом, нитробензолом, анилином, хлорбензолом, метилизо-
бутилкетоном и метилизобутилкарбинолом. Смешение диэтилен-
гликоля с рядом веществ, например с водой и бензолом, сопрово-
126
Таблица 40. Зависимость равновесной концентрации дпэтиленгликоля
от относительной влажности воздуха и температуры
Равновесная концентрация при относительной влажности, %
Температура, °C 10% 20% 30% 40% 50% 60% 70% 80% 90%
-5 97,8 95,1 92 89 86 83 78 68 52
5 97,7 95,0 92 89 86 82 77 67 50
15 97,7 95,0 92 89 86 82 76 66 48
25 97,6 94,9 92 89 85 81 75 65 47
35 97,6 94,8 92 89 85 81 74 64 46
45 97,6 94,8 92 89 85 80 73 63 45
ждается выделением теплоты, количество которой зависит от мольной
концентрации диэтиленгликоля и температуры [20, с. 74, 166].
Так, при смешении с водой выделяется следующее количество те-
плоты (в кал/моль * раствора):
Концентра- ция ДЭГ, % (мол.) При 25 °C При 35 °С^ При;45 »с
3,7 -103 -92,5 -89,3
6,9 —202 -187 —182
15,9 —282 -255 —242
27,4 -360 —274 —269
48,6 —272 -239 —211
60,4 —239 —203 -178
80,0 -153 -127 -115
Теплота же смешения диэтиленгликоля с бензолом равна
(в кал/моль * раствора):
Концентрация
ДЭГ, % (мол.)
65,2
70,1
75,8
82,4
90,4
При 25 °C
-30,6
—27,3
-20,1
-15,6
При 35 °C
-3,8
-3,2
—3,6
-3,1
-2,8
При 45 °C
27,6
25,4
16,3
1,7
6,8
В диэтиленгликоле почти нерастворимы минеральные масла
и масла растительного и животного происхождения: кокосовое,
льняное, лярд, оливковое, соевое, спермацетовое, тунговое, хлоп-
ковое. Растворимость различных веществ в диэтиленгликоле при
25 °C приведена в Приложении, табл. 8, стр. 357 [1, р. 161; 15, р. 9].
* 1 кал/моль = 4,19 Дж/моль.
127
Растворимость хлористого кальция и хлористого натрия в 80%-
ном водном растворе диэтиленгликоля меняется с повышением тем-
пературы следующим образом [21]:
20 °C
СаС12..................8,0
NaCi ..................6,8
40 °C 60 °C 80 °C 100 °C 120 °C
9,7 11,8 14,6 17,8 21,3
8,0 10,4 13,0 15,7 18,0
Растворимость иода в водных растворах диэтиленгликоля при
25 °C составляет [22, с. 40]:
Концентрация
ДЭГ, % (об.) 15 30 45 60 75 90 .100
Растворимость
иода, г/л . . 0,609 1,189 3,234 10,67 39,86 137,2 174,9
В табл. 41 дана зависимость растворимости олеата натрия в сме-
сях диэтиленгликоля с различными растворителями при 25 °C от
концентрации диэтиленгликоля [22, с. 265—274].
Критическая температура растворения (К. Т. Р.) диэтиленгли-
коля, при которой достигается неограниченная взаимная смешива-
емость диэтиленгликоля с некоторыми соединениями, ограниченно
растворимыми при других температурах, приводится ниже [23,
р. 70—71]:
Компонент К.Т.Р., °C
в/пор-Амилбензол . . . 210
Аценафтен ............... 136
Бензол ................. 88,5
втор-Бутилбензол ... 191
тро/га-Бутилбепзол ... 189
Гексаэтилбензол .... 258
Дибензил ................ 160
Ди-тре/п.-бутилпафталип 231
Диизопропилбензол . . 219
Дифенил ................. 126
Диэтилбензол ............ 193
Изопропилбензол .... 178
n-Изопропилтолуол (ци-
мол) .................. 194
Компонент К.Т.Р., °C
Изосафрол .... . . 84,2
о-Ксилол . . 153
1-Метилнафталин . . 126
Нафталин . . 85
«-Нитротолуол . . . . 48,5
о-Нитрофенол . . . . . 42
га-Нитр охлорбензол . . 76
Стирол . . 111
Тимол . . 33
Толуол . . 134
Фенантрен .... . . 128
Флуорен . . . ^ . . . . 138
Этилбензол .... . . 155
С рядом веществ диэтиленгликоль образует азеотропные смеси;
состав и температура кипения некоторых из них приведены в При-
ложении, табл. 7, стр. 356 [24, с. 416—420].
Фазовое равновесие жидкость — пар для системы диэтилен-
гликоль — вода при атмосферном давлении и температура кипения
128
Таблица 41. Зависимость растворимости олеата натрия в смесях
дпэтпленгликоль— растворитель от концентрации ДЭГ
Растворимость олеата натрия в смеси ДЭГ—растворитель, %
Концентраци; ДЭГ, % , четыреххлО' 1 р истый углерод хлороформ хлористый метилен дихлорэтан этиловый спирт пропиловый спирт бутиловый спирт амиловый спирт
20 14,26 15,28 12,79 9,40 7,52 9,47
40 19,83 20,03 19,25 14,69 11,04 13,81 16,15 16,86
60 20,47 20,95 19,13 16,44 13,89 16,61 18,85 19,85
80 17,67 16,76 16,24 13,04 12,97 15,78 17,53 18,21
100 7,45 7,45 7,45 7,45 7,45 7,45 7,45 7,45
растворов характеризуется данными, приведенными ниже [14, v. 4,
р. 2481:
Концентрация ДЭГ,
%
В жидкости . 33 61 72 78 91 98 99 99—100 99—100
в парах , . , о 0 0 0 0 4 18 49 77
Температура, °C 46 49 51 54 64 92 108 132 154
Таблица 42. Фазовое равновесие жидкость — пар для системы
дпэтпленгликоль — то луо л
Концентрация толуола, % (мол.) Давление, мм рт. Ст. * Концентрация толуола, % (мол.) давление, мм рт. ст. *
жидкость пар ЖИДКОСТЬ пар
При 50 °C При 100 °C
6,62 99,82 55,8 11,57 99,46 388,3
8,88 99,84 63,9 15,80 99,47 448,8
10,45 99,85 67,6 19,77 99,62 494,8
12,69 99,88 74,2 21,49 99,69 488,8
14,69 99,88 82,3 При 130 °C
15,33 99,88 83,3 1,61 96,13 274,2
При 100 °C 2,52 96,86 325,3
1,84 98,34 120,7 3,44 97,22 392,3
4,24 99,12 193,1 5,94 98,11 529,6
8,66 99,41 321,3 11,35 98,48 632,3
* 1 март. ст. = 133,32 Па.
9 Заказ 972
129
Для этой же системы при 80 °C фазовое равновесие характери-
зуется следующими данными [25, с. 332]:
Содержание воды, % (мол.)
в жидкости .................. 8,7 14,7 23,2 37,7 77,7
в парах....................... 87,3 91,5 95,8 98,2 99,7
Фазовый состав системы диэтиленгликоль — вода при различном
давлении показан на рис. 42, для системы диэтиленгликоль — три-
Рис. 42. Фазовый состав системы диэтиленгликоль — вода:
130
этиленгликоль при 0,4 кПа (3 мм рт. ст.) — на рис. 43 [1, п 157
10, с. 533].
В табл. 42 и 43 приведено фазовое равновесие жидкость — пар
для систем диэтиленгликоль с толуолом и о-крезолом, а на рис. 44 —
с .w-крезолом и п-крезолом [25, с. 738].
Равновесие в трехкомпонентных системах, включающих диэти-
ленгликоль, представлено графически в виде треугольника Гиббса
на рис. 45. В работах [22, 23, 26—29] изучены также фазовые
Рис. 44. Фазовое равновесие жидкость — пар для систем диэтиленгликоль —
.и-крезол (а) и диэтиленгликоль — re-крезол (б) при атмосферном давлении.
Таблица"43. Фазовое равновесие жидкость — пар для системы
диэтпленглико ль — о-ксилол
Концентрация о-ксилола, % (мол.) Давление, мм рт. ст. * Концентрация о-ксилола, % (мол.) Давление, мм рт. ст.*
жидкость пар ЖИДКОСТЬ пар
При 100 °C При 125 °C 283,4 313,1
2,4 96,36 54,9 9,1 97,39
5,4 97,70 86,8 11,0 97,89
7,4 98,19 - 127,0 При 150 °C
8,2 98,32 136,9 2,2 83,40 170,5
10,9 98,44 155,6 3,8 90,01 261,2
При 125 °C 6,8 92,78 362,8
3,0 93,98 127,9 8,0 94,03 94,98 450,9
6,0 95,71 187,4 10,8 525,9
7,7 96,49 242,3 11,8 95,22 577,4
I мм рт. ст. = 133,32 Па.
9*
131
равновесия ряда других тройных систем, содержащих диэтиленгли-
коль, а именно: вода — бензол, вода — гептан, бензол — гептан, сти-
рол — этилбензол, декан и спирты (н-бутиловый, w-амиловый, к-геп-
тиловый, «-октиловый, «-нониловый, «-дециловый и «-додециловый);
Рис. 45. Диаграмма равновесия в трех- и четырехкомпонентных системах,
включающих дпэтиленгликоль (ДЭГ).
Условные обозначения: Ди-вторбб—ди-втор-бутилбензол; ж — жидкость; НД___________
о-нигродифенил; МН — i-метилнафталин; т — твердая фаза; ------ ноды;________изо-
пикны.
нониловый спирт — гептан, нониловый спирт — додекан, бензол —
тиофен, газовый конденсат — метиловый спирт, бензол — «-гексан.
Данные по фазовому равновесию в четырехкомпонентных систе-
мах, содержащих кроме диэтиленгликоля н-декан — «-октиловый
спирт — н-нониловый спирт, н-декан — «-нониловый спирт, «-деци-
ловый спирт и гептан — бензол — вода, приведены в [22, 30].
132
г
Получение диэтиленгликоля
Диэтиленгликоль впервые был получен в 1859 г. двумя различными
методами [31]:
1) при взаимодействии окиси этилена с водой и этиленгликолем
2Н3С—СН2 + Н2О —> СН2ОНСН2—О—СН2СП2ОН
\)/
Н2С—СН2+СН2ОНСН2ОН —> СН2ОИСН2—О-СН2СН2ОН
44 о
2) при взаимодействии этиленгликоля с бромистым этиленом
ЗСН2ОНСН2ОН+С2Н4Вг2—> СН2ОНСН2—О—СН2СН2ОН+2С2НвОВг+Н2О
Диэтиленгликоль может быть получен также из этиленгликоля
и этиленбромгидрина по реакции
СН2ОНСН2ОН+С2И5ОВг —> СН2ОНСН2—О—СН2СН2ОН+НВг
или из этиленгликольацетата и гликолята натрия по реакции [32]’
CH2OHCH2OCOCH3 + NaOCH2CH2OH —>
—> СН2ОНСН2—О—СН2СН2ОН-|-CH3COONa
По реакции гидролиза [5,[5'-дихлордиэтилового эфира (хлорекса)
содой
CH2ClCH2-O-CH2CH2Cl+H2O + Na2CO3 —>
—> CH2OHCH2-O-CH2CH2OH + 2NaCl+COa
получали небольшие количества диэтиленгликоля в Германии во
время второй мировой войны [33, с. 93].
Известны и другие методы получения диэтиленгликоля [34]:
гидролиз диацетата диэтиленгликоля при его нагревании в мета-
нольном растворе хлористоводородной кислоты, синтез из этилен-
гликоля при его нагревании в токе двуокиси углерода или
в присутствии щелочи, получение диэтиленгликоля при нагревании
слабокислых водных растворов (pH 5—7) высших гликолей до 200—
300 °C:
СН2ОНСП2-О-СН2СН2-О-СН2СН2ОН + Н2О —>
—► СН2ОНСН2-О—СН2СН2ОН + СН2ОНСН2ОН
В настоящее время в промышленных условиях диэтиленгликоль
получается как побочный продукт в производстве этиленгликоля
гидратацией окиси этилена или же он синтезируется (как основной
продукт) из окиси этилена, воды и этиленгликоля. Количество
получающегося в качестве опбочного подукта диэтиленгликоля
зависит от соотношения между водой и окисью этилена в шихте,
поступающей на гидратацию. Так, при мольном соотношении
вода : окись этилена, равном 10,5 : 1, на получение диэтиленгликоля
используется 12,7% окиси этилена и 82,3% — на этиленгликоль;
133
а при соотношении, равном 2,1 : 1, расход окиси этилена на диэтилен-
гликоль повышается до 34,5%, а на этиленгликоль снижается до
47,2%. Дальнейшее снижение соотношения воды и окиси этилена
приводит к уменьшению выхода диэтиленгликоля за счет увеличения
выхода три- и тетраэтиленгликолей.
При производстве этиленгликоля по схеме, описанной в гл. II
(мольное отношение вода : окись этилена 16 : 1), на 1 т товарного
этиленгликоля выделяется 105 кг, или 10,5%, диэтиленгликоля.
При снижении отношения воды и окиси этилена до 14 : 1 выход
дизтпленгликоля повышается до 20% [35, р. 401.
При целевом синтезе диэтиленгликоля из окиси этилена и воды
образующийся этиленгликоль после выделения из смеси гликолей
возвращается в процесс. В качестве побочных продуктов образуются
три- и тетраэтиленгликоль, а также незначительное количество еще
более высокомолекулярных гликолей.
Диэтиленгликоль с высоким выходом получается оксиэтилиро-
ванием этиленгликоля при достаточно большом мольном отношении
этиленгликоля и окиси этилена в исходной смеси. Как и при реакции
окиси этилена с водой, состав продуктов оксиэтилирования этилен-
гликоля зависит от отношения окиси этилена к этиленгликолю в исход-
ной смеси и констант скоростей протекающих последовательно-
параллельных реакций образования ди-, три- и тетраэтиленгликоля
и других полигликолей (см. гл.IV, стр.161) [36].
Процесс получения диэтиленгликоля включает следующие ста-
дии: приготовление шихты — водного раствора окиси этилена и эти-
ленгликоля; взаимодействие окиси этилена с этиленгликолем; выпа-
ривание водного раствора гликолей и ректификация концентриро-
ванного раствора гликолей. Технологическая схема процесса
аналогична схеме процесса получения этиленгликоля (см. рис. 28,
стр. 85), на одном и том же оборудовании могут производиться
оба продукта [35, р. 41].
Режим получения диэтиленгликоля такой же, как при получении
этиленгликоля: температура 180—200 °C и давление 1,47—2,06 МПа
(15—21 кгс/см2) на одних заводах [35, р. 34] и 160—180 °C и 1,08—
1,28 МПа (И—13 кгс/см2) на других [33, с. 73]. Время пребывания
шихты в реакционной зоне около 40 мин.
Однако при производстве диэтиленгликоля в состав шихты входит
3 компонента (окись этилена, вода и этиленгликоль), а при полу-
чении этиленгликоля — два (окись этилена и вода). Для пригото-
вления шихты использовали конденсат вторичного пара после II
и III корпусов трехкорпусной выпарной установки (конденсат I кор-
пуса, содержащий заметное количество легкокипящих органических
примесей, выбрасывали), а также паровой конденсат или воду,
обессоленную с помощью ионитов.
Порядок приготовления шихты был различным; периодическое
приготовление водно-гликолевого раствора и непрерывное его сме-
шение с окисью этилена в смесителе, непрерывное смешение воды
и этиленгликоля в трубопроводе и затем непрерывное же смешение
134
полученного раствора с окисью этилена, непрерывное смешение всех
трех компонентов в смесителе. Состав шихты, поступающей после
смесителя в реактор, таков: 12,3% окиси этилена, 41,5% воды и 46,2%
этиленгликоля. Мольное отношение компонентов в шихте составляло
1 : 8, 2 : 2,65. Практиковались и другие соотношения окиси этилена,
воды и этиленгликоля, а именно: 1 : 8,2 : 1,9 (на более новых заво-
дах) и 1 : 6; 8 : 2,8 (на старых заводах) [33, с. 70]. Реакционная
масса выпаривалась с 35—49 до 8—18% воды.
Отгонка остаточной воды и этиленгликоля на различных заводах
производилась по-разному. На одних заводах, так же как при полу-
чении этиленгликоля (см. рис. 28, стр. 85), вода отгонялась в ко-
лонне 14, а этиленгликоль — в колонне 15. После смешения их
направляли в смеситель 3 для получения шихты. Кубовый остаток
из колонны 15 поступал в колонну 16 для получения товарного ди-
этиленгликоля .
На других заводах колонны 14 и 15 работали параллельно, без
флегмы, и на них отгонялась смесь воды и этиленгликоля, которая
направлялась на смешение с окисью этилена в смеситель 3. В связи
с тем что при получении диэтиленгликоля образуется триэтилен-
гликоля почти в 8 раз больше, чем при получении этиленгликоля,
насадочные колонны 18 для выделения триэтиленгликоля работали
непрерывно. Режим работы систем выпаривания гликолей и их
ректификации при производстве диэтиленгликоля такой же, как
при получении этиленгликоля.
Суммарный выход ди- и триэтиленгликоля достигал 92—96%
от теоретического, в том числе диэтиленгликоля от 76 до 85% и три-
этиленгликоля от 10 до 19% в зависимости от соотношения окись
этилена : вода : этиленгликоль в исходной шихте. В том случае,
когда требовался преимущественно диэтиленгликоль, увеличивали
содержание воды в шихте и снижали содержание этиленгликоля.
Когда же наряду с диэтиленгликолем необходимы были значительные
количества триэтиленгликоля, поступали наоборот — содержание
воды в шихте уменьшали, а этиленгликоля — повышали. Это при-
водило к увеличению выхода не только триэтиленгликоля, но и
тетраэтиленгликоля, а также более высокомолекулярных гликолей.
Расходные коэффициенты на 1 т диэтиленгликоля и выход побоч-
ного продукта триэтиленгликоля приведены ниже [33, с. 77; 35,
Р. 44]:
Окись этилена, т ................1,00—1,14
Пар, т .............................. 6—9
Вода, м3 .......................... 300—500
Электроэнергия, кВч.............. 50—65
Триэтиленгликоль, кг ............ 150—250
В технологическую схему получения диэтиленгликоля, описан-
ную выше, был внесен ряд усовершенствований, значительная часть
которых направлена на улучшение качества продукта. В результате
135
замены аппаратуры из углеродистой стали на аппаратуру из высоко-
легированной стали получается диэтиленгликоль с ничтожным
содержанием железа. Установка на выпарных аппаратах колонн
позволяет отогнать воду, не содержащую заметных количеств эти-
ленгликоля; эта вода выводится из системы, таким образом исклю-
чается накопление различных примесей, отгоняющихся вместе с во-
дой (ранее она возвращалась в процесс для получения шихты).
В этом случае шихта готовится на чистой обессоленной воде.
Применение пленочных или роторных испарителей и эффектив-
ных ректификационных колонн с улучшенной конструкцией тарелок
позволяет проводить ректификацию при более низкой температуре
и получать продукт более высокого качества. Для устранения не-
приятного запаха и повышения термической стойкости ди- и три-
этиленгликоля смесь гликолей, от которой предварительно отогнан
этиленгликоль, перегоняется при пониженном давлении. К получен-
ному дистилляту добавляется 5—£0% воды и на следующей колонне
при пониженном давлении и температуре в кубе не выше 190 °C
из него отгоняются примеси. Кубовая жидкость последней колонны
подвергается ректификации в вакууме при температуре не выше
190—200 °C. Полученный диэтиленгликоль не имеет неприятного
запаха, его цветность равна 5, она не изменяется при нагревании
с соляной кислотой при 200 °C. При разделении смеси гликолей без
добавки воды цветность диэтиленгликоля равна 15 и достигает
200 при нагревании в течение 2 ч с соляной кислотой при
200 °C [37].
Для получения диэтиленгликоля с цветностью по шкале Pt — Со
не более 10 рекомендуется следующий режим ректификации [38]:
давление вверху колонны 0,67 кПа (5 мм рт. ст.), температура верха
колонны 121 °C, низа — 180 СС, pH гликолей, поступающих на
ректификацию, 6,5. Цветность диэтиленгликоля можно улучшить,
пропуская его при 80—100 °C через слой сильнокислого катионита
(леватит 5-115) [391.
Отработанный диэтиленгликоль можно очищать с помощью ионо-
обменных смол. Например, диэтиленгликоль, применяемый для
осушки природного газа, очищают от накапливающихся в^нем при-
месей соли и окислов железа на песочных фильтрах (для отделения
механических примесей), катионите и анионите. Регенерацию ионитов
проводят 1%-ным раствором H2SO4 и 4%-ным раствором NaOH [40].
Очистка диэтиленгликоля от ионов хлора возможна с помощью
сильноосновных анионитов (77L4-400, АВ-17-8) [41].
Применение дпэтиленгликоля
Области применения диэтиленгликоля и этиленгликоля весьма
сходны, однако вследствие меньшей летучести, более высокой темпе-
ратуры кипения и вязкости использование диэтиленгликоля для
некоторых целей предпочтительнее. В 1970 г. в США общее по-
требление диэтиленгликоля составило около 150 тыс. т. Из них 30%
136
Израсходовано на производство полиуретанов и смол из ненасыщен-
ных сложных эфиров, 13% использовано на получение триэтилен-
гликоля, 12% — на текстильные вспомогательные вещества, 7% —
на пластификаторы и поверхностно-активные вещества, 7% — в ка-
честве экстрагирующего агента ароматических углеводородов и
10% — на экспорт. Остальное количество использовалось в различ-
ных отраслях промышленности [42, р. 85].
Используя большую гигроскопичность диэтиленгликоля, его
применяют для поддержания необходимой влажности табака, табач-
ных изделий и бумаги. Диэтиленгликоль применяется в качестве
пластификатора целлофана и других пленочных материалов, проб-
ковых изделий, клеев, набивочных и уплотняющих материалов
газопроводов. Он также нашел применение как растворитель, умяг-
чающий и увлажняющий агент на различных стадиях производства
натуральных и химических волокон, получения пряжи и тканей,
а также для производства печатных красок.
При взаимодействии диэтиленгликоля с ненасыщенными кисло-
тами (малеиновой, фумаровой и др.) получаются сложные эфиры.
При полимеризации последних с мономерами, имеющими винильные
группы (винилацетат, метилметакрилат, стирол), получаются твердые
прозрачные смолы, которые обладают хорошими оптическими свой-
ствами и пригодны для получения стеклопластиков, клеет/ Мягкие
смолы, применяемые в нитроцеллюлозных лаках, эмалям и клеях,
образуются при этерификации диэтиленгликоля смоляными кисло-
тами. Модифицированные алкидные смолы, обладающие большей
щелочестойкостью, получаются при частичной замене глицерина
диэтиленгликолем. Сложные эфиры диэтиленгликоля и жирных
кислот служат пластификаторами и клеями, а эфиры ароматических
кислот или смесей ароматических и жирных кислот являются пла-
стификаторами поливинилхлорида [1, р. 164].
Диэтиленгликоль предлагается применять как усилитель напол-
нителей в светлых резинах. При введении его в резиновые смеси
на основе бутадиенстирольного каучука СКС-30, содержащих белую
сажу, предел прочности резин при разрыве повышается на 20%
и снижается время вулканизации [43].
Стирольный раствор ненасыщенного полиэфира диэтиленгликоля
И малеинового ангидрида, содержащий катализатор и ускоритель
полимеризации, служит покрытием для плит из синтетических
смол [44]. Термостойкие полиуретаны, применяемые в качестве
лаков для покрытия металлических поверхностей, получаются из
диэтиленгликоля и лабильных уретанов, производных диизоциана-
тов и фенолов [45]. Электроизоляционный лак, стойкий к нагрева-
нию до 155 °C и устойчивый к влаге и химическим реагентам, обла-
дающий хорошей адгезией и эластичностью, получается при этери-
фикации терефталевой кислоты смесью ди- и этиленгликоля, а затем
глицерином [46]. Клеи и герметики получаются при конденсации
полиэфира на основе диэтиленгликоля и адипиновой кислоты с эп-
оксидной смолой [47].
137
Диэтиленгликоль используется при синтезе фосфорсодержащих
полиэфиров. Огнестойкие полиуретаны образуются из полиэфиров
фосфорной кислоты, получаемых переэтерификацией диметилфосфата
смесью диэтпленгликоля и триметилолэтана или триметилолпро-
пана [48]. Трудно возгорающиеся и мгновенно гаснущие при удале-
нии пламени линейные полиэфиры с фосфором в основной цепи
получаются поликонденсацией диэтпленгликоля с дихлоридом ал-
лилфосфорной кислоты [49]. Маслянистые жидкости, являющиеся
хорошими отвердителями для смол, получаются этерификацией
диэтпленгликоля хлорокисью фосфора и последующей обработкой
образовавшегося тетрахлорфосфата этилен- или пропиленими-
ном [50].
В результате конденсации диэтпленгликоля с пиромелитовым
диангидридом образуется продукт, употребляемый в качестве отвер-
ждающего агента для эпоксидных смол [51]. Стабилизаторами для
синтетических смол, антиоксидантами, ускорителями вулканизации
и полимеризации являются соединения, полученные при взаимо-
действии оловоорганических дигалогенидов с диэтиленглико-
лем [52]. Диэтиленгликоль предложен в качестве инертной среды
при синтезе бензооксазолинона и Ni-фенолятов диоксиалкилдифенил-
сульфонов, являющихся стабилизаторами полиолефинов [53]. Ди-
этиленгликоль применяется для синтеза лекарственных препаратов,
например алкилстероидов, обладающих многофункциональным дей-
ствием [54], для повышения эффективности инсектицидов и гер-
бицидов [55].
При реакции диэтпленгликоля с трихлорметильными аромати-
ческими соединениями образуются хлоралкиловые эфиры аромати-
ческих кислот, которые могут быть использованы в качестве про-
межуточных продуктов, растворителей и сельскохозяйственных
химикатов [56].
Для получения ряда материалов используется смесь диэтилен-
гликоля с этиленгликолем, например для получения антифриза,
гидравлических и гидротормозных жидкостей, электролита
для электролитических конденсаторов. Диэтиленгликоль при-
меняется для получения диэтиленгликольдинитрата (динитро-
гликоля), сложного эфира диэтпленгликоля и азотной кислоты:
O2NOGH2CH2OGH2GH2ONO2.
Во время второй мировой войны в Германии из-за нехватки
глицерина в большом количестве выпускались нитродигликолевые
пороха и динамиты; эти взрывчатые вещества изготовлялись также
и в других странах.
Диэтиленгликольдинитрат имеет ряд преимуществ по сравнению
с нитроглицерином: у него большая способность пластифицировать
нитроцеллюлозу, что позволяет вводить его в меньших количествах;
пороховая масса обрабатывается на вальцах лучше, стойкость поро-
хов выше и их использование повышает срок службы орудий. Смеси
динитрогликоля и нитроглицерина образуют труднозамерзающие
динамиты, производство и применение которых в качестве взрывча-
138
тых веществ менее опасно, чем полученных на основе одного нитро-
глицерина [57, с. 624].
Способность диэтиленгликоля хорошо растворять ароматические
углеводороды и плохая растворимость в нем парафиновых и нафте-
новых углеводородов обусловили его широкое использование в ка-
честве селективного растворителя для выделения ароматических
углеводородов (бензола, толуола, ксилолов) из продуктов катали-
тического риформинга, пиролиза и других фракций. Как селективный
растворитель диэтиленгликоль предпочтительнее этиленгликоля, так
Рис. 46. Технологическая схема для выделения ароматических углеводородов
экстракцией диэтиленгликолем:
1 — экстракционная колонна; 2 — промывная колонна; 3 — отпарная колонна5, 9,
11, 13 — емкости; 6 — печь; 7 — колонна для очистки глиной; 3 — бензольная колонна;
10 —толуольная колонна; 12 — ксилольная колонна.
как в нем лучше растворяются ароматические углеводороды, а в связи
с большей разностью температур кипения между углеводородами
а растворителем значительно упрощается их выделение из экстракта,
т. е. обеспечивается получение продуктов высокой степени чистоты.
На рис. 46 дана технологическая схема установки для выделения
ароматических углеводородов из продуктов платформинга бензина
[58, с. 93]. В результате экстракции, очистки и ректификации полу-
чаются продукты с содержанием основного вещества: бензол —
99,9%, толуол — 99,8 и ксилол — 99,8% [59].
Диэтиленгликоль широко используется для осушки газов в неф-
тяной, газовой, нефтехимической и других отраслях промышленности.
В 1966 г. в США на осушку природного газа было израсходовано
31% произведенного диэтиленгликоля [42, 2 Ed., р. 72]. При осушке
139
газа диэтиленгликолем практически исключается образование водя-
ного конденсата, ледяных пробок и кристаллогидратов в газопро-
водах, а также снижается коррозия труб. Степень осушки зависит
от давления и температуры газа и от концентрации диэтиленгликоля,
подаваемого на осушку. Чем выше концентрация диэтиленгликоля,
тем выше степень осушки и соответственно ниже точка росы газа
после осушки. При концентрации диэтиленгликоля 97—98% точка
росы осушенного газа снижается на 33—36 °C, а при концентрации
99,5% точку росы можно снизить на 42—58 °C [60, с. 64]. Техноло-
гическая схема установки осушки газа диэтиленгликолем приведена
на рис. 47 [10, 536].
После осушки газа диэтиленгликоль концентрируется в вакууме,
что позволяет снизить температуру в кубе отпарной колонны и тем
Рис. 47. Технологическая схема установки осушки диэтиленгликолем:
1 — водоотделитель; 2 — абсорбер; з — холодильник; 4 — промежуточная емкость; 5 —
теплообменник; в — выпарная колонна; 7 — кипятильник; 8 — конденсатор; 9 — сборная
емкость.
самым уменьшить потери диэтиленгликоля за счет его разложения.
Для получения 99,5—99,8%-ного диэтиленгликоля регенерацию
проводят с применением отпарного газа, т. е. подачей небольшого
количества сухого газа в отпарную колонну [611.
Диэтиленгликоль в смеси с этаноламинами применяется для
очистки углеводородных газов от таких примесей, как H2S, СО2.
Требования, предъявляемые к качеству
диэтиленгликоля
Диэтиленгликоль выпускается промышленностью различных сортов
(марок) в зависимости от требований, предъявляемых в нему по-
требителями. Диэтиленгликоль наиболее высокого качества тре-
буется для получения полиэфиров, полиэфируретанов и некоторых
других продуктов. Наличие в диэтиленгликоле воды, кислот, железа
и других примесей может привести к понижению молекулярной •
массы полиэфира, ухудшению его структуры и некоторых важных^
показателей, а также к повышению расхода катализаторов. ]
В Советском Союзе диэтиленгликоль производится по ГОСТ]
10136—62 и по ГОСТ 5.2266—75 с требованиями к качеству аттесто-^
ванной продукции. Согласно ГОСТ 10136—62 диэтиленгликоль
140
Таблица 44. Технические требования на диэтиленгликоль
по ГОСТ 10136-62
Показатели Нормы для марок
ДП ДН дг
Внешний вид J , в . Цвет по Pt — Со шка- Прозрачная, без механиче- ских примесей жидкость Прозрачная, бес- цветная или слабо- окрашенная в жел- тый цвет жидкость без механических примесей и посто- ронних запахов Прозрачная, бес- цветная или слабо- окрашенная в жел- тый цвет жидкость без механических примесей
ле, не более , . . . Плотность при 20 °C, 30 Не определяют
г/смэ Кислотность .... Число омыления, мг КОН на 1 г диэти- ленгликоля, не бо- 1,1158-1,1163 О т с у 1,1158-1,1163 т с т в и е 1,1158-1,1163
лее 0,1 . 0,4 0,4
Растворимость в воде Содержание, % диэтиленгликоля, О т с у т с г в и е мути Не определяют
не менее .... этиленгликоля. 98,7 96,5 96,5
не более . . . . восставав лива- 0,2 1,0 Не определяют
ющих веществ Не определяют Отсутствие Не определяют
воды, не более 0,1 0,4 0,4
хлор-иона . . . Температурные преде- лы перегонки (при 0,1 МПа), °C начало кипения, Не определяют Отсутствие Не определяют
не ппже .... температура пос- ле отгона 90 мл дистиллята, не 244,0 241,0 240,0
не выше .... конец кипения, не Не определяют 246,5 246,5
Выше Количество отгона в указанных преде- 247,5 250,0 250,0
лах, мл, не менее . . . 98,0 96,0 96,0
Должен соответствовать показателям и нормам, приведенным
в табл. 44.
В соответствии с ГОСТ 5.2266—75 цвет продукта не выше 15,
содержание в нем диэтиленгликоля не менее 99,3%, органических
примесей не более 0,6% (в том числе не более 0,15% этиленгликоля),
конец кипения не выше 249 °C. Остальные показатели соответствуют
требованиям на диэтиленгликоль марки ДП по ГОСТ 10136—62.
141
Таблица 45. Спецификации на диэтиленгликоль иностранных фирм [62, 63]
Наименование фирмы (страна)
Показататели IG1 (Англия) Shell (Англия) Kuhlmann (Франция) Celene (Италия) Shell Chem. Со. (США)
Цвет, не более 15 20 5 15 15
Относительная плотность . . . 1,120- 1,118- 1,116- 1,1170— 1,118—
Кислотность (в расчете на уксус- 1,121 * 1,121 1,117 1,1200 1,121
нуго кислоту), % , не более Содержание, % , не более 0,005 0,005 0,002 0,01 0,005
ВОДЫ 0,2 0,3 0,1 0,2 0,2
золы Температурные пределы пере- гонки при 0,1 МПа, °C 0,005 0,010 — 0,005 0,005
начало кипения, не ниже 240 235 242 235 242
конец кипения, не выше 255 255 246 255 '250
* Относительная плотность при 15,5° С.
Спецификации на диэтиленгликоль некоторых иностранных фирм
приводятся в табл. 45.
Фактические показатели, как правило, лучше, чем указываемые
в спецификациях. Например, для диэтиленгликоля, выпускаемого
фирмой 1CI, они таковы [64]:
Кислотность, % ...................0,002
Содержание воды, % ................ 0,08
Пределы кипения при 0,1 МПа
при 243 °C выкипает, % (об.) ... 5
при 247 °C выкипает, % (об.) ... 95
ЛИТЕРАТУРА
1. Glycols. Ed. by G. О. Curme, F. Johnston. New York, Reinhold Publ. Corp.,
1953. 389 p.
2. Справочник химика. T. 1—3. Изд. 2-е. М.—Л., «Химия», 1963—1966.
3. Vanderkool W. N., Hlldenbrand D. L., Stull D. R., J. Chem. Data, 1967,
v. 12, № 3, p. 377—379; Ганиев Ю. А., ЖФХ, 1969, т. 43, вып. l,c. 239-240.
4. Gallaugher A. F., Hibbert H., J. Am. Chem. Soc., 1937, v. 59, № 12, p. 2514—
2521.
5. Gallant R. W., Hydrocarb. Process, 1967, v. 46, № 4, p. 183—196.
6. M arenas M., Douheret G„ C. r., Ser. C., 1971, v. 272, № 12, p. 1060—1062;
Ахадов Я. Ю. Диэлектрические свойства чистых жидкостей. М., изд. Стан-
дартов, 1972. 412 с.
7. Koizumi N., Hanai Т., J. Phys. Chem., 1956, v. 11, p. 1496—1500.
8. Сталл Д., Вестрам Э., Зинке Г. Химическая термодинамика органических
соединений. Пер. с англ. М., «Мир», 1971. 807 с.
9. Dykyt I., Kuska V., Seprakova М., «Chemicke Zvesti», 1956, v. 10, № 4,
p. 193—203.
142
10. Катц Д. Л. и др. Производство по добыче, транспорту и переработке при-
родного газа. Пер. с англ. Под ред. Ю. П. Коротаева и Г. В. Пономарева.
М., «Недра», 1965. 676 с.
11. Сталл Д. Р. Таблицы давления паров индивидуальных веществ. Пер. с англ.
Под ред. С. В. Горбачева и В. В. Михайлова. М., Издатинлит, 1949.
72 с.
12. Миронов К. Е., Дерябина Л. Д., ЖПХ, 1962, т. 35, № 6, с. 1338—1342.
13. Коулъ А. Л., Ризенфелъд Ф. С. Очистка газа. Изд. 2-е. Пер. с англ. Под ред.
И. И. Абрамсона. М., «Недра», 1968. 392 с.
14. Timmermans I. The Physice-Chemical Constants of Binary Systems in Con-
centrated Solutions. New York — London, Interscience Publ. Inc., 1959—
1960. V. 2 — 1272 p.; v. 4 — 1332 p.
15. Glycols. Properties and Uses. Midlands (Michigan), The Dow Chem. Co.,
1961. 64 p.
16. Расторгуев Ю. Л., Газдиев M. А., Инж.-физ. ж., 1969, т. 17, вып. 1, с.
72—79;
Расторгуев Ю. Л., Газдиев М. А., ЖФХ, 1970, т. 44, вып. 12, с. 3092—3095;
Ганиев Ю. А. В кн.: Теплофизические свойства жидкостей. М., «Наука»,
1970, с. 95—98; Газдиев М. А., Расторгуев Ю. Л., ЖФХ, 1971, т. 45, вып. 3,
с. 692-693.
17. Клименко А. П. и др., Газ. пром., 1967, № 11, с. 47—48.
18. Prey V., Kretschek Е., Berbalk Н., Allgemeine u. praktische Chemie, 1971,
Bd. 22, Heft 6, S. 153-156.
19. Rao K. S., Rao B. R., J. Acoust. Soc. Am., 1959, v. 31, № 4, p. 439—441.
20. Белоусов В. П., Морачевский А. Г. Теплоты смешения жидкостей. Л.,
«Химия», 1970. 256 с.
21. Могильный В. И., Клименко А. П., Чалюк Г. И., Хим. техн., 1972,
№ 1 (61), с. 42.
22. Справочник по растворимости. Т. 2. Под ред. В. В. Кафарова. М., изд.
АН СССР, 1963. 2068 с.
23. Francis A. W. Critical Solution Temperatures. Washington, Am. Chem.
Soc., D. C., 1961. 264 p.
24. Огородников С. К., Лестева T. M., Коган В. В. Азеотропные смеси. Спра-
вочник. Л., «Химия», 1971. 848 с.
25. Коган В. В., Фридман В. М., Кафаров В. В. Равновесие между жид-
костью и паром. М., «Наука», 1966. 1426 с.
26. Francis A. W., J. Chem. Eng. Data, 1965, v. 10, № 1, p. 45—49; № 2,
p. 145—151; № 3, p. 260-264.
27. Ициксон Л. В., ЖФХ, 1967, т. 41, вып. 2, с. 409—413; Ициксон Л. Б.,
Борухова М. С., ЖПХ, 1969, т. 42, вып. 6, с. 1363—1369; Ициксон Л. Б.,
«Труды ВНИИ НП», 1970, вып. 13, с. 200—208.
28. Александрова М. В. и др., Изв. вузов. Сер. Химия и хпм. технол., 1967,
т. 10, № 3, с. 335—337; Зарецкий М. И., ЖПХ, 1970, т. 43, вып. 10,
с. 2269—2274.
29. Dojcansky I., «Chemicke Zvesti», 1969, v. 23, № 4, p. 254—261; Галеева К. Г.,
Минаков В. В., Никифорова Л. И., «Нефть и газ Тюмени», 1969, вып. 3,
с. 56—60.
30. Александрова М. В. и др., ЖПХ, 1966, т. 39, вып. 5, с. 1041—1049; Алек-
сандрова М. В. Кандидатская диссертация. М„ МИТХТ им. М. В. Ломоно-
сова, 1966.
31. Wurtz A., Ann., 1860, Bd. ИЗ, Heft 2, S. 255—256; Lourenco M. A. — V.,
Ann. chim., Ser. 3, 1863, v. 67, № 3, p. 257—339.
32. Mohs B., Chem. zbl. 1866., s. 865.
33. Коган Л. M. Производство и переработка окиси этилена в Германии.
М.—Л., Госхимиздат, 1947. 121 с.
34. Gretcher L. И., Pittenger W. И., J. Am. Chem. Soc., 1925, v. 47, № 1,
р. 163—166; Дмитриев П. И., Догадкина Л. А., ЖПХ, 1941, т. 14, вып. 1,
с 110—119; пат. ФРГ 898894 (1953).
33- Final Report № 1059. British Intelligence Objectives Subcommittee. London,
H. M. Stationery Office, 1947. 55 p.
143
36. Швец В. Ф. Докторская диссертация. М., МХТИ им. Д. И. Менделеева,
1974. 317 с.
37. Пат. СССР 281284 (1970); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1970, № 28; яп. пат. 3607 (1973); РЖХим, 1973, 22Н39.
38. Пат. США 3847754 (1974); РЖХим, 1975, 16Н27.
39. Пат. ФРГ 1668052 (1975); РЖХим, 1975, 21Н23.
40. Martynek М., Sebastianka S., Gorczyca J., «Nafta» (PRL), 1972, v. 28, № 12,
p. 554—555.
41. Соловьев С. И., Михелъман А. И., Резуненко В. II., Изв. вузов. Сер. Нефть
и газ, 1969, № 11, с. 59—62.
42. Waddams A. L. Chemicals from Petroleum. London, John Murray. Ed. 2nd,
1968, 244 p.; Ed. 3rd, 1973, 326 p.
43. Теверовская E. H. и др., «Кожевенно-обувная промышленность», 1970,
№ 10, с. 59—60.
44. Яп. пат. 3989 (1959); РЖХим, 1962, 9П322.
45. Англ. пат. 1075039 (1967); РЖХим, 1968, 15Н174.
46. Польск. пат. 43451 (1960); РЖХим, 1962, 13П260.
47. Wynstra I., «SPE Journal». 1961, v. 17, № 5, р. 481—483.
48. Кирилович В. И. и др., «Пластические массы», 1966, № 2, с. 10—11.
49. Sorokin М. F., Manoviciu I., Mater, plast., 1965, v. 2, № 5, p. 266—268.
50. Пат. США 3244697 (1964); РЖХим, 1968, 14Н141.
51. Пат. США 3225065 (1965); РЖХим, 1967, ЗН170.
52. Пат. США 278994 (1957); РЖХим, 1959, 39597.
53. Яп. пат. 21820(1967); РЖХим, 1968, 15Н151; Фаддеева М. И., Баскаков А.Ю.,
Авт. свид. 261384 (1970); Открытия. Изобр. Пром, образцы. Товарн. знаки,
1970, № 5, с. 23.
54. Англ. пат. 1051186 (1966); РЖХим, 1967, 20Н313. й
55. Пат. ФРГ 1003497 (1958); РЖХим, 1959, 43593; пат. США 2974030 (1961);|
РЖХим, 1962, 6Л425; пат. США 2970080 (1961); РЖХим, 1062, ЗЛ406;|
пат. США 3231398 (1966); РЖХим, 1967, 16Н750. I
56. Пат. США 3050549 (1962); РЖХим, 1964, 8Н154.
57. Орлова Е. Ю. Химия и технология бризантных взрывчатых веществ. Л.,
«Химия», 1973. 688 с.
58. Паушкин Я. М., Адельсон С. В., Вишнякова Т. П. Технология нефтехими-
ческого синтеза. Т. 1. М., «Химия», 1973. 448 с,
59. Hydrocarb. Process., 1974, v. 53, № 9, р. 193.
60. Гликоли и опыт их применения в нефтяной и газовой промышленности.
Серия «Газовое дело». М., ВНИИОЭНГ, 1970. 152 с.
61. Халиф А. Л. и др.. Газ. пром., 1974, № 2, с. 43—45; Кузьмина А. С., там же,
№ 5, с. 37—38; Юзифович В. И. и др., там же, № 11, с. 41—43.
62. Ethylene and Propylene Glycols. Birmingham, Imperial Chemical Indu-
stries, 33 p.
63. Химические продукты и пластические массы. (Celene). Милан, Челене, 66 с.
64. Чернышев А. К. и др. Справочный материал по качеству химической про-
дукции. М., ГИАП, 1972. 492 с.; Shell. Diethylene Glycol. Datasheet 2FOO2;
Diethylene Glycol. PBO2—1. Service Publicite Kuhlmann.
Глава IV
Триэтиленгликоль и тетраэтиленгликоль
Физические свойства триэтиленгликоля
Триэтиленгликоль (тригликоль, этиленгликольдиоксидиэтиловый
эфир, бис(оксиэтил)гликолевый эфир; ди-|3-оксиэтоксиэтан)
СН2ОНСН2ОСН2СН2ОСН2СН2ОН — бесцветная, прозрачная, вяз-
кая, гигроскопичная жидкость, практически без запаха. По многим
свойствам и областям применения он похож на диэтиленгликоль.
Молекулярная масса триэтиленгликоля 150,18. Основные физические
характеристики триэтиленгликоля приведены ниже [1, р. 170] *:
Плотность (истинная) при 20 °C, г/см3 . . . 1,1243 [3]
Относительная плотность dV'l, ............ 1,1254
Температура, °C
кипения при 101,3 кПа (760 мм рт. ст.) 287,4
замерзания ...................... . —7,2
Давление насыщенного пара при 20 °C
Па ..................................... <1,33
мм рт. ст. ........................... <0,01
Коэффициент преломления . g s » . . 1,4559
Молекулярная рефракция
наблюдаемая . . . . ........................ 36,36 [6]
вычисленная ........................... 36,24
Поверхностное натяжение при 25 °C, мН/м
(дин/см).................................... 45,2
Парахор
наблюдаемый . . .'.......................... 350,9 [3]
вычисленный............................ 348,2
Удельная теплоемкость при 20 °C
Дж/(г-К) ............................... 2,198
кал/(г-°С).......................... 0,525
Теплопроводность при 20 °C
Вт/(м-К) . .............................. 0,23
кал/(см-с-°С) ........................ 0,00055 [7]
Вязкость при 20 °C, мПа (сПз) .............. 47,8
Диэлектрическая проницаемость при 20 °C . 23,69 [8]
Электропроводность при 20 °C, Ом-1-см'1 . 8,4-10“8 [8]
* В некоторых источниках приводятся показатели, отличающиеся от ука-
занных ниже: так, т. кип. 278,3 [2, р. 115; 3] и 285,5 °C [4, р. 2]; теплопровод-
яость 0,19 Вт/(м-К) или 0,00047 кал/(см-с-°C) [5].
10 Заказ 972 .
Дипольный момент при 25 °C, D ...........2,99 12, а. 2400
Константа Трутона ....................... 30,95 [3]
Константа диссоциации водного раствора 14,5 [9]
Скорость звука в триэтиленгликоле при 25 °C,
м/с........................................ 1608 [12]
Критические параметры, являющиеся расчетными, так как три-
этиленгликоль начинает разлагаться ниже температуры кипения
(температура начала его разложения 206,5 °C), приведены ниже [7]:
Критическая температура, °C
Критическое давление
мПа .....................
кгс/см2 .................
Критическая плотность, г/см3
437 ± 5
3.4 + 0,4
34,2 + 4,2
0,337 + 0,01
О термодинамических свойствах триэтиленгликоля можно судить
по следующим данным [1, 10—12]:
Теплота, кДж/моль
(ккал/моль)
образования при 25 °C
сгорания жидкости
при постоянном да-
влении .............
плавления ....
испарения при 101,33
кПа (760 мм рт. ст.)
-801,4 (-191,4)
3565,1 (851,5)
18,2 + 0,4 (4,3 + 0,1)
62,6 (14,9)
Зависимость теплоты испарения триэтиленгликоля от темпера-
туры показана на рис. 30 (см. стр. 119).
Плотность триэтиленгликоля в температурном интервале 20—
150 °C рассчитывается по формуле [3]:
р = —0,00080г+ 1,1403
Таблица 46. Зависимость плотности триэтиленгликоля (ТЭГ)
и его водных растворов от температуры
Температу- ра, °C Плотность при различной концентрации ТЭГ, г/см3
20% 40% 60% 80% 100%
100 0,985 1,012 1,037 1,055 1,064
120 0,970 0,994 1,020 1,038 1,048
140 0,953 0,978 1,002 1,022 1,033
160 0,936 0,961 0,974 1,005 1,017
180 0,920 0,944 0,967 0,988 1,002
146
Для триэтиленгликоля и его водных растворов зависимость
плотности от температуры в интервале от —40 до +80 °C дана на
рис. 48 [13, с. 534], а при более высоких температурах —
в табл. 46 [7]. Относительная плотность смесей триэтиленгликоля и
диэтиленгликоля приведена на рис. 32 (см. стр. 120). Температура
Рис. 48. Зависимость
плотности водных рас-
творов триэтиленгликоля
(ТЭГ) от температуры.
кипения триэтиленгликоля при различном давлении меняется
следующим образом [14, с. 20]:
Давление, Т. кип., Давление, Т. кип.
мм рт. ст. * °C мм рт. ст. °C
1 114,0 60 201,5
5 144,0 100 214,6
10 158,1 200 235,2
20 174,0 400 256,6
40 191,3 760 287,3
* 1 мы рт. ст. = 133,32 Па.
Температурный коэффициент давления насыщенного пара три-
этиленгликоля в интервале 56,4—57,8 кПа (740—760 мм рт. ст.)
составляет 0,054 °C на 0,133 кПа (1 мм рт. ст.) [1, р. 170]. Зависи-
мость температуры кипения водных растворов триэтиленгликоля
от концентрации при атмосферном давлении показана на рис. 33
(см. стр. 120).
Температура замерзания водных растворов зависит от концен-
трации триэтиленгликоля, что иллюстрируется данными табл. 47 [10].
Эвтектическая смесь триэтиленгликоля с водой содержит 64—
66% триэтиленгликоля и замерзает при температуре от —49 до
~~54 °C. По данным [15], в эвтектической точке содержание три-
этиленгликоля 69%, а температура замерзания составляет —67 °C.
Диаграмма температур замерзания водных растворов триэтилен-
гликоля, по данным [10, 15], приведена на рис. 49. Значительное
Ю* 147
Таблица 47. Зависимость температуры замерзания водных растворов
триэтиленгликоля (ТЭГ) от его концентрации *
Концентра- ция ТЭГ, % Температура замерзания, °C Концентра- ция ТЭГ, % Температура замерзания, °C Концентра- ция ТЭГ, % Температура замерзания, °C
2 —0,2 40 -13,7 80 —42
6 -0,8 44 —16,9 82 —40
10 —1,6 48 -20,9 84 —37,4
14 -2,4 50 -23,6 86 -34,5
18 -3,4 54 (-29,5) 88 —31,2
20 -4,0 58 (-36) 90 —27,5
24 -5,2 60 (-40) 92 —23,5
28 —6,8 70 (-48) 94 —19,1
30 —7,7 74 —46 96 —14,5
34 -9,9 76 -45 98 —9,0
38 -12,3 78 -43,5
* Цифры в скобках получены экстраполяцией кривых.
различие температур замерзания объясняется не только способ-
ностью растворов триэтиленгликоля к переохлаждению и очень
большой вязкостью при низкой температуре, но и наличием в три-
этиленгликоле примесей. Так, по данным [10], она составляет
—4,4 °C, а по данным [15] —9,4 °C. Температура замерзания смесей
триэтиленгликоля с диэтиленгликолем дана в табл. 37 (см. стр. 121).
Давление насыщенного пара триэтиленгликоля (р, мм рт. ст. *)
изменяется с температурой (71) в соответствии с формулами [2, р. 73]:
3726,2
в интервале 140—210°С lg;j = 9,6396---—
в интервале 20—29 °C 1g/> = 7 758 -
Зависимость давления пара триэтиленгликоля от температуры
представлена на рис. 1 (см. стр. 20), а его водных растворов —
на рис. 50 [16, с. 255].
Коэффициент преломления водных растворов триэтпленгликоля
зависит от его концентрации (см. рис. 36, стр. 122). В интервале
20— 30 °C при изменении температуры на 1 °C коэффициент пре-
ломления триэтиленгликоля изменяется на 0,00031 [1, р. 170].
Поверхностное натяжение триэтпленгликоля (о, мН/м) в темпе-
ратурном интервале 20—150 °C рассчитывается по уравнению [3]:
о = - 0,088г 4-47,33
Зависимость поверхностного натяжения триэтиленгликоля от
температуры и его водных растворов при 25 °C от концентрации
представлена на рис. 8 (см. стр. 46).
* 1 мм рт. ст. = 133,32 Па.
148
Рис. 49. Диаграмма тем-
ператур плавления вод-
ных растворов трпэти-
ленглпколя (О — по
данным [15], • — по
данным [10]).
Рис. 50. Зависимость давления насыщенного пара водных растворов триэти'
ленгликоля (ТЭГ) от температуры.
Рис. 51. Зависимость теплоемкости триэтиленгликоля и его водных растворов
°т температуры.
Рис. 52. Зависимость теплопроводности триэтиленгликоля и его водных раство-
ров от температуры.
Зависимость теплоемкости триэтиленгликоля и его водных рас-
творов от температуры иллюстрируется данными табл. 48 [4, р. 38].
Таблица 48. Зависимость теплоемкости триэтпленгликоля (ТЭГ)
и его водных растворов от температуры
Теплоемкость при различной концентрации ТЭГ, кал/(г/°C)*
Температура, СС ю% 2 0% 40 6 0% 80% 10 0%
20 0,979 0,936 0,868 0,752 0,641 0,529
30 0,980 0,939 0,874 0,761 0,651 0,536
' 40 0,981 0,941 0,879 0,770 0,661 0,542
50 0,981 0,944 0,885 0,778 0,670 0,551
60 0,982 0,946 0,890 0,787 0,680 0,562
70 0,983 0,949 0,895 0,795 0,689 0,569
80 0,984 0,951 0,900 0,804 0,699 0,575
90 0,985 0,954 0,905 0,812 0,708 0,584
100 0,985 0,956 0,911 0,821 0,718 0,592
110 0,986 0,958 0,916 0,829 0,727 0,600
1 кал/(г-°С — 4,19 Дя:/(г-К).
Теплоемкость триэтиленгликоля и его водных растворов в более
широком интервале температур приведена на рис. 51 [7]. Изменение
теплоемкости триэтиленгликоля с температурой до 240 °C дано
в работе [17, р. 292].
Теплопроводность водных растворов трпэтиленгликоля неодно-
значно меняется с повышением температуры и зависит от концентра-
ции триэтиленгликоля (рис. 52); теплопроводность паров триэтилен-
гликоля показана на рис. 10, б (см. стр. 48). По данным [5, 18],
изменение теплопроводности водных растворов триэтиленгликоля
с температурой представлено в табл. 49.
Таблица 49. Зависимость теплопроводности триэтиленгликоля (ТЭГ)
и его водных растворов от температуры
Температура, °C Теплопроводность при различной концентрации ТЭг (в мольных долях), кал/(см-с - °C) - 103 *
1,0 0,8 0,6 0,4 0,2 о,1
40 0,468 0,483 0,511 0,564 0,679 0,825
60 0,466 0,487 0,519 0,574 0,693 0,875
80 0,464 0,487 0,523 0,581 0,705 0,884
100 0,464 0,485 0,521 0,581 0,705 0,899
120 0,461 0,483 0,521 0,583 0,705 0,908
140 0,459 0,478 0,514 0,581 0,712 0,908
160 0,456 0,476 0,511 0,583 0,712 0,908
* 1 кал/(см-с-°С) = 418,68 Вт/(м;К).
150
Номограмма для определения теплопроводности и теплоемкости
водных растворов триэтиленгликоля в зависимости от температуры
приведена в Приложении (стр. 366 и 367) [19].
Зависимость вязкости триэтиленгликоля и его водных растворов
от температуры приведена в табл. 50 [20] и на рис. 53 [7].
Для паров триэтиленгликоля, а также для смеси триэтиленгли-
коля с этиленгликолем, взятых в соотношении 1:1, зависимости
вязкости от температуры представлены на рис. И и 12 (см.
стр. 49).
Рис. 53. Зависимость
вязкости триэтиленгли-
коля и его водных рас-
творов от температуры.
Диэлектрическая проницаемость триэтиленгликоля и его вод-
ных растворов зависит от температуры и при 15, 25 и 35 °C она
равна [21]:
Концент- Концент-
рация тэг, % При 15 ° С При 25 °C При 35 °C рация тэг, % При 15 с ’С При 25 е ’С При 35
0 81,95 78,3 74,8 . 60 54,3 52,1 49,3
10 78,0 74,8 71,2 70 48,2 46,0 43,9
20 73,9 71,0 ' 67,4 80 41,4 39,4 37,6
30 69,5 66,8 , 63,4 90 33,5 31,8 30,3'
40 64,9 62,4 59,1 100 24,0 23,0 21,8
50 59,8 57,5 54,5
Таблица 50. Зависимость вязкости триэтиленгликоля (ТЭГ)
и его водных растворов от температуры
Темпера- тура, °C Вязкость при различной концентрации ТЭГ, мПа-с
9,93% 20,44% 29,60% 38,57% 59,89% 80,06% 100%
5 2,180 3,282 4,768 7,044 20,70 60,76 119,8
10 1,843 2,729 3,901 5,709 15,89 44,35 85,94
20 1,378 1,981 2,758 3,930 10,01 25,23 48,15
40 0,8611 1,183 1,581 2,143 4,777 10,56 19,06
60 0,6005 0,7961 1,034 1,353 2,727 5,456 9,478
80 0,4472 0,5782 0,7366 0,9382 1,736 — 5,429
151
Для жидкого и переохлажденного триэтиленгликоля изменение
диэлектрической проницаемости в зависимости от температуру соста-
вляет [8]:
6 0 °C 50 °C 20 °C 5е С —5 °C -20 °C
£ . . . 19,2 20,1 23,7 25,6 27,0 29,4
Гигроскопичность. Триэтилепгликоль очень гигроскопичен. Рав-
новесная концентрация его с воздухом в зависимости от влажности
воздуха (или другого гдза) и температуры контакта дана в табл. 51
[4, р. 131.
Таблица 51. Зависимость равновесной концентрации триэтиленгликоля
от относительной влажности воздуха и температуры
Равновесная концентрация при относительной влажности, %
ра, °C 10% 20% 30% 40% 50% «0% 70% ' 80% 90%
-5 98,5 96,8 94,0 91,1 89 83 78 68 52
5 98,4 96,5 93,8 91,0 88 83 77 65 51
15 98,2 96,2 93,6 90.8 86 82 77 65 50
25 98,1 96,0 93,4 - 90,7 85 82 76 64 50
35 98,0 95,7 93,2 90,6 84 82 76 64 49
45 97,8 95,4 93,0 90,5 83 82 75 63 49
Зависимость точки росы газа при его осушке от концентрации
триэтиленгликоля в водном растворе и температуры контакта пока-
зана на рис. 54 [22, с. 61].
Растворимость. Триэтиленгликоль полностью смешивается с во-
дой, а также с рядом органических соединений, содержащих кисло-
род, азот и серу. При смешении триэтиленгликоля со многими веще-
ствами, например с водой и бензолом, выделяется теплота, количество
которой зависит от концентрации триэтиленгликоля и темпера-
туры [23, с. 77, 186]. Так, при смешении триэтиленгликоля с водой
выделяется (в кал/моль * раствора)!
/ Концентрация
тэг, % (мол.) При 25 °C При 35 °C При 45 °C
2,6 —122 —107 —97,6
6,1 —247 —207 —222
11,5 —379 —341 —316
20,6 —453 -409 —393
39,3 —409 —392 —345
52,1 —428 -361 —307
69,5 -342 —228 — 183
1 кал/моль ~ 4,19 Дж/моль.
152
Количество же теплоты, выделяющейся при смешении триэтилен-
гликоля с бензолом, равно (в кал/моль раствора):
Концентрация ТЭГ, % (мол.) При 25 °C При 3 5 °C При 45
6,1 35,1 — 72,8
11,4 46,4 67,4 94,2
24,3 52,2 74,8 107
33,9 56,0 — —
39,0 58,1 85,7 119
56,2 36,6 61,5 105
76,2 14,2 23,2 53,0
При обычных условиях триэтиленгликоль [4, р. 9; 24, р. 58—59]
полностью смешивается со следующими органическими соедине-
ниями:
одноатомными спиртами (метиловый,- этиловый, этиленхлорги-
дрин, триметиленхлоргидрин, метилизобутилкарбинол)
Рис. 54. Зависимость
точки росы газа от тем-
пературы контакта с вод-
ными растворами триэти-
ленгликоля.
многоатомными спиртами (этилен-, диэтилен-, тетраэтиленгли-
коль, пропилен- и дипропиленгликоль, 1,3- и 2,3-бутиленглпколь,
2-хлор-1,2-пропиленгликоль
карбонильными соединениями (ацетон, анисовый и коричный
альдегиды, метилизопропилкетон, метилпзобутилкетон, салициловый
альдегид)
кислотами (диэтилуксусная, 4-метил-н-валериановая, этиленди-
ацетат)
аминами (ди-н-бутиламин, диизопропиламин, диэтилентриамин,
изопропиламин, а-метилбензил- и а-метилбензилдиметиламин,
153
2-метил-5-этилпиридин, пиридин, триэтилентетрамин, о-фенетидин,
2-фенилэтиламин)
аминоспиртами (этанол- и диэтаноламин, метилбензилдиэтанол-
амин и метилбензилэтаноламин)
простыми эфирами этилен- и диэтиленгликоля (бутиловый, мети-
ловый, этиловый, этилбутиловый)
сложными эфирами (бромэтилацетат, 3-н-бутилфосфат, триэтил-
фосфат, этилбензоат, этилфенилацетат, этилхлорацетат, этилцин-
намат,
а также с другими органическими соединениями (диметилформ-
амид, н-амилцианид, о-крезол, хлороформ, тиофен).
В три- и тетраэтиленгликолях при 25 °C плохо растворимы
(менее 1%) смолы и масла (растительного и животного происхо-
ждения). Растворимость различных соединений при 25 °C приведена
в Приложении, табл. 9, стр. 357.
Со многими соединениями триэтиленгликоль неограниченно сме-
шивается при критической температуре растворения (К. Т. Р.),
например [25, с. 174—175]:
к.т.р., °с
К.т.р., °с
Бензол ...................... 22
втоо-1>утплбепзпл ...........156
.утплбепзол............153
Дибензпл ....................115
Ди-ето/ьбутилбензол .... 222
Дипзопропплбензол ...........191
Дифенил ..................... 65
Диэтилбензол ................160
Изопропилбензол..............137
п-Изопропилтолуол (цимол) . 161
Инден ...................... 53
.w-Ксилол ...................120
Лимонен......................170
1-Метилнафталин ............ 62
Метилэтилбензол .............138
Псевдокумол ................152
Стирол ..................... 37
Тетралин ....................132
Толуол ...................... 90
Этилбензол .................115
С рядом соединений триэтиленгликоль образует азеотропные
смеси, состав и температура кипения которых даны в Приложении,
табл. 10, стр. 358 [26, с. 343 и след.[.
Фазовое равновесие жидкость — пар для системы триэтилен-
гликоль — вода при 25,03 °C и атмосферном давлении характери-
зуется следующими данными [27, с. 3561:
Концентрация ТЭГ, %
(мол.)
в жидкости .... 32,6 38,2 42,9 . 50,1 57,7 64,4 69,7 74,3 78,3
в пара?:, с-103 . . 1,98 2,94 3,64 5,98 9,19 13,37 18,78 25,84 34,12
Для этой же системы, но при других давлениях фазовый состав
представлен на рис. 55 [13, с. 5331, а равновесие жидкость — пар
для системы дйэтиленгликоль — триэтиленгликоль приведено на
рис. 43 (см. стр. 130).
154
Рис. 55. Фазовый состав
водных растворов трп-
этилепгликоля при
80 кПа, пли 600 мм рт.
ст. (1), 40 кПа, пли
300 мм рт. ст. (2) и
13,3 кПа, пли 100 мм рт.
ст. (3).
Рис. 56. Диаграмма рав-
новесия в трех- и четы-
рехкомпонентных систе-
мах, включающих три-
этпленгликоль (ТЭГ).
Условные обозна-
чения: G;Fi, — пер-
фтор-и-гептан; СцР.,, — пер-
фгорметилнафталин; ж —
жидкость; Л. С. П — линия
сдвоенных коэффициентов
преломления; т — твердая
фаза.
Диаграммы равновесия в трехкомпонентных системах, включа-
ющих триэтиленгликоль, представлены на рис. 56 [28]. Данные
по фазовому равновесию трехкомпонентных систем, содержащих
кроме триэтиленгликоля и-гептан — бензол, w-гептан — толуол,
н-гептан — этилбензол, высшие спирты — углеводороды, вода —
каприлат натрия, а также четырехкомпонентных систем (триэтилен-
гликоль — вода — гептан — бензол) приведены в работах [29—31].
Изучена взаимная растворимость и состав равновесных фаз следу-
ющих систем: ТЭГ — а-пиколин — бензол, ТЭГ — а-пиколин —
метилциклогексан, ТЭГ — а-пиколин — гептан, ТЭГ — а-пиколин —
диизобутилен, ТЭГ — фенол — смесь углеводородов и ТЭГ — фе-
нол — вода — смесь углеводородов [32].
Физические свойства тетразтиленгликоля
Тетраэтиленгликоль (тетрагликоль) СН2ОН — (СН2 ОСН2)3 — СН2ОН
имеет молекулярную массу 194,22. При обычных условиях — это
бесцветная, прозрачная, вязкая, гигроскопичная жидкость. Основные
физические характеристики тетраэтиленгликоля приведены ниже:
Плотность при 25 °C, г/см3 ........ 1,120 [4,р. 2]
Относительная плотность <^20 1,1247 [1,р. 4]
Температура, °C
кипения при 101,3 кПа (760 мм рт. ст.) 327,3 [1]
замерзания . ...................... —6,2 [1]
Давление насыщенного пара прп 20 °C
Па ................................... <1,33 [4]
мм рт. ст................................ <0,01
Коэффициент преломления ............... 1,4598 [1]
Поверхностное натяжение прп 25 °C, мН/м
(дин/см) ............................. 45 [4]
Парахор
наблюдаемый ........................... 453,7 [3]
вычисленный............................. 446,2
Удельная теплоемкость при 25 °C
Дж/(г-К) ................................... 2,18 [4]
кал/(г-°С).............................. 0,52
Вязкость при 20 °C, мПа (сПз) ............... 61,3 [20]
Диэлектрическая проницаемость прп 20 °C 20,44 [8]
Электропроводность при 20 °C, Ом^-см-1 4,7 [8]
Дипольный момент при 25 °C ............ 3,25 [12]
Константа Трутона .......................... 36,66 [3]
Констапта Рамзая — ПТпльдса
наблюдаемая ........................... 2,31 [3]
вычисленная .......................... 2,27
Скорость звука в тетр аэтпленг диколе при
25 °C, м/с .................................. 1586 [12]
Примечание. По другим источникам, т. кип. равна 314 [4, р. 2] и 307,8 °C
[14, с. 25], т. замерз. —9,4 °C и п™ 1,4593 [6].
156
Ниже приведены термодинамические характеристики тетраэти-
ленгликоля по данным [3, И, 12]:
Теплота, кДж/моль (ккал/моль)
образования (стандартная) .................—978 (—233,6)
сгорания (жидкости) при постоянном да-
влении ............................. 4748,7 (1134,2)
испарения прп 101,33 кПа-(760 мм рт. ст.) 89,21 (21,31)
Температура кипения тетраэтиленгликоля в зависимости от
давления меняется следующим образом [14, с. 25]:
Давление, Т. кип., Давление, Т. кип.
мм рт. ст. * °C мм рт. ст. * °C
1 153,9 60 237,8
5 183,7 100 250,0
10 197,1 200 268,4
20 212,3 400 288,0
40 228,0 760 307,8
* 1 мм рт. ст. = 133,32 Па.
В интервале температур 190—235 °C давление насыщенного пара
(р, мм рт. ст.) может быть рассчитано по уравнению [2, р. 74; 3]:
, 4649.4
1g р= 10,886 ---у—
где Т — температура, К. Изменение давления насыщенного пара
с температурой показано на рис. 1 (см. стр. 20).
Вязкость тетраэтиленгликоля г] при повышенных температурах
имеет следующие значения [34]:
Температура, °C .... 10 20 30 40 50 60 70 80 115
Вязкость т], мПа-с . . .101,8 61,3 37,5 24,7 17,4 12,3 8,8 6,5 3,2
Зависимость кинематической вязкости смеси тетраэтиленгликоля
с этиленгликолем, взятых в соотношении 1:1, представлена на
рис. 12 (см. стр. 49).
С понижением температуры диэлектрическая проницаемость е
Жидкого или переохлажденного тетраэтиленгликоля меняется таким
образом [8]:
60 °C 50 °C 35 °C 20 °C 5° С —5 °C —20 °C
. . . . 16,5 17,3 18,8 20,4 22,2 23,6 25,5
Зависимость точки росы газа при осушке водными растворами
тетраэтиленгликоля от температуры контакта показана на рис. 5/
[22, с. 146].
157
Растворимость. Тетраэтиленгликоль полностью смешивается с во-
дой и со многими органическими соединениями: бензолом, стиролом,
скипидаром, дибутилфталатом, моно- и диэтаноламином, дихлорди-
этиловым эфиром, четыреххлористым углеродом, хлорбензолом,
о-дихлорбензолом, метиловым и этиловым спиртами, метилизобутил-
карбинолом, фенолом, метилизобутилкетоном, другими гликолями
и их эфирами [4, р. 9]. Растворимость различных соединений в
тетраэтиленгликоле приведена в Приложении, табл. 9, стр. 357.
Рис. 57. Зависимость
точки росы газа от тем-
пературы контакта с вод-
ными растворами три-
этиленгликоля.
По сравнению с другими этиленгликолями в тетраэтиленгликоле рас-
творяется большее количество ароматических и хлорзамещенных угле-
водородов.
Получение три- и тетраэтиленгликолей
Триэтиленгликоль и тетраэтиленгликоль впервые были получены
в|1859 г. двумя различными способами [35]:
1) при взаимодействии окиси этилена с водой или этиленгликолем
ЗН3С—СН2 + Н2О > СН2ОН—(СН2СН2О)2—СН2ОН
о
ЗН2С—СН2 + СН2ОНСН2ОН —> СН2ОН—(СН2СН2О)з-сн2он
о
2) при взаимодействии этиленгликоля с бромистым этиленом
4CH2OHCH2OIIJ-C2H4Br2 —>
—► СНгОН—(СН2СН2О)2-СН2ОН-г2С2Н3ОВг + 2Н2О
158
Тетраэтиленгликоль наряду с три- и пентаэтиленгликолем можно
получить при нагревании диэтиленгликоля до 190 °C в присутствии
иода как катализатора, а как индивидуальный продукт он получается
|по реакции |3,|3'-дихлордиэтилового эфира с моногликолятом на-
трия [36]:
CH2ClCH2OCH2CH2Cl+2CH2OHCH2ONa —>
I --> СН2ОН-(СН2СН2О)2-СН3ОН +2NaCl
Так можно синтезировать и ряд других индивидуальных поли-
этиленгликолей по схеме:
CH2Cl(CH2OCH2)„CH2Cl+2CH2OII(CH2OCH2)mONa —>
--> CH2OH(CH2OCH2)(rv+2ma)CH2OH + 2NaCl
В небольших количествах три-- и тетраэтиленгликоли полу-
чаются при нагревании этиленгликоля с серной кислотой \[36].
В промышленности триэтиленгликоль получают как побочный про-
дукт в производстве этилен- и диэтиленгликоля: на 1 т этиленгликоля
выделяется 25 кг, а на 1 т диэтиленгликоля — от 150 до 250 кг
триэтплэнгликоля [39]. Три- и тетраэтиленгликоль получают также
оксиэтилированием этилен- и диэтиленгликоля; тетраэтиленгликоль
можно получать оксиэтилированием триэтиленгликоля.
Количество образующихся три- или тетраэтиленгликоля зависит
от соотношения между водойили гликолем и окисью этилена в исход-
ной смеси. В табл. 23 (см. стр. 56) показано, что заметные количества
трпэтиленг лпколя образуются при мольном отношении воды и окиси
этилена, равном 4,2, а тетраэтиленгликоля — при их отношении 2,1.
По мере уменьшения отношения воды и окиси этилена выход три-
и тетраэтиленгликоля повышается за счет непрерывного снижения
выхода этилен- и диэтиленгликоля. При отношении реагентов,
равном 1,4 и ниже, образуются также высшие полигликоли. Так,
при отношении воды и окиси этилена в исходной смеси, равном 0,61,
выход этиленгликоля, ди-, три- и тетраэтиленгликолей, а также
высших полигликолей составляет 15,7, 26,0, 19,8, 19,0 и 14,5%
соответственно. При щелочном катализе выход три- и тетраэтилен-
гликоля выше, чем при кислотном или в отсутствие катализатора,
и возрастает с увеличением концентрации щелочи в воде (см. рис. 24,
стр. 78 и табл. 30, стр. 80).
Состав продуктов реакции окиси этилена с гликолями (как и реак-
ции окиси этилена с водой) можно рассчитать, зная коэффициент
Распределения Cz, равный отношению констант скоростей последо-
вательно-параллельных реакций оксиэтилирования исходного и обра-
зующихся гликолей. Этот коэффициент связан с относительной
кислотностью гликолей уравнением [37, с. 200]:
/К,
159
где Ко, К; — кислотность исходного и образующегося гликоля,
a g и а — коэффициенты, зависящие от типа реакции (их значения
приведены на стр. 81).
Вычисленные значения коэффициентов распределения в реакциях
оксиэтилирования гликолей одной, двумя и тремя молекулами
окиси этилена С1, С2, Cs приведены в табл. 52.
Таблица 52. Коэффициенты распределения в реакциях
оксиэтилирования гликолей
Исходный гликоль Тип реакции * Коэффициенты распределения
С1 сг Сз
Этиленгликоль .... (-) 0,936 0,835 0,826
(НС 0,964 0,900 0,893
(ОН-) 0,872 0,590 0,567
Диэтиленгликоль .... (-) 0,876 0,936 —
(НС 0,945 0,920 —
(ОН-) 0,691 0,685 —
Триэтиленгликоль . . . (-) 1 1 —
(НС 1 1 —
(ОН-) 1 1 —
* (—) — некаталитическая реакция; (Н+) — кислотный катализ; (ОН-) — основной
катализ.
Содержание в смеси продуктов и выход три- и тетраэтиленгликоля
при реакции окиси этилена с гликолями (в присутствии щелочного
катализатора и без него) в зависимости от соотношения исходных
реагентов приведены в табл. 53 [37, с. 204]. Из таблицы видно,
что при малом отношении окиси этилена к гликолю в исходной
смеси выход три- и тетраэтиленгликолей достаточно высокий,
однако с увеличением этого отношения выход их снижается за
счет образования полигликолей.
Выделение чистых три- и особенно тетраэтиленгликоля ректифи-
кацией затруднено в связи с их высокими температурами кипения,
склонностью к разложению (триэтиленгликоль начинает разлагаться
при 206,5 °C, а тетраэтиленгликоль — при 237,9 °C [3]) и большой
вязкостью как целевых продуктов, так и полигликолей, которые
остаются в виде кубового остатка. Поэтому ректификацию проводят
при малом остаточном давлении и при минимальных перепадах
давления в колонне и времени пребывания продуктов в аппарате.
Для выделения три- и тетрагликолей применяют ректификацион-
ные колонны различных типов: с насадкой из колец Рагаига или
Палля, роторные и с плоско-параллельной насадкой из металличе-
ской сетки, уложенной под углом к вертикальной оси колонны.
Применяются также тарельчатые колонны с минимальным коэффи-
160
Таблица 63. Содержание в продуктах и выход три- (ТЭГ) и тетраэтиленгликоля (Тетра)
в условиях щелочного катализа при взаимодействии окиси этилена с гликолями
Без катализатора.
циентом сопротивления тарелок. При ректификации на насадочных
колоннах с кольцами Рашига использовали:
а) схему из одной колонны с высотой насадки 6 м, работающей
при остаточном давлении 0,4 кПа (3 мм рт. ст.) и температуре 190—
195 °C (состав сырья — 8% ДЭГ, 80% ТЭГ, 9% тетраэтиленгликоля
и 3% — высшие полигликоли);
б) схему из двух последовательно работающих колонн с кольцами
Рашига 50 X 50 мм, заполняющими каждую колонну на высоту
3,5 м.
По второй схеме (см. гл. II, рис. 28, стр. 85) дистиллят первой
колонны 18-1, содержащий смесь ди- и триэтиленгликоля) возвра-
щают в сборник концентрированного гликоля-сырца, а кубовые
остатки направляют в выпарной аппарат 17-11 для отгонки три-
этиленгликоля от тяжелокипящих продуктов. Пары из этого аппа-
рата поступают во вторую колонну 18-II, дистиллят которой яв-
ляется товарным триэтиленгликолем, а кубовые остатки — смесь
тетраэтиленгликоля и высших гликолей, используемой как мягчи-
тель.
