/































































































































































































Текст
<5 5 Н /
В ИИ9
__ В. Вилков Ю.А.Пентин
ФИЗИЧЕСКИЕ
МЕТОДЫ
ИССЛЕДОВАНИЯ
ХИМИИ
Ьуктурные методы
и оптическая
спектроскопия
СВОДНАЯ СХЕМА ХАРАКТЕРИСТИЧЕСКИХ ИНФРАКРАСНЫХ ГРУППОВЫХ ЧАСТОТ
too
2000
1800
1600
1000
2500
1400
1200
3000
600
3500
4000
АЛКАНЫ
метиленовая грума
7Г
сл.
ПРОСТЫЕ ЭФИРЫ
АМИНЫ
3500
4000 см-’
АРОМАТИЧЕСКИЕ
СОЕДИНЕНИЯ
ем •
4оа
CH, -NH
>CH-NH
O-NH,
(широкая) амиды ----- ---
монозамещ. амиды
дизамещ. амиды -
ОСН, —
с-сн-
иормальиыс ангидриды —
циклические ангидриды —
алиф. кетоны —
аром, кетоны —
CH, -С метильная группа
сн,-(оо) - — |-
первичные амины-----
ср.
"ср”
ср
Ф-
ср.
с-со-о-со-с
— о=с-о-с=о
(резкая)
— (высокая у-----
сно -
•О-СО-С
2000
мкм
(у свободных О-И понижайся)
(у свободных О-Н понижается)
(v t в 'бедных О-Н понижается)
(у свободных О-Н пеиижается)
— (связанная группа О-Н) —
— (широкая)
АЛКЕНЫ
АЛКИНЫ
СПИРТЫ
(несвязанная группа О-Н) (резкая) ср.
ср.
КИСЛОТЫ
1 1
СЛОЖНЫЕ ЭФИРЫ
- СН;
-ОС—Н
- СН2 -<С=О),-СН2 -(ON)
сн 1--------1 — г--
этильная группа ]--
н-пропильная группа
изопропильная группа—
трет-бутилытая группа--
винильная | |
группа -СН=€Н
^С=С„ (транс)
|----Г_— (цис)
монозамещенные бензола
орто-дизамещ.
мста-ди зам ещ.
пара-дизамещ.
1, 2, 3 - трязамещ.
1.2,4 - трязамещ.
1, 3, 5 - тризамещ.
- ph------нафталины
-----СО" Д-наФ**лияь*
Алифатические эфиры —
Ароматические эфиры —
-СНг-СН2-СНг-СН.
(сопряж.) —
(со пряж.)
(сопряж.)
(сопряж.)
(сопряж.)
.— сн. -о-сн
. <>-О-СН,
АЛЬДЕГИДЫ
1 1
КЕТОНЫ
1 1 1
АНГИДРИДЫ
АМИДЫ
ср.
первичные спирты
вторичные—I-----
третичные — —
ароматические---
- СН2-ОН
- СН-ОН
- С-ОН
О-он —
(широкая)
3000
, — карбоновые кислоты —
ион карбоксила (соли, цвиттерионы и т д.) —
формиаты I-
ацетат ы f-
пропиондты
бутираты и выше
акрилагы
фумараш
малеат ы
бензоаты^фталаты —
-----алиф аладегиды---------
-----аром, альдегиды - —
2500
— СООН
iera-ззмсщ)
----(широкая)
I I I I I
(отсутствует у мономера)
H-CO-O-R
-C1LCOO-R
—CH.-CO-O-R
—CH.-CO-O-R
=CH-CO-O-R
=СН—СО—O-R
CH-CO-OR
-СО-МН
-CO-NH-R
-CO-NR
(широкая-жидкие амины!
400
Л.В.Вилков Ю.А.Пентин
ФИЗИЧЕСКИЕ
МЕТОДЫ
ИССЛЕДОВАНИЯ
В ХИМИИ
Структурные методы
и оптическая
спектроскопия
Допущено
Министерством высшего и среднего
специального образования СССР
в качестве учебника
для студентов химических специальностей
высших учебных заведений
МОСКВА «ВЫСШАЯ ШКОЛА» 1987
ББК 24.5
В44
УДК 541.1
ПРЕДИСЛОВИЕ
р Всемерное ускорение научно-технического прогресса, усиление
ецензенты. роли науки в качественном преобразовании производительных сил
кафедра физической и коллоидной химии Ростовского университетам повышении эффективности общественного производства ЯВЛЯЮТ-
нов гИвановУски°й т!*е«рой Пр°*" В' А' к-°(ан^; ПР°Ф- К. С. Крас- я одной Из главных задач, поставленных в Основных направлениях
нов (Ивановский химико-технологический институт) и дон Н И Типичен- ид шос шил
(Ивановский государственный университет им. первого в России Иваново-экономического И социального развития СССР на 1986—1990 ГОДЫ
Вознесенского общегородского Совета рабочих депутатов)
и на период до 2000 года.
Для решения этой задачи предусматривается широкое внедре-
ние автоматизированных систем в различные сферы хозяйственной
деятельности, в частности в химическую промышленность, создание
новых конструкционных материалов и химических продуктов с за-
ранее заданными свойствами. Особое внимание уделяется развитию
научного приборостроения, оснащению научно-исследовательских и
технологических организаций современными приборами и средства-
ИБХ РАН
Вилков Л. В., Пентин Ю. А.
Физические методы исследования в химии. Структурные
методы и оптическая спектроскопия: Учеб, для хим. спец, ву-
зов. — М.: Высш, шк., 1987. — 367 с.; ил.
тика их тямот и общая классификация физических методов и характерис-
™ и &братных задач. Изложены теоретические основы, техника экспе-
пччкД г»- "Ри“енение масс-спектрометрии, диэлькометрии л метода молекулярных
электроныуафии, методов вращательной (микроволновой и комбииа-
рассеяния), колебательной (ИК и КР) и электронной (УФ) спектроскопии.
Рассмотрены возможности их использования в химических исследованиях.
В
1805000000(4309000000) — 383
001(01)—87
142—87
ББК 24.5
541
© Издательство «Высшая школа», 1987
ми автоматизации.
В связи с этим ставится также задача подготовки и переподго-
товки специалистов, в частности химиков, владеющих современны-
ми инструментальными методами исследования, контроля и управ-
ления технологическими процессами, способных творчески их при-
менять.
Развитие химии невозможно без широкого использования в хи-
мических исследованиях достижений физики и ее новых методов
исследования вещества. Взаимопроникновение наук химии и физи-
ки имеет большое значение для методологии науки и способствует
их взаимному обогащению. Это ни в коем случае не означает заме-
ны химии физикой. Химия с ее особыми понятиями и законами не
сводится к чисто физическим представлениям о веществе. Так, по-
нятие о химической связи существенно углубляется при использова-
нии таких физических характеристик, как межъядерное расстояние
(длина связи), частота колебаний, дипольный момент и т. п.
Арсенал современных физических методов в химии настолько
обширен, а применение их столь разнообразно, что требуется систе-
матическое изучение теоретических принципов, технического вопло-
щения, а главное, возможностей их практического использования.
Среди представленных в учебнике методов в практике химиче-
ских исследований наиболее широко используются методы ИК, УФ
спектроскопии и масс-спектрометрии. Часть методов не имеет ши-
рокого распространения, но их результаты крайне важны для хи-
мии. Это относится, например, к методам микроволновой спектро-
скопии, газовой электронографии и некоторым другим.
Все физические методы можно классифицировать как по ха-
рактеру взаимодействия вещества с полем, излучением или пото-
ком частиц, так и по тем свойствам вещества или параметрам
з
молекулы, которые могут этими методами определяться. По пер.
вому признаку классифицируют методы оптической и радиоспек-
троскопии, дифракционные, электрические, ионизационные и т. д.
а при классификации по второму признаку говорят о методах оп-
ределения геометрического строения молекул, определения элек-
трических дипольных моментов, электронных колебательных ц
вращательных энергетических состояний и спектров молекул и т.д
Названия разделов учебника следуют второй классификации (пс
изучаемым свойствам), а в тексте используется, конечно, вся приня-
тая в литературе терминология.
Кроме определения основных характеристик и свойств молекул,
перечисленных выше, а также симметрии, силовых полей, потенциа-
лов внутреннего вращения, энергий ионизации и т. д. многие из
рассматриваемых методов используют также в аналитических це-
лях, при изучении различных равновесий, кинетики и механизмов
химических реакций и т. п.
Измеряемые характеристики и величины в одних случаях необ-
ходимы для установления закономерностей, связывающих физиче-
ские и химические свойства с химическим строением молекул, а в
других — для оптимизации технологических процессов.
По физическим методам исследования в химии уже издаваласг
как общая, так и специальная литература, например многотомная
серия «Физические методы в органической химии» под редакцией
А. Вайсбергера, выпущенная в 50-х годах и уже существенно уста-
ревшая. Переведены на русский язык также два издания (1967 и
1981) книги Р. Драго «Физические методы в химии». Вышло учеб-
ное пособие «Физические методы определения строения органиче-
ских соединений» Иоффе Б. В., Костикова Р. Р., Разина В. В. и ряд
других пособий.
По отдельным методам имеются также практические руководст-
ва и монографии, например по методам оптической спектроскопии
(УФ, ИК и КР), масс-спектрометрии, дипольным моментам и др.
(см. литературу к соответствующим главам).
В то же время, как убедились авторы, читая в течение ряда лет
данный лекционный курс, назрела необходимость издания учебника
в соответствии с программой курса.
Предисловие и введение написаны авторами совместно, пер-
вый—третий разделы — Л. В. Вилковым, четвертый и пятый —
Ю. А. Пентиным.
Авторы считают своим долгом выразить искреннюю благодар-
ность рецензентам Н. И. Гиричевой, К. С. Краснову, В. А. Когану
и О. А. Осипову за внимательное прочтение рукописи и ряд цен-
ных указаний, а также Ю. А. Устынюку, Н. М. Сергееву, Л. Н. Го-
рохову, Л. Н. Сидорову, М. В. Коробову, В. И. Тюлину и
Н. Е. Кузьменко за сделанные ими по отдельным главам замеча-
ния.
Авторы
ВВЕДЕНИЕ
ОБЩАЯ ХАРАКТЕРИСТИКА ФИЗИЧЕСКИХ
МЕТОДОВ
1. ПРЯМАЯ И ОБРАТНАЯ ЗАДАЧИ МЕТОДОВ
Одной из главных проблем в химии являются идентификация и
установление химического строения молекул веществ. Если в прош-
лом это делалось лишь химическими методами, то в настоящее
время для этого используют в основном физические методы. Обыч-
но химик начинает с элементного анализа и определения брутто-
формулы и лишь затем пытается установить строение молекулы.
Если изучается химический процесс, то возникают задачи определе-
ния качественного состава реакционной смеси и идентификации
ее компонентов в определенный момент времени иди на определен-
ном этапе процесса, а также количественных характеристик.
Кроме того, интерес представляет не только состав вещества и
химическое строение его молекул, но практически все физико-хими-
ческие свойства вещества, в свою очередь, связанные с химическим
строением и способствующие его установлению. Изучение физиче-
ских свойств веществ и молекул в методическом отношении представ-
ляет особый раздел науки, основанный на теории взаимодействия
поля, излучения или потока частиц с исследуемым веществом, при
котором проявляются те или иные свойства вещества и его молекул.
Определение изменений излучения, поля или потока частиц пос-
ле взаимодействия с веществом, обладающим совокупностью физи-
ческих свойств, называют прямой задачей физического мето-
да. Однако главным является обычно решение обратной зада-
чи— определение физических свойств вещества или параметров
молекулы на основе указанных изменений, т. е. данных эксперимен-
та, полученных физическим методом. Именно с этой точки зрения
характеризуют возможности метода, его чувствительность, точность,
доступность, практичность.
Не все параметры исследуемого объекта, включенные в прямую
задачу метода, могут быть определены, как оказывается, при реше-
нии обратной задачи. При математическом рассмотрении различа-
ют два типа задач — корректно и некорректно постав-
ленные.
Рассмотрим следующее уравнение:
Ах = и, (1)
где А — непрерывный оператор, зависящий от характера взаимо-
действия вещества с излучением (полем, потоком частиц); х
совокупность характеристик вещества (параметров молекулы); если
и — измеряемый результат взаимодействия (характеристика видо-
измененного излучения, распределение частиц по энергиям и т. п.);
х<=Х и ii^U, X и U—метрические пространства. Таким образом,
математическая формулировка прямой задачи состоит в том, что в
результате действия оператора А на множество х получается мно-
жество и.
Обратная задача состоит в определении х по экспериментально
найденному множеству и, если известен оператор А, т. е. существу-
ет решение;
х — /?(«).
Задача определения х является корректно поставленной,
удовлетворяются следующие требования:
1) существование решения х для всякого элемента u^U;
2) однозначность решения (единственность решения);
3) устойчивость решения к малым изменениям исходных
ных.
(2)
если
дан-
Существование решения и его единственность определяются
в общем случае существованием оператора А~1, обратного опера-
тору А. Если число уравнений недостаточно, чтобы построить об-
ратный оператор, т. е. получить решение, то задача поставлена
некорректно. Это будет иметь место, например, при попытке опре-
деления геометрических параметров сложных молекул на основе
трех экспериментально определяемых вращательных постоянных
или при нахождении в колебательной спектроскопии силовых по-
стоянных, когда оказывается, что их число больше, чем число час-
тот нормальных колебаний, и т. д.
Устойчивость решения определяется малым влиянием неточнос-
тей в измеряемых величинах и на определяемые величины х. Так,
для малых отклонений 8и A-1 (w-f-dw) = A-!w4-A-16u = x4-6x, где
6х—малая величина. Невыполнение условия устойчивости реше-
ния обусловливает невозможность найти непрерывный обратный
оператор А-1 и, следовательно, определить параметры х.
Обратные задачи физических методов в основном являются не-
корректно поставленными. Поскольку в результате эксперимента
получают приближенные значения величин и', то может оказаться,
что А-1г/ не являются решениями х, т. е. не выполняется первое
условие корректности. Особые проблемы возникают при определе-
нии единственности и устойчивости решения. На практике решение
некорректных по второму условию задач находим с исполь-
зованием дополнительной априорной информации или как предел
решений последовательности соответствующих корректных задач,
поставленных для конкретных условий.
Общее решение относительно легко выразить для системы ли-
п
нейных уравнений, например а^х
, я). Однако
Рис. 1. Задача нахож-
дения точки пересече-
ния прямых:
1—хорошо обуслов-
ленная система
(а)+=а2, 6i=+62);
f pi = aiXi + bi
I 1/2 = 02X3 + 62
2 — плохо обусловлен-
ная система
(а3»а4, 63«64)
I уз = азХз+Ь3
( 1/4 = 04X4+64
даже в этом случае может возникнуть малая устойчивость решения,
данная система уравнений плохо обусловлена, т. е. наблюда-
ется сильная зависимость решения хг от ва-
риаций свободного члена щ, а также от оши-
бок коэффициентов и от ошибок расчета.
Например, такой системой линейных урав-
нений описываются две прямые, пересекаю-
щиеся под малым углом. Точку пересечения,
координаты которой являются решениями
указанной системы двух линейных уравнений,
в этом случае трудно определить, так как ее
положение существенно зависит от малых из-
менений в коэффициентах (рис. 1).
Неустойчивость обратных задач, обуслов-
ленная приближенностью экспериментальных
данных, приводит к неединственности реше-
ния в рамках заданной точности и требует
формулировки принципов отбора приближен-
ного решения среди множества возможных
решений. В настоящее время разработаны
эффективные методы и алгоритмы прибли-
женного решения некорректно поставленных
задач с неточными исходными данными, на-
пример метод регуляризации А. Н. Тихонова.
Регуляризируюшие алгоритмы обладают ус-
тойчивостью по отношению к ошибкам вход-
ных данных и программно реализуются на
современных ЭВМ. Использование этих алгоритмов особенно важ-
но при создании автоматизированных систем обработки результа-
тов экспериментальных измерений.
2. СПЕКТРОСКОПИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
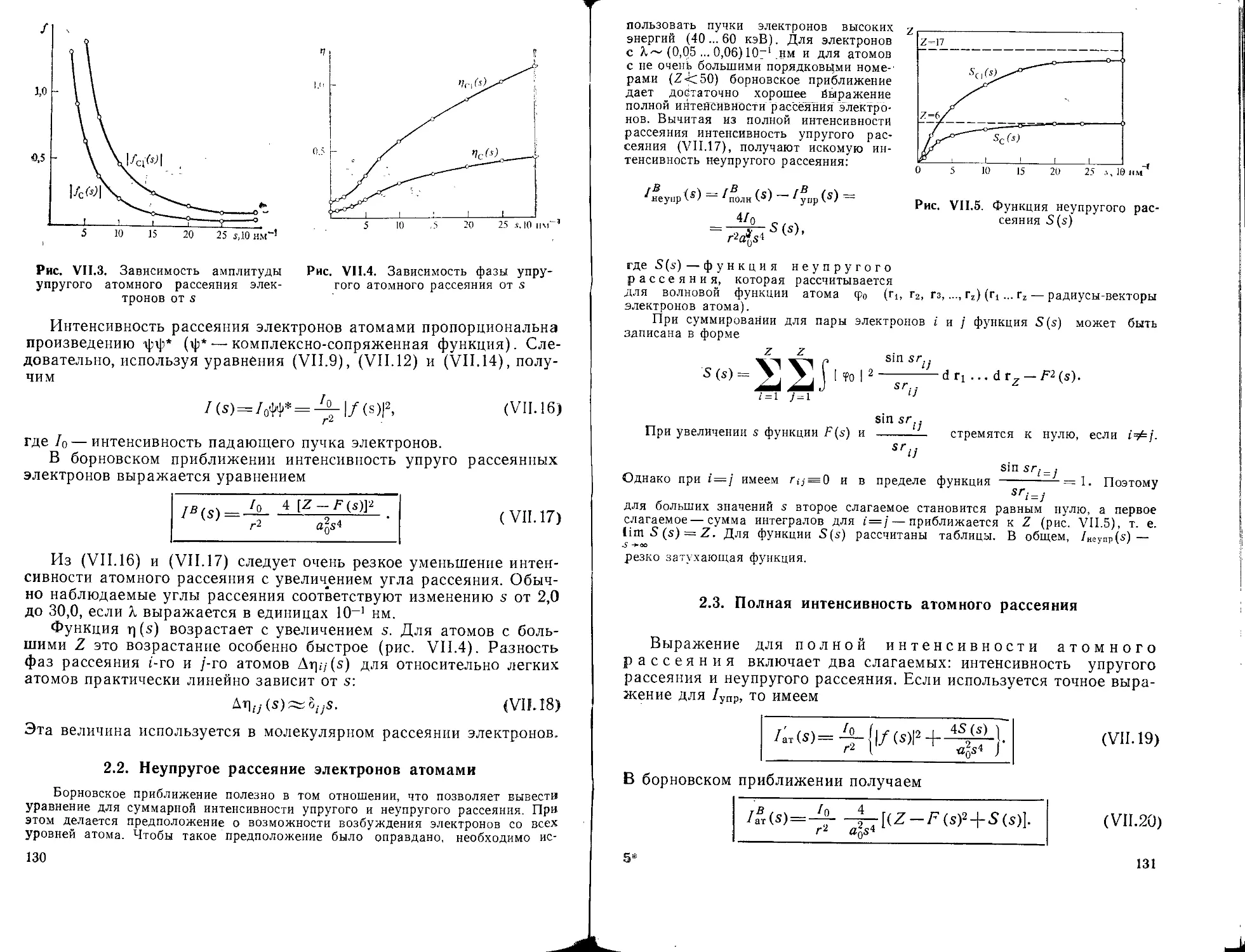
В спектроскопических методах исследуют зависимость интенсив-
ности поглощения или испускания излучения от частоты или длины
волны. Эти методы позволяют изучать энергетические состояния
атомов и молекул, определять разность энергетических уровней по
определяемой частоте перехода
v = (£f-£;)//? = AF,-,-,7г
и оценивать вероятность перехода по интенсивности полосы, а на
их основе также многие другие характеристики молекул (симмет-
рию, геометрию, электрические свойства и т. п.). Интервалы изме-
ряемых энергий ХЕц отличаются для разных методов на много по-
рядков (табл. 1). Это означает, что спектроскопические исследова-
ния охватывают самые различные типы переходов — электронные,
колебательные, вращательные, а также переходы, связанные с изме-
нением направления магнитного момента электронов и ядер и т. д.
Таблица 1. Диапазоны значений частот и длин волн в спектроскопии
Спектры Частоты, Гц Длины волн Единицы измерений
ЯГР 1018... 1021 — 3 пм S
Рентгеновские 10”... 1018 3 нм ... 3 пм эВ
Фотоэлектронные 10” ... 1016 3 ... 700 нм эВ
Электронные 10”... Ю16 3 ... 700 нм
Колебательные 1012... 1014 3 мкм ... 3 мм
Вращательные 1010... 1012 3 см~0,03 мм МГц
ЭПР 109 ... 10” ~3 см МГц
ЯМР 107 ... 108 ~5 м МГц
ЯКР 10е ... ю9 30 ... 300 м МГц
В зависимости от условий
получения
различают
три вида
спектров: поглощения, испускания и рассеяния
(рассеянное излучение наблюдается под каким-либо углом к пада-
ющему). Интенсивность линий или полос спектра пропорциональ-
на прежде всего числу молекул на исходном уровне — нижнем при
поглощении и верхнем при испускании. В условиях теплового рав-
новесия заселенность состояний или уровней определяется распре-
делением Больцмана:
Д-^21
У1 gl
(3)
где N? и N\ — число молекул на верхнем и нижнем уровнях соот-
ветственно, а g2 и g\ — весовые множители верхнего и нижнего
уровней соответственно; \Е%\— разность энергий; k — постоянная
Больцмана; Т — абсолютная температура.
Поскольку для электронных и большинства колебательных пе-
реходов AE^>feT, при обычных температурах в спектрах поглоще-
ния такие переходы имеют место главным образом из основного
состояния (с нулевого уровня). Вращательные переходы возмож-
ны при этом уже для многих нижних состояний.
Значительная энергия, подводимая падающим излучением в
методе УФ спектроскопии, вызывает возбуждение не только элек-
тронных, но также колебательных и вращательных переходов. УФ
спектры сложных молекул характеризуются широкими полосами
с малым разрешением их структуры. У колебательных спектров
сложных молекул в газовой фазе существует вращательная струк-
тура полос, чаще всего плохо разрешенная.
Интенсивность спектров пропорциональна вероятностям пере-
ходов, осуществляющихся в соответствии с квантово-механически-
ми правилами отбора. Вероятность любого перехода определяется
соответствующим моментом — электрическим (дипольным, квад-
рупольным) или магнитным.
Частота и интенсивности, а также ширина и форма спектраль-
ных полос и линий связаны с большой совокупностью молекуляр-
ных параметров и свойств веществ, которые можно исследовать,
решая обратные задачи спектроскопических методов.
3. ДИФРАКЦИОННЫЕ МЕТОДЫ
| Дифракционные методы основаны на рассеянии излучения или
jпотока частиц без изменения их энергии, т. е. на упругом рассел-
ении. Дифракционная картина рассеяния обусловлена волновыми
Двойствами излучения и частиц.
Длина волны электромагнитного
излучения связана с его энергией:
- с с ,
Л = —=---п,
v Е
(1)
где Ev — энергия фотона с часто-
той v; с — скорость света.
Частицам с массой т, движу-
щимся со скоростью v, соответству-
ет волна (уравнение де Бройля):
Рис. 2. Схема дифракции падаю-
щей волны /0 на двух центрах
рассеяния А и В, расстояние меж-
ду которыми равно г:
отрезки ВО и АО' — проекции АВ иа
направление падающего /о и рассеян-
ного /р лучей; R — расстояние от цент-
ра А до плоскости регистрации ди-
фракционной картины; 0 — угол рассея-
ния; С — точка регистрации
(5)
Основное условие дифракции со-
стоит , в том, что X должна быть
близка или меньше расстояний ме-
жду атомами рассеивающего веще-
ства. Это является следствием то-
го, что в общей геометрической
теории дифракции результат интер-
ференции — сложения волн — зави-
сит от разности хода рассеянных лучей (рис. 2):
Д = ДО'— BO = r(sin а2 — sin aj.
(6)
Так как расстояние до точки регистрации значительно больше, чем
расстояние между центрами рассеяния (| R | | г |), то рассеянные
лучи от Л до В, попадающие в точку С, практически параллельны.
Волны суммируются, если Д=пХ, и гасят друг друга, если Д =
— -П-^~ -Х(п —целое число). Поэтому
1 2,
Х = —(sin а2— sin aj),
п
т. е. .
X
(7)
так как (sin «2—sinai)/n<l.
Наибольшее применение имеют три дифракционных метода:
рентгенография, электронография и нейтроно-
графия. В рентгенографии Хр~ 10_ 1 нм, в электронографии для
быстрых электронов, ускоренных в поле высокого напряжения 40...
60 кВ, Хэ~5-10~3 нм и в нейтронографии для тепловых нейтронов
лн~ 10~' нм. Наиболее доступными являются источники рентгенов-
ского излучения и пучков электронов. Нейтронные пучки для ди-
Рис. 3. Качественное
представление зависи-
мости амплитуды рас-
сеяния рентгеновских
лучей Д (—), элек-
тронов /\, (-----) и
нейтронов (О) от
порядкового номера
элемента 2
фракции получают замедлением быстрых нейтронов, выводимых и;
ядерного реактора.
В структурных исследованиях измеряют интенсивность рассея.
ния в зависимости от угла рассеяния. Распределение интенсивности
зависит от структурных параметров. Однако имеется определенна}
специфика в использовании дифракционных методов, обусловлен
ная различной природой взаимодействия рентеновского излучения
электронов и нейтронов с веществом. Рентгеновские лучи рассеива.
ются электронами атомов и молекул, пучвд
электронов — электрическим полем, создавае-
мым ядрами и электронами, а пучки нейтро-
нов— ядерными силами. Интенсивность рас-
сеяния, просуммированная по всем направле-
ниям, характеризует рассеивающую способ-
ность атома к данному виду излучения. Для
рассеивающих способностей атомов в рентге-
нографии, электронографии и нейтроногра-
фии имеет место следующее соотношение:
/р : 7Э :1 : 106: Ю-2. Максимальное рассея
ние, характерное для пучка электронов, объ-
ясняет широкое использование электроногра-
фии для изучения тонких пленок толщиной
10“6...10 5 см и строения молекул в газовой
фазе, а также обусловливает относительно
малые экспозиции. Рентгеновские лучи и пуч-
ки нейтронов используют для исследования
конденсированной фазы веществ, т. е. мак-
роскопических объектов, толщиной в доли
миллиметров в рентгенографии и несколько миллиметров в нейт-
ронографии.
В связи с различным характером взаимодействия излучения и
пучков частиц с веществом наблюдается и различная зависимость
их рассеяния от атомного номера элемента Z рассеивающего ато-
ма. Количественно рассеивающую способность атома определяют
атомной амплитудой рассеяния /(0), где 0 — угол рассеяния. Ве-
личина |f(0)|2 пропорциональна интенсивности излучения /р(0),
рассеянного атомом под углом 0. Амплитуда рассеяния рентгенов-
ских лучей /р(0) при малых углах рассеяния пропорциональна Z,
а при больших углах — Z'y В электронографии в среднем
/a(0)~Z2A Амплитуды рассеяния нейтронов /н не зависят от уг-
ла рассеяния (сферически симметричное рассеяние) из-за малого
размера ядра и явно не зависят от Z (рис. 3). Для ряда ядер
/н<0-
Указанные свойства амплитуд рассеяния показывают, что в
рентгенографии затруднительно определить координаты легки?
атомов в присутствии тяжелых, рассеяние от которых максимально.
Несколько лучше обстоит дело в электронографии. Методом нейтро-
нографии с большой точностью находят координаты атома водо-
рода.
Более подробно методы рентгенографии и нейтронографии рас-
сматривают в курсе кристаллохимии, а в данном учебнике будет
изложен лишь метод газовой электронографии.
4. ОПТИЧЕСКИЕ И ДРУГИЕ МЕТОДЫ
Ряд оптических методов близко примыкает к спектроскопиче-
ским, но они имеют то отличие, что одной из основных изучаемых
в них характеристик излучения является его состояние поляриза-
ции или некоторые вторичные эффекты взаимодействия излучения
с веществом и т. п.
В масс-спектрометрии изучают распределение по массам ионов,
образованных при ионизации молекул различными методами, т. е.
изучается вторичный эффект взаимодействия с полем или излуче-
нием, а не изменение последних при взаимодействии с веществом.
В фотоэлектронной спектроскопии, по существу, также исследуют
вторичный эффект взаимодействия рентгеновского излучения или
излучения в УФ области с веществом, анализируя распределение
выбитых электронов по энергии.
Классические методы определения дипольных моментов основа-
ны на измерениях диэлектрической проницаемости, т. е. изменений
электрического поля под влиянием вещества. В то же время в ме-
тоде молекулярных пучков изучается эффект взаимодействия моле-
кул с электрическим полем, а не изменения последнего.
5. ХАРАКТЕРИСТИЧЕСКОЕ ВРЕМЯ МЕТОДА
Акт взаимодействия излучения или потока частиц с веществом
происходит за определенный промежуток времени. Если изучаемая
система за этот промежуток времени претерпевает изменения, то
результат взаимодействия усредняется по нескольким состояниям
системы. Например, в электронографическом исследовании молеку-
лы PF5 найдена тригональнобипирамидальная конфигурация свя-
зей (рис. 4). Установлено, что длины экваториальных (е) и
аксиальных (а) связей различаются на ~5-10“3 нм. Однако
при изучении спектра ЯМР PF5 для ядер 19F наблюдается синглет-
ный сигнал, т. е. v(Fa)=v(Fe). Это объясняется тем, что в ЯМР
спектроскопии время взаимодействия радиоизлучения с ядерным
магнитным моментом фтора значительно больше, чем время изме-
нения конфигурации молекулы PFs, которая претерпевает так на-
зываемое псевдовращение Берри — внутримолекулярный пе-
реход от одной бипирамидальной конфигурации к другой с обменом
атомов F местами в аксиальном и экваториальном положениях
(рис. 4). Отсюда очевидна необходимость введения понятия харак-
теристического времени физического метода, которое можно сопо-
ставлять со средним временем жизни тех или иных форм и состоя-
ний изучаемых систем.
Формулировка этого понятия вытекает из принципа неопреде-
ленности Гейзенберга. Действительно, если АД и А/ — измеряемые
интервалы соответственно энергии (например, разность энергия
двух состояний системы) и времени, то
(8)
где h—h/2n.
Учитывая, что &E=hv, где v — частота перехода или излучения,
взаимодействующего с системой, имеем
Д/>—. (9)
ДЕ Av 2«v
Таким образом, характеристическое время метода (хар можно
определить как величину, обратно пропорциональную частоте
Рис. 4. Схема псевдовращения Берри для молекул,
имеющих конфигурацию тригональной бипирамиды:
цифры — порядковые номера атомов
v (Гц) квантовых переходов системы, которые могут этим физиче-
ским методом исследоваться.
Поскольку v = c/Z (с — скорость света), можно получить другое
выражение:
Д/----L. . (Ю)
2лс
Для пучка частиц, используя уравнение де Бройля (5), имеем
М------= = . (И)
Е mv^/2 rtv
Эти соотношения можно представить упрощенной картиной
прохождения волн излучения через молекулярную систему. Поло-
жительная полуволна передает свою энергию системе, отрицатель-
ная полуволна характеризует вынужденное испускание и возвра-
щение системы в исходное состояние.
Для потока частиц время взаимодействия с молекулярной си-
стемой соответствует времени прохождения через нее. При дифрак-
ции быстрых электронов используемый пучок имеет X меньше раз-
меров молекулы, а длина пути электронов в молекуле на два по-
рядка больше.
Характеристическое время метода может быть найдено по дан-
ным табл. 1 и уравнениям (9) и (11).
Важную информацию о времени жизни молекулярных форм и
состояний молекул, об обменных, релаксационных процессах
и т. д. содержат ширина и форма спектральных линий. По средне-
му времени жизни состояний молекулы определяется константа
скорости превращения, как для мономолекулярной реакции ki =
= 1/т. Энергию состояния, среднее время жизни которого т, можно
охарактеризовать интервалом значений ЬЕ^И/х, обусловливающим
ширину линии §v = §Elh для перехода из данного состояния. Если
квантовые переходы двух взаимопревращающихся форм молекулы
1 и 2 характеризуются частотами vi и v2, разность которых Av
меньше, чем средняя ширина линии 6v = 1/2(6vi + 6v2), т. е.
Av = Vj—v.2<(8v, (12)
то линии сольются в одну. Отсюда можно записать условие слия-
ния линий в виде
», BE h I 1 /Ю\
« =—--------=----->Дм или т <”------ . (13)
h hr 2лт 2«Av
Так, например, при медленном обмене протонов в системах
А—Н...В щгА...Н—В сигналы ПМР протонов На и Нв полностью
разделяются (т> l/2nAv). Если обмен становится быстрым
(т< l/2nAv), то различные положения протонов нельзя отличить
в спектре ПМР: пики сливаются. Однако различные положения
протона в таком быстром процессе могут быть обнаружены таки-
ми методами, как ИК или УФ спектроскопия.
Методы КР и ИК спектроскопии позволяют исследовать кон-
формационные равновесия при высоте потенциальных барьеров
несколько кДж/моль, так как каждый конформер в этом случае
за время существования дает свой колебательный спектр, тогда
как, например, сигналы ЯМР разных конформеров при этом сли-
ваются. Время существования конформера, определяемое перио-
дом внутреннего вращения, больше, чем время колебательного
перехода.
6. ЗНАЧЕНИЕ ФИЗИЧЕСКИХ МЕТОДОВ
ДЛЯ ТЕОРЕТИЧЕСКОЙ ХИМИИ
Использование физических методов позволяет исследовать ос-
новные вопросы теории химического строения, такие, как последо-
вательность и кратность химических связей, структурная, оптиче-
ская и конформационная изомерия, координационное число
•атомов, взаимное влияние атомов и групп атомов в молекуле, внут-
реннее вращение молекул и другие движения с большими амплиту-
дами, энергетические, электрические и другие молекулярные харак-
теристики, промежуточные продукты и механизмы реакций,
•структура конденсированных фаз и т. д.
Получаемые физическими методами данные, т. е. определяем^
физические характеристики и параметры, обычно связываются с(
строением молекул. Но даже определение геометрии молекулы са
мо по себе еще не позволяет установить наличие или отсутствщ
химических связей между атомами, т. е. их распределение. Косвенщ
можно решить эту задачу, сопоставляя соответствующие межъядер
ные расстояния в исследуемых молекулах с длинами связей в моле
кулах, химическое строение которых не вызывает сомнений. Данные
получаемые некоторыми методами, например определение диполь
ных моментов, относятся к молекуле в целом. В методах УФ, ИЦ
спектроскопии, ЯМР и других влияние характера связей межд}
атомами на определяемые параметры оценивается из определен-
ных модельных представлений.
Получаемые при использовании физических методов данные t
ряде случаев ставят перед химиками новые вопросы, решение ко-
торых, безусловно, способствует развитию теории химической
строения. Так, например, по данным колебательных спектров i
масс-спектрометрии, энергия разрыва связи в молекуле Н2 больше,
чем в ионе Ht, что согласуется с представлениями о больше!
прочности связи, образуемой двумя электронами с антипарал-
лельными спинами. Между тем в других молекулах и соответст-
вующих ионах (Li2 и Li?, Na2 и Nas, К2 и Кг") соотношение ве-
личин энергий разрыва связей обратное. Таких примеров можно
привести больше и на них в книге будет обращено специальное
внимание.
В общем, количественные опорные данные для современной
квантовой и теоретической химии получают в основном с помощью
физических методов исследования.
7. СОВРЕМЕННЫЙ УРОВЕНЬ И ПЕРСПЕКТИВЫ
РАЗВИТИЯ ФИЗИЧЕСКИХ МЕТОДОВ
Широкое использование физических методов способствует их
развитию, повышению технического уровня и точности определяе-
мых параметров.
Известен большой вклад советских ученых в разработку тео-
рии и экспериментальной техники современных физических мето-
дов. Основоположниками квантовой электроники и создателями
первых оптических квантовых генераторов (лазеров) являются
лауреаты Ленинской премии Н. Г. Басов и А. М. Прохоров, удо-
стоенные вместе с американским ученым У. Таунсом "также Нобе-
левской премии. Открытие советскими учеными Л. И. Мандель-
штамом и Г. С. Ландсбергом одновременно с индийскими учеными
Ч. Раманом и К. Кришнаном эффекта комбинационного рассеяния
света привело к созданию метода спектроскопии КР. Явление ре-
зонансного КР открыто П. П. Шорыгиным. Одним из создателей
методов нелинейной оптики является Р. В. Хохлов. Приоритет В
разработке теории колебательных спектров молекул принадлежит
14
м А Ельяшевичу. Много внесли в развитие молекулярной спек-
тпоскопии и фотохимии И. В. Обреимов, А. Н. Теренин, Б. И. Сте-
панов А С. Давыдов и др. За разработку метода высокотемпера-
ТУОНОЙ газовой электронографии группа советских ученых удостое-
на Государственной премии СССР. Необходимо также отметить
значительный вклад советских ученых в развитие и приложения
масс-спектрометрии для исследования реакций свободных радика-
лов и ионов, получения термодинамической информации в области
высокотемпературной неорганической химии. Так В. Л. Тальрозе
обнаружил реакции ионов органических соединений с молекулами
в газовой фазе; им был открыт устойчивый ион метония.
Важной тенденцией в развитии методов является их комплекс-
ное использование особенно в целях идентификации веществ и ус-
тановления их химического строения. Наиболее широко для этого
применяют четыре метода: ИК, УФ, ЯМР спектроскопию и масс-
спектрометрию. Для полного решения задачи установления хими-
ческого строения молекул веществ необходимы данные возможно
большего числа методов, поскольку, как уже отмечалось, сущест-
вует проблема некорректности обратных задач.
Интеграция различных методов увеличивает их возможности в
определении физических параметров. Самый простой пример — со-
четание ИК спектроскопии и спектроскопии комбинационного рас-
сеяния света при решении колебательной задачи. Оно необходимо
уже потому, что не все колебания могут проявляться в ИК спектрах
или отдельно в спектрах КР.
Для более надежного определения геометрии молекул и даже
иногда единственно возможным способом решения этой задачи яв-
ляется использование трех методов: газовой электронографии, вра-
щательной и колебательной спектроскопии.
Накопление данных различных физических методов позволяет
выявлять закономерности и устанавливать корреляции между раз-
личными физическими характеристиками. Так для ряда соединений
установлены зависимости, связывающие длины связей и силовые
постоянные, длины связей и частоты ЯКР одного из атомов связи
[например, г(Р—-С1) и v(35С1)] и т. п.
Широта использования физических методов существенно раз-
лична. Но ценность получаемой некоторыми методами информа-
ции бывает уникальна. Так, например, в методе дефокусировки
молекулярного пучка в неоднородном электрическом поле было
показано отсутствие электрического дипольного момента у моле-
кулы Cs2SO4, что не соответствует классической структурной фор-
°Х/°
муле строения Cs—О — S — О—- Cs.
Развитие современной техники, экспериментальной физики и
физической теории постоянно ведет к повышению чувствительно-
сти, разрешающей способности и других характеристик то одного,
то другого физического метода, к появлению новых возможностей,
открытию новых явлений и разработке на их основе принципиаль-
ножовых методов исследования. Среди последних таких достиже-
ний можно назвать методы нелинейной оптики, которые начинают
постепенно внедряться в химические исследования. Одной из важ-
нейших проблем всегда остается оптимальный выбор метода или
группы методов для решения тех или иных конкретных задач с
учетом не только их возможностей, но также доступности и эконо-
мичности.
Контрольные вопросы
Введение
1. Дайте определение прямой и обратной задачи физического метода.
2. Сформулируйте условия корректно поставленных задач.
3. Приведите примеры корректно и некорректно поставленных задач. __
4. Назовите наиболее важные характеристики спектроскопических методов.?^
5. Укажите области применения методов рентгенографии, электронографии^»,
и нейтронографии. Эффективно ли использование метода рентгенографии для
исследования газов и пленок?
7. Как можно определить характеристическое время метода? Ys
8. Какова роль физических методов в химии?
9. В чем выражается интеграция физических методов исследования?
МЕТОДЫ МАСС-СПЕКТРОМЕТРИИ
Методы масс-спектрометрии являются методами разделения положитель-
ных и отрицательных ионов, образованных при ионизации исследуемого веще-
ства, и измерения их масс (более точно — отношения масс к заряду). На основе
масс-спектрометрии можно изучать многие физические и физико-химические
характеристики вещества: состав, строение молекул и их потенциал ионизации,
определять давление, термодинамические характеристики равновесных процес-
сов— теплоты сублимации, испарения, диссоциации, константы равновесия га-
зофазных и гетерогенных реакций и теплоты этих реакций и т. п.
БИБЛИОТЕКА
ИБХ РАН
Методы масс-спектрометрии являются методами
получения спектров масс ионов. Схема масс-
спектрометров относительно проста и включает
три главных элемента — ионный источник, анали-
затор и детектор. При использовании различных
методов ионизации веществ в ионном источнике
создаются пучки ионов как положительных, так и
отрицательных в зависимости от поставленной
задачи, а иногда те и другие одновременно. Эти
пучки ионов, содержащие ионы различных масс, направляются
далее в анализатор, где под влиянием полей различной природы
формируются пучки ионов определенной массы. Регистрация пучка
ионов в коллекторе ионов позволяет получить спектр масс ионов.
К ионизации вещества в методах масс-спектрометрии прибегают
потому, что существуют эффективные методы управления пучками
заряженных частиц с помощью магнитных и электрических полей.
Теоретические и экспериментальные основы масс-спектрометрии
были заложены еще Д. Д. Томсоном, который впервые в 1912 г. соз-
дал прибор для получения спектра масс положительных ионов. Од-
нако его прибор имел низкое разрешение, т. е. не очень хорошее
разделение ионов, различных по массе. Его ученик Ф. Астон в
1918 г. существенно повысил разрешение за счет лучшей фокуси-
ровки ионного пучка и на своем масс-спектрографе впервые открыл
изотопы элементов. Масс-спектрографы используют для точного
определения атомных масс.
Практически одновременно с Ф. Астоном в Чикаго А. Демпстер
сконструировал первый масс-спектрометр, в котором анализатором
служило поперечное магнитное поле и ионные токи измерялись
электрическими методами. Именно этот тип масс-спектрометров
имеет широкое применение в химии, так как сочетает в себе воз-
можность достаточно точного определения масс ионов и их количе-
ства, т. е. ионного тока. Существенное улучшение разрешения масс-
спектра было получено в 50-х годах в приборах с двойной фоку-
сировкой, т. е. с использованием в анализаторе электрического и
магнитного статических полей.
Наряду со статическими полями для получения масс-спектров
используют переменные электрические поля в динамических масс-
спектрометрах. Это позволяет исключить из конструкции масс-
спектрометров громоздкие магниты. Наряду с чисто техническими
проблемами решаются также проблемы новых приложений масс-
спектрометрии при исследовании различных процессов, в том числе
быстрых.
Методы масс-спектрометрии используются для идентификации
веществ, определения брутто-формул веществ и их химического
строения. Важными для химии являются такие физические харак-
теристики, как потенциал ионизации и энергия разрыва химических
связей. Измерения количества ионов того или другого типа могут
быть связаны с термодинамическими свойствами веществ — пар-
циальным давлением, теплотой сублимации и т. д. Исключительное
значение приобрели методы масс-спектрометрии в изучении меха-
низмов химических реакций.
ГЛАВА I
ПРОЦЕССЫ ИОНИЗАЦИИ И ПРИНЦИПИАЛЬНЫЕ
СХЕМЫ МАСС-СПЕКТРОМЕТРОВ
1. ИОНИЗАЦИЯ АТОМОВ И МОЛЕКУЛ
В методах масс-спектрометрии используют ионизацию вещества,
так как существуют эффективные методы управления пучками за-
ряженных частиц с помощью магнитных и электрических полей.
Большая часть исследований ведется с пучками положительных
ионов.
Образование положительных ионов является результатом взаи-
модействия молекулы, атома или радикала в газовой фазе (М) с
электроном, фотоном, ионом или быстрой молекулой (X), а также
с макроскопическим телом, обладающим электрическим полем с
высоким градиентом.
Схематически процесс ионизации с образованием положи-
тельных ионов можно представить следующим образом:
БСЭ, (1.1)
пМ пХ 111 nz
где им, Их, и и2 — число частиц в единице объема; Ем, Ех, Е},
Е2— энергия соответствующих частиц; q — степень ионизации, в
большинстве случаев равная единице и поэтому п1=п2.
Частица М обычно находится в термическом равновесии с ее
окружением. Однако частица X должна иметь энергию выше неко-
торого минимального значения, соответствующего энергии связи
электрона на самой низкоэнергетической орбитали частицы М, т. е.
Ех должна быть больше потенциала ионизации М. Избы-
ток энергии частицы X над потенциалом ионизации после иониза-
ции распределяется между ионом М®+ с энергией £j и эмиттирован-
ным электроном с энергией Е2.
Ионизацию в основном ведут как непрерывный процесс, так что
«м и пх сохраняются постоянными.
2. ПРОЦЕСС ИОНИЗАЦИИ И ТИПЫ ИОНОВ
Наиболее прост процесс ионизации атомов, хотя он может про-
исходить различными путями. В большинстве случаев однозарядные
ионы образуются при выбивании электрона из наружной валентной
оболочки атома. Образующийся ион может находиться как в основ-
кривые для
различных э.
Рис. 1.1. Потенциальные
иых молекул и их ионов в
состояниях:
а — ионизация без диссоциации; б — ионизация с 1
тичной диссоциацией; в—ионизация с полной диссоц
цией; г — ионизация с образованием возбужденного и<
ном, так и в возбужденном состоянии:
А-{-е_ = А+-|-2е- или А4~е~ = А+*~г2е~ (1.2)
Особенности поведения молекул при ионизации обусловлены
наличием у молекул внутренних степеней свободы. В простейшем
случае, т. е. для двухатомных молекул, возможны четыре вида про-
цессов (рис. 1.1).
Электронное воз-
гв буждение и и о н и -
f3 а ц и я молекул
" подчиняются прин-
ципу Франка —•
Кондона — за вре-
мя электронного пере-
хода межъядерное
расстояние не изменя-
ется. На диаграмме
—потенциальной энер-
гии в форме кривых
Морзе для определен-
ного электронного со-
стояния такой переход
изображается верти-
кальной линией.
Если минимум по-
тенциальной кривой
ионного состояния сме-
щен вправо относи-
тельно минимума кри-
вой молекулы АВ
(рис. 1.1, а), то верти-
кальный переход (по-
казан стрелкой) на бо-
лее высокий по энер-
ом- гии электронный уро-
1ЫХ вень приводит к обра-
зованию молеку-
лярного иона в
она различных возбужден-
ных колебательных
состояниях. Если энергия иона АВ+ больше его энергии диссо-
циации Do и потенциал ионизации атома А меньше В, то воз-
можна диссоциация иона АВ+ с образованием атомного иона А+
(рис. 1.1, б). Процесс называют диссоциативной иониза-
ц и е й, так как ионизация приводит к диссоциации. Следова-
тельно, при осуществлении этого процесса в масс-спектре будут
присутствовать молекулярный ион АВ+, атомный ион А+ и атом В.
На рис. 1.1, в иллюстрируется процесс ионизации с переходом в
антисвязывающее состояние, которое приводит к образованию
ная А+ и атома В. Масс-спектр при таком процессе не содержит
молекулярного иона. Как правило, процессы диссоциативной ио-
низации двухатомных молекул проходят за время порядка 1(Г 13 с —
порядка периода колебания молекулярного иона.
Нижнее электронное состояние иона называется основным, дру-
гие__электронно-возбужденными. Вертикальный переход может
также привести к электронно-возбужденному состоянию АВ+*
(рис. 1.1, а).
Процессы ионизации и диссоциативной ионизации многоатом-
ных молекул более сложны. При удалении одного электрона обра-
зуется молекулярный ион М'. Точка под знаком + озна-
чает, что образовался катион-радикал. Для многоатомных
молекул двухмерные кривые Морзе следует заменить потенциаль-
ными поверхностями в пространстве [ЗА—6(5)]+1 координат (где
N— число атомов в молекуле). Если поверхности потенциальных
энергий для различных электронных состояний молекулярного иона
пересекаются, то возможны безызлучательные переходы с перерас-
пределением колебательной энергии. Молекулярный ион устойчив,
если такое перераспределение энергии не приводит к диссоциации
по какой-либо связи. В противном случае происходит фрагментация
иона. Распад иона является экзотермическим процессом, поскольку
колебательная энергия исходного иона переходит частично в посту-
пательную энергию фрагментов иона.
Таким образом, ионизация многоатомных молекул приводит к
сложным процессам образования нескольких типов ионов.
Молекулярные ионы. Это такие ионы, масса которых равна мас-
се ионизируемой молекулы. К сожалению, до настоящего времени
нет прямых методов определения структуры ионов. Поэтому в масс-
спектральном анализе часто используют предположение о тождест-
венности структуры иона М+ и молекулы М. Вероятность образо-
вания молекулярного иона больше для простых, малых молекул.
С увеличением числа атомов в молекуле увеличивается вероятность
фрагментации молекулярного иона. Так, углеводороды с малой от-
носительной молекулярной массой образуют с большей вероятно-
стью молекулярный ион, чем углеводороды с большой молекуляр-
ной массой. Такие устойчивые группировки, как бензольное кольцо,
способствуют образованию молекулярного иона. Поэтому в аро-
матических углеводородах вероятность образования молекулярного
иона выше, чем в неароматических.
Повышение стабильности молекулярного иона может быть осу-
ществлено различными путями, например введением в молекулу
групп атомов с низким потенциалом ионизации или заменой деста-
билизирующих молекулярный ион групп такими, которые повыша-
ют его устойчивость, например С6Н5-, СН3О-, (СН3) 2М-группы и т. п.
Такие группы и атомы, как NO2, О—N = O, ONO2, Cl, Br, I, умень-
шают стабильность молекулярного иона М+. Увеличение темпера-
туры образца перед ионизацией может существенно изменить масс-
спектр в связи с повышением внутренней энергии молекулярного
иона, способного к фрагментации. Например, ионизация алифати-
ческих соединений при различных температурах дает заметно раз-
личный масс-спектр. Так, с увеличением температуры образца
уменьшается интенсивность пика молекулярного иона. Однако для
ароматических соединений этот эффект мал.
Осколочные ионы. Молекулярный ион может претерпевать фраг-
ментацию в различных направлениях, обусловленных строением
исходной молекулы и методом ионизации. Это мономолекулярный
процесс. В масс-спектре некоторых соединений практически отсут-
ствует молекулярный ион. Так, при ионизации CCU получают ионы
СОз", CClf, СС1+, С1+, С+.Увеличение энергии ионизирующих ча-
стиц приводит к более глубокому распаду молекулярного иона. Ил-
люстрацией влияния строения молекул на масс-спектр могут слу-
жить, например, изомерные нитроанилины. В области высоких мас-
совых чисел масс-спектров пара- и яега-нитроанилина характерно
появление иона (М—16)+, т. е. иона, который на 16 относительных
массовых единиц меньше массы молекулярного иона. В то же вре-
мя в спектре орто-нитроанилина имеется значительный пик для ио-
на (М—17)+.
Экспериментальные данные для многих классов соединений мо-
гут быть описаны на основе ряда закономерностей фрагментации.
Общая строгая теория распада молекулярного иона, к сожалению,
не разработана. Для описания фрагментации в каждом случае тре-
буется знание электронных состояний ионов различных типов и ве-
роятностей переходов между этими состояниями. Качественные по-
луэмпирические представления о направлениях фрагментации ис-
пользуют принципы сохранения структуры молекулы при ее иони-
зации и минимума структурных изменений в результате разрыва
связей при фрагментации. Например, появление иона С6Н5СО+
в спектре эфира бензойной кислоты С6Н5СООСНз объясняют про-
стым разрывом связи С—О.
Перегруппировочные ионы. Однако такие простые представле-
ния о диссоциативной ионизации наталкиваются на определенные
трудности в связи с фактами, показывающими, что процесс иониза-
ции сопровождается значительными перестройками в молекуле.
Так, в масс-спектре оксида таллия, имеющего химическое стпоение
Т1+ +Т1 присутствует ион Th и отсутствует ион Т1О+.
Ионизация молекулы и фрагментация образовавшегося молеку-
лярного иона, как установлено в довольно большом числе случаев,
сопровождается разрывом одних и образованием других связей.
Так, классическим примером перегруппировочного иона является
ион тропилия, который образуется по схеме
Этот же ион наблюдается во многих производных толуола. Цикли-
еское строение, приводящее к равноценности всех атомов углеро-
да доказано с помощью изотопного замещения.
Д ’ Церегруппировка атомов, не включающих атом водорода, назы-
вается скелетной перегруппировкой. Примером перегруппировки с
участием атома водорода может служить фрагментация иона нео-
пентана.
СН3 -1
I
СН3-С-СН3
сн3 _
-»[СН3СН2] +
Представления о механизме образования перегруппировочных ио-
нов основаны на различных допущениях, требующих более строгого
теоретического и экспериментального подтверждения.
Метастабильные ионы. В процессе ионизации образуются неус-
тойчивые (метастабильные) ионы. Если время распада иона состав-
ляет ~10 + с, то это близко к времени нахождения иона в камере
масс-спектрометра на пути от ионного источника до анализатора.
В этом эксперименте будут регистрироваться ионы распада. Однако
пики этих ионов в масс-спектре имеют диффузный характер. Пояс-
нения даются ниже при описании схемы эксперимента.
Отрицательные ионы. Они образуются в результате:
резонансного захвата электрона:
АВН-е--> АВ~
диссоцнативного резонансного захвата:
АВ-]-е-—А- + В-
ион- молекулярной реакции:
AB-I-C-—ABC-
при распаде молекулы на пару ионов:
АВ^А~4-В+
Резонансный захват электрона можно определить как присое-
динение к молекуле электрона, обладающего энергией в узком ин-
тервале значений, соответствующих устойчивости отрицательного
иона (до нескольких электрон-вольт).
Вероятность образования отрицательных ионов при электронном
Ударе очень мала и составляет примерно ~ Ю 7 на одно столкно-
вение. Учитывая,- что вероятность образования положительных ио-
нов значительно выше (~10~4), имеем различие в вероятностях
образования отрицательных и положительных ионов в несколько
порядков. Масс-спектр отрицательных ионов много беднее, чем по-
ложительных, но может дать важную информацию о молекулах.
Так, масс-спектр отрицательных ионов значительно более чувстви-
телен к строению молекулы.
Многозарядные ионы. В экспериментальных условиях вероят-
ность получения многозарядных ионов невелика, хотя для некото-
рых классов соединений, таких, как ароматические, образование
двухзарядных ионов представляет довольно частое явление. Усло-
вием стабилизации двухзарядного иона является максимальное раз-
деление зарядов, которое происходит, например, в молекулах кон-
денсированных ароматических соединений и т. п. Разделение заря-
дов показывают следующим образом:
C6H5N—-------Si(CH3)2
или
(CH,),Si------NC6H5
Двухзарядные ионы также могут претерпевать фрагментацию.
3. МЕТОДЫ ИОНИЗАЦИИ
Ионизация молекул должна проводиться в таких условиях, при
которых образовавшийся ион вне зависимости от метода иониза-
ции не претерпевал бы никаких столкновений с другими молекула-
ми или ионами. Это необходимо для установления взаимосвязи
между свойствами иона и молекулы. Экспериментально поток мо-
лекул без столкновений можно получить при молекулярном течении
газа и в молекулярном пучке.
Одним из основных условий молекулярного течения является
истечение газа (или пара) через отверстие, диаметр d которого зна-
чительно меньше длины свободного пробега молекул Хм, т. е.
или 3(W^/.m. Поскольку длина свободного пробега обратно про-
порциональна давлению, для оптимальных условий работы, при ко-
торых диаметр отверстия изменяется в пределах от нескольких мик-
рометров до десятых долей миллиметра, давление газа составляет
не более 10 Па.
Ионизация может проводиться различными методами.
Метод ионизации электронным ударом. Это наиболее распрост-
раненный метод получения ионов в связи с простотой и доступно-
стью источников электронов и их высокой эффективностью. Энер-
гия ионизирующих электронов должна превышать энергию иони-
зации молекулы (порядка 10 эВ). Обычно используют электроны
с энергией 50 ... 100 эВ, так как для этого интервала энергий вероят-
ность ионизации многих молекул разных классов соединений имеет
максимальное значение.
Число ионов, образующихся в единицу времени при ионизации
электронным ударом, определяет ионный ток, уравнение для
которого имеет вид
Ij=Ienj<3jl, (1.3)
где Л — ионный ток ионов типа /; 1е — электронный ток; п,— число
ионизируемых атомов или молекул типа j в единице объема; I —
длина пути электронов в ионизируемом газе; о/— сечение ио-
низации молекулы, зависящее от энергии электронов в ио-
низирующем пучке.
Вероятность ионизации молекул одним электроном на пути I
равна:
= (1.4)
' е
Из соотношения (1.4) следует, что размерность о,- равна площади,
т. е. L2. Следовательно, упрощенно, чем больше размеры атомов и
молекул, тем больше о,. Поскольку ионизация может привести к
образованию одно- и многозарядных ионов, то в общем случае се-
чение ионизации является суммарным, хотя, конечно, преобладаю-
щим будет сечение однозарядной ионизации. При диссоциативной
ионизации ток осколочных ионов типа t из молекул типа / будет
выражаться уравнением
I и — (1.5)
где оц — парциальное сечение диссоциативной ио-
низации.
Сечения ионизации атомов могут быть вычислены в относительно
хорошем приближении, а также измерены экспериментально. В на-
стоящее время расчет величин щ и вц для молекул представляет
значительные трудности. Упрощенным методом расчета молекуляр-
ных сечений ионизации является аддитивный метод:
амол = 2а*’ (L6>
k
где Ой — сечение ионизации атома k.
Однако ошибки такого расчета могут оказаться значительными.
Иногда рассчитанные значения отличаются от экспериментальных
в 2—3 раза.
Сечение ионизации характеризует вероятность ионизации моле-
кул и зависит от типа молекул и используемых энергий ионизирую-
щих электронов. Форма кривых зависимости сечений ионизации от
энергии электронов (кривые эффективности ионизации)
имеет сходный вид для различных молекул (рис. 1.2). Эта функция
близка к нулю в области энергии ионизации, затем достигает мак-
симума и снова уменьшается. Увеличение сечения ионизации с
Ростом энергии электронов объясняется увеличением вероятности
неупругого рассеяния, но дальнейшее повышение энергии электро-
нов уменьшает время взаимодействия их с электронами молекулы
и> как следствие, снижает вероятность ионизации. Минимальная
энергия электронов, при которой появляется ион, называется потен-
циалом появления иона и обозначается ПП. Она соответствует точ-
ке пересечения кривой o(V) с осью абсцисс. Максимум o(V) для
многих молекул находится в области 70 В. Для ионизации исполь-
зуют ускоряющие напряжения от 5 до 100 В. При низких энергиях
электронов, близких к потенциалу ионизации молекулы, масс-
спектр содержит в основном молекулярный ион. Увеличение энер-
гии электронов приводит к диссоциативной ионизации и к относи-
тельному уменьшению выхода молекулярных ионов.
Рис. 1.3. Принципиальная
схема ионного источ-
ника:
Рис. 1.2. Зависимость сечения
ионизации от ускоряющего по-
тенциала для ионизирующих
электронов
1 — напускной канал; 2 —
ионизационная камера; 3 —
электронная пушка; 4 — вы-
тягивающая линза; 5 — фо-
кусирующая линза; 6 — ион-
ный пучок
Схема ионного источника дана на рис. 1.3. Газообразные и лег-
колетучие вещества поступают в источник из системы напуска.
Труднолетучие и термически неустойчивые вещества испаряют не-
посредственно в источнике и в виде молекулярного пучка на-
правляют в ионизационную камеру. Положительные ионы,
образовавшиеся в ионизационной камере, вытягиваются и ускоря-
ются электрическим полем электродов, находящихся под напряже-
нием 1000... 3000 В. Давление в камере ~ 10~3 Па. Основным недо-
статком метода является неполная монохроматичность ионизирую-
щих электронов, что обусловливает смещение и отклонение от ли-
нейной кривой о(Г) в области потенциала ионизации или появле-
ния иона.
Метод электронного удара позволяет получать и отрицательные
ионы. В ионизационном источнике изменяют полярность ускоряю-
щего, вытягивающего и фокусирующего потенциалов. Интенсивность
отрицательных ионов на 3...4 порядка ниже, чем положитель-
ных.
Метод фотоионизации. Энергия ионизирующего излучения со-
ставляет 7... 15 эВ, длина волны заключена в интервале (800...
1200) 10~10 м. Зависимость эффективности ионизации от энергии
фотонов в области энергии ионизации носит ступенчатый характер,
что, безусловно, обеспечивает большую точность в оценке потенциа-
ла появления иона (рис. 1.4). Кривая интенсивности ионного тока
26
Рис. 1.4. Зависимость сечения
ионизации Оф(Е) от энергии
фотонов Е (очень схематично)
сильного
может иметь несколько ступеней, связанных с переходами на раз-
личные колебательные уровни иона, например, переходами 0—0.
0__1, о—2 и т. д. Принципиальная схема ионизационной камеры
такая же, как и при электронном ударе. Для получения монохро-
матического ионизирующего светового потока используют ультра-
фиолетовое излучение разряда в благородных газах и дифракцион-
ную решетку как монохроматор.
фотоионизационные источники ионов обладают более высокой
монохроматичностью излучения (до ~0,01 эВ), чем в методике
электронного удара. Масс-спектры с
использованием фотоионизации имеют
значительно меньшее число линий.
Преимущества этого метода реализу-
ются при преодолении больших техни-
ческих трудностей. Поэтому метод фо-
тоионизации не имеет еще столь ши-
рокого распространения, как иониза-
ция электронным ударом.
Ионизация электрическим полем.
Ионизация этим методом достигается
на электродах в виде острия или тон-
кой проволоки при градиенте поля
'~1О7...1О8 В/см. Под влиянием тако
поля происходит туннельный переход электрона от молекулы к
аноду за 10’12 с и образуется положительный ион-радикал, кото-
рый выталкивается этим полем. Обычно при ионизации электри-
ческим полем не происходит значительной фрагментации, и наб-
людают в основном молекулярные ионы. Увеличение напряжения
приводит к диссоциативной ионизации. Недостатками метода яв-
ляются низкое значение ионного тока и плохая воспроизводимость
масс-спектра.
Химическая ионизация. Этот вид ионизации осуществляется при
столкновении иона газа-реагента с исследуемой молекулой. Ионы
газа-реагента получают в ионизационной камере электронным уда-
ром. При химической ионизации также понижена фрагментация
молекулярного иона. В качестве газа реагента используют СН4,
СН3СН2СНз, (СНз)зСН. Из метана получают реактивные ионы
CH.-L и С2Н(", из пропана СгН^ и СдН/’ а из изобутана С3Н*
и С4Ну. Эти ионы являются сильными кислотами Льюиса. В ион-
молекулярных реакциях они либо присоединяют протон к молекуле
с образованием иона (М-(-Н)+, либо отщепляют гидрид-ион с
образованием иона (М—Н)+. Имеются технические трудности при
создании значительного перепада давлений в ионизационной камере
и вне ее.
Поверхностная ионизация. Ионный поток можно получить путем
эмиссии положительных ионов с поверхности, нагретой до высоких
температур. В качестве «рабочего» металла (материал для нагре-
ва) обычно используют вольфрам или оксидированный вольфрам.
Температурная зависимость ионных токов в этом методе позволяет!
определять потенциалы ионизации атомов, молекул и радикалов..
Комбинированные методы ионизации. Для увеличения эффек->
тивности исследования различных молекулярных систем конструи-
руют источники, сочетающие два вида ионизации: электронным
ударом и электрическим полем или электронным ударом и химиче-
ской ионизацией. Спектры, полученные двумя разными методами,
могут оказаться более информативными в структурных исследова-
ниях.
В случае малолетучих соединений, которые невозможно переве-
сти в пар при нагревании из-за разложения, например сложные
органические соединения, используют дополнительные методы ио-
низации или электрическим полем с высоким градиентом около
поверхности исследуемого вещества (полевая десорбция), или вто-
ричной ионизацией. В последнем методе потоки первичных ионов,
например Аг+, направляются на вещество, нанесенное на чистую
поверхность серебряной пластинки. Эта поверхность является ис-
точником вторичных ионов изучаемого вещества.
4. ПРИНЦИПИАЛЬНЫЕ СХЕМЫ МАСС-СПЕКТРОМЕТРОВ
Разделение и регистрация ионов осуществляются несколькими
путями. Использование для регистрации фотопластинок отличает
масс-спектрограф от масс-спектрометров, в которых ионные токи
измеряются электрическими методами. Масс-спектрографы приме-
няют для точного определения относительных атомных масс (Астон,
1919). Широкое использование в химии имеют масс-спектрометры,;
так как позволяют с большей точностью определять отношение ион-;
ных токов. Используются два класса масс-спектрометров: статиче-
ские и динамические. В первом типе масс-спектрометров для раз-;
деления и фокусировки ионов применяют статические электрические
или магнитные поля, а во втором типе—переменные электрические
поля.
4.1. Магнитный масс-спектрометр
Принципы разделения ионов. Первый магнитный масс-спектро-
метр был сконструирован в 1918 г. Демпстером. Схема его исполь-
зуется и в современных приборах (рис. 1.5). В ионном источнике
формируется пучок моноэнергетических ионов в поле ускоряющего
напряжения V с энергией:
2
где v — скорость иона; т — его масса; е — единичный заряд иона.
Направленный перпендикулярно магнитному полю пучок ионов
испытывает действие силы Лоренца F.
F = e[vxB| (1.8)
где В — индукция магнитного поля.
В магнитном поле ионы движутся по окружностям разного ра-
диуса. Для положительных ионов это движение подчиняется пра-
вилу левой руки: ладонь направлена вдоль v и в нее входят маг-
нитные силовые линии В; большой палец показывает направление
силы F. В связи с взаимной перпендикулярностью векторов уравне-
ние для силы имеет простой вид согласно уравнению (1.9):
F = evB. (1.9)
Динамическое выражение силы Лоренца можно приравнять цен-
тростремительной силе в кинемати-
ческой форме:
mv2lr = evB, (1-10)
или
r = mv!(eB), (1.11)
где г — радиус кривизны траектории.
В постоянном магнитном полеВ
не изменяется г и, следовательно,
траектория движения иона являет-
ся окружностью. Комбинируя урав-
нения (1.7) и (I.H), исключаем v
и получаем для однозарядных ионов
Рис. 1.5. Принципиальная схема
масс-спектрометра:
ИИ — ионный источник; Д — детектор'
ионов; Si — выходная и $2 — входная
щели; В— магнитное поле, направлен-
ное перпендикулярно плоскости рисун-
ка; О], О, Ог — центры и и, г, г2 — ра-
диусы окружностей, по которым дви-
жутся ионы м 1 и М 2
Для заданных величин г, В и V можно измерить ток ионов
массой т. Изменяя ускоряющий потенциал V (электростатическая
развертка) или индукцию магнитного поля В (магнитная разверт-
ка), получают ионный масс-спектр. Более широко применяют маг-
нитную развертку. Эксперименты по изучению зависимости ионного-
тока от энергии ионизирующих электронов Е проводят при фикси-
рованных В и V.
Метастабильный ион Мо" диссоциирует на пути до входа в ана-
лизатор на две частицы: ион и нейтральную частицу (Мо—MJ,
т. е.
М? = М1Ь + (МО —МД (1.13)
Предполагают, что в этом процессе выделяется незначительное ко-
личество внутренней энергии. Это означает, что первоначальный ион
Диссоциирует так, что осколки продолжают двигаться с той же ско-
ростью V, ЧТО И ИОН Мо-, т. е.
= (Wo-W1)t,2
2 2 "Г 2 ’ V • /
гДе m0, nil — массы ионов Мо- и Mf.
Следовательно, кинетическая"энергия иона М* может быть вы-
ражена через кинетическую энергию иона М* как — eV. Радиус
т0
окружности, по которой движется ион Мо", непосредственно связан
с массой и энергией иона выражением, вытекающим из уравнения
(1.12):
г0 = 1<2^. (1.15)
еВ
Для осколочного иона М* имеем по аналогии
тр
(1.16)
(1.17)|
или из уравнения (1.12)
2
r2B2 _ т* mi
2V е ето
Из уравнения (1.17) следует, что осколочный ион появится на плос- |
кости коллектора в том же месте, где и нормальный ион с эффек- I
тивной массой т* — тутй. |
Так, в масс-спектре «-бутана наблюдают диффузный пик с от- |
носительной молекулярной массой 31,9, что объясняют процессом:
СМ-СзН^ + СНз I
58 43 15 i
так как 432/58 = 31,9. •
Характеристики масс-спектрометра. Важными характеристика- j
ми масс-спектрометров являются: качество фокусировки п у ч • |
ка ионов, разрешающая сила и чувствительность.)
Магнитное поле обладает особенностью осуществлять фоку-)
сировку ионов по направлению. На рис. 1.6 представле-)
на идеальная картина выхода ионов из ионного источника через
щель s в направлении, параллельном ускоряющему электрическому
полю. Часть ионов вылетает под некоторым небольшим углом к на-
правлению поля, т. е. имеет место расходимость пучка ионов. Так,
ион, вылетевший вертикально вверх, опишет полуокружность диа-
метром SA = ^mv-=2r. Другой ион, вылетевший под малым углом
еВ
а к направлению первого, будет двигаться по окружности, которая
пересечет окружность для первого иона в точке С, а диаметр SX —
в точке В. Таким образом, полная фокусировка по направлению осу-
ществляется в точке С. Но на линии SO расхождение траекторий
невелико. Из рисунка видно, что AB = SA—SB = 2r(l—cos а) «rd2.
Отрезок АВ определяет ошибку фокусировки, составляющую малое
значение из-за квадратичной зависимости от а при малых а (по-
рядка 1... 2°). Формирование изображения источника и уменьшение )
расходимости потока ионов в магнитном поле называется фокуси-
ровкой по направлению. Хорошая фокусировка важна для увели-
чения чувствительности и разрешающей способности (силы) при-
Разрешающая сила R спектрометра определяет возможность
азделить два соседних пика — для ионов с наибольшей массой m
и массой т + Апг — и выражается уравнением
= =,
(1.18)
где Sj — ширина выходной щели; s2—ширина
уширение ионного пучка из-за несовершенства
неоднородности магнитного поля, не-
стабильности V и других причин).
Так, например, разрешение 250 оз-
начает, что два одинаковых пика для
ионов с относительными массами 251
и 250 разделены на ленте самописца
и в минимуме между ними интенсив-
ность тока не превышает 10% полно-
го ионного тока. Радиус кривизны г
входной щели; б-—
фокусировки (из-за
в различных приборах составляет от
0,10 до нескольких метров и, конечно,
определяет размеры прибора.
Приборы относятся к классу с
низким разрешением, если R не пре-
вышает 2000, и с высоким, если R бо-
лее 10 000. Разрешающая сила масс-
Рис. 1.6. Фокусировка по на-
правлению в магнитном поле
масс-спектрометра:
три окружности радиуса г пересе-
каются в S; ±а — углы вылета ио-
нов из щели S; SO=OA = r; SB —
=SD cos a
спектрометров существенно зависит от
вида и качества фокусировки.
Для уменьшения влияния магнитного поля на ионы в ионном
источнике и коллекторе ионов широко используют масс-спектро-
метры секторного типа. В этих приборах магнитное поле создается
между полюсными башмаками секторной формы, которые могут
иметь любой секторный угол. В приборе Демпстера этот угол со-
ставляет 180°. На практике часто используется секторный угол, рав-
ный 90° (рис. 1.7). Было показано, что если расходящийся пучок
ионов входит и выходит из однородного магнитного поля перпен-
дикулярно к его границам, то он фокусируется на прямой, прохо-
дящей через выходную щель ионного источника и центр кривизны
траекторий ионов, который совпадает с вершиной сектора. На
рис. 1.7 показана фокусировка по направлению. Разрешающая спо-
собность не зависит от секторного угла. В секторных спектрометрах
уменьшены размеры магнита и ионный источник и коллектор вы-
несены из магнитного поля. Правда, это приводит к увеличению пу-
ти ионов, что обусловливает эффекты рассеяния иона на этом пути.
Масса магнита пропорциональна г3. Поэтому с секторным магнитом
можно достигнуть значительного разрешения при меньшей массе
магнита.
Существенным недостатком магнитной фокусировки и разделе-
ния ионов является невозможность достигнуть максимального раз-
решения спектра из-за разброса ионов по энергиям. Для увеличе-
ния разрешения масс-спектра применяют двойную фокусировку
(рис. 1.8). Ионы из ионного источника проходят через цилиндри-
ческий конденсатор с радиальным электрическим полем, в котором
происходит фильтрация ионов по энергии и фокусировка по направ-
лению для ионов одинаковой энергии. Затем пучок ионов входит в
поперечное магнитное поле, на выходе из которого получают масс-
спектр. Двойная фокусировка увеличивает разрешающую способ-
ность практически на порядок. Так, только с одним магнитным ана-
Рис. 1.7. Масс-спектрометр сек-
торного типа:
Si — выходная и $2 — входная щели;
В — секторное магнитное поле; <р=
— 90°, О — центр сектора
Рис. 1.8. Масс-спектрометр с
двойной фокусировкой:
ИИ — ионный источник; А — элек-
ростатический анализатор; В — маг-
нитный анализатор; Д — детектор
ионов
лизатором разрешение составляет в различных приборах от 300
до 3000. Двойная фокусировка увеличивает разрешение до 30 000,
а в специальных приборах — до 10е.
Чувствительностью прибора называют минимальное определяе-
мое давление паров изучаемого вещества или минимальную массу
этого вещества. Для создания условий образования ионов и их раз-
деления требуются малые давления паров веществ и высокий ва-
куум в спектрометре (до 10~6 Па), что определяет малые ионные
токи (10—8... 10-14 А), регистрируемые на коллекторе, и малые рас-
ходы исследуемого вещества. Чувствительность масс-спектрометров
достигает 10~14 Па по давлению или при определении микропри-
месей до 10-7% (более реально 10~3... 10-4%). Для получения масс-
спектра достаточно нескольких микро- или нанограммов вещества.
Результаты масс-спектрометрического эксперимента представ-
ляют в виде графика зависимости ионного тока от m/е (рис. 1.9)
либо в виде таблиц (табл. 1.1), в которых за 100% принят пик для
максимального ионного тока. В общем случае удается без труда
обнаружить пик с интенсивностью, равной 1% от суммарного ион-
ного тока.
При получении масс-спектра отрицательных ионов методика
работы такая же, за исключением изменения полярности электро-
магнита и ускоряющих потенциалов в ионном источнике.
Новые возможности перед масс-спектрометрией открылись при
совместном использовании масс-спектрометра и хроматографа и
созданием таким образом хроматом асс-спектром етр ни.
Таблица 1.1.Масс-спектр паров NaF (1000К)
—-— 1 Иои Массовое * числю Относи- тельная ин- тенсивность, % Ион Массовое * число Относи- тельная ин* твнсивность, %
F+ 19 1 NaF+ 42 5
23 100 Na2F+ 65 30
* Целочисленное значение относительной молекулярной массы
Основная задача, решаемая этим
фикация и количественный анализ
трометры имеют широ-
кое применение в различ- 7Л"
ных областях науки и
техники, однако они име-
ют и определенные недо- g0 _
статки, в частности отно-
сительно низкую скорость 60 _
измерений, большие га- ф|_
бариты, массу и ряд дру-
гих. 20 -
4.2. Динамические масс- о
спектрометры
методом,— разделение, иденти-
веществ. Магнитные масс-спек-
N;iF(i)^.NaF(n)
В настоящее время
разработаны методы по-
лучения масс-спектров в
Рис. 1.9. Масс-спектр паров фторида натрия
переменных электрических полях. Рас-
смотрим два типа динамических масс-спектрометров.
Времяпролетный масс-спектрометр. Принципиальная схема при-
боров относительно проста (рис. 1.10). Ионный источник испускает
короткие импульсы ионов, поскольку электронная пушка работает
в импульсном режиме (несколько микросекунд). Если все ионы
начали движение из источника в момент ускоряющего импульса
0,01 мкс, то все они приобрели одинаковую энергию eV. Из равен-
ства mv2/2 = eV следует, что скорость иона v= 1/. Тогда вре-
мя движения через участок дрейфа L равно:
/= —
v
(1.19)
2еУ ’
т. е. ионы разной массы пролетают участок дрейфа с разным вре-
менем. Если детектор делается чувствительным только на короткий
промежуток времени, то точное измерение времени между момен-
том активации источника и отпиранием детектора дает информа-
цию о массе регистрируемых ионов. В некоторых схемах подклю-
чают осциллограф, на экране которого можно наблюдать последо-
вательность пиков (массовый спектр) с амплитудами, пропорцио-
2—949 33
нальными числу ионов с определенной массой т в потоке. Период
следования импульсов значительно превышает время пролета са-
мых тяжелых ионов, что позволяет избежать наложения спектра
от различных импульсов. Весь спектр можно получить за 10~3 с.
Диапазон массовых чисел
им
практически неограничен.
Существенным недостатком
этой методики было огра-
ниченное разрешение. В на-
Рис. 1.10. Схема времяпролетного масс-
спектрометра:
стоящее время имеются
приборы с разрешением в
условно показано разделение нонов с массами
и ЛЬ; L — участок дрейфа; ИИ—ионный источ-
ник; Д -- детектор ионов
несколько тысяч.
Квадрупольный масс-
спектрометр. На четыре
М, Ml
парно
оси х
подается переменное
и —(J7-j-Vocos tai) на
электрода квадруполя по-
напряжение в форме П-}-V0cosco£ на
оси у (рис. 1.11). Принцип работы
такого анализатора основан на том, что при прохождении ионов
через квадруполь часть ионов может иметь ограниченную ампли-
туду колебаний, в то время как амплитуда колебаний другой ча-
Рис. 1.11. Схема квадрупольного масс-спектрометра:
а — вид в плоскости ху, б — вид вдоль оси г; ИИ — ионный источник; Д —
детектор ионов
сти ионов неограниченно возрастает со временем. Первая часть
ионов достигает детектора, а вторая часть нейтрализуется на по-
верхностях электродов. Режим работы анализатора выбирают
так, чтобы он действовал как фильтр масс.
Прибор может работать при относительно высоких давлениях
газа до 0,1 Па. Все это обусловлено тем, что уравнения движения
иона в квадруполе задаются такими параметрами, как U, Уо, <м, Пь
е и т, но не содержат энергию иона. Разрешающую способность
квадрупольного масс-спектрометра (до нескольких тысяч) в сущест-
венной степени определяет точность изготовления электродов и их
установки порядка микрометра. Длина квадруполя составляет от
0,2 до 1,0 м.
4.3. Спектрометр ион-циклотронного резонанса
В магнитном поле, перпендикулярном траектории иона, послед-
ний движется по окружности (уравнение 1.10). При этом период
обращения Т не зависит от энергии частицы, так как
Т = 2лг/ф. (1-20)
Подставляя выражение г из уравнения 1.10, получаем
Т==2пшКеВ). (1-21)
Рис. 1.12. Схема спектрометра ион-циклотронного резо-
нанса:
А/ r S —полюса магнита; ИИ — ионный источник, Д—детек-
тор ионов; А — анализатор; траектория иона показана в виде
спирали, а направление переменного электрического поля —
стрелкой
Частота вращения
шс = 2л/Т=еВ/пг.
(1.22)
Таким образом, для данного типа ионов период и частота зави-
сят только от индукции магнитного поля и массы ионов. В связи
с тем, что ионы из ионного источника вылетают под различными
углами к магнитному полю, их траектория имеет форму цилиндри-
ческой спирали (рис. 1.12). Это увеличивает время движения иона
в анализаторе до 5... 10 мс, в то время как при движении по пря-
мой это составляло бы доли микросекунды. Если теперь приложить
переменное электрическое поле перпендикулярно магнитному полю,
то на частотах «> = «с будет происходить поглощение энергии, так
как будет иметь место циклотронный эффект ускорения движения
иона с раскручиванием спирали. Функция поглощения энергии от
частоты дает масс-спектр.
Для спектрометра ион-циклотронного резонанса не требуется
низкое давление, поскольку частота циклотронного резонанса иона
не зависит от его скорости, случайные столкновения с молекулами
газа не влияют на определяемую частоту. Поэтому ион-циклотрон-
НЫЙ резонанс может быть использован для изучения реакций ионов
и нейтральных частиц.
Отрицательные ионы, образованные при столкновении с медлен-
ными электронами, движутся по спирали с противоположным на-
2 35
правлением вращения. Они также могут быть зарегистрированы при
частоте, соответствующей их массе.
Первый прибор ион-циклотронного резонанса был сконструиро-
ван в 1958 г.
ГЛАВА II
ПРИМЕНЕНИЕ МАСС-СПЕКТРОМЕТРИИ
1. ИДЕНТИФИКАЦИЯ И УСТАНОВЛЕНИЕ СТРОЕНИЯ ВЕЩЕСТВ
Точное значение относительной молекулярной массы, получен-
ной из измерения в масс-спектре тока молекулярных ионов, необ-
ходимо для установления молекулярной формулы вещества. В на-
стоящее время относительная молекулярная масса измеряется в
атомных единицах массы (а.е.м.), которая составляет ’/i2 массы
изотопа 12С = 12,000000. В этой шкале атом водорода Н имеет мас-
су 1,007825, а кислорода 16О— 15,994914. Таким образом, масса
атома, измеренная с высокой точностью, отличается от массового
числа. Так, для СОг и СзН8 оно равно 44, но их точные относитель-
ные молекулярные массы равны соответственно 43,989828 и
44,062600, т. е. разница составляет 0,072772 а.е.м. Масс-спектро-
метр с разрешением 1000 позволяет разделить пучки ионов СОз] и
СзН8+, когда они получаются одновременно. Значительно боль-
шее разрешение необходимо для разделения дублета СО+ и N? -ио-
нов, так как их массы равны соответственно 27,9949 и 28,0062 а.е.м.
На масс-спектрометре с разрешением в 100 000 однозначно опреде-
лена, например, молекулярная формула адреналина C9H13O3N.
Определение атомного состава по точному значению массы про-
водится с использованием таблиц точных масс для различных со-
отношений числа атомов С, Н, О и N как наиболее распространен-
ных элементов. Точное измерение масс не заменяет элементного
анализа. Оба метода взаимно дополняют друг друга. Элементный
анализ обеспечивает независимую проверку молекулярной форму-
лы и чистоты вещества.
При исследовании с невысоким разрешением дополнительно к
определению типа молекулярного иона с массовым числом М изме-
ряют пики для изотопных ионов, включающих более легкие или бо-
лее тяжелые изотопы (с массовыми числами М+1, М±2 и т. п.).
Одновременное присутствие нескольких изотопов в молекуле мало-
вероятно, так как естественная распространенность, например, бо-
лее тяжелых изотопов С, Н, О и N незначительна и отношение рас-
пространенностей мало: I3C : 12С= 1 • 10~2; 2Н : 1Н= 1,6-10-4; 15N:
:14N=4-10-3; 18О:17О:16О=2-103:4-10~4:1. Однако это отношение
для хлора 35С1:37С1 = 3:1 и для брома 79Вг:81Вг=1:1. Молекуляр-
ный ион с массовым числом М+1 включает все возможные ком-
бинации изотопов элементов молекулы, например 12Cw-i,3CHxOyN2;
“CwH^-iDOjzNz; 12CwHxI6Oy-i17ONz; !2CwHxOy14Nz_i,5N. Следова-
36
тельно, в масс-спектре наряду с ионом М+ будет присутствовать
ион (М+1) + с интенсивностью, пропорциональной распространен-
ности изотопов. Малая вероятность присутствия в молекуле двух,
например, тяжелых изотопов одного вида или различных атомов,
приводит к меньшей интенсивности пика для ионов (М + 2)+.
В широко используемых справочных таблицах приводятся обыч-
но соотношения интенсивностей пиков молекулярных ионов с мас-
совыми числами М+1 и М + 2. В качестве минимального значения
отношение I (М + 2) // (М) для экспериментального исследования
принято 0,01. Табл. II.1 иллюстрирует соотношение интенсивно-
стей пиков молекулярных ионов с различным содержанием изото-
пов. Использование естественного распределения изотопов при
анализе массы молекулярного иона ограничено в справочных таб-
лицах значением М»250 из-за малых различий в отношениях ин-
тенсивностей ионов М+, (М+1)+ и (М + 2)+ при значительном чис-
ле комбинаций атомов.
Таблица П.1. Отношение интенсивностей пиков ионов М+, (М+1)+ и (М + 2ф+
Массовое число М /(М+1)//(М)
М=15
NH 0,40 <0,01
СН3* 1,13 <0,01
М=30
NO* 0,42 0,20
N2H2 0,79 <0,01
СН2О 1,15 0,20
CHiN* 1,53 0,01 1
СгНе 2,26 0,01
М=31
NOH 0,44 0,20
N2Hs 0,81 <0,01
СН3О* 1,17 0,20
CHSN 1,54 <0,01
* Наиболее распространенные фрагментарные ионы.
При значительных энергиях ионизирующих электронов
(~70 эВ) в масс-спектре может практически отсутствовать моле-
кулярный ион. Тогда проводят эксперимент при уменьшении энер-
гии ионизирующих электронов или используют отрицательные ио-
ны, или изменяют метод ионизации, применяя химическую иониза-
цию или ионизацию неоднородным электрическим полем. Если ука-
занные приемы не эффективны, то проводят химическую модифика-
цию соединения, т. е. получают производное исходного вещества,
Молекулярный ион которого стабилен. Для этого вводят в молеку-
лу группировки с низкими потенциалами ионизации или удаляют
Из молекулы группу, дестабилизирующую молекулярный ион.
Установив молекулярную формулу вещества, можно определять
химическое строение молекулы. На основе брутто-формулы для ор-
ганического соединения вычисляют фактор непредельности, кото-
рый состоит в том, что, замещая галоген на СН3, а кислород или
серу на СН2 и азот на СН-группу, получают формулу углеводоро-
да. Сопоставление с СпН2п+2 дает число кратных связей или насы-
щенных циклов. Такая альтернатива разрешается анализом дан-
ных ИК, УФ и ЯМР спектроскопии.
Дальнейший ход расшифровки масс-спектра основывается на
закономерностях фрагментации. Спектр, содержащий много оско-
лочных ионов, интенсивность пиков которых увеличивается с
уменьшением т/е, как правило, соответствует алифатическому уг-
леводороду или его производному. Как уже указывалось, аромати-
ческие соединения имеют интенсивный пик молекулярного иона.
Главные осколочные ионы позволяют выяснить основные направ-
ления диссоциативной ионизации. Предполагаемый путь фрагмен-
тации проверяют анализом положений пиков метастабильных ио-
нов.
Примером установленных закономерностей фрагментации яв-
ляются следующие схемы разрыва связей.
1. В разветвленных углеводородах наиболее вероятен разрыв
связей у места ветвления:
сн3
сн,
2. Вероятность разрыва связи С—Н уменьшается с увеличением
длины цепи углеводорода.
3. В ароматических производных наиболее вероятен разрыв
P-связи с образованием перегруппировочного тропилиевого иона
С7Н7+:
«Рыболовный крючок» означает предполагаемый переход элект-
рона при фрагментации.
4. В масс-спектрах спиртов характерными осколочными иона-
ми являются ионы с массами 31 и М.—1, которые соответствует сле-
дующей схеме разрыва связей:
R-^-CH—
Предполагают, что при образовании иона происходит стабилизация
за счет образования кратной связи СН2 = О+Н (31) илиКСН = О+Н
(М—1)- Такие же схемы возможны для разветвленных спиртов.
5. Для аминов H2N—СН2—R характерен разрыв а-связи
h2n—сн2-у-с—с
с образованием иона NH2—СН2 (30). Значительным также явля-
ется пик H2N=CHR (М— 1).
X
6. Масс-спектр кетонов /С=О содержит ионы R!—С=Ог и
R/
R2—С = О+. Если алкильная цепь содержит три и более атомов
углерода, происходит разрыв p-связи и далее перегруппировка с
участием атома водорода:
В этом случае кроме иона образуется устойчивая нейтральная мо-
лекула олефина.
7. Металлорганические соединения обладают крайне низкой
энергией связи металл — углерод, что приводит к резкому умень-
шению интенсивности молекулярного иона в масс-спектре.
8. Алифатические фториды, хлориды и бромиды дают малую
интенсивность молекулярного ионного тока, но иодиды имеют зна-
чительный пик молекулярного иона.
Подобные и более детальные закономерности установлены для
многих классов соединений. Они позволяют в определенной мере
объяснить и предсказать фрагментацию молекулярных ионов и,
обратно, на основании масс-спектра установить химическое строе-
ние молекул. Трудность состоит в том, что отсутствуют расчеты
Фрагментации молекул и данные о строении ионов. Степень досто-
верности, с которой определенная структура может быть приписа-
на тем или иным ионам, весьма ограничена. Практически редко
Удается установить полную структуру соединения на основе толь-
ко масс-спектра. Наиболее эффективно совместное использование
нескольких физико-химических методов.
Широкое применение для установления структуры молекул (хи-
мического строения) масс-спектрометрия нашла в органической
кетмии, поскольку гомологические ряды позволяют выявить законов,
мерности, связывающие химическое строение и масс-спектр.
2. ОПРЕДЕЛЕНИЕ ПОТЕНЦИАЛОВ ИОНИЗАЦИИ
МОЛЕКУЛ И ПОЯВЛЕНИЯ ИОНОВ
Расчет энергий разрыва химических связей. В связи с тем, что
согласно принципу Франка — Кондона при ионизации переход от
молекулы к иону происходит без изменения геометрии ядерной
конфигурации, т. е. вертикально, то в результате в большинстве
случаев
так как
.1(E)
получают ион в возбужденном колебательном состоянии,
потенциальная кривая или поверхность иона несколько
сдвинута по отношению к нейтральной мо-
лекуле.
Потенциал ионизации, соответствующий
переходу с нулевого колебательного уров-
ня молекулы, на нулевой колебательный
уровень основного электронного состояния
Рис. ПЛ. Начальные
участки кривых иони-
зации для исследуе-
мого объекта и стан-
дартного вещества
(например, Аг):
ПИ (А) и ПИ (В) — по-
тенциалы ионизации мо-
лекул А и В
Т,эВ
ПН(А) ПШВ)
иона, называется адиабатическим потен-
циалом ионизации. В эксперименте при
ионизации электронным ударом в основ-
ном осуществляется вертикальный
переход на верхние колебательные
уровни с образованием возбужденного ио-
на. Следовательно, измерения приводят не
к адиабатическим, а к вертикальным
потенциалам ионизации, которые
всегда больше первых. Задача эксперимен-
татора — приблизить условия ионизации к
адиабатическим. Адиабатический потен-
циал ионизации может быть получен
спектроскопическим путем. Так, для Н2 адиабатический потенциал
ионизации равен 15,427 эВ, а вертикальный 16,07 эВ, т. е. разни-
ца составляет около 0,4 эВ.
Для сложных многоатомных молекул потенциальные поверхно-
сти многомерны, однако для качественной характеристики процес-
сов ионизации модель двухмерных потенциальных кривых может
быть сохранена. Для этого удобно рассматривать многоатомную
молекулу как состоящую из двух частей — радикалов Ri и R2.
При определении потенциалов ионизации проводят регистрацию
ионного тока в зависимости от напряжения, ускоряющего ионизи-
рующие электроны, т. е. от их энергии Е. При этом получают так
называемые кривые эффективности ионизации 1(E),
соответствующие кривым о(Д) (рис. II.1). В связи с немонохрома-
тичностью ионизирующих электронов (разброс их по энергии до-
стигает 0,5...1,0 эВ) и распределением ионов по колебательно-вра-
щательным уровням энергии начальный участок кривой 1(E) име-
ет пологий подъем («хвост» кривой ионизации), а затем переходит
в линейную область. Поэтому экстраполяция линейного участка до
пересечения с осью абсцисс дает завышенные- значения потенциа-
лов ионизации, являющиеся их верхним пределом. Для увеличения
точности определения потенциалов ионизации методом...электронно-
го уДаРа применяют специальные методы, увеличивающие моно-
энергетичность электронов. Съемка кривых эффективности иони-
зации веществ, молекулы (или атомы) которых имеют известные
потенциалы ионизации и одинаковую линейную часть этих.кривых,
позволяет скорректировать определение потенциала ионизации ис-
следуемого вещества (рис. П.1). Использование такого стандарта
более надежно, если ионизационные потенциалы объекта и стан-
дарта имеют близкие значения. Достигаемая точность определения
составляет 0,1 эВ.
Фотоионизация приводит к повышению точности на порядок (до
~0,01 эВ), так как кривая ионизации имеет ступенчатый характер
(см. рис. 1.4).
Если использовать источник радикалов, например, при разло-
жении РЬ(СгН5)4 и т. п., то теми же методами определяют потен-
циалы ионизации радикалов.
При диссоциативной ионизации наблюдают осколочные ионы,
для которых могут быть получены кривые эффективности иониза-
ции. Для. этого проводят измерение ионного тока исследуемого ио-
на, варьируя Е. Потенциал появления иона (ПП) при диссоциатив>
ной ионизации молекулы на ион Ri' и радикал R2 запишется в виде
/7/7(Rf)=D(R1-R2)-|-^(R1)4-7'(R]b, R2) + E(Rf, R2), (Н.1)
где ПП (Ri')—потенциал появления иона R( = M—R2; D(Ri—R2) —-
энергия диссоциации молекулы R1R2; T'lRi’, R2)— кинетическая
энергия осколков Ri' и R2; Z?(Ri', R2) — энергия возбуждения Ri’
и R2.
Обычно считают, что потенциал появления иона соответствует
минимальной энергии возбуждения Е, которую принимают равной
нулю. Часто также не учитывается кинетическая энергия Ri+h R2»
которая составляет малое значение по сравнению с ПП и ПИ.
В более тщательных экспериментах Г (Ri') измеряется, например-,
методом отклоняющего поля. Зная кинетическую энергию T'(Ri')*
можно вычислить Т (R2) = — -^2’1 Т (Ri').
m (Ri)
Измерив ПП (Ri'), рассчитывают энергию диссоциации:
П^-^ — ПП^-ПИ^). (Д-2)
С другой стороны, это уравнение позволяет определять потен-
циал ионизации радикалов:
ПИ (Я^ПП (Ri+)-£)(R1-R2). (П.З)
Данные по значениям ПИ для ряда соединений позволяют про-
вести, например, следующее сопоставление.
Молекула ПИ, зВ Молекула ПИ, эВ Радикал ПИ, эВ
СНз—СН2—СНз 11,21 сн2=сн2 10,51 СНз 9,95
сн2=сн—сн3 9,73 сн2=сн—сн=сн2 9,06 СН2 = СН—СН2 8,16
СН^С—СНз сн2—сн2 сн2 10,36 10л06 сн2=сн—сн= =сн—сн=сн2 8,26 С6Н5-СН2 7,76
Значения ПИ уменьшаются для молекул, имеющих кратные
связи и сопряженные кратные связи, что согласуется с представле-
ниями об отрыве внешнего электрона с верхней занятой молеку-
лярной орбитали (МО). При наличии кратных связей и сопряжен-
ных связей уменьшается разность энергий между верхней связы-
вающей МО и разрыхляющей МО.
В алканах ПИ уменьшается с увеличением числа атомов угле-
рода от 12,98 эВ СН4 до 10,08 эВ н-гептана. Этот факт указывает
на то, что МО охватывает всю молекулу, а не локализована на
связи.
Найдена линейная корреляция между ПИ аминов и эфиров,
аминов и алкильных радикалов. Значения ПИ важны для исследо-
вания комплексов с переносом заряда, термохимических расчетов,
строения ионов и т. п.
Интересные и важные результаты получают при определении
энергий разрыва химических связей.
Молекула В, кДж/моль Молекула D, кДж/моль Молекула 1 । D, кДж/моль
СНз—Н 428 СНз—С1 338 h2n—nh2 252
С2Н5—н 407 СН2 = СНСН2—С1 254 o2n—no2 57
СН2=СНСН2—н 323 С6Н5СН2—С1 254 f2n—nf2 80
С6Н5—н .428 (С6Н5)зС-С1 202
Значительное уменьшение энергии разрыва связей С—Н и С—С1 в
аллильных и бензильных производных подтверждает представле-
ние о сопряжении в соответствующих радикалах. Энергия разрыва
N—N-связи также зависит от заместителей и валентного состояния
атома N.
В отличие от средней энергии связей, характеризующих энер-
гию атомизации молекулы, энергии разрыва связей изменяются по
мере отрыва атомов. Так, Р(НО—Н) =495,6 кДж/моль, но
42
D(0—H) =425,0 кДж/моль, при средней энергии связи £ (О—Н) =
=461 кДж/моль.
С точки зрения теории химического строения большой интерес
представляют данные для энергий диссоциации двухатомных мо-
лекул и ионов элементов первой группы. Так, Z?(H2)^>jD(H2’)(433,9
и 256,6 кДж/моль), что соответствует ослаблению двухцентровой
связи при удалении одного спаренного электрона. Однако простая
концепция спаренных электронов, осуществляющих связь, не со-
гласуется с данными для других молекул и ионов:
Молекула..................L,j2 LiJ N’a2 Na.j" K3 Kf
О, кДж/моль.............. 79,5 121,8 71,4 94,5 53,8 88
3. МАСС-СПЕКТРАЛЬНЫЕ ТЕРМОДИНАМИЧЕСКИЕ
ИССЛЕДОВАНИЯ
Определение парциального давления паров веществ. Если чис-
тый газ направляют в ионизационную камеру через капилляр, при-
чем скорость откачки и другие условия течения газа сохраняются
постоянными, то интенсивность ионного тока для какого-либо иона
в масс-спектре соединения пропорциональна давлению газа внут-
ри ионного источника, которое определяет частоту столкновений
при ионизации. Давление в ионном источнике в свою очередь про-
порционально давлению пара (газа) в системе напуска, причем мо-
лекулярное истечение газа не меняет этого давления.
В соответствии с уравнением (1.3) для ионного тока можно вы-
разить nf через Pj — парциальное давление в напускной системе —
следующим образом. Интенсивность пучка молекул при молеку-
лярном течении через отверстие с малой площадью 6S определяет-
ся соотношением
= (II.4)
где n°j —число молекул в единице объема напускной системы;
— средняя скорость молекул.
В ионном источнике п, пропорциональна т. е. отношению
где k — постоянная Больцмана; Т — абсолютная температура;
ni~PjlkT — уравнение идеального газа. Из уравнения
(1-3) получаем прямую зависимость // от рр
Ii=HiIe°jl=K’ , (II.6)
где /£— приборная постоянная.
Из уравнения (II.6) получим выражение для р,:
Р)=К^Т=*-1,Т,
И
(П.7)
где K.j — коэффициент чувствительности для ионов типа /; К— кон-
станта чувствительности прибора.
Парциальное давление вещества можно определять, измеряя
f.ij. При этом уравнение (II.7) запишется в виде
aij
(П. 8)
где Кц — коэффициент чувствительности; вц— сечение ионизации
молекулы типа / с образованием ионов типа i, т. е. парциальное
сечение диссоциативной ионизации. Если измеряют интенсивность
всех пиков масс-спектра, то
р. = А_Г2/,7. (II.9)
Постоянную пропорциональности легко определить, используя
стандартное вещество с известным давлением и сечением иониза-
ции, например кадмий, серебро, золото, платина и др.
Определение теплоты сублимации. Зависимость давления от
температуры позволяет рассчитать теплоты сублимации &HSj, если
воспользоваться уравнением Клаузиуса — Клапейрона и уравне-
нием (II.7):
кН si кН si
= или ln/7T = -^+C. (И. 10)
Так, графит при 7=2400 К имеет давление около 4-10~3Па. При
этом в парах присутствуют атомы углерода (Cj) и молекулы Сг и
Сз. Соответственно найдены теплоты сублимации AHsi = 726;
A7/s2 = 85I и A//s3 = 803 кДж/моль.
Определение константы равновесия химической реакции и теп-
лоты реакции. Общее выражение для константы равновесия КР мо-
жет быть преобразовано следующим образом:
Пр"1 Пк"^1
is 1 'рЪп1—Ъп.] i
р Пр? ПК?/? ’
J J
где t — продукты реакции; /— исходные вещества; КР — безразмер-
ная функция.
Введем величину Ki и выразим ее через КР'.
П П кр
К,= = const Кр Т*пГ*п1 (II. 12)
П // П к"1
j i
или
1п^/=1п/<р+(2п; —S^pinr-j-B, (11.13)
где В — постоянная величина.
Зависимость Ki от теплоты реакции &Н выражается уравне-
нием
ЬН°Г
ln/Q =---^+С + (2^-Хцг)1пГ. (11.14)
к/ I
Таким образом, при измерении Ki в относительно узком интер-
вале температур, предполагая величины Д//?и С постоянными,
можно определить теплоту реакции.
Если определены парциальные давления при температуре Т
и найдена Кр, а также рассчитано изменение энтропии процесса
по третьему закону термодинамики, можно также вычислить Д//г
реакции на основе уравнения
-RTlnKp = ^H°T-T^S°T (П.15)
или
kHr=TbSr-RT\nKp. (П.16)
В отличие от органической химии в области неорганической хи-
мии масс-спектрометрия используется в основном (исключая ана-
литические применения) для термодинамических исследований.
Этот метод дал возможность изучать важнейшие процессы, проис-
ходящие при испарении веществ. Так, при исследовании системы
La—La2O3 оказалось, например, что пар содержит La и LaO. Из
масс-спектра определены AHs(La), A/7s(LaO) и P(LaO). В систе-
ме Ва—ВаО в парах найдены молекулы ВаО, Ва2О, Ва2О2, Ва2О3.
Расшифровка масс-спектра. При сложном составе пара систе-
мы, в которой имеет место химическое равновесие, возможно вза-
имное наложение масс-спектров отдельных компонентов пара за
счет диссоциативной ионизации. Такое наложение практически ис-
ключает возможность определения чувствительности прибора по
отношению к компонентам пара с меньшей относительной молеку-
лярной массой. Например, в парах хлорида натрия присутствуют
молекулы NaCl, Na2Cl2 и Na3Cl3. Ионные токи Na+ и NaCl+ обус-
ловлены фрагментацией всех трех молекул. Задача расшифровки
Масс-спектра состоит в следующем:
1) определить молекулярный предшественник иона;
2) найти парциальный ионный ток и рассчитать 7y = V/0;
3) определить коэффициенты чувствительности К/ или K.ij.
