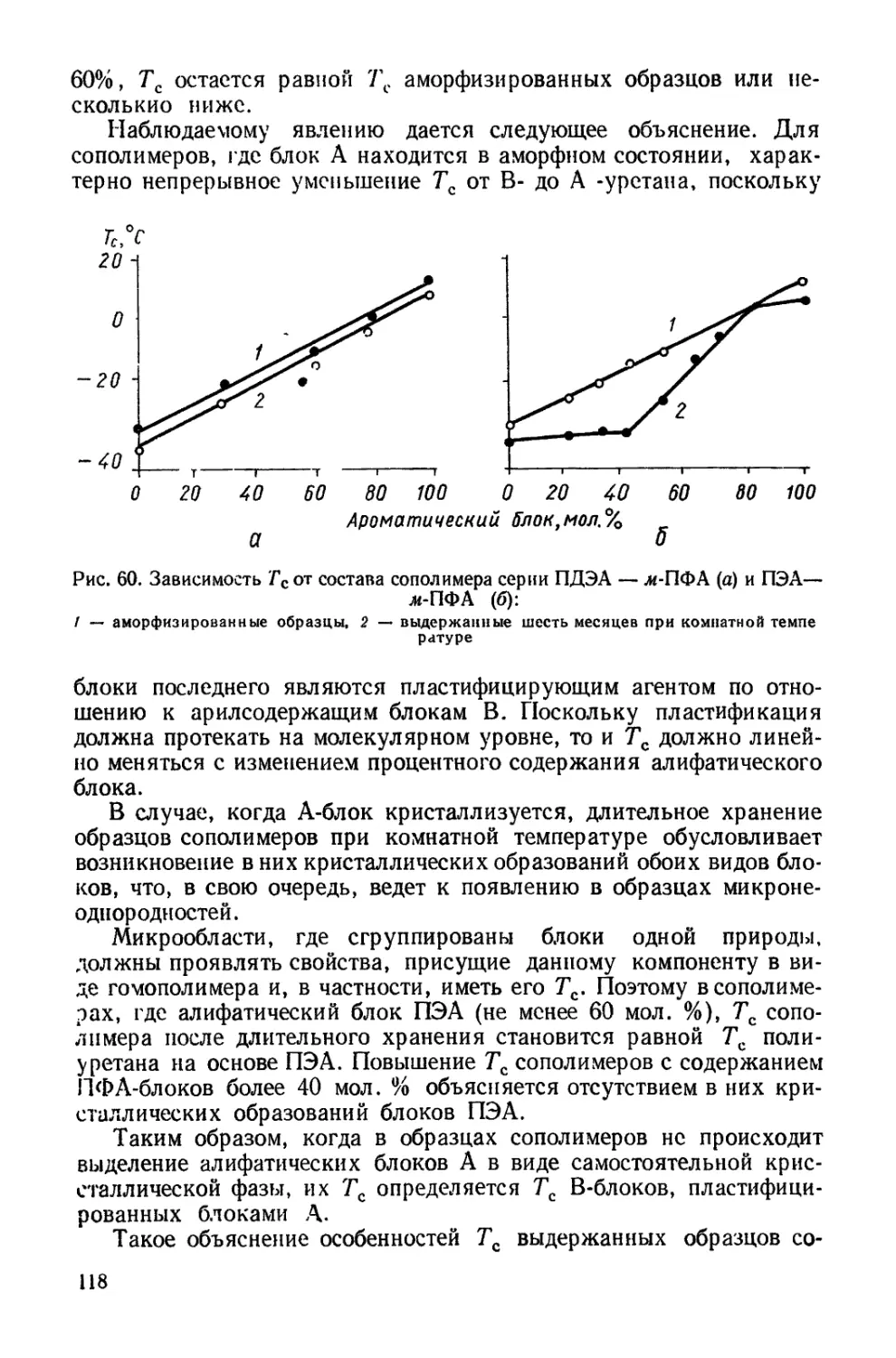
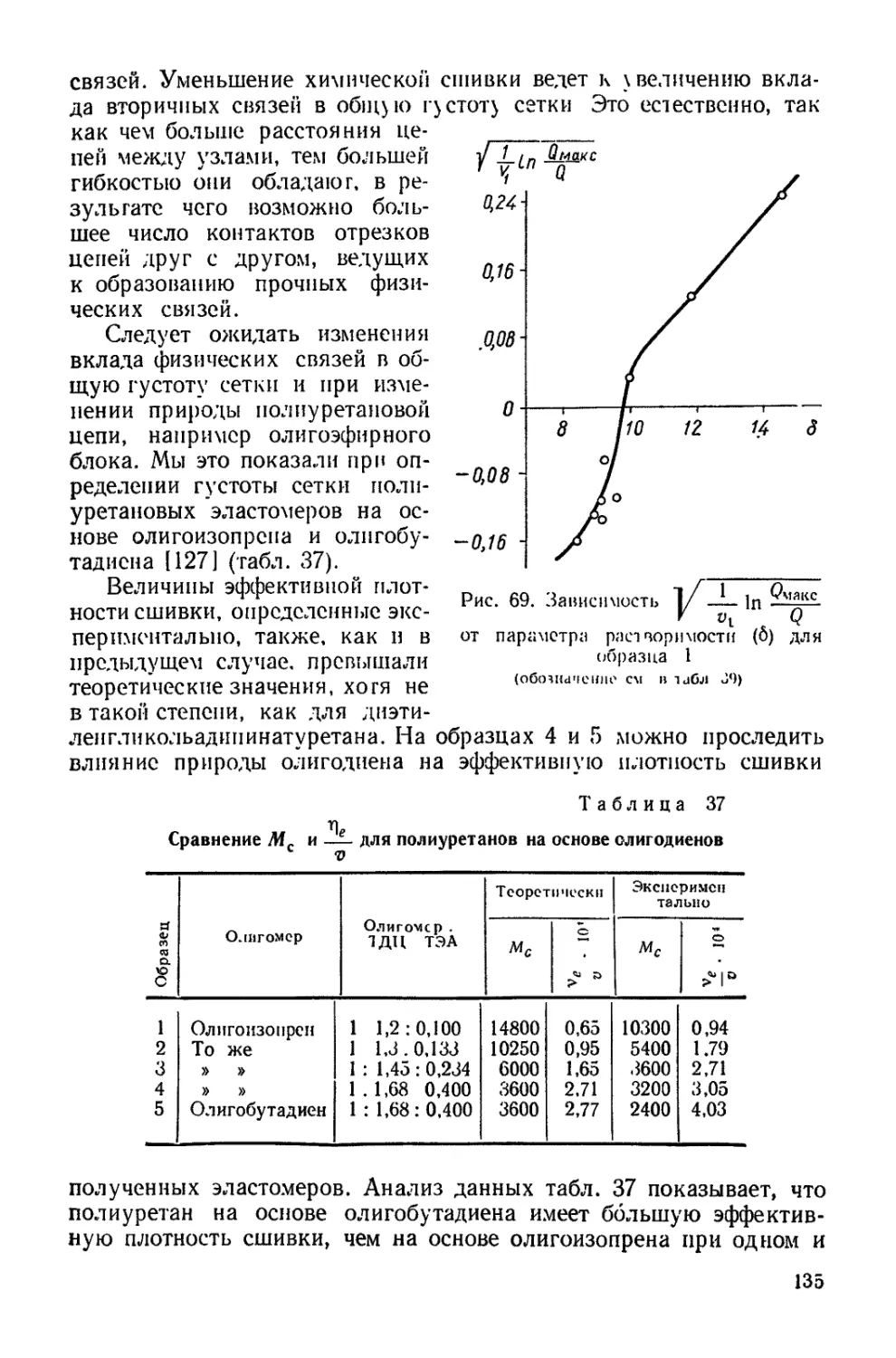
/


































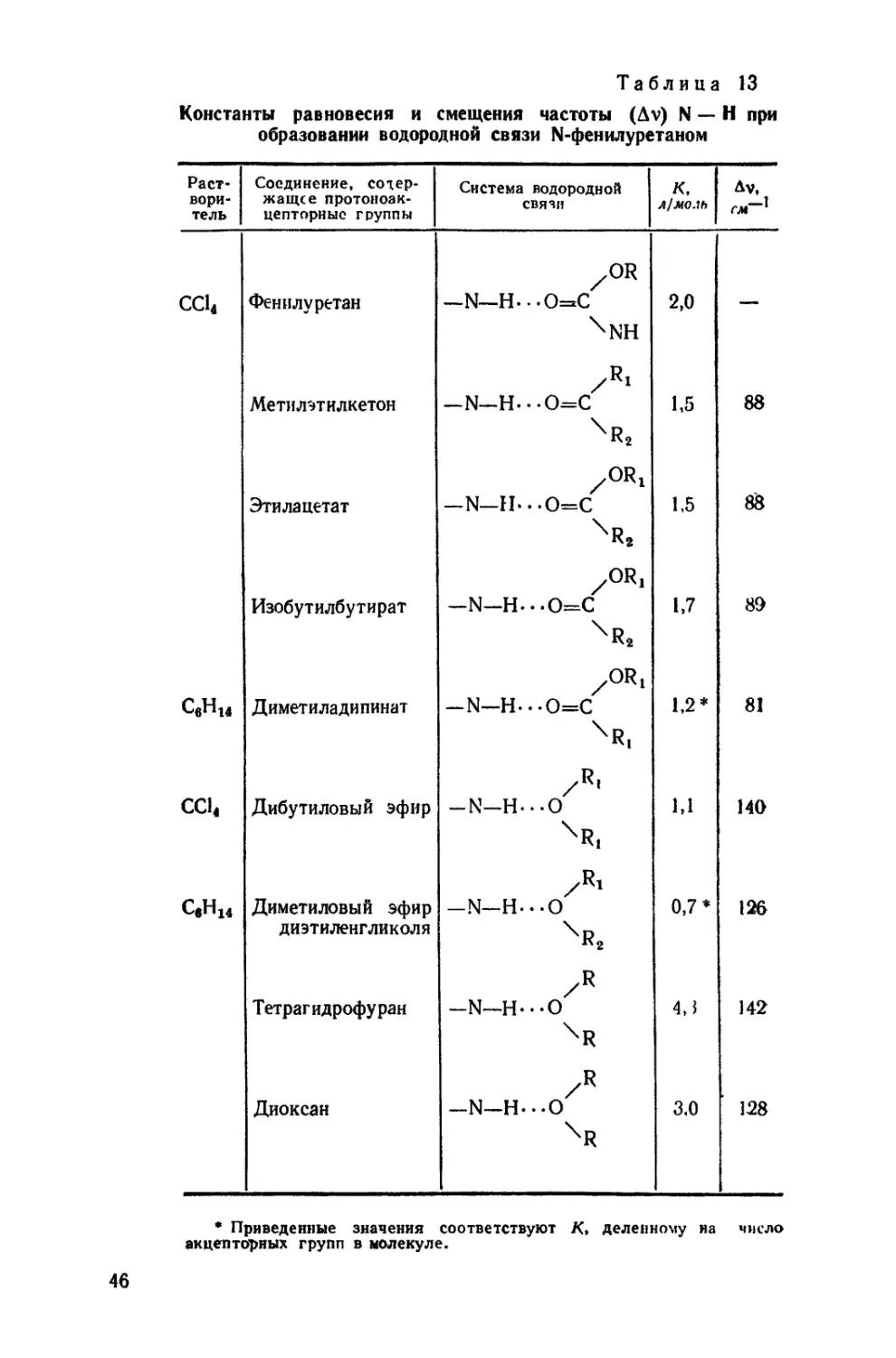



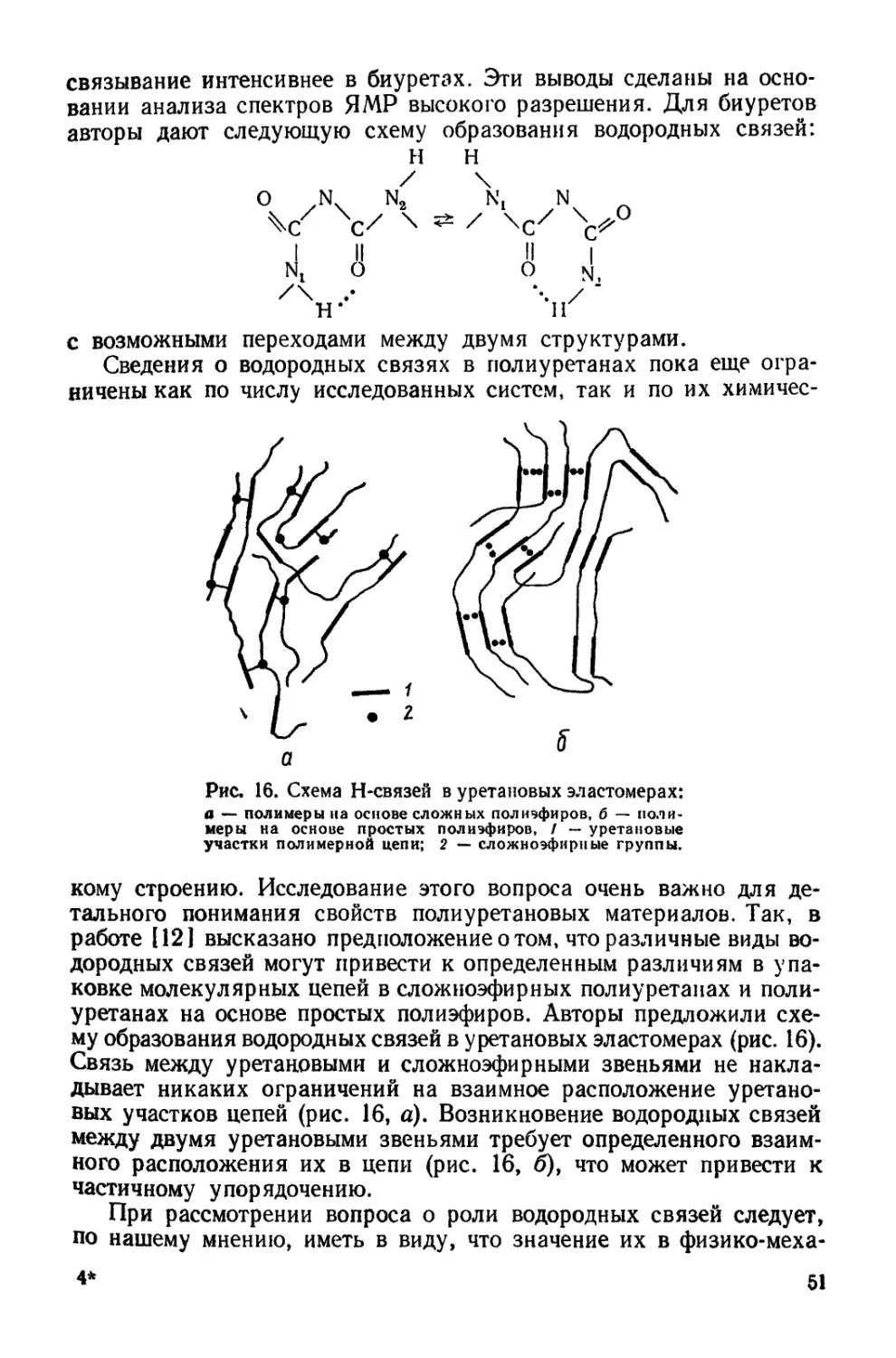




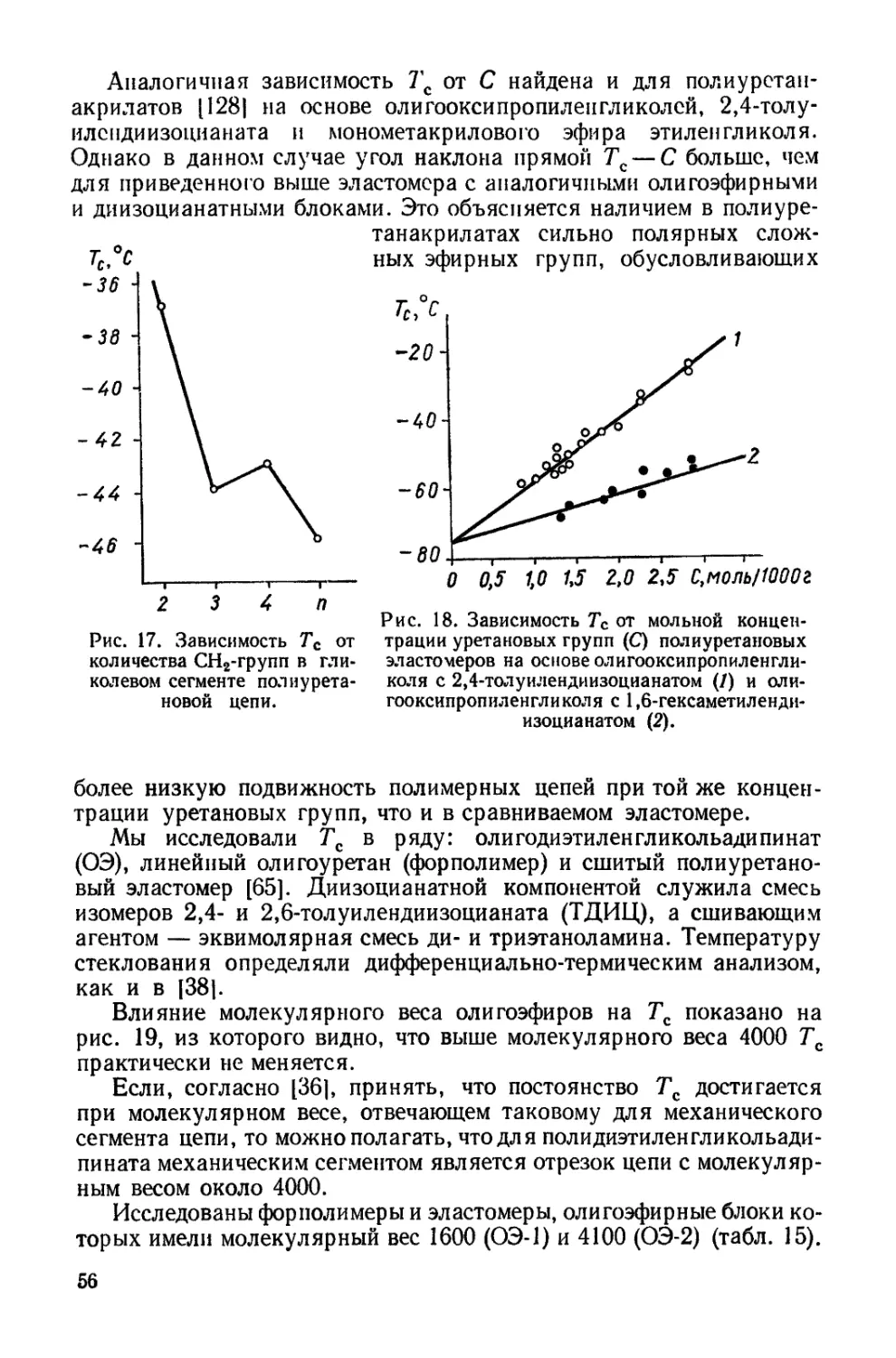











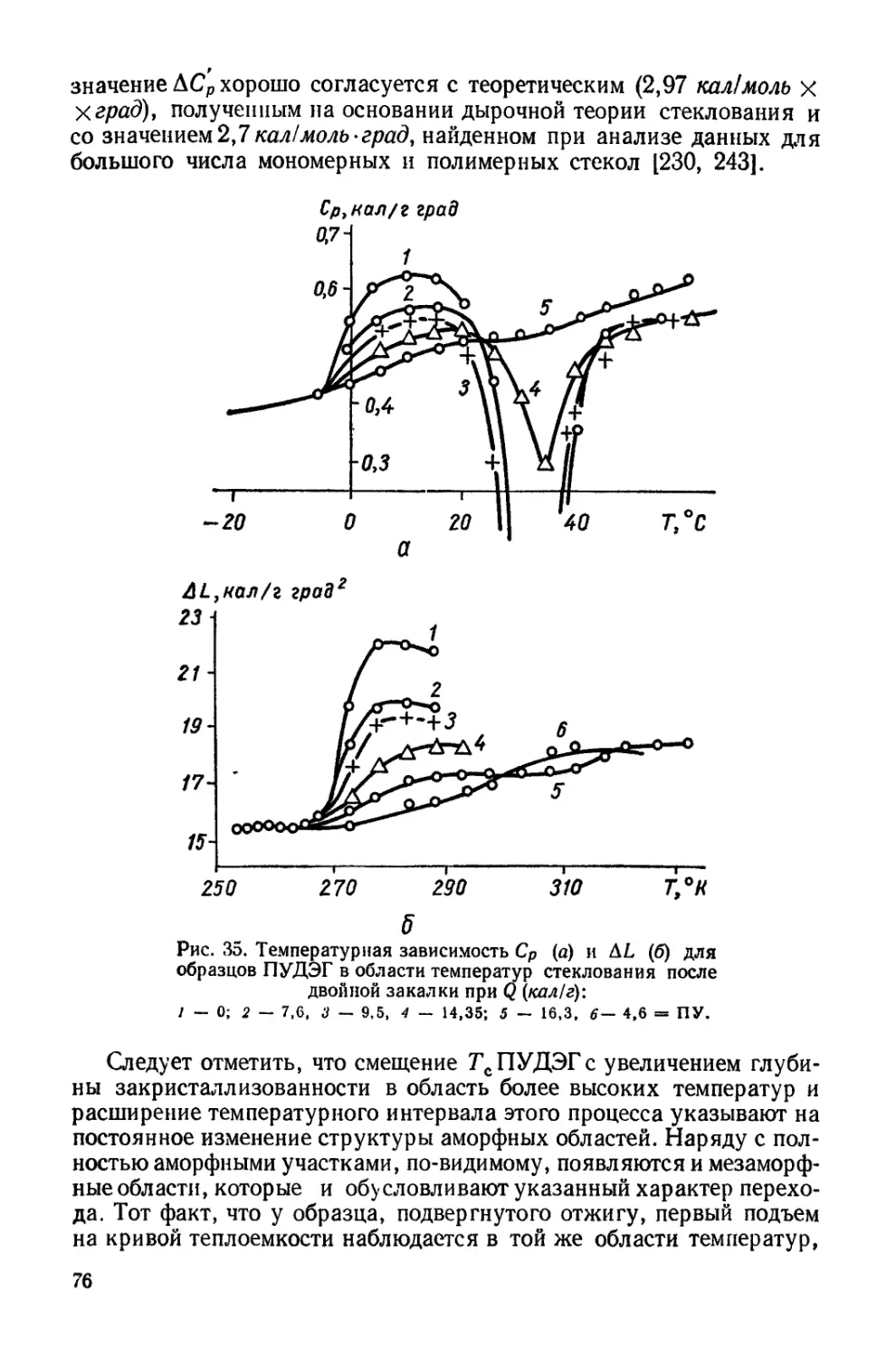

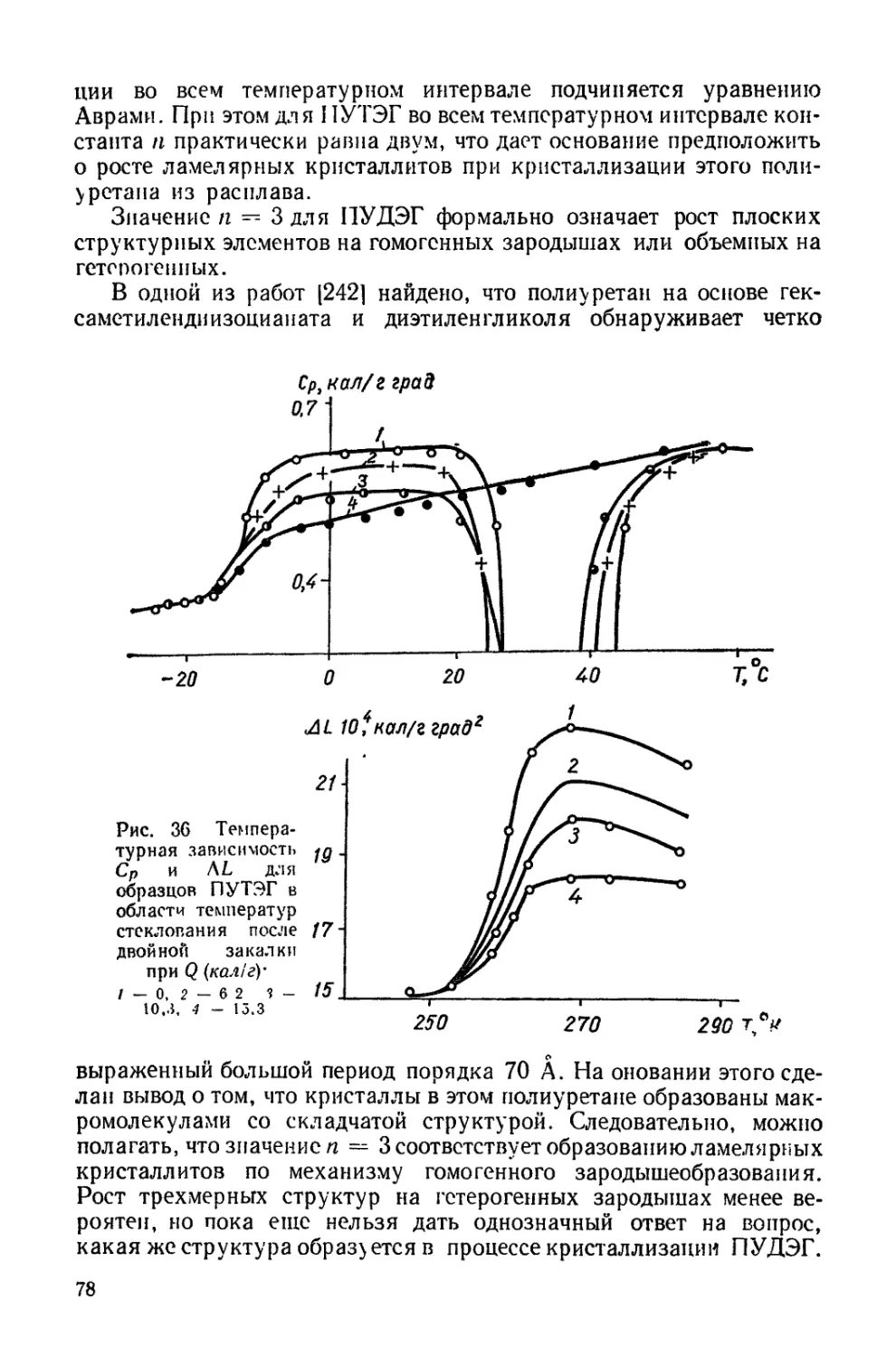
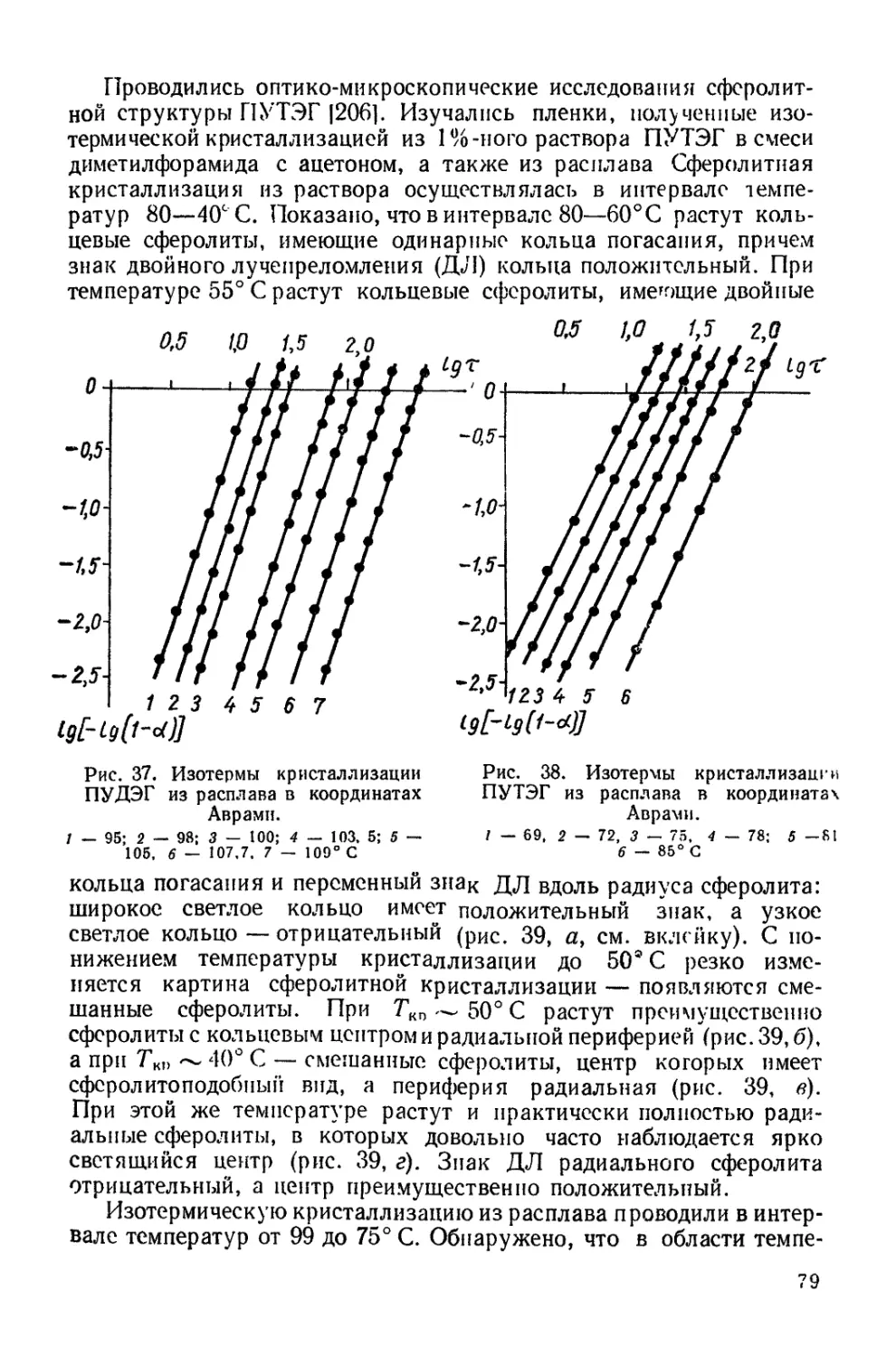




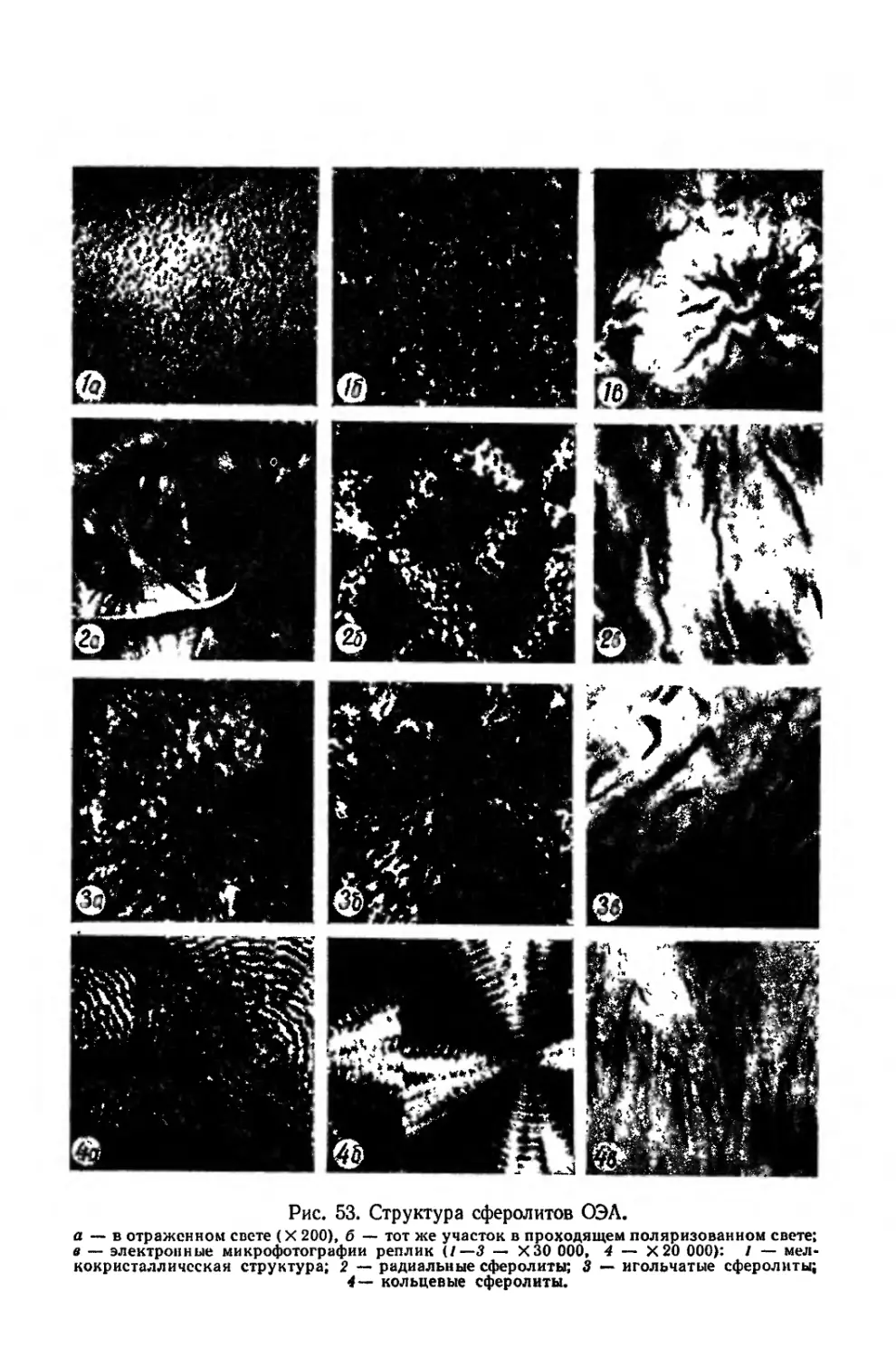


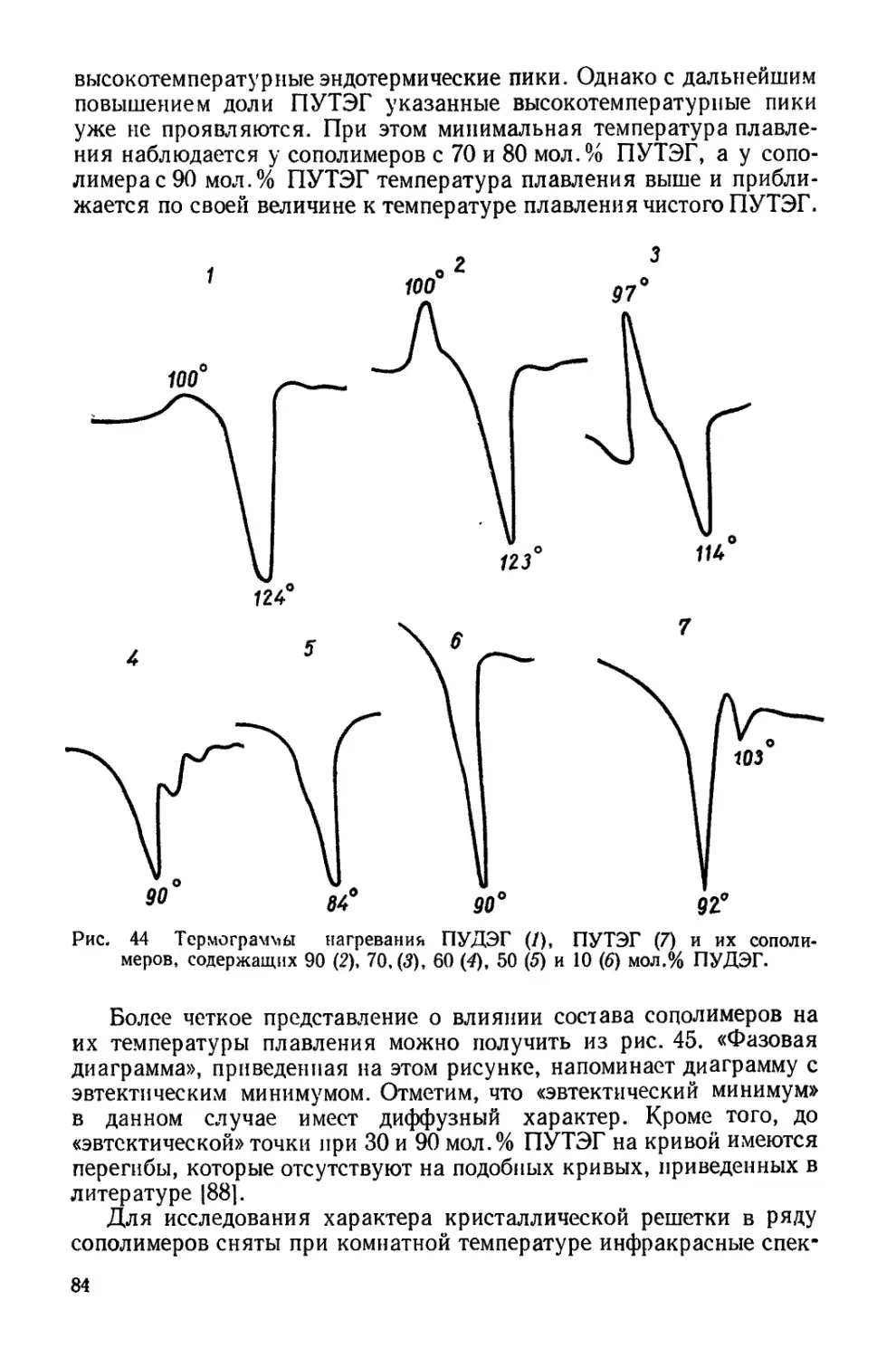
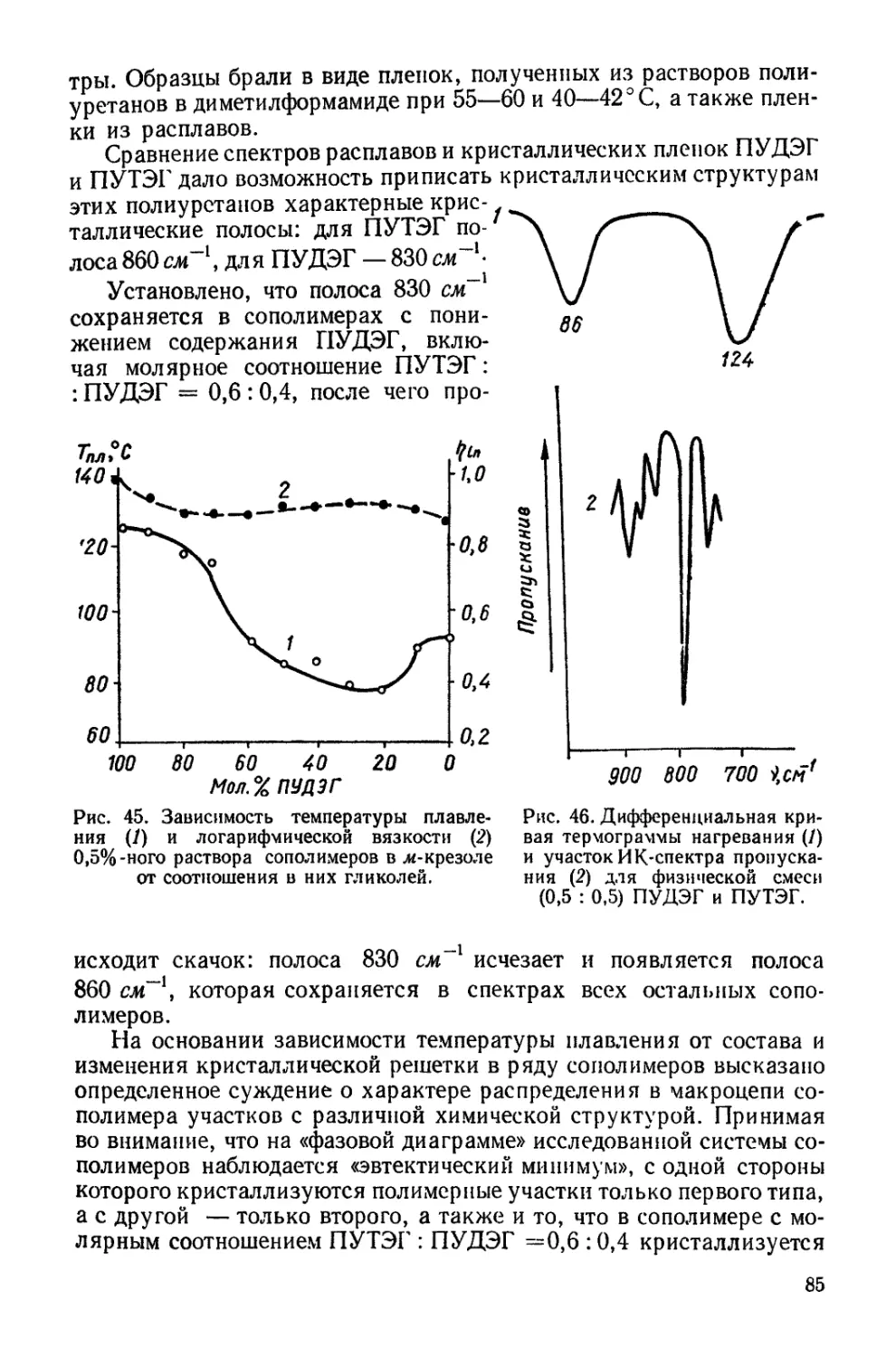

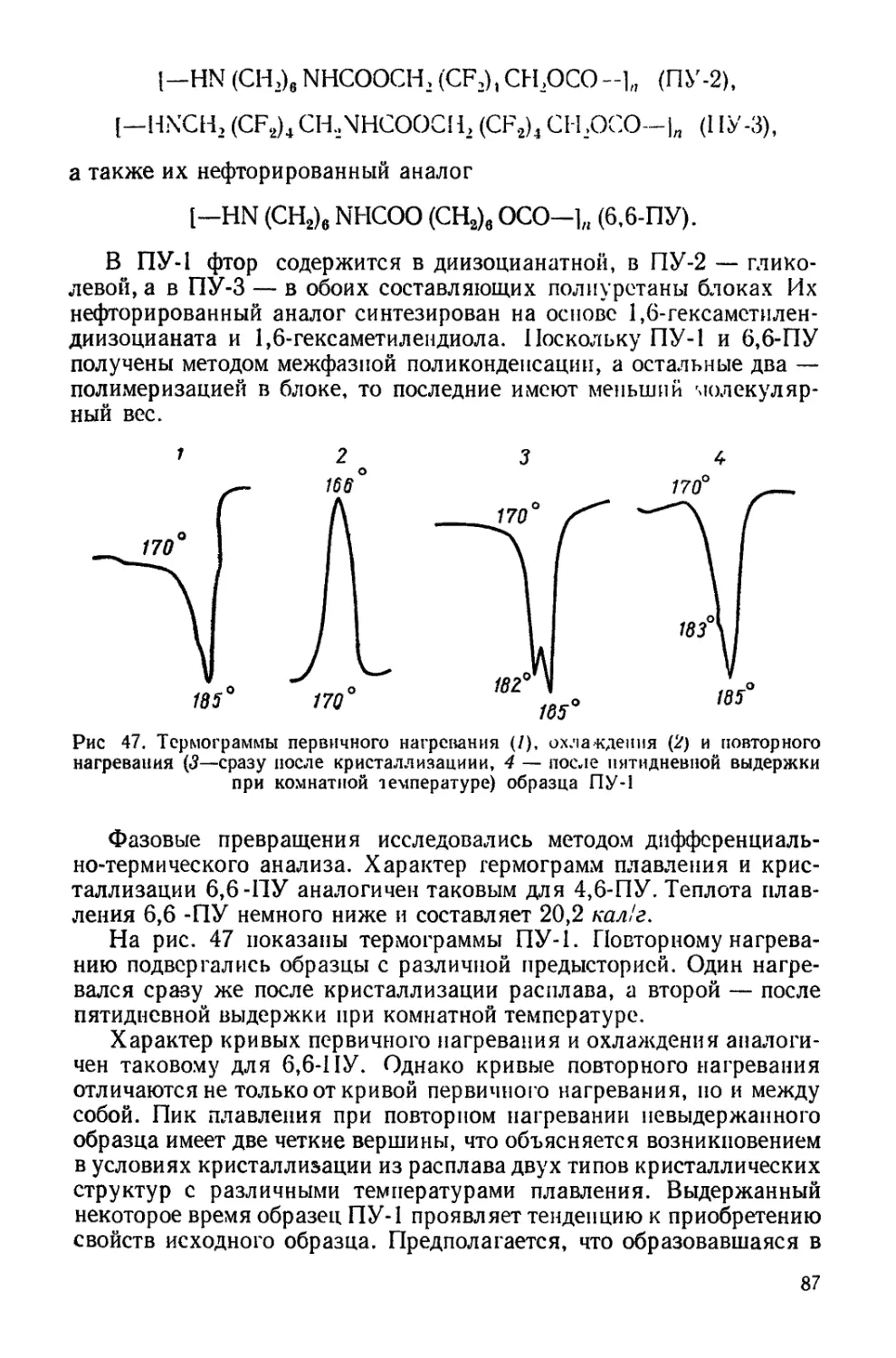
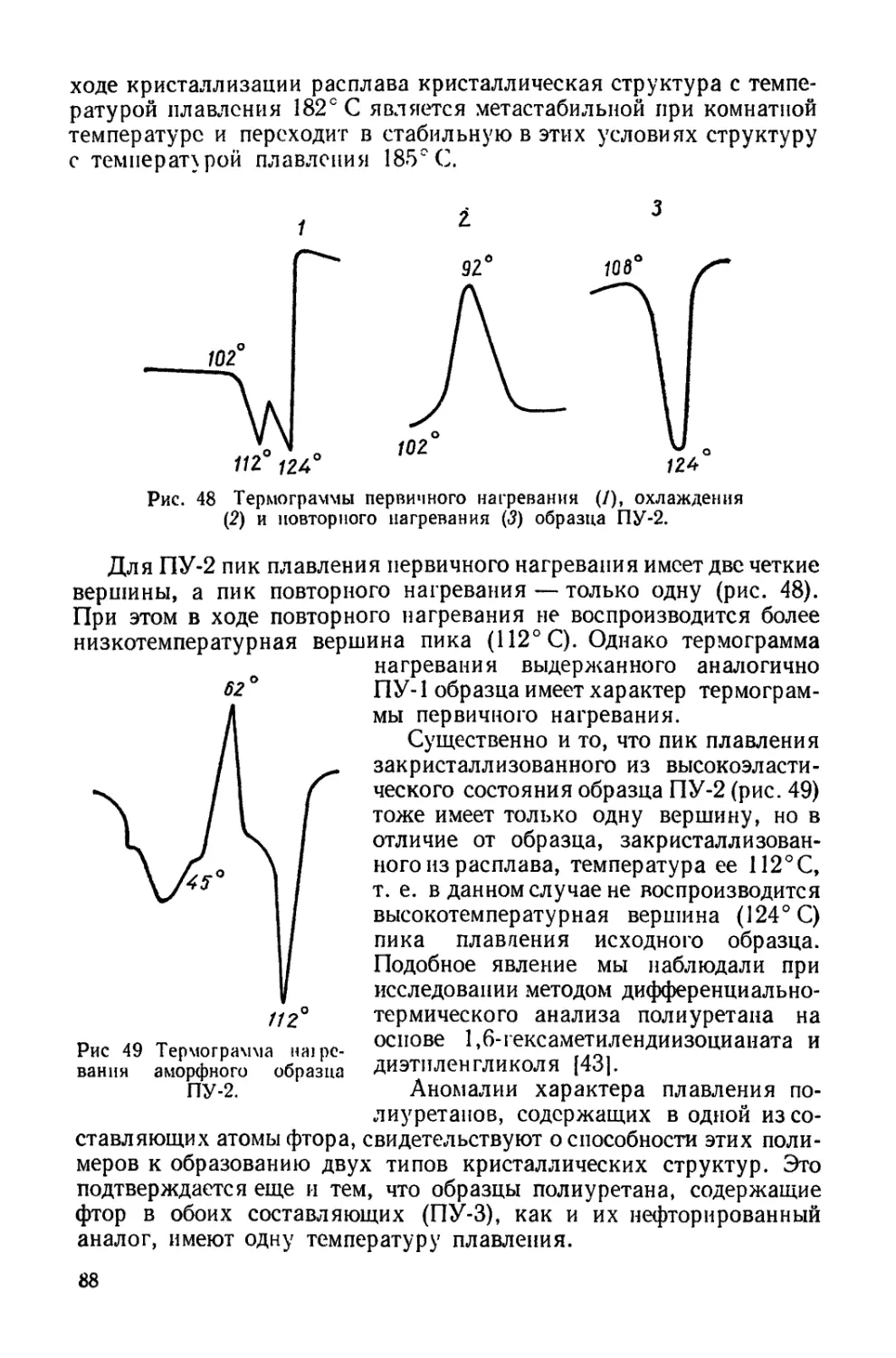

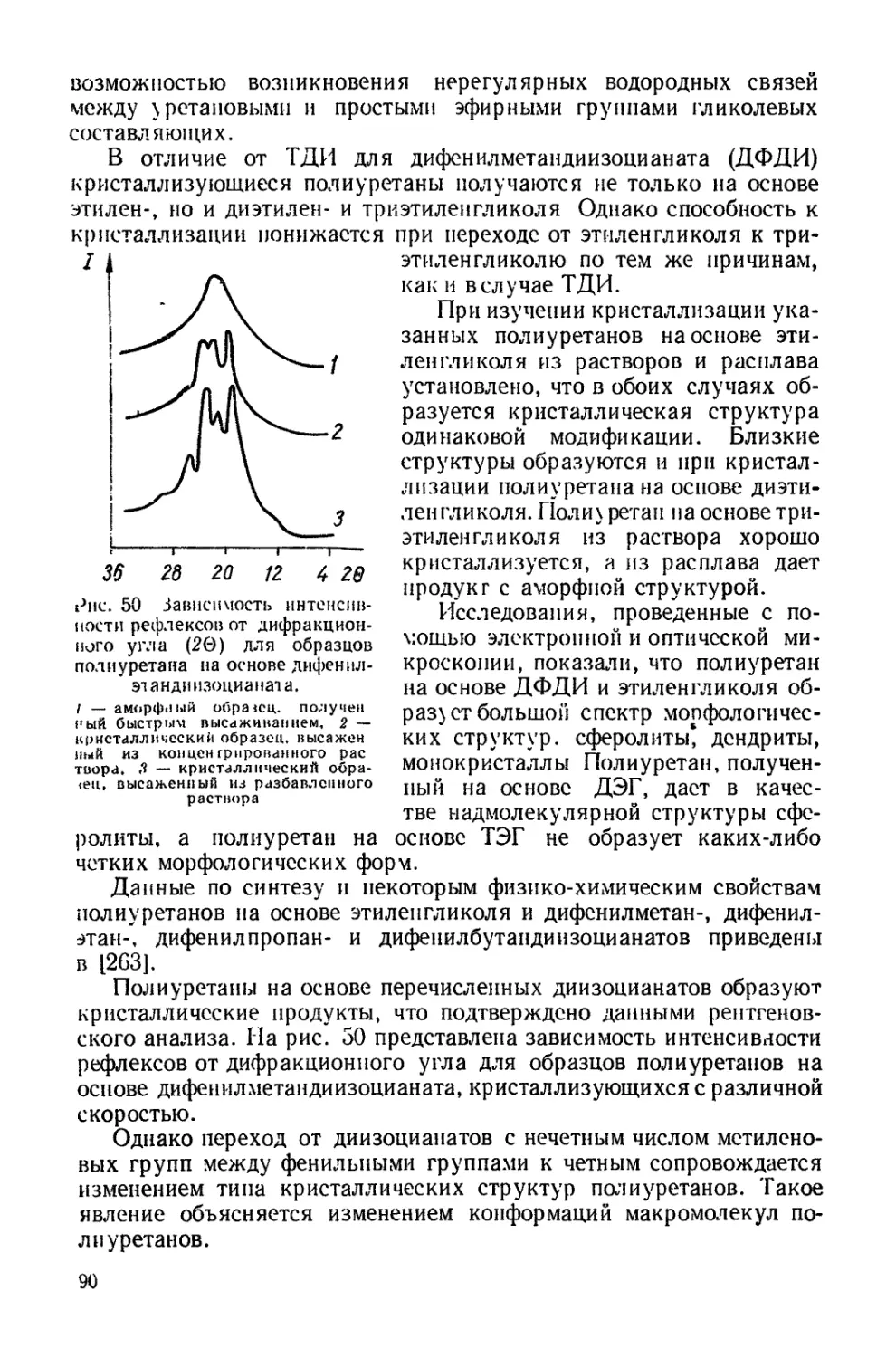

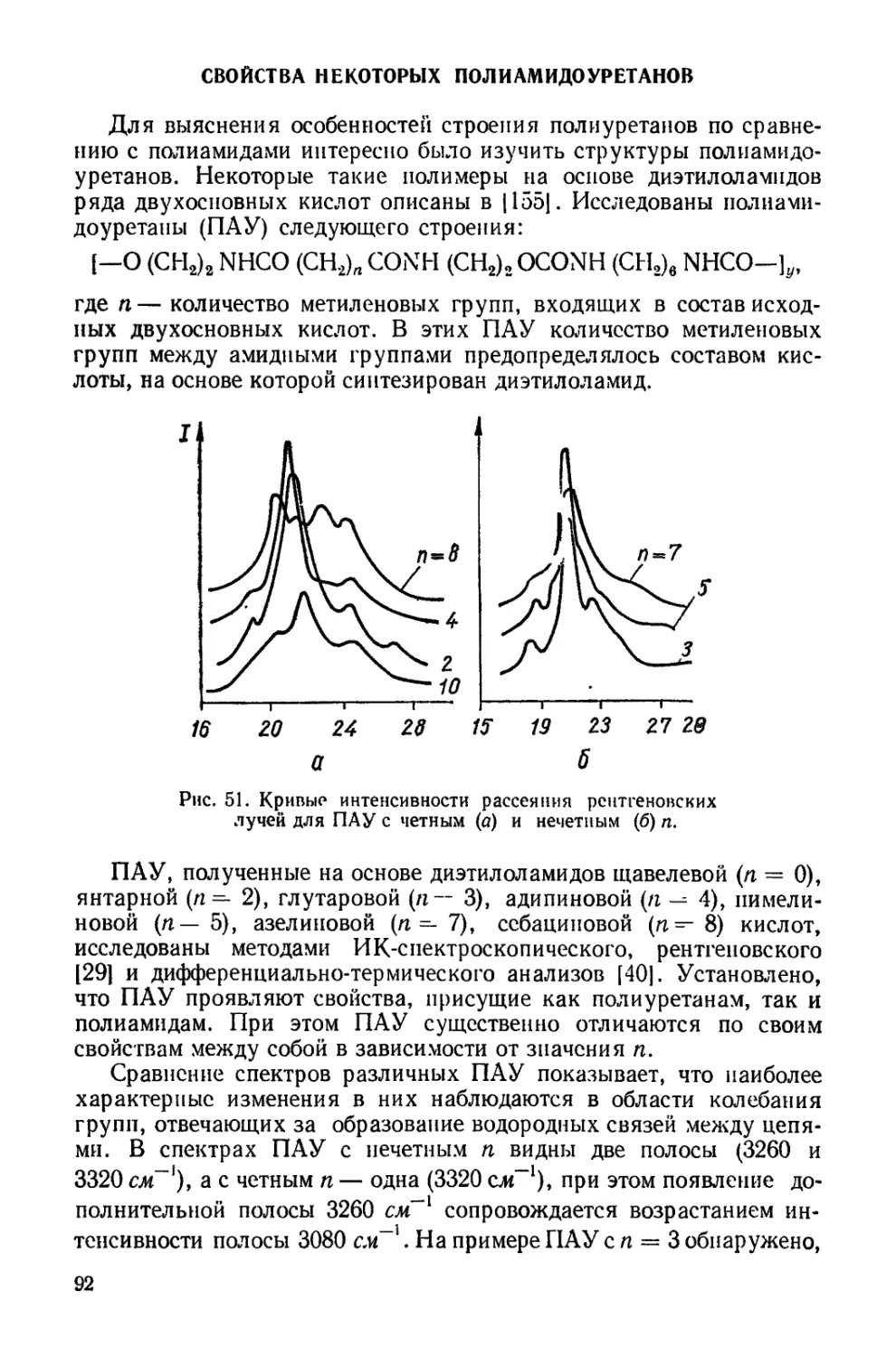
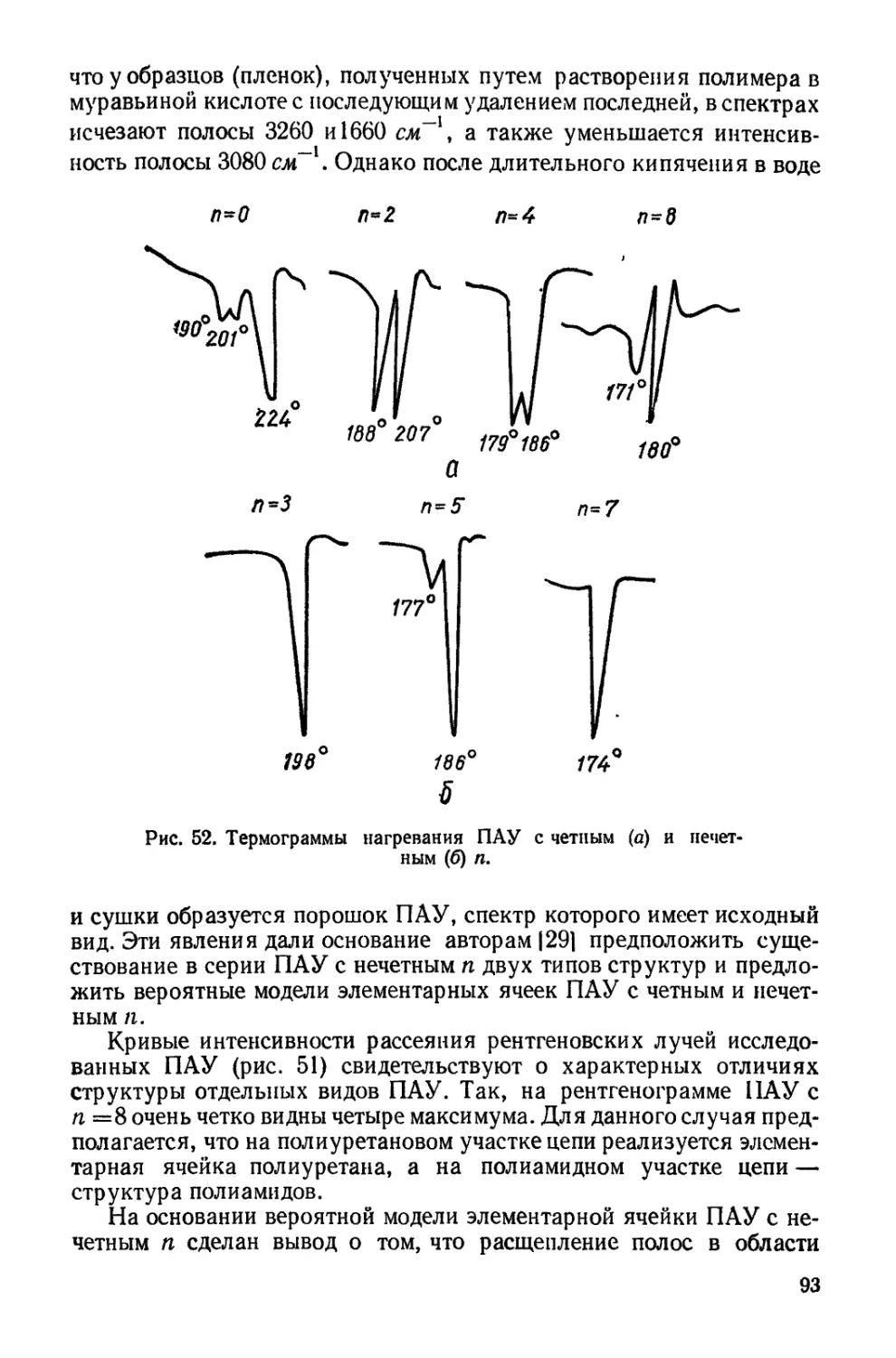








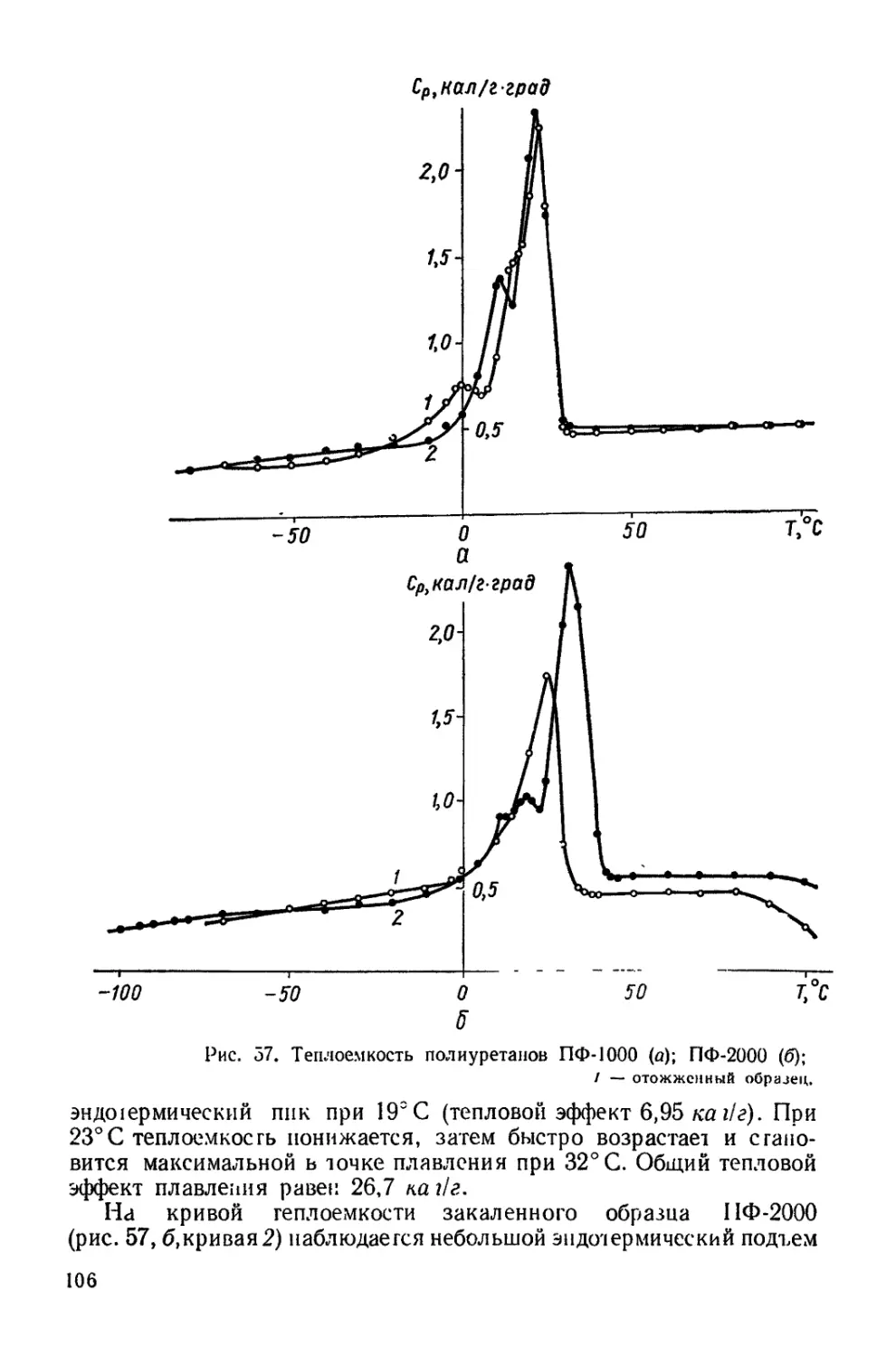
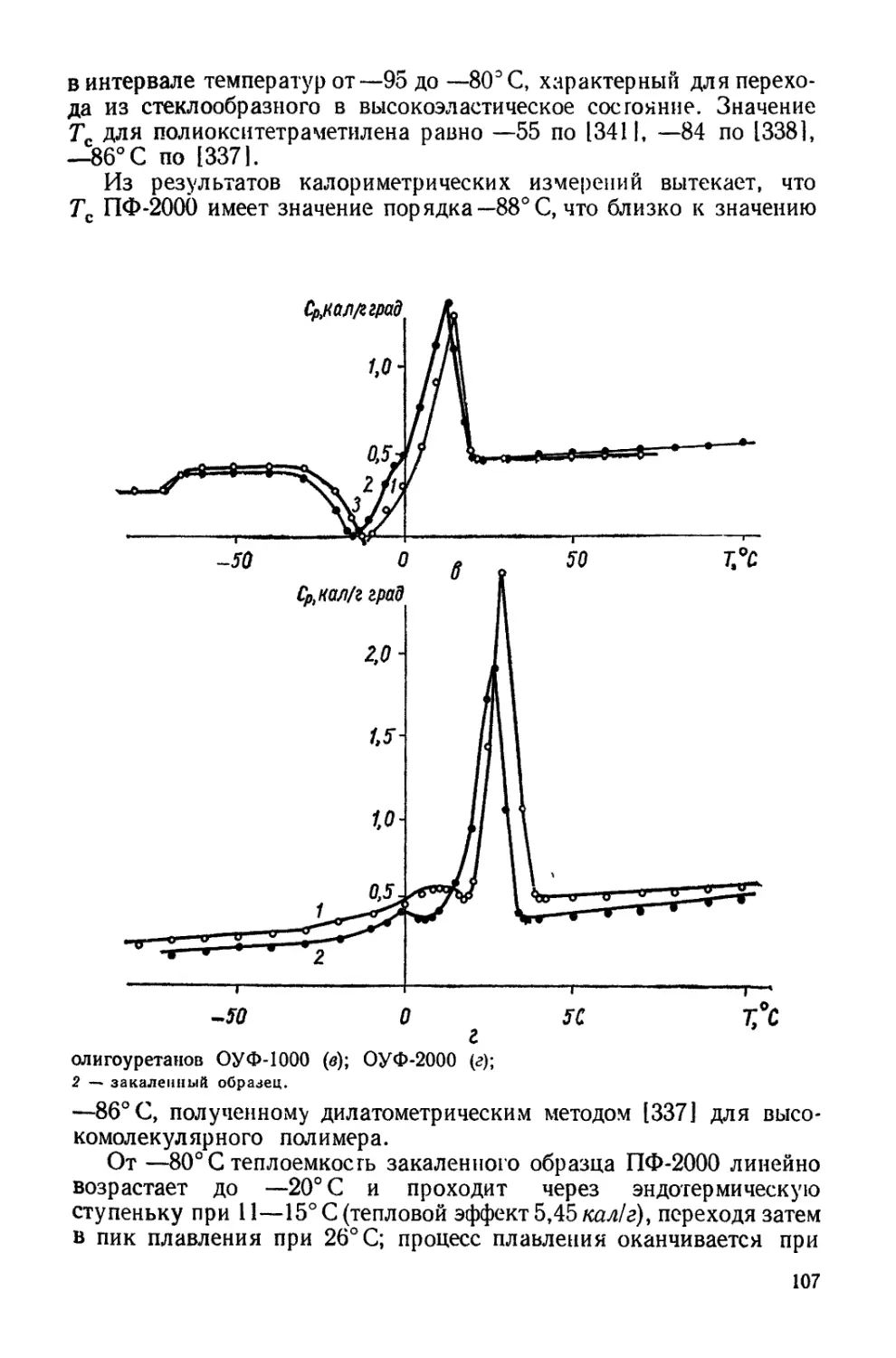
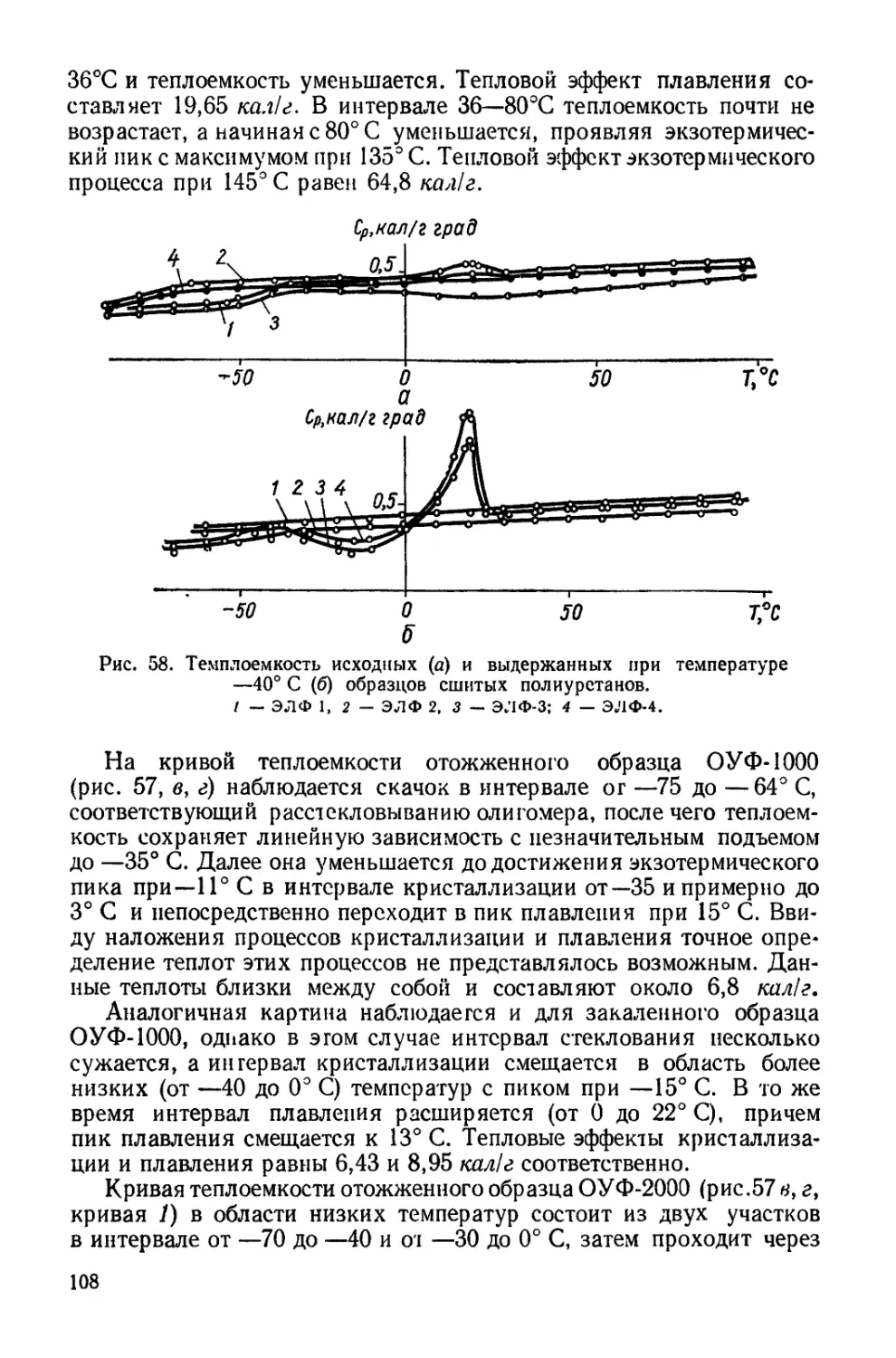
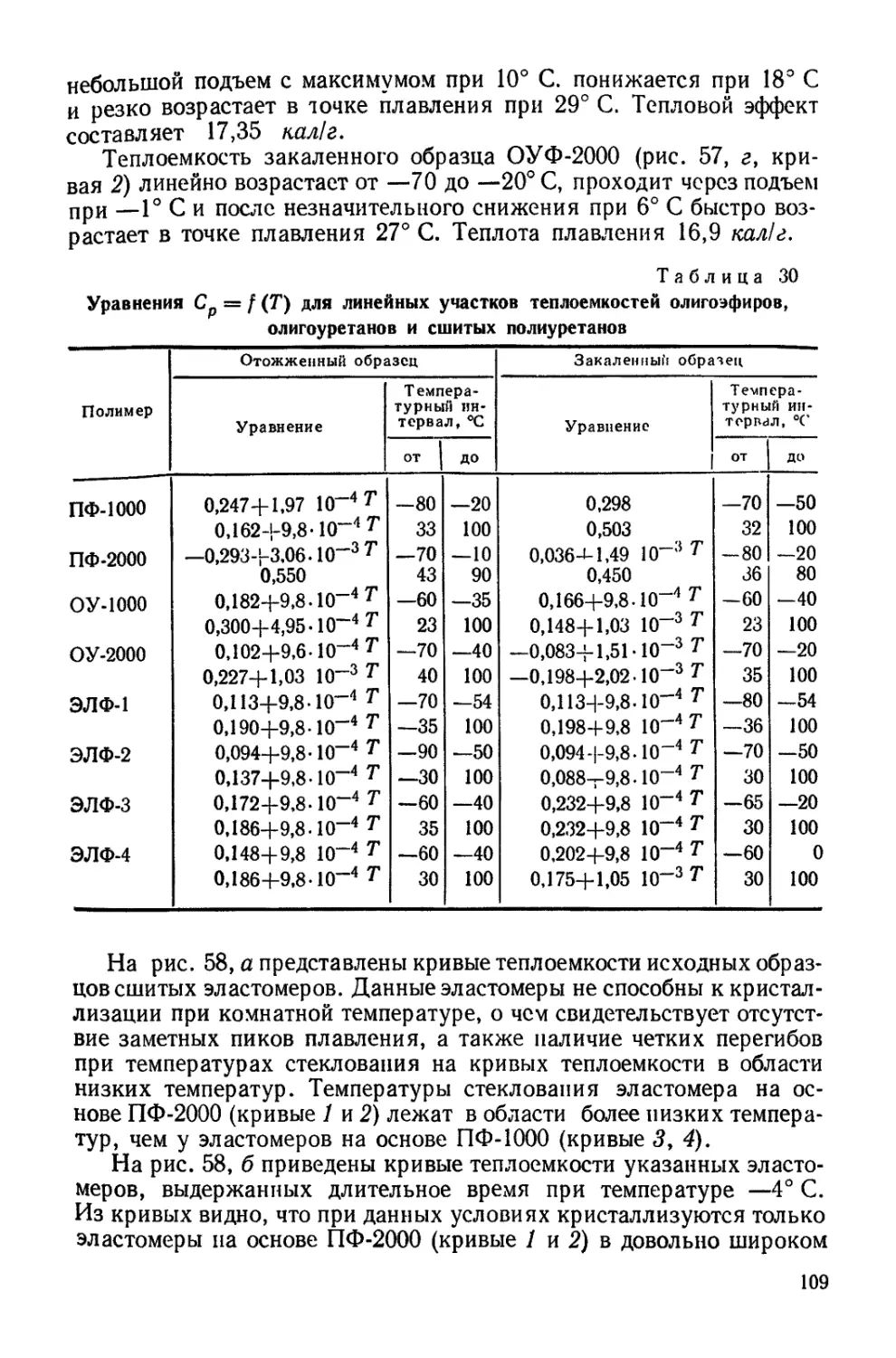
























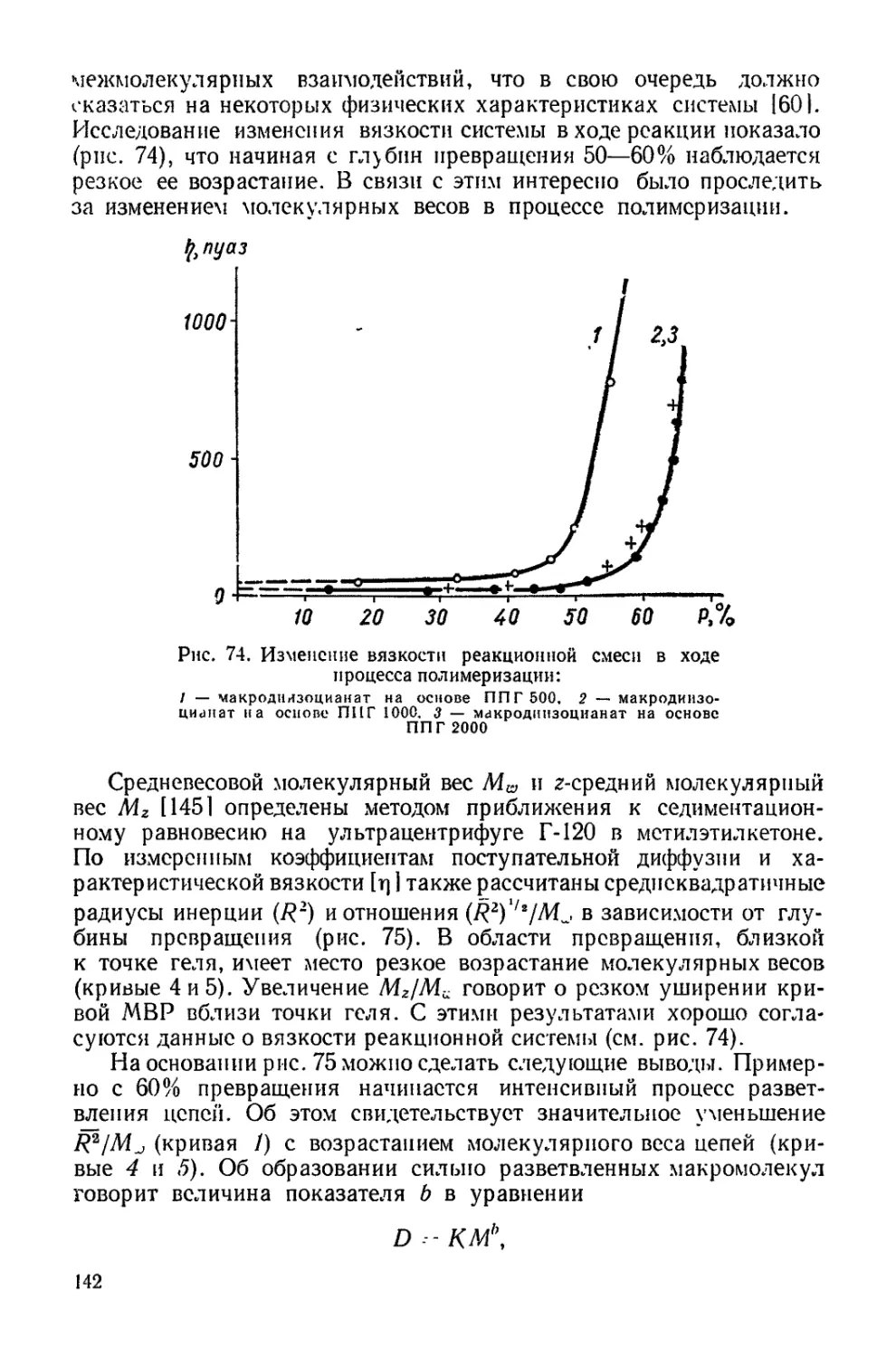
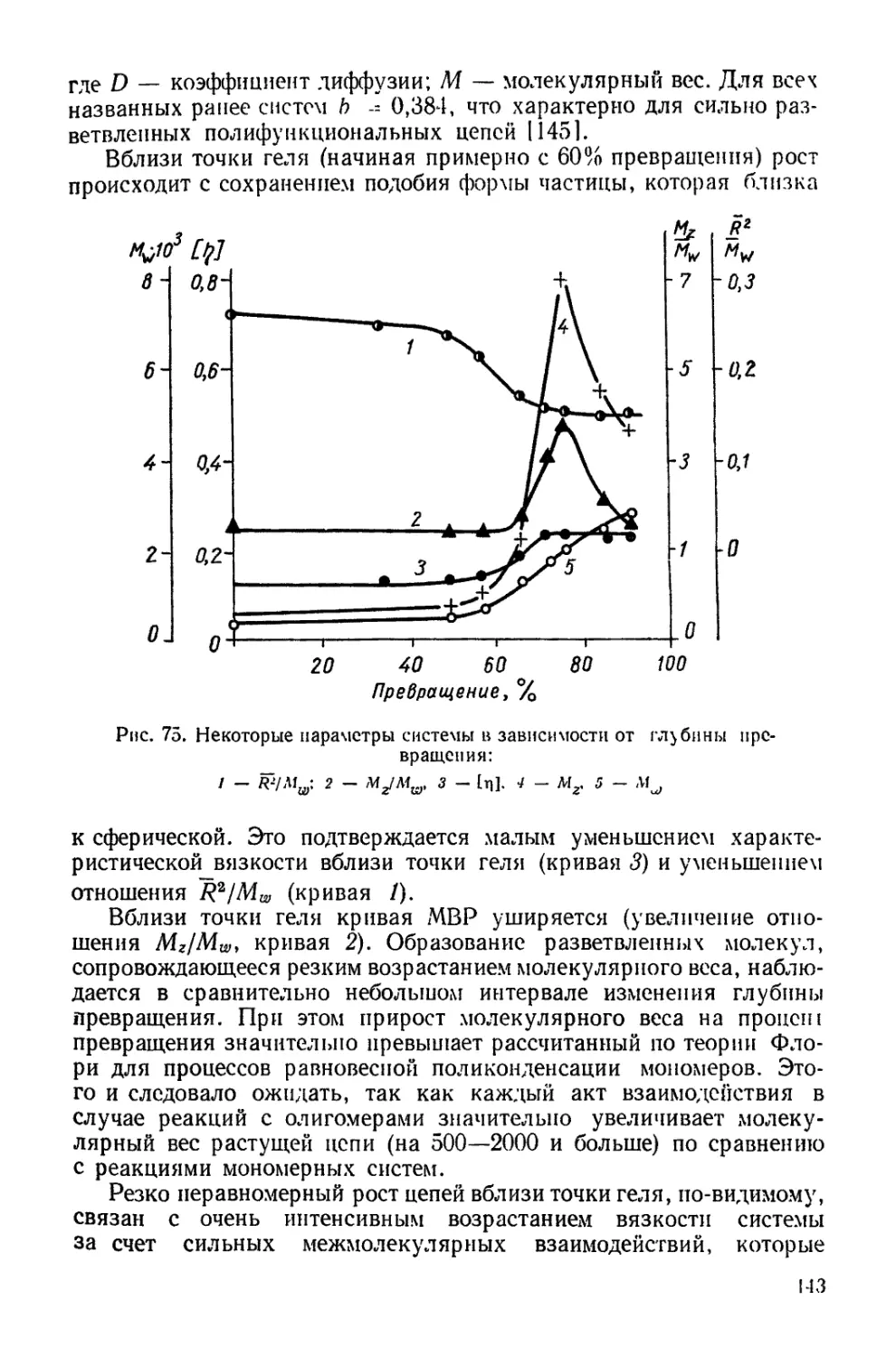

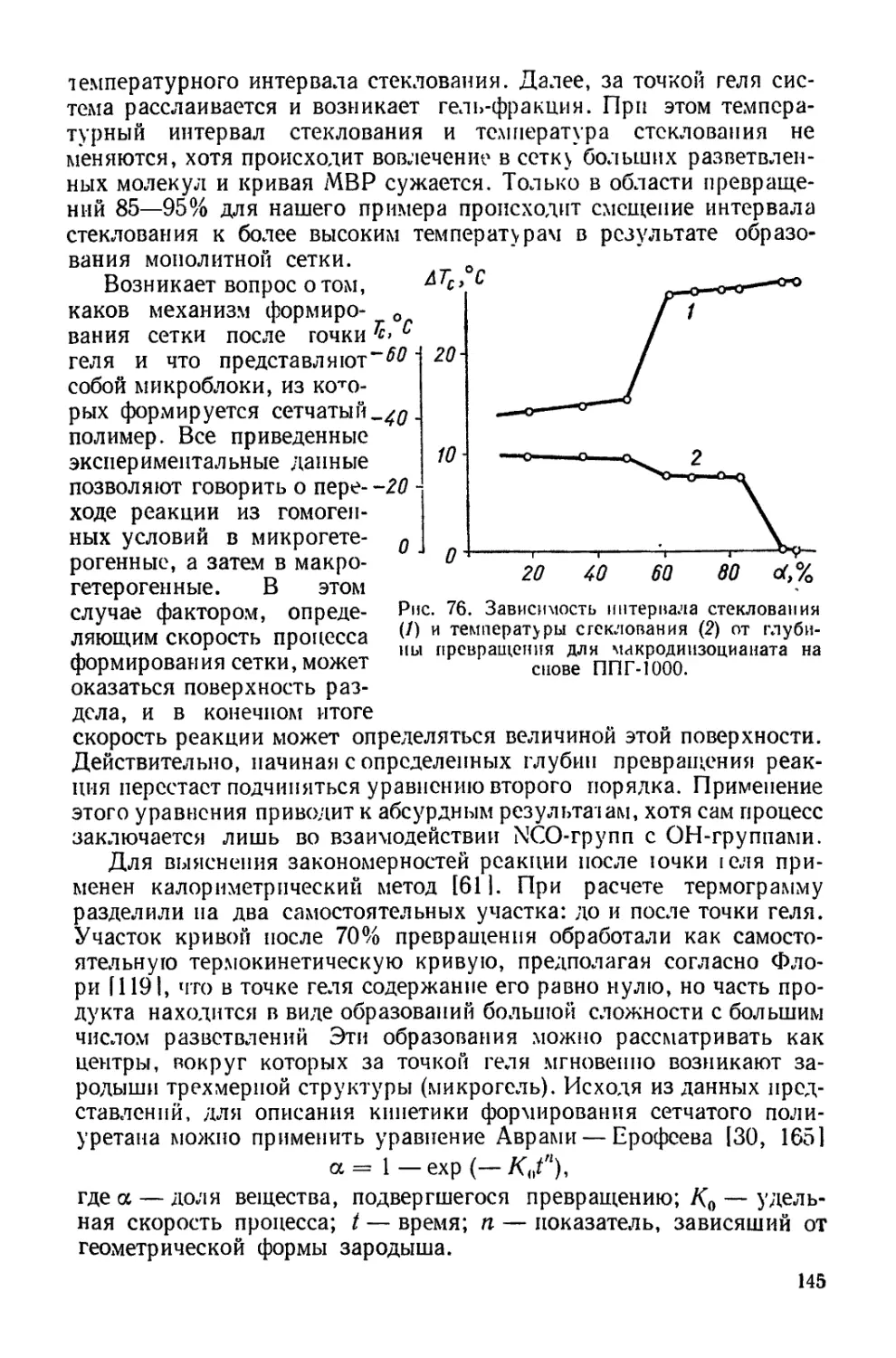
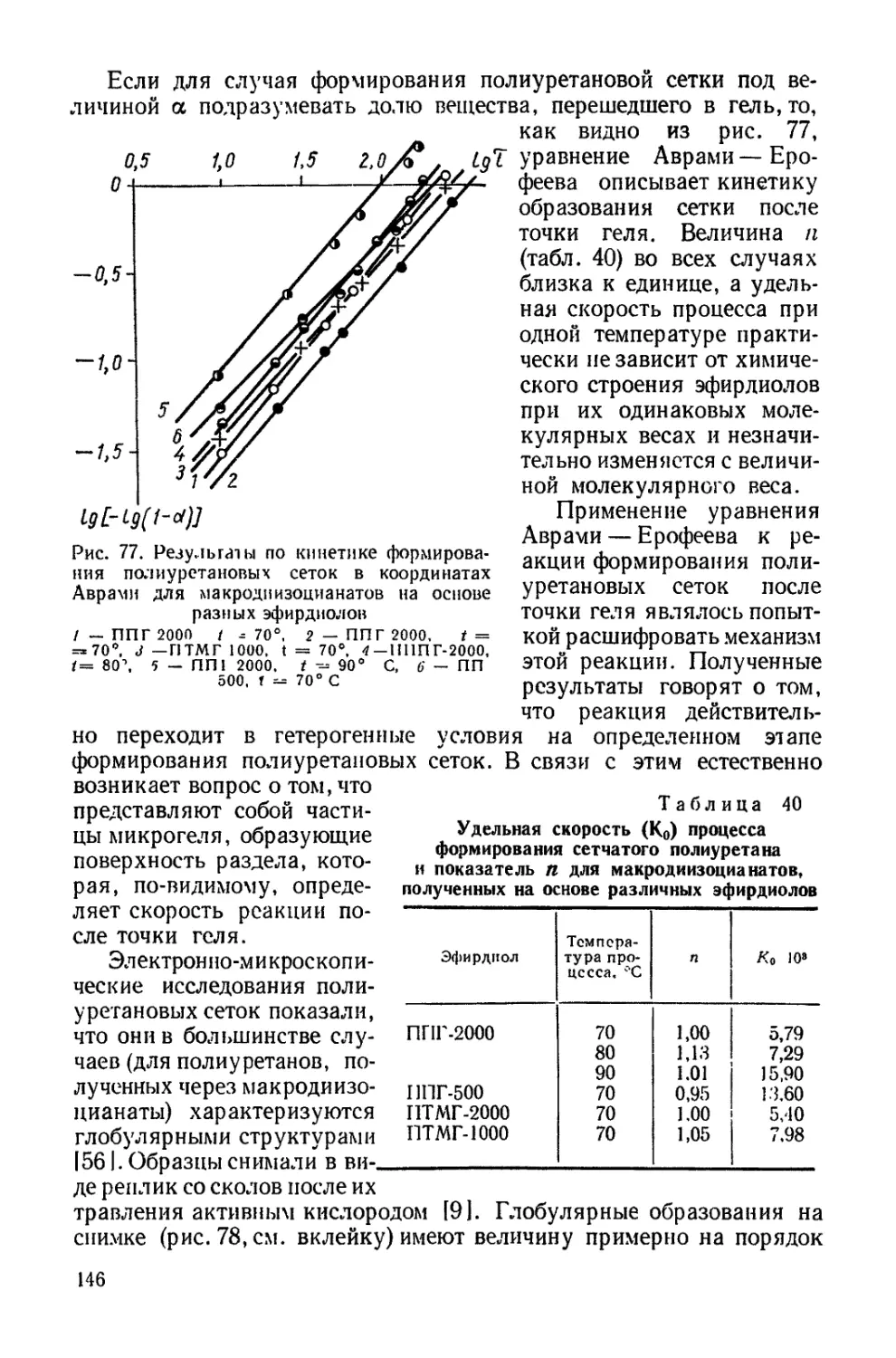













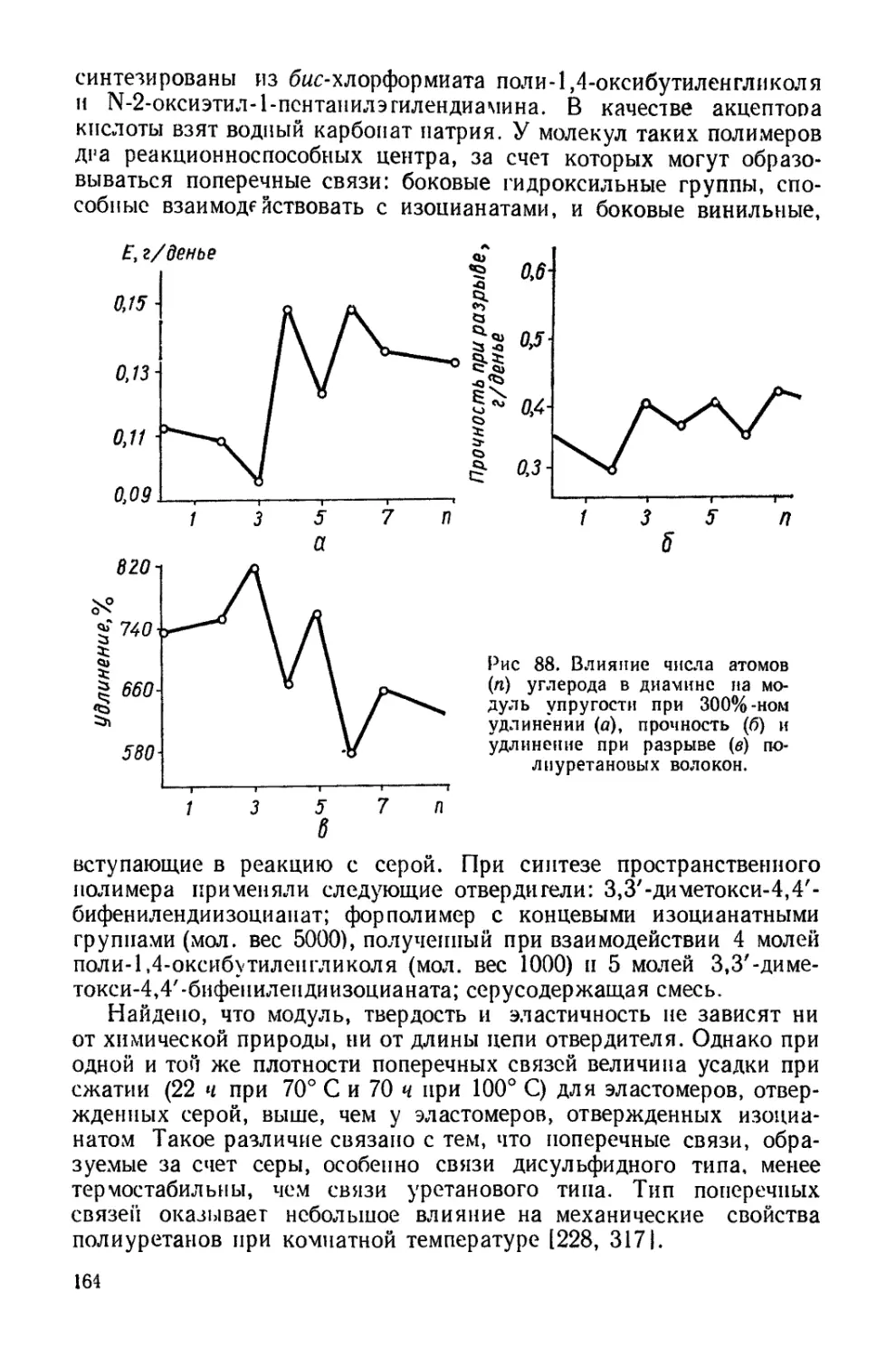
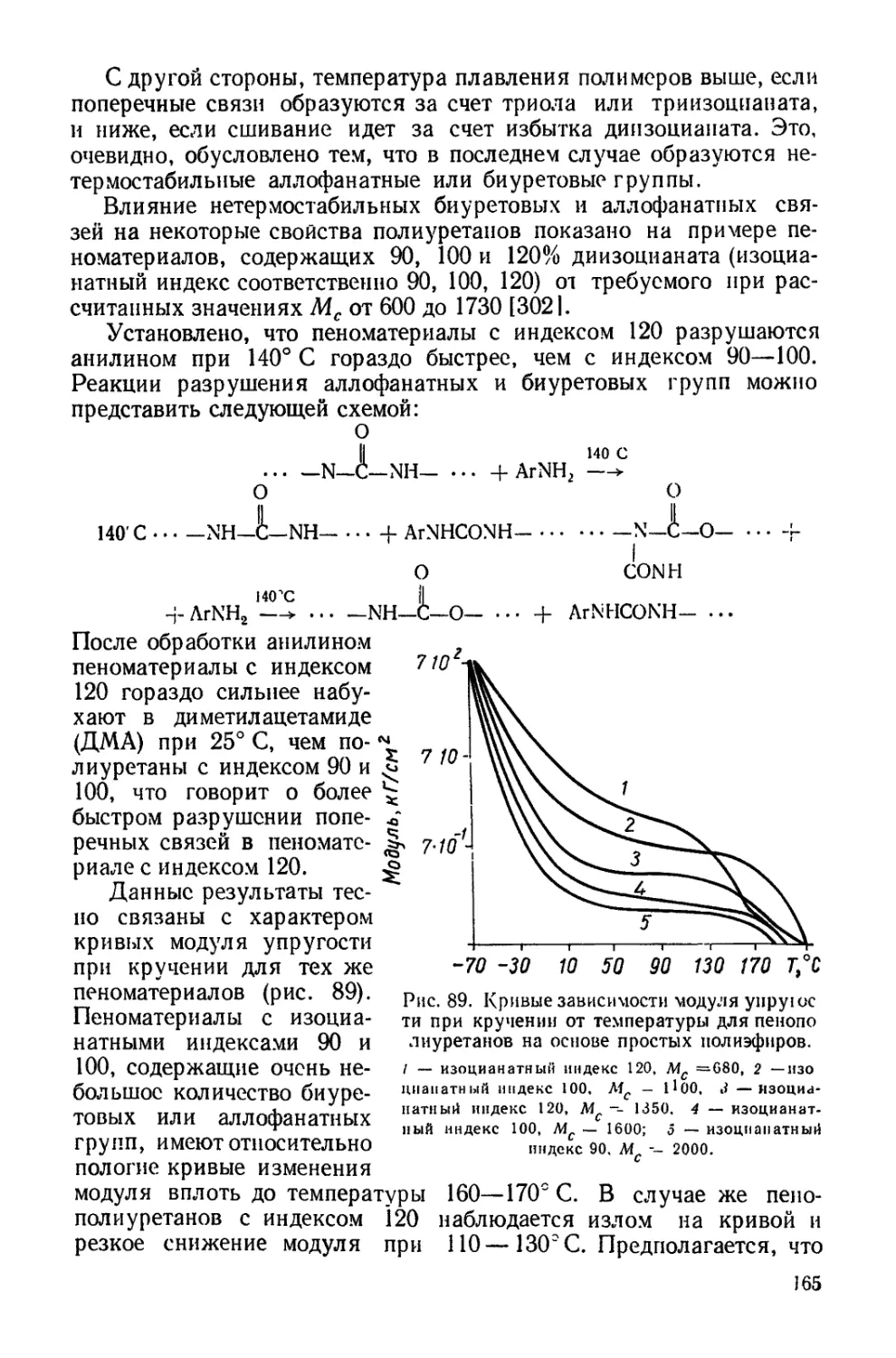
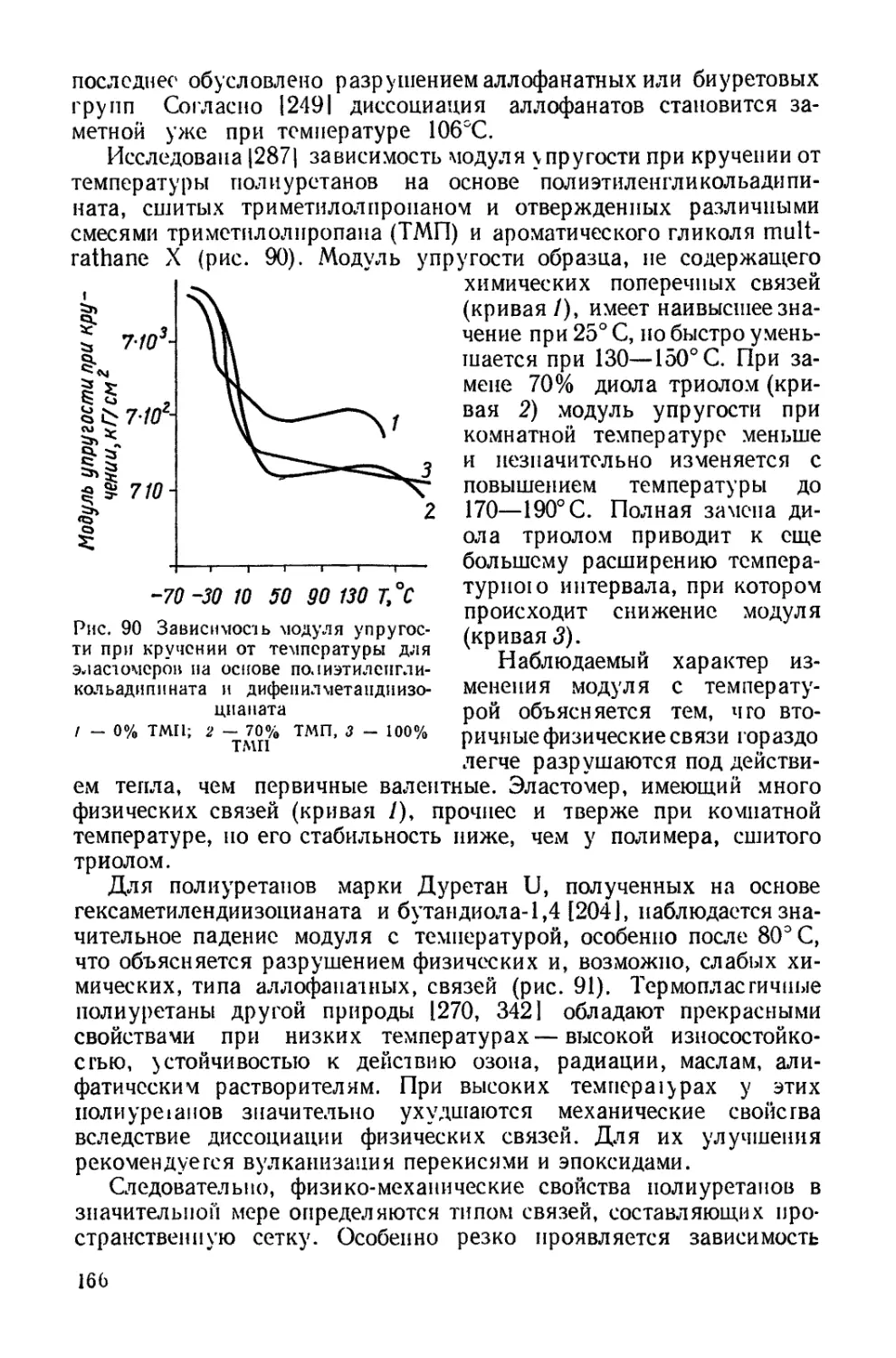
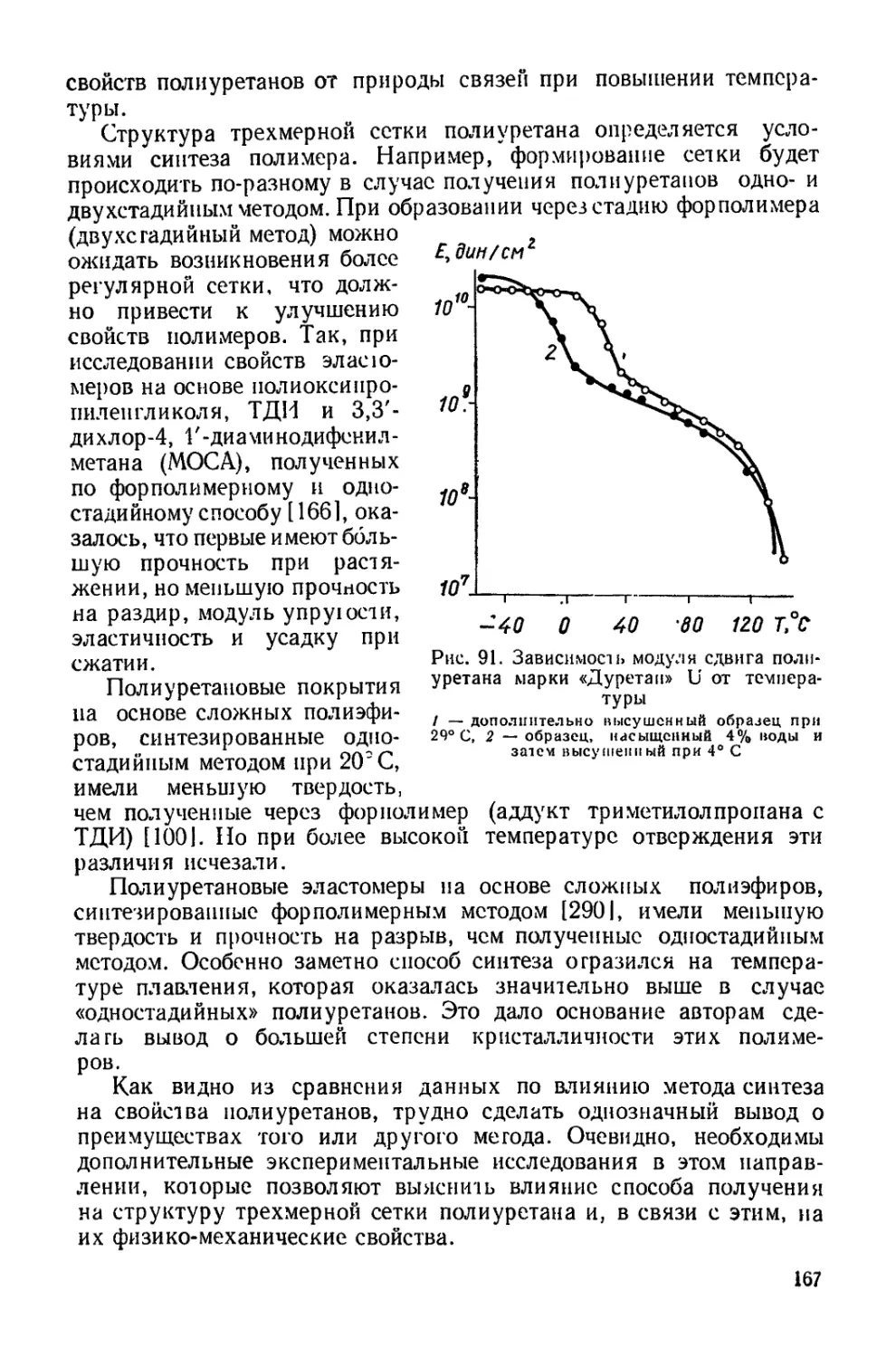


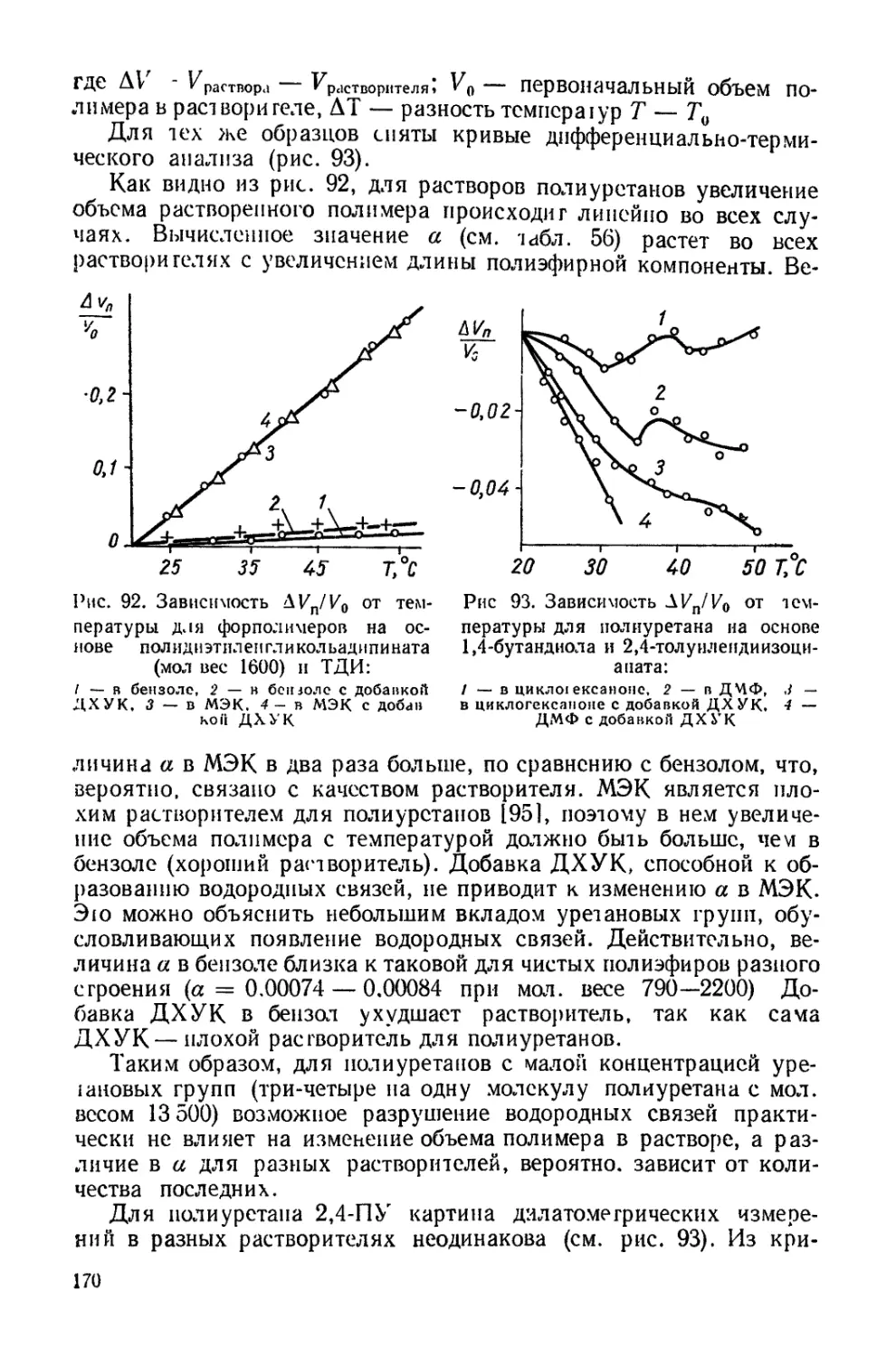


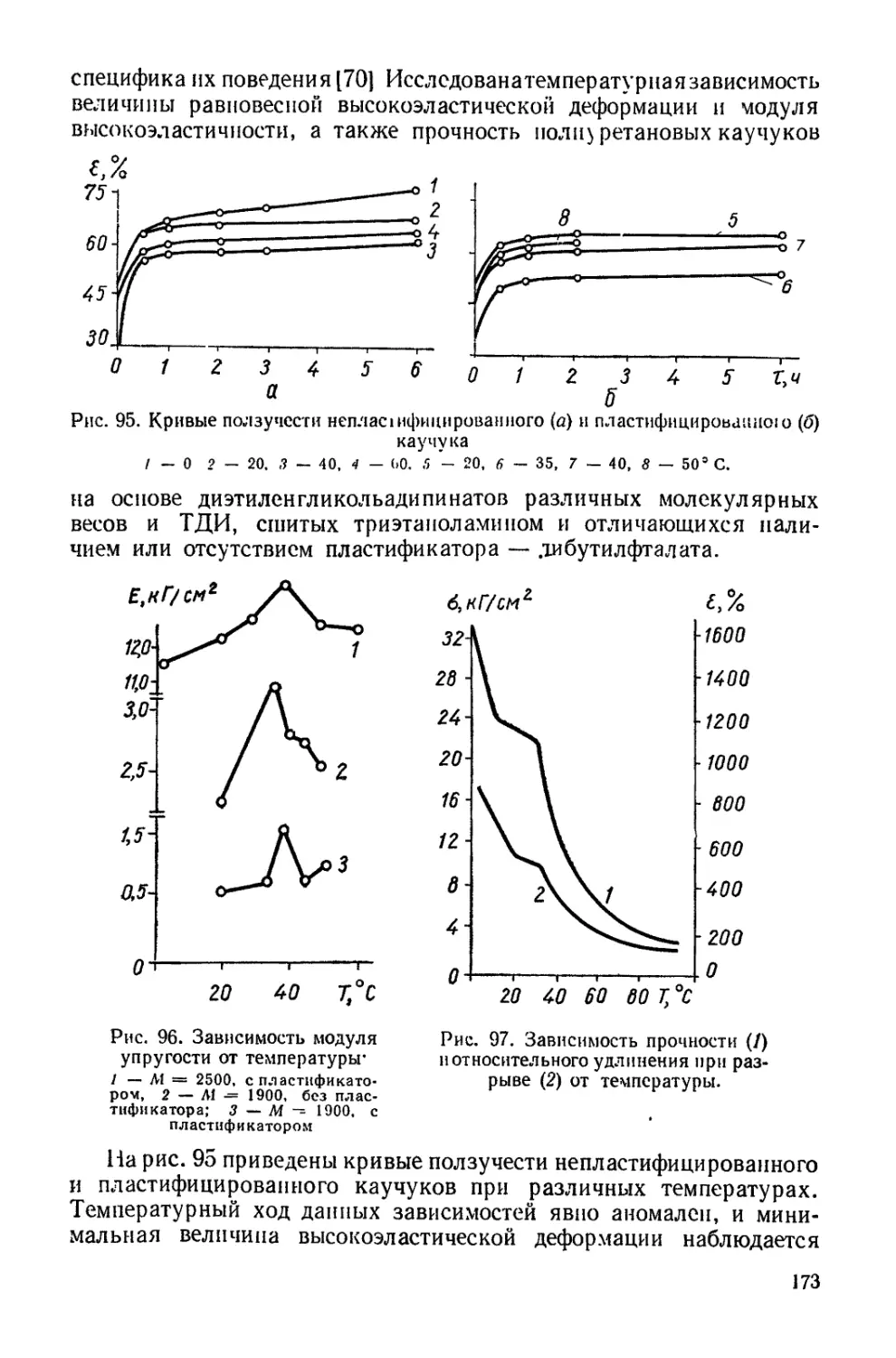
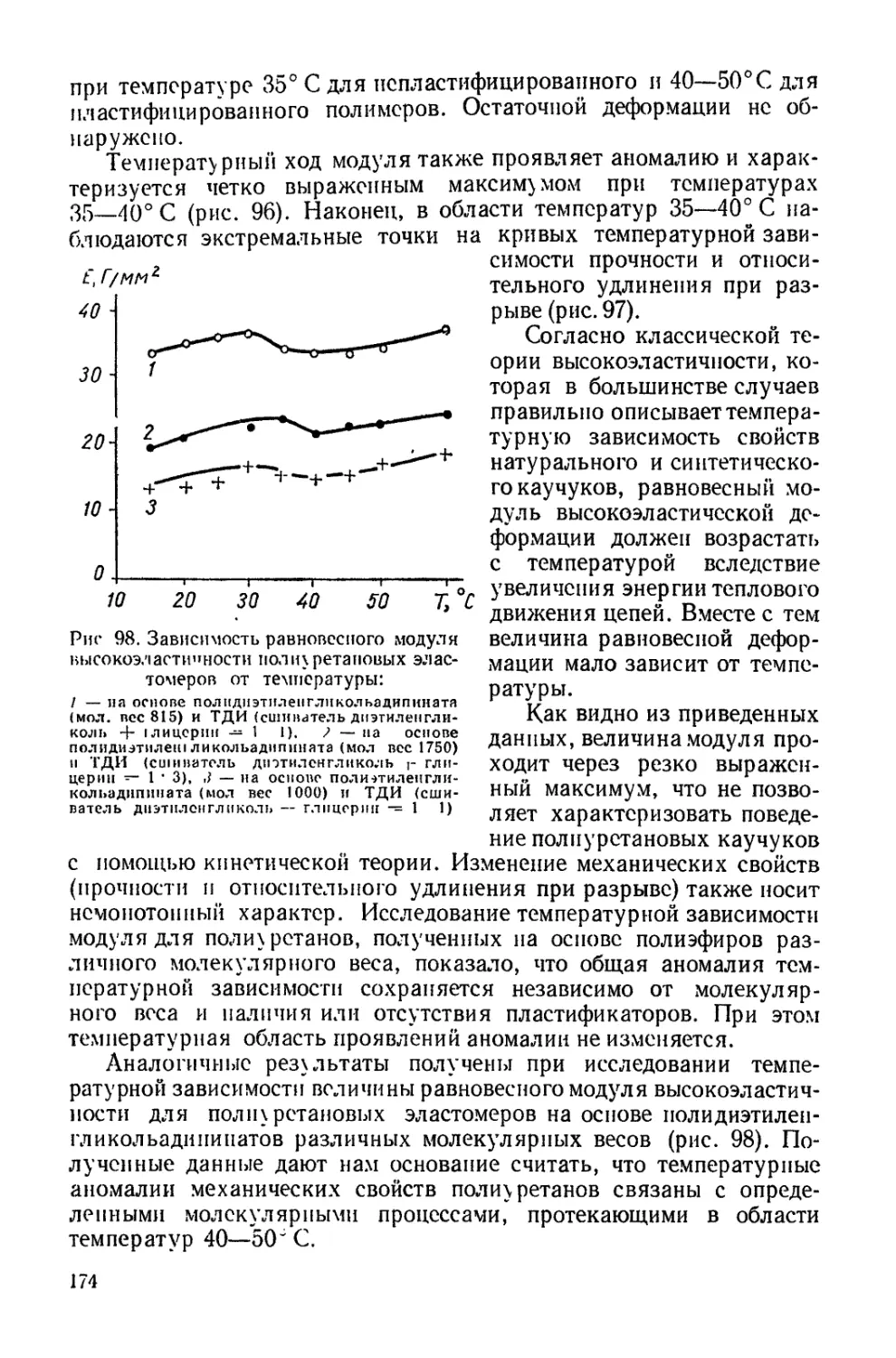
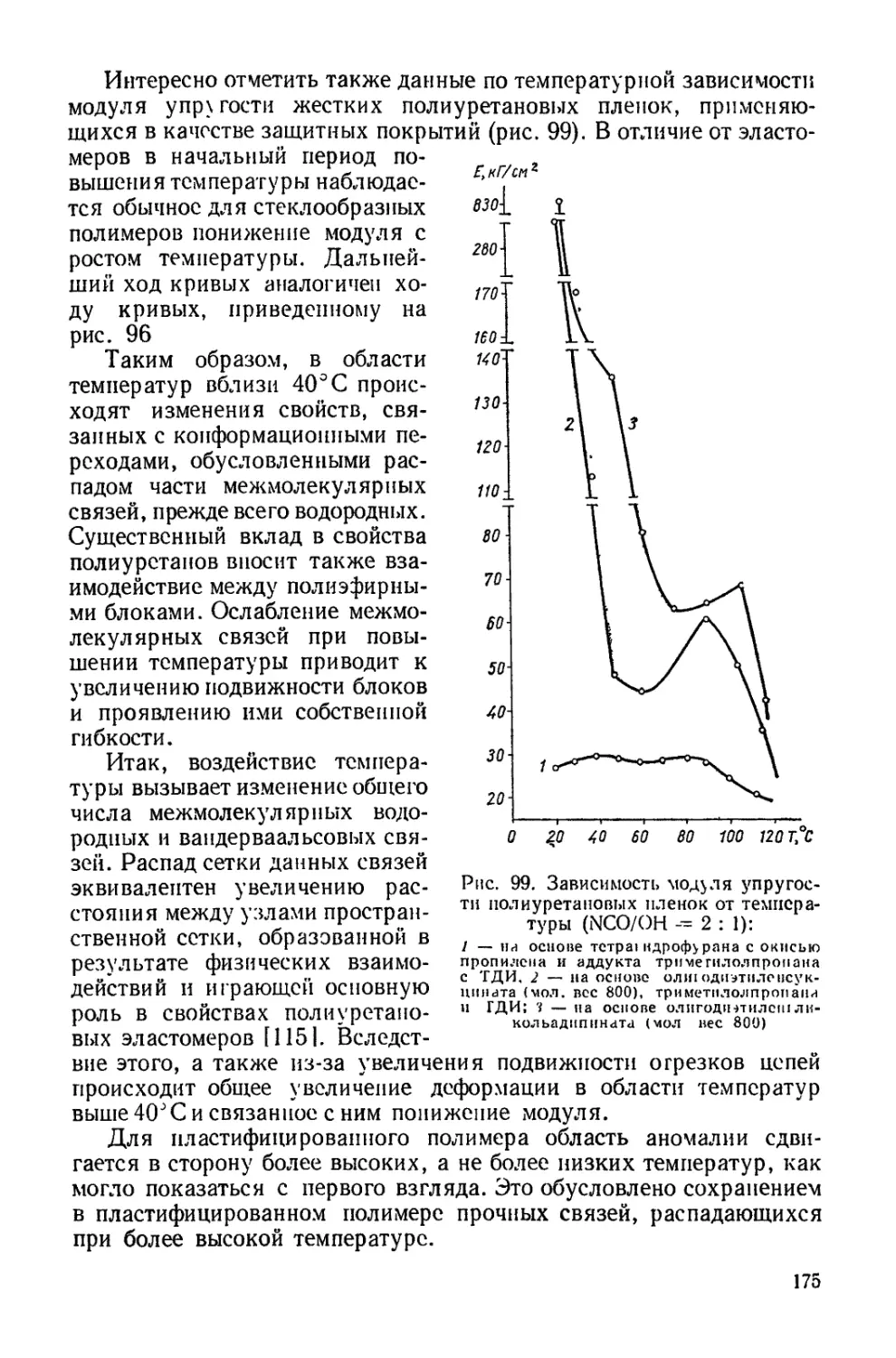