Более эффективными для выделения три- и тетрагликолей яв-
ляются роторные колонны. Так, триэтиленгликоль с температурой
кипения, соответствующей чистому продукту, был получен на ротор-
ной колонне диаметром 1170 мм и высотой 4200 мм [39, с. 89]. По
высоте колонны расположены кольцеобразные тарелки (21 шт.)
с концентрическим вырезом диаметром 720 мм. На вращающемся
валу в центре колонны закреплен 21 диск диаметром 700 мм. Та-
релки и диски снабжены направляющими лопастями для распре-
деления и стока жидкости. Эффективность такой колонны опре-.
деляется интенсивным массообменом между жидкостью, распыляемой •
вращающимися дисками ротора, и парами, движущимися снизу
вверх, навстречу потоку жидкости, которая поступает на верхний
диск.
Для разделения смеси ди- и триэтиленгликоля весьма эффективны
колонны с плоско-параллельной насадкой [38].
Основные характеристики применяемой в них насадки в сопоста-
влении с соответствующими данными для других типов колонн
следующие:
Тип колонны
ВЭТТ *,
мм
Перепад давления
на ВЭТТ, Па
(мм рт. ст.)
Плоско-параллельная насад-
ка...................... 150-200
Пленочные тарелки .... 450 —750
Насадка Палля............ 450—1220
Спиральная насадка . . . 200—500
13,3-40,0 (0,1—0,3)
133—266 (1-2)
67—340 (0,5—2,5)
26,6-66,7 (0,2-0,5)
* Высота, эквивалентная одной теоретической тарелке.
При разделении смеси, содержащей 80% ди- и 20% триэтилен-
гликоля, на промышленной колонне с плоско-параллельной насадкой
162 '
диаметром 1650 см с высотой укрепляющей части 1016 мм и исчер-
пывающей части 1700 мм при флегмовом числе 1 получен 99,1 %-шый
диэтиленгликоль; содержание его в кубовом продукте составляло
0,07%.
Для получения дистиллята с концентрацией диэтиленгликоля
99,9?о и кубового продукта, содержащего 99,9% триэтиленгликоля,
требуется 18—20 теоретических тарелок при содержании ДЭГ на
питающей тарелке 50%. Температура в кубе колонны не должна
превышать 190 °C.
Колонны с указанной насадкой могут быть применены также
для разделения смесей три- и тетраэтиленгликоля. При этом среда
должна быть близка к нейтральной, так как гликоли в кислой и ще-
лочной средах способны к реакциям отщепления воды с образованием
более высоко- или низкомолекулярных гликолей, а также изменению
цвета. Так, триэтиленгликоль, полученный ректификацией (темпе-
ратура низа колонны 190 °C. верха 135 °C, остаточное давление
0,66 кПа, или 5 мм рт. ст.) при pH 6,5 имеет цвет по Pt — Со шкале
sgl5, а при тех же условиях, но других значениях pH цвет достигает
40—70 (40]. Цвет три- и тетраэтиленгликолей можно улучшить,
пропуская их через слой сильнокислого катионита (леватит 2У-115)
при 80-100 °C [41].
Применение триэтиленгликоля
Хорошая растворяющая способность, высокая температура кипения,
малая летучесть и низкая токсичность триэтиленгликоля определили
его широкое применение в качестве экстрагента, растворителя п пла-
стификатора для лакокрасочных изделий, клеев, печатных красок.
Повышенная гигроскопичность позволяет эффективно использовать
триэтпленгликоль в качестве осушающего агента для газов. Он
является также исходным сырьем для синтеза пластификаторов,
смол п каучуконодобных материалов.
В США в 1970 г. 30% произведенного триэтиленгликоля было
израсходовано на осушку газов, 17% — для увлажнения табака,
15% — в качестве растворителя, 12% — для получения пластифи-
каторов. 8% — в производстве полиэфирных и полиуретановых
смол п частично для экстракции ароматических углеводоро-
дов [42].
Триэтиленгликоль, как и другие гликоли, при взаимодействии
С одно-, двух- и многоосновными кислотами и их ангидридами,
образует сложные эфиры, широко применяемые в различных отрас-
лях химической промышленности: лакокрасочной, пластических
масс, синтетического каучука и др. Например, триэтиленгликоль-
Дикаприлат совмещается с такими полимерными веществами, как
поливинилхлорид, поливинилацетат, поливинилбутираль, нитро-
целлюлоза, этилцеллюлоза, хлоркаучук, и применяется для их
пластификации [43. .49]. Хорошими пластификаторами этих же
полимерных материалов являются эфиры триэтиленгликоля и
11* 163
адипиновой, 2-этилмасляной, 2-этилгексановой, фталевой, а также
смесей жирных кислот. В качестве пластификаторов описаны сме-
шанные сложные эфиры: дикаприлат-бис-(триэтиленгликоль)фталат,
дикаприлат-бис-(триэтиленгликоль)адиппнат, дикаприлат-бпс-(три-
этиленгликоль)себацинат [44]. Эфиры триэтиленгликоля и ненасы-
щенных кислот применяются в производстве слоистых пластиков [1,
р. 171].
Органическое стекло, получаемое при сополимеризации три-
этиленгликольдиметакрилата с метилметакрилатом, обладает повы-
шенной поверхностной твердостью, теплостойкостью и эластично-
стью [45, с. 524]. Прессованные и литые изделия, устойчивые к по-
вреждениям покрытия для стекол, линз и тому подобных изделий,
получаются из ненасыщенной полиэфирной смолы на основе три-
этиленгликоля и фумаровой кислоты, а также этилакрилата, аллил-
метакрилата, триаллилцианурата и метакриловой кислоты [46].
Из кислых эфиров триэтиленгиколя и трехосновных кислот при
их нагревании с ненасыщенным одноатомным спиртом и мономером,
содержащим 1—3 винильные группы, в присутствии органической
перекиси получается твердый материал, обладающий хорошими
механическими и электрическими свойствами, водостойкостью и
стабильностью при хранении [47]. Линейные полиэфиры с фосфором
в основной цепи, обладающие высокой огнестойкостью, образуются
поликонденсацией триэтиленгликоля с дихлорпдом аллилфосфорной
кислоты [48]. Оловосодержащие полимеры, являющиеся ускорите-
лями вулканизации и полимеризации, антиоксидантами и стабили-
заторами для синтетических смол, получаются при реакции т^и-
этиленгликоля с оловоорганическими дигалогенами в присутствии
акцепторов галогенов. Из полиацеталей триэтиленгликоля и форм-
альдегида, эфиров виниловых спиртов с акриловой или метакриловой
кислотой синтезируют блоксополимеры, на основе которых получают
пенопласты. Полиэфир триэтиленгликоля и у-кетопимелиновой кис-
лоты является вязкой смолой [49].
При окислении триэтиленгликоля азотной кислотой образуется
этилендигликолевая кислота, а при действии на него треххлористого
фосфора и затем хлористого водорода получается а,(о-дихлортри-
этиленгликоль [50]. При действии на триэтиленгликоль в присут-
ствии Ni-Ренея аминирующих агентов (аммиак, первичные или
вторичные амины С! — Св) образуются алифатические амины.
Например, при 200 СС и 13 МПа (133 кгс/см2) получается
HOC2H4OC2H4OC2H4NH2, а при 225 °C и 15,9 МПа (162 кгс/см2)
H2NC2H4OC2H4OC2H4NH2 [51]. Продукты взаимодействия триэти-
ленгликоля с трихлорметильными производными ароматических
соединений могут быть использованы как промежуточные соедине-
ния, растворители и сельскохозяйственные химикаты [52]. При
реакции триэтиленгликоля с поли- или пергалогенированными поли-
фенилами в присутствии акцептора галогеноводородной кислоты
получаются соединения, которые, взаимодействуя с насыщенными
и ненасыщенными кислотами, образуют полиэфиры с повышенной
164
стойкостью к действию химических реагентов и хорошими механи-
ческими свойствами при повышенной температуре и на холоду [53].
При действии на триэтиленгликоль тионилхлорида, а затем хлора
в присутствии катализаторов образуются полупродукты для синтеза
красителей, пластических и лекарственных препаратов [54]. Три-
этиленгликоль служит одним из видов сырья для получения депрес-
сантов центральной нервной системы, обладающих обезболива-
ющим, антихолинергическим и антигистаминным действием, а также
для синтеза 3,5-дииодоксиалкилпиридонов, которые используют
в рентгеноскопии как контрастные вещества [55].
Трпэтиленгликоль, как и другие гликоли, обладает бактерицид-
ными свойствами и применяется для дезинфекции воздуха. При
комнатной температуре и влажности воздуха 20— 50 % содержание
в нем 5-10“3% (об.) триэтиленгликоля, по лабораторным данным,
приводит к мгновенной гибели 85% бактерий, а остальные 15%
погибают в течение двух минут [1, р. 171]. Смесь триэтиленгликоля
и трихлорэтана предложена в качестве бактерицидного средства
для предотвращения порчи плодов: через 7 дней после обработки
указанной смесью испортился 1% персиков по сравнению с 80%
без обработки. Бактерицидной активностью обладает также эфир
триэтлленгликоля и сульфометилфенилкарбаминовой кислоты [56].
Триэтиленгликоль предложен для стабилизации дитиофосфатов,
являющихся инсектицидными препаратами, а также некоторых
других пестицидов [57].
Триэтиленгликоль применяется в качестве среды при получении
ряда продуктов, например при синтезе 100%-ного у-гексахлорцикло-
гексана, Ni-фенолятов диоксиалкилдифенилсульфонов, являющихся
стабилизаторами полиолефинов [58].
Динитрат триэтиленгликоля, обладающий малой летучестью,
хорошей стабильностью и стойкостью к желатинизации, применялся
в Германии в качестве компонента порохов и как ракетное топливо.
Он рекомендуется в качестве компонента твердого ракетного топлива
и в настоящее время [59]. Триэтиленгликоль применяется также как
высокотемпературный теплоноситель [1, р. 171].
Триэтиленгликоль находит все возрастающее применение для
осушки природных и нефтяных газов, а также воздуха. Его преиму-
щества перед диэтиленгликолем определяются более низким давле-
нием паров над раствором и более высокой температурой начала
разложения. Меньшее давление паров ведет к снижению потерь
абсорбента с газом: в равных условиях потери с 1000 м3 газа три-
этиленгликоля составляют 5 г по сравнению с 20 г диэтилен-
гликоля [60].
С триэтиленгликолем осушку газа можно вести при более высокой
температуре, чем с диэтиленгликолем; удается повысить температуру
Регенерации абсорбента, уменьшить содержание в нем воды и тем
самым дополнительно понизить точку росы осушаемого газа. Потери
триэтиленгликоля в производственных условиях (на испарение,
механический унос, утечки, растворение и т. п.) составляют 0,33 л
165
или 0.4 кг на 100 тыс. м3 осушенного газа [13, с. 536]. При замене
диэтиленгликоля на триэтиленгликоль себестоимость осушки при-
родного газа снижается на 21% [60].
Триэтиленгликоль предложен также для осушки газов, получа-
емых при синтезе хлорвинила из ацетилена и хлористого водо-
рода [61]. Для многих веществ триэтиленгликоль является более
эффективным растворителем, чем диэтиленгликоль. Например, бен-
зол в нем растворяется полностью, а в диэтиленгликоле только
частично; в триэтиленгликоле растворяется в полтора раза больше
толуола, чем в диэтиленгликоле. В связи с этим триэтиленгликоль
нашел широкое применение в качестве селективного растворителя
для экстракции ароматических углеводородов из продуктов катали-
тического риформинга или других продуктов, содержащих аромата-
Ри.с. 58. Распределение бензола между
экстрактом и рафинатом при приме-
нении различных гликолей в ка-
честве экстрагентов:
1 — тетраэтиленгликоль с содержанием
3,9% (об.) воды, 100 °C; 2 — триэтплен-
гликоль с содержанием 5,0% (об.) воды,
121,5 °C; 3 — дпэтиленгликоль с содержа-
нием 8,0% (об.) воды, 125 °C.
ческие углеводороды, превосходя для этой цели диэтиленгликоль.
Экстракцию проводят триэтиленгликолем, содержащим 5% (об.)
воды, так как при этом растворитель кипит при 140 °C, что облегчает
отгонку поглощенных ароматических углеводородов.
На рис. 58 показано распределение бензола между экстрактом
и рафинатом при применении различных гликолей [31]. Коэффи-
циент распределения бензола — отношение концентраций бензола
в % (об.) в экстракте и рафинате — при применении триэтиленгли-
коля значительно выше, чем при использовании диэтиленгликоля.
Толуол и ксилолы экстрагируются триэтиленгликолем еще лучше.
При деароматизации газойля каталитического крекинга серни-
стых нефтей триэтиленгликолем происходит также его значительное
обессеривание, что позволяет получать малосернистое дизельное
топливо [62]. Технологическая схема выделения ароматических
углеводородов триэтиленгликолем такая же, как и диэтиленгликолём
(см. гл. III, рис. 46, стр. 139), однако производительность установки
выше. Так, при переводе блока экстракции ароматических угле-
водородов с диэтиленгликоля на триэтиленгликоль производитель-
ность возросла на 40%, расход пара на отпаривание снизился на
17%, расход экстрагента на 1 т перерабатываемого сырья умень-
шился, несколько возросла глубина извлечения ароматических
углеводородов [63].
166
Скорость окисления триэтиленгликоля при высокой температуре
его регенерации может быть снижена добавкой к нему 0,5% ди-
этаноламина или 0,1% топанола [64].
Применение тетраэтпленгликоля
Литературные данные о применении тетраэтиленгликоля весьма
ограничены: он менее доступен и дороже, чем другие гликоли, так как
высокая температура кипения тетраэтиленгликоля и особенно со-
путствующих полиэтиленгликолей чрезвычайно осложняет процесс
разделения реакционных смесей. В США, например, цена тетра-
этиленгликоля в 1973 г. была в 1,7 раза выше, чем цена диэтилен-
гликоля [33].
Тем не менее применение тетраэтиленгликоля вместо других
гликолей в ряде случаев приводит к достижению заметного эконо-
мического эффекта. Так, в последние годы проявляется большой
интерес к тетраэтиленгликолю как селективному растворителю
для экстракции ароматических углеводородов из продуктов катали-
тического риформинга бензинов. Из рис. 58 (см. стр. 166) видно,
что при применении тетраэтиленгликоля коэффициент распределения
бензола между экстрактом и рафинатом в 1,5 раза выше, чем для
триэтиленгликоля, и в 2,5 раза выше, чем для диэтиленгликоля.
Значительным преимуществом тетраэтиленгликоля является также
его высокая термическая стабильность.
При замене ди- или триэтиленгликоля тетраэтиленгликолем
существенно возрастает производительность установок по выделению
ароматических углеводородов из продуктов риформинга, повы-
шается степень извлечения и качество ароматических углеводо-
родов и сокращается расход пара, воды и электроэнергии [31,
33, 65].
Тетраэтиленгликоль применяется также как пластификатор и рас-
творитель в случаях, когда особо важны его высокая температура
кипения и малая летучесть. Как и другие гликоли, он является
исходным сырьем для синтеза различных пластификаторов, текстиль-
ных вспомогательных веществ, смол и ряда других соедине-
ний [4, р. 4].
Из продукта взаимодействия полигалогенированного полифенила
с избытком тетраэтиленгликоля при последующей реакции с органи-
ческими кислотами получают полиэфиры с высоким содержанием
галогенов, обладающих повышенной стойкостью к химическим
реагентам и хорошими механическими свойствами в широком диапа-
зоне температур [53].
При взаимодействии тетра этиленгликоля с треххлористым фос-
фором образуется а,со-дихлортетраэтиленгликоль [50]. При окисле-
нии тетраэтиленгликоля высшими окислами кобальта получается
триэтоксидикарбоновая кислота [66].
Тетраэтиленгликоль предложен в качестве среды для дегидра-
тации вторичных и третичных спиртов. При диспергировании в
167
тетраэтиленгликоле 5% H2SO4 выход циклогексена из циклогексанола
при 134 °C превышает 98% [67]. Тетраэтиленгликоль может также
служить средой для синтеза Ni-фенолятов диоксиалкилдифенил-
сульфонов, являющихся стабилизаторами полиолефинов [581.
Требования, предъявляемые к качеству
три- и тетраэтиленгликолей
Триэтиленгликоль выпускается в СССР в соответствии
с ТУ 6-01-864—73 (с изменениями, введенными в 1975 г.) двух марок
с государственным знаком качества и марки Б. Технические требо-
вания на триэтиленгликоль по нормам в СССР и некоторых ино-
странных фирм приведены в табл. 54 [68—74].
Таблица 54. Технические требования на триэтиленгликоль
Показатели Нормы
СССР Kuhl- mann (Фран- ция) Celene * (Италия) GAF (США)
с госу- дарствен- ным знаком качества марка Б
Цвет по Pt — Со шкале, не более Плотность при 20 °C, г/смэ . . Коэффициент преломления при 25 100 25
1,121— 1,124 Не ниже 1,118 1,112- 1,124 1,124— 1,126 ** 1,124— 1,126
20 °C Содержание, % , не более 1,4555- 1,4565 1,4550- 1,4580 — — —
диэтиленгликоля .... 3,0 — — — —
гидроксильных групп . . 22,5— 23,1 21,9— 23,5 — — —
воды . перекисных соединений (в пересчете на 12), %, — — 0,20 0,1 0,1
не более Кислотность в пересчете на ук- 0,005 — — — —
сусную кислоту, % , не более Температурные пределы пере- гонки (при 0,1 МПа), °C 0,0025 — 0,005 0,01 0,01
начало кипения, не ниже . 278 275 275 278 283
конец кипения, не выше количество отгона в указан- ных пределах, мл, не ме- 295 295 295 300 300
нее 97 85 — 95 (до 295 °C) —
* Приведены фактические показатели продукта.
** Относительная плотность при 20 °C.
168
Технические требования на тетраэтиленгликоль, выпускаемый
в СССР по ТУ 6-02-371—73, приведены ниже;
Цвет по Pt—Со шкале, не более........... 60
Плотность при 20 °C, г/смэ ............. 1,1240—1,1260
Кислотность в пересчете на уксусную кисло-
ту, % (мол.), не более ................. . 0,01
Содержание гидроксильных групп, % . . . 17,0—18,0
Содержание перекисных соединении в пере-
счете на 12, % (мол.), не более......... 0,05
Содержание альдегидов .................. Отсутствие
ЛИТЕРАТУРА
1. Glycols. Ed. by G. О. Curine, F. Johnston. New York, Reinhold Publ. Corp.,
1953. 389 p.
2. Iordan T. E. Vapour Pressure of Organic Compounds. New York — London,
Interscience Publ. Inc., 1954, 266 p.
3. Gallaugher A. F., Hibbert H., J. Am. Chem. Soc., 1937, v. 59, -V 12, p. 2514—
2525.
4. Glvcols. Properties and Uses. Midland (Michigan), The Dow Chem. Co.,
1961. 64 *p.
5. Ганиев Ю. А., ЖФХ, 1969, t. 43, № 1, c. 239—240.
6. Gallaugher A. F., Hibbert 1J.. J. Am. Chem. Soc., 1936, v. 58, № 5,
p. 813-816.
7. Gallant R. W., Hydrocarb. Process., 1967, v. 46, № 4, p. 183—196.
8. Koizumi N., Hanai T., J. Phys. Chem., 1956, v. 60, № 11. p. 1496—1500.
, 9. Woolley E. M„ Glorge R. E., j. Solut. Chemistry, 1974, v. 3, p. 119 — 126.
10. Dykyi I., Kuska V., Seprdkova M., «Chemicke Zvesti», 1956, v. 10, № 4,
p. 193—203.
И. Сталл Д., Вестрам Э,. Зинке Г. Химическая термодинамика органических
соединений. Пер. с англ. М., «Мир». 1971. 807 с.
12. Beilsteins Handbuch der organischen Chemie. Bd. 1. Teil 2. Aufl. 4. Ber-
lin — Heidelberg, Springer Verlag, 1974. 3014 S.
13. Катц Д. Л. и др. Руководство по добыче, транспорту и переработке природ-
ного газа. Пер. с англ. Под ред. Ю. П. Коротаева и Г. В. Пономарева. М.,
«Недра», 1965. 676 с.
14. Сталл Д. Р. Таблицы давления паров индивидуальных веществ. Пер. с англ.
С. В. Горбачева п В. В. Михайлова. М., Издатиплит, 1949. 72 с.
15. Миронов К. Е., Дерябина Л. Д.. ЖПХ, 1962, т. 35. № 6, с. 1338—1342.
16. Коулъ А . ЛРизенфелъд Ф. С. Очистка газа. Изд. 2-е. Пер. с англ. Под
ред. И. И. Абрамсона. М., «Недра», 1968. 394 с.
17. Monick I. A. Alcohols. New York — London. Reinhold Book Corp., 1968.
594 p.
18. Расторгуев Ю. Л., Газдиев M. А., Инж.-фпз. ж., 1969, т. 17, № 1, с. 72—79;
Ганиев Ю. А. В кн.: Теплофизическпе свойства жидкостей. М., «Наука»,
1970, с. 95—98; Расторгуев Ю. Л., Газдиев М. А., ЖФХ, 1970, т. 44, № 12,
с. 3092—3095; Газдиев М. А., Расторгуев Ю. Л., ЖФХ, 1971, т. 45, № 3,
с. 692—693.
19. Davis D. S., Ind. Chem., 1961, v. 37, № 442, p. 590.
20- Teramoto A. e. a., Makromol. Chem., 1966, Bd. 90, S. 78—90.
21. Morenas M., Douheret G., C. r., Ser. C, 1971, v. 272, № 12, p. 1060—1062.
22. Гликоли и опыт их применения в нефтяной и газовой промышленности.
(Обзор зарубежной литературы). Сер. «Газовое дело». М., ВНИИОЭНГ,
1970. 152 с.
43. Белоусов В. П., Морачевский А. Г. Теплоты смешения жидкостей. Л., «Хи-
мия», 1970. 256 с.
169
24. Mellan I. Compatibility and Solubility. London, Noyes Develop. Corp.,
1968. 304 p.
25. Francis A. W. Critical Solution Temperatures, Washington, Am. Chem. Soc.,
D. C., 1961. 246 p.
26. Огородников С. К., Лестева T. M., Коган В. Б. Азеотропные смеси. Спра-
вочник. Л., «Химия», 1971. 848 с.
27. Коган Б. В., Фридман В. М., Кафаров В. В. Равновесие между жидкостью
п паром. М., «Наука», 1966. 1426 с.
28. Francis A. W., J. Chem. Eng., Data, 1'965, v. 10, № 2, p. 145—151; № 3,
p. 260—264.
29. El Rifai M., Riv. Combust, 1957, v. 11, № 12, p. 811—828; Graham H. L.
J. Chem. Eng., Data, 1962, v. 7, № 2, p. 214—217.
30. Ekwall Per. e. a., J. Colloid a. Interface Sei., 1968, v. 28, № 2, p. 219—226;
Александрова M. В. и др., Изв. вузов. Сер. Химпя и хим. технол., 1967,
т. 10, № 5, с. 559—562.
31. SomekhG. S., Friedlander В. О. In: Refining Petroleum for Chemicals. Advan-
ces in Chemistry Series 97. Washington, Am, Chem. Soc., D. C., 1970,
p. 228—241.
32. Справочник по растворимости. T. 2. Под ред. В. В. Кафарова. М.—Л.,
изд. АН СССР, 1963. 2068 с.
33. Kubec D. J., Soinekh G. S., Oil a. Gas, 1974, v. 72, № 1, р. 93—96.
34. Teramoto A., Fuita Н., Makromol. Chem., 196a, Bd. 85, S. 261—272.
35. Wurtz A., C. r., 1859, v. 49, № 21, p. 813—815; Lourenco А. Г. C. r., 1859,
v. 49. № 19, p. 619—620
36. Пат. США 2056830 (1934); Perry S. L., Hibbert H., Can. J. Research, Ser. B,
1936, v. 14, № 3, p. 77-83.
37. Швец В. Ф. Докторская диссертация. М., МХТИ им. Д. II. Менделеева, 1974.
38. Luboviez В. Е., Reich Р., Chem. Eng. Progr., 1971, v. 67, № 3, p. 59—64.
39. Коган Л. M. Производство и переработка окиси этилена в Германии. М.—Л.,
Госхимиздат, 1947. 121 с.
40. Пат. США 3847754 (1974); РЖХпм, 1975, 16Н27.
41. Пат. ФРГ 1668052 (1975); РЖХим, 1975, 21Н23.
42. Austin G. Т., Chem. Eng., 1974, v. 81, № 16. р. 96—100.
43. Краткая химическая энциклопедия. Т. 4. Под ред. И. Л. Кнунянца. М.,
«Советская энциклопедия», 1965. 1182 с.
44. Берлин А. А.. Белянина Е. Т,, Хим. пром., 1958, № 6, с. 340—342.
45. Лосев И. П., Тростяиская Е. В. Химпя синтетических полимеров. М., Гос-
химиздат, 1960. 574 с.
46. Пат. США 3294867 (1966); РЖХим, 1968, 17С314.
47. Пат. США 2936297 (1960); РЖХим, 1962, 16П180.
48. Sorokin М. F.. Manoviciu I., Mater, plast.. 1965, v. 2, № 5, p. 266—268.
49. Пат. США 278994 (1957); РЖХим, 1959, 39597; пат. ФРГ 1161423 (1967);
РЖХим, 1968, 7С229; Лопатин Д. В. и др. Изв. АН БССР. Сер. хим. наук,
1967, № 3, с. 109 — 116,
50. Пат. США 3294847 (1966); РЖХим, 1968, 11Н46; Николайчук Е. Д. и др.
Авт. свид. № 241426 (1968); Изобр., пром, образцы. Товарн. знаки № 14,
1969 с. 19.
51. Пат.’США 3347926 (1967); РЖХим, 1968, Н89.
52. Пат. США 3050549 (1962); РЖХпм, 1964, 8Н154.
53. Франц, пат. 92074 (1968).
54. Пат. ФРГ 1008724 (1957); РЖХим, 1958, 74851.
55. Пат. США 3252985 (1966); РЖХим, 1967, 22Н360; англ. пат. 1117806 (1968);
РЖХпм, 1969, 10Н352.
56. Пат. США 2755188 (1956); РЖХим, 1958, 30731; пат. США 2827470 (1958);
РЖХим, 1959, 83406.
57. Пат. США 2970080 (1961); РЖХим, 1962, ЗЛ406; пат. США 2967127 (1961);
РЖХим, 1962, 8Л380; пат. США 3231398 (1966); РЖХим, 1967, 16Н750.
58. Пат. ГДР 13441 (1957); РЖХим, 1958, 33662; яп. пат. 21820 (1967); РЖХим,
1968, 15Н151.
59. Пат. США 3711343 (1973); РЖХим, 1974, 1П203.
170
«О Бадалян Ш. М. и др., «Газовое дело», 1969, № 3, с. 32—34.
б1. Пат. ЧССР 124314 (1967); РЖХим, 1969, 24Н31.
62. Гуревич И. Л. и др. Новости нефт. и газ. техники. Серия «Нефтепереработка
п нефтехимия», 1962, № 2, с. 10—13.
03, Гальперин Б. М. и др. «Нефтепереработка п нефтехимия», 1975, № 8
с. 22—23.
64. Жаке Л. Ю. и др., «Химия и технология топлив и масел», 1966, № 12,
с. 27—28.
65. Sornekh G. S., Friedlander В. О., Hydrocarb. Process., 1969, v. 48, № 12,
р. 127—130; Jones W. T., Payne Vic. Ibid., 1973, v. 52, № 3, p. 91—92.
66. Англ. пат. 1114705 (1968); РЖХим, 1969, 12H59.
67. Пат. США 3275698 (1966).
68. Химические продукты и пластические массы (Celene), Милан. Челене, 66 с.
69. Чернышев А. К. и др. Справочный материал по качеству химической про-
дукции. М., ГПАП, 1972. 492 с. Triethylene glycol. Service Publicite Kuhl-
mann. Paris. RB—03—1.
Глава V
Пропиленгликоль, дипропиленгликоль
и трипропиленгликоль
Пропиленгликоль
Пропиленгликоль известен в виде двух изомеров: 1,2-пропилен-
гликоль СН3СНОН—СН2ОН и 1,3-пропиленгликоль (триметилен-
гликоль) СН2ОН—СН2—СН2ОН. Некоторые физические показа-
тели пропиленгликолей приведены в табл. 8 (см. стр. 18). В этой
главе будут описаны свойства, методы получения и области при-
менения 1,2-пропиленгликоля.
Вследствие наличия асимметрического атома углерода имеются
два оптических изомера: один, вращающий плоскость поляризации
света влево, (—)-форма и другой — вправо, (+)-форма. Приводятся
следующие значения угла вращения плоскости для оптических
изомеров: ]a]2D4’4 : -14,9 °C; [а]ь4 1 : -28,6 °C (СНС13); [a]2D412 : +30°
(СНС13) [1, 5. 2468].
Пропиленгликоль, получаемый в промышленности гидратацией
окиси пропилена, представляет собой рацемическую смесь обоих
оптических изомеров (рацемат). В дальнейшем этот (±)-1,2-пропи-
ленгликоль будет именоваться просто пропиленгликолем.
Оптические изомеры пропиленгликоля могут быть получены
из лево- и правовращающей окиси пропилена. Кроме того, лево-
вращающий изомер получают при восстановлении левовращающего
эфира молочной кислоты, а правовращающий — при гидрировании
над никелем левовращающего 3-иод-1,2-пропиленгликоля [2, с. 356].
Оптические изомеры пропиленгликоля можно также получить из
рацемата при фракционировании циклических кеталей пропилен-
гликоля и 1-ментона и последующем кислотном гидролизе индиви-
дуальных кеталей [3].
Предложен способ выделения (—)- и (+)-пропиленгликолей
из раствора рацемата в органическом растворителе (и-пропаноле,
emop-бутаноле, ацетоне и его смеси с этиловыми эфиром или этил-
ацетатом и др.) путем «затравки» кристаллическим пропиленглико-
лем раствора, охлажденного до температуры ниже —29 °C [4].
172
физические свойства
Пропиленгликоль (1,2-пропандиол, 1,2-диоксипропан)
НОСН2СНОНСН3 имеет молекулярную массу 76,09 и при обычных
условиях является бесцветной прозрачной, вязкой жидкостью без
запаха и со сладковатым вкусом. Основные физические характерис-
тики пропиленгликоля приведены ниже [5, р. 211]:
Плотность при 20 °C, г/см3 .................... 1,0363
Относительная плотность rZ|J] ................. 1,0381
Температура,°C
кипения при 101,3 кПа (760 мм рт. ст.) . . . 187,4
замерзания (застывания) ................... —60
Коэффициент преломления nJ,0 .................. 1,4326
Поверхностное натяжение при 25 °C, мН/м (дин/см) 36,5
Удельная теплоемкость при 20 °C
Дж/(г-К) ..................................... 2,483
кал/(г-°С).................................... 0,593
Теплопроводность при 20 °C
Вт/(м-К) ..................................... 0,218
кал/(см-с-°С) ............................... 0,00052
Вязкость при 20 °C, МПа • с (сПз) ................ 56,0
Диэлектрическая проницаемость при 20 °C .... 32,0
Дипольный момент, D
при 20 °C (жидкого) .......................... 3,63
[6, с. 144]
при 25 °C (в диоксане) ....................... 2,25
Константа Верде, мин/(Гс • см)
при 25 °C п 546,1 нм (5461 А) ............. 0,01540
при 25 °C и 589,3 нм (5893 А) ............. 0,01291
Константа
диссоциации, рКа, при 25 °C ............... 14,8 [7]
автопротолиза, рКs, при 20 °C ............... 17,76
[8, с. 417]
Скорость звука в пропиленгликоле, м/с .’.... 1523 [9]
Примечание. В других источниках приводятся показатели,
отличающиеся от указанных выше. Например, т. кип. 188,2,
188— 18!) °C [10, т. 1, с. 624; т. 2, с. 905]; Пд 1,4329 [И, р. 649].
Критические параметры, являющиеся расчетными, так как про-
пиленгликоль подобно другим гликолям разлагается при температуре
значительно ниже критической, приведены ниже [12]:
^Ритическая температура, °C .... 351
критическое давление, мПа (кгс/см2) 6,1 (62,0)
критическая плотность, г/см3 . . . . 0,321
-ИК-спектр абсорбции пропиленгликоля при 26 °C (длина кюветы
>0155 мм) показан на рис. 59 [5].
173
Значения стандартных (298,15 К) термодинамических величин
пропиленгликоля по различным данным следующие:
Теплота, кДж/моль,
(ккал/моль)
образования (жидко-
сти) ................
образования (газа)
сгорания (жидкости)
испарения . , , .
-486,1 ± 2,5 (-116,1 + 0,6) [13]
—499,3 (—119,3) [14]
—421,6 + 3,3 (—100,7 + 0,8) [13]
1839,3 + 2,3 (439,31 + 0,54)
64,5+ 2.1 (15,4 ± 0,5) [13]
Значения этих величин при 20 °C таковы [5]:
Теплота, кДж/моль (ккал/моль)
образования........................ —500,3 (—119,5)
сгорания при постоянном объеме 1825,0 (435,9)
Теплота испарения пропиленгликоля в зависимости от-темпера-
туры иллюстрируется следующими цифрами [5, р. 218]:
Температура, °C . . . . 40 60 80 100 120 140 160 180 187
Теплота испарения
Дж/г . . 871 854 837 816 795 779 754 724 712
кал/г . . 208 204 200 195 190 186 180 173 170
Зависимость теплоты испарения пропиленгликоля от температуры
в интервале 60—350 °C, по данным [12], показана на рис. 60.
Плотность пропиленгликоля и его водных растворов в интер'
вале температур от —15 до 25 °C приведена в Приложении, табл. И
[5, р. 214]. В более широком диапазоне температур она показана
174
ва рис. 61 [12], а изменение абсолютной и относительной плотности
в зависимости от температуры приведено в табл. 55.
При указанных в таблице температурах с изменением t на 1 °C
плотность практически изменяется на постоянную величину, рав-
ную 0,00073.
• По относительной плотности водных растворов пропиленгликоля
можно определить их концентрацию (см. Приложение, табл. 12) [5].
Плотность водных растворов пропиленгликоля с повышением кон-
центрации до 70% возрастает, а затем вновь уменьшается. Таким
образом ряду значений относительной плотности соответствуют
Рис. 60. Зависимость теплоты испарения пропиленгликоля (/) и дипропплен-
гликоля (2) от температуры.
Рис. 61. Зависимость плотности пропиленгликоля и его водных растворов от
температуры:
1 — 20% пропиленгликоля; 2 — 100% пропиленгликоля; 3 — 60 и 80% пропиленгликоля.
два значения концентрации раствора. Максимальное значение плот-
ности соответствует гидрату состава С3Нв(ОН)2 • 2Н2О.
Уменьшение объема (сжатие) водных растворов пропиленгликоля
при 20 °C составляет [26, р. 8]:
Гликоль, % (об.)
Сжатие, % (об.)
20 40 60 80
0,83 1,84 2,08 1,47
Плотность растворов пропиленгликоля и этиленгликоля является
линейной функцией их состава (см. [стр. 41). Относительная
Таблица 55. Зависимость плотности пропиленгликоля от температуры
?е“°еРатура, ь (1) Плотность, г/см3 Относительная плотность
л d4 d25
0 1,0508 1,0509 1,0519 1,0528 1,0540
10 1,0435 1,0436 1,0445 1,0454 1,0466
20 1,0363 1,0363 1,0372 1,0381 1,0393
30 1,0290 1,0290 1,0299 1,0308 1,0320
40 1,0217 1,0217 1,0226 1,0235 1,0247
175
плотность смеси пропиленгликоля с дипропиленгликолем является
также линейной функцией состава: снижение относительной плотности
на 0,0005 соответствует увеличению содержания дипропиленгликоля
на 0,39%. Для смесей пропиленгликоля с дипропиленгликолем
относительная плотность приведена в Приложении, табл. 13 [5,
р. 215].
Температура кипения пропиленгликоля с повышением давления
меняется следующим образом [15, с. 12]:
Давление, Температура, Давление, Температура,
мм рт. ст. * °C ММ рт. Ст. * °C
1 45,5 60 119,2
5 70,8 100 132
10 83,2 200 149,7
20 96,4 400 168,1
40 111,2 760 188,2
* 1 мм рт. ст. — 133,3 Па.
В интервале от 98 до 101 кПа (740—760 мм рт. ст.) при изменении
давления на 0,133 кПа (1 мм рт. ст.) температура кипения пропилен-
гликоля повышается на 0,042 °C.
Определение температуры замерзания пропп лонг лик о ля "и его
водных растворов затруднено из-за их высокой вязкости при низких
температурах и способности к переохлаждению. Указывается, что
растворы, содержащие более 60% пропиленгликоля, имеют темпера-
туру замерзания ниже —70 °C, а для чистого пропиленгликоля
найдены значения от —60 до —77 °C [5, р. 217]. При охлаждении
растворов с концентрацией до 60% в твердую фазу выпадает лед.
Температура замерзания (начала кристаллизации) водных растворов
пропиленгликоля приведена ниже [16, S. 393].
Для водных растворов пропиленгликоля зависимость давления
насыщенных паров от температуры приведена в Приложении, табл. 14
[5,р. 2361.
Коэффициент преломления пропиленгликоля в интервале от 20
до 30 °C уменьшается на 0,0003 при повышении температуры на
i °C. Большая разница коэффициентов преломления пропилен-
гликоля (пд = 1,4326) и воды (пр = 1,3330) позволяет быстро
Л точно определять по коэффициенту преломления концентрацию
пропиленгликоля в его смеси с водой (см. Приложение, табл. 15,
[5,р. 223], а также рис. 36, стр. 122). Коэффициент преломления смесей
этилен- и пропиленгликоля является линейной функцией их состава.
Рис. 62. Зависимость поверхностного натяжения пропиленгликоля (7) и ди-
пропиленгликоля (2) от температуры.
Рис. 63. Зависимость поверхностного натяжения водных растворов пропилен-
гликоля (1) и дипропиленгликоля (2) от концентрации при 25 °C.
Поверхностное натяжение пропиленгликоля и его водных рас-
творов в зависимости от температуры и концентрации показано на
рис. 62 и 63 [12].
Таблица 56. Зависимость давления насыщенного пара пропиленгликоля
от температуры
Пропиленгли-
коль, % (об.) . <0
- Температура, °C
начала кри-
сталлизации —2,5
образования
хлопьев . . —3
20 30 40 50
-7 -13 -23 -35
-8 -13 -25 -42
60 70 80 90 100'
-50 - _ _ _
-69 —66 -63 —61 -59
Давление насыщенного пара пропиленгликоля для различных
температурных интервалов (табл. 56) может быть рассчитано по
следующим уравнениям [5, р. 235; 10, т. 1, с. 699; 17, р. 104]:
в интервале 80—130 ° С 1g р — 9,5157--
в интервале 130—187.4 °C lg/? = 9,2317—
где t — температура, °C; р — давление, мм рт. ст.
'Темпера- тура, °с Давление, мм рт. ст. * Темпера- тура, °C Давление, ммрт. ст. * Темпера- тура, °C Давление, гм рт. ст. * Темпера- тура, °C Давление, ммрт. ст. *
0 0,01 80 7,40 125 76 170 420
10 0,03 85 9,95 130 93 175 505
20 0,08 90 13,4 135 114 180 590
30 0,20 95 17,8 140 140 185 695
40 0,46 100 23,1 145 170 187,4 760
50 0,99 105 30,3 150 205 190 825
60 2,03 ПО 39,3 155 245 195 960
70 3,98 115 49 160 299 200 1110
75 5,46 120 62 165 353
1 им рт. ст. = 133,3 Па.
^2 Заказ 972 177
176
Теплоемкость пропиленгликоля при низких температурах харак-
теризуется следующими цифрами * [5, р. 229]:
Температура,
°C
—70
-80
__90 **
—100 **
—110 **
—120 **
Теплоемкость,
кал/(г -°C)
0,47
0,46
0,46
0,46
0,48
0,29
Температура,
°C
—130
—140
-150
—160
—170
-180
Теплоемкость,
кал/(г- °C)
0,23
0,21
0,20
0,19
0,18
0,16
** Область перехода из жидкого в стеклообразное состояние.
При более высоких температурах теплоемкость составляет *;
Температура, °C.............. 130 140 150 160 170 180 187,4
Теплоемкость, кал/(г-°C) . . . 0,747 0,761 0,775 0,788 0,803 0,817 0,827
* 1 кал/(г-°С) = 4,19 Дж/(г-К).
Зависимость теплоемкости пропиленгликоля и его водных рас-
творов от температуры приведена в табл. 57 [5, р. 228]:
Таблица 57. Зависимость теплоемкости пропиленгликоля
и его водных растворов от температуры
Температу- ра, °C Теплоемкость при различной концентрации пропиленгликоля, кал/(г. °C)*
10% 20% 30% 40% 50% 60% 70% 80% 90% 100%
-60 . 0,760 0,695 0,615 0,545 0,482
-50 — — — — — 0,762 0,698 0,626 0,557 0,496
—40 — — — — — 0,766 0,705 0,636 0,570 0,509
—30 .— — — — 0,823 0,772 0,711 0,647 0,583 0,523
-20 — — — 0,887 0,828 0,778 0,718 0,657 0,597 0,537
-10 — — 0,934 0,887 0,833 0,785 0,727 0,668 0,610 0,552
0 0,994 0,968 0,934 0,890 0,838 0,792 0,735 0,680 0,623 0,565
10 0,990 0,965 0,934 0,893 0,845 0,799 0,745 0,692 0,636 0,579
20 0,989 0,963 0,935 0,897 0,852 0,807 0,754 0,703 0,648 0,593
30 0,989 0.963 0,937 0,902 0,860 0,817 0,765 0,716 0,662 0,607
40 0,989 0,965 0,940 0,907 0,868 0,827 0,777 0,728 0,675 0,622
50 0,990 0,968 0,944 0,918 0,877 0,837 0,788 0,741 0,688 0,635
60 0,992 0,973 0,949 0,918 0,885 0,847 0,801 0,753 0,702 0,649
70 0,994 0,977 0,955 0,926 0,894 0,857 0,813 0,766 0,715 0,663
80 0,997 0,981 0,961 0,934 0,903 0,867 0,825 0,779 0,728 0,678
90 0,998 0,985 0,968 0,942 0,913 0,878 0,837 0,793 0,748 0,692
100 1,002 0,990 0,975 0,951 0,923 0,888 0,851 0,806 0,754 0,705
но .— — — — — 0,863 0,818 0,767 0,719
120 — — — — — — 0,781 0,733
« 1 кал/(г-°C)—4,19 Дж/(г-К)
178
Зависимость теплоемкости паров пропиленгликоля от температуры
доказана на рис. 64 [12].
Теплопроводность пропиленгликоля А, [(Вт/(м-К)] в интервале
температур 18—98 °C и при давлении 0,7 МПа (7,5 кгс/см2) может
быть рассчитана по уравнению [18]:
Л = 0,2028 - 0,084- 10-з«
Изменение теплопроводности пропиленгликоля, его паров и вод-
ных растворов в более широком диапазоне температур показано
в табл. 58 [5, р. 231], а также на рис. 65 и 66 [12].
Таблица 58. Зависимость теплопроводности пропиленгликоля
и его водных растворов от температуры
Температура, °C Теплопроводность при различной концентрации пропиленгликоля, кал/(см-с-°C)
20% 40% 6 0% 80% 10 0%
0 0,00117 0,00100 0,00083 0,00068 0,00054
20 0,00121 0,00100 0,00082 0,00066 0,00052
40 0,00125 0,00101 0,00080 0,00064 0,00050
60 0,00129 0,00102 0,00079 0,00062 0,00048
80 0,00133 0,00102 0,00078 0,00060 0,00045
100 0,00137 0,00103 0,00077 0,00058 0,00043
120 0,00141 0,00104 0,00075 0,00056 0,00041
* 1 кал/(см- с. °С)—1,19-ю2 Вт/(м-К)
Зависимость вязкости пропиленгликоля и его водных растворов
от температуры приведена в табл. 59 [5, р. 238], а вязкости паров
пропиленгликоля — на рис. 67 [12].
Таблица 59. Зависимость вязкости пропиленгликоля и его водных
растворов от температуры
Температура, °C Вязкость при различной концентрации пропиленгликоля, мПа* с ,
20% 40% 6 0% 80% 100%
—40 1100 4100 22 900
-30 — 340 1100 6600
-20 46 140 400 1750
-10 — 20 59 160 585
0 4,2 12,5 29 72 243
10 2,9 7,2 16 34 111
20 2,1 4,4 9,3 20 56
30 1,6 2,9 5,6 12,5 30,3
40 1,20 2,2 3,9 7,4 18,0
50 0,98 1,7 2,9 5,3 11,3
60 0,79 1,3 2,2 3,9 7,7
70 0,68 1,10 1,7 2,9 5,5 1
80 0,58 0,88 1,3 2,0 4,2
90 0,49 0,73 1,1 1,8 3,3
100 0,43 0,58 0,83 1,4 2,7
12*
179
Вязкость смесей пропиленгликоля с этиленгликолем, взятых
в соотношении 1:1, при температурах от —50 до +20 °C приведена
на рис. 11 (см. стр. 49), а смесей пропиленгликоля с этиловым
и пропиловым спиртами, содержащих 40 и 60% воды, — на рис. 12
и 13 (см. стр. 50). Вязкость бинарных смесей пропиленгликоль —
Рис. 64. Зависимость теплоемкости паров пропиленгликоля (?) и дипропилен-
гликоля (2) от температуры.
Рис. 65. Зависимость теплопроводности пропиленгликоля и его водных раство-
ров от температуры.
!00 200 000 Р00 500
Температура., °C
Рис. 66. Зависимость теплопроводности паров пропиленгликоля (7) и дипропи»
ленгликоля (2) от температуры.
Рис. 67. Зависимость вязкости паров пропиленгликоля (1) и дипропнленгликоля
(2) от температуры.
н-бутанол и пропиленгликоль — н-бутилацетат, а также тройной
смеси пропиленгликоль — н-бутанол — н-бутилацетат при 40 °C
приведена в работе [19].
Диэлектрическая проницаемость пропиленгликоля в интервале
температур от —40 до —90 °C и частот от 1 до 100 кГц показана
на рис. 68 [5, р. 216]. Изучена также диэлектрическая проница-
емость растворов пропиленгликоля в воде и диоксане [20, 21]. Для
растворов пропиленгликоля с концентрацией 0,1 мол/л в воде, ди-
180
метилформамиде и пиридине электропроводность при 20 °C равна 73
33 и 6,8-10_8 Ом-1-см-1 соответственно [22]. ’
Дипольный момент смесей пропиленгликоля с водой составляет
при концентрации пропиленгликоля от 0 до 50% — 4,04, от 50 до
85% - 3,85 и от 85 до 100% - 3,81 D [21].
Рис. 68. Диэлектрическая проницаемость пропиленгликоля (цифры на линиях —
килогерцы).
Сжимаемость пропиленгликоля в зависимости от температуры
и давления характеризуется цифрами, приведенными в табл. 60 [5,
р. 221]:
Таблица 60. Сжимаемость пропиленгликоля
Давление, МПа (кгс/см2) Относительный объем
при 0 °C при 50 °C при 95 °C
0 1,0000
4р,0 (500) 0,9819 0,0129 —
98,1 (1000) 0,9664 # 0,9947 1,0201
196,1 (2000) 0,9432 0,9659 0,9877
392,3 (4000) 0,9070 0,9266 0,9430
588,4 (6000) 0,8809 0,8989 0,9110
784,5 (8000) 0,8569 0,8750 0,8867
980,7 (10 000) 0,8380 0,8563 0,8672
1176,8 (12 000) 0,8219 0,8400 0,8514
—
Гигроскопичность пропиленгликоля почти такая же, как гли-
церина, но меныпая, чем этиленгликоля [5, р. 219]. Состав водных
Рестворов пропиленгликоля, находящихся в равновесии с влагой
181
воздуха при определенной его относительной влажности и темпера'
туре приведен в табл. 61 [23, р. 13]:
Таблица 61. Равновесная концентрация пропиленгликоля
с влагой воздуха
Температура воздуха, °C Равновесная концентрация при относительной влажности, %
10% 20% 30% 40% 50% 6 0%
—5 96,8 91,4 ..90,0 84,6 77 73
5 97,0 92,3 90,2 85,2 78 74
15 97,1 92,9 90,4 85,8 80 74
25 97,1 93,5 90,5 86,3 81 75
35 97,2 93,9 90,6 86,6 82 75
50 97,2 94,3 90,7 86,7 83 76
При относительной влажности воздуха выше 70% концентрация
пропиленгликоля не зависит от температуры контакта и составляет:
Относительная влажность, % .............. 70 80 90
Концентрация пропиленгликоля, % .... 68 55 40
Точка росы воздуха в зависимости от концентрации пропилен-
гликоля и температуры приведена на рис. 69 [5, р. 220].
Растворимость. Пропиленгликоль полностью смешивается с во-
дой и при смешении выделяется теплота, количество которой зависит
от соотношения взятых компонентов и температуры [24, с. 70].
Теплота смешения пропиленгликоля с водой при 25 °C равна:
Концентрация
пропиленгликоля,
% (мол.) .... 10 20 30 40 50 60
. Теплота смешения
на 1 г/моль рас-
твора
Дж............—624 —821 —804 —725 —632 -532
кал...........-149 —196 -192 —173 -151 -127
Теплота смешения пропиленгликоля с метиловым, этиловым,
н-пропиловым, бутиловым и октиловым спиртами определена в ра-
боте [25].
Пропиленгликоль является хорошим растворителем для различ-
ного класса соединений. С ним полностью смешивается большинство
низкомолекулярных органических соединений, содержащих кисло-
род и азот:
одноатомные спирты (метиловый, этиловый, изопропиловый, н-бу-
тиловый, н-амиловый, фенилэтиловый)
182
этилен- и пропиленгликоли и пх эфиры
кислоты (уксусная, диэтилуксусная, 4-метил-н-валериановая и
олеиновая)
альдегиды (анисовый, бензойный, горчичный и салициловый,
фурфурол, цитраль) и кетоны (ацетон, а-ионон, метилизопропил-
и метилизобутилкетоны)
сложные эфиры (этил-, этилхлор-, н-бутил-, амил- и изоамил-
ацетаты, три-н-бутилфосфат, н-бутиллактон)
Рис. 69. Зависимость точки росы газа от температуры контакта при осушке
с водными растворами пропиленгликоля.
амины и другие азотсодержащие соединения (а-метилбензил-
этанол- и диэтаноламины, диизопропил-, ди-н-бутил-, ди-н-амил-,
e-метилбензил-, а-метилбензи л диметил-, 2-фенилэтиламины, ди-
этилентриамин, триэтилентетрамин, пиридин, 2-метил-5-этил-
пиридин, диэтилформамид, о-фенетедин, н-амилцианид).
Пропиленгликоль также смешивается с диэтиловым эфиром,
этиленхлоргидрином, хлороформом, кассиевым и гвоздичным мас-
лами, скипидаром, метилантранилатом [5, р. 225; 26, р. 50; 23,
Р- 9; 27, р. 58].
Взаимная растворимость пропиленгликоля и бензола при раз-
личных температурах приведена в работе [28, с. 1300]. Растворимость
183
в пропиленгликоле различных соединений дана в Приложении,
см. стр. 361 —363. Растворимость других синтетических веществ
(душистых веществ, масел и смол) в водных растворах пропилен-
гликоля приведена в работе [26, р. 68].
Азеотропные смеси. Пропиленгликоль образует с рядом органи-
ческих соединений азеотропные смеси. Состав и температура кипения
этих смесей при атмосферном давлении даны в Приложении, стр. 363
[29, с. 340; 10, т. 3, с. 406].
Равновесие между жидкостью и паром водных растворов пропп-
ленгликоля при 101,3 кПа (760 мм рт. ст.) и 18,7 кПа (140 мм рт. ст.)
характеризуется следующими данными [5, р. 233]:
Содержание воды, % мол.
в жидкости .... 10 20 30
в паре при 101,3 кПа 58,5 83,8 91,7
в паре при 18,7 кПа 72,6 87,1 93,8
40 50 60 70 80 90
95,7 97,6 98,3 98,8 99,3 99,7
96,4 97,7 98,5 98,8 99,3 99,7
фазовое равновесие и температура кипения системы пропилен-
гликоль — вода при 101,3 кПа приведены ниже:
Содержание пропилен-
гликоля,% 95,2 96,7 98,0
в жидкости .... 58,5 82,7 89,5 93,0 99,0 100
в паре 3,8 10,0 18,3 29,0 41,2 55,0 70,4 87,0 100
Температура, °C .... ПО 120 130 140 150 160 170 180 187,4
Фазовое равновесие жидкость — пар этой же системы при других
давлениях даны в табл. 62 [30], а также на рис. 70 [26, р. 64].
фазовое равновесие жидкость — пар для системы пропилен-
гликоль — дипропиленгликоль при 1,33 кПа (10 мм рт. ст.) харак-
теризуется следующими цифрами [5]:
Содержание пропилен-
гликоля, % (мол.)
в жидкости .... 10 20 30
вйаре ............ 37,5 57,4 70,3
40 50 60 70 80 90
78,8 85,0 89,2 92,6 95,3 97,7
Таблица 62. Фазовое равновесие системы жидкость—пар для водных
растворов пропиленгликоля
Содержание воды, % (мол.) Температура, 'С Содержание воды, % (мол.) Температура, °C
жидкость пар жидкость пар
Давление 1,33 кПа (10 мм рт. ст.) Давление 3,33 кПа (25 мм рт. ст.)
1,0 35,9 81,1 1,2 36,4 98,2
4,8 76,0 70,0 6,0 77,5 83,5 -
10,2 89,2 62,2 13,3 91,6 69,0
19,5 95,79 49,9 20,6 95,5 57,1
26,6 97,83 38,3 31,9 97,85 48,2
42,7 99,16 31,4 42,8 98,84 42,0 "
51,0 99,47 23,2 51,4 99,22 38,6
60,5 99,67 20,5 60,5 99,50 35,8
68,1 99,78 18,1 70,0 99,68 33,5
Давление 6,67 кПа (50 мм рт. ст.) Давление 13,3 кПа (100 мм рт. ст.)
4,8 66,8 98,5 10,6 82,7 100,8
11,0 85,4 83,8 16,7 89,6 91,1
20,9 85,4 71,4 25,8 94,7 81,3
29,4 96,6 64,0 36,3 97,0 73,4
38,2 98,0 58,3 42,7 97,9 70,3
47,5 98,71 53,4 51,2 98,75 66,5
56,0 99,14 50,1 60,6 99,04 63,0
64,3 99,41 47,1 67,0 99,28 61,1
72,2 99,61 46,0 72,8 99,44 58,4
81,5 99,78 43,5 76,2 99,55 57,5
87,5 99,87 42,1 82,8 99,70 55,7
86,6 99,79 54,3
Рис. 71. Равновесие
в трехкомпонентиых си-
стемах, содержащих про-
пиленгликоль (ПГ).
Условные обозна-
чения: ж — жидкость;
т — твердая фаза.
Рис. 70. Фазовый состав
для системы пропилен-
гликоль — вода:
1 — при 13,3 (100 мм рт.
ст.); 2 — при 40 кПа
(300 мм рт. ст.); 3 — при
80 кПа (В00 мм рт. ст.).
184
185
Данные по фазовому равновесию жидкость — пар для системы
пропиленгликоль — этиленгликоль приведены в табл. 25 (см.
стр. 60). Изучено также равновесие жидкость — пар для систем,
содержащих кроме пропиленгликоля «-бутиловый спирт, «-бутил-
ацетат, глицерин [30, 31].
Равновесие в тройных системах, включающих пропиленгликоль.
представлено графически в треугольной диаграмме Гиббса на
рис. 71 [32].
Состав тройных смесей, содержащих наряду с пропиленгликолем
и олеатом натрия спирты (этиловый, аллиловый, пропиловый и изо-
пропиловый, бутиловый, emop-бутиловый, изобутиловый, mpem-бу-
тиловый и mpem-хлорбутиловый, амиловый, emop-амиловый и изо-
амиловый, гексиловый и циклогексиловый, октиловый и изоактило-
вый, лауриловый, цетиловый, бензиловый), хлорорганические
соединения (этиленхлоргидрин, триметиленхлоргидрин, четырех-
хлористый углерод, хлороформ, хлористый метилен, дихлорэтан,
хлористый бутил), а также ацетон, метилэтилкетон, этилацетат,
бензол, фенол, приведены в работе [28]. Там же приведен состав
тройных систем пропиленгликоль — бензол с лауратом, миристатом,
пальмитатом, стеаратом натрия и пальмитатом калия, пропилен-
гликоль — вода с подом, этиловым эфиром, сахарозой, пропилен-
гликоль — бензоат натрия — хлороформ.
Равновесие жидкость — жидкость для тройных систем пропилен-
гликоль — этанол с гексаном, циклогексаном и бензолом, пропилен-
гликоль — вода с бутиловым и изоамиловым спиртами, изобутил-
кетоном, этил-, н-бутил- и изоамилацетатом изучено в работах [33].
Дипропиленгликоль и трипропиленгликоль
Дипропиленглийель (СвН14О3) и трипропиленгликоль (С9Н20О4)
являются бесцветными, вязкими, умеренно гигроскопичными жидко-
стями. Молекулярная масса дипропиленгликоля — 134,17, а три-
пропиленгликоля — 192,25.
В промышленных условиях получают дипропиленгликоль, явля-
ющийся смесью трех изомеров:
СН3 СН3
НОСН2СН-О-СНСН2ОН
ди-первичный
дипропиленгликоль
СН3 СН3
НОСНСН2—О—CHgCHOH
ддавторичный
дипропиленгликоль
СН3 СН3
НОСН2СН—О—СН2СНОН
первично-вторичный
дипропиленгликоль
186
Соотношение изомеров непостоянно и зависит от природы и кон-
центрации катализатора, применяемого при гидратации окиси про-
пилена. При гидратации в слабощелочной среде содержание изомеров
следующее: 2% ди-первичного, 65—70% ди-вторичного и 30—35%
первично-вторичного. При гидратации же в присутствии серной
кислоты дипропиленгликоль содержит 10—15% ди-первичного, 30—
35% ди-вторичного и 50—60% первично-вторичного изомера [34,
с. 12]. В товарном дипропиленгликоле содержится 4% дипервичного
изомера, 43% дивторичного и 53% первично-вторичного [И, р. 651].
Трипропиленгликоль получается в небольших количествах, как
побочный продукт, в производстве пропиленгликоля и может содер-
жать 7 изомеров, в том числе:
СН3 СН3 СНз
I I I
НОСН2СН-О—СН2СН-О-СНСН2ОН
диепервичный трипропиленгликоль
СНз СНз СН3
НОСНСН2—О—CHgCH—О—СН2СНОН
диввторичный трипропиленгликоль
СНз СНз СНз
НОСН2СН—О-СН2СН-О-СН2СНОН
первичновторичный трипропиленгликоль
Физические свойства
Основные физические характеристики ди- и трипропиленгликоля
приведены ниже (при 20 °C [35, р. 4], при 25 °C [23, р. 2]):
Дипропилен- гликоль Трипропилен- гликоль
Плотность, при 25 °C, г/см® , , , f s 1,023 1,016
Относительная плотность d~g . . . . Температура,°C кипения при 101,3 кПа (760 мм рт. 1,024 —
ст.) 231,8 268
замерзания (потеря текучести) Давление насыщенного пара —38,9 —41,1
при 20 °C, Па (шт рт. ст.) . . . <1,33 (0,01) —
при 25 °C, Па (мм рт. ст.) . . . Коэффициент преломления п 1,д 4Д) (0,03) <1,33 (0,01)
при 20 °C 1,4411 —
при 25 °C Поверхностное натяжение, мН/м (дин/см) 1,439 1 >442
при 20 °C 32,8 —
при 25 °C 33 34
187
Удельная теплоемкость при 20 С°
Дж/(г-К)........................... 2,39
кал/(г-°С)...................... 0,57
Удельная теплоемкость при 25 °C
Дж/(г-К) ....................... 2,43
кал/(г.°С)...................... 0,58
Теплопроводность при 20 °C
Вт/(м-К) ........................... 0,172
кал/(см-с-°С) .................. 0,00041
Вязкость, мПа • с (сПз)
при 20 °C .......................... 106
при 25 °C ...................... 72,5
при 60 °C ...................... 11,1
2,13
0,51
55,1
9,8
Примечание. По другим источникам температура кипения ди-
пропиленгликоля равна 229,2, а трипропиленгликоля — 267,2 °C
[10, т. 1, с. 653—654], коэффициент преломления дипропиленгли-
20
коля п2д—1,Ш0 [5, р, 275] и 1,4407 [36, р. 351], а его теплоем-
кость—2,72 Дж/(г-К)или 0,65 кал/(г-°С) [36].
Критические параметры дипропиленгликоля, являющиеся рас-
четными, так как температура начала его разложения значительно
ниже критической, следующие [12J:
Критическая температура, °C .... 378
Критическое давление, мПа (кгс/см2) 3,6 (36,6)
Критическая плотность, г/см8 .... 0,321
Теплота испарения дипропиленгликоля при 101,3 кПа (760 мм рт.
ст.) равна 400,3 Дж/г или 95,6 кал/г. В интервале 60—380 °C теплота
испарения дипропиленгликоля приведена на рис. 60 (см. стр. 175).
Некоторые физические свойства изомеров дипропиленгликоля при-
ведены в табл. 63 [37].
Таблица 63. Некоторые физические свойства изомеров дипропиленгликоля
Изомер дипропилен- гликоля Температура, °C Относительная плотность Коэффициент преломления
кипения * плавления п60 п4 я25 nD я60 nD
Ди-первич- ный 225,7 (760) 102 (5) Ниже —40 1,0316 1,0018 1,4410 1,4296
Ди-вторич- ный 222,2 (760) 113—114 (10) 45-46,5 — 0,9878 — 1,4284
Первично- вторичный 224 (760) 96—96,5 (4) Ниже —40 1,0229 0,9925 1,4391 1,4283
* В скобках указано давление в мм рт. ст.; 1 мм рт. ст. = 133,32 Па.
188
Плотность дипропиленгликоля (ДПГ) и его водных растворов
с повышением температуры меняется следующим образом, г/см3 [12]:
Температура, °C 100% ДПГ 80% п 60% ДПГ 40% ДПГ
-20 1,05 1,064 —
0 1,038 1,054 1,045
20 1,025 1,040 1,036
40 1,010 1,025 1,023
60 0,996 1,008 1,006
80 0,992 0,992 0,992
100 0,965 0,976 0,976
120 0,950 0,958 0,960
140 0,936 0,943 0,943
160 0,920 0,926 0,930
180 0,906 0,910 0,912
Плотность трипропиленгликоля и его водных растворов при
различной температуре приведена на рис. 72 [23, р. 46]. Плотность
водных растворов ди- и трипропиленгликоля выше плотности чистых
соединений, т. е. при смешении их с водой происходит сжатие рас-
твора.
Температуры кипения дипропиленгликоля (ДПГ) и трипропилен-
гликоля (ТПГ) при различном давлении характеризуются следу-
ющими данными [15, с. 20; 17, р. 81].
Давление, Температура Давление, Температура
мм рт. ст.* кипения, °C мм рт. ст.* кипения, °C
Дпг ТПГ дпг ТПГ
1 73,8 96 60 156,5 184,6
5 102,1 125,7 100 169,9 199
10 116,2 140,5 200 189,9 220,2
20 131,3 155,8 400 210,5 244,3
40 147,4 173,7 760 231,8 267,2
* 1 мм рт. ст. — 133,32 Па.
Для водных растворов дипропиленгликоля на рис. 3 (см. стр. 20)
приведена температура замерзания.
Зависимость давления насыщенных паров ди- и трипропиленгли-
коля от температуры показана на рис. 1, а давления паров водных
Растворов дипропиленгликоля на рис. 73 [23, р. 22]. Приведены
также зависимости коэффициента преломления водных растворов
Дипропиленгликоля при 25 °C от концентрации (см. рис. 36, стр. 122),
поверхностного натяжения дипропиленгликоля от температуры и его
иодных растворов от концентрации при 25 °C (см. рис. 62 и 63,
стр. 177).
Удельная теплоемкость дипропиленгликоля и его водных рас-
творов практически прямолинейно меняется с повышением
189
температуры, что подтверждается данными табл. 64 [23, р. 38] и на
рис. 74 [12]. При более высоких температурах теплоемкость равна [12]:
Температура, °C . . . . 120 140 160 180
Теплоемкость, Дж/(г • К) 2,89 2,97 3,06 3,14
кал/(г-°С) . 0,69 0,71 0,73 0,75
Теплоемкость паров дипропиленгликоля в температурном интер-
вале 0—500 °C приведена на рис. 64 (см. стр. 180).
Теплопроводность дипропиленгликоля, по данным [12], является
линейной функцией температуры и повышается с 1,72-10“3 (0,41 X
X 10“3) при 20 °C до 1,93-10"3 Дж/(см-с-К) [0,46-Ю"3 (кал/(см X
X с-°C)] при 200 °C. Зависимость теплопроводности дипропилен-
Рис. 72. Зависимость
плотности трипропилен-
гликоля (ТПГ) и его вод-
ных растворов от тем-
пературы.
Рис. 73. Зависимость да-
вления пара дипропи-
ленгликоля (ДПГ) и его
водных растворов от
температуры.
190
Таблица 64. Зависимость удельной теплоемкости дипропиленгликоля
и его водных растворов от температуры
Температура, °C Теплоемкость при различной концентрации дипропилен- гликоля, кал/(г-°C)* *
10% 20% 4 0% 60% 80% 100%
20 0,985 0,968 0,902 0,805 0,691 0,575
30 0,987 0,970 0,906 0,813 0,701 0,585
40 0,989 0,972 0,911 0,821 0,710 0,596
50 0,990 0,974 0,916 0,829 0,719 0,607
60 0,991 0,976 0,920 0,836 0,728 0,618
70 0,993 0,978 0,924 0,844 0,738 0,630
80 0,995 0,980 0,928 0,852 0,747 0,642
90 0,996 0,982 0,932 0,860 0,757 0,653
100 0,997 0,984 0,937 0,868 0,766 0,664
НО 0,998 0,986 0,941 0,876 0,776 0,674
* 1 кал/(г-°С) =4,19 Дж/(г-К).
гликоля и его водных растворов [Л,- 10s, кал/(см-с • °C) * от темпера-
туры характеризуется следующими данными [38]:
Температура, °C 20% 40% 6 0% 80% 100%
0 1,09 0,87 0,68 0,52 0,38
50 1,23 0,97 0,74 0,55 0,39
100 1,30 1,02 0,77 0,57 0,39
Теплопроводность паров пропиленгликоля в интервале 0—500 °C
показана на рис. 66 (см. стр. 180).
Зависимость вязкости дипропиленгликоля, трипропиленгликоля
и их водных растворов от температуры приведена на рис. 75 и 76 [12]
и [23, р. 35], а паров дипропиленгликоля — на рис. 67 [см. стр. 180).
При более высоких температурах вязкость дипропиленгликоля
составляет [12]:
Температура, °C .... 120 140 160 180
Вязкость, мПа • с ... 2 1,6 1,4 1,2
Гигроскопичность. Дипропиленгликоль, как и остальные гли-
коли, является гигроскопичным веществом, хотя его гигроскопич-
ность заметно ниже, чем у этилен-, пропилен- и диэтиленгликоля,
и близка, но все же несколько меньше, чем у триэтиленгликоля.
В табл. 65 [23, р. 13] приводится равновесная концентрация водных
* 1 кал/(см-с-°С) = 419 Вт/(м-К).
191
растворов дипропиленгликоля при определенной влажности и тем-
пературе воздуха.
При относительной влажности воздуха выше 50% равновесная
концентрация дипропиленгликоля не зависит от температуры кон-
такта и составляет:
Относительная влажность, % . 50 60 70 80 90
Концентрация ДПГ, % ... 89 85 79 67 51
Рис. 74. Зависимость теплоемкости
дипропиленгликоля- (ДПГ) и его
водных растворов от температуры.
Рис. 75. Зависимость вязкости ди-
проппленгликоля (ДПГ) и его водных
растворов от температуры.
Точка росы газа в зависимости от концентрации и температуры
раствора дипропиленгликоля приведены на рис. 78 [23, р. 18].
Растворимость. Дипропиленгликоль полностью смешивается с во-
дой и при этом выделяется теплота, количество которой зависит'от
Таблица 65. Равновесная концентрация дипропиленгликоля
с, влагой воздуха
Температура воздуха, °C Равновесная концентрация при относительной влажности воздуха, %
10% ' 20% 30% 40%
5 98,5 97,0 95,1 92,6
5 98,4 96,9 95,0 92,5
15 98,4 96,8 94,8 92,4
25 98,3 96,7 94,7 92,3
35 98,3 96,6 94,6 92,1
50 98,2 96,5 94,5 92,0
соотношения взятых компонентов. При 25 °C теплота смешения
дипропиленгликоля с водой равна [24, с. 77]:
Концентрация ДПГ, % (мол.) 10 20 30 40 50
Теплота смешения на г/моль
раствора,
Дж .—837 —904 —804 —682 —569
кал .—200 —216 —192 —163 —136
Температура, °C
Рис. 76. Зависимость вязкости трипропиленглпколя (ТПГ) и его водных рас-
творов от температуры.
Рис. 77. Фазовый состав для системы дипропиленгликоль — вода:
1 — при 13,3 кПа (100 мм рт. от.); 2— при 40 кПа (300 мм рт. ст.); 3 — при 80 кПа
(600 мм рт. ст.).
Дипропиленгликоль полностью смешивается с гликолями и их
простыми эфирами, полиэтиленгликолями с молекулярной массой
200—2000 и следующими органическими соединениями:
спиртами (метиловый, этиловый, метилизобутилкарбинол, 3-геп-
танол)
кислотами (диэтилуксусной, 4-метил-н-валериановой)
альдегидами (анисовым, коричным и салициловым)
кетонами (ацетоном, метилизопропилкетоном, метилизобутил-
кетоном, диизобутилкетоном, 2,6,8-три-метил-4-нонаном)
эфирами кислот (этилхлорацетатом, изоамилацетатом, и-гептил-
ацетатом, этилфенилацетатом, этилциннаматом, 3-и-бутилфосфатом,
Дибутилфталатом)
аминами (диизопропил- и ди-н-пропиламинами, ди-и-бутил-, ди-
н-амил аминами, диэтилентриамином, триэтилентетрамином, а-ме-
тилбензиламином, а-метилбензилдиметиламином, 2-фенил-этил-
амином)
1 • „ 0,193
1 Ззкзз 9 7-2
192
аминоспиртами (моно- и диэтаноламинами, изоиропаноламином,
а-метилбензилэтаноламином, а-метилбензилдиэтаноламином).
Дипропиленгликоль полностью смешивается с этиловыми, ди-
хлордиэтиловым и бензиловым эфирами, фенолом, о-крезолом, амил-
цианидом, касторовым, сосновым, талловым маслами, он также
смешивается с диэтилформамидом, пиридином, 2-метил-5-этилпири-
дином [23, р. 9; 27, р. 59].
Дипропиленгликоль полностью смешивается с рядом веществ,
которые ограниченно растворимы в других гликолях, а именно:
бензолом, толуолом, стиролом, монохлорбензолом, о-дихлорбензо-
лом, а также хлороформом, четыреххлористым углеродом и тетра-
хлорэтиленом.
Трипропиленгликоль является наиболее эффективным раствори-
телем из гликолей. Он полностью смешивается с водой, другими
гликолями и их простыми эфирами, бензолом, толуолом, стиролом,
монохлорбензолом, о-дихлорбензолом, четыреххлористым угле-
родом, перхлорэтиленом, метиловым и этиловым спиртами, метил-
изобутилкарбинолом, а также с метилизобутилкетоном, моно- и ди-
этаноламином, дибутилфталатом, этиловым и дихлорэтиловым
эфиром, фенолом, касторовым, сосновым и талловым маслами [23,
р- 9].
Растворимость ряда веществ в ди- и трипропиленгликоле соста-
вляет (в %):
Декстрин............
Животный клей (сухой)
Карбамид............
Лаковый бензин . . .
Ланолин водный . . .
Лярд................
Масла
ализариновое . .
кокосовое . . . .
льняное ........
оливковое . . . .
ойтиковое . . . .
парафиновое . .
соевое .........
спермацетовое . .
тунговое . . . .
хлопковое . . . .
Смола
даммаровая . . .
каури ..........
Шеллак..............
12
3
1
1,4
0,7
<1
1
1
1
1
1
<0,5
<0,5
<0,5
В дппропп-
ленгликоле
Равновесие жидкость — пар для системы вода — дипропилен
гликоль показано на рис. 77 [23, р. 28].
194
Азеотропные смеси. Дипропиленгликоль образует с рядом соеди-
нений азеотропные смеси, состав и температура кипения которых
приводятся ниже [29, с. 475 и след , 10, т. 3, с. 438]:
Анетол ...........................
о-Броманизол . . . ...............
П-Бромфенетол ................ .
Дифениловый эфир ...............
Метилсалицилат....................
Метиловый эфир а -терпинеола . . .
Метиловый эфир тимола ............
Нафталин..........................
о-Нитротолуол ....................
п-Нитротолуол ....................
п-Нитрохлорбензол ................
Пирокатехин.......................
Сафрол . » .......................
Хинолин ..........................
Этплсалицилат ....................
Т. кип., Содержание
°C дипропилен-
гликоля, %
221,5 48,0
212 30,0
221,0 45,0
<228,0 <77,0
213,0 35,0
<211,5 >24,0
<211,1 30,0
142,9 12,4
216,9 >21,0
225,0 62,0
<228,3 <11
253,0 ' & 12
222,0 50,0
<228,0 <72,0
218,2 55,0
Рис. 78. Зависимость точки росы газа при его осушке от температуры контакта
с водными растворами дипропиленгликоля.
Методы получения пропиленгликолей
Пропиленгликоль и дипропиленгликоль получаются в основном
теми же методами, как этилен- и диэтиленгликоль. Впервые пропи-
ленгликоль был получен Вюрцем в 1859 г. гидролизом пропилен-
гдикольдиацетата, синтезированного из дибромпропана при
13*
195
взаимодействии с ацетатом серебра. Пропиленгликоль был также по-
лучен в результате гидролиза дибром- или дихлорпропана при их
нагревании в течение 4—5 ч с водой и в присутствии окиси свинца.
Наряду с пропиленгликолем образуются ацетон и пропионовый
альдегид.
Гидролиз дихлорпропана может проводиться также водными
растворами карбоната или бикарбоната натрия при температуре
около 200 °C и давлении от 4,9 до 8,1 МПа (50—83 кгс/см2), причем
при использовании карбоната выход пропилена составляет 48%,
а бикарбоната — 60—65% [39]. Пропиленгликоль можно получить
непосредственным окислением пропилена.
При гидроксилировании пропилена безводной перекисью водо-
рода в растворе mpem-бутилового спирта в присутствии четырех-
окиси осмия как катализатора при температуре 0 °C выход про-
пиленгликоля достигает 68% [40, т. 1, с. 128]:
С113--СИСНз+НгО3 —> СН2ОНСНОНСНз
Однако способ этот весьма опасен и характеризуется большим рас-
ходом перекиси водорода (1,5 моль на 1 моль образовавшегося про-
пиленгликоля).
Пропиленгликоль, его сложные эфиры и окись пропилена обра-
зуются при окислении пропилена в растворе карбоновых кислот
надуксусной кислотой или водным раствором перекиси водорода
при умеренной температуре (около 60 °C) и небольшом давлении
(около 0,17 МПа, или 1,7 кгс/см2). Вместо перекиси водорода можно
использовать ее аддукт с карбамидом [41].
Пропиленгликоль получается при окислении пропилена кисло-
родом (воздухом) в щелочном водном растворе, содержащем:
0,1 моль/л КОН, 0,1—0,2 моль/л K3Fe(CN)6 и 1,6 • 10-4 моль/л OsO4.
Процесс включает следующие стадии [42]:
окисление пропилена восъмивалентным осмием
[ОзО4(ОН)3]»--|-СН2=СНСН3+2Н3О —> СН30НСН0НСН3-|-[ОаО2(ОН)412-
окисление образовавшегося шестивалентного осмия ферроцианидом
калия
[OsO3(OH)4]2-+2Fe(CN)J“+2OH- -> [OsO4(OH)2]2- + 2Fe(CN)*-+2H3O
электрохимическое окислениеi феррицианид а калия в ферроцианид
4Fe(CN)j-+O3 + 2H2O —> 4Fe(CN)®"+401I-
В оптимальных условиях выход пропиленгликоля достигает 99%
от теоретического, но концентрация его в растворе весьма низка —
196
3,6 г/л. Особенным недостатком процесса является высокая токсич-
ность четырехокиси осмия.