Решение этой задачи позволяет провести термодинамическое ис-
следование системы. При изучении газов или относительно легко-
летучих веществ используют внешние напускные системы. Для
исследования труднолетучих веществ, требующих нагревания до
высоких температур, применяют специальные камеры.
Испарители с эффузионной ячейкой Кнудсена. Эффузионная
ячейка нашла широкое применение
для изучения термодинамического рав-
новесия конденсированная фаза — пар.
Она представляет собой изотермический
сосуд, в котором, несмотря на удаление
части вещества в процессе эффузии (ис-
течения), пар в камере остается насы-
щенным (рис. II.2). Эффузионное отвер-
стие имеет форму и размеры, обеспечи-
вающие молекулярный характер истече-
ния. Материал камеры не должен взаи-
модействовать с исследуемым вещест-
Рис. П.2. Испаритель с эф-
фузионной ячейкой Кнудсе-
на:
1 — ячейка с держателем; 2 —
экран; 3 — катоды-нагреватели
вом, если это не предполагается заранее.
Разработана специальная теория эф-
фузионного метода исследования при
давлениях вещества ~10 Па. Если про-
изводится полное изотермическое испа-
рение заданной навески вещества, то>
возможно определение коэффициентов чувствительности Кц из
уравнения Герца — Кнудсена:
(ПЛ7)
где П/ — количество вещества (моль), испарившегося за время Д
65 — площадь эффузионного отверстия; L — коэффициент Клау-
зинга, зависящий от формы отверстия и табулированный для раз-
ных форм отверстий; М, — молярная масса.
Уравнение (П.17) позволяет произвести калибровку прибора
без использования стандартного вещества. Далее, если опреде-
лить константу чувствительности прибора К по стандарту, зная К,
или можно рассчитать сечение ионизации молекулы о, или сц;,.
соответственно, по уравнениям (II.7) и (II.8).
Применение двухтемпературной двойной эффузионной камеры для одноком-
понентных систем. Если однокомпонентные системы содержат в парах кроме
мономеров димеры, тримеры и другие типы ассоциатов, то масс-спектр этих
систем содержит сложные пики, являющиеся результатом наложения масс-спект-
ров компонентов пара. Так, было показано, что пары галогенидов щелочных
металлов в значительной степени ассоциированы. Наибольшая степень ассоциа-
ции наблюдается для галогенидов Li, а наименьшая — для галогенидов Cs.
Определение состава пара, соотношения парциальных давлений и констант
равновесия возможно при использовании двойной эффузионной ячейки (рис.
II.3). Нижняя камера 1 соединена каналом 3 с верхней камерой 2. Диаметр сое-
динительного канала значительно больше диаметра эффузионного отверстия.
Этот канал позволяет задавать различные температуры камер (Т (1) и Т (2)
соответственно).
Рассмотрим относительно простой пример равновесия мономер — димер:
1/2D^=tM, для которого константа равновесия KP = pi/p12'z2, где Pi и р2 — парци-
альные давления мономера и димера. Если диаметр соедини-
тельного канала значительно больше длины свободного пробега,
то условие стационарности потока соответствует равенству дав-
лений в камерах 1 и 2:
Р1 (О + р2 (1) = Pi (2) + р2 (2).
При 7(1) =7(2) состав и давление пара в обеих камерах оди-
наковы, но при 7(2) >7(1) пар в нижней камере остается на-
сыщенным, а в верхней — перегретым, ненасыщенным. Степень
диссоциации при этом увеличивается. Эксперимент проводят для
двух вариантов соотношения температур камер 1 и 2.
Первый вариант эксперимента: 7(2)= const;
7(2)>Т(1); изменяется температура камеры 1. Если в масс-
спектре наблюдают две линии, одна из которых определяет, на-
пример, ионный ток димера 1г (базисная линия), а вторая
является двойной Ik = Iki+lk2, где Iki и Кг— токи ионов типа k
соответственно из мономера и димера, то для расшифровки
пика Ik достаточно получить масс-спектр системы при двух тем-
пературах первой камеры 7(1) и 7'(1). Тогда будем иметь си-
стему двух уравнений для Л и / й . Удобно эти уравнения вы-
2
Рис. П.З.
разить через коэффициенты масс-спектра, например
Поскольку 7(2)= const, то Лл2(7(1)) = аА2(7/(1))-
разом, получаем систему:
h ~ hi + akii2l
I к = hi + ak<zh-
Разделив на Z^2 и (Z2)1,/2, соответственно, находим
вм = /м//2. Схема двух-
Таким об- температур-
ной двойной
(II 18) ЭФФУЗИОГ
Iй* 1О/ нои ячей-
ки:
1 — нижняя
камера; 2—
верхняя ка-
ра; 3 — соеди-
(II ка-
4
Z’/2
— Ki +
и
7*
^i/2- = K/+^2 (hyp, (П.20)
где Ki = 7*i/7217“b камере 2 не зависит от 7(1).
Таким образом, получаем два уравнения с двумя неизвестными Ki и ай2-
•Остальные величины измеряются в эксперименте. Определение аьг приводит к
расшифровке двойной линии, так как
7^1 = h —
(П.21)
Проведение измерений при нескольких температурах второй камеры позволяет
Рассчитать теплоты димеризации и сублимации компонентов пара в соответствии
с уравнениями (11.10) и (11.14). Однако при этом не определено соотношение
Давлений мономера и димера в парах камеры 2.
Второй вариант эксперимента: 7(l)=const; 7(2)>7(1); изме-
няется температура камеры 2. Равенство давлений в камерах 1 и 2 будет сохра-
няться при Т' (2) =И=7 (2), т. е. при различных температурах камеры 2:
А(2)-Нр2(2) =л(2) + р2.(2). (11.22)
Выражения для парциальных давлений через ионные токи уравнения (II.7) »
(II.8) запишутся в виде системы равенств:
Р1(2) = ^17Л17’(2); р2(2) = /С2/2Г(2);
Pi (2) = KnIkiT' (2); р'2 (2) = K2l'2T' (2).
Используя систему из пяти уравнений, можно; найти искомое соотношение пар-
циальных давлений: - -
Р2(2) = 4
Pl (2) 41 Z27'(2)-/2r'(2) ’
Таким образом, расшифровка масс-спектра и определение относительного
состава проведены без привлечения .предположений о сечении ионизации. При
более сложном составе пара эксперимент осуществляют при нескольких темпера-
турах. Расчеты несколько изменяются для молекулярного (невязкого) течения
газа.
Интересным примером является исследование хлорида натрия при 1002 К,
Данные по масс-спектру приведены ниже.
Ион Na+ NaCl+ Na2Cl+ NasClJ
Массовое число 23 58 81 139
Относительная интенсивность 100,0 45,3 39,2 0,5
Потенциал появления, эВ . . 9,54 9,24 9,63 —
В результате расшифровки установлено, что в парах хлорида натрия содержат-
ся молекулы NaCl, Na2Cl2 и NasCls. Ранее не предполагали столь сложнога
состава пара. Проведены расчеты различных термодинамических характеристик,
хлорида натрия:
ДЯ„(NaCl) = 216,7 кДж/моль;
A/7S (Na2Cl2) = 227,6 кДж/моль;
Д/75 (Na3CI3) = 284 кДж/моль;
— = 0,36; — =0,01;
Pl Р1
Na2C!2-*2NaCl, ДЯу.= —197,5 кДж/моль;
Na3Cl3->Na2C’l2 + NaCl, Д//£ = — 155,4 кДж/моль.
Подобные исследования проведены для хлоридов лития и цезия. Для хло-
рида лития p2/|Oi = 3,5±0,3 и ps/pi = 0,32, а для хлорида цезия p2/pi = 0,06+0,02,
тримеры не обнаружены.
Применение метода изотермического испарения к двухкомпонеитным систе-
мам. Этот метод применяется для двухкомпонентных систем со сложным соста-
вом пара. Испарение сопровождается изменением состава конденсированной фа-
зы (т), т. е. протекает инконгруэнтно. Общий случай равновесия в такой .системе
можно записать в виде
[Я (А) +Я(В)](т)21[Я(А„Вт) + Я(А/гВ/)+ ...](г), (11.25)
где AnBm, AjB; и т. п. — молекулы комплексного соединения в парах; А (А),
А(В), A(AnBm) и т. д. — молярные доли соединений А, В, AnBm т. д. Рассмот-
рим простой случай равновесия в парах:
АВ^А + В (11.26)
Предположим, что при ионизации наблюдаются только ионы А+ и В+, т. е.
АВ + е— А+ + В ц- 2е~
А + е--*А+ 4- 2е— (11.27)
В + е~ ->В+ + 2е~
Следовательно, в масс-спектре будем иметь только два пика: / + = /д+ А 4-
+ /А+,АВ И Zb+ = Zb+,b’ ГДе Za+,a~tok ионов А+ из A’Za+,Ab~T0K ионоВ
А+ из АВ и I . —ток ионов В+ из В. Первой задачей данного эксперимента
является расшифровка сложного пика двойной линии. Методика эксперимента
состоит в испарении из эффузионной камеры Кнудсена при постоянной темпера-
туре навески известного начального состава в течение некоторого интервала вре-
мени i0—t. Основное требование к эксперименту сводится к тому, чтобы процесс
изотермического испарения протекал достаточно медленно. Это позволяет рас-
сматривать процесс испарения как последовательность равновесных состояний.
Одновременно регистрируется интенсивность ионного тока как функция времени
т. е. в данном примере/д+(/) и / +(/) (рис. II.4). Измерения заканчива-
ются, когда исчезает пик 7д+ в связи с полным испарением компонента А.
Для расшифровки пика / + удобно использовать
уравнение Гиббса — Дю-
гема, которое описывает равновесие двухкомпонентных систем:
d In Ра _ _2в_
d In рв ~ пк'
(П.28)
где Ра, рв — парциальные давления веществ А и В; пд и пв — количество веще-
ства А и В, моль.
Подставляя вместо рд и рв их выражения из уравнений (II.7) и (II.8), по-
лучаем
dlnZA+,A
d!nZB+
nB
«А ’
которое можно переписать в виде
d/A+,A dln/B+ пА а+,а (11.30)
Поскольку имеет место равновесие ^аРв и Кр — , то из уравнения (11.28)
Рав
d In Рав __ _ «в ~ ”а
dlnpB ид
Тогда по аналогии с уравнением (II.30), получим
dZA+,AB _ ИВ - ИА z
dlnZB+ ~ а+>ав‘
Сложив уравнения (11.30) и (11.32), найдем
А! . „
А+ __ _ "В
din7 . “ лА а+ + а+,ав’
ВТ А
(11.29)
(И.31)
(11.32)
(11.33)
откуда
nB
Za+,ab = '^”Za+ +
dZA+
dlnZB+ '
(11.34)
Если известно, что в точке to состав системы равен «д/пв, то измеренные зависи-
мости / + (/) и /в+ (t) дают возможность расшифровать пик в этой точке со-
гласно уравнению (11.34) в связи с тем, что
(11.35)
Za+,a Za+ Za+,ab'
Рис. 11.4. Зависимость
ионного тока 1А± и /в+
от времени:
п g— количество вещества
(моль) В в начальный мо-
мент времени fo
Рис. II.5. Зависимость
парциальных ионных то-
ков
'л+л’О-
и Zb+b
от времени как резуль-
тат расшифровки двой-
ной линии
Поскольку процесс испарения ведется изотермически, то константа равновесия
ле зависит от времени и является постоянной. Запишем это условие для Кг.
/*о /»о / f .
= a+,a- -
a+,ab za+,ab
Из уравнения (11.36) имеем
_ ,aZb+
Za+,ab“ /q
м, преобразовав
получим
Za+
Za+,ab(Z) zb+(0-
Следовательно,
i , (f)~i j. (r) — z . (Z).
A + AB k ’ a+- A + AB ’
Пик Z +(/) расшифрован (рис. II.5).
(11.36)
(И.37)
(11.38)
(11.39)
Теперь необходимо найти коэффициенты чувствительности ионов и выявить
зависимость парциальных давлений от состава конденсированной фазы. Для это-
го используют уравнение (11.17). Исходя из начального состава на основе за-
висимостей 1 , (t) и / . (^определяют количество вещества пА в момент
А , А А , Л О
/ по уравнению
«А = 4 “ °А/<а+ , A J ZA+ ,А (О - D^Ka+ ( ав J /а+ )АВ (О d t. (II. 40>
Подобное уравнение может быть написано для ив- Уравнение (11.40) позволяет
составить систему уравнений для расчета коэффициентов чувствительности, так.
как
dlnZA+,A
dln/B+
»В
«А
л°в-/)в/<в+рв+ (Odl
= -------------------t----------2------------;------------. (11.41)
"А - °А*а+ , A j ZA+ ,А &t ~ ^а+ , АВ j ZA + , АВ (О V
Зная Кд+ д, ав 11 ^в+> а также зависимости nA = nA(t) и Пв =
= Пв(0, получают функции:
^A = Z<a+,AZa+,A(ZVb)^ рв = /<в+7'в+;В(Лгв)7'; Рлв =
ZZA+,abZa+,Ab (jVb)7'>
выраженные через молярные доли Ав. Положение максимума кривой рлв(Ав)
(рис. II.6) определяет состав комплексной молекулы. В данном примере <VBaKC =
= 0,5. Для двухкомпонентных систем с более сложным составом пара задача
соответственно усложняется в связи с увеличением числа различных молекул
в паре. Возможна расшифровка не только двойных линий, но и тройных.
Этим методом изучены многие системы типа LiF-BeF2; LiF-AlFs; NaF-AlFs.
и др. В парах найдены такие молекулы, как LiBeF3, Li2BeF4, LiAlF4, NaAlF4„
(NaAlFi)2. Определены теплоты реакций комплексообразования.
Ион-молекулярные равновесия. Для ря-
да неорганических систем, включающих со-
единения щелочных элементов, можно дос-
тигнуть температур, при которых в газовой
фазе будет иметь место равновесие с участи-
ем ионов, т. е. ионизация происходит в ре-
зультате теплового возбуждения молекул:
АВ(т) = Д+(г) + В-(г)-|-АВ(г)+АВ2(г)4-..
Концентрация ионов, как правило, на
четыре-пять порядков ниже концентра-
ции нейтральных молекул. Это позволяет
пренебречь влиянием кулоновских сил и
объемных зарядов и рассматривать газ,
Находящийся внутри эффузионной ячейки
Рис. П.6. Зависимость
парциальных давлений
Рл, рв и рАВ от моляр-
ной доли N в
как идеальный. В результате появляется возможнсть расчета кон-
стант ион-молекулярных равновесий типа
^в-
Р /’АВ
(11.42)
В эксперименте по изучению ион-молекулярных равновесий
проводится определение парциальных давлений положительных и
«отрицательных ионов и нейтральных молекул на основе измере-
ний соответствующих ионных токов. При этом следует отметить,
что равновесные ионы в ячейке вытягиваются прямо в анализатор
электродами соответствующей полярности. Нейтральные молекулы
подвергаются ионизации и затем направляются в анализатор. Все
измерения проводятся в одном эксперименте, но в разных режимах
работы ионного источника.
На основе значений К(> ион-молекулярных равновесий рассчиты-
вают важные для химии характеристики — энергии образования
ионов, энергии разрыва химических связей, величины сродства к
электрону и потенциалы ионизации молекул. Так, при измерении
температурной зависимости констант ион-молекулярных равнове-
сий для хлоридов натрия, калия и рубидия и бромида калия было
установлено, что энергии разрыва связей М+—MX (где М — атом
металла, X — атом галогена) составляют 150... 200 кДж/моль,
уменьшаясь от натрия к рубидию и от хлора к брому.
Мерой стабильности ионов A1F~ и A12F? следует считать срод-
ство молекул A1F3 и A12F6 к фторид-иону F-, т. е. энергию разрыва
связей A1F3—F~ и Al2Fe—F-. Из данных по ион-молекулярным рав-
новесиям получены следующие значения: £>о(А1Рз—F_)=480 и
£>o(A12F6—F-) =510 кДж/моль. Сопоставление c,Do(A1F2—F) —
= 610 кДж/моль показывает, что сродство молекул AIF3 и А12Рб к
иону F- лишь немного уступает'энергии разрыва очень прочной
связи A1F2—F.
Исследование ион-молекулярных реакций методом химической
ионизации и ион-циклотронного резонанса дает возможность уста-
навливать различие в строении одинаковых,по составу ионов, из-
мерять относительную основность соединений путем обменных ре-
акций типа
AH+ + B5t А4-ВН+
или относительную кислотность протонных кислот для процесса
хн->х-+н+
Так, в реакции ион-радикала СН^ с метаном
CHt+CH4—СН^+СНз
образуется устойчивый ион метония СН*. Реакция экзотермична.
Доказано, что основность аминов в газовой фазе убывает в ряду
(С Н3)3 N > (С Н3)2 NH > CH3NH2
который отличается для растворов в воде:
(СН3)2 N Н > (С Н3)3 N > С H3NH2
что объясняется сольватацией.
Неожиданный ряд кислотности найден для спиртов:
(С Н3)3 СС Н2О Н > (СН3)3 СОН > (С Н3)2 сно н >
> С Н3С Н2ОН > С Н3О Н > Н2О
Он означает, что алкильные группы стабилизируют отрицательный
заряд. В газовой фазе толуол оказался более сильной кислотой,
чем СН3ОН и Н2О, хотя в водном растворе имеется обратное соот-
ношение.
4. МАСС-СПЕКТРОМЕТРИЯ В ХИМИЧЕСКОЙ КИНЕТИКЕ
Общий принцип применения метода масс-спектрометрии в кине-
тических исследованиях основан на возможности идентификации и
определения количества веществ во времени, т. е. на аналитических
приложениях метода. Константы скоростей элементарных процес-
сов в условиях равновесного энергетического распределения участ-
вующих в химической реакции частиц по степеням свободы опреде-
ляют из измерений концентрации реагирующих веществ и продук-
тов реакции. Таким образом были определены константы очень
быстрых реакций, атомов Н с молекулами F2, Cl2, О2, С2Н4, СзН6,
радикалами СН3, НО2 и т. п.
Наиболее распространенным способом изучения механизма ре-
акции оказался метод изотопной метки, которая представ-
ляет, например, более тяжелый изотоп элемента. Так, для реакции
бензойной кислоты с метанолом, меченным кислородом-18:
с6н.с/° +снГои->с.,н5с/0 +н2о
'ОН \1°ОСН3
найдено, что именно кислород спирта становится эфирным кисло-
родом. Реакция этерификации происходит с отщеплением группы
ОН от кислоты и атома Н от спирта. Гидролиз эфира протекает по
обратной реакции:
Ск лО
1р0Н-+ ;C-OR^ H18O-Cf 4~0R-
R'/ \R'
4-ROH
Изотопная метка активно используется в масс-спектрометрии
для исследования механизма фрагментации и установления струк-
туры ионов. Использование дейтерированного метанола позволило
предположить, что при ионизации отщепление водорода осущест-
вляется в основном от метильной группы
СН3О Н 4-е- ->[СН3О]+4-Н- + 26-
ил и
CD3O Н Ц- е- [CD2OH ]+ + D• + 2е~
Однако показано, что упрощенные представления о разрыве
связей в исходной молекуле при образовании осколочных ионов
неверны. Например, найдено, что отщепление радикала 13СН3 от
молекулы СНз—13СН2—СН3 происходит с участием центрального
атома. Изотопная метка использовалась для доказательства тро-
пилиевой структуры иона С7 Некоторый образуется при иониза-
ции толуола. Многочисленны применения этого метода при иссле-
довании перегруппировок в ионах.
Метод ион-циклотронного резонанса (ИЦР) позволил опреде-
лять такие основные кинетические характеристики, как порядок и
молекулярность реакции, константы скорости химических реакций,
а также изучать механизм химических реакций.
Исследование классической реакции нуклеофильного замеще-
ния в газовой фазе методом ИЦР, например
F- + С1СН3 —*-
CH3F +. Cl
н-‘
показало, что скорость этого процесса очень большая и константа:
скорости &2 —48-1013 см3-моль-1-с-1. Это значительно превышает
константу скорости реакции, например, в водном растворе: /г2 =
= 1,4-10~5 см3-моль-1-с-1 (25°С). Столь большая разница в ско-
ростях реакций объясняется совершенно различным их механиз-
мом.
В газовой фазе образование активированного комплекса проис-
ходит без преодоления барьера активации (рис. II.7, 1), наоборот,
энергия его ниже, чем исходной системы. Затем происходит пере-
группировка активированного комплекса с небольшой энергией ак-
тивации и затем распад на свободные частицы CH3F и С1_.
При реакции в растворе вслед-
ствие сольватации иона энергия ак-
тивированного комплекса выше,
чем энергия исходной системы
(рис. II. 7, б).
* *
*
Методы масс-спектрометрии име-
ют очень широкое применение в
различных областях науки и прак-
тики. Основные направления раз-
вития аппаратуры естественно
идут по пути повышения разреша-
ющей способности, чувствительно-
сти и автоматизации на основе ис-
пользования ЭВМ, а также увели-
Рис. II.7. Энергетическая диаграм-
ма пути реакции в газовой фазе
(/) и в растворе (2)
чения интервала измеряемых масс
ионов, разработки новых методов
ионизации. Совершенствование ап-
паратуры расширяет возможности
применения методов. Они являются
перспективными в области термодинамических исследований, а
масс-спектрометрия высоких давлений важна для изучения тех-
нологических процессов.
Структурные исследования в существенной мере опираются на
приближенные представления об образовании ионов и их фрагмен-
тации. Дальнейшее их развитие должно сопровождаться большим
привлечением квантово-химических расчетов структур молекул и
ионов.
Значительная роль принадлежит масс-спектрометрии в изуче-
нии кинетики и механизмов химических реакций, особенно элемен-
тарных химических актов, в том числе ион-молекулярных, процес-
сов возбуждения, ионизации, фрагментации и перестройки моле-
кул.
Огромная информация, полученная при изучении масс-спектров,
систематизирована в справочниках. Для ее использования разрабо-
таны соответствующие алгоритмы. Так, в Институте высоких тем-
ператур АН СССР создан банк данных по термодинамическим
свойствам веществ, называемый ИВТАНТЕРМО. Располагая тер-
модинамическими функциями и термохимическими характеристи-
ками веществ, можно рассчитать равновесия разнообразных про-
цессов. Результаты этих расчетов используются в различных обла-
стях науки и техники: при оптимизации металлургических процес-
сов, для выяснения поведения ионизирующейся присадки в М.ГД-
генераторах, для обработки режимов транспортных реакций при
Получении полупроводниковых материалов и т. п.
Контрольные вопросы
! . Г л а в а I
1. Какие методы' ионизации используют в масс-спектрометрии? Почему ис-
пользуют различные методы ионизации?
2. Какир типы ионов наблюдают в масс-спектре? В каких условиях и для
какого типа"молекул мала вероятность образования молекулярного иона?
3. Дайте определение понятия сечения ионизации. Будет ли и как сечение
ионизации зависеть от энергии ионизирующих электронов?
4. В чем состоит принципиальная схема масс-спектрометра?
5. Назовите особенности статических масс-спектрометров. Есть ли ограниче-
ние по массе иона?
6. Назовите типы динамических масс-спектрометров.
7. В чем состоит фокусирующее действие магнитного поля анализатора в
масс-спектрометре?
8. Что называется разрешающей силой масс-спектрометра и чем она опреде-
ляется? Каковы пути ее увеличения?
9. Что называется чувствительностью масс-спектрометра и чем она опре-
деляется?
10; Каковы принципы работы спектрометра ион-циклотронного резонанса?
Глава II
11. На чем основана идентификация ионов в масс-спектре?
12. Как устанавливается брутто-формула вещества?
13. Приведите примеры закономерностей диссоциативной ионизации органи-
ческих соединений.
14. Как определяются потенциалы ионизации молекул? Почему при фотоио-
низации точность определения потенциалов ионизации наивысшая?
15: В чем состоит .различие вертикальных и адиабатических; потенциалов
ионизации?
16. Как определяются энергии разрыва химических связей? Какие данные
нужны для их определения?
17. Каковы экспериментальные условия проведения масс-спектроскопического
термодинамического эксперимента?
18. Укажите условия для определения парциальных давлений паров веществ,
теплот сублимаций и теплот реакций.
19. Назовите методы исследования ион-молекулярных реакций?
20. Как используется изотопная метка для изучения механизма химических
реакций?
21. Назовите области применения спектроскопии ион-циклотронного резо-
нанса.
МЕТОДЫ ОПРЕДЕЛЕНИЯ ЭЛЕКТРИЧЕСКИХ
ДИПОЛЬНЫХ МОМЕНТОВ МОЛЕКУЛ
Собственные электрические дипольные моменты молекул являются мерой
полярности молекул. Они относятся к главным факторам, определяю-
щим межмолекулярные взаимодействия в веществе. В теории химического
строения полярность молекул объясняется на основе представления о поляр-
ности химических связей.
Несмотря на относительно давнюю историю классического метода Дебая
(1912) для определения электрического дипольного момента, он до сих пор
является одним из важнейших и широко используемых. Наряду с этим разра-
ботаны новые методы определения дипольных моментов.
Данные по дипольным моментам используются в химии для установления
симметрии и строения молекул. Особую ценность представляют результаты
определения структуры молекул этим методом для растворенных и неустой-
чивых в газовой фазе веществ. Прямые структурные методы, к сожалению,
не могут быть непосредственно применены для жидкой фазы.
В этом разделе будут представлены методы определения электрических
Дипольных моментов, основанные на уравнении Дебая, и новые методы моле-
кулярных пучков.
Различный характер распределения электриче-
ского заряда в молекулах позволяет разделить
их на два основных класса — полярные и непо-
лярные. К полярным молекулам относятся моле-
кулы, обладающие важной электрической харак-
теристикой— дипольным моментом. В 1912 г.
П. Дебай впервые ввел представление о диполь-
ном моменте как о величине, определяющей раз-
деление положительных и отрицательных заря-
дов в молекуле. Им создана теория поляризации диэлектриков в
электрическом поле и разработаны экспериментальные методы из-
мерения дипольных моментов .молекул в газовой фазе и в раство-
ре. До сих пор эти методы являются основными для определения
дипольных моментов. Однако методы Дебая имеют некоторые ог-
раничения, так как не позволяют изучать труднолетучие соедине-
ния, например соли и оксиды металлов, или неустойчивые соеди-
нения.
В настоящее время созданы новые методы, основанные на ис-
пользовании молекулярных пучков. В молекулярном пучке моле-
кулы не претерпевают столкновений друг с другом и движутся па-
раллельно.
При воздействии внешнего неоднородного электрического поля
молекулы пучка частично ориентируются в этом поле и отклоняют-
ся от первоначальной траектории.
В целом при воздействии такого поля происходит расфокуси-
ровка и смещение молекулярного пучка, что позволяет опреде-
лять наличие или отсутствие собственного дипольного момента мо-
лекул и определять его значение.
Квантово-механическая теория открыла новые возможности в
определении дипольных моментов на основе эффекта Штарка, ко-
торый заключается в расщеплении вращательных уровней энергии
полярной молекулы в электрическом поле. Степень расщепления
энергетических уровней зависит от напряженности электрического
поля.
В спектре поглощения этот эффект проявляется в увеличении
числа линий вращательных переходов.
Значение дипольного момента можно определить по значениям
частот вращательных переходов. Одновременно при этом опреде-
ляют вращательные постоянные, прямо связанные с геометрией
молекулы.
Дипольные моменты нашли широкое применение в химии для
оценки полярности молекул, для расчета дипольных моментов хи-
мических связей в соответствии с аддитивной схемой и на основе
знаний геометрии молекулы. Использование важного свойства—•
приближенного постоянства дипольного момента той или иной хи-
мической связи в разных молекулах позволяет решать структур-
ные задачи химии.
ГЛАВА III
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДОВ
1. ЭЛЕКТРИЧЕСКИЙ ДИПОЛЬНЫЙ МОМЕНТ МОЛЕКУЛЫ
Различный характер распределения электрического заряда в
молекуле позволяет разделить все молекулы на два основных
класса — полярные и неполярные. К полярным молекулам
относятся молекулы, обладающие электрическим дипольным
моментом.
Дипольный момент молекулы ц является вектором
Р = 2 eiri’ (Ш-Ь
I
где ei — заряды частиц в молекуле; гЕ— их радиусы-векторы.
Для непрерывного распределения заряда в молекуле с плот-
ностью р(х, у, г) вектор ц задается тремя компонентами (проек-
циями) первого момента электрического заряда цх, и цг, где,
например,
= У, z)dx(iy<lz.
Вектор ц нейтральных молекул не зависит от выбора начала
координат. Абсолютное значение ц рассчитывается на основе проек-
ций цх, и цг в соответствии с уравнением
И2=Нх + ^ + иЬ (III.2)
Рассматривая отдельно ядра и электроны, можно представить
молекулу в виде диполя:
ц = 71, (III.3)
где q = e "У zt — сумма положительных зарядов; 1 — вектор, на-
I
правленный, как принято в химии, от центра тяжести положитель-
ных зарядов к центру тяжести отрицательных зарядов (/ — рас-
стояние между этими центрами). Возможность такой замены обус-
ловлена тем, что электрическое поле такого диполя на больших
расстояниях по сравнению с I эквивалентно полю соответствую-
щей молекулы. Размерность ц выражается произведением заряд —
длина; в СИ единицей величины ц является Кл-м; внесистемная
единица — дебай £>(10=3,34- 10~30 Кл-м).
Полярные молекулы характеризуются симметрией Cs, Сп или
Сяо. У молекул, имеющих ось симметрии, дипольный момент на-
правлен вдоль этой оси. Если в молекуле есть плоскость симмет-
рии, то вектор ц лежит в этой плоскости. Молекула не имеет ди-
польного момента (ц=0), если имеется зеркально-поворотная ось
S/j любого порядка, например ось S2 в молекуле Н2 или в линейной
Молекуле С02 (центр симметрии соответствует оси S2), ось 5$ в
тетраэдрической молекуле CPU и в октаэдрической молекуле SF6>
ось S3 в плоской молекуле ВС1з и т. п. О симметрии молекул
см. гл. IX.
Дипольный момент молекулы является ее важной физической
характеристикой, которая непосредственно связана с ее строением
и определяет взаимодействие полярных молекул, а также их ориен-
тацию во внешнем электрическом поле, что в свою очередь обус-
ловливает диэлектрические свойства вещества.
Дипольный момент изолированной молекулы называется соб-
ственным или постоянным дипольным моментом
молекулы (р,0). При поляризации молекул в электрическом поле
высокой частоты возникает индуцированный дипольный
момент молекулы (ц%), который обусловлен смещением
электронного облака молекул. В статических полях или полях с
относительно низкими частотами колебаний происходит поляриза-
ция вещества в целом, включающая небольшое смещение ядер и
преимущественную ориентацию молекул вдоль поля. Индуциро-
ванный ориентационный дипольный момент мо-
лекул (р,г) зависит от постоянного дипольного момента цо- Экс-
периментально измеряемые в электрическом поле величины связа-
ны с индуцированными дипольными моментами, из которых можно
определить постоянный дипольный момент молекулы цо-
2. ЭНЕРГИЯ МОЛЕКУЛЫ ВО ВНЕШНЕМ
ЭЛЕКТРИЧЕСКОМ ПОЛЕ
Энергия молекулы как системы электрических зарядов ei во
внешнем электрическом поле может быть представлена суммой
потенциальных энергий зарядов в этом поле:
<ш-4>
где ф(г/) —потенциал в точке г,-.
Если электрический потенциал <р(г) мало изменяется на рас-
стоянии, соответствующем размеру молекулы, то его можно разло-
жить в ряд Маклорена и ограничиться двумя первыми членами
этого разложения:
Т(Г)_»(О)+У
/о 2 /0
\=x,y,z е т)
~?(0) + 3\ 5 = Т (°) + г - \/ф, (Ш.5)
н
где '\7tP = grad<p=—<£’o—напряженность электрического поля для
г—0.
Тогда для нейтральных молекул Sa,=0 и
и=2 2
или, так как ц,
Ц=^-^й.
(Ш.7>
Во внешнем однородном поле (<F=const) на молекулу, обладаю-
щую дипольным моментом, с одной стороны, действует пара сил,,
которая стремится повернуть ось диполя в направлении поля, так
как на равные по значению положительные и отрицательные заря-
ды действуют одинаковые силы в противоположных направлениях
(рис. III. 1). С другой стороны, внешнее поле смещает заряды про-
тивоположных знаков, т. е. поле поляри-
зует молекулу. Поэтому энергия моле-
кулы в электрическом поле определяет-
ся двумя составляющими — работой по-
ворота молекулы и работой поляризации.
Энергия поворота для жесткого дипо-
ля цо задается уравнением
Рис. III.1. Пара сил, дей-
ствующая на диполь в
однородном электричес-
ком поле
Ur= cos 6,
(Ш.8)
где 0 — угол между <£ и ц0-
Энергия поляризации молекулы оп-
ределяется поляризуемостью мо-
лекулы, которая в общем анизотропна
и представляется тензором ЦацЦ.
Поскольку лишь очень малая часть молекул во внешнем отно-
сительно слабом поле ориентирована вдоль этого поля, то в удов-
летворительном приближении для решения поставленной задачи
определения индуцированного дипольного момента достаточно рас-
сматривать произвольно ориентированную молекулу со средней
Л__ « / Cti -f~ СС.2 CCq
статической поляризуемостью a a=—---——\ где ai, as, аз —
главные значения статической поляризуемости). Такое приближе-
ние вполне удовлетворительно для диэлектрических измерений, в.
которых преобладающую роль играет ориентационная по-
ляризация. Влияние ориентации молекул в электрическом поле
на электронную поляризуемость учитывается в методе эффекта
Керра.
Поляризация молекулы в статическом поле <S выражается в
появлении индуцированного дипольного момента, пропорциональ-
ного <F, за счет смещения электронов и ядер:
p.=(a<?4-aa)<£’ = aod?, (Ш.9>
гДе ае — электронная поляризуемость; аа — атомная
Поляризуемость; ав — суммарная деформационная
Поляризуемость в статическом поле.
Энергия поляризации (деформации) молекулы будет выражать-
ся интегралом
8 8
Z7O== — J p.d<^= — J aD<^d<F==—у-^2- (III.10)
о о
Уравнение для полной энергии молекулы в электрическом поле
можно представить в виде суммы уравнений (III.8) и (III. 10), т. е.
U ^=UT-^UD=-^ cos В-----Ц°2--- • (Ш.И)
3. ОРИЕНТАЦИОННАЯ ПОЛЯРИЗАЦИЯ
МОЛЕКУЛ
Характеристики молекул газа подчиняются статистическим за-
конам. Действие электрического поля на молекулы газа не приво-
дит к полной ориентации молекул в связи с их тепловым движе-
нием.
П. Дебай использовал статистическую теорию ориентации, ко-
торую разработал П. Ланжевен для постоянных магнитных момен-
тов парамагнитных тел, и создал теорию полярных диэлек-
триков.
Эта теория основана на следующих условиях для газа в элек-
трическом поле:
1) плотность газа настолько низка, что энергия диполь-диполь-
яого взаимодействия молекул мала по сравнению с тепловой энер-
гией (~kT) и ею можно пренебречь;
2) поле & не оказывает возмущающего действия на дипольный
момент молекулы;
3) энергия молекулы в поле мала по сравнению со средней
тепловой энергией молекулы:
^<^kT. (Ш.12)
Поляризация газа как диэлектрика будет определяться средним
дипольным моментом молекулы в поле &, т. е. усреднением суммы
уравнений (III.8) и (III.9):
|л = р.о cos 6-j-ap<^, (III.13)
где 0— угол между <S и р,0 — меняется от 0 до л.
Как уже говорилось, в условиях рассматриваемого эксперимен-
та можно принять ощ=ад. Согласно статистической механике при
тепловом равновесии
и
$е krdQ
где dQ — элемент телесного угла 2n;sin0d0 (конуса).
62
Тогда, используя уравнение (III.8), получим
cos 0 exp
Ро <5 cos 0\
. ЙГ у
sin 0d9
cosS =
(Ш.15)
sin fidfl
Уравнение (III. 15) для условия можно упростить и
провести интегрирование. Так как
& cos в \ । , pg % cos 8
ехр kT
то, вводя новую переменную x=cos 0, перепишем уравнение (III. 15)
и получим значение cos 0 в явном виде:
cos 9
0 4-2
4о<У
3kT
3kT
(Ш.16)
2 + 0
Таким образом, у, можно рассматривать как индуцированный ди-
польный момент ц:
2
р=2^_|_а^=(Л. (Ш.17)
Из уравнения (III. 17) следует, что можно формально ввести ориен-
тационную поляризуемость молекулы:
а, = ро/(3£Г).
(Ш.18)
Эта величина не является молекулярной постоянной, так как зави-
сит от Т. Индуцированный дипольный момент является суммой:
p = (aD4-az)^ = p.r-|-pD . (III.19)
Полезно оценить относительную величину у, для <§Г= 105 В/см и
3,34-IO-30 Кл-м при 7=300 К:
Рг __ ~ 10~2...10-3.
jj-q 3^7
^алое значение цг оправдывает использование приближенного ра-
венства aD = aD, так как доля ориентированных молекул в приме-
няемых полях действительно мала. Кроме того, ar>ao для ц0>
3,34-IO'30 Кл-м. Так можно оценить порядок а, из уравнения
(III.18) при T=300K:
О
а.=3 • 10-з9
ЗйГ 3-1,38-10-23-3.102
Кл.м2-В-1.
Суммарная поляризуемость молекулы в электрическом поле
будет равна:
a = ae + a<i+a/" (Ш.20)
4. ЭФФЕКТ ШТАРКА И КВАНТОВО-МЕХАНИЧЕСКИЙ
ПОДХОД К ВЫВОДУ ОРИЕНТАЦИОННОЙ
ПОЛЯРИЗАЦИИ МОЛЕКУЛ
Если однородное электрическое поле & г направлено вдоль оси г, то класси-
ческая функция Гамильтона для молекулы в этом поле в соответствии с уравне-
ниями (III.6) или (III.7) будет иметь вид
Н = г, (III.21)
где Zi — координата i-ro заряда; Но— функция Гамильтона для молекулы в от-
сутствие внешнего поля.
В связи с тем, что <5г = const, преобразуем уравнение (Ш.21) следующим
образом:
H = HQ- S z^ е.г. = Н0-^ 2. (III. 22)
Перейдем теперь к стационарному нерелятивистскому уравнению Шредингера
= (Ш. 23)
где Й— оператор Гамильтона, равный Йо—,и.ге?2; фи—ортонормированная вол-
новая функция молекулы для состояния п; Еп—энергия этого состояния.
Умножив уравнение (III.23) на комплексно-сопряженную функцию и интег-
рируя это произведение, получаем
J^/fy„dr=£„f^„dr = £„. (III.24)
Поскольку выражение = Е°п не зависит от S г, то
г J<)>*M«dT| = = -<^>«- (HI.25)
Здесь и далее <> есть символ среднего. Следовательно,
( &Z > п — дЕп
Это есть квантово-механическое определение проекции индуцированного ди-
польного момента молекулы на направление электрического поля. Таким обра-
зом, дипольный момент молекулы является средним для данного состояния фп,
обладающего энергией Еп в поле S2.
Находим выражение Еп в электрическом поле <S г на примере линейной moj
лекулы. В отсутствие поля энергия вращения (жесткий ротатор) для линейной
молекулы дается уравнением
£° = ЛВ/(/ + 1), (III.27)
где В = /г/(8л2/)—вращательная постоянная; I — момент инерции; / — враща-
тельное квантовое число.
Волновая функция вращения линейной молекулы ф/м(9, ср) определяется
двумя квантовыми числами J и М (М — определяет проекцию момента количе-
ства движения на ось z и изменяется от —J до +/), а также зависит от двух
переменных — угла 0 (угол между осью z и осью молекулы) и угла ср (азиму-
тальный угол). Она имеет следующий вид:
Ьм (в, 4) = NJMP\M\ (соз0)егЛ\ (III.28)
I / 2/ + I (/ - | Af | ) ! V/2
где Njm = —2---- (/ + | М | ) ! / “ НОРМИРОВОЧНЫИ множитель;
(cos 0)—присоединенные полиномы Лежандра, которые
являются действительными функциями 0.
Поскольку можно рассматривать внешнее поле ё z как небольшое возмуще-
ние, то вполне обоснованно использовать теорию возмущений. Тогда вра-
щательная энергия молекулы в поле ёг будет представлена рядом
En = E\±E'n+E"n, (III.29)
где Е'п ’^Ё'п — э и е р г и и возмущения первого и второго п о р я д-
к а соответственно.
В соответствии с уравнением (III.22) возмущение функции Гамильтона сле-
дует представить в форме
Н' = — }>-гё г = — ^ё cos 9. (III.30)
Тогда для линейной молекулы
Е' = J dr = — р.ог? z f cos Odr = 0. (III.31)
Это тождество является следствием того, что 'рум есть четная функция cos 0, а
произведение ф2 M-cos 0 — нечетная функция. Интегрирование нечетной функции
в полном пространстве тождественно дает нуль.
Энергия возмущения второго порядка выражается уравнением
(Ш.32)
т
где Нпт ~ J 'ХГ-^'Ф^т и ф^, ф^—волновые функции; Е^ — энергии не-
возмущенной молекулы в состояниях гейт. Для Н'тп производится перестановка
сомножителей, т. е. под интегралом имеем ф^А/’ф^.
Для вращательного движения линейных молекул в поле ёг можно записать
матричный элемент Нпт в следующей форме:
Н пт Н J MJ’ М' = J Ф/АГ ( г cos 0) Ф/'ЛГ' dt, (III. 33)
где n=JM, а т — ]'М'. При этом Нпт = Нтп.
Далее, учитывая рекуррентное соотношение
cos 9Р । л*' I = -Г ~ 1 1 + 1 р\ М' [ , Г + । MLL pIM'i /ill 34)
7 2/'+1 ^'+1 + 2/'+1 7'-1 (Ш,34)
и условие ортонормированности
+ 1 р при
С Pj м I р\,м | d cos (J 2 + (111.35)
-i 1уТТ(7ЩйЯ"р’/-7'
3—949
можно показать, что Нj#=0 только при М=М' и J — Это прави-
ла отбора для J и М.
Следовательно,
| J .М; 7-1,М | 2 1 Нj,m-, j+i,m I
т 7 1 'V T? 0 TjO * 4 11 • OU f
где
1 f,' I 2 2 2 f (j + A4) (7 — AI) ) (27 + 0(27-1) f (III.37)
И Im' | 2 _ „2 «. 2 ((/ + Л4 + 1)(7 — A4 + 1) । + ~N#z[ (2/ + 1)(2/+3) г
Было рассмотрено направление поля вдоль оси г. Если для данной системы
координат поле направлено вдоль х или у, то
и
:== sin 6 cos у
Ру = р0 sin 0 sin у,
(Ш.38)
a 7-0 при Л4' = Л1±1 и 7' = 7±1, т. е. правила отбора изменяются.
Продолжим рассмотрение поля Поскольку вращательная энергия невоз-
мущенного состояния известна [уравнение (III.27)], то на его основе получаем
разности:
Е° - E°j_j = hBJ (J + 1) - hB (J - 1) J = 2hBJ (III.39)
И
£0 - E° +1 = hBJ (J + 1) - hB (J + 1) (7 + 2) = - 2hB (7 + 1). (III.40)
Теперь по уравнениям (III.36), (III.37), (III.39), (III.40) найдем энергию
возмущения второго порядка отдельно для 7=0 и Л4=0, т. е.
17" 1 ..2 op 2 <0 ® z
Лоо — 2hB 3
и для
p” ЗЛ42-/(/+1) ।
— 2hB 7(7+0(27- l)(27 + 3) Г
Эти уравнения и определяют эффект Штарка — расщепление вращательных .
энергетических уровней полярных молекул в поле Sг — и дают возможность
определить дипольный момент для определенного вращательного состояния:
dlg z
Для состояния 7=0, Л7=0 имеем
г
= ЗАВ ’
(Ш.43)
(Ш.44)
а для 7^=0— соответственно
_ z f ЗМ2 _/(/ + !) 1
hB I/(/+ I)(2/~ 1)(2/+3)/' (
Найдем значение дипольного момента для 7=И=0, усредненное для различных М,
т. е.
Е
M = + J .JM
2 kr
Ъг = -----р---------• (П1.46)
M=+J JM
е kT
M = -J
Однако (e’jm — Еj<g. kT. Поэтому можно упростить уравнение (III.46) и
принять, что
ж = + /
2 ^Л12/^ + Г)- (III.47)
M=-J
При проведении суммирования SAP получим
M=+J
Л42 = 2(12 + 22 + ... + /2) = , (111.48)
Следовательно,
М=+/
2 [ЗЛ42 - 7 (7 + D] = 7 (/ + 1) (2/ + 1) - (2/ + 1) / (/ + 1) = 0.
Al-.—J
Таким образом, pJz = 0 для 7У=0.
В итоге оказалось, что средний дипольный момент по всем вращательным
состояниям будет определяться молекулами на уровне с 7 = 0:
EJ
<^> (ш-49>
2^/'A-r ‘2^/“
где gj — кратность вырождения уровня Ej.
В статистической термодинамике для вращения молекул сумму по J аппрокси-
мируют интегралом
У gjt-TT- \ (2/ + 1)й kT dJ = ——. (III.50)
2 hB
J о
Окончательно имеем
z hB z
P-z ) ~------- ----=---------•
z 3hB kT 3kT
Считывая, что
(III.51)
( Р'г ) — z
йаходим полное согласие с уравнением (III. 18).
3*
Таким образом, уравнения для ориентационной поляризуемости, полученные
на основе квантово-механического и классического рассмотрения задачи, совпа-
ли. Это обусловлено тем, что разности уровней энергии вращения малы по срав-
нению с тепловой энергией молекул. Но квантово-механический подход открыва-
ет возможность определения дипольных моментов, если наблюдать переходы
между различными уровнями энергии, реализуемую в методах электрического
резонанса (см. гл. IV, 4) и микроволновой спектроскопии (см. гл. V, 5)
5. ДИЭЛЕКТРИК В ЭЛЕКТРИЧЕСКОМ ПОЛЕ
Вещества, состоящие из полярных и неполярных молекул, в
основном, являются диэлектриками. Если диэлектрик поместить в
электрическое поле конденсатора, то происходит поляризация
диэлектрика, которая обусловливает изменение электрического
поля конденсатора. Пусть поверхностная плотность за-
рядов на пластинах конденсатора равна + ps и —ps соответст-
венно. Тогда внутри конденсатора, пластины которого удалены в
вакууме на расстояние I, а линейные размеры пластин значительно
больше I, создается однородное электрическое поле, равное по мо-
дулю
^o=Ps/so> (III.52)
где ес—электрическая постоянная.
Уравнение (III.52) следует из законов электростатики.
Заполнение пространства между пластинами конденсатора ди-
электриком приводит к уменьшению напряженности поля по мо-
дулю в е раз’-
(Ш.53)
е ее0
где е — статическая диэлектрическая проницае-
мость диэлектрика.
Уменьшение напряженности электрического поля в конденсаторе
вызвано поляризацией диэлектрика, т. е. накоплением отрицатель-
ных зарядов на поверхности диэлектрика вблизи положительно
заряженной пластины и положительных зарядов вблизи отрица-
тельно заряженной пластины. В объеме диэлектрика индуцирован-
ные заряды компенсируются.
Поверхностная плотность зарядов диэлектрика ps может быть
выражена через среднюю величину индуцированных вдоль поля
молекулярных диполей |<р,>| и число молекул п в 1 м3 вещест-
ва, т. е.
Ps = /z|(ft)[. (Ш.54)
Средний дипольный момент <ц> пропорционален эффективному
полю , действующему на молекулу, и равен
(р)=а^', (III.55)
где а — суммарная поляризуемость.
В связи с тем, что реальный диэлектрик, например, в газообраз-
ном состоянии не представляет собой непрерывную среду, необхо-
димо рассматривать суммарное поле, которое включает поле сво-
бодных зарядов на обкладке конденсатора So и поле всех осталь-
ных молекул диэлектрика. С увеличением расстояния от
рассматриваемой молекулы суммарное поле остальных молекул,
находящихся за сферой радиуса г0, в которой находится молекула,
может быть представлено полем непрерыв-
ной среды (рис. III.2).
Кроме того, на отдельную молекулу дей-
ствует локальное поле индуцированных ею
же зарядов на поверхности сферы радиуса г0
(рис. III.2).
На основе электростатики можно полу-
чить выражение для локального поля в цент.
ре сферы через поверхностную плотность за-
ряда ps'.
S' = S-^~^s- (Ш.56)
Зе0
В приближении слабого взаимодействия мо-
лекул поле молекул внутри сферы компенси-
руется и равно нулю. Поскольку ps-связана
с поляризуемостью молекул уравнениями
(III.54) и (III.55), то
ft'=3 -)—— по.£'.
Зе0
Отсюда находим, что
Рис. III.2. Модель
распределения заря-
дов в диэлектрике
для расчета локаль-
ного поля в центре
сферы с радиусом го
(Ш.57)
(Ш.58)
I - — па
Зе0
Теперь можно найти связь между диэлектрической проницаемостью
вещества е и поляризуемостью молекул а, используя уравнение
для электрической индукции (поле свободных зарядов
на обкладке конденсатора) по модулю
D = = s0<F 4~Ps = So<^-\-tiaS'. (III.59)
Подставляя S' из уравнения (III.58), получим
1 — -— па
3sq
Откуда
s$o = so +---, (Ш.60)
1 — —— па
Зе0
Выразим сначала величину е—1 в виде
е-1 = - ,3^ , . (Ш.бОа)
Зе0 — па
Затем к правой и левой частям этого уравнения прибавим число 3:
£_l2==3J ^._ =—(III.606)
Зе0 г ct 3sq — not
Разделив уравнение (Ш.60 а) на (111.60 б), получим у р а в н ен и е
Клаузиуса — Моссотти:
Число молекул п в 1 м3 может быть выражено через плотность р,
молярную массу7 М и постоянную Авогадро NA:
n=^NA. (111.62)
Л4
Перепишем уравнение (III.61) в традиционной форме:
— (Ш.63)
б у. 2 р 3&q
Уравнение (III.63) применимо к неполярным веществам, для кото-
рых а=ар — деформационная поляризуемость.
Для полярных молекул уравнение (Ш.63) переходит в урав-
нение Дебая:
(Ш.64)
Левая часть уравнения называется молярной поляризацией
вещества.
Для неполярных диэлектриков в полях переменной частоты v,
при которой проявляется только электронная поляризуемость мо-
лекул, уравнение Клаузиуса — Моссотти может быть заменено
уравнением молярной рефракции Лорентца—
Лоренца, так как, e(v) = п2(у):
n2 + 2 p 3eo
(111.65)
где n—-показатель преломления; b — электронная поляризуемость
в переменном электрическом поле высокой частоты.
Обычно эксперименты проводят в поле световой волны желтой
линии натрия. Излучение этой длины волны веществом, как прави-
ло, не поглощается. Эту спектральную характеристику обозначают
индексом D справа внизу, например nD, RD и т. п. Поскольку плот-
ность вещества зависит от Т, молярная рефракция также зависит
от Т, на что указывает верхний правый индекс, например Лд, Rd
и т. п.
Поскольку п зависит от длины волны излучения, то, используя
экстраполяцию п(Х) для Х=<х>, получают п<х>. Величина /?<» позво-
ляет вычислить статическую электронную молярную
поляризацию
. A=J_ Woe = ±2Vxae. (Ш.66)
+2 р 3«о Зво
ГЛАВА IV
ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДИКИ И ПРИМЕНЕНИЕ
ДАННЫХ ПО ЭЛЕКТРИЧЕСКИМ ДИПОЛЬНЫМ
МОМЕНТАМ МОЛЕКУЛ В ХИМИИ
1. ПЕРВЫЙ МЕТОД ДЕБАЯ — ОПРЕДЕЛЕНИЕ
ЭЛЕКТРИЧЕСКОГО ДИПОЛЬНОГО МОМЕНТА
МОЛЕКУЛ ПАРОВ ВЕЩЕСТВ
Согласно уравнению (III.64) измерения статической диэлектри-
ческой проницаемости е газов или паров в зависимости от Т позво-
ляют определить дипольный момент ц0. Уравнение Дебая может
быть преобразовано к виду
/ 2 \
Рт=— Л=— уХт — ) = А + — , (IV.1)
m s + 2 Р 3sq \ ° 1 * * * s 3kT ] Т
1 Ро
где В — — Na. —. Если определено В, то
Гео k
11^1 = }/^ (IV.2)
Следовательно, для экспериментального определения В необходи-
мо изучить температурную зависимость молярной поляризации ве-
щества. Диэлектрическую проницаемость вычисляют на основе из-
мерений емкости конденсатора:
s = C/C0, (IV. 3)
гДе С» — емкость конденсатора в вакууме; С — емкость конденса-
тора с диэлектриком.
При работе с газами или парами требуется очень высокая точ-
ность термостатирования (до ~0,02°С). Температурный интервал
пРи исследовании должен составлять 100. ..150°. Необходимо иметь
оптимальное давление ларов р, так как при низких давлениях
(~400. ..500 Па) затруднено установление температурного равно-
весия. Плотность пара вычисляют по уравнению
p=Mp/(kT). (IV.4)
Если невозможно варьировать температуру паров, то для рас-
чета дипольного момента используют разность между молярной
поляризацией и деформационной поляризацией, вычисляемой на
основе молярной рефракции. При этом не учитывается атомная
поляризация, так как молярная рефракция дает только электрон-
ную поляризацию. Следовательно, в этом эксперименте
Pm-RD = Pr=^-- (IV.5)
<lkT Eq
Таким методом определяют относительно большие дипольные
моменты порядка (6.. .30) 10~30, измеряемые с точностью около
0,3-10-30 Кл-м.
Пренебрежение атомной поляризацией не вносит значительной
ошибки в конечный результат при больших значениях дипольных
моментов, так как имеет место следующее приближенное соотно-
шение между величинами ае, аа, аг:
— (IV.6)
аг ае
Атомную поляризацию можно вычислить по температурной за-
висимости Pm(UT) или из данных поляризации веществ при низ-
ких температурах, при которых отсутствует ориентационная поля-
ризация.
2. ВТОРОЙ МЕТОД ДЕБАЯ — ОПРЕДЕЛЕНИЕ
ЭЛЕКТРИЧЕСКИХ ДИПОЛЬНЫХ МОМЕНТОВ МОЛЕКУЛ
ВЕЩЕСТВ В РАЗБАВЛЕННЫХ РАСТВОРАХ
В связи с рядом трудностей изучения поляризации газов и па-
ров широкое применение нашел так называемый второй метод Де-
бая, который состоит в использовании уравнения Дебая для раз-
бавленных растворов полярных веществ в неполярных растворите-
лях. В первом приближении обоснованием этого метода служит
предположение об отсутствии значительных взаимодействий между
растворителем (1) и растворенным веществом (2), что выражается
в аддитивности свойств раствора.
Поляризация такого раствора будет аддитивной, т. е. в соответ-
ствии с уравнением Клаузиуса — Моссотти (III.61) запишется в
виде
f (IV.7)
®1,2 "Т" ^е0
где et,2 — диэлектрическая проницаемость раствора; л?, «2-число
72
молекул (1) и (2) в единице объема раствора; а2— поляризуе-
мость растворителя и растворенного вещества соответственно.
Введем обозначение общего числа молекул в единице объема
nb2=n?-|-’i2, Умножив обе части равенства (IV.7) на множитель
Nполучим
81,2 + ^ nl<2 nl,2 ds0 '’е0 Je0
(IV.8)
где Wi=#iMi,2 и N2 = n2/nb2~ молярные доли растворителя и
растворенного вещества.
Уравнение (IV.8) можно записать в более компактной форме,
используя выражение для молярных поляризаций: =— -Vacij и
3s0
р.2~—NАа2. Имеем
Зе0
212Z1L ^k = jV1/ni + jv2p2. (IV.9)
«1,2 + 2 nb2
Преобразуем множитель NAlnii2, выражающий молярный объем
раствора, в уравнение
JVjAli -\~N2M2
Pl,2
где pi,2 — плотность раствора.
Тогда уравнение (IV.9) преобразуется к виду
Р1|2==.-еь2--г .1... .У 1^1 + .^2. =4-^Р2, (IV. 10)
е1,2 + 2 Р1,2
где Р1,2 — молярная поляризация раствора. Уравнение (IV.10) яв-
ляется основным при определении дипольных моментов молекул
растворенных веществ. Решение задачи состоит в вычислении Р2
из измерений Pi,2 и заданных Pi и <N2 (7Vi= 1— N2), так как из
уравнения (IV.10)
(IV. П)
У2
В связи с приближенным характером уравнения (IV.10) для
растворов стремятся получить молярную поляризацию растворен-
ного вещества при 'V2->0, а именно
P” = limP2 = lim d], (IV.12)
Л'2^0 лг,->о\ У2 J
Далее вычисляют ориентационную поляризацию при бесконечном
разбавлении:
р2~ = р2»_р2е_р2а.
Однако в связи с отсутствием данных по Р2а ограничиваются вычи-г
танием молярной рефракции (для D-линии натрия):
' P2r~PT-P2e~P2-Rw- ((V.13)
Используя уравнение (IV. 13), вычисляют дипольный момент:
(ЛО= ——J(P2 —Т?2о) • IV. 14)
Ал J
V-Неточности, связанные с пренебрежением атомной поляриза-
цией, не. играют существенной роли и сравнимы с различием ди-
польных моментов молекул, полученных при использовании раз-
личных растворителей. Например, для нитробензола имеются сле-
дующие данные:
Растворитель Бензол (19°С) Толуол (19°С) Гексан (25°С) Диоксан (25°С)
go. IO"30 Кл-м 13,3 12,8 13,4 13,1
Для молекул в газовой фазе p0(C6H5NO2) = 14,0-10-30 К.л/м.
Из сравнения данных величин видны некоторые различия в
значениях цо (СбН5МО2), измеренных в растворах и газе.
3. ОТКЛОНЕНИЕ МОЛЕКУЛЯРНОГО ПУЧКА
В НЕОДНОРОДНОМ ЭЛЕКТРИЧЕСКОМ ПОЛЕ
Ранее обращалось внимание только на то, что в однородном
электрическом поле полярная молекула испытывает крутильные
воздействия. В аналитической форме можно получить выражение
для силы, действующей на молекулу. Если поле направлено вдоль
оси z, то сила вдоль этой оси
(IV. 15)
dz
где U — потенциальная энергия молекулы в поле <SZ, равная
Преобразовав уравнение (IV. 15) и учитывая зависимость U от
ptz, получим
р-------= . (IV. 16)
г d8z dz dz
Следовательно, в неоднородном поле —на молекулу
\ dz /
действует сила, перемещающая ее в пространстве. Направление и
перемещение молекулы зависят от знака и величины при задан-
dS г
ном градиенте —- .
дг
Эксперименты по отклонению молекул в неоднородном элек-
трическом поле должны проводиться при отсутствии столкновений
между молекулами. Такие условия обеспечивает молекулярный
пучок, т. е. пучок молекул, которые движутся в одном направлении,
не испытывая взаимных столкновений.
Молекулярные пучки начали использоваться в физических
экспериментах с 1919 г. в работах О. Штерна. Так, О. Штерном и
В. Герлахом в первых опытах по отклонению атомных пучков в
неоднородном магнитном поле было доказано пространственное
квантование. В дальнейшем молекулярные пучки нашли примене-
ние для изучения распределения скоростей молекул в газе, сечений
взаимодействия атомов и молекул друг с другом, а также с поверх-
ностью твердых тел, для исследования явлений возбуждения и
ионизации и т. д.
Формирование молекулярного пучка возможно при -истечении
молекул газа из отверстия, диаметр которого d значительно мень-
ше длины свободного пробега
молекул лм в ампуле с веще-
ством (рис. IV. 1). В этих ус-
ловиях вылетевшие молекулы
имеют непересекающиеся тра-
ектории. Для выделения тра-
екторий молекул, близких к
параллельным, в направлении
распространения молекул ус-
танавливают диафрагмы и об-
разевавшиеся отсеки соединя-
ют с откачивающей системой.
Давление газа в области рас-
пространения молекулярного
пучка составляет величину по-
рядка 10-5 Па. Давление в
источнике газа должно быть
II 1=3
1 | AB V B *
2 /+\
Рис. IV.l. Отклонение полярных молекул
в неоднородном электрическом поле:
/ — источник молекул; .2 — система диафрагм;
3 — область неоднородного электрического по-
ля, создаваемого электродами, имеющими зна-
чительную и различную кривизну поверхно-
сти; 4 — детектор.
Показана траектория двух противополож-
но ориентированных молекул типа (А) и (В).
Дополнительный индуцированный дипольный
момент направлен по типу (В)
меньше 100 Па. Выходное отверстие обычно имеет форму пря-
моугольной щели, ширина которой равна -~0,01 мм, а высота —
порядка 10 мм. Таким образом, молекулярный пучок представ-
ляет по форме «ленту».
Неоднородное электрическое поле формируется в зазоре между
электродами с различной кривизной поверхности, на которые пода-
ется напряжение в несколько киловольт.
Для регистрации нейтральных молекул используют различные
приемы: поверхностную ионизацию, ионизацию электронной бом-
бардировкой с дальнейшим масс-спектральным анализом ионного
пучка и т. п.
Полярные молекулы, ориентированные вдоль поля и против
поля, будут отклоняться в разные стороны. В целом молекулярный
пучок после прохождения через неоднородное поле будет расши-
ряться (дефокусироваться) и смещаться в направлении поля из-за
того, что число молекул, ориентированных вдоль поля, будет пре-
обладать. Смещение пучка может быть определено из простого
соотношения ;
s — ai2/2,
(IV. 17)
где а — ускорение молекул в направлении смещения, a=Fz/m-,
т — масса молекулы; t=Lfva, L — длина пути молекул; иа=
, no 1 /~ 4kT а
= 1,22 I/ ---- —наиболее вероятная скорость в молекулярном
' т
пучке.
ч-r с d ^0 & z
Поскольку Fz = ^z-^~ и = , то
$=((* JL.. (IV. 18)
V dz ) 2m \ ЫТ dz ) 2m
Это позволяет рассчитать ц0-
Изложенная методика применима при исследовании молекул с
большими значениями р0 как в галогенидах щелочных элементов.
Например, для К.С1 цо = 26,7-10-30 Кл-м, а для CsI ц0 =
= 34,1-10~30 Кл-м. В связи с трудностями точного определения <ог
ds, .
и —— ' а также из-за значительного распределения скоростей мо-
дг
лекул количественные расчеты в этом методе недостаточно на-
дежны.
Для молекул с малыми ц0 существенную роль играет отклоне-
ние молекул из-за индуцированного дипольного момента за счет
электронной и атомной поляризации. Дипольный момент ориенти-
рован в основном вдоль поля. Использование двух последователь-
ных квадрупольных линз, в которых электрическое поле имеет
радиальную симметрию, т. е. определяется только расстоянием от
оси квадруполя, позволило так формировать пучки, что неполяр-
ные и полярные молекулы той же ориентации, что и индуцирован-
ный момент, не попадают в детектор, расположенный на оси
квадруполя, т. е. дефокусируются, а полярные молекулы с противо-
положной ориентацией, наоборот, фокусируются на детектор.
Этим методом в работах У. Клемперера было показано, что мо-
лекулы галогенидов второй группы, как, например, BaF2, ВаС12,
ВаВг2, Bal2, SrF2, SiCl2 и CaF2, имеют постоянный дипольный мо-
мент (цо=#О), т. е. молекулы нелинейные, симметрии Civ. В этих
же экспериментах было найдено, что цо = О для СаС12, СаВг2,
SrBr2 и Srl2, т. е. молекулы линейные.
Полученные результаты имеют важное значение для стереохи-
мии, так как показывают, что направление связей атомов элемен-
тов второй группы зависит от положения их в группе и вида при-
соединенных атомов галогенов.
4. МЕТОД ЭЛЕКТРИЧЕСКОГО РЕЗОНАНСА
Чтобы не учитывать значений скорости молекул, напряженности
поля и его градиента, в расчетах дипольного момента используется
принцип построения траектории частиц, который
впервые предложил и применил И. А. Раби для определения маг-
нитных моментов ядер, атомов и молекул. Схема метода приведена
на рис. IV.2. Для формирования S-образной траектории молекул
пучок направляется в поле А с напряженностью <$ а и градиентом
- А-. Поле А отклоняет пучок в направление поля В с напряжен-
ностью
Между
ностью
менное
меняет
s= „ д&в .
<з в и градиентом —-—, которым фокусируется на детектор,
полями А и В расположено однородное поле С напряжен-
нее
с?о и =0. Перпендикулярно полю направлено пере-
поле <Sv, которое не изменяет траекторию молекул, но из-
их вращательную энергию Ег в соответствии с эффектом
Рис. IV.2. Схема метода электрического резонанса:
/ — источник молекул; 2--диафрагмы; 3 — неоднородное поле А; 4 — одно-
родное поле С; 5 — неоднородное поле В; 6 — детектор.
Перпендикулярно полю С приложено переменное поле . Показаны две
траектории молекул, имеющие различные вращательные энергии
Штарка (см. гл. III, § 4). Поля А и В формируют траекторию мо-
лекул с одинаковой энергией, так как выражение для отклоняю-
щей силы в поле 8г будет иметь вид
д^г дЕ"г дЕ"г Qg
Р, = ^г---- =--------------------------- .
dz dz d$z dz
(IV. 19)
Так, для линейных молекул энергия вращательного состояния
является функцией молекулярных постоянных ц0 (постоянный ди-
польный момент), I (момент инерции) и квантовых чисел J и М,
а также поля 8 (см. уравнение III.42), т. е. '
В=В(р0, /, J, М, <SZ).
Поэтому траектория молекул будет определяться только этими па-
раметрами. В сильных полях необходимо учитывать члены в раз-
ложении Е=Е(8г'} более высокого порядка, чем второй. Это при-
водит к тому, что индуцированный дипольный момент р будет
нелинейной функцией 8г- На рис. IV.3 качественно показана зави-
симость вращательной энергии линейной молекулы для /=0, 1 и 2
от величины поля. Условие фокусировки молекулярного пучка по-
лями А и В обычно выполняется для одного какого-либо враща-
Рис. IV.3. Расщепление вра-
щательных уровней линей-
ной молекулы в электричес-
ком поле S и зависимость
вращательной энергии от
напряженности поля
тельного состояния молекулы и не зависит от ее скорости, а моле-
кулы в других состояниях при этом не попадают на детектор.
В поле С, т. е. на пути молекул из А в В, происходит расщеп-
ление энергетических уровней и при включении поля <£v, перпен-
дикулярного &с, в резонансных условиях часть молекул поглощает
или индуцирование испускает энергию и поэтому изменяет свое
первоначальное состояние. Для этих мо-
лекул нарушаются условия фокусировки
и интенсивность детектируемого сигнала
уменьшается.
Так, например, при исследовании мо-
лекулярных пучков линейных молекул
фокусировали состояния J, М — (1,0)-
При-этом резонанс соответствовал пе-
реходу в состояние J, Л1Д-1= (1,1), т. е.
переходу в состояние с меньшей энер-
гией путем индуцированного излучения
(рис. IV.3). Частота перехода (1,0)^-
->(1,1) задается уравнением (Ш.42)'
как v=AE"/h. Поскольку известны чис-
ла I и М, то для ряда величин <gz ре-
шают системы уравнений и определяют
дипольный момент рю и вращательную
постоянную В. Можно использовать бо-
лее высокие порядки теории возмуще-
ний. Точность измерений определяется
степенью однородности поля С.
Этим методом получают не только
и другие молекулярные постоянные, та-
постоянные, моменты инерции, межъ-
ядерные расстояния и др. Например, получены следующие дан-
ные для галогенидов щелочных элементов.
дипольные моменты, но
кие, как вращательные
Молекула................
Цо, 10-30 Кл-м . . . .
Ге, 10-1 нм.............
39К35С1
37,4
2,66
39К?9Вг
35,1
2,82
133Cswf
26,3
2,34
I33Cs35Cl
34,9
2,88
Важно заметить, что для ионного строения молекулы Cs F~ рас-
чет дает цо= (1,6-10-19) (2,34-10~10) =37,4-10-30 Кл-м, что больше
найденного значения 26,3-10—30 Кл-м. Следовательно, представле-
ние о чисто ионном строении молекулы не является точным.
5. ИСПОЛЬЗОВАНИЕ ДАННЫХ ПО ДИПОЛЬНЫМ
МОМЕНТАМ В ХИМИИ
Следует отметить, что ни один из изложенных выше методов не
позволяет определить направление дипольного момента молекулы,
2 -
так как в уравнения входит или p-о или его абсолютное значение.
Для выяснения этого вопроса используют представления об элек-
троноакцепторных свойствах атомов в молекуле или результаты
квантово-химических расчетов.
Наличие дипольного момента свидетельствует о том, что моле-
кула принадлежит к одной из точечных групп симметрии: Cs, С>,
или Cnv Даже столь ограниченная информация дает некоторое
представление о химическом строении молекул. Как уже упомина-
лось (см. Введение), у молекулы CS2SO4 Цо==О, что не соответст-
вует классической формуле строения.
Дипольные моменты используют для установления химического
строения на1 основе векторной аддитивной схемы:
(1V,2O)
где juz—дипольный момент i-й связи.
Под свойством аддитивности подразумевается сохранение по-
лярных свойств фрагментов молекул — дипольных моментов свя-
зей или групп атомов. Если известны дипольные моменты связей и
измерен дипольный момент ц0, то в относительно простых молеку-
лах можно вычислить валентные углы.
Вращение группы атомов в молекуле симметрии Сга0 вокруг оси
симметрии не изменяет дипольного момента молекулы, как, напри-
мер, вращение групп —СНз, —CF3, —NO2, —СбН5, —FeCsHs и т. п.
Такие группы называют регулярными. Группы атомов, не
о /С1
имеющие осей симметрии, как — NH2, —РН2, —, — С—С1,
н \н
— и т- п-> называются нерегулярными группами.
Их вращение вокруг связи, являющейся осью вращения, вызывает
изменение дипольного момента молекулы.
Предполагая известными валентные углы на основании законо-
мерностей в геометрическом строении молекул, можно рассчиты-
вать дипольные моменты моделей различных конформаций моле-
кул. Согласование рассчитанного дипольного момента одной из
моделей при изменении угла внутреннего вращения с эксперимен-
тальным дает возможность определить конформацию молекулы
(при отсутствии динамической изомерии).
Для определения дипольных моментов связей необходимы дан-
ные или предположения о дипольном моменте какой-либо связи,
например С — Н. Затем на основе подходящих рядов соединений
последовательно оценивают дипольные моменты других связей.
Дипольные моменты связей не являются, условно выражаясь, «аб-
солютными», но зависят от принимаемой аддитивной схемы. Они
даже могут значительно различаться по значению и знаку в раз-
ных схемах, но внутри схемы должны быть внутренне согласованы,
Так, учет изменения валентного состояния атома углерода от тет-
раэдрического типа —С— к плоскому — и далее к линейному
=С— существенно влияет на дипольные моменты связей угле-
род — элемент.
Изучение взаимного влияния атомов в молекуле основано на
отклонениях от аддитивности. Этим путем были выявлены эффек-
ты индукционного влияния, сопряжения, образования координа-
ционных связей и др.
При равновесии двух молекулярных форм измеряемый средний
квадрат дипольного момента
Р-Д + [-1 В ДЛ
p.2=ArAf*l-J-ArBf*|=--------"±_, (IV.21)
, Nb
где цл и цв — дипольные моменты форм А и В; АЛ и Nb— моляр-
ные доли (Aa+Nb=1).
Если ца=#цв и положение равновесия зависит от температуры,
то наблюдаемый дипольный момент также будет, очевидно, функ-
цией температуры ц(Т’). Изучая эту зависимость, можно определить
разность энергий молекулярных форм и константу равновесия
А+*В. Действительно, для этого равновесия
,, Nb ?в ( &Е \ I &Е \ , оо.
KN =------=п-----ехр-------=(Оехр ——- , (IV.22)
N Nk fA \ RI') RT )
где n=~gvlgA— отношение величин статистических весов для форм
В и A; fs и fA—колебательные и вращательные суммы по состоя-
ниям (их отношение во многих случаях, например для упомянутых
поворотных изомеров, близко к единице). Используя уравнение
(IV.22), выражение (IV.21) перепишем в виде
„ Нд+Рв<°ехр( —Л£/ЛГ)
IV ------------------------
1 + ш exp ( — ^.E/RT)
(IV. 2$).
Если рассчитать значения цА и цв по аддитивной схеме (IV.20) и
экспериментально определить ц2(Т), то можно найти Д£=£в—£д,
а при известном множителе w также и Kn — N-b/Na. Так, например,
для 1,2-дихлорэтана было найдено: &Е=Егош—Етранс =5,01
кДж/моль, что хорошо согласуется с данными других методов (см.
гл. XI).
С пбвышением температуры содержание менее устойчивой и бо-
лее полярной гош-формы возрастает, т. е. измеряемый сред-
ний дипольный момент вещества ц увеличивается. Поскольку ди-
польный момент транс-изомера 1,2-дихлорэтана по симмет-
рии (С2/г) должен быть равен нулю, формула (IV.23) позволяет
найти значение дипольного момента гош-изомера ц SOui
тально, что и было сделано: цгоШ = 10,8-10-ЗС1 Кл-м.
эксперимен-
Подобный подход используется также при изучении комплексо-
образования
лдА -)-/гвВ^ А„аВ„в,
где «а и «в — стехиометрические коэффициенты.
Возможно определение молярной поляризации комплекса и
его дипольного момента с одной стороны, а с другой стороны, при
известных дипольных моментах цА и рв, — изучение этого равно-
весия.
Микроволновые исследования с использованием молекулярных
пучков дали возможность определить дипольный момент и некото-
рые другие параметры так называемых ван-дер-ваальсовых моле-
кул типа Ar-BF3, CO-BF3, N2-BF3. В этих молекулах найдена сим-
метрия С3,;, а также следующие параметры (в скобках указаны
ошибки эксперимента):
XBF3............
По, Ю-30 Кл-м...
г(В—X), 10-1 нм ... ,
ZXBF,° ........
ArBFs
0,588
3,325(10)
90k5(5)
OCBFa
1,977
2,886(5)
90,65(25)
N2BFs
1,189
2,875(20)
90,5(5)
Из данных следует, что у молекул Ar-BF3 и N2-BF3 появляется
дипольный момент, а связи В—X характеризуются большими межъ-
ядерными расстояниями.
*
*
Электрический дипольный момент молекулы является важной
характеристикой химического соединения, поскольку дает пред-
ставление о распределении зарядов и электронной плотности в мо-
лекуле, т. е. о полярности самой молекулы и ее химических связей.
На основе экспериментальных значений дипольных моментов моле-
кул и принципа векторной аддитивности дипольных моментов свя-
зей возможно определение симметрии и некоторых структурных
параметров молекул, например валентных углов. Методы изучения
дипольных моментов сыграли значительную роль в развитии уче-
ния о внутреннем вращении, поворотной изомерии и конформациях
молекул.
Особая роль принадлежит дипольным моментам в развитии уче-
ния о химической связи, об электроноакцепторных и электронодо-
норных характеристиках атомов и групп атомов, в развитии пред-
ставлений о взаимном влиянии атомов и групп атомов, об индуктив-
ном эффекте и эффекте сопряжения, об участии неподеленных
электронных пар во внутри- и межмолекулярных взаимодействиях,
в развитии теории комплексных соединений и т. п. В теории хими-
ческой реакционной способности веществ в растворах активно ис-
пользуются представления о диполь-дипольных взаимодействиях,
обусловливающих сольватацию молекул и энергию активации. Дан-
ные о дипольных моментах имеются во многих справочниках (см.
Рекомендованную литературу).
Методы Дебая для определения электрических дипольных мо-
ментов молекул появились несколько десятков лет назад. В настоя-
щее время они стали в каком-то смысле рутинными лабораторными
методами, так как диэлькометры для изучения жидкостей серийно
выпускаются промышленностью и техника работы достаточно прос-
та. Прогресс в этой области исследований существенно зависит от
развития теории жидкости.
Новые экспериментальные методы с молекулярными пучками
имеют более сложную аппаратуру. Этими методами получают очень
важную новую информацию о дипольных моментах и в ряде случа-
ев о структуре малоустойчивых молекул, таких, например, как ван-
дер-ваальсовы молекулы.
Дальнейшее продвижение в этом направлении позволит углу-
бить фундаментальные знания о природе химической связи и меха-
низме химических реакций. В настоящее время один из наиболее
распространенных методов определения дипольных моментов осно-
ван на изучении микроволновых вращательных спектров молекул,
хотя этот метод является, по существу, структурным и рассмотрен
в следующем разделе.
Контрольные вопросы
Глава III
1. При каких точечных группах симметрии молекулы имеют собственный
дипольный момент?
2. При каких точечных группах симметрии молекулы не имеют собственного
дипольного момента?
3. Напишите уравнение энергии молекулы в электрическом поле. Какие виды
поляризуемости можно ввести для молекул?
4. Может ли рассматриваться величина ориентационной поляризуемости в
качестве молекулярной характеристики?
5. В чем состоит эффект Штарка для полярных молекул?
6. Докажите эквивалентность выражений для среднего значения проекции
дипольного момента молекул на направление электрического поля при классиче-
ском и квантово-механическом подходах к решению задачи.
7. Напишите уравнение Клаузиуса — Моссотти. Объясните физический смысл
величин, входящих в это уравнение.
8. Напишите уравнение Лорентца — Лоренца. Объясните физический смысл
величин, входящих в это уравнение.
9. Напишите уравнение Дебая и объясните физический смысл величин, вхо-
дящих в него.
Глава IV
10. Что такое первый метод Дебая определения дипольных моментов мо-
лекул?
И. Что такое второй метод Дебая? Зависит ли определяемое значение ди-
польного момента молекулы от вида растворителя?
12. В чем состоит действие неоднородного электрического поля на молеку-
лярный пучок? Как при этом определяют дипольные моменты молекул?
13. Каковы возможности метода электрического резонанса для определения
дипольных моментов?
14. Можно ли определить направление дипольного момента молекулы рас-
смотренными методами?
15. Каковы проблемы при использовании аддитивной схемы для расчета ди-
польных моментов молекул?
16. Как используются данные по дипольным моментам молекул при изучении
поворотной изомерии?
МЕТОДЫ ОПРЕДЕЛЕНИЯ ГЕОМЕТРИЧЕСКОГО
СТРОЕНИЯ МОЛЕКУЛ
Геометрия молекул является одной из основных характеристик химических
соединений. С ней непосредственно связаны как физические, так и химические
свойства молекул и веществ. Особый интерес представляет геометрия свобод-
ных молекул, т. е. молекул, не подверженных воздействию других молекул или
полей. Ярким примером различия строения молекул в парах при малых давле-
ниях и в кристалле могут служить данные для пентахлорида фосфора РС15.
Молекула этого соединения в парах имеет конфигурацию тригональной
бипирамиды (см. Введение, рис. 4). Однако в кристалле найдена ионная струк-
тура с парой ионов РС1+ (тетраэдр) и РС1 — (октаэдр).
Данные о геометрии молекул необходимы для развития теории строения
веществ, а также для термодинамических расчетов и для развития теории
химической реакционной способности и механизмов химических реакций.
Экспериментальные методы определения геометрического строения сво-
бодных молекул основываются на чисто вращательных спектрах поглощения
в микроволновой области и комбинационного рассеяния света, а также дифрак-
ции быстрых электронов на струе пара.
Геометрическое строение свободных молекул
имеет особое значение для химии, так как состав-
ляет основу теории химического строения моле-
кул.
В настоящем разделе представлены три глав-
ных метода исследования геометрии молекул.
Два первых—метод микроволновой спектроско-
пии и чисто вращательных спектров комбинаци-
онного рассеяния являются спектроскопическими
и основаны на получении и изучении вращатель-
ных спектров молекул. Третий — метод газовой электронографии —
относится к дифракционным методам.
Микроволновая вращательная спектроскопия возникла во вто-
рой половине 40-х годов, поскольку в это время был создан источ-
ник радиоволн в диапазоне частот 10...40 ГГц. Именно в этой об-
ласти расположен чисто вращательный спектр свободных молекул.
Основные условия для получения микроволнового вращательного
спектра состоят в том, чтобы молекулы имели собственный диполь-
ный момент, не равный нулю, и правила отбора разрешали соот-
ветствующие переходы между вращательными уровнями энергий.
Из спектроскопических методов микроволновая спектроскопия
используется наиболее широко. Существенным ограничением этого
метода является относительно малое число возможных определяе-
мых геометрических параметров, т. е. исследование ограничено
лишь относительно простыми молекулами. Важной особенностью
метода микроволновой вращательной спектроскопии является воз-
можность определять дипольные моменты молекул и барьеры по-
тенциалов внутреннего вращения и инверсии молекул.
Чисто вращательные спектры комбинационного рассеяния имеют
более ограниченное применение, но важны для химии, так как по-
зволяют изучать геометрию неполярных молекул, например
CH3CdCH3, СНзСНз, СН2 = СН2 НСз=СН, С6Нз и т. п.
Газовая электронография возникла в 30-х годах. Методика ис-
следования и теория метода существенно изменились к настояще-
му времени. При этом достигнута высокая точность определения
геометрических параметров, сравнимая с точностью микроволновой
спектроскопии. Современная теория строения молекул и методов
их исследования позволяет выявить физический смысл определяе-
мых параметров. В подавляющем большинстве случаев из экспе-
римента находят не равновесную геометрическую конфигурацию,
а некоторую эффективную относительно близкую к равновесной.
Это обусловлено влиянием колебаний молекул, которые по-разно-
му проявляются при расшифровке вращательных спектров или
электронограмм.
В настоящее время высокий уровень эксперимента и теории по-
зволяет проводить совместную обработку данных обоих методов,
что существенно повышает возможности исследования свободных
молекул.
Полученные структурные данные для сотен соединений различ-
ных классов имеют важное значение для химии, так как на их ос-
нове установлены закономерности, связывающие геометрию моле-
кул и их химическое строение, а также введены такие новые поня-
тия теоретической химии, как многоцентровая химическая связь,
полиэдрическое строение молекул, ван-дер-ваальсовы молекулы.
ГЛАВА V
МИКРОВОЛНОВОЙ МЕТОД ИССЛЕДОВАНИЯ
ВРАЩАТЕЛЬНЫХ СПЕКТРОВ МОЛЕКУЛ
1. ВРАЩАТЕЛЬНЫЕ СПЕКТРЫ ПОГЛОЩЕНИЯ МОЛЕКУЛ
Микроволновые спектры изучаются в основном как спектры
поглощения. Во вращательном спектре молекулы появляется ли-
ния поглощения, если при взаимодействии молекулы с электро-
магнитным излучением происходит переход между двумя враща-
тельными энергетическими состояниями. Уравнение для частоты
перехода имеет вид
где £/ и Ei — соответственно энергии верхнего и нижнего состоя-
ний.
Поглощение микроволнового излучения образцом может быть
описано законом Бугера — Ламберта — Бера:
(V.2)
где /qv — интенсивность излучения, поступающего в поглощающую
ячейку длиной /; Iv— интенсивность излучения, выходящего из ячей-
ки; yv — коэффициент поглощения; с — концентрация.
Выражение для коэффициента поглощения получают на основе
квантовой теории излучения. Для микроволновой области оно пред-
ставляет сложную функцию, зависящую от квадрата частоты пере-
хода, формы линии, температуры, числа молекул на нижнем
энергетическом уровне и квадрата матричного элемента дипольно-
го момента перехода. Поскольку yv пропорционален квадрату
матричного элемента дипольного момента, имеем
Ъ-КФ/1 р I Ф/> 12= I н I Ъ= I I I I ?/+ 1 I ?/. (V.3)
где ф; и ф/ — волновые функции молекулы в состояниях i и j; ц —
оператор дипольного момента; рх, цу и цг— проекции дипольного
Момента молекулы на оси лабораторной системы координат.
Из выражения (V.3) следует, что для молекул, не имеющих
Дипольного момента (ц = 0), отсутствует поглощение. Поэтому
Молекулы типа кислорода О2, азота N2, метана СН$, этана С2Не,
бензола CgH6 и т. п., т. е. молекулы, имеющие центр инверсии или
в более общей форме зеркально-поворотную ось Sn (п^2), не
дают чисто вращательного спектра поглощения в микроволновой
области. Дипольным моментом обладают молекулы с симметрией
Сп, Cnv, Cs и Coov (см. гл. III и IV).
С другой стороны, даже когда р,о=#=О, не для всех пар враща-
тельных состояний переходы возможны, т. е. из-за свойств симмет-
рии волновых функций величины у могут быть равными нулю.
Условия, при которых у = 0 или у=^0, определяют правила отбора.
На первом этапе развития метода микроволновой спектроско-
пии исследовались простейшие молекулы с одним или тремя гео-
метрическими параметрами. Для более сложных молекул значения
ряда параметров принимались на основе стереохимических законо-
мерностей. При решении структурных задач значительным продви-
жением вперед было использование, по предложению К. Костейна
(1958), уравнений Д. Крейчмена (1953) для координат атомов, вы-
раженных через разности моментов инерции изотопозамещенных
молекул.
Важным этапом в развитии метода микроволновой спектроско-
пии стали работы Д. Хершбаха, В. Лаури, К. Кучицу, И. Морино и
Т. Ока (1962), направленные на выяснение влияния колебаний мо-
лекулы на определяемые параметры. Они предложили метод расче-
та средних величин геометрических параметров молекул на опреде-
ленном колебательном уровне. Это привело к возможности более
точного сопоставления микроволновых и электронографических
данных и к возможности их совместных исследований (1968).
Вращательная энергия молекул. Ее выделяют из полной энергии
на основе приближения Борна-Оппенгеймера и разде-
ления колебательного и вращательного движений в молекуле, в со-
ответствии с которым полная волновая функция молекулы ф есть
произведение волновых функций электронного фе, колебательного
ф„ и вращательного фд состояний:
Ф==ФЛЛ< (V.4)
а полная энергия:
E = Ee-1rEv-\-Er, (V.5)
где Ее, Ev и Ег—электронная, колебательная и вращательная энер-
гии соответственно.
Вращательная энергия молекулы представляет часть ее кине-
тической энергии, т. е. энергию вращения в пространстве вокруг
ее центра масс. Такое вращение молекулы может быть представ-
лено в произвольной декартовой системе координат с началом в
центре масс как независимое вращение относительно каждой из
трех осей. Однако наиболее удобной и в большинстве случаев ес-
тественной является система главных осей инерции, обозначаемых
буквами а, b и с (вместо х, у и г).
В этой системе координат для У-атомной молекулы имеют мес-
то следующие равенства:
(V.6)
где mi — масса i-го атома молекулы; at, bi, ct— координаты i-го
атома в системе главных осей инерции.
Тогда кинетическая энергия вращения жесткой молекулы (не
испытывающей колебаний и растяжений) Тг выражается в относи-
тельно простой форме:
д2 р2 р2
у. ___ । ГЬ । *_с
г~ 21а 1 2/й 2/с ’
(V.7)
где Ра, Рь, Рс — проекции полного (углового) момента количества
движения Р; Р2==р^ + р^р
д 1а, 1Ь, 1с — моменты инерции отно-
сительно осей а, b и с.
При этом
N N
/a = ^fn!rli:=^ml{b2i-\-c2i'); (V.8)
где ria, rib, ric— расстояния i-го атома от осей, а, Ь, с соответст-
венно.
Уравнение (V.7) может быть переписано в операторном виде.
Введем оператор Tr = flr. Тогда
Нг=АРа + ВР2ь + СР2с, | (V.9)
где А~----, В и С — вращательные постоянные.
8л2/д
Решение уравнения Шредингера
Hri>r=E^r (V.10)
обусловлено типом молекул и зависит от соотношения величин 1а,
1ь и 1С. Принято выбирать оси так, что 1а
Рассмотрим различные типы молекул.
Двухатомные и линейные молекулы симметрии Cxv. Для этого
типа молекул /а = 0, а 1ь = 1с. Оси b и с перпендикулярны оси моле-
кулы. Тогда
Er — hBJ (J 4-1),
(V.11)
где J — вращательное квантовое число, принимающее значения
О, 1 и т. д.
Правила отбора для спектра поглощения определяются равен-
ством А/= +1 [см. уравнение (IIL33)]. Это приводит к простому
виду спектра
+ 1)(Z + + 1)/ =2а(У+1)| (V.I2)
Рис. V.l. Схематическое расположе-
ние уровней энергии молекулы типа
асимметричного волчка:
К+1 и —квантовые числа К соответ-
ственно сплюснутого и вытяиутото волч-
ков. Вращательная постоянная В умень-
шается слева направо
где v — частота вращательного
перехода, Гц; В — вращательная
постоянная двухатомной молеку-
лы, Гц; J — квантовое число>
нижнего уровня энергии.
Переход записывается следу-
ющим образом, например 7 = 0-*-
-И или 7= 1-^-2 и т. д.
Молекулы типа симметрично-
го волчка. К ним относятся мо-
лекулы, имеющие одну ось Сп
при л^З. Например, СН3С1(С3с),
СН3-С= С-Н (С31>), СС13Н (Сзо),
С5Н5ВеН(С’51)) и т. п. В этих мо-
лекулах имеют место следующие
соотношения:
1) 1а<.1ь=1с И А>В = С (для
СН3С1 и CH3Ces ОН)—вытя-
нутый волчок, ось а совпа-
дает с осью Сп',
2) Ia = Ib<Jc и А=В>С
(для СС13Н и CsHsBeH) -—
сплюснутый волчок. Ось с
совпадает с осью симметрии Сп.
Вращательная энергия зада-
ется двумя квантовыми числами
J и К. Число # определяет про-
екцию полного момента количе-
ства движения молекулы на ось
Сп и изменяется от —I до
(ось
Таким образом, для вытянутого симметричного волчка
Сп вдоль оси а)
Л'Г = A [BJ (J + 1)+(А - В) №].
Для сплюснутого волчка нужно заменить А на С, т. е.
(V.13)
Поскольку квантовое число /( соответствует моменту вращения
молекулы вокруг оси Сп, а это вращение не вызывает изменения ди-
польного момента, правило отбора для К есть Д/( = 0. Для J сохраня-
ется прежнее правило отбора: Д7= + 1. Поэтому вращательные
спектры будут определяться уравнением (V. 12), т. е. переходами
между вращательными состояниями относительно оси, например Ь,
перпендикулярной оси Сп молекулы. При таком вращении изменя-
ется направление дипольного момента молекулы в пространстве.
Система энергетических уровней Е"га; и Е^пЛ, задаваемых кван-
товыми числами 7 и К, отличается тем, что с увеличением К энер-
гия £’,ыт повышается, так как (А—В)>0, а £'гпл — уменьшается,
так как (С—В) <0. Этот характер изменения Z+bIT и £,'пл использу-
ется для классификации уровней энергии асимметричного волчка
(рис. V.1).
Молекулы типа асимметричного волчка. Все три момента инер-
ции молекулы этого типа различны: 1а<-1ь<1с и соответственно
А>В>С. Характер асимметрии молекулы может быть выражен
количественно через параметр асимметрии:
2В-А-С
X —-----
А—С
(V.14)
Для предельных случаев: 1) сплюснутого волчка х= + 1 и 2) вытя-
нутого волчка х =—1. Наибольшей асимметрией обладают молеку-
лы с х = 0. При этом В = -^~ , т. е. вращательная постоянная В
2 -
является средней междуД~и С.
С помощью параметра асимметрии х уравнение (V.9) преобра-
зуется в уравнение с квадратом оператора полного момента количе-
ства движения: Р2 — Ра-\~Рь-{-Рс:
где = +
Данному гамильтониану Йг соответствуют энергетические уров-
ни асимметричного волчка:
ЕГ=-(А-^С) V(J-H)+ (-~^ ^(х), (V. 16)
где £^(х) — функция только параметра асимметрии х. Для данно-
го J имеется 27+1 функций El(х), , задаваемых числом т, которое
изменяется от —/ до + /, т. е. номер уровня энергии можно задать
в форме /т. Величины f/(x) табулированы для /х и х.
Следовательно, число уровней энергии асимметричного волчка
больше, чем для линейных молекул и молекул типа симметричного
волчка. Наряду с нумерацией подуровней по числу т в настоящее
время используют также другие обозначения на основе корреляции
уровней энергии асимметричного волчка и уровней энергии симмет-
ричных волчков — вытянутого и сплюснутого.
На рис. V. 1 изображены слева уровни и подуровни сплюснутого
волчка, а справа — вытянутого. Для симметричных волчков имеет
место двухкратное вырождение по квантовому числу ТС Однако при
переходе к асимметричному волчку происходит расщепление каж-
дого уровня на два подуровня, что показано на рис. V.I. Для асим-
метричного волчка при изменении В от А = В к В = С (Х>С) или
при изменении х от +1 до —1 подуровни энергии качественно бу-
дут следовать линиям, соединяющим соответствующие подуровни
симметричных волчков. При этом используется правило непе-
ресечения линий подуровней с одинаковым J, но уровни
с разными значениями J могут пересекаться. Для данного J число
линий подуровней равно 27 + 1. Нумерация уровней начинается сни-
зу. Первое число в индексе указывает квантовое число вытянутого
волчка, а второе — сплюснутого. Например, 20г означает, что 7 = 2
и что этот уровень в предельных случаях совпадает с уровнями
Л_]=0 или Л+1 = 2. Индексы —1 и +1 являются параметрами х
Рис. V.2. Схема уровней энергии асимметричного волчка:
показаны переходы для А/ = 0 (Q-ветвь), 7 = 4-1 (Я-ветвь) и Х7 = — 1
(Р-ветвь)
соответственно вытянутого и сплюснутого волчков. Полное обозна-
чение подуровня задается в виде В новых обозначениях
т=ЛД1-Л+!.
Правила отбора для асимметричных волчков определяют изме-
нение Д7 = 0, ±1 и изменения ДК-i и AK+i, которые носят более
сложный характер. Если дипольный момент не совпадает по на-
правлению ни с одной из главных осей, то разрешены все возмож-
ные изменения ДК-i и ДЛ+i, кроме случая, когда оба эти измене-
ния четны. Любой переход обусловлен только одной проекцией
дипольного момента р,а, ць или цс.
Качественная картина уровней асимметричного волчка для
7=0, 1, 2 и 3 дана на рис. V.2. Переходы между подуровнями для
одного J (Д7 = 0) называются Q-ветвью, для Д7 = 1—R-ветвью и
Д7=— 1—Р-ветвью. Рис. V.2 иллюстрирует переходы =
= 1о1_>1ц, 1ц—*-2]2 И Зоз~*2ц.
Эффект Штарка. В однородном электрическом поле наблюдает-
ся расщепление вращательных линий в связи с тем, что частично
или полностью снимается вырождение в направлении поля (см.
гл. III). Вращательная энергия в этом случае зависит также от
магнитного квантового числа М, принимающего значения от —7 до
+ 7. Она является функцией дипольного момента молекулы ц0 или
его проекций на оси а, Ь, с и от напряженности приложенного по-
ля (go.
Для двухатомных и линейных молекул (см. гл. III)
£r(^0) = ABJ(J + l)
р-pgp 3W _J (У + 1)
2AB/(/+ 1) (27-1) (2/ +3)
Второе слагаемое обусловлено энергией взаимодействия диполя ц
€ полем <S. Правила отбора по 1 сохраняются, т. е. Д7 = + 1. Если
поле <о параллельно линейно поляризованному поглощаемому ра-
диоизлучению, то ДЛ1 = 0 (рис. V.3).
Рис. V.4. Прецессия вектора полного
момента количества движения Pj во-
круг направления электрического по-
ля ё (конус прецессии Р/) и прецес-
сия оси симметричного волчка вокруг
вектора Р/ (конус прецессии Рк); на
рисунке векторы обозначены стрел-
ками
V.3. Расщепление
Рис.
уровней вращательной
энергии в электрическом
поле на составляю-
щие для линейных, моле-
кул или молекул типа
асимметричного волчка:
стрелками показаны перехо-
ды для Д/ = 1 и Д.И = 0
Для симметричного волчка классическая картина вращения
представляет прецессию оси волчка около направления
полного момента количества движения Pj (рис. V.4). При наложе-
нии электрического поля S вектор Pj начинает прецессировать во-
круг направления этого поля. Если угол прецессии около Pj равен
а, а около <£ составляет р, то можно получить уравнения для проек-
ции дипольного момента молекулы на направление поля <8.
Поскольку дипольный момент молекулы типа симметричного
волчка направлен вдоль оси молекулы, то при прецессии проекция
ц на Pj является постоянной величиной и равна pocosa. Проекция
этой составляющей на направление поля <8§ в свою очередь дает
компонент [л ^,=цо cos a cos р. Величины cos а и cos р удобно выра-
зить через соотношения
Pr = Pj COS <l=4lK И Рм= PjCOi, $—hM,
где Рк и Рм — проекции Pj на ось молекулы и на направление по-
ля К и М— квантовые числа для проекций вектора Pj;
= =
Дальнейшие преобразования с использованием условий кванто-
вания Pj, Рк и Рм позволяют получить конечный результат:
—и Рк рм - — км
° Pj Pj J (J + 1)
(V. 17)
Поскольку энергия диполя в электрическом поле пропорциональ-
на проекции диполя, то
^8 ~ — Pg — ~ Р'О^’оу -
(К. 18)
В линейных молекулах а = л/2 и cosa = 0. Это объясняет отсут-
ствие линейного слагаемого в уравнении эффекта Штарка для ли-
нейных молекул.
Для молекул типа симметричного волчка имеют место следую-
щие правила отбора: Д7= + 1; ЛК=0; ДЛ4=0. Поскольку для этого
типа молекул энергия взаимодействия с полем пропорциональна
первому порядку произведения цо^о, то в соответствии с правилами
отбора легко выразить частоту перехода (поглощения):
‘2МК?-о 8 о
/(/+ 1)(/ Ч-2)Л
(V. 19)
У асимметричных волчков эффект Штарка обычно бывает вто-
рого порядка. Энергия взаимодействия такого волчка с полем в об-
общенной форме может быть представлена в виде суммы:
Ej^p=#20 2 АМ
g = а. Ь ,с
(V.20)
где pg — составляющие дипольного момента молекулы по главным
осям а, Ь, с\ Лг(%, J., M2j) — коэффициенты как функции парамет-
ров х, Jх и M2j-
Эффект Штарка используется для отнесения линий в
спектре и определения дипольных моментов молекул.
Влияние колебаний молекул на вращательные постоянные. Ре-
альные молекулы не являются жесткими ротаторами. Нежесткость
проявляется в центробежном растяжении, увеличивающем моменты
инерции, а следовательно уменьшающем вращательную постоян-
ную. Однако если исследуются спектры поглощения для малых /
(до 4 или 5), то центробежным растяжением можно пренебречь.
Кроме того, вращательные постоянные непосредственно связаны с
колебательным состоянием, задаваемым квантовым числом и. На-
пример, для постоянной В имеем
в=ву=ве^аь
(V.21>
где Be — вращательная постоянная равновесной конфигурации; ин-
декс s соответствует s-му нормальному колебанию; а?— постоянные
колебательно-вращательного взаимодействия; ds — степень вырож-
дения; vs — квантовое число s-ro нормального колебания.
Такие же выражения могут быть написаны для вращательных
постоянных А и С.
Постоянные ctf (g=a, b, с) в свою очередь являются суммой
двух слагаемых, представляющих так называемые гармоническую
af (гарм) и ангармоническую сД (ангарм) составляющие, т. е.
a® = af (rapM)4-af (ангарм). (V.22)
Следовательно, колебательное возбуждение молекул приводит
к увеличению числа линий в чисто вращательных спектрах, так как
каждому v будет соответствовать свой вращательный спектр. Отно-
шение интенсивностей в таком спектре определяется больцманов-
ским распределением по колебательным состояниям.
Значения af (гарм) для многоатомных молекул могут быть
вычислены для заданных масс атомов, геометрии и гармоническо-
го силового поля молекулы.
Величина af (ангарм) содержит ангармонические постоянные
потенциальной функции, которые известны только для ряда очень
простых молекул.
Для плоской жесткой молекулы (для равновесной конфигура-
ции плоской молекулы) имеет место равенство
(V.23)
где ось с перпендикулярна плоскости.
Оно легко доказывается на основе уравнения (V.8); так как
с,.=0, то /а = ^т^, Jb = ^mpi и = = 4 + Д
Однако эффективные моменты инерции из-за наличия колебаний
в молекуле приводят к так называемому дефекту инерции:
A = | (V.24)
Величина А — положительная и значительно меньше 1а, 1ь, 1С. Уже
трехатомные молекулы обладают дефектом инерции. Дефект инер-
ции плоских молекул не зависит от ангармонических постоянных
и определяется в основном кориолисовым взаимодействием, неплос-
кими колебаниями и постоянными центробежного искажения. Ко-
лебания в плоскости дают положительный вклад в А, а неплоские
Колебания — отрицательный. Первые имеют обычно большее значе-
ние, поэтому А>0. Однако в случае одного или нескольких низко-
частотных неплоских колебаний, например, в циклах может ока-
заться, что Д<0. Приближенно оценить А дает возможность соотно-
шение
A^/z/(2n2v), (V.25)
где v — самая низкая частота колебаний в плоскости молекулы.
2. МЕТОДИКА ЭКСПЕРИМЕНТА В МИКРОВОЛНОВОЙ
ВРАЩАТЕЛЬНОЙ СПЕКТРОСКОПИИ
Изобретение источников электромагнитного излучения в области
частот 10 000 ... 40 000 МГц послужило основой развития микровол-
новой спектроскопии. Таким источником явился клистрон — элек-
тронный прибор для генерирования и усиления СВЧ-колебаний, в
котором поток электронов, сформированный в сгустки, создает в
резонаторе монохроматическую линейно поляризованную электро-
магнитную волну. Размеры резонатора изменяются механически,
что приводит к изменению частоты излучения на 15%, а для получе-
ния полного спектра используют обычно несколько клистронов.
В настоящее время более широко применяется отражательный
клистрон или лампа обратной волны.
Стабилизированное и линейно поляризованное радиоизлучение
источника пропускают через поглощающую ячейку со штарков-
с к и м электродом (рис. V.5). Ячейка, как и волноводы, имеет
прямоугольное сечение и изготавливается из латуни. В центре ячей-
ки на изоляторах устанавливается штарковский электрод. Вектор
напряженности подаваемого линейно поляризованного излучения
и электрическое поле штарковского электрода параллельны.
Длина поглощающей ячейки 3...5 м. На штарковский электрод по-
дается потенциал прямоугольной формы с частотой от 5 до 100 кГц
и напряжением от 600...3000 В, который позволяет осуществлять
принцип молекулярной модуляции — изменение интенсивности
спектра из-за расщепления линий в электрическом поле.
Детектирование сигнала осуществляется с использованием по-
лупроводниковых кристаллов кремния, германия и ряда других.
Частоты поглощения измеряются с точностью около 10 кГц, т. е.
при 10 000 МГц относительная точность составляет 10~6.