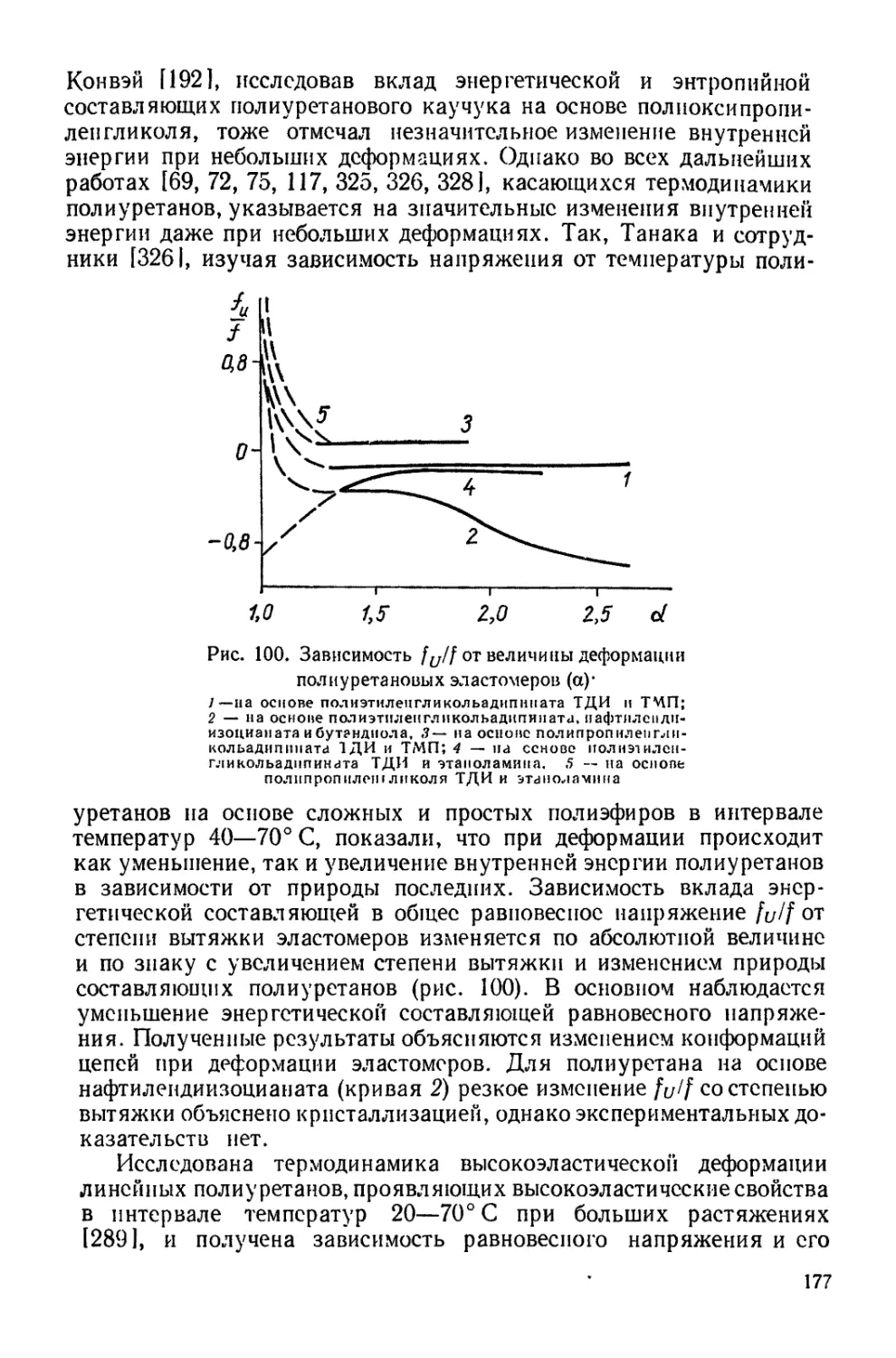
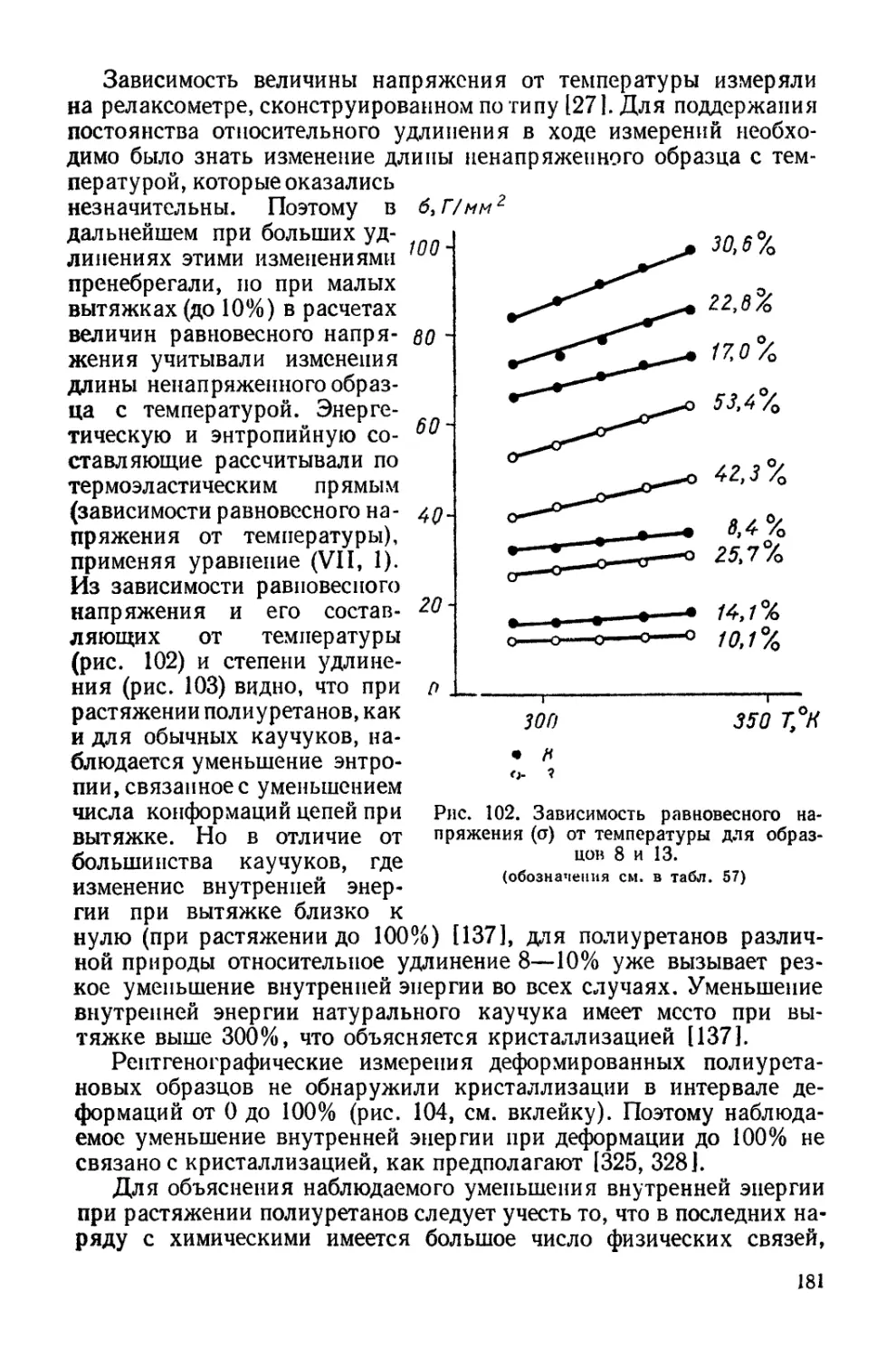
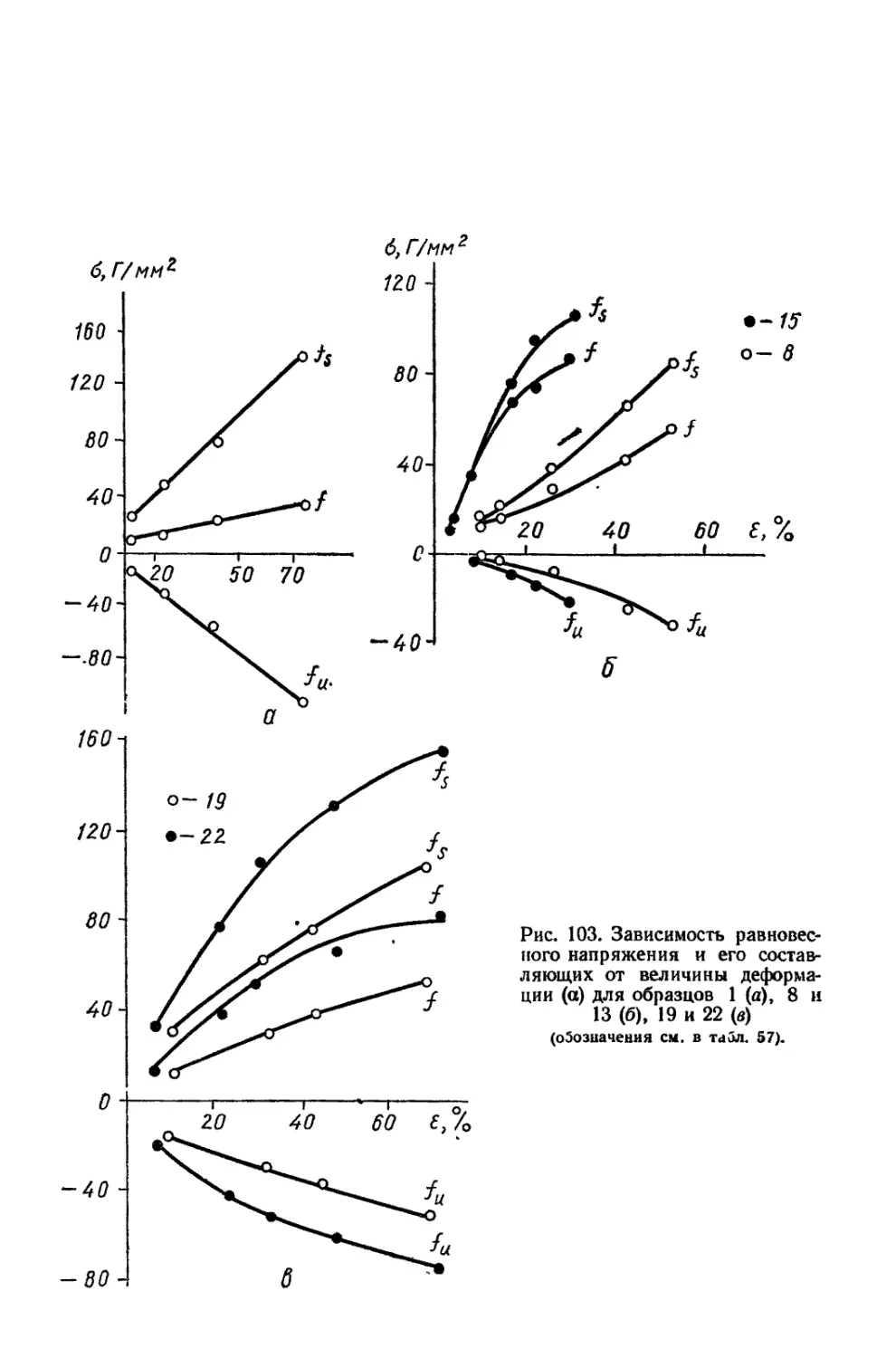
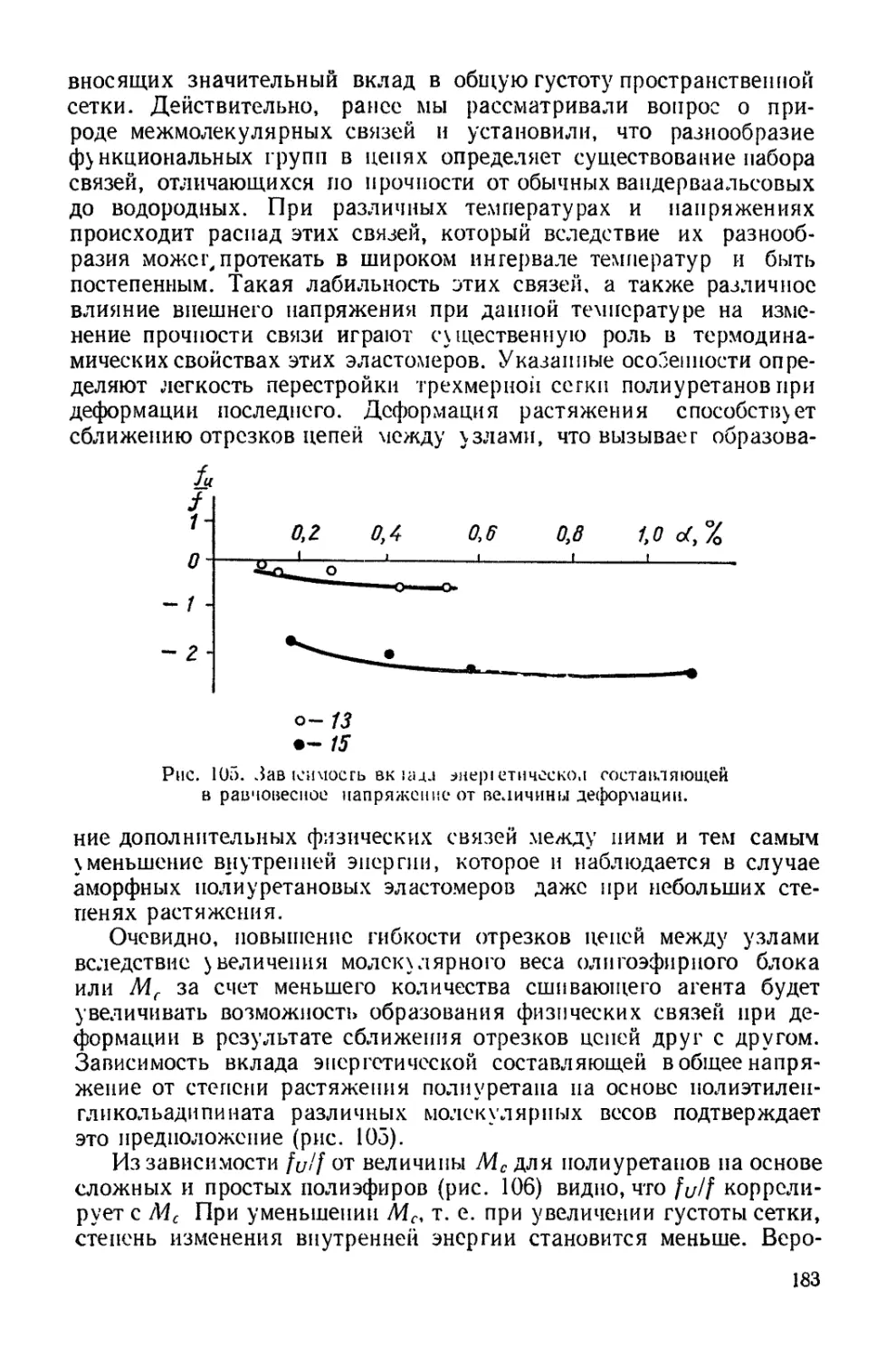
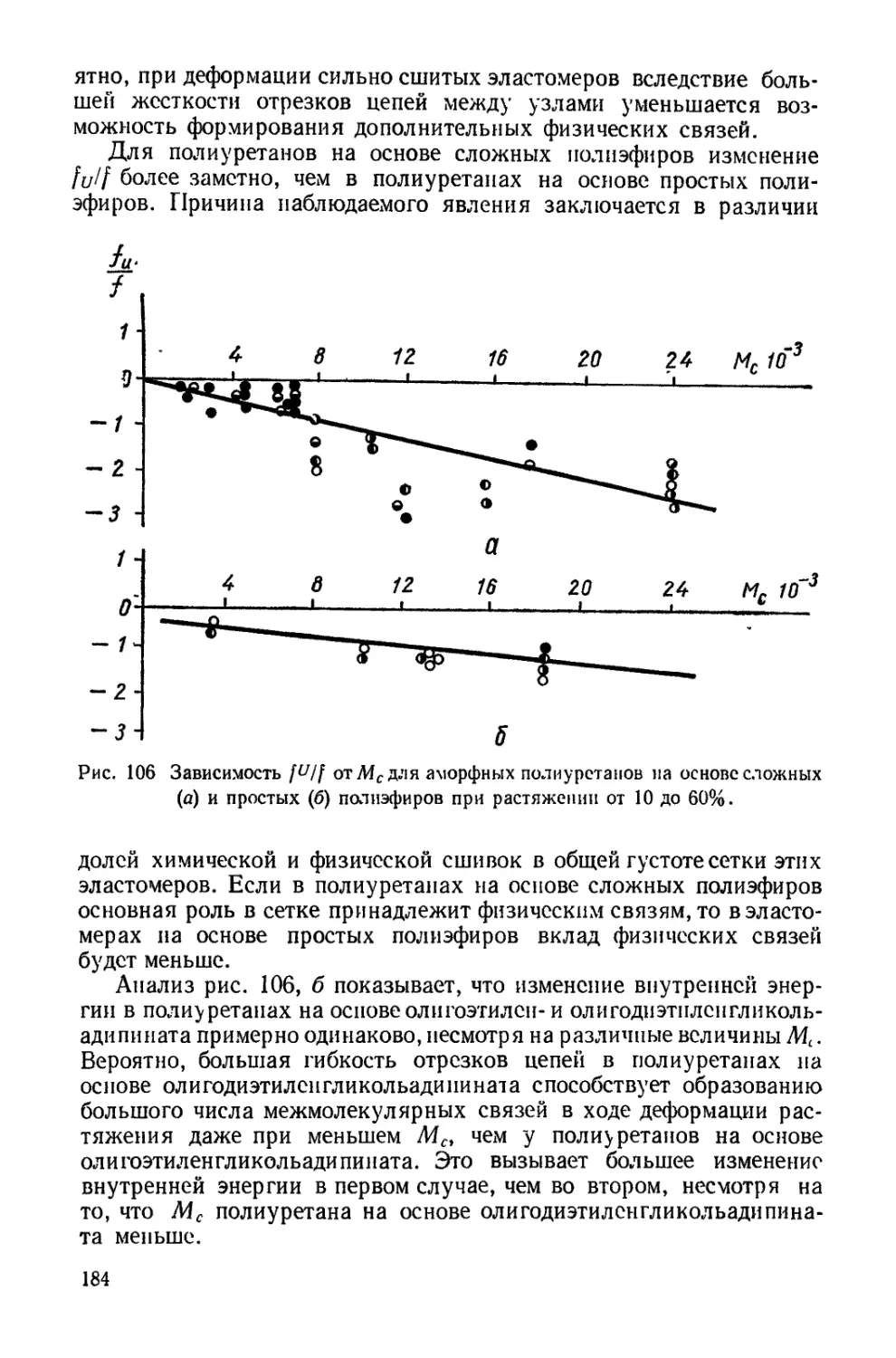
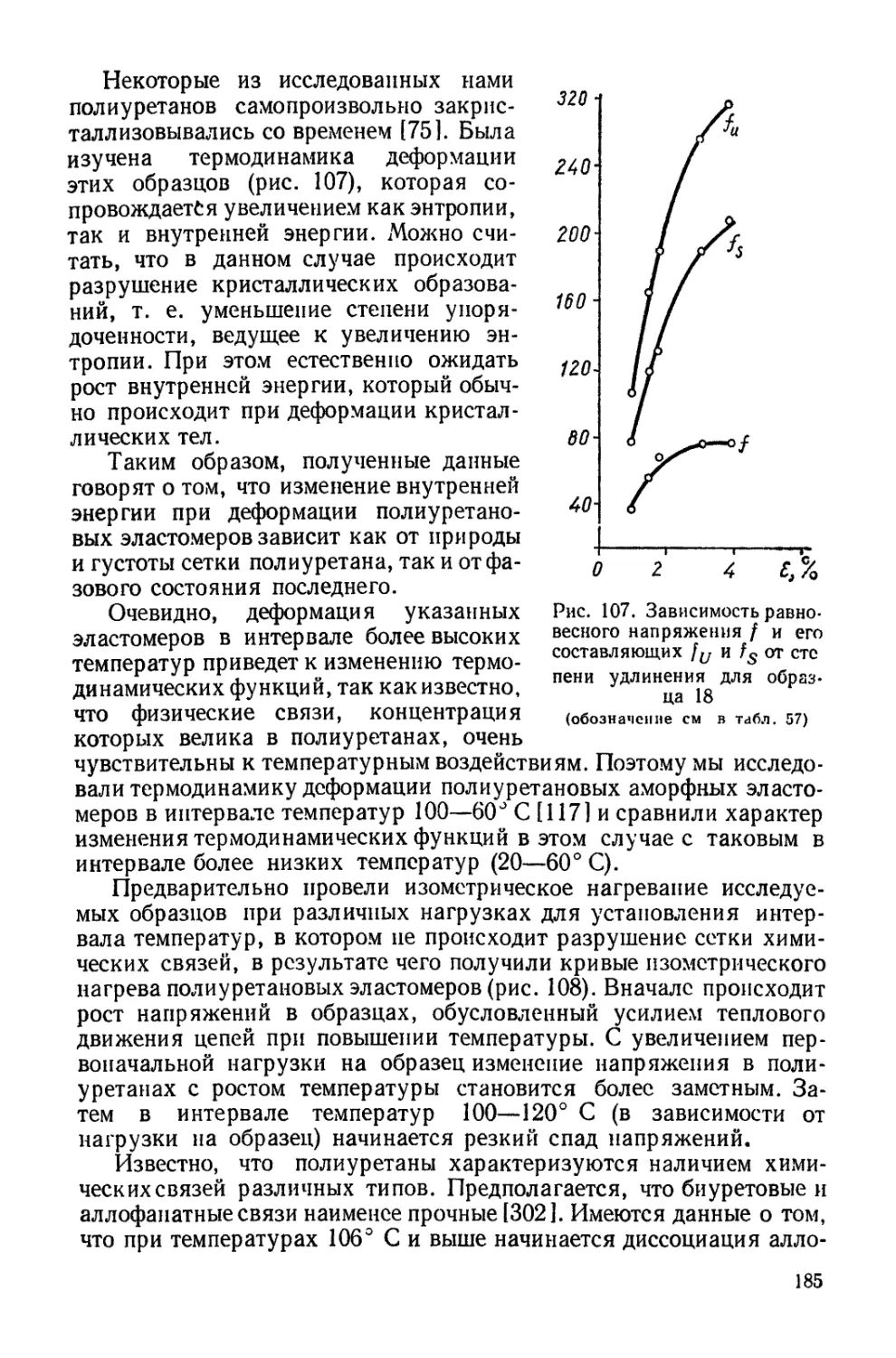
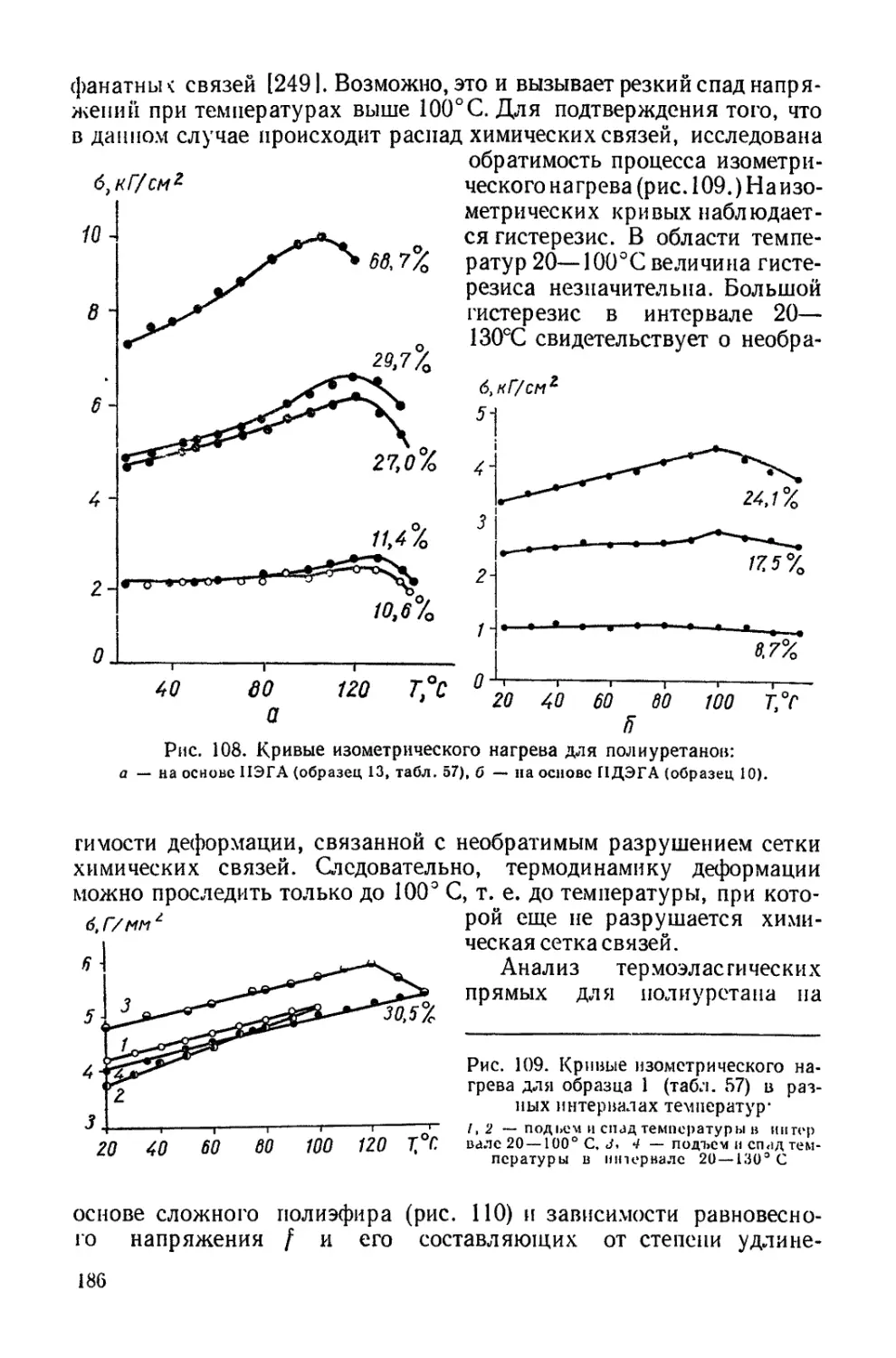



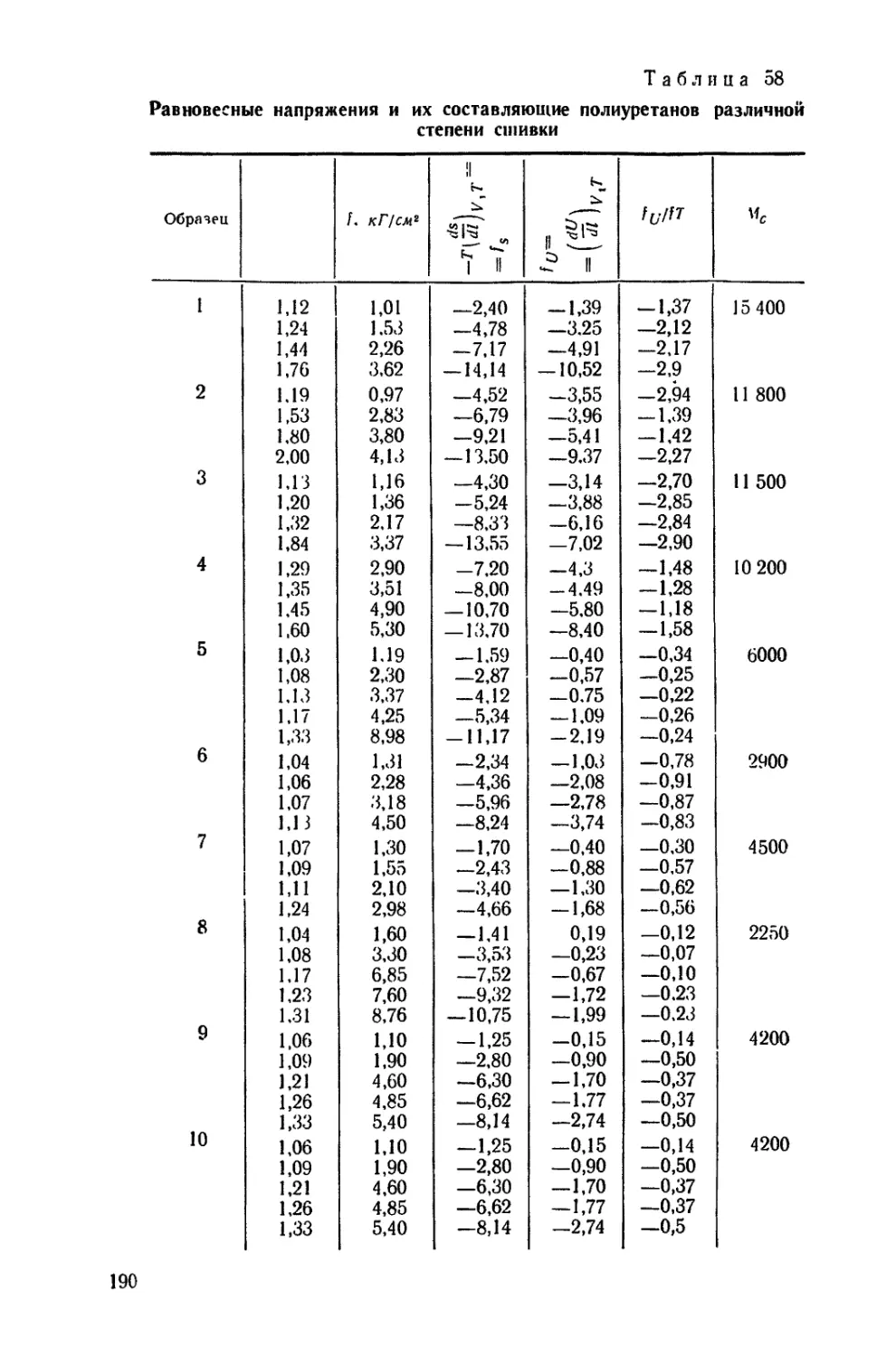
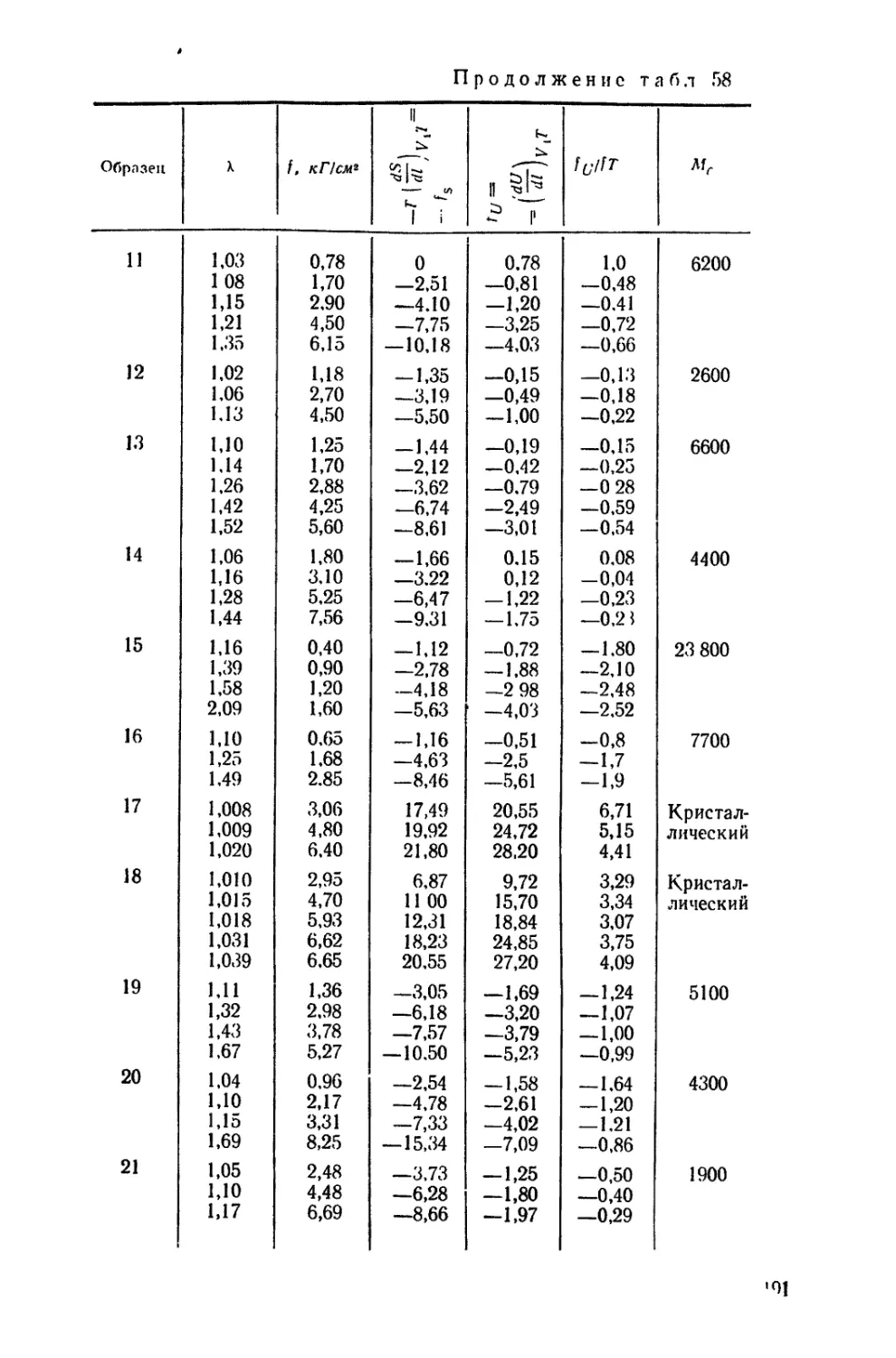


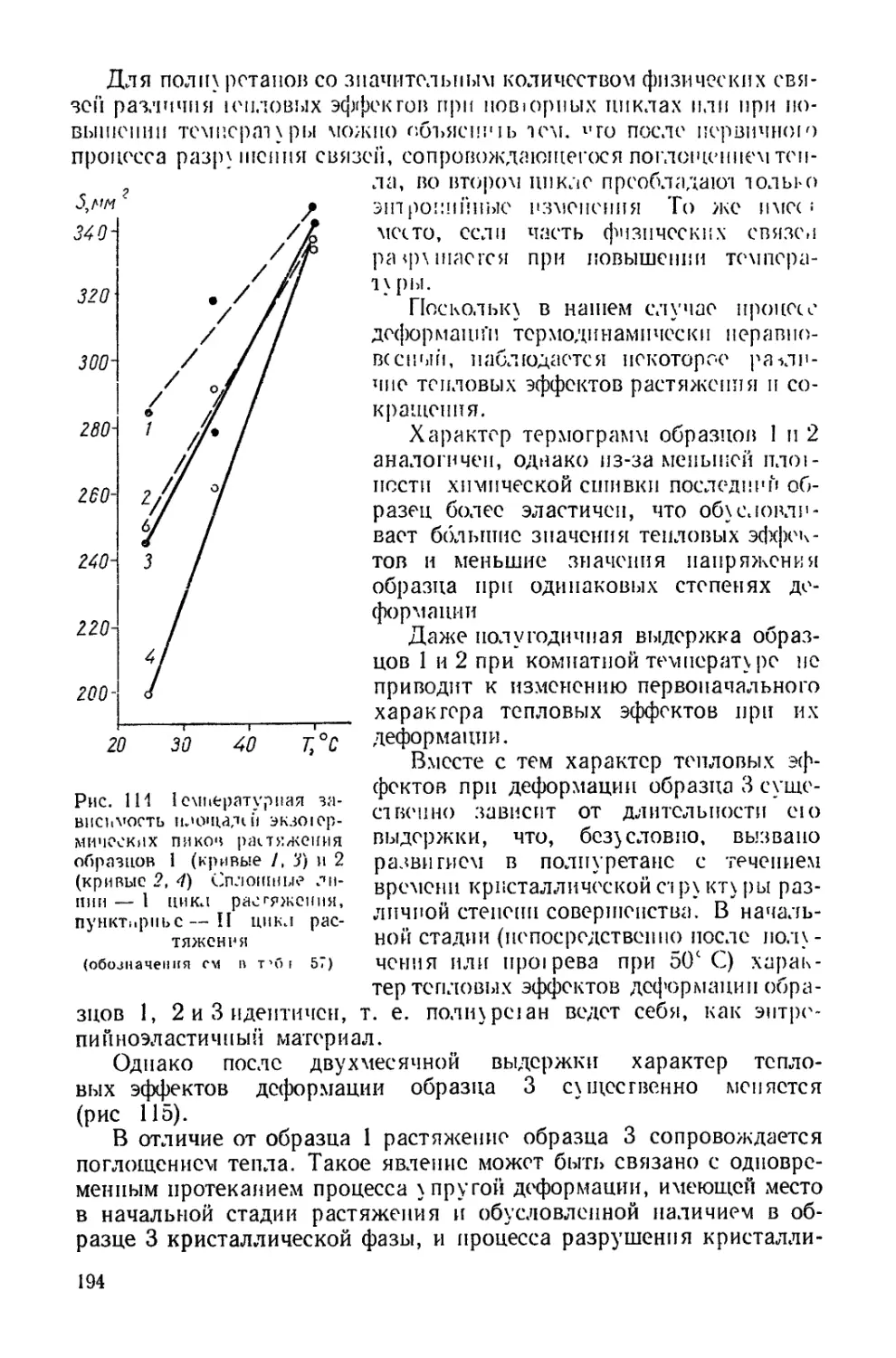




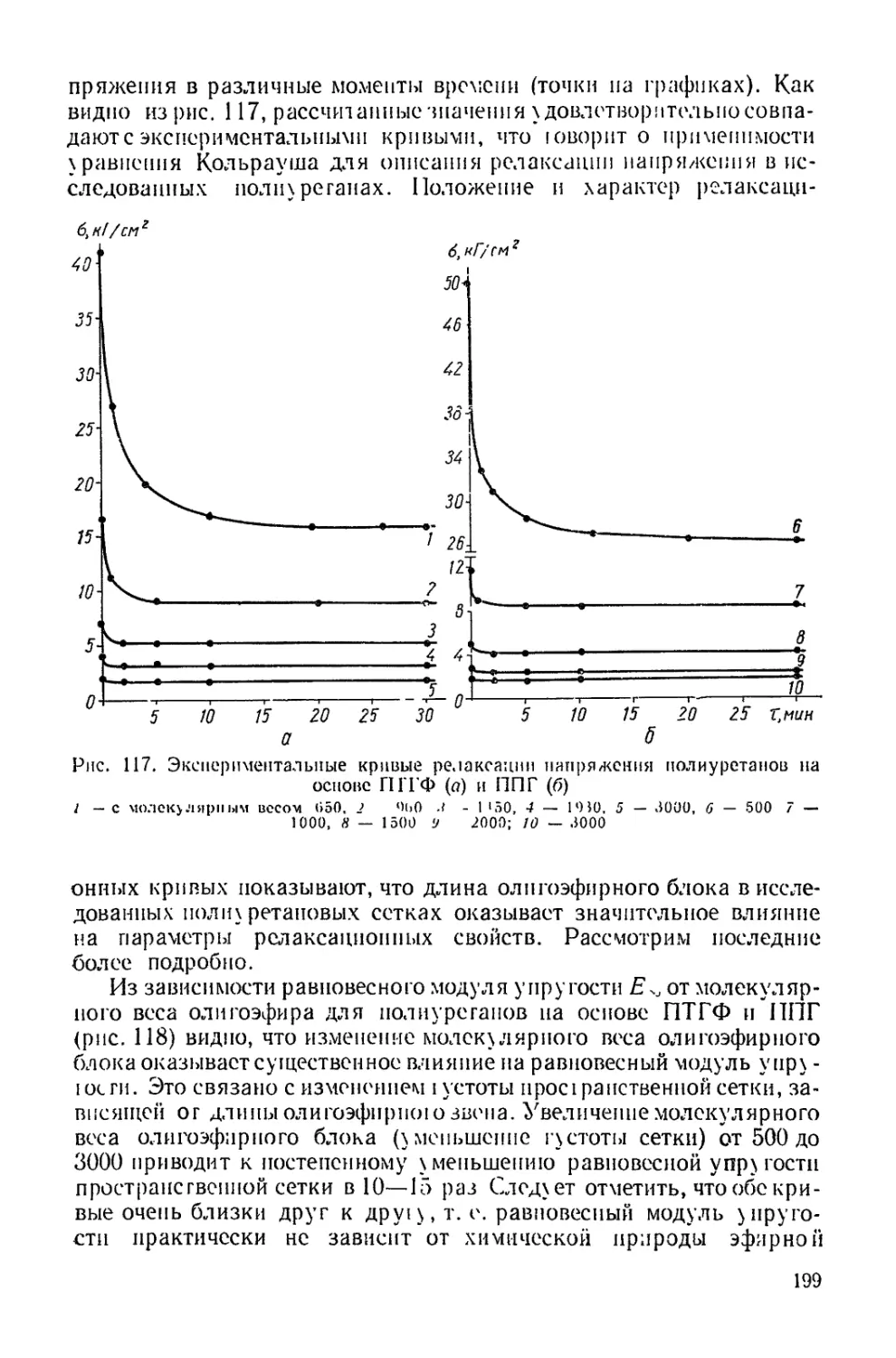
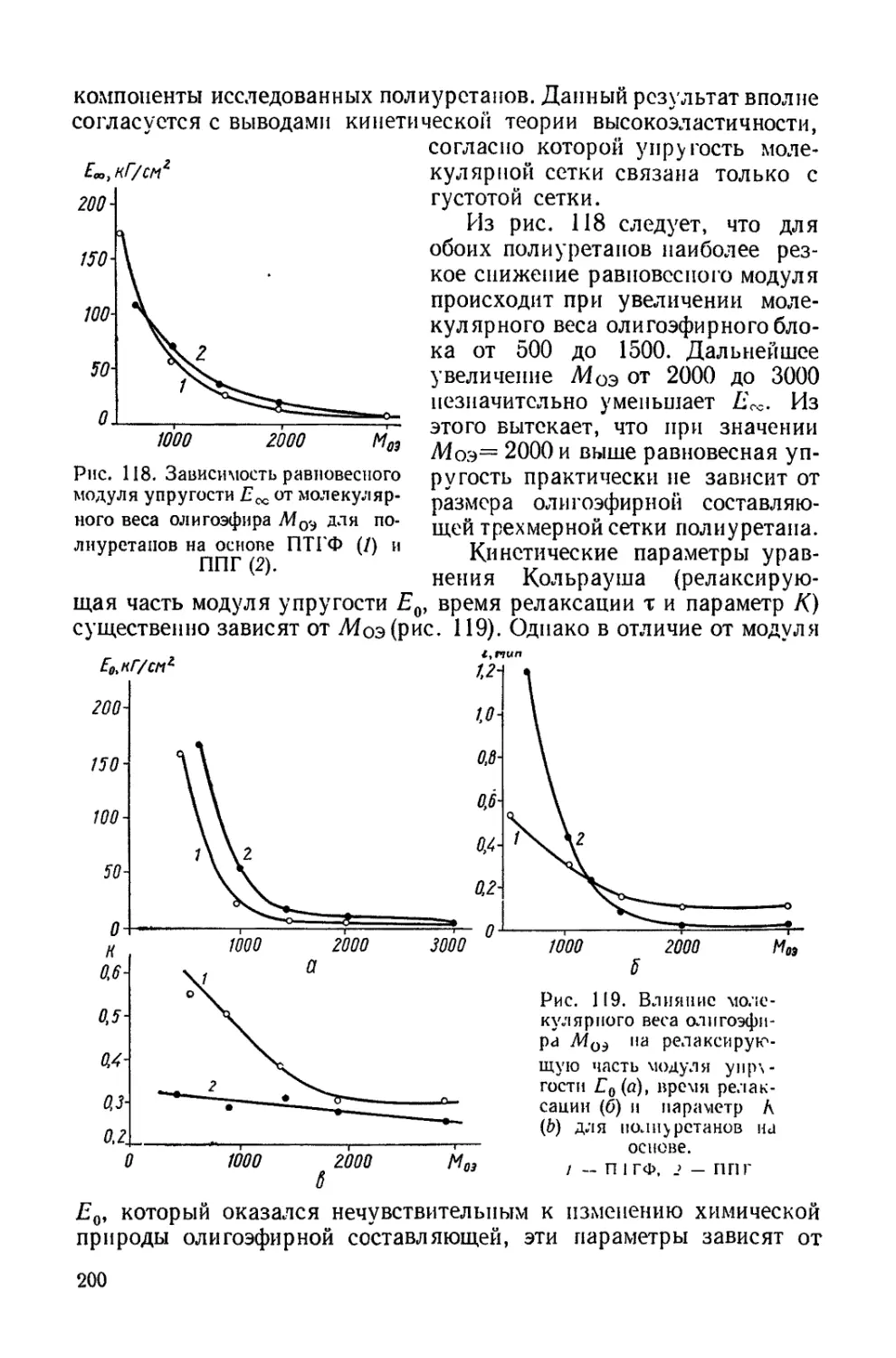
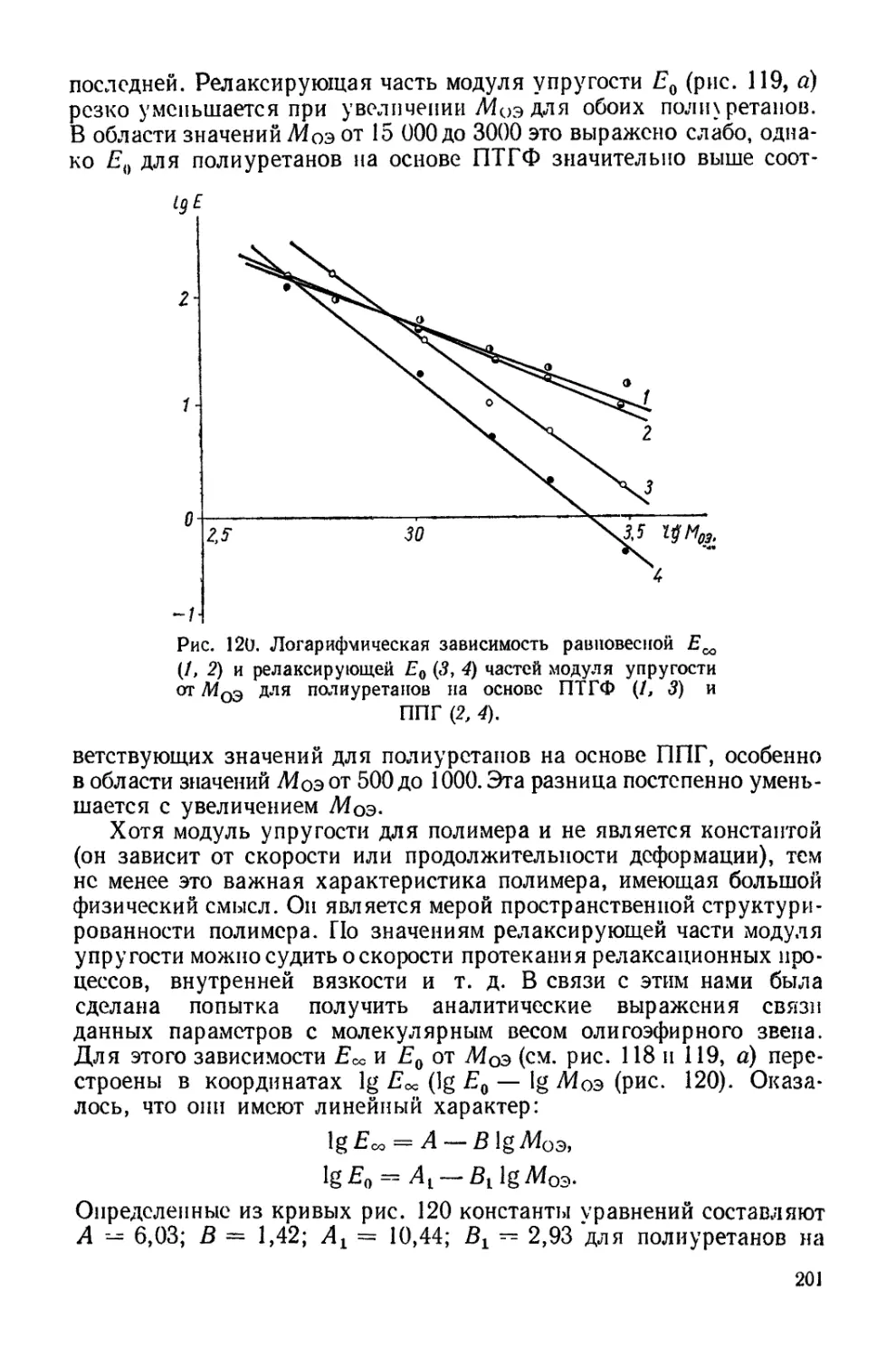


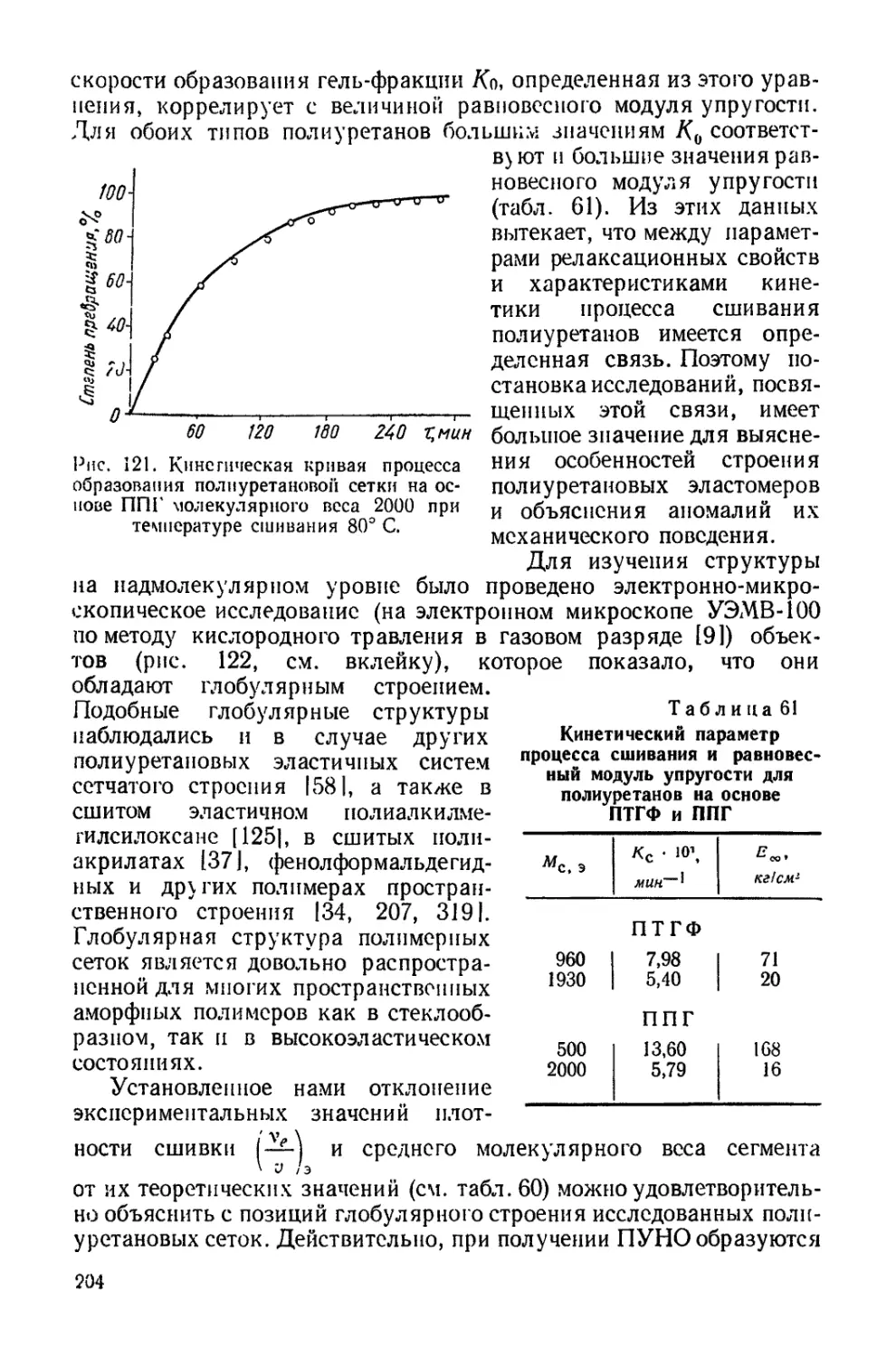




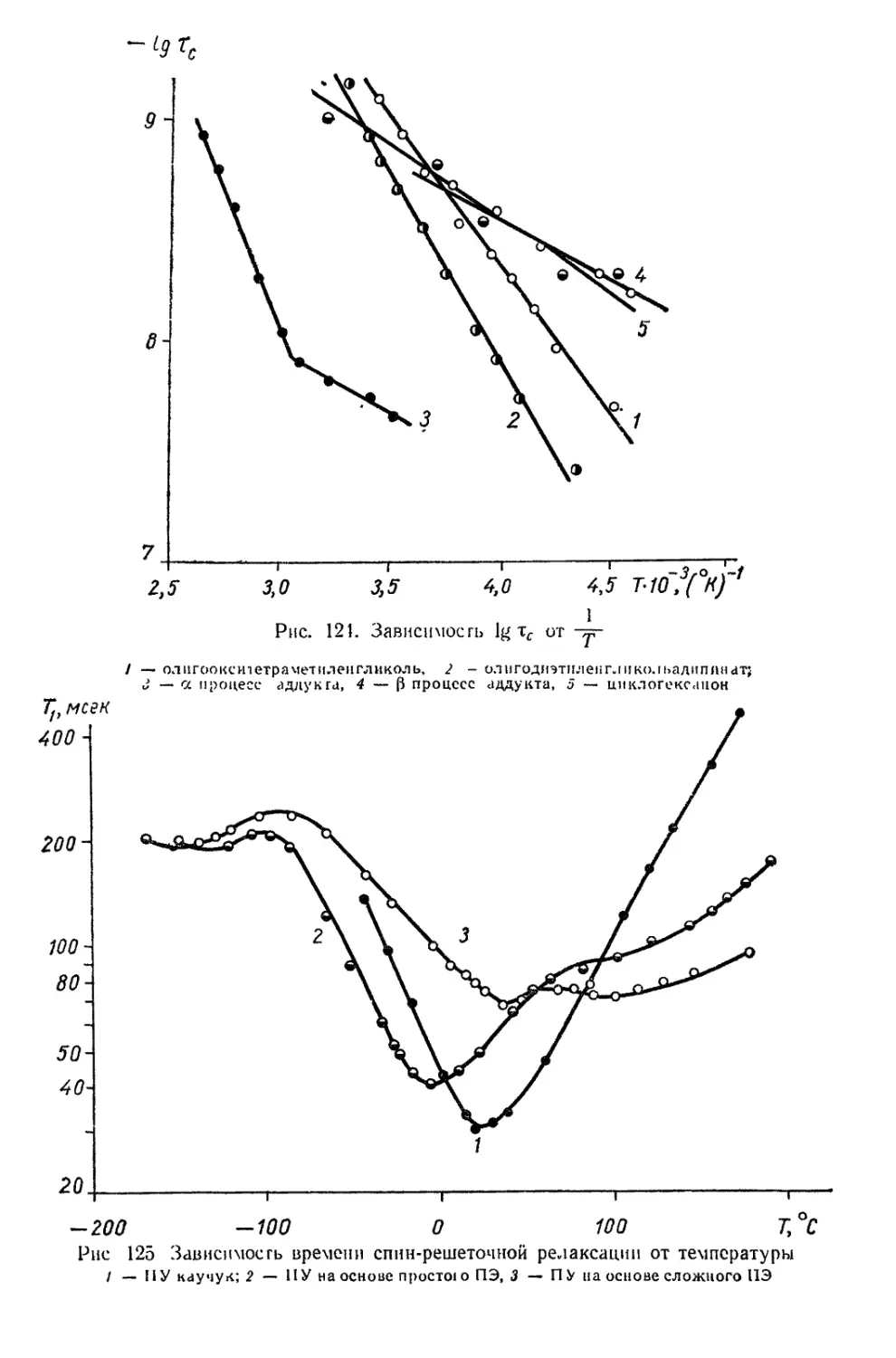


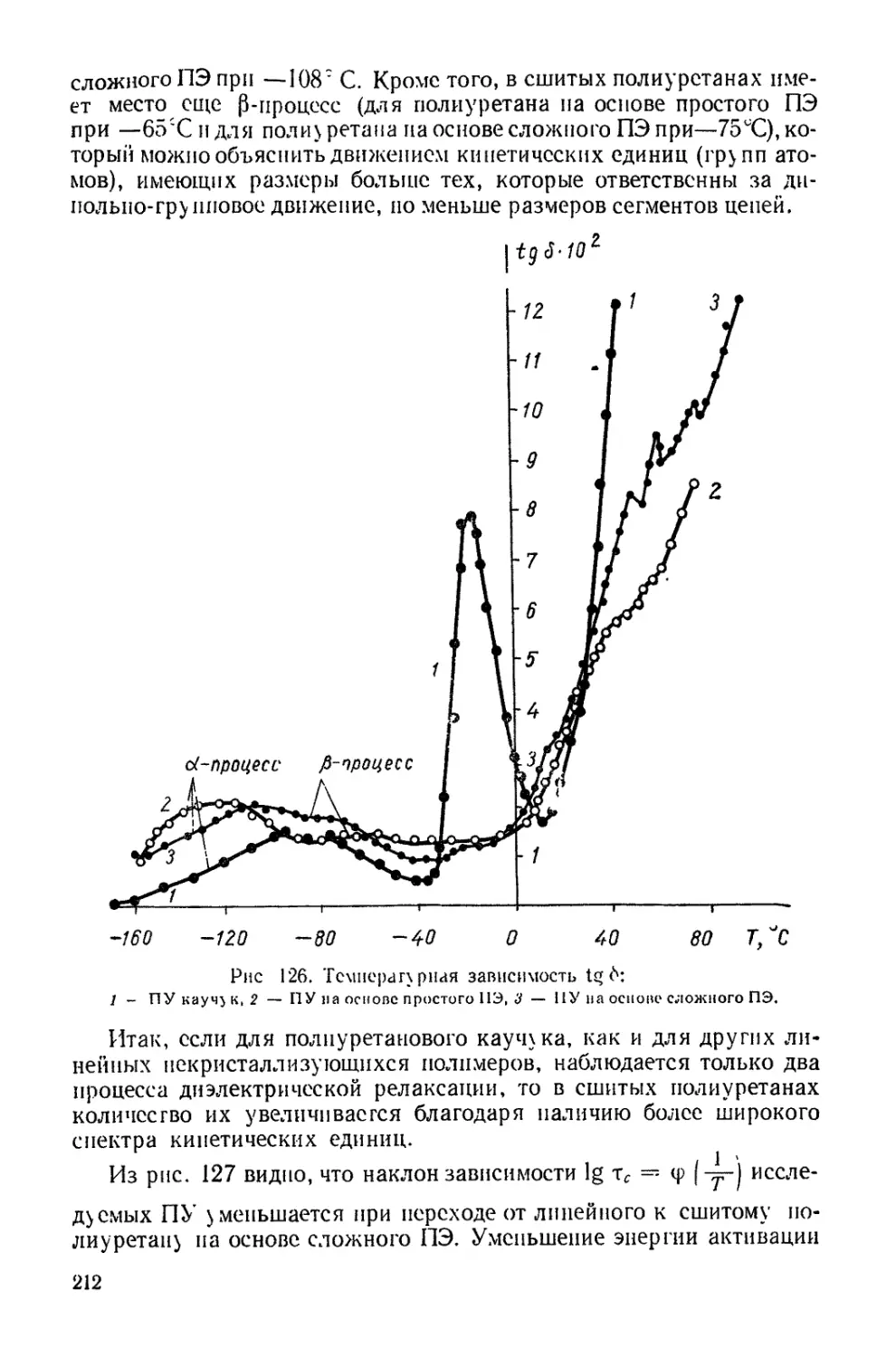
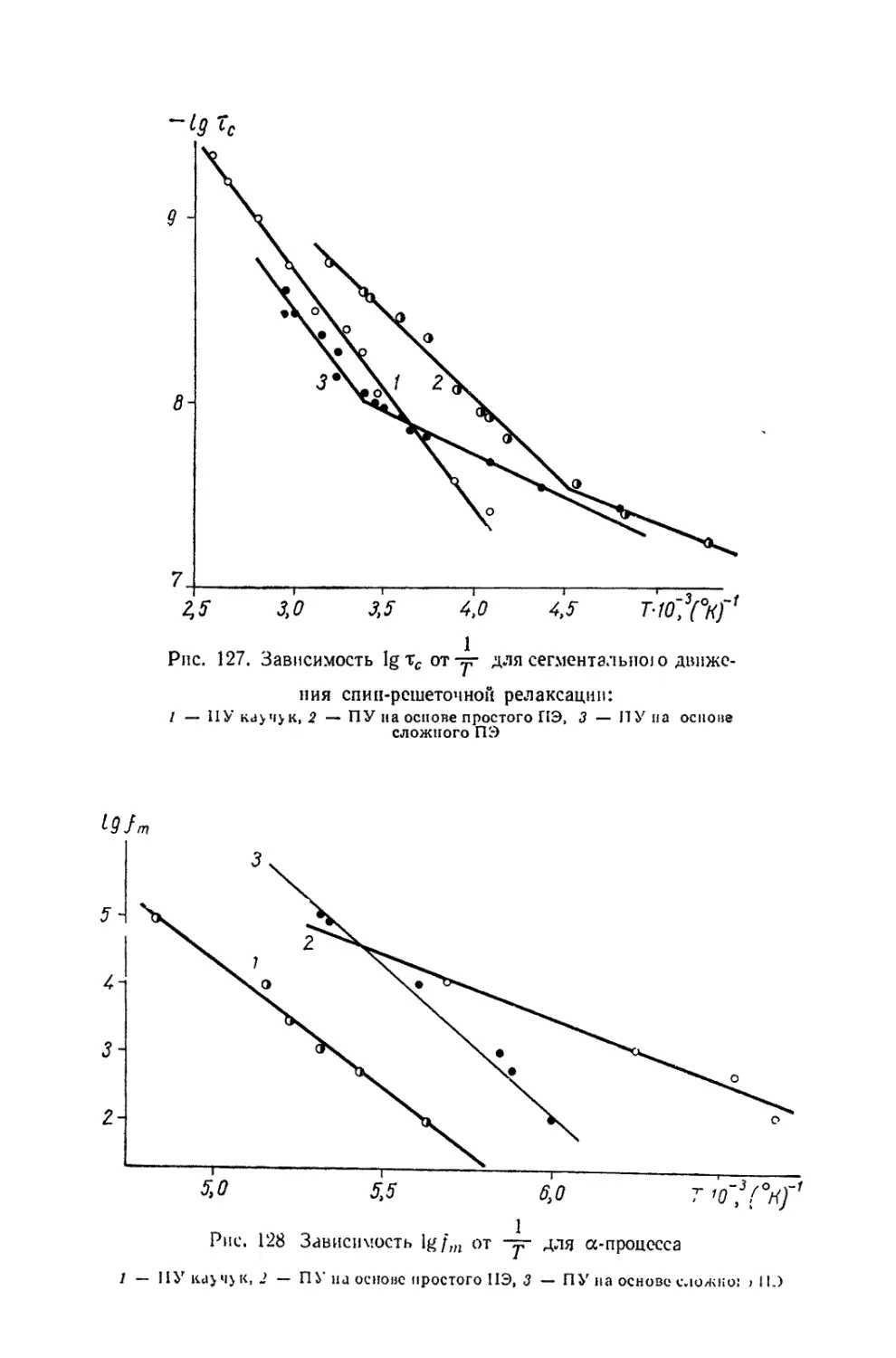
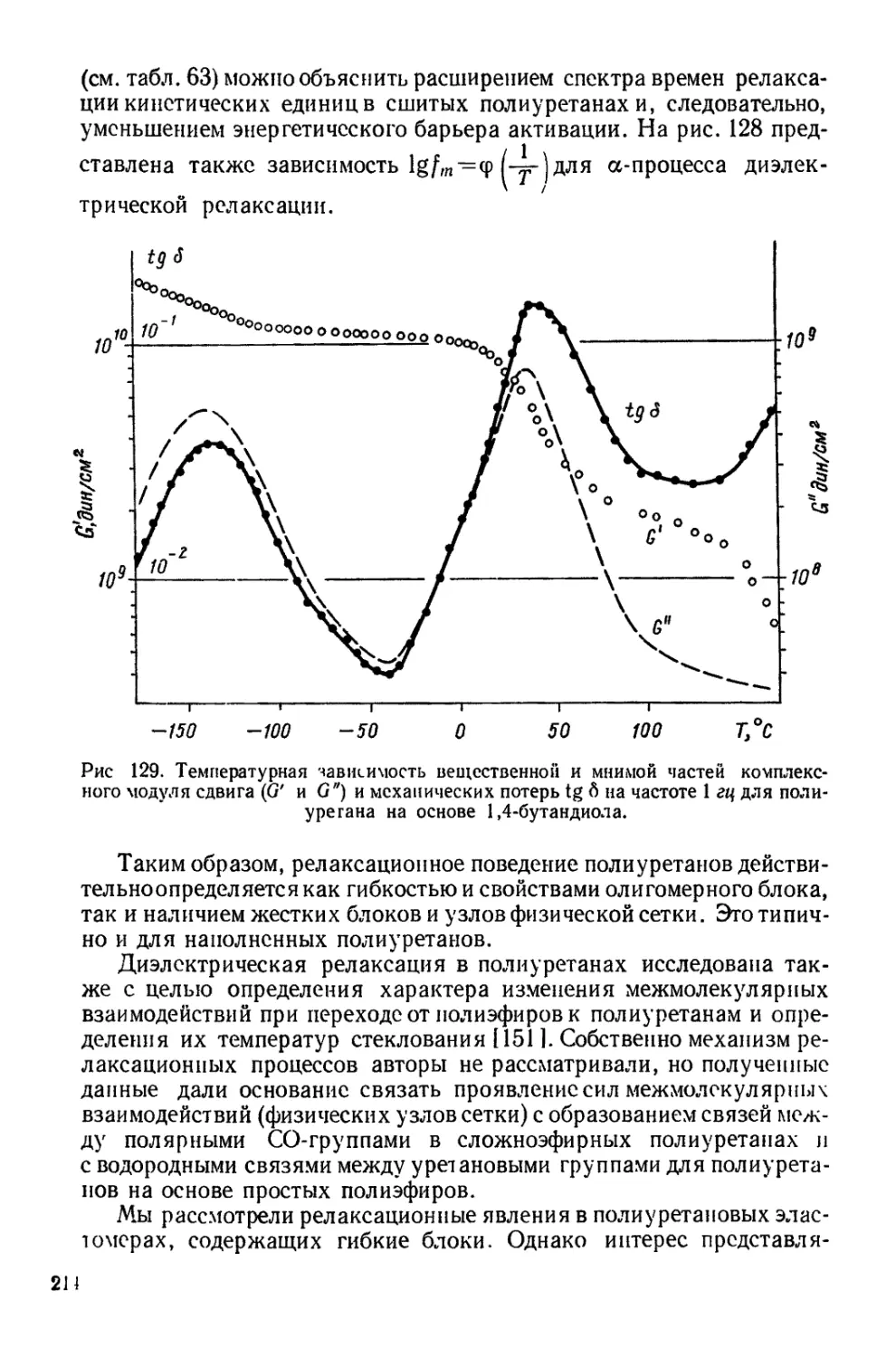
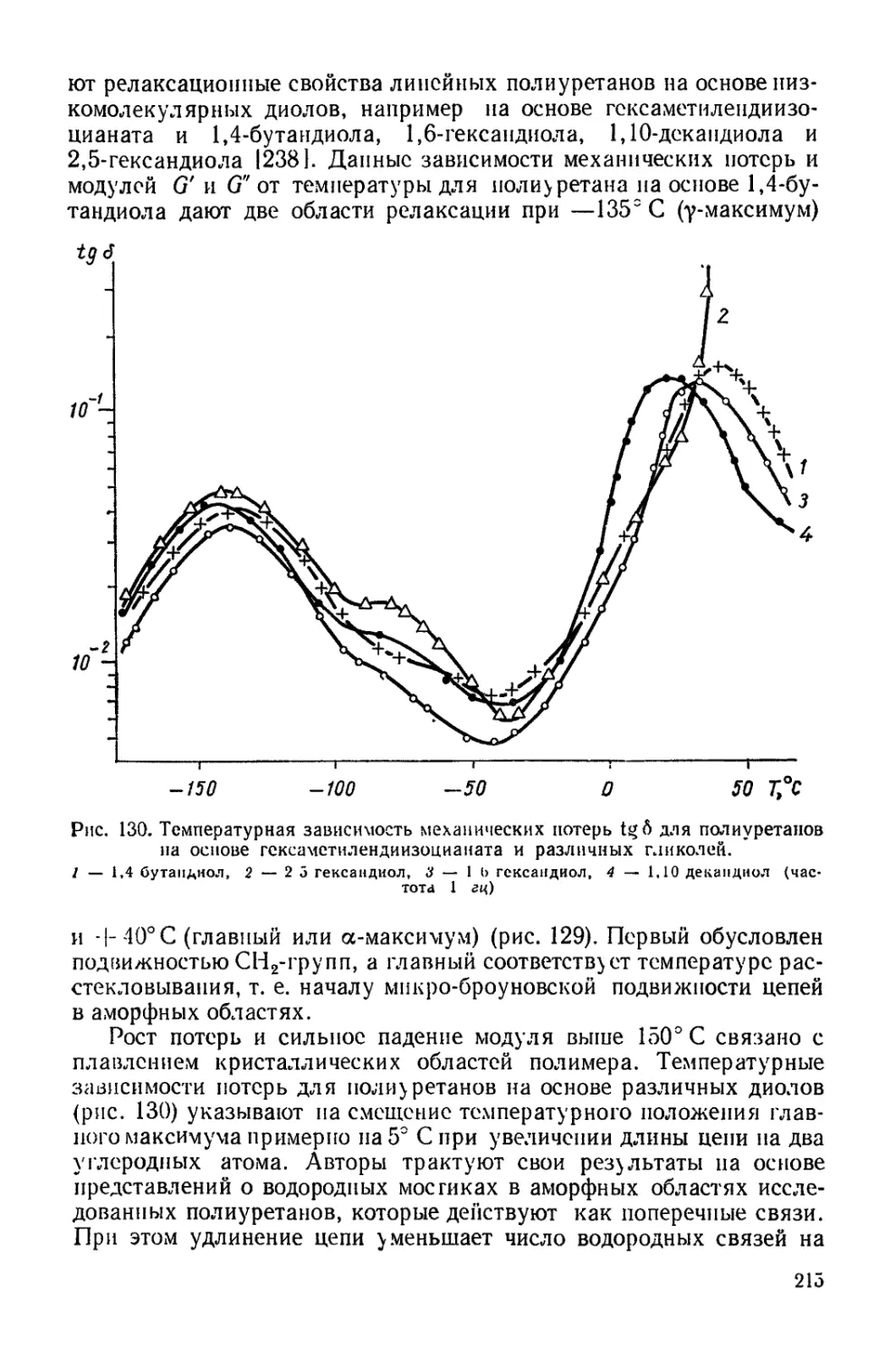

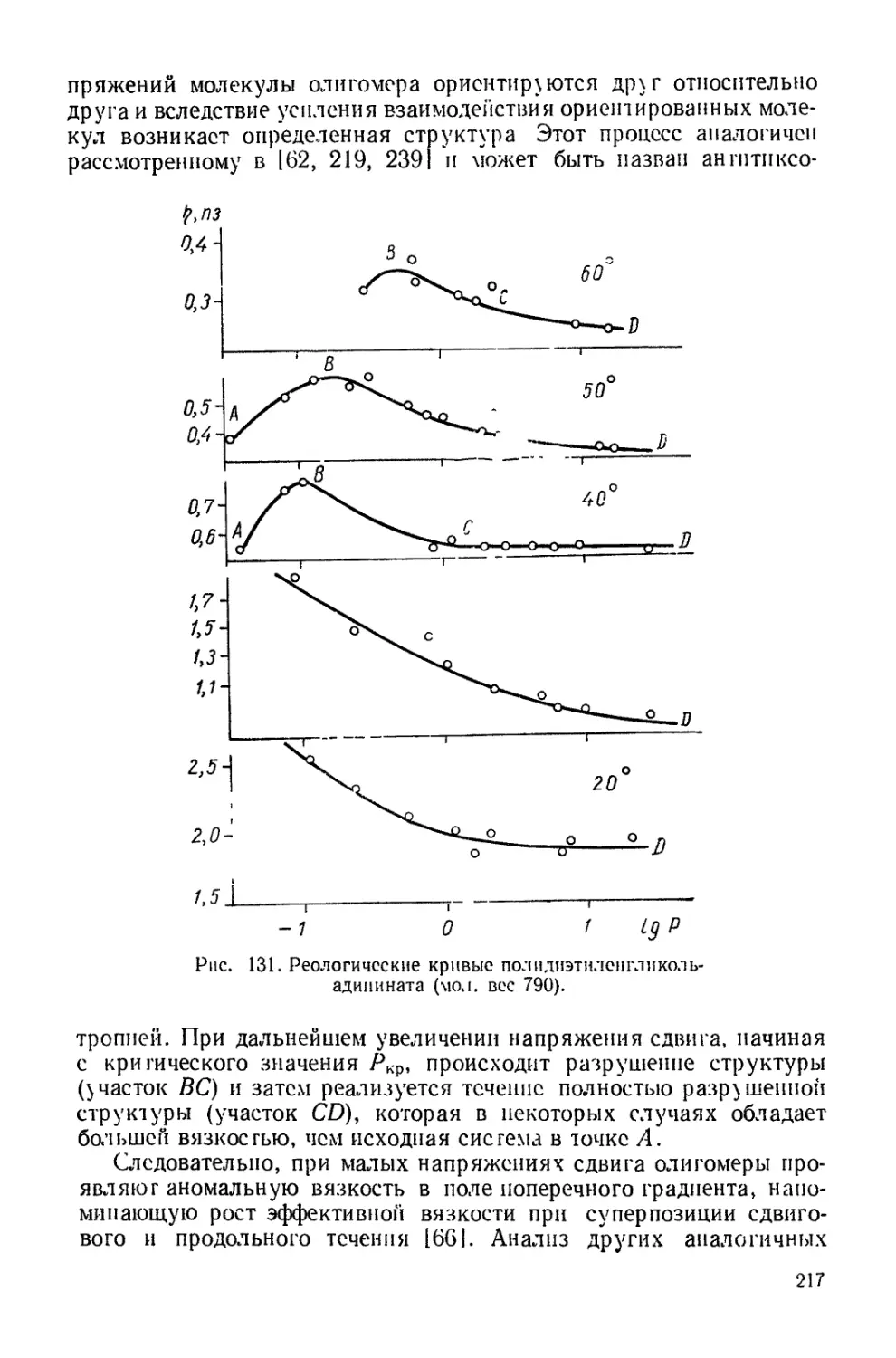

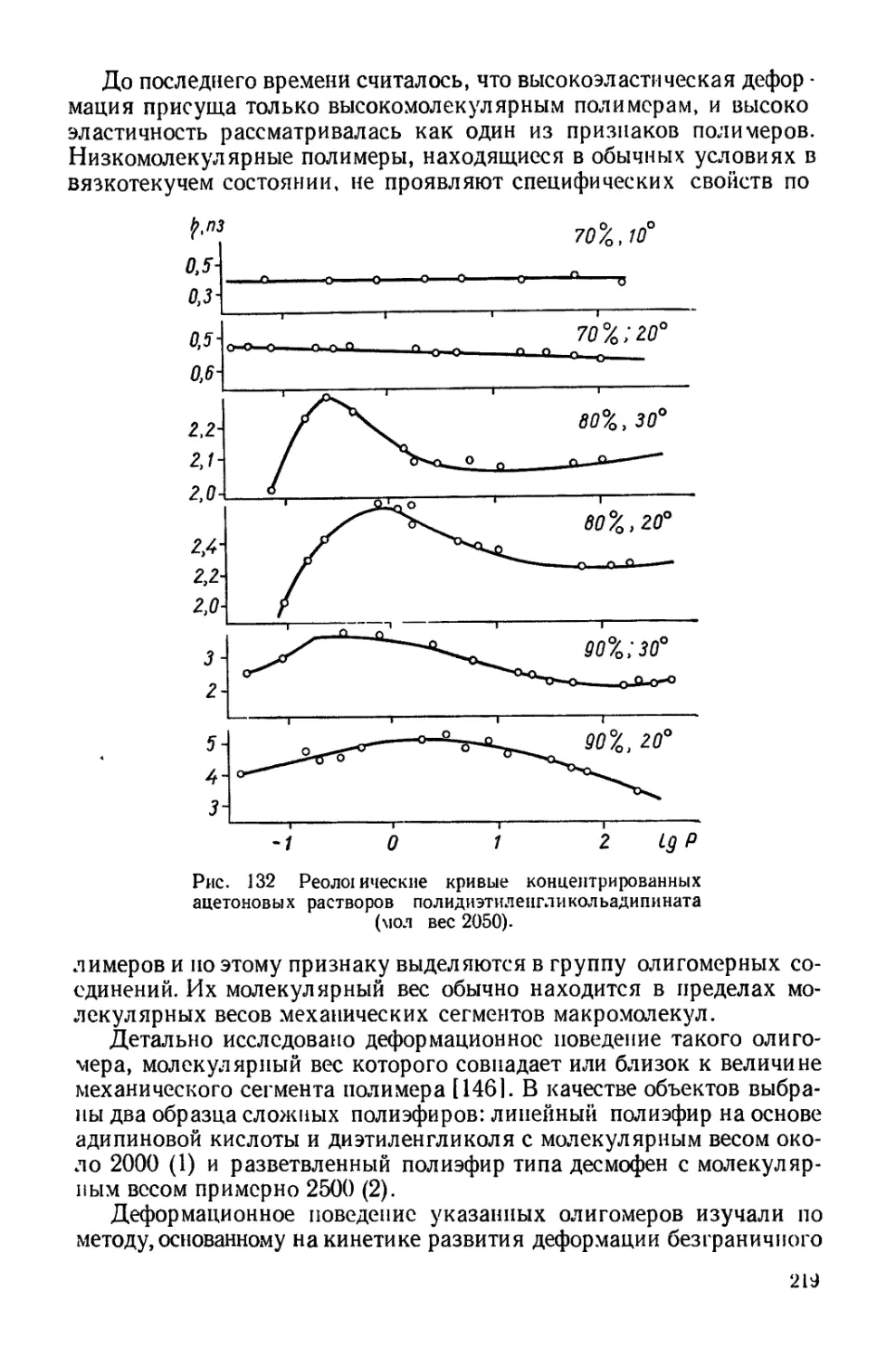
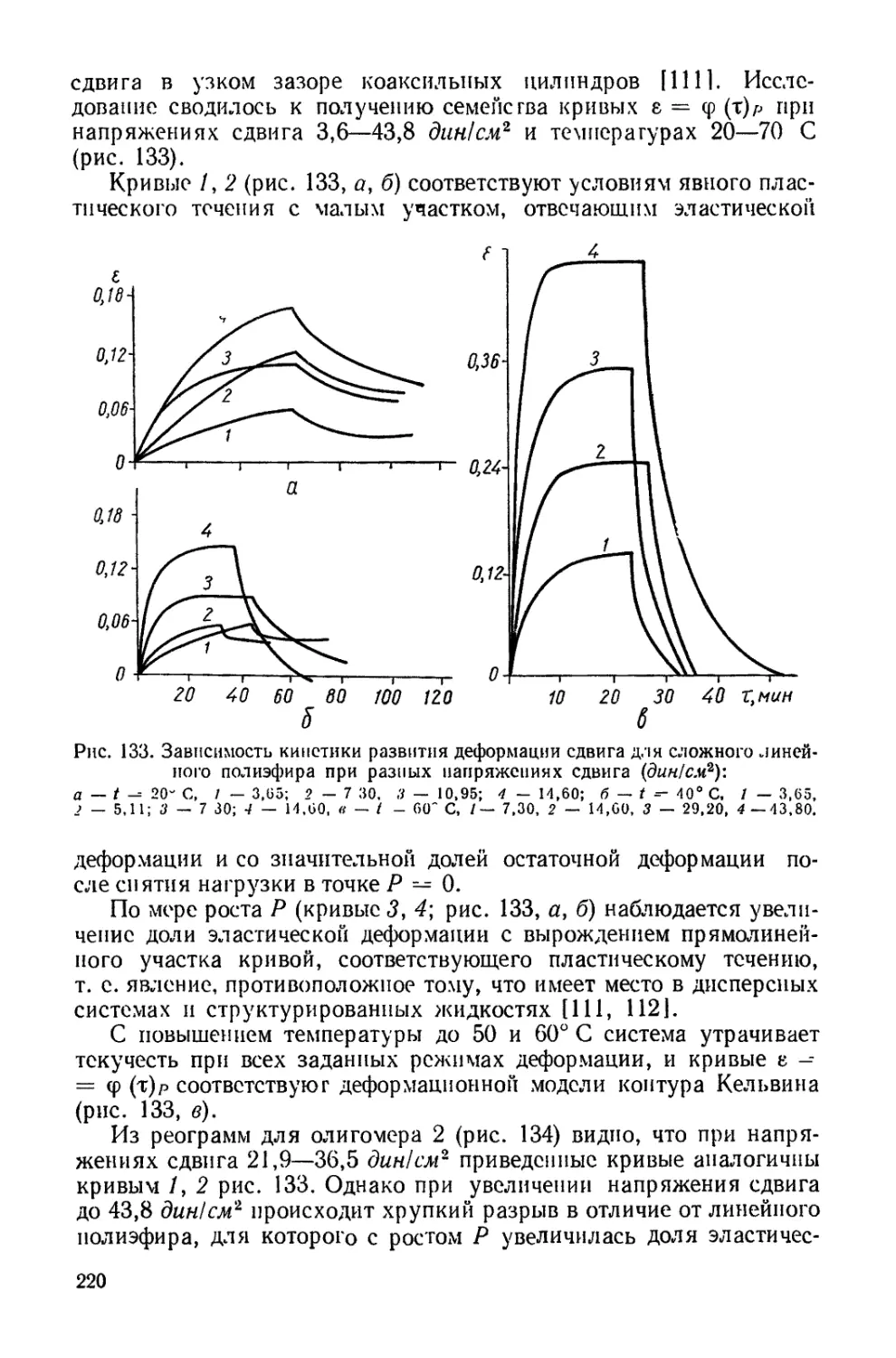
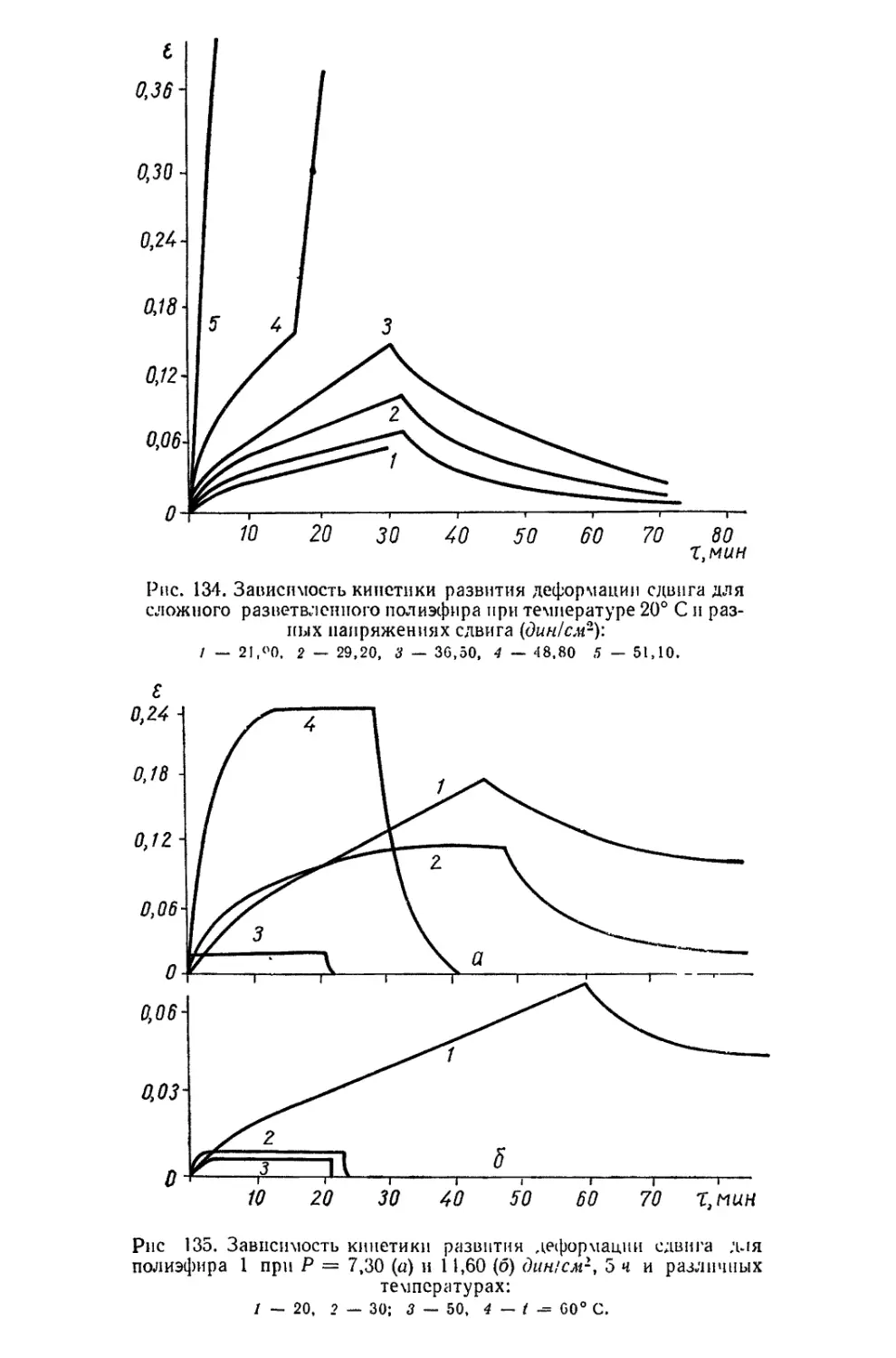
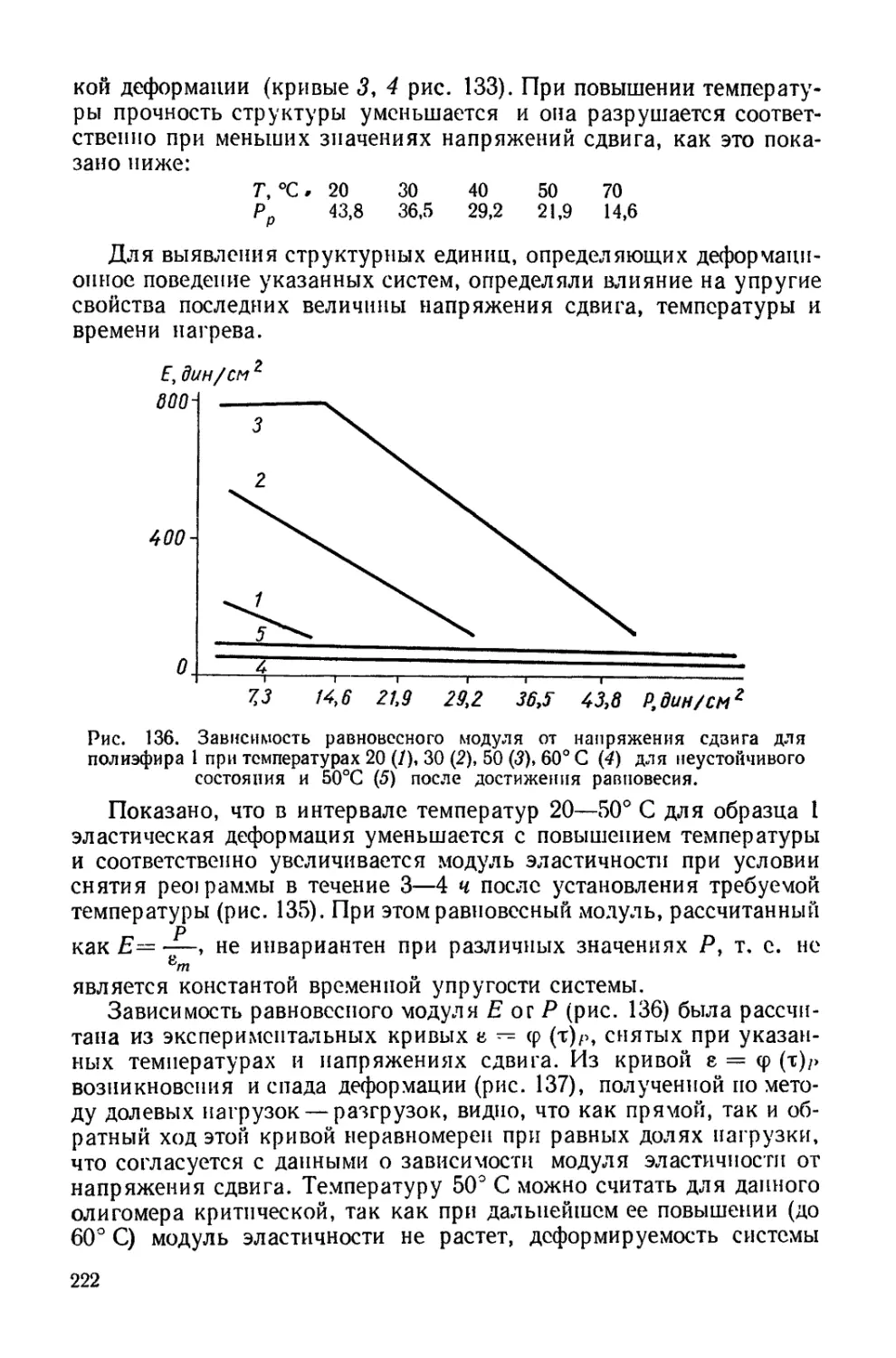
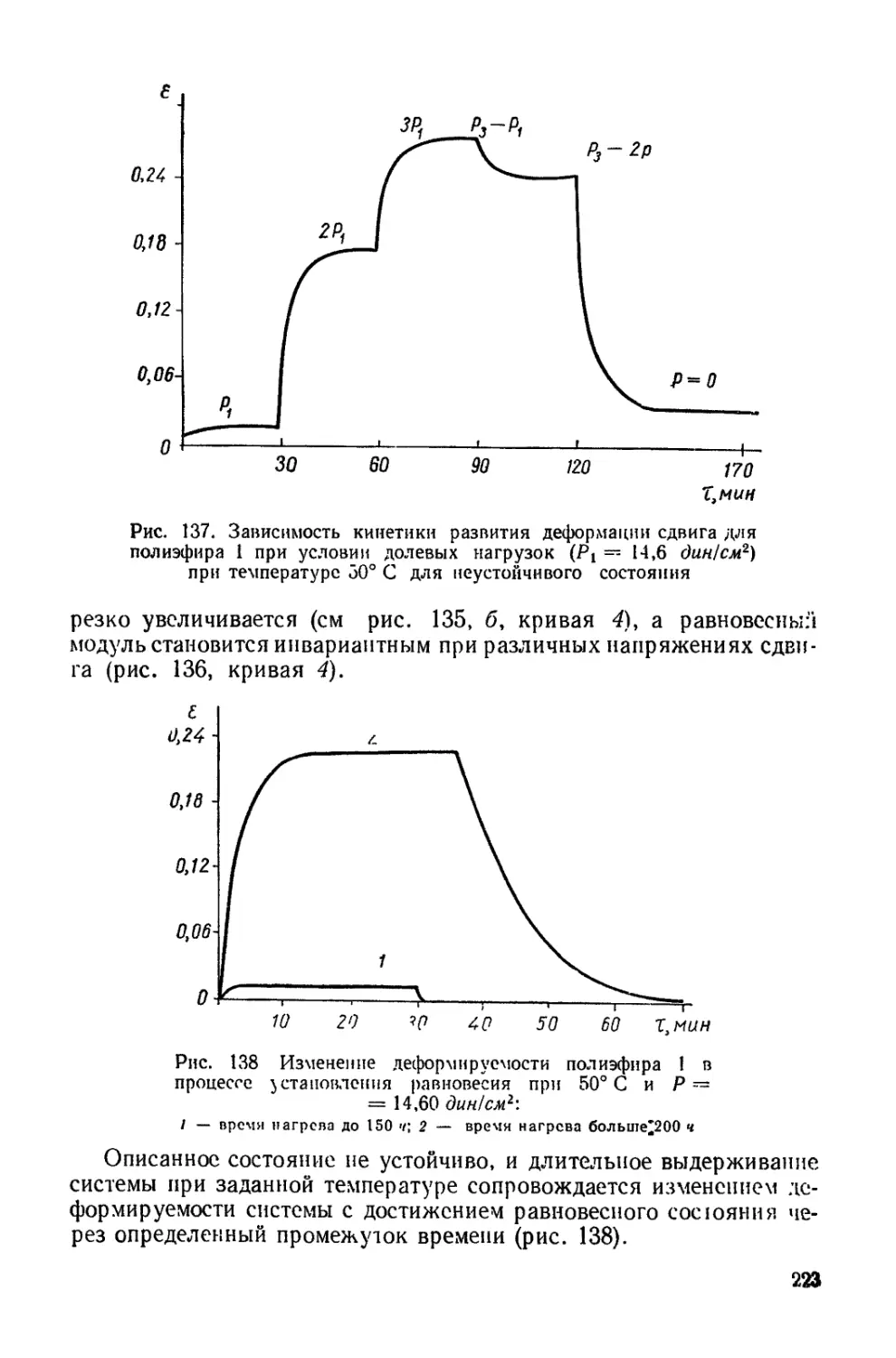

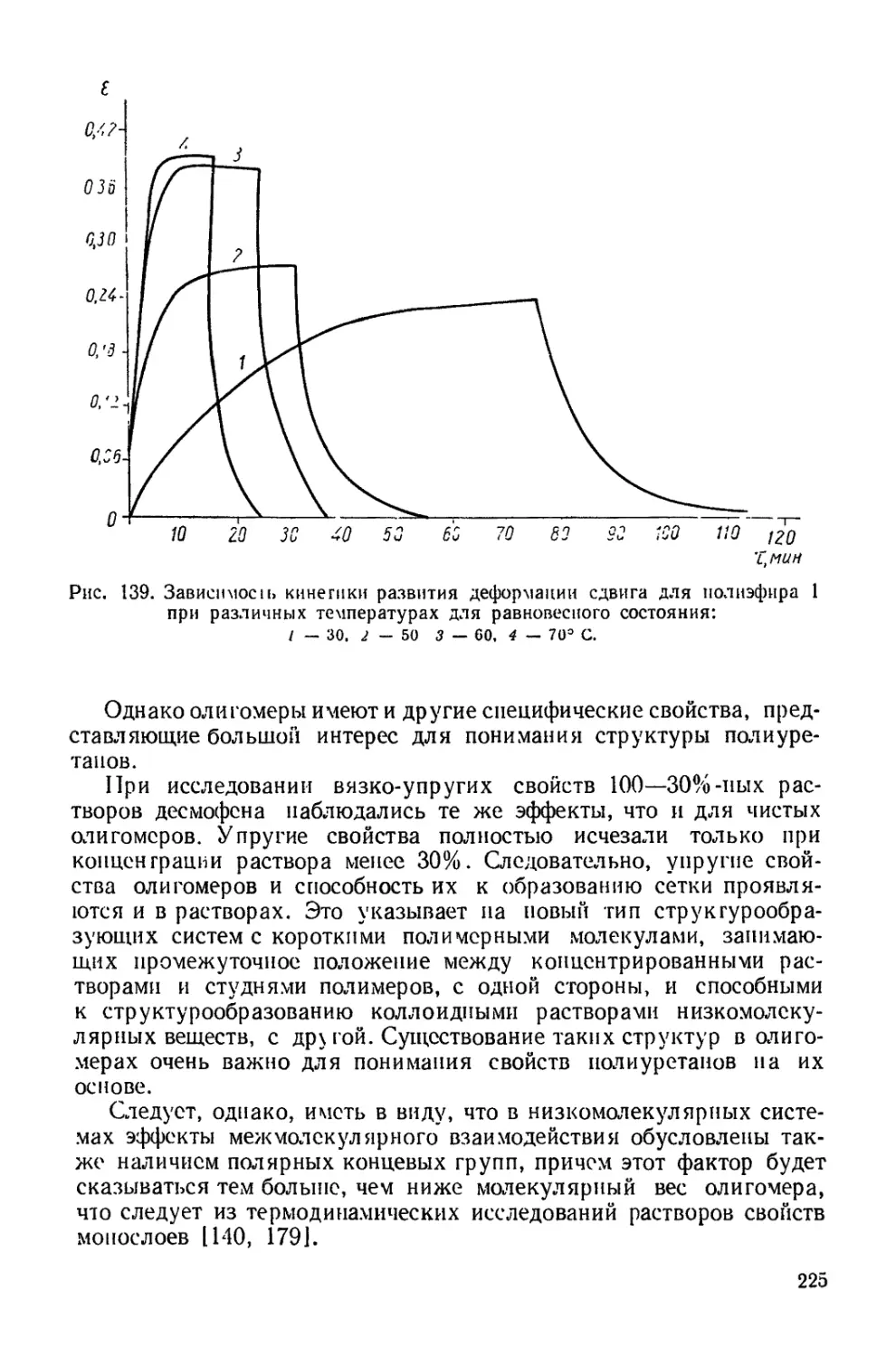
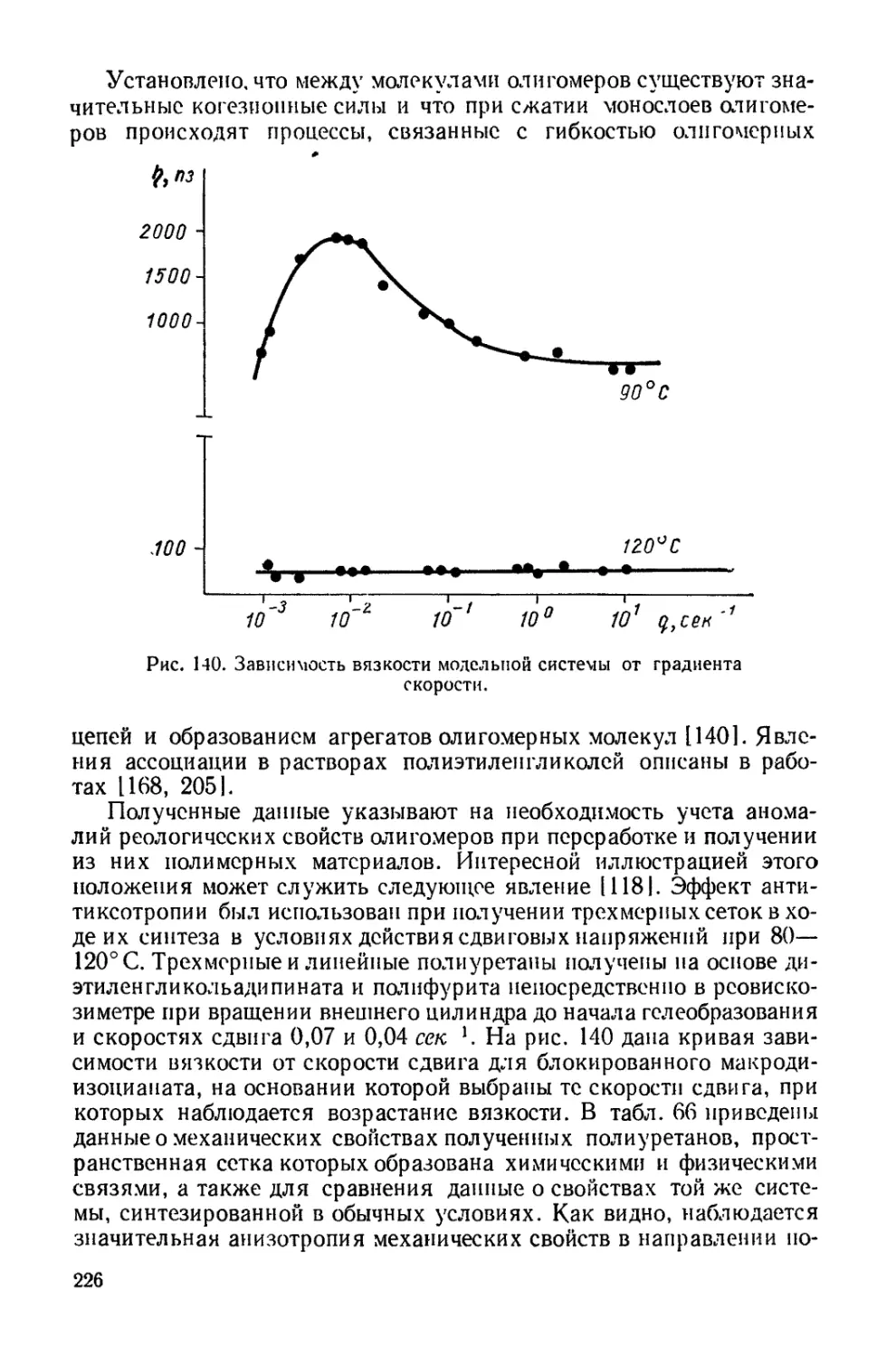

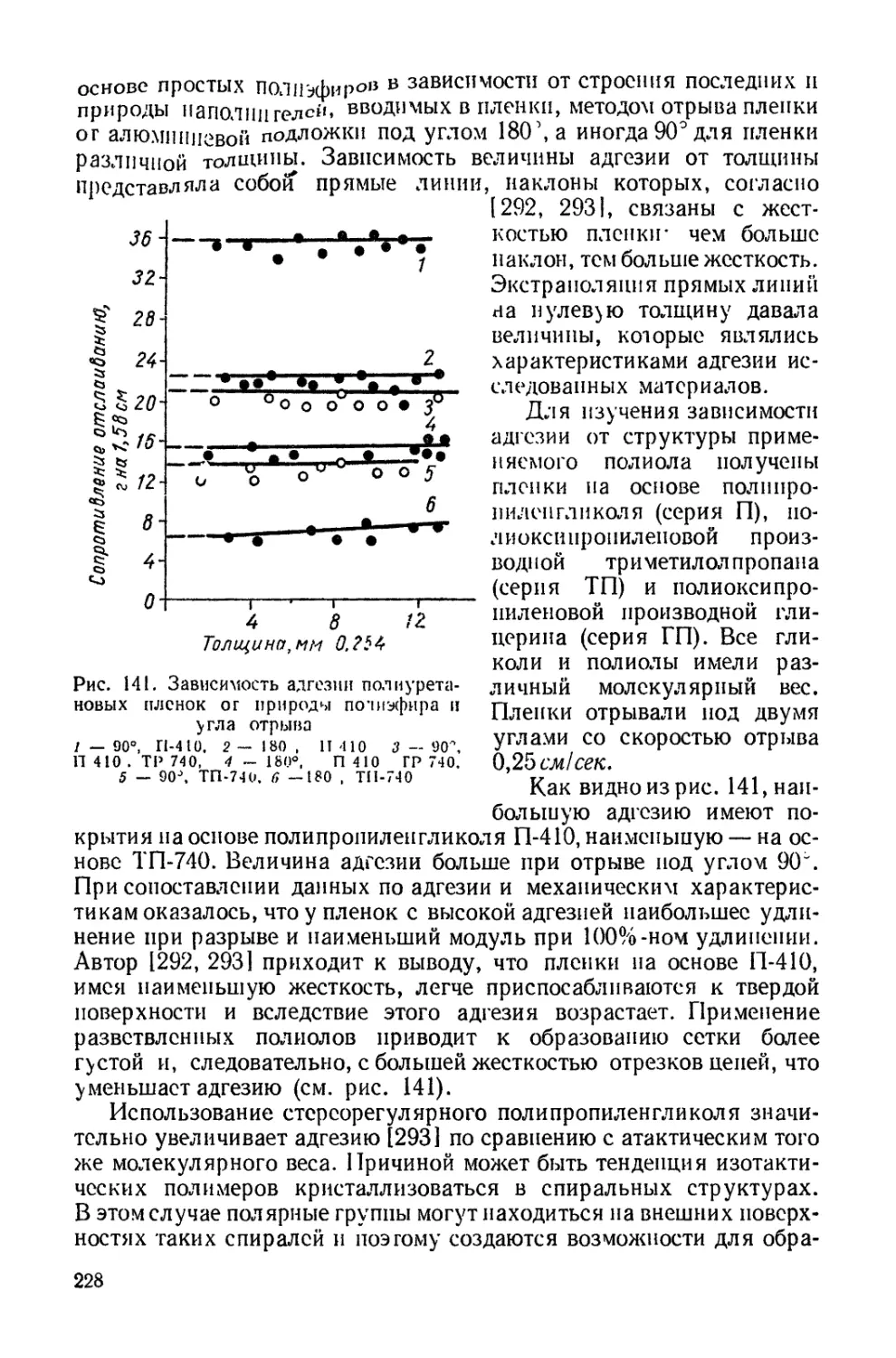

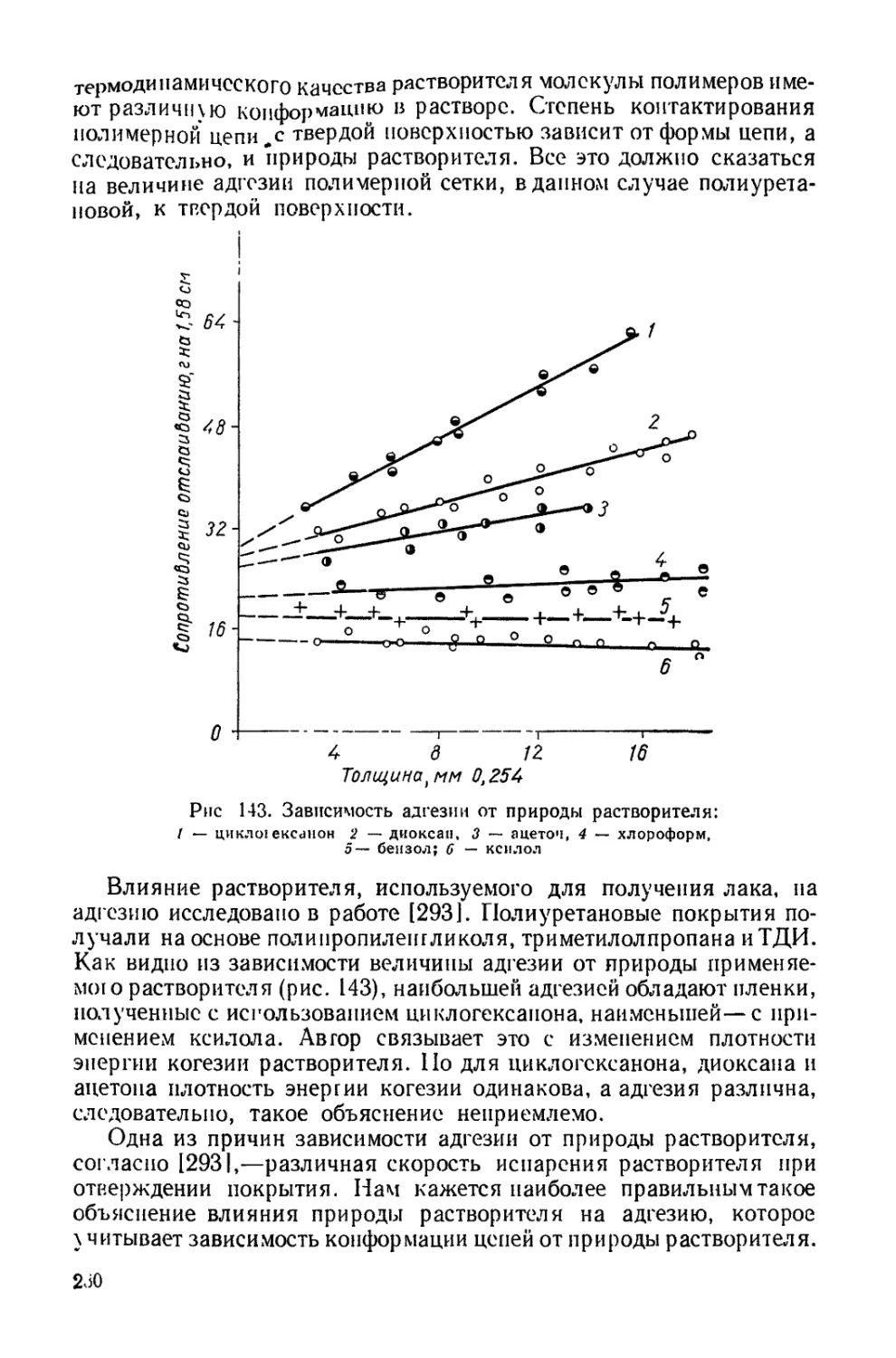





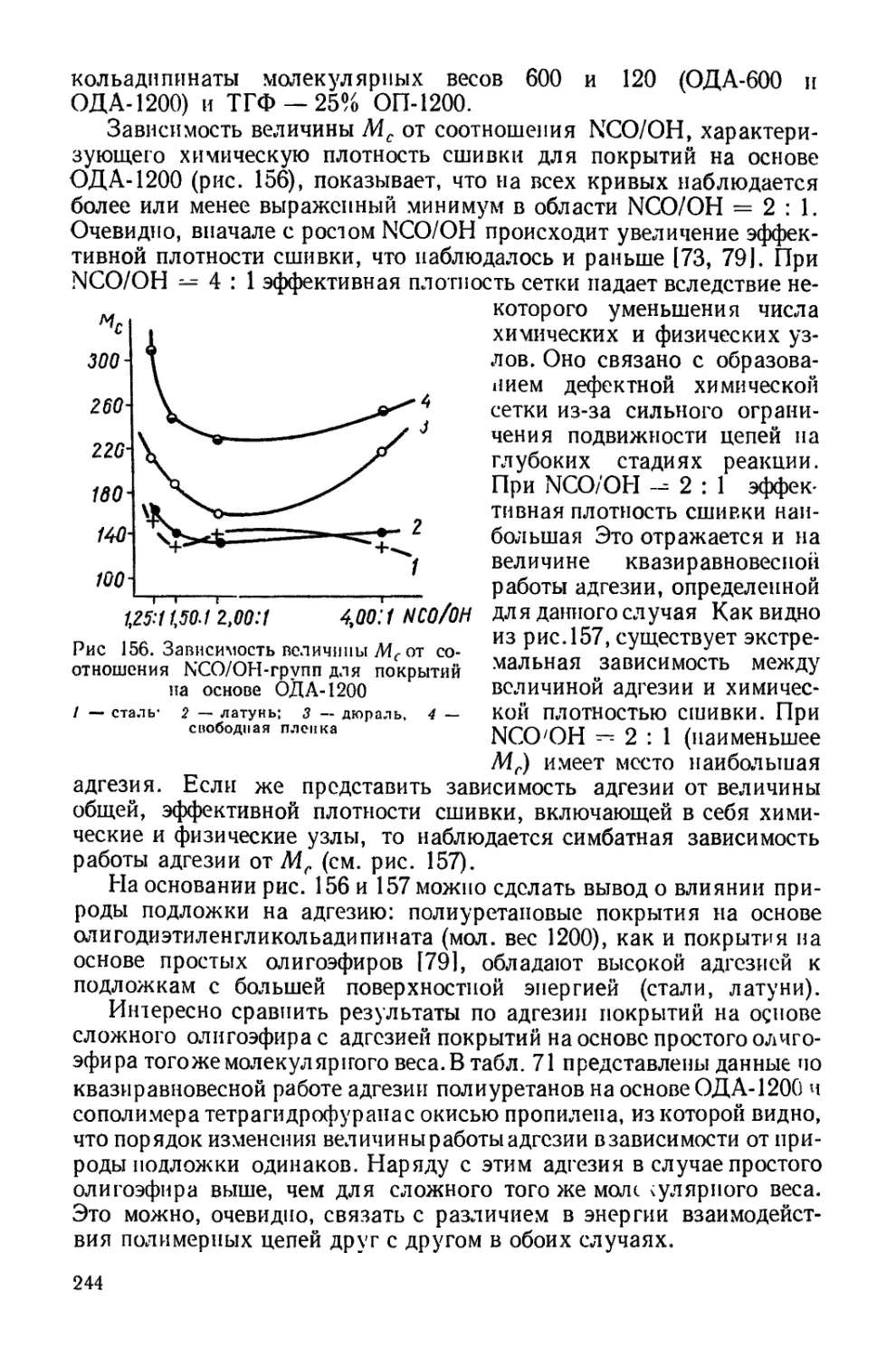
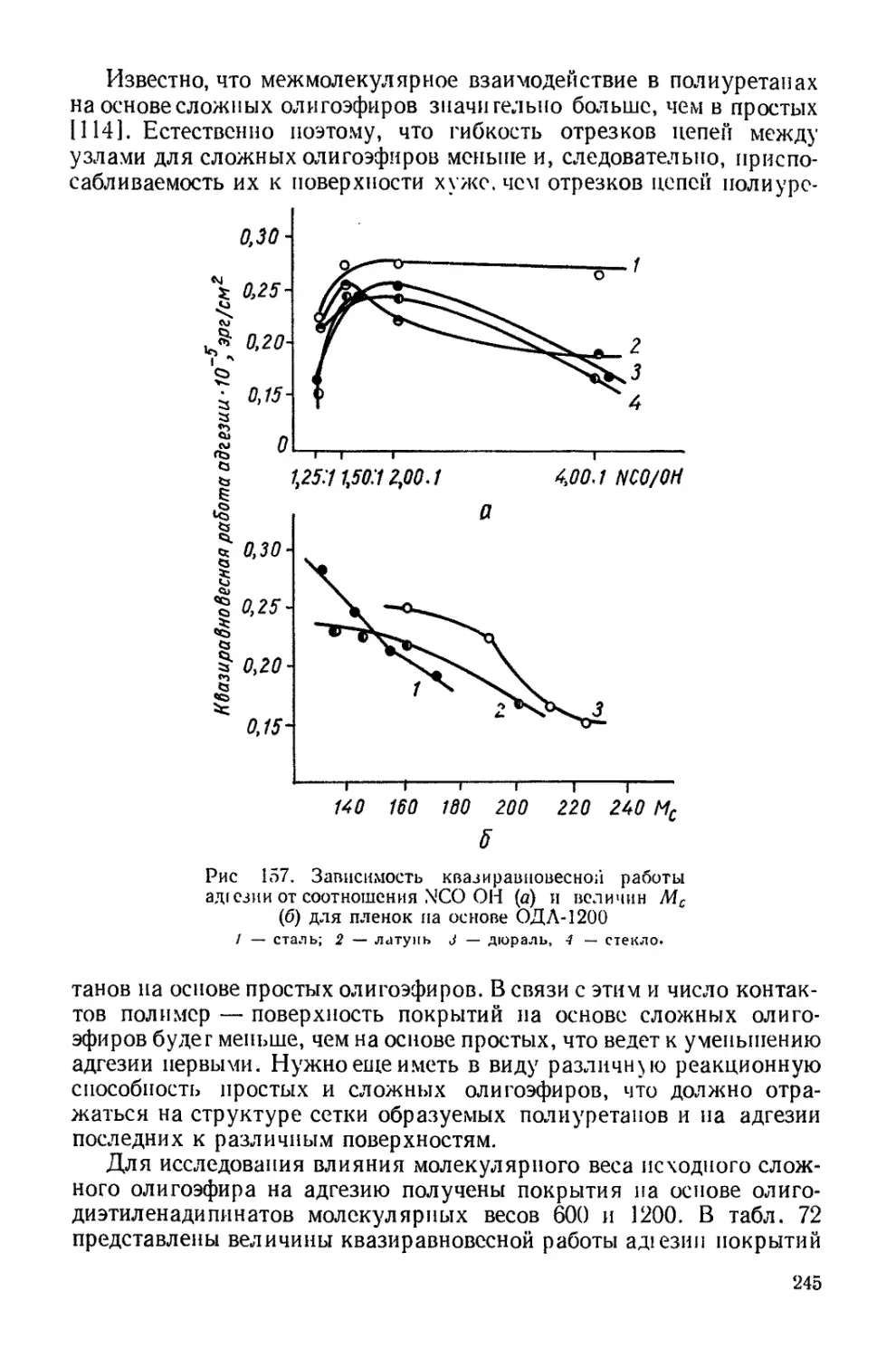

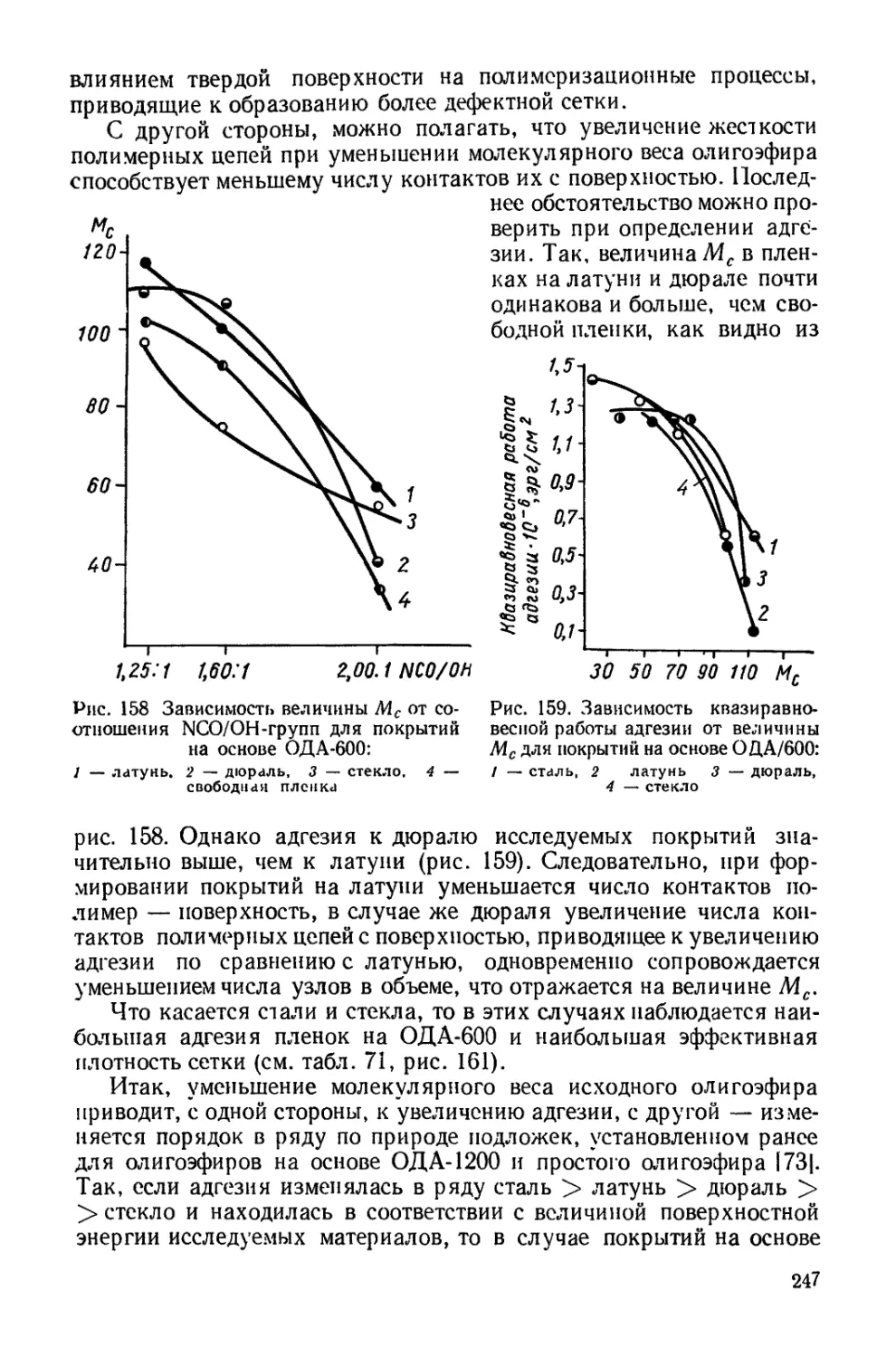


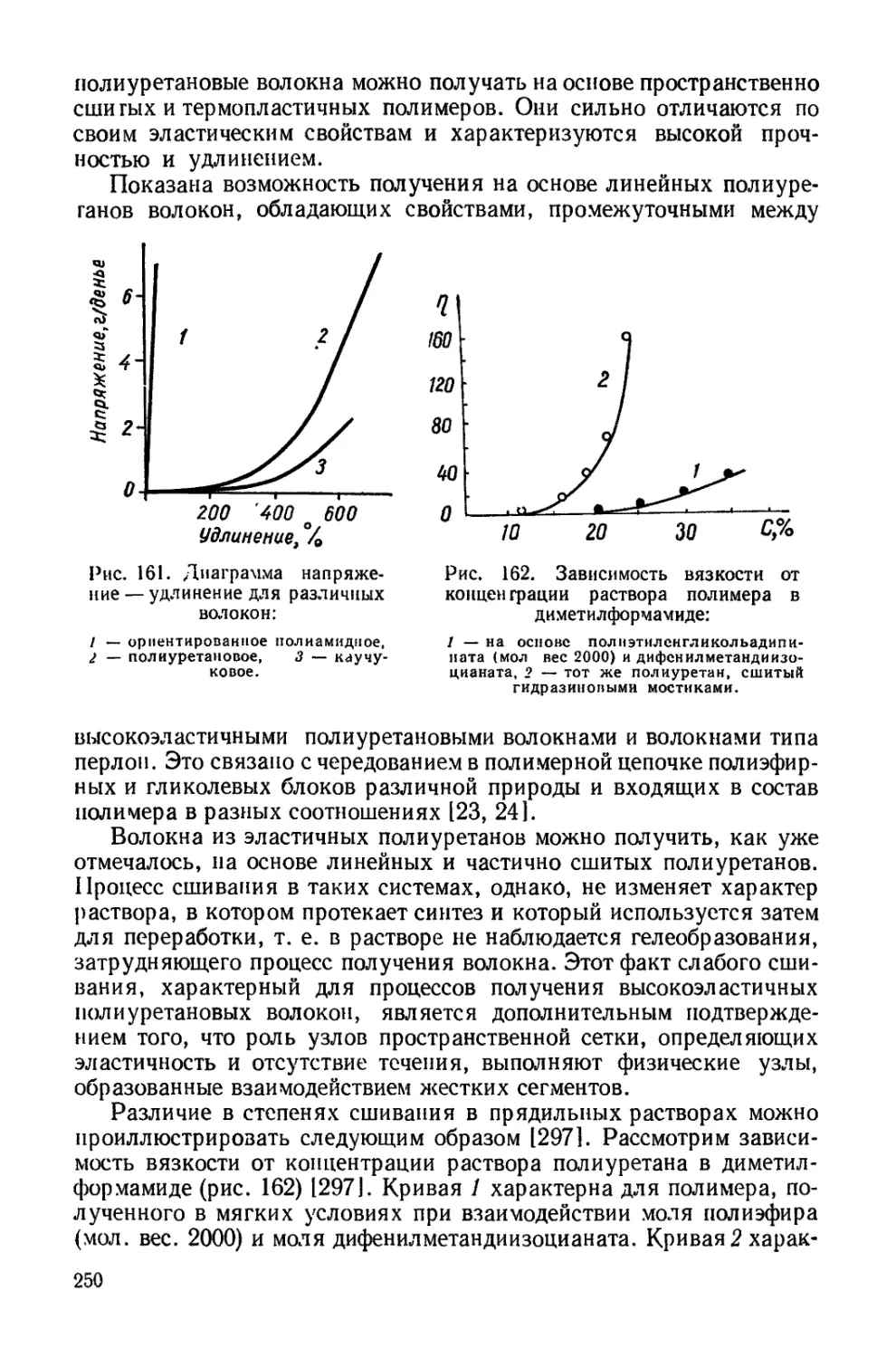


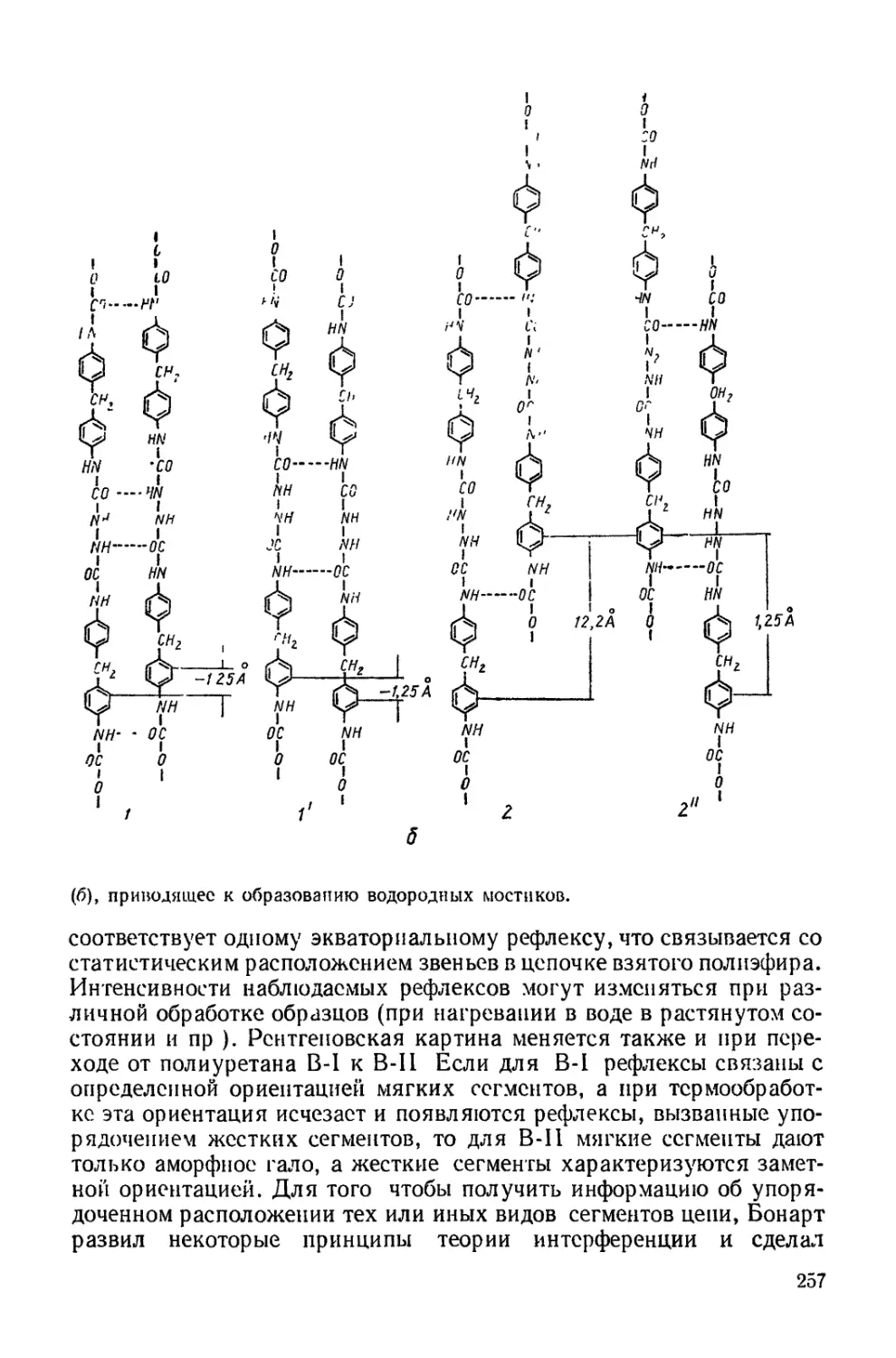
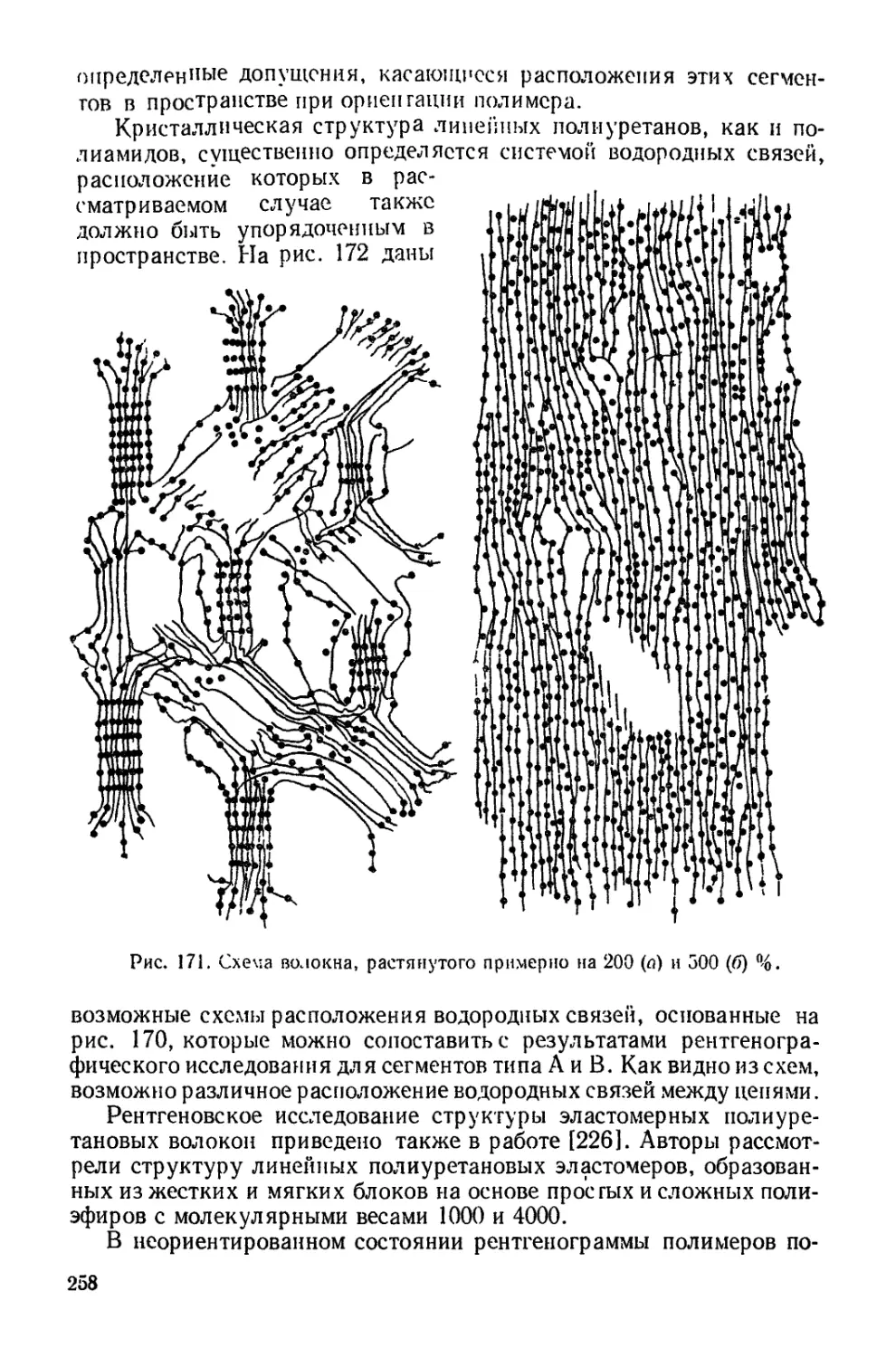