Пропиленгликоль предложено получать из пропилена через
пропиленгликольацетат (или пропиленгликольдиацетат) [43]:
СН3СН = СН2-|-0,5О2+СНзСООН >
—> СН3СНОНСН2ОСОСН3 +li-2-—-> СН3СНОНСН2ОН+СН3СООН
Гидролиз пропиленгликольацетата ведется при давлении, близ-
ком к нормальному. Для полноты гидролиза воду дают в избытке.
Гидролизат подвергается ректификации для выделения пропилен-
гликоля и концентрированной уксусной кислоты, которая возвра-
щается в процесс на получение пропиленгликольацетата. Свежая
уксусная кислота требуется только для возмещения ее потерь на
обеих стадиях синтеза.
В зависимости от применяемого катализатора и условий синтеза
наряду с пропиленгликольацетатом образуются значительные коли-
чества дипропиленгликольацетата, при гидролизе которого также
с хорошим выходом получается пропиленгликоль. Селективность
процессов получения пропиленгликольацетата и его гидролиза
весьма высокая и достигает 95% на каждой стадии.
Пропиленгликоль с выходом около 50% на исходный пропилен
получается при окислении пропилена кислородом воздуха в инертном
растворителе, например в бензоле, в присутствии ацетата марганца.
При этом получается и ряд других кислородсодержащих продуктов.
Процесс проводят при 210—230 °C и 5,5—5,9 МПа (56—60 кгс/см2),
одновременно образующиеся водорастворимые продукты экстраги-
руют водой [44].
Пропиленгликоль совместно с метиловым спиртом образуется
при гидрировании пропиленкарбоната [45]:
СНз—НС—СН.2 + ЗН2 —> СН3СНОНСН2ОН + СН3ОН
В оптимальных условиях (давление 29,4 МПа, или 300 кгс/см2,
температура 220 °C и в присутствии 2,5—10% меднохромового
ка: ализатора) выход пропиленгликоля достигает 95%, а метилового
спирта — 100% при полной конверсии пропиленкарбоната. Полу-
ченный гидрогенизат содержит 64—66% пропиленгликоля, 29—31%
метилового спирта и 4—5% пропиловых спиртов, дипропиленгли-
коля, продуктов конденсации. При снижении давления водорода
Уменьшается селективность процесса.
197
Пропиленкарбонат почти с количественным выходом (98—99,5%)
получается из окиси пропилена и двуокиси углерода [46, 47]:
СНз-НС---СНа + СО2 -> СНз-НС—сн2
\ / II
О 0 0
II
о
Пропиленгликоль можно получить гидрогенолизом высших по-
лиолов (глюкозы, ксилита), образующихся при гидролизе расти-
Рис. 79. Технологическая схема разделения продуктов гидрогенолиза сахарозы:
1 — выпарной аппарат; 2 — колонна для доупаривания гидрогенизата; 3 — колонна пери-
одического действия для выделения гликолевой и глицериновой фракций; 4 — колонна для
разделения гликолей; 5, 6, 7. 8, 9, 10 — колонны для очистки продуктов от примесей.
тельного сырья (древесины, кукурузной кочерыжки, хлопковой
шелухи и др.). Наряду с пропиленгликолем образуются другие
многоатомные спирты:
2СН2ОНСНОНСН2ОН
ЗСН2ОНСН2ОН
CgH12O(;--
+знг
С5Н10О5+Н2 —> СН2ОНСНОНСН2ОН+СН2ОНСН8ОН
CeH10O5+2H2 —► СН2ОНСНОНСНОНСН2ОП + СН3ОН
СН2ОНСНОНСН2ОН + Н2 —> СН3СНОНСН2ОН + Н2О
^Соотношение получающихся глицерина, этилен- и пропилен-
гликолей, а также более тяжелых полиолов, зависит от условий
198
процесса [48]. Жесткие условия — повышенная температура и малая
объемная скорость — способствуют повышению выходов этилен-
и пропиленгликолей. Так, при гидрогенолизе ксилита при 19,6 МПа
(200 кгс/см2), 240 °C и объемной скорости 0,91 я-1 получается 33,8%
этиленгликоля, 14,6% пропиленгликоля и 40,4% глицерина. При
том же давлении, но при повышении температуры до 250 °C и сни-
жении объемной скорости до 0,56 ч-1 выход продуктов составляет
35,6% пропиленгликоля, 39,7% этиленгликоля и 9,6% глицерина.
Выход пропиленгликоля может быть повышен до 43—44% при
возврате глицерина на повторный гидрогенолиз вместе с непро-
реагировавшим ксилитом.
В период второй мировой войны гидрогенолиз сахаров осуше-
ствлялся в промышленных условиях. Получаемая смесь много-
атомных спиртов (40% глицерина, 40% пропиленгликоля и 20%
шестпатомного спирта), называемая «глицерогеном», использовалась
как заменитель глицерина [5, р. 206].
Для разделения продуктов гидрогенолиза пентитов и гекситов
предложена следующая схема. Гидрогенизат после отгонки основной
массы воды в вакуум-выпарном аппарате подается на ректифика-
ционную колонну, сверху которой отбирается смесь этилен- и про-
пиленгликолей, а снизу — смесь глицерина и ксилита с небольшой
примесью этиленгликоля. Верхний продукт подвергается ректифи-
кации для выделения чистых этилен- и пропиленгликолей, а из
нижнего продукта в токе водяного пара отгоняется глицерин с эти-
ленгликолем. Полученный водный раствор глицерина концентри-
руется в пленочном вакуум-аппарате, при этом вместе с водой отго-
няется и этиленгликоль [49]. Схема разделения продуктов гидро-
генолиза сахарозы приведена на рис. 79 [50].
Известны и ряд других методов получения пропиленгликоля,
например, восстановление ацетола (оксиацетона) или глицидного
спирта, гидратация аллилового спирта, гидрогенолиз глицеридов
кокосового масла, гидрирование бутиллактата и некоторые другие.
Эти методы не имеют практического значения, и поэтому их описание
не приводится.
Получение пропиленгликоля гидратацией
окиси пропилена
Процессы получения пропиленгликоля и этиленгликоля имеют
очень много сходного, и закономерности, установленные при гидра-
тации окиси этилена, практически полностью распространяются
на процесс гидратации окиси пропилена. Однако в связи с наличием
в молекуле окиси пропилена замещенного атома углерода ее реак-
ционная способность выше, и процесс протекает с большей скоростью.
В разбавленном водном растворе щавелевой кислоты, взятой
в качестве катализатора, при 20 °C и отношении окиси к воде, равном
1 : 40, скорость гидратации окиси пропилена приблизительно в 5 раз
больше скорости гидратации окиси этилена. Константы скорости
199
гидратации в зависимости от концентрации щавелевой кислоты
в реакционном растворе приведены в табл. 66 [51].
Таблица 66. Зависимость константы скорости гидратации
окиси пропилена и окиси этилена от концентрации
щавелевой кислоты
Концентрация СгН2О4-2Н2О PH feC3H«,'10’« МОЛЬ/(л - МИН) feC2H.O-1°’. моль/(л - мин)
ОД 2,22 10 2
0,3 1,82 32 6
0,5 1,64 52 10
0,7 1,54 73 13
Константы скорости удовлетворяют уравнению для реакции
первого порядка
2,303 а
к = —-— 1г----
т ° а —
где т — время реакции, мин; а — начальная концентрация окиси
пропилена, моль/л; а — х — концентрация окиси пропилена в мо-
мент т, моль/л. Константа скорости гидратации окиси пропилена
при 0 °C в присутствии хлорной кислоты также в 5—5,5 раза больше,
чем таковая для окиси этилена [52, 53]. Первый порядок реакции
наблюдается как по концентрации окиси пропилена, так и по кон-
центрации кислоты.
Давление практически не влияет на скорость гидратации окиси
пропилена. При его повышении от 0,1 до 101,3 МПа (от 1
до 1034 кгс/см2) константа скорости гидратации повысилась
лишь в 1,4 раза [53].
Состав продуктов гидратации окиси пропилена, как и при гидра-
тации окиси этилена, определяется отношением воды и окиси про-
пилена в реакционной смеси. В табл. 67 даны выход и состав продук-
тов гидратации окиси пропилена при 20 °C в присутствии щавелевой
кислоты в зависимости от мольного отношения воды и окиси про-
пилена [51].
Таблица 67. Выход и состав продуктов гидратации окиси пропилена
Вода : окись пропилена Степень конверсии окиси пропилена в пропиленгликоли, % Содержание пропиленгли- колей, %
моно- ДИ- три- моно- ДИ- три-
25 90,0 9,0 1,0 91,0 8,0 1,0
20 ' 87,0 12,0 1,0 88,5 10,5 1,0
12 79,0 18,0 3,0 81,0 16,5 2,5
9 73,5 21,0 5,5 76,0 19,0 5,0
5 60,0 26,0 14,0 63,5 24,0 12,5
200
Для получения максимального выхода пропиленгликоля гидра-
тацию окиси пропилена проводят в нейтральной или кислой среде
при достаточно большом соотношении воды к окиси пропилена.
Гидратация окиси пропилена в присутствии щелочей, как и гидра-
тация окиси этилена, приводит к уменьшению выхода пропилен-
гликоля и увеличению выхода полипропиленгликолей [54].
При гидратации окиси пропилена в присутствии солей слабых
кислот и сильных оснований, например карбонатов и бикарбонатов
щелочных металлов, хотя и протекает реакция щелочного гидролиза,
однако решающее значение имеет реакция, катализируемая ионами
карбоната, и особенно бикарбоната, которая позволяет получать
высокий выход пропиленгликоля при сравнительно малом отношении
между водой и окисью пропилена в исходной смеси [55]. Так, при
концентрации бикарбоната натрия 0,25 моль/л и окиси пропилена
3,1 моль/л (мольное отношение воды к окиси пропилена равно 14 : 1)
выход пропиленгликоля составляет 91%; при концентрации окиси
пропилена 4,4 моль/л (мольное отношение реагентов равно 8,7 : 1)
выход пропиленгликоля остается достаточно высоким (88%).
Гидратация окиси пропилена в присутствии небольших коли-
честв серной кислоты описана в работе [56]. Смесь окиси пропилена
и воды в соотношении, равном 1 : 4 (по объему), при температуре
ниже 35 °C подается в реактор колонного типа с насадкой. В верх-
нюю часть реактора поступает подкисленная серной кислотой вода
в таком количестве, чтобы при практически полной конверсии окиси
пропилена образовался 10%-ный раствор пропиленгликоля. При
таких условиях (мольное отношение воды и окиси пропилена при-
близительно равно 40 : 1) получается небольшое количество ди-
пропиленгликоля. Пары окиси пропилена и воды, образующиеся
при повышении температуры раствора за счет теплоты реакции,
конденсируются в выйосном конденсаторе и возвращаются в реактор.
Раствор пропиленгликоля, содержащий 0,1—0,5 H2SO4, после ней-
трализации щелочью подвергается ректификации в вакууме.
В качестве катализатора предложен также сернистый газ [57].
При его концентрации в воде 0,1% (pH раствора 2,2), мольном отно-
шении воды и окиси пропилена, равном 12 : 1, температуре 30 °C
в начале реакции и 75 °C в конце выход продуктов составляет: 91%
пропиленгликоля, 7,9% дипропиленгликоля и 1,1% трипропилен-
гликоля. Полученный водный раствор гликолей нейтрализуется
известью и подвергается ректификации.
Катализаторами процесса гидратации окиси пропилена являются
также Н-катиониты, причем скорость гидратации зависит от при-
роды катионита, его кислотности, структуры, размера зерен и коли-
чества катионита [58].
Сильнокислый катионит КУ-2 является более эффективным
катализатором, чем фосфорнокислый катионит КФ-1; с наименьшей
скоростью гидратация протекает в присутствии слабокислого кати-
онита СГ-1. Основным недостатком Н-катионитов при гидратации
«-окисей является быстрая потеря их каталитической активности.
201
Например, при температуре реакции 35 °C степень конверсии окиси
пропилена составила 98% при однократном использовании кати-
онита КУ-2, 80% при двукратном и 32?4 при трехкратном, а для
Н-катионита КФ-1 соответственно 80, 55 и 43%.
Разработан селективный процесс получения пропиленгликоля
при почти эквимольном соотношении окиси пропилена и воды [47,
59]. Гидратация окиси пропилена проводится в присутствии дву-
окиси углерода и катализатора, состоящего из галогенида щелоч-
ного металла, галогенсодержащей соли аминов и карбонатов, по
следующей реакции:
СН3-НС—СН2 + Н2О + со2
''о7'
80-120 °с
СН3СНОНСН2ОН ь со2
—> сн3-нс—сн2
о о
о
-ь
160-22 0 °C |
Оптимальные условия процесса: давление 2—3,9 МПа (20—
40 кгс/см2), температура 120—180 °C, 0,3—1,5% катализатора,
мольное отношение вода : окись пропилена = 1,1 : 1. При 80—
120 °C реакция получения пропиленкарбоната протекает со значи-
тельно большей скоростью, чем гидратация окиси пропилена, по-
этому пропиленгликоль в основном образуется за счет разложения
пропиленкарбоната при 160—220 °C с выходом 93—95% на израсхо-
дованную окись пропилена.
При непрерывном процессе в первом реакторе, в нижнюю часть
которого подаются окись пропилена, вода, катализатор и двуокись
углерода, температура 80—120 °C и время контакта около 30 мин.
С верха первого реактора реакционная масса поступает в низ вто-
рого, в котором температура равна 160—220 °C и время пребыва-
ния — около 40 мин. Давление в системе 1,5—3,9 МПа (15—
40 кгс/см2), Процесс может проводиться и в одном реакторе с диф-
ференцированным температурным режимом по высоте реактора.
Основным преимуществом процесса является то, что он осуще-
ствляется почти при стехиометрическом соотношении воды и окиси
пропилена, поэтому не требуется многокорпусных выпарных уста-
новок для концентрирования раствора гликолей, существенно сни-
жаются энергетические затраты. В то же время в этом процессе
требуется дополнительное оборудование для компримирования и
циркуляции двуокиси углерода, приготовления катализатора и вы-
деления его из раствора пропиленгликоля. В промышленности этот
процесс пока не осуществлен.
Гидратацию окиси пропилена можно проводить в паровой фазе.
В присутствии катализатора (трехзамещенного фосфата кальция),
атмосферном давлении, температуре 232 °C, времени контакта 9,4 с,
мольном отношении воды и окиси пропилена, равном 19 : 1, выход
202
пропиленгликоля составляет 91—93% на прореагировавшую окись
пропилена и 55—57% на исходную [601.
В качестве катализатора парофазной гидратации окиси пропи-
лена предложены фториды многовалентных металлов [61]. При
давлении, близком к атмосферному, температуре 256—272 °C, моль-
ном отношении воды и окиси пропилена, равном 7 : 1, в присутствии
трехфтористого алюминия выход пропиленгликоля составляет 90%
на исходную окись пропилена. Наблюдаются образование неболь-
шого количества продуктов изомеризации окиси пропилена (спиртов
и карбонильных соединений) и адсорбция катализатором продуктов
реакции.
Парофазная гидратация окиси пропилена осуществляется и над
Н-катионитами при 115—200 °C, давлении 0,25—6,4 МПа (2,5—
65 кгс/см2) и мольном отношении паров воды и окиси пропилена,
равном (5—20) : 1 [62]. Из-за малой конверсии окиси пропилена,
недостаточной производительности и стабильности катализаторов
процессы парофазной гидратации окиси пропилена не нашли про-
мышленного применения.
Получение пропиленгликолей
в промышленных условиях
В промышленных условиях пропиленгликоля получаются терми-
ческой (некаталитической) гидратацией окиси пропилена при тем-
пературе от 160 до 200 °C и при давлении около 1,6 МПа (16 кгс/см2)
[16,5. 343; 63, р. 287]. Мольное отношение воды и окиси пропилена
в смеси, поступающей на гидратацию, составляет около 15 : 1 (17,7%
окиси пропилена). При этих условиях получается в виде товарных
продуктов 85,5% пропиленгликоля, 13% дипропиленгликоля и 1,5%
трипропиленгликоля. Технологическая схема производства про-
пиленгликоля аналогична схеме получения этиленгликоля.
Гидратация осуществляется непрерывно в трубчатом реакторе
с рубашкой для снятия тейлоты реакции. Продолжительность пре-
бывания реакционной смеси в гидрататоре определяется температу-
рой реакции. При 190—200 °C и давлении до 1,5 МПа (15 кгс/см2)
время реакции составляет 30 мин при проведении процесса в непре-
рывных условиях [65]. Вытекающий из реактора раствор гликолей
с концентрацией пропиленгликоля около 20% упаривается при
пониженном давлении. Полученный паровой конденсат, содержащий
небольшое количество гликоля, смешивается со свежей водой и
окисью пропилена для составления реакционной шихты, а концен-
трированный раствор пропиленгликолей поступает на систему раз-
деления. На первой колонне полностью отгоняется вода, а на осталь-
ных последовательно выделяется пропиленгликоль, дипропилен-
тликоль и трипропиленгликоль. Выделение гликолей производится
в вакууме. Цветность пропиленгликоля можно улучшить, пропуская
его при 80—100 °C через слой сильнокислой катионообменной смолы
(леватит 5-115) [66].
203
Применение пропиленгликоля, дипропиленгликоля
и трипропиленгликоля
Пропиленгликоль применяется в тех же областях, где и этилен-
гликоль, так как их свойства весьма близки. Он используется в ка-
честве растворителя, пластификатора, как компонент для изгото-
вления низкозамерзающих, антиобледенительных, гидравлических
и гидротормозных жидкостей, как исходное сырье для получения
новых материалов, применяемых в промышленности пластических
масс, лаков и красок, пестицидов и т. д.
Основное количество пропиленгликоля расходуется на произ-
водство полиэфирных смол; например, в 1970 г. в США расходова-
лось около 42%, в Японии — около 58%. Другие области при-
менения пропиленгликоля в США и Японии следующие (в %) [67,
68, р. 140]:
В США
Целлофан....................10
Фармацевтические препараты,
косметические средства и пи-
щевые продукты............10
Для увлажнения табака ... 8
Пластификаторы........... 7
Гидравлические жидкости . . 4
Экспорт..................19
В Японии
Пищевые продукты . . . . . 9,8
Косметические средства ... 6,7
Для увлажнения табака ... 5,3
Зубные пасты ...............3,1
Фармацевтические препараты . 2,2
Прочие ....................14,3
В отличие от других гликолей пропиленгликоль практически
не токсичен, поэтому он употребляется в пищевой, фармацевтиче-
ской, парфюмерной и других отраслях промышленности, в которых
этиленгликоль применять нельзя [5, р. 243; 23, р. 5]. Водные рас-
творы пропиленгликоля используются как хладоноситель в холо-
дильных установках и как теплоноситель на предприятиях, свя-
занных с производством и хранением пищевых продуктов. В пищевой
промышленности пропиленгликоль применяется для приготовления
приправ, экстракции специй из природных продуктов (ванильных
бобов, кофе, какао), как растворитель душистых веществ, эфирных
масел. В некоторых случаях растворы этих веществ в пропилен-
гликоле могут разбавляться водой без нарушения их однородности.
Благодаря сладковатому вкусу пропиленгликоль может исполь-
зоваться вместо глицерина и сахара. Гигроскопичность пропилен-
гликоля позволяет пищевым продуктам и табаку сохранять необ-
ходимую влажность при длительном хранении. Применению
пропиленгликоля в пищевой промышленности также способствуют
его консервирующие, стерилизующие и бактерицидные свойства.
Аэрозоли водных растворов пропиленгликоля обладают бактери-
цидными свойствами, поэтому пропиленгликоль применяется для
очистки воздуха, особенно на предприятиях пищевой промышлен-
ности.
204
Пропиленгликоль применяется как пластификатор смол, пласти-
ческих масс и пленок, изделия из которых имеют контакт с пище-
выми продуктами, для смазки и консервации упаковочных маптин
в пищевой, фармацевтической и косметической промышленности.
Как хороший растворитель природных и синтетических материалов,
он нашел широкое применение в фармацевтической промышленности
для приготовления различных тинктур, растворов, для инъекций,
мазей и притираний. Для указанных целей немаловажное значение
имеют также бактерицидные и фунгицидные свойства пропилен-
гликоля. В косметике пропиленгликоль используется для при-
готовления эликсиров, лосьонов, шампуней, эмульсий, паст, кремов,
помад и других препаратов.
Пропиленгликоль является исходным материалом для получения
ряда соединений. При дегидрировании пропиленгликоля в паровой
фазе над медьцинкхромовым катализатором образуется ацетол (окси-
ацетон) с выходом 76%. При окислительном дегидрировании про-
пиленгликоля в присутствии паров воды над катализатором (серебро
на "-окиси алюминия, промотированное окисью бериллия) обра-
зуется метилглиоксаль. При окислении пропиленгликоля над пла-
тиновой чернью образуется молочная кислота (69].
Из пропиленгликоля и аммиака под давлением водорода 17,2 МПа
(175 кгс/см2) и температуре около 300 °C в присутствии никеля или
кобальта получается диметилпиперазин с выходом 52% [70]. При
взаимодействии пропиленгликоля с трифенилфосфитом образуются
фосфорсодержащие соединения, которые употребляются в качестве
стабилизаторов полимеров и являются исходным сырьем для полу-
чения негорючих полиуретанов [71].
Большое промышленное значение имеют линейные полиэфиры,
полученные из пропиленгликоля и дикарбоновых кислот, содер-
жащих несколько метиленовых групп или ароматическую группу
в цепи [63, р. 289]. В зависимости от того, берется ли в избытке
гликоль или кислота, полученный полиэфир имеет по концам гидр-
оксильные или карбоксильные группы.
Особое значение имеют полиэфиры ненасыщенных кислот или
смесей насыщенных и ненасыщенных кислот, которые затем сши-
ваются различными винильными соединениями. Ненасыщенные поли-
эфиры широко применяются для различных покрытий и получения
армированных пластических масс, в частности стеклопластиков.
Например, при взаимодействии пропиленгликоля с изофталевой
Или малеиновой кислотой получаются ненасыщенные полиэфиры,
Которые после отверждения сшивающими агентами, состоящими
Из смеси стирола или а-метилстирола с акрилонитрилом или метак-
рилонитрилом, образуют термореактивные полиэфирные смолы с вы-
сокой теплостойкостью и адгезией к металлу и стеклу [72].
Полиэфирные смолы с хорошими механическими свойствами при
Повышенной температуре и на холоду, а также высокой химической
Стойкостью получаются при взаимодействии пропиленгликоля с
Полигалогенидными полифенплами и образовавшегося соединения
205
с органическими кислотами [73]. Линейные 4-карбоксиполиэфирные
смолы образуются при гидролизе полиэфиров, являющихся продук-
тами реакции пропиленгликоля с безводной солью 4-винилгемимел-
литовой кислоты и эпоксидными соединениями [74].
Пропиленгликоль служит одним из исходных веществ для полу-
чения лекарственных препаратов. Так, эфир пропиленгликоля и
сульфаметилфенилкарбаминовой кислоты обладает бактерицидной
активностью [75]. Сложные эфиры пропиленгликоля и салициловой
кислоты или ее производных обладают противолихорадочным, про-
тивовоспалительным и анальгетическим действием [76].
Пропиленгликоль входит в противовоспалительные и бактери-
цидные составы для лечения заболеваний носовой полости, свищей,
сенной лихорадки и др., а также в составы для заживления ран после
глубоких ожогов или действия химических продуктов [77]. Бактери-
цидным и спермицидным действием обладают стероидные трет-
эфиры пропиленгликоля [78]. Пропиленгликоль входит в состав
препаратов успокоительного действия, применяемых в ветерина-
рии [79]. Поверхностно-активные вещества, которые являются
продуктами конденсации пропиленгликоля с окисью этилена, пред-
лагаются для рассасывания отеков [80].
Пропиленгликоль предложено применять в количествах OH-
IO % для стабилизации акарицидных дустов: нестабилизированные
17%-ные дусты разлагались за 25 дней на 67—88%, а с добавлением
2—5% гликоля только на 0,5—6,4% [81]. Он входит в состав битум-
ных противокорневых препаратов, содержащих гербициды и слу-
жащих для дорожных покрытий, изоляции труб и уплотнения труб-
ных соединений [82].
При реакции пропиленгликоля с ароматическими трихлорметиль-
ными соединениями получаются продукты, которые могут быть
использованы в качестве промежуточных веществ, растворителей,
а также как ядохимикаты [83].
Стабилизаторы синтетических смол, ускорители полимеризации
и вулканизации, антиоксиданты получаются при взаимодействии
пропиленгликоля с оловоорганическими дигалогенидами [84].
Пропиленгликоль используется и как реакционная среда в раз-
личных синтезах. Например, Ni-феноляты диоксиалкилдифенил-
сульфонов, используемые как стабилизаторы полиолефинов, полу-
чаются из исходных материалов в среде пропиленгликоля [85]-
Пропиленгликоль рекомендован в качестве азеотропообразователя
для разделения азеотропной смеси н-бутанол — н-бутилацетат [31],
а также для азеотропного разделения нормальных алифатических
спиртов С8 — С18 и углеводородов С12 — Си [5, р. 246].
Дипропиленгликоль может применяться во многих областях,
где используются и другие гликоли. Однако благодаря исключи-
тельно высокой растворяющей способности и большой вязкости его
применение в некоторых случаях является предпочтительным [23,
р. 5]. В США в 1972 г. 50% дипропиленгликоля расходовалось ва
производство полиэфирных смол, 33% — на получение пластифИ'
206
каюров, остальное — на производство алкидных смол, для экстрак-
ции ароматических углеводородов и других целей [86].
Одним из преимуществ дипропиленгликоля как растворителя
является то, что в нем могут растворяться как лиофильные, так
и лиофобные вещества. В связи с этим он нашел широкое применение
для растворения смол, при приготовлении печатных красок, чернил
для авторучек.
Добавки дипропиленгликоля к другим гликолям улучшают
свойства последних как селективных растворителей для экстракции
ароматических углеводородов из их смесей с парафиновыми и наф-
теновыми. Так, показатели процесса экстракции ароматических
углеводородов улучшаются, если применяется смесь диэтиленгли-
коля с дипропиленгликолем. Эта смесь практически не уступает
триэтиленгликолю, который является весьма эффективным селектив-
ным экстрагентом ароматических углеводородов [87]. Для этих же
целей предложена смесь, содержащая 65% дипропиленгликоля
и 35% этиленгликоля [5, р. 276].
Дипропиленгликоль широко используется как компонент водно-
гликолевых негорючих гидравлических жидкостей [35, р. 17; 88,
89], а также гидравлических жидкостей на базе касторового масла
Смесь дипропилен- и диэтиленгликоля применяется как низко-
замерзающая жидкость для гидроприводов и как теплоноситель [89].
Дипропиленгликоль употребляется в качестве антиобледенительной
присадки к топливу: добавка 0,05% дипропиленгликоля к бензину
исключает образование льда в карбюраторах при отрицательной
температуре [90].
Вследствие малой летучести дипропиленгликоль используется
в качестве среды для реакций, которые проводятся при высокой
температуре, например при декарбоксилировании хинолиновой кис-
лоты в никотиновую. Его можно использовать в процессе формо-
вания акриловых волокон из растворов в этиленкарбонате. Дипро-
пиленгликоль используется в различных композициях аэрозолей
с бактерицидными свойствами, лучше всего в смеси с изопропиловым
спиртом [89]. Дипропиленгликоль служит стабилизатором дустов,
резко уменьшая их разложение при хранении [81], а в смеси с мети-
ловым эфиром n-оксибензойной кислоты является эффективным
ингибитором роста микробов [91].
Дипропиленгликоль является исходным сырьем для синтеза
некоторых соединений, в том числе простых и сложных эфиров,
смол, полупродуктов. При пропускании смеси паров дипропилен-
гликоля, воды, водорода и аммиака над катализатором (никель на
кизельгуре) при 160 °C получается смесь 2,5- и 3,5-диметилморфо-
лина с выходом 80% на прореагировавший дипропиленгликоль [92].
При нагревании дипропиленгликоля в присутствии дегидратиру-
ющих катализаторов (например, серной кислоты) образуется смесь
Циклических эфиров и диметилдиоксанов.
Сложные полиэфиры на основе дипропиленгликоля являются
Ченее кристаллизующимися и более гибкими, чем полученные из
207
моногликолей. При полимеризации диэфиров ненасыщенных карбо-
новых кислот и дипропиленгликоля получаются смолы, пригодные
для покрытия бумаги, тканей и металлов. Сложные эфиры дипро-
пиленгликоля такие, как дипропионат и диизобутират, являются
пластификаторами производных целлюлозы, а дибензоат служит
пластификатором виниловых покрытий для полов. Некоторые слож-
ные эфиры, полученные с дипропиленгликолем, обладают инсекти-
цидными свойствами. Дипропиленгликолевый эфир рицинолевой
кислоты используется как компонент гидравлических жидкостей.
Динитрат дипропиленгликоля предложен в качестве ускорителя
воспламенения дизельного топлива [89].
Полиэфирные смолы, обладающие хорошими механическими свой-
ствами при высокой температуре и на холоду, а также повышенной
стойкостью к химическим реагентам, получаются при взаимодействии
дипропиленгликоля с полигалогенидными полифенилами и образо-
вавшегося продукта с насыщенными и ненасыщенными кис-
лотами [73]. Продукты взаимодействия дипропиленгликоля с три-
фенилфосфитом используются для получения негорючих полиурета-
нов, а также в качестве стабилизаторов полимеров [71].
Дипропиленгликоль является исходным сырьем для получения
тетраоксидифосфитов, применяемых для стабилизации полихлор-
виниловых смол [93].
При этерификации гексахлорбициклогептендикарбоновой кис-
лоты дипропиленгликолем и триметилолпропаном образуются вы-
соковязкие полиолы, применяемые для получения огнестойких
поли- и пенополиуретанов [94].
Применение трипроппленгликоля определяется тем, что он
является отличным растворителем, превосходящим остальные гли-
коли по способности растворять различные органические соединения.
Ряд веществ, которые вообще плохо растворяются в низших гликолях,
в трипропиленгликоле растворяются лучше. Например, по сравне-
нию с таким хорошим растворителем, как дипропиленгликоль,
в трипропиленгликоле значительно лучше растворяются масла:
кокосовое — в 3 раза, ализариновое — в 1,3 раза, льняное — в
• 1,8 раза, оливковое — в 2,1 раза, а специальный бензин (раствори-
тель для лакокрасочной промышленности) — в 1,4 раза. В трипро-
пиленгликоле растворяется 16% смолы каури, т. е. столько же,
сколько и в триэтиленгликоле, что в десятки раз больше, чем в
других гликолях (см. стр. 55 и 194).
Именно поэтому трипропиленгликоль применяется в качестве
растворителя в тех случаях, когда другие гликоли не дают достаточно
хороших результатов и когда имеют значение малая летучесть и вы-
\ сокая температура кипения этого гликоля, а также возможность
\ получения с его участием водорастворимых композиций (например,
текстильных мыл, смазок, смазочно-охлаждающих эмульсий, соста-
вов для снятия чернильных пятен) [23, р. 5]. Трипропиленгликоль,
подобно другим гликолям, может применяться для получения жидко-
стей для гидротормозных систем, а также для получения сложных
208
Таблица 68. Технические требования к качеству пропиленгликоля
14
Заказ 972
Кроме перечисленных показателей пропиленгликоль должен отвечать требованиям фармакопеи но содержанию тяжелых
, мышьяка, хлоридов, золы.
Содержание тяжелых металлов (0,0005%) и мышьяка (0,0001%).
Полностью выкипает в пределах 5°С, включая 187,3 °C-
эфиров насыщенных и ненасыщенных органических кислот, которые
применяются как пластификаторы и полупродукты для производства
лаков, пластических масс, инсектицидов и др.
Требования, предъявляемые к качеству
пропиленгликоля и дипропиленгликоля
Промышленностью выпускается несколько марок пропиленгликоля
в зависимости от его назначения. Union Carbide (США) производит
4 марки пропиленгликоля: стандартный, фармацевтический, для
очистки воздуха и специальный; Imperial Chemical Industries (Анг-
лия), Kuhlmann (Франция) и Celanese (США) выпускают 2 марки
пропиленгликоля: стандартный (технический) и фармацевтический.
Все они — бесцветные прозрачные жидкости без осадка. Специфика-
ции на стандартный и фармацевтический пропиленгликоль указанных
фирм, а также требования по техническим условиям СССР
(6-01-929—74) и Японскому стандарту (JISK 1530—1964) приво-
дятся в табл. 68 [26. р. 49; 35, р. 13; 95, с. 339].
Спецификация на дипропиленгликоль приводится в табл. 69 [35,
р. 17; 95, с. 143; 96, с. 41; 97[.
Таблица 69. Технические требования к качеству дипропиленгликоля
Показатели Нормы
ICI Kuhlmann Celene Celanese (США)
Цвет по Pt — Со шкале, не более 15 20 15 15
Плотность при 20 °C, г/см3 . — 1,024—1,028 — —
Относительная плотность А/, 1,022 — 1.028 — 1,020—1,025 1,022—1,026
Кислотность (в пересчете на уксусную кислоту), %, не более 0,005 0,005 0,01 0,005 -
Содержание, % , не более воды 0,3 0,20 0,1 0,2
золы 0,005 — — —
железа —- — 0,00005 —
Пределы кипения прп 0,1 МПа, °C начало кипения, не ниже 222 225 222 222
конец кипения, не выше 238 240 238 236
ЛИТЕРАТУРА
1. Beilsteins Handbuch der organischen Chemie. Aufl. 4. Bd. 1. Teil 4, Ergiinz.
4. Berlin — Heidelberg — New York, Springer Verlag, 1974. 3014 S.
2. Краткая химическая энциклопедия. T. 4. Под ред. И. Л. Кнунянца. М->
«Советская энциклопедия», 1965, 1182с.
3. Пат. США 3491152 (1970); РЖХим, 1971, 6Н47.
210
4. Howard W. L., J. Chem. a. Eng. Data, 1969, v. 14, № 1, p. 129- пат США
3632657 (1972).
5. Glycols. Ed. by G. 0. Gurme, F. Jblmston. New York, Reinhold Publ.
Corp., 1953. 389 p.
6. Осипов О. А., Минкин В. И.. Гарновский А. Д. Справочник по диполь-
ным моментам. Изд. 3-е. М., «Высшая школа». 1971. 416 с.
7. Woolley Е. М., George В. Е., J. Solut. Chem., 1974, v. 3, № 2, р. 119—126.
8. Крешков А. 11. Основы аналитической химии. Т. 3. М., «Химпя», 1970. 472 с.
9 Litovitz Т. A., Higgs В., Meister В., J. Chem. Phys., 1954, v. 22, К» 8,
р. 1281 — 1283.
10. Справочник химика. Изд. 2-е. Т. 1—3. М.—Л., «Химия», 1963—1966.
11. Kirk-Othmer. Encyclopedia of Chemical Technology. Ed. 2nd. V. 10. New
York, Interscience Inc., 1966, 338p.
12. Gallant B. W., Hydrocarb. Process.. 1967, v. 46. № 5, p. 201—215.
13. Gardner P. I., Hussain K. S., J. Chem. Thermodyn., 1972, v. 4, № 6,
p. 819—827.
14. Сталл Д., Becmpa.it Э., Зинке Г. Химическая термодинамика органических
соединений. Пер. с англ. М., «Мир», 1971. 807 е.
15. Сталл Д. Р. Таблицы давления паров индивидуальных веществ. Пер.
с англ. Под ред. С. В. Горбачева и В. В. Михайлова. М., Издатинлит, 1949.
72 с.
16. Ullmans Enzyklopiidie der technischen Chemie. Bd. 14. Aufl. 3. Munchen —
Berlin. Urban u. Schwarzenberg, 1963. 810 S.
17. Jordan T. E. Vapour Pressure of Organic Compounds. New York — London,
Interscience Publ. Inc., 1954. 266 p.
18. Malian G. M., Michaelian M. S., Lockhart F.. J. Chem. Eng. a. Data, 1972,
№ 4, p. 412—415.
19. Nageshwar G. D., Mene P. S.. Indian Chem. Eng. Trans. Indian Inst. Chem.
Eng., 1968, v. 10, № 3, p. 118—120.
20. Kato J. «Якугаку дзассп» (J. Pharm. Soc. Japan). 1958, v. 78, № .6, p. 565—
567; РЖХпм, 1959, 37855; Bao V. M., Trans. Faraday Soc., 1962, v. 58,
№ 11, p. 2139—2143.
21. Nogami H., Kato J., «Якугаку дзассп» (J. Pharm. Soc. Japan), 1961, v. 81,
№ 3, p. 389—393; РЖХпм, 1962, 4Б123.
22. Prey V., Kretschek E., Berbalk H., Allg. u. prakt. Chem., 1971, Bd. 22, № 6,
S. 153-156.
23. Glycols. Properties and Uses. Midland (Michigan), The Dow Chem. Co.,
1961. 64 p.
24. Белоусов В. П., Морачевский А. Г. Теплоты смешения жидкостей. Л., «Хи-
мия», 1970. 256 с.
25. Kasly А. К., Nageshwar G. D., Mene Р. S., Indian J. Technol., 1974, v. 12,
№ 2, р. 77—78.
26. Mellan I. Polyhydric Alcohols. Washington, Spartan Books, 1962. 208 p.
27. Mellan I. Compatibility and Solubility. London, Noyes Develop. Corp.,
1968. 304 p.
28. Справочник по растворимости. Под ред. В. В. Кафарова. Т. 2. М.—Л.,
изд. АН СССР, 1963. 2068 с.
29. Огородников С. К., Лестева Т. М., Коган В. Б. Азеотропные смеси. Л.,
«Химия», 1971. 848 с.
30. Соколов Н. М., Цыганкова А. Н., Жаворонков Н. М.. Теорет. основы хим.
технол., 1971, т. 5, № 6, с. 900—904; Соколов Н. М. и др., Хим. пром.,
1972, № 7, с. 499—501; 1973, № 5, с. 13—16.
31. Nageshwar G. D., Mene Р. S., J. A ppi. Chem., 1969, v. 19, № 7, р. 195—200;
Nageshwar G. D., Mene P. S., Indian Chem. Eng., 1970, v. 11, № 3,
p. 106—107.
32. Francis A. W., J. Chem. Eng. Data, 1965, v. 10, № 1, p. 45—49; № 3,
n p. 260—264.
33. Degaleesan T. E., Laddha G. S„ J. Appl. Chem., 1962, v. 12, №, 3, p. Ill—
113; Degaleesan T. E., Laddha G. J. Madras Univ., 1960, v. 30, № 2—3,
p. 133—147.
11*
211
34. Чудное А. Ф. Кандидатская диссертация. Томск, Томский политехнический
институт им. С. М. Кирова, 1970.
35. Ethylene and Propylene Glycols. Heavy Organic Chemicals Division. Bir-
mingham, Imperial Chemical Industries Lim. 33 p.
36. Monick I. A. Alcohols. New York — London, Reinhold Book Corp., 1968
594 p.
37. Sexton A. R., Britton E. C., J. Am. Chem. Soc., 1953, v. 75, № 77, p. 4357—
4358.
38. Vanderkool W.N., Hildenbrand D. L., Stull D. R., J. Chem. Eng. Data, 1967,
v. 12, № 3, p. 377—379.
39. Wurzt A., Ann. chim. Phys., Ser. 3, 1859, v. 55, p. 400—478; Элътеков А. П.,
ЖРХО, 1878, t. 10, вып. 5, c. 211 — 222; Клебанский А. Л., Долгополъ-
ский И. M., ЖПХ, 1934, т. 7, № 7, с. 1181—1191.
40. Успехи органической химии. Пер. с англ. Под ред. И. Л. Кнунянца. М.,
Издатпнлит, 1963. 398 с.
41. Англ. пат. 1217000 (1970); РЖХим, 1971, 14Н55.
42. Mayell J. S., I and ЕС, Prod. Research and Develop., 1968, v. ", № 2,
p. 129—136.
43. Mitsutani A., Chem. Economy and Eng. Rev., 1973, v. 5, № 3, p. 32—35.
44. Пат. США 2769846 (1959); РЖХпм, 1959, 87486.
45. Шапиро А. Л., Гуревич Г. С., Левин С. 3. Авт. свпд. № 269925 (1970);
Открытия. Изобр. Пром, образцы. Товарн. знаки, 1970, № 16, с. 21; Грин-
берг Д. И., Шапиро А. Л., Левин С. 3., ЖПХ, 1970, т. 43, № И, с. 2584-
2586; 1971, т. 44, с. 380—385; Гринберг Д. Н. Кандидатская диссертация.
Л., ВНИИнефтехпм, 1971.
46. Левин С. 3. и др. Авт. свид. № 165694 (1962); Билл, изобр. и товарных
знаков, 1964, № 20, с. И; Левин С. 3., Шапиро А. Л., «Нефтехимия», 1965,
т. 5, № 3, с. 388—393.
47. Шапиро А. Л. и др., «Химия и технология топлив и масел», 1971, № 5,
с. 14-18.
48. Васюнина Н. А. и др., Хим. пром., 1962, № 2, с. 82—86; Васюнина Н. А.,
Баландина А. А., Маматов Ю. Л., «Кинетика и катализ», 1963, т. 4, № 1,
с. 156—162.
49. Балахонцева В. И., Гельперин Н. И., Хим. пром., 1962, № 2, с. 86—88.
50. Соколов Н. М., Штром М. И., Жаворонков Н. М., Хим. пром., 1975, № 7,
с. 495—497.
51. Ferrero М. Р., Flamme L. R., Fourez А. М., Ind. chim. Belg., 1954, v. 9,
№ 2, p. 113-119.
52. Pritchard I. G., Long A. F., J. Am. Chem. Soc., 1956, v. 78, № 12, p. 2667—
2670.
53. Koskikallio I., Wholley E., Trans. Faraday Soc., 1959, v. 55, № 5,
p. 815-823.
54. Maget H. I. R., J. Polymer Sci., part A, 1964, v. 2, № 3, p. 1281—1290.
55. Швец В. Ф. Докторская диссертация. М., МХТИ им. Д. И. Менделеева,
1974; Лебедев П. Н. и др. В кн.: Всесоюзное совещание по проблеме меха-
низмы гетеролитическпх реакций. Л., 1974, с. 114—115; Лебедев Н. Н. и др.
Авт. свид. № 418464 (1974); Открытия. Изобр. Пром, образцы. Товарн.
знаки. 1974, № 9, с. 79—80.
56. Пат. США 2255411 (1941).
57. Пат. ФРГ 1043308 (1959).
58. Полянский Н. Г., Потудина Н. Л., «Нефтехимия», 1963, т. 3, № 5, с. 706—
712; Полянский И. Г. Катализ ионитами. М., «Химия», 1973, 214 с.
59. Англ. пат. 1177877 (1970); пат. США 3629343 (1971).
60. Фрейдлин Л. X., Шарф В. 3., Авт. свид. № 114336 (1958).
61. Пат. США 2547766 (1951).
62. Пат. США 3091647 (1963).
63. Propylene and its Industrial Derivatives. Ed. by E. G. Hancock. London,
Ernest Benn Lim., 1973. 517 p. ?
64. Scherwood W., Ind. Chemist, 1960, v. 36, № 429, p. 542—546; Ftvie A. C.,
Chem. a. Ind., 1964, № 10, p. 384—388.
212
g5. Myszkowski J., Zielincki A. Z., Scislewska U., Przem. Chem 1966 № 11
p. 619—620. ’ ’ ’
66. Пат. ФРГ 1668052 (1975); РЖХим, 1975, 21H23.
67. «Oil. Paint and Drug Reporter'), 1971, v. 200, № 10, p. 9; Производство
олефинов (этилена, пропилена п их производных) в основных капиталисти-
ческих странах. М., ЦНИИТЭнефтехим, 1970. 82 с.
68. Waddams A. L. Chemicals from Petroleum. Ed. 3rd. London, John Mur-
ray, 1973. 326 p.
69. MacheH G., Ind. Chemist, 1959, v. 35, № 412, p. 283—290; пат. ЧССР 119683
(1966); РЖХпм, 1968, 11H68; пат. США 3360567 (1967); РЖХим, 1969,
9Н43.
70. Пат. США 3068232 (1962); РЖХпм, 1970, 14Н182.
71. Пат. США 346837 (1969); РЖХпм, 1970, 18Н122.
72. Англ. пат. 894139 (1962); РЖХим, 1964, 7С279.
73. Франц, пат. 92074 (1968); РЖХим, 1970, 4Н192.
74. Пат. США 3264267 (1966); РЖХим, 1967, 24Н72.
75. Пат. США 2827470 (1958); РЖХим, 1959, 83406.
76. Яп. пат. 3291 (1968); РЖХпм, 1969, 10НЗЗЗ.
77. Пат. США 2989437, 2995493 (1961); РЖХим, 1962, 14Л364, 15Л25.
78. Пат. США 3318918 (1967); РЖХим, 1968, 23Н153.
79. Пат. США 3196073 (1965); РЖХпм, 1967, 9Н414.
80. Франц, пат. 1116М (1962); РЖХим, 1964, 4Н257.
81. Пат. США 2832716 (1958); РЖХим, 1959, 75961.
82. Пат. США 3231398 (1966); РЖХпм, 1967, 16Н750.
83. Пат. США 3050549 (1962); РЖХим, 1964, 8Н154.
84. Пат. США 278994 (1957); РЖХим, 1959, 39597.
85. Яп. пат. 21820 (1967); РЖХим, 15Н151.’
86. «Chemical Marketing Reporter», 1973, v. 203, № 9, p. 9.
87. Франц, пат. 1509242 (1967); РЖХим, 1969, 6Н153; Hoover Т. S., Hydro-
carb. Process., 1969, v. 48, № 12, p. 131; Jones W. Payne Vic., Ibid., 1973,
v. 52, № 3, p. 91—92.
88. Пат. ФРГ 1220069 (1967).
89. Dipropylene Glycol. Imperial Chemical Industries. Heavy Organic Chemicals
Division. H/P92E/54/2C.
90. Dipropylene Glycol as a Petrol anti-icing Additive. Imperial Chemical Indu-
stries. Heavy Organic Chemicals Division. H/C29ER/870/GR.
91. Poprzan Johanna, de Navara Maisong, J. Soc. Cosmet. Chem., 1961, v. 12,
№ 5, p. 280—284.
92. Longdon W. К. e. a., I and EC, Prod. Research and Develop., 1965, v. 5, № 3,
p. 268—272.
93. Пат. США 3457331 (1969); РЖХим, 1970, 16H152.
94. Пат. США 3531415 (1970); РЖХим, 1971, 13Н128.
95. Чернышев А. К. и др. Справочный материал по качеству химической про-
дукции. М., ГИАП, 1972. 492 с.
96. Химические продукты и пластмассы (Celene). Милан, Челене. 66 с.
97. Monopropylene Glycol. Service Publicite Kuhlmann. PR 05—1; Dipro-
pylene Glycol. Service Publicite Kuhlmann. PR 06—1.
Глава VI
Полимеры окиси этилена и окиси пропилена
Основные методы полимеризации а-окисей
(Х-Окиси обладают высокой реакционной способностью в процессах
различного характера, однако в силу значительной поляризации
углерод-кислородной связи они более склонны к реакциям по ион-
ному механизму. В связи с этим, а также из-за наличия большого
числа осложняющих процессов радикальная полимеризация окисей
не приводит к практически важным результатам [4]. Ионная поли-
меризация может развиваться по анионному и катионному механизму,
возможны также промежуточные варианты.
Катионная полимеризация
Химия полимерных продуктов на основе а-окисей представляет
сейчас обширную область и имеет уже более чем столетнюю историю.
Современный этап развития химии полиэпоксидов открывается фун-
даментальными работами Штаудингера в 20—30-х годах, и с той
поры интерес к этим продуктам непрерывно возрастает; широко
осваивается технология получения мономеров, а также ряда про-
дуктов на их основе, в частности гликолей, алканоламинов и т. п.
Это направление развития химии полиэпоксидов привело в насто-
ящее время к широкому спектру продуктов, нашедших важное
применение во многих отраслях техники.
Еще один качественный скачок в этой области произошел в 50-ые
годы, когда были получены высокомолекулярный стереорегулярный
полипропиленоксид (Пруит, Баггет, 1955 г.) и ряд исключительно
высокомолекулярных полимеров окиси этилена (Бэйли и др., 1958 г.),
известных теперь под маркой «полиокс». Эти продукты обладают
рядом принципиально новых свойств, и их можно рассматривать
как отдельный класс полимеров. В последнее время полимеры этого
типа вызывают особый интерес.
Можно выделить еще одно направление в развитии полимерной
химии а-окисей, в котором полиалкилепоксиды играют роль реак-
ционноспособных промежуточных продуктов. Значительное место
здесь занимает синтез полиуретанов на основе олигомерных простых
полиэфиров, и по сей день являющийся сильнейшим стимулятором
всей этой области [1]. Развивается и другой метод создания поли-
алкиленоксидных каучуков — введение периферических двойных
связей с последующей радикальной вулканизацией [2]. Ряд фирм
уделяют этому большое внимание. И, наконец, разрабатываются
способы прямой вулканизации и разнообразная химическая моди-
фикация гомополимеров [3], способная обеспечить возможность их
непосредственного практического использования, основным пре-
пятствием которому являются низкие температуры плавления.
Таков в основных чертах спектр полиэпоксидов. В данной главе
будут рассмотрены методы синтеза, свойства и применение некоторых
наиболее типичных и важных из этих продуктов.
214
Полимеризация, которая по тем или иным признакам относится
к этому типу процессов, инициируется протонными кислотами (НС1,
НС1О4), кислотами Льюиса (BF3, SnCl4, PF5 и др.), металлооргани-
ческими соединениями [(C2IIB)2Zn, (С2Н6)3А1] либо более сложными
композициями. Здесь часто используют сокатализатор, функцию
которого может выполнять и сам мономер.
Активный центр катионной полимеризации может, в принципе,
представлять собой карбониевый (I) или оксониевый (II) ионы, часто
находящиеся в равновесии, которое регулирует их относительные
концентрации в системе:
I <\^СП2 + О
сн.,-о
1 п
В случае напряженных циклов, таких, как эпоксиды, стабиль-
ность оксониевой формы должна быть весьма низкой, и она вряд ли
играет здесь существенную роль. Компромиссным решением этого
вопроса является принятие для активных центров карбоксониевой
структуры. Во всяком случае сополимеризация а-окисей со сти-
ролом — типичным «карбониевым» мономером — указывает на карбо-
ниевый характер активных центров [5].
Неоднократно выдвигавшийся в литературе цвиттерионный меха-
низм полимеризации под действием кислот Льюиса
BF3-f- \ > F3BOCH2CH2
о
F3B-[-OCH2CH2-]„-OCII2CII2
ш
До настоящего времени не получил сколько-нибудь серьезных экспе-
риментальных подтверждений, хотя возможно в стадии иницииро-
вания цвиттерионные формы III играют существенную роль.
Противоионом в катионных процессах является, как правило,
Комплексный анион (SbF3, С104, BFj, ROBF3 и т. п.), стабиль-
ность которого гарантирует отсутствие реакций обрыва:
сн2 bf4-> ^CH2F + BF3
215
В связи с этим большой интерес представляют исследования
полимеризации под действием солей стабильных карбониевых (три-
фенилметил, тропиллий, пириллий и т. п.) и оксониевых ионов,
где возможно более детальное изучение механизма роста цепи [6].
Особенностью катионной полимеризации кислородсодержащих
циклов, в частности а-окисей, является их равновесный характер,
обусловленный образованием фрагментов полимерной цепи в виде
циклов:
'xzxzCHgO—[—СН2СН2О—]„—СН2СН2 4^
—[—СН2СН2О—
1----си,сн2-----
Размер циклов и их равновесные концентрации определяются
скорее всего термодинамическими факторами. В случае окиси эти-
лена, например, преобладают наиболее устойчивые 1,4-диоксан
и 2-метил-1,3-диоксолан, тогда как при полимеризации окиси про-
пилена, окиси бутена-1 и эпихлоргидрина под действием BF3 или
(C2H5)3OBF4 образуются тетрамерные 12-членные циклы с выходом
30—40% [7, 8].
Циклизация в катионной полимеризации является наиболее
серьезным и практически непреодолимым препятствием для синтеза
высокомолекулярных полимеров, хотя использовать чрезвычайно
высокую скорость катионных процессов заманчиво. Применение
более сложных катионных инициаторов позволяет несколько снизить
скорость реакций, ограничивающих рост цепи, и других побочных
процессов и поднять молекулярные массы до сотен тысяч.
Близки по свойствам к катионным и системы на основе металло-
органических соединений (C2H5)2Zn, (СН3)6А12, (С2Н5)3А1 и т. и.
По ряду признаков — образованию макроциклов, аномальному на-
правлению раскрытия цикла, частичной рацемизации оптически
активных асимметрических эпоксидов, а также способности к поли-
меризации больших циклов (тетрагидрофуран, 3,3'-бисхлорметил-
оксациклобутан и др.) эти системы могут характеризоваться как
катионные и, более того, непосредственно проявляют кислотные
свойства.
Катионные центры не являются здесь, однако, единственными-
Наряду с ними в реакционной смеси образуется небюлыпая доля
центров (структура которых еще недостаточно ясна), генерирующих
высокомолекулярный полимер. Относительные концентрации актив-
ных центров разного типа зависят от природы и количества соката-
лизатора, способа формирования каталитической системы, темпера-
туры и ряда других, часто весьма тонких факторов. В связи с этим
свойства образующегося полимерного продукта сильно зависят
от многих обстоятельств. Полимеры содержат обычно более или менее
выраженные низко- и высокомолекулярные фракции, часто с при-
месью макроциклов, и, следовательно, чрезвычайно полидисперсны.
Несмотря на указанные осложнения, катионная полимеризация
является наиболее универсальным методом синтеза полимеров орга-
нических окисей. Реализация присущих ей высоких скоростей даже
на стерически затрудненных и мало реакционноспособных эпоксидах
весьма привлекательна и продолжает быть предметом поисков. Еще
перспективнее катионная сополимеризация эпоксидов с мономерами
других типов.
Анионная полимеризация
Более регулируемым и относительно свободным от осложнений
методом полимеризации эпоксидов является анионная полимериза-
ция. Этот классический тип процессов широко распространен в химии
и технологии полиэпоксидов и используется в синтезе полигликолей,
полиоксиэтилированных спиртов, фенолов, меркаптанов, аминов
и т. и.
Полимеризацию по анионному механизму можно осуществить
практически без обрыва цепи, и в этом случае среднечисленная
молекулярная масса полимера Мп определяется соотношением
М„ = Мо----i-- (1)
Ср
где Мо — молекулярная масса мономера; и См — начальная
и текущая концентрации мономера, Ср — полная концентрация
растущих цепей, которая тождественна концентрации инициатора
или суммы инициаторов.
Если гарантирована соответствующая чистота реакционной
среды, то при низких концентрациях инициатора могут быть полу-
чены весьма длинные цепи. Например, при С* = 10“ 4 моль/л и С", =
= 10 моль/л молекулярная масса при полном превращении мономера
достигнет почти 5 миллионов. Такие величины действительно достиг-
нуты при использовании щелочных солей нафталина в качестве
инициаторов в исключительно безводных условиях [9].
Основной реакцией ограничения роста цепи при анионной поли-
меризации а-окисей является перенос протона
<-vzvCH2CH2O- + XII > 'х/’оСН2СН2ОН + Х-
причем образующийся Х“ чаще всего начинает новую цепь, т. е.
эта реакция представляет собой передачу цепи. Она может осуще-
ствляться передачей на мономер, как в случае окиси пропи-
лена [10]
'4z\,cri2CH(CH3)O- + CH3—НС—СН2 —>
---► п^СП2СН(СН3)ОН+ сн2=сн—сн2о-
217
216
на растворитель [И]
^CH2CII,O- + CH3SOCH3 —> ^CH2CH2OH+CH3SOCH2
либо на любой другой реагент с достаточно подвижным водородом,
в частности спирт:
'\zx,CH2CII2O-+BOJI -xz^CH2CH2OII + RO-
Очевидно, передача цепи увеличивает число растущих цепей
и понижает молекулярную массу полимера. При достаточно быстром
Рис. 80. Соотношение между молекулярными массами, вычисленной (Мвычисл)
и наблюдаемой (МНабл), пРи анионной полимеризации окиси этилена в тетра-
гидрофуране (а) и в диоксане с добавками метилового спирта (б).
обмене, например в случае спиртов, С* = [RO~ ] + [ROH], т. е.
все исходные алкокси-анионы могут с равной вероятностью уча-
ствовать в росте цепи. Это обстоятельство широко используется для
регулирования длины цепи при анионной полимеризации. На рис. 80
показана зависимость молекулярной массы полиоксиэтилена, наблю-
даемой прп анионной полимеризации, от рассчитанной по уравне-
нию (1) [9, 10].
В качестве инициаторов анионной полимеризации а-окисей чаще
всего используют производные щелочных и щелочноземельных
металлов — гидроокиси, алкоголяты, амиды, аммиакаты и т. п.,
а также амины. Инициатор в начальной стадии процесса образует
активные центры роста цепи, которые практически всегда предста-
вляют собой алкокси-анионы:
R- Ме+ + Н2С—СН2-----> RCH2CII2O- Ме+
О
Инициатор должен обладать достаточно высокой нуклеофиль-
ностью, иначе стадия инициирования становится продолжительной,
218
что приводит к осложнениям, в частности к увеличению полидис-
персности.
В случае аминов инициирование также приводит к алкокси-
аниону, хотя здесь нет единого мнения о механизме и обсуждаются
два варианта:
R
+ I
R3NCH2CHO-
R'O- R3NCH2CHOH
R
Второй вариант экспериментально более обоснован [12], причем
предполагается, что спирт может образовываться в результате ката-
литической изомеризации эпоксида. Неустойчивость противоиона
в обоих вариантах является, видимо, препятствием для образования
высокомолекулярных полимеров.
Образующийся в стадии инициирования алкокси-анион ведет
в дальнейшем полимеризацию. Его активность зависит от таких
факторов, как характер и энергия связи с противоионом, природа
противоиона, состав среды и, конечно, температура. Существование
свободных анионов и значительная степень диссоциации ионной
пары
-ч^СН2СН2О- Ме+ ДЛГ -v^CH,CII,O- |-Ме+
являются в этих системах большой редкостью [13]. Свободные ани-
оны образуются в значительных количествах лишь в высокополяр-
ных средах (спиртах, диметилсульфоксиде) и в какой-то степени
в гексаметилфосфортриамиде [14].
Чаще активным центром полимеризации эпоксидов является
ионная пара, активность которой зависит от природы катиона.
В тетрагидрофуране, например, активность ионных пар растет от
натрия к цезию [9]. Серьезную и сложную роль играет в полимери-
зации окисей сольватация активных центров, в которой может уча-
ствовать и образующийся полимер. Спирты, вводимые в систему,
с одной стороны, понижают нуклеофильность алкоголятных ионных
пар за счет их сольватации, по, с другой, при более высоких кон-
центрациях, могут благоприятствовать росту цепи по так называ-
емому пуш-пульному * механизму, перераспределяя электронную
плотность в молекуле эпоксида в результате образования водородной
«вязи:
_8+
R° в 5+
[ J^O<HOR
СН2
* От английского слова push-pull.
219
Весьма существенна в анионных системах межмолекулярная
ассоциация активных центров, обусловленная их ярко выраженным
ионным характером и приводящая к понижению истинной концен-
трации активных центров [13]. Склонность к такой ассоциации
растет в ряду щелочных катионов в сторону Li, чем обусловлена
низкая активность катализаторов на основе этого металла. Истинная
производительность активных центров известна лишь для немногих
анионных систем; константы скорости роста цепи при анионной
полимеризации окиси этилена даны в табл. 70 [9—11, 14].
Таблица 70. Константы скорости роста цепи (Лр)
при анионной полимеризации окиси этилена
Противо- ион Растворитель Тип активного центра йр> л/(моль-с)
При температуре 70 °C
к+ Тетрагидрофуран Ионная пара 0,94
Cs+ Тетрагидрофуран То же 3,5
К+ 1,2-Диметоксиэтан » 0,71
К+ Диметиловый эфир диэтиленглпколя » 0,80
Na+ Диметплсульфокспд » 0,041
Na+ Диметилсульфоксид Анион 0,084
При температуре 40 °C
К+ Гексаметил фосфортриамид Ионная пара 2
К+ Г ексаметилфосфортриамид Анион 33
Na+ Диоксан * Ионная пара 7,6—10-5
С добавкой «1 моль/л метилового спирта.
Эти величины представляют принципиальный интерес, поскольку
именно ими определяется скорость роста индивидуальной полимер-
ной цепи
М п = к^М
что особенно важно при синтезе высокомолекулярных полимеров.
Создание высокой концентрации центров роста цепи и наиболее
полная реализация их активности являются непременными усло-
виями производительного синтеза полиэпоксидов.
Полимеризация, координационного типа
Во многих случаях разрыву эпоксидного цикла предшествует стадия
комплексообразования мономера с активным центром, фрагментом
катализатора или противоионом. Такие процессы относят к коорди-
национному типу, который охватывает практически все разнообразие
каталитических систем, не сводимых к более простым механизмам.
Достаточно полные обзоры катализаторов этого типа имеются в лите-
ратуре [15, 16].
Катионная полимеризация эпоксидов протекает через стадию
координирования мономера
^СН2 + О
-4zx>CH2:O
-4z\>CH2OCH2CH2
что равносильно оксониевому механизму.
При анионной полимеризации координация мономера, вероятно,
имеет место уже в системах с щелочными противоионами [17]:
—> ^z^CH2OCH2CH2O- Me4-
И в том, и другом случае координация не создает какой-либо
преимущественной ориентации и лишь дополнительно активирует
мономер.
Рис. 81. Соотношение
между выходом полимера
и характеристической
вязкостью [р] при поли-
меризации окиси пропи-
лена под действием
(C2H5)2Zn в присутствии
сокатализаторов:
1 — фенол: 2 — этиловый
спирт; 3 — метиловый спирт;
4 — этиленгликоль; 5 —
1,3-пропиленгликоль; 6 —
глицерин; 7 — вода; 8 — мо-
нопальмитат сахарозы; 9 —
триэтаноламин.
В сложных металлоорганических системах, где активный центр
является, как правило, многоядерным, такая координация приводит
к серьезным стереохимическим последствиям. Наиболее ярко это
проявляется при полимеризации асимметрических эпоксидов, при-
водящей к образованию стереорегулярных полимеров. Каталитиче-
ские центры способны в этом случае выбирать только один изомер
(В или L) из рацемической смеси, образуя регулярную или частично
регулярную цепь [18]. Более выгодные условия координации одного
из оптических изомеров являются, скорее всего, истинной причиной
стереоспецифичности.
С другой стороны, металлоорганические катализаторы координа-
ционного типа часто приводят к более высокомолекулярным
221
220
полимерам, чем чисто ионные. Например, модифицированные цинк-
органпческие катализаторы позволяют получить полипропиленоксид
с молекулярной массой до 4—5 млн., в то время как «потолком»
анионной полимеризации в связи с наличием передачи цепи на моно-
мер являются молекулярные массы 4—6 тыс.
Можно допустить, что создание благоприятной стереохимической
ситуации и увеличение скорости роста цепи лежит в основе отме-
ченного эффекта. В пользу этого говорит, в частности, параллельное
изменение активности и молекулярной массы, наблюдаемое при
введении сокаталпзаторов. В качестве примера на рис. 81 приведены
данные по полимеризации окиси пропилена в присутствии (C2H5)2Zn
с различными сокатализаторамп [19]. При полимеризации окиси
этилена высокую активность металлоорганических центров в реак-
ции роста используют в синтезе высокомолекулярных полимеров.
Реакционная способность эпоксидных мономеров
Вопрос об относительной реакционной способности эпоксидов пред-
ставляет интерес во многих отношениях, однако его постановка
имеет смысл лишь применительно к определенному механизму реак-
ции. В катионной полимеризации внедрение очередного мономера
в полимерную цепь часто проходит через равновесную стадию ком-
плексообразования с активным центром, в которой значительную
роль играет донорность кислорода. В связи с зтим основность моно-
меров определяет в этих системах скорость процесса, состав продук-
тов сополимеризации и т. и. [20, 21]. На рис. 82, а показана кор-
реляция констант сополимеризации различных эпоксидов и 3,3'-бис-
хлорметилоксетана с их основностью [21].
РКв
Рис. 82. Корреляция реакционной способности эпоксидов в катионной полиме'
рпзации с их основностью рКв (а) и с вычисленной по эмпирическим уравнениям
с учетом напряженности цикла (б).
Другой крайний вариант механизма имеет место в анионной
полимеризации, где элементарный акт роста цепи представляет собой
нуклеофильное присоединение. Здесь решающими оказываются сте-
222
рцческпй эффект заместителя и его полярное влияние, приводящее
К вариации электронной плотности у замещаемого углерода.
Напряженность эпоксидов является еще одним важным фактором
их реакционной способности и играет серьезную роль, например,
в радикальных превращениях эпоксидов.
Предпринято несколько попыток качественно охарактеризовать
влияние различных факторов на активность эпоксидов при полимери-
зации по разным -механизмам. Для катионной сополимеризации
различных циклических эфиров и, в частности, эпоксидов получены
корреляционные уравнения, учитывающие влияние напряженности
циклов и их основности на реакционную способность мономеров,
Рис. 83. Анализ относи-
тельных реакционных
способностей эпоксидов
при сополимеризации
в рамках уравнения
Тафта:
1 — анионная полимериза-
дня, катализатор C6H6oNa
(р = 4-2,07, д = 0,154);
2 — катионная полимериза-
ция, катализатор (С2Н5)2О’
•ВГ3 (р = —1,07, 6 = 0);
3 — координационно-анион-
ная полимеризация, ката-
лизатор (С2Н5)зА1—Н2О
(р —1,82, 6 = 0,267).
за меру которой приняты значения констант сополимеризации =
= где k11 и к12—.константы скорости присоединения моно-
меров 1 и 2 к центру типа 1, принятому за стандарт; на рис. 82, б
показана соответствующая зависимость, причем относительные актив-
ности вычислены по уравнениям, в которых учет напряженности
и основности выполнен в рамках того или иного эмпирического
подхода [21].
Использование корреляционных уравнений Гаммета — Тафта
применительно к полимеризации эпоксидов приводит в случае ка-
тионных систем к значениям р << 0, характерным для электрофиль-
ных реакций, икр >0 — в случае анионных; па рис. 83 даны при-
меры таких соотношений [22]. Все остальное многообразие процес-
сов, где перекрывается действие многих факторов, занимает здесь
промежуточное положение. Однако, как видно из рисунка, при
сополимеризации эпоксидов под действием типичной координа-
Ционно-аииопной каталитической системы (С2Н5)3А1 — Н2О (1 : 1)
электрофильное влияние выражено даже более резко, чем в катион-
ной полимеризации, — значение более отрицательно. Это указывает
На более сильную координацию эпоксидов с атомом металла, чем
с ионом карбония при оксониевом механизме.
Корреляционный подход является, таким образом, одним из
возможных путей качественной идентификации механизма поли-
меризации.
223
Олигомерные продукты на основе а-окисей
Скачок в производстве эпоксидных мономеров в 40—50-ые годы
привел к значительному развитию смежных областей, особенно
неионогенных поверхностно-активных веществ и полимерных про-
дуктов на основе а-окисей. Неионогенные поверхностно-активные
вещества занимают второе место после этиленгликоля в общем ба-
лансе производных окиси этилена.
Ассортимент поверхностно-активных веществ неионогенного типа
включает в себя продукты каталитического полиоксиэтилирования
различных веществ, содержащих подвижный водород: алкилфенолов,
высших спиртов, алкиламинов, меркаптанов, алкиламидов, карбоно-
вых кислот и других соединений. Природа и размер алкильного
радикала стартового вещества регулируют гидрофобный характер,
а длина полиоксиэтиленовой цепи — гидрофильность образующегося
поверхностно-активного агента. Гидрофобную функцию в таких
веществах могут выполнять также полимерные фрагменты других
а-окисей — окиси пропилена, бутилена и т. п.
Большое химическое разнообразие неионогенных поверхностно-
активных веществ, перекрывающих широчайший диапазон требова-
ний соответствующих отраслей техники, их специфические свойства—
способность к мицсллообразованию, термодинамика сорбции, их
активность в процессах солюбилизации, эмульгирования, вспенива-
ния и моющая способность рассмотрены в монографиях [23, 24].
Не меньшее распространение получили в* последнее время гидр-
оксилсодержащие би- или полифункциональные олигомерные про-
дукты на основе а-окисей, находящие непосредственное применение,
а также способные к дальнейшим превращениям с участием функ-
циональных групп. Ниже описаны их основные представители.
П олиэтиленгликоли
Полиоксиэтиленгликоль или, как принято его называть полиэтилен-
гликоль (ПЭГ), имеет следующее строение полимерной молекулы:
Н-[-ОСН2СН2-]„-ОН
Сейчас известны и производятся в промышленном масштабе
продукты такого типа с га от двух (диэтиленгликоль) до »=> 103
(ПЭГ-40 ООО). Методы получения более высокомолекулярных членов
этого ряда не дают, как правило, гарантии бифункционалыюсти,
т. е. наличия двух ОН-групп на концах макромолекулы, и эти про-
дукты называют обычно полиэтиленоксидом или полиоксиэтиленом-
Синтез ПЭГ осуществляют присоединением определенного ко-
личества окиси этилена в присутствии катализаторов основного,
реже кислотного типа к стартовым веществам — воде пли гликолям.
Использование гликолей предпочтительнее, так как позволяет полу-
чить продукты с узким молекулярно-массовым распределением,
приближающиеся к индивидуальным веществам. Механизм при-
224
соединения [25] предполагает равенство констант скорости всех
последовательных стадий роста:
I Н-[ОС2Н4-]п-ОН-нГ —Н-[-ОС2Н4-]—он
' А-р = const Ф f (п)
Катализаторами синтеза ПЭГ являются алкоголяты, гидроокиси
и реже карбонаты щелочных и щелочноземельных металлов в коли-
честве 0,1—2% от массы стартового вещества. Применение гидро-
окисей более целесообразно, поскольку инициирование в этом случае
прямо ведет к бифункциональному активному центру
Н2С—СН2+КОН -----> НОСП2СН2ОК
^0
в отличие от алкоголятов, которые генерируют монофункциональные
макромолекулы:
Н,С—CH2-LROK —> ROCH2CH2OK
Используя щелочи, с другой стороны, необходимо учитывать
содержащуюся в них обычно воду при расчетах длины цепи полимер-
ных продуктов. Что касается активности различных катализаторов,
то значительно большую активность проявляют соединения калия,
однако при температурах выше 100 °C это различие полностью
нивелируется. Некоторые закономерности влияния катализаторов
на эти процессы можно проследить на примере присоединения окиси
этилена к спирту С13 (рис. 84) [26].
В отличие от низших гликолей (моно-, ди-, триэтиленгликоля,
см. гл. II—IV), синтез которых осуществляют непрерывным путем,
для большинства ПЭГ принят полупериодический вариант. На
рис. 85 представлена схема установки, используемой для полиокси-
этилирования различных веществ [27, 28]. Она состоит из реактора
с мешалкой, емкостей для окиси этилена, систем регулирования
температуры, подвода инертного газа, вакуума, коммуникаций.
Термостатирующая линия автоматически связана с подачей моно-
мера, чем обеспечивается заданный температурный режим. Давление
в емкостях мономера всегда выше, чем в зоне реакции, где оно редко
превышает 0,5 МПа (5 кгс/см2); невозможность проникания реагентов
в эти емкости дополнительно гарантируется запорными вентилями.
Процедура полимеризации состоит в следующем [23, 27]. В реак-
тор вводят расчетное количество стартового вещества (гликоль)
и катализатора, повышают температуру до 150—180 °C и систему
вакуумируют. Реактор тщательно продувают азотом и вводят порцию
окиси этилена. После начала реакции подачу окиси этилена регу-
лируют таким образом, чтобы парциальное давление ее в реакторе
был0 ниже взрывного предела для данной температуры. С другой
Ь Заказ 972
стороны, скорость подачи ограничивается возможностями тепло-
отвода и скоростью реакции. Поскольку процесс полимеризации
экзотермичен (более 84 кДж/моль пли 20 ккал/моль), область актив-
ного протекания процесса не требует дополнительного подвода
тепла, в ряде случаев необходимо даже сильное охлаждение. Не-
которые процессы предполагают программированный рост темпе-
ратуры.
По достижении заданного расхода окиси этилена, определяемого
длиной цепи, подачу прекращают и процесс ведут до полного исчер-
Рис. 84. Влияние катализатора и температуры на кинетику океиэтилирования
спирта Cis’.
а) 1 — Na, NaOCH., NaOC2Hs, КОН; 2 — NaOH; 3 — К2СО3, Na2CO3> без катализатора,
135—140 °C; б) 1 — Na, NaOCH3, NaOC2H5, КОН, NaOH; 2 — K2CO3; 3 — Na2CO3; 4 —
без катализатора, 195—200 °C.
пания мономера. Затем реактор охлаждают, реакционную смесь
нейтрализуют кислотами, фильтруют и, если требуется, обесцвечи-
вают с помощью перекиси водорода, активированного угля или
специального катализатора.
Мощность такой установки ограничена в основном низкими воз-
можностями периодической схемы в отводе реакционного тепла.
Более производительные установки работают по схеме с циркуляцией
реакционной смеси через теплообменник либо в непрерывном ре-
жиме [29]. В последнем случае используют реакторы трубчатого
типа; например, для синтеза ПЭГ разработана схема, включающая
спираль (Z = 1200 м) с двадцатью точками ввода мономера [30]-
Для улучшения контакта реагентов предложено также распыление
жидкой компоненты в парах окиси этилена. При синтезе высоко-
молекулярных ПЭГ (10—40 тыс.) такой метод потребует крайве
малых количеств стартового вещества, в связи с чем в качестве исхоД'
226
ньгх здесь целесообразно использовать ПЭГ промежуточных молеку-
лярных масс.
Основной сложностью при синтезе высших ПЭГ является необ-
ходимость строгого учета всех протонсодержащих компонентов
(в основном воды), присутствующих в мономере, катализаторе,
инертном газе, технологических коммуникациях, поскольку их
вклад в общую концентрацию растущих молекул в знаменателе
выражения (1, см. стр. 217) значителен. Так, при синтезе
ПЭГ-40 ООО полимеризацией в блоке концентрация растущих би-
Рис. 85. Технологическая схема полпокспэтилированпя различных веществ:
1 — смеситель; 2 — термодатчики; з — ротаметр; 4 — предохранительные клапаны; 5 —
реактор с якорной мешалкой; 6 — манометры; 7 — вентили; 8 — емкости окиси этилена.
функциональных молекул должна составлять около 0,04 моль/л
с такой точностью, с какой необходимо получить заданную длину
Цепи. Как правило, нужны специальные меры по удалению влаги
из системы, в частности влаги, образующейся из стартового гликоля
и катализаторов, например:
110—R—OH-j-2KOH ДЛ? КО -И--ОК -- 211,0
НО-К-ОП + К2СОз АЛЛ ко-н-ок + н2о + со2
Удаление влаги способствует протеканию этих реакций в напра-
влении алкоголята, чего достигают тщательным вакуумированием
Или продувкой системы, а также повышением температуры при этих
операциях. Более строгим решением вопроса является проведение
стадии подготовки каталитической системы в среде растворителя
(бензол, хлорбензол, толуол), образующего азеотропную смесь с во-
Дой, которая затем отгоняется [31]. Активность полученных таким
образом каталитических систем выше, и они приводят к однородным
полимерным продуктам.
Кроме того, вода, как и спирты, вступая в комплексообразование
с алкоголятными центрами, ингибирует полимеризацию. Устано-
влено [32], что при приготовлении безводной гидроокиси калия по
реакции калий-нафталпна с водой продукт реакции с более чем
стехиометрическим количеством воды не активен в полимеризации
окиси этилена при 30 °C. При высоких температурах этот эффект,
видимо, выражен менее резко в связи с понижением стабильности
комплексов.