Общая блок-схема радиоспектрометра дана на рис. V.6. Изуче-
ние микроволновых спектров поглощения проводится при давлении
пара образца 0,1...10 Па. Поэтому в определенных случаях необхо-
димо или нагревание образца, или охлаждение. Повышение давле-
ния в ячейке приводит к увеличению чувствительности, но уменьша-
ет разрешающую способность. Это обусловлено тем, что с повыше-
нием давления увеличивается полуширина линий за счет столкнове--
ний между молекулами, со стенками ячейки, за счет эффекта
Доплера и т. д. Уменьшение давления, с одной стороны, повышает
разрешающую способность, но сопровождается эффектом на-
сыщения, поскольку большая плотность радиации изменяет
больцмановское распределение, увеличивая заселенность верхних
«4
уровней. Эффект насыщения вызывает уширение линий и уменьше-
ние их интенсивности. Для регулировки плотности излучения в схе-
му радиоспектроскопа введены аттеньюаторы, ослабляющие мощ-
ность излучения.
Амплитудная молекулярная модуляция с помощью штарковско-
го электрода уменьшает влияние низкочастотных шумов и увеличи-
вает чувствительность спектрометра.
Рис. V.6. Блок-схема радиоспектроскопа:
/ — генератор СВЧ; 2 — волномер; 3— волновод;
4 — слюдяное окно; 5 — поглощающая ячейка с
нагревателем; 6 — штарковский электрод; 7 — сис-
тема создания вакуума н ввода газообразного об-
разца; 8 — генератор прямоугольных импульсов;
9— детектор; 10 — усилитель; 11 — индикатор
'\ 2\
Рис. V.5. Сечение
поглощающей
ячейки радиоспект-
рометра
ковским
дом 1,
ным на
со штар-
электро-
укреплен-
изоляторе
2
Радиоспектрометры обладают высокой чувствительностью и по-
зволяют измерять вращательные спектры многих изотопозамещен-
ных молекул в их естественной распространенности (без дополни-
тельного обогащения) и определять вращательные постоянные для
низколежащих возбужденных колебательных состояний. Макси-
мальная чувствительность спектрометров соответствует определе-
нию сигнала на фоне шума с соотношением 1 : 1. При этом у в
уравнении (V.3) составляет 10~8 м-1, что соответствует изменению
в измеряемой мощности поглощения 10~6% для ячейки длиной 1 м.
Однако относительная неточность в определении интенсивности ли-
нии достигает нескольких процентов.
Исследования неустойчивых молекул, в частности ван-дер-вааль-
совых молекул типа Кг-НО, Ат-СЩЮ (фуран) и т. п., поставили
перед микроволновым экспериментом следующие задачи: получе-
ние больших концентраций молекул такого типа, увеличение ско-
рости записи спектра, чувствительности и разрешения спектро-
метра.
Первая задача решалась при использовании импульсного источ-
ника газообразного объекта исследования в виде сопла специаль-
ной конструкции (сверхзвуковое сопло) с прерывающимся истече-
нием газа. Пульсирующий режим позволяет использовать большие
Давления (до 50 ... 200 кПа) исследуемого вещества перед выпус-
ком из сопла. В результате увеличивается концентрация молеку-
лярных комплексов на несколько порядков по сравнению с режи-
мом непрерывного истечения.
Быстрое расширение струи газа в вакуум приводит к резкому
его охлаждению. Эффективная температура для вращательной сте-
пени свободы молекулы с аргоновым носителем падает до 2 ... ЮК,
что значительно повышает чувствительность спектрометра.
Вторая задача решалась методами импульсной фурье-спектро-
скопии в микроволновой области. Адиабатически расширяющийся
газ проходит через резонатор Фабри — Перо со сферическими зер-
калами радиуса ~35 см и расстоянием ~75 см между ними. Вре-
мя прохождения газа через' резонатор составляет 100 мкс ... 1 мс.
Под прямым углом к струе газа направляется микроволновое из-
лучение в виде импульса длительностью ~2 мкс и частоты
~ 10 ГГц (область поглощения).
После затухания микроволнового импульса открывается волно-
вод, выводящий когерентное излучение эмиссии возбужденных мо-
лекул, которое после некоторых преобразований ~й "усиленияреги-
стрируется как функция мощности эмиссии от времени. Этот сигнал
затем подвергается фурье-преобразованию, которое приводит к
получению искомого частотного спектра.
Один короткий микроволновой импульс при фиксированной гео-
метрии резонатора дает возможность измерить область частот
~ 1 МГц за ~40 мкс. Чтобы получить спектр во всей области (по-
рядка 10 ГГц), нужно последовательно изменять расстояние между
зеркалами резонатора с малым шагом (~41 мкм) и с соответст-
вующей перестройкой СВЧ-генераторов с шагом ~500 кГц. Экспе-
римент автоматизирован с помощью мини-ЭВМ.
Этот метод микроволнового исследования не позволяет опреде-
лять дипольные моменты молекул в отличие от метода электриче-
ского резонанса с использованием также пульсирующего сверхзву-
кового сопла.
Импульсная микроволновая вращательная фурье-спектроскопия
еще не приобрела столь широкого распространения, как, например,
ИК (см. гл. XII) или ЯМР спектроскопия, но имеет все данные для
этого.
Импульсная фурье-спектроскопия, как общий метод спектроско-
пического исследования, позволяет существенно ускорить измере-
ние спектра, так как каждый импульс дает информацию о доволь-
но большом интервале частот. Увеличение чувствительности об-
условлено увеличением соотношения сигнал — шум, которое дости-
гается благодаря большей мощности падающего излучения (нет
столь жесткого ограничения падающего излучения щелями спектро-
метра) и тем, что все частоты регистрируются одновременно. Высо-
кое разрешение этой методики связано с использованием интерфе-
рометров.
Фурье-преобразование и спектральный анализ сводятся к рас-
четам определенных интегралов. Интерферограмма представляет
когерентный спад свободной индукции для всех частот перехода
как функцию I от разности хода интерферируемых лучей А, а прак-
тически от времени \t, так как разность хода изменяется с постоян-
ной скоростью перемещения зеркал. На основе теории гармониче-
ского спектрального анализа можно выразить функцию спада ин-
дукции от времени I (£) через функцию интенсивности, в частотной
области S(v), которая характеризует возбуждаемый эмиссионный
спектр молекулы (системы в общем). Такое соотношение в общем
виде дается интегралом или преобразованием Фурье:
I (t)= 1Z — 5 (v) cos 2nv/ dv.
r rt J
о
Замечательным свойством этого уравнения является обратный пе-
реход к функции S (v):
S (v)=2 у^2л JI (/) cos 2лч/ d/.
о
Функции /(/) и 5(v) называют парой косинус-преобразований
Фурье.
Для отдельного перехода с частотой Vo и полушириной линии
Av интерферограммы представляют затухающую косинусоиду с по-
стоянной затухания, обратно пропорциональной Av, и промежутка-
ми между максимумами по оси расстояний, т. е. разности хода лу-
чей, равного длине волны Zo=c/vo. При большом числе переходов
имеет место наложение этих функций.
3. МЕТОДЫ РАСЧЕТА ГЕОМЕТРИЧЕСКИХ
ПАРАМЕТРОВ МОЛЕКУЛ
Определение геометрических параметров из вращательных по-
стоянных является обратной задачей метода микроволновой спект-
роскопии. Решение этой задачи часто может быть неоднозначным,
так как для данного изотопного состава молекулы по спектру опре-
деляются максимум три момента инерции (в зависимости от типа
молекул). Но действительное число определяемых параметров ока-
зывается большим, чем число независимых геометрических пара-
метров. Это понятно из уравнения (V.21) для Bv, поскольку оно
включает также постоянные as. Поэтому общая задача определения
геометрических параметров разделяется на две: определение рав-
новесных геометрических параметров (ге-структура) и, если невоз-
можна оценка полного вклада колебаний, эффективных геометри-
ческих параметров (г0-, rj- и г2-структуры).
Определение равновесной структуры молекул. гв - Структу-
ра. В соответствии с уравнением (V.21) для определения Ве необ-
ходимо решить систему уравнений для различных Bv и vs. Экспе-
риментально эта задача решена для небольшого числа простых мо-
лекул, поскольку возбужденные колебательные состояния имеют
Малую заселенность, и определение вращательных спектров погло-
4-949 97
щения для возбужденных колебательных состояний представляет
очень трудную проблему, особенно для валентных колебаний.
Так, линейная трехатомная молекула XYZ имеет четыре нор-
мальных колебания. Но два деформационных колебания вырожде-
ны, так как совершаются с одинаковой частотой в перпендикуляр-
ных плоскостях. Поэтому наблюдаются три частоты колебаний: vt
и V3 — валентные колебания, V2 (v2a и V2z>) —дважды вырожденное
деформационное колебание. Тогда уравнение для Bv будет иметь
следующий вид:
=Ве - И1 +-у) - а2 (ц2 4- 1) - а3 |ц3Д--у) .
Измерения Ви для основного состояния (0, 0, 0) и для первых
возбужденных колебательных состояний (1, 0, 0), (0, 1,0) и (0, 0, 1)
позволяют определить Ве, си, аг и аз. Такие же измерения необхо-
димо проделать для изотопически замещенной молекулы и опре-
делить В1е, а{, аг и аз- Из двух уравнений для Ве и В1е можно
рассчитать ге(Х—У) и re(Y—Z). Для нелинейных молекул XY2 та-
кие измерения дают сразу три момента инерции (из трех враща-
тельных постоянных Ае, Ве, Се). Так как молекулы плоские, то
1с=1а + 1ь. Но даже двух моментов инерции в случае молекул XY2
достаточно, чтобы определить два равновесных параметра ге(Х—Y)
и <YXY. Данные по ге-структуре имеются для таких молекул, как
Н2О, H2S, H2Se, SO2, OF2, O3, SeO2, SiF2, GeF2, HCN, C1CN,NH3,
NF3, CH4 и др.
Однако наиболее распространенными являются микроволновые
исследования по определению эффективных геометрических пара-
метров.
г0 - Структур а. Обычно определяются вращательные посто-
янные для нулевого колебательного уровня Ао, Во, Со. Для моле-
кул типа Н2О, H2S, SO2 и т. п. этих данных достаточно, чтобы рас-
считать геометрические параметры, если предполагают, что, напри-
мер,
Во-—6^ 1 2 =—(V.26)
8л2 \ S mirtb /о 8л2/»
Значение вращательной постоянной Во выражается через эф-
фективный момент инерции /*, который в свою очередь опреде-
ляется через эффективные расстояния го/ь i-ro атома от оси b (а
также в случае осей а или с), т. е., например,
Эффективные величины Гога, roib, Гоге дают возможность рас-
считать независимые геометрические параметры, определяющие
так называемую г0-стРУктУРУ-
Рассмотрим пример трехатомной молекулы XYZ симметрии Czv. В качестве
независимых параметров удобно принять длину связи г0(Х—Y) и валентный угол
6= ZYXY. Задача состоит в том, чтобы на основе экспериментальных данных
по /д и найти г0 и 0 (далее !а =1а и /£=/ь).
В координатах главных осей инерции для XYZ (рис. V.7) атомы имеют следу-
ющие координаты: Х(ах, 0), Yi(—Яу, by) и Y2(—aY, —by), откуда получаем, что
1а=2туЬ^ и = (т-щ и ту— массы атомов X и Y). Для
2туйу
центра масс по оси а имеем уравнение /ихах==2туау и «х =--------------.
тх
Из свойств равнобедренного треугольника XY2 следует, что Ьу=Го sin 0/2 и ах4-
2mY
-Г Лу — cos 0/2 = Лу 4-
mx
mx
Поэтому Лу = —— r0 cos 0/2 и ax =
2in
a-
\ тх )
rQ cos 0/2.
М
Таким образом, все координаты выраже-
ны через независимые геометрические пара-
г0 и 0. Подстановка их в уравнения
метры
ДЛЯ 1а
и 1ь дает:
/а = 2туГц sin2 в/2
и
2mxmy
ro2cos29/2.
Отношение 1а]1ь позволяет определить 0 из
уравнения
'а
lb
М
----tg20/2, откуда tg9/2 =
mx
<Zy — 2mу -j- тх).
m,. 1 Л
Рис.
= (тх М
\ М I ь)
Затем, умножив /а на тх/М и сложив с 1ъ, получаем
V.7. Главные оси инерции
в молекуле XY«
М
Для Го уравнение имеет вид
2mxWy
.2
о*
М
М
тх
1
2m у
г* =
Задача решена.
Для сложных молекул необходимое число уравнений получают
на основе вращательных постоянных изотопозамещенных молекул,
Предполагая, что эффективные величины геометрических парамет-
ров одинаковы в молекулах разного изотопного состава.
Наиболее общим методом для определения г0-структуры яв-
ляется минимизация квадратичного функционала для моментов
инерции:
/g'reop)2 = min,
< g=a,b,c
(V.28)
где I°g3Kcn, I°gteop —экспериментальные и теоретические моменты
инерции для i-го изотопического замещения и для g=a, b, с —
главных осей инерции; <ог — статистический вес.
Однако влияние колебаний на нулевом уровне приводит к тому,
что предположение о равенстве параметров не является обоснован-
ным. Различия в определяемых параметрах достигают ~0,01Х
ХЮ-1 нм (0,01 в относительных единицах), что значительно пре-
вышает неточность определения эффективных моментов инерции
(или вращательных постоянных), составляющей ~10-5 в относи-
тельных единицах. Так, два изотопных набора линейных молекул
OCS, достаточных для определения длин связей С = О и C = S,
приводят к заметной разнице в этих параметрах (10-1 нм):
Изотопный набор Гд (С=О), 10-1 НМ ro(C=S), 10-1 нм
ieQ12Q32S ieQ12Q34S 1,1647 1,5576
16Q12C32S 13Q12C32S 1,1552 1,5653
Анализ показывает, что только для двухатомных молекул г©
имеют определенный физический смысл:
h
8л2^В0
(V.29)
/ 1 \-1/2 /
го=хТ/ =
\ г2 /о \
Если представить г=ге+х, где х — малая величина, то разложе-
ние в ряд дает:
го=<(ги-г2)Л^|4-^-+3-^-Г1/2
ге ге ге '
3 <х2>0
— \х/о -----------------
£ •е
(V.30)
Потенциальную энергию в ангармоническом приближении задают
часто в упрощенной форме:
1/(л)=-|-(л:2 — ах3), (V.31)
где k — гармоническая силовая постоянная; а — ангармоническая
постоянная. Поскольку молекулы рассматриваются в стационарном
состоянии, то среднее значение силы /------= Это равенство
\ dx /
можно пояснить также выражением усредненного значения силы
через второй закон Ньютона:
_/Л.\=от d2<<2_^0, (V.32)
\ dx / d/2
так как —0 для стационарного состояния. Подставив в урав-
нение (V.32) выражение (V.31), получим
/—\ = /kx —- kax2^ == О
\ dx / \ 2 /
или для нулевого уровня
<-^)о=-|-« Wo-
(V.33J
Следовательно, в соответствии с уравнением (V.30) имеем
Эффективная величина г0 практически всегда больше ге, на
меньше, чем среднее значение гг:
rz=(r)0^re-\-~a {х2)0. (V.35)
Величину rz вводят для обозначения среднего значения межъядер-
ного расстояния на нулевом колебательном уровне (индекс z—от
англ, zero — нуль), которое имеет более ясный физический смысл,
чем г0.
г г - С т р у к т у р а. Из уравнения (V.34) следует, что г0 вклю-
чает две поправки к ге: ангармоническую —а (х2)0 и гармониче-
скую -|- (х2)0.
Вычитание гармонической поправки приводит к гг-
параметру (см. уравнение V.35).
Для многоатомных молекул Д. Хершбах и В. Лаури показали.
что введение гармонических поправок в выражения для вращатель-
ных постоянных До, Во, Со дает возможность получить вращатель-
ные постоянные Аг, Вг, Сг. Так, из уравнений (V.21) и (V.22) с
учетом af (гарм) имеем
5г=50-|-4- б5а*(гарм).
(V.36}
Таким образом, расчет Вг основан на анализе нормальных коле-
баний молекул. Для расчета af(гарм) необходимо знать частоты
нормальных колебаний, коэффициенты линейного преобразования
изменений декартовых координат в нормальные координаты, а так-
же постоянные кориолисова взаимодействия.
Вращательные постоянные Д2, Вг, Сг выражаются в хорошем
приближении через моменты инерции, связанные со средними зна-
чениями координат атомов <с«>о, например
(V.37)
Z
Для Ig (g=a, Ь, с) плоских молекул отсутствует дефект инерции.
Подобные уравнения могут быть выведены и для возбужденных
Колебательных состояний.
Структура молекулы, определяемая минимизацией функцио-
нала
= 2 («г(/|эксп —/^reop)2=min, (V.38)
g-a.b.c
носит название гг-структуры. Физический смысл получаемых пара-
метров соответствует среднему значению координат атомов для ну-
левого колебательного уровня.
Для относительно сложных молекул необходимо рассчитывать
Az, Вz, Cz для нескольких изотопозамещенных. Однако существуют
ограничения при использовании изотопного замещения для опреде-
ления ^-структуры, так как точный расчет требует знания ангар-
монических постоянных.
rs - Структур а. В 1958 г. К. Костейном показано, что коле-
бательные эффекты значительно уменьшаются при использовании
уравнений Крейчмена, которые позволяют рассчитывать положения
атомов в молекуле, если известны моменты инерции молекулы для
двух ее изотопных замещений атомов. При этом Д. Крейчменом
предполагалось, что молекулы являются жесткими, длины связей
и валентные углы не изменяются при изотопном замещении, т. е.
эти уравнения строго справедливы для равновесной конфигурации
ядер.
Рассмотрим сначала пример линейной молекулы. Момент инер-
ции линейной молекулы выражается через расстояния ядер атомов
от центра масс молекулы:
(V.39)
Условие для центра масс т;гг^=0. При изотопном замещении,
например, первого атома, т. е. при замене mi на mi + Ami, центр
масс смещается:
(т1 + Дт1)(г1 + Дг1) + т2(г24-Дг1)4-тз(г34-Дг1)-|-...=0.
Раскрывая скобки и учитывая, что^mzrz = 0 и mz = M, находим
Дг1 =----(V.40)
М + Ami
Момент инерции при таком изотопном замещении выражается
уравнением
lV = (mi + kmi)(ri + kri)2 + m2(r2+kri)2-{-... (V.41)
Преобразование уравнения (V.41) и подстановка в (V.39) дает в
^орошем приближении
+ (V.42)
М + Ami
Такие соотношения могут быть выведены для координаты любого
102
атома. Следовательно, получаем простое уравнение для координа-
ты i-ro атома:
М&т; р-г
(У.43)
где и., = - ^—т.‘-можно условно назвать приведенной массой.
М + \mi
Из уравнения (V.43) следует, что ошибка в определении рас-
стояния гг- дается уравнением
= —-------8Д/#, (V.44)
2рг|^г|
где 8Д/&— ошибка в определении разности моментов инерции.
Уравнение (V.44) показывает, что для атомов, близких к цент-
ру масс молекулы, возникает существенная неопределенность в ко-
ординате ri.
Более сложные выражения имеют место для асимметричных волчков-.. Для
плоских молекул
Для неплоского асимметричного волчка
д2 ЬРь( \РС \[ ХРа I
el~ IP 11+ 1еь-1ес Д1-Г 1еь-!а /’ ! (V‘46)
где ХРа- '—Л/$ + 4-и т. д. в соответствии с циклическими пере-
становками.
Расстояние замещенного атома от центра масс выражается уравнением
4' = Щ + Л/*) (V-47)
Следовательно, ошибка в определении п так же зависит от п, как и для линей-
ных молекул:
8keil^ 4ut-|re,-j + + (V.48)
Из (V.48) следует, что координаты атомов, близко расположенных к осям, опое-
Делаются с малой точностью. г
Для симметричных волчков при замещении атомов на оси Сп
используются уравнения линейных молекул, а вне оси Сп — асим-
метричных волчков.
Число необходимых изотопных замещений определяется числом
неэквивалентных атомов. Координаты атомов, близкие к центру
масс, несколько лучше определяются из соотношения для центра
N
масс: ^^;г,6 = 0.для нескольких атомов, близких к центру масс,
i
используется техника двойного замещения.
К. Костейн предложил использовать уравнения Д. Крейчмена
для экспериментальных /а, Гь и полученных для различных на-
боров изотопозамещенных. Он показал, что несогласованность па-
раметров определяемой таким путем /'s-структуры меньше, чем г0-
структуры. (Индекс s является первой буквой англ, слова substitu-
tion— замещение.) Для двухатомных молекул было доказано, что
— (re + ro)> т- е- rs — величина ближе к равновесному значе-
нию. При исследовании ряда многоатомных молекул было найдено,
что re<Zrs<Zr0. Однако в некоторых случаях наблюдались соотно-
шения rs<Z.re или r0<.rs. Для плоских молекул наличие дефекта
инерции меньше сказывается на ^-структуре, чем на г0-структуре.
Таким образом, различие Го- и /^-структур состоит лишь в раз-
личном подходе к решению обратной задачи, когда исходят, по
существу, из одного и того же ряда экспериментальных величин:
^а, Л и 1°с, т. е. г8-структура рассчитывается из уравнений Крейч-
мена для Д/д, Д/£, Д/р, а Го — из уравнений для /а, 1ь, 1°с-
В качестве примера приведены данные для молекулы SO2:
Тип структуры .... Ге
/(5 = 0), 10-1 нм ... . 1,4308
.ZOSO, 0.............. 119Т9'
ГО rz rs
1,4336 1,4349 1,4312
119°25' 119°21' 119°30'
Оценка ошибок в определении параметров, полученных методом
микроволновой спектроскопии, основана на обычной теории оши-
бок. Однако уравнения для ошибок не включают колебательно-
вращательные эффекты, которые обусловливают отсутствие полно-
ты согласования параметров, полученных в различных методах
расчета: с использованием полного набора изотопозамещенных или
дополнительного использования условий для главных осей инерции,
или минимизацией квадратичного функционала для разностей в
моментах инерции и т. п. В целом неточности в расстояниях могут
составлять от нескольких тысячных до нескольких сотых единицы
10-1 нм, а неточности в углах — от десятых долей до нескольких
градусов. Следовательно, высокая точность определения враща-
тельных постоянных теряется при расчетах эффективных геометри-
ческих параметров молекул из-за нарушения согласованности урав-
нений колебательно-вращательными взаимодействиями.
Если не все неэквивалентные атомы замещены их изотопами,
но общее число изотопозамещенных достаточно для определения
геометрических параметров, то проводят расчет г0-структуры.
4. ОПРЕДЕЛЕНИЕ ЭЛЕКТРИЧЕСКИХ ДИПОЛЬНЫХ
МОМЕНТОВ МОЛЕКУЛ
В 1927 г. П. Дебай впервые предложил использовать эффект
Штарка первого порядка для измерения дипольных моментов. Од-
нако эти эксперименты стали возможны только с появлением ми-
кроволновой спектроскопии. Поскольку расщепление линий враща-
тельных переходов пропорцио-
нально произведению проекции
дипольного момента молекулы
на напряженность электрическо-
го поля (цйс?0)п (лг=1,2), сте-
пень зависит от типа молекул,
то для измерений дипольных мо-
ментов необходимо создание до-
статочно однородного электри-
ческого поля. Для калибровки
радиоспектрометра со штарков-
ской модуляцией используют из-
мерения расщеплений в спектре
соединения OCS, дипольный мо-
мент молекулы которого опреде-
лен с высокой точностью
[po(OCS) = 2.3888± 0,0006 • 10-30
Рис. V.8. Зависимость отщепления
вращательной линии поглощения /=.
= 1—-2 от напряженности штарков»
ского поля для молекулы OCS
Кл-м].
Если известна напряженность
электрического поля с?0 и надеж-
но идентифицирована линия пере-
хода, то проводят измерения «отщеплений» или смещение линии при
различных значениях ^0. «Отщепление» ±Av означает разность
v—vq, где v — частота перехода, появляющаяся при наложении по-
ля g’o. Для линейных молекул достаточно наблюдать смещение од-
ной линии в зависимости от Тогда получают линейную зависи-
мость |Av| от й’о (рис. V.8):
A2v0
' ЗМ? (8/2 + 16/ + 5) — 4/ (/ 4- 1)2 (/ + 2) 1
. J (J +2) (2/ - 1) (2/ + I) (27 +3)(2/ +5)]
(V.49)
_ ММ
15A2v0
и
(Д^)/=о
где J — квантовое число нижнего вращательного уровня.
Уравнения (V.49) получены из (III.42) для Д/=1 и Д7И=(5.
Правило отбора для М обусловлено тем, что векторы <FV и
&о параллельны (см. рис. V.5). Уравнение (V.49) показывает, что
при Л4=0 и при |Л4| =#0 прямые зависимости Av от <$о будут
иметь различный наклон.
Так, для перехода J— 1 -> 2 молекулы OCS наблюдается дублет.
Это обусловлено тем, что в электрическом поле уровень с 7=1
расщепляется на два уровня с М=0 и |Л4|=1, а уровень с /=2
расщепляется на три уровня сЛ1=0, | М | = 1 и |Л1|=2. Но пере-
ходы возможны только для А/=1 и ДЛ1 = 0 Следователь-
но, имеем только две частоты (ср. с переходами 2-> 3, см. рис. V.3).
Если |Л4| == 1, то Av положительна и увеличивается с ростом &0, а
при М—Ь Av — отрицательна и уменьшается с увеличением Sо (см.
рис. V.8).
Для асимметричных волчков, дипольный момент которых опре-
деляется в общем тремя проекциями ца, ц* и рс на главные оси
инерции, изучают зависимость расщепления нескольких линий. Так,
в молекуле винилгермана СНг=СНСеНз определялись штарков-
ские коэффициенты Дм/<?о для переходов 212 -> З13 и 2ц->312 при
|Л1| = 1 и 2. Для плоских молекул определяют, естественно, только
Рис. V.9. Расположение молекул винилгер-
мана CH2=CHGeH'3 (а) и 1, 2, 4-триазо-
ла (б) в плоскости главных осей а и Ь
две проекции: || и |и*
(цс = 0).
Важно отметить, что
экспериментально возмож-
но измерять лишь абсолют-
ные величины проекций ди-
польного момента на глав-
ные оси, т. е. | ца |, ] Иь 1 и
| Цс | • Полный дипольный
момент рассчитывается из
соотношения
|Рполн1 = ((Ла -ЬРйЧ-р'с)172-
Однако направление векто-
ра Цполн из микроволнового
эксперимента не определя-
ется. Чтобы найти его на-
правление, обычно исполь-
зуют векторную схему ди-
польных моментов по свя-
зям или квантово-механи-
ческие расчеты.
Например, в молеку-
ле винилгермана СН2 =
= CHGeH3 направление
главных осей таково, что
атом германия практически
лежит на осип (рис.V.9,а).
Значения проекций и пол-
ного дипольного момента
составляют |р,а| = 1,63Х
XIО-30 Кл-м, |ц6| =0,40-10-30 Кл-м и |цПолн| = 1,66-10~30 Кл-м.
Направление вектора полного дипольного момента отклоняется от
оси а на 14 + 3°. При этом возможны два значения угла между
связью Ge—С и цПолн 31 ±3° и 3+3°.
Для молекулы же 1,2,4-триазола наряду с экспериментальным
определением | р,а| =2,7±0,3-10-30 Кл-м, | р6| = 8,6+0,2- 1О-30 Кл-м
и |Цполн| = 9,0+0,3• 10~30 Кл-м был проведен квантово-химический
расчет, который позволил выбрать направление цПОлн (рис. V.9, б)).
Дипольный момент может быть измерен микроволновым мето-
дом для различных колебательных и электронных состояний. Инте-
ресно отметить различие в дипольных моментах тиофена
C4H4S[ 1,757+17] и дейтеротиофена C4D4S[1,897+18-10~30 Кл-м],
которое объясняется колебательными эффектами.
5. ИССЛЕДОВАНИЕ ВНУТРЕННЕГО ВРАЩЕНИЯ
И ИНВЕРСИИ МОЛЕКУЛ
Метод микроволновой спектроскопии позволяет определять па-
раметры относительно простых функций потенциальной энергии
заторможенного внутреннего вращения молекул. При рассмотрении
внутреннего вращения исходят из модели молекулы, в которой
группы атомов типа СНз, CF3, NO2 и т. п. представляют жесткую»
систему (внутренние волчки). С)собый тип их колебаний представ-
ляют крутильные колебания.
Внутреннее вращение может быть отделено от вращения моле-
кулы в целом и остальных форм колебаний в случае малых частот
крутильных колебаний и достаточно высоких барьеров. Потенци-
альную энергию обычно предполагают в относительно простой
форме:
V (?)=_~ (1 ~ cos п'г
(V.50)
где Ко — барьер вращения; <р — азимутальный угол поворота внут-
реннего вращения; п — кратность потенциала, например, для
СНз — CF3 п—3, для NO2C6H5 п=2, для CH3NO2 п—6.
Поскольку для такой модели движение атомов, находящихся на
расстоянии г от оси вращения, происходит в одной плоскости (на-
пример, атомов Н в плоскости трех атомов водорода группы СНз))„
перпендикулярной оси вращения, т. е. полярный угол 0 = 90°, то
при упрощенном рассмотрении полезна модель плоского ротатора,
в которой изменяется только ф при г—const и 0 = const (sin 0= l)i.
В этом случае оператор кинетической энергии упрощается. Из трех
слагаемых для г, 0 и ф остается лишь одно — для ф:
~ R2 Д2 ^2 Д2 02
1 =------V =-------------=-----------,
2/п 2mr2 д<р 2/ ду2
где т — масса ротатора; г — радиус вращения; I — момент инер-
ции.
Уравнение Шредингера для двух плоских ротаторов аналогично
уравнению для двухатомной молекулы:
Г Д2 d'i d2 . . 1 ,
L 2/j drf 2/2 <*р2 J
Для симметричных групп типа СХ3 уравнение приводится к виду
Г Й-2 ^2 fj.2 (^2 । тл/ J j E*t /\Т С1 \
--------5----------кУ(ср) ф = (V.51)
[ 2/м 2/пр d'f2
где <р=<Р1 —ср2; I ——1111—приведенный момент инерции
11 +/2
. т[т2 \ .
(ср. приведенную массу в двухатомных молекулах р. =--1--- ;
ГП\ + «2 )
lu—h + Iz — полный момент инерции относительно оси вращения;
/1 и Л — моменты инерций внутренних волчков около центральной
связи; ?м= .
I м
Для молекул типа симметричного волчка можно с наибольшей
точностью выделить кинетическую энергию внутреннего вращения
из кинетической энергии вращения в целом.
Если V(ф)=Аф2/2 (k — силовая постоянная), т. е. У(ф) опре-
деляет гармонические крутильные колебания, то частота крутиль-
ного колебания легко выражается через Др’-
При малых изменениях ф потенциал У(ф) в уравнении (V.50)
может быть разложен в ряд. Ограничиваясь двумя членами разло-
жения cos п ф, получаем
tzz х V’o z, И0 Л II nV \ _ И)Я2?2
И(<?)=-^-(1 —cos#<p) —-y-i 1—1-}----7“--- —
Для такого выражения У(ф) силовая постоянная £=Уоя2/2. По-
этому
(V.53)
Частоту крутильного колебания можно определить, если изме-
рено отношение интенсивностей линий поглощения, соответствую-
щих вращательным переходам для двух ближайших уровней кру-
тильного колебания. Поскольку отношение интенсивностей равно
отношению числа молекул на уровнях, например, оКр=0 и окр=1,
Ркр-» __ -^1 __ gl --w f
Л>кр-0 SO
(V.54)
где gi и g2 — статистические веса; ДЕ—разница энергий двух
уровней.
Таким образом, определяется vKp=AE//i, если оценены g0 и gj.
Барьер вращения легко выразить через vKp:
__ ^^кр /;?2
~ «2 /М
(V.55)
Изложенный метод определения Vq применим при относительно
больших барьерах вращения, когда в потенциальной яме имеется
несколько уровней крутильных колебаний и можно использовать
гармоническое приближение.
V(x)
Рис. V.10. Вид потенциальной функ-
ции внутреннего вращения и уровни
энергии для этаноподобных молекул:
показано расщепление уровней энергий на
А — невырожденный и Е — двукратновы-
рожденный
Рис. V.11. Функция потенциальной
энергии инверсии молекул типа NHs,
РНз и т. п.:
показано расщепление деформационных
колебательных уровней на симметричные
(+) и антисимметричные (—)
Более точным является метод измерения расщепления враща-
тельных линий, связанных с расщеплением крутильных колебатель-
ных уровней (рис. V.10) в сочетании с решением уравнения Шре-
дингера для вращения молекулы в целом и внутреннего вращения.
Такое расщепление вызвано наличием нескольких эквивалентных
минимумов потенциала вращения. Для больших барьеров враще-
ния нижние уровни крутильных колебаний почти не расщеплены
и расщепление линий вращательных спектров практически не раз-
решается. Поэтому для определения барьера вращения чаще ис-
пользуют первый метод.
Измерение низких частот в микроволновой спектроскопии позво-
ляет определять барьеры инверсии молекул типа NH3, РН3, AsH3
и др., а также барьеры инверсии и сгибания циклов. Потенциаль-
ная кривая инверсии, например аммиака (рис. V.11), соответствует
изменению энергии молекулы при прохождении атома азота через
плоскость трех атомов водорода, т. е. симметричному деформаци-
онному колебанию. Два эквивалентных минимума разделены барье-
ром. Такой потенциал может быть описан очень приближенно урав-
нением V(x) = axi— bx2, гд$ х — высота пирамиды. Это уравнение
используется и для уплощенных молекул типа H2NCHO, H2NCN,
.СН2
СН2< и т. п. В области минимума колебательные уровни
ХСН/
расщеплены на дублеты. Измеряют переходы между уровнями дуб-
лета и на основе задаваемой функции потенциальной энергии рас-
считывают барьеры инверсии. В более общем случае изучают вра-
щательно-инверсионные спектры.
6. НЕКОТОРЫЕ РЕЗУЛЬТАТЫ МИКРОВОЛНОВЫХ
ИССЛЕДОВАНИЙ
К настоящему времени изучена геометрия свыше 1000 молекул.
Для ряда молекул получены очень точные равновесные парамет-
ры: для двухатомных молекул, трехатомных — Н2О, SO2 и т. п.»
четырехатомных — NH3 и т. п.
На основе систематизации структурных данных установлены
важные стереохимические закономерности. Так, например, для гид-
ридов элементов шестой группы наблюдается уменьшение валент-
ного угла:
Молекула............... Н2О H2S H2Se
Угол................... 104°30' 92°6' 90°55'
Валентный угол при атоме кислорода изменяется в очень широком
интервале значений:
Молекула ...... F2O Н2О С12О КОН RbOH CsOH
Угол.............. , , 103°4' 104°30' 110°50' 180° 180° 180°
Похожая картина наблюдается для двухкоординационного трех-
валентного азота, валентный угол при котором —N= изменяется
следующим образом:
Молекула . . . .
Угол............
Н—N=N=N
114°
Н—N=C=O
128°
HsSi—N = C = O
160°
В области стереохимии соединений азота важными являются
данные для аммиака и простейших производных аминов. Для ва-
лентных углов при атоме азота имеем:
Молекула NH3
Угол 107°3'
Для фосфинов:
Молекула................... РН3
Угол..................... 93О27'
NF3 NC13 N(CH3)3
102°22' 107°(ГЭ)* 108,7° (ГЭ)
PF3 РС13 Р(СН3)3
102° 100°6' 99,1°(ГЭ)
* ГЭ — газовая электронография.
Таким образом, наблюдается некоторое отличие в характере
изменения валентных углов атомов азота и фосфора. В случае ато-
ма азота атомы фтора уменьшают валентный угол, а СН3-группы
немного увеличивают его по сравнению с NH3. Валентный угол ато-
ма фосфора последовательно увеличивается под влиянием атомов
фтора и СН3-групп. Плоское расположение связей атома азота най-
^\NH
дено в пирроле ; и близкое к плоскому — в молекуле фор-
мамида, что обычно объясняют эффектом сопряжения.
Микроволновые исследования дают возможность установить
формулу химического строения молекул, т. е. порядок располо-
жения связей в молекуле. Так, молекула N2O3 имеет строение не
°\ /° °Ч ,^°
О—N ,а N — при этом длина связи N — Nb
N2O3 значительно превышает (на «0,4-10-1 нм) сумму ковалент-
ных радиусов и длину связи в N2H4. Доказано, что молекула
^N”CH
1,2,4-триазола обладает несимметричным строением НС\ ||
Н
N-N
а не строением с симметрией С2г, НС\ || .
'N —СН
Н
Интересная проблема поставлена данными для F2O2 и F2S2, в
соответствии с которыми г (ОО) = 1,217 и г (SS) —1,89-10-1 нм,
что на ~0,2-10-1 нм меньше, чем обычно наблюдают для одинар-
ных связей О— О (1,47) hS — S (2,05).
уСН2
Изучение инверсии триметиленоксида H2CZ /О привело к
\сн/
/СН2\
выводу, что в отличие от молекулы циклобутана Н2С\ /СН2
хсн/
цикл триметиленоксида имеет строение, близкое к плоскому.
В комплексных соединениях X3PBY3 устойчивость к диссоциа-
ции не коррелирует прямо с длиной связи (Д. Дюриг, 1975):
Соединение . , , , . (СНз)з-Р-ВНз F3P-BH3 Н3Р-ВН3 H3P-BF3
л(Р—В), 10-1 нм . . 1,901(7) 1,836(6) 1,937(5) 1,921(7)
ZYBY, ° 113,5(0,5) 115,1(0,5) 114,6(0,2) 112,1(0,3)
ZXPX,0 105,0(0,4) 99,8(1,0) 101,2(0,2) 101
|Цполн|, 10~30 Кл-М . . 16,7(7) 5,48(7) 13,36(10) 12,5(10)
По-видимому, кроме образования координационной связи опреде-
ленную роль играет, судя по изменениям <YBY и <ХРХ, пере-
стройка исходных молекул. В ряду этих молекул понятна малая
величина дипольного момента F3P-BH3, но на основе представлений
об индуктивном эффекте можно было ожидать значительно боль*
шее значение p(H3P-BF3), чем цЭКсп.
Практически в каждом микроволновом исследовании опреде-
ляется дипольный момент молекулы, даже если в распоряжении
исследователя нет достаточного количества изотопозамещенных
для проведения структурного анализа. Некоторые вопросы исполь-
зования данных по дипольным моментам в химии уже обсужда-
лись в гл. IV.
Величины барьеров вращения важны для оценки взаимодейст-
вия несвязанных атомов в молекуле.
Уменьшение барьера вращения при переходе от СН3 — СН3 (тер-
модинамические данные) к СН3—SiH3, СН3—GeH3 и CH3SnH3 обус-
ловлено в основном, по-видимому, увеличением длины центральной
связи:
Соединение . , .
Уз, кДж/моль . ,
г(С—Э), 10-1 нм .
СНз—СНз
12,2
1,536
СНз—SiHs
6,94
1,867
СНз—СеНз
5,18
1,945
СН3—SnHs
2,72
2,143
Увеличение кратности потенциала вращения до п=6 в СН3МО2,
СвНяСНз, CH3BF2 и т. п. приводит практически к свободному вра-
щению. Это обусловлено тем, что в этих молекулах (симметрия
Сз« — для СН3 группы и С2о — для другой группы) при повороте
на 60° конформация молекулы повторяется.
Если число СН3 групп, присоединенных к одному центральному
атому, например углероду, увеличивается, то и барьер вращения
этих групп увеличивается:
Соединение .
Уз, кДж/моль
СНз—СНз СНз—СН2(СНз) СНз—СН(СНз)г
12,2 13,6 16,3
Энергия инверсии молекул характеризует изменение валентного
состояния атома элемента пятой группы при переходе от пирами-
дальной конфигурации связей к плоской:
Соединение................КНз
Барьер инверсии,
кДж/моль................,24,8
РНз AsH3 HN(CH2)2H2NCHO H2NCN
72,7 134 ,75 1,6 3,0
Относительно невысокий барьер инверсии в NH3 объясняет отсут-
ствие оптической изомерии в аминопроизводных. Однако при пе-
реходе к этиленимину резко увеличивается этот барьер. Доказано,
что этилениминопроизводные могут обладать оптической актив-
ностью.
Уменьшение барьера инверсии в формамиде и цианамиде сви-
детельствует о существенном взаимном влиянии аминогруппы и при-
соединенных групп. Обычно оно рассматривается как проявление
сопряжения.
ГЛАВА VI
ЧИСТО ВРАЩАТЕЛЬНЫЕ СПЕКТРЫ
КОМБИНАЦИОННОГО РАССЕЯНИЯ
1. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ МЕТОДА
В связи с невозможностью исследовать неполярные молекулы
микроволновым методом, а также существенными ограничениями
этого метода при изучении симметричных волчков, приобретает
важное значение исследование спектров комбинационного рассея-
ния (КР). Для определения геометрической структуры средних и
больших молекул необходимо точное измерение положения линий
вращательных переходов, расстояние между которыми составляет
десятки тысяч МГц (менее 0,5 см-1). Поэтому изучение чисто вра-
щательных спектров возможно только на приборах высокой разре-
шающей силы. Обычно используемые КР спектрометры имеют раз-
решение порядка тысяч МГц (нескольких см-1). Однако специаль-
но разработанные методики позволяют повысить разрешение до де-
сятков МГц (~0,001 см-1). В этих случаях вращательные постоян-
ные могут быть измерены с точностью до ~0,5 МГц (~2Х
ХЮ-5 см-1).
После открытия эффекта комбинационного рассеяния света
(1928) спектральная техника позволяла исследовать практически
лишь двухатомные молекулы типа Ог, N2 и т. п. В 50-х годах уда-
лось получить разрешенные чисто вращательные спектры ряда сим-
метричных молекул с числом атомов до 8—12 (Б. Стоичев). В на-
стоящее время использование лазерной техники привело к новому
этапу в развитии метода комбинационного рассеяния света для
определения вращательных постоянных и геометрического строения
молекул.
В этом параграфе, а также в четвертом разделе рассматрива-
ются наиболее важные моменты теории спектров комбинационного
рассеяния молекул, так как общая теория изложена достаточно
подробно в курсах физики и строения молекул. Основная часть рас-
сеянного молекулами излучения сохраняет частоту падающего из-
лучения и называется релеевским рассеянием. В то же
время энергия падающего монохроматического излучения частично
изменяется при рассеянии на молекулах, т. е. наблюдается н е -
упругое рассеяние, что обусловлено изменением энергети-
ческого состояния рассеивающей молекулы. Если молекула перехо-
дит под воздействием излучения на более высокий энергетический
Уровень, то частота рассеянного излучения уменьшается. Эти пере-
ходы называются стоксовыми (рис. VI. 1). И наоборот, часто-
та рассеянного излучения увеличивается, если молекула переходит
в более низкое энергетическое состояние. Такие переходы называ-
ются антистоксовыми. Поскольку вращательные уровни
расположены на небольших (в шкале энергий) расстояниях, то
вероятности переходов в верхние и нижние состояния практически
одинаковы (в отличие от колеба-
тельных спектров комбинационного
рассеяния). Это определяется тем,
что в соответствии с больцманов-
ским распределением при малой
разности энергий между вращатель-
ными уровнями верхние состояния
заселены в достаточной степени.
Поэтому чисто вращательный
спектр КР представляет систему ли-
ний, расположенных симметрично
относительно возбуждающей линии
с симметричным распределением
интенсивности этих линий.
При комбинационном рассеянии
промежуточные возбужденные так
называемые виртуальные состояния
с энегией hvo+E" или hvo-\-E' не яв-
ляются собственными состояниями
молекулы. Если такие состояния
собственные (например, возбужденное электронное состояние), то
наблюдают явление флуоресценции.
Частоты вращательного перехода будут определяться разностью
между частотой наблюдаемой линии vj комбинационного рассея-
ния и частотой релеевской линии рассеяния vq. Эта разность частот
Avoy соответствует уравнению
Рис. VI.1. Схема виртуальных
переходов в спектрах комбина-
ционного рассеяния двух уров-
ней энергии Е" и Е'
. Ej-E0
А^0/ =---7---, (VI. 1)
1 h
где Ej — энергия возбужденного уровня; Eq — энергия основного
уровня.
В общем случае для произвольного уровня i имеем
. (VI.2)
J h
Вероятность перехода из состояния i в состояние / пропорциональ-
на квадрату электрического дипольного момента перехода
№;)|2, где — оператор дипольного момента индуциро-
ванного полем световой волны.
Если частота падающего излучения vo меньше, чем частота са-
мого низкого электронного перехода уе, то при комбинационном
рассеянии можно учитывать только основное электронное состояние
молекулы. Комбинационное рассеяние в хорошом приближении
можно рассматривать на основе представлений о поляризуемости
молекул, если v0»v® и ve—vo>vr (yv — частота колебаний молеку-
лы). Вектор индуцированного дипольного момента ц# под влия-
нием слабого поля <5 в этом приближении выражается уравнением
114
(см. гл. Ill)
gg. =aS.
(VI.3>
В уравнении (VI.3) a — величина тензорная и представляется
квадратной симметричной матрицей для произвольной системы ко-
ординат x'y'z' в виде
а —
йх’х'
dy'X'
az'x'
dx'y’ O-x'z'
ay'y' V-y’Z’
di'y' dz'z'
(VI.4>
При этом ax>y> = ay>x,. Проекции индуцированного дипольного мо-
мента на оси Х\, у' и z' задаются следующими равенствами:
g х'==ах'х'(^’х' '^Г(1х'У'<^У' (lx'z'(^’z''-l
Iх <8 у‘ ~ аУ'х'^х' 4“ ау’у'$у' 4“ ay'z'$Z'\
(VI.5>
Pg z'~az’x'^X' -\-&г'у’$у’ -\-^z'z'Sz'-
Матрица a приводится к диагональному виду в системе главных
осей эллипсоида поляризуемости молекулы х, у, z\
a =
<Lxx
О
О
О
Ъуу
О
о
о
Vzz
(VI .6)
Для молекул, обладающих хотя бы одной осью симметрии С2
(точечные группы Сг, C2v, C2h, D2 D2h и др.), оси эллипсоида!
поляризуемости совпадают с главными осями моментов инер-
ции. Кроме того, главные оси или совпадают с какой-то
осью симметрии, или лежат в одной из плоскостей сим-
метрии. Для сферических волчков (Ту, Oh) главные оси имеют про-
извольные, но взаимно перпендикулярные направления, пересекаю-
щиеся в центре масс (эллипсоид вырождается в сферу).
Матричный элемент электрического дипольного момента пере-
хода связан с интегрированием в лабораторной системе координат
X, У, Z. Тензор поляризуемости молекулы преобразуется при этом
следующим образом.
Выражение индуцированного дипольного момента в лаборатор-
ной системе координат figxrz получают с использованием ортого-
нальной матрицы В:
В=
cos (Хх)
cos (Ух)
cos (Zx)
cos (Ху) cos(Xz)
cos (Ку) cos (Кг)
cos(Zy) cos(Zz)
(VI.7)
для которой B-' = Btp и |В| = 1. Введем обозначение Q = X, У, Z.
и q=x, у, z, тогда
Pg xyz~P<8 q = B (ц g ?) = B (ав^9)=Ва9В~1В^’9 = ар^’р, * (Vl-8)
где <^q=B<^9 =
> №q и S’q — векторы напряженности элек-
z
трического поля в координатах q = x, у, z и Q = X, У, Z;
ахх 0 0
aQ=Ba?B-1=B 0 0 В-1
0 0 a**
(VI.9)
Проекции р^$на оси лабораторной системы выражаются урав-
нениями
P’g’ x = = 2 (VI.10)
Q
и т. д. При этом элементы матрицы oxq имеют следующий вид:
<ixY—axx соэХ’л cos Yx-^-agy cos Ху cos Yy-}-агг cos Xz cos Y z,
<ixx=axx cos2 Xx-{-ayy cos2 Xy -j- a22 cos2 Xz, (VI. 11)
или в обобщенной форме
<iXQ = aQX=axx созХх cosQx-j-a^, cos Qy-\-axx cos Xz cos Qz~
— 2 a<z? cos Cos Qq‘ (VI. 12)
9
После проведенных преобразований можно записать выражения
для проекции индуцированного дипольного момента на оси лабора-
торной системы координат:
^’<?2°«cosX'^ cosQq;
Q 9
Y =2 2 C0S C0S Qq' (V1,13)
Q 9
v&z =2 2 awcos Zq cos Q?-
Q q
Квадрат матричного элемента индуцированного дипольного момен-
та перехода можно теперь получить как сумму квадратов проекций:
| j |2=21 f Q^dr Г '
Q
(VI. 14)
В приближении разделения колебательного (ф«) и вращатель-
ного (фг) движений а Функции зависят
от нормальных координат, а фг— только от углов, определяющих
ориентацию молекулы в лабораторной системе координат. Опера-
торами в уравнении (VI.14) являются р^из (VI.13). Величины
<Fq, как постоянные, выходят за знак интеграла, а получающиеся
интегралы произведений типа j 'ha99 cos Xq cosQgAjdt распадают-
ся на два сомножителя:
’ Ф*а99 cos Xq cos Q^dr= <|va99<|vd,rl, cos Xq cos Q^<pr»dTr .
(VI.15)
Колебательные правила отбора определяются пер-
вым интегралом, а вращательные — вторым. Учитывая зави-
симость поляризуемости от 3N—6 нормальных координат Qi, пред-
ставленную рядом
3JV-6
У Qi+-, (Vi.i6>
V dQi /о
первый интеграл в уравнении (VI. 15) можно переписать в форме
3.V—6
<|»Z'a99<|>l,.dTp=a99 f + У [(^Q^-dr,,'. (VI. 17)
J \ dQi JO J
I u\jiQq
= ±1) и хотя бы одна производная I----------
Вследствие ортогональности функций фи для гармонического ос-
циллятора первый член в уравнении (VI. 17) всегда равен нулю,
кроме случая, когда v'=v". Тогда этот член недаетвклад в интенсив-
ность колебательных переходов, но определяет интенсивность вра-
щательного спектра на колебательном уровне v (в частности, когда
о = 0). Второй член будет равен нулю всегда, кроме случая, когда
хотя бы одно квантовое число о, изменяется на единицу (т. е. Ду =
^0.
о '
Правила отбора во вращательном спектре комбинационного
рассеяния определяются свойствами тензора поляризуемости и вол-
новых функций фг, а также характером колебательного уровня мо-
лекулы. Важным следствием уравнений (VI.15) и (VI.17) является
то, что комбинационное рассеяние не связано с наличием у молеку-
лы собственного дипольного момента.
Для линейных молекул в невырожденном колебательном состоя-
нии разрешены переходы с Д/=0 (Q-ветвь) и Д/=±2 (О- и 5-вет-
ви) (рис. VI.2). В чисто вращательном спектре КР-переходы для
Д7 = 0 соответствуют релеевской линии, а в случае Д/= + |2| урав-
нение для частоты перехода из одного вращательного состояния в
Другое имеет следующий вид:
|Av|=4-(£ СМ-2)- - (1У)~В (У+2) (J4-3)-BJ(/+ l)=45(J-j-3/2).
h
(VI. 18)
4S,4B,4B| ЬВ |“ 6B |4Я|4Д|4Д |
о)
Рис. VI.2. Схема переходов
между вращательными уровня-
ми линейных молекул для пра-
вил отбора Д7 = ±2 (а) и по-
ложение линий в спектре КР
(б)
Обозначение |Av| вводится потому,
что отсчет частот ведется от релеев-
ской линии как в сторону больших, так
и в сторону меньших частот. Изменение
Д7=±2 в спектрах КР по сравнению
с Д7=±1 в спектрах поглощения
обусловлено усреднением в уравнении
(VI. 15) произведения двух косинусов
[ср. с уравнением (V.33)].
Для модели жесткой линейной мо-
лекулы расстояние между линиями 43
(рис. VI.2), первая линия отстоит от
v0 на расстоянии 63(|Av| при 7 = 0).
При существенной нежесткости ре-
альных молекул расстояние между ли-
ниями не остается постоянным, а по-
степенно уменьшается с ростом J.
В отличие от микроволновых спект-
ров чисто вращательные спектры ком-
бинационного рассеяния получают при
переходе с 7, достигающих больших
значений (порядка нескольких десят-
ков). Это приводит к необходимости
учитывать поправки на центробежное
растяжение, которое уменьшает вра-
щательную постоянную на величину, пропорциональную квадрату
полного момента количества движения, т. е. 7(7+1). С учетом цен-
тробежного растяжения для линейных молекул BVJ = BV—DvJ(I-\-
+ 1) и при правиле отбора Д7=2 имеем
|Av|=(4BV - 6DV) (J + з/2) - 8DV (J + з/2)з.
(VI. 19)
Величина DV<^BV. Таким образом, с увеличением 7 интервал между
линиями уменьшается.
В чисто вращательном спектре комбинационного рассеяния ли-
нейных молекул симметрии Е+л, например N2, Н2, О2, СО2, CS2,
НС=СН, N=+-C^N, НС^С-С^СН и т. п., наблюдается
чередование интенсивности линий вплоть до исчезновения ряда ли-
ний. Это явление обусловлено свойствами симметрии вращательных
волновых функций и симметрией волновой функции электронного
состояния по отношению к инверсии координат всех частиц.
Вращательные волновые функции (или уровни) называются по-
ложительными и обозначаются знаком « + », если знак функции при
инверсии системы координат не изменяется. Если при этом знак
волновой функции меняется, то такие функции (уровни) называют-
ся отрицательными «—». У молекул симметрии Dxh имеется центр
симметрии и к указанным свойствам волновых функций добавляет-
ся свойство перестановочной симметрии. Если при пе-
рестановке одинаковых ядер
знак волновой функции не,
изменяется, то такая функ-
ция является симметрич-
ной и ее обозначают симво-
лом s. Антисимметрич-
ные волновые функции а
при перестановке одинако-
вых ядер меняют знак. Пе-
реходы между уровнями s и
а запрещены. Статистиче-
ские веса симметричных s и
антисимметричных а уров-
ней различны и определяют-
ся спинами и статистикой
ядер.
Если спины ядер равны
нулю (возможно исключение
для ядра в центре симмет-
рии), то антисимметричные
уровни отсутствуют. Так,
для электронных состояний
(см. раздел пятый) мо- '
лекул СО2, CS2 отсутствуют
линии с нечетными J, а для
молекул типа О2 в электрон-
ном состоянии отсутст-
вуют уровни с четными J.
Таким образом, во враща-
тельных спектрах комбина-
J-5
со2 ’S*
8В6В6В8В
IUULlI IJ1IIHL
vo
85 105 105 85
LLLLLLIШШ
б)
Рис. VI.3. Система уровней вращательной
энергии для молекул симметрии Dx!, с
различным нижним электронным состояни-
ем: 12/для С°2 и32г для °2 и п0'
ложение линий в спектре КР (б):
пунктиром изображены отсутствующие уровни и
линии в спектре комбинационного рассеяния
ционного рассеяния молекул
СО2, CS2 и О2 расстояние между линиями увеличено вдвое, т. е.
до 8В, из-за чередования линий (рис. VI.3). Однако расстояние
между первыми линиями (от релеевской) различное. Для СО2 и
CS2 оно равно 12В, а для О2 — 20В.
Если в молекуле симметрии D^t, имеются пары ядер со спином
/=И=0, то присутствуют все вращательные уровни, но с различными
статистическими весами. Так, соотношение чередующихся интен-
сивностей линий в спектре 14N2 равно 2: 1, в спектре 12С|Н2—3: 1,
в спектре 12С4Н2—3:1, в спектре I2C24N2 — 2:1.
Для симметричных волчков правила отбора разрешают перехо-
ды при А7 = 0, ±1, ±2 и Д2<=0, +2.
Для сферического волчка (ажж=ау?/=аи) поляризуемость не из-
меняется при вращении молекулы. Поэтому у молекул типа SFg,
СН4 или СС14 и т. п. нет чисто вращательного спектра комбина-
ционного рассеяния.
Спектры молекул типа асимметричного волчка имеют сложную
структуру. В ряде изученных молекул этого типа спектры подобны
Рис. VI.4. Блок-схема спектро-
метра КР:
/ — осветитель; 2 —кювета; 3 — мо-
нохроматор; 4 — приемник
пы. При этом добивались
спектрам молекул типа симметричного
волчка, что и используется при реше-
нии обратной задачи.
2. МЕТОДИКА ЭКСПЕРИМЕНТА
ВРАЩАТЕЛЬНОЙ
СПЕКТРОСКОПИИ КР
Рассеянный свет составляет очень
малую долю от падающего (~10~7).
Поэтому до появления лазеров для по-
лучения спектров КР высокого разре-
шения основное внимание было уделе-
но созданию интенсивных источников
света на основе дуговой ртутной лам-
аксимальной концентрации света, рас-
сеянного исследуемым веществом. Для анализа рассеянного излу-
чения использовались светосильные спектрографы высокой разре-
шающей силы.
Схема упрощенной экспериментальной установки для изучения
спектров КР дана на рис. VI.4. Дуговые ртутные лампы располо-
жены вокруг цилиндрической газовой кюветы с исследуемым веще-
ством и образуют своего рода «оптическую печь». Для увеличения
светового потока, направляемого в спектрограф, внутри газовой кю-
веты смонтированы вогнутые сферические зеркала. Объем кюветы
составляет несколько литров, диаметр — несколько сантиметров
(например, около 5 см). В специальных исследованиях объем кю-
веты увеличивали до 100 л. Газ в кювете находится при давлении
~ 105 Па.
С помощью конденсирующих линз рассеянный свет направля-
ется на щель спектрографа с вогнутой или плоской дифракционной
решеткой. Регистрация спектра производится на фотопластинку.
Экспозиция составляет от 1 до 20 ... 30 ч.
Обычно для возбуждения спектра КР используется линия ртути
Х = 435,8 нм. Ее полуширина равна ~0,2 см'1, что и определяет в
основном разрешение спектра (~0,3 ...0,4 см"1).
На основе этой техники были получены структурные данные
приблизительно для сорока молекул, в том числе для молекул эта-
на, этилена, бензола, бутадиена, бутатриена, циклопропана и т. д.
Описанная методика получения чисто вращательных спектров
КР имеет два существенных ограничения.
1. Недостаточное разрешение не позволяет проводить исследо-
вания сложных молекул или молекул, имеющих большие моменты
инерции, а следовательно, малые значения вращательных постоян-
ных. Так, даже в относительно простой молекуле SCSe разность Av
для соседних линий равна 0,1 см-1, что значительно меньше дости-
гаемого разрешения 0,3 ...0,4 см-1.
2. Значительное количество вещества, необходимое для прове-
дения исследования в больших объемах и при высоких давлениях.
Поскольку для решения структурной задачи приходится использо-
вать изотопозамещенные, то это создает значительные трудности.
Появление в 60-х годах лазерных источников возбуждения мож-
но назвать революцией в спектроскопии КР. Для лазерного излу-
чения характерны высокая интенсивность, малая ширина и малая
расходимость пучка, практически полная его поляризация. Может
быть достигнута ширина линии возбуждения ~ 0,001 см-1. Эффек-
тивный объем исследуемого вещества уменьшается до долей см3
вместо нескольких литров. Фотоэлектрическая регистрация спект-
ров КР позволяет резко увеличить чувствительность. Современная
техника колебательной спектроскопии КР подробно рассматрива-
ется в гл. XII.
3. ОПРЕДЕЛЕНИЕ ГЕОМЕТРИИ МОЛЕКУЛ
При решении обратной задачи обычно находят г0-структуру.
Чтобы определить Ва, для линейных молекул и молекул типа симмет-
ричного волчка строят график зависимости | Av|/(7+3/2) от (/-НА)2-
Согласно уравнению (VI.19) пересечение прямой с осью ординат дает
разность 4/?о—6£>0, которая практически равна 4В0, а тангенс угла
наклона — 8£>0. В соответствии с уравнениями для момента инер-
т h 2
ции /0 = -——=[*Го при известной ji легко вычисляют г0 Для двух-
атомных молекул, а также для линейных трехатомных молекул ти-
па СО2, CS2 и др. Так, для ряда изученных молекул получены сле-
дующие данные:
Молекула . . Н2 HD D5 F2 N2 СО2(С = О) CS2(C = S)
Во, см-‘ . . . 59,3392 44,6678 29,91О5 0,8828 1,9897з О,39О4о 0,10910
Го, Ю-1 нм . . 0,75105 0,74973 0,74820 1,418 1,1000в 1,162 1,5545
При исследовании структуры молекулы ацетилена изучались два
вида молекул: С2Н2 и C2D2. Предполагая равенство длин связей
С = С и С—Н (D) в обеих молекулах, они были найдены из двух
значений Во для этих молекул, равными, соответственно, 1,207-10-1
и 1,061-10-1 нм. Таким же образом проводилось определение гео-
метрических параметров для молекул бензола С6Н6, этилена
СН2=СН2, циклопропана С3Нб, аллена Н2С = С = СН2, диацетилена
НС = С—С = СН, бутатриена Н2С = С = С = СН2 и т. д.
Одним из важных стереохимических результатов явилось уста-
новление укорочения связи С = С в аллене (1,308-10-1 нм) и цен-
тральной связи С = С в бутатриене (1,284-10-1 нм) по сравнению со
связью С = С в этилене (1,339-10-1 нм). Однако при исследовании
структуры более сложных молекул только дейтеропроизводных ока-
зывается недостаточно для полного определения параметров. По-
этому приходится принимать часть параметров известными и не
варьировать их. Так, в молекуле дициана C2N2 в предположении,
что r(C==N) = 1,157-10~1 нм, найдена длина связи С—С=1,380х
X10-1 нм.
Г Л AB A VII
МЕТОД ГАЗОВОЙ ЭЛЕКТРОНОГРАФИИ
1. ОСНОВНЫЕ ЭТАПЫ РАЗВИТИЯ ГАЗОВОЙ
ЭЛЕКТРОНОГРАФИИ
Метод газовой электронографии разработан в 1930 г. в Германии Г. Мар-
ком и Р. Вирлем. Они сконструировали первый электронограф для исследования
строения простых молекул и провели исследование нескольких десятков соеди-
нений. Полученные результаты позволили сделать ряд важных стереохимических
выводов.
Методика газовой электронографии первого этапа развития (до 50-х годов)
основывалась на визуальной оценке интенсивности рассеянных электронов. При
расшифровке электронограмм использовалось упрощенное уравнение рассеяния
электронов только на ядрах атомов жестких молекул. Эта методика позволяла
определять лишь ограниченное число геометрических параметров с относительно!
невысокой точностью: для связей—(0,02 ... 0,03) I01 нм, а для валентных уг-
лов—2... 4°. Однако большое число исследованных молекул, особенно Л. О. Брок-
веем, уже позволило на основании полученных данных установить важные зако-
номерности в геометрии молекул.
Последующее совершенствование экспериментальной техники и развитие тео-
рии как прямой, так и обратной задачи метода газовой электронографии сущест-
венно повысило точность определения геометрических параметров молекул и
расширило возможности метода при исследовании относительно сложных моле-
кул. Принципиальным изменением в газовой электронографии был переход в
50-х годах на сектор-микрофотометрическую методику. Использование враща-
ющегося перед фотопластинкой сектора уменьшило резкое затухание полной ин-
тенсивности рассеяния, что позволило микрофотометрировать электронограммы.
Без использования сектора происходит столь резкое затухание интенсивности
рассеяния (на несколько порядков от центра рассеяния до периферии фото-
пластинки), что невозможно правильно оценить интенсивность, измеряя плотность
почернения электронограмм микрофотометрированием.
Одновременно с введением сектор-микрофотометрической методики в газовой
электронографии начали применять более точную теорию рассеяния электронов
на молекулах, которая учитывает функцию плотности вероятности рас-
пределения пар атомов в молекуле. При этом молекула рассматривается в виде
модели, в которой свободные сферически симметричные атомы находятся на рас-
стояниях, соответствующих реальной молекуле. Такая модель не учитывает ре-
ального распределения электронной плотности в молекуле. Однако, поскольку
рассеяние падающих электронов в основном происходит на ядрах атомов и не-
валентных электронных оболочках атомов, то такая модель молекулы хорошо
соответствует электронографическому эксперименту. Небольшие отклонения на-
блюдаются лишь в области малых углов рассеяния, интенсивность в которой
практически не измеряется. Лишь в специально поставленных экспериментах по
исследованию молекул, например Н2, НаО, СН«, МНз, выявлены эффекты химиче-
ских связей, т. е. изменения в дифракционной картине, вызванные изменением
в распределении электронов в молекуле по сравнению с ее моделью как суммы
атомов.
В газовой электронографии успешно используется приближение ма-
лых колебаний атомов в молекуле. В особых случаях, например внутрен-
него вращения или деформационных колебаний низкой частоты, в уравнения ин-
тенсивности молекулярного рассеяния вводятся параметры соответствующих по-
тенциалов.
Возможности современной газовой электронографии при уточнении опреде-
ляемых параметров методом наименьших квадратов существенно расширились
с применением ЭВМ большого быстродействия.
Таким образом, прямая задача метода газовой электроногра-
фии рассматривает рассеяние на молекуле как системе атомов-
122
Поэтому изложение материала начинается с задачи рассеяния элек-
тронов атомами.
2. РАССЕЯНИЕ ЭЛЕКТРОНОВ АТОМАМИ
Постановка задачи. Считают, что при рассеянии электронов
атомом поток падающих электронов на большом расстоянии от
атома описывается плоской волной. Вся задача рассматривается
как стационарная, т. е. вне зависимости от времени, поскольку в
экспериментах по- рассеянию используются стационарные потоки
электронов, и дифракционная картина не изменяется во времени.
Для свободного электрона, движущегося вдоль оси г, уравнение
Шредингера не содержит члена V(z) (потенциальная энергия).
Поэтому стационарное уравнение Шредингера
(VII.1)
переходит в уравнение
(VII.2)
где h = hJ2x (h— постоянная Планка); т — масса покоя электрона;
Е — полная энергия электрона.
Учитывая соотношение де Бройля \ = 1г1тУ, (VII.3)
получим выражение для волнового вектора k , 2л тл р х h ft ’ (VII.4)
где X — длина волны электронов; v — скорость электронов; р — им-
пульс электрона (положительное значение k соответствует р>0, т. е.
движению электронов в сторону положительных величин г).
Для свободного электрона полная энергия электрона равна его
кинетической энергии:
(VII.5)
2 2т
Тогда уравнение (VII. 1) имеет решение в виде
ф = (VII.6)
Уравнение (VII.6) описывает плоскую в о л н у, распространяю-
щуюся вдоль оси z.
. Результат взаимодействия падающего электрона и атома всег-
да наблюдается в точке, удаленной от атома на расстояние, кото-
рое значительно больше, чем размер атома. Поэтому в теории атом-
ного рассеяния ищут асимптотическое решение, т. е. решение за-
дачи рассеяния для больших расстояний рассеянного электрона от
ядра атома г, на которых отсутствует какое-либо взаимодействие
атома и рассеянного электрона (рис. VII.Г).
Рис. VII. 1. Схема рассеяния электро-
нов атомом:
радиус окружности г0=а соответствует
У(с)-0 (e<r):
АА — плоскость регистрации рассеянных
электронов; 1о — интенсивность падающе-
го; /(0) — рассеянного потока электронов
Если рассеянный электрон
только изменил направление дви-
жения и не изменил своей кине-
тической энергии (при этом, ес-
тественно, и атом также сохранил
состояние до рассеяния), то про-
цесс рассеяния произошел упру-
го (упругое рассеяние).
Неупругое рассеяние
возникает при изменении энергии
как рассеянного электрона, так и
рассеивающего атома. При рас-
сеянии пучка электронов на ато-
мах имеются оба вида рассеяния.
Однако наиболее важной состав-
ляющей для структурных иссле-
дований является упругое рассея-
ние электронов, которое обеспечивает постоянство длины волны
рассеиваемых электронов. Неупругое рассеяние электронов имеет
меньший вклад при используемых высоких ускоряющих напряже-
ниях 40... 60 кВ.
2.1. Упругое рассеяние электронов атомами
Поскольку при упругом рассеянии энергетическое состояние ато-
ма не изменяется и сохраняется кинетическая энергия электрона
р2/2т, вся задача о столкновении электрона с атомом сводится к
рассмотрению состояния лишь падающего и рассеянного электрона.
Полное решение задачи. Будем исходить из того, что изолиро-
ванный атом обладает сферической симметрией электростатическо-
го потенциала, т. е. l/(r) = V(r) (г — вектор, направленный от ядра
атома до точки рассеяния). Практически всегда рассматривается
рассеяние на большом числе атомов. В случае отсутствия сфериче-
ской симметрии электронной плотности индивидуального атома
усреднение по ориентации атомов в пространстве приводит к эф-
фективной сферической симметрии потенциала атома.
Состояние рассеиваемого электрона описывается стационарным уравнением
Шредингера:
й.2
_ ?2ф(г) + Г(г)ф(г) = £ф(г), (VII.7)
где ф (г) —волновая функция электрона в точке г; V2 — оператор Лапласа; Е —
полная энергия рассеянного электрона, равная при |г|—>оо кинетической энергии
[см. уравнение (VII.5)].
В полярных координатах г, 0, <р решение уравнения (VII.7) не зависит от <р
в связи с цилиндрической симметрией картины рассеяния (см. рис. VII.1). Опе-
ратор Лапласа в данном случае представляет сумму операторов по г и 0, т. е-
2 1
V2=V2+-
Vj. Учитывая зависимость потенциала атома только от г, реше-
ние уравнения (VII.7) можно выразить через произведение решений радиального
и углового уравнений Шредингера, т. е. ip(r)~/?fti(r) К;(0), где k — абсолютное
значение волнового вектора; I — соответствует моменту количества движения
рассеиваемого электрона.
Функции Уг(0) и Rki(r) удовлетворяют уравнениям, возникающим в резуль-
тате разделения переменных в (VII.7):
Id/ d \ 1
Г / rl* J
Величины /(/+1) являются собственными значениями функций К;(0), кото-
рые сводятся к полиномам Лежандра Pi (cos 0). Второе уравнение при V(r)=O
приводится к уравнению Бесселя, решения которого выражаются через сфериче-
ские функции Бесселя ji(kr) и Неймана ni(kr)-.
и
> / d V cos х
Л/(М = М*) = (-1) — •
\ у л
Из условий рассеяния следует, что при fer—►оо асимптотические решения имеют
вид
и
nf (kr)
1 / \
— cos kr — —— .
kr \ 2 )
Для радиальной волновой функции при V (/)=£ О и г->оо асимптотическое выра-
жение содержит фазу рассеяния 6ц
Rakdr)^~^7 sin (kr
lit
T
+ Ь;
Полное решение уравнения (VII.7) является бесконечным рядом по I для данно-
го k:
Фй(Г, 0)=2С!^ (О Г*(0)>
I
где Ci — коэффициенты разложения в ряд.
Асимптотическое выражение для ф принимает вид
(VII.8>
ОО
Фа(г> 9)= -^7 sin [kr — + 8zjpz(cos 9).
z"
С другой стороны, волновая функция рассеянных электронов на больших рас-
стояниях от центра рассеяния может быть представлена как расходящаяся
сферическая волна:
ф«(г, 0) = /(0)
(VII.9>
где f (0) — амплитуда рассеянной сферической волны.
В отличие от плоской волны полная амплитуда этой волны обратно пропор-
циональна г.
Наиболее общее решение задачи рассеяния включает также пло-
скую волну (условие излучения Зоммерфельда), т. е.
(VII. 10)
В связи с тем, что падающий пучок электронов узкий, практи-
чески исключается интерференция падающего и рассеянного пуч-
ков. Уравнение для ф“ позволяет получить выражение для f(0):
ОО
У(6)=-1_ У (2Z + 1)(<?21^ — 1)PZ(COS0).
1 = 0
(VII.11)
Уравнение (VII.11) впервые выведено Г. Факсеном и И. Хольцмар-
жом (1927). Оно показывает, что /(0)—комплексная величина. Для
удобства расчетов /(0) представляют в виде
/(б)=1/(е)|^(в),
(VII. 12)
где |/(0)|—модуль; т](0) — фаза амплитуды рассеяния
/(0).
Основные трудности расчетов /(0) состоят в вычислении 6/ из
радиального дифференциального уравнения для Rhi(r). Естест-
венно, что для У (г) =0 имеет место 6г —0, т. е. тогда ф. есть плоская
волна в сферических координатах.
Несмотря на то что уравнение (VII.11) было предложено отно-
сительно давно, его стали использовать в полной мере лишь с конца
<60-х годов благодаря возможностям быстродействующих ЭВМ.
В настоящее время имеются таблицы величин |f (0) | и ц (0) почти
для всех элементов периодической системы. Расчеты /(0) и т] (0)
выполнены на основе аналитических выражений статических потен-
циалов Хартри — Фока для нейтральных атомов с порядковыми но-
мерами Z от 2 до 18. Для более тяжелых элементов использованы
численные таблицы релятивистских хартри-фок-слейтеровских по-
тенциалов.
Первое борновское приближение. При малых 6/ разложение
в ряд Тейлора по 6/ приводит к первому борновскому при-
ближению для f(Q) (6; мало при малом У (г) по сравнению с
/В(0)=ув(5)=2т£2_ Z~F(s)=2^z_F{s^ (VII. 13)
где Z — порядковый номер элемента; F(s) — атомный форм
фактор для рентгеновского излучения; а0 =---------боровский ра-
те2
диус (е — заряд электрона), s — новый аргумент, связанный с уг-
лом рассеяния 0 следующим уравнением:
4л . 9
s=------sin----.
X 2
(VII. 14>
Слагаемые уравнения (VII. 13) соответствуют рассеянию на яд-
ре и электронной оболочке атома. Функция fB(s) — действительная
функция и для легких атомов дает хорошее приближение. Ранее
это уравнение широко использовалось в газовой электронографии и
сохранило до сих пор значение как относительно простое явное вы-
ражение /(0) через Z, Л и 0 или, несколько даже удобнее, через
Z и s.
Для относительно легких атомов при больших 5 функция F(s)
быстро уменьшается. Поэтому определяющим становится слагае-
мое ядерного рассеяния:
2те2 Z _ 2Z
J п \s) — rj ~; г
Й2 s2 a^s2
(VII.15>
Из выражения (VII.15) следует, что амплитуда рассеяния элек-
j"i 2/zzt?2 2 г~. г4 / \
трона на электроне j е =--------—--------.Следовательно, F(s) в
Й2з2 аоз2
уравнении (VII.13) есть отношение второго слагаемого /еВ>(£)(бор-
новской амплитуды рассеяния электронов на электронной оболочке
атомов) к f\. Это поясняет широко используемый термин для
F (s)—атомный форм-фактор как фактор, зависящий от распреде-
ления электронов в атоме.
Более наглядным является следующий вывод уравнения амплитуды рассея-
ния в борновском приближении. Уравнение Шредингера перепишем в форме
/ Й2 \ й2
V2+ £ ф = И(г)ф = —— (V2 + *2) ф.
\2т ) 2т
Вводя U (г) = —г- V (г), имеем неоднородное дифференциальное уравнение
(v2+A2)6(r)==C(rH(r),
решение которого можно получить в интегральной форме с использованием:
функции Грина G(r, г'):
Ф (г) = f G (г, г') U (г') ф (г') dr'.
Функция Грина удовлетворяет уравнению
(V2 + ^2)G(r, Г')=8(Г —г')
и для оператора V2+£2 выражается расходящимися сферическими волнами
I г'/
G (г, г') = О (Г — Г') = — —-------— .
4л |г — г |:
Для условий рассеяния |г|»|г'| (рис. VII.2). В знаменателе функции Гри-
на можно принять [г—г'|~г. Однако в показателе экспоненты, который, опреде-
Рис. VII.2. Рассеяние потенциалом атома в
точке, определяемой вектором г' (на ри-
сунке векторы обозначены стрелками)
ляет фазу волны, следует при-
нять, что |г—г'| «г—иг', где и —
единичный вектор вдоль г; иг' —
проекция г' на г. Это равенство
связано с тем, что два вектора:
г и г—г' — практически параллель-
ны.
Переходя к асимптотическим
условиям, имеем
C?(r,r') = -~й—e~iknr'.
v 4ЛГ
Для волновой функции ф(г)
получаем
е1кг (*
(г) = J e~!fcar'u (г’) ф (г') dr'.
Из уравнения (VI 1.9) следует, что
/(8)= - fe-‘'ftnr7Z(r'H(r')<ir'.
4Л J
Приближение Борна состоит в том, что принимают равенство
ф(г') = ехр [/£пог'],
которое основано на предположении о малом значении потенциала рассеяния по
•сравнению с энергией падающих электронов.
Тогда амплитуда рассеяния в борновском приближении
/в(в)=_-/- £ n)r'iZ(r')dr'.
Для равнобедренного треугольника, построенного на единичных векторах п0 и п,
имеем |по—п| =2 sin 0/2 (рис. VII.2), введем новый вектор s=£(n0—и), для ко-
торого (VII.14)
|s| = 2k sin 6/2 = —— sin 6/2.
л
<C использованием вектора s получаем
/в ($) = --/- ftZ(r')e‘(sr/)dr'.
4л J
Если элемент объема dr' задать в форме (r')2dr' sin adad£, где a — угол между
векторами s и г', а £ — азимутальный угол, то
2л it оо
fB(s) = — —— Г dp f \ U’(r')eisr' cosa(r')2 sin a dadr'.
4л J J J
о oo
Проведем интегрирование по £ и по а, сделав замену перемененных isr' cos a=x,
2л те —1 +isr'
—— С dp eisr' ZQ*a sin ada = —( elsr'cos a d cos a = —-— = S*n S~ .
4л J J 2 J 2Zsr' sr'
0 0 1 -isr'
В результате находим, что
ОО
« f „ ч sin sr' ,
/B(S) = - U(r’)(r'P------— dr’.
J sr
0
Убеждаемся, что fB (s) — действительная величина.
Введем кулоновский потенциал взаимодействия падающих электронов н
ядра атома УЯд =—Ze21г'. Тогда умножив и разделив на s, имеем
в 2m Ze2 (* . 2т Z& С .
= J sinSr'd(5r') = -^- — J smydy.
о о
В такой форме интеграл дает неопределенность. Реально потенциал атома V(r)
спадает быстрее, чем по кулоновскому закону. Используем следующий прием:
ОО ОО
I sin ydy = lim \ е~ау sin ydy = lim —-7=1.
J a+0 J a->0 a2 4-1
о 0
Следовательно, для рассеяния на ядре атома получаем
R 2т Z& 2Z
' —=^г-
Для потенциала электронной оболочки в точке г' запишем
KM(r') = J
е2р(г")
|г'-г"|
dr",
где р(г") —электронная плотность в точке г".
Подстановка Кэл (г') в fB(s) дает
fB (s)_ _2sl CC p|r,'Lg/(sr')d ,d
/эл(5) ft2 4Л J J |r'- r"|e drd
Используя соотношение Бете
. г 4л isr
dr' = ——eisr
$2
приходим к уравнению
ОО
С sin sr"
где F(s) = 4 л \ р (г")------------(rzz)2 dr" — форм-фактор.
J sr"
о
Полная амплитуда рассеяния электронов на атоме в борновском приближе-
нии имеет вид
f^} = -Z-lZ-F(s)].
a0s^
Комплексная амплитуда рассеяния может быть также записана как функция
аргумента s:
f (S) = |/ (S)| e1^.
Функция |f(s)l резко уменьшается с увеличением s (рис. VII.3).
5—949 129
Рис. VI 1.3. Зависимость амплитуды
упругого атомного рассеяния элек-
тронов от s
Рис. VI 1.4. Зависимость фазы упру-
гого атомного рассеяния от s
Интенсивность рассеяния электронов атомами пропорциональна
произведению фф* (ф* — комплексно-сопряженная функция). Сле-
довательно, используя уравнения (VII.9), (VII. 12) и (VII. 14), полу-
чим
I ($) = /офф* = \f (S)|2, (VII. 16)
г2
где /0—интенсивность падающего пучка электронов.
В борцовском приближении интенсивность упруго рассеянных
электронов выражается уравнением
Г2
—
Из (VII.16) и (VII.17) следует очень резкое уменьшение интен-
сивности атомного рассеяния с увеличением угла рассеяния. Обыч-
но наблюдаемые углы рассеяния соответствуют изменению 5 от 2,0
до 30,0, если X выражается в единицах 10-1 нм.
Функция ц (s) возрастает с увеличением 5. Для атомов с боль-
шими Z это возрастание особенно быстрое (рис. VII.4). Разность
фаз рассеяния г-го и /-го атомов At](/(s) для относительно легких
атомов практически линейно зависит от 5:
Дт]г;(«)^А;«. (VII. 18)
Эта величина используется в молекулярном рассеянии электронов.
2.2. Неупругое рассеяние электронов атомами
Борновское приближение полезно в том отношении, что позволяет вывести
уравнение для суммарной интенсивности упругого и неупругого рассеяния. При
этом делается предположение о возможности возбуждения электронов со всех
уровней атома. Чтобы такое предположение было оправдано, необходимо ис-
130
пользовать пучки электронов высоких
энергий (40... 60 кэВ). Для электронов
с (0,05 ... 0,06) Юу1 .нм и для атомов
с не очень большими порядковыми номе-
рами (Z<50) борновское приближение
дает достаточно хорошее Выражение
полной интенсивности рассеяния электро-
нов. Вычитая из полной интенсивности
рассеяния интенсивность упругого рас-
сеяния (VII.17), получают искомую ин-
тенсивность неупругого рассеяния:
1 Дупр («) = Лю ли («) - 1 fnp («) =
4Z0
r2a^s4
S(s),
Рис. VII.5. Функция неупругого рас-
сеяния S(s)
где S(s) — функция неупругого
рассеяния, которая рассчитывается
для волновой функции атома Фо (П, г2, гз,..., rz) (п ... rz — радиусы-векторы
электронов атома).
При суммировании для пары электронов i и j функция S(s) может быть
записана в форме
р sin sr j
S (s) = \ \ | % | 2------— d n ... d rz — Д2 (s).
H J Sr'li
sin sr .
При увеличении s функции F(s) и --------- стремятся к нулю, если i^j.
sr..
sin srt_ j
Однако при i=j имеем Г{, = 0 и в пределе функция --------------= 1. Поэтому
sri=j
для больших значений s второе слагаемое становится равным нулю, а первое
слагаемое — сумма интегралов для i=j — приближается к Z (рис. VII.5), т. е.
lim 5 (s) = Z. Для функции S(s) рассчитаны таблицы. В общем, /Heynp(s) —
резко затухающая функция.
2.3. Полная интенсивность атомного рассеяния
Выражение для полной интенсивности атомного
рассеяния включает два слагаемых: интенсивность упругого
рассеяния и неупругого рассеяния. Если используется точное выра-
жение для /упр, то имеем
(VII. 19)
В борновском приближении получаем
/аВ * *т(«) = -^- EW + S(S)].
Г2 AqS4
(VII.20)
При больших s имеем простую
зависимость от Z и s:
В /0 4
[Z2+Z]. (VII.21)
3. РАССЕЯНИЕ ЭЛЕКТРОНОВ
МОЛЕКУЛАМИ
Исходя из модели молекулы
как системы сферически симмет-
ричных атомов, расположенных
Рис. VII.6. Схема рассеяния электро- на расстояниях, соответствующих
нов z-м атомом в молекуле равновесным межъядерным рас-
стояниям реальной молекулы,
можно получить в хорошем приближении (классическом) уравне-
ние для интенсивности рассеяния электронов молекулами. Такая
модель позволяет избежать решения уравнения Шредингера для
системы падающий электрон и молекула, как это приходится делать
при атомном рассеянии. Кроме того, в теории молекулярного рас-
сеяния рассматриваемая модель не учитывает неупругое молеку-
лярное рассеяние.
3.1. Молекулярная составляющая интенсивности рассеяния
Зададим в молекуле радиусы-векторы атомов г, (рис. VII.6).
Тогда расходящуюся сферическую волну для рассеянного электро-
на в наблюдаемой точке экрана С на расстоянии R (|R|^>|rJ) от
начала координат можно записать в виде
Z*|R-rJ
Ф/=Л(0)-~-------7-е1*г‘, (VII.22)
[R - rz|
где f/(0) — амплитуда рассеяния i-го атома под углом 0; |R—rz| —
расстояние от i-го атома до точки С; zi— проекция п на ось г,
которая совпадает с направлением нерассеянного пучка электронов.
Фактор elkZi учитывает разность фаз между фронтом плоской
волны в начале координат и в точке rz. Векторы R и R—г, практи-
чески параллельны. Поэтому уравнение (VII.22) может быть пре-
образовано таким же образом, как при рассеянии на атоме (см.
гл. VII, § 1). Так как | R| |п|, то в знаменателе заменяем | R—г<|
на |R| =R, Фаза волны вдоль вектора R—п практически равна
фазе волны вдоль вектора R в точках п и (rzn)n соответственно, где
л — единичный вектор вдоль R. Поэтому
ei*IR-rz| ^[Я-(г.п)] ==eikRe~ik^i^t
Используя n0 — единичный вектор вдоль 2, выразим проекцию Zi
в виде скалярного произведения ггп0. Тогда уравнение (VII.22) пе-
реписывается по форме
Фг=//(9)-^-^(П-П)-
А
Введем вектор s=fe(no—п) (рис. VII. 6). Окончательно имеем
(VII.23)
В соответствии с принципом суперпозиции волн для молекулы име-
ем
<VIL24>
Для интенсивности рассеяния электронов на молекуле получаема
/ОР=/оФ„олФмол = 7о 2 X А(5)/Н5)Л’ <VIE25>
i J
где /ор — интенсивность электронов, рассеянных фиксированными в
пространстве молекулами; г^=гг-—г,-.
Однако условия эксперимента в газовой электронографии тако-
вы, что электронный пучок пересекает струю пара, в которой моле-
кулы ориентированы произвольно. Поэтому выражение (VII.25) не-
обходимо усреднить по соответствующим углам в полярной системе
координат между векторами s и П/. Такое усреднение приводит к
уравнению (см. гл. VII, § 1)
(VIL26)
Дальнейшее усреднение по изменениям межъядерных расстояний
гц с плотностью вероятности Рц(г) дает наиболее общее выраже-
ние для интенсивности молекулярного рассеяния:
X j p.y(r) -^dr.
И о
При i=j получаем слагаемое атомного упругого рассеяния
/аТ(5)=А-
i
Учитывая неупругое атомное рассеяние, полную интенсивность мож-
но представить суммой двух слагаемых /ат($) и 7Мол(^):
I ($) — /ат (<$) -|~ /мол (<$) — -^полн ($)«
(VIL27>
При этом
(VII.28)
где /мол(«)—молекулярная составляющая интенсивности рассея-
ния.
Суммирование по i и j в выражении с учетом
уравнения (VII.12) приводит к
У I/; (S)| \fi (S)| cos (т]г (s) — 71; ($)).
i+j
Более удобной характеристикой молекулярного рассеяния яв-
ляется функция приведенной молекулярной состав-
ляющей М (s), умноженная на $:
sM(s)=sZm^(s)-=
/а г (S)
= —VlW/WMVs)-4j(sy) f sin srdr. (VII.29)
/ат//2 wtaU < r
i*J 9
Функция sAf(s), по существу, представляет собой сумму затуха-
ющих синусоид, в чем мы убедимся несколько ниже. Деление на
/ат(з) уменьшает затухание M(s) по сравнению с /Мол(з). Явное
выражение для sAf(s) получается при проведении интегрирования
в правой части уравнения (VII.29) для явного вида Рц(г).
Функция Pij(r) является частной функцией плотно-
сти вероятности для пары атомов i и j, т. е. интегралом от
полной функции вероятности по изменениям всех п межъядерных
расстояний, кроме г,,-го:
Рц(г)= р..уР(Г1,--, Gjdrp..., dr„.
7Z~1
Методы расчета P(ri, ..., гп) и, следовательно, Р,Дг) основаны на
рассмотрении малых колебаний в гармоническом или ангармони-
ческом приближении.
3.2. Преобразование Фурье в газовой электронографии
При относительно больших значениях $ (практически при s>
>5-10 нм-"1) рассеяние обусловлено в основном полем ядер и вы-
полняется борновское приближение. Для молекул с Zi^Zj, ис-
пользуя уравнения (VII.17) и (VII.21), получаем
/о \fl (з)| Iff (s)| cos Би (s) - rif (s)J _ ZtZj
Г P2 " (VlLoU)
A
Поэтому можно рассмотреть уравнение (VII.29) в форме
v-. рР-(г) “
sMc (s)== у i Си j ~ sinsrdr=^ -------- sin srdr. (VII.31)
l+j О о
Используя свойства синус-преобразования Фурье
/Т Г /Т р
/(х)=|/ — \ F (и) sip uxdu и F(u)=]/ — f (х) sin uxdx,
о о
получаем
Cij—’у ) "=-|~ s/Wc(s) sin srds. (VII.32)
i^j О
Функция D (г) С и —— называется функцией ради-
ального распределения и может быть вычислена, если
известна функция sAfc(s). При рассеянии на реальных молекулах
получают экспериментальную кривую sAf(s) в ограниченном ин-
тервале величин s: smn—smax. Однако даже в области smln коэффи-
циент перед интегралом в уравнении (VII.29) зависит от s. Поэто-
му для уменьшения влияния этих факторов экспериментальную кри-
вую sMc(s) составляют из двух отрезков кривых. В области s от О
ДО Smin рассчитывают теоретическую кривую sAfc(s) для лучшей
модели молекулы (по согласованию эксперимента и теории). В экс-
периментальную кривую sMaKCn(s) вносят поправку на рассеяние
электронной оболочкой атомов:
sMc'cn (s)=sM3Kcn (s) - AsM (s),
где AsAf (s) = s/WTeop(s) — sAfcep(s)-
В связи с тем что экспериментально не реализуется верхний
предел интегрирования в уравнении (VII.32), т. е. имеет место об-
рыв кривой sAf-,;ciI(s), для уменьшения эффекта обрыва вводят за-
тухающую функцию ехр (—bs2) и вычисляют экспериментальную
кривую радиального распределения:
уэксп(Г) = 2- J sМс^(s)e~bs2 sin srds-f-
0
+ — j sA4'cKCT(s)e~bs‘ sin srds.
vuiin
Соответствующая ей теоретическая кривая радиального распреде-
ления рассчитывается по уравнению
jFTeop(r) = _2_C sMc0p(s)e~l>s‘ sin srds = -|-ysMcop(s)e_fe’ sin srds.
о 0
(VII.336)
—^2
Величину b оценивают из соотношения e max=0,l. Введение функ-
ции exp (—bs2) несколько уширяет пики на кривой f(r), но практи-
чески не смещает их положения.
ФДг) = [2vv '! Ул]
3.3. Двухатомные молекулы
Для двухатомных молекул достаточно хорошо разработаны ме-
тоды расчета Pij(r) =Р(г). Функцию Р(г) получают усреднением
функций P-v(r) по всем колебательным уровням, характеризуемым
квантовым числом и: „
= Р*(Г)еХР[~^]/2 eXpI~E^kT^ (VII.34)
V v
где Pv(r) — l^v(r) |2, ф«(г)—волновая функция для состояния с
квантовым числом и; Ev — колебательная энергия; k — постоянная
Больцмана; Т—абсолютная температура.
Для гармонических колебаний имеем
(VII.35а)
и
(Г - г,)2] , (VII.356)
л J
где Hv(r) — полином Эрмита; ге— равновесное межъядерное рас-
стояние; ц— приведенная масса; v — частота колебаний.
Суммирование в уравнении (VII.34) для гармонического осцил-
лятора приводит е относительно простому выражению Р(г) в виде
гауссовой функции:
-('-ге)г
1 2Z2
Р(г)=—L -е , (VII.36)
где lh — амплитуда колебаний двухатомных молекул, характеризу-
ющая полуширину гауссова пика; индекс h соответствует гармони-
ческому приближению.
Величина 12н называется среднеквадратичной ампли-
тудой и выражается уравнением
,2 / / чох Л it IlV
((г —г )2) =----------cth-----.
е ' 8n2^v 4kT
(VII.37)
Относительно легко можно получить выражение для среднеквадратичной
амплитуды колебаний гармонического осциллятора следующим путем. Средняя
энергия такого осциллятора задается уравнением
V / V
Суммирование проводится с использованием правил о геометрической прогршг-
сии. Сначала необходимо упростить выражение:
/ hv (у 4-
hv (v + 1/2) exp —--------—
\
V
Л v (t/ + 1 /2)\
kT J
V
V
Суммирование в знаменателе первой дроби правой части уравнения дает
_ , -ho/kT
Если ввести обозначение q=e , то числитель первого слагаемого можня
представить как производную Q' по q, умноженную на q, т. е.:
»=1
Р-1 dQ
Vq nq —
vq
Дифференцируя выражение для Q, найдем:
dQ
4 dq ~
hvq
Первое слагаемое будет равно ----—.
hvq
(1-7)2
Следовательно,
. I Ч * 1
< Е > = &v ---------+ —
\1 — q -2
1 + exp —-----
hv 1 kT
2 -f - hv
1 — exp — ——
1 \ kT
Преобразование последнего выражения с использованием е Л’/Йг = е-
v o—hipkT „
X е ‘ дает
ел»/2йт + е-й,/2йт ftv ftv
<Е> = 2 eh^*T - е~^Т = 2 Cth 2«
Поскольку <£> есть сумма средних величин кинетической Т и потенциальной U
энергий
< Е > = < Т ) + < U > , ' '
то, используя для гармонического осциллятора равенство <7'> = <С7>, имеем
< £ > = 2 < С7 > = 2 (Г~2ГгУ / = k{{r- reY > = kl2a,
где k — силовая постоянная.
1
Из соотношения v = — у получаем A = 4n2v2ji. Следовательно,
р _ < Е>h. Av
Л 4n2v2p. 8n2(*v 2kT ’
2
т. е. получили искомое выражение для Zft. Однако в этом выводе отсутствует
доказательство гауссовой формы распределения Р(Р).
Таким образом, функция плотности распределения ядер двух-
атомной молекулы в гармоническом приближении определяется ве-
личинами ге, ц, v и Т. При Т->0 cth стремится к единице и
2 h
liin/A =-----. С другой стороны, при увеличении Т среднеквадра-
r->o
тичная амплитуда также увеличивается. При kTz>hv или при
>(о, где co=v/c— волновое число, с — скорость света, в относи-
тельно хорошем приближении из (VII.37) следует, что
4(^0 Тг
Т2
(VII.38)
Для сравнительно жестких молекул (О2, N2 и т. п.) условие
соответствует Т>1000 К, для менее жестких молекул (Na2, К.2, Rb2
и т. п.) даже комнатная температура удовлетворяет этому усло-
вию.
Важным следствием из уравнения (VII.37) являются также со-
отношения для среднеквадратичных амплитуд колебаний разных
молекул.
При низких температурах, когда cth 1, для молекул 1 и 2
имеем
H2V2 . У1
^й(2) mVi v2 ki
(V1I.39)
где ki и k2 — силовые постоянные (£=4n2v2ji).
Для более высоких температур получаем
'й2(Р _ fe2
4(2) ‘
(VII.40)
Уравнения (VII.39) и (VII.40) показывают, что отношение ам-
плитуд колебаний 1н относительно мало изменяется даже для су-
щественно различных молекул:
Молекула . . . Nfc О». S2 СО CS С12 12
lh, i0‘ 1 нм . . . 0,0319 0,0о<-5 0,0393 0,0337 0,0389 0,0441 0,0510
re, IO-1 нм . . . . 1,0976 1,2074 1,887 1,128 1,5349 1,988 2,667
Эти результаты интересны и важны с точки зрения эксперимен-
тальных исследований, так как значения /д малы по сравнению с
наблюдаемыми ге, а ошибки определения довольно высоки
(3...10%). Уравнения (VII.29) для sM (5) и (VII.36) для Р(г) по-
зволяют получить явное выражение для sM (s) двухатомной моле-
кулы в гармоническом приближении.
Если коэффициент перед интегралом в уравнении (VII.29) обо-
значить через
gl2 =
2 |/1 (s)l I/2^)1 (n (s) — t].2(s))
। / / м2 _i_ 1 f / м2 л. 4 I5'
I/ (*)l2 + 1Л (s)l2 +------------------------------
aos
то можно переписать
S/W(S) = — (
/2л/Л .)
О
Ограничиваясь Двумя членами
имеем
(г —re)2 1 sin sr
2/2 J г
в разложении 1/г и вводя х=г—ге,
sin sr ( 1
2
е ' е
(1 X \
я- j (sin sre cos sx ф- cos sre sin sx).
re rle J
Рассмотрим отдельно четыре интеграла в соответствии с числом слагаемых
под интегралом уравнения для sM(s). При этом следует заметить, что нижний
предел интегрирования можно принять равным минус бесконечности, так как в
приближении малых колебаний lh<^.re и Р(0)=0. Тогда два интеграла тожде-
ственно равны нулю:
х2 __х_
+ “> 2/2 + ~ 2/2
У хе А cos sxdx = 0 = j е ft sin sxdx.
-- ОО -—ОО
Два других интеграла будем анализировать отдельно. Используя для
косинус-преобразования Фурье гауссовой функции уравнение
1
Vlrt
_ г2 _ Ц
е 2 cos ztit ~е 2
(VII.41)
принимая г=хЦк и t=slh, найдем
ffl2
lh /2л
___д-2
212
е й cos sxd х
-*'ll
re
sin sre.
Последний интеграл берется по частям udv = d(uv)—vdu, где
—X* —X*
’ 2/2 2/2
sv = —/дб'1 Л sin sr и — vdu ~ sl2he ftcossxdx,
g12 cos'sre
lh /2л r2e
cos sre
2/2 .
xe sin sxdx =
Первое слагаемое тождественно равно нулю. Повторно используя уравнение
((VII.41), получаем
+“ -±~ ~s4h
— gl2 COS Г 2/д' . , gl2 ,2 ~Г~
—т=------г— I хе "sinsxdx= ——s- slhe cos sr,
/A/2n r2 J г2 Л
— 00
Окончательно имеем
— s!Z2
4 ‘л
e 2 ( sl2h \
sM (s) = gj2 (s)------I sin sre —----cos sre 1. (VI 1.42)
re \ re /
„ s/1
Поскольку —f то можно принять
s/^ s/д S/д
---- « sin и cos « 1. тогда
re--re---------re
e 2
sM (S) = g!2 (s) ----------Sin s
(VII.43)
I2
Величина fe —— оказывается равной среднему значению межъядерного
Р{г)
расстояния для плотности вероятности ------, т, е,
зад
В числителе использовано условие нормировки j Р (г) dr = 1. Для проведения
о
интегрирования в знаменателе перейдем к переменной х=г—ге и разложим в
ряд множитель 1/г, ограничиваясь тремя членами. Тогда имеем
«о 4-во
fP(r)d, - !L С (-L
J г IhV^ J \ Ге
О —оо
Окончательно получаем
1 3
rg G) = ~-------72“ » ге ~ — = Гв,
1 ‘Л ге
где g—от англ, gravity — сила тяжести; (1)—степень аргумента г в знаме-
Р (г)
нателе видоизмененной плотности вероятности ------, равная единице. Часто
используется также обозначение г«(1)=га. Тогда уравнение для sM(s) можно
переписать в форме
sM (s) = 2 8Ш ЗГа
(VII.44)
Для ангармонического приближения колебаний двухатомной
молекулы плотность распределения может быть задана уравнени-
ем
-(r-rg)»
I 2/2 I \
* (ч-р.!"/)-
n
Коэффициенты cn зависят от температуры и от параметров потен-
циальной функции для ангармонического осциллятора. В связи с
малой асимметрией функции плотности распределения Ра(г) обыч-
но ограничиваются малым числом членов полинома.
Для ангармонического осциллятора общее выражение для
sM(s) изменяется лишь немного за счет сдвига фазы синусоиды:
—/а5»
е 2
sM(s)=g12(s)-------sin s(re —xs2).
(VII.45)
Величина l практически равна /д. Новый физический смысл при-
обрела га‘-
ra = rg(^) = rg ~ >
(VII.46)
где ге — среднее межъядерное расстояние, т. е.
°* Г! °°
rg=rg(0)=$ rPa(r)dr \Pa(r)dr.
о / о
Если использовать потенциальную функцию ангармонического
осциллятора в виде уравнения (V.31) и условие стационарности со-
/ dV \ „
стояния молекулы ' --- у=0, то можно относительно просто наи-
\ дх /
тк выражение (V.35):
гг=ге+-^-а^.
(VII.47)
Функция Ра (г), полученная на основе потенциальной функции
Морзе Е(х) =£)[1—ехр (—ах)]2, дает для rg такое же уравнение.
Изменение фазы синусоиды обусловлено величиной х, равной
Так как /4 очень малое число, то в ряде случаев удовлетворитель-
ным приближением является х=0.
3.4. Кривые радиального распределения
Явное выражение функции sMc(s) на основании уравнений
(VII.336) и (VII.44) позволяет провести интегрирование и получить
уравнение для кривой радиального распределения:
оо оо 2Z>-f-Z2 я
f(r)= — f sMc(s)e~~bsI sin srds=—12 Ce 2 sin sr, sin srds.
л J лге J
о 0
Представляя произведения синусов как разность косинусов
sin sra sin sr = -i- COS S(r — ra)------COS S(r/ra)
и используя уравнение для косинус-преобразования Фурье гауссо-
вой функции (VII.41) при z==s(2b+Z2)‘/2 и t = —г ~ r“i/2 , получим
(26 -1- (2)
~ 2»+р
f(r)——\e 2 cos s (г — ra) ds---------- \ e cos s(r-|-re)ds =
лге .1 itre j
о 0
________^12______
re /2л /26 + /2
exp
(Г-Га)2
2 (2b +l2)
+exp
(r + ray 1)
2(26 4-/2)
Второй интеграл дает экспоненту с аргументом (г+га)2, который
делает ее практически равной нулю, поскольку га^>/26//2. По-
этому с достаточной точностью
/(Г)=
_______£12_______
ге /2л /26 4-/2
(т-'аГ
2(26+1 = ) _
(VII. 48)
е
Таким образом, кривая радиального распределения j?(r) есть
гауссов пик, который определяется положением при г=га и полу-
шириной /26+Z2. Если разность фаз атомных амплитуд мала и
пропорциональна s согласно уравнению (VIГ; 18), то cos An]12(s)~ 1 —
5?2«2
— —-— , что в свою очередь может быть представлено как
ехр-------— Поэтому увеличение разности в порядковых номе-
рах атомов приводит к уширению пика кривой f(r) и даже к рас-
щеплению такого пика на два.
3.5. Многоатомные молекулы
В многоатомной молекуле из N атомов общее число межъядер-
ных расстояний равно N(N—1) /2. Наличие у молекулы элементов
симметрии приводит к появлению эквивалентных расстояний, так
что число параметров, подлежащих определению, бывает сущест-
венно меньше.
Наиболее удобной системой координат для представления како-
го-либо межъядерного расстояния гц является такая система, для
которой ось z, например, соединяет равновесные положения ядер
г и j, а оси х и у расположены соответственно в перпендикулярной
плоскости (рис. VII.7). В этой системе координат мгновенное межъ-
ядерное расстояние выражается уравнением
Гц = {(reij + Дг,7)2 + Дл?у + Д (VII.49)
где ге<; — равновесное межъядерное расстояние; Azij—Azt—Azy,
Ахц—Axi—Ах, и Ayn=Ayi—Ay у, Az((/>, Ахщ), Аущ) — изменения в
координатах i-ro или /-го атомов.