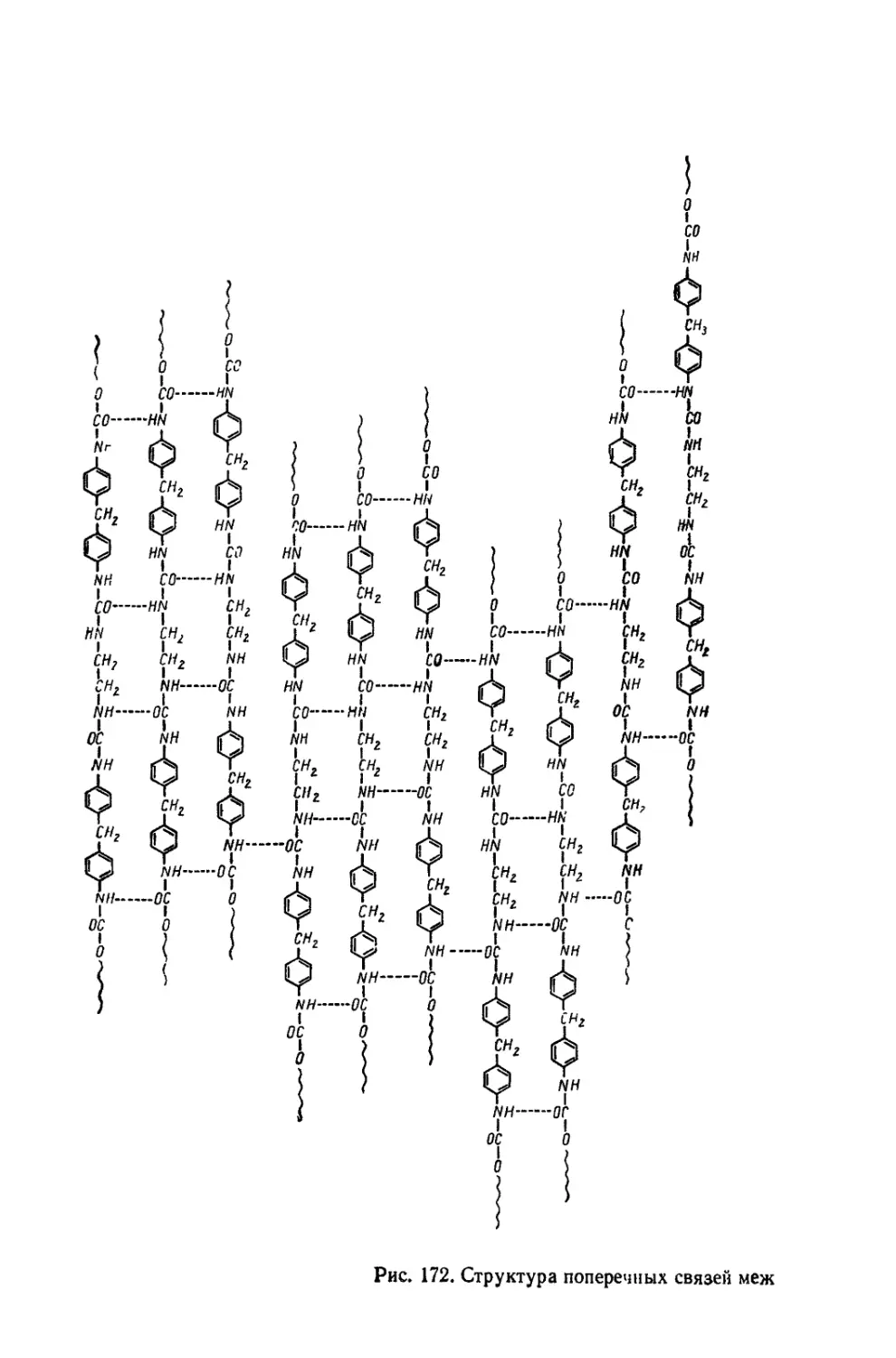
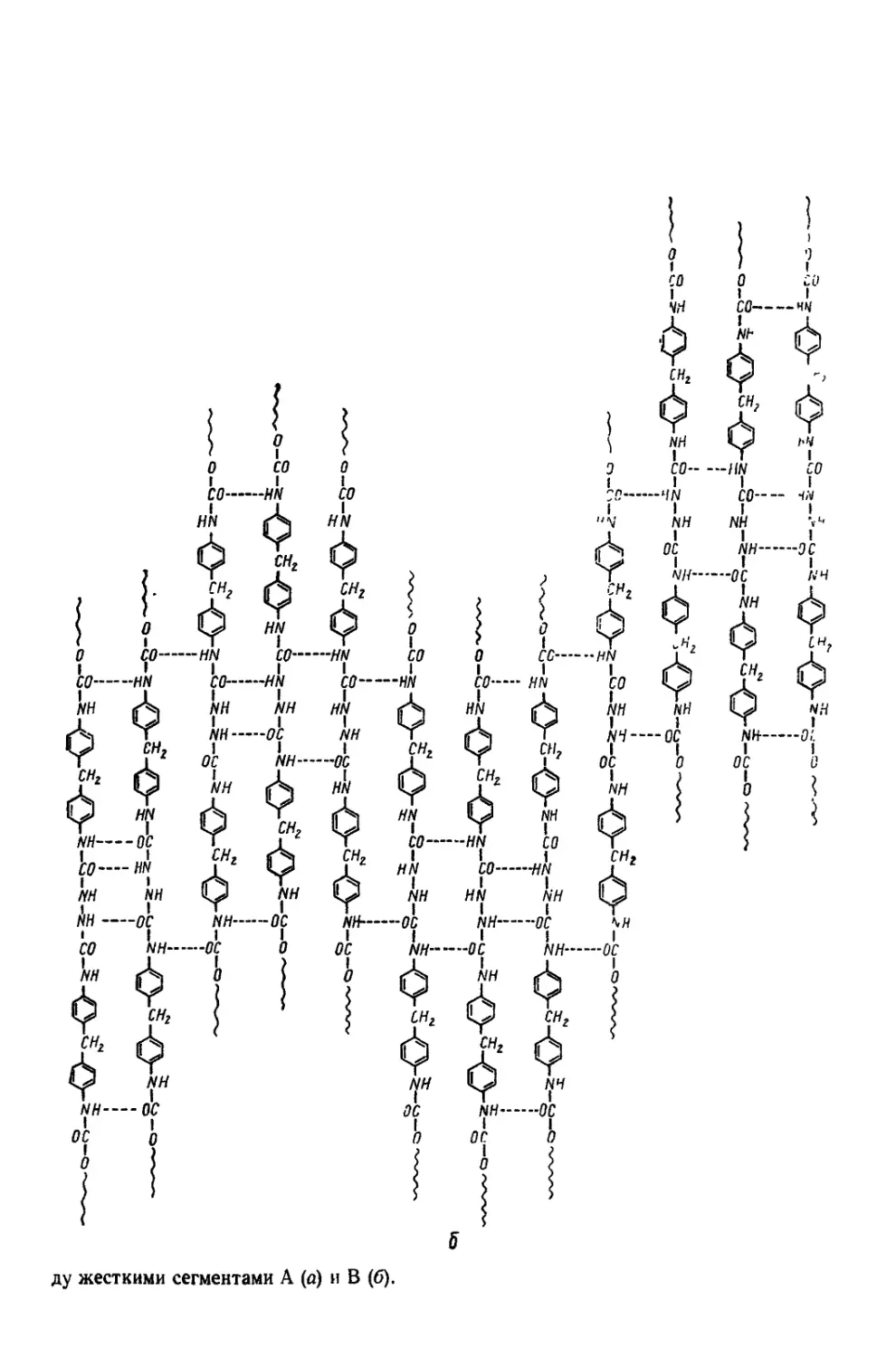






