Имеются мнения о существовании принципиальных препятствий
в синтезе высокомолекулярных ПЭГ. В качестве одного из них ука-
зывают на изомеризацию окиси этилена под действием щелочного-
катализатора в ацетальдегид, который при последующем иницииро-
вании образует неконтролируемое количество новых цепей, понижая
тем самым молекулярную массу. Согласно расчетам на примере
ПЭГ-6000 [24], при изомеризации 0,2% мономера молекулярная
масса не превышает 70% от расчетной.
Отмечается также возможность регенерации щелочи по схеме
CII2CII2ONa —> ^zvCH=CH2 + NaOH
с чем связывали ранние неудачи в синтезе высших ПЭГ анионным
путем, хотя экспериментальные обоснования такой реакции не
опубликованы.
Получение высокомолекулярного бифункционального полиэти-
лепоксида под действием безводной КОН [32] или щелочных солей
нафталина [9] показывает, что эти возможности не реализуются,
а неудачи в прямом синтезе высших ПЭГ обусловлены, скорее всего,
неконтролируемыми примесями, генерирующими дополнительные
цепи, т. е. несогласованным с требованиями синтеза уровнем чистоты
реагентов.
Более серьезные отрицательные последствия в синтезе высоко-
молекулярных ПЭГ может иметь обнаруженное недавно [33] рас-
щепление простых полиэфиров, в том числе полиэтиленоксида, под
действием агентов основного характера типа бутиллития, щелочей,
алкоголятов и т. п. Например, высокомолекулярный полиэтилен-
оксид (молекулярная масса выше 100 тыс.) разлагается под действием
бутиллития при 30 °C до полиэтиленгликоля с Мп около 1500. Ана-
логичным образом, но менее активно действуют соединения натрия
и калия. Нетрудно предположить, что активность соединений щелоч-
ных металлов должна падать с увеличением радиуса и понижением
электрофильности катиона, т. е. от Li+ к Cs+, поскольку первона-
чально должен образовываться комплекс иона металла с кислородом.
Роль таких реакций разрыва цепи непосредственно в ходе полимери-
зациопного процесса анионного типа в настоящее время не ясна.
Таким образом, с точки зрения механизма полимеризации и
вклада различных реакций не существует достаточно серьезных
препятствий для синтеза полиэтиленглпколей с молекулярными
массами вплоть до 10% хотя такая задача встречает значительные
трудности технического порядка.
228
Свойства ПЭГ определяются их молекулярной массой и сте-
пенью кристалличности. Они представляют собой при комнатной
температуре вязкие жидкости (ПЭГ 200—400), воскообразные веще-
ства — до молекулярной массы 6000 и даже твердые кристалли-
ческие продукты. Ряд свойств ПЭГ характеризуются более или менее
аддитивным вкладом мономерных звеньев и концевых групп. Сюда
относятся, в частности, коэффициент преломления, рефракция,
температура плавления, теплота сгорания, поверхностное натяжение
Рис. 86. Температура
плавления полиоксиэти-
ленгликолей как функция
молекулярной массы.
Рис. 87. Зависимость
коэффициента преломле-
ния полиоксиэтиленгли-
колей от молекулярной
массы.
и т. п. [34, 35]. На рис. 86 и 87 показаны в качестве примера зависи-
мости некоторых характеристик ПЭГ от молекулярной массы. Тем-
пература плавления, в частности, описывается широко известным
Уравнением Флори:
_1___1 _ 2»
Т 7’о А7/г
где Т и То — температуры плавления полимеров со степенью поли-
меризации, равной х и сл соответственно; ДЯ — теплота плавления.
Плотность ПЭГ, вопреки обыкновению, не подчиняется адди-
тивному закону, так как она определяется кристалличностью поли-
меров, в свою очередь зависящей от молекулярной массы; наиболее
Высокую плотность имеют полимеры с молекулярной массой 6—
Ю тыс. Еще более аномально ведет себя плотность расплава, не
Зависящая от длины цепи, начиная с тримера (d±a = 1,08) [35].
22»
В табл. 71 приведены свойства некоторых стандартных ПЭГ.
Для сравнения в последней строке таблицы приведены данные для
высокомолекулярного полимера.
Таблица 71. Свойства стандартных полиэтнленгликолей [27]
ПЭГ Молекулярные массы * IO 1О •К * Т. пл., °C | Вязкость при 100 еС, мм2/с Теплота сгорания Растворимость в воде при 25“С, 1 %
С Б if 1^Э к кДж/КГ 1
200 228 240 1,05 1,124 4,3 5656 23 600 100
300 — — — 1,125 1,4459 -15 ч- ~ —8 5,8 6017 25 200 100
400 424 445 1,05 1,125 1,4478 4—10 7,3 6145 25 650 100
600 670 690 1,03 1,126 1,4496 20—25 10,5 6183 25 850 100
1 000 970 1 000 1,03 1,12— 1,16 1,4512 38-41 17 6250 26 100 >--70
1 540 — — — 1,15 1,4523 43-47 25—32 6289 26 300 73
2 000 2 000 2 100 1,05 1,21 — 47-53 — — -—
4 000 3 470 3 820 1,10 1,20— 1,21 1,4535 53-56 78-85 6306 26 400 62
6 000 6 750 7 500 1,14 1,21 1,4536 60-63 700— 900 6328 26 500 «ДО
20 000 17 000 19 000 1,12 — — 64 «^50
40 000 32 350 37 200 1,15 .— — .—. — — —
— — 4 млн. — 1,21 1,470 63-67 105 6380 26 600 100
* По данным [36]. ** По данным [351.
Примечание. Теплоемкости ПЭГ составляют 2,10 — 2,48 кДж/(кг-К) или 0,50—
0,59 кал/(г-°С), теплоты плавления 14,65 —19,15 кДж/кг (35 — 46 кал/г), поверхностнее
натяжение жидких ПЭГ 43 — 45- 10~3 Н'м (43 — 45 дик/см).
Экстраполяция теилот сгорания ПЭГ к бесконечной молекуляр-
ной массе приводит в расчете на мономерное звено к величине
1,18 МДж/моль (281 ккал/моль) и теплоте полимеризации жидкой
окиси этилена в твердый полимер, равной 111,5 кДж/моль
(27,0 ккал/моль). С этой величиной широко согласуется опытное
значение теплоты полимеризации газообразного мономера, равное
—140,0 кДж/моль (—33,5 ккал/моль) [37]. Расчет по средним зна-
чениям энергий связи приводит к величинам \Н„ (газ, газ) =
= —112,1 кДж/моль (—27,3 ккал/моль) [38], калориметрия при
полимеризации мономера в блоке — к ДЯП (ж., раств.), равной
—87,5 кДж/моль (—20,9 ккал/моль) [39]. Высокие значения теплоты
полимеризации выдвигают серьезную проблему эффективного тепло-
обмена в технологии полиэтиленоксида.
ПЭГ хорошо растворимы в большинстве органических раствори-
телей, исключая парафины. Низшие ПЭГ смешиваются во всех
230
отношениях, а высшие — ограниченно, с водой, образуя растворы
с широко варьируемой вязкостью.
Для определения молекулярных масс ПЭГ используют методы
анализа концевых групп [24]. Здесь нашли применение классические
методы — ацетилирование, изоцианатный [40], ИК-спектроско-
пия [41]. На рис. 88 приведены спектры поглощения моно- и три-
этиленгликоля и двух полимерных образцов, а также корреляция
Рис. 88. ПК-спектры
этиленгликоля (а), трп-
этпленгликоля (б),
ПЭГ-300 (в) и ПЭГ-400
(г) и зависимость отно-
сительной интенсив-
ности полосы ОН-групп
(А’ = Аззво/А|)45) от сред-
ней степени полимери-
зации.
Степень полимеризации
интенсивностей полос СН
между отношением (R) интегральных
(2945 см-1) и ОН (3360 см-1) колебаний и степенью полимеризации
(хп), позволяющая рассчитывать молекулярные массы полимеров
До 8000 с точностью около 3% по уравнению:
R = 0.81 + 6,12.
Заслуживают внимания более новые приемы анализа — три-
Фюрацетилирование концевых ОН-групп с последующим анализом
образующихся трифторацетатов методом ЯМР на ядрах 19F [42],
а также ЯМР — спектральное определение молекулярных масс
йо интенсивности пиков предконцевых звеньев [43]. Чувствительные
При современной технике ядерного магнитного резонанса до моле-
кулярных масс около 100 тыс., эти методы свободны от осложне-
ний. обусловленных ассоциацией концевых ОН-групп. Последнее
обстоятельство приводит к аномалиям при эбуллиоскопии и криоско-
пии и ограничивает применение этих методов к полиэтиленгликолям.
Прецизионными вискозиметрическими измерениями показано [44],
что ассоциация увеличивается с понижением полярности раствори-
теля. Оценка констант ассоциации концевых групп полиэтилен-
гликолей приводит к таким величинам: 1,0 (ацетонитрил), 1,7 (ди-
оксан), 4,1 (бензол) и 11,7 л/моль (четыреххлористый углерод) при
25 °C. В воде, метиловом спирте и диметплформамиде ассоциация
ПЭГ пренебрежимо мала.
Ассоциация концевых групп полиэтиленгликоля может быть
и внутрицепной, что установлено методами ИК- и ЯМР-спектро-
скопии [45]. Образование ассоциатов между низко-и высокомолеку-
лярными фракциями полиэтиленгликолей при их фракционировании
может быть причиной серьезных ошибок при определении молеку-
лярной массы по ОН-группам [46].
Широко распространены вискозиметрические методы оценки мо-
лекулярных масс ПЭГ [47]. Для полиэтиленгликолей с молекуляр-
ными массами не выше 30 000 в некоторых распространенных рас-
творителях значения констант А и а в соотношении (2) характери-
стической вязкости * и молекулярной массы (соотношение
Марка — Хоувинка — Куна)
[ц] = ХМ“ (2)
приведены ниже:
Растворитель К- 10S дл/г а
Бензол (при 20 °C) 4,85 0,68
Вода (при 20 °C) 1,6 0,76
Вода (при 25 °C) 2,4 0,73
Метиловый спирт (при 20 °C) 3,3 0,72
Дпоксан (при 20 °C) 3,5 0,71
Четыреххлорпстый углерод (при 20 °C) 6,9 0,61
Диметилформамид (при 25 °C) 2,4 0,73
Молекулярно-массовое распределение (ММР) в продуктах поли-
меризации окиси этилена было предметом многих исследований.
Механизм полимеризации предполагает в этом случае образование
практически монодисперсных продуктов, что легко реализуется
в лабораторном масштабе.
Товарные низкомолекулярные ПЭГ имеют близкие к ожидаемым
факторы полидисперсности, полученные измерением различных сред-
них молекулярных масс (табл. 71). Небольшие отклонения обусло-
влены технологическими причинами.
* Характеристическая вязкость имеет размерность, обратную концентрации;
принятая для растворов полимеров размерность концентрации г/100 мл (г/дл)
сохранена в этой главе, она эквивалентна 10 кг/м'1 в системе СИ.
232
Функции ММР безусловно более информационны, с одной сто-
роны, как тонкий инструмент при анализе механизма, с другой,
как один из наиболее чувствительных параметров при выборе техно-
логической схемы процесса, числа реакторов, их типа, условий
полимеризации и т. и. Для получения функции ММР полиэтилен-
глнколей в последнее время успешно применяются различные виды
хроматографии.
Ассоциация концевых групп значительно понижает летучесть
полиэтиленгликолей, в результате чего возможности их газохрома-
тографического определения довольно быстро приходт в конфликт
с термической стабильностью. Заменой ОН-групп на другие, менее
склонные к ассоциации (CHSO, СН3СОО, (CH3)3Si, СеН5О, C6H5S,
Cl и т. п.), можно значительно повысить летучесть и расширить
возможность газохроматографического анализа полиэтиленглико-
лей [48]. На рис. 89 приведены в качестве примера хроматограммы
различных производных полиэтиленгликолей и полученные на их
основе функции распределения. В зависимости от способа замены
концевых групп функции ММР испытывают лишь незначительные
вариации. Однако, даже несмотря на эту модификацию, газовая
хроматография вряд ли применима к анализу полиэтиленгликолей
с молекулярными массами выше 1000.
Использование различных вариантов жидкостной хроматогра-
фии [49] позволяет несколько расширить эти рамки. Здесь также
наблюдается достаточно четкое разделение на индивидуальные ком-
поненты, причем замена концевых групп позволяет исключить
специфические эффекты адсорбции.
Наиболее универсальна для высокомолекулярных полимеров
гельпроникающая хроматография, где разделение осуществляется
по ситовому принципу, т. е. по размеру макромолекул. Использо-
вание специальной препаративной техники позволяет осуществить
строгий анализ функций ММР полиэтиленгликолей до 2—3 тыс. [50].
На рис. 90 приведены гельхроматограммы полиэтиленгликолей
и рассчитанные на их основе функции ММР. Как видно, полученные
кривые хорошо согласуются с распределением Пуассона, а неболь-
шие отличия в области низких молекулярных масс могут быть свя-
заны с потерей этих фракций при обработке полимера.
В области молекулярных масс до 600 все виды хроматографии
Дают хорошие результаты. Расчет функций ММР более высоко-
молекулярных полимеров из данных аналитической гельхроматогра-
фии наталкивается на ряд трудностей. В настоящее время корректно
Полученные функции ММР и показатели полидисперсности этих
полимеров в литературе отсутствуют.
Конформация полиоксиэтиленовой цепи исследована различными
Методами и ее можно считать надежно установленной. В кристал-
лическом состоянии, как это следует из рентгенографических и
ИК-спектральных измерений [51], она имеет форму спирали 7/2
(семь мономерных единиц на два витка спирали) с периодом повторе-
ния 1,93 нм (19,3 А) и углами внутреннего вращения 64° 58' (по
233-
С—С-связи) и 188° 15' (по С—О-связи). Элементарная кристалли-
ческая ячейка моноклинного типа с параметрами а = 0,816, Ъ =
= 1,299, с = 1,930 нм, р = 126° 05' включает четыре цепи. На
основании этих данных принята траис-конформация углеродных
и гогы-копформация для кислородных атомов в цепи (TTG). На рис. 91
показано строение индивидуальной цепи полиоксиэтилена. Ана-
логичные данные о структуре получены методом ЯМР высокого
Т
I
Рис. 89. Газовая хроматография полиоксиэтиленгликолей с модифицирован-
ными концевыми группами:
а — хроматограмма ПЭГ-400 с фенилсульфидными концевыми группами (цифрами указаве
число звеньев); б — функции молекулярно-массового распределения полиоксиэтиленглико-
лей 300 (7), 400 (2) и 600 (3) с концевыми С„Н5О-группами; в — то же, с концевыми CsH6b
группами.
234
разрешения из температурной зависимости константы спин-спинового
взаимодействия [52].
Транстрансгош-кояформаряя является наиболее термодина-
мически выгодной и сохраняется в растворе. Данные вискози-
метрии [53, 54[ и расчеты по теории вращательной изомерии [55]
согласуются с такой конформацией цепи. Среднеквадратичный не-
возмущенный размер (Гц/М)*-'2 для ПЭО составляет 0,084—0,093 ям.
Рис. 90. Гельхроматографический
анализ полиоксиэтиленгликолей:
а — гельхроматограммы ПЭГ-300 (7) и
ПЭГ-400 (2) (степень полимеризации п ука-
зана цифрами над пиком); б — кривая
молекулярно-массового распределения
ПЭГ-400 в сравнении с распределением
Пуассона = 446, Мп = 429,
= 1,04, по данным ГПХ).
а его отпошение к размеру свободно сочлененной цепи 2 =
== 1,55. Эти данные свидетельствуют о большой гибкости полиокси-
этиленовой цепи, характерной и для других простых полиэфиров.
Гибкость полиоксиэтиленовой цепи проявляется также в низких
температурах стеклования, которые для относительно высокомоле-
кулярных полимеров лежат в области от —50 до —55 °C. Зависи-
мость температуры стеклования от молекулярной массы [63] в дан-
ном случае резко аномальна (рис. 92) [56] и обусловлена эффективной
кристаллизацией полимеров в области молекулярных масс 6—
Ю тыс., сильно ограничивающей подвижность.
Химические свойства ПЭГ определяются двумя обстоятель-
ствами — реакционной способностью концевых гидроксильных
гРупп и возможностью разрыва основной цепи под действием раз-
личных агентов. Все ПЭГ, особенно низкомолекулярные, вступают
в реакции, характерные для спиртов. Это позволяет широко-
235
г
варьировать концевые группы ПЭГ, получая почти индивидуальные
бифункциональные олигомеры, которые могут отверждаться затем
методами поликонденсации или радикальной полимеризации, либо
использоваться для блоксОполимеризации с другими мономерами.
Регулирование концевых групп можно проводить также на стадии
синтеза путем дезактивации «живого» полимера соответствующими
агентами. Имеется множество примеров такого рода превращений.
Рис. 91. Конформация
цепи полпоксиэтиленгли-
коля.
Рис. 92. Температура
стеклования полиокси-
этиленгликолей как
функция молекулярной
массы:
1,5 2 — экспериментальные
данные; 3 — нормальный
вид функции.
Процессы деструкции более сильно проявляются у высокомолеку-
лярных полимеров окиси этилена, где они представлены во всем
многообразии. Для ПЭГ, как и для низкомолекулярных гликолей,
характерно автоокисление, приводящее к появлению у ПЭГ и их
растворов корродирующей способности. Автоокисление ускоряется i
на свету, а также добавками переходных металлов [57, 58]. Деструк-
цию такого типа можно ингибировать небольшими добавками спир-
тов, например изопропилового. Этот метод стабилизации с успехом
используется и для высокомолекулярных полимеров.
В литературе рассмотрены некоторые вопросы биохимии ПЭГ
и родственных соединений: физиологическая активность, токсию
ность, биохимическое окисление и биодеградация. Токсичность 'тП
невелика, доза ЛОБ0 для крыс составляет 35—45 г/кг у жидких
ПЭГ *
236
0 50—60 г/кг — у твердых [24, 59]. При такой токсичности допу-
скается применение ПЭГ в пищевых и фармацевтических целях.
Применение ПЭГ [27, 56, 59, 60]. ПЭГ широко используются
в различных областях техники. Высокая смачиваемость и практи-
чески полная водорастворимость делают чрезвычайно эффективным
применение ПЭГ в различных операциях формования — натуральных
и синтетических каучуков, керамики, волокон и даже металлов.
Будучи легко совместимыми с разнообразными препаратами, они
используются в косметических и фармацевтических рецептурах
в качестве материала для таблеток, паст, мазей и т. п. В текстильной
промышленности ПЭГ находят применение в качестве вспомога-
тельных веществ в мокром прядении, для распыления красителей,
а также как мягчители и антистатические агенты. Аэрозольные
композиции на основе ПЭГ применяются также в агротехнике.
В производстве полиуретанов полиэтиленгликоли находят огра-
ниченное применение в основном из-за склонности вулканизата
к кристаллизации. Более распространены здесь полимеры и сополи-
меры окиси пропилена. Прочные жесткие полиуретаны получают
на основе низкомолекулярных полиэтиленгликолей от 200 до 1500.
Известно множество мелких областей применения полиэтилен-
гликолей — от покрытия электрических контактов и мелования
бумаги до полировки металлов. В практике научно-исследователь-
ских лабораторий они часто используются как эталонные вещества
при молекулярно-массовых измерениях, а также в качестве жидкой
фазы в хроматографии.
. Значительная доля полиэтиленгликолей расходуется на получе-
ние их эфиров с жирными кислотами, являющихся поверхностно-
активными веществами. Их синтез аналогичен синтезу производных
низкомолекулярных гликолей прямой реакцией с соответствующими
кислотами при 120—130 °C. Чаще используют лауриновую, сте-
ариновую или олеиновую кислоты. Температуры плавления воз-
растают от гликоля к моно-, а затем к диэфиру. Ненасыщенные
кислоты образуют более низкоплавкие и более растворимые эфиры,
чем соответствующие насыщенные. Растворимость эфиров в воде
возрастает с молекулярной массой исходного полиэтиленгликоля.
Полиоксипропиленполиолы
Синтез олигомерных продуктов из окиси пропилена почти целиком
Подчинен требованиям химии полиуретанов. Их самостоятельное
значение весьма невелико — они используются как вспомогательные
Вещества, выполняя функции, сходные с ПЭГ.
Методы получения полимеров на основе окиси пропилена, со-
держащих на концах цепи реакционноспособные гидроксильные
гРуппы, аналогичны методам, применяемым в синтезе ПЭГ [27,
с. 221—230; 56, с. 145—148]. Здесь также используются щелочные
11 Щелочноземельные катализаторы; процесс ведут чаще всего при
Постепенной подаче мономера, что позволяет регулировать скорость
237
реакции. Температуру варьируют от 80 до 150 °C, причем верхний
предел обусловлен протеканием побочных процессов, в частности
передачей цепи. Это же обстоятельство предъявляет повышенные
требования к методам выделения полимера, удаления мономера [61]
ит. п., хотя в целом опп аналогичны применяемым в случае ПЭГ.
Более совершенен в технологическом отношении непрерывный
вариант процесса, развиваемый в последнее время. Технологическая
схема синтеза полипроппленглпколей (рпс. 93) [62] состоит из четы-
рех реакторов, в каждом из которых молекулярная масса достигает
Рис. 93. Технологическая схема синтеза полипропиленгликолей:
1,2 — смесители; 3,4 — холодильники; 5 — реакторы; 6 — дозаторы.
заданной величины: 700, 1200, 1800 и 2400. Процесс осуществляется
в аппаратах колонного типа, в которые непрерывно вводят стартовое
вещество с катализатором либо продукт предыдущей стадии, а также
мономер, всегда присутствующий в избытке и конденсирующийся
в выносном холодильнике. Газообразная окись пропилена, двигаясь
вверх по колонне, перемешивает реакционную массу.
Процесс проводят следующим образом. Инициатор — гидроксил-
содержащее вещество — смешивают с окисью пропилена сначала
в смесителе 1, а затем в смесителе 2, куда из холодильника 4 посту-
пает возвратный мономер. На выходе из смесителя 2 отношение
мономер : инициатор составляет 6—12. Охлажденная до требуемой
температуры в холодильнике 3 смесь насосом подается в нижнюю
часть первого реактора 5. Из дозатора 6 вверх реактора вводится
диспергированный катализатор, общее количество которого соста-
вляет 0,3—0,6% на всю жидкую смесь в реакторе. Реактор предста-
вляет собой вертикальный аппарат с соотношением высоты к диа'
метру, равным 30—40, что необходимо для обеспечения соответствУ'
ющего времени контакта.
238
Температура в реакторе поддерживается автоматически на задан-
ном уровне в интервале 100—150 °C. Предусмотрен дополнительный
ввод окпси пропилена в смеситель 2 для регулирования состава
реакционной смеси.
Реакционный продукт из реактора 5 выводят па окончательную
обработку либо, предварительно смешав с мономером, подают в ниж-
нюю часть следующего реактора. Объем каждого последующего
реактора больше предыдущего в 1,5—2,5 раза, а отношение моно-
цер : инициатор, определяющее конечную длину цепи, увеличивается
в каждой стадии на 6—12 моль/моль. Готовый продукт с максималь-
ной молекулярной массой, полученный на выходе последнего реак-
тора, подают на очистку.
Непрерывный способ синтеза полиэфиров более выгоден эконо-
мически и, что не менее важно, приводит к получению продуктов
высокого качества, в частности по полидисперсности. Число стадий
и реакторов определяется требованиями к ассортименту полимеров
и объемом производства. Предложен одностадийный вариант непре-
рывного синтеза полиоксипропиленполиолов в барботажном реак-
торе, где избыточный мономер также используется для перемешива-
ния реакционной массы [63].
Существенным отличием от синтеза ПЭГ является использование
при получении олигомеров из окиси пропилена большего ассорти-
мента стартовых веществ. Для создания сшитых полиуретанов
Таблица 72. Свойства продуктов полпприсоединенпя окпси пропилена
к спиртам различной функциональности [27, 56]
Стартовое ве- щество Молекуляр- ная масса Гидроксиль- ное число Вязкость при 25 °C, мПа • с 1 Плотность . при 25 °C, г/см3 Торговые ** наименования
Пропилспгли- 150 750 55 1,025 * Niax Diol PPG, Pluracol
КОЛЬ 4000 28 1114 1,002 * P, Voranol P
Глицерин 700 241 199 1,03 * Niax Triol LG, Pluracol
5250 32 790 1,017 GP, Voranol CP
Триметплолпр о- 300 561 — 1,027 Pluracol TP Triol
пан 4100 42 670 1,004
1>2,6-Гексан- 700 240 384 * 1,022 * Niax Triol LHT
триол 6000 28 1467 * 1,005 *
Пентаэритрит 400 560 2800 — Pluracol PEP Tetrol
600 374 1140 —
Сорбит 500 . Pluracol SP Hexol, ’
5000 — — — Niax Hexol LS
р * При 2О'°С. ** Продукты фирм: Union Carbide Chem. Со. (Niax), Wyandotte Chem.
J°rP. (Pluracol), Dow Chem. Co. (Voranol).
239
R- + СНз-НС—СН2-
'''о'
с широкой вариацией параметров пространственной сетки требуется
набор олигомеров с различной функциональностью и длиной инди-
видуальной цепи. В связи с этим в качестве инициаторов используют
бифункциональные (моно- и дипропиленгликоль, бутандиолы), три-
функциональные (глицерин, 1,2,6-гексантриол, триметплолпро-
пап), а также этилендиамин и пентаэритрит (тетра-), диэтилептри-
амин (пента-), сорбит, маннит (гекса-), сахароза (окта-функциональ-
ные). Аналогичным образом получают производные диглицерина,
крахмала, целлюлозы, глюкозы и т. п.
Общий объем производства олигомерных полиоксипроппленполи-
олов быстро возрастает. Например, в США в 1972 г. их произведено
160 тыс. т против 66 тыс. т в 1964 г. [56]. Около 85% расходуется
на нужды полиуретановых производств. Свойства основных групп
олигомеров, получаемых из окиси пропилена, представлены в табл. 72
(в таблице указан диапазон изменения соответствующих свойств)
и в Приложении.
Полимеризация окиси пропилена, простейшего из замещенных
эпоксидов, осложнена по сравнению с полимеризацией окиси этилена
некоторыми дополнительными обстоятельствами, и их необходимо
учитывать при направленном синтезе полимеров.
Наличие заместителя приводит к возможности раскрытия цикла
с разрывом а- или |3-С—О-связи, т. е.
---> RCII—СН2О- (а)
I
СН3
-—> RCII2—СНО- (3)
С Н3
причем относительная доля а- и ^-продуктов зависит от катализа-
тора, сокатализатора, температуры процесса, растворителя и т. п.
Только в чисто анионных системах аномальное (по
а-С—0-связи) раскрытие цикла практически исключено: полимери-
зация протекает в основном в результате атаки незамещенного атома
углерода, и доля нормальных ^-структур достигает 97—99%. В кис-
лотных и некоторых металлоорганических системах раскрытие цикла
протекает хаотически, что имеет аналогии и в мономерной химии
а-окисей [13].
При анализе структуры и последовательности чередования нор-
мальных и аномальных звеньев в полипропиленоксиде представляют
интерес разработанные в последнее время методы деструкции поли-
мера озоном или литийбутилом [33, 64] с последующим хромато-
графическим определением состава образующихся дипропиленгли-
колей. Эти методы будут рассмотрены применительно к стерео-
регулярному полипропиленоксиду.
С другой стороны, при полимеризации окиси пропилена возникает
вопрос о взаимной ориентации двух соседних заместителей в цеп0
с образованием изо- или синдиотактических структур и о характер6
240
чередования этих структур в макромолекуле. Обычная окись про-
пилена представляет собой, как известно, рацемическую смесь
оптических изомеров.
Принято считать [65], что при анионной полимеризации при-
соединение любого мономера (D или L) равновероятно, и образуется
атактический полимер. Недавно методами ЯМР показано [66—67],
что эти полимеры действительно содержат примерно равные коли-
чества изо- и синдиотактических диад, т. е. образующаяся полиокси-
нропиленовая цепь представляет собой нерегулярную в стерео-
химическом смысле последовательность звеньев нормальной §-струк-
туры.
И, наконец, еще одна важная проблема синтезе полиоксипро-
пиленгликолей состоит в наличии интенсивной передачи цепи на
мономер, приводящей к появлению ненасыщенности в образующемся
полимере.
На основании кинетических данных, влияния изотопного заме-
щения в мономере на длину цепи и исследования полимеризации
других мономеров предложена [68, 69] схема, включающая образо-
вание аллильной ненасыщенности
СН2—СНО- К++СН3—НС—сн2 ->
СНз
о
СН2СНОН+СН2=СН-СН2О- к-
СНз
ее изомеризацию при последующем росте цепи:
сн2=сн—сн2о- к+
пН2С--СН-СНз
—> СН2=СН—СН2—Г—ОСН2—СН—1—о- к+
СНз Jn
СН2=СН-СН2-Г-ОСН2-СН-1-О- к- ->
СН3 п
СНз—СН=СН—Г—ОСН2—CH—1—О- К+
СНз
п
Спектральный и химический анализы ненасыщенности показы-
®ают, что Скорость изомеризации аллильных групп в г^ис-пропениль-
жЫе зависит от длины цепи и максимальна в случае тримера (п = 3).
16 Заказ 972 241
Возможно, это связано с образованием промежуточного циклического
комплекса.
Исследование кинетики передачи цепи при полимеризации окиси
пропилена в условиях, близких к технологическим, показывает,
что относительная константа скорости передачи kjk? обратно про-
порциональна концентрации ОН-групп [70]. Этот факт может быть
объяснен только участием ОН-групп в акте роста цепи, хотя кине-
тические исследования полимеризации в тех же условиях не обна-
руживают этого. Несомненно, образование двойных связей является
отрицательным фактором, поскольку препятствует образованию по-
лиоксипропиленполиолов строго заданной функциональности.
Введение регуляторов концевых групп, более активных чем моно-
мер, в передаче протона, позволяет несколько снизить ненасыщен-
ность, но такой путь приведет к полимерам ограниченной молеку-
лярной массы. Предпочтительнее использовать катализаторы, актив-
ные при низких температурах, поскольку энергия активации пере-
дачи цепи значительно выше, чем в стадии роста. Рационально,
в частности, использование CsOH„ вместо КОН, поскольку, как
показано в работе [71], при прочих равных условиях ненасыщен-
ность снижается с 0,11 до 0,07 ммоль/г.
Существуют пути ликвидации ненасыщенности на стадии гото-
вого полимера. Предложена [72] для этой цели основно-каталити-
ческая изомеризация в сочетании с кислотным гидролизом по схеме:
^zvCH2OCH2CH=CIl2 [0—— ^zvCH2OCH=CHCH3 'НгО[н+|>
—> ^vCII2OH4-CII3CH2CHO
В результате тех или иных мер содержание ненасыщенности
в полипропиленгликолях удается снизить до 0,01 ммоль/г (мол.
масса до 1200) и до 0,1 ммоль/г — у высокомолекулярных образцов.
Свойства полиоксипропиленполиолов. Полиоксипропиленполполы
представляют собой бесцветные вязкие жидкости с крайне низким
давлением насыщенных паров. Они хорошо растворимы в кетонах,
спиртах, сложных эфирах, большинстве хлорированных раствори-
телей, ароматических и некоторых парафиновых углеводородах.
Растворимость в воде резко падает с молекулярной массой и слабо
зависит от функциональности. На рис. 94 приведены диаграммы
растворимости полиоксипропилендиолов и триолов [73]. Характер
изменения основных свойств полиоксипропиленгликолей с молеку-
лярной массой дан в табл. 73.
Температуры замерзания полиоксипропиленгликолей составляют
—60, —50 и —45 °C для молекулярных масс 400, 1000 и 2000 соот-
ветственно.
При детальном исследовании закономерностей изменения вяз-
кости и плотности полиоксипропиленполиолов с молекулярной мас-
сой и температурой [73] обнаружены аномалии, обусловленные
ассоциацией молекул, которая может быть межцепной и внутри'
242
Концентрация полимера, %
Рис. 94. Диаграммы взаимной растворимости в системах вода — полиоксипро-
пилендиолы (сплошные линии) и триолы (пунктир) с молекулярными массами:
1 — 465—480; 2 — 680—720; 3 — 750; 4 — 1000; S — 1500; 6 — 1660; 7 — 2000; 8 — 2500;
S — 3000; 10 — 6000.
Таблица 73. Свойства полиоксипропиленгликолей * [27, 56]
Молекуляр- ная масса Гидроксиль- ное число d20 а20 Вязкость при 100 °C, мПа - с Температура вспышки, °C Раствори- мость в воде при 20 °C, %
fc. 150 750 1,025 3,0 121 а
Ь" 250 450 1,011 3,4 168 а
Е; 400 280 1,008 5,0 199 а
В 750 150 1,006 7,6 215 15,25 * **
№ Ю00 112 1,005 11,5 227 1,5
1200 93 1,004 13,7 232 1,96 **
В 1850 61 1,003 232 —
2000 56 1,003 23,0 232 0,15
₽ 3000 37 1,002 48,0 232 —
4000 28 1,002 74,0 232 —
* Полпоксипропиленгликоли фирм Union Carbide, Dow Chein. Co., Wyandotte Chem.
Corp.
** При 25 °C.
a—смешиваются во всех отношениях.
16*
цепной. Наиболее сильно влияние ассоциации проявляется при
молекулярных массах ниже 1500, причем ИК-спектральные изме-
рения в области валентных колебаний ОН-групп непосредственно
подтверждают наличие ассоциатов. С повышением температуры
влияние функциональности и молекулярной массы на вязкость
и плотность нивелируется.
Молекулярные массы полиоксипропиленполиолов могут быть
охарактеризованы способами, принятыми для полиэтиленгликолей.
Достаточно надежны методы анализа концевых групп, особенно
ИК-спектральные [74].
Важной аналитической проблемой в случае полиоксипропилен-
полиолов, а также сополимеров окиси пропилена является опре-
деление содержания первичных и вторичных концевых ОН-групп.
Их доля определяется условиями протекания реакции п механизмом
раскрытия цикла, а также наличием реакций передачи цепи протон-
содержащими примесями и исходным инициатором.
Наибольшее распространение получили кинетические методы,
основанные на различной реакционной способности первичной и
вторичной ОН-групп в реакции с уксусным ангидридом или фенил-
изоцианатом [75, 76]. Особый интерес представляет спектрофото-
метрический вариант этого метода, позволяющий измерять содер-
жание первичных ОН-групп в результате анализа кинетики рас-
ходования фенилизоцианата [76]. Реакцию фенилизоцианата
с исследуемым гликолем ускоряют добавками аминов или олово-
органических соединений.
Еще более перспективен метод определения содержания различ-
ных ОН-групп, основанный на их превращении в трифторацетаты
по реакции
-^zx,CHOII + CF3COOCOCF3 -> -^zx,CHOCOCF3 + CF3COOH
R R
с последующим определением доли первичных и вторичных трифтор-
ацетатов методом ЯМР — 19F [42, 67]. Этот метод, как отмечалось
выше, чувствителен и к стереохимии концевых мономерных звеньев.
Структура концевых гидроксильных групп полиоксипропилен-
гликолей и полиолов имеет важное значение в последующем синтезе
полиуретанов на их основе. Первичные ОН-группы обладают более
высокой реакционной способностью в этом процессе, в связи с чем, '
регулируя условия полимеризации (катализатор, гидроксилсодер-
жащее стартовое вещество, температура), стремятся повысить их
долю. Этого можно достичь также введением небольших количеств
окиси этилена [77]. С другой стороны, подбором катализаторов
можно нивелировать различия в реакционной способности различ-
ных гидроксильных групп на стадии уретанообразования. На прак-
тике используют оба подхода.
Для определения молекулярных масс полиоксипропиленполиолов
используют вискозиметрию; ниже приведены коэффициенты в урав-
244
нении Марка — Хувинка — Куна (2) для полиоксипропилендиолов
н триолов при 20 °C в разных растворителях [78].
' К • 1 о *,
дл /г а
Полиоксипропилендиолы (молекулярные мас-
сы 500—3300)
Метиловый спирт ................ 4,06 0,64
Тетрагидрофуран ........... . . , 5,50 0,62
Бензол .............„........... 1,11 0,79
Толуол ......................... 2,08 0,72
Пол и оксипропилентри олы (молекулярные
массы не выше 5000)
Толуол ......................... 2,05 0,71
Метиловый спирт ................ 5,55 0,56
Фактор полидисперсности MwiMn составляет 1,14—1,25 для
полидиолов и не превышает 1,17 для триолов на основе триметилол-
пропана [78].
Молекулярно-массовое распределение в полиоксипропиленовых
олигомерах определяется многими факторами. Оно уширяется с ро-
стом молекулярной массы в связи с более сильным влиянием пере-
дачи цепи, однако в присутствии гидроксилсодержащих инициаторов
распределение сужается, практически совпадая с пуассоновым [79].
Отсутствие стартовых веществ и медленное инициирование, напротив,
уширяют ММР. Такая картина наблюдается, в частности, прп поли-
меризации на твердой КОН [69]; на рис. 95 даны экспериментальные
кривые распределения по молекулярным массам в сопоставлении
с рассчитанными по Флори.
При анализе молекулярно-массового распределения полиокси-
пропиленполиолов в последнее время широко используют хромато-
графические методы. Гельхроматография является наиболее экс-
прессным способом для получения кривых ММР и дает результаты,
Удовлетворительно согласующиеся с данными препаративного фрак-
ционирования ([80], рис. 95).
Необходимо отметить здесь одно важное обстоятельство, общее
Для химии олигомеров с большим числом функциональных групп.
В синтезе этих веществ в результате действия вторичных процессов
(например, при передаче цепи на мономер при полимеризации окиси
пропилена) возникает набор макромолекул с функциональностью
меньшей, чем заданная. Так, триол может содержать молекулы
с двумя, одной функциональными группами или вообще безгидр-
°ксильпые молекулы. Возникает понятие распределения по типам
Функциональности, и реальный образец представляет собой, таким
°бразом, суперпозицию двух распределений — по длине и по функ-
циональности макромолекул. Для решения этих проблем исполь-
3Уют комбинированное фракционирование и хроматографию, что
Позволяет характеризовать оба распределения раздельно [81].
245
Теплоемкости полипропиленгликолей зависят от молекулярной
массы и составляют, например, для полиоксипропиленгликолей 400,
1200 и 2000 при 20 °C соответственно 1,97, 0,444 и 0,428 кДж/(моль х
X К) или 0,471, 1,86 и 1,78 кал/(г-°С). Их зависимость от темпера-
туры линейна, и при 100 °C ППГ-2000 имеет 2,185 кДж/(моль • К.)
или 0,522 кал/(г-°С).
Теплота полимеризации, найденная из теплот сгорания, АНП
(ж., тв.) составляет —109,5 кДж/моль (—26,2 ккал/моль) [39],
а величина, рассчитанная для газовой фазы по данным [38], А//п
(газ, газ) равна —110,0 кДж/моль (—26,4 ккал/моль).
Рис. 95. Молекулярно-массовое распределение в полимерах окиси пропилена;
а — товарный полиоксипропиленгликоль 1600 по данным гельпроникающей (1) и осадитель-
ной (2) хроматографии; б — при полимеризации окиси пропилена на твердой КОН в сравне-
нии с распределением Пуассона (сплошная кривая).
Температура стеклования полипропиленгликолей от димера до
ППГ-4000, измеренная методом механических потерь [82], составляет
—60 ± 2 °C; по данным [83], она может достигать даже —92 °C.
Хотя полипропиленгликоли в силу нерегулярности структуры тне
способны к кристаллизации, присутствие полярных групп вызывает
их ориентацию при добавках активных наполнителей. Так, введение
в ППГ-2025 25% перхлората лития приводит к повышению его
температуры стеклования от —65 до +40 °C [84], причем механи-
ческие свойства указывают на трехмерный характер системы. Ана-
логичные эффекты структурирования могут иметь место и в напол-
ненных полиуретанах.
В связи с широким применением полиоксипропиленполиолов
в полиуретанах и других типах каучуков детально исследовались
их релаксационные свойства как в линейных, так и в сшитых систе-
мах. Здесь использовапы методы измерения диэлектрических, меха-
нических потерь и ядерный магнитный резонанс, приводящие к со-
гласующимся результатам [82, 85].
Высокие динамо-механические и прочностные показатели, потен-
циально заложенные в полиоксипропиленовых цепях, наиболее
246
полно реализуются в различных эластомерах и вулканизатах на их
основе. Рассмотрим лишь самые общие принципы создания полимер-
ных материалов на основе различных полиоксипропиленовых олиго-
меров.
Полиуретановый вариант является наиболее перспективным
и разработанным. Реакцией полиоксипропиленполиолов различной
функциональности с диизоцианатами
'xzvCH-j-CH-OH + OCN—R'-NCO+HO-CH-CH.'xzv —>
Ц; СНз СН3
BL ----> лл,СН2-СН—О—СО—NH—R'—NH— СО—О—CH—CIL'xzv,
СНз СНз
при дополнительном использовании низкомолекулярных полиспир-
тов в качестве разветвляющих агентов удается получить исключи-
тельно широкий спектр продуктов с богатым диапазоном свойств [27,
86]. Химия полиуретанов представляет собой в настоящее время
одну из крупнейших областей полимерной науки и технологии,
сильнейшим образом воздействующую на развитие ассортимента
и объема производства полиоксипропиленовых олигомеров.
Дополнительной возможностью тонкой вариации свойств поли-
уретанов, как многоцелевого продукта, является использование
в качестве олигомеров продуктов сополимеризации окисп пропилена,
например с тетрагидрофураном, окисью этилена или мономерами
других классов. Характеристики некоторых отечественных продук-
тов, являющихся олигомерами в синтезе уретанов п имеющих само-
стоятельные области применения, приведены в Приложении (см.
стр. 364). Разрабатываются различные модификации, исключающие
конечную стадию образования уретановых групп. Одним из пер-
спективных путей является синтез олигомерных ди- или полиизо-
цианатов реакцией полиолов с большим избытком диизоцианата
'v^CH2-CH-OI-I4-OCN-R'-NCO —>
I
R
--> 'Х^СЩ-СН-О-СО—NII-R'-NCO
I
R
и их последующее отверждение при каталитической тримеризации!
О
С
-xzvR'-N/ \N-R'-X7Xz
. 3-xzvR'—NCO > | |
O = C\ /.c = o
tN
I
247
Образующиеся триизоциануратные циклы обладают более высо-
кой по сравнению с уретановой группой стойкостью к некоторым
вторичным взаимодействиям. В этой области также накоплен боль-
шой экспериментальный материал [87] и имеются перспективы
практического использования.
Принципиально иным путем является синтез двойных или трой-
ных сополимеров окиси пропилена с ненасыщенными эпоксидами,
которые отверждаются затем обычной серной вулканизацией [2, 88].
В синтезе промежуточного полимера используют различные металло-
органические катализаторы, а в качестве сшивающего компонента —
моноокись бутадиена, аллилглицидиловый эфир, глицидилакрилат,
моноокись винилциклогексена и др., добавленные в количестве не-
скольких процентов. Разработаны рецептуры отверждения при
150 °C. Склонность готовых каучуков к кристаллизации может быть
понижена за счет отделения стереорегулярной фракции на стадии
олигомера либо введением в сополимер третьего мономера, на-
пример окиси этилена, разупорядочивающего регулярные после-
довательности.
Каучуки этого типа обладают высокой маслостойкостыо, пре-
красными температурными и динамическими характеристиками.
Их прочность в наполненном состоянии может достигать 58,8 МПа
(600 кгс/см2), удлинение 1000—1100% [88]. По ряду других свойств,
в частности озоностойкости и стойкости к истиранию, полипропилен-
оксидные каучуки, например Dynagen ХР-139, превосходят другие
синтетические и натуральные каучуки.
Наилучшие результаты в качестве наполнителя каучуков этого
тица дает коллоидная кремнекислота в силу взаимодействия с по-
лярными группами полиоксипропиленовой цепи, а также различные
образцы мелкой сажи. Модуль наполненного каучука лежит в пре-
делах 4,9—19,6 МПа (50—200 кгс/см2). Прочностные характери-
стики варьируют, изменяя содержание наполнителя и применяя
пластификаторы.
Разрабатываются также методы, комбинирующие оба рассмотрен-
ных подхода [3, 89]. Так, используя в качестве стартовых веществ
при синтезе полипропиленгликоля ненасыщенные спирты, например
2-(оксиметил)-2-метил-4-пентенол, можно получить олигомеры,
способные к отверждению по обоим механизмам, что затем исполь-
зуется при вулканизации. Описано также получение ненасыщенных
полигликолей (с мол. массой от 250 до 16 000) анионной сополимери-
зацией простых эпоксидов с моноокисью бутадиена.
И, наконец, используются методы прямой вулканизации поли-
ексипропилена, главным образом высокомолекулярного. При со-
вместном применении органической перекиси и серы получаются
вулканизаты удовлетворительного качества, тогда как в отсутствие
серы идет деструкция полимера [90].
248
Сополимеры окиси этилена и окиси пропилена
Сополимеры окисей этилена и пропилена друг с другом и с моно-
мерами других типов занимают значительное место в балансе поли-
мерных продуктов на основе эпоксидов, причем в дальнейшем
видимо, их роль будет возрастать. Можно выделить несколько глав-
ных направлений в синтезе эпоксидных сополимеров [3; 24, с. 300_
371; 27, с. 231—237; 56]. Одно йз них — синтез статистических или
блочных сополимеров с концевыми ОН-группами — целиком напра-
влено на решение задач химии полиуретанов. Наиболее успешно
применяются в этой области сополимеры окиси пропилена с окисью
этилена или тетрагидрофураном.
Второе направление — модификация некоторых виниловых
полимеров введением статистически распределенных по цепи звеньев
или блоков из эпоксидов. Целью такой модификации может быть
улучшение релаксационных, низкотемпературных свойств, поляр-
ности, гидрофильности и других характеристик виниловых поли-
меров. Аналогичной модификации поддаются и природные поли-
меры .
Наконец, синтез низкомолекулярных блок- или статистических
сополимеров окисей этилена и пропилена направлен на создание
неионогенных поверхностно-активных веществ. Это — наиболее
разработанная область в химии сополимеров из эпоксидов.
Основные функции блоксополимеров — высокая моющая и сма-
чивающая способность, поверхностно-активные добавки в различных
процессах, а также полупродукты для уретанов. С точки зрения
физико-химических свойств введение окиси этилена повышает рас-
творимость полиоксипропиленполиолов в воде, что в ряде случаев
необходимо. С другой стороны, добавка окиси пропилена разупорядо-
чивает цепи ПЭГ, делая эти продукты жидкими. Комбинация обоих
мономеров внутри цепи позволяет тонко варьировать поверхностно-
активные свойства этих веществ, а также качество уретановых вул-
канизатов.
Сополимеры такого типа можно классифицировать по типу функ-
циональности и характеру строения полиалкиленоксидной цепи.
По первому признаку можно выделить моно-, би-, три-, тетра- и более
функциональные сополимеры, и в этом отношении они полностью
идентичны полиоксипропиленполиолам. С другой стороны, индиви-
дуальные цепи могут быть статистическими или блоксополимерами
либо содержать чередующиеся участки того и другого типа.
Синтез сополимеров в большинстве случаев идентичен описан-
ному выше для полиэтиленгликолей. В случае блоксополимеров
подачу мономеров чередуют таким образом, чтобы получить блоки
необходимой длины. Статистические сополимеры получают совме-
стной полимеризацией окиси этилена и окиси пропилена, взятых
8 соотношении, обеспечивающем требуемое качество продукта.
Среди блоксополимеров окисей этилена и пропилена наиболее
Широко известны продукты, имеющие фирменные наименования
^249
Pluronic и Tetronic * [91]. Полимеры типа Pluronic получают кон-
тролируемым присоединением окиси пропилена к пропиленгликолю
при 120 °C в присутствии КОН. После завершения этой стадии уда-
ляют остатки окиси пропилена и вводят в реактор окись этилена
до достижения нужной длины цепи. Интервал молекулярных масс
этих продуктов составляет 1000—20 000 при молекулярной массе
Последняя цисрра. числового индекса
Рис. 96. Классификационная сетка полимеров окиси этилена и окиси пропилена
типа Pluronic.
Условные обозначения: L — жидкий; Р — пастообразный; F — твердый;
молекулярная масса полиоксипропиленового блока и содержание окиси этилена в сополи-
мере зашифрованы соответственно двумя первыми и последней цифрой числового индекса.
полиоксипропиленовой части не ниже 900 и содержании окиси эти-
лена 20—90%. На рис. 96 приведена классификационная таблица
сополимеров типа Pluronic.
Растворимость этих сополимеров в воде варьируется в широких
пределах. Они растворимы также в ароматических и хлорированных
углеводородах, спиртах, кетонах. Некоторые свойства отдельных
представителей этой группы сополимеров даны в табл. 74.
Известны аналогичные продукты, в которых внутренний блок
представляет собой полиоксиэтиленовую цепь; они отличаются по
свойствам от Pluronic.
* Отечественными (аналогами этих продуктов ^являются проксанолы 11
проксамины.
250
Таблица 74. Свойства некоторых товарйых сополимеров
окиси этилена и окиси пропилена [27, 56]
Toi-говое наименование Фупкцио- Hc'l льпость Молекуляр-' пая масса Вязкость при 25 °C, мПа- с Плотность, г/см3, при 25 °C 1— Точка мут- ности при 20 “С (в 10%-ном растворе)
£ л о к с о полимеры
pluronic L-31 II 1050 200 1,02 29
Pluronic L-35 » 1900 320 1,06 81
Pluronic L-44 » 2000 400 1,05 71 *
Pluronic L-61 » 1950 300 1,02 17
Pluronic L-64 » 2920 550 1,05 59
Pluronic P-84 » 3750 672 * — 73
Pluronic P-104 » 5400 620 ** — 70
Tetronic 304 IV 840 450 1,06 73
Tetronic 701 » 2800 575 1,02 17
Tetronic 704 » 4200 1 110 1,04 68
Tetronic 904 » 6000 4 200 1,04 66
Статистические сополимеры ***
Ucon-B-65 I — 11 0,963 —
Uco п-В-1715 » — 373 1,002 —
Ucun-50-HB-55 » 9 1,002 —
Ucon-50-HB-5100 » 1 180 1,063 —
Ucon-75-H-90 000 » 21 500 1,095 —
iPolyglycol-15-ЮО III 1000 112 1,070 —
Polyglvcol-15-200 » 2600 220 1,063 —
PolyglycoI-15-500 » 5000 514 1,052 —
Polyglycol-15-1000 » 9000 1 010 1,053 —
* При 35 °C.
** При 5 0 °C.
*** Вязкость при 3 °C.
Сополимеры типа Tetronic основаны на этилендиамине как старто-
вом веществе, и их строение можно представить формулой:
[II(C2H4O)i/(C3HeO)x],N(CH2)2N[(C3n6O)x(C2H4O)i,H]2
Они обладают высокой эмульгирующей способностью и особенно
эффективны в жесткой воде при пониженных температурах [23, 24].
В этих сополимерах блоки окиси пропилена, являющиеся носите-
лями гидрофобных свойств, имеют молекулярную массу от 200
До 6000, а содержание окиси этилена в них — от 20 до 90% по массе.
Некоторые свойства сополимеров Tetronic также представлены
в табл, 74.
Обе рассмотренные группы продуктов широко употребляются
в качестве поверхностно-активных веществ в агротехнике, текстиль-
ной, нефтяной, косметической и других отраслях промышленности,
а также при производстве каучуков. В последнее время их исполн-
яют и в качестве полупродуктов для полиуретанов.
251
Статистические сополимеры получили пока меньшее распростра-
нение. Среди товарных продуктов этого типа известны монофунк-
циональные производные спиртов и диолы (серия продуктов Ucon
[92]), триолы на основе глицерина Polyglycol-15 [93] и высокомоле-
кулярные продукты типа Pluracol-U [91]. Эти вещества применяются
как гидравлические жидкости, устойчивые к сдвиговым и терми-
ческим воздействиям, смачиватели для специальных целей, раствори-
тели, теплоносители и т. д.
Дополнительные возможности тонкой вариации свойств поверх-
ностно-активных веществ на основе простых эпоксидов возникают
при переходе от чисто блочных сополимеров к продуктам, содержа-
щим статистические блоки разного состава. Так, монофункциональ-
ные сополимеры Tergitol [94] содержат наряду с гидрофильной
полиоксиэтиленовой частью (44—55% по массе) статистический блок,
сильно обогащенный окисью пропилена (до 85—95%). Эти продукты
представляют собой бесцветные, водорастворимые вещества с тем-
пературой плавления 28—40 °C и высокой эмульгирующей способ-
ностью. Сополимеры Pluradot, синтез которых основан на па-
тенте [95], содержат два статистических блока: один преимущест-
венно полиоксипропиленовый с молекулярной массой 1500—3700
и соотношением ОП/ОЭ от 90 : 10 до 70 : 30 и другой — преимуще-
ственно полиоксиэтиленовый (25—45% по массе, ОЭ/ОП 90 : 10—
70 : 30).
Анализ сополимеров указанных типов включает определение
содержания звеньев той и другой структуры методами газовой хрома-
тографии с предварительным разрушением полимера [96] либо, что
более предпочтительно, методами ИК- и ЯМР-спектроскопии, кото-
рая дает надежные результаты. Измерение относительных интенсив-
ностей полос 2870 и 2975 см*1 в ИК-спектре раствора сополимера
в СС14 позволяет определять состав с точностью 5%. независимо от
молекулярной массы [97]. Предложен метод ЯМР для определения
состава, результаты которого удовлетворительно согласуются с дан-
ными химических методов анализа [98].
Несомненный интерес представляет анализ распределения моно-
мерных звеньев в цепи, так как физические свойства сополимеров,
а также продуктов на их основе, например полиуретанов, в сильной
степени зависят от характера чередования сомономеров. Для оценки
средней блочности сополимеров по концентрациям стыковых звеньев
используют ИК-спектроскопию при 10,3 мкм [99]. Использование
спектроскопии ЯМР — 1SC позволяет охарактеризовать последова-
тельность звеньев в сополимерах окиси этилена и окиси пропилена
набором соответствующих триад [100]. Имеются теоретические пред-
посылки создания ИК-спектрального метода, чувствительного к еще
более длинным последовательностям звеньев и основанного на
зависимости частоты или интенсивности некоторых полос в колеба-
тельном спектре от длины регулярного отрезка цепи [101].
В последнее время развиваются методы синтеза сополимеров
окисей с виниловыми мономерами [102]. В большинстве случаев
252
Г
не удается осуществить прямую статистическую сополимеризацию
в связи с тем, что механизмы полимеризации этих мономеров суще-
ственно различны. Более определенных результатов удается достичь
при синтезе блоксополимеров по анионному механизму на основе
«живых» полимеров в двухстадийном процессе или из полимеров,
содержащих функциональные группы, способные к активации под
действием анионных агентов. Это направление еще не вышло из
стадии исследований, но может представить интерес, помимо отме-
ченных выше причин, как способ создания полимерных композиций
непосредственно полимеризацией.
Отметим в этой связи синтез блоксополимеров «живого» поли-
стирола с окисью этилена [103]:
^Л,СН3—СН + (тг+1)Н2С СН2 >
--> <^СП2—СИ—[—СН2СН2О-]„-СН2СН2О-
Длины соответствующих блоков легко регулируются изменением
количества мономеров, поскольку передача цепи для этих систем
нехарактерна. Аналогичным образом могут быть, видимо, получены
сополимеры окисей с диеновыми полимерами.
Графтсополимеры (привитые сополимеры) окиси этилена полу-
чены на основе винилароматических полимеров: поливинилнафта-
лина, поливинилантрацена [104]. На первой стадии ароматические
группы восстанавливают щелочным металлом, а образующиеся
анион-радикалы присоединяют эпоксид
Подобным же образом, за счет превращения функциональных
(в основном ОН-групп) в алкоголятные, получены графтсополимеры
253
окиси этилена с крахмалом, кератином шерсти, фиброином шелка
и другими природными полимерами, что можно рассматривать как
перспективный путь модификации последних [105].
Наконец, весьма фундаментально развивается направление по
синтезу сополимерных диолов для полиуретановых целей, главным
образом на основе окиси пропилена. Введение окиси этилена, тетра-
гидрофурана и других сополимеров позволяет улучшить совмести-
мость олигомеров с другими компонентами при синтезе полиуретанов,
а также увеличить содержание более реакционноспособных первич-
ных ОН-групп.
Высокомолекулярные полимеры а-окисей
Синтез высокомолекулярных полимеров и их исследование пред-
ставляют собой второе крупное направление в полимерной химии
эпоксидов, развивающееся параллельно с олигомерным. Высоко-
молекулярные полиэпоксиды непосредственно проявляют тот соб-
ственно полимерный комплекс свойств, который может быть реали-
зован олигомерным путем, и имеют самостоятельные области
применения. Высокомолекулярные полиэпоксиды известны с сере-
дины 50-х годов, когда успехи в координационном катализе поз-
волили осуществить полимеризацию окиси этилена и окиси пропи-
лена в длинноцепные полимеры (молекулярные массы 105 и более),
тогда как предпринятые ранее такие попытки не дали результата.
В дальнейшем эти полимеры были детально исследованы как типич-
ные представители класса простых полиэфиров, а изучение про-
цессов их образования позволило значительно расширить возмож-
ности полимерной химии и привело к синтезу новых полимеров,
таких, как поли-2,3-эпоксибутан и ряд других. Практическое при-
менение полимеров рассматриваемого типа непрерывно расширяется.
Стереорегулярный полиоксипропилен
Полимеризация окиси пропилена в присутствии некоторых металло-
органических катализаторов, как отмечалось выше, ведет к стерео-
регулярным полимерам. В продуктах полимеризации содержатся
фракции, обладающие способностью к кристаллизации и предста-
вляющие собой изотактический полипропиленоксид, т. е. полимер,
в цепи которого мономерные звенья имеют одинаковую стереохими-
ческую конфигурацию. Относительная доля такого полимера в сум-
марном продукте широко варьируется подбором катализатора и
сокатализатора, их соотношением, температурой синтеза, средой
ит. п. Эти же факторы существенно влияют на другие свойства
полимеров, в частности на молекулярно-массовое распределение.
При синтезе полиоксипропилена наиболее широко применяются
каталитические системы на основе диалкилцинка и триалкилалюми-
ния, соединения железа, причем необходимым условием высокой
активности и стереоспецифичности является присутствие сокаталй'
254
заторов (вода, спирты, кислород, кетоны, сульфоксиды, хелатные
соединения и т. и.) [3, 15, 16]. Взаимодействие катализатора и со-
катализатора, как правило, многостадийное, отличается сложным
химизмом. Например, взаимодействие диэтилцинка с водой протекает
по схеме [106]:
(CaH5)2Zn + H3O —> C.,H5ZnOH + C2He
nC2H5ZnOH—> C2H5(ZnO)nH + (n-l)C2He
C2H5(ZnO)nZnC2H3 4- C2He HO(ZuO)nH + C2He
Продукты, получающиеся при разных соотношениях реагентов,
были выделены, идентифицированы и исследованы в качестве ката-
лизаторов полимеризации окиси пропилена. «Спектр» активных
продуктов может быть несколько сужен удалением избыточного
(C2H5)2Zn из системы; это приводит к более однородному продукту
полимеризации.
Аналогичные процессы происходят при взаимодействии спиртов
и других протонсодержащих сокатализаторов с (C2H5)2Zn, (С2Н5)3А1
и др. В табл. 75 представлены данные по полимеризации окиси про-
пилена в присутствии некоторых каталитических систем.
Таблица 75. Катализаторы стереоспецифической полимеризации
окиси пропилена [16, 107, 108]
Каталитическая система Концентрация ка- тализатора, % (мол.) 1 Растворитель (температура) Время полимериза- ции, ч Степень конвер- сии Характеристиче- ская вязкость при 25 °C (в бен- золе)
катализатор (I) сокатализа- тор (II) соотношение
(С2Н6)3А1 H.2O 0,5 : 1 0,88 5,8
(C4Hq)2^1 acac 1 : 1 1,о Бензол (50 °C) 20
H2O 1 : 1 1,0 То же 20 0,94 2,7
(C2H5)2Zn CHgSOCHg 1 : 1 0,2 Толуол (70 °C) 40 1,00 12,6
(C2H5)2Zn CH3NO2 0,6 : 1 1,0 То же 50 0,43 20,7
(C2H5)2Zn no"2 0,5 : 1 1,0 В массе (73 °C) Бензол (50 °C) 24 0,83 22,0
(C2H5)2Zn Гидразин 0,7 : 1 1,0 24 0,90 25,0
(C2H5)2Bc H2O 0,5 : 1 5,4 Гептан (70 °C) 20 0,81 7,0
((C4H90)2A10[2Zn — — 5,0 Гептан (50 °C) 0,25 0,50 10,0
* Лцетилацетонат.
** В расчете на мономер.
255
Представления о механизме стереоспецифической полимеризации
окиси пропилена имеют в качестве отправной точки ставшие уже
классическими работы Прайса с сотр. [65, 109], в которых показано,
что образующиеся макромолекулы должны состоять только из моно-
мерных звеньев одного знака (D или L). Эту точку зрения в какой-то
степени подтвердили позднее рентгеноструктурные исследования
полипропиленоксида [126] и, особенно, разделение рацемического
полимера на оптически-активные фракции [127].
В количественном отношении приближенность такой трактовки
очевидна. Следующим логичным шагом должно быть признание раз-
личной способности каталитических центров к отбору энантиомеров
в стадии роста и построение статистической модели полимеризации.
Такая модель — модель энантиоморфно спаренных центров раз-
работана [18], однако ее количественная проверка на примере поли-
оксипропилена наталкивается на ряд трудностей.
Одна из них — методическая, в принципе разрешена. ИК- и
ЯМР-методами удалось количественно охарактеризовать стерео-
регулярность полиоксипропилена, хотя неуспех ранних попыток
породил определенный скептицизм.
Методы магнитного резонанса на ядрах 1Н и В * * * * 13С [66] позволяют
оценить содержание структурных и стереохимических дефектов
в полимерной цепи. В табл. 76 приведены некоторые результаты
этих работ.
Таблица 76. Микротактичность полимеров окиси пропилена [66]
Катализатор Фракция полимера р, ₽** Из них
изо СИНДИО
кон Не фракционирован 0,91 0,53 0,47
[(С2Н5)2А1]2О Кристаллическая * 0,95 0,95 0,05 ,
Аморфная 0,65 0,70 0,30
Не фракционирован 0,68 0,84 0,16
(C2H5)2Zn — Н,0 Кристаллическая * 0,89 1,00 0,00
Аморфная 0,62 0,79 0,21
* Кристаллизация из ацетона при 0 °C.
** Доля диад с «нормальным» раскрытием цикла.
В ИК-спектроскопии для той же цели используется чувствитель-
ность некоторых полос поглощения к длине регулярных отрезков
цепи. Разработанный на этой основе метод анализа распределения
по длинам регулярных блоков предполагает измерение спектров
полимеров в растворе при низких температурах, когда макромоле-
кулы конформационно упорядочены. Получаемая информация
256
характеризует распределение регулярных блоков в полимере
по длине [112].
Более серьезным препятствием развитию количественной схемы
стереоспецифической полимеризации эпоксидов является резкая
структурная неоднородность полимеров и крайне широкое молеку-
лярно-массовое распределение. Об этом, в частности, свидетель-
ствуют данные по фракционированию [113], вынуждающие принять
почти непрерывное распределение каталитических центров по стерео-
специфичности, для чего нет достаточных химических и структурных
оснований.
Рис. 97. Структура поли-
мерных цепей изотакти-
ческого полиоксппропи-
лена и их расположение
в элементарной кристал-
лической ячейке.
Стереорегулярный полипропиленоксид имеет изотактическую
структуру, в которой метильные группы двух соседних звеньев
в отличие от виниловых полимеров расположены по разные стороны
плоскости цепи, что предполагает одинаковую конфигурацию асим-
метрического атома углерода. Полимер кристаллизуется в ячейке
орторомбического типа с параметрами а = 1,040, Ъ = 0,464, с =
= 0,692 нм [110]; элементарная ячейка относится к пространствен-
ной группе симметрии Р212121 и включает по два звена из двух
молекул (рис. 97).
Свойства полиоксипропилена определяются, способом и усло-
виями его синтеза. В табл. 77 приведены характеристики некоторых
стереорегулярных полимеров, полученных непосредственным син-
тезом либо методом фракционирования.
Для оценки степени кристалличности полиоксипропилена кроме
рентгеновских методов используют измерения плотности (плотности
кристаллического и аморфного полимеров равны 1,157 и 1,002 г/см3
соответственно) [83]. Стереорегулярность полимеров оценивают
также по температуре плавления [83, 114], исходя из уравнения
17 Заказ 972
Таблица 77. Свойства полимеров окиси пропилена [83, 114]
Катализатор Молекуляр- ная масса d?° -i Степень кристал- личности T. пл., °C р по формуле (3) ^ИЗО
CH3ONa 2000 1,002 -72
CH3ONa 2000 1,002 — -63 — —
КОН 3200 1,002 — —. — —
SrCO3 104—IO5 1,060 39-41 66,5 0,93 14
FeCl3-4C3HeO — — 49 75 0,992 120
(CnH5)2Zn—Н,0 — -— 34 68 0,93 14
(C2H5)2Zn-H2O* — — 15 66 0,92 12,5
(C.,H5)oZn-HoO ** 106 1,090 61 73 0,98 50
To же 1,8.10е 1,043 30 71 0,97 35
>> 1,3-10’ 1,021 14 66 0,93 14
1,1-10’ 1,015 10 58 0,86 7
* С добавкой аминов.
** Приведены данные для фракций полимера.
Флори, которое применительно к частично стереорегулярному поли-
меру принимает вид:
1 1 , в ,
Г То — ~ А// 111 р
где Т, То — температуры плавления реального и идеально изотакти-
ческого полимеров; АН — теплота плавления; р — вероятность изо-
тактического присоединения (или доля изотактических звеньев
в цепи). Этот метод, результаты расчета вероятностей и средних
длин изотактических последовательностей 1ИЗО = 1/(1 — р) согласно
которому приведены в табл. 77, дает качественную характеристику
полимеров.
Для расчета молекулярных масс полиоксипропилена по харак-
теристической вязкости (в дл/г) используют следующие значения К
и а в соотношении (2): 1,97 - КГ4 и 0,67 (гексан при 46 °C), 1,12-10'4
и 0,77 (бензол при 25 °C). 1,29>10"4 и 0,75 (толуол при 25 °C) [115].
Указанные значения коэффициентов справедливы в интервале моле-
кулярных масс от 30 тыс. до 5 млн.
Стереорегулярный полиоксипропилен растворим в ароматических
и хлорированных углеводородах, некоторых эфирах, спиртах. При
охлаждении его растворов в ацетоне, изопропиловом спирте, «-ге-
ксане, изооктане происходит последовательное выделение стерео-
регулярных фракций, что используется для разделения полимеров.
Полиоксипропилен подвержен различным видам деструкции:
термической и термоокислительной [116], под действием озона[64],
металлоорганических соединений [33], перекисей [90]. Деструкция
протекает по радикальному механизму с разрывом основной цепи
и резким падением молекулярной массы. Для предотвращения де-
струкции предложен ряд стабилизаторов, среди которых наиболее
эффективны фенолы и амины [116].
258
Направленная деструкция высокомолекулярного полиоксипро-
пилена под действием активных химических агентов может явиться
путем создания стереорегулярных олигомерных полипропиленокси-
дов. Наиболее интересны в этом отношении специфические
агенты, способные разрушать цепь по местам структурных или
стереохимических дефектов, поскольку в этом случае образующиеся
низкомолекулярные продукты будут в большей степени свободны
от нарушений регулярности, чем исходные макромолекулы. Такой
способ представляет несомненный практический интерес.
В настоящее время деструкция под действием некоторых агентов:
озона, литийбутила, галогенов используется для анализа структуры
цепи полиоксипропилена. Деструктирующий агент дозируют таким
образом, чтобы образовавшиеся при деструкции фрагменты, чаще
всего дигликоли, поддавались хроматографическому определению.
Например, при деструкции литийбутилом полиоксипропиленовой
цепи, содержащей структурные и стереохимические дефекты, обра-
зуется набор дигликолей (в скобках указаны возможные стерео-
химические комбинации, причем индексами R и S в схеме и табл. 78
обозначены абсолютные конфигурации асимметрических атомов угле-
рода в мономерах и дипропиленгликолях):
О —СН—СИ,—о—СН2 —СН—O-XZV
СНз
Нз: СНз
но-сн2-сн—о-сн2-сн-он
но—сн2—сн—о—сн—сн2—он
С Нз СНз
I (SS, RS-)
СНз
II (RS-)
I
но-сн-сн2-о-сн,-сн-он
СНз
Ш (RX-)
Результаты хроматографического анализа приведены в табл. 78,
где указаны данные параллельных опытов.
Информация, получаемая при анализе продуктов деструкции,
так же как ЯМР высокого разрешения, исчерпывающим образом
характеризует структуру цепи полимера. В сопоставлении с данными
ио оптической активности эти результаты свидетельствуют о том,
что нарушение чередования метильных групп в цепи, как правило,
совпадает с инверсией конфигурации. Этот результат более согла-
суется с энантиоморфным механизмом.
Изотактический полиоксипропилен существует также в оптически
активной форме, которая может быть получена тремя различными
17* 259
Таблица 78. Продукты деструкции аморфных фракций
полиоксипропилена литийбутилом [18]
Конфигура- ция моно- мера Каталитическая система Содержание дигликолей в смеси, %
I И III
Рацемиче- ский (C2H5).,Zn - Н2О 81,0 10,3 8,9
То же XC2H5)2Zn-H2O 69,7 15,3 15,0
» (C,H5)2Zn - Н,0 98,0 1,0 1,0
» FeCl3-4C3H6O 70,0 14,7 15,2
R (C„H5).2Zn — НоО 66,0 18,0 16,0
R (C,H5),Zn - Н,0 61,5 20,1 18,3
S (C,H5)»Zn — НоО 80,8 10,4 8,8
S FeCl3-4C3HeO 69,1 17,2 13,7
Ji FeCl3.4C3HeO 60,8 19,0 20,2
методами. Полимеризация, чистых оптических изомеров окиси про-
пилена на катализаторах анионного или координационно-анион-
ного типа приводит к наиболее оптически чистым молекулам. Так,
из Z-окиси пропилена в присутствии КОН или катализатора на основе
FeCl3 (катализатор Пруита — Баггета) получены практически иден-
тичные кристаллические полимеры: [а]р в хлороформе равна
+25 °C [65]. Аналогичные продукты получены и в присутствии
других катализаторов: Mg(C2H5)2, Zn(C2H5)2 — Н2О, (мзо-
С3Н7О)3А1 — ZnCl2 [109, 117]. Наивысшее значение оптической
активности, достигнутое при катализе КОН, составляет +24° в че-
тыреххлористом углероде и —34° в бензоле; этому полимеру при-
писывается абсолютная /^-конфигурация.
Полимеры различной степени изотактичности и оптической актив-
ности могут быть получены полимеризацией смесей оптических
изомеров. При этом оптические активности непрореагировавшего
мономера и образующегося полимера линейно зависят от активности
исходной смеси [117] (рис. 98).
Такая закономерность в рамках обычного кинетического урав-
нения сополимеризации D- и L-мономеров, очевидно, эквива-
лентна соблюдению условия:
г [Д>*]-т-[Z,*]
[£>*] + '[£*]
(4)
где D* и L* — центры соответствующей конфигурации-, г — кон-
станта сополимеризации (г = kDD/kDt = kLLlkLD).
Соотношение (4) возможно, если [D* ] = [L*] или г — 1. Ви-
димо, для всех металлоорганических катализаторов справедлив
первый случай, т. е. действительно образуется симметричная энан-
тиоморфная система активных центров. В случае КОН, напротив,
реализуется второе условие, т. е. катализ нестереоспецифичен.
В соответствии с этим различием оптическая активность щелочного
26 С
полимера является реальной мерой степени изотактичности каждой
цепочки, тогда как во всех остальных случаях она количественно
характеризует относительную долю макромолекул разной конфигу-
рации в суммарном продукте. Полимеризация оптических изомеров
осложняется частичной рацемизацией на центрах кислотного типа,
присутствующих в металлоорганических системах.
Второй метод синтеза оптически активных полимеров связан
с использованием асимметрических катализаторов, которые полу-
чают введением оптически активных добавок. В этом случае ката-
литическая система преимущественно содержит центры одной кон-
Рис. 98. Оптическая активность |а|у, непрореагировавшего мономера и обра-
зовавшегося полимера при сополимеризации D- и /.-окиси пропилена при
катализе (C2H5)2Zn — о, (C2H5)2Mg — • и (CH3O)2Zn — Д.
фигурации, что приводит к большему связыванию в цепь одного
из энантиомеров. При полимеризации рацемической смеси обра-
зуется оптически активный полимер, а в непрореагировавшем моно-
мере накапливается другой энантиомер [118]. Эффект разделения
изомеров в данном случае зависит от применяемого катализатора,
сокатализатора и глубины полимеризации. Так, используя диэтил-
Цинк в сочетании с d-борнеолом, авторы получили кристаллический
полимер с активностью -4-6,1—7,4° в хлороформе и мономер с [а]Ь° =
= —0,98°. Эти данные свидетельствуют о большом вхождении в цепь
^-мономера. Найти какую-либо связь между конфигурацией асим-
метрического сокатализатора и его способностью вовлекать в поли-
меризацию тот или иной энантиомер, однако, не удалось.
Третий способ синтеза состоит в непосредственном разделении
Полимера. Естественно, такое разделение возможно лишь в случае
Рацемической смеси изотактических макромолекул, которые обра-
зуются на металлоорганических катализаторах. Полимеры подобного
261
типа действительно удается разделить на противоположно враща-
ющие фракции, причем максимальное вращение полимера, полу-
ченного при использовании в качестве асимметрического сорбента
поли-(—)-ментилглицидилового эфира, составляло +6,4° в хлоро-
форме и —6,7° в бензоле [111],
По многим физическим показателям (температура плавления,
кристаллическая структура, плотность) оптически активные поли-
меры абсолютно идентичны своим рацемическим аналогам. Кристал-
лическая структура не препятствует упаковке цепей противо-
положной конфигурации в одной элементарной ячейке.
Высокомолекулярный, полиоксиэтилен
Полимеры окиси этилена с молекулярными массами в диапазоне
1—10 млн. впервые синтезированы в 1957 г. в лабораториях фирмы
Union Carbide [119], которая и сейчас является основным постав-
щиком и пропагандистом этого относительно молодого полимера.
В настоящее время высокомолекулярные полимеры окиси этилена,
известные под наименованием полиокс *, хотя и не являются крупно-
тоннажным продуктом, имеют прочные позиции в ряде областей
техники. Они служат эффективными загустителями, связующими,
хорошо совмещаются с рядом пластиков. Большой интерес при-
влекает высокая коагулирующая способность полиокса по отноше-
нию к минеральным суспензиям различной природы. Здесь он
успешно применяется наряду с полиакриламидом, иногда в компо-
зициях с ним.
Уникальные реологические свойства полиокса в водных раство-
рах привлекают все большее внимание в последнее время. Следует
специально отметить его способность резко снижать гидродинами-
ческое сопротивление, что уже находит широкое применение в прак-
тике. Ряд направлений в использовании полиокса только наме-
чается, но также сулит большие перспективы. Это, в частности,
касается водорастворимых пленок на его основе, а также комплексов
с полимерами и низкомолекулярными веществами.
В настоящем разделе рассмотрены специальные вопросы синтеза
полиокса, его свойства, а также вопросы применения.
Синтез высокомолекулярных полимеров окиси этилена требует
применения специальных катализаторов, в большинстве своем при-
надлежащих к координационно-анионному типу. В качестве актив-
ных катализаторов полимеризации первоначально было предложено
большое число веществ, главным образом производных металлов
II группы (Be, Mg, Са, Sr, Ва): карбонаты, окиси, гидроокиси, суль-
фаты, сульфиды, карбоновые соли [3, 13, 15].
Специфичным для этой группы катализаторов являются гетеро-
генный характер, высокие температуры полимеризации, необходи-
мость в адсорбционной активации поверхности добавками И?-'
* Polyox — торговая марка фирмы Union Carbide.
2,62
или С02, высокие требования к чистоте мономера. Ниже приведено
описание лабораторного варианта полимеризации окиси этилена
в присутствии SrCO3 [120].
Активный карбонат стронция получают осаждением из 23%-ного
водного раствора чистой Sr(OH)2 при 90 °C током СО2. Осадок от-
фильтровывают, промывают и сушат до содержания влаги 0,5—
1,0?6. Примеси некоторых анионов, например нитрата, делают ката-
лизатор неактивным. Активность катализатора понижается также,
если осаждение проводить при более низкой температуре и кон-
центрацци. Катализатор (0,2 г) помещают в стеклянную ампулу,
конденсируют туда же 50 мл чистой окиси этилена, после чего ампулу
запаивают и помещают в термостат при 50 °C. Индукционный период,
составляющий около 90 мин, сменяется быстро протекающей поли-
меризацией, которая полностью завершается за 2 ч. В некоторых
случаях реакция протекает со взрывом и требует специальных мер
предосторожности.