Рассматривая молекулы с малыми колебаниями атомов, можно
Гц представить рядом
о 1 2
Гц = relj + Д*,/ -I--------— + . • •
eij
Усреднение по всем изменениям гг7 при данной температуре дает
/Дх?/>7. -+-/ДгЛЛт
rgij=rei) + (Дг(7) т + х li/T У11/т +... (VII.50)
zr eij
Рис. VII.7. Мгновенное межъядерное расстояние г,,
в системе координат, которая связана с линией, сое-
диняющей равновесные положения ядер (ось z)
Величины и называются перпендикулярны-
ми среднеквадратичными амплитудами колебаний и
обусловлены гармоническими составляющими потенциальной функ-
ции колебаний ядер. Среднеквадратичная амплитуда колебаний
пар атомов i—j в хорошем приближении равна среднеквадратичной
проекции rtj—reij на ось z:
<(С7 —г^)2>т=((Д4/)г + О((АгДл2)г, (ДгДг/2)г...)^
(Дгг-у) j—lij.
(VII.51)
Частная функция плотности распределения пары атомов для
малых колебаний многоатомных молекул достаточно удовлетвори-
тельно описывается слегка асимметричной функцией, как и для
двухатомных молекул. Поэтому в случае многоатомной молекулы
молекулярная составляющая интенсивности рассеяния является
суммой термов для каждого межъядерного расстояния в соответст-
вии с функцией для двухатомных молекул:
«2/2
sM (s)
exp
gij(S)----
sin s (raU — xifs2),
(VII.52)
2
где
\ft (s)l \ fi (S)| cos (^ (S) — («))
(VII.53)
[|/»(s)l2 +
(s) ~l
a2s4 J
Явное выражение для кривой радиального распределения при
%/;=0 и при gtj=Cij согласно уравнению (VII.30) имеет вид сум-
мы гауссовых пиков:
(VII.54)
Динамические параметры равные (Az?;)r,a также перпен-
дикулярные среднеквадратичные амплитуды колебаний (Дл/;)г
и в виДе поправки
Ki]= <ДхЬ/г+<А^/г. (VII.55)
могут быть вычислены из данных по силовому полю молекул для
заданной температуры. Такая возможность определяется линейны-
ми преобразованиями, связывающими изменения декартовых ко-
ординат атомов с нормальными:
з^
Axz = ^ azzQz, (VII.56>
1=1
где Ах,—изменение обобщенной декартовой координаты в произ-
вольной системе осей; a-tj— коэффициенты, определяющие форму
колебаний в декартовой системе координат; Qi — нормальная коор-
дината.
Расчет величин ац проводится на основе анализа нормальных
колебаний для заданной геометрической конфигурации и силового
поля молекулы.
В многоатомных молекулах так же, как и в двухатомных, на-
блюдаются относительно малые изменения амплитуд колебаний
для связей между атомами различных элементов, а также и для не-
связанных пар атомов (табл. VII.2), что можно считать общей за-
кономерностью. Найдена практически линейная зависимость вели-
чин hj от ги на примере пар атомов CH, СС, ХН (Х=Ве, N, О, F,
Al, Si, Р, Cl, In).
Таблица VII.2. Межъядерные расстояния и амплитуды колебаний для
некоторых многоатомных молекул
XY„ r(X-Y), 10-’ нм Z(X-Y), 10—1 нм r(Y ... Y), 10-1 нм /(Y-Y), 10—1 нм Т, К
СС14 1,767(2)** 0,049(2) 2,888(2) 0,070(1) 293
в (снз); 1,576(3) 0,054(3) 2,722(6) 0,076(6) 293
РС1з 2,039(1) 0,050(1) 3,130(3) 0,083 (2) 300
♦ X=B; Y=C.
** В скобках указаны ошибки эксперимента.
В молекулах ароматических или конденсированных полицикли-
ческих систем, а также в линейных молекулах с жесткой системой
связей (например, в ацетиленовых, нитрильных группах и др.) ве-
личины 1ц изменяются в относительно малом интервале значений:
до 0,1-10-1 нм. С другой стороны, наличие крутильных колебаний
и внутреннего вращения или низких частот деформационных коле-
баний приводит к увеличению амплитуд колебаний пар атомов до
(0,15 ... 0,25) -10-1 нм.
В случае внутреннего вращения может оказаться недостаточным
приближение малых колебаний. Тогда используют классическую
функцию распределения для терма функции sM(s), обусловленно-
го внутренним вращением:
(VII.57)
где У(<р)—потенциал внутреннего вращения; ф-^-угол вращения;
— амплитуда колебаний остова.
Для простых молекул типа С2Не, C6HsNO2, CH3NO2 и т. п., как
уже указывалось выше, потенциал вращения задается в виде
Vr(?) = -y-(l — cos гаер),
где Уо — потенциальный барьер; п — кратность потенциала.
Амплитуды колебаний остова 1д/(ф) можно рассчитать на осно-
ве геометрии и силового поля остова.
4 . МЕТОДИКА ЭКСПЕРИМЕНТА В ГАЗОВОЙ
ЭЛЕКТРОНОГРАФИИ
Задачей эксперимента в газовой электронографии является по-
лучение интенсивности рассеяния / (s) в максимально возможном
интервале углов рассеяния 0min—0Шах- Поскольку /(s) является
непрерывной функцией, то ее представляют в виде таблицы интен-
сивностей I при рассчитанных значениях s. В свою очередь s явля-
ется функцией нескольких аргументов:
s =sin Г—— arc ftg ——, (VII.58)
л I 2 \ /J
где t — расстояние от центра дифракционной картины до точки, в
которой измеряется /; Ro— расстояние сопло — пластинка; к—
длина волны электронов, определяемая по электронограммам стан-
дартных веществ, структура молекул которых определена с высо-
кой точностью.
4.1. Принципиальная схема электронографа
Практически все исследователи работают с электронографиче-
ской аппаратурой, созданной самими экспериментаторами, и по-
этому имеющей свои особенности.
Несмотря на разнообразие экспериментальных методик, общая
принципиальная схема электронографа остается единой
(рис. VII.8).
Общая схема прибора. Электронограф — электровакуумный
прибор, предназначенный для получения и регистрации дифракци-
онной картины в результате рассеяния монохроматического потока
электронов на струе пара исследуемого вещества. Электронная
пушка является источником пучка электронов, ускоренного в элек-
трическом поле, напряжением 40—60 кВ. Магнитные линзы фоку-
сируют электронный пучок на флуоресцентный экран, который ус-
танавливается вблизи плоскости фотопластинки и убирается во
время съемки. Впуск пара производится через сопло испарителя,
напротив которого обычно монтируется ловушка для выморажива-
ния этого пара. Вымораживание необходимо для поддержания об-
146
щего высокого вакуума с давлением ос-
таточного газа не более 10~2 Па. Резкое
падение интенсивности рассеяния, про-
порциональное s'-4 (VII.21), компенсиру-
ется быстро вращающимся сектором.
В центре сектора укреплена ловушка не-
отклоненного пучка, сила тока которого
измеряется в ходе съемок.
Испарители. Формирование струи па-
ра производится с помощью испарителя.
Рис. VII.8. Принципиальная схема
электронографа:
1 — катод; 2 — анод; 3 — пучок электронов;
4— электромагнитная линза; 5 — ловушка
паров; 6 — соплю испарителя; 7 — ловушка
пучка; 8 — сектор; 9— фотопластинка
Рис. VII.9. Схема испарителя:
1 — сопло; 2—печь; 3 — боек для надло-
ма ампул; 4— игла; 5 — вентиль; б — бен-
зол; 7 — исследуемое вещество; 8 — термо-
пара
Испарители бывают различных типов. На рис. VII.9 показана схе-
ма испарителя для исследования веществ при нагревании от ком-
натной температуры до ~600 К.
Особенностью этого испарителя является возможность в одной
съемке получать электронограммы исследуемого и стандартного
вещества, например СО2, CS2 или С6Н6. Стандартное вещество на-
ходится вне прибора в разборной внешней камере, исследуемое ве-
щество, запаянное в стеклянные ампулы внутри прибора,— во внут-
ренней металлической камере. Нагрев внутренней камеры и ампулы
производится электропечью с бифилярной обмоткой. Материал ка-
меры— никель, сопла — латунь. Диаметр отверстия сопла ~0,2 мм.
Дозирующая игла используется одновременно для вскрытия стек-
лянных ампул специальным диском, укрепленным на ней. Возмож-
ность вскрывать ампулы внутри прибора позволяет исследовать
гигроскопичные и разлагающиеся на воздухе вещества.
Для исследования газов применяют специальные приставки.
Изучение труднолетучих неорганических соединений проводится
при использовании небольших открытых камер, нагреваемых тепло-
вым излучением раскаленной вольфрамовой проволоки или элек-
тронной бомбардировкой. Такие испарители обеспечивают нагрев
до 2000 ...2500°С.
Рис. VII.10. Секторы двухлепестко-
вый (а) и однолепестковый (б)
Оптимальная упругость пара
для многих веществ составляет
(2... 8) 103 Па при токе в элект-
ронном пучке ~0,1 мкА. Специ-
альные методики позволяют по-
лучать электронограммы при дав-
лениях ~ 10 Па.
Сектор. В большинстве иссле-
дований вырез лепестка сектора
задается следующими функци-
ями: 1) для двухлепесткового
(рис. VII.10, а)
Pi (?)=ро — ;
Л
2) для однолепесткового (рис. VI 1.10, б)
з, . з v
Р2(?) = Р0 ~ ,
ZJt
где ро — максимальный радиус; <р — угол раскрытия сектора.
Два лепестка первого сектора дают такое же раскрытие, как
второй сектор. Монтирование двухлепесткового сектора на держа-
теле труднее, чем однолепесткового. Однако периметр и, следова-
тельно, рассеяние на краях больше для однолепесткового. Обычно
лепестки изготавливаются из фосфористой бронзы или дюралю-
миния толщиной 0,2 ...0,5 мм.
В связи с тем, что необходима высокая точность при изготов-
лении сектора, возникают трудности обработки его внутренней ча-
сти при малых <р [или р(ф)]. В этой части сектора ширина щели
сравнима с его толщиной. Поэтому достаточно высокая надежность
может быть обеспечена лишь с некоторого ртщ, составляющего до
15...20% от ро- Расстояние между лепестком сектора и фотопла-
стинкой не должно быть большим (от 2 до 5 мм).
Обычно используемые скорости вращения сектора составляют
300... 600 об/мин.
Дифракционная камера. Дифракционная камера — часть колон-
ны электронографа, в которой располагаются испаритель, ловушка
для вымораживания паров, секторное устройство, фотокамера и со-
единения с вакуумным насосом. Поскольку сектор не позволяет
получать на одной электронограмме максимально возможный ин-
тервал углов рассеяния, проводят съемки при двух или трех рас-
стояниях сопло — фотопластинка. При съемках на большом рас-
стоянии получают дифракционную картину, начинающуюся при
минимальном угле рассеяния (0=0mta). Максимальные углы рас-
сеяния достигаются только при съемках на коротком расстоянии.
Фотокамера обеспечивает необходимое число фотопластинок
для съемок. Конструкция фотокамеры предусматривает точную
фиксацию фотопластинок в одном и том же положении, чтобы во
148
время съемки сохранялось расстояние сопло — фотопластинка. Экс-
позиции варьируются от долей минуты до нескольких минут.
Определение К. Определение X в большинстве исследований про-
водится с использованием поликристаллического стандарта —
NH4CI, ZnO, Т1С1; газового стандарта — СО2, CS2, С6Н6 или непо-
средственным измерением напряжения с использованием делителя
напряжения, откалиброванного, например, по газовому стандарту.
Проводить такие измерения при каждой съемке нет необходимости.
Требования к веществу. Электронографический метод предна-
значен для исследования индивидуальных веществ. Поэтому жела-
тельна высокая степень чистоты веществ. Однако следует сказать,
что если известна примесь и ее доля в исследуемом веществе, то,
вообще говоря, возможно учесть это при расшифровке электроно-
грамм. Если примесь рассеивает значительно слабее, чем исследу-
емое вещество, то ее небольшое присутствие не сыграет существен-
ной роли. Вместе с тем в ряде работ ставится задача исследования
систем со сложным составом пара: конформационных равновесий
или равновесных систем, в которых имеет место диссоциация моле-
кул или, наоборот, ассоциация.
4.2. Микрофотометрирование
Микрофотометр — оптический прибор, предназначенный
для измерения оптических плотностей фотоматериалов (фотопла-
стинок или фотопленок) на малых участках, в частности электро-
нограмм. Полученные электронограммы микрофотометрируют на
микрофотометрах разных типов, приспособленных для измерения
плотности почернений дифракционных картин, обладающих ради-
альным распределением интенсивности. Аналоговую систему (си-
стему построения графика) заменяют выводом сигнала на цифро-
печать или прямо на перфорацию. Используется также прямой
ввод информации в управляющую ЭВМ с дальнейшей числовой об-
работкой информации. Приэтом измерения производят с заданным
шагом (0,4 ... 0,5 мм) с остановкой или в непрерывном режиме
трансляционного движения электронограммы.
При микрофотометрировании измеряется интенсивность свето-
вого потока, который прошел через фотопластинку (фотопленку),
т. е. измеряется функция пропускания, которая преобразуется в
функцию плотности почернения
а
где а0— разница в величинах микрофотометрической записи для
темновой (при закрытом ФЭУ) и световой (при записи вуали) ли-
ний; а — разница в величинах записи пропускания и темновой ли-
нии.
Для пересчета плотности почернения в интенсивность рассеяния
строят зависимости этих величин в форме калибровочной кривой.
Часто такая зависимость выражается двухчленным рядом:
J = у (D -]~cD2),
где с^О, 1; у — коэффициент пропорциональности. Однако при D<.
<1,0 для диапозитивных пластинок удовлетворительным соотноше-
нием будет простое равенство I=yD.
4.3. Выделение молекулярной составляющей
интенсивности рассеяния
Рассчитанная из микрофотограмм интенсивность рассеяния
/сект (з) представляет сложную функцию полной интенсивности рас-
сеяния молекулой /полн(з) (VII.27), так как последняя изменяется
вращающимся сектором пропорционально углу раскрытия сектора
<р(з)/2л и, кроме того, может добавляться постороннее рассея-
ние B(s). Далее, поскольку дифракционная картина регистрирует-
ся плоской фотопластинкой, расстояние сопло — точка регистрации
будет определяться соотношением R=R0/cosQ, где /?0—расстояние
сопло — фотопластинка; 9 — угол рассеяния. При этом нужно
учесть, что электронный пучок падает на фотопластинку не под
прямым углом, а под углом 9 к нормали, т. е. согласно закону ос-
вещенности вводится множитель cos 9. Удобно учесть зависимость
всей дифракционной картины от угла 9 множителем cos3 9. Тогда
имеем
/сект («)= -^~ C°S3 «н" («) + В (3),
где <p(s) —угол раскрытия сектора как функция з.
Если каким-либо способом вычитают постороннее рассеяние или
если В (s) «0, то
/полн (з) ==-~-- sec3 9/ eKT(s). (VII.59)
? (з)
Поскольку функция <p(s) известна недостаточно точно и коэффици-
ент у не определен, появляется некоторая неопределенность в коэф-
фициенте пропорциональности k(s), связывающем /„олн и сумму
/ат+/МОЛ (VII.27), т. е.
/ голн (3) = k (S) [/ат (з) / мол(з)]. (VII. 60)
Следует отметить, что после умножения /пин на cos3 9 в уравнениях
для /ат(з) и /Мол(«) величины R переходят в Ro.
Разделив обе части уравнения (VII.60) на /ат(з)
/ЭКСП/ ч
_^2=^(s) + ^(s)A/(s),
4т (з)
можно выделить функцию М (s):
MSKCn(3)
г =КСП /
1 ПОЛИ V6/
k (s)/a.e(s)
1.
Функция k(s) представляет
линию фона, проведенного^
гладким образом*, на рассчи-
танной кривой /пмн(5)//ат(5).
Обычно эту операцию прово-
дят графически.
Упрощенно функцию
Mwcn(s) рассчитывают из
уравнения
Рис. VII.11. Кривые полной интенсивности
и линии фона /фон азетидина C3HSNH для
длинного (/) и короткого (2) расстояний
сопло — пластинка
^эксп(^)= "I,
/фон(«)
(VH.61)
где /фон — обобщенная ли-
ния фона, включающая в
качестве сомножителей все поправки (рис. VII.11). Гладкость функ-
ции /фОн($) относительна по сравнению с явно негладкими функ-
циями /полн(з) .
Неточности в оценке линии фона не очень сильно сказываются
на величинах основных определяемых параметров га и I. Последо-
вательное уточнение параметров позволяет исправлять линию фо-
на. Процедура исправления линии фона может быть автоматизи-
рована.
5. РАСШИФРОВКА ЭЛЕКТРОНОГРАММ
Определение геометрических параметров молекул ratj и ампли-
туд колебаний 1ц представляет обратную задачу газовой электроно-
графии, т. е. расшифровку электронограмм. Общим методом ее ре-
шения является метод наименьших квадратов, в котором формули-
руется поиск минимума квадратичного функционала:
а
Q.M = y KS/^эксп (Si) — ^5,/Итеор (S;)]2=min,
(VII.62)
где coi — статистический вес измерения $M(s) в точке s>; kM— мас-
штабный множитель.
Минимум функции многих переменных соответствует системе
уравнений, называемых нормальными:
ovk o^k
(VII.63)
где 9а — обобщенный параметр, включающий гог/, 1ц, км-
Однако эта система из-за нелинейности выражения для sAfTeOp
dsMieop (s) r-r
и ----——-----не имеет решения в общем виде. Поскольку часто мно-
гие параметры молекул могут быть оценены в хорошем приближе-
нии, возможно использование разложения в ряд sAfTeOp(s) по Д9&
с ограничением этого ряда только линейными членами:
т
^теор(5)-5ЛГ0 + У Д9Й,
\ Jo
*=1
(V1I.64)
где sAfo(s)—функция sM(s) для выбранного начального прибли-
жения параметров Qok; Дй^ —приращения искомых параметров.
В качестве параметров могут быть выбраны не только гац и 1ц,
но также валентные углы и углы внутреннего вращения или какие-
либо другие параметры, накладывающие геометрическую связь на
общий набор межъядерных расстояний в данной молекуле, а также
поворотно-изомерный состав.
Подстановка уравнений (VII.64) в уравнение (VII.63) дает си-
стему линейных уравнений:
эксп (5<) (Sz)
dsiM (si)
дв6
х fdstM ($<) \ _q
\ /о
(VII.65)
Решение этой системы приводит к определению искомых парамет-
ров.
Практически более удобным функционалом для минимизации
является /?-фактор, имеющий смысл относительной ошибки в
зЛ1 (s) для всего интервала s и вычисляемый на основе функциона-
ла. Qm-
(VII.66)
Величина R— зависит от степени сходимости теоретической моде-
ли и эксперимента, а также от характера изменения sAI (s). Наи-
более часто получающиеся значения R лежат в области 0,05 ...0,15.
Иллюстрацией сходимости теоретических и экспериментальных
кривых sM(s) и f(r) являются приводимые на рисунках кривые
раЗНОСТИ sAJaKcn('S)—^М^Л1теор (s) И f зксп fтеор (рИС. VII.12 И
рис. VII.13).
Очень важен выбор нулевого приближения решения задачи.
При поиске минимума (или R), по существу, проводится его
уточнение.
Для этого привлекаются данные по химическому строению, по-
лученные другими физико-химическими методами, используются
закономерности в геометрическом строении молекул и, наконец^
спектр межъядерных рас-
стояний из рассчитанных
кривых радиального рас-
пределения f(r). Для про-
стых молекул типа CCh,
ВС13, РС13> CF3CI, NF2CJ.
и т. п. кривые радиального
распределения дают хоро-
шо разрешенный спектр
межъядерных расстояний
(см. для NF2C1 рис.
VII. 13, а). Для более
сложных молекул кривые
f(r) могут быть полезны
для оценки лишь ряда
5 10 15 20 25 5,10 нм"1
Рис. VII.12. Кривая sM(s) для азетидина
СзН6МН
параметров в связи с тем,
что пики для близких межъядерных расстояний перекрываются
(рис. VII.13, б). Однако даже такие кривые f(r) со сложными
пиками дают определенную информацию о геометрии молекулы.
В том случае, если возможно несколько моделей молекул, не-
обходима проверка всех предположений о строении молекулы. Так,
г /СН2\
например, у молекулы азетидина СН2/ ^NH два главных
ХСН2'
пика f(r) для связей С—С и С—N, а также для С...С и C...N явля-
ются сложными (рис. VII.13, б). Следовательно, нельзя не только
выделить отдельные пики, но и оценить, какое из расстояний (СС
или CN) в каждом из этих сложных пиков больше. Стереохимиче-
ские закономерности позволяют предполагать, что г (С—С) всегда
больше г (С—N) для подобного типа связей. Это и было использо-
вано в работе, а при уточнении найдено соответственно 1,553(9) и
1,482(6) 10-1 нм. Уточнение двух моделей с различными диагона-
лями в четырехчленном цикле г (С...С) и r(C...N) привело к близким
значениям Л-факторов (0,06 и 0,066). Решение задачи оказалось
неоднозначным.
При небольших различиях межъядерных расстояний, меньших
0,05-10-1 нм, не всегда удается обнаружить это различие. Поэтому
наличие элементов симметрии в молекуле, которые определяют ра-
венство ряда межъядерных расстояний, например равенство С—С1
в СС14, Р—С1 в РС13 и т. п., существенно упрощает задачу расшиф-
ровки. Обнаружить электронографическим методом небольшие раз-
личия длин каких-либо связей, валентных углов и т. д. может ока-
заться весьма затруднительным или просто невозможным.
При наличии поворотной изомерии приходится исследовать гео-
метрию различных конформеров и изомерный состав, что сущест-
венно усложняет расшифровку.
Надежность определения параметров в молекуле обусловлена
вкладом в рассеяние соответствующих расстояний. Такой вклад
задается фактором
153
L
Рис. VII.13. Кривые f(r) для F2NCI (а) и C3HeNH (б)
nijZiZj
А7=——-exp —-
м/ L z J
где пц — число одинаковых расстояний ij (i и j относятся к ато-
мам). В случае легких атомов в молекуле при малых пц вклад в
рассеяние для параметров, включающих эти атомы, также мал.
Так, в простейшей молекуле этана С2Н6 электронографически хо-
рошо определяются г (С—Н), г (С—С) и Z.CCH. Но конформация
не определяется, так как вклад в рассеяние для расстояний Н...Н,
характеризующих конформацию, практически отсутствует из-за
малых значений факторов Z,, Z/, пц и больших 12ц.
Метод наименьших квадратов позволяет вычислять ошибки оп-
ределения параметров. Точность определения длин связей в благо-
приятных случаях достигает 0,001-IO-10 м, а валентных углов —
десятые доли градуса.
6. ВЛИЯНИЕ ВНУТРИМОЛЕКУЛЯРНЫХ КОЛЕБАНИЙ
НА КОНФИГУРАЦИЮ МОЛЕКУЛ, ОПРЕДЕЛЯЕМУЮ МЕТОДОМ
ГАЗОВОЙ ЭЛЕКТРОНОГРАФИИ
Эффект сокращения. В связи с высокой точностью определения
основных хорошо разрешенных параметров (длин связей, несвязных
межъядерных расстояний *) возникает необходимость учета эф-
фектов внутримолекулярных колебаний. Так, даже для rg- и
Га-структур простейшей молекулы СО2 наблюдаются отклонения от
равновесной конфигурации, выходящие за пределы ошибок экспе-
римента.
Тип структуры.....................
г (С = О), 10-1 нм................
г (О... О), 10-' нм...............
СОСО, °...........................
Ге ге Га
1,1600 1,1652 1,1646
2,3200 2,3261 2,3244
180,0 172,27 172,64
Из этих данных следует, что rg(O...O)<2rg(C=O), хотя для
равновесной конфигурации соблюдается равенство ге(О...О) =
= 2ге(С = О). Величина га очень мало отличается от rg. Этот эффект
уменьшения rg для несвязных межъядерных расстояний линейной
молекулы по сравнению с суммой rg для длин связей открыт
в 1959 г. О. Бастиансеном при исследовании структуры аллена
СН2=С = СН2, в 1962 г. объяснен И. Морино и в настоящее время
называется эффектом сокращения Бастиансена —
Морино. В общем случае для линейных молекул эффект сокра-
щения 6g выражается в виде
N—1
~\=rgiN — '^i rgi’
(VII.67)
где rgiN — несвязное межъядерное расстояние между 1-м и У-м
атомами; rgI- — длина связи i.
Преимущественное использование величин rg объясняется их
наглядным физическим смыслом:
rg— (г)т — re-j- (Az)r-|-Kr,
(VII.68)
где Кт— поправка на перпендикулярные колебания (VII.58). Ве-
личины Кт составляют 10“3... 10-4 нм.
Для линейных молекул подстановка уравнения (VII.68) в урав-
нение (VI 1.67). приводит к тому, что все (Az>r взаимно уничтожа-
ются и остается только алгебраическая сумма поправок на перпен-
дикулярные колебания:
N-1
— ^g—К IN
(VII.69)
* Несвязное межъядерное расстояние — расстояние между атомами, не свя-
занными химической связью.
Рис. VII.14. Деформационное
колебание СО2, приводящее к
увеличению г (С=О) при г
(О ... О) = const
Форма колебаний жестких линей-
ных молекул такова, что, например,
для молекулы СО2 величина К (О ...О)
равна нулю, поскольку при деформа-
ционном колебании атомы кислорода
движутся без изменения расстояния
г (О... О) в направлении, перпендику-
лярном линии О—С—О (рис. VII.14).
Поэтому для СО2 (и подобных моле-
кул):
5 =2/Сг(С=О)= <Д*2(СО)>г+ < Д</2 (СО) > т-_ . (VII.70)
В Ге
Таким образом, конфигурация молекулы, полученная из элек-
тронографического эксперимента, отличается от равновесной.
В частности, в молекуле СО2 такое отклонение от равновесной кон-
фигурации обусловлено в большей степени эффективным увеличе-
нием длины связи СО по сравнению с равновесным. В молекулах
с тройной связью С = С и C = N га-конфигурации соответствуют
нелинейным фрагментам =С—. При малых частотах деформаци-
онных колебаний эффекты сокращения увеличиваются.
В случае молекул, имеющих в равновесной конфигурации, на-
пример, плоскость симметрии, существуют внутримолекулярные ко-
лебания, которые антисимметричны по отношению к плоскости и
приводят к эквивалентным конфигурациям при отклонении от
этой плоскости в ту или другую сторону, например при |<pd | =
= |<р;| (рис. VII.15). Так, например, в молекулах C2F6 и Si2F6 кру-
тильные колебания приводят к тому, что средняя конфигурация
утрачивает плоскость симметрии, а экспериментально наблюдаемые
средние конфигурации отличаются от равновесной конфигурации
симметрии D3d на 7,3 и 25,4° для угла ср.
Рис. VII.15. Ньюменовская проекция для молекул А2Х6
вдоль связи А—А:
показано отклонение от равновесной конфигурации симметрии
D3d при Фе=0; ф — угол вращения вокруг связи А—А
Учитывая геометрическую эквивалентность конфигурации мо-
лекул при положительных и отрицательных отклонениях от плос-
кости симметрии, усреднение межъядерных расстояний, зависящих
от <р, реализуется в интервале углов <р с одним знаком. Это и при-
водит к тому, что средняя конфигурация не обладает плоскостью
симметрии.
Поскольку поправки на перпендикулярные колебания не удов-
летворяют определенным геометрическим условиям, совокупность
rg для длин связей и несвязных межъядерных расстояний молеку-
лы (гё-структура) может оказаться геометрически не согласован-
ной. Структурой геометрически согласованной можно назвать сово-
купность геометрических параметров, в которой через любые неза-
висимые параметры получают все остальные. В качестве незави-
симых параметров обычно выбирают длины связей, валентные и
двугранные углы. Если в качестве независимых параметров, на-
пример, в тетраэдрической молекуле СС1< выбираются равновесные
ге(С—С1) и 2_еС1СС1= 109°28', то эти параметры полностью опи-
шут геометрию молекулы. Из экспериментальных данных для СС1$
получают rg(C—Cl) и rg(Cl...Cl), которые должны отличаться от
равновесных значений ге(С—С1) и ге(С1...С1). Следовательно, и ва-
лентный угол Z_rg (С1СС1) будет отличаться от угла для равновесной
конфигурации (109°28'). Поэтому на основе данных по rg(C—Cl)
и rg(Cl...Cl) можно построить пирамиду для группы СС13, но не пра-
вильный тетраэдр CCh.
Другим примером может служить молекула А12(СН3)6, для ко-
торой остов А12Сб характеризуется равновесной симметрией £)2л-
СН3\ >(СН3)мост\ /СНз
>А1< >А1<
СН3/ \(СН3)мост/ \сн3
Экспериментально определены все значения rg остова. Если задать
в качестве независимых параметров длины связей rg(Al—С) и
гё(А1—С)мост, а также валентные углы Z.CA1C и Z_A1CMoctA1, то для
заданной симметрии D2h легко вычислить все межъядерные рас-
стояния остова А12Сб. При этом оказывается, что рассчитанные
значения величин несвязных межъядерных расстояний для даль-
них С...С значительно отличаются от экспериментальных. Естест-
венно, что таких примеров существует большое число.
Таким образом, совокупность величин rg представляет набор от-
резков, из которых не всегда можно построить точную геометриче-
скую фигуру. Равновесная конфигурация (ге-структура) является
геометрически согласованной, так как она описывает заданное рас-
положение точек в пространстве. Для ге-структуры положение ядер
в пространстве задается координатами минимума полной энергии
молекулы в многомерном пространстве.
Чтобы в электронографическом эксперименте получать данные,,
более близкие к равновесной конфигурации, необходимо вводить
поправки на колебательные эффекты. Однако теория колебаний и
имеющиеся экспериментальные данные позволяют вводить лишь
поправки на перпендикулярные колебания, так как они не вклю-
чают практически неизвестные для многоатомных молекул ангар-
монические постоянные.
Если имеются данные по структуре и силовому полю молекулы,
до возможно рассчитать поправки Кт, которые используются для
перехода от гй-структуры к га-структуре, задаваемой уравнением
rtt^rg-Kr^re4-\Az)rT| (VII.71)
Уравнение (VII.71) представляет хорошее приближение для
ге-структуры. Оно соответствует средней проекции межъядерного
расстояния на линию, соединяющую ядра. Межъядерные расстоя-
ния го практически образуют геометрически согласованную сово-
купность параметров так же, как и rz (см. гл. V), поскольку эти
расстояния описывают заданное расположение точек в простран-
стве.
Для га-структуры геометрия молекулы задается средними зна-
чениями координат атомов в системе координат <Xz>r, {у^т, <£г)т.
Если направить ось z вдоль линии, соединяющей равновесные по-
ложения ядер (см. рис. VII.7), то межъядерное расстояние raij лег-
ко выразить, как это было сделано ранее для гг\
razy = 5(r ez/+ (Дг<7)т)2+ ^Уц}т\1/2,
где <Л?1[>т, — разность средних значений координат
4-го и /-го атомов в молекуле, измеренных при температуре Т.
Разложение в ряд дает уравнение, сходное с таковым для г/.
(VII. 72)
Третий член разложения значительно меньше второго, так как сна-
чала усредняются величины разного знака, а затем эта средняя воз-
водится в квадрат, поэтому третьим членом можно пренебречь.
Следовательно, уравнение (VII.71) в хорошем приближении соот-
ветствует гац и представляет геометрически согласованную систему.
Дальнейшим шагом к уменьшению влияния колебаний на опре-
деляемую структуру является переход от параметров, усредненных
при температуре Т, к параметрам молекул при Т=0, т. е. переход
от га- к Га-структуре:
= (Мо = ^-
Эта величина может быть рассчитана лишь на основе предпо-
ложения о постоянной ангармоничности а. Постоянные ангармо-
ничности для несвязных расстояний малы и ими часто пренебрега-
ют. Тогда приближенно оценивают для связей
rg-r0g=^r}T-^r}0^-^-a[l2T~K0\. (VII.73)
J58
Определив rg и рассчитав <Дх2>0 и <Ду2>0, находят г °, которое
практически близко rz:
г°=г°-^Ах->0-^-<Ау2>0 . (-VII.74)
2г е
Так, для молекулы бензола' .СвНб r° (С—С) = 1,3959-10-1 нм и
г° (С—Н) = 1,091 • 10-1 нм. Эти величины несколько меньше; чем в
/^-структуре (1,399-1О-1 нм и 1,099-1нм). Экспериментальные
данные показывают, что валентные углы, рассчитанные для га- и
-структур, не .отличаются .в.пределах ошибки эксперимента.
Для представления окончательных данных'целесообразно дли-
ны связей давать в ^-параметрах, так какэФи величины соответст-
вуют полному усреднению. С другой стороны, валентные углы и
углы внутреннего вращения в га-структуре будут ближе к равновес-
ным, поскольку они включают поправки на перпендикулярные коле-
бания.
Взаимосвязь геометрических Параметров молекул. К. Кучину
и С. Сивин предложили очень удобную схему, показывающую вза-
имосвязь всех геометрических параметров, определяемых спектро-
скопически и электронографически (рис. VII. 16). Влияние колеба-
ний таково, что г0- и ^-структуры не могут быть приведены друг к
другу или к rz и rg с использованием явных соотношений
(рис. VII.16). . . .
На примере данных для молекулы бензола СвН6 можно сопо-
ставить rg-, г°-, гг- и Го- структуры:
Тип структуры • г.2 . г0
r(C___C), 10-* нм . . . 1,399(1) 1,395g (19) 1,3967(09) 1,397(1)
г(С—Н), 10-1 нм . . . 1,101(5) 1,091(12) 1,083(6) 1,084(5)
Наибольшие отличия наблюдаются для г (С—Н). В г°-структуре
по сравнению с rg имеет место увеличение ошибки параметров, ко-
торое вызвано введением поправок.
Поскольку наибольшее количество данных, представленных в
литературе, относится к гй-структуре (или га-структуре) из электро-
нографических исследований и г8-структуре из микроволновых ис-
следований, представляется важным сопоставить эти величины.
Такой анализ был проведен К. Кучицу и С. Сивином. Авторы пре-
достерегают от слишком детального анализа различий, меньших
чем 10~3 нм.
Разница в длинах связей одной и той же молекулы для rg- и-
г8-структуры составляет 0,01+0,01 • 10-1 нм. При этом не наблюда-
ется систематических отклонений. Действительно, как предполагает-
К- Костейн ^-структура ближе к ге-структуре. Следовательно, мож-
но ожидать, что для одних и тех же связей rs<rg. Однако имеют-
место отклонения rs от rg как в большую, так и в меньшую сторо-
ну. Например, , ,
t
Рис. VII.16. Схематическая связь определяемых в газовой электронографии и
спектроскопии величин:
символы И означают изотопическое замещение; Г — гармонические поправки; АНГ— ан-
гармонические поправки; Э — экстраполяция по уравнениям (VII.81) и (VII.82)
Молекула . . . . . СНзСН2СНз (СН3)2С = О (СН3)3СС1
Связь . . С—С С = О С—С С—С1 ... С—С
rs, Ю-1 нм . . . . 1,526(2) 1,222(3) 1,507(3) 1,803(2) 1,530(2)
4-g, Ю'1 НМ . . . . 1,532(3) 1,212(4) 1,518(3) 1,828(5) 1,528(2)
гг, 10-1 нм . . . . 1,533(7) 1,209(4) 1,515(3) 1,827(5) 1,525(3)
7. ВОЗМОЖНОСТИ МЕТОДА ГАЗОВОЙ ЭЛЕКТРОНОГРАФИИ
Газовая электронография в .основном применяется для опреде-
ления геометрического строения молекул. Для двухатомных и отно-
сительно простых многоатомных молекул (имеются в виду моле-
кулы с высокой симметрией и небольшим числом параметров) мо-
жет быть достигнута высокая точность определения — до 10~4 нм.
При этом подразумевается, что индивидуальное вещество дает оп-
тимальную упругость пара в условиях эксперимента —(2...8)Х
У 103Па. В случае сложных молекул различные параметры в прин-
ципе определяются с неодинаковой точностью. Поэтому при иссле-
довании ряда молекул некоторые параметры найдены с очень вы-
сокой точностью, например г (С—Н), г (С—С) и Z.CCH в НзС—СН3,
но конформация молекулы, т. е. относительное расположение
СНз-групп, практически не может быть установлена из-за малого
вклада в рассеяние электронов для расстояний между атомами во-
дорода различных метильных групп.
В протяженной цепи с хорошей точностью определяются длины
связей и валентные углы. Однако установить конфигурацию такой
цепи затруднительно. Тем более, что для больших расстояний воз-
можны большие амплитуды колебаний пар атомов. Для изучения
внутреннего вращения в производных бутина-2 специально иссле-
давался 1,4^дибромбутиН‘2 BrCH2—С=С—СН2Вг, так как рЯссеи-
вающая'способйость пары атомов Вт... Вг достаточно велика.
Важным достоинством газовой электронографии является воз-
можность изучения молекул,''не имеющих дипольного момента.''
Поскольку интенсивность рассеяния пропорциональна концен-
трации конформеров, метод газовой электронографии позволяет
найти в благоприятных случаях'конформационный состав, а также
при изменении температуры йсс'ледуемого вещества — термодина-
мические параметры: разности энтальпий и энтропий изомёров. ’
На основе амплитуд колебаний пар атомов, полученных элёк-
тронографически, и частот колебаний, найденных спектроскопиче-
ски, можно уточнить силовое поле молекул,
В последнее время В. П. Спиридоновым показана возможность
для простейших молекул: определять из электронографических и
спектроскопических данных равновесную геометрию и силовое
поле молекул. Если известно силовое поле для остова молекулы,
обладающей внутренним вращением с потенциалом, задаваемым в
параметрической форме, то возможно найти и параметры этого по-
тенциала. Так, для молекулы C2F6 был найден барьер вращения,
равный 15,7±3,4 кДж/моль. Барьеры вращения рассчитываются
также из амплитуд колебаний для межъядерных расстояний, зави-
сящих от вращения.
Наибольшие трудности при электронографическом исследова-
нии вызывает установление различий в межъядерных расстояниях,
меньших 10-2 нм. Решить такую задачу можно, если в молекуле
больше межъядерных расстояний, чем независимых параметров.
В общем же случае для молекул, в которых различия в межъядер-
ных расстояниях малы, находят средневзвешенное расстояние.
Эта проблема легко иллюстрируется на примерах кривых ради-
ального распределения f(r). Если в одном пике кривой f(r) пере-
крываются два практически одинаковых пика для расстояний и и
г2, то ошибка определения каждого из них велика. Уменьшая ам-
плитуды колебаний 1\ и 12, можно увеличить’: разницу г2—и по-
лучить ту же полную кривую, f (г). Однако среднее значение и и
г2 ( — । практически не будет изменяться. Поэтому, переходя к
новым переменным
—— — и г2—можно с большей точностью оп-
ределить среднее и независимо найти г2—п; однако с большой
ошибкой.
Все сказанное приводит к тому, что электронографически за-
труднительно определять такие небольшие отклонения от предпола-
гаемой симметрии, как: а) отклонения от линейности молекулы или
ее фрагмента; б) отклонения от плоского строения молекулы или
ее фрагмента; в) различие, например, симметрии C2v и Cs и т. п.
Однако найденные небольшие отклонения от предполагаемой
равновесной конфигурации заданной симметрии еще не доказывают
того, что сделанное предположение ошибочно. Это объясняется тем,
что в электронографическом эксперименте определяются величины
га или rg, которые несколько отличаются от равновесных и зависят
от колебаний в молекуле (см. гл. VII, 5). Более того, rg соответст-
вует усреднению по всем колебательным уровням и поэтому имеет
место некоторая зависимость rg от температуры.
Необходимо иметь в виду, что кроме принципиальной возмож-
ности определения структуры молекул методом газовой электроно-
графии важным фактором является время проведения исследова-
ния. Для относительно простых веществ полное исследование мо-
жет быть завершено за несколько недель. Изучение сложных
молекул требует иногда времени больше года.
8. ОПРЕДЕЛЕНИЕ ГЕОМЕТРИИ МОЛЕКУЛ
ПРИ СОВМЕСТНОМ ИСПОЛЬЗОВАНИИ ЭЛЕКТРОНОГРАФИЧЕСКИХ
И СПЕКТРОСКОПИЧЕСКИХ ДАННЫХ
Установленные связи между геометрическими параметрами, оп-
ределяемыми электронографически и спектроскопически, позволя-
ют использовать экспериментальные молекулярные составляющие
интенсивности sM(s) и вращательные постоянные ЛоВоСо для сов-
местного уточнения искомых параметров. Такая возможность при-
водит к решению структурных задач, которые возникают, напри-
мер, в газовой электронографии при наличии в молекуле близких
расстояний, а также в спектроскопических исследованиях враща-
тельных переходов при отсутствии достаточного числа изотопоза-
мещенных или при малых расстояниях атомов от центра масс или
оси инерции.
Совместное использование данных для определения геометри-
ческих параметров основано на минимизации суммарного квадра-
тичного функционала:
k g = a,b>c
где юм(«й) и (»ig —статистические веса соответственно для элек-
тронографических данных и моментов инерции;
А» [$Л4 ($)]== (Sft)3Kcn kMSkM (Sfe).reop]2 и Ag (/z) = (/g5Kcn /g-геор) -
Уточняемыми параметрами являются г° в sM(s)теор и rz в /хев4>
или rav — в среднем между г ° и rz.
При совместном анализе необходимо введение гармонических
поправок в rg и Во. Следовательно, в таком исследовании исполь-
зуются также результаты анализа нормальных колебаний. Поэто-
му спектроскопические данные включают в себя как совокупность
вращательных постоянных, так и силовое поле исследуемых моле-
кул.
Для иллюстрации применения совместного анализа интересно
привести результаты структурного определения молекулы фосфа-
162
бензола 6 Ь полученные методами газовой электронографии
\з_2/
(ГЭ) и при совместном анализе (ГЭ-|-МВ):
Метод . . . . ГЭ гэ+мв
гср (С—Н), 10-‘ нм . . . . 1,120(15) 1,124(15)
г (Р—С), IO-1 нм . . . . 1,732(3) 1 733(3)
г (С2—С3), 10-1 нм . . . . 1,413(41) 1,413(10)
т (С3—С4), 10-1 нм . . . . 1,384(56) 1,384(12)
r<-v (С С) 10-1 нм . . . . . . 1,398(3) 1 399(3)
Аг (С С) 10-1 нм . . . . . . . . 0,029(98) 0 029(23)
<СРС, ° . . . . 101,0(1,8) 101,1(3)
<РСС, ° . . . . 124,4(3,3) 124,4(6)
<СзСзС4, 0 . . . . 123,7(12,7) 123,7(7)
<СзС4Сз, 0 . . . . 122,5(3,6) 122,8(8)
Совместный анализ позволяет судить о том, что есть различие
в длинах связей С2—С3 и С3—С4 молекулы фосфабензола, хотя оно
находится на грани ошибки эксперимента. Только электроногра-
фическое исследование приводит к очень большим ошибкам в оп-
ределении Ar (С—С). Существенно повышается точность определе-
ния валентных углов, особенно Z_C2C3C4.
Говоря о совместном анализе данных электронографии и спек-
троскопии, следует отметить, что даже частичная спектроскопиче-
ская информация очень полезна при электронографических иссле-
дованиях. Данные колебательной и вращательной спектроскопии
дают возможность определять симметрию молекул — линейность,
плоскостность, наличие центра симметрии и др. Это существенно
помогает при электронографических исследованиях. Уже упомина-
лось, что знание гармонического силового поля позволяет рассчи-
тывать амплитуды колебаний и вводить поправки на эффект со-
кращения. Наличие вращательных постоянных помогает сделать
выбор из ряда моделей, удовлетворяющих электронографическому
эксперименту.
9. НЕКОТОРЫЕ СТЕРЕОХИМИЧЕСКИЕ РЕЗУЛЬТАТЫ
ЭЛЕКТРОНОГРАФИЧЕСКИХ ИССЛЕДОВАНИЙ
В настоящее время имеются данные для более чем 1000 молекул
различных классов соединений, изученных методом газовой элек-
тронографии. В области неорганических соединений получены ос-
новные геометрические параметры для молекул галогенидов, окси-
дов, оксигалогенидов, солей кислородных кислот и комплексных
соединений. Эти данные позволили выявить важные закономерно-
сти, связывающие положение элемента в периодической системе и
валентные состояния атома элемента, которые определяют конфи-
гурацию связей атома. В галогенидах первой группы найдена
структура димеров. Так, в молекуле Li2Cl2 определены расстояния
г (Li—Cl) и r(Cl---Cl). В предположении плоской конфигурации
(конфигурации ромба) можно вычислить все валентные углы.
Галогениды бора ВХ3 (X=F, Cl, Br, I) имеют плоскую конфигура-
цию. Однако в галогенидах алюминия, галлия и индия преобладают
димеры А2Х6, обладающие симметрией D27l. Атомы X и А об-
разуют цикл
Исследование А12С16 при повышен-
ных температурах показало, что в парах присутствовал преимуще-
ственно мономер А1С13 симметрии D3h- Галогениды четвертой груп-
пы имеют тетраэдрическую конфигурацию молекул типа АХ4, пятой
группы — пирамидальную для тригалогенидов типа АХ3 и триго-
нальнобипирамидальную для пентагалогенидов типа АХ5, шестой
группы — угловую для молекул типа АХ2, неправильного тетраэдра
для АХ4 и правильного октаэдра для АХ6, седьмой группы — плос-
кую Т-образную для молекул типа АХ3 и тетрагональнопирамидаль-
ную для АХ5. Изучены также тримеры и тетрамеры некоторых га-
логенидов.
При изучении фторидов ксенона найдено, что молекула XeF2 —
линейная, XeF4 — плоская, с квадратным расположением атомов
фтора, XeF6 — неправильный октаэдр.
Оксиды элементов в парах могут содержать мономеры, димеры,
тримеры и другие полимеры. Валентное состояние двухкоординиро-
ванного атома кислорода в простейших оксидах характеризуется
валентным углом, изменяющимся от 104,5° в Н2О до 118° в С12О и
С1О3—О—С1О3 и далее до ~140° в А12О, 1п2О и Т12О. Полимерные
оксиды образуют циклы. Так, в (WO3)3 шестичленный цикл (WO)3
имеет конфигурацию «кресло», в молекуле (МоО3)3 шестичленный
цикл (МоО)3 практически плоский.
Важным стереохимическим результатом являются данные для
солей кислородных кислот типа T12SO4, Cs2SO4, Cu(NO3)2, T1NO3
и др. В этих молекулах, по-видимому, атомы металлов участвуют
в образовании циклов:
В комплексных соединениях изменяются валентные состояния
атомов донора и акцептора. Например, рассмотрим данные для
комплексов (CH3)3N-A (A=BF3 и ВС13):
Молекула ....
В—X, 10-1 нм .
ZXBX, ° . . .
N—С, 10~* нм .
ZCNC, °. . . .
В—N, 10-1 нм .
(CH3)3N-BF3 (CH3)3N-BC13 BF3 ВС13 N(CH3)3
1,354(6) 1,839(4) 1,313(1) 1,742(4)
113,1(0,9) 110,8(0,3) 120 120 —.
1,468(10) 1,495(4) — — 1,454(2)
108,5(0,7) 108,7(0,5) — — 110,6(0,6)
1,664(11) 1,659(6) — — —
Наиболее существенные изменения при комплексообразовании кос-
нулись группы ВХ3: уменьшен угол ZLXBX и удлинена связь В—X.
В области органических и элементорганических соединений с по-
мощью электронографических исследований выявлены эффекты
взаимного влияния атомов в молекуле. В углеводородах наблюда-
лось значительное увеличение длины связи С—С под влиянием сте-
рических взаимодействий:
Молекула......... Н3С—СН3 (СН3)3С—С(СН3)3 (СН3)3-СН[С(СНя)312
гС— С, 10*> нм . . 1,534 1,58 1,61
В сопряженных углеводородах найдено
бутадиен
2
акролеин
увеличение связи
0^1,526 (1) g/H
н/
глиоксаль.
Длина связи углерод — галоген зависит от числа атомов галогена,
связанных с данным атомом углерода. Так, при переходе от CF4
к (CH3)3CF г(С—F) увеличивается от 1,319(5) до 1,425(8) • 10-1 нм.
Имеется большое число структурных данных, которые позволя-
ют установить закономерности в изменении длин связей углерод —
элемент в зависимости от валентного состояния атома углерода.
Однако при этом найдены факты, которые трудно объяснить на ос-
нове широко распространенных концепций. Так, для связи С—С1
получены следующие данные:
О
Молекула.............
г (С—Cl), 10-1 нм ... .
СН3СН2—О
1,78
СН2=СН—С1
1,73
СН3—С—С1
1,80
Увеличение г (С—О) в ацетилхлориде противоречит представлени-
ям о сопряжении.
Интересна проблема конформаций молекул. В молекулах «-бу-
тана СНзСНгСНгСНз и 1,2-дифторэтана FCH2CH2F транс-конфи-
гурация не является абсолютно преобладающей. В парах обоих
веществ транс-конформеры присутствуют в количествах соответ-
ственно ~53,5 и -~10%. Определение состава позволило оценить
AG0 (гош-транс) =2,08(0,92) кДж/моль для н-бутана, а на основе
температурной зависимости состава 1,2-дифторэтана -рассчитаны
ЛЕ (гош-транс) =—0,398 кДж/моль и AS0 = 5,87 Дж/(моль-К).
Методом газовой электронографии определены геометрические
параметры нового класса соединений — карборанов общей формулы
С2ВпН„+2 (п=3, 4, 8, 10). В молекулах этих соединений атомы уг-
лерода и бора имеют необычно высокие координационные числа (до
шести в С2ВюН12). Остов этих молекул построен в форме полиэд-
ра (рис. VII.17). Сопоставление данных для молекул пара-карбора-
нов показывает, что увеличение координационных чисел (КЧ) ато-
мов С и В сопровождается относительно закономерным удлинением
связи В—С. Однако для связей В—В такая закономерность отсут-
ствует. Связь В—В в основании пирамиды с атомом С в вершине,
т. е. В—В (основ.), при переходе от п=3 к п=4 значительно уко-
рачивается от 1,853 до 1,722-К)-1 нм, а затем снова удлиняется при
п=8 и укорачивается при п=10.
пара - СН(ВН)„СН
В-С. 1СГ1... 1,556 (2) 1,635 (4)
В-В, 10"1... 1,853 (2) 1,725 (8)
1,811 (3) 1,772 (13)
(основ.)
В-В, 10“'нм -
(пояс.)
Рис. VII.17. Структура карборанов пара-СН(ВН)ПСН
* * *
Методы микроволновой вращательной спектроскопии, чисто вра-
щательных спектров комбинационного рассеяния (КР) и газовой
электронографии являются в основном методами определения гео-
метрического строения свободных молекул, т. е. молекул в газовой
фазе при относительно малом давлении.
Некоторое исключение составляет метод КР, поскольку исполь-
зуемые давления паров веществ иногда составляют до 105 Па. Тем
не менее структурную информацию, получаемую этими методами,
можно отнести к молекулам, взаимодействие между которыми пре-
небрежимо мало.
Наибольшее применение имеют методы микроволновой враща-
тельной спектроскопии и газовой электронографии. Ограничения ме-
тодов состоят в том, что эффективно исследование лишь относи-
тельно простых молекул, т. е. молекул с относительно небольшим
числом атомов и геометрических параметров. Методом микровол-
новой спектроскопии возможно исследование лишь полярных мо-
лекул (цо#=О) при достаточном для решения структурной задачи
числе изотопозамещенных производных. В методе газовой электро-
нографии трудности определения структуры молекулы возникают в
том случае, если в молекуле различные независимые расстояния
близки между собой или если вклад в рассеяние каких-либо пар
атомов слишком мал.
В настоящее время развиваются совместные электронографиче-
ские и спектроскопические исследования, в которых используются
данные колебательной спектроскопии о силовом поле для расчета
поправок к данным микроволновой вращательной спектроскопии и
газовой электронографии. При совместном исследовании преодоле-
вается ряд перечисленных трудностей и получают более надежные
результаты.
Структурные данные используются для выявления закономер-
ностей, связывающих геометрию молекул и их химическое строение.
На этой основе возможны проверка и дальнейшее развитие пред-
ставлений о строении вещества.
Контрольные вопросы
Глава V
1. Почему одним из основных условий получения микроволнового враща-
тельного спектра молекул является наличие электрического дипольного мо-
мента?
2. Как уравнения для энергии вращения линейных молекул и молекул
типа симметричного волчка зависят от вращательных постоянных и квантовых
чисел, характеризующих вращение молекул?
3. Как вводится классификация уровней энергии для молекул типа сим-
метричного волчка?
4. Опишите эффект Штарка для линейных молекул, для молекул типов сим-
метричного волчка и асимметричного волчка.
5. В чем состоит принципиальная схема радиоспектрометра? Какие условия
проведения эксперимента? Что дает введение Штарковского электрода?
6. Как используется изотопное замещение при определении геометрического
строения?
7. Выведите уравнения Крейчмена для линейных молекул.
8. Каковы особенности определения координат атомов, находящихся на
близком расстоянии от центра масс или главной оси инерции?
9. В чем выражается взаимосвязь вращения и колебания молекул?
10. В чем состоит физический смысл определяемых величин re, r0, rs и rz?
II. Как определяют значения проекций электрического дипольного момента
молекул методом микроволновой спектроскопии? Как определяют направление
дипольного момента молекул?
12. Как определяют барьеры внутреннего вращения метильных групп?
Глава VI
13. Назовите условия изменения величин поляризуемости молекул в мето-
де КР.
14. Изобразите схематично энергетические переходы в молекуле при ком-
бинационном рассеянии света. В чем состоит отличие чисто вращательных
спектров КР от колебательно-вращательных спектров КР?
15. Опишите схему эксперимента и условие его проведения при изучении
чисто вращательных спектров КР- Какова роль лазеров в эксперименте КР?
16. Каковы правила отбора в чисто вращательных спектрах КР? Каково их
отличие от вращательных спектров в ИК и микроволновой областях?
17. Каковы особенности чисто вращательных спектров КР для молекул
симметрии D., h?
18. Приведите примеры исследованных молекул.
Глава VII
19. Назовите условия задачи рассеяния пучка электронов молекулой.
20. Чем определяется значение атомной амплитуды рассеяния?
21. На какие составляющие можно разделить полную интенсивность рас-
сеяния молекулой?
22. Как используется преобразование Фурье в методе газовой электроно-
графии?
23. Каков характер зависимости приведенной молекулярной составляющей
интенсивности рассеяния от геометрических и динамических параметров, т. е. от
межъядерных расстояний и средних амплитуд колебаний пар атомов?
24. Как зависит амплитуда колебаний от температуры?
25. Каков физический смысл определяемых параметров межъядерных рас-
стояний в уравнении для кривых sAf(s)?
26. Сопоставьте параметры межъядерных расстояний в молекулах, полу-
чаемые методами вращательной спектроскопии и газовой электронографии.
27. Приведите схему эксперимента и перечислите условия его проведения
в методе газовой электронографии.
28. Перечислите возможности и ограничения в определении структуры мо-
лекул методом газовой электронографии.
МЕТОДЫ КОЛЕБАТЕЛЬНОЙ ИК И КР
СПЕКТРОСКОПИИ
Колебательные спектры молекул, наблюдаемые как ИК спектры и спектры
комбинационного рассеяния света, являются такой же специфической харак-
теристикой вещества, как отпечатки пальцев человека. По этим спектрам ве-
щество может быть идентифицировано, если его колебательный спектр уже из-
вестен. По ИК и КР спектрам определяют симметрию и структуру неизученных
молекул. Частоты основных колебаний, находимые из спектров, необходимы
для расчетов термодинамических свойств веществ. Измерение интенсивности
полос в спектрах позволяет проводить количественный анализ, изучать химиче-
ские равновесия и кинетику химических реакций, контролировать ход техноло-
гических процессов. Дальнейшее развитие методов колебательной спектроскопии
и расширение их применения в науке, технике и производстве — непреложное
требование ускорения научно-технического прогресса.
Колебательные спектры молекул эксперимен-
тально изучаются методами инфракрасной (ИК)
спектроскопии и спектроскопии комбинационно-
го рассеяния (КР) света. Эти спектры связаны с
переходами между колебательными энергетиче-
скими состояниями или, в классической интер-
претации, с колебаниями атомных ядер относи-
тельно равновесных положений и определяются
строением молекулы. Число и частоты полос за-
висят, во-первых, от числа образующих молекулу
атомов, масс атомных ядер, геометрии и симметрии равновесной
ядерной конфигурации и, во-вторых, от потенциального поля внут-
римолекулярных сил. Что касается распределения интенсивности
в спектре, то оно определяется электрическими свойствами моле-
кулы: электрическим дипольным моментом ц и поляризуемостью а,
а также их изменением в процессе колебаний.
Таким образом, колебательные спектры являются чрезвычайно
специфическими и чувствительными характеристиками молекул, чем
и объясняется широкое применение их в химических исследованиях.
Колебательные частоты имеют порядок 1012... 1014 Гц и совпадают
с ИК диапазоном частот электромагнитного излучения. Этими
частотами может модулироваться также рассеянное веществом
электромагнитное излучение, например, в видимой или ультрафио-
летовой (УФ) областях, что и дает эффект КР света.
Тепловое ИК излучение было открыто У. Гершелем еще в кон-
це XVIII в., а ИК спектры поглощения молекул впервые были по-
лучены лишь в начале XX в. Эффект комбинационного рассеяния
света веществом был сначала предсказан теоретически А. Смека-
лем, а экспериментально открыт Л. И. Мандельштамом и
Г. С. Ландсбергом в СССР и независимо индийскими учеными
Ч. В. Раманом и К. С. Кришнаном в 1928 г. * Оба метода особенно
успешно стали развиваться в середине нашего века: ИК спектро-
скопия— в конце 40-х, начале 50-х годов, благодаря достижениям
в создании необходимых оптических материалов и развитии элек-
тронной техники, а спектроскопия КР — в 60-х годах, в результате
появления лазерных источников возбуждения этих спектров.
Современные методы колебательной спектроскопии получили
очень широкое распространение и, например, без стандартного
ИК спектрометра не обходится в настоящее время практически ни
одна химическая лаборатория. В силу исключительно высокой спе-
цифичности ИК и КР спектров, которые сравнивают даже с дакти-
лоскопическими отпечатками, они служат незаменимым средством
идентификации соединений и используются в аналитических целях.
По колебательным спектрам проводят структурные исследования,
определяют симметрию молекул, наличие тех или иных функцио-
нальных групп, получают другие сведения о строении и внутримо-
* В зарубежной литературе явление КР света и спектры КР называют Р а-
м а н - эффект и Раман- спектры.
лекулярных силах. Полный набор основных частот колебаний сво-
бодной молекулы необходим для статистического расчета термо-
динамических функций вещества и констант равновесия реакций в
газовой фазе. Поскольку спектр чувствителен по отношению к ма-
лейшим изменениям структуры и силового поля молекулы, методы
ИК и КР спектроскопии позволяют изучать также различные меж-
молекулярные взаимодействия.
Интенсивность спектра зависит от концентрации вещества, что
дает основу для проведения количественного анализа по ИК и КР
спектрам. Качественные и количественные определения этими ме-
тодами возможны как в статике, так и в динамике. Поэтому методы
колебательной спектроскопии широко используют в исследованиях
различного рода равновесий, в том числе очень быстрых, и кине-
тики реакций. Диапазон частот или характеристическое время ме-
тодов колебательной спектроскопии таковы, что они позволяют фик-
сировать и изучать находящиеся в равновесии молекулярные со-
стояния, время жизни которых составляет >10-12 с. Это относится
к быстрым обменным процессам, конформационным равновесиям
и т. п.
Большие возможности для изучения кинетики и механизмов ре-
акций открывают современные скоростные спектрометры, некото-
рые из них позволяют регистрировать полные спектры за время
10~7... 10-8 с.
Достоинством методов колебательной спектроскопии является
то, что они допускают исследование практически любого неоргани-
ческого или органического вещества в любом агрегатном состоя-
нии— газе, жидкости, растворах, кристаллах или аморфной фазе.
Для получения спектра требуются миллиграммовые образцы, при-
чем вещество обычно полностью сохраняется в неизменном состоя-
нии. Имеются методики и приспособления, позволяющие получать
колебательные спектры микро- и макрообъектов без отбора и при-
готовления образцов. По нижнему пределу количественного опре-
деления методы ИК и КР спектроскопии в обычном аппаратурном
оформлении уступают некоторым другим физическим методам, но
использование новейших фурье-спектрометров позволяет повысить
концентрационную чувствительность во много раз.
ГЛАВА VIII
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ КОЛЕБАТЕЛЬНОЙ
спектроскопии
1. КВАНТОВО-МЕХАНИЧЕСКОЕ ПРЕДСТАВЛЕНИЕ
КОЛЕБАТЕЛЬНЫХ СПЕКТРОВ
Применения методов колебательной спектроскопии в химии тре-
буют разной глубины знания теории колебательных спектров, кото-
рая разработана достаточно хорошо. Современная электронно-вы-
числительная техника позволяет проводить теоретический расчгет
колебательного спектра и ряда молекулярных параметров даже для
сравнительно сложных молекул. Владение теорией и техникой рас-
четов особенно важно в структурных исследованиях и для строгой
интерпретации спектров, но при использовании методов колеба-
тельной спектроскопии в прикладных целях, как чисто аналитиче-
ских, также необходимо понимание их теоретических основ.
Колебательные энергетические уровни молекулы можно полу-
чить решением соответствующей квантово-механической задачи.
Вообще говоря, состояния молекулы как единой системы из ядер
и электронов описываются полной волновой функцией Т. Квантово-
механический оператор Й (гамильтониан) в волновом уравнении
Шредингера
HW=E^, (VIII. 1)
определяющем энергии Е стационарных состояний, включает чле-
ны, связанные с орбитальным и спиновым движением электронов:
колебательным и вращательным движением ядерного скелета (по-
ступательным движением можно пренебречь).
В приближении Борна — Оппенгеймера, учитывающем большое
различие масс и подвижностей электронов и ядер, возможно, преж-
де всего, разделение электронного и ядерного движений, причем
для волновых функций — с точностью до (m/M)Vs, а для энергий —
с точностью до (m/Л!)1''4, где т — масса электронов, а М — масса
ядер. Тогда можно записать: Чг=ЧгеЧгп и Й=Йе-{-Йп, где и Йе
относятся только к электронному движению, а и Йп— только
к движению ядер. В достаточно хорошем для многих целей прибли-
жении можно разделить также колебательное и вращательное дви-
жения ядерного скелета, т. е. представить Чг=ЧгеЧгг!Чгг и Й=Йе-\-
что дает возможность решать три уравнения вида
(VIII.1) отдельно и находить соответственно энергии электронных,
колебательных и вращательных состояний молекулы, составля-
ющих полную энергию E=Ee-\-Ev-[-Er.
Нас будет интересовать здесь результат решения только коле-
бательного уравнения
Н Ф =£ Ф
2 * ® * г»
(VIII.2)
Для рассмотрения колебательного движения требуется ввести
координаты, которые описывали бы только относительные смеще-
ния ядер. Поступательное движение молекулы может быть описано
тремя координатами, характеризующими положение центра масс,
а вращательное движение — тремя координатами, в качестве кото-
рых могут быть выбраны углы Эйлера, описывающие ориентацию
молекулы относительно внешней системы координат с началом в
центре масс. В случае линейной молекулы ее ориентация пол-
ностью характеризуется двумя независимыми координатами.
У «двухатомной молекулы имеется лишь одна колебательная сте-
пень свободы (п=3-2—5, см. ниже), т. е. только одна координата
Q = Ar, представляющая изменение межъядерного расстояния, от
которой зависит колебательная волновая функция. Для V-атомной
молекулы можно ввести n=3V—6 (для линейной 5V—5) таких
координат Qk, что каждая из них полностью характеризует смеще-
ние ядер относительно равновесных положений в каком-то одном
основном колебательном состоянии. Это так называемые нор-
мальные координаты, определение которых будет рассмот-
рено позднее.
При использовании нормальных координат колебательную вол-
новую функцию и колебательную энергию М-атомной молекулы
можно представить соответственно как произведение:
п п
^=П ^(Qfe) и сумму:
*=1 й=1
В гармоническом приближении ЧД и Ek — собствен-
ные функции и собственные значения операторов Hv^ зависящих
только от одной координаты Q*:
А.,-—<vm.3>
где h=hl2n', h — постоянная Планка; J2, -vfe — в с~Ч
Этот оператор приводит к уравнению Шредингера для гармони-
ческого осциллятора, решение которого известно. Его собственные
значения энергии:
£*(»*)=Чй (^+v) ’
(VIII.4)
где vk — 0, 1, 2, 3, ... — колебательное квантовое число, a vek — ко-
лебательная постоянная.
Для двухатомных молекул индекс k везде можно опустить, счи-
тая Q—Аг, а постоянная v£>=(k/2n)'j/Ae/|x, т. е. совпадает с выраже-
нием классической частоты гармонических колебаний. Здесь
। т\т.-2
11*=---—------приведенная масса двухатомной молекулы с ядра-
тх + т2
ми, имеющими массы т.\ и m2; ke — гармоническая силовая по-
стоянная, представляющая вторую производную функции по-
, (<PU \
тенциальнои энергии в точке равновесия, т. е. ke = (-- или,
\ dr2 J r=re
что то же, . Потенциальная кривая для реальной
двухатомной молекулы приближенно аппроксимируется функцией
Морзе:
tZ(r)=E>Jl-e“p(r-r*))2, (VIII.5
где De и р — постоянные.
Рис. VIII.1. Качествен-
ная потенциальная кри-
вая U (г) двухатомной
молекулы с системой ко-
лебательных энергетиче-
ских уровней и парабо-
лическая функция
V(Q) = ’/2&eQ2 гармони-
ческого осциллятора
(пунктир)
Вместе с параболической функцией гар-
монического осциллятора эта функция по-
казана на рис. VIII. 1. Система уровней энер-
гий реально является сходящейся к диссо-
ционному пределу, а не системой равноот-
стоящих уровней, согласно формуле (VIII.4),
получающейся в гармоническом приближе-
нии.
Колебательная энергия многоатомной мо-
лекулы в гармоническом приближении яв-
ляется функцией п колебательных кванто-
вых чисел vk:
Ev(vx, v2, v3,
(VIII.6)
где dk — степень вырождения (может рав-
няться 1, 2, 3) колебательного состояния,
которое считается один раз, т. е. при нали-
чии вырождений n<3V—6 (или 3N—5).
Волновая колебательная функция многоатомной молекулы как
произведение волновых функций гармонического осциллятора име-
ет вид
п
Lft = i
1
2ft
где Nv — нормировочный множитель; Hvk — полином Эрмита;
Вырожденные состояния совпадают по энергии, но характери-
зуются разными волновыми функциями (разных координат).
Таким образом, у многоатомной молекулы имеется набор ко-
лебательных состояний с определенной энергией, зависящей от п
колебательных квантовых чисел vk, характеризуемых колебатель-
ной волновой функцией, зависящей от всех 3N—6 (или 3N—5) ко-
лебательных координат.
Большая сложность такой системы не должна пугать. Во-пер-
вых, не все мыслимые уровни, например, с высокими квантовыми
числами реально существуют, т. е. отвечают недиссоциированной
молекуле. Во-вторых, интерес представляют только уровни, лежа-
щие в энергетическом диапазоне, переходы в котором попадают в
исследуемую область спектра (ИК и КР). В-третьих, не все суще-
ствующие в этом диапазоне уровни и переходы оказываются оди-
наково важными для практического рассмотрения и использования
Рис. VHI.2. Нижние энергетические термы E(vi, и2,
«з)//гс (см-1), типы возможных переходов и основ-
ные частоты со; (см-1) молекулы Н2О (смещения
ядер, описываемые нормальными координатами Qft,
показаны на рис. IX.2)
колебательных спектров, так что возможна определенная их клас-
сификация на главные и второстепенные.
В качестве примера на рис. VIII.2 показана система достаточно
низколежащих колебательных уровней энергии, а точнее
термов в единицах волновых чисел (см-1), получающихся при
делении энергии на he (с — скорость света), для молекулы воды.
Эта трехатомная молекула имеет три колебательные степени сво-
боды и ее энергетические уровни зависят от трех квантовых чисел:
Vi, v2, v3. Когда все vk равны нулю, имеется уровень нулевой коле-
бательной энергии, которая, однако, согласно уравнению (VIII.6)
не равна нулю. Нулевой уровень молекулы воды
з
£Д0, О, 0)=±У^,
что соответствует ~4502 см-1.
Уровни, характеризуемые набором квантовых чисел, из которых
только одно имеет значение 1, а остальные равны нулю, называют-
ся главными или фундаментальными. Если какое-то од-
но квантовое число vk имеет значение >1, а остальные равны ну-
лю, то уровень называется обертонным (первым обертонным
при vk = 2, вторым при щ = 3 и т. д.). Наконец, когда два или бо-
лее квантовых чисел, характеризующих уровень, отличны от нуля,
уровень называют составным или комбинированным.
Колебательный спектр вещества наблюдается при поглощении
им ИК излучения или комбинационном рассеянии света, когда в
результате взаимодействия молекул с фотонами /iv происходят из-
Рис. VI11.3. Схема возникновения
менения колебательных состояний,
т. е. молекулы переходят на другие
уровни энергии. Разность энергий
Состояний, между которыми проис-
ходит переход, равна согласно соот-
ношению Бора
A£’=/zv = Ac«>,
где v — частота поглощаемого (или
испускаемого)- излучения.
Разные типы возможных перехо-
дов также показаны для молекулы
Н2О на рис. VIII.2. Волновое число
поглощенного ИК излучения
(см-1) при каком-тошереходе выра-
жается просто как разность соответ-
ствующих термов — более высокого
E'l(hc) и более низкого E"l(hc)-.
Е’ — Е"
ш=--------- .
he
линий релеевского и комбинаци- На рис. VIII.2 указаны, например,
онного рассеяния волновые числа основных переходов
Ий молекулы Н2О.
В спектрах КР (см. гл. VI) волновое число со, соответствующее
разности колебательных термов, измеряют как абсолютную величи-
ну разности волновых чисел возбуждающего излучения соо и рас-
сеянного излучения сор, происходящего по схеме, показанной на
рис. VIII,3:
Е' — Е" v
ш =----------=----
he с
1 “о — “р I
Согласно этой схеме молекула, взаимодействуя с фотоном моно-
хроматического излучения /ivq=/zccoo, сначала возбуждается до како-
го-то неустойчивого, так называемого виртуального, состоя-
ния. Затем она может отдавать этот фотон, не обмениваясь с ним
энергией, т. е. возвращаясь в исходное состояние,— это релеев-
ское рассеяние света. Возможно, однако, заимствование мо-
лекулой части энергии фотона, т. е. отдается фотон меньшей энер-
гии, а мрлекула переходит на более высокий по сравнению с исход-
ным. энергетический уровень Е',— это - стоксово КР. Если
молекула уже находилась в возбужденном состоянии Ё', то при
взаимодействии с фотоном она может отдавать часть своей энергии,
рассеивая фотон большей энергии и переходя на более низкий энер-
гетический уровень Е",— это антистоксово КР. В связи с
меньшей заселенностью более высоких уровней в соответствии с
тепловым распределением молекул антистоксовых переходов в еди-
ницу времени происходит меньше, чем стоксовых, так что интенсив-
ность стоксовых линий КР намного больше и обычно регистрируют
именно такие спектры КР. Они расположены с красной стороны,
т. е. со стороны больших длин волн или меньших частот от релеев-
ской линии.
В принципе, не обязательно возможны все мыслимые переходы
между различными уровнями. Правила отбора разрешенных пе-
реходов, как и интенсивность соответствующих им полос в спектре,
определяются свойствами волновых функций 4%, характеризующих
состояния, между которыми происходит переход, и квантово-меха-
ничеркими операторами собственного или наведенного дипольного
момента, которые совпадают с классическими, выражениями этих
электрических моментов.
В ИК спектре поглощения интенсивность пропорциональна веро-
ятности, т. е. и квадрату момента перехода | |2:
(VIII.7)
где у— оператор электрического дипольного момента, являющий-
ся функцией колебательных координат.
Имея в виду связь момента с его проекциями на оси координат
I М I 2= [Мх I 2+ | /И, I 2-1- | Mz I 2,
можно записать также составляющие
мх=\ dr,
7My = J<VXdr, (VIII.8)
которые являются определенными интегралами по всему коорди-
натному, пространству, т. е. числами, и не меняются при выполне-
нии операций симметрии.
Чтобы переход между состояниями Чгг1 и Т» был возможен, по
крайней мере одна из составляющих Мх, Му, Мх должна быть от-
лична от нуля. Интенсивность же зависит от значения этих состав-
ляющих. Если в приближении малых колебаний (смещений) про-
вести разложение у,ж (у.^, yz) в ряд по степеням Qk около положе-
ния равновесия, то, опуская выкладки и пренебрегая высшими
членами разложения, для перехода между состояниями ^(Q^) и
имеем
(viii.9)
\ dQk Jo J
(и аналогичные выражения для Му и Mz). Таким образом, чтобы
177
переход был возможен, хотя бы одна из производных:
ди-х \ ду-у \ / \
dQk Jo \ dQk )q к dQk /о
должна быть не равна нулю.
В зависимости от того, какая из проекций Мх, Му или Afz=^O
или какая является наибольшей, говорят о поляризации перехода
вдоль соответствующей оси. Это дает иногда возможность выде-
лять так называемые параллельные и перпендикуляр-
ные переходы или классифицировать их по типам Л, В и С в
соответствии с поляризацией вдоль главных осей молекулы.
В спектрах КР интенсивность также пропорциональна вероятно-
сти перехода, но она зависит от оператора наведенного дипольного
момента. В этом случае
M„=AE,
где Е — напряженность поля возбуждающего электромагнитного
излучения, а матрица
Кг л Kry ®xz
Чух Чуу ауг
-azx ^zy ^zz _
Матричные элементы момента перехода будут включать интегралы
вида
Ацл— J
(т, n=ix, у, z).
(VIII. 10)
Поскольку для компонентов тензора поляризуемости справедливо
равенство amn = anm, имеется 6 независимых величин Атп. Чтобы
переход в КР был разрешен, по крайней мере один из этих 6 мат-
ричных элементов должен быть отличен от нуля. В приближении
малых смещений можно прийти к выражению для Атп, аналогич-
ному (VIII.9):
= ?<(Qfe)QM(Qft)dQft (VIII.11)
\ CVS Jo J
и, чтобы переход между уровнями с (Qft) и (Qft) оказался воз-
можен, хотя бы одна из шести производных /'dKnn.'j должна отли-
\ dQk Jo
чаться от нуля.
Для всех точечных групп симметрии, к которым могут относить-
ся молекулы, отличные от нуля компоненты Mi (1 = х, у, г) и Атп
(т, п = х, у, г) найдены и указаны для каждого типа симметрии
колебаний в таблицах характеров точечных групп (см. ниже).
При решении квантово-механической задачи в гармоническом
приближении подстановка и анализ полиномов Эрмита в подынте-
тральных выражениях (VIII.9) и (VIII.11) дает правило о т б о-
р а дипольных переходов с учетом симметрии волновых функций:
Д^=1.
Важно заметить, что, как и в двухатомной молекуле, при таком
правиле отбора частота Vk перехода между уровнями Vk и Vk+ 1 сов-
падает с колебательной постоянной уек:
__ Е (vk + \) — Е (ук) _ hvek (vfe + 1 + l/2) — h\'ek (vk + У2) _
k h h
(VIII. 12>
t. e. с классической частотой гармонического колебания, описывае-
мого координатой Qh.
Для реальных молекул из-за ангармоничности колебаний такого
совпадения нет и возможны также другие переходы: при До = 2, 3,...
или одновременном изменении сразу нескольких квантовых чисел
Vi, Vj, однако их интенсивность обычно много слабее основных пе-
реходов.
На рис. VIII.2 кроме системы колебательных уровней энергии
стрелками показаны возможные в принципе типы переходов. Пере-
ходы с нулевого уровня энергии на уровни с одним из v = l (осталь-
ные v = 0) называются основными и дают в спектре основные
или фундаментальные частоты. Переходы с нулевого
уровня на уровни с одним из v — 2, 3, ... (остальные v = 0) называют
обертоннь/лш и дают в спектре более слабые полосы оберто-
нов (первого, второго и т. д.) основных частот (приблизительно с
удвоенным, утроенным и т. д. значениями). Переходы на уровни с
несколькими v, отличными от нуля, называются составными или
комбинированными и дают составные (комбинирован-
ные) частоты также, обычно, с малой интенсивностью. Нако-
нец, переходы с уровней, у которых одно (или несколько) V>0, на
еще более высокие уровни называют «горячими» и дают в спектре
«горячие полос ы».
Относительная интенсивность полос спектра для различных ти-
пов переходов зависит от заселенности колебательных состояний
или уровней. Согласно больцмановскому распределению
Л’(т') = /гД/exp (— E(v)lkT), (VIII. 13)
и при комнатной температуре наиболее заселенным является уро-
вень нулевой колебательной энергии. Соответственно этому наибо-
лее интенсивны в спектре полосы основных или фундаментальных
частот. Однако велика бывает также заселенность низких по энер-
гии возбужденных уровней (ниже -~300 см-1), например крутиль-
ных колебаний и некоторых других, и соответствующие «горячие»
полосы переходов между ними имеют достаточно высокую интен-
сивность (см. гл. XI; 4).
Рис. VIII.4. Спектры комбинационного рассеяния —
вверху и ИК поглощения — внизу жидкого индена
Таким образом, колебательный спектр многоатомной молекулы
представляет набор основных частот va (в общем случае в числе
3V—6 или менее, если какие-то основные переходы запрещены) с
определенным распределением интенсивности и наложением спект-
ра обертонов nvk, составных частот nvk±tnvi (т, п = 0, 1, 2, 3, ...) и
-«горячих» полос.
На рис. VIII.4 приведены примеры сложных по виду ИК и КР
спектров жидкого индена в диапазоне частот, доступном для иссле-
дования на обычных серийных спектрометрах средней разрешаю-
щей силы.
Чаще всего применения колебательной спектроскопии связаны
с использованием и исследованием спектра основных колебательных
частот, но иногда особый интерес могут представлять и обертоны
или составные частоты, например, для использования в аналитиче-
ских целях, для идентификации типа замещения бензольных колец
(по характерной картине спектра обертонов и составных частот в
области 1650...2000 см-1, см. гл. X; 4).
Колебательно-вращательные спектры молекул в газовой фазе
при достаточно высоком разрешении могут иметь развитую вра-
щательную структуру, изучение которой может представ-
лять как самостоятельный интерес (получение информации, анало-
гичной той, которая извлекается из чисто вращательных спектров,
см. гл. V и VI), особенно для сравнительно простых молекул, так и
служить для интерпретации самих колебательных спектров. Теория
колебательных спектров молекул в конденсированных фазах ус-
ложняется межмолекулярным взаимодействием и влиянием крис-
таллического поля.
2. ОСНОВЫ КЛАССИЧЕСКОЙ ТЕОРИИ
КОЛЕБАТЕЛЬНЫХ СПЕКТРОВ
Молекулы, строго подчиняясь законам квантовой механики,
относятся к таким системам, что некоторые аспекты их поведения
достаточно хорошо описываются в классическом приближении. Это
относится, в частности, к колебаниям их ядерного скелета. В преды-
дущем разделе уже отмечалось, что как у двухатомных, так и у мно-
гоатомных молекул частоты основных колебательных переходов
между квантовыми уровнями, полученными в гармоническом при-
ближении, совпадают с частотами колебаний, получающимися
в классической теории малых колебаний. Вводимые при квантово-
.механическом рассмотрении колебательной задачи для многоатом-
ной молекулы нормальные координаты представляют линейные
комбинации изменений равновесных геометрических параметров
(внутренних координат) молекулы или смещений атомов относи-
тельно положений равновесия в декартовой системе координат.
Коэффициенты соответствующих линейных преобразований ко-
ординат, т. е. линейные комбинации смещений в явном виде, пред-
ставляющие формы нормальных колебаний, могут быть получены
наряду с колебательными частотами путем решения классической
задачи о малых колебаниях.
В настоящее время по классической схеме широко проводятся
расчеты не только частот и форм колебаний — прямая колебатель-
ная задача, но и силовых постоянных (коэффициентов функции по-
тенциальной энергии) — обратная колебательная задача, а также
интенсивности колебательных спектров и электрооптических пара-
метров — соответственно прямая и обратная электрооптические
задачи.
Для решения колебательной задачи в качестве физической мо-
дели берут систему точечных масс, связанных между собой упру-
гими силами, и используют какое-либо известное из теоретической
физики уравнение движения в частных производных (в форме Лаг-
ранжа или Гамильтона). Составление уравнения требует знания
выражений кинетической энергии Т и потенциальной энергии V че-
рез обобщенные координаты или сопряженные с ними импульсы
(или обобщенные силы).
Возьмем систему из Л’-связанных материальных точек. Для опи-
сания ее внутренних колебаний вводится и = ЗМ—6 (или 3N—5) об-
общенных, например, естественных координат qit t'=l, 2, ..., п.
В этих координатах
п
т =" 2
Z,'2=1 (VIII. 14)
u=i
где tn — коэффициенты кинетической энергии, являющиеся функ-
циями масс и геометрических параметров равновесной конфигура-
ции системы; fa — силовые постоянные, равные вторым производ-
I d'2V \
ным потенциальной энергии д д~\ в точке равновесия, т. е. пер-
вые неравные нулю коэффициенты разложения функции
потенциальной энергии в ряд по степеням малых смещений около
положения равновесия (гармоническое приближение).
Можно представить выражения кинетической и потенциальной
энергии (VIII. 14) также в матричной записи:
(VIII. 15)
ИР ни-
где фигурные скобки { } представляют строчную матрицу; прямые
скобки || ||—столбцовую матрицу; Т = [ЛУ] и F=[fi;] — квадратные
симметрические матрицы коэффициентов кинетической и потенци-
альной энергии соответственно.
Теперь из выражений (VIII. 14) или (VIII. 15) можно составить
функцию Лагранжа
L=T-V
и подставить ее в уравнение движения в форме Лагранжа
d dL dL g
d t dq. dq.
что приводит к системе n-линейных дифференциальных уравнений
второго порядка:
7=1
Приняв известное решение дифференциального уравнения гар-
монических колебаний:
qj = Ij cos (j^Lt —|— s) ,
где lj—амплитуда, ]/T=2;rtv,v — частота, t — время, 6 — фаза, по-
лучают в интегральной форме систему уравнений:
2(Л;-Ц7)/7=0.
7 = 1
или в матричной записи:
(F —XT) || Z || — О, (VIII. 16)
где || 1|| матрица — столбец амплитуд изменения координат q,.
182
Чтобы эта однородная система линейных уравнений имела не-
тривиальные решения относительно амплитуд ||Z||, необходимо и до-
статочно, чтобы ее определитель равнялся нулю, т. е.
|F— XT |==0, (VIII. 17)
или в развернутом виде:
/11 ^/1 /12~^12"- /1Я №1п
f 2\ ^21 f 22~'^22--- fin ^2я
=0.
/я1 ^я1 /я2 ^я2’ • • f пп *'f пп
Раскрывая этот определитель, получим уравнение я-го порядка от-
носительно X, решение которого дает п корней Хк=4л2>л (k =
= 1, 2, ..., я), т. е. квадраты всех частот колебаний выбранной
модели. Подстановка каждого корня /./> в (VIII. 16) и решение си-
стемы линейных уравнений дает й-й набор амплитуд Z, (/=1, 2, ...
..., га) или ||Z||k, т. е. смещения или форму колебания с частотой vs.
Такое колебание, при котором все смещения атомов или изменение
всех га естественных координат q{ происходят с одной частотой Vk
и в определенной фазе dk, называется нормальным колебанием. Для
описания каждого нормального колебания можно, в принципе, ввес-
ти одну новую — нормальную координату Qk.
Систему (VIII. 16) можно переписать в виде уравнения
F||Z|| =ХТ Н I || .
Составим из всех столбцов ||Z||(ft> (£=1, 2, ..., п) квадратную мат-
рицу L, а из всех корней /./,— диагональную матрицу Л, тогда име-
ем полное матричное уравнение :
FL = TLA. (VIII. 18)
Если провести нормировку каждого набора амплитуд ||Z||<fe> с по-
мощью множителя
Nk = 1 /-----—г-- ,
* у {ZJ F ||Z||
то составленная после этого полная матрица нормированных форм
колебаний L представляет матрицу линейного преобразования нор-
мальных координат:
II (7 II =L||Q|| , (VIII. 19)
или в развернутом виде :
<71 — Л1Ф1Ч-Л2(?2_|“-”_|-Ля(?л
<?2 = ^21Q1-H22Q2 + -" +^2«Qn (VIII. 20)
Чп. == I nfffc • • • ~\~^nnQn‘
При умножении уравнения (VIII.18) после- нормировки всех на транс-
понированную матрицу L слева
LFL = LTLA,
обе матрицы — Т и F—одновременно диагонализируются:
LTL = Е
(Е — единичная матрица) и
LFL = Л.
Выражения кинетической и потенциальной энергии приводят в нормальных
координатах к каноническому виду:
п п
или в матричной записи:
Т = -j- {Q} Е[Д| и К = у{(г}АЙ,
и каждая координата Qk описывает одно нормальное колебание с частотой v^.
Для нахождения нормальных координат необходимо, таким образом, знать мат-
рицу L нормированных форм колебаний.
Итак, решение прямой колебательной задачи заключается в рас-
чете частот нормальных колебаний, т. е. нахождения диагональной
матрицы Л, и форм нормальных колебаний, т. е. определение мат-
рицы L, при заданных матрицах кинетической и потенциальной
энергии. При этом на практике оказывается более удобным нахо-
дить и использовать не матрицу кинетической энергии в координат-
ном представлении Т, введенную выше, а обратную ей матрицу
Т-1, обозначаемую и называемую также G-матрицей. В теории коле-
баний, используя преобразования квадратичных форм, показыва-
ют, что это матрица коэффициентов кинетической энергии в им-
п
пульсном представлении: Т = — ^uPiPj или в матричной
записи
? = у {/?}G|| д || , где О = [тй а
импульсы р сопряжены
обобщенным координатам q. При умножении векового уравнения
(VIII. 17) слева на эту обратную матрицу оно преобразуется в
| GF —ХЕ | =0,
(VIII.21)
или в развернутом виде:
й(ц X dn ...........d\n
d%\ й?22 — .....d^n
dn\ dn2 • • • dnn X
где = 2 xibfM~ элементы матрицы произведения D = GF, а
\ входит только в диагональные члены.
Полное колебательное уравнение (VIII. 18) преобразуется к
виду __________
GFLLA. (VIII.22)
При заданной геометрической модели системы точечных масс эле-
менты матрицы G=[t,j] вычисляются точно. Если также известна
или просто выбрана в численном виде какая-то матрица F, то пря-
мая колебательная задача решается однозначно.
Необходимо сразу заметить, что нахождение матрицы F встре-
чает принципиальные трудности (см. гл. XI). Обратная колебатель-
ная задача состоит в определении элементов матрицы F, т. е. сило-
вых постоянных fij при известных матрицах G и А, первую из кото-
рых рассчитывают, а вторую составляют по экспериментальным
значениям колебательных частот. При этом матрица форм колеба-
ний L заранее неизвестна и элементы ее из эксперимента прямо не
определяются, а могут быть только рассчитаны.
3. ПРАКТИЧЕСКИЙ РАСЧЕТ КОЛЕБАТЕЛЬНЫХ СПЕКТРОВ
Для расчета частот и форм нормальных колебаний по классиче-
ской схеме, как и при анализе колебательных спектров на основа-
нии экспериментальных данных (см. гл. X), требуется в зависимо-
сти от конкретной задачи исследования либо знать заранее геомет-
рическую конфигурацию, либо выбрать какую-то геометрическую
модель (модели) молекулы.
При нормально-координатном анализе используют известные по
электронографическим или микроволновым данным геометрические
параметры. Если данное конкретное соединение этими методами
экспериментально не исследовалось, то геометрические параметры
выбирают, исходя из общих закономерностей и переноса их из род-
ственных молекул. При этом принимается, что геометрия опреде-
ленных структурных элементов и групп атомов остается в разных
молекулах практически неизменной.
После того как геометрическая конфигурация модели молекулы
задана, вводятся так называемые естественные внутрен-
ние координаты с учетом специфики внутримолекулярных
сил и наличия химических связей между атомами. В валентно-
силовой схеме используют следующие виды естественных коор-
динат:
1) изменения межатомных расстояний по химическим связям
(длин связей):
?z = Д'/
2) изменения валентных углов:
3) изменения двухгранных углов:
При введении всех возможных для выбранной модели коорди-
нат указанных видов (полная система естественных координат) их
число, вообще говоря, может оказаться избыточным, т. е. превы-
сить n = 3N—6. Это связано с тем, что в плоских, объемных (типа
тетраэдра, октаэдра и т. п.) и циклических структурах некоторые
координаты являются линейно зависимыми. Например, в структуре
сумма изменений валентных углов а, равна нулю: Да1 + Да2 + Даз =
= 0 и т. п.
Существуют методы решения колебательной задачи в полной
системе естественных координат, когда по числу «лишних» коорди-
нат получают просто нулевые значения частот, но решают задачу
и в независимой системе координат, заранее исключая лишние ко-
ординаты.
Для решения колебательной задачи необходимо далее найти в
численном виде матрицу кинетической энергии.
Легче всего кинетическая энергия колебаний системы связанных точечных
масс может быть представлена как функция скоростей смещений атомов в де-
картовых координатах:
п
= 2 mk (х1 + Ук + гй)
ft -1
Координаты атома, смещенного из положения равновесия, могут быть представ-
лены как:
Xk = XOk + ^xk,
У к = Уок 4* д Ук.
Zk = 2йк + Ь?к,
где индекс 0 обозначает равновесное значение координаты; Д — смещение.
Тогда, вводя для /г-го атома трехмерный вектор Г/, с компонентами Ахл,
Лук, Дгь, можно записать
N
2Т = ^ ткг2к
Й = 1
или в матричном виде
2Г= (г) М||г||, (VIII.23)
где М — диагональная матрица кинетической энергии, элементами которой яв-
ляются массы атомов ть.
При малых колебаниях каждое смещение атома в декартовой системе коор-
динат можно представить как функцию естественных координат, используя раз-
ложение в степенной ряд и отбрасывая высшие члены, тогда
Д*А = 2 axi4i,
I
^Ук = ^а^Ч1,
i
^Zk^^a^qi,
i
.. к (dxk\ (дук\ к (дгк\
где коэффициенты а*, = —— , aff! = ~— ,а_, == -— . Вводя трехмер-
\ д<П /о yl k dqi /0 г1 \dqtJo
ь ъ ъ
ные векторы с компонентами ayl, az[t можно составить матрицу
А= [aft<] линейного преобразования естественных координат:
М = А М или {г) = [q] А. (VIII.24)
Подстановка (VIII.24) в (VIII.23) и сравнение с выражением кинетической энер-
гии из (VIII.15):
2Т = {г} М И = {?} АМД И = {?} Т Й
показывает, что
Т = АМА,
а элементы матрицы Т= имеют вид
А
tlj — 2 тк {.лк1УЛк]} •
к=1
Выше указывалось, что на практике решают уравнения
(VIII.21), (VIII.22), в которые входит матрица G = T-1. Для ее
получения не обязательно находить элементы (у, т. е. векторы
Дад/), и затем проводить обращение матрицы Т, Оказывается, го-
раздо проще сразу искать элементы матрицы называемый
коэффициентами кинематического взаимодей-
ствия:
т0=2е‘(Ь«хЬ^’ (VI1L25)
й = 1
для чего необходимы трехмерные векторы Ьадь, являющиеся эле-
ментами матрицы В линейного преобразования декартовых коор-
динат смещений:
II q || =В || г || или |<7} = {г}В. (VIII.26)
Аналитические выражения векторов b для всех основных видов ес-
тественных координат q известны и приведены в специальной лите-
ратуре по теории и расчету колебательных спектров молекул (см.
список литературы), а величины е* в (VIII.25) представляют об-
ратные массы атомов, т. е. е*=1/7Пь. Более того, выведены и табу-
лированы в явном виде аналитические выражения самих коэффи-
циентов кинематического взаимодействия (VIII.25) для пар
(i, /) основных видов естественных координат, так что составление
матрицы G в численном виде при заданной геометрической модели
молекулы не встречает принципиальных затруднений (теперь обыч-
но выполняется ЭВМ).
Как уже отмечалось выше и подробнее рассмотрено в гл. XI,
принципиальные трудности встречаются при составлении матрицы
F в численном виде, т. е. нахождении силовых постоянных fa, назы-
ваемых также коэффициентами динамического взаи-
модействия. В нулевом приближении их обычно подбирают по
литературным данным, перенося силовые постоянные для сходных
структурных элементов из найденных ранее силовых полей других
молекул. Так или иначе при составленных в численном виде матри-
цах G и F можно провести, решая уравнения (VIII.21) и (VIII.22);
модельный расчет, который дает набор частот нормальных колеба-
ний молекулы сопоставляемых далее с частотами эксперимен-
тально наблюдаемого колебательного спектра данного вещества, и
относительные амплитуды изменения естественных координат при
каждом нормальном колебании ||/|ИЧ т. е. формы колебаний.
Отнесение наблюдаемых экспериментально основных колеба-
тельных частот представляет сопоставление каждой из них какой-то
форме колебания молекулы. В некоторых случаях можно выделить
при этом какие-то естественные координаты, амплитуды изменения
которых при данном нормальном колебании велики по сравнению
с другими, так что колебание можно условно приписать только к
этим координатам (связям, валентным углам и т. д., см. о характе-
ристических или групповых частотах гл. X; 4). В других случаях
этого сделать нельзя, т. е. форма колебания является сложной,
включающей сравнимые по значениям амплитуды изменения мно-
гих координат. Это, например, колебания всего скелета молекулы.
Графически формы колебаний представляют наиболее наглядно
188
стрелочками относительных смещений атомов в декартовой системе
координат. Поскольку эти смещения связаны с естественными ко-
ординатами (VIII.24), (VIII.26), то их относительные величины
могут быть рассчитаны.
Отнесению частот помогает также расчет распределения
колебательной энергии по естественным координатам с
использованием нормированных форм колебаний. Но окончатель-
ная интерпретация колебательного спектра обязательно должна
опираться на использование и анализ экспериментальных данных
по ИК и КР спектрам и сопоставление их в рядах родственных сое-
динений, т. е. имеются в виду: 1) активность колебаний и относи-
тельная интенсивность полос в ИК и КР спектрах, которые связаны
со свойствами симметрии молекул, 2) степени деполяризации ли-
ний КР; 3) типы контуров вращательной структуры полос в ИК
спектрах газов, 4) данные по спектрам изотопных разновидностей
молекул, 5) данные по групповым частотам и некоторые другие.
Формы колебаний, как уже отмечалось, нужны и для решения
прямой и обратной электрооптических задач. Согласно классиче-
ской теории электромагнетизма при изменении электрического ди-
польного момента системы с частотой v может излучаться или по-
глощаться электромагнитное излучение данной частоты (длины
волн). Собственный дипольный момент молекулы р или его проек-
ции могут быть представлены при малых колебаниях в виде разло-
жения в степенной ряд по нормальной координате Qk. Если отбро-
сить высшие члены разложения, то можно говорить об изменения
собственного дипольного момента с частотой vk, т. е. о появлении
и зависимости интенсивности полос поглощения ИК излучения дан-
u v / д it \
нои частоты от значения первой производной ---- в точке равно-
\ dQk /о
весия. Изменение индуцированного световой волной дипольного
момента Др происходит с частотой колебаний молекулы vs, моду-
лированной частотой монохроматического излучения vo, что и при-
водит, согласно классической теории, к комбинационному рассея-
нию света. Интенсивность КР зависит от значения первой производ-
„ / да \
нои поляризуемости ----- , если иметь в виду, что поляризуемость
V dQk Jo
а (ее компоненты) также можно представить степенным рядом и
высшие члены отбросить.
Как при квантово-механическом, так и при классическом подхо-
де к рассмотрению интенсивностей и расчете их по любой схеме не^
обходимо знать указанные производные. Эти производные могут
быть представлены, как и сами нормальные координаты, в виде
линейных комбинаций, но тоже производных р (или а) по естест-
венным координатам:
/др \ _ (д? \ / dv-\ f I д_/ д р \ .
\dQk /о \dqi Jo \dq2 Jo \dqn Jo
где lki — амплитуды, т. e. элементы k-vo столбца матрицы L
форм колебаний.
В матричной записи
|| I [| (*). (VIII.27)
\ dQk /о l\<fyz/o) |
{ д ;i \ ( да \
Элементы и \dq~j могут быть приняты в качестве парамет-
ров, общих для всех нормальных колебаний молекулы, и производ-
ные (-^-1 будут отличаться только по форме колебания ||/||(ft). ।
\ dQk /о
Практическое нахождение указанных производных основывается,
обычно, на так называемой валентно-оптической схеме.
В ней собственный дипольный момент молекулы представляется
как аддитивная векторная величина, складывающаяся из диполь-
ных моментов р/ отдельных химических связей в молекуле
Н = [
j
где е/ — направляющий вектор /-й связи.
Подстановка этого выражения дипольного момента в формулу
/ \
(VIII.27) приводит к появлению производных I I по естествен-
ным координатам, которые вместе с р/ образуют систему элек-
трооптических параметров, входящих в линейные урав-
нения функциональной (F) зависимости производных:
/'JjL') || z || (S)\ (VIII.28)
dQk /о V д<Ц /
Аналогично, но несколько сложнее можно подойти к выражению
производных Если набор электрооптических параметров
' dQk /о
установлен и рассчитаны формы колебаний, то можно находить
соответствующие производные по нормальным координатам, кото-
рыми определяются абсолютные интенсивности полос основных
тонов нормальных колебаний.
Обратная электроопическая задача состоит в
расчете электрооптических параметров по экспериментальным зна-
чениям интенсивностей полос в ИК и КР спектрах и рассчитанным
формам нормальных колебаний. Эти параметры находятся с точно-
стью до знака, так как интенсивность пропорциональна квадрату
модуля соответствующих производных (см. гл. VIII; 1).
Степень приближения при решении прямой задачи сильно зави-
сит от выбора и определения электрооптических параметров. Чем
детальнее будет классификация химических связей с учетом их типа
и окружения, т. е. чем тщательнее развита аддитивная схема для
представления ц и а, тем в лучшем приближении будет решение.
Можно проиллюстрировать, например, на связях С—Н сильную
зависимость электрооптических параметров рс-н и производных по
растяжению самой связи {--и соседней С—Н связи I——1 от
\dqijo \dq2r
структуры группы, включающей эту связь:
Группы........................... =СН
Ис-н, Ю-30 Кл-м (D).............. 3,67(1,1)
/ ди. \ о
I—----- >Ю-1 2» Кл (£>/А) . . . 3,00(0,9)
\ д<7! Jo
/ дц \ 0
= СН2 —СН3
1,97(0,59) —0,93 (—0,28)
2,10(0,63) 2,47(0,74)
0,30(0,09) 1,03(0,31)
-— , 10-20 Кл (D/A)
д<12 Jo
Следует также помнить, что результаты решения электроопти-
ческих задач зависят, как очевидно, от выбора матрицы силовых
постоянных F (см. гл. XI) при анализе нормальных координат, т. е.
нахождении форм колебаний.
При всей приближенности расчетов спектральных кривых путем
решения прямых, колебательной (частоты) и электрооптической
(интенсивности) задач показана уже их большая практическая
польза при идентификации соединений и установлении деталей
строения молекул.
ГЛАВА IX
СИММЕТРИЯ МОЛЕКУЛ И НОРМАЛЬНЫХ КОЛЕБАНИЙ
1. ОБЩИЕ ПРЕДСТАВЛЕНИЯ О СИММЕТРИИ МОЛЕКУЛ
Важное значение при анализе нормальных колебаний имеет их
симметрия, которая определяется свойствами симметрии равновес-
ной ядерной конфигурации молекулы. Учет этих свойств, с одной
стороны, облегчает расчет колебательных спектров, так как позво-
ляет проводить разделение колебательной задачи. С другой сторо-
ны, симметрия нормальных колебаний определяет их активность
(правила отбора) в ИК и КР спектрах, и сопоставление этих экс-
периментально наблюдаемых спектров позволяет определить сим-
метрию молекулы.
Под симметрией какого-либо предмета понимается вся совокуп-
ность имеющихся у него элементов симметрии. Элементам
симметрии соответствуют операции симметрии, переводя-
щие предмет сам в себя. Возможные комбинации операций симмет-
рии, оставляющих без изменения хотя бы одну точку (в частности,
центр масс), называются точечными группами симметрии. Суще-
ствуют следующие элементы и операции симметрии.
I. Сп — поворотная ось симметрии порядка п; со-
ответствующие ей операции симметрии — повороты на угол р(2л/и),
где р может принимать значения от 1 до п—1. Специфической оси
Соо соответствует операция поворота на сколь угодно малый угол.
Рис. IX.1. Схема определения точечной группы симметрии
2. а — плоскость симметрии; соответствующая ей опера-
ция симметрии — отражение в плоскости.
3. i—центр симметрии; операция симметрии — инверсия
в центре (отражение в точке).
4. Sn — зеркально-поворотная ось симметрии
порядка и; операции симметрии — повороты на угол р(2л/п) с
одновременным отражением в плоскости, перпендикулярной оси
(р от 1 до п—1).
5. I (или Ci) — вводимый для общности тождественный
элемент симметрии; соответствующая операция симмет-
рии— операция идентичности или тождественного преобразова-
ния — оставляет предмет в покое.
Любая молекула относится к какой-либо точечной группе сим-
метрии, если не исключать и тривиальную группу Ci, не имеющую
элементов симметрии и операций, кроме тождественного преобра-
зования. Во всех остальных случаях точечные группы включают
несколько операций симметрии. Для определения точечной груп-
пы симметрии молекулы можно воспользоваться схемой, представ-
ленной на рис. IX. 1, которая предусматривает следующую последо-
вательность действий.
1. Определяют, имеет ли молекула форму, близкую к сферо-
идальной, т. е. с несколькими пересекающимися осями Сп (п>2).
Если нет, то переходят ко второму действию. Если да, то она отно-
сится к одной из точечных групп высшей симметрии: Та, Oh, 1к
(точечные группы Т, Th, О, I среди молекул не встречаются) .
2. Находят поворотную ось симметрии Сп. Если есть, то перехо-
дят к третьему действию. Если осей нет, определяют, есть ли плос-
кость о или центр г, определяющие группы Cs и Ci\ если их тоже
нет, то имеем тривиальную группу С\.