Текст
АКАДЕМИЯ НАУК УКРАИНСКОЙ ССР
ИНСТИТУТ ХИМИИ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
Ю. С. ЛИПАТОВ, Ю. Ю. КЕРЧА, Л. М. СЕРГЕЕВА
СТРУКТУРА и СВОЙСТВА
ПОЛИУРЕТАНОВ
«НАУКОВА ДУМКА», КИЕВ-1970
517
Л61
УДК 541.64+ 678.5»
В книге рассматриваются основные свойства класса полиуре-
танов, связанные со спецификой построения полимерных молекул
и их межмолекулярных взаимодействий. Данная книга—первая в
мировой литературе, посвященная структуре и свойствам одного
из наиболее важных классов высокомолекулярных соединений —
полиуретанов. Анализ особенностей гибкости цепей, природы
связей, влияния химической природы цепей на фазовые и физи-
ческие переходы, физико-механические и другие свойства поз-
воляет установить основные причины проявления полиуретана-
ми того комплекса свойств, который определяет их широкое
практическое применение в виде каучуков и резин, покрытий,
волокон и других материалов.
Монография рассчитана на научных работников, аспирантов
и работников промышленности, специализирующихся в области
химии, физико-химии и технологии полимерных материалов.
Рецензенты:
докт. хим. наук Э, М. Натансон
и канд. хим наук Т. М. Гриценко-
2-5-3
284—70 М
ПРЕДИСЛОВИЕ
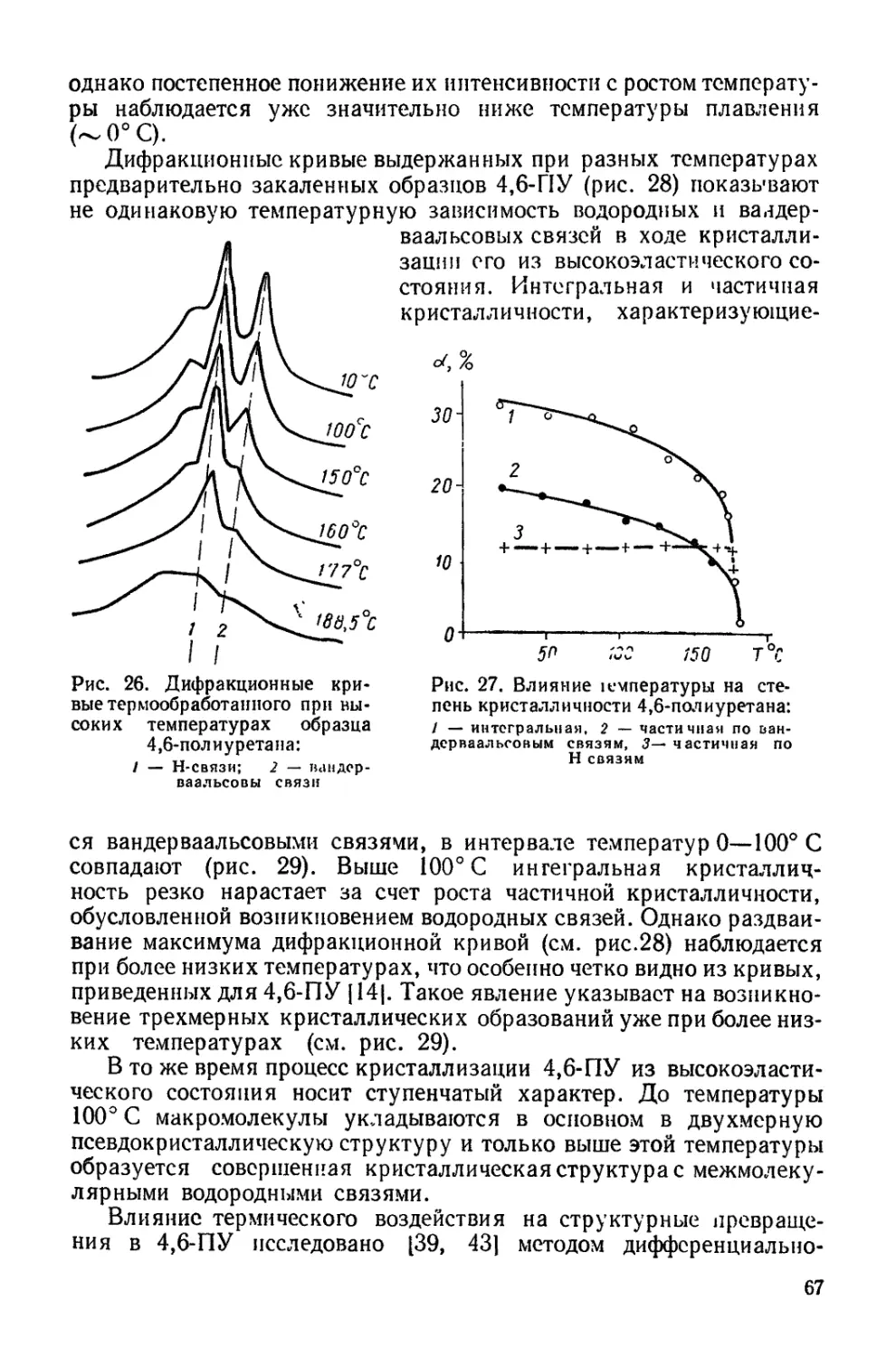
Среди большого числа полимерных материалов, использу-
емых в народном хозяйстве, особое место занимают полиу-
ретаны. Это определяется весьма ценным и специфичным ком-
плексом свойств, проявляемых полимерами, что делает возмож-
ным их применение в самых разнообразных отраслях промы-
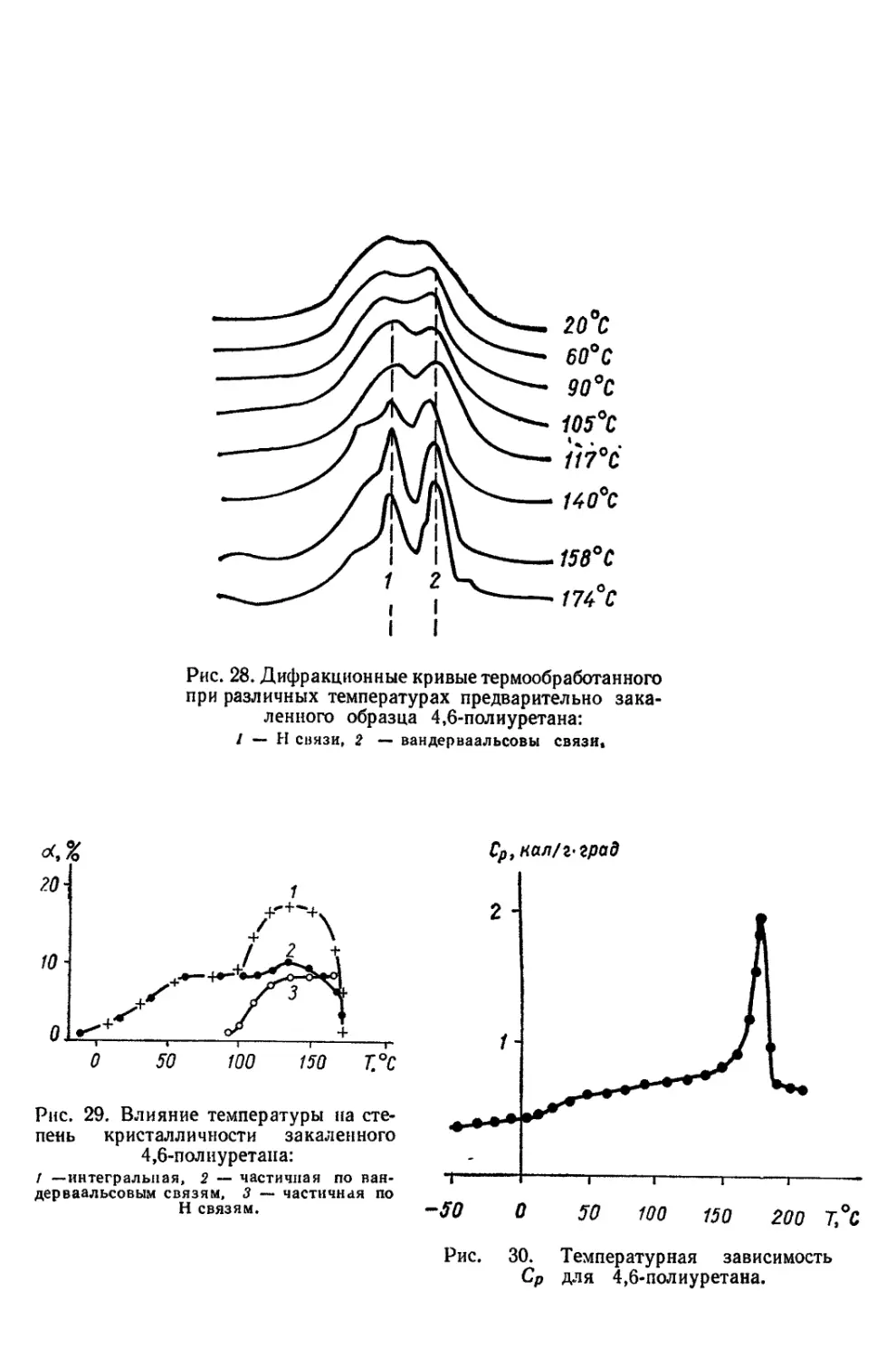
шленности и в быту. Действительно, мы не знаем другого класса
полимеров, на базе которого можно получить практически все
технически ценные полимерные материалы — каучуки и резины»
герметики и заливочные компаунды, жесткие и эластичные син-
тетические волокна, клеи и покрытия, пенопласты и многие
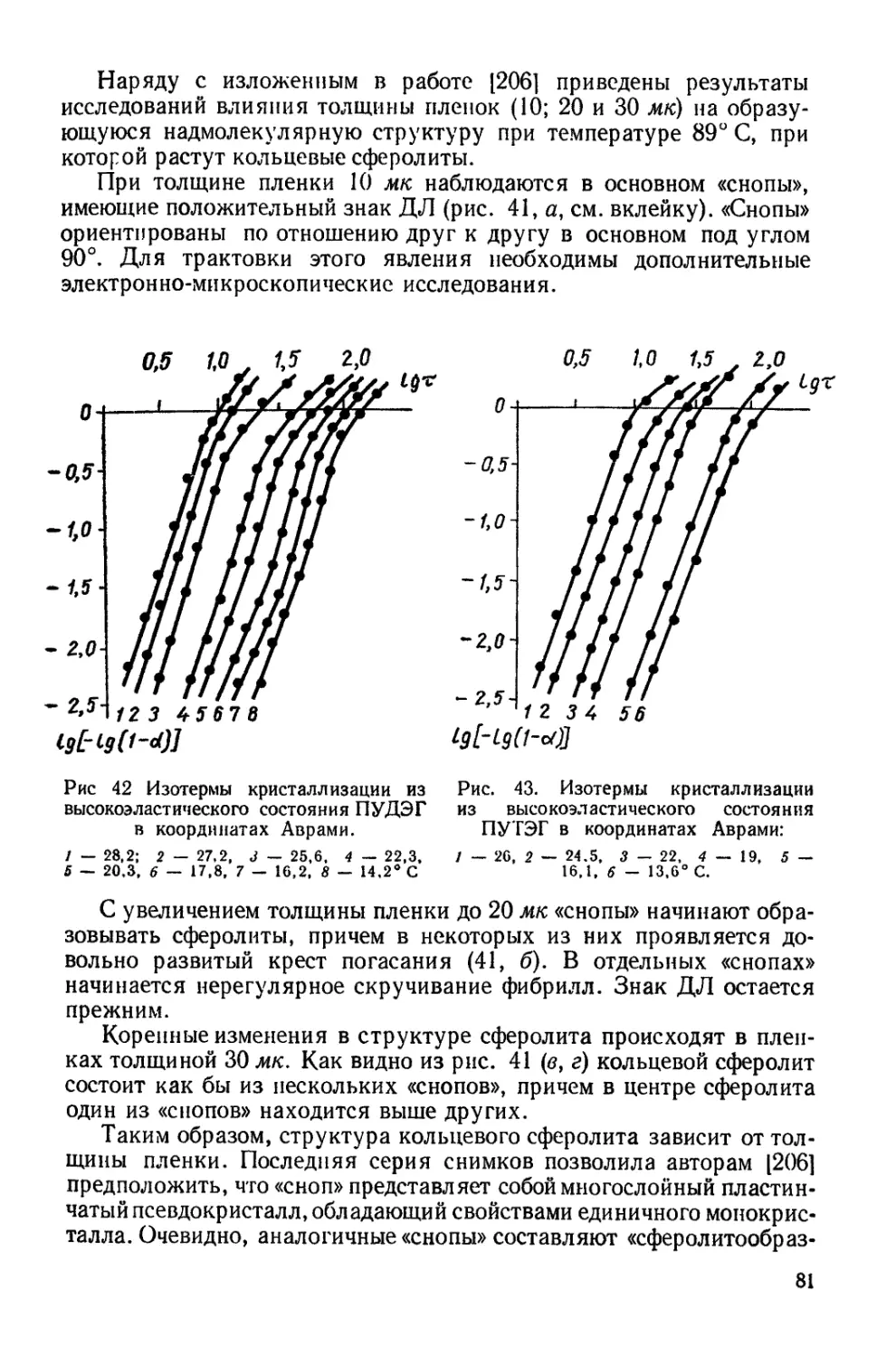
другие. Возможности получения столь разнообразных материа-
лов заложены в с собенностях химического строения полиурета-
нов и неограниченны возможностях регулирования их стру-
ктуры.
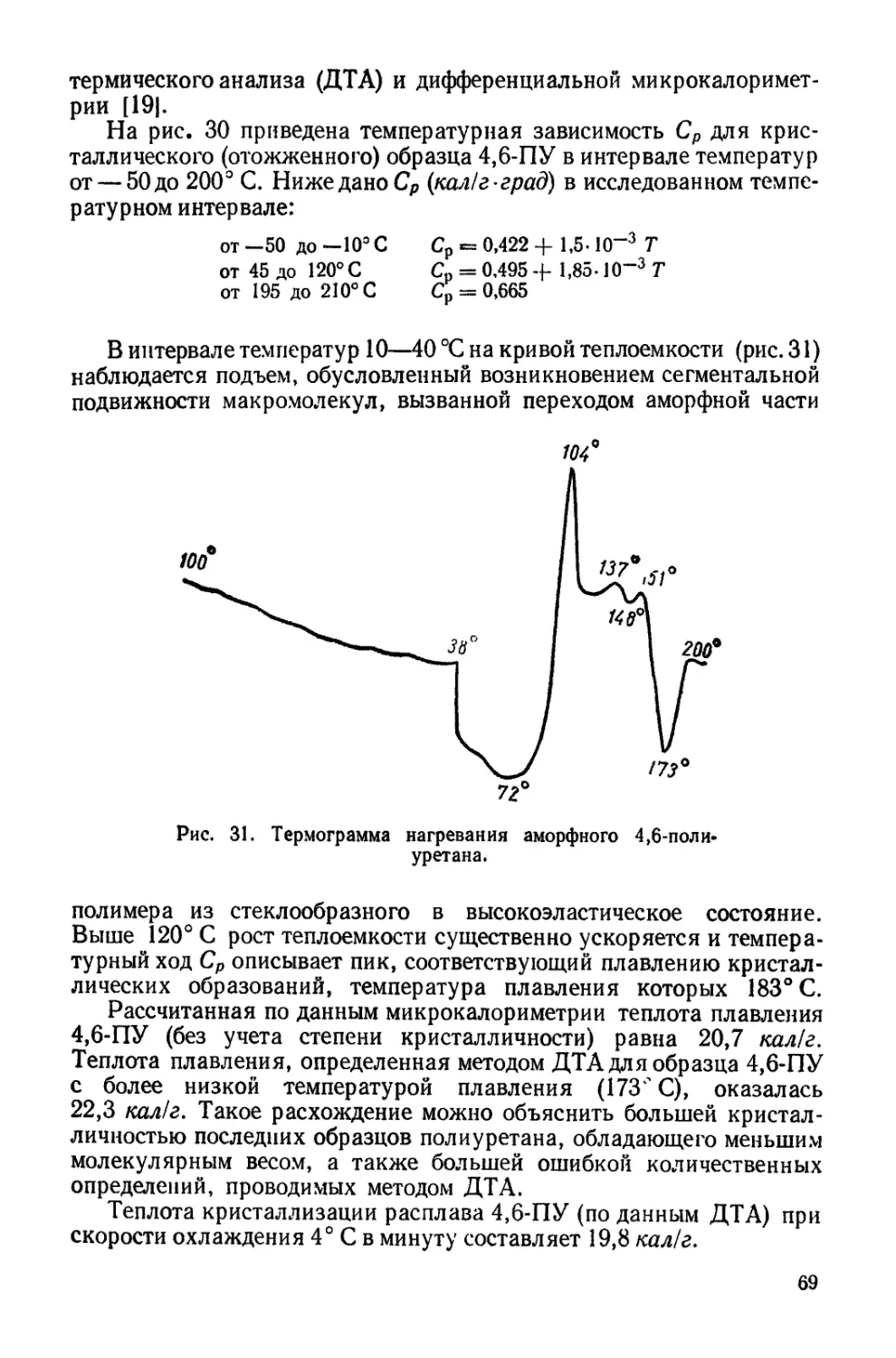
В мировой литературе известно большое число исследований,
в которых рассматриваются вопросы синтеза, технологии и пере-
работки полиуретанов. Эти работы обобщены в монографии Саун-
дерса и Фриша, переведенной на русский язык.
В результате проведенных исследований созданы десятки и
сотни полиуретановых соединений и многочисленные технически
ценные материалы на их основе. Однако до сих пор мы распола-
гаем весьма ограниченными сведениями о связи между химической
структурой и свойствами полиуретанов, а некоторые важнейшие
особенности полиуретанов, которые опред ляют особую ценность
этих соединений, до сих пор исследованы очень мало. В литера-
туре нет обобщающих данных по соотношениям между структу-
рой и свойствами полиуретанов, а большинство исследований фи-
зико-химического и физического характера посвящено свойствам
пенопластов на основе полиуретанов. По отдельным вопросам
физико-химии полиуретанов (гибкость цепей, свойства растворов
и расплавов и пр.) имеется очень мало исследований по сравне-
нию с данными для других классов полимеров.
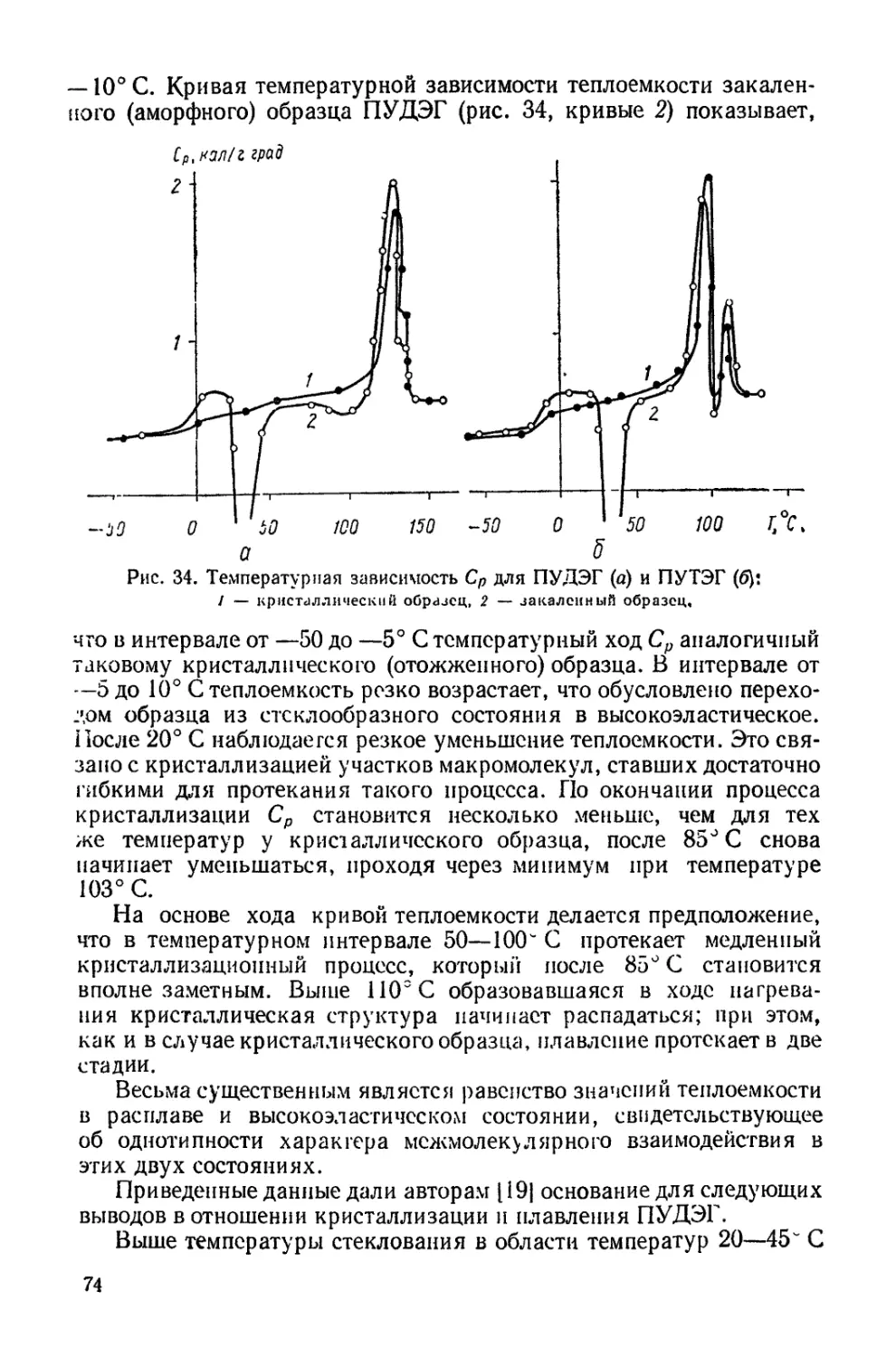
Все изложенное и определило ту задачу, которую авторы по-
ставили перед собой в настоящей монографии. Она заключает-
ся в обобщении имеющихся сведений о структуре и свойствах
полиуретанов и выяснении на этой основе причин специфических
свойств данного класса полимерных материалов, что является
весьма существенным как с точки зрения их использования в
практике, так и с точки зрения нахождения правильных путей
синтеза полимеров с заранее заданным комплексом физико-хи-
мических свойств.
Главная задача физики и физико-химии полиуретанов —
установление основных закономерностей, связывающих свойства
и строение с их специфическими особенностями и определяющих
пути их применения. Рассмотрению этих вопросов посвящена дан-
ная работа.
В основу монографии положены данные, опубликованные в
мировой литературе, и результаты исследований, проводимых в
отделе физико-химии полимеров Института химии высокомо-
лекулярных соединении АН УССР авторами и их сотрудниками.
Главы I — III написаны Ю. С. Липатовым, глава IV —
Ю. Ю Керчой, глава V—Л. М Сергеевой, глава VII— Ю. С. Ли-
патовым и Л. М. Сергеевой
Проблемы связи между условиями реакции синтеза полиу-
ретанов и надмолекулярными структурами, возникающими при
синтезе, изучались?. Э. Липатовой, которая написала главу VI.
Глава I. ОСНОВЫ ХИМИИ ПОЛИУРЕТАНОВ
Физические и химические свойства полиуретанов определяются
наличием в полимерных цепях различных типов химических свя-
зей и функциональных групп. Это обусловлено тем, что для синте-
за полиуретанов и полиуретановых материалов используется значи-
тельно больше исходных соединений, чем для других классов полиме-
ров. Поэтому прежде всего необходимо рассмотреть основные методы
синтеза полиуретанов и получения на их основе различных техни-
ческих материалов, с тем чтобы связать их свойства с условиями
синтеза и переработки.
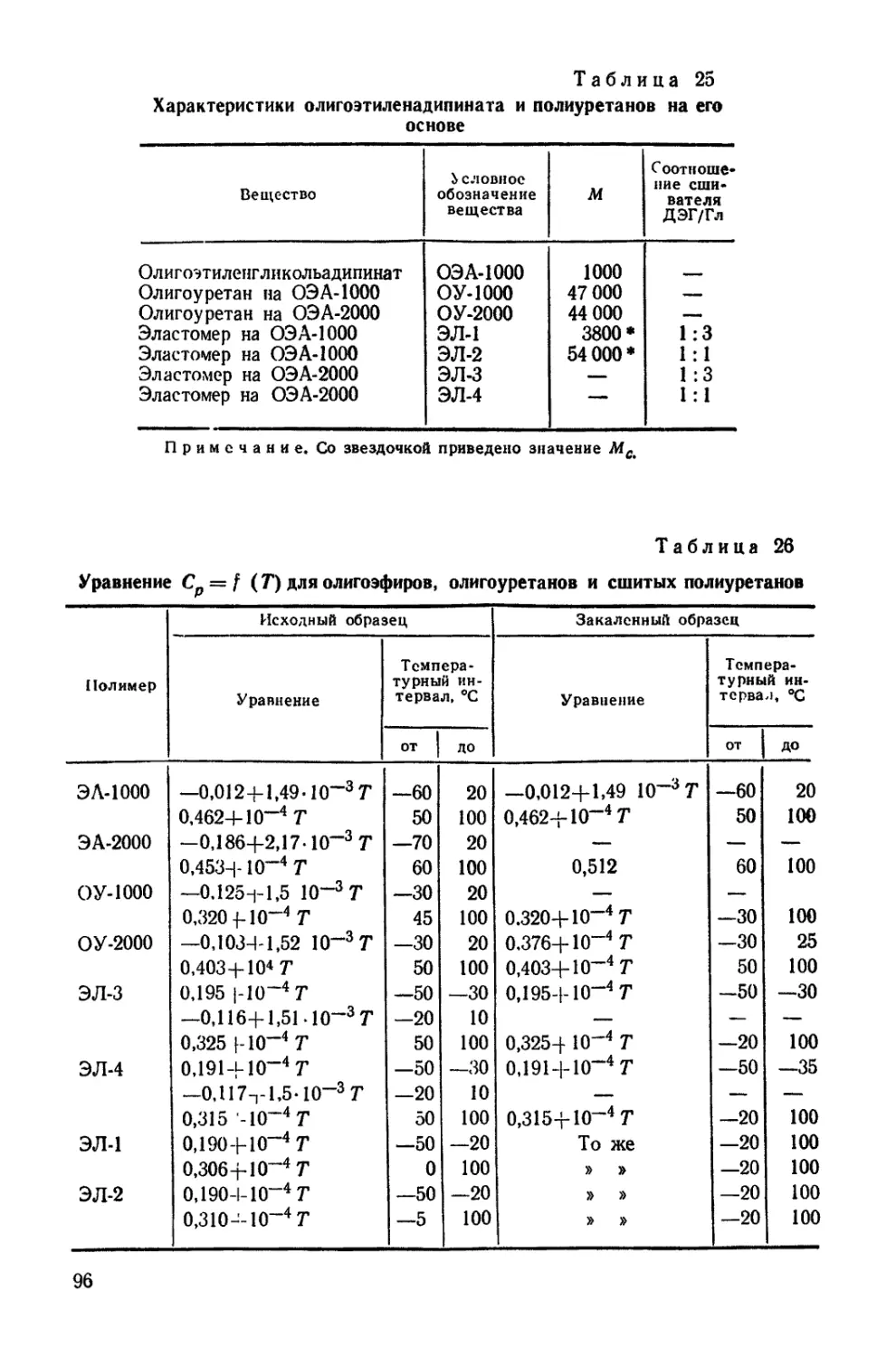
ОБЩИЕ ПРИНЦИПЫ СИНТЕЗА ПОЛИУРЕТАНОВ
В основе получения полиуретанов лежит реакция ступенчатой
полимеризации, под которой понимают реакцию, в результате кото-
рой при присоединении ди- или полифу пкциональных участников
реакции образуются макромолекулы без oti цеп лен и я осколков реаги-
рующих групп. Для этого типа реакций характерна миграция ато-
ма водорода на каждой ступени, что дает основание называть реак-
цию также миграционной полимеризацией [47].
Образование уретанов связано с реакцией присоединения изо-
цианатов, описанной еще Вюрцем [3451,
R—Х-=С-^ОН R'-OH -> R-X-C-OR'
I •
н о
Для уретанов характерна группировка атомов
О -С-Х-
Метод получения полиуретанов разработан в Германии [286] и
США [2851 одновременно и основан на взаимодействии изоцианатов
с гликолями, в результате которого происходит образование линей-
ного полиуретана по общей схеме
пНО—R—ОН 4- пО = С - X—R'—N = С = О ->
ОН НО
II I I II
О—R—О—С—X—R'-—N—С—)л.
5
Полимеры такой структуры имеют много общих свойств с по-
лиамидами и другими линейными полимерами, что предопределяет
потенциальные возможности их применения. С помощью подбора
компонентов (диизоцианатов и гидроксилсодержащих соединений)
можно в широких пределах варьировать их свойства. Именно воз-
можность активного регулирования структуры полимерной цепи
позволяет получать на основе полиуретанов материалы с самыми
разнообразными свойствами.
Для получения высокомолекулярных продуктов необходимо брать
исходные компоненты высокой степени чистоты в строго эквимолеку-
лярных количествах. Если один из компонентов используется в
избытке, это приводит, как и в случае поликонденсации, к умень-
шению молекулярного веса. Компонент, взятый в избытке, образует
преимущественно концевые группы. Высокая реакционность диизо-
цианатов позволяет проводить реакции при невысоких температурах
(в случае катализируемых реакций — при комнатных). В мягких
условиях протекает меньше побочных реакций, что позволяет из-
бежать разветвлений вследствие взаимодействия диизоцианатов с
образовавшимися уретановыми группами.
Рассмотренный выше метод получения полиуретанов наиболее
распространен и применяется в промышленном масштабе. Сущест-
вует также много других способов их синтеза, один из которых ос-
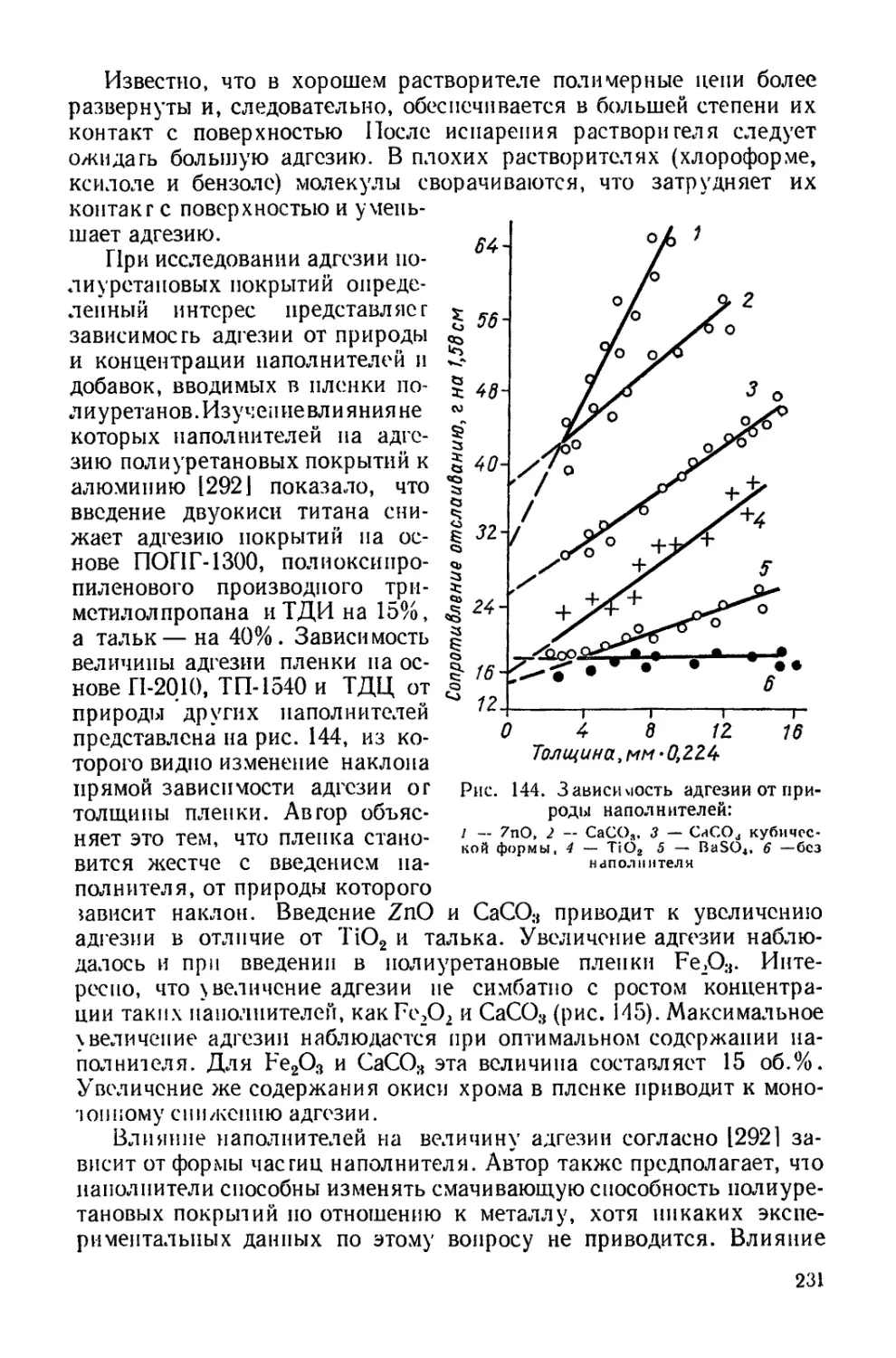
нован на реакции поликонденсации дихлор угольных эфиров гли-
колей с диаминами [2671:
nCICOOR—ООСС1 + nNH2—R'—NH2
[—R-OOCNH—Rff—NH-COO-h-hHCl.
Дихлоругольный эфир гликоля получается при взаимодействии
с фосгеном. Таким образом, данный метод включает взаимодейст-
вие какого-то компонента с фосгеном, как и получение диизоциана-
тов, основанное на реакции аминов с фосгеном, но он не нашел
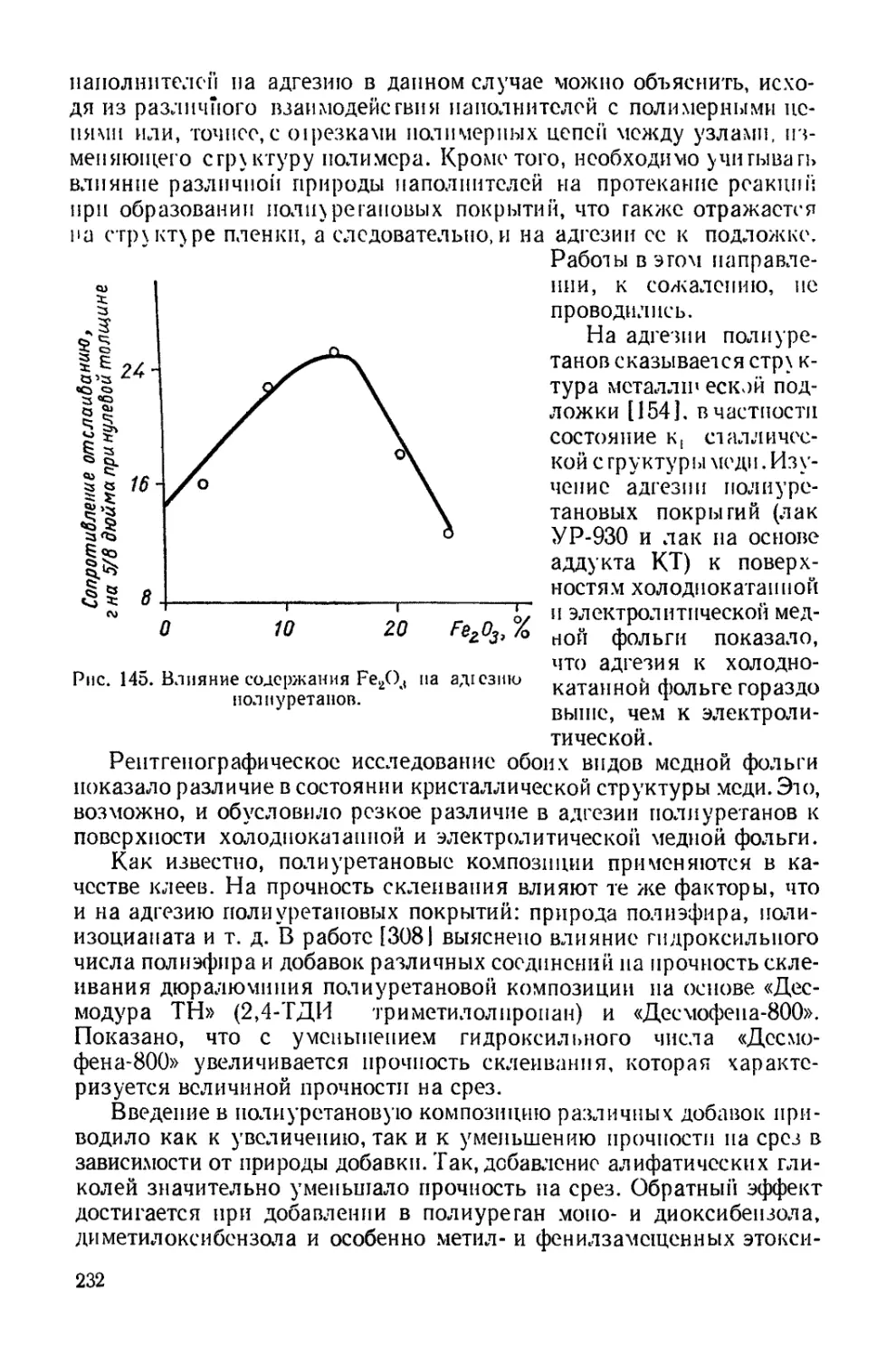
широкого распространения. Необходимо отметить, что поиски пу-
тей синтеза полиуретанов без применения изоцианатов и синтеза
изоцианатов без участия фосгена представляют большой практи-
ческий интерес, однако в настоящее время эти способы не могут
конкурировать с основным методом синтеза полиуретанов, изло-
женным выше.
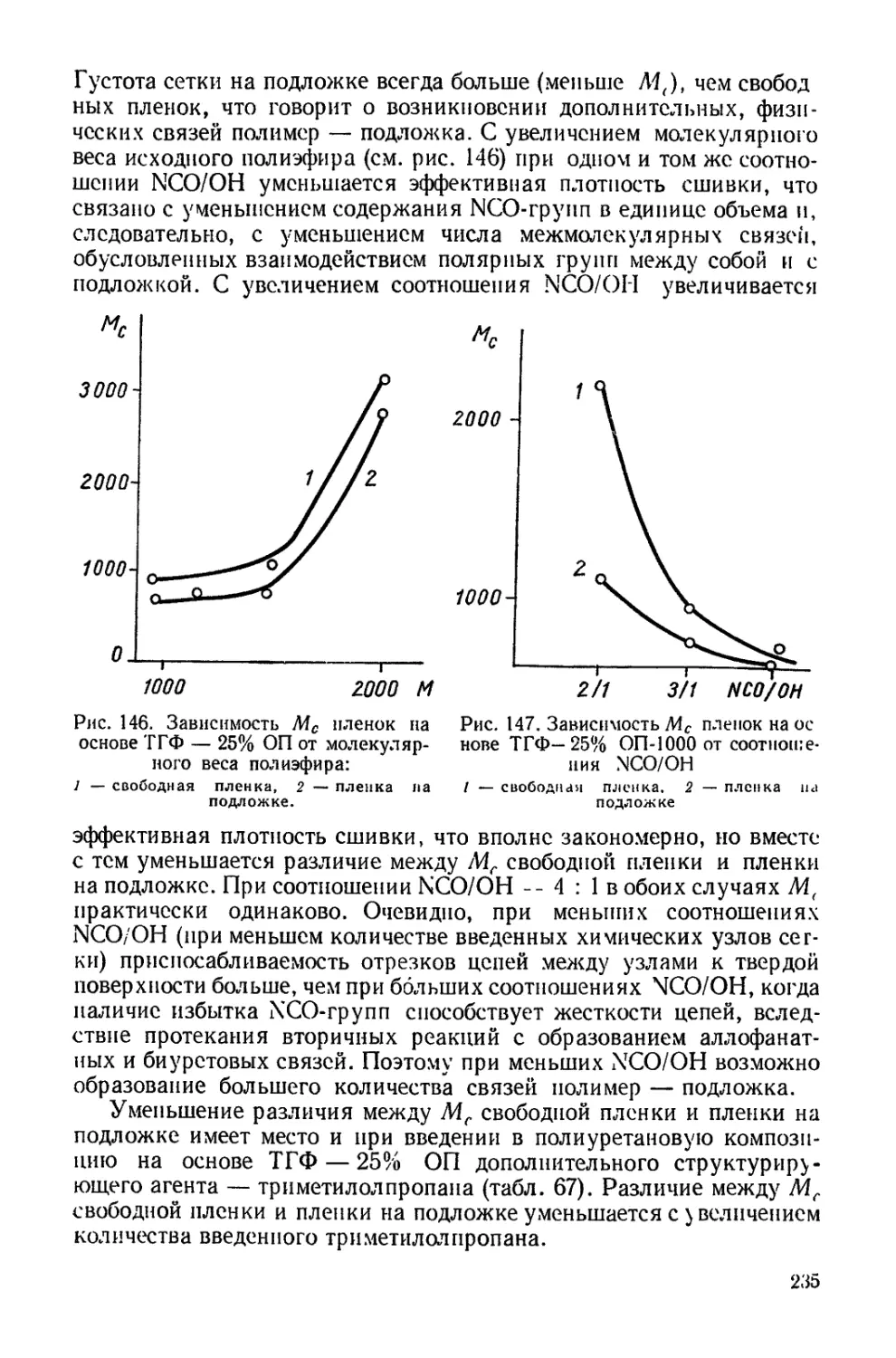
Рассмотренные реакции лежат в основе синтеза линейных поли-
уретанов.
Существенным отличием полиуретанов от всех других полиме-
ров является наличие в полиуретановых цепях также и иных ти-
пов химических связей. Именно многообразие последних во многом
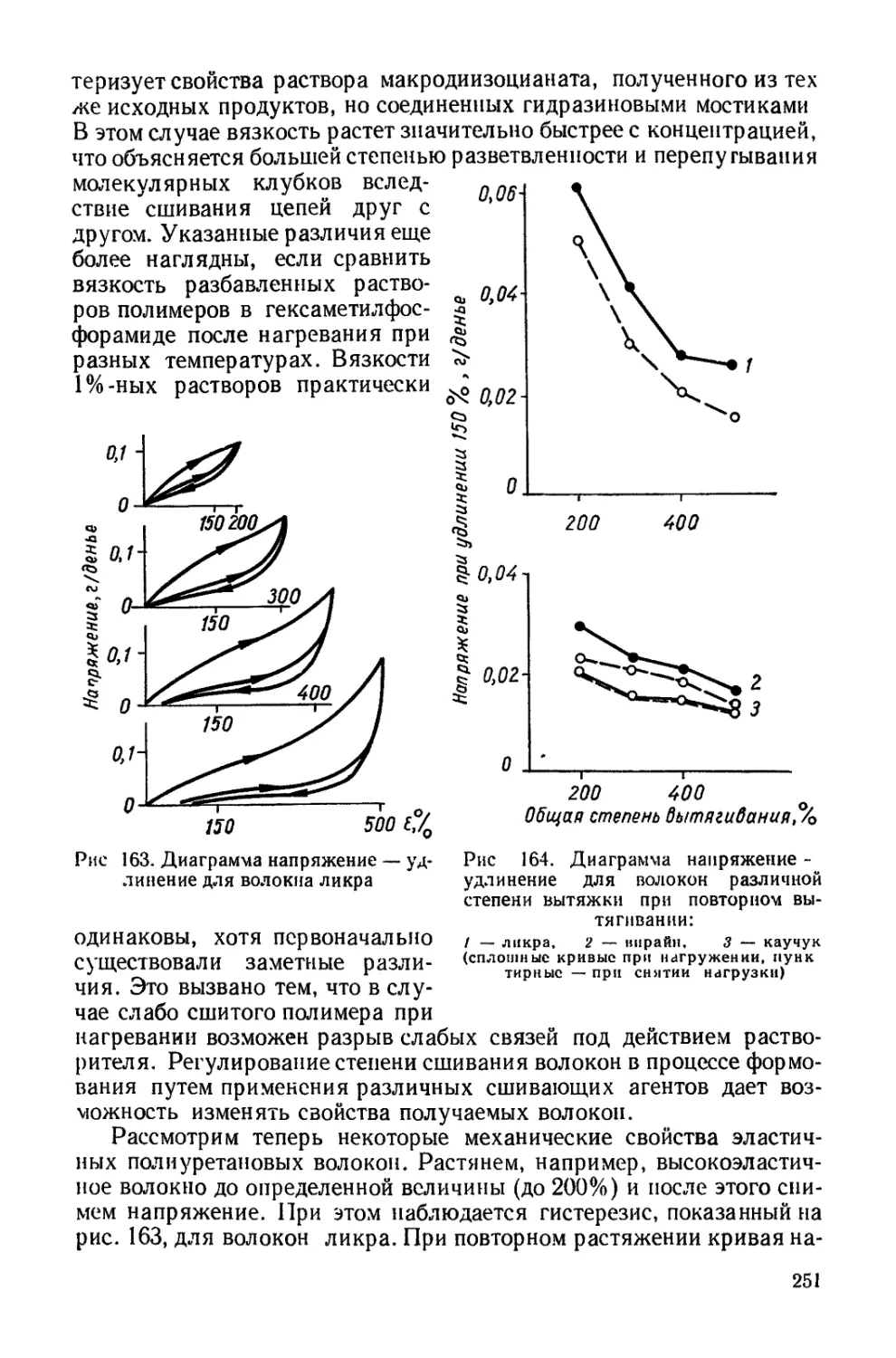
определяет химические и физические свойства полиуретанов и их
структуру.
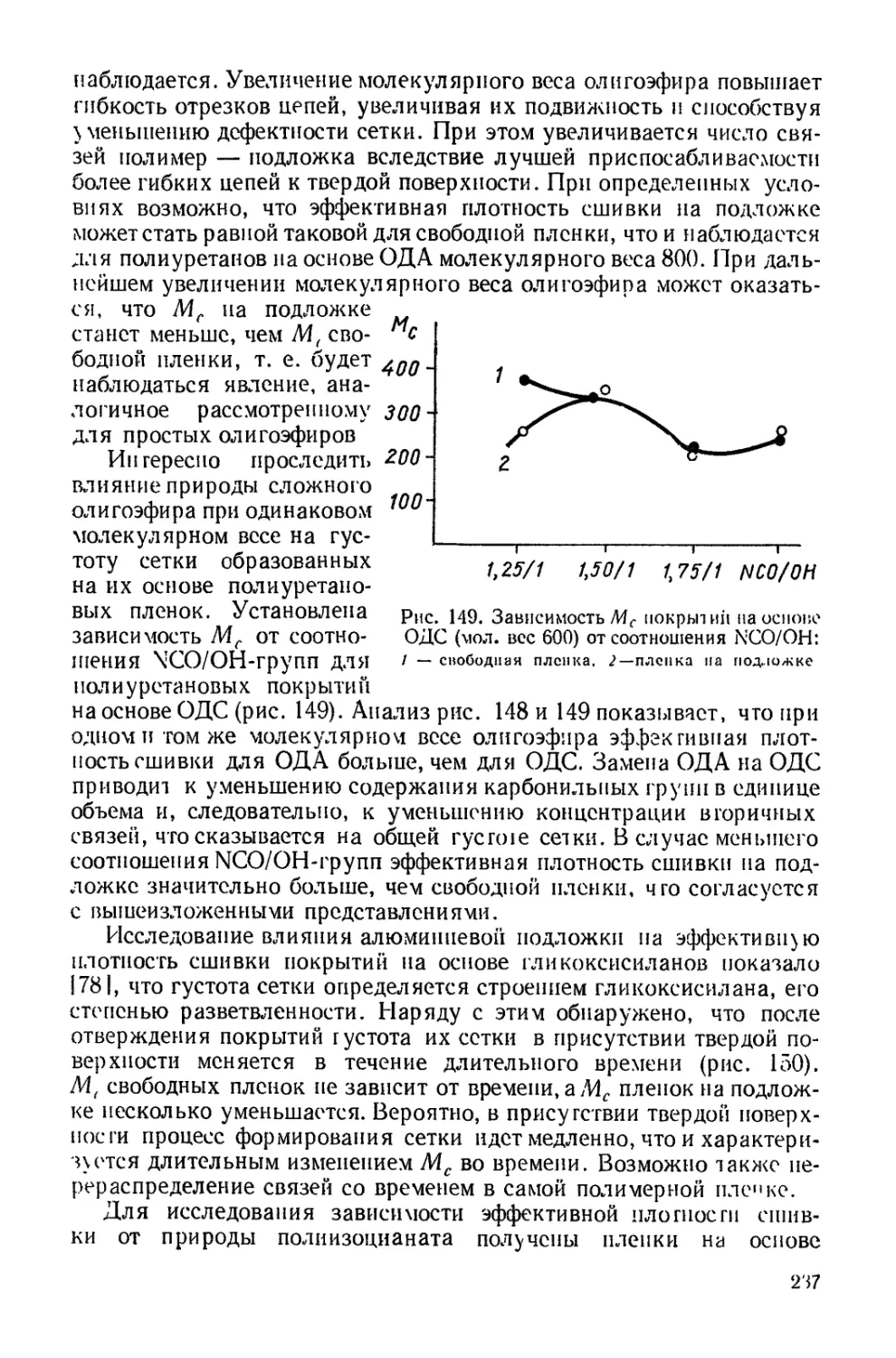
Полиуретановая цепочка в зависимости от мольного соотношения
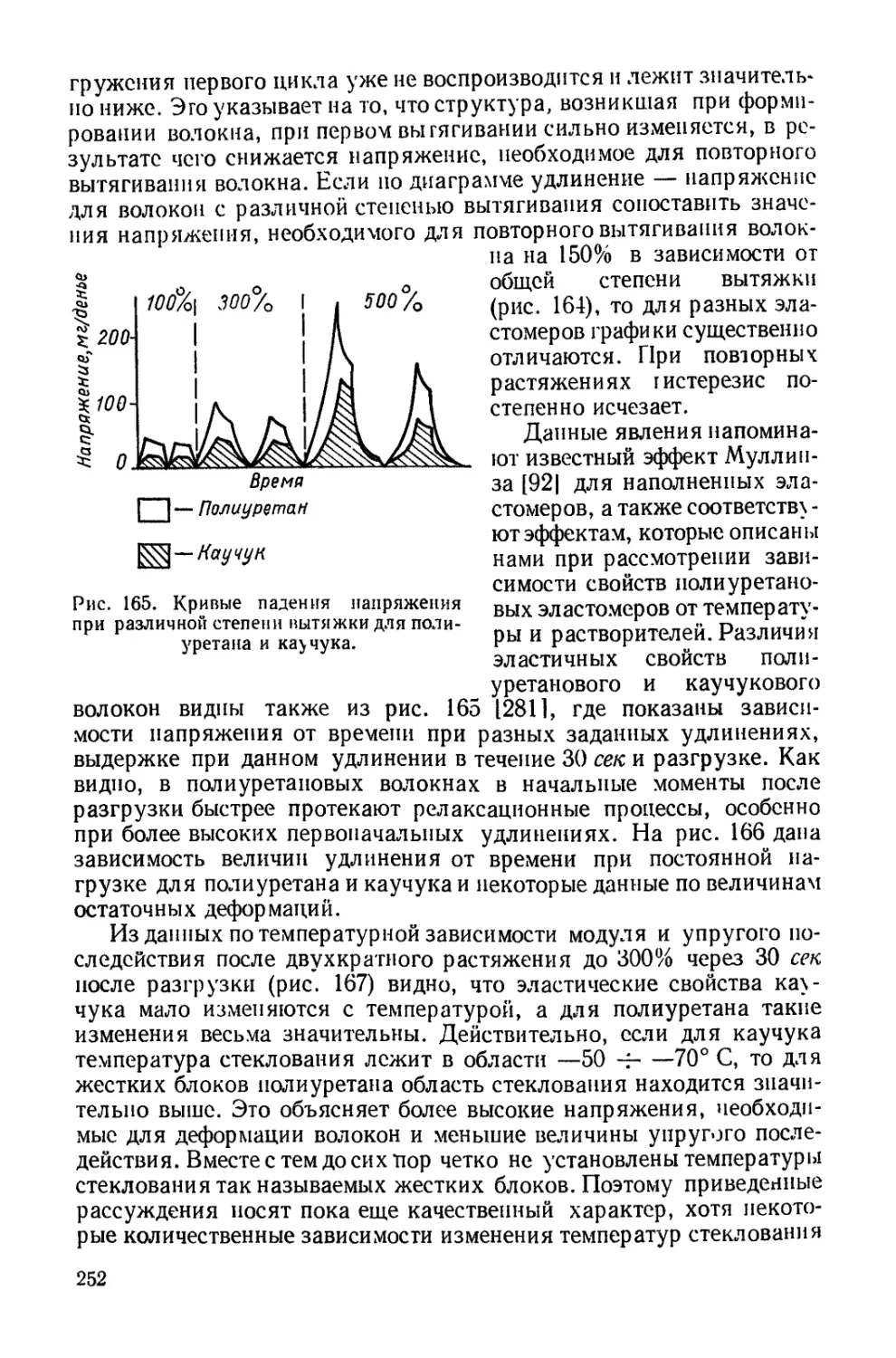
компонентов, взятых для синтеза, может иметь различные конце-
вые реакционноспособные группы. На их реакционности основаны
6
методы удлинения цепей или получения блок-сополимеров. Так,
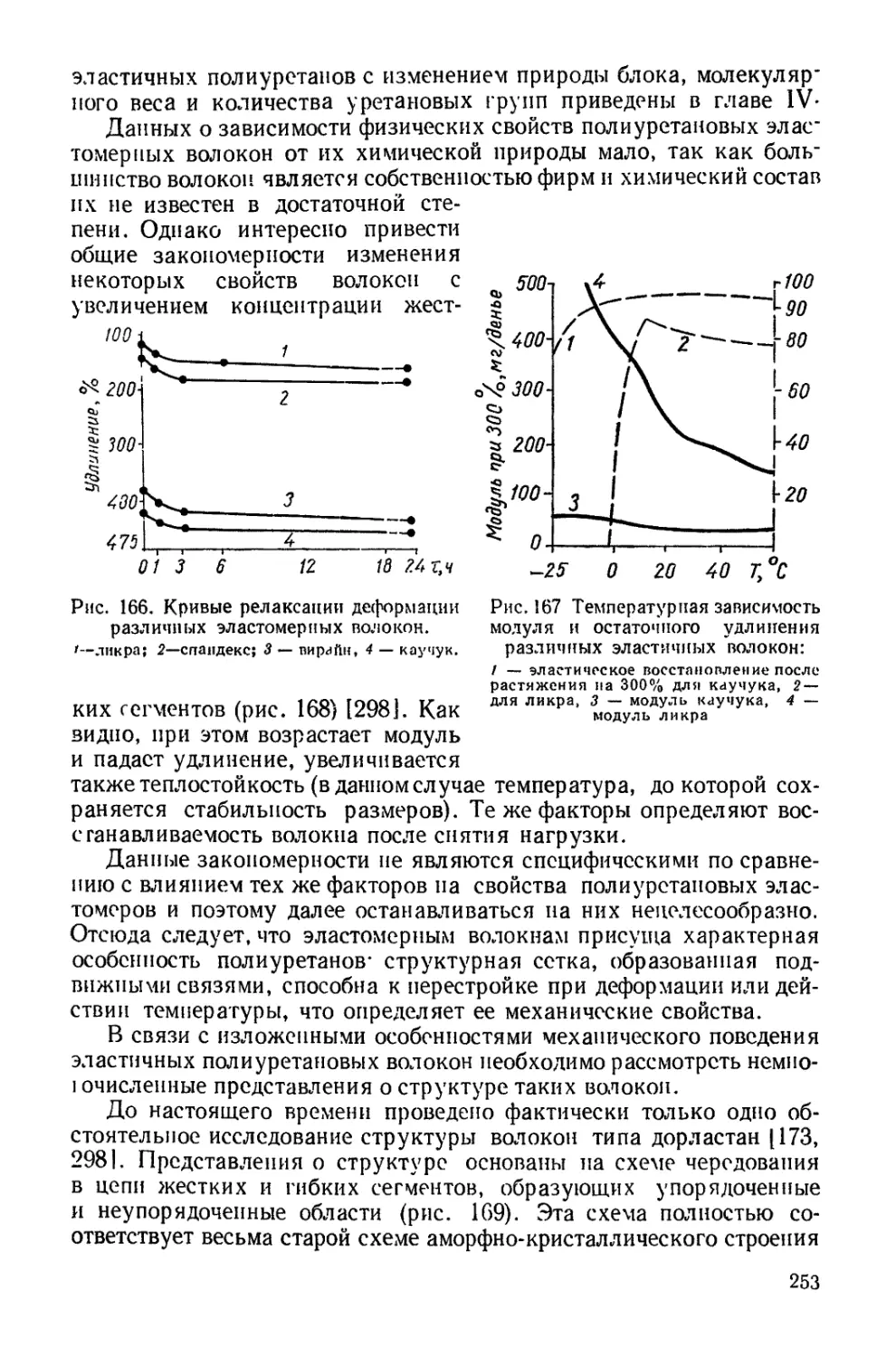
при взаимодействии двух молекул полиуретанов, полученных при
избытке диизоцианата и имеющих концевые изоцианатные группы,
с водой происходит удлинение цепи и возникновение мочевинной
связи
2OCN—NCO + НОН -> OCN—NH-CO—NH-—NCO СО,.
Аналогичное удлинение цепей с образованием мочевинной груп-
пировки происходит при взаимодействии такого полиуретана с диа-
минами
OCN—NCO + H2NRNH2 + OCN—NCO -> OCN—NHCONH-R-
—NHCONH—NCO.
Так получают высокомолекулярные полиуретаны, в основной
цепи которых чередуются уретановые и мочевинные группировки.
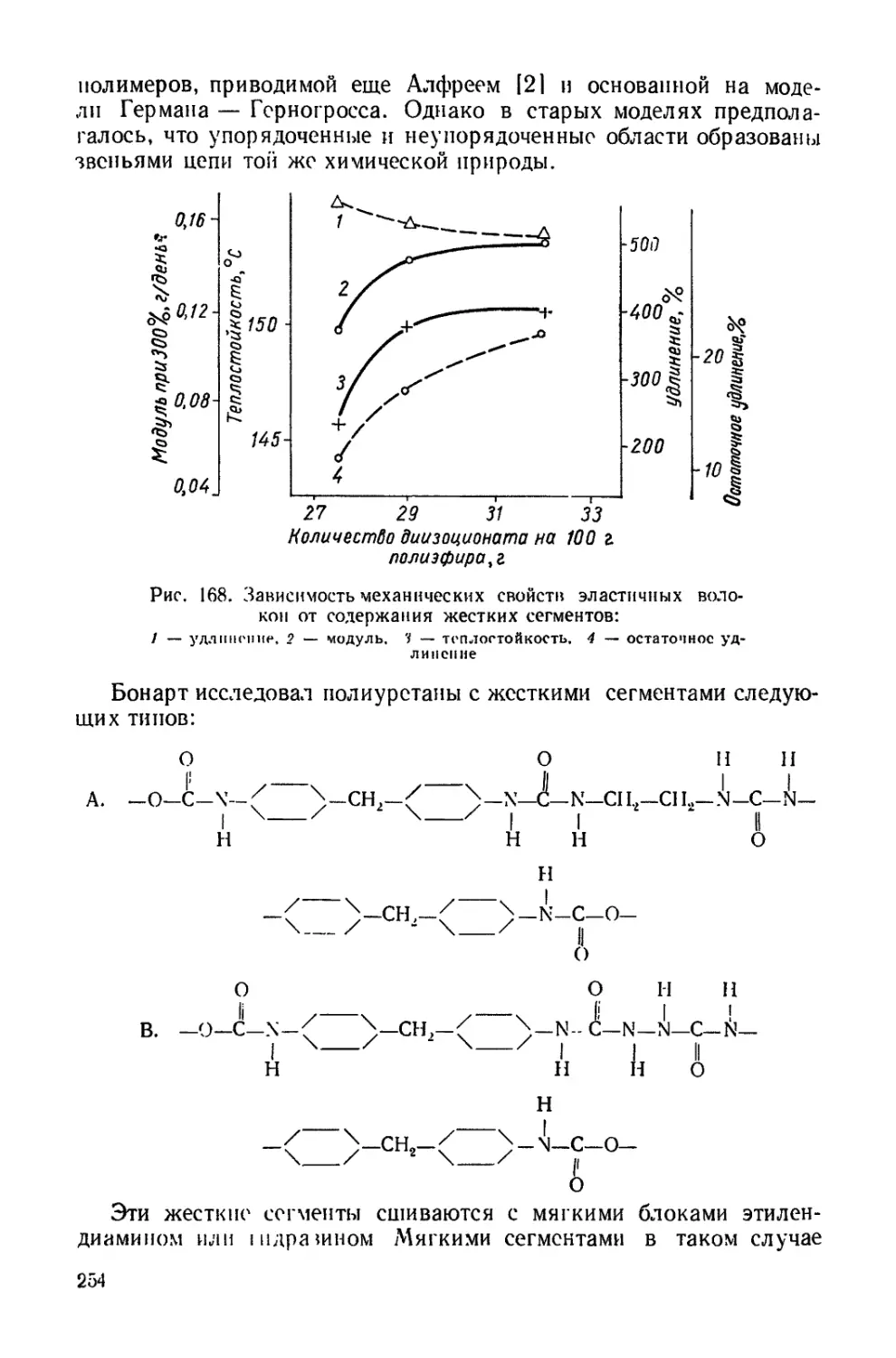
При получении конкретных полиуретановых материалов, особен-
но трехмерного строения, в основной цепи полимера могут присут-
ствовать иные типы связей. Эго объясняется тем, что при образова-
нии полиуретанов протекают и другие реакции, имеющие техничес-
кое значение
R- \ С = 0+
R'NH, R-NH-CO-NH-R'
R'COOH -> R—NH—СО—R' Ь СО2
НОН RNH-COOH RNH2 -г СО, ->
R-N = С = О
-------R—NH-CO-NH—R.
Первичные продукты присоединения имеют в мочевинных, уре-
тановых, амидных и других группах реакционноспособные атомы
водорода, которые при повышенных температурах взаимодействуют
с изоцианатами с образованием новых группировок
R-NH—СО—OR уретан -> -N-CO—0 R 1 i i СО—NH— :—R' аллофапатиая группа
RNH—CO—NH—R мочевина + R'N = С = 0 R N—СО—NH— —R' , CO-NH— R биуретовая группа
R -NH-CO-R амид -> R N-CO——R' . 1 1 CO-NH—:—R'
ацилмочевинная группа
7
Дальнейшее разнообразие вносят реакции димеризации диизоциа-
натов, приводящие к образованию урстдионового кольца
Таким образом, полиуретаны в отличие от других классов поли-
меров нс являются соединениями, в цепи которых имеется только
один характерный тип связей. В ряде случаев концентрацию урета-
новых связей в полимере можно сопоставить с концентрацией свя-
зей других типов. Несмотря на это, основные характеристики дан-
ного класса полимеров определяются участием изоцианатов в реак-
циях синтеза, и поэтому все полимеры данного вида относятся к
классу полиуретанов.
Для синтеза полиуретанов трехмерного строе ня используются
трифункциональныс соединения, содержащие либо гри гидроксиль-
ные группы (например, глицерин), либо триизоцианаты. Эги методы
широко применяются при получении полимерных материалов на
основе полиуретанов.
При изучении структуры полиуретанов необходимо иметь в ви-
ду кинетические особенности реакции. Их связь со структурой мы
рассмотрим отдельно. Однако следует отметить, что диизоцианаты
в зависимости от их химического строения обладают различной
реакционной способностью — с наименьшей скоростью вступают в
реакцию алифатические диизоцианаты, в то время как ароматичес-
кие, особенно содержащие электроноакцепторные заместители (нит-
ро-, нитрильные, галоидные группы), обладают повышенной реак-
ционностью.
Скорость реакции изоцианатов со спиртами падает при переходе
от первичных ко вторичным гидроксильным группам и от насыщен-
ных к ненасыщенным алифатическим гликолям. Алифатические пер-
вичные и вторичные амины реагируют с изоцианатами практически
мгновенно. Если при температурах 20—80° С в основном проте-
кают реакции диизоцианатов с гликолями, водой и диаминами, то
при температурах выше 100° С имеют место побочные реакции,
ведущие к образованию разветвлений и сшивок. Скорость этих реак-
ций возрастает с ростом температуры в большей степени, чем основ-
ных.
На скорость различных реакций, ведущих к образованию поли-
уретанов, влияют многочисленные катализаторы — органические
основания, диамины, пиперидин, пиперазин, гидроокиси щелочных
металлов, ацетил ацетон аты меди, бериллия и ванадия, нафтенат
свинца и кобальта, трибутилолово и многие другие. Действие ка-
тализаторов распространяется не только на основную реакцию
диизониапатов с гликолями, но и на побочные реакции, связанные
8
с образованием мочевинных, аллофанатных, биуретовых групп, с
полимеризацией изоцианатов в изоциапураты и пр, 13331. Все это
ведет к большому разнообразию продуктов реакции.
Из краткого обзора основных принципов синтеза полиуретанов
видно, что он основан на применении соединений с изоцианатными
группами, отличающимися уникальной реакционностью и способ-
ностью ко многим химическим реакциям. Химия высокомолекуляр-
ных соединений не знает других мономеров, кроме изоцианаюв,
способных к такому большому числу разнообразных химических
реакций. Данная специфика основного мономера определяет много-
образие типов химических связей в цепях и химических превраще-
ний полиуретанов. Это и создает возможность в пределах одною
класса соединений — полиуретанов — получать материалы с самыми
разнообразными свойствами. Более детальные сведения о химии
изоцианатов и механизме образования полиуретанов представлены
в работах 1114, 262, 3331.
ПОЛУЧЕНИЕ ПОЛИУРЕТАНОВЫХ КАУЧУКОВ И РЕЗИН
Уретановые каучуки весьма перспективны в области практичес-
кого использования полиуретанов. Полиуретановые эластомеры
характеризуются уникальными свойствами — исключительно высо-
кой прочностью на раздир и истирание, высокой прочностью и элас-
тичностью.
Полиуретановые эластомеры получают на основе олигомерных
полиэфиров, содержащих концевые гидроксильные группы, и диизо-
цианатов. Термин «олигомер» обозначает низкомолекулярный поли-
мер, получаемый при реакциях радикальной теломеризации или при
поликонденсации, проводимой с большим избытком одного из ком-
понентов. Олигомеры представляют собой вязкие жидкости, и их
молекулы являются основным блоком линейной высокомолекуляр-
ной цепи. Обычный молекулярный вес олигомерных полиэфиров,
применяемых для синтеза полиуретанов, составляет 1000—2000.
Для синтеза полиуретановых эластомеров можно использовать
как простые, так и сложные полиэфиры. В случае простых полиэфи-
ров широко применяют различные сополимеры — окиси этилена и
пропилена, их сополимеры с тетрагидрофураном и пр. Из сложных
полиэфиров наиболее распространены полиэфиры на основании раз-
личных гликолей (этиленгликоля, диэтиленгликоля и пр.) и ади-
пиновой кислоты. Основными диизоцианатами, применяющимися для
синтеза, являются 2.4-толуилендиизоцианат (2,4-ТДИ), 1,5-нафти-
лендиизоцианат, 4,4'-дифенилметандиизоцианат, 1,6-гексаметилеи-
диизоцианат (ГМДИ) и др.
Синтез полиуретановых эластомеров протекает в две стадии
1297J. На первой стадии из 2 молей олигомерного полиэфира и
9
3 молей диизоцианата получают так называемый макродиизоцианат,
или форполимер:
OCN-R—NCO+HO—ОН +OCN—R—NCO+HO—OH+OCN— R—NCO
I
OCN-R—NH-C0-0—OCONH-R—NHCOO—OCONH—R—NCO,
где волнистая линия означает молекулу олигоэфира или олигоурета-
на (макродиизоцианата).
Форполимер — вязкая жидкость или легко размягчающееся твер-
дое тело. Концевые изоцианатные группы позволяют удлинять
цепь с помощью диаминов или гликолей (бутандиола, триэтиленгли-
коля и др.).
При взаимодействии макродиизоцианатов и диаминов вначале
получают линейные полиурета"мочсвины
OCN—NCO + H2N—R—NIL -J- OCN—NCO + H2N-R-NH2
Ф
—OC-MI—NH— СО—IINR—NH-CO—HN—Nil—СО—NH—R—NH—
Избыток макродиизоцианата, не вошедшего в реакцию, вызывает
сшивание цепей. При таком сшивании макродиизоцианат присоеди-
няется но предварительно образовавшимся мочевинным связям,
которые достаточно реакционны по отношению к диизоцианатным
группам. В результате этого в сшитых каучуках появляется еще
один тип связей — биуретовых:
-ОС—HN—NH—СО—NH—R—NH-CO—HN—NH —СО—HN—R—NfI—
4-
NCO
i
NCO
-I-
—ОС—HN"" NH—СО—NH-R—NH—СО—HN—NH—СО—HN—R—NH—
Ф
—ОС—HN—NH—СО—N—R—NH—СО—HN'—NH—СО—HN—R—NH—
I ----------
CO
I
NH
i —биуретовые
j^H связи
I
CO
I 4---------
—ОС—HN—NH—CO—N—R—NH—CO—HN-—NH—CO—HN—R—NH-
Один и тот же макродиизоцианат служит как для построения
цепей, так и для их сшивания, что определяет, в частности, почти
одинаковые размеры ячеек сетки. В этом заключается, очевидно,
одна из причин высокой прочности и малой истираемости полиуре-
танов.
10
Эластомеры с аналогичными свойствами образуются также и в
том случае, когда вместо диамина для сшивания берется вода. При
отщеплении СО2 вначале образуется линейная полиуретанмочевина,
которая, взаимодействуя с избытком макродиизоцианата, приво-
дит к удлинению цепей и их сшиванию с образованием биуретовых
связей.
При получении высокомолекулярных продуктов для сшивания
может быть также использована реакция взаимодействия макро-
диизоцианатов с двухатомными спиртами, например бугандиолом,
OCN—NCO 4- НО—R—ОН OCN—\СО + HO-R-OH
—-ОС—HN—NH—СО—О—R—О—СО— СО—О—R—О—
+
NCO
NCO
+
—OC-HN—NH—СО—О—R—О—СО—СО—O-R-O-
Ф
—N—СО—О—R—
io"
I
NH
*
I — аллофанатные
NH связи
io
—.4—СО—О—R—
Гликоля в данном случае берется несколько меньше, чем тре-
буется для полного насыщения диизоцианата. Избыток макродиизо-
цианата сшивает полимерные цепи за счет взаимодействия с атомами
водорода уретановых групп. При этом возникает еще один тип свя-
зей в цепях — аллофанатный.
Сочетание рассмотренных методов дает возможность широко
варьировать структуры сетки каучуков и, таким образом, изме-
нять их физико-механические характеристики, особенно если учесть,
что в качестве переменных, определяющих свойства, можно выби-
рать олигомеры различного химического строения и молекуляр-
ного веса и различные диизоцианаты.
Существует также и другая возможность получения сшитых по-
лиуретановых эластомеров по одностадийному методу. Она заклю-
чается в том, что в олигоэфир при его синтезе вводится небольшое
количество трехатомного спирта, например триметилолпропана. При
этом образуется разветвленный полиэфир. В ходе взаимодействия
его с диизоцианатами одновременно с ростом цепи происходит
11
сшивание по свободным гидроксильным группам введенного треха-
томного спирта.
Из изложенного видно, что химия полиуретановых эластоме-
ров имеет практически неограниченные возможности изменения хи-
мической структуры полиуретановых эластомеров. Общим для всех,
однако, остается наличие четырех основных типов связен (урета-
новых, мочевинных, биуретовых и аллофанатных) и чередование
в цепи блоков олигомерной компоненты — олигоэфиров или других
гидроксилсодержащих соединений — и блоков, вводимых ди изо-
цианатной компонентой.
ПОЛИУРЕТАНОВЫЕ ВОЛОКНА
Волокна на основе полиуретанов разделяются на два основных
класса. К первому относятся волокна, аналогичные другим термо-
пластичным волокнам типа полиамидных, полиэфирных и др. Это
волокна на основе линейных термопластичных кристаллизующихся
полиуретанов. Химия получения таких волокон заключается в син-
тезе линейных продуктов при взаимодействии дигидроксилсодер-
жащих короткоцепных соединений (этиленгликоля, ди-, триэтилеп-
гликоля, 1,4-бутапдиола) с различными диизоцианатами. Наиболее
распространенный тип полиуретановых волокон — перлон U —по-
лучается при взаимодействии бутандиол а-1,4 с гексаметилендиизо-
цианатом. Варьируя компоненты, применяемые для синтеза
волокнообразующих полимеров, можно изменять температуры
плавления полимеров и их физико-механические характеристики
1231.
Ко второму классу полиуретановых волокон относятся эластич-
ные волокна, обладающие линейной или трехмерной пространствен-
ной структурой Эта группа волокон уникальна по своим свойствам
и не имеет аналогов среди других классов полимерных соединений.
Эластичные волокна, в состав которых входит не менее 85% поли-
уретанов, впервые были выпущены в США в конце 1960 г. Благода-
ря своему химическому строению они обладают высоким разрывным
удлинением (500—800%), низким модулем упругрсти и высокой
эластической деформацией.
Химизм образования эластичных трехмерных полиуретановых
волокон и трехмерных полиуретановых резин аналогичен. В на-
стоящее время существуют различные виды полиуретановых эластич-
ных волокон, известные под разными фирменными наименованиями
(ликра, спандекс, внрайп) [481. Для их получения применяются
различные полиэфиры с концевыми гидроксильными группами (прос-
тыми и сложными) и диизоцианаты — толуилендиизоциапат, ди-
фенилметан-4,4'-диизоцианат, 1,5-нафгалиндиизоцианат и пр., т. е.
соединения, используемые также при получении полиуретановых
12
резин и каучуков. Отличия в способах получения носят не столько
химический, сколько технологический характер и связаны с особы-
ми условиями формования волокна, которое может быть осуществ-
лено мокрым и сухим способом. Высокоэластичные полиуретановые
волокна в зависимости от типа реакций, используемых для их по-
лучения, имеют трехмерную и линейную структуру. Принципиаль-
ное отличие этих волокон, как и полиуретановых эластомеров, от
других каучуков заключается в большой роли физических узлов
в сетке.
Основной особенностью строения полиуретановых эластомерных
волокон является чередование блоков различной химической при-
роды, что позволяет придать волокну необходимые текстильные
и механические свойства. Подробно вопросы химии эластомер-
ных полиуретановых волокон рассмотрены в работах [48, 298,
303].
Образование водородных связей между группами СО и NH сосед-
них цепей в некоторой степени заменяет химическое сшивание. Жест-
кие сегменты, содержащие уретановые связи, являются узлами
трехмерной физической сетки, эластические свойства которой опре-
деляются чистыми полиэфирными блоками.
ПОЛИУРЕТАНОВЫЕ ПОКРЫТИЯ
Путем подбора компонентов, применяемых для синтеза полиуре-
танов, им можно придать самые разнообразные свойства. Из трех-
мерных полиуретанов, в которых блоки гидроксилсодержащей ком-
поненты достаточно жесткие по сравнению с таковыми в каучуках,
можно получить трехмерные полиуретановые пленки и покрытия.
Наличие большого числа полярных групп обеспечивает высокую
адгезию покрытий к поверхностям, а специфические свойства поли-
уретанов — высокие физико-механические свойства покрытий. Ос-
новной принцип получения покрытий основан на использовании по-
лифункциональных соединений, обеспечивающих образование трех-
мерной пространственной сетки. В качестве полифункциональных
соединений применяются трех- и четырехатомные спирты, разветвлен-
ные полиэфиры и их комбинации. При образовании полиуретановых
покрытий имеют место практически те же реакции, что при получе-
нии резин, т. е. в продуктах реакции имеются кроме уретановых так-
же мочевинные, биуретовые и аллофанатные связи. С химической
точки зрения весьма важны реакции с водой, так как отвержде-
ние лаковых покрытий на основе полиуретанов обычно происхо-
дит в условиях контакта с влагой воздуха. Так как полифункцио-
нальность исходных соединений является необходимым условием
получения трехмерных покрытий, то в качестве одного из компо-
13
центов .реакции применяется аддукт толуилендиизоцианата с триме-
тилол пропаном и диэтиленгликолем
NCO
CH.OCONH—^—СН3
/ =Лчсо
C2H6 -С^—CH2OCONH-^ сн8
\ ==/NCO
ch2ocomi—^>—сн3
и
OCN
СН3-^ ^-NH—COO—Cl I2—сн2—о—сн2—сн2—
“ NCO
—О—СО—NH—^>—СН3
В настоящее время известно большое число самых разнообраз-
ных композиций, применяемых для получения полиуретановых по-
крытий, позволяющих получать покрытия, отвечающие самым раз-
личным требованиям. Однако в основе всех методов лежат одни и
те же химические принципы получения трехмерных структур.
Получение полиуретановых клеящих композиций практически
ничем не отличается от принципов получения покрытий. В каждом
отдельном случае химическая природа компонентов и условия от-
верждения композиции подбирают так, чтобы обеспечить необхо-
димую жизнеспособность композиций, температурные режимы от-
верждения, скорости процессов и т. п.
Из изложенного видно, что разнообразие химического строения
полиуретанов определяется применением для их синтеза соединений
самых различных классов, начиная с низкомолекулярных гликолей
и кончая многочисленными олигоэфирами и другими олигомерными
соединениями с концевыми гидроксильными группами.
Наличие в цепях различных типов связей не позволяет уже счи-
тать полиуретаны единым классом полимеров, как, например, по-
лиамиды или полиакрилаты. Действительно, единственным общим
признаком, дающим основание отнести полимеры к полиуретанам,
во многих случаях может служить только присутствие уретановой
группировки.
Таким образом, особенности химической структуры полиуре-
танов и многообразие их свойств определяются тем, что для синтеза
применяются исходные соединения, относящиеся к различным клас-
сам. В этом смысле нет проблемы полиуретанов как таковых, но
существует проблема исходных соединений для синтеза полимеров,
содержащих уретановые группы.
Глава II. ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ
ПОЛИУРЕТАНОВ
При рассмотрении особенностей физической и молекулярной
структур полиуретанов прежде всего необходимо остановиться на
гибкости молекулярных цепей. Гибкость цепей как характеристика,
присущая только высокомолекулярным соединениям, определяет ос-
новные физико-механические и физические свойства полимеров.
Сведения о гибкости можно получить либо при исследовании свойств
разбавленных растворов полимеров, либо из исследований сорбции
паров полимерами, либо из данных по механическим и релаксацион-
ным свойствам.
СВОЙСТВА РАЗБАВЛЕННЫХ РАСТВОРОВ ПОЛИУРЕТАНОВ
Специфической чертой строения полиуретанов, обусловленной
химизмом их образования, является чередование в полимерной
цепочке участков различной химической природы — остатков мо-
лекул гликоля или полиэфира и диизоцианата. Таким образом, в
линейной полимерной цепи находятся гетеросвязи различной при-
роды, кроме углерод-углеродных, связи типа С—О—С в гликоле
и простых полиэфирах, связи С—О— в сложных полиэфирах, связи
II
О
—NH—СО—О— и пр. Разнообразие гетеросвязей должно существен-
ным образом сказаться на гибкости полиуретановой цепи. Гибкость
цепи определяет основные физико-химические и физико-механические
свойства полимеров. Известно, что причиной гибкости цепи являет-
ся наличие известной свободы вращения вокруг валентных связей
атомов, образующих молекулы. Это вращение в большей или мень-
шей степени заторможено как внутримолекулярными, так и межмо-
лекулярными силами. Потенциальный барьер вращения зависит
от типа валентной связи и природы заместителей у соответствующих
атомов полимерной цепи. Рассмотрение данных по внутреннему
вращению в органических молекулах [16] показывает, в частности,
что внутреннее вращение вокруг связи С—О облегчено по сравнению
с вращением вокруг связи С—С. Поэтому можно полагать, что поли-
мерные цепи с гетеросвязями типа простых полиэфиров будут об-
ладать повышенной гибкостью. С другой стороны, наличие сильно
взаимодействующих полярных групп в цепи должно было бы
15
вызвать увеличение барьеров внутреннего вращения и ожестчение
цепи. Сочетание в одной полимерной молекуле различных типов
связей определяет сложный характер зависимости гибкости цепей
полиуретанов от их химической природы.
На основании данных о размерах и геометрической форме макро-
молекул в растворах может быть определена термодинамическая
(равновесная) гибкость полимерной цепи, которая характеризуется
отношением невозмущенных взаимодействием с растворителем раз-
меров цепей (Ло)'/г к размерам, которые имел бы полимерный клубок
при полностью свободном вращении всех звеньев (Ьсв)2. Для опреде-
ления этой величины необходимо знать размеры цепи в идеальном
(невзаимодействующем) растворителе. Величина (й?в)7* обычно оп-
ределяется расчетным
ных связей, длиной /
свободным вращением,
путем и для цепи, состоящей из п валент-
с постоянным валентным углом л — 0 и
72 to 1 ! COS0
ЙСв = tlP i.
1 — cos О
Непосредственное определение этих величин для полиуретанов зат-
руднено по двум причинам. Как правило, линейные полиуретаны
сравнительно низкомолекулярные полимеры, что ограничивает воз-
можность применения метода светорассеяния для определения раз-
меров цепи. Они обладают ограниченной растворимостью во многих
органических растворителях, которая уменьшается при повышении
молекулярного веса, что создает трудности при подборе идеального
растворителя.
С другой стороны, размер цепи при условии свободного враще-
ния не может быть определен из уравнения, учитывающего только
С—С-связи в главной цепи. Расчет размеров цепей, в которых чере-
дуются два или значительно больше типов связей, как в полиуре-
танах, является более сложным [16]. Отсутствие в ряде случаев
точных данных о порядке чередования и количестве гетеросвязей
в цепи делает такие расчеты трудно осуществимыми. Поэтому для
получения сведений о гибкости полиуретановых цепей необходимо
применять косвенные методы.
Рассмотрим этот вопрос на примере исследования форполимеров
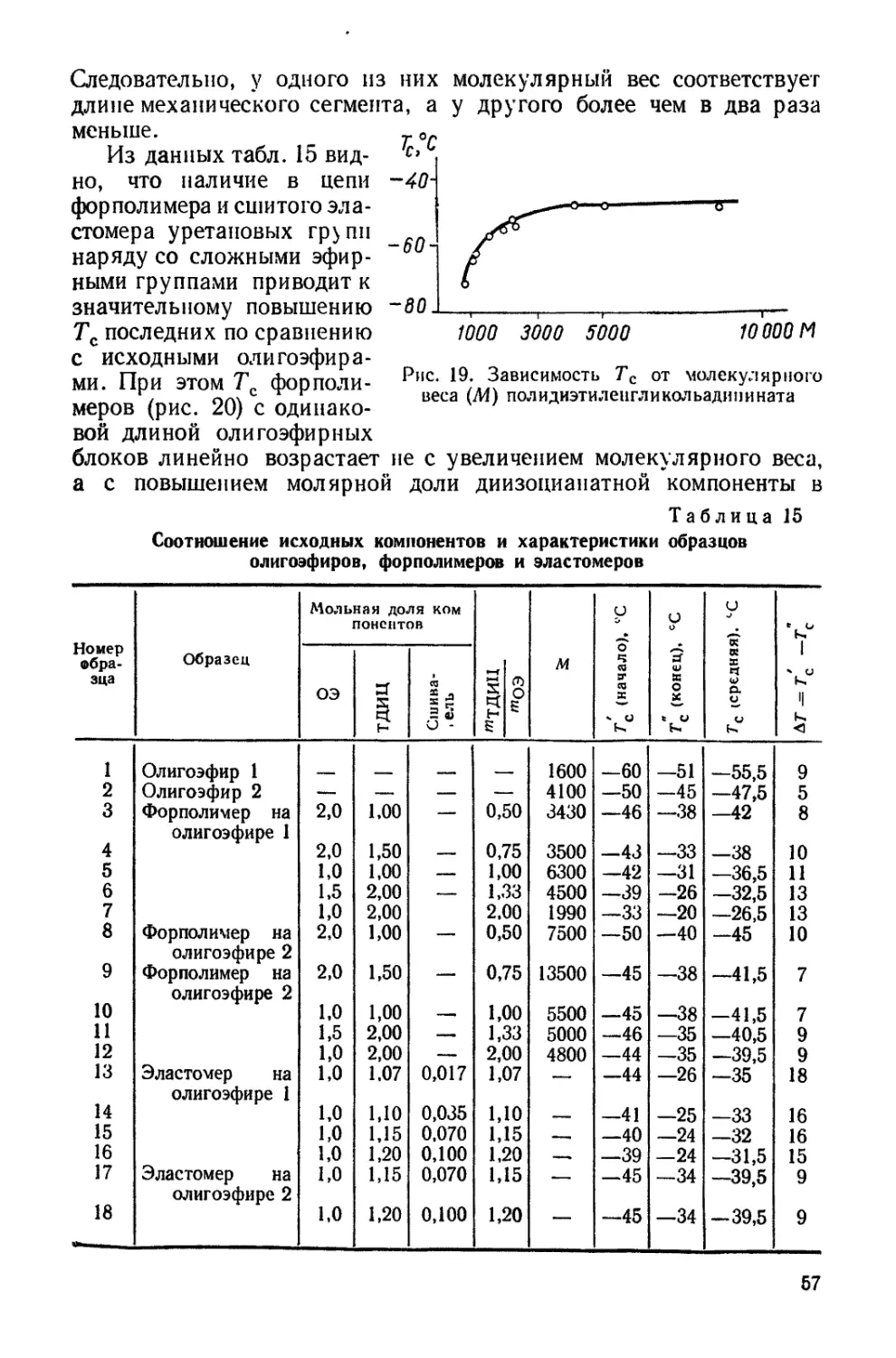
на основе полидиэтиленгликольадипината (молекулярный вес 4100)
и толуилендиизоцианата, а также полиуретана на основе бутан-
диола и толуилендиизоцианата [96]. х
Для низкомолекулярных форполимеров непосредственно опре-
делить величину (й2)*'8 нельзя, поэтому был применен графический ме-
тод, предложенный Штокмайером и Фиксманом [3231. Они показа-
ли, что известное уравнение Флори, связывающее характеристичес-
кую вязкость с молекулярным весом и размерами цепи,
fo] = ф №)*'чм
16
легко преобразуется:
[Л] = ф (hl/Mf’M'1’ + 0.51ФВЛ1,
В =Ъ2 (1 — 2x1)/V17V^.
где v — парциальный удельный объем полимера в растворе, V\ —
мольный объем растворителя, Xi—термодинамический параметр
взаимодействия.
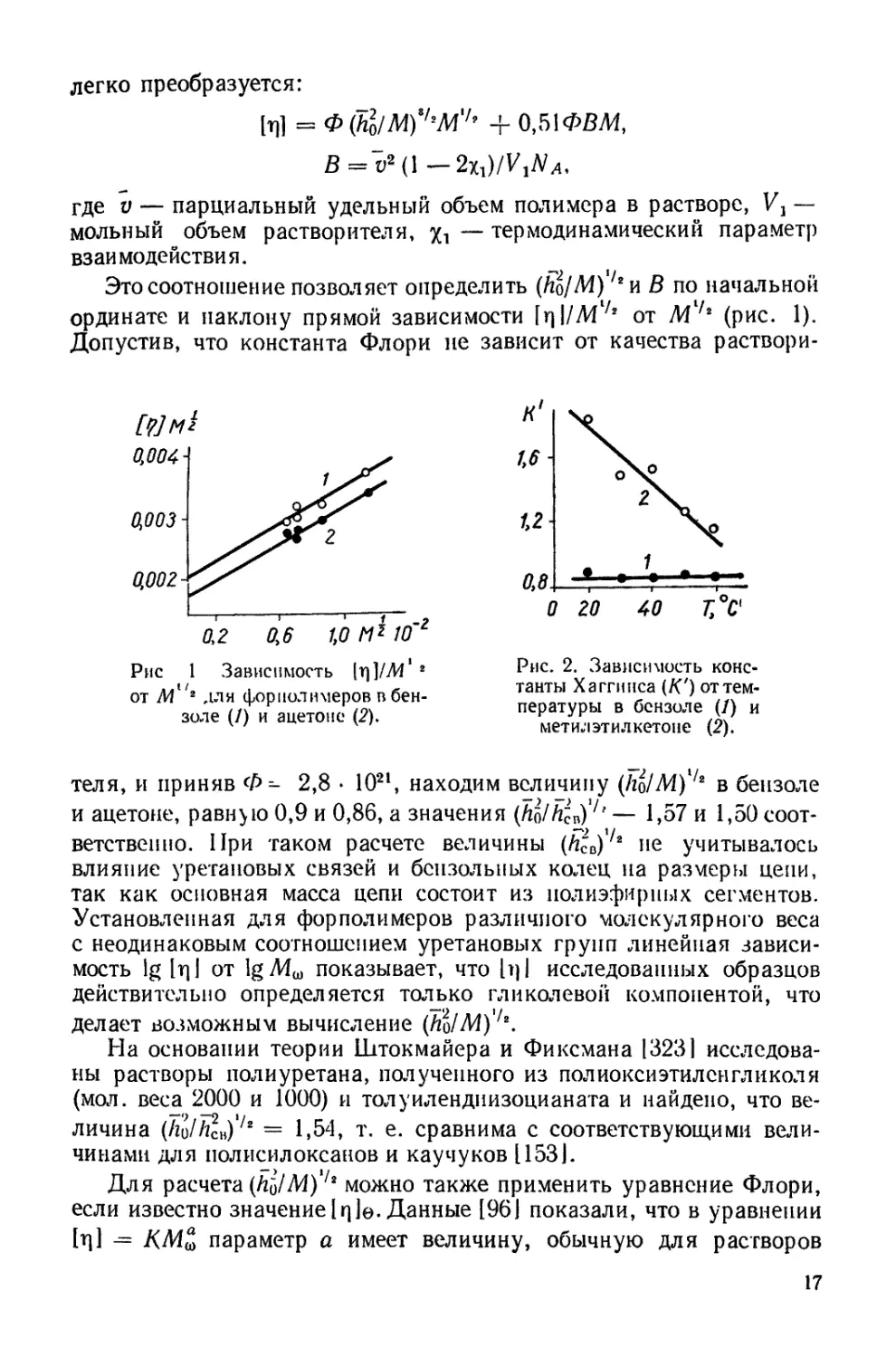
Это соотношение позволяет определить (йо/Л4),/2и В по начальной
ординате и наклону прямой зависимости [rj \/M'/s от (рис. 1).
Допустив, что константа Флори не зависит от качества раствори-
Рис 1 Зависимость (т|]/Л11 2
от М'‘2 для форполимеров в бен-
золе (/) и ацетоне (2).
Рис. 2. Зависимость конс-
танты Хаггинса (/С) от тем-
пературы в бензоле (/) и
метил этил кетоне (2).
теля, и приняв Ф- 2,8 • 1021, находим величину (Iiq/M)4* в бензоле
и ацетоне, равную 0,9 и 0,86, а значения (Ло/йсв)7' — 1,57 и 1,50 соот-
ветственно. При таком расчете величины (йсв)/2 не учитывалось
влияние уретановых связей и бензольных колец на размеры цени,
так как основная масса цепи состоит из полиэфирных сегментов.
Установленная для форполимеров различного молекулярного веса
с неодинаковым соотношением уретановых групп линейная зависи-
мость 1g [т| 1 от 1g Мц, показывает, что h) I исследованных образцов
действительно определяется только гликолевой компонентой, что
делает возможным вычисление (Ло/7И)’/г.
На основании теории Штокмайера и Фиксмана [323] исследова-
ны растворы полиуретана, полученного из полиоксиэтиленгликоля
(мол. веса 2000 и 1000) и толуилендиизоцианата и найдено, что ве-
личина (Аи//7св) /г = 1,54, т. е. сравнима с соответствующими вели-
чинами для полисилоксанов и каучуков [153].
Для расчета (Лб/А4)‘/2 можно также применить уравнение Флори,
если известно значение IqJe- Данные [96] показали, что в уравнении
[т}1 параметр а имеет величину, обычную для растворов
17
гибких макромолекул в хороших растворителях (0,5 < а < 0,8)
2371. Вычисленная по наклону прямых 1г\]1М',‘= f и извест-
ным значениям v - 0,900 (для бензола) и 0,887 смЧг (для ацетона)
величина составляет 0,163 и 0,228 соответственно. Так как [цI ~
~ M4t 1 Je/! [2111 (е — параметр, связанный с объемными эффек-
тами), то степень набухания а можно определить по уравнению
8 = (а2 — 1) (5а2 — 3).
В нашем случае для обоих растворителей е — 0,133, что при-
водит к а 1,33. Зная а. можно рассчитать [257] [г)]е и, следова-
тельно, (ho/M)l/l из отношений
« = (tnl/lnle)2’43,
w = ЛГ'-.
Вычисления дают — 0,88 и 0,84, что хоро шо согласуется
величиной (Ло/Лсв)7*.
Таким образом, приведенное значение (йо/йсв)1'* показывает, что
гибкость цепочек низкомолекулярных форполимеров превосходит
гибкость винильных полимеров и сравнима с гибкостью полисилок-
санов и каучуков [257].
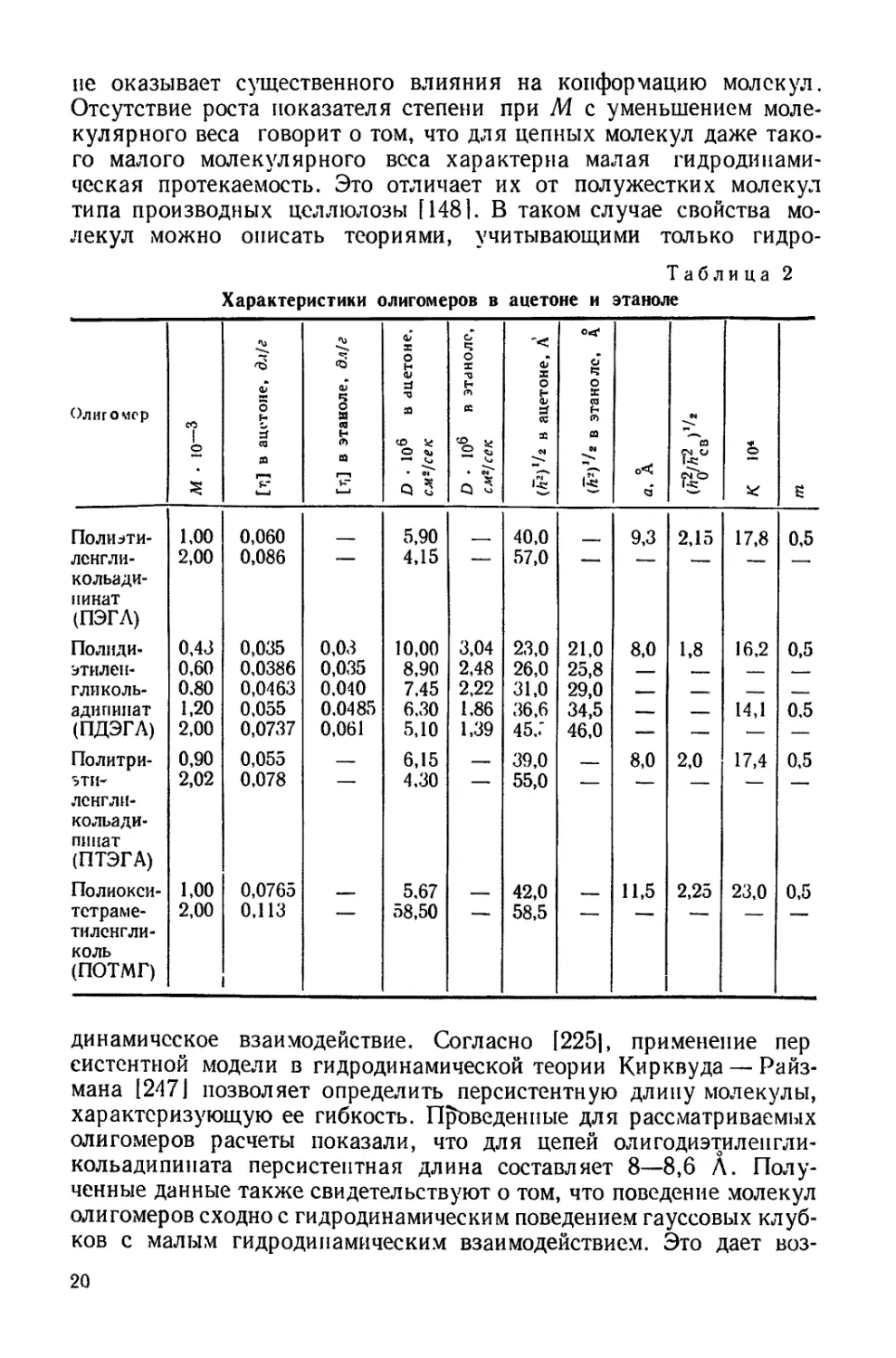
Для форполимера с Мю — 1,35 • 104 также измерена темпера-
турная зависимость характеристической вязкости в бензоле и ме-
тил этилкетоне. Как видно из рис. 2, характеристическая вязкость
форполимера в бензоле падает с повышением температуры. Значе-
ние температурного коэффициента = — 1,09 • 10~3 ха-
рактерно для полимеров в хороших растворителях, где термодина-
мическое взаимодействие полимера с растворителем незначительно
меняется с температурой, а основное влияние на вязкость оказывает
изменение близкодействия. В метил этил кетоне [ц 1 растет с повы-
шением температуры и — 4,16 • 10-3. Очевидно, что метил-
этилкетои является плохим растворителем. Обращает на себя вни-
мание также и большая величина константы Хаггинса К (рис. 2),
которая в бензоле в исследованном температурном интервале прак-
тически не меняется, а в мети лэти л кетоне резко падает с ростом
температуры.
Переход к линейным полиуретанам, в которых гликолевая ком-
понента имеет малый размер, должен, очевидно, привести к повы-
шению жесткости цепи. Для линейных полиуретанов на основе
бутандиола и толуилеидиизоцианата (мол. вес 0,2—0,96 -104) этим же
методом определены значения равнаресной гибкости цепей. Найдено,
что (Л^/Л4)72 -- 1,03, а соответствующее ей значение (hllh^) — 2,0.
18
Это указывает на то, что повышение концентрации уретановых
групп в цепи приводит к значительному снижению ее гибкости,
которая в данном случае сравнима с гибкостью цепей обычных ви-
ниловых полимеров.
Приведенные данные действительно позволяют считать, что гиб-
кость полиуретановых цепей характеризуется в основном гибкостью
олигомерной полиэфирной компоненты. В связи с этим встает вопрос
о гибкости собственно олигомерных молекул. Изучение поведения
олигомеров в растворах является весьма важным, так как дает
возможность судить также о свойствах и гибкости цепей тех трехмер-
ных полимеров, которые получаются на их основе. С помощью гид-
родинамических характеристик олигомеров можно определить их
размер, конфигурацию и взаимодействие с растворителем.
Таблица 1
Гидродинамические характеристики олигомеров
Ь]. йл/г D 1G6. см21сек (А2)1/, . А W/< ю’°, град
м X X Ацетон Г7
Ацетон Этанол О < о «5 й о о X я й Этано
430 0,0360 0,0300 10,0 3,04 23,0 21,0 2,78 2,89
600 0,0386 0,0350 8,9 2,48 26,0 25,8 2,85 2,78
800 0,0463 0,0400 7,45 2,22 31,0 29,0 2,80 2,85
1200 0,0550 0,0485 6,3 1,86 36,6 34,5 2,86 2,92
2000 0,0737 0,0610 5,1 1.39 45,5 46,0 3,03 2,80
Примечание. А9 = (АЦг,]) ^3* где т0 — вязкость рас гвори геля.
Работ по исследованию диффузии и вязкости олигомеров очень
мало, а сведения о свойствах олигомеров сложных эфиров, представ-
ляющих наибольший интерес для синтеза полиуретанов, отсутст-
вуют. В связи с этим целесообразно остановиться на проведенных
Нестеровым и Липатовым исследованиях диффузии и вязкости оли-
гомеров на основе диэтиленгликоля и адипиновой кислоты с моле-
кулярным весом от 2000 до 430 [971. Кривые зависимости коэффи-
циентов диффузии и характеристической вязкости от молекуляр-
ного веса имеют наклон, близкий к 0,5. Соответствующие данные
представлены в табл. 1, из которой видно, что абсолютные значе-
ния вязкости в ацетоне несколько больше, чем в этаноле. Это, ве-
роятно, обусловлено различным влиянием растворителей на ске-
летную жесткость (близкодействие) цепей, а не на объемные эффекты
(дальнодействие). Значения а, близкие к 0,5, указывают на то, что
параметр набухания а приближается к единице. Вероятно, при
низком молекулярном весе взаимодействие полимер — растворитель
19
не оказывает существенного влияния на конформацию молекул.
Отсутствие роста показателя степени при М с уменьшением моле-
кулярного веса говорит о том, что для цепных молекул даже тако-
го малого молекулярного веса характерна малая гидродинами-
ческая протекаемость. Это отличает их от полужестких молекул
типа производных целлюлозы [1481. В таком случае свойства мо-
лекул можно описать теориями, учитывающими только гидро-
Таблица 2
Характеристики олигомеров в ацетоне и этаноле
Олигомер со 1 О [т;] в ацетоне, дл/г [Т]] в этаноле, дл/г D • 10® в ацетоне, см-/сек D • 10® в этаноле, смг/сск (й2)1/* в ацетоне, Л (Л2)1/» в этаноле, 4 <3 о * g
Полиэти- 1,00 0,060 5,90 40,0 9,3 2,15 17,8 0,5
ленгли- кольади- нинат (ПЭГА) 2,00 0,086 4,15 57,0 — —
Полиди- 0,43 0,035 0,03 10,00 3,04 23,0 21,0 8,0 1,8 16,2 0,5
этил ен- 0,60 0,0386 0,035 8,90 2,48 26,0 25,8 — -
гликоль- 0.80 0,0463 0,040 7,45 2,22 31,0 29,0 — — : —
адипипат 1,20 0,055 0,0485 6,30 1.86 36,6 34,5 — — 14,1 0,5
(ПДЭГА) 2,00 0,0737 0,061 5,10 1,39 45.: 46,0 — — — —
Политри- 0,90 0,055 — 6,15 — 39,0 — 8,0 2,0 17,4 0,5
эти- ленгли- кольади- пииат (ПТЭГА) 2,02 0,078 4,30 55,0
Полиокси- 1,00 0,0765 5.67 — 42,0 11,5 2,25 23,0 0,5
тстраме- тиленгли- коль (ПОТМГ) 2,00 0,113 1 58,50 58,5
динамическое взаимодействие. Согласно [225], применение пер
систентной модели в гидродинамической теории Кирквуда — Райз-
мана [2471 позволяет определить персистентную длину молекулы,
характеризующую ее гибкость. Проведенные для рассматриваемых
олигомеров расчеты показали, что для цепей олигодиэтиленгли-
кольадипипата персистентная длина составляет 8—8,6 А. Полу-
ченные данные также свидетельствуют о том, что поведение молекул
олигомеров сходно с гидродинамическим поведением гауссовых клуб-
ков с малым гидродинамическим взаимодействием. Это дает воз-
20
и этаноле и 1,
2.6 3,0 3,4 3,8 4,2 4,6 LgM
Рис. 3 Зависимость 1g [т] ] от lg М для рас-
творов олигомеров окситетраметиленгликоля
в ацетоне (/) и метил этил кето не (2); ПЭГА в
ацетоне (5), ПТЭГА в ацетоне (4); ПДЭГА в
ацетоне (5) и эталоне (6); полиуретана на
основе диэтиленгликоля и 2,4-ТДИ в ацето-
не (7) и бензоле (5); полиуретана на основе
ПОПГ и 2,4-ТДИ в бензоле (9)н метаноле (10),
ПОПГ (11) и полиуретана (12) в 6-раство-
рителе.
можность использовать соотношения, связывающие коэффициент
диффузии и размеры диффундирующих частиц
для определения размеров молекул олигомеров. Вычисленные зна-
чения (/?)1/2 для олигомеров диэтиленгликольадипината представ-
лены в табл. 1. Величина (Я2)’'2 в этаноле немного меньше, чем в
ацетоне, что согласуется с различиями вязкости. Отношение (йо/йсв)7*
равное соответственно в ан
значениям, получаемым для
обычных высокомолекуляр-
ных каучуков.
Таким образом, иссле-
дование диффузии и харак-
теристической вязкости оли-
гомеров показали, что гид-
родинамические характе-
ристики этих молекул
хорошо согласуются с та-
ковыми для модели непро-
текаемых гауссовых клуб-
ков, хотя это и не гауссовы
клубки в их обычном виде
из-за недостаточного числа
звеньев в молекулярной
цепи.
В работе [98] предпри-
нято более детальное иссле-
дование свойств различных
олигомеров и низкомолеку-
лярных полиуретанов, по-
лученных на их основе, методами диффузии и характеристической
вязкости. Были изучены олигомеры этиленгликолъадипината, диэти-
ленгликольадипината, триэтиленгликольадипината, окситетра-
метилснгликоля и полиуретаны на основе этих олигомеров и 2,4-то-
луилендиизоцианата. В табл. 2 п 3 представлены характеристики
исследованных объектов и полученные результаты. Из зависи-
мости 1g [i|J от lgМ (рис. 3) видно, что наклоны прямых для всех
олигомеров равны 0,5. Для сравнения на этом же рисунке приве-
дена зависимость 1g [т] I — lg М для полиуретана на основе диэтилен-
гликольадипината н 2,4-ТДИ.
Значения характеристической вязкости олигомеров зависят от
природы растворителя. Это, вероятно, обусловлено влиянием рас-
творителя на скелетную жесткость молекулярной цепи, а не объем-
ными эффектами, поскольку значение экспоненты в уравнении типа
21
fyj = KMa (равное 0,5) показывает, что параметр термодинамичес-
кого набухания а — 1, и свидетельствует о малой гидродинами-
ческой протекаемости всех исследованных молекул. Из расчетов
персистентной длины (981 следует, что гибкость цепи олигомеров
имеет тенденцию к росту с увеличением числа кислородных атомов
в цепи в соответствии с определением размеров цепей по коэффи-
циентам диффузии (см. табл. 2 и 3).
Таблица 3
Характеристики растворов полиуретанов в ацетоне и бензоле
Полимер М олшомсра М полимера [х] в ацетоне, дл/г [т(] в бензоле, дл/г D 10е' в ацетоне, см2! се к о< % NCO . ОН К * 104 т
ПЭГА-1 ТЛИЦ 1000 4700 0,243 2 70 88,0 1,1
2000 4400 0,234 — 2,80 84,5 1,1 — —
ПДЭГА 4000 13 500 0,390 0,440 — — 0,75 5,05 0,7
-1-ТДИЦ 4000 7500 0,260 0,280 0,5 (ацетон)
800 6600 0,234 — 2,44 98,0 1,1 — —
1700 5200 0,200 — 2,80 82,5 1,1 5,5 0,7
4000 5100 0,190 0,230 1,1 (бензол)
4000 5000 0,186 0,218 — 1,33 — —
4000 4350 0,185 0,195 — — 2,0 —
ПТЭГА + 900 5000 0.230 — 2,90 82,0 1,1 — —
+ ТДИЦ 2020 4700 0,200 — 3,32 70,0 1,1 — —
потмг 2000 15 600 0,630 .— 1,31 181,0 1.1 — —
-I- тдиц 1000 6700 0,380 — 2,02 118,0 1,1 — —
При переходе к полиуретанам (см. рис. 3) изменение характерис-
тической вязкости целиком зависит от строения и свойств олигомер-
ной компоненты. Величина показателя степени при М в уравнении
Марка — Хувинка больше 0,5, что обусловлено влиянием объем-
ных эффектов на характеристическую вязкость, а не увеличением
частичной протекаемости клубков при переходе от олигомера к
полиуретану. Точки зависимости lg (rjj от lg М для олигомеров и
полиуретанов в О-растворителе укладываются на одну прямую
(см. рис. 3). Это подтверждает отсутствие влияния уретановых групп
на термодинамическую гибкость цепи для случая полиуретанов на
основе олигомеров. Термодинамическую гибкость цепей определя-
ли по методу Штокмайера — Фиксмана, значения (йо/Лсв)/2 приве-
дены в табл. 3. При расчете (ЛсВ) /г пренебрегали вкладом уретановых
групп на основании того, что точки на прямой 7 рис. 3 получены
для полиуретана из диэтиленгликольадипината и 2,4-ТДИ, оли-
22
log fl
Рис. 4. Зависимость размеров клубков
пол ипропиленгли колей от молекулярного
веса
I — на основе 2,3 пропиленгликоля в тетра
гидрофуране, 2 — пропиленгликоля и гли
церина в тетрагидрофуране, ,3 - 2,3 пропи-
ленгликоля в толуоле
гомерные компоненты которого имеют разный молекулярный вес
(от 800 до 4000) и тем не менее хорошо укладываются на одну
прямую. Другим подтверждением служит то, что зависимость lg [т)|
от lgМ для полиуретана на основе полиоксипропиленгликоля и
2,4-ТДИ описывается одной прямой линией для олигомера и поли-
уретана.
Метод Штокмайера — Фиксмана применен и для оценки невоз-
мущенных размеров для линейного ПУ на основе этиленгликоля и
4,4'-дифенилметандиизоциапа-
та по данным вискозиметрии
в 0-точке, которая создава-
лась с помощью бинарного
растворителя [172]. В этом
случае для девяти фракций
найдено соотношение [г] । —
= 3,64 • 10-Ж71, что очень
близко к результатам, изло-
женным выше. Показатель
а = 0,62—0,66 для линейных
ПУ на основе 4,4'-дифенил-
метандиизоцианата и поли-
капролактона определен в
[298]. Результаты исследова-
ния разветвленных полиуре-
танов [304] согласуются с об-
щими закономерностями свой-
ств растворов ПУ.
Гибкость полиуретановой
цепи определяется в основном
свойствами блоков — олиго-
эфиров, и поэтому изучение
поведения их в растворах
и в массе имеет важное значение для понимания структуры ПУ.
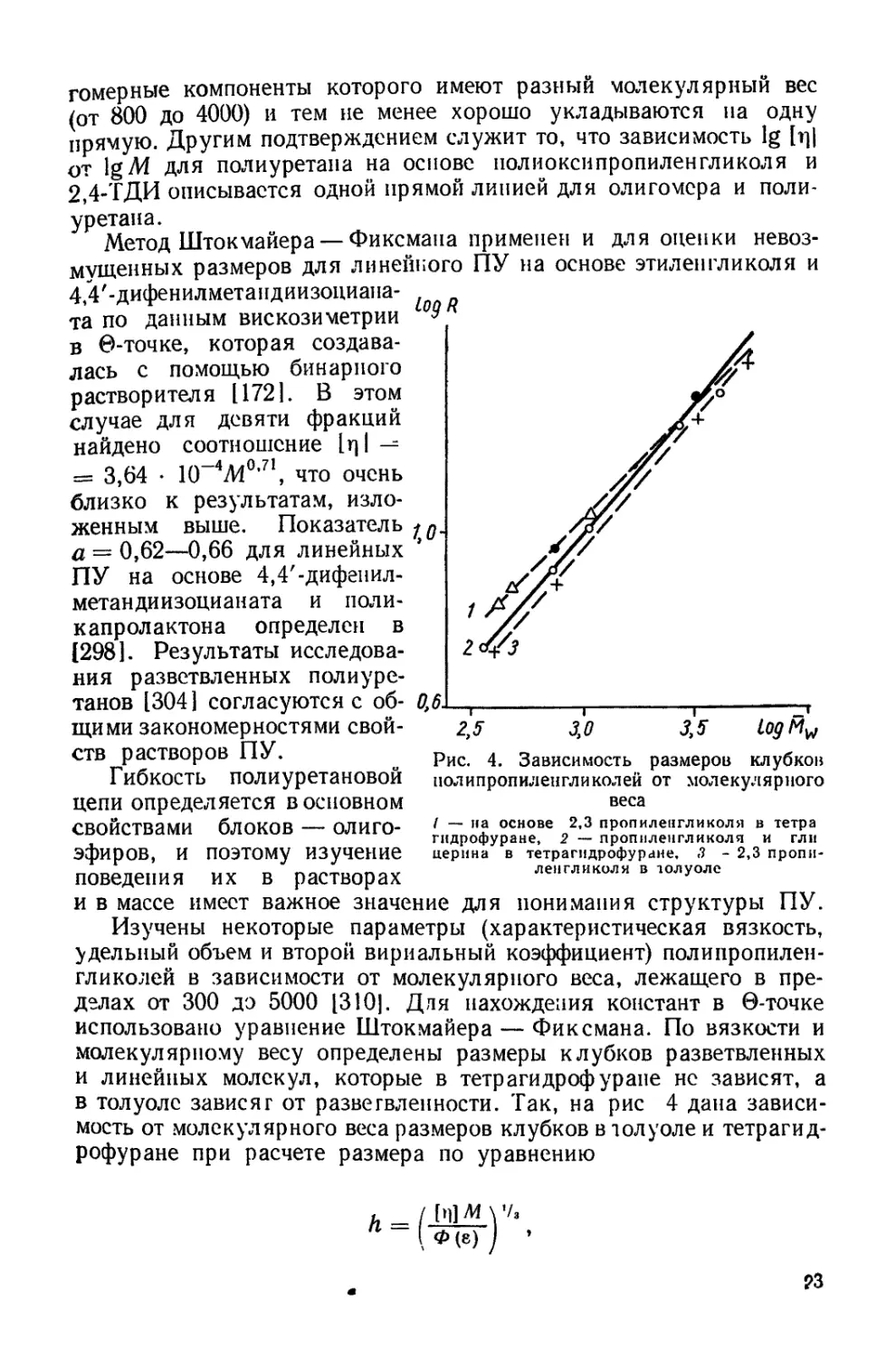
Изучены некоторые параметры (характеристическая вязкость,
удельный объем и второй вириальный коэффициент) полипропилен-
гликолей в зависимости от молекулярного веса, лежащего в пре-
делах от 300 до 5000 [310]. Для нахождения констант в 0-точке
использовано уравнение Штокмайера — Фиксмана. По вязкости и
молекулярному весу определены размеры клубков разветвленных
и линейных молекул, которые в тетр аги дроф уране не зависят, а
в толуоле зависят от разветвленности. Так, на рис 4 дана зависи-
мость от молекулярного веса размеров клубков в толуоле и тетрагид-
рофуране при расчете размера по уравнению
в котором
Ф = ф0 (1 — 2,678 4- 2,86г2), Ф = 2,84 • 1023, г = 2/3 (а — 0,5),
где а— экспонент уравнения Марка — Хувинка. При этом бы-
ло обнаружено, что при переходе от низко- к высокомолекулярным
продуктам происходит изменение конформации молекул. Весьма
важна, с нашей точки зрения, ассоциация молекул в толуоле при
молекулярном весе меньше 800, которая отразилась на зависимос-
тях констант Хаггинса от степени ассоциации. Показана также
возможность описания поведения низкомолекулярных олигомеров
с точки зрения теорий, развитых для высокомолекулярных цепей.
Для точного учета конформаций цепей следовало бы провести строгий
расчет размеров цепей, при условии свободного вращения, с точной
оценкой всех типов связей как в олигомерном блоке, так и в диизо-
цианатном. Такие расчеты довольно трудны. Рассмотрение конфигу-
рационной статистики полиамидных цепей, более простых по сравне-
нию с полиуретанами, которые, однако, содержат большое число
различных связей внутри каждого мономерного звена, показало
вклад этих связей в размеры цепей [2161. В дальнейшем целесооб-
разно перенести предложенные в данной работе методы расчета на
полиуретаны. Приведенные данные позволяют считать, что
для получения сведений о свойствах олигомеров и форполиме-
ров можно использовать существующие статистические теории, раз-
работанные для гибких макромолекул. Эго показывает, что сравни-
тельно низкомолекулярные блоки обладают собственной гибкостью,
как и линейные высокомолекулярные цепи. Отсюда, проявление
гибкости макромолекул в сетках полиуретановых эластомеров на
основе форполимеров не только результат сшивания их в длинную
последовательность сравнительно коротких олигомерных отрезков,
но и следствие собственной гибкости молекул олигомеров. Гибкость
полиуретановой цепи действительно определяется в основном гли-
колевой компонентой, поскольку её свойства могут быть хорошо
объяснены независимо от числа коротких жестких блоков в моле-
кулах теориями, разработанными для линейных цепей.
СОРБЦИОННЫЕ СВОЙСТВА ПОЛИУРЕТАНОВ
Олигомерные молекулы со сравнительно невысоким молекуляр-
ным весом обладают собственной гибкостью, и, следовательно, гиб-
кость полиуретановой цепи определяется тремя основными фак-
торами— собственной гибкостью олигомерных блоков, их концент-
рацией в цепи и числом уретановых групп в цепях. Последние
влияют на равновесную гибкость цепи и в большей степени — на
подвижность макромолекул в конденсированной фазе, так как гиб-
кость макромолекул зависит не только от внутримолекулярного, но
и от межмолекулярного взаимодействия, которое не может быть
94
учтено при оценке гибкости по данным о свойствах разбавленных
растворов.
Возможен также и термодинамический подход к оценке гибкости
цепей, который заключается в расчете величины сегментов цепной мо-
лекулы. Под величиной сегмента понимают значение молекулярно-
го веса, которое должны иметь мо-
лекулы полимера, чтобы полимер-
ная система подчинялась обычно-
му для низкомолекулярных тел
термодинамическому закону 11321.
Нахомэняе величины сегмента из
термодинамических данных осно-
вано на измерении сорбции паров
растворителей полимерами и оп-
ределении по закону Рауля эффек-
тивной (кажущейся) молярной до-
ли полимера в растворе, откуда
вычисляется эффективный молеку-
лярный вес полимера или сег-
мента. А. А. Тагер и сотрудники для
полиуретанов различной степени
сетчатоеги, но с одинаковой кон-
центрацией уретановых групп изу-
чили ч изотермы сорбции диок-
сана, в котором исследованные
полиуретаны растворялись или на-
бухали атермически 1131]. Полу-
ченные данные показали, что сорб-
ционная способность исследован-
ных полиуретанов и термодинами-
ческое сродство к ним раствори-
теля зависят только от химической
Рис. 5. Зависимость энтропии сме-
шения от состава раствора
/ — полиуретансукцинат. 2—пол и уре-
танади пин ат 3 — пол и уретан себацинат
природы, а не от степени сетчатости, которую регулировали та-
ким образом, что молекулярный вес отрезка цепи между узлами
сетки изменялся от 2000 до 27 000.
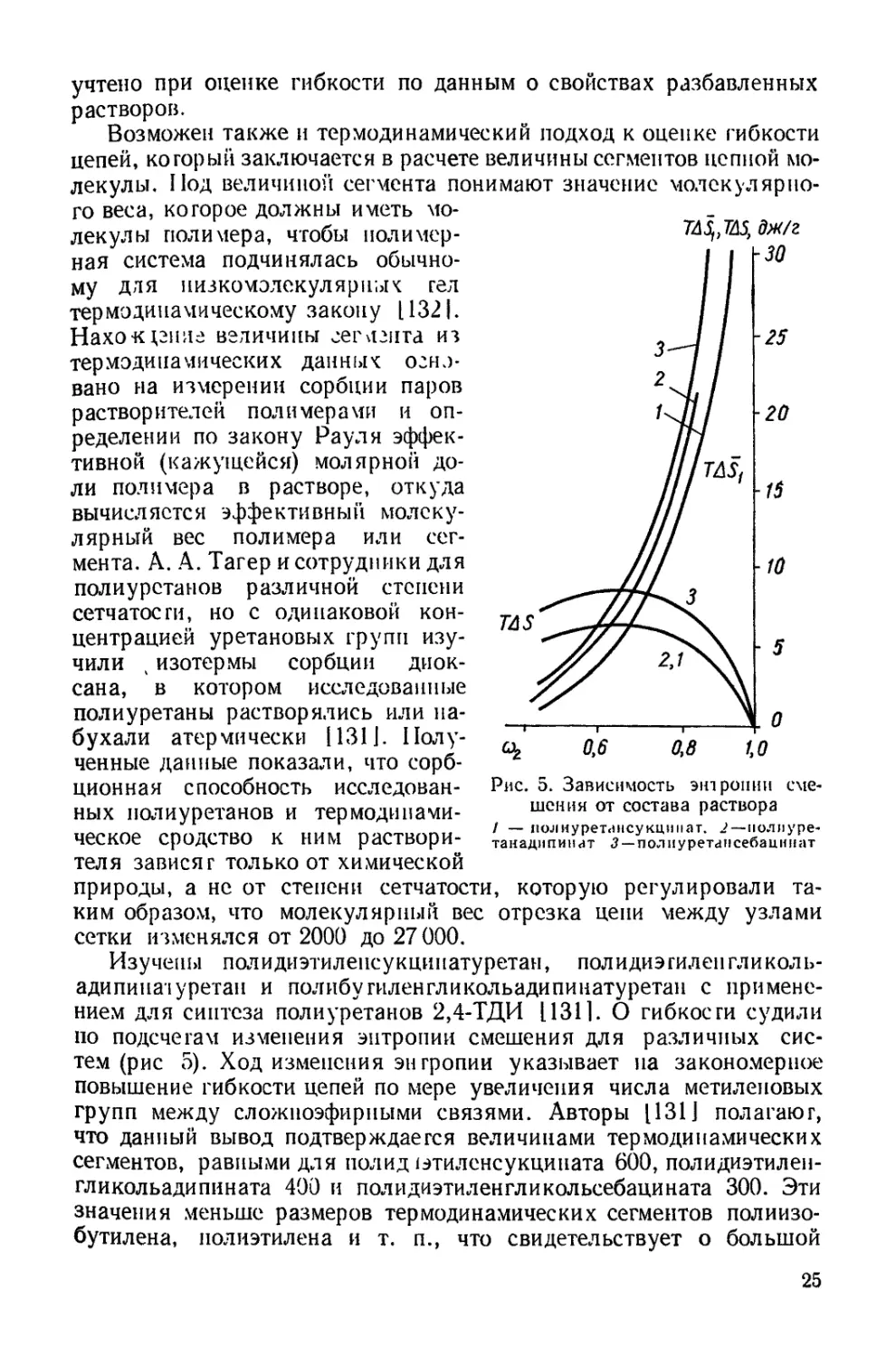
Изучены полидиэтилеисукцинатуретан, полидиэгиленгликоль-
адипиназуретан и полибугиленгликольадипинатуретан с примене-
нием для синтеза полиуретанов 2,4-ТДИ 1131]. О гибкости судили
по подсчетам изменения энтропии смешения для различных сис-
тем (рис 5). Ход изменения энтропии указывает на закономерное
повышение гибкости цепей по мере увеличения числа метиленовых
групп между сложноэфирными связями. Авторы 1131] полагают,
что данный вывод подтверждается величинами термодинамических
сегментов, равными для пол ид 1этиленсукцината 600, полидиэтилен-
гликольадипината 400 и полидиэтиленгликольсебацината 300. Эти
значения меньше размеров термодинамических сегментов полиизо-
бутилена, полиэтилена и т. п., что свидетельствует о большой
25
гибкости цепей полиуретанов, обусловленной наличием гибких
связей С—О—С.
Термодинамические параметры сеток не зависят от их густоты,
и авторы это связывают с малым значением термодинамического сег-
мента, поскольку минимальное расстояние между узлами сетки
составляло 2300, что намного превышало размеры термодинамичес-
кого сегмента. Однако они не использовали своих данных для
вычисления величины сегмента цепи в сетке, а фактически провели
корреляцию данных по термодинамике сорбции и гибкости цепей
полиэфиров, входящих в сетку.
Таблица 4
Характеристики полиуретанов, исследованных сорбционным методом
Образец Потиэфир С ъивающий агент Мольное соотношение Мс экс- перимен- тальное
1 Диэтилснгликольадипи- нат-1600 Триэтанола- мин + диэта- ноламин (1,5:1) 1 . 1, 1 : 0,035 3700
2 Диэтиленгликольадипи- нат-1600 То же 1 : 1,2:0,100 2600
3 Ол игобу та д иенд ио л-1600 Триэтаноламин 1 . 1,68.0,4 2400
4 0лигоизопрсндиол-2000 То же 1 1,68 0,4 3200
Таким образом, представления о гибкости цепей линейных по-
лиэфиров необоснованно перенесены на гибкость цепей полиуретанов
в сетке. Кроме того, хорошо известно, что специфической чертой строе-
ния полиуретанов является большой вклад физических взаимо-
действий в эффективную плотность сетки, в результате чего эффек-
тивная плотность сетки, вычисленная физическими методами, всег-
да намного больше той, которая определяется из стехиометрического
соотношения компонентов, взятых для синтеза. Поэтому резуль-
таты данной работы нельзя считать достаточно достоверными.
Между тем, представляет интерес именно гибкость полимерных
цепей в трехмерной сетке и изменение гибкости цепи при ее вхож-
дении в сетку, так как гибкость зависит от межмолекулярных взаи-
модействий и связи между цепями.
Для выяснения этого вопроса исследованы процессы сорбции
паров полиуретанами на основе различных олигомеров и толуилен-
диизоцианата (табл. 4) [71].
Густоту сетки оценивали в отличие от [1311 на основании данных
по равновесному модулю эластичгГости, что позволило учитывать
химические и физические узлы сетки. Как видно из рис. 6, изотер-
мы сорбции диоксана изученными полиуретанами имеют S-образный
вид и отличаются от полученных в работе [1311. Анализ изотерм по-
26
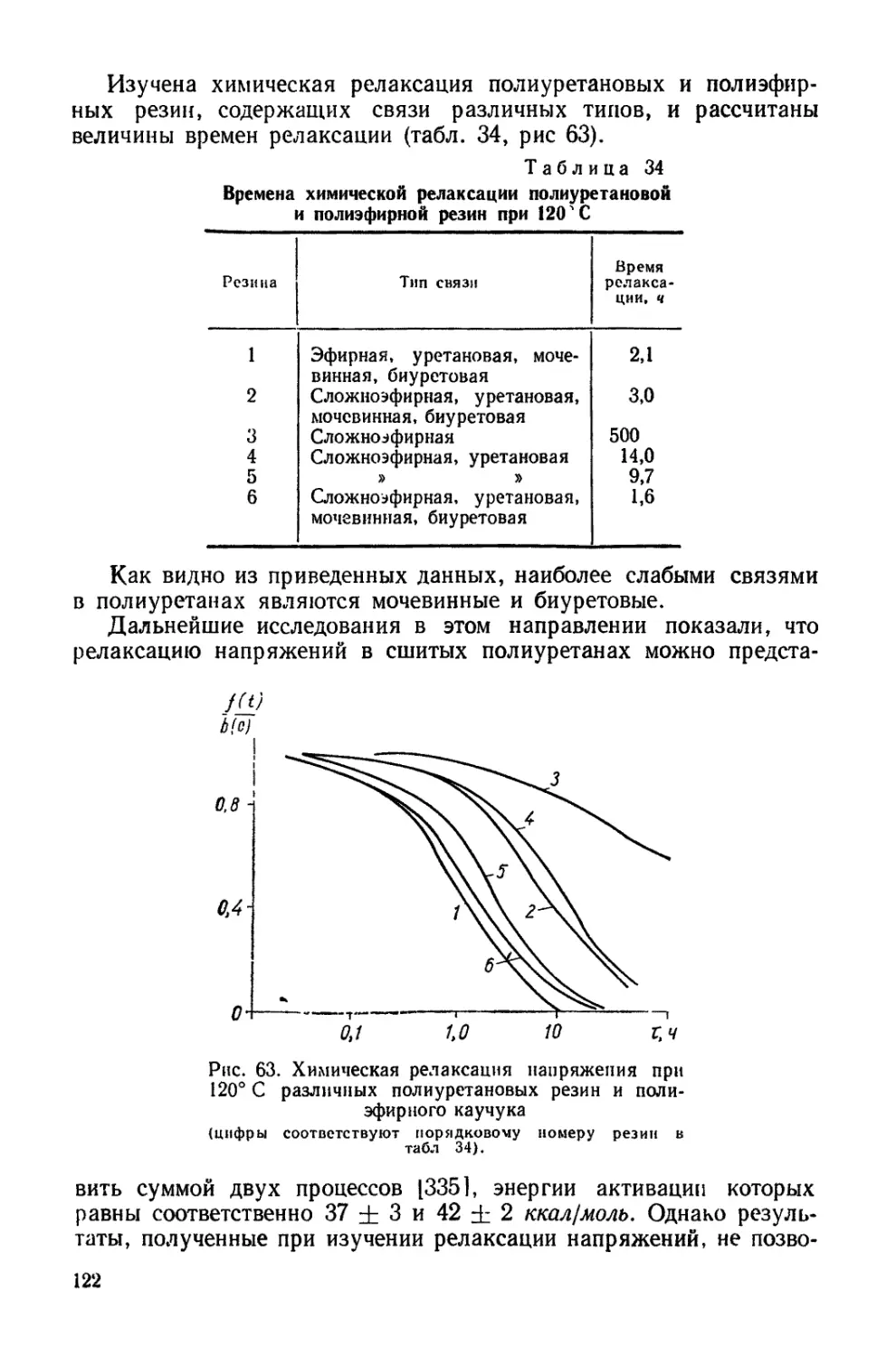
Рис. 6. Изотермы сорбции диоксана
полиуретана на основе ДЭГА и
ТДИ (/,2), олигобута диендиола и ТДИ
(3) и олигоизопрендиола (4) при 25 - С
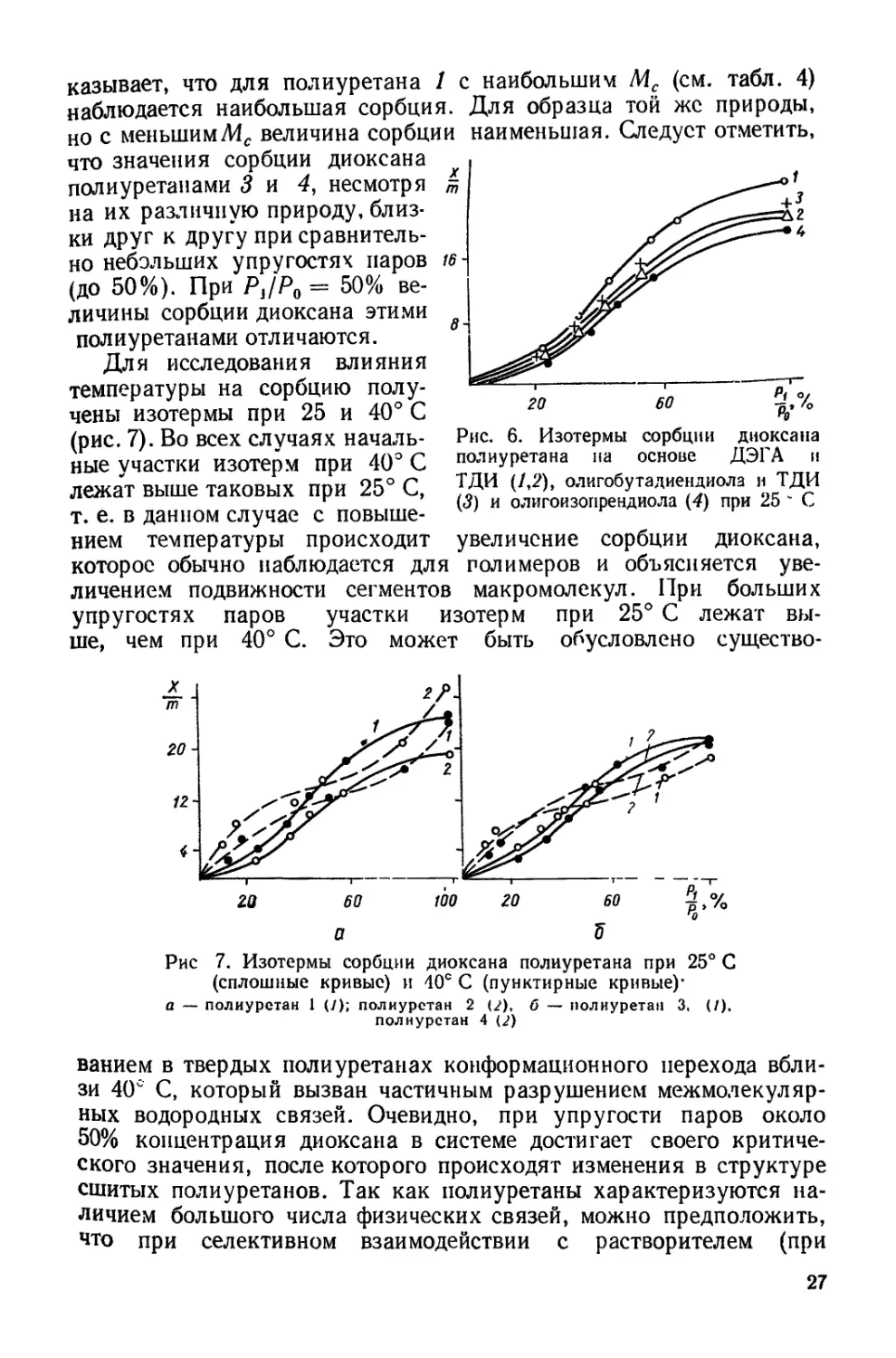
увеличение сорбции диоксана,
казывает, что для полиуретана / с наибольшим М? (см. табл. 4)
наблюдается наибольшая сорбция. Для образца той же природы,
но с меньшимЛ4с величина сорбции наименьшая. Следует отметить,
что значения сорбции диоксана
полиуретанами 3 и 4, несмотря
на их различную природу, близ-
ки друг к другу при сравнитель-
но небольших упругостях паров
(до 50%). При Р,/7>0 = 50% ве-
личины сорбции диоксана этими
полиуретанами отличаются.
Для исследования влияния
температуры на сорбцию полу-
чены изотермы при 25 и 40° С
(рис. 7). Во всех случаях началь-
ные участки изотерм при 40° С
лежат выше таковых при 25° С,
т. е. в данном случае с повыше-
нием температуры происходит
которое обычно наблюдается для полимеров и объясняется уве-
личением подвижности сегментов макромолекул. При больших
упругостях паров участки изотерм при 25° С лежат вы-
ше, чем при 40° С. Это может быть обусловлено существо-
Рис 7. Изотермы сорбции диоксана полиуретана при 25° С
(сплошные кривые) и 40е С (пунктирные кривые)’
а ~ полиуретан 1 (/); полиуретан 2 (2), б ~ полиуретан 3. (/),
полиуретан 4 (2)
ванием в твердых полиуретанах конформационного перехода вбли-
зи 40е С, который вызван частичным разрушением межмолекуляр-
ных водородных связей. Очевидно, при упругости паров около
50% концентрация диоксана в системе достигает своего критиче-
ского значения, после которого происходят изменения в структуре
сшитых полиуретанов. Так как полиуретаны характеризуются на-
личием большого числа физических связей, можно предположить,
что при селективном взаимодействии с растворителем (при
27
определенной его концентрации) происходит перераспределение фи-
зических связей в пространственной сетке полиуретана. Это приводит
к появлению структуры, отличающейся от исходной, и изменению
ее поведения при сорбции. Такая подвижность и легкость пере-
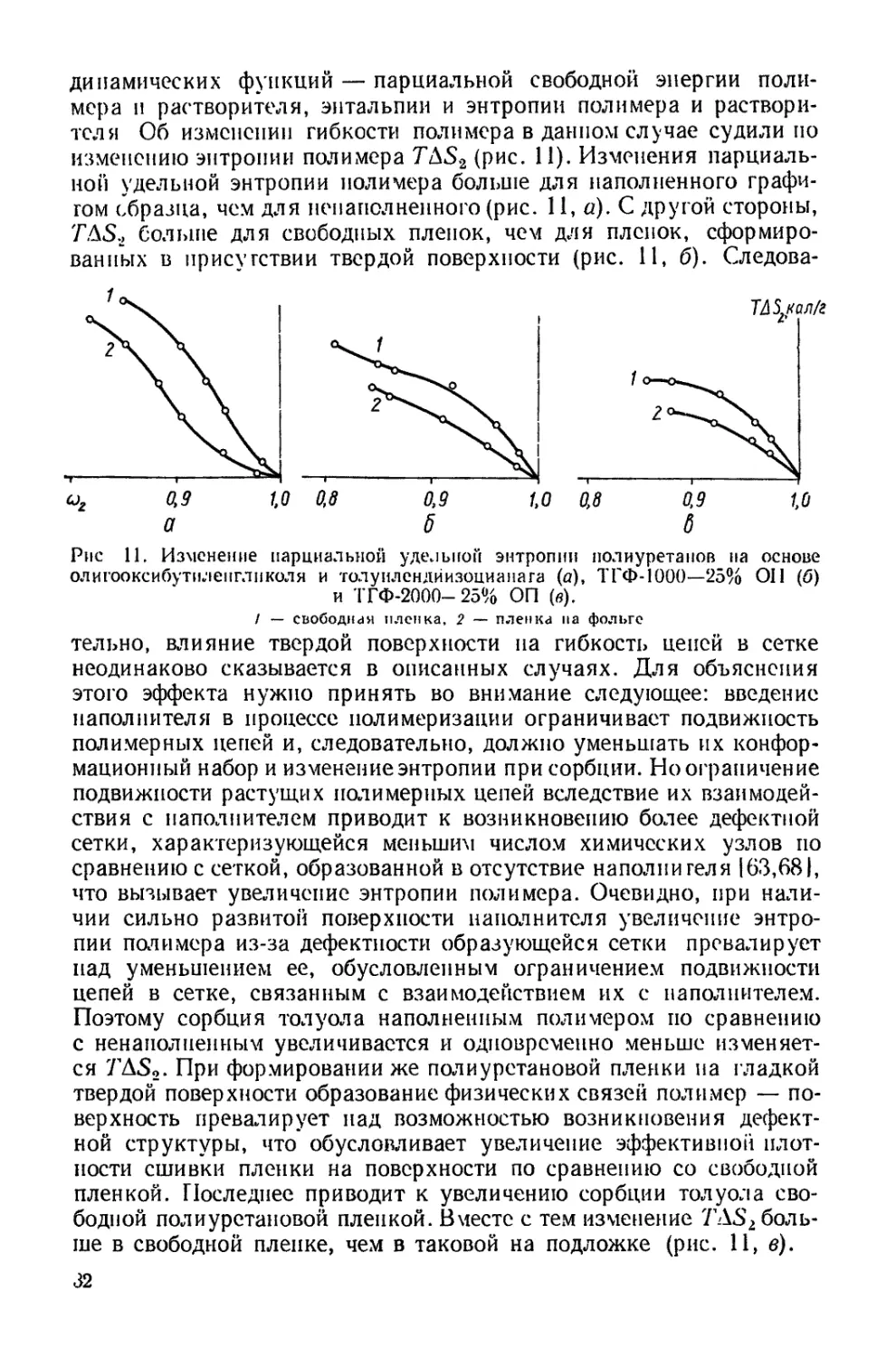
Рис. 8. Изменение парциальной
удельной энтропии полиурета-
нов при сорбции
(обозначения см. в табл. 3)
стройки пространственной сетки —
одна из специфических особенностей
строения трехмерных полиуретанов.
Таким образом, при сорбции паров
растворителя, как и при набухании
полиуретанов [701, густота сетки фи-
зических связей меняется. Поэтому
для расчетов термодинамических функ-
ций использованы только участки изо-
терм до 50% относительной упругости
паров, соответствующей началу из-
менения структуры [711 (табл. 5).
В связи с тем, что изменение эн-
тальпии в исследованных случаях зна-
чительно по своей величине, опреде-
ление сегмента, характеризующего
гибкость цепи, из сорбционных дан-
ных [132] неверно, так как величина
сорбции здесь определяется не только
по изменению числа конформаций
цепей, но и по изменению энергетичес-
кого барьера вращения отрезков цепей
в сетке, отражающемся на А/7. По-
этому об изменении гибкости цепей можно судить по изменению пар-
циальной удельной энтропии полимера T/\S2 (рис. 8). Для поли-
уретана на основе олигодиэтиленгликольадипината изменение энтро-
пии зависит от степени сшивки: TAS2 больше для полиуретана с
меньшей степенью сшивки. Эго указывает на увеличение гибкости
отрезков цепей в сетке при уменьшении эффективной плотности
сшивки. Зависимость гибкости от природы олигомера можно про-
Изменения термодинамических функций
х/т ‘уд ка 1/г &F> . ^УД
1 2 1 1 з 1 1 4 1 2 | 3 । 4 | 1 1 2
0,025 0,975 0,976 0,975 0,976 — 12,82 — 11,23 — 12.39 — 13,80 —0,03 —0,05
0,050 0,953 0,952 0,952 0,953 —9,37 —7,92 —8,85 —9.37 —0,16 —0,12
0,075 0,930 0,930 0,930 0,930 —7,7(Г —6.56 —6,89 —7,10 —0,27 —0,21
0,100 0,910 0,909 0,909 0,908 —6.53 —5,38 —5,78 —4,94 —0,37 —0,31
0,125 0,881 0,889 0,889 0,888 —5,86 —4,42 —4,69 —4,82 —0,45 —0,46
0,150 0,870 — — — —4,83 — — — —0,56 —
28
следить на примере полиуретанов на основе олигодиендиолов, ко-
торые отличаются только природой олигомеров. Изменение TAS2
для полиуретанов на основе олигоизопрендиола меньше, чем для
олигобутандиендиола, т. е гибкость цепи меньше для простран-
ственной сетки полиуретана на основе олигоизопрена. Это можно
объяснить наличием в цепи олигоизопрена метильной группы, кото-
рая чисто геометрически ограничивает гибкость цепи. Однако вли-
яние природы олигодиендиола на степень гибкости сказывается
не так сильно, как изменение степени сшивки для полиуретанов
на основе сложных эфиров.
Таким образом, проведенные исследования показали, что гиб-
кость отрезков цепей в пространственной сетке полиуретана оп-
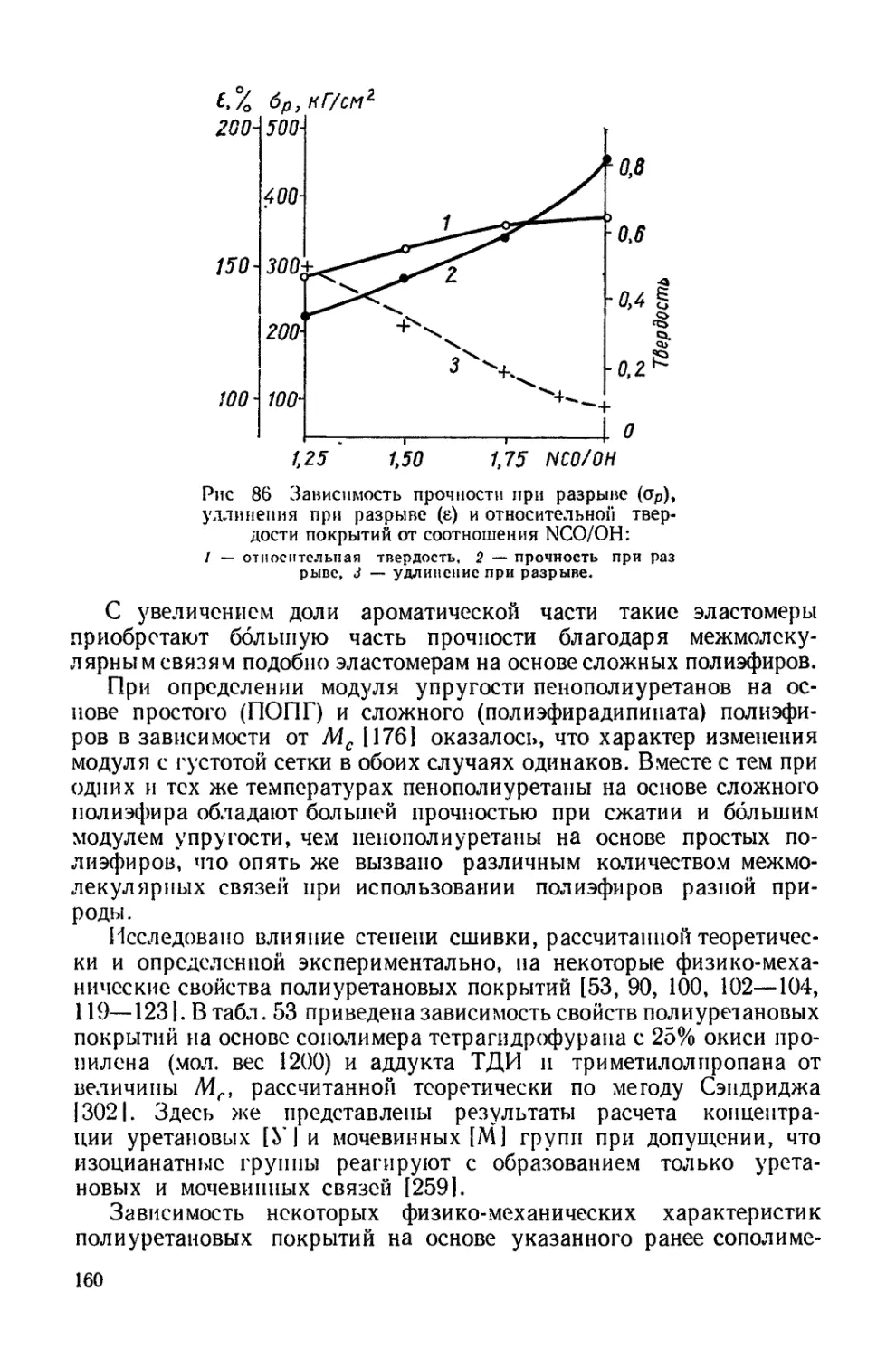
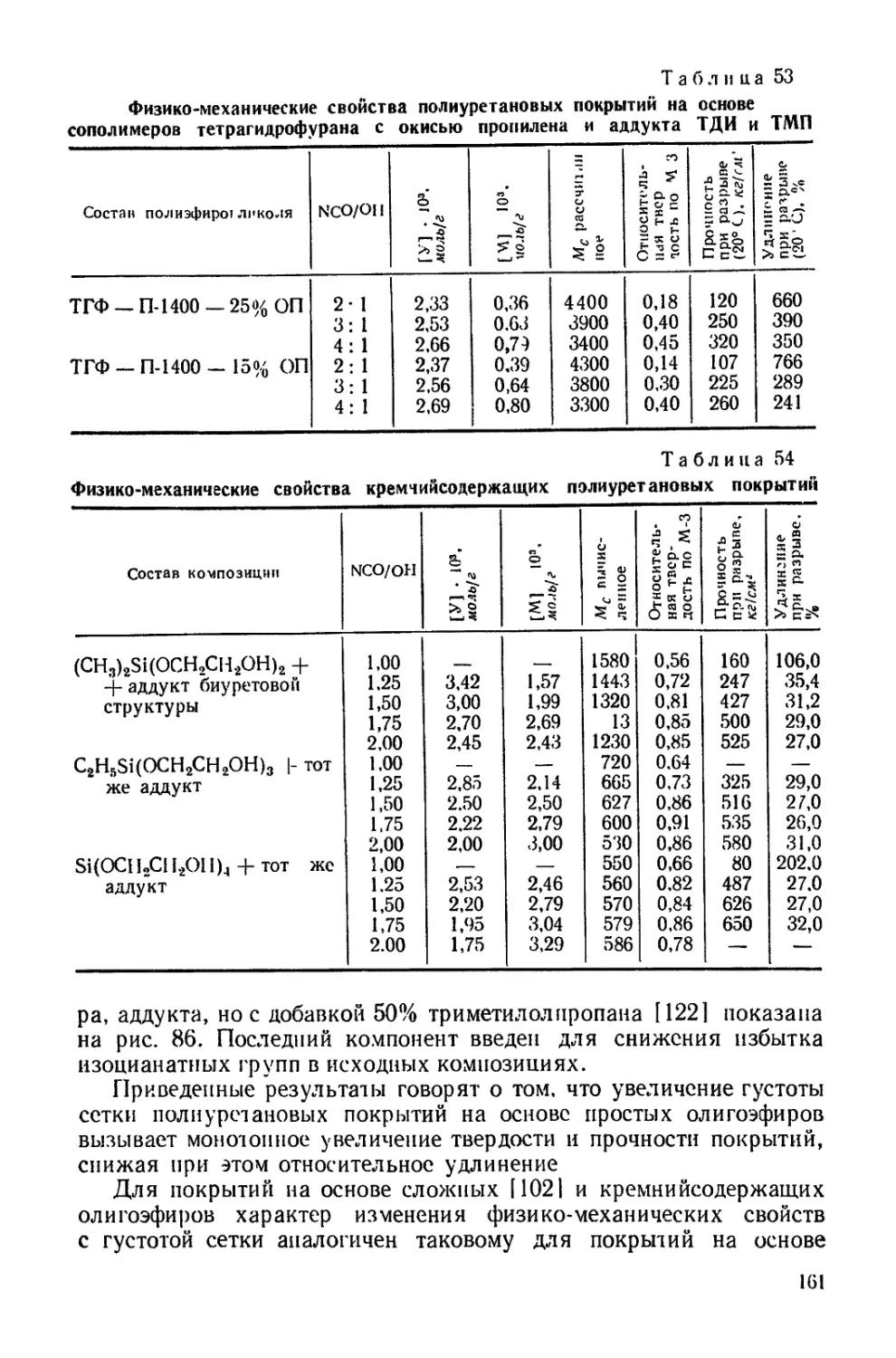
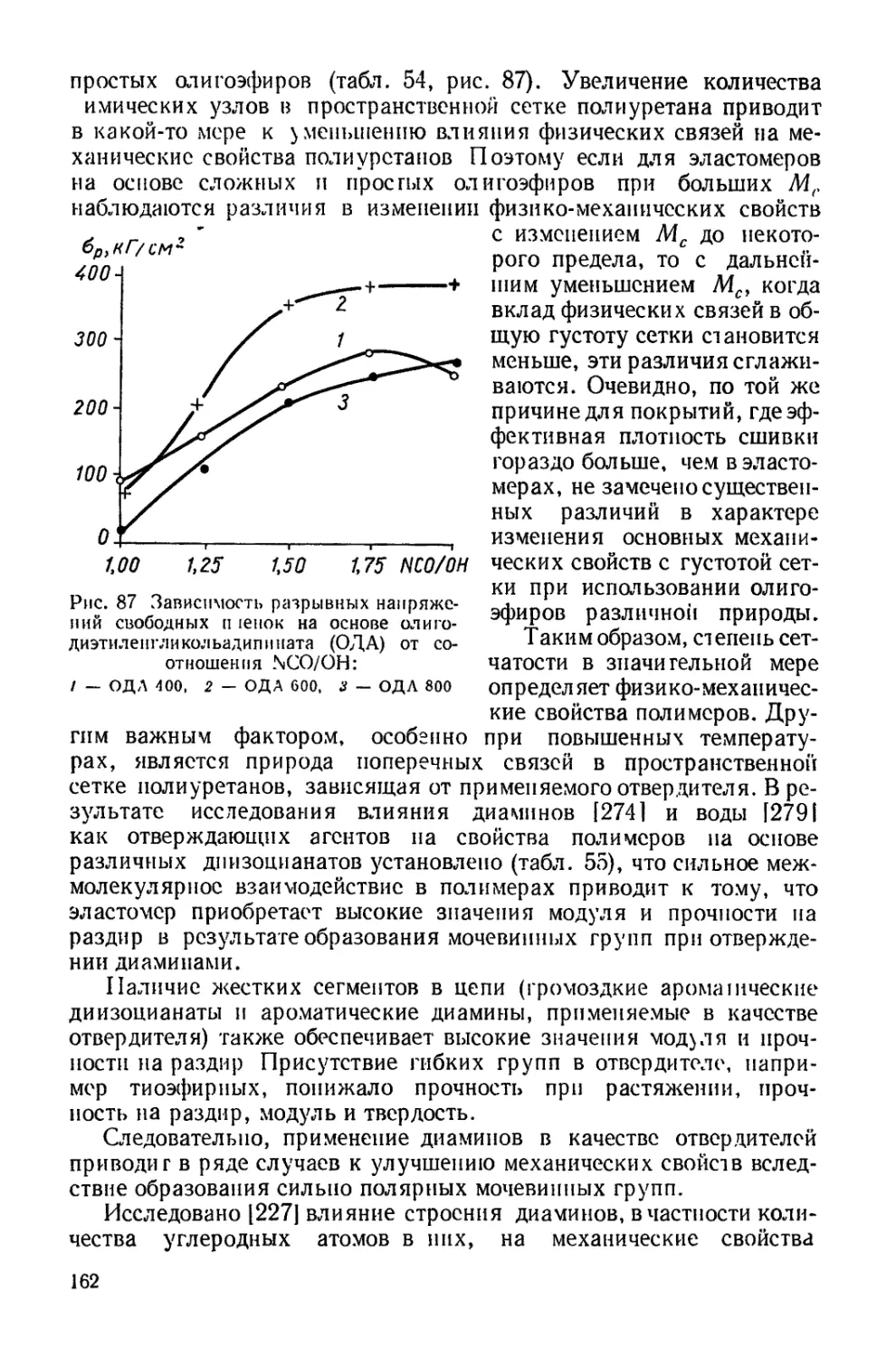
ределяется как химической природой блока, так и степенью сшивки
сетки. Отсюда следует, что оценку гибкости цепей, произведен-
ную для линейных молекул, нельзя перенести на системы, в кото-
рых молекулы сшиты и образуют трехмерную сетку. Действительно,
чем большее число конформаций принимает цепь, тем более она
гибкая. Естественно, что при вхождении в сетку число возможных
конформаций ограничивается. В рассматриваемом случае наличие
межмолекулярных взаимодействий, зависящих от химической гус-
тоты сетки, изменяет потенциальный барьер вращения и, соот-
ветственно, гибкость отрезков цепей между узлами даже тогда, когда
расстояние между ними превосходит величину термодинамического
сегмента, найденного для несвязанных в сетку высокомолекуляр-
ных цепей. Подразумевается, что цепочка как бы однородна в от-
ношении гибкости, т. с. не рассматривается раздельное влияние
на сорбцию жестких и гибких блоков. Такой подход допустим для
получения общей картины поведения полиуретанов при исследо-
вании сорбции веществ, являющихся растворителями для обоих
блоков цепи.
Более строго следовало бы рассматривать и разграничивать
сорбцию раздельно жесткими и гибкими участками. Такие данные
можно получить при изучении диффузии газов или проницаемос-
ти паров. Например, при диффузии СО2 в полиуретанах |178|
Таблица 5
при сорбции паров полиуретанами
кал/г Д// — о^Д/У, кал/г ТД32, кал/г
3 4 2 3 4 1 1 2 3 4
—0,07 —0,18 —0,26 —0,39 -0,54 —0.06 —0,24 —0,34 —0,46 —0,61 3,71 4,89 5,33 5,21 5,68 3,73 6.64 9,48 12,05 14,23 2,73 6,57 6,30 6,90 3,40 5,97 8,64 10,71 1,09 2,67 6,64 11,20 16,27 19,19 0,96 2,53 4,70 0,74 3,56 4,08 6,29 1,09 2,87 6,70 9,18
29
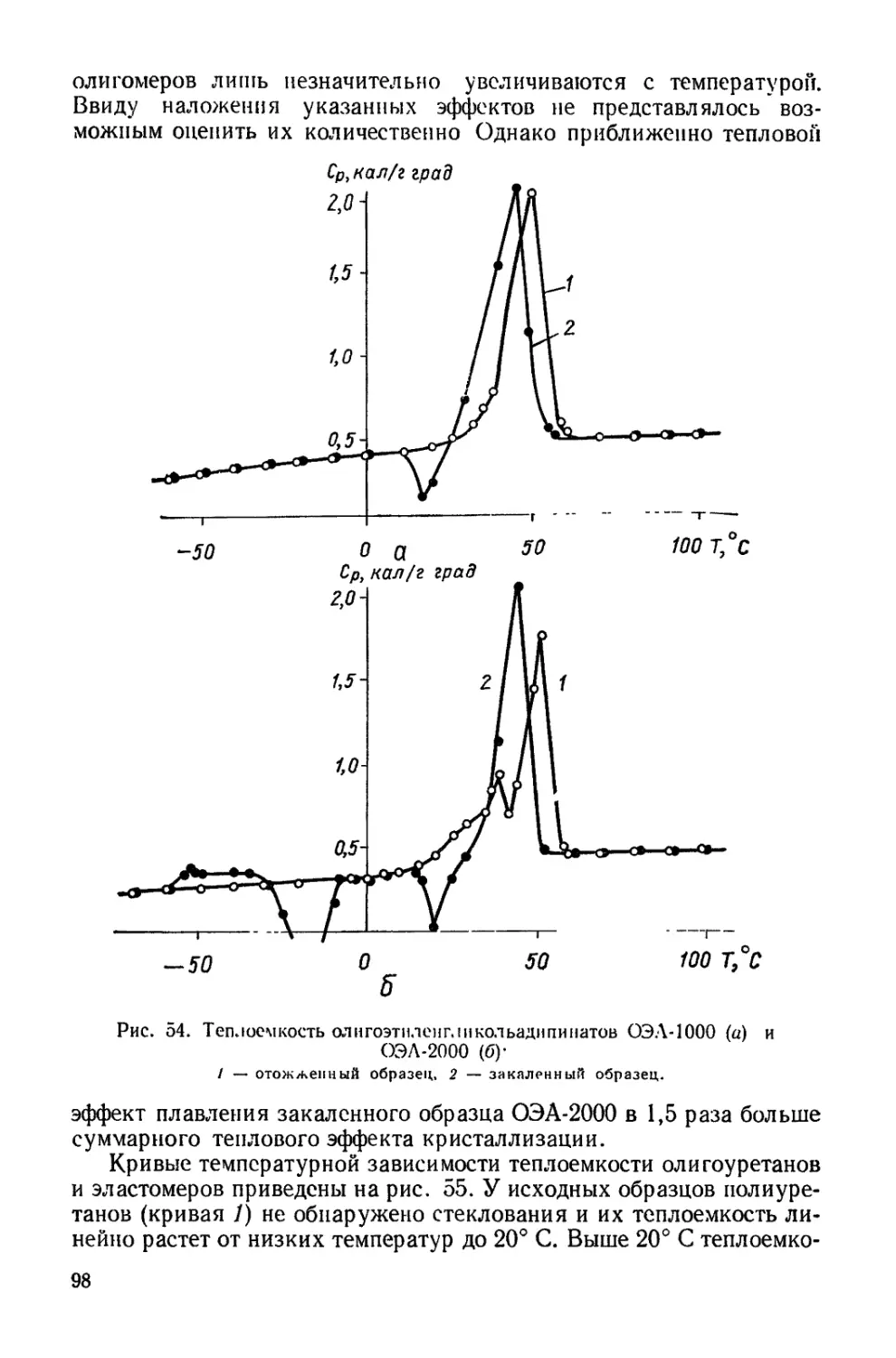
установлено, что коэффициенты диффузии в них значительно ниже,
чем в других каучуках. Это связано с тем, что, несмотря на значи-
Таблица 6
Характеристики некоторых полиуретанов
Олигоэфир( ЛИКО '1Ь Сшивающий агент Тип твердой поверхности
Олигооксибутиленгликоль 3,3'-Дихлор- 4,4'-диами- нофенилен То же 20% коллоид- ного графи- та 260
ПТГФ-1000 + 25% ОП Триметилолпропан » Алюминиевая подложка 460 300
ПТГФ-2000 I- 25% ОП » » Алюминиевая подложка 800 700
* Величина Мс определена по набуханию.
тельную гибкость отдельных участков цепей, сильное межмолеку-
лярное взаимодействие в результате образования водородных связей
приводит к более плотной упаковке цепей. Данный эффект прева-
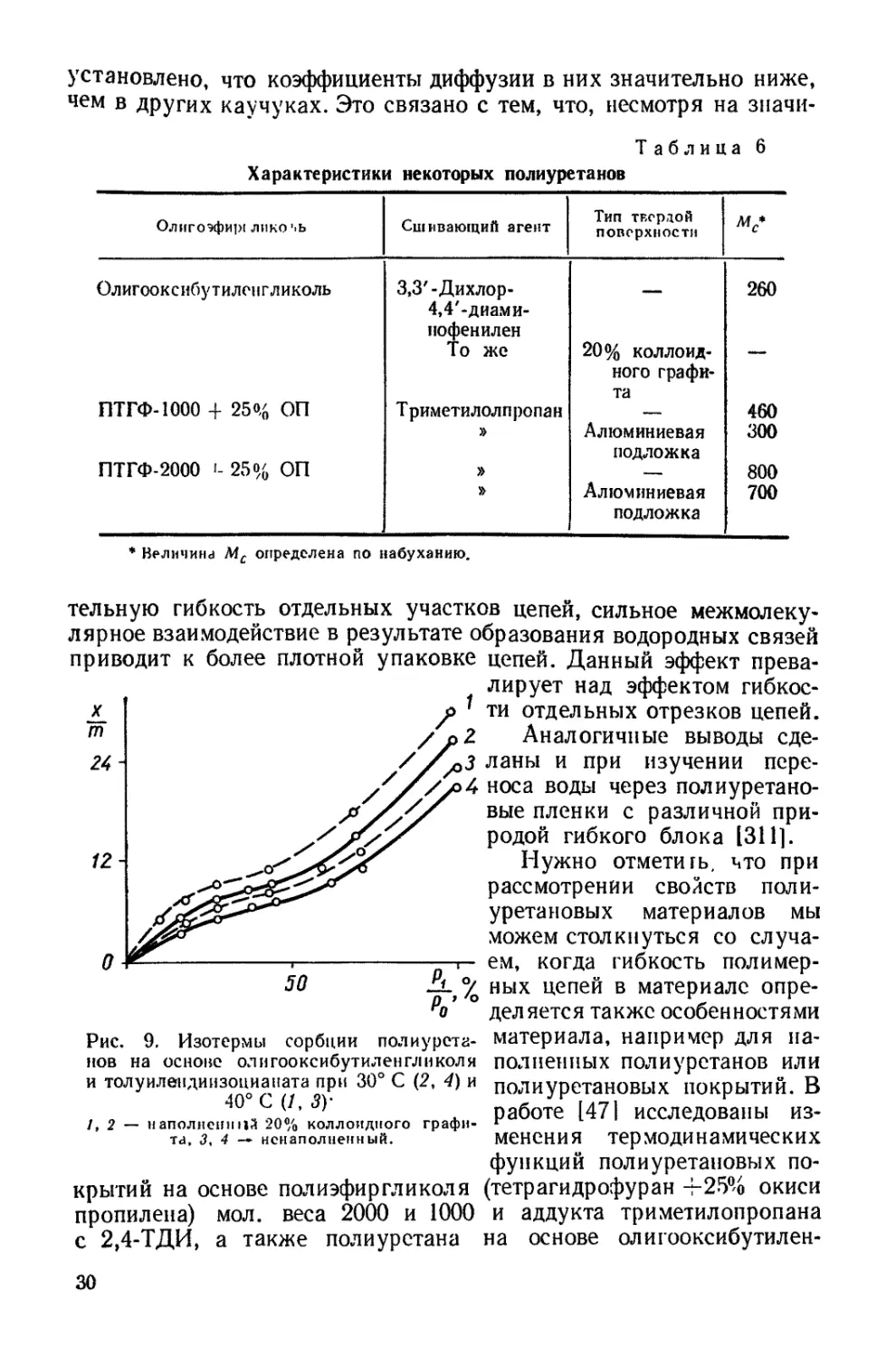
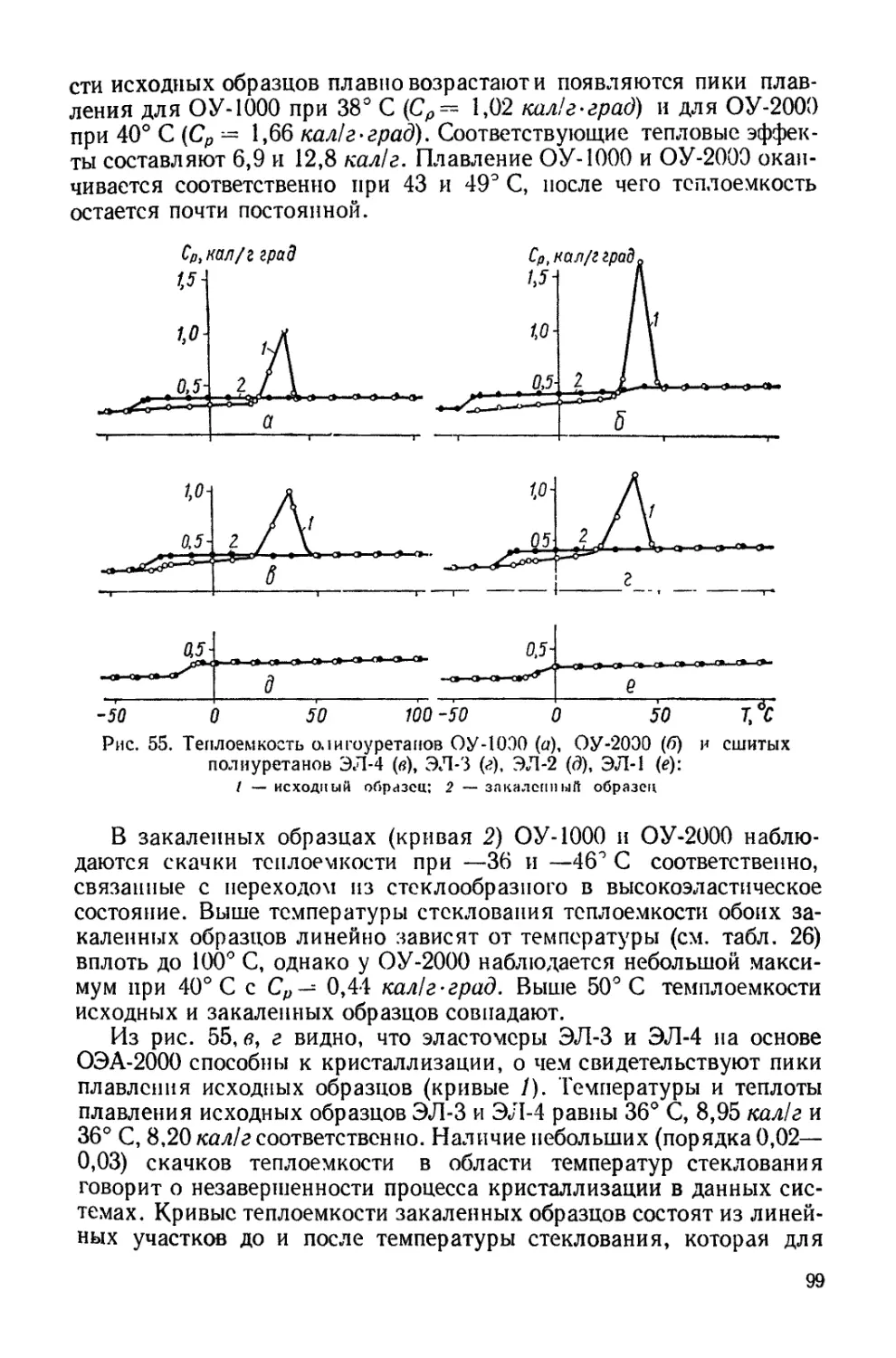
Рис. 9. Изотермы сорбции полиурета-
нов на основе олигооксибутилен гл и кол я
и толуилендиизоцианата при 30° С (2, 4) и
40° С (/, ЗУ
1,2 — наполненной 20% коллоидного графи-
та, 3, 4 — нснаполненный.
лирует над эффектом гибкос-
ти отдельных отрезков цепей.
Аналогичные выводы сде-
ланы и при изучении пере-
носа воды через полиуретано-
вые пленки с различной при-
родой гибкого блока [311].
Нужно отметить, что при
рассмотрении свойств поли-
уретановых материалов мы
можем столкнуться со случа-
ем, когда гибкость полимер-
ных цепей в материале опре-
деляется также особенностями
материала, например для на-
полненных полиуретанов или
полиуретановых покрытий. В
работе [471 исследованы из-
менения термо ди нами чес ких
функций полиуретановых по-
крытий на основе полиэфиргликоля (тетрагидрофуран -г25% окиси
пропилена) мол. веса 2000 и 1000 и аддукта триметилопропана
с 2,4-ТДИ, а также полиуретана на основе олигооксибутилен-
30
Гликоля и ТДИ, полученного при взаимодействии ТДИ с тетра-
гидрофураном и наполненного 20% коллоидного графита (табл. 6).
Изотермы сорбции толуола изученными полиуретанами пред-
ставлены на рис. 9. Изотермы, характеризующие сорбцию при 40° С,
лежат во всем интервале
относительных упругостей
паров выше аналогичных
изотерм при 30° С. Подоб-
ное возрастание сорбции с
повышением температуры
характерно для полимеров
и обусловлено увеличением
подвижности сегментов ма-
кромолекул.
Мы отмечали, что при
сорбции паров диоксана не-
котор ыми пол и уретан ами
при разных температурах
наблюдалось пересечение
изотерм при 40 и 25° С при
определенной упругости
паров диоксана. Учитывая
то, что диоксан способен
образовывать водородные
связи с уретановыми груп-
пами [12), сделано пред-
положение о перераспреде-
лении физических связей в
пространственной сетке по-
лиуретана вследствие вза-
имодействия с растворите-
лем при определенной его
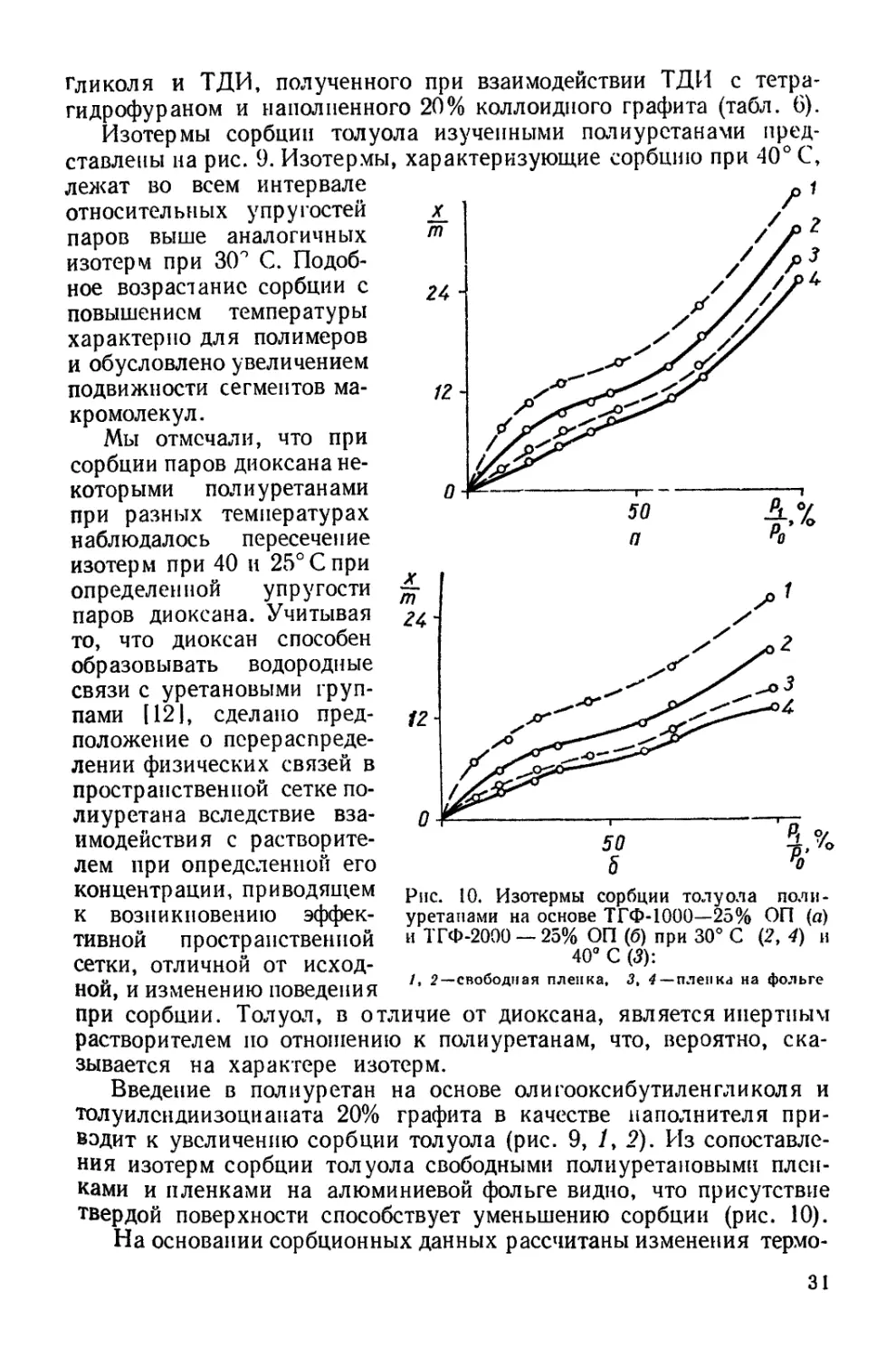
Рис. 10. Изотермы сорбции толуола поли-
уретанами на основе ТГФ-1000—25% ОП (а)
и ТГФ-2000 — 25% ОП (б) при 30° С (2, 4) и
40° С (3):
/, 2—свободная пленка, 3t 4 — пленка на фольге
концентрации, приводящем
к возникновению эффек-
тивной пространственной
сетки, отличной от исход-
ной, и изменению поведения
при сорбции. Толуол, в отличие от диоксана, является инертным
растворителем по отношению к полиуретанам, что, вероятно, ска-
зывается на характере изотерм.
Введение в полиуретан на основе олигооксибутиленгликоля и
толуилендиизоцианата 20% графита в качестве наполнителя при-
водит к увеличению сорбции толуола (рис. 9, /, 2). Из сопоставле-
ния изотерм сорбции толуола свободными полиуретановыми плен-
ками и пленками на алюминиевой фольге видно, что присутствие
твердой поверхности способствует уменьшению сорбции (рис. 10).
На основании сорбционных данных рассчитаны изменения термо-
31
динамических функций — парциальной свободной энергии поли-
мера п растворителя, энтальпии и энтропии полимера и раствори-
теля Об изменении гибкости полимера в данном случае судили по
изменению энтропии полимера T&S.2 (рис. 11). Изменения парциаль-
ной удельной энтропии полимера больше для наполненного графи-
том образца, чем для ненаполненного (рис. 11, а). С другой стороны,
T&S., больше для свободных пленок, чем для пленок, сформиро-
ванных в присутствии твердой поверхности (рис. И, б). Следова-
Рис 11. Изменение парциальной удельной энтропии полиуретанов на основе
олигооксибутиленглпколя и толуилендйизоцианага (д), ТГФ-1000—25% 011 (6)
и ТГФ-2000— 25% ОП (/?).
/ — свободная пленка, 2 — пленка на фольге
тельно, влияние твердой поверхности па гибкость ценой в сетке
неодинаково сказывается в описанных случаях. Для объяснения
этого эффекта нужно принять во внимание следующее: введение
наполнителя в процессе полимеризации ограничивает подвижность
полимерных цепей и, следовательно, должно уменьшать их конфор-
мационный набор и изменение энтропии при сорбции. Ноограпичение
подвижности растущих полимерных цепей вследствие их взаимодей-
ствия с наполнителем приводит к возникновению более дефектной
сетки, характеризующейся меньшим числом химических узлов по
сравнению с сеткой, образованной в отсутствие наполнителя [63,681,
что вызывает увеличение энтропии полимера. Очевидно, при нали-
чии сильно развитой поверхности наполнителя увеличение энтро-
пии полимера из-за дефектности образующейся сетки превалирует
над уменьшением ее, обусловленным ограничением подвижности
цепей в сетке, связанным с взаимодействием их с наполнителем.
Поэтому сорбция толуола наполненным полимером по сравнению
с ненаполненным увеличивается и одновременно меньше изменяет-
ся 7AS2. При формировании же полиуретановой пленки на гладкой
твердой поверхности образование физических связей полимер — по-
верхность превалирует над возможностью возникновения дефект-
ной структуры, что обусловливает увеличение эффективной плот-
ности сшивки пленки на поверхности по сравнению со свободной
пленкой. Последнее приводит к увеличению сорбции толуола сво-
бодной полиуретановой пленкой. Вместе с тем изменение ТAS2 боль-
ше в свободной пленке, чем в таковой на подложке (рис. 11, в).
32
Полученные данные говорят о том, что присутствие твердой по-
верхности при формировании полиуретановой трехмерной ссгки
сказывается на структуре последней и обусловливает изменение
гибкости отрезков цепей между узлами сетки. Следовательно, ме-
ханические свойства наполненных полиуретанов следует сопостав-
лять с гибкостью цепей, определенной в присутствии твердой по-
верхности, и нельзя переносить данные о гибкости, полученные
для ненаполненных полимеров, на наполненные.
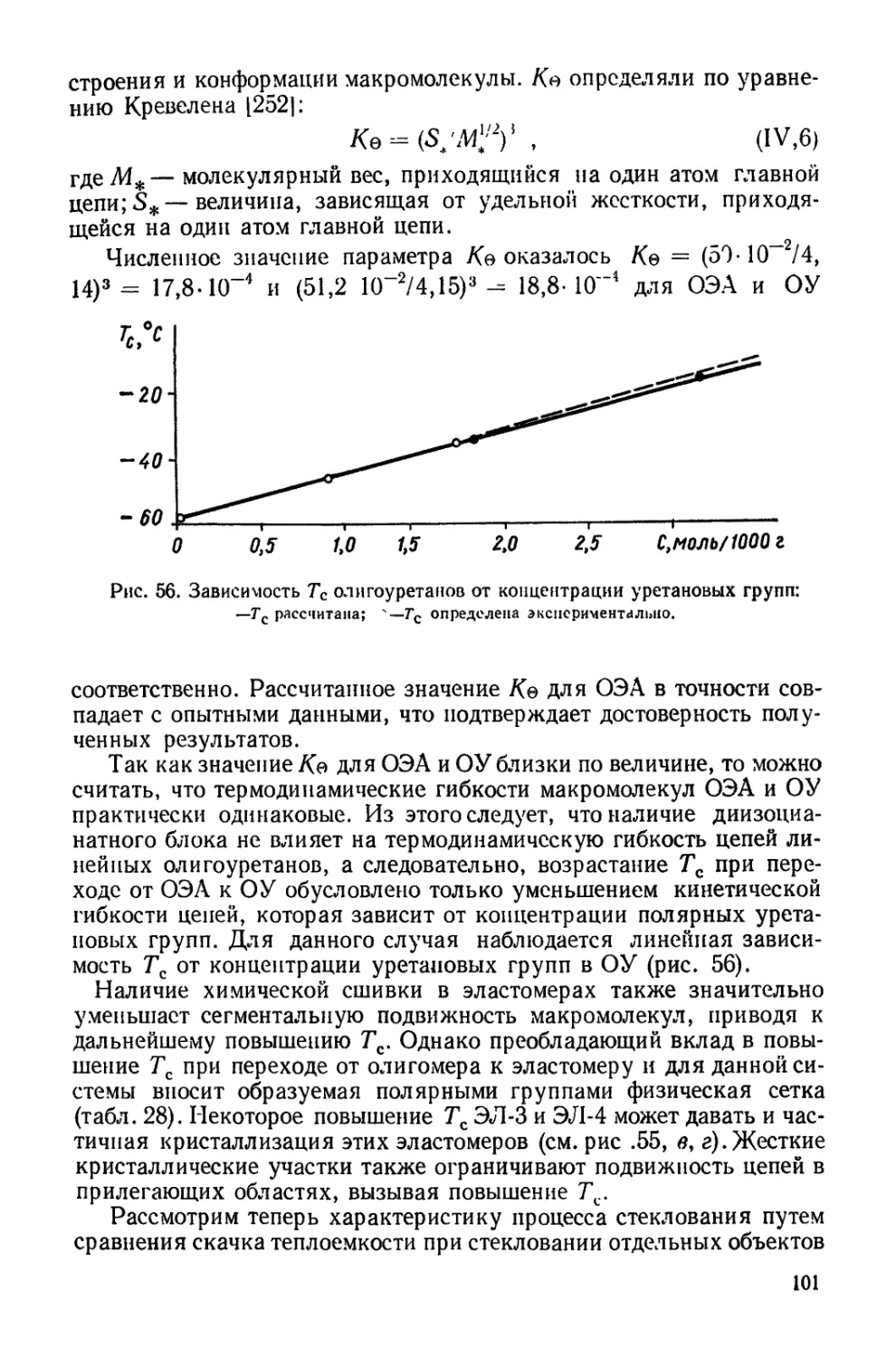
НЕКОТОРЫЕ СВОЙСТВА МОНОСЛОЕВ ОЛИГОМЕРОВ И ПОЛИУРЕТАНОВ
В настоящее время известно, что из данных о свойствах монослоев
полимеров на жидких подложках можно получить существенные све-
дения о строении цепей, их гибкости и характере упаковки 1991.
Это положено в основу теории Сингера [3,41, которая рассматри-
вает состояние макромолекул на границе раздела фаз, исходя
из теории растворов полимеров Хаггинса [2331. Сингером выведено
уравнение
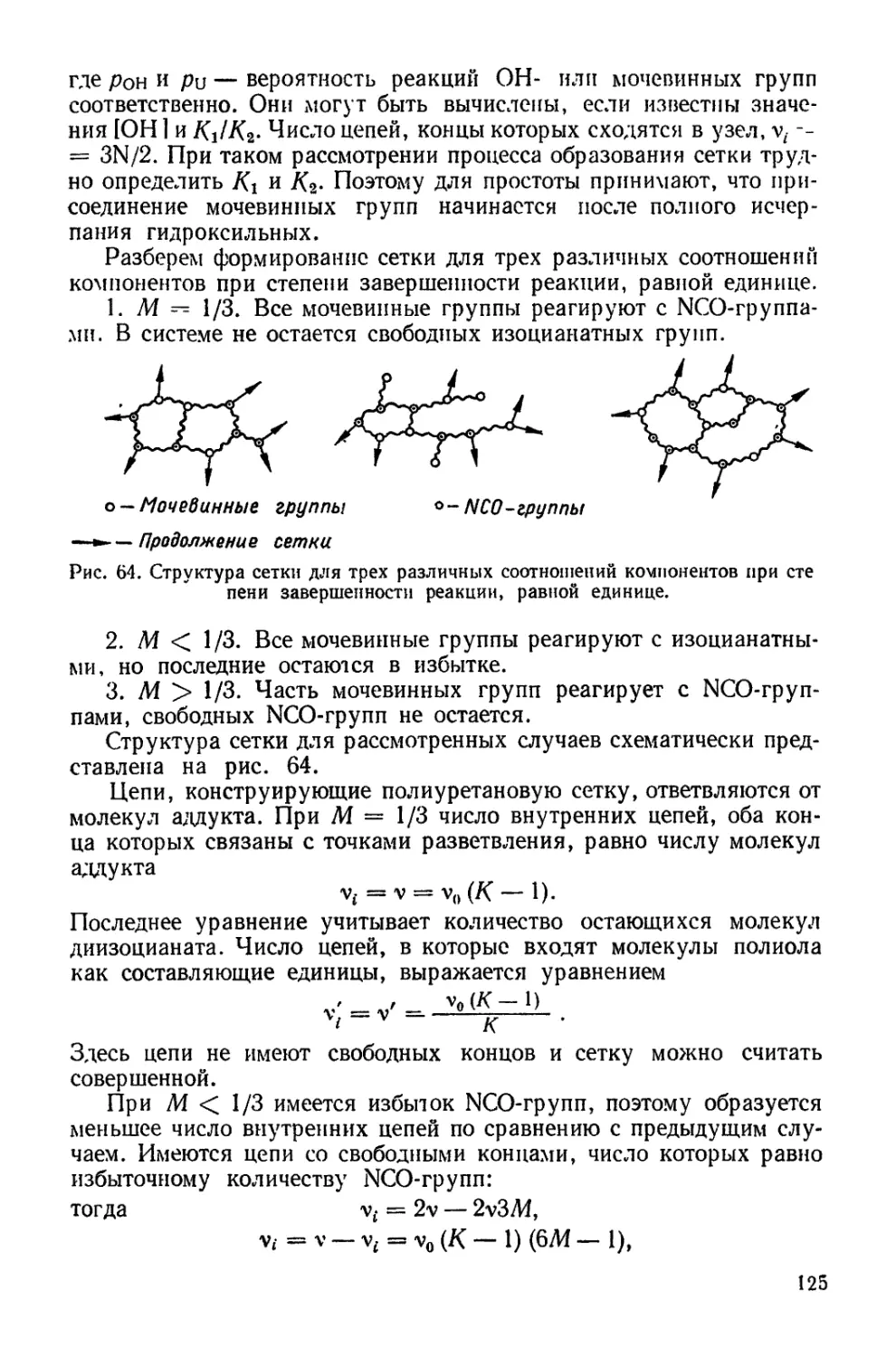
хКТ li /1 4» \ / х — 1 \ Z , /« 240 \ I
я=-лг||п(1-^)-(—2т)|’
где/— координационное число мономерных звеньев в цепи; Ао —
величина удельной поверхности мопомолекулярного слоя при пуле-
вом давлении; х — степень полимеризации; А — площадь поверх-
ности при поверхностном давлении л. Уравнение модифицировано
Девисом [200], который ввел в него условную характеристику гиб-
кости цепи со, принимающую значения от 0 до 2 при переходе от
жесткой к полностью гибкой цепи:
л = — ф41п(1- Ф) — —- In fl---То-Ф) •
у40[ \ А I х 2 со + 2 А I
Метод Девиса использован при оценке гибкости цепей полимеров
[296], а также при характеристике гибкости цепей олигомеров
и полиуретанов на границе раздела фаз и выяснении ее зави-
симости от взаимодействия с подложкой [140].
Для исследования были взяты низкомолекулярные полидиэтилен-
гликольадининаты разных молекулярных весов и полиуретаны,
полученные при взаимодействии этих олигомеров с 2,4-ТДИ.
В таких цепях содержатся группировки, способные к образо-
ванию водородных связей. Свойства монослоев изучены на че-
тырех типах подложек: дистиллированной воде (подложка 1),
3-н. растворе НО (подложка 2), 40%-ном растворе (NH4)2SO4
(подложка 3) и смешанном растворе из 30% сернокислого аммония
и 10% НО (подложка 4). Выбор подложек связан с тем, что подложка
2 может образовывать сильные водородные связи с монослоем при
наличии в полимерной цепи соответствующих групп [3461; на под-
ложке 3 повышается общее адгезионное взаимодействие из-за уве-
"личения поверхностного натяжения раствора [1571, и, наконец,
33
Площадь на одну сложнозфирную связь, А?
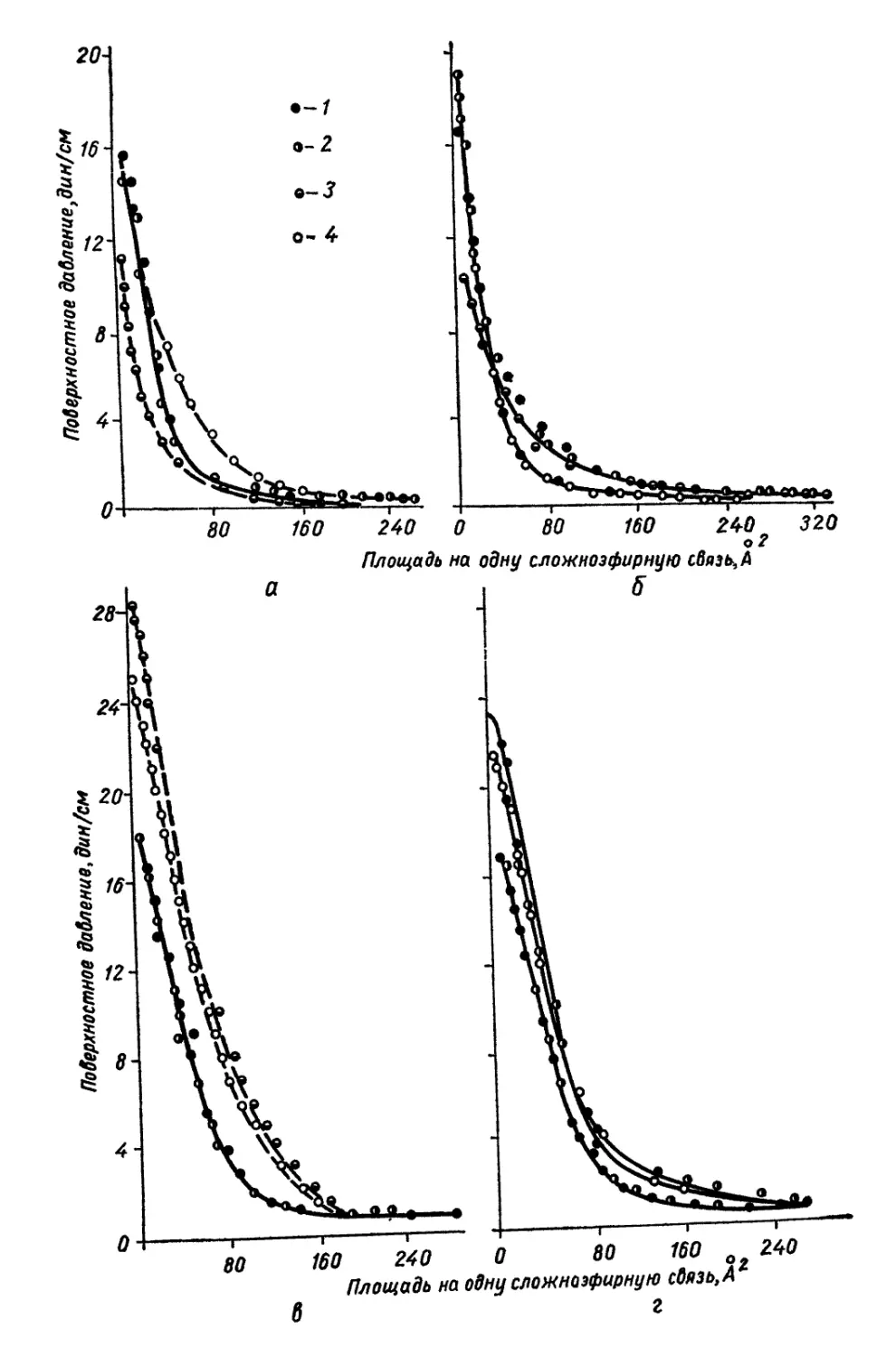
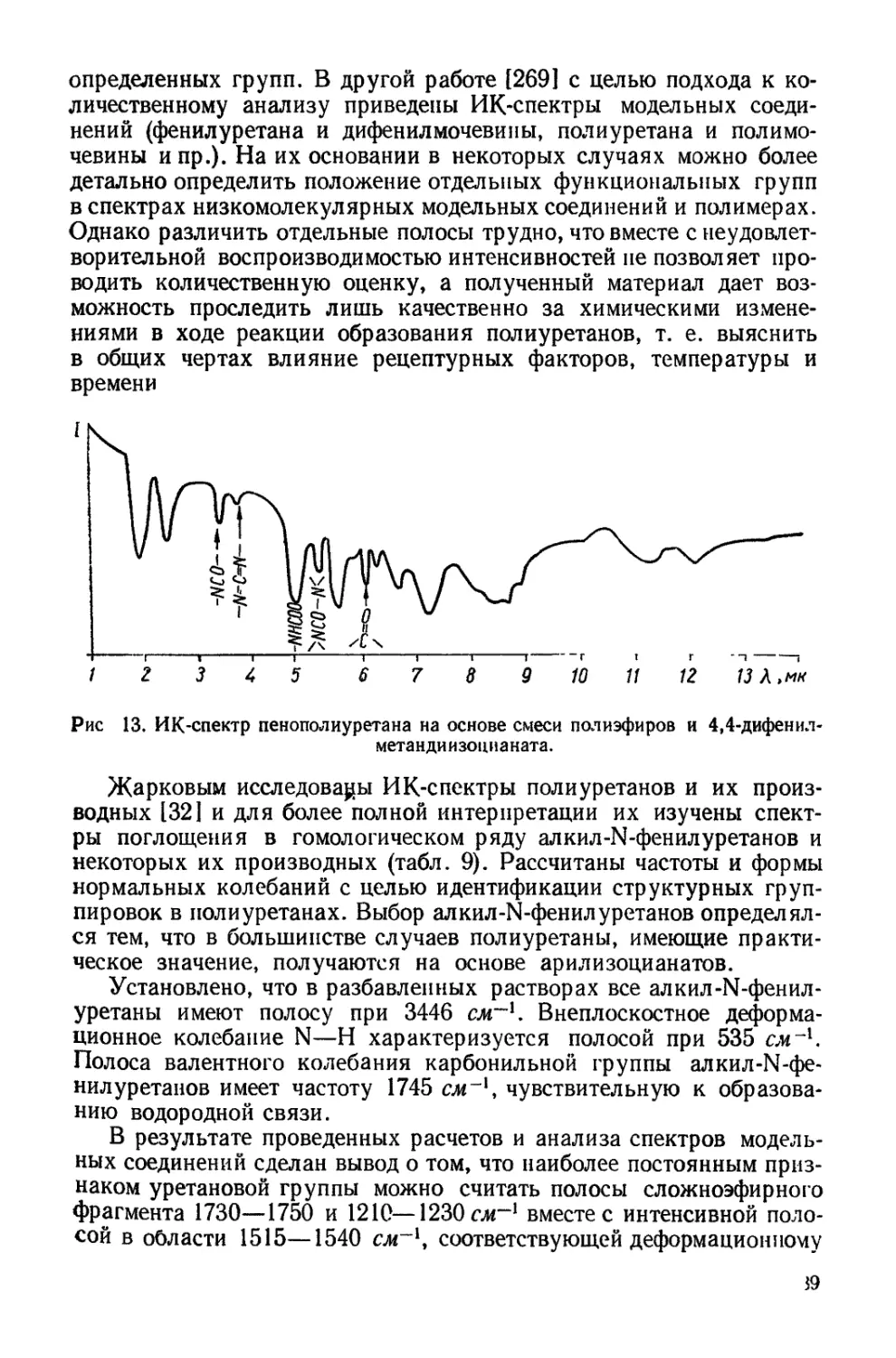
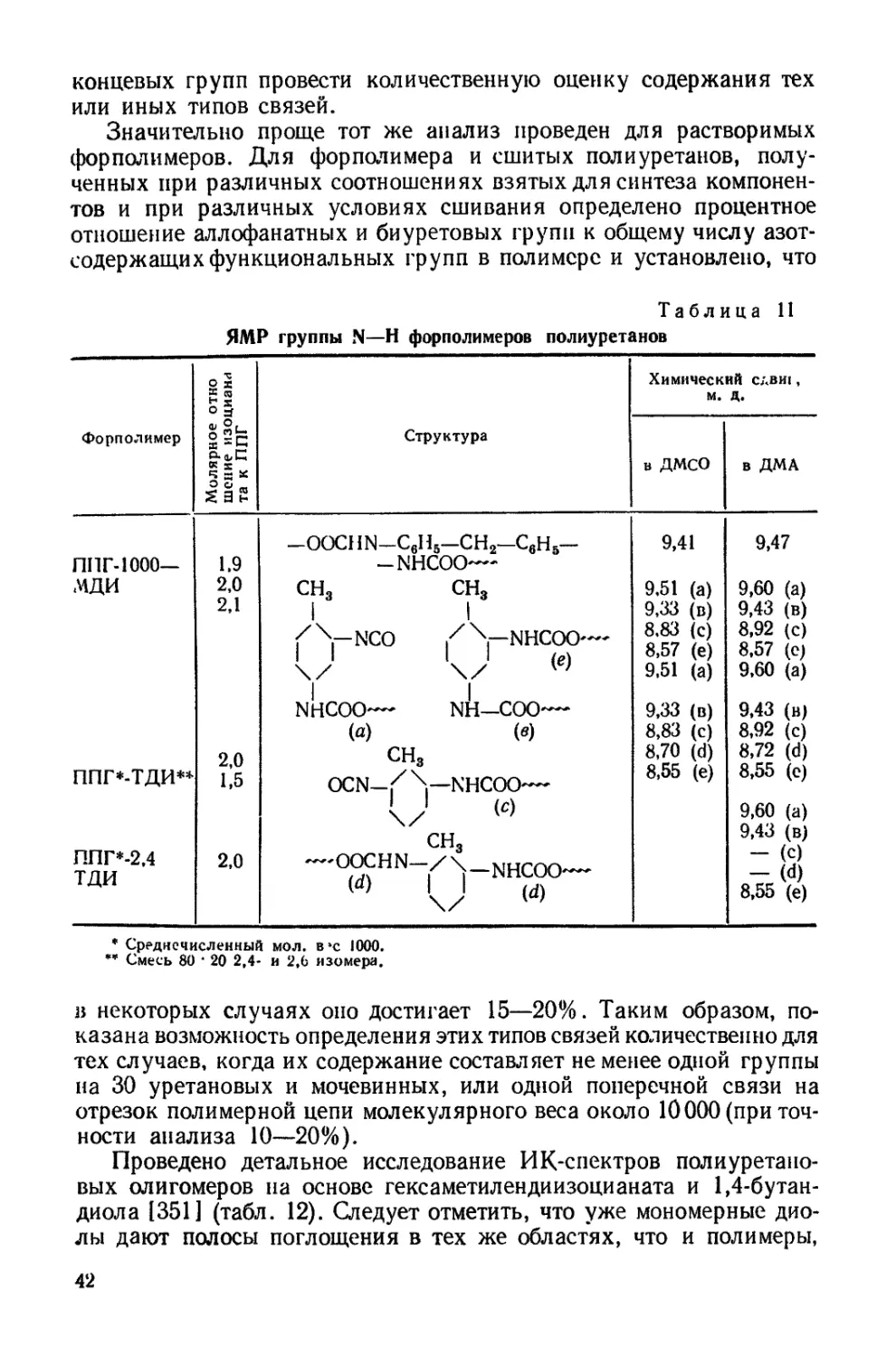
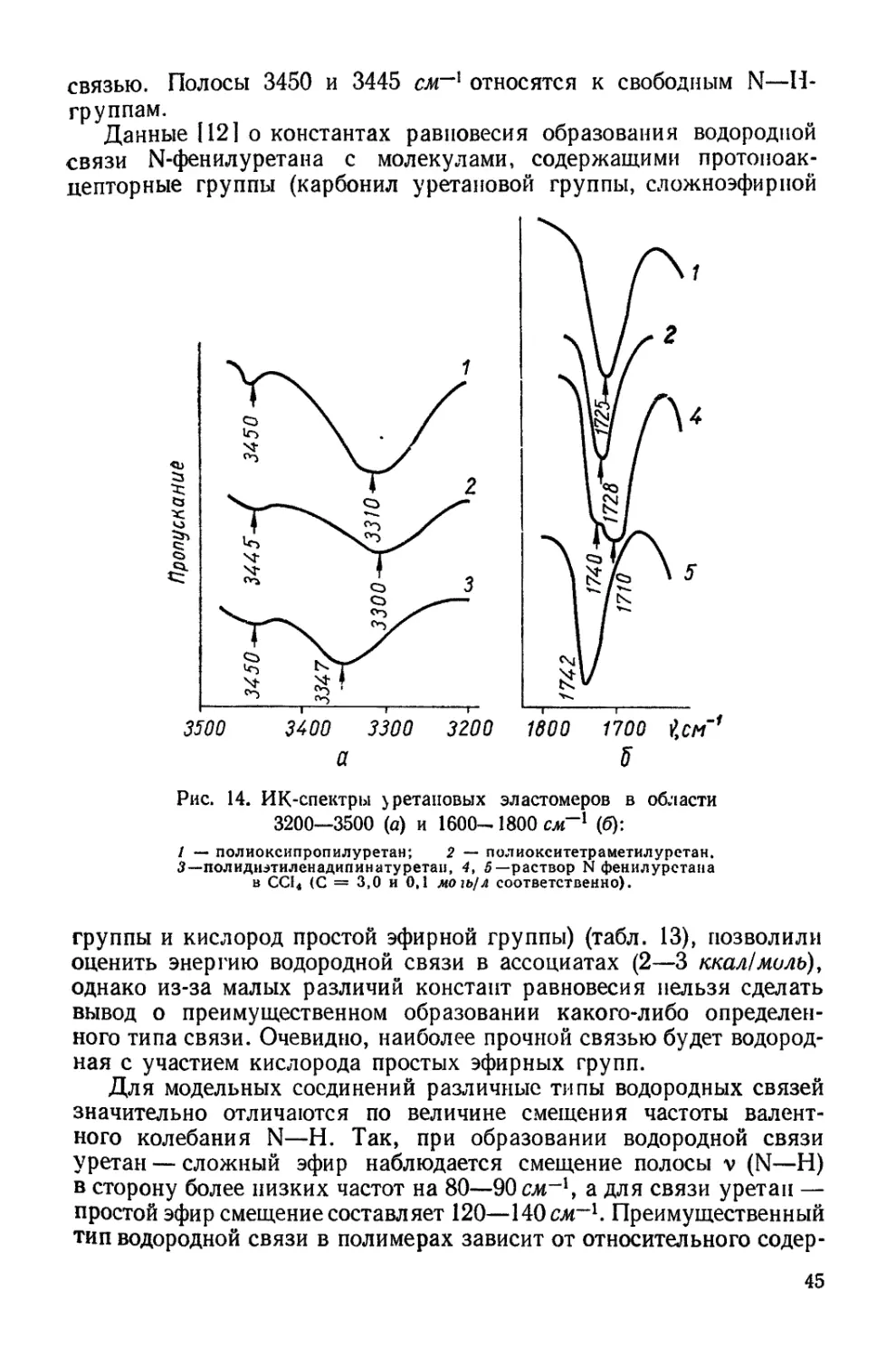
0 4-------,-------,------,----- Ч---------1-------1-------г-
80 160 240 0 80 160 о 240
Площадь на одну сложнозфирную связь,А*
6 г
подложка 4 должна вызвать суммарное воздействие подложек 3 и 4
(рис. 12, табл. 7). Монослои полиуретанов независимо от длины
полиэфирного блока дают идентичные изотермы сжатия, и лишь на
подложке 4 эти полиуретаны имеют несколько различные изотермы.
В тоже время изотермы сжатия монослоев полиэфиров на всех под-
ложках весьма отличаются при изменении длины цепи [141]. При
сопоставлении изотерм сжатия монослоев полиуретанов на подлож-
ках, содержащих соляную кислоту (подложки 2 и 4), и бескислот-
ных (подложки 1 и 3) видно, что в первом случае образуются более
растянутые монослои.
Таблица 7
Плотность упаковки и гибкость цепей полиуретанов и полиэфиров на
различных подложках
Характеристика цепей в моно- слоях Образец Поверхность
Н,О 3-н. НС1 40%-ный (NH4)2SO4 30% "ЙО-
Площадь плот- Полиуретан 1 25,5 25 40 40
ной.. упаков- Полиуретан 2 25,5 25 40 38
ки, А3 Полиэфир 1 10,0 25 37 22
Полиэфир ’2 40,0 40 46 38
Гибкость цепи Полиуретан 1 0,12 0,28 0,28 0,56
Полиуретан 2 0,12 0,28 0,28 0,88
Полиэфир 1 — — 1,00 0
Полиэфир 2 0,20 0,30 1,00 0,47
Идентичность изотерм сжатия монослоев полиуретанов, так же
как и меньшая растянутость по сравнению с таковыми для моно-
слоев исходных полиэфиров, объясняется, очевидно,тем, что длина
полиэфирного блока не оказывает влияния на плотность упаковки
молекулы в монослое. Увеличение взаимодействия между цепями
обнаруживается также и путем прямого измерения поверхностного
натяжения полиуретанов методом висячей капли [158], так как эти
продукты представляют собой высоковязкие жидкости. Измерения
показали, что если поверхностное натяжение полиэфиров состав-
ляет величину порядка 40—42 дин!см [1421, то полиуретанов —
46—47 дин!см, несмотря на малую концентрацию уретановых груп-
пировок в цепи (одна уретановая связь на 7,4 сложноэфирных
связей для полиэфирного блока с Мп = 800 и одна на 15,7 для
блока с Л4Я = 1700).
Рис. 12. Изотермы сжатия монослоев полимеров на воде (в), 3-н. соляной кис-
лоте (б), 40%-ном растворе сернокислого аммония (в) и на смешанном растворе
из 30% сернокислого аммония и 10% соляной кислоты (г):
I — полиуретан 1 (Afw = 12 200); 2 — полиуретан 2_(Afffl = 6400); 3 — полиэфир 1 (М„=*
= 640), 4 —- полиэфир 2 {Мп =» 2500).
35
Растянутость монослоев полиуретанов на подложках, содер-
жащих кислоту, позволяет однозначно установить, что сильное взаи-
модействие между цепями, вызванное появлением уретановых груп-
пировок, представляет взаимодействие типа водородной связи [268J.
Особенно четко эти закономерности проявляются при рассмотре-
нии плотности упаковки и гибкости цепи полиуретанов на различ-
ных подложках. Монослои полиуретанов имеют разную плотность
упаковки на подложках со слабым и сильным взаимодействием
(см. табл. 6), причем плотность упаковки меняется почти вдвое,
хотя такое различие для полиуретанов с разной длиной полиэфирно-
го блока отсутствует. В то же время плотность упаковки монослоев
полиэфиров меняется как от длины цени, так и от типа подложки.
Эти изменения так же, как и изменения гибкости цепи, объ-
ясняются следующим образом. Повышение гибкости полиурета-
новой цепи на подложке 2 связано с тем, что сильные водородные
связи подложки частично исключают подобное взаимодействие
между цепями в монослое. При этом имеется в виду боковая ко-
гезия между молекулами полимера и гибкость этой молекулы в
плоскости монослоя. Некоторое повышение гибкости полиэфира
обусловлено незначительным увеличением адгезионной связи с
концевыми группами полимера. Повышение адгезии монослоя по-
лиуретана к подложке 3 резко уменьшает плотность упаковки,
однако наличие водородной связи между цепями приводит к тому,
что гибкость цепи остается такой же, как и на подложке 2. В то
же время гибкость цепи полиэфиров, где отсутствуют подобные
связи, возрастает весьма значительно. Отсюда вытекает, что моно-
слои полиуретанов па подложках 1 и 2 имеют более высокую упо-
рядоченность, которая вызвана комплексным взаимодействием всех
видов вапдерваальсовых связей. При этом ожестчение цепи из-за
наличия уретановой группировки настолько сильно, что на упаков-
ку молекул в монослое нс влияет различная длина полиэфирного
блока. Лишь па смешанной подложке 4 гибкость полиуретановых
цепей возрастает настолько, что обнаруживается различное пове-
дение молекул в зависимости от длины полиэфирного блока: цепи
с меньшей концентрацией уретановых групп более гибки, так как
водородные связи подложки исключают взаимодействие между це-
пями в монослое. Отсутствие этой связи у полиэфиров приводит
к иному поведению их монослоев на подложке 4: падает гибкость,
возрастает плотность упаковки из-за уменьшения адгезионной свя-
зи монослой — подложка.
Следовательно, плотность упаковки и гибкость полимерной цепи
в поверхностном слое можно существенно изменять, подбирая раз-
личные подложки. Энергетический фактор оказывает сильное влия-
ние на конформацию полимерной цепи в поверхностном слое,
причем на гибкость и плотность упаковки полимерной молекулы
влияет не только величина взаимодействия на границе раздела
фаз, но и природа этого взаимодействия.
36
Глава III. ПРИРОДА ХИМИЧЕСКИХ И ФИЗИЧЕСКИХ
МЕЖМОЛЕКУЛЯРНЫХ СВЯЗЕЙ В ПОЛИУРЕТАНАХ
Гибкость цепей полиуретанов — один из факторов, определяю-
щих основные характеристики полимера. Однако необходимо учи-
тывать при изучении свойств полимеров природу межмолекуляр-
ных связей между полимерными цепями, которые определяют
физическое и фазовое состояние полимера и его основные механичес-
кие свойства. Многообразие функциональных групп в цепи поли-
уретанов создает широкие возможности для возникновения меж-
молекулярных связей различной энергии и химической природы —
от водородных до вапдерваальсовых. Роль межмолекулярных взаимо-
действий особенно важно учитывать при рассмотрении трехмерных
сетчатых полиуретанов, в которых, в отличие от большинства дру-
гих сетчатых каучукоподобных материалов, эти связи имеют первосте-
пенное значение. Они определяют также возможности кристаллиза-
ции полиуретанов, температурные области плавления или перехо-
дов из одного физического состояния в другое и т. п.
В настоящей главе мы рассмотрим существующие данные по
природе химических и физических связей в полиуретанах, имея в
виду, что участие тех или иных групп цепи в образовании химичес-
ких поперечных связей ограничивает их участие в формировании
физических связей.
ТИПЫ ХИМИЧЕСКИХ СВЯЗЕЙ В ЦЕПЯХ
При взаимодействии получающихся на первых стадиях реакции
уретановых, мочевинных и амидных групп с изоцианатами возмож-
но образование биуретовых (1), аллофанатных (II) и ацилмоче-
винных группировок (III)
-NH—СО—NH— + —1ХСО — X—CO-NH—
\о—NH-
I
-NH-COO - —NCO -> -Х-СОО—
СО—\Н—
II
—NH—СО— + —NCO —X—СО—
\o-NH-
Ш
37
Наиболее удобно идентифицировать различные типы связей в
сшитых полиуретанах с помощью инфракрасной спектроскопии.
Этот метод применен для анализа типов химических связей в пено-
полиуретанах различного химического строения [268].
Различные структурные группировки в полиуретанах дают в
ИК-спектрах характеристические частоты колебаний, известные для
соответствующих низкомолекулярных аналогов. Поэтому путем вы-
бора подходящих низкомолекулярных соединений (например,
М,Ы'-дифенилмочевины, Ы,1Ч',1Ч"-трифенилизоцианурата и пр.)
можно получить сведения о характеристических полосах. Для ха-
рактеристики соответствующих групп следует подбирать только та-
кие характеристические полосы, которые обладают достаточной
интенсивностью и наименее подвергаются влиянию других групп.
Авторы [268] рассматривают полосы, которые даны в табл. 8 и при-
Таблица 8
Характеристические полосы структурных групп в полиуретанах
Структурная группа х, мк V, см~~1
—N=C=O (кумулированные двойные связи) —N=C=N— (кумулированные двойные свя- 4,40—4,46 2270—2240
зи) /со\ —N N— Уретдионовое кольцо (кар- 4,72 2120
бонил) NH—СО—О Сложноэфирная группа (карбо- 5,62—5,72 1780-1755
нил) Уретановая группа, изоциануратное кольцо, 5,71—5,83 1750—1717
биуретовая группа (карбонил) 5,81—5,92 1720—1690
Аллофанатная группа (карбонил) 5,71—6.06 1750-1658
Мочевинная и амидная группы (карбонил) 6,10—6.20 1640-1610
Уретдионовое кольцо (колебания кольца) 7,04—7,14 1420—1410
Изоциануратное кольцо (колебания кольца) 7,00—7,11 1428-1406
водят типичный спектр отвержденного пенополиуретана на основе
десмофена и десмодура с добавкой других компонентов (рис, 13).
Эти данные указывают на сложность проведения количествен-
ного анализа полиуретанов по интенсивностям характеристических
частот. Действительно, если первые две группы идентифицировать
достаточно легко, то отличить сложноэфирные, уретановые, алло-
фанатные и другие связи по колебаниям карбонила практически
невозможно, так как одновременное наличие различных групп при-
водит к расширению полосы колебаний карбонильной группы. По-
этому результаты работы [268] можно применить лишь для установ-
ления наличия тех или иных групп только в том случае, когда на
основании химического состава композиции ожидается появление
38
определенных групп. В другой работе [269] с целью подхода к ко-
личественному анализу приведены ИК-спектры модельных соеди-
нений (фенилуретана и дифенилмочевины, полиуретана и полимо-
чевины и пр.). На их основании в некоторых случаях можно более
детально определить положение отдельных функциональных групп
в спектрах низкомолекулярных модельных соединений и полимерах.
Однако различить отдельные полосы трудно, что вместе с неудовлет-
ворительной воспроизводимостью интенсивностей не позволяет про-
водить количественную оценку, а полученный материал дает воз-
можность проследить лишь качественно за химическими измене-
ниями в ходе реакции образования полиуретанов, т. е. выяснить
в общих чертах влияние рецептурных факторов, температуры и
времени
Рис 13. ИК-спектр пенополиуретана на основе смеси полиэфиров и 4,4-дифенил-
метандиизоцианата.
Жарковым исследованы ИК-спектры полиуретанов и их произ-
водных [32] и для более полной интерпретации их изучены спект-
ры поглощения в гомологическом ряду алкил-М-фенилуретанов и
некоторых их производных (табл. 9). Рассчитаны частоты и формы
нормальных колебаний с целью идентификации структурных груп-
пировок в полиуретанах. Выбор ал кил-N-фенил уретанов определял-
ся тем, что в большинстве случаев полиуретаны, имеющие практи-
ческое значение, получаются на основе арилизоцианатов.
Установлено, что в разбавленных растворах все ал кил-N-фенил-
уретаны имеют полосу при 3446 см~~\ Внеплоскостное деформа-
ционное колебание N—Н характеризуется полосой при 535 см~{.
Полоса валентного колебания карбонильной группы алкил-И-фе-
нилуретанов имеет частоту 1745 см~\ чувствительную к образова-
нию водородной связи.
В результате проведенных расчетов и анализа спектров модель-
ных соединений сделан вывод о том, что наиболее постоянным приз-
наком уретановой группы можно считать полосы сложноэфирного
фрагмента 1730—1750 и 1210—1230 см~х вместе с интенсивной поло-
сой в области 1515—1540 ел/”1, соответствующей деформационному
$9
колебанию группы N—Н. Выбраны аналитические полосы для
идентификации аллофанатных и биуретовых групп. В спектрах
полимеров аллофанатные структуры маскируются связанными
NH-группами уретана. На модельных аллофанатах показано [49],
что их спектры отличны от спектров соответствующих уретанов.
Появление аллофанатов в спектрах полиуретанов связано с по-
лосами 1280, 1310 и 1965 см~{.
Таблица 9
Основные полосы поглощения, отнесенные к колебаниям
уретановой группы в алкил-Ь’-фенилуретанах (Ph—НСОО—R)
Индекс полосы Частота колебаний, см *
-сн2 “С2Н5 -СН(СП,), —С(СН3),
vo 3446 3446 3446 3446
vl' 1750 1745 1743 1742
v2 1529 1582 1528 1520
v3 1450 1445 1443 1443
v4 1312 1312 1311 1312
V5 1213 1210 1212 1222
V6 1068 1068, 1097* 1048, 1113* 1055, 1171*
V7 770 770 765 770
Vn 538 537 540 545
* Полоса растеплена за счет взаимодействия со скелетными колебания
ми алкильного радикала.
Таким образом, на модельных соединениях удается идентифици-
ровать различные типы связей, но их определение в полимерах
остается еще не решенной проблемой.
Значительно легче определить методом ИК-спектроскопии изо-
цианатную группу, что позволяет по ее исчезновению в ходе реак-
ции исследовать кинетику процесса образования полиуретанов. Дан-
ная реакция исследовалась по полосе поглощения изоцианатной
группы 2270 см-1 [169, 279J и уретановой 6750 см~1 [2491, а также
по исчезновению гидроксильных групп [501. Механизм реакций
образования полиуретановых эластомеров методом ИК-спектро-
скопии изучен также в работе [59], а полиуретановых покрытий—в
работе [101].
До сих пор метод ИК-спектроскопии не дает возможности коли-
чественно определить соотношение типов связей в сшитых полиурета-
нах. С этой точки зрения большой интерес представляют работы
40
[283, 3241, в которых проведено детальное исследование резонансной
области протонов для азотсодержащих связей в полиуретанах ме-
тодом ЯМР высокого разрешения. Установлено, что все соединения,
имеющие такие связи, дают резкие сигналы ЯМР в полярных раст-
ворителях типа диметилсульфоксида (ДхМСО), ди метил ацетамида
(ДМА), пиридина и др. Синтезирован ряд специальных соединений,
содержащих такие типы связей, которые могут присутствовать в
полиуретанах, и проведен их детальный анализ методом ЯМР. Для
модельных соединений (табл. 10) напряженности поля, при которых
Таблица 10
Ядерный ма1нитный резонанс группы N—Н
в модельных соединениях
Связь Химический сдвиг, м д.
в ДМСО в ДМА
Мочевинная 5,70-8,58 5,93—8,77
Уретановая 8.60—9,70 8,57—9,88
Биуретовая 9,60-10,25 9,85—10,55
Аллофанат на я 10,62-10,67 10,70-10,87
появляются сигналы ЯМР, сильно отличаются для различных типов
связей. Это позволяет использовать метод для количественного опре-
деления связей в полиуретанах. Действительно, анализ форполи-
меров, полученных из полипропиленгликоля и дифен ил мета иди изо-
цианата, показал, что наблюдается хорошее соответствие между
спектрами ЯМР модельных веществ и исследованных форполимеров
(табл. 11). При сравнении данных для форполимеров на основе поли-
пропиленгликоля и толуилендиизоцианата с данными для модель-
ных соединений (см. табл. 11) видно, что в спектрах такого форполи-
мера имеются три сигнала, отвечающие группам N—Н, два из которых
(9,43 и 8,55 м. д.) относятся к N—Н-протону 2,4-диуретана, а третий—
к протонам 4-уретана (а). Относительные интенсивности сигналов
зависят от условий реакции синтеза форполимера.
Для анализа типов связей авторы [283, 3241 использовали
метод, основанный на деструкции сшитых полиуретанов при по-
мощи аминов. Исследовались полиуретаны, полученные па основе
полипропиленгликоля (мол. вес 1000) и дифеп ил метан-4, ь4'-диизо-
цианата.
Изучены также модельные соединения, содержащие замещенные
уретаны, мочевины, аллофанаты и биуреты. Авторы при этом ис-
ходили из того, что при воздействии аминов на сшитые полиурета-
ны аллофанатные и биуретовые связи разрушаются значительно
быстрее мочевинных или уретановых. Это позволяет перевести сши-
тый полимер в растворимое состояние и по анализу образовавшихся
41
концевых групп провести количественную оценку содержания тех
или иных типов связей.
Значительно проще тот же анализ проведен для растворимых
форполимеров. Для форполимера и сшитых полиуретанов, полу-
ченных при различных соотношениях взятых для синтеза компонен-
тов и при различных условиях сшивания определено процентное
отношение аллофанатных и биуретовых групп к общему числу азот-
содержащих функциональных групп в полимере и установлено, что
Таблица 11
ЯМР группы N—Н форполимеров полиуретанов
Форполимер
Структура
Химический сдви1, м. д.
в ДМСО в ДМА
ППГ-1000—
мди
ППГ*-ТДИ**
ППГ*-2,4
ТДИ
1.9
2,0
2,1
2,0
1,5
2,0
- OOCHN-С6115—СН2—СвН5—
-NHCOO—
СН3
/\-NCO
NHCOO—
(Ф
СН3
—NHCOO—
(с)
СН3
—OOCHN—/\
(Ф I I
СН3
/Х—NHCOO—
\/ (е)
NH—СОО—
(в)
—NHCOO—
(Ф
9,41 9,47
9,51 (а) 9,60 (а)
9,33 (в) 9,43 (в)
8.83 (с) 8,92 (с)
8,57 (е) 8,57 (с)
9,51 (а) 9.60 (а)
9,33 (в) 9,43 (в)
8,83 (с) 8,92 (с)
8,70 (d) 8,72 (d)
8,55 (е) 8,55 (е)
9,60 (а)
9,43 (в)
- (О
- (Ф
8,55 (е)
* Среднечисленный мол. в»с 1000.
** Смесь 80 4 20 2,4- и 2,0 изомера.
в некоторых случаях оно достигает 15—20%. Таким образом, по-
казана возможность определения этих типов связей количественно для
тех случаев, когда их содержание составляет не менее одной группы
на 30 уретановых и мочевинных, или одной поперечной связи на
отрезок полимерной цепи молекулярного веса около 10000 (при точ-
ности анализа 10—20%).
Проведено детальное исследование ИК-спектров полиуретано-
вых олигомеров на основе гексаметилен диизоцианата и 1,4-бутан-
диола [351] (табл. 12). Следует отметить, что уже мономерные дио-
лы дают полосы поглощения в тех же областях, что и полимеры,
42
отличаясь только наличием характеристических полос, относящихся
к концевым группам. Следовательно, методом инфракрасной спект-
роскопии пока можно только качественно анализировать структу-
Таблица 12
Отнесение частот в олигоуретанах
V» Интенсивность Колебания
3325 Сильная N—Н-валентные
3060 Очень слабая Амид 11 в резонансе Ферми с Nrl-группой
2945 Сильная Антисимметричные СН2-валент- ные
2860 Средняя Симметричные СН2
1690 Очень сильная Амид I
1540 » » . Амид II
1474—1414 Средняя до сла- бой Средняя СН 2“ деформационные
1340 ОС-деформационные Амид III
1265 Очень сильная
1244 Сильная Антисимметричные СОС-валент- ные
1180 Очень слабая Симметричные СОС-валентные
1137 Сильная С N-валентные
1065 —'
995 Средняя С—С-валентные
783 » С—О-деформационные
748 Слабая СН2-маятниковые, цепи (СН2)Л
730 » СН2-маятниковые. цепи (СН2)6
ру трехмерных полиуретанов и количественно определять измене-
ния концентрации уретановых групп при сшивании и других хими-
ческих превращениях. Однако метод не пригоден для установления
соотношения между структурой и свойствами полиуретанов, а также
числом и типом поперечных связей.
ВОДОРОДНАЯ СВЯЗЬ В ПОЛИУРЕТАНАХ
Химическое строение полиуретановых цепей и наличие различ-
ных функциональных групп определяют большие возможности
для образования в полиуретанах межмолекулярных связей, в том
числе водородных. Хорошо известно, что внутри- и межмолекуляр-
ные связи в полимерах оказывают заметное влияние на структуру
и весь комплекс физико-механических свойств. Особенно велика
роль водородных связей в полиуретанах, так как протонодонорная
группировка N—-Н в уретановой группе и протоноакцепторные ато-
мы кислорода (в виде эфирного кислорода в простой эфирной связи
и в карбонильной группе) определяют возможности водородного
43
связывания. Схематически три основных типа водородных связей
можно представить следующим образом:
Н
К °ч 'с = о
О-С С = О.....Н\’^
ХО......H-NZ
Z I
О
'о.....H-NZ N—Н.......O = cf
/ \ I
II III
Как видно, это связи между уретановыми группами или уретано-
вой и сложноэфирной группой и простым эфирным кислородом смеж-
ных цепей.
Осуществление в полиуретанах того или иного типа водородных
связей зависит от жесткости молекулы, определяемой длиной оли-
гомерного блока или цепочки гликоля, концентрации уретановых
групп в цепи, густоты пространственной сетки, ограничивающей
подвижность отрезков цепей между узлами химической сетки. Жар-
ков 132] полагает, что поскольку эластомерные полиуретаны мож-
но рассматривать как своеобразный «раствор» уретановых групп
в полиэфире, то преобладающим типом водородных связей будет
форма II.
Для исследования водородных связей применяется метод ИК-
спектроскопии, так как при образовании водородных связей частоты
валентных колебаний групп N—Н и С=О сдвигаются в сторону
более низких частот. Число работ в этом направлении невелико.
С помощью ИК-спектроскопии изучены водородные связи в уре-
тановых эластомерах, полученных на основе сложных и простых
полиэфиров, а также на модельном соединении — N-фенилуре-
тане [12].
Для оценки вероятности образования того или иного типа свя-
зи определялась относительная электронодонорная способность
протоноакцепторных групп при сравнении констант ассоциации
молекул N-монозамещенных уретанов, моделирующих уретановое
звено полимера, с молекулами соединений, содержащих протоно-
акцепторные группы, подобные таковым в полимерных цепях (метил-
этилкетон, этилацетат, изобутилбутират, тетрагидрофуран, диок-
сан и пр.).
Из ИК-спектров уретановых эластомеров в области 3200—3500
и 1600—1800 см"1 (рис. 14) видно, что для всех эластомеров наблю-
даются широкие полосы поглощения (3300, 3310 и 3347 cjh-1), отно-
сящиеся к поглощению группы N—Н, возмущенной водородной
44
связью. Полосы 3450 и 3445 см~' относятся к свободным N—Н-
группам.
Данные (121 о константах равновесия образования водородной
связи N-фенилуретана с молекулами, содержащими протоноак-
цепторные группы (карбонил уретановой группы, сложноэфирной
Рис. 14. ИК-спектры уретановых эластомеров в области
3200—3500 (а) и 1600—1800 см~1 (б):
1 — полиоксипропилуретан; 2 — полиокситетраметилуретан.
<3—полидиэтиленадипин ату ретаи, 4, 5—раствор N фен ил уретан а
в CCI4 (С = 3,0 и ОД MOib/A соответственно).
группы и кислород простой эфирной группы) (табл. 13), позволили
оценить энергию водородной связи в ассоциатах (2—3 ккал/моль),
однако из-за малых различий констант равновесия нельзя сделать
вывод о преимущественном образовании какого-либо определен-
ного типа связи. Очевидно, наиболее прочной связью будет водород-
ная с участием кислорода простых эфирных групп.
Для модельных соединений различные типы водородных связей
значительно отличаются по величине смещения частоты валент-
ного колебания N—Н. Так, при образовании водородной связи
уретан — сложный эфир наблюдается смещение полосы v (N—Н)
в сторону более низких частот на 80—90 см~х, а для связи уретан —
простой эфир смещение составляет 120—140 см~х. Преимущественный
тип водородной связи в полимерах зависит от относительного содер-
45
Таблица 13
Константы равновесия и смещения частоты (Av) N — Н при
образовании водородной связи N-фенилуретаном
Раст- вори- тель Соединение, содер- жащее протоноак- цепторные группы Система водородной связи X, Л/МОЛЬ Av, cm—1
СС14 Фенилу ретан о Z 11 О X .OR NH 2,0 —
Мети л этил кетон о 7 II о X 1 .R, Ra 1,5 88
Этилацетат _N-H...O=C OR, 'R* 1,5 88
Изобутилбутират 1 Z X о 11 OR, r2 1,7 89
Свни Диметиладипинат 1 Z 1 X о II о .OR, R. 1,2* 81
СС14 Дибутиловый эфир XR, 1,1 140
с«н14 Диметиловый эфир диэтиленгликоля 0? оГ oZ X Z 1 0,7* 126
Тетрагидрофуран СИ СИ Хо Z X Z 1 4 J 142
Диоксан 1 Z X д яз я 3,0 128
♦ Приведенные значения соответствуют К, деленному на число
акцепторных групп в молекуле.
46
жания различных протоноакцепторных групп и их пространствен-
ного расположения в цепи.
Сравнение спектров уретанов на основе простых и сложных
полиэфиров показывает (рис. 14, а) большое различие в частотах
поглощения N—Н-групп, возмущенных водородной связью, что
указывает на различные виды связей в этих системах. В сложно-
эфирном полиуретане, очевидно, основным видом водородной связи
является связь с карбонилом сложноэфирной группы. Образова-
ние водородной связи сопровождается также смещением полосы
С=О в область низких частот, что можно использовать для оценки
участия группы С=О в водородной связи. Эта полоса имеет максиму-
мы 1725 и 1728 см-1 (полиокси-пропиленуретан и полиокситетраме-
тиленуретан). Смещение максимума в область более низких частот
указывает на участие С=О-групп в водородных связях. Таким об-
разом, можно сделать заключение о преимущественном образова-
нии в сложноэфирных полиуретанах водородной связи между N—Н
уретанового звена и сложноэфирной группой, а в эластомерах на
основе простых полиэфиров — между N—Н и С=О уретановых
звеньев. Небольшая роль связей N—Н с С=О уретановых групп
объясняется их малым содержанием, а незначительная доля
связей с простым эфирным кислородом — стерическими факто-
рами.
Исследование водородных связей в полиуретанах, полученных
на основе диизоцианатов со смесями высоко- и низкомолекулярных
диолов [32], позволило установить еще одну интересную особенность
этих связей в полиуретанах. Участие диолов в реакции нарушает
регулярное расположение уретановых групп в цепях получающего-
ся полимера. Эффективное изменение концентрации этих групп
сопровождается не только изменением общего количества, но и
видов водородных связей, образующихся при взаимодействии про-
тона NH—COO-групп с включенными в полимерную цепь прото-
ноакцепторными группами. Показано [32], что при больших кон-
центрациях уретановых групп специфические взаимодействия меж-
ду ними отличаются от взаимодействий в полимерах с меньшим
содержанием таких групп, т. е. в зависимости от условий уретановые
группы могут образовывать два вида комплексов с водородными
связями, соотношение которых может зависеть от химической при-
роды полимера. Этот факт еще раз подчеркивает разнообразие типов
водородных связей в полиуретанах.
С целью получения сведений о роли водородной связи в поли-
уретановых эластомерах [326] методом ИК-спектроскопии исследо-
вано взаимодействие между N-фенилуретаном (I) и дифениламином (II)
как донорами протонов и ди-н-бутиловым эфиром и полиоксиэтилен-
гликолем (мол. вес 6000) как акцепторами протонов в растворе
СС14. С помощью метода Вильсона — Уэллса [339] определены
интегральные коэффициенты поглощения свободных N—Н-групп в
растворах разных концентраций для обоих компонентов. Образование
47
водородной связи сдвигает полосу поглощения деформационных
колебаний N-—Н в сторону низких частот.
На основании экспериментальных спектров с использованием
интегральных коэффициентов поглощения вычислены концентрации
свободных N—Н-групп во всех исследованных системах в зависи-
мости от суммарной и раздельной концентрации исследованных
компонентов. Был также рассмотрен вопрос о равновесии в раст-
ворах между связанными и свободными N—Н-группами и опре-
делены кажущиеся константы равновесия. Истинные константы
равновесия не могут быть определены из-за различных степеней
ассоциации молекул при связывании водородной связью.
Установлено [3261, что N—Н в I и II может образовывать водо-
родную связь с простым эфирным кослородом, которая, однако,
характеризуется невысоким значением константы равновесия и по-
этому слаба по сравнению со связью N—Н с карбонильной группой.
Водородное связывание в системе I — простой эфир осложняется
наличием двух типов акцепторов: —О— и С=О. Полученные дан-
ные показали, что в разбавленных растворах образуется водо-
родная связь с простым эфирным кислородом, если его концентра-
ция в системе достаточно велика. С ростом концентрации уретана
становится возможной ассоциация его молекул по С=О-группам,
а при высокой концентрации уретана реализуется только послед-
ний тип взаимодействия. На этом основании делается вывод о том,
что в полиэфируретапах имеет место сложное равновесие водород-
ных связей между N—Н и С=О уретановых ipynn и —О— поли-
эфирной цепи. Если концентрация N—Н в полиуретанах мала,
большая часть их образует водородные связи с эфирным кислородом.
В таком случае вторичные межцепные связи рассеяны по всему объе-
му полимера. Если концентрация N—Н-групп у низкомолекуляр-
ного полиэфира велика, то локализуются связи типа N—Н • • С=О.
Отсюда создается возможность изменения конфигурации цепей в
сетке при изменении концентрации уретановых групп. В сложно-
эфирных системах преобладает сильный акцептор С=О и здесь
снова появляется вероятность того, что связи NH • • С=О нелокализо-
ваны в объеме даже при высокой концентрации уретатовых групп.
С целью исследования природы водородной связи в полиуре-
танах некоторые проявления такой связи изучены на модели, в
качестве которой взят N-этилуретан 1181]. На основании анали-
за ИК-спектров N-этилуретана и его растворов в растворителях,
содержащих простые и сложноэфирпые связи, а также исследова-
ния вязкости и диэлектрической поляризации сделан вывод о том,
что в полиуретанах вследствие высокого содержания простых и
сложных эфирных групп процессы взаимодействия определяются
преимущественно взаимодействием уретановых групп с эфирными,
а не друг с другом.
Для полиуретанов методом ИК-спектроскопии изучено погло-
щение в области колебаний группы N — H 1327]. Оно имеет место
48
в области А = 3 лк, что интерпретируется как поглощение N—Н-гру н-
пами, связанными водородными связями, и свободными группа-
ми N—Н. Большая часть N—Н-групп участвует в водородных
связях независимо от структуры самого полиуретана (эфирно- или
сложноэфирного типа, линейных или сшитых).
Исследовано также влияние растворителей на спектры погло-
щения линейных полиуретанов и установлено, что при разбавлении
происходит диссоциация водородных связей. Для сшитых полиуре-
танов незначительная диссоциация водородных связей отмечена в
интервале температур 20—100° С.
Частичная диссоциация наблюдалась
и при растяжении образцов.
В связи с тем, что многие свой-
ства полиуретанов объясняются имен-
но различием в степени водородного
связывания макромолекул друг с
другом, этот вопрос был специально
исследован 1220].
Изучены ряды полиуретанов об-
щей формулы
[- (CH2)i-OCONH- (СН2)в -
—NHCOO—]„
при у = 4 — 10, включая линейные
и сшитые полимеры, а также их мо-
дели — диуретаны. Свободные коле-
бания N — Н-групп имеют место, как
было указано выше, при 2,9 мк
(3448 см~'). Типичный спектр в этой
на приведен на рис. 15. Спектры сняты в интервале температур
20—300 С.
Рис. 15. Спектр полиуретана в
области колебаний N—Н при
22° С (/) и 100 °C (2).
области для 6,8-полиурета
Интенсивность колебаний (рис. 15) свободных N—Н-групп воз-
растает с ростом температуры (спектр 2). Однако авторы не нашли
различий в степени водородного связывания для четных и нечет-
ных полиуретанов, а также полиамидов. При комнатных темпера-
турах у линейных полиуретанов менее 1% свободных N—Н-групп
(0,8% для 6,8- и 6,9-гомологов и 1,5% для 6,6- и 6,10-сополиурета-
нов). Интересно, что сшитые полиуретаны не показывают большей
интенсивности колебаний свободных N—Н-групп, т. е. даже в
нерегулярных системах наблюдается практически полное связыва-
ние. Для 6,8-полиуретана при 175°С содержание несвязанных групп
составляет всего 15%. Таким образом, объяснение различия свойств
полиуретанов разной степенью водородного связывания не может
быть приемлемым вследствие того, что практически во всех слу-
чаях свободные 1\'Н-группы в системе отсутствуют.
Для рассмотрения роли водородной связи в полиурета-
нах большой интерес представляет вопрос о возможности
4 407
49
о 1енки энергии этой связи Для такой оценки можно воспользовать-
ся поглощением групп N—Н. Соответствующие расчеты приведены
в [32]. Константа равновесия между несвязанной и связанной во-
дородной связью группой N—Н определяется выражением
тг _ Уд
К “7“ ‘ ’
где ta и tm — молярные коэффициенты поглощения, Da и —оп-
тические плотности полос поглощения связанной и свободной
N—Н-групп. Рассчитанная из величин К при двух различных темпе-
ратурах, энергия водородной связи (ДН) в полиуретанах составила
4,8± 1 ккал! моль. Согласно [2201, эта величина для связи N—Н • -ОС
равна 8,36-Ь 1,6 ккал! моль, Интересно отметить, что в полиуретанах
на основе 2,4- и 2,6-толуилендиизоцианата уретановые группы рас-
положены неэквивалентно по отношению к арильному радикалу,
в результате чего образуемые этими уретановыми группами водо-
родные связи должны различаться по своей энергии. Полоса погло-
щения (N—Н) с частотой 3444 см 1 относится к уретановой группе,
пространственное расположение которой способствует образова-
нию водородной связи, а полоса 3460—3463 см~х отвечает уретано-
вой группе, пространственное расположение которой затрудняет
образование таких связей. Вследствие этого в полиуретановых элас-
томерах могут существовать несвязанные водородной связью N—FI-
группы. Рассмотрение вопроса об ассоциации в результате возникно-
вения водородных связей в модельных уретанах и некоторых поли-
уретанах привело к выводу о том, что самоассоциация уретановых
групп не может быть определяющим фактором в образовании водо-
родных связей в полиуретанах на основе простых полиэфиров [321.
Большое значение имеет связь между уретановой группой и эфир-
ным кислородом.
В реальных полиуретановых эластомерных системах должна
наблюдаться более сложная картина. С одной стороны, наличие
только мочевинных групп приводит к возможности образования
водородных связей между мочевинными группами
Н—N О.....Н—N
\ = О
с/
Образование аллофанатных и других групп еще больше изменяет
условия и возможности образования водородных связей.
С другой стороны, их возникновение существенно зависит от
химической природы полиуретана в том отношении, что длина гли-
колевых блоков, тип диизоцианата и прочие химические факторы
влияют на взаимное расположение групп, способных образовывать
водородные связи, и на их подвижность. Так, в работе [324] показана
возможность образования внутримолекулярных водородных свя-
зей для аллофанатов и биуретов и установлено, что водородное
50
связывание интенсивнее в биуретах. Эти выводы сделаны на осно-
вании анализа спектров ЯМР высокого разрешения. Для биуретов
авторы дают следующую схему образования водородных связей:
Н Н
с возможными переходами между двумя структурами.
Сведения о водородных связях в полиуретанах пока еще огра-
ничены как по числу исследованных систем, так и по их химичес-
Рис. 16. Схема Н-связей в уретановых эластомерах:
а — полимеры на основе сложных полиэфиров, б — поли-
меры на основе простых полиэфиров, / — уретановые
участки полимерной цепи; 2 — сложноэфнрные группы.
кому строению. Исследование этого вопроса очень важно для де-
тального понимания свойств полиуретановых материалов. Так, в
работе [12) высказано предположение о том, что различные виды во-
дородных связей могут привести к определенным различиям в упа-
ковке молекулярных цепей в сложноэфирных полиуретанах и поли-
уретанах на основе простых полиэфиров. Авторы предложили схе-
му образования водородных связей в уретановых эластомерах (рис. 16).
Связь между уретановыми и сложноэфирными звеньями не накла-
дывает никаких ограничений на взаимное расположение уретано-
вых участков цепей (рис. 16, а). Возникновение водородных связей
между двумя уретановыми звеньями требует определенного взаим-
ного расположения их в цепи (рис. 16, б), что может привести к
частичному упорядочению.
При рассмотрении вопроса о роли водородных связей следует,
по нашему мнению, иметь в виду, что значение их в физико-меха-
4* 51
нических свойствах полиуретановых эластомеров невелико. Это свя-
зано с тем, что в эластомерных системах относительная концентра-
ция способных к образованию водородных связей уретановых групп
мала и снижается с ростом молекулярного веса полиэфира. Вместе с
тем, общий вклад физических взаимодействий в эффективную плот-
ность сетки полиуретановых эластомеров существенен. Это обуслов-
ленотем, что наличие многих функциональных групп в полиуретано-
вой цепи приводит к значительным вандерваальсовым взаимодейст-
виям между цепями, значительно большим, чем в случае обычных
(неполярных) каучуков. Поэтому большой интерес представляет
исследование межмолекулярного взаимодействия между составляю-
щими блоками полиуретановой цепи и обычных вандерваальсовых
взаимодействий. Приведенные нами данные о реологических свой-
ствах олигомеров, применяющихся для синтеза полиуретанов, от-
четливо показывают, какую важную роль в общем комплексе свойств
играют обычные вандерваальсовые взаимодействия (см. стр. 216).
Другая картина наблюдается для кристаллических полиурета-
нов Образование водородных связей имеет существенное значение
для кристаллизующихся полиуретанов, особенно на основе низко-
молекулярных блоков. Эти связи определяют как кристаллическую
решетку, так и другие свойства полимеров. Однако в отличие от
полиамидов, для кристаллических полиуретанов исследований по
водородным связям проведено мало.
ИК-спектры полиэфиров и полиуретанов описаны в работах
[81, 821. Данные объекты способны существовать в двух кристал-
лических формах, если в цепочке структуры
О О
II II
[—С— (СН2)4 —С—О— (СН2), -О-Аг-О- (СН2)2 -О-]„
Аг—м- или n-фенилен. Для разных полимеров определены по-
лосы кристалличности в разных модификациях. В зависимости от
кристаллической формы может образоваться водородная связь. По-
явление водородных связей способно стабилизировать кристалли-
ческие модификации. Эти работы указывают на значение водород-
ных связей в закристаллизованных полимерах уретанового ряда.
Для способных к кристаллизации поли амидоуретанов ИК-
спектры сопоставлены с рентгенографическими исследованиями
[29]. Учитывая, что водородные связи между отдельными молекула-
ми влияют как на конформации самих молекул, так и на их упаковку
в кристаллических структурах, и что помимо принципа плотной
упаковки должен соблюдаться принцип максимального насыщения
водородных связей, предложена вероятная модель расположения
макромолекул в кристаллических областях полиамидуретанов.
В литературе имеются некоторые данные по межмолекулярным
взаимодействиямвсоединепиях, являющихся компонентами полиуре-
танов. Исследованы внутри- и межмолекулярные водородные связи
в гликолях [255, 256], с помощью спектроскопии изучены внутримо-
52
лекулярные связи в некоторых полиоксиалкилендиолах [311. Для
полиоксипропиленгликоля и политетрагидрофурана полосы погло-
щения 3480 и 3457 си-1 отнесены к колебаниям ОН-групп, включен-
ных в водородную связь, а 3640 см~' — к свободной ОН-грунпе в по-
литетрагидрофуране. Из температурной зависимости коэффициентов
поглощения для полос‘свободных и связанных водородной связью
ОН-групп вычислена энергия внутримолекулярной водородной
связи по методу [33]. Установлено, что эта энергия для исследован-
ных систем составляет 2,1—2,8 ккал!моль. Однако надо иметь в виду,
что эти взаимодействия определяются в основном концевыми груп-
пами.
Таким образом, применение спектроскопических методов к ис-
следованию полиуретанов позволяет дать качественные характерис-
тики как химического строения сетки, так и некоторых типов межмо-
лекулярных взаимодействий в полиуретанах аморфного и кристал-
лического строения. Тем не менее, создание стройной теории свойств
полиуретанов, основывающейся на детальном качественном и коли-
чественном спектральном анализе, остается делом будущего.
Глава IV. СТЕКЛОВАНИЕ И ФАЗОВЫЕ ПРЕВРАЩЕНИЯ
В ЛИНЕЙНЫХ И ТРЕХМЕРНЫХ ПОЛИУРЕТАНАХ
СТЕКЛОВАНИЕ АМОРФНЫХ ПОЛИУРЕТАНОВ
Известно, что температура стеклования (TJ полимеров опре-
деляется как внутренней подвижностью или гибкостью полимерных
цепей, так и силами межмолекулярного взаимодействия, в значи-
тельной степени зависящими от наличия в цепях полярных групп.
Таким образом, Тс полимеров зависит от длины, природы и структу-
ры их цепей (речь идет об аморфных не модифицированных наполни-
телями или пластифика-
торами полимерных си-
стемах).
Все указанные фак-
торы для полиуретанов
определяются свойства-
Таблица 14
Температуры переходов полиуретановых
эластомеров
Полимер Тс гмин
ми их исходных состав-
ляющих, т. е. свойствами
гликолей (эфироглико-
лей) и диизоцианатов.
Следовательно, свойства
исходных составляющих
и их соотношение в поли-
Полидиэтпленадипииатуретан —39 —13
Папитриэтиленадипинатуретан —42 — 16
Полигексаэтиленадипинатуретан —44 -18
Примечание. Для всех полимеров ДГ = Тс —
~ ^мии =“ 23' С
мерной цепи в основном и должны определять Тс полиуретанов.
Изучена зависимость Тс полиэфируретановых эластомеров от
структуры исходных гликолей, в частности, от наличия простых
эфирных связей в основной цепи и ее боковых подвесках, а также
от числа метиленовых групп в гликоле (93].
Наряду с Тс, которую определяли на приборе, описанном в [113]
на маятниковом эластомере КС [52], исследовали эластичность
полиуретановых эластомеров на основе сложных олигоэфиров и
2,4-толу илендиизоцианата со средним молекулярным весом около
20 000. По минимумам кривых температурной зависимости эластич-
ности вычисляли температуру минимума эластичности (ТМИ11)
(табл. 14).
Из данных табл. 14 видно, что возрастание концентрации
С—О—С- групп в основной цепи полимера приводит к последова-
тельному понижению Тс. Постоянная для данного ряда полимеров
разность межд\ Тс и Тмин свидетельствует о четко выраженной коли-
чественной связи между этими двумя величинами.
£4
Наблюдаемый характер изменения Тс объясняется ослаблением
межмолекулярного взаимодействия вследствие уменьшения концент-
рации сильно полярных сложных эфирных групп в полимерной
цепи по мере увеличения длины мономерного звена исходного гли-
коля.
На примере полидиэтилен- и полипентаметиленадипинатурета-
нов, отличающихся лишь наличием в гликолевых звеньях цепи
СН.2-группы вместо эфирного кислорода, показано, что отсутствие
С—О—С-групп приводит к понижению Тс. Это объясняется возмож-
ностью связывания эфирного кислорода водородными связями,
приводящего к понижению подвижности цепей.
В то же время для полипропил- и полимстоксиметилэтиленадипи-
натуретанов, отличающихся лишь структурой боковых групп
----NHCO- [О-СН—СН,ОСН2ОСО (СН,)4 СО— J,,-,
I
сн,
I
сн2
I
сн.,
—-NHCO- [О—СН—СН2ОСН2ОСО (СН2)4 СО—]„——,
I
СН2
о
дн
отсутствие в боковой цепи эфирного кислорода не сказывается на
температуре стеклования, которая для обоих полимеров одинакова.
Изучение Тс полиуретанов, полученных на основе гликолей с
четной й нечетной последовательностями метиленовых групп с общей
формулой
—NHCO-— (О (СН2)„ОСО (СН2)4СО]Х---------,
где п = 2^ 3,4, 5, показало, что полимеры с нечетным числом п имеют
более низкую Тс, чем с четным (рис. 17).
Исследована зависимость Тс полиуретановых эластомеров на ос-
нове полиоксипропиленгликоля и 2,4-толуилендиизоцианата или
гексаметилендиизоцианата от мольной концентрации уретановых
групп [317| (рис. 18).
Как видно из рис. 18, Тс для обоих полиуретанов линейно растет
с ростом концентрации уретановых групп. Для эластомера на основе
2,4-толуилендиизоцианата угол наклона прямой больше, чем для
эластомера на основе гексаметилендиизоцианата, что вызвано одно-