Активность карбоната стронция в значительной мере опреде-
ляется степенью заполнения поверхности катализатора адсорбиро-
ванной водой. Кинетика процесса носит сложный характер: период
резкого начального ускорения подвержен влиянию примесей и по-
лярных добавок; стационарная полимеризация протекает по первому
порядку относительно мономера. Концентрация активных центров
составляет примерно 1011 на 1 см2, что, скорее всего, отвечает кон-
центрации поверхностных дефектов. В пользу большой неоднород-
ности активной поверхности свидетельствуют также исследования
закономерностей активации катализатора при адсорбции
воды [121, 122].
Закономерности формирования активной поверхности катализа-
торов, а также механизм полимеризации окиси этилена подробно
исследованы на примере окисей и гидроокисей металлов II группы,
в основном Be и Mg [123]. Установлено, что каталитическая актив-
ность гидроокисей Be, Mg и Са в полимеризации существенно воз-
растает по мере их термической дегидратации, достигая максимума
вблизи состава, отвечающего чистой окиси соответствующего ме-
талла. Предложенный механизм, основанный на изучении активности
и спектров катализаторов, предполагает наличие на поверхности
Центров координации и активации мономера (ионы металла), обна-
жающихся лишь при сильной дегидратации образца. Вторая группа
Центров, которыми являются структурные ОН-группы поверхности,
Участвует лишь в адсорбции мономера, но не в реакции.
Гидроокиси Be и Mg, а также катализаторы, полученные при
Их термическом разложении в вакууме, обладают заметной актив-
ностью и при комнатной температуре, так как энергия активации
Полимеризации в этом случае ниже обычной. Тем не менее боль-
шинство рассмотренных катализаторов проявляют свою активность
лишь при повышенных температурах (80—100 °C). Полимеризация
в массе мономера или в растворе в таких условиях практически
г₽Удно осуществима, она сопровождается резким нарастанием
26 а
вязкости реакционной смеси, что значительно ухудшает условия
теплоотвода и гомогенизации системы. Блочный продукт с тру-
дом поддается дальнейшей переработке. Такой вариант процесса не
нашел, видимо, практического выхода.
В связи с этим предприняты поиски катализаторов, способных
эффективно вести полимеризацию при низких температурах (О—
30 °C), что позволило - бы осуществить процесс в суспензионном
режиме ниже температуры плавления полимера, используя в каче-
стве среды различные осадители. Преимущества такого технологи-
ческого варианта очевидны: разрешаются все трудности блочного
процесса, не требуется дополнительная переработка продукта, так
как он получается в виде мелких гранул или порошка. Его очистку
от катализатора можно осуществить экстракцией минеральной кис-
лотой в среде осадителя, используемого при полимеризации [124].
В качестве среды для проведения суспензионной полимеризации
наиболее предпочтительны парафиновые углеводороды (гептан, ге-
ксан), циклогексан, некоторые хлорсодержащие растворители [125].
Основным методом синтеза катализаторов для суспензионной
полимеризации являются реакции в жидком аммиаке как наиболее
универсальный способ получения органических производных ще-
лочноземельных металлов [126]. Так, в частности, синтезируют
алкоголяты и амидалкоголяты металлов, являющиеся активными
катализаторами полимеризации окиси этилена [127]. Способ обла-
дает рядом преимуществ перед прямым синтезом алкоголятов из
спиртов, однако и он не приводит к абсолютно чистым и однородным
продуктам, так как разложение гексааммиаката металла в растворе
до амида всегда сопутствует основному процессу. Гексааммиакаты
и амиды металлов также активны при полимеризации окиси эти-
лена [128]. Все эти катализаторы дополнительно активируются
такими добавками, как нитрилы, сульфоксиды, трифенилметан,
флуорен и рядом других, позволяющими одновременно регулировать
молекулярную массу полимера [3, 129].
Суспензионная полимеризация является в настоящее время
основным способом синтеза высокомолекулярных полимеров окпси
этилена, хотя какие-либо детали технологического осуществления
процесса в литературе отсутствуют. Процесс имеет и ряд сложно-
стей, одной из которых является стабилизация растущей суспензии
против коагуляции и сохранение достаточно высокой дисперсности
полимерного продукта до конца полимеризации. С другой стороны,
немаловажным обстоятельством является необходимость диффузии
мономера к активной поверхности через пленку образующегося
полимера, толщина которой непрерывно увеличивается. Подобные
проблемы возникают в синтезе полиолефинов.
Кинетические исследования суспензионной полимеризации окиси
этилена под действием амидалкоголята кальция или системы три-
изобутилалюминий — диметилглиоксим [130] показывают, что
скорость полимеризации резко падает в ходе процесса, в результате
чего общая производительность не превышает 60—70 г полимера
264
на 1 г Са; полимеризация проводится в бензине при 30 °C с образо-
ванием порошкообразного полимера с молекулярной массой 2—
5 млн.
Из других катализаторов следует отметить различные алюминий-
органические соединения, модифицированные количественным гидро-
лизом, добавками хелатов, а также нанесенные на поверхность
твердых носителей (А1,О3, силикагель и т. п.). Цинк- и магний-
органические соединения как катализаторы высокомолекулярной
полимеризации используются в практических целях [15, 16, 131].
Они обладают весьма высокой активностью, но, сохраняя даже
после модификации сокатализатором кислотный характер, парал-
лельно генерируют низкомолекулярные фракции либо приводят
к реакциям разрыва цепи. В табл. 79 приведены данные по поли-
меризации окиси этилена на катализаторах различного типа.
Таблица 79. Катализаторы высокомолекулярной полимеризации
окиси этилена [107, 130—132]
Каталитическая система Концентрация, % (мол.) Растворитель (температура, °C) Время реакции, ч Выход полиме- ра, % Характеристи- ческая вязкость, дл/г * (в ацето- нитриле при 30° С) Молекулярная масса, млн
NH2CaOC2H5 1,1 Бензин * (17) 20 61 3,0
Ca(NH2)2 (u?o-C4H9)8A1 — диметплгли- оксим (1 : 1) 1,1 Бензин * (10) 7 35 — 10,0
2,1 Бензин * (30) 20 95 — 5,0
Zn(C2H5)2 - (CH3)2SO (1 : 1) 1.0 Толуол (70) 15 68 22,7 —
Zn(C2Hj), - CH3NO„ (0,5 :1) 1,0 Толуол (70) 21 68 37,5 —
To же 1,0 Гептан * (70) 17,5 100 30,0 —
Zn(C2H5)2-CeH5NO (1 : 1) 0,5 Толуол (30) 193 100 59,0 —
(mpem-C4H9O)2Zn 0,5 Бензол (60) 24 23 19,4 ** 6,9
* Полимер не растворим, полимеризация в суспензии.
** В бензоле при 25 °C.
Таким образом, несмотря на обилие каталитических систем,
лишь немногие вполне соответствуют требованиям технологии по
активности или производительности. Что касается фундаментальных
йаучных проблем синтеза высокомолекулярного полиоксиэтилена,
io эти вопросы в литературе практически не рассмотрены.
Свойства полиоксиэтилена. Полиоксиэтилен приобретает харак-
терные для высокомолекулярных полимеров пластические и реоло-
гические свойства в области молекулярных масс порядка нескольких
сотен тысяч. Именно к этим полимерам применимо понятие «поли-
окс», тогда как термин «полиоксиэтиленгликоль» относится к поли-
мерам с молекулярной массой не выше 60 тыс. и гарантированной
бифуцкциональностью по гидроксильным группам. В химическом
265
отношении оба понятия идентичны. В табл. 80 приведены молеку-
лярная масса и вязкость известных в настоящее время марок высоко-
молекулярных полимеров окиси этилена [133].
Таблица 80. Свойства некоторых высокомолекулярных полимеров
окиси этилена [133]
Марка полимера Молекулярная масса, тыс Вязкость 5%-ного водного раствора при 25 °C, Па - с
Polyox (Union Carbide, США)
WSR-N-10 100 10—20
WSR-N-80' 200 55-95
WSR-N-750 300 550-900
WSR-N-300 600 2 250-3 350
WSR-35 200 520-900
WSR-205 600 4 100-8 000
WSR-301 4000 1 500-3 500 **
Coagulant >5000 >4000 **
FRA * — —
Alcox (Meisei Chem., Япония)
.Е-30
Е-4.5
Е-60
900—3 000
4 000—10 000
10 000-40 000
* Friction reduction agent.
** В 1%-ном водном растворе.
Полиокс представляет собой порошкообразный или мелкограну-
лированный продукт белого цвета. Он содержит небольшие количе-
ства катализатора — не более 1% в расчете на СаО. В условиях
совершенной технологии и высокоэффективного катализа зольность
полимера может быть снижена до уровня, не требующего дополни-
тельной обработки полимера. Как отмечалось, остатки катализатора
извлекают из полимера кислотной экстракцией в среде использу-
емого для полимеризации осадителя.
Полиокс является термопластичным полимером, температура
плавления 65—67 °C, плотность около 1,21—1,22 г/см3, кристал-
личность в обычных условиях до 95%. Высокая кристалличность
обусловливает низкую гигроскопичность полиокса; при относитель-
ной влажности до 60% содержание влаги в полимере не превышает
2% и лишь выше 80%-ной влажности полиокс становится гигро-
скопичным и резко теряет прочность в связи с поглощением влаги-
Полиокс хорошо растворим во многих растворителях. Растворы
полимеров с молекулярной массой около 4 млн. в воде, ацетонитриле,
этилендихлориде, четыреххлористом углероде, трихлорэтилене
устойчивы до 0 °C. Критические температуры растворения полиокса
266
VFSI2-3O1 при 1%-ной концентрации в других распространенных
растворителях приведены ниже [133]:
Температура выпадения при охлажде- нии Температура растворения при нагрева- нии выше 25 °C Температура выпадения при охлажде- нии Температу ра растворения при нагрева- нии выше 25 °C
Бензол , . . 2 — 1,4-Диоксан 4 40
Диметилформ- Этилацетат . . 25 40
амид .... 12 — Диметилцелло-
Тетрагидрофу- зольв .... 27 40
ран .... 18 — Бутиловый
Этиленкарбонат, спирт .... 33 50
метиловый Изопропило-
спирт, метпл- вый спирт 37 50
этилкетон . . 20 — Метилпзобутил-
Толуол,ксилол 20 30 кетон .... 40 50
Ацетон . . , 20 35 Диэтилцелло-
Анизол 0 40 зольв . . . 46 50
Высокомолекулярный полиоксиэтилен не растворим в парафино-
вых углеводородах и полиспиртах — этиленгликоле, диэтилен-
гликоле, 1,3-бутандиоле, глицерине и т. п.
Растворимость полиокса в кетонах, спиртах, гликолях и их
эфирах заметно повышается при добавках воды. Растворы в пара-
финовых углеводородах могут быть полуиены введением до 10%
третьего компонента — ароматических и хлорсодержащих соедине-
ний, нитрилов. Добавки метилового спирта, в свою очередь, повы-
шают растворимость полиокса в бензоле при низких температурах.
В водных растворах для полиокса характерно наличие верхней
предельной температуры растворимости, выше которой полимер
выпадает из раствора в виде хлоньев или объемистого геля. Предель-
ная температура зависит от концентрации и молекулярной массы
полимера, а также присутствия в растворе электролитов, высалива-
ющих полимер (рис. 99). Такое поведение согласуется с мнением, ч
что растворимость полиоксиэтилена в воде обусловлена образова-
нием водородных связей, которые удерживают макромолекулу в ли-
лейном состоянии.
Повышение температуры, межмолекулярные взаимодействия, на-
пример комплексообразование, а также введение солей разрушают
водородные связи, и макромолекулы переходят в состояние клубка,
а затем образуют новую фазу. Об этом очень ярко свидетельствует,
в частности, параллельное изменение предельной температуры и
вязкости растворов под влиянием электролитов (рис. 100).
Ограниченная растворимость некоторых полиэтиленгликолей
в воде обусловлена существованием еще одной критической темпе-
ратуры в области высоких концентраций [134]. Очевидно, полимер
выпадает в виде кристаллической фазы. Существование этой ветви
Фазовой диаграммы у высокомолекулярных полимеров не уста-
вовлено.
267
Термодинамика водных растворов полиокса детально не исследо-
валась. Измерения теплот смешения полиокса с хлороформом и ме-
тиленхлоридом позволили рассчитать теплоту плавления полимера
Рис. 99. Зависимость температуры выпадения полиокса из водного раствора:
а) от концентрации полимера для различных молекулярных масс; б) от концен-
трации солей для полиокса И5А-301, 0,5%.
в растворе, которая составляет в обоих случаях около 239 Дж/Г
(57 кал/г). Теплоты разбавления водных растворов приводят к прин'
ципиально иной величине, равной 74,7 Дж/г (17,8 кал/г). С учетов
различного состояния полимера в этих растворителях найдена те-
268
плота перехода спираль — клубок для полиоксиэтилена, равная
164 Дж/г (39,2 кал/г) [135].
Исследования растворов полиокса указывают на существование
в них надмолекулярных частиц, что может быть причиной некоторых
аномалий в свойствах. Так, электронномикроскопическое изучение
частиц, полученных лиофильной сушкой распыленных разбавленных
растворов полиокса (молекулярная масса около 200 тыс.) в воде
и диметилформамиде, приводит к размерам частиц до 100 нм [136],
что в 5—10 раз превышает оценки размера индивидуальных макро-
молекул. К аналогичным выводам приводят исследования свето-
рассеяния растворов, которым приписывается микрогетерогенный
характер [137]. Видимо, межцепная ассоциация является причиной
Рис. 100. Корреляция
между температурой вы-
падения полиокса и его
характеристической вяз-
костью [г]] в воде при
35 °C с добавкой различ-
ных солей (точки отно-
сятся к различным кон-
центрациям и типам со-
лей).
этих эффектов, причем полиокс может вступать в подобные взаимо-
действия с другими макромолекулами, образуя прочные комплексы.
Аномально высокие значения вязкостей растворов полиокса
являются основой многих применений полимера. На рис. 101 пред-
ставлена концентрационная зависимость вязкости растворов разных
марок полиокса. По вязкости растворов полиокс намного превышает
Другие водорастворимые полимеры [60]. Концентрированные рас-
творы полиокса представляют собой гели, а при концентрации более
5% — твердые растворы. Вязкость растворов полиокса понижается
при введении солей, но в интервале pH от 2 до 12 практически не
Изменяется.
Растворы высокомолекулярного полиокса при любых концентра-
циях имеют ярко выраженный псевдопластический характер, об-
условленный большими линейными размерами макромолекул или
0Х ассоциатов, а также их высокой гибкостью. Типичным проявле-
нием этого является зависимость вязкости от скорости сдвига, кото-
Рая становится более сильной с увеличением концентрации и моле-
Улярной массы полимера.
Сдвиговая зависимость вязкости сохраняется у растворов поли-
са вплоть до самых низких концентраций, и ее следует иметь в виду
269
при измерениях молекулярной массы вискозиметрическиц
методом [133, 138]. В связи с этим рекомендуется применение рота-
ционных или капиллярных вискозиметров, допускающих экстра-
поляцию к нулевой скорости сдвига, а также использование при
расчетах коэффициентов, полученных с учетом сдвиговой зависи-
мости; такие коэффициенты в соотношении (2) между характеристи-
ке. 101. Зависимость
вязкости водных раство-
ров различных марок
полиокса при 25 °C от
их концентрации:
1 — WSR-1V-10; г —
WSR-jV-80; 3—VFSR--V-750,
WSR-35; 4 -- WSR-jV-3000;
5 — WSR-205; 6 — WSR-
301; 7 — Coagylant.
ческой вязкостью и молекулярной массой для полиокса, по дан-
ным [53, 138—140], приведены ниже:
Растворитель
К • 104, дл/г
Вода
при 25 °C .............................. 1,03 0,78
при 30 °C .............................. 1,25 0,78
при 35 °C .............................. 0,64 0,82
при 45 °C .............................. 0,69 0,81
0,45 М водный раствор K2SO4 при 35 °C . . 13,0 0,50
Бензол при 25 °C ........................... 3,97 0,686
Для характеристики молекулярной массы полиокса используют
также светорассеяние [138, 1401 и седиментацию [139, 141]; соотно-
шение седиментационной константы в водном растворе и молекуляр'
ной массы имеет вид:
S0=l,26- 10-15М0Л1
Молекулярно-массовое распределение высокомолекулярно10
полиоксиэтилена систематически не исследовано. Согласно ДаН'
ным [141], полученным методом скоростной седиментации в водн*1*
растворах, ширина ММР лежит в пределах 1,18—2,19, причем не
возможно установить какой-либо корреляции с условиями синтез3-
270
Методика получения и анализа функций ММР полиокса еще,
по существу, не отработана. Применение традиционных методов
препаративного фракционирования в различных системах раствори-
тель — осадитель либо при понижении температуры [142] наталки-
вается на ряд серьезных трудностей, связанных со спецификой
выделения кристаллизующихся полимеров. Лишь в условиях жидко-
фазного разделения выше точки плавления полимера возможно
получение правдоподобных результатов. На базе этого в последнее
время создан новый метод получения функций ММР — осаждение
полимера из водно-солевых смесей при повышении темпера-
туры [143].
Интерес к функциям ММР полиоксиэтилена несомненен, по-
скольку они являются ключом к механизму процессов синтеза и де-
струкции полимера.
Пластические свойства полпокса привлекли значительный инте-
рес сразу после первой публикации, несмотря на то, что трудно
было ожидать серьезных прямых применений полимера в связи
с его низкой температурой плавления и высокой растворимостью.
Полиокс характеризуется высоким удлинением, гибкостью и тенден-
цией к ориентации под нагрузкой. Ниже приведены основные проч-
ностные характеристики высокомолекулярного полиоксиэтилена на
примере nz57?-301 [56. 133]:
Разрушающее напряжение при растя-
жении, МПа (кгс/см2) ..............
Относительное удлинении при разрыве
%.............................
Предел текучести, МПа (кгс/см) . .
Упругое удлинение, % ..............
Модуль упругости, МПа (кгс/см2) . .
Модуль кристаллической решетки* *,
МПа (кгс/см2) .....................
Ударная вязкость (по Изоду) ....
Твердость (по Шору)................
Температура, °C
плавления ........................
хрупкости .....................
12,8-16,7 (130-170)
700-1200
6,9—9,8 (70—100)
5-30
196—490 (2000—5000)
9,8-10® (105)
Без разрушения
99 (шкала А)
66
—50
* Найдено из деформации кристаллической решетки в направле-
иии основной цепи по данным [144].
По своим механическим свойствам высокомолекулярный поли-
оксиэтилен подобен полиэтилену среднего давления, но имеет зна-
чительно более ярко выраженную термопластичность. В области
температур 65—70 °C полиокс резко теряет прочность, однако и
8Ыше температуры плавления сохраняет каучукоподобные свойства
® исключительно высокие значения вязкости: величины порядка
*0* Па • с (105 Пз) являются нормальными для полиокса в расплаве
цРи температурах на 100—150 °C выше точки плавления.
На рис. 102 приведены данные разных авторов [56, 145] по вяз-
°сти полимеров окиси этилена в широком интервале молекулярных
271
масс при температурах 80 и 125 °C. Эта зависимость, в частности
предельные ее наклоны в области низких и высоких молекулярных
масс (0,88 и 3,4 соответственно), хорошо согласуется с теорией,
однако точка перегиба, отвечающая началу перепутывания цепей,
наступает у полиоксиэтилена слишком рано — при длине цепи
в 100 звеньев.
Расплавы полиокса псевдопластичны и характеризуются резкой
зависимостью вязкости от скорости сдвига. Температурная зависи-
мость вязкости несколько нивелируется с ростом молекулярной
экструзией, литьем, каландрованием рекомендуют область характе-
ристических вязкостей 2,5—3,0 дл/г, где, повышая температуру,
еще можно достичь некоторого снижения вязкости.
В твердом состоянии полиокс имеет, по данным ЯМР, измере-
ниям плотности и рентгеновского рассеяния, кристалличность до
95%. Кристаллическая структура и конформация цепи полиокса
полностью идентичны наблюдаемым для полиэтиленгликолей
(см. стр. 233) [146].
Были исследованы закономерности кристаллизации высокомоле-
кулярного полиоксиэтилена из расплава, в том числе в присутствии
разбавителей, и из растворов, а также образующиеся при этом над-
молекулярные структуры [34, 147—149]. Кинетика кристаллизации
полиоксиэтилена описывается, как обычно, уравнением Аврами:
X~\ — e~ktn
где X — измеряемая каким-либо методом степень кристалличности!
t — время от начала кристаллизации. Показатель степени и, опр8'
деляемый механизмом кристаллизации и типом растущих структур’
варьируется для полиоксиэтилена от 1,5 до 4 в зависимости от прг
272
роды образцов, их молекулярной массы, полидисперсности и т. п.
В случае кристаллизации узких фракций из этилового спирта п = 4,
что характерно для невозмущенного роста трехмерных сферолитов.
Морфологически полиоксиэтилен состоит обычно из крупных
сферолитов, складывающихся из ламелларных образований, т. е.
крупных ассоциатов параллельно расположенных цепей. Низко-
молекулярные полимеры образуют ламеллы из вытянутых линейных
макромолекул, но с ростом молекулярной массы в области 6—10 тыс.
цепи начинают складываться. В этой же области наблюдается резкий
минимум скорости кристаллизации.
Фибриллярные кристаллы полиоксиэтилена образуются при кри-
сталлизации из перемешиваемых растворов в этиловом спирте при
температурах 31 — 37 °C [149]. Макромолекулы в этом случае ориен-
тируются в гидродинамическом поле, причем важную роль играет
их высокая гибкость. Температуры плавления фибриллярных кри-
сталлов, как правило, ниже, чем у полученных из расплава, и воз-
растают с температурой кристаллизации.
Релаксационные свойства полиокса исследованы методами ЯМР,
диэлектрических и механических потерь [150, 151]. В низкотемпе-
ратурной области все методы приводят к близким значениям для
максимума потерь около —55 °C при частоте 1 Гц [150]; частота
этого максимума растет с температурой, как обычно, экспоненци-
ально, энергия активации составляет 250—290 кДж/моль (60—
70 ккал/моль).
Другой максимум потерь наблюдается в области плавления
полиокса, но его трактовка менее однозначна. Предполагается,
что оп обусловлен подвижностью сегментов в аморфных областях,
участвующих в процессе рекристаллизации. При температуре пла-
вления, согласно динамомеханическим испытаниям, резко падает
сдвиговый модуль и растет механический индекс затухания.
При измерении электропроводности полиоксиэтилена на воздухе
ив вакууме получены значения 10"9 Ом"1-см"1, т. е. на 9—10 поряд-
ков превышающие аналогичные величины других валентпо насыщен-
ных полимеров — полиоксиметилена, полиэтилена, полипропилена
и т. п. [152]. Предполагается, что проводимость обусловлена про-
тонсодержащими примесями (например, водой), прочно удержива-
емыми полимерной цепью и транспортируемыми эстафетным
механизмом. Значительный вклад здесь опять-таки вносит высокая
Подвижность сегментов цепи; в области температуры плавления
Проводимость возрастает в 30—40 раз. Аналогичные свойства
отмечены у политриметиленоксида, близкого к полиоксу по свой-
ствам.
Химические свойства высокомолекулярного полиоксиэтилена
в основном идентичны свойствам низкомолекулярных аналогов,
Исключая реакции по концевым группам. Носителями химической
ирироды являются простые эфирные группы, с одной стороны, склон-
Jne к разрыву по различным механизмам, с другой, проявляющие
Псктронодонорные свойства.
18 Заказ 972 273
| Образование комплексов полпокса с различными низкомолекуляр-
ными соединениями и полимерами является характерным продолже-
нием их склонности к различного рода меж- и внутрпцепной ассо-
циации, образованию водородных связей. Комплексы полиокси-
этилена являются объектом пристального внимания как возможный
метод модификации и синтеза новых полимерных веществ. Найден
ряд интересных применений для этих продуктов.
Хорошо изучены закономерности образования и свойства ассо-
циатов полиокса с HgCl2 [153, 154], карбамидом и тиокарбами-
дом [155], акриловой и метакриловой кислотами [156]. Способы
получения комплексов различны — соосаждение из растворов,
выдерживание пленки полиокса в растворе другого реагента, на-
пример HgCl2, прокатывание на вальцах и т. п.
Комплекс полиокса с карбамидом содержит две молекулы карб-
амида на мономерное звено, не растворим в бензоле, имеет высокую
кристалличность и температуру плавления около 145 °C. Комплекс
с тиокарбамидом того же состава образуется более медленно. Оба
комплекса растворимы в воде. Исследование ИК-спектров ком-
плексов позволяет предположить изменение конформации полимера
при комплексообразовании, о чем свидетельствуют также изменения
вязкости.
Более детально структура комплексов исследована на примере
HgCl2. Комплекс состава [—СН2СН2О—]4 • HgCl2 кристаллизуется
в ячейке ортобромического типа с параметрами а = 1,355, b =0,858,
с = 1,175 нм, содержащей 4 молекулы HgCl2 и 16 мономерных
звеньев из 4 цепей [153]. Таким образом, каждая молекула HgCI2
координирована с четырьмя кислородами из разных макромолекул.
Конформация цепи полимера в этом комплексе TfiT\G, где G и G
обозначают правую и левую гомл-конформации. Исходный полимер,
как известно, имеет ГГС-конформацию.
Второй кристаллический комплекс также имеет ячейку орто-
ромбического типа (а = 0,775, b = 1,209, с = 0,588 нм), в которую
входят 4 молекулы HgCl2 и 4 звена от двух макромолекул [154].
Конформация цепи полимера в этом случае TG.,TG2. Структуры
обоих комплексов подтверждены измерениями плотности и расчетом
ИК-спектров. Комплексообразование с HgCl2 значительно понижает
гибкость макромолекул полиоксиэтилена, увеличивая барьеры вну-
треннего вращения; образующиеся пленки хрупки, комплексы не-
растворимы в воде.
Образование комплексов за счет ассоциации макромолекул поли-
окса с поликислотамп представляет собой удобный объект для из-
учения подобных кооперативных взаимодействий в растворе. На
рис. 103 показаны закономерности изменения вязкости бинарны*
систем полиокс — полиакриловая кислота (а) и полиокс — полИ"
метакриловая кислота (б) [156]. Как видно, в обоих случаях состав
близок к стехиометрическому (1 : 1), т. е. комплексы образуются
при взаимодействии карбоксильных групп с эфирным кислороде51,
274
Характер образования комплексов практически не зависит от моле-
кулярной массы, но чувствителен к pH в связи с его влиянием на
диссоциацию карбоксильных групп и конформацию молекул
(рис. 103, в).
Поликомплексы являются перспективной формой практического
использования полиокса. Обсуждаются возможности применения
поликомплексов в медицине в качестве инплантантов, в агротехнике,
для разделения суспензий, т. е., по существу, в тех же областях,
Концентрация полиокса., %
Рис. 103. Изменение вязкости
при образовании комплексов
полиокса в водных растворах:
а — полиакриловая кислота, 30 °C,
pH = 4, концентрации 0,05 (1),
0,1 (2), 0,15 (3) п 0,2% (4); б —
полиметакриловая кислота, 30 °C,
pH — 5,3; в—комплекс с полиакри-
ловой кислотой, 1:1, 25 °C. Ско-
рость сдвига при измерениях вяз-
кости 0,1 с-1.
где применяется собственно полиокс. Возможности вариации рас-
творимости и скорости растворения полиокса за счет комплексо-
образования, совмещения с другими пластиками, а также облучения
несомненно расширяют перспективы его применения.
Деструкция с разрывом цепи по эфирным связям — другой
характерный для полиокса тип химических процессов. Такой разрыв
происходит под действием большого числа физических и химических
факторов. Пиролиз полиоксиэтилена заметно происходит при темпе-
ратурах выше 300 °C. Согласно [157], 30-минутная экспозиция
в вакууме приводит к потере полимером 7,3% массы при 324 °C,
52% — при 347 °C и 98,6% — при 363 °C. Кинетическая кривая
имеет индукционный период; начальные скорости пиролиза 0,33%
в мин при 320 °C и 0,86% при 335 °C. Энергия активации, найденная
по скоростям, составляет около 192 кДж/моль (46 ккал/моль). В га-
зообразных продуктах разложения полиоксиэтилена обнаружены
18* 275
формальдегид, этиловый спирт, СО2, Н2О и низкомолекулярные
фрагменты цепи.
Кинетические данные по деструкции полиокса в кислороде при
70—160 °C свидетельствуют о радикальноцепном механизме с вы-
рожденным разветвлением [116, 158]. Процесс протекает автоуско-
ренно с промежуточным образованием гидроперекисей. Действие
антиоксидантов на окисление характерно для подобных процессов;
выше некоторой предельной концентрации окисление происходит
стационарно и крайне медленно, ниже — автоускоренно. В качестве
антиоксидантов наиболее эффективны 2,2'-метилен-бис(4-метил-6-
третбутилфенол), пропилгаллат, ]\’-фенил-К-циклогекспл-тг-фенилен-
диамин, стабильные радикалы.
Основными продуктами окисления являются Н2О, СО2, форм-
альдегид, ацетальдегид и формиаты. Стадийная схема окисления
дополнительно подтверждена опытами с изотопом 18О; в частности,
показано, что ацетальдегид в основном содержит кислород цепи,
тогда как СН2О и СО2 — из газовой фазы. Исследовано изменение
ИК-спектров полимера в ходе окисления [158].
Предложен большой ряд антиоксидантов, надежно защищающих
полиокс от окислительной деструкции [158, 159]. Их основная
функция состоит в предотвращении образования перекисей, которые,
как показано [160], являются затем источником разрыва цепи даже
в отсутствие кислорода: аналитически определенное количество
перекисей хорошо соответствовало числу разрывов цепи, найден-
ному из вязкости. Распределение перекисных групп по цепи, видимо,
носит случайный характер , поскольку после деструкции полимер
имеет ММР, характерное для статистического разрыва цепи
= 1,8 —2,0).
Более важным С точки зрения основных применений полиокса
являются процессы деструкции в растворах. Полиокс разрушается
в водном растворе с потерей вязкости под действием кислорода,
озона, надкислот, перекисей, С12, Вг2, перманганатов, персульфатов
и других окислителей [58, 161]. Деструкция ускоряется в кислых
средах. Данные по потере вязкости растворов полиокса под дей-
ствием различных агентов, а также по влиянию некоторых стабили-
заторов приведены ниже [58]:
Химический агент* Потеря
вязкости,
(I/
/О
Надуксусная кислота ... 99
КМпО4 ..................... 97
Н2О, ...................... 96
Вг2 "..............» . . • 92
FeSO4 ..................... 96
CuCl2 ..................... 74
Боз добавок................ 10
Стабилизатор Потеря
вязкости,
%
Аллиловый спирт............ 7,6
Изопропиловый спирт . . . 10.0
Этиловый спирт .......... 12,5
Этиленгликоль ............ 20,0
Метиловый спирт.......... 50
трспг-Бутиловый спирт . . 67
Без добавок.............. 64
* 0,01% агента, 1 г полиокса WSH-301, 100 г НгО, 2 дня, комнатная температура.
276
г
Спирты стабилизируют полиокс также от воздействия Н2О2,
надкислот, света. Ультрафиолетовое облучение ускоряет деструкцию
полиоксиэтилена. Особенно резкий эффект наблюдается в присут-
ствии некоторых ионов. Например, характеристическая вязкость
полимера в воде падает за 24 ч от 1,60 до 0,3 дл/г в присутствии
4,7 ммоль/л Fe3+, а при дополнительном облучении до 0,13 дл/г [161].
В случае Сг3+ эффект ускорения светом выражен менее резко. Ком-
бинация ионов переменной валентности с Н2О2 или С12 также при-
водит к сильной деградации.
В качестве дополнительных мер по стабилизации растворов
полиокса против окисления рекомендуется использование деиони-
зованной воды, буферных растворов с pH = 8 и более, введение
деактивирующих катионов типа Мн2+, образующих комплекс с поли-
мером. Фотохимическую стабильность повышают введением сали-
цилатов, меркаптанов, дитиокарбаматов, ксантатов, фосфитов и дру-
гих известных светостабилизаторов [162].
Радиационная деструкция полимеров окиси этилена под дей-
ствием у-лучей [163] идет с частичным разрывом цепи и образова-
нием гель-фракции. Отношение вероятностей разрыва и сшивки
близко к 0,6 и зависит от температуры. Высокомолекулярные поли-
меры образуют гель-фракцию при более низких дозах. Несомненно,
облучение может быть наряду с другими способами перспективным
методом модификации полиокса, в частности, повышения его тепло-
стойкости и снижения водорастворимости.
Механохимическая деструкция является, пожалуй, наиболее
характерным для полиокса процессом снижения молекулярной
массы в растворах, а также самым серьезным препятствием или
ограничением для его успешного применения в ряде важных отраслей
техники [164, 165]. Деструкция полимера в сдвиговом поле различ-
ной конфигурации обусловлена большими линейными размерами
макромолекул, причем в наибольшей степени затрагивает самую
высокомолекулярную часть полимера.
Деструкцию полимера наблюдают в реовискозиметрах с коакси-
альными цилиндрами, при истечении через капилляры при Re =
= 8500—13 000, в условиях высокоскоростного перемешивания
(30 000 об/мин), в центробежных насосах и т. п. Процесс осущест-
вляется, видимо, по радикально-цепному механизму, поскольку
в условиях мехаподеструкцип в бензоле полиокс вызывает полимери-
зацию метилметакрилата [164]. Радикальные ингибиторы, однако,
не влияют на интенсивность деструкции.
Молекулярная масса полиокса падает при механодеструкции
По закону первого порядка независимо от концентрации и исходной
длины цепи [24, с. 805—807] (рис. 104) деструкция подчиняется
Уравнению:
1 1
. где М, Мо — молекулярные массы; ц — вязкость среды; D — гра-
диент сдвига, t — время деструкции; ИМ эквивалентно числу
277
разрывов цепи. Скорость процесса зависит только от общего подвода
энергии в системе. ц
Высокая чувствительность полиокса к сдвиговым воздействиям
вынуждает применять специальные методы для приготовления рас-
творов [133]. Рекомендуется, например, предварительное пригото-
вление паст илп суспензий полимера в «плохих» растворителях —
изопропиловом или этиловом спиртах, глицерине, концентрирован-
ных растворах солей — и их последующее смешение с водой. При
этом используют специальные конструкции безградиентных мешалок.
Предварительное набухание полимера является, видимо, основным
фактором в таком способе, поскольку позволяет значительно снизить
давление набухания внутри полимерной глобулы, способное при-
водить к деструкции непосредственно в стадии растворения.
Рис. 104. Изменение мо-
лекулярной массы поли-
оксиэтилена при механо-
деструкции в водном
растворе (концентрации
полимеров указаны на
графике).
Деградация полиоксиэтилена в сдвиговом поле более сильная,
чем у других подобных полимеров (полиакриламид, гуаровая смола,
полисахариды), имеет особенно заметные отрицательные последствия
в условиях применения полиокса для снижения гидродинамического
сопротивления [165, 166].
Не считая возможного ингибирования вторичных процессов,
единственным способом борьбы против механического разрыва связей
в полимере является уменьшение размеров макромолекулы. В случае
полиокса этого можно достичь введением солей, осадителей, повы-
шением температуры, переводящих полиокс в состояние клубка.
Однако большинство функций полимера — повышение вязкости,
снижение сопротивления, флокуляция — обусловлены именно боль-
шими линейными размерами молекул. Эти факторы вступают в про-
тиворечие друг с другом, и в каждом случае должен быть найден
соответствующий компромисс, удовлетворяющий всем требованиям-
Применение высокомолекулярного полиоксиэтилена. Несмотря
на относительную молодость, полиокс быстро нашел свое место средн
водорастворимых полимеров, и диапазон областей его применения
непрерывно расширяется [56, 60, 133]. Вместе с другими синтети-
ческими водорастворимыми полимерами полиокс вытесняет нату-
ральные полимеры из различных сфер, хотя относительно более
высокая стоимость синтетических полимеров пока несколько ограни-
чивает их распространение.
278
Одним из наиболее рекламируемых применений полиокса
является его использование для шлихтования тканей в текстильной
промышленности. По меньшей мере три обстоятельства делают эту
область перспективной для полиокса — применимость к широкому
ассортименту волокон от синтетических до хлопчатобумажных,
легкая растворимость в воде после сушки и, наконец, чрезвычайно
низкие требования к биологическому кислороду, что крайне облег-
чает проблему сброса вод. Физические качества пленок, образуемых
полиоксом па поверхностях нитей, также весьма удовлетвори-
тельны.
Полиокс вообще широко используется в различных операциях
формования стеклянных и керамических изделий, металлов, бумаги,
каучуков и пластиков, строительных элементов и т. п. Полиокс
легко удаляется растворением либо обжигом. Он может выполнять
также функции связующего благодаря своей высокой адгезии к раз-
личным поверхностям.
Фармацевтическая промышленность занимает одно из первых
мест в сфере применений полиокса. Он широко используется в каче-
стве связующего для таблеток, паст, мазей, в стоматологических
составах и т. п. Аналогичные функции выполняет полиокс в косме-
тике, где он употребляется дополнительно в аэрозольных компози-
циях, лосьонах, пудрах. Низкая токсичность полиокса чрезвычайно
благоприятствует расширению этих областей применения [167].
Сфера медицинского применения полиокса еще, пожалуй, не
оформилась. Отмечается возможность создания инплантантов на базе
полиокса, причем их рассасываемость широко регулируется за счет
совмещения полиокса с другими пластиками или комплексообразо-
вания .
Высокая загущающая способность полиокса используется в ряде
областей, например при создании гидравлических жидкостей для
автомобильной и авиационной промышленности на базе полиокса,
хотя его склонность к сдвиговой деструкции вряд ли благоприятна
для этой сферы применения.
Флокуляция и коагуляция — традиционные области использова-
ния водорастворимых полимеров. Широкое применение нашли здесь,
в частности, поливиниловый спирт и полиакриламид. Высокомоле-
кулярный полиоксиэтилен быстро занял одно из ведущих мест в этой
сфере техники, и сейчас его используют для осветления вод, сгущения
осадков, обогащения руд различного типа, в промышленности фос-
фатных и калийных удобрений. Его активность как флокулянта
очень высока, причем фирма Union Carbide выпускает специальную
марку Polyox Coagulant с высокой молекулярной массой. В табл. 81
приведены данные по осаждению суспензий глины [168] полимерами
окиси этилена. В качестве коагулянта полиокс дает наибольшие
Плотности осадка, высокую прочность флокул и полноту осаждения
при очень низких концентрациях и имеет слабую чувствительность
к pH среды. В ряде случаев к наилучшим результатам приводит
комбинация полиокса с полиакриламидом.
279
Коагулирующая способность полимеров обусловлена их высокой
молекулярной массой и способностью адсорбироваться на поверх-
ностях твердых частиц. Наличие донорных групп в молекуле, высо-
кая гибкость и большие линейные размеры цепи делают полиокс
одним из наиболее универсальных флотационных реагентов.
В последние годы особенный интерес привлекает способность
полиокса и ряда других аналогичных полимеров резко снижать
гидродинамическое сопротивление в водных растворах [168—170].
Феноменологическая сторона этого явления, известного как эффект
Томса, состоит в том, что малые добавки полимеров вызывают значи-
тельное повышение скорости потока жидкости или снижение сопро-
тивления в условиях турбулентного течения. На рис. 105 показана
концентрационная зависимость эффекта снижения сопротивле-
ния (концентрация выражена в млн-1) [168].
концентрация полимера, мольн. Воли
Рис. 105. Снижение гид-
родинамического сопро-
тивления в водном рас-
творе полиокса Ж9Д-301
в зависимости от кон-
центрации.
Оставляя в стороне методологию и многие физические аспекты
этого сложного явления, отметим, что эффективность полимеров
возрастает с молекулярной массой (табл. 82) и падает при введении
солей, «ухудшении» растворителя, повышении температуры [170].
Так, повышение концентрации MgSO4 в воде до 0,65 моль/л пол-
Таблица 81. Флокуляция суспензий глины полиоксиэтиленом [168]
Флотационный реагент Содержание флокулянта, % * Время седимента- ции, с Пропуска- ние спета через 20 ч, %
В отсутствие реагента — Нет границы 22,5
Carbowax-6000 осадка
5,00 То же 9,5
Polyox-WSR-35 1,00 » 57,0
Polyox WSR-301 0,10 355 97,5
Polyox Coagulant 0,05 90 100,0 **
Polyox Coagulant 0,10 10 100,0
* В расчете на твердое вещество суспензии.
** Через 5 мин после введения реагента.
280
ностью снимает эффект снижения сопротивления. Влияние солей
на величину эффекта аналогично их влиянию на температуру выпа-
дения полиокса и вязкость его растворов, откуда можно заключить,
что в максимально развернутой форме полимер имеет наивысшую
гидродинамическую активность.
Таблица 82. Снижение гидродинамического сопротивления (Е)
в водных растворах полимеров [169]
Марка полимера £max’ % Концентрации, 10-* %
при 1/2 Emax при Emax
Polyox WSR-N-10 33,4 800 10 000
Polyox WSR-N-80 53,5 280 5 000
Polyox WSR-N-750 65 32 500
Polyox WSR-N-3000 ...... 64 30 500
Polyox WSR-35 65 40 400
Polyox WSR-205 66,5 20 150
Polyox WSR-301 * 66—67 2-4 20—30
Polyox WSR-701 66,7 2 15
Polyox FRA ** 67,8 1 15
Гуаровая смола * 67—69 30—40 200-300
Полиакриламид * 62,0—69,4 2-8 14—100
ДНК* 64,0-68,4 16—23 200
* Различные партии.
** Специальная марка: „ Friction Reduction Agent" (Union Carbide).
Попытки установления строгой корреляции между величиной
эффекта или характерной концентрацией полимера и его молекуляр-
ной массой были неудачны, хотя очевидно, что наиболее высоко-
молекулярный «хвост» распределения отвечает за величину эффекта
и его чувствительность к сдвиговой деградации.
Есть две точки зрепия на прпроду гашения турбулентности малыми добав-
ками полимеров. Одна из них, наиболее распространенная, предполагает, что
активность обусловлена вязкоэластичными клубками индивидуальных макро-
молекул, вытянутых в гидродинамическом поле до 5—10 мкм или несколько
выше. В качественном отношении такая точка зрения удовлетворительно согла-
суется с большинством экспериментальных фактов. Установлена некоторая
корреляция между размерами турбулентных вихрей и макромолекул в рас-
творе.
Более детальный подход к гидродинамическому поведению растворов такого
рода полимеров вынуждает принять существование надмолекулярных частиц
больших размеров, вплоть до 1 мм, участвующих в гашении турбулентных
вихрей жидкости. Ярким проявлением «суспензионной» природы разбавленных
водных растворов полиокса является, в частности, способность разрл’шать
металлические пластины при ударе о их поверхность, тогда как вода не оказы-
вает подобного действия [171].
281
Способность полиокса к снижению гидродинамического сопро-
тивления обусловливает его разнообразное применение: увеличение
производительности трубопроводов жидкостей и пульп, мощности
насосных систем, гидравлических пушек при добыче полезных
ископаемых, брандспойтов в пожарном деле. Даже крайне небольшие
добавки полиокса позволяют повысить на 10—15% содержание
твердого вещества в транспортируемой суспензии без изменения
мощности насосной системы. Наконец, известно о применении поли-
окса для снижения давления при инъекциях и введении крове-
заменителей [169]. Трудно предвидеть возможные области исполь-
зования этого эффекта в будущем, но несомненно здесь имеется
огромное поле деятельности.
Рассмотренные области применения полиоксиэтилена основаны
главным образом на свойствах растворов этого полимера. Сфера
применения полиокса как пластика гораздо более узка, но также
полна перспектив. Пленки из полиокса * имеют прекрасные физико-
механические свойства, высокую прочность, прозрачны, хорошо
окрашиваются, растворяются в воде за 20—100 с при толщине 1 мм.
Их получают с использованием литьевой техники, применяемой
для полиэтилена. Пленки рекомендуются как упаковочный материал
для любых бытовых и технических препаратов, предназначенных
для растворения в воде,— чернил, красок, пигментов, латексов,
моющих и агрохимических средств, химических реагентов для
обработки воды и т. п.
В качестве защитных покрытий пленки полиокса наносят на
фрукты, корневую систему саженцев, посевной материал. Рекла-
мируется так называемая семялента из полиокса, содержащая посев-
ной материал с интервалами, оптимальными для посадки данной
культуры; лента укладывается непосредственно в борозду в почве.
Применение полиокса в агротехнике пока несколько ограничи-
вается стоимостью продукта. Привлекает интерес возможность кон-
диционирования и гранулирования удобрений с помощью полиокса
для пролонгирования их действия. Скорость рассасывания гранул
можно регулировать, совмещая полиокс с другими пластиками,
например полиэтиленом, сополимером винилхлорида с винилацета-
том. Предполагается возможность структурирования почвы вве-
дением полиокса, поскольку механизм этого эффекта аналогичен
процессам коагуляции.
Хорошая совместимость полиокса с многими пластиками и высо-
кая термодинамическая гибкость его цепей позволяют использовать
его в качестве пластификатора, в частности для повышения проч-
ности и эластичности желатиновых слоев в современных фото- и
кинопленках. Будучи относительно новым материалом, полиокс
далеко не исчерпал возможностей своего практического исполь-
зования.
Торговое наименование Radel-Film.
ЛИТЕРАТУРА
1. Саундерс Д. X., Фриш К. К. Химия полиуретанов. Пер. с англ. Под ред.
С. Г. Энтелиса. М., «Химия», 1968. 470 с.
2. Madge Е. С., Chem. a. Ind., 1962, № 10, р. 1806—1810; Hendrickson J. G.
е. a., I and ЕС, Prod. Research and Develop., 1963, v. 2, № 3, p. 199—204.
3. Gurgiolo A. E., J. Macromol. Sci., part D, 1966, v. 1, № 1, p. 39—190.
4. Мелешевич А. П., Усп. хим., 1970, т. 39, вып. 3, с. 444—470.
5. Aoki S. е. a., Bull. Chem. Soc. Japan, 1966, v. 39, № 4, p. 729—733.
6. Sakai S., Asai J., Ishii J., «Когё кагаку дзассп» (J. Chem. Soc. Japan, Ind.
Chem. Sec.), 1967, v. 70, № 11, p. 2036—2042; Hornof V., Gabra G., Blan-
chard L. P., J. Polym. Sci., Polym. Chem. Ed., 1973, v. 11, № 8, p. 1825—
1839.
7. Истхэм A. M. В кн.: Катионная полимеризация. Под ред. П. Плеша. Пер.
с англ. Под ред. С. С. Медведева. М., «Мпр», 1966, с. 341—365.
8. Kern R. G., J. Org. Chem., 1968, v. 33, № 1, p. 388—390; Kondo S., Blan-
chard L. P., J. Polym. Sci., part B, 1969, v. 7, № 8, p. 621—623.
9. Соловьиное А. А., Казанский К. С., Высокомол. соед., А, 1970, т. 12, № 9,
с. 2114—2124; 1972, т. 14, № 5, с. 1063—1071.
10. Gee G. е. a., J. Chem. Soc., 1961, р. 4298—4303.
11. Baum С. Е. Н. е. a., «Polymer», 1967, v. 8, № 9, р. 484—487; 1969, v. 10,
№ 8, р. 653—659.
12. Сорокин М. Ф., Шоде Л. Г., Штейнпресс А. В., Высокомол. соед., А, 1971,
т. 13, Xs 4. с. 747—752; Tanaka Y., J. Macromol. Sci., part A, 1967, v. 1,
№ 3, p. 471—491.
13. Энтелис С. Г., Казанский К. С. В кн.: Успехи химии и физики полимеров.
М., «Химия», 1970, с. 324—385.
14. Figueruelo J. Е., Worsfold D. J., Europ. Polym. J., 1968, v. 4, № 4, p. 439—
444; Nenna S., Figueruelo J. E., «Anales de la R. Sociedad Espanola de fi-
sika у quimica», $er. B, 1973, v. 69, № 1, p. 25—35.
15. Фурукава Д., Саегуса T. Полимеризация альдегидов и окисей. Пер. с англ.
Под ред. Н. С. Ениколопяна и В. А. Кабанова. М., «Мир», 1965. 479 с.
16. Ishii У., Sakai S. In: Ring-opening Polymerization. Ed. by К. C. Frisch
and S. L. Reegen. New York — London, M. Dekker Publ., 1969,
p. 13—109.
17. Соловьиное А. А., Казанский К. С., Высокомол. соед., А, 1974, т. 16, № 3,
с_ 595_____599.
18. Tsuruta Т., J. Macromol. Sci., part. D, 1972, v. 6, № 1, p. 179—250.
19. Furukawa J. e. a., Makromol. Chem., 1959, Bd. 32, № 1, S. 90—94; Mat-
sui U. e. а., «Когё кагаку дзассп» (J. Chem. Soc. Japan, Ind. Chem. Sec.),
1966, v. 69, Xs 9, p. 1375—1382.
20. Iwatsuki S. e. a., J. Polymer Sci., part B, 1964,’ v. 2, № 5, p. 549—552;
Tanaka I., J. Macromol. Sci., part A, 1967, v. 1, X» 6, p. 1059—1068.
21. Aoki S. e. a., J. Polymer Sci., part A-l, 1968, v. 6, Xs 9, p. 2885—2890;
Kagiya T., Sumida Y., Inoue T., Polymer J., 1970, v. 1, Xs 3, p. 312 — 321.
22. Пономаренко В. А. и др., Изв. АН СССР. Сер. хим., 1969, вып. 2, с. 454—
455; Высокомол. соед., А, 1971, т. 13, X» 7, с. 1546—1556; Геллер Н. М.,
Кропачёв В. А., Высокомол. соед., Б, 1974, т. 16, Xs 2, с. 103—105.
23. Шенфельд Н. Неионогенные моющие средства. Пер. с нем. Под ред. А. И. Гер-
шеновича. М., «Химия», 1965. 488 с.
24. Nonionic Surfactants. Ed. by M. J. Schick. New York, M. Dekker, 1967.
1085 p.
25. Flory P. J., J. Am. Chem. Soc., 1940, v. 62, Xs 6, p. 1561—1565.
26. Satkowsky W. D„ Hsu C. G., Ind. Eng. Chem., 1957, v. 49, X» 11, p. 1875 —
1878.
27. Polyalkylene Oxides and Other Polyethers. Ed. by N. Gaylord. New York,
Interscience, 1963. 491 p.
28. Malkemus J. D., J. Am. Oil Chem. Soc., 1956, v. 33, Xs 2, p. 571 — 579.
29. Герм. пат. 735418 (1943); С. A., 1944, v. 38, 2662.
30. Пат. США 2988572 (1961); С'. А., 1961, v. 55, 21704g.
283
31. Англ. пат. 1040783 (1966); франц, пат. 1388588 (1966); С. А., 1966, V. 64,
838b.
32. Bar-Ilah A., ZilkhaA., J. Macromol. Sci., part A, 1970, v. 4, № 8, p. 1727—
1741.
33. Vandenberg E. J., J. Polym. Sci., Polym. Chem. Ed., 1973 v. 10 № 10
p. 2887—2902.
34. Годовский Ю. К., Слонимский Г. Л., Гарбер II. М., Высокомол. соед.. А,
1973, т. 15, № 4, с. 813-827.
35. Ingham J. D., Lawson D. D., J. Polym. Sci., part A, 1965, v. 3, № 7
p. 2707—2710; Bender G. W., Le Grand D. G., Gaines G. L., «Macromolecules»,
1969, v. 2, № 6, p. 681—682.
36. Ellias H.—G., Makromol. Chem., 1967, Bd. 103, S. 214—229.
37. Островский В. E., Ходжемиров В. А., Баркова А. П., ДАН СССР, 1970,
т. 191, № 5, с. 1095—1098.
38. Сох J. D., «Tetrahedron», 1963, v. 19, № 7, р. 1175—1184; Pell A. S., Pil-
cher G., Trans. Faraday Soc., 1965, v. 61, № 1, p. 71—77.
39. Казанский К. С., Энтелис С. Г., Изв. АН СССР Сер. хим., 1965, вып. 6,
с. 1089—1091.
40. Oley F. II., Zagoren В. L., Mehltretter С. L., J. Appl. Polym. Sci., 1964.
v. 8, № 5, p. 1985—1989.
41. Magnasco V., Bossi M., J. Polym. Sci., 1962, v. 62, № 174, p. 286—289.
42. Mohall S. L. e. a., Anal. Chem., 1966, v. 38. № 8, p. 1063—1065.
43. Liu K.—J., Makromol. Chem., 1968, Bd. 116, S. 146—151; Tanner J. E..
Liu K.—J., Ibid., 1971, Bd. 142, S. 309—312.
44. Ellias H.—G., Lus H., Makromol. Chem., 1966, Bd. 92, S. 1—24; Bd. 96.
S. 64-82.
45. Liu K.—J., Parsons J. L., «Macromolecules», 1969, v. 2, № 5, p. 529—
533.
46. Delmas G., J. Арр]. Polym. Sci., 1968, v. 12, № 4, p. 839—851.
47. Sadron С., Rempp P., J. Polym. Sci., 1958, v. 29, № 1, p. 127—140; Rit-
cher T. A., Ellias II,—G., Makromol. Chem., 1959, Bd. 30, S. 48—80; Ina-
gaki H., Tanaka M.. Ibid., 1964, Bd. 74, S. 145—157; Ring IF., Cantow II. J.,
Holtrup IV., Europ. Polym. J., 1966, v. 2, № 2, p. 151—162.
48. Fletcher J. P., Persinger H. F., J. Polym. Sci., part A-l, 1968, v. 6, № 4,
p. 1025—1032; Calzolarn C., Favretto L., Stancher B., J. Chromatogr., 1969.
v. 39, № 3, p. 318—321.
49. Favretto L. e. a.. J. Chromatogr., 1970, v. 50, № 3, p. 304—310.
50. Heitz IV., Borner B., Uliner H., Makromol. Chem., 1969, Bd. 121, S. 102—116.
51. Miyazawa T., Fukushima K., Ideguchi Y., J. Chem. Phys., 1962, v. 37, № 12,
p. 2764—2776; Tadokoro II. e. a., Makromol. Chem., 1964, Bd. 73,
S. 109—127.
52. Connor T. M., .'[cLaughlan K. A., J. Phys. Chem., 1966, v. 69, № 6,
p. 1888—1893.
53. Bailey F. E., Callard R. IF., J. Appl. Polym. Sci., 1959, v. 1, № 1, p. 56—71.
54. Allen G. e. a., «Polymer», 1967, v. 8, № 8, p. 391—397; Beech D. R., Bo-
oth C., J. Polym. Sci., part A-2, 1969, v. 7, № 3, p. 575—586.
55. Mark J. E., Flory P. J., J. Am. Chem. Soc., 1965, v. 87, № 7, p. 1415—1423:
Gorin S., Monnerle L., J. chim. phys., et phys.-chim. biol., 1968, v. 65,
№ 11—12, p. 2076—2083.
56. Encyclopedia of Polymer Science and Technology. V. 6. Ed. by H. Mark.
New York, John Wiley, 1967. 818 p.
57. Lloid W. G., J. Am. Chem. Soc., 1956, v. 78, № 1, p. 72—75; J. Polymer
Sci., part A, 1963, v. 1, № 8, p. 2551—2563; Vink H., Makromol. Chem.,
1963, Bd. 67, S. 105—123.
58. McGary C. IF., J. Polym. Sci., 1960, v. 46, № 147, p. 51—58.
59. Glycols. Ed. by G. Curme, F. Johnston. New York, Reinhold Publ., 1952.
389 p.
60. McClelland С. P., Rector P. R. In: Encyclopedia of Chemical Technology-
V. 7. Ed. by R. E. Kirk and D. F. Othmer. New York, Interscience Inc.,
1966, p. 238—263; Harding R. II., Rose J. K. In: Water-Soluble ResinS-
284
Ed. 2nd. Ed. by R. L. Davidson and M. Sittig. New York, Van Nostrand-
Reinhold, 1968, p. 191—226.
61. Гладковский Г. А., Веденеева Г. Ф., Лебедев В. С., Пласт, массы, 1971,
№ 2, с. 3-4.
62. Пат. США 3117998 (1964); С. А., 1964, v. 60, 8214f.
63. Гриценко Т. М., Семенидо А. В., Карцовник В. И. Авт. свпд. № 244612
(1969); Изобр., пром, образцы. Товарн. знаки, 1969, т. 46, № 18, с. 96.
64. Price С. С., Spector R., J. Am. Chem. Soc., 1965, v. 87, № 9, p. 2069—2070;
Price С. C.. Spector R., Tumolo A. L., J. Polym. Sci., part A-l, 1967, v. 5,
№ 1, p. 175-181; № 3, p. 407—416.
65. Price С. C., Osgan M., J. Am. Chem. Soc., 1956, v. 78, № 18, p. 4787—4792.
66. Tant H., Oguni N., Watanabe Sh., J. Polym. Sci., part B, 1968, v. 6, № 7,
p. 577—580; Schaeffer J., «Macromolecules», 1969, v. 2, № 4, p. 533—537.
67. Mathias A., Anal. Chim. Acta, 1964, v. 31, № 3, p. 598—601; Ing-
ham J. D. e. a., J. Macromol. Sci., part A, 1966, v. 1, № 1, p. 75—83.
68. Simons D. M., Verbanc J. J., J. Polym. Sci., 1960, v. 44, № 144, p. 303—311;
Snyder W. H. e. a., Trans. N. Y. Acad. Sci., Ser. 2, 1962, v. 24, № 4,
p. 341—343.
69. Steiner E. L.. Pelletier R. R., Trucks R. O., J. Am. Chem. Soc., 1964, v. 86,
№ 21, p. 4678—4686.
70. Гладковский Г. А. и др., Высокомол. соед., А, 1972, т. 14, № 5, с. 1174—
1179; 1973, т. 15, № 6, с. 1221 — 1226.
71. Пат. США 3393243 (1968); С. А., 1968, v. 69, 44286b.
72. Пат. США 2996550 (1961).
73. Кузнецов В. Н., Коган В. Б., Вилесова М. С., Высокомол. соед., А, 1969,
т. 11, № 6, с. 1330—1342.
74. Burns F. А., Мигаса В. F., Anal. Chem., 1959, v. 31, № 3, р. 397—399;
Blanchard L. P., Baijal M. D., J. Polym. Sci., part B, 1966, v. 4, № 11,
p. 837—841.
75. Hanna G. J., Siggia S., J. Polym. Sci., 1962, v. 56, № 164, p. 297—304;
Марк Г. Ф., Рехниц Г. Кинетика в аналитической химии. Пер. с англ.
Под ред. К. Б. Яцпмирского. М., «Мир», 1972. 368 с.
76. Willerboordze F. G. е. a., Anal. Chem., 1964, v. 36, № 12, р. 2270—2274;
1966, v. 38. № 7, р. 854-858.
77. Гладковский Г. А. Кандидатская диссертация. М., НИИПМ, 1974.
78. Sholtan W., Lie S. J., Makromol. Chem., 1967, Bd. 108, S. 104—128.
79. Case L. C., J. Phys. Chem., 1958, v. 62, № 7, p. 895—896.
80. Bajal M. D., Blanchard L. P., J. Appl. Polym. Sci., 1968, v. 12, № 2,
p. 169—175.
81. Эптелис С. Г., Евреинов В. В., Кузаев А. И. В кн: Успехи химии п физики
полимеров. М., «Химия», 1973, с. 201—238.
82. Faucher A., J. Polym. Sci., part В, 1965, v. 3, № 2, р. 143—145.
83. Allen G. e. a., «Polymer», 1964, v. 5, № 11, p. 547—552.
84. Moacanin JCuddihy E. F., J. Polym. Sci., part C, 1966, № 14, p. 313—322.
85. Stockmayer W. H., Burke J. J., «Macromolecules», 1969, v. 2, № 6, p. 647—
650; Connor T. M. e. a., «Polymer», 1968, v. 9, № 11, p. 591—595.
86. Convay В. E., J. Polym. Sci., 1960, v. 46, № 147, p. 113—127, 129—138;
Havlik A. J., Smith T. L., J. Polym. Sci., part A, 1964, v. 2, № 2,
p. 539—586.
87. Тизер P. П., Сарапина Л. И., Энтелис С. Г., Усп. хим., 1972, т. 41, вып. 9,
с. 1672—1695.
88. Gruber Е. Е, е. a., I and ЕС, Prod. Research and Develop., 1964, v. 3, № 3,
p. 194—200; Allen G., Crossley II. G., «Polymer», 1964, v. 5, № 11, p. 553
557; Vaugham G. e. a.. Europ. Polym. J., 1968, v. 5, № 11, p. 207—226.
89. Пат. США 3042725 (1962); С. A., 1963, v. 58, 7011h; пат. США 3133906 (1964).
90. Франц, пат. 1305274 (1962); Rowe Р. D., Thomas D. К., J. Appl. Polym.
Sci., 1963, v. 7, № 2, p. 461—468.
91. Wyandotte Key Chemicals for Industry. Technical Bulletin. Wyandotte
Chemicals Corp. S. C., 1966.
92. Millet W. H., Ind. Eng. Chem., 1950, v. 42, № 12, p. 2436—2441.
285
93. Dow Polyglycol 15. Technical Bulletin, TSD-6. Midland (Michigan), The
Dow Chem. Co., 1956.
94. Physical Properties Synthetic Organic Chemicals. Technical Bulletin
F—6136S. New York, Union Carbide Chem. Co., 1965.
95. Пат. США 3101374 (1963); С. A., 1963, v. 59, 10350g.
96. Rosen M. J., Goldsmith H. A. Systematic Analysis of Surface Active Agents.
New York — London, Interscience Publ., 1960. 422 p.; Waszeciak P., Na-
deau H. G., Anal. Chem., 1964, v. 36, № 4, p. 764—767.
97. Uno T., Miyajima K., Miyajima Y., Chem. Pharm. Bull., 1967, v. 15, № 1,
p. 77—82.
98. Mathias A., Mellor N., Anal. Chem., 1966, v. 38, № 3, p. 472—477.
99. Gibson U. II., Quick Q., J. Appl. Polym.‘Sci., 1971, v. 15, № 11, p. 2667—
2676.
100. Whipple E. B., Green P. J., «Macromolecules», 1973, v. 6, № 1, p. 38—42.
101. Кумпаненко II. В., Каванский К. С. В кн.: Успехи химии и физики поли-
меров. М., «Химпя», 1973, с. 64—96.
102. Patslga R. О., J. Macromol. Sci., part С, 1967, v. 1, № 2, р. 223—274.
103. Richards D. H., Szwarc M., Trans. Faraday Soc., 1959, v. 55, № 441,
p. 1644—1650; Finaz G., Rempp P., Parrod J., Bull. Soc. chim. France,
1962, № 2, p. 262—266.
104. Rembaum A., Moacanin J., Cuddihy E., J. Polym. Sci., part C, 1963, № 4,
p. 529—549.
105. Tahan M., Zilkha A., J. Polym. Sci., part A-l, 1969, v. 7, № 6, p. 1815—
1824, 1839—1860; Shiozaki H., Tanaka Y., Makromol. Chem., 1970, Bd. 138,
S. 215-221; 1971, Bd. 143, S. 25—45.
106. Ishimory M. e. a., Makromol. Chem., 1963, Bd. 64, S. 190—207; 1968,
Bd. 115, S. 103—118; Nakaniwa M. e. a., Ibid., 1970, Bd. 138, S. 197—208.
107. Calderon N., Scott K. W., .1. Polym. Sci., part A-l, 1967, v. 5, № 4, p. 917—
919; Kuntz I., Kroll IF. R., Ibid., 1970, v. 8, № 7, p. 1601 — 1621; Naka-
niwa M. e. a., J. Polym. Sci., part B, 1970, v. 8, № 2, p. 131—135.
108. Chang E. J. C., J. Appl. Polym. Sci., 1970, v. 14, № 12, p. 3137—3141.
109. Price С. С. e. a., J. Am. Chem. Soc., 1956, v. 78, № 3, p. 690—691; ShuN.Ch.,
Price С. C., J. Polym. Sci., part A, 1963, v. 1, № 11, p. 1105—1113.
110. Stanley E., Litt M., J. Polym. Sci., 1960, v. 43, № 142, p. 453—458; Ma-
. gill J. H., Makromol. Chem., 1965, Bd. 86, S. 283—288.
111. Tsuruta T., Inoue S., Tsukuma I., Makromol. Chem., 1965. Bd. 84, S. 298—
299; Furukawa J. e. a., Ibid., 1966, Bd. 94. S. 68—82.
112. Кумпаненко И. В. и др., Высокомол. соед., А, 1973, т. 15, № 2, с. 594—601;
Птицына Н. В., Кумпаненко И. В., Казанский К. С., там же, Б, 1975,
т. 17, № 8, с. 617—620.
ИЗ. Allen G., Booth С., Jones М. N., «Polymer», 1964, v. 5, № 5, р. 257—264;
Powell Е., Ibid., 1967, v. 8, № 4, p. 211—216.
114. Aggarval S. L. e. a. In: Advances in Chemistry Series № 52. Washington,
Am. Chem. Soc., 1966, p. 88—99.
115. Allen G., Booth C., Jones M. N., «Polymer», 1964, v. 5, №4, p. 195—199.
116. Noshay II., Price С. С., J. Org. Chem., 1958, v. 23, № 4, p. 647—648;
Гоглее P. С.. Нейман M. В., Высокомол. соед., A 1967, т. 9, № 10,
с. 2083—2093.
117. Tsuruta T. e. a., Makromol. Chemi, 1965, Bd. 81, S. 191 — 197; Unoue S.
e. a., Ibid., 1967, Bd. 103, S. 151—163; Tsuruta T. e. a., Ibid.,S. 164—174.
118. Inoue S. e. a., Makromol. Chem., 1962, Bd. 53, S. 215—218; 1966, Bd..9O,
S. 131—138.
119. Bailey F. E. e. a., Ind. Eng. Chem., 1958, v. 50, № 1, p. 5—16.
120. Sorenson W. R-, Campbell T. IF., Preparative Methods of Polymer Chenh"
stry. New York — London, Interscienco Publ., 1961. 337 p.
121. Казанский К. С., Еереинов В. В., Энтелис С. Г., Изв. АН СССР. Сер-
хим., 1964, вып.>2, с. 274—281; Казанской К. С., Тарасов А. Н., Шарма-
нова Т. Г. В кн.: Доклады юбилейной сессии по высокомолекулярны5:
соединениям (сборник препринтов). М., ИХФ АН СССР, 1970, с. 68—74.
122. Казанский К. С. и др., Изв. АН СССР. Сер. хим., 1964, вып. 4, с. 759—761-
286
123. Крылов О. В., Синяк 10. Е., Высокомол. соед., 1961, т. 3, № 6, с. 898—
900; «Нефтехимия», 1962, т. 2, № 5, с. 688—696; Крылов О. В., Кушне-
рев М. Я., Фокина Е. А., там же, 1962, т. 2, № 5, с. 697—704.
124. Пат. США 2941963 (1960); С. А. 1960, v. 54, 20347е.
125. Англ. пат. 869114 (1961); С. А., 1962, v. 56, 596а.
126. Watt G. W.. Chem. Rev., 1950, v. 46, № 2, p. 317—352.
127. Пат. США 2971988 (1961); С. А., 1961, v. 55, 216621; пат. США 3100750
(1963); С. А., 1964, v. 60, 14630L
128. Англ. пат. 869116 (1961); С. А., 1962, V. 57, 2411d.
129. Пат. США 3037943 (1962); С. А., 1962, v. 57, 3630с; англ. пат. 926243 (1963);
С. А., 1963, v. 59, 5282а; англ. пат. 926346 (1963); С. А., 1963, v. 59, 4118b;
англ. пат. 926860 (1963); С. А., 1963, v. 59, 5282с.
130. Тарноруцкий М. М. и др., Высокомол. соед., А, 1972, т. 14, № 7, с. 1459—
1461; № И, с. 2371—2375.
131. Furukawa J., Kawabata N., Kato A., J. Polym. Sci., part A-l, 1967, № 12,
p. 3139—3150; Nakaniwa M. e. a., Makromol. Chem., 1972, Bd. 155 S. 185—
196.
132. Bruce M. J., Rabagliati F. M., «Polymer», 1974, v. 8, № 7, p. 361 —
367.
133. Ucar Polyox WSR-N Resins. Technical Bulletin F—41013A. New York,
Union Carbide Chem. Co., 1963; Alkox. Meisei Chem. Works Ltd., Kyoto,
1964; Polyox Water-Soluble Resins. Technical Bulletin F—44029A, New
York, Union Carbide Chem. Co., 1973.
134. Malkolm J. N., Rowlinson J. S., Trans. Faraday Soc., 1957, v. 53, part 7,
p. 921—931.
135. Maron S.H., Filisko F.F., J. Macromol. Sci., part B, 1972, v. 6, № 1,
p. 79—92.
136. Cuniberti C., Fernanda R.. «Polymer», 1972, v. 13, № 8. p. 379—384.
137. Straztelle C., Makromol. Chem., 1968, Bd. 119, S. 50—63; Hert M., Stra-
zielle C., Europ. Polym. J., 1973, v. 9, № 6, p. 543 — 551.
138. Merill E. K. e. a., Trans. Soc. Rheol., 1966, v. 10, Л» 1, p. 335—351.
139. Bailey F. E., Kucera J. L., Imhof L. G., J. Polym. Sci., 1958, v. 32,
№ 125, p. 517—518.
140. Allen G. e. a., «Polymer», 1967, v. 8, № 8, p. 391 — 397.
141. Карасёва H. H. и др., Высокомол. соед., Б, 1973, т. 15, № 11, с. 787—789.
142. Booth С., Price С., «Polymer», 1966, v. 7, № 2, р. 85—90.
143. Дубровский С. А. и др., Высокомол. соед., А, 1975, т. 17, № 12, с. 2733—
2741.
144. Sakurada I., Kaji К., J. Polymer Sci., part С, 1970, № 31, p. 57 — 76.
145. Teramoto A., Fujita H., Makromol. Chem., 1965, Bd. 85, S. 261—272.
146. Richards J. R., Dissert. Abstr., 1961, v. 22, p. 1029.
147. Hay J. N. e. a., «Polymer», 1969, v. 10, № 3, p. 187—211; Jain N. L.
e. a., Makromol. Chem., 1969, Bd. 185, S. 192—200; 1971, Bd. 142, S. 293—
301.
148. Beech D. R. e. a., Europ. Polym. J., 1972, v. 8, № 6, p. 799—807; J. Polym.
Sci., part A-2, 1972, v. 10, № 8, p. 1555—1564; «Polymer», 1972, v. 13,
№ 7, p. 355—359.
149. Pelzbauer Z., Manley R. St.-J., J. Macromol. Sci., part B, 1970, v. 4,
№ 4, p. 761—774; RijkeA. M., McCoy S., J. Polym. Sci., part A-2, 1972,
v. 10, № 9, p. 1845—1848.
150. Connor T. M., Read В. E., Williams G., J. Appl. Polym. Sci., 1964, v. 14,
№ 2, p. 75—91.
151. Wetton R. E., Allen G„ «Polymer», 1966, v. 7, № 7, p. 331—365; Hart-
mann B., Ibid., 1972, v. 13, № 9, p. 460—464.
152. Binks A. E., Sharpies A., J. Polvm. Sci., part A-2, 1968, v. 6, № 2, p. 407—
420.
153. Blumberg A. A., Pollack S. S., Moeve С. A., J. Polym. Sci., part A, 1968,
v. 2, № 8, p. 2499—2502.
154. Iwamoto R. e. a., J. Polym. Sci., part A-2, 1968, v. 6, № 8, p. 1509—1525;
Yokohama M. e. a., «Macromolecules», 1968, v. 2, № 2, p. 184—192.
287
155. Bailey F. E., France H. G., J. Polym. Sci., 1961, v. 49, № 152, p. 397-
406; Tadoroko H. e. a., J. Polym. Sci., part B, 1964, v. 2, № 4, p. 363—368.
156. Bailey F. E., Lundberg B. D., Callard B. W., J. Polym. Sci., part A, 1964
v. 2, № 2, p. 845—851.
157. Madorsky S. L., Straus S., J. Polym. Sci., 1959, v. 36, № 130, p. 183—194.
158. Моисеев В. Д. и др., «Труды по химии и химической технологии» (Горь-
кий), 1962, вып. 2, с. 459—464; Гоглев Р. С., Нейман М. Б. В кн.: Высо-
комолекулярные соединения. Химические свойства и модификация поли-
меров. М., «Наука», 1964, с. 156—168.
159. Татевосян Е. Л. и Ор., Высокомол. соед., Б, 1974, т. 16, №4, с. 284—287.
160. Affifi-Afjat А. М., Hay J. М., Europ. Polym. I., 1972, v. 8, № 2, р. 289—
297.
161. Stability of Polyox Resins. Technical Bulletin № 45—T—2. Geneva, Union
Carbide Europe, 1969.
162. Salovey B., Dammont F. В., J. Polym. Sci., part A, 1963, v. 1, № 6
p. 2155—2162.
163. Schnabel W., Makromol. Chem., 1970, Bd. 131, S. 287—293.
164. Nakano A., Minoura J., J. Appl. Polym. Sci., 1971, v. 15, № 4, p. 927—
936.
165. Kenis P. В., J. Appl. Polym. Sci., 1971, v. 15, № 3, p. 607—618; Fischer D. H.,
Bodriguez F., Ibid., № 12, p. 2975—2985.
166. Gordon B. J., Balakrishman C., J. Appl. Polym. Sci., 1972, v. 16, № 7,
p. 1629—1639.
167. Smith H. F. e. a., Toxicol. Appl. Pharmacol., 1970, v. 16, № 2, p. 442 —450.
168. Mansfield B., «Process Biochemistry», 1970, v. 5, № 2, p. 28—30.
169. Hoyt J. W., J. Polym. Sci., part B, 1972, v. 9, № 11, p. 858—862.
170. Little В. С. e. a., I. and EC, Fundamentals, 1969, v. 8. № 3, p. 557—561:
3125PP1’ P°lym‘ Sci" 1971’ V’ 15’ № U’ P’ 2623“2637; № 12’ P- 3117--
171. Власов С. А., Кандидатская диссертация. M., ИПМ АН СССР, 1973.
172. Простые олцгоэфиры и полиуретановые материалы на пх основе. Каталог.
М., НИИТЭхим, 1975. 18 с."
Глава VII
Эфиры гликолей
Проявляя характерные свойства спиртов, гликоли образуют простые
и сложные эфиры, при этом из-за наличия двух гидроксильных групп
возможны полные и неполные замещенные гликоля:
R(O—CR'R"—CR'R'%OH
R(O—CR'R"— CR'R")nOR П "
где R — алкильные, арильные радикалы или их производные,
винильные, ацильные группы, кислотные остатки неорганических
кислот; R', R" — водород, алкильные, арильные радикалы.
Полные эфиры могут быть простыми, сложными или со смешан-
ными функциями. Наличие двух ОН-групп у соседних атомов угле-
рода является причиной образования циклических эфиров гликолей
типа:
Н2С — С Но
of х;о
Н2С-СН2
о
СНз-Нс/ СН2
\ ZCII2
oz
1,4-диоксап 2-метил-1,3-диоксолан
Приведенные общие формулы эфиров гликолей объединяют прак-
тически неограниченное число соединений, которые могут иметь
самые разнообразные физико-химические свойства. После разра-
ботки технологии синтеза эфиров гликолей из а-окисей в 20-е годы
нашего века они за короткий срок стали одними из самых доступных
И распространенных химических продуктов [1, с. 560; 2, р. 114].
В настоящее время во всех индустриальных странах отмечается
значительный рост производства и потребления эфиров гликолей.
Физические свойства эфиров гликолей
Простые ациклические эфиры
На основании общей формулы простых эфиров гликолей можно
Полагать, что их свойства зависят от четырех основных факторов:
иРироды оксиалкиленовой и эфирной групп, числа оксиалкиленовых
19 Заказ 972 , 189
групп и степени замещения гликоля. Эфиры гликолей относятся
к ассоциированным жидкостям, образующим внутри- и межмолеку-
лярные водородные связи [3, с. 29; 4; 5].