3. При наличии оси или осей симметрии выбирается ось высше-
го порядка (п>2), а если ее нет, то одна из осей — С2 или единст-
венная ось Czn, ориентируемая вертикально и совпадающая с глав-
ной осью молекулы Z.
4. Определяют, есть ли зеркально-поворотная ось симметрии.
Если есть только ось S2n, колинеарная оси Czn, а кроме нее и, воз-
можно, центра i других элементов симметрии нет, то это группы
52п. Если кроме Sn (п>2) есть другие элементы симметрии или
вообще нет оси Sn, то переходят к пятому действию.
5. Находят набор п осей С2, перпендикулярных оси Сгп. Если
таковые есть, то переходят к шестому действию, т. е. точечным
группам ряда Dn\ если нет, то переходят к седьмому действию,
т. е. точечным группам ряда Сп.
6. Смотрят, есть ли горизонтальная плоскость Uh (перпендику-
лярная Сд), если есть, то это будет точечная группа Dnh, если ее
нет, но имеется п диагональных (вертикальных) плоскостей под,
то — Dna, если их тоже нет — Dn.
7. В другом ряду групп при наличии од группы символизируют-
ся как-Cnh\ при отсутствии о/ь но наличии вертикальных плоскостей
— Сп?, а при отсутствии плоскостей — Сп.
Полные совокупности операций симметрии точечных групп даны
в таблицах типов симметрии и характеров представлений (напри-
мер, табл. IX.1).
Таблица IX. 1. Таблицы типов симметрии и характеров неприводимых
представлений групп C2v, C2h, D2h, C3v, D3h
I cz C2 <3v(xz)
At 1 1 1 1 Тг XX, yy, zz
A2 1 1 —1 —1 xy
Bi 1 — 1 1 —1 Tx Rv xz
B2 1 — 1 —1 1 Tv yz
П родолжение табл. 1Х.1
^2 h I cz с2- oh(xy) 1
Ag 1 1 1 1 Rz XX, yy, ZZ, X#
А и 1 1 —1 —1 tz
Bg 1 —1 —1 1 RX, Ry xz, yz
Ви 1 —1 1 —1 Ху '1 у
D2h^v I cz L 2 СУ2 c 2 i G(XZ) a(yz)
Ag Au Big Bzg Bag Blu Biu Bau . I 1 1 1 1 1 1 1 1 1 1 —1 —1 1 -1 —1 1 1 — 1 1 — 1 >—1 1 — 1 1 1 —1 —1 1 —1 —1 1 1 —1 1 1 1 —1 —1. —1 1 —1 1 —1 —1 —1 1 1 1 — 1 — 1 1 — 1 1 — 1 1 1 —1 —1 —1 1 1 1 —1 Rz Ry Rx Tz Ty Tx XX, yy, ZZ xy xz yz
cav I 2C3 3av
Ai A2 E 1 1 2 1 1 — 1 1 — 1 0 Tz (Tx, Ty) Rz (Rx, Ry) xx+yy, ZZ (xx—yy, xy); (yz, xz)
D3h I 2C3 2.S3 3C2 ЗЩ
^2 E' E" 1 1 1 1 2 2 1 1 1 1 —1 —1 1 — 1 1 — 1 — 1 1 1 1 —1 —1 0 0 1 —1 1 —1 2 —2 1 — 1 — 1 1 0 0 Tz Tx, Ty Rz Rx, R у xx + yy, ZZ XX—yy, xy xz, yz
2.
КАЧЕСТВЕННЫЕ ПРЕДСТАВЛЕНИЯ О СИММЕТРИИ
КОЛЕБАНИЙ
При колебаниях молекулы возможны только определенные ком-
бинации свойств симметрии смещенной (от равновесной) ядерной
конфигурации, если иметь в виду ее отношение к операциям симмет-
рии точечной группы. Эти комбинации и представляют типы сим-
метрии, по которым классифицируются нормальные колебания.
Нормальное колебание называется симметричным (s) по отно-
шению к данной операции симметрии, если при ее выполнении все
амплитуды естественных координат или векторы смещений атомов
не меняют знака и абсолютного значения (умножаются на +1).
Колебание антисимметрично (as) относительно операции симмет-
рии, если при ее выполнении знак смещений меняется на обратный
(умножение на —1). Нормальное колебание, симметричное относи-
тельно всех операций симметрии, образующих точечную группу, к
которой принадлежит молекула, называется полносимметричным.
Все остальные типы нормальных колебаний неполносимметричные
или дважды (£), или трижды (F) вырожденные. При вырожденных
колебаниях операция симметрии переводит одну форму колебаний
в другую, т. е. векторы смещений умножаются на числа не все рав-
ные 1 или все неравные 1.
Полная характеристика типа симметрии нормального колебания
описывается его отношением ко всем операциям симметрии данной
точечной группы. Невырожденные типы симметрии обозначаются
символами А и В. При этом А используется для обозначения коле-
баний, симметричных относительно выделенной главной оси, ориен-
тируемой вертикально, и колебаний при отсутствии осей симмет-
рии— группы Cs (/ио), Ci (I и i), а В — антисимметричных отно-
сительно такой оси. Подстрочные индексы g и и при А и В
обозначают соответственно симметричное и антисимметричное ко-
лебания по отношению к операции инверсии в центре i. Подстроч-
ные цифровые индексы 1, 2 обозначают симметричный и антисим-
метричный типы по отношению к операции отражения в вертикаль-
ной плоскости ov, в которой лежит ось, или по отношению к
повороту вокруг оси второго порядка С2, перпендикулярной глав-
ной оси. Надстрочные индексы — один штрих' или два штриха" при .
прописных буквах — обозначают симметричный и антисимметрич-
ный типы колебаний относительно отражения в горизонтальной
плоскости Oh, перпендикулярной оси симметрии, и в точечной груп-
пе Cs. Цифровые индексы 1, 2, 3 используются также при символах
вырожденных колебаний Е и F, но не имеют того же смысла, что
для невырожденных колебаний. Символика типов симметрии ко-
лебаний для линейных молекул (точечные группы симметрии
Coov и Осой) заимствуется из обозначений электронных состояний
в зависимости от абсолютного значения проекции электронного
орбитального момента на ось молекулы (см. гл. XIII; 2).
На рис. IX.2 схематически представлены формы нормальных
колебаний простейших многоатомных молекул: нелинейной и ли-
нейной трехатомных молекул ХУ2. Нелинейная молекула имеет
одну ось симметрии второго порядка С2, две пересекающиеся на
ней под прямым углом плоскости симметрии dj и о2 и относится к
точечной группе симметрии С2г>. Такими являются, например, мо-
лекулы Н2О, SO2 и др., у которых одна из плоскостей симметрии
совпадает с плоскостью самой молекулы, а ось симметрии прохо-
7* 195
Рис. IX.2. Нормальные колебания нелинейной (а) и линейной (б)
трехатомной молекулы
26—2
дит по биссектрисе валентного угла (рис. IX.3). Колебания с час-
тотами (01 и соз сопровождаются в основном растяжением связей в
фазе и не в фазе и называются соответственно симметричным
и антисимметричным vas валентными колебаниями. Пер-
вое сох полносимметрично—тип Ль а второе — соз — антисиммет-
рично относительно оси С2 и плоскости симметрии, перпендикуляр-
ной плоскости молекулы, — тип В2.
Вместо эквивалентных естественных координат q\ и q2 растяже-
ния связей X—Y можно ввести новые координаты — координа- (
ты симметрии qAi и q\ каждая из которых описывает пове-
дение пары и 92 при нормальных колебаниях по отношению ко
всем операциям симметрии. Новые и старые координаты связаны
между собой с учетом требования нормировки 2* =2(qs)2 соот-
ношениями:
?л,==т75"(^1+^2); ^1=-т7=-(?л’Я-7В2);
V V (IX. 1)
7в’=-р=-(?1-72); <72=-4--(^Л1-7В2)-
Колебание с частотой <в2 в основном сопровождается изменение
ем валентного угла и называется деформационным б. Оно '
полносимметрично — тип Ai, т. е. естественная координата а сама
является координатой симметрии типа Аг
а = ал>. (1Х.2>
Рис. IX.3. Расположение ядер не-
линейной трехатомной молекулы
и ее главных осей относительно
элементов симметрии точечной
группы C2v
Таким образом, для нелинейной молекулы ХУ2 можно ввести
две координаты симметрии типа Ai—qA* и ал* и одну координату
симметрии типа В2—qB*- Поскольку потенциальная и кинетиче*-
ская, а следовательно, и полная энергия не зависят от выбора ко-
ординат, а, с другой стороны, инвариантны по отношению к операн
циям симметрии для смещенной конфигурации, происходив
частичное разделение колебательной задачи. Это значит, что она
может решаться отдельно для каждого типа симметрии, надо толь-
ко матрицы G и F привести по симметрии с использованием коэф-
фициентов линейного преобразования координат (IX.I) и (IX.2).
Отвечающая колебанию с частотой со3 координата qB*, как единст-
венная данного типа симметрии, является и нормальной координа-
той Q3, координаты же qAl и aAi не являются нормальными коор-
динатами Q] и Q2, а связаны с ними линейным преобразованием:
qA1 — “h
аЛ1== AxiQi “ЬАхгФг»
(IX.3)
где столбцы коэффициентов амплитуд представляют форму
&-го нормального колебания в координатах симметрии.
Поэтому-то разделение колебательной задачи и является не пол-
ным (по нормальным координатам), а частичным. Для молекулы
Н2О частоты колебаний coi, сог, (о3 или, согласно квантовой механи-
ке, основных колебательных переходов в состояния, описываемые
волновыми функциями ^(Qi), ^?(Q2), (Q3), указаны на рис.
VIII.2 в гл. VIII, где рассмотрена схема колебательных энергетиче-
ских уровней этой молекулы. Для молекулы SO2 соответствующие
фундаментальные частоты имеют значения: coi = 1151 см-1, <й2=
= 519 см-1, со3= 1361 см-1. Каждое из нормальных колебаний этих
молекул сопровождается такими изменениями собственного диполь-
ного момента и поляризуемости, что их производные по соответст-
вующей координате в точке равновесия отличны от нуля, и все ко-
лебания активны как в ИК спектре, так и в спектре КР.
Линейные молекулы ХУ2 имеют ось симметрии Соо, совпадаю-
щую с осью молекулы, центр симметрии i на ядре атома X, т. е.
в точке пересечения оси Соо с плоскостью симметрии и бесконеч-
Рис. IX.4. Качественные кривые зависимости
дипольного момента (а) и поляризуемости (б)
от нормальной координаты Qk (k= 1, 2, 3)
для линейной трехатомной молекулы XY2
ное число плоскостей <з-о, пересекающихся на оси, точечная группа
симметрии Dooh- Формы нормальных колебаний молекул ХУ2 пред-
ставлены на рис. IX.2, а в табл. IX.2 приведены частоты, их обо-
значения по форме и типы симметрии для некоторых конкретных
молекул и молекулярных ионов. В этом случае деформационное
колебание ба дважды вырождено, поэтому экспериментально на-
блюдается не четыре (для линейной молекулы число нормальных
колебаний n=3N—5, N — число атомов), а три частоты, причем
две из них в ИК спектре, а одна в спектре КР, что также указано
в табл. IX.2. Это можно понять, если для описания трех колебаний
молекулы ХУ2 ввести соответственно нормальные координаты Qi,
Q2, Q3 и рассмотреть производные по этим координатам собствен-
ного дипольного момента ц и поляризуемости а. Здесь рассмот-
рение будет приведено несколько упрощенно, но на качественных
результатах это не отражается. Как уже говорилось, если для дан-
ного нормального колебания производная
то колеба-
I да \ _г_п
ние активно в ИК спектре, если производная то
колебание активно в спектре КР- На рис. IX.4 иллюстрируются
кривые зависимости дипольного момента и поляризуемости от
координат Qk (й=1, 2, 3). У центросимметричной молекулы соб-
ственный дипольный момент ц=0 и при полносимметричном ко-
лебании Mi он не меняется (прямая 1 на рис. IX.4, а), т. е.
(-J* ) =0, и колебание неактивно в ИК спектре. При деформа-
\ dQi /Q1 = a
ционном со2 и антисимметричном валентном соз колебаниях (кри-
вые 2(3) на рис. IX.4, а) в каждой фазе колебания молекула те-
ряет исходную симметрию и у нее появляется дипольный момент,
причем с изменением фазы колебания ц меняет свой знак, так что
кривые зависимости ц от Qk проходят в точке равновесия с каким-
Таблица IX.2. Значения частот (см-1), отнесение, типы симметрии
и активность в ИК и КР спектрах колебаний некоторых линейных молекул,
XY2 (симметрия Dooh)
Частота Отнесение (тип симметрии) Спектр Молекула (ион)
СО2 uof+ XeF2 hf2
<01 V.(S+) КР (-1340) 860 515 675
(02 Sd (Пц) ИК 667 210 213 1200
(03 Vas(S+) ИК 2349 930 558 1550
то наклоном через 0, а „ / д [1 \ производные в этой точке \dQ-i /Q И = 0
БМ #0,т.е.оба эти колебания проявляются в ИК спектре.
W3 /бз = о
Что касается поляризуемости, то она всегда отлична от нуля и
имеет положительное значение; для равновесной конфигурации оно
соответствует точке на ординате (рис. IX.4, б). При полносиммет-
ричном колебании coi поляризуемость («объем эллипсоида поляри-
зуемости») плавно меняется от какого-то минимального значения
в одной фазе до какого-то максимального значения в другой, про-
ходя через точку равновесного значения при Qi = 0 (кривые 7 на
рис. IX.4, б), так что f-——) ф0, и колебание активно в спектре
WC?! /Q, = o
КР. Наоборот, при колебаниях ®2 и ®3 (см. рис. IX.2) в обеих фа-
зах поляризуемость (эллипсоид поляризуемости) меняется одина-
ково по сравнению с равновесной, т. е. кривая ее зависимости от
Q* проходит в точке равновесия через экстремум (кривые 2 (3) на
рис. IX.4, б), где =0 и =0, т. е. эти колебания
\dQi 'q2=o \<?<?з/Сз=о
не активны в спектре КР.
Таким образом, для молекул ХУ2 можно установить, является
молекула линейной или нелинейной, просто сравнивая спектры КР
и ИК- Необходимо отметить, что для линейной молекулы симмет-
рии £>оол колебания, активные в ИК спектре, не наблюдаются в
спектре КР и, наоборот, активные в КР не активны в ИК спектре.
Это частный случай проявления правила взаимоисключения частот
(альтернативного запрета) для центросимметричных молекул, бо-
лее общая формулировка которого будет дана ниже.
3. РЕЗУЛЬТАТЫ ТЕОРЕТИКО-ГРУППОВОГО
АНАЛИЗА КОЛЕБАНИИ
Строго и последовательно симметрия нормальных колебаний и
правила отбора для ИК и КР спектров рассматриваются с по-
мощью математической теории групп. При анализе, интерпретации
и практическом использовании колебательных спектров важно
зиять и уметь пользоваться следующими основными результатами
такого рассмотрения.
В терминах теории групп, применительно к точечным группам
симметрии, принадлежность нормального колебания к какому-то
типу симметрии соответствует принадлежности к определенному
неприводимому представлению данной группы. Вообще представ-
лением группы называется совокупность линейных преобразований
координат, соответствующих операциям симметрии точечной груп-
пы. Оно является приводимым, если для него можно выбрать коор-
динаты так, чтобы они преобразовывались раздельно. Представле-
ние называется неприводимым, если его нельзя дальше упростить,
т. е. нельзя выбрать новые координаты, которые преобразовыва-
лись бы раздельно.
Если, например, рассматривать отношение смещений ядер мо-
лекулы из положений равновесия к операциям симметрии в декар-
товой системе координат или в системе внутренних естественных
координат, как это было сделано выше для нелинейной трехатом-
ной молекулы ХУ2, то можно получить приводимые представления
точечной группы симметрии.
Вводимые для описания колебаний молекулы естественные ко-
ординаты разбиваются на совокупности симметрично
эквивалентных координат, переводимых операциями сим-
метрии при равновесной конфигурации друг в друга. Например, у
молекулы ХУ2 это две совокупности: одна — и Цг, а другая — а.
Для смещенных конфигураций молекулы при нормальных колеба-
ниях координаты в этих совокупностях преобразуются операциями
симметрии по-разному, но для разных совокупностей симметрично
эквивалентных координат могут существовать преобразова-
ния, происходящие одинаково в отношении каждой из опе-
раций симметрии. Именно поэтому возможно введение координат
симметрии, например qAi, аА' и qB* для нелинейной молекулы XY2.
Это значит, что координаты симметрии разбиваются по типам
симметрии нормальных колебаний или неприводимым представле-
ниям группы, т. е. преобразуются раздельно. Каждая из них опре-
деляет поведение всех эквивалентных естественных координат
соответствующей совокупности по отношению ко всем операциям
симметрии.
Общее число координат симметрии равно общему числу есте-
ственных координат, а для определения числа координат симмет-
рии данного неприводимого представления, т. е. для подсчета чис-
ла нормальных колебаний каждого типа симметрии, необходимо
знать свойства приводимых и неприводимых представлений. Это
так называемые характеры представлений, т. е. суммы
диагональных элементов (следы) матриц преобразований коорди-
нат. В теории групп выводится следующая формула:
(IX.4)
где ns — число колебаний типа симметрии s, в которое могут вхо-
дить вместе с настоящими и ненастоящие колебания
(трансляции, вращения); h — число операций симметрии в
группе; hi — число операций в i-м классе * (при отсутствии в моле-
куле осей симметрии выше второго порядка все й(=1); %«(/?)—
характер приводимого представления для операции R (в i-м клас-
се); у! (R)—характер неприводимого представления для опера-
ции R (в i-м классе). Суммирование ведется по всем классам, при
всех hi= 1 по всем операциям симметрии группы.
Характеры приводимых представлений молекулы для различных
операций симметрии находятся по формулам:
Х(1)=ЗЛС
Х(3) = ДГО,
х(/)=-ЗЛГ;, (1Х,5)
x(C£) = ^cp[1 + 2cos[^)],
X (Spn) = Ns J( - 1 )₽ + 2 cos Y| ,
n j. \ n ji
где N — число атомов в молекуле; Na, Nt, Nc.p и NSP—числа ато-
n n
мов молекулы, не меняющих своего положения при выполнении
соответствующих операций симметрии, т. е. лежащих соответствен-
но в данной плоскости симметрии (о), в центре симметрии (Nt
может быть равно 0 или 1) и на осях симметрии (С„ или S„).
Характеры неприводимых представлений по операциям симмет-
рии или типы симметрии колебаний даны для всех точечных групп
в таблицах, которые приводятся в учебниках и монографиях по
симметрии молекул и кристаллов, молекулярной спектроскопии и
теории групп. В качестве примеров приведены таблицы характеров
(типов симметрии) для пяти точечных груцп симметрии: С2с, С2л,
D2h, c3v и D3h (табл. IX.1). В таких таблицах кроме операций сим-
метрии, образующих данную точечную группу, и характеров при-
водятся и правила отбора для ИК и КР спектров, а также указы-
вается, к какому типу симметрии относятся трансляции и вращения
относительно системы главный осей.
Зная, к какой точечной группе симметрии относится молекула
или выбранная модель молекулы, можно с помощью формул
(IX.4), (IX.5) и табл. IX.1 определить: 1) число колебаний (вместе
с трансляциями и вращениями), относящихся к каждому типу
симметрии; 2) типы симметрии трансляций и вращений для вычи-
тания их и получения истинного числа нормальных колебаний каж-
дого типа симметрии; 3) сколько и каких нормальных колебаний
должно проявляться в ИК спектре; 4) сколько и каких нормальных
* Классы операций симметрии образуются из операций, соответствующих
осям порядка га>2.
^колебаний должно проявляться в спектре КР; 5) сколько линий
КР и для каких нормальных колебаний должно быть поляризова-
но или деполяризовано (см. гл. X; 2).
Против того типа симметрии в таблице характеров, к которому
относится трансляция или вращение, ставится, соответственно,
один из символов Тх, Ту, Тг (иногда просто х, у, г) и Rx, Ry, Rz.
Не представляет большого труда определить это и без таблиц.
Достаточно задать направления главных осей (X, У, Z) при из-
вестной точечной группе симметрии и, смещая в направлениях
осей или поворачивая относительно их молекулу, определить, как
будут меняться знаки координат ядер в этой системе при выпол-
нении каждой операции симметрии, т. е. определить характер каж-
дого преобразования координат. Например, для нелинейной моле-
кулы ХУ2 направления главных осей, проходящих через центр
масс, показаны на рис. IX.3, и легко видеть, что типы симметрии
смещений молекулы по осям и поворотов вокруг осей именно те,
которые указаны для точечной группы С2а в табл. IX. 1.
Как уже говорилось, для проявления в ИК спектре правила
отбора требуют, чтобы была отлична от нуля хотя бы одна из
проекций электрического момента данного перехода Мх, Му,
Mz или производная хотя бы одной проекции цх, j.iy, цг собственно-
го дипольного момента молекулы по нормальной координате в
точке равновесия. Для этого достаточно, чтобы тип симметрии
нормального колебания совпадал с типом симметрии трансляций
в направлении одной из декартовых координат (в системе главных
осей молекулы). Таким образом, нужно найти, в каких строках
таблицы характеров неприводимых представлений точечной груп-
пы стоят координаты х, у, z или символы* с этими подстрочными
индексами (Д, Mi, Pi и т. п., i=x, у, г), обозначающие трансляцию
или проекцию электрического дипольного момента перехода.
Для определения правил отбора в спектре КР надо знать, к
каким типам симметрии относятся компоненты тензора поляризуе-
мости ац (i, /=х, у, z).
Если выбрать направление электрического поля вдоль оси х,
то компоненты (т. е. проекции на оси координат) индуцированного
этим полем электрического дипольного момента определяются по
формулам-’
^хх^х’ Р'у=:=а'ХуЕх, р.г = а^-гЕ'г.
Тип симметрии Ех, как и цж, совпадает с типом симметрии са-
мой координаты х (или трансляции Тх). Если при выполнении
операции симметрии координата х меняет знак, то меняют знак и
Ех, и ух, но не ахх=ух/Ех, т. е. ажж относится к тому же типу сим-
метрии **, что и х\х = х2. Вообще тип симметрии компоненты а<>
* В литературе, в которой можно найти таблицы характеров точечных групп,
используют разную символику: Т — трансляция, М и Р — электрические диполь-
ные моменты перехода и др.
"* Знак X символизирует прямое произведение.
определяется прямым произведением типов симметрии i-й
и j-н координат. Это значит, что перемножаются характеры непри-
водимых представлений двух строк, в которых проставлены i-я и
/-я координаты, для каждой операции симметрии и определяется,
к какому типу симметрии относится полученная комбинация про-
изведений. В таблицах характеров в строках типов симметрии ко-
лебаний, активных в спектре КР, обычно указываются какие-то
из компонент ац или их комбинации, или произведения, или ком-
бинации произведений координат (см. табл. IX.1).
Для несимметричных молекул (Ci) практически все колебания
активны в ИК и КР спектрах, но из-за приближенной или локаль-
ной симметрии отдельных групп интенсивности полос могут сильно
различаться. В зависимости от симметрии частоты колебаний мо-
гут частично или все повторяться в ИК и КР спектрах при разной
или сравнимой интенсивности полос или совсем не повторяться в
этих спектрах. Рассматривая колебания линейных трехатомных
молекул ХУ2, мы уже встретились, например, с так называемым
правилом альтернативного запрета или взаимоисклю-
чения частот. Формулируется оно следующим образом: у всех мо-
лекул, имеющих центр симметрии, и молекул, относящихся к то-
чечным группам Du, C5h, D5il и D6d, нормальное колебание может
быть активно или только в ИК спектре, или только в спектре КР
или неактивно ни в том, ни в другом спектре, но никогда не может
быть активно в обоих этих спектрах одновременно. Это правило
легко можно переформулировать, говоря вместо активности коле-
баний о разрешенное™ или запрещенное™ основных квантовых
колебательных переходов в ИК и КР спектрах.
У центросимметричных молекул, принадлежащих, например, к
точечной группе симметрии Д2л, возможен тип симметрии Аи, кото-
рому не принадлежат ни одна из проекций электрического диполь-
ного момента и ни одна из компонент тензора поляризуемости
(см. табл. IX.1), т. е. нормальные колебания этого типа не должны
проявляться ни в ИК спектре, ни в спектре КР. Так, по формуле
(IX.4) можно определить, что у молекулы этилена Н2С = СН2 из
12 нормальных колебаний 6 (из них 3 — Ag, 1 — Bg, 2 — Bsg) ак-
тивны в спектре КР, 5 (из них 2 — В1и, 2 — Biu, 1 — Biu) —в ИК
спектре и одно колебание (Ли) запрещено в обоих спектрах. Это
крутильное (или торсионное) колебание, характеризующееся по
форме смещениями четырех атомов водорода из плоскости молеку-
лы в одну и другую сторону в шахматном порядке:
z11
,с=с'
Hz
У молекулярного иона транс-[Р1С12Вг2]2-, относящегося к той
же точечной группе симметрии, что и молекула этилена, из 9 нор-
4000 3000 2000 1500 1000 500 см-1
Рис. IX.5. ИК и КР спектры бензола
мальных колебаний 6 актив-
ны в ИК спектре (2 — Bi„,
2— В2и, 2— Взи), 3 — в
спектре КР (2—7Ц, 1—•
В^) и нет колебаний типа
Аи, запрещенных в том и
другом спектрах. У рассмот-
ренных соединений, как и у
всех центросимметричных
молекул, нет повторяющих-
ся в ИК и КР спектрах час-
тот.
Неактивно в обоих спек-
трах крутильное колебание
этана, гексахлорэтана и по-
добных молекул. На рис.
IX.5 видно, как дополняют друг друга ИК и КР спектры центросим-
метричной молекулы бензола (точечная группа симметрии £>бл),
у которого из 30 нормальных колебаний 8 запрещены в ИК и КР
спектрах.
4. РЕЗОНАНС ФЕРМИ
Частота полносимметричного колебания молекулы СО2 в табл.
IX.2 указана в скобках потому, что фактически в спектре КР вме-
сто одной полосы наблюдается дублет с компонентами примерно
равной интенсивности: 1286 и 1388 см-1. Это частный случай доста-
точно общего явления, состоящего в следующем. Если частота
какого-то основного колебания оказывается близкой к частоте
какого-то обертона или составной частоте, т. е. имеет место слу-
чайное вырождение соответствующих энергетических уровней, то
обе частоты смещаются в разные стороны друг от друга, а менее
интенсивная полоса (обертон или составная) «заимствует» интен-
сивность из более сильной основной полосы, так что компоненты
дублета оказываются сравнимыми по интенсивности. У молекулы
СО2 частота <щ (~ 1340 см-1) близка к первому обертону дефор-
мационного колебания ~2ы2 (~ 2x667= 1334 см'1)- Это явление
впервые было объяснено Э. Ферми как раз для данной молекулы
и получило название резонанса Ферми. Положение невозму-
щенных резонансом частот можно оценить по формуле
1 £ 1а+‘Ь
где (оо — невозмущенная частота, имеющая два значения (при зна-
ках + или — в формуле); соа и ш — наблюдаемые частоты; 1а и
h — наблюдаемые интенсивности.
Наличие резонанса Ферми определяется разными способами, в
частности исследованием поведения пары полос (предполагаемых
компонентов дублета) при изотопозамещении, изменении раствори-
204
теля или агрегатного состояния вещества и т. д., хотя идентифици-
ровать его удается не всегда легко.
Необходимо отметить, что, хотя сама основная частота
667 см-1 (Пм) в спектре КР не наблюдается, ее первый обертон
вступает в резонанс Ферми с частотой vs и его волновое число мо-
жет быть оценено по формуле (IX.6), а по нему и частота ба-
5. ЭФФЕКТЫ КРИСТАЛЛИЧНОСТИ
До сих пор речь шла о колебаниях изолированных молекул,
т. е. рассмотренный материал может использоваться при анализе
и интерпретации спектров веществ в газовой фазе и растворах, но
с осторожностью в случае жидкостей и может быть совершенно
недостаточным при детальном рассмотрении спектров кристалли-
ческих веществ.
При грубо приближенном рассмотрении молекулярного кри-
сталла как «ориентированного газа», т. е. упорядоченного распо-
ложения определенным образом ориентированных, но не взаимо-
действующих частиц, уже можно объяснить отсутствие вращатель-
ной структуры и сужение полос, а также разную их поляризацию
в зависимости от ориентации кристалла. При учете взаимодействия
молекул в реальном кристалле в первом приближении имеются в
виду два эффекта. Во-первых, статический эффект поля
кристалла на отдельную молекулу, который может приводить
к смещению, расщеплению и появлению новых полос из-за сдвига
энергетических уровней, снятия их вырождений и запретов на пере-
ходы. Во-вторых, динамический эффект резонансного
взаимодействия молекул, находящихся в одной элементар-
ной ячейке, называемый также эффектом Давыдова, когда
происходит расщепление энергетического уровня (даже не вырож-
денного) изолированной молекулой и в спектре вместо одной поло-
сы наблюдается мультиплет.
Правила отбора для ИК и КР спектров зависят не только от
симметрии молекулы (молекулярного иона), но и от симметрии ее
позиции в кристалле и от структуры и симметрии самого кристал-
ла. Совокупность операций симметрии, соответствующих элемен-
там симметрии, на которых лежит центр масс молекулы (иона),
образует так называемую местную или сайт-группу. Она, как пра-
вило, является подгруппой точечной группы симметрии
изолированной частицы, т. е. в кристалле симметрия частицы пони-
жается, что и приводит к расщеплению вырожденных колебаний и
снятию запретов, т. е. проявлению статического эффекта поля кри-
сталла. Для определения сайт-группы нужно знать пространствен-
ную группу кристалла и число частиц в элементарной ячейке, что
возможно по данным рентгеноструктурного анализа.
Динамический резонансный эффект наблюдается обычно в ИК
спектрах и является результатом диполь-диполного взаимодейст-
вия колеблющихся молекул. Для определения числа компонент
давыдовского расщепления полосы нужно знать число частиц в
элементарной ячейке и ее симметрию, т. е. так называемую фак-
тор-группу. Последняя является подгруппой пространственной
группы симметрии кристалла и представляет совокупность опера-
ций симметрии, получающуюся после замены всех простых транс-
ляций в пространственной группе операцией идентичности (тожде-
ственного преобразования).
Как сайт-групповой анализ, т. е. определение местной
группы симметрии, так ифактор-групповой анализ, позво-
ляющие подсчитать и объяснить число наблюдаемых частот в коле-
бательных спектрах молекулярных кристаллов, проводят с по-
мощью таблиц, приводимых в специальной литературе. Они сущест-
венно усложнены по сравнению с табл. IX. 1 для точечных групп
симметрии, и требуют кристаллографических знаний. Оценка рас-
щепления колебательных уровней в кристаллах — еще более слож-
ная задача, и можно только отметить тот экспериментальный факт,
что она обычно не очень велика (10.. .30 см-1).
В спектрах кристаллов наблюдаются также так называемые
внешние колебания или колебания решетки. Они связаны с
отсутствием у молекул в кристалле поступательных и вращатель-
ных степеней свободы и называются соответственно трансля-
ционными и ориентационными. Полосы колебаний ре-
шетки лежат обычно в довольно низкочастотной области (ниже
200 см-1) и могут быть достаточно интенсивны как в ИК, так и в
КР спектрах.
ГЛАВА X
АНАЛИЗ И ИНТЕРПРЕТАЦИЯ СПЕКТРОВ.
ОПРЕДЕЛЕНИЕ СИММЕТРИИ И СТРУКТУРЫ МОЛЕКУЛ
1. ВЫВОДЫ ИЗ СОПОСТАВЛЕНИЯ ИК и КР СПЕКТРОВ
Знание симметрии нормальных колебаний и правил отбора для
ИК и КР спектров позволяет, сравнивая эти экспериментально
полученные спектры, решать обратную задачу, т. е. определять
симметрию равновесной геометрической конфигурации молекулы.
Это уже иллюстрировалось в гл. IX на простейшем примере нели-
нейных (Сго) и линейных (Ооой) трехатомных молекул ХУ2, кото-
рые легко идентифицируются по числу наблюдаемых частот в ИК
спектре (3 — для симметрии С2о, 2 — для Dxh) и спектре КР (3 —
ДЛЯ 1 ДЛЯ Dooh) .
Если известна брутто-формула химической частицы, то можно
представить несколько геометрических моделей равновесной ядер-
ной конфигурации, имеющих разную симметрию. Для каждой мо-
дели, пользуясь формулами (IX.5) и таблицами характеров (типа
табл. IX. 1), можно найти характеры приводимых и неприводимых
представлений, а по формуле (IX.4) определить число нормальных
колебаний каждого типа симметрии данной точечной группы.
Определив по тем же таблицам, какие типы симметрии разре-
шены в ИК и КР спектрах, можно для каждой модели подсчитать
число фундаментальных частот, которые должны наблюдаться в
том и другом спектрах.
Часто оказывается достаточным сравнить полученные для раз-
ных моделей результаты такого подсчета с экспериментально на-
блюдаемыми спектрами, чтобы по числу полос в ИК спектре и спек-
тре КР сделать вывод о том, какой из моделей соответствует реаль-
ная молекула.
Поясним сказанное, рассматривая молекулу XY3. Эта молекула
может быть неплоской (пирамидальной), относясь к точечной
группе симметрии С3а, или плоской — точечная группа D3h- В этих
случаях из 6 нормальных колебаний два колебания являются дваж-
ды вырожденными, т. е. должно наблюдаться 4 фундаментальных
частоты. На рис. Х.1 представлена корреляционная схема, на кото-
рой показано, как распределяются колебания по типам симметрии,
формы этих колебаний и указано (см. также табл. IX.1), какие из
них активны в ИК спектре, а какие — в спектре КР- Выбор между
двумя моделями может быть сделан, очевидно, на основании того,
что для пирамидальной молекулы все четыре колебания должны
проявляться в обоих спектрах, а для плоской молекулы как в ИК,
так и в КР спектре активны по три колебания, причем две наблю-
даемые частоты (вырожденных колебаний) должны совпадать в
этих спектрах, а одна частота в каждом из спектров не повторяет-
ся в другом.
При рассмотрении экспериментальных спектров должна учиты-
ваться возможность проявления некоторых обертонов и составных
частот. Для уверенного отнесения наблюдаемых частот по типам
симметрии основных переходов необходимы дополнительные дан-
ные. Некоторые из них, в частности о поляризации линий КР и
контурах ИК полос в газовой фазе, также приведены на корреля-
ционной схеме (рис. Х.1) и будут разъяснены ниже (см. гл. X;
2 и 3).
Иногда у частицы XY3 можно предположить также такую кон-
фигурацию, что она будет относиться к точечной группе симметрии
C2v, например j "-1" Т . На корреляционной схеме
(рис. Х.1) видно также, как вырожденные колебания групп С3а и
D3h распадаются по невырожденным типам симметрии группы С2а
и что все шесть нормальных колебаний должны проявляться как в
ИК спектре, так и в спектре КР-
В качестве примеров частиц типа XY3 можно рассмотреть моле-
кулы РН3, BF3, ВС13 или молекулярный ион МОГ и т. д. У первой
молекулы, как и у дейтерированного аналога, в ИК и КР спектрах
наблюдаются совпадающие четыре фундаментальные частоты
(см-1):
vs (Л1) vas(£) МЛ) баз(£>
РН3 2327 2421 991 1121
PD3 1694 1698 730 806
т. е. эти молекулы имеют пирамидальную конфигурацию (Сзи)>
причем предложенное отнесение согласуется с данными по поляри-
зации линий КР и контурам ИК полос (см. рис. Х.1). Для молекул
BF3, ВС13, как и для молекулярного иона NO7, спектроскопиче-
ские данные доказывают плоское строение симметрии D3h- В табл*
Рис. Х.1. Корреляционная диаграмма типов симметрии колебаний, фор-
мы колебаний, активность в ИК и КР спектрах и другие характерис-
тики для молекул ХУз разной симметрии
Таблица Х.1. Частоты (см-1), отнесение, типы симметрии и активность в ИК
и КР спектрах колебаний некоторых молекул симметрии РЗЛ
Молекула Отнесение, тип симметрии, спектр, поляризация
₽(A? vaS<£'> Ъ(Е')
КР, р ик ИК; КР, dp ИК; КР, dp
nBF3 888 681 1446 480
10BF3 888 718 1505 482
ИВС13 471 460 956 243
10ВС13 471 480 995 244
NO7 . -1050 -820 -1400 -720
Х.1 приведены значения и отнесение частот для изотопных разно-
видностей BF3 и ВС13. Для иона NO? можно было предположить
также в качестве возможной конфигурацию симметрии С2и. Одна-
ко экспериментально в ИК и КР спектрах наблюдали по три фун-
даментальных частоты, причем единственная поляризованная ли-
ния КР не имела аналога в ИК спектре, что вместе с другими
данными приводит к отнесению частот, указанному в табл. Х.1, и
однозначному выводу о симметрии D3h- По колебательным спект-
рам, согласно корреляционной схеме (рис. Х.1), для свободных ра-
дикалов CF3, СНз, CD3 в матрицах из инертных газов (см. гл. XII;
1.3) установлено, что первый (CF3) имеет пирамидальное строение
(Сзи), а два других — плоское (£>зл).
Еще один пример определения точечной группы симметрии, в
частности для молекулы IF5, представлен в табл. Х.2, в которой
приведены результаты теоретических подсчетов для разных моде-
лей и экспериментальные данные об общем числе фундаменталь-
ных частот, о числах колебаний, наблюдаемых в ИК и КР спектрах,,
поляризованных линий КР, полос, совпадающих в ИК и КР спект-
рах и из них поляризованных в спектре КР. При сопоставлении
видно, что экспериментальные данные лучше всего соответствуют
точечной группе симметрии молекулы т. е. конфигурации че-
тырехгранной пирамиды.
Аналогичным образом доказано, например, что скелет молекулы
N2(SiH3)4 относится к точечной группе симметрии
’ а
молекула N2O4 — к точечной группе d2
4 в газовой
фазе имеет конфигурацию молекул симметрии D2a, а в твердой_
D2h. По спектрам можно отличить, является ли четырехчленный
цикл плоским или неплоским; в случае одинаковых атомов в цикле
это соответственно точечные группы Д4/г и D2d-
Н
цис- и транс-дизамещенных этилена С2Н2Х2:
Разная симметрия
/Н
/С=С\ (C2v) и
II ХХ - - -
(С’гл)» или 2ош- и анти-(s-транс) поворотных изомеров
1,2-дизамещенньгх этана СН2ХСН2Х, изображаемых ньюменев-
ским и проекциями:
в скобках указаны их точечные группы симметрии — приводит к
существенным различиям их ИК и КР спектров, по которым- они и
могут быть идентифицированы.
Иногда для определения конфигурации молекулы, комплекса
и т. д. бывает достаточно рассматривать не полный набор фундамен-
тальных частот, а только некоторые из них, относящиеся к коле-
баниям группы скелетных атомов, с учетом локальной симметрии
этой группы. В табл. Х.З представлены, например, данные, позво-
лившие определить конфигурацию комплекса OsO4Py (Ру — пири-
дин) только по частотам валентных колебаний Os — О в области
900 см-1. Это оказалась конфигурация симметрии Cs, показанная
в последней строке табл. Х.З. По ИК и КР спектрам показано, что,
например, молекула XeF6 не является октаэдрической, т. е. имеет
более низкую симметрию, чем О/..
Для иллюстрации влияния структуры кристалла на колебатель-
ный спектр, образующих его молекул или молекулярных ионов
рассмотрим, например молекулярный ион NO-Г, фундаментальные
частоты которого в отсутствие сильных межмолекулярных взаимо-
действий и его симметрия указаны в табл. Х.1. Колебательный
спектр кристалла нитрата натрия в отличие от изолированных
ионов NOF состоит уже из шести фундаментальных частот, причем
три из них наблюдаются только в ИК спектре, три — только в
спектре КР. В кристалле NaNO3 происходит расщепление дважды
вырожденных колебаний иона NOF и активность компонентов
расщепления оказывается различной, т. е. по одной мы наблюдаем
в ИК спектре и по одной — в спектре КР- Разобраться в этом
позволяет фактор-групповой анализ. Из рентгеноструктурных дан-
ных известно, что пространственная группа кристалла нитрата нат-
рия Did (R3C), а в его элементарной ячейке содержатся две фор-
210
Рис. Х.2. Корреляционная схема расщепления колебательных
уровней и частоты ио:-:а NO^" в ИК и КР спектрах кристалла нит-
рата натрия
Таблица Х.2. Теоретическое число фундаментальных частот в ИК и КР
спектрах для возможных конфигураций молекулы XY5 и экспериментальные
данные для 1F5
Точечная группа Общее число частот ИК КР р Совпадают в ИК и КР спектрах
всего из них р
Dsh 7 3 3 1 0 0
Dsh 8 5 6 2 3 0
C$v 7 4 7 2 4 2
Civ 9 6 9 3 6 3
Сз„ 8 8 8 4 8 4
Cz-o 12 12 12 5 12 5
c, 12 12 12 8 12 8
Экспериментальные данные для IF5 9 4 8 3 3 3
* Вывод: молекула IFs имеет симметрию C^v, незначительные расхождения экспери-
мента с теорией могут быть связаны с малой интенсивностью некоторых полос и другими
экспериментальными причинами.
мульные единицы. Порядок фактор-группы кристалла соответст-
вует Dm, а симметрия иона Озн, при этом симметрия позиции иона
в кристалле (сайт-группа) D3, т. е. можно построить корреляцион-
ную схему, представленную на рис. Х.2, на которой указано и пред-
ложенное отнесение экспериментально наблюдаемых частот.
Таблица Х.З. Число частот валентных колебаний Os—О для различных
моделей комплекса OsO4Py (Ру — пиридин) и экспериментальные данные
по ИК и КР спектрам
Конфигурация Точеч- ная группа Числю частот (0s—О) ИК КР Р Совпадают в ИК и КР спектрах
всего из них р
?/° Ру—Os^ Сги 4 4 4 2 4 2
1 о 1 0—0—0 сл Оз© 4 3 3 2 3 2
0 0 /Os\ oz 0 4 2 3 1 2 1
0 ° о /Os\ Ру 0 с. 4 4 > 4 3 4 3
Найдено: ИК, V, см-1... 926, 915, 908, 885
КР, v, см-1... 928 (р), 916 (р), 907 (р), 886 (dp)
Вывод: комплекс имеет симметрию С,.
2. ПОЛЯРИЗАЦИЯ ПОЛОС В СПЕКТРАХ КР
Как уже отмечалось, полосы спектра комбинационного рассея-
ния, относящиеся к различным нормальным колебаниям, в зависи-
мости от типа симметрии колебаний по-разному поляризованы. По-
этому исследование поляризации полос КР позволяет делать за-
ключения об отнесении частот по некоторым типам симметрии
колебаний и помогает в определении симметрии молекул.
На рис. Х.З схематично показано, как для какой-то заданной
частоты рассеянного молекулой света экспериментально определя-
ется его поляризация. Пусть молекула находится в начале системы
координат. Электрический вектор падающего на молекулу в на-
правлении оси х электромагнитного излучения <УП осциллирует,
как показано на рис. Х.З, параллельно оси z, т. е. излучение плоско
поляризовано, как это имеет место в современных схемах возбуж-
дения спектров КР с помощью лазеров. Рассеянное под прямым
углом (в направлении оси у) излучение <FP направляется в щель S
212
спектрометра. На пути этого излуче-
ния помещается поляризатор Р, поз-
воляющий измерять интенсивность
рассеянного излучения, поляризо-
ванного в направлениях, перпенди-
кулярном /j_ и параллельном I w
направлению поляризации электри-
ческого вектора падающего излуче-
ния. Измеряемое экспериментально
отношение рп = /д//ц называется
степенью деполяризации
рассеянного излучения данной час-
тоты.
Если перейти к системе главных
осей молекулы, являющихся осями
эллипсоида поляризуемости а, Ъ, с
и совпадающих с осями внешней сис-
Рис. Х.З. Схема экспериментально-
го изучения поляризации рассеян-
ного света
темы х, у, z, в начало которой помещена молекула, то проекции
наведенного дипольного момента на оси
—аа^а>
= ab#b’
—аХс’
где аа, аь, ас — главные значения поляризуемости.
Для изотропной молекулы, обладающей сферической симмет-
рией, аа=аб = ас=а. Для анизотропной молекулы выделяют, как
известно, сферическую часть или среднее значение поляризуемости:
а=-£-(а°"Ьа*+а<!)
и анизотропию поляризуемости:
= у Каа + а*)2 4-(аь — ас)2(ас — аа)2].
Очевидно, что для изотропной молекулы |32 = О. В другом предель-
ном случае — максимальной (полной) анизотропии (при аа =
= аь = 0, ас=#=0) а = ас/3, |32 = а2.
Из физики известно, что для степени деполяризации релеевско-
го рассеяния света при плоской поляризации падающего
излучения теория дает следующее выражение:
Таким образом, для релеевского рассеяния изотропных молекул
рп = 0 (/± = 0), а при максимальной анизотропии рп='/з.
При комбинационном рассеянии света, когда меняется колеба-
тельное состояние молекулы, интенсивность рассеянного излучения
данной частоты vk зависит, как уже отмечалось, от производных
элементов тензора поляризуемости по соответствующей нормаль-
ной координате Q* в точке равновесия. Используя производные
(да/ )
~dQ^)Q -о (^==а’ с>’ М0ЖН(>
вместо средней поляризуемости а и анизотропии |32.ввести величи-
ны а* и р? и преобразовать формулу (Х.1), записанную для ре-
леевского рассеяния, в форму для степени деполяризации комби-
национного рассеяния данной частоты
Рпй ,2 ,2 •
45aft
(Х.2)
Но, если сама средняя поляризуемость любой молекулы никогда
не может быть равна нулю: а=#0, то as в точке равновесия может
быть равна или неравна нулю в зависимости от типа симмет-
рии нормального колебания. Так, для любого неполносимметрич-
ного нормального колебания а*=0. Действительно, поляризуемо-
сти в точке равновесия соответствует экстремум, так как противо-
положные фазы неполносимметричного колебания приводят к
одинаковой деформации эллипсоида поляризуемости, как уже от-
мечалось, например, при рассмотрении колебаний vas и линей-
ной трехатомной молекулы ХУ2 (см. рис. IX.2 и IX.4). Тогда для
всех неполносимметричных колебаний согласно (Х.2) степень де-
поляризации рпА=3/4, и говорят, что полосы этих колебаний пол-
ностью деполяризованы (символ dp).
Полносимметричные колебания (Л, Ai, A', Ag,...) приводят к
монотонному изменению эллипсоида поляризуемости от поворотной
точки одной фазы до поворотной точки другой фазы (см. рис. IX.2,
IX.3) так, что в точке равновесия аД=£0. Степень деполяризации
таких колебаний лежит в пределах 0<СРпа<С—» и говорят, что
соответствующие полосы КР полностью (при рп/г = 0) или частично
при 0<^рпй<^ —) поляризованы (символ р). Первое воз-
можно только для молекул сферической симметрии, когда (3* =0
и 7j_=0.
Следует отметить, что при возбуждении спектров КР неполя-
ризованным излучением, например светом ртутной лампы,
как это было на практике до появления лазеров, выражения для
степени деполяризации р рассеянного излучения отличаются по
виду от (Х.1) и (Х.2). Имеется следующее соотношение величин:
р=_^£п—
1+ Ри
(Х.3>
п , , 6
а следовательно, 0 р -% — , причем для полностью деполяризо-
ванных неполносимметричных колебаний р = 6/7.
Таким образом, экспериментальные определения числа деполя-
ризованных и поляризованных полос в спектре КР и сопоставление
этих данных с теоретически ожидаемым числом неполносимметрич-
ных и полносимметричных колебаний для моделей молекулы той
или иной симметрии позволяют наряду с анализом правил отбора
в ИК и КР спектрах и другими рассматриваемыми данными де-
лать обоснованный вывод о симметрии молекулы и относить коле-
бательные частоты.
Наиболее точные экспериментальные измерения степеней депо-
ляризации полос КР осуществляются по интегральным интенсивно-
стям в спектрах паров (газовой фазы). При использовании совре-
менных спектрометров с мощными аргоновыми и другими лазера-
ми получение спектров КР газов не составляет больших трудностей.
В то же время достаточно надежно можно различать деполяризо-
ванные и поляризованные полосы в большинстве случаев также в
спектрах КР жидкостей и растворов, а до 70-х годов в литературе
вообще существовали в основном только такие данные.
Измеряемые степени деполяризации могут содержать ряд оши-
бок, которые надо учитывать (так называемые ошибки конечных
аппертур, ошибки, связанные с разными потерями интенсивности
в приборе для компонент Iц и и различной шириной линий,
ошибки фотометрии). Для приведения наблюдаемой степени депо-
ляризации рнабл к истинному значению рИСт можно пользоваться
калибровочным графиком, построенным для данного прибора по
известным величинам рИСт и измеряемым на приборе значениям
Рнабл для эталонных веществ. В табл. Х.4 приведены достаточно
Таблица Х.4. Степень деполяризации рп некоторых линий
комбинационного рассеяния
СС14 СеНа CeHi2
V, см — 1 Рп V, см-1 Рп V, см-1 Рп
217 0,72±0,01 606 0,75±0,02 802 0,04
313 0,72±0,02 992 0,04±0,01 1028 0,65
459 0,01+0,01 3063 0,21 ±0,02
надежно определенные (с нужными поправками) значения степеней
деполяризации некоторых линий для жидких веществ (тетрахло-
рида углерода, бензола и циклогексана), которые могут служить
эталонами измерений рп в спектрах КР жидкостей.
3. КОНТУРЫ ВРАЩАТЕЛЬНОЙ СТРУКТУРЫ ПОЛОС
В газовой фазе веществ молекулы имеют возможность достаточ-
но свободного вращения, а иногда вращение молекул, хотя и за-
трудненное, возможно также в конденсированных фазах. При на-
личии вращательных степеней свободы экспериментально наблюда-
ются не чисто колебательные, а колебательно-вращательные пере-
ходы.
Для полярных двухатомных молекул при колебательном пере-
ходе правило отбора для вращательного квантового числа I'—J"—
=А/ = ±1. Это приводит к появлению частот вращательной струк-
туры колебательно-вращательной полосы, образующих ее Р-ветвь,
если Д/= + 1, и Р-ветвь, если А/=—1. Соответствующие переходы
схематично показаны на рис. Х.4. Частоты R- и Р-ветвей в прибли-
жении жесткого ротатора и отсутствия колебательно-вращательно-
Рис. Х.4. Переходы между враща-
тельными подуровнями колебатель-
ных состояний двухатомной молеку-
лы, дающие Р- и /?-ветви частот ко-
лебательно-вращательной полосы в
ИК спектре поглощения
го взаимодействия выражаются
формулами:
<0д=(0о4-5[(/"-|-1)(Л + 2)-
- J"(/" + 1)] = «>O + 25(J" + 1)
(J"=0, 1, 2, 3,...),
Wp = <uo-)- В \ J’ (J'-]- 1)-(/' -j-
+ 1)(У' + 2)] = (о0-25(J'+ 1)
(J'=0, 1, 2, 3,...), (Х.4)
или общей формулой
ш=(о0-]-25/п (/п= + 1, ±2,
+ 3,...), (Х.5)
где coo — начало полосы (или фак-
тически отсутствующая в спектре
частота чисто колебательного пе-
рехода); В — вращательная пос-
тоянная (см. гл. V), лг=/+1, при
Д7 = 4-1 /и>0 (Р-ветвь), при
Д/ =— 1 m<Z0 (Р-ветвь). Неболь-
шим различием вращательных по-
стоянных В для разных колебательных состояний пренебрегаем.
Распределение интенсивности между компонентами вращатель-
ной структуры определяется заселенностью вращательных под-
уровней колебательного состояния, которая по закону Больцмана
и с учетом того, что каждый вращательный уровень имеет степень-
вырождения 2/4-1, может быть выражена как Р;=Ро(2/+1)Х
Хехр(—EjIkT), где энергия /-го уровня Ej = hcB](J + 1). Макси-
мально заселенным при температуре Т является уровень с кванто-
вым числом Jмакс==1/ЛkT/2hcB i-, что легко получить, продиф-
ференцировав Ni по / и приравняв dNt/dJ нулю. Тогда, учитывая
уравнение (Х.5) и что т=/ + 1, для частот максимумов интенсив-
ности в R- и Р-ветвях можно записать:
“Жмакс)—“о 4*25 (VkT!2hcB .
“Жмакс)— “О - 2-S
Расстояние между максимумами:
Дшл р=В! he 25.
(Х.6)
Таким образом, грубо приближенное значение вращательной
постоянной В, а следовательно, момента инерции и межатомного
расстояния, можно получить даже при плохом
разрешении колебательно-вращательного спек-
тра по положению максимумов Р/?-контура
полосы. Так, например, на рис. Х.5 показан
контур основной колебательно-вращательной
полосы СО, полученный в ИК спектре с очень
низким разрешением при Т~300 К. Расстоя-
ние между максимумами Аыд-р~50 см-1. От-
сюда В ~ 1,7 см-1, что достаточно близко к зна-
чению 5—1,915 см-1, полученному из анализа
разрешенной вращательной структуры R- и
Р-ветвей той же полосы (точное значение по
микроволновым данным 5 = 1,92118 см-1).
2100 2200 см-'
Рис. Х.5. Контур
вращательной
структуры ИК по-
лосы поглощения
СО при низком
разрешении
Исследование контуров вращательной стру-
ктуры полос в ИК спектрах многоатомных мо-
лекул может быть полезным для отнесения ко-
лебательных частот. Как и в случае чисто вра-
щательных спектров (см. гл. V), рассмотрим
разные типы молекулярных волчков. Для ли-
нейных молекул и симметричных волчков мож-
но различать два типа колебательных переходов или нормальных
колебаний: параллельный || и перпендикулярный ±. При первом (||)
происходит изменение компоненты электрического дипольного мо-
мента в направлении главной оси вращения, совпадающей с осью
симметрии высшего порядка — у линейной молекулы и Сп> где
л>2, — у симметричного волчка), т. е.
dp-g \ / q. \ q
. dQk /о ’ \ dQk )q
При втором (_1_) изменение компоненты дипольного момента про-
исходит в направлении, перпендикулярном оси а:
=0. / / о
dQk \ dQk /о
При параллельных и перпендикулярных переходах различен
характер колебательно-вращательного взаимодействия и правила
отбора для вращательных переходов, что приводит к разному виду
6) II
Рис. Х.6. Примерные контуры вращательной структуры ко-
лебательно-вращательных полос в ИК спектрах:
а— линейных молекул; б — молекул типа симметричного волчка;
в — молекул типа асимметричного волчка
контуров параллельных (||) и перпендикулярных (_L) полос в ИК
спектрах поглощения.
У линейных молекул правила отбора колебательно-вращатель-
ных переходов следующие:
для Ц-полос:
Д® = + 1, Д/= + 1
для ±-полос:
Дг>=± 1, Д/ = 0, + 1,
а примерный вид контуров полос показан на рис. Х.6, а. Например,
в колебательном спектре КгРг из трех основных частот: o)i = 449,
0)2 = 233 и о)з = 288 см~* — первая наблюдается только в спектре
КР и поляризована, а две другие — только в ИК спектре, причем
полоса о)2 имеет Рф/?-структуру, т. е. является перпендикулярной,
а у полосы о)з имеются только Р- и 7?-ветви, т. е. она является
параллельной. Это позволяет, во-первых, сделать вывод, что моле-
кула линейна и принадлежит точечной группе симметрии Dxh, и,
во-вторых, отнести частоту он к полносимметричному валентному
колебанию vs (Sg) . частоту ы2 —к дважды вырожденному дефор-
мационному колебанию б (Пы), а частоту соз — к антисимметрично-
му .валентному колебаний (2а) (см> рис. IX.2K
’ ' Выражение вращательной энергии молекул-типа'симметричного
волчка зависит (см. гл. V) от дв'ух квантовых чисел J и К, а в за-
висимости от соотношения вращательных постоянных А и В разли-
чают вытянутый волчок (А>В), например молекула СНзВг, и
сплющенный волчок (Л<В), например молекула СвНе. Для основ-
ных колебательно-вращательных переходов Ду = ±1, при этом пра-
вила отбора
для ||-полос:
Д/=0, + 1; Д/С = О, если К =f~ О,
Д/=± 1; Д/С =0, если /С=0,
для ±-полос:
Д/ = 0, ± 1; Д^=+ 1.
Примерный вид контуров полос вращательной структуры показан
на рис. Х.6, б, где даны также для молекулы СН3Вг примеры па-
раллельных и перпендикулярных колебаний. На рис. Х.1 для моле-
кул ХУ3 симметрии С3а и D3ft, представляющих сплющенные сим-
метричные волчки, также указано, какие полосы в ИК спектре яв-
ляются параллельными, а какие перпендикулярными.
Для молекул типа асимметричного волчка, когда все главные
моменты инерции и, следовательно, вращательные постоянные раз-
личны (обозначаются в следующем порядке: 1а<1ь<1с, А>В>С),
правила отбора для Д/=0, ±1, а для ДК меняются в зависимости
от поляризации переходов в направлении разных главных осей.
В принципе можно различать три типа контуров вращательной
структуры полос асимметричных волчков: А, В и С (рис. Х.6, в).
Молекулы типа сферического волчка, у которых все главные
моменты инерции равны, для активных в ИК спектре колебаний
правила отбора Д/=0, ±1 и нет каких-либо характерных разли-
чий контуров вращательной структуры полос, как для других типов
волчков.
Оценка расстояний между максимумами Р-, Q-, /^-структуры ко-
лебательно-вращательных полос поглощения многоатомных моле-
кул, хотя и более сложна и приближенна, чем у двухатомных мо-
лекул, также может быть полезной. Следует заметить, что до сих
пор данные о контурах вращательной структуры колебательно-вра-
щательных полос получали в основном из ИК спектров. Однако
использование мощных лазеров для возбуждения спектров КР об-
легчает получение аналогичных данных о колебательно-вращатель-
ных полосах в спектрах газов и методом КР света. Особенности
вращательной структуры в колебательных спектрах КР не рассмат-
риваются, так как ее изучение методом спектроскопии КР пока
еще проводилось мало.
4. ГРУППОВЫЕ ИЛИ ХАРАКТЕРИСТИЧЕСКИЕ ЧАСТОТЫ
В химии, особенно органической, уже давно и широко вошел в
практику структурно-групповой анализ по колебатель-
ным спектрам. Он базируется прежде всего на концепции так на-
зываемых групповых или характеристических ча-
стот, правда, в последнее время все больше внимания обращают
также на интенсивности полос. Вообще в спектроскопии различа-
ют колебания характеристические по частоте или
по форме, или по интенсивности, или вполне харак-
теристические, но этот специальный вопрос здесь не детали-
зуется.
Хотя, по определению, каждое нормальное колебание—это ко-
лебание всей молекулы в целом (см. выше), некоторые из них мо-
гут быть в большей или меньшей степени локализованы на каких-
то отдельных связях, структурных фрагментах или группах атомов.
При этом оказывается, что для некоторых фрагментов или связей,
в каких бы молекулах они ни находились, характерны свои более
или менее узкие интервалы групповых частот, а часто и относи-
тельные интенсивности полос. Так, например, при наличии в моле-
куле карбонильной группы в ИК спектре всегда наблюда-
ется очень сильная полоса поглощения в области ~ 1700 см-’.
В общем случае для всего множества классов соединений, содер-
жащих эту группу, интервал частот колебаний группы С = О весь-
ма широк, но при рассмотрении отдельных классов соединений с
учетом окружения данной группы характерные для них интервалы
частот v (С—О) сильно сужаются (табл. Х.5).
Рассмотрим линейное преобразование координат, обратное
представленному уравнениями (VIII. 19), (VIII.20), в развернутом
виде:
Q1 = 6191 4“ Лг9г 4"6з9з 4" •••
Q2 = ^2l^l 4*^2292 4" ^2з9з 4“ ••• 4“^2л9л» v _
(Х.7)
СЬ~61914*6292 4-6'з9з4' 4~6«9zi>
Qn ~ 4- 4г9г + ^з9з + • • • 4- ^nQn-
Предположим, что нормальная координата Q, соответствует
колебанию, характеризуемому групповой частотой vc=o~ 1700 см-1,
а внутренняя естественная координата, введенная как изменение
длины связи С = О, <7з = Агс=о-
Тогда в уравнении (Х.7) для Q; наибольшее значение имеет
коэффициент линейного преобразования /гз, т. е. с наибольшей ам-
плитудой меняется. именно естественная координата q3, и i-e нор-
мальное колебание локализовано в основном на связи С = О. Дру-
гие коэффициенты /г;- (/=1, 2, 4, 5,...) могут быть при этом равны
220
внутреннее
валентные деформационное
внешние деформационные'
антисим-
симметричное метричное
V5(A})
(СН2: —2870 -2960
Vas(B2)
-1300
крутильное маятниковое
-1200 -720 см"1)
деформационные
валентные
симметричное
симмет-
антисим- ричное антисим-
метричное зонтиковое метричное
<СНГ' 2940—2860 3040-2940 1380-1200 1470-1400
маятниковое крутильное.
г(Е) Г("Л2)
1200—900 250-200 с»-')
Рис. Х.7. Колебания групп —АХ2 и —АХз и значения соответствующих частот
для метиленовой и метильной групп
или не равны нулю. Чем больше значения коэффициентов, отлич-
ных от нуля, тем больше меняются при t-м нормальном колебании,
соответствующие естественные координаты или, как иногда гово-
рят, тем больше данное колебание (карбонильной группы) сме-
шано с другими колебаниями, относимыми по максимальным амп-
литудам к каким-то иным связям или группам.
Примеры характеристических частот валентных колебаний ря-
да кратных связей приведены в табл. Х.5, а, например, в одной из.
первых корреляционных диаграмм, составленной Колтупом (см.
форзацы), указаны интервалы групповых частот для многих клас-
сов соединений. Более подробные корреляционные диаграммы к
таблицы групповых колебательных частот приводятся во многих:
специальных монографиях и справочниках. Имеются также систе-
мы поиска на ЭВМ с соответствующими банками данных. При
анализе спектров с использованием групповых частот следует на-
ходить по возможности все характерные для данной группы часто-
ты, учитывая при этом также ее локальную симметрию и
относительные интенсивности полос в ИК и КР спектрах.
Так, например, у группы типа—АХ2— локальная симметрия
С2а и имеется шесть основных колебательных частот (приближен-
но считаем, что атом А привязан к бесконечно большой массе, т. е.
рассматриваем систему из четырех масс), а у группы—АХ3 —
локальная симметрия CSv и девять колебательных степеней свобо-
ды (пять масс), но наблюдаются также шесть частот, так как три
из них соответствуют (каждая) дважды вырожденным колебаниям..
T a 6}jiиц а Х.5. Характеристические частоты валентных колебаний
для некоторых кратных связей
V, см—1
v, см-1
с=с
Сложные эфиры
Алкены
^ис-Замещенные
транс-Замещенные
Циклические
Сопряженные
1680... 1620
1665... 1635
1675... 1660
1650 ... 1550
1660... 1580
алифатические 1750... 1735
непредельные 1730... 1710
виниловые и арома- 1800 ... 1770
тические
-С = С = С—
Лактоны
Ангидриды
1850... 1720
Аллены
f vas 1970 ... 1940
I vs 1070... 1060
—С Бе-
ллкины
—CsC—Н
2270 ... 2190
2140... 2100
В открытой цепи
а, p-Непредельные 1
В цикле J
—C^N
1690 ... 1620
1660... 1480
Кетоны алифатические 1725 .. .. 1700
непредельные 1690 .. .. 1660
арилкетоны 1700 .. .. 1680
диарилкетоны 1670 .. .. 1660
циклические 1780 .. . 1700
а 1730 .. . 1710
дикетоны | 0 1640 .. . 1535
Альдегиды алифатиче- 1740... 1720
ские
непредельные 1705... 1650
ароматические 1715... 1685
Нитрилы предельные
а, [З-Непредельные
Арилнитрилы
Изонитрилы
2265 ... 2240
2240 ... 2215
2185... 2120
Q4Q
Карбоновые кислоты
мономер 1760
димер 1725... 1700
непредельные 1715... 1680
ароматические 1700 ... 1680
—О—N=O
С—N = O
N—N = O
N = O
1680.. .. 1610
1600.. . 1500
1500.. . 1440
—N = N—
Азосоединения 1600. .. 1400
Азиды-Ns 2160.. .2120
^>C = S 1250 .. . 1020
—s=o 1070 .. . 1010
—Р = О 1350 .. . 1100
C = N
Обозначения колебаний и типы их симметрии (согласно локаль-
ной симметрии) для таких групп указаны на рис. Х.7, где приве-
дены также приблизительные значения или интервалы соответст-
вующих частот для метиленовой и метильной групп.
Колебания фрагментов молекулы, разделенные тяжелыми ато- ;
мами (Si, Ge, Hg и т. п.), мало смешаны и могут рассматриваться
отдельно. Это так называемый барьерный эффект тяжелого атома.
Колебания же центрального фрагмента, образуемого самим таким
атомом и его ближайшим окружением, надо рассматривать в це-
лом с учетом его локальной симметрии. Так, например, при после- ।
довательном изменении углерод-кислородного окружения атома
кремния в кремнийорганических соединениях могут быть представ-
лены фрагменты, принадлежащие следующим точечным группам
симметрии:
Без учета симметрии нельзя правильно охарактеризовать ни ва-
лентные, ни деформационные колебания каждого из этих фрагмен-
тов, и неправильно будет просто указывать, например, одну какую-
то частоту валентного колебания Si—О или Si—С. На аналогич-
ном примере комплекса OsO4Py уже отмечалось, что в зависимо-
сти от симметрии он мог бы иметь две, три или четыре частоты
валентных колебаний Os—О (см. табл. Х.З).
Пользуясь концепцией групповых или характеристических час-
тот, необходимо помнить о ее ограничениях. Отсутствие в молеку-
ле какой-то связи или фрагмента доказывается на основании от-
сутствия в ИК и КР спектрах соответствующих характеристиче-
ских или групповых частот более надежно, чем наличие в молеку-
ле тех или иных связей или фрагментов по наблюдаемым в спект-
рах полосам, предположительно принимаемым за характеристиче-
ские.
Дело в том, что полоса может быть обусловлена какими-то сов-
сем другими колебаниями молекулы. При использовании характе-
ристических или групповых частот надо рассматривать достаточно
широкий интервал возможных для данной группы частот и учиты-
вать относительную интенсивность полос как в ИК, так и в КР
спектрах. Так, например, полоса валентных колебаний этиленовой
связи vc=c в области 1600 см-1 интенсивна в спектре КР, тогда как
в ИК спектре она слабая, а при центросимметричном замещении
совсем не наблюдается. Наоборот, карбонильная полоса очень
интенсивна в ИК спектре и гораздо менее интенсивна в спек-
трах КР.
Обнаружение в каком-то интервале частот полосы, которую
можно отнести к определенной структурной группе, само по себе
не доказывает наличия данной группы в молекуле, так как, вооб-
ще говоря, наблюдаемая полоса может не иметь к ней никакого
отношения. Нужно искать несколько характерных для данной груп-
пы полос как в ИК, та и в КР спектрах.
Иногда для идентификации соединения или, по крайней мере,
какого-то фрагмента определенного строения, или типа замещения
используют не только характерные полосы, а целые участки спект-
ра, имеющие характерный вид с учетом всех полос и распределе-
ния интенсивности в данной области. Это может относиться как к
фундаментальным частотам, так и к обертонам и составным часто-
там. Например, при различных типах замещения бензольного коль-
ца внеплоскостные колебания незамещенных атомов водорода да-
ют весьма характерные наборы основных частот в области ~700...
...900 см-1 (см. форзацы), а в области 1650...2000 см ~' наблюдаются
1900 1700 1900 1700 1900________1700 см'
моно 1,2-ди 1,3-ди
1,4-,in 1,2,3 —три 1,2,4-три
Г
1,3,5-гри 1,2,3,4-1 е гра 1,2,3,5-тетра
Y 1 1 i 1 1——1— 1
1,2,4,5-ieipa пента гекса
Рис. Х.8. Характерные спектральные кривые ИК
поглощения для разных типов замещения бен-
зольного кольца в области 1650 ... 2000 см-1
очень характерные сочетания полос ИК спектров поглощения, обу-
словленные обертонами и составными частотами. По таким спект-
рам, представленным на рис. Х.8, достаточно надежно определяет-
ся тип замещения бензольного кольца.
Чем меньше различаются массы атомов в молекуле, тем силь-
нее происходит смешение колебаний и труднее выделить какие-то
трупповые частоты. Особенно это касается многих неорганических
соединений. В молекулах органических соединений мало характе-
ристичны или совсем нехарактеристичны скелетные, прежде всего
.деформационные колебания углеродного скелета, включающего и
другие атомы (О, N и др.). В то же время установлены многие до-
статочно тонкие закономерности изменения частот колебаний фраг-
ментов молекул даже в зависимости от их конформации. Напри-
мер, у четырехатомного углеродного или кремний-углеродного ске-
лета с'атомом галогена С-С-С-Гал или C-C-Si-Гал возможны,
как известно, поворотные изомеры
характеризующиеся изменением двухгранного угла на ~120°. Это-
го оказывается достаточно, чтобы некоторые частоты колебаний
фрагмента существенно отличались. Так, частота, условно назы-
ваемая частотой валентного колебания vc-Гал, у транс-конформе-
ра выше (примерно на 100 см-1), чем у гош-конформера, а, напри-
мер, частота деформационного колебания d^ccsi транс-конформе-
ра ниже (примерно на 50 см-1), чем гош-конформера. Имеются со-
ответственно различия групповых частот при аксиальном а и эква-
ториальном е замещении циклогексана и т. д. Данные такого рода
по групповым частотам помогают делать выводы об устойчивых
конформациях молекул.
Рассмотрим интересный пример изучения поворотной изомерии 1,4-дибром-
бутана. В этой молекуле имеется три связи С—С, при заторможенном внут-
реннем вращении вокруг которых возможна поворотная изомерия, т. е. транс-
(Г) или гош- (Г) расположение двух атомов С или С и Вг. Всего можно пред-
ставить 10 шахматных конформаций молекулы Вг—СН2—СН2—СН2—СН2—Вг:
(1) ТТТ, (2) ГТ Г, (3) ГТ Г', (4) ТТГ, (5) ТГТ, (6) ТГГ, (7) ТГГ', (8) ГГГ,
(9) ГГТ, (10) ГГГ', три из них (1, 2, 3) показаны на рис. Х.9. Теоретически
все они должны иметь отличающиеся колебательные спектры, но практически
многие частоты каждого конформера будут совпадать с какими-то частотами
некоторых других конформеров. Так, например, валентные колебания С—Вг
двух групп СН2Вг практически оказываются мало связаны друг с другом, т. е.
частоты vc-вг синфазных и антифазных колебаний близки и зависят
в основном от конформации фрагмента —С—СН2—СН2—Вг.
Таким образом, в указанном выше ряду для (1) и (5) конформеров должны
наблюдаться только частоты vc-вг, характерные для транс-конформации данно-
го фрагмента, для конформеров (2), (3), (8), (9) и (10)—только частоты
д’с-вг, характерные для гош-конформации фрагмента, а для (4), (6) и (7) кон-
формеров— те и другие частоты*. В ИК спектре жидкого вещества (рис.
Х.10, а) наблюдаются полосы с максимумами ~646 и 555 см-1, относимые к
валентным колебаниям С—Вг соответственно транс- и гош-конформаций фраг-
ментов С—СН2—СН2—Вг. Спектр жидкости по числу наблюдаемых в нем (во
всей области) частот и его температурной зависимости свидетельствует о нали-
чии равновесной смеси конформеров.
Конечно, не все конформации указанного ряда одинаково устойчивы и,
возможно, не все даже существуют реально в сколько-нибудь заметных кон-
центрациях. Некоторые качественные оценки можно сделать на моделях, отвергая
стерически затрудненные конформации. Надежную идентификацию всех реально
существующих в парах и жидкости конформаций осуществить по колебательным
спектрам, к сожалению, не представляется возможным. Что касается кристалли-
ческой фазы, то для нее это сделать проще. На рис. Х.9, б мы видим впечатля-
ющее упрощение спектра вещества при его замораживании.
* Имеются в виду, по крайней мере, спектры невысокого разрешения.
Рис. Х.9. Некоторые из устойчивых поворотных изо-
меров 1,4-дибромбутана
Рис. Х.10. ИК спектр жидкого (1) и кристаллического (2) 1,4-дибромбу-
тана
Сравнение со спектром КР кристаллической фазы показало, кроме того,
выполнение правила альтернативного запрета. Это однозначно свидетельствует,
что в твердом состоянии 1,4-дибромбутана остается только один, причем центро-
симметричный конформер. На рис. Х.10 показаны два конформера (1) и (2),
относящиеся к точечным группам С2Й и С; соответственно, у которых имеется
центр симметрии. Выбор между ними можно сделать, основываясь на том,
что в спектре «вымораживается» полоса 646 см-1, характерная для транс-
конформации, и остается полоса 555 см"' гош-конформации фрагмента
С—СН2—СН2—Вг, что согласуется также с отнесением ряда других полос.
У конформера (3) (рис. Х.9), как и других менее устойчивых конформеров с гош-
ориентацией обоих атомов Вг, нет центра симметрии.
Таким образом, делается вполне определенный вывод о том, что в кристал-
лической модификации 1,4-дибромбутана, полученной при равновесном замора-
живании жидкости, остается только конформер (2) ГТ Г симметрии С,.
Еще один пример касается изучения полиморфизма органических соедине-
ний, связанного с конформационной изомерией.
В зависимости от условий кристаллизации образцов, а также при вариации
температуры и давления могут получаться различные полиморфные модифика-
ции веществ прямо в специальных кюветах спектрометров.
Модельные колебательные расчеты и анализ закономерностей в групповых
частотах фтор-хлорзамещенных алканов позволили, в частности, обнаружить
следующие весьма интересные явления. При охлаждении фреона — 1,1,1-триф-
тор-3-хлорпропана сначала при равновесной кристаллизации получается моди-
фикация кристалл-!, устойчивая в интервале от температуры плавления
(—93,8°С) до —103,4°С, в которой находятся в равновесии оба поворотных изо-
мера: транс- и гош-, что фиксируется по колебательному спектру. Затем проис-
ходит обратимый фазовый переход в модификацию кристалл-!!, устойчивую при
температуре плавления ниже — 103,4°С, в которой остается только транс-пово-
ротный изомер, о чем свидетельствует «вымораживание» полос, относимых по
последним данным эксперимента и расчета к гош-изомеру.
Эта фаза может быть получена также нагреванием метастабильной моди-
фикации, получающейся при быстром замораживании фреона жидким азотом
(—196°С). В спектре метастабильной фазы также видны полосы обоих кон-
формеров, а при необратимом переходе в кристалл-!! (при ~ —157°С) полосы
гош-изомера исчезают. В метастабильной модификации заторможенного внут-
реннего вращения молекул не происходит и изомеры не находятся в равнове-
сии. Эти результаты подтверждены термохимическими данными об энтальпиях
и энтропиях фазовых переходов, полученными методом низкотемпературной
калориметрии.
5. ИЗОТОПНЫЕ ЭФФЕКТЫ
Исследование колебательных спектров изотопных разновидно-
стей молекул может существенно облегчить отнесение полос, т. е.
интерпретацию спектров, помогает в решении обратной колеба-
тельной задачи, т. е. нахождении силового поля молекулы.
В адиабатическом приближении предполагается, что при
изотопозамещении распределение электронной плотности, равно-
весные межъядерные расстояния, функция потенциальной энергии
и силовые постоянные (матрица F), через которые она выражает-
ся, остаются неизменными. Различия в массах ядер приводят лишь
к изменению кинетической энергии, т. е. коэффициентов кинемати-
ческого взаимодействия (матрица G), чем и обусловливаются раз-
личия колебательных частот изотопных разновидностей молекул.
Эти различия, вообще говоря, должны быть тем значительнее, чем
больше отношение масс изотопов пг1/пг (индексом i обозначены ве-
личины, относящиеся к более тяжелому изотопу). Поэтому наи-
больший изотопный эффект дает, например, замещение атома во-
дорода (протия) на тритий и дейтерий. Для двухатомных молекул
X—Н (или, приближенно, для такой связи в многоатомной моле-
куле), исходя из выражения для гармонической частоты
1 -1 Г ke
® = —— |/ — , легко получить соотношение
(Х.8)
Таблица Х.6. Частоты колебаний (см-1) молекул изотопных разновидностей
синильной кислоты
Молекула XCN vc_x б VC=N
HCN 3311 712 2097
DCN 2630 569 1925
TCN 2460 513 1724
При тх>тц замена водорода на дейтерий приводит к пониже-
нию частоты в V~2 раз, а на тритий — в ]/Зраз («x-d^^x-h/F^;
«>х—т~мх-н/)' 3) • Аналогично меняются и частоты деформацион-
ных колебаний, например, в молекулах (или группах) DXD по срав-
нению с НХН (в}/'2 раз). У многоатомных молекул это прибли-
женно выполняется только для строго характеристических колеба-
ний, а вообще смещение частоты колебания с участием ядра тяже-
лого изотопа меньше оцениваемого по (Х.8), и при изотопозамеще-
нии могут меняться также другие частоты. В табл. Х.6 видно, на-
пример, что у цианистоводородной (синильной) кислоты частоты
vc-d и vc-т понижаются по сравнению с vc-н не в ]/^2 или J3 раз,
а значительно меньше (приблизительно b]F1,6 и]/1,8раз),причем
сильно меняются (понижаются) также частоты vc=n и 6, т. е. коле-
бание vc = n не является характеристичным. С другой стороны, на-
пример, для молекул РН3 и PD3 (см. гл. X; 1) частоты всех четы-
рех колебаний у дейтерированной молекулы ниже, чем у легкой,
почти точно в |/"2 раз.
Дейтерирование широко применяется при определении силовых
постоянных, так как из-за сильного изменения частот (до несколь-
ких сотен и даже 1000 см-1) удается получить существенно разные
уравнения. Что касается использования изотопных сдвигов полос
в спектрах для отнесения частот, то выгоднее даже изотопозаме-
щение с небольшим изменением массы, например 10В, ПВ, 12С, 13С,
14N, 15N, 1бО, 18О и т. д. Сдвиг частот при этом невелик (до не-
скольких десятков см-1), но легко регистрируется и сильно не ис-
кажает вида спектра, позволяя легко сопоставлять полосы изотоп-
ных разновидностей молекул. Уже приводился пример того, как
меняются частоты в случае плоских четырехатомных молекул с
изотопами 10В, НВ (см. табл. Х.1).
Существует ряд важных соотношений и правил для частот изо-
топных разновидностей молекул, облегчающих отнесение частот и
позволяющих даже по известным частотам одних изотопных раз-
новидностей предсказывать частоты других еще не изученных раз-
новидностей молекул. В приближении гармонических колебаний
справедливо правило: при замене одного или нескольких атомов
молекулы атомами более тяжелых изотопов частоты нормальных
колебаний уменьшаются или, по крайней мере, не увеличиваются.
228
Другое правило, называемое правилом произведений,
может быть сформулировано следующим образом: отношение про-
изведения фундаментальных частот одного изотопного вида моле-
кулы к произведению частот другого вида зависит только от маек
атомов и геометрической структуры молекулы. Его можно записать
в виде
п! /«Т (Х.ЭХ
11 \ det G /
й
Частный случай этого правила для двухатомной молекулы был.
уже представлен соотношением (Х.8).
Если изотопные виды молекулы имеют одинаковую симметрию,,
то это правило выполняется для каждого типа симметрии в отдель-
ности. Если при частичном изотопозамещении симметрия молеку-
лы понижается, то правило надо применять для частот с учетом
распадения типов симметрии точечных групп, т. е. корреляционной
схемы типа представленной, например, на рис. Х.1 для точечных
групп Рзл и С2о, если в плоской молекуле XY3 один из атомов. Y
замещается его изотопом.
Правило сумм для квадратов частот Л. можно сформулиро-
вать так: если совокупность изотопных разновидностей молекул раз-
бивается на группы I и II так, что в группах имеется по одинако-
вому числу эквивалентных атомов каждого изотопного вида, то
сумма квадратов фундаментальных частот всех молекул одной
группы (I) равна соответствующей сумме для другой группы (II):
п m
22х<=22х>- <хло>
I 1 = 1 II 7 = 1
Например, для конкретных рядов изотопных разновидностей моле-
кул воды и аммиака, выбранных как показано ниже, можно обра-
зовать следующие группы:
I II
HOD + DOH = 2HOD НОН-(-DOD
14N Н3 4- 15ND3 14ND3 + 15NH3
Тогда для молекул воды из (Х.10) следует
2 2 Z‘-(H0D) = 2 (НОН) + V Ху (DOD),
z=i 7=1 7=1
а для изотопных разновидностей молекул аммиака, имеющих оди-
наковую симметрию, правило (Х.10) должно выполняться для
квадратов частот колебаний каждого типа симметрии (Л1 и Е) в
отдельности:
2 Х^ (14N Н3) + 2 V1 (15ND3) = J X/1 (14ND3) + J Xf - (I5N H3)
1 = 1 i = l j=l j = l
и аналогично для частот дважды вырожденных колебаний.
Существуют и другие соотношения и правила, рассматриваемые
в специальной литературе. Все эти правила для наблюдаемых фун-
даментальных частот выполняются приближенно, но и при этом
позволяют вычислять частоты одних изотопных разовидностей мо-
лекул по другим с точностью до 10...20 см-1.
ГЛАВА XI
ДРУГИЕ ПРИМЕНЕНИЯ КОЛЕБАТЕЛЬНЫХ
СПЕКТРОВ
1. ОПРЕДЕЛЕНИЕ СИЛОВЫХ ПОЛЕЙ МОЛЕКУЛ
Нахождение силового поля молекулы связано с решением об-
ратной колебательной задачи, которая, как большинство обратных
задач в физике, относится'к разряду некорректных в мате-
матическом понимании. Только для двухатомных молекул сущест-
вует взаимнооднозначная связь между гармонической частотой йе
и силовой постоянной ke:
ke = ^c'1uie^, (XI. 1)
где ц— приведенная масса.
Для двухатомных молекул легче также получить эксперимен-
тальные данные из вращательных, колебательно-вращательных и
электронно-колебательно-вращательных спектров, необходимые для
более точного воспроизведения функции потенциальной энергии.
Если эта функция представлена степенным рядом:
l/ = l/° + ^-^(Ar)2 + g(Ar)3 + /(Ar)H-> (XI.2)
то для нахождения следующих за ke коэффициентов требуется
кроме йе знать ряд других молекулярных постоянных: равновесное
межъядерное расстояние ге, вращательную постоянную Ве, посто-
янную колебательно-вращательного взаимодействия ае, постоянные
ангармоничности а>ехе, ыеуе, центробежного возмущения D и др.:
g=^(6^ + aA)/1252ere,
/ = (15g2Sere?- 2k2^eXe)l6keBer2e.
В случае многоатомных молекул обратная колебательная зада-
ча однозначно не решается, а при ее решении приближенными ме-
тодами обычно ограничиваются гармоническим силовым
полем.
Простейшим приближением при выборе силового поля много-
атомной молекулы является модель валентных сил, отражающая
специфику химической и пространственной структуры молекулы.
В этом приближении рассматриваются силы, противодействующие
растяжению и сокращению валентных химических связей, дефор-
мации валентных и двухгранных углов, кручению групп атомов во-
круг связей, т. е. изменениям равновесных геометрических пара-
метров, которые образуют систему внутренних естественных коор-
динат. Силовые постоянные или элементы матрицы потенциальной,
энергии представляют вторые производные потенциальной энергии
по естественным координатам в положении равновесия'.
Д = °
Это следует из разложения функции потенциальной энергии V в
степенной ряд при ограничении гармоническим членом.
Поскольку потенциальная энергия молекулы при любом откло-
нении от равновесной конфигурации может только увеличиваться,
все диагональные элементы матрицы F положительны: ft/Х). Не-
диагональные элементы в общем случае могут быть больше, мень-
ше или равны нулю: А/^О(г=/=/).
В приближении простого валентно-силового поля при-
нимаются неравными нулю только диагональные силовые постоян-
ные, но это очень грубое приближение, дающее большую ошибку
в оценке параметров колебательных спектров. Оно может служить,
однако, исходным при решении обратной колебательной задачи,
т. е. нахождении какими-то методами более удовлетворительного
обобщенного валентно-силового поля, в котором наряду с диаго-
нальными элементами матрицы потенциальной энергии появляется
ряд отличных от нуля недиагональных элементов.
Порядок величины силовых постоянных растяжения связей мо-
жет быть известен уже из данных по двухатомным молекулам.
Для двухатомных молекул силовые постоянные ke (Н-см1) и
равновесные межъядерные расстояния ге связаны, согласно Бэд-
жеру, эмпирическим соотношением
(re — bij'P
(XI.3)
где а,/, Ьц—постоянные для молекул, образованных атомами эле-
ментов i-го и /-го периодов. Так, например, для гидридов элемен-
тов второго периода это соотношение имеет вид
k - 2’52
е (/> — 0,30)3 ’
а соотношение
= —,
{re-bl}Y
где ^22 = 0,68, &2з = 0,90, Ьзз = 1,18, можно использовать для оцен-
ки силовых постоянных двухатомных молекул, образованных ато-
мами второго (индекс 2) и третьего (индекс 3) периодов.
Самая грубая оценка некоторых диагональных силовых посто-
янных при выборе исходного нулевого приближения может прово-
диться по экспериментально наблюдаемым колебательным часто-
там при известном их отнесении. Для этого нужно квадраты частот
я р о сто поделить на соответствующие диагональные элементы мат-
рицы кинетической энергии (G): Это понятно из анало-
гии с гармоническим осциллятором или колебанием двухатомной
молекулы, где силовая постоянная дается выражением (XI.1).
С приведенной массой ц в уравнении (XI. 1) можно сопоставить об-
ратный коэффициент та, а Хг = 4л2с2оц .
В общем случае силовое поле молекулы можно искать путем ре-
шения обратной колебательной задачи, используя матричное урав-
нение (VI11.22):
GFL = LA.
Если формы колебаний нормированы, можно записать:
LGFL = A (XI.4)
При известных значениях экспериментальных колебательных час-
тот, т. е. при известной диагональной матрице Л и найденной рас-
четом матрице коэффициентом кинематического взаимодействия G,
можно пытаться найти матрицу F. Однако решение этой задачи
встречает принципиальные трудности. Для нелинейной трехатом-
ной молекулы, например С1—N = O, имеющей всего три фундамен-
тальных частоты, число независимых силовых постоянных равно
шести, а при симметрии С2о, например, у молекулы Н2О — четы-
рем. Если использовать для этой молекулы координаты симметрии
Si = qA\ .s2 = a‘1' и s3 = 7S2, то можно решать задачу отдельно для
типов симметрии At и В2. После приведения по симметрии для ти-
па At имеем блоки матриц:
G= Гп
т21
^12 . р_____ /11
Т22 /21
/12 . Лг . ^1 О
/221 ^21 ^22 IP Х2
и из матричного уравнения (XI.4) получается система двух урав-
нений с тремя неизвестными:
/11Т11 f 22Т22~Ь ^/12r i2— М 4~^-2’
(/11/22 '/12) (иД22 /12) =
(XI.5)
которую однозначно решить относительно неизвестных ftt, /12, /12 =
—fit нельзя.
В общем случае несимметричной многоатомной молекулы при
«-колебательных степенях свободы число независимых силовых по-
п ( п + 1)
стоянных равно , а для симметричной молекулы, имеющей
по ps координат симметрии каждого типа симметрии S, это число
VI Ps(ps + A
меньше и равно сумме Zj 2 по всем типам. Реально для
s
больших молекул в приближении обобщенного валентно-
силового поля число искомых силовых постоянных может
быть, конечно, существенно меньше, так как многими далекими
взаимодействиями можно пренебречь, т. е. соответствующие не-
диагональные элементы матрицы F приравниваются нулю. Но во
всяком случае для решения обратной колебательной задачи необ-
ходимы дополнительные данные, а методы ее решения приближен-
ные и не позволяют найти истинное силовое поле.
Одним из основных источников дополнительной информации
служит использование изотопных разновидностей молекул, для ко-
торых в адиабатическом приближении принимается неизменность
силовых полей. В первую очередь это относится к использованию
дейтерирования. Так, например, у молекулы тяжелой воды D2O си-
ловые постоянные остаются теми же, что и у легкой воды, т. е. в
блоке симметрии At все три значения fij сохраняются, тогда как
из-за отличия элементов т°, т?>, тР2 (индекс D означает принад-
лежность к тяжелой изотопной разновидности молекулы) колеба-
тельные частоты, т. е. л? и л°, будут другими. Таким образом, для
нахождения появляются еще два уравнения вида (XI.5), т. е. име-
ем избыточную, несовместную систему уравнений, в данном случае
четырех уравнений с тремя неизвестными. Часто ищут решение
(вернее псевдорешение) такой системы, например, методом наи-
меньших квадратов, но оно всегда неоднозначно, и существует
проблема критериев отбора.
В последнее время эта проблема решается на основе более
строгой математической постановки обратной колебательной зада-
чи и применения регуляризирующих алгоритмов.
С точки зрения химика разумен выбор силового поля молекулы,
наилучшим образом согласующегося с представлениями теории
химического строения. За нулевое приближение поэтому выбирают,
например, валентно-силовое поле. Существуют различные другие
методы выбора силовых полей, включая полуэмпирические и не-
эмпирические квантово-механические расчеты. Для уточнения ре-
шения задачи используются дополнительные экспериментальные
данные, например, по среднеквадратичным амплиту-
дам колебаний атомов, находимым методом газовой электроно-
графии, по кориолисовым постоянным, постоянным
центробежного искажения, интенсивности полос и т. д.
В специальных исследованиях проводят учет ангармоничности, од,-
нако для химиков интерес представляют прежде всего гармониче-
ские силовые постоянные.
В настоящее время в результате многочисленных колебатель-
ных расчетов, включающих и решение обратной задачи, накоплены
обширные данные по силовым полям молекул, хотя и не всегда со-
гласующиеся между собой. Имеющиеся наборы силовых постоян-
ных позволяют проводить модельные расчеты и анализ нормальных
колебаний огромного числа молекул, главным образом органиче-
ских и элементорганических соединений. Параллельно теперь про-
водится, как правило, уточнение силового поля. Достоинством мо-
дели обобщенного валентно-силового поля является возможность
рассмотрения отдельных достаточно независимых фрагментов мо-
лекул с учетом их локальной симметрии, т. е. возможность сведе-
ния задачи о нахождении силового поля молекулы к задаче иссле-
дования и использования силовых постоянных отдельных структур-
ных элементов.
Строго говоря, силовые постоянные не обладают свойством пе-
реносимости, так как зависят от выбора всех естественных коорди-
нат, т. е. от строения молекулы в целом. Однако эта зависимость
между отдельными фрагментами молекулы в количественном вы-
ражении может быть мала. На этом основывается использование
наборов силовых постоянных для определенных структурных эле-
ментов. Важно только всегда помнить о приближенном характере
модельных расчетов и, по возможности, использовать силовые по-
стоянные, найденные на базе одной модели и по единой схеме
расчета, в частности, модели обобщенного валентно-сило-
вого поля и, например, в координатах локальной симметрии по
фрагментам.
Рассмотрим типы и размерности силовых постоянных для мо-
дели валентных сил. Сами естественные координаты имеют при ис-
пользовании такой модели разные размерности: одни — размер-
ность длины, а другие — размерность угла. Поэтому и элементы
матрицы F, соответствующие разным координатам, имеют разные
размерности. Различаются три типа силовых постоянных: 1) диа-
гональный (fa) или недиагональный (ftj, i=^=j) элемент матрицы F,
относящийся к паре координат изменения длин связей — fq, выра-
жаемый в единицах СИ Н-м-1 (103 дин-см-1); 2) диагональный
или недиагональный элемент матрицы F, относящийся к паре ко-
ординат изменения каких-то углов fa, выражаемый в единицах
Н-м (107 дин-см); 3) недиагональный элемент матрицы F, отно-
сящийся к координате изменения длины связи и координате изме-
нения какого-то угла — fqa, измеряемый единицами Н (105 дин).
Нередко при колебательных расчетах для удобства делают
внутренние естественные координаты безразмерными. При этом из-
менения длин связей выражают, выбрав в качестве единицы изме-
рения длину какой-то связи (в частности, С—Н, т. е. го=1,О9Х
ХЮ~8 см), а изменения углов выражают в радианах. При исполь-
зовании таких безразмерных геометрических параметров элементы
матрицы кинетической энергии G также делают безразмерными,
выражая массы атомов через массу какого-то атома, условно при-
нятую за единицу (в частности, массу атома водорода що = 1,ОО8Х
X 1.66-10-24 г или иногда так называемую спектроскопическую
массу водорода, равную 1,088 а.е.м.). Тогда удобной единицей всех
силовых постоянных в колебательных расчетах является 106 см-2,
а рассчитываемые частоты получаются в 103 см-1. Диапазон же ко-
. лебательных частот (волновых чисел) молекул составляет, как из-
вестно, от десятков до тысяч см-1. Численные значения как безраз-
мерных элементов т,/ матрицы G, так и силовых постоянных /Д—
элементов матрицы F в 106 см-2, зависят от выбора единиц длины
Го и массы т0.
Существует следующая связь между значениями этих силовых
постоянных и постоянных в СИ (численные коэффициенты даны
при указанном выше выборе г0 и т0):
/J(105 см-2)=----1----10-6/„(Н-м-1) = 1,68-IO-8Л,(Н-м-1);
4.т2с-/«0
/НЮ’ см-2)=—j 10-е/а(Н-м)= 1,43-104/а(Н-м);
4л2с2т0Гд
fq>. (103 см2) = —J---10-%а(Н)=:1,54.10-2/9а(Н).
4л2с2тсг0
По силовым постоянным молекул имеется справочный материал,
который служит основой для построения приближенных матриц
потенциальной энергии F в колебательных расчетах многих рядов
многоатомных молекул.
2. КОРРЕЛЯЦИИ СИЛОВЫХ ПОСТОЯННЫХ МОЛЕКУЛ
С ДРУГИМИ СВОЙСТВАМИ
Химиков всегда интересовала возможность использования ко-
лебательных спектров и вычисляемых из экспериментальных коле-
бательных частот силовых постоянных для суждения о характере,
химических связей и различных электронных эффектах. Силовые
постоянные в модели валентно-силового поля, являющиеся, в част-
ности, диагональными элементами матрицы F, которые относятся к
координатам растяжения связей, коррелируют с другими парамет-
рами, характеризующими химическую связь. Это не удивительно,
так как свойства химических связей между атомами в молекуле
обусловлены характером распределения электронной плотности.
Силовые постоянные могут помочь выявлению некоторых зако-
номерностей и позволяют косвенно оценивать существенные раз-
личия электронной структуры молекул. Валентные силовые посто-
янные коррелируют с энергией связи, закономерно растут с увели-
чением кратности связи и уменьшением ее длины и т. д.
Все эти характеристики между собой связаны, но следует рас-
смотреть некоторые примеры отдельно.
Как правило, хотя и не всегда, имеет место симбатная зависи-
мость между силовой постоянной растяжения связи и энергией свя-
зи, что на примере двухатомных молекул водорода и галогеноводо-
родов иллюстрирует рис. XI. 1. Больше всего силовая постоянная
зависит от кратности (порядка) связи. Так, для молекул с различ-
ной кратностью связи азот-—азот имеем следующее соотношение
наблюдаемых частот валентных колебаний и межатомных расстоя-
ний с рассчитанными силовыми постоянными /nn.‘
h2n—nh2 hn=nh n=n
vnn, cm-1 ........... 1100 1552 2331
tnn, 10—1 нм ..... . . 1,45 1,24 1,097
H-CM-1........... 3,4 10,0 19,64
Рис. XI.1. Зависимость между энтальпией и силовой
постоянной для некоторых двухатомных молекул
В ряду молекул с меняющейся кратностью связей азот — кислород
силовая постоянная fNo меняется следующим образом:
fe, Н-см-1...........
H2N—ОН О —N —О C1N = O N = O+
4,0 8,0 14,1 23,9
а в ряду связей С—С, С = С, С^С силовая постоянная fee изме-
няется примерно в последовательности ~5, ~10 и ~15 Н-см-1.
Следует подчеркнуть, что нужно обратить внимание только на тен-
денцию относительного изменения величин. Придавать большое
значение абсолютным значениям силовых постоянных нельзя, так
как рассчитанные частоты колебаний могут удовлетворительно со-
гласовываться с экспериментальными и при сильно различающих-
ся матрицах потенциальной энергии F. Например, рассчитанная
частота валентного колебания С = О в молекуле ацетона может
быть близка к экспериментальной при использовании в расчете
значений валентной силовой постоянной fc=o от 8 до 12 Н-см-1 при
вариации также других силовых постоянных.
-Об изменении силовой постоянной, сопровождающем изменение
кратности связи, судят иногда по смещению частот валентных ко-
лебаний. Так, например, при исследовании с помощью спектров КР
процессов, протекающих при добавлении солей серебра к этилено-
вым и ацетиленовым углеводородам, было обнаружено понижение
частот валентных колебаний С = С и С=С соответственно на ~60
и ~ 120 см-1. На основании этого был сделан вывод об образова-
нии координационных соединений — л-комплексов с понижением
кратности ненасыщенных связей. В ИК спектре комплекса с про-
пиленом К.[PtCl3(С3Нб)]-Н2О частота v(C=C) сильно уменьша-
ется, а в комплексе с этиленом, имеющим центр симметрии,
K[PtCl3-(C2H4)]-Н2О полосу v(C = C) наблюдать не удается. Это
позволило сделать вывод о сохранении высокой симметрии моле-
кулы этилена в л-комплексе и предложить следующую структуру:
доказанную впоследствии рентгеноструктурными исследованиями.
В данном случае изменение распределения электронной плотности
на этиленовой связи, а значит, и силовой постоянной связи оче-
видно.
Однако вообще связывать изменение частоты валентного коле-
бания какой-то связи при комплексообразовании только с измене-
нием силовой постоянной связи и делать на основании этого выво-
ды о месте координации и т. п. без дополнительных данных некор-
ректно.
Дело в том, что в многоатомных системах (молекула, комплекс,
лиганд) не все частоты достаточно характеристичны, и изменение
кинематических факторов (введение новых масс, изменение гео-
метрической структуры) может влиять на их значения не меньше,
чем динамических (силовых постоянных).
При единой схеме решения обратной колебательной задачи про-
слеживаются закономерности относительного изменения силовых
постоянных и в результате достаточно тонких электронных эффек-
тов. Валентная силовая постоянная одинарной связи С—С, как и
ее длина, закономерно зависит от валентных состояний связанных
атомов С (типа гибридизации их атомных орбиталей) и увеличива-
ется по ряду молекул: этан, пропилен, метилацетилен, бутадиен,
винилацетилен, диацетилен в общей сложности в полтора раза —
от ~ 4,17 Н-см-1 (~7 -106 см-2) до ~6,25 Н см-1 (~ 10,5-106 см-2),
коррелируя с уменьшением межатомного расстояния гс-с от '-—-1,54
до ~1,37-10-1 нм. Аналогично силовая постоянная связи С—Н
(Н-см-1) несколько возрастает в ряду СН3СНз(5,0), СНгСНг^Д),
СНСН(5,9).
Можно проследить и зависимость силовой постоянной растяже-
ния какой-либо связи от ее окружения. Закономерный ход измене-
ния силовой постоянной для связи Si—N виден, например, в ряду
(CH3)3Si—NH(CH3) [CH3)3Si]3NH (H3Si)3N (Cl3Si)3N
fj, Н-см-* . . . 3,04 3,35 3,63 11,83
а силовая постоянная внутримолекулярной координационной свя-
зи Si<— N в силатране RSi[—ОСН2СН2—]3N оценивается, пример-
но, значением 0,5...0,6 Н-см-1.
В химических работах нередко можно встретить самые различ-
ные и далеко идущие корреляции силовых постоянных с многими
другими характеристиками молекул и соединений, например с ин-
дексами реакционной способности, постоянными Гаммета, Тафта
и т. д. Не всегда такие корреляции одинаково надежны, и к ним
нужно относиться критически, помня о приближенности и неодно-
значности расчетов силовых постоянных. Существует даже такая
крайняя точка зрения, что рассчитываемые силовые постоянные
представляют собой набор некоторых колебательных параметров,
физическая значимость которых близка к нулю. Во всяком случае
любые их сравнения и применения для приближенной оценки дру-
гих физических свойств молекул следует проводить лишь в рядах
родственных молекул, придавая смысл только относительным из-
менениям значений силовых постоянных, полученных единообраз-
но, известным методом. Необходимо подчеркнуть, что речь все вре-
мя идет о квадратичных силовых постоянных, определяемых в гар-
моническом приближении, что по-разному огрубляет разные типы
постоянных.
Следует также иметь в виду, что обратная колебательная зада-
ча решается часто с использованием частот колебаний молекул не
в газовой, а в конденсированных фазах (жидкость, твердое вещест-
во). При этом вообще получаются некоторые эффективные
параметры силового поля, включающего не только внут-
римолекулярные, но и межмолекулярные взаимодействия.
Таким образом, хотя силовые поля и несут информацию об эле-
ктронной структуре молекул и характере химических связей, к ее
извлечению, анализу и использованию нужно относиться с большой
осторожностью.
3. КРУТИЛЬНЫЕ КОЛЕБАНИЯ И ПОТЕНЦИАЛЬНЫЕ
БАРЬЕРЫ ВНУТРЕННЕГО ВРАЩЕНИЯ
Большой интерес для химиков представляет, как уже отмеча-
лось в гл. V, явление заторможенного внутреннего вращения моле-
кул вокруг одинарных связей. Это движение, приводящее к измене-
нию конформации молекул и переходу от одной устойчивой, т. е.
отвечающей минимуму энергии, конформации к другой, сопровож-
дается преодолением потенциального барьера. Потенциаль-
ная функция внутреннего вращения (ПФВВ) мо-
жет иметь несколько разных по высоте максимумов и разных по
глубине минимумов.
Высота максимума относительно соседнего с ним минимума
представляет собой потенциальный барьер Vo для пере-
хода данного устойчивого конформера в другой, а разность глубин
минимумов соответствует разности энергий конформе-
ров ДУ, которую иногда приближенно считают равной ДЯ (или:
Д£) (рис. XI.2).