временным ростом концентрации как уретановых, так и фенилено-
вых групп; последние также увеличивают жесткость цепей. Однако
экстраполяция кривой Тс — С к С=0для обоих эластомеров дает
Гс, соответствующую таковой для высокомолекулярного полиокси-
пропилен гл и кол я.
55
Аналогичная зависимость Тс от С найдена и для полиуретан-
акрилатов [128| на основе олигооксипропиленгликолсй, 2,4-толу-
илендиизоцианата и монометакрилового эфира этиленгликоля.
Однако в данном случае угол наклона прямой Тс — С больше, чем
для приведенного выше эластомера с аналогичными олигоэфирными
и диизоцианатными блоками. Это объясняется наличием в полиуре-
танакрилатах сильно полярных слож-
Тс°С ных эфирных групп, обусловливающих
Рис. 18. Зависимость Тс от мольной концен-
Рис. 17. Зависимость Тс от трации уретановых групп (С) полиуретановых
количества СН2-групп в гли- эластомеров на основе олигооксипропиленгли-
колевом сегменте полиурета- коля с 2,4-тол у илендиизоцианатом (/) и оли-
новой цепи. гооксипропиленгликоля с 1,6-гексаметиленди-
изоцианатом (2).
более низкую подвижность полимерных цепей при той же концен-
трации уретановых групп, что и в сравниваемом эластомере.
Мы исследовали Тс в ряду: олигодиэтилен гл икольадипинат
(ОЭ), линейный олигоуретан (форполимер) и сшитый полиуретано-
вый эластомер [65]. Диизоцианатной компонентой служила смесь
изомеров 2,4- и 2,6-толуилендиизоцианата (ТДИЦ), а сшивающим
агентом — эквимолярная смесь ди- и триэтаноламина. Температуру
стеклования определяли дифференциально-термическим анализом,
как и в [38].
Влияние молекулярного веса олигоэфиров на Тс показано на
рис. 19, из которого видно, что выше молекулярного веса 4000 Тс
практически не меняется.
Если, согласно [36], принять, что постоянство Тс достигается
при молекулярном весе, отвечающем таковому для механического
сегмента цепи, то можно полагать, что для полидиэтиленгликольади-
пината механическим сегментом является отрезок цепи с молекуляр-
ным весом около 4000.
Исследованы форполимеры и эластомеры, олигоэфирные блоки ко-
торых имели молекулярный вес 1600 (ОЭ-1) и 4100 (ОЭ-2) (табл. 15).
56
Следовательно, у одного из них молекулярный вес соответствует
длине механического сегмента, а у другого более чем в два раза
меньше.
Из данных табл. 15 вид- с>
но, что наличие в цепи
форполимера и сшитого эла-
стомера уретановых rpjnn
наряду со сложными эфир-
ными группами приводит к
значительному повышению
Тс последних по сравнению
с исходными олигоэфира-
ми. При этом Тс форполи-
меров (рис. 20) с одинако-
вой длиной олигоэфирных
блоков линейно возрастает
Рис. 19. Зависимость Тс от молекулярного
веса (М) полидиэтиленгликольадипината
не с увеличением молекулярного веса,
а с повышением молярной доли диизоцианатной компоненты в
Таблица 15
Соотношение исходных компонентов и характеристики образцов
олигоэфиров, форполимеров и эластомеров
Номер обра- зца Образец Мольная доля ком понентов S 5 I шоэ I м 1 Гс (начало), °C Тс (конец), СС Тс (средняя). СС || <
оэ ТДИЦ Сшива- . ель
1 Олигоэфир 1 1600 —60 —51 —55,5 9
2 Олигоэфир 2 — —- — 4100 —50 -45 —47,5 5
3 Форполимер на 2,0 1,00 — 0,50 3430 —46 —38 —42 8
4 олигоэфире 1 2,0 1,50 0,75 3500 —43 —33 —38 10
5 1,0 1,00 — 1,00 6300 —42 —31 —36,5 11
6 1,5 2,00 — 1,33 4500 —39 -26 —32,5 13
7 8 Форполимер на 1,0 2,0 2,00 1,00 2,00 0,50 1990 7500 —33 —50 —20 -40 —26,5 -45 13 10
9 олигоэфире 2 Форполимер на 2,0 1,50 0,75 13500 —45 —38 —41,5 7
10 олигоэфире 2 1,0 1,00 1,00 5500 —45 —38 —41,5 7
11 1,5 2,00 1,33 5000 —46 —35 —40,5 9
12 1,0 2,00 — 2,00 4800 —44 —35 —39,5 9
13 Эластомер на 1,0 1,07 0,017 1,07 — —44 —26 —35 18
14 олигоэфире 1 1,0 1,10 0,035 1,10 —41 —25 —33 16
15 1,0 1,15 0,070 1,15 — —40 —24 —32 16
16 1,0 1,20 0,100 1,20 —. —39 —24 —31,5 15
17 Эластомер на 1,0 1,15 0,070 1,15 — —45 —34 —39,5 9
18 олигоэфире 2 1,0 1,20 0,100 1,20 — -45 —34 —39,5 9
57
молекуле форполимера. В то же время кривая / лежит выше и имеет
больший угол наклона, чем кривая 2. Такое явление объясняется
Рис. 20. Зависимость Тс (I и 2) и молекуляр-
ного веса (3 и 4) от мольного соотношения
олигоэфира и диизоцианата для форполиме-
ров на основе олигоэфиров с 44=1600 (/, 3)
и 4100 (2, 4).
более высокой концентра-
цией уретановых групп в
единице объема форполи-
мера, содержащего олиго-
эфир ные блоки меньшего
молекулярного веса, и бо-
лее значительным ростом
концентрации этих групп
в этом же форполимере по
мере роста доли диизоци-
аната.
Существенно, что экст-
раполяция обоих кривых к
нулевому значению моль-
ной доли диизоцианата дает
Тс = — 47е С, которая соот-
ветствует таковой меха-
нического сегмента ПОЛИ-
диэтиленгликольадипината
(см. рис. 19).
Кривая зависимости Тс
от мольной концентрации
уретановых групп для фор-
полимеров (рис. 21) имеет
тот же характер, что и аналогичные кривые для полиуретанов иной
природы (см. рис. 18). При этом данные обоих форполимеров ло-
жатся на одну и ту же кривую, что говорит об основной роли кон-
центрации уретановых
ipynn в изменении Те
полиэфируретанов.
В свою очередь пере-
ход от форполимера к
сшитому эластомеру не
сопровождается таким
значительным повыше-
нием Тс> как переход от
олигоэфира к форполи-
меру (см. табл. 15). От-
сюда следует вывод о
Гс,°С
60 J---^Гс^оль/ЮООг
Рис. 21. Зависимость Тс от мольной концентра-
ции уретановых групп (С) форполимеров на
основе ОЭ с М = 4100 (/) и 1600 (2).
том, что переход от оли-
гоэфира к форполимеру приводит к резкому качественному изме-
нению свойств молекулы, в которой свойства исходных блоков
как бы частично утрачиваются. Новая молекула (молекула фор-
полимера) представляет собой самостоятельную кинетическую еди-
ницу, характеризующуюся своей собственной подвижностью.
58
В противном случае потеря подвижности такой системой проис-
ходила бы в две стадии—сначала подвижность падала за счет свя-
зывания участков цепей по полярным уретановым группам, но с
сохранением подвижности олигомерного блока; далее при более низ-
кой температуре происходила потеря подвижности олигомерных
блоков. При этом мы должны наблюдать две Тс, как и для блок-сопо-
лимеров [159].
Таблица 16
Вклад диизоцианата и сшивающего агента в повышение температуры
стеклования олигоэфира
Номер образца ттдиц ™оэ Количе ство сши- вателя Тс фор- полимера (рассчи- тано), °C Тс элас- томера. °C дг тдиц ДГ сшива- теля
13 1,07 0,017 —36,0 -35,0 19,5 1,0
14 1,10 0,035 —35,5 —33,0 20,0 1,5
15 1,15 0,070 —35,0 —32,0 20,5 3,0
16 1,20 0,100 —34,5 —31,5 21,0 3,0
17 1,15 0,070 —41,5 —39,5 6,0 2,0
18 1,20 0,100 —41,0 —39,5 6,5 5,0
Из величины разности между Тс олигоэфира и форполимера,
форполимера и эластомера (табл. 16) видно, что основную
роль в повышении Тс, а значит в понижении подвижности олиго-
эфирных блоков в эластомере, играет не сшивающий агент, т. е. не
поперечные химические связи, а полярные уретановые группы или
физические поперечные связи. Следовательно, основной вклад в
пространственную сетку полиуретановых эластомеров вносят не хи-
мические, а физические поперечные связи 1115].
Увеличение в форполимерах молекулярного веса олигоэфирных
блоковом, табл. 15) сопровождается наряду с понижением Тс суже-
нием температурного интервала этого процесса, что объясняется
уменьшением среднего времени релаксации полимерных цепей вслед-
ствие повышения их подвижности.
К аналогичным выводам о Тс приводят и термомеханические ис-
следования указанных систем (рис. 22). Как видно из характера тер-
момеханических кривых, ни олигоэфир, ни форполимер не обнару-
живают высокоэластической деформации и выше Тс находятся в
вязко-текучем состоянии. Лишь сшивание в пространственную
сетку придает полученным системам высокоэластические свойства.
Сопоставление термомеханических кривых эластомеров на основе
олигоэфиров с различными молекулярными весами показывает, что
для эластомера на основе олигоэфира с большим молекулярным
весом наблюдается наряду с высокоэластической и некоторая пласти-
ческая деформация, которая объясняется меньшей концентрацией
химических и физических поперечных связей в образце.
50
Совсем иную картину мы наблюдаем в случае полиуретановых
эластомеров с карбоцеппыми (олигоизопреновыми и олигобутадие-
Рис. 22. Термомеханические кривые олигоэфиров с /И — 1600 (а) и 4100 (б) и син-
тезированных на их основе форполимеров и эластомеров*
/ — общая деформация, II — необратимая деформация (номера соответствуют номерам
образцов табл. 15)
новыми) олигомерными блоками [127] (табл. 17). При синтезе указан-
ных эластомеров в качестве диизоцианатной компоненты использо-
вался толуилендиизоцианат, а сшивающим агентом служил триэтано-
ламин.
Таблица 17
Температурная характеристика процессов стеклования полиуретановых
эластомеров и их олигомерных составляющих
Соотношение исход-
ных компонентов
Вещество
Олигоизопрен-1 (М — 2000) —60 -50 10
Эластомер на ОИ-1 1 1,68 0,4 —60 —34 26
Олигоизопрен-2 (/И = 5500) — — — —61 —50 11
Эластомер на ОИ-2 1 1,68 0,4 —60 —45 15
Олигобутадиен (А4 = 2000) — — — -80 —61 18
Эластомер на ОБ 1 1,68 0,4 —80 —60 20
Весьма существенно, что эластомеры, синтезированные как на
олигоизопрене, так и на олигобутадиене, начинают расстекловывать-
ся при той же температуре, что и составляющие их олигомеры, толь-
ко в более широком интервале температур.
Сопоставление данных табл. 15 и 17 показывает, что замена в
эластомере олигомерного блока сложноэфирной природы на углево-
60
дородный приводит к существенному отличию характера стеклования
этих видов полиуретановых эластомеров. Отличие заключается в
скачкообразном повышении температуры стеклования при переходе
от эфирогликолей к полиуретановым эластомерам, что свидетельству^
ет о резком понижении подвижности олигоэфирного блока в составе
эластомера. Переход же от олигодисновых гликолей к полиуретано-
вому эластомеру не сопровождается скачкообразным изменением
подвижности олигомерных блоков, о чем говорят одинаковые темпе-
ратуры начала расстекловывания олигодиеновых гликолей и эласто-
меров.
Увеличение интервала стеклования исследованных эластомеров
по отношению к исходным олигогликолям объясняется наличием в
них жестких отрезков полимерной цепи, образованных диизоцианат-
ной компонентой, которые, взаимодействуя др> г с другом, увеличи-
вают жесткость системы, затрудняя протекание релаксационных
процессов. Все это в конечном счете сказывается и на времени релак-
сации полимерных цепей. Увеличение времени релаксации в свою
очередь приводит к расширению интервала стеклования эластомера.
Такое объяснение повышения интервала стеклования у эластомеров
подтверждается и тем, что интервал стеклования у эластомеров по-
нижается с увеличением длины олигомерного блока, т. е. с пониже-
нием доли диизоцианатной компоненты в единице объема эластомера
(см. табл. 17).
Особенности поведения полиуретановых эластомеров на основе
олигодиеновых гликолей следует объяснить неполяркостью олиго-
диеновых блоков. Отсутствие полярных групп в цепи олигодиеновых
блоков не позволяет им вступать в сильное взаимодействие с диизо-
цианатными блоками и друг с другом. В результате возможность об-
разования устойчивых физических связей незначительна. Поэтому
резкого понижения подвижности олигомерного блока в составе элас-
томера, как это имело место для случая сложноэфирной природы
блока, не наблюдается.
Сопоставление температурных интервалов стеклования эласто-
меров, полученных на олигоизопренах с молекулярными весами
2600 и 5500, показывает, что эластомер с блоками большего мо-
лекулярного веса имеет меньший температурный интервал расстек-
ловывания.
Переход из стеклообразного состояния в высокоэластическое
для полиуретановых эластомеров с олигодиеновыми блоками проте-
кает в более широком температурном интервале (см. табл. 17), чем
в эластомерах с блоками сложноэфирной природы (см. табл. 15).
Это, очевидно, можно объяснить однородностью основных физиче-
ских связей в эластомерах, содержащих блоки сложноэфирной приро-
ды, и неоднородностью таковых в эластомерах с блоками углеводо-
родной природы.
У первых межмолекулярное взаимодействие благодаря наличию
полярных сложных эфирных групп определяется главным образом
61
водородными связями, которые вследствие одинаковой интенсивности
на всем протяжении макроцепей диссоциируют в довольно узком
температурном интервале. У вторых из-за неполярной природы бло-
ков межмолекулярное взаимодействие осуществляется в основном
дисперсионными силами.
Широкий набор расстояний между атомами соседних макромоле-
кул, обусловленный их относительно малой полярностью, вызывает
образование между цепями полиуретана дисперсионных связей раз-
личной интенсивности, разрушение которых должно протекать в зна-
чительном температурном интервале. В связи с этим мы полагаем,
что различия в изменении Тс при переходе от олигомерных блоков
к сшитым эластомерам для полиуретановых эластомеров со сложно-
эфирной и углеводородной природой объясняются различием харак-
тера физической сетки, образующейся в полиуретановом эластомере.
В свою очередь, в полиуретановых эластомерах со сложноэфир-
ной природой олигомерных блоков преобладающий вклад в эффекти-
вную плотность физической сетки должны вносить водородные
связи.
Таким образом, Тс аморфных полиуретанов предопределяется как
химической природой, структурой и молярной долей составляющих
полиуретаны диизоцианатов и гликолей, так и молекулярным весом
последних. Основной вклад в повышение Тс сшитых полиуретанов
по сравнению с исходными олигогликолями вносит не простран-
ственное строение полиуретанов, а наличие в них уретановых групп,
играющих основную роль в образовании вторичных физических
связей.
ФАЗОВЫЕ ПРЕВРАЩЕНИЯ
И СТЕКЛОВАНИЕ ЛИНЕЙНЫХ АЛИФАТИЧЕСКИХ ПОЛИУРЕТАНОВ
НА ОСНОВЕ НИЗКОМОЛЕКУЛЯРНЫХ ГЛИКОЛЕЙ
Свойства полиуретанов существенно зависят от того, будет ли
его гликолевая составляющая мономером или олигомером. В случае
мономерных гликолей образующиеся полиуретаны в основном крис-
таллизуются, тогда как способ-
ность к кристаллизации полиу-
ретанов с олигогликолевыми бло-
ками не является необходимым
следствием химической природы
цепи. Поэтому целесообразно
рассмотреть вопросы, связанные
Рис. 23. Зависимость температуры
плавления полиуретанов на основе бу-
тандиола и линейных алифатических
диизоцианатов от числа углеродных
атомов в цепи (л) последних.
62
Таблица 18
Некоторые характеристики линейных полиуретанов
Диизоцианат Гликоль ?ПЛ’ £ Свойства
—(СН,)*— 1,4-Бутандиол 193 Очень хорошо
вытягивается в нить
1,6-Гександиол 180 То же
1,10-Декандиол 171 » »
1,3-Пропандиол 167 » »
1,5-Пентандиол 151 » »
1,9-Нонандиол 147 » »
ОНСН.СН,—О-СН2СН2ОН 120 » »
ОН (СН2)*—0—(СН2)4ОН 124 » »
СН3-СН-СН2СН2-СН-СН3 104 » »
Ан 6н
ОН(СН2)3-^~> (СН2)3ОН 158 » »
OH(CH2)2S (СН2)2ОН 129-134 » »
OH(CH2)*S (СН2)*ОН 120-125 » »
—(СНа)е— 1,4-Бутандиол 160 » »
1-3-Бутандиол 77-82 Рогоподобный
1,6-Гександиол 153 Очень хорошо
вытягивает- ся в нить
ОН—/ ОН (транс, цис) 215-220 Хорошо вытя-
гивается в нить, хруп- кий
ОН—ОН (транс) 250—255 Разлагается
ОНСН2—У~СН2ОН 168 Плохо вытяги-
вается в нить
ОН(СН2)2-^ V-(CH2)2OH 208—212 Вытягивается в
нить
-(СНЖ- 1,4-Бутандиол 136-140 Очень хорошо
вытягивает- ся в нить
-(СН»)и- 1,4-Бутандиол 121—125 То же
1,5-Пентандиол 143—146 » »
•жа» «мма 1,4-Бутандиол 260 Разлагается
—(СН2)12— 1,12-Додекандиол 128 Очень хорошо
вытягивает- ся в нить
1,10-Декандиол 215-219 Волокнообразу-
ющий, хруп-
сн3 Хсн, кий
-(СН2)3 S(CH2)3 - 1,4-Бутандиол 126-133 То же
63
с фазовыми превращениями, для каждого вида полиуретанов от-
дельно.
Линейные полиуретаны на основе низкомолекулярных гликолей
имеют много общего с аналогичными полиамидами. Так, этот вид по-
лиуретанов обладает способностью вытягиваться в нить, и поэтому
представляет интерес как волокнообразующий материал. Для поли-
уретанов так же, как и для полиамидов, наблюдается зигзагообраз-
ное изменение температуры плавления с изменением числа метилено-
вых групп между полярными группами цепи (рис. 23). Температу-
ра плавления полиуретанов с четным числом метиленовых групп
выше, чем у их ближайших полимергомологов, содержащих нечет-
ное число метиленовых групп в углеводородной цепочке.
Свойства линейных полиуретанов сильно зависят от природы и
структуры их составляющих, о чем можно судить по данным табл. 18
1170].
Остановимся на изложении имеющихся в литературе сведений,
касающихся отдельных алифатических полиуретанов.
Полиуретан на основе 1,6-гексаметилендиизоцианата
и 1,4-тетраметиленгликоля (перлон-U)
Известно значительное количество кристаллизующихся алифа-
тических полиуретанов, но основательно изучена структура полиуре-
тана на основе 1,6-гексаметилендиизоцианата и 1,4-бутандиола
(перлон-U или 4,6-ПУ). Это объясняется тем, что данный полиуретан
уже нашел практическое применение как волокнообразующий мате-
риал.
Впервые структуру 4,6-ПУ описал Брилл ]177], затем Цан и
Винтер 1349, 350], а также Борхерт 1174].
Схематическое изображение строения элементарной ячейки
4,6-ПУ, предложенное Бриллом, приведено на рис. 24. Указанные
авторы приписывают данному полиуретану триклинную решетку
со следующими параметрами элементарной ячейки:
а. А ь, А е, А а°. V0»
9,5 19,10 8,4 90 63—65 77 [349, 350]
4,95 10,10 8,23 90 60,2 73 1174)
9,56 19,10 8,23 90 63 73 [243, 244]
Триклинная кристаллическая решетка 4,6-ПУ состоит из «плос-
ких сеток» 1147, 170], соседние макромолекулы которых соединены
через регулярно повторяющиеся участки Н-мостиками (рис. 25,
пунктирные линии). В свою очередь, «плоские сетки», накладываясь
друг на друга, образуют трехмерную решетку.
Таким образом, 4,6-ПУ упаковывается по типу слоевой решетки.
При этом в направлении цепей (ось в) лежат главные валентные
64
связи, в «плоской сетке» (ось а) действуют водородные связи. Между
«плоскими сетками» связь осуществляется вандерваальсовыми сила-
ми (ось с).
Рис. 24. Схема части элементарной ячейки 4,6-поли-
уретана.
Обычно кристаллизующиеся полимеры не образуют идеальных
кристаллических систем и всегда содержат участки с одно-, двух-
и трехмерным нарушениями кристаллической решетки. Кристалли-
ческие структуры с одно- и двухмерным нарушениями решетки назы-
вают псевдо- или паракристаллическими. Подобные структуры могут
образовываться и в полиуретанах, особенно в начальной (первичной)
стадии кристаллизации.
5 407
65
Для определения степени упорядоченности (кристалличности)
в 4,6-ПУ по данным рентгеновских исследований Килиан и Енкель
(243, 244( рассчитали интегральную степень кристалличности
4,95 А
Рис. 25. Строение «плоской
4,6-пол и уретана
а -yf/Ж (IV,!)
в,
частичную кристалличность, харак-
теризующую водородные связи
а.. = у f lctd&, (IV,2)
20»
и частичную кристалличность, ха-
рактеризующую вандерваальсовы
связи
= 2_- С lCtdQ, (IV,3)
I / J
<! (002)
й! где 7 — общая интенсивность ин-
I терференции; 0! и 02— углы отра-
женин интерференции (200) и (002)
. соответственно; /Г1 и 1(2 — интен-
сивность интерференции кристал-
лической части спектра при и 02.
Дифракционные кривые 4,6-ПУ
(рис. 26) показывают, что с рос-
том температуры интенсивностьван-
д_ дсрваальсовых связей понижается
и характеризующий их пик прак-
тически исчезает при температуре
плавления полимера. Интенсив-
сеткн» ность жс водородных связей прак-
Р тически не меняется вплоть до тем-
пературы плавления.
Это явление весьма наглядно отображает характер кривых за-
висимости интегральной и частичной степеней кристалличности
4,6-ПУ от температхры (рис. 27), рассчитанных по формулам (IV, 1;
IV,2 и IV,3).
Таким образом, наблюдается различная температурная зависи-
мость интенсивности интерференции (200), соответствующая ван-
дерваальсовым связям. Данная зависимость свидетельствует о проч-
ности «плоских сеток» и о том, что кристаллическая структура, обу-
словленная водородными связями, остается неизменной вплоть до
температуры плавления.
Одновременно с разрушением «плоских сеток» происходит и
спонтанное разрешение межплоскостных вандерваальсовых связей,
66
однако постепенное понижение их интенсивности с ростом температу-
ры наблюдается уже значительно ниже температуры плавления
(~0°С).
Дифракционные кривые выдержанных при разных температурах
предварительно закаленных образцов 4,6-ПУ (рис. 28) показывают
не одинаковую температурную зависимость водородных и ваядер-
ваальсовых связей в ходе кристалли-
зации его из высокоэластнческого со-
стояния. Интегральная и частичная
к р истал л и ч ности, ха р актер изу ющи е-
Рис. 26. Дифракционные кри-
вые термообработанного при вы-
соких температурах образца
4,6-полиуретана:
/ — Н-связи; 2 — вандср-
ваальсовы связи
Рис. 27. Влияние 1емпературы на сте-
пень кристалличности 4,6-полиуретана:
/ — интегральная, 2 — частичная по ван-
дер ва ал ьсо в ым связям, 5— частичная по
Н связям
ся вандерваальсовыми связями, в интервале температур 0—100° С
совпадают (рис. 29). Выше 100° С интегральная кристаллич-
ность резко нарастает за счет роста частичной кристалличности,
обусловленной возникновением водородных связей. Однако раздваи-
вание максимума дифракционной кривой (см. рис.28) наблюдается
при более низких температурах, что особенно четко видно из кривых,
приведенных для 4,6-ПУ [ 14|. Такое явление указывает на возникно-
вение трехмерных кристаллических образований уже при более низ-
ких температурах (см. рис. 29).
В то же время процесс кристаллизации 4,6-ПУ из высокоэласти-
ческого состояния носит ступенчатый характер. До температуры
100° С макромолекулы укладываются в основном в двухмерную
псевдокристаллическую структуру и только выше этой температуры
образуется совершенная кристаллическая структура с межмолеку-
лярными водородными связями.
Влияние термического воздействия на структурные превраще-
ния в 4,6-ПУ исследовано [39, 43J методом дифференциалыю-
67
Рис. 28. Дифракционные кривые термообработанного
при различных температурах предварительно зака-
ленного образца 4,6-полиуретана:
I — И связи, 2 — вандерваальсовы связи.
Рис. 29. Влияние температуры на сте-
пень кристалличности закаленного
4,6-пол иуретапа:
/ —интегральная, 2 — частичная по ван-
дерваальсовым связям, 3 ~ частичная по
Н связям.
Рис. 30. Температурная зависимость
Ср для 4,6-полиуретана.
термического анализа (ДТА) и дифференциальной микрокалоримет-
рии [19].
На рис. 30 приведена температурная зависимость Ср для крис-
таллического (отожженного) образца 4,6-ПУ в интервале температур
от — 50 до 200° С. Ниже дано Ср (кал/гград) в исследованном темпе-
ратурном интервале:
от-50 до—10эС
от 45 до 120° С
от 195 до 210°С
Ср = 0,422 + 1,5-10~3 Т
Со = 0,495 4- 1,85-10~3 Т
Ср = 0,665
В интервале температур 10—40 °C на кривой теплоемкости (рис. 31)
наблюдается подъем, обусловленный возникновением сегментальной
подвижности макромолекул, вызванной переходом аморфной части
Рис. 31. Термограмма нагревания аморфного 4,6-поли-
уретана.
полимера из стеклообразного в высокоэластическое состояние.
Выше 120° С рост теплоемкости существенно ускоряется и темпера-
турный ход Ср описывает пик, соответствующий плавлению кристал-
лических образований, температура плавления которых 183°С.
Рассчитанная по данным микрокалориметрии теплота плавления
4,6-ПУ (без учета степени кристалличности) равна 20,7 кал/г.
Теплота плавления, определенная методом ДТА для образца 4,6-ПУ
с более низкой температурой плавления (173'' С), оказалась
22,3 кал/г. Такое расхождение можно объяснить большей кристал-
личностью последних образцов полиуретана, обладающего меньшим
молекулярным весом, а также большей ошибкой количественных
определений, проводимых методом ДТА.
Теплота кристаллизации расплава 4,6-ПУ (по данным ДТА) при
скорости охлаждения 4° С в минуту составляет 19,8 кал/г.
69
О термическом поведении аморфного образца 4,6-ПУ, полученно-
го закалкой .малых количеств (0,05 г) расплава жидким азотом, мож-
но судить и по гермограмме (рис 31).
При температуре 38' С на дифференциальной кривой обнаружива-
ется скачок в сторону эндотермических эффектов, свидетельствую-
щий о резком возрастании теплоемкости полимера вследствие появ-
ления сегментальной подвижности полимерных цепей (расстекловы-
вание) Дальнейшее повышение температуры придает оптимальную
для кристаллизации подвижность, и они кристаллизуются, о чем
свидетельствует наличие на кривой четкого экзотермического пика
с вершиной при 104° С. Плавление образовавшейся кристаллической
структуры характеризуется в основном последующим эндотермиче-
ским пиком с вершиной при 173'С. Предшествующий ему небольшой
эндотермический пик с вершиной при 148° С тоже характеризует
процесс плавления, но менее совершенных кристаллических образо-
ваний, возникновение которых обусловлено жесткими условиями
кристаллизации. На это явление указывает как соизмеримость
площадей экзотермического ника кристаллизации с таковыми обоих
эндотермических пиков плавления, так и совпадение температур
начала малого пика (137° С) с температурой начала плавления ис-
ходного кристаллического образна, имеющего такой же характер,
как и пик (см. рис. 30) |39|. Следовательно, данные микрокалори-
метрии и ДТА хороню согласуются с данными рентгеновского ана-
лиза. Так, для кристаллического образца 4,6-ПУ в области 130 —
160° С теплоемкость вначале относительно медленно, а затем
резко повышается (см. рис. 30). Такое поведение объясняется ани-
зотропным характером кристаллической решетки, для которой пер-
вая область повышения (^соответствует постепенному исчезновению
вандерваальсовых связей, а вторая — относительно быстрому раз-
рушению водородных связей, соединяющих макромолекулы в
«плоские сетки».
Рис. 31 тоже хорошо показывает механизм кристаллизации
4,6-ПУ из высокоэластпческого состояния. Четкий экзотермический
максимум выше 100е С отображает образование совершенной крис-
таллической структуры с межмолекулярными водородными связями.
Начало максимума характеризует возникновение псевдокристалл и че-
ских структур, часть которых не успевает организоваться в трехмер-
ную структуру и последующее плавление которой фиксируется
эндотермическим пиком при 148е С.
Однако все термограммы нагревания указывают на более высокие
температуры стеклования 4,6-11У, чем это вытекает из рентгеновских
данных, в частности из рис. 29.
Кинетика кристаллизации 4,6-полиуретана. Существенный ин-
терес представляют данные по кинетике изотермической кристалли-
зации 4,6-ПУ, которые позволяют сделать выводы как о температур-
ной зависимости скорости кристаллизации, так и о характере струк-
турообразования в полимере.
70
Кинетика кристаллизации многих гомополимеров вполне удов-
летворительно описывается уравнением Аврами [88|
а - 1 — (IV,4)
где а — доля вещества, подвергшегося фазовому превращению за
время /; п — константа, характеризующая для данного вещества
ти I зародышеобразования и тип растущих структур, принимает
значение от 1 до 4; /(0—константа скорости.
Кинетику изотермической кристаллизации 4,6-ПУ из расплава
изучали методом взвешивания «весами плотности» [300) и дифферен-
Рис 32. Зависимость степе-
ни завершенности теплового
эффекта кристаллизации 4,6-
пол иуретана от логарифма
времени кристаллизации при
температурах (°C):
1 — Ь2, 2 — 163. 3 - 1ь5, 4 —
167, 5 - 169
Рис 33 Изотермы кристаллизации
4,6-полпуретаиа в координатах Аврами
при темпераrvрах (°C):
/ ~ 162, 2 — 1()3, 3 - 165; 4 — 167;
5 — Ю9
циальной микрокалориметрии |21]. Методика микрокалоримстри-
ческих исследований кинетики кристаллизации полимеров описана в
[17, 18]. На рис. 32 в полу логарифмических координатах представ-
лены изотермы кристаллизации 4,6-ПУ, построенные по данным
микрокалориметрии.
Однако, чтобы судить о кинетике и типе растущих структур,
следует определить Ао и п. Последние определяются путем представ-
ления экспериментальных результатов в предложенных Аврами
координатах
lg I— lg(l — а)| — IgL
71
После двойного логарифмирования уравнения (IV, 4) имеем
lg I— 1g (!— а)1 = lg 0,434/<0 + п lg t.
По наклону к оси абсцисс получающейся в указанных координа-
тах прямой легко определить значение п, а по отрезку отсекаемому
ею на оси ординат — значение Ло.
Кинетика кристаллизации 4,6-ПУ описывается уравнением
(IV, 4) только в области значений а не выше 0,25, после которых
наблюдаются систематические отклонения от теорий. Действительно,
наблюдаемые степени превращения меньше теоретических (рис. 33).
Значения п, определенные на основе прямолинейных участков
изотерм, близки к двум, что согласно [88] соответствует образованию
либо плоских структурных элементов на гетерогенных зародышах,
либо линейных структур на гомогенных зародышах (табл. 19).
Таблица 19
Характеристики кристаллизации для 4,6-полиуретана
Гкр. °C г* - пл Ткр 1g Ко п ТО,5» мин
162 21 —1,51 1,95 6,0
163 20 — 1,99 1,88 10,5
165 18 —2,60 2,00 18,5
167 16 —3,10 2,06 27,5
169 14 -3,61 2,08 59,0
* Температура плавления 4,6-ПУ 183J С.
На основе современных представлений о лямелярном характере
кристаллитов в полимерах и возможности существования в расплаве
4,6-ПУ гетерогенных образований (отдельных участков пачек мак-
ромолекул) можно полагать, что значение п — 2 отвечает протека-
нию процесса кристаллизации с образованием лямелярных кристал-
литов. Это хорошо согласуется с электронно-микроскопическими
[206] и рентгеновскими исследованиями [245], из которых следует,
что кристаллиты в 4,6-полиуретане действительно плоские.
В результате исследования кинетики кристаллизации 4,6-ПУ
при помощи взвешивания авторы [300] приходят к заключению,
что п = 2,3, что противоречит другим данным [21].
Полиуретаны на основе гексаметилендиизоцианата
и этилен-, диэтилен- и триэтиленгликолей
Вследствие своих волокнообразующих свойств представляют ин-
терес и линейные алифатические полиуретаны на основе эти-
ленгликолей.
72
Так, рентгеноструктурным анализом исследованы процессы плав-
ления и кристаллизации полиуретанов на основе гексаметиленди-
изоцианата и этилен-, диэтилен-, триэтиленгликолей [14|.
Авторы [14] приходят к заключению, что изменения природы
и структуры гликоля не меняют характер процесса плавления и
кристаллизации полиуретанов на их основе, т. е. в данных полиуре-
танах, как и в 4,6-ПУ, процесс плавления и кристаллизации двух-
ступенчатый. Различны только температурные интервалы этих про-
цессов, которые зависят от природы и длины составляющих полиуре-
таны блоков. Однако в ]14] данные о характере кристаллической
структуры отдельных видов полиуретанов не приведены; им по ана-
логии с 4,6-ПУ приписана триклинная решетка.
Эти же авторы в [15] отмечают, что в зависимости от условий
кристаллизации полиуретаны на основе этилен- и диэтиленгликолей
способны образовывать различные кристаллические модификации.
О характерных особенностях кристаллизации и плавления полиуре-
танов на основе диэтилен- и триэтиленгликолей свидетельствуют
данные дифференциально-термического анализа [43, 45] и микрока-
лориметрии [19—20].
В отличие от 4,6-ПУ для полиуретанов на основе диэтилен-
(ПУДЭГ) и триэтиленгликолей (ПУТЭГ) в области плавления наблю-
дается сложный ход Ср, выражающийся в наличии двух максимумов
(рис. 34). Такой вид кривой Ср в области плавления указывает на
протекание двух последовательных процессов, каждому из которых
соответствует определенный тепловой эффект. В случае ПУДЭГ эти
тепловые эффекты разделить не удалось, и в сумме они равны
19,8 кал! г. Сумма тепловых эффектов плавления ПУТЭГ 15,3 кал /г,
из которых 12,8 кал/г отвечает первому, а 2,5 кал/г — второму теп-
ловому эффекту.
Теплоты были рассчитаны по данным теплоемкости, которые для
кристаллических отожженных образцов изменялись в пределах:
Для ПУДЭГ
от -50 до —5° С Ср = 0,422 + 1,5 • 10“3 Т,
от 50 до 100° С Ср = 0,512 + 1,5 • 10~3 Т,
от 140 до 160° С Ср = 0,623,
Для ПУТЭГ
от -50 до -20° С Ср = 0,422 + 1,5- 10~3 Г;
от 0 до 60° С Ср = 0,308 -г 2 • 10~3 Т,
от 120 до 135° С Ср = 0,631.
Следует отметить, что на кривой температурной зависимости
теплоемкости отожженного образца ПУДЭГ в температурной области
стеклования видны два отчетливо выраженных подъема: первый в
интервале температур от —5 до +10°, второй от 30 до 45° С
Для ПУТЭГ наблюдается только один подъем в области от —15 де
73
—10° С. Кривая температурной зависимости теплоемкости закален-
ного (аморфного) образца ПУДЭГ (рис. 34, кривые 2) показывает,
Рис. 34. Температурная зависимость Ср для ПУДЭГ (а) и ПУТЭГ (б):
/ — кристаллический образец, 2 —- закаленный образец,
что в интервале от —50 до —5° С температурный ход Ср аналогичный
таковому кристаллического (отожженного) образца. В интервале от
—5 до 10° С теплоемкость резко возрастает, что обусловлено перехо-
дом образца из стеклообразного состояния в высокоэластическое.
После 20° С наблюдается резкое уменьшение теплоемкости. Это свя-
зано с кристаллизацией участков макромолекул, ставших достаточно
гибкими для протекания такого процесса. По окончании процесса
кристаллизации Ср становится несколько меньше, чем для тех
же температур у кристаллического образца, после 85° С снова
начинает уменьшаться, проходя через минимум при температуре
103° С.
На основе хода кривой теплоемкости делается предположение,
что в температурном интервале 50—100" С протекает медленный
кристаллизационный процесс, который после 85° С становится
вполне заметным. Выше 110° С образовавшаяся в ходе нагрева-
ния кристаллическая структура начинает распадаться; при этом,
как и в случае кристаллического образца, плавление протекает в две
стадии.
Весьма существенным является равенство значений теплоемкости
в расплаве и высокоэластическом состоянии, свидетельствующее
об однотипности характера межмолекулярного взаимодействия в
этих двух состояниях.
Приведенные данные дали авторам [ 19| основание для следующих
выводов в отношении кристаллизации и плавления ПУДЭГ.
Выше температуры стеклования в области температур 20—45" С
74
кристаллическая структура образуется в основном за счет возникно-
вения водородных связей в плоскостях (возникновение псевдокрис-
таллов). Выше 50° С порядок в псевдокристаллах увеличивается, и
в интервале 85—110° С кристаллизуются эфирные звенья, которые
к этому моменту приобрели достаточную подвижность. Одновременно
начинается постепенный распад кристаллической решетки. Первый
пик на кривой плавления рис. 34 соответствует распаду основной
массы кристаллов, в то время как второй связан с распадом кристал-
лических участков, образованных эфирными звеньями.
Сложный характер кристаллизации и плавления можно объяс-
нить склонностью ПУДЭГ к образованию полиморфной кристалли-
ческой структуры, как это предположено в работе [43]. Однако
для окончательных выводов нужны детальные структурные ис-
следования.
Представляют интерес результаты по исследованию влияния
глубины закристаллизованное™ ПУДЭГ на характер изменения
теплоемкости в области стеклования. Образцы с разной глубиной
кристаллизации получены следующим образом. Расплав ПУДЭГ
закаливали, затем при температуре 29J С кристаллизовали в
течение определенного времени в изотермических условиях и снова
закаливали.
При анализе процесса стеклования образцов, подвергнутых двой-
ной закалке, кроме температурной зависимости теплоемкости
(рис. 35, а) рассмотрено изменение с температурой и функции CDjT
(рис. 35, б), для которой Дол и сотрудники [156] предложили на-
звание энкратии и обозначили через L. Значения Q указывают на ве-
личины теплового эффекта (кал/г), зафиксированные перед второ f
закалкой.
В табл. 20 даны максимальные значения изменения энкратии в
области стеклования AL, а также Тс, которые определяли как тем-
пературы, при которых энкратия принимала половину своего мак-
симального значения и ДСР при стекловании.
Из данных табл. 20 видно, что по мере увеличения глубины за-
кристаллизованное™ ПУДЭГ температура стеклования его несколь-
ко смещается в область более высоких значений, а температурный
интервал стеклования расширяется. Для отожженного образца
(см. табл. 19) получены результаты отдельно для первого и второго
подъемов на температурной зависимости теплоемкости (см. рис. 34.)
Для полностью аморфного образца, у которого Q - 0, рассчитана ве-
личина изменения теплоемкости при стекловании на 1 моль звеньев
в соответствии с [343]. В рассматриваемом случае Ср ври стекловании
составляло 0,196 кал/г-град. Молекулярный вес повторяющегося
участка макромолекулы ПУДЭГ равен 274,3. Согласно [343|, этот
участок содержит 17 кинетических звеньев, откуда ЛСР = (M/17) х
X &СР — М -АСР — 16,3 • 0,196 — 3,16 кал моль • град, где М.—
средний молекулярный вес кинетического звена. Рассчитанное
75
значение АСр хорошо согласуется с теоретическим (2,97 кал!моль х
-/.град), полученным па основании дырочной теории стеклования и
со значением2,7кал!моль-град, найденном при анализе данных для
большого числа мономерных и полимерных стекол [230, 243].
5
Рис. 35. Температурная зависимость Ср (а) и Д£ (6) для
образцов ПУДЭГ в области температур стеклования после
двойной закалки при Q (кал/г):
I — О; 2 — 7,6, 3 — 9,5, 4 - 14,35; 5 - 16,3, 6— 4,6 = ПУ.
Следует отметить, что смещение Тс ПУДЭГс увеличением глуби-
ны закристаллизованности в область более высоких температур и
расширение температурного интервала этого процесса указывают на
постоянное изменение структуры аморфных областей. Наряду с пол-
ностью аморфными участками, по-видимому, появляются и мезаморф-
ные области, которые и обусловливают указанный характер перехо-
да. Тот факт, что у образца, подвергнутого отжигу, первый подъем
на кривой теплоемкости наблюдается в той же области температур,
76
что и у закаленного, свидетельствует о наличии в нем некристалли-
ческих областей. Второй подъем на кривой теплоемкости отожжен-
ного образца в области 30—45° С объясняется дополнительной под-
вижностью отдельных участков макромолекул ПУДЭГ. Поскольку
Таблица 20
Характеристики стеклования ПУДЭГ
Q, кал! г Гс, °C я кал/г гра<)
0 —1 6,6 0,196
7,60 —1 4,6 0,144
9,50 0 4,1 0.131
14.35 2 3,1 0.108
16,30 4 1,9 0,091
43 1,1 0,054
Таблица 21
Характеристики стеклования ПУТЭГ
Q, кал/г ?с, °C AL- 10*. кам! г град^ дср. кал/г •град
0 -13 7,3 0,215
6,2 -13 5,8 0,178
10,3 -13 4,9 0,153
15,3 — 13 3,15 0,107
у 4,6-ПУ наблюдается один подъем примерно в этом же интервале
температур, то сделано предположение о том, что двухстадийный
характер стеклования ПУДЭГ каким-то образом связан с наличием
в его макромолекулах эфирного кислорода.
Экспериментальные данные, полученные для ПУТЭГ, также ука-
зывают на раздельную кристаллизацию диизоцианатных и эфирогли-
колевых участков полимерных цепей. Однако увеличение длины эфи-
рогликолевого блока вызывает значительно большую разницу между
температурами максимумов пиков на кривых плавления и несколько
меняет соотношение температур максимумов (см. рис. 34). Для
ПУТЭГ это соотношение зависит от термической обработки об-
разца.
Температурная зависимость теплоемкости и энкратии в облас-
ти температур стеклования подвергнутых двойной закалке образ-
цов ПУТЭГ приведена на рис. 36. Основные количественные дан-
ные, характеризующие процесс стеклования ПУТЭГ, даны в табл. 21.
Рассчитанное значение ДСрсоставляло дляПУТЭГЗ,42кал/ж>ль-град.
Для образцов ПУТЭГ, полученных из расплава, в отличие от
ПУДЭГ на кривой температурной зависимости теплоемкости в обла-
сти температур стеклования (см. рис. 34) наблюдается лишь один
подъем. Этот факт пока не получил удовлетворительного объяснения.
Изотермическая кристаллизация из расплава и раствора. Изотер-
мы кристаллизации расплавов ПУДЭГ и ПУТЭГ в координатах
Аврами представлены на рис. 37, 38. Рассчитанные на их основе зна-
чения 1g /(0 и п, приведены в табл. 22. Из характера изотерм и дан-
ных табл. 22 видно, что для обоих полимеров кинетика кристаллиза-
77
ции во всем температурном интервале подчиняется уравнению
Аврами. При этом для НУТЭГ во всем температурном интервале кон-
станта п практически равна двум, что дает основание предположить
о росте ламелярных кристаллитов при кристаллизации этого поли-
уретана из расплава.
Значение п Здля ПУДЭГ формально означает рост плоских
структурных элементов на гомогенных зародышах или объемных на
гетерогенных.
В одной из работ [242] найдено, что полиуретан на основе гек-
самстилендиизоцианата и диэтиленгликоля обнаруживает четко
Рис. 36 Темпера-
турная зависимость
Ср и AL для
образцов ПУ ТЭГ в
области температур
сте к лов а н и я п осл е
двойной закалки
при Q (кал/г)’
/ — 0, 2 — 6 2 3 ~
10,3, 4 - 15.3
выраженный большой период порядка 70 А. На оновании этого сде-
лан вывод о том, что кристаллы в этом полиуретане образованы мак-
ромолекулами со складчатой структурой. Следовательно, можно
полагать, что значение п — 3 соответствует образованию ламелярных
кристаллитов по механизму гомогенного зародышеобразования.
Рост трехмерных структур на гетерогенных зародышах менее ве-
роятен, но пока еще нельзя дать однозначный ответ на вопрос,
какая же структура образуется в процессе кристаллизации ПУДЭГ.
78
Проводились оптико-микроскопические исследования сферолит-
ной структуры ПУТЭГ |206]. Изучались пленки, полученные изо-
термической кристаллизацией из 1 % -кого раствора ПУТЭГ в смеси
диметилфорамида с ацетоном, а также из расплава Сферолитная
кристаллизация из раствора осуществлялась в интервале темпе-
ратур 80—40е С. Показано, что в интервале 80—60°С растут коль-
цевые сферолиты, имеющие одинарные кольца погасания, причем
знак двойного лучепреломления (ДЛ) кольца положительный. При
температуре 55° С растут кольцевые сферолиты, имеющие двойные
1 — 95; 2 — 98; 3 — 100; 4 — 103, 5; 5 —
105, 6 — 107,7. 7 — 109° С
I — 69, 2 — 72, 3 — 75, 4 — 78; 5 —81
6 — 85° С
кольца погасания и переменный знак ДЛ вдоль радиуса сферолита:
широкое светлое кольцо имеет положительный знак, а узкое
светлое кольцо — отрицательный (рис. 39, а, см. вклейку). С по-
нижением температуры кристаллизации до 50s С резко изме-
няется картина сферолитной кристаллизации — появляются сме-
шанные сферолиты. При Ткп— 50° С растут преимущественно
сферолиты с кольцевым центром и радиальной периферией (рис. 39, б),
а при Тк„ ~ 40° С — смешанные сферолиты, центр которых имеет
сферолитоподобный вид, а периферия радиальная (рис. 39, в).
При этой же температуре растут и практически полностью ради-
альные сферолиты, в которых довольно часто наблюдается ярко
светящийся центр (рис. 39, г). Знак ДЛ радиального сферолита
отрицательный, а центр преимущественно положительный.
Изотермическую кристаллизацию из расплава проводили в интер-
вале температур от 99 до 75° С. Обнаружено, что в области темпе-
79
ратур от 99 до 93° С растут сферолиты, не имеющие креста погасания
(рис. 40, а, см. вклейку). У элементов, направленных по радиусу
сферолита, положительный или отрицательный знак ДЛ почти по
всей своей длине. Закономерного расположения радиальных струк-
турных элементов не наблюдается.
Резкие изменения в морфологии сферолитов происходят при
дальнейшем понижении температуры кристаллизации всего на не-
сколько градусов. При этом имеют место следующие два явления:
Таблица 22
Характеристики кристаллизации ПУДЭГ и ПУ ТЭГ
из расплава
ГКр. °C дг=4- гкр 18 К0 п ТО,5» мин
ПУДЭГ
95 38 —3,24 3,02 10,5
98 35 —3,82 3,00 16,0
100 33 —4,17 2,97 23,0
103,5 29,5 —5,20 2,95 51,0
105 28 -5,90 3,00 81,0
107 26 —6 73 3,02 148,0
109 24 —7,54 3,00 288,0
ПУТЭГ
69 44 — 1,86 2,02 7,0
72 41 —2,13 1,97 10,5
75 38 —2,51 2,00 15,5
78 35 —2,84 2,00 22,5
81 32 -3,33 2,04 36,0
85 28 —3,79 1,99 71,0
* Температура плавления ПУДЭГ 133, ПУТЭГ 113° С.
во-первых, структурные элементы сферолитов как бы начинают со-
бираться в «снопы»; во-вторых, ДЛ сферолитов становится преиму-
щественно положительным. Наиболее четко это проявляется при
Ткр ~ 90° С (рис. 40, б).
Понижение температуры кристаллизации до 89° С приводит к
зарождению колец погасания и появления креста. Более регулярно
кольца погасания в сферолитах наблюдаются при температурах
кристаллизации от 85 до 80° С. Крест погасания в этих сферолитах
имеет зигзагообразную форму (рис. 40, в). При 75° С кольца пога-
сания, по-видимому, настолько узки, что не разрешаются в оптичес-
ком микроскопе, а крест погасания становится прямым. По своему
внешнему виду эти сферолиты (рис. 40, г) похожи на радиальные,
полученные кристаллизацией из раствора, но в отличие от последних
имеют положительный знак ДЛ.
80
Рис. 39. Надмолекулярные структуры в тонких пленках ПУТЭГ, полученных из
раствора при температуре кристаллизации;
а — 55; б — 40. в, г — 50е С (Х750).
Рис 40 Надмолекулярные структуры в тонких пленках ПУТЭГ, полученных из
расплава при температуре кристаллизации:
а — 93; б — 90; § — 89—80; г — 75° С (Х280).
Рис. 41. Надмолекулярные структуры в тонких пленках ПУ ГЭГ, полученных при
температуре кристаллизации 89° С в зависимости от толщины пленки.
а — 10; б — 20; в — 30 (Х280), г — 30 як, (Х750).
Рис. 53. Структура сферолитов ОЭА.
а — в отраженном свете (X 200), б — тот же участок в проходящем поляризованном свете;
в — электронные микрофотографии реплик (1—3 — ХЗО ООО, 4 — Х20 ООО): 1 — мел-
кокристаллическая структура; 2 — радиальные сферолиты; 3 — игольчатые сферолиты;
4— кольцевые сферолиты.
Наряду с изложенным в работе [206] приведены результаты
исследований влияния толщины пленок (10; 20 и 30 мк) на образу-
ющуюся надмолекулярную структуру при температуре 89° С, при
которой растут кольцевые сферолиты.
При толщине пленки 10 мк наблюдаются в основном «снопы»,
имеющие положительный знак ДЛ (рис. 41, а, см. вклейку). «Снопы»
ориентированы по отношению друг к другу в основном под углом
90°. Для трактовки этого явления необходимы дополнительные
электронно-микроскопические исследования.
Рис 42 Изотермы кристаллизации из
высокоэластического состояния ПУДЭГ
в координатах Аврами.
/ — 28,2; 2 — 27,2, 3 — 25.6. 4 — 22,3.
5 — 20.3, 6 ~ 17,8, 7 — 16,2, 8 — 14.2° С
Рис. 43. Изотермы кристаллизации
из высокоэластического состояния
ПУТЭГ в координатах Аврами:
/ __ 26, 2 — 24.5, 3 — 22, 4 — 19, 5 —
16,1, б - 13,6° С.
С увеличением толщины пленки до 20 мк «снопы» начинают обра-
зовывать сферолиты, причем в некоторых из них проявляется до-
вольно развитый крест погасания (41, б). В отдельных «снопах»
начинается нерегулярное скручивание фибрилл. Знак ДЛ остается
прежним.
Коренные изменения в структуре сферолита происходят в плен-
ках толщиной 30 мк. Как видно из рис. 41 (в, г) кольцевой сферолит
состоит как бы из нескольких «снопов», причем в центре сферолита
один из «снопов» находится выше других.
Таким образом, структура кольцевого сферолита зависит от тол-
щины пленки. Последняя серия снимков позволила авторам [206]
предположить, что «сноп» представляет собой многослойный пластин-
чатый псевдокристалл, обладающий свойствами единичного монокрис-
талла. Очевидно, аналогичные «снопы» составляют «сферолитообраз-
81
ные» центры радиальных сферолитов» выросших из раствора (см. рис.
39)» и положительные по знаку ДЛ радиальные элементы сферолитов,
полученных из расплава в области температур 94—93° С (рис. 40, а).
Отрицательные по знаку ДЛ радиальные элементы структуры этих
же сферолитов являются теми же «снопами», но ориентированные
так, что плоскость водородных связей примерно параллельна плос-
кости образца. С понижением температуры кристаллизации преобла-
дают структуры, положительные по знаку ДЛ, и, наконец, при опре-
Таблица 23
Характеристики кристаллизации ПУДЭГ и ПУТЭГ
из высокоэластического состояния
7' О ' 1 кр’ ' О 1 1 (Г* 1 1е к0 п ?0,5’ мин
ПУДЭГ
28,2 24,2 —2,40 2,82 5,5
27,2 23,2 —2,72 2,80 7.2
25,6 21,6 —3,20 2,86 10
22.3 18,3 —33,79 2,44 22
20,3 16,3 -4,00 2,38 32,4
17,8 13,8 —4,32 2,31 50
16.2 12,2 —4,77 2,36 66
14.2 10,2 —5,07 2,38 93
ПУТЭГ
26,0 39 —2.498 2,86 6,5
24,5 37,5 —2,81 2,62 10
22.5 35 -3.48 2,78 16
19,0 32 -4.04 2,78 22
16,0 29,1 —4,61 2.64 49
13,6 26,6 -4,79 2,55 66
* Температура стоклорания ПУДЭГ—4, ПУТЭГ—13е С.
деленных условиях возникают более или менее правильные кольце-
вые сферолиты с зигзагообразным крестом погасания. Результаты
калориметрических и оптико-микроскопических исследований нахо-
дятся в хорошем согласии, и можно с уверенностью говорить о том,
что при кристаллизации ПУТЭГ из расплава происходит рост ламе-
лярных кристаллитов.
Изотермическая кристаллизация из высокоэластического состоя-
ния, Согласие теории с экспериментом в случае изотермической
кристаллизации ПУДЭГ и ПУТЭГ из высокоэластического состояния
значительно хуже, чем для кристаллизации из расплава (табл. 23).
Так для ПУДЭГ кристаллизация протекает в три стадии (рис. 42).
В начальных участках изотермы описываются уравнением Аврами с
дробными значениями (в интервале от двух до трех) константы п.
При а = 0,05 происходит ускорение кристаллизации, а после того,
82
как а достигает значения 0,7, наступает замедление процесса, сте-
пень которого существенно зависит от температуры. Если при отно-
сительно высоких температурах, достаточно удаленных от темпера-
туры стеклования, замедление не такое резкое, то по мере понижения
температуры оно становится значительным. Это свидетельствует о
том, что кристаллизация в данном случае протекает гораздо сложнее,
чем это предусмотрено теорией.
Аналогичный характер кинетики кристаллизации наблюдается и
для ПУТЭГ (рис. 43), однако систематическое отклонение от теории
выражено гораздо слабее, так что наблюдаемый механизм кристалли-
зации в какой-то мере обусловлен спецификой молекулярного строе-
ния данных полиуретанов, содержащих в полимерной цепи участки
различной гибкости. Это касается в первую очередь резкого замедле-
ния скорости кристаллизации, которое, вероятно, связано с тем, что
в указанном температурном интервале не кристаллизуются эфиро-
гликолевые участки цепей исследуемых полиуретанов.
Сополиуретаны на основе гексаметилендиизоцианата,
диэтилен- и триэтиленгликоля
Сополимеры на основе гексаметилендиизоцианата, диэтилен- и
триэтиленгликоля, т. е. такие, исходными гомополимерами которых
являются ПУДЭГ и ПУТЭГ, были исследованы дифференциально-
термическим, термомеханическим и ИК-спектроскопическим мето-
дами |46].
Термограммы получены для образцов исходных гомополимсров и
некоторых сополимеров (рис. 44), закристаллизованных из распла-
ва путем охлаждения со скоростью 1°С в минуту.
Термограммы ПУДЭГ и ПУТЭГ имеют тот же характер, что и
термограммы уже описанных гомополимеров [19, 20, 43|. В данном
случае для ПУДЭГ наблюдается один пик плавления, которому пред-
шествует экзотермический пик, характеризующий кристаллизацию
перед плавлением. Для ПУТЭГ нет четко выраженного эффекта крис-
таллизации перед плавлением, однако проявляется сложный харак-
тер плавления: на термограмме имеется два эндотермических пика в
области плавления полимера.
В характере термограмм сополимеров проявляются свои особен-
ности, которые зависят от соотношения гомополимеров в сополимере.
Как видно из дифференциальных кривых 2 и 3 (см. рис. 44). введение в
ПУДЭГ до 30 мол. % ПУТЭГ приводит к резкому увеличению экзо-
термического пика кристаллизации перед плавлением сополимера.
Одновременно наблюдается понижение температур и площадей пи-
ков плавления. Увеличение доли ПУТЭГ до 40 мол.% приводит к
резкому изменению характера термограмм и понижению температу-
ры и площади основного пика плавления сополимера. Термограмма
имеет такой же характер, как и термограмма исходного ПУТЭГ:
экзотермический пик кристаллизации до плавления отсутствует;
кроме основного пика плавления присутствуют другие, более
83
высокотемпературные эндотермические пики. Однако с дальнейшим
повышением доли ПУТЭГ указанные высокотемпературные пики
уже не проявляются. При этом минимальная температура плавле-
ния наблюдается у сополимеров с 70 и 80 мол. % ПУТЭГ, а у сопо-
лимера с 90 мол.% ПУТЭГ температура плавления выше и прибли-
жается по своей величине к температуре плавления чистого ПУТЭГ.
Рис. 44 Термограммы нагревания ПУДЭГ (/), ПУТЭГ (7) и их сополи-
меров, содержащих 90 (2), 70,(3), 60 (4), 50 (5) и 10 (5) мол.% ПУДЭГ.
Более четкое представление о влиянии состава сополимеров на
их температуры плавления можно получить из рис. 45. «Фазовая
диаграмма», приведенная на этом рисунке, напоминает диаграмму с
эвтектическим минимумом. Отметим, что «эвтектический минимум»
в данном случае имеет диффузный характер. Кроме того, до
«эвтектической» точки при 30 и 90 мол.% ПУТЭГ на кривой имеются
перегибы, которые отсутствуют на подобных кривых, приведенных в
литературе |88].
Для исследования характера кристаллической решетки в ряду
сополимеров сняты при комнатной температуре инфракрасные спек*
84
тры. Образцы брали в виде пленок, полученных из растворов поли-
уретанов в диметилформамиде при 55—60 и 40—42°С, а также плен-
ки из расплавов.
Сравнение спектров расплавов и кристаллических пленок ПУДЭГ
и ПУТЭГ дало возможность приписать кристаллическим структурам
этих полиуретанов характерные крис-
таллические полосы: для ПУТЭГ по-
лоса 860 см~1, дл я ПУДЭГ — 830 см •
Установлено, что полоса 830 см~
сохраняется в сополимерах с пони-
жением содержания ПУДЭГ, вклю-
чая молярное соотношение ПУТЭГ:
: ПУДЭГ = 0,6:0,4, после чего про-
Мол.% ПУДЭГ
Рис. 45. Зависимость температуры плавле-
ния (/) и логарифмической вязкости (2)
0,5%-ного раствора сополимеров в м-крезоле
от соотношения в них гликолей.
—!-----!-----!----
900 800 700 9,CMf
Рис. 46. Дифференциальная кри-
вая термограммы нагревания (/)
и участок И К’Спектра пропуска-
ния (2) для физической смеси
(0,5 : 0,5) ПУДЭГ и ПУТЭГ.
исходит скачок: полоса 830 см 1 исчезает и появляется полоса
860 см~~\ которая сохраняется в спектрах всех остальных сопо-
лимеров.
На основании зависимости температуры плавления от состава и
изменения кристаллической решетки в ряду сополимеров высказано
определенное суждение о характере распределения в макроцепи со-
полимера участков с различной химической структурой. Принимая
во внимание, что на «фазовой диаграмме» исследованной системы со-
полимеров наблюдается «эвтектический минимум», с одной стороны
которого кристаллизуются полимерные участки только первого типа,
а с другой — только второго, а также и то, что в сополимере с мо-
лярным соотношением ПУТЭГ : ПУДЭГ =0,6 :0,4 кристаллизуется
85
второй компонент, сделано предположение о приближении поведе-
ния системы в области средних составов к поведению статистического
сополимера В подобных случаях обычно «эвтектическая» точка
смещена в сторону компонента, обладающего более низкой темпера-
турой плавления, что и наблюдается в указанной системе.
Скорость кристаллизации чистого ПУДЭГ больше, чем ПУТЭГ.
Полагая, что это верно и для входящих в состав макроцепей сополи-
мера участков ПУДЭГ и ПУТЭГ, авторы [21] объясняют отсутствие
«кристаллических» полос ПУТЭГ в сополимере, содержащем 0,6 моль-
ных долей последнего, пространственными затруднениями для крис-
таллизации второго компонента, которые создают образовавшиеся
кристаллические области ПУДЭГ.
Полученные сополимеры не относятся к блок-сополимерам с
длинными блоками обеих химических структур, способных кристал-
лизоваться одновременно и независимо друг от друга, что подтверж-
дают спектры и термограммы физической смеси ПУДЭГ и ПУТЭГ
(рис. 46). Термограммы и спектры указывают на наличие двух крис-
таллических структур и после совместной кристаллизации.
По мере приближения к эвтектическому минимуму процесс крис-
таллизации все более затрудняется, и в результате нарушения регу-
лярного распределения межмолекулярных водородных связей воз-
растает дефектность кристаллической структуры сополимеров.
Перегибы, которые наблюдаются на кривой / (см. рис. 45) при удале-
нии от центрального участка к краям, возникают, очевидно, в резуль-
тате изменений в характере кристаллических образований, обуслов-
ленных изменением молекулярной структуры цепи сополимера. При
малом содержании одного из компонентов сополимера он статисти-
чески распределяется по цепи в виде мономерных участков между
блоками компонента другой химической структуры. Поэтому глико-
левые блоки оказывают пластифицирующее действие по отношению
к кристаллическим областям, образованным вторым, преобладающим
в составе сополимера компонентом. Это отражается на температурах
плавления сополимеров, однако не в такой степени, как для сополи-
меров со средним составом обоих компонентов, где должны статис-
тически распределяться уже небольшие блоки ПУДЭГ и ПУТЭГ.
Таким образом, упомянутые перегибы авторы [46] связывали с
изменениями в характере распределения и длинах составляющих
сополимер участков ПУДЭГ и ПУТЭГ.
Особенности кристаллизации и плавления
алифатических фторсодержащих полиуретанов
В работах (44, 84—86] приведены сведения о синтезе и особеннос-
тях фазовых превращений ряда фторсодержащих алифатических по-
лиуретанов. В частности, исследовались полиуретаны следующей
химической структуры*
(-HNCH, (CF2)4 CILNHCOO (СН»)вОСО--|я (ПУ-1),
86
HN (CH,)e NHCOOCH, (CF,),CH.OCO(ПУ-2),
[—H.X'CH,(CF2)4СНЛНСООСН,(CF2)4CH.OCO--U (11У-3),
а также их нефторированный аналог
[—HN (CH2)e NHCOO (СН2)в ОСО—]„ (6,6-ПУ).
В ПУ-1 фтор содержится в диизоцианатной, в ПУ-2 — глико-
левой, а в ПУ-3 — в обоих составляющих полиуретаны блоках Их
нефторированный аналог синтезирован на основе 1,6-гексамстилен-
диизоцианата и 1,6-гексаметилендиола. Поскольку ПУ-1 и 6,6-ПУ
получены методом межфазной поликонденсации, а остальные два —
полимеризацией в блоке, то последние имеют меньший молекуляр-
ный вес.
Рис 47. Термограммы первичного нагревания (/), ох.чакдения (2) и повторного
нагревания (3—сразу после кристаллизациии, 4 — после пятидневной выдержки
при комнатной температуре) образца ПУ-1
Фазовые превращения исследовались методом дифференциаль-
но-термического анализа. Характер гермограмм плавления и крис-
таллизации 6,6-ПУ аналогичен таковым для 4,6-ПУ. Теплота плав-
ления 6,6 -ПУ немного ниже и составляет 20,2 кал 1г.
На рис. 47 показаны термограммы ПУ-1. Повторному нагрева-
нию подвергались образцы с различной предысторией. Один нагре-
вался сразу же после кристаллизации расплава, а второй — после
пятидневной выдержки при комнатной температуре.
Характер кривых первичного нагревания и охлаждения аналоги-
чен таковому для 6,6-ПУ. Однако кривые повторного нагревания
отличаются не только от кривой первичного нагревания, по и между
собой. Пик плавления при повторном нагревании невыдержанного
образца имеет две четкие вершины, что объясняется возникновением
в условиях кристаллизации из расплава двух типов кристаллических
структур с различными температурами плавления. Выдержанный
некоторое время образец ПУ-1 проявляет тенденцию к приобретению
свойств исходного образца. Предполагается, что образовавшаяся в
87
ходе кристаллизации расплава кристаллическая структура с темпе-
ратурой плавления 182е С является метастабильной при комнатной
температуре и переходит в стабильную в этих условиях структуру
с температурой плавления 185° С.
Рис. 48 Термограммы первичного нагревания (/), охлаждения
(2) и повторного нагревания («?) образца ПУ-2.
Для ПУ-2 пик плавления первичного нагревания имеет две четкие
вершины, а пик повторного нагревания — только одну (рис. 48).
При этом в ходе повторного нагревания не воспроизводится более
низкотемпературная вершина пика (112° С). Однако термограмма
Рис 49 Термограмма наде-
вания аморфного образца
ПУ-2.
нагревания выдержанного аналогично
ПУ-1 образца имеет характер термограм-
мы первичного нагревания.
Существенно и то, что пик плавления
закристаллизованного из высокоэласти-
ческого состояния образца ПУ-2 (рис. 49)
тоже имеет только одну вершину, но в
отличие от образца, закристаллизован-
ного из расплава, температура ее 112°С,
т. е. в данном случае не воспроизводится
высокотемпературная вершина (124° С)
пика плавдения исходного образца.
Подобное явление мы наблюдали при
исследовании методом дифференциально-
термического анализа полиуретана на
основе 1,6-гексаметилендиизоцианата и
диэтиленгликоля [43].
Аномалии характера плавления по-
лиуретанов, содержащих в одной из со-
ставляющих атомы фтора, свидетельствуют о способности этих поли-
меров к образованию двух типов кристаллических структур. Это
подтверждается еще и тем, что образцы полиуретана, содержащие
фтор в обоих составляющих (ПУ-3), как и их нефторированный
аналог, имеют одну температуру плавления.
88
Можно предположить, что введение в макромолекулы полиуре-
танов атомов фтора, являющееся фактически заменой этиленовых
звеньев тетрафгорэтиленовыми, обусловливает неоднородность гео-
метрической структуры (переход от зигзагообразной конформации
к спиралевидной) макроцепей и способствует образованию различ-
ных типов кристаллических структур. Особенно четко это проявляет-
ся при введении фтора в более подвижный гликолевый блок.
ЛИНЕЙНЫЕ ПОЛИУРЕТАНЫ
НА ОСНОВЕ НИЗКОМОЛЕКУЛЯРНЫХ ГЛИКОЛЕЙ
И АРОМАТИЧЕСКИХ ДИИЗОЦИАНАТОВ
В промышленности широко применяются полиуретаны на основе
толуилендиизоцианата (ТДИ). Обычно используется смесь 2,4- и
2,6-изомеров ТДИ в соотношении 80:20 и 65:35. При таких соотно-
шениях изомеров ТДИ полиуретаны на основе низкомолекуляр-
ных гликолей не кристаллизуются и являются аморфными вещес-
твами.
Исследование образцов полиуретанов на основе этиленгликоля и
ТДИ с соотношением 2,4- и 2,6-изомеров 100 :0; 80:20; 65:35; 40:60;
15:85 (15] показало, что образцы полиуретанов, содержащие в
смеси свыше 50% 2,6-изомера, способны кристаллизоваться. Степень
кристалличности растет с увеличением содержания 2,6-изомера. Та-
кое явление объясняется более симметричным строением диизоциа-
натного звена полиуретана на основе 2,6-изомера по сравнению с
2,4-изомером. 2,6-ТДИ может находиться в полимерной цепи только
в двух, а 2,4-ТДИ — в восьми различных конфигурациях.
Изучение процесса кристаллизации с помощью электронного
микроскопа показывает, что полиуретан на основе 2,4-ТДИ и эти-
ленгликоля дает аморфную пленку. Увеличение содержания 2,6-ТДИ
в смеси изомеров приводит к образованию более упорядоченных
структур. Так, полиуретан с соотношением 2,4- и 2,6-изомеров
ТДИ 80 :20 и 65 :35 образуют глобулы и пачки аморфного строения.
Полиуретан на основе 2,6-ТДИ, имея молекулы более регулярного
строения, дает в качестве первичных структур пачки с трехмерной
кристаллической упорядоченностью. Из пачек формируются сферо-
литы и дендриты. Если полиуретан на основе смеси изомеров ТДИ
15:85 и этиленгликоля при испарении разбавленных растворов
дает сферолиты, то для полиуретана на основе ТДИ 5 : 95 и этилен-
гликоля упорядочение идет дальше и наряду со сферолитами обра-
зуются дендриты, основой которых являются моноплоскости фибрил-
лярного строения.
В отличие от гексаметилендиизоцианата полиуретаны на ос-
нове 2,6-ТДИ, диэтилен- и триэтиленгликоля не кристаллизуются
из-за более низкой стереорегулярности 2,6-ТДИ. Эю объясняется
89
‘-----1----1----1---г—
36 2д 20 12 4 28
Рис. 50 Зависимость интенсив-
ности рефлексов от дифракцион-
ного угла (20) для образцов
полиуретана на основе дифенил-
этандиизоцианат а.
I — аморфный образец, получен
ныЙ быстрым высаживанием, 2 —
кристаллический образец, высажен
ный из концентрированного рас
твора, 3 — кристаллический обра-
зец, высаженный из разбавленного
раствора
ролиты, а полиуретан на
возможностью возникновения нерегулярных водородных связей
между уретановыми и простыми эфирными группами гликолевых
составляющих.
В отличие от ТДИ для дифенилметандиизоцианата (ДФДИ)
кристаллизующиеся полиуретаны получаются не только на основе
этилен-, но и диэтилен- и три этилен гл и кол я Однако способность к
кристаллизации понижается при переходе от этиленгликоля к три-
этиленгликолю по тем же причинам,
как и в случае ТДИ.
При изучении кристаллизации ука-
занных полиуретанов на основе эти-
ленгликоля из растворов и расплава
установлено, что в обоих случаях об-
разуется кристаллическая структура
одинаковой мод и фи к а ци и. Близкие
структуры образуются и при кристал-
лизации полиуретана на основе диэти-
ленгликоля. Г1оли\ ретаи на основе три-
этиленгликоля из раствора хорошо
кристаллизуется, а из расплава дает
продукт с аморфной структурой.
Исследования, проведенные с по-
мощью электронной и оптической ми-
кроскопии, показали, что полиуретан
на основе ДФДИ и этиленгликоля об-
разу ет большой спектр мопфологичес-
ких структур, сферолиты* дендриты,
монокристаллы Полиуретан, получен-
ный на основе ДЭГ, даст в качес-
тве надмолекулярной структуры сфс-
основс ТЭГ не образует каких-либо
четких морфологических форм.
Данные по синтезу и некоторым физико-химическим свойствам
полиуретанов на основе этиленгликоля и дифснилметан-, дифенил-
этан-, дифенилпропан- и дифенилбутандиизоцианатов приведены
в L2G3J.
Полиуретаны на основе перечисленных диизоцианатов образуют
кристаллические продукты, что подтверждено данными рентгенов-
ского анализа. На рис. 50 представлена зависимость интенсивности
рефлексов от дифракционного угла для образцов полиуретанов на
основе дифенилметандиизоцианата, кристаллизующихся с различной
скоростью.
Однако переход от диизоцианатов с нечетным числом метилено-
вых групп между фенильными группами к четным сопровождается
изменением типа кристаллических структур полиуретанов. Такое
явление объясняется изменением конформаций макромолекул по-
лиуретанов.
90
В табл. 24 даны некоторые свойства указанных полиуретанов.
Отметим, что величина отношения Тс /Тпл не зависит ог химического
строения макромолекул, а определяется типом кристаллической
структуры полиуретана.
В работе (240] приведены данные по исследованию кристалли-
ческой структуры полиуретана на основе 4,4'-дифенилмстандиизо-
Таблица 24
Структура и свойства полиуретанов на основе этиленгликоля и ароматических
диизоцианатов
Структура полиуретана
Логарифми
ческая вяз-
кость * **
Лиг
°C
Плот
я ость
при
30° С,
г/см'
СН,—
—NHCOOCI12СН2ОСО-)„
(СН2>2-\Г/-
- NHCOOCH2CH2OCO-)„
(—NH—(СН2)4— \_/ —
-NHc6oCH2CH2OCO--)~
(NH-^_ ^-(CH2)4-^ V.
—NHCOOCH2CH2OCO—)„
1,01 в ДМФА 239 92,7 0,72 1.3233
0,70 в H2SO4 312 116,9 0,67 1,3055
0,36 в ДМФА 207 74,1 0,72 —
0,46 в ДМФА 274 105.7 0,69
* Вязкость 0,5% ного раствора при 30° С.
** Значения Тс и Ггь1 даны в °К.
цианата и тетраметиленгликоля. Показано, что этот полиуретан
образует а-,Р-и у-кристаллические формы. а-Форма плохо поддается
ориентации, а при растяжении более чем на 300% разрушается. Она
возникает в результате отжига при снятии с образца напряжения.
Формы р и у легко образуются, когда молекулярные цепи полиурета-
на ориентированы. Высказано предположение, что a-форма имеет
складчатую структуру, ар- иу-формы — бахромчатуюмицелярную
Синтез полиуретанов на основе фторированного и нефторирован-
ного 1,4-бис- (Р-оксиэтокси)-бензола, а также исследование их неко-
торых физико-химических свойств описаны в [87]. Эти полиуретаны
следует отнести к плохо кристаллизующимся полимерам. Темпера-
туры плавления и кристаллизации фторсодержащих полиуретанов
значительно ниже, чем у их нефторированных аналогов.
91
СВОЙСТВА НЕКОТОРЫХ ПОЛ И AM ИДО УРЕТАНОВ
Для выяснения особенностей строения полиуретанов по сравне-
нию с полиамидами интересно было изучить структуры полиамидо-
уретанов. Некоторые такие полимеры на основе диэтилоламндов
ряда двухосновных кислот описаны в 1155]. Исследованы полиами-
доуретаны (ПАУ) следующего строения:
(—О (СН2)2 NHCO (CH2)rt CONK (СН2)2 OCONH (CH2)e NHCO-]^,
где п— количество метиленовых групп, входящих в состав исход-
ных двухосновных кислот. В этих ПАУ количество метиленовых
групп между амидными группами предопределялось составом кис-
лоты, на основе которой синтезирован диэтилоламид.
Рис. 51. Кривые интенсивности рассеяния рентгеновских
лучей для ПАУ с четным (а) и нечетным (б) п.
ПАУ, полученные на основе диэтилоламидов щавелевой (п = 0),
янтарной (п ~ 2), глутаровой (п~~ 3), адипиновой (п 4), пимели-
новой (п-5), азелиновой (п =- 7), ссбациновой 8) кислот,
исследованы методами ИК-спектроскопического, рентгеновского
[29] и дифференциально-термического анализов [40]. Установлено,
что ПАУ проявляют свойства, присущие как полиуретанам, так и
полиамидам. При этом ПАУ существенно отличаются по своим
свойствам между собой в зависимости от значения п.
Сравнение спектров различных ПАУ показывает, что наиболее
характерные изменения в них наблюдаются в области колебания
групп, отвечающих за образование водородных связей между цепя-
ми. В спектрах ПАУ с нечетным п видны две полосы (3260 и
3320 а с четным п — одна (3320 слС1), при этом появление до-
полнительной полосы 3260 смГ{ сопровождается возрастанием ин-
тенсивности полосы 3080 см~}. На примере ПАУ с п — 3 обнаружено,
92
что у образцов (пленок), полученных путем растворения полимера в
муравьиной кислоте с последующим удалением последней, в спектрах
исчезают полосы 3260 и 1660 см~1, а также уменьшается интенсив-
ность полосы 3080 см~х. Однако после длительного кипячения в воде
п~0 п~2 п-4
п=8
Рис. 52. Термограммы нагревания ПАУ с четным (а) и нечет-
ным (б) п.
и сушки образуется порошок ПАУ, спектр которого имеет исходный
вид. Эти явления дали основание авторам |29] предположить суще-
ствование в серии ПАУ с нечетным п двух типов структур и предло-
жить вероятные модели элементарных ячеек ПАУ с четным и нечет-
ным п.
Кривые интенсивности рассеяния рентгеновских лучей исследо-
ванных ПАУ (рис. 51) свидетельствуют о характерных отличиях
структуры отдельных видов ПАУ. Так, на рентгенограмме ПАУ с
п =8 очень четко видны четыре максимума. Для данного случая пред-
полагается, что на полиуретановом участке цепи реализуется элемен-
тарная ячейка полиуретана, а на полиамидном участке цепи —
структура полиамидов.
На основании вероятной модели элементарной ячейки ПАУ с не-
четным п сделан вывод о том, что расщепление полос в области
93
связанных NH- и С=0- колебаний обусловлено наличием в этом виде
ПЛУ напряженной водородной связи.
В отличие от [29], где фактически изучалась структура исходных
образцов ПАУ, в ]40] исследовалось влияние термического воз-
действия на структуру и структурные превращения в ПАУ (рис. 52).
У всех ПАУ с четным числом п пики плавления имеют сложный
характер и отличаются для отдельных ПАУ. Для ПАУ с нечетным и
только 11АУ с /1~-5 имеет сложный пик плавления. На этом основании
сделано предположение о способности ПАУ к образованию поли-
морфных кристаллических структур, особенно для ПАУ с четным п.
ФАЗОВЫЕ ПРЕВРАЩЕНИЯ
И СТЕКЛОВАНИЕ ЛИНЕЙНЫХ И ТРЕХМЕРНЫХ ПОЛИУРЕТАНОВ
С ОЛИГОЭФИРОГЛИКОЛЕВЫМИ БЛОКАМИ
Способность полиуретанов кристаллизоваться зависит не только
от структуры составляющих полиуретан диизоцианатных и гликоле-
вых (эфирогликолевых) блоков, но и от длины последних (4, 15].
Так, на примере полиуретанов с олигооксиэтиленовыми блоками
различного молекулярного веса установлено [15], что способность
кристаллизоваться проходит через минимум с ростом длины олиго-
эфирного блока.
Найдено также, что строение кристаллической решетки для по-
лиуретанов с олигоэфирными блоками в отличие от полиуретанов с
низкомолекулярными гликолевыми блоками не зависит от строения
диизоцианата, а определяется строением и природой олигоэфиро-
гликоля. Это обусловлено тем, что с ростом эфирного блока возрас-
тает вероятность образования водородных связей между водородом
уретановой группы и кислородом эфирной группы эфирного блока,
которая приводит к дефектам в кристаллической структуре и увели-
чивает долю аморфной структуры. Однако при дальнейшем росте оли-
гоэфирного блока уретановые звенья начинают играть только роль
дефектных участков, которые затрудняют кристаллизацию, но не
могут полностью воспрепятствовать ей. В этом случае кристаллиза-
ция идет только по олигоэфирпым звеньям и строение кристалличес-
кой решетки полиj ретана становится достаточно близким к строению
решетки олигоэфира.
В связи с изложенным целесообразно рассмотреть наряду со
свойствами кристаллизующихся полиуретанов и свойства составля-
ющих их олигоэфирогликолей.
В практике широко применяются сшитые полиуретаны с такими
кристаллизующимися олигоэфирогликолевыми блоками, как олиго-
этиленгликольадипипат и олигоокситетраметиленгликоль (полифу-
рит). Поэтому мы основное внимание уделим этим олигомерам, а
также линейным и сшитым полиуретанам на их основе.
94
Свойства олигоэтиленгликольадипинатов
и синтезированных на их основе
линейных и сшитых полиуретанов
Процесс кристаллизации в олигоэтиленгликольадипинате (ОЭА) с
М == 2000 обстоятельно исследован [ 1341 методами дифференциально-
термического и рентгеноструктурного анализов, ИК-спектроскопип,
оптической и электронной микроскопии (рис. 53, см. вклейку).
Для образца, закристаллизованного при —8° С, наблюдается
мелкокристаллическая структура (рис. 53, 1а, б); сферолит -
ный характер кристаллических зерен хорошо раскрывается лишь
при электронно-микроскопических увеличениях (рис. 53, 1а). В об-
ласти температур от 0 до + 20° С кристаллизуются радиальные сфе-
ролиты (рис. 53, 2а, б), для которых электронная микроскопия
выявляет ламслярпую структуру (рис. 53, 2в). Выше этой тем-
пературы образуются сферолиты кольцевого типа (рис. 53, 4а, б).
В свою очередь до достижения 40° С сферолиты приобретают иголь-
чатую структуру (рис. 53, За, б), которой на электронной микро-
фотографии (рис. 53, Зе) соответствует поверхность с большим чис-
лом как бы торчащих из нее выступов.
По данным рентгеноструктурного анализа, ДТА и ИК-спектро-
скопии установлено, что ОЭА может существовать в двух кристалли-
ческих модификациях — а- и p-формах. В области температур от
0 до 4-20° С стабильна p-форма, а выше 40° С — a-форма; в
промежуточной температурной области сосуществуют обе формы.
Таким образом, p-форма предопределяет образование радиаль-
ных, а-форма— игольчатых, а смесь этих форм — кольцевых сфе-
ролитов.
В связи с этим высказано предположение о том, что своеобразная
структура кольцевых сферолитов обусловлена ритмической кристал-
лизацией попеременно а- и p-форм ОЭА. Однако при нагревании
сферслптов, сформированных при низких температурах, в них на-
блюдаются полиморфные превращения, фиксируемые рентгеновски-
ми и спектральными исследованиями, но нет видимых изменений
форм сферолитов вплоть до их плавления. Это свидетельствует о
стабильности надмолекулярных структур высшего порядка, какими
являются сферолиты, не изменяющихся даже при перестройке пер-
вичных структур, определяющих характер укладки молекулярных
цепей.
Калориметрическим путем изучены ОЭА, а также линейные и
сшитые полиуретаны на ею основе 11091 • Исследования данного сис-
тематического ряда позволили оценить влияние таких параметров,
как гибкостьцепи олигоэфира, густота физической и химической сеток,
на некоторые связанные с подвижностью макроцепей свойства сши-
тых полиуретановых эластомеров. В табл. 25 приведены исходные
характеристики исследованных веществ. Диизоцианатной компо-
нентой при синтезе полиуретанов служил 2,4-толуилендиизоцианат.
95
Таблица 25
Характеристики олигоэтиленадипината и полиуретанов на его
основе
Вещество 5 словное обозначение вещества м С оотноше- ние сши- вателя ДЭГ/Гл
Олигоэтиленгликольадипинат ОЭА-ЮОО 1000
Олигоуретан на ОЭА-ЮОО ОУ-ЮОО 47 000 —
Олигоуретан на ОЭА-2000 ОУ-2000 44 000
Эластомер на ОЭА-ЮОО ЭЛ-1 3800* 1:3
Эластомер на ОЭА-ЮОО ЭЛ-2 54 000* 1:1
Эластомер на ОЭА-2000 ЭЛ-3 — 1:3
Эластомер на ОЭА-2000 ЭЛ-4 —- 1:1
Примечание. Со звездочкой приведено значение MCt
Таблица 26
Уравнение Ср = f (Г) для олигоэфиров, олигоуретанов и сшитых полиуретанов
Полимер Исходный образец Закаленный образец
Уравнение Темпера- турный ин- тервал, °C Уравнение Темпера- турный ин- тервал, °C
от | до от ДО
ЭЛ-1000 —0,012-Ь1,49- Ю-37 —60 20 —0,0124-1,49 Ю“37 —60 20
0,4624-10-4 Т 50 100 0,4624-Ю-4 7 50 100
ЭА-2000 —0,1864-2,17-10—3 7 —70 20 — — —
0,4534- Ю-4 Т 60 100 0,512 60 100
ОУ-1000 —0.125-1-1,5 Ю-37 -30 20 — —
0,320-f-10-4 7 45 100 0.3204-Ю-4 7 —30 100
ОУ-2000 —0,1034-1,52 Ю-37 -30 20 0.3764-10“4 7 -30 25
0,403 4-10“ Т 50 100 0,4034-10-4 7 50 100
ЭЛ-3 0,195 |-10“47 —50 —30 0,1954-10-4 7 -50 —30
—0,1164-1,51- Ю-37 —20 10 — — —
0,325 f-10-4 7 50 100 0,3254-10“4 7 —20 100
ЭЛ-4 0,191-Ь 10—4 Г —50 —30 0,1914-Ю-4 7 —50 —35
-0.ll7-i-l.5-Ю-3 Т —20 10 — — —
0,315 -Ю-47 50 100 0,3154-Ю-4 7 —20 100
ЭЛ-1 0,1904-10“47 —50 —20 То же —20 100
0,3064-10^7 0 100 » » —20 100
ЭЛ-2 0,1904-10“4 Т —50 —20 » » —20 100
0,310-Ю“4 7 —5 100 » » —20 100
96
Соотношение NCO : ОН в форполимерах (олигоуретанах) равнялось
1:1. Сшитые полиуретановые эластомеры получали через стадию
форполимера при соотношении NCO :ОН =2:1; сшивка произво-
дилась смесью диэтиленгликоля (ДЭГ) с глицерином (Гл). Общее
соотношение NCO : ОН =1:1.
Кривые температурной зависимости теплоемкости образцов всех
указанных видов полимеров приведены на рис. 54 и 55. Как видно
из рисунков, кривые теплоемкости Ср/Т состоят из нескольких линей-
ных участков, соответствующих областям стеклообразного, высоко-
эластического, кристаллического и вязкотекучего состояний поли-
меров, а также участков с аномальными отклонениями от линейности
в областях стеклования и фазовых переходов (табл. 26).
Ход температурной зависимости теплоемкости для кристалли-
ческих (отожженных) образцов ОЭА-ЮОО и ОЭА-2000 (рис. 54) в
широком интервале температур, лежащем ниже температуры начала
фазовых превращений, является линейным. Начиная с 20° С на
кривых / обоих олигомеров наблюдается подъем, который для
ОЭА-ЮОО, пройдя через точку перегиба при 40° С, переходит в пик
плавления при 50° С с Ср = 1,95 кал!г-град. Для ОЭА-2000 этот
подъем оканчивается эндотермическим пиком при 40° С с Ср —
= 0,94 кал/г-град, после которого значение теплоемкости резко
падает до 0,76 при 43° С и вновь возрастает, переходя во второй
эндотермический пик (плавления) при 53° С с Ср - 1,76 кал/г-град.
Полученные значения температуры плавления ОЭА-2000 совпадают
со значением для высокомолекулярного полиэтиленгликольади-
пината 1221].
По окончании процесса плавления (60° С) теплоемкость распла-
вов обоих олигомеров почти не изменяется. Тепловые эффекты плав-
ления отожженных образцов ОЭА-ЮОО и ОЭА-2000 равны соответ-
ственно 18,7 и 17,8 кал/г.
Более существенные различия в ходе теплоемкости наблюдают-
ся для образцов, подвергнутых закалке. Как видно из рис. 54, а,
образец ОЭА-ЮОО не подвергается закалке, о чем свидетельствует
отсутствие характерного перегиба кривой 2 при стекловании и ли-
нейная зависимость теплоемкости от температуры в интервале от —60
до 5° С, совпадающая с таковой для отожженного образца. Однако
при 16° С на кривой 2 имеется экзотермический пик кристаллизации,
после которого теплоемкость непрерывно возрастает, переходя в пик
плавления при 45° С с Ср = 1,98 кал/г-град, и уменьшается до
0,493 при 55° С.
При нагревании закаленного образца ОЭА-2000 (рис. 54, б,
кривая 2) наблюдается скачок теплоемкости в интервале от—62 до
—54° С, обусловленный переходом олигомера из стеклообразного в
вязкотекучсе состояние, и два экзотермических пика кристал-
лизации при —16 и 20° С, после чего теплоемкость непрерывно воз-
растает до 0,46 кал/г-град при 53° С. Как и в случае отожженных
образцов, теплоемкости расплавов закаленных образцов обоих
7 407
97
олигомеров лишь незначительно увеличиваются с температурой.
Ввиду наложения указанных эффектов не представлялось воз-
можным оценить их количественно Однако приближенно тепловой
CPi кал/г град
Рис. 54. Теплоемкость атигоэтиленгликольадипинатов ОЭЛ-ЮОО (а) и
ОЭЛ-2000
/ —• отожженный образец. 2 — закаленный образец.
эффект плавления закаленного образца ОЭА-2000 в 1,5 раза больше
суммарного теплового эффекта кристаллизации.
Кривые температурной зависимости теплоемкости олигоуретанов
и эластомеров приведены на рис. 55. У исходных образцов полиуре-
танов (кривая /) не обнаружено стеклования и их теплоемкость ли-
нейно растет от низких температур до 20° С. Выше 20° С теплоемко-
98
ста исходных образцов плавно возрастают и появляются пики плав-
ления для ОУ-1000 при 38° С (Ср = 1,02 кал/г-град) и для ОУ-2000
при 40° С (Ср — 1,66 кал/г-град). Соответствующие тепловые эффек-
ты составляют 6,9 и 12,8 кал/г. Плавление ОУ-1000 и ОУ-200Э окан-
чивается соответственно при 43 и 49э С, после чего теплоемкость
остается почти постоянной.
-50 0 50 100-50 0 50 Т^С
Рис. 55. Теплоемкость олигоуретанов ОУ-10Э0 (а), ОУ-20Э0 (б) и сшитых
полиуретанов ЭЛ-4 (в), ЭЛ-3 (г), ЭЛ-2 (б), ЭЛ-1 (е):
/ — исходный образен: 2 — закаленный образец
В закаленных образцах (кривая 2) ОУ-ЮОО и ОУ-2000 наблю-
даются скачки теплоемкости при —36 и —46° С соответственно,
связанные с переходом из стеклообразного в высокоэластическое
состояние. Выше температуры стеклования теплоемкости обоих за-
каленных образцов линейно зависят от температуры (см. табл. 26)
вплоть до 100° С, однако у ОУ-2000 наблюдается небольшой макси-
мум при 40° С с Ср — 0,44 кал!г-град. Выше 50° С темплоемкости
исходных и закаленных образцов совпадают.
Из рис. 55, в, г видно, что эластомеры ЭЛ-3 и ЭЛ-4 на основе
ОЭА-2000 способны к кристаллизации, о чем свидетельствуют пики
плавления исходных образцов (кривые /). Температуры и теплоты
плавления исходных образцов ЭЛ-3 и ЭЛ-4 равны 36° С, 8,95 кал!г и
36° С, 8,20 кал!г соответственно. Наличие небольших (порядка 0,02—
0,03) скачков теплоемкости в области температур стеклования
говорит о незавершенности процесса кристаллизации в данных сис-
темах. Кривые теплоемкости закаленных образцов состоят из линей-
ных участков до и после температуры стеклования, которая для
99
ЭЛ-3 и ЭЛ-4 равна соответственно—27 и—29° С. Эластомеры ЭЛ-1
и ЭЛ-2 не способны к кристаллизации (рис. 55, d, е). Кривые исход-
ных и закаленных образцов для этих эластомеров совпадают и сос-
тоят из линейных участков, разделенных интервалами перегибов,
соответствующих температурам стеклования (—7° С для ЭЛ-1 и
”-14° С для ЭЛ-2).
Все исследованные образцы, за исключением ОЭЛ-ЮОО, подвер-
гались закалке, и для них определены значения некоторых величин,
характеризующих процесс стеклования (табл. 27). В данном случае,
Таблица 27
Характеристики процесса стеклования систем на основе
олигоэтилен! ликольадипината
Вещество ка 1/ гХ X град ДГ*. °C * * тс , °C м кал/моль X X град
ОЭА-1000 0,108 8 —57 17,2 1,86
ОУ-1000 0,132 8 -36 22,4 2,96
ОУ-2000 0,130 8 -46 22,4 2,91
ЭЛ-4 0,127 9 —29 22,4 2,85
ЭЛ-3 0,135 8 —27 22,4 3,02
ЭЛ-2 0,126 12 —14 22,4 2,82
ЭЛ-1 0,120 15 —7 22,4 2,69
* ДТС — интервал стеклования.
** Тс — определялась как температура, при которой инкремент тепло-
емкости Ср при стекловании достигал половины своею значения.
как и для систем с олигодиэтиленгликолевыми блоками, при пере-
ходе от олигомера к сшитому эластомеру Тс закономерно повы-
шается.
Поскольку Тс полимеров зависит от молекулярного веса, регу-
лярности строения, гибкости цепи, наличия и расположения функ-
циональных групп, густоты сетки химических сшивок и прочее, то
повышение Тс линейных олигоуретанов обусловлено либо большей
по сравнению с исходным олигоэфиром жесткостью цепей, определя-
емой термодинамической гибкостью, либо наличием в последних
полярных уретановых групп, влияющих на кинетическую гибкость
(подвижность) цепей. Для систем с олигодиэтиленгликолевыми бло-
ками уже показано бесспорное влияние второго фактора.
В данном случае сравнение термодинамических гибкостей олиго-
эфиров и олигоуретанов проведено исходя из эквивалентности формы
и размеров макромолекул в ©-растворителе и неупорядоченном
состоянии. В качестве меры термодинамической гибкости выбран
параметр До из уравнения Куна — Марка — Хувинка для вязкости
полимеров в ©-растворителях, поскольку До зависит только от
100
строения и конформации макромолекулы. Ке определяли по уравне-
нию Кревелена [252]:
Ke = (S/X'V . (IV,6)
гдеАЦ—молекулярный вес, приходящийся па один атом главной
цепщЗ*—величина, зависящая от удельной жесткости, приходя-
щейся на один атом главной цепи.
Численное значение параметра К& оказалось Ке = (59-10-2/4,
14)3 = 17,8-10—4 и (51,2 10-2/4,15)3 — 18,8- 10”4 для ОЭА и ОУ
Рис» 56. Зависимость Тс олигоуретанов от концентрации уретановых групп:
—Тс рассчитана; Гс определена экспериментально.
соответственно. Рассчитанное значение Ке для ОЭА в точности сов-
падает с опытными данными, что подтверждает достоверность полу-
ченных результатов.
Так как значение К® для ОЭА и ОУ близки по величине, то можно
считать, что термодинамические гибкости макромолекул ОЭА и ОУ
практически одинаковые. Из этого следует, что наличие диизоциа-
натного блока не влияет на термодинамическую гибкость цепей ли-
нейных олигоуретанов, а следовательно, возрастание Тс при пере-
ходе от ОЭА к ОУ обусловлено только уменьшением кинетической
гибкости цепей, которая зависит от концентрации полярных урета-
новых групп. Для данного случая наблюдается линейная зависи-
мость Тс от концентрации уретановых групп в ОУ (рис. 56).
Наличие химической сшивки в эластомерах также значительно
уменьшает сегментальную подвижность макромолекул, приводя к
дальнейшему повышению Тс. Однако преобладающий вклад в повы-
шение Тс при переходе от олигомера к эластомеру и для данной си-
стемы вносит образуемая полярными группами физическая сетка
(табл. 28). Некоторое повышение Тс ЭЛ-3 и ЭЛ-4 может давать и час-
тичная кристаллизация этих эластомеров (см. рис .55, в, г). Жесткие
кристаллические участки также ограничивают подвижность цепей в
прилегающих областях, вызывая повышение Tv.
Рассмотрим теперь характеристику процесса стеклования путем
сравнения скачка теплоемкости при стекловании отдельных объектов
101
исследованной системы с учетом правила Вундерлиха [343]. Согласно
этому правилу инкремент теплоемкости при стекловании в расчете
на 1 моль структурных «бусинок» молекулы является постоянной
величиной, равной 2,97 кал!молъ град. В табл. 27 приведены зна-
чения АСР для исследованного ряда полимеров, полученные по фор-
муле
\с'р - мср,
где М — средний молекулярный вес, приходящийся на одну «бусин-
ку»; ДСР — наблюдаемый в эксперименте скачок теплоемкости при
стекловании.
Значения А Ср для всех исследованных систем (см. табл. 27),
за исключением ОЭА-2000, близки к теоретическому. Несколь-
ко низкое значение Л Ср для ОЭА-2000 объясняется частичной кри-
Таблица 28
Вклад диизоцианата и химической сшивки в повышении Тс
олигодиэтиленгликольадипината
Вещество С. ио и, 1000 г тс ОУ (расечя таннля), °C тс, °C дгс ТДИЦ ДГС сши- вателя
ОЭА-2000 —57
ЭЛ-1 1,8 — 19 —7 38 12
ЭЛ-2 1 8 — 19 — 14 38 5
ЭЛ-3 3,2 -35 -27 22 8
ЭЛ-4 3,2 1 —35 —29 22 6
сталлизацией олигомера при закалке. На это указывав! и то, что
отношение теоретического инкремента теплоемкости при с мило-
вании к экспериментальному, которое может служить мерой глу-
бины закристаллизовапности полимера, для ОЭА-2000 близко
к значению (1,5) соотношения его теплот плавления и крис!ал-
лизации.
Согласно [343], Д Ср пропорционально удельному объему энер-
гии образования новых дырок и обратно пропорционально Тс. Из-
вестно [3431, что сшивка полимеров сопровождается уменьшением
удельного обьема. В го же время при переходе от ОУ к сшитым
эластомерам Д Ср остается неизменной, а Тс возрастает (см. табл. 27).
Отсюда, наличие в молекулярной структуре полиуретанов сетки
физических и химических связей повышает энергию образования
новых дырок тем больше, чем больше густота сетки.
Следует отметить, что абсолютное значение, а также скорость
роста теплоемкости в области низких температур при переходе от
102
олигоэфиров к сшитым каучукам понижается (см. табл. 26). Это
можно объяснить уменшснием числа колебательных степеней свободы
системы благодаря наличию сильного взаимодействия между поляр-
ными группами в олигоуретанах, а в сшитых эластомерах — нали-
чию сетки химических связей.
Из сравнения кривых темпсра1урной зависимости теплоемко-
сти отожженных и закаленных образцов ОЭА-1000 и ОЭА-2000
(см. рис. 54, а, б) видно, что максимумы пиков плавления закален-
ных образцов обоих олигомеров смещены в область более низких тем-
ператур Кроме того, для закаленного образца ОЭА-2000 наблюдает-
ся два экзоермических пика кристаллизации. Обнаруженное явле-
ние объясняется тем, что в зависимости от режима термообработки
в образцах ОЭА образуются различные кристаллические модифи-
кации.
Небольшая площадь пика кристаллизации высокотемператур-
ной формы (при 16°С) и отсутс!вие пика кристаллизации низко-
температурной формы в ОЭА-1000 указывают на его чрезвычайно
высокую скорость кристаллизации, которую нельзя полностью
подавить даже закалкой в жидком азоте. Сложный характер за-
висимости теплоемкости обоих олигоэфиров в инюрвале 20—40° С
(см. рис. 54, кривая 1) объясняется перестройкой и распадом над-
молекулярных структур.
Как видно из рис. 55, переход от ОЭА к ОУ сопровождается
заметным уменьшением способности кристаллизоваться, о чем сви-
детельствуют значительное уменьшение тепловых эффектов плав-
ления исходных образцов и отсутствие кристаллизации в закален-
ных образцах ОУ. ОУ кристаллизуются лишь во времени, причем
ОУ-2000 кристаллизуется значительно быстрее, чем ОУ 1000.
Процесс кристаллизации полимеров можно разделить на две
стадии: образование метасiабильного двухмерно-упорядоченного
состояния (без изменения межмолекул яркого взаимодействия) и
спонтанный переход в конечное состояние с фехмерным поряд-
ком, сопровождающийся увеличением межмолекулярного взаимо-
действия. Для полимеров, не имеющих полярных функциональных
групп, способность к кристаллизации определяется в основном
термодинамической гибкостью цепей, и поэтому для них указанные
стадии кристаллизации экспериментально разделить невозможно.
Однако свойства макромолекул ОУ определяются нетолько термоди-
намической, но и кинетической гибкостью, зависящей от концентра-
ции уретановых ipynn. В этом случае межмолекулярные водородные
связи, которые образуются па первой стадии кристаллизации, замет-
но стабилизируют двухмерно-упорядоченное псевдокристалл ичес-
кое состояние, в результат чею в указанных температурных ус-
ловиях вторая стадия кристаллизации протекает в течение длитель-
ного времени [276, 2661.
Поскольку концентрация уретановых групп в ОУ-2000 меньше,
чем в ОУ-ЮОО, способность к кристаллизации, т. е. меньшая про-
103
должительность первой стадии кристаллизации, у ОУ-2000
больше. С другой стороны, хотя плотность энергии когезии в ОУ
выше благодаря наличию уретановых групп, чем у соответствующих
ОЭЛ, температура и теплота плавления ниже. Это объясняется значи-
тельно большей дефектностью кристаллической решетки ОУ, обус-
ловленной асимметрией молекулярного строения звеньев изомера
2,4-ТДИ. По этой же причине тепловой эффект плавления ОУ-1000
значительно меньше, чем у ОУ-2000. Так как концентрация блоков
2,4-ТДИ в ОУ-ЮОО больше, чем в ОУ-2000, можно ожидать, что
понижение температуры плавления у ОУ-ЮОО будет больше. Одна-
ко разность температур плавления ОЭА и соответствующих ОУ при-
мерно одинакова. Предполагается, что относительно высокая
температура плавления ОУ-ЮОО вызвана небольшой энтропией
плавления, вследствие повышения энтропии в кристалличес-
ком состоянии благодаря разупорядочивающему действию звеньев
2,4-ТДИ.
Переход к сшитым эластомерам сопровождается дальнейшим
понижением способности к кристаллизации в результате стеричес-
ких ограничений, накладываемых на подвижность цепей узлами
химической сетки. Способность к кристаллизации сохраняют толь-
ко ЭЛ-4 и ЭЛ-3 (см. рис. 55), густота сетки в которых значительно
меньше, чем в ЭЛ-1 и ЭЛ-2.
Согласно теории плавления сетчатых полимеров [88], сшивка
статистических цепей должна приводить к понижению температуры
плавления, пропорциональному густоте сетки. Незначительное
(порядка 2° С) понижение температуры плавления ЭЛ-3 и ЭЛ-2
объясняется настолько малой i устотой сетки, что исключение сши-
тых участков цепей из кристаллической решетки приводит лишь к
незначительному увеличению энтропии плавления и почти не влия-
ет на температуру плавления.
Абсолютная величина теплоемкости расплавов исследованных
ОЭА и ОУ в интервале температур до 100' С почти не возрастает.
В соответствии с теорией свободного обьема [2311 теплоемкость
жидкостей складывается из «колебательной?/ и «дырочной» (свя-
занной с образованием новых дырок) частей. Если принять, что
процесс плавления сводится к образованию и обогащению расплава
дырками [1431, io наблюдаемый ход теплоемкости расплавов ис-
следуемых олигомеров следует, очевидно, объяснить нулевым вкла-
дом одного из слагаемых общей теплоемкости, а именно «дырочной»
части. Это говорит о том, что энергия образования новых дырок
превосходит энергию теплового движения молекул в расплавах ОЭА
и ОУ.
104
Свойства олигоокситетраметиленгликоле й
и синтезированных на их основе
линейных и сшитых полиуретанов
Некоторые свойства полиуретанов па основе олигоокситегра-
метиленгликоля (полифурита) исследованы калориметрическим ме-
тодом [77] (1абл. 29). Соотношение OH/NCO для олиюуретанов
и эластомеров было таким же, как и для 1аковых на основе ОЭА.
Как видно на рис. 57, а, теплоемкость отожженного образца
ПФ-1000 (кривая 1) в интервале от —70 до —50сС имеет постоян-
ное значение и затем плавно возрастает, проявляя небольшой
Таблица 29
Исходная характеристика олигоокситетраметиленгликоля
и полиуретанов на его основе
Вещество
Полифурит
Олигоурстан на ПФ-1000
Олигоуретан на ПФ-2000
Эластомер на ПФ-1000
Эластомер на ПФ-1000
Эластомер на ПФ-2000
Эластомер на ПФ-2000
ПФ-1000 960 —
ОУФ-Ю00 6700 —
ОУФ-2000 15 600 —
ЭЛФ-1 4600* 1.1
ЭЛФ-2 2540* 1:3
ЭЛФ-3 7950 * 1:1
ЭЛФ-4 4360 * 1:3
* Приведены значения Мс
эндотермический пик (тепловой эффект 5,81 кал/г) с максимумом
при 0°С, после чего понижаекя при 6° С и затем резко возрастает
при 13° С. В интервале 13—18° С рост теплоемкости несколько за-
медляется, а затем возникает в основной пик плавления при 23° С.
Плавление оканчивается при 33° С, после чею значение теплоемкос-
ти до 100°С почти не меняется. Общий тепловой эффект плавления
составляет 28,65 кал'г.
При нагревании закаленного образца ПФ-1000 (рис. 57, а, кри-
вая 2 ) теплоемкость растет линейно в интервале температур от
—80 до —20°С, после которою теплоемкость быстро возрастает,
переходя в эндотермический пик с максимумом при 11°С и тепло-
вым эффектом 10,6 кал!г. После уменьшения при 15°С теплоемкость
вновь быстро возрастает в точке пл авления при 22° С и уменьшается
вплоть до 32°С. Суммарный тепловой эффект плавления составляет
27,95 кал/г.
Теплоемкость отожженною образца ПФ-2000 (рис. 57, б. кривая
1) линейно возрастает от —70 до —10° С и затем плавно возникает
105
Ср9кал/гград
Рис. 57. Теплоемкость полиуретанов ПФ-1000 (а); ПФ-2000 (б);
/ — отожженный образец.
эндо1ермический пик при 19° С (тепловой эффект 6,95 ка i/г). При
23°С теплоемкость понижается, затем быстро возрастает и стано-
вится максимальной ь точке плавления при 32°С. Общий тепловой
эффект плавления равен 26,7 ка ?/г.
На кривой теплоемкости закаленного образца ПФ-2000
(рис. 57, б,кривая2) наблюдается небольшой эндотермический подъем
106
в интервале температур от—95 до —803С, характерный для перехо-
да из стеклообразного в высокоэластическое состояние. Значение
Тс для полиокситетраметилена равно —55 по 13411, —84 по 13381,
—86° С по 13371.
Из результатов калориметрических измерений вытекает, что
Тс ПФ-2000 имеет значение порядка—88°С, что близко к значению
2 —> закаленный образец.
—86° С, полученному дилатометрическим методом [337] для высо-
комолекулярного полимера.
От —80° С теплоемкость закаленного образца ПФ-2000 линейно
возрастает до —20° С и проходит через эндотермическую
ступеньку при 11—15° С (тепловой эффект 5,45 кал!г), переходя затем
в пик плавления при 26° С; процесс плавления оканчивается при
107
36°С и теплоемкость уменьшается. Тепловой эффект плавления со-
ставляет 19,65 кал!г. В интервале 36—80°С теплоемкость почти не
возрастает, а начиная с 80° С уменьшается, проявляя экзотермичес-
кий пик с максимумом при 135° С. Тепловой эффект экзотермического
процесса при 145° С равен 64,8 кал!г.
I — ЭЛФ 1,2- ЭЛФ 2, 3 — ЭЛФ-3; 4 — ЭЛФ-4.
На кривой теплоемкости отожженного образца ОУФ-1000
(рис. 57, в, г) наблюдается скачок в интервале ог —75 до —64° С,
соответствующий расстекловыванию олигомера, после чего теплоем-
кость сохраняет линейную зависимость с незначительным подъемом
до —35° С. Далее она уменьшается до достижения экзотермического
пика при—11° С в интервале кристаллизации от—35 и примерно до
3° С и непосредственно переходит в пик плавления при 15° С. Вви-
ду наложения процессов кристаллизации и плавления точное опре-
деление теплот этих процессов не представлялось возможным. Дан-
ные теплоты близки между собой и составляют около 6,8 кал!г.
Аналогичная картина наблюдается и для закаленного образца
ОУФ-1000, однако в этом случае интервал стеклования несколько
сужается, а интервал кристаллизации смещается в область более
низких (от —40 до 0° С) температур с пиком при —15° С. В то же
время интервал плавления расширяется (от 0 до 22° С), причем
пик плавления смещается к 13° С. Тепловые эффекты кристаллиза-
ции и плавления равны 6,43 и 8,95 кал!г соответственно.
Кривая теплоемкости отожженного образца ОУФ-2000 (рис.57 в, г,
кривая /) в области низких температур состоит из двух участков
в интервале от —70 до —40 и от —30 до 0° С, затем проходит через
108
небольшой подъем с максимумом при 10° С. понижается при 18° С
и резко возрастает в точке плавления при 29° С. Тепловой эффект
составляет 17,35 кал!г.
Теплоемкость закаленного образца ОУФ-2000 (рис. 57, г, кри-
вая 2) линейно возрастает от —70 до —20° С, проходит через подъем
при —1° С и после незначительного снижения при 6° С быстро воз-
растает в точке плавления 27° С. Теплота плавления 16,9 кал!г.
Таблица 30
Уравнения Ср = f (Т) для линейных участков теплоемкостей олигоэфиров,
олигоуретанов и сшитых полиуретанов
Полимер Отожженный образец Закаленный образец
Уравнение Темпера- турный ин- тервал, °C Уравнение 1 Темпера- турный ин- тервал, °C
от до от до
ПФ-1000 0,247+1,97 1(Г4Т -80 —20 0,298 —70 —50
0,162+9,8-10-4 т 33 100 0,503 32 100
ПФ-2000 -0,293-1-3.06.10~3 т —70 —10 0,0364-1,49 10~'лт -80 -20
0,550 43 90 0,450 36 80
ОУ-ЮОО 0,182+9,8.10~4Т —60 —35 0,166+9,8-10-4 т -60 —40
0,300+4,95-10~4 т 23 100 0,148+1,03 10-3 т 23 100
ОУ-2000 0,102+9,6- 10~4Т —70 —40 -0,083+1,51-10~3 Т —70 —20
0,227+1,03 10“3 т 40 100 —0,198+2,02 -10“3 Т 35 100
ЭЛФ-1 0,113+9,8-Ю-4 т —70 —54 0,113+9,8-10-4 т —80 -54
0,190+9,8-10-4 т —35 100 0,198+9,8 10"47, -36 100
ЭЛФ-2 0,094+9,8-10-4 Т —90 —50 0,094-1-9,8-10-4 Т —70 —50
0,137+9,8-10-4 т —30 100 0,088—9,8-10-4 Т 30 100
ЭЛФ-3 0,172+9,8-10~4 Т -60 —40 0,232+9,8 10~4 т -65 —20
0,186+9,8-10-4 т 35 100 0,232+9,8 10~4 Т 30 100
ЭЛФ-4 0,148+9,8 10~4 т —60 —40 0,202+9,8 10“4 т —60 0
0,186+9,8-10—4 т 30 100 0,175+1,05 10-3 Т 30 100
На рис. 58, а представлены кривые теплоемкости исходных образ-
цов сшитых эластомеров. Данные эластомеры не способны к кристал-
лизации при комнатной температуре, о чем свидетельствует отсутст-
вие заметных пиков плавления, а также наличие четких перегибов
при температурах стеклования на кривых теплоемкости в области
низких температур. Температуры стеклования эластомера на ос-
нове ПФ-2000 (кривые 1 и 2) лежат в области более низких темпера-
тур, чем у эластомеров на основе ПФ-1000 (кривые 3, 4).
На рис. 58, б приведены кривые теплоемкости указанных эласто-
меров, выдержанных длительное время при температуре —4° С.
Из кривых видно, что при данных условиях кристаллизуются только
эластомеры на основе ПФ-2000 (кривые 1 и 2) в довольно широком
109
температурном интервале от —40 до 5° С. Плавление этих образцов
сопровождается появлением четких максимумов теплоемкости при
20° С. Теплоты кристаллизации и плавления составляют соответст-
венно 5,06 и 5,83 кал/г для ЭЛФ-3 и 2,93 и 3,66 кал/г ддя ЭЛФ-4.
В табл. 30 даны параметры уравнения Cp=f (Т) для линейных
участков приведенных кривых, рассчитанные по методу наименьших
квадратов. Табл. 31 содержит значения величин, характеризующих
переход исследованных объектов из стеклообразного в высокоэла-
стическое состояние. Константа Ket рассчитанная по формуле
Таблица 31
Характеристики процесса стеклования полифурита и полиуретанов
на его основе
Образец кал/ гХ Хград ДТС, °C тс, °C М ч кал^мольХ X град
ПФ-2000 —88 0,062 14,4 0,89
ОУФ-ЮОО 6 —67 0,116 23,3 2,71
ЭЛФ-1 23 —43 0,104 23,3 2,42
ЭЛФ-2 21 —39 0,098 23,3 2,28
ЭЛФ-3 20 —77 0,113 23,3 2,63
ЭЛФ-4 20 -80 0,106 23,3 2,47
(IV, 6), при переходе от олигоэфира к олигоуретану при этом практи-
чески не изменяется. Значения ее для ПФ и ОУФ равны 19,1-КП4
и 19,4 • 10“4 соответственно. Следовательно, термодинамическая гиб-
кость макромолекул и этих олигомеров приблизительно одинакова,
т. е. наличие блоков 2,4-толуилендиизоцианата не отражается на
гибкости цепи и Тс для данных систем предопределяется глав-
ным образом изменением кинетической гибкости макроцепи. Это
соответствует данным, приведенным в гл. II.
Наличие сетки химических связей также приводит к некоторому
повышению Тс. Однако основное влияние поперечные связи оказы-
вают на величину интервала стеклования, причем наблюдается тен-
денция к расширению интервала стеклования при увеличении степе-
ни сшивки и уменьшении молекулярного веса исходного олигоэфира.
Так как стеклование является релаксационным процессом, увели-
чение интервала стеклования можно объяснить расширением спектра
времен релаксации в результате локализации свободного объема
системы в местах сшивки 12461, а также частичной диссоциацией
относительно слабых водородных связей в полиуретанах на основе
простых олигоэфиров.
Значения инкремента теплоемкости при стекловании А Ср для
исследованного ряда полимеров (табл. 31) близки к значениям, рас-
считанным для полимерных стекол [343]. Исключение составляет ПФ-
2000, что объясняется частичной его кристаллизацией при закалке.
ПО
Величина инкремента теплоемкости при стекловании пропор-
циональна энергии образования новых дырок и обратно пропор-
циональна температуре стеклования. Отсюда можно сделать вывод»
что и для данного ряда полимеров энергия образования новых дырок
возрастает при переходе от исходного олигоэфира к сшитому
эластомеру.
В связи со сказанным интересно сравнить параметры процесса
стеклования полимеров обоих исследованных рядов. Так как Тс
полиуретанов на основе простых полиэфиров (полифуритов) зна-
чительно ниже, чем у полиуретанов на основе сложных полиэфиров
(олигоэтиленадипинатов), то энергия образования дырок в послед-
них соответственно выше. Поскольку полиуретаны на основе прос-
тых полиэфиров отличаются от полиуретанов на основе сложных
полиэфиров только характером межмолекулярных водородныхсвязей,
естественно предположить, что водородные связи между кислоро-
дом сложных эфирных групп и водородом конпевых гидроксиль-
ных групп или NH-групп уретанового звена более прочные, чем
связи с участием кислорода простых эфирных групп. Это подтвер-
ждается и литературными данными [13, 327, 3321.
Для закаленных и отожженных образцов ПФ-1000 и ПФ-2000
наблюдается ступенчатый характер плавления (см. рис. 57). Фор-
мально такой характер распада кристаллической структуры на-
поминает картину плавления отожженного образца ОЭА-2000
(см. рис. 54), который в кристаллическом состоянии может существо-
вать в двух модификациях. Однако результаты рентгеноструктур-
ного анализа полиокситетраметилена [183,2361 позволяют считать,
что для него характерна только одна кристаллическая структура и
что наблюдаемый ход кривой плавления обусловлен кинетическими
условиями кристаллизации полифурита. При этом отмечаются две
особенности. Во-первых, плавление закаленных и отожженных
образцов имеет аналогичный характер, свидетельствующий о том,
что механизм зародышеобразования при кристаллизации этих оли-
гомеров не зависит от степени переохлаждения и скорости охлажде-
ния расплавов. Отсюда, образование зародышей кристаллической
фазы имеет атермический характер, обусловленный существова-
нием микрогетеро!снностей в расплавах ПФ-1000 и ПФ-2000, нагре-
тых до температуры 100° С. Такими микрогетерогенностями, по-ви-
димому, являются ассоциаты из макромолекул, образующиеся
благодаря водородным связям между эфирным кислородом и кон-
цевыми гидроксильными группами.
Во-вторых, в соответствии с термодинамической теорией Флори
[2101 температура плавления полимерных кристаллов определяется
их размерами. В случае равномерного распределения кристаллов
по размерам кривая плавления полимера должна быть плавной и
иметь один максимум при температуре, соответствующей плавлению
основной массы кристаллов. На полученных кривых теплоемкости
ПФ-1000 и ПФ-2000 есть несколько максимумов в интервале
Ш
плавления (см. рис. 57, а, б). Поэтому предполагается, что при кри-
сталлизации этих олигомеров происходит образование кристаллов
нескольких дискретных размеров, которые плавятся при температу-
рах, соответствующих максимумам на кривых теплоемкости. Это
согласуется с результатами рентгенографического и дилатометри-
ческого исследований сходных по строению низкомолекулярных
полиоксиэтиленов [1611, для которых обнаружено явление фракцио-
нирования в блоке плоских кристаллов в зависимости от их высоты.
В свою очередь, высота кристаллита в низкомолекулярных по-
лиэфирах определяется длш.ой макромолекулы [1611. Таким обра-
зом, наблюдаемый характер плавления может свидетельствовать
одновременно о пол и дисперсности низкомолекулярных ПФ-1000
и ПФ-2000, которая в зависимости от условия синтеза может до-
стигнуть значительной величины [83, 3341.
Таблица 32
Термодинамические параметры плавления
Полимер Образец ГПЛ’ °с Д^лл* кал/г Д^пл» кал/ моль Д-^пл» кал/молъХ Хград
ПФ-1000 Отожженный 23 28,65 2060 6,98
Закаленный 22 27,95 2010 6,83
ПФ-2000 Отожженный 32 26,7 1920 6,40
Закаленный 26 19,65 1415 4,74
ОУ-1000 Отожженный 15 6,8 557 1,93
Закаленный 13 8,95 735 2,56
ОУ-2000 Отожженный 29 17,35 1330 4,40
Закаленный 27 16,90 1300 4,33
ЭЛФ-3 Отожженный 20 5,83 448 1,52
ЭЛФ-4 20 3,66 232 0,96
’Энтропию плавления рассчитывали по формуле
Д5пл = д^пл/?пл-
Низкие температуры плавления ПФ-1000 и ПФ-2000 (табл. 32)
объясняются большими значениями энтропии плавления. Поскольку
энтропия плавления сильно зависит от конформации макромолекул
в расплавленном состоянии, ниже приводится оценка вклада конфор-
мационной энтропии этих олигомеров при переходе из кристалли-
ческого в жидкое состояние.
Энтропию плавления полимеров можно представить в виде
следующего выражения [881:
А5Пл — ASy т (ASn<n)vs
где A Sy— вклад в энтропию, вызванный изменением объема при
плавлении А Кпл; А5ПЛ — вклад, вызванный изменением конформа-
ции цепи при плавлении при постоянном объеме. Величину ASy опре-
деляли по формуле
Д$у = _-2-ДУпл,
р
112
где а — коэффициент объемного термического расширения; (J — ко-
эффициент изотермической сжимаемости. Значения аир взягы из
[313 J. Так как ПФ-1000 и ПФ-2000 при 20сС находятся в частично рас-
плавленном состоянии, то для расчета ЛИпл нельзя воспользоваться
экспериментальными значениями плотности при 20°C(o2ft). Поэтому
использовали значения р16 и р71 для ПФ-3000 из [313], экстраполируя
их до соответствующих температур плавления. Полученные значения
приращения объема при плавлении одинаковы для ПФ-1000 и ПФ-2000
и составляют 0,08 ои3/г=5,76 см3! моль, а ДЗу = —(7,29- 10“4/6,85х
Х10'5) • 5,76-- —61,4 см3 атм1град-моль^= —1,48 кал!град*моль.
Так как вклад изменения объема в энтропию плавления нс бо-
лее 21—23%, то высокое значение энтропии плавления ПФ-1000
и ПФ-2000 обусловлено главным образом увеличением числа кон-
формаций макромолекул при плавлении. Таким образом, макромо-
лекулы ПФ в расплаве должны иметь более или менее свернутые
конформации по сравнению с конформацией плоского зигзага в кри-
сталлическом состоянии [236, 1831.
Для линейных олигоуретанов, особенно для ОУФ-1000, наблю-
даются уменьшение теплот плавления и смещение температур плав-
ления в область более низких температур по сравнению с исходны-
ми олигоэфирами. Это объясняется тем, что в ОУФ-1000 концен-
трация звеньев 2,4-ТДИ, наличие которых в цепи приводит к
дефектам кристаллической решетки [15], выше, чем в ОУФ-2000. По
той же причине энтропия плавления ОУФ-ЮОО меньше энтропии
плавления ОУФ-2000 (табл. 32).
Существенно, что наличие диизоцианатных блоков практически
не сказывается на способности олигоуретанов на основе полифурита
(ОУФ) кристаллизоваться, в то время как ОУ на основе сложных
олигоэфиров способны кристаллизоваться лишь во времени. Такое
явление связано с большей регулярностью молекулярного строе-
ния полифурита и наличием в расплавах ОУ на основе сложных
олигоэфиров более прочных межмолекулярных водородных связей,
препятствующих образованию трехмерной кристаллической струк-
туры. Подтверждением служит тот факт, что теплоемкость распла-
вов ОУФ возрастает быстрее, чем у ОУ, вероятно, вследствие боль-
шой энергии образования дырок в последних.
Как и для эластомеров на основе олигоэтилепгликольадипинага,