Строение эфирной группы, в частности, для метил-, этил- и бути-
ловых эфиров этиленгликоля мало влияет на внутримолекулярную
связь. Сдвиг внутримолекулярной водородной связи в гликолях
и моноэфирах гликолей составляет [4, 51:
Сдвиг колеба-
Соединение нийОНвсм-1
НОСН»СН,ОН .... 32
СН3ОСН.,СН,ОН . . . 30 .35
С,Н5 ОСН.,СН2ОН ... 33
С]Н9ОСН,СН,ОН ... 33
СН3СН(ОН)СН2ОН . . 45
Сдвиг колеи:
Соединение ний ОН в с.м-
НОСН2СН2СН,ОН . . 78
СН3ОСН2СН,СН2ОН 86—93
СН3СН(ОН)СН(ОН)СН3 . 39
НО(СН2ЦОН............. 156
СН3О(СН,)4ОН.......180—184
Молекула простого эфира образует в растворе четыреххлористог >
углерода пятичленный цикл или находится в равновесии с диме-
ром [5]:
НОСН2СН2ОСН3
О
Щс/ \
I ,н
Н2СХ
о
СН3
Для метилцеллозольва энергия перехода между слабозаселен-
ным транс-конформером и более стабильным гош-конформером
составляет 7,5 ± 0,96 кДж/моль (1,8 ± 0.23 ккал/моль) [6]. В ин-
тервале 0—60 °C внутримолекулярная связь ОН-группы простого
эфира зтиленгликоля меньше подвержена влиянию температуры, чем
межмолекулярная.
Внутримолекулярная водородная связь является преобладающей
только для разбавленных растворов 0,05 М) эфиров. По мер6
увеличения концентрации возрастают межмолекулярные водородные
связи эфиров. Количественное изучение этой связи чрезвычайно
затруднено из-за наличия в объеме жидкости единой сетки водород-
ных связей. Приходится пользоваться косвенными методами, кото-
рые основаны на измерении влияния добавок, разрушающих'или
упрочняющих водородные связи эфирных растворов: плотность
и вязкость [7], теплота смешения и изменение температуры замерза-
ния [8], изменение диэлектрической проницаемости и релакса-
ции [9], рассеяние света и электронная парамагнитная релакса-
ция [10].
290
Остановимся для примера на методе теплот смешения. Теплота смешения
простого монобутилового эфира этиленгликоля и воды при различных темпера-
турах составляет (в кал/моль *) [11, с. 77]:
Содержание
афира, % (мол.) При 20 °C При 40 °C При 60 °C При 80 °C При 100 °C
28 —117 —73,6 -20,8 41,2 —
49,6 -85,8 —51,3 4,7 52,8 116
73,6 —41,8 —20,4 7,5 38,5 —
При 20 и 40 °C по всей области концентраций наблюдается выделение тепла;
максимум дает >-==30%-ный раствор эфира. С ростом температуры вследствие
ослабления межмолекулярной водородной связи как для воды, так и для эфира
наблюдается изменение знака энтальпии. При растворении эфиров гликолей,
подобных бутиловому, в частности 2-этоксиэтанола, в октане и этилбензоле
происходит поглощение тепла.
Замещение двух ОН-групп гликоля усиливает акцепторные
свойства атома кислорода и экзотермический эффект растворения
диметилового зфира этиленгликоля в воде достигает 1,35 кДж/моль
(320 кал/моль) уже для 15,5%-ного раствора. Но смешение с водой
при концентрации зфира более 75% сопровождается поглощением
тепла [11, с. 74].
Учитывая электростатическую природу водородной связи, можно
предполагать значительное влияние стерических затруднений на
межмолекулярную водородную связь и, как следствие, на физи-
ческие свойства эфиров гликолей. В табл. 83 приведены .свойства
простых этиловых эфиров гликолей, полученных из различных
окисей. С увеличением молекулярной массы окиси возрастают темпе-
ратура кипения и коэффициент преломления эфиров. При более
высокой температуре кипит изомер с первичной ОН-группой.
Таблица 83. Свойства моноэтпловых эфиров гликолей [2, 12]
Исходная окись олефина '.Формула эфира ткип, °C 2 0 d 20 nD
Окись этилена С2Н5—О—СН2СН,ОН 135,1 0,9290 1,4076
Окись пропилена Окись пропилена С,Н5—О—СН,СНЮН)СН3 130—131 0,8960 1,4060
С2Н5—О—СН(СН3)СН,ОН 137—138 0,9080 1,4100
Окись изобутилена С2Н5—О—СН2С(ОН)(СН3)2 129—130 0,8783 1,4062
Окись изобутилена С2Н5-О—С(СН3)2СН2ОН 146—148 0,9071 1,4190
Окись амилена —.—- С2Н5-О-С5Н10ОН 158—162 0,8877 1,4149
Физические свойства эфиров этилен- и пропиленгликолей прежде
®сего зависят от молекулярной массы эфирной группы. С ее ростом
Увеличиваются температура кипения и коэффициент преломления,
Ьо уменьшаются плотность и растворимость эфира гликоля (табл. 84,
* 1 кал/моль = 4,19 Дж/моль.
19* 291'
Таблица 84. Физические свойства моноэфиров этиленгликоля
[2, 13—15, 17, 19]
Моноэфир этиленгликоля 20 d го 2 0 nD Т. кии. , °C Т. пл., °C Т. всп., °C Растворимое! при 20°С, г/11
при 0 ,1 МПа при давлении (мм рт. ст. )* в воде I воды в
Метиловый . . . 0,9663 1,4021 124,5 27 (10) -85,1 43,4 ОС
Этиловый .... 0,9311 1,4076 135,6 35 (10) —70 48,9 сю ОС
Пропиловый . . 0,9120 1,4136 149,0 58,3 (20) 50 (17) — — — —
Изопропиловый — 1,4072 140,7 — — — —
и-Бутпловып . , 0,9019 1,4193 171,2 61 (Ю) 57 (13) — 69,5 оо ОС
Пзобутиловып . . 0,893 *’ 1,4135 160,0 — 59,4 — —
emop-Бутиловый . — 1,4173 — 68,6 (40) — — —
2-Этилбутиловып 0,8954 1,4304 196,7 84 (10) -90 82,3 1,2 10,0
и-Октиловый . . — 1,4363 — 109 (4) — — •— —
2-Этилгексиловый 0,8850 1,4363 229,0 -70 из 0,09 5,3
Фениловый . . . 1,1094 1,5386 244,9 118 14,0 121 2,7 10,8
Амплфениловый , 1,0180 1,5190 297,0 (10) -55 — — —
* I мм рт. ст. = 133,32 Пл.
“ При 1:> °C..
85). Все моноэфиры низших спиртов — легкоподвпжные жидкости.
Если исходный спирт твердое тело, эфир, как правило, пасто
образен.
Введение в молекулу эфира моногликоля дополнительных окси-
алкиленовых групп, как видно из табл. 86 и 87, сопровождается
заметрым изменением физических свойств. Это позволяет для отно-
сительно низкомолекулярных производных окиси этилена и окиси
пропилена установить корреляцию состава с коэффициентом пре-
ломления [16—18], удерживаемым объемом [13, 20] и молекулярной
абсорбцией [21]. По мере увеличения молекулярной массы эфира
за счет его оксиалкиленовой группы происходит нивелирование
многих физических показателей, что может быть объяснено особен-
ностью строения его молекулы.
С помощью- рептгеноструктурного анализа установлено, что
полиоксиэтиленовая цепь не является линейной] а имеет извилистую
структуру с длиной оксиэтиленовой группы примерно 0,2 нм. Изви-
листая цепь образует спираль со звеном из 9—11 оксиэтильны^
292
Таблица 85. Физические свойства моноэфиров
пропиленгликоля [2, 15, 16, 19]
Радикал (R) Плотность, г/см3 2 0 nD Т. кип., °C
при 0,1 МПа при давлении (мм рт. ст.) *
Первичные эфиры НОСН2СНОНСН3
Метил .... 0,9207 1,4039 122,5
Этил 0,8941 1,4071 131,8 43,0 (20)
/(-Пропил . . 0,8847 1,4119 148,7 57,5 (20)
Изопропил . . 0,9059 1,4070 142,0 .—
и-Бутил . . . 0,8789 1,4180 170,0 72,8 (20)
трет-'З утил 0,8707 — 152,0 —
2-Этплгекспл 0,8710 ** 1,4327 —. 101,7 (10)
Фенпл .... 1,0622 1,5232 — 130 (21)
Вторичные эфиры HOCH2CHORCH3
Метпл .... 0,9365 1,4072
Этил 0,9044 1,4097
н-Пропил . . 0,8925 —
Изопропил . . 0,885 ** 1,4094
я-Бутпл . . . 0,8880 1,4192
* 1 мм рт. ст. = 133,32 Па.
** При 15,6 °C.
131,0
140,5
150,0
143,5
78,2 (20)'
групп и может быть свернута в клубок. Из-за наличия пространствен-
ных модификаций один и тот же оксиэтилированный спирт может
иметь различное строение и не обладает строго воспроизводимыми
свойствами. Водные растворы эфиров полигликолей образуют жидко-
кристаллические системы (пластинчатая структура), в которых
вода за счет водородных связей стягивает слой полимера.
Для высокомолекулярных полиэфиров наиболее важным свой-
ством является поверхностная активность, определяемая гидро-
фильно-гидрофобным балансом и способностью к мицелло-
образованию [22, 23].
Этерификация второй гидроксильной группы гликоля приводит-
к получению простых дпэфиров, свойства которых (табл. 88) значи-
тельио отличаются от мопозамещенных. Но и в этом случае физико-
химические показатели зависят от молекулярной массы. Увеличивая
^Исло оксиэтильных групп, можно регулировать такие важные
Показатели диэфиров гликолей, как температура кипения и по-
лярность [24].
293
Таблица 86. Физические свойства моноэфиров
этиленгликолей [13, 15, 17, 19]
Радикал эфира R (ОС2Н4)„ОН п Плот- ность, г/см3 2 0 nD Т. кип., °C Т. всп °C
при 0,1 МПа при дав- лен ИИ (ЙШ
рт. с т.)
Метил . , . s s. , , » 2 1,0211 * 1,4263 194 105 (40) 93,3
Метил 3 1,0494* 1,4370 241 121 (5) 118,4
Метил . . 4 — 1,4452 — 162 (5) —
Этил 2 0,9890 * 1,4273 201,9 99,6 (20) 96
Этил 3 1,020 1,4380 254,4 125 (Ю) —
Этил 4 1,045 •1,4450 305 140 (3) —
«-Пропил 2 0,9685 1,4288 — 110,3 (20) —
«-Пропил . , ; 3 — 1,4386 — 129 (5) —
и-Проппл 4 — 1,4452 — 165 (5) —
Изопропил 2 — 1,4267 — 110 (40) —
Изопропил 3 — 1,4362 — 125 (5) —
и-Бутил ....... 2 0,9536 * 1,4316 230,6 122,5 (20) 115
«-Бутил 3 — 1,4400 142 (5) —
и-Бутил 4 — — 173- -177 —
(13)
Изобутил 2 — 1,4289 100 (8) —
Изобутил 3 — 1,4357 140 (5) •—
втор-Бутил 2 — 1,4300 95 (8) —
^тмор-Бутил 3 — 1,4300 138 (5) —
и-Октил 2 — 1,4417 147 (4) —
н-Октил 3 — 1,4454 179 (4) —
* Относительная плотность при 20 °C.
Простые эфиры гликолей с близкой молекулярной массой обла-
дают схожими свойствами. Замещение водорода гидроксильной
группы гликоля уменьшает вероятность межмолекулярной водород-
ной связи, поэтому моно- и особенно диэфиры гликолей кипят прп
более низкой температуре и имеют меньшую плотность, чем сами
гликоли.
Для моноэфиров гликолей есть несколько отклонений зависимости
температуры кипения от молекулярной массы. Так, метиловый
и этиловый эфиры пропиленгликоля, имеющие вторичную гидр-
оксильную группу, кипят при температуре ниже соответствующих
эфиров этиленгликолей с первичной ОН-группой. Этот факт хорош0
объясняется затрудненностью образования межмолекулярных свя-
зей для эфиров пропиленгликолей с вторичной ОН-группой. С уве-
личением молекулярной массы различия в свойствах многих эфироВ
выравниваются. По-видимому, из моиоэфиров ди-, три- и тетра-
алкиленгликолей всегда можно подобрать близкие по свойства^
соединения.
294
Таблица 87. Физические свойства технических моноэфпров
пропиленгликолей [15, 16, 19]
Эфир Плот- ность, г/см3 20 "d Т. кип., °C Т, всп., °C
при 0,1 МПа при давле- нии (мм рт. ст.)
Метиловый эфир дипропилен- гликоля 0,950 * 1,4190 189,9 87,8 (20) 82,3
Метиловый эфир трипропи- ленгликоля 1,4297 100—101 (2)
Метиловый эфир тетрапро- пиленгликоля 1,4348 124—125 (2)
Метиловый эфир пентапропи- ленгликоля 1,4383 151—155 (2)
Этиловый эфир дипропилен- глпколя 0,930 * 1,4210 197,7 95,0 (20) 90,7
н-Пропиловып эфир дипропп- ленгликоля 0,919 1,4229 213,4 107,7 (20) 119 (20)
н-Бутпловый эфир дипропи- 0,913 * 1,4250 231,0 113,0
ленгликоля ** н-Бутиловый эфир трипро- 0,936 1,4332 108-110 (3)
пиленгликоля
* Относительная плотность при 25 °C.
** Растворимость в воде 4,3 г/100 г.
Таблица 88. Физические свойства простых диэфиров гликолей [2, 19]
Эфир
Плот- ность, г / см3 20 nD Т. кип., °C Т. пл., °C Т. всп., °C
при 0,1 МПа при 10 мм рт. ст.
Диэфир моноэтиленгликоля
Диметиловый , 8 0,8692 85 14 —71,0 4,40 оо со
Диэтиловый . . 0,8417 1,3922 121 20 —74,0 35,0 21,0 3,4
Ди-и-бутиловый е 0.8374 1,4131 203 84 —69,1 84,4 0,2 0,6
Д и эфир![ ди этиленгликоля
Диметпловый . . 0,9514 1,4097 161 — — ОО СО
Диэтиловый . . 0,9082 1,4115 189 — -44,3 82,3 оо оо
Дибутиловый . . 0,8853 1,4233 256 — —60,2 118,0 0,3 1,4
—
295
Интересным проявлением межмолекулярной водородной связи
эфиров гликолей является образование азеотропных смесей типа
моногликоль — моноэфир дигликоля и дигликоль — моноэфир три-
гликоля [25]. Свойства азеотропной смеси этилкарбитол — этилен-
гликоль приведены ниже:
Содержание гликоля, % . 46,0 32,5 28,8 27,1 23,1 20,5
Температура кипения, °C . 194 135 123 114 97,4 85,4
Давление, мм рт. ст. * . 760 100 60 40 20 10
Плотность, г/см3 .... . 1,0480 1,0301 1,0267 1,0246 — 1,0166
* 1 мм рт. ст. = 1 33,32 Па.
Основные физические и физико-химические показатели наиболее
широко применяемых эфиров гликолей — этилделлозольва и этил-
карбитола, а также циклического эфира 1,4-диоксана приведены
ниже [2, 14]:
Этилцел- лозольв Этилкарби- тол 1,4-Диоксап
Молекулярная масса >.... 90,12 134,03 88,10
Плотность при 20 °C, г/смэ . . . 0,9300 0,9870 * 1,0337
Относительная плотность . . 0,9311 0,9880 1,0356
Температурный коэффициент относительной
плотности bd/kt 0,00090 0,000887 0,00112
Температура кипения прп 101,3 кПа, °C
(760 мм рт. ст.) । . 135,1 201,9 101,3 .
замерзания —70,0 —45,0 11,8
вспышки 44,4 94,4 18,33
Температурный коэффициент давления пара
Дг/Ар,°С/мм 0,042 0,049 0,043
Давление насыщенного пара прп 20 °C . .
Па 505 13,3 3860
мм рт. ст 29 3,8 0,1
Коэффициент преломления Пд 1,4076 '1,4273 1,4224
Удельная теплоемкость при 20 °C
Дж/(кг - К) 2,32 2,31 1,71
кал/(г - °C) 0,555 0,552 0,410
Вязкость при 20 °C, мПа - с (сПз) 2,1 4,5 1,3
Теплота испарения при т. кип.
Дж/г 403 412
кал/г 130 96,2 98,4
Область воспламенения, % (об.) 2,6-15,7 — 1,97—22,5
—
Этилцеллозольв и этилкарбитол при обычных условиях — доста-
точно подвижные бесцветные жидкости со слабым «гликолевым»
запахом. Этилцеллозольв полностью смешивается с водой и со всеми
используемыми в технике растворителями. По растворяющей спо-
собности этилкарбитол близок к этилцеллозольву, но в пем ограни-
ченно растворяются углеводороды, особенно при пониженных
температурах.
296
Для водных растворов этилцеллозольва и этилкарбитола харак-
терны уменьшение объема и низкие температуры замерзания. Так,
50%-ный раствор этилкарбитола замерзает при температуре ниже
—55 °C. Растворы указанных эфиров в этилен- и диэтиленгликолях
замерзают при более низкой температуре, чем чистые вещества,
плотности этих растворов подчиняются правилу аддитивности.
Простые циклические эфиры
Молекула 1,4-диоксана представляет собой симметричный шести-
членный цикл и является самым термодинамически стойким поли-
мером окиси этилена. Свойства 1,4-диоксана схожи со свойствами
простых эфиров этиленгликоля, но наличие цикла значительно
усиливает акцепторную способность атома кислородй. Сопряжение
находящихся в a-положении неспаренных электронов кислорода
приводит к усилению заряда на одном из этих атомов. 1,4-Диоксан
весьма склонен к образованию водородных связей [2, р. 119; 26,
с. 109; 27, с. 33].
При обычных условиях 1,4-диоксап — бесцветная прозрачная
жидкость со слабым запахом. Он смешивается с водой, с большин-
ством органических растворителей, масел и смол. С водой 1,4-ди-
оксаи образует при атмосферном давлении азеотропную смесь,
содержащую 81,6% основного вещества и кипящую при 87,8 °C;
прп 34,8 кПа (260 мм рт. ст.) она кипит при 60 °C и содержит 84,6%
1,4-диоксана [2, с. 120]. Основные свойства 1,4-диоксана даны
на стр. 296.
Гомологи 1,4-диоксана описаны в литературе очень кратко.
Продукт, синтезированный при нагревании пропиленгликоля с сер-
ной кислотой, кипит при 122—125 °C. Чистый 2,5-диметил-1,4-ди-
оксап имеет: т. кип. 120—121 °C, = 0,9244 и = 1,4169 [28].
Температуры кипения метил-1,4-диоксана и 2,2,5,5-тетраметил-1,4-
диоксана равны 111,5 и 138 СС, а плавления — минус 67 и минус
105 °C соответственно.
Более подробно в литературе представлены 1,3-диоксоланы,
особенно замещенные в положение 2 [2, р. 125]:
1,3-Диоксолан..............................
2-Метил-!.,3-диоксолаи ....................
2-Этил-! ,3-диоксолан......................
2-Пропил-1,3-диоксолап.....................
2-Этокси-1,3-диоксолан.....................
2,2-Диметил-1,3-диоксолан .................
2-Метил, 2-бутил-1,3-диоксолан.............
2-Метил, 2-фепил-1,3-диоксолан.............
Плотность, Г/СМ3 Т. иип., °C п20 nD
1,063 * 74-75
0,988 * 81—82 _—
0,954 * 105 —
0,943 130—131 —
1,053 120—123 1,4000
0,9417 92,5 1,4000
0,922 62—63 (20 мм) 1,4232
0,897 88-89 (11 мм) 1,4200
* Относительная плотность dgg.
297
1,3-Диоксолан полностью смешивается с водой и большинством
органических веществ. С водой он образует азеотропную смес:,
первого рода, содержащую 6,7% воды. При замещении алкилом
в положение 2 по мере роста молекулярной массы увеличивается
температура кипения диоксолана, но снижаются его плотность
и растворимость в воде.
Аналогичная зависимость показателей от молекулярной массы
обнаруживается и для диоксоланов, замещенных в положении 4.
Их получают в основном из замещенных <х-окисей. Все 1,3-диоксо-
ланы вследствие малой способности к образованию межмолекуляр-
ных водородных связей кипят при более низкой температуре, чем
соответствующие им гликоли или алициклические эфиры с одинако-
вой молекулярной массой.
Сложные эфиры
Известны многочисленные сложные эфиры органических и неорга-
нических кислот для «-гликолей. Сложные эфиры имеют более высо-
кую температуру кипения и плавления, плотность и коэффициент
преломления, чем простые; у сложных диэфиров эти показатели
выше, чем у смешанных. Свойства нескольких низкомолекулярных
эфиров моцокарбоновых кислот этилен- и пропиленгликолей при-
ведены в табл. 89.
Таблица 89. Физические свойства сложных эфиров
моноэтилен- и пропиленгликолей [2]
Эфир 2 0 Плотность а 20 2 0 п D Т. кип, °C ***
этилен- гликоля пропилен- гликоля этилен- гликвля пропилен- гликоля этилен- гликоля пропилен- гликоля
Моноформиат . . 1,0890 * 179,7
Моноацетат . . . 1,1060 1,065 1,4224 1,4197 182,0 182,5
Монобутират . . — — — 1,4246 ** 220,0 56-57 (1
Монобензоат . . 1,0937 * — — — 173 (21) 142 (8)
Диформиат . . . 1,2277 1,136 1,4153 1,4130** 177,1 116 (111
Дпацетат .... 1,1063 1,059 1,4159 1,4173 190,5 190—191
Днбутират . . . 1,0240 * — — — 240,0 — •
* Относительная плотность при 15 °C.
** При 25 °C.
*** в скобках указано давление (мм рт. ст.).
Сложные эфиры пропиленгликоля мало отличаются от соответ-
ствующих производных этиленгликоля по температуре кипения-
плотности и коэффициенту преломления. Различия свойств ациЛ'
замещенных первичных и вторичных ОН-групп пропиленгликоля
298 '
весьма незначительны [2, р. 252; 29], поэтому точный изомерный
состав сложных моноэфиров пропиленгликоля часто не установлен.
Моноформиат и ацетат этиленгликоля смешиваются с водой
и большинством органических соединений, но уже диацетат этилен-
гликоля ограниченно растворяется в воде. Заметные изменения
свойств сложных эфиров гликолей достигаются при введении в их
молекулу гетероатомов или арильных радикалов, например, синте-
зированных из замещенных а-окисей и галогенангидридов органи-
ческих кислот [12, с. 229].
Из сложных эфиров гликолей и неорганических кислот (нитра-
тов, сульфатов, боратов, карбонатов и др.) наиболее подробно опи-
саны этилен и диэтиленгликольдинитраты. Они полностью смеши-
ваются с диэтиловым эфиром, ацетоном, бензолом, метиловым
спиртом, нитроглицерином, хлороформом и ограниченно с водой [30].
Весьма интересной разновидностью сложных эфиров гликолей
являются алкиленкарбонаты — циклические эфиры угольной
кислоты [31, 32]. Вследствие большой полярности эти эфиры сме-
шиваются со многими органическими высокомолекулярными веще-
ствами и водой. Особенно удобен в работе пропиленкарбонат, кото-
рый в отличие от кристаллического этиленкарбоната при обычных
условиях является достаточно подвижной жидкостью (т. пл. минус
49,2 СС).
Из всех сложных эфиров гликолей наибольшее распространение
имеют эфиры высших карбоновых кислот, которые обладают свой-
ствами неионогенных ПАВ и прекрасно совмещаются со многими
полимерными материалами [33, 34]. Если сложные эфиры этих
кислот и моноэтплен- и пропиленгликолей — кристаллические
вещества (табл. 90), то продукты, содержащие несколько оксиалки-
леновых групп, пастообразны. Поверхностная активность эфиров
Таблица 90. Свойства некоторых моно- и диэфиров гликолей
и высших карбоновых кислот [2]
Эфир Плот- ность эфира этилен- гликоля при т. пл. 20 Пд эфира Т. пл. эфира, °C
этилен- гликоля пропи- лен- гликоля этилен- гликоля пропи- лен- гликоля
Монолаурат 1,4440 27,5 14
Моноолеат 1,4600* 1 6
Монопальмитат 0,8786 1,4411 1,4405 50 54,3
Моностеарат 0,8780 1,4310 1,4424 57 40
Дилаурат — — — 52 35
Дипальмитат 0,8594 1,4378 1,4364 70 68,8
Дистеарат 0,8581 1,4385 1,4366 75,5 72,3
При 27 °C.
299
характеризуется соотношением доли лиофильных и гидрофильных
групп [35]. При взаимодействии а-гликолей с многоосновными
карбоновыми кислотами образуются сложные полиэфиры, явля-
ющиеся смесью веществ различной молекулярной массы. Как пра-
вило, это твердые полярные вещества с мелкокристаллической
структурой, обладающие высокой избирательной способностью при
сорбции паров и газов [36].
Смешанные сложные и простые эфиры
Смешанные эфиры гликолей, включающие в свой состав простую
и сложную эфирные группы, по свойствам занимают промежуточное
положение между простыми и сложными эфирами. Сочетание простой
и сложно-эфирной групп в одной молекуле способствует растворя-
ющей способности эфира. В табл. 91 приведены некоторые свойства
используемых в промышленности смешанных эфиров моно- и ди-
этиленгликолей.
Таблица 91. Физические свойства ацетатов простых эфиров
моно- и диэтиленгликолей
Радикал R Плот- ность 20 d 29 2 0 nD Т. кип., °C Т. пл., °C Т. всп., °C Раствори при 20 °( « в воде мость 3, г/100 г воды в
Эфир этиленгликольацетата
Метил 1,0067 1,4019 145,0 —65,1 57,3 ОО оо
Этил 0,9748 1,4058 156,0 -61,7 65,4 23,0 6,5
Бутил 0,9424 1,4138 192,3 -63,5 87,8 1 >5 1,7
Фенил 1,1084 1,5076 259,2 —2,7 143 0,17 0,92
Эфир ди этиленгликольацетата
Метил 1,0412 1,4209 209,1 -99,3 110 ОО оо
Этил 1,0114 1,4213 217,4 -25,0 ПО оо оо
Бутил 0,9810 1,4262 246,7 —32,0 115 6,3 3,7
Химические свойства эфиров гликолей
Химическую активность эфиров гликолей целесообразно рассмо-
треть в зависимости от тех же факторов, которые определяют их
физические свойства: степени замещения исходного гликоля, строения
и числа окспалкиленовых групп, природы эфирной связи (простая
или сложная), а также от свойств заместителей.
Ациклические монозамещенные эфиры гликолей имеют одн)
гидроксильную группу и обладают многими свойствами спиртов-
.390
Как простые, так и сложные эфиры гликолей являются слабыми
кислотами, диссоциирующими с выделением протона:
R—[—О—СН2СН2—]п—ОН R—[—О—СН2СН2—]n—О_-]-Н+
Их кислотность зависит от строения радикала и числа окси-
этиленовых групп [17]. Наиболее значительное изменение кислот-
ности отмечается при переходе от спирта к простым эфирам моно-
и диэтиленгликоля. Водород гидроксильной группы легко вытес-
няется щелочными металлами и их гидроокисями, но в случае слож-
ных эфиров гликолей предпочитают металлы, чтобы избежать омы-
ления эфирной связи [37].
В отличие от гликолей некоторые их моноэфиры реагируют с алю-
минием даже в мягких условиях. Так, метилцеллозольв растворяет
алюминий уже при комнатной температуре. Технический алюминий
реагирует при этом активнее химически чистого, а выделившийся
водород содержит 99,8% основного вещества. При отгонке в вакууме
непрореагировавшего метилцеллозольва в кубе остается жидкий
однородный продукт светло-коричневого цвета. Попадание влаги
вызывает быстрый гидролиз алкоголята алюминия.
Замещение водорода ОН-группы достигается также в реакции
этерификации моноэфиров гликолей с помощью карбоновых кислот
и их сложных эфиров, сульфирующих и нитрующих агентов [35, 38]:
R-O-CH2CII2OH-|-R'COOH --> R-O-CH2CH2-O—СО—R'4-H2o
R-о—СН2СН2ОН-|-АсОС2Н5 > R—О—CH2CH2OAc4-C2H5OI-I
R—О—(С2Н4О)3Н +S0"; -X'iC)IT > R—О—(C2H4O)3SO3Na-f-H2O
Указанным способом этерифицируют также моноэфиры поли-
гликолей.
Наибольшее практическое применение для гликолевых моно-
эфиров нашла реакция p-оксиалкилирования, осуществляемая прп
небольшом давлении и 120—200 °C в присутствии катализаторов,
обычно щелочного характера [39, 40]:
R-O-CH2CH2OH-J
r-(-ocii,ch2-\—он
' >П+1
Ас—О—СН2СН2ОН +
Ас—(—О—СН2СН2—\—он
' /п-1
Наряду с целевыми продуктами образуется некоторое количество
Полигликолей, реакционная масса часто бывает окрашенной и тре-
бует дополнительной очистки.
Катализаторы типа BF3 позволяют работать при более низкой
температуре (50—75 °C), дают бесцветные продукты, но 10—20%
окиси превращается в 1,4-диоксан, 1,3-диоксолан и другие низко-
молекулярные соединения [35]. Когда используют смеси окиси
этилена и окиси пропилена, образуются эфиры нерегулярного
301
строения, в которых отмечается некоторое чередование оксиэтил!
новых и оксипропиленовых групп.
Эфиры гликолей, синтезированные из высших непредельны
спиртов и кислот, являются весьма стойкими соединениями и широк
используются в технике. Но виниловый эфир этиленгликоля вслед
ствие сопряжения ОН-группы и двойной связи может изомеризо
ваться со взрывом до метилдиоксолана [41]:
О
сн2=сн-о-сн2сн2он —> сп3-нс(^
\ Л На.
(У
Спиртовая группа моноэфира может окисляться до карбоксиль-
ной с образованием оксизамещенных монокарбоновых кислот. Реак-
ция идет при повышенной температуре в щелочной среде в присут-
ствии перекиси никеля или солей кобальта [42]:
R-O-CJI2CII2-O-CH2CH2OH4-NaOCl ^н2о
--> R—О—СН2СН2—О—СН2СООН
Максимальное число оксиалкиленовых звеньев в эфире гликоля,
не приводящее к разрушению молекулы, равно 7 для производных
полиэтиленгликолей и 4 для пропиленгликолей,
Довольно жесткие условия требуются для аминирования простых
эфиров алкиленгликолей: температура 150—250 °C, повышенное
давление и наличие катализатора гидрирования — дегидрирова-
ния [43]
R(OCR'HCR"H)„OH+NH3 ——* [R(OCR'HCR"H)„]mNH(>_m)
где R — алкил Сх — С8; R' и R" — Н или СН3, С2Н5; п = 1—3;
т — 1—3.
Гидроксильные группы эфиров гликолей устойчивы к замещению
на хлор большинством хлорирующих агентов. Эту реакцию можно
осуществить с небольшим выходом только при действии хлористого
фосфора или хлористого тионила [44].
При нагревании и под действием облучения простые и сложные
эфиры гликолей распадаются с образованием легкокипящих и смо-
листых продуктов. Разложение ускоряется в кислой среде и может
сопровождаться реакцией диспропорционирования. В присутствии
хлорного железа из монометилового эфира этиленгликоля обра-
зуется диметиловый эфир этиленгликоля и этиленгликоль: •
2СН3—О—СН2СН2ОП —► СН3—О—СН2СН2—О-СП34-СН2ОНСН2ОН
Облучение способствует разрыву С—С-связи, и в продукта-
реакции обнаруживается метан, этан и формальдегид [45]. ПоД
действием HF и BF3 из диметилового эфира этиленгликоля оора'
зуется диметиловый эфир, 1,4-диоксан и следы метилового спирта
302
Пятифтористый фосфор ускоряет превращение диэфира диэтилен-
гликоля в 1,4-диоксан и простой диэфир [46]. При кипячении эти-
лового эфира диэтиленгликоля в присутствии серной кислоты обра-
зуются этиловый спирт, этилцеллозольв и ряд других соеди-
нений [47], но основным продуктом является 1,4-диоксан.
Нагревание монометил- и моноэтиловых эфиров этиленгликоля
над катализаторами дегидратации — дегидрирования приводит
к образованию альдегидов С2 и С4, спиртов Сх и С2, алкокспальдеги-
дов и др. [48]. Механизм реакции может быть как свободнорадикаль-
ный, так и ионный, так как в спектрах названных эфиров после
пропускания над катализатором обнаружены и свободные радикалы
и ионы.
В обычных условиях простые эфиры гликолей не подвергаются
гидролизу и достаточно стабильны. Нуклеофильные свойства этих
соединений проявляются в образовании комплексов с кислотами
Льюиса, фосфорномолибденовой кислотой, кремневольфрамовой и
железистосинеродистыми кислотами, отличающимися характерной
окраской [49]. Особенно сильно проявляется электронодонорная
способность простых эфиров гликоля у 1,4-диоксана. Он образует
оксониевые соединения с бромом и серным ангидридом, которые
легко выделяются в чистом виде, а также склонен к окислению [1,
с. 603; 2, р. 120; 50, с. 158].
Окисление и самоокисление простых эфиров гликолей является
автокаталитическим процессом, который идет уже при комнатной
температуре и ускоряется при нагревании или повышении давления
кислорода. Во всех случаях промежуточными продуктами являются
перекиси, распадающиеся с выделением формальдегида, формиатов
и других кислородсодержащих соединений [51].
В слабощелочной среде простые эфиры гликолей не способны
образовывать новые связи и поэтому могут длительное время хра-
ниться без изменений [52]; увеличение концентрации щелочи более
1% и нагревание способствуют их осмолению.
В работе [53] рассмотрена ионизация и распад 1,3-диоксолана
и 1,4-диоксана методом электронного удара. Потенциал ионизации
для первого соединения составил 1010 кДж/моль (241,06 ккал/моль)
и для второго 925 кДж/моль (220,7 ккал/моль). Для 1,3-диоксолана
характерен распад с выделением атома водорода, а у 1,4-диоксана
он начинается с разрушения циклической структуры.
Метилен, полученный фотолизом диазометана, внедряется в мо-
лекулу 1,3-диоксолана преимущественно по С—Н-связи, хотя воз-
можна реакция и по связи С—О [54]:
О о
тт с/ Х(рН2-1-ён _ Н С НС^ ХСН2
| "Г '^П-2 " XIS'-1—।
\ .СН2 \ си2
ОХ 0х
303
При действии магнийорганических соединений на 2-алкоксп
1,3-диоксолан получен 2-алкил-1,3-диоксолан [55]. Этот пример,
как и приведенные в работе [54], подтверждает большую (по срав-
нению с линейной) прочность циклической простой эфирной связи.
Средняя энергия напряжения цикла в 1,3-диоксолане, вычисленная
по теплоте сгорания, составляет всего 26,4 кДж/моль (6,3 ккал/моль).
Под действием кислых катализаторов (H2SO4, BF3, толуолсуль-
фокислоты, галогенидов металлов) 1,3-диоксолан полимеризуется,
а также образует продукты полиприсоединения с аминами, спиртами
и карбоновыми кислотами [55, с. 348].
По стабильности сложные эфиры гликолей значительно усту-
пают простым главным образом из-за способности к гидролизу и
переэтерификации. Этиленгликольдпформиат медленно гидроли-
зуется уже при обычных условиях:
11СООСП2СН2ООСИ-р 112О —> 2CHOOII-I- СН2ОНСН2ОН
Конверсия этиленгликоль диацетата в гликоль при температуре
около 100 °C и при избытке воды в течение 1—2 ч составляет выше
99%. Гидролиз [З-оксинитроэфиров в присутствии кислот сопрово-
ждается циклизацией с образованием диоксанонов [56]:
R— СН—СП2—О—CII2CII2NO,
ОН
О
+н+: +Н£О^ Н2С C1I2 +NIl2()H
RHC^ уС=О
О
Из низкомолекулярных сложных эфиров гликолей относительно
стойки к гидролизу алкпленкарбонаты, в частности пропиленкарбо-
нат. С водой он образует три типа соединений, распадающихся прп
умеренном нагревании до исходных веществ [32]:
СН3—нс—СН2
0 9
С
Нх I!
ХО-Н...О
СИз-НС—С112
О О
' \ Z ' и
С \
При кипячении пропиленкарбоната с водой при атмосферном
давлении получается пропиленгликоль.
Реакция переэтерификации при синтезе моноэфиров карбоновых
кислот приводит к образованию гликолей [35]:
2RCOO(C2H4O)nH - +~ И" -> RCOO(C2IJ4O)„OCR + IIO(C2H4O)nH
В основном за счет внутримолекулярной переэтерификации
объясняют также преобладание замещенных первичной гидроксиль-
ной группы при ацилировании пропиленгликоля [29, 38]. В некото-
рых случаях эта реакция используется для специальных синтезов
304
диэфиров. Так, диолеат этиленгликоля готовят из моноолеата,
пропуская его (в растворе легкой нафтеновой фракции) над активи-
рованной окисью алюминия. Иногда химическая активность эфиров
гликолей определяется функциональной группой, примыкающей
к эфирной связи.
Моно- и дивиниловые эфиры этилен- и диэтиленгликолей легко
полимеризуются, образуя прозрачные блоки [41]. Аналогично ведет
себя диметакриловый эфир триэтиленгликоля. Окисление моно-
и диолеата этиленгликоля в токе кислорода (катализатор олеат
урана) приводит к образованию вязких окрашенных жидкостей
с незначительным содержанием гидроперекисей [57].
Токсические свойства эфиров гликолей
Многочисленные эксперименты на животных, а также системати-
ческое наблюдение за людьми, занятыми производством и исполь-
зованием эфиров гликолей, позволили установить, что характер
действия на организм эфиров гликолей и самих гликолей в общем
одинаков [2, р. 305; 58, с. 347]. При длительном вдыхании паров,
при попадании через кожу или рот наблюдается головная боль,
общая депрессия, апатия, психическое расстройство, изменение
состава крови, поражение печени и почек. При больших дозах воз-
можна гематурия (кровь в моче), а также кровотечение в пищевари-
тельном тракте. Отравление может быть обнаружено по анализу
крови.
Токсичность эфиров уменьшается по мере снижения давления
их паров. Для некоторых веществ установлены предельно допусти-
мые концентрации в воздухе производственных помещений (мг/м3)
[58; 59, с. 352]:
Метилцеллозольв ............... 80 Монометиловый эфир пропилен-
Этилцеллозольв ............... 200 гликоля ....................600
Вутилцеллозольв .............. 240 1,4-Диоксан.................... 10
Для этилцеллозольва ПДК в воде водоемов равно 1,0 мг/л [60,
с. И].
Некоторые эфиры гликолей, используемые как ПАВ, обладают
кумулятивным действием. Клиническая картина отравления не-
ионогенными (стереокс-6, препарат ОС-20) и катионоактивными
(выравниватель А и алкомон ОС-2) ПАВ свидетельствует об обще-
токсичном характере их действия с преимущественным поражением
Нервной системы. ПДК в воде водоемов составляет для стереокса-6,
алкомона ОС-2, выравнивателя А и препарата ОС-20 соответственно
1,0, 0,5, 0,3 и 0,1 мг/л [61].
Альгинаты этиленгликолей нетоксичны и употребляются для
Приготовления пищевых эмульгаторов и загустителей [62, с. 189].
Сложные высокомолекулярные эфиры пропиленгликолей также без-
вредны для людей, их используют при консервировании продуктов,
20 Заказ 972 305
как добавки к сливочному маслу и в тесто [28, 63]. В целом считается
что неионогенные ПАВ безвредны при приеме с кормом животных
в количестве до 1 %; однако доза выше 15% смертельна [64].
Из имеющихся результатов можно сделать несколько обобщений
по токсичности эфиров гликолей:
наиболее ядовитыми являются циклические простые эфиры типа
1,4-диоксан и 1,3-диоксолан;
простые ациклические эфиры более токсичны, чем сложные;
при переходе от производных этиленгликоля к соответствующим
производным пропиленгликоля происходит снижение физиологи-
ческой активности;
для одного гомологического ряда токсичность уменьшается по
мере роста молекулярной массы.
Можно полагать, что чем более активно эфир гликоля (например,
1,4-диоксан или метилцеллозольв) вступает в химическое взаимодей-
ствие, начиная с комплексообразования, и обладает большей рас-
творяющей способностью, тем выше его токсикологическая актив-
ность.
Последствия легкого отравления эфирами гликолей обычно исче-
зают при исключении контакта с ядом. Результаты работ по биоло-
гическому разложению оксиалкилированных продуктов приведены
в [59, с. 316; 64]. ’
Методы получения эфиров гликолей
Синтезы на основе окисей олефинов
Для получения эфиров гликолей используют главным образом окиси
олефинов, непосредственно олефины, а-гликоли и их галогенпроиз-
водные. Наиболее распространенным методом синтеза в настоящее
время является оксиалкилирование спиртов, фенолов и карбоновых
кислот по реакциям [39, с. 389; 65]:
RCH2OH + nC2H4O---> RCH2O(CH2CH2O)„H
R
--O(CH2CH2O)nH
RCOOH + nC2H4O
।-> RCOO—(—CH3CH2O—)n—H
I-> RCOO—(~CH2CH2O—)n—OCR+ H2O
Они протекают по общему механизму, который предполагает
разрыв окисного цикла при совместном воздействии нуклеофильного
и протонодонорного агентов:
н2с—сн2+па+а- —>
’А-..-Н2С—сн
ПА - • •
--> НОСН2СН2А + А-
о
306
В отсутствие катализаторов эту двойную функцию выполняет
одна молекула реагента:
Н,С—CH2+RCOOH —>
RC
^О---Н.2С---СН2’
\он • • — о
RCOOCH2CH2OH
Так как в следующем разделе подробно описаны кинетика и меха-
низм взаимодействия окисей олефинов со спиртами, здесь рассмо-
трены общие приемы синтеза сложных и простых эфпров, а также
их принципиальные отличия.
Состав продуктов реакции в нейтральных или слабощелочных
средах будет определяться нуклеофильностью реагентов, содержащих
активный атом водорода. Анионы кислоты являются более сильными
нуклеофилами, чем сложные эфиры гликолей, и поэтому кислоты
полностью связываются в эфиры моногликолей. Только после рас-
ходования всей кислоты начинается образование эфиров ди- и поли-
гликолей. Аналогичным образом ведут себя фенолы.
При получении простых эфиров нуклеофильная активность окси-
этилированных производных обычно выше, чем у исходных спиртов.
По этой причине всегда образуется смесь эфиров моно-, ди- и поли-
алкиленгликолей. Для обеспечения 85—90%-ного выхода эфира
моногликоля требуется 8—10-кратный избыток спирта.
Избыток кислоты при щелочном катализе [66] не влпяет на
выход сложного эфира моногликоля. В кислой среде избыток кислоты
приводит к образованию смеси моно- и диэфира. Повышению выхода
диэфиров, в частности диацетата этиленгликоля, способствует при-
менение ангидридов кислот [67]. В случае кислотного катализатора
при этерификации низших спиртов обнаруживаются также диэфиры,
но их содержание, как правило, невелико [2, р. 118].
При обычных условиях взаимодействие окисей олефинов с карбо-
новыми кислотами и особенно спиртами идет очень медленно. Про-
стой нагрев оказывается малоэффективным. Взаимодействие в паро-
вой фазе, например, для окиси этилена, метилового или бутилового
спиртов не начинается вплоть до 350 °C [27, с. 64].
Для ускорения реакции оксиэтилирования карбоновых кислот
и спиртов предложено множество различных веществ кислотного
я основного характера, цеолитов и других полярных веществ [68,
69]. С помощью больших доз катализаторов (10% тиоэфира, 2%
КОН) часто удается синтезировать эфиры гликолей даже при ком-
натной температуре [70].
В промышленности применение высоких концентраций катализа-
тора осложняет очистку и выделение продуктов реакции. Добавки
катализаторов стараются свести к минимуму за счет повышения тем-
пературы и давления.
В процессах при давлении используют жидкие окиси олефинов,
Кто приводит к увеличению скорости оксиалкилирования и сокра-
щению объема реакционных аппаратов. По мере увеличения
Температуры кипения исходной кислоты или спирта рабочее давление
20* 307
снижается. Для получения сложных и простых эфиров из кислот
и спиртов С1? — С?п достаточно давления 0,15—0,5 МПа (1,5—
5 кгс/см*) [69, р. 172].
Рассмотрим для примера взаимодействие окиси этилена и олеиновой кис-
лоты в присутствии едкого кали или аминов [66]. Процесс осуществляется
при 95 ± 3 °C. В автоклав загружается 2 моль олеиновой кислоты, 0,03 моль
КОН или 0,01 моль триэтиламина и при перемешивании вводится под давлением
2,6 моль окиси этилена. В присутствии КОН получено 72,9% моноэфира, 9,3%
диэфпра и 18% этиленгликоля. В случае катализа трйэтиламином получено
91,2% моноэфира, 2,4% диэфира и 6,5% этиленгликоля.
Реакция в присутствии КОН имеет первый порядок по кислоте, окиси
и катализатору. Катализ же амином дает общий второй порядок и первый по
окиси этилена и катализатору; реакция идет при этом в 4 раза быстрее, чем
в присутствии едкого кали.
Оксиэтилирование стеариновой кислоты проводилось при темпе-
ратуре до 150 °C, давлении до 0,52 МПа (5,2 кгс/см2) и концентрации
NaOH — 0,2% от содержания кислоты [35].
При оксиэтилировании низкомолекулярных кислот и особенно
спиртов приходится ужесточать условия реакции: если при реакции
с кислотами Сх — С4 давление не превышает 1 МПа (10 кгс/см2),
а температура 150 °C, то взаимодействие окисей с соответствующими
спиртами проводится при давлении до 4 МПа (40 кгс/см2) и темпера-
туре до 220 °C. ,
Синтезы через галогенпроизводные
Самым старым способом получения ациклических эфиров гликолей
является алкилирование галогенсодержащими веществами соедине-
ний с активным атомом водорода, замещенным на металл. Действуя
иодистыми алкилами на моногликолят натрия, можно получить
простые моноэфиры этиленгликоля с выходом до 90% [27, с. 204]:
CH2OlICH2ONa-[-Rl —> CH2OHCH2OR + NaI
Простые моноэфиры образуются также при действии олефин-
хлоргидрина на алкоголят натрия:
RONa-J-СН2ОНСН2С1 ---> CH2OHCH2OR -j-NaCl
Выход эфиров нормальных спиртов составляет 60—65%, а для
изоспиртов он снижается до 30—35%. Наряду с эфирами моноэти-
ленгликоля образуются эфиры диэтиленгликоля, выход которых
составляет 10—15% [71].
Хлоргидрины три- и тетраэтиленгликолей конденсируются с бу-
тилатом натрия, при этом получаются соответствующие простые
бутиловые эфиры этиленгликолей [72]. С меньшим выходом реаги-
рует хлоргидрин диэтиленгликоля, который под действием едкого
натра, а также разбавленных алкоголятов натрия этилового и бути-
лового спиртов переходит в 1,4-диоксан. Этот циклический эфир
получается и при взаимодействии едкого натра и дихлордиэтилового
эфира [1, с. 601].
308
Этиленхлоргидрин также легко реагирует с фенолятом натрия
п солями карбоновых кислот с образованием простых и сложных
эфиров [50, с. 348].
Оксиалкильные сложные эфиры этиленгликоля общей фор-
мулы RCHOHCOOCH2CH2OOCR' получены из RCHOHGOONa
и CICH2CH2OOCR' [73]. Использование бифункционального ре-
агента позволяет получать сразу диэфиры [74]:
Na0CH2CH20Na-|-RCl--> ROCH2CH2OR + NaCl
2RCOONa+CH2BrCH2Rr----> RCOOCH2CH2OOCR+ 2NaBr
С целью повышения выхода обычно предпочитают последова-
тельное введение эфирных групп. Диметиловый эфир этиленгликоля
получается из метилцеллозольва, металлического натрия и хлори-
стого метила с выходом 78% [50, с. 334]. Сложный моноэфир пере-
водят в диэфир в присутствии кислотного катализатора за счет
реакции переэтерификации [57, 75].
Моно- и диэфиры гликолей можно синтезировать без специаль-
ного получения алкоголятов или солей карбоновых кислот. Так,
из этиленгликоля и алкилгалогенида получают 2-алкоксиэтиловые
спирты, нагревая их в растворе едкого натра. Из этиленхлорги-
дрина, жирной пли нафтеновой кислот в присутствии 40%-ного
раствора NaOH с высоким выходом образуются сложные моноэфиры
гликолей. 2-Метоксиэтилхлоргидрин и спирт при наличии гидро-
окиси металла превращается в диэфир гликоля. Таким образом
получаются соединения общего типа:
CH3-O-CH2CH2-(-OCH2CII2-)„-O-R
где R = Cj — С4, п = 0 3.
Из монохлоруксусной кислоты и спирта получают смешанный
сложный и простой эфиры этиленгликоля, сначала нагревая их
со щелочью и затем с серной кислотой или серным ангидридом.
Выход мет- и этоксиацетатов этиленгликоля по этому способу дости-
гает 90-95% [76].
Синтезы на основе гликолей
Образование простых и сложных эфиров при взаимодействии глико-
лей с диэтилсульфатом, кислотами и их ангидридами, ацетиленом
и некоторыми другими соединениями, а также в результате превра-
щения гликолей в присутствии ряда катализаторов рассмотрены
в гл. 1 (см. стр. 22). Ниже описаны некоторые особенности этих
синтезов.
Катализаторами реакции этиленгликоля с диэтилсульфатом на-
ряду со щелочью являются сульфаты ципка, никеля и хрома [77].
Для получения сложных эфиров используют серную, соляную,
фосфорную и арилсульфоновые кислоты, кислые соли, катиониты
и окись цинка. Сильные минеральные кислоты вызывают образова-
ние побочных веществ, и поэтому в ряде случаев удобнее работать
309
с катионитами [78]. Избыток гликоля приводит к образованию
мопоэфира. Для удаления реакционной воды используют другие
вещества, образующие с водой азеотропные смеси (толуол, бутило-
вый спирт).
Этерификации карбоновыми кислотами подвергаются не только
гликоли, но и их простые эфиры. Сложный эфир циклогексилцелло-
зольва получается из моноциклогексилового эфира этиленгликоля
и монокарбоновой кислоты в присутствии каталитических количеств
серной кислоты в среде толуола при температуре кипения послед-
него [79]. В присутствии окиси цинка к этилкарбитолу присоеди-
няются жирные кислоты С4 — С6 с образованием смешанного слож-
ного и простого эфира диэтиленгликоля:
Са115(ОСН2СН)2ОН%С4Н9СООН о - С2Н5(ОСН2СН2)2ООСС4Н9
Реакции гликолей с дикарбоновыми кислотами осуществляют
при 180—230 °C. Они ускоряются катализаторами этерификации
монокарбоновых кислот. Продукты реакции — сложные полиэфиры,
как правило, высокомолекулярные соединения и не могут быть
очищены перегонкой. Поэтому количество катализатора стараются
свести к минимуму, часто применяют катионит, который легко уда-
лить фильтрованием. При получении этиленсукцината использовали
смолу КУ-2 в количестве 2% от сырья. Выход эфира равен 94% [80].
Ароматические дикарбоновые кислоты конденсируются с гли-
колями в присутствии солей олова при 290—320 °C. Фталаты гли-
колей с выходом более 90% получают при 270 °C и давлении
0,6 МПа (6 кгс/см2) в присутствии 0,01—0,1% триэтаноламин —
ацетата кобальта [81].
а-Гликоли способны конденсироваться с некоторыми непредель-
ными соединениями. В присутствии серной кислоты вторичные
и третичные бутилены образуют соответственно вторичные и третич-
ные эфиры этиленгликоля. В щелочной среде моно- и диэтиленгли-
коль присоединяют ацетилен, давая виниловые эфиры [82].
Виниловый эфир этиленгликоля может быть получен также при
нагревании винилхлорида с гликолятом натрия.
С альдегидами и кетонами «-гликоли в присутствии кислотного
катализатора образуют производные 1,3-диоксолана [2, р. 123;
12, с. 181]. Циклические эфиры гликолей типа 1,4-диоксан или его
гомологи образуются при дегидратации «-гликолей [83].
Синтезы на основе олефинов
Весьма перспективным способом получения сложных эфиров гли-
колей является сопряженное присоединение монокарбоновой кис-
лоты к олефину в процессе его каталитического окисления:
.—► CH3COOCH2CIbOH-t-H20
СП3СООИ+СоН4-Ь02 -
1—> СН3СООСН2СН2ООССНз + Н20
310
Реакция ведется в жидкой фазе в присутствии окислительно-
восстановительного катализатора, включающего окись селена или
соли двухвалентного палладия, а также галогениды щелочных ме-
таллов, нитраты лития, меди и железа. Скорость реакции, катали-
зируемой двухвалентным палладием и нитратом лития, описывается
уравнением [84]:
—kPC2H4CPdX
где к — константа скорости; Рс2н« — давление этилена; СРЙХ —
концентрация соли двухвалентного палладия.
Выход моноацетата этиленгликоля зависит от избытка уксусной
кислоты, температуры, давления, концентрации катализатора и
в оптимальных условиях достигает 95%. Наряду с ацетатами этилен-
гликоля образуется некоторое количество ацетальдегида, винил-
ацетата и этилидендиацетата.
В растворе пропионовой и к-масляной кислот из этилена обра-
зуются соответственно монопропионат и моно-к-бутират этилен-
гликоля. Аналогичным образом синтезируются моноацетаты про-
пиленгликоля и а-бутиленгликоля из пропилена и а-бутилена.
Реакцию этилена, карбоновой кислоты С2 — С6 и кислорода можно
проводить с преимущественным выходом соответствующего ди-
эфира [56].
Другой тип каталитической системы позволяет получать р-окси-
алкиловые эфиры малеиновой кислоты взаимодействием высших
а-олефинов, малеинового ангидрида и перекиси карбамида:
I Со
I H2O2-H2NCONH2ARCH=CH2 + --►
В нсх /
СО
I ---► RCHCH2OOCCH=CHCOOH4-H2NCONH2
I I
I он
Реакция проводится в среде ацетонитрила при 45—55 °C и моль-
ном соотношении реагентов, блпзком к единице. Выход моноэфира
достигает 90—95% [85].
Простые эфиры гликолей образуются из олефина, первичного
алкоголя и органической гидроперекиси в присутствии катализатора
(титана, вольфрама, хрома, рения, урана или их соединений) [86]:
2RCH=CH2-bR'OOH-:-R"OII —> RCH2CII2OH + RCHOHCH2OR''-rR'OII
Алкоксилирование олефинов можно проводить также электро-
химическим методом, например:
Cell5CH=CH24-2CII3OH CeII5CH(OCH3)CH2(OCH3)
Выход диметоксифенилэтана составляет около 20% [87].
311
Получение эфиров гликолей
из а-окисей и спиртов
Теоретические основы
Взаимодействие а-окисей со спиртами относится к типу реакцп
окисного цикла с активным водородом. Реакция является последов^
тельно-параллельной, так как первичные продукты сами обладают
активным водородом и взаимодействуют с окисью. Она во многом
схожа с реакцией гидратации окиси этилена, особенно в отношении
активации окисного цикла, по различия в свойствах спиртов и воды
(кислотность и растворимость, специфическая сольватация и стери-
ческие затруднения) не позволяют полностью принять механизм
гидратации.
Некаталитический синтез. В отсутствие катализаторов раскрытие
цикла окиси при реакции со спиртами идет за счет нагревания.
Так как энергия активации в этом случае несколько больше, чем
в присутствии кислот и оснований [88, 89], температура реакции
на 30—80 °C выше, чем в каталитических процессах. Предполагается,
что Р-оксиалкилирование спиртов протекает в результате внутри-
молекулярных превращений комплекса окиси алкилена и спирта,
образованного за счет водородных связей в первой, быстрой стадии
реакции: ''
Н2С—CTio ROTJ —> Н2С—СТ12 —>•
o' ^O^-HOR
'Н2С—сн2
OR
--> HOCH2C1I2OR
Реакция имеет второй порядок. Для отдельных структурных
групп спиртов, например для насыщенных незамещенных спиртов
и целлозольвов, между реакционной способностью и кислотностью
наблюдается зависимость типа Бренстеда [89]:
I. й-0,22
КНОН '1(0ПЛК0!1
&roh — константа скорости реакции; Кк0н — константа иониза-
ции спирта; Groh — постоянная для одного ряда.
Это уравнение позволяет рассчитать относительную реакционную
активность (г) различных спиртов одного ряда. Для насыщенных
незамещенных спиртов R'OH и R"OH
г==(АК'он/Ак'оп)0’22
При сравнении целлозольвов с соответствующими спиртами или
спиртов с водой вводят поправочные коэффициенты 0,59 и 0,53:
Гцелл. =0,59 (ХВДС2Н1Он/^ВОн)0’22
312
Для различных по строению спиртов при одинаковой кислот-
ности наблюдается ряд активности, который однозначен и для. основ-
ного катализа:
ROH > ROC2H4OH > Н2О
СН2=СНСН2О~ > RO- > ROC2H4O- > он-
Скорость реакции несколько возрастает при увеличении ди-
электрической проницаемости среды, например при значительном
избытке спирта. Наиболее полно изучен некаталитический синтез
моноэфиров этиленгликоля [90, 91]. Исследована зависимость вы-
хода моноэфиров от соотношения спирт : окись, времени контакта,
температуры, давления и чистоты исходных веществ. Оптимальные
условия синтеза целлозольва таковы: температура 200—210 °C,
давление 3—3,6 МПа (30—36 кгс/см2) и время реакции 1—2 ч. Пре-
имущественное образование эфиров моногликолей достигается только
при 7—8-кратпом избытке спирта. Но даже в этих условиях реак-
ционные продукты всегда содержат эфиры ди- и триэтиленгликолей.
Из окиси этилена и метилового спирта, взятых в объемном отношении
1 : 7,5, образуется 83,5% эфира моно-, 11,4% эфира ди- и 0,8%
эфира триэтиленгликоля.
При получении этилцеллозольва наряду с целевым продуктом
идентифицированы этилкарбитол, этиленгликоль, диэтилацеталь ацет-
альдегида, ацетальдегид и диэтиловый эфир. Причиной образования
гликолей является вода, содержащаяся в спирте. Диэтиловый эфир
и ацетальдегид могут попасть в продукты реакции с сырьем, а также
образоваться при повышении температуры. Количество ацеталей
возрастает пропорционально содержанию альдегидов в сырье, но
в присутствии воды их выход снижается.
Известно, что раскрытие цикла окиси пропилена в реакции с на-
сыщенными спиртами идет по двум направлениям:
.—> СН3ОСН2СП(ОН)СН3
СН3ОН + СН3-НС—СН2 -
\ / 1—г СН3ОСН(СН3)СН»ОП
о
Были поставлены специальные опыты метанолиза окиси пропи-
лена в ампулах при 100 °C и с обратным холодильником при кипении
реакционной жидкости. В обоих случаях получали метиловый эфир
монопропиленгликоля, состоящий из 66,8% соединения с вторичной
ОН-группой и 33,2% — с первичной, хотя реакция при атмосфер-
ном давлении идет намного медленнее, чем в ампуле.
Основной катализ. При взаимодействии эпоксидов со спиртами
в качестве щелочных катализаторов используют гидроокиси метал-
лов, алкоголяты, карбонат натрия, третичные амины и другие соеди-
нения.
В случае катализа алкоголятами и гидроокисями металлов не-
которые авторы [92, 931 предполагали бимолекулярный механикум
Реакции по типу и SNi. Но показано, что наряду с анионом
313
в реакции участвует молекула спирта и реакция имеет общий третий
порядок [89, 94]:
—> НОСН2СН2А4-А-
Константа скорости реакции окиси этилена со спиртами и их
оксиэтильными производными в основном катализе также выра-
жается уравнением Бренстеда с показателем степени 0,65. Для учета
специфической сольватации кислорода окисного цикла и сопряжен-
ного основания в показатель степени вводят дополнительно а2:
1 __г-
^ROH-^ROH^ROH
где т а2 = 0,65.
Для бинарных смесей метилцеллозольв — метиловый спирт,
этилцеллозольв — этиловый спирт, этилкарбитол — этиловый спирт
наблюдается хорошее совпадение расчетных и экспериментальных
значений состава продуктов реакции. Коэффициент распределения
окиси этилена между эфиром гликоля и спиртом равен соответ-
ственно 0,7, 2 и 3,1 [89].
В реакции оксипропилирования октадеканола реакционная, спо-
собность эфиров ниже и составляет только 0,37—0,52 от реакционной
активности спирта [92]. По-видимому, найденные коэффициенты
справедливы только для довольно узкой области концентраций
катализатора. Прп кислотности спиртов, равной кислотности воды,
реакционная способность различных анионов и гидроксил-иона
относятся между собой как [89]:
RO- : ВОСоЩО- : СНГ = 1,00 : 0,48 : 0,33
Малая 'активность воды приводит к концентрированию ее в реак-
ционной системе, хотя часть воды при этом связывается в моно-
ди- и триэтиленгликоли [95].
Существует большая группа спиртов С! — С8, скорости окси-
этилирования которых соизмеримы и не зависят от кислотности
и строения спирта [96]. С повышением температуры увеличивается
скорость реакции. Полное взаимодействие окиси этилена и этилового
спирта при концентрации едкого натра 0,016 мол/л при 150, 120
и 90 СС протекает соответственно за 32, 80 и 130 мин [97].
Ускорение этерификации спиртов третичными аминами проходит
через стадию амфотерного иона и определяется скоростями его
об разования и разложения;
н2с —сп2 r3Kch2ch2o- _ВС)Н-> R3NCH2CH2OH + RO-
о
Реакция в этом случае идет медленнее, чем при катализе гидро
окисиями металлов [96].
314
Типичные условия синтеза простых эфиров гликолей в щелочной
среде описаны при оксиэтилировании вторичных спиртов
С10 — С20 [40]. Реакция осуществлялась при 150—160 °C в при-
сутствии измельченного едкого кали. По окончании подачи расчет-
ного количества окиси этилена реакционную массу выдерживали
при 140 °C до тех пор, пока давление в реакторе не переставало
снижаться, охлаждали, продували азотом, нейтрализовали уксусной
кислотой и фильтровали. При получении эфиров, содержащих
12 оксиэтильных групп, целевое использование окиси составляет
64,6%; остальное ее количество расходуется на образование поли-
этиленгликолей.
Характерным для реакции в щелочной среде является увеличение
выхода высокомолекулярных соединений. Так, из этилового спирта-
ректификата при некаталитическом синтезе этилцеллозольва полу-
чено 10,1% этиленгликоля и 5,7% этплкарбитола, а в присутствии
едкого натра (0,016 М) их получено соответственно 3,6 и 26% [97].
В основном катализе при взаимодействии окиси пропилена со
спиртами соотношение изомерных эфиров гликолей не зависит от.
природы спирта. Выход изомера, отвечающего правилу Красуского,
достигает 97 и даже 100% [98].
Кислотный катализ. При этерификации спиртов окисями выде-
ляют три разновидности кислотного катализа: действие протона,
протонных и апротонных кислот. В присутствии водородных ионов,
например, при катализе Н-катионитами
форма окиси этилена, к которой легко
фильный реагент:
образуется оксониевая
присоединяется нуклео-
К
+н*
Н2С—сн2 ----* Н2С—сн2
но-
Ч-ROH
--► ГН2С—СН2-О-Рр-->- HOCH2CH2OR4-H
d-н й
t Сильные кислородсодержащие кислоты образуют с окисями
эфиры, которые медленно подвергаются алкоголизу;
2с—сн2—па (быстро) -> носн2сн2а вон_(^дленцо) носи2сы2ок
-НА
О
При этом вполне вероятно циклическое переходное состояние,
например:
Н,С—СН2 + НОСЮ3 " Н,С——СН,—О;
V
/ :;сю2
О—Н—(Z
| Реакция имеет нулевой порядок по окиси и приблизительно
второй по спирту.
315
Для случая использования апротонных кислот типа галогенидов
металлов получено следующее кинетическое уравнение [98]:
dx ( к 1
дБ ~ + [roh]J IBFs] [СН2СН2О]
Катализатор в растворе спирта находится в виде диалкоголята
ROBF3H2OR, который активирует окись.
Хотя кинетика катализа протонными кислотами и галогенидами
металлов заметно отличается, механизм раскрытия цикла, по-види-
мому, аналогичен. Об этом, в частности, свидетельствует почти
равное образование «нормальных» (т. е. образующихся в соответ-
ствии с правилом Красуского) и «анормальных» продуктов при-
соединения метилового и этилового спиртов к окиси пропилена в при-
сутствии BF3 и НС1О4. Этерификация спиртов окисью изобутилена
при катализе трехфтористым бором приводит к преимущественному
образованию изомера (CH3)2C(OR)CH2OH [99].
Для кислотного катализа показатель степени в уравнении Брен-
стеда составляет 0,13, что свидетельствует о малой избирательности
реакции оксиалкилирования по отношению к различным спиртам.
Изучено распределение продуктов при метанолизе окиси про-
пилена в присутствии серной и соляной кислот. Оказалось, что
серная кислота действует так же, как хлорная кислота и трех-
фтористый бор: в продуктах реакции содержится 50%
СН3ОСН2СН(ОН)СН3. В присутствии соляной кислоты получено
88% этого эфира. Скорость реакции при этом мало отличается от
скорости некаталитического процесса. В продуктах реакции обна-
ружен также пропиленхлоргидрин. При взаимодействии окиси
пропилена и этиленгликоля в присутствии НС1 идет только «нор-
мальное» раскрытие цикла, а в присутствии H2SO4 образуются
два изомера в равных количествах.
Таким образом, раскрытие цикла окиси олефина при взаимодей-
ствии со спиртами зависит прежде всего от протонодонорной актив-
ности реагента и нуклеофильной активности реакционной среды.
Решающее значение на скорость присоединения имеет сопряженная
система кислота — основание; по этой причине независимо от исполь-
зования кислотного или основного ускорителя реакции порядок по
катализатору остается первым.
Как в некаталитических реакциях, так и при использовании
кислотных и основных катализаторов наиболее важным фактором,
влияющим на выход оксиалкилированных продуктов, является
соотношение спирт : окись. Даже при 8—10-кратном избытке спирта
наряду с эфиром моногликоля образуется эфир дигликоля. Если
учесть, что в настоящее время все эфиры полигликолей находят
широкое применение, выбор способа синтеза будет определяться
не столько образованием этих полиэфиров, сколько скоростью реак-
ции и наличием «посторонних» (альдегиды, 1,4-диоксан, диэфиры)
примесей. Б этом смысле предпочтение отдается нейтральному
и слабощелочному синтезам.
316
Технология получения простых
эфиров гликолей
Промышленный синтез эфиров гликолей, освоенный в США с 30-х го-
дов, к 1972 г. достиг 270 тыс. т зфиров этиленгликолей, в том
числе 208 тыс. т эфиров моноэтиленгликоля (целлозольва) и 36 тыс. т
эфиров диэтиленгликоля. В СССР технология получения простых
эфиров гликолей была разработана также в 30-ые годы; крупное же
производство организовано после Великой Отечественной войны [27,
с. 8].
В промышленности США предпочитали работать без катализатора
при давлении 3—4 МПа (30—40 кгс/см2) и температуре 200 °C [100,
с. 97]. Наиболее благоприятными считали эти условия и немецкие
ОкиСь
этилена
Спирт
Рецикл
спирта.
Катализа^
тор
Монозрир
диэтален-
г л и. коля
Моноэфар
моноэгпи.лен-
гликоля
Монозсрир
траэтилен-
г лакол я
| Остаток
Рис. 106. Блок'схема получения простых моноэфиров этиленгликоля:
1 — колонна синтеза; 2, 3, 4, 5 — ректификационные агрегаты.
химики, но ими были проделаны опыты по синтезу этилцеллозольва
в присутствии едких щелочей при 1,4—1,5 МПа (14—15 кгс/см2).
К некаталитическим процессам в Германии относили также синтезы
этилцеллозольва с добавкой в шихту малых количеств щелочи (до
0,01%), что позволяло иметь после реактора практически нейтраль-
ную среду. Использование кислоты, например серной, в качестве
катализатора считалось нецелесообразным из-за коррозии обору-
дования.
В последнее время наметилась тенденция к унификации техно-
логии и строительству комбинированных установок для последова-
тельного получения эфиров гликолей из различных спиртов в зави-
симости от спроса. Хотя такая установка имеет большую стоимость,
нем отдельное производство, но в целом экономические показатели
Получения нескольких эфиров гликолей возрастают [101].
Считается, что переработку окиси этилена в этиленгликоль и гли-
колевые эфиры целесообразно организовывать на месте ее произ-
водства [102]. На рис. 106 представлена схема получения простых
эфиров гликолей, предусматривающая выделение моноэфиров моно-,
Ди- и триэтиленгликоля [19, v. 10, у. 645] (отдельные узлы этой
Лхемы подробно рассмотрены ниже).
317
Производство этилцеллозольва
Для производства этилцеллозольва используют этиловый спирт
и окись этилена. Как правило, качество сырья мало сказывается
на технологических параметрах и схеме производства, но влияет на
качество товарного этилцеллозольва и способ переработки его кубо-
вых остатков. Хорошим сырьем для производства этилцеллозольва
является окись этилена, получаемая прямым окислением этилена.
В ней содержится более 99,5% основного вещества, а примеси аце-
тальдегида п воды не превышают 0,05%.
Сложнее обстоит дело с этиловым спиртом. Наибольшими пре-
имуществами обладает абсолютный этиловый спирт, но он весьма
Рис. 107. Технологическая схема производства этилцеллозольва:
1 — емкость; 2, 4, 5 — теплообменники; з — смеситель; в — этерификатор; " — дроссе.и
8 — сборник; 9. 10 — конденсаторы-холодильники; 11,1.;, is— ректификационные .к"
лонны; 12, 14, 1в — дефлегматоры; 17 — паро-эжекционный вакуум-насос.
дорог. Этиловый же спирт-ректификат содержит около 94% основ-
ного вещества и около 6% (масс.) или 14% (мол.) воды. Так как
для связывания одной молекулы воды необходима одна молекула
окиси этилена, то как минимум 14% (мол.) окиси будет расходо-
ваться на образование гликолей. Выгоднее использовать синтети-
ческий этиловый спирт, обезвоженный с помощью этиленглико-
лей [103]. Содержание влаги в таком спирте составляет 0,5—1,2%
и в нем, в отличпе от спирта, который обезвоживается бензолом,
нет никаких посторонних примесей |90].
На рис. 107 изображена технологическая схема производства
этилцеллозольва с использованием 99% этилового спирта и окиси
этилена прямого окисления. Шихта содержит 10—15% .окиси эти-
лена, 85—90% этилового спирта и 0,02—0,1 г/л едкого натра. Готовят
шихту в смесителе 3, куда одновременно подают спирт (смесь све-
жего и возвратного), окись этилена и 2—5%-ный раствор NaOn
в этиловом спирте.
Во избежание вскипания окиси этилена поступающий в смеситель
спирт охлаждается в теплообменнике 2 до 20—30 °C. Каталитический
раствор готовят в отдельной емкости из 40%-ного раствора ХаОп
и спирта.
318
Из смесителя 3 шихта с помощью насоса высокого давления по-
дается через теплообменник-рекуператор 4 и обогреваемый паром
высокого давления теплообменник 5 в нижнюю часть этерифика-
тора 6. Режим работы этерификатора следующий:
Давление, МПа (кгс/см2) ...........
Температура, °C
низа .............................
верха ........................
Время реакции, ч ..................
Степень конверсии окиси этилена, % .
Содержание NaOH на выходе, г/л . ,
3,0-3,5 (30-35)
160—170
190-200
1
—98
0,001-0,05
Наряду с этилцеллозольвом образуются этиловые эфиры ди-
и триэтиленгликоля, а также моно- и диэтиленгликоли. Кроме них
в небольшом количестве обнаруживаются этиловый эфир тетра-
этиленгликоля и триэтиленгликоль, которые накапливаются в кубо-
вых остатках этилцеллозольва.
После этерификатора реакционная жидкость поступает в тепло-
обменник 4, где отдает часть тепла шихте, затем дросселируется
вентилем 7 до давления 0,1—0,2 МПа (1—2 кгс/см2) и сливается
в сборник 8. Парогазовая фаза из сборника 8 конденсируется в водя-
ном конденсаторе 9 и конденсаторе 10, охлаждаемом антифризом
с температурой —15 °C. Жидкая фаза после конденсатора 10, содер-
жащая некоторое количество окиси этилена, возвращается на при-
готовление шихты.
Разделение реакционной жидкости осуществляется последова-
тельно на трех ректификационных колоннах. Вначале при атмосфер-
ном давлении отгоняется непрореагировавший этиловый спирт в ко-
лонне 11 (температура куба колонны 140—150 °C, флегмовое число
от 0,5 до 1).
Ректификация этилцеллозольва ведется на тарельчатой ко-
лонне 13 при остаточном давлении 6,66—10,6 кПа (50—80 мм рт. ст.);
температура куба 140—150 °C, верха 75—80 СС и флегмовое число,
близкое к двум. Дистиллят содержит 98,0—99,8% основного веще-
ства. Кубовые остатки этилцеллозольва на 70—80% состоят иэ
этилкарбитола (см. стр. 320), из них вакуумной ректификацией
На колонне 15 выделяют технический этилкарбитол.
Производство этилцеллозольва из синтетического 88—90%-ного
этилового спирта и 99 % -ной хлоргидринной окиси этилена сопряжено
с рядом затруднений [95]. Кроме воды (содержание которой соста-
вляет 10%), в нем имеются диэтиловый эфир, ацетальдегид, ацетон,
Изопропиловый спирт, непредельные и полимерные соединения.
Наиболее значительными примесями хлоргидринной окиси этилена
Являются ацетальдегид, вода и этиленхлоргидрин.
ML В продуктах реакции содержится значительное количество гли-
Ирлей, но вода не полностью взаимодействует и накапливается
H/Возвратном спирте (до 18%). Наряду с водой в этом спирте увели-
319
чивается (до 9% и более) концентрация диэтилового эфира. Возврат
ный спирт постепенно обогащается ацеталями и полуацеталями,
образующимися из ацетальдегида. Они попадают в товарный продук-
и снижают его качество по показателю «смешиваемость с водой»
Такой же отрицательный эффект дают полимеры, содержащиеся
в исходном этиловом спирте. Возникает необходимость в периоди-
ческом удалении возвратного спирта, и производственный процесс
становится фактически полунепрерывным.
Хлор из хлорорганических соединений окиси этилена в процессе
синтеза связывается щелочью в хлористый натрий, который отла-
гается в теплообменниках, и для его удаления требуется периоди
ческая промывка водой. Избыток щелочи приводит к разложению
многих продуктов реакции и, как следствие, к окрашиванию этил-
целлозольва-ректификата. Окраска также усиливается за счет кор-
розии, что вызывает необходимость использовать специальные марки
сталей.
Щелочная среда препятствует гидролизу ацеталей и таким обра-
зом способствует увеличению количества водонерастворимых при-
месей. Даже прп незначительной кислотности в этерификаторе
образуются сложные эфиры и повышается число омыления, поэтому
в некоторых случаях перед ректификацией в реакционную массу
добавляют небольшое количество щелочи.
На основании производственного опыта установлено, что для
получения этилцеллозольва высокого качества требуется: исполь-
зование концентрированного сырья, обеспечение слабощелочной
среды на всех стадиях производства; четкая ректификация реак-
ционных продуктов; использование коррозионностойких материалов
для аппаратуры.
I
Производство этилкарбитола
Хотя в литературе имеются указания на специальное получение
этилкарбитола, значительная часть его производится совместно
с этилцеллозольвом. В состав кубовых остатков этилцеллозольва
наряду с этилкарбитолом входят простые этиловые эфиры три-
и тетраэтиленгликолей, а также моно-, ди- и триэтиленгликоли.
Содержание компонентов приведено ниже (в %):
Компонент Из 99%-ного этилового спирта Из 90%-ного этилового спирта
Этилкарбитол . 72—79 25-33
Этиловый эфир триэтиленгликоля . . 10-13 2-3
Этиловый эфир тетраэтиленгликоля . 2-2,5 —
Этиленгликоль 5-12 45-50
Диэтиленгликоль . 1,5—3 14-18
Триэтпленгликоль . 0,3—0,9 3-4
По мере разгонки кубовых остатков этилцеллозольва вначале
отбирается азеотропная смесь этилкарбитол — этиленгликоль,
320
свойства которой приведены на стр. 296, затем этилкарбитол и азео-
тропная смесь этиловый эфир триэтиленгликоля — диэтиленгликоЛь.