В самом общем виде зависимость потенциальной энергии от ес-
тественной координаты — двухгранного или торсионного угла <р,
характеризующего относительную ориентацию, т. е. поворот двух
связанных между собой частей молекулы, может быть представле-
на как периодическая функция У(<р) разложением в ряд
= cosm?)+-i-j? (XI.6),
т = 1 т—\
Если функция симметрична относи-
тельно точки ср = О, что обычно и бы-
вает, то остается только первая сум-
ма:
V (?) = у- УКД1 —cos^r)-
т~ 1
(XI.7)
При достаточно глубоких потен-
циальных ямах (высота барьеров
порядка или больше kT) в них мо-
жет быть несколько (около десятка
и более) уровней энергии крутиль-
ных колебаний. Анализ ПФВВ мож-
Рис. XI.2. Общий вид потенциаль-
ной функции внутреннего враще-
ния (ПФВВ) для асимметричных
групп
но в таких случаях проводить по экс-
периментальным данным, получаемым, в частности, методами ко-
лебательной спектроскопии, основываясь на зависимости располо-
жения энергетических уровней Et (i — индекс, указывающий но-
мер ямы, т. е. устойчивого конформера) от параметров ПФВВ Vm-
Чем больше найдено коэффициентов Фурье, тем лучше воспроиз-
водится ПФВВ. Для этого используются, например, следующие со-
отношения (без вывода) между Vm и такими экспериментальными
данными, как (oei — гармонические крутильные частоты; Хщ— по-
стоянные ангармоничности; XHoi — разности энтальпий i-ro кон-
формера и того, для которого i-О, т. е. <р = 0 (см. рис. XI.2); Fj=
=/г/(8л2с/пР)—вращательная постоянная; /пр— приведенный мо-
мент инерции волчка и остова молекулы (предполагается извест-
ным из геометрии распределения масс):
H(?z) = y dm(l — cosmtpz) = A7/01.4-^-(<ue0 — <ueZ),
т
(XI.8)
т
-^X^F] и др.,
т
где Е"(фг) и VIV (ф>)—соответственно вторая и четвертая произ-
водные от функции в точке i-ro минимума; все величины берут в
см '1 (1 см “’ = 11,97 Дж/моль).
При простой форме потенциала, когда все максимумы и мини-
мумы одинаковы и его можно аппроксимировать одной косинус-
гармоникой ряда (XI.7), т. е.
V (<р)==-^-(1 — cos Дф),
(XI.9)
существует взаимооднозначное соответствие положения уровней
крутильного колебания и единственного параметра барьера Vп=
— Vo. В гармоническом приближении для оценки потенциального
барьера по частоте крутильного колебания при этом используют
формулы (V.53), (V.55) или
_ п 1/Ур
““Р 2лс У 2/„р ’
(XI. 10)
где п — кратность барьера, т. е. число минимумов (максимумов)
при изменении ср от 0 до 2л, а приведенный момент инерции в про-
стейшем случае можно представить как /^=/1/2/(Л + Л) (Л и
1а— моменты инерции поворачивающихся друг относительно друга
групп).
У молекулы пероксида водорода Н2О2, например, барьер дву-
кратный (га = 2), у этана С2Н6 и ацетальдегида СН3СНО— трех-
кратный (га = 3), у метилдифторборана CH3BF2— шестикратный
(п=6) и т. д. На рис. V.10 в качестве примера показан вид потен-
циальной кривой внутреннего вращения для этаноподобных моле-
кул, описываемой функцией
V(?)=^-(l-cos3<?). (XI.11)
На рисунке стрелкой показан переход с частотой <акр.
Формула (XI.10) следует из решения соответствующего кванто-
во-механического уравнения (см. гл. V) при использовании выра-
жения потенциальной энергии в виде (XI.9), когда находят набор
энергетических уровней крутильного гармонического осциллятора:
£,(г,) = ‘°е(кр)^ + -у) ’
где v — колебательное квантовое число (I, 2, 3,...); <ае(Кр) — соот-
ветствующая колебательная постоянная, численно равная прибли-
зительно крутильной частоте а>Кр-
Строго говоря, при заторможенном внутреннем вращении для.
каждого значения и появляются подуровни энергии. Общий харак-
тер картины расположения уровней для трехкратного барьера в за-
висимости от его высоты Vo виден на рис. V.10. При больших Vo и
малых v расщеплением на подуровни А—Е, отражающим враща-
тельный характер движения, можно пренебречь. Роль вращения:
(туннелирования через барьер) возрастает по мере приближения к
вершине барьера, а выше полностью преобладает вращение.
Итак, для оценки потенциальных барьеров внутреннего враще-
ния необходимы значения частот крутильных колебаний, которые
лежат обычно в области ниже 300...200 см-1, т. е. могут быть най-
дены методами длинноволновой ИК спектроскопии и в спектрах
КР, если соответствующие переходы разрешены правилами отбора.
Например, у этана или гексахлорэтана крутильные колебания не-
активны как в ИК, так и в КР спектрах, и для определения высо-
240
Таблица XI.1. Частоты крутильных колебаний и потенциальные барьеры
бензальдегида и его пара-замещенных
Соединение <0газ> см 1 Vo, кДж/моль “жидк- Vo, кДж/моль
CJUCHO 111,0 19,5 133 28,0
n=FC6H4CHO 93,5 15,0 113 21,9
п=С1С6Н4СНО 81,5 11,7 104 19,2
п=ВгС6Н4СНО 73,5 9,9 92 15,6
п = СНзСв1ЬСНО 89,5 14,5 114 23,5
ты барьера использовались другие методы. Идентификация частот
крутильных колебаний бывает осложнена наличием в той же об-
ласти полос других колебаний, например скелетных деформацион-
ных, особенно с участием тяжелых атомов, составных (разностных)
частот и т. д. Поэтому требуются исследования низкочастотной об-
ласти с использованием различных приемов. Существенную по-
мощь могут оказать «горячие» полосы, а также обертоны крутиль-
ных колебаний в спектрах КР, иногда полезным для отделения ча-
стот других колебаний является изотопозамещение.
На значение крутильной частоты, а следовательно, и высоту по-
тенциального барьера оказывают сильное влияние различные фи-
зические и химические факторы. Точнее находятся значения внут-
римолекулярных потенциальных барьеров по частотам крутильных
колебаний молекул в газовой фазе. Эти частоты обычно ниже, чем
в спектрах конденсированной фазы, где большую роль играют меж-
молекулярные взаимодействия. У хлорзамещенных этанов разли-
чие крутильных частот для газовой и жидкой фазы составляет 5...
10 см-1, например, для СН2С1—СС1з в газе щкр=108 см-1, а в жид-
кости иКр=116 см-1. Вообще же это различие бывает до 20...25%,
если в конденсированной фазе не возникает каких-либо сильных
специфических взаимодействий типа водородных связей, когда оно
становится больше.
Пропорциональность барьера Vo квадрату крутильной частоты
(V.55) указывает на его большую чувствительность к изменению
агрегатного состояния вещества. Это хорошо иллюстрируется на
примере частот крутильных колебаний и высоты двукратных барь-
еров внутреннего вращения в бензальдегиде и его яара-замещен-
ных (табл. XI. 1). Приведенные данные показывают также, что вы-
сота барьера зависит не только от стерических и электронных эф-
фектов ближайшего окружения связи, вокруг которой происходит
вращение, но и от распределения электронной плотности по всей
молекуле, так как в ряду приведенных соединений меняется только1
заместитель X в пара-положении, не оказывающий какого-либо
стерического влияния на поворот альдегидной группы относительно
бензольного кольца:
Потенциальные барьеры при заторможенном внутреннем вра-
щении в свободных молекулах относительно одинарных связей до-
стигают нескольких десятков кДж/моль. Чем выше кратность (по-
рядок) связи, тем выше потенциальный барьер. При обычных тем-
пературах не происходит, например, внутреннего вращения вокруг
двойной связи С = С, так как барьер составляет около
200 кДж/моль.
В кристаллах внутреннее вращение молекул относительно оди-
нарных связей, как правило, отсутствует, хотя встречаются исклю-
чения, т. е. твердые фазы, в которых оно возможно, и осуществля-
ются переходы между разными устойчивыми конформациями, на-
пример в некоторых галогензамещенных углеводородах.
При наличии поворотной изомерии молекул, т. е. потенциальной
кривой типа, представленной на рис. XI.2, когда имеется несколько
разных по глубине минимумов, отвечающих различным конформе-
рам, можно наблюдать соответствующее число частот крутильных
колебаний, которые могут использоваться для приближенной оцен-
ки эффективного барьера или вместе с другими экспериментальны-
ми данными для более детального анализа ПФВВ, например, с по-
мощью соотношений типа (XI.8).
4. ИСПОЛЬЗОВАНИЕ ФУНДАМЕНТАЛЬНЫХ ЧАСТОТ
ДЛЯ РАСЧЕТА КОЛЕБАТЕЛЬНЫХ ВКЛАДОВ
В ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ
Расчет термодинамических функций газов методами статисти-
ческой термодинамики связан, как известно, с нахождением стати-
стических сумм для поступательного движения и внутренних дви-
жений молекул. К числу последних относятся колебания молекул,
и для расчета колебательной статистической суммы и колебатель-
ного вклада в термодинамические функции необходимо знание пол-
ного набора основных колебательных частот многоатомной молеку-
лы в газовой фазе.
В приближении гармонических колебаний вклады в стандарт-
ные термодинамические функции в зависимости от величины
/ a.he \
= =1,4388о>,-/Г,
где ю,-— волновое число (гармоническая частота в см-1), вычисля-
ют по следующим, выводимым в статистической термодинамике
формулам:
C°p = R V Xt exp xt (ехрхг —1)“2,
(Н° — Н°о) Т~1 = % xL(expxt — I)-1,
i
(G° — Hq) = In [ 1 — exp( — xz)],
S°=[(H°—Ho)T-i—(G°—Ho)T~1] ,
где Cp—теплоемкость; Но —энтальпия; G° — свободная энергия
Гиббса; S0 — энтропия.
С уменьшением волнового числа колебательный вклад в значе-
ние всех функций возрастает и особенно сильно в энтропию и при-
веденный термодинамический потенциал: Ф* = — (G° —£/о) Т~г.Ес-
ли учесть, что от полного значения некоторой термодинамической
функции SF:
& 3" пост 4~ЭЛ 4“ # кол 4“ &вр>
колебательный вклад 2ГК0Л обычно составляет около 30%, а наи-
большую роль играют низкие частоты, то становится очевидным
важность правильного отнесения и точного определения именно
этих частот. В то же время для низкочастотных колебаний обычно
бывает высока ангармоничность, т. е. оценки термодинамических
функций, основанные на приближении гармонического осциллято-
ра, могут требовать существенных поправок.
Кроме того, малая частота, т. е. малое значение характери-
стической температуры 0КОл = 1,4388со, соответствует по-
вышенной заселенности более высоких колебательных уровней в
соответствии с тепловым распределением Больцмана (VIII.13), пе-
реписанным в виде
“еХр(
kT
где N2 и Ni — число молекул
на энергетических уровнях,
разность энергий между кото-
рыми А£=Е2—E\ = hv (прини-
маем, что оба уровня невы-
рождены, т. е. n=g2lgx= 1).
Действительно, даже при обыч-
ных температурах (~300 К) и
частотах ниже 200 см-1 отно-
шение N2/N1 достаточно вели-
ко, что хорошо видно на рис.
XI.3, где представлены кривые
зависимости этого отношения
(/) — от частоты при Г=300 К
и (2)—от температуры при
частоте 50 см-1. Это означает,
1,4388о>\
Т ) ’
Рис. XI.3. Огносигельная заселенность
колебательных уровней в зависимости от
температуры и разности энергий (волно-
вого числа перехода)
что переходы с более высоких колебательных уровней имеют боль-
шую вероятность, чем в высокочастотной области, и даже при уме-
ренных и низких температурах будут наблюдаться горячие полосы,
смещенные относительно частоты основного перехода из-за ангар-
моничности и запутывающие картину спектра.
Определение полного набора фундаментальных колебательных
частот многоатомной молекулы для точного расчета термодинами-
ческих функций вещества, как уже отмечалось, является делом не-
простым. Обоснованное отнесение наблюдаемых в ИК и КР спект-
рах частот включает использование всех доступных данных экс-
перимента и расчета. Случается, что некоторые из основных частот
приходится оценивать только расчетом или находить из экспери-
мента косвенно (из составных частот, из колебательной структуры
электронных спектров и т. д.), если соответствующие переходы за-
прещены и не проявляются в ИК и КР спектрах.
5. ИДЕНТИФИКАЦИЯ СОЕДИНЕНИЯ И КАЧЕСТВЕННЫЙ
АНАЛИЗ СМЕСЕЙ
Колебательные спектры обладают очень высокой специфично-
стью и являются уникальной физической характеристикой вещест-
ва. Поэтому ИК и КР спектры широко используются для иденти-
фикации индивидуальных химических соединений. Каждому соеди-
нению соответствует свой спектр, и нет двух таких веществ, кото-
рые имели бы абсолютно одинаковые ИК и КР спектры. Более то-
го, даже очень мало отличающиеся по другим свойствам изомеры,
например поворотные изомеры или конформеры (но не оптические
изомеры), имеют разные спектры. Не случайно поэтому колеба-
тельный спектр сравнивают с дактилоскопическим отпечатком.
Иногда как область «отпечатков пальцев» выделяют область час-
тот в ИК и КР спектрах ниже 1500 см-1, в которой общая картина
спектра наиболее чувствительна к малейшим изменениям в струк-
туре молекулы и вещества.
Идентификация неизвестного соединения по ИК спектру или
спектру КР, или по тому и другому одновременно, если такое сое-
динение было уже раньше исследовано, может осуществляться
сравнением его спектра с эталонными спектрами. Для этого необ-
ходима обширная картотека эталонных спектров, и при этом важ-
нейшим для возможности сравнения спектров является стандарт-
ность условий их регистрации. Как для эталонных спектров разных
классов точности, так и для любых других спектров должны обя-
зательно указываться спектрометр, условия регистрации, способ
подготовки образца и все другие важные параметры.
В настоящее время число известных химических веществ уже порядка
10 млн., и единой библиотеки такого количества спектров не существует. Однако
имеются многочисленные атласы ИК спектров поглощения для различных клас-
сов органических, элементорганических и неорганических соединений, несколько
меньше пока атласов спектров КР. Очень полезны для работы создаваемые в
лабораториях собственные картотеки эталонных спектров. Но если даже иметь
достаточно большую коллекцию спектров для работы в какой-то области химии,
244
биохимии, геохимии и т. д., то это еще не решает проблемы отыскания нужного
спектра в данной коллекции. Теперь на базе современной электронно-вычисли-
тельной техники создаются для этой цели информационно-поисковые
системы, например, такие, как система в Научно-информационном центре по
молекулярной спектроскопии Сибирского отделения АН СССР, разработанная
под руководством акад. В. А. Коптюга. Эти системы имеют и более широкое
назначение для одновременного использования данных различных методов при
идентификации соединений.
Идентификация вещества по ИК (или КР) спектру является
полностью достоверной только при точном совпадении со спектром
эталона по положению (частоте), форме и относительной интен-
сивности всех полос, т. е. всей спектральной кривой. Наличие
в спектре идентифицируемого вещества большего числа полос по
сравнению со спектром эталона может быть вызвано как разли-
чием веществ, спектры которых сравниваются, так и загрязнения-
ми, отсутствие же каких-то полос в спектре, сравнимаемом с
эталонным, однозначно указывает, что это различные соединения.
При сравнении спектров с целью идентификации нужно быть
достаточно уверенным в чистоте образца. Кроме того, следует
иметь в виду, что спектры, снятые на различных спектрометрах
или при разных условиях регистрации, могут отличаться; они
могут также зависеть от температуры, концентрации, раствори-
теля и особенно от агрегатного состояния вещества. Таким обра-
зом, даже отождествление по колебательному спектру индиви-
дуального вещества, которое ранее уже было известно и исследо-
вано, является задачей непростой и трудоемкой. Еще более
сложной и в то же время более актуальной является задача
идентификации неизвестных и ранее не изученных химических
веществ. Ежегодно синтезируется более 100 тыс. новых веществ,
каждое из которых требует идентификации.
Проблема идентификации неизвестных примесей и загрязнений
как в продукции различных производств химической, пищевой,
медицинской и других отраслей промышленности, так и окружаю-
щей среды не менее важна.
Хотя методы колебательной ИК и КР спектроскопии широко
применяются для решения этих задач, однако далеко не всегда,
а в случае достаточно сложных соединений весьма редко удается,
используя только эти спектры, полностью идентифицировать не-
известное вещество.
Кроме каталогов спектров и информационно-поисковых систем, которые
заведомо не могут «узнать» новые, отсутствующие в памяти ЭВМ соединения,
для идентификации служат известные структурно-спектральные корреляции.
Выше уже была рассмотрена концепция групповых или характеристических час-
тот, на которой базируются такие корреляции. В настоящее время появилось
новое направление, называемое методом искусственного интеллек-
та в аналитической спектроскопии, в котором используются мето-
ды математической логики и вычислительная техника для моделирования спо-
соба рассуждения специалиста при идентификации вещества на основе структур-
но-спектральных корреляций и расчета спектральных кривых. Весьма перспек-
тивно объединение этого метода с информационно-поисковой системой и созда-
ние автоматизированных спектрально-аналитических комплексов, сочетающих
современное спектральное оборудование и ЭВМ. Такие успешно работающие
-системы уже существуют, общая схема одной из них, в частности, разработанной
под руководством Л. А. Грибова, приведена на рис. XI.4. Очень эффективны
системы идентификации веществ, основывающиеся на совместном использовании
данных разных методов, и чаще всего с ИК и КР спектроскопией сочетаются
методы УФ спектроскопии, ЯМР. масс-спектрометрии, рентгеноструктурного ана-
лиза. Но так или иначе важнейшая аналитическая задача надежной идентифи-
кации неизвестного вещества является наиболее сложной и трудоемкой.
Методы колебательной спектроскопии используются и для ка-
чественного анализа смесей или частично извест-
ного состава или полностью неизвестного состава.
Если известен, например, основной компонент и нужно опреде-
лить примеси, то можно на двухлучевом ИК спектрометре, по-
местив в канал сравнения кювету переменной толщины с извест-
ным компонентом и отрегулировав толщину слоя, скомпенсиро-
вать-.поглощение этого компонента в образце. Компенсационный
метод имеет свои ограничения, и может успешно применяться
только в спектральных областях, где исключаемый компонент
не имеет сильного поглощения. При наличии в смеси нескольких
известных или предполагаемых компонентов можно последова-
тельно вычитать из спектра смеси спектры этих компонентов. Этот
прием особенно просто осуществляется на современных фурье-
спектрометрах со встроенной ЭВМ. В обоих указанных выше слу-
чаях задача сводится в конечном счете к анализу смеси неизвестно-
го состава. В принципе к этому же можно прийти, проводя качест-
венный анализ смесей частично известного состава и по спектрам
КР.
В качественном анализе смесей неизвестного состава важно
возможно полное разделение смеси на отдельные ее составляю-
щие. Для этого используются такие физические методы, как
фракционная перегонка, жидкостная и газовая хроматография,
пропускание через твердый адсорбент, экстракция, осаждение от-
дельных компонентов и т. д. Об идентификации индивидуальных
компонентов по колебательным спектрам сказано выше. При ана-
лизе смеси нескольких компонентов для получения их спектров
в чистом виде при современной автоматизации эксперимента и
математизации обработки спектральных данных с использованием
ЭВМ можно ограничиться осуществлением лишь частичного раз-
деления смеси каким-то из способов (частичное испарение смеси,
частичная экстракция и т. д.). При этом анализируется отноше-
ние спектров исходной смеси и после частичного ее разделения,
и по указанному отношению с использованием специальной тех-
ники и программ воссоздаются спектры отдельных компонентов
смеси.
Методы ИК и КР спектроскопии могут применяться не только
в статических условиях, но и для обнаружения и идентификации
неустойчивых промежуточных частиц в химических реакциях, т. е.
могут служить для изучения механизмов химических превраще-
ний. При этом используют различные специальные методики ре-
гистрации спектров (струевые условия, матричная изоляция
и т. п.).
Общую характеристику чувствительности методов колебатель-
ной спектроскопии дать трудно. При малоинтенсивных полосах,
перекрывающихся к тому же полосами основного компонента
смеси, трудно обнаружить даже значительные количества приме-
сей. Наоборот, если примеси имеют интенсивные полосы, непере-
крывающиеся с полосами основного компонента, чувствительность
методов колебаний спектроскопии может быть очень высока.
6. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
В ИК спектроскопии, как и в абсорбционной спектроскопии
в видимой и УФ областях (см. гл. XIV; 2.2), количественный
анализ по спектрам основывается на законах светопоглощения.
Объединенный закон Бугера — Ламберта — Бера имеет
вид
т т
(XI.12)
где к— интенсивность излучения с длиной волны %, прошедшего
через вещество; 10к — интенсивность излучения с той же длиной
волны X, входящего в исследуемое вещество; щ— коэффициент
поглощения для данной длины волны, л-моль-1-см-1; с — кон-
центрация вещества, моль-л-1; 1 — толщина поглощающего слоя, см.
Б логарифмической форме, переходя к десятичным логарифмам
и опуская подстрочный индекс X, получаем
4 = lgA = lg-L = £CZ, (XI-13)
где А — оптическая плотность или погашение (в тер-
минологии, рекомендуемой IUPAC — «absorbance»); T=I/I0 — про-
пускание или прозрачность (обычно выражается как Т=
= (///о) • 100%); 8 = 0,434а— коэффициент погашения или
экстинкции (иногда называют молекулярный или моляр-
ный коэффициент поглощения). Величину (1—7")-100%
называют процент поглощения. Интегральная ин-
тенсивность (коэффициент поглощения) равна площади по-
лосы и, если в качестве координат выбраны е и со — волновое
число (см-1), она выражается интегралом: 3= 2,304 j edw. На
ш,
многих ИК спектрометрах спектральные кривые представляют за-
висимость поглощения или пропускания (в процентах) от длины
волны, а на некоторых может регистрироваться непосредственно
и зависимость оптической плотности от длины волны (волнового
числа).
Логарифмическая форма закона светопоглощения практически
более удобна, так как представляет линейную зависимость опти-
ческой плотности от величин, входящих в правую часть уравне-
ния (XI.13). Коэффициент 8 является молекулярной характерис-
тикой, не зависящей от концентрации и толщины поглощающего
слоя. Интегральная интенсивность полосы поглощения <S, а приб-
лиженно и величина щ в максимуме полосы непосредственно
связываются с вероятностями соответствующих квантовых пере-
ходов.
Если закон Бугера — Ламберта — Бера выполняется, что бы-
вает далеко не всегда, то при фиксированной толщине слоя опти-
ческая плотность линейно зависит от концентрации вещества,
что и позволяет легко проводить количественный анализ. Откло-
нения от линейной зависимости бывают связаны или с межмоле-
кулярными взаимодействиями компонентов смеси (раствор), вклю-
чая специфические (ассоциация, водородная связь) и химические
248
взаимодействия, или с инструментальными причинами. Последние
возникают, например, когда щель монохроматора из-за низкой
интенсивности ИК излучения или малой чувствительности прием-
ника должна быть шире собственной ширины колебательной
полосы поглощения. Играют роль также эффекты отражения,
рассеяния излучения и т. д. Поэтому всегда проводится проверка
выполнения закона светопоглощения и чаще всего для проведения
количественного анализа строятся градуировочные графики по эта-
лонам.
Интегральная интенсивность меньше зависит от разрешающей
силы прибора, чем интенсивность в максимуме, поэтому она вос-
производится лучше. Часто для описания зависимости оптической
плотности от частоты по контуру полосы в предположении, что
полоса симметрична относительно максимума и не перекрывается
с другими полосами, используют функцию Лоренца:
Лш=а/[(ш-«>макс)2+^], (XI. В)
где а и b — постоянные.
Обычно чем шире полоса, тем лучше она описывается этой
функцией. Если это приближение удовлетворительно, то площадь,
ограниченная кривой (XI. 14) и горизонтальной нулевой линией
(Д = 0), можно вычислить по формуле:
5 = у-Ди)‘лЛакс, (XI. 15)
где Дс01/2—так называемая полуширина полосы, опреде-
ляемая между точками кривой, в которых оптическая плотность
равна половине максимальной.
При обычной ширине колебательных полос в спектрах жидко-
стей (порядка 10...20 см-1) и значениях спектральной ширины
щели спектрометра (3...5 см' ') значения S и абсолютной инте-
гральной интенсивности ё отличаются в среднем на 5%, что укла-
дывается в пределы ошибок эксперимента средней точности. При
необходимости для перехода от найденной по уравнению (XI.15)
площади S к величине ё находят и вводят поправочный множи-
тель, учитывающий инструментальные эффекты.
Для снижения ошибок количественных измерений рекомен-
дуется работать с пропусканием в пределах 20...60% (оптическая
плотность в пределах ~0,1...1,0) или, по крайней мере, не выхо-
дить за пределы 10...80% пропускания, когда ошибки резко воз-
растают. При большом поглощении необходимо уменьшать либо
толщину слоя, либо концентрацию.
Методом ИК спектроскопии, как и многими другими методами,
могут проводиться анализы, различные по точности, в зависимо-
сти от предъявляемых требований, с одной стороны, и реальных
возможностей, с другой. При полуколичественной обработке спек-
тров можно анализировать смеси или загрязнения с относительной
точностью ±10% и даже лучшей. Для измерения интенсивности
фона (7о) при длине волны пика полосы (7) простейшим является
а) б)
Рис. XI.5. Проведение
базовой линии:
а — для изолированной по-
лосы; б — в случае перекры-
вания с другими полосами
так называемый метод базовой линии,
широко применяемый в аналитической
практике.
Прямая «базовая» линия проводится
для аналитической полосы через ее край-
ние точки, где уже нет поглощения, или
как касательная к спектральной кривой
по краям полосы, если она попадает на
плечо другой полосы, как показано на
рис. XI.5. Хотя выбор точек, через кото-
рые проводят прямую, в известной степе-
ни произволен, это не имеет большого
значения, если в спектрах эталонов для
построения калибровочного графика и
при выполнении анализов эти точки все-
гда выбираются одинаково и нет нало-
полое. Систематические ошибки компеп-
жения новых мешающих
сируются при этом калибровочным графиком. При анализе смесей
из-за неоднозначного проведения базовой линии могут возникать
и весьма существенные ошибки.
Для повышения точности анализа очень важен правильный
выбор аналитических полос. Желательно, чтобы эти полосы были
изолированными или с минимальным перекрыванием, и они не
должны быть самыми сильными в спектре. Важны также воспро-
изводимая установка нуля и линия фона. Регулировка спектро-
метра при выполнении всей серии измерений должна оставаться
неизменной. Увеличить отношение сигнала s к шуму п можно
расширением щелей согласно зависимости
sin — bt'/vw2,
где b — константа; t — время сканирования; w — ширина щели.
Это может привести к некоторому ухудшению разрешения и кон-
тура полос, т. е., возможно, и к отклонениям от закона Бугера —
Ламберта — Бера, но важнее снизить влияние шумов, а остальное
компенсирует калибровка. Достаточно медленно должна вестись
также запись полосы поглощения. Скорость сканирования подби-
рается так, чтобы не происходило занижения оптической плот-
ности в максимуме полосы (т. е. очередное замедление записи при
подборе скорости не должно приводить к увеличению интенсив-
ности).
Если в смеси есть компонент, концентрация которого остается
постоянной, то можно пользоваться при анализе отношением оп-
тических плотностей пар полос вместо обычных калибровочных
кривых. При высокой точности анализа, достигаемой исключи-
тельно тщательной работой с эталонами и образцами, прецизион-
ным определением толщины слоев, применением разностной или
дифференциальной методики спектроскопии и других специальных
приемов, ошибка определений может быть порядка 0,1%. Как в
любых аналитических методах, результаты измерений должны
250
подвергаться обычной статистической обработке с оценкой стан-
дартного отклонения и других величин статистического анализа.
Современная фурье-спектроскопия позволяет успешно решать
проблемы, связанные с охраной окружающей среды, в частности
с определением загрязнений воды и атмосферы. Возможности этого
метода неоценимы как в изучении микрообъектов, свойств поверх-
ности твердых тел, тонких адсорбционных слоев, так и в атмосфер*
пых, астрофизических и космических исследованиях.
В настоящее время существуют аппаратура и методики, поз-
воляющие определять следовые количества примесей. В частно-
сти, использование многоходовых газовых кювет с длиной опти-
ческого пути несколько десятков метров дает возможность обна-
руживать в атмосфере миллионные доли загрязняющих ее газов,
таких, как H2S, НС1, СО, О3, SO2, NO2, СН4 и др., а некоторые
пока еще уникальные методики позволяют довести пределы опре-
деления до миллиардных долей.
Количественный анализ по спектрам КР основывается на
прямой пропорциональности интенсивности I линий КР числу мо-
лекул 'V в единице объема, т. е. на зависимости вида
I = ikN, (XI. 16)
где i — интенсивность рассеиваемого света на одну молекулу;
k — коэффициент, зависящий от условий эксперимента (оптиче-
ских характеристик прибора, спектральной чувствительности
приемника и т. д.).
Использование этой зависимости на практике затруднено преж-
де всего сложностью нахождения k. Учитывая, что для всех ли-
ний спектров КР, полученных в одних и тех же условиях экспери-
мента на одной и той же установке, коэффициент k остается
постоянным, реализуют возможность определения относительных
величин.
Для аналитических целей используют или интегральную
интенсивность линий КР I™, или интенсивность в
максимуме линий 10. Первая имеет однозначный физический
смысл и определяется путем измерения площади, ограниченной
контуром полосы.
Интенсивность в максимуме полосы 10 выражают обычно либо
в произвольной относительной шкале, либо в условной, например,
циклогексановой шкале, принимая интенсивность линии 802 см-1
циклогексана CeHi2 равной 250 единицам. При фотоэлектрической
регистрации спектров КР интенсивность в этой шкале для еди-
ницы объема
/ о = 25О (/„//802) (^ш/^802)> (XI. 17)
где (Д/Дог)—отношение наблюдаемых интенсивностей в макси-
мумах (высот пиков) для полосы со исследуемого вещества и ли-
нии 802 см-1 циклогексана; k,„— спектральная чувствительность
прибора при частоте со исследуемой полосы; й802 — то же, при
частоте линии циклогексана.
Иногда в качестве эталона выбирают линию 313 см-1 тетра-
хлорида углерода CCU, интенсивность которой принимают за 100.
При количественном анализе смесей может проводиться после-
довательная регистрация спектров в одинаковых условиях и срав-
нение интенсивностей линий чистых эталонных образцов. Это пре-
дусматривает, во-первых, знание состава смеси, т. е. все ее ком-
поненты должны быть идентифицированы, и, во-вторых, наличие
соответствующих эталонов. Если известна интенсивность линий
всех индивидуальных веществ в единой шкале, то достаточно срав-
нить интенсивность линий компонентов в смеси со спектром только
одного эталона. Сравнение можно осуществить и по методу вну-
треннего стандарта, когда в анализируемую смесь добавляют
определенное количество эталонного вещества. Оба метода имеют
свои преимущества и недостатки.
Обычно при регистрации колебательных спектров КР много-
атомных молекул ширина щели прибора меньше ширины наиболее
узкой полосы в спектре и контур полосы близок к идеальному,
т. е. отношение измеряемых интегральных интенсивностей не зави-
сит от аппаратной функции. Прецизионное же измерение самих
/«> требует учета влияния ряда искажающих факторов, рассматри-
ваемых в специальной литературе. Измеряемая величина /0 зави-
сит от отношения ширины щели прибора к ширине изучаемой
полосы.
Современная вычислительная техника позволяет разрабаты-
вать и реализовать на практике расчетные методы количествен-
ного анализа по спектроскопическим данным для многокомпонент-
ных систем как известного состава, так и неполностью известного
и даже неизвестного состава.
7. ИССЛЕДОВАНИЕ РАВНОВЕСИЙ
Экспериментальное исследование равновесий методами моле-
кулярной спектроскопии основывается на изучении температурной
зависимости относительной интенсивности полос, принадлежащих
спектрам участвующих в нем частиц. Для простоты рассмотрим
равновесие А^В каких-то двух молекулярных форм, идентифи-
цируемых по колебательным спектрам. Их молярные доли соот-
ветственно Ад и NB, молярные интенсивности Д и iB, наблюдаемые
интенсивности 7А и 7В. Здесь имеются в виду или интегральные
интенсивности полос, или, что грубее, иногда используемые интен-
сивности полос в максимумах. В случае ИК спектров это отно-
сится к оптической плотности A, a i, согласно уравнению (XI. 13),
соответствует el (при постоянстве I далее сокращается). Имея в
виду соотношение I = iN, для константы равновесия имеем
К = Nа/Nb = (Ib/к) <Лк1в) = С (/а/Iв)=СК'- (XI. 18)
Предполагается, что C=iB/iA не зависит или практически не зави-
сит от температуры Т, по крайней мере, для выбранных пар поло?
в заданном интервале температур при данном агрегатном состоя-
252
нии вещества (газ, раствор или чистая жидкость). Тогда, исполь-
зуя известное из химической термодинамики уравнение изобары
(или изохоры) Вант-Гоффа для температурной зависимости кон-
станты равновесия
dln/Cp д/7
dT — RT2 '
или в интегральной форме
1п^=-4т-+5’ (Х1Л9>
по наклону прямой зависимости In К' от обратной температуры
можно графически (или расчетом по точкам) определить тепловой
эффект реакции или разность энтальпий (внутренних энергий)
молекулярных форм, усредненную по некоторому температурному
интервалу (от Т\ до Т2).
В константу интегрирования уравнения (XI. 19) входит, как
известно, разность энтропий AS, форм А и В. В принципе разра-
ботаны методики определения и этой величины, т. е. и свободной
энергии Гиббса AG, и истинной константы равновесия, согласно
уравнению
-/?Г1пМ/?==ДОо=ДЯо-ГД5°. (XI.20)
Одна из таких предложенных методик основывается на гра-
фическом определении С=1В/й из уравнения прямой:
/b=42-/a + Zb^, (XI.21)
'а
где постоянная величина N = = Кроме предполо-
жения о независимости отношения С от температуры предпо-
лагается независимость от Г и величин iA и iB по отдельности.
Если найдено С, то из уравнения (XI. 18) определяется истинная
К?, а при известной А Я0 — также AG° и AS0 по (XI.20). В кон-
кретных исследованиях могут рассматриваться либо концентрации
с, либо парциальные давления р находящихся в равновесии ве-
ществ, так как по уравнению Клапейрона — Менделеева
p=2LRT = c^T.
Сказанное применимо, в основном, к газовой фазе, но приб-
лиженно с соблюдением известной осторожности и некоторыми
поправками такие исследования можно проводить также в жид-
кой фазе.
В несколько более сложном случае равновесия А+±2В, напри-
мер гомогенной газовой реакции
N2O4^2NO2
представляющей классический пример обратимой химической
реакции, равновесие которой в сильной степени зависит от темпе-
ратуры и давления, вместо уравнения (XI.18) записывают
ZBZA J________С* ZA
Р~~ ръ ZAZB РТ ~ Т
(XI.22)
где С
‘к
и для определения среднего теплового эф-
фекта реакции строится график зависимости
lg fот 1/7\
\ 1 'в /
Рассмотренный метод широко применяется в исследованиях
быстро устанавливающихся подвижных равновесий различных
молекулярных форм, таких, как таутомерные, конформационные
или поворотно-изомерные, адсорбционные, координационные при
различных процессах ассоциации и комплексообразования, вклю-
чая водородные связи, и т. д. Как уже отмечалось, характеристи-
ческое время методов колебательной спектроскопии таково, что
при времени жизни какого-то молекулярного объекта (состояния)
больше 10“11 с этот объект может данными методами идентифи-
цироваться и изучаться. Такими быстрыми переходами характе-
ризуются, например, поворотные изомеры при высоте потенциаль-
ного барьера заторможенного внутреннего вращения порядка
нескольких или даже десятков кДж/моль.
Именно указанным методом температурной зависимости интенсивности полос
в колебательных спектрах определены разности энергий (энтальпий) поворотных
изомеров и конформеров (рис. XI.2) сотен органических и элементорганических
соединений. Так, например, по зависимости от температуры относительной интен-
сивности полос гош- и транс(анти)-изомеров молекул 1,2-дихлорэтана и 1,2-ди-
бромэтана разности энтальпий ДЯ поворотных изомеров этих соединений в га-
зовой фазе найдены равными, соответственно, 4,9±0,4 и 7,4±0,6 кДж/моль (бо-
лее устойчив транс-изомер):
Для монохлорциклогексана, например, разность энтальпий аксиальной а и
экваториальной е конформаций
как в газовой, так и в жидкой фазах найдена в пределах 1,2... 1,7 кДж/моль.
254
г
В таких исследованиях немаловажен выбор полос для изуче-
ния температурной зависимости их относительных интенсивностей.
Желательно, чтобы они относились к близким по форме колеба-
ниям молекул разных конформеров. Но и при этом неправильно
считать, что молярные интенсивности выбранных полос конфор-
меров близки или даже равны, как это иногда делают, и, пред-
полагая, что 1’вЛ’а=1, оценивают константу конформационного
равновесия по наблюдаемому отношению /АДв без изучения его
температурной зависимости. Это ведет к грубым ошибкам, так
как обычно 1В/1а^=1; например, отношение молярных интенсив-
ностей полос валентных колебаний С—Гал для конформеров моно-
замещенных циклогексанов ie/ia может быть больше 2.
8. КОМПЛЕКСЫ С ВОДОРОДНЫМИ связями
По температурным зависимостям спектров, совершенно анало-
гично рассмотренному в § 7, определяют энтальпии образования
комплексов или ассоциатов, например, при образовании водород-
ных или каких-то других межмолекулярных связей. Водородная
связь возникает, как известно, между группой — донором протона
(О—Н, N—Н и др.) и акцептором протона (атомы с неподелен-
ными парами электронов: О, N, С1 и др.), т. е. образуется система
вида
R' —А —Н...В —R"
Для нее характерны собственные колебательные частоты, а кроме
того, она меняет ряд частот колебаний связанных частей ком-
плекса. Что касается первых, то можно условно говорить о часто-
тах валентного колебания vh в и деформационных колебаний
А—Н...В.
Например, для димеров карбоновых кислот
//O...H-Ох
R-Cf )C-R
ЧО-Н...О^
это частоты валентных колебаний водородной связи О...Н и час-
тоты плоских и неплоских деформационных колебаний О—Н...О,
лежащие в длинноволновой области. Теоретически в сумме долж-
но быть шесть таких частот. Но, в частности, у муравьиной кисло-
ты в ИК спектре наблюдаются три частоты: 248 (Ви), 170 (Лм)
и 68 (Ли) см-1, отнесенные по типам симметрии точечной группы
С2н- Для уксусной кислоты частота валентных колебаний О...Н
(~190 см-1) ниже, чем у муравьиной. В результате образования
водородной связи понижаются частоты валентных колебаний
va-h и Vr’-b, меняется частота деформационного колебания у
(R'—А—Н) и т. д. Так, например, узкая полоса свободной гидро-
ксильной группы в разбавленных растворах спиртов наблюдается
в области 3625 см-1. При увеличении концентрации появляется
широкая полоса, смещенная в область более низких частот и от-
носящаяся к ассоциированным группам. При слабой водородной
связи частота vo-н понижается на 300 ... 400 см-1, а при сильной —
до 1000 см-1 и более.
Установлена корреляционная зависимость энтальпии образова-
ния водородной связи и увеличения интенсивности ИК полосы
vo-н («правило интенсивности» Иогансена*) для большого ряда
Н-комплексов с разными основаниями и в широком диапазоне
энергий Н-связей:
- Д//= 12,14ДЛ'Ч (XI.23)
где ДД1/1==Дко2мпл —Лстоб! ЛСВОб — интегральная интенсивность по-
лосы vo-н в разбавленном растворе (ССК, н-С6Н14).
Выявлена также взаимосвязь усиления и смещения полос
•vo-н различных спиртов:
ДД^^О.ЮСДш)1/», (XI.24)
ГДе А® — СОсвоб—СОкомпл-
Из уравнений (XI.23) и (XI.24) следует корреляционное соот-
ношение
-Д//=1,25(Д<о)’/., (XI.25)
.которое, как и соотношение (XI.24), справедливо, по крайней
мере, для диапазона смещений 1000 см-1^А®^100 см-1. Анало-
гичные корреляции установлены и для других рядов Н-комплек-
сов. Необходимо отметить, что полоса поглощения связанных
групп А—Н обычно очень широкая, например у карбоновых
кислот, и имеет сложный контур. Интегральная интенсивность
ее должна измеряться без накладывающихся посторонних острых
пиков, а частота измеряется для «центра тяжести» такой, обычно
.асимметричной полосы (в точке, где перпендикуляр к оси абсцисс
делит полосу на две равные по площади части).
Частоты колебаний групп — акцепторов протонов, например
vc=o, понижаются в меньшей степени, но и они успешно исполь-
зуются при изучении ассоциатов с водородными связями.
Если в одной и той же молекуле имеются как группы — доноры
протонов, так и акцепторные группы, то при подходящей прост-
ранственной структуре возможно образование внутримолекуляр-
ных водородных связей, например в салициловой кислоте:
Правило записано при ДЯ в кДж/моль; А — в 10~4 см/ммоль.
Такие внутримолекулярные комплексы также имеют свои харак-
терные спектральные проявления. Отличить их от межмолекуляр-
иых ассоциатов можно, изучая концентрационную зависимость
спектров растворов или зависимость от давления паров вещества.
При разбавлении относительная интенсивность широких полос,
характерных для межмолекулярных связей, уменьшается и в конце
концов эти полосы исчезают, в то же время полосы внутримоле-
кулярных связей от разбавления не зависят.
9. КИНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ
Использование методов колебательной спектроскопии в кине-
тических исследованиях основывается на возможности контролиро-
вать протекание химических реакций по интенсивности полос
исходных веществ и конечных, а иногда и промежуточных продук-
тов. Изменение интенсивности полос, т. е. концентраций соответ-
ствующих продуктов, во времени позволяет получить такие харак-
теристики реакций, как ее порядок, константа скорости и энергия
активации.
Существуют различные методики изучения кинетики реакций
по спектрам. Для очень медленно идущих превращений можно
проводить через определенные промежутки времени отбор проб
из реакционной смеси для регистрации спектра. Если скорость
реакции велика, то при необходимости реакцию можно прекратить
в отобранной пробе охлаждением, разбавлением или добавкой
соответствующего дезактиватора.
Другая методика предусматривает проведение реакции непо-
средственно в кювете спектрометра и многократную регистрацию
спектра (или его. части) через определенные промежутки времени.
В некоторых случаях можно установить спектрометр на фиксиро-
ванную частоту какой-то представляющей интерес полосы погло-
щения и наблюдать зависимость оптической плотности от времени,
т. е. непосредственно записывать на диаграммной ленте прибора
кинетическую кривую. При этом необходимо контролировать тем-
пературу образца, который должен термостатироваться.
Прием, облегчающий отнесение ИК полос поглощения и наб-
людение за ними, состоит в том, что в пучке сравнения помещают
кювету с реакционной смесью, в которой реакция закончилась.
Линию 100%-кого пропускания выводят на середину диаграммной
ленты. При этом в спектре анализируемого образца полосы исход-
ных веществ будут направлены вниз, а полосы продуктов реак-
ции— вверх от указанной линии. По ходу реакции интенсивность
всех полос будет падать, а после окончания реакции они все ис-
чезнут.
Как пример изучения методом спектроскопии КР кинетики
медленной реакции можно привести гидролиз ацетонитрила, ката-
лизируемый перхлоратом ртути. В спектре КР реакционной смеси
кроме исходных веществ и конечных продуктов обнаруживаются
полосы, характерные для молекул ацетонитрила, связанных в
комплекс с ионами Hg2+, а также полосы комплекса ацетамида
с ионами Hg2+. Идентификация этих промежуточных продуктов
позволила предложить следующий механизм реакции:
CH3CN + Hg2+ CH3CN • Hg2+—-CH3CONH2-Hg2+^CH3CONH2 +
+Hg2+
а по изменению интенсивности полос изучить ее кинетику. Изуче-
ние кинетики достаточно медленной реакции окисления вторичных
спиртов до кетонов методом ИК спектроскопии проводится по
постепенно исчезающей полосе поглощения гидроксильной группы
при ~3570 см-1 и появляющейся полосе поглощения карбониль-
ной группы при ^—-1720 см-1.
Для исследования быстро протекающих реакций и короткожи-
вущих промежуточных частиц необходимо применять спектро-
метры с быстрым сканированием, в частности ИК интерферометры
(фурье-спектрометры), с выдачей спектра на дисплей или на
запись. С высокими скоростями позволяют регистрировать спектры
также современные спектрометры КР с лазерным возбуждением,
особенно мощными импульсными лазерами. Созданы уникальные
приборы, на которых достигают очень высокого разрешения по
времени (^мкс).
10. КОЛЕБАТЕЛЬНАЯ СПЕКТРОСКОПИЯ
ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
Методы колебательной спектроскопии применяются в исследо-
ваниях самых различных по своему характеру высокомолекуляр-
ных соединений как природных, так и синтетических, как органи-
ческого происхождения, так и неорганических. Для всех таких
систем характерно большое разнообразие структурных особен-
ностей. Многие из них можно отнести к полимерам, макромоле-
кулы которых состоят из повторяющихся структурных фрагментов.
По ИК и КР спектрам можно идентифицировать тип полимера,
судить о структуре и конформации цепей их ориентации и упаков-
ке, осуществлять контроль за изменением структурных параметров
под влиянием внешних воздействий. В этих целях ИК спектроско-
пия использовалась раньше и до сих пор применяется пока чаще,
хотя современная лазерная спектроскопия КР имеет ряд своих
преимуществ. Это, в частности, возможность получения спектра
без такой трудоемкой подготовки некоторых образцов, какая тре-
буется в ИК спектроскопии, отсутствие сильных искажений спектра
КР наполнителями и др. Однако, как и для низкомолекулярных
соединений, нужно по возможности сочетать эти взаимодополняю-
щие методы или, зная их отдельные преимущества, при решении
каких-то конкретных задач использовать соответствующий метод.
В структурных исследованиях макромолекул также исполь-
зуется концепция групповых частот, так как для некоторых нор-
мальных колебаний частоты сравнимы с групповыми частотами
258
более простых молекул (например, для групп С = О, О—Н, N—Н,
СНз и т. п.). Полоса валентного колебания С = С характеристична
и имеет высокую интенсивность в спектрах КР как низкомолеку-
лярных, так и высокомолекулярных соединений. Это дает возмож-
ность определения типа замещения при двойной связи в полиме-
рах, степени сшивки, например, по уменьшению интенсивности
полосы С = С сшивающего агента (аллилового спирта), степени
отверждения полиэфирных смол на основе сополимеров полисти-
рола и т. д.
Рассмотрение же спектра полимера только с точки зрения иден-
тификации групповых частот совершенно недостаточно.
Теория колебательных спектров макромолекулярных систем, в
частности полимеров, имеет много общего с теорией колебаний
кристаллов, которые в каком-то смысле тоже представляют собой
макромолекулярные системы. Полимерные цепи могут быть стерео-
химически и конформационно упорядоченными, имея, например,
вид вытянутой спирали. При этом бесконечная последовательность
однородных фрагментов может рассматриваться как идеальный
одномерный кристалл. Такие цепи упаковываются в бездефектную
кристаллическую решетку, т. е. образуют идеальный трехмерный
кристалл. Хотя в действительности в реальных полимерах всегда
имеются какие-либо дефекты (химической структуры, стереохими-
ческая и (или) конформационная неупорядоченность, дефекты
решетки), можно все-таки говорить о регулярной, т. е. кристал-
лической, структуре упорядоченных полимеров и отдельно
рассматривать неупорядоченные полимеры. Методы рас-
смотрения колебательных спектров в этих двух случаях будут
различны.
Нормальные колебания бесконечной полиэтиленовой цепи в
зигзагообразной плоской конформации могут быть охарактеризо-
ваны разностью фаз 6 для соседних метиленовых групп. Расчет
колебаний для значений б, меняющихся в диапазоне 0—л, дает
девять ветвей (дисперсионных кривых) частот, с которыми в общих
чертах согласуются экспериментальные спектры н-алканов и по-
лиэтилена.
Для одномерно и трехмерно упорядоченных полимеров строгое
определение числа колебаний, активных в ИК и КР спектрах,
может проводиться с использованием теоретико-группового ана-
лиза аналогично тому, как это делается для простых многоатом-
ных молекул и, особенно, как для молекулярных кристаллов.
Полимеры на основе монозамещенных этиленов (поливиниль-
ные) могут быть стереорегулярными, т. е. синдиотактическими или
изотактическими в плоской или спиральной конформации, а также
атактическими в беспорядочной конформации. Правила отбора
для ИК и КР спектров в каждом случае свои и в принципе можно
для любого неизвестного образца винильного полимера однознач-
но определить по спектрам его строение.
По колебательным спектрам идентифицируются, например, син-
диотактический полипропилен (ПП) в форме двойной спирали
и изотактический ПП, имеющий спиральную форму с тремя моно-
мерными единицами в витке (31).
Рассмотрим идеальный поливинилхлорид (ПВХ), цепь которого
состоит из последовательности звеньев —СНС1—СН2—, соединен-
ных по типу «голова к хвосту». Синдиотактическая спираль мо-
жет отвечать или последовательности транс-...ТТТТ... поворотных
форм, или последовательности: ...ТТГГТТГГ..., где Г — гош-форма.
В изотактическом полимере спирали 31 отвечает последователь-
ность ...ТГТГТГ... . Для каждой из трех моделей характерны свои
правила отбора в ИК и КР спектрах. Предполагается, что наи-
более вероятна первая модель, с которой связываются сильные
ик полосы поглощения синфазных и антифазных колебаний
С—С1 соответственно при 640 и 604 см-1.
Реальный ПВХ содержит разные химические, конфигурацион-
ные и конформационные дефекты, проявляющиеся в спектре. Так,
для химического дефекта образования связей типа «голова к
голове», т. е. структуры —СН2—СН2—, характерна полоса 763 см~1
маятниковых колебаний двух рядом расположенных групп СН2.
При появлении дефектов любого из перечисленных типов возни-
кают колебания с частотами выше обычных для валентных коле-
баний С—CI и т. д. В результате взаимодействия дефектов могут
наблюдаться расщепления характерных для них полос, величина
которых зависит от последовательности дефектов и расстояния
между ними в цепи.
Теория и расчет колебаний упорядоченных полимеров основаны
на наличии у них трансляционной симметрии в одном из трех
направлений. Силовое поле при расчетах составляется в первом
приближении из силовых постоянных, найденных для простых мо-
лекул сходного с фрагментами полимера строения, а затем может
варьироваться или каким-то образом уточняться, например, с
использованием координат локальной симметрии.
Но если и для кристаллических структур полимеров теория
и интерпретация колебательных спектров является сложной, то
еще сложнее дело обстоит в случае неупорядоченных полимеров.
Отсутствие периодичности не позволяет вводить фазовую харак-
теристику колебаний и заставляет рассматривать колебания очень
большого числа беспорядочно расположенных в пространстве
атомов. Из сказанного выше о спектрах одномерных и трехмер-
ных упорядоченных структур и неупорядоченных полимеров оче-
видно, что колебательные спектры позволяют отличать регуляр-
ность и кристалличность от нерегулярности и аморфности.
Полосы, которые связаны с наличием трехмерного упорядоче-
ния и межмолекулярных взаимодействий в кристалле, называют
«кристалличными». Примером «кристалличной» полосы является
один из компонентов дублета около 725 см-1 в ИК спектре поли-
этилена, исчезающий в спектре расплава.
Использование «кристалличных» полос для определения сте-
пени кристалличности полимера встречает те трудности, что пря-
мое измерение коэффициента поглощения для такой полосы не
260
предствляется возможным, так как получить полностью кристал-
лический полимер обычно нельзя. Этот коэффициент определяется
по образцу, для которого степень кристалличности известна по
каким-то другим данным, например, по измерениям плотности
после определения аморфной доли по «аморфной» полосе
(см. ниже) и т. д.
При кристаллизации полимера макромолекулярная цепь дол-
жна много раз сложиться, чтобы образовать отдельный кристал-
лит. Поэтому даже в монокристалле полимера неизбежно содер-
жится какая-то доля «аморфных», неупорядоченных структур.
Казалось бы, что из-за чрезвычайной сложности спектра аморф-
ного полимера, обусловленной нарушением закономерностей в
соотношениях фаз колебаний одинаковых групп и правил отбора
при всевозможных поворотах цепи, он должен резко отличаться
от спектра того же полимера в кристаллическом состоянии. На
практике такого резкого отличия этих спектров не наблюдается,
хотя у некоторых полимеров в аморфном состоянии, действитель-
но, появляется ряд новых, «аморфных» полос. Нечеткость различия
спектров аморфной и кристаллической формы полимеров застав-
ляет предполагать, что аморфные по рентгеноструктурным данным
полимеры содержат все-таки достаточно упорядоченные участки
цепей, у которых некоторые формы колебаний, как и в кристал-
лических полимерах, неактивны, а полосы ряда других колебаний
очень мало интенсивны.
Б настоящее время начали различать так называемые «регу-
лярные» полосы, которые обусловлены одномерной регулярной
геометрией (конформацией) полимерной цепи, например спираль-
ной. Такие полосы могут наблюдаться не только в спектрах твер-
дых полимеров, так как регулярные макромолекулярные цепи мо-
гут существовать также в аморфных и жидких образцах.
Для определения аморфной доли в твердом полимере исполь-
зуют «аморфные» полосы после измерения их интенсивности в
спектре эталона — полностью аморфного образца, получение кото-
рого принципиальных трудностей не вызывает. Эти полосы обычно
слабые, и для измерений могут использоваться достаточно толстые
пленки.
Поскольку многие химические и физические свойства поли-
мерных материалов зависят от типа и концентрации неупорядочен-
ных образований, то использование колебательной спектроскопии
для их определения имеет большое практическое значение.
Например, в ИК спектре ПЭ полосы 1350 и 1304 см-1 относятся
к гош-поворотно-изомерной форме, которая приводит к разупоря-
дочению, и по ним можно оценивать степень скрученности цепи.
Наличие таких конформационных дефектов поддается определе-
нию, если их содержание превышает 5%.
При аморфной доле полиэтилена от 0,1 до 0,9 по указанной по-
лосе веерного колебания СН2 1304 см-1, рассматриваемой в качест-
ве «аморфной», получили хорошее согласие с рентгеноструктурны-
ми данными.
В спектрах КР ПЭ «кристалличным» являются полосы 1170
и 1296 см-1, а «аморфной» — полоса 1081 см-1, соответствующая
гош-поворотно-изомерной форме.
Необходим правильный выбор и отнесение «кристалличных»,
«регулярных» и «аморфных» полос, так как при ошибочном от-
несении неверно трактуется и природа полимера.
Низкочастотные спектры КР позволяют получать и такую ин-
формацию о структуре полимеров, которую метод ИК спектро-
скопии не дает. В частности, в спектре КР активно продольное
акустическое (LA) колебание зигзагообразной цепи, похожее
по форме на растяжение мехов гармони
являющееся, однако, полносимметричным валентным колебанием
С—С, смешанным с деформационным колебанием С—С—С. Соот-
ветствующая полоса наблюдалась в спектрах ПЭ, ПП и полиэти-
леноксида. Частота этого колебания обратно пропорциональна
длине цепи с полностью транс-конформацией и, например, в за-
висимости от обработки образца у кристаллического ПЭ меняется
в диапазоне 10...40 см'1. Значение этой частоты использовалось
даже для определения длины вытянутых цепей, причем получен-
ные результаты оказывались согласующимися с рентгеноструктур-
ными данными. Для участка зигзагообразной цепи из двух десят-
ков углеродных атомов частота КЛ-колебания выше 100 см-1, а
при длине порядка 100 атомов ~20...30 см-1.
На частоту акустических колебаний оказывают возмущающее
влияние концевые группы. Нарушение регулярности цепи приводит
к появлению вытянутых участков различной длины, так что в
реальном спектре может наблюдаться ряд полос акустических ко-
лебаний, отражающих и число разных участков.
Поскольку частоты акустических колебаний зависят от степени
кристалличности или аморфной доли в полимере, низкочастотная
спектроскопия КР также позволяет изучать морфологические
эффекты в полимерах, их сферолитную и ламелярную
структуру.
Б исследованиях колебательных спектров ориентированных об-
разцов, например, полученных механической обработкой (прокат-
кой, волочением) полимерных материалов в виде пленок, волокон
и т. п. или кристаллов важную дополнительную информацию мо-
жет дать использование поляризованного излучения. Измерения
поляризации линий КР в спектрах полимеров хотя и сильно за-
труднены, но при тщательном приготовлении образцов и отработке
методик возможны и в последнее время начали проводиться.
У полимеров нерегулярного строения, когда все колебания актив-
ны в ИК й КР спектрах, все линии КР поляризованы. При регу-
лярном строении данные о структуре, конформации цепей и
морфологических эффектах получают не только изучая и сопо-
ставляя сами по себе ИК и КР спектры, но также из поляриза-
ционных измерений.
Поляризованное ИК излучение в спектроскопии ориентирован-
ных образцов применяется уже давно. Оно позволяет получать
структурную информацию из данных по так называемому дихроич-
ному отношению. Для любой полосы поглощения дихроичное от-
ношение определяется как отношение ее интенсивностей, измерен-
ных в излучении, поляризованном параллельно и перпендикулярно
заданному направлению, например направлению растяжения:
R = IJI±. (XI.26)
Оно может в принципе меняться от 0 до к>, а чаще всего наблю-
даемое отношение лежит в пределах от 0,1 до 10. При 1
полосу называют перпендикулярной, при R> 1 — параллельной * —
это если определение R, как в уравнении (XI.26), а не обратное,
которое тоже используют в литературе.
Для использования и интерпретации R предложены разные
модели и теоретические подходы. Согласно одной из простых мо-
делей рассматривается зависимость дихроичного отношения от
двух параметров — степени ориентации f (или доли полимера,
строго ориентированного в одном направлении, когда остальная
его часть 1—f полностью беспорядочна) и угла между направле-
нием момента перехода и осью цепи а, согласно которой:
/ // fcos^a+ 1(1 —f)/3)]
11 ((/sin2 а)/2] + [(1 —/)/3]
В другом простейшем приближении вводится понятие усред-
ненного угла ориентации 0, которое образуется осью цепи поли-
мера с направлением оси волокна (кристаллита и т. п.). В этой
модели для параллельной полосы
а для перпендикулярной полосы
г ,т 2 sin2 9
' 2-sin2 9
Обе модели связаны соотношением
/= 1 — (3 sin2 6)/2.
Решая обратную задачу, по дихроичному отношению судят о
степени упорядоченности, ориентации цепей в образце полимера
и направлениях моментов перехода для различных полос погло-
* Нельзя путать с перпендикулярными и параллельными полосами в ИК
спектрах газов, где терминология связана с контурами вращательной структуры
полос.
щеяия. При интерпретации дихроичного отношения для полос,
относимых к колебаниям каких-то фрагментов или групп атомов,
всегда должен анализироваться вопрос, насколько эти колебания
характеристичны и могут ли они рассматриваться независимо от
колебаний других групп. Иногда при высокой характеристичности
оказывается возможным оценить угол, образуемый какими-то
связями с направлением цепи или волокна. Например, направле-
ния моментов перехода для валентных колебаний N—Н, О—Н
и т. п. примерно совпадают с направлением самих валентных свя-
зей. По дихроичному отношению определяют также направление
моментов перехода, а следовательно, ориентацию связей относи-
тельно заданной оси для амидной группы. Когда можно пред-
сказать, какие полосы поглощения должны быть параллельны,
а какие перпендикулярны, дихроичное отношение помогает в от-
несении частот колебаний..
К числу объектов, успешно изучаемых методами колебатель-
ной спектроскопии с применением тех или иных описанных под-
ходов и приемов, относятся и такие высокомолекулярные соеди-
нения, как белки, нуклеотиды, нуклеиновые кислоты, а также дру-
гие биологические и синтетические макромолекулярные системы.
Каждый раз может появляться какая-то специфика, но обычно
она не имеет принципиального значения,
ГЛАВА XII
ПРИБОРЫ И ЭКСПЕРИМЕНТАЛЬНАЯ ТЕХНИКА
1. ТЕХНИКА И МЕТОДИКИ ИК СПЕКТРОСКОПИИ
ИК область спектра охватывает длины волн от границы види-
мой области, т. е. от 0,7, до 1000 мкм, что соответствует 10 см-1,
т. е. нижнему пределу колебательных частот молекул. Вся ИК об-
ласть условно делится на ближнюю, среднюю и даль-
нюю, или длинноволновую. Такое подразделение возникло
в связи со свойствами оптических материалов (прозрачностью и
линейной дисперсией), применявшихся, в частности, для изготов-
ления призм. Если границей между ближней и средней областью
принято считать ~2 мкм (-~5000 см1), то граница между сред-
ней и длинноволновой областью связывалась с длинноволновым
пределом рабочего диапазона призмы из кристалла КВг — 25 мкм
(400 с\г!). В связи с созданием, с одной стороны, призм из бро-
мида и иодида цезия, а с другой, ИК спектрометров с дифрак-
ционными решетками и интерферометров Международным сою-
зом по чистой и прикладной химии (IUPAC) было рекомендовано
называть длинноволновой область ниже 200 см-1 (низкочастот-
ный предел рабочего диапазона призмы CsI, соответствующий
длине волны 50 мкм). Принципиальных различий между интер-
валами 10...200 см-1 и 10...400 см-1, как и областью выше
400 см'1, конечно, нет, но аппаратура и методики имеют свою
специфику для каждой из областей. Спектральный интервал ниже
10 см-1 (Х> 1000 мкм) обычно исследуется методами микровол-
новой и радиоспектроскопии.
Экспериментальные установки и технику измерений и анализа
в ИК спектроскопии можно разделить на традиционные, которые
активно разрабатывались до 60-х годов и, продолжая совершенст-
воваться, широко применяются в химических лабораториях, и
новейшие, обеспечивающие другое качество и совершенно иной
уровень.информации.
1.1. Принципы устройства и действия ИК спектрометров
При множестве различных типов современных ИК спектро-
метров по общим принципам устройства их можно разделить на
две основные группы. Первая включает приборы с последова-
тельным сканированием и регистрацией спектра с помощью одно-
канального приемника, а вторая — спектрометры, в которых на
приемник попадает сразу излучение всего изучаемого спектраль-
ного диапазона, но сигналы преобразуются и расшифровываются
так, что получается информация о каждом отдельном участке и
регистрируется полный спектр во всем диапазоне. Приборы и той,
и другой групп могут быть диспергирующие и недиспергирующие.
Диспергирующие приборы первой группы — это наиболее
распространенные сканирующие спектрометры, а недис-
пергирующие — очень перспективные, но пока еще редкие приборы,
например с лазерами на красителях, в которых возможна плав-
кая перестройка длины волны монохроматического излучения
источника. К недиспергирующим приборам второй группы отно-
сятся фурье-спектрометры, а к диспергирующим — разра-
батываемые в самое последнее время приборы, основанные на
преобразовании Адамара.
Сканирующие диспергирующие ИК спектрометры по схеме ос-
вещения бывают одно лучевыми и двухлучевыми. При
однолучевой схеме спектр поглощения исследуемого объекта ре-
гистрируется на спадающей с длиной волны кривой интенсивности
излучения источника вместе с фоновым поглощением. Чтобы по-
лучить спектр (в процентах пропускания), нужно зарегистрировать
также кривую интенсивности испускания источника (фоновый
спектр). Принимая интенсивность при каждой X в этих спектрах
соответственно как Д и 10к, находят значение пропускания
= (Л//ол) • 100% и строят по точкам спектральную кривую в за-
висимости от Z, (или со) или аналогично для поглощения
(1—7’)-100%, или в шкале оптической плотности A = lg (1/Г).
Теперь чаще используется двухлучевая схема, которая позво-
ляет выравнивать фон, т. е. линию полного пропускания, и ком-
пенсировать поглощение атмосферных паров НгО и СО2, а также
ослабление пучков окнами кюветы и, если необходимо, поглоще-
ние растворителей.
Рис. XII.1. Блок-схема двухлучевого
сканирующего ИК спектрометра:
----—ИК излучение; —.—.— электричес-
кий сигнал;--------механическая связь;
1 — источник ИК излучения; 2— система
зеркал; 3 — рабочий пучок и образец; 4—
пучок сравнения и компенсатор фона; 5 —
прерыватель-модулятор; 5 — входная щель
монохроматора; 7 — диспергирующий эле-
мент (дифракционная решетка или при'зма
с зеркалом Литтрова); 8 — выходная щель
монохроматора; 9 — приемник; 10 — усили-
тель; // — мотор отработки; /2 —фотомет-
рический клин; 13— самописец; 14 — мотор
развертки
Блок-схема (двухлучевогу ска-
нирующего ИК“......спектрометра
представлена на рис. XII.1. Реги-
страция спектра осуществляется
следующим образом. ИК излуче-
ние от источника 7 делится на
два пучка. Рабочий пучок прохо-
дит через образец, а пучок срав-
нения— через какой-то компенса-
тор (кювета с растворителем, ок-
но и т. п.). С помощью прерыва-
теля 5 пучки поочередно направ-
ляются на входную щель 6 моно-
хроматора и через нее на диспер-
гирующий элемент 7. При мед-
ленном его повороте (или поворо-
те зеркала Литтрова за призмой),
осуществляемом мотором раз-
вертки 14, через выходную щель
8 монохроматора на приемник 9
последовательно проходят выре-
заемые щелью узкие по интерва-
лу длин волн, в идеале монохро-
матические, лучи. Если излучение данной длины волны в рабочем
пучке и пучке сравнения имеет разную интенсивность, например
ослаблено в рабочем пучке поглощением образца, то на приемнике
возникает переменный электрический сигнал. После усиления и
преобразования этот сигнал поступает на мотор отработки 11, ко-
торый приводит в движение фотометрический клин 12 (диафрагму)
до уравнивания потоков излучения (метод оптического нуля). Дви-
жение фотометрического клина связано с движением "пера само-
писца 13 по ординате, а поворот диспергирующего элемента — с
протяжкой бумажной ленты или движением держателя пера по
абсциссе. Таким образом, в зависимости от градуировки в процес-
се сканирования может регистрироваться спектральная кривая
зависимости пропускания (поглощения) в процентах или оптиче-
ской плотности образца от волнового числа (или длины волны).
В нашей стране промышленностью выпускаются как двухлучевые сканиру-
ющие спектрометры (например, ИКС-30 и ИКС-29), так и однолучевые приборы
(ИКС-33) — все с дифракционными решетками.
В настоящее время спектрометры все чаще сочетаются с ЭВМ, спектр
регистрируется в цифровом виде и после обработки может быть по желанию
вызван из памяти в виде обычной спектральной кривой.
Принципиальной частью сканирующих спектрометров является
монохроматор. В качестве диспергирующего устрой-
ств а в нем могут служить призмы из прозрачных в ИК области
материалов (см. табл. XII. 1) с подходящей дисперсией или диф-
ракционные решетки — эшелетты. Поскольку дисперсия
266
Таблица XII. 1. Некоторые оптические материалы для ИК спектроскопии
Материал Область пропус- кания, мкм Показатель преломле- ния Примечание
Стекло 0,35 ... 2 1,5... 1,9
Кварц SiO2 0,2 ... 4 и после 50 1,43
LiF 0,2 ... 7 и после 200 1,39
Сапфир А120з 0.2... 6 1,77 Твердый, нехрупкий Поликристаллический
Иртран-1 MgF2 2... 8 1,3
Флюорит CaF2 0,2 ... 10 1,40
Иртран-3 CaF2 0,2... И 1,34 Поликристаллический, нехрупкий
Иртран-2 Zn2S 1 ... 14 2,24 Нерастворим в боль- шинстве растворителей
Каменная соль NaCl 0,2 ... 16 1,52 Г игроскопичен
AgCl 0,4... 20 2,0 Очень мягкий, разлага- ется на свету и в кон- такте с металлами
Сильвин КС1 0,3 ... 21 1,49 Г игроскопичен
КВг 0,2... 27 1,53 »
CsBr 0,3 ... 40 1,66 Гигроскопичен, мягкий
CsI 0,3 ... 50 1,74 То же
KRS-5 (TiBr+ 1 ... 40 2,38 Токсичен, мягкий
+ T1I, 42 и 58%)
Германий Ge 2 ... 20 4,0 —
Кремний Si 2 ... 6 и 40 ... 300 3,5 Длинноволновая гра- ница зависит от чистоты материала
Полиэтилен (—СН2—СН2—)п 20... 200 1,52 Очень мягкий, полоса поглощения 72 см-1
Иртран-4 Zn2Se 1 ...21 2,5 —
KRS-6 (Т1Вг+ 0,4 ...25 2,2 —
+ Т1С1, 40 и 60%)
Алмаз (тип II) Вся ИК область Исключительно прочен, есть полосы поглощения
материалов является наибольшей у длинноволнового предела их
прозрачности и быстро падает с уменьшением длины волны, в
средней ИК области используют обычно сменные призмы, изго-
товленные из монокристаллов LiF, NaCl, КВг, а для области
200...400 см-1 — из CsI.
В настоящее время призмы все более заменяются дифракцион-
ными решетками. Но решетка, на порядок лучше пропуская лу-
чистую энергию по сравнению с призмой, имеет тот недостаток,
что дает наложение спектров высших порядков. Он не устраняется
наличием в спектрометре нескольких сменных решеток для раз-
ных спектральных областей и требует, особенно в длинноволновой
области, где интенсивность излучения очень мала, хороших спект-
ральных фильтров.
В качестве источников непрерывного ИК из луче-''
ния используются обычно силитовый стержень — «г л об ар»
j _£штифт из карбида кремния) или штифт Н е р н с т а (из оксидов
редкоземельных элементов). Кривая интенсивности излучения этих
источников, нагреваемых током до высоких температур, имеет вид
кривой излучения абсолютно черного тела. Так, например, у гло-
бара при температуре ~ 1300°С максимум интенсивности излуче-
ния приходится на область ~ 5000 см-1 (~2 мкм), а в области
-~600 см-1 (16,7 мкм) интенсивность падает примерно в 600 раз.
Хорошие и с т о ч н и к и излучения в длинноволновой
ИК области вообще отсутствуют. Основная доля теплового из-
лучения нагретых твердых тел или излучения газового разряда
приходится на видимую и ближнюю ИК область спектра, а в
длинноволновой части мощность излучения этих источников со-
ставляет ничтожную долю общей мощности. Например, дуговая
лампа при полной мощности излучения 1 кВт дает здесь мощность
всего 10"1 Вт. До низкочастотного предела 200 см-1 используются
обычно указанные выше тепловые источники ИК излучения, но
они являются очень слабыми даже в интервале 400...200 см-1, где
кривая интенсивности /(%) имеет далекий от максимума склон.
Ниже 200 ему1 в качестве источника служит обычно ртутная
лампа высокого давления. В верхней части ее рабочего
диапазона используется в основном тепловое излучение нагретых
стенок, а ниже — поток излучения ртутной дуги и плазменная
эмиссия.
В связи со слабостью источников, как уже указывалось, воз-
никает проблема фильтрации полезного излучения. Используются
четыре основных типа фильтров: поглощающие (кристалли-
ческий кварц, кристаллы галогенидов щелочных металлов, плас-
тинки из некоторых полупроводниковых материалов, например
Ge, InAs, InSb и т. д.), отражающие (ионные кристаллы-
фильтры остаточных лучей, решетки-эшелетты), рассеиваю-
щие (металлические сетки, алюминиевые пластинки с шерохо-
ватой поверхностью и др.), интерференционные (плоскопа-
раллельные слои из прозрачных материалов с разными
показателями преломления).
Как приемники излучения в спектрометрах для средней ИК
области используются ч у в ст в и т е л ь н ы е термопары («тер-
мостолбики») или болометры, построенные по принципу термо-
метров сопротивления. Площадки тепловых приемников порядка
от десятых до целых мм2 для большей эффективности покрываются
чернью, и приемник помещается в эвакуированный корпус с ок-
ном, прозрачным для ИК излучения (KBr, CsI).
К тепловым приемникам относится также пневматический
или оптико-акустический приемник. (я ч е й к а Го ле я),
в котором под действием излучения происходит тепловое расши-
рение газа. Газ помещается в зачерненной камере с гибкой
стенкой, имеющей зеркальное внешнее покрытие. Движение отра-
женного зеркалом светового луча регистрируется фотоэлементом.
Этот приемник изготовляется обычно для длинноволновой ИК
268
Рис. XII.2. Принципиальная опти-
ческая схема интерферометра
Майкельсона (без коллимации
пучков)
области, где используется также
другая группа приемников: кван-
товые или фотонные, например,
такие п р и е м н и к и с фотопро-
водимостью, как Ge, легирован-
ный бором или сурьмой, с низкоча-
стотным пределом 100 см-1, или
InSb — ниже 50 см-1, и др.
В фурье-спектроскопии использу-
ются в основном три типа и нте р-
ферометров: Фабр и- Пер о,
Майкельсона и ламелляр-
ный интерферометр. Не останавли-
ваясь на особенностях и устройстве
отдельных типов интерферометров,
следует лишь отметить, что в основу конструкции всех этих прибо-
ров положено явление интерференции волн электромагнитного из-
лучения. Принципы их действия также существенно не отличаются
(см. гл. V).
На рис. XII.2 в качестве примера представлена принципиальная
оптическая схема фурье-спектрометра, построенного по принципу
Майкельсона. Поток ИК излучения от источника 1, модулирован-
ный прерывателем 2, делится светоделителем 4 на два пучка. Один
из них направляется на зеркало 3, которое (6вязано.=микрщметри-
ческон передачей с двигателем. гф может поступательно переме-
щаться с определенной длиной пробега и возвращаться в исход-
ное положение. Отраженный от этого зеркала пучок^'интерфери-
рует,(Имея заданную зеркалом 3 разность х'одй} с пучком, отражен-
ным от закрепленного зеркала 5? Дальше излучение>фокусируется
линзами 6 на приемнике 8’г проходя "через “исследуемый образец,
помещенный в кюветное отделение 7. При движении зеркала З и
интерференции пучков с (изменяющейся'- разностью ходЗ) происхо-
дит сканирование в определенном спектральном диапазоне.
““ Регистрируемая интерферограмма представляет зависи-
мость сигнала от разности хода пучков и является функцией
энергии источника, видоизмененной поглощением образца. Фурье-
преобразование полученной интерферограммы, проводимое по за-
данной программе мини-ЭВМ, входящей в комплект современных
фурье-спектрометров, дает результирующий спектр поглощения
исследуемого образца. 1Спектральный интервал, который доступен
для изучения, определяется используемым светоделителем. Чтобы
охватить всю ИК область, необходимо несколько сменных свето-
делителей, которые бывают в виде металлических сеток, пленок и
диэлектрических покрытий на твердых подложках. Для длинновол-
новой ИК области светоделителем может служить, например, плен-
ка из лавсана (полиэтилентерефталата), в области средних частот
в качестве подложек для нанесения светоделительных покрытий
из материалов с высоким показателем преломления (например,
германия) используются пластинки из кристаллов CsI и КВг, а в
ближней ИК области — флюорит CaF2 с покрытием из Fe2O3. По-
лученный на приборе спектр остается в памяти ЭВМ и по команде
может быть в нужном виде выдан или на экран дисплея, или для
записи на бумаге.
Фурье-спектроскопия имеет ряд больших достоинств. Два глав-
ных преимущества интерферометров перед обычными спектроме-
I трами заключаются в следующем. Во-первых, это выигрыш в энер-
гии за счет того, что при сканировании в каждый момент времени
на приемник попадает излучение всего исследуемого спектраль-
ДюгсГдйапазона длин волн, а не узкий его участок, определяемый
в' монохроматоре обычного прибора диспергирующей системой и
щелями. Иными словами, в интерферометре в течение всего вре-
мени сканирования получается информация одновременно обо всем
исследуемом спектральном диапазоне, а в обычном спектрометре
в разные моменты времени получается информация только об
узких спектральных полосах исследуемого диапазона. Данное преи-
мущество интерферометров особенно важно в длинноволновой об-
ласти, где интенсивность излучения источника мала и Отношение
сигнала к шуму является лимитирующим'фактором.
Во-вторых, большой выигрыш дает возможность повышения
разрешающей силы интерферометра без уменьшения потока лучи-
стой энергии. Разрешающая способность фурье-спектрометра про-
порциональна максимальной разности хода пучков и, чтобы по- (
‘ высить, например, вдвое разрешение спектра, нужно просто удвоить !
длину перемещения зеркала, т. е. X время регистрацииГНё’накла-"
дываёт больших" ограничений'нД-разрешение апертура, и можно
использовать большие телесные углы как у источника, так и у
приемника. У дифракционных спектрометров разрешение находится
в обратно "пропорциональной зависимости от ширины щелеД,' а
энергия, попадающая на приемник, пропорциональна квадрату
площади двух одинаковых щелей. Если вдвое уменьшить ширину
щелей (для повышения разрешения), то сигнал уменьшится в
четыре раза и для сохранения отношения сигнала к шуму время
регистрации необходимо увеличить в 16 раз.
В интерферометрах проще, чем в дифракционных спектроме-
трах, осуществляется фильтрация излучения нужного спектраль-
ного диапазона, т. е. значительно упрощается проблема устра-
нения паразитного или рассеянного света.
Указанные преимущества обеспечивают такие достоинства
фурье-спектроскопии, как очень высокая чувствительность и точ-
ность измерений интенсивности, особенно при многократном ска-
нировании и накоплении сигнала# очень высокое разрешение (до'
10~-’ см-1) "ивысокая точность определения волновых чисел; быст-
родействие, т. е. возможность быстрого исследования широкой
, спектральной области (время сканирования для интервала в не-
• сколько сотен см 1 составляет менее 1 с), и др. Эти достоинства и
"определяют большие, зачастую уникальные возможности фурье-
спектроскопии, которая уже находит широкое применение в раз-
личных направлениях науки и техники. Производятся приборы,
270
охватывающие диапазон от видимой области до миллиметровых
длин волн.
Примером многоканального диспергирующего спектрометра
является прибор с преобразованием Адамара. Его
принципиальное отличие от обычного монохроматора с дифрак-
ционной решеткой связано с тем, что вместо выходной щели в
фокальной плоскости, где излучение разложено в спектр, имеется
полевая диафрагма и так называемая маска или матрица Ада-
мара с множеством вертикальных щелей. Через диафрагму и маску
проходит весь изучаемый интервал длин волн, и этим обеспечи-
вается большой выгрыш в энергии, попадающей на приемник,
по сравнению с обычным сканирующим спектрометром. Маска
перемещается в горизонтальной плоскости шаговым двигателем
и происходит кодирование спектра по Адамару. Сигнал с прием-
ника, соответствующий суммарной интенсивности для всех длин
волн, усиливается, поступает в ЭВМ, декодируется по преобразо-
ванию Адамара и выдается в виде спектральной кривой обычного
вида. Для всестороннего сравнения этого типа приборов с другими
типами, например, фурье-спектрометрами, пока еще опубликова-
но недостаточно данных.
Если обычные сканирующие спектрометры только в послед-
нее время стали сочетаться с ЭВМ, но долго использовались и
по-прежнему применяются для получения спектров и без ЭВМ, то
появление и применение серийных спектрометров с преобразова-
ниями Фурье и Адамара стало возможным только благодаря сов-
ременной вычислительной и микропроцессорной технике, без кото-
рых они немыслимы. Сложность и прецизионность конструкции
этих приборов влечет возможность появления многих неисправ-
ностей при неумелом и неосторожном обращении, т. е. требует
более высокой квалификации и ответственности операторов.
1.2. Характер и подготовка образцов
ИК спектроскопия выгодно отличается от многих других физи-
ческих методов исследования веществ большим разнообразием
доступных для изучения объектов и методик подготовки образцов.
Почти нет никаких ограничений, связанных с физическим состоя-
нием вещества, а образец при необходимости может быть под-
лостью сохранен в неизменном виде.
Газы и пары исследуют в специальных газовых кюве-
тах. Обычная кювета представляет цилиндр длиной ~ 10 см с
прозрачными окнами на торцах и вакуумными кранами для от-
качки и заполнения исследуемым газом. Имеются также много-
ходовые газовые кюветы, в которых с помощью системы
зеркал обеспечивается многократное прохождение пучка излуче-
ния через слой газа и общая длина пути лучей через исследуемый
газ может достигать в зависимости от конструкции кюветы от
одного до десятков метров. Применение таких кювет необходимо
для анализа следовых количеств газов и при исследованиях труд-
нолетучих веществ с малой упругостью паров. Как обычные, так
и многоходовые газовые кюветы могут иметь систему обогрева
и термостатирования. Получение спектров при высоких темпера-
турах требует специальных печей.
Количественные измерения интегральных интенсивностей полос
в колебательно-вращательных ИК спектрах проводят с добавкой
к исследуемому газу постороннего инертного газа (N2, Аг и т. и.),
стандартизуя образцы по давлению, например, доводя его до ат-
мосферного или выше.
Для исследования спектров «изолированных» молекул исполь-
зуется также техника матричной изоляции при низких
температурах, когда вращательная структура полос отсутствует.
Жидкие образцы исследуют или в виде раствора, или в
чистом виде в специальных жидкостных кюветах с задан-
ной толщиной слоя (от долей мм до нескольких мкм), или в виде
пленки, получаемой при сжимании капель образца между пластин-
ками из прозрачного материала. Толщина пленки при сжатии
1...10 мг образца составляет несколько мкм. Легко летучие жидко-
сти исследуются в герметичных кюветах. Жидкостные кюветы с
постоянной толщиной слоя бывают неразборные и разборные.
Толщина слоя задается прокладкой или, в разборных кюветах,
стаканом с вкладышем, зажимаемым между окнами. '
Имеются также кюветы переменной толщины с мик-
рометрическим винтовым устройством, позволяющим плавно ме-
нять толщину слоя. Эти кюветы особенно полезны при изучении
растворов, когда необходимо точно скомпенсировать поглощение
растворителя. Исследуемый раствор (обычно с невысокой кон-
центрацией растворенного вещества) помещают в кювету постоян-
ной толщины (0,1...1 мм и более), а в пучке сравнения ставят
для компенсации поглощения растворителя такую же кювету или,
что лучше, кювету переменной толщины с растворителем.
Наиболее широко применяемые в ИК спектроскопии раствори-
тели и области их собственного поглощения указаны на рис.
XI 1.3. На обычных сканирующих спектрометрах, даже двухлуче-
вых, невозможно получить спектр растворенного вещества в тех
интервалах длин волн, где имеется сильное собственное поглоще-
ние растворителя. Поэтому для регистрации полного спектра ра-
створенного образца чаще всего приходится готовить несколько
растворов в разных растворителях.
Вода является мало удобным для ИК спектроскопии раство-
рителем, во-первых, из-за того, что в ней устойчивы лишь не-
многие из прозрачных в ИК области материалов (см. табл. XII. 1),
и, во-вторых, из-за очень сильного собственного поглощения. В то
же время использование воды в качестве растворителя бывает
иногда совершенно необходимым, например в биологических ис-
следованиях. В значительной области (от 900 до 3100 см-1) при
толщине слоя 10...25 мкм водные растворы можно успешно иссле-
довать, за исключением узкого интервала вблизи ~ 1650 см-1,
где расположена полоса поглощения деформационных колебаний
272
Рис. XII.3. Растворители, используемые в колебательной спектроскопии:
зачерненные участки показывают полностью маскируемые поглощением или рассеянием. са-
мого растворителя области спектра (КР или ПК); участки, обведенные рамкой, — частично-
маскируемые области
воды. Этот интервал, как и область валентных колебаний Н2О^
можно изучать, используя раствор в тяжелой воде D2O. В облас-
тях ниже ~900 см-1 и выше ~3100 с'.г-1 исследовать водные
растворы возможно лишь при трудно получаемых и воспроизво-
димых толщинах слоев порядка ~2,5 мкм и менее. Поглощение
образцов при таких толщинах и обычных концентрациях, конечно,,
очень слабое.
При сочетании ИК спектрометра с ЭВМ. и накоплении сигнала при много-
кратном сканировании спектра, а особенно на фурье-спектрометрах оказываете»:
возможным регистрировать ничтожные доли излучения источника, попадающего»
на приемник. Даже при достаточно большой толщине слоя раствора, когда
поглощением растворителя (например, Н2О) перекрыт какой-то широкий интер-
вал спектра, но пропускание составляет .ходя,, бы сотые доли процента, в этом
интервале на фурье-спектрометре все-таки можно получить четкий спектр рас-
творенного даже в небольшой концентрации вещества над кривой поглощения
растворителя по однолучевой или по двухлучевой схеме. Этот спектр растяги-
вается по ординате (интенсивность) на всю шкалу, возможность чего на совре-
менных приборах предусмотрена (рис. XII.4).
Выбирая растворитель, необходимо иметь в виду не только»
его прозрачность, но также инертность по отношению к образцу,
растворимость в нем исследуемого вещества, токсичность (напри-
мер, пары СС14 и особенно CS2 весьма токсичны) и т. д.
При разбавлении и изменении растворителей спектры могут
сильно меняться. Поэтому важно стандартизировать условия реги-
страции спектров, а каждый приводимый где-либо спектр должен
сопровождаться указанием всех условий, при которых был полу-
чен.
Иногда при исследовании жидких веществ для приготовления
образцов применяют методику с набуханием полимер-
ных пленок. Например, в длинноволновой ИК области для
этих целей используют полиэтилен.
A
Рис. XII,4. ИК спектры НПВО, получен-
ные на фурье-спектрометре с жидкост-
ной кюветой и элементом из KBS-5:
1 — спектр водного раствора неомицина; 2 —
спектр воды; 3 — спектр неомицина после вы-
читания спектра поглощения воды и растяж-
ки шкалы оптической плотности
Из-за сильного поглощения
веществ в ИК области и соот-
ветственно малых толщин сло-
ев в жидкостных кюветах и
малых концентраций растворов
при количественном анализе
предъявляются достаточно вы-
сокие требования к качеству
кювет и точности измерений
толщины слоя. Должна быть
обеспечена плоскопараллель-
ность как самих окон кюветы,
так и относительного их рас-
положения, окна должны быть
хорошо отполированы и перио-
дически проверяться на нали-
чие поверхностных загрязне-
ний записью спектра. Плоско-
стность, т. е. отсутствие вогну-
тости или выпуклости поверх-
ности пластинки,при переполи-
ровке проверяется по интерфе-
ренционной картине в монохро-
матическом свете, например
натриевой лампы. Должно на-
блюдаться несколько (3 ... 4)
интерференционных колец пра-
вильной формы.
Толщину кювет, как и тон-
ких пленок, в интервале от сотых до нескольких десятых мм точнее
всего определяют также по интерференционной картине. Интерфе-
рограмму получают при записи спектра пустой кюветы или пленки
в виде характерной гребенки чередующихся максимумов и миниму-
мов, которые возникают из-за интерференции (усиления и ослаб-
ления) отраженного от разных поверхностей (внутренних в пустой
кювете) излучения. Толщина слоя точно рассчитывается по фор-
муле
п 10
2 <0^ — о>2
где I — толщина, мм; сщ и ®2— волновые числа двух удаленных
друг от друга интерференционных максимумов, см-1; п — число
максимумов от ®i до ®2 при нумерации с нуля.
Другой, менее точный способ определения толщины, если нельзя
использовать первый, может быть основан на измерении оптической
плотности стандартных жидкостей (СбН12, СС1Д при наличии ка-
либровки по кюветам с известной толщиной слоев. Наконец, можно
с помощью микрометра определить толщину прокладки, как и лю-
бой пленки, или толщину вкладыша и высоту стакана сборных кю-
вет, но это может удовлетворять при слоях от нескольких деся-
тых мм и больше.
Специфика подготовки жидких образцов для исследования в
длинноволновой ИК области состоит только в отличии прозрачных
в этой области материалов для окон кювет (см. табл. XII.1). Что-
касается растворителей, то кроме представленных на рис. XII.3 при-
меняются так же, как и в других областях, ацетон, диоксан, бен-
зол, дихлорэтан, нитрометан, бромоформ и др.
Твердые образцы могут готовиться для исследования ИК.
спектров различными способами. Наиболее распространенными яв-
ляются: прессование таблеток с КВг или другими напол-
нителями, приготовление суспензий в вазелиновом мас-
ле или других иммерсионных средах. Иногда исследуются также
пленки, получаемые осаждением из растворов на про-
зрачной подложке, и тонкие пленки расплавов, например,
удерживаемых на металлических сетках силами поверхностного
натяжения. Удобными объектами являются пластичные материалы,
например полимерные пленки. При наличии дополнительных уст-
ройств могут исследоваться маленькие монокристаллы, волокна и
другие микрообразцы.
Тонкие покрытия (~0,01 мм) на гладких зеркальных поверх-
ностях (металлических), полученные, например, напылением при
возгонке, можно изучать по принципу зеркального отражения. При
очень тонких слоях (меньше длины волны ИК излучения) исполь-
зуется скользящее падение и многократное отражение. В послед-
нее время в ближней ИК области начали применять также технику
диффузного отражения.
Об исследованиях растворов в жидкостных кюветах уже говори-
лось выше, а готовится ли раствор твердого, жидкого вещества или
газа — принципиальных отличий нет. При исследовании пленок раз-
ной природы следует иметь в виду, что на получаемый от них
спектр поглощения может накладываться, по крайней мере, в неко-
торых областях (в зависимости от толщины и плоскопараллельно-
сти пленки) мешающая интерференционная картина (см. выше).
Для съемки ИК спектров порошкообразных и кристаллических
веществ используют в основном методики прессования таблеток и
приготовления взвесей. В обеих методиках образец измельчается
или в специальной мельнице, например шаровой, или растирается
пестиком в агатовой (яшмовой) ступке. Получить доброкачествен-
ный спектр поглощения мелкодисперсного порошка не представ-
ляется возможным из-за сильного рассеяния излучения микроча-
стицами. Поэтому требуется использование иммерсионных сред, за-
полняющих пустоты между частичками исследуемого вещества.
Желательно, чтобы среда имела близкий с образцом показатель
преломления. В качестве твердой иммерсионной среды использу-
ются, например, КВг и другие соли, а для длинноволновой обла-
сти — порошкообразный полиэтилен или его смесь с парафином
и т. д. Смесь твердого образца и соли в соотношении примерно
1 : 100 (например, 2—3 мг вещества и 200—300 мг соли) тщатель-
ко растирается и перемешивается, причем иногда лучшего резуль-
тата можно достигнуть добавкой небольшого количества легко ле-
тучего нейтрального растворителя. Смесь переносят затем в спе-
циальную пресс-форму и под большим давлением (~109 Па),
иногда при откачке вакуумным насосом, прессуют таблетку (круг-
лой или прямоугольной формы). Это, как правило, прочные, про-
зрачные не только для ИК, но и для видимого и УФ излучения
пластинки, позволяющие получить высококачественный спектр по-
глощения исследуемого вещества, если нет взаимодействия его с
иммерсионной средой.
Суспензии готовят обычно с вазелиновым маслом, представляю-
щим высококипящую нефтяную фракцию насыщенных углеводоро-
дов (называют также «нуйол» или минеральное масло). Его недо-
статком является сильное поглощение в области колебаний групп,
-содержащих СН-связи (2700 ... 3000 и 1340 ... 1500 см-1). Исполь-
зование в области частот выше 1340 см-1, например хлорированных
масел (гексахлорбутадиен и др.), снимает эти трудности. Растер-
тый в агатовой ступке в течение 5 ... 10 мин образец массой
5 ... 20 мг (размер частиц при этом должен быть меньше длины
волны ИК излучения) смачивается 1 ... 2 каплями иммерсионной
.жидкости. После этого растирание продолжается до получения од-
нородной массы. Полученная суспензия (паста) наносится на соле-
вое окно и прижимается другим окном до равномерного распреде-
ления без образования пузырьков.
1.3. Дополнительные приспособления.
Исследования специфических образцов
В ИК спектроскопии в зависимости от поставленных задач и
«объектов исследования применяется множество разнообразных до-
полнительных устройств и приспособлений.
Для изучения температурной зависимости спектров, различных
.агрегатных состояний веществ и полиморфизма используются раз-
личные конструкции криостатов, печей и термостатирующих си-
стем.
Низкотемпературные исследования связаны с необходимостью
изоляции кюветы или подложки с образцом от атмосферной влаги,
т. е. возможности ее конденсации на окнах (образце), что может
достигаться вакуумированием внешнего сосуда криостата с соле-
выми окнами или помещением охлаждаемой кюветы в пенопласто-
вую рубашку с подогреваемыми окнами.
Для термостатирования при низких температурах наряду с
хладагентом (жидкие N2 или Не) в криостате предусматривается
наличие системы электрообогрева с соответствующим регулировоч-
ным реле. Имеются также системы с термостатированием при по-
мощи регулируемого по скорости подачи потока холодных паров
сжиженных газов. Термостатирование при комнатной и близких к
ней температурах может обеспечиваться как системой электрообо-
276
грева, так и рубашкой с циркулирующей от термостата жид-
костью.
Высокотемпературные исследования ИК спектров требуют весь-
ма сложных конструкций печей, изоляции их от спектрометра во
избежание его нагрева и устранения эффектов, связанных с тем,
что сам образец при сильном нагревании является источником до-
статочно интенсивного для создания помех ИК излучения.
Важным при исследованиях ИК спектров неустойчивых частиц
(молекул, молекулярных ионов, радикалов, комплексов) является
метод матричной изоляции, суть которого заключается
в следующем. Газообразный образец, нагретый до высоких темпе-
ратур, из печки намораживается на охлажденное обычно жидким
гелием окошко вместе с большим количеством инертного газа (N2,
Аг и т. д.). Исследуемые молекулы в образуемой таким путем
инертной матрице находятся в очень низкой концентрации и не вза-
имодействуют между собой. С другой стороны, в отличие от газо-
вой фазы в ИК спектрах матрично-изолированных молекул отсут-
ствует, как уже отмечалось, вращательная структура полос.
Аналогичным образом могут намораживаться в матрице слож-
ные газовые смеси для идентификации отдельных компонентов, изу-
чения продуктов реакционных смесей низкотемпературной плазмы
и т. п.
Специальная техника для исследования ИК спектров микрооб-
разцов производится серийно, а иногда конструируется самими
экспериментаторами. К таким специальным устройствам относятся
микрокюветы (газовые — малого объема и жидкостные), микро-
пресс-формы для изготовления таблеток, микроскопы и микрокон-
денсоры для фокусировки и уменьшения площади сечения пучка
излучения в месте расположения микрообразца и т. д. Уникальные
возможности исследования микрообъектов практически без всяких
дополнительных приспособлений предоставляет фурье-спектроско-
пия. Поскольку отношение сигнала к шуму в фурье-спектрометрах
очень высоко, можно регистрировать хорошие спектры при очень
малых отверстиях — до десятых долей мм. Пучок, проходящий че-
рез микрообразец, просто диафрагмируется до размеров образца.
При этом из-за быстрого сканирования нет проблем, связанных с
разогреванием образца пучком излучения, которые возникают при
использовании микроконденсоров. Успешно исследуются таким об-
разом спектры микрокристаллов, волокон, образцов в специальных
микрокюветах и т. д.
Большой интерес для некоторых целей, например, при изучении
фазовых диаграмм, равновесий и т. п. представляют исследования
ИК спектров при высоких давлениях (до 109 ... 1010 Па). В каче-
стве материалов для изготовления окон (наковален) кювет высо-
кого давления могут служить технические алмазы (безазотные ти-
па П-а), прозрачные практически во всей ИК области, сапфир —
с ограниченной областью пропускания (см. табл. XII.1) и некото-
рые другие. Диаметр отверстия таких кювет обычно мал — порядка
одного или долей мм (определяется, например, размером алма-
зов), что также требует использования или микротехники, или
фурье-спектроскопии.
Для проведения поляризационных измерений в ИК спектроско-
пии, т. е. определения дихроичного отношения, например, при ис-
следовании ориентированных образцов используются поляризато-
ры ИК излучения. Это обычно пропускающие поляризаторы, со-
стоящие из нескольких пластинок селена или, чаще, хлорида се-
ребра, которые располагаются по отношению к направлению па-
дающего излучения под углом Брюстера а:
tga=/z,
где п — показатель преломления материала пластинок. В слу-
чае AgCl а=63,5°, а степень поляризации излучения при шести та-
ких пластинках в поляризаторе составляет 95 ... 98%. При поля-
ризационных измерениях необходимо учитывать также приборную
поляризацию, а если возможно, образец не следует помещать меж-
ду поляризатором и монохроматором, а лучше до поляризатора
(или после выходной щели).
2. НАРУШЕННОЕ ПОЛНОЕ ВНУТРЕННЕЕ ОТРАЖЕНИЕ (НПВО)
Разработанный сравнительно недавно метод спектроско-
пии нарушенного полного внутреннего отраже-
ния (НПВО) значительно расширил круг объектов, доступных
для исследования спектров в оптической области. Известно, что лу-
чи света, падая на границу раздела двух сред с показателями пре-
ломления П\ и П2 (^i>/i2), будут частично отражаться от этой гра-
ницы, а частично проходить через нее, как показано на рис. XII.5
(луч а). Коэффициент отражения 7? = ///0, где /, /0 — интенсивно-
сти света, отраженного и падающего на границу раздела соответ-
ственно. Преломленный луч подчиняется закону Снеллиуса:
sin 6 = я2 sin ср, (XII. 1)
sin 0/sin ср = zz.2/zz.1 == zz.219
где /г21 — относительный показатель преломления.
При увеличении угла падения 0 наступит момент, когда угол ср
станет равным 90°, т. е. преломленный луч будет распространяться
вдоль границы раздела двух сред (рис. XII.5, луч б). Такой угол
падения называется критическим, и из (XII. 1), если sin ф= 1„
sin0Kp = n2i. При всех 0>0кр преломленный луч отсутствует, R = 1
и наблюдается полное внутреннее отражение (ПВО), как показано
на рис. XII.5 (луч в).
Согласно электромагнитной теории света оказывается, что при
явлении ПВО электромагнитное поле излучения возникает и в
оптически менее плотной среде (/г2). Это подтверждается и экспе-
риментально. Если, например, варьировать зазор между двумя
призмами, то можно наблюдать изменение соотношения отражен-
ного и пропущенного излучения, как показано на рис. XII.6.
Глубина проникновения све-
та при ПВО в оптически менее
плотную среду dn имеет поря-
док длины волны и определяет-
ся как расстояние от границы
Рис. ХП.6. Схема эксперимента Шефера
и Гросса с внутренним отражением
Рис. XI 1.5. Ход лучей в области
границы раздела двух сред (ni>.
>п2)
раздела, на котором амплитуда колебаний электрического векто-
ра световой волны уменьшается в е раз; она зависит от угла паде-
ния 0 и относительного показателя преломления п2ь
________Н_________
2л; (sin2 0 — п^)172
(XII.2)
где — длина волны излучения в оптически более плотной среде.
Если оптически менее плотная среда обладает селективным по-
глощением (некоторых длин волн), то спектральный состав света,
падающего на границу раздела и претерпевающего полное внут-
реннее отражение, будет различен. Это явление и получило назва-
ние нарушенного полного внутреннего отраже-
ния (НПВО). В 1960 г. был предложен (Фаренфорт и Харрик)
основанный на этом явлении новый метод оптической
спектроскопии НПВО *. Если сплошное излучение, прошед-
шее через более плотную среду и претерпевшее НПВО на границе
раздела с поглощающей оптически менее плотной средой, напра-
вить в щель спектрометра, то можно зарегистрировать спектр, по-
чти идентичный обычному спектру пропускания менее плотной сре-
ды изучаемого образца (рис. XII.7).
* В зарубежной литературе приняты названия ослабленное ATR
(attenuated total reflection) и расстроенное FTR (frustrated total reflec-
tion) полное внутреннее отражение.
л
2 „4 6 8 10 12 14 -16 A,MKNf
Рис. XII.7. ИК спектры бензола:
/— спектр поглощения (Z=25 мкм); 2 — спектр МНПВО (призма KRS-5, 0 = 52°, W = 20)
Важно отметить, что при показателе поглощения образца
в спектроскопии НПВО применим закон, аналогичный закону Буге-
ра — Ламберта — Бера:
где 10К и I},— интенсивности падающего и отраженного лучей; k\—
бугеровский показатель поглощения (4nvx2); с — концентрация
поглощающих частиц; М —число отражений, (ДГ=1 соответствует
однократному отражению, т. е. просто методу НПВО; при М>1
имеем многократное отражение, т. е. метод МНПВО —
многократного НПВО, обычно УУ=20 ... 50); б/Эф х— эффективная
толщина слоя, эквивалентная по физическому смыслу толщине по-
глощающего слоя в абсорбционной спектроскопии. При геометри-
ческой толщине I образца намного больше длины волны к величи-
на йэФг растет с увеличением К (как и rfn); она зависит также от
поляризации падающего излучения (с?эф11>^эф-Ь). Таким образом,
спектроскопия НПВО может применяться и для количественного
анализа.
Для получения спектров НПВО и МНПВО на серийных спектро-
метрах производятся специальные приставки, помещаемые в кю-
ветное отделение. Основная часть приставки — оптический элемент
НПВО, прозрачный в нужной области спектра и создающий необ-
ходимые условия для получения спектра НПВО. Значения показа-
телей преломления типичных объектов исследования лежат в пре-
делах 1,35 ... 1,55. Некоторые используемые для изготовления эле-
ментов НПВО материалы и их характеристики представлены в
табл. XII.1.
Рис. XII.8. Некоторые типы оптических элементов:
а — твердотельный элемент НПВО с переменным углом падения (/ — образец): б — жидко-
стной элемент НПВО с конфигурацией призмы Дове (/ — образец; 2 — жидкость; 3 — кюве-
та); в — твердотельный элемент МНПВО однократного прохождения с переменным, углом
падения (/ — образец); г — твердотельный элемент МНПВО двойного прохождения с посто-
янным углом падения, погруженный в исследуемую среду (/ — образец)
Элементы НПВО однократного отражения представляют обыч-
но призму или полуцилиндр (реже полусферу). Полуцилиндриче-
ский элемент позволяет плавно менять угол падения, а следова-
тельно, и глубину проникновения dn (XI 1.2) излучения для дости-
жения оптимальных условий получения спектров. Для увеличения
интенсивности и повышения контрастности спектров используют ме-
тод спектроскопии МНПВО. Некоторые из элементов НПВО и
МНПВО показаны на рис. XII.8. Существуют элементы с однократ-
ным, двойным и многократным прохождением лучей. Преимуще-
ством элемента двойного прохождения типа, показанного на
рис. XII.8, г, является наличие свободного конца, который можно
погрузить, например, в жидкость или мелкий порошок и получить
спектр без специальной подготовки образца. Элемент такого типа
в принципе может быть изготовлен в виде иглы для введения в жи-
вые ткани и изучения их спектров, например, с применением гиб-
ких волокон-световодов.
Кроме твердотельных элементов НПВО иногда применяются их
жидкостные аналоги, достоинство которых состоит в том, что они
позволяют получать спектры образцов с шероховатой поверх-
ностью. При регистрации спектров НПВО контакт между объектом
исследования и элементом НПВО играет большую роль. Оптималь-
ными являются условия, когда достигается оптический контакт.
При его отсутствии может происходить заметное искажение спект-
ров и исключается возможность проведения количественных изме-
рений. Рекомендуется поэтому для исследования жидких и пластич-
ных образцов применять твердотельные элементы НПВО, а в
случае твердых образцов — или жидкостные элементы, или твердо-
тельные, но при тщательной обработке поверхности, дополнитель-
ном поджатии или использовании высокопреломляющих иммерси-
онных жидкостей в качестве интермедиатов (прослоек).