способность к кристаллизации проявляют только эластомеры с
полифуритными блоками с молекулярным весом 2000 (ЭЛФ-3 и
ЛФ-4), хотя, как видно из табл. 29, густота сегки химических сши-
вок ЭЛФ-1 и ЭЛФ-4 (эластомер на основе ПФ-1000) примерно оди-
накова. Из этого следует, что основное влияние па способность поли-
уретановых эластомеров к кристаллизации оказывает густота
сетки не химических, а физических (водородных) связей, число ко-
торых в ЭЛФ-3 и ЭЛФ-4 меньше, чем в ЭЛФ-1 и ЭЛФ-2 из-за кон-
центрации уретановых групп. Низкое значение энтропии плавле-
113
ния ЭЛФ-3 и ЭЛФ-4 объясняется небольшим вкладом конфигура-
ционной энтропии при плавлении.
Сравнительная характеристика
процесса стеклования сшитых полиуретанов
на основе олигоэтиленгликольадипинатов
и олигоокситетраметиленгликолей
Для сшитых эластомеров обоих рядов произведена полуколи-
чественная оценка вклада меж- и внутримолекулярного взаимо-
действия в их кинетические свойства (1101.
В соответствии с [2311, процесс перехода из стеклообразного
состояния в жидкое сопровождается образованием новых дырок,
характеризующихся мольным объемом Vh и мольной избыточной
энергией (по сравнению с исходным «бездырочным» состоянием) е/2.
Энергия ел связана с приращением теплоемкости приближенным
соотношением [88, 2311
хг_____Ёс. . _еА Д
Тс #ТС ехр \ RTC / ’
Ес— мольная внутренняя остаточная теплота парообразования
(энергия когезии) при температуре стеклования Тс. Величину £с
можно определить из эмпирической зависимости [182]
Ес-Е _ 1 7Ч V~Vc
С ус ’
где Е — мольная энергия когезии; V — мольный удельный объем
при температуре кипения; Vc — мольный удельный объем при Тс.
Е и V рассчитывали из значений соответствующих приращений для
функциональных групп [182].
Величины Vc определяли экстраполяцией У20доТс, для чего ис-
пользовали значения коэффициента объемного термического расшире-
нияв жидком состоянии аж, определенного по эмпирическому соотно-
шению Тс*аж= 0,164 [313]. Поскольку макромолекулы исследо-
ванных полимеров представляют собой чередование очень длинных
олигоэфирных и коротких диизоцианатных участков, то в дальней-
ших расчетах /И, М и соответствующие мольные величины для по-
вторяющихся структурных элементов исследованных полиурета-
нов определяли, считая полиуретаны блок-сополимерами (например,
повторяющийся элемент цепи макромолекулы ЭЛ-1 принимали сос-
тоящим из 86% звена ОЭА-ЮОО и 14?6 звена 2,4-ТДИ). Значение Vu
рассчитывали по формуле [343]
V с
vh — •
L-C
Известно, что теоретические обоснования процесса стеклования
в терминах свободного объема основываются на предположении
114
Таблица 33
Термодинамические параметры стеклования
Эластомер 1 1 И, СМ3/М0 1Ь © © 3 Г, кал/мол' кал/моль X © S к £ £ © 5? 5 •St S ^3 Эо
ЭЛ-1 173 190,0 141,0 138,0 19900 17980 21.40 885 6,80 130,0 — 7
ЭЛ-2 178 190,0 141,0 137,0 10900 18200 22,40 845 6,35 151,5 -14
ЭЛ-3 175 189,5 140,5 136,0 10450 17600 23,60 770 5,93 129,5 —27
ЭЛ-4 175 189,5 140,5 135,5 10450 17800 22,20 845 6,40 132,0 —29
ЭЛФ-1 81 101,0 73,0 70,0 4800 8480 8,17 1080 8,90 121,5 —43
ЭЛФ-2 81 101,0 73,0 70,0 4800 8480 7,95 1100 9,06 121,3 -39
ЭЛФ-3 77 99,0 73,0 66,5 4363 8055 8,70 945 7 80 121,0 -77
ЭЛФ-4 77 99,0 73,0 66,5 4365 8065 8,15 975 8,05 121,0 —80
о том, что переход из стеклообразного состояния происходит при
температуре, при которой величина свободного объема становится
большей некоторого критического значения. Очевидно, этот крити-
ческий свободный объем зависит от величины структурных элемен-
тов, принимающих участие в переходе [64]. В свою очередь, про-
цесс образования свободного объема (дырок) в низкомолекулярных
стеклах сопровождается увеличением среднего расстояния между
отдельными молекулами, имеющим статистический характер. При
этом размер образующихся дырок соизмерим с размерами молекул.
При расстекловании полимерных стекол помимо увеличен и я среднего
межмолекулярного расстояния будет изменяться также конформация
цепей врезультате приобретения макромолекулами сегментальной по-
движности. В этом случае размер дырок определяется уже величиной
сегмента цепи. Так как величина сегмента характеризует гибкость
цепи, то, следовательно, меньшими значениями дырочного объема
(т. е. Vh) обладают более гибкие цепи. Таким образом, процесс
образования дырок в полимерах связан с величиной как меж-, так
и внутримолекулярного взаимодействия. Поскольку величина тепло-
емкости определяется суммарным вкладом всех видов молекуляр-
ной подвижности, рассчитанные по Д Ср значения ел (см. табл. 33)
также будут зависеть от величины меж- и внутримолекулярных сил.
Из табл. 33 видно, что величины мольных дырочных объемов для
полиуретанов на основе сложных олигоэфиров (ЭЛ-1—ЭЛ-4) мень-
ше, чем для полиуретанов на основе простых олигоэфиров (ЭЛФ-1 —
ЭЛФ-4). Следовательно, термодинамическая гибкость макромолекул
полиуретана со сложноэфирными блоками выше, чем у макромолекул
полиуретана с простыми эфирными блоками. Аналогичный вывод
сделан в результате исследований разбавленных растворов линей-
115
иых полиуретанов идентичной природы 198]. Значение для
ЭЛ-1 — ЭЛ-4 также выше, чем для ЭЛФ-1 — ЭЛФ-4 . Сле-
довательно, основной вклад в энергию «дырочного» состояния
исследованных полиуретанов вносит внутримолекулярное взаимо-
действие.
Несколько неожиданно, что температуры стеклования сложно-
эфирных полиуретанов выше, чем у соответствующих полиуретанов на
основе простых олигоэфиров (табл. 33). Такое отсутствие корреляции
между термодинамической гибкостью цепи и температурой стекло-
вания еще раз говорит о том, что последняя зависит главным обра-
зом не от термодинамической, а от кинетической гибкости цепей,
которая определяется концентрацией полярных групп, способных к
образованию вторичных связей, а также относительной прочностью
(интенсивностью) последних. Так как концентрация уретановых
групп в соответствующих полиуретанах практически одинакова,
то более высокие температуры стеклования сложноэфирных полиуре-
танов свидетельствует о большей интенсивности межмолекулярного
взаимодействия. Это видно также из сравнения значений ehIVh
(табл. 33). Отношение которое можно назвать «дырочной»
плотностью энергии когезии, является характеристикой объемных
свойств полимера, т. е. чувствительно к молекулярным связям.
Сравнение значений «дырочных» плотностей энергии когезии по-
лиуретанов подтверждает предположение о том, что интенсив-
ность межмолекулярного взаимодействия больше в сложноэфир-
ных полиуретанах.
Полиуретаны на основе
других олигоэфирогликолевых блоков
Исследование влияния химического строения полиэфир уретанов
на их способность к кристаллизации [94] показало, что полиме-
ры, синтезированные на основе эфирогликолей (ди-, три- и гекса-
этиленадипинатуретапы), а также полимеры с боковыми группами
(метоксиметил- и пропиленэтиленадипинатуретаны) являются аморф-
ными. В то же время полимеры на основе гликолей полиметилено-
вого ряда (этилен-, триметилен-, тетраметилен- и пентаметилен-
адипинатуретаны) представляют собой кристаллизующиеся поли-
меры.
Из сопоставления высот максимумов кривых температурной за-
висимости динамического модуля указанных кристаллизующихся
полимеров видно, что более высокий максимум у политетрамети-
ленади пинатуретана (рис. 59). На основании этого делается вывод
о том, что указанный полиуретан обладает максимальной скоро-
стью кристаллизации, что подтверждается дилатометрическими дан-
ными [941.
116
Е,кГ/смг
Рис. 39. Температур-
ный ход динамического
модуля:
/ — полиэтиленгл иколь
адипинатуретан, 2
политри метиленгликолъ -
адипинатуретан, 3 — по-
литетраметилен гликоль
адипинатуретан, 4 — по-
липе нтаметилен гликоль-
адипипатуретан
Изучена зависимость Тс блок-сополимеров как от природы, мо-
лекулярного веса, мольного содержания блоков, так и наличия
в блок-сополимерах кристаллической фазы, образуемой кристалли-
зующимися блоками 125, 26, 160]. В качестве
объекта выбраны уретановые блок-сополимеры
следующей общей формулы:
(—A—OCONHRNHCOO—)i_,( (—В—
—OCONHRNCOO—)„,
где А — блок алифатического сложного
пол иэфи р а: пол иэтиленгл икол ьади пи на га
(ПЭА) или полидиэтиленгликольадипината
(ПДЭА); В — блок сложного эфира на осно-
ве о-, м-, и n-бис (р-гидроксиэтокси)-фепи-
ленадипиповой кислоты
|-ОС2Н4ОСвН4ОС,Н4ОСОС4Н8СО—]„
обозначаемых в дальнейшем о-, м- и н-ПФА.
Исследовались серии блок-сополимеров на
основе следующих пар полиэфиров: ПЭА —
n-ПФА, ПЭА — л-ПФА, ПЭА — о-ПФА,
ПДЭА —jw-ПФА, ПДЭА—о-ПФА. Для каж-
дой серии сополимеров п менялось от 0 до 1.
Значение Тс определяли термомеханичес-
ким методом.
Установлено, что характер зависимости Тс блок-сополимеров
от мольного соотношения блоков не меняется при переходе от од-
ного изомера ПФА к другому и только переход от ПЭА к ПДЭА вы-
зывает изменения для образцов, выдержанных в течение шести
месяцев при комнатной температуре.
Зависимость Тс для сополимеров ПДЭА — лс-ПФА от мольного
соотношения блоков показывает, что 7’с для всех сополимеров хо-
рошо ложатся на прямую, соединяющую значения Тс для гомо-
полиуретанов на основе ПДЭА и л-ПФА (рис. 60, а).
На основе наблюдаемого линейного понижения Тс сополимеров
с повышением содержания блоков ПДЭА, делается вывод о пласти-
фицирующем действии последних на блоки л-ПФА. При этом учи-
тывается, что блоки ПДЭА не способны к кристаллизации, а блоки
.м-ПФА легко образуют кристаллическую фазу.
В связи с этим рассматривается и серия сополимеров ПЭА —
jh-ПФА, в которых обоим блокам свойственно образование кристал-
лической фазы (рис. 60, б), и в этом случае только для аморфизи-
рованных образцов характерно линейное увеличение Тс с ростом
содержания ПФА. Однако для выдержанных во времени образцов
картина меняется. Так, Тс сополимеров с преобладающим количеством
ПЭА (не менее 60%) становится равной Тс полиуретана на осно-
ве ПЭА, тогда как для сополимеров, где содержание ПЭА ниже
117
60%, Тс остается равной 7’с аморфизированных образцов или ие-
сколькио ниже.
Наблюдаемому явлению дается следующее объяснение. Для
сополимеров, где блок А находится в аморфном состоянии, харак-
терно непрерывное уменьшение Тс от В- до А -уретана, поскольку
Рис. 60. Зависимость Гсот состава сополимера серии ПДЭА — лг-ПФА (а) и ПЭА—
л-ПФА (б):
/ — аморфизированные образцы, 2 — выдержанные шесть месяцев при комнатной темпе
ратуре
блоки последнего являются пластифицирующим агентом по отно-
шению к ар ил содержащим блокам В. Поскольку пластификация
должна протекать на молекулярном уровне, то и Тс должно линей-
но меняться с изменением процентного содержания алифатического
блока.
В случае, когда A-блок кристаллизуется, длительное хранение
образцов сополимеров при комнатной температуре обусловливает
возникновение в них кристаллических образований обоих видов бло-
ков, что, в свою очередь, ведет к появлению в образцах микроне-
однородностей.
Микрообласти, где сгруппированы блоки одной природы,
должны проявлять свойства, присущие данному компоненту в ви-
де гомополимера и, в частности, иметь его Тс. Поэтому в сополиме-
рах, где алифатический блок ПЭА (не менее 60 мол. %), Тс сопо-
лимера после длительного хранения становится равной Тс поли-
уретана на основе ПЭА. Повышение Тс сополимеров с содержанием
ПФА-блоков более 40 мол. % объясняется отсутствием в них кри-
сталлических образований блоков ПЭА.
Таким образом, когда в образцах сополимеров не происходит
выделение алифатических блоков А в виде самостоятельной крис-
сталлической фазы, их Тс определяется Тс В-блоков, пластифици-
рованных блоками А.
Такое объяснение особенностей Тс выдержанных образцов со-
118
полимеров серии ПЭА — л-ПФА объясняет и тот факт, что длитель-
ное выдерживание образцов сополимеров серии ПДЭА — лг-ПФА
не приводит к изменению
их Тс по сравнению с ис-
ходными.
Зависимость Тс п-ПФА
и гомополиэфир уретанов на
их основе от молекуляр-
ного веса n-ПФА показы-
вает, что антибатность из-
менения Тс в ряду полиэфи-
ров и гомополиэфирурета-
-50
------------------1-------------1—
О 1000 Мполиэфира
Рис. 61. Зависимость Тс для п-ПФА (/) раз-
личною молекулярного веса и полиэфируре-
танов (2) на их основе.
нов хорошо согласуется с
приведенными выше данны-
ми и еще раз свидетельст-
вует об определяющей роли
концентрации уретановых
групп в изменении Тс по-
лиэфируретанов (рис. 61).
Существенно, что даже
блоков, равном 2000, блок-сополимер ни одной из исследованных
серий не обнаружил двух Тс. Это, очевидно, объясняется более вы-
при молекулярном весе полиэфирных
сокими значениями молекулярных весов механических сегментов
полиэфиров. Как видно из рис. 62,
экспериментальные значения Тс
хорошо согласуются с Тс, вычислен-
ными по формуле Фокса 1218] для
сополимеров
Тс = ТСАТСВ/(<ОД ТСВ + ТСА),
Рис. 62. Зависимость Тс от состава
сополимеров серии ПЭА — п-ПФА
при молекулярном весе блоков 2000.
I — экспериментальные данные для
аморфизированных образцов, 2 — дан-
ные. рассчитанные по формуле Фокса
где ТСА и ТСВ — температуры
стеклования гомополимеров А и В;
о) а и со в—- их весовые доли в соот-
ветствующих сополимерах.
Таким образом, Тс полиэфир-
уретанов характеризуется, с одной
стороны, природой и концентра-
цией диизоцианатной компоненты,
а с другой — природой и молеку-
лярным весом гликолевых блоков,
определяющих склонность к крис-
таллизации и концентрацию полярных групп, обусловливающих
возникновение между макроцепями полиуретана физических связей
различной степени интенсивности.
Показано [261, что отдельные блоки сополимера ПЭА — п-ПФА
способны образовывать самостоятельные кристаллические фазы, и
что эта способность падает в ряду сополимеров серий 2000, 1000 и
119
600. Для сополимеров серии 2000 постоянство Тпл ПФА-блоков со-
храняется вплоть до содержания последних порядка 50 вес.%.
В области средних составов наблюдается плавление двух типов
кристаллических образований, соответствующих кристаллизации
каждого из видов блоков. У сополимеров серий 1000 и 600 происхо-
дит изменение напоминающее диаграммы эвтектического типа,
и только сополимеры с преобладающим содержанием ПЭА имеют
Тпл, равную 7'„л гомополимера. Уменьшение величины ПФА-блока
сопровождается возникновением менее совершенных кристалли-
ческих структур и сужением области независимости Тс от состава
сополимера. Таким образом, уменьшение длины составляющих
блок-сополимеров полиэфирных блоков вызывает появление свойств
статистического сополимера.
Исследовались некоторые мономерные и олигомерные гликоли,
которые могут быть использованы при синтезе полиуретанов 1411.
В частности, на основании термограммы нагревания сополимеров тег-
рагидрофурана и окиси пропилена с различным содержанием послед-
нею найдено, что способность к кристаллизации понижается с ростом
содержания окиси пропилена. Однако даже при 25% -номсодержании
окиси пропилена сополимер еще способен кристаллизоваться.
В то же время для сшитых полиуретанов на основе смеси 2,4- и
2,6-ТДИ и сополимера тетрагидрофурана с различным содержа-
нием окиси пропилена, но с одной и той же степенью химической
сшивки, установлено, что полиуретан теряет способность кристал-
лизоваться при более низких содержаниях окиси пропилена, т. е.
при 12% [151. Это говорит о том, что диизоцианатная составляю-
щая препятствует протеканию процесса кристаллизации в полиу-
ретановых эластомерах.
Исследование полиуретановых эластомеров на основе толуилен-
диизоцианата с различным соотношением 2,4- и 2,6-ТДИ и сопо-
лимера с 10% окиси пропилена показало, что переход от 2,4- к 2,6-
ТДИ не меняет строения кристаллической решетки полиуретана,
но увеличивает скорость кристаллизации и конечную степень кри-
сталличности полиуретанового эластомера
Из изложенного вытекает, что способность к кристаллизации
проявляют только те полиуретановые эластомеры, олигомерные сос-
тавляющие которых также кристаллизуются. При этом молекулярный
вес олигомерных блоков должен превышать некоторое критическое
значение. Кристаллическая структура полиуретановых эластомеров
образуется вследствие кристаллизации олигоэфирных блоков, а дии-
зоцианатные звенья играют только роль дефективных участков в
структуре.
Если строение кристаллической решетки зависит только от при-
роды и строения олигомерного блока, го склонность к кристаллиза-
ции и степень кристалличности определяются строением как диизо-
цианата, так и олигомерного блока. Последние параметры связаны
также со степенью химической сшивки эластомера.
Глава V. СТРУКТУРА СЕТКИ АМОРФНЫХ ПОЛИУРЕТАНОВ
ХАРАКТЕРИСТИКА СЕТОК ПРОСТРАНСТВЕННЫХ ПОЛИУРЕТАНОВ
Получение полиуретанов с использованием полифункциональ-
ных соединений приводит к образованию полимеров с пространст-
венной структурой, или так называемых трехмерных полимеров.
Простейший критерий образования полимера с трехмерной сет-
кой — нерастворимость его в ряде растворителей. Точки соедине-
ния сегментов цепей называются узлами или точками разветвления.
Особенности химического строения полиуретанов определяют
возможность существования в них самых различных по строению
химических поперечных связей. Наряду с биуретовой и аллофана-
тной поперечными связями могут образоваться сшивающие узлы,
представляющие остаток полиола (I, II), или разветвленный изо-
цианат (III, IV)
СН2
—Н2С—CII—СН.— —Н СНГ'~
I I
сн2 сн2
I и
III
IV
Таким образом, в полиуретанах образуется целый набор различных
типов химических связей, обладающих различной прочностью.
Тобольский и сотрудники [188, 3301 провели исследования ре-
лаксации напряжений в сшитых полиуретановых эластомерах, что
позволило получить некоторые сведения о прочности связей в сетке.
Они установили, что при повышенных температурах в сшитых поли-
уретанах наблюдается так называемое химическое течение, свиде-
тельствующее о разрыве химических связей.
121
Изучена химическая релаксация полиуретановых и полиэфир-
ных резин, содержащих связи различных типов, и рассчитаны
величины времен релаксации (табл. 34, рис 63).
Таблица 34
Времена химической релаксации полиуретановой
и полиэфирной резин при 120"С
Резина Тип связи Время релакса- ции, ч
1 Эфирная, уретановая, моче- винная, биуретовая 2,1
2 Сложноэфирная, уретановая, мочевинная, биуретовая 3,0
3 Сложноэфирная 500
4 Сложноэфирная, уретановая 14,0
5 » » 9,7
6 Сложноэфирная, уретановая, мочевинная, биуретовая 1,6
Как видно из приведенных данных, наиболее слабыми связями
в полиуретанах являются мочевинные и биуретовые.
Дальнейшие исследования в этом направлении показали, что
релаксацию напряжений в сшитых полиуретанах можно предста-
Рис. 63. Химическая релаксация напряжения при
120° С различных полиуретановых резин и поли-
эфирного каучука
(цифры соответствуют порядковому номеру резин в
табл 34).
вить суммой двух процессов [335], энергии активации которых
равны соответственно 37 ± 3 и 42 ± 2 ккал/молъ. Однако резуль-
таты, полученные при изучении релаксации напряжений, не позво-
122
лили Тобольскому дать химическую трактовку обнаруженным
процессам. Можно говорить только о различной прочности хими-
ческих связей, составляющих сетку полиуретановых эластомеров.
Наличие в полиуретанах разнообразных химических связей и
большого числа полярных групп приводит к образованию в них
наряду с химическими прочных физических связей (водородных
и вандерваальсовых), обусловленных взаимодействием полярных
групп друг с другом.
Исследование структуры сетки полиуретанов чрезвычайно
сложная задача, необходимая для выяснения количественного со-
отношения между структурой и физическими свойствами.
Наиболее детально изучили структуры сетчатых полиуретанов
Танака и Йокояма [325, 326, 347, 348] на системе полиол — ди-
изоцианат — этаноламин. Схема образования сетчатого продукта
в данном случае, ^следующая:
НО—011 4- OCN-R— NCO
полиол диизоцианат
I
OCN—OCONH—NCO
форполимер
I HOCH2CH,NH2
Ф этаноламин
—NH-CO-NHCH2CH2OCONH'—
n£o
ф
™NCONH—
СО
NH
NH-CO—NCONH—
Молярное соотношение компонентов представлено таким образом*
мольное содержание NCO-групп в диизоцианате
мольное содержание ОН-групп в полиоле
_______________________число молей этанола мина_
“ мольное содержание NCO-групп в аддукте ’
Прежде чем подойти к описанию трехмерной сетки, авторы 13481
рассматривают получение форполимера, принимая во внимание
уравнения, описывающие реакции поликонденсации. Средняя сте-
пень полимеризации
X - 1
Х'1 2(1 - р) + (К - 1) ’
121
где р — степень завершенности реакции. При р = 1
Лп к — 1 ’
Средняя степень полимеризации хп показывает сколько молекхл
полиола и днизоциапата образуют одну молекулу форполимера.
Распределение по степеням полимеризации описывается уравнением
где х—степень полимеризации. Доля молекул диизоцианата, ко-
торая остается в аддукте после завершения реакции Р19 а доля
молекул, имеющих в своем составе полиол, 1 — pt = ]/{(. Число
молекул форполимера при р = 1
V-VO(K-1),
где v0 — число молекул полиола. Тогда число молекул форполи-
мера, имеющих в своем составе полиол,
V- VQ W ~
М
Предполагается, что выведенные уравнения справедливы и при
распределен и" молекул по длинам, так как принимается во внима-
ние числи молекул.
Пос \ введения в форполимер этаноламина молекулы форполи-
мера растут за счет присоединения к ним амино- и гидроксильных
групп, которые образуют мочевинные и уретановые при вза-
имодействии с избытком изоцианатных групп. Следовательно, эта-
полампп можно рассматривать как трехфункциональный агент.
В ]ервый момент реакции справедливо соотношение [ОН ]0
- IJI = начальной концентрации этаноламина, где [U] — концен-
трация мочевинных групп. Константы скорости реакций образова-
ния уретановых и мочевинных групп, протекающих одновременно,
обозначим и К2:
—NCO Н------ОН -> мочевинная,
/г8
—NCO4-------NHCONH--------- биуретовая.
Tci да
|ОН| = [LT|
|ОН|о [U]o-
Так как точкой разветвления, или узлом сетки, является молекула
этаноламина, все реакционные группы которой реагируют, то чис-
ло узлов
N = (число молекул этаноламина) роири,
124
где рон и ри — вероятность реакций ОН- или мочевинных групп
соответственно. Они могут быть вычислены, если известны значе-
ния [ОН 1 и KJKz- Число цепей, концы которых сходятся в узел, v( --
= 3N/2. При таком рассмотрении процесса образования сетки труд-
но определить и /(2. Поэтому для простоты принимают, что при-
соединение мочевинных групп начинается после полного исчер-
пания гидроксильных.
Разберем формирование сетки для трех различных соотношений
компонентов при степени завершенности реакции, равной единице.
1. М — 1/3. Все мочевинные группы реагируют с NCO-rpynna-
ми. В системе не остается свободных изоцианатных групп.
о — Мочевинные группы °-NCO-группы
—*- — Продолжение сетки
Рис. 64. Структура сетки для трех различных соотношений компонентов при сте
пени завершенности реакции, равной единице.
2. М < 1/3. Все мочевинные группы реагируют с изоцианатны-
ми, но последние остаются в избытке.
3. М > 1/3. Часть мочевинных групп реагирует с NCO-груп-
пами, свободных NCO-групп не остается.
Структура сетки для рассмотренных случаев схематически пред-
ставлена на рис. 64.
Цепи, конструирующие полиуретановую сетку, ответвляются от
молекул аддукта. При М = 1/3 число внутренних цепей, оба кон-
ца которых связаны с точками разветвления, равно числу молекул
а;щукта
v( = v = v0 (К — 1).
Последнее уравнение учитывает количество остающихся молекул
диизоцианата. Число цепей, в которые входят молекулы полиола
как составляющие единицы, выражается уравнением
Здесь цепи не имеют свободных концов и сетку можно считать
совершенной.
При М < 1/3 имеется избыток NCO-групп, поэтому образуется
меньшее число внутренних цепей по сравнению с предыдущим слу-
чаем. Имеются цепи со свободными концами, число которых равно
избыточному количеству NCO-rpynn:
тогда Vi = 2v — 2v3Al,
Vi ^v — Vi = vQ(K— 1) (6Л1 — 1),
125
v _ v„(K-l)(6M-l)
‘ ~ К
’ Если М > 1/3, число концов молекул, которые образуются при
реакции удлинения аддуктом,
2v —2- 2vM = 2v(l — 2Л4),
а число молекул — v (1—2Л4). На этой стадии число мочевинных
групп равно числу молекул этаноламина, т. е. 2vM.
Когда свободные концы цепей (NCO-группы) присоединяются
к мочевинным группам, находящимся в цепи, увеличение количе-
ства цепей равно числу присоединений. Тогда число внутренних
цепей
v, = v (1 — 2/И) + 2v (1 - 2М) - 3v0 (К — 1) (1 — 2(И).
Сетка в данном случае будет совершенной.
ВеличинаV; вычисляется несколько иначе. Доля молекул, кото-
рые не присоединяются к этаноламину, -- 1—2Л4. Количество
молекул диизоцианата, находящихся в аддукте,
V — v' = v0(K— 1) р--.
Число молекул диизоцианата в форполимере, не взаимодействую-
щих с этаноламином, равно
v0(K - 1) (1 - -L) (1 - 2А4),
откуда
vi = 3v0(K-l)(l-2/4)-v0(K-l)p--bj(l-2М) =
= v0(K-l) (2 + -Lj (1 - 2М).
Авторы считают, что величина V/, т. е. число внутренних цепей
или отрезков цепей между точками разветвления, недостаточно точ-
но характеризует истинную картину образования сетки, так как
не все точки разветвления являются узлами трехмерной сетки. Иног-
да на конце цепи, отходящей от точки разветвления, находится
свободная N’CO-группа, но точка разветвления не представляет
собой узел сетки. Поэтому авторы вводят понятие «число эластич-
ных элементов» (vj или число эффсктивносшитых цепей но Флори
[211], которое наиболее полно характеризует трехмерную прост-
ранственную сетку
Величина ve для случая М > 1/3 выражается уравнением
ve = 2 [2v — (v j- 2vM — 1)] - 2v(l — 2M) - 2v0 (/< — 1) (1 — 2M).
При М< ИЗ
v„ - 2 [6vM — (v + 2vM — 1)| -- 2v (4М — 1) = 2v0 (/< — 1) (4Л4 — 1).
126
Таким образом, Танака и Йокояма вывели уравнения, по которым
можно рассчитать концентрации сшитых цепей в трехмерных поли-
уретанах для различных мольных соотношений сшивающего аген-
та и изоцианатных групп в аддукте Они считают, что предлагае-
мый способ расчета ve упрощен и не учитывает некоторых факто-
ров, например перепутанности цепей.
Для проверки приведенных уравнений проведено эксперимен-
тальное определение концентрации эффективносшитых цепей в по-
лиуретанах на основе полио-
лов, толуилендиизоцианата и
этаноламина. В качестве полио-
лов использованы полиэтилен-
гликоль, полипропиленгликоль,
полиэтиленадипинат, полиэти-
ленсебацинат и др. Экспери-
ментальное определение ve прово-
дилось по уравнению кинети-
ческой теории высокоэластич-
ности
т = и,
Рис 65. Зависимость aS от у для поли-
уретана на основе полиэтилен гл и кол я
(М = 1540)*
/ - К = 2.45. 2 — К ~ 1.98. 5 — К —
= 1 51; 4 - К = 1,21.
где т—напряжение, г/см2\^~ —
концентрация эффективносши-
тых цепей в единице объема;
а—степень растяжения обра-
зца. Данное уравнение можно
переписать в форме
(V.1)
где Е — модуль высокоэластичности.
Уравнение (V, 1) выведено только для равновесных систем при сле-
дующих допущениях [254,3311: 1)цепи,образующие сетку,имеютоди-
наковую полную длину; 2) распределение расстояний между концами
цепей в недеформированном состоянии представлено формулой Гаусса;
3) объем при деформации остается постоянным; 4) изменения в проек-
циях расстояния между концами каждой цепи при деформации по-
добны изменениям размеров деформированного образца каучука.
Хотя данные условия в основном соблюдаются только для нату-
рального каучука, для сравнительной характеристики степени сшив-
ки трехмерных сеток уравнения (V, 1) применяют ко многим другим
системам, в том числе и к полиуретанам [3261. Известно, что если
зависимость aS от у (а -- -j-, где S — напряжение на еди-
ницу площади при деформации) дает прямую линию, то это явля-
ется подтверждением равновесности состояния. Наклон прямой дол-
жен быть равен единице.
12"
Зависимость a S от у для полиуретана на основе полиэтилен-
гликоля молекулярного при разных значениях К 1961 представлена
на рис. 65. Наклон log у от log yS составляет примерно единицу
(рис. 66). Следовательно, условие равновесия соблюдается в дан-
Рис. 66. Зависимость log aS от
logy полиуретана на основе по-
лиэтилен гл и кол я
(обозначения те же, что и на
рис. 65).
Рис. 67. Сравнение теоретических и экспе-
ve
риментальных величин— полиуретанов на
основе 2,2-ТДИ и различных гликолей при
М = 1/3:
1 — ПОПГ- 545; 2 — ПОПГ 1950, 3 — ПЭГ 1540,
4 — ПЭГ 4000, 5 — ПЭГ 4 000 (М = 0,3) (обозна-
чение см в абл.36)
ном случае, что дает возможность использовать результаты опре-
деления модуля для экспериментального нахождения эффективной
плотности сшивки.
На рис. 67 представлено соотношение между вычисленным по
уравнениям Танака и Йокояма, и ^-, наблюдаемым для полиуре-
танов на основе простых полиэфиров. Почти во всех случаях-^- вы-
численное больше наблюдаемого, причем расхождение наибольшее
для полиуретанов на основе полипропиленглпколя. Для полиуре-
тана на основе полиэтиленгликоля с молекулярным весом 4000 при
М-1/3 значения наблюдаемое и вычисленное, почти одинаковы.
Несколько больше расхождение в случае полиуретанов на основе
полиэфиров меньшего молекулярного веса. Схождение эксперимента-
льных прямых в начале координат авторы считают доказательством
128
правильности предлагаемого метода расчета При М < 1 3
д! _ концентрация этаноламина
число NCO-групп в аддукте
наблюдаемое становится больше вычисленного Танака связыва-
ет это с тем, что свободные концевые изоцианатные группы, при-
соединяются к мочевинным и уретановым и увеличивают экспери-
ментальное ~ по сравнению с вычисленным.
СООТНОШЕНИЕ МЕЖДУ ХИМИЧЕСКИМИ И ФИЗИЧЕСКИМИ УЗЛАМИ
В СЕТКЕ ПОЛИУРЕТАНОВ
Если при выводе уравнений для расчета эффективной плот-
ности сетки полиуретанов ие учитывается возможность образова-
ния физических связей, которые существуют в такой специфичной
сетке, нельзя искать совпадения расчетной и вычислительной ве-
личины Совпадение их может быть только случайным. Как пра-
вило, - экспериментальное будет больше теоретического в силу
приведенных выше причин. Экспериментальные результаты, полу-
ченью некоторыми исследователями, подтверждают это [317, 1981.
Теоретический расчет — чаще всего проводят на основании
предположения о том, что при использовании трехфункционального
сшивающего агента каждый моль последнего дает 1,5 моль сшитых
цепей, т. е.
% = 3/2СГ, (V.2)
где С г — концентрация трехфункционального сшивающего агента,
моль [133, 1981. Параллельно экспериментально определяютпо
модулю высокоэластичности или по методу Флори — Ренера [211].
Проведенное сравнение величин определенных по модулю
высокоэластичности и рассчитанных по уравнению (V, 1) для
полиуретанов на основе полиоксипропиленгликоля и толуилен-
диизоцианата, показало, что (™-)э может быть близко по ве-
личине к теоретическому, но может значительно превосходить по-
следнее. Все это зависит от эффективности отверждения исследо-
ванных полиуретанов, которая зависит от природы применяемых
катализаторов и условий получения. Иными словами, чем меньше
эффективность отверждения полиуретанов, т. е. чем дефектнее
сетка, тем более эксперимен алыюе отличалось от вычислен-
129
ного экспериментальное теоретического!. При повыше-
нии эффективности отверждения за счет изменения условий полу-
чения полимеров (введение катализатора и пр.) эксперименталь-
ное значение —г приближалось к теоретическому или превосходило
его в пять-шесть раз. Таким образом, сравнение эффективной плот-
ности сшивки, найденной экспериментально и вычисленной тео-
ретически, показывает, что экспериментальное, как правило,
больше теоретического. Расхождение можно объяснить только тем,
что в полиуретановой сетке наряду с химическими поперечными
связями имеется большое число поперечных физических связей,
приводящих к увеличению значения эффективной плотности сшив-
ки, определяемого экспериментально
Часто густоту сеток вычисляют по методу Флори — Ренера [2111,
который удобен и прост в экспериментальном отношении. В дан-
ном случае используется уравнение вида [1981
1 ' ! - \
— [In (1 — V2) V2 Н- ~ t’l (-?-1 ( V-2 — Ф'/ ,
\ \ i J
где — объемная доля полимера в набухшем геле; ЧГ1 — пара-
метр взаимодействия полимер — растворитель, — моляр-
ный объем растворителя; f — функциональность сшивающего
агента. Уравнение выведено на основании термодинамического
рассмотрения набухания слабовулкапизованного натурального
каучука в термодинамически хороших растворителях. Границы
применения этого уравнения для полимеров разной степени хими-
ческой сшивки при использовании различных растворителей не
установлены. Флори экспериментально показал, что будучи от-
носительным, данный способ удобнее для определения густоты сетки
путем изучения набухания одного и того же полимера в одном
и том же растворителе Другие исследователи применяют его
для определения степени сшивки полимеров различной природы
[191, 258] и лаже для сильносшитых эпоксидных смол [258]. Оце-
нить соответствие экспериментально найденных значений ис-
тинной плотности сшивки трудно, так как в настоящее время нет
еще способа получения сетчатых полимеров с точно заданной стру-
ктурой.
Метод Флори основан на исследовании набухания полимеров,
поэтому интересно проследить, как влияет качество растворителя
па определение эффективной плотности сетки. В таких полиме-
рах, как натуральный каучук, где кроме вводимых при вулкани-
зации химических поперечных связей существуют переплетения
цепей, степень сшивки не должна сильно отличаться при измене-
нии термодинамического качества растворителя, тем более, что
130
в уравнение Флори — Ренера введен параметр учитывающий
взаимодействие полимера с изучаемым растворителем К сожа-
лению, исследования в этом направлении не проводились. Эффсктив
ную плотность сшивки обычно определяют в одном растворителе,
который выбирают чисто эмпирически. Иногда плотносп, сшивки,
определенная при набухании в двух разных растворителях, оказы-
валась близкой [19].
Влияние растворителя на результаты определения плотности
сшивки исследовано только в одной работе 13121 для наполненного на-
турального каучука. В качестве раст! орнтелей служили спирты,
кетоны, ароматические углеводороды. Оказалось, что внутри раст-
ворителей одного класса плотность сшивки увеличивалась с умень-
шением полярности растворитетя. В то же время не наблюдалось
корреляции между плотностями сшивок и физическими характе-
ристиками растворителей различных классов.
В данном случае, очевидно, необходимо учитывать наличие
большого числа физических связей между каучуком и наполните-
лем. Диссоциация связей под действием растворителей происходит
по-разному в зависимости от природы растворителей. Поэюму плот-
ность сшивки, определенная в разных растворителях, оказывает-
ся неодинаковой.
Для определения плотности сшивки в полиуретанах по методу
Флори — Ренера нужно принимать во внимание специфику послед-
них. Наличие в цепях полиуретанов участков различной полярнос-
ти и способность полиуретанов к образованию водородных связей
приводит к возникновению в полиуретанах наряду с химической
прочной физической сетки, на которую растворители в зависимости
от природы будут действовать по-разному. Некоторые раствори-
тели способны давать комплексы при взаимодействии с уретановой
группой [3, 5]. Прочность таких комплексов различна. Наиболее
прочные комплексы образуются при набухании полиуретанов в
диоксане и тетрагидрофуране. Возможно, в данном случае при
набухании получается новая сетчатая структура, отличная от
исходной. В растворителях типа диоксана при повышении темпера-
туры происходит необратимое набухание, т. е. очевидно, происхо-
дит разрыв некоторых сравнительно слабых химических связей.
Таким образом, определение плотности сшивки полиуретанов
по Флори — Ренеру с помощью растворителей, вступающих в спе-
цифическое взаимодействие с функциональными группами поли-
уретанов, не имеет смысла. Невозможность использования таких
растворителей связана и с тем, что нельзя определить существую-
щими методами истинную величину взаимодействия полимер —
растворитель (lF) из-за неприменимости теории к необратимым про-
цессам.
Однако можно получить некоторое представление о густоте сет-
ки полиуретанов по методу Флори — Ренера с помощью раствори-
телей, не взаимодействующих с функциональными группами
131
полихретанов. Спектральные исследования показали, [5) что для
полиуретанов таким растворителем будет бензол; вероятно, и то-
луол пригоден для этой цели.
При исследовании набухания полиуретана на основе олигоди-
этиленгликольадипината в толуоле при разных температурах мы
обнаружили, что набухание в данном случае обратимо (рис. 68).
Такая же картина наблюдалась и для бензола [51. В ацетоне и цик-
ло! сксане, которые способны вступать в специфическое взаимодей-
vz
0,6-
0,4-
0,2-
Ацетон
°------—
о-«-----------
Циклогексанон
10 20 30 Т^С
Рис. 68. Зависимость V2 от темпе-
ратуры для полиуретана на основе
ол и годи этил е н ги кол ьа ди п и ната.
ствие с полиуретанами, набуха-
ние необратимо.
Следовательно, косвенным кри-
терием отсутствия взаимодействия
является обратимость набухания
при изменении температуры. Но
даже при использовании таких
растворителей трудно предполо-
жить, что они не действуют на
межмолекулярные связи в полиуре-
танах. Поэтому метод Флори — Ре-
нера даег только относительную
информацию о густоте сетки, но
тем не менее он очень удобен для
определения изменения густоты
сетки полиуретана при воздейст-
вии таких факторов, как темпера-
тура, влага воздуха, среда.
Таким образом, существующие методы оценки густоты сетки
дают лишь приблизительные результаты при применении их к вы-
числению степени сшивки полиуретанов. Степень приближения
к истинной величине густоты сетки можно оценить лишь при на-
личии образцов полиуретанов с точно известной химической сте-
пенью сшивки, что представляет трудную задачу, учитывая слож-
ность процессов, протекающих при синтезе полиуретанов. Однако
расчет густоты сетки на основании стехиометрического состава и
сравнение полученных результатов с экспериментальными могут
дать полезную информацию о природе трехмерной сетки полиуре-
танов.
Мы провели [1151 сравнение результатов определения плотнос-
ти сшивки полиуретановых эластомеров с помощью уравнений (V,
1 и V, 2), а также по методу Клаффа — Гл един га [1851 (табл. 35),
в основе которого лежит уравнение
_ Р
V 3AqRT~~ Мс '
где Ло — площадь поперечного сечения образца; R — газовая по-
стоянная; — высота недеформированного ненабухшего образ-
ца; S —наклон прямой, представляющей собой зависимость вели-
чины деформации от нагрузки.
132
Эластомеры получены на основе олигодиэтиленгликольадипи-
ната (мол. вес 2000) и ТДЦ. Сшивающим агентом служила смесь
ди- и триэтаноламина в мольном соотношении 1 : 1,5.
Как видно из табл. 35, значения вычисленные на основании
стехиометрических соотношений, самые большие, а из равновес-
ного модуля — самые малые. Превалирование величин эффектив-
ной плотности сшивки, определенной экспериментально, над плот-
ностью, найденной теоретически, объясняется существованием по-
мимо химических вторичных физических связей, определяемых
взаимодействием цепей друг с другом.
Таблица 35
v
Сравнение значений — и ЛС, определенных различными
v с
методами
ТДЦ. МОЛЬ Сшивающий ai снт. моль Стехиометри- ческий метод Сжатие на- бухших образ цов Растяжение ненабухших образцов
- 10* V Мс ve — 10* V Мс ve - .10* V Мс
1,10 0,035 0,35 33800 0,30 33000 3,2 3700
1,15 0,070 0,50 24000 0,49 25000 3,4 3300
2,20 0,100 0,70 20000 0,51 24000 4,7 2600
На первый взгляд кажется удивительным несоответствие эффек-
тивной плотности сшивки, определенной из данных по растяжению
ненабухших образцов и сжатию набухших. Оба эти метода осно-
ваны на определении величины равновесного модуля высокоэлас-
тичности и обычно дают одинаковые результаты [185]. Однако чаще
всего это относится к случаю, когда физические связи образованы
захлестами и переплетениями цепей, а не межмолемулярными свя-
зями. Согласно [3121 расхождение в величинах эффективной плот-
ности сшивки, определенной различными методами, объясняется
присутствием растворителя, который способен разрушить вторич-
ные физические связи. Можно полагать, что в нашем случае при
проведении испытаний на сжатие набухших образцов растворитель
ослабля т физические связи, что является предпосылкой для их
разрушения при деформации. При этом нужно учитывать специ-
фику строения полиуретановой цепи, в которой чередуются участ-
ки различной полярности. Применяемые растворители по-разному
взаимодействуют с различными участками полимерных цепей, воз-
действуя на разные физические связи.
На основании данных по эффективной плотности сшивки, опре-
деленной теоретически и экспериментально, можно приблизительно
оценить вклад тех и других связей в общую густоту сетки (табл. 36).
Величина эффективной плотности сшивки в полиуретанах данной
133
природы определяется главным образом физическими вторичными
связями, образующимися при взаимодействии цепей друг с другом.
Известно, что критерием такого взаимодействия является плот-
ность энергии когезии, которую мы определили по методу Джи
Таблиц а 36
Структурные составляющие эффективной плотности сшивки
для диэтиленгликольадипинатуретана
Образец vL, Be тмина у- 10’ Сшивка, %
химичес- кая фи шчес кая общая химииес кая физичес- кая
1 0,33 2,85 3,2 11 89
2 0,50 2,90 3,4 15 85
3 0,70 4,00 4,7 16 84
[28], основанному на исследовании набухания полимеров в ряду
растворителей, различающихся по параметру растворимости в широ-
ком интервале. На рис. 69 представлена зависимость
от 6, где — величина набухания (г/г) в лучшем из исследо-
ванных растворителей, Q — величина набухания для остальных
растворителей; 6 — параметр растворимости растворителя. Вели-
чина параметра растворимости для полиуретанов на основе диэти-
леигликольадипината составляет примерно 9,8. Этой величине
соответствует плотность энергии когезии около 96 кал!см\ Такая
высокая плотность говорит о сильном взаимодействии отрезков
полимерных цепей в сетке, связанным с наличием разнообразных
физических связей.
Для приблизительной оценки доли физических связей в общей
густоте сетки [335] использованы результаты зависимости модуля
от температуры. Предполагалось, что с увеличением температуры
физические связи диссоциируют, химические остаются неизменными
в некотором интервале температур. Модуль упругости вычисляли
по уравнению
g = ! v Н- Т-; ~ ARTe~E™l,iT + (RT,
где g— модуль упругости; -------концентрация вторичных свя-
зей; ---------------------------концентрация первичных, или химических, связей; ЕаКт —
энергия активации связи. Установлено, что в полиуретанах на
основе простых и сложных полиэфиров содержится в зависимости
от степени химической сшивки эластомеров от 20 до 57% вторичных
131
образца 1
(обозначение см в табл J9)
связей. Уменьшение химической сшивки ведет к \величению вкла-
да вторичных связей в общ^ю ijctotj сетки Это естественно, так
как чем больше расстояния це-
пей между узлами, тем большей
гибкостью они обладают, в ре-
зультате чего возможно боль-
шее число контактов отрезков
цепей друг с другом, ведущих
к образованию прочных физи-
ческих связей.
Следует ожидать изменения
вклада физических связей в об-
щую густоту сетки и при изме-
нении природы полиуретановой
цепи, например олигоэфирного
блока. Мы это показали при оп-
ределении густоты сетки поли-
уретановых эластомеров на ос-
нове олигоизопрена и олигобу-
тадиена [127] (табл. 37).
Величины эффективной плот-
ности сшивки, определенные экс-
периментально, также, как в в
предыдущем случае, превышали
теоретические значения, хотя не
в такой степени, как для днэти-
ленгликольадипинатуретана. На образцах 4 и 5 можно проследить
влияние природы олигодиена на эффективную плотность сшивки
Таблица 37
Т|
Сравнение и —— для полиуретанов на основе олигодиенов
v
1 Образец Олигомер Олигомер. ТДЦ ТЭА Теоретически Эксиеримсн тально
мс Мс о
1 Олигоизопрен 1 1,2:0,100 14800 0,65 10300 0,94
2 То же 1 1,3.0,133 10250 0,95 5400 1.79
3 » » 1 : 1,45 : 0,234 6000 1,65 3600 2,71
4 » » 1 . 1,68 0,400 3600 2,71 3200 3,05
5 Олигобутадиен 1 : 1,68:0,400 3600 2,77 2400 4,03
полученных эластомеров. Анализ данных табл. 37 показывает, что
полиуретан на основе олигобутадиена имеет большую эффектив-
ную плотность сшивки, чем на основе олигоизопрена при одном и
135
том же количестве введенного сшивающего агента. Очевидно,
наличие в цепи олигоизопрена метильной группы чисто геометричес-
ки препятствует образованию такого же количества вторичных
связей, как в потиуретане на основе олигобутадисиа.
Таблица 38
Структурные составляющие эффективной плотности сшивки
для полиуретанов на основе олигодиенэв
Образец vc Величина ♦ 1М V Сшивка, %
химиче- ская физиче- ская общая химиче- ская физиче- ская
1 0,65 0,29 0,94 69 31
2 0,95 0,84 1,79 53 47
3 1,65 1,06 2,71 61 39
4 2,71 0,34 3,05 89 11
5 2,77 1,26 4,03 67 31
Физические вторичные связи вносят значительный вклад в об-
щую густоту сетки, но основная роль в данном случае принадле-
жит химическим связям (табл. 38). Это отличает данные эластоме-
ры от эластомеров на основе сложных
олигоэфиров, в которых основной
вклад в общую густоту сетки вно-
сят вторичные связи. Очевидно,
наличие в сложноэфирном блоке кар-
бонильной группы приводит к возник-
новению в полиуретанах на основе
сложных олигоэфиров большого числа
физических связей вследствие взаимо-
действия полярной С=О-группы с
другими функциональными группами
полимерных цепей.
Из рис. 70 видно, что величина от-
клопений --- экспериментальных от
теоретических зависит от количества
вводимого сшивающего агента. При
наименьшем и наибольшем содержа-
ниях триэтаноламина эксперименталь-
ное значение приближается к те-
оретическому. При содержании три-
0,234 моль наблюдаются наибольшие
12 3 4
10 4(теорет) моль/см
Рис. 70 Сравнение теоретичес-
ких (7) и экспериментальных (2)
величин —- для полиуретана на
v
основе олшоизопрена.
этаноламина от 0,133 до
отклонения. Очевидно, при таком содержании сшивающего аген-
та создаются наиболее благоприятные условия для образования
136
физических связей в трехмерной сетке такого типа. Но все же
в полиуретановых эластомерах на основе сложных олигоэфиров
роль физических связей гораздо больше, чем в полиуретанах
на основе олигодиенов. Возможно, этим обусловлены значе-
ния других параметров, например для полиуретанов на осно-
ве олигодиенов плотность энергии когезии составляет 81 кал/см*.
Величина плотности энергии когезии для данных образцов меньше,
чем для полиуретанов на основе сложных полиэфиров, но больше,
чем, например, для полибутадиена и натурального каучука
(66 кал/cjH3). Разрывные прочности колебались в пределах 10—
20 кг!см\ удлинение при разрыве — около 30%, в то время как для
Таблица 39
Структурные составляющие эффективной плотности сетки
полиуретанакрилатов
Мпопг ve Величина V . 10* Сшивка, %
химиче- ская физиче- ская общая химичс екая физиче- ская
400 20,4 234,8 255,2 8 92
400 23,1 331 354 7 93
400 41,5 419,5 461 9 91
1000 14,6 37,3 51,9 28 72
1000 20,7 42,2 62,9 33 67
1000 25,7 54,8 80,5 32 68
2000 11,1 5,8 16,9 65 35
2000 16,1 7,0 23,1 70 30
2000 20 15 35 57 43
Примечание. AfПОПГ — молекулярный вес полиоксипро-
пиленгликоля.
полиуретанов на сложных олигоэфирах оно на порядок выше (до
700%). Возможно, ухудшение характеристик полиуретанов на
основе олигодиенов связано с меньшим количеством физических
связей в этих эластомерах по сравнению с полидиэгиленгликоль-
адипинатуретанами. Довольно быстрое перераспределение физичес-
ких связей в процессе деформации способствует, с одной стороны,
большей прочности, а с другой — увеличению максимальной дли-
ны при разрыве полиуретанов на сложных олигоэфирах.
В работе 1128 ] исследована структура сетки нового типа полиуре-
танов—полиуретанакрилатов, получаемых на основе олигоэфиров,
диизоцианатов и монометакрилового эфира этиленгликоля. Резуль-
таты расчетов вклада химических и физических связей в общую
густоту сетки полиуретанов на основе полиоксипропиленгликоля
различного молекулярного веса представлены в табл. 39. С увели-
чением молекулярного веса исходного олигоэфира вклад физичес-
137
ких связей в общую густоту сетки уменьшается» Это связано,
очевидно, с уменьшением концентрации полярных уретановых групп
в единице объема и, следовательно, с уменьшением количества
физических связей
Некоторые исследователи, отмечая большую роль вторичных
физических связей в полиуретанах, считают, что основное значе-
ние в этих полимерах принадлежит водородным связям 1330, 3351.
Последние работы Тобольского и сотрудников [189, 1901 по вязко-
у пругим свойствам полиуретанов различной природы и сополиме-
ров бутадиена со стиролом показали, что роль водородных связей в
полиуретанах сильно преувеличена, во всяком случае, при объяс-
нении температурных переходов. Полиуретаны, по мнению авто-
ров, следует рассматривать как блок-сополимеры, причем при изуче-
нии структурных переходов в области низких температур нужно
учитывать взаимодействие гибких олигомерных блоков, а в области
высоких температур — жестких. Очевидно, авторы отдают предпоч-
тение вандерваальсовым связям. В направлении идентификации
физических связей необходимы дальнейшие исследования, хотя
задача эта чрезвычайно трудная.
Таким образом, особенность трехмерной сетки полиуретанов
заключается в том, что она содержит большое число физических
связей наряду с химическими поперечными, вводимыми при син-
тезе. Иногда эффективная плотность сшивки полиуретановых элас-
томеров определяется в основном физическими связями. Такая
специфика строения сетки предопределяет основные как физико-хи-
мические, так и механические свойства полиуретанов.
Глава VI. О СВЯЗИ МЕЖДУ КИНЕТИКОЙ РЕАКЦИИ
ОБРАЗОВАНИЯ, СТРУКТУРОЙ И НЕКОТОРЫМИ
СВОЙСТВАМИ ПОЛИУРЕТАНОВ
Теперь уже не вызывает сомнений, что существует тесная связь
между характером надмолекулярных структур и физико-механи-
ческими свойствами полимеров. Отсюда, естественно, возникает
задача научиться управлять получением того или иного типа струк-
тур с целью влияния на комплекс физико-механических свойств
полимерных материалов.
Прежде чем останавливаться на вопросах о формировании над-
молекулярных структур в сетчатых полиуретанах, необходимо разо
брать механизм образования самой полиуретановой сетки. Работ,
посвященных решению этой задачи, практически нет. Поэтому мы
изложим результаты исследований в этом направлении, проведен
ные под руководством Т. Э. Липатовой.
Особенностью процесса образования поли)ретановых сеток ив
ляется то, что они формируются из олигомерных молекул, которые
характеризуются большими силами межмолекулярных взаимодей-
ствий (водородные связи и другие виды физических взаимодейст-
вий). Формирование надмолекулярных структур, если оно имеет
место, должно происходить в условиях сильных межмолскулярных
взаимодействий одновременно с протеканием химической реакции,
которая приводит в конечном итоге к образованию сетчатого по-
лимера. Берлиным с сотрудниками [70] при изучении трехмерной
полимеризации олигоэфиракрилатов установлен ряд кинетических
эффектов, вызываемых предварительной упорядоченностью цепей
кристаллизующихся олигоэфиров и охарактеризован тип надмо-
лекулярных структур полимерного продукта. В литературе поч-
ти нет сведений о взаимосвязи между кинетическими условиями
образования полимеров при реакциях поликонденсации или сту-
пенчатой полимеризации и характером надмолекулярных структур.
Прежде всего возникает вопрос о зарождении надмолекуляр-
ных структур при образовании полимеров. В работах Платэ, Кар-
гина и других [35, 108, 159] был поставлен вопрос, является ли
образование упорядоченных агрегатов макромолекул в процессе
полимеризации общей закономерностью и каковы условия, способ-
ствующие структурообразованию полимеров. С этой целью было про-
ведено электронно-микроскопическое исследование продуктов ра-
диационной полимеризации кристаллических мономеров.
Показано, что одновременно с процессом полимеризации происхо-
дит формирование надмолекулярной структуры полимерного вещест-
139
ва. Обычно это структуры фибриллярного типа с преимущественным
направлением по отношению к какому-либо геометрическому пара-
метру исходного кристалла. Уже с первых секунд реакции
наблюдается формирование фибриллярных надмолекуля рных
структур полимеров. Это, а также установленная авторами за-
висимость образования определенных типов структур от усло-
вия протекания процесса позволило сделать вывод о том, что хими-
Рис 71. Кинетические кривые взаимодействия макродиизоциана-
тов на основе ППГ-2000 и 4,4'-дифенилметандиизоцианата с три-
метилол upon а ном при различных температурах:
/ — 100; 2 — 80; 3 — 60° С
ческая реакция полимеризации и структурообразование полиме-
ров являются единым процессом и что при твердофазной полиме-
ризации изучаемых мономеров предпочтительнее происходит рост
не отдельных макромолекул, а сразу пучков цепей.
В данных работах сделано еще одно интересное наблюдение.
Надмолекулярные структуры полимеров, образующихся непо-
средственно в ходе полимеризации, очень близки (имеют место мно-
гоступенчатые агрегаты тех же морфологических форм) структурам
тех же полимеров, полученных из растворов. Это показывает, что
условия формирования структур в момент, когда макромолекулы
сами строятся в процессе химической реакции, весьма благоприят-
ны для образования упорядоченных надмолекулярных структур.
Внимание исследователей было прежде всего обращено на кине-
тику формирования полимеров и надмолекулярных структур в них
для кристаллизующихся исходных объектов, как мономерных так и
олигомерных. Однако самый большой интерес представляют амо-
рфные сетчатые полимеры, но для них связь кинетики и надмо-
лекулярных структур еще не исследована. Мы рассмотрим это на
примере механизма формирования полиуретановых сеток, получен-
ных на основе макродиизоцианатов, в состав которых входили
140
гликоли двух видов — полиоксипропилеигликоли (ППГ) и поли-
тетраметиленгликоли (ПТМГ) различных молекулярных весов
(500, 1000 и 2000). Во всех случаях для синтеза макродиизоциа-
Рис. 72 Кинетические кривые взаимодействия макроди изоциана-
тов с триметилолпропаном при температуре 80° С на разных основах:
/ - П11Г 500. 2 — 11ПI 2000; 3, 4 — ППГ 1000 и ПТМГ 1000
натов применялся 4,4'-дифенилметандиизоцианат; триметилолпро-
пан служил сшивающим агентом.
Кратко остановимся на результатах исследования кинетики про
цесса образования сетчатого
полиуретана до степеней пре-
вращения, близких к точке
геля [591.
Из рис. 71 и 72 видно, что
кинетические кривые имеют
вид, характерный для реак-
ции второго порядка. В резуль-
тате обработки кинетических
данных в предположении би-
Рис. 73. Зависимость удельной скорости
(же NCO/1000 г • мин) реакции взаимодей-
ствия макродиизоцианатов на основе раз-
личных эфирдиолов стриметилолпропаном
от глубины превращения (t == 80° С);
/ — ППГ-500, 2 — ППГ-1000 <3 —ППГ 2000.
молекулярного механизма ре-
акции установлено, что удель-
ная скорость реакции незна-
чительно или практически сов-
еем не меняется в зависимости
от длины и химической приро-
ды эфирдиола до глубины превращения около 70% (рис. 73).
В некоторых работах [12, 57] показано, что в процессе протека-
ния реакции образования полиуретанов происходит перераспреде-
ление водородных связей. Это говорит об изменении в ходе реакции
141
межмолекулярных взаимодействии, что в свою очередь должно
сказаться на некоторых физических характеристиках системы 1601.
Исследование изменения вязкости системы в ходе реакции показало
(рис. 74), что начиная с глубин превращения 50—60% наблюдается
резкое ее возрастание. В связи с этим интересно было проследить
за изменением молекулярных весов в процессе полимеризации.
Рис. 74. Изменение вязкости реакционной смеси в ходе
процесса полимеризации:
/ — макродиизоцианат на основе ППГ 500, 2 —• макродиизо-
цианат на основе ППГ 1000, 3 — макродиизоцианат на основе
ППГ 2000
Средневесовой молекулярный вес М& и z-средний молекулярный
вес Mz [1451 определены методом приближения к седиментацион-
ному равновесию на ультрацентрифуге Г-120 в мстилэтилкетоне.
По измеренным коэффициентам поступательной диффузии и ха-
рактеристической вязкости [т] ] также рассчитаны среднеквадратичные
радиусы инерции (AJ2) и отношения (Д2)'/2/М^ в зависимости от глу-
бины превращения (рис. 75). В области превращения, близкой
к точке геля, имеет место резкое возрастание молекулярных весов
(кривые 4 и 5). Увеличение Mz/M^ говорит о резком уширении кри-
вой МВР вблизи точки геля. С этими результатами хорошо согла-
суются данные о вязкости реакционной системы (см. рис. 74).
На основании рис. 75 можно сделать следующие выводы. Пример-
но с 60% превращения начинается интенсивный процесс развет-
вления цепей. Об этом свидетельствует значительное уменьшение
R2IM^ (кривая /) с возрастанием молекулярного веса цепей (кри-
вые 4 и 5). Об образовании сильно разветвленных макромолекул
говорит величина показателя b в уравнении
D - КМ”,
142
где D — коэффициент диффузии; М — молекулярный вес. Для всех
названных ранее систем b -- 0,384, что характерно для сильно раз-
ветвленных полифункциональных цепей 1145].
Вблизи точки геля (начиная примерно с 60% превращения) рост
происходит с сохранением подобия формы частицы, которая близка
Рис. 75. Некоторые параметры системы в зависимости от глубины пре-
вращения:
/ - ^/л^: 2 - м2/л^, з - IriL - м2. 5 -
к сферической. Это подтверждается малым уменьшением характе-
ристической вязкости вблизи точки геля (кривая 3) и уменьшением
отношения (кривая /).
Вблизи точки геля кривая МБР уширяется (увеличение отно-
шения Mz/Mw, кривая 2). Образование разветвленных молекул,
сопровождающееся резким возрастанием молекулярного веса, наблю-
дается в сравнительно небольшом интервале изменения глубины
превращения. При этом прирост молекулярного веса на пропен i
превращения значительно превышает рассчитанный по теории Фло-
ри для процессов равновесной поликонденсации мономеров. Это-
го и следовало ожидать, так как каждый акт взаимодействия в
случае реакций с олигомерами значительно увеличивает молеку-
лярный вес растущей цепи (на 500—2000 и больше) по сравнению
с реакциями мономерных систем.
Резко неравномерный рост цепей вблизи точки геля, по-видимому,
связан с очень интенсивным возрастанием вязкости системы
за счет сильных межмолекулярных взаимодействий, которые
143
характерны для полиу ретанов. Отсюда сетка, возникающая сразу за
точкой геля, должна характеризоваться большой неравномерностью
расстояний между узлами. Чтобы проверить эти предположения,
необходимо исследовать образцы после точки геля.
Для приведенных систем точка геля лежит в области 71—73%
глубины превращения и удовлетворительно согласуется с величиной,
рассчитанной по Флори. Точки максимума на некоторых кривых
рис 75 соответствуют точке геля. При экстракции образцов после
точки геля даже при 93% превращения весовая доля золь-фракции
составляет 58%. При этом гель-фракция представляет не монолит-
ную систему, а микроблоки. Полностью сшитый продукт (при пол-
ном отверждении весовая доля золь-фракции составляет около 4,5%)
образуется в основном на самых последних стадиях реакции в ре-
зультате редких химических сшивок между микроблоками. Резуль-
таты исследования системы после точки гелия (см. рис. 75) ука-
зывают на сужение кривой МВР после гелеобразования. Отно-
шение Mz/Мл практически становится равным исходному (кривые
2 и 4). Следовательно, в гель-фракцию вовлекаются преимущест-
венно сильно разветвленные большие молекулы. При этом продол-
жается рост молекул золь-фракции (увеличение кривая 5) с
сохранением подобия формы. Об этом свидетельствуют неизмен-
ность величин [q I (кривая 3) и (кривая /).
Естественно ожидать, что изменение молекулярной структуры
реакционной системы в ходе полимеризации и появление в ней эле-
ментов сетки должно сказаться на ее релаксационных характерис-
тиках.
Действительно, при переходе от линейного полимера к сшито-
му наблюдается повышение температуры стеклования, а также ха-
рактерное расширение интервала стеклования 1160]. В связи с этим
измерены параметры стеклования реакционной смеси в процессе
ее отверждения, которые позволили выявить некоторые изменения
релаксационных свойств системы. Установлено, что на кривых
теплоемкости в интервале перехода из стеклообразного в жидкое
состояние наблюдается появление излома. Кроме того, имеет место
расширение температурного интервала стеклования в пределах
определенных глубин превращения, а затем он практически не
меняется до более глубоких стадий (рис. 76).
В области превращений 50—60% наблюдается значительное
расширение температурного интервала стеклования, который за-
тем практически не меняется, хотя область стеклования смещается
в сторону более высоких температур. Это смещение для макроди-
изоцианата на основе ППГ-1000 происходит в пределах превращения
85-95%.
Полученные результаты хорошо согласуются с характеристикой
системы по молекулярным весам и вязкости. Область превращений
50—60% характеризуется резким увеличением молекулярного
веса и расширением кривой МВР, что проявляйся в расширении
144
(/) и температуры стеклования (2) от глуби-
ны превращения для макродиизоцианата на
снове ППГ-1000.
температурного интервала стеклования. Далее, за точкой геля сис-
тема расслаивается и возникает гель-фракция. Прп этом темпера-
турный интервал стеклования и температура стеклования не
меняются, хотя происходит вовлечение в сетку больших разветвлен-
ных молекул и кривая МВР сужается. Только в области превраще-
ний 85—95% для нашего примера происходит смещение интервала
стеклования к более высоким температурам в результате образо-
вания монолитной сетки.
Возникает вопрос о том,
каков механизм формиро-
вания сетки после точкиJ
геля и что представляют’
собой микроблоки, из кото-
рых формируется сетчатый,
полимер. Все приведенные
экспериментальные данные
позволяют говорить о пере-«
ходе реакции из гомоген-
ных условий в микрогете-
рогенные, а затем в макро-
гетерогенные. В этом
случае фактором, опреде-
ляющим скорость процесса
формирования сетки, может
оказаться поверхность раз-
дела, и в конечном итоге
скорость реакции может определяться величиной этой поверхности.
Действительно, начиная с определенных глубин превращения реак-
ция перестает подчиняться уравнению второго порядка. Применение
этого уравнения приводит к абсурдным результатам, хотя сам процесс
заключается лишь во взаимодействии NCO-групп с ОН-группами.
Для выяснения закономерностей реакции после ючки 1еля при-
менен калориметрический метод [61]. При расчете термограмму
разделили на два самостоятельных участка: до и после точки геля.
Участок кривой после 70% превращения обработали как самосто-
ятельную термокинетическую кривую, предполагая согласно Фло-
ри [1191, что в точке геля содержание его равно нулю, но часть про-
дукта находится в виде образований большой сложности с большим
числом разветвлений Эти образования можно рассматривать как
центры, вокруг которых за точкой геля мгновенно возникают за-
родыши трехмерной структуры (микрогель). Исходя из данных пред-
ставлений, для описания кинетики формирования сетчатого поли-
уретана можно применить уравнение Аврами — Ерофеева [30, 1651
а= 1-ехр
где а — доля вещества, подвергшегося превращению;/^— удель-
ная скорость процесса; t — время; п — показатель, зависящий от
геометрической формы зародыша.
145
Если для случая формирования полиуретановой сетки под ве-
личиной а подразумевать долю вещества, перешедшего в гель, то,
Рис. 77. Резу.льга1ы по кинетике формирова-
ния полиуретановых сеток в координатах
Аврами для макродиизоцианатов на основе
разных эфирдиолов
/— ППГ 2000 / - 70°, 2 — ППГ 2000, t =
=х70°, J — ПТМГ 1000, t = 70°, 4— 11ППГ-2000,
Г= 80\ 5 — ПП1 2000, t =* 90° С, 6 - ПП
500, ? - 70°С
но переходит в гетерогенные условия на определенном этапе
формирования полиуретановых сеток. В связи с этим естественно
возникает вопрос о том, что
представляют собой части-
цы микрогеля, образующие
поверхность раздела, кото-
рая, по-видимому, опреде-
ляет скорость реакции по-
сле точки геля.
Электронно-микроскопи-
ческие исследования поли-
уретановых сеток показали,
что они в большинстве слу-
чаев (для полиуретанов, по-
лученных через макродиизо-
цианаты) характеризуются
глобулярными структурами
1561. Образцы снимали в ви-
как видно из рис. 77,
уравнение Аврами — Еро-
феева описывает кинетику
образования сетки после
точки геля. Величина п
(табл. 40) во всех случаях
близка к единице, а удель-
ная скорость процесса при
одной температуре практи-
чески не зависит от химиче-
ского строения эфирдиолов
при их одинаковых моле-
кулярных весах и незначи-
тельно изменяется с величи-
ной молекулярного веса.
Применение уравнения
Аврами — Ерофеева к ре-
акции формирования поли-
уретановых сеток после
точки геля являлось попыт-
кой расшифровать механизм
этой реакции. Полученные
результаты говорят о том,
что реакция действитель-
Таблица 40
Удельная скорость (Ко) процесса
формирования сетчатого полиуретана
и показатель п для макродиизоцианатов,
полученных на основе различных эфирдиолов
Эфирдпол Темпера- тура про- цесса, °C п Ко 10»
ППГ-2000 70 1,00 5,79
80 1,13 7,29
90 1.01 15.90
ППГ-500 70 0,95 13,60
ПТМГ-2000 70 1,00 5,40
ПТМГ-1000 70 1,05 7.98
де реплик со сколов после их
травления активным кислородом [9]. Глобулярные образования на
снимке (рис. 78, см. вклейку) имеют величину примерно на порядок
146
больше, чем величина частиц, рассчитанная методом диффузии в ходе
реакции. Это может быть связано с агрегацией исходных частиц в
более крупные глобулярные образования [371.
Из всех приведенных данных следует, что полиуретановые сетки,
полученные на основе макродиизоцианатов, представляют собой
глобулярные образования, сшитые редкими химическими связями.
Теперь остановимся на результатах электронно-микроскопиче-
ских исследований полиуретановых сеток, полученных при разных
молекулярных весах исходных диолов и различной их химической
природе. На рис. 79, а, б (см. вклейку) представлены электронно-ми-
кроскопические снимки полиуретана на основе макродиизоцианата,
полученного из ППГ-2000 и4,4'-дифенилметандиизоцианата итриме-
тилолпропана при 60 и 125°С. На них ясно видны беспорядочно распо-
ложенные глобулярные образования и их редко расположенные агре-
гаты. На рис. 79, б концентрация агрегатов и их размеры больше, чем
на рис. 79, а. Таблица 41
Некоторые механические характеристики образцов
полиуретанов
Олигоэфирдиол задавае- мое COOT ношением компонен TOR аразр, кг 1см2 Удлине ние % Темпера- тура от- вержде- ния/ С
ППГ-500 1000 484 29 80
ППГ-1000 1500 54,3 186,0 80
ППГ-2000 2500 9,6 85 60
2500 14,0 139 80
2500 17,0 183,7 125
ППГ-3000 3500 8,2 109 80
ПТМГ-1000 1500 48,3 214,0 80
ПТМГ-1000+1,4- бу- тандиол 40 000 60,3 984,0 80
По-видимому,повышениетемпературы способствует агрегации гло-
бул и, возможно, также их химическому связыванию, что должно при-
водить к улучшению механических показателей. Это действительно
имеет место, как видно из результатов прочности на разрыв (табл. 41).
С повышением температуры отверждения прочность образцов рас-
тет, оставаясь, однако, достаточно низкой, что объясняется их гло-
булярной структурой. Релаксационные свойства сшитых полиурета-
нов также хорошо объясняются с точки зрения образования глобу-
лярных структур.
При электронно-микроскопическом исследовании полиурета-
новых образцов, полученных в одинаковых условиях, но на ос-
нове эфирдиолов различного химического строения и одинакового
молекулярного веса, видно (рис. 80, см. вклейку), что их структу-
ры похожи и близки размеры глобулярных образований.
147
Кинетические кривые реакций, приводящих к их получению, также
очень похожи (см. рис. 71). По-видимому, при одних и тех же молеку-
лярных весах эфирдиолов, ио при различной их химической приро-
де сетчатые полиуретаны имеют очень близкую структуру.
При сравнении структур образцов полиуретанов (электронно-
микроскопические фотографии) на основе эфирдиолов различных
молекулярных весов (рис. 81, см. вклейку) обращает внимание то,
что образец на основе диола с большим молекулярным весом
(рис. 81, а) практически не имеет никакой структуры. У образца на
основе ППГ-2000 ярко выражены структуры в виде деформиро-
ванных сферических тел, которые состоят из агрегированных гло-
бул (рис. 81, б). По механическим показателям «бесструктурный»
образец значительно лучше образца с четко выраженной струк-
турой (см. табл. 41).
Способ получения сетчатых полиуретанов также оказывает вли-
яние на их надмолекулярную структуру. Полиуретаны получают по
двухстадийному способу, который заключается в том, что сначала син-
тезируют макродиизоцианат на основе диизоцианата и эфирдиола.
Макродиизоцианат — продукт присоединения двух молекул диизо-
цианата по двум гидроксильным группам эфирдиола. Затем макро-
диизоцианат трехатомным спиртом связывается в пространствен-
ную сетку. Этим способом мы получали все образцы. При односта-
дийном способе смешивают в соответствующих соотношениях
одновременно все три компоненты.
Принципиальной разницы между кинетикой протекания реак-
ции при одно- и двухстадийном способах нет [171, однако в структу-
рах таких образцов заметно некоторое отличие (рис. 82. см. вклей-
ку). Полиуретаны в обоих случаях синтезированы при одной темпе-
ратуре 80° С на основе ППГ-2000. В качестве диизоцианата взят
4,4'-дифенилметандиизоцианат, сшивающего агента — триметилол-
пропан. У образца, полученного одностадийным способом, прак-
тически отсутствуют признаки структуры, тогда как образец, полу-
ченный по двухстадийному способу, имеет ярко выраженную глобу-
лярную структуру. На рис. 81, б хорошо видны агрегаты глобул,
плотно расположенные друг относительно друга. По механичес-
ким показателям «одностадийный» образец лучше «двухстадийно-
го» (см. табл. 41).
Таким образом, данные электронно-микроскопических иссле-
дований согласуются с результатами, полученными при изучении
молекулярных весов и формы макромолекул в ходе полимери-
зации. При формировании сетчатых полиуретанов непосредственно
в ходе ступенчатой полимеризации, вероятно вблизи точки геля,
возникают глобулярные образования, которые агрегируются в
зависимости от условий полимеризации в более крупные структур-
ные формы. При агрегации может происходитьредкая химическая сши-
вка образований. Это определяет малуюмеханическуюпрочностьобра-
зцов, полученных по двухстадийному способу, а, возможно, также
148
и зависимость механических показателей от температуры реакции
и расстояния между узлами сшивки A1f.
При протекании реакции по одно- и двухстадийному способам
формирование надмолекулярных структу р происходит одновременно
с ростом молекулярных цепей и образованием микрогеля. Оба про-
цесса взаимосвязаны, как и для кристаллизующихся мономеров [35,
108|. В двухстадийном процессе в начале формирования сетки си-
стема, вероятно, более упорядоченная, чем в одностадийном.
Образование структур в полиуретановых системах обусловлено
сильными межмолекулярными взаимодействиями, которые харак-
терны для этих систем. При двухстадийном способе получения поли-
уретанов условия для реализации межмолекулярных взаимодейст-
вий более благоприятны, чем при одностадийном. Это связано с тем,
что при двухстадийном способе формирование начинается в систе-
ме, более однородной по молекулярным весам, чем при одностадий-
ном. Поэтому при двухстадийном способе, вероятно, имеется боль-
шая упорядоченность в системе, чем при одностадийном, что опре-
деляет различия в конечных структурах.
Приведенные результаты ставят своей целью показать, что в
полиуретанах, как и в других полимерных системах, кинетические
условия формирования полимерных молекул в известной мере оп-
ределяют процессы формирования надмолекулярных структур.
Образование полиуретановой сетки происходит из олигомерных
молекул, что вносит некоторую специфику в этот процесс.
На основании всего изложенного ранее можно сформулировать
ряд задач, решение которых весьма существенно как с точки зрения
развития теории связи механизма образования макромолекул и над-
структур, так и для управления надмолекулярной структурой по-
лимерного материала.
Мы здесь отмечаем три из них.
1. Изучение процесса межглобулярного сшивания, которое
происходит, по-видимому, на глубоких стадиях реакции. Решениеэтой
задачиважно в связис тем, что межглобулярное сшивание должно при-
водить к повышению механической прочности полимерного материа-
ла. До сих пор этот процесс практически не изучен.
2. Установление возможности сшивания внутри глобулярных
структур, которое, по нашему мнению, должно обязательно про-
исходить, а также изучение этого процесса. Структура глобуляр-
ных образований и их прочность должны играть большую роль при
деформировании метериала.
3. Поиск условий, при которых олигомерные молекулы перед
сшиванием были бы развернуты, а затем фиксированы химическими
связями в виде сетчатой структуры. Кинетика реакции в «ориен-
тированном» состоянии и формирование надструктур в этих усло-
виях до сих пор практически не исследованы, хотя это важно для
полимерной химии в целом.
149
Глава VII. ФИЗИКО-МЕХАНИЧЕСКИЕ СВОЙСТВА
ПОЛИУРЕТАНОВ
связь основных физико-механических свойств
ПОЛИУРЕТАНОВ СО СТРУКТУРОЙ
Интерес к полиуретанам определяется прежде всего теми фи-
зико-механическими показателями, которыми обладают материалы
на их основе. Высокое сопротивление истиранию и значительная
механическая прочность позволяют использовать резины из урета-
новых каучуков в машиностроении и других отраслях промышлен-
ности в качестве конструкционного материала. Хорошо зареко-
мендовали себя полиуретановые клеи и покрытия, пенополиурета-
ны, волокна.
Получение материалов на основе полиуретанов с таким широким
диапазоном свойств обусловлено применением при их синтезе
химических соединений различного класса. Поэтому вопросам
связи между структурой полиуретанов и их основными физико-ме-
ханическими свойствами посвящено большое число работ и обзоров
[167, 175, 287, 290, 305, 306, 317].
Так как эти вопросы подробно освещены в монографии Саун-
дерса и Фриша [ 114], то мы лишь в основных чертах рассмотрим вли-
яние природы составляющих компонентов на основные физико-
механические свойства полиуретанов, используя данные, опубли-
кованные в последние годы.
Влияние природы полиэфира
и его молекулярного веса
Свойства полиуретановых эластомеров в значительной степени
зависят от строения и молекулярного веса основного компонента —
полиэфира. Изучено влияние строения сложного полиэфира на свой-
ства эластомеров на основе 1,5-нафтилендиизоцианата [175, 305]
(табл. 42) и полиуретанов на основе дифенил метандиизоцианата
и 1,4-бутандиола [2871 (табл. 43).
Как видно из приведенных данных, на механических свойствах
полиуретанов сказываются увеличение количества метиленовых
ipynn в полиэфире и наличие боковых цепей в гликольной компо-
ненте, что в большой степени снижает прочность. Ухудшение неко-
торых механических характеристик в этих случаях связано с пони-
жением концентрации сложноэфирных групп, что уменьшает
количество межмолекулярных связей, обусловленных взаимодей-
150
Таблица 42
Зависимость свойств полиуретановых эластомеров от строения сложных
полиэфиров
Гликоль Кислота Предел прочнос- ти при растяже иии, кг!смг Относи- тельное удлине ние при разрыве, Предел прочности на раздир, кг/см,-
Этиленгликоль Янтарная 273 625 119
Адипиновая 350 640 158
Дигликолевая 266 570 147
1,2-Пропилепгл и коль Адипиновая 217 480 91
2,3-Бутиленгликоль » 179 630 91
ствием этих групп с другими полярными группами в цепях поли-
уретанов.
Аналогично понижаются физико-механические свойства поли-
уретанов при замене сложного полиэфира на простой, что, как по-
казал Смит и Магнуссон [3181, связано с отсутствием сложноэфирпых
Таблица 43
Свойства полиуретановых эластомеров на основе сложных полиэфирдиолов
Сложный полиэфир Предел проч ности при растяжении, кг; см* Относитель- ное удлине- ние при раз рыве, °6 Остаточное удлинение, % Модуль уп РУ1 ости при 3 0% ном удли нении, кг!см- Предел проч ности на раз- дир, кг; см2 ; Твердость по Шору (шкала В)
11олиэтиленадипииат 483 590 25 108,5 43,2 60
Поли-1,4-бутилеиадипинат Поли-1,5-пен1аметмленадипи- 420 510 15 133 48,6 70
нат 441 450 10 126 10,8 60
Поли-1,3-бутиленади пинат 224 520 15 77 18,0 58
Полиэтиленсукцинат 476 420 40 224 36,0 75
Поли-2,3-бутилснсукцинат 245 380 105 — 93,6 85
Полинеопентилсх кцкнат 182 400 70 140 41,4 67
групп в полиуретанах на основе простых полиэфиров. Они изу-
чали эластомеры на основе простых и сложных полиэфиров, 2,4-то-
луилендиизоциапата и триметилолпропана. В качестве простого
полиэфира брали ППГ, сложным служил полиэфир, полученный
при сополимеризации мегил-е- и е-капролактона. При сравне-
нии некоторых характеристик обоих полиуретанов оказалось, что
если модуль упругости полиуретана на основе ППГ составлял
9,1—12,2 кг!см2, то при замене ППГ на сложный полиэфир он по-
вышался до 18,5—24,3 кг! см2.
151
Согласно [2081, полиуретановые эластомеры на основе сложных
полиэфиров (полиэтиленгликольадипината) обладают большими
прочностью, износостойкостью, устойчивостью к действию раствори-
телей, чем полиуретаны на основе простых полиэфиров (политетра-
метилен гликоля и нолиоксипропиленгликоля). Это говорит о боль-
шом влиянии сложноэфирных групп на механические свойства
полиуретанов. Прочность межмолекулярных связей, образованных
сложноэфирньши группами с другими группами полимерных цепей,
настолько велика, что часто удается получить линейные полиуре-
тановые эластомеры, обладающие прекрасными механическими
свойствами при комнатной температуре 13061.
Таблица 44
Зависимость свойств полиуретановых эластомеров от молекулярного веса
полиэфира и количества диизоцианата
м полиэфира Количество диизоциана- та, г/100 м з полиэфира Предел проч- ности при растяжении, кг/см2 Относитоль ное удлиие ние при раз- рыве, ?0 Модуль уп- ругости при 300% НОМ удли- i нении, кг!см2 Предел проч- ности на раз дир, кг/ см2 Эластич- ность, % Твердость по Шору (шкала А)
2290 15,2 392 680 93 189 58 65—71
3500 11,9 346 560 130 249 65 67—73
3100 12,6 305 610 111 172 59 68-73
2440 14,6 350 645 112 186 59 71—76
2080 16,2 385 635 130 186 60 71—76
1385 21,6 371 645 109 125 50 74—77
7
Изменение молекулярного веса сложного полиэфира незначи-
тельно влияет на начальные свойства полиуретанов, как показано
на примере полиэтиленгликольадипината 1175, 3051 (табл. 44).
При хранении эластомеры на основе полиэфиров с большим моле-
кулярным весом медленно кристаллизуются. При наименьшем
из изученных молекулярных весов (1385) наблюдалась наимень-
шая способность к кристаллизации, но прочность на раздир и
эластичность были малы. Изменение молекулярного веса от 2160
до 4680 практически не влияло на механические свойства полиме-
ров на их основе, а от 1180 до 2160 увеличивало прочность на раз-
дир и удлинение.
В соответствии с полученными данными, оптимальным моле-
кулярным весом полиэтиленгликольадипината, применяемого для
синтеза полиуретанов, признан молекулярный вес порядка 2000.
Изучение физико-механических свойств полиуретана при раз-
личных молекулярных весах пол и изофталата, представляющего
смесь поли-1,4-аминобутиленадипината (ПБА), поли-1,5-амилен-
изофталата (ПАИ), полиэтнленизофгалата (НЭИ) [1961 показало,
чго молекулярный вес сложного полиэфира значительно изменяет
физические свойства полиуретанов (табл.45).
152
Таблица 45
Зависимость физических свойств полиуретанового форполимера от
средне числового молекулярного веса полиизофталата
М поли- изофтала- та Компонента, % Проч ность на разрыв, кг 1см* Удлине- ние, % Твердость по Уэлласу
ПБА ПАИ пэи
600 40 10 50 398 575 50
600 40 20 40 454 533 57
600 40 30 30 244 650 53
600 30 10 60 245 400 77
600 30 20 50 362 457 68
600 30 35 35 238 288 66
600 30 50 20 517 463 59
600 20 50 50 348 10 94
2000 60 10 30 232 567 52
2000 60 20 20 139 603 48
2000 50 10 40 194 573 46
2000 50 25 25 381 513 60
2000 50 40 10 111 585 51
2000 40 10 50 227 445 86
2000 40 20 40 226 428 68
2000 35 15 50 293 450 62
Образцы с наибольшей прочностью на разрыв получены на основе
полиизофталата с молекулярным весом 600. Увеличение молеку-
лярного веса до 2000 уменьшает прочность на разрыв полиурета-
нов.
В работе[266| исследовано влияние молекулярного веса сложно: о
полиэфира адипиновой кислоты и смеси этилен-и проииленгликоля
(молекулярноесоотношение 70:30) на величину модуля полиуретанов.
Показано, что с увеличением молекулярного веса полиэфира от
1200 до 3200 величина модуля при 100, 200 и 300%-ном удлинении
понижается.
Влияние молекулярного веса простого полиэфира на механи-
ческие свойства полиуретанов [1671 изучено на форполимерах
из 2,4-ТДИ и ППГ разного молекулярного веса с соотношением
NCO : ОН—2 : 1; отвердитель — метилен-бг/с-о-хлоранилин. Уста-
новлено, что прочность при растяжении, модуль и некоторые дру-
гие характеристики улучшаются при меньшем молекулярном весе
(табл. 46). С увеличением молекулярного веса ППГ снижается ис-
тираемость и растет эластичность полиуретанов.
Полиуретанакрилаты (полиуретановые материалы на основе
олигоэфиров, диизоцианатов и монометакрилового эфира этилен-
гликоля) обладают лучшей прочностью на растяжение при малых
молекулярных весах как простых, так и сложных олигоэфиров [861
(табл. 47). С увеличением молекулярного веса прочность уменьшает-
ся, но растет относительное разрывное удлинение.
153
Таблица 46
Механические свойства эластомеров на основе ППГ и 2,4-ТДИ
Свойство мппг
1000 | 1250 1500 2000
Предел прочности при растяжении, кг 1см1 353 315 245 84
Относительное удлинение при разрыве, % — 860 — ——
Модуль упрхгости при 300%-ном удлинении, кг 'см2 147 70 42 28
Прочность на раздир (по Грейвсу), кг! см1 56 43 40 22
Твердость по Шору (шкала А) 88 77 67 60
1 ютсря массы при истирании через 1000 циклов (по Табор}), мг 85 60 25 8
Температура хрупкости (по Тиниусу— Олсену), ГС —40 -50 —55 —56
Эластичность по отскоку (по Башору), % 24 20 22 38
Таблица 47
Некоторые физико-механические свойства полиуретанакрилатов
О Я [ ОэфН рО! .7И КО."Ь Моэг Диизоцианат Предел прочности при рас тяжешш кг/см* Относи- тельное удлине- ние при разрыве, % л с рассчитан ное
Пол иокс и пр о 11 иленгл и- 400 4,4'-Дифенил- 650 12 386
кэль 1000 мета ндиизоци- анаг То же 130 112 585
2000 » » 60 120 920
Адипиновая кислота и 630 2,4-ТДЦ 200 60 413
этиленгликоль 1000 » 80 140 535
1360 » 90 145 656
1600 » 80 130 672
При исследовании влияния молекулярного веса на механические
свойства пол и мочевин уретанов на основе ТДИ и нолиоксиэтилен-
iдикол ей (ПЭГ) рассчитано содержание мочевинных и уретановых
ipynn [2291. Найдено, что увеличение молекулярного веса олиго-
эфира сопровождается уменьшением содержания уретановых и мо-
чевинных групп, что вызывает понижение прочности, но увеличи-
вает эластичность и удлинение (табл» 48).
Аналогичное влияние молекулярного веса олигоэфиров на ме-
ханические свойства наблюдается и для полиуретановых покрытий
1102, 103, 119, 120, 1231.
Таким образом, изменение молекулярного веса исходного
слигоэфира позволяет варьировать свойства полиуретановых ма-
154
Таблица 48
Характеристики полимочевинуретановых пленок на основе ТДИ
и простых полиэфиров
1 Свойство Полиол
ПЭГ-200 ПЭГ 400 ПЭГ 1000
Содержание уретановых групп, % 21,4 15,6 8,8
Содержание мочевинных групп, % Содержание ароматических групп 10,5 ; 7,7 4.3
(CgHg), % Максимальная прочность при рас- 27,2 19,9 11,1
тяжении, кг/см- 262,0 210,0 21,0
Максимальное удлинение, % 0 400,0 1000,0
териалов, что связано с различной концентрацией полярных групп
при различной длине олигоэфирного блока.
Величина оптимального молекулярного веса исходного поли-
эфира зависит от природы олигоэфира и определяется главным об-
разом гибкостью последнего: чем большей гибкостью обладает
молекула олигоэфира, тем при меньшем молекулярном весе можно
получить полиуретаны с оптимальными свойствами.
Строение диизоцианата
Свойства полиуретанов зависят от природы изоцианата, ис-
пользуемого при синтезе. В табл. 49 показано влияние строения ди-
изоцианатов на свойства эластомеров на основе полиэтиленгликоль-
адипината молекулярного веса 2000 [175], отвердителем служила
вода.
Таблица 49
Влияние строения изоцианатов на свойства
полиуретановых эластомеров
Диизо цианат Предел прочности при рас- тяжении, кг} см2 Относи- тельное удлине- ние при разрыве, % Предел прочности на раздир, кг! см*
2,4-Т ол у и лен ди изоцианат 200—249 730 83
1,5-Нафтилендиизоцианат 308 765 166
2,7-Флуорендиизоциапат 434 660 141
Найдено, что громоздкие ароматические группы диизоцианатов
в значительной степени способствуют повышению прочности эласто-
меров. Эти результаты согласуются с данными [287] (табл. 50).
Модуль, прочность на раздир и твердость увеличиваются как при
введении жестких ароматических структур, так и в случае диизо-
цианатов без метиленовых заместителей.
155
Механические свойства эластомеров определяются не только
природой диизоцианата, но и соотношением в нем изомеров [290].
Исследовано влияние соотношения 4,4'-и 2,4'-дифенилметапди-
изоцианата на механические свойства полиуретанов на основе по-
Таблица 50
Влияние строения диизоцианатов на свойства полиуретановых эластомеров на
основе сложных полиэфиров (отвердитель гликоль)
Диизоцианат Предел проч- ности при растяжении, кг/ см- Относитель- ное удлине- ние при раз- рыве, % Остаточное удлинение, % Модуль уп- ругости при 30096 ном уд- линении, кг! см2 Предел проч- ности на раз- дир, кг} см2 Твердость по Шору
Нафтилендиизоцианат 301,0 500 85 210,0 36,0 80
Фенилендиизоцианаг 447,5 600 25 161,0 54,0 72
2,4-Толу илевдиизоцианат 322,0 600 1 24,5 27,0 40
4,4'-Дифенилметандиизоциа- нат 552,5 600 10 112,0 48.6 61
3,3'-Димстил-4,4'-дифенилмс- тандиизоцианат 371,0 500 0 42,0 7,2 47
4,4'-Дифенилизопропилди- изоцианат 245,0 700 10 21,0 16,2 56
3,3' - Диметил-4,4' - толу иле! i- диизоцианат 280,0 400 10 161,0 32,4 70
ликапролактона (мол. вес 1050) и бутандиола (рис. 83) и установ-
лено, что основные характеристики ухудшаются при увеличении
содержания 2,4'-изомера. Данное явление связано с изменением
симметрии полимерных цепей при изменении изомерного состава
диизоцианата.
Влияние природы структуры
сетки и типов поперечных
связей на свойства полиуретанов
Рассматривая влияние природы составляющих на механические
свойства полиуретанов, следует прежде всего исследовать их роль
в структуре сетки полимеров, так как природа поперечных связей
в полиуретанах, их количество, регулярность построения трех-
мерной сетки являются главными факторами, обусловливающими
физико-механические свойства полимеров.
Многие исследователи изучали влияние густоты сетки, рассчи-
танной теоретически или определенной экспериментально, на ме-
ханические свойства полиуретанов [130, 186, 224, 287, 305, 3171.
Например, представлена зависимость некоторых механических
свойств полиуретанового каучука СКУ-В (па основе сложного
полифункционал иного полиэфира П-В и 2,4-ТДИ) от соотношения
156
ТДИ: полиэфир. При увеличении соотношения ТДИ: полиэфир от
0,8 до 1,1 величина Мс, определенная по равновесному модулю,
изменялась от 41 500 до 1650, сопротивление разрыву увеличивалось,
а относительное удлинение непрерывно падало (рис. 84).
В других случаях характер зависимости механических свойств
от густоты сетки более сложный [287, 1351. В табл. 51 приведены ре-
зультаты изучения влияния гус-
тоты сетки на основные физико-
механические свойства эластоме-
ров на основе полиэтиленгли-
кольадипината, дифенил метанди-
2,4'~МДИ,мол 7О
Рис. 83. Зависимость изомерного
состава и физических свойств
этастомеров на основе поли-
капролактона, бутандиола, 4,4'-
МДИ и 2,4'-МДИ:
1 — прочность на разрыв, кГ}см2
(X 70) 2 — модуль при 100%-ном
удлинении, (кГ[смъ (Х7); 3 — уд-
линение при разрыве, % (X 100).
Рис 84. Зависимость физико-механи-
ческих свойств полиуретана СКУ-В от
исходного соотношения ТДЦ/поли-
эфир:
1 — прочность при разрыве, 2 — относи-
тельное удлинение
изоцианата и триметилол пропана 1287]. Величина Мс рассчита-
на теоретически из стехиометрического состава.
Уменьшение Мс от 21000 до 5300 приводит к понижению твер-
дости, прочности при расстяжеиии, удлинения, модуля и прочности
на раздир, в то же время увеличивается эластичность. Согласно
1287], при увеличении степени сшивания за счет первичных химичес-
ких связей затрудняется наиболее выгодное для межмолекулярного
взаимодействия расположение цепей в пространстве, что уменьша-
ет эффективность межмолекулярного взаимодействия. Следователь-
но, большое влияние на механические свойства полиуретанов ока-
зывают вторичные межмолекулярные связи.
Дальнейшее увеличение густоты сшивки (уменьшение Мс от
5300 до 2100) приводит к увеличению модуля, что свидетельствует
157
Таблица 51
Влияние степени сшивания на свойства полиуретановых
эластомеров на основе сложных полиэфиров
Ме эл а с томера Предел проч- ное! и при растяжении, ; кг!см* Относитель- ное удлине ние при раз- рыве, % Остаточное удлинение, % Модуль уп- ругости при 100% ном уд- . мнении, КЗ/СМ* Предел проч ности на раз дир, кг/см* Твердость по Шору (шкала В)
2100 126.0 170 0 38,9 5,4 57
3100 122.5 200 0 29,4 4,5 53
4300 101,5 280 0 21,0 5,4 49
5300 196,0 350 0 18,9 5,4 46
7100 315,0 410 0 23,1 7,2 51
10 900 392.0 490 5 32,2 10,8 55
21 000 385 0 510 10 35,0 25,2 56
о доминирующем влиянии первичных поперечных связей на изме-
нение механических свойств. При изучении механических свойств
^4
— -10, моль/см3
Рис 85. Зависимость разрывного
напряжения от степени сстчатостн.
/ — полидиэтиленсукцинатуретана, 2 —
полидиэтиленадипинатуретапа, 3 —по
лидиэтиленсебацинатуретана.
межмолекулярных связей (та
полиуретанов на основе сложных
полиэфиров, ТДИ и триметилол-
пропана наблюдалась экстремаль-
ная зависимость разрывного на-
пряжения от степени сетчатости,
определенной экспериментально
11301, (рис. 85) , что согласуется с
другими данными [1, 118].
Мы считаем, что, объясняя экс-
тремальную зависимость механи-
ческих свойств от густоты сетки,
нужно учитывать изменение гиб-
кости цепей от густоты сетки. Так,
при больших Ме9 когда гибкость от-
резков между узлами сетки велика,
возможно сближение отрезков це-
пей и образование большого чис-
ла межмолекулярных связей бла-
годаря наличию в полиуретанах
полярных групп различных типов.
Очевидно, при больших Мс, когда
вклад вторичных связей в общую
густоту сетки полиуретанов на
основе сложных полиэфиров велик
[118], механические свойства этих
полиуретанов будут определяться
главным образом концентрацией
I. 52). Уменьшение Мс приводит
к понижению гибкости отрезков цепей и падению числа вто-
158
ричных связей в общей густоте сетки. Дальнейшее увеличение
числа узлов в сетке за счет введения трехфункционалыюго сши-
вающего агента настолько уменьшает гибкость отрезков межд}
узлами, что количество вторичных поперечных связей очень пада-
ет. Основная роль с этого момента принадлежит химическим попе-
речным связям, увеличение количества которых способствует ростх
модуля [287].
Сравнение данных табл. 51 и 52 показывает, что величина М
полиуретанов на основе различных полиэфиров неодинаково влия-
Таблица 52
Влияние Мс на механические свойства эластомеров на основе форполимеров
из поли-1,4-оксибутиленгликоля и диизоцианата с молекулярным весом
около 2000, отвержденных смесями диола и триола
Количество отверди- теля на 1 г экв форполимера мс Предел прочности при рас- тяжении, кг! см"1 Относи- тельное удлине- ние при разрыве, % Модуль упругости при сдви ге при 100%-ной деформа- ции, кг/см2 Твердость по Шору (шкала А) Прочность на раздир по Грейсу, кг!см>
Гексантриол 1,0 2090 33,2 235 17,50 55 8,1
Полиоксипропилен- триол (М -700) 1,0 3700 38,8 380 11 90 43 7,2
ПТ 0,6 + пентанди- ол 0,4 5800 66,6 645 7,35 38 15,1
ПТ 0,6 -f- полиок- сипропиленгли- коль (М - 425) 0,4 6150 93,8 625 5,60 37 11,7
ет на их механические свойства. Так, зависимость от Мс модуля
твердости, относительного удлинения при разрыве в интервале
от 2000 до 5000—6000 одинакова для простых и сложных поли-
эфиров. Вместе с тем, если в случае сложных полиэфиров предел
прочности при растяжении с изменением Мс от 2000 до 3000 умень-
шается, то для простых полиэфиров — приводит к увеличению
предела прочности при растяжении. Это может быть связано с тем,
что в эластомерах на основе простых полиэфиров межмолекуляр-
ные силы не играют такой роли, как в эластомерах на основе
сложных полиэфиров, в которых наличие карбонильной группы
способствует возникновению большого количества межмолекуляр-
ных связей.
Иногда зависимость между структурой и свойствами полиуре-
танов на основе простых полиэфиров такая же, как для сложных
полиэфиров, что наблюдалось при изучении взаимосвязи структу-
ры и свойств эластомеров из форполимера на основе поли-1,4-оксибу-
тиленгликоля, отвержденных ароматическими диаминами [163, 164].
159
Рис 86 Зависимость прочности при разрыве (Ор),
удлинения при разрыве (е) и относительной твер-
дости покрытий от соотношения NCO/OH:
/ — относительная твердость, 2 — прочность при раз
рыве, 3 — удлинение при разрыве.
С увеличением доли ароматической части такие эластомеры
приобретают большую часть прочности благодаря межмолску-
лярным связям подобно эластомерам на основе сложных полиэфиров.
При определении модуля упругости пенополиуретанов на ос-
нове простого (ПОПГ) и сложного (полиэфирадипината) полиэфи-
ров в зависимости от Мс [1761 оказалось, что характер изменения
модуля с густотой сетки в обоих случаях одинаков. Вместе с тем при
одних и тех же температурах пенополиуретаны на основе сложного
полиэфира обладают большей прочностью при сжатии и большим
модулем упругости, чем пенополиуретаны на основе простых по-
лиэфиров, чю опять же вызвано различным количеством межмо-
лекулярных связей при использовании полиэфиров разной при-
роды.
Исследовано влияние степени сшивки, рассчитанной теоретичес-
ки и определенной экспериментально, на некоторые физико-меха-
нические свойства полиуретановых покрытий [53, 90, 100, 102—104,
119—1231. В табл. 53 приведена зависимость свойств полиуретановых
покрытий па основе сополимера тетрагидрофурана с 25% окиси про-
пилена (мол. вес 1200) и аддукта ТДИ и триметилолпропана от
величины Ме, рассчитанной теоретически по методу Сэндриджа
13021. Здесь же представлены результаты расчета концентра-
ции уретановых [У| и мочевинных [М] групп при допущении, что
изоцианатные группы реагируют с образованием только урета-
новых и мочевинных связей [259].
Зависимость некоторых физико-механических характеристик
полиуретановых покрытий на основе указанного ранее сополиме-
160
Рис. 78. Структура сетчатого полиуретана, полу-
ченного из макроди изоцианата на основе ППГ-500
при 80° С.
Рис. 79. Структура сетчатого полиуретана на основе ППГ-2000
при 60° (а) 125° С (б).
Рис. 80. Структура образцов сетчатого полиуретана на основе ППГ-ЮОО
(а) и ПТМГ-1000 (б) при 80° С.
Рис. 81. Структура сетчатых полиуретанов на основ? эрирдиолов различного
молекулярного веса (Z = 80е С):
а — М *>40000, б — М * 1000.
Рис. 82. Структура сетчатых полиуретанов, полученных одностадийным
(а) и двухстадийным способом (б).
Рис. 104. Рентгенограмма полиуретана на основе полидиэтмлснгликоль-
адипината:
а - до растяжения; б — после растяжения до 100%.
Таблица 53
Физико-механические свойства полиуретановых покрытий на основе
сополимеров тетрагидрофурана с окисью пропилена и аддукта ТДИ и ТМП
Состав полиэфироглрколя NCO/OH [У] ♦ J03, ( моль!г о 5? 2 - Мс рассчиын вое Относитель- ная твср тость no М 3 Прочность при разрыве (20° С), кг!см' Удлинение при разрыве (20 ‘ С), %
ТГФ — П-1400 — 25% ОП 2- 1 2,33 0,36 4400 0,18 120 660
3: 1 2,53 0,63 3900 0,40 250 390
4: 1 2,66 0,79 3400 0,45 320 350
ТГФ — П-1400 - 15% ОП 2: 1 2,37 0.39 4300 0,14 107 766
3:1 2,56 0,64 3800 0,30 225 289
4: 1 2,69 0,80 3300 0,40 260 241
Т а б л и и а 54
Физико-механические свойства кремчийсодержащих полиуретановых покрытий
Состав композиции NCO/OH [У] . 103, моль/г [М] Ю3, молъ!г Мс вычис- ленное Относитель- ная твер- дость по М-3 £ з о t tr с к S 25* Сс« Удлинение при разрыве. %
(CH3)2SI(OCH2CH2OH)2 + 1,00 1580 0,56 160 106,0
+ аддукт биуретовой 1,25 3,42 1,57 1443 0,72 247 35,4
структуры 1,50 3,00 1,99 1320 0,81 427 31,2
1,75 2,70 2,69 13 0,85 500 29,0
2.00 2,45 2,43 1230 0,85 525 27,0
C2H5Si(OCH2CH2OH)3 |- тот 1,00 — — 720 0.64 — —
же аддукт 1,25 2,85 2,14 665 0,73 325 29,0
1,50 2,50 2,50 627 0,86 516 27,0
1.75 2.22 2,79 600 0,91 535 26,0
2,00 2,00 3,00 530 0,86 580 31,0
Si (OCILCI 12ОН)4 + тот же 1,00 — — 550 0,66 80 202.0
аддукт 1.25 2,53 2,46 560 0,82 487 27.0
1,50 2,20 2,79 570 0,84 626 27,0
1,75 1,95 3,04 579 0,86 650 32,0
2.00 1,75 3,29 586 0,78 — —
ра, аддукта, нос добавкой 50% триметилол пропана [122] показана
на рис. 86. Последний компонент введен для снижения избытка
изоцианатных групп в исходных композициях.
Приведенные результаты говорят о том. что увеличение густоты
сетки полиуретановых покрытий на основе простых олигоэфиров
вызывает монотонное увеличение твердости и прочности покрытий,
снижая при этом относительное удлинение
Для покрытий на основе сложных [1021 и кремнийсодержащих
олигоэфиров характер изменения физико-механических свойств
с густотой сетки аналогичен таковому для покрытий на основе
161
простых олигоэфиров (табл. 54, рис. 87). Увеличение количества
имических узлов в пространственной сетке полиуретана приводит
в какой-то мере к уменьшению влияния физических связей на ме-
ханические свойства полиуретанов Поэтому если для эластомеров
на основе сложных и простых ол
наблюдаются различия в изменении
Рис. 87 Зависимость разрывных напряже-
ний свободных п ienoK на основе олиго-
диэтиленгликольадипината (ОДА) от со-
отношения NCO/OH:
/ — ОДЛ 400, 2 - ОДА 600, 3 — ОДЛ 800
гим важным фактором, особенно
игоэфиров при больших Ме
физико-механических свойств
с изменением Мс до некото-
рого предела, то с дальней-
шим уменьшением Мс, когда
вклад физических связей в об-
щую густоту сетки становится
меньше, эти различия сглажи-
ваются. Очевидно, по той же
причине для покрытий, где эф-
фективная плотность сшивки
гораздо больше, чем в эласто-
мерах, не замечено существен-
ных различий в характере
изменения основных механи-
ческих свойств с густотой сет-
ки при использовании олиго-
эфиров различной природы.
Таким образом, степеиьсет-
чатости в значительной мере
определяет физико-механичес-
кие свойства полимеров. Дру-
при повышенных температу-
рах, является природа поперечных связей в пространственной
сетке полиуретанов, зависящая от применяемого отвердителя. В ре-
зультате исследования влияния диаминов [274] и воды [2791
как отверждающих агентов на свойства полимеров на основе
различных дпизоцианатов установлено (табл. 55), что сильное меж-
молекулярное взаимодействие в полимерах приводит к тому, что
эластомер приобретает высокие значения модуля и прочности на
раздир в результате образования мочевинных групп при отвержде-
нии диаминами.
Наличие жестких сегментов в цепи (громоздкие аромашческие
диизоцианаты и ароматические диамины, применяемые в качестве
отвердителя) также обеспечивает высокие значения модуля и проч-
ности на раздир Присутствие гибких групп в отвердителе, напри-
мер тиоэфирных, понижало прочность при растяжении, проч-
ность на раздир, модуль и твердость.
Следовательно, применение диаминов в качестве отвердителей
приводит в ряде случаев к улучшению механических свойств вслед-
ствие образования сильно полярных мочевинных групп.
Исследовано [227] влияние строения диаминов, в частности коли-
чества углеродных атомов в них, на механические свойства
162
пол и эфир уретановых волокон, полученных на основе сложного оли-
гоэфира (олигоэтиленгликольадипината с мол весом 2000) и
дифенилметандиизоцианата. Волокна проявляли кауч\коподобные
свойства, несмотря на отсутствие химических поперечных связей,
что обусловлено образованием достаточно прочной сетки физических
связей. Волокна, полученные на основе диаминов с нечетным числом
1 а б л и ц а 55
Влияние отвердителей на свойства полиуретановых эластомеров
Отвердитель Предел прочно- сти при растяже- нии, кг/см2 Относитель- ное удлине- ние при раз- рыве, % 1 Модуль уп- ругости при 300%-ном уд- линении, кг/см2 Предел проч- ности на раз- дир, кг/ см2 Эластич- ность, % Твердость по Шору (шкала А)
2,4-Т о л у и л е и д и и w цианат
Дианизидин 273,0 635 31,5 182,0 37 58
2,4-Толуилендиамин 280.0 720 35,0 143,5 51 60
Вода 199,5—248,5 730 — 82,5 — —
Гексаметилен ди изоцианат
4,4'-Диаминодифе- нилметан 269,5 680 80,5 210,0 53 90
Дианизидин 217,0 650 50,3 101,5 53 77
2,4-Толуилсндиа- мин 266,0 710 39,9 143,5 57 69
2,4-ТДА 126,0 707 25,9 112,0 54 70
Вода 234,5 1000 — 57,5 2 —
Примечание. Последние два полимера получены на основе поли-1,2 пропиленади
пината, остальные — па основе полиэтиленадипин<ла, молекулярный все обоих сложных
полиэфиров равен 2000.
атомов углерода (рис. 88), имеют более высокую прочность при раз-
рыве и большее удлинение; обратная картина наблюдается для мо-
дуля. Можно предположить, что такое резкое изменение свойств
полиуретанов с содержанием числа атомов углерода связано с об-
разованием различного числа вторичных физических связей в
исследуемых полимерах в случае четного и нечетного количеств
углеродных атомов в диамине. Для доказательства этого не-
обходимы дальнейшие исследования с привлечением более тонких
физико-химических методов изучения структуры полимеров
Клафф и Глединг [187] исследовали влияние типа поперечных
связей на свойства уретановых эластомеров на основе простых
олигоэфиров. Изученные линейные полиуретаны
О 0 -1
— (ОСН8СН,СНгСН4),„—О—X—СН2СНг—N—(!—
СН? (СН »)3
сн2он in = сп„ А
163
синтезированы из бмс-хлорформиата поли-1,4-оксибутиленгликоля
и N-2-оксиэтил-Ьпентапилэгилендиамина. В качестве акцептора
кислоты взят водный карбонат натрия. У молекул таких полимеров
дга реакционноспособных центра, за счет которых могут образо-
вываться поперечные связи: боковые гидроксильные группы, спо-
собные взаимодействовать с изоцианатами, и боковые винильные,
-г--1-—т-1-г
/357л
6
Рис 88. Влияние числа атомов
(п) углерода в диамине на мо-
дуль упругости при 300%-ном
удлинении (а), прочность (б) и
удлинение при разрыве (в) по-
лиуретановых волокон.
вступающие в реакцию с серой. При синтезе пространственного
полимера применяли следующие отвердители: 3,3'-диметокси-4,4'-
бифенилендиизоцианат; форполимер с концевыми изоцианатными
группами (мол. вес 5000), полученный при взаимодействии 4 молей
поли-1,4-оксибутиленгликоля (мол. вес 1000) и 5 молей 3,3'-диме-
токси-4,4'-бифенилепдиизоцианата; серусодержащая смесь.
Найдено, что модуль, твердость и эластичность не зависят ни
от химической природы, ни от длины цепи отвердителя. Однако при
одной и той же плотности поперечных связей величина усадки при
сжатии (22 ч при 70° С и 70 ч при 100° С) для эластомеров, отвер-
жденных серой, выше, чем у эластомеров, отвержденных изоциа-
натом Такое различие связано с тем, что поперечные связи, обра-
зуемые за счет серы, особенно связи дисульфидного типа, менее
термостабильны, чем связи уретанового типа. Тип поперечных
связен оказывает небольшое влияние на механические свойства
полиуретанов при комнатной температуре [228, 3171.
164
С другой стороны, температура плавления полимеров выше, если
поперечные связи образуются за счет триата или триизоцианата,
и ниже, если сшивание идет за счет избытка диизоциапата. Это,
очевидно, обусловлено тем, что в последнем случае образуются не-
термостабильные аллофанатные или биуретовые группы.
Влияние нетермостабильных биуретовых и аллофанатных свя-
зей на некоторые свойства полиуретанов показано на примере пе-
номатериалов, содержащих 90, 100 и 120% диизоцианата (изоциа-
натный индекс соответственно 90, 100, 120) от требуемого при рас-
считанных значениях Мс от 600 до 1730 [3021.
Установлено, что пеноматериалы с индексом 120 разрушаются
анилином при 140° С гораздо быстрее, чем с индексом 90—100.
Реакции разрушения аллофанатных и биуретовых групп можно
представить следующей схемой:
О
| 140 С
... -N-C-NH- ••• + ArNH2 —>
О О
140' С--NH—LNH-------1- ArNHCONH—....—N-Jj-O- • • • -U
О CONH
140 ‘‘С Я
-j-ArNH2 —> ... —NH—С—О— ... + ArNHCONH— ...
После обработки анилином
пеноматериалы с индексом
120 гораздо сильнее набу-
хают в ди метил ацетамиде
(ДМА) при 25° С, чем по-
лиуретаны с индексом 90 и
100, что говорит о более
быстром разрушении попе-
речных связей в пеноматс-
риале с индексом 120.
Данные результаты тес-
но связаны с характером
кривых модуля упругости
при кручении для тех же
пеноматериалов (рис. 89).
Пеноматериалы с изоциа-
натными индексами 90 и
Рис. 89. Кривые зависимости модуля упруюс
ти при кручении от температуры для пенопо
лиуретанов на основе простых полиэфиров.
100, содержащие очень не-
большое количество биуре-
товых или аллофанатных
групп, имеют относительно
пологие кривые изменения
/ — изоцианатный индекс 120, Мс =680, 2 —изо
цианатный индекс 100, Мс - ПОО, 3 —изоциа-
натный индекс 120, Мс 1350, 4 — изоцианат-
ный индекс 100, Мс — 1600; 5 — изоцианатный
индекс 90, Мс -- 2000.
модуля вплоть до температуры
полиуретанов с индексом 120
резкое снижение модуля при
160—170е С. В случае же пено-
наблюдается излом на кривой и
НО—130“ С. Предполагается, что
165
последнее обусловлено разрушением аллофанатных или биуретовых
групп Согласно 12491 диссоциация аллофанатов становится за-
метной уже при температуре 106°С.
Исследована [287| зависимость модуля упругости при кручении от
температуры полиуретанов на основе полиэтиленгликольадипи-
ната, сшитых триметилолпропаном и отвержденных различными
смесями триметилолпропана (ТМП) и ароматического гликоля mult-
rathane X (рис. 90). Модуль упругости образца, не содержащего
Зависимость модуля упругое-
Рис. 90
ти при кручении от температуры для
эластомеров на основе полиэтилепгли-
кольадипината и дифен ил мета иди изо-
цианата
/ - 0% ТМП; 2 - 70% ТМП, 3 - 100%
ТМП
химических поперечных связей
(кривая/), имеет наивысшее зна-
чение при 25° С, но быстро умень-
шается при 130—150° С. При за-
мене 70% диола триолом (кри-
вая 2) модуль упругости при
комнатной температуре меньше
и незначительно изменяется с
повышением температуры до
170—190° С. Полная замена ди-
ола триолом приводит к еще
большему расширению темпера-
турною интервала, при котором
происходит снижение модуля
(кривая 3).
Наблюдаемый характер из-
менения модуля с температу-
рой объясняется тем, чго вто-
ричные физические связи гораздо
легче разрушаются под действи-
ем тепла, чем первичные валентные. Эластомер, имеющий много
физических связей (кривая /), прочнее и тверже при комнатной
температуре, но его стабильность ниже, чем у полимера, сшитого
триолом.
Для полиуретанов марки Дуретан U, полученных на основе
гексаметилендиизоцианата и бутан диол а-1,4 [204], наблюдается зна-
чительное падение модуля с температурой, особенно после 80° С,
что объясняется разрушением физических и, возможно, слабых хи-
мических, типа аллофанатных, связей (рис. 91). Термопластичные
полиуретаны другой природы [270, 342] обладают прекрасными
свойствами при низких температурах—высокой износостойко-
стью, устойчивостью к действию озона, радиации, маслам, али-
фатическим растворителям. При высоких темпера!урах у этих
полиуре1анов значительно ухудшаются механические свойства
вследствие диссоциации физических связей. Для их улучшения
рекомендуется вулканизация перекисями и эпоксидами.
Следовательно, физико-механические свойства полиуретанов в
значительной мере определяются типом связей, составляющих про-
странственную сетку. Особенно резко проявляется зависимость
166
свойств полиуретанов от природы связей при повышении темпера-
туры.
Структура трехмерной сетки полиуретана определяется усло-
виями синтеза полимера. Например, формирование сетки будет
происходить по-разному в случае получения полиуретанов одно- и
двухстадийиым методом. При образовании через стадию форполимера
(двухсгадийный метод) можно
ожидать возникновения более
регулярной сетки, что долж-
но привести к улучшению
свойств полимеров. Так, при
исследовании свойств эласю-
меров на основе полиоксипро-
пилепгликоля, ТДИ и 3,3'-
дихлор-4, 1'-диаминодифенил-
метана (МОСА), полученных
по форполимерному и одно-
стадийному способу [1661, ока-
залось, что первые имеют боль-
шую прочность при растя-
жении, но меньшую прочность
на раздир, модуль упруюсти,
эластичность и усадку при
сжатии.
Полиуретановые покрытия
па основе сложных полиэфи-
ров, синтезированные одно-
стадийным методом при 20° С,
Рис. 91. Зависимость модуля сдвига поли-
уретана марки «Дуретан» U от темпера-
туры
/ — дополнительно высушенный образец при
29° С, 2 — образец, насыщенный 4% воды и
затем высушенный при 4° С
имели меньшую твердость,
чем полученные через форполимер
ТДИ) [1001. Но при более высокой
(аддукт триметилолпропана с
температуре отверждения эти
различия исчезали.
Полиуретановые эластомеры на основе сложных полиэфиров,