В результате однократной ректификации получается 88—93%-ный
продукт. Повторная ректификация позволяет получить этилкарби-
тол с содержанием основного вещества до 99,8%.
В том случае, когда в кубовых остатках этилцеллозольва содер-
жится большое количество этиленгликоля и мало этилкарбитола,
метод обычной ректификации для извлечения этилкарбитола непри-
годен. Весь этилкарбитол расходуется на образование азеотропной
смеси с этиленгликолем, и за одну операцию его концентрация
повышается только до 60—70%. Дальнейшее концентрирование
этилкарбитола можно проводить несколькими способами.
Предлагают ректификацию при переменном давлении. Вначале
при остаточном давлении 1,33 кПа (10 мм рт. ст.) отбирают азео-
тропную смесь, содержащую 79,5% этилкарбитола и 20,5% этилен-
гликоля. Затем ее перегоняют при атмосферном давлении, и во вновь
полученной азеотропной смеси содержится 46% этиленгликоля.
Этот продукт полностью отбирается в дистиллят, а этилкарбитол
остается в кубе [104]. Серьезным недостатком способа является
разложение этилкарбитола при атмосферной разгонке, сопровожда-
ющееся появлением запаха, окраски и водонерастворимых примесей
в готовом продукте.
Разделение смеси этиленгликоля с этилкарбитолом достигается
при разгонке с ксилолом. Метод, основанный на образовании азео-
тропной смеси этиленгликоль — ксилол [105], позволяет получать
пригодный для технического применения продукт с ксилольным
запахом.
Этиленгликоль больше, чем этилкарбитол, склонен к образованию
водородных связей. Эта его способность использована при экстрак-
тивной ректификации азеотропной смеси с этилкарбитолом в при-
сутствии диэтиленгликоля. Концентрация этилкарбитола в дистил-
ляте пе превышает 94%, и для получения чистого продукта требуется
дополнительная вакуумная ректификация. Достоинством способа
является отсутствие в продукте посторонних примесей, он не имеет
запаха и цвета [106].
В промышленности для выделения этилкарбитола из бедных
смесей с этиленгликолем используется экстракция с толуолом.
В товарном продукте содержится до 96% основного вещества.
Целевой синтез этилкарбитола осуществляется оксиэтилирова-
нием смеси этанола и этилцеллозольва (при объемном соотношении,
равном 1 : 1) в присутствии едкого натра. После ректификации
реакционной массы этилцеллозольв возвращается на приготовление
шихты и его количество в производственном цикле остается постоян-
ным. Содержание окиси этилена в шихте составляет 9—13%. Прак-
тически полная конверсия окиси этилена достигается при давлении
3,0—3,5 МПа (30—35 кгс/см2) и температуре 180—200 °C в течение
1 ч. Как и при получении целлозольва, примесь воды в этиловом
спирте приводит к образованию гликолей, которые затрудняют
21 Заказ 972
321
последующую ректификацию зтилкарбитола. Наряду с этилкарби-
толом при его производстве вырабатывают достаточно концентри-
рованный этиловый эфир тризтиленгликоля.
Применение эфиров гликолей
Особенно ценным качеством эфиров гликолей является их сродство
как к гидрофильным, так и к гидрофобным веществам, обусловленное
наличием в молекуле зфира гликоля биполярных групп. Такие
соединения имеют высокую поверхностную активность. Это и пред-
определило их использование главным образом в качестве раствори-
телей, пластификаторов, солюбилизаторов, моющих и очищающих
средств.
Для удобства рассмотрения областей применения зфиров гли-
колей они разделены на три большие группы:
1) низкомолекулярные эфиры моно- и дигликолей, содержащие
от С3 до С10 и растворимые в воде;
2) эфиры со средней молекулярной массой, включающие от 1
до 6 оксиалкиленовых групп и содержащие С1о — С25;
3) эфиры полигликолей, в состав которых входит более 8 окси-
алкиленовых групп.
По характеру применения необходимо различать использование
эфиров гликолей в массе, как гидротропные добавки и, наконец,
как поверхностно-активные вещества. В какой-то мере разделение
зфиров гликолей по молекулярной массе и характеру применения
совпадают.
Эфиры низкомолекулярных гликолей
Низшие простые зфиры этиленгликоля известны в химической прак-
тике под названием целлозольвов, так как вначале их использовали
для растворения эфиров целлюлозы. Наиболее распространены
метил-, зтил- и бутилцеллозольвы, на долю которых приходится
большая часть окиси этилена, перерабатываемой в эфиры гликолей.
В настоящее время целлозольвы и их аналоги из пропиленгли-
коля включают в состав различных лаков и змалей не только на
основе эфиров целлюлозы, но также многих природных и синтети-
ческих смол. В лакокрасочной промышленности они заменяют ски-
пидар и, кроме того, позволяют придавать лакам и краскам прин-
ципиально новые качества: водорастворимость и стабильность при
хранении [107, с. 377; 108]. Особенно ценным оказался бутилцелло-
зольв, имеющий наибольшую совместимость с алкидными смолами:
он снижает вязкость лаковых растворов, улучшает водораствори-
мость и позволяет наносить алкидный лак по нитроцеллюлозному
покрытию.
Простые эфиры моноэтилен- и пропиленгликолей используются
в чистящих средствах, при крашении и печатании, в производстве
кино- и фотопленки [2, р. 117 и 264]. Добавка 50 г/л этилцелло-
зольва в красильную ванну улучшает окраску полиамидного во-
322
л окна и существенно сокращает время операции. Этот эффект дости-
гается за счет набухания волокна и более равномерного распределе-
ния красителя по поперечному срезу [109].
Этилцеллозольв и другие родственные соединения используются |
как добавки в моторных и реактивных топливах для предотвращения ’
выделения п замерзания растворенной в топливах воды [110]. !
Фотографический проявитель с 3-пиразолидином в растворе мети-
лового пли зтилового эфира этиленгликоля хранится в течение года
и при надобности легко разбавляется водой до нужной концентра-
ции [111], С помощью зфиров извлекают кристаллизационную воду
из некоторых гидратов и затем количественно определяют ее методом
газовой хроматографии. Этилцеллозольв служит для идентификации
набухающих глин методом рентгеноструктурного анализа [112].
Целлозольвы и 1,4-диоксан являются средой для неводного титро-
вания [26, с. 105]. Метил-, этил- и бутилцеллозольвы используют
как азеотропные добавки при разделении различных углеводородов
и спиртов [ИЗ].
Простые эфиры дпгликолей по физико-химическим свойствам
мало отличаются от зфиров моногликолей. Области их применения
часто схожи. Наиболее распространен в практике этиловый эфир
диэтиленгликоля — зтилкарбптол. Благодаря низкой температуре
замерзания и малой летучести он входит в состав гидротормозных
жидкостей, антифризов, абсорбентов при сушке газов [114, 115].
Из него изготавливают растворимые в воде масла, а особо чистый
продукт используется как растворитель зфирного масла и для за-
мены алкоголя в косметической и парфюмерной промышленности.
В последнее время его стали включать в состав антиперсперантов
и дезодорантов в качестве бактерицидного вещества.
Фенилгликолевый и бутилгликолевые эфиры почти не обладают
запахом и очень труднолетучи, их употребляют в парфюмерии в ка-
честве фиксаторов запаха. Для этой же цели используют диацетат
этиленгликоля.
Высокая избирательная сорбция эфиров дигликолей делает их
ценными разделяющими агентами в экстрактивной ректификации,
а также в экстракции. С добавкой этилкарбитола разделяют метил-
ацетат и метиловый спирт. Метилацетат выводят через верх первой
колонны, а метиловый спирт и этилкарбитол разделяют во вто-
рой [116].
В некоторых случаях использование индивидуальных эфиров
гликолей не дает преимуществ перед техническими смесями, и потому
для охлаждения моторов предложен водный раствор метиловых
эфиров пропиленгликолей, включающих от 1 до 4 оксипропильных
групп. Для очистки карбюраторов используют сложную углеводо-
родную смесь, содержащую бутилцеллозольв и изобутилкарби-
тол [117]. Универсальное средство для мойки поверхностей, покры-
тых краской, наряду с этилцеллозольвом включает оксиэтилиро-
ванные жирные кислоты и другие неионогенные поверхностно-
активные вещества.
21*
323
Особенно сложно снять старую краску с автомобиля и удалить
нагар в духовках и печах. Нужен поистине универсальный раствори-
тель и для его изготовления используют различные эфиры гликолей.
В состав для чистки печей кроме сырого парафина, хлор- и фтор-
углеводородов входят этилцеллозольв, целлозольвацетат и этил-
карбитол [107, с. 339].
В последнее время все большее развитие получают простые ди-
эфиры гликолей. Они весьма стабильны и позволяют создавать
реакционную среду с регулируемой диэлектрической проницаемостью
и вязкостью [24, 114, с. 304]. Особенно известны моноглим и диглим
(диметиловый эфир моно- и диэтиленгликоля), применяемые как
растворители в синтезе металлоорганических соединений, при полу-
чении лекарственных препаратов и в других областях [118]. Аналог
моноглима пз пропиленгликоля включен в состав для размягчения
старой краски.
Диметиловый эфир триэтиленгликоля является важной состав-
ной частью реакционной жидкости при получении стабилизаторов
против термического, окислительного и другого старения [119].
Простые циклические эфиры гликолей 1,4-диоксан и 1,3-диоксо-
лан обладают прекрасной растворяющей способностью и часто
используются в качестве среды для химических реакций и физико-
химических измерений. 1,4-Диоксан является настолько сильным
акцептором протонов, что полностью разрушает структуру' воды
и изменяет растворимость в ней многих соединений [120]. 1,3-Ди-
оксолан менее устойчив, чем 1,4-диоксан, в присутствии разбавлен-
ных кислот он гидролизуется с выделением исходных компонентов.
Работать с ними нужно в щелочной среде или со стабилизирующими
добавками (гидрохинон, тг-аминофенол и дифениламин) [2, р. 125;
121].
Склонность к гидролизу ограничивает применение сложных
эфиров гликолей, полученных из окисей олефинов и низших кислот.
Этиленгликольдиформиат предложено даже использовать в реакциях
и процессах, где желательно постепенное образование муравьиной
кислоты. В отсутствие воды сложные эфиры могут служить раствори-
телями. Наиболее распространен диацетат этиленгликоля, как вы-
сококипящий, медленно испаряющийся растворитель эфиров цел-
люлозы и фторированных углеводородов. В смесях с другими раство-
рителями он употребляется для очистки смазочных масел от свобод-
ных жирных кислот [2, р. 128].
Из сложных эфиров гликолей, относящихся к первой группе,
наиболее перспективны как растворители и гидротропные вещества
алкиленкарбонаты. Этилен- и пропилеикарбонаты в количестве
100—200 г/л интенсифицируют процесс крашения за счет увеличения
содержания фиксированного красителя и снижения нагрева рабо-
чего раствора. При этом они активны как в водной, так и спиртовой
средах [122]. Пропиленкарбонат оказался пригодным для избира-
тельной сорбции воды и двуокиси углерода при очистке промышлен-
ного и природного газов [31]. Динитрат диэтиленгликоля при-
324
меняется в составе баллистических порохов и промышленных взрыв-
чатых веществ как в чистом виде, так и в смесях с нитроглицери-
ном [30]. Сложные эфиры гликолей, например ацетаты
этиленгликоля, стали важнейшими промежуточными продуктами
при получении а-гликолей из а-олефинов [56].
Эфиры гликолей средней молекулярной массы
К этой группе эфиров гликолей относится большое число соединений,
занимающих промежуточное положение между растворителями и
истинными неионогенными поверхностно-активными веществами. Как
правило, это — жидкие или пастообразные вещества с высокой
температурой кипения. По классификации Ребиндера [23], они
относятся ко 2-ой группе поверхностно-активных веществ и прежде
всего являются диспергаторами.
Монододециловый эфир этиленгликоля или аналогичный моно-
эфир третичного бутилциклогексилового спирта пригоден для пла-
стифицирования карбамидных смол, целлюлозы и каучука. Дифени-
ловый эфир этиленгликоля смешивается с эфирами полиметакриловой
кислоты. Как составную часть поливинилацетатных эмульсий и по-
крытий на основе поливинилхлорида используют дибутилцелло-
зольвфталат и дибутилцеллозольвадипат. В качестве пластификато-
ров предложено использовать многие из смешанных сложно-простых
эфиров [2, р. 139; 34, с. 584].
Простые моноэфиры гликолей активно модифицируют поверх-
ности раздела. С их помощью регулируют обогащение полезных
ископаемых и массообмен в гетерогенных процессах. Специфиче-
скими пенообразователями для полиметаллических руд являются
отечественные флотореагенты Э-1, ОПСБ и ОПСМ (метиловые и бу-
тиловые эфиры три-, тетра- и пентагликолей) [123, с. 76]. Метиловый
эфир полипропиленгликолей используется для интенсификации вы-
щелачивания КС1 из сильвинита [124]. Пленка октадецилового
эфира этиленгликоля в виде монослоя снижает испарение воды
в водоемах и крупных резервуарах [125].
В присутствии гликолей поверхностная активность их простых
эфиров заметно падает, поэтому очисткой от гликолей можно повы-
сить эффективность многих технических препаратов [126]. С по-
мощью сложных эфиров этиленгликолей и нормальных жирных
кислот в технике готовят эмульгаторы и диспергаторы для системы
вода — масло, а также регулируют вязкость гидравлических жидко-
стей [33, 43, 127].
Эфиры полигликолей
Наибольшее распространение эфиры полигликолей получили как
неионогенные поверхностно-активные вещества. Достаточно высо-
кая молекулярная масса и широкий интервал гидрофйльпо-лиофиль-
ного баланса позволяют получать гелеобразную структуру как
325
в адсорбционном слое, так и в растворе воды и углеводородов. Рас-
сматриваемые эфиры относятся к поверхностно-активным веществам
3-ей и 4-ой групп [23]. С их помощью получают стабилизированные
суспензии, эмульсии и пены, смачиватели и гидрофобизаторы поверх-
ности, адсорбционные пластификаторы и другие вспомогательные
вещества, применяемые в разнообразных процессах техники
и быта [23, 128, с. 178].
Моющие и очищающие средства, содержащие продукты присоеди-
нения окисей этилена и пропилена к высшим спиртам, алкилфенолам
и карбоновым кислотам позволяют стирать в мягкой, жесткой и даже
морской воде, хорошо совмещаются с мылом и другими моющими
средствами [22, с. 272].
В последнее время эфиры полигликолей начали широко исполь-
зовать в нефтедобыче. Хорошими деэмульгаторами в системе нефть —
вода наряду с проксанолами являются различные неионогенные
ПАВ, содержащие 20—30 оксиэтильных групп [33. с. 205; 129].
Оксиэтилированные и-октилфенолы типа ОП-Ю увеличивают нефте-
отдачу пласта, сокращают сроки разработки залежей и снижают
расход закачиваемой в пласт воды. При бурении нефтяных и газовых
скважин они пластифицируют глинистые растворы.
Эфиры гликолей пригодны для адсорбционной пластификации
смазочных масел и бетона. При введении поверхностно-активного
вещества в цементный раствор снижается его вязкость и повышается
морозостойкость, а прочность бетона возрастает в 1,5—2 раза. Не-
ионогенные ПАВ применяются при обработке металлов прежде всего
в составе смазочно-охлаждающих жидкостей и как средство для
очистки поверхности от жиров, окисных пленок, масел [22, с. 3381.
Оксиэтилированные алкилфенолы ОП-4, ОП-7, ОП-10, а также
простые и сложные эфиры полиэтиленгликолей внедрены в сельское
хозяйство для повышения активности удобрений и химических
средств защиты растений. При этом они, как правило, не действуют
на ткани растений. Показано влияние ПАВ, в том числе оксиэтили-
рованных спиртов и фенолов, на прорастание, корневую систему
и рост огурцов, редиса и хлопчатника. Добавка монолаурата поли-
этиленгликоля способствует лучшему усвоению кормов и прибавке
в весе цыплят. Оксиэтилированные алкилфенолы, внесенные в корм
для молочного скота, повышают в нем содержание мелассы и спо-
собствуют удоям. Запатентованы добавки оксиэтилированной жир-
ной кислоты для получения мороженого, глазури, кремов и хлебо-
булочных изделий (однако федеральное ведомство пищевой про-
мышленности в США запретило ее применение в качестве добавок
к хлебу) [22, с. 332; 64].
Некоторые сложные эфиры этилен- и пропиленгликолей, обра-
зованные салициловой, бензойной и ацетилбензойной кислотами,
обладают терапевтическими свойствами [22, с. 328]. Метоксиэтил-
никотин снижает содержание холестерина в крови. Полигликоли
и их эфиры используются в медицине для приготовления легко-
проникающих в кожу мазевых основ, в которые добавляются лекар-
326
ственные препараты, тонизирующие вещества и травы. В них наряду
с высокомолекулярными веществами иногда включают этилцелло-
эольв и этилкарбитол. Для усиления гидрофобного характера мази
предпочитают брать диэфиры, например дистеарат или дилаурат
полиэтиленгликоля 400. Твин-80 (полиоксиэтиленсорбитанмоноолеат)
и ряд других эфиров полигликолей применяются для регулирования
роста микроорганизмов [130].
Требования, предъявляемые к качеству
эфиров гликолей
Эфиры гликолей применяют во многих отраслях народного хозяйства
для самых разнообразных целей. Поэтому при разработке техни-
ческих условий на товарные продукты неизбежно возникает ряд
трудностей, связанных со спецификой их использования. Казалось
бы, можно добиться единого подхода при введении такого показа-
теля, как содержание основного вещества. Но многие даже низко-
молекулярные эфиры гликолей (особенно производные пропилен-
гликолей) являются смесями нескольких изомеров или родственных
соединений, и их определение весьма трудно.
В аналитической практике применяют метплцеллозольв, содер-
жащий 99,8% целевого продукта, в то время как для технических
целей пригоден 98%-ный эфир. Технический этилцеллозольв содер-
жит 97—99%, а технический агилкарбитол 93—96% основного
вещества (табл. 92).
При повышении молекулярной массы эфира гликоля доля основ-
ного вещества еще больше снижается. В пенообразователе ОПСМ сум-
ма фракций монометилового эфира три- и тетрапропиленгликолей
(активное начало) для I сорта должна быть не менее 80%, а для II —
ве менее 73%. Судя по фракционному составу, пенообразователь
ОПСБ имеет еще меньше активных компонентов [123, с. 76].
Для эфиров полигликолей основное вещество обычно не норми-
руется. Во многих случаях пригодность эфира полигликоля оцени-
вается по соответствию целевому назначению, например снижению
поверхностного натяжения.
Технические требования на моющие оксиэтилированные алкил-
фенолы ОП-7 и ОП-Ю приведены ниже [132, с. 3791:
ОП-7 оп-ю
pH водного раствора (10 г/л) .... Температура, при которой осветляется водный раствор, °C 6-8 6-8
при концентрапии 10 г/л .... ' — 80—90
при концентрации 20 г/л .... Поверхностное натяжение водного рас- 55—65 —
твора (5 г/л), мН/м (дин/см), не более 35 37
Содержание влаги, % , не более , . . 0,5 0,5
327
О
Содержа- ние воды, %, пе бол. S 1 1 1 1 0,25 0,3 0,2 0,1
Кислотность 1 (СНзСООН), %, пе более ( ( i ( 1 О О 0,008 0,01 1
CO
Темпера- 1 тура пе- регонки, °C 123—126 124—125 123-125 lQ 00 co co Ш oo co 190-203 190—202 225—233 227-232
5 , о К i и S-o°S О СО О о CO co
Сод, НИС П0В1 веще' % СО Ci Ci Ci Ci Ci cd о ь- Ci Ci Ci S 1 1 1
г/см3 ,966 ,971 066‘ л Ь-С £ CO Ci 00 LQ LO CC Ю Ci Ci
? 1 ? ? 1 1 ?? d d i i
о с 0,964- 0,965- 0,929- 1 1 i о LC Ci LQ Ci CO o 1 I LQ OJ LQ LQ Ci_ Ci
Цвет, пе более 1 1 1 I 5 (Pt-Co)' 1 15 (APHA) 10 (Pt-Co)
Фирма (страна), стандарт Merck (ФРГ) * Uinon Carbide (США) МРТУ 6-09-6347-69 Ferak (ФРГ) Enjay Chem. Co (США) ГОСТ 8313—60 Hiils (ФРГ) МРТУ 6-09-3911—67 BDH (Англия) Shell Chem. Co (США)
Продукт M етилцеллозольн Этилцеллозольв Этилкарбитол Бутплкарбитол
Коэффициент преломления п.. = 1,4010 —1,4030.
328
Важным показателем качества является плотность, которая
определяется практически для всех жидких веществ. Но плотность,
как и показатель преломления, является аддитивной величиной
и надежно характеризует качество только при совпадении других
физико-химических величин.
На стабильность продукта, а также его пригодность к исполь-
зованию в стальной таре указывает показатель «кислотность», пере-
считываемый обычно на уксусную кислоту. Кислотность опреде-
ляется как для жидких, так и для пастообразных продуктов и со-
ставляет не более 0,02%. В некоторых случаях оценка среды произ-
водится по pH водной вытяжки. При наличии ингибиторов
в спецификации отражается их содержание и предельная кислот-
ность. Так, в 1,3-диоксолан добавляют 2—3% метилового спирта —
стабилизатора при кислотности не выше 0,01% СН3СООН [132,
с. 316].
К общим методам оценки качества нужно отнести такие пока-
затели, как нелетучий остаток, цветность продукта и наличие меха-
нических примесей. Более специфическим показателем является
запах, учитывающийся при изготовлении фармацевтических и кос-
метических препаратов. В некоторых случаях регламентируется
содержание железа, тяжелых металлов, хлоридов или сульфатов.
Такие требования, в частности, предъявляются к метилцеллозольву,
используемому в производстве электролитических конденсаторов.
При хранении и транспортировании эфиров гликолей возникает
необходимость в устранении контакта с воздухом, который способ-
ствует окислению простых и гидролизу сложных эфиров. Поэтому
в большинстве технических условий оговаривается транспортирова-
ние в герметичной таре с весьма высокой степенью заполнения. Тре-
бования к таре такие же, как и для гликолей (гл. I, стр. 34), но
для хранения целлозольвов не пригоден алюминий.
ЛИТЕРАТУРА
1. Эллис К., Химия углеводородов нефти и их производных. Т. 1. Пер. с англ.
Под ред. Н. Д. Зелинского. М., ОНТИ, 1936. 634 с.
2. Glycols Ed. by G. О. Curme, F. Johnston. New York, Reinhold Publ. Corp.,
1953. 389 p.
3. Пиментел Дж., Мак-Клеллан О. Водородная связь. Пер. с англ. Под ред.
В. М. Чулановского. М., «Мир», 1964. 462 с.
4. Kuhn L. Р., J. Am. Chem. Soc., 1958, v. 80, № 22, р. 5950—5954.
5. Yonezawa Т. е. a., Bull. Chem. Soc. Japan, 1965, v. 38, № 9, p. 1431 —1353;
Kuhn L. P., Wires R. A., J. Am. Chem. Soc., 1964, v. 86, № 11, p. 2161 —
2165; Saito H. e. a., Bull. Chem. Soc. Japan, 1966, v. 39, № 5, p. 989—994.
6. Krueger P. J., Mettee II. D., J. Mol. Spectroscopy, 1965, v. 18, № 2, p. 131 —
140.
7. Reineschneider G. P., Monatsch. Chem., 1959, v. 90, № 6, p. 783 — 786;
Tseng M., Thompson A. R., J. Chem. Eng. Data, 1962, v. 7, № 4, p. 483—
485; Schott H., Ibid., 1966, v. 11, № 3, p. 417—418.
8. Белоусов В. П., Макарова Н. Л., Вести. ЛГУ, 1970, № 22, с. 101 —107;
Железный Б. В., Ж. структ. хим., 1970, т. 11, № 4, с. 595—599.
9. Pearce Р. J., Strauss W., Austr. J. Chem., 1968, v. 21, № 8, p. 2127—2129;
Buttner D., Heydtmann II., Z. phys. Chem., 1969, Bd. 63, № 5, S. 316—318;
Pottel R., Ber. Bundesges. phys. Chem., 1969, Bd. 73, № 5, S. 437—446.
329
Таблица 92. Требования к качеству целлозольвов и карбитолов [131]
Содержа- ние воды, %, не более 1 1 о 0,25 0,3 о о
Кислотность , (СНзСООП), %, не более I 1 i 1 wo‘o 1 CO 11 0,01
1 Темпера- тура пе- регонки, °C 123—126 124—125 123—125 оо Ю СО СО СО 177 со ^'3 со 190—203 190—202 225—233 227—232
Содержа- ние ос- ! новиого вещества, % 98,0 99,8 99,0 О О со со г-7 Оi Oj Oj 94,7 1 1
Плотность, г/см3 § сл о о" 1 1 1 << LQ СС 50 о о 0,929-0,930 0,990—0,995 0,9855-0,9885 0,955—0,965 0,952—0,955
Цвет, не более 1 ! 1 5 (Pt-Со) 15 (ЛР/7Л) 10 (Pt—Со)
Фирма (страна), стандарт Merch (ФРГ) * Uinoii Carbide (США) МРТУ 6-09-6347- 69 Ferak (ФРГ) Enjay Chem. Co (США) ГОСТ 8313—60 Huis (ФРГ) МРТУ 6-09-3911—67 BDH (Англия) Shell Chem. Со (США)
Продукт Метилцеллозолъв Этилцеллозольв Этилкарбитол | Бутплкарбитол
Коэффициент преломления п,. = 1,4010 — 1,4030.
328
Важным показателем качества является плотность, которая
определяется практически для всех жидких веществ. Но плотность,
как и показатель преломления, является аддитивной величиной
и надежно характеризует качество только при совпадении других
физико-химических величин.
На стабильность продукта, а также его пригодность к исполь-
зованию в стальной таре указывает показатель «кислотность», пере-
считываемый обычно на уксусную кислоту. Кислотность опреде-
ляется как для жидких, так и для пастообразных продуктов и со-
ставляет не более 0,02%. В некоторых случаях оценка среды произ-
водится по pH водной вытяжки. При наличии ингибиторов
в спецификации отражается их содержание и предельная кислот-
ность. Так, в 1,3-диоксолан добавляют 2—3% метилового спирта —
стабилизатора при кислотности не выше 0,01% СН3СООН [132,
с. 316].
К общим методам оценки качества нужно отнести такие пока-
затели, как нелетучий остаток, цветность продукта и наличие меха-
нических примесей. Более специфическим показателем является
запах, учитывающийся при изготовлении фармацевтических и кос-
метических препаратов. В некоторых случаях регламентируется
содержание железа, тяжелых металлов, хлоридов или сульфатов.
Такие требования, в частности, предъявляются к метилцеллозольву,
используемому в производстве электролитических конденсаторов.
При хранении и транспортировании эфиров гликолей возникает
необходимость в устранении контакта с воздухом, который способ-
ствует окислению простых и гидролизу сложных эфиров. Поэтому
в большинстве технических условий оговаривается транспортирова-
ние в герметичной таре с весьма высокой степенью заполнения. Тре-
бования к таре такие же, как и для гликолей (гл. I, стр. 34), но
для хранения целлозольвов не пригоден алюминий.
ЛИТЕРАТУРА
1. Эллис К., Химия углеводородов нефти и их производных. Т. 1. Пер. с англ.
Под ред. Н. Д. Зелинского. М., ОНТИ, 1936. 634 с.
2. Glycols Ed. by G. О. Curme, F. Johnston. New York, Reinhold Publ. Corp.,
1953. 389 p.
3. Пиментел Дж., Мак-Клеллан О. Водородпая связь. Пер. с англ. Под ред.
В. М. Чулановского. М., «Мир», 1964. 462 с.
4. Kuhn L. Р., J. Am. Chem. Soc., 1958, v. 80, № 22, р. 5950—5954.
5. Yonezawa Т. е. a., Bull. Chem. Soc. Japan, 1965, v. 38, № 9, p. 1431 —1353;
Kuhn L. P., Wires R. A., J. Am. Chem. Soc., 1964, v. 86, № 11, p. 2161 —
2165; Saito II. e. a., Bull. Chem. Soc. Japan, 1966, v. 39, № 5, p. 989—994.
6. Krueger P. J., Mettee II. D., J. Mol. Spectroscopy, 1965, v. 18, № 2, p. 131 —
140.
7. Reineschneider G. P., Monatsch. Chem., 1959, v. 90, № 6, p. 783—786;
Tseng M., Thompson A. R., J. Chem. Eng. Data, 1962, v. 7, № 4, p. 483—
485; Schott II., Ibid., 1966, v. 11, № 3, p. 417—418.
8. Белоусов В. И., Макарова II. Л., Вести. ЛГУ, 1970, № 22, с. 101 —107;
Железный Б. В., Ж. структ. хим., 1970, т. 11, № 4, с. 595—599.
9. Pearce Р. J., Strauss W., Austr. J. Chem., 1968, v. 21, № 8, p. 2127—2129;
Buttner D., Ileydtmann H., Z. phys. Chem., 1969, Bd. 63, A? 5, S. 316—318;
Pottel R., Ber. Bundesges. phys. Chem., 1969, Bd. 73, № 5, S. 437—446.
329
10. Сучков В. А., Вести. ЛГУ, 1964, Ns 16, с. 53—59; Armitage J. М. е. а.,
J. Magn. Reson., 1974, v. 15, № 1, р. 142—149.
11. Белоусов В. П., Морачевский А. Г. Теплота смешения жидкостей. Л.,
«Химия», 1970. 252 с.
12. Малиновский М. С. Окиси олефинов и их производные. М., Госхимпздат,
1961.553 с.
13. Singlar М., Dykyi J., Coll. Czech. Chem. Commun., 1969, v. 34, № 3. p. 767—
775.
14. Вайсбергер А. и др. Органические растворители. Пер. с англ. Под ред.
Я. М. Варшавского. М., Издатинлит, 1958. 518 с.
15. Комарова Л. Ф., Гарбер Ю. И., Ж. орг. хим., 1971, т. 7, № 12, с. 2507—
2509.
16. Гурвич С. М., ЖОХ, 1955, т. 25, № 9. с. 1713-1716; ЖПХ, 1959, т. 32,
№ 7, с. 1640—1642.
17. Сигэмацу X., «Когё кагаку дзасси», (J. Chem. Soc. Japan, Ind. Chem. Sec.),
1962, v. 65, № 6, p. 945—949; РЖХим, 1963, 8Ж7.
18. Weimer D. R., Lindemuth L. D., Groves W. L.. J. Am. Oil Chemi Soc.,
1967, v. 44, № 3, p. 171—174.
19. Kirk-Othmer. Encyclopedia of Chemical Technology. Ed. 2nd. V. 8, 10. New
York, Interscience Inc., 1965 —1966.
20. Haken J. K., Khemangkorn V., J. Chromatogr. Sci., 1972, v. 10, № 1. p. 41 —
47.
21. Селицкая H. Д., Корецкий А. Ф.. Изв. CO АН СССР. Сер. хим., 1967, т. 6,
№ 14, с. 48—57.
22. Шенфельд Н. Неионогенные моющие средства. Пер. с нем. Под ред. А. И. Гер-
шеновпча. М., «Химия», 1965. 488 с.
23. Ребиндер И. А., Жури. ВХО пм. Д. И. Менделеева, 1959, т. 4, № 5, с. 554—’
565; 1966, т. 11, № 4, с. 362—370.
24. Katsunosuke М., Spectrochim. Acta, 1964, v. 20, № 12, р. 1865 —1873;
Kimura К., Fujishiro R., Bull. Chem. Soc. Japan. 1966, v. 39, № 3, p. 608—
610; Agemi Claude A., Bull. Soc. chim. France, 1968, № 11, p. 4467—4470.
25. Чудное А. Ф., Мирошников A. M., Техническая и экономическая информа-
ция. Сер. Методы анализа и контроля производства в химической промыш-
ленности. 1969, Ai 3, с. 3—5; Комарова Л. Ф. Кандидатская диссертация.
Томск, ТПП. 1969; Чудное А. Ф., Мирошников А. М., ЖФХ, 1969, т. 43,
№ 10, с. 2631 — 2632.
26. Денеш И. Титрование в неводных средах. Пер. с англ. Под ред. И. П. Белец-
кой. М., «Мир», 1971. 414 с.
27. Окись этилена. Под ред. П. В. Зимакова и О. Н. Дымепта. М., «Химия»,
1967. 320 с.; Зимаков П. В., Окись этилена. М., ГХИ, 1946. 240 с.
28. Bigeon J., Ind. chim. Beige, 1953, v. 40, № 433, p. 221—229; № 434,
p. 249—258; № 435, p. 281—286; Altona C., Veek A., «Tetrahedron», 1968,
v. 24, № 12, p. 4377—4391; Sable H., Ritchey W., «Spectroscopy Letters»,
1969, v. 2, № 8, p. 239—245; Pihlaja Kalevi, Acta chem. Scand., 1969, v. 23,
Ns 3, p. 1053—1055.
29. Петров А. А., Ж0Х, 1940, t. 10, № 11, c. 981—986; BrandnerJ. D., J. Am.
Oil Chem. Soc., 1964, v. 41, № 5, p. 367-370.
30. Nichols P. L., Magnusson Ir., Ingham I. D., J. Am. Chem. Soc., 1953, v. 75,
№ 17, p. 4255—4259; Орлова E. Ю. Химия и технология взрывчатых ве-
ществ. Изд. 2-е. Л., «Химия», 1973. 688 с.
31. Справочник азотчика. Т. 1. Под ред. Е. Я. Мельникова. М., «Химия», 1967.
492 с.; Дин Вэй и др., Газ. пром., 1969, № 2, с. 40—42; яп. пат. 3759JL (1971);
Gosse В., Denat А., С. г. Acad. Sci., 1974, v. 279, № 6, р. 249—25Г.
32. Cogley D. R., Falk J. Phys. Chem., 1972, v. 76, № 6, p. 855—861.
33. Goldsmith H. A., Chem. Rest., 1943, v. 33, p. 234—239; Файнголъд С. И.
Синтетические моющие средства из нефтяного и сланцевого сырья. Л-,
«Недра», 1964. 287 с.; Chem. Labor und Betrieb, 1966, Bd. 17, Ns 2, S. 73—74.
34. Тиниус К. Пластификаторы. Пер. с нем. Под ред. Е. Б. Тростянскои-
М.—Л., «Химия», 1964. 915 с.
35. Malkemus J.D., J. Am. Oil Chem. Soc*, 1956, v. 33, N 11, p. 571—574.
330
йЗб. Лейбниц Е., Труппе Г., Руководство по газовой хроматографии. Пер,
В- с нем. Под ред. А. А. Жуховицкого. М., «Мир», 1969. 504 с.; Dufka О. е.
Chem. prumysl., 1970, v. 20, № 10, р. 489—492.
37. Мамедов Ш. Простые эфиры гликолей. Баку, изд. АН и АзССР, 1961. 212 с.;
Филатов П. Г. и др., «Химия и технология топлив и масел», 1972, № 12
с. 46—49.
38. Яп. пат. 6122 (1968).
39. Лебедев Н. Н. Химия и технология основного органического и нефтехими-
ческого синтеза. М., «Химия», 1975. 734 с.
40. Береснев В. Н., ЖПХ, 1965, т. 38, № 12, с. 2655; Резников И. Г. и др.,
Хим. пром., 1973, № 7, с. 495—598.
41. Zimmermann R. L., Jones G. D., Nummy W. В., I and EC, Prod. Research,
and Develop., 1963, v. 2, № 4, p. 294—303.
42. Англ. пат. 1089134 (1967); 1114705 (1968).
43. Пат. ФРГ 127570 (1969).
44. Пат. США 3325548 (1967).
45. Inoue Н. е. a., Bull. Chem. Soc. Japan. 1966, v. 39, А» 7, p. 1577—1583.
46. Kasadinos J. V., Hazdra J. J., J. Org. Chem., 1962, v. 27, № 12, p. 4253—
4255; Kern R. J., J. Org. Chem., 1966, v. 31, № 4, p. 1274—1275.
47. Долгополов H. H., Мельников II. H., Наметкин С. С., ЖПХ, 1947, T. 20,
№ 6, c. 486—493.
48. Долгов Б. H., Ивановский В. И., Вести. ЛГУ, 1960, № 10, с. 152—159.
49. Гурьянова Е. II., Гольдштейн II. П.. ЖОХ, 1966, т. 36, № 10, с. 1822—
1826; Krygielova W., Szmaloiva II., Wasowicz J., Przem. Chem., 1966, v. 45,
№ 9, p. 516—519; Неволин Ф. В., Журн. ВХО им. Д. И. Менделеева, 1966,
т. 11, № 4, с. 446—449.
50. Вейганд-Хилъгетаг. Методы эксперимента в органическом химии. Пер.
с нем. Под ред. Н. Н. Суворова. М., «Химия», 1968. 994 с.
51. Чудное А. Ф., Мирошников А. М., Швец Д. Л., Техническая информация
Сер. Методы анализа и контроля в химической промышленности, 1967, № 1,
с. 37-43; Lloyd W.G., J. Am. Chem. Soc., 1956, v. 78, № 1, p. 72-75;
Griffiths T. R., Can. Spectroscopy, 1968, v. 13, Al 5, p. 131—133, 144; РЖХлм
1974, 8Ж85.
52. Пат. США 303511 (1962); Eastoe J. E., Chem. and Ind., 1966, № 1, p. 25;
Мирошников A. M. и др. Авт. свид. Al 201363 (1966); Изобр. пром, образцы.
Товарн. знаки, 1967, № 18.
53. Collin J. Е., Conde G. L., Bull, classe sci. Acad. R. Belg., 1966, v. 52, № 7,
p. 978—1009.
54. Kirmse W., Buschhoff M., Chem. Ber., 1969, Bd. 102, № 4, S. 1087—1097.
55. Фуракава Дж., Саегуса T. Полимеризация альдегидов п окисей. Пер.
с англ. Под ред. II. С. Ениколопяна и В. А. Кабанова. М., «Мир», 1965.
479 с.; Tanimoto Sigeo е. а., «Юки госэи кагаку кёкай си» (J. Soc. Org.
Synth. Chem.), 1968, v. 26, № 5, p. 435—438; РЖХим, 1969, 7C315.
56. Пат. США 3647892 (1972); Тезисы докладов 2 Всесоюзной конференции
по эпоксидным мономерам и эпоксидным смолам. Министерство высшего
и среднего образования. Днепропетровск, изд. Госуниверситета, 1974. 310 с.
57. Skellon J. Н., Whiteley Р., J. Appl. Chem., 1964, v. 14, № 10, р. 448—449.
58. Вредные вещества в промышленности. Т. 1. Под ред. Н. В. Лазарева. Л.,
«Химия», 1971. 832 с.
59. Предельно допустимые концентрации вредных веществ в воздухе и воде.
Справочное пособие ГИГХ. Л., «Химия», 1972. 376 с.
60. Carpenter С. Р. е. a., Arch. Ind. Heath, 1956, v. 14, А"» 2, p. 114—131; Пре-
дельно допустимые концентрации вредных веществ в воде водоемов сани-
тарно-бытового водопользования. М., Министерство здравоохранения
СССР, 1973. 26 с.
61. Кордыш Э. А., Ратпан М. М., «Гигиена и санитария», 1974, № 10, с. 16—
62. Неволин Ф. В. Химия и технология синтетических моющих средств. М.,
«Пищевая промышленность», 1971. 424 с.
331
63. Пат. США 3034897 (1962); Kapsabis J. G. e. a., Dairy Sci., 1963, v. 46, № 2,
p. 107 — 113.
64. Волков Ю. M., Журн. ВХО им. Д. И. Менделеева, 1966, т. 11, № 4, с. 427 —
432; Лукиных Я. А., там же, с. 432—437; Лукиных И. А. Очистка сточных
вод, содержащих синтетические поверхностно-активные соединения. М.,
Стройиздат, 1972. 96 с.
65. Лебедев И. И., В кн.: Строение, реакционная способность и механизм
превращений соединений с кратными связями и малыми циклами. Л., «Хи-
мия», 1967, с. 137; Швец В. Ф., Докторская диссертация. М., МХТИ им.
Д. И. Менделеева, 1974.
66. Кюма Тацуо и др., «Когё кагаку дзасси» (J. Chem. Soc. Japan, Ind. Chem.
Soc.), 1967, v. 70, № 2, p. 169—172; РЖХим, 1967, 24Ж109.
67. Wurtz A., Ann., 1860, Bd. 113, S. 255; Bd. 116, S. 249.
68. Зимаков П.В., Чураков А., Промышленность органической химии», 1936,
№ 6, с. 329—336; Петров А. А., ЖОХ, 1944, т. 14, № И, с. 1038—1043;
пат. США 2748171 (1956); 3328467 (1967); шведск. пат. 166364 (1959); яп.
пат. 4858 (1963). , '
69. Пат. США 3398201 (1968); Polyethers. Ed. by Е. N. Gaylord. New York,
Interscience Publ., 1963. 404 p.
70. Корнева Э. Д., Рыбникова A. H., ЖПХ, 1961, t. 34, № 8, c. 1875—1878;
пат. ФРГ 1248660 (1968).
71. Савоськин В. М., Лысяк В. Т., Авт. свид. № 162163 (1963); Бюлл. изобр.
и товарн. знаков, 1964, № 9, Миханпгьев Б. И., «Труды проблемной лабора-
тории химии ВМС Воронежского университета», 1966, № 4, с. 42—43.
72. Баранова Г. Ф., Григорьев Ф. П., Гурвич С. М., ЖПХ, 1972, т. 45, № 7,
с. 1553—1556.
73. Зейналов Б. К. и др., Авт. свид. № 242870 (1968). Изобр., пром, образцы,
товарн. знаки, 1969, № 16.
74. Пат. ФРГ 1129147 (1962).
75. Пат. ФРГ 804683 (1951). .
76. Яп. пат. 26613 (1967); 9207 (1968); Зейналов Б. К. и др., Азерб. нефт. хоз.,
1967. № 3, с. 41—42; пат. ГДР 65594 (1969).
77. Пат. США 1614863 (1927).
78. Hradil J., Malek J., Coll. Czech. Chem. Common., 1969, v. 34, № 12,
p. 3959—3966; Полянский H. Г. и др., Хим. пром., 1970, № 2,
с. 87—92.
79. Зейналов Б. К. Авт. свид. № 232226 (1967); Изобр., пром, образцы. Товарн.
знаки, 1969, № 1.
80. Рохманкулов Д. И. и др., «Доклады нефтехимической секции Башкирского
правления ВХО им. Д. И. Менделеева», 1968, № 4, с. 24—26.
81. Франц, пат. 1583867 (1969); пат. США 3491143 (1970).
82. Пат. США 1968033 (1934); 2067385 (1937); Фаворский А. Е.. Шостаков-
ский М. Ф., ЖОХ. 1943, т. 13, № 1, с. 1—20.
83. Фаворский А. Е., ЖРФХО, 1906, т. 38, с. 741.
84. Пат. США 2497408 (1950); 3262969 (1966), 3393225 (1968); пат. США 3427348
(1969); Тамура Масухико, Яуси Тэруо, «Когё кагаку дзасси» (J. Chem. Soc.
Japan, Ind. Chem. Sec.), 1969, v. 72, № 2, p. 578—585; РЖХим, 1969,
19Б1020.
85. Маркевич В. С., Ульянова В. И., Хим. пром., 1973, № 8, с. 580—581.
86. Пат. ФРГ 1276621 (1969).
87. Фиошин М. Я., Миркинд Л. А., Журиков М. Ж., Усп. хим., 1973, т. 42,
•М 4, с. 677—695.
88. Olavi /., Virtanen Р., Suomen Kem., 1965, v. 38, № 7, p. 135 — 142.
89. Лебедев H. H. и др., Теорет. и эксперим. химия, 1968, т. 4, № 2, с. 203—
206; Ж. орг. хим., 1969, т. 5, № 9, с. 1542—1548.
90. Леонов М. Р., Коршунов И. А., ЖПХ, 1962, т. 35, № 10, с. 2324-2328,
ЖОХ, 1962, т. 32, № 1, с. 208—212; Леонов М. Р., Маленева И. Г., «ТрУДЫ
по химии и химической технологии» (Горький), 1962. т. 2, с. 480—48-,
1963, т. 1, с. 186 — 187; Леонов М. Р., Коршунов И. А., там же, 1964, т. 3,
С. 515—520; Леонов М. Р., Маленева И. Г., Коршунова И. А., ЖПХ, 196 ,
332
т. 38, № 6, с. 1367—1373; Власов Г. М. и др., Хим. пром., 1975, К» 3
с. 30-31.
91. Repas М. е. a., Chem.. Prumysl, 1961, v. 11. № 7, р. 345—348; № 9, р. 469_
472.
92. Wrigley A. N., Smith F.D., Stirton A. J., J. Am. Oil Chem. Soc., 1957
v. 34, № 1, p. 39—44. ’
93. Gee G. e. a., J. Chem. Soc., Sec. B, 1959, № 4, p. 1338—1352.
94. Parker R. E., Isaacs N. S., Chem. Rev., 1959, v. 59, № 4, p. 737 — 799.
95. Мирошников A. M., Чудное А. Ф., В кн.: Физико-химическпе методы ана-
лиза. Кемерово, Кемеров. кн. изд., 1970, с. 141—148.
96. Laird R. М., Parker R. Е., J. Chem. Soc., Sec. В, 1969, № 9, р. 1062—1068.
97. Лебедев Н. II., Савелъянов В. П., Антипова II. В., Изв. вузов. Сер. Химия
и хим. технол., 1967, т. 10, № 4, с. 461—464.
98. Козлов В. М., Лебедев II. II., «Труды МХТИ им. Д. И. Менделеева», 1965,
вып. 48, с. 68—72; Козлов В. М., Лебедев Н. II., «Кинетика и катализ»,
1967, т. 8, №. 1, с. 18—23; Лебедев И. II. и др., Ж. орг. хим., 1969, т. 5
№ 4, с. 608—612.
99. Сэкигути С. и др., «Когё кагаку дзасси» (J. Chem Soc. Japan, Ind. Chem
Sec.). 1967, v. 70, № 1, p. 49-52; 1968; v. 71, № 11, p. 1943-1945; РЖХим
1969, 15Ж81.
100. Коган Л. M., Производство и переработка окиси этилена в Германии.
М., Госхимиздат, 1947. 121 с.
101. Woodcock С. A., Chem. in Canada, 1963, v. 15, № 5, p. 17—22.
102. Stobaugh R. e. a.. Hydrocarb. Process., 1970, v. 49, № 10, p. 105—115;
1973, v. 52, № 1, p. 99—109; Химическая промышленность США. T. 2.
Под ред. Г. А. Коренькова. М., НИИТЭхим, 1972. 611 с.
103. Мирошников А. М., Чудное А. Ф., Шигарова 3. Г. Авт. свид.
№ 424854 (1972); Открытия. Изобр. Пром, образцы. Товарн. знаки, 1974
№ 4.
104. Гарбер Ю. Н., Комарова Л. Ф., ЖПХ, 1966, т. 39, № 6, с. 1366—1372.
105. Азингер Ф. Химия п технология моноолефинов. Пер. с нем. Под ред.
В. И. Исагулянца. М., Гостехиздат, 1960. 739 с.; Азеотропные смеси. Спра-
вочник. Под ред. В. Б. Когана. Л., «Химия». 1971. 848 с.
106. Чудное А. Ф., Мирошников А. М., Авт. свид. № 331054 (1969); Открытия.
Изобр. Пром, образцы. Товарн. знаки, 1972, № 9.
107. Чалмерс Л. Химические средства в быту и промышленности. Пер. с англ.
Под ред. Л. С. Эфроса. Л., «Химия», 1969. 528 с.
108. Беляева К. П. и др., Лакокрасочные материалы и их применение, 1966,
№ 3, с. 28—3(1; англ. пат. 1229779 (1971).
109. Мельникова Б. И., Блиничева И. Б., Журн. ВХО им. Д. М. Менделеева,
1970, т. 15, № 3, С. 263—271.
110. Энглин Б. А., Применение моторных топлив при низких температурах.
М., «Химия», 1968. 164 с.; Gardner L., J. Inst. Petrol., 1969, v. 55, № 546,
p. 418-421.
111. Пат. США 3038801 (1962).
112. Bnrriel-Marli F., «Afinidad», 1969, v. 26, № 269, p. 505 — 515; Reynolds R.C.,
Am. Mineralogist, 1969. v. 54, № 3, p. 562—567.
113. Пат. США 2559519 (1951); Гарбер IO. H., Бовкун П. А., ЖПХ, 1968, т. 41,
№ 2, с. 318—324.
114. Хаттон Р. Е. Жидкости для гидравлических систем. Пер. с англ. Под
, ред. В. В. Вайнштока. М., «Химия», 1965. 362 с.
;115. Англ. пат. 1055641 (1967); Бедарев Н. Г. и др., Авт. свид. № 318620 (1969).
• Изобр.. пром, образцы. Товарн. знаки, 1971, № 32; Гликоли и опыт их
; применения в нефтяной и газовой промышленности. Сер. «Газовое дело»,
! М., ВНИИОЭНГ, 1970. 212 с.
116. Пат. ФРГ 1070165 (1960).
'117. Пат. США 2952637 (1960); яп. пат. 24406 (1969).
118. Пат. США 2983574 (1961); англ. пат. 1235997 (1971); Крумголъз Б. С-,
Ж. структ. химии. 1972, т. 13, № 5, с. 774—779.
119. Пат. США 3455994 (1969).
333
120. Амис Э. Влияние растворителя на скорость и механизм химических реак-
ций. Пер. с англ. Под ред. М. И. Кабачника. М., «Мир», 1968. 328 с.;,
Крестов Г. А. и др., ИЗв. вузов. Сер. Химия и хим. технол., 1970, т. 13,
с. 169—171; «Труды Ивановского химико-технологического института»
1971, № 12, с. 53—55; Campanella L., «Plating», 1972, v. 59, № 11, р. 1043—
1047.
121. Пат. ФРГ 1234196 (1967); Barbat A., Sandru D., Rev. chem. (RSR), 1968,
v. 19, № 2, p. 78—81.
122. Кричевский Г. E., Росинская У. Я., Коваль В. В., Журн. ВХО им.
Д. И. Менделеева, 1970, т. 15, № 3, с. 300—306.
123. Дуденков С. В., Основы теории и практики применения флотационных ре-
агентов. М., «Недра», 1969. 320 с.
124. Пат. США 2923408 (1960).
125. Shukla R. N., е. a., J. Appl. Chem., 1964, v. 14, № 6, р. 236—239.
126. Князькова Т. Ф. и др., Техническая и экономическая информация. Сер.
Методы анализа и контроля производства в химической промышленности,
1967, № 4, с. 8—12.
127. Dohogne A., Rev. techn. md. cuir., 1963, v. 55, № 4, p. 104—107.
128. Штюпелъ Г. Синтетические моющие и очищающие средства. Пер. с нем.
Под ред. А. И. Гершеновича. М., Госхимиздат, 1960. 672 с.
129. Петров А. А., Журн. ВХО им. Д. И. Менделеева, 1966. т. 11, с. 398—
401; Бабалян Г. А., Кравченко И. И., там же, с. 405—407.
130. Яп. пат. 28476 (1965); 25404 (1967); 515 (1968); англ. пат. 113380 (1968);
Осе И. К., В кн.: Биосинтетические и физиологические свойства микро-
организмов. Под ред. М. Е. Беккер. Рига, «Зинатне», 1975, с. 41—45.
131. Чернышев А. К., Справочный материал по качеству химической продукции.
М., ГИАП, 1972. 492 с.
132. Химические товары. Т. 2. Справочник. М., «Химия», 1969. 512 с.
Глава VIII
Методы анализа гликолей и их эфиров
Химические методы анализа
Качественный анализ
Качественные методы определения гликолей и их эфиров основаны
главным образом на колориметрических измерениях и в меньшей
мере — на получении нерастворимых соединений (осадков). Описано
несколько цветных реакций для открытия алифатических спиртов,
пригодных и для определения диолов и их эфиров. Этиленгликоль
в растворе и-диазобензолсульфокислоты в присутствии гидразо-
бензола дает оранжевую окраску, переходящую в красную, диэти-
ленгликоль — бледнооранжевую. Раствор 2,4-динитротолуола в ще-
лочной среде в присутствии этиленгликоля окрашивается в оранже-
вый цвет, а в присутствии диэтиленгликоля — в зеленый. Наличие
этиленгликоля придает динитробензолу в щелочной среде фиолето-
вый цвет. Алифатические спирты при этом также окрашиваются,
поэтому определению мешают [1].
Соли четырехвалентного церия, например желтый концентриро-
ванный раствор гексанитроцерата, при добавлении этиленгликоля
окрашиваются в красный цвет. Но метод малочувствителен и не-
селективен, так как такую же реакцию дают алифатические спирты
п простые эфиры гликолей. Альдегиды, кетоны, органические кис-
лоты и сложные эфиры не мешают определению [2, с. 291]. Этот
колориметрический метод применяют для определения в реактивном
топливе метилового эфира этиленгликоля, являющегося присадкой,
предотвращающей образование льда [3].
Предложен полумикрометод идентификации спиртов, основанный
на образовании красной окраски при растворении в спирте продукта
взаимодействия 5,7-дихлор-2-метил-8-оксихинолина с пятивалент-
ным ванадием. Окрашивание отмечено для 30 одноатомных спиртов,
8 двухатомных гликолей, метилового и уксусного эфиров, карбо-
ваксов 1500 и 6000, глицерина. Однако цветную реакцию дают и дру-
гие классы веществ — кетоны, альдегиды, эфиры, кислоты и амиды.
Метод применим для капельных проб [4].
Некоторые гликоли, например пропиленгликоль, дают с арома-
тическими альдегидами в присутствии концентрированной серной
335
кислоты цветные реакции (так называемые реакции Комаровского),
что позволяет определить примесь пропиленгликоля в этиленгли-
коле, который не окрашивает раствор. Аналогично определяются
2,3-бутиленглпколь, пентаметиленгликоль и некоторые другие гли-
коли [2, с. 191].
Специфические реакции на эфиры гликолей, по-впдимому, отсут-
ствуют. Простейшей качественной пробой на эфир гликоля, а именно
на ОН-группу, является реакция с металлическим натрием. Опре-
делению мешают вода, спирты и сложные эфиры оксикислот. В слу-
чае сложной смеси рекомендуется вначале провести разделение
на индивидуальные компоненты, например, ректификацией. Вы-
деленные простые моноэфиры гликолей можно идентифицировать
по температурам плавления соответствующих эфиров аллофановой
кислоты или инструментальными методами [5, с. 941]. Эта реакция
используется для определения простых моноэфиров гликолей коло-
риметрическим методом, например метилового эфира этиленгликоля,
применяемого в качестве присадки, предотвращающей образование
льда [5].
Для качественного анализа гликолей можно использовать спе-
цифические только для данного диола нерастворимые сложные
и простые эфиры, которые образуются при взаимодействии гликолей
с некоторыми ароматическими соединениями. Это — сложные ди-
эфиры бензойной, тг-нитробензойной и 3,5-динитробензойной кислот;
сложные диэфиры N-замещенных карбаминовых кислот (уретаны);
бис(трифенилметил)эфиры [6].
Гликоли легко окисляются иодной кислотой или ее солями по
реакции:
RCIIOHCHOIIR' + IOI--> Г.( НО R'CIIO IO- Н.,о
При этом из этиленгликоля образуются две молекулы формальде-
гида, из пропиленгликоля — формальдегид и ацетальдегид, из
глицерина — две молекулы формальдегида и одна молекула му-
равьиной кислоты. Продукты реакции — иодат и особенно альде-
гиды — довольно легко обнаруживаются многочисленными
качественными химическими реакциями и инструментальными мето-
дами .
Количественный анализ
Количественный анализ гликолей химическими методами основы-
вается на характерных реакциях гидроксильных групп — ацилиро-
вании, окислении и расщеплении. Наибольшее распространение
получили два метода ацилирования, которые основаны на реакции
уксусного и фталевого ангидридов со спиртами в среде пиридина
по следующей схеме:
взаимодействие уксусного ангидрида с пиридином
(CII3CO)2O + C5H5N —> C5II5N • (СН3СО)2О
336
реакция пиридинийацетата с гликолем'.
СН2ОН СН-ОСОСНз
| +2C5H5N-(CH3CO)2O---->1 " +2C5H5N • СН3СООН' '
СНоОН С1Т.,ОСОСН3
разложение избытка реагента водой
C5H3N.(CH3CO)2O-H2O —> C5H5N СН3СООН-4-СН3СООП
Выделившаяся свободная кислота титруется щелочью *.
По Критчфилду аналогичное определение проводится в плотно
закупоренных бутылках, выдерживающих нагревание до 100 °C
под давлением. Для большей безопасности бутылки помещают в за-
щитные мешки. В таких условиях установлено необходимое для
данной реакции время (в мин): для этиленгликоля — 30, для про-
пиленгликоля — 120, для диэтиленгликоля — 30, для дипропилеи-
гликоля — 150, для глицерина — 30 и для бутилцеллозольва —
30 [7, с. 104].
Спирты, амины и фенолы также взаимодействуют с уксусным
ангидридом, поэтому определению мешают. Искажают результаты
и альдегиды, меркаптаны, эпоксиды, легко гидролизующиеся слож-
ные эфиры. Вода в небольших количествах не мешает определению,
а при значительных концентрациях на воду вводится поправка,
однако точность определения при этом намного снижается.
Реакции ацетилирования катализируют минеральные кислоты.
Разработано два метода ацетилировайия в присутствии хлорной
кислоты в среде пиридина или этилацетата. Реакции хорошо про-
текают уже при комнатной температуре, причем в этилацетате более
активно, чем в пиридине [7, с. 107]. Важным преимуществом мето-
дики является проведение анализа при комнатной температуре
и возможность определения пространственно экранированных
ОН-групп. Мешают определению те же соединения, что и при ацети-
лировании без катализатора.
В этилацетате в присутствии хлорной кислоты реакция про-
текает по следующей схеме:
/° /° '
сн3сг cn3cf
)О + Н+ —> /ОН СН3С+4-СНзСООН
сн3(Г сн3сГ 1
\о
CH3C+ + ROH --> CH3COR + H+
II II
о о
Это наиболее быстрая реакция ацетилирования, известная в на-
стоящее время. Для большинства гликолей реакция проходит до
* Методика определения этиленгликоля этим методом приведена в ГОСТ
11033—64, Этиленгликоль. . ,
22 Заказ 972
337
конца при комнатной температуре в течение 10 мин. Однако вслед-
ствие высокой активности реагента возникают многочисленные
помехи. Альдегиды, кетоны, олефины, некоторые простые эфиры,
нмиды, гидразиды, третичные спирты и эпоксиды мешают определе-
нию. Ацетиленовые соединения и сложные эфиры не влияют на
точность анализа. Концевые ОН-группы полиэтилен- и полипропи-
ленгликолей не могут быть определены этим методом из-за побочной
реакции с полиэфирами [7, с. 109].
Следующий наиболее распространенный метод макроопределения
1,2-гликолей основан на взаимодействии диолов с фталевым анги-
дридом в среде пиридина [8]:
Предполагается образование промежуточного продукта взаимо-
действия ангидрида с пиридином:
Определение с фталевым и уксусным ангидридами проводится
аналогичным образом [7, с. 112].
Преимуществом метода является его специфичность на первичные
и вторичные ОН-группы и применимость к водным растворам. Ке-
тоны, альдегиды, сложные эфиры и фенолы не мешают определению.
Точность определения ± 1% (отн.) при содержании гликоля до 0,3%.
Гликоли, содержащие третичные ОН-группы, количественно этим
методом не определяются. Амины, меркаптаны, эпоксиды и легко
гидролизующиеся сложные эфиры (например, формиаты) определе-
нию мешают.
В качестве ацилирующих агентов можно использовать ангидриды
других кислот: пропионовый ангидрид — для полиэтиленглико-
лей [9], пиромеллитовый диангидрид — для низших диолов [6,
р. 587], а также изопропенилацетат, трифторуксусная кислота,
трифенилхлорметан [10]. Важное в теоретическом и практическом
отношении содержание первичных и вторичных гидроксильных
групп в диолах может быть установлено по реакции гликоля с фенил-
изоцианатом или ацетангидридом. Принцип метода основан на раз-
личной скорости взаимодействия ОН-групп с ацетилирующим аген-
том: первичные. реагируют быстрее, чем вторичные. Этим методом
изучено соотношение рервичных и вторичных гидроксильных групп
в полипропиленгликолях [11]. Присутствие других гидроксил-
338
содержащих веществ мешает определению, методика применима для
анализа чистого продукта и не позволяет установить изомерный
состав.
Разработан метод определения концевых ОН-групп в гликолях
и полигликолях, основанный на их реакции с фенилизоцианатом
в присутствии октоата олова II (катализатор). Избыток фенил-
изоцианата определялся газохроматографически (колонка 100 X
X 0,4 см, 10% силикона GI7100V на стерхамоле, температура
75 °C) [12].
Самостоятельное значение имеют способы, основанные на окис-
лении (расщеплении) 1,2-гликолей иодной кислотой и ее солями.
Различают три метода [7, с. 114]: кислотно-основной метод с исполь-
зованием NaIO4; окислительно-восстановительный метод с исполь-
зованием иодной кислоты и колориметрическое определение основ-
ного продукта реакции — формальдегида.
Определение избытка NaIO4 основано па установлении равно-
весия в водной среде:
NaIO4-2II,O ч-д_- NaH4fO6
Кислая соль определяется титрованием едким натром. Образу-
ющийся после расщепления гликоля иодат натрия нейтрален по
отношению к индикатору. Следовательно, снижение кислотности
служит мерой расщепления гликоля реагентом. Для повышения
четкости определения титрование проводят при пониженной до
1 С температуре. Этим методом успешно определяется большинство
диолов. Время реакции при комнатной температуре обычно не пре-
вышает 20 мин. Соединения кислотного и основного характера ме-
шают определению.
В окислительно-восстановительном методе непрореагировавшая
иодная кислота взаимодействует с избытком арсенита натрия, избы-
ток последнего титруют стандартным раствором иода. Метод может
быть использован для определения большинства гликолей.
Колориметрический метод основан на расщеплении гликоля
перйодатом натрия с последующим определением образующегося
формальдегида цветной реакцией, чаще всего с хромотроповой
кислотой. Он особенно удобен для определения следовых концен-
траций (^ 1 млн-1) и имеет много модификаций [2, 7, 13]. Метод
не применим для анализа гликолей типа RCHOHCHOHR, где R —
алкил. Мешают определению органические примеси, альдегиды
и вицинальные соединения, которые при взаимодействии с иодно-
кислым натрием дают формальдегид. Кроме колориметрирования,
полученные при расщеплении (окислении) диолов альдегиды могут
быть определены как химическими, так и инструментальными мето-
дами.
Перйодатный метод положен в основу анализа сложных смесей
гликолей. Принцип метода основан на последовательном определе-
нии продуктов окисления, характерных для определенного ди-
ола [4, р. 348]. Этим же методом определяют примесь этиленгликоля
22*
339
в диэтиленгликоле. Реже используют для расщепления гликолей
другие реагенты — тетраацетат свинца [14], бромистый водо-
род [7, с. 118].
В ряде методик для анализа гликолей применены реакции ком-
плексообразования. Например, при образовании комплекса эти-
ленгликоля с вольфраматом натрия выделившаяся щелочь опре-
деляется титриметрическим методом [15]. Полиэтиленгликоли и их
производные определяются прямым потенциометрическим титрова-
нием тетрафенилборатом натрия в присутствии ионов бария. Поли-
этиленгликоли молекулярной массы 600—4000 реагируют стехио-
метрически с образованием осадка — комплекса, содержащего
2 моль тетрафенилбората и 10,4 ± 0,2 моль этиленоксида на 1 моль
бария. Приблизительная молекулярная масса неизвестного поли-
этиленгликоля может быть определена по ИКС осадка. В аналогич-
ной методике полиэтиленгликоли осаждали тетрафенилборатом
в присутствии ионов бария; избыток бария оттитровывали этилен-
диаминтетрауксусной кислотой, индикатором служил метилтимол
синий [16].
Смесь цис- п тпраис-изомеров циклических диолов проанализи-
рована на основе реакции с борной кислотой — последняя реагирует
только с цис-гидроксильными группами [17]. Количественное опре-
деление простых моноэфиров гликолей основано главным образом
на реакциях ацилирования и осуществляется так же, как и анализ
гликолей. Сложные эфиры гликолей определяют качественно и коли-
чественно гидролизом щелочами. После гидрориза раствор титруют
соляной кислотой в присутствии фенолфталеина; уменьшение содер-
жания щелочи пропорционально количеству сложного эфира в пробе.
Параллельно проводят холостой опыт [4, р. 981].
В целом химические методы анализа гликолей и их производных
характеризуются длительнбстью определения, малой точностью
в случае сложных смесей и не различают гомологов. Поэтому в по-
следнее время предпочтение отдается физико-химическим методам
анализа, в частности, газохроматографическому методу.
Хроматографические методы анализа
Гликоли являются сильно полярными ассоциированными в жидком
состоянии высококипящими соединениями. Газохроматографический
анализ таких веществ осуществляется либо непосредственно (прямой
анализ), либо после предварительной этерификации. Прямой анализ
проводится для сравнительно низкокипящих смесей, как правило,
на колонках, заполненных инертным носителем с небольшим коли-
чеством малополярной неподвижной фазы. Более высококипящие
полиолы и их эфиры с низким давлением пара и недостаточной для
прямого хроматографирования термостабильностью переводят в бо-
лее легкокипящие соединения, обычно превращением концевых
ОН-групп в эфирные. Эфиры менее полярны, имеют более низкую
температуру кипения, достаточно стабильны для хроматографиро-
вания и легко синтезируются.
340
В последние годы газохроматографический метод анализа стал
наиболее распространенным и универсальным для определения
гликолей и их производных. Определение первых членов гомологи-
ческого ряда этиленгликолей предложено производить на колонке
60x0,6 см с полиэтиленгликолем молекулярной массы 1000
(ПЭГ-1000) при 150 °C [18], на колонке с вакуумной смазкой при
175 °C [19] и на апиезоне [20]. Смесь 1,2-пропиленгликоля и эти-
ленгликоля, содержащая 90% воды, анализировалась при 105 °C
на колонке 200x0,6 см, заполненной хромосорбом с 5% тетра-
оксиэтилендиамина в качестве неподвижной фазы [21].
Предложен способ определения многоатомных спиртов в алкид-
ных смолах, полученных на основе фталевой кислоты [22]. Проба
алкидной смолы разлагалась в среде бутиламина, выделенные много-
атомные спирты ацетилировались уксусным ангидридом. Хромато-
графический анализ полученных таким образом ацетатов 1,2-про-
пиленгликоля, этиленгликоля, диэтиленгликоля, маннита, сор-
бита, глицерина, триметилолпропана, триметилолэтана и пен-
таэритрита производился при программируемом повышении
температуры от 50 до 225 °C со скоростью 7,9 °C в 1 мин на колонке
122x0,6 см, заполненной хромосорбом с 10% карбовакса 20М.
Полное разделение девяти изученных многоатомных спиртов про-
должается 50 мин. Для сокращения времени анализа до 25 мин
используют колонку с неполярной неподвижной фазой — 20%
силиконового масла на том же носителе с программированием темпе-
ратуры от 50 до 275 °C. При этом ацетаты 1,2-пропиленгликоля
и этиленгликоля, а также маннит и сорбит не разделяются.
Методика была усовершенствована (заменили бутиламин), после
чего стало возможным в тех же условиям определять еще четыре
спирта — бутандиол-1,4, дипропиленгликоль, триэтиленгликоль
и неопентилгликоль. Для идентификации многоатомных спиртов
в полиэфирах их переводили в соответствующие ацетаты в результате
переэтерификации избытком уксусной кислоты в присутствии 1%
серной кислоты. Полученные ацетаты этиленгликоля, 1,2-пропилен-
гликоля, 1,3-, 2,3- и 1,4-бутиленгликолей, диэтиленгликоля и гли-
церина разделили при 100 °C на колонке 120x0,5 см с 5% ПЭГ
на эмбацеле [23].
Сложная смесь полиолов природного происхождения, получа-
ющаяся при каталитическом гидрогенолизе моносахаридов и име-
ющая широкий интервал температур кипения и пониженную термо-
стойкость высокомолекулярных компонентов, анализировалась
в виде ацетильных производных при 200 °C на колонке 200x0,6 см,
заполненной стерхамолом с 15% полидиэтиленгликольсукцината.
Время анализа — 70 мин [24]. Основные компоненты указанной
смеси (этиленгликоль, 1,2-пропиленгликоль и глицерин) предложено
анализировать прямым хроматографированием в два этапа: вначале
определяют этиленгликоль и 1,2-пропиленгликоль на колонке
с ПЭГ-10 000, нанесенным на целит-545 при 135—140 °C; затем
«аходят сумму этих даолов и глицерин на колонке с ацетилирован-
341
ным декстрином (25% по массе) на том же носителе [25]. При этом
этиленгликоль и 1,2-пропиленгликоль не делятся, а глицерин вы-
ходит асимметричным пиком, по-видимому, из-за остаточной адсорб-
ции его на носителе.
Полное разделение и количественный анализ смеси этиленгли-
коля, 1,2- и 1,3-пропиленгликоля, 1,2-, 1,3-, 1,4-, мезо-2,3-бутилен-
гликолей пентандиола-1,5, гександиола-1,6 и глицерина получено
на колонке 145 X 0,3 см, наполненной хромосорбом с 10% поли-
диэтиленгликольсукцината LAC-3 77-728 при программируемой тем-
пературе от 80 до 185 °C со скоростью 4 °C в мин (детектор — пла-
менно-ионизационный) [26].
Диолы С2 — С4 и глицерин, содержащиеся в липидных гидро-
лизатах, переведенные в ацетаты или триметилсцлиловые эфиры,
анализируются газохроматографическим методом в изотермическом
режиме: ацетаты при 140 °C на колонке 200 X 0,3 см, 10% поли-
этиленгликольсукцината или реоплекса-400 на хромосорбе; три-
метилсилиловые эфиры при 100 °C на колонке 120 X 0,4 см, 10%
силикона СКТ-В на том же носителе.
Весьма важным является не только селективность неподвижной
фазы, но и инертность носителя. Остаточная адсорбция на носителе
является одной из причин асимметрии пиков, что довольно часто
встречается в хроматографии особенно сильнополярных веществ —
воды, спиртов, диолов и кислот. Поэтому для анализа соединений
подобного рода рекомендуется брать адсорбционно-инертные носи-
тели — силанизированные хромосорбы, тефлон, хлорид натрия,
кварц, металл, стеклянные шарики с небольшим количеством не-
подвижной фазы.
В последнее время для анализа полярных веществ предложено
использовать высокопористые сетчатые сополимеры дивинилбензола
со стиролом — пор опаки и полисорбы. Они, в отличие от обычных
носителей, не адсорбируют полярные вещества, даже вода и спирты
имеют малое время удерживания и выходят симметричными пиками.
Их можно применять как в газоадсорбционном варианте хроматогра-
фии, так и с небольшим количеством неподвижной фазы — в газо-
жидкостном. На колонке 120 X 0,4 см, заполненной поропаком,
при 200 °C получено хорошее разделение 8 низших гликолей и их
эфиров, выходящих в такой последовательности: метилцеллозольв,
этиленгликоль, этилцеллозольв, н-пентанол (внутренний стандарт),
изопропилцеллозольв, н-бутилцеллозольв, метилкарбитол, 2-ме-
тилпентан-2,4-диол и этплкарбитол. В течение месяца работы колонка
давала полностью воспроизводимые результаты [27].
На колонке с хромосорбом-102 при 215—220 °C проанализиро-
вана смесь: этиленгликоль, 1,2-пропиленгликоль, глицерин и вода
с диэтиленгликолем или 1,4-бутиленгликолем в качестве внутрен-
него стандарта за 10—12 мин. Хромосорб-102 брался как немодифи-
цированвый, так и модифицированный 2,5% ПЭГ-40 000. В последнем
случае разделение улучшается [28]. Эта же смесь анализировалась
газоадсорбционной хроматографией при 178 °C на колонке 100 X
342
X 0,4 см с полисорбом-1, время анализа составило 8 мин. Хромато-
грамма смеси представлена на рис. 108 («Цвет-4», скорость газа-
носителя 175 мл/мин, детектор — катарометр).
Свойство такого типа сорбентов давать симметричные пики поляр-
ных веществ использовано для определения примеси воды от 0,3
до 12% в исследованных многоатомных спиртах как в смеси, так
и в отдельных компонентах [29]. В обоих последних методиках
.легко достигается полное разделение «роковой» пары этиленгли-
коль — 1,2-пропиленгликоль.
Рис. 108. Хроматограмма
смеси гликолей и глице-
рина при 178 °C:
1 — вода: 2 — этиленгли-
коль; 3 — 1,2-пропиленгли-
коль; 4 — глицерин.
мин
Хроматографический анализ примесей этилен- и диэтиленгли-
коля в триэтиленгликоле проводился при 175 °C на колонке
с ПЭГ-6000 [30]. Для понижения температуры анализ проводили
при пониженном давлении, однако значительного снижения темпе-
ратуры не достигнуто. Методика с вакуумированием сложна для
серийных анализов. Такого же эффекта добиваются применением
коротких колонок, снижением количества неподвижной фазы, а
также уменьшением полярности фазы и разделяемых днолов. На ко-
лонке 85 X 0,6 см, наполненной целитом-545 с 15% полиэтилен-
гликольадипата при 170 °C, внутренний стандарт трибутилфосфат,
определялась чистота этилен- и триэтиленгликоля. Найдено 10 2%
примеси диэтиленгликоля в этиленгликоле, этилен- и диэтиленгли-
колей в триэтиленгликоле с точностью ±10% [31].
Технический этиленгликоль анализировался на колонке 166 X
X 0,6 см, с 5% карбовакса М при 100 °C, внутренний стандарт —
бутилкарбитол; детектор пламенно-ионизационный, газ-носитель —
азот. Продукт содержал 0,02% ацетальдегида, 0,1% 2-метил-1,3-
диоксолана, 2,5% диэтпленгликоля и 0,01% триэтиленгликоля
[32]. Применение в качестве неподвижной фазы воды и насыщение
^Водяными парами газа-носителя позволило разделить смесь
343
этиленгликоля, 1,2- и 1,3-пропиленгликолей, бутандиолы и спирты
Сх — Св в порядке их полярности [33].
Анализ пропилен- и бутиленгликолей осложняется наличием
изомеров — геометрических и оптических. Дипропиленгликоль
анализировался на колонке 279 X 0,6 см, заполненной флуоро-
паком-80 с 5% карбовакса 20М при 175 °C. На этой же неподвижной
фазе, нанесенной в количестве 10% на хромосорб (колонка 180 X
X 0,3 см) при 95 °C разделены диастероизомеры 2,3-бутиленгли-
коля [34]. Хроматографирование на неподвижных фазах различной
природы (сквалан, полиэтиленгликольадипинат, ПЭГ-400, тетра-
цианэтилпентаэритрит) показало, что диастереоизомеры 2,3-бутилен-
гликоля элюируют в соответствии с их температурами кипения:
рацемат — 176,5 °C, мезоформа — 181 °C. Интересно, что диацетаты
этих изомеров имеют обратный порядок температур кипения: ди-
ацетат мезо — 189 °C, диацетат рацемат — 192 °C; в этой же после-
довательности они и выходят из колонки [35].
Динитропроизводные этилен-, диэтилен- и триэтиленгликолей,
1,2-пропиленгликоля и пентадиола-1,5 анализировались на колонках
35 — 50 X 0,5 см, заполненных целитом С-22 с 10% моноэтилен-
гликольсукцината при 145—150 °C, газ-носитель — гелий [36].
Аналитический контроль технологического процесса получения
простых моноэфиров гликолей реакцией окисей алкиленов со спир-
тами может быть полностью проведен газохроматографическим
методом. Не вступивший в реакцию этиловый спирт в синтезе этило-
вого эфира этиленгликоля (так называемый возвратный спирт)
анализируется при 90 °C на колонке 150 X 0,4 см, наполненной
тефлоном, модифицированном триэтиленгликолем; детектор — ката-
рометр. Компоненты выходят в следующем порядке: диэтиловый
эфир, окись этилена, вода и этилцеллозольв [37]. Интересно, что
окись этилена (т. кип. 10,7 °C) удерживается на триэтиленгликоле
сильнее диэтилового эфира (т. кип. 34,6 °C), что можно объяснить
образованием весьма прочной водородной связи циклического
эфира — окиси с триэтиленгликолем.