Поскольку интенсивность в спектрах НПВО зависит как от
свойств образца, так и, особенно, от условий эксперимента, очень
важен правильный выбор последних. При исследовании слабо по-
глощающих веществ (х2<0,1) следует выбирать элемент МНПВО
из материалов с относительно невысоким показателем преломле-
ния («1 = 2,0 ... 2,4), работать при небольших углах падения (0 =
= 30 ... 45°), с числом отражений А=20 ... 50. При сильно погло-
щающих образцах целесообразно использовать элементы однократ-
ного отражения («1>2,4) и большие углы падения (0 = 50 ... 70°).
Конкретные области науки и техники, в которых спектроскопия
МПВО может успешно применяться и уже применяется, весьма
разнообразны. Это одна из новых экспериментальных методик ко-
лебательной (ИК) и электронной (УФ) спектроскопии, так что
принципиальные задачи, решаемые этими методами, остаются теми
же, что в обычных условиях эксперимента. Важны новые возмож-
ности, открываемые в отношении исследования и анализа объектов,
работа с которыми в обычной абсорбционной спектроскопии за-
труднена или даже вообще невозможна. Это касается исследова-
ния массивных тел, живых тканей (in vivo), получения ИК
спектров водных и агрессивных растворов, вязких и пластичных ма-
териалов и т. д.
Методические возможности позволяют использовать спектроско-
пию НПВО не только в научно-исследовательской работе, но и в
различных отраслях промышленности — химической, пищевой, фар-
мацевтической и др. — для контроля качества продукции без раз-
рушения образцов.
Аналитические преимущества спектроскопии МНПВО проявля-
ются при обнаружении очень малого содержания токсичных ве-
ществ в воздухе и сточных водах, что делает методику перспектив-
ной для работ, связанных с проблемой охраны окружающей среды.
Если покрыть рабочие поверхности элемента МНПВО тонким сло-
ем платины (20 ... 30 нм), которая служит адсорбентом и не ме-
шает прохождению излучения за границу раздела двух сред, то в
воздухе легко обнаруживаются, например, фосфорорганические
соединения при концентрации 10-7 г/л. Адсорбированное вещество
затем очень просто удаляется с поверхности (например, продув-
кой), и анализатор может использоваться многократно.
Метод НПВО неоценим при проведении различного рода экс-
пертиз, применяется в криминалистике, клинических исследовани-
ях, анализе косметических изделий и т. п.
Представление о принципах спектроскопии НПВО, а в значи-
тельной мере и фантазия экспериментатора могут определить еще
очень много новых творческих применений этого перспективного
метода.
3. ТЕХНИКА СПЕКТРОСКОПИИ КР
3.1. Спектральная аппаратура и образцы
Интенсивность спектров обычного (спонтанного) ком-
бинационного рассеяния света относительно невысока, и для их
изучения требуются приборы, обладающие большой светосилой.
Если раньше часто использовались призменные спектрографы с
фотографической регистрацией спектров или призменные спектро-
метры с фотоэлектрической регистрацией, то теперь применяются
обычно дифракционные спектрометры с двойной или даже тройной
монохроматизацией. Примером двойного монохроматора с дифрак-
ционными решетками являются отечественный спектрометр ДФС-
24, имеющий относительное отверстие 1 : 5,3 и обратную линейную
дисперсию 0,5 нм/мм, и последняя усовершенствованная модель
ДФС-52. Типичная блок-схема сканирующего прибора для получе-
ния спектров КР показана на рис. XII.9.
Важнейшим элементом в спектроскопии КР является достаточ-
но мощный источник монохроматического излуче-
ния, поскольку интенсивность линий КР зависит от интенсивно-
сти возбуждающего света. Долгое время в качестве источника воз-
буждающего излучения использовали, а иногда используют и сей-
час ртутные лампы цилиндрической формы в специальном
осветителе для образца или спиральной формы (типа «Торонто»).
Рис. ХП.9. Блок-схема прибора для изучения спектров КР:
/ — лазер; 2 — зеркала; 3 — образец в осветителе с конденсором; 4 — анализатор; 5 — вход-
ная, промежуточная и выходная щели двойного монохроматора; 6 — объективы; 7 — дифрак-
ционные решетки; 8 — фотоэлектронный умножитель; 9 — усилитель; 10— регистрирующее
устройство (или счет фотонов)
С помощью фильтров из ртутного спектра выделяют одну из ли-
ний, обычно с длиной волны К 435,8 нм (синяя), а иногда 546,1 нм
(зеленая), в частности, для окрашенных в желтый цвет веществ.
Фильтры изготовляются из специальных стекол или ими служат
концентрированные водные растворы нитрита натрия — для синей
линии и хромата калия — для зеленой линии.
Настоящую революцию в спектроскопии КР произвело приме-
нение оптических квантовых генераторов — лазеров*. Давая
мощное, монохроматическое, когерентное и поляризованное излу-
чение, лазер явился почти идеальным источником для возбуждения
спектров КР. Из газовых лазеров непрерывного действия в спект-
роскопии КР первым стал применяться гелий-неоновый лазер (со-
отношение газов Не — Ne в смеси 1 : 7) с длиной волны 632,8 нм.
Уже на его примере можно проиллюстрировать огромное преиму-
щество перед ртутной лампой: при мощности этого лазера 30 мВт
(мощность Не — Ne-лазера достигает до 200 мВт) он дает интен-
сивность КР такую же, какую можно получить от ртутной лампы
с мощностью 2 кВт. Ширина линии лазера не превышает 0,1 см-1,
а расхождение луча составляет ничтожные доли градуса.
В дальнейшем нашли широкое применение аргоновый (Аг+, 7 =
= 488,0 нм; 514,5 нм и др.) и криптоновый (Кг+, 7=530,9 нм;
647,1 нм) лазеры, дающие большой выигрыш в интенсивности по
сравнению с Не — Ne-лазером. Во-первых, интенсивность растет
обратно пропорционально 74, во-вторых, мощность этих лазеров
может достигать нескольких ватт. В последнее время появились и
начинают применяться импульсные лазеры, в том числе — на кра-
сителях с плавно перестраиваемой длиной волны, которые особен-
но перспективны для изучения резонансного КР света.
Рассеянное излучение обычно собирается конденсором и направ-
ляется в щель монохроматора под углом 90° к падающему на об-
разец лучу, как на рис. XII.9, хотя в принципе могут использовать-
ся схемы, работающие под углами 180 или 45°, а также на про-
свет (0°). В спектроскопии КР большое значение имеет устранение
паразитного рассеянного излучения и флуоресценции образцов.
Отчасти проблема решается применением фильтров и двойной или
большей монохроматизацией с помощью нескольких, иногда смен-
ных дифракционных решеток. В перспективе значительное увеличе-
ние отношения сигнала к шуму может быть достигнуто использо-
ванием последней новинки в технике спектроскопии КР — гологра-
фических решеток.
Регистрация светового потока производится обычно по однолу-
чевой схеме с фотоэлектронным умножителем (ФЭУ) и усилителем
на самопишущем потенциометре. На некоторых приборах преду-
смотрена возможность компенсации и устранения флуктуаций фо-
на источника по двухлучевой схеме. Многие современные спектро-
* Название «лазер» произошло от англ.: Light Amplification by Stimulated
Emission of Radiation, что может быть переведено как усиление света вынужден-
ным испусканием радиации.
Рис. XII.10. Некоторые типы кювет в лазерной спектро-
скопии КР:
а — многоходовая газовая кювета; б — капилляр для жидких и
порошкообразных образцов; в —держатель для твердых микро-
образцов и расплавов
метры могут работать также в режиме счета фотонов, что вместе
с охлаждением ФЭУ значительно повышает чувствительность мето-
да. Если же учесть, что теперь спектрометры сочетаются с мини-
ЭВМ и микропроцессорами, то, помимо усиления сигналов путем
их накопления на счетчике фотонов, оказывается возможным усред-
нение их (сглаживание спектральных кривых) за счет исключения
флуктуаций и устранение фона флуоресценции. Это достигается при-
цифровой обработке измерений и фурье-преобразовании спектра
КР.
При использовании в качестве источника возбуждающего излу-
чения ртутных ламп круг объектов, которые могут изучаться мето-
дом спектроскопии КР, довольно ограничен. Это связано прежде-
всего с необходимостью использования больших количеств образ-
цов и трудностями работы с газами и твердыми образцами. Жид-
кости и растворы в объеме нескольких миллилитров помещаются
при этом в кювету в виде стеклянной трубочки с приклеенным к
торцу плоским окном и зачерненным загнутым другим концом.
В редких исследованиях спектров КР газов при таком возбужде-
нии конструировались как специальные, большого размера и мощ-
ности ртутные лампы, так и объемные газовые кюветы. Спектры
КР кристаллов и порошков удавалось получить с помощью кону-
сообразных кювет и некоторых других специальных устройств.
Высокая мощность и другие указанные достоинства лазерных,
источников позволяют легко исследовать спектры КР веществ в
любых агрегатных состояниях и в микроколичествах. Газы могут
изучаться, например, при возбуждении спектра аргоновым, крип-
тоновым и другими лазерами с использованием многоходовой кю-
веты (рис. XII.10) весьма небольшого объема (от одного до не-
скольких см3). Микроколичества образцов в конденсированных фа-
зах— твердых, порошкообразных, мелкокристаллических (до-
~10-4 г) или жидких (до ~10~4 см3) исследуются в капиллярах
(рис. XII.10) или в открытом виде, например маленькие монокри-
сталлы, волокна и т. и. Не составляет трудностей получение спект-
ров КР водных и других растворов. Некоторые из обычных раство-
рителей с указанием их собственных полос КР представлены на
рис. XII.3.
Следует, однако, иметь в виду, что при мощности лазерного
луча в несколько ватт (иногда даже до 10 и более) возникает
угроза разложения образцов, что часто и происходит, если не при-
нимать специальных мер предосторожности, например вращение
кюветы (образца) вокруг своей оси, исследование жидкости в по-
токе и др. Выбирая для возбуждения спектров КР лазеры с подхо-
дящими длинами волн или меняя X для перестраиваемых лазеров,
можно исследовать всевозможные окрашенные вещества. Значи-
тельные затруднения встречаются лишь при изучении черных или
глубоко окрашенных веществ и соединений, дающих сильную флуо-
ресценцию.
Сочетание лазера с оптическим микроскопом и КР спектромет-
ром, а также с телевизионной техникой для регистрации спектров
и получения изображения привело к разработке такой новинки,
как микрозонд и метод лазерной микрографии для изучения спект-
ров КР микронных образцов и получения изображений объектов в
•свете выбранной линии КР. Это позволяет получать наглядную
картину распределения тех или иных компонентов (веществ) в об-
разце.
Использование мощных импульсных лазеров, позволяющих по-
лучать спектры КР от одного импульса, привело к развитию мето-
дов скоростной спектроскопии КР, которые неоценимы, например,
в кинетических исследованиях (см. гл. XI, 9).
3.2. Резонансное и инверсное КР
Явление резонансного КР (РКР) света, открытое совет-
ским ученым П. П. Шорыгиным в начале 50-х годов, заключается
в следующем. Если частота возбуждающего излучения v0 оказы-
вается близкой к частоте собственного поглощения молекулы,
связанного с разрешенным электронным переходом, то наблюдает-
ся резкое усиление некоторых линий в спектре КР, причем интен-
сивность их может возрасти в тысячи и даже десятки тысяч раз.
Повышение за счет этого чувствительности имеет огромное значение
для аналитических применений метода.
При возбуждении с частотами, близкими к разным полосам по-
глощения вещества, может наблюдаться усиление разных линий КР
и в разной степени. На рис. XII.11 видно, как усиливаются при пе-
реходе от криптонового лазера к аргоновому линии 183 и 247 см-1
полносимметричных колебаний Со — Со в спектре резонансного
КР порошкообразного карбонила кобальта. В резонансных спектрах
КР действуют другие правила отбора по сравнению с обычным
(спонтанным) КР. Сопоставление этих спектров позволяет получать
важные данные для изучения электронно-колебательного взаимо-
действия и структуры молекул в различных электронных состоя-
ниях.
Так, например, методом РКР было
показано, что если в основном элект-
ронном состоянии для молекулы мети-
ламина CH3NH2 в равновесной конфи-
гурации характерна пирамидальная
система связей при атоме азота, то в
первом (самом низком) возбужденном
электронном состоянии равновесная
конфигурация аминогруппы плоская.
В перспективе для количественного
анализа может быть использовано так-
же явление инверсного КР. Оно
наблюдается при одновременном облу-
чении образца источником белого (не-
прерывного) света и лазером с мощ-
ным монохроматическим излучением с
частотой vq. В сплошном спектре пер-
вого источника происходит поглощение
света с частотами vo±vz-, где V/ — ко-
лебательные частоты молекул, актив-
ные в КР- Как и в обычных спектрах
поглощения, при этом действуют зако-
ны светопоглощения, в частности
закон Бугера — Ламберта — Бера
(XI.12).
300 200 100 счГ1
Рис. XII.11. Изменение
спектра КР карбонила ко-
бальта Со4(СО)12 при резо-
нансном возбуждении:
1 — возбуждение линией
647,1 нм Кг-лазера; 2 — возбуж-
дение линией 514,5 нм Аг-лазе-
ра; 3 — возбуждение линией
488,0 нм Аг-лазера
3.3. Методы нелинейной спектроскопии КР
С появлением и внедрением в практику спектроскопии КР мощ-
ных оптических генераторов (лазеров) были открыты явления и
стали развиваться соответствующие методы, основывающиеся на
эффектах нелинейности. Дело в том, что наведенный ди-
польный момент молекулы, возникающий под действием электри-
ческого поля световой волны напряженностью ё, можно предста-
вить в виде следующего (несколько упрощенно) разложения в ряд:
!г^ = а(1)^ + а(2/2 + с!(з)СГз + ... . (XII.3)
В обычном КР света, как уже рассматривалось, играет роль
только первый член разложения, описывающий линейную зависи-
мость поляризации от напряженности поля. Здесь ац) — поляризуе-
мость молекулы. Каждый последующий коэффициент, а именно:
а(2) (называемый гиперполяризуемостью), а<з> (воспри-
имчивость третьего порядка) ит. д. — уменьшается по
сравнению с предыдущим в 1010 раз, поэтому из-за малости нели-
нейных членов ряда ими пренебрегают.
Однако интенсивность КР пропорциональна мощности лазера
только до определенного предела. При очень высокой интенсивно-
сти возбуждения, когда напряженность поля ё становится больше
109 В-м-1, начинают давать существенный вклад и нелинейные чле-
Рис. XII.12. Взаимное
расположение волновых
векторов в случае согла-
сованности фаз (Дк=0)
ны. Вклад второго члена (а®) обуслов-
ливает эффект гиперкомбинацион-
ного рассеяния, когда в спектре КР
появляются частоты 2vo±v(-, где vi — ко-
лебательные частоты молекулы.
Правила отбора в спектре гипер-КР
отличаются от обычного КР. В частно-
сти, в нем проявляются все колебания,
активные в ИК спектре, включая низко-
частотные. Это, в принципе, открывает
возможность получения полных колеба-
тельных спектров на одном приборе и от-
каза от необходимости исследования образца методами .ИК спект-
роскопии. Однако пока практическое использование гипер-КР ог-
раничено из-за трудностей наблюдения и регистрации спектра, так
как интенсивность его чрезвычайно мала и составляет ~ 10~13 от
интенсивности источника.
С эффектами третьего члена ряда (XII.3), т. е. сцз), связано по-
явление так называемой когерентной антистоксовой раман-спектро-
скопии (КАРС) *. Суть метода заключается в следующем. Если
образец облучается одновременно двумя лазерами с частотами vi
;и V2 и выполняется некоторое условие согласованности фаз, для
чего лучи лазеров должны пересекаться в образце под определен-
ным углом 9, то в результате взаимодействия пучков между собой
и со средой может генерироваться направленное когерентное лазе-
роподобное излучение с частотой v = 2vi — v2- Интенсивность сла-
бого излучения, генерируемого таким образом в любой среде, за-
висит от угла 0 и максимальна при точном фазовом синхронизме
{полной согласованности фаз).
Для этого должно выполняться следующее соотношение волно-
вых векторов для лазерных (kt и к2) и генерируемого (к) лучей
(рис. XII.12):
Дк=2к1 — к2 — к=0.
Фактор несогласованности Дк, особенно при исследовании газов,
может быть и несколько отличен от нуля. Для большинства жид-
костей угол 0 составляет 1 ... 3°.
Если возникают такие резонансные условия, что частота како-
го-то энергетического молекулярного перехода, например колеба-
тельного vK, совпадает с разностью частот лазеров, т. е. vK=Vi — V2,
то происходит резкое увеличение интенсивности генерируемого из-
лучения. Спектр КАРС можно получить, используя лазер с плавно
перестраиваемой частотой V2 и измеряя интенсивность v как функ-
цию vi — v2. Малая пока распространенность метода КАРС связа-
на с дороговизной приборного оборудования, прежде всего лазеров
* Иногда расшифровывают как когерентное антистоксово рас-
сеяние света.
на красителях, а также трудностью перестройки частот во всей об-
ласти колебательного спектра.
Однако при тех же правилах отбора, что и в обычных спектрах
КР, метод КАРС очень перспективен из-за ряда больших преиму-
ществ. К их числу относятся: высокая интенсивность, примерно в
105 раз превосходящая интенсивность КР, высокое разрешение (со-
тые доли см-1), легкость регистрации из-за направленности излу-
чения, отсутствие мешающей флуоресценции из-за работы в анти-
стоксовой области и возможность получения спектров при сильном
постороннем фоне излучения (плазма, газовый разряд, фотохими-
ческие процессы и т. д.) и, наконец, монохроматичность генерируе-
мого излучения, т. е. отсутствие необходимости в использовании
монохроматоров. Все это может быть охарактеризовано как каче-
ственно новый и более высокий уровень эксперимента в спектро-
скопии КР.
При изучении поверхностей и поверхностных явлений — адсорб-
ционных, каталитических и др. — может привести к успеху также
очень интересный, но пока еще недостаточно понятый эффект уси-
ленного поверхностью КР света. Наблюдавшееся повыше-
ние интенсивности КР в результате этого эффекта достигало по-
рядка 106 раз (это явление называют также гигантским КР)-
* * *
Современное состояние теории и технического воплощения ме-
тодов колебательной спектроскопии позволяет решать множество
задач структурной и прикладной химии, а также возникающих в
других областях науки и техники. Понимание основ теории колеба-
тельных спектров, кратко излагаемых в гл. VIII, IX, практических
возможностей методов ИК и КР спектроскопии, представление о
которых дают гл. X, XI, и знание особенностей эксперимента —
гл. XII, совершенно необходимы для успешного использования этих
методов в научно-исследовательской работе и внедрения их в про-
изводственную практику.
Многообещающи перспективы развития методов колебательной
спектроскопии, открываемые совершенствованием спектральной ап-
паратуры и появлением принципиально новой экспериментальной
техники. Имеется в виду прежде всего увеличение спектрального
разрешения приборов, повышение чувствительности методов и воз-
можность автоматизации получения и обработки данных в единой
системе с ЭВМ.
Приборами будущего в ИК спектроскопии являются уже су-
ществующие интерферометры (фурье-спектрометры) высокого раз-
решения, а также некоторые типы приборов, разрабатываемые на
совершенно новых принципах. Использование лазеров в спектро-
скопии КР привело не только к качественному скачку в традицион-
ных применениях этого метода, но и к появлению новых методов,
основывающихся на нелинейных оптических эффектах. Хотя такие
методы, как, например, КАРС, гигантское КР и др., пока еще хи-
миками почти не применяются, в будущем они, несомненно, найдут
свое место в химических лабораториях.
Колебательные спектры являются настолько уникальной и спе-
цифической характеристикой веществ и содержат такую богатую
информацию, что интерес к их изучению и практическому исполь-
зованию не только не ослабевает, а постоянно растет. Возможность
получения новых, недоступных ранее экспериментальных данных,
имея огромное значение для практики, стимулирует в то же время
дальнейшее развитие теории колебательно-вращательных спектров
и их связи со структурой вещества.
Контрольные вопросы
Глава VIII
1. Сколько колебательных степеней свободы имеется у молекул ацетилена,
бензола, гексафторида урана?
2. ’ Как приближенно соотносятся с основными колебательными частотами:
Vi, Vj, Vh,... обертона и составные или комбинированные частоты?
3, Чем определяются правила отбора переходов в ИК и КР спектрах?
(Дайте также классическую трактовку активности колебаний в этих спектрах.)
4. Что такое нормальные колебания и нормальные координаты?
5. Какие естественные внутренние координаты можно ввести для молекул
BF3, СН2=СН2, СН3С1?
6. Как связаны нормальные.координаты с: естественными?
(ДТЖак определяются частоты нормальных колебаний и от каких парамет-
ров молекулы они зависят?
С^Что такое форма нормального колебания?
ПЗ/От каких электрооптических параметров зависят интенсивности в ИК и
КР спектрах?
Глава IX
. 10. Какие существуют элементы и операции симметрии для точечных групп
симметрии?
И. Определите, к каким точечным группам относятся молекулы SF6, С6Н12
(в конформации «кресло»), СН2=СН2, BF3,
(двугран-
ный угол, образуемый плоскостями циклов, ~40°); для каких из этих молекул
действует правило альтернативного запрета в ИК и КР спектрах?
12. Какие совокупности симметрично-эквивалентных естественных координат
можно выделить у молекулы BF3 и какие можно ввести для нее координаты
симметрии? (Используйте ответы на вопросы 5, И и табл. IX. 1.)
13. Чему равны характеры приводимых представлений для всех операций
симметрии молекулы этилена?
14. Определите число нормальных колебаний разных типов симметрии для
молекулы этилена по формуле (IX.4), используя ответы на вопросы 5, 11, 13
и табл. IX.1. Какие из этих колебаний по табл. IX.1 активны в ИК и КР спект-
рах? (Сравните с указанием на с. 203)
15. Что такое резонанс Ферми и для каких определений он может быть
полезен?
Глава X
16. Как по ИК и КР спектрам вещества можно определить, к какой точеч-
ной группе симметрии относится молекула?
17. Для какого агрегатного состояния вещества из сопоставления ИК и КР
спектров получают наиболее надежные данные о симметрии молекулы и почему?
18. Если для линии спектра КР, возбуждаемого плоскополяризованным
светом, интенсивность при перпендикулярной поляризации равна 6 условным
единицам, а при параллельной поляризации—11, то чему равна степень деполя-
ризации линии рп? Чему' она была бы равна при возбуждении спектра неполя-
ризованным излучением? Как такую линию называют и к какому типу симмет-
рии принадлежит колебание, к которому данная линия относится?
19. Какое из двух колебаний молекулы бензола:
будет давать в ИК спектре параллельную и какое перпендикулярную полосу,
какие v этих полос будут контуры вращательной структуры?
с 20? В чем состоит кажущееся противоречие определения нормального коле-
бания молекулы и понятия характеристической или групповой частоты и как это
противоречие снимается?
21. Какие названия, используемые при условном отнесении колебательных
частот, приняты для основных форм колебаний атомных групп?
22. Как групповые частоты связаны с локальной симметрией фрагментов
молекулы?
23. Для каких исследований и определений используется концепция груп-
повых или характеристических частот?
<Д^“"2Т~Если частота валентного колебания гидроксильной группы в некотором
соединении равна 3600 см-1, то приблизительно какая она должна быть при
дейтерозамещении этой группы?
Глава XI
25. Проведите оценку силовых постоянных ke (Н-см-1) молекул N2, СО,
NO, CN, О2, CF по соотношению Беджера, используя, соответственно, следующие
значения равновесных межатомных расстояний: 1,10; 1,13; 1,15; 1,17; 1,21;
1,27 (10-1 нм).
26. Что такое обобщенное гармоническое валентно-силовое поле молекулы?
Чем обусловлена неоднозначность решения обратной колебательной задачи?
27. Сколько основных частот колебаний и сколько независимых силовых по-
стоянных у молекул С.Н.СГ. МЙ2—NH2, CH3HgOH, без учета и с учетом сим-
метрии?
28. Как по колебательным ИК и КР спектрам могут определяться потенци-
альные барьеры и другие параметры потенциальной функции заторможенного
внутреннего вращения?
29. Какие колебания дают наибольший вклад в термодинамические функ-
ции веществ, в какие из функций колебательный вклад является наибольшим?
30. Как используют колебательные спектры для качественного анализа сме-
сей частично известного и полностью неизвестного состава?
31. Что такое оптическая плотность и как она зависит от концентрации при
выполнении закона Бугера — Ламберта — Бера, как она используется для коли-
чественного анализа при отклонениях поглощения от этого закона?
32. Что позволяет определить исследование температурной зависимости
относительной интенсивности полос колебательных спектров, относящихся к раз-
ным молекулярным формам, которые находятся в равновесии?
г'” 3?.'>Как можно оценить по колебательным спектрам энергию образования
водородной связи? Как можно различить полосы гидроксильных групп, обра-
зующих внутримолекулярную и межмолекулярную водородную связь?
34. Что такое дихроичное отношение в ИК спектрах и как оно используется
при изучении высокомолекулярных соединений?
Глава XII
35. Перечислите основные типы ИК спектрометров. В чем преимущества
фурье-спектроскопии?
36. Как можно подготовить образец для исследования ИК спектра твер-
дого вещества?
37. В чем суть метода матричной изоляции? Какие преимущества он дает
при исследовании колебательных спектров изолированных молекул по сравнению
с изучением их в газах и парах?
38. В чем состоит отличие спектров НПВО от обычных спектров пропус-
кания?
39. Какие возможности открывает применение лазеров для возбуждения
спектров КР? Какие предосторожности нужно соблюдать при их использовании?
40. В чем суть метода КАРС и каковы перспективы его применения?
МЕТОДЫ электронной УФ спектроскопии
Спектры поглощения и спектры люминесценции в видимой и УФ областях
позволяют изучать основное и возбужденные электронные состояния молекул
и переходы между ними. Эти спектры используются для идентификации соеди-
нений, определения их структуры, они дают также информацию о распреде-
лении электронной плотности в молекуле и ее изменениях. Эти данные явля-
ются опорными для теоретической химии и квантово-химических расчетов.
Абсорбционная и люминесцентная спектроскопия широко применяются как
высокочувствительные аналитические методы, особенно пси исследовании рас-
творов, они могут успешно служить для автоматизации контроля и упразпения
производственными процессами в химической и других отраслях промышлен-
ности.
Группа методов электронной УФ спектроскопии
охватывает оптические спектры не только в ульт-
рафиолетовой (УФ), но и в видимой (ВИ) и са-
мой ближней ИК областях, связанные с перехо-
дами между различными электронными состоя-
ниями атомов и молекул. Электронные переходы
атомов и связанные с ними спектры в указанных
областях являются основой атомного эмиссион-
ного и абсорбционного спектрального анализа.
Высокотемпературный нагрев вещества, например, в вольтовой дуге
или искровом разряде, как это делается при эмиссионном спект-
ральном анализе, переводит образец в парообразное, обычно ато-
марное состояние, причем атомы химических элементов, входящих
в состав вещества, возбуждаются. Излучение, возникающее при
переходах атомов в основное электронное состояние, и дает линей-
чатый спектр, используемый для качественного и количественного
элементного анализа, который, как и вся группа связанных с ним
спектральных методов, здесь рассматриваться не будет.
Если говорить о молекулярных электронных спект-
рах, то такие эмиссионные спектры при высокотемпературном воз-
буждении могут быть получены в основном только для достаточно
прочных простых молекул. Электронные спектры многоатомных
молекул исследуются обычно как спектры поглощения или спектры
люминесценции; первые возникают в результате переходов из ос-
новного (вообще более низкого по энергии) электронного состояния
в возбужденные за счет поглощения квантов электромагнитного
излучения из сплошного спектра источника, а вторые — в результа-
те перехода молекулы из возбужденного состояния в основное с
испусканием электромагнитного излучения. Этот релаксационный
процесс предусматривает, очевидно, предварительный перевод мо-
лекул в возбужденное состояние, например, облучением вещества,
т. е. при поглощении молекулами квантов излучения. В принципе
существует также метод спектроскопии электронного комбинацион-
ного рассеяния света, но он пока мало исследован, а его применение
весьма ограничено и не получило распространения.
Электронные спектры поглощения молекул и ионов в УФ и ви-
димой областях используются химиками уже более 100 лет. Клас-
сическими являются применения абсорбционной УФ спектроскопии
для качественного и количественного анализов. Хотя по сравнению
с некоторыми другими спектрами, например ИК, КР или ЯМР,
электронные спектры поглощения менее специфичны, УФ спектро-
скопия в сочетании с этими методами, а также масс-спектрометрией
продолжает использоваться для идентификации и определения
структуры химических соединений. Этим методом изучаются равно-
весия и кинетика химических реакций, различного рода комплексы
и межмолекулярные взаимодействия и т. д.
Методы электронной спектроскопии относятся к основным экс-
периментальным методам, на которые опирается теоретическая и
квантовая химия. Именно этими методами получают данные об
294
электронных состояниях молекул, в частности об изменениях энер-
гии, геометрической конфигурации, распределения электронной
плотности и других молекулярных характеристик, при переходе из
основного электронного состояния в возбужденные. Прецизионные
определения многих молекулярных постоянных по электронно-коле-
бательно-вращательным спектрам возможны только для двухатом-
ных и некоторых простейших многоатомных молекул в газовой
фазе. Но и для достаточно сложных соединений возможны более
или менее полная интерпретация электронных спектров с отнесе-
нием полос к определенным электронно-колебательным переходам
и получение интересующей химика информации. Этому служат уже
сами экспериментальные данные о положении, интенсивности полос
и зависимости их от различных факторов (например, полярности
растворителя и т. д.). Сопоставление же таких данных с результа-
тами приближенных квантово-химических расчетов частот и веро-
ятностей переходов может, с одной стороны, облегчить интерпрета-
цию экспериментальных спектров, а с другой — явиться критери-
ем правильности расчетов, т. е. достаточно ли используемое в них
’ приближение.
ГЛАВА XIII
ОСНОВЫ ТЕОРИИ ЭЛЕКТРОННЫХ
СПЕКТРОВ МОЛЕКУЛ
1. ОБЩАЯ ХАРАКТЕРИСТИКА СВОЙСТВ
ЭЛЕКТРОННЫХ СОСТОЯНИЙ
Электронные состояния молекул определяются при разделении
электронного и ядерного движения в адиабатическом приближении
Борна —Опенгеймера (см. гл. V и гл. VIII) электронным уравне-
нием Шредингера:
HeWe=Ee4e, (XIII.1)
где оператор Гамильтона Йе и волновая функция Фе описывают
состояния электронов молекулы в поле закрепленных ядер, напри-
мер, в равновесной конфигурации (в общем случае это не обяза-
тельно), а Ее—собственное значение электронной энергии (пусть
в данном случае для равновесной конфигурации ядер). Конкрет-
ный вид оператора Не и выражение волновой функции здесь не
рассматриваются. Необходимо отметить, что ЧС может быть выра-
жена с помощью координатных и спиновых множителей, характе-
ризующих соответственно орбитальное и спиновое движения элект-
ронов. Строгое решение уравнения (XIII.1) для многоэлектронных
молекулярных систем невозможно и существуют лишь приближен-
ные методы решения электронного уравнения, в частности молеку-
лярных орбиталей (МО), валентных схем (ВС) и др. Некоторые из
них могут приводить для достаточно простых молекул к решениям
высокой точности (например, метод Хартри — Фока — Рутана).
Даже для двухатомных молекул нет общего аналитического выра-
жения для энергии электронного состояния Ее или электронного
терма (см-1): Тэл = Ее!(he) как функции каких-то квантовых чисел
в отличие от колебательного и вращательного движений ядерного
скелета молекулы.
К основным характеристикам каждого электронного состояния
молекулы относятся энергия, волновая функция, степень вырожде-
ния, мультиплетность и время жизни.
Энергия молекулы в заданном состоянии является функ-
цией ядерной конфигурации, но обычно за энергию электронного
состояния принимают ее минимальное значение, соответствующее
равновесной ядерной конфигурации: Ее — Е (Re), где Re— совокуп-
ность равновесных значений ядерных координат. Электронное со-
стояние, для которого Ее имеет наименьшее значение, называется
основным, а последующие — первым, вторым и т. д., воз-
бужденными электронными состояниями:
Е(е0} < < Е^ < ... < Е(ет} < ... . (XIII.2)
Основное состояние принято обозначать буквой X, а остальные — в
порядке латинского алфавита: А, В, С и т. д.
Для каждого электронного состояния характерна своя функция
потенциальной энергии. У двухатомных молекул она может быть
аппроксимирована, например, функцией Морзе (см. рис. VIII.1 и
формулу VIII.5), т. е. имеется зависимость энергии только от од-
ной координаты — межъядерного расстояния г. На рис. XIII.1 схе-
матично показано возможное относительное расположение потен-
циальных кривых нескольких электронных состояний двухатомной
молекулы.
Функция потенциальной энергии многоатомной молекулы пред-
ставляется гиперповерхностью n-го порядка (n = 3V—6 или у ли-
нейных молекул n=3N—5, где N — число атомов) в (п+ 1)-мерномЕ
пространстве. Наглядно представить такую функцию невозможно^
но в этом и нет необходимости. Можно, если требуется, рассматри-
вать графически зависимость потенциальной энергии от одной или
двух каких-то нормальных координат, координат симметрии или
естественных координат. Например, для линейной молекулы ХУг
на рис. XIII.2, а, б приведены качественные кривые зависимости по-
тенциальной энергии от какой-либо одной из нормальных коорди-
нат, описывающих смещение ядер при колебаниях, формы которых
были рассмотрены в гл. IX (см. рис. IX.2).
На рис. XIII.2, в дана контурная диаграмма зависимости потен-
циальной энергии от изменения двух межъядерных расстояний
Дг1=^! и Ar2 = q2 (т. е. естественных координат), на которой через
определенные интервалы соединены между собой точки с одинако-
вой энергией. Такая диаграмма дает представление о потенциаль-
ной поверхности в трехмерном пространстве, которую можно изго-
товить из подходящего материала (гипса, пластилина и т. п.). Оче-
296
Рис, ХП1.1, Потенциальные кри-
вые для электронных состояний
двухатомной молекулы YZ:
О — для основного состояния (X); 1 и
2 — для устойчивых возбужденных со-
стояний (А и В); 3 — отталкивательная
кривая
видно, что сечение поверхности
по биссектрисе координатного
угла даст потенциальную кри-
вую, представленную на рис.
XIII.2, а для полносимметрич-
ного колебания, а сечение по
Рис. XIII.2. Качественные зависимо-
сти потенциальной энергии линейной.
молекулы XY2:
а — от нормальной координаты Qi полно-
симметричного колебания; б — от нормаль-
ной координаты Qs антисимметричного ва-
лентного колебания (или Qs — вырожден-
ного деформационного колебания): в — от
межатомных расстояний Tj и или есте-
ственных координат ^! = ДГ] и (кон-
турная диаграмма)
линии, проходящей через минимум энергии перпендикулярно бис-
сектрисе,— кривую, представленную на рис. XIII.2, б для антисим-
метричного колебания.
Вопросы, связанные с функциями потенциальной энергии моле-
кул, более подробно уже рассмотрены в гл. XI. Все, что сказано
в ней о колебаниях молекул, может быть отнесено к различным
электронным состояниям, для каждого из которых характерен свой
набор колебательных уровней энергии.
Волновая функция ЧЦ представляющая решений элект-
ронного уравнения Шредингера (XIII.1), является функцией про-
странственных и спиновых координат электронов и описывает эле-
ктронное состояние молекулы, т. е. распределение электронной
плотности. Ряду возможных значений электронной энергии Ее: ос-
новного, первого возбужденного и т. д. состояний (XIII.2)—соот-
ветствует ряд электронных волновых функций:
Ч'е'Ц...
(ХШ.З)
Хотя сами волновые функции, как правило, не могут быть строго
найдены, но обычно известны некоторые их важные свойства, в
частности свойства симметрии и др. Каждое электронное состоя-
ние описывается, таким образом, своей волновой функцией, кото-
рой, в конечном счете, определяются физические, а в какой-то сте-
пени и химические свойства молекулы в данном электронном со-
стоянии.
Различным электронным состояниям обычно соответствуют, на-
пример, различные равновесные ядерные конфигурации молекулы.
'Так, если в основном электронном состоянии молекула этилена
Нч „ /Н
—плоская, а молекула ацетилена Н—С^С—Н — ли-
нейная, то в возбужденном состоянии у первой молекулы груп-
пы— СН2 оказываются во взаимно-перпендикулярных плоскостях,
а вторая становится нелинейной. Кроме того, длина связи СС аце-
тилена в основном состоянии составляет гсс—1,208-10~‘ нм, а в
первом возбужденном — гСс— 1,388-10-1 нм.
Каждому электронному состоянию молекулы соответствует не
только, как уже говорилось, своя функция потенциальной энергии
и набор колебательно-вращательных состояний, но и свой набор
практически всех молекулярных постоянных и характеристик. Это
касается и собственного дипольного момента, который может ме-
няться даже в несколько раз, и поляризуемости, и магнитных
свойств, и констант некоторых используемых химиками корреляци-
онных соотношений, и т. д. Конечно, не у всех соединений происхо-
дящие при электронном возбуждении молекул изменения свойств
одинаково существенны; они бывают относительно меньше, напри-
мер, с увеличением размеров молекул.
Электронное состояние может быть невырожденным или
вырожденным, если одному значению энергии Ее соответст-
вуют одна или несколько разных электронных волновых функций
а степень вырождения состояния равна числу таких
функций. Следует отметить, что вырожденные электронные (как и
колебательные) состояния встречаются только у молекул средней
и высшей симметрии, т. е. имеющих одну или несколько осей сим-
метрии порядка выше второго.
На энергию электронного состояния оказывает влияние суммар-
ный спин электронов, и состояние характеризуется мульти-
п легкостью. Результирующий электронный спин представляет
векторную сумму собственных моментов количества движения эле-
ктронов:
S = ][s,., (XIII.4)
а спиновое квантовое число S может быть или равно 0, или прини-
мать полуцелочисленные и целочисленные значения.
Спин-орбитальное взаимодействие, т. е. связь спина с орбиталь-
ным моментом количества движения электронов, может приводить
298
к расщеплению электронного состояния на 2S+1 компонентов. Эта
величина и есть мультиплетность состояния. При S = 0 мультиплет-
ность равна 1 и состояние называют синглетным, при S=1
(мультиплетность равна 3) состояние называется триплетным
и т. д.
Как и для атомных состояний, электронное состояние молекулы
может быть охарактеризовано средним временем жизни
тп, представляющим среднюю продолжительность нахождения мо-
лекулы в данном n-м состоянии. Эта величина обратна полной ве-
роятности спонтанного испускания Ап или вероятности перехода из
данного электронного состояния на более низкие уровни:
т
п
1
Ап
1
2 Апт
т
1.5)
Для основного электронного состояния т0=оо, для долгоживу-
щих (метастабильных) состояний порядок т„Д>10-4 с, а для ко-
роткоживущих состояний, когда наблюдаются интенсивные элект-
рические дипольные переходы, тп имеет порядок 10-7...10~9 с.
2. НОМЕНКЛАТУРА И СИМВОЛИКА ЭЛЕКТРОННЫХ
СОСТОЯНИЙ
Полное описание возможных энергетических состояний моле-
кулы дает полная волновая функция, но здесь будут в принятом
приближении рассмотрены отдельно только электронные состояния
и в какой-то степени электронно-колебательные (вибронные) со-
стояния.
Электронные состояния классифицируются по свойствам элект-
ронных волновых функций и в соответствии с тем, какие из свойств
и в каком приближении берутся при этом за основу, в литературе
существуют различные системы классификации, номенклатуры и
символики состояний. Прежде всего необходимо рассмотреть учет
свойств симметрии (см. гл. IX; 1). Симметрия ядерной конфигура-
ции определяет симметрию всей молекулы в целом, т. е. и симмет-
рию распределения электронной плотности. У симметричных моле-
кул (или приближенно симметричных), т. е. принадлежащих к ка-
кой-либо точечной группе симметрии, исключая тривиальную (С)),
при классификации электронных состояний и выводе правил отбо-
ра для переходов между ними нет необходимости находить сами
волновые функции, а важно определить только их свойства сим-
метрии. Электронная волновая функция (как и колебательная)
может принадлежать только к одному из типов симметрии точеч-
ной группы, к которой относится молекула. Таким образом, и эле-
ктронным состояниям приписываются соответствующие типы сим-
метрии с использованием для их обозначения принятых символов:
А, В, Е, F и т. д. (см. табл. IX.I).
При характеристике электронных состояний, как уже отмеча-
лось, необходимо рассмотрение орбитального и спинового момен-
тов количества движения электронов. В общем случае квадрат мо-
дуля суммарного орбитального момента выражается формулой
L2 = ft2Z (Z.-ф 1), (ХШ.6>
где L — квантовое число, принимающее значение 0, 1, 2, 3,..., а
сам вектор L представляют как векторную сумму орбитальных мо-
ментов отдельных электронов: XL; h = h/2n,.
I
Если все электроны в молекуле спарены, то L = 0; орбитальные
моменты неспаренных электронов могут быть различны и соответ-
ственно различен их вклад в суммарный момент. В зависимости от
этого вклада для электронов условно приняты следующие обозна-
чения:
L................... О 1 2
Состояние........... о л 6 ...
При наличии какого-то выделенного направления (высшей оси
симметрии, вектора напряженности поля и т. д.) важна проекция
момента количества движения на это направление:
Lz = hML, (XIII.7)
где ML — квантовое число, которое может иметь значения —L,
—L+ 1,..., 0, ..., L—2, L—1, L (всего 2L+ 1 значений Ml).
У молекул с осью симметрии бесконечного порядка Соо f т. е. у
двухатомных и линейных многоатомных молекул, относящихся к
точечным группам и DXh, в основу классификации электрон-
ных состояний положен модуль проекции суммарного орбитально-
го момента количества движения электронов на ось молекулы:
|Л4£|=Л, так как энергия состояния зависит именно от этой вели-
чины. Электронные состояния молекулы обозначаются в соответст-
вии со значениями Л следующими символами:
Л....................... О, 1, 2, 3,
Символ.................. S, П, Д, - Ф,
Очевидно, что все состояния, кроме 2, вырождены, так как од-
но значение Л отвечает двум (±) ML.
Спиновой момент количества движения, представляющий век-
торную сумму электронных спинов (XIII.4), также характеризует-
ся квадратом модуля
52 = /г25(5-ф1), (XIII.8)
(квантовое число 5 = 0, ’/2, 1, 3/2, 2, 5/2,...) и проекцией на выде-
ленное направление
Sz = fWs, (XIII.9)
(квантовое число Afs = —5, —5+ 1,..., 0,..., 5—2, S—1, 5).
При осевой симметрии молекулы для классификации важна
проекция S, для которой принято обозначение 2т (оно эквивалент-
но Sz (XIII.9) и его не надо путать с обозначением состояния 2
при Л=0), принимающая (2S+1) значений.
Расщепление уровней энергии в зависимости от спина происхо-
дит под влиянием магнитного поля. В невырожденном состоянии
орбитальное движение электронов не создает магнитного поля, и
энергия от спина не зависит. При вырожденных состояниях (П,
Д,...) в молекуле имеется внутреннее магнитное поле, пропорцио-
нальное А=^0. Тогда для характеристики электронного состояния
линейной молекулы рассматривают полный момент количества дви-
жения:
S2 = A-|-2m.
При заданных значениях Л и S полный момент, как и проекция
2т, принимает (2S+1) значений. Это и характеризует число ком-
понент, на которые расщепляется электронное состояние в зависи-
мости от спина, т. е. мультиплетность, которая указывается верх-
ним индексом перед символом состояния; значение полного момен-
та Q может быть указано справа от символа нижним индексом:
2S+1Aa. Например, при Л=1 и спиновом квантовом числе S=V2,
т. е. значениях проекции Sm= + 1/2 и — '/г, возможны два состоя-
ния: 2П3/2 и 2П'/2.
При символах состояний дополнительными индексами может
быть охарактеризовано также отношение волновой функции к опе-
рациям симметрии. Так, например, если при отражении в плоско-
сти симметрии, проходящей через ось молекулы, функция Те не ме-
няет знак, то ставится верхний индекс плюс (2+), если меняет знак,
то минус (S-); если при инверсии в центре симметрии (точечная
группа Hoo/i) ЧЛ. не меняет знак, то ставится нижний индекс g: 2g,
если меняет знак, то и: Su, откуда и следуют обозначения типов сим-
метрии, использованные на рис. IX.2 и в табл. IX.2. Буквы X, А, В,
С, показывающие относительное расположение (последователь-
ность по шкале энергии) электронных состояний, ставятся слева,
например Х1Х+, А3Х~, В2П и т. п.
Для нелинейных многоатомных молекул, когда какое-либо вы-
деленное направление, как правило, отсутствует, провести класси-
фикацию электронных состояний с использованием векторной мо-
дели нельзя, так как говорить о проекциях электронного орбиталь-
ного или спинового моментов не имеет смысла. Номенклатура эле-
ктронных состояний многоатомных молекул носит или эмпириче-
ский характер, или следует из рассмотрения свойств симметрии
электронных волновых функций с использованием теории групп.
Если молекула принадлежит к некоторой точечной группе сим-
метрии, то ее электронные состояния классифицируются по типам
симметрии данной точечной группы и обозначаются соответствую-
щими символами с указанием мультиплетности. Относительное
расположение состояний показывают теми же буквами: X, А, В, но,
чтобы не путать с символами типов симметрии, над ними ставится
волнистая линия (тильда), например: X'Ag, А1Ви, АДг, B2Bi и т. п.
При рассмотрении электронной волновой функции молекулы
Для многих целей оказывается достаточным так называемое одно-
электронное приближение, результатами использования которого
будем ограничиваться, тем более, что усложнение модели, как по-
казано, не меняет основных качественных выводов, в частности, ка-
сающихся свойств симметрии. Электронная волновая функция, за-
висящая, как уже упоминалось, от пространственных и спиновых
координат, приближенно может быть представлена как произведе-
ние одноэлектронных функций — молекулярных орбиталей
(МО) или спин-орбиталей:
^ = П V/O/o % (XIII. 10)
i
где qi-—совокупность пространственных координат; о, — спиновые
координаты.
В свою очередь МО приближенно записывают в виде линейной
комбинации каких-то функций /Р:
Ъ=^е1р1р. (ХШ.И)
р
Хотя чаще всего в качестве этих функций принимают атомные
функции или орбитали АО (метод МО ЛКА.О), однако это
совсем не обязательно. Можно воспользоваться линейной комбина-
цией и других функций, но обычно этим функциям Хр приписывают
свойства локальности, т. е. считают, что каждая из них определена
в основном в пределах какой-то одной части многоатомной моле-
кулы. В связи с возможностью больших различий в значениях ко-'
эффициентов С[Р можно в определенном приближении рассматри-
вать МО как локализованные на одном атоме или группе атомов.
Построение МО приближенно относимой, например, к какой-то
химической связи, т. е. центрированной на двух ядрах химически
связанных атомов, как в двухатомных молекулах, требует, чтобы
обе функции, из которых она строится (АО), имели одну и ту же
симметрию относительно межъядерной оси и отвечали мало отли-
чающимся энергиям. Главное условие то, что всякая возможная
МО должна преобразовываться в соответствии с неприводимыми
представлениями точечной группы симметрии, к которой относится
молекула.
Типы симметрии АО и МО обозначают строчными буквами:
Ь2, е и т. п. Различают также связывающие орбитали <т
(сигма) и л (пи) с объединением атомов (символика Каша) и а н-
тисвязывающие или разрыхляющие орбитали о* и
л* с разъединением атомов (в простейшем случае, по аналогии с
s и р АО, первая должна быть симметрична относительно оси и
плоскости, в которой она лежит, а вторая — антисимметрична). Не
участвующие в образовании химических связей электроны неподе-
ленных пар образуют несвязывающие орбитали п.
Поясним построение и символику МО на примере молекулы
формальдегида Н2СО, которая относится к точечной группе С2г>
(см. табл. IX.1). Систему координат ориентируем относительно мо-
лекулы, как показано на рис. XIII.3. Базисные АО, из которых мо-
302
Рис. ХП1.3. Система коор-
динат и элементы симмет-
рии молекулы формальдеги-
да (плоскость симметрии
axz показана не полностью)
гут быть построены МО, преобразуются
при выполнении операций симметрии, как
указано в табл. XIII. 1 (предполагается,
что s-орбитали обладают сферической
симметрией, а р-орбитали имеют в нача-
ле координат узловую точку, где меня-
ют знак, имея вид объемной восьмерки).
На рис. XIII.4 показано относительное
расположение уровней энергии *, сопо-
ставляемых молекулярным орбиталям
формальдегида, тип симметрии и прибли-
зительное объемное представление МО,
относящихся в основном к карбонильной
группе С = О. Самыми низкими являются
три связывающие о-орбитали, а самыми
высокими — три антисвязывающие <т-ор-
битали. Имеются также связывающая и
антисвязывающая л-орбитали, а также
две несвязывающие n-орбитали неподе-
ленных пар электронов атома О. Надо
сказать, что три орбитали одного типа симметрии tZi, вообще гово-
ря, только условно могут быть отнесены, как указано на рис.
XIII.4, так как являются смешанными.
У молекулы в основном электронном состоянии все связываю-
щие и несвязывающие орбитали заполнены, т. е. каждой из них от-
вечают два электрона. Из всех заполненных орбиталей высшей
оказывается несвязывающая n-орбиталь симметрии Ь2.
Если полная электронная волновая функция представлена как
произведение МО (XIII.10) и известна симметрия всех МО, то
можно определить, к какому типу симметрии относится она сама, а
значит, и описать электронное состояние. Для этого образуют так
называемое прямое произведение, т. е. перемножают ха-
рактеры неприводимых представлений (каждой операции симмет-
рии) соответствующих типов симметрии (строк) таблицы характе-
ров (см. табл. IX.1). Рассматривая полученный ряд значений как
Таблица XIII.1. Симметрия АО для валентных электронов формальдегида
^2v / Сг СТ1 СГ2 Тип ^•20 I Сг Oi cr2 Тип
2H(ls) 2 0 0 2 Я1 + У O(2s) 1 1 1 1 «1
С (2s) 1 1 1 ' 1 01 О(2рх) 1 — 1 1 —1 bl
С(2рх) 1 —1 1 —1 &1 О(2ра) 1 — 1 —1 1 bz
С(2р„) 1 —1 —I 1 I&2 O(2pz) 1 1 1 1 Cli
С(2рг) 1 1 1 - 1 «1
* Как и в случае схем расположения АО, энергия связи электронов в моле-
кулах растет по МО сверху вниз.
3b2
><т*(СН2)
Рис. XIII.4. Схема относительного расположения МО фор-
мальдегида по энергиям:
цифрами указаны порядковые номера уровней данного типа сим-
метрии; на схематических изображениях МО сплошные и пунктир-
ные контуры отвечают разным знакам волновой функции, а боль-
ший и меньший размеры контура — объемам над и под плоскостью
рисунка (yz)
строку характеров представлений данной точечной группы, одно-
значно определяют тип симметрии прямого произведения.
При заполнении МО электронами со спаренными спинами (спи-
новые волновые функции взаимно ортогональны) электронное со-
стояние будет характеризоваться суммарным спином S=0, т. е. бу-
дет синглетным, а соответствующая ему полная волновая функция
будет относиться к полносимметричному типу. В случае формаль-
дегида для волновой функции основного состояния можно записать
304
Таблица XIII.2. Прямые произведения типов симметрии точечной группы Сг
C2v Ai A2 Bi to to
Ai At A2 Bi B2
А2 A2 Ai to N to
Bl Bl B2 Ai A2
tn N B2 to A2 Ai
прямое произведение (ограничиваясь орбиталями карбонильной
группы):
WQ==alxbixbl.. = A1,
т. е. это состояние имеет символ Mi. Здесь al, b\
мне произведения вида aiXai или, например:
и т. д.—уже пря-
Z С2 Hi о2
felXbi=(+i)x (-1)х(-1) = +i (+i)x(4-i)=+1 1-1)x(-i)=+i=ai
X (+!)=+!
и т. д., что и приводит к конечному Ai.
Рассмотрим два возбужденных элект-
ронных состояния, схематично показан-
ных на рис. XIII.5. Для одного из них
волновая функция может быть представ-
лена прямым произведением:
1®rA=ai XbiXb2x Ь{ = А2,
а состояние может быть обозначено как
*Д2; для другого
4rs=ai xbl х b2
состояние характеризуется двумя элект-
ронами с параллельными спинами (сум-
марный спин 5 = 1) и обозначается 3В2
(триплетное).
Прямые произведения функций нуж-
ны не только для классификации элект-
ронных состояний, но и для вывода пра-
вил отбора переходов между ними, и для
решения ряда других задач, в частности
квантовой химии. На практике удобно
пользоваться составленными для точеч-
Рис. XI П.5. Некоторые при-
меры заполнения МО (кар-
бонильной группы) при од-
ноэлектронном возбужде-
нии, симметрия волновой
функции и обозначение со-
ответствующих электронных
состояний
ных групп симметрии таблицами прямых произведений типов сим-
метрии вида табл. XIII.2.
3. КЛАССИФИКАЦИЯ ЭЛЕКТРОННЫХ ПЕРЕХОДОВ,
ИХ ОТНОСИТЕЛЬНОЕ ПОЛОЖЕНИЕ
Происхождение любых молекулярных спектров: вращательных,
колебательных, электронных — с точки зрения квантовой механи-
ки в принципе объясняется на основе рассмотрения переходов мо-
лекул между соответствующими энергетическими состояниями.
Правила отбора и интенсивность переходов связаны с их вероят-
ностью, которая определяется квадратом момента перехода | М21, а
М = f 'Р,*ц4г"0т, (XIII. 12)
где 4f/ и Чг"— волновые функции верхнего и нижнего состояний;
р—оператор электрического момента; dr — элемент фазового про-
странства.
При всей общности такого подхода в каждом методе молеку-
лярной спектроскопии существует своя специфика описания спект-
ров, а также классификация переходов и принятая терминология.
Это касается и электронных спектров молекул, тем более, что не-
которые запреты на электрические дипольные переходы наруша-
ются, некоторые переходы оказываются характеристичными груп-
повыми переходами, они бывают по-разному поляризованы, могут
называться с учетом мультиплетности состояний, между которыми
они происходят, и т. д.
Исторически первой при описании электронных спектров погло-
щения возникла концепция хромофоров и ауксохромов,
связанная с экспериментальными исследованиями спектров в до-
ступном для обычных серийных спектрофотометров диапазоне длин
волн УФ и видимой области. Многочисленные данные показывали,
что для определенных рядов соединений, содержащих одни и те
же структурные фрагменты, в указанной области наблюдаются ха-
рактерные полосы поглощения. Такие фрагменты, т. е. группиров-
ки атомов с сохраняющимся для ряда молекул электронным спект-
ром, и были названы хромофорами.
К хромофорам относятся, например, различные группы, содер-
жащие изолированные и сопряженные кратные связи, радикалы и
атомы с неподеленными электронными парами, ароматические цик-
лы и т. д. Иногда различают так называемые /(-хромофоры (от
нем. konjugiert— сопряженный) и /(-хромофоры (от нем. radical —
радикал), а также хромофоры ароматического типа, дающие ха-
рактерное бензольное поглощение. Полосы поглощения приписы-
вали различным хромофорам и соответственно различали /(-поло-
сы— относительно более длинноволновые и наиболее интенсивные
(е^ 10 000), /(-полосы — относительно коротковолновые и слабые
(е=С100) и В-полосы— имеющие промежуточные положение и ин-
тенсивность (е = 250...3000).
Ауксохромами были названы группировки атомов, присоедине-
ние которых к хромофорной системе ведет к смещению характер-
ной для хромофора полосы поглощения в сторону больших длин
волн. В качестве примеров могут быть названы гидроксильная ОН
и амино.NH2 группы, алкильные радикалы, галогены и.т. д. Разли-
чие между ауксохромами и хромофорами чисто условно и даже ис-
кусственно. Ауксохромы сами могут поглощать при относительно
более коротких длинах волн, т. е. выступать в роли хромофоров
(например, 7?-типа).
Концепции хромофоров и ауксохромов можно дать некоторое
обоснование, ограничиваясь при рассмотрении электронных состоя-
ний и переходов одноэлектронным приближением. Представление
о локализации электронного перехода на' какой-то атомной груп-
пировке (хромофоре) согласуется с представлением о возможной
локализации MQ, отражающим свойства аддитивности частей мно-
гоатомной молекулы. Условиями же сохранения характеристик
электронного перехода для данного хромофора в разных молеку-
лах является однотипность по симметрии тех МО, между которы-
ми происходит переход, и примерное постоянство разности энергий
между соответствующими уровнями.
Симметрия разных молекул, содержащих данный хромофор,
редко может быть строго одинакова, поэтому указанные условия
выполняются приближенно, но речь и идет о. приблизител fa-
il о одинаковых положении и интенсивности полос поглощения, ха-
рактерных для данного хромофора. Так, например, для карбониль-
ного хромофора Q)C=0j система «уровней» (МО) будет всегда
похожа, т. е. включать а-, л-, п-, л*- и <т*-орбитали, аналогично тому,
как это было рассмотрено для молекул формальдегида (см. рис.
XIII.4 и XIII.5), но никогда не будет совпадать точно у молекул
разных соединений.
Р. С. Малликен ввел представление о классах электронных пе-
реходов внутри валентной оболочки, названных N-+V и N-+Q в от-
личие от ридберговских переходов N^-R с электронным воз-
буждением до более высокой оболочки (символ N везде соответст-
вует «нормальному», т. е. основному электронному состоянию).
К классу N^-V относятся переходы между связывающими и раз-
рыхляющими (антисвязывающими) орбиталями, т. е. типов л—
о—мг*, о->-а* и т. п. При нескольких возбужденных К-состояниях
соответствующие переходы индексируются порядковыми номерами
этих состояний: А->-К2 и т. д. Эти переходы дают полосы
поглощения средней и высокой интенсивности в относительно ко-
ротковолновой области, хотя А—>-/?-переходы еще больше смещены
в область коротких длин волн (так называемую вакуумную УФ об-
ласть) и обычно более интенсивны.
В класс A->Q входят переходы с несвязывающих орбиталей на
разрыхляющие орбитали, т. е. типов га-э-л*, п-*-в* и т. п. Обычно
этим переходам соответствуют слабые полосы в относительно бо-
лее длинноволновой области. Как правило, A-^-Q-переходы поляри-
11* 307
I
I
I
a*
я*
n
it
a
Рис. XIII.6. Типы электронных
переходов для карбонильного
хромофора
зованы перпендикулярно направлению
связи хромофора (его оси) в отличие
от Л'->]/-переходов, поляризованных
обычно параллельно.
При отнесении переходов к какому-
то классу или типу, если возможно,
указываются символы типов симмет-
рии и мультиплетное™ электронных
состояний, между которыми осуществ-
ляется переход.
На рис. XIII.6 показаны переходы
различных типов на примере карбо-
нильного хромофора, а на рис.
XIII.7 — относительное расположение,
относительные интенсивности и сдвиг
при увеличении полярности раствори-
теля или замещающей группы в окру-
жении хромофора для полос поглоще-
ния разного типа. Легко видеть, что
наименьшему изменению энергии соот-
ветствует переход п->л* типа V->Q.
Относимая к этому переходу слабая полоса (е~20) в области
270...300 нм характерна для всех алифатических альдегидов и ке-
тонов. Положение этой полосы меняется в зависимости как от
внутримолекулярного окружения карбонильной группы, так и от
растворителя. Например, в ряду соединений: СН3СН, СН3ССН3,
II II
О
О
СН3СС1 (в гексане), т. е. с увеличением электроотрицательности
заместителя, наблюдается сдвиг полосы в сторону меньших длин
волн — синий, или гипсохромный, сдвиг, а именно: ХМакс =
=293,4; 279,0; 235,0 нм и некоторое повышение интенсивности —
гиперхромный эффект: еМакс=И,8; 14,8; 53,0. Для длинно-
волнового смещения приняты термины красный, или бато-
хром н ы й, сдвиг, а для понижения интенсивности — гипохром-
ный эффект.
Влияние растворителя можно проиллюстрировать на примере
ацетона СН3СОСН3: Лмакс (нм) полосы п-^-л* перехода меняется от
279 в гексане до 277 в хлороформе, 272 в этаноле, 270 в метаноле и
264,5 в воде, т. е. с увеличением полярности растворителя здесь
опять-таки наблюдается гипсохромный сдвиг. Это объясняют ста-
билизацией неподеленной электронной пары, т. е. понижением «-ор-
битали в основном электронном состоянии из-за сольватации или,
в частности, при образовании водородной связи с молекулами раст-
ворителя. Чем сильнее такое межмолекулярное взаимодействие,
тем больше разность энергий АД состояний и выше частота пере-
хода с л-орбитали на разрыхляющую орбиталь; рассмотренные эф-
Хромофоры;
очень
сильная
К В
R
средняя различной интенсивности
с колеб. т
структурой
по Малликену:
N-+-Q
средняя
сильная очень
-— сильная
п-»~а*
по Каша: п^*-П* сРел^няя
'слабая средняя сильная
* сильная
___________1_____________________—_______________1__________________
/ 1000 180 нм-
______________I_________________________________________I_____________
оз 104 103 см^г
Рис. XIII.7. Относительное расположение и другие ха-
рактеристики полос переходов различных типов:
стрелки под символами перехода указывают сдвиги (синий —
-> или красный — **-) при увеличении полярности растворителя
или замещающей группы в окружении хромофора
фекты характерны и для других переходов N-+Q, т. е. типа п->о*
и др.
Область частот переходов л->-л* и может совпадать, т. е.
они близки по энергии, но для полос л—>-л*, относящихся к классу
N—+V, характерна высокая интенсивность и батохромный сдвиг под
влиянием полярного растворителя или электроотрицательной груп-
пы в окружении хромофора. Для изолированных хромофоров по-
лосы этих переходов, как и перехода о-мт*, часто лежат за корот-
коволновым пределом рабочего диапазона обычных серийных
спектрофотомеров, а переход <т-(N^V) может быть близок по
частоте к ридберговским переходам N—>~R, т. е. проявляется лишь
в вакуумной УФ области.
Для систем сопряженных кратных связей (Х-хромофоры), как
отмечалось, наблюдается повышение интенсивности и смещение по-
глощения в сторону больших длин волн; эти батохромный и гипер-
хромный эффекты тем больше, чем длиннее цепь сопряжения, т. е.
чем большее число кратных связей участвует в сопряжении. Это
объясняют сближением связывающих (л) и разрыхляющих (л*)
орбиталей или несвязывающих (л) и разрыхляющих (л*) орбита-
лей, когда частота переходов л->л* и га->л* понижается по схеме,
показанной на рис. XIII.8. Иногда описание указанных эффектов
дают в терминах делокализации электронов в сопряженных систе-
мах.
Весьма характерны по виду полосы бензольного поглощения
(В-полосы). В л-электронном приближении орбитальная схема
бензола представлена на рис. XIII.9, а. Для ароматического цикла
Рис. Х1П.8. Схема л-электронных уровней:
а — сопряженного диена; б — а,(3-иепредельиого карбонильного со-
единения с включением п-орбитали
Рис. XIII.11. Схема наи-
более вероятных виброн-
ных переходов на приме-
ре двухатомной молеку-
лы, иллюстрирующая
принцип Франка —
Кондона:
при электронном переходе
r~rconst и г-гмакс или
гмин
Рис. XIII.10. Спектры
поглощения (В-поло-
сы):
1 — бензола (в циклогек-
сане) и 2 — пиридина (в
спирте)
пиридина с гетероатомом азота, имеющим неподеленную электрон-
ную пару в системе уровней, представленной на рис. XIII.9, б, при-
бавляется несвязывающая (и) орбиталь, а все л-орбитали невы-
рождены и энергия уровней меняется. Казалось бы, можно ожидать
очень большого отличия спектра пиридина в области В-полосы
(~250 нм) от спектра бензола, однако полосы поглощения у обе-
их молекул оказываются очень похожи (рис. XIII.10). По-видимо-
му, у пиридина интенсивная полоса перехода л3—»Л4 перекрывает
слабую полосу перехода п Л4, так как уровни Лз и п почти вы-
рождены.
. В-полоса обычно имеет хорошо
•------ 71*
(/2и) ' -------71*
----------------------------------Л’,
------- ---------712,^3 ।
----------------------------------[ '
-------- Я,
(«2^ ---------
-------
а) б)
Рис. XIII.9. Сравнение системы уровней
бензола (а) и пиридина (б) в n-элек-
тронном приближении:
Символы 7t и п указывают лишь тип орбита-
ли, но не орбитальную симметрию
выраженную колебательную
структуру, как это видно на
рис. XIII. 10. В принципе любые
электронные переходы сопро-
вождаются изменением не
только электронного, но и ко-
лебательного состояний моле-
кулы, т. е. являются электрон-
но-колебательными, и называ-
ются вибронными пере-
ходами, как и соответствую-
щие им полосы называются
вибронными полосами.
Проявление колебательной
структуры в электронных
спектрах многоатомных моле-
кул сильно зависит от агрегат-
ного состояния вещества. Для
детального вибронного анали-
за спектра требуется обычно
исследовать вещество в газовой фазе (в парах) при достаточно
хорошем разрешении. Колебательная структура становится четче
и лучше разрешается также при низких температурах (жидкого
азота и, еще лучше, жидкого гелия), а также в неполярных рас-
творителях.
Переход между уровнями нулевой колебательной энергии основ-
ного электронного состояния (все колебательные квантовые числа
^* = 0) и возбужденного электронного состояния (все т4=0) на-
зывается «ноль — ноль» (0—0)-переходом и является чисто элект-
ронным переходом. Ему соответствует в спектре поглощения 0—
0-полоса, имеющая наинизшую частоту. Вибронные переходы
включают изменение кватовых чисел vk. Если нормальные коле-
бания многоатомной молекулы или фундаментальные частоты пе-
реходов пронумерованы, как и квантовые числа vk (индекс k меня-
ется от 1 до 3N—6 или до 3N—5 для линейной yV-атомной молеку-
лы), то вибронный переход обозначается цифрами так: kvт. е.,
vk
например, 1о (переход с нулевого колебательного уровня, хц =0,
основного электронного состояния на первый возбужденный коле-
бательный уровень, '01 = 1, возбужденного электронного состояния,
для колебания k= 1), аналогично расшифровываются символы:
1о, 1о, 8о, 13о и т. п.
Поскольку частота электронного перехода гораздо выше коле7
бательных частот, то, в соответствии с принципом Франка — Кон-
дона, выше относительная интенсивность тех полос, которые соот-
ветствуют переходам без изменения геометрии молекулы, являю-
щимся наиболее вероятными. Наиболее вероятная геометрия рас-
положения ядер в неравновесной конфигурации и не при всех Vk~
=0 соответствует поворотным точкам, т. е. точкам пересечения ко-
лебательного уровня с потенциальной поверхностью. В нормаль-
ных координатах наиболее вероятная конфигурация отвечает мак-
симумам их абсолютной величины. На рис. XIII.11 вероятные виб-
ронные переходы схематично показаны вертикальными стрелками.
В электронной спектроскопии выделяют особые, так называе-
мые переходы с переносом заряда (ПЗ). К таковым от-
носится любой переход электрона с орбитали, локализованной в
'одной части молекулы, на орбиталь, локализованную в другой час-
ти. Как и в других случаях, орбиталь, с которой удаляется элект-
рон, называют донорной, а орбиталь, на которую переходит элект-
рон,— акцепторной. Различают переходы с внутримолекулярным
переносом заряда (ВПЗ) и переходы в комплексах с переносом за-
ряда (КПЗ). В качестве перехода первого типа (ВПЗ) рассматри-
вают, например, перенос заряда с аминогруппы на разрыхляющую
МО кольца в ароматических аминах. Такой переход обозначают
(по Каша) 1-+ая, где / — орбиталь неподеленной пары электронов
атома азота, а а„— разрыхляющая л-орбиталь кольца. ВПЗ на-
блюдается в таких молекулярных ионах, какСЮГ, SO4-. МпОГ>
СгО2Г и др. Электрон с несвязывающей орбитали кислорода (или
связывающей л-орбитали) может переходить, например, на орби-
тали Мп или Сг с восстановлением металла в возбужденном элект-
ронном состоянии.
В КПЗ две обычные в классическом понимании химические час-
тицы или молекулы образуют как бы одно целое, т. е. комплекс
или новую частицу, в которой происходит переход электрона с ор-
битали одной из взаимодействующих молекул на орбиталь другой.
Например, в комплексе бензол-иод переход с ПЗ можно предста-
вить следующим образом:
С6Н6.12±ЩЗ)^С6Н6+.12-
В комплексах пиридина с металлами или в солях Л^-алкилпири-
диния C5H5NCH3 с галогенами также наблюдаются переходы с ПЗ
соответственно от металла на л*-орбиталь пиридина или с несвя-
зывающей орбитали азота к галогену (п^-а*).
Переходы КПЗ характерны, например, для октаэдрических
комплексов переходных металлов типа MLe (М— металл, L — ли-
ганд). Электрон с заполненной о-орбитали (или л-орбитали) ли-
ганда может перейти на свободную с?-орбиталь металла (L-^-M-ne-
реход КПЗ, высокочастотный). Для некоторых лигандов возможны
переходы КПЗ типа M->L, а самыми низкочастотными (обычно в
видимой области) и малоинтенсивными в таких комплексах явля-
312
Р ются так называемые d—d-переходы между связывающими и раз-
t рыхляющими d-орбиталями металла.
Для полос, соответствующих большинству переходов с перено-
сом заряда, характерна высокая интенсивность (saslO4).
4. ПРАВИЛА ОТБОРА И ИНТЕНСИВНОСТЬ ПЕРЕХОДОВ
Переходы между различными электронными состояниями мо-
гут сопровождаться спонтанным, т. е. самопроизвольным или вы-
нужденным (при воздействии излучения), испусканием и всегда
вынужденным поглощением электромагнитного излучения. Наибо-
лее важными являются электрические дипольные переходы, сопро-
вождающиеся изменением электрического дипольного момента. Ин-
тенсивность в спектрах испускания и поглощения связана с веро-
ятностью соответствующих переходов. Число фотонов Z, испущен-
ных или поглощенных за единицу времени, пропорционально чис-
лу молекул N на уровне, с которого совершается переход. При
спонтанном испускании (переход с n-го на m-й уровень)
Z СПОЯТ я Т т
пт = п<
где Апт — вероятность такого испускания (т. е. число фотонов
на одну возбужденную молекулу) или так называемый коэффиь
циент Эйнштейна для спонтанного испускания.
Предполагается, что элементарный процесс испускания фотона
осуществляется мгновенно и независимо от других процессов тако-
го же типа. При вынужденном испускании или поглощении:
zB„THcn=^P(v)^,
Z^=Bmn?b)Nm,
где Впт и Втп — коэффициенты Эйнштейна для вынуж-
денного испускания и поглощения, представляющие
вероятность на одну молекулу (в состоянии соответственно п и т)
и на единицу плотности излучения p(v).
Связь между коэффициентами Апт, Впт, Втп строго может
быть получена в рамках квантовой электродинамики или, что
проще, из рассмотрения термодинамического равновесия вещества
и излучения, когда число фотонов Znm, испускаемых в единице объ-
ема в единицу времени, равно числу поглощаемых фотонов; Zma
(излучение черного тела):
7 __ 7СПОИТ I уВЫН.ИСП 7
пт. — пт пт -—
или после подстановки выражений для Z;
ИПт+В
Пт? =
Тогда
Mt _ ДтпР(У)
№ т Апт + ВПт$ (v)
йо в соответствии с законом распределения Больцмана
Nn gnexp(—En/kT)
gmexp(—Em/kT)
-^-exp(-h»nm/kT),
gm
где gn и gm — степени вырождения уровней En и Em. Из сравнения
этих двух выражений имеем
(gm!gп) Вщп exp (hvтп/kT') —' Епт
Плотность излучения абсолютно черного тела определяется по фор-
муле Планка:
8rtftv3
Р0) =------
и при 7->оо p(v)->oo, т. е. gmBmn=gnBпт- Окончательно получим *
V3 п- vs
Апт=М Qnfl
c3 gtl c3
Коэффициент Anm, как уже видели (XIII.5), имеет размерность,
обратную времени, а коэффициенты Впт и Втп выражаются в
Дж-]-с-2-м3.
Вероятность электронного перехода пропорциональна квадра-
ту момента перехода и, в частности, показано, что коэффициент
Эйнштейна для поглощения (как и для вынужденного испускания
Впт) связан с ним следующим образом:
= (Х111.13)
Матричный элемент момента перехода (XIII. 12) конкретно из
состояния п в состояние т является векторной величиной с компо-
ллпт мпт ллпт
нентами Мх , Му , Mz , которые определяются интегралами
вида
(XIII. 14)
и аналогично для Л4ут и Mzm, где щ— квантово-механиче-
ские операторы компонентов (проекции) электрического дипольно-
го момента; и Тт — волновые функции n-го и m-го состояний.
При этом
I + (ХШ.15)
Интенсивность поглощения (поглощенная энергия) равна:
/Tn*=BmnP(y)Nmfomn, (XIII. 16)
где hvmn — энергия поглощаемых фотонов.
* Есть два способа определения коэффициентов Эйнштейна, и если использу-
ется не плотность излучения, а удельная интенсивность, то получаются другие
соотношения между Апт и Втп, а также силой осциллятора fnm, рассматривае-
мой ниже.
Учитывая уравнения (XIII.13) — (ХШ.15), I будет отлична от
нуля если хотя бы один из компонентовМут, 714^” не равен
нулю.
Интеграл вида (XIII.14) не обращается в нуль только в том
случае, когда подынтегральное выражение полносимметрично, а
для этого необходимо, чтобы прямое произведение типов симмет-
рии 'K.X'F,,, совпадало с типом симметрии щ (i=x, у, z). Этим и.
определяются правила отбора для разрешенных по симметрии пе:
реходов.
Если переход разрешен, то количественно вероятность перехо-
да и интенсивность соответствующей полосы можно было бы вы-
числить, только зная точные волновые функции состояний, между
которыми происходит переход, и проекции Цх, цу, щ. Обычно они
неизвестны и приходится рассматривать вопрос приближенно. Ес-
ли известна связь экспериментально наблюдаемой интенсивности
с вероятностью перехода, то можно решать обратную задачу, т. е.
по измеренным в спектре интенсивностям определять вероятности
переходов и, сопоставляя их с результатами теоретических оценок,
относить полосы спектра по типам переходов.
В прикладной абсорбционной спектроскопии часто определяют
молярный коэффициент экстинкции (или погашения) в максимуме
полосы поглощения еМакс, хотя более строгой мерой является интег-
рал от функции s(v) по всей полосе или системе полос поглощения,
называемый интегральным коэффициентом погашения. Этот интег-
рал как раз и можно сравнивать с выражением интенсивности по-
глощения (XIII.16), т. е. для единичной плотности излучения p(v):
f (XIII.17)
Помимо квантово-механической вероятности перехода, в электрон-
ной спектроскопии также используют понятие осцилляторной
силы перехода, или «силы осциллятора»,/, основанное
на простой классической модели осциллирующего заряда, который
совершает затухающие колебания данной частоты. Связь си-
лы осциллятора с вероятностью перехода выражается соотноше^
нием
(XIII. 18)
где т — масса электрона и е — его заряд.
Учитывая (XIII.17) и переходя к волновым числам, силу осцил-
лятора можно определять непосредственно по интегральной интен-
сивности системы полос электронного перехода:
<хшл9>
N— число молекул в единице объема (см3).
mh.vmn R
,, Dmtv>
ле2
При введении поправки на влияние среды (1,02-10-1) *
/ = 4,31-10-9 Je (со) dco, (XIII.20)
где и — волновое число.
Величина f безразмерна и обычно нормируется к единице, т. е.
для полностью разрешенного перехода, для которого коэффициент
погашения е=105 л-М-'-см-1, f— 1.
Интенсивность электронных переходов, измеряемая в абсорб-
ционных спектрах, меняется в пределах десяти порядков, т. е. 8 от
ГО5 до IO-5, a f от 1 до 10~9, и иногда для удобства пользуются их
десятичными логарифмами (1g е от 5 до —5, lg f от 0 до —9).
Говоря о переходах между двумя электронно-колебательными
состояниями, мы пока не делали некоторой детализации, которая
необходима при рассмотрении интенсивности и правил отбора. При
электронно-колебательном переходе оператор электрического ди-
польного момента ц в (XIII.12) можно разделить на две состав-
ляющие, одна из которых зависит только от ядерных координат
а другая — только от электронных (р,е). Если представить
также волновые функции как произведения электронной и колеба-
тельной Ч^Ч^-Хъ то уравнение (XIII.12) можно переписать в
виде
м= [ СХ’ (ц„+Ю 4^XdT=J CK*.uXX-dT-|-
+ j <X‘£XX-dT=J Cs^dT, J 4C(lXdT„ +
+J K,K.dr„ J (Xin.21)
Электронные волновые функции ортогональны, поэтому первое
слагаемое последней суммы равно нулю. Первый интеграл второ-
го слагаемого в последней сумме не равен нулю, так как колеба-
тельные волновые функции относятся к разным электронным со-
стояниям и поэтому не должны быть ортогональны.
Электронную волновую функцию можно записать отдельно для
пространственных Чге и спиновых 4fs координат. Оператор р,е на
спиновую составляющую не действует, и окончательно имеем сле-
дующее выражение момента перехода:
М = J <X’dr„ J Ф; pX’dr, f C^dr„ (XIII.22)
которое и определяет правила отбора электронно-колебательных
переходов. Квадрат первого интеграла в (XIII.22) называется фак-
тором Франка — Кондона, второго — орбитальным фактором или
моментом собственно электронного перехода, а третьего — спино-
вым фактором. Переход формально запрещен, если | Мпт |2 стре-
* Поправка найдена относительно газа при 0°С и атмосферном давлении,
когда множитель перед интегралом равен 4,2-10~®.
мится к нулю, и можно отдельно рассматривать в (XIII.22) каж-
дый интеграл, хотя это рассмотрение всегда приближенно, и полу-
чаемые правила отбора не являются поэтому совершенно стро-
гими.
Прежде всего рассматриваются как разрешенные переходы, при
которых происходит возбуждение не более, чем одного электрона.
Относительно самым строгим при этом является правило от-
бора по спину, согласно которому разрешены переходы без
изменения спинового квантового числа: AS=0, т. е. только между
электронными состояниями одинаковой мультиплетности. При этом
последний интеграл в (XIII.22) не равен нулю.
Если переход запрещен спиновым правилом отбора (J 1I,/W‘idTs=
=0) ,то интенсивность соответствующих полос мала и характери-
зуется значениями е от 1 до 10-s (л-М-'-см^1), если даже первые
два интеграла в (ХШ.22) отличны от нуля. Появление этих сла-
бых полос в спектре обусловлено так называемыми интерком-
бинационными переходами между состояниями различной
мультиплетности. Они возможны в результате спин-орбиталь-
ной связи, которая особенно сильна в молекулах с атомами тя-
желых элементов. У комплексов тяжелых переходных металлов,
например, коэффициент погашения для полос «запрещенных по
спину» интеркомбинационных переходов может даже иметь поря-
док величины е~102.
Правило отбора по симметрии, называемое также
орбитальным, определяется вторым интегралом в (ХШ.22). Оно
аналогично правилу отбора для колебательных переходов, и к нему
относится все, что было уже сказано относительно матричного эле-
мента перехода Мпт и его компонентов вида (XIII.14). Согласно
этому правилу отбора электронный переход разрешен, если трой-
ное прямое произведение типов симметрии X х относится
к полносимметричному типу точечной группы, которой принадле-
жит молекула. Как уже говорилось, должен быть отличен от нуля
интеграл вида (XIII.14) хотя бы для одной проекции цг- (i=x, у,
г”). Правило отбора для линейных молекул, например, требует
ДЛ=0, ±1, при этом возможны переходы: +<-»• + , ——, а для
точечной группы D^h и g++u-, переходы + «—/—>—, g g,
и <—/—> и —запрещены.
Рассмотрим электронные переходы из основного состояния мо-
лекулы формальдегида М] в возбужденные состояния, представ-
ленные на рис. XIII.5, т. е. переходы и Mi->-3B2. Первый из
них запрещен орбитальным правилом отбора, так как прямое про-
изведение типов симметрии электронных волновых функций Д1Х
ХД2=Д2 (см. табл. XIII.2) не совпадает с типом симметрии ни од-
ной из проекций щ (см. табл. IX.1), т. е. тройное прямое произве-
дение типов симметрии х fie X 'К- не будет полносимметричным.
Второй переход разрешен по симметрии, так как прямое произ-
ведение АгХВ2=В2 совпадает с типом симметрии и тройное
прямое произведение ' AiXB2xB2=Ai— полносимметрично. Но
этот переход, как синглет-триплетный, запрещен по спину. Из од-
ноэлектронных переходов, показанных на рис. XIII.6 стрелками,
все переходы разрешены по симметрии, кроме перехода п->
->-л* (Mi->M2). Вообще из полносимметричного основного элект-
ронного состояния орбитальным правилом отбора разрешены пере-
ходы только в те электронные состояния, для которых тип симмет-
рии волновой функции совпадает с типом симметрии хотя бы одной
из проекций электрического дипольного момента.
Переходы, которые разрешены как спиновым, так и орбиталь-
ным правилами отбора, называются полностью разрешенными и
дают наиболее интенсивные полосы поглощения, характеризуемые
величинами 8~ 103...105. Интенсивность разрешенных по спину, но
запрещенных по симметрии переходов лежит в пределах 8~ 1...103,
т. е. все-такй достаточно высока, что обусловлено следующим. По-
ка рассматривались правила отбора для чисто электронного (0->0)
перехода, когда Ч^^,, и Ч^полносимметричны, или по крайней мере
не обращая внимания на первый интеграл в уравнении (XIII.22),
который отличен от нуля, если прямое произведение типов симмет-
рии х 4> также полносимметрично.
Этим интегралом и определяется при разрешенном электронном
переходе колебательная структура, т. е. между какими колебатель-
ными подуровнями двух электронных состояний переходы наиболее
вероятны. Однако в общем случае колебательные волновые функ-
ции и прямое произведение их типов симметрии не обязательно
должны быть полносимметричными, и для определения правила от-
бора вибронных переходов нужно рассматривать электронно-коле-
бательное взаимодействие. Выражение (XIII.22) можно преобразо-
вать к виду
М = j СчСцЛЖ'<Ки J (XIII.23)
Первый интеграл будет не равен нулю, т. е. вибронный переход
будет разрешен, если прямое произведение типов симметрии элект-
ронных и колебательных волновых функций и хотя бы одной из
проекций щ полносимметрично. При соответствующей симметрии
колебательных волновых функций это возможно, даже когда элект-
ронный переход по симметрии запрещен, т. е. тройное прямое про-
изведение типов симметрии Ч> X рг X Ч’г неполносимметрично и в
(ХШ.22) второй интеграл равен нулю. Если орбитальным прави-
лом отбора переход запрещен, а так называемым вибронным
правилом отбора разрешен, то он и соответствует по интен-
сивности указанному выше интервалу значений 8 для разрешенных
по спину и запрещенных по симметрии электронных переходов.
Когда электронный переход запрещен по спину и по симметрии,
но наблюдается вибронный, то соответствующая полоса имеет
318
J
Таблица XIII.3. Шкала коэффициента погашения и силы осциллятора
для электронных переходов
Электрические
дипольные переходы
1. С переносом заряда; полностью +
разрешенные по спину (синглет-синг- |
летные и др.) и по симметрии 4.
2. Разрешенные по спину, запре- Т
гценные по симметрии, вибронные 1
3. Запрещенные по спину (синглет-
триплетные и др.), интеркомбинаци-
онные (разрешенные и запрещенные
по симметрии)
lg е 1g, f
5 О
3 —2
1 —4
О —5
— 1 —6
—2 —7
—8
—9
—10
Магнитные диполь-
ные переходы
Электрические
квадрупольные пере-
ходы
4 —1
очень малую интенсивность. К таким переходам относятся, в част-
ности, d—d-переходы в комплексах переходных металлов симмет-
рии Oh, коэффициент экстинкции которых е«5...25. Например, все
комплексы Мп(II)—электронная конфигурация (/5—-при отсутст-
вии электронно-колебательного взаимодействия должны быть бес-
цветны. В действительности же ион гексагидрата Мп(II) имеет
слабо-розовую окраску из-за наличия полос поглощения вибронно-
го перехода.
Для тетраэдрических (Та) комплексов переходных металлов за-
прет d—(/-переходов является менее жестким, и интенсивность со-
ответствующих полос выше (е~ 102...103).
Следует подчеркнуть, что здесь были рассмотрены только эле-
ктрические дипольные переходы, а кроме них возможны также
магнитные дипольные и электрические квадрупольные переходы.
Если момент перехода определяется оператором магнитного ди-
польного момента, интенсивность на 5 порядков ниже, а если опе-
ратором является электрический квадрупольный момент, интенсив-
ность на 8 порядков ниже, чем для полностью разрешенного эле-
ктрического дипольного перехода. Учитывая, что для последних ин-
тенсивность меняется в пределах десяти порядков, нельзя полно-
стью игнорировать возможность вклада указанных переходов,
правда, лишь при рассмотрении запрещенных электрических ди-
польных переходов.
Необходимо отметить также, что под воздействием внешних
электрических и магнитных полей, в частности, возникающих в ре-
зультате межмолекулярных взаимодействий, возможны так назы-
ваемые индуцированные переходы. Эти переходы дают, например,
слабые полосы в спектрах жидкостей, которые не наблюдаются в
парах, что связано с частичным снятием запрета по симметрии из-
за искажения электронной оболочки молекул в конденсированной
фазе.
Примерные интервалы значений коэффициента погашения и си-
лы осциллятора для различных типов переходов указаны в табл.
Вращательная структура электронно-колебательных переходов
может наблюдаться и исследоваться только для достаточно легких,
в частности, двухатомных молекул в газовой фазе вещества, на
приборах высокого разрешения и обычно в эмиссионной спектро-
скопии, поэтому здесь она не рассматривается.
ГЛАВА XIV
ПРИМЕНЕНИЕ ЭЛЕКТРОННЫХ СПЕКТРОВ
1. СТРУКТУРНО-СПЕКТРАЛЬНЫЕ КОРРЕЛЯЦИИ
Электронные спектры поглощения различных органических и
неорганических соединений могут лежать не только в ближней ИК,
видимой и ближней УФ областях, но и в далекой УФ области,
вплоть до энергий излучения, приводящих к ионизации молекул.
Изучены переходы электронов валентных оболочек в молекулах,
ионах и комплексах самых разных типов, для чего использовались
как стандартные для химических лабораторий спектрофотометры,
так и более сложные вакуумные спектрометры высокого разреше-
ния. Для целей идентификации соединений и решения структурных
проблем обычно используются характерные полосы поглощения
электронных спектров в области ~ 180...8000 нм, которые только и
приводятся в большинстве справочников и руководств по абсорб-
ционной УФ спектроскопии. Если в этой области вещество не по-
глощает, то его обычно называют прозрачным, хотя в далекой УФ
области оно может иметь свой спектр поглощения.
1.1. Органические соединения
В далекой УФ области поглощают алканы, цикланы и очень
многие другие предельные соединения, являющиеся прозрачными
в основной исследуемой области спектра. Самыми коротковолно-
выми, как уже говорилось, являются <т—►о*-переходы, имеющие ме-
сто в этих молекулах. При наличии в насыщенном соединении ге-
тероатомов, таких, как кислород, азот, сера, галогены, содержащих
неподеленные электронные пары (спирты, эфиры, тиоэфиры, ами-
ны, алкилгалогениды и т. д.), слабые или средние по интенсивно-
сти полосы поглощения п->-сг*-переходов могут наблюдаться выше
коротковолнового предела средней УФ области, т. е. в интервале
180...220 нм и выше.
Простые изолированные хромофоры, т. е. такие л-электронные
системы, как изолированные кратные связи, также поглощают час-
то в далекой УФ области и прозрачны в ближней УФ области. Ес-
320
Г ли же группа содержит как л-, так и п-электроны, то могут наблю-
даться более длинноволновые, но слабые полосы запрещенных п—>-
; ->л*-переходов. Полосы поглощения некоторых изолированных
i хромофоров приведены в табл. XIV. 1, причем значения ^макс И
смаке указаны в большинстве случаев для конкретных соединений
(дана формула или название в скобках).
Как уже говорилось (см. гл. XIII,3) в электронных спектрах
ярко проявляется эффект так называемого л—л-сопряжения. Если
две какие-то ненасыщенные хромофорные группы А и В, например,
такие, как —С = С—, '\С=О, —СООН, ^C = N—, —N — N —
—С6Н5 и т. п., разделены в цепи молекулы двумя и более атомами
углерода (А—(СН2)П—В; и>2), то наблюдается практически ад-
дитивный спектр хромофоров А и В (как спектр AR + BR', где R—
предельные радикалы). Для сопряженной системы А—В, т. е. при
отсутствии промежуточных метиленовых или каких-то других групп,
спектр резко отличается от спектра смеси AR + BR'. Полосы погло-
щения наблюдаются при больших длинах волн (батохромный эф-
фект) и сильно возрастает их интенсивность (гиперхромный эф-
фект). Например, если полоса поглощения этиленового хромофора
лежит обычно ниже 200 нм (см. табл. XIV.1), то бутадиен-1,3 име-
ет интенсивную полосу поглощения Хмакс=217 нм (емакс =21 000).
Чем длиннее цепь сопряжения, тем больше оба эти эффекта, и
наблюдается практически линейная зависимость квадрата ^Макс от
числа сопряженных двойных связей, например в дифенилполиенах
(рис. XIV.1). Сильные батохромный и гиперхромный эффекты на-
блюдаются для карбоцианиновых красителей и порфириновых пиг-
ментов с сопряженными двойными связями С = С и C=N в откры-
той цепи у первых и в сложной циклической системе у вторых. Для
длинных цепей сопряженных связей интенсивность поглощения
возрастает настолько, что сила осциллятора полосы, смещенной в
видимую область, превышает единицу, доходя до 2 и более.
Для системы сопряженных двойных связей характерно обычно
плоское строение скелета в s-транс-
или, хотя и
в мень-
конформации. При этом для
первой
шей степени, s-цис- j,
интенсивность полосы поглощения (молярный коэффициент пога-
шения) выше, чем для второй.
В частности, у бутадиена-1,3 полоса 217 нм относится к преоб-
ладающей х-транс-конформации. Объяснение более высокой интен-
сивности этой полосы может быть дано из рассмотрения компонен-
тов момента перехода с учетом принадлежности тра«с-конформа-
Ции к точечной группе С2ь, а цис-конформации — к C2v при исполь-
зовании метода МО ЛКАО. Нарушение плоского строения системы
сопряженных связей по каким-либо причинам, например из-за
Таблица XIV.1. УФ полосы поглощения некоторых изолированных
хромофоров
Хромофорная группа ^макс’ нм ®макс Переход Раствори- тель
Этиленовая СН2 = СН2 rch=ch2 RCH = CHR-rpaHc -цис 165 193 177 180 183 15 000 10 000 л->л* Газ
Ацетиленовая НС = СН 173 6 000 л->л* »
Карбонильная (СНз)2С = О 190 280 1 900 15 л->л* п->л* «Тексан
СН3СН=О 290 16 м—>л* Гептан
Карбоксильная СНзСООН 204 60 п->л* Вода
Азометиновая >C = N— 190 5 000 л->л* »
(ацетоксим)
Нитрильная —C = N <160 — —— —
(ацетонитрил)
Азо —N = N— 347 4,5 п->л* Диоксан
(азометан) Нитрозо —N = O (нитрозобутан)
300 665 100 20 Эфир
Нитратная —ONO2 270 12 п—»-л* Диоксан
(этилнитрат)
Нитро —NO2 271 19 п->л* Спирт
(нитрометан)
Нитритная —ONO 218,5 1120 л—>-л* Петролей-
(амилнитрит) 346,5 (широ- кая со струк- турой) п->л* ный эфир
Сульфоксидная >S = O 210 1500 Спирт
( циклогексилметилсульфоксид)
Сульфоновая >SO2 (диметил сульфон) <180
стерических затруднений, приводит к снятию эффектов сопряжения
и характерному изменению электронного спектра поглощения.
Это явление наблюдается, в частности, и при переходе от ди-
фенила к 2, 6, 2', б'-тетраметилдифенилу (рис. XIV.2): первый в
конденсированной фазе имеет плоское или почти плоское строение,
а заместители в орто-положениях создают такие пространственные
затруднения, что бензольные кольца становятся некопланарны и
их сопряжение нарушается.
Для оценки положения максимума полосы поглощения диенов,
в которых сопряженные двойные связи находятся в открытой цепи,
в разных циклах (гетероаннулярный диен) или в одном цикле (го-
моаннулярный диен), Р. Б. Вудворд и Л. Ф. Физер на основании
анализа многочисленных экспериментальных данных установили
ряд эмпирических правил, суммированных в табл. XIV.2.
Рис. XIV.1. Зависимость по-
ложения полосы поглощения
системы сопряженных двой-
ных связей в дифенилполи-
ене от ее длины
Рис. XIV.2. УФ спектры по-
глощения незамещенного
(/) и 2, 6, 2', 6-тетразаме-
щенного (2) дифенила
Поясним использование этой таблицы на одном примере. В УФ
спектре эргостерина:
максимум наиболее интенсивной полосы поглощения наблюдается
при Хмакс = 282 нм. Оценка по правилам Вудворда — Физера дает:
^рассч=253 (исходный гомоядерный диен)+20 (четыре связи цик-
лов а, Ь, с, d при хромофоре) +10 (каждая из двойных связей хро-
мофора является экзоциклической: одна — к циклу А, другая — к
циклу С) =283 нм, что хорошо согласуется с экспериментом.
Правила оценки положения полос поглощения, аналогичные
представленным в табл. XIV.2, найдены также для енонов и диено-
нов с учетом влияния растворителей. В УФ спектрах простых а, p-не-
насыщенных карбонильных соединений (хромофор ^С=С — С = О)
наблюдается интенсивная полоса поглощения (е ~ 10 000...
Таблица XIV.2. Оценка величины Хмакс для полосы поглощения сопряженных
диеновых систем (емакс = 6000 ... 35 000) в спиртовом растворе
Система Исходные значения X, нм
Гетероаннулярный диен или открытая цепь со- 214
«ряжения
Гомоаннулярный диен * 253
Дополнения Инкремеаты AZ-, нм
Двойная связь, продолжающая цепь сопряже- +30
ЛИЯ
Алкильный заместитель или связь С—С цикла + 5
при хромофоре или хромофорах С = С—С = С
Экзоциклическая двойная связь (относительно + 5
любого цикла)
Ауксохромные группы: OR +6
ОАс 0
SR +30
N(R)2 +60
Cl, Вг + 5
Храссч = общая сумма
* Если в системе присутствует как гетероаннулярный диен, так и гомоаниулярный дней.
например в j I II , то за исходное значение берут к для гомоаниулярного диена.
15 000) в области 220...260 нм, относимая к л->л*-переходу, и сла-
бая полоса (е~ 50... 100) п->л*-перехода в области 310...330 нм.
У сложных, например стероидных, систем интервал возможных
значений длин волн для этих полос существенно расширяется.
В частности, для п->л*-перехода ХМакс~300...380 нм и положение
полосы чувствительно к конформации. Так, в системе при аксиаль-
ном положении атома С1 ^макс на 11,5 нм выше, чем для экватори-
альной конформации.
У р-дикарбонильных соединений возможна енолизация:
—С—СН—С——С=С—С— , и это равновесие между кетонной и
О
О ОН О
енольной формами также может прослеживаться и изучаться мето-
дом УФ абсорбционной спектроскопии. а-Дикарбонильные соедине-
ния характеризуются двумя или тремя полосами низкой интенсив-
ности, наиболее характерная полоса п->л*-перехода лежит в обла-
сти 300...500 нм, что придает этим соединениям светло-желтую ок-
раску. Полосы средней и высокой интенсивности в области 210...
230 нм наблюдаются у систем с сопряженными ацетиленовыми свя-
зями.
Весьма характерны УФ спектры поглощения ароматических
соединений. Бензол в гексановом растворе имеет три полосы погло-
щения: %макс — 184 нм (8=60 000), %макс=203,5 нм (8=7400) и
254 нм (е=204). Длинноволновая полоса с ярко выраженной коле-
бательной структурой (бензольное поглощение — В-полоса) при за-
мещении претерпевает батохромный сдвиг и растет по интенсивно-
сти, как и с увеличением числа конденсированных циклов.
Полоса поглощения стирола 7Макс=248 нм (е= 14 000) при ор-
то-замещении метильными группами уменьшается по интенсивно-
сти (е=7000) и несколько смещается в сторону меньших длин
волн (Z=245 нм) из-за нарушения копланарности вследствие сте-
рических эффектов, например, в
I
СН3
Цис- и транс-стильбены также характеризуются различными по по
ложению и интенсивности полосами:
Амакс—224 нм (£-24000)
Умакс НМ (£=10500)
Лмакс =228 нм (£=16400)
Л-маю^Ж5 нм (£“29000)
(в спиртовом растворе)
Они также чувствительны к орто-замещению. Замещение аромати-
ческого цикла ауксохромными группами приводит к появлению но-
вых более длинноволновых полос, связанных, в частности, с нали-
чием неподеленных электронных пар и новых типов переходов, ха-
рактерных для каждого класса соединений.
Характерное поглощение наблюдается для таких гетероцикли-
ческих соединений, как пиррол, фуран, тиофен и др. Изоэлектрон-
ный бензолу пиридин (см. гл. XIII, § 3) имеет, например, три поло-
сы: 2.макс=195 нм (е = 7500), 251 нм (е=2000) и 270 нм (е =
= 450) —раствор в гексане, причем последнюю условно относят к
п->л*-переходу. 4-Оксипиридин может существовать в двух тауто-
мерных формах: енольной, которая преобладает при pH 13, и амид-
ной, преобладающей при pH 7. Они характеризуются следующими
полосами поглощения, по которым их можно идентифицировать и
определять относительное содержание:
хмакс = 239 нм (© = 14 100)
/-макс = 260 нм (г = 2200)
^Макс = 253 НМ ($ = 14 800)
Приведенные примеры показывают, что УФ спектры поглощения
могут успешно использоваться для изучения структурной изомерии,
как статической, например, цис-транс (см. о стильбенах), так и ди-
намической, т. е., например, таутомерии, а также конформационной
изомерии (выше был рассмотрен соответствующий пример акси-
ально-экваториальной изомерии в стероидных системах).
Характерные изменения наблюдаются в электронных спектрах
при образовании внутри- и межмолекулярных водородных связей,
как и в результате других специфических взаимодействий, напри-
мер внутрикомплексной координации или в координационных сое-
динениях и т. д. Межмолекулярная водородная связь приводит к
синему сдвигу полос п->л*- и п->о*-переходов и к красному сдвигу
полос л->л*-переходов, так что сравнение спектров в инертном и,
например, гидроксилсодержащем растворителе способствует, как
уже отмечалось, отнесению полос. Красным сдвигом полос л->л*-
переходов характеризуется также образование внутримолекуляр-
ной водородной связи. Интенсивность полос этого типа обычно
возрастает при образовании как внутри-, так и межмолекулярной
водородной связи, а интенсивность полос переходов с участием
несвязывающих п электронов уменьшается вплоть до полного
исчезновения полос. Это понятно, так как именно эти электроны
ответственны за образование водородных связей. Еще одним
проявлением водородной связи в электронных спектрах может
быть размытие колебательной структуры. Следует отметить, что
несколько более чувствительны к образованию водородной связи:
могут быть спектры люминесценции (см. гл. XV; 4).
1.2. Неорганические и комплексные соединения
Электронные спектры используются в целях идентификации и
при решении структурных и некоторых других химических проб-
лем в неорганической химии и химии координационных соедине-
ний.
Растворы неорганических соединений элементов первых перио-
дов системы Д. Н. Менделеева, как правило, не поглощают в
видимой и УФ областях. Исключение составляют некоторые анио-
ны, тогда как аквокомплексы катионов обычно «бесцветны» (ка-
вычки указывают, что речь идет и об отсутствии поглощения не
только в видимой, но и в УФ области).
В табл. XIV.3 приведены примеры характеристических макси-
мумов поглощения некоторых неорганических анионов. В опреде-
ленных степенях окисления некоторые элементы образуют сильно
окрашенные соединения, например ионы СгО^ (Сг2О7^) в водном
растворе, марганцовая кислота, соединения ванадия и т. д. Для
простых анионов галогенов (С1_, Вг~, Г~) в растворах характерны
полосы поглощения, обусловленные переходами с переносом
заряда.
Окраска аквокомплексов катионов тяжелых элементов связана
€ заполнением d-орбиталей. При полностью занятых d-орбиталях,
например у цинка и кадмия, растворы солей бесцветны (см. табл.
XIV.4), а при незанятых d-орбиталях, например, у кобальта, ни-
келя, меди и др. элементов,— окрашены (цвет раствора является
дополнительным к цвету поглощенного излучения).
Как уже упоминалось, строение комплексных соединений пере-
ходных металлов описывается с учетом частично или полностью
заполненных d-орбиталей. Простейшая модель может быть пред-
ставлена теорией поля лигандов или несколько менее сложной
теорией кристаллического поля. Основной результат такого рас-
смотрения состоит в выяснении, как расщепляются d-электронные
уровни центрального иона из-за снятия вырождения под воздей-
ствием окружающих его лигандов. Так, например, у иона Си2+
пятикратно вырожденный 3d-ypoBeHb энергии под влиянием поля
Таблица XIV.3. Полосы поглощения некоторых неорганических анионов
в водных и спиртовых растворах
Анион ^макс емак с Анион ^макс емакс
ci- 1810 10 000 s2o|~ 2200 4 000
Вг- 1995 11 000
1900 12 000 NOT 3546 23
I- 2260 12 600 2100 5 380
1940 12 600 2870 9
он- 1870 5 000 NOT 3025 7
SH- 2300 8 000 1936 8 800
Таблица XIV.4. Окраска аквокатионов некоторых элементов
и заполнение d-орбиталей
Катион Число d-электронов Число неспаренных электронов Цвет
Мп2+ 5 2 Слабо-розовый
Со2+ 7 3 Розовый
Ni2+ 8 2 Зеленый
Cu2+ 9 1 Синий
Zn2+ 10 —. Бесцветный
Cd2+ 10 — »
лигандов в зависимости от образуемой ими конфигурации может
расщепляться по-разному.
При тетраэдрическом, кубическом и октаэдрическом окружении
расщепление приводит к появлению двух, а при тетрагональном
(квадратном и квадратно-пирамидальном) окружении — четырех
подуровней, т. е. в первом случае (высокосимметричное окруже-
ние) в спектре поглощения должна наблюдаться одна полоса, а
во втором —три полосы. Таким образом, уже по числу полос
можно делать некоторые заключения о структуре, скажем, соль-
ватированного иона. Более определенные выводы делаются на
основе приближенных расчетов значений длин волн полос погло-
щения и сопоставления их с экспериментальными данными. Так,
по наиболее коротковолновым полосам акво- и аминокомплексов
Си2"' был сделан вывод о их тетрагональной конфигурации.