синтезированные форполимерным методом [2901, имели меныпую
твердость и прочность на разрыв, чем полученные одностадийным
методом. Особенно заметно способ синтеза отразился на темпера-
туре плавления, которая оказалась значительно выше в случае
«одностадийных» полиуретанов. Это дало основание авторам сде-
лать вывод о большей степени кристалличности этих полиме-
ров.
Как видно из сравнения данных по влиянию метода синтеза
на свойства полиуретанов, трудно сделать однозначный вывод о
преимуществах того или другого метода. Очевидно, необходимы
дополнительные экспериментальные исследования в этом направ-
лении, которые позволяют выяснить влияние способа получения
на структуру трехмерной сетки полиуретана и, в связи с этим, на
их физико-механические свойства.
167
Результаты исследования влияния природы составляющих
на структуру и свойства полиуретанов указывают на то, что фи-
зико-химические свойства этих ма!ериалов, также как и других
полимеров, определяются молекулярным весом исходных компонен-
тов, эффективностью межмолекулярного взаимодействия, жесткостью
сегментов, составляющих полимерные цепи. Полиуретаны можно
рассматривав как блок-сополимеры |307| с мя1кими сегментами
простого или сложного полиэфира и жесткими сегментами, обра-
зованными уретановыми или мочевиноуретановыми блоками.
Мягкие сегменты обусловливают увеличение эласшчности и уд-
линение при разрыве. Увеличение концентрации жестких сегментов
спссэбствуег усилению межмолскулярного взаимодействия и
поэтому повышает твердость полиуретанов, их прочность на раз-
рыв, температуры плавления и стеклования, но уменьшает эла-
стичность и удлинение при разрыве. Изменение природы мягких
и жестких сегментов и типа поперечных связей позволяет получать
полиуретановые материалы с заданными свойствами.
ТЕМПЕРАТУРНАЯ ЗАВИСИМОСТЬ
НЕКОТОРЫХ СВОЙСТВ ПОЛИУРЕТАНОВ
Рассмотрение некоторых физико-механических свойств поли-
уретановых эластомеров и других материалов показало, что
они проявляют ряд специфических свойств, отличающих их от дру-
гих полимеров. Однако причины специфичности до сих пор иссле-
дованы очень мало. С этой точки зрения интересно изучить темпе-
ратурную зависимость некоторых свойств полиуретанов в растворе
и блоке. Важно также выяснить вопрос о том, какие свойства изо-
лированных макромолекул передаются полимерному телу и как
это происходит.
Обнаружено, чго в полимерах помимо основных перехо-
дов, связанных со стеклованием, существуют дополнительные тем-
пературные переходы, проявляющиеся в виде аномалий темпера-
турной зависимости некоторых свойств [138, 139, 152, 197, 209,
295, 301, 331, 342J. Подобного рода переходы обнаружены как в
чистых полимерах, так и в растворах 1139, 152, 209, 2951. Так,
дополнительный конформационный переход полистирола в блоке об-
наруживается в виде размытого максимума около 50 С на кривой
дифференциально-термического анализа 13441; четкого падения
внутреннего давления (т. е. энергии когезии) 13011; увеличения
брегговских расстояний между фенильными группами 12461; излома
на дилатометрических прямых [197]. В растворах аналогичные пере-
ходы обнаружены в видеаномалыюйзависимостихарактерисгической
вязкости и размеров клубков [152, 2951; тангенса учла диэлектри-
ческих потерь [138, 139J; падения анизотропии цепи с температурой
11521; скачка поверхностного натяжения [2091.
168
Для эфиров целлюлозы найден набор температур перехода
и сделан вывод о том, что последние обусловлены изменением мо-
лекулярного механизма движения макромолекул [1971. Так, низшей
температуре перехода отвечаег минимальное число возможных
конформаций, и по мере повышения температуры число конформаций
и свободный объем увеличиваются кооперативным способом, т е.
эти переходы могут рассматриваться как конформационные.
Таблица 56
Коэффициенты объемного расширения (а) полиуретанов
Растворитель М полиэ- фирной компонен- ты Растворитель М полиэ фирной ко мнойен ты а
Бензол 1600 0,00065 МЭК 4100 0,0107
4100 0,00100 МЭК + ДХУК 1600 0,0077
Бензол +ДХУК 1600 0,00084 4100 0,0095
4100 0,00193 ДХУК 1600 0,0026
МЭК 1600 0,00770 4100 0.0047
Сделана попытка связать температурные переходы в разбав-
ленных растворах, отражающие конформационные превращения
в изолированных макромолекулах, с температурными переходами
в полимерном блоке, носящими кооперативный характер [951. Авто-
ры пытались обнаружить конформационные переходы для полиуре-
танов разного типа. Предполагалось, что наличие в полиуретановой
цепи двух типов участков (гибкой эфирной или гликолевой компо-
ненты и жестких уретановых групп), способных образовывать
внутри-и межмолекулярные водородные связи, должно определен-
ным образом сказаться на температурной зависимости некоторых
свойств полимеров. Проведены дилатометрические измерения, диф-
ференциально-термический анализ, а также измерена характеристи-
ческая вязкость растворов некоторых полиуроанов.
Исследованы два образца форполимеров на основе пол иди -
этилепгликольадипината (мол. веса 1600 и 4100) и смеси изомеров
2,4- и 2,6-ТДИ (соответствующий мол. вес полиуретанов 3200 и
13 500), а также полиуретан на основе 1,4-бутандиола и 2,4-ТДИ
(2,4-11У, мол. все 9000). Измерены коэффициенты объемного рас-
ширения для первых двух образцов в бензоле, мет ил эти л кетоне
(МЭК) и в этих же растворителях с добавкой дихлоруксусной кисло-
ты /ДХУК/, а также в чистой ДХУК (рис. 92, табл. 56). Для 2,4-ПУ
в качестве растворителей взяты цикло! ексанон, диметилформамид
(ДМФ) и их смеси с ДХУК (рис. 93). Коэффициенты объемного
расширения рассчитывали по формуле
a-AV/V0AT, (VII, 1)
169
ГДС AV ’ ^раствора ^растворителя! — ПерВОНЗЧЗЛЬНЫЙ объем ПО*
димера в растворителе, ДТ — разность темпера тур Т — Т1}
Для тех же образцов сняты кривые дифференциально-терми-
ческого анализа (рис. 93).
Как видно из рис. 92, для растворов полиуретанов увеличение
объема растворенного полимера происходит линейно во всех слу-
чаях. Вычисленное значение а (см. табл. 56) растет во всех
растворителях с увеличением длины полиэфирной компоненты. Ве-
Рис. 92. Зависимость AVn/V0 от тем-
пературы для форполимеров на ос-
нове полидиэтллеигликольадипината
(мат вес 1600) и ТДИ:
I — в бензоле, 2 — в бензоле с добавкой
ДХУК, 3 — в МЭК. 4 - в МЭК с добав
кой ДХУК
20 30 40 50 Г,°C
Рис 93. Зависимость Д/п/|/0 от тем-
пературы для полиуретана на основе
1,4-бутандиола и 2,4-толуилендиизоци-
апата:
/ — в цикло! ексанонс, 2 — в ДМФ, 3 —
в циклогексаноне с добавкой ДХУК, 4 —
ДМФ с добавкой ДХУК
личина а в МЭК в два раза больше, по сравнению с бензолом, что,
вероятно, связано с качеством растворителя. МЭК является пло-
хим растворителем для полиуретанов [951, поэтому в нем увеличе-
ние объема полимера с температурой должно быть больше, nevi в
бензоле (хороший растворитель). Добавка ДХУК, способной к об-
разованию водородных связей, не приводит к изменению а в МЭК.
Эю можно объяснить небольшим вкладом уретановых групп, обу-
словливающих появление водородных связей. Действительно, ве-
личина а в бензоле близка к таковой для чистых полиэфиров разного
строения (а = 0.00074 — 0.00084 при мол. весе 790—2200) До-
бавка ДХУК в бензол ухудшает растворитель, так как сама
ДХУК—’Плохой растворитель для полиуретанов.
Таким образом, для полиуретанов с малой концентрацией уре-
тановых групп (три-четыре на одну молекулу полиуретана с мол.
весом 13 500) возможное разрушение водородных связей практи-
чески не влияет на изменение объема полимера в растворе, а раз-
личие в а для разных растворителей, вероятно, зависит от коли-
чества последних.
Для полиуретана 2,4-ПУ картина дилатометрических измере-
ний в разных растворителях неодинакова (см. рис. 93). Из кри-
170
вой AV/AV0 от Т в циклогексаноне и ДМФ виден явный
конформационный переход около 40 ° С. В данном случае кон-
центрация уретановых групп в цепи значительно выше, чем в пре-
дыдущем, поэтому их влияние на свойства полиуретана должно
быть значительным. Поскольку добавка в циклогексанон и ДМФ
дихлоруксусной кислоты, способной к образованию водородной
связи, приводит к исчезновению конформационного перехода, су-
ществующего в чистых растворителях при 40° С, можно предполо-
жить, что он обусловлен разрывом
водородных связей. Данные диф-
ференциально-термического анали-
за (рис. 94) указывают также на
конформационный переход в облас-
ти 40° С во всех случаях для этого
полимера, в то время как для чис-
тых полиэфиров такового не наблю-
дается. Характерно, что анало1ич-
ный переход наблюдается и в блоч-
ных полимерах (рис. 94, кривые
7, S).
Термомеханическое исследова-
ние эластомеров на основе полиэти-
лепгликольадипината с длиной цепи
большей, чем их механический сег-
мент, и толуилендиизоцианата, так-
же показывает, что при Т ~ — 40сС
начинается необратимая деформа-
ция. Зависимость характеристичес-
кой вязкости [ц| 2,4-ПУ от темпе-
Рис. 91. Термограммы нагревания*
J — 2,4-ПУ в циклогексаноне, 2—2,4 ПУ
в ДМФ, 3 — 2,4-ПУ в циклогексаноне
с добавкой ДХУК: 4 — ПУ в бензоле,
5 — ПДЭГЛ, 6 — ПУ 7 - 2,4-ПУ. и
термограмма охлаждения 2,4-ПУ в цик-
логексаноне (цифры на кривых соответ
ствуют температурам переходов)
ратуры в циклогексаноне показывает, что в области 40—50е С вяз-
кость резко понижается, и это, вероятно, связано с разрывом
внутримолекулярных водородных связей. Увеличение цуд/с при
разбавлении, согласно [3271, связано с диссоциацией водородных
связей.
Из сказанного вытекает, что конформационный переход в области
40—50° С обусловлен разрывом водородных связей, имеющим место
в разбавленных растворах и блоке.
Как в растворе, так и в блоке помимо перехода в области около
40° С для 2,4-ПУ наблюдаются дополнительные максимумы в области
10—12° и 20°С. Однако в чистом ДМФ и в циклогексаноне с до-
бавкой ДХУК переход при 20°С исчезает, в то время как при 10—
12 ° С он сохраняется. Для растворов ПУ в бензоле при 25е С также
имеет место переход, но в чистом ПУ он отсутствует. Наличие этих
переходов, вероятно, обусловлено проявлением подвижности других
компонентов \ретановой цепи (помимо уретановых групп).
Дифференциальная кривая охлаждения (см. рис. 94, кривая 2) для
2,4-ПУ в циклогексаноне показывает, что все процессы — как
171
разрыв водородных связей, так и другие конформационные перехо-
ды — обратимы.
Итак, анализ экспериментальных данных позволяет сделать
заключение, что аномалия температурной зависимости некоторых
свойств полимеров в блочном состоянии тесно связана с конформа-
ционными переходами, существующими в разбавленных растворах,
и обусловлена свойствами индивидуальных макромолекул.
Очень интересно сопоставить изложенные выше результаты
сданными непосредственного исследования температх рной зависи-
мости механических свойств.
Из классической теории высокоэластичности известно, что для
гибких цепей, между которыми отсутствуют специфические взаимо-
действия, равновесный модуль высокоэластичности Е^ связан с мо-
лекулярным весом отрезка цепи между узлами сетки Мс и темпера-
турой следующим соотношением:
£оо == 3/?Tp/A1f,
где R — газовая постоянная, р — плотность. Была исследована
применимость этого уравнения для описания свойств некоторых
полиуретановых эластомеров 17]. В достаточно широком интервале
температур найдена линейная зависимость модуля от температуры,
однако при экстраполяции экспериментальных прямых до пересе-
чения с осью температур они не проходят через начало координат,
как это следует из уравнения кинетической теории, а пересекают
ось абсцисс при температурах, далеких от абсолютного нуля. Сле-
довательно, экстраполяция в область низких температур оказы-
вается необоснованной для полиуретанов. В общем виде это можно
объяснить тем, что ниже температуры стеклования характер тепло-
вых движений в полимере аналогичен характеру тепловых дви-
жений в обычных твердых телах и не связан с гибкостью цепей.
В данной области температур деформация носит уже не высокоэлас-
тический, а истинно упругий характер и, следовательно, не может
быть описана кинетической теорией эластичности. Возможно, что
определяемая по пересечению с осью абсцисс температура соответ-
ствует температуре хрупкости эластомера и может служить до-
волнительной характеристикой. На основании этого в уравнение
можно ввести поправку, учитывающую результаты экстраполяции
экспериментальной прямой
£м==^Г(т-т0).
Полученное уравнение находится в лучшем согласии с экспе-
риментом, а параметр То можно связать с температурой хрупко-
сти, поскольку последняя наступает тогда, когда полностью пре-
кращаются все сегментальные движения.
Установлены некоторые температурные аномалии механических
свойств полиуретановых каучуков, в которых отчетливо проявляется
172
специфика их поведения [70] Исследованатемпературнаязависимость
величины равновесной высокоэластической деформации и модуля
высокоэластичности, а также прочность поли) ретановых каучуков
Рис. 95. Кривые ползучести неплас! «филированного (а) и пластифицированною (б)
каучука
/ — 0 2 — 20. 3 — 40, 4 — GO. 5 — 20, 6 — 35, 7 — 40, 8 — 50° С.
на основе диэтиленгликольадипинатов различных молекулярных
весов и ТДИ, сшитых триэтаноламином и отличающихся нали-
чием или отсутствием пластификатора — дибутилфталата.
Рис. 96. Зависимость модуля
упругости от температуры*
/ — Л1 = 2500, с пл астификато-
ром, 2 — Л1 — 1900, без плас-
тификатора; 3 — М — 1900, с
пластификатором
Рис. 97. Зависимость прочности (/)
и относительного удлинения при раз-
рыве (2) от температуры.
На рис. 95 приведены кривые ползучести непластифицированного
и пластифицированного каучуков при различных температурах.
Температурный ход данных зависимостей явно аномален, и мини-
мальная величина высокоэластической деформации наблюдается
173
при температуре 35° С для непластифицировапного и 40—50°C для
пластифицированного полимеров. Остаточной деформации нс об-
наружено.
Температурный ход модуля также проявляет аномалию и харак-
теризуется четко выраженным максимумом при температурах
35—40° С (рис. 96). Наконец, в области температур 35—40° С на-
блюдаются экстремальные точки на
t, Г/ммг
40 -
1
30-
2
20-
,+-V--+-+
10-
0 -------,------1------1------1 — "|—
10 20 30 40 50 Т, °C
Рис 98. Зависимость равновесного модуля
высокоэластичности полиуретановых элас-
томеров от температуры:
/ — на основе полндиэтиленглнкольадипнната
(мол. вес 815) и ТДИ (сшиватель диэтиленгли-
коль 4- (лицерин — 1 I). 2 — на основе
полиднэтилет ликольадипината (мол вес 1750)
и ТДИ (сшиватель дпэтиленглнколь г гли-
церин — 1*3), 3 — на основе полиэтиленгли-
кольадипината (мол вес 1000) и ТДИ (сши-
ватель дпэтиленглнколь — глицерин — 1 1)
кривых температурной зави-
симости прочности и относи-
тельного удлинения при раз-
рыве (рис. 97).
Согласно классической те-
ории высокоэластичности, ко-
торая в большинстве случаев
правильно описывает темпера-
турную зависимость свойств
натурального и синтетическо-
го каучуков, равновесный мо-
дуль высокоэластичсской де-
формации должен возрастать
с температурой вследствие
увеличения энергии теплового
движения цепей. Вместе с тем
величина равновесной дефор-
мации мало зависит от темпе-
ратуры.
Как видно из приведенных
данных, величина модуля про-
ходит через резко выражен-
ный максимум, что не позво-
ляет характеризовать поведе-
ние полиуретановых каучуков
с помощью кинетической теории. Изменение механических свойств
(прочности и относительного удлинения при разрыве) также носит
немонотонный характер. Исследование температурной зависимости
модуля для полиуретанов, полученных на основе полиэфиров раз-
личного молекулярного веса, показало, что общая аномалия тем-
пературной зависимости сохраняется независимо от молекуляр-
ного веса и наличия или отсутствия пластификаторов. При этом
температурная область проявлений аномалии не изменяется.
Аналогичные результаты получены при исследовании темпе-
ратурной зависимости величины равновесного модуля высокоэластич-
ности для полиуретановых эластомеров на основе полидиэтилен-
гликольадипипатов различных молекулярных весов (рис. 98). По-
лученные данные дают нам основание считать, что температурные
аномалии механических свойств полиуретанов связаны с опреде-
ленными молекулярными процессами, протекающими в области
температур 40—5(V С.
174
Интересно отметить также данные по температурной зависимости
модуля упр\гости жестких полиуретановых пленок, применяю-
щихся в качестве защитных покрытий (рис. 99). В отличие от эласто-
меров в начальный период по-
вышения температуры наблюдае-
тся обычное для стеклообразных
полимеров понижение модуля с
ростом температуры. Дальней-
ший ход кривых аналогичен хо-
ду кривых, приведенному на
рис. 96
Таким образом, в области
температур вблизи 40°С проис-
ходят изменения свойств, свя-
занных с конформационными пе-
реходами, обусловленными рас-
падом части межмолекулярных
связей, прежде всего водородных.
Существенный вклад в свойства
полиуретанов вносит также вза-
имодействие между полиэфирны-
ми блоками. Ослабление межмо-
лекулярных связей при повы-
шении температуры приводит к
увеличению подвижности блоков
и проявлению ими собственной
гибкости.
Итак, воздействие темпера-
туры вызывает изменение общего
числа межмолекулярных водо-
родных и вандерваальсовых свя-
зей. Распад сетки данных связей
эквивалентен увеличению рас-
стояния между узлами простран-
ственной сетки, образованной в
результате физических взаимо-
действий и играющей основную
роль в свойствах полиуретано-
вых эластомеров [1151. Вследст-
Е.кГ/см2-
Рис. 99. Зависимость модуля упругос-
ти полиуретановых пленок от темпера-
туры (NCO/OH --2:1):
/ — на основе тетра» ндрофу рана с окисью
пропилена и аддукта тримегилолпропана
с ТДИ, 2 — на основе оли1 одиэтилевсук-
цината (мол. вес 800), трнметнлолпронана
и ТДИ; 7 — на основе олигодиггилсшлн-
кольадипината (мол вес 800)
вне этого, а также из-за увеличения подвижности отрезков цепей
происходит общее увеличение деформации в области температур
выше 40JC и связанное с ним понижение модуля.
Для пластифицированного полимера область аномалии сдви-
гается в сторону более высоких, а не более низких температур, как
могло показаться с первого взгляда. Это обусловлено сохранением
в пластифицированном полимере прочных связей, распадающихся
при более высокой температуре.
175
Таким образом, процесс распада сетки физических связей и вы-
званное этим изменение гибкости отрезков полимерных цепей, т. с.
фактическая перестройка структуры сетки с температурой, опреде-
ляют аномалии физико-механических свойств, составляющие суще-
ственную особенность полиуретановых эластомеров. Вполне воз-
можно, что такая подвижность структурной сетки и легкость ее
перестройки под влиянием внешних воздействий определяют мно-
гие специфические свойства полиуретанов.
ТЕРМОДИНАМИКА ВЫСОКОЭЛАСТИЧЕСКОЙ ДЕФОРМАЦИИ
ПОЛИУРЕТАНОВЫХ ЭЛАСТОМЕРОВ
При выяснении связи механизма деформации со структурой
полимера особое значение имеют случаи, когда сама структура
тела претерпевает изменения при деформации.
Характерные особенности упругих свойств и поведения поли-
уретанов при деформации, выражающихся в способности к само-
залечиванию дефектов при развитии трещин и высокой сопротив-
ляемости напряжениям, во многом определяются типом пространст-
венной сетки. Она отличается от обычных вулканизационных тем,
что вклад физических взаимодействий в эффективную плотность сет-
ки чрезвычайно велик. Возможность перераспределения межмо-
лекулярных связей в полиуретановых каучуках при деформации —
одна из причин проявления этими полимерами особых свойств.
Поэтому интересно исследовать термодинамику высокоэласти-
ческой деформации полиуретановых эластомеров и установить
вклад внутренней энергии и энтропии в упругую силу, возника-
ющую при деформации эластомера. В этом направлении известно
только несколько работ [69, 72, 75, 117, 192, 325, 326, 328], проведен-
ных, в основном, в последние годы.
Как известно, расчеты энтропийной и энергетической составля-
ющих равновесного напряжения, прилагаемого к образцу каучука
при его растяжении, проводят по прямым зависимости равновесного
напряжения от температуры при различных удлинениях, называ-
емых термоэластическими. При этом используют общее уравнение
кинетической теории эластичности каучуков [137, 2111
। dU , гр /_____\
I dT Vtl~ , dl 'TtV ' 1 \dT lpiK'
где [/ — внутренняя энергия; S — энтропия; / — длина образца;
f — растягивающая сила; fu — упругая составляющая равновесной
силы; fs—энтропийная составляющая равновесной силы.
Изменения внутренней энергии резин обычно весьма незначи-
тельны и если проявляются, то при больших величинах деформации.
176
Конвэй [192], исследовав вклад энергетической и энтропийной
составляющих полиуретанового каучука на основе полиоксипропи-
лен гликоля, тоже отмечал незначительное изменение внутренней
энергии при небольших деформациях. Однако во всех дальнейших
работах [69, 72, 75, 117, 325, 326, 3281, касающихся термодинамики
полиуретанов, указывается на значительные изменения внутренней
энергии даже при небольших деформациях. Так, Танака и сотруд-
ники [3261, изучая зависимость напряжения от температуры поли-
Рис. 100. Зависимость от величины деформации
полиуретановых эластомеров (а)*
У—на основе полиэтнленгликольадипината ТДИ и ТМП;
2 — на основе полиэтнленгликольадипината, нафтнлепди-
изоцианата ибутанднола, 3 — на основе полипропнлеигли-
кольадипината 1ДИ и ТМП; 4 — на основе иолнэтилен-
гликольаднпината ТДИ и этаноламина. <5 — на основе
полипропилен!ликоля ТДИ и этаноламина
уретанов на основе сложных и простых полиэфиров в интервале
температур 40—70° С, показали, что при деформации происходит
как уменьшение, так и увеличение внутренней энергии полиуретанов
в зависимости от природы последних. Зависимость вклада энер-
гетической составляющей в общее равновесное напряжение /{///от
степени вытяжки эластомеров изменяется по абсолютной величине
и по знаку с увеличением степени вытяжки и изменением природы
составляющих полиуретанов (рис. 100). В основном наблюдается
уменьшение энергетической составляющей равновесного напряже-
ния. Полученные результаты объясняются изменением конформаций
цепей при деформации эластомеров. Для полиуретана на основе
нафтилендиизоцианата (кривая 2) резкое изменение /{/// со степенью
вытяжки объяснено кристаллизацией, однако экспериментальных до-
казательств нет.
Исследована термодинамика высокоэластической деформации
линейных полиуретанов, проявляющих высокоэластичсскиесвойства
в интервале температур 20—70° С при больших растяжениях
[289], и получена зависимость равновесного напряжения и его
177
Таблица 57
Состав исследованных полиуретанов
Образец Полиэфир Изоцианат Сшивающий агент Мольное соотноше- ние
1 ПДЭГА (мол вес 1600) 2,4-ТДИ Полиол * — 15100
2 То же То же ДЭА 4- ТЭЛ (1 ♦ 1,5) 1,0: 1,2 :0,1 11 800
3 » » » » То же 1,0 1,3:0,13 11 500
4 » » » » » » 1,0 1,4’0,20 10 200
5 ПДЭГА (мол. вес 800) 2,4 -ТДИ -4- ГМДИ (20:80) Полиол * 1,0. 1,4.0,20 6000
6 ПДЭГА (мол. вес 1600) То же Глицерин -| ДЭГ (3’1) 1,0 1,4.0,20 2900
7 Разветвленный ДЭГА 2,4-ТДИ — 1.0 0,85 4500
8 ПДЭГА (мол вес 900) 2,4-ТДИ Глицерин ДЭГ (1 1) —• 2250
9 То же То же Глицерин + ДЭГ (3:1) — 4200
10 ПДЭГА (мол вес 1730) » » То же — 4200
И ПДЭГА (мол. вес 900) » » Глицерин + ДЭГ (1.1) — 6200
12 ПДЭГА (мол вес 900) 2,4-ТДИ Глицерин ДЭГ (3.1) — 2600
13 ПЭГА (мол. вес 1000) То же Глицерин 4- ДЭГ (1 .1) — 6600
14 То же » » Глицерин 4-ДЭГ (3:1) ч —- 4400
15 ПЭГА (мол. вес 2000) » » Глицерин 4- ДЭГ (1:1) 23 800
16 То же » » Глицерин 4- ДЭГ (3:1) — 7700
17* ** » Глицерин 4- ДЭГ (1 : 1) —- —
18** » » Глицерин 4- ДЭГ (3: 1) — —
19 птмд (мол вес 1000) » » ТМП 1:2:1 5100
20 То же дмди То же 1.2:1 4300
21 ПТМД (мол вес 1540) 2,4 -ТДИ 1,4-Бутандиол -4 ТМП 1 2:1 1900
22 птмд (мол вес 960) То же Глицерин 4-ДЭГ (1 1) — 10 060
23 То же » » Глицерин 4- ДЭГ (3:1) — 3350
24 попд (мол. вес 1010) » » Глицерин i ДЭГ (1:3) __ 13 000
25 ПОПД (мол. вес 2050) » » То же — 18 300
* Полиол на основе адипиновой кислоты, глицерина и диэтиленгликоля
*• Мс нс определяли из-за кристаллизации образцов.
составляющих со степенью растяжения для некоторых полиуре-
танов [325] (рис. 101). В этом случае наблюдается как увеличение
внутренней энергии при растяжении исследованных образцов
(рис 101, б), так и уменьшение (рис. 101, а). Последний факт связан
с кристаллизацией при деформации, подтвержденной рентгеновским
Рис. 101. Изменение равновесного напряжения и его составляющих от
степени растяжения для полиуретанов на основе поликапролактондиола
и л/-ксилилендиамина (а); поликарбонатдиола и этилендиамина (б).
методом. При растяжении полиуретана на основе поликарбонатди-
ола, когда имеет место увеличение внутренней энергии при дефор-
мации, кристаллизация не была замечена даже при растяжении
до 500%.
Мы изучили термодинамику высокоэластической деформации по-
лиуретановых эластомеров на основе простых и сложных полиэфи-
ров [69, 72, 75, 1171. В качестве сложных полиэфиров взяты линей-
ный и разветвленный полидиэтиленгликольадипинат (ПДЭГА) и
полиэтиленглнкольадипинат ((ПЭГА), в качестве простых — поли-
окситетраметилендиол (ПТМД) и полиоксипропилсндиол (ПОПД);
использованы такие диизоцианаты, как смесь 2,4- и 2,6-ТДЦ (Т-102),
гексаметилендиизоцианат (Г-102), дифеннлметандиизоцианат
(ДМИ). Сшивающими агентами были трнметилолпропан (ТМП),
смесь 1,4-бутандиола с ТМП, полиол на основе адипиновой кислоты,
диэтиленгликоля и глицерина, смесь триэтаноламина и диэтанолами-
на (ТЭА + ДЭА). Характеристики исследованных образцов, а так-
же величины средних молекулярных весов между узлами сетки
(М(), рассчитанные по величине равновесного модуля высокоэластич-
иости, приведены в табл. 57.
180
Зависимость величины напряжения от температуры измеряли
на релаксометре, сконструированном по типу 127 ]. Для поддержания
постоянства относительного удлинения в ходе измерений необхо-
димо было знать изменение длины ненапряженного образца с тем-
пературой, которые оказались
незначительны. Поэтому в
дальнейшем при больших уд-
линениях этими изменениями
пренебрегали, по при малых
вытяжках (до 10%) в расчетах
величин равновесного напря-
жения учитывали изменения
длины ненапряженного образ-
ца с температурой. Энерге-
тическую и энтропийную со-
ставляющие рассчитывали по
термоэластическим прямым
(зависимости равновесного на-
пряжения от температуры),
применяя уравнение (VII, 1).
Из зависимости равновесного
напряжения и его состав-
ляющих от температуры
(рис. 102) и степени удлине-
ния (рис. 103) видно, что при
растяжении полиуретанов, как
и для обычных каучуков, на-
блюдается уменьшение энтро-
пии, связанное с уменьшением
300 350 Т°К
• О
<)- ?
Рис. 102. Зависимость равновесного на-
пряжения (о) от температуры для образ-
цов 8 и 13.
(обозначения см. в табл. 57)
числа конформаций цепей при
вытяжке. Но в отличие от
большинства каучуков, где
изменение внутренней энер-
гии при вытяжке близко к
нулю (при растяжении до 100%) [137], для полиуретанов различ-
ной природы относительное удлинение 8—10% уже вызывает рез-
кое уменьшение внутренней энергии во всех случаях. Уменьшение
внутренней энергии натурального каучука имеет место при вы-
тяжке выше 300%, что объясняется кристаллизацией [137].
Рентгенографические измерения деформированных полиурета-
новых образцов не обнаружили кристаллизации в интервале де-
формаций от 0 до 100% (рис. 104, см. вклейку). Поэтому наблюда-
емое уменьшение внутренней энергии при деформации до 100% не
связано с кристаллизацией, как предполагают [325, 328].
Для объяснения наблюдаемого уменьшения внутренней энергии
при растяжении полиуретанов следует учесть то, что в последних на-
ряду с химическими имеется большое число физических связей,
181
б, Г/мм*
6, Г/мм2
1Z0 -
160 -
О
/20 -
ВО-
40-
BO-
40-
л
20
40
'S
f
То
С
60
• -15
о— В
-404
-80-
-40->
и*
160 -|
0
120-
80-
40-
О- /9
•-22
Рис. 103. Зависимость равновес-
ного напряжения и его состав-
ляющих от величины деформа-
ции (а) для образцов 1 (а), 8 и
13 (б), 19 и 22 (в)
(обозначения см. в табл. 57).
вносящих значительный вклад в общую густоту пространственной
сетки. Действительно, ранее мы рассматривали вопрос о при-
роде межмолекулярных связей и установили, что разнообразие
функциональных групп в цепях определяет существование набора
связей, отличающихся по прочности от обычных вандерваальсовых
до водородных. При различных температурах и напряжениях
происходит распад этих связей, который вследствие их разнооб-
разия может, протекать в широком интервале температур и быть
постепенным. Такая лабильность этих связей, а также различное
влияние внешнего напряжения при данной температуре на изме-
нение прочности связи играют существенную роль в термодина-
мических свойствах этих эластомеров. Указанные особенности опре-
деляют легкость перестройки трехмерной сетки полиуретанов при
деформации последнего. Деформация растяжения способствует
сближению отрезков цепей между узлами, что вызывает образова-
0-/5
15
Рис. 105. Зав 1си\юсгь вк низ ^неритической составляющей
в равновесное напряжение от величины деформации.
ние дополнительных физических связей между ними и тем самым
уменьшение внутренней энергии, которое и наблюдается в случае
аморфных полиуретановых эластомеров даже при небольших сте-
пенях растяжения.
Очевидно, повышение гибкости отрезков цепей между узлами
вследствие увеличения молекулярного веса олигоэфирного блока
или Мг за счет меньшего количества сшивающего агента будет
увеличивать возможность образования физических связей при де-
формации в результате сближения отрезков цепей друг с другом.
Зависимость вклада энергетической составляющей в общее напря-
жение от степени растяжения полиуретана на основе полиэтилен-
гликольадипината различных молекулярных весов подтверждает
это предположение (рис. 103).
Из зависимости fu/f от величины Мс для полиуретанов на основе
сложных и простых полиэфиров (рис. 106) видно, что fu/f коррели-
рует с Мс При уменьшении Мс, т. е. при увеличении густоты сетки,
степень изменения внутренней энергии становится меньше. Веро-
183
ятно, при деформации сильно сшитых эластомеров вследствие боль-
шей жесткости отрезков цепей между узлами уменьшается воз-
можность формирования дополнительных физических связей.
Для полиуретанов на основе сложных полиэфиров изменение
fu/f более заметно, чем в полиуретанах на основе простых поли-
эфиров. Причина наблюдаемого явления заключается в различии
Рис. 106 Зависимость fu/f от Л4С для аморфных полиуретанов на основе сложных
(а) и простых (6) полиэфиров при растяжении от 10 до 60%.
долей химической и физической сшивок в общей густоте сетки этих
эластомеров. Если в полиуретанах на основе сложных полиэфиров
основная роль в сетке принадлежит физическим связям, то в эласто-
мерах па основе простых полиэфиров вклад физических связей
будет меньше.
Анализ рис. 106, б показывает, что изменение внутренней энер-
гии в полиуретанах на основе олигоэтилен-и олигодиэтпленгликоль-
адипината примерно одинаково, несмотря на различные величины Мс.
Вероятно, большая гибкость отрезков цепей в полиуретанах на
основе олигодиэтилеигликольадииината способствует образованию
большого числа межмолекулярных связей в ходе деформации рас-
тяжения даже при меньшем Mct чем у полиуретанов на основе
олигоэтиленгликольадипината. Это вызывает большее изменение
внутренней энергии в первом случае, чем во втором, несмотря на
то, что Мс полиуретана на основе олигодиэтиленгликольадипина-
та меньше.
184
Некоторые из исследованных нами
полиуретанов самопроизвольно закрис-
таллизовывались со временем [75]. Была
изучена термодинамика деформации
этих образцов (рис. 107), которая со-
провождается увеличением как энтропии,
так и внутренней энергии. Можно счи-
тать, что в данном случае происходит
разрушение кристаллических образова-
ний, т. е. уменьшение степени упоря-
доченности, ведущее к увеличению эн-
тропии. При этом естественно ожидать
рост внутренней энергии, который обыч-
но происходит при деформации кристал-
лических тел.
Таким образом, полученные данные
говорят о том, что изменение внутренней
энергии при деформации полиуретано-
вых эластомеров зависит как от природы
и густоты сетки полиуретана, так и от фа-
зового состояния последнего.
Рис. 107. Зависимость равно-
веского напряжения / и его
составляющих [у и fs от сто
пени удлинения для образ-
ца 18
(обозначение см в табл. 57)
Очевидно, деформация указанных
эластомеров в интервале более высоких
температур приведет к изменению термо-
динамических функций, так как известно,
что физические связи, концентрация
которых велика в полиуретанах, очень
чувствительны к температурным воздействиям. Поэтому мы исследо-
вали термодинамику деформации полиуретановых аморфных эласто-
меров в интервале температур 100—60° С [117] и сравнили характер
изменения термодинамических функций в этом случае с таковым в
интервале более низких температур (20—60° С).
Предварительно провели изометрическое нагревание исследуе-
мых образцов при различных нагрузках для установления интер-
вала температур, в котором не происходит разрушение сетки хими-
ческих связей, в результате чего получили кривые изометрического
нагрева полиуретановых эластомеров (рис. 108). Вначале происходит
рост напряжений в образцах, обусловленный усилием теплового
движения цепей при повышении температуры. С увеличением пер-
воначальной нагрузки на образец изменение напряжения в поли-
уретанах с ростом температуры становится более заметным. За-
тем в интервале температур 100—120° С (в зависимости от
нагрузки на образец) начинается резкий спад напряжений.
Известно, что полиуретаны характеризуются наличием хими-
ческихсвязей различных типов. Предполагается, что биуретовые и
аллофанатные связи наименее прочные [302]. Имеются данные о том,
что при температурах 106° С и выше начинается диссоциация алло-
185
фанатны< связей [2491. Возможно, это и вызывает резкий спад напря-
жений при температурах выше 100°С. Для подтверждения того, что
в данном случае происходит распад химических связей, исследована
обратимость процесса изометри-
ческого нагрева (рис.109.)Наизо-
метрических кривых наблюдает-
ся гистерезис. В области темпе-
ратур 20—100°С величина гисте-
резиса незначительна. Большой
гистерезис в интервале 20—
130°С свидетельствует о необра-
Рнс. 108. Кривые изометрического нагрева для полиуретанов:
а — на основе ПЭГА (образец 13, табл. 57), б — на основе ПДЭГА (образец 10).
гимости деформации, связанной с необратимым разрушением сетки
химических связей. Спсдовательно, термодинамику деформации
можно проследить только до 100° С, т. е. до температуры, при кото-
рой еще не разрушается хими-
ческая сетка связей.
Анализ термоэлас ги чески х
прямых для полиуретана на
Рис. 109. Кривые изометрического на-
грева для образца 1 (табл. 57) в раз-
ных интервалах температур-
/, 2 — под нем и спад температуры в нигер
вале 20 — 100° С. 4 — подъем н спад тем-
пературы в ши ер вале 20 —130° С
основе сложного полиэфира (рис. ПО) и зависимости равновесно-
го напряжения f и его составляющих от степени удлине-
186
ния (рис. Ill) показывает, что при деформации полиуретановых
эластомеров происходит уменьшение как энтропии, так и внутренней
энергии.
Рис. ПО. Термоэластические прямые для образца
14 (табл 57) в разных температурных интер-
валах
С другой стороны, термоэластические прямые в интервале темпе-
ратур 100 — 60° С лежат ниже, чем при 60 —20° С. Очевидно, на-
гревание образца до 100° С приводит к диссоциации большей части
физических связей. Иначе, мы имеем дело с пространственной сет-
кой, обладающей меньшей концентрацией физических связей, что
Рис. 111 Зависимость равновесною
напряжения и его составляющих от
величины деформации (образец 14,
табл 57):
1 2 ? /. /s и 1Ц в облг-сги 60 — 100° С,
<5 Ь — /. |'s и fu в области 20—60° С.
ведет к уменьшению эффективной
плотности сшивки и, следователь-
но, к увеличению эффективной гиб-
кости цепей между химическими
узлами сетки.
Мы отмечали, что при деформа-
ции полиуретановых эластомеров
с мсныпей плотностью сшивки из-
менение внутренней энергии при
деформации значительно больше,
чем в случае эластомеров с боль-
шой плотностью сшивки. Анало-
гично, деформация эластомеров при
высоких температурах сопровож-
дается большим изменением внут-
ренней энергии, чем при низких
температурах (рис. 111, 112 а). Это
объясняется тем,что при вытягива-
нии образца в области высоких тем-
ператур, когда часть физическихсвя-
зей разрушена и отрезки между уз-
187
лами обладают большей гибкостью, создается большая возможность
восстановления старых и образования новых физических связей
при сближении отрезков цепей, а энергетическая составляющая
равновесного напряжения возрастает.
При деформации полиуретановых эластомеров на основе простых
полиэфиров при повышенных температурах изменение внутренней
энергии также больше, чем в интервале низких температур (рис. 112,
а, б). Более резкое различие между значениями fu/f в разных тем-
пературных интервалах при деформации полиуретана с меньшей
' 0,2 0,4
В- — - —у
•'•-20-60°С
о,о-бО-ЮО°С
Рис. 112. Зависимость fu/f от степени вытяжки % для полиуретанов на основе слож-
ного (а) и простого полиэфиров (б, в):
а— образец, 14, б — образец 23; « — образец 22 (обозн шения см. в табл. 57)
эффективной плотностью сшивки (рис. 112, б, в) естественно в свете
соображений об изменении гибкости отрезков цепей при разных
плотностях сшивки трехмерной сетки.
Известно, что измерение зависимости равновесного напряжения
растянутого полимерного образца от температуры лежит в основе
метода расчета температурного коэффициента размеров свободной
полимерной цепи в невозмущенном состоянии 1212—214]. Метод
основан на теории упругости каучуков, предполагающей незави-
симость межмолекулярного взаимодействия от деформации, и на
принципе аддитивности [215] вкладов свободной энергии отдельных
цепей в общую свободную энергию системы. Расчеты основаны на
уравнении
fu/fT = - [d In (f/T)/dT\vl = d In FJIdT.
Если измерения напряжение—температура проводятся при
постоянном давлении, данное уравнение записывается в виде
1- d In (f/T) dT\P l = d InffldT + , (VII,2)
гдсб/1пг02/бГГ —температурный коэффициент среднеквадратичного
расстояния между концами цепи; [3— кубический коэффициент
расширения эластомера; % — степень вытяжки эластомера.
Согласно [212—214, 184, 265], fu/fT не зависит от удлинения,
а также от степени сшивки натурального каучука, что полностью
совпадает с теорией.
188
Обработка [69] полученных по уравнению (У11,2) данных показала
(табл. 58), что значения величины fu/fTотрицательны во всех случа-
ях,что говорит об уменьшении размеров невозмущенной полимерной
цепи с температурой. Вместе с тем величины fulfT сильно разли-
чаются по абсолютной величине, что казалось бы вполне естествен-
ным, если учесть различный состав исследованных полиуретанов.
Но эти различия сохраняются и в случае одного и того же по каче-
ственному составу полиуретана (образцы 14, 15) с разной степенью
сшивки. Зависимость fu/fT от степени сшивки наблюдалась и тогда,
когда объектами изучения были полиэтилен, полипропилен и их
сополимеры [284].
Метод расчета температурного коэффициента размеров свобод-
ной полимерной цепи основан на предположении, что изменение
внутренней энергии при деформации эластомера обусловлено из-
менением только внутримолекулярного взаимодействия; изменение
межмолекулярного взаимодействия не учитывается. Между тем
нельзя допустить, что значительное изменение внутренней энергии,
наблюдаемое при растяжении полиуретанов, вызвано увеличением
внутримолекулярного взаимодействия, так как вытягивание от-
резков полимерных цепей способствует удалению полярных групп
в цепи друг от друга и снижает вероятность их внутримолекулярного
взаимодействия. Вероятно, в процессе вытяжки полиуретанов про-
исходит сближение отрезков цепей между узлами, приводящее к
образованию дополнительных физических узлов в сетке из-за вза-
имодействия полярных групп соседних цепей. Таким образом, умень-
шение внутреннейэнергии при вытяжке полиуретановых эластомеров
обусловлено, в первую очередь, изменением межмолекулярного вза-
имодействия отрезков полимерных цепей. Изменение внутренней
энергии тем больше, чем больше расстояние между узлами в сетке
В связи с изложенным для полиуретанов нельзя получить при-
емлемые величины изменений среднеквадратичного расстояния меж-
ду концами свободной цепи изданных напряжение — температура.
Проведенные исследования показывают, что наличие в полиуре-
танах физических связей различных типов приводит к иному пове-
дению при деформациях и температурных воздействиях этого
класса каучуков по сравнению с натуральным и другими каучу-
ками [72, 325, 326, 3281 Данные особенности связаны с наличием
подвижной сетки физических связей и с ее перестройкой в ходе де-
формации и температурных воздействий, при которой возникает
новая структура, отвечающая деформированному состоянию. В тех
случаях, когда скорости деформации и перестройки сетки будут
сопоставимы, устанавливается новое равновесие в системе, что
очень важно при рассмотрении механизма деформации полиуре-
тановых эластомеров. Действительно, при наличии подвижной сетки
на каждой стадии деформации необходимо дальнейшее разрушение
сетки в отличие от других эластомеров, где деформация не со-
провождается восстановлением первоначально разр} шенных
189
Таблица 58
Равновесные напряжения и их составляющие полиуретанов различной
степени сшивки
Образец f. кГ/см2 II "г 7 II II
1 1,12 1,01 —2,40 -1,39 -1,37 15 400
1,24 1.53 —4,78 —3,25 —2,12
1,44 2,26 -7,17 —4,91 -2,17
1,76 3,62 — 14,14 -10,52 -2,9
2 1.19 0,97 —4,52 -3,55 —2,94 11 800
1,53 2,83 —6,79 —3,96 -1,39
1.80 3,80 —9,21 —5,41 — 1,42
2,00 4,13 — 13,50 —9,37 —2,27
3 1,13 1,16 —4,30 —3,14 —2,70 11 500
1,20 1,36 -5,24 —3,88 —2,85
1,32 2,17 —8.33 -6,16 —2,84
1,84 3,37 — 13,55 -7,02 —2,90
4 1,29 2,90 -7,20 -4,3 —1,48 10 200
1,35 3,51 -8,00 -4.49 —1,28
1,45 4,90 — 10,70 -5.80 -1,18
1.60 5,30 — 13.70 —8,40 —1,58
5 1,0.3 1.19 — 1.59 -0,40 —0,34 6000
1,08 2,30 —2,87 -0,57 —0,25
1.13 3,37 -4,12 -0.75 —0,22
1,17 4,25 —5,34 — 1,09 -0,26
1,33 8,98 -11,17 -2.19 —0,24
6 1,04 1,31 -2,34 — 1,03 —0,78 2900
1,06 2,28 —4,36 —2,08 -0,91
1,07 3,18 —5,96 —2,78 —0,87
1,13 4,50 —8,24 -3,74 -0,83
7 1,07 1,30 — 1,70 —0,40 —0,30 4500
1,09 1,55 —2,43 -0,88 —0,57
1,11 2,10 —3,40 —1,30 —0,62
1,24 2,98 -4,66 -1,68 -0,56
8 1,04 1,60 — 1,41 0,19 —0,12 2250
1,08 3,30 -3,53 —0,23 —0,07
1,17 6,85 —7,52 -0,67 —0,10
1,23 7,60 —9,32 —1,72 —0.23
1.31 8,76 — 10,75 — 1,99 —0.23
9 1,06 1,10 — 1,25 -0,15 —0,14 4200
1,09 1,90 —2,80 —0,90 —0,50
1,21 4,60 —6,30 — 1,70 —0,37
1,26 4,85 —6,62 —1,77 —0,37
1,33 5,40 —8,14 —2,74 —0,50
10 1,06 1,10 —1,25 -0,15 —0,14 4200
1,09 1,90 —2,80 —0,90 —0,50
1.21 4,60 —6,30 —1,70 —0,37
1,26 4,85 —6,62 — 1,77 —0,37
1,33 5,40 —8,14 —2,74 -0,5
190
Продолжение табл 58
Образен X f, кПсм* II — "Г i и уЬ •? г full?
11 1,03 0,78 0 0.78 1,0 6200
1 08 1,70 —2,51 —0,81 —0,48
1,15 2,90 —4.10 —1,20 —0.41
1,21 4,50 —7,75 —3,25 —0,72
1,35 6,15 —10,18 —4,03 —0,66
12 1,02 1,18 -1,35 —0,15 —0,13 2600
1,06 2,70 —3,19 —0,49 —0,18
1,13 4,50 —5,50 -1,00 —0,22
13 1,10 1,25 — 1,44 —0,19 —0,15 6600
1,14 1,70 —2,12 -0,42 —0,25
1,26 2,88 -3,62 —0.79 —0 28
1,42 4,25 —6,74 -2,49 -0,59
1,52 5,60 —8,61 —3,01 -0,54
14 1,06 1,80 — 1,66 0.15 0.08 4400
1,16 3,10 —3.22 0,12 -0,04
1,28 5,25 —6,47 — 1,22 —0,23
1,44 7,56 —9,31 — 1,75 —0,2$
15 1,16 0,40 — 1,12 —0,72 — 1.80 23 800
1,39 0,90 —2,78 — 1,88 —2,10
1,58 1,20 -4,18 -2 98 -2,48
2,09 1,60 —5,63 -4,03 —2,52
16 1,10 0,65 -1,16 -0,51 -0,8 7700
1,25 1,68 —4,63 -2,5 — 1,7
1,49 2,85 —8,46 -5,61 -1,9
17 1,008 3,06 17,49 20,55 6,71 Кристал-
1,009 4,80 19,92 24,72 5,15 лический
1,020 6,40 21,80 28.20 4,41
18 1,010 2,95 6,87 9,72 3,29 Кристал-
1,015 4,70 11 00 15,70 3,34 лический
1,018 5,93 12,31 18,84 3,07
1,031 6,62 18,23 24,85 3,75
1,039 6,65 20,55 27,20 4,09
19 1,11 1,36 —3,05 —1,69 — 1,24 5100
1,32 2,98 —6,18 —3,20 -1,07
1,43 3,78 —7,57 —3,79 — 1,00
1,67 5,27 — 10.50 —5,23 —0,99
20 1,04 0,96 —2,54 — 1,58 — 1.64 4300
1,10 2,17 —4,78 -2,61 — 1,20
1,15 3,31 —7,33 -4,02 — 1.21
1,69 8,25 — 15,34 -7,09 —0,86
21 1,05 2,48 —3.73 — 1,25 —0,50 1900
1,10 4,48 —6,28 —1,80 —0,40
1,17 6,69 —8,66 —1,97 —0,29
’01
П р о д о л ж с н и е т а б л. 68
Образец X I, кГ/см* <л II *т л - II fu/fT мс
22 1,07 1,23 —3,2 —1,98 —1,60 10 060
1,22 3,85 —7,24 —3,39 —0,88
1,30 5,30 — 11,15 —5,85 — 1,10
1,47 6,00 — 13,07 -6,33 —0,96
1,71 8,20 —15.82 —7,62 —0,93
23 1,04 1,85 —1,36 0,49 0,26
1,09 3,55 —4 56 —1,01 —0,28
1,16 6,50 —9,56 —3,06 —0.47
1,21 9,80 -14,70 —4.80 —0,49
24 1,20 1,35 —2,90 —1,55 —1,15 13 000
1,22 1,82 —3,67 — 1,86 —1,02
1,30 2.55 —5 64 —3,09 -1,21
1,38 3,60 -8.08 —4,48 — 1,24
25 1.14 0,65 — 1,29 -0,64 —0,92 18 300
1,33 1,30 —2,96 —1.66 —1,30
1,39 1,65 —3,90 —2,25 —1.40
связей. Мы полагаем, что сравнительная легкость разрушения и
восстановления сетки объясняет способность полиуретанов к зале-
чиванию дефектов при деформации, которая может рассматриваться
как процесс тиксотропного изменения пространственной сетки.
ТЕПЛОВЫЕ ЭФФЕКТЫ ПРИ ДЕФОРМАЦИИ ПОЛИУРЕТАНОВ
Известно, что механическая деформация полимеров неизменно
сопровождается тепловыми эффектами, знак которых зависит от
вида деформации, фазового и физического состояний деформируемою
полимера.
Исследование тепловых эффектов деформации представляет
существенный интерес, поскольку дает важную информацию о тер-
модинамике и молекулярной природе этого процесса. Однако работ
в данном направлении мало, что обусловлено трудностями коли-
чественного определения тепловых эффектов, сопровождающих про-
цесс деформации, потому что величины их весьма незначительны.
Первые существенные работы в этом направлении проделаны
Мюллером и сотрудниками [99, 2751, разработавшими газонапол-
ненный дифференциальный микрокалориметр.
Нами проведено определение тепловых эффектов, сопровождающих
деформацию полиуретановых эластомеров [47, 751, на специальном
192
блоке, позволяющем одновременно записывать дифференциальную
кривую температур и изменение напряжения при деформации и
релаксации образца. Объектами исследования служили полиуретаны
на основе 2,4-ДТИ и полиэтнленгликольадипината (ПЭГА) различ-
ного молекулярного ве-
са (табл. 59).
ПЭГА легко кристал-
лизуется, что может при-
вести к кристаллизации
и полиуретанов на их
основе. Однако, как от-
мечалось в [15], само-
произвольная кристал-
лизаци я полиуретанов,
Таблица 59
Характеристика исследованных полиуретанов
Обрати Полиэфир (. Н1МВНК>1ЦИЙ a ip ir (Д 31 — глицерин)
1 ПЭГ VI 000 1 3 3800
2 ПЭГА-1000 1 I 5400
3 ПЭГА-2000 1 1 23 800
содержащих кристалли-
зующиеся олигоэфирные блоки, протекает лишь при достаточной
длине олигоэфирпых блоков, когда жесткие диизоцианатные бло-
ки макроцепи уже не могут воспрепятствовать кристаллизации.
Действительно, даже после полугодичной выдержки при комнат-
ной температуре процесс самопроизвольной кристаллизации на-
ной
блюдается только для об-
разца 3 (см. табл. 57). Об
этом свидетельствует ха-
рактер термограмм [42, 751.
Характер тепловых эф-
фектов деформации образца
1 (рис. 113) аналогичен та-
ковому для высокоэласти-
ческой деформации других
полимерных систем, т. е.
растяжение образца сопро-
вождается выделением теп-
ла. Это объясняется умень-
Рис. 113 Термограмма растяжения и сокра- шеиием энтропии системы,
щепля образца 1 (табл. 57). происходящим вследствие
уменьшения числа конфор-
маций макромолекул в полимере в ходе процесса. Снятие напряже-
ния и сокращения образца сопровождается поглощением тепла.
Следовательно, исследованный поли\ретап при деформации про-
являет свойства энтропийно-эластичного материала.
На рис. 114 приведена количественная зависимость площадей
пиков тепловых эффектов растяжения от температуры в двух пос-
ледовательных циклах деформации для образцов 1 и 2.
При повторном цикле деформации растяжение обоих образцов
сопровождается большим экзотермическим эффектом, чем при пер-
вичном растяжении; возрастают значения тепловых эффектов и с
ростом тем пер ату р ы.
193
Рис. 1И 1 емнературная за-
висимость п.»о»цали‘| экзо1 ер-
нических пиков ргктяжеиия
образцов 1 (кривые / Л) и 2
(кривые 2, 4) Сплошные ли-
нии — 1 цикл растяжения,
пунктмрньс—II цикл рас-
тяжения
(обозначения см п т'б( 57)
зцов 1, 2 и 3 идентичен, т
Для полях ретанов со значительным количеством физических свя-
зен различия юпловых эффектов при новюрных циклах или при по-
вышении температуры можно объясни н> тем. 1,го после первичною
процесса разр) шснпя связей, сопровождающегося поглощением теп-
ла, во втором цикле преобладают толью
энтропийные нзменения То же имей
место, если часть физических связен
ра ф\ шасгся при повышении темпсра-
т у ры.
Поскольку в нашем случае процесс
деформаций термодинамически неравно-
вссный, наблюдается некоторое ра->лн-
чие тепловых эффектов растяжения и со-
кращения.
Характер термограмм образцов I и 2
аналогичен, однако из-за меньшой плон
пости химической сшивки последний об-
разец более эластичен, что обусловли-
вает большие значения тепловых эффек-
тов и меньшие значения напряжения
образца при одинаковых степенях де-
формации
Даже полугодичная выдержка образ-
цов 1 и 2 при комнатной температуре не
приводит к изменению первоначального
характера тепловых эффектов при их
деформации.
Вместе с тем характер тепловых эф-
фектов при деформации образца 3 суще-
ственно зависит от длительности ею
выдержки, что, безусловно, вызвано
развитием в полиуретане с течением
времени кристаллической структуры раз-
личной степени совершенства. В началь-
ной стадии (непосредственно после полу-
чения или upoiрева при 50е С) харак-
тер тепловых эффектов деформации обр а -
. е. полиурщан ведет себя, как энтре-
п и й ноэл асти ч и ы й матер и ал.
Однако после двухмесячной выдержки характер тепло-
вых эффектов деформации образца 3 существенно меняется
(рис 115).
В отличие от образца 1 растяжение образца 3 сопровождается
поглощением тепла. Такое явление может быть связано с одновре-
менным протеканием процесса упругой деформации, имеющей место
в начальной стадии растяжения и обусловленной наличием в об-
разце 3 кристаллической фазы, и процесса разрушения кристалли-
194
ческой фазы, который может протекать при растяжении и тоже со-
провождается поглощением тепла.
При этом в отличие от образцов 1 и 2 после быстрого растяженп5(
образца 3 наблюдается релаксация напряжения, которая сопро-
вождается выделением тепла, о чем говорит широкий экзотерми-
ческий максимум на дифференциальной кривой температуры. Эю
указывает на то, чго наряду с
описанными процессами в ходе рас-
тяжения развивается, по мере роста
содержания аморфной фазы, обыч-
ный процесс высокоэластическои
деформации, теплота которого сна-
чала полностью комп('1'<’ир\ ется теп-
Рис. 115 Термограмма еформа-
ни и образца 3, выдержанного
два месяца при комнатной тем-
исрату ре.
Рис 116. Термограмма деформации
образца 3, выдержанного четыре
месяца.
лотами указанных выше двух процессов, являющихся превалиру-
ющими в начальной стадии деформации.
Однако процесс разрушения кристалл и чекой фазы со временем
приостанавливается, а развитие высокоэластической деформации
продолжается, что и отражается на характере фиксируемого термо-
граммой теплового эффекта.
Следовательно, образец 3 при растяжении ведет себя одновремен-
но как энерго-, так и энтропийно-эластичный материал.
Однако сокращение деформированного образца 3, а также об-
разцов 1 и 2 сопровождается поглощением тепла (рис. 116), т. е.
после заданной деформации, приводящей к разрушеииюкрпсталли-
ческой фазы, полиуретан ведет себя как энтропийно-эластичны!
материал.
Интересно, что более длительная выдержка (четыре месяца)
образца 3 приводит к новому изменению характера тепловых эф-
фектов его деформации (рис. 116). При этом, как и для образцов с
двухмесячной выдержкой, растяжение тоже сопровождается погло-
щением тепла, однако сокращение идет уже, наоборот, с выделением
тепла Такое явление обусловлено дальнейшим ростом степени кри-
сталличности и совершенства кристаллической фазы, приводящей
195
к тому, что тепловые эффекты деформации полиуретана определяют-
ся уже преимущественно наличием кристаллической фазы.
Таким образом, основным процессом, вызывающим эндотерми-
ческий эффект при деформации, является процесс упругой деформа-
ции. Несоответствие площадей пиков при растяжении и сокращении
связано с сокращением частично амортизированного образца. Сле-
дует отметить, что на более высокую и совершенную кристалличность
образца 3, выдержанного в течение четырех месяцев, указывают
повышенные значения напряжений, соответствующие такой же сте-
пени растяжения, что и в образце, выдержанном в течение двух
месяцев.
Поскольку при данной степени растяжения процесс разрушения
кристаллической фазы протекает нс столь глубоко, то естественно,
что она ограничивает развитие высокоэластической деформации. Это
вытекает из отсутствия явно выраженного экзотермического эффек-
та после растяжения, а также из превалирующего протекания экзо-
термического процесса обратимой упругой деформации при сокраще-
нии образца. Итак, образец 3 после четырехмесячной выдержки
при комнатной температуре ведет себя при растяжении и сокраще-
нии преимущественно как энерго-эластичный материал.
Из изложенного вытекает, что увеличение в сшитых полиэтилен-
гликольадипипатуретанах длины олигоэфирного блока, обуслов-
ливая протекание в нем кристаллизационных процессов, приводит к
изменению характера их деформации и молекулярного механизма
этого процесса. Характер и механизм деформации для полиурета-
нов с достаточно длинными олигоэфирными блоками сильно зави-
сит от глубины кристаллизационных процессов и совершенства об-
разовавшейся кристаллической фазы.
РЕЛАКСАЦИОННЫЕ СВОЙСТВА ПОЛИУРЕТАНОВ
Хотя многие специфические особенности полиуретанов связаны
с их релаксационным поведением, работ в этой области мало. Релак-
сационные процессы в полиуретановых эластомерах по трем сле-
дующим причинам должны отличаться от таковых в других эласто-
мерных системах :большой вклад физических взаимодействий в эффек-
тивную плотность сетки; возможность перестройки структуры сетки
при деформации; особенности строения полиуретановой цепи, явля-
ющейся по существу молекулой блоксополимера.
В многочисленных работах по механическим свойствам полиуре-
танов приводятся отдельные сведения о релаксации напряжения в
тех или иных материалах, по эти результаты не дают еще представ-
ления об особенностях релаксационного поведения и зависимости
его от напряжения, эффективной плотности сетки, температуры,
длины и природы олигомерного блока и т. п. Между тем, рассмотре-
ние этого вопроса важно не только для понимания специфики поли-
196
уретанов, но и с общих позиций теории вязкоупругости перестраи-
вающихся сеток [135]. Процессы химической релаксации полиурета-
новых резип связаны с распадом химических связей, образующих
пространственную сетку, и с обусловленной этим фактором релак-
сацией напряжений [188, 282|. Так, для шести типов резин, в кото-
рых реализуются различные связи, получены кривые релаксации,
из которых вычислены времена химической релаксации (см. рис. 63,
табл. 34). Эти данные показывают, что наиболее слабыми связями в
полиуретановых резинах должны быть связи типа дизамещенной
мочевины или биуретовые. Возрастание времени химической релак-
сации при 120° С в десять раз может быть достигнуто при замене
этих связей на другие. Найдено, что релаксация напряжений имеет
одинаковую скорость как у эластомеров на основе простых поли-
эфиров, так и у эластомеров на основе сложных полиэфиров, т. е.
сложноэфирные группы не оказывают влияния па скорость релак-
сации напряжений [188, 282].
На низкомодульной полиуретановой композиции с высокой про-
зрачностью и двулучепреломляющей чувствительностью проведено
сопоставление процессов механической и оптической релаксации,
основанное на применении принципа температурно-временной супер-
позиции и теории Вильямса — Ланделла — Ферри (ВЛФ) [1621.
Для такой специфичной и нехарактерной в целом для полиурета-
нов системы рассчитаны величины релаксационного модуля, факто-
ра приведения ат и другие характеристики, определяемые теорией.
Результаты сопоставления экспериментальных данных совпали с
рассчитанными по теории ВЛФ. Следовательно, в работе показана
применимость теории ВЛФ к системам полиуретанового ряда, но
общие закономерности или особенности не установлены.
Некоторые динамические механические свойства исследованы
для полиуретанов на основе поли-е-капролактона [2021. Изучение
механических потерь при разных температурах позволило свя-
зать их с молекулярным весом полиэфирного блока. Интересно,
что при этом наблюдалась своеобразная зависимость механичес-
ких потерь от температуры (рост и достижение максимума при Тс,
дальнейшее уменьшение и вновь рост). В области выше температуры
стеклования полиэфиров потери растут с уменьшением молекуляр-
ного веса.
Приготовлены полиуретаны в виде гомологической серии в от-
ношении концентрации полярных групп и плотности узлов, и в об-
ласти перехода от стеклообразного к эластическому состоянию ис-
следованы их вязкоупругие свойства. Установлено, что температура
стеклования и пологость области вязкоупругой дисперсии умень-
шаются с увеличением концентрации уретановых и биуретовых
связей и плотности сшивки. В каждом случае обнаружено большое
расхождение между значениями и коэффициентами расширения
свободного объема, вычисленными по теории ВЛФ и уравнению
Дуллитла (параметр В принят за единицу). Для объяснения данных
197
расхождений можно предположить, что В уменьшается с температу-
рой вследствие термической диссоциации вторичных связей между по-
лярными группами, снижающей величины элемента цепи, участвую-
щего в молекулярном движении.
В работе 12801 впервые изучены релаксационные свойства сетча-
тых полиу ротанов на основе простых полиэфиров с целью связать их
с химической природой и молекулярным весом полиэфира, а так-
же с условиями реакции синтеза, в ходе которой закладывается
структура полиуретана. В качестве объектов выбраны сшитые
полиуретаны на основе простых олнгоэфиров различной химической
природы, 4,4'-дифенилметандиизоцианата и триметилолпропана
(TMII)
Провесе получения полиуретанов можно разделить на две ста-
дии: синтез макродиизоцианата и образование трехмерной сетки в
результате взаимодействия МДИ с ТМП. Для синтеза МДИ исполь-
зовали ПНГ (мол. веса 500, 1000, 1500, 2000 и 3000), а также поли-
окситетраметиленглпколь (ПТГФ, мол. веса 645, 960, 1450, 1930,
3000) и 1. Г-дифенилметандиизоцианат, взятые в соотношении 1 : 2
12801. Сшивку полиуретанов проводили в результате взаимодейст-
вия МДИ и ТМП в молярном соотношении NCO/OH--1.
О релаксационных свойствах сшитых полиуретанов судили по
данным измерения релаксации напряжения пленочных образцов
при постоянной деформации одноосного растяжения и комнатной
температуре. Для этой цели снимали релаксационные кривые, по
которым рассчитывали параметры релаксационного поведения по
уравнению Кольрауша [2501
а(/)=.аоэ4_а0е 1 4 гЕ^е ’ (VII,3)
где (Е...) — равновесная часть напряжения (модуля упругости);
о0 (Eft) — релаксирующая часть напряжения (модуля упругости);
т — время релаксации; f —относительная деформация образцов;
К — константа материала.
Измерения проводили на релаксометре с жестким динамомет-
ром, обеспечивающим постоянство деформации в течение опыта, и
автоматической регистрацией измеряемых величин. Эксперименталь-
ные значения Мс^ и РМ определяли из уравнения (V, 1) по значе-
\ v 'э
ниям равновесного модуля упругости Еоо, теоретические значения
Л4с>т и -j-j рассчитывали по уравнению (V, 2).
Из экспериментальных кривых релаксации напряжения при
постоянной деформации для двух серий образцов сшитых поли-
уретанов с различной густотой сетки (рис. 117) по методике
[105, 121J вычислены параметры уравнения Кольрауша. По най-
денным значениям этих параметров получены величины на-
198
пряжения в различные моменты времени (точки на графиках). Как
видно из рис. 117, рассчитанные значения у довлетворнтелыю совпа-
дают с экспериментальными кривыми, что творит о применимости
уравнения Кольрауша для описания релаксации напряжения в ис-
следованных полиуретанах. Положение и характер релаксаци-
Рис. 117. Экспериментальные кривые релаксации напряжения полиуретанов на
основе И Г ГФ (а) и ППГ (б)
I — с молекулярным весом 650. 2 МьО Л - 1 <50, 4 — ПВО, 5 — 3000, б — 500 7 —
1000, 8 — 1500 9 2000; 10 — 3000
онных кривых показывают, что длина олнгоэфирного блока в иссле-
дованных полиуретановых сетках оказывает значительное влияние
на параметры релаксационных свойств. Рассмотрим последние
более подробно.
Из зависимости равновесного модуля упругости Б,, от молекуляр-
ного веса олигоэфира для полиуретанов па основе ПТГФ и ППГ
(рис. 118) видно, что изменение молекулярного веса олнгоэфирного
блока оказывает существен ное влияние на равновесный модуль упру -
юсги. Это связано с изменением i устоты прос1ранственной сетки, за-
висящей от длины олигоэфнрпо! о звена. Увеличение молекулярного
веса олнгоэфирного блока (уменьшение густоты сетки) от 500 до
3000 приводит к постепенному уменьшению равновесной упругости
пространственной сетки в 10—15 раз Следует отметить, что обе кри-
вые очень близки друг к друц, т. е. равновесный модуль упруго-
сти практически нс зависит от химической природы эфирной
199
компоненты исследованных полиуретанов. Данный результат вполне
согласуется с выводами кинетической теории высокоэластичности,
Рис. 118. Зависимость равновесного
модуля упругости Гос от молекуляр-
ного веса олигоэфира Моз для по-
лиуретанов на основе ПТГФ (/) и
ППГ (2).
согласно которой упругость моле-
кулярной сетки связана только с
густотой сетки.
Из рис. 118 следует, что для
обоих полиуретанов наиболее рез-
кое снижение равновесного модуля
происходит при увеличении моле-
кулярного веса олигоэфир но го бло-
ка от 500 до 1500. Дальнейшее
увеличение Л1оэ от 2000 до 3000
незначительно уменьшает ЕСс. Из
этого вытекает, что при значении
7Иоэ= 2000 и выше равновесная уп-
ругость практически не зависит от
размера олигоэфирной составляю-
щей трехмерной сетки полиуретана.
Кинетические параметры урав-
нения Кольрауша (релаксирую-
щая часть модуля упругости EQt время релаксации т и параметр К)
существенно зависят от Л4оэ(рис. 119). Однако в отличие от модуля
Ео.кГ/си2'
1.21
200-
150-
100-
50-
0,5-
0,4-
0.3-
01
к
0,6-
1000
1000
2000
6
2000
to-
о.д-
о,б-
0,4-
0,2]
0
3000
1000
2000
Рис. 119. Влияние моле-
кулярного веса олигоэфи-
ра иа релаксирую-
щую часть модуля упр\-
гости Eq (а), время релак-
сации (б) и параметр Л
(Ь) для полиуретанов иа
основе.
/ - п 1 ГФ, 2 - ппг
м03
0,2.~.
0
М03
Eq, который оказался нечувствительным к изменению химической
природы олигоэфирной составляющей, эти параметры зависят от
200
последней. Релаксирующая часть модуля упругости £0 (рис. 119, а)
резко уменьшается при увеличении М^э для обоих полихретанов.
В области значений Моэ от 15 000 до 3000 это выражено слабо, одна-
ко Ео для полиуретанов на основе ПТГФ значительно выше соот-
Рис. 120. Логарифмическая зависимость равновесной
(/, 2} и релаксирующей EQ (<?, 4) частей модуля упругости
от Л1оэ для полиуретанов на основе ПТГФ (/, 3) и
ППГ (2, 4).
ветствующих значений для полиуретанов на основе ППГ, особенно
в области значений /Иоэот 500 до 1000. Эта разница постепенно умень-
шается с увеличением Л4Оэ.
Хотя модуль упругости для полимера и не является константой
(он зависит от скорости или продолжительности деформации), тем
нс менее это важная характеристика полимера, имеющая большой
физический смысл. Он является мерой пространственной структури-
рованности полимера. По значениям релаксирующей части модуля
упругости можно судить о скорости протекания релаксационных про-
цессов, внутренней вязкости и т. д. В связи с этим нами была
сделана попытка получить аналитические выражения связи
данных параметров с молекулярным весом олнгоэфирного звена.
Для этого зависимости £сс и £0 от 7Иоэ (см. рис. 118 и 119, а) пере-
строены в координатах lg Е^ (1g £0 — 1g /ИОэ (рис. 120). Оказа-
лось, что они имеют линейный характер:
lg£oo = A — BlgAfos,
lg £0 = At — BllgMO3.
Определенные из кривых рис. 120 константы уравнений составляют
А — 6,03; В = 1,42; Лх = 10,44; — 2,93 для полиуретанов на
20J
основе ПТГФ н А =- 6,62; В = 1,63; /Ц — 10,7; В{ = 3,15 для полн-
уретанов на основе ППГ
Молекулярный вес олигоэфира влияет и на время релаксации т,
которое являете я характеристикой скорости релаксационных процес-
сов (см. рис. 119, б) С у величснием Моэ время релаксации для обоих
Таблица 60
Экспериментальные и теоретические значения среднего
молекулярного веса сегмента (Л4С э и Мс т) и эффективной
плотности сшивки полиуретанов на основе ПТГФ и ППГ
л’оэ '^е, э (-1 • 10*. ' и э ^с, т (-) 10’. 'U /т «0 1Ь/СЧ* 1-1 / -) V э/ V 1 т
11 Т Г Ф
650 774 1 гзо 1195 9,28 1,54
960 1320 8,05 1505 7,07 1.14
1450 2120 4,79 1995 5,10 0.91
1910 1465 2,86 2475 4,01 0,71
3000 6070 1,61 «1545 2,77 0,58
ППГ
500 508 21.0 1045 11,20 2,05
1000 1453 7.5 1545 7,07 1 Об
1500 2690 3,8 2045 5,02 0,80
2000 4470 1,22 2545 3,92 0.57
5000 G085 1,6 3545 2,78 0,58
волну ретанов резко снижается в области значений Л4Оэ - 500—1500,
приближаясь к равновесному при дальнейшем увеличении Л4Оэ* При
Л/оэ, близком к 1200, время релаксации, а следовательно, и скорость
релаксационных процессов для обоих полиуретанов одинаковы;
ниже этого значения Л4оэвремя релаксации имеет большее значение
для полиуретана на основе ПТГФ, а выше его — для полиуретана
на основе ППГ. По-видимому, при молекулярном весе олигоэфп-
ра более 1200 становится возможной упорядоченность участков
макромолекул между узлами для полиуретановых сеток па основе
ПТГФ вследствие более регулярного химического строения послед-
него.
Параметр К уравнения обнаруживает более резкую зависимость
ог Моэ для полиуретанов па основе ПТГФ, монотонно снижаясь от
0,55 до 0,29 при увеличении ТИоэ от 645до 3000 Его значения для ПУ
на основе ППГ уменьшаются линейно от 0,32 до 0,24 при увеличении
Л4оэ от 500 до 3000.
Из экспериментальных значений равновесного модуля и плот-
ности р рассчитан молекулярный вес сегмента Л4оэ и эффективная
202
плотность сшивки (4г) (табл. 60). Для сравнения представлены
теоретические значения /Ис,т и (-—) , определенные но уравнению
(V, 2), на основании стехиометрических соотношений исходных
продуктов, взятых для синтеза полиуретанов
Как видно,для обоих полиуретанов с малыми 7Иоэ (500—1000)
экспериментальные значения Л4С,Э и |4rj оказались соответствен но
\ v' 'э
меньшими и большими, чем теоретические, а для полиуретанов с
большим УИоэ (2000—3000)—наоборот. Такая зависимость, по-видп-
мому, связана либо с изменением характера дефектности химической
сетки (например, переходом от захлестывания пеней в ПУНО
действие которых аналогично действию химических связей, к боль-
шому количеству несшитых участков и концов цепей для НУ ВО”
которые ничего нс вносят в упруюсть молекулярной сетки), либо
с особенностями надмолекулярного строения этих полиуретанов
Обнаруженная аномалия удовлетворительно объясняется с позиций
надмолекулярных структур исследованных полиуретанов, что при-
водит к необходимости рассмотрения их механического поведения
в тесной связи с условиями синтеза и кинетикой процесса сшивания.
Рассматривая полученный результат с такой позиции, можно кон-
статировать, что поведение исследованных полиуретанов с большой
длиной олиюэфирного блока связано с незавершенностью формиро-
вания химической сетки. Очевидно, в этом случае условия и кинети-
ческие особенности процесса сшивания таковы, что невозможна реа-
лизация полного отверждения МДИ в соответствии со стехиометрией
исходных продуктов. Поэтому дефектность сетки для данных поли-
уретанов характеризуется меньшим числом сшивок в единице объе-
ма, чем следует из теоретических соображений, и большим числом
свободных концов цепей.
Для выяснения причин обнаруженной аномалии проведено кало-
риметрическое исследование кинетики процесса сшивания, которое
показало, что скорость сшивания МДИ уменьшается с увеличением
34оэ Однако даже в случае высокомолекулярных олигоэфиров про-
должительность отверждения не превышает 6 ч, что хорошо иллю-
стрирует кривая зависимости параметра завершенности процесса
сшивания по тепловому эффекту реакции от времени (рис. 121). От-
сюда следует, что выбранное нами время для отверждения полиуре-
танов вполне достаточно и не является причиной незавершенности
формирования сетки
Т. Э. Липатовой и С. А. Зубко показано [61], что процесс форми-
рования сетки после точки гелеобразования описывается уравне-
нием Аврами 11651. Интересно отмстить, что величина удельной
ПУНО п ПУВО—полиуретаны на основе низкомолекулярного и высоко-
молекулярно, о олиюэфиров соответственно.
203
скорости образования гель-фракции Ко, определенная из этого урав-
нения, коррелирует с величиной равновесного модуля упругости.
Для обоих типов полиуретанов большим значениям Ко соответст-
Рнс. 121. Кинетическая кривая процесса
образования полиуретановой сетки на ос-
нове ППГ молекулярного веса 2000 при
температуре сшивания 80° С.
вуют и большие значения рав-
новесного модуля упругости
(табл. 61). Из этих данных
вытекает, что между парамет-
рами релаксационных свойств
и характеристиками кине-
тики процесса сшивания
полиуретанов имеется опре-
деленная связь. Поэтому по-
становка исследований, посвя-
щенных этой связи, имеет
большое значение для выясне-
ния особенностей строения
полиуретановых эластомеров
и объяснения аномалий их
механического поведения.
Для изучения структуры
проведено электронно-микро-
на надмолекулярном уровне было
скопическое исследование (на электронном микроскопе УЭхМВ-iOO
по методу кислородного травления в газовом разряде 191) объек-
тов (рис.
122, см. вклейку), которое показало, что они
обладают глобулярным строением.
Подобные глобулярные структуры
наблюдались и в случае других
полиуретановых эластичных систем
сетчатого строения 1581, а также в
сшитом эластичном полиалкилме-
гилсилоксане [125], в сшитых поли-
акрилатах [371, фенолформальдегид-
ных и других полимерах простран-
ственного строения 134, 207, 3191.
Глобулярная структура полимерных
сеток является довольно распростра-
ненной для многих пространственных
аморфных полимеров как в стеклооб-
разном, так и в высокоэластическом
состояниях.
Установленное нами отклонение
экспериментальных значений плот-
Таблица 61
Кинетический параметр
процесса сшивания и равновес-
ный модуль упругости для
полиуретанов на основе
ПТГФ и ППГ
э Кс • Ю’, лик-1 KelcM1
ПТГФ
960 | 7,98 I 1 71
1930 1 1 5,40 1 1 20
ППГ
500 13,60 168
2000 5,79 16
ности сшивки [ — | и среднего молекулярного веса сегмента
\ V /э
от их теоретических значений (см. табл. 60) можно удовлетворитель-
но объяснить с позиций глобулярного строения исследованных поли-
уретановых сеток. Действительно, при получении ПУНО образуются
204
глобулы меньших размеров, чем в случае I1VBO, что подтверждаю'!
электронно-микроскопические наблюдения Для малых глобхл по
сравнению с крупноглобулярной структурой имеется большая ве-
роятность, что NCO-группы, участвующие в реакции сшивания, рас-
положены на их поверхности или вблизи нее. В крупноглобулярной
структуре расположение реакционноспособных NCO-rp\nn более
вероятно внутри объема глобул, чем на их поверхности, вследствие
больших размеров. Это приводит к образованию дефектной сетки с
большим количеством непрореагировавших функциональных групп,
а следовательно, с большим числом свободных концов цепей сетки.
Таким образом, неполное отверждение и незавершенность формиро-
вания химической сетки в ПУВО есть следствие их глобулярного
строения.
И наконец, при рассмотрении особенностей строения исследован-
ных нами сеток и их связи с механическим поведением необходи-
мо принимать во внимание положение уретановых групп в сетке,
которые являются главной причиной сильного физического взаимо-
действия. Очевидно, уретановые группы, расположенные в хими-
ческих узлах сетки, имеют меньшую возможность для образования
водородных связей, так как их подвижность подавлена сильными
химическими связями. Однако степень участия этих групп в обра-
зовании физических связей меняется при переходе от НУНО к
ПУВО Действительно, в случае применения для синтеза ьизке мо-
лекулярного олигоэфира образуется полиуретан мелкоглобуляр-
ной структуры, которая имеет более развитую поверхность раздела,
что обусловливает более сильное физическое взаимодействие между
глобулами при максимально возможной (для данного размера гло-
бул) густоте химической сетки, приближающейся к теоретической.
Несмотря на то что уретановые группы «закрепощены» в химичес-
ких узлах сетки, их возможности в образовании водородных свя-
зей в определенной степени возрастают благодаря большому коли-
честву соседних мелких глобул. Это, по-видимому, и приводит к боль-
шим экспериментальным значениям равновесных модулей упругости,
эффективной плотности сшивки и меньшим значениям среднею мо-
лекхлярного веса сегмента, по сравнению с теоретическими для
ПУНО.
При синтезе ПУВО образуется химическая сетка с более круп-
ными глобулами, обладающая гораздо меньшей поверхностыо раз-
дела. В связи с этим происходит неполное отверждение полиурета-
нов и формирование более дефектной химической сетки с меньшим
количеством химических связей в единице объема. Кроме того, ве-
личина физического взаимодействия минимальна из-за меньшего
количества физических контактов между более крупными глоб\ ла-
мп и меньших возможностей для образования водородных связей,
«закрепощенными» в химических узлах уретановыми группами.
Следовательно, в этом случае вклад и химического, и физическо-
го взаимодействия в равновесную упругость минимален, чем и
205
объясняются полученные нами значительно более низкие экспери-
ментальные значения равновесного модуля упру гости, эффективной
плотности сшивки и более высокие значения среднего молекуляр-
ного веса сегмента по сравнению с теоретическими.
Химическая природа олигоэфир по го блока практически не влия-
ет на равновесную упругость (см. рис. 118, табл. 60), а также на
средний молекулярный вес сегмента и эффективную плотность сшив-
ки, однако оказывает заметное вчнянне на кинетику релаксацион-
ною процесса (см. рис. 119).
Таким образом, изучение релаксационных свойств сетчатых по-
лиуретанов на основе простых полиэфиров в связи сих молекулярной
и надмолекулярной структурами и условиями получения позволило
установить некоторые особенности строения и механических свойств.
При увеличении длины олнгоэфирного блока уменьшается химичес-
кое и физическое взаимодействия, а следовательно, и равновесная
упругость, что обусловлено глобулярным строением полиуретано-
вых сеток.
Полученные результаты показывают, что химическая сетка ис-
следованных полиуретанов характеризуется значительной структур-
ной неоднородностью, оказывающей существенное влияние на меха-
ническое поведение полимерного тела. Поэтому нужно пересмотреть
механическое проведение таких систем на основе новых представле-
ний о их глобулярном строении.
Результаты данного исследования относятся к полиуретанам, в
которых роль физических связей относительно невелика. Между
тем, интерес представляют процессы релаксации, связанные также с
распадом и восстановлением физических узлов сетки. Такие процессия
до некоторой степени аналогичны процессам химической релаксации.
С этой точки зрения важно изучепнелинейных аморфных «сегменти-
рованных» эластомеров, в молекулах которых существует последова-
тельность гибких и жестких сегментов 11931. У таких эластомеров
большая прочность и высокий модуль упругости выше температуры
стеклования где другие эластомеры имели бы свойства вязких
жидкостей. Авторы [1931 предположили, что жесткие сегменты игра-
ют роль наполнителей и поперечных связей. Область повышенных
модулей упру гости между Tg и температу рой стеклования связанных
жестких сегментов Т ' обусловлена ассоциацией стеклообразных
блоков эластомеров. На примере полиэфируретана, содержащего
пластификатор (полиэтиленглнколь с мол. весом 200), показано, что
последний сольватирует преимущественно сегменты полиэфира и
снижает не уменьшая модуль унруюсти выше 7\. Если брать
диметилсульфоксид, то он сольватирует жесткие сегменты,понижая
модуль между и Тс. Для таких систем были исследованы вязко-
упругие свойства и температурная зависимость модуля.
В свое время Бартенев и Вишницкая [81 исследовали релакса-
ционные свойства наполненных резин и установили, что релакса-
ция напряжений в них определяется тремя процессами — релакса-
206
цией цепей, релаксацией, связанной с отрывом цепей кахчука ог
частиц наполнителя, и перегруппировкой самих частиц наполни-
теля Возвращаясь к полиуретанам, можно полагать, что в них дейст-
вительно должны осуществляться процессы, связанные с релакса-
Рис 123. Зависимость времени спин-решеточной релаксации oi
температуры
1 — о«1И1 оокснтстра метилен гликоль 2 - олигоди этпленгл нкольвдипииа г,
J — аддукт на основе тримети^олпропана и ТДИ, 4 — циклогексанон
цией физических узлов (эквивалентных связям каучук — наполни-
тель в обычных резинах). Такие процессы действительно обнаружены
при изучении диэлектрической и спин-решеточной релаксации в
олигомерах и полиуретанах [761. В качестве объектов выбраны олн-
гоокситетраметиленгликоль, олигодиэтилеигликольадипинат и ад-
дукт на основе триметилолпропана и ТДИ, а также полиуретаны на
основе олигодиэгиленгликольадипината и димера ТДИ, олигоокен-
тетраметилепгликоля и олигодиэтиленглпколя и упомянутого выше
аддчкта.
На рис. 123 представлена температурная зависимость времени
спин-решеточной релаксации протонов для олигоокситетраметилен-
20 Z
гликоля, олигодиэтиленгликольадипината и аддукта на основе три-
мстилолпропана и толуилендиизоцианата, а также для чистого цик-
логексанона, так как в исследуемом аддукте его содержалось око-
ло 30%. Как видно, для полиэфиров наблюдается один релакса-
ционный процесс: для простого полиэфира при —25° С, для сложно-
го — при —8е С. Известно, 1961, что в олигомерных молекулах про-
является собственная гибкость цепи. Так как исследуемые полиэфи-
ры линейны и не содержат боковых и СН3-групп, то очевидно, что
существующий релаксационный процесс связан только с движением
сегментов молекулярных цепей; их подвижность больше для олиго-
окситетраметиленгликоля, чем для олигодиэтиленгликольадипината.
Заторможенность движения молекул сложного полиэфира объясняе-
1ся наличием в цепи дополнительных полярных групп, приводящих
к более прочной структурной физической сетке. В табл. 62»приведены
значения энергий активации релаксационного процесса.
Таблица 62
Значения энергии активации (ккал!моль)
исходных компонентов
Компонент Движение сегментов а Процесс ТМИН» °C
Простой ПЭ 6,10 —25,5
Сложный ПЭ 7,34 — —8,0
Аддукт — 13,75 (2,57) —70 0
—48,0
Циклогексанон —• 2,95 —52
Примем ание Энергия активации (3 процесса ад-
дукта равна 2,4 ккал!моль
Аддукт характеризуется двумя (а и Р) релаксационными процес-
сами (рис. 123, кривая 3) при 70 и —48° С, вызванными движени-
ем ассоциатов двух типов, проявляющих релаксацию при более
высоких и низких температурах. Для a-процесса наблюдается из-
лом на кривой зависимости тс — ср(1 /Т), причем прямая с меньшим
наклоном практически параллельна соответствующей прямой для
p-перехода и мало отличается от наклона прямой для циклогек-
санона (рис. 124), что дает практически одинаковые энергии акти-
вации (2,4; 2,57; 2,95 ккал/моль, табл. 62). Если учесть, что р-пере-
ход аддукта и процесс релаксации циклогексанона протекают почти
при одинаковых температурах (—48 и-------52° С), то очевидно, что
p-процесс и зависимость 1g = ср с меньшим наклоном обус-
ловлены наличием в аддукте циклогексанона, а a-процесс вызван
молекулярной подвижностью ассоциатов, образованных за счет на-
личия в аддукте большого количества уретановых и других групп.
Из температурной зависимости времени спин-решеточной релак-
сации 7\ (рис. 125) видно, что для исследованных полиуретанов на-
208
~19ГС
т———।--------1-------1------1——_» о г_7
2,5 3,0 3,5 4,0 4,5 Т-10,( К)
Рис. 121. Зависимость тс от -^г
Рис 125 Зависимость времени спнн-решеточной ре,1аксацип от температуры
/—НУ каучук; 2 — НУ на основе просто! о ПЭ, 3 — П У на основе сложного ПЭ
блюдается три релаксационных процесса: низкотемпературный (от
—138 до —141 ' С), связанный с движением СН3-групп; процесс, обу-
словленный сегментальной подвижностью цепей для полиуретаново-
го каучука (20е С), полиуретана на основе простого ПЭ (—10° С) и
полиуретана на основе сложного ПЭ (35е С), а также процесс при
более высоких температурах, который представляет для полиурета-
новых систем особый интерес» Если для полиуретанового каучука
(рис. 125, кривая /) этого перехода не наблюдается, то уже для
ПУ на основе простого ПЭ (кривая 2) существует перегиб — слабо
выраженный минимум релаксации, который хорошо виден для поли-
уретанов па основе сложного ПЭ (кривая 3). Энергия активации
данного процесса в два раза превышает таковую для сегментально-
Таблица 63
Значения энергии активации (ккалf моль) полиуретанов
Полиуретан Движение групп Движе- ние сег- ментов а-Процесс 6-Процесс
ПУ каучук ПУ на основе просто- — 5,95 15,00 —
го ПЭ ПУ на основе слож- 0.8 4,38 8,24 13,25
ною ПЭ 1,1 2,05 18,80 21,0
При м еч я н и е. Энергия активации движения узлов для полиуре-
тана на ос. ове сложного полиэфира равна 4,3 ккач/моль
го движения и составляет 4,29 ккал!моль (табл. 63). Этот процесс
может быть обусловлен перераспределением межмолекулярных свя-
зей и подвижностью узлов физической трехмерной сетки, что можно
представить следующим образом: при повышении температуры в
поле наложенных возмущающих нагрузок часть сегментов цепей, свя-
занных узлами трехмерной физической сетки, освобождается, вслед-
ствие чего образуются новые узлы и развивается подвижность доста-
точно больших структурных единиц.
По-видимому, этот процесс нельзя отнести к разрушению или воз-
можной перестройке 11141 биуретовых, аллофанатных и мочевинных
групп, так как для их перестройки требуется энергия активации
от 20 до 50 ккал моль, а, согласно [254 I, диссоциация биуретов ста-
новится заметной лишь при 120, аллофанатов — при 106 ~ С. По
данным [294 I, биуреты реагируют с аминами при 150° С. Результаты
исследования диэлектрической релаксации (рис. 126) показыва-
ют, что процесс релаксации узлов физической трехмерной сетки при
частоте 10 гц происходит при 40—50° С (кривая 2) для полиуретанов
на основе простого ПЭ, 45—75е С (кривая 3) для полиуретанов на
основе сложного ПЭ и не проявляется для полиуретанового кау-
чука (кривая /).
210
Для полиуретанового каучука имеют место два процесса диэлект-
рической релаксации: дипольно-сегментальный при —20эС и ди-
польно-групповой при —95° С. Известно, что область релаксации
сегментов и цепей (а-поглощение) для полиуретанов наблюдается
примерно при —40° С [150, 234, 2351. При переходе от полиэфира к
соответствующему полиуретану имеет место смещение диэлектриче-
ской релаксации в сторону положительных температур (в данном
случае —20° С). Аналогично для спин-решеточной релаксации поли-
уретанового каучука (см. рис. 125, кривая /) смещение составляет
31° . Для сшитых полиуретанов также происходит смещение спин-
решеточной релаксации сегментов в сторону высоких температур
по сравнению с переходом для полиэфиров, но это смещение не на-
много превышает температуру высокотемпературного релаксаци-
онного процесса аддукта (табл. 64).
Таблица 64
Смещение минимумов спин-решеточных релаксационных
процессов при переходе от полиэфиров
к соответствующим полиуретанам
Наименование Тмин • Смещение по отно- шению к полиэфи- ру, град Смещение по отно шению к аддукту, град
Простой ПЭ ПУ на основе просто- 247,5 — —
го ПЭ 263 15,5 80
Сложный ПЭ ПУ на основе сложно- 265 — —
го ПЭ 308 43,0 35
Таким образом, релаксация сегментов в полиуретанах проявляе-
тся при температурах, превышающих температурную область релак-
сации соответствующих исходных полиэфиров, но не превышает тако-
вую высокотемпературного релаксационного процесса аддукта. Это
связано с уменьшением подвижности вследствие сшивания гибких
олигомерных цепей жесткими ассоциатами аддукта, изменением вели-
чины кинетического сегмспга и, в основном, с образованием структур-
ной физической сетки, так как смещение области релаксации для по-
лиуретанов на основе сложного ПЭ почти в три раза больше смеще-
ния для полиуретанов на основе простого ПЭ (табл. 64).
Рассматривая теперь процессы диэлектрической релаксации для
сшитых полиуретанов (рис. 126, кривая 2 и 3), можно сказать, что
дипольно-сегментальный процесс должен проявиться в области тем-
ператур выше —40° С при частотах ниже 100 гц. Из результатов
исследования a-процесса видно, что для полиуретанового ка-
учука переход наблюдается при —95° С, для полиуретанов на
основе простого ПЭ при—120° С и для полиуретанов на основе
211
сложного ПЭ при —108 ’ С. Кроме того, в сшитых полиуретанах име-
ет место еще p-процесс (для полиуретана на основе простого ПЭ
при —65'Сндля полиуретана на основе сложного ПЭ при—75СС),ко-
торый можно объяснить движением кинетических единиц (групп ато-
мов), имеющих размеры больше тех, которые ответственны за ди-
полыю-групповое движение, по меньше размеров сегментов цепей.
Рис 126. Температурная зависимость tg <s:
1 - ПУ кауч> к, 2 — ПУ на основе простого ПЭ, 3 — ПУ на основе сложного ПЭ.
Итак, если для полиуретанового каучука, как и для других ли-
нейных пекристаллизующихся полимеров, наблюдается только два
процесса диэлектрической релаксации, то в сшитых полиуретанах
количество их увеличивается благодаря наличию более широкого
спектра кинетических единиц.
Из рис. 127 видно, что наклон зависимости 1g те — <р (иссле-
дуемых ПУ уменьшается при переходе от линейного к сшитому по-
лиуретану па основе сложного ПЭ. Уменьшение энергии активации
212
^9
ния спин-решеточной релаксации:
/ — ПУ каучук, 2 — ПУ иа основе простого ПЭ» 3 — ПУ на основе
сложного ПЭ
1 — НУ каучук, 2 — ПУ на основе простого НЭ, 3 — ПУ на основе сложно: > ПЭ
(см. табл. 63) можно объяснить расширением спектра времен релакса-
ции кинетических единиц в сшитых полиуретанах и, следовательно,
уменьшением энергетического барьера активации. На рис. 128 пред-
ставлена также зависимость 1g Дп-(р для a-процесса диэлек-
трической релаксации.
Рис 129. Температурная чавнеимость вещественной и мнимой частей комплекс-
ного модуля сдвига (G' и G") и механических потерь tg б на частоте 1 гц для поли-
уретана на основе 1,4-бутандиола.
Таким образом, релаксационное поведение полиуретанов действи-
тельноопределяетсякак гибкостью и свойствами олигомерного блока,
так и наличием жестких блоков и узлов физической сетки. Это типич-
но и для наполненных полиуретанов.
Диэлектрическая релаксация в полиуретанах исследована так-
же с целью определения характера изменения межмолекулярных
взаимодействий при переходе от полиэфиров к полиуретанам и опре-
деления их температур стеклования 1151 ]. Собственно механизм ре-
лаксационных процессов авторы не рассматривали, но полученные
данные дали основание связать проявление сил межмолекулярных
взаимодействий (физических узлов сетки) с образованием связей меж-
ду полярными СО-группами в сложноэфирных полиуретанах и
с водородными связями между уретановыми группами для полиурета-
нов на основе простых полиэфиров.
Мы рассмотрели релаксационные явления в полиуретановых элас-
томерах, содержащих гибкие блоки. Однако интерес прсдставля-
211
ют релаксационные свойства линейных полиуретанов на основе низ-
комолекулярных диолов, например иа основе гексаметилендиизо-
цианата и 1,4-бутандиола, 1,6-гександиола, 1,10-дскандиола и
2,5-гександиола 1238]. Данные зависимости механических потерь и
модулей Gf и G” от температуры для полиуретана иа основе 1,4-бу-
тандиола дают две области релаксации при —135е С (у-максимум)
Рис. 130. Температурная зависимость механических потерь tg 6 для полиуретанов
на основе гсксаметилендиизоцианата и различных гликолей.
1 — 1,4 бутаиднол, 2 — 2 5 гександиол, 3 — 1 ь гександиол, 4 — 1,10 декапднол (час-
тота 1 гц)
и -|-40° С (главный или a-максимум) (рис. 129). Первый обусловлен
подвижностью СН2-групп, а главный соответствует температуре рас-
стекловывания, т. е. началу микро-броуновской подвижности цепей
в аморфных областях.
Рост потерь и сильное падение модуля выше 150° С связано с
плавлением кристаллических областей полимера. Температурные
зависимости потерь для полиуретанов на основе различных диолов
(рис. 130) указывают на смещение температурного положения глав-
ного максимума примерно на 5° С при увеличении длины цепи на два
углеродных атома. Авторы трактуют свои результаты на основе
представлений о водородных мосгиках в аморфных областях иссле-
дованных полиуретанов, которые действуют как поперечные связи.
При этом удлинение цепи уменьшает число водородных связей на
215
единицу длины цепи, вследствие чего возрастает гибкость и изменя-
ется релаксационное поведение.
Исследовано влияние термической предыстории на положение
максимума потерь и его временную зависимость при постоянной тем-
пературе 12381. Изменения, которые при этом имели место, связаны с
процессами кристаллизации, что, в частности, позволило рассмот-
реть также процессы изотермической кристаллизации в терминах
уравнения Аврами из зависимости lg tg 6 от времени.
РЕОЛОГИЧЕСКИЕ СВОЙСТВА ОЛИГОМЕРОВ
Многие важнейшие свойства полиуретанов определяются та-
ковыми олиюмерных блоков (см., например, данные по стеклова-
нию, гибкости и пр.). Этим объясняется интерес к изучению олиго-
мерных систем. Существенный вклад в понимание поведения поли-
уретанов могли бы внести реологические исследования олигомеров.
Эта область разработана очень мало. В данном разделе мы попыта-
емся показать взаимосвязь между реологическими свойствами оли-
гомеров и свойствами полиуретанов.
Ю. С. «Липатов и сотрудники впервые установили некоторые су-
щественные аномалии реологического поведения олигомеров [66]. Они
исследовали реологические свойства некоторых олигомерных поли-
эфиров, применяющихся для синтеза полиуретановых эластомеров.
В качестве объектов выбраны политетрагидрофураноксипропилеп-
гликоли с молекулярным весом 1000 и 2000 (содержание окиси
пропилена в сополимере 20—25%) и сложные полиэфиры— полпди-
эгиленглнкольадипинаты с молекулярными весами 790 и 2050, а
также полидиэтиленгликольадипинаттриметилопроиан (десмофен) с
молекулярным весом 2200. Кроме того, изучены концентрированные
растворы указанных олигомеров в ацетоне (концентрации 70, 80 и
90%). Вязкости олигомеров измеряли в интервале температур 20—
60° С, а их растворов — при 20—30° С на ротационном вискози-
метре в интервале скоростей сдвига от 4 * 10‘4 до 1,5 • К)2 сек~{
(рис. 131).
Анализируя ход кривых вязкости, можно установить, что все
системы характеризуются аналогичным ходом реологической кривой,
на которой можно выделить три участка. ЛВ — восходящая ветвь
антптиксофопнн пли структурирования, ВС — область разрушения
структуры и в некоторых случаях — ветвь CD — область течения
при разрушенной структуре с постоянной вязкостью. В зависимоеги
от типа олигомера и температуры опыт отдельные участки кривых
выражены менее или более четко. В некоторых случаях часть участ-
ков смещается по шкале напряжений сдвига за пределы исследован-
ной области.
Обнаруженный аномальный ход кривых вязкости обьясияется
следующим образом. На участке АВ под действием сдвпювых на-
216
пряжений молекулы олигомера ориентируются др\г относительно
друга и вследствие усиления взаимодействия ориентированных моле-
кул возникает определенная структура Этот процесс аналогичен
рассмотренному в [62, 219, 2391 и может быть назван ангптпкео-
— 1 0 1 Lg Р
Рис. 131. Реологические кривые полидиэтиленгликоль-
адипината (мои. вес 790).
тропией. При дальнейшем увеличении напряжения сдвига, начиная
с критического значения Ркр, происходит разрушение структуры
(участок ВС) и затем реализуется течение полностью разрешенной
структуры (участок CD), которая в некоторых случаях обладает
большей вязкостью» чем исходная система в точке А.
Следовательно, при малых напряжениях сдвига олигомеры про-
являют аномальную вязкость в поле поперечного градиента, напо-
минающую рост эффективной вязкости при суперпозиции сдвиго-
вого и продольного течения [661. Анализ других аналогичных
217
кривых показывает, что эффекты аномалии вязкости выражены более
резко для оли!омеров меньшего молекулярного веса. Это, возмож-
но, связано с повышенной жесткостью и большей подвижностью
более коротких цепей. Для низкомолекулярных систем стр у ктуро-
образование происходит по мере повышения температуры, начиная
с более высоких напряжений сдвига, как и следовало ожидать, при-
нимая во внимание тепловое движение молекул, а разрушение
структуры начинается при более высоких значениях РКр (например,
на рис. 131 при 20 и 30° ветвь АВ не проявляется, а есть лишь учас-
ток BCD).
В системе с большим молекулярным весом ветвь АВ появляется
5же при комнатной температуре. Изменение молекулярного веса
олигомера приводит к изменению зависимости Ркр от температуры.
Для низкомолекулярного олигомера РКр растете увеличением тем-
пературы, а для более высокомолекулярного — падает. Аналогич-
ная картина наблюдается для двух сложных полиэфиров разных мо-
лекулярных весов. Наконец, для разветвленного олигомера — де-
смофена — рост температуры приводит к уменьшению РКр.
Такие изменения Ркр с температурой могут быть связаны с раз-
личиями в ориентации и гибкости малых и больших молекул олиго-
меров. Вполне вероятно, что в первом случае имеют место только ори-
ентационные эффекты, тогда как для более высокомолекулярных
образцов уже проявляются и деформационные [149].
Остановимся теперь на вязкости концентрированных ацетоновых
растворов олигомеров (рис. 132). В более концентрированных раст-
ворах аномалии вязкости сохраняются, в то время как понижение
концентрации или повышение температуры приводит к их вырожде-
нию (вплоть до перехода к ньютоновскому течению). Для более вы-
сокомолекулярных образцов аномалии вязкости сохраняются при
больших разбавлениях.
Очевидно, аномалии вязкости связаны не просто с ориентацией
молекул в потоке, по и с сопутствующим усилением взаимодействия
между ними. Полученные данные нельзя объяснить разворачиванием
цепей в поле продольною градиента, как это наблюдалось в [62,
2191, из-за того, что возможности изменения конформаций в моле-
кулах олигомеров значительно меньше, чем в высокомолекулярных
полимерах. Кроме того, если бы аномалии были связаны с про-
цессами деформации олигомерных молекул, они были бы ярч^ выра-
жены для более высокомолекулярных образцов.
Эти результаты указывают на необходимость учета аномалий рео-
логических свойств олигомеров при переработке и синтезе полиме-
ров. Из них также следует, что межмолекулярные взаимодействия
между олигомерными блоками оказывают существенное влияние на
свойства полимеров, полученных на их основе. Возможно, это позво-
лит объяснить некоторые свойства полиуретанов, для коюрых обыч-
но учитываются только межмолекулярные взаимодействия, опреде-
ляемые образованием водородных связей.
218
До последнего времени считалось, что высокоэластическая дефор •
мация присуща только высокомолекулярным полимерам, и высоко
эластичность рассматривалась как один из признаков полимеров.
Низкомолекулярные полимеры, находящиеся в обычных условиях в
вязкотекучем состоянии, не проявляют специфических свойств по
Рис. 132 Реоло! ические кривые концентрированных
ацетоновых растворов полидиэтиленгликольадипината
(мол вес 2050).
лимеров и поэтому признаку выделяются в группу олигомерных со-
единений. Их молекулярный вес обычно находится в пределах мо-
лекулярных весов механических сегментов макромолекул.
Детально исследовано деформационное поведение такого олиго-
мера, молекулярный вес которого совпадает или близок к величине
механического сегмента полимера [1461. В качестве объектов выбра-
ны два образца сложных полиэфиров: линейный полиэфир на основе
адипиновой кислоты и диэтиленгликоля с молекулярным весом око-
ло 2000 (1) и разветвленный полиэфир типа десмофен с молекуляр-
ным весом примерно 2500 (2).
Деформационное поведение указанных олигомеров изучали по
методу, основанному на кинетике развития деформации безграничного
219
сдвига в узком зазоре коаксильпых цилиндров [1111. Иссле-
дование сводилось к получению семейства кривых е = <р (т)р при
напряжениях сдвига 3,6—43,8 дин!см2 и температурах 20—70 С
(рис. 133).
Кривые /, 2 (рис. 133, а, б) соответствуют условиям явного плас-
тического течения с малым участком, отвечающим эластической
Рис. 133. Зависимость кинетики развития деформации сдвига для сложного линей-
ного полиэфира при разных напряжениях сдвига (дин/см2):
а _ t - 20- С, / - 3,65; 2- 7 30. .7 — 10,95; 4 - 14,60; б — t =- 40° С, /—3,65,
2 — 5.11; 3 — 7 30; 4 — 14,60, в — t - 60" С, /— 7,30, 2 — 14,60, 3 — 29,20, 4—43,80.
деформации и со значительной долей остаточной деформации по-
сле снятия нагрузки в точке Р — 0.
По мере роста Р (кривые 3, 4\ рис. 133, а, б) наблюдается увели-
чение доли эластической деформации с вырождением прямолиней-
ного участка кривой, соответствующего пластическому течению,
т. с. явление, противоположное тому, что имеет место в дисперсных
системах и структурированных жидкостях [111, 1121.
С повышением температуры до 50 и 60° С система утрачивает
текучесть при всех заданных режимах деформации, и кривые е --
= <р (т)р соответствуют деформационной модели контура Кельвина
(рис. 133, в).
Из реограмм для олигомера 2 (рис. 134) видно, что при напря-
жениях сдвига 21,9—36,5 дин!см2 приведенные кривые аналогичны
кривым /, 2 рис. 133. Однако при увеличении напряжения сдвига
до 43,8 дин!см2 происходит хрупкий разрыв в отличие от линейного
полиэфира, для которого с ростом Р увеличилась доля эластичес-
220
Рис. 134. Зависимость кинетики развития деформации сдвига для
сложного разветвленного полиэфира при температуре 20° С и раз-
ных напряжениях сдвига (дин/см'2):
/ — 21,°0, 2 — 29,20, 3 — 30,50, 4 — 48.80 5 — 51,10.
Рис 135. Зависимость кинетики развития деформации сдвига для
полиэфира 1 при Р = 7,30 (а) и 11,60 (б) дин,'см2, 5 ч и различных
температурах:
/ — 20, 2 — 30; 5 — 50, 4 — t -= 60° С.
кой деформации (кривые 3, 4 рис. 133). При повышении температу-
ры прочность структуры уменьшается и она разрушается соответ-
ственно при меньших значениях напряжений сдвига, как это пока-
зано ниже:
Т, °C, 20 30 40 50 70
Рр 43,8 36,5 29,2 21,9 14,6
Для выявления структурных единиц, определяющих деформаци-
онное поведение указанных систем, определяли влияние на упругие
свойства последних величины напряжения сдвига, температуры и
времени нагрева.
Рис. 136. Зависимость равновесного модуля от напряжения едзига для
полиэфира 1 при температурах 20 (/), 30 (2), 50 (3), 60° С (4) для неустойчивого
состояния и 50°С (5) после достижения равновесия.
Показано, что в интервале температур 20—50° С для образца I
эластическая деформация уменьшается с повышением температуры
и соответственно увеличивается модуль эластичности при условии
снятия рео1раммы в течение 3—4 ч после установления требуемой
температуры (рис. 135). При этом равновесный модуль, рассчитанный
р
как Е=—, не инвариантен при различных значениях Р, т. с. не
является константой временной упругости системы.
Зависимость равновесного модуля Е or Р (рис. 136) была рассчи-
тана из экспериментальных кривых в ср (т)Р, снятых при указан-
ных температурах и напряжениях сдвига. Из кривой е = <р (т)/>
возникновения и спада деформации (рис. 137), полученной по мето-
ду долевых нагрузок — разгрузок, видно, что как прямой, так и об-
ратный ход этой кривой неравномерен при равных долях нагрузки,
что согласуется с данными о зависимости модуля эластичности от
напряжения сдвига. Температуру 50° С можно считать для данного
олигомера критической, так как при дальнейшем ее повышении (до
60° С) модуль эластичности не растет, деформируемость системы
222
Рис. 137. Зависимость кинетики развития деформации сдвига для
полиэфира 1 при условии долевых нагрузок (Pt ~ 14,6 дин/см2)
при температуре 30° С для неустойчивого состояния
резко увеличивается (см рис. 135, б, кривая 4), а равновесный
модуль становится инвариантным при различных напряжениях сдви-
га (рис. 136, кривая 4).
Рис. 138 Изменение деформируемости полиэфира 1 в
процессе 3становления равновесия при 50° С и Р =
= 14,60 дин/см2'.
I — время нагрева до 150 ч\ 2 — время нагрева больпте’200 ч
Описанное состояние не устойчиво, и длительное выдерживание
системы при заданной температуре сопровождается изменением де-
формируемости системы с достижением равновесного сосюяния че-
рез определенный промежуток времени (рис. 138).
223
В табл. 65 представлены соответствующие значения равновесно-
го модуля Е, рассчитанные из кривых г <[ (т)Р при различных на-
пряжениях сдвига (от 7,3 до 43,8 дин!смг) и времени нагрева (от
30 до 400 ч) для температуры 50° С.
Таблица 65
Зависимость модуля сдвига от напряжения
/- (дин1 см2) при напряжениях, дин с i2
т. г 7,3 | 14.6 | 20, > | 36,5 43,8
30 610 810 1
60 6090 2030 277 320 259,0
80 — 2440 286 289 —
150 — 2440 216 — 159,0
200 47,2 68,0 129 — 135,0
400 58,6 95,2 103 87 97,5
Температурная зависимость кинетики развития деформации сдвп-
ia (рис. 139) была исследована также в условиях достижения равно-
весного состояния при каждой заданной температуре, в отличие от
данных рис. 135, где кривые 8 — ср (т)р снимались сразу же после
установления соответствующей температуры.
Таким образом, исследование упругих свойств вязко-текучего
олигомера позволило установить, что олигомерные жидкости являют-
ся тиксотропно-структурированными системами, в которых возмож-
но развитие у пругих обратимых деформаций Эти данные и результаты
изучения свойств мономолекулярных слоев олигомеров 1140) и
свойств разбавленных растворов [971 позволяют считать, что оли-
гомерные молекулы обладают собственной гибкостью и способностью
к изменениям формы под действием напряжений.
Зависимость величины модуля от напряжения (см. рис. 136)
можно истолковав как розу л ы ат существования в олигомере флукту-
ационной аморфной сети с временными контактами, обусловленны-
ми наличием единичных или множественных вандерваальсовских
взаимодейовнй 11411. Падение модуля сростом напряжения типич-
но для структурированных систем и связано с тиксотропным
разрушением флуктуационной сетки под действием напряжений
сдвига.
При температуре 60° С или в условиях длительного выдержива-
ния при более низких температурах, когда максимально разрушают-
ся или разрыхляются флуктуационные структуры, на первый план
выступают деформационные свойства гибких молекул олигомера,
что проявляется в отчетливом возникновении и последующем спаде
после разгрузки эластической временной деформации. В этом случае
вязко-текучий полиэфир по реологическим свойствам подобен
нетекучему образцу эластомера, деформируемого нафузками, нс
вызывающими пластического течения образца.
224
Рис. 139. Зависимоеib кинетики развития деформации сдвига для полиэфира 1
при различных температурах для равновесного состояния:
/ — 30. 2 - 50 3 — 60, 4 — 70° С.
Однако олигомеры имеют и другие специфические свойства, пред-
ставляющие большой интерес для понимания структуры полиуре-
танов.
При исследовании вязко-упругих свойств 100—30%-пых рас-
творов десмофсна наблюдались те же эффекты, что и для чистых
олигомеров. Упругие свойства полностью исчезали только при
концентрации раствора менее 30%. Следовательно, упругие свой-
ства олигомеров и способность их к образованию сетки проявля-
ются и в растворах. Это указывает на новый тип структурообра-
зующих систем с короткими полимерными молекулами, занимаю-
щих промежуточное положение между концентрированными рас-
творами и студнями полимеров, с одной стороны, и способными
к структурообразованию коллоидными растворами низкомолеку-
лярных веществ, с др\ гой. Существование таких структур в олиго-
мерах очень важно для понимания свойств полиуретанов на их
основе.
Следует, однако, иметь в виду, что в низкомолекулярных систе-
мах эффекты межмолскулярного взаимодействия обусловлены так-
же наличием полярных концевых групп, причем этот фактор будет
сказываться тем больше, чем ниже молекулярный вес олигомера,
чю следует из термодинамических исследований растворов свойств
монослоев 1140, 179].
225
Установлено, что между молекулами олигомеров существуют зна-
чительные когезионные силы и что при сжатии монослоев олигоме-
ров происходят процессы, связанные с гибкостью олигомерных
2000 -
/5/7/?-
1000-
.100 -
120°С
Ю'3 1O'Z W"~' 10* $,сек '1
Рис. 140. Зависимость вязкости модельной системы от градиента
скорости.
цепей и образованием агрегатов олигомерных молекул [140]. Явле-
ния ассоциации в растворах полиэтиленгликолей описаны в рабо-
тах [168, 205].
Полученные данные указывают на необходимость учета анома-
лий реологических свойств олигомеров при переработке и получении
из них полимерных материалов. Интересной иллюстрацией этого
положения может служить следующее явление [1181. Эффект анти-
тиксотропии был использован при получении трехмерных сеток в хо-
де их синтеза в условиях действия сдвиговых напряжений при 80—
120° С. Трехмерные и линейные полиуретаны получены на основе ди-
этиленгликольадипината и полифурита непосредственно в рсовиско-
зиметре при вращении внешнего цилиндра до начала гелеобразования
и скоростях сдвига 0,07 и 0,04 сек L На рис. 140 дана кривая зави-
симости вязкости от скорости сдвига для блокированного макроди-
изоцианата, на основании которой выбраны тс скорости сдвига, при
которых наблюдается возрастание вязкости. В табл. 66 приведены
данные о механических свойствах полученных полиуретанов, прост-
ранственная сетка которых образована химическими и физическими
связями, а также для сравнения данные о свойствах той же систе-
мы, синтезированной в обычных условиях. Как видно, наблюдается
значительная анизотропия механических свойств в направлении по-
226
тока и перпендикулярно к нему. Такая анизотропия не проявляется,
если синтез вести при более высоких градиентах скорости, отвечаю-
щих ниспадающей ветви на кривой рис. 140. Это указывает на то,
что вследствие ориентации олигомерных молекул в поле сдвиговых
Таблица 66
Прочность на разрыв образцов полимеров {кг см1), синтезированных
в поле сдвиювьп напряжений
Состав композиции Градиент скорости сдвт а, сек~~\
0 0,'ИИ о.опз
Олигодиэтиленгликольадипипат-2050. Т -65 : сши- 6,82 ф 20,7 >•
ватель - 1 1,15 0,08 6,82^ 10,7 v
Олигофурит-1680.2,4 - ТДИ. МОСА =11:1 76,0 Г 133.0 t 177,01
76,0-> 181,0 » 105,0->
Примечание. Стрелками указано направление высечки образцов из полимера
(t вдоль оси цилиндров, -+ вдоль потока).
напряжений формирование поперечных связей (химических и физи-
ческих) происходит преимущественно в направлении, перпендику-
лярном оси молекулы, где вероятность образования таких связей
больше, чем в направлении течения. Данный эффект приводит к
анизотропии получаемой сетки.
АДГЕЗИЯ ПОЛИУРЕТАНОВ К ТВЕРДЫМ ПОВЕРХНОСТЯМ
Полиуретаны широко применяются в качестве клеев, а также для
покрытия дерева, металлов, бумаги, тканей, стекла, кожи [11, 55,
126, 171 180,194,199,201,248, 320—322, 336, 340]. Полиуретановые
покрытия обладают хорошей адгезией к различным поверхностям,
свсто- и атмосферостойки, хорошо сохраняют блеск, обладают
хорошими электрическими свойствами и низкой газопроницаемо-
стью, а также устойчивы к действию растворителей. Клеи использу-
ются для склеивания стекла, дерева, пластмасс, резин, полиэти-
лена, шерсти, волокон, металлов, кожи [80, 223, 232, 241, 261, 277,
278, 288, 315, 3161.
В связи с большим практическим применением полиуретанов воз-
никает необходимость исследования процессов, происходящих при
формировании полиуретановой пленки на поверхности, влияния на
эти процессы природы и соотношения компонентов, составляющих
покрытие, температуры, присутствия добавок и некоторых других
факторов. В данном направлении известно только несколько работ,
касающихся исследования адгезии полиуретанов к твердой поверх-
ности 1133,291, 292|. Изучена адгезия полиуретановых покрытий на
227
основе простых полиэфиров в зависимости от строения последних и
природы наполни гелей, вводимых в пленки, методом отрыва пленки
ог алюминиевой подложки под углом 180’, а иногда 90° для пленки
различной толщины. Зависимость величины адгезии от толщины
представляла собой прямые линии, наклоны которых, согласно
[292, 2931, связаны с жест-