Наилучшие результаты анализа технологических смесей синтеза
этилцеллозольва получены при 160 °C на колонке 100 X 0,4 см,
наполненной тефлоном с 5% трикрезилфосфата. Компоненты выходят
в такой последовательности: вода, этилцеллозольв, этиленгликоль,
карбитол, ди- и триэтиленгликоль, однако на хроматограммах тех-
нологических смесей вместо триэтиленгликоля должен выходить
его этиловый эфир, а уже за ним — триэтиленгликоль. Именно
такая последовательность получена на колонке 50 X 0,4 см с ука-
занными носителем и неподвижной фазой при анализе кубовых
остатков синтеза этилцеллозольва при 160 °C и при 150 °C (рис. 109,
Perkin-Elmer, скорость газа-носителя 25 мл/мин, пламенно-иони-
зационный детектор) [38].
Применение капиллярной хроматографии для анализа этих про-
дуктов позволяет значительно снизить температуру определения
и получить хорошее разделение с симметричными пиками [39, 40L
344
Использовались медные капиллярные колонки длиной 36—38 м
и диаметром 0,3—0,35 мм. Особое внимание уделяется чистоте и
однородности внутренней поверхности капилляра, так как от этого
зависит толщина и равномерность пленки неподвижной фазы и в ко-
нечном счете — эффективность разделения. На колонке с поли-
пропиленгликолем 425 (ППГ-425), а также на силиконе Е-301 ком-
поненты кубового остатка синтеза этилцеллозольва выходили в соот-
ветствии с температурами кипения, как на трикрезилфосфате [38].
На колонке с ПЭГ-400 или ПЭГ-1000 порядок выхода меняется,
юльва:
— этиленгликоль; 2 — этилкарбитол; 3 — диэтиленгликоль; 4 — этиловый эфир три-
^гиленгликоля; 5 — триэтиленгликоль.
?мс. ПО. Хроматограмма товарного дипропиленгликоля (ДПГ):
' — ди-вторичный ДПГ; 2 — первично-вторичный ДПГ; 3 — ди-первичный ДПГ.
i именно: этиловый эфир диэтиленгликоля, этиленгликоль, этиловый
|фир триэтиленгликоля и диэтиленгликоль.
Сравнение результатов хроматографирования указанных компо-
нентов на разных неподвижных фазах позволило установить пары
своеобразных хроматографических аналогов типа гликоль — эфир
дигликоля, дигликоль — эфир тригликоля. Изменения порядка
выхода объясняются различной способностью примененных непо-
движных фаз образовывать водородную связь с разделяемыми ком-
понентами. Это явление использовано для четкого определения
примеси одного компонента перед пиком основного вещества другого,
для идентификации компонентов смесей гликолей и их эфиров.
Указанная выше капиллярная колонка с ПЭГ-400 или
ПЭГ-1000 оказалась универсальной для анализа целого ряда гли-
колей и их производных. На ней при 130 °C проанализированы
изомеры дипропиленгликоля, полученные в присутствии щелочного
катализатора (рис. 110, колонка 37 000 X 0,3 мм, 150 °C, пламенно-
345
ионизационный детектор), этилен- и пропиленгликоль, изомеры окиси
бутилена, установлены соотношения изомеров простых моноэфиров
моно- и дипропиленгликолей, полностью разделены смеси моно-
бутиленгликолей и ряд их эфиров, анализировались водные растворы
этиленхлоргидрина с пропилен- и бутиленхлоргидринами [39—41].
По-видимому, ПЭГ является наиболее селективной и достаточно
термостойкой неподвижной фазой для анализа гликолей и их произ-
водных. Он включен в отечественные и зарубежные стандарты (в оте-
чественном ГОСТ 5.594—70, количество основного вещества в эти-
ленгликоле определяется газохроматографическим методом на ко-
лонке с ПЭГ-4000).
По спецификациям США газохроматографический анализ моно-,
ди-, триэтилен- и пропиленгликолей осуществляется на колонке
120 X 0,6 см, неподвижная фаза — карбовакс 20М (ПЭГ-2000),
на галопорте или другом тетрафторэтиленовом полимере; темпера-
тура испарителя 215 °C (в случае присутствия тетраэтиленгликоля —
230 °C), температура детектора 270 °C, газ-носитель — гелий,
75 мл/мин, детектор по теплопроводности.
Выбор температуры колонки зависит от состава пробы. Если
проба содержит этилен-, диэтилен- и триэтиленгликоль, предлагается
температура 170 °C; при наличии в пробе тетраэтиленгликоля —
200 °C. Для пропиленгликолей рекомендована температура 190 °C.
Во всех трех случаях анализ возможен при программируемой темпе-
ратуре, скорость программирования в интервале 150—225 °C соста-
вляет 5,6 °C в мин.
Количественный расчет хроматограмм проводится методом вну-
тренней нормализации с учетом поправочных коэффициентов [6].
Полипропилен- и полибутиленгликоли (ППГ и ПБГ) менее полярны,
но более селективны для разделения небольших молекул. На
ППГ-400 и ППГ-2000 основан газохроматографический анализ смеси
низших окисей олефинов — этилена и изомеров бутилена с альдеги-
дами и кетонами [41]. Установлено, что степень разделения смеси
окись этилена — ацетальдегид растет в ряду ПЭГ — ППГ —
ПБГ [42].
Для более крупных молекул — спиртов, диолов, их эфиров —
время удерживания на ППГ и ПБГ меньше, чем на ПЭГ, что по-
зволяет анализировать более высококипящие вещества. Например,
на колонке 200 X 0,4 см с поли-2,3-бутиленгликольсукцинатом при
150 °C проведен анализ многокомпонентной смеси дипервичных ди-
олов С4 — С1о с первичными одноатомными спиртами С9 — С13 [43].
Уменьшение полярности полигликолей этерификацией гидроксиль-
ных групп (чаще всего адипиновой и янтарной кислотами) широко
используется в практике. Большое значение в газохроматографи-
ческом анализе диолов имеют пространственные эффекты и водород-
ная связь.
Низкомолекулярные простые монометиловые эфиры пропилен-
гликолей (легкокипящие фракции промышленного флотореагента
ОПСМ) анализировали при 205—210 °C на колонке 100 X 0,4 см,
346
наполненной тефлоном с 5% эластомера Е-301, детектирование по
теплопроводности [44]. В этих условиях выходят пять пиков моно-
метиловых эфиров моно-, ди-, три-, тетра- и пентапропиленгликолей;
внутренний стандарт — дибутилфталат. Методика не дает возмож-
ности определять изомерный состав эфиров, примеси гликолей
п эфиров бутиленглико лей. На этой же колонке и режиме проана-
лизированы флотореагенты ОПСБ и Э-1 (соответственно пять пиков
монобутиловых эфиров моно-, ди-, три-, тетра-, пентапропилен-
гликолей и пять пиков монобутиловых эфиров моно-, ди-, три-,
тетра-, пентаэтиленгликолей [42, с. 5—6].
Монометпловые, этиловые, пропиловые и бутиловые простые
эфиры моно-, ди-, три- и тетраэтиленгликолей анализировали при
140 СС на колонке 240 X 0,3 см с изменением количества неподвиж-
ной фазы. Первая треть колонки содержала 2% фазы, вторая треть —
6% и последняя — 10%. В качестве неподвижной фазы применен
полинеопентилгликольсукцинат молекулярной массы 1740 на хромо-
сорбе W [45]. На примере почти 40 простых эфиров моно-, ди- и три-
этиленгликолей и их ацетатов показана возможность идентификации
этих соединений газохроматографическим методом при 150 °C на
колонках 360 X 0,6 см, заполненных 10% OV-1, 07-25 и 07-210
соответственно на силиконизированном целите-545; индексы удер-
живания исследованных веществ возрастают с увеличением поляр-
ности неподвижной фазы. Изучены также аналогичные производные
моно- и дипропиленгликолей [46].
Более высококипящие эфиры гликолей можно проанализировать
газохроматографическим методом лишь после превращения в более
легколетучие производные, чаще всего в ацетильные или триметил-
силильные. Именно в виде последних анализировались продукты
оксиэтилировапия додеканола-1 с молекулярной массой 300—500
при программировании температуры от 100 °C со скоростью 10 °C
в мин на колонке 60 X 0,6 см с хромосорбом и с 2% высоковакуум-
ной смазки Dow Corning [47]. На этой же колонке определялись
полиэтиленгликоли молекулярной массы 100—400 в виде их три-
метилсилильпых производных при программируемой температуре.
Температура испарителя — 430 °C, неподвижная фаза оставалась
стабильной при 350 °C.
Другие методы анализа гликолей
и их производных
Из оптических методов анализа рефрактометрия является, по-види-
мому, наиболее доступным и приемлемым в практике лабораторного
и цехового контроля спиртов и неионогенных ПАВ на основе а-оки-
сей. Метод, основанный на существовании корреляции между содер-
жанием гидроксильных групп и коэффициентом преломления, поз-
воляет определить окись этилена в оксиэтилировэнных продуктах
и может заменить известный метод анализа по точке помутнения
1%-ного раствора. Характер распределения аддуктов окиси этилена
на показания не влияет [48].
347
Коэффициент преломления для низших этилен- и пропилен-
гликолей находится в линейной зависимости от их молекулярной
массы [49, 50]. Состав бинарных смесей достаточно точно в ряде
случаев определяется по коэффициенту рефракции или по плотности.
Молекулярная масса гликолей определяется из ИК-спектров
на основании корреляции «отношение полос поглощения ОН/СН
колебаний — молекулярная масса» [50, 51]. С помощью ИКС опре-
деляли соотношение окись этилена : окись пропилена в сополимерах
типа Pluronic, причем использовались частоты колебаний метильных
и метиленовых групп при 2975 и 2870 см-1 соответственно, калиб-
ровка проведена по смесям ПЭГ и ППГ [52]. По ИК-спектру можно
определить ненасыщенные эфирные группы — аллиловые и цис-аро-
пениловые в полипропиленгликолях [53], соотношения первичных
п вторичных ОН-групп в дипропиленгликолях [54], присутствие
карбонильных и сложноэфирных соединений в низших глико-
лях [55].
Комбинирование ИКС-метода с газохроматографией позволяет
по составу конечных продуктов судить о механизме раскрытия
цикла а-окисей, в частности, окиси пропилена при получении ди-
пропиленгликолей или эфиров пропиленгликолей [55]. Весьма удоб-
ным и быстрым является метод ИКС для определения структуры
и строения ПАВ на основе а-окисей, их молекулярной массы, числа
оксиэтильных и оксипропильных групп [56].
Полярография применяется в исследовании гликолей в двух
аспектах — анализ полярографически активных компонентов и ис-
следование адсорбционных явлений. Формальдегид и ацетальдегид
как продукты окисления (или самоокисления) могут определяться
не только цветными реакциями, описанными в разделе химических
методов определения, но и классическим полярографическим мето-
дом, как это делалось в исследованиях по самоокислению диэтилен-
гликоля [57], анализе смеси этиленгликоль — пропиленгликоль
перйодатным методом и др. В полипропиленгликолях на полярографе
определяли перекиси и альдегиды [58].
Гликоли и их эфиры, адсорбируясь на ртутной капле, приводят
к уменьшению полярографических максимумов металлов и кисло-
рода. Это явление предложено использовать для установления
молекулярной массы полиэтиленгликолей и для определения их
концентрации в водных растворах [59]. Указанным методом исследо-
валась поверхностная активность этилен-, пропилен- и некоторых
бутиленгликолей, отдельных простых моноэфиров моно-, ди-, три-
и тетрагликолей и товарные флотореагенты ОПСМ, ОПСБ, Э-1.
Установлено, что низшие гликоли почти не влияют на величину
максимума. Активность эфиров в общем случае возрастает с увели-
чением молекулярной массы алкила в ряду метил — этил — про-
пил — бутил — алкил, с увеличением числа оксиалкиленовых фраг-
ментов от моно- до тетра- и увеличением их молекулярной массы
в ряду этилен--пропилен — бутиленгликолевые производные [60].
343
ЛИТЕРАТУРА
1. Legradi L., Microchim. Acta, 1970, v. 27, № 1, p. 33—38; РЖХим 1970
14Г205. ’
2. Коренман И. M. Методы определения органических соединений. М., «Хи-
мия», 1970. 343 с.
3. Gardner L., J. Inst. Petrol., 1969, v. 55, № 46, p. 418—421; РЖХим 1970
ЮП291.
4. Izguirdo A., Lacort G., Inform, quim. anal., 1972, v. 26, № 4, p 192—196'
РЖХим, 1973, ЗГ155.
5. Рубен-Вейль. Методы органической химии. Пер. с нем. Под ред. Е. Мюл-
лера. М., «Химия», 1967. 1032 с.
6. Budke С., Banerjee D. In: Encyclopedia of Industrial Chemical Analysis.
V. 13. New York, Interscience Publ., 1971, p. 533—607.
7. Критчфилд Ф. Анализ основных функциональных групп в органических
соединениях. Пер. с англ. Под ред. М. А. Володиной. М., «Мио», 1965.
208 с.
8. Elving Р., Warshovsky В., Anal. Chem., 1947, v. 19, р. 1006—1011.
9. Sezerat A., Ann. pharm. France, 1955, v. 13, p. 516—520.
10. Fijolka P., «Plaste und Kautschuk», 1971, Bd. 18, № 6, S. 341—345; Воло-
дина M. А., Каранти И. В., Вести. МГУ, 1971, т. 12, № 4, с. 504—505;
Климова В. А., Чан Мань Винь, Шерман Ф. Б., Изв. АН СССР. Сер. хим.,
1962, № 3,с. 588—590.
11. Hanna J., Siggta S., J. Polym. Sci., 1962, v. 56, № 164, p. 297—304.
12. Gnayck R., Fijolka P., «Plaste und Kautschuk», 1971, Bd. 18, № 7, S. 498—
500; РЖХим, 1972, 2Г211.
13. Афанасьев A. M. и др., ЖАХ, 1971, t. 26, № 1, c. 189 — 190.
14. Benson D., Fletcher N., «Taianta», 1966, v. 13, p. 1207—1209; РЖХим,
1967, 2Г197.
15. Plsko E., «Cemick6 Zvesti», 1958, v. 12, p. 312—314.
16. Levins R., Ikeda R., Anal. Chem., 1965, v. 37, p. 671—673; Maros L. e. a.,
Chem. Abstr., 1966, v. 65, p. 8008h.
17. Общий практикум по органической химии. Пер. с нем. Под ред. А. Н. Коста.
М., «Мир». 1965. 174 с.
18. Ring R. Gas Chromatography. New York, Academic Press, 1958. 148 p.
19. Dal Nogare S.. Safranowski L., Anal. Chem., 1958, v. 30, p. 894—897.
20. Ginsburg L., Ibid, 1959, v. 31, p. 1822—1824.
21. Nadey H., Oaks D., Ibid., 1960, v. 32, p. 1760—1763.
22. Esposito G., Swann M., Ibid., 1961, v. 33, p. 1854—1858; 1962, v. 34, p. 1173.
23. Mleinek O., Chem. prumysl, 1963, v. 13, № 2, p. 105—107.
24. Анжеле П. Г., В кн.: Молекулярная хроматография. М., «Наука», 1964,
с. 64—68.
25. Балахонцева В. Н., Полтинина Р. М., ЖАХ, 1964, т. 19, № 6, с 757—760;
1965, т. 20. № 6, с. 739—742.
26. Вавер В. А. и др., Изв. АН СССР. Сер. хим., 1967, № 6, с. 1187—1192;
1 1968, № 2, с. 400—402.
>'27. Palframan Y., Walker Е., «Analyst», 1967, v. 92, № 1097, р. 535—536.
28. Филатова Т.Н. и др., Изв. АН СССР. Сер. хим., 1969, № 11, с. 2583.
29. Уманская А. А. и др., ЖАХ, 1970, т. 25, № 6. с. 1211—1214.
30. Комиссаров П. Ф. и др., «Газовая хроматография», 1965, вып. 1, с. 99—107.
31. Переплетчикова Е. М.,Лазарис А. Л., ЖАХ, 1966, т. 21, № 10, с. 1280—
1281.
32. Singliar М.. Rendkowa I., Chem. Prumysl, 1971, v. 21, № 5, p. 229—232.
33. Phifer L., Plumer H., Anal. Chem., 1966, v. 38, p. 1652—1656.
34. Gross F., Assoc. Offic. Agr. Chem., 1965, v. 48, № 3, p. 647; Maniys G. e. a.,
J. Chromatogr. Sci., 1971, v. 9, № 6, p. 367—369.
35. Насибуллина P. К. и др., Изв. АН СССР. Сер хим., 1973, № 4, c. 800—802.
36. Camera E., Prarisani D., Anal. Chem., 1964, v. 36, p. 2108.
37. Косенко H. H., Галузо Л. В., Зав. иаб., 1965, т. 31, № 3, с. 285—287;
ЖАХ, 1968, т. 23, № 3, с. 432—435.
349
38. Мисак А. Е. и др., «Химия и технология топлив и масел», 1971, № 3, с. 49—
53.
39. Чудное А. Ф., Мирошников А. М. Техническая и экономическая информа-
ция. Сер. Методы анализа и контроля производства в химической промышлен-
ности. 1967. вып. 2, с. 15—16; вып. 5, с. 23—24; 1969, вып. 3, с. 3—5.
40. Чудное А. Ф., Мирошников А. М., То же, 1974, вып. 5, с. 5—8.
41. Чудное А. Ф. и др. В кн.: Физико-химические методы анализа. Кемеров-
ское книжное изд., 1970, с. 141—154.
42. Мирошников А. М. и др., В кн.: Физико-химические методы анализа. Те-
зисы научно-технической конференции, изд. Кемеровского ЦНТИ, 1970,
с. 10.
43. Прокопенко Н. А. и др., ЖАХ, 1968, т. 23, № 4, с. 642—644.
44. Косенко Н. Н., Баранова Г. Ф., Гурвич С. М., ЖПХ, 1968, т. 41, № 2,
с. 324—328.
45. Singliar М., Dykjj J., Coll. Czech. Chem. Commun., 1969, v. 34, № 3,
p. 767—775.
46. HakenJ., Khemangkorn V., J. Oil and Colour Chem. Assoc., 1971, v. 54, № 8,
p. 764—773; Haken J., Khemangkorn V., J. Chromatogr. Sci., 1972, v. 10,
№ 1, p. 41—47.
47. Tornquist J., Acta Chem. Scand., 1967, v. 21, № 8, p. 2095—2103; РЖХим,
1968, 11Г193; Tornquist J., Acta Chem. Scand., 1969, v. 23, № 6. p. 1935—
1942; РЖХим, 1970, 7Г245.
48. Weimer D., J. Am. Oil Chem. Soc., 1967, v. 44, № 3, p. 171—174.
49. Ingham I., Lawson D., J. Polym. Sci., 1965, A, v. 3, № 7, p. 2707—2710.
50. Чудное А. Ф-, Швец Д. П., Мирошников A. M. Техническая и экономи-
ческая информация. Сер. Методы анализа и контроля в химической промыш-
ленности, 1967, вып. 2, с. 23—26.
51. Magnasco V., Rossi М., J. Polym. Sci., 1962, v. 62, № 174, p. 173—175.
52. Uno T., Chem. and Pharm. Bull. (Japan), 1967, v. 15, № 1, p. 77—79.
53. Dege L., J. Am. Chem. Soc., 1959, v. 81, p. 3374—3378; Simons D., Ver-
banc J., J. Polym. Sci., 1960, v. 44, p. 303—308.
54. Sexton A., Britton E., J. Am. Chem. Soc., 1953, v. 75, № 17, p. 4357—4358.
55. Чудное А. Ф-, Мирошников A. M., Швец Д. П. Техническая и экономиче-
ская информация. Сер. Методы анализа и контроля в химической промыш-
ленности, 1967, вып. 1, с. 37—43.
56. Селицкая Н.Д., Корецкий А. Ф., Изв. СО АН СССР. Сер. хим., 1967,
№ 6. с. 48—54.
57. Lloyd W., J. Am. Chem. Soc., 1956, v. 78, p. 72—80.
58. Булыгин Б. M., ЖАХ, 1968, t. 23, № 3, c. 425—431.
59. Badinand A., Bouchere A., Bull. Soc. chim. France, 1967, № 8/9, p. 1021 —
1024.
60. Князькова T. Ф. и др., Техническая и экономическая информация. Сер.
методы анализа и контроля производства в химической промышленности,
1967, вып. 4, с. 8—11s
ПРИЛОЖЕНИЯ
Таблица 1. Плотность этиленгликоля и его водных растворов (в г/см8)
при различных температурах
Темпера- тура, °C Концентрация этиленгликоля, %
10 20 30 40 50 ^0 70 80 9 0 100
—45 __ . . 1,110 1,125 1,137
—40 —. — —- — 1,108 1,122 1,134 — —.
—35 —_ — •— 1,087 •1,105 1,120 1,131 — —
—30 —. — 1,086 1,103 1,118 1,129 — —.
—25 — — — 1,085 1,101 1,115 1,126 1,136 —.
—20 —_ —. 1,067 1,083 1,098 1,112 1,123 1,133 —.
—15 —. — 1,066 1,081 1,096 1,109 1,120 1,130 - -
—10 —_ 1,048 1,064 1,079 1,094 1,107 1,117 1,127 1,134
—5 —. 1,047 1,062 1,077 1,091 1,104 1,114 1,123 1,131
0 1,015 1,030 1,046 1,061 1,075 1,088 1,101 1,111 1,120 1,127
5 1,014 1,029 1,044 1,059 1,073 1,085 1,097 1,108 1,116 1,124
10 1,013 1,027 1,042 1,056 1,070 1,083 1,094 1,105 1,113 1,120
15 1,012 1,026 1,041 1,054 1,067 1,080 1,091 1,102 1,110 1,117
20 1,011 1,024 1,038 1,052 1,064 1,077 1,088 1,098 1,106 1,113
25 1,009 1,022 1,036 1,050 1,062 1,074 1,084 1,094 1,102 1,110
30 1,007 1,021 1,034 1,047 1,059 1,071 1,081 1,091 1,099 1,106
35 1,006 1,019 1,032 1,045 1,056 1,067 1,078 1,087 1,092 1,103
40 1,004 1,017 1,029 1,041 1,053 1,064 1,074 1,084 1,022 1,099
45 1,002 1,014 1,026 1,038 1,049 1,060 1,071 1,081 1,089 1,096
50 0,999 1,012 1,023 1,035 1,046 1,057 1,067 1,077 1,085 1,093
55 0,996 1,009 1,021 1,032 1,043 1,054 1,064 1,073 1,082 1,089
60 0,994 1,006 1,018 1,029 1,040 1,051 1,060 1,069 1,078 1,085
65 0,991 1,003 1,014 1,026 1,037 1,047 1,057 1,065 1,074 1,081
70 0,988 1,000 1,011 1,023 1,033 1,044 1,053 1,062 1,<|70 1,078
75 0,986 0,997 1,008 1,019 1,029 1,040 1,050 1,058 1,066 1,074
80 0,983 0,994 1,005 1,016 1,026 1,036 1,046 1,054 1,063 1,070
85 0,979 0,990 1,001 1,012 1,022 1,032 1,042 1,050 1,059 1,066
90 0,976 0,987 0,997 1,008 1,018 1,028 1,038 1,046 1,055 1,063
95 0,973 0,983 0,993 1,004 1,014 1,024 1,034 1,043 1,051 1,059
100 0,969 0,980 0,990 1,000 1,010 1,020 1,030 1,039 1,047 1,055
ПО 0,961 0,972 0,982 0,992 1,003 1,012 1,022 1,030 1,039 1,047
120 0,953 0,964 0,974 0,984 0,994 1,004 1,013 1,022 1,031 1,039-
130 0,945 0,955 0,965 0,975 0,985 0,995 1,005 1,014 1,024 1,031
140 0,937 0,947 0,956 0,966 0,976 0,986 0,996 1,005 1,014 1,022
150 0,927 0,937 0,947 0,957 0,967 0,977 0,987 0,996 1,005 1.014
160 0,917 0,928 0,938 0,948 0,958 0,968 0,977 0,987 0,996 1,005
170 0,907 0,918 0,928 0,938 0,948 0,957 0,968 0,978 0,987 0,995
Примечание. Плотность этиленгликоля при 180, 185, 190, 195 и 290 °C Обставляет
соответственно 0,982ъ 0,977, 0,972 и 0,907 г/см3.
351
Таблица 2. Относительная плотность водных растворов этиленгликоля
d™ 20 Концент- рация гликоля, % d2° 20 Концент- рация гликоля, % d2° 20 Концент- рация гликоля, %
1,001 0,80. 1,024 18,26 1,047 35,01
1,002 1,59 1,025 18,99 1,048 35,75
1,003 2,38 1,026 19,72 1,049 36,49
1,004 3,17 1,027 20,44 1,050 37,24
1,005 3,95 1,028 21,18 1,051 37,98
1,006 4,73 ’ 1,029 21,92 1,052 38,73
1,007 5,51 1,030 22,65 1,053 39,47
1,008 6,29 1,031 23,39 1,054 40,23
1,009 7,07 1,032 24,12 1,055 40,98
1,010 7,85 1,033 24,85 1,056 -41,73
1,011 8,62 1,034 25,58 1,057 42,48
1,012 9,39 1,035 26,30 1,058 43,23
1,013 10,15 1,036 27,02 1,059 43,98
1,014 10,91 1,037 27,74 28,46 1,060 44,74
1,015 11,65 1,038 1,061 45,52
. 1,016 12,40 1,039 29,17 1,062 46,28
1,017 13,15 1,040 29,88 1,063 47,05
1,018 13,90 1,041 30,60 1,064 47,82
1,019 14,63 1,042 31,33 1,065 48,59
1,020 15,36 1,043 32,07 1,066 49,38
1,021 16,09 1,044 32,80 1,067 50,18
1,022 16,81 1,045 33,53 1,068 50,98
1,023 17,54 1,046 34,27 1,069 51,78
1,070 52,60 1,086 66,26 1,102 82,38
1,071 53,43 1,087 ‘ 67,19 1,103 83,55
1,072 54,25 1,088 68,13 1,104 84,73
1,073 55,08 1,089 69,08 1,105 85,90
1,074 55,90 1,090 70,03 1,106 87,10
1,075 56,72 1,091 70,98 1,107 88,33
1,076 57,54 1,092 71,95 1,108 89,59
1,077 58,37 1,093 72,93 1,109 90,90
1,078 59,21 1,094 73,92 1,110 92,24
1,079 60,07 1,095 74,92 1,111 93,62 '
1,080 60,93 1,096 75,93 1,112 95,01
1,081 61,80 1,097 76,95 1,113 96,43
1,082 62,27 1,098 77,99 1,114 97,88
1,083 1,084 1,085 63,56 64,45 65,35 1,099 1,100 1,101 79,05 80,12 81,23 1,115 99,34
Таблица 3. Давление насыщенных паров водных растворов
этиленгликоля (в мм рт. ст. *)
Темпера- тура, °C Концентрация этиленгликоля, %
2 0 40 60 70 8 0 90 95
60 71 82 139 229 363 125 206 326 105 172 272 147 234 115 183 70 112 40 65
352
Продолжение табл. 3
Темпера- тура °C Концентрация этиленгликоля, %
20 40 60 70 80 90 95
88 451 406 339 291 228 140 81
99 682 614 513 442 346 213 125
110 1004 905 757 652 512 317 188
121 1441 1300 1088 938 738 460 . 275
132 2023 1826 1531 1321 1042 653 395
138 2378 2147 1801 1555 1228 772 470
149 3240 2927 2458 2125 1682 1065 656
160 4337 3920 3297 2853 2264 1443 899
166 4986 4508 3794 3286 2611 1670 1046
171 5711 5165 4350 3770 2999 1925 1213
177 6517 5896 4969 4309 3432 2211 1401
1 мм рт. ст. = 133,32 Па.
Таблица 4. Растворимость органических и неорганических соединений
в этиленгликоле при 259С
Соединение Раствори- • мость в эти- ленгликоле, г/100 г Соединение Раствори- мость в эти- ленгликоле, г/100 г
Бензол втор-Бутиламин .... Гексиловый эфир . . . Дибутилфталат о-Дихлорбензол .... Дихлордиэтиловый эфир Диэтил амин Изобутиламин Камфора Карбамид . Метилизобутилкетон . . Метилоранж Стирол Тетрахлорэтилен . . . Толуол Фениловый эфир ". . . . Хлорбензол Четыреххлористый угле- род Этиловый эфир .... Натрий ацетат 6,0 39,7 * 0,09 0,5 4,7 11,8 37,1 * 38,1 * 4 ** 44,0 0,7 1,8 3 4** 0J ** 3,1 1,7 6,0 6,6 8.5 22,78 Барий хлористый . . . Калий бромистый . . . Калий иодистый .... Калий перхлорат . . . Калий сернокислый . . Калий углекислый . . . Калий хлористый . . . Калий хлорат Кальций хлористый . . Литий бромистый . . . Литий хлористый . . . Медь сернокислая (CuSO4-5H2O) .... Натрий бромистый . . . Натрий иодистый .... Натрий перхлорат . . . Натрий сернокислый . . Натрий хлорат .... Натрий хлористый . . . Ртуть уксуснокислая [Hg(C2H3O2)2] . . . Свинец хлористый . . . 3,6 15,5 49,9 1,03 0,0 34,4 5,18 1,21 20,5 39,4 14,3 46,9 35,4 107,4 75,5 0,54 *** 16,0 7,15 17,8 0,85 (гл. II, с. 36)
Натрий бензоат .... Натрий лаурат .... 19,92 ' 10,65 Серебро азотнокислое 93,4
23 Заказ 972
353
Продолжение табл. 4.
Соединение Раствори- мость в эти- ленгликоле, г/100 г Соединение Раствори- мость в эти- ленгликоле, г/100 г
Натрий миристат . . . 3,84 Натрий стеарат .... 0,48
Натрий олеат 14,11 Стронций хлористый . . (гл. II, сс. 37)
Натрий пальмитат . . , 1,18 Таллий хлористый . . . 36,4 (гл. II, сс. 36) 0,13
* При 4,5 °C в % (масс.)
** В % (мол.).
*** Этиленгликоль содержит 0,8% воды.
Таблица 5. Критическая температура растворения (К. Т. Р.) этиленгликоля
для различных соединений
Компонент К. Т. Р., °C Компонент К. Т. Р., °C
Адиподинитрил .... 27 Метилацетат 26,8
Анизол 134,5 Метилбензоат 107,5
Ацетонитрил —13,5 Метилгексилкетон . . . 66
Ацетонилацетон .... >180,5 2-Метилгептанол .... <0
Ацетофенон 114,5 Метилгептенон .... 65
Бензил ацетат 100 Метилмалеат 47,8
Бензол 180 Метилмеркаптан .... >13,5
Борнеол <99 Метилсалицилат .... 143
Бромбензол >150,2 Нафталин 195
Бромистый ВИПИЛ . . . 15,7 Нитробензол 120,2
Бромистый водород . . <45 Нитрометан 39,7
Бромоформ 142 о-Нитротолуол .... 142
Бромтолуол ...... >168 о-Нитрофенол >189,3
и-Бутилацетат 178 га-Нитрохлорбензол . . 136,5
и-Гептан 80 Нитроэтан ...... 68
2-Гептанон >25 н-Октиловый спирт . . <0
«-Гептиловый спирт . . <о Пентахлорэтан .... >154,5
«-Дециловый спирт . . 60 Пропилбензоат .... 164
НД-Дпметиланилин . . 171,4 Тетралин 213
Дифенил 217 1,1,2,2-Тетрахлорэтан 88,5
о-Длхлорбензол .... '-Дихлордиэтиловый >165,8 о-Толуидин -55
115 Толуол 210
эфир 1,3,5-Трихлорбензол . . >181
ХД-Дпэтиланилин . . «-Додециловый спирт >183,4 135 Фенантрен 1 225
Изоамилацетат .... 26 Хлорбензол >130,8
Изоамилкарбонат . . . >188,5 1-Хлорнафталин .... >193,1
Камфен >152,5 о-Хлортолуол >152,5
N-Метил анилин .... 70 N-Этиланилин 126,5
354
Продолжение табл. 5
i * Компонент К. Т. Р., °C Компонент К. Т. Р., °C
: ; Камфора L о-Крезолметиловый эфир || Ментол >117 156 >17,6 Этилацетат Этилбензоат 56,5 136
И Таблица 6. Температуры кипения азеотропных смесей ! при атмосферном давлении и содержание в них этиленгликоля
* Компонент Т. кип. смеси, ®С Содержание этилен- гликоля, %
Il Амилацетат II Анизол || Анилин If Ацетофенон Г. Бензальдегид 1 Бензиловый спирт Бензилхлорид Борнеол :' Бромоформ । Бутилбутират Гептан 1-Гептанол Декан . 1-Деканол Диацетат этиленгликоля ' 2-Диизопропиламиноэтанол N Д-Диметиланилин Дифенил Дифениловый эфир Дифенилметан р,р '-Дихлорэтиловый эфир N .N-Диэтиланилин Диэтиловый эфир диэтпленгликоля . . . Диэтилмалеат Изоамилацетат Изопропилбензол Камфен ; Камфора N-Метиланилин Метиловый эфир диэтиленгликоля . . . Метиловый эфир трипропиленглпколя . . 1-Метилнафталин Моноацетат этиленгликоля Монобутиловый эфир диэтиленгликоля 1 о-Крезол , ; о-Ксилол Нафталин Нитробензол о-Нитротолуол 1 t о-Нитрофенол : f о-Нитрохлорбензол 147,6 150,4 180,5 185,6 <173,5 193,1 — 167,0 189,2 145,45 146,75 160,6 98,3 174,1 161,0 191,8-193,0 <179,5 74,0 (10) * 178,55 192,0—192,25 192,3 193,3 171,05 183,4 178,0 ' 193,1 141,95 147,0 152,5 186,1 181,6 192,0 192,0 190,25 184,75 196,2 189,5 135,7 183,6-183,9 185,9 188,5 189,5 193,5 6,0 10,5 24,0 52,0 >15,0 56,0 — 30,0 54,2 6,5 . 16,0 —3,0 17,0—20,0 ' 23,0 ' 67,0 <24,0 10,0 33,5 64,0—66,5 64,5 68,5 21,0 33,0 26,1 55,0 — 3,0 18,0 20,0 40,0 40,2 30,0 82,0 60,0 25,0 72,5 26,0 6,9 46,0—51,0 59,0 48,5 49,0 68,0
; г | 2.3* 355 i к
Продолжение табл. 6
Компонент Т. кип. смеси, °C Содержание этилен ГЛИКОЛЯ, %
Октан 123,5 11,5
1-Октанол 184,3 36,5
2-Октанол 175,0 =«12,0
2-Октанон 168,0 =«79,0
Стирол 139,5 16,5
Тетрахлорэтилен 119,1 6,0
Толуол 110,1—110,2 2,3-6,5
1 ,2,3-Трихлорпропап 160,8 ==«13,0
2-Фенилэтанол 194,4 69,0
фенол 199,0 78,0
Хлорбензол 130,0 5,6
o-Хлортолуол 152,5 13,0-
N-Этилапилин 183,7 43,0
Этилбензол 133,0 13,5
Этиленкарбонат 88,0 (10) * 13,9
Этиловый эфир диэтилснглпколя .... 192,0—194,0 . 45,5-46,0
* При 10 мм рт. ст. Данные для других давлений приведены в работе [5], см. гл. I.
Таблица 7. Температуры кипения азеотропных смееей
при атмосферном давлении и содержание в нпх диэтиленгликоля (ДЭГ)
Компонент Т. кип., °C Содержание ДЭГ, %
Аценафтен 239,6 62,0
Бутилбензоат 232,2 43,0
Дигексиловый эфир 129,9 (50) * 15,5
Дифенил 232,65 48,0
Дифениловый эфир 234,4 49,5
Дпфенилметан 236,0 52,0
1,2-Дифенилэтан 241,0 66,0
'-Дихлорэтиловый эфир 174,6 8,0
Изоамилбензоат 236,55 52,5
1-Метилнафталин 227,0 45,0
Метиловый эфир а -терпинеола 210,5 20,0
Метилсалицилат 220,55 16,0
Монометиловып эфир триэтиленгликоля 245,0 22,0
Нафталин 212,6 22,0
Нитробензол 210,0 10,0
o-Нитротолуол 218,2 17,5
о-Нитрофенол 216,0 10,5
о-Нитрохлорбензол 233,3—233,5 41,0
Фенантрен 146,0 (20) 95,7
1-Хлорнафталин 234,1 47,0
Этиловый эфир триэтпленгликоля 87,0 (2) 43,0
Этилсалицилат 225,15 30,0
* Цифры в скобках обозначают давление в мм рт. ст.
356
Таблица 8. Растворимость веществ в диэтиленгликоле при 25 °C
Соединение Растворимость в ДЭГ, г/100 г
по данным [15, р. 9] । по данным [1, р. 161]
Бензол 45,5 45,5
Декстрин < 1 Почти нерастворим
Декстрин (10%-нып раствор
в воде) — Растворим
Дибутилфталат 11,9 11,8
о-Дихлорбензол 93,8 93,6
Канифоль — <2,0
Карбамид 42,8 24,0
Масла
ализариновое <1 6,3
касторовое <0,5 0,1
ланолиновое <0,5 Почти нерастворимо
ойтиковое <1 —
сосновое Смешивается
талловое <1 3,1
Метилоранж 4,2
Нитроцеллюлоза >25 Растворима
Смолы -
дамарова <0,5 Почти нерастворима
каурп <0,5 »
шеллак <0,5 »
Стирол 36 —
Тетрахлорметан 35,5 35,5
Тетрахлорэтилен 12,0 12,0
Толуол 20,7 20,7
Этиленгликольстеарат . . — Почти нерастворим
Этиловый эфир 19,5 19,5
Таблица 9. Растворимость веществ в три- и тетраэтиленгликоле
Соединение Растворимость в триэтилен- гликоле Раствори- мость в тет- раэтилене, % [4]
% [4] г/ 100 мл [24]
Ацетат целлюлозы — Растворим .—
Гептан — Очень мало рас- творим —
Декстрин <1 Нерастворим <1
Декстрин (10%-ный раствор в воде) — Растворим —
Дибутилфталат 16,5 19,8 оо
Диэтиловый эфир 16,9 20,4 20
Канифоль — 5.0 —
Карбамид 37,0 31,0 28
Нитроцеллюлоза — Растворима —
Тетрахлорметан 33,6 40,6 62
Тетрахлорэтилен 15 17,7 19
Толуол 24,8 33,0 89
Этиленгликольстеарат Масло — Очень малорас- творим ' —
ализариновое 1 12,3 1
талловое <1 10,1 <1
Смола каури <0,5 Малорастворима .<16
357
Таблица 10. Температуры кипения азеотропных смесей
при атмосферном давлении и содержание в них
триэтиленгликоля
Компонент Т. кип., °C Содержание триэтиленгли- коля, %
Аценафтен 271,5 35
1-Бромнафталин ' 273,4 33
Глицерин 285,1 63
Дибензиловый эфир — 28*
Диметилфталат 277,0 33
Дифенил 255,3 10
Дифенилметан 263,0 20
1,2-Дифенилэтан 275,5 42
Дифениловый эфир 258,7 3
Диэтилфталат <285,5 >58
Изоамилбензоат - 261,4 14
Изоамилсалицилат 269,0 30
Стильбен 284,5 60
Фенилбензоат 286,0 80
Фенилбензпловый эфир 280,0 40
1-Хлорнафталин . . . 261,5 33
* При остаточном давлении 6,6 кПа (5 мм рт. ст.).
Таблица 11. Плотность пропиленгликоля и его водных растворов (в г/см3)
при различных температурах
Температура, °C Концентрация пропиленгликоля, %
10 20 30 40 50 во 70 80 90 1 о 0
25 1.0037 1.0115 1,0195 1,0271 1,0322 1,0375 1,0397 1,0395 1,0373 1,0326
20 1,0058 1,0146 1 0234 1,0319 1,0382 1,0423 1.0439 10435 1,0410 1,0363
15 1,0078 1,0165 1,0255 1,0344 1,0416 1.0464 1,0485 1,0481 1,0453 1,0399
10 1,0082 1,0178 1 0282 1,0376 1.0445 1,0490 .— — — 1,0435
0 1,0092 1,0196 1,0327 1,0442 1,0520 1,0559 .— — — 1,0508
—5 — 1,0203 1,0354 1,0469 1.0547 1,0593 — — — 1,0544
—10 1,0216 1,0374 1,0498 1,0582 1,0624 — — — 1 0589
-15 — — — 1.0517 1,0606 1,0650 — — — 1,0616
Плотность пропиленгликоля при —20, —25 и —30 °C равна 1,0652, 1,0688 и 1,0727
г/см3 соответственно.
358
L Таблица 12. Относительная плотность водных растворов пропиленгликоля
20 d 20 Концентрация гликоля, % 20 d 20 Концентрация 1 гликоля, % 20 Й20 Концентрация гликоля, % 20 d 20 Концентрация гликоля, %
1,001 1,42 1,015 18,55 1,029 34,38 1,043 56,9
1,002 2,83 1,016 19,68 1,030 35,58 1,044 59,7
1,003 4,17 1,017 20,80 1,031 36,78 1,045 63,5
1,004 5,48 1,018 21,92 1,032 38,04 1,045 66,3
1,005 6,78 1,019 23,04 1,033 39,32 1,0459 72,95
1,006 8,05 1,020 24,16 1,034 40,65 1,045 79,9
1,007 9,28 1,021 25,28 1,035 42,01 1,045 82,8
1,008 10,48 1,022 26,40 1,036 43,43 1,044 86,6
1,009 11,65 1,023 27,52 1,037 44,95 1,043 89,5
1,010 12,83 1,024 28,63 1,038 46,58 1,042 92,0
1,011 14,00 1,025 29,74 1,039 48,37 1,041 94,3
1,012 15,14 1,026 30,90 1,040 50,3 1,040 96,4
1,013 16,28 1,027 32,05 1,041 52,3 1,039 98,4
1,014 17,41 1,028 33,21 1,042 54,4 1,038 100,0
Таблица 13. Относительная плотность смесей пропиленгликоля
и дипропиленгликоля
20 d 2 0 Концентра- ция дипропи- ленглико- ЛЯ, % 20 Концентра- ция дипро- пиленгли- коля, % 2 0 d 20 Концентра- ция дипропи- ленглико- ля, %
1,03805 0,31 1,03760 3,80 1,03715 7,29
1’03800 0,70 1,03755 4,19 1,03710 7,67
1,03795 1,09 1,03750 4,57 1,03705 8,06
1,03790 1,47 1,03745 4,96 1,03700 8,45
1,03785 1,86 1,03740 5,35 1,03695 8,84
1,03780 2,25 1,03735 5,74 1,03690 9,22
1.03775 2,64 1,03730 6,12 1,03685 9,61
1,03770 3,02 1,03725 6,51 1,03680 10,0
1,03765 3,41 1,03720 6,90 . .
Таблица 14. Давление насыщенных паров водных растворов
пропиленгликоля (в мм рт. ст.*)
Концентрация пропиленгликоля, % (об.)
Температура, °C 30 50 70 80 9 0 9 7
20 17 13,6 11,5 10,2 5,6 —
30 30,3 24,8 21 18,2 10,2 —
40 51,5 42,7 36,3 31,3 18,0 5,4
50 85 72 61 51 30,1 9,4
359
Продолжение табл. 14
Температура, °C Концентрация цропиленгликоля, % (об.)
30 50 70 80 90 9 7
60 137 117 98 84 49 16,0
70 212 185 155 131 76 26,2
80 322 285 236 202 118 42,2
90 ( 475 425 353 298 177 63,0
100 690 615 520 433 257 95,0
110 980 890 740 615 373 143
120 — — — 855 525 207
130 — — — — 720 295
140 —- — — — 985 408
150 — — — — — 560
160 — — — — — 755
* 1 мм рт. ст. = 133,32 Па
Таблица 15. Коэффициенты преломления водных растворов
пропиленгликоля при 25° С
26 HD Концент- рация гликоля, % 26 nD Концент- рация гликоля, % 28 nD Концент- рация гликоля, % 2 5 nD Концент- рация гликоля, %
1,333 0,4 1,358 23,1 1,383 45,7 1,408 70,6
1,334 1,3 1,359 24,0 1,384 46,6 1,409 71,7
1,335 2,3 1,360 24,9 1,385 47,5 1,410 72,8
1,336 3,2 1,361 25,8 1,386 48,5 1,411 74,0
1,337 4,1 1,362 26,7 1,387 49,4 1,412 75,2
1,338 5,0 1,363 27,6 1,388 50,3 1,413 76,3
1,339 5,9 1,364 28,6 1,389 51,3 1,414 77,5
1,340 6,8 1,365 29,5 1,390 52,3 1,415 78,7
1,341 7,7 1,366 30,4 1,391 53,3 1,416 79,9
1,342 8,6 1,367 31,3 1,392 54,3 1,417 81,1
1,343 9,5 1,368 32,2 1,393 55,2 1,418 82,3
1,344 10,4 1,369 33,1 1,394 56,2 1,419 83,6 .
1,345 11,3 1,370 34,0 1,395 57,2 1,420 84,9
1,346 12,2 1,371 34,9 1,396 58,2 1,421 86,2
1,347 13,1 1,372 35,8 1,397 59,2 1,422 87,5
1,348 14,0 1,373 36,7 1,398 60,2 1,423 88,8
1,349 14,9 1,374 37,6 1,399 61,2 1,424 90,1
1,350 15,8 1,375 38,5 1,400 62,2 1,425 91,4 '
1,351 16,7 1,376 39,4 1,401 63,2 1,426 92,7
1,352 17,6 1,377 40,3 1,402 64,2 1,427 94,0
1,353 18,5 1,378 41,2 1,403 65,2 1,428 95,4
1,354 19,4 1,379 42,1 1,404 66,3 1,429 96,8
1,355 20,3 1,380 43,0 1,405 67,4 1,430 98,3
1,356 21,2 1,381 43,9 1,406 68,5 1,431 99,7
1,357 22,2 1,382 44,8 1,407 69,5 1,432 100,0
360
и
I Таблица 16. Растворимость различных соединений
в пропиленгликоле при 259С
Соединение
Растворимость, г/100 г
Растворимость чистых соединений [5, р. 224]
Антипирин ........................................
Аравийская камедь ................................
Бензиловый спирт .................................
Висмут тиогликолят ...............................
Гексилрезорцин ...................................
Гидроокись висмута ...............................
Гликольстеарат ...................................
Глицерофосфат кальция.............................
Дезоксикортикостеронацетат .......................
Дибутилфталат.....................................
р,р '-Дихлордиэтиловый эфир.......................
Дубильная кислота ................................
Йодвисмутит натрия ...............................
Лимоннокислый натрий .............................
Метилсалицилат....................................
Пектин ...........................................
Перхлорэтилен ....................................
Сульфапиридин ....................................
Тимол ............................................
Толуол ...........................................
Фенотиазин .......................................
Хлорбензол........................................
Цетиловый спирт...................................
Четыреххлористый углерод..........................
25
10,4
Растворим
Растворима
Слаборастворим
<0,07
Растворим
8,8
144
>45,20
6,4
0,23
24,7 ’
Нерастворим
11,7
3,1
Растворим
14.0
<1,15
29,0
0,23
30,5
Растворимость материалов технической квалифи-
кации [5, р. 236; 20, р. 5, 26, р. 9]
Ацетат целлюлозы..................................
Бензол ...........................................
Гликольдистеарат .................................
Декстрин .........................................
Декстрин (10% -ный раствор в воде) ...............
о-Дихлорбензол . . ...............................
Канифоль..........................................
Карбамид..........................................
Каучук ...........................................
Клей животный ....................................
Ланолин водный..............'.....................
Масла
ализариновое .................................
касторовое ...................................
льняное ......................................
талло ое......................•...............
кокосовое, оливковое, парафиновое, соевое, спермаце-
товое, тунговое, хлопковое ...................
Метилоранж .......................................
Нитроцеллюлоза ...................................
Олеомаргарин......................................
Нерастворим
23,8
Очень слаборас-
творим
Слаборастворим
Полностью растворим
24,1
Слаборастворима
26,0
Нерастворим
Очень слаборас-
творим
Слаборастворим
3,7
0,8
Слаборастворимо
9,9
Нерастворимы
0,6
Нерастворима
Нерастворим
361
Продолжение табл. 16
Соединение Растворимость, г/100г
Смолы бакелитовая, винилитовая, каури . . . даммаровая Стирол Судан Ш . . . Шеллак Нерастворимы Слаборастворима 17,6 Слаборастворим Очень слаборас- творим
Таблица 17. Растворимость веществ, применяемых в пищевой
промышленности, в пропиленгликоле
и его водных растворах при 25 °C
Растворимость при различной концентрации про-
пиленгликоля , %______________________________
Соединение 100% (об.) 80% (об.) 60% (об.) 40% (об.) 20% (об.)
Амилацетат СО СО ОС 1,48 1,34
Бензальдегид со 18,97 4,62 1,80 0,82
Винилип 20,20 20,10 12 60 5,85 2,09
Изоамилформпат ОС 5,22 4,51 1,68 1,53
Кумарин > . . . . 7,70 5,05 2,30 0,50 0,32
Масла
апельсиновое 0,26 0,13 0,08 0,06 0,03
гвоздичное СО 1,19 0,26 0,24 0,12
кассиевое со 3,13 0,85 0,69 0,21
лимонное 0,81 0,52 0,32 0,13 0,03
мускатное 1,53 0,34 0,17 0,14 0,11
сассафрасовое 2,02 1,21 0,20 0,12 0,08
Метплантранилат СО СО ОО — —
Фенилэтиловый спирт .... со со со 18,95 3,11
Цитраль со 0,35 0,17 0,10 0,04
Эвкалиптол 19,90 4,75 1,73 0,35 0,25
Этилацетат СО СО ОО 11,65 8,09
Этилванилин 14,20 10,80 5,20 1,84 0,79
Этилформиат СО оо ОО — 17,45
Таблица 18. Растворимость фармацевтических
препаратов в пропиленгликоле
Соединение Раствори- мость, % Соединение Раствори- мость, %
Алоин Антипирин Антифебрин (ацетанилид) Ацетарсон (осарсол) , . Гексаметилентетрамин 4,37 >55,00 2,09 0,52 11,22 Гексилрезорцин .... Камфора Ментол Мертиолат Метафен >80,00 9,80 >50,00 29,00 <0,27
362
Продолжение табл. 18
Соединение Раствори- мость, % Соединение Раствори- мость, %
Глицин Карбамид Кофеин Люминал (фенобарбитал) Оксибензиловый спирт Паральдегид Пепсин Резорцин Сульфадиазин Сульфаниламид .... Сульфатиазол Терпингидрат Фенацетин Хлоралгидрат Этилкарбамат Анестезин (бензокаин) Бензиловый спирт . . . <0,45 22,20 0,77 >49,00 44,10 оо <0,08 55,70 0,3 7,25 1,71 18,20 2,10 >89,00 >57,00 12,20 оо Диотан Салициловый спирт . . . Салол Трихлор-ирет-бутанол Хлортимол Цинковая соль фенолсуль- фокислоты Аскорбиновая кислота Витамин А (12% в масле) Витамин А Витамин В, (тиампнгид- рохлорид) Витамин В2 (рибофлавин) Витамин Д Никотиновая кислота Пиридоксингидрохлорид 4 10,50 >60,00 70,00 >39,00 8,16 Нераство- рим Растворим [5] 5,14 <0,006 Растворим 0,86 2,73
Таблица 19. Температуры кипения азеотропных смесей при атмосферном
давлении и содержание в них пропиленгликоля
Компонент Т кип. смеси, °C Содержан ие пропилен- гликоля, %
Анилин 179,5 43
Ацетофенон <183,5 —
Бис (2-бромэтиловый) эфир 176,0-180,0 —
Бис(2-этилгексиловый) эфир 84,0 —
Дибутиловый эфир 136,0 —
Н,И-Диметиланилин 177,0 45,0
Н,И-Диметил-о-толуидин <174,0 37,0
Н.И-Диметил-п-толуидин 178,0 60,0
Додекан 175,0 67,0
Камфора <185,0
о-Ксилол 135,8 10,0
Ментон <185,0 <85,0
N-Метиланилин <181,0 >46,0
п-Метоксифенол 242,2 ж 7,0
Монобутиловый эфир дипропиленгликоля .... 83,1 (10) * 90,44 **
Монометиловый эфир дипропиленгликоля .... 183,8 40,38 **
о-Нитрофенол <186 >62
2-октанон <169,5 —
Тетрадекан 179,0 76,0
Толуол 110,5 1,5
* Цифры в скобках показывают давление в мм рт. ст. (1 мм рт. ст. = 133,3 Па).
** В мольных процентах.
363
S Габм1<а 20 Физико-химические свойства простых олигоэфиров на основе окиси пропилена [см сс. 172, гл. VI].
п.льность я га в число, не более IO, е более : (пэ); ИС К га № ф S О'"- Температура, °C Ь В -ВО- %
Наименование Функцией: Молекуляр масса Содержание ОН-групп, % Кислотное 1 мг КОН г, Подное 4i!c; г12/100 г, н I ВЯЗКОСТЬ П] 2 5 °C, мПа - с Плотность п 20 °C, г 'см3 Коэффициент преломления дЩ □ я о . Щ ф-2 И Я S о Ф 2г самовоспла- менения J вспышки Растворимост де при 20 °C,
Лапрол 2G3 Лапрамол 294 ... . Лапрол 3 73 Лапрол 502 . . . ’ ’ Лапрол 503 . , . ’ Лапрол 805 . . . . Лапрол 1002 .... Лапрол 1003С .... Лапрол 2002 .... Лапрол 3002 .... Лапрол 3003 .... Стат Лапрамол 375-2-20 . Лапрол 1601-2-50 А Лапрол 3 503-2Б-6 . Лапрол-5003-2Б-10 . 3 4 3 2 3 5 2 3 2 2 3 иста 5 1 2 3 266 292 370 500 500 800 1000 100 0 2000 3000 3000 чески 370 1600 3500 5000 П 18,2—20,4 21-24 13,1-14,2 6,0 —7,0 9,6 — 11,0 10,0-11,5 3,05-3,50 4,85-5,55 1,5-1,72 1,0—1,30 1,5-1,7 е и блок- 21—24 1,00—1,20 1,4—1,6 0,95—1,1 о л пощ 0,3 0,3 0,1 0,5 0,5 0,1 0,20 0,05 0,2 0,5 С О ПОЛ I 0,1 0,1 0,1 и про 1,0 1,0. 1,0 1,0 1,0 2,0 2,0 меры ОД 1,8 2,0 11 и лс н п 800—900 40 000— 50 000 400-550 60—90* 250—350 2500— 4000 120—200 600-750 300-400 400—600* 450-600 окиси 45 осо- бо Ооо 16 0—23 0 500—650 780 — 900 ЭЛИО л ы 1,08* 1,0365 1,0 6** 1,02 1,065** 1,003— 1,007 1,024** 1,0047 1,0 пропил 1,06 1,040— 1,055 1,01 14512 1,450 н а и Qi 1,4560 — 1,4609 1,451 33,0 33,1 35,0 31,6 31,0 и с II Э Т I 34,5 32,0 33,1 370-380 344 370—380 392 I л е н а 377 400 370 I 24 5 235 245 235 202 204 206 215 246 237 195 1 210 195 226 а а а а а а 6,0 0,1 0,5 1,0 0,17 0,8
Лапрол 1102-4-80 . . I Лапрол 3203-4-80 . . ’ 1 С о п О 1100 1 3200 л и м е р ы 2,8—3,6 1,5 —1,7 о 1 с и с и п 0,1 1 0,1 ропилена и ~ 1 350—500 1 — 1450-2200 е т р а г и 1,0 05 | 0,92 Д Р О ф у р 1.469 н а 400 | дроксильн льзуются псионоген оме указа! твора. 1
- Примечание. * Вязкость в /с !сСт) ** При 25° С - £ Х°рошо Раст?орим в воде и органических растворителя т веществ (ЛапролыГим^полиаминм»“(Ji”?рамольпе^™ч?сттеТеакц3аЦИеЙ °КИСИ пропилена в присутствии полиги Z X О | ых стартовых синтезе раз- юго типа, за- ных в табли-
Номограмма для определения теплоемкости в интервале 15,6—60 °C и теплопро-
водности в интервале 10—107 °C водных растворов этиленгликоля при разных
температурах (1 кал = 4,19 Дж).
Номограмма для определения теплопроводности водных растворов триэтилеи-
гликоля при разных температурах [1 Вт/(м• К) = 2,39• 10~3 кал/(с• см °C)].
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азеотропные смеси
с дипропиленгликолем
195, 363 сл.
с диэтиленгликолем 128, 356 сл.
с пропиленгликолем 184
с трпэтиленгликолем 154, 358
с этиленгликолем 55 сл., 296,
355 сл.
с этилкарбитолом 320 сл.
Алканоламины 9
Алкомон ОС-2 305
Амберлит 72, 76, 94, 96
Амилфениловый эфир этиленгликоля
292
Антифризы 8, И, 14, 31, 40, 42,
65, 98 сл., 101 сл., 105 сл.,
108, 138, 323
ТОСОЛ-А 104 сл.
ТОСОЛ-А40 104 сл.
ТОСОЛ-А65 104 сл.
этиленгликоль 104 сл.
Бис(оксиэтил)гликолевый эфир см.
Триэтиленгликоль
Бис-2-оксиэтиловый эфир см. Ди-
этиленгликоль
Бутиленгликоль (2-Метил-1,2-про-
пандиол) 16 сл., 342, 344
Бутилкарбитол 328
Бутиловый эфир дипропиленгликоля
295, 363
Бутиловый эфир диэтиленгликоль-
диацетата 300
н-Бутиловый эфир пропиленгликоля
293
трета-Бутиловый эфир пропиленгли-
коля 293
н-Бутиловый эфир трппропиленгли-
коля 295
Бутиловый эфир этиленгликоля 300
и-Бутиловый эфир этиленгликоля
(Бутилцеллозольв) 290 сл.,
305, 322, 337
emop-Бутиловый эфир этиленглпколя
292
Виниловый эфир этиленгликоля 302
310
Выравниватель А 305
Гидравлические жидкости 106 сл.,
138, 204, 207
Гидратация окиси пропилена 199 сл.
Гидратация окиси этилена 68 сл.,
200
катализ катионитами 72 сл.,
95 сл.
— кислотный 69 сл., 92 сл.
— основной 70, 73 сл.
кинетика реакции 68 сл.
константа скорости 72 сл., 199 сл.
моделирование процесса 82 сл.
некаталитическая 71, 84 сл.
парофазная 96 сл.
тепловой эффект 84 сл.
технологическая схема 84 сл.
Гликоли
ассоциация 26 сл.
аутоокисление 29 сл.
конформеры 26 сл.
методы анализа 335 сл.
радиолиз 28 сл.
свойства 16 с л.
техника безопасности 33 сл.
токсические свойства 30 сл.
транспортирование 34 сл.
физические свойства 17 сл.
368
Гликоли
фотолиз 28 сл.
химические свойства 21 сл.
хранение 34 сл.
эфиры 8, 289 сл.
Глиоксаль 99
Глицерин 32, 98, 102, 198 сл., 240,
336 сл., 341 сл.
Глпцероген 198
Графтсополпмеры окиси этилена
253 сл.
Дакрон’ (Лавсан, Терилен) 105 сл.
Дпбутиловый эфир диэтиленгликоля
295
Ди-и-бутиловый эфир этиленгликоля
295
Дигликоль см. Диэтиленглпколь
Диглим см. Диметиловый эфир ди-
этиленгликоля
2,5- Диметил-1,4-диоксан 297
Диметиловын эфир диэтиленгли-
коля (Диглим) 295, 323
Диметиловый эфир триэтиленгли-
коля 324
Диметиловый эфир этиленгликоля
(Моноглим) 295, 309, 324
Динитрогликоль (Диэтиленгликоль-
динитрат) 138
1,4- Диоксан 24, 216, 289, 296 сл.,
301, 305 сл., 308, 310,
323 сл.
1,4- Диоксан-2,3-дион 22
Диоксанон 304
2,2' -Диоксидиэтиловый эфир см. Ди-
этиленгликоль
1,2-, 1,3-Диоксипропан см. Про-
пиленгликоль
1,2-Диоксиэтан см. Этиленгликоль
1,3-Диоксолан 99, 297 сл., 301 сл.,
303 сл., 306, 329
Диоксоланы 23
Дипропиленгликоль (1,1 '-Оксиди-2-
пропанол) 186 сл., 337
азеотропные смеси 195, 363 сл.
анализ 345 сл.
изомеры 186 сл.
качество 210
получение 195 сл.
применение 206 сл.
производство 12
растворимость 192 сл., 208
токсические свойства 30 сл.
физические свойства 18 сл.,
185 сл.
эфиры 8, 12, 207 сл., 295, 300 сл.,
363
Р,Р'-Дихлордиэтиловый эфир 159
а,со-Дихлортетраэтиленгликоль 167
24 заказ 972
Диэтиленгликоль (Бис-2-оксиэтило-
вый эфир, Дигликоль,
2,2'- Диоксидпэтиловый
эфир, 2,2'-Оксидиэтанол)
8 сл., 16 сл., 74 сл., 84, 87
азеотропные смеси 128, 356 сл.
анализ 335 сл.
качество 140 сл.
очистка 136 сл.
получение
гидратацией окиси этилена 133
гидролизом диацетата диэти-
ленгликоля 133
— Р,Р'-дихлорэтилового эфи-
ра 133
из окиси этилена 133
из этиленгликольацетата 133
из этиленгликоля 133
оксиэтилированием этилен-
гликоля 134
применение 136 сл.
производство 12 сл.
растворимость 126 сл., 139. 166,
297, 356 сл.
токсические свойства 30 сл.
физические свойства 18 сл.,
117 сл.
эфиры (карбитолы) 8, 12, 295,
300 сл.
Диэтиленгликольдинитрат (Дини-
трогликоль) 138
Диэтиловый эфир диэтиленгликоля
295, 355
Диэтиловый эфир этиленгликоля 295
Изобутиловый эфир этиленгликоля
292 сл.
Изопропиловый эфир пропиленгли-
коля 298
Изопропиловый эфир этиленгликоля
292 сл.
Карбитолы (Эфиры диэтиленглико-
ля) 8, 12, 295, 300 сл.
Карбоваксы (Полимеры окиси эти-
лена) 9, 214 сл., 280, 344
Катиониты 72, 95 сл.
Лавсан (Дакрон, Терилен) 105 сл.
Лапрамол 364
Лапрол 364
Леватит 90, 136, 163, 203
Метил-1,4-диоксан 297
Метилдиоксолан 289, 302
2-Метил-1,3-диоксолан 28, 216
36»
4-Метил-1,3-диоксолан 23
2-Метил-2-оксиметил-1,3-диоксолан
99
Метиловый эфир дипропиленгликоля
295, 363
Метиловый эфир диэтиленгликоль-
ацетата 300
Метиловый эфир пентапропиленгли-
коля 295
Метиловый эфир пропиленгликоля
293, 305, 313, 323
Метиловый эфир тетрапропиленгли-
коля 295
Метиловый эфир трипропиленгли-
коля 295, 355 сл.
Метиловый эфир этиленгликольаце-
тата 300
Метиловый эфир этиленгликоля (Ме-
тилцеллозольв) 290 ел.,
301 сл., 305 сл., 327 сл.,
336, 342
2-Метил-1,2-пропандиол см. Бути-
ленгликоль
Моноглим см. Диметиловый эфир
этиленглпколя
Нитрогликоль (Этиленгликольдини-
трат) 98
Нитроглицерин 98, 138
а-Окси 214 сл., 312 сл.
Окись пропилена 9 сл., 199 сл.,
313
гидратация 199 сл., 203 сл.
полимеры 9, 237 сл., 214 сл.,
254 сл.
производство 12 сл.
сополимеры с окисью этилена 9,
107, 249 сл.
Окись этилена 9 сл.
в синтезах 134 сл.
гидратация 68 сл.
полимеризация 263 сл.
полимеры 9, 262 сл., 214 сл.,
253 сл.
производство 12 сл.
сополимеры с окисью пропилена
9, 107, 249 сл.
1,1'-Оксиди-2-пропанол см. Дипро-
пиленгликоль
2,2'-Оксидиэтанол см. Диэтиленгли-
коль
Оксипропилирование спиртов 314 сл.
Оксиэтилирование гликолей 159 сл.
Оксиэтилирование спиртов 315
н-Октиловый эфир этиленгликоля 292
370
Пеноуретаны 9
Пентаэтиленгликоль 159
Пинаконы 17
Поверхностно-активные вещества
(ПАВ) 8 сл., 224, 236,
299, 305 сл., 322 сл., 325 сл.
Полиакриламид 279, 281
Полигликоли 16 сл., 217 сл.
Полимеризация а-окисей
анионная 217 сл., 222 сл., 241
катионная 215 сл., 222 сл.
координационного типа 220 сл.
Полимеры
окиси пропилена 9, 214 сл.,
237 сл., 249 сл.
окиси этилена (карбоваксы) 9,
214 сл., 249 сл., 344
Полиокс (Полиоксиэтилен высоко-
молекулярный) 9, 214,
262 сл., 266, 280 сл.
Полиоксипропилен стереорегуляр-
ный 254 сл.
Полиоксипропиленполиолы см. По-
липр опиленглпколь
Полиоксиэтилен (ПолиэтиленоксиД)
9, 107, 224
высокомолекулярный 262 сл.
синтез 262 сл.
пластические свойства 271 сл.
применение 278 сл.
растворимость 267
физические свойства 265 сл.
химические свойства 273 сл.
Полиоксиэтиленгликоль см. По-
лиэтиленгликоль
Полипропиленгликоль (Полиокси-
пропилен. Полиоксипро-
ппленполиолы, Полипро-
ппленоксид) 214, 222, 237,
338, 346
производство 12
синтез 238 сл.
физические свойства 242 сл.
Полисорб 137 сл., 214, 237, 246 сл.
Полиэпоксиды 214, 254 сл.
Полиэтиленгликоль (Полиокспэти-
лен, Полиоксиэтиленгли-
коль, Полиэтиленоксид,
ПЭГ) 9,107, 159 сл., 224 сл.,
302, 338, 340, 346
применение 227
производство 12 сл.
синтез 224 сл.
токсические свойства 236 сл.
физические свойства 229 сл.
химические свойства 235 сл.
Полпэтилентерефталат 90. 105, 108
Полиэфирные волокна 105 сл.
Полиэфиры на основе окиси пропи-
лена 9
Поропак 342
Препарат ОС-20 305
Проксанол 326
Пропиленгликоль (1,2-, 1,3 диок-
сипропан, 1,2-Пропанди-
ол, 1,2-, 1,3-Пропилен-
гликоль, Трпметиленгли-
коль) 8 сл., 16 сл., 172 сл.
азеотропные смеси 184
анализ 335 сл., 342 сл.
изомеры 172
качество 209 сл.
моноэфпры 291, 293 сл.. 305,
313, 323. 327, 346 сл.
получение 195 сл.
гидратацией окиси пропилена
199 сл.
гидрированием пропиленкар-
боната 197 сл.
гидрогенолизом высших по-
дполов 198 сл.
гидроксилированием пропи-
лена 196
гидролизом дихлормроиана
196
из пропилена 197
некаталитической гидрата-
цией окиси пропилена
203 сл.
при окислении пропана 196
при окислении пропилена
196 сл.
применение 204 сл.
производство 12 сл.
растворимость 182 сл., 361 сл.
сложные эфиры 298 сл.
токсические свойства 30 сл.
физические свойства 18 сл.,
173 сл.
Пропиленгликольацетат 197. 298
Пропиленгликольбензоат 298
Пропиленгликольдиацетат 298
Пропиленглпкольдибутират 298
Иропиленгликольдилаурат 299
Пропиленгликольдипальмитат 299
Препиленгликольдистеарат 299
Нропиленгликольдиформиат 298
Пропиленгликольлаурат 299
Пропиленгликольолеат 299
Пропиленгликольпалымитат 299
Пропиленгликольстеарат 299
Пропиленгликольформпат 298
Пропиленкарбонат 197 сл., 304
м-Пропиловый эфир дипропилен-
гликоля 295
н-Пропиловып эфир пропиленглико-
ля 293
Пропиловый эфир этиленгликоля
292 сл.
ПЭГ см. Полиэтиленгликолп
Растворимость
в дипропиленгликоле 192 сл.,
208
в диэтиленгликоле 126 сл.,
139, 166, 297, 356 сл.
в проппленгликоле 182 сл.,
361 сл.
в тетраэтпленгликоле 158,
166, 357 Сл.
в трипропиленгликоле 194,
в триэтиленгликоле 152 сл.,
166, 357 сл.
в этиленгликоле 52 сл., 297,
353 сл.
диэтиленгликоля 139
полиоксиэтилена 267
полипропиленгликолей 242 сл.
полиэтиленглпколей 230 сл.
сополимеров окисей этилена
и пропилена 250
этиленгликоля 55
Сополимеры окиси этилена и про-
пилена 9, 107, 249 сл.
Стереокс-6 305
Терилен (Дакрон, Лавсан) 105 сл.
Тетрагликоль см. Тетраэтилен-
гликоль
2,2',5,5'-Тетраметил-1,4-диоксан 294
Тетраэтиленгликоль (Тетрагликоль)
74 сл.
качество 169
получение 158 сл.
применение 167 сл.
растворимость 158, 166, 357 сл.
токсические свойства 30 сл.
физические свойства 18 сл.,
156 сл.
Технологическая схема
выделения ароматических угле-
водородов экстракцией
• диэтпленгликолем 139 сл.
некаталитической гидратации
окпси этилена 84 сл.
осушки диэтиленгликолем 140
получения диэтиленгликоля
134 сл.
— простых эфиров этиленгли-
коля 317 сл.
— этиленгликоля из метанола 68
— этилцеллозольва 318 сл.
разделения продуктов гидроге-
нолиза сахарозы 198 сл.^
синтеза полипропиленгликолей
238
установки полиоксиэтилирова-
. нин 225, 227
24*
371
Токсические свойства
гликолей 30 сл.
полиэтиленгликоля 236 сл.
эфиров гликолей 305 сл.
ТОСОЛ-А 104 сл.
ТОСОЛ-А40 104 сл.
ТОСОЛ-А65 104 сл.
Тригликоль см. Триэтиленгликоль
Трпметилгликоль см. Пропилен-
гликоль
Трипропиленгликоль 184 сл.
изомеры 185
применение 208 сл.
растворимость 194, 208
физические свойства 185 сл.
эфиры 295, 355 сл.
Триэтиленгликоль (Бис(оксиэтил)
гликолевый эфир, Три-
гликоль, Этиленгликоль-
диокспдиэтиловый эфир]
16 сл., 74 сл., 84, 87,
134 сл.
азеотропные смеси 154, 358
анализ 343
качество 168
получение 158 сл.
применение 163 сл.
производство 12 сл.
растворимость 152 сл., 166,
357 сл.
токсические свойства 30 сл.
физические свойства 18 сл.,
145 сл.
эфиры 12
Фениловый эфир пропиленгли-
коля 293
Фениловый эфир этиленгликоля
292, 300
Физические свойства
ацетатов простых эфиров 300
бутиленгликоля 18 сл.
гликолей 17 сл.
дипропиленгликоля 18 сл., 187 сл.
диэтиленгликоля 18 сл., 117 сл.
диэфиров диэтпленглпколя 295
— этиленгликоля 295
моноэтиловых эфиров 291
моноэфиров пропиленгликоля
293, 295
— этиленгликоля 292, 294
полиоксипропилена 258
полиоксиэтилена 265 сл.
полипропиленгликолей 242 сл.
полиэтилепгликолей 229 сл.
1,2-пропиленгликоля 18 сл.,
173 сл.
1,3-пропиленгликоля 18 сл.
простых олигоэфиров окиси
пропилена 364
физические свойства
сополимеров окисей этилена
и пропилена 250
тетра этиленгликоля 18 сл.,
156 сл.
трипропиленгликоля 18 сл.,
187 сл.
триэтиленгликоля 18 сл., 145 сл.
этиленгликоля 18 сл., 38 сл.
эфиров гликолей 289 сл.
Флокуляция 279 сл.
Целлозольвы (Эфиры этиленгли-
коля) 8, 12, 23, 290,
292 сл., 301 сл., 305 сл.,
313, 317 сл., 322 сл.,
327 сл., 336, 342
Эпоксиды 313
1,2-Этандиол см. Этиленгликоль
Этаноламины 9 сл., 12 сл.
2-Этилбутиловып эфир этилен-
гликоля 292
2-Этилгексиловый эфир пропилен-
гликоля 293
2 Этилгексиловып эфир этилен-
гликоля 292
Этиленгликоль (1,2-Диоксиэтан,
1,2-Этандиол) 8 сл.,
16 сл., 313
азеотропные смеси 55 сл., 296,
355 сл.
анализ 335 сл.. 342 сл.
антифриз 104 сл.
диэфиры 295. 299 сл., 323
качество 108 сл.
моноэфиры 289 сл., 301 сл.,
305 сл., 313, 317 сл.,
322 сл.
очистка 90 сл.
получение
гидратацией окиси этилена
68 сл.
гидрогенолизом многоатом-
ных спиртов 66 сл.
гидролизом дигалогенидов эти-
лена 60 сл.
— этиленгликольацетатов
62 сл.
— этиленхлоргидрина 61 сл.
из дибромэтана 60 сл.
из ди-трет-бутила 66
из метанола 67 сл.
из окиси углерода и водорода
67
из формальдегида 66, 68
из этиленкарбоната 64 сл.
372
Этиленгликоль, получение
кислотно-каталитической гид-
ратацией окиси этилена
92 сл.
некаталитической гидратаци-
ей окиси этилена 84 сл.
окислением этилена 63 сл.
радиационно-химическим ме-
тодом 66
электрохимическим окисле-
нием этилена 64
применение 98 сл.
производство 12 сл.
токсические свойства 30 сл.
физические свойства 18 сл.,
38 сл.
эфиры см. Целлозольвы
Этиленгликольацетат 100, 298, 304,
311
Этиленгликольбовзоат 298
Этиленгликольбутират 298, 311
Этпленгликольдиапетат 998,300, 304,
323
Этилевглпкольдинитрат (Нитрогли-
коль) 98
Этиленгликольдилаурат 299
Этиленгликольдиоксидиэтиловый
эфир см. Триэтиленгли-
коль
Этиленгликольдиолеат 305
Эгиленгликольдипальмитат 299
Этиленгликольдистеараг 299
Этиленгликольдиформиат 298, 304,
324
Этиленгликольлаурат 299
Этиленглпкольолеат 299, 305
Этиленгликольпальмитат 299
Этиленгликольпропиоват 312
Этиленгликольстеарат 299
Этиленгликольтерефталат 22
Этиленгликольформиат 298
Этиленкарбонат 22, 64 сл.
Этиленсукцинат 310
Этилкарбитол (Этиловый эфир
диэтиленгликоля) 296 сл.,
310, 313, 319 сл., 323,
о 327 сл., 356
Этиловый эфир дипропиленгликоля
295
Этиловый эфир диэтиленгликоль-
ацетата 300
Этиловый эфир диэтиленгликоля
(Этилкарбитол) 296 сл.,
310, 313, 319 сл., 323,
327 сл., 356
Этиловый эфир пропиленгликоля 293
Этиловый эфир тетраэтиленгли-
коля 319
Этиловый эфпр триэтиленгликоля
319, 322, 356
Этиловый эфир этиленгликольаце-
тата 300
Этиловый эфир этиленгликоля
(Этилцеллозольв) 290 сл.,
296 сл., 305, 312 сл.,
318 сл., 327 сл., 344 сл.
Эфиры гликолей 8, 289 сл.
анализ 335 сл.
бутпленгликоля 291
дипропиленгликоля 8, 12,
207 сл.. 295, 300 сл., 363
диэтиленгликоля (карбитолы) 8,
12, 295, 300 сл.
качество 327 сл.
низкомолекулярные 322 сл.
певтапропиленгликоля 295
полигликолей 322, 325 сл.
получение 312 сл.
применение 322 сл.
пропиленгликоля 290 сл.,
293 сл., 298 сл., 305, 313,
323, 327, 346 сл.
простые 311
— ациклические 289 сл.
— циклические 297 сл., 317 сл.,
324 сл.. 344
синтезы на основе гликолей 309 сл.
----- окисей олефинов 306 сл.
—— олефинов ЗЮ сл.
— через галогенпроизводные
308 сл.
сложные 298 сл., 310 сл., 340
смешанные 299, 325
средней молекулярной массы 322,
325
тетрапропиленгликоля 295
токсические свойства 305 сл.
трипроппленгликоля 295. 355 сл.
триэтиленгликоля 12
физические свойства 289
химические свойства 300 сл.
этиленгликоля см. Целлозольвы