Молекулярный иод склонен образовывать комплексы с пере-
носом заряда, выступая в качестве акцептора электронов. Опре-
деление энтальпий ассоциации для систем донор — /2 спектро-
фотометрическим методом позволило получить данные о донорных
свойствах непредельных и ароматических молекул, сопоставить
донорные свойства карбонильных и сульфоксидных соединений и
показать, что у двойной связи углерод — кислород эти свойства
выражены сильнее, чем у л-связи сера — кислород.
Проведена также относительная оценка донорных свойств дру-
гих оснований, в частности, первичных, вторичных и третичных
аминов. Комплексы с переносом заряда образуют также некоторые
другие льюисовские кислоты, и методом абсорбционной УФ
спектроскопии оценены относительные кислотности Вг2, I2, IC1, SO2
и С6Н5ОН с использованием в качестве донора N, N-диметилацета-
мида.
Большой интерес представляют спектры поглощения растворов
солей редкоземельных элементов и актинидов. Эти спектры, связан-
ные с наличием заполняющихся /-орбиталей при занятых более вы-
соких s- и р-подуровнях, весьма сложны и сохраняют дискретную
структуру, в какой-то мере сходную со структурой спектров ато-
марных газов. Различия в электронной конфигурации отдельных
элементов обусловливают высокую индивидуальность спектров
растворов их солей.
2. АНАЛИТИЧЕСКИЕ ПРИМЕНЕНИЯ
Возможности аналитических применений электронных спектров
с очевидностью вытекают, как и в случае других спектральных
методов, из непосредственной связи спектра со строением хими-
ческих частиц и характером их взаимодействия между собой и
со средой вообще, а также с концентрацией вещества.
2.1. Качественный анализ и идентификация веществ
Обнаружение определенных химических соединений, установле-
ние качественного состава смесей и строения химических частиц —
молекул, ионов и т. д., основывается на сравнении полученных
спектральных кривых со стандартными спектрами и на структур-
но-спектральных корреляциях. При этом используются каталоги
спектров, таблицы характерных для различных соединений или
хромофоров полос поглощения с указанием их положения и ин-
тенсивности в максимуме или, наконец, соответствующие банки
данных в информационно-поисковых системах на ЭВМ.
Как уже было сказано, во-первых, в пределах обычно иссле-
дуемой на серийных УФ спектрометрах спектральной области (А.
выше 180 нм) поглощают далеко не все химические соединения,
а, во-вторых, наблюдаемое для ряда органических и координацион-
ных соединений поглощение связано с наличием в них каких-то
хромофорных групп, т. е. не очень специфично. Поэтому очевидна
большая ограниченность указанных применений абсорбционной
УФ спектроскопии в молекулярном спектральном анализе по
сравнению, например, с гораздо более избирательными колеба-
тельными спектрами.
Проводить идентификацию какого-то неизвестного вещества,
опираясь только на его спектр в видимой и УФ областях, по
меньшей мере рискованно и, строго говоря, невозможно. В то же
время метод абсорбционной УФ спектроскопии часто может слу-
жить хорошим дополнением и в сочетании с другими методами,
например ИК спектроскопии, ЯМР и масс-спектрометрии, способ-
ствовать надежной идентификации и установлению строения ис-
следуемых веществ. Для анализа и идентификации некоторых
ионов, их аквокомплексов, в частности лантанидов и актинидов,
рассматриваемый метод может играть решающую роль.
При качественном анализе необходимо исследовать спектры
пробы в широкой области длин волн и для растворов различной
концентрации, а иногда и в различных растворителях. При этом
определяют не только положение максимумов всех полос 2.макс с
точностью не менее 1 нм, но и интенсивности в максимумах хотя
бы по порядку величины еМакс или £1°см . Полосы поглощения
различных хромофоров могут иметь близкие значения Лмакс, но
сильно отличаться по интенсивности. Задача определения состава
многокомпонентных смесей по УФ спектрам поглощения является
весьма сложной и требует применения специальных математиче-
ских методов количественной обработки спектральных кривых.
Для ее упрощения необходимо возможно более полное разделение
смеси.
При сравнении спектра исследуемого образца со спектрами,
имеющимися в каталогах, для идентификации необходимо совпа-
дение положения и интенсивности всей совокупности полос погло-
щения в сравниваемых спектрах, снятых в одном и том же раст-
ворителе и при одинаковых других внешних условиях.
Если в спектре кроме полос предполагаемого основного ком-
понента наблюдаются «лишние» полосы или при совпадении числа
и положения полос существенно отличаются интенсивности, то это
свидетельствует о наличии примесей, которые требуется отделить
и идентифицировать. Когда примесь сама не поглощает в .иссле-
дуемой области спектра, ее наличие проявляется в кажущемся
уменьшении коэффициента экстинкции полос основного поглощаю-
щего компонента. Это же происходит с полосами поглощения
хромофора при увеличении молекулярной массы за счет групп
атомов в молекуле, не являющихся хромофорами и не проявляю-
щих ауксохромных свойств. Иными словами, удельный коэффи-
циент поглощения зависит от молекулярной массы. На этом
основывается один из методов ее определения по соотношению
(XIV.2).
Без каких либо предварительных предположений, химических
сведений и данных других методов о составе и строении компо-
нентов смеси ее качественный анализ по УФ спектрам погло-
щения— дело чрезвычайно трудное, если не сказать бесперспек-
тивное.
2.2. Количественный анализ
Измерение интенсивности в электронных спектрах поглощения
в видимой и УФ области, как и в ИК спектрах, основывается на
законе Бугера — Ламберта — Бера (XI.12), который в логариф-
мической форме имеет вид (XI.13)
Дх = гхс/
и рассмотрен в гл. XI вместе с рядом вопросов, являющихся об-
щими для абсорбционной спектрометрии в любом спектральном
диапазоне. Это касается, например, метода базовой линии при
измерении интегральных интенсивностей и т. п. Здесь рассмотрение
будет продолжено с учетом некоторой специфики практической
УФ спектрометрии, которая по ряду причин шире используется
химиками в целях количественного анализа, чем ИК спектроско-
пия. Прежде всего это обусловлено сочетанием высокой чувстви-
тельности метода с возможностью исследования сильно разбав-
ленных растворов при большой толщине слоя, так как существует
много растворителей, включая воду, прозрачных в видимой и УФ
областях.
При неизвестной относительной молекулярной массе анализи-
руемого вещества, когда раствор определяют в массовых долях
(%), вместо молярного коэффициента экстинкции в абсорбцион-
ной спектроскопии используют оптическую плотность для 1%-ноГо
раствора при толщине слоя 10 мм: АЙ , для которой принято
также обозначение £1см. Связь этой величины с относительной
молекулярной массой и коэффициентом экстинкции выражается
соотношением
£Й=10г/Л1. (XIV.1)
Если концентрацию вещества в растворе п выразить в г/см3, то
наблюдаемая оптическая плотность
А = 1000neZ/M. (XIV.2)
Если известен молярный коэффициент экстинкции хромофора е
(постоянен), а удельное, поглощение зависит от молекулярной
массы М поглощающего вещества, то, как уже отмечалось, соот-
ношение (XIV.2) при известных п, е и I может использоваться
для определения М по измеренной оптической плотности А.
Возможные отклонения от закона Бугера — Ламберта — Бера
могут быть связаны с высокой мощностью источника излучения,
с высокой концентрацией растворов, химическими или сильными
специфическими межмолекулярными взаимодействиями (ассоциа-
ция, водородные связи) и т. д. При таких отклонениях, т. е. от-
сутствии линейной зависимости оптической плотности от концен-
трации, строят градуировочные графики или применяют другие
специальные методы анализа с использованием ЭВМ.
Для многокомпонентных систем при выполнении закона Бу-
гера — Ламберта — Бера
A = zVe,.cz, (XIV.3)
i
(суммирование по всем компонентам), т. е. соблюдается аддитив-
ность поглощения. В частности, в случае двух поглощающих ве-
ществ X и Y для определения их концентраций в растворе нужно
решать систему двух уравнений вида (XIV.3), индекс i=X или У,
записанных для двух длин волн М и Лг. Обязательным условием
возможности решения системы является, как известно, неравен-
ство нулю ее определителя, что в данном конкретном случае при-
водит к неравенству
£x7®y ^sx/£y >
т. е. для анализа необходимо выбирать участки спектров, где ход
кривых поглощения компонентов смеси, возможно, более разли-
чен.
В, аналитической практике часто имеют дело со смесями пос-
тоянной суммарной концентрации двух веществ или постоянной
полной концентрации при равно-
весии двух форм (X=s=tY):
с=Сх-\-Су-
В этом случае, согласно
(XIV.3),
А = &хС\1 -}- ~\С\1
и 2
Л// = гхСх4"®у(£— сх)
или
All=e^c — Cx) + ^yCy,
Рис. XIV. 3. Изобестические точки
при ~234 и 272 нм для спектров по-
глощения водных растворов цитози-
на при различных pH:
кривые 1 и 4 (pH 6,9 и 13,0) соответствуют
протонированной и непротонированной
формам; кривые 2 и 3 (pH 12,0 и 12,6) —
смесям обеих форм
а отсюда
{А/1) —
Сх—------------
®Х ~ *Y
{АЦ) - ехС
И Су —-------------
eY — SX
(XIV.4)
т. е. если известна суммарная концентрация с, то для определе-
ния концентрации одного из компонентов достаточно провести
измерение А при одной длине волны.
Если при какой-то длине волны молярные коэффициенты экс-
тинкции компонентов одинаковы: е^=е^, то при этой длине
волны образуется так называемая изобестическая точка, в кото-
рой происходит пересечение спектральных кривых, получающихся
для различных соотношений компонентов смеси, как показано на
рис. XIV.3. В этой точке суммарная оптическая плотность не
будет зависеть от концентрации каждого из компонентов в отдель-
ности. Изобестические точки могут появляться в сериях спектраль-
ных кривых, регистрируемых для изучаемой системы при измене-
нии самых различных внешних параметров: температуры, давления,
pH, концентрации добавляемого к раствору вещества, раствори-
теля, времени протекания реакции и т. д.
По значениям оптической плотности Л‘ и Л при двух длинах
волн соответственно: изобестической точки Л, и другой произволь-
ной Л можно найти суммарную концентрацию и концентрацию
обоих компонентов. В изобестической точке:
А1=г‘с1 И С =--------,
ъ11
а из (XIV.4) следует
А _Л'(еу/е‘')
Сх-----77-----7—’
I (SX— sY)
откуда вытекает следующая линейная зависимость:
сх = Л-Лг(»у/В'~) = А________еу
с (sx Sy> А‘/s1' ех Sy ех — еу
Таким образом, можно, зная ех, еу и ъ1, определить долю каж-
дого компонента по значениям оптической плотности А‘ и А и,
наоборот, из зависимости отношения оптических плотностей от
состава смеси (по эталонам) можно найти значения коэффициен-
тов экстинкции чистых компонентов (методом наименьших квад-
ратов, например, или графически).
Обычно ошибка измерения концентраций спектрофотометриче-
ским методом в оптимальном диапазоне оптических плотностей
(~ 0,3...0,8) составляет ~3%. С увеличением числа компонентов
в смеси точность анализа быстро понижается.
Методом абсорбционной спектрофотометрии могут количест-
венно определяться, например, аквоионы различных неорганиче-
ских солей (кобальта, никеля, меди, редкоземельных и других
элементов), но чувствительность метода мала, так как молярный
коэффициент погашения растворов аквокомплексов обычно не-
выше ~102. В таких случаях часто оказывается недостаточным
получить раствор анализируемого образца. Если определяемое-
вещество поглощает очень слабо, то проводят реакцию, которая
переводит его в соединение, обладающее достаточно сильным
поглощением в каком-то участке спектра. Одной из важнейших
таких реакций является реакция комплексообразования со спе-
циальными органическими или неорганическими реагентами. Ком-
плексные или внутрикомплексные (хелатные) соединения ионов
металлов или анионов с органическими лигандами обычно раство-
римы в воде и дают интенсивные полосы поглощения (е« 104...105).
Органические вещества, образующие такие комплексы с метал-
лами, называют, в частности, метал лохромн ы м и комплек-
сонометрическими индикаторами. Существуют также
комплексы, образующие с катионами металлов интенсивно флуо-
ресцирующие хелаты.
Соединения с неорганическими лигандами (анионы неоргани-
ческих кислот, аммиак, пероксид водорода и т. д.) обладают менее
интенсивным поглощением (е'лЛО3). Например, KSCN образует
с Ее (III) слабо окрашенный комплекс, так что чувствительность
определения металлов с такими индикаторами не очень велика.
Для спектрофотометрического определения некоторых элементов
(Si, Р, As, Се, Zr) используют гетерополисоединения.
ГЛАВА XV
ТЕХНИКА И МЕТОДИКИ ЭЛЕКТРОННОЙ
спектроскопии
1. АППАРАТУРА АБСОРБЦИОННОЙ СПЕКТРОСКОПИИ
Оптическая область электромагнитного спектра, в которой наб-
людаются электронные переходы, охватывает интервал длин волн
от ~ 120 нм, т. е. от жесткого УФ излучения, до ~ 1200 нм, т. е.
Рис. XV.1. Блок-схема УФ спектрофо-
тометра:
1 — блок сменных источников излучения
•с осветительным устройством; 2 — моно-
хроматор с входной и выходной щелями
и диспергирующим устройством (кварце-
вая призма или дифракционная решетка);
3 — кюветное отделение (кювета с обра-
зом, кювета сравнения); 4 — сменные при-
емники; 5 —усилитель; 6 — регистрирую-
щее устройство или блок связи с ЭВМ
никает необходимость
исследования
теплового излучения ближней
ИК области; это соответ-
ствует волновым числам от
~ 80 000 см-1 до ~8000 см-1
(энергиям от ~ 10 до ~1 эВ).
Границей видимой и УФ обла-
сти считают 400 нм, а видимой
и ИК области — 750 нм. Ниже
180 нм лежит далекая или ва-
куумная УФ область, так как
при этой длине волны начи-
нается поглощение излучения
атмосферным кислородом, а
ниже 160 нм — и азотом, и воз-
спектрометрах. Вообще для
в каждой ее
в вакуумных
всей указанной области необходима
части своя техника эксперимента, включая различные источники
и приемники излучения, прозрачные материалы и т. д.
Что касается принципов устройства и действия спектрофото-
метров, то они во многом являются общими с теми, которые рас-
смотрены в гл. XII, где речь шла о приборах для исследования
ИК спектров поглощения, хотя имеется специфика, обсуждаемая
ниже. Основные узлы спектрофотометра представлены на блок-
схеме (рис. XV.1).
Источники излучения. Для УФ области спектра в качестве источ-
ника излучения используется водородная лампа с дуговым разрядом
или в 3—5 раз более мощная дейтериевая лампа, дающая сплош-
ной спектр излучения в области 180...400 нм при мощности
30...60 Вт. В видимой области у этой лампы имеются линии 486,0
и 656,1 нм, которые могут служить для калибровочных целей.
Для изучения спектров люминесценции и в некоторых случаях для
аналитических целей в абсорбционной спектроскопии используются
такие мощные источники излучения, как ртутная или ксеноновая
лампы высокого давления. Последняя имеет непрерывный спектр
в области 190...750 нм, а ртутная лампа дает над сплошным фо-
ном ряд сильных линий в области от 240 до 700 нм и далее непре-
рывный спектр до 2,6 мкм. При работе с источниками УФ излу-
чения необходимы их хорошая экранировка для предохранения
ют прямого воздействия излучения на глаза и кожу, а также
исключение возможности появления образующегося в результате
фотохимической реакции крайне токсичного озона.
Обычным источником видимого излучения от 360 нм до ближ-
ней ИК области является лампа накаливания с вольфрамовой
нитью. Ее рабочая температура -~2900 К, а максимум интенсив-
ности излучения лежит при ~ 1000 нм. С уменьшением длины
волны интенсивность быстро падает, но источник может исполь-
зоваться во всем видимом диапазоне. В последнее время появились
более мощные вольфрам-галогеновые лампы и другие источники,
в том числе импульсные. Изучаются также принципиально новые
возможности, открываемые применением в качестве источников
излучения лазеров, в частности, на красителях — с плавно пере-
страиваемой длиной волны — для бездисперсионной спектроско-
пии.
Монохроматоры. Монохроматор или двойной монохроматор
имеет входную и выходную щели и призмы или дифракционные
решетки, плоские или вогнутые, в качестве диспергирующих эле-
ментов. Для уменьшения паразитного (рассеянного) излучения
могут использоваться различные фильтры, этому же способствует
двойная монохроматизация.
К числу оптических элементов, которые могут находиться в
приборе в зависимости от его конструкции и принципов действия
(однолучевая или двухлучевая схема и т. п.), относятся линзы,
зеркала, окна, а также делители пучков, прерыватели и т. д.
Прозрачными материалами служат в видимой и УФ областях оп-
тическое стекло, кварц, LiF, CaF2, КС1 и др.
Для абсорбционной спектроскопии в УФ и видимой областях спектра суще-
ствуют следующие отечественные спектрофотометры. Однолучевой спектрофото-
метр СФ-26 с рабочим диапазоном 186... 1100 нм и его новая модификация
СФ-46. Модель двухлучевого спектрофотометра СФ-20 (сменившая модель-
СФ-8) имеет рабочий диапазон 190... 2500 нм и позволяет проводить измерения
в шкале пропускания или шкале оптической плотности. В монохроматоре уста-
новлены две сменные дифракционные решетки, смена которых в процессе ска-
нирования спектра осуществляется автоматически, как и раскрытие щели
(последнее с обеспечением постоянства энергии на выходе системы). Модель
СФ-39 (190... 750 нм)—с автоматизацией на мини-ЭВМ. Производится также
специально для исследований в видимой области спектрофотометр СФ-18, пред-
ставляющий собой двойной монохроматор с рабочим диапазоном 400 ... 750 нм_
Кюветное отделение обычно делается в приборах достаточно
просторным, чтобы в нем могли размещаться газовые кюветы,
термостатирующие и другие специальные устройства.
Кюветы. Нормальные жидкостные кюветы имеют объем 2 мл
и толщину слоя 10 мм. Обычно они прямоугольной формы, хотя
бывают и цилиндрические, изготавливаются из стекла или кварца,
а иногда из прозрачного в области до 300 нм органического стек-
ла. Кювета закрывается притертой крышкой или притертой проб-
кой. Используются также полумикро- и микрокюветы: к первым
относятся кюветы с объемом раствора от 0,5 мл и выше (до
нормального, см. выше) и толщиной слоя около 4 мм, а ко вто-
рым— с объемом меньше 0,2 мл и толщиной слоя меньше 2 мм.
При работе с газами или очень разбавленными растворами
на некоторых спектрофотометрах предусмотрено использование
кювет с толщиной слоя 100 мм. Для специальных целей, в част-
ности для непрерывного анализа в потоке, применяют проточные
кюветы. Иногда используются разборные кюветы, как проточные,
так и обычные.
При количественных измерениях интенсивности поглощения
необходимо термостатирование образцов или, по крайней мере,
температурный контроль. Измеряемая оптическая плотность раст-
вора обычно зависит от температуры, что связано прежде всего
•с температурной зависимостью плотности растворителя. Так, напри-
мер, при температуре, близкой к комнатной, плотность диоксана
изменяется на 0,11 % при изменении температуры на 1°С. Это
значит, что нагревание кюветного отделения спектрометра на 10°С,
включенного в холодном помещении, приведет к изменению из-
меряемой оптической плотности А диоксанового раствора на 1,1%.
-С изменением температуры может происходить также смещение
равновесия каких-то существующих в растворе молекулярных
•форм, разложение растворенного образца или какая-то химическая
реакция, что приведет к изменению измеряемой при какой-то
длине волны оптической плотности А>, и кажущегося коэффициен-
та погашения аг..
Прецизионные измерения интенсивности должны проводиться
поэтому при определенной температуре, которая должна поддер-
живаться постоянной в пределах ±0,5°С. При изучении кинетики
каких-то реакций точность регулировки и измерения температуры
образца должна быть еще выше (±0,05°С). Это обеспечивается
различными термостатирующими устройствами или для всего кю-
ветного отделения, или для держателя кювет, или самих кювет.
Для работы при низких температурах существуют криостаты, а
простейшим устройством может служить прозрачный сосуд Дьюа-
ра (без зеркального покрытия), заполняемый каким-либо проз-
рачным хладагентом, например жидким азотом, в который погру-
жается исследуемый образец.
В некоторых современных спектрофотометрах имеются устрой-
ства для автоматической смены кювет с временным реле, что об-
легчает, например, кинетические измерения. При таких измере-
ниях используют также специальные маленькие механические или
магнитные мешалки, не препятствующие прохождению через
кювету светового пучка. Обычно к спектрофотометрам придаются
парные кюветы, подобранные по пропусканию во всем рабочем
диапазоне, одна из которых заполняется исследуемым раствором,
д другая используется как кювета сравнения.
При проведении массовых анализов или в непрерывном кон-
троле на производстве с помощью абсорбционной УФ спектроско-
пии используются различные устройства, обеспечивающие автома-
тический отбор и смену образцов, или проточные системы.
Приемники. В качестве приемников в УФ и видимой областях
используют вакуумные фотоэлементы и фотоэлектронные умножи-
тели (ФЭУ), а также твердотельные фотоэлементы и фотопла-
стинки. Спектральная чувствительность фотокатода вакуумных
элементов определяется его покрытием, в качестве которого ис-
пользуется тонкий слой щелочного металла, чаще всего цезия.
Для УФ области (150...400 нм) приемником служит фотоэлемент
с сурьмяно-цезиевым фотокатодом, иногда с включением серебра
или его оксида, в видимой и близкой ИК областях (до 1200 нм)
применяют элементы с кислородно-цезиевым катодом. Могут быть
и другие покрытия. В ФЭУ происходит многокаскадное усиление
336
слабого фототока. В твердотельных фотоэлементах применяют
полупроводниковые покрытия, в частности, для видимой области
это может быть тонкий слой селена между тонким железным или
алюминиевым электродом и напыленным покрытием из серебра
или золота (освещаемая поверхность). В качестве чувствительных
приемников для УФ и видимой области используют также крем-
ниевые диоды и видиконовые трубки, которые в сочетании с ЭВМ
могут применяться как многоканальные анализаторы на нескани-
рующих спектрометрах.
Иногда при исследовании УФ и видимых электронных спектров
для их регистрации используют специальные фотопластинки или
фотопленки с соответствующей спектральной чувствительностью.
•Фотографический способ регистрации спектров оставляет за собой
право на существование, позволяя получать достаточно высокое
разрешение и обеспечивая высокую точность определения длин
волн (частот), например, полос колебательной, а для легких, в
частности, двухатомных молекул и линий вращательной струк-
туры спектров.
Регистрирующие системы. Связанные с приемником излучения
электронный усилитель и самописец обеспечивают на обычных
спектрометрах регистрацию спектра в виде кривой зависимости
интенсивности (оптической плотности или пропускания) от длины
волны. Автоматизация эксперимента и обработки результатов из-
мерений требует сочетания спектрофотометра с ЭВМ, это же не-
обходимо и для внедрения и эффективного использования новых
прогрессивных методик спектроскопии (некоторые из них будут
рассмотрены).
Для сочетания ЭВМ со спектрометрами (любого типа, как и
с другими исследовательскими приборами и измерительными уст-
ройствами) существуют разного типа интерфейсы. Они выполняют
две основные задачи: 1) обеспечивают преобразование данных,
выдаваемых одним из партнеров, к виду, который может воспри-
ниматься другим, и 2) передают информацию от одного партнера
другому и обратно в оптимальном режиме. В частности, спектро-
метр дает сигнал в аналоговой форме, а ЭВМ требует его преоб-
разования в цифровую форму. Обратно, запрошенный из памяти
ЭВМ спектр может быть получен на самописце спектрометра в гра-
фической форме. Режим передачи информации должен обеспечи-
вать наиболее рациональную загрузку ЭВМ. Существуют как стан-
дартные интерфейсы, производимые для некоторых типов устройств,
так и создаваемые специалистами в лабораторных условиях ин-
терфейсы разного уровня сложности.
2. ПОДГОТОВКА ОБРАЗЦОВ
При исследовании газов и жидкостей требуется лишь выбрать
соответствующую кювету с нужной толщиной слоя, а для газа —
также подобрать необходимое давление.
Приготовление растворов. Растворитель для приготовления
растворов выбирается с учетом свойств как растворяемого веще-
12—949 337
Таблица XV. 1. Некоторые растворители для УФ спектроскопии
Растворитель * кн ** S X u W о 03 OJ LO « £(20. . . 25° С) 1 О 2 в S X Плотность, г/см3 (20°С)
Вода 195 1,3330 78,5 0 100 0,9982
Метиловый спирт 210 1,3288 32,6 —97,8 64,6 0,7913
Этиловый спирт ,(абс.) 207 1,3610 24,3 —117,3 78,3 0,7893
Хлороформ 246 1,4460 4,8 —63,5 61,2 1,489
Ацетон 331 1,3591 20,7 —94,8 56,2 0,791
Диоксан 215 1,4224 2,2 11,8 101,3 1,0337
Бензол 280 1,5011 2,3 5,5 80,1 0,8790
Гексан 199 1,3749 1,9 —95,3 68,7 0,649
Циклогексан 211 1,4263 2,0 6,6 80,9 0,7787
* Гексан и циклогексан нерастворимы, бензол малорастворим (0,07) в воде, раствори-
мость остальных бесконечна, кроме хлороформа (0,82).
** Коротковолновый предел, гд,е при толщине слоя 10 мм пропускание У=0,25.
ства, так и самого растворителя. Свойства некоторых раствори-
телей, используемых в УФ спектроскопии, указаны в табл. XV.1.
Хорошо известно, что полярные вещества легче растворяются в
полярных растворителях, а неполярные — в неполярных. Для
подавляющего большинства низкомолекулярных соединений прак-
тически всегда можно найти подходящий растворитель, тогда как
высокомолекулярные соединения в любом растворителе раство-
ряются с большим трудом и не образуют истинных растворов.
При выборе растворителя необходимо учитывать также возмож-
ность взаимодействия его с растворяемым веществом, область
прозрачности и вызываемые неспецифическими межмолекуляр-
ными взаимодействиями смещения максимумов полос поглощения.
Концентрация растворов подбирается так, чтобы значение оп-
тической плотности в максимуме полосы попадало в оптимальный
интервал фотометрических измерений, который для больщинства
современных двухлучевых приборов с фотоумножителем в каче-
стве приемника составляет 0,3...1,5, а на однолучевых приборах
с фотоэлементами его пределы лежат несколько ниже
( ~ 0,15...0,8). Растворы готовят обычным образом в мерных кол-
бах, беря, насколько возможно, точные навески образца. При
использовании кювет с толщиной слоя 10 мм и значении коэф-
фициента экстинкции 8 порядка 1О3...1О4 обычные концентрации
растворов составляют ~ 10~2 г-л-1 (или ~1СН м-л~‘), т. е. для
приготовления 10 мл раствора требуются менее чем миллиграм-
мовые массы образца (при молекулярной массе порядка 102).
При необходимости получения очень разбавленных растворов
используют метод последовательных разведений. Особое внима-
ние должно обращаться на чистоту и аккуратность заполнения
кювет.
Твердые образцы. Абсорбционные спектры
монокристаллов, стекол, полимерных образ-
цов, если они имеют плоскопараллельные гра-
ни, измеряют просто, помещая образец в пу-
чок излучения, если необходимо, то с диафраг-
мированием. Если образец имеет неправиль-
ную форму, то его можно погрузить в иммер-
сионную жидкость с близким показателем пре-
ломления, которая не поглощает в исследуе-
мой области спектра. Используют также тех-
нику прессования таблеток из смеси исследуе-
мого вещества с галогенидами щелочных ме-
таллов или технику приготовления взвесей,
как для ИК спектроскопии (см. гл. XII;
1.2). Как твердые, так и жидкие образцы,
включая расплавы, могут исследоваться также
методом НПВО или МНПВО (см. гл. XII; 2).
3. СПЕКТРОСКОПИЯ с ДИФФЕРЕНЦИРОВАНИЕМ,
РАЗНОСТНАЯ СПЕКТРОСКОПИЯ
И ДВУХВОЛНОВАЯ СПЕКТРОСКОПИЯ
Для увеличения «контрастности» спектров,
т. е. лучшего выявления всех характерных осо-
бенностей спектральных кривых, может про-
водиться их преобразование диффе-
ренцированием. Тогда спектр представ-
ляет собой не функцию оптической плотности
от длины волны: А = /(л), а кривую зависимо-
сти от А первой или второй и т. д. производной
оптической плотности по длине волны:
dA/dX=f (7.), d2A/dV—f"(A.), — • Для первой
и вообще нечетных производных вместо обыч-
ной полосы поглощения получаются кривые
вида дисперсионной функции. Эти производ-
ные позволяют легче выявить и определить по-
ложение точек перегиба и замаскированных
пиков (в максимумах поглощения dA/dA=0).
На рис. XV.2 показано, как выглядят про-
изводные функции Гаусса, а на рис. XV.3 в ка-
честве примера приведены спектр поглощения
антрацена до дифференцирования и его первая
Рис. XV.2. Функ-
ция Гаусса, аппро-
ксимирующая кон-
тур полосы погло-
щения, и ее произ-
водные (/—IV)
производная по Z.
Вторая и последующие четные производные дают пики, сов-
падающие по положению с максимумом полосы поглощения
(рис. ХУД). Эти пики резче, чем исходная полоса, за счет чего
Рис. XV.3. Спектр поглощения ан-
трацена (1) и кривая зависимости
первой производной спектра от
длины волны (2):
горизонтальная пунктирная линия —
нуль производной
может быть получено более высокое
разрешение, хотя из-за сателлитных
пиков меньшей интенсивности кар-
тина несколько усложняется.
Если выполняется закон Буге-
ра— Ламберта—Бера, т. е. при за-
данной длине волны Ai=eid, то ли-
нейная зависимость от концентра-
ции при данной X сохраняется и для
производных:
dM/dX« = (dne/dk'!)c/.
Регистрация спектра с диффе-
ренцированием * кривых поглоще-
ния может осуществляться на раз-
ных приборах по-разному. Иногда
используют оптико-механический
метод с модуляцией длины волны, но более перспективным, особен-
но для получения второй и высших производных, является приме-
нение электронных аналоговых или цифровых вычислительных
машин.
В известном смысле абсорбционная спектроскопия практиче-
ски почти всегда является разностной, так как из спектра погло-
щения, скажем, раствора всегда вычитают поглощение раствори-
теля и ослабление пучка излучения парной кюветой, т. е измеряют
ДА = (ЯОбР4-Лр4-Лк) — (АР4-Ак) — Асбр,
где Аобр — оптическая плотность растворенного образца; Ар —
оптическая плотность растворителя; Ак — эффективная оптическая
плотность кюветы.
Однако имеются и особые приемы, относящиеся к разност-
ной спектроскопии, в частности следующие. Допустим,
что требуется провести определение некоторого компонента в жид-
кой среде, которую назовем матрицей, имеющей свой накладываю-
щийся спектр поглощения. Если можно каким-то образом изме-
нять физические или химические свойства этого компонента, т. е.
и его спектр, а спектр матрицы при этом изменяться не будет, то
этот компонент поддается определению в данной матрице. Напри-
мер, поглощение определяемого компонента при данной длине
волны может зависеть от pH, Т или какого-то другого параметра,
а поглощение матрицы не зависит. Тогда для двух значений изме-
няемого параметра значения измеряемой оптической плотности
* Иногда в литературе можно встретить термин дифференциальная
спектроскопия, использования которого следует избегать, так как он вно-
сит путаницу между спектроскопией с дифференцированием и
разностной спектроскопией.
выражаются следующими формулами:
Д — еобр^обр^ ~Ьематр£матр^’
А' = еобр£обр^ 4“ ®матр^матр^’
а их разность:
ДД = | Л — Д' | = I еСбр — ®обр 1 собр^ — Аеобрсобр^-
При выполнении закона Бера при аналитической длине волны и
соответствующей калибровке концентрацию определяемого компо-
нента (образца) можно найти из соотношения
△Лобр/А-^этал = ^сбр/^Эчта.т
Следует иметь в виду, что с увеличением коэффициента экстинк-
ции матрицы ематр относительно коэффициента экстинкции образ-
ца растет систематическая ошибка, связанная с неопределен-
ностью концентрации матрицы сматф.
Другой прием разностной спектроскопии, применяемый при
точных определениях сильно поглощающих растворов, заключает-
ся в вычитании спектра стандартного раствора того же вещества
несколько меньшей концентрации, чем в анализируемом образце.
При выполнении закона Бера разность оптических плотностей
анализируемого и стандартного растворов будет
АД = Добр Дэтал == ® (^Сбр ^этал) tLcl.
На практике обычно строят калибровочные кривые зависимости
ДД от Дс, что уменьшает ошибки, связанные с рассеянным светом,
шумами и нелинейностью фотометрической шкалы.
В количественном анализе по разностным спектрам может
применяться, как указывалось, их преобразование дифференциро-
ванием с получением первой или второй производной.
Существует также более сложный в инструментальном отно-
шении и менее распространенный метод двухвол новой
спектроскопии, в котором сравниваются два независимо
монохроматизованных пучка при двух разных длинах волн, одно-
временно проходящих через одну и ту же кювету, т. е.. измеряется
разность оптических плотностей
ДД = ДХа — дЧ
где Ха — аналитическая длина волны; Хс — длина волны пучка
сравнения.
Этот метод позволяет исключить поглощение матрицы, анали-
зировать многокомпонентные смеси, причем точные измерения им
возможны как для сильно поглощающих, так и для слабо погло-
щающих систем.
В двухволновом спектрометре вместо одного монохроматора
используются два независимых монохроматора, выделяющих две
любые выбираемые длины волны, на которых измеряют и сравни-
вают поглощение пучков в одной и той же кювете.
Двухволновая спектрофотометрия позволяет „на порядок по-
высить чувствительность по сравнению с обычной двухлучевой
спектрофотометрией и измерять очень малые изменения оптиче-
ский плотности (до 10~3). Поскольку измерения обычно проводят-
ся при близких значениях длин волн ?.а и Л,с, легче исключается
паразитное рассеянное излучение, которое, будучи примерно оди-
наковым при этих длинах волн, взаимно компенсируется. На двух-
волновом спектрометре легко записать первую производную
спектра, просто беря постоянную маленькую разность ДЛ.(~1...
2 нм) и проводя сканирование. Возможно изучение двух различ-
ных процессов, одновременно протекающих в одной и той же кю-
вете, если регистрацию изменения оптической плотности при двух
длинах волн проводить на отдельных самописцах.
Удобно и продуктивно применение двухволновой спектрометрии
при изучении равновесий и кинетики химических реакций.
Если, например, при аналитической длине волны ?.а
71 71 об:, Т^матр’
а длина волны пучка сравнения Хс выбрана так, что при ней
Ао6ср=°> а Аисатр=Амаатр’ то ДЛ = Аха-Лхс = Лохар. Это можно Рас-
сматривать как один из специальных случаев разностной спектро-
скопии. При изучении комплексообразования и равновесий моле-
кулярных форм в качестве Хс может использоваться изобестиче-
ская точка.
4. СПЕКТРЫ ЛЮМИНЕСЦЕНЦИИ
4.1. Теоретические основы
Экспериментальные методы исследования спектров люминес-
ценции основываются на релаксационных процессах, происходя-
щих при электронном возбуждении частиц вещества. Перевод мо-
лекул в возбужденное состояние может осуществляться различны-
ми путями, как и расходование приобретенной при этом молеку-
лами энергии. Свечение вещества, возникающее при переходе
молекул из возбужденного состояния в основное, называют люминес-
ценцией, а если возбуждение происходило за счет поглощения
электромагнитного излучения в оптической области, то испуска-
ние излучения в процессе релаксации называют фотолюминесцен-
цией в отличие, например, от рентгенолюминесценции,
термо- и электролюминесценции, хемилюминес-
ценции ит. д.
Когда прямой и обратный переходы, происходящие одновре-
менно, осуществляются между одними и теми же фиксированны-
ми энергетическими уровнями, частоты эмиссионного спектра точ-
но совпадают с частотами спектра поглощения. Если бы заселен-
ности нижнего и верхнего состояний были одинаковы, то не на-
342
блюдалось бы ни поглощения, ни
испускания электромагнитного
излучения. Поскольку наиболее
заселенным является основное
электронное состояние, а еще
точнее.— его нулевой колебатель-
ный уровень энергии, то при об-
лучении наблюдаются полосы по-
глощения, связанные с перехода-
ми молекул с этого уровня на ко-
лебательные подуровни того или
иного возбужденного электронно-
го состояния, как это схематично
показано на рис. XV.4. При этом
может возникать неравновесная
заселенность колебательных уров-
ней.
В возбужденном электронно-
колебательном состоянии избы-
Рис. XV.4. Схема низших энергетиче-
ских уровней молекулы и переходов
между ними при фотолюминесценции
ток колебательной энергии может теряться в результате межмоле-
кулярных столкновений или каких-то других безызлучательных
колебательных переходов. Иными словами, происходит колебатель-
ная релаксация (к. рел), представляющая переход от неравновесно-
го распределения по колебательным уровням, энергии в данном
электронном состоянии к равновесному тепловому распределению.
В конечном счете наиболее заселенным опять-таки оказывается ну-
левой (все щ = 0) колебательный уровень (см. рис. XV.4).
В зависимости от характера электронного состояния, из кото-
рого молекулы переходят в основное состояние с испусканием
электромагнитного излучения, фотолюминесценция подразделяет-
ся на флуоресценцию и фосфоресценцию. Флуорес-
ценция наблюдается при переходе между состояниями, имеющими
одинаковую мультиплетность, обычно между синглетными первым
возбужденным и основным Si->S0. Возможные безызлучательные
переходы между различными электронными состояниями одной и
той же мультиплетности называют внутренней конверс и-
е й (вн. к) и обозначают, как любые безызлучательные переходы,
волнистой стрелкой, в частности $2-----—. Среднее время
жизни молекулы в возбужденном синглетном состоянии мало, и яв-
ление флуоресценции связано с высокой вероятностью спонтанного
перехода молекул в основное синглетное состояние, т. е. характери-
зуется отсутствием длительного «послесвечения» (после
прекращения облучения, приводящего к электронному возбужде-
нию молекул).
Затухание флуоресценции происходит по экспоненциальному
закону:
Z = Zoe-^, (XV. 1)
где I — интенсивность флуоресценции через время t после прекра-
щения облучения; /0 — постоянная интенсивность флуоресценции
во время облучения; т — среднее время затухания, за которое ин-
тенсивность флуоресценции уменьшается от исходной величины в
е раз (обычно имеет порядок 10~4... 10~-9 с).
Если спонтанный радиационный переход представляет единст-
венный процесс, путем которого дезактивируется возбужденное
состояние, то естественное среднее время жизни этого состояния
совпадает со средним временем затухания т в (XV. 1) и обратно
пропорционально константе скорости естественного затухания
фотолюминесценции &ф.л:
Т°=1/^ф.л.
Рассмотренная в гл. XIII; 4 сила осциллятора f, характеризую-
щая вероятность спонтанного перехода из более высокого в более
низкое электронное состояние, связана с естественным средним
временем жизни возбужденного состояния равенством
/=150—---------—, (XV.2)
J g" О)2т0
где gz и g" — мультиплетности соответственно верхнего и нижнего
состояний.
Это равенство получено применительно к атомным переходам,
а для молекулярных переходов его можно использовать лишь с
корректировкой, но и при этом, особенно в случае малых интен-
сивностей переходов, возможны лишь грубые оценки только по
порядкам величин.
Как возможны с малой вероятностью радиационные (излуча-
тельные) переходы между электронными состояниями разной
мультиплетности (см. гл. XIII; 4), также возможны и безызлуча-
тельные переходы между ними, называемые интеркомбина-
ционной конверсией (ин. к). Если, например, первое воз-
бужденное синглетное состояние близко по энергии к первому
триплетному состоянию, то может осуществляться безызлучатель-
ный переход , индуцируемый «столкновениями»
молекул, находящихся на колебательных подуровнях этих состоя-
ний вблизи «пересечения» их потенциальных гиперповерхностей.
В триплетном состоянии происходит колебательная релаксация, т. е.
достигается равновесное распределение с наибольшей заселенно-
стью нулевого колебательного уровня.
Оптические переходы между электронными состояниями разной
мультиплетности, обычно нижним возбужденным триплетным и
344
основным синглетным T^So, приводят к явлению фосфоресцен-
ции. В связи с большим естественным временем жизни триплет-
ного состояния и очень малой вероятностью указанного перехода
константа скорости затухания фосфоресценции мала, что и обу-
словливает длительное послесвечение «фосфоров».
Реальное время жизни люминесцирующего возбужденного со-
стояния, как и среднее время затухания т, зависит не только от
вероятности радиационных переходов или величины &ф.л, но и от
констант скорости (или вероятностей) всех других процессов:
внутренней (&вн.к) и интеркомбинационной (&ин.к) конверсий и
фотохимических реакций (£ф.р), т. е.
1
т —---------------------.
^ф.л Н“ ^вн.к 4“ ^ин.к ^ф.р
На общей схеме (рис. XV.4) энергетических уровней и перехо-
дов между ними для молекул люминесцирующего вещества запре-
щенные по спину переходы с поглощением и фосфоресценцией
указаны пунктирными стрелками, а разрешенные (поглощение,
флуоресценция) — сплошными стрелками; волнистые стрелки соот-
ветствуют безызлучательным переходам (вн. к и ин. к), показана
также колебательная релаксация (к. рел). Жирные горизонталь-
ные линии, обозначающие электронные состояния, можно считать
нулевыми колебательными уровнями, энергия которых включает
сумму нулевых колебательных энергий по всем нормальным коор-
динатам (см. гл. VIII), а остальные уровни (горизонтальные ли-
нии) соответствуют, как обычно, одному или нескольким отлич-
ным от нуля колебательным квантовым числам vk-
В конденсированных системах для высоковозбужденных моле-
кул наиболее вероятным является процесс внутренней конверсии,
и радиационный переход с флуоресценцией обычно происходит с
самого нижнего возбужденного уровня данной мультиплетности.
Поэтому, во-первых, полоса флуоресценции, как правило, смещена
в область более низких частот по сравнению с полосами поглоще-
ния, и, во-вторых, ее положение не зависит от длины волны воз-
буждающего излучения, соответствующей какому-то электронно-
му переходу с поглощением.
Первая особенность: Упогл>Тф.л— получила название законна
Стокса — Ломмеля, а длинноволновое (красное) смещение
полосы испускания относительно поглощаемой частоты названа
стоксовым. Иногда этот закон нарушается и наблюдают' ком-
поненты полосы фотолюминесценции со стороны более высоких
частот (меньших длин волн) относительно частоты 0—0-полосы
поглощения, называемые антистоксовыми*.
* Отсюда термины «стоксовы» и «антистоксовы» были механически' перене-
сены на длинноволновые и коротковолновые относительно релеевского рассеяния
сателлиты спектра КР (см. гл. VIII), не имеющие по своей природе никакого от-,
ношения к спектрам фотолюминесценции.
Если сопоставить прямой и обратный переходы между одними
и теми же электронными состояниями, т. е., например, процессы
поглощения Si<-S0 и флуоресценции Si^S0 (см. рис. XV.4), то
легко видеть следующую характерную черту. Самая низкая часто-
та вибронного перехода в спектре поглощения совпадает с самой
высокой частотой в спектре флуоресценции, так как обе отвечают
О—0-полосе. Предположим, что соединенные с нулевым уровнем
стрелками ненулевые колебательные подуровни электронных со-
стояний So и Si характеризуются последовательно одинаковыми
квантовыми числами т?" и v'k, а соответствующие колебательные
кванты при А^=1 и &v'k =1 (индекс k один и тот же) равны или
приблизительно равны. Тогда полосы спектра флуоресценции с
низкочастотной стороны 0—0-полосы будут расположены зеркаль-
но симметрично соответствующим полосам спектра поглощения,
расположенным с высокочастотной стороны 0—0-полосы, т. е.
происходит инверсия колебательной структуры. Из сказанного еще
не следует, что относительные интенсивности полос спектров по-
глощения и испускания тоже будут зеркально симметричны. Одна-
ко явление приближенной зеркальной симметрии спектров погло-
щения и люминесценции, называемое по имени его первого иссле-
дователя правилом Левшина, наблюдается экспериментально
(рис. XV.5). В то же время полного зеркального соответствия нет
никогда, и отклонения от него связаны, во-первых, с отличиями
функций потенциальной энергии в основном и возбужденном
электронных состояниях и, во-вторых, с разной частотной зависи-
мостью интенсивности спонтанного процесса испускания и вынуж-
денного процесса поглощения. С перекрыванием колебательной
структуры полос спектров поглощения и испускания может быть
связано и появление антистоксовых компонентов в спектре фото-
люминесценции.
Величину, определяющую долю радиационных переходов по
отношению ко всем процессам, приводящим к уменьшению засе-
ленности возбужденного электронного состояния, называют в ы-
ходом люминесценции. Он характеризует эффективность
преобразования возбуждающего излучения исследуемым вещест-
вом в свет фотолюминесценции. Различают энергетический
и квантовый выходы. Первый представляет собой отно-
шение излучаемой при люминесценции энергии Ещт к поглошенной
энергии излучения £Погл, равное при стационарном режиме непре-
рывного облучения и испускания отношению соответствующих ин-
тенсивностей:
Г = £исп. = 2исп . (XV.3)
Епогл Люгл
Квантовым выходом у называют отношение числа квантов лю-
минесценции, испускаемых единицей объема вещества в единицу
времени Nwni, к числу поглощенных фотонов возбуждающего излу-
Чвния Л^погл**
У = ЛГисгЖ,огл- (XV.4)
Его можно определить так-
же через константы скоро-
сти затухания фотолюмине-
сценции и различных безыз-
лучательных процессов сле-
дующим образом:
j). л
у —-----------------------
^ф.л + ^ВЧ.К + ^ин.к + ^ф.р*
Энергетический и квантовый
выходы связаны соотноше-
нием
Г = Y ,
^’возб
где Уф.л — средняя частота
(«центр тяжести») полосы
фотолюминесценции; vBO36 —
Рис. XV.5. Правило зеркальной симметрии
Левшина:
а — общая схема зеркальной симметрии спектров
поглощения (пунктирная кривая) и флуоресцен-
ции; б — приближенная зеркальная симметрия по-
лос переходов <S0->Si (пунктирная кривая) и
5’i->S0 с инверсией колебательной структуры для
антрацена
частота возбуждающего из-
лучения.
Выход люминесценции
чувствителен к внешним воз-
действиям, которые могут
приводить ктушению све-
чения. Возможно тушение
люминесценции посторонними примесями (тушителями), темпера-
турное, концентрационное (при высоких концентрациях), известны
и другие виды тушения. Механизм тушения обычно связан с раз-
витием безызлучательных процессов деградации энергии возбуж-
дения за счет увеличения числа столкновений частиц или образо-
вания нелюминесцирующих ассоциатов, или ионизации, диссоциа-
ции и т. д. Учет тушения, влияющего на интенсивность люмине-
сценции, является очень важным в люминесцентном анализе.
4.2. Практическое применение
и техника люминесцентной спектроскопии
Спектры люминесценции, как и абсорбционные электронные
спектры, применяются для качественного и количественного ана-
лиза, в структурных исследованиях, изучения электронно-колеба-
тельных состояний молекул, физико-химических свойств растворов,
газообразных, жидких и твердых образцов. Это понятно из преды-
дущего рассмотрения природы этих спектров, и нет каких-либо
принципиальных отличий в извлекаемой из них информации по
сравнению со спектрами поглощения.
Таблица XV.2. Выход флуоресценции некоторых ароматических
углеводородов в гексановом растворе при возбуждении линией 253,7 нм, 20°С
Соединение Выход Г Диапазон длин волн, нм Соединение Выход Г Диапазон длин волн, нм
.Бензол 0,11 270 ... 310 Фенантрен 0,27 380 ... 470
Тексаметил бензол 0,04 280 ... 330 Флуорен 1,00 300 ...370
Нафталин 0,38 300... 360 Дифенил 0,23 290 ... 360
Антрацен 0,46 370... 460 Аценафтен 0,70 330 ... 400
К особенностям качественного люминесцентного анализа, поз-
воляющего по спектру свечения обнаруживать и идентифициро-
вать вещество или группу вещества, можно отнести еще большую
узость круга поддающихся такому анализу объектов, чем в аб-
сорбционной спектроскопии. Четкими, имеющими характерную
структуру спектрами люминесценции обладают немногие вещест-
ва: соединения редкоземельных элементов, соли уранила, арома-
тические соединения, порфирины и некоторые другие. В большин-
стве случаев наблюдаются широкие, лишенные структуры полосы,
которые для смеси флуоресцирующих веществ часто перекрывают-
ся и трудно разделимы.
В основе структурного анализа по спектрам люминесценции
лежит не только зависимость положения полосы, но и квантового
выхода от структурных особенностей молекулы.
Предельные углеводороды и непредельные углеводороды с изо-
лированными кратными связями, соответствующие спирты, кисло-
ты, эфиры, галогенопроизводные и т. д. не флуоресцируют в види-
мой и УФ областях, тогда как, например, альдегиды и кетоны
дают видимую люминесценцию. Флуоресценция бензола, нафтали-
на и т. д. происходит в УФ области спектра, других ароматиче-
ских полициклических соединений — частично или полностью в
видимой области (табл. XV.2).
Сильно флуоресцируют многие кислород- и азотсодержащие
гетероциклические соединения ароматического ряда, металлорга-
нические соединения. Вообще отмечено, что жесткая, в частности,
замкнутая планарная структура способствует интенсивной флуо-
ресценции, тогда как структура, допускающая внутреннюю пере-
ориентацию (движение) больших фрагментов молекулы, препят-
ствует ее появлению. По интенсивности флуоресценции можно
различать пространственные цис-транс-изомеры и конформации,
например, 1,4-дифенилбутадиена и т. п. Вещества, отличающиеся
по «жесткости» структуры, но имеющие сходные спектры погло-
щения, могут часто сильно различаться по интенсивности флуорес-
ценции. Выводы о структуре на основании спектров флуоресцен-
ции носят весьма общий характер, но вместе с другими данными
иногда могут облегчить решение тех или иных структурных проб-
лем.
Если в спектрах флуоресценции молекул в растворе или в га-
зовой фазе (при низком давлении) наблюдается хорошо разрешен-
ие
ная колебательная структура, то можно определить колебатель-
ные частоты в основном (для газообразного состояния иногда и
в возбужденном) электронном состоянии.
Положение, форма полосы и интенсивность флуоресценции для
соединений, способных к ионизации и диссоциации в какой-то сре-
де, могут зависеть от pH. Это относится, например, к нафтолсуль-
фоновым кислотам, а- или р-нафтиламинам. Так, в нейтральном
или щелочном растворе флуоресценция последних наблюдается в
видимой области, а в кислом растворе — только в УФ области.
Кислотность и основность могут, в принципе, различаться для
основного и возбужденного электронных состояний молекул, по-
этому не всегда наблюдается даже приблизительное соответствие
изменений спектров поглощения и флуоресценции в зависимости
от pH.
Явление фосфоресценции используется для изучения метаста-
бильных, возбужденных триплетных фосфоресцентных состояний
молекул, парамагнетизма, возникающего при облучении, в связи
с особенностями строения как органических, так и неорганических
соединений. Спектры фосфоресценции обычно исследуют для твер-
дых растворов веществ, при низких температурах или в очень вяз-
ких жидкостях, что позволяет предотвратить электронную дезакти-
вацию молекул, ограничивая их движение, хотя иногда фосфорес-
ценция наблюдается даже у паров, например диацетила.
В случае неорганических «примесных» фосфоров они
вместе с основным веществом составляют обычно единую фосфо-
ресцирующую систему, тогда как фосфоресценция органических
соединений — свойство, присущее самим молекулам. В первом слу-
чае длина волны возбуждающего излучения определяется часто
спектром поглощения среды, а не примесных центров свечения, у
органических же фосфоров спектр флуоресценции в разных средах
практически одинаков, так как возбуждается полосой излучения,
поглощаемого самим фосфоресцирующим веществом, и может
лишь слегка зависеть от межмолекулярных взаимодействий.
Количественный анализ основывается на зависимости интенсив-
ности фотолюминесценции от концентрации люминесцирующего
вещества в растворе. Из соотношения (XV.3) следует
•^исп~ ^погл^1’ (XV.5)
а по закону Бугера — Ламберта — Бера
Логл = /0-/ = /0(1-е-2>3^),
где 70 — интенсивность падающего, возбуждающего излучения;
1 — интенсивность прошедшего непоглощенного излучения.
После разложения экспоненты в ряд и подстановки 7ПОгл в
(XV.5) получается соотношение, связывающее 7ИОП с концентра-
цией флуоресцирующего вещества, которое для разбавленных
растворов (ес/<0,1) представляет линейную зависимость вида
Люп=2, Зе cZ/q Г.
6
Рис. XV.6. Блок-схема
установки для получения
и измерения спектров
фотолюминесценции:
освещение образца: а — сбо-
ку; б —спереди; в — на про-
свет; 1 — источник излуче-
ння; 2 —линзы; 3, 3' — све-
тофильтры; 4, 4' — поляри-
заторы; 5 — образец; 6 — мо-
нохроматор; 7 —приемник;
8 — усилительно-регист-
рирующее устройство
Интенсивность свечения прямо пропорциональна интенсивности
возбуждающего излучения /0, и, увеличивая последнюю, можно
добиваться очень большой чувствительности спектрофлуориметри-
ческих измерений. Надежно регистрируется, например, свечение
растворов с концентрацией флуоресцирующего вещества порядка
~!0~п моль/л, что превышает чувствительность метода абсорб-
ционной спектрофотометрии.
Растворы для исследования спектров флуоресценции должны
приготовляться с использованием растворителей особо высокой
«флуоресцентной» чистоты, т. е. очищенных с помощью специаль-
ных методов от микропримесей, которые сами могут флуоресци-
ровать.
В связи с высокой чувствительностью химический люминесцент-
ный анализ широко используется в различных областях промыш-
ленности, включая химическую и нефтехимическую, в биологии,
медицине и геологии, а так называемый люминесцентный анализ
обнаружения — также в пищевой промышленности, сельскохозяй-
ственном производстве, дефектоскопии, археологии, криминали-
стике и т. д.
Обычная схема получения и измерения спектров фотолюмине-
сценции показана на рис. XV.6. Фотовозбуждение осуществляется
источником УФ или видимого излучения 1 по трем вариантам его
расположения относительно образца и оптической оси входного
коллиматора прибора (рис. XV.6). Первый вариант применяется
чаще всего для измерения спектров флуоресценции растворов,
второй — твердых образцов, третий — применяется обычно при
изучении спектров фосфоресценции.
В качестве источников могут служить ртутная, ксеноновая, во-
дородная и другие лампы, широко применяются также лазеры.
Свечение фокусируется на входную щель монохроматора 6 и раз-
350
ложенное в спектр попадает через выходную щель на приемник 7,
связанный с регистрирующим устройством 8 (см. о приемниках
для видимой и УФ спектрофотометрии). При широких полосах
испускания, которые характерны для спектров флуоресценции
растворов, для возбуждения используется излучение с частотой
Увозб, лежащей в пределах полосы поглощения образца, но вне
пределов его полосы испускания.
Для устранения возможности попадания на приемник возбуж-
дающего излучения применяют метод скрещенных фильтров. Это
значит, что после источника 1 устанавливают светофильтр 3 (или
светосильный монохроматор), выделяющий линию увозб (в случае
лазерного источника необходимость в них отпадает), а между об-
разцом 5 и монохроматором 6 помещают светофильтр 3', погло-
щающий возбуждающее излучение (уВозб). В спектрофлуоримет-
рах предусматривается также возможность установки поляризато-
ров 4 и 4'\ система линз 2 обеспечивает соответствующую фокуси-
ровку и освещение.
Кюветы для исследования спектров флуоресценции растворов
представляют собой прямоугольные или треугольные в сечении
стаканы с прозрачными стенками, иногда используются цилиндри-
ческие кюветы или капилляры типа применяемых в спектроскопии
КР. В настоящее время широко применяются полумикро- и микро-
кюветы, используются высоко- и низкотемпературные термостати-
руемые устройства и проточные кюветы. Для регистрации спект-
ров фосфоресценции применяют специальные криостаты с различ-
ными хладагентами (жидкий азот, сухой лед в ацетоне или спирте
и т. д.). В качестве твердых растворов для наблюдения фосфорес-
ценции используют застывающие при комнатной температуре
расплавы, например с борной кислотой, а вязкой жидкостью для
получения раствора может служить, например, глицерин.
* * *
Рассмотренные в разделе методы электронной спектроскопии
в видимой и УФ областях являются основными источниками экспе-
риментальных данных об энергетических состояниях молекул, свя-
занных с электронным возбуждением. Интерпретация электронных
спектров молекул основывается на использовании представлений
приближенных методов квантовой химии. С другой стороны, по-
лучаемые из спектров данные могут служить критерием пригод-
ности и ограничений тех или иных приближений в описании элек-
тронных состояний и переходов, стимулирующим развитие и уточ-
нение теоретических представлений.
Установлено достаточно много закономерностей и корреляций,
связывающих строение и электронные спектры, а также спектры
и некоторые другие физико-химические свойства. В то же время
используемые химиками в структурных исследованиях электрон-
ные спектры не столь специфичны, как, например, колебательные
спектры молекул. Далеко не все вещества вообще имеют электрон-
ные спектры поглощения в видимой и ближней УФ областях.
Как абсорбционная УФ спектроскопия, так и флюориметрия
относятся к мощным аналитическим методам, обладающим высо-
кой чувствительностью. Это обусловливает их широкое примене-
ние как в научных исследованиях, так и в самых различных от-
раслях народного хозяйства для количественного контроля, а так-
же управления технологическими процессами. Помимо давно при-
меняемых рутинных методик абсорбционного и люминесцентного
анализа, разрабатываются и внедряются в практику новые анали-
тические методики, например НПВО и некоторые другие. Даль-
нейшее повышение эффективности применения методов электрон-
ной спектроскопии непосредственно связано с развитием их теории
и экспериментальной техники.
Контрольные вопросы
Глава XIII
1. Какими основными свойствами характеризуются электронные состояния
молекул?
2. Какова мультиплетность состояния молекулы, при котором спины двух
электронов не спарены (параллельны)?
3. Объясните, что означают следующие символы электронных состояний
линейных молекул: X'St; В3П2; С2Л5^ и нелинейных молекул: ХМц Ч3Ви;
в1Ав.
4. Составьте таблицу прямых произведений типов симметрии точечной груп-
пы C2/i, аналогичную табл. XIII.2 (используйте табл. IX.1).
5. По каким признакам можно идентифицировать в УФ спектре полосу
п->-л*-перехода? Чем объясняются сдвиги этой полосы при изменении поляр-
ности растворителя?
6. Как формулируется правило отбора электронных переходов по спину
и в результате чего оно может нарушаться?
7. Почему для вибронных переходов запрет по орбитальному правилу от-
бора может быть не строгим?
8. Как формулируется принцип Франка — Кондона для вероятности элект-
ронных переходов?
Глава XIV
9. Как влияет сопряжение хромофорных групп на их электронный спектр
поглощения? Как отражается на интенсивности л->л*-полосы поглощения из-
менение конформации сопряженной системы двойных связей (5-цис-5-транс- и
плоская—неплоская)?
10. Оцените по табл. XIV.2 Хмакс для соединения
(сравните с экспериментальным значением Хмакс = 355 нм).
11. Как отражается образование межмолекулярной и внутримолекулярной
водородной связи на положении полос л-*л*-переходов?
12. С чем и как связана окраска аквокомплексов катионов тяжелых эле-
ментов?
-13. Каков смысл величины Е 1с% и для чего она используется?
14. Что такое изобестическая точка, для чего и как она используется?
. Глава XV
15. Запишите закон поглощения света Бугера — Ламберта — Бера и объяс-
ните смысл входящих в него величин.
16. Какие преимущества дает преобразование спектральных кривых диффе-
ренцированием (первая, вторая и последующие производные оптической плот-
ности по длине волны)?
17. В чем отличие явлений флуоресценции и фосфоресценции?
18. Что такое интеркомбинационная конверсия и почему она возможна?
19. В чем заключается правило зеркальной симметрии спектров поглощения
и люминесценции (Левшина)? Почему оно является приближенным?
20. Что такое энергетический и квантовый выходы люминесценции?
21. На чем основывается количественный люминесцентный анализ и какова
его чувствительность сравнительно с абсорбционной спектрофотометрией?
Рассмотрение теоретических основ, практических применений
и техники эксперимента представленной в учебнике группы физи-
ческих методов, применяемых в химии, показывает их большие и
полностью еще не используемые возможности в структурных, ана-
литических, термодинамических, кинетических и других исследова-
ниях, а некоторых из них и в промышленном производстве. По-
следнее относится, в частности, к методам оптической спектроско-
пии (абсорбционной УФ спектрофотометрии, люминесцентному
анализу, ИК спектроскопии) и отчасти к масс-спектрометрии.
Практически каждый метод в чем-то уникален и имеет свою
специфику — у одних это возможность количественного определе-
ния геометрических параметров молекул (газовая электроногра-
фия, методы вращательной спектроскопии), у других — определе-
ния электрических свойств (дипольных моментов и поляризуемости
молекул), у третьих—-энергетических состояний или спектральных
характеристик и т. д. Применения некоторых методов очень раз-
нообразны (например, спектральных), а других — более узкие;
одни данные, получаемые тем или иным методом, являются вполне
достоверными, а другие — оценочными или косвенными. Во мно-
гих случаях для повышения надежности результатов требуется
комплексное применение нескольких физических методов исследо-
вания. Так, например, при установлении структуры сложных соеди-
нений необходимо совместное использование масс-спектрометрии,
ИК, КР, УФ спектроскопии, ЯМР и других методов. Все они вхо-
дят в арсенал современной инструментальной химии.
Совершенствование экспериментальной техники исследований,
повышение качества и новизна характера получаемых данных рас-
ширяют круг тех задач, которые могут ставиться и решаться при
использовании того или другого метода, что, в свою очередь, сти-
мулирует развитие теории физических методов исследования. Раз-
работка, серийный выпуск и широкое внедрение принци-
пиально новой, сочетающейся с ЭВМ, высоко производительной
аппаратуры, удовлетворяющей требованиям будущего, являются
важнейшими условиями ускорения научно-технического прогресса.
Только на этой базе может обеспечиваться как автоматизация и
ускорение научного эксперимента до выдачи обработанных конеч-
ных результатов исследования, так и автоматизация контроля и
управления технологическими процессами.
В соответствии с требованием опережающего развития высшей
школы по сравнению с научно-технической реконструкцией народ-
ного хозяйства будущие специалисты уже сейчас должны быть
готовы к использованию новейшей аппаратуры и применению пер-
спективных физических методов. Эта задача может быть решена
только при намечаемом материально-техническом переоснащении
высшей школы и прочном усвоении фундамента тех знаний, кото-
рые можно получить при изучении курса «Физические методы
исследования в химии».
ПРИЛОЖЕНИЕ
1. Фундаментальные физические константы
Физическая величина Обозначение Численное значение Единицы (СИ)
Скорость света в ва- кууме С 2,99792458(1,2) 108 м-с-1
Магнитная постоянная Цо 4л = 12,5663706144 10-7 Г-м-1
Электрическая постоян- ная Со 8,854187818(71) 10~12 Ф м-1
Элементарный заряд е 1,6021892(46) 10-19 Кл
Постоянная Планка h 6,626176(36) 10-34 Дж-с
Постоянная Авогадро Na 6,022045(31) 1023 моль-1
Атомная единица мас- сы а.е.м. 1,6605655(86) 10-27 кг
Масса покоя электрона те 9,109534(47) 10-31 кг
Масса покоя протона 1ПР 1,6726485(86) 10-27 кг
Боровский радиус aQ 5,2917706(44) IO-11 м
Объем моля идеально- го газа при нормальных условиях vm 22,41383(70) 10-3 м3-моль-'
Универсальная моляр- ная газовая постоянная R 8,31441(26) Дж-моль-1 К-1
Постоянная Больцмана k 1,380662(44) IO-23 Дж-К-1
Гравитационная посто- янная G 6,6720(41) м3-с-2-кг-1
[
2. Множители и приставки для образования кратных и дольных единиц
Множи- тель Наименование Обозначение (лат.) Множи- тель Наименование Обозначение (лат.)
ю18 экса Э (£) 10-' деци Д (d)
ю15 пэта П (Р) 10-2 санти с (с)
ю12 тер а Т (7) ю-3 милли м (т)
ю9 гига Г (G) ю-6 микро мк (ц)
10е мега М (А1) 10-9 нано н (п)
103 кило К (k) ю-12 пико п (р)
102 гекто г (/г) 10-ls фетто Ф (D
10* дека да (da) 10-18 атто а (а)
ЛИТЕРАТУРА
Общая
Бабушкин А. А., Бажулин П. А., Королев Ф. А., Левшин Л. В., Прокофь-
ев В. К., Стриганов А. Р. Методы спектрального анализа. — М.: Изд-во МГУ.
1962.
Внутреннее вращение молекул/Под ред. В. Дж. Орвилл-Томаса. — М.: Мир,
1977.
Водородная связь/Под ред. Н. Д. Соколова. — М.г Наука, 1981.
Герцберг Г. Спектры и строение двухатомных молекул. — М.: Издатин-
лит, 1949.
Грибов Л. А. Введение в молекулярную спектроскопию. — М.: Наука, 1976.
Драго Р. Физические методы в химии. — М.: Мир, 1981, т. 1, 2.
Ельяшевич М. А. Атомная и молекулярная спектроскопия. — М.: Физмат-
тиз, 1962.
Зоркий П. М. Симметрия молекул и кристаллических структур. — М.:
Изд-во МГУ, 1986.
Иоффе Б. В., Костиков Р. Р., Разин В, В. Физические методы определения
строения органических соединений. — М.: Высшая школа, 1984.
Казицина Л. А., Куплетская Н. Б. Применение УФ, ИК и ЯМР спектроско-
пии в органической химии. — М.: Высшая школа, 1971.
Мальцев А. А. Молекулярная спектроскопия. — М.: Изд-во МГУ, 1980.
Минкин В. Н., Симкин Б. Я., Минаев Р. М. Теория строения молекул. —
М.: Высшая школа, 1979.
Новиков Г. И. Физические методы в неорганической химии. — Минск:
Вышэйшая школа, 1975.
Пентин Ю. А., Тарасевич Б. Н. Новые методы спектроскопии в химии. — М.:
Знание, 1975.
Татевский В. М. Строение молекул. — М.: Химия, 1977.
Саидов Г. В,, Свердлова О. В. Практическое руководство по молекулярной
спектроскопии. — Л.: Изд-во ЛГУ, 1980.
Сильверстейн Р., Басслер Г., Моррил Т. Спектрометрическая идентификация
срганических соединений. — М.: Мир, 1977.
Уитли П. Определение молекулярной структуры. — М.: Мир, 1970.
Харрик Н. Спектроскопия внутреннего отражения. — М.: Мир, 1970.
Шрайнер Р., Фьюзон Р., Кертин Д., Моррил Т. Идентификация органических
соединений. — М.: Мир, 1983.
Экспериментальные методы химической кинетики/Под ред. Н. М. Эмануэля
и Г. Б. Сергеева. — /И.: Высшая школа, 1980.
Эляшберг М. Е., Грибов Л. А., Серов В. В. Молекулярный спектральный
анализ и ЭВМ. — М.: Наука, 1980.
К первому разделу
Бейнон Дж. Масс-спектрометрия и ее применение в органической химии. —
М.: Мир, 1964.
Горохов Л. Н. Масс-спектрометрия в неорганической химии —М.: Зна-
ние, 1984.
Леман Т., Берси М. Спектроскопия ионного циклотронного резонанса. — М.:
Мир, 1980.
Семенов Г. А., Николаев Е. Н., Францева К. Е. Применение масс-спектро-
метрии в неорганической химии. •—Л.: Химия, 1976.
Сидоров Л. Н., Коробов М. В., Журавлева Л. В. Масс-спектральные тер-
модинамические исследования. — М.: Изд-во МГУ, 1985.
Сысоев А. А., Чупахин М. С. Введение в масс-спектрометрию. — М.: Атом-
издат, 1977.
Терентьев П. Б. Масс-спектрометрия.в органической химии. — М.: Высшая
школа, 1979.
Ко второму разделу
Дипольные моменты в химии комплексных соединений/Под ред. О. А. Оси-
пова.— Ростов: Изд-во Ростовского ун-та, 1976.
Минкин В. И., Осипов О. А., Жданов Ю. А. Дипольные моменты в органи-
ческой химии. — Л.: Химия, 1968.
Осипов О. А., Минкин В. И. Справочник по дипольным моментам.. — М.:
Высшая школа, 1965.
Рамсей Н. Молекулярные пучки —М.: ИЛ, 1960.
Смит К. Ф. Молекулярные пучки. — М.: Физматгиз, 1959.
К третьему разделу
Вилков Л. В., Анашкин М. Г., Засорин Е. 3. и др. Теоретические основы
газовой электронографии. — М.: Изд-во МГУ, 1974.
Вилков Л. В., Мастрюков В. С., Садова Н. И. Определение геометрического
строения свободных молекул. —Л.: Химия, 1978.
Вебер А. Спектроскопия комбинационного рассеяния высокого разрешения
газов. — В кн.: Применение спектров комбинационного рассеяния/Под ред.
А. Андерсена. — М.: Мир, 1977.
Горди В., Смит В., Трамбаруло Р. Радиоспектроскопия. — М.: Гостехиздат,
1955.
Спиридонов В. П., Акишин П. А., Засорин Е. 3. Высокотемпературная газо-
вая электронография в Московском университете. — В кн.: Современные проб-
лемы физической химии. —М.: Изд-во МГУ, 1979. С. 3—58.
Сивин С. Колебания молекул и среднеквадратичные амплитуды. — М.:
Мир, 1971.
Стоичев Б. Спектры комбинационного рассеяния газообразных веществ, по-
лученные на приборах высокой разрешающей силы. — В кн.: Успехи спектро-
скопии/Под ред. Г. У. Томпсона. — М.: ИЛ, 1963.
Таунс Ч., Шавлов А. Радиоспектроскопия.—М.: ИЛ, 1959.
Харгиттаи М., Харгиттаи И. Геометрия молекул координационных соедине-
ний в паровой фазе. — М.: Мир, 1976.
К четвертому разделу
Беллами Л. Инфракрасные спектры сложных молекул. — М.: Издатинлит,
1963.
Беллами Л. Новые данные по ИК спектрам сложных молекул. — М.:
Мир, 1971.
Белл Р. Дж. Введение в фурье-спектроскопию. — М.: Мир, 1975.
Волькенштейн М. В., Грибов Л. А., Ельяшевич М. А., Степанов Б. И. Коле-
бания молекул.— М.: Наука, 1972.
Герцберг Г. Колебательные и вращательные спектры многоатомных моле-
кул.— М.: Издатинлит, 1949.
Гилсон Т., Хендра П. Лазерная спектроскопия КР в химии.— М.: Мир, 1973.
Граселли Дж., Снейвили М., Балкин Б. Применение спектроскопии КР в
химии.— М.: Мир, 1984.
Григорьев А. И. Введение в колебательную спектроскопию неорганических
соединений.— М.: Изд-во МГУ, 1977.
Коптев Г. С., Пентин Ю. А. Расчет колебаний молекул —М.: Изд-во
МГУ, 1977.
Наканиси К. Инфракрасные спектры и строение органических соединений.—
М.: Мир, 1965.
Накамото К. Инфракрасные спектры неорганических и координационных со-
единений.— М.: Мир, 1966.
Современные проблемы спектроскопии комбинационного рассеяния света/
Под ред. М. М. Сущинского.— М.: Наука, 1978.
Сущинский М. М. Спектры комбинационного рассеяния молекул и кристал-
лов.— М.: Наука, 1969.
Смит А. Прикладная ИК спектроскопия.— М.: Мир, 1982.
Свердлов Л. М„ Ковнер М. А., Крайнов Е. П. Колебательные спектры мно-
гоатомных молекул.— М.: Наука, 1960.
Финч А., Гейтс П., Редклиф К., Диксон Ф., Бентли Ф. Применение длинно-
волновой, ИК спектроскопии в химии. — М.: Мир, 1973.
Эллиот АЛ Инфракрасные спектры и структура полимеров. — М.: Мир, 1972.
Harris D. C., Bertolucci М. D. Symmetry and Spectroscopy. An Introduction
to Vibrational and Electronic Spectroscopy.— N.-Y.: Oxford Un. Press, 1978.
К пятому разделу
Бернштейн И. Я., Каминский Ю. Л. Спектрофотометрический анализ в орга-
нической химии.— Л.: Химия, 1975.
Герцберг Г. Электронные спектры и строение многоатомных молекул. — М.:
Мир, 1969.
Коряжкин В. А. Электронные спектры молекул.— М.: Изд-во МГУ, 1984.
Кузьменко Н. Е., Кузнецова Л. А., Кузяков Ю. Я. Факторы Франка — Кон-
дона двухатомных молекул.— М.: Изд-во МГУ, 1984.
Мак-Глинн С„ Адзуми Т., Киносита М. Молекулярная спектроскопия три-
плетного состояния.— М.: Мир, 1972.
Пешкова В. М., Громова М. И. Методы абсорбционной спектроскопии в ана-
литической химии.— М.: Высшая школа, 1976.
Свердлова О. В. Электронные спектры в органической химии—.Л.: Хи-
мия, 1973.
Штерн Э., Тиммонс К. Электронная абсорбционная спектроскопия в органи-
ческой химии,—М.: Мир, 1974.
Practical Absorption Spectrometry ed. by A. Knowles and C. Burgess..— L.—
N.-Y.: Chapman and Hall, 1984.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Авогадро постоянная 70
Аддитивность электрических диполь-
ных моментов связей 79
Адиабатическое приближение 172, 227
Азимутальное квантовое число 125
Альтернативного запрета правило 199,
203
Амплитуда атомного рассеяния 10,
126
Амплитуды колебаний пар атомов
---------среднеквадратичные 136
---------перпендикулярные 144
Ангармонические колебания 141
Анизотропия поляризуемости 213
Асимметрии параметр 89
Асимметричный волчок 89
Атомное рассеяние электронов 123
Ауксохром 307
Базовой линии мет.од 250
Барьер внутреннего вращения 107, 238
Барьер инверсии 109
Барьерный эффект тяжелого атома
222
Батохромный эффект 308
Бензольное поглощение 306, 325
Берри псевдовращение 11
Бете соотношение 129
Больцмана распределение 8, 179
Бора соотношение 176
Борна—-Оппенгеймера приближение
85, 248, 330
Бугера — Ламберта — Бера закон 85,
248
Валентно-оптическая схема 190
Валентно-силовое поле 231
----обобщенное 231
Ван-дер-ваальсовы молекулы 81
Вероятности переходов
----вращательных 78, 114
•---колебательных 177
----электронных 314
Взаимоисключения частот правило 203
Вибронные переходы 310, 318
Виртуальные уровни (состояния) 114,
176
Внутреннее вращение 107
Внутренние координаты 185
Внутренняя конверсия 343
Возмущения теория
•--- поправки первого порядка 65
---- поправки второго порядка 65
Возмущения теория, энергия и волно-
вая функция нулевого приближения
65
Волновой вектор 123
Волновое число 8
Волновые функции
—— антисимметричные 119
----вращательного состояния 65
---- колебательного состояния 174
----отрицательные 118
----положительные 118
----симметричные 119
---электронного состояния 297
Вращательные постоянные 87
Вращательные спектры 85
Вращательная структура 215, 217
Вудворда—Фишера правила 322
Вырождение
— вращательных состояний 67
— колебательных состояний 174, 198
— электронных состояний 298
Вытянутый волчок 88
Газовая постоянная 44
Гармонические колебания 136
Гармонический осциллятор 173, 174
Гармонические частоты 227
Гармоническое приближение 173
----силовое поле 230
Гаусса распределение 136
---- функция 339
Геометрическая структура молекул
143
Геометрические параметры молекул 97
Герца — Кнудсена уравнение 46
Гигантское КР 289
Гипсохромный эффект 308
Гиперхромный эффект 308
Гипохромный эффект 308
Главные значения (эллипсоида) инер-
ции молекул 87
Главные значения (эллипсоида) поля-
ризуемости молекул 61, 115
Главные оси (эллипсоида) инерции
молекул 86
Галея приемник 268
Горячие частоты (переходы) 179
Грина функция 127
Групповые частоты 220
Давыдова эффект 205
Двухатомные молекулы
--------вращательные постоянные 100
----рассеяние электронов 136
----колебания 173
— — электронные состояния 300
Двухволновая спектроскопия 341
Дебая первый метод 71
Дебая второй метод 72
Дебая уравнение 70
Де Бройля уравнение 9
Деполяризации степень в КР 213
Дефект инерции 93
Деформационные колебания 196, 198
— — внешние 221
----внутренние 221
Динамического взаимодействия коэф-
фициенты 188
Дипольные-переходы 85, 116
Дипольный момент молекул 59
-------собственный 59
------- индуцированный 60
•------поворотных изомеров 80
Диссоциации энергия 41
Дифракционные методы 9
Дифференциальная спектроскопия 340
Дихроичное отношение 263
Диэлектрическая проницаемость 68
Длина свободного пробега молекул
24
Длина электромагнитных волн 9
Доплера эффект 94
Естественные координаты 185
Жесткие молекулы 87, 93, 122, 138
Жесткий ротатор 87, 107
Зависимые координаты 186
Заторможенное вращение 107, 238
Зеркальной симметрии правило 346
Зоммерфельда условие излучения 126
Измерения масс ионов 36
Изобестическая точка 332
Изомерия
— геометрическая 111
— поворотная 80
Изотопические правила 229
Изотопная метка в масс-спектромет-
рии 53
Изотопные эффекты
----во вращательных спектрах 102
---- в колебательных спектрах 227
Инверсия
— аммиака 109
— фосфина 109
Инерции дефект (см. дефект инерции)
Интеграция методов 15
Интенсивность рассеяния электронов
атомами 123
Интенсивность спектральных полос
-------абсолютная 248
-------в инфракрасной спектроско-
пии 177
•-----в максимуме 248, 251
------ в микроволновой спектроско-
пии 85
------в электронной спектроскопии
314
------комбинационного рассеяния
120, 178
Интеркомбинационная
— конверсия 344
— переходы 317
Интерферометры 269
Информ ационно.поисковые системы
идентификации веществ 245
Ионизация атомов и молекул 19
Ионизация
— диссоциативная 20
•— поверхностная 27
—• электрическим полем 27
•— электронным ударом 24
Ион-молекулярные равновесия 51
•---реакции 52
Ион-циклотронный резонанс 35
Ионы
— метастабильные 23
— многозарядные 24
— молекулярные 21
— осколочные 22
— отрицательные 23
— перегруппировочные 22
— положительные 19
Искусственного интеллекта метод 245
Источники излучения
— — видимого 334
--УФ 334
--ИК 267
----в КР спектроскопии 284
КАРС — когерентное антистоксово
рассеяние света 288
Катион — радикал 21
Кинематического взаимодействия ко-
эффициенты 188
Кинетики изучения 53, 257
Кинетическая энергия колебаний 184,
196
Классификация колебаний по типам
симметрии 195
Классификация уровней энергии
------асимметричного волчка 88
— симметричного волчка 88
Классификация электронных перехо-
дов 306
------по Малликену 307, 308
------по Каша 309
Клаузиуса — Моссоти уравнение 70
Клистрон 94
Клаузинга коэффициент 46
Кнудсена ячейка 46
---- двойная 47
Колебания
— валентные 183
— вырожденные 195
—‘деформационные 196
— нормальные 183
— акустические 262
•— решеточные 206
— трансляционные 206
— ориентационные 206
— крутильные 108, 221, 240
Колебательная релаксация 343
Колебательный вклад в термодина-
мические функции 242
Колебательная задача
---- прямая 181
----обратная 181, 230
Колебательные частоты (переходы)
----фундаментальные (основные)
179
------обертонные 179
— —составные (комбинированные)
179
----«горячие» 179
Комбинационное рассеяние света
—:----спонтанное 283
------ резонансное 286
------ инверсное 287
------гигантское 289
------ когерентное антистоксово
(КАРС) 288
------стоксово 113, 176
------антистоксово 113, 176, 345
Комплексонометрические индикаторы
333
Комплексные соединения 111, 327
Константы равновесия химической ре-
акции 44, 252
Координаты
—внутренние 185 :
— естественные 185
— нормальные 173
— симметрии 196, 200
Корректно поставленная задача 5
Кристалличности эффекты 205
Коэффициент поглощения 85
Коэффициент чувствительности масс-
спектрометра .44
Крейчмена уравнения 102
Кривые эффективности ионизации 25
Крутильные
— колебания 108
— частоты 221, 240
Лампа обратной волны 94
Лазер 121
— применение в спектроскопии КР
284
Лежандра полиномы 65, 125
Левшина правило 346
Линейные молекулы
----вращение 87, 118
----колебания 197, 199
----колебательно-вращательные спек-
тры 218
----электронные состояния 300
Лорентца — Лоренца уравнение мо-
лярной рефракции 70
Лоренца сила 28
Лоренца функция 249
Люминесценция 342
Люминесценции выход
----квантовый 346
----энергетический 346
Люминесцентный анализ 348
Майкельсона интерферометр 269
Масс-спектрометры
— статические 28
— динамические 33
— магнитные 28
— времяпролетные 33
— квадрупольные 34
— с двойной фокусировкой 32
— ион-циклотронного резонанса 35
Масс-спектрометрия высокого разре-
шения 31
Матрица компонентов (тензора) по-
ляризуемости молекулы 115. 178
Матричной изоляции метод 172, 277
Матричные элементы 314
Метод наименьших квадратов в га-
зовой электронографии 151
Микрофотометр 149
Молекулярная формула вещества 36
Момент инерции 87, 99, 102
Момент перехода 177, 306
Морзе функция 142, 173, 236
Молекулярный пучок 74
?4ультиплетность 299, 301
Направляющие косинусы 115
Напряженность электрического поля
градиент 60
Напряженность электрического поля
60
Нежесткие молекулы 118
Нейтронография 9
Некорректно поставленная задача 5
Неопределенности принцип 12
Непересечения правило 90
Неприводимые представления 200
Нерегулярные группы атомов 79
Нернста штифт 268
Несвязывающие орбитали 302
Неупругого рассеяния функция 131
Нормальные
— колебания 183
— координаты 173
Нормированные формы колебания 183
Нулевой уровень колебательной энер-
гии 175, 179
НПВО (нарушенного полного внут-
реннего отражения)
— метод 278
— оптические элементы 282
Обертоны 179
Обобщенное валентно-силовое поле
231, 233
Обратная задача
--- метода 5
---колебательная 181, 230, 232
--электрооптическая 181, 190
Определение длины волны
------электронов 149
Оптическая плотность 149, 248
Орбитали атомные и молекулярные
302
Орбитальный фактор 316
Осцилляторная сила 315
Отклонение молекулярного пучка в
неоднородном электрическом поле
74
Отнесение линий в микроволновой
спектроскопии 92
Относительная интенсивность ионного
тока 32
Отношение сигнала к шуму 250
Отщепление линий в микроволновой
спектроскопии 105
Парциальный ионный ток 44
Парциальное сечение ионизации 25
Перестановочная симметрия 118
Переходы с переносом заряда 312
Плоская волна 123
Плотность почернения 149
Погашение 148
Поглощения
— процент 240
— коэффициент 248
Полосы
— «аморфные» 261
— вибронные 310
— «горячие» 179
— деполяризованные 214
— «кристаллические» 260
— параллельные 217, 263
—перпендикулярные 217, 263
— поляризованные 214
Поляризуемость диэлектрика 68
Полуширина полосы 249
Поляризация молярная 70
— ориентационная 62
Поляризуемость молекулы
- атомная 61
---деформационная 61
--- ориентационная 63
---электронная 61
Постоянная колебательно-вращатель-
ного взаимодействия 93
Послесвечение 343
Потенциал ионизации 40, 42
--- адиабатический 40
---вертикальный 40
---ионизации 19, 40
— появления ионов 25. 41
Потенциальная функция внутреннего
вращения 109, 238
Потенциальная энергия
----ангармонического осциллятора
100
----гармонического осциллятора 100
Потенциальный барьер
----внутреннего вращения (см. Барь-
ер внутреннего вращения)
---- инверсии (см. Барьер инверсии)
Правила отбора
---во вращательных спектрах 86,
117
----в колебательных спектрах 177,
179
----колебательно-вращательных пе-
реходов 218
Представление группы 200
Приближение малых колебаний 122
Приведенная масса 100, 103, 108
Приведенный мометт инерции 240
Приведенная молекулярная составля-
ющая интенсивности рассеяния элек-
тронов 132
Приемники
— ИК 268
— КР 284
— УФ 336
Принцип Франка — Кондона (см.
Франка—Кондона принцип)
Пропускание (прозрачность) 248
Прозрачные оптические материалы
-------ИК 267
-------УФ 335
Производных спектроскопия (с диф-
ференцированием) 339
Пропускание функция 149
Прямая задача
---- колебательная 181
----электрооптическая 181, 189
Прямое произведение 303
Псевдовращепие Берри (см. Берри
псевдопревращение)
Путь реакции в газах 55
Равновесное межатомное расстояние
(см. ге-структура)
Равновесий изучение 252
Радиального распределения кривая
135
Радиального распределения функция
135
Разностная спектроскопия 340
Разрешающая сила масс-спектрометра
31
Раман-эффект 170
— спектры 170
Распределение колебательной энергии
189
Рассеяние электронов атомами 123
•------упругое 124, 131
-------неупругое 124, 131
Рассеяние электронов молекулами
двухатомными 136
Растворители
— для ИК и КР 273
— для УФ 338
Расшифровка
— масс-спектра 45
— электронограмм 151
Регулярные группы атомов 79
Резонансное КР 286
Релеевское рассеяние 113, 176
Рентгенография 9
Рефракция молярная 70
Ридберговские переходы 307, 309
«Рыболовный крючок» 38
Ряд кислотности для спиртов 53
Ряд основности аминов 53
Сайт-группа 205
Свободное вращение групп в молеку-
ле 112
Сечение ионизации 25
Силовое поле молекулы 144, 230
Силовые постоянные 100, 173, 230
---эффективные 238
Симметрии
— элементы 191
— оси 191, 192
—плоскость 192
— центр 192
— операции 191, 192
— точечная группа 191
•—типы колебаний 195
Симметричный волчок 88
Синглетные состояния 299
Составные (комбинированные) пере-
ходы и частоты 179
Спектрометры
— сканирующие 265, 266, 283
— однолучевые 265, 283, 334
— двухлучевые 265, 266, 334
— Фурье 265, 269
•—Адамара 265, 271
— диспергирующие 265
— многоканальные 271
— одноканальные 265
— недиспергирующие 265, 269
Спектроскопические методы, общая
характеристика 7
Сплюснутый волчок 88
Спиновый фактор 316
Сродство к иону 52
Сродство к электрону 52
Степень вырождения колебаний 174
------деполяризации 213
Статический эффект поля кристалла
205
Стокса — Ломмеля закон 345
Стоксово КР ИЗ, 176, 345
--испускание (люминесценции)
345
Структура
— геометрически согласованная 157
— несогласованная 157
Структурно-групповой анализ 220
Сферическая волна 125
Сферический волчок 119
Сферические функции
---• Бесселя 125
---Неймана 125
Тензор поляризуемости 175, 178
Теплоты
— диссоциации 43 j
— реакции 45 '
•— сублимации 44
Термостолбик (термопары) 268
Термы 175
Типы симметрии колебаний 195
Тихонова метод регуляризации 7
Точечная группа симметрии 191
Триплетные состояния 299
Тушение люминесценции 347
Угловой момент количества движе-
ния 87
Уравнение ионного тока 24
Уровни энергии
---вращательной 85, 113
---колебательной 174, 175
---электронной 296
Условие дифракции на молекулах 9
Фабри-Перо интерферометр 96
Фаза рассеяния 125, 126
Факсена — Хольцмана уравнение 126
Фактор-группа 206
Ферми-резонанс 204
Фильтры-ИК 268, 269
— в КР 284
Флуоресценция 114, 343
Фокусировка ионов по направлению
30
Фона линия 150
Формы колебаний 183
Форм-фактор атомный 126, 129
Фосфоресценция 345
Фосфоры 345
• — «примесные» 349
Фотоионизация молекул 26
Фотолюминесценция 342
Франка-Кондона принцип 20, 311
• фактор 316
Фундаментальные
— уровни 175
— частоты 179
Функция плотности вероятности
------ неупругого рассеяния 131
------радиального распределения
135
------частная 134
Фурье преобразование в газовой элек-
тронографии 135
Характеристики масс-спектрометра 30
Характеристическая температура 243
Характеристические частоты 220
Характеристическое время метода 11
Характеры представлений групп
-------приводимых 201
-------неприводимых 193, 194, 201
Химическая ионизация 27
Хроматомасс-спектрометрия 32
Хромофор 306
Центр
— масс молекулы 86
— симметрии 192
Центробежное растяжение 118
Частоты переходов
---вращательных 85
---колебательных 178
---электронных 309
Чувствительность масс-спектрометра
32
Ширина линии 94
Шкала интенсивностей КР
-------тетрахлорида 252
-------циклогексановая 251
Шредингера уравнение 64, 172, 295
Штарка эффект 64
Щели
— источника молекулярного пучка 75
— масс-спектрометра 29
Эйнштейна коэффициент 313
Экстинкции коэффициент 248
Электрический дипольный момент мо-
лекулы 59
Электрического резонанса метод 76
Электронная поляризуемость 61
Электронограф 146
— дифракционная камера 148
— испаритель 147
— сектор 148
Электрооптическая задача
— —прямая 181, 189
— — обратная 181, 190
Электрооптические параметры 190
Элементы симметрии 191
Энергия молекулы 296
Энергия разрыва химической связи 41
Энергия средняя гармонического ос-
циллятора 137
Эрмита полиномы 136
Эффективности ионизации кривые 40
Эффективности параметры силового
поля 238
Эффекты взаимного влияния 165
— насыщения 94
— сокращения 155
Ячейка Кнудсена (см. Кнудсена ячей-
ка)
Ячейка со штарковским электродом
94
ОГЛАВЛЕНИЕ
Предисловие . .......................................................... 3
Введение. Общая характеристика физических методов....................... 5
1. Прямая и обратная задачи методов................................. 5
2. Спектроскопические методы исследования.......................... 7
3. Дифракционные методы............................................ 9
4. Оптические и другие методы..................................... 11
5. Характеристическое время метода................................ 11
6. Значение физических методов для теоретической химии............ 13
7. Современный уровень и перспективы развития физических методов 14
Контрольные вопросы................................................ 16
Первый раздел. Методы масс-спектрометрии............................... 17
Глава I. Процессы ионизации и принципиальные схемы масс-спектромет-
ров ............................................................... 19
1. Ионизация атомов и молекул.................................... 19
2. Процесс ионизации и типы ионов................................. 19
3. Методы ионизации................................................ 24
4. Принципиальные схемы масс-спектрометров......................... 28
4.1. Магнитный масс-спектрометр.................................. 28
4.2. Динамические масс-спектрометры.............................. 33
4.3. Спектрометр ион-циклотронного резонанса .................... 35
Глава II. Применение масс-спектрометрии.............................. 36
1. Идентификация и установление строения веществ................. 36
2. Определение потенциалов ионизации молекул и появления иоиов . 40
3. Масс-спектральные термодинамические исследования............. 43
4. Масс-спектрометрия в химической кинетике..................... 53
Контрольные вопросы................................................ 56
Второй раздел. Методы определения электрических дипольных моментов
молекул............................................................ 57
Глава III. Теоретические основы методов................................ 59
1. Электрический дипольный момент молекулы....................... 59
2. Энергия молекулы во внешнем электрическом поле................. 69
3. Ориентационная поляризация молекул............................ 62'
4. Эффект Штарка и квантово-механический подход к выводу ориен-
тационной поляризации молекул .................................. 64
5. Диэлектрик в электрическом поле................................ 68
Глава IV. Экспериментальные методики и применение данных по элект-
рическим дипольным моментам молекул в химии....................... 71'
1. Первый метод Дебая—определение электрического дипольного
момента молекул паров веществ................................... 71
2. Второй метод Дебая — определение электрических дипольных мо-
ментов молекул веществ в разбавленных растворах................... 72
3. Отклонение молекулярного пучка в неоднородном электрическом
поле.............................................................. 74
4. Метод электрического резонанса................................. 76
5. Использование данных по дипольным моментам в химии .... 78
Контрольные вопросы................................................ 82
Третий раздел. Методы определения геометрического строения молекул 83
Глава V. Микроволновой метод исследования вращательных спектров мо-
лекул ............................................................. 85
1. Вращательные спектры поглощения молекул......................... 85
2. Методика эксперимента в микроволновой вращательной спектро-
скопии ........................................................... 94
3. Методы расчета геометрических параметров молекул ....... 97
4. Определение электрических дипольных моментов молекул........... 105
5. Исследование внутреннего вращения и инверсии молекул........... 107
6. Некоторые результаты микроволновых исследований................. ПО
Глава VI. Чисто вращательные спектры комбинационного рассеяния ... 113
1. Теоретические основы метода.................................... 113
2. Методика эксперимента вращательной спектроскопии КР............ 120
3. Определение геометрии молекул.................................. 121
Г лава VII. Метод газовой электронографии............................. 122
1. Основные этапы развития газовой электронографии................ 122
2. Рассеяние электронов атомами................................... 123
2.1. Упругое рассеяние электронов атомами......................... 124
2.2. Неупругое рассеяние электронов атомами....................... 130
2.3. Полная интенсивность атомного рассеяния...................... 131
3. Рассеяние электронов молекулами............................... 132
3.1. Молекулярная составляющая интенсивности рассеяния............ 132
3.2. Преобразование Фурье в газовой электронографии............... 134
3.3. Двухатомные молекулы......................................... 136
3.4. Кривые радиального распределения............................. 142
3.5. Многоатомные молекулы........................................ 143
4. Методика эксперимента в газовой электронографии............... 146
4.1. Принципиальная схема электронографа.......................... 146
4.2. Микрофотометрирование........................................ 149
4.3. Выделение молекулярной составляющей интенсивности рассеяния 150
5. Расшифровка электронограмм.................................... 151
6. Влияние внутримолекулярных колебаний на конфигурацию моле-
кул, определяемую методом газовой электронографии................ 155
7. Возможности метода газовой электронографии.................... 160
8. Определение геометрии молекул при совместном использовании
электронографических и спектроскопических данных.............. 162
9. Некоторые стереохимические результаты электронографических ис-
следований ...................................................... 163
Контрольные вопросы............................................... 167
^Четвертый раздеХ'''Методы колебательной ИК и КР спектроскопии . . . ч 169
Глава VIII. Теоретические основы колебательной спектроскопии............ 171
1. Квантово-механическое представление колебательных спектров ... 171
2. Основы классической теории колебательных спектров.............. 181
3. Практический расчет колебательных спектров..................... 185
Глава IX. Симметрия молекул и нормальных колебаний...................... 191
1. Общие представления о симметрии молекул........................ 191
2. Качественные представления о симметрии колебаний............... 194
3. Результаты теоретико-группового анализа колебаний.............. 199
4. Резонанс Ферми................................................. 204
5. Эффекты кристалличности........................................ 205
Глава X. Анализ и интерпретация спектров. Определение симметрии и
структуры молекул.................................................. 206
1. Выводы из сопоставления ИК и КР спектров....................... 206
2. Поляризация полос в спектрах КР................................ 212
3, Контуры вращательной структуры полос........................... 215
4. Групповые или характеристические частоты...................... 220
5. Изотопные эффекты.............................................. 227
Глава XI. Другие применения колебательных спектров.................... 230
1. Определение силовых полей молекул............................. 230
2. Корреляции силовых постоянных молекул с другими свойствами . 235
3. Крутильные колебания и потенциальные барьеры внутреннего вра-
щения ............................................................ 238
4. Использование фундаментальных частот для расчета колебатель-
ных вкладов в термодинамические функции........................... 242
5. Идентификация соединения и качественный анализ смесей........ 244
6. Количественный анализ.......................................... 247
7. Исследование равновесий........................................ 252
8. Комплексы с водородными связями................................ 255
9, Кинетические исследования...................................... 257
10. Колебательная спектроскопия высокомолекулярных соединений . . 258
Глава XII. Приборы и экспериментальная техника........................ 264
1. Техника и методики ИК спектроскопии............................ 264
1.1. Принципы устройства и действия ИК спектрометров.............. 265
1.2. Характер и подготовка образцов............................... 271
1.3. Дополнительные приспособления. Исследования специфических
образцов......................................................... 276
2. Нарушенное полное внутреннее отражение (НПВО)................. 278
3. Техника спектроскопии КР..................................... 283
3.1. Спектральная аппаратура и образцы............................ 283
3.2. Резонансное и инверсное КР................................. 286
3.3. Методы нелинейной спектроскопии КР.........................__ 2&Д
Контрольные вопросы.............................................^^290
Пятый раздел. Методы электронной УФ спектроскопии................... 293
Глава XIII. Основы теории электронных спектров молекул................ 295
1. Общая характеристика свойств электронных состояний............ 295
2. Номенклатура и символика электронных состояний............... 299s
3. Классификация электронных переходов, их относительное положе-
ние .............................................................. 306
4. Правила отбора и интенсивность переходов...................... 313
Глава XIV. Применение электронных спектров............................ 320
1. Структурно-спектральные корреляции............................. 320
1.1. Органические соединения...................................... 320
1.2. Неорганические и комплексные соединения...................... 327
2. Аналитические применения....................................... 329
2.1. Качественный анализ и идентификация веществ.................. 329
2.2. Количественный анализ........................................ 330
Глава XV. Техника и методики электронной спектроскопии................ 333
1. Аппаратура абсорбционной спектроскопии ........................ 333
2. Подготовка образцов............................................ 337
3. Спектроскопия с дифференцированием, разностная спектроскопия
и двухволновая спектроскопия ..................................... 339
4. Спектры люминесценции......................................... 342
4.1. Теоретические основы......................................... 342
4.2. Практическое применение и техника люминесцентной спектро-
скопии .......................................................... 347
Контрольные вопросы............................................... 352
Заключение............................................................ 354
Приложение............................................................ 355
Литература....................-....................................... 356
Предметный указатель.................................................. 359
Учебное издание
Лев Васильевич Вилков
Юрий Андреевич Пентин
ФИЗИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЯ В ХИМИИ.
СТРУКТУРНЫЕ МЕТОДЫ
И ОПТИЧЕСКАЯ СПЕКТРОСКОПИЯ
Зав. редакцией С. Ф. Кондрашкова. Редактор В. Н. Бораненкова. Мл. редакторы
С. М. Ерохина, Л. С. Макаркина. Художественный редактор Т. В. Скворцова. Художник
Ю. Д. Федичкин. Технический редактор Е. И. Герасимова. Корректор С. К. Завьялова
И Б № 6242
Изд. Хим-822. Сдано в набор 06.02.87. Подп. в печать 25.06.87. Формат бОХЭО’Лв. Бум. тип.
№ 1. Гарнитура литературная. Печать высокая. Объем 23 усл. печ. л. +0,25 усл. печ. л.
форзац. 23,25 усл. кр.-отт. 24,68 уч.-изд. л. +0,30 уч. изд. л. форзац. Тираж 8 400 экз.
Зак. № 949. Цена 1 р. 20 к.
Издательство «Высшая школа», 101430, Москва, ГСП-4, Неглинная ул., д. 29/14.
Московская типография № 8 Союзполиграфпрома при Государственном комитете СССР
по делам издательств, полиграфии и книжной торговли, 101898, Москва, Центр,
Хохловский пер., 7.
г
СВОДНАЯ СХЕМА ХАРАКТЕРИСТИЧЕСКИХ ИНФРАКРАСНЫХ ГРУППОВЫХ ЧАСТОТ
1500
3000
2500
4000__________
АМИНЫ
(продолжение)
имины
НИТРИЛЫ
гидрахлориды
вторичные амины
имины ,_____
замещ. имины
вторичные амины
:за-_ЕЕ;
третичные амины
c-nh;
C = NH
-C=N-C
ПРОЧИЕ ГРУППЫ
нитрилы
мзоцианиды
-CaN
№С
группы, содержащие серу
фосфор
кремний
фтор
фтор
фтор
хлор
хлор
бром~“
НЕОРГАНИЧЕСКИЕ СОЛИ
И ИХ ПРОИЗВОДНЫЕ
ОТНЕСЕНИЯ
4000 см'1
SH
Соединения,
содержащие О и S
соединения,
содержащие С и О
соединения,
содержащие P и О
валент. ОН и NH
налет. CH
сл.
3500
см'1
400
2000
1800
1600
1400
1 000
800
600
Л
SH
Si-С
(SO.
С = О хлорокарбонатов
С=О хлорангидридов
со.иса,
—< СП (алиф.)
(NO, Г--
r„O-NO.
R-NO.
l.CH.-S-CH,
.„I.,. । p=s
СВг, и СВгл—.
СВг (алиф.)
ср.
СЩ-О-СЗцРилиЗ)
ср.
SiH
СИ. О!’>О
R-NO
иалент. С~Х
вален r.C-O
валент.С-К
‘ валент C-C
деформ. СН
' 4 деформ ОН
маятнСН
маятн NH |
CHj-NH-CH
CH-NH-CH
ONH-R
(CHJjN
O-n-r,
(при сопряжении понижается)
Х=С=Х (изоцианаты, 1,2 - диеноидные-------
системы и т.д.)
Н .... И С=О напряженных диолов (/J-лактамы)
ион сульфата
ионы сульфона гоп
сульфоновые кислоты
ковалентно связанная сульфатная группа
ковалентно связанная сульфонатная гру ППЗ
сульфонамиды
сульфоны |
сульфоокиси I
ион фосфата •
ковалентно связанная фосфатная группа
ион карбоната | 111
ковалентно связанная карбонатная группа
иминокарбонаты I | I | I
соединения,
содержащие N и О
ион аммония NIlJ*
3000
( эпоксидиклы
г C-F (ненасыщ.)
C-F (насъ’щ)
(СОД
O=C(O-R)X'
HN=C(O-R)
ковалентно связанная
f | ни гро-группа
нитрососдинеиня
нитросоединения
ковалентно связанная
I нитритная группа
нитрозосоединения
R-O-NO L
2500
2000
(800
R-SO
R-SOjH
R-O-SOj
R-O-SO2
R-SOj-N
R-SO2-R
R-SO-R
(P°4')3'__
(RO), P
(нссопряж )
(сопряж.)
ср. 1
валент. С -О
—— ч валент. C^N
— -4 валент. C~C
деформ. Mi
1600
1400
(200
(1)00
ьоо
600
400
I----------------1____________I__________I__________f t I_______________________f I_________________I_____________I_________I_________1________I_______i___________I________I_______1______1______I---1---1______________!__________1
2,50 2,75 3,00 3,25 3,50 3.75 4,00 4.5 5.0 5,5 6.0 6.5 7.0 7.5 8.0 9.0 10 II 12 13 14 (5 20 25
икм
i