55-
32-
28-
24-
20-
Толщина,мм 0,254
Рис. 141. Зависимость адгезии полиурета-
новых пленок ог природы полиэфира и
угла отрыва
1 — 90°, П-410, 2—180, 11410 3 — 90",
П 410 . ТР 740, 4 — 180°, П410 ГР 740,
5 __ 90°, ТП-740. С> —180 , ТП-740
костью пленки* чем больше
наклон, тем больше жесткость.
Экстраполяция прямых линий
на нулевую толщину давала
величины, которые являлись
характеристиками адгезии ис-
следованных материалов.
Для изучения зависимости
адгезии от структуры приме-
няемого полиола получены
пленки на основе полипро-
пиленгликоля (серия П), по-
лиоксинропилеповой произ-
водной триметилолпропана
(серия ТП) и полиоксипро-
пиленовой производной гли-
церина (серия ГП). Все гли-
коли и полиолы имели раз-
личный молекулярный вес.
Пленки отрывали иод двумя
углами со скоростью отрыва
0,25 см! сек.
Как видно из рис. 141, наи-
большую адгезию имеют по-
крытия на основе полипропиленгликоля П-410, наименьшую — на ос-
нове ТП-740. Величина адгезии больше при отрыве под углом 90\
При сопоставлении данных по адгезии и механическим характерис-
тикам оказалось, что у пленок с высокой адгезией наибольшее удли-
нение при разрыве и наименьший модуль при 100%-ном удлинении.
Автор [292, 2931 приходит к выводу, что пленки на основе П-410,
имея наименьшую жесткость, легче приспосабливаются к твердой
поверхности и вследствие этого адгезия возрастает. Применение
разветвленных полиолов приводит к образованию сетки более
густой и, следовательно, с большей жесткостью отрезков цепей, что
уменьшает адгезию (см. рис. 141).
Использование стерсорегулярного полипропиленгликоля значи-
тельно увеличивает адгезию [2931 по сравнению с атактическим того
же молекулярного веса. Причиной может быть тенденция изотакти-
ческих полимеров кристаллизоваться в спиральных структурах.
В этом случае полярные группы могут находиться на внешних поверх-
ностях таких спиралей и поэтому создаются возможности для обра-
228
зования большего количества вапдерваальсовых и водородных свя-
зей с окисной пленкой па поверхности алюминия.
Подобный эффект наблюдался для полимегилметакрилата [222,
2721. Полиолы, содержание фтор, снижают адгезию, несмотря на
малые различия в жесткости пленок на основе фюрсодержащпх
полиолов и обычно применяемых полиолов.
Исследовано влияние природы диизоцианата на адгеипо покры-
тий на основе ксилилендпизоцианата, ТДИ и дифенил метан ди изо-
цианата [2931 и установлено, что по
крыгия на основе дифенил мегандиию
цианата, несмотря на их большую
жесткость, обладают наибольшей ад-
гезией к алюминиевой подложке.
Объяснения этому эффекту авторы не
дают.
Исследование зависимости адгезии
от соотношения ХСО/ОН-груии на
покрытиях, полученных на основе по
лиола ТП-700 с различным содержа
нием ТДИ показало [2941, что с увеличе-
нием соотношения NCO/OH-групп ад-
гезия уменьшается, но растет модуль
при 100%-ном удлинении и прочности
па разрыв. Уменьшение адгезии с
ростом соотношения NCO/OH, а сле-
довательно, и густоты сетки связывае-
тся с увеличением жесткости поли-
уретановой пленки.
С другой стороны, обнаружено, что
Рис 142. Влияние молекуляр-
ного веса гликоля на адгезию*
1 — II 2010 2 — П 1310, 5 —П-410
увеличение содержания уретановых и, особенно, мочевинных групп
приводит к повышению адгезии, несмотря на возрастание плотности
сшивки.
Для установления влияния на адгезию полиуретанов обоих фак-
торов одновременно изучена адгезия пленок на основе гликолей и
разветвленных полиолов разного молекулярного веса. Увеличе-
ние молекулярного веса полиола приводит к уменьшению содер-
жания уретановых групп и плотности сетки одновременно.
Такая же зависимость получена для гликолей (рис. 142) и раз-
ветвленных полиолов. Согласно 1292], в данном случае увеличение
адгезии в результате роста содержания уретановых групп значи-
тельно перекрывается се снижением за счет возрастания жесткости
пленки, обусловленной повышением плотности сшивки. Если это так,
то адгезия достигнет максимальной величины при каком-то среднем
содержании уретановых групп, обеспечивающем оптимальную элас-
тичность пленки.
При получении полиуретановых покрытий применяются различ-
ные по своей природе растворители. Известно, что в зависимости от
229
термодинамического качества растворителя молекулы полимеров име-
ют различную конформацию и растворе. Степень контактирования
полимерной цепи .с твердой поверхностью зависит от формы цепи, а
следовательно, и природы растворителя. Все это должно сказаться
па величине адгезии полимерной сетки, в данном случае полиурета-
новой, к твердой поверхности.
Рис 143. Зависимость адгезии от природы растворителя:
/ — циклен ексанон 2 — диоксан, 3 — ацетон, 4 — хлороформ,
5— бензол; 6 — ксилол
Влияние растворителя, используемого для получения лака, па
адгезию исследовано в работе [293]. Полиуретановые покрытия по-
лучали на основе полипропиленгликоля, триметилолпропана и ТДИ.
Как видно из зависимости величины адгезии от природы применяе-
мою растворителя (рис. 143), наибольшей адгезией обладают пленки,
полученные с использованием циклогексанона, наименьшей—с при-
менен ием ксилола. Автор связывает это с изменением плотности
энергии когезии растворителя. Но для циклогексанона, диоксана и
ацетона плотность энергии когезии одинакова, а адгезия различна,
следовательно, такое объяснение неприемлемо.
Одна из причин зависимости адгезии от природы растворителя,
согласно 12931,—различная скорость испарения растворителя при
отверждении покрытия. Нам кажется наиболее правильным такое
объяснение влияния природы растворителя на адгезию, которое
\ читывает зависимость конформации цепей от природы растворителя.
230
Известно, что в хорошем растворителе полимерные цепи более
развернуты и, следовательно, обеспечивается в большей степени их
контакт с поверхностью После испарения растворителя следует
ожидать большую адгезию. В плохих растворителях (хлороформе,
ксилоле и бензоле) молекулы сворачиваются, что затрудняет их
контакт с поверхностью и умень-
шает адгезию.
При исследовании адгезии по-
лиуретановых покрытий опреде-
ленный интерес представляет
зависимость адгезии от природы
и концентрации наполнителей и
добавок, вводимых в пленки по-
лиуретанов. Изучепиевлиянияне
которых наполнителей на адге-
зию полиуретановых покрытий к
алюминию [292] показало, что
введение двуокиси титана сни-
жает адгезию покрытий на ос-
нове ПОПГ-1300, полиоксипро-
пиленового производного три-
метилол пропана и ТДИ на 15%,
а тальк—на 40%. Зависимость
величины адгезии пленки па ос-
нове П-2010, ТП-1540 и ТДЦ от
природы других наполнителей
представлена на рис. 144, из ко-
торого видно изменение наклона
прямой зависимости адгезии от
толщины пленки. Автор объяс-
няет это тем, что пленка стано-
вится жестче с введением па-
Рис. 144. Зависимость адгезии от при-
роды наполнителей:
/ — 7пО, 2 — СаСОя, 3 — CaCOd кубичеС’
кой формы, 4 — ТЮ2 5 — BaSO4, 6 —без
наполнителя
полнителя, от природы которого
зависит наклон. Введение ZnO и СаСО3 приводит к увеличению
адгезии в отличие от ТЮ2 и талька. Увеличение адгезии наблю-
далось и при введении в полиуретановые пленки Fe2O3. Инте-
ресно, что увеличение адгезии не симбатно с ростом концентра-
ции таких наполнителей, какГе2О2 и СаСО3 (рис. 145). Максимальное
увеличение адгезии наблюдается при оптимальном содержании на-
полнителя. Для Fe2O3 и СаСО3 эта величина составляет 15 об.%.
Увеличение же содержания окиси хрома в пленке приводит к моно-
топному снижению адгезии.
Влияние наполнителей на величину адгезии согласно [292] за-
висит от формы часгиц наполнителя. Автор также предполагает, что
наполнители способны изменять смачивающую способность полиуре-
тановых покрытий по отношению к металлу, хотя никаких экспе-
риментальных данных по этому вопросу не приводится. Влияние
231
наполнителей на адгезию в данном случае можно объяснить, исхо-
дя из различного взаимодействия наполнителей с полимерными пе-
нями или, точнее, с оiрезками полимерных цепей между узлами, из-
меняющего структуру полимера. Кроме того, необходимо учитывать
влияние различной природы наполнителей на протекание реакций
при образовании полиуретановых покрытий, что также отражается
на структуре пленки, а следовательно,и на адгезии ее к подложке.
Работы в этом направле-
нии, к сожалению, не
проводились.
На адгезии полиуре-
танов сказывается стру к-
тура металл ш еской под-
ложки [154]. в частности
состояние к, сталличес-
кой с груктуры меди. Изу-
чение адгезии полиуре-
тановых покрытий (лак
УР-930 и лак на основе
аддукта КТ) к поверх-
ностям холоднокатанпой
и электролитической мед-
ной фольги показало,
что адгезия к холодно-
катанной фольге гораздо
выше, чем к электроли-
тической.
Рис. 145. Влияние содержания Fe2O3 на адгезию
полиуретанов.
Рентгенографическое исследование обоих видов медной фольги
показало различие в состоянии кристаллической структуры меди. Эю,
возможно, и обусловило резкое различие в адгезии полиуретанов к
поверхности холоднокат анной и электролитической медной фольги.
Как известно, полиуретановые композиции применяются в ка-
честве клеев. На прочность склеивания влияют те же факторы, что
и на адгезию полиуретановых покрытий: природа полиэфира, поли-
изоцианата и т. д. В работе [3081 выяснено влияние гидроксильного
числа полиэфира и добавок различных соединений на прочность скле-
ивания дюралюминия полиуретановой композиции на основе «Дес-
модура ТН» (2,4-ТДИ триметилолнропан) и «Десмофена-800».
Показано, что с уменьшением гидроксильного числа «Десмо-
фена-800» увеличивается прочность склеивания, которая характе-
ризуется величиной прочности на срез.
Введение в полиуретановую композицию различных добавок при-
водило как к увеличению, так и к уменьшению прочности па срез в
зависимости от природы добавки. Так, добавление алифатических гли-
колей значительно уменьшало прочность па срез. Обратный эффект
достигается при добавлении в полиуретан моно- и диоксибензола,
ди метил оксибензол а и особенно метил- и фенилзамещенных этокси-
232
силана. Влияние добавок на прочность склеивания авторы объяс-
няют каталитическим действием некоторых из них.
Изучение зависимости прочности склеивания дюралюминия по-
лиуретаном от температуры показало, что с увеличением температу-
ры, при которой проводят склеивание, увеличивается прочность на
срез, достигая максимального значения при 130 С. Исследованные
клеевые соединения неустойчивы к действию многих органичес-
ких растворителей и воды. Так, в ацетоне прочность клеевого сое-
динения падает на 100, в бензоле — на 70, .м-к-резоле и воде — на 60
и в бензине — на 7%. Эти результаты противоположны данным,
относящимся к прочности прилипания пенополиуретана к металлам
и зависимости ее от температуры и действия воды [ 1331.
Таким образом, применение полиуретановых композиций в ка-
честве покрытий и клеев дает хорошие результаты и зависит от ряда
факторов. Основное значение имеет наличие полярных уретановых
групп. Этот вывод напрашивается при рассмотрении исследований,
касающихся улучшения адгезии синтетических и натуральных кау-
чуков к металлу и древесине при добавлении к ним изоцианатов 12531.
Хорошие результаты получаются при обработке склеиваемых пред-
метов (из металла и древесины) триизоциана том и последующим
нанесением клея из каучука. Адгезия некоторых полимеров к различ-
ным поверхностям улучшается при смешивании их с полиизоциана-
тами [251, 299, 309], особенно при оптимальной примеси триизоциа-
ната. Очевидно, изоцианатные группы в небольшом количестве
образуют недостаточную концентрацию связей между поверхностью
и клеем, но, с другой стороны, применение больших количеств изо-
цианата ожесточает композицию, уменьшает гибкость полимерных
цепей и тем самым ухудшает приспосабливаемость последних к
твердой поверхности. Поэтому лучшие результаты по адгезии будут
всегда наблюдаться при каком-то оптимальном содержании изо-
цианата, применяемого в качестве примеси к другим клеям [2531 и
полиуретановым композициям.
Обзор работ по адгезии полиуретановых покрытий и клеев носит
поверхностный характер. При рассмотрении влияния составляющих
(полиэфира, диизоцианата, концентрации различных групп) на ад-
1езию ни в одном случае не изучалось влияние этих составляющих
на структуру полиуретанов. Между темструкту ра трехмерной сетки,
возникающая при отверждении полиуретанов на твердой поверх-
ности и определяемая как природой составляющих полиуретанов,
таки природой твердой поверхности, на которой идет формирование
покрытия, является основным фактором, определяющим адгезию.
Поэтому при исследовании адгеши полиуретановых покрытий к
различным поверхностям следует подходить к этому вопросу с точ-
ки зрения зависимости адгезии от структуры трехмерной сетки.
Прежде всего необходимо исследовать влияние твердой поверхности
на структуру трехмерной сетки. Поверхность оказывает заметное
влияние на характер формирующейся сетки вследствие того, что
233
отрезки цепей между химическими узлами сетки, обладая заметной
гибкостью, способны приспосабливаться к поверхности и взаимодей-
ствовать с ней. При этом образуются дополнительные физические
связи полимер -— подложка, концентрация которых зависит от при-
роды и соотношения составляющих.
Для исследования изменения структуры сетки полиуретанов в
присутствии твердой поверхности изучали эффективную плотность
пленок, находящихся на подложке, и свободных пленок, полученных
в аналогичных условиях отверждения [67, 1161. Объектами иссле-
дования служили полиуретановые покрытия на основе сополимера
тетрагидрофурана с 25% окиси пропилена (ТГФ — 25% ОП) раз-
ного молекулярного веса, олигодиэтиленгликольадипината (ОДА),
олигодиэтиленгликольсебацината (ОДС), а также кремнийсодержа-
щих полиолов. В качестве изоцианатной компоненты брали аддукт
триметилолпропана с ТДИ (аддукт 1) и полиизоцианат биуретовой
структуры (аддукт 2).
Упрощенные формулы аддуктов следующие:
О /пз
CI I2—NH—С—О— / У - NCO
/ О /СНз
СН3—СН ,-С----СН„- NH -(!—О— \—NCO
\ о ~:сн3
\ II /—(
ci INI1-С-О-/____\ _ NCO
Аддукт 1
NH (СН>)в-NCO
0= с/
N— (CIIJ.-NCO
О - cf
xNII(CH,)e—NCO
Аддукт 2
Свободные пленки полиуретанов получены путем огливки компо-
зиции на тефлоне или стекле, предварительно обработанном гак,
чтобы свести адгезию пленки к поверхности до минимума. Для опре-
деления густоты сетки в присутствии твердой поверхности покрытия
наносили на алюминиевую фольгу толщиной 14 мк. Величину эф-
фективной плотности сшивки характеризовали величиной среднего
молекулярного веса между узлами сетки Ме.
Получены зависимости Мс свободных пленок и пленок на под-
ложке полиуретановых покрытий на основе ТГФ — 25% 011 от моле-
кулярного веса исходного полиэфира при соотношении NCO/OH =
-~3:1 (рис. 146), а также от соотношения NCO/OH-групп при мо-
лекулярном весе указанного выше полиэфира, равном ЮбЭ (рис. 147).
234
Густота сетки на подложке всегда больше (меньше Л1(), чем свобод
ных пленок, что говорит о возникновении дополнительных, физи-
ческих связей полимер — подложка. С увеличением молекулярного
веса исходного полиэфира (см. рис. 146) при одном и том же соотно-
шении NCO/OH уменьшается эффективная плотность сшивки, что
связано с уменьшением содержания NCO-грунп в единице объема и,
следовательно, с уменьшением числа межмолекулярных связей,
обусловленных взаимодействием полярных групп между собой и с
подложкой. С увеличением соотношения NCO/OH увеличивается
Рис. 146. Зависимость Мс пленок на Рис. 147. Зависимость Мс пленок на ос
основе ТГФ — 25% ОП от молекул яр- нове ТГФ— 25% ОП-1000 от соотноше-
ного веса полиэфира: иия NCO/OH
1 — свободная пленка, 2 — пленка на / — свободная пленка. 2 — пленка па
подложке. подложке
эффективная плотность сшивки, что вполне закономерно, но вместе
с тем уменьшается различие между Ме свободной пленки и пленки
на подложке. При соотношении NCO/OH -- 4 : 1 в обоих случаях М.(
практически одинаково. Очевидно, при меныпнх соотношениях
NCO/OH (при меньшем количестве введенных химических узлов сет-
ки) приспосабливаемость отрезков цепей между узлами к твердой
поверхности больше, чем при больших соотношениях NCO/OH, когда
наличие избытка NCO-групп способствует жесткости цепей, вслед-
ствие протекания вторичных реакций с образованием аллофанат-
ных и биуретовых связей. Поэтому при меньших NCO/OH возможно
образование большего количества связей полимер — подложка.
Уменьшение различия между Мс свободной пленки и пленки на
подложке имеет место и при введении в полиуретановую компози-
цию на основе ТГФ — 25% ОП дополнительного структуриру-
ющего агента — триметилол пропана (табл. 67). Различие между Ме
свободной пленки и пленки на подложке уменьшается с у величением
количества введенного триметилолпропана.
235
Несколько иначе сказывается влияние подложки на густоте
сетки полиуретановых пленок на основе сложных олигоэфир гл и ко-
лей. Введение последних в
сетку должно приводить к увеличению
числа межмолскулярных физических
связей из-за наличия карбонильной
iруппы, способной ко взаимодействию
с уретановыми группами через водо-
родные связи.
Зависимость Мг пленок от соотно-
шения NCO/OH-rpynn для полиурета-
нов па основе ОДЛ различного моле-
кулярного веса (рис. 148) показывает,
что для полиуретана на основе ОДА
молекулярного веса 600 густота сетки
свободной пленки больше, чем на
подложке.
11ри увеличении молекулярного
веса ОДЛ до 800 эффективная плот-
ность сшивки свободной пленки уменьшается, что закономерно,
но величины Мг свободных пленок и пленок на подложке одинаковые.
Это связано, очевидно, с 1ем, что более сильное взаимодействие с
поверхностью исходных олигомеров изменяет условия формирования
сетки.
С одной стороны, при
формировании пленкина
поверхности, достаточно
сильно с ней взаимодей-
ствующей, происходи г
образование физических
связей полимер — позерх
ность, вносящих опреде-
ленный вклад в общую
густоту сетки. С другой
стороны, взаимодействие
олигомеров и растущих
цепей с поверхностью
приводит к ограничению
их подвижности. При
этом возможна дезактивация реакционных центров и возраста-
ние скорости обрыва реакционных цепей на поверхности, в
результате чего густота образующейся сетки уменьшается и
последняя станет более дефектной. Наличие дефектов в сетке
естественно зависит в значительной мерс от гибкости блоков в поли-
уретанах. Так, при меныпем молекулярном весе олигоэфира гиб-
кость цепи между узлами в сетке уменьшается и следует ожидать
большей дефектности сетки. Поэтому густота сетки в пленке на под-
ложке может оказаться меньше, чем в свободной пленке, что и
Т а 6 л п н а 67
Влияние добавки триметилол-
лропана на эффективную
плотность сшивки полиуретанов
на основе сополимера тетрагид-
рофурана с 25% окиси пропилена
гмгь % сво- бодной пленки пленки на под. ож- ке
0 2230 1100
25 600 460
50 450 300
Me
200 ’
100 -
о
1,25/1 1,50/1 1,75/1 NC0/0H
Рис. 1 18 Зависимость Мс пленок на основе ОДХ
от соотношения NCO/OH:
/ — свободная пленка на ОДА (мол вес 600), 2 —
пле1 ка па подложке (мол вес 600), 3 — пленка п;<
подложке и свободная пленка па ОДЛ (мол вес 800)
236
Рис. 149. Зависимость Ме покрыт nit на основе
ОДС (мол. вес 600) от соотношения NCO/OH:
/ —• свободная пленка, 2—пленка на подложке
наблюдается. Увеличение молекулярного веса олигоэфира повышает
гибкость отрезков цепей, увеличивая их подвижность и способствуя
уменьшению дефектности сетки. При этом увеличивается число свя-
зей полимер — подложка вследствие лучшей приспосабливаемое™
более гибких цепей к твердой поверхности. При определенных усло-
виях возможно, что эффективная плотность сшивки на подложке
может стать равной таковой для свободной пленки, что и наблюдается
для полиуретанов на основе ОДА молекулярного веса 800. При даль-
нейшем увеличении молекулярного веса олигоэфира может оказать-
ся, что Мс на подложке
станет меньше, чем М( сво-
бодной пленки, т. е. будет
наблюдаться явление, ана-
логичное рассмотренному
для простых олигоэфиров
Ин гереспо проследить
влияние природы сложного
олигоэфира при одинаковом
молекулярном весе на гус-
тоту сетки образованных
на их основе полиуретано-
вых пленок. Установлена
зависимость Мс от соотно-
шения \СО/ОН-групп для
полиуретановых покрытий
на основе ОДС (рис. 149). Анализ рис. 148 и 149 показывает, что при
одном и том же молекулярном весе олигоэфира эффективная плот-
ность сшивки для ОДА больше, чем для ОДС. Замена ОДА на ОДС
приводит к уменьшению содержания карбонильных групп в единице
объема и, следовательно, к уменьшению концентрации вторичных
связей, что сказывается на общей rycroie сетки. В случае меньшего
соотношения NCO/OH-групп эффективная плотность сшивки па под-
ложке значительно больше, чем свободной пленки, что согласуется
с вышеизложенными представлениями.
Исследование влияния алюминиевой подложки па эффективную
плотность сшивки покрытий па основе гликоксисиланов показало
1781, что густота сетки определяется строением гликоксисилана, его
степенью разветвленности. Наряду с этим обнаружено, что после
отверждения покрытий густота их сетки в присутствии твердой по-
верхности меняется в течение длительного времени (рис. 150).
М( свободных пленок не зависит от времени, а Ме пленок на подлож-
ке несколько уменьшается. Вероятно, в присутствии твердой поверх-
ности процесс формирования сетки идет медленно, что и характери-
зуется длительным изменением Л4е во времени. Возможно также пе-
рераспределение связей со временем в самой полимерной пленке.
Для исследования зависимости эффективной плотности сшив-
ки от природы полиизоцианата получены пленки на основе
237
1еградиэтиленгликоксисилана с использованием аддуктов 1 и 2.
Показано, что абсолютные величины Мс пленок на основе аддукта 1
больше, чем на основе аддукта 2. Это связано с меньшим содержа-
нием NCO-групп в единице объема в первом случае. С другой сторо-
ны, если в пленках на основе аддукта 1 Мс на подложке больше, чем
свободных пленок, то в покрытиях на основе аддукта 2 наблюдает-
ся обратная картина. Возможно, наличие в молекуле аддукта 2
метиленовых групп приводит к образованию более гибких отрезков
200-
100-
200-~
100
200-
100-_
100-
0 п--------1 । । 1 I
20 40 60 80 Т, сутки
Рис 150 Изменение Мс во времени для полиуретановых
покрытий на основе димстилдибутиленгликоксисилана при
различном соотношении NCO/OH:
/ — 1 5 1 2 — 1.75 : 1, 3 — 2,00 : 1 (О — свободная пленка;
Ф — пленка на подложке)
цепей в пространственной сетке, что обеспечивает большее количест-
во связей полимер — подложка. Наличие же ароматических групп
в аддукте 1 способствует большей жесткости цепей в сетке, что умень-
шает возможность образования связей полимер — подложка п
обусловливает формирование более дефектной сетки.
Таким образом, эффективная плотность сшивки полиуретановых
покрытий в присутствии подложки определяется природой олиго-
эфира, его молекулярным весом, природой используемого изоциа-
ната и соотношением NCO/OH-i рупп. Указанные фак горы влияют на
гибкость цепей полиуретанов, определяя тем самым соотношение
физических и химических узлов в сетке.
При формировании сегкн на поверхности следует учитывать од-
новременное протекание двух процессов: образование физических
связей полимер — твердая поверхность, которые должны увеличи-
вать густоту сетки; ограничение подвижности растущих цепей вслед-
ствие взаимодействия их с твердой поверхностью, которое приводит
к образованию более дефектной сетки, характеризующейся меньшим
числом узлов, чем таковая свободной пленки. В случае превалиро-
238
вания первого процесса над вторым мы получаем Мс пленки, образо-
ванной в присутствии твердой поверхности меньшим, чем свободной
пленки, что имело место для покрытий на основе простых оли-
гоэфиров. Возрастание жесткости цепей за счет увеличения соотно-
шения NCO/OH и введения олигоэфирных блоков с группами, спо-
собными к взаимодействию с уретановыми (например, сложноэфир-
ных блоков), содействует протеканию второго процесса, т. е. обра-
зованию более дефектной сетки и обусловливает увеличение Мс плен-
Рис. 151. Изменение усилия отслаивания во вре-
мени при заданной скорости отслаивания.
толщины покрытия, скорости его отслаи-
ки в присутствии под-
ложки по сравнению со
свободной пленкой.
Изменение структуры
пленки под влиянием
твердой поверхности ска-
зывается на ее адгезии.
Нами изучена зависи-
мость адгезии полиурета-
нов от густоты сетки и
природы подложки[791 и
сделана попытка опреде-
лить величину адгезии,
которая не зависела бы
от побочных факторов:
вания и в связи с этим электростатических явлений |3|.
Исследовалась адгезия покрытий на основе ТТФ — 25% ОП
молекулярного веса 1200 и аддукта 1 к алюминиевой, латунной,
стальной и стеклянной подложкам [791. Работу адгезии определяли
на адгезиометре АЗС-2 [51], у которого изменена силоизмеритель-
ная часть и место крепления испытательного ролика находится па
динамометре. Исследуемое покрытие наносилось из раствора на
испытательный ролик при его вращении и отверждалось при темпе-
ратуре 80° С. Испытательные ролики были изготовлены из металла
и стекла и имели свободное вращение, благодаря которому угол от-
рыва всегда оставался постоянным и равным 90°. Положение грани-
цы отрыва также постоянно относительно отслаивающего усилия.
При испытании край полоски исследуемого покрытия укрепляли в
зажиме, перемещающемся с заданной скоростью, которую можно
изменять в пределах 0,025—0,3 см!сек.
При испытании с заданной скоростью отслаивания происходит
плавное увеличение фиксируемого усилия (рис. 151, А Б), а затем
усилие отрыва остается почти постоянным (рис. 151, БВ). Это усилие
соответствует напряжению отслаивания при известной скорости от-
рыва Из усредненной величины напряжения при отрыве опреде-
ляли работу адгезии покрытия при данной скорости отслаивания,
отнесенную к единице ширины пленки.
Если после определения усилия отрыва с известной скоростью
отслаивания остановить перемещение зажима, то отслаивание пленки
239
продолжается вследствие релак-
сации напряжений в пленке, веду-
щей к сокращению последней. От-
слаивание происходит до тех пор,
пока напряжения в пленке не урав-
новесятся силами взаимодействия
исследуемого покрытия с поверх-
ностью ролика — силами адгезии
(рис. 151, ВГ), после чего отсла-
ивание прекращается (рис. 151,
5 точка Г). Величину остаточного
2 напряжения, отнесенную к единице
7 ширины пленки, авторы называют
5 квазиравповесной работой адгс-
2 зии. На рис. 152 приведены типич-
| ныс кривые релаксации напряже-
§ ний в пленках, по которым рассчи-
тывали величины квазиравновесной
5 работы адгезии (табл. 68).
§ Работа адгезии убывает в ряду
S сталь > латунь > дюраль > стек-
лои при увеличении густоты сетки
(увеличение NCO/OH, уменьшение
Ме) возрастает во всех случаях.
Введение в покрытия и клеи
§ соединений, содержащих изоциа-
5 натные группы, обычно приводит
| к увеличению адгезии [251, 299,
г 3091. Поэтому возрастание адгезии
° сростом NCO/OH, а следователь-
§ но, и густоты сетки является ло-
| гичным результатом. Для нагляд-
S ностп представлена зависимость
работы адгезии от гусюты сетки
! (рис. 153). Для покрытий с боль-
§ шей густотой сетки наблюдается
§ резкая зависимость адгезии от при-
? роды подложки, несмотря на то,
ю что изменение густоты сетки ч при
~ изменении типа твердой поверх-
- ности невелико. Возможно, резкое
= увеличение адгезии с изменением
2 природы поверхности можно объ-
яснить различным взаимодействием
« подложек с исследованными покры-
с тиями из-за неодинакового харак-
тера связывания функциональных
240
групп, находящихся на поверхности металлов, с полиуретанами
При объяснении зависимости адгезии от природы подложки нуж-
но учесть следующее. Известно, что поверхностное натяжение мате-
риалов исследуемых под-
ложек убывает в том же
порядке, что и работа
адгезии, определенная
нами для этих подложек
[129, 2731 (табл. 69). Ве-
щества с большим повер-
хностным натяжением
обладают большей повер-
хностной энергией, что,
возможно, и обусловли-
вает более сильное вза-
и модействие исследуемых
полимеров с металлами
(в частности, со сталью),
чем со стеклом. Анализ
Рис. 152. Изменение усилия отслаивания во вре-
мени за счет напряжений, возникших в пленке
при ее отслаивании, со скоростью 0,025 см! сек
для покрытия с соотношением NCO/OII — 2’1
и с добавкой 5О°6 ТМП*
результатов табл. 69
/ — сталь, 2 — дюраль, 3 — стекло
показывает, что работа
адгезии зависит от скорости отслаивания. Кроме того, с увеличе-
нием толщины покрытия растет работа адгезии (рис. 153, кривая /).
Увеличение работы отрыва с ростом толщины наблюдается обычно
при использовании метода отслаивания [31 и особенно характерно
для эластичных покрытий. Это яв-
ление связанос напряжениями, воз-
никающими при изгибе, а также с
деформацией пленки по толщине
при отрыве ее от подложки. Работа,
затрачиваемая на деформацию плен-
ки, увеличивается с ростом скорос-
ти деформации и толщиной пленки,
влияя тем самым на работу от-
рыва покрытия.
Таким образом, для получения
сравнимых результатов по вели-
Рис. 153 Зависимость квази равновесной
работы адгезии от густоты сетки:
/ — латунь 2 *— дюраль
чине работы адгезии необходимо исследовать покрытия одинаковой
толщины при одной и той же скорости отслаивания. Методика полу-
чения пленок одинаковой толщины очень сложная. Кроме того, для
оценки адгезии покрытий одной природы, но разной густоты сетки
даже одинаковая толщина пленок не всегда может обеспечить
241
получение сравнимых результатов, а так как механические свойства
этих пленок не одинаковы, то и работа, которая идет на их деформа-
цию при отрыве, будет раз-
Таблица 69 лична.
Величины квазиравновес-
Величины поверхностного натяжения
некоторых металлов и стекла
Вещество 1емпср । тур 1 опрс деления. сс V, дин/см Граница раздела
Железо 1267 936 Но
1310 917 Hi
Алюминии 700 840 Воздух
706 494 Пары алюминия
Стекло 550 135-170 Воздух
ной работы адгезии не зави-
сят от толщины покрытий и
начальной скорости отрыва
(рис. 154,табл. 70), что являет-
ся вполне закономерным ре-
зультатом, так как характер
и прочность связывания поли-
мер — твердая поверхность
о п р е дел яетс я структурой
сравнительно тонкого слоя
полимера, прилегающего к по-
верхности 164]. Поэтому ад-
гезия определяется не толщиной покрытия, а природой подложки и
структурой полимера.
По абсолютному значению величина «равновесной» работы адге-
зии, как видно из табл. 68, составляет 10—40% работы адгезии,
определяемой при конечных
скоростях отрыва, что согласу-
ется с выводом [911 о решаю-
щем вкладе работы деформации
в общий баланс работы отсла-
ивания.
Исходя из изложенного,
можно ожидать, что величина
квазиравновссной работы ад-
гезии, при определении кото-
рой снимаются эффекты, свя-
занные с толщиной покрытия,
деформацией его, скоростью
отслаивания и в связи с этим
электростатическими явления-
ми, более правильно характе-
Таблица 70
Изменение работы адгезии со
скоростью отслаивания
Подложка Скорость отслаива ния. см/сек Рдбгта ад езии X X Ю“5, эрг см‘г Квазирав- новесная работа ад- 1езии X х io-5, эрг, см
Сталь 0,300 6,84 0,56
0,100 4,68 0,60
0,025 3,73 0,54
Латунь 0,300 5,98 0,97
0,100 5,10 0,98
0,025 3,04 0,88
ризуст величину межмолекулярного взаимодействия* на границе
раздела, чем определяемая обычно работа отслаивания поли-
мерных покрытий от твердой поверхности.
Отмечалось, что твердая поверхность оказывает существенное
влияние на эффективную плотность сшивки сетчатых полиуретанов.
Интересно выяснить, на какую глубину простирается действие под-
ложки в исследованном случае. С этой целью проведено определение
эффективной плотности сшивки как свободных пленок (полученных
путем налива на пленку из фторопласта), так и пленок на материалах
различной природы [54]. Толщина пленок изменялась от 10 до 300 лк
242
Как видно из зависимости величины Мс от толщины пленки на
основе ОДА-1200 (рис. 155), эффективная плотность сшивки свобод-
ной пленки, т. е. пленки,
полученной практически
при отсутствии взаимодей-
ствия полимер — твердая
поверхность, в исследуемом
интервале не зависит от
толщины. Величина М.с пле-
нок на подложках в значи-
тельной степени определя-
ется не только наличием
твердой поверхности, но
и природой последней.
В пленках на стали и лату-
ни, адгезия полиуретановых
покрытий к которым выше»
чем к алюминию, различие
в значении Мс со свободной
пленкой несколько выше,
чем для алюминия. Это,
Рис. 154. Зависимость работы адгезии от
толщины покрытия подложки из стали при
NCO/OH-2 1:
/ — скорость отрыва 0,025 см!сек, 2 — квазмрав
новесная работа адгезии
очевидно, связано с тем, что в случае латуни и стали образу-
ется большее число контактов полимерных цепей с поверхно-
стью, что уменьшает Мс.
По мере увеличения толщины покрытий различие между Ме
свободных пленок и пленок на подложке уменьшается и при толщи-
Мс не 120 мк и выше Мс в
300 J обоих случаях становят-
ся одинаковыми.
Эти результаты под-
тверждают выводы о том,
что влияние твердой по-
верхности существенно
отражается на структуре
как линейных, так и
сетчатых полимеров и
простирается на значи-
тельную глубину поли-
ера в зависимости or
прягоды и физического
состоя ния последнего
[64]. Поэтому нами
-----1,-------1----j---1----1---1—
0 40 80 120 160 200 240 280
Толщина-103 мм
Рис. 155. Зависимость Мс от толщины пленки на
основе ОДА-1200:
/ — свободная пленка; 2 — на стали, 3 — на лату
ни; 4 — на дюрале,
для получения сравнимых результатов при определении Мс
пленок на подложке использовались пленки толщиной до 40 мк
[54, 781. Исследована зависимость адгезии полиуретановых покры-
тий от природы олигоэфира, а также его молекулярного веса и гус-
тоты сетки [781. В качестве олигоэфиров брали олигодиэтиленгли-
243
кольаднпинаты молекулярных весов 600 и 120 (ОДА-600 и
ОДА-1200) и ТГФ —25% ОП-1200.
Зависимость величины Мс от соотношения NCO/OH, характери-
зующего химическую плотность сшивки для покрытий на основе
ОДА-1200 (рис. 156), показывает, что на всех кривых наблюдается
более или менее выраженный минимум в области NCO/OH = 2:1.
Очевидно, вначале с ростом NCO/OH происходит увеличение эффек-
тивной плотности сшивки, что наблюдалось и раньше [73, 79]. При
NCO/OH = 4:1 эффективная плотность сетки падает вследствие не-
Рис 156. Зависимость величины Ме от со-
отношения NCO/OH-групп для покрытий
на основе ОДА-1200
которого уменьшения числа
химических и физических уз-
лов. Оно связано с образова-
нием дефектной химической
сетки из-за сильного ограни-
чения подвижности цепей на
глубоких стадиях реакции.
При NCO/OH = 2:1 эффек-
тивная плотность сшивки наи-
большая Это отражается и на
величине квазиравновесной
работы адгезии, определенной
для данного случая Как видно
из рис. 157, существует экстре-
мальная зависимость между
величиной адгезии и химичес-
/ — сталь* 2 - латунь; 3 - дюраль, 4 - КОЙ ПЛОТНОСТЬЮ СШИВКИ. При
свободная пленка NCO'OH -2:1 (наименьшее
Мс) имеет место наибольшая
адгезия. Если же представить зависимость адгезии от величины
общей, эффективной плотности сшивки, включающей в себя хими-
ческие и физические узлы, то наблюдается симбатная зависимость
работы адгезии от Ме (см. рис. 157).
На основании рис. 156 и 157 можно сделать вывод о влиянии при-
роды подложки на адгезию: полиуретановые покрытия на основе
олигодиэтиленгликольадипината (мол. вес 1200), как и покрытия на
основе простых олигоэфиров [791, обладают высокой адгезией к
подложкам с большей поверхностной энергией (стали, латуни).
Интересно сравнить результаты по адгезии покрытий на основе
сложного олигоэфира с адгезией покрытий на основе простого олиго-
эфира того же молекул яр ио го веса.В табл. 71 представлены данные по
квазиравновесной работе адгезии полиуретанов на основе ОДА-1200 и
сополимера тетрагидрофуранас окисью пропилена, из которой видно,
что порядок изменения величины работы адгезии взависимости от при-
роды подложки одинаков. Наряду с этим адгезия в случае простого
олигоэфира выше, чем для сложного того же моле <улярного веса.
Это можно, очевидно, связать с различием в энергии взаимодейст-
вия полимерных цепей друг с другом в обоих случаях.
244
Известно, что межмолекулярное взаимодействие в полиуретанах
на основе сложных олигоэфиров значительно больше, чем в простых
|114]. Естественно поэтому, что гибкость отрезков цепей между
узлами для сложных олигоэфиров меньше и, следовательно, приспо-
сабливаемость их к поверхности хуже, чем отрезков цепей полиуре-
5
Рис 157. Зависимость квазиравновесной работы
ад|езии от соотношения NCO ОН (а) и величин Мс
(б) для пленок на основе ОДА-1200
/ — сталь; 2 — латунь J — дюраль, 4 — стекло.
танов па основе простых олигоэфиров. В связи с этим и число контак-
тов полимер — поверхность покрытий па основе сложных олиго-
эфиров будет меньше, чем на основе простых, что ведет к уменьшению
адгезии первыми. Нужно еще иметь в виду различимо реакционную
способность простых и сложных олигоэфиров, что должно отра-
жаться на структуре сетки образуемых полиуретанов и на адгезии
последних к различным поверхностям.
Для исследования влияния молекулярного веса исходного слож-
ного олигоэфира на адгезию получены покрытия на основе олиго-
диэтиленадипинатов молекулярных весов 600 и 1200. В табл. 72
представлены величины квазиравновесной работы ащезии покрытий
245
на основе ОДА-600 и ОДА-1200. Работа адгезии для ОДА-600 во
всех случаях больше, чем для покрытий на основе ОДА-1200. Уве-
личение концентрации полярных групп в единице объема за счет
Таблица 71
Зависимость квазиравновесной работы адгезии
(IO*”5 эрг!см2) полиуретановых покрытий
от природы олигоэфира, соотношения NCO/OH
и типа твердой поверхности
Подлож- ка ОДА-1200 ТГФ - 25% ОП 1200
1,25 ‘1 | 1,5 :1 | 2:1 | 4 • 1 2 1 1 4 1
Сталь 0,23 0,28 0,28 0,28 0,47 1,97
Латунь 0,22 0,26 0,23 0,20 0,34 1,50
Дюраль 0,16 0,25 0,25 0,17 0,34 0,75
Стекло 0,17 0,25 0,26 0,18 0,27 0,30
уменьшения молекулярного веса олигоэфира и возрастание возмож-
ности образования связей полимер — поверхность является причи-
ной наблюдаемых различий.
Рассмотрим связь между определяемой экспериментально вели-
чиной и адгезией покрытий на основе ОДА-600.
Изменение экспериментальной величины Мс с ростом NCO/OH
для пленок на основе ОДА-600 в присутствии различных подложек
Таблица 72
Квазиравновесная работа адгезии полиуретановых
покрытий на основе сложных олигоэфиров
Олт оэфир NCO/OH [NCO1 • 10», М0ЛЬ[СМ? Квазиравновесная работа адгезии • 10эрг [см?
Сталь Латунь Дюраль Стекло
ОДА-1200 1,25. 1 1,17 0,23 0,22 0,16 0,17
1,5: 1 1,24 0,28 0,26 0,25 0,25
2: 1 1,50 0,28 0,23 0,25 0,26
4 1 2,16 0,28 0,20 0,18 0,18
ОДА-600 1,25: 1 1,85 0,64 — 0,32 0,68
1,5: 1 2,04 1,24 0,59 1,24 1,18
2,0: 1 2,22 1,45 1,24 1,27 1,33
(рис. 158) показывает, что эффективная плотность сшивки свобод-
ной пленки больше, чем пленок на поверхности латуни и дюраля, но
несколько меньше, чемнастекле и стали.Обуменьшении эффективной
плотности пленок на основе сложных олигоэфиров малых молеку-
лярных весов в присутствии твердой поверхности по сравнению со
свободной пленкой уже сообщалось [671. Эту аномалию связывали с
246
влиянием твердой поверхности на полимсризационные процессы,
приводящие к образованию более дефектной сетки.
С другой стороны, можно полагать, что увеличение жесткости
полимерных цепей при уменьшен и и молекулярного веса олигоэфира
способствует меньшему числу контактов их с поверхностью. Послед-
нее обстоятельство можно про-
Мс верить при определении адге-
зии. Так, величина Мс в плен-
ках на латуни и дюрале почти
одинакова и больше, чем сво-
бодной пленки, как видно из
7,25.7 7,50.7 2tOQ.t NCO/OH 30 50 70 90 110 Мс
Рис. 158 Зависимость величины Мс от со-
отношения NCO/OH-групп для покрытий
на основе ОДА-600:
1 — латунь. 2 — дюраль, 3 — стекло. 4 —
свободная пленка
Рис. 159. Зависимость квазиравно-
весной работы адгезии от величины
Мс для покрытий на основе О ДА/600:
/ — сталь, 2 латунь 3 — дюраль,
4 — стекло
рис. 158. Однако адгезия к дюралю исследуемых покрытий зна-
чительно выше, чем к латуни (рис. 159). Следовательно, при фор-
мировании покрытий на латуни уменьшается число контактов по-
лимер — поверхность, в случае же дюраля увеличение числа кон-
тактов полимерных цепей с поверхностью, приводящее к увеличению
адгезии по сравнению с латунью, одновременно сопровождается
уменьшением числа узлов в объеме, что отражается на величине Мс.
Что касается стали и стекла, то в этих случаях наблюдается наи-
большая адгезия пленок на ОДА-600 и наибольшая эффективная
плотность сетки (см. табл. 71, рис. 161).
Итак, уменьшение молекулярного веса исходного олигоэфира
приводит, с одной стороны, к увеличению адгезии, с другой — изме-
няется порядок в ряду по природе подложек, установленном ранее
для олигоэфиров на основе ОДА-1200 и простого олигоэфира 173|.
Так, если адгезия изменялась в ряду сталь > латунь > дюраль >
> стекло и находилась в соответствии с величиной поверхностной
энергии исследуемых материалов, то в случае покрытий на основе
247
ОДА-600 этот ряд имеет следующий вид: сталь > стекло > дюраль >
> латунь.
Авторы 154] полагают, что такая инверсия в ряду, также как
и зависимость ад1езии от природы исследуемых подложек, в значи-
тельной степени может быть связана с процессами структурообразо-
вания, происходящими на границе раздела с твердой поверхностью,
определяемыми природой последней и строением исходных составля-
ющих полиуретанов.
Таким образом, анализ немногочисленных работ, посвященных
изучению адгезии полиуретанов, говорит о том, что адгезион-
ные свойства исследуемых покрытий определяются, во-первых, их
химическим строением, а именно природой олигомерных блоков,
изоцианата, сшивающего агента; во-вторых, структурой сетки,
образующейся при отверждении покрытий на подложке. Применение
блоков различной гибкости в цепях полиуретанов и подложек раз-
ной природы — все это изменяет структуру сетки, соотношение фи-
зических и химических связей в ней и вместе с тем определяет
адгезионные свойства полиуретановых покрытий.
СВОЙСТВА И СТРУКТУРА
ЭЛАСТОМЕРНЫХ ПОЛИУРЕТАНОВЫХ ВОЛОКОН
В последнее время разрабатываются новые материалы на основе
линейных полимеров, проявляющие при определенных условиях
свойства эластомеров, но при повышенных температурах или воз-
действии растворителей ведущих себя как обычные термопластичные
материалы. Речь идет о полимерах, которые в режиме эксплуатации
проявляют эластичные свойства при отсутствии достаточно прочных
химических связей, объединяющих отдельные макромолекулы в
единую пространственную сетку.
Такая высокоэластичность связана с гибкостью цепей макромо-
лекул, имеющих в своем составе группы, которые в условиях экс-
плуатации находятся как бы при температуре ниже температуры
стеклования этих участков полимерных цепей. Данные системы по-
лучили общее название эластопластов. Типичным примером эласто-
пластов являются линейные и слабо сшитые полиуретаны, которые
служат исходным материалом для получения высокоэластических
полиуретановых волокон. Схематическое строение таких молекул
представлено на рис. 160. Мягкие сегменты, образованные линейны-
ми цепями, например цепями олигоэфиров, входящих в состав поли-
уретановой молекулы, находятся при условиях эксплуатации в вы-
сокоэластическом состоянии. Температура стеклования жестких
сегментов участков изоцианатной компоненты в обычных условиях
значительно выше. Таким образом, жесткие сегменты составляют
узлы физической пространственной сетки, которая в целом способ-
на к высоким обратимым упругим деформациям и не способна к те-
248
чению при обычных температурах. Повышение температуры приво-
дит к распаду физических узлов, а иногда и слабых химических (на-
пример, аллофанатных или биуретовых), и система приобретает спо-
собность к вязкомх течению, что позволяет перерабатывать такой
полимер обычными методами, применяемыми для термопластов.
Направление вытяжки
Рис. 160. Принцип построения сегментированного уретано-
вого эластомера из жестких и мягких сегментов.
Методы получения и некоторые свойства термопластических
полиуретанов с лабильными химическими связями, разрушающими-
ся при высоких температурах и восстанавливающихся при низких,
изложены в работе 1342].
Описанный принцип построения цепи лежит в основе синтеза
линейных или слабо сшитых полиуретановых эластомеров, из ко-
торых методами сухого или мокрого формования получаются волок-
на (типа спандекс, лайкра и др.), обладающие высокой прочностью
(0,6—0,8 г!денье), высокими обратимыми удлинениями (500—800%)
и модулем, в два-1ри раза превышающим модуль упругости каучу-
ковых волокон. Отличие таких волокон от других синтетических, на-
пример полиамидов, заключается в том, что последние после вы-
тяжки приобретают ориентированную устойчивую структуру, не
исчезающую после снятия растягивающего напряжения, в то время
как полиуретановые волокна способны к самопроизвольному обра-
тимому возвращению в исходное состояние.
Для сравнения приведены кривые зависимости напряжения ог
деформации для ориентированного полиамидного, полиуретаново-
го эластичного и каучукового волокон (рис. 161). Эластичные
249
полиуретановые волокна можно получать на основе пространственно
сшигых и термопластичных полимеров. Они сильно отличаются по
своим эластическим свойствам и характеризуются высокой проч-
ностью и удлинением.
Показана возможность получения на основе линейных полиуре-
танов волокон, обладающих свойствами, промежуточными между
Рис. 161. Диаграмма напряже-
ние — удлинение для различных
волокон:
/ — ориентированное полиамидное,
2 — полиуретановое, 3 каучу-
ковое.
Рис. 162. Зависимость вязкости от
концентрации раствора полимера в
диметилформамиде:
1 — на основе полнэтиленгликольадипи-
ната (мол вес 2000) и дифснилметандиизо-
цианата, 2 — тот же полиуретан, сшитый
гидразиновыми мостиками.
высокоэластичными полиуретановыми волокнами и волокнами типа
перлон. Это связано с чередованием в полимерной цепочке полиэфир-
ных и гликолевых блоков различной природы и входящих в состав
полимера в разных соотношениях 123, 24].
Волокна из эластичных полиуретанов можно получить, как уже
отмечалось, па основе линейных и частично сшитых полиуретанов.
Процесс сшивания в таких системах, однако, не изменяет характер
раствора, в котором протекает синтез и который используется затем
для переработки, т. е. в растворе не наблюдается гелеобразования,
затрудняющего процесс получения волокна. Этот факт слабого сши-
вания, характерный для процессов получения высокоэластичных
полиуретановых волокон, является дополнительным подтвержде-
нием того, что роль узлов пространственной сетки, определяющих
эластичность и отсутствие течения, выполняют физические узлы,
образованные взаимодействием жестких сегментов.
Различие в степенях сшивания в прядильных растворах можно
проиллюстрировать следующим образом [297]. Рассмотрим зависи-
мость вязкости от концентрации раствора полиуретана в диметил-
формамиде (рис. 162) [297]. Кривая 1 характерна для полимера, по-
лученного в мягких условиях при взаимодействии моля полиэфира
(мол. вес. 2000) и моля дифенилметандиизоцианата. Кривая 2 харак-
250
теризует свойства раствора макродиизоцианата, полученного из тех
же исходных продуктов, но соединенных гидразиновыми мостиками
В этом случае вязкость растет значительно быстрее с концентрацией,
что объясняется большей степенью разветвленности и перепутывания
молекулярных клубков вслед-
ствие сшивания цепей друг с
другом. Указанные различия еще
более наглядны, если сравнить
вязкость разбавленных раство-
ров полимеров в гексаметилфос-
форамиде после нагревания при
разных температурах. Вязкости
1%-ных растворов практически
Рис 164. Диаграмма напряжение -
удлинение для волокон различной
степени вытяжки при повторном вы-
Рис 163. Диаграмма напряжение — уд-
линение для волокна ликра
тягивании:
/ — ликра, 2 — вирайн, 3 — каучук
(сплошные кривые при нагружении, пунк
тирные — при снятии нагрузки)
одинаковы, хотя первоначально
существовали заметные разли-
чия. Это вызвано тем, что в слу-
чае слабо сшитого полимера при
нагревании возможен разрыв слабых связей под действием раство-
рителя. Регулирование степени сшивания волокон в процессе формо-
вания путем применения различных сшивающих агентов дает воз-
можность изменять свойства получаемых волокон.
Рассмотрим теперь некоторые механические свойства эластич-
ных полиуретановых волокон. Растянем, например, высокоэластич-
ное волокно до определенной величины (до 200%) и после этого сни-
мем напряжение. При этом наблюдается гистерезис, показанный на
рис. 163, для волокон ликра. При повторном растяжении кривая на-
251
гружения первого цикла уже не воспроизводится и лежит значитель-
ио ниже. Эго указывает на то, что структура, возникшая при форми-
ровании волокна, при первом вытягивании сильно изменяется, в ре-
зультате чего снижается напряжение, необходимое для повторного
вытягивания волокна. Если по диаграмме удлинение — напряжение
для волокон с различной степенью вытягивания сопоставить значе-
ния напряжения, необходимого для повторного вытягивания волок-
на на 150% в зависимости от
Каучук
Рис. 165. Кривые падения напряжения
при различной степени вытяжки для поли-
уретана и каучука.
общей степени вытяжки
(рис. 164), то для разных эла-
стомеров графики существенно
отличаются. При повторных
растяжениях гистерезис по-
степенно исчезает.
Данные явления напомина-
ют известный эффект Муллин-
за [92| для наполненных эла-
стомеров, а также соответству -
ют эффектам, которые описаны
нами при рассмотрении зави-
симости свойств полиуретано-
вых эластомеров от температу-
ры и растворителей. Различия
эластичных свойств поли-
уретанового и каучукового
волокон видны также из рис. 165 12811, где показаны зависи-
мости напряжения от времени при разных заданных удлинениях,
выдержке при данном удлинении в течение 30 сек и разгрузке. Как
видно, в полиуретановых волокнах в начальные моменты после
разгрузки быстрее протекают релаксационные процессы, особенно
при более высоких первоначальных удлинениях. На рис. 166 дана
зависимость величин удлинения от времени при постоянной на-
грузке для полиуретана и каучука и некоторые данные по величинам
остаточных деформаций.
Из данных по температурной зависимости модуля и упругого по-
следействия после двухкратного растяжения до 300% через 30 сек
после разгрузки (рис. 167) видно, что эластические свойства кау-
чука мало изменяются с температурой, а для полиуретана такие
изменения весьма значительны. Действительно, если для каучука
температура стеклования лежит в области —50 4----70° С, то для
жестких блоков полиуретана область стеклования находится значи-
тельно выше. Это объясняет более высокие напряжения, необходи-
мые для деформации волокон и меньшие величины упругого после-
действия. Вместе с тем до сих пор четко не установлены температуры
стеклования так называемых жестких блоков. Поэтому приведенные
рассуждения носят пока еще качественный характер, хотя некото-
рые количественные зависимости изменения температур стеклования
252
эластичных полиуретанов с изменением природы блока, молекуляр-
ного веса и количества уретановых групп приведены в главе IV-
Данных о зависимости физических свойств полиуретановых элас-
томерных волокон от их химической природы мало, так как боль-
шинство волокон является собственностью фирм и химический состав
их не известен в достаточной сте-
пени. Однако интересно привести
общие закономерности изменения
некоторых свойств волокон с
увел и чением конце11 тр аци и жест-
Рис. 167 Температурная зависимость
модуля и остаточного удлинения
различных эластичных волокон:
Рис. 166. Кривые релаксации деформации
различных эластомерных волокон.
ликра; 2—спандекс; 3 — вирлйн, 4 — каучук.
ких сегментов (рис. 168) [298]. Как
/ — эластическое восстановление после
растяжения на 300% для каучука, 2 —
для ликра, 3 — модуль каучука, 4 —
модуль ликра
видно, при этом возрастает модуль
и падает удлинение, увеличивается
также теплостойкость (в данном случае температура, до которой сох-
раняется стабильность размеров). Те же факторы определяют вос-
станавливаемость волокна после снятия нагрузки.
Данные закономерности не являются специфическими по сравне-
нию с влиянием тех же факторов па свойства полиуретановых элас-
томеров и поэтому далее останавливаться на них нецелесообразно.
Отсюда следует, что эластомерным волокнам присуща характерная
особенность полиуретанов* структурная сетка, образованная под-
вижными связями, способна к перестройке при деформации или дей-
ствии температуры, что определяет ее механические свойства.
В связи с изложенными особенностями механического поведения
эластичных полиуретановых волокон необходимо рассмотреть немно-
1 очисленные представления о структуре таких волокон.
До настоящего времени проведено фактически только одно об-
стоятельное исследование структуры волокон типа дорластан |173,
2981. Представления о структуре основаны па схеме чередования
в цепи жестких и гибких сегментов, образующих упорядоченные
и неупорядоченные области (рис. 169). Эта схема полностью со-
ответствует весьма старой схеме аморфно-кристаллического строения
253
полимеров, приводимой еще Алфреем [2] и основанной на моде-
ли Германа — Герногросса. Однако в старых моделях предпола-
галось, что упорядоченные и неупорядоченные области образованы
звеньями цепи той же химической природы.
Количество диизоционата на 100 г
полиэфира^
Рис. 168. Зависимость механических свойств эластичных воло-
кон от содержания жестких сегментов:
1 — удлинение. 2 — модуль. 3 — теплостойкость. 4 — остаточное уд-
линение
Бонарт исследовал полиуретаны с жесткими сегментами следую-
щих типов:
О ОНИ
А. _0—С—X—Х-СН.-/ X-N—d—N-CILj-CIL— 51-С— Ж-
I 4-Z 4------Z I I II
н н н О
н
—( Х_сн / \_N_C-O-
х.__/ - х_/ ||
О О II и
II /---\ /---ч II I I
В. —9—С-.Х-( Х_си / \_N-С—N—N—C-N—
I 4 Z ' 4 Z I I II
н и н о
н
—/ X сн2—/ X _ ^„С—О—
X---/ X------/ ||
о
Эти жесткие сегменты сшиваются с мягкими блоками этилен-
диамином или 1идрашном /Мягкими сегментами в таком случае
254
являются смешанные сложные полиэфиры (I) или простые полиэфи-
ры (II). Автор снял рентгенограммы полиуретанов типа A-I, В-I и
В-П, так как полимер типа А-П плохо растворим.
Рентгенограммы невытянутых и не подвер гнутых тер ми чес кой об-
работке образцов показывают широкое аморфное гало с дв^мя ин-
терференциями.
Рис. 169 Схема строения нерастянутого волокна.
При удлинении 500% у полиуретана В-П появляется рентгено-
грамма, характерная для волокна вследствие кристаллизации при
растяжении мягких сегментов цепей. В жестких сегментах (рис. 170)
расположение типа / с четырьмя водородными мостиками энергети-
чески предпочтительнее расположения 1" с двумя мостиками.
В реальной системе может иметь место чередование различных распо-
ложений цепей друг относительно друга (например, 2 и 2'). Такая
же картина возможна для сегментов типа В.
Сложная картина пространственного расположения жестких и
мягких сегментов приводит к тому, что при вытяжке полимера ориен-
тация мягких и жестких групп может быть различна. Бонарт пока-
зал, что при удлинениях до 200—300% жесткие сегменты ориенти-
рованы преимущественно наклонно направлению деформации,
тогда как мягкие — вдоль направления ориентации (рис. 171, а).
Дальнейшее растяжение приводит к переориентации жестких блоков
цепей (рис. 171, б).
Интересно, что при термообработке ориентированного волокна
наблюдаются процессы релаксации, приводящие к почти полной
разориентации мягких сегментов; ориентация жестких сегментов
не нарушается. Термофиксация волокна, очевидно, связана с
255
Рис. 170. Возможное расположение жестких сегментов А (а) и В
разрушением части поперечных связей таким образом, что жесткие
сегменты начинают располагаться вдоль направления ориентации.
Распад ча/ти связей при вытяжке приводит к увеличению удлинения,
сопротивление которому не соответствует большой возвращающей
силе, что связано с известными явлениями размягчения под действи-
ем напряжения, аналогичными гистерезису при нагрузке и разгру-
жении. В этом случае надо учитывать не только водородные связи,
но и возможность образования узлов вследствие взаимодействия л-
электропов бензольных колец. По рентгенографическим данным, па-
раметры решетки при этом а — 5, b = 4, с =- 12,5А, у -- 67°.
Однако на рентгенограммах образцов типа A-I или В-I наблюда-
лось только паракристаллическое расположение мягких сегментов
вместо кристаллизации, вызванной растяжением. Такая картина
256
(б), приводящее к образованию водородных мостиков.
соответствует одному экваториальному рефлексу, что связывается со
статистическим расположением звеньев в цепочке взятого полиэфира.
Интенсивности наблюдаемых рефлексов могут изменяться при раз-
личной обработке образцов (при нагревании в воде в растянутом со-
стоянии и пр ). Рентгеновская картина меняется также и при пере-
ходе от полиуретана В-I к В-И Если для В-I рефлексы связаны с
определенной ориентацией мягких сегментов, а при термообработ-
ке эта ориентация исчезает и появляются рефлексы, вызванные упо-
рядочением жестких сегментов, то для В-П мягкие сегменты дают
только аморфное гало, а жесткие сегменты характеризуются замет-
ной ориентацией. Для того чтобы получить информацию об упоря-
доченном расположении тех или иных видов сегментов цепи, Бонарт
развил некоторые принципы теории интерференции и сделал
257
определенные допущения, касающиеся расположения этих сегмен-
тов в пространстве при ориентации полимера.
Кристаллическая структура линейных полиуретанов, как и по-
лиамидов, существенно определяется системой водородных связей,
расположение которых в рас-
Рис. 171. Схема волокна, растянутого примерно на 200 (а) и 500 (б) %.
возможные схемы расположения водородных связей, основанные на
рис. 170, которые можно сопоставить с результатами рентгеногра-
фического исследования для сегментов типа А и В. Как видно из схем,
возможно различное расположение водородных связей между цепями.
Рентгеновское исследование структуры эластомерных полиуре-
тановых волокон приведено также в работе [226]. Авторы рассмот-
рели структуру линейных полиуретановых эластомеров, образован-
ных из жестких и мягких блоков на основе простых и сложных поли-
эфиров с молекулярными весами 1000 и 4000.
В неориентированном состоянии рентгенограммы полимеров по-
258
называют диффузное гало и отсутствие структуры. При растяжении
появляются дискретные рефлексы, исчезающие при снятии нагрузки
Авторы полагают, что причиной высокой прочности волокна в рас-
тянутом состоянии является кристаллизация при растяжении гиб-
ких сегментов, которые составляют 70—80% всего полимера. На-
помним, что Бонарт наблюдал на аналогичных эластомерах при
удлинении до 200% практически полную кристаллизацию мягких
сегментов и систематическое распределение водородных связей. Ко-
личественная связь между степенями кристаллизации мягких сег-
ментов и вытягивания зависит от химической природы полимера.
Однако авторы считают, что возникающее при синтезе регуляр-
ное расположение мягких и жеенчих сегментов в цепи и возникаю-
щая при растяжении волокна картина позволяют ожидать определен-
ной упорядоченности и в перастяпутом материале. При этом они
предполагают, что при растяжении цепи скользят относительно
друг друга, пока сильные дипольные взаимодействия между жест-
кими сегментами не начинают препятствовать такому сколь-
жению.
Для проверки данной гипотезы проведены рентгеновские иссле-
дования под малыми углами эластомеров на простом (ликра) и на
сложном (дорластан) полиэфирах. Па диаграммах малоуглового
рассеяния волокон с непрерывным рассеянием для иерастянутых
препаратов видны серповидные рефлексы на экваторе, отвечающие
периодам 60 h для дорластана и 47—50 А для ликра. Для дорла-
стана есть еще два широких серпа на меридиане, отвечающие рас-
стоянию 71 А. Несмотря на это, авторы не склонны считать исходные
материалы упорядоченными, так как после их трехчасовой экстрак-
ции метанолом указанные рефлексы исчезают. Они, очевидно, свя-
заны с экстрагируемым веществом. Но экстрагированные препараты
проявляют все же дискретное малоугловое рассеяние. Такие мериди-
ональные рефлексы могут быть связаны с периодическими колеба-
ниями плотности в направлении волокна, средняя периодичность
которых составляет 95 А для дорластана и 130 А для ликра. При
растяжении возникают резкие рефлексы. Это дает авторам основание
заключить, что в полиуретановых эластомерных волокнах есть не-
которые типы структур, зависящие ог состава полимера и степени
его растяжения.
В связи с проблемой эластичных волокон вновь встает вопрос о
водородных связях в полиуретановых эластомерах, которые в боль-
шой мере определяют структуру волокна. Однако следует отметить,
что, например, Ринке |29бI наряду с водородными связями придает
определенное значение и обычным вандерваальсовским взаимо-
действиям ме?кду мягкими сегментами, которые, по его мнению,
определяют эффекты повторной деформации. Он считает, что для
преодоления этих связей, не разрушающихся полностью при де-
формации, затрачивается дополнительное усилие вследствие сохра-
нения некоторой степени структурированности по физическим
259
Рис. 172. Структура поперечных связей меж
со о га
। । i
w co------w
ду жесткими сегментами А (а) и В (б).
6
связям, которые после снятия нагрузки мешают восстановлению
исходных размеров.
Представления Ринке хорошо согласуются с нашими о тиксо-
тропной структурной сетке, которая определяет основные свойства
полиуретанов, и с результатами реолошчсских исследований, ука-
зывающих на важную роль обычных физических взаимодействий в
упругих свойствах олигомеров и полиуретанов. Действительно,
Ринке считает, что при повышении температуры прочность водород-
ных связей падает, причем для жестких сегментов это проявляется
особенно сильно при 130—190° С, вследствие чего физические связи
разрушаются, но сохраняют способность к восстановлению при
понижении температуры. Мы уже отмечали, что это — типичная
особенность эластопластов, которая определяет возможность форми-
рования волокон и термофиксации структур, возникающих при фор-
мовании.
Мы обнаружили также эффекты, связанные с распадом связей в
области 4(ГС, существенно влияющие на свойства эластомеров. Спе-
цифичные свойства полиуретанов (например, высокая стойкость к
истиранию), присущие как каучукам, так и термопластичньш во-
локнам типа перлон, характерны и для эластичных волокон. По
мнению Ринке, высокая стойкость к истиранию и разрушению при
действии напряжений полиуретановых волокон в сравнении с обыч-
ными является результатом дальнейшего сшивания вследствие об-
разования водородных мостиков в жестких сегментах. При действии
механических напряжений возможно частичное разрушение и вос-
становление связей между жесткими сегментами, что невозможно для
химических связей, которые после разрушения уже не восстанавли-
ваются.
Следовательно, образование физической структурной сетки, не-
регулярность которой вследствие строения цепи и структуры жест-
кого сегмента значительно больше, чем в обычных каучуках, оп-
ределяет основные физические свойства полиуретановых элас-
1ИЧНЫХ волокон.
ЗАКЛЮЧЕНИЕ
Задачей, которую ставили себе авторы в данной монографии, яв-
лялось установление физико-химических особенностей поведения
полиуретанов и выяснение причин, отличающих этот класс полиме-
ров от других. Изучение данного вопроса важно прежде всего по-
тому, что полиуретаны представляют собой единственный класс
полимерных соединений, на основе которых можно получить практи-
чески все ценные типы полимерных материалов — каучуки и плас-
тики, обычные и эластомерные волокна, клеи и покрытия, 1ерметикп
и пенопласты и др.
Анализ структуры полиуретанов, выяснение основных законо-
мерностей их образования, исследования механических и физи-
ко-химических свойств линейных и трехмерных полиуретанов и
свойств изолированных цепей позволяют сейчас установить причины,
по которым полиуретаны занимают особое место среди других полиме-
ров. Оно определяется двумя основными факторами: разнообразием
химического строения полиуретановых цепей и специфической
структурой полимерной цепи, определяющей структуру полимеров
в блоке.
Отличительные черты химического строения полиуретанов опре-
деляются прим нением для их синтеза ди изоцианатов, которые уни-
кальны по многообразию химических реакций, в которых могут
принимать участие. Использование диизоцианатов различной хими-
ческой природы (алифатических, ароматических, циклических и пр.)
значительно расширяет возможности варьирования химической
структуры синтезируемых полиуретанов. Применение в качестве
второго компонента полифункциональпых соединений самых различ-
ных классов — от низкомолекулярных гликолей до олигомеров и
сополимеров с гидроксильными концевыми группами — приводит
к такому разнообразию окончательной химической структуры поли-
меров, какого не наблюдается ни в одном из других классов поли-
меров. Вхождение в цепи полиуретанов многих типов функциональ-
ных групп и структурных единиц позволяет широко варьировать
свойства полиуретановых материалов.
Дополнительные возможности изменения свойств полиуретанов
связаны с применением для их сшивания различных по своей хими-
ческой природе соединений, способных взаимодействовать с изо-
цианатными или гидроксильными группами. В результате этого
содержание в конечном продукте реакции уретановых групп может
быть весьма незначительным. С данной точки зрения, полиуретаны
263
вообще нельзя рассматривать как единый класс полимеров (как,
например, полиамиды или пол и акрилаты), так как единственным об-
щим признаком является наличие в цепях уретановой группировки.
Другая особенность химического строения полиуретанов заклю-
чается в том, что при синтезе их используются олшомерные блоки,
которые по существу являются низкомолекулярными полимерами и
самостоятельно обладают комплексом физико-химических свойств,
присущим полимерам. Применение олигомерных блоков различной
химической природы и, следовательно, различной гибкости полимер-
ной цепи дает основание рассматривать полиуретаны как блок-
сополимеры, в цепях которых чередуются гибкие и жесткие блоки.
Чередование блоков различной химической природы позволяет ши-
роко изменять свойства полиуретанов, что и определяет возможность
получения из них всей гаммы полимерных материалов. Между тем,
принцип чередования блоков, специфичный для полиуретанов,
еще мало использован на практике. Недостаточно проведено иссле-
дований но разработке методов синтеза полиблочных полиуретанов,
в которых наряду с чередованием олигомерного и диизоцианатного
блока но цепи осуществлялось еще чередование самих блоков —
простых, сложноэфирных, сополимерных и т п.
Некоторой демонстрацией принципа чередования блоков для ви-
доизменения свойств полиуретанов является введение в полиуретаны
блоков на основе олигодиенов с концевыми гидроксильными груп-
пами, приводящее к получениюэластомеров. Получение олигоуретан-
акрилатов с концевыми двойными связями привело к появлению но-
вого класса полимеров — полиуретанакрилатов, сочетающих свой-
ства полиуретанов и полимеризациопных пластиков.
Таким образом, особенности химического строения и возможность
придания полиуретанам различных свойств определяются, прежде
всего, широким кругом исходных соединений, применяемых для
синтеза полиуретанов и относящихся к различным классам. Можно
сказать, что не существует проблемы полиуретанов как таковых,
сосуществует проблема исходных соединений для синтеза полимеров,
содержащих уг становые группы. Действительно, в силу разнообра-
зия химического строения молекулярных блоков, принадлежащих к
различным классам (полибутадиены, полиэфиры и пр.), полиурета-
ны, очевидно, не могут рассматриваться как единый класс, по долж-
ны рассматриваться шире, как группа полимеров разных классов,
для которой общим является наличие в цепях редких уретановых
групп — NCOO—. Даже участие в синтезе диизоиианатов — не
обязательное условие получения полиуретанов, так как возможны
безизоцианатные методы синтеза.
При рассмотрении особенностей физической и молекулярной
структур полиуретанов прежде всего нужно обратить внимание на
гибкость полимерных цепей и межмолекулярные взаимодействия
между ними. Природа гликолевого и изоцианатного блоков
определяет гибкость отдельных участков цепей и межмолекулярные
264
взаимодействия между ними. Это приводит к проявлению полиурета-
нами свойств эластопластов или термопластов. В случае
полиуретановых эластомеров изменение молекулярного веса поли-
эфирного блока служит одним из основных путей регулирования
свойств трехмерной сетки. Способность блока к кристаллизации, за-
висящая от молекулярного веса и химической природы, в конечном
счете определяет фазовое состояние полимеров и уровень их надмо-
лекулярной организации.
Разнообразие гетеросвязей в цепи полиуретанов существенно
влияет на их гибкость. Рассмотрение данных по внутреннему враще-
нию в органических молекулах показывает, в частности, что внутрен-
нее вращение вокруг С—О-связи облегчено по сравнению с враще-
нием вокруг связи С—С. Поэтому можно полагать, что полимерные
цепи с гетеросвязями типа простых полиэфиров обладают повышен-
ной ибкостью.
С другой стороны, наличие сильно взаимодействующих групп в
цепи должно вызвать увеличение барьеров внутреннего вращения
и ожестчение цепи. Сочетание в одной молекуле различных типов
связей, наиболее типичное для полиуретанов, определяет сложный
характер зависимости гибкости цепей полиуретанов от их химичес-
кой природы.
Большое влияние на способность молекул полиуретанов к изме-
нению конформаций оказывает собственная гибкость олигомерных
блоков, вклад которой в гибкость цепи является преобладающим.
Это очень существенно с той точки зрения, что проявление гибкости
молекул в полиуретановых сетках есть не исключительный резуль-
тат сшивания в длинную последовательность коротких олигомерных
цепей, а результат собственной гибкости олигомеров. Собственная
гибкость олигомерных блоков и возможность сильных межмолеку-
лярных взаимодействий между блоками представляется существен-
ной особенностью полиуретанов.
В пространственной сетке полиуретанов возможны наряду с хи-
мическими и водородными связями между уретановыми и другими
группами обычные вандерваальсовские взаимодействия между от-
резками цепей в сетке. Наличие в сетке таких взаимодействий, сте-
пень которых зависит от химической густоты сетки, изменяет барьер
вращения и, таким образом, гибкость отрезков цепей между узлами
даже тогда, когда расстояние между узлами превосходит величину
термодинамического сегмента, найденного для несвязанных в сетку
высокомолекулярных цепей.
Вопрос о роли межмолекулярных взаимодействий в полиурета-
нах очень важный, но специфика полиуретанов в этом отношении
проявляется только при рассмотрении трехмерных сетчатых поли-
уретанов.
Многообразие функциональных групп в цепи создает широкие
возможности для образования межмолекулярных связей различной
природы и энергии от водородных до вандерваальсовых. Существен-
255
ная роль таких межмолекулярных взаимодействий в трехмерной сет-
ке особенно характерна для полиуретановых эластомеров. Очевидно,
такие взаимодействия не исключаются и для других каучукоподоб-
ных материалов. Следует отметить, что если проблеме межмолеку-
лярных взаимодействий в линейных полимерах уделялось всегда
много внимания, то данные вопросы недостаточно освещались при-
менительно к трехмерным полимерам. Рассмотрение дефектов сет-
ки и образования захлестов и зацеплений цепей в какой-то мере учи-
тывают эти взаимодействия, однако не с точки зрения образования
межмолекулярных связей. Полиуретаны представляют собой систе-
му, в которой роль межмолекулярных взаимодействий в сетке вы-
ступает особенно отчетливо.
Одна из основных особенностей строения сетки трехмерных по-
лиуретанов заключается в том, что в ней главная роль принадлежит
не химическим, а физическим узлам, образованным в результате
межмолекулярного взаимодействия с участием различных функци-
ональных групп молекул.
Эффективная плотность сшивки в полиуретанах определяется
главным образом вторичными физическими связями, образующими-
ся в трехмерной сетке вследствие взаимодействия цепей друг с дру-
гом. Большая роль физических взаимодействий подтверждается опре-
делениями плотности энергии когезии полиуретановых каучуков,
значения которой выше, чем для обычных каучуков.
Сильные меж молекулярные связи, образующие в полиуретанах
непрерывную пространственную сетку, в определенных случаях
приводят к тому, что линейные полиуретаны, «сшитые» только фи-
зическими связями, проявляют свойства, обычно характерные толь-
ко для химических сшитых линейных цепей.
Другая особенность структуры пространственной сетки полиуре-
танов заключается в ее высокой подвижности, т. е. способности к
перестройке при воздействии температуры или механическом воздей-
ствии. Это заключение следует из аномальной температурной зави-
симости эластического модуля таких сеток и из данных по термоди-
намике высокоэластических деформаций. Распад межмолекулярных
связей в сетке при нагревании или деформации приводит к увеличе-
нию расстояний между узлами сетки. Способность сетки к распаду
и перестройке при действии температуры, растворителей и механи-
ческих нагрузок дает основание рассматривать подобную сетку по
аналогии с тиксотропными структурами, хорошо изученными в
коллоидной химии. Такое представление хорошо согласуется с резу-
льтатами исследования реологических свойств олигомеров и мак-
родиизоцианатов, которые подтверждают высокую степень тиксо-
трошюсти уже в олигомерных системах.
Наличие подвижной структурной сетки в полиуретанах обуслов-
ливает многие специфические свойства. Очевидно, сравнительной
легкостью разрушения и последующего восстановления сетки можно
объяснить способность полиуретанов к самозалечиваниюдефектов при
266
деформациях. Действительно, при деформации, которая особенно
интенсивно развивается в местах концентрации напряжений, проис-
ходит заметный распад сетки, в результате чего происходит как бы
тиксотропный переход полимера из высокоэластического в вязко*
текучее состояние. Возможное в условиях действия деформации те-
чение приводит к залечиванию дефектов в наиболее напряженных
местах. При этом пагружа распределяется снова более равномерно
и вновь восстанавливаются разрушенные связи.
Подвижность сетки способствует, очевидно, и сравнительно быст-
рому установлению равновесия и новой структуры, отвечающей
деформированному состоянию. Это будет происходить, если скорости
деформации и перестройки структуры сопоставимы. Тогда в системе
при деформации устанавливается новое равновесие, в результате
которого на каждой стадии деформации нужно будет затрачивать ра-
боту не только на растяжение цепей, но и па разрушение узлов сет-
ки. Это отличает полиуретаны от других эластомеров, где разруше-
ние связано преимущественно с деформацией цепей.
Учет особенностей физико-химического поведения полиурета-
нов позволяет подойти к выяснению причин, приводящих к повышен-
ной прочности и стойкости к истиранию. Мы полагаем, что здесь
путь к решению проблемы лежит через изучение механических и дру-
гих свойств поверхностных слоев полиуретанов. В последнее вре-
мя было установлено [22|, что процессы износа при трении
сопровождаются заметными перестройками структуры поверх-
ностного слоя полимера. В определенных условиях такая перестройка
приводит к структуре, более устойчивой к истиранию. Молеку-
лярная подвижность в поверхностных слоях и структура поверх-
ностных слоев полимеров отличаются ог таковой в объеме 164, 761,
что связано с изотропностью распределения межмолекулярных
связей в объеме и отсутствием ее в поверхностном слое. Эти обстоя-
тельства вместе с учетом особой роли физических взаимодействий в
полиуретановых сетках позволяют представить процессы истирания
полиуретанов как специфические релаксационные процессы в поверх-
ностных слоях, связанные с тиксотропным разрушением и восстанов-
лением структуры поверхностного слоя.
Рассмотренные в данной монографии закономерности изменения
свойств полиуретанов с изменением структуры и выяснение ряда
причин их специфического поведения дают возможность распростра-
нить принципы построения полиуретановых цепей на другие мате-
риалы, а реализация в них ряда характеристик, присущих полиуре-
танам,позволит подойти к созданию широкого круга полимеров и
полимерных материалов различной химической природы, которые
буду? обладать наиболее ценными свойствами, присущими полиуре-ч
танам, и будут лишены их недостатков.
267
ЛИТЕРАТУРА
1. А в е р к о - А н т о н о в п ч Ю. О., Кирпичников П. Я. — Кауч,
и рсз , 1967, 3, 12—14.
2. А л ф р е й Т. Механические свойства полимеров. ИЛ, М., 1948.
3. Андреевская Г. Д. Высокопрочные ориентированные стеклопла-
стики. «Наука», М., 1966.
4. А п у х т и н а Н. П. и др. — Высокомолек. соед., 1965, 7, 6, 1117.
5. Апухтина Н. П., Эренбург Е. Г., Раппопорт Л. Я. —
Высокомолек. соед., 1966, 8, 6, 1057.
6. А п у х т и п а Н. П., К о р о т к и н а Д. Ш., Лившиц К. С.,
Сидорович Е. А. — Кауч, и рез., 1967, 12, 11.
7. Б а б и ч В. Ф., . ипатов Ю. С., Вильчинская А. В.—
В кн.: Синтез и физико-химия полиуретанов. «На кова думка», Киев, 1967,
138.
?. Барте нев Г. М., Вишницкая Л. А. — Коллоид, ж., 1956,
18, 135.
9. Базрук Л. И., Гороховский Г. А., Липатов ГО. С. —
Высокомолек. соед., 1968, 10А, 1434.
10. Берлин А. А., Т во рогов Н. Н., Королев Г. В. — ДАН
СССР , 1966, 170, 1073.
11. Благонравова А. А., Пронина И. А. — Лакокрасочные ма-
териалы, 1961, 2, 3.
12. Боярчук ГО. В., Раппопорт Л. Я., Никитина В. Н.,
Апухтина Н. П. — Высокомолек. соед., 1965, 7, 5, 778.
13. Б о я р ч у к Ю. В., Медведева Г. И., Раппопорт Л. Я.,
Эрлих И. М. — Высокомолек. соед., 1968, Б10, 173.
14. В а с и л ь е в Б. В., Тараканов О. Г. — Высокомолек. соед., 1964,
6, 2189.
15. В а с и л ь е в Б. В., Т а р а к а н о в О. Г., Демина А. И., Ши-
робокова А. И. — Высокомолек. соед., 1966, 8, 938.
16. Вольке н штейн М. В. Конфигурационная статистика полимерных
цепей. Изд-во АН СССР, М.— Л., 1959.
17. Годовский Ю. К-, Барский
8, 395.
18. Г о д о в с к и й Ю. К«, Слонимский
1966, 8, 403.
19. Г о д о в с к и й Ю. К., Липатов
10А, 32.
20. Годовский Ю. К., Липатов
10Б, 323.
21. Годовский Ю. К., Липатов
Ю. П. — Высокомолек. соед., 1966,
Г. Л. — Высокомолек. соед.,
Ю. С. — Высокомолек. соед., 1968,
Ю. С. — Высокомолек. соед., 1968,
Ю. С.'— Высокомолек. соед., 1968,
10А, 741.
22. Гороховский Г. А. Автореф. докт. дисс. ИПП АН УССР, Киев, 1968.
23. Г р и ц е н к о Т. М. Тезисы второго Всесоюзного совещания по физико-
химии полиуретанов. «Наукова думка», Киев, 1968, 21.
24. Г р и ц е н к о Т М., Л и п а т н и к о в Н. А., Попов И. А. —
В кн.- Синтез и физико-химия полимеров (полиуретаны), 7. «Наукова думка»,
Киев, 1970.
268
25. Губанов Э. Ф., Синайский А. Г., Апухтина Н. П.,
Тейтельбаум Б. Я. — ДАН СССР, 1965, 163, 1151
26. Г у б а н о в Э. Ф., Тейтельбаум Б. Я , Апухтина Н. П.,
Синайский А. Г. — В кп.: Синтез и физико-химия полимеров (поли
уретаны), 5. «Наукова думка», Киев, 1968, 168.
27. Г у з е е в В. В., Мал и нс ки й Ю. М. — Зав. лабор , 1963, 29, 11,
1373.
28. Д ж и Г. —В кн.: Химия больших молекул, 1. ИЛ, М., 1948, 138.
29. Е г о р о в Ю. П. и др. — В кн.. Спектроскопия полимеров. «Наукова дум-
ка», Киев, 1968, 43.
30. Ерофеев Б. В. — ДАН СССР, 1946, 52, 515.
31. Жарков В. В.—Ж* прикл. спектр., 1966, 4, 572.
32. Ж а р к о в В. В. Автореф. канд. дисс. Изд. Горьковского университе-
та, 1967.
33. Ж а р к о в В. В., Р у д н е в с к и й Н. Н. — Оптика и спектр., 1968,
7, 848.
34. К а р г и н В. А. Современные проблемы науки о полимерах. Изд. /МГУ
М., 1962.
35. К а р г и н В. А., Аз о р и М., П л а т э Н. А., Б а н д у р я н С. И.—
ДАН СССР, 1964, 154, 1157.
36. К а р г и н В. А., Слонимский Г. Л. Краткие очерки по физико-хи-
мии полимеров. «Химия», М., 1967.
37. К а р г и н В. А , Письменно И. В., Чернова Е. П. — Вы-
сокомолек. соей. 1968, 10А, 4, 846.
38. К е р ч а Ю. Ю., Липатов Ю. С., Ивченко Н. К — Высоко-
молек. соед., 1967, 9А, 798.
39. К е р ч а Ю. Ю., Липатов IO. С. — В кн.: Синтез и физико-химия
полиуретанов. «Наукова думка», Киев, 1967, 193.
40. К е р ч а Ю. Ю., Липатов Ю. С., Я н г о л ь Г. А. — В кн.: Син-
тез и физико-химия полиуретанов. «Наукова думка», Киев, 1967, 126.
41. Кер ч а Ю. Ю , Липатов Ю. С. — В кн.: Синтез и физико-химия
полиуретанов. «Наукова думка», Киев, 1967, 120.
42. К е р ч а Ю. Ю., Л i п а т о в Ю. С., Середа О. В. — ДАН УРСР.
Сер. Б, 1968, 3, 256.
43. К е р ч a JO. JO., Липатов Ю. С. — Укр. хим. ж., 1968, 2, 158.
44. Кер ч а Ю. Ю , Рябоконь Л. И., Маличенко Б. Ф.—
В кн.’ Синтез и физико-химия полимеров (полиуретаны), 5. «Наукова думка»,
Киев, 1968, 198.
45. К е р ч а Ю. Ю , Л и п а т о в Ю. С., Р я б о к о н ь Л. И. — В кн.:
Синтез и физико-химия полимеров (полиуретаны), г. «Наукова думка»,
1968, 193.
46. К е р ч а Ю. Ю., Л и п а то в Ю. С., Лапти й В. С, Ватулев В. Н ,
Липатников Н. А., Гриценко Т. М.— Вкн.: Синтез и физико-химия
полимеров (полиуретаны), 7. «Наукова думка», Киев, 1970.
47. Коваленко Г. Ф., Липатов Ю. С., Сергеева Л. М. — В кн.:
Синтез и физико-химия полимеров (полиуретаны), 6 «Наукова думка», Киев,
1970, 126.
48. К о ш е в о й Л. Г. и др. — Химические волокна, 1967, 2, 6
49. К о п у с о в Л. И., Жарков В. В. — Ж. прикл. спектр, 1966,
5, 1, 125.
50. К о п у с о в Л. И., Жарков В. В. — Ж. прикл. спектр , 1967, 6, 2,
393.
51. Кротова П. А, Карасев В. В, Кириллова Ю. М — В
кн.. Новые методы физико-химических исследований, 6, Изд-во АН СССР,М.,
1957, 52.
52. К у в ш и н с к и й Е. В., Сидорович Е. А.-- ЖТФ, 1956,
26, 878.
269
53. К у з н е ц о в а В. П., Б ел о го л о в и н а Г. Н., Ще п е т ки-
на Н. И. — В кн.: Синтез и физико-химия полимеров (полиуретаны), 7.
«Наукова думка», Киев, 1970.
4. Ку кс и и А. Н., Липатов Ю. С., С е р г е е в а Л. М., К а д у-
р и н а Т. И. — В кн.: Синтез и физико-химия полимеров (полиуретаны),
7. «Наукова думка», Киев, 1970.
55. Л а р к и н а Т. А., Елисеева В. И. — Лакокрасочные материалы
и их применение, 1964, 4, 5—8.
6. Л и п а т о в а Т. Э., Иващенко В. К., Безрук Л. И. — ДАН
СССР, 1968, 178, 1116.
57. Л и п а т о в а Т. Э., Б а к а л о Л. А., Локтионова Р. А. —
Высокомолек. соед., 1968, 10А, 1554.
58. Л и п а т о в а Т. Э. Тезисы второго Всесоюзного совещания по химии и
ф зико-химии полиуретанов. «Наукова думка», Киев, 1968, 6.
59. Л и п а т о в а Т. Э., Иващенко В. К.— В кн.: Синтез и физико-хи-
мия полимеров (полиуретаны), 6. «Наукова думка», Киев, 1970, 73.
60. Л и п а т о в а Т. Э., И в а щ е н к о В. К., Н е с т е р о в А. Е., Ли-
патов Ю. С. — Высокомолек. соед., 1969 11 А, 602.
61. Л и п а т о в а Т. Э., Зубко С. А. — ДАН СССР, 1969, 184, 4, 877.
62. Л и п а т о в Ю. С. Автореф. докт. дисс. Физико-хим. ин-т им. Л. Я. Кар-
пова, М., 1963.
63. Липатов Ю С., Сергеева Л. М. — Высокомолек. соед., 1966,
8, 11, 1895.
64. Липатов IO. С. Физико-химия наполненных полимеров. «Наукова
думка». Киев, 1967.
65. Л и п а то в Ю. С., К е р ч а Ю. IO ., Синявский В. Г., Л и -
пат ников Н. А. — Высокомолек. соед., 1967, 9А, 1340.
66. Л и п а т о в Ю. С., Панов IO. Н., Сушко Л С., Френ-
кель С. Я.—ДАН СССР, 1967, 176, 6, 1341.
67. Л и п а т о в Ю. С., Сергеева Л. М., Бинькевич Н. И.,
Омельченко С. И. — Высокомолек. соед., 1968, 10Б, 11, 816.
68. Л и п а т о в Ю. С.— Высокомолек. соед., 1968, 10А, 12, 2737.
69. Липатов Ю. С., Середа О. В., Се р г е е в а Л. М., А п у х т и н а Н. П.,
Мюллер Б. Е., Мозжухина Л. В — В кн.: Синтез и физико-химия
полимеров (полиуретаны), 5. «Наукова думка», Киев, 1968, 185.
70. Л и п а т о в Ю. С., ЩспеткипаН. И.—ДАН СССР, 1968, 179, 4, 879.
71. Липатов ГО. С., Сергеева Л. М., Коваленко Г. Ф. —
Высокомолек. соед., 1968, 10Б, 3, 205.
72. Л и п а т о в Ю. С., Середа О. В., Сергеева Л. М. — Ме-
ханика полимеров, 1968, 3, 450—453.
73. Л и п а т о в Ю. С., ,К у кс ина А. Н., Сер гее в а Л. М., Сметанки-
на Н П., О п р я В. Я. — В кн.: Адгезия и прочность адгезионных соеди-
нен й, 2. Московск. дом научно-технич. пропаганды им. Ф. Э. Дзержин-
ского, 1968, 143.
74. Л и п а т о в Ю. С., Сушко Л. М., Синявский В. Г., Ба-
рашенков Ю. Ю. — Высокомолек. соед., 1969, 11Б, 4.
75. Л и п а т о в Ю. С., С е р е д а О. В., С е р г е е в а Л. М. К е р ч а Ю. Ю.,
Апухтина Н. П., Мозжухина Л. В.—Высокомолек. соед., 1969, ПА,
4, 682.
76. Л ип атов Ю.С., Ф а б у л я к Ф. Г.—Высокомолек. соед., 1969, 11 \, 7С8.
77. Липа Ю. С., П р и в а л к о В. П., ' К е р ч а Ю. Ю., Мюл-
лер Б. Ё. — В кн.: Синтез и физико-химия полимеров (полиуретаны), 6.
«Наукова думка», Киев, 1970, 98.
78. Сергеева Л. М., Липатов Ю. С., Сав ченко Т. Т., Биньке-
вич Н. И., Кузнецова В. П., Бе л о г о лов и н а Г. Н. — В кн.: Син-
тез и физико-химия полимеров (полиуретаны), 6. «Наукова думка», Киев,
1970, 122.
79. Л и п а т о в Ю. С., К у к с и н А. Н., Сергеева Л. М. — Фи-
зико-хими ’еская механ ка материалов, 1969, 5, 6, 688.
270
80. Л о с е в И. П., Д а ц ке в и ч Л. А. — Авт. свид. СССР, 1364499, 14.
03.1961; Бюлл. 5, 1961.
81. М а к л а к о в Л. И., Коваленко В. И., Апухтина Н. П.,
Синайский А. Г. —В кн.: Спектроскопия полимеров. «Наукова
думка», Киев, 1967, 23.
82. Маклаков Л. И., Коваленко В. И, Апухтина Н. П.,
Синайский А. Г. — Ж. прикл. спектр., 1937, 7, 1, 99—107.
83. М а к л е ц о в а Н. В. и др.— Высокомолек. соед., 1965, 7, 70.
84. Маличенко Б. Ф., Шелудько Е. В., Керча Ю. Ю.—Вы-
сокомолек. соед , 1967, 9А, 2482.
85. М а л и ч е н к о Б. Ф., Шелудько Е. В, Керча Ю. Ю., Сав-
ченко Р. Л. — Высокомолек. соед., 1969, НА, 377.
86. Маличенко Б. Ф., Шелудько Е. В., Керча Ю. Ю., Сав-
ченко Р Л. — Высокомолек. соед., 1969, НА, 1513.
87. М а л и ч е н к о Б. Ф., Комарова Н. А., Керча Ю. Ю., Сав-
ченко Р. Л.—Высокомолек. соед., 1969, НА
88 Мандел ькер н Л. Кристаллизация полимеров. «Химия», М., 19 6.
89. М а с л ю к А. Ф., Магдилец В. В., Р у д ь ко А. Р., Щ е потки-
на Н. И., Спирин Ю. Л. — В кн Синтез и физико-химия полимеров
(полиуретаны), 6. «Наукова думка», Киев, 1970, 57.
90. Матюшова В. Г., Омельченко С И. — В кн.* Синтез и физи-
ко-химия полимеров (полиуретаны), 6. «Наукова думка», Киев, 1970.
91. Москвитин Н. И. Физико-химические основы процессов склеивания
и прилипания. «Лесная промышленность», М., 1964, 52.
92. Му л л и н з Л. — Кауч, и рез , 1968, 7, 10.
93. 14 ю л л е р Б. Е., Апухтина Н. П., К л о б а н с к и й А. П. —
Высокомолек. соед., 1964, 6, 2, 329.
94. Мю л л е р Б. Е., Апухтина Н. П., К л е б а н с к и й А. П. —
Высокомолек. соед., 1964, 6, 1330.
95. Нестеров А. Е , Керча Ю Ю., Липатов Ю. С., Су ш -
к о Л. М., Рябоконь Л. И.— Высокомолек. соед., 1967, 9А, 11, 2459.
96. Н е с т е р о в А. Е., Л и и а т о в Ю. С., Пилюгина Н. А. — Вы-
сокомолек. соед., 1967, 9Б, 9, 695.
97. Н е с т е р о в А. Е., Л и п а т о в Ю. С — В» сокомолек. соед., 1968,
10Б, 277.
98. Н е с т е р о в А. Е, Липатов Ю С , Мюллер Б. Е., Моз
ж у х и н а Л. В. — Высокомолек. соед , 1968, 10Б, 900
99. Новейшие методы исследования полимеров. «Мир», М , 1966, 286.
100. Омельченко С. И, Безменов А. Я., Кадурина Т. И.—
В кн.. Синтез и физико-химия полимеров. «Наукова думка», Киев, 1967, 57.
101. Омельченко С. И., Матюшова В. Г. — Лакокрасочные ма-
териалы и их применение, 1969, 1, 35.
102. Омельченко С. И, Кадурина Т. И — Лакокрасочные ма-
териалы и их применение, 1970
103. Омельченко С. И , Логвинова О. М. — В кн. Синтез и фи-
шко-химия полимеров (полиуретаны), 6. «Наукова думка», Киев, 1970.
104. Омельченко С. И., Кадурина Т. И — В кн.: Синтез и физи-
ко-химия полимеров (полиуретаны), 6. «Наукова думка», Киев, 1970.
105 Павлов В. И., А с к а д с к и й А. А., Слонимский Г. Л. —
Механика полимеров, 1965, 6, 15.
106. Пасечник Ю. В., Кузьмина В. А., Егоров Ю. П. —
В кн. - Синтез и физико-химия полимеров (полиуретаны), 6. «а кова думка»,
Киев, 1970, 112.
107. Плата Н. А., А з о р и М, Матюхи на О. С., Каргин В. Л —
Высокомолек соед., 1966, 8, 1727.
108. Платэ Н А., АзориМ., Рудковская Г. Д., Соколова?. А.,
Каргин В. А — Высокомолек. соед , 1966, 8, 759.
109 Привал ко В. П., Липатов Ю. С, Керча Ю. Ю. — Высо-
комолек. соед., 1969, НА, 1, 237.
271
110. П р и в а л к о В. П., Л i п атов Ю. С., К е р ч а Ю. Ю. — ДАН
УРСР. Сер. Б, 1969, 3, 255.
111. Ребиндер П. А. — Сборник трудов ин-та физической химии АН СССР,
Изд-во АН СССР, М., 1950, 1, 5.
112. Р е б и н д е р П А., ЛА и х а й л о в Н. В. — Коллоид, ж., 1855, 18,
2, 107.
113. Рейх В. Н., Файнберг Б. А. Методы технического контроля
качества синтетических каучуков и латексов. Госхимиздат, М., 1951.
114. Саундерс Д. X., Фриш К. К. Химия полиуретанов. «Химия»,
М., 1968.
115. Сергеева Л. М., Липатов Ю. С., Б и н ь к е в и ч Н. И.— Вкн.:
Синтез и физико-химия полиуретанов. «Наукова думка», Киев, 1967, 131.
116. Сергеева Л. М., Липатов Ю. С., Бинькевич Н. И. —
Высокомолек. соед., 1967, 9А, 12, 2672—2675.
117. Сергеева Л. М., Липатов Ю. С., Середа О. В. — В кн.:
Синтез и физико-химия полимеров (полиуретаны), 7. «Наукова думка», Ки-
ев, 1970.
118. Синявский В. Г., Л и п а т о в Ю. С., С у ш к о Л. М.— Высокомолек.
ссед., 1969, БХ1, 249.
119. С м е т а н к и н а Н. П., А н г е л о в а А. В., Люк ас С. Д.—
В кн.* Синтез и физико-химия полиуретанов. «Наукова думка», Киев, 1967, 49.
120. С м е т а н к и п а Н. П., Омельченко С. И., А н г е л о в а А. В. —
Лакокрасочные материалы и их применение, 1969, 1, 35.
121. С м е т а н к и и а Н. П., О п р я В. Я., Омельченко С. И.,
Щепеткина Н. И. — В кн.: Синтез и физико-химия полимеров, 6.
«Наукова думка», Киев, 1970, 168.
122. С м е т а н к и н а Н. П., О п р я В. Я., О м е л ь ч е н к о С. И., Кри-
вченко Г. Н. — Вкн.: Синтез и физико-химия полимеров, 6. «Наукова
думка», Киев, 1970, 175.
123. СметанкинаН. П., ОпряВ. Я., Омельченко С. И. —
В кн.: Синтез и физико-химия полимеров (полиуретаны), 7. «Наукова дум-
ка», Киев£ 1970.
124. С л о н и м с к и й Г. Л., Роговина Л. 3. —Высокомолек. соед.,
1964, 6, 314.
125. Слонимский Г. Л. и др — Высокомолек. соед., 1966, 8, 1312.
126. С о р о к и и М. Ф. и др. — Лакокрасочные материалы и их применение,
1964, 4, 1—4, 1964, 5, 1—5.
127. С п и р и н Ю. Л., Л и п а т о в Ю. С. и др. — Высокомолек. соед.,
1968, ЮА, 2, 263.
128. С п и р и н Ю. Л., Липатов Ю. С. и др. — Высокомолек. соед.,
1968, ЮА, 9, 2116.
129. Справочник химика, 1, «Химия», М.— Л., 1966.
130. Т а г е р А. А., К а р а с ь Л. Я. — Высокомолек. соед , 1965, 7, 5, 891.
131. Таге р А. А. и др. — ДАН СССР, 1965, 165, 5, 1122.
132. Тагер А. А. Физико-химия полимеров. «Высшая школа», М, 1968.
133. Тараканов О. Г., Демина А. И., Васильев Б. В. —
Пластмассы, 1962, 1, 41
134. Тейтельбаум Б. Я. и др. — Высокомолек. соед , 1967, 9А, 1672.
135. Тобольский А. Свойства и структура полимеров. «Химия», М.,
1964, 148
136. Трапезников А А. Труды конференции по коллоидной химии. Изд-во
АН СССР, Киев, 1952.
137. Трелоар Л. Физика упругости каучука. ИЛ, М., 1952, 52.
138. У р азовский С. С. — Высокомолек. соед ,' 1962, 4, 481
139. Уразовский С. С., Ежик И. И. — Укр. хим. ж., 1962, 28,329.
140. Файнерман А. Е., Липатов Ю. С., М а й стр у к В. К. —
ДАН СССР, 1969, 188, 152.
141. Ф а й н е р м а н А. Е., Майструк В. К-> Липатов Ю. С. —
ДАН СССР, 1968, 178, 5, 1129.
272
142. Файнерман А. Е., Липатов Ю. С., М а й с т р у к В. К. —
Коллоид, ж., 1970, 32.
143. Френкель А. И. Кинетическая теория жидкостей. Изд-во АН СССР,
М., 1959.
144. Френкель С. Я., Б а р а н о в В. Г., П а н о в Ю. И. — Высо
комолек. соед., 1964, 6, 1912.
145. Френкель С. Я. Введение в статистическую теорию полимеризации
«Наука», М., 1965.
146. X а й л е н к о Л. В., Л и п а т о в Ю. С., — Высокомолек. соед., 1969,
AXI, 1202.
147. Хувинк Р., Ставерман Р. Химия и технология полимеров, 1
«Химия», М., 1965.
148. Цветков В. Н., Эскин В. Е , Френкель С. Я* Структура
макромолекул в растворах. «Наука», М., 1964.
149. Цветков В. Н., Гармонов Т. И., Станкевич Р. П. —
Высокомолек. соед., 1966, 8, 890.
150 Эрлих И. М., Апухтина Н. П., Раппопорт Л Я. —В кн..
Промышленность синтетическою каучука, 3, «Химия», М., 1967, 53.
151. Эрлих И. М. и др —Изв высш уч. зав Сер. физ., 1967, 4, 113—116.
152. Эскин В. Е., Сердюк И. Н — Высокомолек. соед., 1966, 8, 1316.
153. Эскин В. Е. — Усн. физ н., 1964, 82, 649.
154. Якубович Д. С., С а п ж а р о в с к и й А. Т., 3 у б о в П. И. —
Лакокрасочные материалы и их применение, 1963, 5, 10.
155. Я н г о л ь Г. А., Корнев К. А. — В кн.. Синтез и физико-химия
полимеров. «Наукова думка», Киев, 1964, 30.
156. Abu-Isa J., Crawford V., Holy A., Dole М.— J. Polymer
Sci., 1959, 34, 721
157. Adamson A. Physical Chemistry of Surfaces, Intersc. Publ. N.Y , 1960,107.
158 AndreasJ.,HanserE .Tucker W.—J. Phys. Chem , 1938, 42, 1001.
159. Angelo R., Ikeda R, Wallach M.— Polymer, 1965, 6, 3, 141.
160 A p u c h t i n a N. P , S i n a j s k i A. G.—Plaste u. Kautschuk, 1966, 14,
4, 212.
161. Arlie J,Spegt P., Skoul ios A.— Compt. rend., 1966, 99, 160.
162. Arenz N., Ferguson C, Williams L.— Exptl. Meeh., 1967,
7, 4, 183.
163 Athey R.— Rubb. Age, 1959, 85, 1, 77.
164 Athey R — Ind. Eng Chem., 1960, 52 , 611.
165 Avrami M — J Chem. Phys., 1939, 7, 1103, 1941, 9, 177.
166 A x e 1 г о о d S. L., II a m i 1 t о n C., F r i s c h K.— Ind. Eng. Chem ,
1961 53 899
167. Axelrood S. L., Frisch K.— Rubb. Age, 1960, 88, 465.
168 В a i j a 1 B.— J. Appl. Pol. Sci , 1968, 12, 1, 169.
169. В a i 1 у M. E.-~ Ind. Eng. Chem., 1956, 48, 754.
170 Bayer O.— Angew. Chem., 1947, A59, 257.
171. Bayer O.— Farbe u. Lack, 1958, 5, 237.
172 В e a c h e 1 1 H., Peterson J.— J. Amer. Chem. Soc. Polymer Preprints,
1967, 8, 1, 456.
173. В о n a r t R.— J. Macromol. Sci. Phys., 1968, B21, 115.
174. Borchert W — Angew. Chem., 1951, 63, 31.
175 Boyer R., M u 1 1 e r E.— Angew. Chem., 1950, 62, 57.
176. Bolin R., S z a b a t 1., Cote R.— J. Chem. a. Eng. Data, 1959, 4, 261.
177. Brill R. Z.— J. Phys Chem., 1939, 53, 61.
178. Brandt W. W., L у m a n D. E., E a s о S.— Koll. Z. u. Z. Polymere,
1964, 199, 2, 104.
179. Bruns W., Mehdorn F.— Koll. Z. u. Z. Polymere, 1968, 224, 1, 17.
180 Bui st J., Smith W.—British pat., 853384 , 9. 11 1960.
181. Bullok A., North A,Shortall J.— Europ. Polymer J., 1964,
4, 5, 587.
182. Bunn C. W.—J. Polymer Sci., 1955, 16, 323.
273
183, Cesari M.— Macromol, Chem., 1965, 83, 196.
184. C i f e r r i A.— Trans. Farad. Soc., 1961, 57, 846.
185, Cluff E., Gladding E., Pariser R.— J. Polymer Sci., 1960,
45, 341.
186 Cluff E., G 1 a d d i n g E., Rogan J.— J. Appl. Pol. Sci., 1961, 5,
80.
187. Cluff E.,Gradding E.— J. Appl. Pol. Sci., 1960, 3, 290.
188. Colo dny P., Tobol sky A,— J. Amer. Chem. Soc., 1957, 79,
4320.
189. Cooper S., T о b о 1 s к у A.— J. Appl. Pol. Sci., 1966, 10, 12, 1837.
190. Cooper S. L., T о b о 1 s к у A.— Rubb. Chem. and Techn., 1967, 40, 4,
1105.
191. Conway В , Tong S.— J. Polymer Sci., I960, 46, 113.
192. Conway B.— J Polymer Sci., 1960, 46, 129.
193. Cooper S,Tobolsky A.— Amer. Chem. Soc. Polymer Preprints,
1967 8 1 52
194. С г о с о С a r 1 t о n.— Pat USA, 2901467, 25.8.1959.
195. Cu ba j a m a K.— Chem. High Polymer (Japan), 1964, 21, 236, 737.
196. Cusano C., Dunigan E., Weiss P.™ J. Polymer Sci., 1963, 4,
743.
197. Deane J., Barker R.— Polymer Letter, 1964, 2, 343.
198. Dannusis A., Ashe W., Frisch K.— J. Appl. Pol. Sci., 1965, 9,
9 2965.
199. Dannusis A, McClellan J., Frisch K.— Ind. Eng. Chem.,
1959, 51, 11, 1387.
200. Davies J.— Biochim. et biophys. acta, 1953, 11, 2, 165.
201. D ass ar gues G — Bull. mens. Centre beige etude et docum., 1960, 109, 9.
202. Dunleavy R.Critchfield F — Rubb. World, 1967, 156, 3, 53.
203. Duzek K.— Collect Czechosl Chem. Communs., 1962, 27, 12, 2841.
204. Ebneth —Plastverarbeiter, 1964, 15, 1, 37; 2, 44.
205. Elias H.— Macromol. Chem , 1966, 99, 291.
206. Eppe R., Fisher E., Stuart H.— J Polymer Sci., 1959, 34, 721.
207. Erath E., Spurr R — J Polymer Sci., 1959, 35, 391.
208. Ferrari R — Rubb. Age, 1967, 99, 2,53
209 Ferroni E, Gabrielli G.~ J. Polymer Sci., 1964, B2, 173.
210. Flory P.— J. Chem. Phys., 1949, 17, 225.
211. Flory P. Principles of Polymer Chemistry N. Y., 1953, 476.
212. Flory P.— J. Amer. Chem. Soc., 1956, 78, 5222.
213. Flory P., Hoeve C.,Ciferri A.— J Polymer Sci , 1959, 34, 337.
214. Flory P., Hoeve C,Ciferri A.— J. Polymer Sci., 1960, 45, 235.
215. Flory P. I.— Trans. Farad. Soc., 1961, 7, 829.
216. Flory P. I., W i 1 1 i a m s A. D.— J. Polymer Sci., 1967, A2, 5, 3, 399.
217. Fox T., Loshaek S.— J. Polymer Sci., 1955, 15, 371.
218. Fox T.— Bull. Amer. Phys. Soc., 1956, 1, 123.
219. Frenkel S. Y.— J. Polymer Sci, 1967, C16, 1655.
220. Frifan D.,Ter enzi J.— J. Polymer Sci., 1958, 28, 117, 443
221. Fuller C., Erikson C.— J. Amer. Chem. Soc., 1937, 59, 314.
222. Glavis F.— Polymer Sci., 1959, 36, 130.
223. Gosnell R., Susman S., Smith M.— Adhesive Age, 1962, 5, 9, 32.
224. G u a n t A.— SPE Journal, 1959, 15, 298.
225. Hearst J., St о kma у er W.— J. Chem. Phys., 1962, 37, 1425.
226. Heidemann J., Jellinek G., Ringens W.— Koi. Z. u. Z.
Polymere, 1967, 221, 119.
227. H e i к e n s D., M e i j e r s A., P e t h P.— Polymer, 1968, 9, 1, 15.
228. Heiss H.—Rubb. Age, 1960, 88, 89.
229. Heiss Н.» Saunders J.— Ind. Eng. Chem., 1954, 46, 1438.
230. Hirai N., Eyr i ng N — J. Appl Pol. Sci., 1958, 29, 810.
231. Hirai N., Eyring H.— J. Polymer Sci., 1959, 37, 51.
232. Holub J.— Bor - es cipotechn., 1960, 10, 3, 70.
274
233. Huggins M. L.—J. Phys. Chem., 1942, 46, 151,
234. I a h i a d a I., Ito M.— J. Polymer Sci., 1956, 3, 87.
235. I c h i d a I.— Koll. Z., 1964, 49, 200.
236. I m a d a K.— Makromol. Chem., 1965, 83, 113.
237. Ivin K., Ende H.— J. Polymer Sci., 1961, 54, 17.
238. Jacobs H., Jenkel E.~ Macromol. Chem., 1961, 43, 132.
239. Joly M.— Disc. Farad. Soc., 1958, 25, 150
240 Kato Y.,Nakamura S.~~ Preprint International Simposium on Macro-
molec. Chemistry. Tokyo — Kyoto, 1966, 46.
241. Keene n V.— Pat. USA, 3007831, 7.11.1961.
242. Kern W., Davidovits J., Rauterkus K — Macromol. Chem.,
1961, 43, 106.
243. Kilian H., Y e n к e 1 E.— Z. Elektrochem., 1959, 63, 951.
244. Kilian H., Jenkel E.— Koll. Z., 1959, 165, 25.
245. Kilian H.— Koll. Z., 1961, 176, 49.
246. Kilian H., В о u e к e K.— J. Polymer Sci., 1962, 58, 311.
247. Kirkwood W., Riseman J.— J. Chem. Phys., 1948, 16, 365.
248. К 1 a u к e E., В a у e r O.— Pat. DBR, 1074179, 21.7.1960.
249. К о g о n J.— J. Org. Chem., 1959, 24, 83, 438
250. Kohlrausch F.— Pogg. Ann., 1863, 119, 337.
251. К о i d e T., К u b о t a T.— J. Soc. Rubb. Ind Japan, 1951, 24, 247.
252. Krevelen D., Hoftyser P.— J. Appl. Pol. Sci., 1967, 11, 1409.
253. Kuessner K.— Plaste u Kautschuk, 1960, 7, 8, 389.
254. Kuhn W.— Koll. Z., 1936, 76, 258.
255. Kuhn L.-~ J. Amer. Chem. Soc., 1952, 74, 2492.
256. Kuhn L., Wires R.— J. Amer. Chem., Soc., 1964, 86, 2181.
257. К u r a t a M., Yamakawa H.— J. Chem. Phys., 1958, 29, 311.
258. К we у T.—J. Polymer Sci., 1963, Al, 9, 2977.
259. Kyoichi S., Mi nekasu К — J. Polymer Sci., 1966, 4, 83
260. Little J., Gregg R.— Rubb. Chem. and Techn., 1966, 39, 4, 1089
261. Lowe A., W о о d J.— British pat., 885084, 20 12. 1961.
262. Lyman D.— Rev. in Macromol. Chem., 1966, 1, 1, 191.
263. Lyman D., Heller J., Barlow N.—Macromol. Chem., 1965, 84,
64.
264. Mason P.~ J. Chem. Phys., 1961, 35, 1523.
265. Mark J., Flory P.— J. Amer. Chem. Soc., 1964, 86, 138.
266. Martin H., M u 1 1 e r E.— Koll. Z., 1960, 42, 991.
267. Marvel C.,Aldrich P.— J. Amer. Chem Soc., 1950, 72, 1978.
268 Merten R., Braun G , L a u r er D.— Kunststoffe, 1965, 55, 4, 249.
269. Merten R., Laurer D., Braun G.—Macromol. Chem., 1967, 101,
337.
270. M i t c h e 11 D — British Plastics, 1967, 40, 5, 105
271.
272.
273.
274.
275.
276.
277.
278.
M о a c a n i n J.— J. Appl. Pol. Sci., 1959, 1, 272.
Morawetz H., Zi mm er i ng P — J. Phys Chem., 1954, 58, 753.
M о r r e y. The properties of glass. Reinhold Publ. Corp , N. Y., 1954.
Muller E., Bayer O.—- Angew. Chem., 1952, 64, 523.
Muller F., Engetter A.— Koll. Z., 1957, 152, 15.
Muller F., M a r t i n H.— Koll. Z., 1960, 42, 991.
Muller H.— Fertigungtechn. u. Betrieb, 1961, 11, 1, 45.
Muller H.— Plaste u. Kautschuk, 1962, 9, 335
279. Murphy E., Neil O.— SPE Journal, 1062, 18, 191.
280. Nagasawa A., Kitano H.— Mem. Fac. Eng. Kyoto Univ., 1963,25, 1.
281. О e г t e 1 H.~ Melliand Textilberichte, 1965, 1, 51.
282. Offenbach J.,Tobolsky A.— J. Coll. Sci , 1956, 11, 39.
283. Okato H.—Macromol. Chem., 1966, 98, 148.
284. Opschoor A., Prins W.— J. Polymer Sci., 1967, C16, 1095.
285. Pat. USA, 228496, 1942.
286. Pat. Germany, 728981, 1937.
287. Pigott K.,Frye B.— J. Chem. Eng Data, 1960, 5, 39L
275
288. Paulin de Meyni s.~ Rev. aluminium, 1960, 37, 274.
289. P u e t t D — J. Polymer Sci., 1967, A2, 5, 839.
290. Rausch R., S a у i g h A.— Ing. Fng. Chem., 1965, 57, 6, 92.
291. R eegen J.— Adhesive Age, 1963, 6, 30.
292. Rec gen J.-—J. Appl. Pol. Sci., 1965, 9, 11, 279.
293. R ее gen J.—J. Appl. Pol. Sci., 1966, 10, 9, 1247.
294. Reilly C., Orchin M.— Paper Am. Chem. Soc. Meeting, Athlantic
City, Sept., 1956.
295. Reiss С., В e n о i t N.— Compt. rend., 1961, 253, 268.
296. R i d e a 1 E.— J. Polymer Sci., 1955, 16, 531.
297. Rinke H.—Chimia, 1962, 16, 4, 93.
298. Rinke H.— Chimia, 1968, 22, 164.
299. R о j c i к G.— Kozarstvi, 1958, 8, 176; Chem. Abs., 1960, 54, 945.
300. Rohlender J., Stuart H.— Macromok Chem., 1960, 41, 110.
301. Rossi C., Bianchi U.— Materie plast et elastomeri, 1964, 30, 386.
302. Sandridge R., Morecrof t A.— J. Chem. a. Eng. Data, i960, 5,
495.
303. Sato Hirosh i.— Bull. Chem. Soc. Japan, 1966, 39, 11, 2335.
304. Sato Hiroshi — Bull. Chem. Soc. Japan, 1966, 39, 11, 2340.
305. Saunders J. H.—- Rubb. Chem. and Techn., 1960, 33, 1259.
306. Saunders J., Steingiser S.— Ind. Eng. Chem. Eng. Ser., 1958,
3, 153.
307. Saunders J.—Journal I. R. I., 1968, 2, 1, 21.
308. Schwaner K.— Plaste u. Kautschuk, 1960, 72, 59.
309. Schulz H.— Sprechsaal, 1952, 85, 581; Chem. Abs., 1953, 47, 2531.
310. Scholtan L., Lie Sie Ying.—Macromol. Chem., 1967, 108, 104.
311. Schneider M., Dussablon L„ Shele E.—Amer. Chem. Soc.
Polymer Preprints, 1968, 9, 2, 1481.
312. Seely R.—J. Appl. Pol. Sci., 1965, 9, 9, 3049.
313. Simha R., Boyer R.— J. Chem. Phys., 1962, 37, 1003.
314. Singer S.— J. Chem. Phys. 1948, 16, 872.
315. Sievers R.— Pat. USA, 2983693, 16.11.1960.
316. Simon E.— Pat. USA, 2929794, 22.03.1960.
317. Smith T.,M agussor A.— J. Polymer Sci., 1960, 42, 391.
318. Smith T., M a g u s s о a A.— J. Appl. Pol. Sci., 1961, 5, 218.
319. Spurr R., Erath L.— Ind. Eng. Chem., 1957, 49, 1839.
320. Speicher W.— J. Soc. Leather Trades Chem., 1961, 1, 48.
321. Spencer D.—Pat. Australian, 231132, 16.11.1960.
322. Stanton J., Roholt D.— Paint Ind. Mag., 1961, 76, 7, 7.
323. Stockmayer W., Fixman M.— J. Polymer Sci., 1963, Cl, 137.
324. Sumi M., C h о к к i J.— Macromol. Chem., 1964, 78, 147.
325. Tanaka T.— Rubb. Chem. and Techn., 1962, 35, 4, 970.
326. Tanaka T. Yokoyama T.— Mem. Fac. Eng. Kyushu Univ., 1962,
21, 2, 106.
327. Tanaka T., Yokoyama T., Kajku U.— Mem. Fac., Eng. Kyushi
Univ., 1963, 23, 2, 113.
328. Tanaka T., Yokoyama T.— Kyushi Daigaki Kogaku Shoho, 1964,
37 2 135
329. Tanaka’ T.— J. Polymer Sci., 1968, Al, 8, 2137.
330. Tobolsky A., Johnson V., McKnight W.— J. Phys. Chem.,
1965, 69, 2, 476.
331. Treloar L.— Trans. Farad. Soc., 1943, 39, 36.
332. Trifan D., Terensi J.— J. Polymer Sci., 1958, 28, 443.
333. Ulmanns Encyklopadie der techn. Chemie. Urban, Sehwarzeuberg, Munchen —
Berlin, 1963, 14, 338.
334. V о f s i D., T о b о 1 s к у A.— J. Polymer Sci., 1965, A3, 3261.
335. Weisfeld L., Little Y., Wo Istenho 1 me K.— J. Polymer,
Sci., 1962, 56, 164, 455.
336. Wells E., Hudson G.— Paint Oil a. Chem. Rev., 1959, 122, 20, 8.
276
337. Wetton R., Williams G — Trans. Farad Soc., 1965, 61, 2132.
338. W e 11 о n R., A I 1 e n G.— Polymer, 1966, 7, 331.
339. Wilson K., W e 1 1 s E.— J. Chem. Phys., 1946, 14, 578.
340. W i n dem u t h E„ В r ache I H., F i n к G.—Pat DBR, 1112286,
8 02 1962
341. Willbourn A.— Trans. Farad. Soc , 1958, 54, 717.
342. Wright R. — Rubb. Chem. and Plast. Age, 1966, 47, 10, 1077.
343. Wuderlich B.— J. Phys. Chem., 1960, 64, 1052.
344. Wunderlich B., Bodily D.— J Polymer Sci., 1964, C6, 137.
345. Wurtz A.— C. R Acad. Sci, 1848, 27, 242
346. Y amashit a T.— Bull. Chem. Soc. Japan, 1965, 38, 31, 430.
347. Yokoyama T.—• J. Chem Soc. Japan, Ind. Chem. Sec , 1960, 63, 11, 2050.
348. Yokoyama T.— J. Chem. Soc. Japan, Jnd. Chem. Sec., 1961, 64, 4, 737.
349. Zahn H.—Meli and Textilber., 1951, 32, 534.
350. Zahn H., W i n t e r U.— Koll. Z., 1952, 128, 142.
351. Z a h n H. D о m i n i n M.— Macromol. Chem., 1961, 44—46, 290.
ОГЛАВЛЕНИЕ
Предисловие ........................................................ 3
Глава I. Основы химии полиуретанов ............................. 5
Общие принципы синтеза полиуретанов ..................... 5
Получение полиуретановых каучуков и резин................ 9
Полиуретановые волокна .....................♦........... 12
Полиуретановые покрытия .................................13
Глава II. Термодинамические свойства растворов полиуретанов .... 15
Свойства разбавленных растворов полиуретанов.............15
Сорбционные свойства полиуретанов........................24
Некоторые свойства монослоев олигомеров и полиуретанов . 33
Глава III. Природа химических и физических межмолекулярных связей
в полиуретанах ................................................... 37
Типы химических связей в цепях...........................37
Водородная связь в полиуретанах..........................43
Глава IV. Стеклование и фазовые превращения в линейных и трехмер-
ных полиуретанах ...................................................54
Стеклование аморфных полиуретанов........................54
Фазовые превращения и стеклование линейных алифатических
полиуретанов на основе низкомолекулярных гликолей ... 62
Полиуретан на основе 1,6-гексаметилендиизоцианатаи 1,4-те-
траметиленгликоля (перлон-U)..........................64
Полиуретаны на основе гексаметилендиизоцианата и этилен-,
диэтилен- и триэтиленгликолей ........................72
Сополиуретаны на основе гексаметилендиизоцианата, днэти-
лен- и триэтиленгликоля ..............................83
Особенности кристаллизации и плавления алифатических
фторсодержащих полиуретанов ..........................86
Линейные полиуретаны на основе низкомолекулярных гликолей
и ароматических ди изоцианатов ..........................89
Свойства некоторых полиамидоуретанов.....................92
Фазовые превращения и стеклование линейных и трехмерных
полиуретанов с олигоэфирогликолевыми блоками ............94
Свойства олигоэтиленгликольадипинатов и синтезированных
на их основе линейных и сшитых полиуретанов ..........95
Свойства олигоокситетраметиленгликолей и синтезирован-
ных на их основе линейных и сшитых полиуретанов . . . 105
Сравнительная характеристика процесса стеклования сши-
тых полиуретанов на основе олигоэтиленгликольадипи-
натов и олигоокситетраметиленгликолей ...............114
Полиуретаны на основе других олигоэфирогликолевых бло-
ков .................................................116
Глава V. Структура сетки аморфных полиуретанов.....................121
Характеристика сеток пространственных полиуретанов ... 121
Соотношение между химическими и физическими узлами в сетке
полиуретанов ...........................................129
278
Глава VI. О связи между кинетикой реакции образования, структурой
и некоторыми свойствами полиуретанов ...........................139
Глава VII. Физико-механические свойства полиуретанов.............150
Связь основных физико-механических свойств полиуретанов со
структурой ..........................................150
Влияние природы полиэфира и его молекулярного веса . . 150
Строение диизоцианата .................................. 155
Влияние природы структуры сетки и типов поперечных свя-
зей на свойства полиуретанов ........................156
Температурная зависимость некоторых свойств полиуретанов 168
Термодинамика высокоэластической деформации полиурета-
новых эластомеров ...................................176
Тепловые эффекты при деформации полиуретанов.............192
Релаксационные свойства полиуретанов................... 196
Реологические свойства олигомеров ..................... 216
Адгезия полиуретанов к твердым поверхностям..............227
Свойства и структура эластомерных полиуретановых волокон 248
Заключение ......................................................263
Литература ......................................................
268